
徐放性微粒子を製造する方法
【課題】徐放性微粒子を製造する方法
【解決手段】徐放性微粒子を製造する方法の様々な実施態様が記載される。一つの実施態様において、本方法は、ポリマー中の活性剤の分散相を形成し、分散相を連続相と混合して微粒子分散系を形成する工程を含む。本方法はさらに微粒子分散系に一定量の希釈組成物を添加する工程を含む。様々な量の希釈組成物を用いた徐放性微粒子を製造するための様々な実施態様が、特定の活性剤に関する徐放性微粒子の放出速度を変更することがここに見出される。
【解決手段】徐放性微粒子を製造する方法の様々な実施態様が記載される。一つの実施態様において、本方法は、ポリマー中の活性剤の分散相を形成し、分散相を連続相と混合して微粒子分散系を形成する工程を含む。本方法はさらに微粒子分散系に一定量の希釈組成物を添加する工程を含む。様々な量の希釈組成物を用いた徐放性微粒子を製造するための様々な実施態様が、特定の活性剤に関する徐放性微粒子の放出速度を変更することがここに見出される。
【発明の詳細な説明】
【技術分野】
【0001】
様々な天然及び合成ポリマー並びに樹脂から形成される微粒子及びミクロスフェア薬剤、診断試薬等の様々な活性剤の一般的な輸送手段となっている。分解可能な徐放性微粒子は、所謂“デポ”製剤における使用のために特に重要であり、長期にわたる活性剤の輸送が求められている。
【背景技術】
【0002】
徐放性微粒子の処方の最適な組成は、特定期間、特定の状態を効果的に治療できる、治療量の生物学的活性剤の有効量を放出する必要がある。一般に、徐放性組成物は、活性剤が微粒子から患者の組織中に放出される速度などの放出プロファイルを有する。微粒子中に封入された特定の活性剤に対する放出速度のコントロールを可能にする形で、徐放性微粒子の放出プロファイルを変更することは、有益であり得る。
【発明の概要】
【課題を解決するための手段】
【0003】
ひとつの実施態様としては、ポリマーに溶解した活性剤で分散相を形成する工程;分散相を連続相と混合して、連続相中で懸濁された微粒子を有する微粒子分散系を形成する工程;そして、微粒子形成後に、微粒子分散系に希釈用組成物の一定量を添加する工程;を含む方法であって、ここで前記希釈用組成物量が、活性剤の放出速度(放出率)を変更するのに十分な量である、徐放性微粒子の形成する方法を包含するものである。
【0004】
本発明の別の実施態様としては、活性剤とポリマーを用いて分散相を形成する工程;分散相と連続相を混合し、連続相中に懸濁された微粒子を有する微粒子分散系を形成する工程;そして、希釈用組成物を微粒子分散系に添加して、未処理の徐放性微粒子の放出速度とは異なる放出速度を有する徐放性微粒子を製造する工程;を含む方法であって、ここで前記希釈用組成物の体積は連続相の体積の少なくとも50%である、徐放性微粒子を形成する方法を包含するものである。
【0005】
さらに別の実施態様としては、ロイプロリドとポリマーを備える分散相を形成する工程を含む徐放性微粒子の放出速度をコントロールする方法を包含するものであって、前記ロイプロリドはポリマー中の溶解されているものである。本方法はさらにミキサー中で分散相を連続相と混合し、微粒子分散系を形成する工程;及び、微粒子形成後に微粒子分散系に希釈用組成物の一定量を添加する工程を含む。希釈用組成物量はまた、特定の活性剤の放出速度を変更させるのに十分であり得る。
【0006】
別の実施態様は、変更された放出速度を有し、特定の活性剤とポリマーを備える分散相を有する徐放性組成物であって、前記分散相は連続相と組み合わせられて微粒子分散系を形成し、微粒子分散系は微粒子形成後に一定量の希釈用組成物に接触させられ、ここで前記希釈用組成物は、特定の薬剤の放出速度を変更させるための連続相を希釈するのに十分であるものである。
【図面の簡単な説明】
【0007】
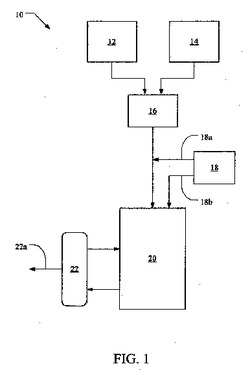
【図1】図1は、本発明の方法の一態様の略図(配置図)である。
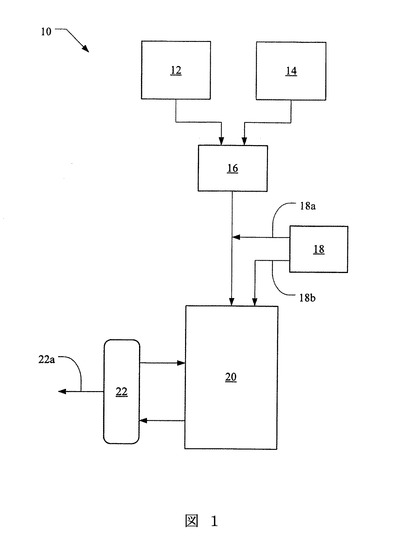
【図2】図2は、本発明の実施態様に従うところの、表3及び表4にて製造され記載された微粒子の、生体内(in vivo)ラット試験における放出特性を示すグラフ図である。
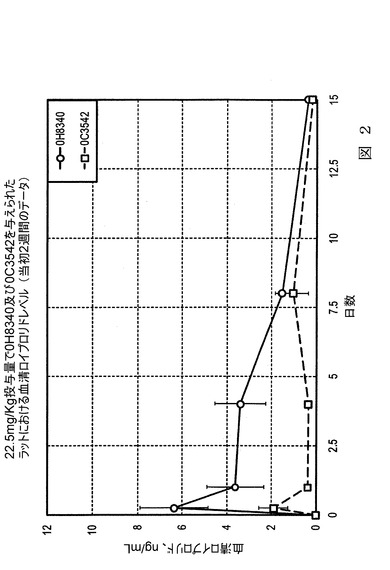
【図3】図3は、本発明の実施態様に従うところの、表3及び表4にて製造され記載されたロイプロリド微粒子の、生体内(in vivo)ラット試験における、放出特性を示すグラフ図である。
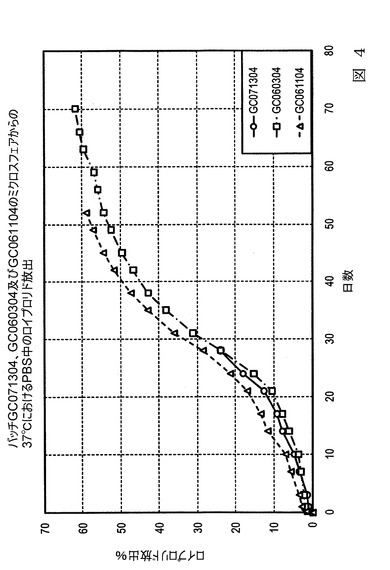
【図4】図4は、本発明の実施態様に従うところの、表5及び表6にて製造され記載された微粒子の、放出特性を示すグラフ図である。
【図5】図5は、本発明の実施態様に従うところの、表7及び表8にて製造され記載された微粒子の、放出特性を示すグラフ図である。
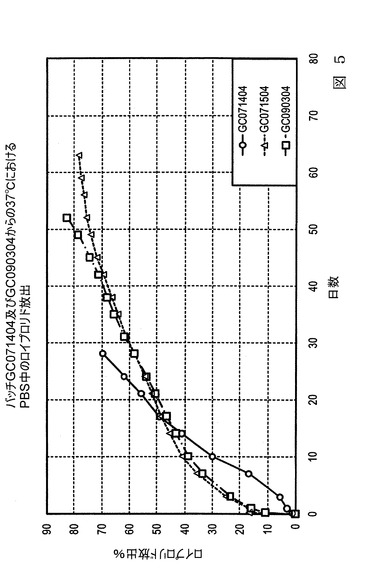
【図6】図6は、本発明の実施態様に従うところの、表5及び表6にて製造され記載された微粒子からの、生体外(in−vitro)の促進放出条件下での、酢酸ロイプロリドの放出を示すグラフ図である。
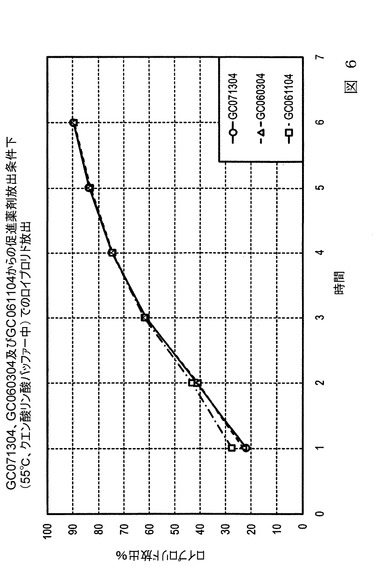
【図7】図7は、本発明の実施態様に従うところの、表7及び表8にて製造され記載された微粒子からの、生体外(in−vitro)の促進放出条件下での、酢酸ロイプロリドの放出速度(放出率)を示すグラフ図である。
【図8】図8は、本発明の実施態様に従うところの、表9及び表10にて製造され記載された2種を与えられたラットにおける、血清ロイプロリド濃度レベルを示すグラフ図である。
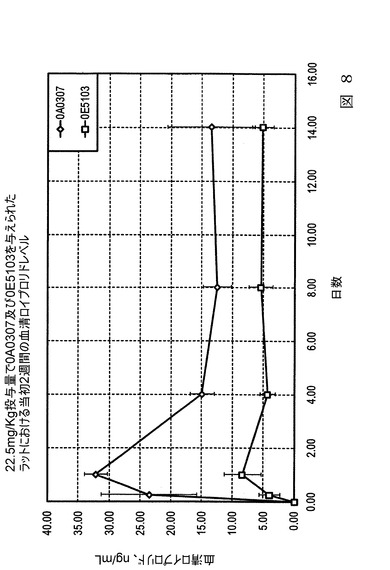
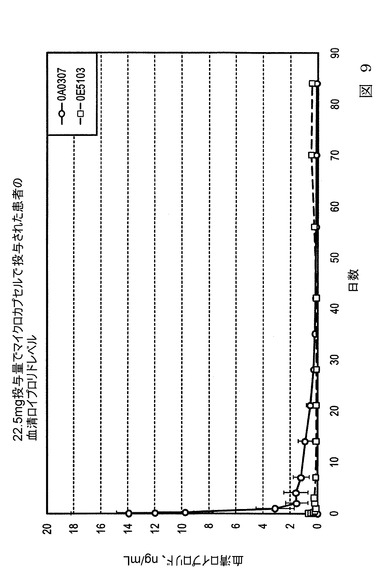
【図9】図9は、本発明の実施態様に従うところの、表9及び表10にて製造され記載された、ヒトにおける、血清ロイプロリド濃度レベルを示すグラフ図である。
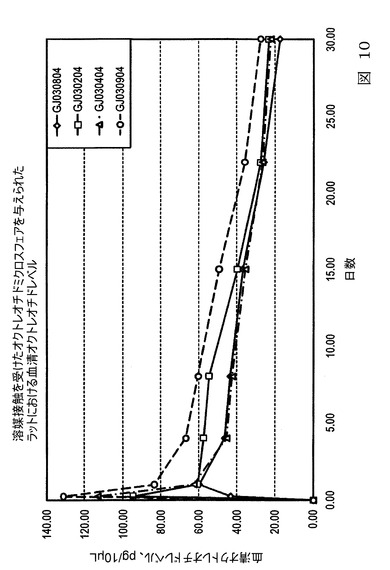
【図10】図10は、本発明の実施態様に従うところの、表11にて製造され記載された微粒子の、ラットにおける生体内(in vivo)オクトレオチド試験における、放出特性を示すグラフ図である。
【発明を実施するための形態】
【0008】
ここに、ヒトを含む温血哺乳類における、徐放性微粒子に封入された生物学的活性剤の放出プロファイルを最適化する為の方法が開示される。また、形成後の微粒子を、添加した希釈用組成物で、連続・無菌工程の一環として処理することによる、微粒子からの生物学的活性剤の放出速度(放出率)を変化させる方法をも開示する。
【0009】
図1に示すように、一つの実施態様は、徐放性微粒子からの活性剤の初期放出速度を最適化する方法10を含む。この実施態様において、微粒子中の酢酸ロイプロリドの放出プロファイルは、形成したての微粒子が接触させられるところの溶媒の濃度を調整することによって、変更され得る。微粒子の放出プロファイルは、乾燥、加熱、又は水でもどす工程の間、残存溶媒不純物又は凝集問題を取り込み得るところの関連する工程なしに、微粒子が室温で水相にとどまる間に、変更し得る。
【0010】
徐放性製品において、組成物からの初期放出される薬剤量は重要である。初期投与時に放出される薬剤、又は活性剤の量は、しばしば、初期バースト又は初期放出と呼ばれる。初期バースト又は初期放出の量は、特定の薬剤及びそれらの使用目的を検討し最適化するために重要である。
【0011】
例えば、酢酸ロイプロリド、いわゆる“ロイプロリド”は、ゴナドトロピン放出ホルモン(GnRH)の既知のアゴニスト類似物である生物学的な活性剤である。GnRHは、黄体形成ホルモン(LH)及び卵胞刺激ホルモン(FSH)の生産を促進(刺激)する。血流中に放出されると、これらホルモンは、男性においてはテストステロンを、女性においてはエストロゲンの生産を引き起こす。それ故、前立腺がん(prostrate cancer)などの疾患において、生産されるテストストロンのレベルをコントールすることは、効果的な治療であることが知られており、GnRH、LH及びFSHの生産を阻害する活性剤は有益であろう。実際には、生産されるテストストロンのレベルをコントロールすることは、去勢手術の必要性を制限し得る。
【0012】
治療的適応において、ロイプロリドは下垂体前葉からLH及びFSHを初期に放出する
。しかしながら、連続使用すると、ロイプロリドは下垂体脱感作、そしてホルモン生産テストステロンの発現低下を引き起こす。ロイプロリドは治療で使用されると、初期上昇の後、2乃至4週間の間、血中性ホルモンレベルを抑制する。よって、ロイプロリドが有効となるために、輸送微粒子からの薬剤の非常に高い初期放出速度が望ましい。米国食品医薬品局によれば、より早い去勢を獲得するために、ロイプロリドに対する高い初期放出が求められる。ロイプロリドを用いた化学的去勢は、95%の患者が28日以内に50ng/dL(0.5ng/mL)以下のテストステロンレベルを保有するもの、と定義される。
【0013】
一方、別の活性剤であるオクトレオチドは、肢端(先端)肥大症の長期間の治療、並びに、悪性カルチノイド腫瘍、血管作動性腸管ペプチド腫瘍、フラッシング及び下痢の患者に使用されるソフトスタチン誘導体である。血清中のオクトレオチドは成長ホルモン(GH)、並びにインシュリン様成長因子−1(IGF−1)をコントロールする。長期間の徐放性デポ製剤におけるオクトレオチドの使用の一般的な不利益は、高いピークレベルのオクトレオチドが、ある血漿濃度で有毒となり得ることである。より遅い初期バーストまたは放出速度が、この状況においては好ましい。
【0014】
図1に示すように、ロイプロリド−含有微粒子製造のために連続工程10が微粒子を製造するために適用される。連続工程10は一般に分散相12と連続相14の混合を含む。分散相12は、活性剤、ポリマー及び適当な溶媒を含む。連続相は、一般に、水溶液を含む。分散相12及び連続相14は、インラインミキサー16中で混合され、微粒子分散系を形成する。微粒子分散系は、一般に、水溶液中に分散された形成したての微粒子を含み得る。
【0015】
本出願の目的において、前立腺がんの治療のための徐放性微粒子の形態にあるロイプロリド製剤は、ポリマー状徐放性微粒子から放出される活性剤の一例として使用される。活性剤は小さなポリマー体へのカプセル化又は散在化が望まれている任意の薬剤にすることができることは、本開示の観点から当業者にとって明らかである。さらに、臨床的効果を改善するために放出速度が最適化され得る活性剤の例としては、ペプチド、ケトチフェン、チオリダジン、オランザピン、リスペリドン、オキシブチニン、オクトレオチド、ナルトレキソン、オルニチド、又は、Woc4D、ベータメゾン(betaqmethasone)、デキサメタゾン、クロニジン、ベラパミル、又は、薬学的に可能なそれらの塩が挙げられる。特に興味深いのは、ロイプロリド、トリプトレリン、ゴセレリン、ナファレリン、ヒストレリン、及びブセレリンなどの黄体形成ホルモン放出ホルモンアゴニスト(LH−RHアゴニスト)である。また特に興味深いものは、ソマトスタチン類似体であって、例えば、オクトレオチド、ヒト、鮭及びウナギのカルシトニン、成長ホルモン、成長ホルモン放出ホルモン、成長ホルモン放出ペプチド、副甲状腺ホルモン及び関連するペプチド、インターフェロン、エリスロポエチン、GM−SSF、G−CSF、サイモシン、アンチトリプシン及び化学療法薬、局所投与の為の抗生物質及び鎮痛剤、並びに、放出速度の調整が徐放製剤の効果を増加し得る他の薬剤、である。
【0016】
活性剤を分散相12に組み込むために、少なくとも1つの溶媒に活性剤を溶解することが通常必要である。活性剤のための溶媒は、薬剤の性質に応じて勿論様々であり得る。活性剤を溶解するための分散相に使用され得る典型的な溶媒としては、但しこれらに限定されないが、水、メタノール、エタノール、ジメチルスルホキシド(DMSO)、ジメチルホルムアミド、ジメチルアセトアミド、ジオキサン、テトラヒドロフラン(THF)、ジクロロメタン(DCM)、塩化エチレン、四塩化炭素、クロロホルム、ジエチルエーテル及びメチルエチルエーテルなどの低級アルキルエステル、ヘキサン、シクロヘキサン、ベンゼン、アセトン、酢酸エチル、メチルエチルケトン、酢酸、又はそれらの混合物が挙げられる。加えて、氷酢酸、乳酸等の酸、脂肪酸、又はアクリル酸が、ポリマー中への活性
剤の溶解とカプセル化を改善するのを促進するプロセスで使用される。与えられた系のための好適な溶媒の選択は、本開示を考慮して技術常識の範囲内事項である。
【0017】
活性剤は、続いて、ポリマーと混合され、分散相12を形成する。当業者に既知であり、一実施態様において有用である、ポリマーの例は、例えば米国特許第4,818,542号明細書、米国特許第4,767,628号明細書、米国特許第3,773,919号明細書、米国特許第3,755,558号明細書、米国特許第5,407,609号明細書に見出され、参照することにより本明細書に組み込まれる。与えられた系における特に好ましいポリマーの選択において、生分解性(例えば放出プロファイル)及び生体適合性といった所望の臨床的特徴を有する生成物を生産する目的のために、多数の因子が考慮される。一旦、ある当業者が、所望の臨床的特徴を提供するポリマー群を選択すると、続いて、該ポリマーは、製造工程を最適化することとなる望ましい特徴が評価される。例えば、ある場合には、微粒子の処理を促進する、薬物搭載量を増進する、分散相からの溶媒の除去を強化する、或いは、分散相から連続相への薬物の移動を抑制するという形で、活性剤と相互に作用するポリマーを選択することが可能である。
【0018】
好適なポリマーの具体例は、乳酸のホモポリマー又は乳酸とグリコール酸のコポリマー、又は、ポリ(ラクチド−コ−グリコリド)、“PLGA”ポリマーである。乳酸残基とグリコール酸残基の比率は様々であり、典型的には25:75乃至85:15の範囲であり、但し、高いラクチド含量は低い粘性及び高い溶解性につながるため、10%のグリコリドであっても使用される。好ましいコポリマーは、少なくとも約50%の乳酸残基を含み、例えば50:50、75:25、又は85:15ポリマーである。ポリ(ラクチド−コ−グリコリド)コポリマーは、多くの供給源から商業的に入手可能であり、そして、従来の合成ルートによって容易に製造される。ベーリンガー インゲルハイム社は、R202H、RG502、RG502H、RG503、RG503H、RG752、RG752H、RG756及びその他の称号にて好適なポリマーを製造している。R202H、RG752H又はRG503HとのLH−RH微粒子は、ある実施態様の分散相において使用され得る。与えられた系に対する好適なポリマーの選択は、本開示を考慮して当業者であれば明らかである。
【0019】
好ましいポリマーの選択における一つの検討事項は、ポリマーの親水性/疎水性である。ポリマーと活性剤の双方が、疎水性又は親水性であり得る。可能であれば、親水性の活性剤と共に使用するために親水性ポリマーを選択すること、そして、疎水性活性剤とともに使用するために疎水性ポリマーを選択することが望ましい。好ましい微粒子において、薬剤とポリマーの親水性カルボキシル基との間のイオン相互作用は、薬物搭載量を増進するとみられている。しかしながら、一般に、親水性薬剤は水に溶解するものの、ポリマーと薬剤の間に親和性がない場合、又は、固化が十分に早くない場合、薬剤搭載量は減少し得る。疎水性ポリマーにおける親水性薬剤の使用もまた可能である。
【0020】
特定のポリマーの選択において、系の残留溶媒へのポリマーの疎水性/親水性の影響もまた考慮する必要がある。親水性ポリマーは、親水性ペプチドといった親水性薬剤では、少ない残留溶媒を生じることが期待される。ロイプロリド微粒子の場合、薬剤は、分散相の液滴から疎水性溶媒を迅速にそして効率的に排除するのを補助する傾向を有する。加えて、より多量の薬剤搭載量は、より低い残留溶媒濃度と相互に関係がある傾向にあることが見出されている。よって、いくつかの系においては、親水性ポリマー内に親水性薬剤が内包されるとき、より少ない残留溶媒による間接的な利点が存在する。しかしながら、残留溶媒における親水性以外の他の影響因子が存在する場合、この効果は、非ペプチド薬剤に対して一様には適用できない。それにも関わらず、分散相の液滴からの溶媒の排除を増進する活性剤が、付随する薬剤損失なしに、優れた生産物を生産することを、理解すべきである。
【0021】
別の検討事項は、ポリマーの分子量である。ポリマーの分子量は、放出速度、放出プロファイルなどの生成物の特徴に対して明らかに影響を及ぼすと同時に、微粒子の製造プロセスにも影響を与える。より高分子量のポリマーは、概してより粘性の高い分散相と関連し、大きな粒子をもたらすか、或いは、小さな粒子を得ることの難しさの増加をもたらし、そして、いくつかの例において、残留溶媒の増加をもたらす。一方、より低分子量のポリマーは、ポリマーがより溶解性である傾向のために、概してより遅い固化と関連する。好ましい系において、より多い残留溶媒、より高い薬剤搭載量及び増進された取り込み効率は、より高分子量ポリマーの使用に由来することが見出されている。本発明の工程のひとつの有利点は、優れた、小さい、少ない残留溶媒である微粒子を、高分子量ポリマーとともに形成し、そしてそれゆえに粘性の分散相を形成することとなる、その能力である。勿論、特定の選択はまた、所望の生成物の特徴に依存する。例えば、より高分子量で、体内でのより長い分解時間、そしてより長い薬剤放出持続時間である。
【0022】
さらに、適用される特定のポリマー濃度は、生成物の形態の観点だけでなく、製造の観点から、系に影響を与える。粘性の分散相は、固化のためのより少ない溶媒を除去する必要があるため、ポリマー濃度の増加は、より多い薬剤搭載量と関連する傾向がある。増加された固化速度は、より高い薬剤保有を引き起こす傾向がある。その上、粘性の分散相は、固化の間に、連続相へのより少ない薬剤拡散をもたらす。いくつかの系において、これはまたより多い残留溶媒をもたらす。好ましい実施態様において、分散相中のポリマー濃度は約5乃至40%であり、そしてより好ましくは約8乃至約30%である。
【0023】
ポリマーのための溶媒は、ポリマーの性質、活性剤、毒性、系中の他の溶媒との相溶性、そしてさらに微粒子が適用される用途などの、多くの因子に応じて適切に選択される。よって、ポリマーの溶解に加えて、溶媒は、液滴を形成するために連続相と混和しないこと、最適な蒸発効率のための高い揮発性、そして望ましくは安全性の理由から不燃性でなければならない。好ましいポリ(乳酸)又はポリ(ラクチド−コ−グリコリド)ポリマーのための好適な溶媒としては、塩化メチレン、クロロホルム、酢酸エチル、置換されたピロリドンなどが挙げられる。場合によっては活性剤のための溶媒は、ポリマーのための溶媒と同じである。一部の薬剤、概して画像解析に使用される放射性無機塩などの診断薬は、有機溶媒に溶解しないか、ほんの少ししか溶解しない。これらの例において、微細なサブ−サブミクロンサイズの粉体が、ポリマー溶液に直接懸濁して、微粒子を形成する。薬物送達において、これを用いることはまれではあるが、診断薬では役立つことが証明されている。本発明のプロセスに従う他の有用な溶媒の選択は、本開示を考慮して、当技術範囲内にある。
【0024】
この実施態様において、ロイプロリド含有ポリ(乳酸)又はポリ(ラクチド−コ−グリコリド)微粒子は、R202Hポリマーに、塩化メチレン、メタノール及び氷酢酸とともにロイプロリド活性剤を溶解して、分散相12を形成することにより、製造される。
分散相は、実に、均質な溶液である。或いは、それぞれ個々の溶媒中にポリマーと活性剤の別々の溶液を準備し、そして続いて混合して分散相12を形成する。場合によっては、活性剤及び/又はポリマーの性質の理由から、分散相12は、エマルションとして形成せざるを得ない。例えば、所定のタンパク質性の薬剤が好適な活性剤の溶媒中に溶解された場合、得られた溶液は、特定のポリマー溶媒中のポリマー溶液とは、完全に混和しない。薬剤とポリマーが比較的均一に散在しているところの、比較的均質である分散相12を提供するために、薬剤及び薬剤溶媒は、ポリマー及びポリマー溶媒と乳化し、分散相エマルションを形成し得る。分散相12の連続相14への導入において、水−油−水、又はW/O/Wエマルションが形成され得る。さらに別の系において、分散相12は、ポリマー溶液中の活性剤の直接的な懸濁液の形成によって製造される。
【0025】
下記に記載される本発明のプロセスに応じて、連続相14中に分散された分散相12の液滴又は内包物を有する微粒子分散系を形成するために、分散相12は連続相14と混合され得る。ここで使用する用語“分散された(dispersed)”とは、広義において、連続相中に散在する分散相の不連続部位を意味するものと意図される。上記内包物は、概して、通常球状の液滴として生じるが、場合によっては、特定の乳化条件に応じて、不規則な内包物と成り得る。分散相12が液滴又は内包物を形成するところの任意の適当な媒体が、特に望ましいとされる分散相12溶媒のための最大溶媒シンクを提供するものと共に、連続相14として使用され得る。
【0026】
しばしば、連続相14は、乳化工程を変更させ影響を与えるところの界面活性剤、安定剤、塩又はその他の添加剤をも含む。典型的な界面活性剤は、ドデシルスルホン酸ナトリウム、ジオクチルスルホコハク酸ナトリウム、スパン、ポリソルベート80、トウィーン80、プルロニクスなどが挙げられる。特定の安定剤としては、タルク、PVA及びコロイダル水酸化マグネシウムが挙げられる。粘度ブースターとしては、ポリアクリルアミド、カルボキシメチルセルロース、ヒドロキシメチルセルロース、メチルセルロースなどが挙げられる。緩衝塩は薬剤安定剤として使用され、一般的な塩でさえも活性剤の連続相14への移動の防止を促進するために使用される。連続相14の塩の飽和に関連する一つの問題は、PVA及びその他の安定剤が、連続相から固体物として沈殿する傾向を有し得ることである。そのような場合、微粒子安定剤が使用され得る。塩化ナトリウムや硫酸ナトリウムなどの適当な塩並びに他の添加剤は、本開示を考慮すると当業者に明白である。
【0027】
ひとつの実施態様において、連続相14は、100−50%の水を含む。水性の連続相14は、安定剤を含み得る。好ましい安定剤は、約0.1%乃至約5.0%量のポリビニルアルコール(PVA)である。より具体的には、PVAは約0.35%量で存在し得る。連続相14への使用のための適当な他の安定剤は、本開示を考慮すると、当業者に明白である。
【0028】
特定のポリマー、溶媒及び連続相の選択は、無論、活性剤並びに所望の生成物の特徴に応じて様々である。一旦、臨床的適用、放出プロファイルなどの所望の生成物の特徴が確立されたとしても、それでもなお、製造プロセスを促進するためのポリマー、溶媒及び連続相の選択には多少の自由度が残されている。
【0029】
分散相12及び連続相14が混合されるとすぐに、形成したての微粒子が、ミキサー16から第一容器20へ連続的に移動させられ得る。この実施態様において、微粒子は一般に十分に固体且つ濾過可能であり、それ故、米国特許第6,270,802号明細書及び米国特許第6,361,798号明細書に開示されるように、中空糸フィルタを使用する微粒子の連続処理に適する。この方法は、分散相と連続相の混合物が、別々の微粒子をすぐに形成しないような、一般的な微粒子形成方法とは区別される。
【0030】
微粒子がさらなる形成手順を必要とする方法とは対象的に、この実施態様の微粒子は約20秒以内に、より好ましくは約10秒未満で、そしてより特に好ましくは約5秒未満で固化する。ロイプロリド−ポリマーの実施態様において、微粒子は、約3秒未満で固化する。本実施態様において、微粒子の固化は連続相への添加時に事実上一瞬であるにしても、形成された微粒子は、それにもかかわらず、更なる残留溶媒を連続相へ引き渡すことに影響を与えやすい。
【0031】
薬剤搭載量は、好ましいロイプロリド/ポリマー微粒子の場合、総固形物をベースとして約12%を目標とし、10乃至21%の範囲である。実際には、約9乃至17、約17%の薬剤搭載量が得られる。勿論、薬剤の性質、所望の放出プロファイル、ポリマーの性質、そして勿論処理方法全てが、所望のそして実際の薬剤搭載量に影響する。典型的なケ
ースにおいて、薬剤とポリマーの混合量をベースとして約5%乃至20%の薬剤搭載量が望ましく、本発明のプロセスによって達成可能なものである。典型的なカプセル化効率は約75%である。
【0032】
有利なことに、一旦所望の薬剤搭載量を得ると、供給量、温度などのパラメーターが算出され、製造レベルのバッチ数を含むより大きなバッチ数のスケールアップは、単により長いプロセスを実施すれば良いだけのこととなる。所望の特徴を有する微粒子を大量に生産するのに、さらなる供給チューブ(管)、乳化剤、羽根車などのものを必要としない。加えて、本発明の連続プロセスの間に製造される微粒子は、プロセスの間にいつ製造されたかにかかわらず、サイズ分布、薬剤搭載量などが並外れて均一である。
【0033】
ひとつの実施態様において、上述したように、微粒子形成の一般的プロセスは、微粒子の溶媒接触を変化させることによって、微粒子バッチの放出特性又は放出プロファイルが変化するように修正され得ることが見出されている。形成後のより低いレベルの溶媒接触は、形成したての微粒子に対して希釈組成物18を加えることによって達成される。希釈組成物18は、水又はポリビニルアルコール溶液であり得る。微粒子形成後の一定量の希釈組成物18の添加は、驚くべきことに、形成したての微粒子の放出速度を変化させることが見出されている。
【0034】
図1に示すように、微粒子が形成され、連続相に分散された後、希釈組成物18が添加され得る。図1中、18aで示される希釈組成物は、第一容器20に移送される時に、微粒子分散系に添加され得る。第一容器20に移送される際の希釈組成物の微粒子分散系への添加は、第一容器20に到達し、中空糸フィルタ22を用いて濾過される前に、微粒子分散系と希釈組成物を完全に混合させることを可能にする。
【0035】
代わりに、18bに示すように、希釈組成物18は、微粒子分散系の前に、又は、分散系と同時に、第一容器20に移送され得る。一旦、第一容器に入ると、微粒子と希釈組成物の混合物は、連続的に撹拌され得る。
【0036】
微粒子分散系に添加される希釈組成物18の量は、哺乳類に注射するために、最終製剤の放出プロファイルを調整するのに十分であるべきである。希釈組成物18は、微粒子中にカプセル化された活性剤の溶解性、微粒子の粒子サイズ、或いは各バッチにおける活性剤のカプセル化効率を逆向きに或いは大幅に変更すべきではない。添加される希釈組成物18の所定量に対応した、所定の溶媒接触レベルは、所望の放出速度に対して計算される。その結果、特定の放出速度が希釈組成物18の添加によって得られる。
【0037】
ひとつの実施態様において、所望の放出プロファイルを備えるロイプロリド微粒子を得るために、連続相中の溶媒の規定濃度が5000ppm未満になるまで、希釈組成物18を加える。規定量及び時間の希釈組成物18の添加によって、放出プロファイルは、予想通りのそして制御可能な方法で、連続プロセスの一環として調整される。
【0038】
形成された微粒子に対して添加する希釈組成物量は、連続相14の元の体積の少なくとも約50%であり得る。ひとつの実施態様において、水溶液中で溶媒への接触の減少は、形成されたロイプロリド微粒子の放出速度を増加させ得る。異なる活性剤の放出速度の制御は、しかしながら、所望の放出プロファイルを獲得するために異なるステップを必要とし得る。本プロセスは、異なる放出プロファイルを備える異なる活性剤の使用に適合するように、容易に調節可能である。
【0039】
ミキサー16からの微粒子分散系と希釈組成物18を、第一容器20で混合し、微粒子/希釈組成物混合物を、中空糸フィルタ22にて濾過する。廃棄物を、ライン22aを介
して、分離された容器に廃棄すると同時に、微粒子を第一容器20に戻す。微粒子が容器20に戻されると、それらは新たな微粒子/希釈組成物混合物と連続的に混合され、フィルタを通して循環させられる。
【0040】
必須組成物及び所定時間で処理された後、所望の放出速度を獲得した微粒子を、その後、幾らか残存する水溶液を除去するために濾過し、そして、プロセス副生成物を除去するために水で洗浄し得る。微粒子が形成されると、患者投与のためにそれらを準備するところの充填容器に対して、微粒子を準備するためになされ得る、多くの最終的な調整があることは、当業者に既に明らかである。
【0041】
本発明による方法は、添付の図面を参照すると共に、以下の限定されない実施例によって例示される。
【実施例】
【0042】
以下の実施例は、ここに主張する方法を実施し評価する方法を当業者に対して詳細な記載と共に説明するものであるが、発明者が自身の発明とみなしているところの範囲を制限することを意図するものではない。特に指定の無い限り、部は質量で、温度は℃である。
【0043】
分子量は、重量平均分子量(Mw)又は数平均分子量(Mn)で示され、そして、ポリスチレン(PC)を分子量標準に用いたゲル浸透クロマトグラフィー(GPC)を用いて測定される。実施例1は、本方法の一態様を示す概略図を提供する。ポリ(d,l−ラクチド)、R202Hは、ベーリンガーインゲルハイム社から市販されている。
【0044】
実施例1
酢酸ロイプロリド微粒子を、下記表1に記載されるように製造した。分散相及び連続相を混合し、約7Lの微粒子分散系を形成することにより、微粒子を形成した。概して、分散相は表1中に記載されたパラメーターによれば、酢酸ロイプロリド(L.A.)、ポリ(d,l−ラクチド)(R202H)及び溶媒 塩化クロリド(DCM)、メタノール(MeOH)及び氷酢酸(A.Acid)の溶液であリ得る。R202Hは、温血動物及びヒトにおける生体内(in−vivo)研究に基づき、3乃至4ヶ月間、ロイプロリドの徐放性を提供する。連続相は、0.0035g/gポリビニルアルコール溶液を用いて製造した。
【0045】
連続相及び分散相をインライン シルバーソン(Silverson)ミキサーに、それぞれ流速2L/分及び38g/分にて同時に添加した。ミキサー中の混合速度は7000rpmであり、微粒子形成期間は3.5分であった。この微粒子分散系を、約40Lの希釈相を含む容器に連続的に供給した。
【0046】
希釈相もまた0.0035g/gポリビニルアルコール水溶液を含み得、但し希釈相は表1に示す溶媒が混合されていた。希釈相中の塩化メチレン量は、各バッチで様々であり、具体的には、微粒子バッチ:GG090303に対して約5000ppmから、微粒子バッチ:GG091003に対して約8500ppmであった。反応容器において、希釈相入りの溶媒との溶媒接触に関する対応時間は、全ての4つ微粒子バッチに対して100分間で一定であった。
【0047】
溶媒接触後、希釈相に懸濁している微粒子を溶媒除去容器(SRV)に移送した。全ての微粒子バッチを、40分間、2 体積変化にて周囲水洗浄、80SLPMのエアスウィ
ープ;80SLPMのエアスウィープしながら、100分間、5 体積変化にて温水洗浄
(39℃)を用いて洗浄し;そして80SLPMでエアスウィープしながら、30分間,1.5 体積交換にて冷却した。洗浄プロセスの間、ヘッドスペース溶媒除去を助けるた
めにエアスウィープを導入した。微粒子をメンブレンフィルタで濾過し、真空下で凍結乾燥した。
【0048】
表1は、活性剤、ポリマー及び分散相の製造に用いた溶媒の濃度、並びに、希釈相の調製に用いた各溶媒の濃度及び実際の量を示す。
【表1】

【0049】
表2は、粒子サイズ、促進放出条件下での薬剤放出、並びに、生理学的な条件下での薬剤放出などの、表1の製造パラメーターにて製造した微粒子の特徴を示す。微粒子の薬剤搭載量は、微粒子中の活性剤の割合として記述される。これは、溶媒(例えばジメチルスルホキシド)中に微粒子を溶解し、微粒子溶液にホスフェートバッファーを添加することによって決定された。濾過された溶液は、その後、キャリブレーション基準を対照としてHPLCによって分析された。
【0050】
粒子サイズ分布(Part.Size Dist.)は、界面活性剤又は希釈剤を含有する水中で、微粒子懸濁液に関して決定された。これは、レーザー光散乱で決定された。粒子サイズ分布を体積分布(CVF)で示した。
【0051】
生理学的条件下(D.R.(Phys.))でのロイプロリドの放出速度を決定するために、薬剤放出速度の算出をリン酸バッファー溶液(PBS)中で実施した。表1に示すように処方された微粒子を、温度37℃で、スクリューキャップの付いたガラスチューブ中、PBSに懸濁させた。少なくとも80%の放出媒体が、標本点において、新たな媒体と置換された。放出媒体についてHPLCを用いて薬剤濃度の測定を行い、放出率を算出した。
【0052】
放出条件を適切に操作し、生体内で獲得される放出速度、或いはPBSへの放出による放出速度を促進させた。ロイプロリド微粒子に対して、使用される放出媒体は、0.1Mクエン酸リン酸塩バッファーであり、温度は55℃であった。170回転/分にて撹拌された、撹拌された水バッチ中で放出を実施した。試験品について各時間ポイントで放出された薬剤の測定を行い、促進条件下(D.R.(Acc.))の放出速度を見出した。
【表2】
【0053】
上述の製造ステップ及び表1に従い形成された微粒子において、同じような粒子サイズ分布及び薬剤搭載量がみられた。しかしながら塩化メチレン溶媒の最も高い接触レベルに接触させられた微粒子バッチは、若干減少した薬剤搭載量を示した。
【0054】
より意味があることは、ロイプロリドの薬剤放出速度は、促進薬剤放出試験(55℃クエン酸リン酸塩バッファー)並びに生理学的条件(リン酸塩バッファー溶液、pH7.4、37℃)下の薬剤放出の双方とも、バッチによって様々であった。促進試験条件下での薬剤放出は、バッチによって放出速度の違いに関して素早い比較を提供した。生理学的条件下での薬剤放出を、臨床適用における徐放性製剤の実際の持続期間をシミュレートするために、最大60日以上のより長い試験期間の間、実施した。最も高いレベルの溶媒(8500ppm)に接触したバッチGG091003の微粒子は、一貫して最も遅い放出速度を有していた。とくに、放出速度の違いは、初期の時点の間でより大きなものであった。一般に、酢酸ロイプロリドで形成され、より高い溶媒濃度に接触させられた微粒子バッチは、減少した放出速度を示した。よって、ロイプロリドを含有する微粒子の放出速度を増加させるためには、微粒子形成後に、溶媒接触を減少させることが望ましい。
【0055】
実施例2
酢酸ロイプロリド微粒子を、下記表3に記載される通りに製造した。微粒子分散相バッチを、酢酸ロイプロリド(L.A.)、塩化メチレン、メタノール及び氷酢酸からなる一貫した組成物を用いて調製した。2つの微粒子バッチ、0C3542及び0H8340は、14kDaの分子量を有するR202Hのポリマーロットを用いて、懸濁液中で、それぞれ0.7kg、0.4kgの固体微粒子をもって製造した。3つの微粒子バッチ、0D4022、0E5103、及び0S1723を、18kDaの分子量を有するR202Hのポリマーロットを用いて、懸濁液中で、それぞれ0.1kg、0.4kg、0.4kgをの固体微粒子をもって製造した。各バッチを、同様の連続相処方及び同様の製造パラメーターを用いて製造した。但し、微粒子形成時間は、バッチサイズを変化させることによって、即ち懸濁液中の微粒子が0.4kgと対照的に0.7kg、並びに、それぞれのバッチの懸濁液を反応容器への分散相の流速を変化させることによって連続相を含む溶媒と接触させる時間を変化させることによって、様々であった。
【0056】
具体的には、微粒子は、それぞれの分散相を、0.35%のポリビニルアルコール水溶液の連続相に添加することによる水中油型プロセスによって形成した。インラインシルバーソンミキサーの混合速度は7000rpmであった。微粒子が形成するまで、分散系の連続相の溶媒濃度は、およそ塩化メチレン 7200ppmとメタノール 1600ppmであった。
【0057】
インラインミキサー中で形成後に微粒子バッチを連続的に溶媒除去容器に移した。微粒子形成時間は、0.4Kgバッチで約40分であり、一方、微粒子形成時間は、0.7kgバッチで約70分であった。微粒子形成の20分後、分散相を連続相と、所定の流速で混合し、溶媒除去容器は40Lの懸濁液量に達した。溶媒除去容器中の懸濁液を、その後、中空糸メンブレンフィルタ(閉鎖系)を通して再循環させ、溶媒除去容器への懸濁液供給と同じ速度(2L/分)で透過物を除去した。よって連続相中の溶媒レベルは、0.4kgバッチで40分間、0.7kgバッチで70分間で一定に維持された。
【0058】
微粒子バッチを、40分間、2 体積変化で周囲水洗浄、80SLPMのエアスウィー
プ;80SLPMのエアスウィープしながら、100分間、5 体積変化で温水洗浄(3
9℃)を用いて洗浄し;そして80SLPMのエアスウィープしながら、30分間、1.5 体積交換で冷却した。洗浄プロセスの間、ヘッドスペース溶媒除去を助けるためにエ
アスウィープを導入した。
【表3】
【0059】
表3の形成されたバッチの微粒子特性を下記表4に示す。
【表4】
【0060】
微粒子サイズはバッチに関して同程度であった。実施例1に関して上述に記載されたように、薬剤放出特性を生理学的条件及び促進条件にて試験した。結果は、より長い溶媒接触期間、即ち、溶媒接触を増加させた微粒子OC3542及びOD4022に関して減少した初期放出を示した。バッチOC3542から、すなわち、低分子量ポリマー(Mw=14kDa)から製造された微粒子は、生理学的条件下及び促進条件下のいずれにおいても、殆どの区間にわたって、最も遅い放出速度を有した。よって、高い溶媒接触において、ポリマーの分子量が小さくなると、ロイプロリドの初期放出が低くなった。
【0061】
表3及び表4に従って製造された微粒子を、ラットの体重1kgあたり酢酸ロイプロリド9mgの懸濁液として注射した。図2は、バッチ0H8340(40分溶媒接触、14kDa)とバッチ0C3542(70分溶媒接触、14kDa)の同様の投与量を受けたラットにおける、血清ロイプロリドを比較する。両物質とも、分子量14kDaを有するポリマーで製造されているものの、0C3542は、0.7kgのバッチサイズで、70分間の溶媒接触にて製造され、一方、0H8340は0.4kgのバッチサイズで40分間の溶媒接触で製造されている。この研究は、0C3542が微粒子からのより遅い初期の薬剤放出のために、血清中で、始めはより低い薬剤レベルを生ずることを示している。約三週間後の第二局面は同程度の薬剤レベルを生じさせた。これは、より長い溶媒接触を受け、より大きなバッチサイズを有するバッチ0C3542からの微粒子のより低い初期放出を確認するものである。
【0062】
図3は、バッチ0D4022(70分溶媒接触、18kDa)とバッチ0E5103(40分溶媒接触、18kDa)の同様の投与量を受けたラットにおける血清ロイプロリドを比較する。両物質とも、分子量18kDaを有するポリマーで製造されているものの、0D4022は0.7kgのバッチサイズで70分間の溶媒接触にて製造され、一方、0E5103は0.4kgのバッチサイズで40分間の溶媒接触で製造されている。再度、より長い溶媒接触期間を有するバッチである0D4022は、血清中で始めはより少ない薬剤レベルを生じ、接触の初期段階でより遅い放出を確認した。
【0063】
実施例3
ロイプロリド微粒子を、固有粘度0.206dl/g、分子量18kDaを有するポリマーで製造した。微粒子バッチを、10gスケールにて、連続処理手順を用いて製造した。分散相組成物、連続相組成物、分散相流速、連続相流速、及び1分あたりのミキサー回転数などの製造パラメーターを、全てのバッチで等しくなるように保った。
【0064】
具体的には、水中油型プロセスを用いて、それぞれの分散相と連続相を混合することにより、微粒子を形成した。連続相は、0.35の%ポリビニルアルコール水溶液から構成されていた。インラインシルバーソンミキサーの混合速度は7000rpmであった。
【0065】
微粒子分散系をミキサーから溶媒除去容器に移送した。各バッチにおいて、希釈組成物量を溶媒除去容器に添加した。各バッチにおける希釈組成物は水であった。分散系をインラインミキサーから溶媒除去容器へ移動させるのと同時に、希釈組成物を溶媒除去容器に添加した。具体的には、バッチGC071304を増加される希釈組成物なしに製造し、バッチGC060404は連続相の体積の50%に相当する希釈組成物にて製造した。バッチGC060404を、シルバーソンミキサーから、約2Kg/分の速度で移送し、懸濁液を室温水(22乃至25℃)で、約1kg/分の同時フローにて、希釈した。
【0066】
バッチGC060304に関して添加された希釈組成物は連続相の体積の約100に%相当し、ここでバッチを2kg/分の速度でミキサーから移送し、希釈組成物を同時に2kg/分の速度で添加した。バッチGC061104に関して2kg/分の懸濁液フローを使用しながら希釈組成物を4kg/分の速度で添加し、懸濁液の連続相の体積を約200%増加させた。混合速度は、全ての試験において、7000rpmで一定とした。
【0067】
最後に、微粒子バッチを、40分間、2 体積変化で周囲水洗浄にて洗浄、80SLP
Mのエアスウィープ;80SLPMのエアスウィープしながら、100分間、5 体積変
化で温水洗浄(39℃)を用いて洗浄し;そして80SLPMのエアスウィープしながら、30分間、1.5 体積交換で冷却した。洗浄工程の間、ヘッドスペース溶媒除去を助
けるためにエアスウィープを導入した。微粒子をメンブレンフィルタで濾過し、真空下で凍結乾燥した。
【表5】
【0068】
表5で製造した微粒子の特性を表6に示す。
【表6】
【0069】
実施例1に関して上述されるように、薬剤放出粗特性を生理学的条件下並びに促進条件下で試験した。異なる組成物から製造した微粒子の薬剤カプセル化及び粒子サイズ特性は
同程度であった。ロイプロリドフリーベースのカプセル化効率は、80乃至82%の範囲であった。結果、薬剤カプセル化は、連続相部分に添加する量を0から200%の範囲に増加させた希釈組成物を有する全てのバッチにおいて類似していることを示した。平均粒子サイズ(50%体積分布下)は、25乃至29μmにおよび、90%体積分布では47乃至50μmにおよんだ。一般に、薬剤カプセル化と粒子サイズ分布は、バッチ間で類似している。
【0070】
生理学的条件下で測定された薬剤放出速度をリアルタイム薬剤放出速度(長期間)の指標として測定した。希釈組成物が増加し、そして連続相がさらに希釈されるにつれて、結果はより早い放出速度を示した。促進放出速度試験条件も連続相の希釈組成物が増加するにつれてわずかに高い初期放出速度を示した。例えば、連続相部分に、最も高い希釈組成物量と、最も低い溶媒濃度を有するバッチGC061104は、全ての時間間隔にて、両放出条件下で最も早い放出を示した。バッチGC071304は希釈組成物の増加をしていない。最も高い溶媒濃度を有する試料として、この試料は3日及びそれ以下の時間間隔に関して、生理学的条件下で、最も遅い放出速度を示した。従って微粒子形成後の溶媒接触の減少は、微粒子からのロイプロリドの放出速度を増加させる。
【0071】
図4は、表5及び6に製造され記載されたバッチGC071304、GC060304、及びGC061104を含む、37℃のリン酸バッファー溶液中のロイプロリド微粒子の放出特性を示すグラフ図である。図4は、異なる希釈組成物レベルにてバッチが製造されたとき、PBS中で37℃で放出されたロイプロリドの割合を表す3つのS−型カーブを示す。全3つのカーブの間に狭い分離が存在するが、図4は、200%の最も多い希釈組成物の添加に対応するバッチGC061104が、特に早い時間段階で、最も高いロイプロリド放出速度(放出率)を有することを示す。
【0072】
図6は、表5及び6に製造され記載されたバッチGC071304、GC060304、及びGC061104を含む、55℃のクエン酸リン酸バッファー溶液中の、促進放出条件下での酢酸ロイプロリド微粒子の放出特性を示すグラフ図である。微粒子バッチは促進放出試験条件によるロイプロリドの放出速度(放出率)を示すようにプロットされ、1時間ごとの時間間隔に対して記録した。最初のデータポイント(1時間及び5時間)に関して、バッチGC061104がわずかに高い放出を有することを示した。
【0073】
実施例4
微粒子バッチをロイプロリド及び固有粘度0.157dL/g、分子量11.5kDaを有するR202Hを用いて製造した。バッチを、約10gスケールで製造した。
【0074】
表7はバッチの製造パラメーターを比較する。製造パラメーターは、添加した希釈組成物量を除いて、全てのバッチで類似している。
【表7】
【0075】
表8は、0.157IVポリマーを用い希釈組成物量を変化させ、表7に示すように製造された、微粒子バッチの特性を比べる。
【表8】
【0076】
希釈組成物の連続相への添加なしに製造された微粒子は、比較的より低い放出速度を有
する。より低分子量のポリマーは、より高分子量のポリマーで製造された微粒子に比べて、溶媒接触並びに微粒子中の薬剤含量に対してより敏感である。粒子サイズ分布は3つのバッチに関して同程度であり、変化の度合は23乃至26μmの範囲の平均粒子径(50%体積分布下)と小さい。希釈組成物を含まないバッチGC071404は、他の2つのバッチのものと比べて、より遅い初期放出を示した。早期放出におけるこの違いは、促進体外(in−vitro)放出にも反映される。
【0077】
図5は0.157の固有粘度を有するポリマーで形成された微粒子に関するリン酸バッファー溶液中の薬剤放出速度(放出率)を比較する。0.157IVポリマーで形成された微粒子は、希釈組成物を含まないバッチにおいて、より遅い初期放出を示した。100及び200%の希釈組成物添加のバッチに関して、0.157IVポリマー微粒子はより高い放出速度を示した。希釈組成物の添加は、形成したての微粒子に対する溶媒接触レベルを最小限に抑え、低IVポリマーに関する放出速度を増加させる。
【0078】
図7は、表7に示す希釈組成物の様々な量を有する3つの試料のグラフ図であって、促進体外(in−vitro)条件下での1時間に対してプロットした放出された酢酸ロイプロリド割合のグラフ図である。増加させた希釈組成物を有するバッチは、生理学的条件下で最初の2週間、促進放出試験条件下で最初の5時間、より高い放出速度を有した。
【0079】
実施例5
実施例5は、連続相への希釈組成物の添加/無添加での大規模微粒子バッチを詳述する。薬物動態(PK)及び有効性データ、そして3つのロイプロリド微粒子バッチ(0E5103、0B6315及び0A0307)に関して、約0.21dL/gの固有粘度を有するR202Hポリマーを用いて製造した。バッチ0E5103及び0A0307を、薬物動態及び有効性を目的として、ヒトへの22.5mg投与量で試験した。薬物動態(PK)群の患者は血中(血清)ロイプロリドレベルとテストステロン抑制に関してモニターし、残りの患者を有効性(テストステロン抑制)のみモニターした。ロイプロリドを用いる化学的去勢を示すためには、95%の患者が28日間以内で50ng/dL(0.5ng/mL)以下のテストステロンレベルを示す必要がある。
【0080】
微粒子バッチ0E5103を、0.206IV(固有粘度)と分子量18kDaを有するR202Hを用い、希釈組成物の添加や希釈なしに製造した。このバッチの塩化メチレン接触は約7200ppmであり、接触期間は約40分間であった。微粒子バッチ0B6315を、0.206IV(固有粘度)と分子量約18kDaを有するポリ(D,L−ラクチド)で製造した。連続相が溶媒除去容器にある間に、希釈組成物を連続相フローに対して水150%の添加により製造した。
【0081】
微粒子バッチ0A0307をバッチ0B6315と同様のIV(固有粘度)と分子量を有するポリ(d,l−ラクチド)を用いて製造した。連続相及び分散相エマルションがインラインミキサーを通って溶媒除去容器へ供給される間に、連続相フローに対して200%の水を添加することにより希釈組成物を製造した。塩化メチレン溶媒レベルは、ガスクロマトグラフィー(GC)をもとにして、連続相中で約1500ppmであった。表9は製造パラメーターを比較し、表10は表9で製造した微粒子の特性を比較する。
【0082】
特に、0A0307に関するバッチサイズは約1Kgスケールであり、反応容器中の微粒子形成時間は100分間であった。バッチ0E5103は0.4kgスケールのバッチサイズを有し、微粒子形成時間は40分間であった。分散相に関する処方は3つのバッチに関して同様であり、0E5103バッチはわずかに増加した活性剤及びポリマー濃度と溶媒(メタノール)及び酢酸濃度が減少していた。
【表9】
【表10】
【0083】
促進体外(in−vitro)放出速度は、増加された希釈組成物が添加され、そしてそれ故、溶媒接触が減少した0B6315及び0A0307が、希釈組成物が添加されていない0E5103と比較して、より高い初期放出を備える微粒子を製造したことを示した。図8は、バッチ0E5103及び0A0307に関して、1kgあたり22.5mgのロイプロリド投与量を与えたラットにおける、2週間の血清ロイプロリド濃度を比較する。もっとも低い溶媒接触であるバッチ0A0307は、0E5103に比べて、より高い早期放出を有し、血清中でより高いロイプロリド濃度となった。
【0084】
図9は、表9に従って製造したロイプロリド微粒子を投与したヒトにおける、血清ロイプロリド濃度レベル(ng/ml)のグラフ図である。微粒子バッチ0E5103及び0A0307は表9に製造され記載された。表10はヒトの血中(血清)ロイプロリド濃度の臨床データを示す。この試料の結果は、希釈組成物(0A0307と)低DCM接触である処方が、より早い放出の為に、最初の一ヶ月間、血中でより高いロイプロリドレベルを有することが示されている。このバッチは、ヒトの組織中において、GnRHアゴニストとしてより効果的に作用し、テストステロンレベルの長期間の減少を獲得する。同じ投与量で0A0307を投与された患者は、血液中のより低いロイプロリドレベルを有し、
薬剤は試験のより遅い段階(一ヶ月後)の間、より早い速度で放出された。
【0085】
バッチ0E5103と0A0307を比較した臨床試験を実施し、微粒子形成後に添加した希釈組成物の様々なレベルに対して、ロイプロリド微粒子投与後の患者のテストステロンレベルを比較した。理想的には95%の患者が28日間で50ng/dLより低いテストステロンレベルを獲得し維持することが望ましい。表11は、2つのロイプロリド微粒子バッチの臨床試験結果を詳述する。
【表11】
【0086】
バッチ0E5103からの遅い初期放出によって、79%の患者のみが28日までに所望のレベルを獲得した。一方、200%の希釈組成物を有するバッチ0A0307を投与された患者は、28日までに集団の98%が所望のレベルを獲得した。この結果、前立腺がんなどの疾病にあり、より高いテストステロンレベルのために不快症状及び痛みを伴う患者をより効果的に治療可能であり、ロイプロリドのより高い初期放出速度を有する徐放性薬剤の投与によって、より早くかれらの痛みを減じることが可能であり
【0087】
実施例6
実施例6に関するバッチを、固有粘度0.183IVを有するR202Hを用いて約10gスケールで製造した。表12はバッチに関する製造パラメーターを比較する。分散相組成物及び連続相組成物は全てのバッチに関して同じとし、目的とする薬剤搭載量を全てのバッチに関して16.5%L.A.とした。連続相及び分散相に関する流速を同じ又は同程度とした。
【表12】
【0088】
表13は、微粒子バッチの特性を比較する。
【表13】
【0089】
微粒子中の薬剤搭載量はバッチに関して同程度であった。粒子サイズは、バッチ間で、
小さく、典型的であり、変化を示した。平均粒子サイズ(50%体積分布下)は22乃至28μmに及び、90%体積分布は40乃至46μmに及んだ。一般に粒子サイズはバッチ間で大きな違いを示さず、明確な傾向はなかった。
【0090】
PBS中、37℃での薬剤放出は3ヶ月を超える薬剤放出が期待される。最少1ヶ月の初期放出をPBS中で追跡した。希釈組成物を含まないバッチ(GC061404)は、希釈組成物を用いて製造したバッチと比較してより低い初期放出を示した。50,100及び200%の希釈組成物を有するバッチは、同程度のより早い放出速度を示した。
【0091】
促進放出法もまた、希釈組成物で製造したバッチに関して、わずかにより高い初期放出(1時間及び5時間)を示した。様々な希釈物で製造されたバッチ間で、放出に関する違いはない。希釈物による初期放出(1及び5時間)の増加は50%希釈で獲得した。さらなる希釈は目立つほど大きくは初期放出を改善しなかった。
【0092】
実施例7
3つの微粒子バッチを、100,200及び300%希釈組成物で、固有粘度0.206IVのR202Hを用いて製造した。GC071204、GC090904、及びGC091004に関する製造パラメーターを表14に示す。製造パラメーターは、意図的に変化させた希釈組成物を除いて、これらバッチに関して同様のものであった。
【表14】
【0093】
表14で製造したバッチの特性を下記表15に示す。最終微粒子は71乃至75%のカプセル化効率を有し、薬剤搭載量はバッチに関して同程度であった。100及び200%
希釈のバッチは同程度の粒子サイズを示した。わずかに早い放出速度(又は同程度の放出速度)が、より多い希釈物で製造したバッチに関して、より早い時点の間、PBS中で観測された。促進放出が同程度の初期放出を示した。
【表15】
【0094】
実施例8
3つの微粒子バッチを、100、200及び300%希釈組成物にて、0.157IV固有粘度を有するR202Hを用い、そしてL.A.目的投与量19%にて製造した。製造パラメーターを表16に示す。製造パラメーターは、意図的に変化させた希釈組成物以外は、バッチに関して同程度であった。
【表16】
【0095】
表17は上記に製造された微粒子バッチの特性を示す。最終バッチは69乃至73%のカプセル化効率を有する微粒子を有していた。薬剤搭載量はおよそ14%で、バッチに関して同程度であった。微粒子サイズはバッチに関して同程度であった。薬剤放出速度はPBS中で全てのバッチに関して同程度であり、促進放出がわずかに高い初期放出を示した。
【表17】
【0096】
実施例9は様々な溶媒接触のオクトレオチド微粒子を比較する。オクトレオチド含有ポリ(d,l−ラクチド−コ−グリコリド)微粒子を、ロイプロリド微粒子の製造手順と類
似の手法で製造した。PLGA(85:15 PLGA、Alkermes社)、オクトレオチド、塩化メチレン、メタノール及び氷酢酸を含む分散相を、米国特許第5,945,126号明細書に例示されるような修正シルバーソンインラインミキサーの分散相注入チューブを介して、1分あたり約18mLで供給した。分散系におけるオクトレオチドの目標搭載量は10%であった。0.35%ポリビニルアルコール水溶液の連続相を、毎分2Lにて、インラインシルバーソンの連続相注入ポートを介して供給した。シルバーソンの回転速度は5500rpmであった。詳細な製造パラメータを下記表18に示す。製造された約2Lの懸濁液を、その後一定に撹拌しながら、15Lの連続相に接触させた。この15Lの連続相は、それぞれ2000、4000又は6000ppmの塩化メチレンを含んでいた。形成したての微粒子を、洗浄前に、100分間、接触させた。コントロールバッチは溶媒接触をせずに直接洗浄した。
【表18】
【0097】
促進体外(in−vitro)試験を実施し、各バッチの微粒子を比較した。試験は、pH4.0、60℃で酢酸バッファー溶液中で実施した。放出速度をもまた生理学的条件下、37℃で、1日間、バッチに関して算出した。
【表19】
【0098】
表19に示すように、より多い量の溶媒に接触した微粒子は、初期の時点において、放出媒体中でより高い初期放出を示した。インシュリン様成長因子−1(IGF−1)がより早い放出速度に対応してあまりにも急激に減少し得るように、より高い初期バーストは安全性の懸念を引き起こし得る。徐放性微粒子処方におけるオクトレオチドに関するより低い初期放出速度が最適であろう。ロイプロリドとは異なり、オクトレオチド微粒子に関して、減少した溶媒接触の処方を用いることによって、放出速度が減少される。
【0099】
図10は、ラットにおけるオクトレオチドの生体内研究において、表18に従って製造された微粒子の放出特性のグラフ図である。ラットはオクトレオチド微粒子バッチ5mgの投与量を与えられた。GJ030804は連続相中で増加した溶媒に接触しておらず、より高濃度の塩化メチレンで製造されたバッチと比較して、より低い初期バースト放出を示した。より高い溶媒接触がより高いバースト放出速度を引き起こすため、より低いバーストの微粒子は好ましい速度に応じて、水希釈によって製造され得ることが推測される。
【0100】
実施例10
実施例10は、希釈組成物が添加/無添加で、微粒子のバッチサイズが様々であってあるときの、微粒子のロイプロリドの薬剤放出速度の効果を比較する。まずバッチGC071304、0E5103及び0D4022を連続相への希釈組成物の添加なしに、様々なバッチサイズで製造した。表20に示すように、8gバッチであるGC071304を、分子量18kDaのR202H、酢酸ロイプロリド、塩化メチレン及びメタノールを含む分散相29gを用いて製造した。GC071304の連続相は、1.5Lの0.35%ポリビニルアルコール水溶液を含んでいた。分散相に関して30−35g/分、連続相に関して2L/分の流速にて、微粒子形成を50秒以内で完了した。バッチをインラインミキサーにて7000rpmで一定に撹拌し、周囲水中及び39℃水中、およそ10 体積変
化にて洗浄した。
【0101】
バッチ0E5103を上述と同じ製造パラメーターを用い、但しバッチサイズを400gに増加させた。微粒子形成時間は40分間であった。バッチサイズを700gに増加させたバッチ0D4022を上述と同じ製造パラメーターを用いて製造した。微粒子形成時間は70分間であった。後述の2つのバッチに使用した分散相及び連続相の量は、検討されたバッチサイズを製造するために、比例して調整した。
【0102】
下記表20に示すように、3つのバッチを0.206IV(固有粘度)ポリマーを用いて製造した。表21に、バッチの特性を比較する。
【表20】
【0103】
表20で形成された微粒子の特性を表21に示す。
【表21】
【0104】
生理学的条件下で試験された放出速度は、pH7.4、37℃でPBSに対して接触させられた。50秒という最も早い微粒子形成時間と、対応する最も短い溶媒接触時間で製造された8gバッチは、400gバッチ及び700gバッチと比較して、最も早い初期放出を有した。最も長い溶媒接触時間(微粒子形成時間:70分間)の最大の700gバッチは、最も遅い初期放出を有していた。よって、生理学的条件及び促進条件下で、バッチサイズの増加、又はバッチのスケールアップは、全ての他の因子がほぼ同じである、薬剤の放出速度に反転効果を与える。
【0105】
表22に詳述されるように、微粒子を10,100及び1000gのスケールバッチで製造した。100gスケールでは試料の再現性を示すために3つのバッチを製造した。
バッチは、分散相に関して0.183IV(固有粘度)、分子量14kDaを有するR202Hポリマー、ロイプロリド、DCM、MeOH及び氷酢酸を用いて、連続相に関して水とポリビニルアルコールを用いて製造した。また、約1500、±500mlの希釈組成物を連続相に加えた。具体的には希釈組成物、或いは水を、微粒子形成時に溶媒除去容器に対して2乃至4L/分で連続的に添加した。製造パラメーターはバッチに関して同じであり、微粒子製造時間は、10gバッチで1分間、100gバッチで10分間、1000gバッチで100分間であった。100gスケールにて再現性を示すために3つのバッチを製造した。
【表22】
【表23】
【0106】
薬剤カプセル化効率は10、100及び1000gバッチで75乃至79%の範囲の値で同程度である。薬剤搭載量は12.8乃至13.7%と正常変動である。粒子サイズの結果は同程度のサイズ分布データを示す。薬剤放出速度は10,100及び1000gバッチで類似している。10、100及び1000gで製造したバッチは全て非常に低い残存溶媒を示した。よって、連続相に希釈組成物を添加することによって、上記測定された放出プロファイルに関しては、バッチサイズは小さなファクターになる。連続相の溶媒レベルが閾値下に減少すると、希釈組成物の添加なしに製造されたバッチがそうであったように、微粒子の放出特性はバッチサイズに依存しない。
【0107】
実施例11
実施例11は、希釈組成物を用いて製造されたリスペリドン微粒子の放出プロファイルを詳述する。まず、微粒子を生体適合性及び生体分解性ポリマーとして、27.5gのポリ(D,L−ラクチド−コ−グリコリド)、すなわちラクチド:グリコリド比が85:15であり、固有粘度が0.61g/dLであるPLGAを用いて製造した。バッチを27
.5gのリスペリドンを用いて製造した。リスペリドン及びポリマーを220gのジクロロメタンに溶解した。ポリマー、生物学的活性剤及び溶媒のこの混合有機溶液が分散相を構成した。別に、連続相を供給するために、87.5gのポリビニルアルコール及び69gのリン酸二ナトリウム七水和物を25kgの精製水中に溶解した。分散相及び連続相を同時に、それぞれ38.5ml/分及び4000mL/分で、シルバーソンインラインミキサーに注入し、約5分間4000rpmで混合した。
【0108】
分散相を連続相と混合する際、得られた混合物をシルバーソンミキサーにてリスペリドン搭載PLGA微粒子に固化させた。リスペリドン微粒子懸濁液を、希釈組成物を微粒子分散液に室温で添加するところの溶媒除去容器(SRVに)排出し、希釈組成物をおよそ5分間、流速2000mL/分で加えた。SRV中で、室温及び33−37℃の水で微粒子を洗浄し、有機溶媒を除去した。洗浄及び溶媒除去後、微粒子を濾過にて回収し、凍結乾燥した。微粒子は、微粒子1gあたり0.39gのリスペリドンの薬剤搭載量を有すると決定された。PLGA微粒子のリスペリドン搭載効率は77.5%であった。微粒子の放出特性を評価した。pH7バッファー中24時間以内で、1%未満の薬剤の初期放出が検出された。
【0109】
実施例12
実施例12は水希釈なしに微粒子にカプセル化されたリスペリドンの放出プロファイルを詳述する。微粒子を27.5gのポリ(D,L−ラクチド−コ−グリコリド)、すなわち、固有粘度0.61g/dL、ラクチド:グリコリド比=85:15のPLGAを用いて製造した。リスペリドン27.5gを分散相としての役割を果たす有機溶液の一部としてのジクロロメタン220gに溶解した。別に、ポリビニルアルコール87.5g、リン酸二ナトリウム七水和物69gを精製水25kgに溶解することで連続相を調製した。分散相と連続相を、それぞれ38.5ml/分、4000mL/分にて同時にシルバーソンインラインミキサーに注入した。混合懸濁液を4000rpmで混合した。分散相を連続相と混合し、得られた混合物をシルバーソンミキサーでリスペリドン搭載PLGA微粒子に固化させた。リスペリドン微粒子懸濁液を溶媒除去容器(SRV)に排出した。この実施例では希釈組成物を用いなかった。SRV中で、室温及び33−37℃水で微粒子を洗浄し有機溶媒を除去した。洗浄及び溶媒除去工程後、微粒子を濾過にて回収し、凍結乾燥した。微粒子は、微粒子1gあたりリスペリドン0.33gの含量を有すると決定された。PLGA中のリスペリドン搭載効率は66.2%であった。後の放出プロファイル試験で、およそ80−82%のリスペリドンがpH7バッファー中24時間以内で微粒子から放出された。
【表24】
【0110】
希釈組成物の添加なしに製造されたバッチに関して、微粒子からの放出速度(放出率)はリスペリドン80−82%であった。2000ml/分で希釈組成物にて希釈したバッチは、初期投与において放出速度(放出率)が1%未満であった。
【符号の説明】
【0111】
10・・・連続工程
12・・・分散相
14・・・連続相
16・・・インラインミキサー
18・・・希釈組成物
18a・・・希釈組成物
18b・・・希釈組成物
20・・・第一容器
22・・・中空糸フィルタ
22a・・・ライン
【先行技術文献】
【特許文献】
【0112】
【特許文献1】米国特許第4,818,542号明細書
【特許文献2】米国特許第4,767,628号明細書
【特許文献3】米国特許第3,773,919号明細書
【特許文献4】米国特許第3,755,558号明細書
【特許文献5】米国特許第5,407,609号明細書
【特許文献6】米国特許第6,270,802号明細書
【特許文献7】米国特許第6,361,798号明細書
【特許文献8】米国特許第5,945,126号明細書
【技術分野】
【0001】
様々な天然及び合成ポリマー並びに樹脂から形成される微粒子及びミクロスフェア薬剤、診断試薬等の様々な活性剤の一般的な輸送手段となっている。分解可能な徐放性微粒子は、所謂“デポ”製剤における使用のために特に重要であり、長期にわたる活性剤の輸送が求められている。
【背景技術】
【0002】
徐放性微粒子の処方の最適な組成は、特定期間、特定の状態を効果的に治療できる、治療量の生物学的活性剤の有効量を放出する必要がある。一般に、徐放性組成物は、活性剤が微粒子から患者の組織中に放出される速度などの放出プロファイルを有する。微粒子中に封入された特定の活性剤に対する放出速度のコントロールを可能にする形で、徐放性微粒子の放出プロファイルを変更することは、有益であり得る。
【発明の概要】
【課題を解決するための手段】
【0003】
ひとつの実施態様としては、ポリマーに溶解した活性剤で分散相を形成する工程;分散相を連続相と混合して、連続相中で懸濁された微粒子を有する微粒子分散系を形成する工程;そして、微粒子形成後に、微粒子分散系に希釈用組成物の一定量を添加する工程;を含む方法であって、ここで前記希釈用組成物量が、活性剤の放出速度(放出率)を変更するのに十分な量である、徐放性微粒子の形成する方法を包含するものである。
【0004】
本発明の別の実施態様としては、活性剤とポリマーを用いて分散相を形成する工程;分散相と連続相を混合し、連続相中に懸濁された微粒子を有する微粒子分散系を形成する工程;そして、希釈用組成物を微粒子分散系に添加して、未処理の徐放性微粒子の放出速度とは異なる放出速度を有する徐放性微粒子を製造する工程;を含む方法であって、ここで前記希釈用組成物の体積は連続相の体積の少なくとも50%である、徐放性微粒子を形成する方法を包含するものである。
【0005】
さらに別の実施態様としては、ロイプロリドとポリマーを備える分散相を形成する工程を含む徐放性微粒子の放出速度をコントロールする方法を包含するものであって、前記ロイプロリドはポリマー中の溶解されているものである。本方法はさらにミキサー中で分散相を連続相と混合し、微粒子分散系を形成する工程;及び、微粒子形成後に微粒子分散系に希釈用組成物の一定量を添加する工程を含む。希釈用組成物量はまた、特定の活性剤の放出速度を変更させるのに十分であり得る。
【0006】
別の実施態様は、変更された放出速度を有し、特定の活性剤とポリマーを備える分散相を有する徐放性組成物であって、前記分散相は連続相と組み合わせられて微粒子分散系を形成し、微粒子分散系は微粒子形成後に一定量の希釈用組成物に接触させられ、ここで前記希釈用組成物は、特定の薬剤の放出速度を変更させるための連続相を希釈するのに十分であるものである。
【図面の簡単な説明】
【0007】
【図1】図1は、本発明の方法の一態様の略図(配置図)である。
【図2】図2は、本発明の実施態様に従うところの、表3及び表4にて製造され記載された微粒子の、生体内(in vivo)ラット試験における放出特性を示すグラフ図である。
【図3】図3は、本発明の実施態様に従うところの、表3及び表4にて製造され記載されたロイプロリド微粒子の、生体内(in vivo)ラット試験における、放出特性を示すグラフ図である。
【図4】図4は、本発明の実施態様に従うところの、表5及び表6にて製造され記載された微粒子の、放出特性を示すグラフ図である。
【図5】図5は、本発明の実施態様に従うところの、表7及び表8にて製造され記載された微粒子の、放出特性を示すグラフ図である。
【図6】図6は、本発明の実施態様に従うところの、表5及び表6にて製造され記載された微粒子からの、生体外(in−vitro)の促進放出条件下での、酢酸ロイプロリドの放出を示すグラフ図である。
【図7】図7は、本発明の実施態様に従うところの、表7及び表8にて製造され記載された微粒子からの、生体外(in−vitro)の促進放出条件下での、酢酸ロイプロリドの放出速度(放出率)を示すグラフ図である。
【図8】図8は、本発明の実施態様に従うところの、表9及び表10にて製造され記載された2種を与えられたラットにおける、血清ロイプロリド濃度レベルを示すグラフ図である。
【図9】図9は、本発明の実施態様に従うところの、表9及び表10にて製造され記載された、ヒトにおける、血清ロイプロリド濃度レベルを示すグラフ図である。
【図10】図10は、本発明の実施態様に従うところの、表11にて製造され記載された微粒子の、ラットにおける生体内(in vivo)オクトレオチド試験における、放出特性を示すグラフ図である。
【発明を実施するための形態】
【0008】
ここに、ヒトを含む温血哺乳類における、徐放性微粒子に封入された生物学的活性剤の放出プロファイルを最適化する為の方法が開示される。また、形成後の微粒子を、添加した希釈用組成物で、連続・無菌工程の一環として処理することによる、微粒子からの生物学的活性剤の放出速度(放出率)を変化させる方法をも開示する。
【0009】
図1に示すように、一つの実施態様は、徐放性微粒子からの活性剤の初期放出速度を最適化する方法10を含む。この実施態様において、微粒子中の酢酸ロイプロリドの放出プロファイルは、形成したての微粒子が接触させられるところの溶媒の濃度を調整することによって、変更され得る。微粒子の放出プロファイルは、乾燥、加熱、又は水でもどす工程の間、残存溶媒不純物又は凝集問題を取り込み得るところの関連する工程なしに、微粒子が室温で水相にとどまる間に、変更し得る。
【0010】
徐放性製品において、組成物からの初期放出される薬剤量は重要である。初期投与時に放出される薬剤、又は活性剤の量は、しばしば、初期バースト又は初期放出と呼ばれる。初期バースト又は初期放出の量は、特定の薬剤及びそれらの使用目的を検討し最適化するために重要である。
【0011】
例えば、酢酸ロイプロリド、いわゆる“ロイプロリド”は、ゴナドトロピン放出ホルモン(GnRH)の既知のアゴニスト類似物である生物学的な活性剤である。GnRHは、黄体形成ホルモン(LH)及び卵胞刺激ホルモン(FSH)の生産を促進(刺激)する。血流中に放出されると、これらホルモンは、男性においてはテストステロンを、女性においてはエストロゲンの生産を引き起こす。それ故、前立腺がん(prostrate cancer)などの疾患において、生産されるテストストロンのレベルをコントールすることは、効果的な治療であることが知られており、GnRH、LH及びFSHの生産を阻害する活性剤は有益であろう。実際には、生産されるテストストロンのレベルをコントロールすることは、去勢手術の必要性を制限し得る。
【0012】
治療的適応において、ロイプロリドは下垂体前葉からLH及びFSHを初期に放出する
。しかしながら、連続使用すると、ロイプロリドは下垂体脱感作、そしてホルモン生産テストステロンの発現低下を引き起こす。ロイプロリドは治療で使用されると、初期上昇の後、2乃至4週間の間、血中性ホルモンレベルを抑制する。よって、ロイプロリドが有効となるために、輸送微粒子からの薬剤の非常に高い初期放出速度が望ましい。米国食品医薬品局によれば、より早い去勢を獲得するために、ロイプロリドに対する高い初期放出が求められる。ロイプロリドを用いた化学的去勢は、95%の患者が28日以内に50ng/dL(0.5ng/mL)以下のテストステロンレベルを保有するもの、と定義される。
【0013】
一方、別の活性剤であるオクトレオチドは、肢端(先端)肥大症の長期間の治療、並びに、悪性カルチノイド腫瘍、血管作動性腸管ペプチド腫瘍、フラッシング及び下痢の患者に使用されるソフトスタチン誘導体である。血清中のオクトレオチドは成長ホルモン(GH)、並びにインシュリン様成長因子−1(IGF−1)をコントロールする。長期間の徐放性デポ製剤におけるオクトレオチドの使用の一般的な不利益は、高いピークレベルのオクトレオチドが、ある血漿濃度で有毒となり得ることである。より遅い初期バーストまたは放出速度が、この状況においては好ましい。
【0014】
図1に示すように、ロイプロリド−含有微粒子製造のために連続工程10が微粒子を製造するために適用される。連続工程10は一般に分散相12と連続相14の混合を含む。分散相12は、活性剤、ポリマー及び適当な溶媒を含む。連続相は、一般に、水溶液を含む。分散相12及び連続相14は、インラインミキサー16中で混合され、微粒子分散系を形成する。微粒子分散系は、一般に、水溶液中に分散された形成したての微粒子を含み得る。
【0015】
本出願の目的において、前立腺がんの治療のための徐放性微粒子の形態にあるロイプロリド製剤は、ポリマー状徐放性微粒子から放出される活性剤の一例として使用される。活性剤は小さなポリマー体へのカプセル化又は散在化が望まれている任意の薬剤にすることができることは、本開示の観点から当業者にとって明らかである。さらに、臨床的効果を改善するために放出速度が最適化され得る活性剤の例としては、ペプチド、ケトチフェン、チオリダジン、オランザピン、リスペリドン、オキシブチニン、オクトレオチド、ナルトレキソン、オルニチド、又は、Woc4D、ベータメゾン(betaqmethasone)、デキサメタゾン、クロニジン、ベラパミル、又は、薬学的に可能なそれらの塩が挙げられる。特に興味深いのは、ロイプロリド、トリプトレリン、ゴセレリン、ナファレリン、ヒストレリン、及びブセレリンなどの黄体形成ホルモン放出ホルモンアゴニスト(LH−RHアゴニスト)である。また特に興味深いものは、ソマトスタチン類似体であって、例えば、オクトレオチド、ヒト、鮭及びウナギのカルシトニン、成長ホルモン、成長ホルモン放出ホルモン、成長ホルモン放出ペプチド、副甲状腺ホルモン及び関連するペプチド、インターフェロン、エリスロポエチン、GM−SSF、G−CSF、サイモシン、アンチトリプシン及び化学療法薬、局所投与の為の抗生物質及び鎮痛剤、並びに、放出速度の調整が徐放製剤の効果を増加し得る他の薬剤、である。
【0016】
活性剤を分散相12に組み込むために、少なくとも1つの溶媒に活性剤を溶解することが通常必要である。活性剤のための溶媒は、薬剤の性質に応じて勿論様々であり得る。活性剤を溶解するための分散相に使用され得る典型的な溶媒としては、但しこれらに限定されないが、水、メタノール、エタノール、ジメチルスルホキシド(DMSO)、ジメチルホルムアミド、ジメチルアセトアミド、ジオキサン、テトラヒドロフラン(THF)、ジクロロメタン(DCM)、塩化エチレン、四塩化炭素、クロロホルム、ジエチルエーテル及びメチルエチルエーテルなどの低級アルキルエステル、ヘキサン、シクロヘキサン、ベンゼン、アセトン、酢酸エチル、メチルエチルケトン、酢酸、又はそれらの混合物が挙げられる。加えて、氷酢酸、乳酸等の酸、脂肪酸、又はアクリル酸が、ポリマー中への活性
剤の溶解とカプセル化を改善するのを促進するプロセスで使用される。与えられた系のための好適な溶媒の選択は、本開示を考慮して技術常識の範囲内事項である。
【0017】
活性剤は、続いて、ポリマーと混合され、分散相12を形成する。当業者に既知であり、一実施態様において有用である、ポリマーの例は、例えば米国特許第4,818,542号明細書、米国特許第4,767,628号明細書、米国特許第3,773,919号明細書、米国特許第3,755,558号明細書、米国特許第5,407,609号明細書に見出され、参照することにより本明細書に組み込まれる。与えられた系における特に好ましいポリマーの選択において、生分解性(例えば放出プロファイル)及び生体適合性といった所望の臨床的特徴を有する生成物を生産する目的のために、多数の因子が考慮される。一旦、ある当業者が、所望の臨床的特徴を提供するポリマー群を選択すると、続いて、該ポリマーは、製造工程を最適化することとなる望ましい特徴が評価される。例えば、ある場合には、微粒子の処理を促進する、薬物搭載量を増進する、分散相からの溶媒の除去を強化する、或いは、分散相から連続相への薬物の移動を抑制するという形で、活性剤と相互に作用するポリマーを選択することが可能である。
【0018】
好適なポリマーの具体例は、乳酸のホモポリマー又は乳酸とグリコール酸のコポリマー、又は、ポリ(ラクチド−コ−グリコリド)、“PLGA”ポリマーである。乳酸残基とグリコール酸残基の比率は様々であり、典型的には25:75乃至85:15の範囲であり、但し、高いラクチド含量は低い粘性及び高い溶解性につながるため、10%のグリコリドであっても使用される。好ましいコポリマーは、少なくとも約50%の乳酸残基を含み、例えば50:50、75:25、又は85:15ポリマーである。ポリ(ラクチド−コ−グリコリド)コポリマーは、多くの供給源から商業的に入手可能であり、そして、従来の合成ルートによって容易に製造される。ベーリンガー インゲルハイム社は、R202H、RG502、RG502H、RG503、RG503H、RG752、RG752H、RG756及びその他の称号にて好適なポリマーを製造している。R202H、RG752H又はRG503HとのLH−RH微粒子は、ある実施態様の分散相において使用され得る。与えられた系に対する好適なポリマーの選択は、本開示を考慮して当業者であれば明らかである。
【0019】
好ましいポリマーの選択における一つの検討事項は、ポリマーの親水性/疎水性である。ポリマーと活性剤の双方が、疎水性又は親水性であり得る。可能であれば、親水性の活性剤と共に使用するために親水性ポリマーを選択すること、そして、疎水性活性剤とともに使用するために疎水性ポリマーを選択することが望ましい。好ましい微粒子において、薬剤とポリマーの親水性カルボキシル基との間のイオン相互作用は、薬物搭載量を増進するとみられている。しかしながら、一般に、親水性薬剤は水に溶解するものの、ポリマーと薬剤の間に親和性がない場合、又は、固化が十分に早くない場合、薬剤搭載量は減少し得る。疎水性ポリマーにおける親水性薬剤の使用もまた可能である。
【0020】
特定のポリマーの選択において、系の残留溶媒へのポリマーの疎水性/親水性の影響もまた考慮する必要がある。親水性ポリマーは、親水性ペプチドといった親水性薬剤では、少ない残留溶媒を生じることが期待される。ロイプロリド微粒子の場合、薬剤は、分散相の液滴から疎水性溶媒を迅速にそして効率的に排除するのを補助する傾向を有する。加えて、より多量の薬剤搭載量は、より低い残留溶媒濃度と相互に関係がある傾向にあることが見出されている。よって、いくつかの系においては、親水性ポリマー内に親水性薬剤が内包されるとき、より少ない残留溶媒による間接的な利点が存在する。しかしながら、残留溶媒における親水性以外の他の影響因子が存在する場合、この効果は、非ペプチド薬剤に対して一様には適用できない。それにも関わらず、分散相の液滴からの溶媒の排除を増進する活性剤が、付随する薬剤損失なしに、優れた生産物を生産することを、理解すべきである。
【0021】
別の検討事項は、ポリマーの分子量である。ポリマーの分子量は、放出速度、放出プロファイルなどの生成物の特徴に対して明らかに影響を及ぼすと同時に、微粒子の製造プロセスにも影響を与える。より高分子量のポリマーは、概してより粘性の高い分散相と関連し、大きな粒子をもたらすか、或いは、小さな粒子を得ることの難しさの増加をもたらし、そして、いくつかの例において、残留溶媒の増加をもたらす。一方、より低分子量のポリマーは、ポリマーがより溶解性である傾向のために、概してより遅い固化と関連する。好ましい系において、より多い残留溶媒、より高い薬剤搭載量及び増進された取り込み効率は、より高分子量ポリマーの使用に由来することが見出されている。本発明の工程のひとつの有利点は、優れた、小さい、少ない残留溶媒である微粒子を、高分子量ポリマーとともに形成し、そしてそれゆえに粘性の分散相を形成することとなる、その能力である。勿論、特定の選択はまた、所望の生成物の特徴に依存する。例えば、より高分子量で、体内でのより長い分解時間、そしてより長い薬剤放出持続時間である。
【0022】
さらに、適用される特定のポリマー濃度は、生成物の形態の観点だけでなく、製造の観点から、系に影響を与える。粘性の分散相は、固化のためのより少ない溶媒を除去する必要があるため、ポリマー濃度の増加は、より多い薬剤搭載量と関連する傾向がある。増加された固化速度は、より高い薬剤保有を引き起こす傾向がある。その上、粘性の分散相は、固化の間に、連続相へのより少ない薬剤拡散をもたらす。いくつかの系において、これはまたより多い残留溶媒をもたらす。好ましい実施態様において、分散相中のポリマー濃度は約5乃至40%であり、そしてより好ましくは約8乃至約30%である。
【0023】
ポリマーのための溶媒は、ポリマーの性質、活性剤、毒性、系中の他の溶媒との相溶性、そしてさらに微粒子が適用される用途などの、多くの因子に応じて適切に選択される。よって、ポリマーの溶解に加えて、溶媒は、液滴を形成するために連続相と混和しないこと、最適な蒸発効率のための高い揮発性、そして望ましくは安全性の理由から不燃性でなければならない。好ましいポリ(乳酸)又はポリ(ラクチド−コ−グリコリド)ポリマーのための好適な溶媒としては、塩化メチレン、クロロホルム、酢酸エチル、置換されたピロリドンなどが挙げられる。場合によっては活性剤のための溶媒は、ポリマーのための溶媒と同じである。一部の薬剤、概して画像解析に使用される放射性無機塩などの診断薬は、有機溶媒に溶解しないか、ほんの少ししか溶解しない。これらの例において、微細なサブ−サブミクロンサイズの粉体が、ポリマー溶液に直接懸濁して、微粒子を形成する。薬物送達において、これを用いることはまれではあるが、診断薬では役立つことが証明されている。本発明のプロセスに従う他の有用な溶媒の選択は、本開示を考慮して、当技術範囲内にある。
【0024】
この実施態様において、ロイプロリド含有ポリ(乳酸)又はポリ(ラクチド−コ−グリコリド)微粒子は、R202Hポリマーに、塩化メチレン、メタノール及び氷酢酸とともにロイプロリド活性剤を溶解して、分散相12を形成することにより、製造される。
分散相は、実に、均質な溶液である。或いは、それぞれ個々の溶媒中にポリマーと活性剤の別々の溶液を準備し、そして続いて混合して分散相12を形成する。場合によっては、活性剤及び/又はポリマーの性質の理由から、分散相12は、エマルションとして形成せざるを得ない。例えば、所定のタンパク質性の薬剤が好適な活性剤の溶媒中に溶解された場合、得られた溶液は、特定のポリマー溶媒中のポリマー溶液とは、完全に混和しない。薬剤とポリマーが比較的均一に散在しているところの、比較的均質である分散相12を提供するために、薬剤及び薬剤溶媒は、ポリマー及びポリマー溶媒と乳化し、分散相エマルションを形成し得る。分散相12の連続相14への導入において、水−油−水、又はW/O/Wエマルションが形成され得る。さらに別の系において、分散相12は、ポリマー溶液中の活性剤の直接的な懸濁液の形成によって製造される。
【0025】
下記に記載される本発明のプロセスに応じて、連続相14中に分散された分散相12の液滴又は内包物を有する微粒子分散系を形成するために、分散相12は連続相14と混合され得る。ここで使用する用語“分散された(dispersed)”とは、広義において、連続相中に散在する分散相の不連続部位を意味するものと意図される。上記内包物は、概して、通常球状の液滴として生じるが、場合によっては、特定の乳化条件に応じて、不規則な内包物と成り得る。分散相12が液滴又は内包物を形成するところの任意の適当な媒体が、特に望ましいとされる分散相12溶媒のための最大溶媒シンクを提供するものと共に、連続相14として使用され得る。
【0026】
しばしば、連続相14は、乳化工程を変更させ影響を与えるところの界面活性剤、安定剤、塩又はその他の添加剤をも含む。典型的な界面活性剤は、ドデシルスルホン酸ナトリウム、ジオクチルスルホコハク酸ナトリウム、スパン、ポリソルベート80、トウィーン80、プルロニクスなどが挙げられる。特定の安定剤としては、タルク、PVA及びコロイダル水酸化マグネシウムが挙げられる。粘度ブースターとしては、ポリアクリルアミド、カルボキシメチルセルロース、ヒドロキシメチルセルロース、メチルセルロースなどが挙げられる。緩衝塩は薬剤安定剤として使用され、一般的な塩でさえも活性剤の連続相14への移動の防止を促進するために使用される。連続相14の塩の飽和に関連する一つの問題は、PVA及びその他の安定剤が、連続相から固体物として沈殿する傾向を有し得ることである。そのような場合、微粒子安定剤が使用され得る。塩化ナトリウムや硫酸ナトリウムなどの適当な塩並びに他の添加剤は、本開示を考慮すると当業者に明白である。
【0027】
ひとつの実施態様において、連続相14は、100−50%の水を含む。水性の連続相14は、安定剤を含み得る。好ましい安定剤は、約0.1%乃至約5.0%量のポリビニルアルコール(PVA)である。より具体的には、PVAは約0.35%量で存在し得る。連続相14への使用のための適当な他の安定剤は、本開示を考慮すると、当業者に明白である。
【0028】
特定のポリマー、溶媒及び連続相の選択は、無論、活性剤並びに所望の生成物の特徴に応じて様々である。一旦、臨床的適用、放出プロファイルなどの所望の生成物の特徴が確立されたとしても、それでもなお、製造プロセスを促進するためのポリマー、溶媒及び連続相の選択には多少の自由度が残されている。
【0029】
分散相12及び連続相14が混合されるとすぐに、形成したての微粒子が、ミキサー16から第一容器20へ連続的に移動させられ得る。この実施態様において、微粒子は一般に十分に固体且つ濾過可能であり、それ故、米国特許第6,270,802号明細書及び米国特許第6,361,798号明細書に開示されるように、中空糸フィルタを使用する微粒子の連続処理に適する。この方法は、分散相と連続相の混合物が、別々の微粒子をすぐに形成しないような、一般的な微粒子形成方法とは区別される。
【0030】
微粒子がさらなる形成手順を必要とする方法とは対象的に、この実施態様の微粒子は約20秒以内に、より好ましくは約10秒未満で、そしてより特に好ましくは約5秒未満で固化する。ロイプロリド−ポリマーの実施態様において、微粒子は、約3秒未満で固化する。本実施態様において、微粒子の固化は連続相への添加時に事実上一瞬であるにしても、形成された微粒子は、それにもかかわらず、更なる残留溶媒を連続相へ引き渡すことに影響を与えやすい。
【0031】
薬剤搭載量は、好ましいロイプロリド/ポリマー微粒子の場合、総固形物をベースとして約12%を目標とし、10乃至21%の範囲である。実際には、約9乃至17、約17%の薬剤搭載量が得られる。勿論、薬剤の性質、所望の放出プロファイル、ポリマーの性質、そして勿論処理方法全てが、所望のそして実際の薬剤搭載量に影響する。典型的なケ
ースにおいて、薬剤とポリマーの混合量をベースとして約5%乃至20%の薬剤搭載量が望ましく、本発明のプロセスによって達成可能なものである。典型的なカプセル化効率は約75%である。
【0032】
有利なことに、一旦所望の薬剤搭載量を得ると、供給量、温度などのパラメーターが算出され、製造レベルのバッチ数を含むより大きなバッチ数のスケールアップは、単により長いプロセスを実施すれば良いだけのこととなる。所望の特徴を有する微粒子を大量に生産するのに、さらなる供給チューブ(管)、乳化剤、羽根車などのものを必要としない。加えて、本発明の連続プロセスの間に製造される微粒子は、プロセスの間にいつ製造されたかにかかわらず、サイズ分布、薬剤搭載量などが並外れて均一である。
【0033】
ひとつの実施態様において、上述したように、微粒子形成の一般的プロセスは、微粒子の溶媒接触を変化させることによって、微粒子バッチの放出特性又は放出プロファイルが変化するように修正され得ることが見出されている。形成後のより低いレベルの溶媒接触は、形成したての微粒子に対して希釈組成物18を加えることによって達成される。希釈組成物18は、水又はポリビニルアルコール溶液であり得る。微粒子形成後の一定量の希釈組成物18の添加は、驚くべきことに、形成したての微粒子の放出速度を変化させることが見出されている。
【0034】
図1に示すように、微粒子が形成され、連続相に分散された後、希釈組成物18が添加され得る。図1中、18aで示される希釈組成物は、第一容器20に移送される時に、微粒子分散系に添加され得る。第一容器20に移送される際の希釈組成物の微粒子分散系への添加は、第一容器20に到達し、中空糸フィルタ22を用いて濾過される前に、微粒子分散系と希釈組成物を完全に混合させることを可能にする。
【0035】
代わりに、18bに示すように、希釈組成物18は、微粒子分散系の前に、又は、分散系と同時に、第一容器20に移送され得る。一旦、第一容器に入ると、微粒子と希釈組成物の混合物は、連続的に撹拌され得る。
【0036】
微粒子分散系に添加される希釈組成物18の量は、哺乳類に注射するために、最終製剤の放出プロファイルを調整するのに十分であるべきである。希釈組成物18は、微粒子中にカプセル化された活性剤の溶解性、微粒子の粒子サイズ、或いは各バッチにおける活性剤のカプセル化効率を逆向きに或いは大幅に変更すべきではない。添加される希釈組成物18の所定量に対応した、所定の溶媒接触レベルは、所望の放出速度に対して計算される。その結果、特定の放出速度が希釈組成物18の添加によって得られる。
【0037】
ひとつの実施態様において、所望の放出プロファイルを備えるロイプロリド微粒子を得るために、連続相中の溶媒の規定濃度が5000ppm未満になるまで、希釈組成物18を加える。規定量及び時間の希釈組成物18の添加によって、放出プロファイルは、予想通りのそして制御可能な方法で、連続プロセスの一環として調整される。
【0038】
形成された微粒子に対して添加する希釈組成物量は、連続相14の元の体積の少なくとも約50%であり得る。ひとつの実施態様において、水溶液中で溶媒への接触の減少は、形成されたロイプロリド微粒子の放出速度を増加させ得る。異なる活性剤の放出速度の制御は、しかしながら、所望の放出プロファイルを獲得するために異なるステップを必要とし得る。本プロセスは、異なる放出プロファイルを備える異なる活性剤の使用に適合するように、容易に調節可能である。
【0039】
ミキサー16からの微粒子分散系と希釈組成物18を、第一容器20で混合し、微粒子/希釈組成物混合物を、中空糸フィルタ22にて濾過する。廃棄物を、ライン22aを介
して、分離された容器に廃棄すると同時に、微粒子を第一容器20に戻す。微粒子が容器20に戻されると、それらは新たな微粒子/希釈組成物混合物と連続的に混合され、フィルタを通して循環させられる。
【0040】
必須組成物及び所定時間で処理された後、所望の放出速度を獲得した微粒子を、その後、幾らか残存する水溶液を除去するために濾過し、そして、プロセス副生成物を除去するために水で洗浄し得る。微粒子が形成されると、患者投与のためにそれらを準備するところの充填容器に対して、微粒子を準備するためになされ得る、多くの最終的な調整があることは、当業者に既に明らかである。
【0041】
本発明による方法は、添付の図面を参照すると共に、以下の限定されない実施例によって例示される。
【実施例】
【0042】
以下の実施例は、ここに主張する方法を実施し評価する方法を当業者に対して詳細な記載と共に説明するものであるが、発明者が自身の発明とみなしているところの範囲を制限することを意図するものではない。特に指定の無い限り、部は質量で、温度は℃である。
【0043】
分子量は、重量平均分子量(Mw)又は数平均分子量(Mn)で示され、そして、ポリスチレン(PC)を分子量標準に用いたゲル浸透クロマトグラフィー(GPC)を用いて測定される。実施例1は、本方法の一態様を示す概略図を提供する。ポリ(d,l−ラクチド)、R202Hは、ベーリンガーインゲルハイム社から市販されている。
【0044】
実施例1
酢酸ロイプロリド微粒子を、下記表1に記載されるように製造した。分散相及び連続相を混合し、約7Lの微粒子分散系を形成することにより、微粒子を形成した。概して、分散相は表1中に記載されたパラメーターによれば、酢酸ロイプロリド(L.A.)、ポリ(d,l−ラクチド)(R202H)及び溶媒 塩化クロリド(DCM)、メタノール(MeOH)及び氷酢酸(A.Acid)の溶液であリ得る。R202Hは、温血動物及びヒトにおける生体内(in−vivo)研究に基づき、3乃至4ヶ月間、ロイプロリドの徐放性を提供する。連続相は、0.0035g/gポリビニルアルコール溶液を用いて製造した。
【0045】
連続相及び分散相をインライン シルバーソン(Silverson)ミキサーに、それぞれ流速2L/分及び38g/分にて同時に添加した。ミキサー中の混合速度は7000rpmであり、微粒子形成期間は3.5分であった。この微粒子分散系を、約40Lの希釈相を含む容器に連続的に供給した。
【0046】
希釈相もまた0.0035g/gポリビニルアルコール水溶液を含み得、但し希釈相は表1に示す溶媒が混合されていた。希釈相中の塩化メチレン量は、各バッチで様々であり、具体的には、微粒子バッチ:GG090303に対して約5000ppmから、微粒子バッチ:GG091003に対して約8500ppmであった。反応容器において、希釈相入りの溶媒との溶媒接触に関する対応時間は、全ての4つ微粒子バッチに対して100分間で一定であった。
【0047】
溶媒接触後、希釈相に懸濁している微粒子を溶媒除去容器(SRV)に移送した。全ての微粒子バッチを、40分間、2 体積変化にて周囲水洗浄、80SLPMのエアスウィ
ープ;80SLPMのエアスウィープしながら、100分間、5 体積変化にて温水洗浄
(39℃)を用いて洗浄し;そして80SLPMでエアスウィープしながら、30分間,1.5 体積交換にて冷却した。洗浄プロセスの間、ヘッドスペース溶媒除去を助けるた
めにエアスウィープを導入した。微粒子をメンブレンフィルタで濾過し、真空下で凍結乾燥した。
【0048】
表1は、活性剤、ポリマー及び分散相の製造に用いた溶媒の濃度、並びに、希釈相の調製に用いた各溶媒の濃度及び実際の量を示す。
【表1】
【0049】
表2は、粒子サイズ、促進放出条件下での薬剤放出、並びに、生理学的な条件下での薬剤放出などの、表1の製造パラメーターにて製造した微粒子の特徴を示す。微粒子の薬剤搭載量は、微粒子中の活性剤の割合として記述される。これは、溶媒(例えばジメチルスルホキシド)中に微粒子を溶解し、微粒子溶液にホスフェートバッファーを添加することによって決定された。濾過された溶液は、その後、キャリブレーション基準を対照としてHPLCによって分析された。
【0050】
粒子サイズ分布(Part.Size Dist.)は、界面活性剤又は希釈剤を含有する水中で、微粒子懸濁液に関して決定された。これは、レーザー光散乱で決定された。粒子サイズ分布を体積分布(CVF)で示した。
【0051】
生理学的条件下(D.R.(Phys.))でのロイプロリドの放出速度を決定するために、薬剤放出速度の算出をリン酸バッファー溶液(PBS)中で実施した。表1に示すように処方された微粒子を、温度37℃で、スクリューキャップの付いたガラスチューブ中、PBSに懸濁させた。少なくとも80%の放出媒体が、標本点において、新たな媒体と置換された。放出媒体についてHPLCを用いて薬剤濃度の測定を行い、放出率を算出した。
【0052】
放出条件を適切に操作し、生体内で獲得される放出速度、或いはPBSへの放出による放出速度を促進させた。ロイプロリド微粒子に対して、使用される放出媒体は、0.1Mクエン酸リン酸塩バッファーであり、温度は55℃であった。170回転/分にて撹拌された、撹拌された水バッチ中で放出を実施した。試験品について各時間ポイントで放出された薬剤の測定を行い、促進条件下(D.R.(Acc.))の放出速度を見出した。
【表2】
【0053】
上述の製造ステップ及び表1に従い形成された微粒子において、同じような粒子サイズ分布及び薬剤搭載量がみられた。しかしながら塩化メチレン溶媒の最も高い接触レベルに接触させられた微粒子バッチは、若干減少した薬剤搭載量を示した。
【0054】
より意味があることは、ロイプロリドの薬剤放出速度は、促進薬剤放出試験(55℃クエン酸リン酸塩バッファー)並びに生理学的条件(リン酸塩バッファー溶液、pH7.4、37℃)下の薬剤放出の双方とも、バッチによって様々であった。促進試験条件下での薬剤放出は、バッチによって放出速度の違いに関して素早い比較を提供した。生理学的条件下での薬剤放出を、臨床適用における徐放性製剤の実際の持続期間をシミュレートするために、最大60日以上のより長い試験期間の間、実施した。最も高いレベルの溶媒(8500ppm)に接触したバッチGG091003の微粒子は、一貫して最も遅い放出速度を有していた。とくに、放出速度の違いは、初期の時点の間でより大きなものであった。一般に、酢酸ロイプロリドで形成され、より高い溶媒濃度に接触させられた微粒子バッチは、減少した放出速度を示した。よって、ロイプロリドを含有する微粒子の放出速度を増加させるためには、微粒子形成後に、溶媒接触を減少させることが望ましい。
【0055】
実施例2
酢酸ロイプロリド微粒子を、下記表3に記載される通りに製造した。微粒子分散相バッチを、酢酸ロイプロリド(L.A.)、塩化メチレン、メタノール及び氷酢酸からなる一貫した組成物を用いて調製した。2つの微粒子バッチ、0C3542及び0H8340は、14kDaの分子量を有するR202Hのポリマーロットを用いて、懸濁液中で、それぞれ0.7kg、0.4kgの固体微粒子をもって製造した。3つの微粒子バッチ、0D4022、0E5103、及び0S1723を、18kDaの分子量を有するR202Hのポリマーロットを用いて、懸濁液中で、それぞれ0.1kg、0.4kg、0.4kgをの固体微粒子をもって製造した。各バッチを、同様の連続相処方及び同様の製造パラメーターを用いて製造した。但し、微粒子形成時間は、バッチサイズを変化させることによって、即ち懸濁液中の微粒子が0.4kgと対照的に0.7kg、並びに、それぞれのバッチの懸濁液を反応容器への分散相の流速を変化させることによって連続相を含む溶媒と接触させる時間を変化させることによって、様々であった。
【0056】
具体的には、微粒子は、それぞれの分散相を、0.35%のポリビニルアルコール水溶液の連続相に添加することによる水中油型プロセスによって形成した。インラインシルバーソンミキサーの混合速度は7000rpmであった。微粒子が形成するまで、分散系の連続相の溶媒濃度は、およそ塩化メチレン 7200ppmとメタノール 1600ppmであった。
【0057】
インラインミキサー中で形成後に微粒子バッチを連続的に溶媒除去容器に移した。微粒子形成時間は、0.4Kgバッチで約40分であり、一方、微粒子形成時間は、0.7kgバッチで約70分であった。微粒子形成の20分後、分散相を連続相と、所定の流速で混合し、溶媒除去容器は40Lの懸濁液量に達した。溶媒除去容器中の懸濁液を、その後、中空糸メンブレンフィルタ(閉鎖系)を通して再循環させ、溶媒除去容器への懸濁液供給と同じ速度(2L/分)で透過物を除去した。よって連続相中の溶媒レベルは、0.4kgバッチで40分間、0.7kgバッチで70分間で一定に維持された。
【0058】
微粒子バッチを、40分間、2 体積変化で周囲水洗浄、80SLPMのエアスウィー
プ;80SLPMのエアスウィープしながら、100分間、5 体積変化で温水洗浄(3
9℃)を用いて洗浄し;そして80SLPMのエアスウィープしながら、30分間、1.5 体積交換で冷却した。洗浄プロセスの間、ヘッドスペース溶媒除去を助けるためにエ
アスウィープを導入した。
【表3】
【0059】
表3の形成されたバッチの微粒子特性を下記表4に示す。
【表4】
【0060】
微粒子サイズはバッチに関して同程度であった。実施例1に関して上述に記載されたように、薬剤放出特性を生理学的条件及び促進条件にて試験した。結果は、より長い溶媒接触期間、即ち、溶媒接触を増加させた微粒子OC3542及びOD4022に関して減少した初期放出を示した。バッチOC3542から、すなわち、低分子量ポリマー(Mw=14kDa)から製造された微粒子は、生理学的条件下及び促進条件下のいずれにおいても、殆どの区間にわたって、最も遅い放出速度を有した。よって、高い溶媒接触において、ポリマーの分子量が小さくなると、ロイプロリドの初期放出が低くなった。
【0061】
表3及び表4に従って製造された微粒子を、ラットの体重1kgあたり酢酸ロイプロリド9mgの懸濁液として注射した。図2は、バッチ0H8340(40分溶媒接触、14kDa)とバッチ0C3542(70分溶媒接触、14kDa)の同様の投与量を受けたラットにおける、血清ロイプロリドを比較する。両物質とも、分子量14kDaを有するポリマーで製造されているものの、0C3542は、0.7kgのバッチサイズで、70分間の溶媒接触にて製造され、一方、0H8340は0.4kgのバッチサイズで40分間の溶媒接触で製造されている。この研究は、0C3542が微粒子からのより遅い初期の薬剤放出のために、血清中で、始めはより低い薬剤レベルを生ずることを示している。約三週間後の第二局面は同程度の薬剤レベルを生じさせた。これは、より長い溶媒接触を受け、より大きなバッチサイズを有するバッチ0C3542からの微粒子のより低い初期放出を確認するものである。
【0062】
図3は、バッチ0D4022(70分溶媒接触、18kDa)とバッチ0E5103(40分溶媒接触、18kDa)の同様の投与量を受けたラットにおける血清ロイプロリドを比較する。両物質とも、分子量18kDaを有するポリマーで製造されているものの、0D4022は0.7kgのバッチサイズで70分間の溶媒接触にて製造され、一方、0E5103は0.4kgのバッチサイズで40分間の溶媒接触で製造されている。再度、より長い溶媒接触期間を有するバッチである0D4022は、血清中で始めはより少ない薬剤レベルを生じ、接触の初期段階でより遅い放出を確認した。
【0063】
実施例3
ロイプロリド微粒子を、固有粘度0.206dl/g、分子量18kDaを有するポリマーで製造した。微粒子バッチを、10gスケールにて、連続処理手順を用いて製造した。分散相組成物、連続相組成物、分散相流速、連続相流速、及び1分あたりのミキサー回転数などの製造パラメーターを、全てのバッチで等しくなるように保った。
【0064】
具体的には、水中油型プロセスを用いて、それぞれの分散相と連続相を混合することにより、微粒子を形成した。連続相は、0.35の%ポリビニルアルコール水溶液から構成されていた。インラインシルバーソンミキサーの混合速度は7000rpmであった。
【0065】
微粒子分散系をミキサーから溶媒除去容器に移送した。各バッチにおいて、希釈組成物量を溶媒除去容器に添加した。各バッチにおける希釈組成物は水であった。分散系をインラインミキサーから溶媒除去容器へ移動させるのと同時に、希釈組成物を溶媒除去容器に添加した。具体的には、バッチGC071304を増加される希釈組成物なしに製造し、バッチGC060404は連続相の体積の50%に相当する希釈組成物にて製造した。バッチGC060404を、シルバーソンミキサーから、約2Kg/分の速度で移送し、懸濁液を室温水(22乃至25℃)で、約1kg/分の同時フローにて、希釈した。
【0066】
バッチGC060304に関して添加された希釈組成物は連続相の体積の約100に%相当し、ここでバッチを2kg/分の速度でミキサーから移送し、希釈組成物を同時に2kg/分の速度で添加した。バッチGC061104に関して2kg/分の懸濁液フローを使用しながら希釈組成物を4kg/分の速度で添加し、懸濁液の連続相の体積を約200%増加させた。混合速度は、全ての試験において、7000rpmで一定とした。
【0067】
最後に、微粒子バッチを、40分間、2 体積変化で周囲水洗浄にて洗浄、80SLP
Mのエアスウィープ;80SLPMのエアスウィープしながら、100分間、5 体積変
化で温水洗浄(39℃)を用いて洗浄し;そして80SLPMのエアスウィープしながら、30分間、1.5 体積交換で冷却した。洗浄工程の間、ヘッドスペース溶媒除去を助
けるためにエアスウィープを導入した。微粒子をメンブレンフィルタで濾過し、真空下で凍結乾燥した。
【表5】
【0068】
表5で製造した微粒子の特性を表6に示す。
【表6】
【0069】
実施例1に関して上述されるように、薬剤放出粗特性を生理学的条件下並びに促進条件下で試験した。異なる組成物から製造した微粒子の薬剤カプセル化及び粒子サイズ特性は
同程度であった。ロイプロリドフリーベースのカプセル化効率は、80乃至82%の範囲であった。結果、薬剤カプセル化は、連続相部分に添加する量を0から200%の範囲に増加させた希釈組成物を有する全てのバッチにおいて類似していることを示した。平均粒子サイズ(50%体積分布下)は、25乃至29μmにおよび、90%体積分布では47乃至50μmにおよんだ。一般に、薬剤カプセル化と粒子サイズ分布は、バッチ間で類似している。
【0070】
生理学的条件下で測定された薬剤放出速度をリアルタイム薬剤放出速度(長期間)の指標として測定した。希釈組成物が増加し、そして連続相がさらに希釈されるにつれて、結果はより早い放出速度を示した。促進放出速度試験条件も連続相の希釈組成物が増加するにつれてわずかに高い初期放出速度を示した。例えば、連続相部分に、最も高い希釈組成物量と、最も低い溶媒濃度を有するバッチGC061104は、全ての時間間隔にて、両放出条件下で最も早い放出を示した。バッチGC071304は希釈組成物の増加をしていない。最も高い溶媒濃度を有する試料として、この試料は3日及びそれ以下の時間間隔に関して、生理学的条件下で、最も遅い放出速度を示した。従って微粒子形成後の溶媒接触の減少は、微粒子からのロイプロリドの放出速度を増加させる。
【0071】
図4は、表5及び6に製造され記載されたバッチGC071304、GC060304、及びGC061104を含む、37℃のリン酸バッファー溶液中のロイプロリド微粒子の放出特性を示すグラフ図である。図4は、異なる希釈組成物レベルにてバッチが製造されたとき、PBS中で37℃で放出されたロイプロリドの割合を表す3つのS−型カーブを示す。全3つのカーブの間に狭い分離が存在するが、図4は、200%の最も多い希釈組成物の添加に対応するバッチGC061104が、特に早い時間段階で、最も高いロイプロリド放出速度(放出率)を有することを示す。
【0072】
図6は、表5及び6に製造され記載されたバッチGC071304、GC060304、及びGC061104を含む、55℃のクエン酸リン酸バッファー溶液中の、促進放出条件下での酢酸ロイプロリド微粒子の放出特性を示すグラフ図である。微粒子バッチは促進放出試験条件によるロイプロリドの放出速度(放出率)を示すようにプロットされ、1時間ごとの時間間隔に対して記録した。最初のデータポイント(1時間及び5時間)に関して、バッチGC061104がわずかに高い放出を有することを示した。
【0073】
実施例4
微粒子バッチをロイプロリド及び固有粘度0.157dL/g、分子量11.5kDaを有するR202Hを用いて製造した。バッチを、約10gスケールで製造した。
【0074】
表7はバッチの製造パラメーターを比較する。製造パラメーターは、添加した希釈組成物量を除いて、全てのバッチで類似している。
【表7】
【0075】
表8は、0.157IVポリマーを用い希釈組成物量を変化させ、表7に示すように製造された、微粒子バッチの特性を比べる。
【表8】
【0076】
希釈組成物の連続相への添加なしに製造された微粒子は、比較的より低い放出速度を有
する。より低分子量のポリマーは、より高分子量のポリマーで製造された微粒子に比べて、溶媒接触並びに微粒子中の薬剤含量に対してより敏感である。粒子サイズ分布は3つのバッチに関して同程度であり、変化の度合は23乃至26μmの範囲の平均粒子径(50%体積分布下)と小さい。希釈組成物を含まないバッチGC071404は、他の2つのバッチのものと比べて、より遅い初期放出を示した。早期放出におけるこの違いは、促進体外(in−vitro)放出にも反映される。
【0077】
図5は0.157の固有粘度を有するポリマーで形成された微粒子に関するリン酸バッファー溶液中の薬剤放出速度(放出率)を比較する。0.157IVポリマーで形成された微粒子は、希釈組成物を含まないバッチにおいて、より遅い初期放出を示した。100及び200%の希釈組成物添加のバッチに関して、0.157IVポリマー微粒子はより高い放出速度を示した。希釈組成物の添加は、形成したての微粒子に対する溶媒接触レベルを最小限に抑え、低IVポリマーに関する放出速度を増加させる。
【0078】
図7は、表7に示す希釈組成物の様々な量を有する3つの試料のグラフ図であって、促進体外(in−vitro)条件下での1時間に対してプロットした放出された酢酸ロイプロリド割合のグラフ図である。増加させた希釈組成物を有するバッチは、生理学的条件下で最初の2週間、促進放出試験条件下で最初の5時間、より高い放出速度を有した。
【0079】
実施例5
実施例5は、連続相への希釈組成物の添加/無添加での大規模微粒子バッチを詳述する。薬物動態(PK)及び有効性データ、そして3つのロイプロリド微粒子バッチ(0E5103、0B6315及び0A0307)に関して、約0.21dL/gの固有粘度を有するR202Hポリマーを用いて製造した。バッチ0E5103及び0A0307を、薬物動態及び有効性を目的として、ヒトへの22.5mg投与量で試験した。薬物動態(PK)群の患者は血中(血清)ロイプロリドレベルとテストステロン抑制に関してモニターし、残りの患者を有効性(テストステロン抑制)のみモニターした。ロイプロリドを用いる化学的去勢を示すためには、95%の患者が28日間以内で50ng/dL(0.5ng/mL)以下のテストステロンレベルを示す必要がある。
【0080】
微粒子バッチ0E5103を、0.206IV(固有粘度)と分子量18kDaを有するR202Hを用い、希釈組成物の添加や希釈なしに製造した。このバッチの塩化メチレン接触は約7200ppmであり、接触期間は約40分間であった。微粒子バッチ0B6315を、0.206IV(固有粘度)と分子量約18kDaを有するポリ(D,L−ラクチド)で製造した。連続相が溶媒除去容器にある間に、希釈組成物を連続相フローに対して水150%の添加により製造した。
【0081】
微粒子バッチ0A0307をバッチ0B6315と同様のIV(固有粘度)と分子量を有するポリ(d,l−ラクチド)を用いて製造した。連続相及び分散相エマルションがインラインミキサーを通って溶媒除去容器へ供給される間に、連続相フローに対して200%の水を添加することにより希釈組成物を製造した。塩化メチレン溶媒レベルは、ガスクロマトグラフィー(GC)をもとにして、連続相中で約1500ppmであった。表9は製造パラメーターを比較し、表10は表9で製造した微粒子の特性を比較する。
【0082】
特に、0A0307に関するバッチサイズは約1Kgスケールであり、反応容器中の微粒子形成時間は100分間であった。バッチ0E5103は0.4kgスケールのバッチサイズを有し、微粒子形成時間は40分間であった。分散相に関する処方は3つのバッチに関して同様であり、0E5103バッチはわずかに増加した活性剤及びポリマー濃度と溶媒(メタノール)及び酢酸濃度が減少していた。
【表9】
【表10】
【0083】
促進体外(in−vitro)放出速度は、増加された希釈組成物が添加され、そしてそれ故、溶媒接触が減少した0B6315及び0A0307が、希釈組成物が添加されていない0E5103と比較して、より高い初期放出を備える微粒子を製造したことを示した。図8は、バッチ0E5103及び0A0307に関して、1kgあたり22.5mgのロイプロリド投与量を与えたラットにおける、2週間の血清ロイプロリド濃度を比較する。もっとも低い溶媒接触であるバッチ0A0307は、0E5103に比べて、より高い早期放出を有し、血清中でより高いロイプロリド濃度となった。
【0084】
図9は、表9に従って製造したロイプロリド微粒子を投与したヒトにおける、血清ロイプロリド濃度レベル(ng/ml)のグラフ図である。微粒子バッチ0E5103及び0A0307は表9に製造され記載された。表10はヒトの血中(血清)ロイプロリド濃度の臨床データを示す。この試料の結果は、希釈組成物(0A0307と)低DCM接触である処方が、より早い放出の為に、最初の一ヶ月間、血中でより高いロイプロリドレベルを有することが示されている。このバッチは、ヒトの組織中において、GnRHアゴニストとしてより効果的に作用し、テストステロンレベルの長期間の減少を獲得する。同じ投与量で0A0307を投与された患者は、血液中のより低いロイプロリドレベルを有し、
薬剤は試験のより遅い段階(一ヶ月後)の間、より早い速度で放出された。
【0085】
バッチ0E5103と0A0307を比較した臨床試験を実施し、微粒子形成後に添加した希釈組成物の様々なレベルに対して、ロイプロリド微粒子投与後の患者のテストステロンレベルを比較した。理想的には95%の患者が28日間で50ng/dLより低いテストステロンレベルを獲得し維持することが望ましい。表11は、2つのロイプロリド微粒子バッチの臨床試験結果を詳述する。
【表11】
【0086】
バッチ0E5103からの遅い初期放出によって、79%の患者のみが28日までに所望のレベルを獲得した。一方、200%の希釈組成物を有するバッチ0A0307を投与された患者は、28日までに集団の98%が所望のレベルを獲得した。この結果、前立腺がんなどの疾病にあり、より高いテストステロンレベルのために不快症状及び痛みを伴う患者をより効果的に治療可能であり、ロイプロリドのより高い初期放出速度を有する徐放性薬剤の投与によって、より早くかれらの痛みを減じることが可能であり
【0087】
実施例6
実施例6に関するバッチを、固有粘度0.183IVを有するR202Hを用いて約10gスケールで製造した。表12はバッチに関する製造パラメーターを比較する。分散相組成物及び連続相組成物は全てのバッチに関して同じとし、目的とする薬剤搭載量を全てのバッチに関して16.5%L.A.とした。連続相及び分散相に関する流速を同じ又は同程度とした。
【表12】
【0088】
表13は、微粒子バッチの特性を比較する。
【表13】
【0089】
微粒子中の薬剤搭載量はバッチに関して同程度であった。粒子サイズは、バッチ間で、
小さく、典型的であり、変化を示した。平均粒子サイズ(50%体積分布下)は22乃至28μmに及び、90%体積分布は40乃至46μmに及んだ。一般に粒子サイズはバッチ間で大きな違いを示さず、明確な傾向はなかった。
【0090】
PBS中、37℃での薬剤放出は3ヶ月を超える薬剤放出が期待される。最少1ヶ月の初期放出をPBS中で追跡した。希釈組成物を含まないバッチ(GC061404)は、希釈組成物を用いて製造したバッチと比較してより低い初期放出を示した。50,100及び200%の希釈組成物を有するバッチは、同程度のより早い放出速度を示した。
【0091】
促進放出法もまた、希釈組成物で製造したバッチに関して、わずかにより高い初期放出(1時間及び5時間)を示した。様々な希釈物で製造されたバッチ間で、放出に関する違いはない。希釈物による初期放出(1及び5時間)の増加は50%希釈で獲得した。さらなる希釈は目立つほど大きくは初期放出を改善しなかった。
【0092】
実施例7
3つの微粒子バッチを、100,200及び300%希釈組成物で、固有粘度0.206IVのR202Hを用いて製造した。GC071204、GC090904、及びGC091004に関する製造パラメーターを表14に示す。製造パラメーターは、意図的に変化させた希釈組成物を除いて、これらバッチに関して同様のものであった。
【表14】
【0093】
表14で製造したバッチの特性を下記表15に示す。最終微粒子は71乃至75%のカプセル化効率を有し、薬剤搭載量はバッチに関して同程度であった。100及び200%
希釈のバッチは同程度の粒子サイズを示した。わずかに早い放出速度(又は同程度の放出速度)が、より多い希釈物で製造したバッチに関して、より早い時点の間、PBS中で観測された。促進放出が同程度の初期放出を示した。
【表15】
【0094】
実施例8
3つの微粒子バッチを、100、200及び300%希釈組成物にて、0.157IV固有粘度を有するR202Hを用い、そしてL.A.目的投与量19%にて製造した。製造パラメーターを表16に示す。製造パラメーターは、意図的に変化させた希釈組成物以外は、バッチに関して同程度であった。
【表16】
【0095】
表17は上記に製造された微粒子バッチの特性を示す。最終バッチは69乃至73%のカプセル化効率を有する微粒子を有していた。薬剤搭載量はおよそ14%で、バッチに関して同程度であった。微粒子サイズはバッチに関して同程度であった。薬剤放出速度はPBS中で全てのバッチに関して同程度であり、促進放出がわずかに高い初期放出を示した。
【表17】
【0096】
実施例9は様々な溶媒接触のオクトレオチド微粒子を比較する。オクトレオチド含有ポリ(d,l−ラクチド−コ−グリコリド)微粒子を、ロイプロリド微粒子の製造手順と類
似の手法で製造した。PLGA(85:15 PLGA、Alkermes社)、オクトレオチド、塩化メチレン、メタノール及び氷酢酸を含む分散相を、米国特許第5,945,126号明細書に例示されるような修正シルバーソンインラインミキサーの分散相注入チューブを介して、1分あたり約18mLで供給した。分散系におけるオクトレオチドの目標搭載量は10%であった。0.35%ポリビニルアルコール水溶液の連続相を、毎分2Lにて、インラインシルバーソンの連続相注入ポートを介して供給した。シルバーソンの回転速度は5500rpmであった。詳細な製造パラメータを下記表18に示す。製造された約2Lの懸濁液を、その後一定に撹拌しながら、15Lの連続相に接触させた。この15Lの連続相は、それぞれ2000、4000又は6000ppmの塩化メチレンを含んでいた。形成したての微粒子を、洗浄前に、100分間、接触させた。コントロールバッチは溶媒接触をせずに直接洗浄した。
【表18】
【0097】
促進体外(in−vitro)試験を実施し、各バッチの微粒子を比較した。試験は、pH4.0、60℃で酢酸バッファー溶液中で実施した。放出速度をもまた生理学的条件下、37℃で、1日間、バッチに関して算出した。
【表19】
【0098】
表19に示すように、より多い量の溶媒に接触した微粒子は、初期の時点において、放出媒体中でより高い初期放出を示した。インシュリン様成長因子−1(IGF−1)がより早い放出速度に対応してあまりにも急激に減少し得るように、より高い初期バーストは安全性の懸念を引き起こし得る。徐放性微粒子処方におけるオクトレオチドに関するより低い初期放出速度が最適であろう。ロイプロリドとは異なり、オクトレオチド微粒子に関して、減少した溶媒接触の処方を用いることによって、放出速度が減少される。
【0099】
図10は、ラットにおけるオクトレオチドの生体内研究において、表18に従って製造された微粒子の放出特性のグラフ図である。ラットはオクトレオチド微粒子バッチ5mgの投与量を与えられた。GJ030804は連続相中で増加した溶媒に接触しておらず、より高濃度の塩化メチレンで製造されたバッチと比較して、より低い初期バースト放出を示した。より高い溶媒接触がより高いバースト放出速度を引き起こすため、より低いバーストの微粒子は好ましい速度に応じて、水希釈によって製造され得ることが推測される。
【0100】
実施例10
実施例10は、希釈組成物が添加/無添加で、微粒子のバッチサイズが様々であってあるときの、微粒子のロイプロリドの薬剤放出速度の効果を比較する。まずバッチGC071304、0E5103及び0D4022を連続相への希釈組成物の添加なしに、様々なバッチサイズで製造した。表20に示すように、8gバッチであるGC071304を、分子量18kDaのR202H、酢酸ロイプロリド、塩化メチレン及びメタノールを含む分散相29gを用いて製造した。GC071304の連続相は、1.5Lの0.35%ポリビニルアルコール水溶液を含んでいた。分散相に関して30−35g/分、連続相に関して2L/分の流速にて、微粒子形成を50秒以内で完了した。バッチをインラインミキサーにて7000rpmで一定に撹拌し、周囲水中及び39℃水中、およそ10 体積変
化にて洗浄した。
【0101】
バッチ0E5103を上述と同じ製造パラメーターを用い、但しバッチサイズを400gに増加させた。微粒子形成時間は40分間であった。バッチサイズを700gに増加させたバッチ0D4022を上述と同じ製造パラメーターを用いて製造した。微粒子形成時間は70分間であった。後述の2つのバッチに使用した分散相及び連続相の量は、検討されたバッチサイズを製造するために、比例して調整した。
【0102】
下記表20に示すように、3つのバッチを0.206IV(固有粘度)ポリマーを用いて製造した。表21に、バッチの特性を比較する。
【表20】
【0103】
表20で形成された微粒子の特性を表21に示す。
【表21】
【0104】
生理学的条件下で試験された放出速度は、pH7.4、37℃でPBSに対して接触させられた。50秒という最も早い微粒子形成時間と、対応する最も短い溶媒接触時間で製造された8gバッチは、400gバッチ及び700gバッチと比較して、最も早い初期放出を有した。最も長い溶媒接触時間(微粒子形成時間:70分間)の最大の700gバッチは、最も遅い初期放出を有していた。よって、生理学的条件及び促進条件下で、バッチサイズの増加、又はバッチのスケールアップは、全ての他の因子がほぼ同じである、薬剤の放出速度に反転効果を与える。
【0105】
表22に詳述されるように、微粒子を10,100及び1000gのスケールバッチで製造した。100gスケールでは試料の再現性を示すために3つのバッチを製造した。
バッチは、分散相に関して0.183IV(固有粘度)、分子量14kDaを有するR202Hポリマー、ロイプロリド、DCM、MeOH及び氷酢酸を用いて、連続相に関して水とポリビニルアルコールを用いて製造した。また、約1500、±500mlの希釈組成物を連続相に加えた。具体的には希釈組成物、或いは水を、微粒子形成時に溶媒除去容器に対して2乃至4L/分で連続的に添加した。製造パラメーターはバッチに関して同じであり、微粒子製造時間は、10gバッチで1分間、100gバッチで10分間、1000gバッチで100分間であった。100gスケールにて再現性を示すために3つのバッチを製造した。
【表22】
【表23】
【0106】
薬剤カプセル化効率は10、100及び1000gバッチで75乃至79%の範囲の値で同程度である。薬剤搭載量は12.8乃至13.7%と正常変動である。粒子サイズの結果は同程度のサイズ分布データを示す。薬剤放出速度は10,100及び1000gバッチで類似している。10、100及び1000gで製造したバッチは全て非常に低い残存溶媒を示した。よって、連続相に希釈組成物を添加することによって、上記測定された放出プロファイルに関しては、バッチサイズは小さなファクターになる。連続相の溶媒レベルが閾値下に減少すると、希釈組成物の添加なしに製造されたバッチがそうであったように、微粒子の放出特性はバッチサイズに依存しない。
【0107】
実施例11
実施例11は、希釈組成物を用いて製造されたリスペリドン微粒子の放出プロファイルを詳述する。まず、微粒子を生体適合性及び生体分解性ポリマーとして、27.5gのポリ(D,L−ラクチド−コ−グリコリド)、すなわちラクチド:グリコリド比が85:15であり、固有粘度が0.61g/dLであるPLGAを用いて製造した。バッチを27
.5gのリスペリドンを用いて製造した。リスペリドン及びポリマーを220gのジクロロメタンに溶解した。ポリマー、生物学的活性剤及び溶媒のこの混合有機溶液が分散相を構成した。別に、連続相を供給するために、87.5gのポリビニルアルコール及び69gのリン酸二ナトリウム七水和物を25kgの精製水中に溶解した。分散相及び連続相を同時に、それぞれ38.5ml/分及び4000mL/分で、シルバーソンインラインミキサーに注入し、約5分間4000rpmで混合した。
【0108】
分散相を連続相と混合する際、得られた混合物をシルバーソンミキサーにてリスペリドン搭載PLGA微粒子に固化させた。リスペリドン微粒子懸濁液を、希釈組成物を微粒子分散液に室温で添加するところの溶媒除去容器(SRVに)排出し、希釈組成物をおよそ5分間、流速2000mL/分で加えた。SRV中で、室温及び33−37℃の水で微粒子を洗浄し、有機溶媒を除去した。洗浄及び溶媒除去後、微粒子を濾過にて回収し、凍結乾燥した。微粒子は、微粒子1gあたり0.39gのリスペリドンの薬剤搭載量を有すると決定された。PLGA微粒子のリスペリドン搭載効率は77.5%であった。微粒子の放出特性を評価した。pH7バッファー中24時間以内で、1%未満の薬剤の初期放出が検出された。
【0109】
実施例12
実施例12は水希釈なしに微粒子にカプセル化されたリスペリドンの放出プロファイルを詳述する。微粒子を27.5gのポリ(D,L−ラクチド−コ−グリコリド)、すなわち、固有粘度0.61g/dL、ラクチド:グリコリド比=85:15のPLGAを用いて製造した。リスペリドン27.5gを分散相としての役割を果たす有機溶液の一部としてのジクロロメタン220gに溶解した。別に、ポリビニルアルコール87.5g、リン酸二ナトリウム七水和物69gを精製水25kgに溶解することで連続相を調製した。分散相と連続相を、それぞれ38.5ml/分、4000mL/分にて同時にシルバーソンインラインミキサーに注入した。混合懸濁液を4000rpmで混合した。分散相を連続相と混合し、得られた混合物をシルバーソンミキサーでリスペリドン搭載PLGA微粒子に固化させた。リスペリドン微粒子懸濁液を溶媒除去容器(SRV)に排出した。この実施例では希釈組成物を用いなかった。SRV中で、室温及び33−37℃水で微粒子を洗浄し有機溶媒を除去した。洗浄及び溶媒除去工程後、微粒子を濾過にて回収し、凍結乾燥した。微粒子は、微粒子1gあたりリスペリドン0.33gの含量を有すると決定された。PLGA中のリスペリドン搭載効率は66.2%であった。後の放出プロファイル試験で、およそ80−82%のリスペリドンがpH7バッファー中24時間以内で微粒子から放出された。
【表24】
【0110】
希釈組成物の添加なしに製造されたバッチに関して、微粒子からの放出速度(放出率)はリスペリドン80−82%であった。2000ml/分で希釈組成物にて希釈したバッチは、初期投与において放出速度(放出率)が1%未満であった。
【符号の説明】
【0111】
10・・・連続工程
12・・・分散相
14・・・連続相
16・・・インラインミキサー
18・・・希釈組成物
18a・・・希釈組成物
18b・・・希釈組成物
20・・・第一容器
22・・・中空糸フィルタ
22a・・・ライン
【先行技術文献】
【特許文献】
【0112】
【特許文献1】米国特許第4,818,542号明細書
【特許文献2】米国特許第4,767,628号明細書
【特許文献3】米国特許第3,773,919号明細書
【特許文献4】米国特許第3,755,558号明細書
【特許文献5】米国特許第5,407,609号明細書
【特許文献6】米国特許第6,270,802号明細書
【特許文献7】米国特許第6,361,798号明細書
【特許文献8】米国特許第5,945,126号明細書
【特許請求の範囲】
【請求項1】
変更された放出速度を有する徐放性組成物であって:
特定の活性剤及びポリマーを含む分散相を含み、前記分散相は連続相と組み合わせられて微粒子分散系を形成し、前記微粒子分散系は、微粒子が形成された後に一定量の希釈用組成物と接触させられており;前記希釈組成物は、特定の活性剤の放出速度を変更させる連続相を希釈するのに十分であるところの、組成物。
【請求項2】
前記希釈用組成物は、徐放性組成物のバッチサイズの増加の悪影響をなくすところの連続相を希釈するのに十分である、請求項1記載の徐放性組成物。
【請求項3】
前記希釈用組成物は、形成したての微粒子に添加され、その後第一容器に移し替えられる、
請求項1記載の徐放性組成物。
【請求項4】
前記希釈用組成物は、微粒子が第一容器に移し替えられた後に微粒子分散系に添加される、請求項1記載の徐放性組成物。
【請求項5】
請求項1記載の徐放性組成物を形成する方法であって、
ポリマーに溶解した特定の活性剤を含む分散相を形成する工程;
分散相を連続相と共に混合して、連続相中に懸濁している微粒子を含む微粒子分散系を形成させる工程;
一定量の希釈用組成物を、微粒子が形成された後に、微粒子分散系へ添加する工程を含み;
添加された希釈用組成物量が、特定の活性剤の放出速度を変更させるために十分な量である、方法。
【請求項6】
前記方法が、さらに、希釈用組成物を第一容器に添加するのと同時に、ミキサーから第一容器に微粒子分散系を移送する工程を含む、請求項5記載の方法。
【請求項7】
前記希釈用組成物が水であり、前記水は連続相の少なくとも50%乃至約300%量で、微粒子分散系に加えられる、請求項5記載の方法。
【請求項8】
前記希釈用組成物及び前記微粒子分散系が、第一容器に同時に移送される、請求項5記載の方法。
【請求項9】
請求項1記載の徐放性組成物を形成する方法であって、
特定の活性剤及びポリマーを含む分散相を形成する工程;
分散相と連続相を混合して、連続相中に懸濁された微粒子を含む微粒子分散系を形成する工程;そして
微粒子分散系に希釈用組成物を添加して、未処理の徐放性微粒子の放出速度とは異なる放出速度を有する徐放性組微粒子を製造する工程を含み;
希釈用組成物の体積が、連続相の体積の少なくとも50%である、方法。
【請求項10】
前記特定の活性剤がロイプロリドであり、前記ロイプロリドがポリマー中に溶解されており、かつ前記希釈用組成物の体積が連続相の体積の少なくとも200%である、請求項9記載の方法。
【請求項11】
前記特定の活性剤がオクトレオチドであり、前記希釈用組成物の体積が連続相の体積の少なくとも100%である、請求項9記載の方法。
【請求項12】
前記特定の活性剤がリスペリドンである、請求項9記載の方法。
【請求項13】
前記連続相が溶媒を含み、そして溶媒が約5000ppm未満の溶媒濃度になるまで、前記希釈用組成物が連続相の部分に添加される、請求項10記載の方法。
【請求項14】
本方法が連続的に実施される、請求項9記載の方法。
【請求項15】
分散相が連続相と混合された後、微粒子が約2乃至約30秒で形成され、そして、微粒子が一定期間希釈用組成物に接触させられる、請求項9記載の方法。
【請求項16】
希釈用組成物が、徐放性組成物のバッチサイズの増加の悪影響をなくすところの連続相を希釈するのに十分である、請求項9記載の方法。
【請求項17】
請求項1記載の徐放性組成物の放出速度をコントロールする方法であって、
特定の活性剤とポリマーを含む分散相を形成する工程、ここで前記特定の活性剤は、ロイプロリドであり、該ロイプロリドがポリマー中に溶解されてなり;
ミキサー中で分散相を連続相と混合し、微粒子分散系を形成する工程;
微粒子の形成後に、一定量の希釈用組成物を微粒子分散系に添加する工程を含み;そして希釈用組成物量がロイプロリドの放出速度を変更させるのに十分な量である、方法。
【請求項18】
連続相が溶媒を含み、添加された希釈用組成物量が、連続相中の溶媒を約5000ppm未満に希釈するのに十分な量である、請求項17記載の方法。
【請求項19】
微粒子分散系が第一容器に移送される前に、定められた期間の間、ミキサー中で保持される、請求項17記載の方法。
【請求項1】
変更された放出速度を有する徐放性組成物であって:
特定の活性剤及びポリマーを含む分散相を含み、前記分散相は連続相と組み合わせられて微粒子分散系を形成し、前記微粒子分散系は、微粒子が形成された後に一定量の希釈用組成物と接触させられており;前記希釈組成物は、特定の活性剤の放出速度を変更させる連続相を希釈するのに十分であるところの、組成物。
【請求項2】
前記希釈用組成物は、徐放性組成物のバッチサイズの増加の悪影響をなくすところの連続相を希釈するのに十分である、請求項1記載の徐放性組成物。
【請求項3】
前記希釈用組成物は、形成したての微粒子に添加され、その後第一容器に移し替えられる、
請求項1記載の徐放性組成物。
【請求項4】
前記希釈用組成物は、微粒子が第一容器に移し替えられた後に微粒子分散系に添加される、請求項1記載の徐放性組成物。
【請求項5】
請求項1記載の徐放性組成物を形成する方法であって、
ポリマーに溶解した特定の活性剤を含む分散相を形成する工程;
分散相を連続相と共に混合して、連続相中に懸濁している微粒子を含む微粒子分散系を形成させる工程;
一定量の希釈用組成物を、微粒子が形成された後に、微粒子分散系へ添加する工程を含み;
添加された希釈用組成物量が、特定の活性剤の放出速度を変更させるために十分な量である、方法。
【請求項6】
前記方法が、さらに、希釈用組成物を第一容器に添加するのと同時に、ミキサーから第一容器に微粒子分散系を移送する工程を含む、請求項5記載の方法。
【請求項7】
前記希釈用組成物が水であり、前記水は連続相の少なくとも50%乃至約300%量で、微粒子分散系に加えられる、請求項5記載の方法。
【請求項8】
前記希釈用組成物及び前記微粒子分散系が、第一容器に同時に移送される、請求項5記載の方法。
【請求項9】
請求項1記載の徐放性組成物を形成する方法であって、
特定の活性剤及びポリマーを含む分散相を形成する工程;
分散相と連続相を混合して、連続相中に懸濁された微粒子を含む微粒子分散系を形成する工程;そして
微粒子分散系に希釈用組成物を添加して、未処理の徐放性微粒子の放出速度とは異なる放出速度を有する徐放性組微粒子を製造する工程を含み;
希釈用組成物の体積が、連続相の体積の少なくとも50%である、方法。
【請求項10】
前記特定の活性剤がロイプロリドであり、前記ロイプロリドがポリマー中に溶解されており、かつ前記希釈用組成物の体積が連続相の体積の少なくとも200%である、請求項9記載の方法。
【請求項11】
前記特定の活性剤がオクトレオチドであり、前記希釈用組成物の体積が連続相の体積の少なくとも100%である、請求項9記載の方法。
【請求項12】
前記特定の活性剤がリスペリドンである、請求項9記載の方法。
【請求項13】
前記連続相が溶媒を含み、そして溶媒が約5000ppm未満の溶媒濃度になるまで、前記希釈用組成物が連続相の部分に添加される、請求項10記載の方法。
【請求項14】
本方法が連続的に実施される、請求項9記載の方法。
【請求項15】
分散相が連続相と混合された後、微粒子が約2乃至約30秒で形成され、そして、微粒子が一定期間希釈用組成物に接触させられる、請求項9記載の方法。
【請求項16】
希釈用組成物が、徐放性組成物のバッチサイズの増加の悪影響をなくすところの連続相を希釈するのに十分である、請求項9記載の方法。
【請求項17】
請求項1記載の徐放性組成物の放出速度をコントロールする方法であって、
特定の活性剤とポリマーを含む分散相を形成する工程、ここで前記特定の活性剤は、ロイプロリドであり、該ロイプロリドがポリマー中に溶解されてなり;
ミキサー中で分散相を連続相と混合し、微粒子分散系を形成する工程;
微粒子の形成後に、一定量の希釈用組成物を微粒子分散系に添加する工程を含み;そして希釈用組成物量がロイプロリドの放出速度を変更させるのに十分な量である、方法。
【請求項18】
連続相が溶媒を含み、添加された希釈用組成物量が、連続相中の溶媒を約5000ppm未満に希釈するのに十分な量である、請求項17記載の方法。
【請求項19】
微粒子分散系が第一容器に移送される前に、定められた期間の間、ミキサー中で保持される、請求項17記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公表番号】特表2012−511003(P2012−511003A)
【公表日】平成24年5月17日(2012.5.17)
【国際特許分類】
【出願番号】特願2011−539632(P2011−539632)
【出願日】平成21年12月1日(2009.12.1)
【国際出願番号】PCT/US2009/066212
【国際公開番号】WO2010/065513
【国際公開日】平成22年6月10日(2010.6.10)
【出願人】(511135835)オークウッド ラボラトリーズ,エル.エル.シー. (1)
【Fターム(参考)】
【公表日】平成24年5月17日(2012.5.17)
【国際特許分類】
【出願日】平成21年12月1日(2009.12.1)
【国際出願番号】PCT/US2009/066212
【国際公開番号】WO2010/065513
【国際公開日】平成22年6月10日(2010.6.10)
【出願人】(511135835)オークウッド ラボラトリーズ,エル.エル.シー. (1)
【Fターム(参考)】
[ Back to top ]