
微生物のコウジ酸生産性を向上させる方法
【課題】コウジ酸の生産に関与する新たな遺伝子を同定し、麹菌等の微生物のコウジ酸生産性を向上させる新規な手段を提供すること。
【解決手段】本発明に係るコウジ酸生産性を向上させる方法は、麹菌やペニシリウム属菌等のコウジ酸生産能を有する微生物において、Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子並びにこれらにコードされるタンパク質から成る群より選択される少なくとも1つの機能を阻害する工程を含む。該工程は、例えば遺伝子破壊により行なわれる。あるいは、該工程は、HDAC阻害剤等の、上記遺伝子にコードされるタンパク質の阻害剤で微生物を処理することにより行なわれる。
【解決手段】本発明に係るコウジ酸生産性を向上させる方法は、麹菌やペニシリウム属菌等のコウジ酸生産能を有する微生物において、Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子並びにこれらにコードされるタンパク質から成る群より選択される少なくとも1つの機能を阻害する工程を含む。該工程は、例えば遺伝子破壊により行なわれる。あるいは、該工程は、HDAC阻害剤等の、上記遺伝子にコードされるタンパク質の阻害剤で微生物を処理することにより行なわれる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、微生物のコウジ酸生産性を向上させる方法、コウジ酸の製造方法、コウジ酸生産性の高い微生物、及びコウジ酸生産性の高い微生物の育種方法に関する。
【背景技術】
【0002】
コウジ酸(5‐ヒドロキシ‐2(ヒドロキシメチル)-4‐ピロン)は、麹から発見された複素環化合物であり、麹菌(アスペルギルス属菌)、ペニシリウム属菌等の微生物によるいわゆるコウジ酸発酵で生成することが知られている。
【0003】
コウジ酸は、抗酸化活性を有しているほか、メラノサイトに作用し、チロシナーゼの活性や合成を阻害し、メラニンの生成を抑える活性を持っており、美白化粧品(医薬部外品)の有効成分として使用されている。一時発がん性が疑われたが、安全性が証明されたため現在は販売されている。
【0004】
コウジ酸生産性微生物が有するコウジ酸生産に関わる遺伝子として、特許文献1及び非特許文献1には、コウジ酸の生産を正に制御する遺伝子が開示されている。しかしながら、負に制御する遺伝子は知られていない。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2010−246532号公報
【非特許文献】
【0006】
【非特許文献1】Fungal Genetics and Biology 47 (2010) p.953-961
【非特許文献2】Marui et al., "Kojic acid biosynthesis in Aspergillus oryzae is regulated by a Zn(II)2Cys6 transcriptional activator and induced by kojic acid at the transcriptional level" Journal of Bioscience and Bioengineering (2011) in press
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明の目的は、コウジ酸の生産に関与する新たな遺伝子を同定し、麹菌等の微生物のコウジ酸生産性を向上させる新規な手段を提供することにある。
【課題を解決するための手段】
【0008】
本願発明者らは、鋭意研究の結果、麹菌において特定のヒストン脱アセチル化遺伝子やヒストンアセチル基転移酵素のサブユニットが麹菌生産を負に制御しており、該遺伝子の破壊やコードされるタンパク質の阻害などによってコウジ酸の生産量を増大させることができることを見出し、本願発明を完成した。
【0009】
すなわち、本発明は、コウジ酸生産能を有する微生物において、Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子並びにこれらにコードされるタンパク質から成る群より選択される少なくとも1つの機能を阻害する工程を含む、微生物のコウジ酸生産性を向上させる方法を提供する。また、本発明は、上記本発明の方法でコウジ酸の生産性が向上された微生物を培養し、該微生物が生産したコウジ酸を回収することを含む、コウジ酸の製造方法を提供する。さらに、本発明は、Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子から成る群より選択される少なくとも1つの遺伝子の機能が阻害されている、コウジ酸生産性の高い微生物を提供する。さらに、本発明は、コウジ酸生産能を有する微生物をジェノトキシンで処理し、ジェノトキシン感受性が高い菌株を選抜することを含む、コウジ酸生産性の高い微生物の育種方法を提供する。さらに、本発明は、配列番号3に示す塩基配列中の1nt-2000ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号3に示す塩基配列中の3785nt-5142ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なHst4遺伝子を含まないDNA断片を、コウジ酸生産能を有する微生物の細胞に導入し、該DNA断片と微生物ゲノムとの間で相同組換えを生じさせることを含む、コウジ酸生産性が向上した微生物の製造方法を提供する。さらに、本発明は、配列番号6に示す塩基配列中の1nt-2856ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号6に示す塩基配列中の4318nt-5694ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なHos2遺伝子を含まないDNA断片を、コウジ酸生産能を有する微生物の細胞に導入し、該DNA断片と微生物ゲノムとの間で相同組換えを生じさせることを含む、コウジ酸生産性が向上した微生物の製造方法を提供する。さらに、本発明は、配列番号9に示す塩基配列中の1nt-2000ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号9に示す塩基配列中の4299nt-5594ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なSpt3遺伝子を含まないDNA断片を、コウジ酸生産能を有する微生物の細胞に導入し、該DNA断片と微生物ゲノムとの間で相同組換えを生じさせることを含む、コウジ酸生産性が向上した微生物の製造方法を提供する。さらに、本発明は、下記のいずれかのDNA断片を提供する。
(1) 配列番号3に示す塩基配列中の1nt-2000ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号3に示す塩基配列中の3785nt-5142ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なHst4遺伝子を含まないDNA断片。
(2) 配列番号6に示す塩基配列中の1nt-2856ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号6に示す塩基配列中の4318nt-5694ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なHos2遺伝子を含まないDNA断片。
(3) 配列番号9に示す塩基配列中の1nt-2000ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号9に示す塩基配列中の4299nt-5594ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なSpt3遺伝子を含まないDNA断片。
【発明の効果】
【0010】
本発明により、コウジ酸生産を負に制御する遺伝子が新たに同定された。本発明によれば、従来技術と同等又はそれ以上のコウジ酸生産性を得ることができる。負の制御遺伝子は、遺伝子を破壊することにより、コウジ酸生産性をとりわけ顕著に高めることができる。本発明の方法によれば、通常の野生株ではコウジ酸を生産しにくい条件で培養しても、コウジ酸を生産することができる。例えば、Hst4遺伝子破壊麹菌株では、親株ないしはコントロール株と比較して200倍にまでコウジ酸生産量が増大する。その上さらに、特許文献1記載の技術と組み合わせることで、コウジ酸生産性をさらに高めることができると期待される。また、作製した遺伝子破壊株の特性解析の結果に基づき、コウジ酸高生産性の変異株を効率よく取得できる新規な育種方法も提供された。コウジ酸は、一時的に安全性が疑われたものの、現在では安全性も再確認され、美白化粧品の有効成分として商業利用されている。本発明は、コウジ酸の製造に大いに貢献するとともに、コウジ酸生合成経路の研究にも貢献し得る。
【図面の簡単な説明】
【0011】
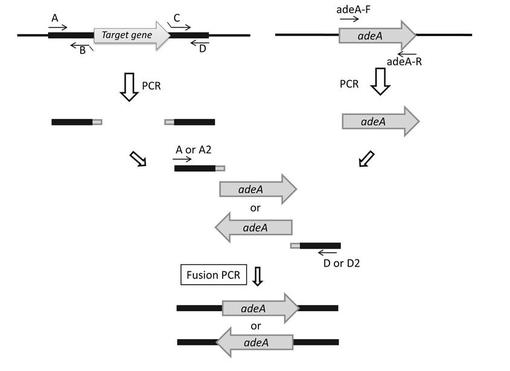
【図1】遺伝子破壊用DNA断片の増幅のためのfusion PCRの概略を説明する図である。A、B、C、D、A2、D2、adeA-F、及びadeA-RはPCRプライマーである。
【図2】実施例で作製した遺伝子破壊株によるコウジ酸生産量を定性的に評価した結果を示す写真である。control株では菌糸の周縁部は赤色着色していないが、遺伝子破壊株では菌糸の外周部にまで赤色着色が確認された。
【図3】実施例で作製した遺伝子破壊株によるコウジ酸生産量を定量した結果を示すグラフである。
【図4】コウジ酸生産に不適な最小培地上で遺伝子破壊株によるコウジ酸生産を検出した結果を示す写真である。
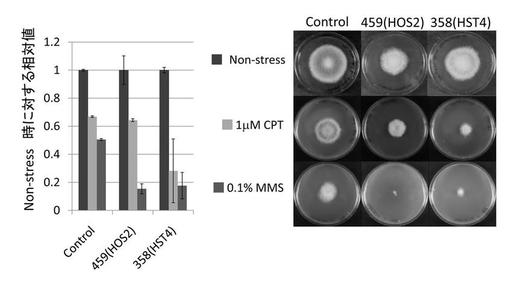
【図5】ジェノトキシン存在下で遺伝子破壊株を培養し、ジェノトキシン非添加条件(Non-stress)での生育と相対評価した結果を示す図である。
【図6】HDAC阻害剤ニコチンアミドを添加したコウジ酸産生培地上でcontrol株を培養し、コウジ酸の生産量を調べた結果を示す写真である。
【発明を実施するための形態】
【0012】
本発明で対象となる微生物は、コウジ酸生産能を有する微生物であれば特に限定されない。そのような微生物の具体例として、ペニシリウム(Penicillium)属菌、アスペルギルス(Aspergillus)属菌等を挙げることができる。アスペルギルス属菌の種類も特に限定されず、麹菌を含む各種アスペルギルス属菌が広く包含され、具体例としては黄麹菌(Aspergillus oryzae)、黒麹菌(Aspergillus nigerなど)、及び白麹菌(Aspergillus usamii、Aspergillus kawachiiなど)等の麹菌や、フラブス菌(Aspergillus flavus)等を挙げることができる。
【0013】
Aspergillus oryzae RIB40ゲノムデータベース(http://nribf2.nrib.go.jp/)にAO080533000358として登録されている遺伝子は、SIR2ファミリー(Sirtuin 5 and related class III sirtuins)のヒストン脱アセチル化酵素(HDAC)をコードしている遺伝子として知られている。酵母が有する該遺伝子のホモログがHst4であることから、本発明ではこのAO080533000358遺伝子を「Hst4」又は「AOhst4」と呼ぶ。配列番号1及び2に示す配列は、上記の麹菌ゲノムデータベースに登録されているRIB40株のHst4遺伝子のcDNA配列及びコードされるHST4タンパク質のアミノ酸配列であり、配列番号3に示す配列は、RIB40株における該遺伝子及びその近傍のゲノム配列である。配列番号3中の2001nt-3784ntの領域がHst4遺伝子のコード領域である。また、RIB40株のHst4遺伝子は、NCBIのGenBankにもXM_001825249で登録されており、この配列を配列番号35(塩基配列)及び配列番号36(アミノ酸配列)に示す。
【0014】
上記データベースにAO080511000459として登録されている遺伝子は、ヒストン脱アセチル化酵素複合体の触媒成分をコードしている遺伝子として知られている。酵母が有する該遺伝子のホモログがHos2であることから、本発明ではこのAO080511000459遺伝子を「Hos2」又は「AOhos2」と呼ぶ。配列番号4及び5に示す配列は、麹菌ゲノムデータベースに登録されているRIB40株のHos2遺伝子のcDNA配列及びコードされるHOS2タンパク質のアミノ酸配列であり、配列番号6に示す配列は、RIB40株における該遺伝子及びその近傍のゲノム配列である。配列番号6中の2857nt-4317ntの領域がHos2遺伝子のコード領域である。また、RIB40株のHos2遺伝子は、NCBIのGenBankにもXM_003189254で登録されている。
【0015】
上記データベースにAO080525000462として登録されている遺伝子は、ヒストンアセチル基転移酵素のサブユニットをコードしている遺伝子として知られている。酵母が有する該遺伝子のホモログがSpt3であることから、本発明ではこのAO080525000462遺伝子を「Spt3」又は「AOspt3」遺伝子と呼ぶ。配列番号7及び8に示す配列は、麹菌ゲノムデータベースに登録されているRIB40株のSpt3遺伝子のcDNA配列及びコードされるSPT3タンパク質のアミノ酸配列であり、配列番号9に示す配列は、RIB40株における該遺伝子及びその近傍のゲノム配列である。配列番号9中の2001nt-4298ntの領域がSpt3遺伝子のコード領域である。また、RIB40株のSpt3遺伝子は、NCBIのGenBankにもXM_001727435で登録されており、この配列を配列番号37(塩基配列)及び配列番号38(アミノ酸配列)に示す。
【0016】
本発明において、「Hst4遺伝子」といった場合、配列番号2又は36と完全に同一のアミノ酸配列からなるタンパク質をコードし、イントロンを除いた塩基配列が配列番号1又は35と完全に同一であり、ゲノム上での配列が配列番号3に示す塩基配列と完全に同一である遺伝子に限定されるものではなく、天然に生じ得る変異を含む変異配列からなり、かつ、ヒストン脱アセチル化酵素活性を有するタンパク質をコードする遺伝子が包含される。「Hos2遺伝子」といった場合も同様であり、配列番号4〜6のみに限定されず、天然に生じ得る変異配列からなり、かつ、ヒストン脱アセチル化酵素活性を有するタンパク質をコードする遺伝子が包含される。「Spt3遺伝子」といった場合もまた同様であり、配列番号7〜9、37、38のみに限定されず、天然に生じ得る変異配列からなり、かつ、ヒストンアセチル基転移酵素のサブユニットとして機能するタンパク質をコードする遺伝子が包含される。以下、配列の一部が本願配列表とは異なるがもとの生理活性を大きく減弱せずに維持している変異配列からなる遺伝子ないしはタンパク質を、単に「天然の変異体」と呼ぶことがある。
【0017】
そのような変異配列は、通常、少数(例えば、1〜20個、1〜15個、1〜10個、又は1〜数個)の塩基ないしはアミノ酸の置換、欠失、挿入又は付加を含み、特に限定されないが、もとの配列との同一性は90%以上、例えば95%以上、あるいは98%以上である。ここで、配列の「同一性」とは、アミノ酸配列の場合であれば、比較すべき2つのアミノ酸配列のアミノ酸残基ができるだけ多く一致するように両アミノ酸配列を整列させ、一致したアミノ酸残基数を、全アミノ酸残基数で除したものを百分率で表したものである。上記整列の際には、必要に応じ、比較する2つの配列の一方又は双方に適宜ギャップを挿入する。このような配列の整列化は、例えばBLAST、FASTA、CLUSTAL W等の周知のプログラムを用いて行なうことができる。ギャップが挿入される場合、上記全アミノ酸残基数は、1つのギャップを1つのアミノ酸残基として数えた残基数となる。このようにして数えた全アミノ酸残基数が、比較する2つの配列間で異なる場合には、同一性(%)は、長い方の配列の全アミノ酸残基数で、一致したアミノ酸残基数を除して算出される。塩基配列の同一性についても同様である。
【0018】
本発明では、コウジ酸生産能を有する微生物において、Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子並びにこれらにコードされるタンパク質から成る群より選択される少なくとも1つの機能を阻害する。「遺伝子の機能を阻害する」とは、その遺伝子が本来コードしているmRNAないしはタンパク質の生成若しくは蓄積を低下若しくは欠失させること、又は、対象とする微生物細胞内の少なくとも一部のゲノム若しくは核において、その遺伝子が本来コードしているmRNAないしはタンパク質を生成できないようにゲノム上の対象遺伝子領域の少なくとも一部を改変することをいう。特定の遺伝子の機能を阻害するための遺伝子改変方法はこの分野で広く知られており、当業者であれば適宜選択して実行できる。具体例としては、アンチセンス法、RNAi、遺伝子破壊法等を挙げることができるが、これらに限定されない。また、タンパク質の機能を阻害するとは、例えば、そのタンパク質の阻害剤で微生物を処理することが挙げられる。
【0019】
例えば、本発明の方法では、上記3つの遺伝子のいずれか1つの機能を阻害するだけでもよいし、任意の2つないしは3つの遺伝子の機能を阻害してもよい。野生型の微生物株をHST4、HOS2及びSPT3タンパク質のうちの少なくとも1つの阻害剤で処理してもよいし、また、いずれかの遺伝子の機能が阻害されるように遺伝的に改変した微生物株をさらに阻害剤で処理してもよい。
【0020】
本発明の方法では、遺伝子の機能を阻害する手段として、上記3遺伝子の少なくともいずれかを破壊する方法が好ましい。「遺伝子を破壊する」とは、微生物ゲノムの少なくとも一部のアリル又は微生物体の少なくとも一部の核において、好ましくは全てのアリル又は核において、微生物ゲノム上の対象遺伝子のコード領域を欠失させること又は正常な遺伝子産物を産生できないように変異させることをいう。対象遺伝子の欠失は、コード領域の全体を欠失させてもよく、また一部を欠失させてもよい。全体を欠失させる場合には、対象遺伝子に隣接する領域もあわせて広く欠失させてもよい。一部を欠失させる場合には、特に限定されないが、対象遺伝子のコード領域の半分以上を欠失させることが好ましい。コード領域の変異により遺伝子を破壊する場合には、例えば、該コード領域の好ましくは中央よりも上流の部位にナンセンス変異又はフレームシフト変異を導入して、本来の生理活性を有しない変異タンパク質や全く無関係なタンパク質の配列とすることができる。対象遺伝子の一部又は全部を他の配列(例えばマーカー遺伝子配列)に置き換えてもよい。
【0021】
各種の微生物について、その微生物に適した遺伝子破壊方法が公知である。例えば、麹菌等の菌類の遺伝子破壊は、相同組換えを利用した手法により実施することができる(北本勝ひこ,丸山潤一:発酵・醸造食品の最新技術と機能性(シーエムシー出版,東京),95-103 (2006)等を参照)。Hst4遺伝子及び麹菌を例に具体的に説明すると、例えば、Hst4遺伝子の上流及び下流のゲノム領域を麹菌ゲノムからPCRにより増幅して上流側相同領域(第1の相同領域)及び下流側相同領域(第2の相同領域)を調製し、正常なHst4遺伝子を含まない配列の両末端に2つの相同領域をそれぞれ連結して遺伝子破壊用DNA断片を調製し、これを麹菌細胞に導入すればよい。麹菌が有する内在の酵素の働きで、相同な領域間で相同組換えが生じ、ゲノム中のHst4遺伝子領域が遺伝子破壊用DNA断片と置き換わるので、ゲノムからHst4遺伝子を欠失させることができる。
【0022】
「正常なHst4(又は、Hos2若しくはSpt3)遺伝子を含まない配列」とは、配列番号2(又は、配列番号5若しくは配列番号8)に示されるアミノ酸配列からなるタンパク質をコードする配列も、該タンパク質の天然の変異体をコードする配列も、いずれも含んでいない配列である。相同領域と連結する「正常なHst4(Hos2、Spt3)遺伝子を含まない配列」は、該遺伝子以外の他の遺伝子配列でもよいし、本来の生理活性を有するタンパク質をコードしないこれらの変異遺伝子(正常なヒストン脱アセチル化酵素として機能するタンパク質をコードしない変異Hst4若しくは変異Hos2遺伝子、又はヒストンアセチル基転移酵素のサブユニットとして正常に機能するタンパク質をコードしない変異Spt3遺伝子)であってもよい。他の遺伝子配列としてマーカー遺伝子を用いることで、形質転換体(遺伝子破壊株)のスクリーニングが容易になる。
【0023】
相同領域としては、通常、微生物ゲノム中の対応する領域と同一の塩基配列からなる断片が用いられるが、対応領域と一部が異なる配列であっても、微生物細胞内で相同組換えが生じる程度の配列同一性を有していれば、相同領域として使用することができる。すなわち、相同領域には、配列番号3、6又は9に示される塩基配列中の対応する部分と同一の塩基配列からなるものの他、該塩基配列において相同組換えが起きる程度に少数(例えば1個又は数個)の塩基が置換し、欠失し、挿入され又は付加された塩基配列からなる断片も包含される。相同領域と配列番号3、6又は9中の対応領域との同一性は90%以上、好ましくは95%以上、より好ましくは98%以上であり、最も好ましくは100%である。本発明において、ある領域と「相同な領域」とは、その領域と上記の通りの同一性を有する塩基配列からなる領域をいう。
【0024】
相同領域のサイズは特に限定されないが、鎖長が長い方が相同組換えの効率が高まり、短くなりすぎると効率が低下することがあるため、通常30塩基以上であり、好ましくは100塩基以上、より好ましくは200塩基以上、さらに好ましくは300塩基以上、特に好ましくは500塩基以上である。サイズの上限は特に限定されないが、DNA合成の便宜から通常は10000塩基以下、好ましくは2500塩基以下である。
【0025】
例えば、配列番号3に示す配列のうち、Hst4遺伝子コード領域は2001nt-3784ntであるから、1nt-2000ntの上流領域のうちの30塩基以上の領域を増幅して上流側相同領域(第1の相同領域)を、3785nt-5142ntの下流領域のうちの30塩基以上の領域を増幅して下流側相同領域(第2の相同領域)をそれぞれ得ることができる。下記実施例では、配列番号3中の620nt-2000ntを増幅して上流側相同領域とし、3789nt-5142ntを増幅して下流側相同領域としているが、これに限定されない。
【0026】
Hos2遺伝子の場合は、配列番号6に示す配列のうちHos2遺伝子のコード領域が2857nt-4317ntであるから、同様にして、1nt-2856ntから上流側相同領域を、4318nt-5694ntから下流側相同領域を増幅して得ることができる。また、Spt3遺伝子の場合は、配列番号9のうちSpt3遺伝子のコード領域が2001nt-4298ntであるから、1nt-2000ntから上流側相同領域を、4299nt-5594ntから下流側相同領域を増幅して得ることができる。下記実施例では、Hos2遺伝子破壊用DNAの作製のため、配列番号6中の1nt-1326ntを増幅して上流側相同領域とし、4320nt-5693ntを増幅して下流側相同領域としており、また、Spt3遺伝子破壊用DNA作製のため、配列番号9中の1170nt-1998ntを増幅して上流側相同領域とし、4668nt-5594ntを増幅して下流側相同領域としているが、これに限定されない。
【0027】
なお、配列番号3、6、9には、各遺伝子破壊用DNA断片の調製に十分と考えられる上下流領域を示しているが、これらよりもさらに上流ないし下流の配列は、上記したAspergillus oryzae RIB40ゲノムデータベース等から入手することができる。
【0028】
遺伝子破壊用DNA断片に適宜含めることができるマーカー遺伝子としては、使用する微生物株の栄養要求性に適合した栄養要求性相補遺伝子や、各種薬剤耐性遺伝子等が挙げられ、適宜選択して用いることができる。栄養要求性マーカーの場合は、遺伝子破壊の親株として、該当するマーカー遺伝子をもともと欠失している微生物株を用いる必要があるが、遺伝子破壊株を用いて製造された食品や化粧品、医薬部外品等に対する消費者の安心を得る観点からは栄養要求性マーカー遺伝子が有利である。一方、薬剤耐性マーカーは、使用する親株の遺伝的背景に制限がなく、より広い範囲の微生物株に対し利用することができ有利である。
【0029】
各種のマーカー遺伝子が公知であり、適宜選択して使用することができる。例えば、麹菌で用いられる栄養要求性マーカー遺伝子としては、adeA遺伝子(アデニン合成に関与し、アデニン要求性を相補)、sC遺伝子(硫酸(硫黄源)資化に関与し、硫酸(硫黄源)資化欠損株の栄養要求性を相補)、argB遺伝子(アルギニン合成に関与し、アルギニン要求性を相補)等が知られている。これら遺伝子の配列は公知であり、前掲のAspergillus oryzae RIB40ゲノムデータベースやNCBIのGenBank等から配列情報を入手できる。下記実施例では、adeAを欠損しアデニン要求性である麹菌株を遺伝子破壊の親株として使用し、マーカー遺伝子としてadeA遺伝子を用いることで、アデニン要求性を指標として遺伝子破壊株をスクリーニングしているが、これに限定されない。adeA遺伝子の配列は、Aspergillus oryzae RIB40ゲノムデータベースにはAO080527000383として、またGenBankにはAB121755のアクセッション番号で登録されている。配列番号34に示す配列は、adeA遺伝子及びその近傍のゲノム配列であり、下記実施例では360nt-2362ntの領域を用いて遺伝子破壊用DNA断片を調製しているが、これに限定されない。麹菌で用いられる薬剤耐性マーカー遺伝子としては、例えば、ptrA遺伝子(チアミンの代謝拮抗アナログであるピリチアミンに対する耐性遺伝子)等が挙げられるが、これに限定されない。
【0030】
上流側相同領域及び下流側相同領域とマーカー遺伝子DNAとの連結はfusion PCR法により行うことができる(図1参照)。上流側相同領域及び下流側相同領域をゲノムから増幅する際、マーカー遺伝子末端の部分領域とハイブリダイズする相補領域をプライマー対の一方(図1ではB及びC)に付加しておき、この相補領域において3つの断片をハイブリダイズさせて1本のDNA断片に融合し、これを鋳型としてPCRを行えば、[上流側相同領域]−[マーカー遺伝子]−[下流側相同領域]が連結した構造の遺伝子破壊用DNA断片を増幅することができる。
【0031】
遺伝子破壊用DNA断片は、適当なベクターに組み込んで微生物細胞に導入することができ、例えば麹菌細胞の場合はpUSC等のプラスミドベクターを用いることができる(Bioscience, Biotechnology, and Biochemistry, 61(8),1367-1369,1997)。あるいは、微生物細胞のプロトプラストを調製し、DNA断片のまま直接該プロトプラストに導入することもできる。麹菌を例にプロトプラスト法を簡単に記載すると、麹菌分生子を液体培地中で24時間程度培養した後、孔サイズ20〜30μm程度の不織布等で濾過して菌体を回収し、これを糸状菌細胞壁溶解酵素(キチナーゼ、キトビアーゼ、β-1,3-グルカナーゼ等)を含むプロトプラスト化溶液中で数時間振とう培養し、次いでガラスフィルター等で濾過して菌体残渣を除去することで、麹菌プロトプラストを得ることができる。プロトプラスト懸濁液と遺伝子破壊用DNAを混合し、数十分氷冷後、ポリエチレングリコールを混合して静置後、寒天選択培地上で培養すればよい。
【0032】
マーカーによるスクリーニング後に得られる形質転換体は、全ての核の対象遺伝子が破壊されたホモカリオンの破壊株か、又は一部の核の対象遺伝子が破壊され、少なくとも1つの核の対象遺伝子が破壊されずに残っているヘテロカリオンの破壊株であるが、遺伝子破壊用DNA断片が異所的に組み込まれた対象遺伝子非破壊株が得られることもある。従って、マーカーによるスクリーニング後、適宜サザン解析やPCR増幅産物の確認、ダイレクトシークエンシング等を行ない、目的通りの位置にDNA断片が挿入され対象遺伝子が破壊されていることを直接確認することが望ましい。
【0033】
本発明では、対象遺伝子の発現を完全に欠失できるホモカリオンの破壊株が好ましい。ホモカリオンの破壊株は、上記した遺伝子破壊方法で直接得ることができるし、また、得られたヘテロカリオンの破壊株について、全ての核の対象遺伝子が破壊されたホモカリオンの株となるまで純化の処理(例えば、選択培地上での植え継ぎ操作)を繰り返し行なってもよい。交配可能な微生物株を用いて遺伝子破壊株を作製した場合には、ヘテロのアレルを有する破壊株については、交配によりホモ破壊株を得ることもできる。
【0034】
本発明の方法をタンパク質の機能の阻害により実施する場合、使用し得るタンパク質阻害剤としては、以下のような阻害剤を挙げることができる。
(1) Sirtuin系(クラスIII)HDAC用阻害剤(Hst4を含むSirtuinの阻害剤)
Nicotinamide
2-Anilino-benzamide
(2) 古典的(クラスI又はクラスII)HDAC用阻害剤(Hos2を含む古典的HDACの阻害剤)
HC-toxin
Trichostatin A
酪酸(n-Butyrate)
【0035】
下記実施例では、麹菌のHst4遺伝子、Hos2遺伝子又はSpt3遺伝子を破壊することで、コウジ酸の生産性が向上することが確認されている。このことは、これら遺伝子を有するコウジ酸生産性微生物株を、HST4、HOS2又はSPT3タンパク質の阻害剤で処理することによっても、該微生物の生産性を高めることができることを示している。
【0036】
コウジ酸生産能を有する微生物において、Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子並びにこれらにコードされるHST4タンパク質、HOS2タンパク質及びSPT3タンパク質のうちの少なくともいずれかの機能を阻害すると、コウジ酸の生産性が高まる。すなわち、正常な各遺伝子を有するもとの親株又は阻害剤で処理しない同一の株と比較して、コウジ酸の生産量が増大する。本発明によるコウジ酸生産性を向上させる方法によれば、親株の30倍〜200倍以上にコウジ酸生産性を高めることも可能である。また、例えば特許文献1記載の公知技術と組み合わせて行なうことで、さらなる生産性の向上が期待される。
【0037】
コウジ酸の生産性の向上は、終濃度が1mM〜5mM程度となるように第二塩化鉄を加えた培地で微生物を培養し、第二塩化鉄とコウジ酸とのキレート化合物の生成による赤色発色の程度で確認することができる。また、微生物を液体培養し、培養液を適宜希釈した試料に1mM〜5mM程度の第二塩化鉄を添加し、495nmにおける吸光度を測定することでコウジ酸の定量も可能である。495nmの吸光度は、0.1〜1.0程度の範囲でコウジ酸濃度に比例する。
【0038】
本発明はまた、上記本発明の方法でコウジ酸の生産性が向上された微生物を培養し、該微生物が生産したコウジ酸を回収することを含む、コウジ酸の製造方法も提供する。該製造方法では、例えば、Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子のうちの少なくとも1つの機能が阻害されるように遺伝的に改変された麹菌等の微生物を、コウジ酸の生成に好ましい条件下で培養し、生成したコウジ酸を回収する。あるいは、コウジ酸生産能を有する微生物(野生型でも遺伝的に改変された微生物でもよい)を、HST4、HOS2又はSPT3タンパク質の阻害剤の共存下で培養し、生成したコウジ酸を回収してもよい。
【0039】
コウジ酸生産のための培地は公知である。コウジ酸の生産に適した培地は、一般に、窒素源として酵母エキス、ペプトン、無機窒素化合物(硝酸やアンモニウム塩)等を約0.1〜2.0%、炭素源としてグルコース、スクロース、デンプン等の糖類を約1〜20%含む。その他、無機塩、ビタミン類や、分散剤、消泡剤等を適宜使用してよい。pHは通常5.0〜6.5程度である。培養温度は、微生物の種類により適宜選択され、麹菌の場合は通常25℃〜35℃程度である。
【0040】
生成したコウジ酸は、公知の手法により回収、精製できる。例えば、再結晶、アフィニティークロマトグラフィー、イオン交換クロマトグラフィー、ゲル濾過等が挙げられる。また、麹菌を用いた商業的なコウジ酸製造法が各種開示されている(特公昭60-36753、特開平5-76378など)。
【0041】
本願発明者らがコウジ酸生産の負の制御因子として同定した遺伝子は、ヒストン脱アセチル化酵素やヒストンアセチル基転移酵素のサブユニットをコードしているが、これら遺伝子が破壊された麹菌はジェノトキシン(genotoxin、遺伝毒性物質)に対する感受性が高まっていることが確認された。自然界から分離したコウジ酸生産性微生物、又は該分離菌若しくは既存の微生物を変異処理したものの中から、ジェノトキシンに対する感受性の高い菌株を選抜することで、コウジ酸生産性の高い微生物株を取得し得ることを示している。すなわち、本発明は、コウジ酸生産能を有する微生物をジェノトキシンで処理し、ジェノトキシン感受性が高い微生物株を選抜することを含む、コウジ酸生産性の高い微生物の育種方法を提供する。
【0042】
使用可能なジェノトキシンは特に限定されず、DNAを損傷する作用があることが知られているいかなる物質であってもよい。例えば、トポイソメラーゼ阻害剤(カンプトテシン等)、DNAアルキル化剤(メチルメタンスルホン酸、エチルメタンスルホン酸、アジ化ナトリウム等)等を挙げることができる。
【0043】
該育種方法に供する微生物としては、上述の通り、自然界から分離された微生物をそのまま用いてもよいし、また、変異処理した微生物であってもよい。変異処理の方法は特に限定されず、例えば放射線処理(γ線、β線、X線、中性子線等)、紫外線処理、化学物質処理(エチルメタンスルホン酸等)等を挙げることができ、いずれの方法であってもよい。
【0044】
高濃度のジェノトキシンは麹菌等の微生物に対し致死的であるが、中程度の濃度では分生子の発芽及び生育を数時間遅らせる。そこで、ジェノトキシンが概ね50%程度生育を阻害する濃度で培地に添加し、該培地に微生物又はその胞子を植え、適当な時間(例えば7時間〜10時間程度)インキュベートすることで、成長度合い(例えば菌糸の成長度合い)によって感受性の高いものと感受性が低いないしは通常レベルであるものとを区別することができる。例えば、麹菌分生子をジェノトキシン添加培地に播種し、一定時間インキュベート後、フィルターを通して出芽伸長したものを取り除くことで、ジェノトキシン感受性の高い株を濃縮することができる。ジェノトキシン処理濃度は、ジェノトキシン非添加の培地とジェノトキシンを各種濃度で添加した培地を調製し、微生物の生育(分生子の発芽や菌糸の進展の度合いなど)を確認して、選抜に適した濃度を適宜設定することができる。
【実施例】
【0045】
以下、本発明を実施例に基づきより具体的に説明する。もっとも、本発明は下記実施例に限定されるものではない。
【0046】
adeAマーカー遺伝子との遺伝子置換により、麹菌のAOhst4遺伝子(AO080533000358)、AOhos2遺伝子(AO080511000459)及びAOspt3遺伝子(AO080525000462)の破壊を試みた。Fusion PCR法及びプロトプラスト法は、北本勝ひこ,丸山潤一:発酵・醸造食品の最新技術と機能性(シーエムシー出版,東京), 95-103 (2006)に記載の方法に準じて行なった。
【0047】
遺伝子破壊用DNA断片の作成
麹菌ゲノムDNAを鋳型としたPCRにより、AOhst4遺伝子、AOhos2遺伝子及びAOspt3遺伝子の5'側上流領域及び3'側下流領域について各1kb程度のDNA領域を増幅した。また、遺伝子破壊株の選択用のマーカーとしてアデニンの合成に関わるadeA栄養要求性マーカー遺伝子(AO080527000383)とその上流及び下流の一部を含む断片もPCRで増幅した。その後、5'側上流領域, adeA, 3'側下流領域を混合してPCRを行い、5'側上流領域(上流側相同領域), adeA, 3'側下流領域(下流側相同領域)の順にDNA断片をつなぎ合わせた遺伝子破壊用DNA断片をFusion PCRにより作製した。尚、Fusion PCRの模式図を図1に記す。また、用いたプライマー配列を表1に示す。表中のプライマーの名称は、図1記載のプライマーに対応する。
【0048】
Fusion PCRに用いる5'側上流領域,3'側下流領域の各DNA断片を作製の際に、Fusion PCRでadeA遺伝子との断片との連結部になる位置にアニールするプライマー(BとC)のテール部に、adeA断片の5'末端配列及び3'末端配列にハイブリダイズする相補配列(表1中の小文字の部分)を付加しプライマーを設計した。なお、AOhst4及びAOhos2の破壊には、5'側上流領域がadeA遺伝子配列の3'末端に連結された遺伝子破壊用断片を使用し、AOspt3の破壊には、5'側上流領域がadeA遺伝子配列の5'末端に連結された遺伝子破壊用断片を使用した。
【0049】
【表1】

【0050】
1st PCR
5'側上流領域, adeA, 3'側下流領域の各DNA断片は、KOD PLUS Version 1(東洋紡)で増幅した。テンプレートは、全てアスぺルギルス・オリゼ野生株RIB40の染色体DNAを用いた(QIAGEN社製のDNeasy Plant Maxi Kitを用いて抽出)。
【0051】
【表2】
【0052】
上記反応液50mlを0.2mlPCRチューブ内で混合してDNA Thermal Cyclerにセットし、以下のような温度設定によりPCRを行った。
94℃、2分 1サイクル
94℃、20秒 55℃、30秒 68℃ 2分 30サイクル
68℃、5分 4℃、O/N 1サイクル
【0053】
PCR反応液50μlをQIAquick PCR purification kit(キアゲン)を用いて、Elution Buffer 50μlをカラムに通し、カラムからDNAの溶出を行った(増幅産物の精製)。
【0054】
Fusion PCR
精製した5'側上流領域, adeA, 3'側下流領域の各DNA断片の濃度を、NanoDrop ND-1000 Spectrophotometerを用いて計測し、それぞれのDNA断片を50 ng/μlになるように調整した後、5'側上流領域断片 : adeA断片 : 3'側下流領域断片=1 : 1.6 :1になるようにして、且つ3つのDNA断片の合計液量が3.6μlになるように混合し、これをTemplateとして用いて、以下の反応液でFusion PCRを行った。
【0055】
【表3】
【0056】
上記反応液50mlを0.2mlPCRチューブ内で混合してDNA Thermal Cyclerにセットし、以下のような温度設定によりPCRを行った。
94℃、2分 1サイクル
94℃、20秒 55℃、30秒 68℃ 5分 30サイクル
68℃、5分 4℃、O/N 1サイクル
【0057】
PCR反応液50μlをQIAquick PCR purification kit(キアゲン)を用いて、Elution Buffer 50μlをカラムに通し、カラムからDNAの溶出を行った。また、得られた精製遺伝子破壊用DNA断片は、アガロースゲル電気泳動により非特異バンドがほとんどないことを確認した。また、上記のPCRを2チューブ以上行う事により、遺伝子破壊用DNA断片を20μg程度作製した。
【0058】
形質転換
アスペルギルス・オリゼ NSR-ΔlD2株を宿主麹菌として利用した。この株の分生子縣濁液を、アデニンを0.05%添加した酵素生産培地(1L中の組成で、グルコース 20g, トリプトン 1g(ディフコ), 酵母エキス 5g (ディフコ), NaNO3 1g, K2HPO4 0.5g, MgSO4・7H2O 0.5g, FeSO4・7H2O 0.01g, pH6.0)に植菌し、30℃で24時間振盪培養した。菌体を、ミラクロスを用いて集菌後、菌体を滅菌0.8 M NaCl/10mM Phosphate buffer (pH6.0)で3回洗浄した。その後、菌体をマイレックスHV(ミリポア)でフィルター滅菌した30mlプロトプラスト化溶液(100mg / 30ml Yatalase(タカラバイオ)になるよう0.8 M NaCl/10mM Phosphate buffer (pH6.0)にYatalaseを溶解)の入った50ml容の滅菌済み遠心チューブに移して懸濁し、30℃、80rpm、3時間振盪しプロトプラスト化反応を行った。滅菌した3G1ガラスフィルターにてろ過し、濾液中のプロトプラストを5 分間遠心分離することで沈殿を得た。30mlの滅菌0.8M NaClにてプロトプラストを洗浄し3,000rpm、4 ℃で5 分間遠心分離することで沈殿を得た。このプロトプラストを滅菌0.8M NaCl 10mlとSolution I (0.8 M NaCl, 10 mM CaCl2, 10 mM Tris-HCl 、pH 8.0) 20mlに懸濁し、懸濁液中のプロトプラスト数をトーマの血球計数機で計測した。計測後、3,000rpm、4 ℃で5 分間遠心分離することで沈殿を得た。
【0059】
このプロトプラストを2.4×108 protoplast / ml Solution IとなるようにSolution Iを加えて懸濁した。プロトプラスト懸濁液50mlを滅菌した2ml容のマイクロチューブに移し、それぞれにSolution II (40% (w/v) PEG4000, 50 mM CaCl2, 10 mM Tris-HCl 、pH8.0)12.5 μlと前述した形質転換用DNA溶液各5μl(DNA量として2μg以上)を加えて優しく混合した後、氷上で30min放置した。この溶液に、Solution II 500μlを加えて優しく混合し、室温で15min放置した。この溶液にSolution I 1mlを加えて転倒混和した。この溶液を0.8MのNaClを添加した選択寒天培地(1L中の組成で、Glucose 20g, NH4Cl 2g, (NH4)2SO4 1g, KCl 0.5g , KH2PO4 1g, MgSO4・7H2O 0.5g, FeSO4・7H2O 0.02g, L-methionine 1.5g、pH5.5)に250μlをスポット植菌し、そこに0.8MのNaClを添加した選択軟寒天培地約10mlを重層した。その後、コロニーを形成するまで30℃で培養した。
【0060】
形質転換後、再生した菌体のうちシングルコロニーを形成した菌体の菌糸を火炎滅菌したピンセットで採取し、選択培地に植え継いだ。その後、30℃で分生子を形成するまで培養した。その後、コロニーの端から菌糸を火炎滅菌したピンセットで選択培地に植え継いだ。この植え継ぎ操作を少なくとも3回以上行うことで核の純化を行った。
【0061】
遺伝子破壊株の確認
形質転換体から遺伝子破壊株の確認は、Fusion PCRに用いたAとDプライマー並びに、欠失破壊の対象とした遺伝子の内500bp程度を増幅できるように設計したFとGプライマー(下記表4)を用いてPCRを行った。各断片の増幅には、KOD FX(東洋紡)を用いた。テンプレートは、滅菌した1.5ml容のマイクロチューブに100μlずつ分注したBufferA(1M KCl、10mM EDTA、100mM Tris-HCl、pH8.5)に各形質転換体の菌糸と胞子をそれぞれ懸濁し、5分間ボルテックスした後に、95℃で10分間煮沸後、さらに5分間ボルテックスし、15000rpm、 4℃で1分間遠心分離して得られた上清1μlを用いた。PCRの結果、欠失破壊用DNA断片に相当する大きさのDNA断片の増幅が認められ、且つFG間の増幅が認められなかったものをホモカリオン欠失破壊株、野生株の場合と同じ大きさのDNA断片の増幅のみが見られ、且つFG間の増幅がみられたものはadeAがectopicに取り込まれた非欠失破壊株、欠失破壊用DNA断片に相当する大きさのDNA断片並びに野生株の場合と同じ大きさのDNA断片の増幅とFG間の増幅がみられたものは、ヘテロカリオン欠失株とした。以後の実験では、ホモカリオン欠失破壊株を使用した。
【0062】
【表4】
【0063】
遺伝子破壊株におけるのコウジ酸産生量の定性的評価
プレート上でのコウジ酸産生への影響は、遺伝子破壊株を、コウジ酸を産生する条件で生育させ、培地に分泌されたコウジ酸と予め培地に混入させた塩化第二鉄とのキレートによる赤色発色により判断した。
【0064】
5mMになるように塩化第二鉄を添加したコウジ酸産生寒天培地(1L中の組成で、glucose 100g, 酵母エキス 1g(ディフコ)、KH2PO4 1g、MgSO4・7H2O 0.5g、pH6.0)を用いた。1×108 遺伝子破壊株胞子/ mlに調整した胞子懸濁液1μlをプレートの中央部に植菌し、30℃の恒温槽中に一週間静置した。
【0065】
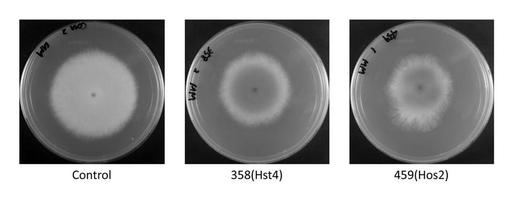
NSR-ΔlD2株のadeA遺伝子のみを相補した株(以下control株とする)を対照とし、AOhst4、AOhos2及びAOspt3欠失破壊変異株における培地の着色度合を比較した結果、control株と比較し、AOhst4、AOhos2及びAOspt3欠失破壊変異株において培地の赤色強度の増加が認められた(図2)。尚図2中には、AOhst4(AO080533000358)及びAOhos2(AO080511000459)遺伝子欠失株をそれぞれ358, 459と表示する。培地の赤色強度の増加は、コウジ酸生産量の増加を意味する。すなわち、上記3つの遺伝子は、コウジ酸の生産を負に制御することを示唆している。
【0066】
遺伝子破壊株におけるコウジ酸生産量の定量
上述のようにコウジ酸は、第二塩化鉄とのキレート化合物の生成による赤色の発色により検出することが可能である。さらにコウジ酸量は、培養液を適宜希釈した試料に最終濃度として1mM〜5mMになるように塩化第二鉄溶液を添加した液の、495nmにおける吸光度を測定することで定量することができる。また、この495nmの吸光度は、0.1〜1.0程度の範囲でコウジ酸濃度に比例する。このような検出法により麹菌培養液を適宜希釈し、そのコウジ酸濃度の定量が可能である。
【0067】
100mlのバッフル付フラスコに20mlの上記コウジ酸生産培地をいれ、遺伝子欠失破壊株あるいは上述のcontrol株胞子を105 conidia/ mlコウジ酸生産培地になるよう植菌した。これを7本用意した。培養条件は、7本とも30℃、120rpmであった。
【0068】
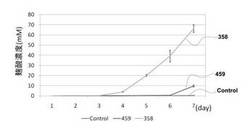
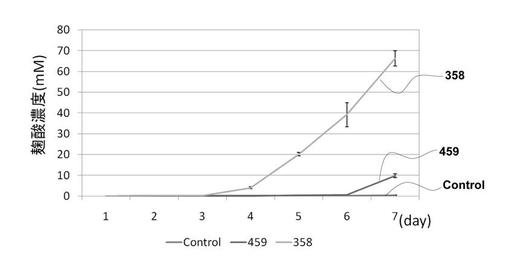
7本の中から1日につき1本の培養液を全てミラクロス(メルク)でろ過し、濾液を得た。この操作を7日間毎日行った。得られた、7つの試料を上述の塩化第二鉄とのキレートによる発色を用いて495nmでの吸収により測定した。尚、この測定では、コウジ酸生産培地をブランクとした。コウジ酸定量結果をグラフで図3に示す。尚、図3中には、AOhst4(AO080533000358)及びAOhos2(AO080511000459)遺伝子欠失株をそれぞれ358, 459と表示する。
【0069】
AOhos2遺伝子破壊株では、control株と比較し30倍のコウジ酸生産量の増加が認められた。また、AOhst4遺伝子破壊株では、control株と比較し200倍のコウジ酸生産量の増加が認められた。
【0070】
上記2つの遺伝子は、それぞれヒストン脱アセチル化遺伝子のモチーフを有している。一般にヒストンのアセチル化は、クロマチン構造を緩和する方向に働き、結果転写因子や転写装置が遺伝子近傍にアクセスできるようになり遺伝子発現を亢進することが知られている。一方で、ヒストン脱アセチル化遺伝子は、上記のアセチル化とは逆で、クロマチンを凝集する方向に働き、結果転写因子や転写装置が遺伝子にアクセスできなくなり遺伝子発現が抑制される。これらの現象は、染色体の幅広い領域で起こっており遺伝子発現に大きな影響を与えることが知られている。すなわち、AOhst4及びAOhos2Aは、上記の結果と同様に遺伝子発現の負の制御因子であるHDACがコウジ酸生産を負に制御していると考えられる。
【0071】
最小培地上でのコウジ酸生産性
下記の組成に5mM になるように塩化第二鉄を添加したM培地(最小培地)を作製し、5*107 conidia/mlに調整した分生子懸濁液を培地の中心に2μl植菌し、30℃、飽和蒸気圧で5日間培養した。
【0072】
【表5】
【0073】
図4は、5日間培養後のプレートの裏面を撮影した写真である。上記の最小培地では通常の麹菌はコウジ酸を生産できない。control株では赤色発色は認められなかったが、一方、欠失変異株ではいずれも赤色発色が認められ、コウジ酸の生産が確認された。
【0074】
ジェノトキシンに対する感受性
上記で得られた遺伝子破壊株について、ジェノトキシン存在下での菌糸の成長を調べた。ジェノトキシンとして、トポイソメラーゼ阻害剤であるカンプトテシン(CPT)及びDNAのアルキル化剤であるメチルメタンスルフォネート(MMS)を用いた。終濃度1μMのCPT又は終濃度0.1%のMMSを加えたM培地(最小培地)の中央に麹菌分生子を播種し、30℃で5日間培養した。円状に繁殖した菌糸の直径を測定し、ジェノトキシン非添加(Non-stress)の培地上での直径と相対評価した。結果を図5に示す。
【0075】
AOhst4破壊株において、CPT及びMMSに対する感受性が上昇していた。また、AOhos2破壊株においてMMSに対する感受性が上昇していた。hos2とhst4は、機構は異なるが共にDNA修復系に関与している可能性が考えられた。
【0076】
ジェノトキシンによるコウジ酸高生産株のスクリーニングの検討
control株の分生子と遺伝子破壊株の分生子を表6に示すInitial conditionで混和し、5μM CPTまたは0.05%MMS存在下で穏やかに振とうしながら30℃、8時間培養した。その後、孔経が5μmのフィルターで濾過し、ろ液を0.3% Triton-X100を含んだプレートに塗布し、30℃で4-5日間培養した。得られたコロニーの形状(分生子の着生が悪いものを破壊株と判断)から各破壊株とコントロール株の比を算出した。
【0077】
【表6】
【0078】
表6に示す通り、CPT処理及びMMS処理のいずれにおいても、コウジ酸高生産性である遺伝子破壊株の割合がinitial conditionよりも増大しており、破壊株の分生子が濃縮されていた。この結果は、変異原処理した野生型麹菌株からジェノトキシンによるスクリーニングでコウジ酸高生産性の変異株を取得できる可能性を示している。
【0079】
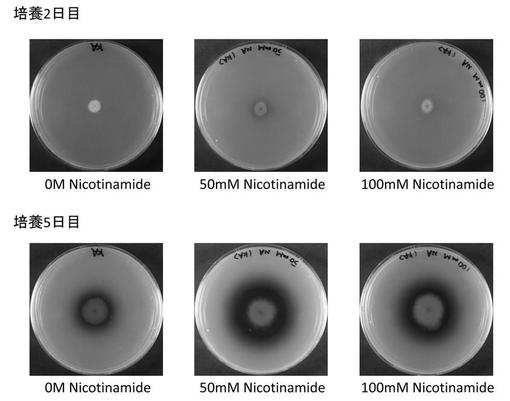
HDAC阻害剤によるコウジ酸生産性の向上
上記のコウジ酸産生培地に0, 50, 100mMのニコチンアミドを添加した培地を作成し、5*107 conidia/ ml に調整したcontrol株の分生子懸濁液を培地の中心に2μl植菌し、30℃、飽和蒸気圧で5日間培養した。
【0080】
図6は2日間と5日間培養後のプレートの裏面を撮影した写真である。ニコチンアミドを添加しないものに比べ、添加したものでは培地の赤色強度の顕著な増加が認められた。これは、コウジ酸生産量の増加を意味する。すなわち、HDAC活性を阻害によりコウジ酸の生産を増加させることを示している。
【技術分野】
【0001】
本発明は、微生物のコウジ酸生産性を向上させる方法、コウジ酸の製造方法、コウジ酸生産性の高い微生物、及びコウジ酸生産性の高い微生物の育種方法に関する。
【背景技術】
【0002】
コウジ酸(5‐ヒドロキシ‐2(ヒドロキシメチル)-4‐ピロン)は、麹から発見された複素環化合物であり、麹菌(アスペルギルス属菌)、ペニシリウム属菌等の微生物によるいわゆるコウジ酸発酵で生成することが知られている。
【0003】
コウジ酸は、抗酸化活性を有しているほか、メラノサイトに作用し、チロシナーゼの活性や合成を阻害し、メラニンの生成を抑える活性を持っており、美白化粧品(医薬部外品)の有効成分として使用されている。一時発がん性が疑われたが、安全性が証明されたため現在は販売されている。
【0004】
コウジ酸生産性微生物が有するコウジ酸生産に関わる遺伝子として、特許文献1及び非特許文献1には、コウジ酸の生産を正に制御する遺伝子が開示されている。しかしながら、負に制御する遺伝子は知られていない。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開2010−246532号公報
【非特許文献】
【0006】
【非特許文献1】Fungal Genetics and Biology 47 (2010) p.953-961
【非特許文献2】Marui et al., "Kojic acid biosynthesis in Aspergillus oryzae is regulated by a Zn(II)2Cys6 transcriptional activator and induced by kojic acid at the transcriptional level" Journal of Bioscience and Bioengineering (2011) in press
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明の目的は、コウジ酸の生産に関与する新たな遺伝子を同定し、麹菌等の微生物のコウジ酸生産性を向上させる新規な手段を提供することにある。
【課題を解決するための手段】
【0008】
本願発明者らは、鋭意研究の結果、麹菌において特定のヒストン脱アセチル化遺伝子やヒストンアセチル基転移酵素のサブユニットが麹菌生産を負に制御しており、該遺伝子の破壊やコードされるタンパク質の阻害などによってコウジ酸の生産量を増大させることができることを見出し、本願発明を完成した。
【0009】
すなわち、本発明は、コウジ酸生産能を有する微生物において、Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子並びにこれらにコードされるタンパク質から成る群より選択される少なくとも1つの機能を阻害する工程を含む、微生物のコウジ酸生産性を向上させる方法を提供する。また、本発明は、上記本発明の方法でコウジ酸の生産性が向上された微生物を培養し、該微生物が生産したコウジ酸を回収することを含む、コウジ酸の製造方法を提供する。さらに、本発明は、Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子から成る群より選択される少なくとも1つの遺伝子の機能が阻害されている、コウジ酸生産性の高い微生物を提供する。さらに、本発明は、コウジ酸生産能を有する微生物をジェノトキシンで処理し、ジェノトキシン感受性が高い菌株を選抜することを含む、コウジ酸生産性の高い微生物の育種方法を提供する。さらに、本発明は、配列番号3に示す塩基配列中の1nt-2000ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号3に示す塩基配列中の3785nt-5142ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なHst4遺伝子を含まないDNA断片を、コウジ酸生産能を有する微生物の細胞に導入し、該DNA断片と微生物ゲノムとの間で相同組換えを生じさせることを含む、コウジ酸生産性が向上した微生物の製造方法を提供する。さらに、本発明は、配列番号6に示す塩基配列中の1nt-2856ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号6に示す塩基配列中の4318nt-5694ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なHos2遺伝子を含まないDNA断片を、コウジ酸生産能を有する微生物の細胞に導入し、該DNA断片と微生物ゲノムとの間で相同組換えを生じさせることを含む、コウジ酸生産性が向上した微生物の製造方法を提供する。さらに、本発明は、配列番号9に示す塩基配列中の1nt-2000ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号9に示す塩基配列中の4299nt-5594ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なSpt3遺伝子を含まないDNA断片を、コウジ酸生産能を有する微生物の細胞に導入し、該DNA断片と微生物ゲノムとの間で相同組換えを生じさせることを含む、コウジ酸生産性が向上した微生物の製造方法を提供する。さらに、本発明は、下記のいずれかのDNA断片を提供する。
(1) 配列番号3に示す塩基配列中の1nt-2000ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号3に示す塩基配列中の3785nt-5142ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なHst4遺伝子を含まないDNA断片。
(2) 配列番号6に示す塩基配列中の1nt-2856ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号6に示す塩基配列中の4318nt-5694ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なHos2遺伝子を含まないDNA断片。
(3) 配列番号9に示す塩基配列中の1nt-2000ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号9に示す塩基配列中の4299nt-5594ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なSpt3遺伝子を含まないDNA断片。
【発明の効果】
【0010】
本発明により、コウジ酸生産を負に制御する遺伝子が新たに同定された。本発明によれば、従来技術と同等又はそれ以上のコウジ酸生産性を得ることができる。負の制御遺伝子は、遺伝子を破壊することにより、コウジ酸生産性をとりわけ顕著に高めることができる。本発明の方法によれば、通常の野生株ではコウジ酸を生産しにくい条件で培養しても、コウジ酸を生産することができる。例えば、Hst4遺伝子破壊麹菌株では、親株ないしはコントロール株と比較して200倍にまでコウジ酸生産量が増大する。その上さらに、特許文献1記載の技術と組み合わせることで、コウジ酸生産性をさらに高めることができると期待される。また、作製した遺伝子破壊株の特性解析の結果に基づき、コウジ酸高生産性の変異株を効率よく取得できる新規な育種方法も提供された。コウジ酸は、一時的に安全性が疑われたものの、現在では安全性も再確認され、美白化粧品の有効成分として商業利用されている。本発明は、コウジ酸の製造に大いに貢献するとともに、コウジ酸生合成経路の研究にも貢献し得る。
【図面の簡単な説明】
【0011】
【図1】遺伝子破壊用DNA断片の増幅のためのfusion PCRの概略を説明する図である。A、B、C、D、A2、D2、adeA-F、及びadeA-RはPCRプライマーである。
【図2】実施例で作製した遺伝子破壊株によるコウジ酸生産量を定性的に評価した結果を示す写真である。control株では菌糸の周縁部は赤色着色していないが、遺伝子破壊株では菌糸の外周部にまで赤色着色が確認された。
【図3】実施例で作製した遺伝子破壊株によるコウジ酸生産量を定量した結果を示すグラフである。
【図4】コウジ酸生産に不適な最小培地上で遺伝子破壊株によるコウジ酸生産を検出した結果を示す写真である。
【図5】ジェノトキシン存在下で遺伝子破壊株を培養し、ジェノトキシン非添加条件(Non-stress)での生育と相対評価した結果を示す図である。
【図6】HDAC阻害剤ニコチンアミドを添加したコウジ酸産生培地上でcontrol株を培養し、コウジ酸の生産量を調べた結果を示す写真である。
【発明を実施するための形態】
【0012】
本発明で対象となる微生物は、コウジ酸生産能を有する微生物であれば特に限定されない。そのような微生物の具体例として、ペニシリウム(Penicillium)属菌、アスペルギルス(Aspergillus)属菌等を挙げることができる。アスペルギルス属菌の種類も特に限定されず、麹菌を含む各種アスペルギルス属菌が広く包含され、具体例としては黄麹菌(Aspergillus oryzae)、黒麹菌(Aspergillus nigerなど)、及び白麹菌(Aspergillus usamii、Aspergillus kawachiiなど)等の麹菌や、フラブス菌(Aspergillus flavus)等を挙げることができる。
【0013】
Aspergillus oryzae RIB40ゲノムデータベース(http://nribf2.nrib.go.jp/)にAO080533000358として登録されている遺伝子は、SIR2ファミリー(Sirtuin 5 and related class III sirtuins)のヒストン脱アセチル化酵素(HDAC)をコードしている遺伝子として知られている。酵母が有する該遺伝子のホモログがHst4であることから、本発明ではこのAO080533000358遺伝子を「Hst4」又は「AOhst4」と呼ぶ。配列番号1及び2に示す配列は、上記の麹菌ゲノムデータベースに登録されているRIB40株のHst4遺伝子のcDNA配列及びコードされるHST4タンパク質のアミノ酸配列であり、配列番号3に示す配列は、RIB40株における該遺伝子及びその近傍のゲノム配列である。配列番号3中の2001nt-3784ntの領域がHst4遺伝子のコード領域である。また、RIB40株のHst4遺伝子は、NCBIのGenBankにもXM_001825249で登録されており、この配列を配列番号35(塩基配列)及び配列番号36(アミノ酸配列)に示す。
【0014】
上記データベースにAO080511000459として登録されている遺伝子は、ヒストン脱アセチル化酵素複合体の触媒成分をコードしている遺伝子として知られている。酵母が有する該遺伝子のホモログがHos2であることから、本発明ではこのAO080511000459遺伝子を「Hos2」又は「AOhos2」と呼ぶ。配列番号4及び5に示す配列は、麹菌ゲノムデータベースに登録されているRIB40株のHos2遺伝子のcDNA配列及びコードされるHOS2タンパク質のアミノ酸配列であり、配列番号6に示す配列は、RIB40株における該遺伝子及びその近傍のゲノム配列である。配列番号6中の2857nt-4317ntの領域がHos2遺伝子のコード領域である。また、RIB40株のHos2遺伝子は、NCBIのGenBankにもXM_003189254で登録されている。
【0015】
上記データベースにAO080525000462として登録されている遺伝子は、ヒストンアセチル基転移酵素のサブユニットをコードしている遺伝子として知られている。酵母が有する該遺伝子のホモログがSpt3であることから、本発明ではこのAO080525000462遺伝子を「Spt3」又は「AOspt3」遺伝子と呼ぶ。配列番号7及び8に示す配列は、麹菌ゲノムデータベースに登録されているRIB40株のSpt3遺伝子のcDNA配列及びコードされるSPT3タンパク質のアミノ酸配列であり、配列番号9に示す配列は、RIB40株における該遺伝子及びその近傍のゲノム配列である。配列番号9中の2001nt-4298ntの領域がSpt3遺伝子のコード領域である。また、RIB40株のSpt3遺伝子は、NCBIのGenBankにもXM_001727435で登録されており、この配列を配列番号37(塩基配列)及び配列番号38(アミノ酸配列)に示す。
【0016】
本発明において、「Hst4遺伝子」といった場合、配列番号2又は36と完全に同一のアミノ酸配列からなるタンパク質をコードし、イントロンを除いた塩基配列が配列番号1又は35と完全に同一であり、ゲノム上での配列が配列番号3に示す塩基配列と完全に同一である遺伝子に限定されるものではなく、天然に生じ得る変異を含む変異配列からなり、かつ、ヒストン脱アセチル化酵素活性を有するタンパク質をコードする遺伝子が包含される。「Hos2遺伝子」といった場合も同様であり、配列番号4〜6のみに限定されず、天然に生じ得る変異配列からなり、かつ、ヒストン脱アセチル化酵素活性を有するタンパク質をコードする遺伝子が包含される。「Spt3遺伝子」といった場合もまた同様であり、配列番号7〜9、37、38のみに限定されず、天然に生じ得る変異配列からなり、かつ、ヒストンアセチル基転移酵素のサブユニットとして機能するタンパク質をコードする遺伝子が包含される。以下、配列の一部が本願配列表とは異なるがもとの生理活性を大きく減弱せずに維持している変異配列からなる遺伝子ないしはタンパク質を、単に「天然の変異体」と呼ぶことがある。
【0017】
そのような変異配列は、通常、少数(例えば、1〜20個、1〜15個、1〜10個、又は1〜数個)の塩基ないしはアミノ酸の置換、欠失、挿入又は付加を含み、特に限定されないが、もとの配列との同一性は90%以上、例えば95%以上、あるいは98%以上である。ここで、配列の「同一性」とは、アミノ酸配列の場合であれば、比較すべき2つのアミノ酸配列のアミノ酸残基ができるだけ多く一致するように両アミノ酸配列を整列させ、一致したアミノ酸残基数を、全アミノ酸残基数で除したものを百分率で表したものである。上記整列の際には、必要に応じ、比較する2つの配列の一方又は双方に適宜ギャップを挿入する。このような配列の整列化は、例えばBLAST、FASTA、CLUSTAL W等の周知のプログラムを用いて行なうことができる。ギャップが挿入される場合、上記全アミノ酸残基数は、1つのギャップを1つのアミノ酸残基として数えた残基数となる。このようにして数えた全アミノ酸残基数が、比較する2つの配列間で異なる場合には、同一性(%)は、長い方の配列の全アミノ酸残基数で、一致したアミノ酸残基数を除して算出される。塩基配列の同一性についても同様である。
【0018】
本発明では、コウジ酸生産能を有する微生物において、Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子並びにこれらにコードされるタンパク質から成る群より選択される少なくとも1つの機能を阻害する。「遺伝子の機能を阻害する」とは、その遺伝子が本来コードしているmRNAないしはタンパク質の生成若しくは蓄積を低下若しくは欠失させること、又は、対象とする微生物細胞内の少なくとも一部のゲノム若しくは核において、その遺伝子が本来コードしているmRNAないしはタンパク質を生成できないようにゲノム上の対象遺伝子領域の少なくとも一部を改変することをいう。特定の遺伝子の機能を阻害するための遺伝子改変方法はこの分野で広く知られており、当業者であれば適宜選択して実行できる。具体例としては、アンチセンス法、RNAi、遺伝子破壊法等を挙げることができるが、これらに限定されない。また、タンパク質の機能を阻害するとは、例えば、そのタンパク質の阻害剤で微生物を処理することが挙げられる。
【0019】
例えば、本発明の方法では、上記3つの遺伝子のいずれか1つの機能を阻害するだけでもよいし、任意の2つないしは3つの遺伝子の機能を阻害してもよい。野生型の微生物株をHST4、HOS2及びSPT3タンパク質のうちの少なくとも1つの阻害剤で処理してもよいし、また、いずれかの遺伝子の機能が阻害されるように遺伝的に改変した微生物株をさらに阻害剤で処理してもよい。
【0020】
本発明の方法では、遺伝子の機能を阻害する手段として、上記3遺伝子の少なくともいずれかを破壊する方法が好ましい。「遺伝子を破壊する」とは、微生物ゲノムの少なくとも一部のアリル又は微生物体の少なくとも一部の核において、好ましくは全てのアリル又は核において、微生物ゲノム上の対象遺伝子のコード領域を欠失させること又は正常な遺伝子産物を産生できないように変異させることをいう。対象遺伝子の欠失は、コード領域の全体を欠失させてもよく、また一部を欠失させてもよい。全体を欠失させる場合には、対象遺伝子に隣接する領域もあわせて広く欠失させてもよい。一部を欠失させる場合には、特に限定されないが、対象遺伝子のコード領域の半分以上を欠失させることが好ましい。コード領域の変異により遺伝子を破壊する場合には、例えば、該コード領域の好ましくは中央よりも上流の部位にナンセンス変異又はフレームシフト変異を導入して、本来の生理活性を有しない変異タンパク質や全く無関係なタンパク質の配列とすることができる。対象遺伝子の一部又は全部を他の配列(例えばマーカー遺伝子配列)に置き換えてもよい。
【0021】
各種の微生物について、その微生物に適した遺伝子破壊方法が公知である。例えば、麹菌等の菌類の遺伝子破壊は、相同組換えを利用した手法により実施することができる(北本勝ひこ,丸山潤一:発酵・醸造食品の最新技術と機能性(シーエムシー出版,東京),95-103 (2006)等を参照)。Hst4遺伝子及び麹菌を例に具体的に説明すると、例えば、Hst4遺伝子の上流及び下流のゲノム領域を麹菌ゲノムからPCRにより増幅して上流側相同領域(第1の相同領域)及び下流側相同領域(第2の相同領域)を調製し、正常なHst4遺伝子を含まない配列の両末端に2つの相同領域をそれぞれ連結して遺伝子破壊用DNA断片を調製し、これを麹菌細胞に導入すればよい。麹菌が有する内在の酵素の働きで、相同な領域間で相同組換えが生じ、ゲノム中のHst4遺伝子領域が遺伝子破壊用DNA断片と置き換わるので、ゲノムからHst4遺伝子を欠失させることができる。
【0022】
「正常なHst4(又は、Hos2若しくはSpt3)遺伝子を含まない配列」とは、配列番号2(又は、配列番号5若しくは配列番号8)に示されるアミノ酸配列からなるタンパク質をコードする配列も、該タンパク質の天然の変異体をコードする配列も、いずれも含んでいない配列である。相同領域と連結する「正常なHst4(Hos2、Spt3)遺伝子を含まない配列」は、該遺伝子以外の他の遺伝子配列でもよいし、本来の生理活性を有するタンパク質をコードしないこれらの変異遺伝子(正常なヒストン脱アセチル化酵素として機能するタンパク質をコードしない変異Hst4若しくは変異Hos2遺伝子、又はヒストンアセチル基転移酵素のサブユニットとして正常に機能するタンパク質をコードしない変異Spt3遺伝子)であってもよい。他の遺伝子配列としてマーカー遺伝子を用いることで、形質転換体(遺伝子破壊株)のスクリーニングが容易になる。
【0023】
相同領域としては、通常、微生物ゲノム中の対応する領域と同一の塩基配列からなる断片が用いられるが、対応領域と一部が異なる配列であっても、微生物細胞内で相同組換えが生じる程度の配列同一性を有していれば、相同領域として使用することができる。すなわち、相同領域には、配列番号3、6又は9に示される塩基配列中の対応する部分と同一の塩基配列からなるものの他、該塩基配列において相同組換えが起きる程度に少数(例えば1個又は数個)の塩基が置換し、欠失し、挿入され又は付加された塩基配列からなる断片も包含される。相同領域と配列番号3、6又は9中の対応領域との同一性は90%以上、好ましくは95%以上、より好ましくは98%以上であり、最も好ましくは100%である。本発明において、ある領域と「相同な領域」とは、その領域と上記の通りの同一性を有する塩基配列からなる領域をいう。
【0024】
相同領域のサイズは特に限定されないが、鎖長が長い方が相同組換えの効率が高まり、短くなりすぎると効率が低下することがあるため、通常30塩基以上であり、好ましくは100塩基以上、より好ましくは200塩基以上、さらに好ましくは300塩基以上、特に好ましくは500塩基以上である。サイズの上限は特に限定されないが、DNA合成の便宜から通常は10000塩基以下、好ましくは2500塩基以下である。
【0025】
例えば、配列番号3に示す配列のうち、Hst4遺伝子コード領域は2001nt-3784ntであるから、1nt-2000ntの上流領域のうちの30塩基以上の領域を増幅して上流側相同領域(第1の相同領域)を、3785nt-5142ntの下流領域のうちの30塩基以上の領域を増幅して下流側相同領域(第2の相同領域)をそれぞれ得ることができる。下記実施例では、配列番号3中の620nt-2000ntを増幅して上流側相同領域とし、3789nt-5142ntを増幅して下流側相同領域としているが、これに限定されない。
【0026】
Hos2遺伝子の場合は、配列番号6に示す配列のうちHos2遺伝子のコード領域が2857nt-4317ntであるから、同様にして、1nt-2856ntから上流側相同領域を、4318nt-5694ntから下流側相同領域を増幅して得ることができる。また、Spt3遺伝子の場合は、配列番号9のうちSpt3遺伝子のコード領域が2001nt-4298ntであるから、1nt-2000ntから上流側相同領域を、4299nt-5594ntから下流側相同領域を増幅して得ることができる。下記実施例では、Hos2遺伝子破壊用DNAの作製のため、配列番号6中の1nt-1326ntを増幅して上流側相同領域とし、4320nt-5693ntを増幅して下流側相同領域としており、また、Spt3遺伝子破壊用DNA作製のため、配列番号9中の1170nt-1998ntを増幅して上流側相同領域とし、4668nt-5594ntを増幅して下流側相同領域としているが、これに限定されない。
【0027】
なお、配列番号3、6、9には、各遺伝子破壊用DNA断片の調製に十分と考えられる上下流領域を示しているが、これらよりもさらに上流ないし下流の配列は、上記したAspergillus oryzae RIB40ゲノムデータベース等から入手することができる。
【0028】
遺伝子破壊用DNA断片に適宜含めることができるマーカー遺伝子としては、使用する微生物株の栄養要求性に適合した栄養要求性相補遺伝子や、各種薬剤耐性遺伝子等が挙げられ、適宜選択して用いることができる。栄養要求性マーカーの場合は、遺伝子破壊の親株として、該当するマーカー遺伝子をもともと欠失している微生物株を用いる必要があるが、遺伝子破壊株を用いて製造された食品や化粧品、医薬部外品等に対する消費者の安心を得る観点からは栄養要求性マーカー遺伝子が有利である。一方、薬剤耐性マーカーは、使用する親株の遺伝的背景に制限がなく、より広い範囲の微生物株に対し利用することができ有利である。
【0029】
各種のマーカー遺伝子が公知であり、適宜選択して使用することができる。例えば、麹菌で用いられる栄養要求性マーカー遺伝子としては、adeA遺伝子(アデニン合成に関与し、アデニン要求性を相補)、sC遺伝子(硫酸(硫黄源)資化に関与し、硫酸(硫黄源)資化欠損株の栄養要求性を相補)、argB遺伝子(アルギニン合成に関与し、アルギニン要求性を相補)等が知られている。これら遺伝子の配列は公知であり、前掲のAspergillus oryzae RIB40ゲノムデータベースやNCBIのGenBank等から配列情報を入手できる。下記実施例では、adeAを欠損しアデニン要求性である麹菌株を遺伝子破壊の親株として使用し、マーカー遺伝子としてadeA遺伝子を用いることで、アデニン要求性を指標として遺伝子破壊株をスクリーニングしているが、これに限定されない。adeA遺伝子の配列は、Aspergillus oryzae RIB40ゲノムデータベースにはAO080527000383として、またGenBankにはAB121755のアクセッション番号で登録されている。配列番号34に示す配列は、adeA遺伝子及びその近傍のゲノム配列であり、下記実施例では360nt-2362ntの領域を用いて遺伝子破壊用DNA断片を調製しているが、これに限定されない。麹菌で用いられる薬剤耐性マーカー遺伝子としては、例えば、ptrA遺伝子(チアミンの代謝拮抗アナログであるピリチアミンに対する耐性遺伝子)等が挙げられるが、これに限定されない。
【0030】
上流側相同領域及び下流側相同領域とマーカー遺伝子DNAとの連結はfusion PCR法により行うことができる(図1参照)。上流側相同領域及び下流側相同領域をゲノムから増幅する際、マーカー遺伝子末端の部分領域とハイブリダイズする相補領域をプライマー対の一方(図1ではB及びC)に付加しておき、この相補領域において3つの断片をハイブリダイズさせて1本のDNA断片に融合し、これを鋳型としてPCRを行えば、[上流側相同領域]−[マーカー遺伝子]−[下流側相同領域]が連結した構造の遺伝子破壊用DNA断片を増幅することができる。
【0031】
遺伝子破壊用DNA断片は、適当なベクターに組み込んで微生物細胞に導入することができ、例えば麹菌細胞の場合はpUSC等のプラスミドベクターを用いることができる(Bioscience, Biotechnology, and Biochemistry, 61(8),1367-1369,1997)。あるいは、微生物細胞のプロトプラストを調製し、DNA断片のまま直接該プロトプラストに導入することもできる。麹菌を例にプロトプラスト法を簡単に記載すると、麹菌分生子を液体培地中で24時間程度培養した後、孔サイズ20〜30μm程度の不織布等で濾過して菌体を回収し、これを糸状菌細胞壁溶解酵素(キチナーゼ、キトビアーゼ、β-1,3-グルカナーゼ等)を含むプロトプラスト化溶液中で数時間振とう培養し、次いでガラスフィルター等で濾過して菌体残渣を除去することで、麹菌プロトプラストを得ることができる。プロトプラスト懸濁液と遺伝子破壊用DNAを混合し、数十分氷冷後、ポリエチレングリコールを混合して静置後、寒天選択培地上で培養すればよい。
【0032】
マーカーによるスクリーニング後に得られる形質転換体は、全ての核の対象遺伝子が破壊されたホモカリオンの破壊株か、又は一部の核の対象遺伝子が破壊され、少なくとも1つの核の対象遺伝子が破壊されずに残っているヘテロカリオンの破壊株であるが、遺伝子破壊用DNA断片が異所的に組み込まれた対象遺伝子非破壊株が得られることもある。従って、マーカーによるスクリーニング後、適宜サザン解析やPCR増幅産物の確認、ダイレクトシークエンシング等を行ない、目的通りの位置にDNA断片が挿入され対象遺伝子が破壊されていることを直接確認することが望ましい。
【0033】
本発明では、対象遺伝子の発現を完全に欠失できるホモカリオンの破壊株が好ましい。ホモカリオンの破壊株は、上記した遺伝子破壊方法で直接得ることができるし、また、得られたヘテロカリオンの破壊株について、全ての核の対象遺伝子が破壊されたホモカリオンの株となるまで純化の処理(例えば、選択培地上での植え継ぎ操作)を繰り返し行なってもよい。交配可能な微生物株を用いて遺伝子破壊株を作製した場合には、ヘテロのアレルを有する破壊株については、交配によりホモ破壊株を得ることもできる。
【0034】
本発明の方法をタンパク質の機能の阻害により実施する場合、使用し得るタンパク質阻害剤としては、以下のような阻害剤を挙げることができる。
(1) Sirtuin系(クラスIII)HDAC用阻害剤(Hst4を含むSirtuinの阻害剤)
Nicotinamide
2-Anilino-benzamide
(2) 古典的(クラスI又はクラスII)HDAC用阻害剤(Hos2を含む古典的HDACの阻害剤)
HC-toxin
Trichostatin A
酪酸(n-Butyrate)
【0035】
下記実施例では、麹菌のHst4遺伝子、Hos2遺伝子又はSpt3遺伝子を破壊することで、コウジ酸の生産性が向上することが確認されている。このことは、これら遺伝子を有するコウジ酸生産性微生物株を、HST4、HOS2又はSPT3タンパク質の阻害剤で処理することによっても、該微生物の生産性を高めることができることを示している。
【0036】
コウジ酸生産能を有する微生物において、Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子並びにこれらにコードされるHST4タンパク質、HOS2タンパク質及びSPT3タンパク質のうちの少なくともいずれかの機能を阻害すると、コウジ酸の生産性が高まる。すなわち、正常な各遺伝子を有するもとの親株又は阻害剤で処理しない同一の株と比較して、コウジ酸の生産量が増大する。本発明によるコウジ酸生産性を向上させる方法によれば、親株の30倍〜200倍以上にコウジ酸生産性を高めることも可能である。また、例えば特許文献1記載の公知技術と組み合わせて行なうことで、さらなる生産性の向上が期待される。
【0037】
コウジ酸の生産性の向上は、終濃度が1mM〜5mM程度となるように第二塩化鉄を加えた培地で微生物を培養し、第二塩化鉄とコウジ酸とのキレート化合物の生成による赤色発色の程度で確認することができる。また、微生物を液体培養し、培養液を適宜希釈した試料に1mM〜5mM程度の第二塩化鉄を添加し、495nmにおける吸光度を測定することでコウジ酸の定量も可能である。495nmの吸光度は、0.1〜1.0程度の範囲でコウジ酸濃度に比例する。
【0038】
本発明はまた、上記本発明の方法でコウジ酸の生産性が向上された微生物を培養し、該微生物が生産したコウジ酸を回収することを含む、コウジ酸の製造方法も提供する。該製造方法では、例えば、Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子のうちの少なくとも1つの機能が阻害されるように遺伝的に改変された麹菌等の微生物を、コウジ酸の生成に好ましい条件下で培養し、生成したコウジ酸を回収する。あるいは、コウジ酸生産能を有する微生物(野生型でも遺伝的に改変された微生物でもよい)を、HST4、HOS2又はSPT3タンパク質の阻害剤の共存下で培養し、生成したコウジ酸を回収してもよい。
【0039】
コウジ酸生産のための培地は公知である。コウジ酸の生産に適した培地は、一般に、窒素源として酵母エキス、ペプトン、無機窒素化合物(硝酸やアンモニウム塩)等を約0.1〜2.0%、炭素源としてグルコース、スクロース、デンプン等の糖類を約1〜20%含む。その他、無機塩、ビタミン類や、分散剤、消泡剤等を適宜使用してよい。pHは通常5.0〜6.5程度である。培養温度は、微生物の種類により適宜選択され、麹菌の場合は通常25℃〜35℃程度である。
【0040】
生成したコウジ酸は、公知の手法により回収、精製できる。例えば、再結晶、アフィニティークロマトグラフィー、イオン交換クロマトグラフィー、ゲル濾過等が挙げられる。また、麹菌を用いた商業的なコウジ酸製造法が各種開示されている(特公昭60-36753、特開平5-76378など)。
【0041】
本願発明者らがコウジ酸生産の負の制御因子として同定した遺伝子は、ヒストン脱アセチル化酵素やヒストンアセチル基転移酵素のサブユニットをコードしているが、これら遺伝子が破壊された麹菌はジェノトキシン(genotoxin、遺伝毒性物質)に対する感受性が高まっていることが確認された。自然界から分離したコウジ酸生産性微生物、又は該分離菌若しくは既存の微生物を変異処理したものの中から、ジェノトキシンに対する感受性の高い菌株を選抜することで、コウジ酸生産性の高い微生物株を取得し得ることを示している。すなわち、本発明は、コウジ酸生産能を有する微生物をジェノトキシンで処理し、ジェノトキシン感受性が高い微生物株を選抜することを含む、コウジ酸生産性の高い微生物の育種方法を提供する。
【0042】
使用可能なジェノトキシンは特に限定されず、DNAを損傷する作用があることが知られているいかなる物質であってもよい。例えば、トポイソメラーゼ阻害剤(カンプトテシン等)、DNAアルキル化剤(メチルメタンスルホン酸、エチルメタンスルホン酸、アジ化ナトリウム等)等を挙げることができる。
【0043】
該育種方法に供する微生物としては、上述の通り、自然界から分離された微生物をそのまま用いてもよいし、また、変異処理した微生物であってもよい。変異処理の方法は特に限定されず、例えば放射線処理(γ線、β線、X線、中性子線等)、紫外線処理、化学物質処理(エチルメタンスルホン酸等)等を挙げることができ、いずれの方法であってもよい。
【0044】
高濃度のジェノトキシンは麹菌等の微生物に対し致死的であるが、中程度の濃度では分生子の発芽及び生育を数時間遅らせる。そこで、ジェノトキシンが概ね50%程度生育を阻害する濃度で培地に添加し、該培地に微生物又はその胞子を植え、適当な時間(例えば7時間〜10時間程度)インキュベートすることで、成長度合い(例えば菌糸の成長度合い)によって感受性の高いものと感受性が低いないしは通常レベルであるものとを区別することができる。例えば、麹菌分生子をジェノトキシン添加培地に播種し、一定時間インキュベート後、フィルターを通して出芽伸長したものを取り除くことで、ジェノトキシン感受性の高い株を濃縮することができる。ジェノトキシン処理濃度は、ジェノトキシン非添加の培地とジェノトキシンを各種濃度で添加した培地を調製し、微生物の生育(分生子の発芽や菌糸の進展の度合いなど)を確認して、選抜に適した濃度を適宜設定することができる。
【実施例】
【0045】
以下、本発明を実施例に基づきより具体的に説明する。もっとも、本発明は下記実施例に限定されるものではない。
【0046】
adeAマーカー遺伝子との遺伝子置換により、麹菌のAOhst4遺伝子(AO080533000358)、AOhos2遺伝子(AO080511000459)及びAOspt3遺伝子(AO080525000462)の破壊を試みた。Fusion PCR法及びプロトプラスト法は、北本勝ひこ,丸山潤一:発酵・醸造食品の最新技術と機能性(シーエムシー出版,東京), 95-103 (2006)に記載の方法に準じて行なった。
【0047】
遺伝子破壊用DNA断片の作成
麹菌ゲノムDNAを鋳型としたPCRにより、AOhst4遺伝子、AOhos2遺伝子及びAOspt3遺伝子の5'側上流領域及び3'側下流領域について各1kb程度のDNA領域を増幅した。また、遺伝子破壊株の選択用のマーカーとしてアデニンの合成に関わるadeA栄養要求性マーカー遺伝子(AO080527000383)とその上流及び下流の一部を含む断片もPCRで増幅した。その後、5'側上流領域, adeA, 3'側下流領域を混合してPCRを行い、5'側上流領域(上流側相同領域), adeA, 3'側下流領域(下流側相同領域)の順にDNA断片をつなぎ合わせた遺伝子破壊用DNA断片をFusion PCRにより作製した。尚、Fusion PCRの模式図を図1に記す。また、用いたプライマー配列を表1に示す。表中のプライマーの名称は、図1記載のプライマーに対応する。
【0048】
Fusion PCRに用いる5'側上流領域,3'側下流領域の各DNA断片を作製の際に、Fusion PCRでadeA遺伝子との断片との連結部になる位置にアニールするプライマー(BとC)のテール部に、adeA断片の5'末端配列及び3'末端配列にハイブリダイズする相補配列(表1中の小文字の部分)を付加しプライマーを設計した。なお、AOhst4及びAOhos2の破壊には、5'側上流領域がadeA遺伝子配列の3'末端に連結された遺伝子破壊用断片を使用し、AOspt3の破壊には、5'側上流領域がadeA遺伝子配列の5'末端に連結された遺伝子破壊用断片を使用した。
【0049】
【表1】
【0050】
1st PCR
5'側上流領域, adeA, 3'側下流領域の各DNA断片は、KOD PLUS Version 1(東洋紡)で増幅した。テンプレートは、全てアスぺルギルス・オリゼ野生株RIB40の染色体DNAを用いた(QIAGEN社製のDNeasy Plant Maxi Kitを用いて抽出)。
【0051】
【表2】
【0052】
上記反応液50mlを0.2mlPCRチューブ内で混合してDNA Thermal Cyclerにセットし、以下のような温度設定によりPCRを行った。
94℃、2分 1サイクル
94℃、20秒 55℃、30秒 68℃ 2分 30サイクル
68℃、5分 4℃、O/N 1サイクル
【0053】
PCR反応液50μlをQIAquick PCR purification kit(キアゲン)を用いて、Elution Buffer 50μlをカラムに通し、カラムからDNAの溶出を行った(増幅産物の精製)。
【0054】
Fusion PCR
精製した5'側上流領域, adeA, 3'側下流領域の各DNA断片の濃度を、NanoDrop ND-1000 Spectrophotometerを用いて計測し、それぞれのDNA断片を50 ng/μlになるように調整した後、5'側上流領域断片 : adeA断片 : 3'側下流領域断片=1 : 1.6 :1になるようにして、且つ3つのDNA断片の合計液量が3.6μlになるように混合し、これをTemplateとして用いて、以下の反応液でFusion PCRを行った。
【0055】
【表3】
【0056】
上記反応液50mlを0.2mlPCRチューブ内で混合してDNA Thermal Cyclerにセットし、以下のような温度設定によりPCRを行った。
94℃、2分 1サイクル
94℃、20秒 55℃、30秒 68℃ 5分 30サイクル
68℃、5分 4℃、O/N 1サイクル
【0057】
PCR反応液50μlをQIAquick PCR purification kit(キアゲン)を用いて、Elution Buffer 50μlをカラムに通し、カラムからDNAの溶出を行った。また、得られた精製遺伝子破壊用DNA断片は、アガロースゲル電気泳動により非特異バンドがほとんどないことを確認した。また、上記のPCRを2チューブ以上行う事により、遺伝子破壊用DNA断片を20μg程度作製した。
【0058】
形質転換
アスペルギルス・オリゼ NSR-ΔlD2株を宿主麹菌として利用した。この株の分生子縣濁液を、アデニンを0.05%添加した酵素生産培地(1L中の組成で、グルコース 20g, トリプトン 1g(ディフコ), 酵母エキス 5g (ディフコ), NaNO3 1g, K2HPO4 0.5g, MgSO4・7H2O 0.5g, FeSO4・7H2O 0.01g, pH6.0)に植菌し、30℃で24時間振盪培養した。菌体を、ミラクロスを用いて集菌後、菌体を滅菌0.8 M NaCl/10mM Phosphate buffer (pH6.0)で3回洗浄した。その後、菌体をマイレックスHV(ミリポア)でフィルター滅菌した30mlプロトプラスト化溶液(100mg / 30ml Yatalase(タカラバイオ)になるよう0.8 M NaCl/10mM Phosphate buffer (pH6.0)にYatalaseを溶解)の入った50ml容の滅菌済み遠心チューブに移して懸濁し、30℃、80rpm、3時間振盪しプロトプラスト化反応を行った。滅菌した3G1ガラスフィルターにてろ過し、濾液中のプロトプラストを5 分間遠心分離することで沈殿を得た。30mlの滅菌0.8M NaClにてプロトプラストを洗浄し3,000rpm、4 ℃で5 分間遠心分離することで沈殿を得た。このプロトプラストを滅菌0.8M NaCl 10mlとSolution I (0.8 M NaCl, 10 mM CaCl2, 10 mM Tris-HCl 、pH 8.0) 20mlに懸濁し、懸濁液中のプロトプラスト数をトーマの血球計数機で計測した。計測後、3,000rpm、4 ℃で5 分間遠心分離することで沈殿を得た。
【0059】
このプロトプラストを2.4×108 protoplast / ml Solution IとなるようにSolution Iを加えて懸濁した。プロトプラスト懸濁液50mlを滅菌した2ml容のマイクロチューブに移し、それぞれにSolution II (40% (w/v) PEG4000, 50 mM CaCl2, 10 mM Tris-HCl 、pH8.0)12.5 μlと前述した形質転換用DNA溶液各5μl(DNA量として2μg以上)を加えて優しく混合した後、氷上で30min放置した。この溶液に、Solution II 500μlを加えて優しく混合し、室温で15min放置した。この溶液にSolution I 1mlを加えて転倒混和した。この溶液を0.8MのNaClを添加した選択寒天培地(1L中の組成で、Glucose 20g, NH4Cl 2g, (NH4)2SO4 1g, KCl 0.5g , KH2PO4 1g, MgSO4・7H2O 0.5g, FeSO4・7H2O 0.02g, L-methionine 1.5g、pH5.5)に250μlをスポット植菌し、そこに0.8MのNaClを添加した選択軟寒天培地約10mlを重層した。その後、コロニーを形成するまで30℃で培養した。
【0060】
形質転換後、再生した菌体のうちシングルコロニーを形成した菌体の菌糸を火炎滅菌したピンセットで採取し、選択培地に植え継いだ。その後、30℃で分生子を形成するまで培養した。その後、コロニーの端から菌糸を火炎滅菌したピンセットで選択培地に植え継いだ。この植え継ぎ操作を少なくとも3回以上行うことで核の純化を行った。
【0061】
遺伝子破壊株の確認
形質転換体から遺伝子破壊株の確認は、Fusion PCRに用いたAとDプライマー並びに、欠失破壊の対象とした遺伝子の内500bp程度を増幅できるように設計したFとGプライマー(下記表4)を用いてPCRを行った。各断片の増幅には、KOD FX(東洋紡)を用いた。テンプレートは、滅菌した1.5ml容のマイクロチューブに100μlずつ分注したBufferA(1M KCl、10mM EDTA、100mM Tris-HCl、pH8.5)に各形質転換体の菌糸と胞子をそれぞれ懸濁し、5分間ボルテックスした後に、95℃で10分間煮沸後、さらに5分間ボルテックスし、15000rpm、 4℃で1分間遠心分離して得られた上清1μlを用いた。PCRの結果、欠失破壊用DNA断片に相当する大きさのDNA断片の増幅が認められ、且つFG間の増幅が認められなかったものをホモカリオン欠失破壊株、野生株の場合と同じ大きさのDNA断片の増幅のみが見られ、且つFG間の増幅がみられたものはadeAがectopicに取り込まれた非欠失破壊株、欠失破壊用DNA断片に相当する大きさのDNA断片並びに野生株の場合と同じ大きさのDNA断片の増幅とFG間の増幅がみられたものは、ヘテロカリオン欠失株とした。以後の実験では、ホモカリオン欠失破壊株を使用した。
【0062】
【表4】
【0063】
遺伝子破壊株におけるのコウジ酸産生量の定性的評価
プレート上でのコウジ酸産生への影響は、遺伝子破壊株を、コウジ酸を産生する条件で生育させ、培地に分泌されたコウジ酸と予め培地に混入させた塩化第二鉄とのキレートによる赤色発色により判断した。
【0064】
5mMになるように塩化第二鉄を添加したコウジ酸産生寒天培地(1L中の組成で、glucose 100g, 酵母エキス 1g(ディフコ)、KH2PO4 1g、MgSO4・7H2O 0.5g、pH6.0)を用いた。1×108 遺伝子破壊株胞子/ mlに調整した胞子懸濁液1μlをプレートの中央部に植菌し、30℃の恒温槽中に一週間静置した。
【0065】
NSR-ΔlD2株のadeA遺伝子のみを相補した株(以下control株とする)を対照とし、AOhst4、AOhos2及びAOspt3欠失破壊変異株における培地の着色度合を比較した結果、control株と比較し、AOhst4、AOhos2及びAOspt3欠失破壊変異株において培地の赤色強度の増加が認められた(図2)。尚図2中には、AOhst4(AO080533000358)及びAOhos2(AO080511000459)遺伝子欠失株をそれぞれ358, 459と表示する。培地の赤色強度の増加は、コウジ酸生産量の増加を意味する。すなわち、上記3つの遺伝子は、コウジ酸の生産を負に制御することを示唆している。
【0066】
遺伝子破壊株におけるコウジ酸生産量の定量
上述のようにコウジ酸は、第二塩化鉄とのキレート化合物の生成による赤色の発色により検出することが可能である。さらにコウジ酸量は、培養液を適宜希釈した試料に最終濃度として1mM〜5mMになるように塩化第二鉄溶液を添加した液の、495nmにおける吸光度を測定することで定量することができる。また、この495nmの吸光度は、0.1〜1.0程度の範囲でコウジ酸濃度に比例する。このような検出法により麹菌培養液を適宜希釈し、そのコウジ酸濃度の定量が可能である。
【0067】
100mlのバッフル付フラスコに20mlの上記コウジ酸生産培地をいれ、遺伝子欠失破壊株あるいは上述のcontrol株胞子を105 conidia/ mlコウジ酸生産培地になるよう植菌した。これを7本用意した。培養条件は、7本とも30℃、120rpmであった。
【0068】
7本の中から1日につき1本の培養液を全てミラクロス(メルク)でろ過し、濾液を得た。この操作を7日間毎日行った。得られた、7つの試料を上述の塩化第二鉄とのキレートによる発色を用いて495nmでの吸収により測定した。尚、この測定では、コウジ酸生産培地をブランクとした。コウジ酸定量結果をグラフで図3に示す。尚、図3中には、AOhst4(AO080533000358)及びAOhos2(AO080511000459)遺伝子欠失株をそれぞれ358, 459と表示する。
【0069】
AOhos2遺伝子破壊株では、control株と比較し30倍のコウジ酸生産量の増加が認められた。また、AOhst4遺伝子破壊株では、control株と比較し200倍のコウジ酸生産量の増加が認められた。
【0070】
上記2つの遺伝子は、それぞれヒストン脱アセチル化遺伝子のモチーフを有している。一般にヒストンのアセチル化は、クロマチン構造を緩和する方向に働き、結果転写因子や転写装置が遺伝子近傍にアクセスできるようになり遺伝子発現を亢進することが知られている。一方で、ヒストン脱アセチル化遺伝子は、上記のアセチル化とは逆で、クロマチンを凝集する方向に働き、結果転写因子や転写装置が遺伝子にアクセスできなくなり遺伝子発現が抑制される。これらの現象は、染色体の幅広い領域で起こっており遺伝子発現に大きな影響を与えることが知られている。すなわち、AOhst4及びAOhos2Aは、上記の結果と同様に遺伝子発現の負の制御因子であるHDACがコウジ酸生産を負に制御していると考えられる。
【0071】
最小培地上でのコウジ酸生産性
下記の組成に5mM になるように塩化第二鉄を添加したM培地(最小培地)を作製し、5*107 conidia/mlに調整した分生子懸濁液を培地の中心に2μl植菌し、30℃、飽和蒸気圧で5日間培養した。
【0072】
【表5】
【0073】
図4は、5日間培養後のプレートの裏面を撮影した写真である。上記の最小培地では通常の麹菌はコウジ酸を生産できない。control株では赤色発色は認められなかったが、一方、欠失変異株ではいずれも赤色発色が認められ、コウジ酸の生産が確認された。
【0074】
ジェノトキシンに対する感受性
上記で得られた遺伝子破壊株について、ジェノトキシン存在下での菌糸の成長を調べた。ジェノトキシンとして、トポイソメラーゼ阻害剤であるカンプトテシン(CPT)及びDNAのアルキル化剤であるメチルメタンスルフォネート(MMS)を用いた。終濃度1μMのCPT又は終濃度0.1%のMMSを加えたM培地(最小培地)の中央に麹菌分生子を播種し、30℃で5日間培養した。円状に繁殖した菌糸の直径を測定し、ジェノトキシン非添加(Non-stress)の培地上での直径と相対評価した。結果を図5に示す。
【0075】
AOhst4破壊株において、CPT及びMMSに対する感受性が上昇していた。また、AOhos2破壊株においてMMSに対する感受性が上昇していた。hos2とhst4は、機構は異なるが共にDNA修復系に関与している可能性が考えられた。
【0076】
ジェノトキシンによるコウジ酸高生産株のスクリーニングの検討
control株の分生子と遺伝子破壊株の分生子を表6に示すInitial conditionで混和し、5μM CPTまたは0.05%MMS存在下で穏やかに振とうしながら30℃、8時間培養した。その後、孔経が5μmのフィルターで濾過し、ろ液を0.3% Triton-X100を含んだプレートに塗布し、30℃で4-5日間培養した。得られたコロニーの形状(分生子の着生が悪いものを破壊株と判断)から各破壊株とコントロール株の比を算出した。
【0077】
【表6】
【0078】
表6に示す通り、CPT処理及びMMS処理のいずれにおいても、コウジ酸高生産性である遺伝子破壊株の割合がinitial conditionよりも増大しており、破壊株の分生子が濃縮されていた。この結果は、変異原処理した野生型麹菌株からジェノトキシンによるスクリーニングでコウジ酸高生産性の変異株を取得できる可能性を示している。
【0079】
HDAC阻害剤によるコウジ酸生産性の向上
上記のコウジ酸産生培地に0, 50, 100mMのニコチンアミドを添加した培地を作成し、5*107 conidia/ ml に調整したcontrol株の分生子懸濁液を培地の中心に2μl植菌し、30℃、飽和蒸気圧で5日間培養した。
【0080】
図6は2日間と5日間培養後のプレートの裏面を撮影した写真である。ニコチンアミドを添加しないものに比べ、添加したものでは培地の赤色強度の顕著な増加が認められた。これは、コウジ酸生産量の増加を意味する。すなわち、HDAC活性を阻害によりコウジ酸の生産を増加させることを示している。
【特許請求の範囲】
【請求項1】
コウジ酸生産能を有する微生物において、Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子並びにこれらにコードされるタンパク質から成る群より選択される少なくとも1つの機能を阻害する工程を含む、微生物のコウジ酸生産性を向上させる方法。
【請求項2】
前記工程が、Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子から成る群より選択される少なくとも1つの遺伝子を破壊することにより行なわれる請求項1記載の方法。
【請求項3】
前記工程が、HST4、HOS2及びSPT3から成る群より選択される少なくとも1つのタンパク質の阻害剤で前記微生物を処理することにより行なわれる請求項1記載の方法。
【請求項4】
前記阻害剤が、サーチュイン阻害剤及び古典的ヒストン脱アセチル化酵素阻害剤から成る群より選択される少なくとも1つである請求項3記載の方法。
【請求項5】
前記サーチュイン阻害剤が、ニコチンアミド及び2-アニリノベンズアミドから成る群より選択される少なくとも1種であり、前記古典的ヒストン脱アセチル化酵素阻害剤が、HC毒素、トリコスタチンA及びn-酪酸から成る群より選択される少なくとも1種である請求項4記載の方法。
【請求項6】
前記微生物が麹菌を含むアスペルギルス属菌又はペニシリウム属菌である請求項1ないし5のいずれか1項に記載の方法。
【請求項7】
前記微生物が麹菌である請求項6記載の方法。
【請求項8】
請求項1ないし7のいずれか1項に記載の方法でコウジ酸の生産性が向上された微生物を培養し、該微生物が生産したコウジ酸を回収することを含む、コウジ酸の製造方法。
【請求項9】
Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子から成る群より選択される少なくとも1つの遺伝子の機能が阻害されている、コウジ酸生産性の高い微生物。
【請求項10】
前記少なくとも1つの遺伝子が破壊されたゲノムを有する請求項9記載の微生物。
【請求項11】
麹菌を含むアスペルギルス属菌又はペニシリウム属菌である請求項9又は10記載の微生物。
【請求項12】
麹菌である請求項11記載の微生物。
【請求項13】
コウジ酸生産能を有する微生物をジェノトキシンで処理し、ジェノトキシン感受性が高い菌株を選抜することを含む、コウジ酸生産性の高い微生物の育種方法。
【請求項14】
変異原処理した微生物をジェノトキシンで処理する請求項13記載の方法。
【請求項15】
前記ジェノトキシンが、トポイソメラーゼ阻害剤又はDNAアルキル化剤である請求項13又は14記載の方法。
【請求項16】
前記微生物が麹菌を含むアスペルギルス属菌又はペニシリウム属菌である請求項13ないし15のいずれか1項に記載の方法。
【請求項17】
前記微生物が麹菌である請求項16記載の方法。
【請求項18】
配列番号3に示す塩基配列中の1nt-2000ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号3に示す塩基配列中の3785nt-5142ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なHst4遺伝子を含まないDNA断片を、コウジ酸生産能を有する微生物の細胞に導入し、該DNA断片と微生物ゲノムとの間で相同組換えを生じさせることを含む、コウジ酸生産性が向上した微生物の製造方法。
【請求項19】
配列番号6に示す塩基配列中の1nt-2856ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号6に示す塩基配列中の4318nt-5694ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なHos2遺伝子を含まないDNA断片を、コウジ酸生産能を有する微生物の細胞に導入し、該DNA断片と微生物ゲノムとの間で相同組換えを生じさせることを含む、コウジ酸生産性が向上した微生物の製造方法。
【請求項20】
配列番号9に示す塩基配列中の1nt-2000ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号9に示す塩基配列中の4299nt-5594ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なSpt3遺伝子を含まないDNA断片を、コウジ酸生産能を有する微生物の細胞に導入し、該DNA断片と微生物ゲノムとの間で相同組換えを生じさせることを含む、コウジ酸生産性が向上した微生物の製造方法。
【請求項21】
前記微生物が麹菌を含むアスペルギルス属菌又はペニシリウム属菌である請求項18ないし20のいずれか1項に記載の方法。
【請求項22】
前記微生物が麹菌である請求項21記載の方法。
【請求項23】
下記のいずれかのDNA断片。
(1) 配列番号3に示す塩基配列中の1nt-2000ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号3に示す塩基配列中の3785nt-5142ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なHst4遺伝子を含まないDNA断片。
(2) 配列番号6に示す塩基配列中の1nt-2856ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号6に示す塩基配列中の4318nt-5694ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なHos2遺伝子を含まないDNA断片。
(3) 配列番号9に示す塩基配列中の1nt-2000ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号9に示す塩基配列中の4299nt-5594ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なSpt3遺伝子を含まないDNA断片。
【請求項1】
コウジ酸生産能を有する微生物において、Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子並びにこれらにコードされるタンパク質から成る群より選択される少なくとも1つの機能を阻害する工程を含む、微生物のコウジ酸生産性を向上させる方法。
【請求項2】
前記工程が、Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子から成る群より選択される少なくとも1つの遺伝子を破壊することにより行なわれる請求項1記載の方法。
【請求項3】
前記工程が、HST4、HOS2及びSPT3から成る群より選択される少なくとも1つのタンパク質の阻害剤で前記微生物を処理することにより行なわれる請求項1記載の方法。
【請求項4】
前記阻害剤が、サーチュイン阻害剤及び古典的ヒストン脱アセチル化酵素阻害剤から成る群より選択される少なくとも1つである請求項3記載の方法。
【請求項5】
前記サーチュイン阻害剤が、ニコチンアミド及び2-アニリノベンズアミドから成る群より選択される少なくとも1種であり、前記古典的ヒストン脱アセチル化酵素阻害剤が、HC毒素、トリコスタチンA及びn-酪酸から成る群より選択される少なくとも1種である請求項4記載の方法。
【請求項6】
前記微生物が麹菌を含むアスペルギルス属菌又はペニシリウム属菌である請求項1ないし5のいずれか1項に記載の方法。
【請求項7】
前記微生物が麹菌である請求項6記載の方法。
【請求項8】
請求項1ないし7のいずれか1項に記載の方法でコウジ酸の生産性が向上された微生物を培養し、該微生物が生産したコウジ酸を回収することを含む、コウジ酸の製造方法。
【請求項9】
Hst4遺伝子、Hos2遺伝子及びSpt3遺伝子から成る群より選択される少なくとも1つの遺伝子の機能が阻害されている、コウジ酸生産性の高い微生物。
【請求項10】
前記少なくとも1つの遺伝子が破壊されたゲノムを有する請求項9記載の微生物。
【請求項11】
麹菌を含むアスペルギルス属菌又はペニシリウム属菌である請求項9又は10記載の微生物。
【請求項12】
麹菌である請求項11記載の微生物。
【請求項13】
コウジ酸生産能を有する微生物をジェノトキシンで処理し、ジェノトキシン感受性が高い菌株を選抜することを含む、コウジ酸生産性の高い微生物の育種方法。
【請求項14】
変異原処理した微生物をジェノトキシンで処理する請求項13記載の方法。
【請求項15】
前記ジェノトキシンが、トポイソメラーゼ阻害剤又はDNAアルキル化剤である請求項13又は14記載の方法。
【請求項16】
前記微生物が麹菌を含むアスペルギルス属菌又はペニシリウム属菌である請求項13ないし15のいずれか1項に記載の方法。
【請求項17】
前記微生物が麹菌である請求項16記載の方法。
【請求項18】
配列番号3に示す塩基配列中の1nt-2000ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号3に示す塩基配列中の3785nt-5142ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なHst4遺伝子を含まないDNA断片を、コウジ酸生産能を有する微生物の細胞に導入し、該DNA断片と微生物ゲノムとの間で相同組換えを生じさせることを含む、コウジ酸生産性が向上した微生物の製造方法。
【請求項19】
配列番号6に示す塩基配列中の1nt-2856ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号6に示す塩基配列中の4318nt-5694ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なHos2遺伝子を含まないDNA断片を、コウジ酸生産能を有する微生物の細胞に導入し、該DNA断片と微生物ゲノムとの間で相同組換えを生じさせることを含む、コウジ酸生産性が向上した微生物の製造方法。
【請求項20】
配列番号9に示す塩基配列中の1nt-2000ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号9に示す塩基配列中の4299nt-5594ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なSpt3遺伝子を含まないDNA断片を、コウジ酸生産能を有する微生物の細胞に導入し、該DNA断片と微生物ゲノムとの間で相同組換えを生じさせることを含む、コウジ酸生産性が向上した微生物の製造方法。
【請求項21】
前記微生物が麹菌を含むアスペルギルス属菌又はペニシリウム属菌である請求項18ないし20のいずれか1項に記載の方法。
【請求項22】
前記微生物が麹菌である請求項21記載の方法。
【請求項23】
下記のいずれかのDNA断片。
(1) 配列番号3に示す塩基配列中の1nt-2000ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号3に示す塩基配列中の3785nt-5142ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なHst4遺伝子を含まないDNA断片。
(2) 配列番号6に示す塩基配列中の1nt-2856ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号6に示す塩基配列中の4318nt-5694ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なHos2遺伝子を含まないDNA断片。
(3) 配列番号9に示す塩基配列中の1nt-2000ntの領域内の連続する30塩基以上の領域と相同な領域からなる第1の相同領域と、配列番号9に示す塩基配列中の4299nt-5594ntの領域内の連続する30塩基以上の領域と相同な領域からなる第2の相同領域とを含み、正常なSpt3遺伝子を含まないDNA断片。
【図1】

【図2】

【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2013−17408(P2013−17408A)
【公開日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願番号】特願2011−151980(P2011−151980)
【出願日】平成23年7月8日(2011.7.8)
【出願人】(301025634)独立行政法人酒類総合研究所 (55)
【Fターム(参考)】
【公開日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願日】平成23年7月8日(2011.7.8)
【出願人】(301025634)独立行政法人酒類総合研究所 (55)
【Fターム(参考)】
[ Back to top ]