
形質転換酵母、それを用いた分析方法および分析キット
【課題】 高検出感度でRXRαへテロ二量体型核内受容体のリガンドの分析が可能な形質転換酵母を提供する。
【解決手段】 本発明の形質転換酵母は、動物核内受容体遺伝子およびレポーター遺伝子が発現可能に導入されており、かつ動物核内受容体リガンドと動物核内受容体との複合体を認識して前記レポーター遺伝子を発現し得る形質転換酵母であって、前記動物核内受容体遺伝子として、RXRα核内受容体遺伝子およびRXRαへテロ二量体型核内受容体遺伝子を発現可能に含み、さらに、RNA不安定化配列および動物転写共役因子遺伝子を含む形質転換酵母である。図1のグラフに示すように、本発明の形質転換酵母を使用すれば、RXRαへテロ二量体型核内受容体のリガンドを高感度で分析可能である。
【解決手段】 本発明の形質転換酵母は、動物核内受容体遺伝子およびレポーター遺伝子が発現可能に導入されており、かつ動物核内受容体リガンドと動物核内受容体との複合体を認識して前記レポーター遺伝子を発現し得る形質転換酵母であって、前記動物核内受容体遺伝子として、RXRα核内受容体遺伝子およびRXRαへテロ二量体型核内受容体遺伝子を発現可能に含み、さらに、RNA不安定化配列および動物転写共役因子遺伝子を含む形質転換酵母である。図1のグラフに示すように、本発明の形質転換酵母を使用すれば、RXRαへテロ二量体型核内受容体のリガンドを高感度で分析可能である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、形質転換酵母、それを用いた分析方法および分析キットに関する。
【背景技術】
【0002】
環境中の化学物質の分析において、従来は機器分析が主流であったが、生物的な影響をも評価できる分析手法として核内受容体遺伝子を宿主細胞に組み込み、その発現の程度で化学物質の分析を行う手法が開発されている。宿主細胞としては、ヒト等の動物細胞および酵母が使用されている。動物細胞を用いた分析方法としては、例えば、アンドロゲンレセプター(AR)遺伝子をヒト由来細胞株に組み込み、テストステロン(T)等を分析する方法がある(非特許文献1)。一方、雌化現象等で注目された環境ホルモン(内分泌撹乱物質)のエストロゲン様活性の分析方法として、ブルーギル由来のエストロゲン受容体遺伝子(特許文献1)、オオマキトカゲ由来エストロゲン受容体遺伝子(特許文献2)、ワニ由来エストロゲン受容体遺伝子(特許文献3)、ファットヘッドミノー由来エストロゲン受容体遺伝子(特許文献4)等の各種野生動物のエストロゲン受容体遺伝子を酵母に組み込み、その発現を測定するという手法が開発されている(特許文献1、特許文献2、特許文献3、特許文献4)。この他、ヒト由来エストロゲン受容体遺伝子を組み込んだ形質転換酵母を用いてアルキルフェノール化合物のエストロゲン様活性を測定する手法が開発されている(非特許文献2)。また、例えば、ダイオキシン等のアリールハイドロカーボン類を測定するために、モルモット由来のアリールハイドロカーボン受容体遺伝子を組み込んだ形質転換酵母を用いた手法が開発されている(特許文献5)。これらの酵母を用いた手法では、各種核内受容体遺伝子が発現し、測定対象となるリガンド(化学物質等)が前記核内受容体に結合して複合体を形成し、この複合体がレポーター遺伝子を発現させる。前記レポーター遺伝子としては、β−ガラクトシダーゼ遺伝子が使用されており、β−ガラクトシダーゼの酵素活性を測定することで、遺伝子の発現を測定している。酵母を用いた分析は、動物細胞を用いた分析方法に比べて、短時間分析が可能であり、操作の簡便性および低コスト等の利点を有する。
【0003】
一方、ヒト核内受容体は、二量体形成後、核内受容体応答配列に結合して、標的遺伝子の転写調節を行う。ヒト核内受容体には、同じ核内受容体と二量体を形成するホモ二量体型核内受容体と、異なる核内受容体と二量体を形成するヘテロ二量体型核内受容体がある。ホモ二量体型核内受容体には、ステロイドホルモン核内受容体等の数種類があるが、ヒト核内受容体の多くは、レチノイン酸受容体α(RXRα受容体)とヘテロ二量体を形成するRXRαヘテロ二量体型核内受容体である。ホモ二量体型核内受容体については、前述のレポーター遺伝子導入酵母を用いたリガンド分析手法が開発されている(特許文献6)。しかし、ヘテロ二量体型核内受容体については、形質転換酵母における分析方法が開発されていない。また、RXRαヘテロ二量体型核内受容体については、酵母ツーハイブリッド法によるリガンド分析方法が報告されている(非特許文献3)。しかしながら、酵母ツーハイブリッド法は、リガンド非依存的な転写活性が生じるため、分析精度が低いという問題がある。
【0004】
【特許文献1】特開2001−197890号公報
【特許文献2】特開2003−274号公報
【特許文献3】特開2002−360273号公報
【特許文献4】特開2001−352992号公報
【特許文献5】特開2005−87077号公報
【特許文献6】特開2007−295806号公報
【非特許文献1】Toxicology、2006年、Vol.220、p.90−103
【非特許文献2】The Journal Of Biological Chemistry、1997年、Vol.272、No.6、p.3280−3288
【非特許文献3】Journal of Environmental Biotechnology、2003年、 Vol.3、No.1、p.37−42
【発明の開示】
【発明が解決しようとする課題】
【0005】
そこで、本発明の目的は、RXRαヘテロ二量体型動物核内受容体リガンドを高感度で分析可能な形質転換酵母、それを用いたRXRαヘテロ二量体型動物核内受容体リガンドの分析方法および分析キットを提供することである。
【課題を解決するための手段】
【0006】
前記目的を達成するために、本発明の形質転換酵母は、動物核内受容体遺伝子およびレポーター遺伝子が発現可能に導入されており、かつ動物核内受容体リガンドと動物核内受容体との複合体を認識して前記レポーター遺伝子を発現し得る形質転換酵母であって、
前記動物核内受容体が、RXRα核内受容体と二量体を形成する、RXRαヘテロ二量体型核内受容体であり、
前記動物核内受容体遺伝子として、RXRα核内受容体遺伝子およびRXRαへテロ二量体型核内受容体遺伝子を発現可能に含み、
前記動物核内受容体に直接結合して前記レポーター遺伝子の転写を活性化する動物転写共役因子遺伝子を発現可能に含み、
さらに、前記動物核内受容体遺伝子の発現量を調節するRNA不安定化配列を含む。
【0007】
本発明の分析方法は、検体に含まれる動物核内受容体リガンドを分析するための分析方法であって、
検体を含む培地中で、動物核内受容体遺伝子およびレポーター遺伝子が発現可能に導入されており、かつ動物核内受容体リガンドと動物核内受容体との複合体を認識して前記レポーター遺伝子を発現し得る形質転換酵母を培養する培養工程と、
前記培養工程の後、前記培地中に産生された前記レポーター遺伝子の発現産物を測定する測定工程とを包含し、
前記形質転換酵母として、本発明の形質転換酵母を使用することを特徴とする。
【0008】
本発明の分析キットは、検体に含まれる動物核内受容体リガンドを分析するための分析キットであって、
(a)培地と、
(b)本発明の形質転換酵母と、
(c)発色基質と
を備える。
【0009】
本発明のベクターは、動物核内受容体遺伝子を含むベクターであって、
前記動物核内受容体遺伝子として、RXRα核内受容体遺伝子およびRXRαへテロ二量体型核内受容体遺伝子を含み、
さらに、前記動物核内受容体遺伝子の発現量を調節するRNA不安定化配列を含む。
【0010】
本発明のベクターセットは、レポーター遺伝子を含むベクター、動物転写共役遺伝子を含むベクターおよび本発明のベクターを含有する。
【発明の効果】
【0011】
本発明者等は、形質転換酵母を用いたRXRαへテロ二量体型核内受容体リガンドの分析方法の感度向上を目的として、一連の研究を行った。その過程で、前記RXRαへテロ二量体型核内受容体の発現量に着目し、その発現量を必要最小限に留めるという着想を得た。すなわち、形質転換酵母で発現させたRXRαへテロ二量体型核内受容体は、リガンド非依存的に、転写活性化されるため、従来は、形質転換酵母でのヘテロ二量体型核内受容体リガンドの分析手法は実現できなかった。そこで、本発明者等は、その原因をつぎのように推察した。リガンド非依存的な転写活性化の原因の一つとして、前記RXRαへテロ二量体型核内受容体の過剰発現により、前記RXRαへテロ二量体型核内受容体の細胞質内保持および核内移行の制御が適切に行われないことが考えられる。また、別の原因として、前記RXRαへテロ二量体型核内受容体とヘテロ二量体を形成するRXRα核内受容体に起因する転写活性化等も考えられる。したがって、RXRαへテロ二量体型核内受容体の発現を必要最小限に留めること、およびRXRα核内受容体に起因する転写活性化を抑制することが、RXRαへテロ二量体型核内受容体リガンドの分析方法の確立に有効であると考えた。そこで、本発明者等は、遺伝子の3′非翻訳領域に存在し、前記遺伝子から転写されたmRNAの分解を促進するRNA不安定化配列に着目し、前記RNA不安定化配列を形質転換酵母に組み込んだ。その結果、前記RNA不安定化配列を組み込んだ形質転換酵母を使用すれば、RXRαへテロ二量体型動物核内受容体リガンドを高感度で分析できることを見出し、本発明に到達した。
したがって、本発明の形質転換酵母を用いれば、RXRαへテロ二量体型動物核内受容体リガンドを、簡単な操作により、低コスト、短時間かつ高感度で分析可能である。また、本発明の分析キットを用いれば、さらに簡便に、RXRαへテロ二量体型動物核内受容体リガンドの分析を実施可能である。
【発明を実施するための最良の形態】
【0012】
本発明の形質転換酵母において、前記RNA不安定化配列が、前記RXRαへテロ二量体型核内受容体遺伝子の発現量を調節するRNA不安定化配列であることが好ましい。前記発現量の調節としては、前記RXRαへテロ二量体型核内受容体遺伝子の発現量を低減することが好ましい。
【0013】
本発明の形質転換酵母において、前記RXRα核内受容体が、C末端のHelix12領域を欠失させたRXRα核内受容体であることが好ましい。
【0014】
本発明の形質転換酵母において、さらに、ターミネーター配列を含むことが好ましい。
【0015】
本発明の形質転換酵母において、前記動物核内受容体が、ヒト核内受容体であり、前記動物転写共役因子がヒト転写共役因子であることが好ましい。
【0016】
本発明の形質転換酵母において、前記ヒト転写共役因子遺伝子が、ヒトSRC1遺伝子であることが好ましい。
【0017】
本発明の形質転換酵母において、前記動物核内受容体遺伝子および前記RNA不安定化配列を常染色体上に有し、前記レポーター遺伝子を第1のプラスミドに有し、前記動物転写共役因子遺伝子を第2のプラスミドに有し、前記第1のプラスミドは、前記レポーター遺伝子と作動的に組み込まれた動物核内受容体応答配列を含むことが好ましい。
【0018】
前記レポーター遺伝子が、β−ガラクトシダーゼ遺伝子であることが好ましい。
【0019】
前記形質転換酵母が、出芽酵母(Saccharomyces cerevisiae)であることが好ましい。
【0020】
本発明の形質転換酵母は、例えば、検体に含まれる動物核内受容体リガンドの分析に使用される。
【0021】
本発明の分析方法において、前記レポーター遺伝子が、β−ガラクトシダーゼ遺伝子であり、前記培地が、炭素源としてガラクトースおよびグルコースを含有することが好ましい。グルコースを含む培地で前記形質転換酵母を培養すれば、例えば、18時間の短時間で分析可能なレベルまで増殖させることができる。本発明の分析方法において、前記ガラクトースおよび前記グルコースの合計に対する前記グルコースの重量割合は、特に制限されないが、例えば、0を超え60重量%以下の範囲、好ましくは、0を超え40重量%以下の範囲、より好ましくは、10〜30重量%の範囲である。
【0022】
本発明の分析方法において、前記形質転換酵母が、グリセロール存在下で保存されていた酵母であることが好ましい。前記グリセロール存在下で保存されていた形質転換酵母は、予め、グルコース単独を炭素源とする培地で培養された酵母であることが好ましい。
【0023】
本発明の分析方法は、例えば、検出、定性分析または定量分析である。また、本発明において、分析は、検出、定性分析および定量分析を含む。
【0024】
本発明の分析キットにおいて、前記培地は、グルコースおよびガラクトースを含む培地であることが好ましい。前記グルコースの重量割合は、前述のとおりである。
【0025】
本発明のベクターにおいて、前記RNA不安定化配列が、前記RXRαへテロ二量体型核内受容体遺伝子の発現量を調節するRNA不安定化配列であることが好ましい。そして、本発明のベクターにおいて、前記RXRα核内受容体が、C末端のHelix12領域を欠失させたRXRα核内受容体であることが好ましい。また、本発明のベクターは、さらに、ターミネーター配列を含むことが好ましい。
【0026】
つぎに、本発明について詳しく説明する。
【0027】
本発明において、前記RXRαへテロ二量体型核内受容体および前記RXRαへテロ二量体型核内受容体リガンドは、特に制限されず、例えば、下記に示すRXRαへテロ二量体型核内受容体および前記受容体に結合可能なリガンドがあげられる。
【0028】
(1)TRα:Thyroid Hormone Receptor alpha
(2)TRβ:Thyroid Hormone Receptor beta
(3)RARα:Retinoic Acid Receptor alpha
(4)RARβ:Retinoic Acid Receptor beta
(5)RARγ:Retinoic Acid Receptor gamma
(6)PPARα:Peroxisome proliferator activated receptor alpha(PPAR−alpha)
(7)PPARδ:Peroxisome proliferator activated receptor delta(PPAR−delta、PPAR−beta、Nuclear hormone receptor 1、NUC1)
(8)PPARγ1:Peroxisome proliferator activated receptor gamma 1(PPAR−gamma1)
(9)PPARγ2:Peroxisome proliferator activated receptor gamma 2(PPAR−gamma2)
(10)LXRβ:Oxysterols receptor LXR−beta (Liver X receptor beta、Nuclear orphan receptor LXR−beta、Ubiquitously−expressed nuclear receptor、Nuclear receptor NER)
(11)LXRα:Oxysterols receptor LXR−alpha(Liver X receptor alpha、Nuclear orphan receptor LXR−alpha)
(12)VDR:Vitamin D3 receptor(VDR、1,25−dihydroxyvitamin D3 receptor)
(13)PXR−1:Orphan nuclear receptor PXR−1(Pregnane X receptor−1、Orphan nuclear receptor PAR−1、Steroid and xenobiotic receptor−1、SXR−1)
(14)PXR−2:Orphan nuclear receptor PXR−2(Pregnane X receptor−2、Orphan nuclear receptor PAR−2、Steroid and xenobiotic receptor−2、SXR−2)
(15)CAR:Orphan nuclear receptor NR1I3(Constitutive androstane receptor、CAR、Orphan nuclear receptor MB67)
(16)COUP−TF I:COUP transcription factor 1(COUP−TF1、COUP−TFα、V−erbA related protein EAR−3)
(17)COUP−TF II:COUP transcription factor 2(COUP−TF2、COUP−TFβ、Apolipoprotein AI regulatory protein−1、ARP−1)
(18)ERRα:Steroid hormone receptor ERR1(Estrogen−related receptor, alpha、ERR−alpha、Estrogen receptor−like 1)
(19)ERRβ:Steroid hormone receptor ERR2(Estrogen−related receptor,beta、ERR−beta、Estrogen receptor−like 2、ERR beta−2)
(20)ERRγ:Estrogen−related receptor gamma(Estrogen receptor related protein 3、ERR gamma−2)
【0029】
これらのRXRαへテロ二量体型核内受容体は、分析対象となるリガンドに応じて、適宜選択すればよい。
【0030】
つぎに、本発明において、動物転写共役因子は特に制限されない。動物転写共役因子としては、例えば、下記表1に示す8種類の転写共役因子が使用可能である。
【0031】
【表1】

【0032】
本発明の形質転換酵母において、導入するRXRαへテロ二量体型核内受容体および動物転写共役因子は、分析の対象となるリガンド(化学物質)の種類により適宜決定できる。例えば、本発明の形質転換酵母は、前記RXRαへテロ二量体型核内受容体として、プレグナンX受容体(PXR)、ビタミンD受容体(VDR)等を導入した態様がある。
【0033】
まず、PXR遺伝子を導入した形質転換酵母について説明する。PXRとしては、特に制限されないが、例えば、ヒトPXRがある。PXRは、リガンド(プロゲステロンもしくはプロゲステロン様化学物質)および転写共役因子と複合体を形成し、これが応答配列(DR−3配列)を認識して結合し、これにより、応答配列の下流に存在するレポーター遺伝子(例えば、β−ガラクトシダーゼ遺伝子)が発現する。レポーター遺伝子の転写は、前記転写共役因子により活性化される。
【0034】
前記ヒトPXRを酵母で発現させるためには、例えば、ヒトPXR遺伝子を前記酵母の染色体遺伝子内に導入すればよい。
【0035】
前記ヒトPXR遺伝子としては、例えば、下記(a)または(b)のポリヌクレオチドを含むことが好ましい。下記(b)のポリヌクレオチドは、下記(a)のポリヌクレオチドとの相同性が、例えば、90%以上であって、好ましくは、93%以上であり、より好ましくは、95%以上であり、さらに好ましくは、98%以上である。また、下記(b)のポリヌクレオチドにおいて、置換、付加、挿入もしくは欠失した塩基の数は、前記相同性に対応した数である。
(a)配列番号1に記載の塩基配列からなるポリヌクレオチド。
(b)配列番号1に記載の塩基配列の少なくとも1つの塩基が置換、付加、挿入もしくは欠失した塩基配列からなるポリヌクレオチドであって、ヒトPXRとして機能するタンパク質をコードするポリヌクレオチド。
【0036】
前記PXR遺伝子は、配列番号1の塩基配列に基づいて、ヒトのトータルRNA等を使用してクローニングしてもよく、また、ホスホルアミダイト(phosphoramidite)法を利用して化学的にDNA合成してもよい。前記クローニングの方法は、特に制限されず、例えば、市販のクローニングキット等を利用して実施できる。
【0037】
PXRに対する転写共役因子としては、特に制限されないが、例えば、SRC1、CBP、P300がある。したがって、これらの転写共役因子の遺伝子を形質転換酵母に組み込めばよい。これらの中で、好ましいのは、SRC1である。転写共役因子遺伝子は、第2のプラスミドに組み込んで形質転換酵母に導入することが好ましい。前記第2のプラスミドは、高コピー型プラスミドであることが好ましい。
【0038】
前記応答配列およびレポーター遺伝子は、作動的に連結して、第1のプラスミドに組み込み、この第1のプラスミドを形質転換細胞に導入することが好ましい。前記DR−3配列は、一配列のみ組み込まれてもよく、繰り返し配列として組み込まれてもよい。また、第1のプラスミドは、低コピー型プラスミドであることが好ましい。
【0039】
つぎに、VDR遺伝子を導入した形質転換酵母について説明する。VDRとしては、特に制限されないが、例えば、ヒトVDRがある。VDRは、リガンド(ビタミンD若しくはビタミンD様化学物質)および転写共役因子と複合体を形成し、これが応答配列(DR−3配列)を認識して結合し、これにより、応答配列の下流に存在するレポーター遺伝子(例えば、β−ガラクトシダーゼ遺伝子)が発現する。レポーター遺伝子の転写は、前記転写共役因子により活性化される。
【0040】
前記ヒトVDRを酵母で発現させるためには、例えば、ヒトVDR遺伝子を前記酵母の染色体遺伝子内に導入すればよい。
【0041】
前記ヒトVDR遺伝子としては、例えば、下記(c)または(d)のポリヌクレオチドを含むことが好ましい。下記(d)のポリヌクレオチドは、下記(c)のポリヌクレオチドとの相同性が、例えば、90%以上であって、好ましくは、93%以上であり、より好ましくは、95%以上であり、さらに好ましくは、98%以上である。また、下記(d)のポリヌクレオチドにおいて、置換、付加、挿入もしくは欠失した塩基の数は、前記相同性に対応した数である。
(c)配列番号2に記載の塩基配列からなるポリヌクレオチド。
(d)配列番号2に記載の塩基配列の少なくとも1つの塩基が置換、付加、挿入もしくは欠失した塩基配列からなるポリヌクレオチドであって、ヒトVDRとして機能するタンパク質をコードするポリヌクレオチド。
【0042】
前記VDR遺伝子は、配列番号2の塩基配列に基づいて、ヒトのトータルRNA等を使用してクローニングしてもよく、また、ホスホルアミダイト(phosphoramidite)法を利用して化学的にDNA合成してもよい。前記クローニングの方法は、特に制限されず、例えば、市販のクローニングキット等を利用して実施できる。
【0043】
VDRに対する転写共役因子としては、特に制限されないが、例えば、SRC1、CBP、P300、AIBがある。したがって、これらの転写共役因子の遺伝子を形質転換酵母に組み込めばよい。これらの中で、好ましいのは、SRC1である。転写共役因子遺伝子は、第2のプラスミドに組み込んで形質転換酵母に導入することが好ましい。前記第2のプラスミドは、高コピー型プラスミドであることが好ましい。
【0044】
前記応答配列およびレポーター遺伝子は、作動的に連結して、第1のプラスミドに組み込み、この第1のプラスミドを形質転換細胞に導入することが好ましい。前記DR−3配列は、例えば、一配列のみ組み込まれてもよく、繰り返し配列として組み込まれてもよい。また、第1のプラスミドは、低コピー型プラスミドであることが好ましい。
【0045】
本発明の形質転換酵母には、前記RXRαへテロ二量体型核内受容体とヘテロ二量体を形成するレチノイン酸X受容体α(RXRα)遺伝子を導入する。RXRαとしては、特に制限されないが、例えば、ヒトRXRαがある。RXRαは、前記RXRαへテロ二量体型核内受容体とヘテロ二量体を形成する。前記RXRαへテロ二量体型核内受容体とそのリガンドが結合すると、前記へテロ二量体は、前記応答配列(DR−3配列)を認識して結合し、これにより、応答配列の下流に存在するレポーター遺伝子(例えば、β−ガラクトシダーゼ遺伝子)が発現する。レポーター遺伝子の転写は、前記転写共役因子により活性化される。
【0046】
前記ヒトRXRαを酵母で発現させるためには、例えば、ヒトRXRα遺伝子を前記酵母の染色体遺伝子内に導入すればよい。
【0047】
前記ヒトRXRα遺伝子としては、例えば、下記(e)または(f)のポリヌクレオチドを含むことが好ましい。下記(f)のポリヌクレオチドは、下記(e)のポリヌクレオチドとの相同性が、例えば、90%以上であって、好ましくは、93%以上であり、より好ましくは、95%以上であり、さらに好ましくは、98%以上である。また、下記(f)のポリヌクレオチドにおいて、置換、付加、挿入もしくは欠失した塩基の数は、前記相同性に対応した数である。
(e)配列番号3に記載の塩基配列からなるポリヌクレオチド。
(f)配列番号3に記載の塩基配列の少なくとも1つの塩基が置換、付加、挿入もしくは欠失した塩基配列からなるポリヌクレオチドであって、ヒトRXRαとして機能するタンパク質をコードするポリヌクレオチド。
【0048】
前記RXRα遺伝子は、配列番号3の塩基配列に基づいて、ヒトのトータルRNA等を使用してクローニングしてもよく、また、ホスホルアミダイト(phosphoramidite)法を利用して化学的にDNA合成してもよい。前記クローニングの方法は、特に制限されず、例えば、市販のクローニングキット等を利用して実施できる。
【0049】
本発明の形質転換酵母には、前記RXRα遺伝子に代えて、例えば、C末端に存在するHelix12領域を欠失させたRXRα(以下、RXRαTとする)遺伝子を導入してもよい。前記Helix12領域は、SRC1との結合の必須領域であるため、前記RXRαT遺伝子の導入により、RXRα核内受容体に起因するリガンド非依存的な転写活性化を抑制できる。
【0050】
前記RXRαTは、例えば、前記RXRαにナンセンス変異を導入して作製できる。前記RXRαTを酵母で発現させるためには、例えば、RXRαT遺伝子を前記酵母の染色体遺伝子内に導入すればよい。
【0051】
前記RXRαT遺伝子としては、例えば、下記(g)または(h)のポリヌクレオチドを含むことが好ましい。下記(h)のポリヌクレオチドは、下記(g)のポリヌクレオチドとの相同性が、例えば、90%以上であって、好ましくは、93%以上であり、より好ましくは、95%以上であり、さらに好ましくは、98%以上である。また、下記(h)のポリヌクレオチドにおいて、置換、付加、挿入もしくは欠失した塩基の数は、前記相同性に対応した数である。
(g)配列番号4に記載の塩基配列からなるポリヌクレオチド。
(h)配列番号4に記載の塩基配列の少なくとも1つの塩基が置換、付加、挿入もしくは欠失した塩基配列からなるポリヌクレオチドであって、ヒトRXRαとして機能するタンパク質をコードするポリヌクレオチド。
【0052】
前記動物転写共役因子である前記ヒトSRC1遺伝子としては、例えば、下記(i)または(j)のポリヌクレオチドを含むことが好ましい。下記(j)のポリヌクレオチドは、下記(i)のポリヌクレオチドとの相同性が、例えば、90%以上であって、好ましくは、93%以上であり、より好ましくは、95%以上であり、さらに好ましくは、98%以上である。また、下記(j)のポリヌクレオチドにおいて、置換、付加、挿入もしくは欠失した塩基の数は、前記相同性に対応した数である。
(i)配列番号5に記載の塩基配列からなるポリヌクレオチド。
(j)配列番号5に記載の塩基配列の少なくとも1つの塩基が置換、付加、挿入もしくは欠失した塩基配列からなるポリヌクレオチドであって、ヒトSRC1として機能するタンパク質をコードするポリヌクレオチド。
【0053】
本発明の形質転換酵母には、前記RNA不安定化配列を導入する。前記RNA不安定化配列としては、特に制限されないが、例えば、HOエンドヌクレアーゼ遺伝子の3′非翻訳領域に存在するRNA不安定化配列がある。
【0054】
本発明の形質転換酵母において、前記RNA不安定化配列は、例えば、前記酵母の染色体遺伝子内に導入すればよい。
【0055】
前記RNA不安定化配列としては、例えば、下記(k)または(l)のポリヌクレオチドを含むことが好ましい。下記(l)のポリヌクレオチドは、下記(k)のポリヌクレオチドとの相同性が、例えば、90%以上であって、好ましくは、93%以上であり、より好ましくは、95%以上であり、さらに好ましくは、98%以上である。また、下記(l)のポリヌクレオチドにおいて、置換、付加、挿入もしくは欠失した塩基の数は、前記相同性に対応した数である。
(k)配列番号6に記載の塩基配列からなるポリヌクレオチド。
(l)配列番号6に記載の塩基配列の少なくとも1つの塩基が置換、付加、挿入もしくは欠失した塩基配列からなるポリヌクレオチドであって、RNA不安定化配列として機能するポリヌクレオチド。
【0056】
本発明の形質転換酵母には、例えば、ターミネーター配列を導入してもよい。前記ターミネーター配列としては、特に制限されないが、例えば、酵母cyc1遺伝子のターミネーター配列、酵母ADH遺伝子のターミネーター配列等がある。
【0057】
本発明の形質転換酵母において、前記ターミネーター配列は、例えば、前記酵母の染色体遺伝子内に導入すればよい。
【0058】
前記酵母cyc1遺伝子のターミネーター配列としては、例えば、下記(m)または(n)のポリヌクレオチドを含むことが好ましい。下記(n)のポリヌクレオチドは、下記(m)のポリヌクレオチドとの相同性が、例えば、90%以上であって、好ましくは、93%以上であり、より好ましくは、95%以上であり、さらに好ましくは、98%以上である。また、下記(n)のポリヌクレオチドにおいて、置換、付加、挿入もしくは欠失した塩基の数は、前記相同性に対応した数である。
(m)配列番号7に記載の塩基配列からなるポリヌクレオチド。
(n)配列番号7に記載の塩基配列の少なくとも1つの塩基が置換、付加、挿入もしくは欠失した塩基配列からなるポリヌクレオチドであって、ターミネーター配列として機能するポリヌクレオチド。
【0059】
前記酵母ADH遺伝子のターミネーター配列としては、例えば、下記(o)または(p)のポリヌクレオチドを含むことが好ましい。下記(p)のポリヌクレオチドは、下記(o)のポリヌクレオチドとの相同性が、例えば、90%以上であって、好ましくは、93%以上であり、より好ましくは、95%以上であり、さらに好ましくは、98%以上である。また、下記(p)のポリヌクレオチドにおいて、置換、付加、挿入もしくは欠失した塩基の数は、前記相同性に対応した数である。
(o)配列番号8に記載の塩基配列からなるポリヌクレオチド。
(p)配列番号8に記載の塩基配列の少なくとも1つの塩基が置換、付加、挿入もしくは欠失した塩基配列からなるポリヌクレオチドであって、ターミネーター配列として機能するポリヌクレオチド。
【0060】
本発明の遺伝子組換え細胞の宿主細胞は、酵母であり、好ましいのは、出芽酵母であることは、前述のとおりである。
【0061】
前記酵母内に、前記核内受容体遺伝子を導入する方法としては、特に制限されず、従来公知の遺伝子導入方法により行うことができる。前記遺伝子導入方法としては、例えば、酢酸リチウム法、リン酸カルシウム法、リポソームを用いた方法、エレクトロポレーション、ウイルスベクターを用いる方法、マイクロピペットインジェクション法等があげられる。本発明における前記核内受容体遺伝子の導入は、そのリガンドの分析に用いることが可能であれば、一過性型の導入でもよく、または、宿主染色体への組込み型もしくは自律複製・分配可能な人工染色体もしくはプラスミド型の導入でもよい。取扱いや保存の容易性およびリガンドの分析の迅速性、正確性、簡便性からは、前述のように、宿主(酵母)染色体への遺伝子導入方法が好ましい。
【0062】
前記酵母内に導入する前記核内受容体遺伝子は、酵母内で恒常的または任意に発現するように、必要な調節配列と作動的に連結されていることが好ましい。前記調節配列とは、宿主細胞(酵母)内において、作動的に連結された前記遺伝子の発現に必要な塩基配列であって、例えば、真核細胞に適した調節配列としては、プロモーター、ポリアデニル化シグナル、エンハンサー等があげられる。本発明において、作動的に連結とは、各構成要素が機能を果たすことができるように並置していることを意味する。
【0063】
前述のように、本発明において、前記核内受容体とリガンドの複合体を検出するためのレポーター遺伝子として、例えば、β−ガラクトシダーゼ遺伝子を使用することが好ましい。前記β−ガラクトシダーゼ遺伝子は、その上流に、前記複合体が認識して結合する応答配列(転写調節配列)と動作的に結合している。前記応答配列(転写調節配列)は、動物核内受容体遺伝子の種類により適宜決定される。前述のように、PXRおよびVDRの場合の応答配列は、DR−3である。これらの応答配列(転写調節配列)およびβ−ガラクトシダーゼ遺伝子は、第1のプラスミドに組み込まれて形質転換酵母に導入されていることが好ましい。前記レポーター遺伝子としては、β−ガラクトシダーゼに限定されず、その他、ルシフェラーゼ遺伝子、蛍光タンパク質遺伝子、β−グルクロニダーゼ遺伝子等が使用できる。
【0064】
前述のように、本発明において、転写共役因子遺伝子は、前記レポーター遺伝子を組み込んだプラスミド(第1のプラスミド)とは別のプラスミド(第2のプラスミド)に組み込んで酵母に導入することが好ましい。
【0065】
つぎに、本発明の分析方法の一例を示す。
【0066】
まず、所定の核内受容体遺伝子、RNA不安定化配列、動物転写共役因子遺伝子、前記動物核内受容体応答配列およびβ−ガラクトシダーゼ遺伝子を導入した形質転換酵母を準備する。そして、この形質転換酵母のグリセロールストックを調製することが好ましい。グリセロールストックを調製するために使用する培地は、特に制限されず、例えば、窒素源、炭素源、アミノ酸、核酸、ビタミン類等を含む培地があげられる。グリセロールストックを調製する培地組成の例を、下記表2に示す。
【0067】
(表2)
成分 配合量
ドロップアウトパウダー (注1) 1.3g
窒素源(Yeast nitrogen base、注2) 1.7g
(NH4)2SO4 5.0g
炭素源(グルコースまたはガラクトース) 20.0g
水 1000mL
(注1)ドロップアウトパウダー組成
成分 配合量 成分 配合量
アデニン 2.5g L−メチオニン 1.2g
L−アルギニン 1.2g L−フェニルアラニン 3.0g
L−アスパラギン酸 6.0g L−セリン 22.5g
L−グルタミン酸 6.0g L−スレオニン 12.0g
L−ヒスチジン 1.2g L−チロシン 1.8g
L−リジン 1.8g L−バリン 9.0g
ウラシル 1.2g
(注2)窒素源:Yeast Nitrogen W/O Amino Acid &
Ammonium Sulfate (Difco社製)
【0068】
前記グリセロールストックを調製するための培養は、前記のような培地で継続的に培養された状態のものを利用する場合には、例えば、前記継続培養後の培養液を50μL分取し、前記表2の培地5000μL中に加え、30℃で12〜24時間程度振とう培養することによって実施する。一方、形質転換酵母として凍結乾燥菌体を利用してグリセロールストックを調製するための培養を行う場合には、例えば、YPD培地等の栄養培地を加え、30℃で24時間培養した培養液を50μL分取し、前記と同様に前記表2に示す培地5000μLに加え、30℃で12〜24時間程度振とう培養する。
【0069】
前記グリセロールストックを調製するための培養により、菌体が十分増殖したら、得られた培養液から10倍濃縮グリセロールストック液を作製する。このグリセロールストック液の組成は、例えば培養液80%、グリセロール20%から成り、−80℃での保存も可能である。そして、このグリセロールストック液から本培養を実施する。前記本培養に使用する培地としては、例えば、前記表2に示す培地において、炭素源として、ガラクトースおよびグルコースを含むものを使用する。ガラクトースとグルコースの比率は、前述のとおりである。また、本培養においては、培地中に、化学物質等の被験物質を添加して行う。前記本培養の温度は、例えば、28〜30℃であり、前記本培養時間は、例えば、10〜48時間、好ましくは、15〜36時間、より好ましくは18〜24時間である。すなわち、前記培地にガラクトースとグルコースを添加することにより、本培養における前記形質転換酵母の増殖を促進させることができるので、短時間の本培養で、被験物質を高感度かつ高信頼性で分析可能である。また、本培養は、振とう培養でもよいし、静置培養でもよい。なお、前記被験物質が固体の場合には、例えば、ジメチルスルホキシド(DMSO)等の形質転換酵母に対して毒性を示さない溶媒を用いて前記被験物質を溶解してから前記培地と混合することが好ましい。
【0070】
つぎに、本培養後の前記形質転換酵母を、公知の手法により溶菌させる。この溶菌には、例えば、下記に示すような、Zバッファーに界面活性剤(サルコシル)を添加した溶菌液等が使用できる。
【0071】
(溶菌液:Zバッファー)
成分 配合量(最終濃度)
Na2HPO4 60mM
NaH2SO4 40mM
MgCl2 1mM
KCl 10mM
ジチオスレイトール(Dithiothreitol) 2mM
サルコシル(注3) 0.20%
(注3)N−Lauroylsarcosine sodium salt
【0072】
そして、前記溶菌後の溶菌液を適量採取し、産生されたβ−ガラクトシダーゼの酵素活性を測定する。β−ガラクトシダーゼの活性は、例えば、ONPG(オルソニトロフェニルガラクトピラノシド)やCPRG(クロロフェノールレッドガラクトピラノシド)等の発色試薬を作用させ、その吸光度を測定すればよい。この測定において、予め、標準物質を用いて検量線を作成しておくことが好ましく、前記検量線は、被験物質の測定と同時に作成することが好ましい。さらに、測定に際しては、用量作用関係の範囲内に納まるように被験物質を適宜希釈することが好ましい。このような分析方法であれば、吸光度を測定するだけでよく、簡便かつ迅速に分析できる。
【0073】
本発明の分析用キットは、本発明の分析方法を簡単に実施するためのキットであって、前述のように、培地、本発明の前記形質転換酵母および発色基質(発色試薬)を含むキットである。前記発色基質(発色試薬)としては、例えば、ONPGやCPRG等があげられる。さらに、本発明の分析キットは、検量線作成のための標準物質等を含んでいてもよい。前記形質転換酵母は、凍結乾燥させて粉末状にしてもよい。
【0074】
本発明のベクターは、前述の遺伝子および配列以外に、他の遺伝子または配列等を含んでいてもよい。前記他の遺伝子または配列は、特に限られず、例えば、前記調節配列が挙げられる。
【0075】
本発明のベクターキットは、前述のベクターを含有すること以外は、特に限定されず、他のベクターを含有してもよい。
【実施例】
【0076】
つぎに、本発明の実施例について比較例と併せて説明する。ただし、本発明は、下記の実施例および比較例に限定されるものではない。
【0077】
(実施例1)
本例では、下記の手順によりヒトPXR遺伝子、ヒトRXRα遺伝子、RNA不安定化配列および酵母cyc1遺伝子のターミネーター配列を組み込んだ形質転換酵母を作製した。
【0078】
(1) レポータープラスミドの作製
まず、酵母チトクロームC遺伝子のプロモーターおよびβ−ガラクトシダーゼ遺伝子を有するプラスミド(pYT−β;BioTechniques(1996)20、568−574)を準備した。一方、PXRの応答配列であるDR−3を含む、下記のリン酸化合成オリゴDNA(配列番号9)およびその相補鎖DNA(配列番号10)を作製した。作製したこれらのオリゴDNAを、94℃で1分間を1サイクルでアニーリングした。アニーリング後、DR−3が1個アニーリングしたものを精製して、前記プラスミドpYT−βのSpeIサイトに挿入した。DR−3が1個挿入されたプラスミドをpYT−β−DR3×1とした。
【0079】
(DR−3)
DR3Nh:5´−ctagcagatgaactttatgaactgttt−3´(配列番号9)
DR3Xb:5´−ctagaaacagttcataaagttcatctg−3´(配列番号10)
【0080】
(2)発現量調節プラスミドの作製
まず、酵母ura3遺伝子、gal1/gal10双方向性プロモーターを有するプラスミドpUdp3を準備した。前記gal1/gal10双方向性プロモーターは、プラスミドYEpLacから、BamHIサイトとSmaIサイトで切り出した。プラスミドpUC19のEcoRIサイトに、前記酵母ura3遺伝子を挿入後、BamHIサイトとSmaIサイトの間に前記gal1/gal10双方向性プロモーターを挿入し、前記プラスミドpUdp3を得た。つぎに、前記プラスミドpUdp3に、酵母cyc1遺伝子のターミネーター配列を挿入したプラスミドpUdp5を作製した。前記酵母cyc1遺伝子のターミネーター配列は、酵母W303aより抽出したゲノムDNAよりPCR法にて増幅した。用いたプライマー(配列番号11および12)およびPCRの条件を以下に示す。
【0081】
(cyc1)
PolAfHd:5´−tatgtcaagcttacattcacgccctc−3´(配列番号11)
PolArBs:5´−cttctcaagctaggtcttcagtataatg−3´(配列番号12)
【0082】
(PCR条件:1×反応バッファー)
1μM MgSO4、0.2μM dNTP、各プライマー各0.3μM、ゲノムDNA 0.5μL、5% DMSO、KOD−Plus−(東洋紡社製)1unitを反応液50μL中に含む。
【0083】
(PCR条件:サイクル)
94℃で40秒、54℃で20秒、68℃で30秒のサイクルを35サイクル実施した。
【0084】
PCR後、増幅した前記酵母cyc1遺伝子のターミネーター配列を含むDNAフラグメントを、制限酵素HindIIIおよびBbsIを用いて切断し、前記プラスミドpUdp3のHindIIIサイトに挿入した。得られたプラスミドをpUdp5とした。
【0085】
さらに、前記プラスミドpUdp5にRNA不安定化配列を挿入した、プラスミドpUdp8を準備した。前記RNA不安定化配列としては、酵母HOエンドヌクレアーゼ遺伝子の3´非翻訳領域に存在するRNA不安定化配列を用いた。前記RNA不安定化配列を含む下記のリン酸化合成オリゴDNA(配列番号13)およびその相補鎖DNA(配列番号14)を作製した。作製したこれらのオリゴDNAを、94℃で1分間を1サイクルでアニーリングした。アニーリング後、アニーリングしたオリゴDNAを、pUdp5の前記酵母cyc1遺伝子ターミネーター配列上流のHindIIIサイトに挿入した。得られたプラスミドをpUdp8とした。
【0086】
(RNA不安定化配列)
HOup:5´−agcttgtatattagtttaaaaagttgtatgtaat−3´(配列番号13)
HOdn:5´−agctattacatacaactttttaaactaatataca−3´(配列番号14)
【0087】
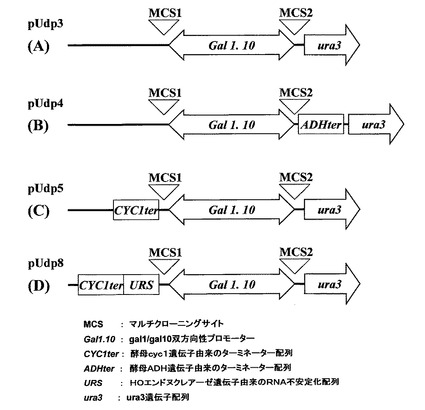
図3に、前記三種類のプラスミドpUdp3、pUdp5、pUdp8、および後述するプラスミドpUdp4のプロモーター周辺の構造を示す。同図において、MCSは、核内受容体のcDNAを挿入するマルチクローニングサイトであり、CYC1terは、前記酵母cyc1遺伝子由来のターミネーター配列であり、ADHterは、前記酵母ADH遺伝子由来のターミネーター配列であり、Gal1.10は、前記gal1/gal10双方向性プロモーターであり、URSは、HOエンドヌクレアーゼ遺伝子由来のRNA不安定化配列である。
【0088】
(3) 動物核内受容体発現プラスミドの作製
ヒトPXRおよびRXRαの各cDNAのオープンリーディングフレーム(ORF)を、ヒト肝がん細胞HepG2から抽出したmRNAを用いて、RT−PCR法により増幅した。用いたプライマー(配列番号15〜18)及びPCRの条件を以下に示す。
【0089】
(PXR)
PXRfRbSp:5´−ttccaactagtaaaacatggaggtgagacccaaag−3´(配列番号15)
PXRrHd:5´−ccttcaagcttctcagctacctgtgatgccgaac−3´(配列番号16)
【0090】
(RXRα)
RXRaRbKpnF:5´−aaaggggtacctcaaaaatggacaccaaacatttcctgcc−3´(配列番号17)
RXRaEcr:5´−cttaagaattctaagtcatttggtgcggc−3´(配列番号18)
【0091】
(PCR条件:1×反応バッファー)
1μM MgSO4、0.2μM dNTP、プライマー各0.3μM、cDNA 1μL、5% DMSO、KOD−Plus−(東洋紡社製)1unitを反応液50μL中に含む。
【0092】
(PCR条件:サイクル)
94℃で40秒、58℃で20秒、68℃で3分のサイクルを40サイクル実施した。
【0093】
つぎに、増幅した各核内受容体cDNAを、制限酵素を用いて切断した。前記プラスミドpUdp8のgal1プロモーター下流に位置する部位に、切断したRXRαのcDNAを挿入し、gal10プロモーター下流に位置する部位に、切断したPXRのcDNAを挿入した。得られたプラスミドを、pUdp8PXR/RXRαとした。なお、クローニングには大腸菌DH5α株を使用した。cDNAおよびプラスミドの切断に用いた制限酵素を以下に示す。
【0094】
(制限酵素)
挿入したcDNA cDNA切断用制限酵素 プラスミド切断用制限酵素
PXR SpeI、HindIII XbaI、HindIII
RXRα KpnI、EcoRI KpnI、EcoRI
【0095】
(4) SRC1プラスミドの作製
SRC1cDNAのオープンリーディングフレーム(ORF)を、ヒト精巣cDNA(クロンテック社製)よりPCR法を用いて増幅した。用いたプライマー(配列番号19および20)及びPCRの条件を以下に示す。
【0096】
hSRC1bgF:5´−tggaactcaagatttgaccatatc−3´(配列番号19)
hSRC1xhR:5´−gagcattcctctagtctgtagtc−3´(配列番号20)
【0097】
(PCR条件:1×反応バッファー)
1μM MgSO4、0.2μM dNTP、プライマー各0.3μM、cDNA 1μL、5% DMSO、KOD−Plus−(東洋紡社製)1unitを反応液50μL中に含む。
【0098】
(PCR条件:サイクル)
94℃で40秒、56℃で20秒、68℃で5分のサイクルを40サイクル実施した。
【0099】
増幅したSRC−1 cDNAを精製し、さらに以下のプライマー(配列番号21および22)を用いたPCR法にてcDNAに制限酵素サイトを付加した。増幅したcDNAを、BglIIおよびXhoIで切断後、pESC−Leuベクターのgal1プロモーター下流に挿入した。本プラスミドDNAをpESC−Leu−hSRC1とした。
【0100】
SRC−1eFbg2:5´−caaagaagatctcccaggtgtgaag−3´(配列番号21)
SRC−1eRxh2:5´−agggccctcgagactctagtctgtag−3´(配列番号22)
【0101】
(PCR条件:1×反応バッファー)
1μM MgSO4、0.2μM dNTP、プライマー各0.3μM、PCR産物(cDNA) 1μL、5% DMSO、KOD−Plus−(東洋紡社製)1unitを反応液50μL中に含む。
【0102】
(PCR条件:サイクル)
94℃で40秒、56℃で20秒、68℃で5分のサイクルを20サイクル実施した。
【0103】
(5) 形質転換酵母の作製
まず、酵母(Saccharomyces cerevisiae)W303a株に、塩化リチウム法にて、前記pYT−β−DR3×1プラスミド DNAを導入した。このときの形質転換酵母選択培地は、後述する前培養培地にロイシンを100mg添加したものである。
【0104】
つぎに、前記形質転換酵母に、pESC−Leu−hSRC1プラスミド DNAを導入した。このときの形質転換酵母選択培地は、後述する前培養培地である。
【0105】
さらに、前記pUdp8PXR/RXRαを、制限酵素EcoRVで処理して直鎖状とし、以下のように前記形質転換酵母へ導入し、染色体への組込みを行った。
【0106】
まず、前記形質転換酵母を、30℃で濁度(Abs660nm、以下、OD660とする)が1〜2になるまでYPD培地中で培養し、ソリューションA(0.1M Lithium acetate、10mM Tris−HCl(pH 7.5)、1mM EDTA いずれも最終濃度)で洗浄後、OD660=150となるようにソリューションAに再懸濁し、1.5mLマイクロチューブに100μLずつ分注して、30℃で1時間インキュベートした。その後、直鎖状の前記の各pAUR101ベクター 5μgと、キャリアーDNA(商品名:SALMON TESTES DNA for hybridization、SIGMA社製)150μg(合計20μL)を加え、さらに、ソリューションB(ソリューションA100mLにポリエチレングリコール4000を溶解したもの)を850μL添加し、緩やかに混合した。
【0107】
つぎに、前記混合液を30℃で30分インキュベートして42℃で15分熱処理した後、さらに、10分室温に放置してから、5000rpm、1分間の遠心分離で集菌し、それらを5〜10mLのYPD培地に懸濁し、30℃で一晩培養した。そして、培養後集菌して洗浄した酵母を、0.9%NaCl溶液に懸濁し、オーレオバシディンA(AureobasidinA、0.5g/mL)を含むYPD選択プレートに100μLずつ塗布して培養した。そして、オーレオバシディンA耐性株をGal1プロモーターとPXRのcDNA領域が染色体上に組込まれた形質転換酵母として選択した。以上の操作により、pYT−β−DR3×1およびpESC−Leu−hSRC1DNAをプラスミドとして保持し、ゲノム中よりヒトRXRαおよびPXRを発現する形質転換酵母を作製した。
【0108】
(6)PXRのmRNA量の測定
本例の形質転換酵母について、PXRのmRNA量を測定した。
【0109】
(形質転換酵母の前培養)
まず、本例の形質転換酵母を、下記組成の前培養用の培地にて18時間前培養を行った。培養の結果、濁度(Abs595nm)が1.0程度に形質転換酵母が増殖した。
【0110】
(前培養用培地の組成)
成分 配合量
ドロップアウトパウダー(前記注1) 1.3g
窒素源(Yeast nitrogen base、前記注2) 1.7g
(NH4)2SO4 5.0g
グルコース 20.0g
水 1L
【0111】
(形質転換酵母の本培養)
つぎに、96穴プレートを用い、1ウェルあたり以下の構成で、リガンド溶液、前培養した菌体液および本培養用培地を混合し、30℃で18時間、静置培養を行った。
【0112】
リガンド溶液:10mM DMSOに溶解したリファンピシン(Rifampicin、RIF)1μL
菌体液(O.D.1〜1.5):5μL
本培養用培地:下記組成の培地100μL
【0113】
(本培養用培地の組成)
成分 配合量
ドロップアウトパウダー(前記注1) 1.3g
窒素源(Yeast nitrogen base、前記注2) 1.7g
(NH4)2SO4 5.0g
グルコース 2.0g
ガラクトース 18.0g
水 1L
【0114】
(RNAの抽出およびcDNA合成)
さらに、前記本培養後の形質転換酵母を、遠心して集菌後、ソルビトールバッファー(1M ソルビトール、0.1M EDTA)に懸濁し、30℃で30分振とうした。振とうした菌体は、再度遠心して集菌後、RNeasy mini kit(Quiagen社製)を用いて、トータルRNA(total RNA)を抽出した。前記トータルRNAの抽出操作は、キット添付の操作書に従って行った。抽出したトータルRNAを用いて、cDNAを合成した。前記cDNAの合成は、以下の条件で行った。
【0115】
(PCR条件:反応液 5×SSIIバッファー)
5mM DTT、0.5mM dNTP、RNase inhibitor 20unit、2.5μM oligo−dT primer(oligo−dT、タカラバイオ社製)または random 6mer(Rd6、タカラバイオ社製)、SuperScriptIII RT(Invitrogen社)200unit、2μg yeast total RNAを反応液20μL中に含む。
【0116】
(PCR条件:サイクル)
25℃で10分、50℃で30分、55℃で30分の条件で反応させた。
【0117】
cDNA合成後、Smart Cycler V2.0(Cepheid社製)を用いてリアルタイムPCRを行った。用いたプライマー(配列番号23および24)リアルタイムPCRの条件を以下に示す。
【0118】
hPXR1008RTf: 5′−tgaggaggagtatgtgctgatgc−3′(配列番号23)
PXRrHd2: 5′−ttctcagctacctgtgatgccgaac−3′(配列番号24)
【0119】
(PCR条件:反応液組成)
1×CYBR Premix Ex Taq(TAKARA社製)、プライマー各0.5μM、1μL cDNA(またはスタンダードとしてプラスミドDNA)を反応液25μL中に含む。
【0120】
(PCR条件:サイクル)
94℃で30秒、60℃で30秒、72℃で30分のサイクルを45サイクル実施した。
【0121】
(リアルタイムPCRによる測定)
mRNA定量における検量線作成用のスタンダードとして、プラスミドpUdp3PXR/RXRαを作製した。前記プラスミドpUdp3PXR/RXRαは、前記プラスミドpUdp8に代えて、前記プラスミドpUdp3を用いたこと以外は、前記プラスミドpUdp8PXR/RXRαと同様にして作製した。前記プラスミドpUdp3PXR/RXRα DNAは、1pg/mL〜1μg/mLの濃度に希釈した。得られたcDNAサンプルおよび前記スタンダードについて、同時に、リアルタイムPCRによる測定を行った。
【0122】
(7)リガンド応答性の測定
本例の形質転換酵母について、リガンドへの応答性を測定した。
【0123】
(形質転換酵母の培養)
本例の形質転換酵母は、前述と同様に、前培養および本培養を行った。
【0124】
(β−ガラクトシダーゼ活性の測定)
前記本培養の後、各ウェルから培養液を5μLずつ採取し、新たな96穴プレートの各ウェルに移し、下記組成の測定試薬を100μL加えた後、37℃で30分間反応させた。反応後、マイクロプレートリーダーを用いて、前記反応液のβ−ガラクトシダーゼ活性および菌体量を測定した。前記β−ガラクトシダーゼ活性は、o−ニトロフェノールの生成量から算出するため、405nmにおける反応液の吸光度を測定した。また、前記菌体量は、620nmにおける吸光度を測定した。分析結果は、405nmの吸光度/620nmの吸光度の比を算出し、リガンド非添加(溶媒である10mM DMSOのみを添加)での値を1とした誘導率で示した。
【0125】
(測定試薬の組成)
成分 配合量(最終濃度)
Na2HPO4(注4) 60mM
NaH2SO4(注4) 40mM
MgSO4 1mM
KCl 10mM
ジチオスレイトール(Dithiothreitol) 2mM
サルコシル(前記注3) 0.40w/v%
ONPG(o−ニトロフェニル−β−D−ガラクトピラノシド) 1mg/mL
(注4)Na2HPO4およびNaH2SO4を混合後、pH7.5に調整
【0126】
(比較例1−1)
(1)レポータープラスミドの作製
本例の形質転換酵母において、レポータープラスミドは、実施例1と同様に作製した。
【0127】
(2)発現量調整プラスミドの作製
本例の形質転換酵母において、発現量調整プラスミドは、前記酵母cyc1遺伝子のターミネーター配列および前記RNA不安定化配列を含まない、前記プラスミドpUdp3を用いた。前記プラスミドpUdp3は、実施例1と同様に作製した。
【0128】
(3)動物核内受容体発現プラスミドの作製
本例の形質転換酵母において、動物核内受容体発現プラスミドとして、前記プラスミドpUdp3PXR/RXRαを用いた。
【0129】
(4)SRC1プラスミドの作製
本例の形質転換酵母において、SRC1プラスミドは、実施例1と同様に作製した。
【0130】
(5)形質転換酵母の作製
本例の形質転換酵母は、前記動物核内受容体発現プラスミドとして、前記プラスミドpUdp8PXR/RXRαに代えて、前記プラスミドpUdp3PXR/RXRαを用いたこと以外は、実施例1と同様に作製した。
【0131】
(6)PXRのmRNA量の測定
本例の形質転換酵母における、PXRのmRNA量測定は、実施例1と同様にして行った。
【0132】
(7)リガンド応答性の測定
本例の形質転換酵母について、実施例1と同様に、リガンド応答性を測定した。
【0133】
(比較例1−2)
(1)レポータープラスミドの作製
本例の形質転換酵母において、レポータープラスミドは、実施例1と同様に作製した。
【0134】
(2)発現量調整プラスミドの作製
本例の形質転換酵母において、発現量調整プラスミドは、前記RNA不安定化配列を含まず、かつ前記酵母cyc1遺伝子のターミネーター配列に代えて、酵母ADH遺伝子のターミネーター配列を挿入した、プラスミドpUdp4を用いた。前記酵母ADH遺伝子のターミネーター配列の挿入は、以下のように行った。
【0135】
前記酵母ADH遺伝子のターミネーター配列の増幅は、用いたプライマー以外は、前記酵母cyc1遺伝子のターミネーター配列と同様にして行った。以下に、用いたプライマー(配列番号25および26)を示す。増幅させた前記酵母ADH遺伝子のターミネーター配列を含むDNAフラグメントは、制限酵素EcoRIおよびMflIを用いて切断し、前記pUdp3のEcoRIサイトに挿入した。
【0136】
(ADH)
scADH3UTRfEc:5´−ctaaataagcgaattccttatgatttatg−3´(配列番号25)
scADH3UTRrMf:5´−tgcaggcaattgctcggcatgccggtag−3´(配列番号26)
【0137】
(3)動物核内受容体発現プラスミドの作製
本例の形質転換酵母において、動物核内受容体発現プラスミドは、前記プラスミドpUdp8に代えて、前記プラスミドpUdp4を用いたこと以外は、実施例1と同様にして作製した。前記プラスミドpUdp4を用いて作製した本例の動物核内受容体発現プラスミドを、プラスミドpUdp4PXR/RXRαとした。
【0138】
(4)SRC1プラスミドの作製
本例の形質転換酵母において、SRC1プラスミドは、実施例1と同様に作製した。
【0139】
(5)形質転換酵母の作製
本例の形質転換酵母は、前記動物核内受容体発現プラスミドとして、前記プラスミドpUdp8PXR/RXRαに代えて、前記プラスミドpUdp4PXR/RXRαを用いたこと以外は、実施例1と同様に作製した。
【0140】
(6)PXRのmRNA量の測定
本例の形質転換酵母において、PXRのmRNA量測定は、実施例1と同様にして行った。
【0141】
(7)リガンド応答性の測定
本例の形質転換酵母について、実施例1と同様に、リガンドへの応答性を測定した。
【0142】
(比較例1−3)
(1)レポータープラスミドの作製
本例の形質転換酵母において、レポータープラスミドは、実施例1と同様に作製した。
【0143】
(2)発現量調整プラスミドの作製
本例の形質転換酵母において、発現量調整プラスミドは、前記RNA不安定化配列を含まない、前記プラスミドpUdp5を用いた。前記プラスミドpUdp5は、実施例1と同様に作製した。
【0144】
(3)動物核内受容体発現プラスミドの作製
本例の形質転換酵母において、動物核内受容体発現プラスミドは、前記プラスミドpUdp8に代えて、前記プラスミドpUdp5を用いたこと以外は、実施例1と同様にして作製した。前記プラスミドpUdp5を用いて作製した本例の動物核内受容体発現プラスミドを、プラスミドpUdp5PXR/RXRαとした。
【0145】
(4)SRC1プラスミドの作製
本例の形質転換酵母において、SRC1プラスミドは、実施例1と同様に作製した。
【0146】
(5)形質転換酵母の作製
本例の形質転換酵母は、前記動物核内受容体発現プラスミドとして、前記プラスミドpUdp8PXR/RXRαに代えて、前記プラスミドpUdp5PXR/RXRαを用いたこと以外は、実施例1と同様に作製した。
【0147】
(6)PXRのmRNA量の測定
本例の形質転換酵母において、PXRのmRNA量測定は、実施例1と同様にして行った。
【0148】
(7)リガンド応答性の測定
本例の形質転換酵母について、実施例1と同様に、リガンド応答性を測定した。
【0149】
前記実施例1および前記比較例1−1〜1−3で作製した形質転換酵母について、PXRのmRNA量を測定した結果を、下記表3に示す。
【0150】
【表3】
【0151】
前記表3に示すように、Rd6を用いてcDNAを作製した場合、実施例1のmRNA量は、比較例1−1〜1−3の約1/70〜1/170であった。また、oligo−dTを用いてcDNAを作製した場合、実施例1のmRNA量は、比較例1−1〜1−3の約1/4〜1/85であった。すなわち、RNA不安定化配列を有する実施例1において、PXRのmRNA量が著しく低いことが示された。また、Rd6を用いてcDNAを作製した場合、比較例1−1〜1−3間のmRNA量に大きな差異はなかった。しかし、oligo−dTを用いてcDNAを作製した場合、酵母cyc1遺伝子のターミネーター配列を持つ比較例1−3が、他の比較例に比べて特に高い値を示した。前記Rd6プライマーは、ランダム配列を有するプライマーであり、前記oligo−dTプライマーは、ポリA tailを持つmRNAの逆転写反応用プライマーである。すなわち、ターミネーター配列の有無および種類等により、ポリAが付加されたPXRのmRNA量が異なることが示された。
【0152】
前記実施例1および前記比較例1−1〜1−3で作製した形質転換酵母について、リガンドへの応答性を測定した結果を、下記表4に示す。
【0153】
【表4】
【0154】
前記表4に示すように、実施例1では、リファンピシンに応答して、β−ガラクトシダーゼが誘導された。実施例1の前記誘導率は、約4倍であった。それに対し、比較例1−1〜1−3では、β−ガラクトシダーゼの誘導は認められなかった。また、リガンド非添加時(陰性対照)におけるβ−ガラクトシダーゼの発現量が、実施例1では低いのに対し、比較例1−1〜1−3では、リガンド添加時の値に近い高値を示した。
【0155】
(実施例2)
(1)レポータープラスミドの作製
本例の形質転換酵母において、レポータープラスミドは、実施例1と同様に作製した。
【0156】
(2)発現量調整プラスミドの作製
本例の形質転換酵母において、発現量調整プラスミドは、実施例1と同様に作製した。
【0157】
(3)動物核内受容体発現プラスミドの作製
本例の形質転換酵母において、動物核内受容体発現プラスミドとしては、前記プラスミドpUdp8PXR/RXRαの、RXRαのC末端のHelix12を欠失させたプラスミドpUdp8PXR/RXRαTを用いた。前記プラスミドpUdp8PXR/RXRαTは、以下のように作製した。
【0158】
まず、QuickChange Site−Directed Mutagenesis Kit(Stratagene社製)を用いて、前記pUdp8PXR/RXRαに含まれるRXR cDNAの443番目のコドンにナンセンス変異を導入し、RXRαのC末端に存在するHelix12領域を欠失させた。用いたプライマー(配列番号27および28)および反応条件を以下に示す。
【0159】
RXR443F: 5′−cttcttcaagctcatcgggtagacacccat−3′(配列番号27)
RXR443R: 5′−ggtgtcaatgggtgtctacccgatgagctt−3′(配列番号28)
【0160】
(PCR条件:1×反応バッファー)
0.25mM dNTP、3μL Quick solution、pfu ultra polymerase 10unit(以上Stratagene社製)、0.25μM 各プライマー、10ng プラスミドDNAを反応液50μL中に含む。
【0161】
(PCR条件:サイクル)
94℃で50秒、60℃で50秒、68℃で7分のサイクルを18サイクル実施した。
【0162】
PCR後、制限酵素DpnIで鋳型プラスミドDNAを消化し、大腸菌に形質転換した。得られたプラスミドDNAをpUdp8PXR/RXRαTとした。
【0163】
(4)SRC1プラスミドの作製
本例の形質転換酵母において、SRC1プラスミドは、実施例1と同様に作製した。
【0164】
(5)形質転換酵母の作製
本例の形質転換酵母は、前記動物核内受容体発現プラスミドとして、前記プラスミドpUdp8PXR/RXRαに代えて、前記プラスミドpUdp8PXR/RXRαTを用いたこと以外は、実施例1と同様に作製した。
【0165】
(6)リガンド応答性の測定
本例の形質転換酵母について、以下のようにしてリガンド応答性を測定した。
【0166】
(形質転換酵母の培養)
本例の形質転換酵母は、実施例1と同様にして、前培養を行った。
【0167】
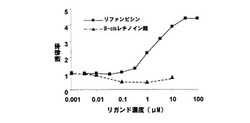
本例の形質転換酵母の本培養は、リファンピシンおよび9−cisレチノイン酸をリガンド溶液として用いたこと以外は、実施例1と同様に行った。なお、前記リファンピシンは、10mM DMSOに溶解させ、下記表5に示すように、異なる10濃度の溶液を調製した。また、前記9−cisレチノイン酸は、10mM DMSOに溶解させ、下記表6に示すように、異なる5濃度の溶液を調製した。
【0168】
(β−ガラクトシダーゼ活性の測定)
本例の形質転換酵母は、実施例1と同様にしてβ−ガラクトシダーゼ活性を測定した。前記測定の結果を、下記表5、表6および図1に示す。表5は、前記リファンピシンをリガンドとして用いた測定結果であり、表6は、前記9−cisレチノイン酸をリガンドとして用いた測定結果である。図1において、横軸は、各リガンドの濃度(μM)であり、縦軸は、リガンド非添加での値を1とした誘導率である。
【0169】
【表5】
【0170】
【表6】
【0171】
前記表5、6および図1に示すように、本例の形質転換酵母において、リファンピシンによるβ−ガラクトシダーゼの誘導率は、約4倍であった。それに対し、本例の形質転換酵母において、9−cisレチノイン酸による誘導は、全く見られなかった。すなわち、本例の形質転換酵母は、RXRαのリガンドには反応せず、PXRのリガンドのみに反応することが示された。
【0172】
(実施例3)
(1)レポータープラスミドの作製
本実施例の形質転換酵母において、レポータープラスミドは、実施例1と同様に作製した。
【0173】
(2)発現量調整プラスミドの作製
本実施例の形質転換酵母において、発現量調整プラスミドは、実施例1と同様に作製した。
【0174】
(3)動物核内受容体発現プラスミドの作製
本実施例の形質転換酵母において、動物核内受容体発現プラスミドは、前記核内受容体遺伝子PXRに代えて、核内受容体遺伝子VDRを発現可能に導入したこと以外は、実施例2と同様に作製した。本実施例における前記動物核内受容体発現プラスミドを、プラスミドpUdp8VDR/RXRαTとした。前記プラスミドpUdp8VDR/RXRαTは、前記ヒトVDRの増幅および導入時に用いたプライマーおよび制限酵素以外は、実施例2と同様に作製した。以下に、前記用いたプライマー(配列番号29および30)および制限酵素を示す。
【0175】
(プライマー)
VDRfXb:5´−ctccttctagaatggaggcaatggcggccagcac−3´(配列番号29)
VDRrSp:5´−gctgtactagtcaggagatctcattgccaaacac−3´(配列番号30)
【0176】
(制限酵素)
挿入したcDNA cDNA切断用制限酵素 プラスミド切断用制限酵素
VDR SpeI、XbaI XbaI
【0177】
(4)SRC1プラスミドの作製
本実施例の形質転換酵母において、SRC1プラスミドは、実施例1と同様に作製した。
【0178】
(5)形質転換酵母の作製
本実施例の形質転換酵母は、前記動物核内受容体発現プラスミドとして、前記プラスミドpUdp8PXR/RXRαに代えて、前記プラスミドpUdp8VDR/RXRαTを用いたこと以外は、実施例1と同様に作製した。
【0179】
(6)リガンド応答性の測定
本例の形質転換酵母について、以下のようにしてリガンド応答性を測定した。
【0180】
(形質転換酵母の培養)
本例の形質転換酵母は、実施例1と同様にして、前培養を行った。
【0181】
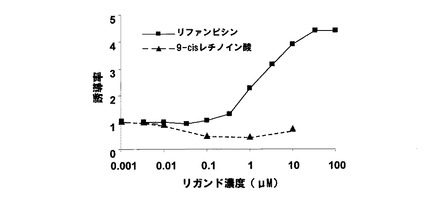
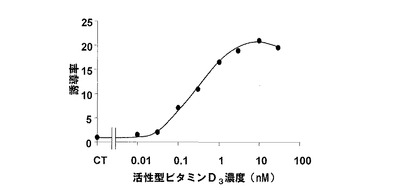
本例の形質転換酵母の本培養は、リガンド溶液として、活性型ビタミンD3(1,25−OH VD3)を用いたこと以外は、実施例1と同様に行った。なお、前記活性型ビタミンD3は、10mM DMSOに溶解させ、下記表7に示すように、異なる9濃度の溶液を調製した。
【0182】
(β−ガラクトシダーゼ活性の測定)
本例の形質転換酵母は、実施例1と同様に、β−ガラクトシダーゼ活性を測定した。前記測定の結果を、下記表7および図2に示す。図2において、横軸は、活性型ビタミンD3の濃度(μM)であり、縦軸は、リガンド非添加での値を1とした誘導率である。
【0183】
【表7】
【0184】
前記表7および図2に示すように、本例の形質転換酵母では、活性型ビタミンD3により、β−ガラクトシダーゼが誘導された。本例の形質転換酵母において、前記誘導率は、最大約20倍であった。
【0185】
これらの結果から、本発明の形質転換酵母を用いることにより、RXRα核内受容体とヘテロ二量体を形成する核内受容体のリガンドを、高感度で分析できる。また、前記核内受容体発現プラスミドpUdp8は、PXRだけでなく、本実施例のVDRのように、RXRαとヘテロ二量体を形成する他の核内受容体にも、利用可能である。さらに、例えば、前記非特許文献3の図1(2)のグラフ(j)において、酵母ツーハイブリッド法を用いた、活性型ビタミンD3誘導によるβ−ガラクトシダーゼ活性の測定結果が示されているが、同法は、本発明の前記実施例3と比べ、活性を示す最低濃度が約10倍大きく、測定感度が低い。このことから、本発明の形質転換酵母を用いた分析方法は、前記酵母ツーハイブリッド法と比べ、高い測定感度を有するといえる。しかも、本発明の形質転換酵母を用いた分析方法は、操作が簡単であり、短時間で分析ができ、低コストであるという利点もある。
【産業上の利用可能性】
【0186】
以上のように、本発明の形質転換酵母を用いれば、高検出感度でRXRα核内受容体とヘテロ二量体を形成する核内受容体のリガンドの分析が可能となる。したがって、本発明の分析方法は、化学物質やタンパク質等のリガンドの分析の分野において有用な方法であり、その用途は制限されず広い。
【図面の簡単な説明】
【0187】
【図1】図1は、本発明の実施例2におけるβ−ガラクトシダーゼ活性の測定結果を示すグラフである。
【図2】図2は、本発明の実施例3におけるβ−ガラクトシダーゼ活性の測定結果を示すグラフである。
【図3】図3は、本発明の実施例および比較例の形質転換酵母に含まれる、核内受容体発現プラスミドのプロモーター周辺の構造図である。図3(A)は、比較例1−1(pUdp3)、図3(B)は、比較例1−2(pUdp4)、図3(C)は、比較例1−3(pUdp5)、図3(D)は、実施例1(pUdp8)に含まれる核内受容体発現プラスミドの前記構造図である。
【技術分野】
【0001】
本発明は、形質転換酵母、それを用いた分析方法および分析キットに関する。
【背景技術】
【0002】
環境中の化学物質の分析において、従来は機器分析が主流であったが、生物的な影響をも評価できる分析手法として核内受容体遺伝子を宿主細胞に組み込み、その発現の程度で化学物質の分析を行う手法が開発されている。宿主細胞としては、ヒト等の動物細胞および酵母が使用されている。動物細胞を用いた分析方法としては、例えば、アンドロゲンレセプター(AR)遺伝子をヒト由来細胞株に組み込み、テストステロン(T)等を分析する方法がある(非特許文献1)。一方、雌化現象等で注目された環境ホルモン(内分泌撹乱物質)のエストロゲン様活性の分析方法として、ブルーギル由来のエストロゲン受容体遺伝子(特許文献1)、オオマキトカゲ由来エストロゲン受容体遺伝子(特許文献2)、ワニ由来エストロゲン受容体遺伝子(特許文献3)、ファットヘッドミノー由来エストロゲン受容体遺伝子(特許文献4)等の各種野生動物のエストロゲン受容体遺伝子を酵母に組み込み、その発現を測定するという手法が開発されている(特許文献1、特許文献2、特許文献3、特許文献4)。この他、ヒト由来エストロゲン受容体遺伝子を組み込んだ形質転換酵母を用いてアルキルフェノール化合物のエストロゲン様活性を測定する手法が開発されている(非特許文献2)。また、例えば、ダイオキシン等のアリールハイドロカーボン類を測定するために、モルモット由来のアリールハイドロカーボン受容体遺伝子を組み込んだ形質転換酵母を用いた手法が開発されている(特許文献5)。これらの酵母を用いた手法では、各種核内受容体遺伝子が発現し、測定対象となるリガンド(化学物質等)が前記核内受容体に結合して複合体を形成し、この複合体がレポーター遺伝子を発現させる。前記レポーター遺伝子としては、β−ガラクトシダーゼ遺伝子が使用されており、β−ガラクトシダーゼの酵素活性を測定することで、遺伝子の発現を測定している。酵母を用いた分析は、動物細胞を用いた分析方法に比べて、短時間分析が可能であり、操作の簡便性および低コスト等の利点を有する。
【0003】
一方、ヒト核内受容体は、二量体形成後、核内受容体応答配列に結合して、標的遺伝子の転写調節を行う。ヒト核内受容体には、同じ核内受容体と二量体を形成するホモ二量体型核内受容体と、異なる核内受容体と二量体を形成するヘテロ二量体型核内受容体がある。ホモ二量体型核内受容体には、ステロイドホルモン核内受容体等の数種類があるが、ヒト核内受容体の多くは、レチノイン酸受容体α(RXRα受容体)とヘテロ二量体を形成するRXRαヘテロ二量体型核内受容体である。ホモ二量体型核内受容体については、前述のレポーター遺伝子導入酵母を用いたリガンド分析手法が開発されている(特許文献6)。しかし、ヘテロ二量体型核内受容体については、形質転換酵母における分析方法が開発されていない。また、RXRαヘテロ二量体型核内受容体については、酵母ツーハイブリッド法によるリガンド分析方法が報告されている(非特許文献3)。しかしながら、酵母ツーハイブリッド法は、リガンド非依存的な転写活性が生じるため、分析精度が低いという問題がある。
【0004】
【特許文献1】特開2001−197890号公報
【特許文献2】特開2003−274号公報
【特許文献3】特開2002−360273号公報
【特許文献4】特開2001−352992号公報
【特許文献5】特開2005−87077号公報
【特許文献6】特開2007−295806号公報
【非特許文献1】Toxicology、2006年、Vol.220、p.90−103
【非特許文献2】The Journal Of Biological Chemistry、1997年、Vol.272、No.6、p.3280−3288
【非特許文献3】Journal of Environmental Biotechnology、2003年、 Vol.3、No.1、p.37−42
【発明の開示】
【発明が解決しようとする課題】
【0005】
そこで、本発明の目的は、RXRαヘテロ二量体型動物核内受容体リガンドを高感度で分析可能な形質転換酵母、それを用いたRXRαヘテロ二量体型動物核内受容体リガンドの分析方法および分析キットを提供することである。
【課題を解決するための手段】
【0006】
前記目的を達成するために、本発明の形質転換酵母は、動物核内受容体遺伝子およびレポーター遺伝子が発現可能に導入されており、かつ動物核内受容体リガンドと動物核内受容体との複合体を認識して前記レポーター遺伝子を発現し得る形質転換酵母であって、
前記動物核内受容体が、RXRα核内受容体と二量体を形成する、RXRαヘテロ二量体型核内受容体であり、
前記動物核内受容体遺伝子として、RXRα核内受容体遺伝子およびRXRαへテロ二量体型核内受容体遺伝子を発現可能に含み、
前記動物核内受容体に直接結合して前記レポーター遺伝子の転写を活性化する動物転写共役因子遺伝子を発現可能に含み、
さらに、前記動物核内受容体遺伝子の発現量を調節するRNA不安定化配列を含む。
【0007】
本発明の分析方法は、検体に含まれる動物核内受容体リガンドを分析するための分析方法であって、
検体を含む培地中で、動物核内受容体遺伝子およびレポーター遺伝子が発現可能に導入されており、かつ動物核内受容体リガンドと動物核内受容体との複合体を認識して前記レポーター遺伝子を発現し得る形質転換酵母を培養する培養工程と、
前記培養工程の後、前記培地中に産生された前記レポーター遺伝子の発現産物を測定する測定工程とを包含し、
前記形質転換酵母として、本発明の形質転換酵母を使用することを特徴とする。
【0008】
本発明の分析キットは、検体に含まれる動物核内受容体リガンドを分析するための分析キットであって、
(a)培地と、
(b)本発明の形質転換酵母と、
(c)発色基質と
を備える。
【0009】
本発明のベクターは、動物核内受容体遺伝子を含むベクターであって、
前記動物核内受容体遺伝子として、RXRα核内受容体遺伝子およびRXRαへテロ二量体型核内受容体遺伝子を含み、
さらに、前記動物核内受容体遺伝子の発現量を調節するRNA不安定化配列を含む。
【0010】
本発明のベクターセットは、レポーター遺伝子を含むベクター、動物転写共役遺伝子を含むベクターおよび本発明のベクターを含有する。
【発明の効果】
【0011】
本発明者等は、形質転換酵母を用いたRXRαへテロ二量体型核内受容体リガンドの分析方法の感度向上を目的として、一連の研究を行った。その過程で、前記RXRαへテロ二量体型核内受容体の発現量に着目し、その発現量を必要最小限に留めるという着想を得た。すなわち、形質転換酵母で発現させたRXRαへテロ二量体型核内受容体は、リガンド非依存的に、転写活性化されるため、従来は、形質転換酵母でのヘテロ二量体型核内受容体リガンドの分析手法は実現できなかった。そこで、本発明者等は、その原因をつぎのように推察した。リガンド非依存的な転写活性化の原因の一つとして、前記RXRαへテロ二量体型核内受容体の過剰発現により、前記RXRαへテロ二量体型核内受容体の細胞質内保持および核内移行の制御が適切に行われないことが考えられる。また、別の原因として、前記RXRαへテロ二量体型核内受容体とヘテロ二量体を形成するRXRα核内受容体に起因する転写活性化等も考えられる。したがって、RXRαへテロ二量体型核内受容体の発現を必要最小限に留めること、およびRXRα核内受容体に起因する転写活性化を抑制することが、RXRαへテロ二量体型核内受容体リガンドの分析方法の確立に有効であると考えた。そこで、本発明者等は、遺伝子の3′非翻訳領域に存在し、前記遺伝子から転写されたmRNAの分解を促進するRNA不安定化配列に着目し、前記RNA不安定化配列を形質転換酵母に組み込んだ。その結果、前記RNA不安定化配列を組み込んだ形質転換酵母を使用すれば、RXRαへテロ二量体型動物核内受容体リガンドを高感度で分析できることを見出し、本発明に到達した。
したがって、本発明の形質転換酵母を用いれば、RXRαへテロ二量体型動物核内受容体リガンドを、簡単な操作により、低コスト、短時間かつ高感度で分析可能である。また、本発明の分析キットを用いれば、さらに簡便に、RXRαへテロ二量体型動物核内受容体リガンドの分析を実施可能である。
【発明を実施するための最良の形態】
【0012】
本発明の形質転換酵母において、前記RNA不安定化配列が、前記RXRαへテロ二量体型核内受容体遺伝子の発現量を調節するRNA不安定化配列であることが好ましい。前記発現量の調節としては、前記RXRαへテロ二量体型核内受容体遺伝子の発現量を低減することが好ましい。
【0013】
本発明の形質転換酵母において、前記RXRα核内受容体が、C末端のHelix12領域を欠失させたRXRα核内受容体であることが好ましい。
【0014】
本発明の形質転換酵母において、さらに、ターミネーター配列を含むことが好ましい。
【0015】
本発明の形質転換酵母において、前記動物核内受容体が、ヒト核内受容体であり、前記動物転写共役因子がヒト転写共役因子であることが好ましい。
【0016】
本発明の形質転換酵母において、前記ヒト転写共役因子遺伝子が、ヒトSRC1遺伝子であることが好ましい。
【0017】
本発明の形質転換酵母において、前記動物核内受容体遺伝子および前記RNA不安定化配列を常染色体上に有し、前記レポーター遺伝子を第1のプラスミドに有し、前記動物転写共役因子遺伝子を第2のプラスミドに有し、前記第1のプラスミドは、前記レポーター遺伝子と作動的に組み込まれた動物核内受容体応答配列を含むことが好ましい。
【0018】
前記レポーター遺伝子が、β−ガラクトシダーゼ遺伝子であることが好ましい。
【0019】
前記形質転換酵母が、出芽酵母(Saccharomyces cerevisiae)であることが好ましい。
【0020】
本発明の形質転換酵母は、例えば、検体に含まれる動物核内受容体リガンドの分析に使用される。
【0021】
本発明の分析方法において、前記レポーター遺伝子が、β−ガラクトシダーゼ遺伝子であり、前記培地が、炭素源としてガラクトースおよびグルコースを含有することが好ましい。グルコースを含む培地で前記形質転換酵母を培養すれば、例えば、18時間の短時間で分析可能なレベルまで増殖させることができる。本発明の分析方法において、前記ガラクトースおよび前記グルコースの合計に対する前記グルコースの重量割合は、特に制限されないが、例えば、0を超え60重量%以下の範囲、好ましくは、0を超え40重量%以下の範囲、より好ましくは、10〜30重量%の範囲である。
【0022】
本発明の分析方法において、前記形質転換酵母が、グリセロール存在下で保存されていた酵母であることが好ましい。前記グリセロール存在下で保存されていた形質転換酵母は、予め、グルコース単独を炭素源とする培地で培養された酵母であることが好ましい。
【0023】
本発明の分析方法は、例えば、検出、定性分析または定量分析である。また、本発明において、分析は、検出、定性分析および定量分析を含む。
【0024】
本発明の分析キットにおいて、前記培地は、グルコースおよびガラクトースを含む培地であることが好ましい。前記グルコースの重量割合は、前述のとおりである。
【0025】
本発明のベクターにおいて、前記RNA不安定化配列が、前記RXRαへテロ二量体型核内受容体遺伝子の発現量を調節するRNA不安定化配列であることが好ましい。そして、本発明のベクターにおいて、前記RXRα核内受容体が、C末端のHelix12領域を欠失させたRXRα核内受容体であることが好ましい。また、本発明のベクターは、さらに、ターミネーター配列を含むことが好ましい。
【0026】
つぎに、本発明について詳しく説明する。
【0027】
本発明において、前記RXRαへテロ二量体型核内受容体および前記RXRαへテロ二量体型核内受容体リガンドは、特に制限されず、例えば、下記に示すRXRαへテロ二量体型核内受容体および前記受容体に結合可能なリガンドがあげられる。
【0028】
(1)TRα:Thyroid Hormone Receptor alpha
(2)TRβ:Thyroid Hormone Receptor beta
(3)RARα:Retinoic Acid Receptor alpha
(4)RARβ:Retinoic Acid Receptor beta
(5)RARγ:Retinoic Acid Receptor gamma
(6)PPARα:Peroxisome proliferator activated receptor alpha(PPAR−alpha)
(7)PPARδ:Peroxisome proliferator activated receptor delta(PPAR−delta、PPAR−beta、Nuclear hormone receptor 1、NUC1)
(8)PPARγ1:Peroxisome proliferator activated receptor gamma 1(PPAR−gamma1)
(9)PPARγ2:Peroxisome proliferator activated receptor gamma 2(PPAR−gamma2)
(10)LXRβ:Oxysterols receptor LXR−beta (Liver X receptor beta、Nuclear orphan receptor LXR−beta、Ubiquitously−expressed nuclear receptor、Nuclear receptor NER)
(11)LXRα:Oxysterols receptor LXR−alpha(Liver X receptor alpha、Nuclear orphan receptor LXR−alpha)
(12)VDR:Vitamin D3 receptor(VDR、1,25−dihydroxyvitamin D3 receptor)
(13)PXR−1:Orphan nuclear receptor PXR−1(Pregnane X receptor−1、Orphan nuclear receptor PAR−1、Steroid and xenobiotic receptor−1、SXR−1)
(14)PXR−2:Orphan nuclear receptor PXR−2(Pregnane X receptor−2、Orphan nuclear receptor PAR−2、Steroid and xenobiotic receptor−2、SXR−2)
(15)CAR:Orphan nuclear receptor NR1I3(Constitutive androstane receptor、CAR、Orphan nuclear receptor MB67)
(16)COUP−TF I:COUP transcription factor 1(COUP−TF1、COUP−TFα、V−erbA related protein EAR−3)
(17)COUP−TF II:COUP transcription factor 2(COUP−TF2、COUP−TFβ、Apolipoprotein AI regulatory protein−1、ARP−1)
(18)ERRα:Steroid hormone receptor ERR1(Estrogen−related receptor, alpha、ERR−alpha、Estrogen receptor−like 1)
(19)ERRβ:Steroid hormone receptor ERR2(Estrogen−related receptor,beta、ERR−beta、Estrogen receptor−like 2、ERR beta−2)
(20)ERRγ:Estrogen−related receptor gamma(Estrogen receptor related protein 3、ERR gamma−2)
【0029】
これらのRXRαへテロ二量体型核内受容体は、分析対象となるリガンドに応じて、適宜選択すればよい。
【0030】
つぎに、本発明において、動物転写共役因子は特に制限されない。動物転写共役因子としては、例えば、下記表1に示す8種類の転写共役因子が使用可能である。
【0031】
【表1】
【0032】
本発明の形質転換酵母において、導入するRXRαへテロ二量体型核内受容体および動物転写共役因子は、分析の対象となるリガンド(化学物質)の種類により適宜決定できる。例えば、本発明の形質転換酵母は、前記RXRαへテロ二量体型核内受容体として、プレグナンX受容体(PXR)、ビタミンD受容体(VDR)等を導入した態様がある。
【0033】
まず、PXR遺伝子を導入した形質転換酵母について説明する。PXRとしては、特に制限されないが、例えば、ヒトPXRがある。PXRは、リガンド(プロゲステロンもしくはプロゲステロン様化学物質)および転写共役因子と複合体を形成し、これが応答配列(DR−3配列)を認識して結合し、これにより、応答配列の下流に存在するレポーター遺伝子(例えば、β−ガラクトシダーゼ遺伝子)が発現する。レポーター遺伝子の転写は、前記転写共役因子により活性化される。
【0034】
前記ヒトPXRを酵母で発現させるためには、例えば、ヒトPXR遺伝子を前記酵母の染色体遺伝子内に導入すればよい。
【0035】
前記ヒトPXR遺伝子としては、例えば、下記(a)または(b)のポリヌクレオチドを含むことが好ましい。下記(b)のポリヌクレオチドは、下記(a)のポリヌクレオチドとの相同性が、例えば、90%以上であって、好ましくは、93%以上であり、より好ましくは、95%以上であり、さらに好ましくは、98%以上である。また、下記(b)のポリヌクレオチドにおいて、置換、付加、挿入もしくは欠失した塩基の数は、前記相同性に対応した数である。
(a)配列番号1に記載の塩基配列からなるポリヌクレオチド。
(b)配列番号1に記載の塩基配列の少なくとも1つの塩基が置換、付加、挿入もしくは欠失した塩基配列からなるポリヌクレオチドであって、ヒトPXRとして機能するタンパク質をコードするポリヌクレオチド。
【0036】
前記PXR遺伝子は、配列番号1の塩基配列に基づいて、ヒトのトータルRNA等を使用してクローニングしてもよく、また、ホスホルアミダイト(phosphoramidite)法を利用して化学的にDNA合成してもよい。前記クローニングの方法は、特に制限されず、例えば、市販のクローニングキット等を利用して実施できる。
【0037】
PXRに対する転写共役因子としては、特に制限されないが、例えば、SRC1、CBP、P300がある。したがって、これらの転写共役因子の遺伝子を形質転換酵母に組み込めばよい。これらの中で、好ましいのは、SRC1である。転写共役因子遺伝子は、第2のプラスミドに組み込んで形質転換酵母に導入することが好ましい。前記第2のプラスミドは、高コピー型プラスミドであることが好ましい。
【0038】
前記応答配列およびレポーター遺伝子は、作動的に連結して、第1のプラスミドに組み込み、この第1のプラスミドを形質転換細胞に導入することが好ましい。前記DR−3配列は、一配列のみ組み込まれてもよく、繰り返し配列として組み込まれてもよい。また、第1のプラスミドは、低コピー型プラスミドであることが好ましい。
【0039】
つぎに、VDR遺伝子を導入した形質転換酵母について説明する。VDRとしては、特に制限されないが、例えば、ヒトVDRがある。VDRは、リガンド(ビタミンD若しくはビタミンD様化学物質)および転写共役因子と複合体を形成し、これが応答配列(DR−3配列)を認識して結合し、これにより、応答配列の下流に存在するレポーター遺伝子(例えば、β−ガラクトシダーゼ遺伝子)が発現する。レポーター遺伝子の転写は、前記転写共役因子により活性化される。
【0040】
前記ヒトVDRを酵母で発現させるためには、例えば、ヒトVDR遺伝子を前記酵母の染色体遺伝子内に導入すればよい。
【0041】
前記ヒトVDR遺伝子としては、例えば、下記(c)または(d)のポリヌクレオチドを含むことが好ましい。下記(d)のポリヌクレオチドは、下記(c)のポリヌクレオチドとの相同性が、例えば、90%以上であって、好ましくは、93%以上であり、より好ましくは、95%以上であり、さらに好ましくは、98%以上である。また、下記(d)のポリヌクレオチドにおいて、置換、付加、挿入もしくは欠失した塩基の数は、前記相同性に対応した数である。
(c)配列番号2に記載の塩基配列からなるポリヌクレオチド。
(d)配列番号2に記載の塩基配列の少なくとも1つの塩基が置換、付加、挿入もしくは欠失した塩基配列からなるポリヌクレオチドであって、ヒトVDRとして機能するタンパク質をコードするポリヌクレオチド。
【0042】
前記VDR遺伝子は、配列番号2の塩基配列に基づいて、ヒトのトータルRNA等を使用してクローニングしてもよく、また、ホスホルアミダイト(phosphoramidite)法を利用して化学的にDNA合成してもよい。前記クローニングの方法は、特に制限されず、例えば、市販のクローニングキット等を利用して実施できる。
【0043】
VDRに対する転写共役因子としては、特に制限されないが、例えば、SRC1、CBP、P300、AIBがある。したがって、これらの転写共役因子の遺伝子を形質転換酵母に組み込めばよい。これらの中で、好ましいのは、SRC1である。転写共役因子遺伝子は、第2のプラスミドに組み込んで形質転換酵母に導入することが好ましい。前記第2のプラスミドは、高コピー型プラスミドであることが好ましい。
【0044】
前記応答配列およびレポーター遺伝子は、作動的に連結して、第1のプラスミドに組み込み、この第1のプラスミドを形質転換細胞に導入することが好ましい。前記DR−3配列は、例えば、一配列のみ組み込まれてもよく、繰り返し配列として組み込まれてもよい。また、第1のプラスミドは、低コピー型プラスミドであることが好ましい。
【0045】
本発明の形質転換酵母には、前記RXRαへテロ二量体型核内受容体とヘテロ二量体を形成するレチノイン酸X受容体α(RXRα)遺伝子を導入する。RXRαとしては、特に制限されないが、例えば、ヒトRXRαがある。RXRαは、前記RXRαへテロ二量体型核内受容体とヘテロ二量体を形成する。前記RXRαへテロ二量体型核内受容体とそのリガンドが結合すると、前記へテロ二量体は、前記応答配列(DR−3配列)を認識して結合し、これにより、応答配列の下流に存在するレポーター遺伝子(例えば、β−ガラクトシダーゼ遺伝子)が発現する。レポーター遺伝子の転写は、前記転写共役因子により活性化される。
【0046】
前記ヒトRXRαを酵母で発現させるためには、例えば、ヒトRXRα遺伝子を前記酵母の染色体遺伝子内に導入すればよい。
【0047】
前記ヒトRXRα遺伝子としては、例えば、下記(e)または(f)のポリヌクレオチドを含むことが好ましい。下記(f)のポリヌクレオチドは、下記(e)のポリヌクレオチドとの相同性が、例えば、90%以上であって、好ましくは、93%以上であり、より好ましくは、95%以上であり、さらに好ましくは、98%以上である。また、下記(f)のポリヌクレオチドにおいて、置換、付加、挿入もしくは欠失した塩基の数は、前記相同性に対応した数である。
(e)配列番号3に記載の塩基配列からなるポリヌクレオチド。
(f)配列番号3に記載の塩基配列の少なくとも1つの塩基が置換、付加、挿入もしくは欠失した塩基配列からなるポリヌクレオチドであって、ヒトRXRαとして機能するタンパク質をコードするポリヌクレオチド。
【0048】
前記RXRα遺伝子は、配列番号3の塩基配列に基づいて、ヒトのトータルRNA等を使用してクローニングしてもよく、また、ホスホルアミダイト(phosphoramidite)法を利用して化学的にDNA合成してもよい。前記クローニングの方法は、特に制限されず、例えば、市販のクローニングキット等を利用して実施できる。
【0049】
本発明の形質転換酵母には、前記RXRα遺伝子に代えて、例えば、C末端に存在するHelix12領域を欠失させたRXRα(以下、RXRαTとする)遺伝子を導入してもよい。前記Helix12領域は、SRC1との結合の必須領域であるため、前記RXRαT遺伝子の導入により、RXRα核内受容体に起因するリガンド非依存的な転写活性化を抑制できる。
【0050】
前記RXRαTは、例えば、前記RXRαにナンセンス変異を導入して作製できる。前記RXRαTを酵母で発現させるためには、例えば、RXRαT遺伝子を前記酵母の染色体遺伝子内に導入すればよい。
【0051】
前記RXRαT遺伝子としては、例えば、下記(g)または(h)のポリヌクレオチドを含むことが好ましい。下記(h)のポリヌクレオチドは、下記(g)のポリヌクレオチドとの相同性が、例えば、90%以上であって、好ましくは、93%以上であり、より好ましくは、95%以上であり、さらに好ましくは、98%以上である。また、下記(h)のポリヌクレオチドにおいて、置換、付加、挿入もしくは欠失した塩基の数は、前記相同性に対応した数である。
(g)配列番号4に記載の塩基配列からなるポリヌクレオチド。
(h)配列番号4に記載の塩基配列の少なくとも1つの塩基が置換、付加、挿入もしくは欠失した塩基配列からなるポリヌクレオチドであって、ヒトRXRαとして機能するタンパク質をコードするポリヌクレオチド。
【0052】
前記動物転写共役因子である前記ヒトSRC1遺伝子としては、例えば、下記(i)または(j)のポリヌクレオチドを含むことが好ましい。下記(j)のポリヌクレオチドは、下記(i)のポリヌクレオチドとの相同性が、例えば、90%以上であって、好ましくは、93%以上であり、より好ましくは、95%以上であり、さらに好ましくは、98%以上である。また、下記(j)のポリヌクレオチドにおいて、置換、付加、挿入もしくは欠失した塩基の数は、前記相同性に対応した数である。
(i)配列番号5に記載の塩基配列からなるポリヌクレオチド。
(j)配列番号5に記載の塩基配列の少なくとも1つの塩基が置換、付加、挿入もしくは欠失した塩基配列からなるポリヌクレオチドであって、ヒトSRC1として機能するタンパク質をコードするポリヌクレオチド。
【0053】
本発明の形質転換酵母には、前記RNA不安定化配列を導入する。前記RNA不安定化配列としては、特に制限されないが、例えば、HOエンドヌクレアーゼ遺伝子の3′非翻訳領域に存在するRNA不安定化配列がある。
【0054】
本発明の形質転換酵母において、前記RNA不安定化配列は、例えば、前記酵母の染色体遺伝子内に導入すればよい。
【0055】
前記RNA不安定化配列としては、例えば、下記(k)または(l)のポリヌクレオチドを含むことが好ましい。下記(l)のポリヌクレオチドは、下記(k)のポリヌクレオチドとの相同性が、例えば、90%以上であって、好ましくは、93%以上であり、より好ましくは、95%以上であり、さらに好ましくは、98%以上である。また、下記(l)のポリヌクレオチドにおいて、置換、付加、挿入もしくは欠失した塩基の数は、前記相同性に対応した数である。
(k)配列番号6に記載の塩基配列からなるポリヌクレオチド。
(l)配列番号6に記載の塩基配列の少なくとも1つの塩基が置換、付加、挿入もしくは欠失した塩基配列からなるポリヌクレオチドであって、RNA不安定化配列として機能するポリヌクレオチド。
【0056】
本発明の形質転換酵母には、例えば、ターミネーター配列を導入してもよい。前記ターミネーター配列としては、特に制限されないが、例えば、酵母cyc1遺伝子のターミネーター配列、酵母ADH遺伝子のターミネーター配列等がある。
【0057】
本発明の形質転換酵母において、前記ターミネーター配列は、例えば、前記酵母の染色体遺伝子内に導入すればよい。
【0058】
前記酵母cyc1遺伝子のターミネーター配列としては、例えば、下記(m)または(n)のポリヌクレオチドを含むことが好ましい。下記(n)のポリヌクレオチドは、下記(m)のポリヌクレオチドとの相同性が、例えば、90%以上であって、好ましくは、93%以上であり、より好ましくは、95%以上であり、さらに好ましくは、98%以上である。また、下記(n)のポリヌクレオチドにおいて、置換、付加、挿入もしくは欠失した塩基の数は、前記相同性に対応した数である。
(m)配列番号7に記載の塩基配列からなるポリヌクレオチド。
(n)配列番号7に記載の塩基配列の少なくとも1つの塩基が置換、付加、挿入もしくは欠失した塩基配列からなるポリヌクレオチドであって、ターミネーター配列として機能するポリヌクレオチド。
【0059】
前記酵母ADH遺伝子のターミネーター配列としては、例えば、下記(o)または(p)のポリヌクレオチドを含むことが好ましい。下記(p)のポリヌクレオチドは、下記(o)のポリヌクレオチドとの相同性が、例えば、90%以上であって、好ましくは、93%以上であり、より好ましくは、95%以上であり、さらに好ましくは、98%以上である。また、下記(p)のポリヌクレオチドにおいて、置換、付加、挿入もしくは欠失した塩基の数は、前記相同性に対応した数である。
(o)配列番号8に記載の塩基配列からなるポリヌクレオチド。
(p)配列番号8に記載の塩基配列の少なくとも1つの塩基が置換、付加、挿入もしくは欠失した塩基配列からなるポリヌクレオチドであって、ターミネーター配列として機能するポリヌクレオチド。
【0060】
本発明の遺伝子組換え細胞の宿主細胞は、酵母であり、好ましいのは、出芽酵母であることは、前述のとおりである。
【0061】
前記酵母内に、前記核内受容体遺伝子を導入する方法としては、特に制限されず、従来公知の遺伝子導入方法により行うことができる。前記遺伝子導入方法としては、例えば、酢酸リチウム法、リン酸カルシウム法、リポソームを用いた方法、エレクトロポレーション、ウイルスベクターを用いる方法、マイクロピペットインジェクション法等があげられる。本発明における前記核内受容体遺伝子の導入は、そのリガンドの分析に用いることが可能であれば、一過性型の導入でもよく、または、宿主染色体への組込み型もしくは自律複製・分配可能な人工染色体もしくはプラスミド型の導入でもよい。取扱いや保存の容易性およびリガンドの分析の迅速性、正確性、簡便性からは、前述のように、宿主(酵母)染色体への遺伝子導入方法が好ましい。
【0062】
前記酵母内に導入する前記核内受容体遺伝子は、酵母内で恒常的または任意に発現するように、必要な調節配列と作動的に連結されていることが好ましい。前記調節配列とは、宿主細胞(酵母)内において、作動的に連結された前記遺伝子の発現に必要な塩基配列であって、例えば、真核細胞に適した調節配列としては、プロモーター、ポリアデニル化シグナル、エンハンサー等があげられる。本発明において、作動的に連結とは、各構成要素が機能を果たすことができるように並置していることを意味する。
【0063】
前述のように、本発明において、前記核内受容体とリガンドの複合体を検出するためのレポーター遺伝子として、例えば、β−ガラクトシダーゼ遺伝子を使用することが好ましい。前記β−ガラクトシダーゼ遺伝子は、その上流に、前記複合体が認識して結合する応答配列(転写調節配列)と動作的に結合している。前記応答配列(転写調節配列)は、動物核内受容体遺伝子の種類により適宜決定される。前述のように、PXRおよびVDRの場合の応答配列は、DR−3である。これらの応答配列(転写調節配列)およびβ−ガラクトシダーゼ遺伝子は、第1のプラスミドに組み込まれて形質転換酵母に導入されていることが好ましい。前記レポーター遺伝子としては、β−ガラクトシダーゼに限定されず、その他、ルシフェラーゼ遺伝子、蛍光タンパク質遺伝子、β−グルクロニダーゼ遺伝子等が使用できる。
【0064】
前述のように、本発明において、転写共役因子遺伝子は、前記レポーター遺伝子を組み込んだプラスミド(第1のプラスミド)とは別のプラスミド(第2のプラスミド)に組み込んで酵母に導入することが好ましい。
【0065】
つぎに、本発明の分析方法の一例を示す。
【0066】
まず、所定の核内受容体遺伝子、RNA不安定化配列、動物転写共役因子遺伝子、前記動物核内受容体応答配列およびβ−ガラクトシダーゼ遺伝子を導入した形質転換酵母を準備する。そして、この形質転換酵母のグリセロールストックを調製することが好ましい。グリセロールストックを調製するために使用する培地は、特に制限されず、例えば、窒素源、炭素源、アミノ酸、核酸、ビタミン類等を含む培地があげられる。グリセロールストックを調製する培地組成の例を、下記表2に示す。
【0067】
(表2)
成分 配合量
ドロップアウトパウダー (注1) 1.3g
窒素源(Yeast nitrogen base、注2) 1.7g
(NH4)2SO4 5.0g
炭素源(グルコースまたはガラクトース) 20.0g
水 1000mL
(注1)ドロップアウトパウダー組成
成分 配合量 成分 配合量
アデニン 2.5g L−メチオニン 1.2g
L−アルギニン 1.2g L−フェニルアラニン 3.0g
L−アスパラギン酸 6.0g L−セリン 22.5g
L−グルタミン酸 6.0g L−スレオニン 12.0g
L−ヒスチジン 1.2g L−チロシン 1.8g
L−リジン 1.8g L−バリン 9.0g
ウラシル 1.2g
(注2)窒素源:Yeast Nitrogen W/O Amino Acid &
Ammonium Sulfate (Difco社製)
【0068】
前記グリセロールストックを調製するための培養は、前記のような培地で継続的に培養された状態のものを利用する場合には、例えば、前記継続培養後の培養液を50μL分取し、前記表2の培地5000μL中に加え、30℃で12〜24時間程度振とう培養することによって実施する。一方、形質転換酵母として凍結乾燥菌体を利用してグリセロールストックを調製するための培養を行う場合には、例えば、YPD培地等の栄養培地を加え、30℃で24時間培養した培養液を50μL分取し、前記と同様に前記表2に示す培地5000μLに加え、30℃で12〜24時間程度振とう培養する。
【0069】
前記グリセロールストックを調製するための培養により、菌体が十分増殖したら、得られた培養液から10倍濃縮グリセロールストック液を作製する。このグリセロールストック液の組成は、例えば培養液80%、グリセロール20%から成り、−80℃での保存も可能である。そして、このグリセロールストック液から本培養を実施する。前記本培養に使用する培地としては、例えば、前記表2に示す培地において、炭素源として、ガラクトースおよびグルコースを含むものを使用する。ガラクトースとグルコースの比率は、前述のとおりである。また、本培養においては、培地中に、化学物質等の被験物質を添加して行う。前記本培養の温度は、例えば、28〜30℃であり、前記本培養時間は、例えば、10〜48時間、好ましくは、15〜36時間、より好ましくは18〜24時間である。すなわち、前記培地にガラクトースとグルコースを添加することにより、本培養における前記形質転換酵母の増殖を促進させることができるので、短時間の本培養で、被験物質を高感度かつ高信頼性で分析可能である。また、本培養は、振とう培養でもよいし、静置培養でもよい。なお、前記被験物質が固体の場合には、例えば、ジメチルスルホキシド(DMSO)等の形質転換酵母に対して毒性を示さない溶媒を用いて前記被験物質を溶解してから前記培地と混合することが好ましい。
【0070】
つぎに、本培養後の前記形質転換酵母を、公知の手法により溶菌させる。この溶菌には、例えば、下記に示すような、Zバッファーに界面活性剤(サルコシル)を添加した溶菌液等が使用できる。
【0071】
(溶菌液:Zバッファー)
成分 配合量(最終濃度)
Na2HPO4 60mM
NaH2SO4 40mM
MgCl2 1mM
KCl 10mM
ジチオスレイトール(Dithiothreitol) 2mM
サルコシル(注3) 0.20%
(注3)N−Lauroylsarcosine sodium salt
【0072】
そして、前記溶菌後の溶菌液を適量採取し、産生されたβ−ガラクトシダーゼの酵素活性を測定する。β−ガラクトシダーゼの活性は、例えば、ONPG(オルソニトロフェニルガラクトピラノシド)やCPRG(クロロフェノールレッドガラクトピラノシド)等の発色試薬を作用させ、その吸光度を測定すればよい。この測定において、予め、標準物質を用いて検量線を作成しておくことが好ましく、前記検量線は、被験物質の測定と同時に作成することが好ましい。さらに、測定に際しては、用量作用関係の範囲内に納まるように被験物質を適宜希釈することが好ましい。このような分析方法であれば、吸光度を測定するだけでよく、簡便かつ迅速に分析できる。
【0073】
本発明の分析用キットは、本発明の分析方法を簡単に実施するためのキットであって、前述のように、培地、本発明の前記形質転換酵母および発色基質(発色試薬)を含むキットである。前記発色基質(発色試薬)としては、例えば、ONPGやCPRG等があげられる。さらに、本発明の分析キットは、検量線作成のための標準物質等を含んでいてもよい。前記形質転換酵母は、凍結乾燥させて粉末状にしてもよい。
【0074】
本発明のベクターは、前述の遺伝子および配列以外に、他の遺伝子または配列等を含んでいてもよい。前記他の遺伝子または配列は、特に限られず、例えば、前記調節配列が挙げられる。
【0075】
本発明のベクターキットは、前述のベクターを含有すること以外は、特に限定されず、他のベクターを含有してもよい。
【実施例】
【0076】
つぎに、本発明の実施例について比較例と併せて説明する。ただし、本発明は、下記の実施例および比較例に限定されるものではない。
【0077】
(実施例1)
本例では、下記の手順によりヒトPXR遺伝子、ヒトRXRα遺伝子、RNA不安定化配列および酵母cyc1遺伝子のターミネーター配列を組み込んだ形質転換酵母を作製した。
【0078】
(1) レポータープラスミドの作製
まず、酵母チトクロームC遺伝子のプロモーターおよびβ−ガラクトシダーゼ遺伝子を有するプラスミド(pYT−β;BioTechniques(1996)20、568−574)を準備した。一方、PXRの応答配列であるDR−3を含む、下記のリン酸化合成オリゴDNA(配列番号9)およびその相補鎖DNA(配列番号10)を作製した。作製したこれらのオリゴDNAを、94℃で1分間を1サイクルでアニーリングした。アニーリング後、DR−3が1個アニーリングしたものを精製して、前記プラスミドpYT−βのSpeIサイトに挿入した。DR−3が1個挿入されたプラスミドをpYT−β−DR3×1とした。
【0079】
(DR−3)
DR3Nh:5´−ctagcagatgaactttatgaactgttt−3´(配列番号9)
DR3Xb:5´−ctagaaacagttcataaagttcatctg−3´(配列番号10)
【0080】
(2)発現量調節プラスミドの作製
まず、酵母ura3遺伝子、gal1/gal10双方向性プロモーターを有するプラスミドpUdp3を準備した。前記gal1/gal10双方向性プロモーターは、プラスミドYEpLacから、BamHIサイトとSmaIサイトで切り出した。プラスミドpUC19のEcoRIサイトに、前記酵母ura3遺伝子を挿入後、BamHIサイトとSmaIサイトの間に前記gal1/gal10双方向性プロモーターを挿入し、前記プラスミドpUdp3を得た。つぎに、前記プラスミドpUdp3に、酵母cyc1遺伝子のターミネーター配列を挿入したプラスミドpUdp5を作製した。前記酵母cyc1遺伝子のターミネーター配列は、酵母W303aより抽出したゲノムDNAよりPCR法にて増幅した。用いたプライマー(配列番号11および12)およびPCRの条件を以下に示す。
【0081】
(cyc1)
PolAfHd:5´−tatgtcaagcttacattcacgccctc−3´(配列番号11)
PolArBs:5´−cttctcaagctaggtcttcagtataatg−3´(配列番号12)
【0082】
(PCR条件:1×反応バッファー)
1μM MgSO4、0.2μM dNTP、各プライマー各0.3μM、ゲノムDNA 0.5μL、5% DMSO、KOD−Plus−(東洋紡社製)1unitを反応液50μL中に含む。
【0083】
(PCR条件:サイクル)
94℃で40秒、54℃で20秒、68℃で30秒のサイクルを35サイクル実施した。
【0084】
PCR後、増幅した前記酵母cyc1遺伝子のターミネーター配列を含むDNAフラグメントを、制限酵素HindIIIおよびBbsIを用いて切断し、前記プラスミドpUdp3のHindIIIサイトに挿入した。得られたプラスミドをpUdp5とした。
【0085】
さらに、前記プラスミドpUdp5にRNA不安定化配列を挿入した、プラスミドpUdp8を準備した。前記RNA不安定化配列としては、酵母HOエンドヌクレアーゼ遺伝子の3´非翻訳領域に存在するRNA不安定化配列を用いた。前記RNA不安定化配列を含む下記のリン酸化合成オリゴDNA(配列番号13)およびその相補鎖DNA(配列番号14)を作製した。作製したこれらのオリゴDNAを、94℃で1分間を1サイクルでアニーリングした。アニーリング後、アニーリングしたオリゴDNAを、pUdp5の前記酵母cyc1遺伝子ターミネーター配列上流のHindIIIサイトに挿入した。得られたプラスミドをpUdp8とした。
【0086】
(RNA不安定化配列)
HOup:5´−agcttgtatattagtttaaaaagttgtatgtaat−3´(配列番号13)
HOdn:5´−agctattacatacaactttttaaactaatataca−3´(配列番号14)
【0087】
図3に、前記三種類のプラスミドpUdp3、pUdp5、pUdp8、および後述するプラスミドpUdp4のプロモーター周辺の構造を示す。同図において、MCSは、核内受容体のcDNAを挿入するマルチクローニングサイトであり、CYC1terは、前記酵母cyc1遺伝子由来のターミネーター配列であり、ADHterは、前記酵母ADH遺伝子由来のターミネーター配列であり、Gal1.10は、前記gal1/gal10双方向性プロモーターであり、URSは、HOエンドヌクレアーゼ遺伝子由来のRNA不安定化配列である。
【0088】
(3) 動物核内受容体発現プラスミドの作製
ヒトPXRおよびRXRαの各cDNAのオープンリーディングフレーム(ORF)を、ヒト肝がん細胞HepG2から抽出したmRNAを用いて、RT−PCR法により増幅した。用いたプライマー(配列番号15〜18)及びPCRの条件を以下に示す。
【0089】
(PXR)
PXRfRbSp:5´−ttccaactagtaaaacatggaggtgagacccaaag−3´(配列番号15)
PXRrHd:5´−ccttcaagcttctcagctacctgtgatgccgaac−3´(配列番号16)
【0090】
(RXRα)
RXRaRbKpnF:5´−aaaggggtacctcaaaaatggacaccaaacatttcctgcc−3´(配列番号17)
RXRaEcr:5´−cttaagaattctaagtcatttggtgcggc−3´(配列番号18)
【0091】
(PCR条件:1×反応バッファー)
1μM MgSO4、0.2μM dNTP、プライマー各0.3μM、cDNA 1μL、5% DMSO、KOD−Plus−(東洋紡社製)1unitを反応液50μL中に含む。
【0092】
(PCR条件:サイクル)
94℃で40秒、58℃で20秒、68℃で3分のサイクルを40サイクル実施した。
【0093】
つぎに、増幅した各核内受容体cDNAを、制限酵素を用いて切断した。前記プラスミドpUdp8のgal1プロモーター下流に位置する部位に、切断したRXRαのcDNAを挿入し、gal10プロモーター下流に位置する部位に、切断したPXRのcDNAを挿入した。得られたプラスミドを、pUdp8PXR/RXRαとした。なお、クローニングには大腸菌DH5α株を使用した。cDNAおよびプラスミドの切断に用いた制限酵素を以下に示す。
【0094】
(制限酵素)
挿入したcDNA cDNA切断用制限酵素 プラスミド切断用制限酵素
PXR SpeI、HindIII XbaI、HindIII
RXRα KpnI、EcoRI KpnI、EcoRI
【0095】
(4) SRC1プラスミドの作製
SRC1cDNAのオープンリーディングフレーム(ORF)を、ヒト精巣cDNA(クロンテック社製)よりPCR法を用いて増幅した。用いたプライマー(配列番号19および20)及びPCRの条件を以下に示す。
【0096】
hSRC1bgF:5´−tggaactcaagatttgaccatatc−3´(配列番号19)
hSRC1xhR:5´−gagcattcctctagtctgtagtc−3´(配列番号20)
【0097】
(PCR条件:1×反応バッファー)
1μM MgSO4、0.2μM dNTP、プライマー各0.3μM、cDNA 1μL、5% DMSO、KOD−Plus−(東洋紡社製)1unitを反応液50μL中に含む。
【0098】
(PCR条件:サイクル)
94℃で40秒、56℃で20秒、68℃で5分のサイクルを40サイクル実施した。
【0099】
増幅したSRC−1 cDNAを精製し、さらに以下のプライマー(配列番号21および22)を用いたPCR法にてcDNAに制限酵素サイトを付加した。増幅したcDNAを、BglIIおよびXhoIで切断後、pESC−Leuベクターのgal1プロモーター下流に挿入した。本プラスミドDNAをpESC−Leu−hSRC1とした。
【0100】
SRC−1eFbg2:5´−caaagaagatctcccaggtgtgaag−3´(配列番号21)
SRC−1eRxh2:5´−agggccctcgagactctagtctgtag−3´(配列番号22)
【0101】
(PCR条件:1×反応バッファー)
1μM MgSO4、0.2μM dNTP、プライマー各0.3μM、PCR産物(cDNA) 1μL、5% DMSO、KOD−Plus−(東洋紡社製)1unitを反応液50μL中に含む。
【0102】
(PCR条件:サイクル)
94℃で40秒、56℃で20秒、68℃で5分のサイクルを20サイクル実施した。
【0103】
(5) 形質転換酵母の作製
まず、酵母(Saccharomyces cerevisiae)W303a株に、塩化リチウム法にて、前記pYT−β−DR3×1プラスミド DNAを導入した。このときの形質転換酵母選択培地は、後述する前培養培地にロイシンを100mg添加したものである。
【0104】
つぎに、前記形質転換酵母に、pESC−Leu−hSRC1プラスミド DNAを導入した。このときの形質転換酵母選択培地は、後述する前培養培地である。
【0105】
さらに、前記pUdp8PXR/RXRαを、制限酵素EcoRVで処理して直鎖状とし、以下のように前記形質転換酵母へ導入し、染色体への組込みを行った。
【0106】
まず、前記形質転換酵母を、30℃で濁度(Abs660nm、以下、OD660とする)が1〜2になるまでYPD培地中で培養し、ソリューションA(0.1M Lithium acetate、10mM Tris−HCl(pH 7.5)、1mM EDTA いずれも最終濃度)で洗浄後、OD660=150となるようにソリューションAに再懸濁し、1.5mLマイクロチューブに100μLずつ分注して、30℃で1時間インキュベートした。その後、直鎖状の前記の各pAUR101ベクター 5μgと、キャリアーDNA(商品名:SALMON TESTES DNA for hybridization、SIGMA社製)150μg(合計20μL)を加え、さらに、ソリューションB(ソリューションA100mLにポリエチレングリコール4000を溶解したもの)を850μL添加し、緩やかに混合した。
【0107】
つぎに、前記混合液を30℃で30分インキュベートして42℃で15分熱処理した後、さらに、10分室温に放置してから、5000rpm、1分間の遠心分離で集菌し、それらを5〜10mLのYPD培地に懸濁し、30℃で一晩培養した。そして、培養後集菌して洗浄した酵母を、0.9%NaCl溶液に懸濁し、オーレオバシディンA(AureobasidinA、0.5g/mL)を含むYPD選択プレートに100μLずつ塗布して培養した。そして、オーレオバシディンA耐性株をGal1プロモーターとPXRのcDNA領域が染色体上に組込まれた形質転換酵母として選択した。以上の操作により、pYT−β−DR3×1およびpESC−Leu−hSRC1DNAをプラスミドとして保持し、ゲノム中よりヒトRXRαおよびPXRを発現する形質転換酵母を作製した。
【0108】
(6)PXRのmRNA量の測定
本例の形質転換酵母について、PXRのmRNA量を測定した。
【0109】
(形質転換酵母の前培養)
まず、本例の形質転換酵母を、下記組成の前培養用の培地にて18時間前培養を行った。培養の結果、濁度(Abs595nm)が1.0程度に形質転換酵母が増殖した。
【0110】
(前培養用培地の組成)
成分 配合量
ドロップアウトパウダー(前記注1) 1.3g
窒素源(Yeast nitrogen base、前記注2) 1.7g
(NH4)2SO4 5.0g
グルコース 20.0g
水 1L
【0111】
(形質転換酵母の本培養)
つぎに、96穴プレートを用い、1ウェルあたり以下の構成で、リガンド溶液、前培養した菌体液および本培養用培地を混合し、30℃で18時間、静置培養を行った。
【0112】
リガンド溶液:10mM DMSOに溶解したリファンピシン(Rifampicin、RIF)1μL
菌体液(O.D.1〜1.5):5μL
本培養用培地:下記組成の培地100μL
【0113】
(本培養用培地の組成)
成分 配合量
ドロップアウトパウダー(前記注1) 1.3g
窒素源(Yeast nitrogen base、前記注2) 1.7g
(NH4)2SO4 5.0g
グルコース 2.0g
ガラクトース 18.0g
水 1L
【0114】
(RNAの抽出およびcDNA合成)
さらに、前記本培養後の形質転換酵母を、遠心して集菌後、ソルビトールバッファー(1M ソルビトール、0.1M EDTA)に懸濁し、30℃で30分振とうした。振とうした菌体は、再度遠心して集菌後、RNeasy mini kit(Quiagen社製)を用いて、トータルRNA(total RNA)を抽出した。前記トータルRNAの抽出操作は、キット添付の操作書に従って行った。抽出したトータルRNAを用いて、cDNAを合成した。前記cDNAの合成は、以下の条件で行った。
【0115】
(PCR条件:反応液 5×SSIIバッファー)
5mM DTT、0.5mM dNTP、RNase inhibitor 20unit、2.5μM oligo−dT primer(oligo−dT、タカラバイオ社製)または random 6mer(Rd6、タカラバイオ社製)、SuperScriptIII RT(Invitrogen社)200unit、2μg yeast total RNAを反応液20μL中に含む。
【0116】
(PCR条件:サイクル)
25℃で10分、50℃で30分、55℃で30分の条件で反応させた。
【0117】
cDNA合成後、Smart Cycler V2.0(Cepheid社製)を用いてリアルタイムPCRを行った。用いたプライマー(配列番号23および24)リアルタイムPCRの条件を以下に示す。
【0118】
hPXR1008RTf: 5′−tgaggaggagtatgtgctgatgc−3′(配列番号23)
PXRrHd2: 5′−ttctcagctacctgtgatgccgaac−3′(配列番号24)
【0119】
(PCR条件:反応液組成)
1×CYBR Premix Ex Taq(TAKARA社製)、プライマー各0.5μM、1μL cDNA(またはスタンダードとしてプラスミドDNA)を反応液25μL中に含む。
【0120】
(PCR条件:サイクル)
94℃で30秒、60℃で30秒、72℃で30分のサイクルを45サイクル実施した。
【0121】
(リアルタイムPCRによる測定)
mRNA定量における検量線作成用のスタンダードとして、プラスミドpUdp3PXR/RXRαを作製した。前記プラスミドpUdp3PXR/RXRαは、前記プラスミドpUdp8に代えて、前記プラスミドpUdp3を用いたこと以外は、前記プラスミドpUdp8PXR/RXRαと同様にして作製した。前記プラスミドpUdp3PXR/RXRα DNAは、1pg/mL〜1μg/mLの濃度に希釈した。得られたcDNAサンプルおよび前記スタンダードについて、同時に、リアルタイムPCRによる測定を行った。
【0122】
(7)リガンド応答性の測定
本例の形質転換酵母について、リガンドへの応答性を測定した。
【0123】
(形質転換酵母の培養)
本例の形質転換酵母は、前述と同様に、前培養および本培養を行った。
【0124】
(β−ガラクトシダーゼ活性の測定)
前記本培養の後、各ウェルから培養液を5μLずつ採取し、新たな96穴プレートの各ウェルに移し、下記組成の測定試薬を100μL加えた後、37℃で30分間反応させた。反応後、マイクロプレートリーダーを用いて、前記反応液のβ−ガラクトシダーゼ活性および菌体量を測定した。前記β−ガラクトシダーゼ活性は、o−ニトロフェノールの生成量から算出するため、405nmにおける反応液の吸光度を測定した。また、前記菌体量は、620nmにおける吸光度を測定した。分析結果は、405nmの吸光度/620nmの吸光度の比を算出し、リガンド非添加(溶媒である10mM DMSOのみを添加)での値を1とした誘導率で示した。
【0125】
(測定試薬の組成)
成分 配合量(最終濃度)
Na2HPO4(注4) 60mM
NaH2SO4(注4) 40mM
MgSO4 1mM
KCl 10mM
ジチオスレイトール(Dithiothreitol) 2mM
サルコシル(前記注3) 0.40w/v%
ONPG(o−ニトロフェニル−β−D−ガラクトピラノシド) 1mg/mL
(注4)Na2HPO4およびNaH2SO4を混合後、pH7.5に調整
【0126】
(比較例1−1)
(1)レポータープラスミドの作製
本例の形質転換酵母において、レポータープラスミドは、実施例1と同様に作製した。
【0127】
(2)発現量調整プラスミドの作製
本例の形質転換酵母において、発現量調整プラスミドは、前記酵母cyc1遺伝子のターミネーター配列および前記RNA不安定化配列を含まない、前記プラスミドpUdp3を用いた。前記プラスミドpUdp3は、実施例1と同様に作製した。
【0128】
(3)動物核内受容体発現プラスミドの作製
本例の形質転換酵母において、動物核内受容体発現プラスミドとして、前記プラスミドpUdp3PXR/RXRαを用いた。
【0129】
(4)SRC1プラスミドの作製
本例の形質転換酵母において、SRC1プラスミドは、実施例1と同様に作製した。
【0130】
(5)形質転換酵母の作製
本例の形質転換酵母は、前記動物核内受容体発現プラスミドとして、前記プラスミドpUdp8PXR/RXRαに代えて、前記プラスミドpUdp3PXR/RXRαを用いたこと以外は、実施例1と同様に作製した。
【0131】
(6)PXRのmRNA量の測定
本例の形質転換酵母における、PXRのmRNA量測定は、実施例1と同様にして行った。
【0132】
(7)リガンド応答性の測定
本例の形質転換酵母について、実施例1と同様に、リガンド応答性を測定した。
【0133】
(比較例1−2)
(1)レポータープラスミドの作製
本例の形質転換酵母において、レポータープラスミドは、実施例1と同様に作製した。
【0134】
(2)発現量調整プラスミドの作製
本例の形質転換酵母において、発現量調整プラスミドは、前記RNA不安定化配列を含まず、かつ前記酵母cyc1遺伝子のターミネーター配列に代えて、酵母ADH遺伝子のターミネーター配列を挿入した、プラスミドpUdp4を用いた。前記酵母ADH遺伝子のターミネーター配列の挿入は、以下のように行った。
【0135】
前記酵母ADH遺伝子のターミネーター配列の増幅は、用いたプライマー以外は、前記酵母cyc1遺伝子のターミネーター配列と同様にして行った。以下に、用いたプライマー(配列番号25および26)を示す。増幅させた前記酵母ADH遺伝子のターミネーター配列を含むDNAフラグメントは、制限酵素EcoRIおよびMflIを用いて切断し、前記pUdp3のEcoRIサイトに挿入した。
【0136】
(ADH)
scADH3UTRfEc:5´−ctaaataagcgaattccttatgatttatg−3´(配列番号25)
scADH3UTRrMf:5´−tgcaggcaattgctcggcatgccggtag−3´(配列番号26)
【0137】
(3)動物核内受容体発現プラスミドの作製
本例の形質転換酵母において、動物核内受容体発現プラスミドは、前記プラスミドpUdp8に代えて、前記プラスミドpUdp4を用いたこと以外は、実施例1と同様にして作製した。前記プラスミドpUdp4を用いて作製した本例の動物核内受容体発現プラスミドを、プラスミドpUdp4PXR/RXRαとした。
【0138】
(4)SRC1プラスミドの作製
本例の形質転換酵母において、SRC1プラスミドは、実施例1と同様に作製した。
【0139】
(5)形質転換酵母の作製
本例の形質転換酵母は、前記動物核内受容体発現プラスミドとして、前記プラスミドpUdp8PXR/RXRαに代えて、前記プラスミドpUdp4PXR/RXRαを用いたこと以外は、実施例1と同様に作製した。
【0140】
(6)PXRのmRNA量の測定
本例の形質転換酵母において、PXRのmRNA量測定は、実施例1と同様にして行った。
【0141】
(7)リガンド応答性の測定
本例の形質転換酵母について、実施例1と同様に、リガンドへの応答性を測定した。
【0142】
(比較例1−3)
(1)レポータープラスミドの作製
本例の形質転換酵母において、レポータープラスミドは、実施例1と同様に作製した。
【0143】
(2)発現量調整プラスミドの作製
本例の形質転換酵母において、発現量調整プラスミドは、前記RNA不安定化配列を含まない、前記プラスミドpUdp5を用いた。前記プラスミドpUdp5は、実施例1と同様に作製した。
【0144】
(3)動物核内受容体発現プラスミドの作製
本例の形質転換酵母において、動物核内受容体発現プラスミドは、前記プラスミドpUdp8に代えて、前記プラスミドpUdp5を用いたこと以外は、実施例1と同様にして作製した。前記プラスミドpUdp5を用いて作製した本例の動物核内受容体発現プラスミドを、プラスミドpUdp5PXR/RXRαとした。
【0145】
(4)SRC1プラスミドの作製
本例の形質転換酵母において、SRC1プラスミドは、実施例1と同様に作製した。
【0146】
(5)形質転換酵母の作製
本例の形質転換酵母は、前記動物核内受容体発現プラスミドとして、前記プラスミドpUdp8PXR/RXRαに代えて、前記プラスミドpUdp5PXR/RXRαを用いたこと以外は、実施例1と同様に作製した。
【0147】
(6)PXRのmRNA量の測定
本例の形質転換酵母において、PXRのmRNA量測定は、実施例1と同様にして行った。
【0148】
(7)リガンド応答性の測定
本例の形質転換酵母について、実施例1と同様に、リガンド応答性を測定した。
【0149】
前記実施例1および前記比較例1−1〜1−3で作製した形質転換酵母について、PXRのmRNA量を測定した結果を、下記表3に示す。
【0150】
【表3】
【0151】
前記表3に示すように、Rd6を用いてcDNAを作製した場合、実施例1のmRNA量は、比較例1−1〜1−3の約1/70〜1/170であった。また、oligo−dTを用いてcDNAを作製した場合、実施例1のmRNA量は、比較例1−1〜1−3の約1/4〜1/85であった。すなわち、RNA不安定化配列を有する実施例1において、PXRのmRNA量が著しく低いことが示された。また、Rd6を用いてcDNAを作製した場合、比較例1−1〜1−3間のmRNA量に大きな差異はなかった。しかし、oligo−dTを用いてcDNAを作製した場合、酵母cyc1遺伝子のターミネーター配列を持つ比較例1−3が、他の比較例に比べて特に高い値を示した。前記Rd6プライマーは、ランダム配列を有するプライマーであり、前記oligo−dTプライマーは、ポリA tailを持つmRNAの逆転写反応用プライマーである。すなわち、ターミネーター配列の有無および種類等により、ポリAが付加されたPXRのmRNA量が異なることが示された。
【0152】
前記実施例1および前記比較例1−1〜1−3で作製した形質転換酵母について、リガンドへの応答性を測定した結果を、下記表4に示す。
【0153】
【表4】
【0154】
前記表4に示すように、実施例1では、リファンピシンに応答して、β−ガラクトシダーゼが誘導された。実施例1の前記誘導率は、約4倍であった。それに対し、比較例1−1〜1−3では、β−ガラクトシダーゼの誘導は認められなかった。また、リガンド非添加時(陰性対照)におけるβ−ガラクトシダーゼの発現量が、実施例1では低いのに対し、比較例1−1〜1−3では、リガンド添加時の値に近い高値を示した。
【0155】
(実施例2)
(1)レポータープラスミドの作製
本例の形質転換酵母において、レポータープラスミドは、実施例1と同様に作製した。
【0156】
(2)発現量調整プラスミドの作製
本例の形質転換酵母において、発現量調整プラスミドは、実施例1と同様に作製した。
【0157】
(3)動物核内受容体発現プラスミドの作製
本例の形質転換酵母において、動物核内受容体発現プラスミドとしては、前記プラスミドpUdp8PXR/RXRαの、RXRαのC末端のHelix12を欠失させたプラスミドpUdp8PXR/RXRαTを用いた。前記プラスミドpUdp8PXR/RXRαTは、以下のように作製した。
【0158】
まず、QuickChange Site−Directed Mutagenesis Kit(Stratagene社製)を用いて、前記pUdp8PXR/RXRαに含まれるRXR cDNAの443番目のコドンにナンセンス変異を導入し、RXRαのC末端に存在するHelix12領域を欠失させた。用いたプライマー(配列番号27および28)および反応条件を以下に示す。
【0159】
RXR443F: 5′−cttcttcaagctcatcgggtagacacccat−3′(配列番号27)
RXR443R: 5′−ggtgtcaatgggtgtctacccgatgagctt−3′(配列番号28)
【0160】
(PCR条件:1×反応バッファー)
0.25mM dNTP、3μL Quick solution、pfu ultra polymerase 10unit(以上Stratagene社製)、0.25μM 各プライマー、10ng プラスミドDNAを反応液50μL中に含む。
【0161】
(PCR条件:サイクル)
94℃で50秒、60℃で50秒、68℃で7分のサイクルを18サイクル実施した。
【0162】
PCR後、制限酵素DpnIで鋳型プラスミドDNAを消化し、大腸菌に形質転換した。得られたプラスミドDNAをpUdp8PXR/RXRαTとした。
【0163】
(4)SRC1プラスミドの作製
本例の形質転換酵母において、SRC1プラスミドは、実施例1と同様に作製した。
【0164】
(5)形質転換酵母の作製
本例の形質転換酵母は、前記動物核内受容体発現プラスミドとして、前記プラスミドpUdp8PXR/RXRαに代えて、前記プラスミドpUdp8PXR/RXRαTを用いたこと以外は、実施例1と同様に作製した。
【0165】
(6)リガンド応答性の測定
本例の形質転換酵母について、以下のようにしてリガンド応答性を測定した。
【0166】
(形質転換酵母の培養)
本例の形質転換酵母は、実施例1と同様にして、前培養を行った。
【0167】
本例の形質転換酵母の本培養は、リファンピシンおよび9−cisレチノイン酸をリガンド溶液として用いたこと以外は、実施例1と同様に行った。なお、前記リファンピシンは、10mM DMSOに溶解させ、下記表5に示すように、異なる10濃度の溶液を調製した。また、前記9−cisレチノイン酸は、10mM DMSOに溶解させ、下記表6に示すように、異なる5濃度の溶液を調製した。
【0168】
(β−ガラクトシダーゼ活性の測定)
本例の形質転換酵母は、実施例1と同様にしてβ−ガラクトシダーゼ活性を測定した。前記測定の結果を、下記表5、表6および図1に示す。表5は、前記リファンピシンをリガンドとして用いた測定結果であり、表6は、前記9−cisレチノイン酸をリガンドとして用いた測定結果である。図1において、横軸は、各リガンドの濃度(μM)であり、縦軸は、リガンド非添加での値を1とした誘導率である。
【0169】
【表5】
【0170】
【表6】
【0171】
前記表5、6および図1に示すように、本例の形質転換酵母において、リファンピシンによるβ−ガラクトシダーゼの誘導率は、約4倍であった。それに対し、本例の形質転換酵母において、9−cisレチノイン酸による誘導は、全く見られなかった。すなわち、本例の形質転換酵母は、RXRαのリガンドには反応せず、PXRのリガンドのみに反応することが示された。
【0172】
(実施例3)
(1)レポータープラスミドの作製
本実施例の形質転換酵母において、レポータープラスミドは、実施例1と同様に作製した。
【0173】
(2)発現量調整プラスミドの作製
本実施例の形質転換酵母において、発現量調整プラスミドは、実施例1と同様に作製した。
【0174】
(3)動物核内受容体発現プラスミドの作製
本実施例の形質転換酵母において、動物核内受容体発現プラスミドは、前記核内受容体遺伝子PXRに代えて、核内受容体遺伝子VDRを発現可能に導入したこと以外は、実施例2と同様に作製した。本実施例における前記動物核内受容体発現プラスミドを、プラスミドpUdp8VDR/RXRαTとした。前記プラスミドpUdp8VDR/RXRαTは、前記ヒトVDRの増幅および導入時に用いたプライマーおよび制限酵素以外は、実施例2と同様に作製した。以下に、前記用いたプライマー(配列番号29および30)および制限酵素を示す。
【0175】
(プライマー)
VDRfXb:5´−ctccttctagaatggaggcaatggcggccagcac−3´(配列番号29)
VDRrSp:5´−gctgtactagtcaggagatctcattgccaaacac−3´(配列番号30)
【0176】
(制限酵素)
挿入したcDNA cDNA切断用制限酵素 プラスミド切断用制限酵素
VDR SpeI、XbaI XbaI
【0177】
(4)SRC1プラスミドの作製
本実施例の形質転換酵母において、SRC1プラスミドは、実施例1と同様に作製した。
【0178】
(5)形質転換酵母の作製
本実施例の形質転換酵母は、前記動物核内受容体発現プラスミドとして、前記プラスミドpUdp8PXR/RXRαに代えて、前記プラスミドpUdp8VDR/RXRαTを用いたこと以外は、実施例1と同様に作製した。
【0179】
(6)リガンド応答性の測定
本例の形質転換酵母について、以下のようにしてリガンド応答性を測定した。
【0180】
(形質転換酵母の培養)
本例の形質転換酵母は、実施例1と同様にして、前培養を行った。
【0181】
本例の形質転換酵母の本培養は、リガンド溶液として、活性型ビタミンD3(1,25−OH VD3)を用いたこと以外は、実施例1と同様に行った。なお、前記活性型ビタミンD3は、10mM DMSOに溶解させ、下記表7に示すように、異なる9濃度の溶液を調製した。
【0182】
(β−ガラクトシダーゼ活性の測定)
本例の形質転換酵母は、実施例1と同様に、β−ガラクトシダーゼ活性を測定した。前記測定の結果を、下記表7および図2に示す。図2において、横軸は、活性型ビタミンD3の濃度(μM)であり、縦軸は、リガンド非添加での値を1とした誘導率である。
【0183】
【表7】
【0184】
前記表7および図2に示すように、本例の形質転換酵母では、活性型ビタミンD3により、β−ガラクトシダーゼが誘導された。本例の形質転換酵母において、前記誘導率は、最大約20倍であった。
【0185】
これらの結果から、本発明の形質転換酵母を用いることにより、RXRα核内受容体とヘテロ二量体を形成する核内受容体のリガンドを、高感度で分析できる。また、前記核内受容体発現プラスミドpUdp8は、PXRだけでなく、本実施例のVDRのように、RXRαとヘテロ二量体を形成する他の核内受容体にも、利用可能である。さらに、例えば、前記非特許文献3の図1(2)のグラフ(j)において、酵母ツーハイブリッド法を用いた、活性型ビタミンD3誘導によるβ−ガラクトシダーゼ活性の測定結果が示されているが、同法は、本発明の前記実施例3と比べ、活性を示す最低濃度が約10倍大きく、測定感度が低い。このことから、本発明の形質転換酵母を用いた分析方法は、前記酵母ツーハイブリッド法と比べ、高い測定感度を有するといえる。しかも、本発明の形質転換酵母を用いた分析方法は、操作が簡単であり、短時間で分析ができ、低コストであるという利点もある。
【産業上の利用可能性】
【0186】
以上のように、本発明の形質転換酵母を用いれば、高検出感度でRXRα核内受容体とヘテロ二量体を形成する核内受容体のリガンドの分析が可能となる。したがって、本発明の分析方法は、化学物質やタンパク質等のリガンドの分析の分野において有用な方法であり、その用途は制限されず広い。
【図面の簡単な説明】
【0187】
【図1】図1は、本発明の実施例2におけるβ−ガラクトシダーゼ活性の測定結果を示すグラフである。
【図2】図2は、本発明の実施例3におけるβ−ガラクトシダーゼ活性の測定結果を示すグラフである。
【図3】図3は、本発明の実施例および比較例の形質転換酵母に含まれる、核内受容体発現プラスミドのプロモーター周辺の構造図である。図3(A)は、比較例1−1(pUdp3)、図3(B)は、比較例1−2(pUdp4)、図3(C)は、比較例1−3(pUdp5)、図3(D)は、実施例1(pUdp8)に含まれる核内受容体発現プラスミドの前記構造図である。
【特許請求の範囲】
【請求項1】
動物核内受容体遺伝子およびレポーター遺伝子が発現可能に導入されており、かつ動物核内受容体リガンドと動物核内受容体との複合体を認識して前記レポーター遺伝子を発現し得る形質転換酵母であって、
前記動物核内受容体が、RXRα核内受容体と二量体を形成する、RXRαヘテロ二量体型核内受容体であり、
前記動物核内受容体遺伝子として、RXRα核内受容体遺伝子およびRXRαへテロ二量体型核内受容体遺伝子を発現可能に含み、
前記動物核内受容体に直接結合して前記レポーター遺伝子の転写を活性化する動物転写共役因子遺伝子を発現可能に含み、
さらに、前記動物核内受容体遺伝子の発現量を調節するRNA不安定化配列を含む形質転換酵母。
【請求項2】
前記RNA不安定化配列が、前記RXRαへテロ二量体型核内受容体遺伝子の発現量を調節するRNA不安定化配列である請求項1記載の形質転換酵母。
【請求項3】
前記RXRα核内受容体が、C末端のHelix12領域を欠失させたRXRα核内受容体である請求項1または2に記載の形質転換酵母。
【請求項4】
さらに、ターミネーター配列を含む請求項1から3のいずれか一項に記載の形質転換酵母。
【請求項5】
前記動物核内受容体が、ヒト核内受容体であり、前記動物転写共役因子がヒト転写共役因子である請求項1から4のいずれか一項に記載の形質転換酵母。
【請求項6】
前記ヒト転写共役因子遺伝子が、ヒトSRC1遺伝子である請求項5記載の形質転換酵母。
【請求項7】
前記動物核内受容体遺伝子および前記RNA不安定化配列を常染色体上に有し、
前記レポーター遺伝子を第1のプラスミドに有し、
前記動物転写共役因子遺伝子を第2のプラスミドに有し、
前記第1のプラスミドは、前記レポーター遺伝子と作動的に組み込まれた動物核内受容体応答配列を含む請求項1から6のいずれか一項に記載の形質転換酵母。
【請求項8】
前記レポーター遺伝子が、β−ガラクトシダーゼ遺伝子である請求項1から7のいずれか一項に記載の形質転換酵母。
【請求項9】
前記形質転換酵母が、出芽酵母(Saccharomyces cerevisiae)である請求項1から8のいずれか一項に記載の形質転換酵母。
【請求項10】
検体に含まれる動物核内受容体リガンドの分析に使用される請求項1から9のいずれか一項に記載の形質転換酵母。
【請求項11】
検体に含まれる動物核内受容体リガンドを分析するための分析方法であって、
検体を含む培地中で、動物核内受容体遺伝子およびレポーター遺伝子が発現可能に導入されており、かつ動物核内受容体リガンドと動物核内受容体との複合体を認識して前記レポーター遺伝子を発現し得る形質転換酵母を培養する培養工程と、
前記培養工程の後、前記培地中に産生された前記レポーター遺伝子の発現産物を測定する測定工程とを包含し、
前記形質転換酵母として、請求項1から10のいずれか一項に記載の形質転換酵母を使用することを特徴とする分析方法。
【請求項12】
前記レポーター遺伝子が、β−ガラクトシダーゼ遺伝子であり、前記培地が、炭素源としてガラクトースおよびグルコースを含有する請求項11記載の分析方法。
【請求項13】
前記ガラクトースおよび前記グルコースの合計に対する前記グルコースの重量割合が、0を超え60重量%以下の範囲である請求項12記載の分析方法。
【請求項14】
前記形質転換酵母が、予め、グリセロール存在下で保存されていた酵母である請求項11から13のいずれか一項に記載の分析方法。
【請求項15】
前記形質転換酵母が、予め、グルコース単独を炭素源とする培地で培養された酵母である請求項11から14のいずれか一項に記載の分析方法。
【請求項16】
検体に含まれる動物核内受容体リガンドを分析するための分析キットであって、
(a)培地と、
(b)請求項1から10のいずれか一項に記載の形質転換酵母と、
(c)発色基質と
を備える分析キット。
【請求項17】
前記培地が、グルコースおよびガラクトースを含む培地である請求項16記載の分析キット。
【請求項18】
動物核内受容体遺伝子を含むベクターであって、
前記動物核内受容体遺伝子として、RXRα核内受容体遺伝子およびRXRαへテロ二量体型核内受容体遺伝子を含み、
さらに、前記動物核内受容体遺伝子の発現量を調節するRNA不安定化配列を含むベクター。
【請求項19】
前記RNA不安定化配列が、前記RXRαへテロ二量体型核内受容体遺伝子の発現量を調節するRNA不安定化配列である請求項18記載のベクター。
【請求項20】
前記RXRα核内受容体が、C末端のHelix12領域を欠失させたRXRα核内受容体である請求項18または19に記載のベクター。
【請求項21】
さらに、ターミネーター配列を含む請求項18から20のいずれか一項に記載のベクター。
【請求項22】
レポーター遺伝子を含むベクター、動物転写共役遺伝子を含むベクターおよび請求項18から21のいずれか一項に記載のベクターを含有するベクターセット。
【請求項1】
動物核内受容体遺伝子およびレポーター遺伝子が発現可能に導入されており、かつ動物核内受容体リガンドと動物核内受容体との複合体を認識して前記レポーター遺伝子を発現し得る形質転換酵母であって、
前記動物核内受容体が、RXRα核内受容体と二量体を形成する、RXRαヘテロ二量体型核内受容体であり、
前記動物核内受容体遺伝子として、RXRα核内受容体遺伝子およびRXRαへテロ二量体型核内受容体遺伝子を発現可能に含み、
前記動物核内受容体に直接結合して前記レポーター遺伝子の転写を活性化する動物転写共役因子遺伝子を発現可能に含み、
さらに、前記動物核内受容体遺伝子の発現量を調節するRNA不安定化配列を含む形質転換酵母。
【請求項2】
前記RNA不安定化配列が、前記RXRαへテロ二量体型核内受容体遺伝子の発現量を調節するRNA不安定化配列である請求項1記載の形質転換酵母。
【請求項3】
前記RXRα核内受容体が、C末端のHelix12領域を欠失させたRXRα核内受容体である請求項1または2に記載の形質転換酵母。
【請求項4】
さらに、ターミネーター配列を含む請求項1から3のいずれか一項に記載の形質転換酵母。
【請求項5】
前記動物核内受容体が、ヒト核内受容体であり、前記動物転写共役因子がヒト転写共役因子である請求項1から4のいずれか一項に記載の形質転換酵母。
【請求項6】
前記ヒト転写共役因子遺伝子が、ヒトSRC1遺伝子である請求項5記載の形質転換酵母。
【請求項7】
前記動物核内受容体遺伝子および前記RNA不安定化配列を常染色体上に有し、
前記レポーター遺伝子を第1のプラスミドに有し、
前記動物転写共役因子遺伝子を第2のプラスミドに有し、
前記第1のプラスミドは、前記レポーター遺伝子と作動的に組み込まれた動物核内受容体応答配列を含む請求項1から6のいずれか一項に記載の形質転換酵母。
【請求項8】
前記レポーター遺伝子が、β−ガラクトシダーゼ遺伝子である請求項1から7のいずれか一項に記載の形質転換酵母。
【請求項9】
前記形質転換酵母が、出芽酵母(Saccharomyces cerevisiae)である請求項1から8のいずれか一項に記載の形質転換酵母。
【請求項10】
検体に含まれる動物核内受容体リガンドの分析に使用される請求項1から9のいずれか一項に記載の形質転換酵母。
【請求項11】
検体に含まれる動物核内受容体リガンドを分析するための分析方法であって、
検体を含む培地中で、動物核内受容体遺伝子およびレポーター遺伝子が発現可能に導入されており、かつ動物核内受容体リガンドと動物核内受容体との複合体を認識して前記レポーター遺伝子を発現し得る形質転換酵母を培養する培養工程と、
前記培養工程の後、前記培地中に産生された前記レポーター遺伝子の発現産物を測定する測定工程とを包含し、
前記形質転換酵母として、請求項1から10のいずれか一項に記載の形質転換酵母を使用することを特徴とする分析方法。
【請求項12】
前記レポーター遺伝子が、β−ガラクトシダーゼ遺伝子であり、前記培地が、炭素源としてガラクトースおよびグルコースを含有する請求項11記載の分析方法。
【請求項13】
前記ガラクトースおよび前記グルコースの合計に対する前記グルコースの重量割合が、0を超え60重量%以下の範囲である請求項12記載の分析方法。
【請求項14】
前記形質転換酵母が、予め、グリセロール存在下で保存されていた酵母である請求項11から13のいずれか一項に記載の分析方法。
【請求項15】
前記形質転換酵母が、予め、グルコース単独を炭素源とする培地で培養された酵母である請求項11から14のいずれか一項に記載の分析方法。
【請求項16】
検体に含まれる動物核内受容体リガンドを分析するための分析キットであって、
(a)培地と、
(b)請求項1から10のいずれか一項に記載の形質転換酵母と、
(c)発色基質と
を備える分析キット。
【請求項17】
前記培地が、グルコースおよびガラクトースを含む培地である請求項16記載の分析キット。
【請求項18】
動物核内受容体遺伝子を含むベクターであって、
前記動物核内受容体遺伝子として、RXRα核内受容体遺伝子およびRXRαへテロ二量体型核内受容体遺伝子を含み、
さらに、前記動物核内受容体遺伝子の発現量を調節するRNA不安定化配列を含むベクター。
【請求項19】
前記RNA不安定化配列が、前記RXRαへテロ二量体型核内受容体遺伝子の発現量を調節するRNA不安定化配列である請求項18記載のベクター。
【請求項20】
前記RXRα核内受容体が、C末端のHelix12領域を欠失させたRXRα核内受容体である請求項18または19に記載のベクター。
【請求項21】
さらに、ターミネーター配列を含む請求項18から20のいずれか一項に記載のベクター。
【請求項22】
レポーター遺伝子を含むベクター、動物転写共役遺伝子を含むベクターおよび請求項18から21のいずれか一項に記載のベクターを含有するベクターセット。
【図1】

【図2】
【図3】
【図2】
【図3】
【公開番号】特開2009−232697(P2009−232697A)
【公開日】平成21年10月15日(2009.10.15)
【国際特許分類】
【出願番号】特願2008−79803(P2008−79803)
【出願日】平成20年3月26日(2008.3.26)
【出願人】(000214272)長瀬産業株式会社 (137)
【出願人】(505127721)公立大学法人大阪府立大学 (688)
【Fターム(参考)】
【公開日】平成21年10月15日(2009.10.15)
【国際特許分類】
【出願日】平成20年3月26日(2008.3.26)
【出願人】(000214272)長瀬産業株式会社 (137)
【出願人】(505127721)公立大学法人大阪府立大学 (688)
【Fターム(参考)】
[ Back to top ]