
弱毒HCMV及びそのワクチンとしての使用
【課題】ワクチンとして有用な弱毒ヒトサイトメガロウイルスを提供すること。
【解決手段】ヒトサイトメガロウイルス(HCMV)のUL79、87又は95遺伝子オープンリーディングフレーム(ORF)中に突然変異を生じさせることにより、これらの遺伝子の1つ若しくは複数からの産生が低下又は消失している、弱毒HCMV。
【解決手段】ヒトサイトメガロウイルス(HCMV)のUL79、87又は95遺伝子オープンリーディングフレーム(ORF)中に突然変異を生じさせることにより、これらの遺伝子の1つ若しくは複数からの産生が低下又は消失している、弱毒HCMV。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、弱毒ヒトサイトメガロウイルス(HCMV)及びそのワクチンとしての使用に関する。
【背景技術】
【0002】
ヒトサイトメガロウイルス(HCMV)は、ベータヘルペスウイルスファミリーのメンバーである。そのウイルスゲノムは240,000bpを有し、少なくとも150個の予測されるオープンリーディングフレーム(ORF)を有する(9、56)。HCMVによる感染は多くの個体で生じるが、通常は無症候性である。このウイルスは免疫抑制条件下で再活性化し、肺炎、肝炎、腎炎、及び消化器疾患の病原となる。このウイルスは、線維芽細胞、上皮細胞及び内皮細胞などの最終的に分化した細胞、ならびに単球由来マクロファージにおいて、産生的に複製される(11、12、18、27、40、41、47)。
【0003】
産生的複製の間、HCMV遺伝子群は、前初期(IE)、初期及び後期と呼ばれる一時的カスケードで発現される。主要IE遺伝子群(MIE)であるUL123/122(IE1/IE2)は、その後のウイルス遺伝子発現及びウイルス複製の効率において決定的な役割を果たす(19、20、23、32−34)。初期ウイルス遺伝子はウイルスDNA複製に必要なタンパク質をコードする(35)。ウイルスDNA複製に続いて、遅延初期及び後期ウイルス遺伝子が発現され、これらはウイルス粒子のための構造タンパク質をコードする。これに関連して、MIE遺伝子プロモーターのシス作用性エレメント(crs)配列の一部に突然変異を有し、これによってウイルス複製が不能ではないが抑制されている弱毒化組換えヒトサイトメガロウイルスが特許文献1に開示されている。
【0004】
真性後期(true late)(γ−2クラス)ウイルス遺伝子の発現は、ウイルスDNA合成の阻害により妨げられる。UL75(gH)及びUL99(pp28)は、真性後期転写産物によりコードされるHCMV遺伝子の2つの例である(25、31、52)。HCMV前初期遺伝子及び初期遺伝子発現の制御は集中的に研究されてきているが、後期遺伝子発現を制御する特異的メカニズムについてはほとんど知られていない。
【0005】
HCMVでは、IE2タンパク質は感染後初期の微小フォーカス(microfoci)中に見出すことができ、直接的なDNA接触により親ウイルスゲノムと会合している。一部の微小フォーカスは拡大して、感染後の後期に複製コンパートメント(RC)を形成する(42)。しかしながら、HCMVのIE遺伝子群単独では真性後期ウイルス遺伝子の活性化に十分ではない。どのHCMV初期遺伝子が後期遺伝子発現に必要とされるのかは知られていない。
【0006】
マウスガンマヘルペスウイルス68(MHV−68)のORF18、24、30及び34は、後期遺伝子転写に必須である(3、53、54)。これらの不存在が初期ウイルス遺伝子発現又はDNA複製にほとんど影響を及ぼさないことから、これらの遺伝子産物は、後期遺伝子のプロモーターを刺激するために特異的に機能すると考えられる。したがって、後期遺伝子発現は、ウイルスDNA複製のみならず、それらのウイルスによりコードされるトランス作用性因子にも依存している。
【0007】
DNAポリメラーゼ進行因子(DNA polymerase processivity factor)(UL44)及び一本鎖DNA結合タンパク質(SSB)(UL57)をはじめとするHCMVウイルスDNA複製タンパク質群は、1型単純ヘルペスウイルス(HSV−1)RCに似た大型の核構造中に局在する(36、38)。HSV感染細胞では、ウイルスDNA合成を阻害剤ホスホノ酢酸(PAA)で阻害した場合、ウイルスによりコードされるSSB(ICP8)タンパク質を含有する小型のウイルス前複製部位又はフォーカスが観察される(6、29、37)。PAAの不存在下では、BrdUがウイルスDNAに取り込まれて、DNAは子ウイルスと共存して見出される(7、57)。HCMVでは、UL44タンパク質は、感染後初期の段階では、SSB(UL57)タンパク質含有フォーカスと常に共存しているわけではない(36)。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2008−092854号公報
【非特許文献】
【0009】
【非特許文献1】Dunn, W., C. Chou, H. Li, R. Hai, D. Patterson, V. Stolc, H. Zhu, and F. Liu. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. U. S. A. 100:14223-14228. (文献9)
【非特許文献2】Yu, D., M. C. Silva, and T. Shenk. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. U. S. A. 100:12396-12401. (文献56)
【非特許文献3】Fish, K. N., W. Britt, and J. A. Nelson. 1996. A novel mechanism for persistence of human cytomegalovirus in macrophages. J. Virol. 70:1855-1862. (文献11)
【非特許文献4】Fish, K. N., A. S. Depto, A. V. Moses, W. Britt, and J. A. Nelson. 1995. Growth kinetics of human cytomegalovirus are altered in monocyte-derived macrophages. J. Virol. 69:3737-3743. (文献12)
【非特許文献5】Ibanez, C. E., R. Schrier, P. Ghazal, C. Wiley, and J. A. Nelson. 1991. Human cytomegalovirus productively infects primary differentiated macrophages. J. Virol. 65:6581-6588. (文献18)
【非特許文献6】Lathey, J. L., and S. A. Spector. 1991. Unrestricted replication of human cytomegalovirus in hydrocortisone-treated macrophages. J. Virol. 65:6371-6375. (文献27)
【非特許文献7】Sinzger, C., A. Grefte, B. Plachter, A. S. H. Gouw, T. Hauw The, and G. Jahn. 1995. Fibroblasts, epithelial cells, endothelial cells, and smooth muscle cells are the major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J. Gen. Virol. 76:741-750. (文献40)
【非特許文献8】Sinzger, C., B. Plachter, A. Grefte, A. S. H. Gouw, T. H. The, and G. Jahn. 1996. Tissue macrophages are infected by human cytomegalovirus. J. Infect. Dis. 173:240-245. (文献41)
【非特許文献9】Taylor-Wiedeman, J., J. G. Sissons, L. K. Borysiewicz, and J. H. Sinclair. 1991. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 72:2059-2064. (文献47)
【非特許文献10】Isomura, H., and M. F. Stinski. 2003. The human cytomegalovirus major immediate-early enhancer determines the efficiency of immediate-early gene transcription and viral replication in permissive cells at low multiplicity of infection. J. Virol. 77:3602-3614. (文献19)
【非特許文献11】Isomura, H., M. F. Stinski, A. Kudoh, T. Daikoku, N. Shirata, and T. Tsurumi. 2005. Two Sp1/Sp3 binding sites in the major immediate-early proximal enhancer of human cytomegalovirus have a significant role in viral replication. J. Virol. 79:9597-9607. (文献20)
【非特許文献12】Isomura, H., T. Tsurumi, and M. F. Stinski. 2004. Role of the proximal enhancer of the major immediate-early promoter in human cytomegalovirus replication. J. Virol. 78:12788-12799. (文献23)
【非特許文献13】Meier, J. L., M. J. Keller, and J. J. McCoy. 2002. Requirement of multiple cis-acting elements in the human cytomegalovirus major immediate-early distal enhancer for viral gene expression and replication. J. Virol. 76:313-326. (文献32)
【非特許文献14】Meier, J. L., and J. A. Pruessner. 2000. The human cytomegalovirus major immediate-early distal enhancer region is required for efficient viral replication and immediate-early gene expression. J. Virol. 74:1602-1613. (文献33)
【非特許文献15】Meier, J. L., and M. F. Stinski. 1997. Effect of a modulator deletion on transcription of the human cytomegalovirus major immediate-early genes in infected undifferentiated and differentiated cells. J. Virol. 71:1246-1255. (文献34)
【非特許文献16】Pari, G. S., and D. G. Anders. 1993. Eleven loci encoding trans-acting factors are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA replication. J. Virol. 67:6979-6988. (文献35)
【非特許文献17】Kohler, C. P., J. A. Kerry, M. Carter, V. P. Muzithras, T. R. Jones, and R. M. Stenberg. 1994. Use of recombinant virus to assess human cytomegalovirus early and late promoters in the context of the viral genome. J. Virol. 68:6589-6597. (文献25)
【非特許文献18】McWatters, B. J., R. M. Stenberg, and J. A. Kerry. 2002. Characterization of the human cytomegalovirus UL75 (glycoprotein H) late gene promoter. Virology. 303:309-316. (文献31)
【非特許文献19】Winkler, M., S. A. Rice, and T. Stamminger. 1994. UL69 of human cytomegalovirus, an open reading frame with homology to ICP27 of herpes simplex virus, encodes a transactivator of gene expression. J. Virol. 68:3943-3954. (文献52)
【非特許文献20】Sourvinos, G., N. Tavalai, A. Berndt, D. A. Spandidos, and T. Stamminger. 2007. Recruitment of human cytomegalovirus immediate-early 2 protein onto parental viral genomes in association with ND10 in live-infected cells. J. Virol. 81:10123-10136. (文献42)
【非特許文献21】Arumugaswami, V., T. T. Wu, D. Martinez-Guzman, Q. Jia, H. Deng, N. Reyes, and R. Sun. 2006. ORF18 is a transfactor that is essential for late gene transcription of a gammaherpesvirus. J. Virol. 80:9730-9740. (文献3)
【非特許文献22】Wong, E., T. T. Wu, N. Reyes, H. Deng, and R. Sun. 2007. Murine gammaherpesvirus 68 open reading frame 24 is required for late gene expression after DNA replication. J. Virol. 81:6761-6764. (文献53)
【非特許文献23】Wu, T. T., T. Park, H. Kim, T. Tran, L. Tong, D. Martinez-Guzman, N. Reyes, H. Deng, and R. Sun. 2009. ORF30 and ORF34 are essential for expression of late genes in murine gammaherpesvirus 68. J. Virol. 83:2265-2273. (文献54)
【非特許文献24】Penfold, M. E., and E. S. Mocarski. 1997. Formation of cytomegalovirus DNA replication compartments defined by localization of viral proteins and DNA synthesis. Virology 239:46-61. (文献36)
【非特許文献25】Sarisky, R. T., and G. S. Hayward. 1996. Evidence that the UL84 gene product of human cytomegalvorus is essential for promoting oriLyt-dependent DNA replication and formation of replication compartments in cotransfection assays. J. Virol. 70:7398-7413. (文献38)
【非特許文献26】de Bruyn Kops, A., and D. M. Knipe. 1988. Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell 55:857-868. (文献6)
【非特許文献27】Lukonis, C. J., and S. K. Weller. 1996. Characterization of nuclear structures in cells infected with herpes simplex virus type 1 in the absence of viral DNA replication. J. Virol. 70:1751-1758. (文献29)
【非特許文献28】Quinlan, M. P., L. B. Chen, and D. M. Knipe. 1984. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell 36:857-868. (文献37)
【非特許文献29】de Bruyn Kops, A., and D. M. Knipe. 1994. Preexisting nuclear architecture defines the intranuclear location of herpesvirus DNA replication structures. J. Virol. 68:3512-3526. (文献7)
【非特許文献30】Zhong, L., and G. S. Hayward. 1997. Assembly of complete, functionally active herpes simplex virus DNA replication compartments and recruitment of associated viral and cellular proteins in transient cotransfection assays. J. Virol. 71:3146-3160. (文献57)
【発明の概要】
【発明が解決しようとする課題】
【0010】
本明細書において、本発明者らは、HCMVのORFであるUL79、87及び95が、後期ウイルスmRNA及び感染性ウイルス産生に必須であるが、ウイルスDNA複製には必須ではないことを示す。UL87タンパク質とUL95タンパク質はウイルスDNA複製の前にウイルスゲノムと組み立てられて、ウイルスのUL44タンパク質及びUL122(IE2)タンパク質ならびに細胞の転写因子TBPを含む微小フォーカスを形成することが明らかとなった。UL79、87又は95ORFに突然変異を導入することにより、HCMVの弱毒ウイルスを作製することを本発明の目的とする。
【課題を解決するための手段】
【0011】
本発明者らは、ヒトサイトメガロウイルス(HCMV)のUL79、87及び95タンパク質が、感染性ウイルス増殖には必要とされるが、ウイルスDNA複製には必要とされないこと、真性後期ウイルスmRNAの産生に必須であることを見出した。これらの知見に基づき、本発明者らは、HCMVのUL79、87又は95遺伝子ORFを突然変異させた弱毒HCMVを作製し、本発明を完成させた。
【0012】
具体的には、本発明は、以下の特徴を有する。
〔1〕ヒトサイトメガロウイルス(HCMV)のUL79、87又は95遺伝子オープンリーディングフレーム(ORF)中に突然変異を生じさせることにより、これらの遺伝子の1つ若しくは複数からの発現が低下又は消失している、弱毒HCMV。
〔2〕前記突然変異が、ORF中への塩基の挿入によるものである、上記〔1〕に記載の弱毒HCMV。
〔3〕前記弱毒HCMVからの感染性ウイルス増殖は野生型ウイルスと比較して低下しているが、ウイルスDNA複製は野生型ウイルスと同等である、上記〔1〕又は〔2〕に記載の弱毒HCMV。
〔4〕後期ウイルスmRNAの発現が野生型ウイルスと比較して低下している、上記〔1〕〜〔3〕のいずれか1つに記載の弱毒HCMV。
〔5〕上記〔1〕〜〔4〕のいずれか1つに記載の弱毒HCMVを有効成分として含む、HCMV感染の予防又は治療用ワクチン。
〔6〕アジュバントをさらに含む、上記〔5〕に記載のワクチン。
〔7〕上記〔1〕〜〔4〕のいずれか1つに記載の弱毒HCMV又は上記〔5〕若しくは〔6〕に記載のワクチンを非ヒト動物に免疫して抗体を作製するステップ、及び該抗体を回収するステップを含む、抗HCMV抗体の作製方法。
〔8〕前記抗体がヒト抗体である、上記〔7〕に記載の方法。
【発明の効果】
【0013】
本発明によって、ウイルス複製が抑制された弱毒化ヒトサイトメガロウイルス(HCMV)を提供することができ、該弱毒化ウイルスは、ワクチンとして使用することができる。
【図面の簡単な説明】
【0014】
【図1】組み換えHCMV BAC DNAの構造を示す図である。(a)wt(野生型)、RdlUL79+F、RdlUL87+F及びRdlUL95+Fの組み換えBAC DNAを示す模式図である。ORFに突然変異を有する組み換えHCMV BAC DNAを構築する場合、FRT配列をORFの中央部に挿入し、次いでFLP媒介組み換えによりKanRを切り出し、34bpのFRT配列のみを残した。(b)UL95及びUL87ORFに融合したエピトープを含む組み換えBAC DNAの模式図である。UL95ORFと融合したHAエピトープを挿入する場合、逆選択を行なって薬剤耐性カセットを除去した。BACUL95Nneo+St又はBACUL95Cneo+Stの構築のために、RpsL遺伝子(ストレプトマイシンに対する感受性を増大させる)及びネオマイシン耐性マーカー(カナマイシン耐性を付与するため)を含有するマーカーカセットをそれぞれUL95ORFのN末端又はC末端に挿入した。中間体BACクローンはカナマイシン耐性に基づいて単離した。相同組み換えの第2ラウンドでは、マーカーカセット全体をオリゴヌクレオチドを用いたカウンター選択によりHAエピトープを含む配列と置き換えた。UL87ORFのC末端に融合したmycエピトープを含む組み換えBACを構築するために、BACHAUL95及びBACUL95HAの構築後に逆選択を繰り返した。(c)wt(親ウイルス)及び組み換えBACのPCR分析を示す。PCR産物は表2に示されるプライマーを用いて増幅し、3.0%アガロースゲルで電気泳動し、臭化エチジウムで染色し、配列決定した。レーン1、2、5、6及び9:RUL95HAUL87myc;レーン3、4、7、8及び10:RHAUL95UL87myc;レーン11:wt。
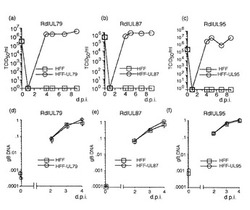
【図2】UL79、87及び95がウイルス増殖に必要とされるが、ウイルスDNA複製には必要とされないことを示す図である。補充タンパク質を発現するか発現しないHFF細胞に3MOIでウイルスを感染させた。ウイルス力価は50%組織培養感染用量(TCID50)アッセイにより決定した。(a〜c)ウイルス増殖曲線を示す。(d〜f)ウイルスDNA複製を示す。ウイルスDNAはgBプライマー及びプローブを用いるリアルタイムPCRにより定量化した。18Sプライマー及びプローブを用いるリアルタイムPCRも、内部対照として用いるために行なった。データは3回の独立した実験の平均である。(a)及び(d):RdlUL79;(b)及び(e):RdlIL87;(c)及び(f):RdlUL95。
【図3a】組み換えウイルス感染後のUL75、99及び44後期mRNAについて示す図である。補充タンパク質を発現するか発現しないHFF細胞に3MOIで突然変異型組み換えウイルスを感染させた。PAAの存在下又は非存在下で細胞質RNAを1、2及び3d.p.i.で回収した。(a〜c)IE1(左パネル)、UL75(gH)(中央パネル)又はUL99(pp28)(右パネル)についてのノザンブロットである。28S及び18SリボソームRNAをRNAロード量の等量の対照として用いた。(a)RdlUL79感染細胞。レーン1、3及び5:HFF細胞;レーン2、4及び6:UL79融合タンパク質を発現するHFF細胞;レーン1及び2:1d.p.i.;レーン3及び4:2d.p.i.;レーン5及び6:3d.p.i.。
【図3b】組み換えウイルス感染後のUL75、99及び44後期mRNAについて示す図である。補充タンパク質を発現するか発現しないHFF細胞に3MOIで突然変異型組み換えウイルスを感染させた。PAAの存在下又は非存在下で細胞質RNAを1、2及び3d.p.i.で回収した。(a〜c)IE1(左パネル)、UL75(gH)(中央パネル)又はUL99(pp28)(右パネル)についてのノザンブロットである。28S及び18SリボソームRNAをRNAロード量の等量の対照として用いた。(b)RdlUL87感染細胞。UL87融合タンパク質を発現するHFF細胞;レーン1及び2:1d.p.i.;レーン3及び4:2d.p.i.;レーン5及び6:3d.p.i.;レーン7及び8:PAAの存在下での2d.p.i.。
【図3c】組み換えウイルス感染後のUL75、99及び44後期mRNAについて示す図である。補充タンパク質を発現するか発現しないHFF細胞に3MOIで突然変異型組み換えウイルスを感染させた。PAAの存在下又は非存在下で細胞質RNAを1、2及び3d.p.i.で回収した。(a〜c)IE1(左パネル)、UL75(gH)(中央パネル)又はUL99(pp28)(右パネル)についてのノザンブロットである。28S及び18SリボソームRNAをRNAロード量の等量の対照として用いた。(c)RdlUL95感染細胞。UL95融合タンパク質を発現するHFF細胞。レーン1及び2:1d.p.i.;レーン3及び4:2d.p.i.;レーン5及び6:PAAの存在下での2d.p.i.;レーン7及び8:3d.p.i.。
【図3d】組み換えウイルス感染後のUL75、99及び44後期mRNAについて示す図である。補充タンパク質を発現するか発現しないHFF細胞に3MOIで突然変異型組み換えウイルスを感染させた。PAAの存在下又は非存在下で細胞質RNAを1、2及び3d.p.i.で回収した。(d)UL44転写産物についてのRNase保護アッセイ。3MOIでのRdlUL79感染後1、2及び3日に細胞質RNAを回収した。20μgのRNAを32P標識アンチセンスUL44プロモータープローブと37℃で一晩ハイブリダイズさせ、続いてRNaseT1で消化した。アンチセンスUL44プローブはすべてのUL44転写産物の転写開始部位の上流配列を含んでいる。保護されたRNA断片を変性6%ポリアクリルアミドゲルで電気泳動した。レーン1:RNaseT1を含まないプローブ;レーン2、4及び6:HFF細胞;レーン3、5及び7:UL79融合タンパク質を発現するHFF細胞;レーン2及び3:1d.p.i.;レーン4及び5:2d.p.i.;レーン6及び7:3d.p.i.。矢印1、2及び3はそれぞれ開始部位1、2又は3から開始する転写産物を示す。
【図4】UL87−myc融合タンパク質及びUL95−HA融合タンパク質が感染後の初期に発現されることを示す図である。ウイルスDNA合成をPAA又はGCVにより阻害した。前初期pIE86及びpIE72(UL122及び123)、HA若しくはmycエピトープ、又はGAPDHに対する抗体を用いて、PAA若しくはGCVの存在下又は非存在下でのRUL95HAUL87mycの感染後の示した時点でウエスタンブロット分析を行なった。GAPDHはロード量対照として用いた。(A)抗HA抗体、(B)抗myc抗体、(C)抗IE72/IE86抗体、抗UL44抗体、及び抗GAPDH抗体。
【図5】UL79、87及び95タンパク質の複製コンパートメントへの動員を示す図である。(A)対応するORFが突然変異した組み換えウイルス感染後のUL79、87又は95−mcherry融合タンパク質とUL44との局在を示す。RdlUL79(a)、RdlUL87(b)又はRdlUL95(c)に感染させた細胞を固定し、処理した。(a)UL79−mcherry及びUL44タンパク質;(b)UL87−mcherry及びUL44タンパク質;(c)UL95−mcherry及びUL44タンパク質。(B)RUL95HAUL87myc感染後のUL95−HA及びUL87−myc融合タンパク質、UL44タンパク質、及びウイルスDNAの局在を示す。RUL95HAUL87mycに感染させた細胞を、示したd.p.i.で回収した。細胞を固定し、抗体を用いて顕微鏡観察のために又はFISH分析のために処理した。(a〜c)UL95−HAタンパク質とUL87−mycタンパク質との共局在;(d)UL95−HAタンパク質とUL44タンパク質との共局在;(e)ウイルスDNAとUL44タンパク質との共局在;(a)1d.p.i.;(b、d及びe)2d.p.i.;(c)3d.p.i.。
【図6−1】PAA若しくはGCVの存在下でのUL95−HA又はUL87−myc融合タンパク質の局在を示す図である。PAA(a、c、e及びg)又はGCV(b、d及びf)の存在下でRUL95HAUL87mycに感染させた細胞を2d.p.i.で処理した。(a及びb)HCMV DNAとUL44タンパク質との共局在;(c及びd)UL95−HAとUL44との共局在;(e及びf)UL87−mycとUL44タンパク質との共局在。
【図6−2】PAA若しくはGCVの存在下でのUL95−HA又はUL87−myc融合タンパク質の局在を示す図である。PAA(a、c、e及びg)又はGCV(b、d及びf)の存在下でRUL95HAUL87mycに感染させた細胞を2d.p.i.で処理した。(g)UL87−mycとUL57タンパク質との共局在。
【図7】ウイルスDNA複製前のUL87とIE2タンパク質及びUL44とTBPタンパク質の局在を示す図である。PAAの非存在下(a及びd)若しくはPAAの存在下(b及びe)又はGCVの存在下(c及びf)で細胞をRUL95HAUL87mycに感染させ、2d.p.i.で回収し、抗体で処理した。(a〜c)UL87−mycとIE2タンパク質との共局在;(d〜f)UL44タンパク質とTBPとの共局在。
【図8】3MOIでのwt、RUL95HAUL87myc及びRHAUL95UL87mycの増殖曲線を示す図である。ウイルス力価は50%組織培養感染用量(TCID50)アッセイにより決定した。
【発明を実施するための形態】
【0015】
前述したように、本発明は、HCMVのORFであるUL79、87及び95が、後期ウイルスmRNA及び感染性ウイルス産生に必須であるが、ウイルスDNA複製には必須ではないとの知見に基づく。具体的には、UL79、87若しくは95 ORFに突然変異を導入することによりこれらのORFの発現を低下又は消失させることによって、ウイルス複製の抑制、ひいてはHCMVの弱毒化を可能にした。ホルマリンなどの化学物質での処理による弱毒化では、外被(エンベロープ)タンパク質の変性などの問題も生じかねないが、本発明の弱毒化ウイルスは天然ウイルスの構造を保持するという利点を有する。
【0016】
<HCMV>
本発明の対象であるヒトサイトメガロウイルス(HCMV)は、すべてのウイルス株を包含するものとする。HCMVは、ヘルペスウイルス科βヘルペスウイルス亜科に属し、その直径が約180nmで約230kbpの塩基数からなる二本鎖DNAウイルスである。HCMVの株には公知の株が含まれ、例えば、AD169株、Towne株、Major株、BT1943株、Davis株、Merlin株、Toledo株、3157株、6397株、3301株、C87株などが知られている(Dolan,A.ら,2004,J.Gen。Virol.85:1301−1312;Chee,M.S.ら,1990,Curr.Top.Microbiol.Immunol.154:125−169)。
【0017】
上記のHCMV株のゲノム配列は、例えばGenBank(NCBI、米国)などに蓄積されたデータから入手可能である。例えば、一般によく知られているAD169株及びTowne株のゲノム配列はそれぞれGenBank登録番号FJ527563、AY315197として登録されている。
【0018】
HCMVは、ヒト線維芽細胞、例えばヒト包皮線維芽(HFF)細胞(ATCC−CC2509)やヒト胎児線維芽細胞、ヒト内皮細胞、例えばヒト毛細血管内皮細胞(HMVEC)(ATCC CC−2527)やヒト網膜色素内皮細胞(RPE)(ATCC C4000−1)などのヒト細胞に感染することができる。したがって、これらの細胞に、HCMV又はそのウイルスベクター、例えばHCMVゲノム配列とBAC(細菌人工染色体)配列から作製されたHCMVBAC、を感染又は導入することによってウイルスDNAを回収することができる(Dunn,W.ら,2003,Proc.Natl.Acad.Sci.USA,100(24):14223−14228)。
【0019】
HCMVのUL79、87及び95ORFは、後期転写のためのウイルストランス作用因子として知られるMHV−68のUL18、24及び34と相同性を有するが、本願以前にはその機能は未知であった。本発明者らは、以下の実施例に示すように、これらのORFが、感染性ウイルス増殖には必要とされるが、ウイルスDNA複製には必要とされないこと、真性後期ウイルスmRNAの産生に必須であることを見出した。
【0020】
例えば、HCMV Towne株(GenBank登録番号:AY315197)のUL79、87及び95ORF配列は、以下の通りである。他のHCMV株のUL79、87及び95ORF配列も、GenBankなどの公知の遺伝子バンク又は文献などから入手可能である。
UL79ORF塩基配列(配列番号1):atgatggcccgcgacgaagagaaccccgccgtcccgcgggtccgcacaggcaaattctcctttacttgcgccaatcatctaatattacagattagcgagaagatgtcgcgcggacagccgctgagctcgctgcgtttggaagaactcaaaatcgtacgcctcatctgcgtcctcctctttcaccgcggtctcgaaacgctgctactgcgcgaaactatgaacaacctgggtgtctcggaccacgccgtgcttagccgcaagacgccgcaaccctactggcctcatctgtaccgcgaactgcgccaggccttcccggggctggactttgaggcggccgtgttcgatgaaacacgcgccgcccgtctcagccagcgcctgtgtcacccgcgcttgagcggcggactgctgacgcgctttgtgcagcgccacaccggcctgccggtcgttttccccgaagacctggcgcgcaacggcaacatcctcttctccctaggcacgctctacggacaccgcctgtttcgtctggcggccttcttcacacgccactggggtgccgaagcgtacgaacccttgattcgcatcatctgtcaaaaaatgtggtacttttatctcatcggcaccggcaagatgcgcattacccccgacgccttcgagatccagcggagtcgacacgaaacgggcatttttacctttattatggaagattacagaacgttcgccggcacgctgtcccggcacccgcaccgtccgcacccacaacagcagcagcaccaccaccccggtcccccccatcctcctctttctcaccctgcctcgtcctgtctcaacccagaggccgtactggccgcccgcgcccttcatatgccgacgctggctaacgacgtgtga
【0021】
UL79ORFアミノ酸配列(配列番号2):MMARDEENPAVPRVRTGKFSFTCANHLILQISEKMSRGQPLSSLRLEELKIVRLICVLLFHRGLETLLLRETMNNLGVSDHAVLSRKTPQPYWPHLYRELRQAFPGLDFEAAVFDETRAARLSQRLCHPRLSGGLLTRFVQRHTGLPVVFPEDLARNGNILFSLGTLYGHRLFRLAAFFTRHWGAEAYEPLIRIICQKMWYFYLIGTGKMRITPDAFEIQRSRHETGIFTFIMEDYRTFAGTLSRHPHRPHPQQQQHHHPGPPHPPLSHPASSCLNPEAVLAARALHMPTLANDV
【0022】
UL87ORF塩基配列(配列番号3):atggccggcgctgcgccgcgccgcctcggctgcgacgctctaatagtcgtcggcggctccgctacgccgcgccgggttttacacgtccccgtgcacgttcgcgcctgcaacctcacccaagagctatcgacgggcgaggacgcccgcttctgtcgtccgcgacccgttaacgtcgaacgggtacgcgctgttttcgcggctctctaccgtgcctgtcccgtacacgcgaggaccgagtccgagcgtgtcaagctggtactgggtcgtctgttgctgggacccgtggccgtaccctgtttttgcgacggtgaagtggagggccacggcgagcatctggtacccacgacgcagttttgtcgcgggccgctgctctacgtgcaccgacgttgttgttgcggatccgtgaccgccgggcgcgcgctgtcctaccacgttctcgaaaaccacgtggccacgcatgtgttacgcggattgctctcgctgacggaatggaatcgagagttgccgggccttttttgcgactgtcctggcagcggtggcgccttgggaaccgaggaacgctacgccatggcctgcttgccgcgcgacctcagcctgcacctggacgactatccttacctgatggtggaaatcggacgcgtactcagcgtcagcgaggtagacgactacgtaaccgccgtctccggctacctgggcgaggccgcggcgccgcgcattcaggttcactacaagctgctctttggactcaacgtgcgtccgcaagcgccgtgcgcgttggacgctacacgcgacttttttctgctggagctgcaaaagctttggctgggcgttgaatatcaccacgaagtcacgtcggagtttttcggtcgcgtactggctcagctgcatcgcgaccgcgcccgcgtcatgatggcacttcgcttgcccgagcagacggtgtgccacctaagcaccttcgttctcagtcgcttcaagcgacaggtactgtacttcaagttacaggtgagctacggcaagtgccggactggccacgctgacagaagtgggggagggggaaacggtggaagtcagggacaccacaacctactgtgttatcgacgtcttagcgtcacgtttgccgacacggacacggtgtggagaaaccttttctacgtttattatgaactagctcgggatctggggtcccatgggacagaggaccgatccgtaagccgcggttacggtgtttcttgcgctccgaggacgtcgcggctaccaccgtcagaaccgacggtggtttcagccaacggacacgcgctgtcttccaccgcgctcccgacgacgagcgcgggtcacaagctgtcgctgccgcgcgacccggccgcagatcgcgttcgacgttacgtgtgcattatctcgcgtctcatgttcgcccggtatggggagagatggcgtaaacaccgtcgacggcggtcggagacgggagaagaggaggaggaagagacggtggaatcgggggagactgacgccacgccgccatttgactttacggggcagcagctgcgccgggcctatcaggaacaccgacgtcgtaaacatctagccgtgcagcgttacgcgccgtgccgtcgtaagctcatcggcgggatggagtttgccgaggtgacgggcgttagtctggaccgcatcgccgtcaacgctttcaacaccaaccgcgttatcaatatgaaggccgcactctcgtccatcgccgcgtcgggtctcggcgtgcgcgcgccgcggcttcccaagaacatgacccacagttttgtgatgtacaagcacaccttcaaggagcccgcttgcaccgtcagcacttttgtttccaacgacgccgtctacatcaactcgctcaacgtcaatattcgcggttcctatcccgagtttctgtactcgctgggcgtgtaccggctgcacgttaatatcgatcacttctttctgccggccgtggtgtgcaacagcaactcctcgctggacgtgcatgggctggaggaccaggcggtgatccgctcggagcgcagcaaggtgtactggaccaccaactttccgtgcatgatctcgcatactaacaacgtcaacgtgggttggttcaaagcggctacggccattgtgccgcgcgtctcgggcgctgacctggaagccattctgctcaaagaactctcgtgcatcaagaacatgcgcgacgtgtgcatcgattacggtctgcatcgcgttttcacgcaactagagctgcgcaattcgtaccagatccccttcctggccaagcagttggtgctgtttctgcgtgcttgcctgctcaagctgcacggtcgagagaagcggctgcagttggaccgcctagtatttgaggcggcacagcggggtctctttgactacagcaagaacctcacggcgcacaccaagatcaagcacacttgcgcgctcatcggcagtcgtctagccaacaacgtgcccaagatcctggcccggaacaaaaaagtcaaattggatcacctgggccggaacgccaacgtgctgacggtgtgtcggcacgtggaagcccacaagatccctcgcacgcgcctcaaagtgttagtcgaggtgctgggcgcgttgcagagtatcagcggtacgccgcacacgcgtgaagtgatccaccagacgttgtttcgattgtgctcggcggccgcagccacctcgggcctgtgttcatcccctcccccattgtgtgtgtcctcatcttcctccgccccttctgtcccaacctccgtcagcgttgacggcagttctgaacccacgtcgccgcgagcgcggtttgcatcacgatga
【0023】
UL87ORFアミノ酸配列(配列番号4):MMEAAAAAAAAFRPEERPTPGWHDAALLMDDGTVREHAFRNGPLSQLIRRVLPPPPDAEDDVVFASELCFYCSGRFNRRSSVFSIYWQKHSDLVYALTGITHCAKLVVECGQLGSGRLRWRDGDVGGEERRGDDDSRDELYDVPGIYMIRVNDGGSTGPRHVIWPGTSVLWAPDVVITTVQRRISAARALVNTFRQYFFLLERRSHEELVLCPPEMEERLAPLLQSATRGDSDMFDGVVASAYHRLRISNIPRSSARLLEHCVGLAGAKKLLLLDVPRLENYFLCQVCLYELDEDEMGEEMLGMLAGKPEDAAVSGASGGFLLHRKTMKLAACLCLLLNSLHLHQEALEALDPPPPRVEENDLVNVVLRRYYRSHGGVQARTLAAARALLADYAETFSPLGSFTRLGYDRLVSADAGVSRRHLVALLRA
【0024】
UL95ORF塩基配列(配列番号5):tgatggcggcggcggtggtgcgagcggaggttaggcggcagcggcgagaggagaggaaaaagatggcggccgcgaggacgacggaggatccacccgaaaaccacgttgttgcggacgtggcttgtgggacgggcgccgtcactcgttcgtcttcgtcgtccctagtggtgtcgtcttcctcggcgtcaggctcggacgaaccttcctccgcctctcctctcagtttccccgtctgctccccctcaactgccgtcaggtctccggggtccgccggggtttcaacgtccctgtgctcggtggaacggatggtcgagctgtcggcgcagtctccggccgccgatttctcggtctccgaggcttggcgcttcgaggaggccgtaaatatggcgctggtggcctgcgaggccgtgtcaccttacgatcgctttcgcctaattgaaacgcccgacgagaatttcttgttggtcaccaacgtaattccgcgcgagtcggccgaggtgccggtgttggatagcagtagcagcggtggcgatagcgggccggaggacaaaaagaaaaacgtcgggaataaaaccgcgggggaaaagaacggcggtgggtctcgggccaaacgccgtcgtagacgacgcgctccgaaaaacgacgccgccacgccgtcttttctacgtcgacacgacgtgctggagcgtttcgcggccgcggctgagcctttgccgtcgctttgtgtgcgtgattatgcgttacgcaatgctgaccgtgttacctacgacggcgaattaatctacggcagttacctgttgtatcgcaaggctcacgtggagctgtcactctccagcaacaaggtgcaacacgtggaagccgtgctgcgacaggtgtacacgccgggcttgttagatcatcacaacgtgtgcgacgtggaggccctgctgtggctgctgtactgcggaccgcgtagcttttgcgcgcgtgacacctgtttcggtcgcgaaaaaaacggttgtcctttccccgcgttgttgcccaaactcttttacgaacccgtgcgggactatatgacctacatgaatctggctgagctgtacgtctttgtttggtatcgcggctacgaattccctgcgccgacgccgcaggcgacgacggcgggtagtggtggcggcggcggggccggcgcttgtgcggtcgagacgagcgcgtcagcaggccgggtcgatgacgccggcgacgaggtgcatttgcctttaaagcccgtctcgctggaccgtctcagagaggtgttgcaggcggtgcgcggccgcttctcggggcgcgaggtgcccgcctggccggcctcgtcgcgcacctgtttgttgtgcgcgctctacagtcagaaccgtctctgtttagatctcgcgcgtgacgaggcgcggaccgtgagttatagccccatcgttatccaagactgcgccgcggctgtcaccgacgtcactttgagccacatcttgcccggccagagcaccgtctcgcttttccccgtctaccacgtcggaaagttgctggacgccctctcgctgaacgacgcgggtctcatcacgttgaatctatga
【0025】
UL95ORFアミノ酸配列(配列番号6):MMAAAVVRAEVRRQRREERKKMAAARTTEDPPENHVVADVACGTGAVTRSSSSSLVVSSSSASGSDEPSSASPLSFPVCSPSTAVRSPGSAGVSTSLCSVERMVELSAQSPAADFSVSEAWRFEEAVNMALVACEAVSPYDRFRLIETPDENFLLVTNVIPRESAEVPVLDSSSSGGDSGPEDKKKNVGNKTAGEKNGGGSRAKRRRRRRAPKNDAATPSFLRRHDVLERFAAAAEPLPSLCVRDYALRNADRVTYDGELIYGSYLLYRKAHVELSLSSNKVQHVEAVLRQVYTPGLLDHHNVCDVEALLWLLYCGPRSFCARDTCFGREKNGCPFPALLPKLFYEPVRDYMTYMNLAELYVFVWYRGYEFPAPTPQATTAGSGGGGGAGACAVETSASAGRVDDAGDEVHLPLKPVSLDRLREVLQAVRGRFSGREVPAWPASSRTCLLCALYSQNRLCLDLARDEARTVSYSPIVIQDCAAAVTDVTLSHILPGQSTVSLFPVYHVGKLLDALSLNDAGLITLNL
【0026】
<弱毒化>
本発明では、HCMVのUL79、87又は95ORFを突然変異させることにより不活性化して、HCMVを弱毒化する。
【0027】
本明細書中で使用される「弱毒化」との用語は、ウイルスの毒性(すなわち、感染性及び病原性)を減弱させることを意味する。
【0028】
ORFの不活性化は、ORFから最終的に翻訳されるタンパク質の発現を阻害すること、又は機能が抑制されたタンパク質を発現させることを意味する。ORFを不活性化させるための突然変異は、例えばORF配列の一部若しくは全部を別のヌクレオチド配列によって置換すること、又はORFの内部での配列の欠失、付加若しくは挿入によって行なうことができる。好ましくは、ORFの内部への別の配列の挿入により、ORFを不活性化する。しかし、ORFの機能を阻害することができれば、突然変異の程度(例えば配列同一性の程度)、突然変異の方法などは限定されないものとする。
【0029】
UL79、87又は95ORFが不活性化されることにより、HCMVの複製が抑制される。本発明により、ORFへの突然変異の導入によって、組み換えHCMVの複製能が約1/10以下、1/50以下、好ましくは1/100以下に抑制される。
【0030】
本発明の組み換えHCMVは、HCMVのUL79、87又は95ORFの一部又は全部に突然変異を導入したゲノムDNAを、適切なベクターを利用して作製し、これを適切な宿主細胞、例えば上記のヒト線維芽細胞やヒト内皮細胞に感染させ、組み換えウイルス粒子を回収することを含む手順によって作製することができる。突然変異は、例えば部位特異的突然変異誘発法、相同組み換え法などの公知の方法によって導入することができる。部位特異的突然変異誘発法は、例えば、野生型HCMVのUL79、87又は95配列の一部又は全部に突然変異を導入し、それに隣接する野生型配列と同一の十分な長さの配列を配置したヌクレオチド配列を合成し、クローニングベクターに導入して大腸菌などの細菌中にサブクローニングすること、野生型HCMVゲノムDNAを含む人工染色体ベクター、例えば細菌人工染色体(BAC、PAC)を作製し、次いで、上記ヌクレオチド配列を人工染色体ベクターにアニーリングし、PCRを行い、目的DNAを増幅することなどを含む。
【0031】
具体的な方法として、例えば大腸菌内でバクテリオファージλ組み換えタンパク質exo、beta及びgamを発現するための迅速相同組み換え系(Ellis,H.M.ら(2001)Proc.Natl.Acad.Sci.USA,98:6742−6746)、HCMVのBAC(細菌人工染色体)DNA(Dunn,W.ら(2003)Proc.Natl.Acad.Sci.USA,100:14223−14228;Lee,E−C.ら(2001)Genomics,73:56−65;Borst,E−M.ら(1999)J.Virol.,73(10):8320−8329)などを使用して突然変異誘発を行なうことができる。
【0032】
組み換えのためのHCMVの二本鎖DNAは、カナマイシン耐性遺伝子などの薬剤耐性遺伝子と70bp程度の相同ウイルスDNA配列を含み、この耐性遺伝子には、34bpの最小FRT部位を連結させることができる(後述の実施例1参照)。UL79、87又は95ORFの突然変異体を作製するために、適切なプライマーを使用してPCRによる増幅を行なう。PCR条件は、例えば、94℃2分の変性を1サイクル、94℃15秒の変性、55℃30秒のアニーリング及び72℃1分の伸長を30サイクル、並びに72℃5分の伸長を1サイクルとするPCRサイクルプログラムからなる。PCR増幅産物をフェノール−クロロホルムで抽出し、95%エタノールで沈殿する。
【0033】
残存する鋳型DNAを除去するため、PCR産物を制限酵素を用いて消化したのち、得られたDNA断片を、HCMV−BAC DNAを含むコンピテント大腸菌へ、エレクトロポレーション、リポフェクション、リン酸カルシウム法などの手法、好ましくはエレクトロポレーションを用いて形質転換する。
【0034】
カナマイシン耐性遺伝子を欠失させるために、組み換えHCMV−BAC DNAを大腸菌に形質転換する。また、リコンビナーゼを発現するプラスミドpCP20(Hahn,W.ら(2003)Virology,307:164−177)を、組み換えHCMV−BAC DNAを含む大腸菌(DH10B)に形質転換する。カナマイシン耐性遺伝子を含まないHCMV BAC DNAを、アンピシリン及びクロラムフェニコール含有LBプレート上で選択する。
【0035】
HFF細胞などの細胞を、リン酸カルシウム沈殿法によって、プラスミドpSVpp71の存在中、上記の各組み換えBACでトランスフェクションする。約10日後にウイルスプラークが出現し、100%細胞変性作用(CPE)の7日後に、細胞外液を回収し、約1:10に希釈したものと未希釈のものを調製し、HFF細胞などの細胞に感染させ、組み換えウイルスを回収する。このとき、ウイルスの選択は、ORFに所望の突然変異が導入されていることを配列決定によって確認すること、及びHFF細胞などの細胞に導入又は感染させてウイルス複製が抑制されていることを確認し、また、野生型の表現型と同一又は本質的に同一であることを確認することによって行なうことができる。
【0036】
<ワクチン>
本発明はさらに、弱毒化HCMVを含むことを特徴とする、HCMV感染症を治療又は予防するためのワクチンを提供する。
【0037】
ワクチンは、一般に生ワクチンと不活性化ワクチンに大別される。特に生ワクチンは安価に製造可能であることや接種回数が少なくても高い効果が期待できるなどの利点を有するが、同時に毒性復帰にリスクを有する。本発明の弱毒化HCMVは、顕著にウイルス複製が抑制されたウイルスであるため、安全性の高い生ワクチンとして使用可能であると考えられる。
【0038】
本発明のワクチンは、AIDS患者、臓器移植患者などの免疫抑制患者、胎児、乳幼児、高齢者、重症患者などの抵抗性が低下したヒトに対し、肺炎、肝炎、網膜炎、胃腸炎などを含む広範な疾患を引き起こす可能性のあるHCMV感染の予防及び治療のために使用可能である。HCMVは、血液、尿、唾液などを介して感染するウイルスであり、上記のヒトに感染すると重症化する場合がある。そのような感染を抑制するために、本発明のワクチンは有効である。
【0039】
本発明のワクチンは、適切な賦形剤、特に希釈剤、例えば滅菌水、生理食塩水、リンゲル液、緩衝液などとともに処方することができる。また、ブドウ糖、トレハロース、エタノール、グリセリンなどの添加剤を含有させることができる。さらにまた、免疫応答を増強させるためのアジュバント、例えば水酸化アルミニウム(alum)、ムラミルジペプチド、サポニン類、リポ多糖、リポゾーム、消化性ポリマー(Gupta,R.K.et al.,Vaccine 13:1263−1276(1995))などをその有効量で含有させてもよい。さらにまた、安定化剤を添加することも可能であり、安定化剤としては、ゼラチン、ヒト血清アルブミンなどのタンパク質、ポリソルベートなどの界面活性剤、スクロース、トレハロースなどの糖類、メチオニン、リジンなどのアミノ酸類などが挙げられる。
【0040】
投与方法は、例えば皮下、筋肉内又は皮内注射、鼻腔内などの粘膜投与を含む。鼻腔内投与の場合、噴霧器による投与が望ましい。投与量は、例えば103〜1011PFUウイルス/kg体重、又は1回あたり10μg〜10mgの用量であるが、この範囲に制限されない。ワクチンの投与量は、患者などの被験体の状態、年齢、体重、性別、重症度などを考慮に入れて、医師の裁量で判断されるべきである。被験体は、1回又は2回以上の投薬を受容でき、複数回投与の場合、1週間〜1ヵ月以上の間隔、例えば2、3、5、12、18又は24ヵ月間隔で投与することができる。
【0041】
<抗体の作製>
本発明はさらに、上記弱毒化HCMVを非ヒト動物に免疫して抗体を生成させ、該抗体を回収することを含む、抗HCMV抗体の作製方法を提供する。
【0042】
抗体は、IgA、IgG、IgD、IgE、IgMのいずれのクラスでもよいし、またIgG1、IgG2、IgG3、IgG4、IgA1、IgA2などのいずれのサブクラスでもよい。さらにまた、抗体は、ポリクローナル抗体、モノクローナル抗体、抗体フラグメント、組み換え抗体のいずれでもよい。組み換え抗体には、例えば単鎖Fv抗体(scFv)、単鎖ダイアボディ(scDb)などが含まれる(浅野竜太郎、生化学、77(12):1497−1500(2005))。抗体フラグメントには、例えばFab、F(ab’)2、Fvなどがある。
【0043】
また、抗体には、ヒト抗体、ヒト化抗体、キメラ抗体なども包含され、好ましい抗体はヒト抗体又はヒト化抗体である。
抗体の作製は、慣用の方法で行なうことができる。
【0044】
ポリクローナル抗体は、弱毒化HCMV粒子を動物に免疫し、抗血清、さらにIgG画分を回収することによって作製できる。
【0045】
モノクローナル抗体は、弱毒化HCMV粒子を免疫した動物(例えばマウス、ラットなど)から脾臓を取り出し、脾臓細胞又はリンパ球とミエローマ細胞とを融合させ、該HCMV粒子抗原と特異的に免疫反応する抗体をスクリーニングすることによって作製できる。
【0046】
ヒト抗体は、ヒト抗体産生非ヒト動物(例えばKMマウス、XenoMouse)を用いて作製することができる。あるいは、弱毒化HCMV粒子を免疫した動物のゲノムから抗体遺伝子を回収し、重鎖可変領域及び軽鎖可変領域の各CDR1〜CDR3コード配列を決定し、ヒト抗体遺伝子の本来のCDR1〜CDR3コード配列を前記CDR1〜CDR3コード配列と置換することを含む遺伝子組み換え技術を用いてヒト化抗体を作製することができる。
上記の抗体は、ポリエチレングリコールなどの分子によって化学修飾することもできる。
【0047】
上記のようにして得られた抗体は、HCMV感染症の治療及び予防のために使用することができ、或いはHCMVの検出のために使用することもできる。ここで、HCMVの検出のために、抗原−抗体反応を利用する手法のいずれを使用することもできる。そのような手法には、例えば均質法、固相法(例えばサンドイッチ法)などが含まれる。
【実施例】
【0048】
以下に実施例を挙げて本発明をさらに具体的に説明するが、本発明の範囲は、該実施例によって制限されないものとする。
【0049】
実施例1
突然変異型若しくはエピトープタグ付けされたUL79、87、又は95ORFを有する組み換えウイルスの構築
ウイルスDNA複製及び後期mRNAに対するHCMV UL79、87、又は95の影響を調べるために、UL79、87又は95のORFを中断する突然変異を有する組み換えウイルスを構築した。
【0050】
<材料と方法>
細胞及びウイルス
初代ヒト包皮線維芽(HFF)細胞を、以前に報告されているように(43)、10%ウシ胎児血清(Sigma,St.Louis,Mo)、ペニシリン(100U/mL)及びストレプトマイシン(100μg/mL)を添加したイーグルの最小必須培地中で、37℃、5%CO2で維持した。ウイルスDNA複製を阻害するために、ホスホノ酢酸(PAA,Sigma,St.Louis,MO)(200μg/mL)、又は9−[(1,3−ジヒドロキシ−2−プロピル)メチル]グアニン(ガンシクロビル[GCV],Calbiochem,Darmstad,Germany)(450μM)を、感染時及び感染して維持する間、培地に添加した。
【0051】
HCMV UL79、87又は95ORFを発現するHFF細胞を作製するために、Nolan博士の研究室から入手したプロトコールに従いレトロウイルス系を使用した(http://www.stanford.edu/group/nolan/protocols/pro_helper_dep.html)。UL79及び95ORFは、それぞれ、XhoIUL79ORFF及びHindIIIUL79ORFR、並びにXhoIUL95ORFF及びHindIIIUL95ORFRのプライマーペアを用いて、HCMV TowneのBAC DNAからPCRにより増幅した。PCRプライマーの配列は表1に示してある。PCR産物を制限エンドヌクレアーゼXhoI及びHindIIIで消化し、pLBC(2)(スタンフォード大学Nolan博士の許可を得て、Fred Hunchinson Cancer Research CenterのKiem博士から供与された)にクローニングし、その結果、ORFのC末端にmcherry(京都大学医学系研究科、松田博士より供与された)が融合された。pLBCは、EBVゲノムのEBNA1 ORF及びOriP配列を有する、組み換えレトロウイルスの構築のためのシャトルベクターである。UL97ORF配列はATG開始コドンから815ヌクレオチド(n.t.)に制限エンドヌクレアーゼHindIII部位を含む。HindIII部位はpLBCへのORFのクローニングに必要である。UL87の1〜814n.t.の領域をまずプライマーXhoIUL87ORFF及びUL87HindIII(815)Rを用いるPCRにより増幅し、pLBCの対応する制限エンドヌクレアーゼ部位にクローニングし、DNA配列決定した(愛知県がんセンター中央研究所)。続いて、815〜2826n.t.の領域を、UL87(815)HindIIIF及びHindIIIUL87ORFRのプライマーペアを用いるPCRにより増幅し、UL87の1〜814n.t.の領域を有するpLBCのHindIII部位にクローニングし、DNA配列決定した。各ORFを含有するシャトルベクターをパッケージング細胞株であるPhoenix−GALV(17)(Nolan博士の許可を得て、Kiem博士から供与された)にトランスフェクションすることにより、レトロウイルスストックを調製した。HFF細胞を組み換えレトロウイルスに感染させ、ピューロマイシン選択下でpUL79−mcherry、pUL87−mcherry又はpUL95−mcherry発現細胞集団を作製した。
【0052】
野生型(wt)HCMV Towne、RHAUL95UL87myc、RUL95HAUL87myc、RdlUL79、RdlUL87、又はRdlUL95のウイルス力価は、HFF細胞又はpUL79、87若しくは95−mcherry発現HFF細胞に対する標準的なプラークアッセイにより、以前に記載されているように決定した(34)。ウイルス力価は、段階希釈で感染させた単層HFF細胞でのGFP蛍光フォーカスによっても決定した。感染後の種々の時点で細胞及び上清を回収し、3回の凍結融解サイクルに供した。ウイルス力価を、HFF細胞又はpUL79、87若しくは95−mcherry発現HFF細胞での50%組織培養感染用量(TCID50)アッセイにより決定した。TCID50を算出するために、Reed−Muench法を用いた。野生型ウイルス及び組み換えウイルスは、置き換え可能な10kb US1−US12領域(US:ユニークショート)をGFP遺伝子で置換されている。
【0053】
ウイルスDNA複製
3MOIでの感染後、2、3及び4d.p.i.で回収した。3個の35mmプレート中の細胞を、50μg/mLプロテアーゼKを含有する溶解バッファー(50mM Tris−HCl,pH8,10mM EDTA,1%SDS及び20μg/mL RNaseA)中に懸濁した。複製したウイルスDNAをHCMV gBプライマー及び以前に記載されたプローブ(20、23)を用いるリアルタイムPCRにより測定した。Applied Biosystems(Foster City,CA)から購入した18Sプライマー及びプローブを用いるリアルタイムPCRは、入力DNAの内部対照としても機能した。データは3回の独立した実験の平均である。
【0054】
HCMA BAC DNAの突然変異誘発
バクテリオファージλ組み換えタンパク質exo、beta及びgamを発現する大腸菌(NIHのCourt博士から供与された)での迅速相同組み換えシステムを、以前に記載されているように用いた(10)。HCMV Towne株のBAC DNAはLiu博士(カリフォルニア大学)から供与された(9)。ORFを欠損した組み換えHCMV BAC DNAの構築に際して、ORFの中央にFRT配列を挿入し、隣接する遺伝子への34bp配列の影響を最小限に抑えた。さらに、隣接遺伝子の調節領域が除去される可能性を除外するために、ヌクレオチドを欠失させることはしなかった。BACdlUL79Kan+FRT、BACdlUL87Kan+FRT及びBACdlUL95Kan+FRT(図1a参照)の組み換えHCMV BAC DNAを作製するために、組み換えのための二本鎖DNAを、プラスミドpACYC177(NEB)を鋳型として、BACdlUL79FRTFKanF及びBACdlUL79FRTRKanR、BACdlUL87FRTFKanF及びBACdlUL87FRTRKanR、並びにBACdlUL95FRTFKanF及びBACdlUL95FRTRKanRのプライマーペアをそれぞれ用いるPCRにより増幅した。プライマー配列は表2に示してある。
【0055】
BACUL95N neo+St及びBACUL95 neo+Stを作製するために、プラスミドpRpsL−neo(Gene Bridges,Dresden,Germany)を鋳型として、BACUL95Nneo+StF及びBACUL95Nneo+StR、並びにBACUL95Cneo+StF及びBACUL95Cneo+StRのプライマーペアをそれぞれ用いるPCRにより二本鎖DNAを増幅した。プライマー配列は表2に示してある。プラスミドpACYC177又はpRpsL−neo(Gene Bridges)は、カナマイシン耐性(KanR)又はKanR及びストレプトマイシン耐性遺伝子をそれぞれ含有する。組み換えのために増幅された二本鎖DNAは、最小FRT部位(5’−GAAGTTCCTATTCTCTAGAAAGTATAGGAACTTC−3’)(15)、又はRpsLneo遺伝子(Gene Bridges)に連結されたKanR遺伝子、及び70bpの相同ウイルスDNA配列を含有していた。DpnIでの37℃、1.5時間の消化の後に、PCR産物をゲル精製し、親HCMV BAC DNAを含有するDY−380に形質転換した。相同組み換え後、KanR及びFRT配列、又はRpsL−neo遺伝子を含有する突然変異型BAC DNAはカナマイシンに対して耐性であった(図1を参照)。
【0056】
FRT配列を含む突然変異型HCMV BAC DNAからKanR配列を切り出すために、FRTを介した組み換えを、以前に記載されているように用いた(23)。プラスミドpCP20(Max von Pettenkofer Institute,Munich,GemanyのHuhn博士から供与された)を組み換えHCMV BAC DNAを含むDH10Bに形質転換した。カナマイシンを含まないHCMV BAC DNAを、アンピシリン及びクロラムフェニコールを含有するLBプレート上で選択した。UL79、87又は95ORFの欠失を有する組み換えBAC DNAを選択するために、UL79detectF及びUL79detectR、UL87BACdetectF及びUL87BACdetectR、並びにUL95BACdetectF及びUL95BACdetectRのプライマーペアをそれぞれ用いてPCR分析を行なった。プライマー配列は表1に示してある。PCR産物をDNA配列決定し、組み換えを確認した(愛知県がんセンター中央研究所)。
【0057】
逆選択を、以前に記載されているように行なった(21、50)。RpsLはストレプトマイシン感受性遺伝子を付与するので、突然変異型BAC DNAを、Counter Selection Modification Kit(Gene Bridges)を用いて増大したストレプトマイシン耐性に基づいて選択した。BACHAUL95又はBACUL95HAを構築するために、オリゴヌクレオチドBAColigoHAUL95又はBACUL95Coligoを用いてそれぞれ逆選択した(図1b参照)。オリゴ配列は表2に示してある。
【0058】
組み換えBACHAUL95UL87myc又はBACUL95HAUL87myc(図1b参照)を構築するために、BACHAUL95又はBACUL95HAの構築後に、逆の手順を繰り返した。BACUL87Cneo+StF及びBACUL87Cneo+StRのプライマーペアを用いるPCRにより、二本鎖DNAを増幅し、BACHAUL95又はBACUL95HAを含有するDY−380に形質転換した。プライマー配列は表2に示してある。BACHAUL95neo+St又はBACUL95HAneo+Stを、カナマイシン含有プレート上で選択した。BACUL87oligoのオリゴヌクレオチドを用いる逆選択を行なった後、BACHAUL95UL87myc又はBACUL95HAUL87mycを上記のように増大したストレプトマイシン耐性に基づいて選択した(図1参照)。
【0059】
HA−UL95、UL95−HA及びUL87−mycを含む組み換えBAC DNAを選択するために、UL95NdetectF及びUL95NdetectR、UL95CdetectF及びUL95CdetectR、並びにUL87CdetectF及びUL87CdetectRのプライマーペアをそれぞれ用いてPCR分析を行なった。プライマー配列は表2に示してある。PCR産物は、組み換え及び切り出しを確認するために配列決定した(愛知県がんセンター中央研究所)。
【0060】
抗体
HA又はmcherryでタグ付けした融合タンパク質を検出するために、ラット又はウサギポリクローナル抗体(3F10,Roche又はPM005,MBL,名古屋)をそれぞれ用いた。mycエピトープを含む融合タンパク質を検出するために、マウスモノクローナル抗体9B11、又はウサギポリクローナル抗体7D10(Cell Signaling,Boston,MA)を用いた。IE1及びIE2タンパク質、IE2、UL44、UL57、TBP又は細胞内GAPDHを検出するための抗体は、NEA−9221(Perkin Elmer,Boston,MA)、12E2(Vancouver Biotech,Ltd.,Vancouver,CA)、CA006(EastCoast Bio,North Berwick,ME)、CH167(Santa Cruz, Santa Cruz,CA)、sc−273(Santa Cruz)、及びMAB374(Chemicon,Temucula,CA)をそれぞれ用いた。
【0061】
ウエスタンブロット分析
細胞を感染後の示した時点で回収し、PBSで洗浄し、以前に記載されているように溶解バッファーで処理した(19、20)。20μgのタンパク質のアリコートを、ドデシル硫酸ナトリウム(SDS)−10%ポリアクリルアミドゲル電気泳動(SDS−PAGE)のために各レーンにロードし、PVDFメンブレンにトランスファーした。増強化学発光検出試薬(GE Healthcare UK Ltd.,Buckinghamshire,UK)及び二次西洋ワサビペルオキシダーゼ標識抗マウス、ウサギ、又はラットIgG抗体(Zymed,San Francisco,CA)を製造業者の取扱説明書に従って用いた。
【0062】
【表1】

【0063】
【表2】
【0064】
<結果>
C末端に蛍光タンパク質mcherryをインフレームで融合させたUL79、87又は95を安定的に発現するHFF細胞を、組み換えレトロウイルスを用いて単離した。これらの細胞を、組み換えHCMVを増殖させるために用いた。組み換えBAC DNAを親BAC DNAから選択するために薬剤耐性遺伝子が必要であったので、34bpの最小FRT部位に連結されたKanRを有する組み換えBAC DNAをまず構築し、続いてFRT−FLP組み換えによりKanRを切り出した。図1aを参照されたい。隣接遺伝子への影響を最小限にするために、FRTはORFの中央に挿入した。
【0065】
本発明者らはまた、UL87又はUL95ORFのN末端又はC末端に融合されたHA又はmycエピトープを有する組み換えウイルスも構築した。以前に報告されているようにUL98遺伝子は必須であり(9、56)、UL95ORFはUL89と重複しているので、UL95のN末端へのHAエピトープの挿入はUL89タンパク質の機能に障害を与える可能性がある。したがって、本発明者らは、UL95ORFのN末端又はC末端に融合されたHAエピトープを有する組み換えウイルスを構築した。
【0066】
UL86は必須であり(10、59)、UL87とは逆方向に転写されるので、mycエピトープをUL87ORFのC末端に挿入した。UL87ORFのTGA停止コドンとUL88のATG開始コドンは重複しているので、mycエピトープはUL87ORFのTGA停止コドンの前に挿入した。ORFのN末端又はC末端にエピトープを挿入するために、逆の手順を行なった。図1bに示されているように、マーカーカセットをRpsL遺伝子(Gene Bridges)で置き換え、ストレプトマイシンに対する感受性を増大させた。中間体BACクローンは、カナマイシンに対する耐性に基づいて選択した。これらのクローンの完全性はHindIIIでの消化によりチェックし、正しい位置でのマーカーカセットの挿入はPCRにより確認した。相同組み換えの第2ラウンドでは、マーカーカセット全体をカウンター選択によりエピトープを含む配列と置き換えた。組み換え構築物を、以前に記載されているように(50)ストレプトマイシンに対する増大した耐性に基づいて単離し、エピトープが挿入された組み換えBACをPCRにより選択した。PCR分析により、組み換えBAC DNA中へのエピトープの挿入が確認された(図1c)。DNA配列決定により組み換えを確認し(データは示していない)、突然変異型BACの完全性をHindIIIによる消化によってチェックした(データは示していない)。3の高MOIで、これら組み換えウイルスの増殖を親ウイルス(wt)と比較した。RUL95HAUL87mycは感染4及び5日後(d.p.i.)で野生型と比較して1/2対数ほど遅く複製したが、7d.p.i.までに感染力価は同等となった。RHAUL95UL87mycは、4、5及び7d.p.i.で野生型と同等に複製した(図8)。
【0067】
実施例2
UL79、87及び95はHCMVの増殖には必要であるが、DNA複製には必要でない
HCMV UL79、87又は95タンパク質がウイルス増殖又はDNA複製に必要であるかを決定するために、補充ウイルスタンパク質を発現するか発現しないHFF細胞に組み換えウイルスを3MOIで感染させた。ウイルス力価を1、4、5、7及び9d.p.i.でアッセイし、同数の感染細胞からのDNAを2、3及び4d.p.i.で回収した。突然変異させたUL79、87又は95ORFを含む組み換えウイルスは、補充ウイルスタンパク質の発現なしではHFF細胞で感染ウイルスを産生しなかった(それぞれ、図2a、b及びc)。対照的に、ウイルスDNAは両方のHFF細胞で同等であった(図2d、e及びf)。これらの結果は、UL79、87及び95がウイルス増殖に必要とされるが、ウイルスDNA複製には必要でないことを示している。
【0068】
実施例3
UL79、87及び95タンパク質は真性後期(γ2)ウイルスmRNAに必要とされる
<材料と方法>
ノザンブロット分析
以前に記載されたように、感染後1、2及び3日で細胞質RNAを単離した(5、16)。20μgの細胞質RNAを2.2Mホルムアルデヒドを含有する1%アガロースゲルでの電気泳動に供し、最大強度のHybond N+(GE Healthcare UK Ltd.)にトランスファーした。IE1、UL75又はUL99DNAプローブを用いるノザンブロット分析を以前に記載されているように実施した(20、22、23)。放射標識プローブは32P−dCTPでの標識により以前に記載されているように作製した(23)。
【0069】
RNase保護アッセイ
32P標識アンチセンスUL44 RNAプローブを以前に記載されているように構築した(23)。以前に記載されているように(19、26)、20μgのRNAを32P標識アンチセンスUL44プロモータープローブに37℃で一晩ハイブリダイズさせ、その後、RNaseT1(100U)で消化した。保護されたRNA断片を変性6%ポリアクリルアミドゲルでの電気泳動に供し、続いてHyperfilmMP(GE Healthcare UK Ltd.)でのオートラジオグラフィーに供した。
【0070】
<結果>
UL79、87又は95タンパク質が真性後期(γ2)mRNAに必要とされるかを決定するために、補充ウイルスタンパク質を発現するか発現しないHFF細胞を、PAAの存在下又は非存在下で、組み換えウイルスに3MOIで感染させた。1、2及び3d.p.i.で細胞質RNAを回収し、IE1、UL75(gH)又はUL99(pp28)DNAプローブのいずれかを用いてノザンブロット分析によって分析した。IE1RNAは、予想される通り、組み換えウイルスを感染させた両方の細胞タイプで1d.p.i.に検出された(図3a〜c、左パネル)。
【0071】
UL75プローブを用いると、UL75、76、77及び78mRNAに対応する3種類のサイズのmRNAが検出された(図3a〜c、中央パネル)。これらのウイルスmRNAは以前に報告されているように異なる開始部位を有する(48)。UL75の転写方向はUL76、77及び78とは逆方向である(52)。初期/後期UL76〜78転写産物はPAAの存在下では著明に減少した(図3b及びc)。UL75転写産物は後期転写産物であり、PAAの存在下又は補充タンパク質の発現なしにはHFF細胞で検出されなかった(図3a〜c、中央パネル)。
【0072】
UL99ORFは一連の3’共同末端転写産物内のウイルスゲノムの複合領域に位置する(51)。すべての転写産物はUL99ORFの共通のポリアデニル化部位下流配列を用いる(51)。pp28テグメントタンパク質は1.6kbのmRNAから翻訳される(1)。以前に報告されているように(1)、UL99プローブを用いるノザンブロット分析は、PAAの存在下で、UL99プロモーターからの1.6kbのmRNAが検出されないこと、したがってUL99は真性後期遺伝子であることを示した(図3b及びc、右パネル)。以前の報告と合致して(51)、PAA処理RNAの分析から、この領域内の可能性があるORFのそれぞれの上流から開始されるmRNAが遅延初期転写産物を含むことが示された(図3b及びC、右パネル)。PAAの存在下、又は補充タンパク質の発現なしには、1.6kb mRNAはHFF細胞で検出されなかった(図3a〜c、右パネル)。これらのことから、HCMV UL79、87及び95は真性後期ウイルスmRNAのために必須であると結論づけられる。
【0073】
HCMV UL44転写ユニットは3つの異なる部位で開始され、それぞれは約50塩基により隔てられ、生産性感染時に別々に調節される。これらの開始部位のうち2つ、遠位及び近位が初期に活性であり、中央開始部位は後期まで不活性である(28)。後期開始部位からの発現はウイルスDNA複製に依存し、後期ウイルス遺伝子発現に影響を有する(22)。本発明者らは、UL44転写に対するUL79タンパク質の影響も調べた。異なる開始部位に由来するすべての転写産物を検出するために、RNase保護アッセイを行なった。以前の報告と合致して(21、22、28)、離れた開始部位から開始される3種類の主要な転写産物が補充細胞では検出された(図3d、レーン3、5及び7)。対照的に、中央の開始部位からの転写産物はRdlUL79感染HFF細胞では検出されず、他の2種類の初期転写産物は検出された(図3d、レーン2、4及び6)。これらのことから、UL44の中央の後期開始部位からの転写産物はUL79ウイルスタンパク質を必要とすることが結論づけられる。
【0074】
実施例4
UL87及び95ORFは初期ウイルスタンパク質である
HCMV ORF79、87及び95はMHV−68 ORF18、24及び34と機能の上で似通っているので、これらの発現時期及びHCMV UL44との共局在について調べた。図1bに示されるエピトープタグ付けされたUL87及びUL95ORFを含む組み換えウイルスを構築した。UL79ORFに融合したエピトープを含む組み換えウイルスは構築しなかった。
【0075】
UL87及びUL95融合タンパク質の発現動態を調べるために、PAA若しくはGCVの存在下又は非存在下で、HFF細胞をRUL95HAUL87myc又はRHAUL95UL87mycに3MOIで感染させた。感染細胞を1、2及び3d.p.i.で回収し、IE1若しくはUL44タンパク質に対する抗体、又はHA若しくはmycエピトープに対する抗体を用いるウエスタンブロット分析によりアッセイした。タグ付けされた融合タンパク質は、1〜3d.p.i.で、またPAA若しくはGCVの存在下又は非存在下で2d.p.i.で検出された(図4A及びB)。UL87及びUL95融合タンパク質の発現動態は、UL44遺伝子の初期ウイルスタンパク質と似通っていた(図4C)。これらの結果は、UL87及びUL95が感染の初期に発現されることを示している。
【0076】
実施例5
UL79、87及び95ウイルスタンパク質は複製コンパートメント(RC)に動員される
<材料と方法>
共焦点顕微鏡観察前のタンパク質のin situ抽出
24ウェルプレート中のコンフルエントのHFF細胞に3MOIで組み換えウイルスを感染させた。以前に記載されているように(55)、感染後の種々の時点で、カバーガラス上で増殖した細胞を50μg/mLのジチオビス(スクシンイミジルプロピオネート)(DSP、Thermo,Rockford,IL)(ホモ二官能性NHSエステル架橋剤)で4℃にて10分間固定した。マトリックスに結合していないタンパク質を核又は細胞質から除去するために、細胞をTNEバッファー(10mM Tris−HCl,pH7.8,1%NP40及び0.15M NaCl)で処理し、2%パラホルムアルデヒドで4℃にて60分間固定した。
【0077】
顕微鏡観察
細胞を高塩濃度TPBS(0.001Mリン酸ナトリウム[pH7.3],0.5M NaCl,0.1%Tween20,及び0.1%TritonX−100)で3回洗浄し、示した一次抗体と共に4℃にて一晩インキュベートした。室温でのTPBSでの3回の洗浄後、細胞をAlexaFluor594若しくは680標識二次抗マウスIgG(Molecular Probes)、AlexaFluor594若しくは680標識抗ウサギIgG(Molecular Probes)、又はAlexaFluor594標識ラットIgG抗体(Molecular Probes)と共に37℃で30分間インキュベートした。感染細胞においてHA又はmyc融合タンパク質を検出する場合には、Signal immunostain(TOYOBO,大阪)を用いて特異的シグナルの強度を増強した。スライドを、4’,6’−ジアミジノ−2−フェニルインドール(DAPI)を含むProLong Goldアンチフェード試薬(Molecular Probes)でマウントした。画像取得は、PlainApo63又は100×1.4NA油浸対物レンズを装着したCarl Zeiss Meta共焦点レーザー走査顕微鏡(Carl Zeiss MicroImaging,Inc.)を用いて行なった。LSM510Meta共焦点顕微鏡にはAlexaFluor594及び680蛍光団を励起するためのHeNeレーザーを装着した。細胞をいずれかの抗原に関して染色した場合、まずタンパク質の局在を調べ、次に同じ条件で別のフィルターを用いて免疫蛍光が検出されないことを確認した。
【0078】
<結果>
HCMV複製タンパク質UL44は約2d.p.i.でRCに局在化する。BrdUをこれらのコンパートメント中のウイルスDNAに取り込ませると、子孫ウイルスで検出される(36、38)。本発明者らは、RdlUL79、RdlUL87又はRdlUL95での感染後の細胞内でその局在を調べるために、HFF細胞で安定に発現させたmcherry蛍光融合ウイルスタンパク質を用いた。3MOIでの感染2日後に細胞を固定し、UL44に対する抗体とインキュベートした。組み換えウイルスはGFPタンパク質を含有しているので、AlexaFluor680標識二次抗マウスIgGを用いてUL44の細胞内局在を検出した。図5A、a〜cに示されているように、UL79、87及び95−mcherry融合ウイルスタンパク質はUL44タンパク質と核で共局在していた。これらの結果は、mcherryと融合させた補充タンパク質UL79、87又は95が、それぞれRdlUL79、RdlUL87又はRdlUL95での組み換えウイルス感染後にRCに動員されることを示している。
【0079】
実施例6
組み換えウイルスから発現させたUL87及び95タンパク質は複製コンパートメントに動員される
ウイルスから発現させたUL87及び95タンパク質も互いに共局在することを示すために、HFF細胞を3MOIでRUL95HAUL87mycに感染させた。1〜3d.p.i.において、細胞を固定し、HAエピトープに対するラットポリクローナル抗体またはmycエピトープに対するマウスモノクローナル抗体と共にインキュベートした。AlexaFluor594標識二次抗ラットIgG及びAlexaFluor680標識抗マウスIgGをそれぞれ用いた。共焦点顕微鏡観察は、1、2及び3d.p.i.でのUL87融合タンパク質とUL95融合タンパク質との共局在を証明した(図5B、a〜c)。
【0080】
共焦点顕微鏡観察はまた、2d.p.i.でのUL95融合タンパク質とUL44タンパク質との共局在も実証した(図5d)。さらに、UL44タンパク質は新規に合成されたウイルスDNAとも共局在していた(図5e)。これらの結果は、UL87及びUL95ウイルスタンパク質が共局在し、UL95がRCに動員されたことを示している。
【0081】
実施例7
UL87及びUL95タンパク質はウイルスDNA複製前にUL44の微小フォーカスに動員される
<材料及び方法>
FISHアッセイ
HCMV BAC DNAを、ニックトランスレーションキット(Abbott Molecular,Abbott Park,Illinois)を製造業者の取扱説明書に従って用いてSpectrumOrangeで標識した。FISH分析は多少の改変を加えて、基本的に以前に記載された方法に基づいて行なった(24)。カバーガラス上で増殖させた細胞を、in situ細胞抽出後に2%パラホルムアルデヒドで4℃にて60分間固定し、RNase(100μg/mL)を含有するブロッキングバッファー(2.5%BSA,0.2Mグリシン,及び0.1%TritonX−100)で1時間透過化し、その後ハイブリダイゼーションに用いた。各カバーガラスを、5μLのハイブリダイゼーションバッファー(2×SSC,10%硫酸デキストラン及び50%ホルムアミド)及びDNAプローブと共にスライドガラス上に逆にふせて置いた。ハイブリダイゼーションバッグ中に密封した後、スライドを85℃の水浴中に5分間浸漬し、続いて37℃で一晩インキュベートした。FISH分析後、抗UL44抗体を用いてUL44抗原を検出した。カバーガラスを、50%ホルムアミドを含有する2×SSCで50℃にて3分間ずつ3回洗浄し、2×SSCで50℃にて3分間ずつ3回洗浄し、0.1×SSCで60℃にて10分間、1回洗浄した。SpectrumOrange蛍光団は560nmの吸収極大と588nmの蛍光発光極大を有し、HeNeレーザーで励起した(http://www.zeiss.de/C12567BE0045ACF1/Contents−Frame/5CDD62C92B112413C1256A140040C831)。
【0082】
<結果>
UL87及びUL95がウイルスDNA複製前にUL44と共局在するかを調べるために、PAA又はGCVの存在下でHFF細胞を3MOIでRUL95HAUL87mycに感染させた。細胞を固定し、HA又はmycエピトープに対するポリクローナル抗体、及びUL44タンパク質に対するモノクローナル抗体で処理した。AlexaFluor594標識二次抗ラット又はウサギIgG及びAlexaFluor680標識抗マウスIgGを用いた。ウイルスDNAを検出するためのFISH分析後に、細胞をUL44タンパク質に対するモノクローナル抗体とインキュベートし、続いてAlexaFluor680標識二次抗マウスIgGとインキュベートした。予想された通り、UL44はウイルスDNA合成阻害剤の存在下でウイルスDNAと共局在した(図6a及びb)。UL87及びUL95はPAA又はGCVの存在下でUL44と共局在した(図6c〜f)。これらの結果は、UL87及び95タンパク質がウイルスDNA複製前にUL44と共局在することを示している。
【0083】
UL87とUL44との共局在の特異性を調べるために、UL57及びUL44の局在を調べた。HCMVでは、UL44タンパク質はウイルスDNA合成前にはUL57タンパク質含有小フォーカスには伴われないことが報告されている(36)。細胞を固定し、UL87と融合したmycエピトープに対する抗体及びUL57タンパク質に対する抗体で処理した。AlexaFluor594標識二次抗ウサギIgG又はAlexaFluor680標識抗マウスIgGをそれぞれ用いた。以前に報告されている通り(38)、PAAの存在下では、UL87融合タンパク質はUL57タンパク質と共局在しなかった(図6g)。UL57タンパク質はウイルスDNA複製開始後にはUL44タンパク質と共局在するようである。併せて考えると、HCMV UL87及び95タンパク質は、ウイルスDNA複製前にはUL44と共局在する。
【0084】
実施例8
IE2タンパク質はウイルスDNA複製前にUL87と共局在する
Sourvinosらによるin situハイブリダイゼーション分析(42)により、IE2タンパク質は12時間p.i.で直接的なDNAとの接触により親ウイルスゲノムに伴われていることが示されている。IEタンパク質は点状構造を形成することができ、これがRCへと発達する。IE2はPAAの存在下でウイルスDNA複製と関連するため、本発明者らは、HCMV後期トランス因子がウイルスDNA複製前にIE2と共局在するかを調べた。細胞を3MOIでRUL95HAUL87mycに感染させ、固定し、IE2、myc、UL44及びTBPに対する抗体で処理した。AlexaFluor594標識抗マウスIgG及びAlexaFluor680標識抗ウサギIgGの二次抗体を用いた。UL87タンパク質はPAA若しくはGCVの存在下又は非存在下でIE2フォーカスと共局在した(図7a〜c)。TATA結合タンパク質(TBP)はIE2タンパク質と直接的に相互作用するので(14)、TBPに対するポリクローナル抗体及びUL44タンパク質に対するモノクローナル抗体を用いて、PAA若しくはGCVの存在下又は非存在下でTBPがUL44微小フォーカスに動員されるかを調べた。図7d〜fに示される通り、PAA若しくはGCVの存在下又は非存在下で、TBPはUL44と共局在し、したがって、予想されたように、RC前駆体に動員された。これらの結果から、UL87タンパク質は、IE2タンパク質及びTBPと同様に、ウイルスDNA複製前にウイルスRC前駆体に動員されると結論づけられる。
【0085】
<考察>
MHV−68のUL18、24及び34は後期転写のためのウイルストランス作用因子である。これらのMHV−68ORFは初期に発現されるが、ウイルスDNA複製には必要とされない。これらのMHV−68ウイルスタンパク質はインジケーター遺伝子の発現を駆動する後期ウイルスプロモーターを活性化させるので、後期ウイルストランス活性化因子であると考えられている。後期ウイルストランス活性化因子がどのようにして後期ウイルスプロモーターを特異的に活性化するのかは未だ明らかになっていない。
【0086】
後期ウイルスプロモーターの活性化についての理解は、ユニークな抗ウイルス剤のための新規なアプローチに寄与するものと思われる。HCMVは深刻な病原ウイルスであり、現在利用可能な抗ウイルス剤は限定された有効性及び副作用を有するので、HCMV後期転写因子の解析は新規な抗ウイルス剤の開発のために重要であろう。したがって、本発明者らは、MHV−68後期ウイルストランス活性化因子に相同なHCMV遺伝子が真性後期(γ2)HCMV遺伝子の転写に影響を及ぼすかを調べた。
【0087】
HCMVでは、プロモーターは単純であり、それらは上流又は下流配列を必要としないコアエレメントを含む(8、25、31)。UL44初期ウイルスプロモーターはカノニカルなTATA配列を有する。対照的に、UL44後期ウイルスプロモーターはノンカノニカルなTATA配列を有する(図3dを参照されたい)。本発明者らは、このノンカノニカルなTATA配列が後期ウイルス遺伝子発現に重要であることを報告している(21)。Serioら(39)もまた、EBVのBcLF1後期プロモーターからの発現が変異型TATAエレメントのみに依存することを報告している。ノンカノニカルなTATA配列へのTBPの弱い結合親和性は、感染後の初期での転写の欠如を部分的に説明するものかもしれない。TBPの後期プロモーターへの結合親和性が、後期遺伝子特異的トランス活性化因子の動員及びウイルスDNA複製中の二重らせんの開放後に強まる可能性はある。これらの後期遺伝子トランス活性化因子が、IE2トランス活性化因子と共に、TBP及びRNAポリメラーゼIIの後期プロモーターへの結合を安定化させるということも考えられる。HCMVでは、初期/後期ウイルス遺伝子発現に影響を与えるIE2遺伝子にはいくつかのアイソマーがある。感染初期には、ウイルスIE2−p86タンパク質が初期ウイルス遺伝子発現に必須である。感染後期には、IE2アイソマーであるIE2−p60及びIE2−p40が後期ウイルス遺伝子発現に多大な影響を及ぼす(49、50)。ウイルスDNAとウイルスIE2トランス活性化因子との複合体は、ウイルスDNAが複製し始める時点で検出され、RCの発達を引き起こす(42)。本発明者らはHCMV UL87がウイルスRCでIE2タンパク質と共局在することを示したが、現状ではこれら2つのウイルスタンパク質が相互作用するという証拠はない。
【0088】
なぜ後期ウイルス遺伝子の転写がウイルスDNA複製を必要とするのかは分かっていない。本発明者らのデータは、HCMV UL79、87及び95がUL75(gH)及びUL99(pp28)後期mRNAに必要であることを示している。これらのウイルスタンパク質はウイルスDNA複製の前に発現され、ウイルスRCに組み込まれる。RCは、部分的にはウイルスDNA鋳型を濃縮することにより、ウイルス遺伝子発現のためのフォーカスとして機能する。UL44タンパク質は、ウイルスDNA複製前にIE蓄積部位に組み込まれた。IE2タンパク質は後期遺伝子転写に十分ではないので、UL44がUL79、87及び95と共にIE2フォーカスに動員されることにより後期遺伝子転写に適した環境を提供したとは考えにくい。UL44タンパク質がUL79、87及び95のタンパク質と直接的に相互作用するという証拠はない。UL44は構造的に真核生物の増殖細胞細胞核抗原(Proliferating Cell Nuclear Antigen;PCNA)と相同であり、PCNAは複数のタンパク質と相互作用する(30)。近年のプロテオーム解析により、UL44タンパク質がウイルス複製関連タンパク質以外の転写因子を含む広範なタンパク質と相互作用することが実証された(13、44〜46)。したがって、UL44タンパク質が後期遺伝子転写のタイミングを最適化していると考えられる。HCMV RCはまた、ヘルペスウイルス複製タンパク質(すなわち、HSV ICP8、UL9、及びヘテロ三量体ヘリカーゼ−プライマーゼ複合体UL5−UL8−UL52)に相同な5種類のウイルスタンパク質も含有する。これらのヘルペスウイルス複製タンパク質がまずウイルスの複製前駆体微小フォーカスに組み込まれ、続いてこれらのフォーカスにポリメラーゼ及びポリメラーゼアクセサリータンパク質が動員される(4)。しかしながら、培地にPAAを添加した場合、HCMV UL87タンパク質はSSB UL57タンパク質と共局在しなかった。HCMVでは、HCMV UL57の微小フォーカスは後期遺伝子転写を実現するための足場としては機能しないように思われるが、ウイルスDNA複製の開始後には重要な役割を果たすようである。
【0089】
本願では、UL79、87及び95タンパク質が後期ウイルスmRNAに必要であり、したがってウイルス増殖に必要とされることが示された。UL79、87若しくは95 ORFに突然変異を導入することによりこれらのORFの発現を低下又は消失させることによって、ウイルス複製を抑制し、弱毒化したHCMVは、ワクチンとして使用する上で、天然ウイルスの構造を保持するという利点を有する。
【産業上の利用可能性】
【0090】
本発明により、HCMVを治療又は予防するためのワクチンが提供される。
参考文献
1. Adam, B. L., T. Y. Jervey, C. P. Kohler, G. L. Wright, Jr., J. A. Nelson, and R. M. Stenberg. 1995. The human cytomegalovirus UL98 gene transcription unit overlaps with the pp28 true late gene (UL99) and encodes a 58-kilodalton early protein. J. Virol. 69:5304-5310.
2. Akatsuka, Y., T. A. Goldberg, E. Kondo, E. G. Martin, Y. Obata, Y. Morishima, T. Takahashi, and J. A. Hansen. 2002. Efficient cloning and expression of HLA class I cDNA in human B-lymphoblastoid cell lines. Tissue Antigens 59:502-511.
3. Arumugaswami, V., T. T. Wu, D. Martinez-Guzman, Q. Jia, H. Deng, N. Reyes, and R. Sun. 2006. ORF18 is a transfactor that is essential for late gene transcription of a gammaherpesvirus. J. Virol. 80:9730-9740.
4. Carrington-Lawrence, S. D., and S. K. Weller. 2003. Recruitment of polymerase to herpes simplex virus type 1 replication foci in cells expressing mutant primase (UL52) proteins. J. Virol. 77:4237-4247.
5. Chang, C.-P., C. L. Malone, and M. F. Stinski. 1989. A human cytomegalovirus early gene has three inducible promoters that are regulated differentially at various times after infection. J. Virol. 63:281-290.
6. de Bruyn Kops, A., and D. M. Knipe. 1988. Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell 55:857-868.
7. de Bruyn Kops, A., and D. M. Knipe. 1994. Preexisting nuclear architecture defines the intranuclear location of herpesvirus DNA replication structures. J. Virol. 68:3512-3526.
8. Depto, A. S., and R. M. Stenberg. 1992. Functional analysis of the true late human cytomegalovirus pp28 upstream promoter: cis-acting elements and viral trans-acting proteins necessary for promoter activation. J. Virol. 66:3241-3246.
9. Dunn, W., C. Chou, H. Li, R. Hai, D. Patterson, V. Stolc, H. Zhu, and F. Liu. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. U. S. A. 100:14223-14228.
10. Ellis, H. M., D. Yu, T. DiTizio, and D. L. Court. 2001. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. U. S. A. 98:6742-6746.
11. Fish, K. N., W. Britt, and J. A. Nelson. 1996. A novel mechanism for persistence of human cytomegalovirus in macrophages. J. Virol. 70:1855-1862.
12. Fish, K. N., A. S. Depto, A. V. Moses, W. Britt, and J. A. Nelson. 1995. Growth kinetics of human cytomegalovirus are altered in monocyte-derived macrophages. J. Virol. 69:3737-3743.
13. Gao, Y., K. Colletti, and G. S. Pari. 2008. Identification of human cytomegalovirus UL84 virus- and cell-encoded binding partners by using proteomics analysis. J. Virol. 82:96-104.
14. Hagemeier, C., S. Walker, R. Caswell, T. Kouzarides, and J. Sinclair. 1992. The human cytomegalovirus 80-kilodalton but not the 72-kilodalton immediate-early protein transactivates heterologous promoters in a TATA box-dependent mechanism and interacts directly with TFIID. J. Virol. 66:4452-4456.
15. Hahn, G., D. Rose, M. Wagner, S. Rhiel, and M. A. McVoy. 2003. Cloning of the genomes of human cytomegalovirus strains Toledo, TownevarRIT3, and Towne long as BACs and site-directed mutagenesis using a PCR-based technique. Virology 307:164-177.
16. Hermiston, T. W., C. L. Malone, P. R. Witte, and M. F. Stinski. 1987. Identification and characterization of the human cytomegalovirus immediate-early region 2 gene that stimulates gene expression from an inducible promoter. J. Virol. 61:3214-3221.
17. Horn, P. A., M. S. Topp, J. C. Morris, S. R. Riddell, and H. P. Kiem. 2002. Highly efficient gene transfer into baboon marrow repopulating cells using GALV-pseudotype oncoretroviral vectors produced by human packaging cells. Blood 100:3960-3967.
18. Ibanez, C. E., R. Schrier, P. Ghazal, C. Wiley, and J. A. Nelson. 1991. Human cytomegalovirus productively infects primary differentiated macrophages. J. Virol. 65:6581-6588.
19. Isomura, H., and M. F. Stinski. 2003. The human cytomegalovirus major immediate-early enhancer determines the efficiency of immediate-early gene transcription and viral replication in permissive cells at low multiplicity of infection. J. Virol. 77:3602-3614.
20. Isomura, H., M. F. Stinski, A. Kudoh, T. Daikoku, N. Shirata, and T. Tsurumi. 2005. Two Sp1/Sp3 binding sites in the major immediate-early proximal enhancer of human cytomegalovirus have a significant role in viral replication. J. Virol. 79:9597-9607.
21. Isomura, H., M. F. Stinski, A. Kudoh, T. Murata, S. Nakayama, Y. Sato, S. Iwahori, and T. Tsurumi. 2008. Noncanonical TATA sequence in the UL44 late promoter of human cytomegalovirus is required for the accumulation of late viral transcripts. J. Virol. 82:1638-1646.
22. Isomura, H., M. F. Stinski, A. Kudoh, S. Nakayama, S. Iwahori, Y. Sato, and T. Tsurumi. 2007. The late promoter of the human cytomegalovirus viral DNA polymerase processivity factor has an impact on delayed early and late viral gene products but not on viral DNA synthesis. J. Virol. 81:6197-6206.
23. Isomura, H., T. Tsurumi, and M. F. Stinski. 2004. Role of the proximal enhancer of the major immediate-early promoter in human cytomegalovirus replication. J. Virol. 78:12788-12799.
24. Kanda, T., M. Kamiya, S. Maruo, D. Iwakiri, and K. Takada. 2007. Symmetrical localization of extrachromosomally replicating viral genomes on sister chromatids. J. Cell Sci. 120:1529-1539.
25. Kohler, C. P., J. A. Kerry, M. Carter, V. P. Muzithras, T. R. Jones, and R. M. Stenberg. 1994. Use of recombinant virus to assess human cytomegalovirus early and late promoters in the context of the viral genome. J. Virol. 68:6589-6597.
26. Lashmit, P. E., M. F. Stinski, E. A. Murphy, and G. C. Bullock. 1998. A cis-repression sequence adjacent to the transcription start site of the human cytomegalovirus US3 gene is required to down regulate gene expression at early and late times after infection. J. Virol. 72:9575-9584.
27. Lathey, J. L., and S. A. Spector. 1991. Unrestricted replication of human cytomegalovirus in hydrocortisone-treated macrophages. J. Virol. 65:6371-6375. 28. Leach, F. S., and E. S. Mocarski. 1989. Regulation of cytomegalovirus late-gene expression: differential use of three start sites in the transcriptional activation of ICP36 gene expression. J. Virol. 63:1783-1791.
29. Lukonis, C. J., and S. K. Weller. 1996. Characterization of nuclear structures in cells infected with herpes simplex virus type 1 in the absence of viral DNA replication. J. Virol. 70:1751-1758.
30. Maga, G., and U. Hubscher. 2003. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J. Cell Sci. 116:3051-3060.
31. McWatters, B. J., R. M. Stenberg, and J. A. Kerry. 2002. Characterization of the human cytomegalovirus UL75 (glycoprotein H) late gene promoter. Virology. 303:309-316.
32. Meier, J. L., M. J. Keller, and J. J. McCoy. 2002. Requirement of multiple cis-acting elements in the human cytomegalovirus major immediate-early distal enhancer for viral gene expression and replication. J. Virol. 76:313-326.
33. Meier, J. L., and J. A. Pruessner. 2000. The human cytomegalovirus major immediate-early distal enhancer region is required for efficient viral replication and immediate-early gene expression. J. Virol. 74:1602-1613.
34. Meier, J. L., and M. F. Stinski. 1997. Effect of a modulator deletion on transcription of the human cytomegalovirus major immediate-early genes in infected undifferentiated and differentiated cells. J. Virol. 71:1246-1255.
35. Pari, G. S., and D. G. Anders. 1993. Eleven loci encoding trans-acting factors are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA replication. J. Virol. 67:6979-6988.
36. Penfold, M. E., and E. S. Mocarski. 1997. Formation of cytomegalovirus DNA replication compartments defined by localization of viral proteins and DNA synthesis. Virology 239:46-61.
37. Quinlan, M. P., L. B. Chen, and D. M. Knipe. 1984. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell 36:857-868.
38. Sarisky, R. T., and G. S. Hayward. 1996. Evidence that the UL84 gene product of human cytomegalvorus is essential for promoting oriLyt-dependent DNA replication and formation of replication compartments in cotransfection assays. J. Virol. 70:7398-7413.
39. Serio, T. R., N. Cahill, M. E. Prout, and G. Miller. 1998. A functionally distinct TATA box required for late progression through the Epstein-Barr virus life cycle. J. Virol. 72:8338-8343.
40. Sinzger, C., A. Grefte, B. Plachter, A. S. H. Gouw, T. Hauw The, and G. Jahn. 1995. Fibroblasts, epithelial cells, endothelial cells, and smooth muscle cells are the major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J. Gen. Virol. 76:741-750.
41. Sinzger, C., B. Plachter, A. Grefte, A. S. H. Gouw, T. H. The, and G. Jahn. 1996. Tissue macrophages are infected by human cytomegalovirus. J. Infect. Dis. 173:240-245.
42. Sourvinos, G., N. Tavalai, A. Berndt, D. A. Spandidos, and T. Stamminger. 2007. Recruitment of human cytomegalovirus immediate-early 2 protein onto parental viral genomes in association with ND10 in live-infected cells. J. Virol. 81:10123-10136.
43. Stinski, M. F. 1978. Sequence of protein synthesis in cells infected by human cytomegalovirus: early and late virus-induced polypeptides. J. Virol. 26:686-701.
44. Strang, B. L., S. Boulant, and D. M. Coen. 2010. Nucleolin associates with the human cytomegalovirus DNA polymerase accessory subunit UL44 and is necessary for efficient viral replication. J. Virol. 84:1771-1784.
45. Strang, B. L., A. P. Geballe, and D. M. Coen. 2010. Association of human cytomegalovirus proteins IRS1 and TRS1 with the viral DNA polymerase accessory subunit UL44. J. Gen. Virol. 91:2167-2175.
46. Strang, B. L., E. Sinigalia, L. A. Silva, D. M. Coen, and A. Loregian. 2009. Analysis of the association of the human cytomegalovirus DNA polymerase subunit UL44 with the viral DNA replication factor UL84. J. Virol. 83:7581-7589.
47. Taylor-Wiedeman, J., J. G. Sissons, L. K. Borysiewicz, and J. H. Sinclair. 1991. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 72:2059-2064.
48. Wang, S. K., C. Y. Duh, and C. W. Wu. 2004. Human cytomegalovirus UL76 encodes a novel virion-associated protein that is able to inhibit viral replication. J. Virol. 78:9750-9762.
49. White, E. A., C. L. Clark, V. Sanchez, and D. H. Spector. 2004. Small internal deletions in the human cytomegalovirus IE2 gene result in nonviable recombinant viruses with differential defects in viral gene expression. J. Virol. 78:1817-1830.
50. White, E. A., C. J. Del Rosario, R. L. Sanders, and D. H. Spector. 2007. The IE2 60-kilodalton and 40-kilodalton proteins are dispensable for human cytomegalovirus replication but are required for efficient delayed early and late gene expression and production of infectious virus. J. Virol. 81:2573-2583.
51. Wing, B. A., and E. S. Huang. 1995. Analysis and mapping of a family of 3'-coterminal transcripts containing coding sequences for human cytomegalovirus open reading frames UL93 through UL99. J. Virol. 69:1521-1531.
52. Winkler, M., S. A. Rice, and T. Stamminger. 1994. UL69 of human cytomegalovirus, an open reading frame with homology to ICP27 of herpes simplex virus, encodes a transactivator of gene expression. J. Virol. 68:3943-3954.
53. Wong, E., T. T. Wu, N. Reyes, H. Deng, and R. Sun. 2007. Murine gammaherpesvirus 68 open reading frame 24 is required for late gene expression after DNA replication. J. Virol. 81:6761-6764.
54. Wu, T. T., T. Park, H. Kim, T. Tran, L. Tong, D. Martinez-Guzman, N. Reyes, H. Deng, and R. Sun. 2009. ORF30 and ORF34 are essential for expression of late genes in murine gammaherpesvirus 68. J. Virol. 83:2265-2273.
55. Xiang, C. C., E. Mezey, M. Chen, S. Key, L. Ma, and M. J. Brownstein. 2004. Using DSP, a reversible cross-linker, to fix tissue sections for immunostaining, microdissection and expression profiling. Nucleic Acids Res. 32:e185.
56. Yu, D., M. C. Silva, and T. Shenk. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. U. S. A. 100:12396-12401.
57. Zhong, L., and G. S. Hayward. 1997. Assembly of complete, functionally active herpes simplex virus DNA replication compartments and recruitment of associated viral and cellular proteins in transient cotransfection assays. J. Virol. 71:3146-3160.
【技術分野】
【0001】
本発明は、弱毒ヒトサイトメガロウイルス(HCMV)及びそのワクチンとしての使用に関する。
【背景技術】
【0002】
ヒトサイトメガロウイルス(HCMV)は、ベータヘルペスウイルスファミリーのメンバーである。そのウイルスゲノムは240,000bpを有し、少なくとも150個の予測されるオープンリーディングフレーム(ORF)を有する(9、56)。HCMVによる感染は多くの個体で生じるが、通常は無症候性である。このウイルスは免疫抑制条件下で再活性化し、肺炎、肝炎、腎炎、及び消化器疾患の病原となる。このウイルスは、線維芽細胞、上皮細胞及び内皮細胞などの最終的に分化した細胞、ならびに単球由来マクロファージにおいて、産生的に複製される(11、12、18、27、40、41、47)。
【0003】
産生的複製の間、HCMV遺伝子群は、前初期(IE)、初期及び後期と呼ばれる一時的カスケードで発現される。主要IE遺伝子群(MIE)であるUL123/122(IE1/IE2)は、その後のウイルス遺伝子発現及びウイルス複製の効率において決定的な役割を果たす(19、20、23、32−34)。初期ウイルス遺伝子はウイルスDNA複製に必要なタンパク質をコードする(35)。ウイルスDNA複製に続いて、遅延初期及び後期ウイルス遺伝子が発現され、これらはウイルス粒子のための構造タンパク質をコードする。これに関連して、MIE遺伝子プロモーターのシス作用性エレメント(crs)配列の一部に突然変異を有し、これによってウイルス複製が不能ではないが抑制されている弱毒化組換えヒトサイトメガロウイルスが特許文献1に開示されている。
【0004】
真性後期(true late)(γ−2クラス)ウイルス遺伝子の発現は、ウイルスDNA合成の阻害により妨げられる。UL75(gH)及びUL99(pp28)は、真性後期転写産物によりコードされるHCMV遺伝子の2つの例である(25、31、52)。HCMV前初期遺伝子及び初期遺伝子発現の制御は集中的に研究されてきているが、後期遺伝子発現を制御する特異的メカニズムについてはほとんど知られていない。
【0005】
HCMVでは、IE2タンパク質は感染後初期の微小フォーカス(microfoci)中に見出すことができ、直接的なDNA接触により親ウイルスゲノムと会合している。一部の微小フォーカスは拡大して、感染後の後期に複製コンパートメント(RC)を形成する(42)。しかしながら、HCMVのIE遺伝子群単独では真性後期ウイルス遺伝子の活性化に十分ではない。どのHCMV初期遺伝子が後期遺伝子発現に必要とされるのかは知られていない。
【0006】
マウスガンマヘルペスウイルス68(MHV−68)のORF18、24、30及び34は、後期遺伝子転写に必須である(3、53、54)。これらの不存在が初期ウイルス遺伝子発現又はDNA複製にほとんど影響を及ぼさないことから、これらの遺伝子産物は、後期遺伝子のプロモーターを刺激するために特異的に機能すると考えられる。したがって、後期遺伝子発現は、ウイルスDNA複製のみならず、それらのウイルスによりコードされるトランス作用性因子にも依存している。
【0007】
DNAポリメラーゼ進行因子(DNA polymerase processivity factor)(UL44)及び一本鎖DNA結合タンパク質(SSB)(UL57)をはじめとするHCMVウイルスDNA複製タンパク質群は、1型単純ヘルペスウイルス(HSV−1)RCに似た大型の核構造中に局在する(36、38)。HSV感染細胞では、ウイルスDNA合成を阻害剤ホスホノ酢酸(PAA)で阻害した場合、ウイルスによりコードされるSSB(ICP8)タンパク質を含有する小型のウイルス前複製部位又はフォーカスが観察される(6、29、37)。PAAの不存在下では、BrdUがウイルスDNAに取り込まれて、DNAは子ウイルスと共存して見出される(7、57)。HCMVでは、UL44タンパク質は、感染後初期の段階では、SSB(UL57)タンパク質含有フォーカスと常に共存しているわけではない(36)。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2008−092854号公報
【非特許文献】
【0009】
【非特許文献1】Dunn, W., C. Chou, H. Li, R. Hai, D. Patterson, V. Stolc, H. Zhu, and F. Liu. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. U. S. A. 100:14223-14228. (文献9)
【非特許文献2】Yu, D., M. C. Silva, and T. Shenk. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. U. S. A. 100:12396-12401. (文献56)
【非特許文献3】Fish, K. N., W. Britt, and J. A. Nelson. 1996. A novel mechanism for persistence of human cytomegalovirus in macrophages. J. Virol. 70:1855-1862. (文献11)
【非特許文献4】Fish, K. N., A. S. Depto, A. V. Moses, W. Britt, and J. A. Nelson. 1995. Growth kinetics of human cytomegalovirus are altered in monocyte-derived macrophages. J. Virol. 69:3737-3743. (文献12)
【非特許文献5】Ibanez, C. E., R. Schrier, P. Ghazal, C. Wiley, and J. A. Nelson. 1991. Human cytomegalovirus productively infects primary differentiated macrophages. J. Virol. 65:6581-6588. (文献18)
【非特許文献6】Lathey, J. L., and S. A. Spector. 1991. Unrestricted replication of human cytomegalovirus in hydrocortisone-treated macrophages. J. Virol. 65:6371-6375. (文献27)
【非特許文献7】Sinzger, C., A. Grefte, B. Plachter, A. S. H. Gouw, T. Hauw The, and G. Jahn. 1995. Fibroblasts, epithelial cells, endothelial cells, and smooth muscle cells are the major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J. Gen. Virol. 76:741-750. (文献40)
【非特許文献8】Sinzger, C., B. Plachter, A. Grefte, A. S. H. Gouw, T. H. The, and G. Jahn. 1996. Tissue macrophages are infected by human cytomegalovirus. J. Infect. Dis. 173:240-245. (文献41)
【非特許文献9】Taylor-Wiedeman, J., J. G. Sissons, L. K. Borysiewicz, and J. H. Sinclair. 1991. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 72:2059-2064. (文献47)
【非特許文献10】Isomura, H., and M. F. Stinski. 2003. The human cytomegalovirus major immediate-early enhancer determines the efficiency of immediate-early gene transcription and viral replication in permissive cells at low multiplicity of infection. J. Virol. 77:3602-3614. (文献19)
【非特許文献11】Isomura, H., M. F. Stinski, A. Kudoh, T. Daikoku, N. Shirata, and T. Tsurumi. 2005. Two Sp1/Sp3 binding sites in the major immediate-early proximal enhancer of human cytomegalovirus have a significant role in viral replication. J. Virol. 79:9597-9607. (文献20)
【非特許文献12】Isomura, H., T. Tsurumi, and M. F. Stinski. 2004. Role of the proximal enhancer of the major immediate-early promoter in human cytomegalovirus replication. J. Virol. 78:12788-12799. (文献23)
【非特許文献13】Meier, J. L., M. J. Keller, and J. J. McCoy. 2002. Requirement of multiple cis-acting elements in the human cytomegalovirus major immediate-early distal enhancer for viral gene expression and replication. J. Virol. 76:313-326. (文献32)
【非特許文献14】Meier, J. L., and J. A. Pruessner. 2000. The human cytomegalovirus major immediate-early distal enhancer region is required for efficient viral replication and immediate-early gene expression. J. Virol. 74:1602-1613. (文献33)
【非特許文献15】Meier, J. L., and M. F. Stinski. 1997. Effect of a modulator deletion on transcription of the human cytomegalovirus major immediate-early genes in infected undifferentiated and differentiated cells. J. Virol. 71:1246-1255. (文献34)
【非特許文献16】Pari, G. S., and D. G. Anders. 1993. Eleven loci encoding trans-acting factors are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA replication. J. Virol. 67:6979-6988. (文献35)
【非特許文献17】Kohler, C. P., J. A. Kerry, M. Carter, V. P. Muzithras, T. R. Jones, and R. M. Stenberg. 1994. Use of recombinant virus to assess human cytomegalovirus early and late promoters in the context of the viral genome. J. Virol. 68:6589-6597. (文献25)
【非特許文献18】McWatters, B. J., R. M. Stenberg, and J. A. Kerry. 2002. Characterization of the human cytomegalovirus UL75 (glycoprotein H) late gene promoter. Virology. 303:309-316. (文献31)
【非特許文献19】Winkler, M., S. A. Rice, and T. Stamminger. 1994. UL69 of human cytomegalovirus, an open reading frame with homology to ICP27 of herpes simplex virus, encodes a transactivator of gene expression. J. Virol. 68:3943-3954. (文献52)
【非特許文献20】Sourvinos, G., N. Tavalai, A. Berndt, D. A. Spandidos, and T. Stamminger. 2007. Recruitment of human cytomegalovirus immediate-early 2 protein onto parental viral genomes in association with ND10 in live-infected cells. J. Virol. 81:10123-10136. (文献42)
【非特許文献21】Arumugaswami, V., T. T. Wu, D. Martinez-Guzman, Q. Jia, H. Deng, N. Reyes, and R. Sun. 2006. ORF18 is a transfactor that is essential for late gene transcription of a gammaherpesvirus. J. Virol. 80:9730-9740. (文献3)
【非特許文献22】Wong, E., T. T. Wu, N. Reyes, H. Deng, and R. Sun. 2007. Murine gammaherpesvirus 68 open reading frame 24 is required for late gene expression after DNA replication. J. Virol. 81:6761-6764. (文献53)
【非特許文献23】Wu, T. T., T. Park, H. Kim, T. Tran, L. Tong, D. Martinez-Guzman, N. Reyes, H. Deng, and R. Sun. 2009. ORF30 and ORF34 are essential for expression of late genes in murine gammaherpesvirus 68. J. Virol. 83:2265-2273. (文献54)
【非特許文献24】Penfold, M. E., and E. S. Mocarski. 1997. Formation of cytomegalovirus DNA replication compartments defined by localization of viral proteins and DNA synthesis. Virology 239:46-61. (文献36)
【非特許文献25】Sarisky, R. T., and G. S. Hayward. 1996. Evidence that the UL84 gene product of human cytomegalvorus is essential for promoting oriLyt-dependent DNA replication and formation of replication compartments in cotransfection assays. J. Virol. 70:7398-7413. (文献38)
【非特許文献26】de Bruyn Kops, A., and D. M. Knipe. 1988. Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell 55:857-868. (文献6)
【非特許文献27】Lukonis, C. J., and S. K. Weller. 1996. Characterization of nuclear structures in cells infected with herpes simplex virus type 1 in the absence of viral DNA replication. J. Virol. 70:1751-1758. (文献29)
【非特許文献28】Quinlan, M. P., L. B. Chen, and D. M. Knipe. 1984. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell 36:857-868. (文献37)
【非特許文献29】de Bruyn Kops, A., and D. M. Knipe. 1994. Preexisting nuclear architecture defines the intranuclear location of herpesvirus DNA replication structures. J. Virol. 68:3512-3526. (文献7)
【非特許文献30】Zhong, L., and G. S. Hayward. 1997. Assembly of complete, functionally active herpes simplex virus DNA replication compartments and recruitment of associated viral and cellular proteins in transient cotransfection assays. J. Virol. 71:3146-3160. (文献57)
【発明の概要】
【発明が解決しようとする課題】
【0010】
本明細書において、本発明者らは、HCMVのORFであるUL79、87及び95が、後期ウイルスmRNA及び感染性ウイルス産生に必須であるが、ウイルスDNA複製には必須ではないことを示す。UL87タンパク質とUL95タンパク質はウイルスDNA複製の前にウイルスゲノムと組み立てられて、ウイルスのUL44タンパク質及びUL122(IE2)タンパク質ならびに細胞の転写因子TBPを含む微小フォーカスを形成することが明らかとなった。UL79、87又は95ORFに突然変異を導入することにより、HCMVの弱毒ウイルスを作製することを本発明の目的とする。
【課題を解決するための手段】
【0011】
本発明者らは、ヒトサイトメガロウイルス(HCMV)のUL79、87及び95タンパク質が、感染性ウイルス増殖には必要とされるが、ウイルスDNA複製には必要とされないこと、真性後期ウイルスmRNAの産生に必須であることを見出した。これらの知見に基づき、本発明者らは、HCMVのUL79、87又は95遺伝子ORFを突然変異させた弱毒HCMVを作製し、本発明を完成させた。
【0012】
具体的には、本発明は、以下の特徴を有する。
〔1〕ヒトサイトメガロウイルス(HCMV)のUL79、87又は95遺伝子オープンリーディングフレーム(ORF)中に突然変異を生じさせることにより、これらの遺伝子の1つ若しくは複数からの発現が低下又は消失している、弱毒HCMV。
〔2〕前記突然変異が、ORF中への塩基の挿入によるものである、上記〔1〕に記載の弱毒HCMV。
〔3〕前記弱毒HCMVからの感染性ウイルス増殖は野生型ウイルスと比較して低下しているが、ウイルスDNA複製は野生型ウイルスと同等である、上記〔1〕又は〔2〕に記載の弱毒HCMV。
〔4〕後期ウイルスmRNAの発現が野生型ウイルスと比較して低下している、上記〔1〕〜〔3〕のいずれか1つに記載の弱毒HCMV。
〔5〕上記〔1〕〜〔4〕のいずれか1つに記載の弱毒HCMVを有効成分として含む、HCMV感染の予防又は治療用ワクチン。
〔6〕アジュバントをさらに含む、上記〔5〕に記載のワクチン。
〔7〕上記〔1〕〜〔4〕のいずれか1つに記載の弱毒HCMV又は上記〔5〕若しくは〔6〕に記載のワクチンを非ヒト動物に免疫して抗体を作製するステップ、及び該抗体を回収するステップを含む、抗HCMV抗体の作製方法。
〔8〕前記抗体がヒト抗体である、上記〔7〕に記載の方法。
【発明の効果】
【0013】
本発明によって、ウイルス複製が抑制された弱毒化ヒトサイトメガロウイルス(HCMV)を提供することができ、該弱毒化ウイルスは、ワクチンとして使用することができる。
【図面の簡単な説明】
【0014】
【図1】組み換えHCMV BAC DNAの構造を示す図である。(a)wt(野生型)、RdlUL79+F、RdlUL87+F及びRdlUL95+Fの組み換えBAC DNAを示す模式図である。ORFに突然変異を有する組み換えHCMV BAC DNAを構築する場合、FRT配列をORFの中央部に挿入し、次いでFLP媒介組み換えによりKanRを切り出し、34bpのFRT配列のみを残した。(b)UL95及びUL87ORFに融合したエピトープを含む組み換えBAC DNAの模式図である。UL95ORFと融合したHAエピトープを挿入する場合、逆選択を行なって薬剤耐性カセットを除去した。BACUL95Nneo+St又はBACUL95Cneo+Stの構築のために、RpsL遺伝子(ストレプトマイシンに対する感受性を増大させる)及びネオマイシン耐性マーカー(カナマイシン耐性を付与するため)を含有するマーカーカセットをそれぞれUL95ORFのN末端又はC末端に挿入した。中間体BACクローンはカナマイシン耐性に基づいて単離した。相同組み換えの第2ラウンドでは、マーカーカセット全体をオリゴヌクレオチドを用いたカウンター選択によりHAエピトープを含む配列と置き換えた。UL87ORFのC末端に融合したmycエピトープを含む組み換えBACを構築するために、BACHAUL95及びBACUL95HAの構築後に逆選択を繰り返した。(c)wt(親ウイルス)及び組み換えBACのPCR分析を示す。PCR産物は表2に示されるプライマーを用いて増幅し、3.0%アガロースゲルで電気泳動し、臭化エチジウムで染色し、配列決定した。レーン1、2、5、6及び9:RUL95HAUL87myc;レーン3、4、7、8及び10:RHAUL95UL87myc;レーン11:wt。
【図2】UL79、87及び95がウイルス増殖に必要とされるが、ウイルスDNA複製には必要とされないことを示す図である。補充タンパク質を発現するか発現しないHFF細胞に3MOIでウイルスを感染させた。ウイルス力価は50%組織培養感染用量(TCID50)アッセイにより決定した。(a〜c)ウイルス増殖曲線を示す。(d〜f)ウイルスDNA複製を示す。ウイルスDNAはgBプライマー及びプローブを用いるリアルタイムPCRにより定量化した。18Sプライマー及びプローブを用いるリアルタイムPCRも、内部対照として用いるために行なった。データは3回の独立した実験の平均である。(a)及び(d):RdlUL79;(b)及び(e):RdlIL87;(c)及び(f):RdlUL95。
【図3a】組み換えウイルス感染後のUL75、99及び44後期mRNAについて示す図である。補充タンパク質を発現するか発現しないHFF細胞に3MOIで突然変異型組み換えウイルスを感染させた。PAAの存在下又は非存在下で細胞質RNAを1、2及び3d.p.i.で回収した。(a〜c)IE1(左パネル)、UL75(gH)(中央パネル)又はUL99(pp28)(右パネル)についてのノザンブロットである。28S及び18SリボソームRNAをRNAロード量の等量の対照として用いた。(a)RdlUL79感染細胞。レーン1、3及び5:HFF細胞;レーン2、4及び6:UL79融合タンパク質を発現するHFF細胞;レーン1及び2:1d.p.i.;レーン3及び4:2d.p.i.;レーン5及び6:3d.p.i.。
【図3b】組み換えウイルス感染後のUL75、99及び44後期mRNAについて示す図である。補充タンパク質を発現するか発現しないHFF細胞に3MOIで突然変異型組み換えウイルスを感染させた。PAAの存在下又は非存在下で細胞質RNAを1、2及び3d.p.i.で回収した。(a〜c)IE1(左パネル)、UL75(gH)(中央パネル)又はUL99(pp28)(右パネル)についてのノザンブロットである。28S及び18SリボソームRNAをRNAロード量の等量の対照として用いた。(b)RdlUL87感染細胞。UL87融合タンパク質を発現するHFF細胞;レーン1及び2:1d.p.i.;レーン3及び4:2d.p.i.;レーン5及び6:3d.p.i.;レーン7及び8:PAAの存在下での2d.p.i.。
【図3c】組み換えウイルス感染後のUL75、99及び44後期mRNAについて示す図である。補充タンパク質を発現するか発現しないHFF細胞に3MOIで突然変異型組み換えウイルスを感染させた。PAAの存在下又は非存在下で細胞質RNAを1、2及び3d.p.i.で回収した。(a〜c)IE1(左パネル)、UL75(gH)(中央パネル)又はUL99(pp28)(右パネル)についてのノザンブロットである。28S及び18SリボソームRNAをRNAロード量の等量の対照として用いた。(c)RdlUL95感染細胞。UL95融合タンパク質を発現するHFF細胞。レーン1及び2:1d.p.i.;レーン3及び4:2d.p.i.;レーン5及び6:PAAの存在下での2d.p.i.;レーン7及び8:3d.p.i.。
【図3d】組み換えウイルス感染後のUL75、99及び44後期mRNAについて示す図である。補充タンパク質を発現するか発現しないHFF細胞に3MOIで突然変異型組み換えウイルスを感染させた。PAAの存在下又は非存在下で細胞質RNAを1、2及び3d.p.i.で回収した。(d)UL44転写産物についてのRNase保護アッセイ。3MOIでのRdlUL79感染後1、2及び3日に細胞質RNAを回収した。20μgのRNAを32P標識アンチセンスUL44プロモータープローブと37℃で一晩ハイブリダイズさせ、続いてRNaseT1で消化した。アンチセンスUL44プローブはすべてのUL44転写産物の転写開始部位の上流配列を含んでいる。保護されたRNA断片を変性6%ポリアクリルアミドゲルで電気泳動した。レーン1:RNaseT1を含まないプローブ;レーン2、4及び6:HFF細胞;レーン3、5及び7:UL79融合タンパク質を発現するHFF細胞;レーン2及び3:1d.p.i.;レーン4及び5:2d.p.i.;レーン6及び7:3d.p.i.。矢印1、2及び3はそれぞれ開始部位1、2又は3から開始する転写産物を示す。
【図4】UL87−myc融合タンパク質及びUL95−HA融合タンパク質が感染後の初期に発現されることを示す図である。ウイルスDNA合成をPAA又はGCVにより阻害した。前初期pIE86及びpIE72(UL122及び123)、HA若しくはmycエピトープ、又はGAPDHに対する抗体を用いて、PAA若しくはGCVの存在下又は非存在下でのRUL95HAUL87mycの感染後の示した時点でウエスタンブロット分析を行なった。GAPDHはロード量対照として用いた。(A)抗HA抗体、(B)抗myc抗体、(C)抗IE72/IE86抗体、抗UL44抗体、及び抗GAPDH抗体。
【図5】UL79、87及び95タンパク質の複製コンパートメントへの動員を示す図である。(A)対応するORFが突然変異した組み換えウイルス感染後のUL79、87又は95−mcherry融合タンパク質とUL44との局在を示す。RdlUL79(a)、RdlUL87(b)又はRdlUL95(c)に感染させた細胞を固定し、処理した。(a)UL79−mcherry及びUL44タンパク質;(b)UL87−mcherry及びUL44タンパク質;(c)UL95−mcherry及びUL44タンパク質。(B)RUL95HAUL87myc感染後のUL95−HA及びUL87−myc融合タンパク質、UL44タンパク質、及びウイルスDNAの局在を示す。RUL95HAUL87mycに感染させた細胞を、示したd.p.i.で回収した。細胞を固定し、抗体を用いて顕微鏡観察のために又はFISH分析のために処理した。(a〜c)UL95−HAタンパク質とUL87−mycタンパク質との共局在;(d)UL95−HAタンパク質とUL44タンパク質との共局在;(e)ウイルスDNAとUL44タンパク質との共局在;(a)1d.p.i.;(b、d及びe)2d.p.i.;(c)3d.p.i.。
【図6−1】PAA若しくはGCVの存在下でのUL95−HA又はUL87−myc融合タンパク質の局在を示す図である。PAA(a、c、e及びg)又はGCV(b、d及びf)の存在下でRUL95HAUL87mycに感染させた細胞を2d.p.i.で処理した。(a及びb)HCMV DNAとUL44タンパク質との共局在;(c及びd)UL95−HAとUL44との共局在;(e及びf)UL87−mycとUL44タンパク質との共局在。
【図6−2】PAA若しくはGCVの存在下でのUL95−HA又はUL87−myc融合タンパク質の局在を示す図である。PAA(a、c、e及びg)又はGCV(b、d及びf)の存在下でRUL95HAUL87mycに感染させた細胞を2d.p.i.で処理した。(g)UL87−mycとUL57タンパク質との共局在。
【図7】ウイルスDNA複製前のUL87とIE2タンパク質及びUL44とTBPタンパク質の局在を示す図である。PAAの非存在下(a及びd)若しくはPAAの存在下(b及びe)又はGCVの存在下(c及びf)で細胞をRUL95HAUL87mycに感染させ、2d.p.i.で回収し、抗体で処理した。(a〜c)UL87−mycとIE2タンパク質との共局在;(d〜f)UL44タンパク質とTBPとの共局在。
【図8】3MOIでのwt、RUL95HAUL87myc及びRHAUL95UL87mycの増殖曲線を示す図である。ウイルス力価は50%組織培養感染用量(TCID50)アッセイにより決定した。
【発明を実施するための形態】
【0015】
前述したように、本発明は、HCMVのORFであるUL79、87及び95が、後期ウイルスmRNA及び感染性ウイルス産生に必須であるが、ウイルスDNA複製には必須ではないとの知見に基づく。具体的には、UL79、87若しくは95 ORFに突然変異を導入することによりこれらのORFの発現を低下又は消失させることによって、ウイルス複製の抑制、ひいてはHCMVの弱毒化を可能にした。ホルマリンなどの化学物質での処理による弱毒化では、外被(エンベロープ)タンパク質の変性などの問題も生じかねないが、本発明の弱毒化ウイルスは天然ウイルスの構造を保持するという利点を有する。
【0016】
<HCMV>
本発明の対象であるヒトサイトメガロウイルス(HCMV)は、すべてのウイルス株を包含するものとする。HCMVは、ヘルペスウイルス科βヘルペスウイルス亜科に属し、その直径が約180nmで約230kbpの塩基数からなる二本鎖DNAウイルスである。HCMVの株には公知の株が含まれ、例えば、AD169株、Towne株、Major株、BT1943株、Davis株、Merlin株、Toledo株、3157株、6397株、3301株、C87株などが知られている(Dolan,A.ら,2004,J.Gen。Virol.85:1301−1312;Chee,M.S.ら,1990,Curr.Top.Microbiol.Immunol.154:125−169)。
【0017】
上記のHCMV株のゲノム配列は、例えばGenBank(NCBI、米国)などに蓄積されたデータから入手可能である。例えば、一般によく知られているAD169株及びTowne株のゲノム配列はそれぞれGenBank登録番号FJ527563、AY315197として登録されている。
【0018】
HCMVは、ヒト線維芽細胞、例えばヒト包皮線維芽(HFF)細胞(ATCC−CC2509)やヒト胎児線維芽細胞、ヒト内皮細胞、例えばヒト毛細血管内皮細胞(HMVEC)(ATCC CC−2527)やヒト網膜色素内皮細胞(RPE)(ATCC C4000−1)などのヒト細胞に感染することができる。したがって、これらの細胞に、HCMV又はそのウイルスベクター、例えばHCMVゲノム配列とBAC(細菌人工染色体)配列から作製されたHCMVBAC、を感染又は導入することによってウイルスDNAを回収することができる(Dunn,W.ら,2003,Proc.Natl.Acad.Sci.USA,100(24):14223−14228)。
【0019】
HCMVのUL79、87及び95ORFは、後期転写のためのウイルストランス作用因子として知られるMHV−68のUL18、24及び34と相同性を有するが、本願以前にはその機能は未知であった。本発明者らは、以下の実施例に示すように、これらのORFが、感染性ウイルス増殖には必要とされるが、ウイルスDNA複製には必要とされないこと、真性後期ウイルスmRNAの産生に必須であることを見出した。
【0020】
例えば、HCMV Towne株(GenBank登録番号:AY315197)のUL79、87及び95ORF配列は、以下の通りである。他のHCMV株のUL79、87及び95ORF配列も、GenBankなどの公知の遺伝子バンク又は文献などから入手可能である。
UL79ORF塩基配列(配列番号1):atgatggcccgcgacgaagagaaccccgccgtcccgcgggtccgcacaggcaaattctcctttacttgcgccaatcatctaatattacagattagcgagaagatgtcgcgcggacagccgctgagctcgctgcgtttggaagaactcaaaatcgtacgcctcatctgcgtcctcctctttcaccgcggtctcgaaacgctgctactgcgcgaaactatgaacaacctgggtgtctcggaccacgccgtgcttagccgcaagacgccgcaaccctactggcctcatctgtaccgcgaactgcgccaggccttcccggggctggactttgaggcggccgtgttcgatgaaacacgcgccgcccgtctcagccagcgcctgtgtcacccgcgcttgagcggcggactgctgacgcgctttgtgcagcgccacaccggcctgccggtcgttttccccgaagacctggcgcgcaacggcaacatcctcttctccctaggcacgctctacggacaccgcctgtttcgtctggcggccttcttcacacgccactggggtgccgaagcgtacgaacccttgattcgcatcatctgtcaaaaaatgtggtacttttatctcatcggcaccggcaagatgcgcattacccccgacgccttcgagatccagcggagtcgacacgaaacgggcatttttacctttattatggaagattacagaacgttcgccggcacgctgtcccggcacccgcaccgtccgcacccacaacagcagcagcaccaccaccccggtcccccccatcctcctctttctcaccctgcctcgtcctgtctcaacccagaggccgtactggccgcccgcgcccttcatatgccgacgctggctaacgacgtgtga
【0021】
UL79ORFアミノ酸配列(配列番号2):MMARDEENPAVPRVRTGKFSFTCANHLILQISEKMSRGQPLSSLRLEELKIVRLICVLLFHRGLETLLLRETMNNLGVSDHAVLSRKTPQPYWPHLYRELRQAFPGLDFEAAVFDETRAARLSQRLCHPRLSGGLLTRFVQRHTGLPVVFPEDLARNGNILFSLGTLYGHRLFRLAAFFTRHWGAEAYEPLIRIICQKMWYFYLIGTGKMRITPDAFEIQRSRHETGIFTFIMEDYRTFAGTLSRHPHRPHPQQQQHHHPGPPHPPLSHPASSCLNPEAVLAARALHMPTLANDV
【0022】
UL87ORF塩基配列(配列番号3):atggccggcgctgcgccgcgccgcctcggctgcgacgctctaatagtcgtcggcggctccgctacgccgcgccgggttttacacgtccccgtgcacgttcgcgcctgcaacctcacccaagagctatcgacgggcgaggacgcccgcttctgtcgtccgcgacccgttaacgtcgaacgggtacgcgctgttttcgcggctctctaccgtgcctgtcccgtacacgcgaggaccgagtccgagcgtgtcaagctggtactgggtcgtctgttgctgggacccgtggccgtaccctgtttttgcgacggtgaagtggagggccacggcgagcatctggtacccacgacgcagttttgtcgcgggccgctgctctacgtgcaccgacgttgttgttgcggatccgtgaccgccgggcgcgcgctgtcctaccacgttctcgaaaaccacgtggccacgcatgtgttacgcggattgctctcgctgacggaatggaatcgagagttgccgggccttttttgcgactgtcctggcagcggtggcgccttgggaaccgaggaacgctacgccatggcctgcttgccgcgcgacctcagcctgcacctggacgactatccttacctgatggtggaaatcggacgcgtactcagcgtcagcgaggtagacgactacgtaaccgccgtctccggctacctgggcgaggccgcggcgccgcgcattcaggttcactacaagctgctctttggactcaacgtgcgtccgcaagcgccgtgcgcgttggacgctacacgcgacttttttctgctggagctgcaaaagctttggctgggcgttgaatatcaccacgaagtcacgtcggagtttttcggtcgcgtactggctcagctgcatcgcgaccgcgcccgcgtcatgatggcacttcgcttgcccgagcagacggtgtgccacctaagcaccttcgttctcagtcgcttcaagcgacaggtactgtacttcaagttacaggtgagctacggcaagtgccggactggccacgctgacagaagtgggggagggggaaacggtggaagtcagggacaccacaacctactgtgttatcgacgtcttagcgtcacgtttgccgacacggacacggtgtggagaaaccttttctacgtttattatgaactagctcgggatctggggtcccatgggacagaggaccgatccgtaagccgcggttacggtgtttcttgcgctccgaggacgtcgcggctaccaccgtcagaaccgacggtggtttcagccaacggacacgcgctgtcttccaccgcgctcccgacgacgagcgcgggtcacaagctgtcgctgccgcgcgacccggccgcagatcgcgttcgacgttacgtgtgcattatctcgcgtctcatgttcgcccggtatggggagagatggcgtaaacaccgtcgacggcggtcggagacgggagaagaggaggaggaagagacggtggaatcgggggagactgacgccacgccgccatttgactttacggggcagcagctgcgccgggcctatcaggaacaccgacgtcgtaaacatctagccgtgcagcgttacgcgccgtgccgtcgtaagctcatcggcgggatggagtttgccgaggtgacgggcgttagtctggaccgcatcgccgtcaacgctttcaacaccaaccgcgttatcaatatgaaggccgcactctcgtccatcgccgcgtcgggtctcggcgtgcgcgcgccgcggcttcccaagaacatgacccacagttttgtgatgtacaagcacaccttcaaggagcccgcttgcaccgtcagcacttttgtttccaacgacgccgtctacatcaactcgctcaacgtcaatattcgcggttcctatcccgagtttctgtactcgctgggcgtgtaccggctgcacgttaatatcgatcacttctttctgccggccgtggtgtgcaacagcaactcctcgctggacgtgcatgggctggaggaccaggcggtgatccgctcggagcgcagcaaggtgtactggaccaccaactttccgtgcatgatctcgcatactaacaacgtcaacgtgggttggttcaaagcggctacggccattgtgccgcgcgtctcgggcgctgacctggaagccattctgctcaaagaactctcgtgcatcaagaacatgcgcgacgtgtgcatcgattacggtctgcatcgcgttttcacgcaactagagctgcgcaattcgtaccagatccccttcctggccaagcagttggtgctgtttctgcgtgcttgcctgctcaagctgcacggtcgagagaagcggctgcagttggaccgcctagtatttgaggcggcacagcggggtctctttgactacagcaagaacctcacggcgcacaccaagatcaagcacacttgcgcgctcatcggcagtcgtctagccaacaacgtgcccaagatcctggcccggaacaaaaaagtcaaattggatcacctgggccggaacgccaacgtgctgacggtgtgtcggcacgtggaagcccacaagatccctcgcacgcgcctcaaagtgttagtcgaggtgctgggcgcgttgcagagtatcagcggtacgccgcacacgcgtgaagtgatccaccagacgttgtttcgattgtgctcggcggccgcagccacctcgggcctgtgttcatcccctcccccattgtgtgtgtcctcatcttcctccgccccttctgtcccaacctccgtcagcgttgacggcagttctgaacccacgtcgccgcgagcgcggtttgcatcacgatga
【0023】
UL87ORFアミノ酸配列(配列番号4):MMEAAAAAAAAFRPEERPTPGWHDAALLMDDGTVREHAFRNGPLSQLIRRVLPPPPDAEDDVVFASELCFYCSGRFNRRSSVFSIYWQKHSDLVYALTGITHCAKLVVECGQLGSGRLRWRDGDVGGEERRGDDDSRDELYDVPGIYMIRVNDGGSTGPRHVIWPGTSVLWAPDVVITTVQRRISAARALVNTFRQYFFLLERRSHEELVLCPPEMEERLAPLLQSATRGDSDMFDGVVASAYHRLRISNIPRSSARLLEHCVGLAGAKKLLLLDVPRLENYFLCQVCLYELDEDEMGEEMLGMLAGKPEDAAVSGASGGFLLHRKTMKLAACLCLLLNSLHLHQEALEALDPPPPRVEENDLVNVVLRRYYRSHGGVQARTLAAARALLADYAETFSPLGSFTRLGYDRLVSADAGVSRRHLVALLRA
【0024】
UL95ORF塩基配列(配列番号5):tgatggcggcggcggtggtgcgagcggaggttaggcggcagcggcgagaggagaggaaaaagatggcggccgcgaggacgacggaggatccacccgaaaaccacgttgttgcggacgtggcttgtgggacgggcgccgtcactcgttcgtcttcgtcgtccctagtggtgtcgtcttcctcggcgtcaggctcggacgaaccttcctccgcctctcctctcagtttccccgtctgctccccctcaactgccgtcaggtctccggggtccgccggggtttcaacgtccctgtgctcggtggaacggatggtcgagctgtcggcgcagtctccggccgccgatttctcggtctccgaggcttggcgcttcgaggaggccgtaaatatggcgctggtggcctgcgaggccgtgtcaccttacgatcgctttcgcctaattgaaacgcccgacgagaatttcttgttggtcaccaacgtaattccgcgcgagtcggccgaggtgccggtgttggatagcagtagcagcggtggcgatagcgggccggaggacaaaaagaaaaacgtcgggaataaaaccgcgggggaaaagaacggcggtgggtctcgggccaaacgccgtcgtagacgacgcgctccgaaaaacgacgccgccacgccgtcttttctacgtcgacacgacgtgctggagcgtttcgcggccgcggctgagcctttgccgtcgctttgtgtgcgtgattatgcgttacgcaatgctgaccgtgttacctacgacggcgaattaatctacggcagttacctgttgtatcgcaaggctcacgtggagctgtcactctccagcaacaaggtgcaacacgtggaagccgtgctgcgacaggtgtacacgccgggcttgttagatcatcacaacgtgtgcgacgtggaggccctgctgtggctgctgtactgcggaccgcgtagcttttgcgcgcgtgacacctgtttcggtcgcgaaaaaaacggttgtcctttccccgcgttgttgcccaaactcttttacgaacccgtgcgggactatatgacctacatgaatctggctgagctgtacgtctttgtttggtatcgcggctacgaattccctgcgccgacgccgcaggcgacgacggcgggtagtggtggcggcggcggggccggcgcttgtgcggtcgagacgagcgcgtcagcaggccgggtcgatgacgccggcgacgaggtgcatttgcctttaaagcccgtctcgctggaccgtctcagagaggtgttgcaggcggtgcgcggccgcttctcggggcgcgaggtgcccgcctggccggcctcgtcgcgcacctgtttgttgtgcgcgctctacagtcagaaccgtctctgtttagatctcgcgcgtgacgaggcgcggaccgtgagttatagccccatcgttatccaagactgcgccgcggctgtcaccgacgtcactttgagccacatcttgcccggccagagcaccgtctcgcttttccccgtctaccacgtcggaaagttgctggacgccctctcgctgaacgacgcgggtctcatcacgttgaatctatga
【0025】
UL95ORFアミノ酸配列(配列番号6):MMAAAVVRAEVRRQRREERKKMAAARTTEDPPENHVVADVACGTGAVTRSSSSSLVVSSSSASGSDEPSSASPLSFPVCSPSTAVRSPGSAGVSTSLCSVERMVELSAQSPAADFSVSEAWRFEEAVNMALVACEAVSPYDRFRLIETPDENFLLVTNVIPRESAEVPVLDSSSSGGDSGPEDKKKNVGNKTAGEKNGGGSRAKRRRRRRAPKNDAATPSFLRRHDVLERFAAAAEPLPSLCVRDYALRNADRVTYDGELIYGSYLLYRKAHVELSLSSNKVQHVEAVLRQVYTPGLLDHHNVCDVEALLWLLYCGPRSFCARDTCFGREKNGCPFPALLPKLFYEPVRDYMTYMNLAELYVFVWYRGYEFPAPTPQATTAGSGGGGGAGACAVETSASAGRVDDAGDEVHLPLKPVSLDRLREVLQAVRGRFSGREVPAWPASSRTCLLCALYSQNRLCLDLARDEARTVSYSPIVIQDCAAAVTDVTLSHILPGQSTVSLFPVYHVGKLLDALSLNDAGLITLNL
【0026】
<弱毒化>
本発明では、HCMVのUL79、87又は95ORFを突然変異させることにより不活性化して、HCMVを弱毒化する。
【0027】
本明細書中で使用される「弱毒化」との用語は、ウイルスの毒性(すなわち、感染性及び病原性)を減弱させることを意味する。
【0028】
ORFの不活性化は、ORFから最終的に翻訳されるタンパク質の発現を阻害すること、又は機能が抑制されたタンパク質を発現させることを意味する。ORFを不活性化させるための突然変異は、例えばORF配列の一部若しくは全部を別のヌクレオチド配列によって置換すること、又はORFの内部での配列の欠失、付加若しくは挿入によって行なうことができる。好ましくは、ORFの内部への別の配列の挿入により、ORFを不活性化する。しかし、ORFの機能を阻害することができれば、突然変異の程度(例えば配列同一性の程度)、突然変異の方法などは限定されないものとする。
【0029】
UL79、87又は95ORFが不活性化されることにより、HCMVの複製が抑制される。本発明により、ORFへの突然変異の導入によって、組み換えHCMVの複製能が約1/10以下、1/50以下、好ましくは1/100以下に抑制される。
【0030】
本発明の組み換えHCMVは、HCMVのUL79、87又は95ORFの一部又は全部に突然変異を導入したゲノムDNAを、適切なベクターを利用して作製し、これを適切な宿主細胞、例えば上記のヒト線維芽細胞やヒト内皮細胞に感染させ、組み換えウイルス粒子を回収することを含む手順によって作製することができる。突然変異は、例えば部位特異的突然変異誘発法、相同組み換え法などの公知の方法によって導入することができる。部位特異的突然変異誘発法は、例えば、野生型HCMVのUL79、87又は95配列の一部又は全部に突然変異を導入し、それに隣接する野生型配列と同一の十分な長さの配列を配置したヌクレオチド配列を合成し、クローニングベクターに導入して大腸菌などの細菌中にサブクローニングすること、野生型HCMVゲノムDNAを含む人工染色体ベクター、例えば細菌人工染色体(BAC、PAC)を作製し、次いで、上記ヌクレオチド配列を人工染色体ベクターにアニーリングし、PCRを行い、目的DNAを増幅することなどを含む。
【0031】
具体的な方法として、例えば大腸菌内でバクテリオファージλ組み換えタンパク質exo、beta及びgamを発現するための迅速相同組み換え系(Ellis,H.M.ら(2001)Proc.Natl.Acad.Sci.USA,98:6742−6746)、HCMVのBAC(細菌人工染色体)DNA(Dunn,W.ら(2003)Proc.Natl.Acad.Sci.USA,100:14223−14228;Lee,E−C.ら(2001)Genomics,73:56−65;Borst,E−M.ら(1999)J.Virol.,73(10):8320−8329)などを使用して突然変異誘発を行なうことができる。
【0032】
組み換えのためのHCMVの二本鎖DNAは、カナマイシン耐性遺伝子などの薬剤耐性遺伝子と70bp程度の相同ウイルスDNA配列を含み、この耐性遺伝子には、34bpの最小FRT部位を連結させることができる(後述の実施例1参照)。UL79、87又は95ORFの突然変異体を作製するために、適切なプライマーを使用してPCRによる増幅を行なう。PCR条件は、例えば、94℃2分の変性を1サイクル、94℃15秒の変性、55℃30秒のアニーリング及び72℃1分の伸長を30サイクル、並びに72℃5分の伸長を1サイクルとするPCRサイクルプログラムからなる。PCR増幅産物をフェノール−クロロホルムで抽出し、95%エタノールで沈殿する。
【0033】
残存する鋳型DNAを除去するため、PCR産物を制限酵素を用いて消化したのち、得られたDNA断片を、HCMV−BAC DNAを含むコンピテント大腸菌へ、エレクトロポレーション、リポフェクション、リン酸カルシウム法などの手法、好ましくはエレクトロポレーションを用いて形質転換する。
【0034】
カナマイシン耐性遺伝子を欠失させるために、組み換えHCMV−BAC DNAを大腸菌に形質転換する。また、リコンビナーゼを発現するプラスミドpCP20(Hahn,W.ら(2003)Virology,307:164−177)を、組み換えHCMV−BAC DNAを含む大腸菌(DH10B)に形質転換する。カナマイシン耐性遺伝子を含まないHCMV BAC DNAを、アンピシリン及びクロラムフェニコール含有LBプレート上で選択する。
【0035】
HFF細胞などの細胞を、リン酸カルシウム沈殿法によって、プラスミドpSVpp71の存在中、上記の各組み換えBACでトランスフェクションする。約10日後にウイルスプラークが出現し、100%細胞変性作用(CPE)の7日後に、細胞外液を回収し、約1:10に希釈したものと未希釈のものを調製し、HFF細胞などの細胞に感染させ、組み換えウイルスを回収する。このとき、ウイルスの選択は、ORFに所望の突然変異が導入されていることを配列決定によって確認すること、及びHFF細胞などの細胞に導入又は感染させてウイルス複製が抑制されていることを確認し、また、野生型の表現型と同一又は本質的に同一であることを確認することによって行なうことができる。
【0036】
<ワクチン>
本発明はさらに、弱毒化HCMVを含むことを特徴とする、HCMV感染症を治療又は予防するためのワクチンを提供する。
【0037】
ワクチンは、一般に生ワクチンと不活性化ワクチンに大別される。特に生ワクチンは安価に製造可能であることや接種回数が少なくても高い効果が期待できるなどの利点を有するが、同時に毒性復帰にリスクを有する。本発明の弱毒化HCMVは、顕著にウイルス複製が抑制されたウイルスであるため、安全性の高い生ワクチンとして使用可能であると考えられる。
【0038】
本発明のワクチンは、AIDS患者、臓器移植患者などの免疫抑制患者、胎児、乳幼児、高齢者、重症患者などの抵抗性が低下したヒトに対し、肺炎、肝炎、網膜炎、胃腸炎などを含む広範な疾患を引き起こす可能性のあるHCMV感染の予防及び治療のために使用可能である。HCMVは、血液、尿、唾液などを介して感染するウイルスであり、上記のヒトに感染すると重症化する場合がある。そのような感染を抑制するために、本発明のワクチンは有効である。
【0039】
本発明のワクチンは、適切な賦形剤、特に希釈剤、例えば滅菌水、生理食塩水、リンゲル液、緩衝液などとともに処方することができる。また、ブドウ糖、トレハロース、エタノール、グリセリンなどの添加剤を含有させることができる。さらにまた、免疫応答を増強させるためのアジュバント、例えば水酸化アルミニウム(alum)、ムラミルジペプチド、サポニン類、リポ多糖、リポゾーム、消化性ポリマー(Gupta,R.K.et al.,Vaccine 13:1263−1276(1995))などをその有効量で含有させてもよい。さらにまた、安定化剤を添加することも可能であり、安定化剤としては、ゼラチン、ヒト血清アルブミンなどのタンパク質、ポリソルベートなどの界面活性剤、スクロース、トレハロースなどの糖類、メチオニン、リジンなどのアミノ酸類などが挙げられる。
【0040】
投与方法は、例えば皮下、筋肉内又は皮内注射、鼻腔内などの粘膜投与を含む。鼻腔内投与の場合、噴霧器による投与が望ましい。投与量は、例えば103〜1011PFUウイルス/kg体重、又は1回あたり10μg〜10mgの用量であるが、この範囲に制限されない。ワクチンの投与量は、患者などの被験体の状態、年齢、体重、性別、重症度などを考慮に入れて、医師の裁量で判断されるべきである。被験体は、1回又は2回以上の投薬を受容でき、複数回投与の場合、1週間〜1ヵ月以上の間隔、例えば2、3、5、12、18又は24ヵ月間隔で投与することができる。
【0041】
<抗体の作製>
本発明はさらに、上記弱毒化HCMVを非ヒト動物に免疫して抗体を生成させ、該抗体を回収することを含む、抗HCMV抗体の作製方法を提供する。
【0042】
抗体は、IgA、IgG、IgD、IgE、IgMのいずれのクラスでもよいし、またIgG1、IgG2、IgG3、IgG4、IgA1、IgA2などのいずれのサブクラスでもよい。さらにまた、抗体は、ポリクローナル抗体、モノクローナル抗体、抗体フラグメント、組み換え抗体のいずれでもよい。組み換え抗体には、例えば単鎖Fv抗体(scFv)、単鎖ダイアボディ(scDb)などが含まれる(浅野竜太郎、生化学、77(12):1497−1500(2005))。抗体フラグメントには、例えばFab、F(ab’)2、Fvなどがある。
【0043】
また、抗体には、ヒト抗体、ヒト化抗体、キメラ抗体なども包含され、好ましい抗体はヒト抗体又はヒト化抗体である。
抗体の作製は、慣用の方法で行なうことができる。
【0044】
ポリクローナル抗体は、弱毒化HCMV粒子を動物に免疫し、抗血清、さらにIgG画分を回収することによって作製できる。
【0045】
モノクローナル抗体は、弱毒化HCMV粒子を免疫した動物(例えばマウス、ラットなど)から脾臓を取り出し、脾臓細胞又はリンパ球とミエローマ細胞とを融合させ、該HCMV粒子抗原と特異的に免疫反応する抗体をスクリーニングすることによって作製できる。
【0046】
ヒト抗体は、ヒト抗体産生非ヒト動物(例えばKMマウス、XenoMouse)を用いて作製することができる。あるいは、弱毒化HCMV粒子を免疫した動物のゲノムから抗体遺伝子を回収し、重鎖可変領域及び軽鎖可変領域の各CDR1〜CDR3コード配列を決定し、ヒト抗体遺伝子の本来のCDR1〜CDR3コード配列を前記CDR1〜CDR3コード配列と置換することを含む遺伝子組み換え技術を用いてヒト化抗体を作製することができる。
上記の抗体は、ポリエチレングリコールなどの分子によって化学修飾することもできる。
【0047】
上記のようにして得られた抗体は、HCMV感染症の治療及び予防のために使用することができ、或いはHCMVの検出のために使用することもできる。ここで、HCMVの検出のために、抗原−抗体反応を利用する手法のいずれを使用することもできる。そのような手法には、例えば均質法、固相法(例えばサンドイッチ法)などが含まれる。
【実施例】
【0048】
以下に実施例を挙げて本発明をさらに具体的に説明するが、本発明の範囲は、該実施例によって制限されないものとする。
【0049】
実施例1
突然変異型若しくはエピトープタグ付けされたUL79、87、又は95ORFを有する組み換えウイルスの構築
ウイルスDNA複製及び後期mRNAに対するHCMV UL79、87、又は95の影響を調べるために、UL79、87又は95のORFを中断する突然変異を有する組み換えウイルスを構築した。
【0050】
<材料と方法>
細胞及びウイルス
初代ヒト包皮線維芽(HFF)細胞を、以前に報告されているように(43)、10%ウシ胎児血清(Sigma,St.Louis,Mo)、ペニシリン(100U/mL)及びストレプトマイシン(100μg/mL)を添加したイーグルの最小必須培地中で、37℃、5%CO2で維持した。ウイルスDNA複製を阻害するために、ホスホノ酢酸(PAA,Sigma,St.Louis,MO)(200μg/mL)、又は9−[(1,3−ジヒドロキシ−2−プロピル)メチル]グアニン(ガンシクロビル[GCV],Calbiochem,Darmstad,Germany)(450μM)を、感染時及び感染して維持する間、培地に添加した。
【0051】
HCMV UL79、87又は95ORFを発現するHFF細胞を作製するために、Nolan博士の研究室から入手したプロトコールに従いレトロウイルス系を使用した(http://www.stanford.edu/group/nolan/protocols/pro_helper_dep.html)。UL79及び95ORFは、それぞれ、XhoIUL79ORFF及びHindIIIUL79ORFR、並びにXhoIUL95ORFF及びHindIIIUL95ORFRのプライマーペアを用いて、HCMV TowneのBAC DNAからPCRにより増幅した。PCRプライマーの配列は表1に示してある。PCR産物を制限エンドヌクレアーゼXhoI及びHindIIIで消化し、pLBC(2)(スタンフォード大学Nolan博士の許可を得て、Fred Hunchinson Cancer Research CenterのKiem博士から供与された)にクローニングし、その結果、ORFのC末端にmcherry(京都大学医学系研究科、松田博士より供与された)が融合された。pLBCは、EBVゲノムのEBNA1 ORF及びOriP配列を有する、組み換えレトロウイルスの構築のためのシャトルベクターである。UL97ORF配列はATG開始コドンから815ヌクレオチド(n.t.)に制限エンドヌクレアーゼHindIII部位を含む。HindIII部位はpLBCへのORFのクローニングに必要である。UL87の1〜814n.t.の領域をまずプライマーXhoIUL87ORFF及びUL87HindIII(815)Rを用いるPCRにより増幅し、pLBCの対応する制限エンドヌクレアーゼ部位にクローニングし、DNA配列決定した(愛知県がんセンター中央研究所)。続いて、815〜2826n.t.の領域を、UL87(815)HindIIIF及びHindIIIUL87ORFRのプライマーペアを用いるPCRにより増幅し、UL87の1〜814n.t.の領域を有するpLBCのHindIII部位にクローニングし、DNA配列決定した。各ORFを含有するシャトルベクターをパッケージング細胞株であるPhoenix−GALV(17)(Nolan博士の許可を得て、Kiem博士から供与された)にトランスフェクションすることにより、レトロウイルスストックを調製した。HFF細胞を組み換えレトロウイルスに感染させ、ピューロマイシン選択下でpUL79−mcherry、pUL87−mcherry又はpUL95−mcherry発現細胞集団を作製した。
【0052】
野生型(wt)HCMV Towne、RHAUL95UL87myc、RUL95HAUL87myc、RdlUL79、RdlUL87、又はRdlUL95のウイルス力価は、HFF細胞又はpUL79、87若しくは95−mcherry発現HFF細胞に対する標準的なプラークアッセイにより、以前に記載されているように決定した(34)。ウイルス力価は、段階希釈で感染させた単層HFF細胞でのGFP蛍光フォーカスによっても決定した。感染後の種々の時点で細胞及び上清を回収し、3回の凍結融解サイクルに供した。ウイルス力価を、HFF細胞又はpUL79、87若しくは95−mcherry発現HFF細胞での50%組織培養感染用量(TCID50)アッセイにより決定した。TCID50を算出するために、Reed−Muench法を用いた。野生型ウイルス及び組み換えウイルスは、置き換え可能な10kb US1−US12領域(US:ユニークショート)をGFP遺伝子で置換されている。
【0053】
ウイルスDNA複製
3MOIでの感染後、2、3及び4d.p.i.で回収した。3個の35mmプレート中の細胞を、50μg/mLプロテアーゼKを含有する溶解バッファー(50mM Tris−HCl,pH8,10mM EDTA,1%SDS及び20μg/mL RNaseA)中に懸濁した。複製したウイルスDNAをHCMV gBプライマー及び以前に記載されたプローブ(20、23)を用いるリアルタイムPCRにより測定した。Applied Biosystems(Foster City,CA)から購入した18Sプライマー及びプローブを用いるリアルタイムPCRは、入力DNAの内部対照としても機能した。データは3回の独立した実験の平均である。
【0054】
HCMA BAC DNAの突然変異誘発
バクテリオファージλ組み換えタンパク質exo、beta及びgamを発現する大腸菌(NIHのCourt博士から供与された)での迅速相同組み換えシステムを、以前に記載されているように用いた(10)。HCMV Towne株のBAC DNAはLiu博士(カリフォルニア大学)から供与された(9)。ORFを欠損した組み換えHCMV BAC DNAの構築に際して、ORFの中央にFRT配列を挿入し、隣接する遺伝子への34bp配列の影響を最小限に抑えた。さらに、隣接遺伝子の調節領域が除去される可能性を除外するために、ヌクレオチドを欠失させることはしなかった。BACdlUL79Kan+FRT、BACdlUL87Kan+FRT及びBACdlUL95Kan+FRT(図1a参照)の組み換えHCMV BAC DNAを作製するために、組み換えのための二本鎖DNAを、プラスミドpACYC177(NEB)を鋳型として、BACdlUL79FRTFKanF及びBACdlUL79FRTRKanR、BACdlUL87FRTFKanF及びBACdlUL87FRTRKanR、並びにBACdlUL95FRTFKanF及びBACdlUL95FRTRKanRのプライマーペアをそれぞれ用いるPCRにより増幅した。プライマー配列は表2に示してある。
【0055】
BACUL95N neo+St及びBACUL95 neo+Stを作製するために、プラスミドpRpsL−neo(Gene Bridges,Dresden,Germany)を鋳型として、BACUL95Nneo+StF及びBACUL95Nneo+StR、並びにBACUL95Cneo+StF及びBACUL95Cneo+StRのプライマーペアをそれぞれ用いるPCRにより二本鎖DNAを増幅した。プライマー配列は表2に示してある。プラスミドpACYC177又はpRpsL−neo(Gene Bridges)は、カナマイシン耐性(KanR)又はKanR及びストレプトマイシン耐性遺伝子をそれぞれ含有する。組み換えのために増幅された二本鎖DNAは、最小FRT部位(5’−GAAGTTCCTATTCTCTAGAAAGTATAGGAACTTC−3’)(15)、又はRpsLneo遺伝子(Gene Bridges)に連結されたKanR遺伝子、及び70bpの相同ウイルスDNA配列を含有していた。DpnIでの37℃、1.5時間の消化の後に、PCR産物をゲル精製し、親HCMV BAC DNAを含有するDY−380に形質転換した。相同組み換え後、KanR及びFRT配列、又はRpsL−neo遺伝子を含有する突然変異型BAC DNAはカナマイシンに対して耐性であった(図1を参照)。
【0056】
FRT配列を含む突然変異型HCMV BAC DNAからKanR配列を切り出すために、FRTを介した組み換えを、以前に記載されているように用いた(23)。プラスミドpCP20(Max von Pettenkofer Institute,Munich,GemanyのHuhn博士から供与された)を組み換えHCMV BAC DNAを含むDH10Bに形質転換した。カナマイシンを含まないHCMV BAC DNAを、アンピシリン及びクロラムフェニコールを含有するLBプレート上で選択した。UL79、87又は95ORFの欠失を有する組み換えBAC DNAを選択するために、UL79detectF及びUL79detectR、UL87BACdetectF及びUL87BACdetectR、並びにUL95BACdetectF及びUL95BACdetectRのプライマーペアをそれぞれ用いてPCR分析を行なった。プライマー配列は表1に示してある。PCR産物をDNA配列決定し、組み換えを確認した(愛知県がんセンター中央研究所)。
【0057】
逆選択を、以前に記載されているように行なった(21、50)。RpsLはストレプトマイシン感受性遺伝子を付与するので、突然変異型BAC DNAを、Counter Selection Modification Kit(Gene Bridges)を用いて増大したストレプトマイシン耐性に基づいて選択した。BACHAUL95又はBACUL95HAを構築するために、オリゴヌクレオチドBAColigoHAUL95又はBACUL95Coligoを用いてそれぞれ逆選択した(図1b参照)。オリゴ配列は表2に示してある。
【0058】
組み換えBACHAUL95UL87myc又はBACUL95HAUL87myc(図1b参照)を構築するために、BACHAUL95又はBACUL95HAの構築後に、逆の手順を繰り返した。BACUL87Cneo+StF及びBACUL87Cneo+StRのプライマーペアを用いるPCRにより、二本鎖DNAを増幅し、BACHAUL95又はBACUL95HAを含有するDY−380に形質転換した。プライマー配列は表2に示してある。BACHAUL95neo+St又はBACUL95HAneo+Stを、カナマイシン含有プレート上で選択した。BACUL87oligoのオリゴヌクレオチドを用いる逆選択を行なった後、BACHAUL95UL87myc又はBACUL95HAUL87mycを上記のように増大したストレプトマイシン耐性に基づいて選択した(図1参照)。
【0059】
HA−UL95、UL95−HA及びUL87−mycを含む組み換えBAC DNAを選択するために、UL95NdetectF及びUL95NdetectR、UL95CdetectF及びUL95CdetectR、並びにUL87CdetectF及びUL87CdetectRのプライマーペアをそれぞれ用いてPCR分析を行なった。プライマー配列は表2に示してある。PCR産物は、組み換え及び切り出しを確認するために配列決定した(愛知県がんセンター中央研究所)。
【0060】
抗体
HA又はmcherryでタグ付けした融合タンパク質を検出するために、ラット又はウサギポリクローナル抗体(3F10,Roche又はPM005,MBL,名古屋)をそれぞれ用いた。mycエピトープを含む融合タンパク質を検出するために、マウスモノクローナル抗体9B11、又はウサギポリクローナル抗体7D10(Cell Signaling,Boston,MA)を用いた。IE1及びIE2タンパク質、IE2、UL44、UL57、TBP又は細胞内GAPDHを検出するための抗体は、NEA−9221(Perkin Elmer,Boston,MA)、12E2(Vancouver Biotech,Ltd.,Vancouver,CA)、CA006(EastCoast Bio,North Berwick,ME)、CH167(Santa Cruz, Santa Cruz,CA)、sc−273(Santa Cruz)、及びMAB374(Chemicon,Temucula,CA)をそれぞれ用いた。
【0061】
ウエスタンブロット分析
細胞を感染後の示した時点で回収し、PBSで洗浄し、以前に記載されているように溶解バッファーで処理した(19、20)。20μgのタンパク質のアリコートを、ドデシル硫酸ナトリウム(SDS)−10%ポリアクリルアミドゲル電気泳動(SDS−PAGE)のために各レーンにロードし、PVDFメンブレンにトランスファーした。増強化学発光検出試薬(GE Healthcare UK Ltd.,Buckinghamshire,UK)及び二次西洋ワサビペルオキシダーゼ標識抗マウス、ウサギ、又はラットIgG抗体(Zymed,San Francisco,CA)を製造業者の取扱説明書に従って用いた。
【0062】
【表1】
【0063】
【表2】
【0064】
<結果>
C末端に蛍光タンパク質mcherryをインフレームで融合させたUL79、87又は95を安定的に発現するHFF細胞を、組み換えレトロウイルスを用いて単離した。これらの細胞を、組み換えHCMVを増殖させるために用いた。組み換えBAC DNAを親BAC DNAから選択するために薬剤耐性遺伝子が必要であったので、34bpの最小FRT部位に連結されたKanRを有する組み換えBAC DNAをまず構築し、続いてFRT−FLP組み換えによりKanRを切り出した。図1aを参照されたい。隣接遺伝子への影響を最小限にするために、FRTはORFの中央に挿入した。
【0065】
本発明者らはまた、UL87又はUL95ORFのN末端又はC末端に融合されたHA又はmycエピトープを有する組み換えウイルスも構築した。以前に報告されているようにUL98遺伝子は必須であり(9、56)、UL95ORFはUL89と重複しているので、UL95のN末端へのHAエピトープの挿入はUL89タンパク質の機能に障害を与える可能性がある。したがって、本発明者らは、UL95ORFのN末端又はC末端に融合されたHAエピトープを有する組み換えウイルスを構築した。
【0066】
UL86は必須であり(10、59)、UL87とは逆方向に転写されるので、mycエピトープをUL87ORFのC末端に挿入した。UL87ORFのTGA停止コドンとUL88のATG開始コドンは重複しているので、mycエピトープはUL87ORFのTGA停止コドンの前に挿入した。ORFのN末端又はC末端にエピトープを挿入するために、逆の手順を行なった。図1bに示されているように、マーカーカセットをRpsL遺伝子(Gene Bridges)で置き換え、ストレプトマイシンに対する感受性を増大させた。中間体BACクローンは、カナマイシンに対する耐性に基づいて選択した。これらのクローンの完全性はHindIIIでの消化によりチェックし、正しい位置でのマーカーカセットの挿入はPCRにより確認した。相同組み換えの第2ラウンドでは、マーカーカセット全体をカウンター選択によりエピトープを含む配列と置き換えた。組み換え構築物を、以前に記載されているように(50)ストレプトマイシンに対する増大した耐性に基づいて単離し、エピトープが挿入された組み換えBACをPCRにより選択した。PCR分析により、組み換えBAC DNA中へのエピトープの挿入が確認された(図1c)。DNA配列決定により組み換えを確認し(データは示していない)、突然変異型BACの完全性をHindIIIによる消化によってチェックした(データは示していない)。3の高MOIで、これら組み換えウイルスの増殖を親ウイルス(wt)と比較した。RUL95HAUL87mycは感染4及び5日後(d.p.i.)で野生型と比較して1/2対数ほど遅く複製したが、7d.p.i.までに感染力価は同等となった。RHAUL95UL87mycは、4、5及び7d.p.i.で野生型と同等に複製した(図8)。
【0067】
実施例2
UL79、87及び95はHCMVの増殖には必要であるが、DNA複製には必要でない
HCMV UL79、87又は95タンパク質がウイルス増殖又はDNA複製に必要であるかを決定するために、補充ウイルスタンパク質を発現するか発現しないHFF細胞に組み換えウイルスを3MOIで感染させた。ウイルス力価を1、4、5、7及び9d.p.i.でアッセイし、同数の感染細胞からのDNAを2、3及び4d.p.i.で回収した。突然変異させたUL79、87又は95ORFを含む組み換えウイルスは、補充ウイルスタンパク質の発現なしではHFF細胞で感染ウイルスを産生しなかった(それぞれ、図2a、b及びc)。対照的に、ウイルスDNAは両方のHFF細胞で同等であった(図2d、e及びf)。これらの結果は、UL79、87及び95がウイルス増殖に必要とされるが、ウイルスDNA複製には必要でないことを示している。
【0068】
実施例3
UL79、87及び95タンパク質は真性後期(γ2)ウイルスmRNAに必要とされる
<材料と方法>
ノザンブロット分析
以前に記載されたように、感染後1、2及び3日で細胞質RNAを単離した(5、16)。20μgの細胞質RNAを2.2Mホルムアルデヒドを含有する1%アガロースゲルでの電気泳動に供し、最大強度のHybond N+(GE Healthcare UK Ltd.)にトランスファーした。IE1、UL75又はUL99DNAプローブを用いるノザンブロット分析を以前に記載されているように実施した(20、22、23)。放射標識プローブは32P−dCTPでの標識により以前に記載されているように作製した(23)。
【0069】
RNase保護アッセイ
32P標識アンチセンスUL44 RNAプローブを以前に記載されているように構築した(23)。以前に記載されているように(19、26)、20μgのRNAを32P標識アンチセンスUL44プロモータープローブに37℃で一晩ハイブリダイズさせ、その後、RNaseT1(100U)で消化した。保護されたRNA断片を変性6%ポリアクリルアミドゲルでの電気泳動に供し、続いてHyperfilmMP(GE Healthcare UK Ltd.)でのオートラジオグラフィーに供した。
【0070】
<結果>
UL79、87又は95タンパク質が真性後期(γ2)mRNAに必要とされるかを決定するために、補充ウイルスタンパク質を発現するか発現しないHFF細胞を、PAAの存在下又は非存在下で、組み換えウイルスに3MOIで感染させた。1、2及び3d.p.i.で細胞質RNAを回収し、IE1、UL75(gH)又はUL99(pp28)DNAプローブのいずれかを用いてノザンブロット分析によって分析した。IE1RNAは、予想される通り、組み換えウイルスを感染させた両方の細胞タイプで1d.p.i.に検出された(図3a〜c、左パネル)。
【0071】
UL75プローブを用いると、UL75、76、77及び78mRNAに対応する3種類のサイズのmRNAが検出された(図3a〜c、中央パネル)。これらのウイルスmRNAは以前に報告されているように異なる開始部位を有する(48)。UL75の転写方向はUL76、77及び78とは逆方向である(52)。初期/後期UL76〜78転写産物はPAAの存在下では著明に減少した(図3b及びc)。UL75転写産物は後期転写産物であり、PAAの存在下又は補充タンパク質の発現なしにはHFF細胞で検出されなかった(図3a〜c、中央パネル)。
【0072】
UL99ORFは一連の3’共同末端転写産物内のウイルスゲノムの複合領域に位置する(51)。すべての転写産物はUL99ORFの共通のポリアデニル化部位下流配列を用いる(51)。pp28テグメントタンパク質は1.6kbのmRNAから翻訳される(1)。以前に報告されているように(1)、UL99プローブを用いるノザンブロット分析は、PAAの存在下で、UL99プロモーターからの1.6kbのmRNAが検出されないこと、したがってUL99は真性後期遺伝子であることを示した(図3b及びc、右パネル)。以前の報告と合致して(51)、PAA処理RNAの分析から、この領域内の可能性があるORFのそれぞれの上流から開始されるmRNAが遅延初期転写産物を含むことが示された(図3b及びC、右パネル)。PAAの存在下、又は補充タンパク質の発現なしには、1.6kb mRNAはHFF細胞で検出されなかった(図3a〜c、右パネル)。これらのことから、HCMV UL79、87及び95は真性後期ウイルスmRNAのために必須であると結論づけられる。
【0073】
HCMV UL44転写ユニットは3つの異なる部位で開始され、それぞれは約50塩基により隔てられ、生産性感染時に別々に調節される。これらの開始部位のうち2つ、遠位及び近位が初期に活性であり、中央開始部位は後期まで不活性である(28)。後期開始部位からの発現はウイルスDNA複製に依存し、後期ウイルス遺伝子発現に影響を有する(22)。本発明者らは、UL44転写に対するUL79タンパク質の影響も調べた。異なる開始部位に由来するすべての転写産物を検出するために、RNase保護アッセイを行なった。以前の報告と合致して(21、22、28)、離れた開始部位から開始される3種類の主要な転写産物が補充細胞では検出された(図3d、レーン3、5及び7)。対照的に、中央の開始部位からの転写産物はRdlUL79感染HFF細胞では検出されず、他の2種類の初期転写産物は検出された(図3d、レーン2、4及び6)。これらのことから、UL44の中央の後期開始部位からの転写産物はUL79ウイルスタンパク質を必要とすることが結論づけられる。
【0074】
実施例4
UL87及び95ORFは初期ウイルスタンパク質である
HCMV ORF79、87及び95はMHV−68 ORF18、24及び34と機能の上で似通っているので、これらの発現時期及びHCMV UL44との共局在について調べた。図1bに示されるエピトープタグ付けされたUL87及びUL95ORFを含む組み換えウイルスを構築した。UL79ORFに融合したエピトープを含む組み換えウイルスは構築しなかった。
【0075】
UL87及びUL95融合タンパク質の発現動態を調べるために、PAA若しくはGCVの存在下又は非存在下で、HFF細胞をRUL95HAUL87myc又はRHAUL95UL87mycに3MOIで感染させた。感染細胞を1、2及び3d.p.i.で回収し、IE1若しくはUL44タンパク質に対する抗体、又はHA若しくはmycエピトープに対する抗体を用いるウエスタンブロット分析によりアッセイした。タグ付けされた融合タンパク質は、1〜3d.p.i.で、またPAA若しくはGCVの存在下又は非存在下で2d.p.i.で検出された(図4A及びB)。UL87及びUL95融合タンパク質の発現動態は、UL44遺伝子の初期ウイルスタンパク質と似通っていた(図4C)。これらの結果は、UL87及びUL95が感染の初期に発現されることを示している。
【0076】
実施例5
UL79、87及び95ウイルスタンパク質は複製コンパートメント(RC)に動員される
<材料と方法>
共焦点顕微鏡観察前のタンパク質のin situ抽出
24ウェルプレート中のコンフルエントのHFF細胞に3MOIで組み換えウイルスを感染させた。以前に記載されているように(55)、感染後の種々の時点で、カバーガラス上で増殖した細胞を50μg/mLのジチオビス(スクシンイミジルプロピオネート)(DSP、Thermo,Rockford,IL)(ホモ二官能性NHSエステル架橋剤)で4℃にて10分間固定した。マトリックスに結合していないタンパク質を核又は細胞質から除去するために、細胞をTNEバッファー(10mM Tris−HCl,pH7.8,1%NP40及び0.15M NaCl)で処理し、2%パラホルムアルデヒドで4℃にて60分間固定した。
【0077】
顕微鏡観察
細胞を高塩濃度TPBS(0.001Mリン酸ナトリウム[pH7.3],0.5M NaCl,0.1%Tween20,及び0.1%TritonX−100)で3回洗浄し、示した一次抗体と共に4℃にて一晩インキュベートした。室温でのTPBSでの3回の洗浄後、細胞をAlexaFluor594若しくは680標識二次抗マウスIgG(Molecular Probes)、AlexaFluor594若しくは680標識抗ウサギIgG(Molecular Probes)、又はAlexaFluor594標識ラットIgG抗体(Molecular Probes)と共に37℃で30分間インキュベートした。感染細胞においてHA又はmyc融合タンパク質を検出する場合には、Signal immunostain(TOYOBO,大阪)を用いて特異的シグナルの強度を増強した。スライドを、4’,6’−ジアミジノ−2−フェニルインドール(DAPI)を含むProLong Goldアンチフェード試薬(Molecular Probes)でマウントした。画像取得は、PlainApo63又は100×1.4NA油浸対物レンズを装着したCarl Zeiss Meta共焦点レーザー走査顕微鏡(Carl Zeiss MicroImaging,Inc.)を用いて行なった。LSM510Meta共焦点顕微鏡にはAlexaFluor594及び680蛍光団を励起するためのHeNeレーザーを装着した。細胞をいずれかの抗原に関して染色した場合、まずタンパク質の局在を調べ、次に同じ条件で別のフィルターを用いて免疫蛍光が検出されないことを確認した。
【0078】
<結果>
HCMV複製タンパク質UL44は約2d.p.i.でRCに局在化する。BrdUをこれらのコンパートメント中のウイルスDNAに取り込ませると、子孫ウイルスで検出される(36、38)。本発明者らは、RdlUL79、RdlUL87又はRdlUL95での感染後の細胞内でその局在を調べるために、HFF細胞で安定に発現させたmcherry蛍光融合ウイルスタンパク質を用いた。3MOIでの感染2日後に細胞を固定し、UL44に対する抗体とインキュベートした。組み換えウイルスはGFPタンパク質を含有しているので、AlexaFluor680標識二次抗マウスIgGを用いてUL44の細胞内局在を検出した。図5A、a〜cに示されているように、UL79、87及び95−mcherry融合ウイルスタンパク質はUL44タンパク質と核で共局在していた。これらの結果は、mcherryと融合させた補充タンパク質UL79、87又は95が、それぞれRdlUL79、RdlUL87又はRdlUL95での組み換えウイルス感染後にRCに動員されることを示している。
【0079】
実施例6
組み換えウイルスから発現させたUL87及び95タンパク質は複製コンパートメントに動員される
ウイルスから発現させたUL87及び95タンパク質も互いに共局在することを示すために、HFF細胞を3MOIでRUL95HAUL87mycに感染させた。1〜3d.p.i.において、細胞を固定し、HAエピトープに対するラットポリクローナル抗体またはmycエピトープに対するマウスモノクローナル抗体と共にインキュベートした。AlexaFluor594標識二次抗ラットIgG及びAlexaFluor680標識抗マウスIgGをそれぞれ用いた。共焦点顕微鏡観察は、1、2及び3d.p.i.でのUL87融合タンパク質とUL95融合タンパク質との共局在を証明した(図5B、a〜c)。
【0080】
共焦点顕微鏡観察はまた、2d.p.i.でのUL95融合タンパク質とUL44タンパク質との共局在も実証した(図5d)。さらに、UL44タンパク質は新規に合成されたウイルスDNAとも共局在していた(図5e)。これらの結果は、UL87及びUL95ウイルスタンパク質が共局在し、UL95がRCに動員されたことを示している。
【0081】
実施例7
UL87及びUL95タンパク質はウイルスDNA複製前にUL44の微小フォーカスに動員される
<材料及び方法>
FISHアッセイ
HCMV BAC DNAを、ニックトランスレーションキット(Abbott Molecular,Abbott Park,Illinois)を製造業者の取扱説明書に従って用いてSpectrumOrangeで標識した。FISH分析は多少の改変を加えて、基本的に以前に記載された方法に基づいて行なった(24)。カバーガラス上で増殖させた細胞を、in situ細胞抽出後に2%パラホルムアルデヒドで4℃にて60分間固定し、RNase(100μg/mL)を含有するブロッキングバッファー(2.5%BSA,0.2Mグリシン,及び0.1%TritonX−100)で1時間透過化し、その後ハイブリダイゼーションに用いた。各カバーガラスを、5μLのハイブリダイゼーションバッファー(2×SSC,10%硫酸デキストラン及び50%ホルムアミド)及びDNAプローブと共にスライドガラス上に逆にふせて置いた。ハイブリダイゼーションバッグ中に密封した後、スライドを85℃の水浴中に5分間浸漬し、続いて37℃で一晩インキュベートした。FISH分析後、抗UL44抗体を用いてUL44抗原を検出した。カバーガラスを、50%ホルムアミドを含有する2×SSCで50℃にて3分間ずつ3回洗浄し、2×SSCで50℃にて3分間ずつ3回洗浄し、0.1×SSCで60℃にて10分間、1回洗浄した。SpectrumOrange蛍光団は560nmの吸収極大と588nmの蛍光発光極大を有し、HeNeレーザーで励起した(http://www.zeiss.de/C12567BE0045ACF1/Contents−Frame/5CDD62C92B112413C1256A140040C831)。
【0082】
<結果>
UL87及びUL95がウイルスDNA複製前にUL44と共局在するかを調べるために、PAA又はGCVの存在下でHFF細胞を3MOIでRUL95HAUL87mycに感染させた。細胞を固定し、HA又はmycエピトープに対するポリクローナル抗体、及びUL44タンパク質に対するモノクローナル抗体で処理した。AlexaFluor594標識二次抗ラット又はウサギIgG及びAlexaFluor680標識抗マウスIgGを用いた。ウイルスDNAを検出するためのFISH分析後に、細胞をUL44タンパク質に対するモノクローナル抗体とインキュベートし、続いてAlexaFluor680標識二次抗マウスIgGとインキュベートした。予想された通り、UL44はウイルスDNA合成阻害剤の存在下でウイルスDNAと共局在した(図6a及びb)。UL87及びUL95はPAA又はGCVの存在下でUL44と共局在した(図6c〜f)。これらの結果は、UL87及び95タンパク質がウイルスDNA複製前にUL44と共局在することを示している。
【0083】
UL87とUL44との共局在の特異性を調べるために、UL57及びUL44の局在を調べた。HCMVでは、UL44タンパク質はウイルスDNA合成前にはUL57タンパク質含有小フォーカスには伴われないことが報告されている(36)。細胞を固定し、UL87と融合したmycエピトープに対する抗体及びUL57タンパク質に対する抗体で処理した。AlexaFluor594標識二次抗ウサギIgG又はAlexaFluor680標識抗マウスIgGをそれぞれ用いた。以前に報告されている通り(38)、PAAの存在下では、UL87融合タンパク質はUL57タンパク質と共局在しなかった(図6g)。UL57タンパク質はウイルスDNA複製開始後にはUL44タンパク質と共局在するようである。併せて考えると、HCMV UL87及び95タンパク質は、ウイルスDNA複製前にはUL44と共局在する。
【0084】
実施例8
IE2タンパク質はウイルスDNA複製前にUL87と共局在する
Sourvinosらによるin situハイブリダイゼーション分析(42)により、IE2タンパク質は12時間p.i.で直接的なDNAとの接触により親ウイルスゲノムに伴われていることが示されている。IEタンパク質は点状構造を形成することができ、これがRCへと発達する。IE2はPAAの存在下でウイルスDNA複製と関連するため、本発明者らは、HCMV後期トランス因子がウイルスDNA複製前にIE2と共局在するかを調べた。細胞を3MOIでRUL95HAUL87mycに感染させ、固定し、IE2、myc、UL44及びTBPに対する抗体で処理した。AlexaFluor594標識抗マウスIgG及びAlexaFluor680標識抗ウサギIgGの二次抗体を用いた。UL87タンパク質はPAA若しくはGCVの存在下又は非存在下でIE2フォーカスと共局在した(図7a〜c)。TATA結合タンパク質(TBP)はIE2タンパク質と直接的に相互作用するので(14)、TBPに対するポリクローナル抗体及びUL44タンパク質に対するモノクローナル抗体を用いて、PAA若しくはGCVの存在下又は非存在下でTBPがUL44微小フォーカスに動員されるかを調べた。図7d〜fに示される通り、PAA若しくはGCVの存在下又は非存在下で、TBPはUL44と共局在し、したがって、予想されたように、RC前駆体に動員された。これらの結果から、UL87タンパク質は、IE2タンパク質及びTBPと同様に、ウイルスDNA複製前にウイルスRC前駆体に動員されると結論づけられる。
【0085】
<考察>
MHV−68のUL18、24及び34は後期転写のためのウイルストランス作用因子である。これらのMHV−68ORFは初期に発現されるが、ウイルスDNA複製には必要とされない。これらのMHV−68ウイルスタンパク質はインジケーター遺伝子の発現を駆動する後期ウイルスプロモーターを活性化させるので、後期ウイルストランス活性化因子であると考えられている。後期ウイルストランス活性化因子がどのようにして後期ウイルスプロモーターを特異的に活性化するのかは未だ明らかになっていない。
【0086】
後期ウイルスプロモーターの活性化についての理解は、ユニークな抗ウイルス剤のための新規なアプローチに寄与するものと思われる。HCMVは深刻な病原ウイルスであり、現在利用可能な抗ウイルス剤は限定された有効性及び副作用を有するので、HCMV後期転写因子の解析は新規な抗ウイルス剤の開発のために重要であろう。したがって、本発明者らは、MHV−68後期ウイルストランス活性化因子に相同なHCMV遺伝子が真性後期(γ2)HCMV遺伝子の転写に影響を及ぼすかを調べた。
【0087】
HCMVでは、プロモーターは単純であり、それらは上流又は下流配列を必要としないコアエレメントを含む(8、25、31)。UL44初期ウイルスプロモーターはカノニカルなTATA配列を有する。対照的に、UL44後期ウイルスプロモーターはノンカノニカルなTATA配列を有する(図3dを参照されたい)。本発明者らは、このノンカノニカルなTATA配列が後期ウイルス遺伝子発現に重要であることを報告している(21)。Serioら(39)もまた、EBVのBcLF1後期プロモーターからの発現が変異型TATAエレメントのみに依存することを報告している。ノンカノニカルなTATA配列へのTBPの弱い結合親和性は、感染後の初期での転写の欠如を部分的に説明するものかもしれない。TBPの後期プロモーターへの結合親和性が、後期遺伝子特異的トランス活性化因子の動員及びウイルスDNA複製中の二重らせんの開放後に強まる可能性はある。これらの後期遺伝子トランス活性化因子が、IE2トランス活性化因子と共に、TBP及びRNAポリメラーゼIIの後期プロモーターへの結合を安定化させるということも考えられる。HCMVでは、初期/後期ウイルス遺伝子発現に影響を与えるIE2遺伝子にはいくつかのアイソマーがある。感染初期には、ウイルスIE2−p86タンパク質が初期ウイルス遺伝子発現に必須である。感染後期には、IE2アイソマーであるIE2−p60及びIE2−p40が後期ウイルス遺伝子発現に多大な影響を及ぼす(49、50)。ウイルスDNAとウイルスIE2トランス活性化因子との複合体は、ウイルスDNAが複製し始める時点で検出され、RCの発達を引き起こす(42)。本発明者らはHCMV UL87がウイルスRCでIE2タンパク質と共局在することを示したが、現状ではこれら2つのウイルスタンパク質が相互作用するという証拠はない。
【0088】
なぜ後期ウイルス遺伝子の転写がウイルスDNA複製を必要とするのかは分かっていない。本発明者らのデータは、HCMV UL79、87及び95がUL75(gH)及びUL99(pp28)後期mRNAに必要であることを示している。これらのウイルスタンパク質はウイルスDNA複製の前に発現され、ウイルスRCに組み込まれる。RCは、部分的にはウイルスDNA鋳型を濃縮することにより、ウイルス遺伝子発現のためのフォーカスとして機能する。UL44タンパク質は、ウイルスDNA複製前にIE蓄積部位に組み込まれた。IE2タンパク質は後期遺伝子転写に十分ではないので、UL44がUL79、87及び95と共にIE2フォーカスに動員されることにより後期遺伝子転写に適した環境を提供したとは考えにくい。UL44タンパク質がUL79、87及び95のタンパク質と直接的に相互作用するという証拠はない。UL44は構造的に真核生物の増殖細胞細胞核抗原(Proliferating Cell Nuclear Antigen;PCNA)と相同であり、PCNAは複数のタンパク質と相互作用する(30)。近年のプロテオーム解析により、UL44タンパク質がウイルス複製関連タンパク質以外の転写因子を含む広範なタンパク質と相互作用することが実証された(13、44〜46)。したがって、UL44タンパク質が後期遺伝子転写のタイミングを最適化していると考えられる。HCMV RCはまた、ヘルペスウイルス複製タンパク質(すなわち、HSV ICP8、UL9、及びヘテロ三量体ヘリカーゼ−プライマーゼ複合体UL5−UL8−UL52)に相同な5種類のウイルスタンパク質も含有する。これらのヘルペスウイルス複製タンパク質がまずウイルスの複製前駆体微小フォーカスに組み込まれ、続いてこれらのフォーカスにポリメラーゼ及びポリメラーゼアクセサリータンパク質が動員される(4)。しかしながら、培地にPAAを添加した場合、HCMV UL87タンパク質はSSB UL57タンパク質と共局在しなかった。HCMVでは、HCMV UL57の微小フォーカスは後期遺伝子転写を実現するための足場としては機能しないように思われるが、ウイルスDNA複製の開始後には重要な役割を果たすようである。
【0089】
本願では、UL79、87及び95タンパク質が後期ウイルスmRNAに必要であり、したがってウイルス増殖に必要とされることが示された。UL79、87若しくは95 ORFに突然変異を導入することによりこれらのORFの発現を低下又は消失させることによって、ウイルス複製を抑制し、弱毒化したHCMVは、ワクチンとして使用する上で、天然ウイルスの構造を保持するという利点を有する。
【産業上の利用可能性】
【0090】
本発明により、HCMVを治療又は予防するためのワクチンが提供される。
参考文献
1. Adam, B. L., T. Y. Jervey, C. P. Kohler, G. L. Wright, Jr., J. A. Nelson, and R. M. Stenberg. 1995. The human cytomegalovirus UL98 gene transcription unit overlaps with the pp28 true late gene (UL99) and encodes a 58-kilodalton early protein. J. Virol. 69:5304-5310.
2. Akatsuka, Y., T. A. Goldberg, E. Kondo, E. G. Martin, Y. Obata, Y. Morishima, T. Takahashi, and J. A. Hansen. 2002. Efficient cloning and expression of HLA class I cDNA in human B-lymphoblastoid cell lines. Tissue Antigens 59:502-511.
3. Arumugaswami, V., T. T. Wu, D. Martinez-Guzman, Q. Jia, H. Deng, N. Reyes, and R. Sun. 2006. ORF18 is a transfactor that is essential for late gene transcription of a gammaherpesvirus. J. Virol. 80:9730-9740.
4. Carrington-Lawrence, S. D., and S. K. Weller. 2003. Recruitment of polymerase to herpes simplex virus type 1 replication foci in cells expressing mutant primase (UL52) proteins. J. Virol. 77:4237-4247.
5. Chang, C.-P., C. L. Malone, and M. F. Stinski. 1989. A human cytomegalovirus early gene has three inducible promoters that are regulated differentially at various times after infection. J. Virol. 63:281-290.
6. de Bruyn Kops, A., and D. M. Knipe. 1988. Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell 55:857-868.
7. de Bruyn Kops, A., and D. M. Knipe. 1994. Preexisting nuclear architecture defines the intranuclear location of herpesvirus DNA replication structures. J. Virol. 68:3512-3526.
8. Depto, A. S., and R. M. Stenberg. 1992. Functional analysis of the true late human cytomegalovirus pp28 upstream promoter: cis-acting elements and viral trans-acting proteins necessary for promoter activation. J. Virol. 66:3241-3246.
9. Dunn, W., C. Chou, H. Li, R. Hai, D. Patterson, V. Stolc, H. Zhu, and F. Liu. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. U. S. A. 100:14223-14228.
10. Ellis, H. M., D. Yu, T. DiTizio, and D. L. Court. 2001. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. U. S. A. 98:6742-6746.
11. Fish, K. N., W. Britt, and J. A. Nelson. 1996. A novel mechanism for persistence of human cytomegalovirus in macrophages. J. Virol. 70:1855-1862.
12. Fish, K. N., A. S. Depto, A. V. Moses, W. Britt, and J. A. Nelson. 1995. Growth kinetics of human cytomegalovirus are altered in monocyte-derived macrophages. J. Virol. 69:3737-3743.
13. Gao, Y., K. Colletti, and G. S. Pari. 2008. Identification of human cytomegalovirus UL84 virus- and cell-encoded binding partners by using proteomics analysis. J. Virol. 82:96-104.
14. Hagemeier, C., S. Walker, R. Caswell, T. Kouzarides, and J. Sinclair. 1992. The human cytomegalovirus 80-kilodalton but not the 72-kilodalton immediate-early protein transactivates heterologous promoters in a TATA box-dependent mechanism and interacts directly with TFIID. J. Virol. 66:4452-4456.
15. Hahn, G., D. Rose, M. Wagner, S. Rhiel, and M. A. McVoy. 2003. Cloning of the genomes of human cytomegalovirus strains Toledo, TownevarRIT3, and Towne long as BACs and site-directed mutagenesis using a PCR-based technique. Virology 307:164-177.
16. Hermiston, T. W., C. L. Malone, P. R. Witte, and M. F. Stinski. 1987. Identification and characterization of the human cytomegalovirus immediate-early region 2 gene that stimulates gene expression from an inducible promoter. J. Virol. 61:3214-3221.
17. Horn, P. A., M. S. Topp, J. C. Morris, S. R. Riddell, and H. P. Kiem. 2002. Highly efficient gene transfer into baboon marrow repopulating cells using GALV-pseudotype oncoretroviral vectors produced by human packaging cells. Blood 100:3960-3967.
18. Ibanez, C. E., R. Schrier, P. Ghazal, C. Wiley, and J. A. Nelson. 1991. Human cytomegalovirus productively infects primary differentiated macrophages. J. Virol. 65:6581-6588.
19. Isomura, H., and M. F. Stinski. 2003. The human cytomegalovirus major immediate-early enhancer determines the efficiency of immediate-early gene transcription and viral replication in permissive cells at low multiplicity of infection. J. Virol. 77:3602-3614.
20. Isomura, H., M. F. Stinski, A. Kudoh, T. Daikoku, N. Shirata, and T. Tsurumi. 2005. Two Sp1/Sp3 binding sites in the major immediate-early proximal enhancer of human cytomegalovirus have a significant role in viral replication. J. Virol. 79:9597-9607.
21. Isomura, H., M. F. Stinski, A. Kudoh, T. Murata, S. Nakayama, Y. Sato, S. Iwahori, and T. Tsurumi. 2008. Noncanonical TATA sequence in the UL44 late promoter of human cytomegalovirus is required for the accumulation of late viral transcripts. J. Virol. 82:1638-1646.
22. Isomura, H., M. F. Stinski, A. Kudoh, S. Nakayama, S. Iwahori, Y. Sato, and T. Tsurumi. 2007. The late promoter of the human cytomegalovirus viral DNA polymerase processivity factor has an impact on delayed early and late viral gene products but not on viral DNA synthesis. J. Virol. 81:6197-6206.
23. Isomura, H., T. Tsurumi, and M. F. Stinski. 2004. Role of the proximal enhancer of the major immediate-early promoter in human cytomegalovirus replication. J. Virol. 78:12788-12799.
24. Kanda, T., M. Kamiya, S. Maruo, D. Iwakiri, and K. Takada. 2007. Symmetrical localization of extrachromosomally replicating viral genomes on sister chromatids. J. Cell Sci. 120:1529-1539.
25. Kohler, C. P., J. A. Kerry, M. Carter, V. P. Muzithras, T. R. Jones, and R. M. Stenberg. 1994. Use of recombinant virus to assess human cytomegalovirus early and late promoters in the context of the viral genome. J. Virol. 68:6589-6597.
26. Lashmit, P. E., M. F. Stinski, E. A. Murphy, and G. C. Bullock. 1998. A cis-repression sequence adjacent to the transcription start site of the human cytomegalovirus US3 gene is required to down regulate gene expression at early and late times after infection. J. Virol. 72:9575-9584.
27. Lathey, J. L., and S. A. Spector. 1991. Unrestricted replication of human cytomegalovirus in hydrocortisone-treated macrophages. J. Virol. 65:6371-6375. 28. Leach, F. S., and E. S. Mocarski. 1989. Regulation of cytomegalovirus late-gene expression: differential use of three start sites in the transcriptional activation of ICP36 gene expression. J. Virol. 63:1783-1791.
29. Lukonis, C. J., and S. K. Weller. 1996. Characterization of nuclear structures in cells infected with herpes simplex virus type 1 in the absence of viral DNA replication. J. Virol. 70:1751-1758.
30. Maga, G., and U. Hubscher. 2003. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J. Cell Sci. 116:3051-3060.
31. McWatters, B. J., R. M. Stenberg, and J. A. Kerry. 2002. Characterization of the human cytomegalovirus UL75 (glycoprotein H) late gene promoter. Virology. 303:309-316.
32. Meier, J. L., M. J. Keller, and J. J. McCoy. 2002. Requirement of multiple cis-acting elements in the human cytomegalovirus major immediate-early distal enhancer for viral gene expression and replication. J. Virol. 76:313-326.
33. Meier, J. L., and J. A. Pruessner. 2000. The human cytomegalovirus major immediate-early distal enhancer region is required for efficient viral replication and immediate-early gene expression. J. Virol. 74:1602-1613.
34. Meier, J. L., and M. F. Stinski. 1997. Effect of a modulator deletion on transcription of the human cytomegalovirus major immediate-early genes in infected undifferentiated and differentiated cells. J. Virol. 71:1246-1255.
35. Pari, G. S., and D. G. Anders. 1993. Eleven loci encoding trans-acting factors are required for transient complementation of human cytomegalovirus oriLyt-dependent DNA replication. J. Virol. 67:6979-6988.
36. Penfold, M. E., and E. S. Mocarski. 1997. Formation of cytomegalovirus DNA replication compartments defined by localization of viral proteins and DNA synthesis. Virology 239:46-61.
37. Quinlan, M. P., L. B. Chen, and D. M. Knipe. 1984. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell 36:857-868.
38. Sarisky, R. T., and G. S. Hayward. 1996. Evidence that the UL84 gene product of human cytomegalvorus is essential for promoting oriLyt-dependent DNA replication and formation of replication compartments in cotransfection assays. J. Virol. 70:7398-7413.
39. Serio, T. R., N. Cahill, M. E. Prout, and G. Miller. 1998. A functionally distinct TATA box required for late progression through the Epstein-Barr virus life cycle. J. Virol. 72:8338-8343.
40. Sinzger, C., A. Grefte, B. Plachter, A. S. H. Gouw, T. Hauw The, and G. Jahn. 1995. Fibroblasts, epithelial cells, endothelial cells, and smooth muscle cells are the major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J. Gen. Virol. 76:741-750.
41. Sinzger, C., B. Plachter, A. Grefte, A. S. H. Gouw, T. H. The, and G. Jahn. 1996. Tissue macrophages are infected by human cytomegalovirus. J. Infect. Dis. 173:240-245.
42. Sourvinos, G., N. Tavalai, A. Berndt, D. A. Spandidos, and T. Stamminger. 2007. Recruitment of human cytomegalovirus immediate-early 2 protein onto parental viral genomes in association with ND10 in live-infected cells. J. Virol. 81:10123-10136.
43. Stinski, M. F. 1978. Sequence of protein synthesis in cells infected by human cytomegalovirus: early and late virus-induced polypeptides. J. Virol. 26:686-701.
44. Strang, B. L., S. Boulant, and D. M. Coen. 2010. Nucleolin associates with the human cytomegalovirus DNA polymerase accessory subunit UL44 and is necessary for efficient viral replication. J. Virol. 84:1771-1784.
45. Strang, B. L., A. P. Geballe, and D. M. Coen. 2010. Association of human cytomegalovirus proteins IRS1 and TRS1 with the viral DNA polymerase accessory subunit UL44. J. Gen. Virol. 91:2167-2175.
46. Strang, B. L., E. Sinigalia, L. A. Silva, D. M. Coen, and A. Loregian. 2009. Analysis of the association of the human cytomegalovirus DNA polymerase subunit UL44 with the viral DNA replication factor UL84. J. Virol. 83:7581-7589.
47. Taylor-Wiedeman, J., J. G. Sissons, L. K. Borysiewicz, and J. H. Sinclair. 1991. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 72:2059-2064.
48. Wang, S. K., C. Y. Duh, and C. W. Wu. 2004. Human cytomegalovirus UL76 encodes a novel virion-associated protein that is able to inhibit viral replication. J. Virol. 78:9750-9762.
49. White, E. A., C. L. Clark, V. Sanchez, and D. H. Spector. 2004. Small internal deletions in the human cytomegalovirus IE2 gene result in nonviable recombinant viruses with differential defects in viral gene expression. J. Virol. 78:1817-1830.
50. White, E. A., C. J. Del Rosario, R. L. Sanders, and D. H. Spector. 2007. The IE2 60-kilodalton and 40-kilodalton proteins are dispensable for human cytomegalovirus replication but are required for efficient delayed early and late gene expression and production of infectious virus. J. Virol. 81:2573-2583.
51. Wing, B. A., and E. S. Huang. 1995. Analysis and mapping of a family of 3'-coterminal transcripts containing coding sequences for human cytomegalovirus open reading frames UL93 through UL99. J. Virol. 69:1521-1531.
52. Winkler, M., S. A. Rice, and T. Stamminger. 1994. UL69 of human cytomegalovirus, an open reading frame with homology to ICP27 of herpes simplex virus, encodes a transactivator of gene expression. J. Virol. 68:3943-3954.
53. Wong, E., T. T. Wu, N. Reyes, H. Deng, and R. Sun. 2007. Murine gammaherpesvirus 68 open reading frame 24 is required for late gene expression after DNA replication. J. Virol. 81:6761-6764.
54. Wu, T. T., T. Park, H. Kim, T. Tran, L. Tong, D. Martinez-Guzman, N. Reyes, H. Deng, and R. Sun. 2009. ORF30 and ORF34 are essential for expression of late genes in murine gammaherpesvirus 68. J. Virol. 83:2265-2273.
55. Xiang, C. C., E. Mezey, M. Chen, S. Key, L. Ma, and M. J. Brownstein. 2004. Using DSP, a reversible cross-linker, to fix tissue sections for immunostaining, microdissection and expression profiling. Nucleic Acids Res. 32:e185.
56. Yu, D., M. C. Silva, and T. Shenk. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. U. S. A. 100:12396-12401.
57. Zhong, L., and G. S. Hayward. 1997. Assembly of complete, functionally active herpes simplex virus DNA replication compartments and recruitment of associated viral and cellular proteins in transient cotransfection assays. J. Virol. 71:3146-3160.
【特許請求の範囲】
【請求項1】
ヒトサイトメガロウイルス(HCMV)のUL79、87又は95遺伝子オープンリーディングフレーム(ORF)中に突然変異を生じさせることにより、これらの遺伝子の1つ若しくは複数からの発現が低下又は消失している、弱毒HCMV。
【請求項2】
前記突然変異が、ORF中への塩基の挿入によるものである、請求項1に記載の弱毒HCMV。
【請求項3】
前記弱毒HCMVからの感染性ウイルス増殖は野生型ウイルスと比較して低下しているが、ウイルスDNA複製は野生型ウイルスと同等である、請求項1又は2に記載の弱毒HCMV。
【請求項4】
後期ウイルスmRNAの発現が野生型ウイルスと比較して低下している、請求項1〜3のいずれか1項に記載の弱毒HCMV。
【請求項5】
請求項1〜4のいずれか1項に記載の弱毒HCMVを有効成分として含む、HCMV感染の予防又は治療用ワクチン。
【請求項6】
アジュバントをさらに含む、請求項5に記載のワクチン。
【請求項7】
請求項1〜4のいずれか1項に記載の弱毒HCMV又は請求項5若しくは6に記載のワクチンを非ヒト動物に免疫して抗体を作製するステップ、及び該抗体を回収するステップを含む、抗HCMV抗体の作製方法。
【請求項8】
前記抗体がヒト抗体である、請求項7に記載の方法。
【請求項1】
ヒトサイトメガロウイルス(HCMV)のUL79、87又は95遺伝子オープンリーディングフレーム(ORF)中に突然変異を生じさせることにより、これらの遺伝子の1つ若しくは複数からの発現が低下又は消失している、弱毒HCMV。
【請求項2】
前記突然変異が、ORF中への塩基の挿入によるものである、請求項1に記載の弱毒HCMV。
【請求項3】
前記弱毒HCMVからの感染性ウイルス増殖は野生型ウイルスと比較して低下しているが、ウイルスDNA複製は野生型ウイルスと同等である、請求項1又は2に記載の弱毒HCMV。
【請求項4】
後期ウイルスmRNAの発現が野生型ウイルスと比較して低下している、請求項1〜3のいずれか1項に記載の弱毒HCMV。
【請求項5】
請求項1〜4のいずれか1項に記載の弱毒HCMVを有効成分として含む、HCMV感染の予防又は治療用ワクチン。
【請求項6】
アジュバントをさらに含む、請求項5に記載のワクチン。
【請求項7】
請求項1〜4のいずれか1項に記載の弱毒HCMV又は請求項5若しくは6に記載のワクチンを非ヒト動物に免疫して抗体を作製するステップ、及び該抗体を回収するステップを含む、抗HCMV抗体の作製方法。
【請求項8】
前記抗体がヒト抗体である、請求項7に記載の方法。
【図1】

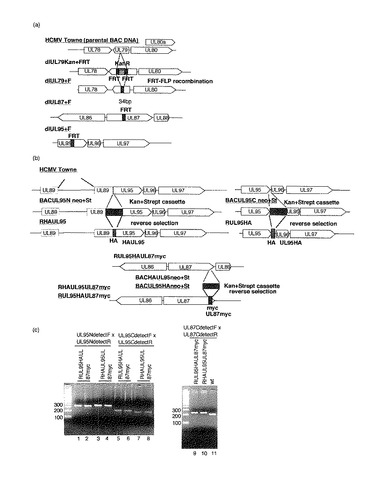
【図2】
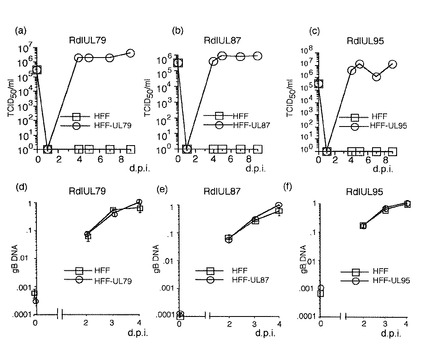
【図3a】
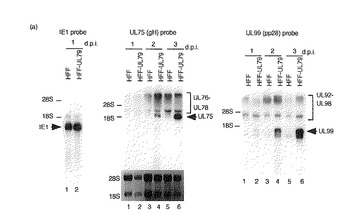
【図3b】
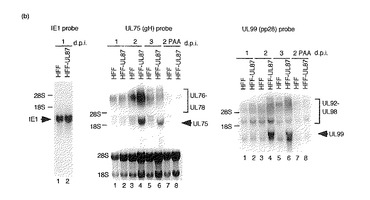
【図3c】
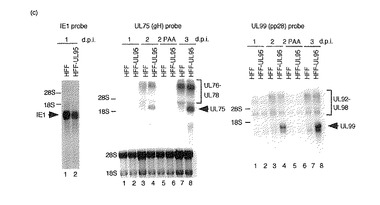
【図3d】
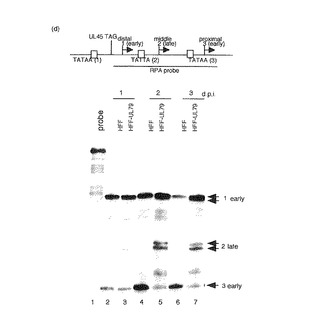
【図4】
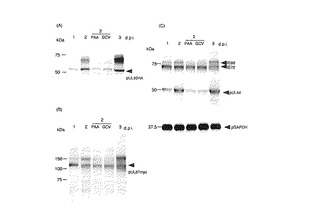
【図5】
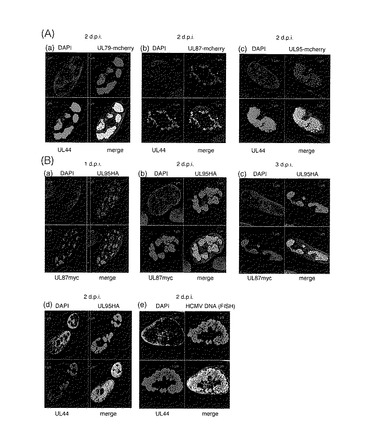
【図6−1】
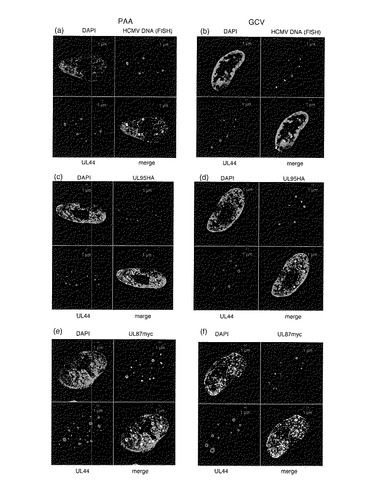
【図6−2】
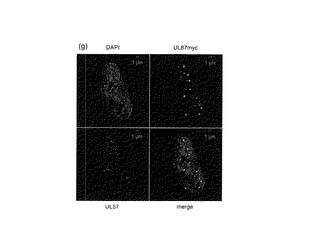
【図7】
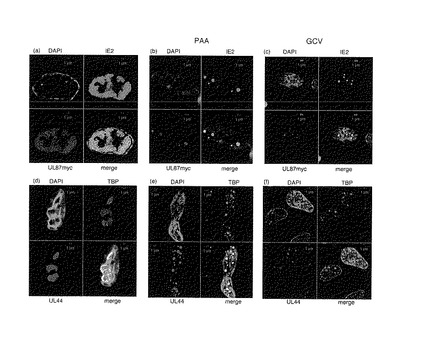
【図8】
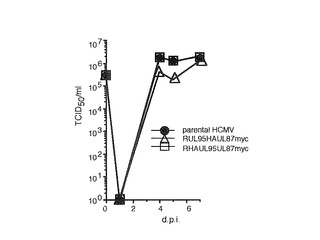
【図2】
【図3a】
【図3b】
【図3c】
【図3d】
【図4】
【図5】
【図6−1】
【図6−2】
【図7】
【図8】
【公開番号】特開2012−152147(P2012−152147A)
【公開日】平成24年8月16日(2012.8.16)
【国際特許分類】
【出願番号】特願2011−14548(P2011−14548)
【出願日】平成23年1月26日(2011.1.26)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 刊行物名 :第69回 日本癌学会学術総会記事 発行日 :平成22年8月23日 発行者 :日本癌学会 講演番号 :P−0307 公開者 :磯村 寛樹、鶴見 達也
【出願人】(304031427)愛知県 (36)
【Fターム(参考)】
【公開日】平成24年8月16日(2012.8.16)
【国際特許分類】
【出願日】平成23年1月26日(2011.1.26)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 刊行物名 :第69回 日本癌学会学術総会記事 発行日 :平成22年8月23日 発行者 :日本癌学会 講演番号 :P−0307 公開者 :磯村 寛樹、鶴見 達也
【出願人】(304031427)愛知県 (36)
【Fターム(参考)】
[ Back to top ]