
微生物蛋白質と、この蛋白質を産生する微生物と、該蛋白質のワクチンおよび結核検出での利用
【課題】ワクチンあるいは結核特異的抗体を検出するために使用することができる蛋白質。
【解決手段】分子量が28,779DaのMycobacterium tuberculosis(結核菌)蛋白質と、この蛋白質の配列の少なくとも一部を含むハイブリッド蛋白質。
【解決手段】分子量が28,779DaのMycobacterium tuberculosis(結核菌)蛋白質と、この蛋白質の配列の少なくとも一部を含むハイブリッド蛋白質。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は微生物蛋白と、この蛋白を産生する微生物とに関するものである。
本発明はさらに、この蛋白のワクチンまたは結核検出での利用に関するものである。
【背景技術】
【0002】
結核は現在でも世界中で公衆衛生上の問題になっている。結核を直接の原因とする年間の死者数は約300 万人で、新たな結核の件数は約1500万人である。この結核による死者の数は先進国でも高く、例えばフランスでは年間1500人に達する。公式な数字と正確な分析結果との差に関するルジョーの評価方法を考慮すると、この数字は2分の1から3分の1に過少評価されている。最近の結核患者数の増加あるいは少なくともこの疾患の発生頻度が減少せず、横ばいである状況は感染性HIV/AIDS疾患の拡大との関連で考える必要がある。結論として、結核はフランスや先進国でも依然として頻度の高い主要な感染症であり、発展途上国では単一の病気による死亡の主要な原因になっている。
【0003】
現在、患者から採取したサンプル中の細菌の培養で判定する診断結果の確度は結核患者の半分以下である。結核患者の80〜90%を占める細菌の検出が最も容易な肺結核の場合でも、喀出物の検査で陽性が示されるのは患者の半数に満たない。
【0004】
PCR(ポリメラーゼチェーンリアクションによる増幅)のような感度の高い方法の開発で常に問題になることはサンプル採取が難しいことである。すなわち、女性や子供は一般に唾を吐くことがなく、幼児のサンプルを得るには特殊な医学的介入(例えば神経節の生検や腰部穿刺による頭脊柱液の採取など)を必要とすることが多い。
しかも、サンプルの起源を管理できないため、サンプルをPCR法で使用することができないといったPCR法そのものに起因する制限が存在する。
【0005】
さらに、顕微鏡検査と培養による伝統的な細菌学的診断法は感度が低い(サンプル中に104〜105オーダーの細菌が必要)ため、既にかなり細菌が繁殖している状態、すなわち、かなり病気が進行した状態でなければならない。
従って、細菌自体の検出が困難または不可能なこの病気の診断では、Mycobacterium tuberculosis(結核菌)に対して特異的な抗体を用いる検出方法が大きな助けになるものと思われる。
研究者達は結核の血清学的診断方法を確立すべく努力してきた。この分野で行われた研究の概論は下記文献に記載されている。
【特許文献1】国際特許PCT WO92/21758号公報
【0006】
大抵の従来法では、生化学的特性を利用して蛋白質を予め単離する。本発明者はこの蛋白を単離した後に初めてこの蛋白の肺結核患者検出能力をテストした。
上記特許文献1(国際特許PCT WO92/21758号公報)には、結核患者または生菌で免疫処理したモルモットに由来する血清を用いて結核感染症の代表的な抗原を確実に選択する方法が開示されている。この方法では従来の試験法とは異なり、分子量44.5〜47.5kDのM.bovis 蛋白質を単離することができる。
これら蛋白質の1つのN末端に位置する17個のアミノ酸は下記のように決定されている:
【化1】

【0007】
下記文献にはこの国際特許に記載された結果の内容が要約されている。
【非特許文献1】ローマン達、1993、Infection and Immunity, 61, 742-750
【0008】
この論文は特に上記45〜47kDの複合蛋白質に対してウサギを免疫処理して得られる兎のポリクロナル免疫血清を用いた競合ELISA 試験について述べたものである。
これと平行して、ヤコブ(JACOBS)達によって結核菌の遺伝子ライブラリーが作られた(下記非特許文献2参照)。このライブラリーは多数の異なるクローンを含む。
【非特許文献2】1991, Method Enzymol., 204, 537-557
【0009】
別のマイコバクテリア種である癩菌(M. leprae) 由来の蛋白質がワイエル(WIELES)達によって同定されている (下記非特許文献3参照)。
【非特許文献3】Infection and Immunity, 62, 252-258, 1994
【0010】
この蛋白は43L とよばれ、ヌクレオチド配列から推定される分子量は約25.5Daである。そのN末端は M. bovis BCG において同定された45〜47kDa の複合蛋白質のN末端と47%の相同性を有し、その17個のアミノ酸配列は上記のとおりである。
既に述べたように、ヒトの医学分野では診断上の観点および治療上の観点では、マイコバクテリア、特に結核菌によって産生される蛋白質の正確な同定が主たる興味の対象である。
すなわち、実際に直面している未解決の課題は多くの疾患に対するワクチンの製造である。
もう1つの課題は、マイコバクテリアによって引き起こされる病気、例えば結核の検出である。
【発明の開示】
【発明が解決しようとする課題】
【0011】
従って、本出願人は、免疫応答において主要な役割を担うと期待される結核菌の蛋白の配列決定を行った。
本出願人は、上記45〜47kDの複合体に相当する蛋白質群が同一の遺伝子によってコードされるということ、そしてプロリン含有率が高いために分子量の計算値がポリアクリルアミドゲル上で推定される分子量と一致しないということを見出した。
【課題を解決するための手段】
【0012】
従って、本発明の対象は下記配列 No.2または配列 No.3のいずれか一方の少なくとも一部を含む蛋白質にある。
【化2】
【化3】
【0013】
本発明はさらに、配列 No.2または配列 No.3の少なくとも一部を含むハイブリッド蛋白質と、人間または動物の体内で免疫応答を誘導可能なペプチドまたは蛋白質の配列に関するものである。
【発明を実施するための最良の形態】
【0014】
抗原決定基は体液性および/または細胞性の応答を誘導可能なものであるのが好ましい。そのような決定基は各種のものにすることができ、特に複数のエピトープに対する抗体の合成を誘導可能な免疫源性組成物を得るために使用される抗原性蛋白断片、好ましくは糖蛋白にすることができる。
【0015】
これらのハイブリッド分子は、配列 No.2または配列 No.3を含む分子と、ジフテリア毒素、破傷風毒素、HBVウィルスのHBS抗原、ポリオウィルスの抗原、その他任意のウィルス毒素あるいは抗原の一部、特にエピトープとを一緒にしたもので構成することができる。
ハイブリッド分子の合成方法は、必要な蛋白質またはペプチド配列をコードしたハイブリッドDNAを製造するために遺伝子工学で用いられている方法にすることができる。
【0016】
本発明はさらに、アミノ酸配列中に配列 No.2と配列 No.3とを含む蛋白質またはこれらの配列の少なくとも一部を含むハイブリッド蛋白質に比べて機能的な変性を与えない二次的な相違点または一定の変化を有する蛋白質に関するものである。
【0017】
本発明は、配列 No.3に相当する蛋白質について算出される分子量 28779Da と、SDSゲル電気泳動で得られる複合体の分子量 45 〜47kDとの間の非常に大きな差を明らかにした点に注意されたい。この差はポリペプチド鎖中のプロリン頻度の高さ(21.7%)によるものと思われる。
【0018】
本発明はさらに他の対象は、上記定義の蛋白質をコードしたオリゴヌクレオチド、RNAまたはDNAにある。このヌクレオチドは下記の配列 No.1の少なくとも一部を含むのが有利である:
【化4】
【0019】
本発明はさらに、上記蛋白質を産生する微生物、特に上記蛋白質を分泌する微生物に関するものである。
【0020】
この微生物は Mycobacterium bovis BCGのような細菌であるのが好ましい。このバクテリアは結核に対する免疫を獲得する目的で既に人間で使用されている。
本発明のハイブリッド蛋白質を M.bovis BCGで製造する利点は、M. bovis BCGがワクチン目的で広く用いられている株であり、人間に無害であることが知られている点にある。この株は人体に注射後、15日〜1ヵ月かけて緩やかに増殖し、生体からの免疫応答の対象となる優れた抗原を発現する。
【0021】
これに対して、人間の癩病の原因物質である Mycobacteriumlepraeについてはほとんど知られていない。この細菌は現在でも培地で培養することができず、M. bovisと比較して成長期間が非常に長い。また、この細菌をワクチン処理に利用しない理由としては、この細菌のもつ病原性がある。
【0022】
配列 No.2または配列 No.3を有する蛋白質は、結核患者の体内に存在する抗体によって認識されるという利点があり、従って、先天的に免疫原性の高い抗原を構成する。
これらの蛋白質は M. bovis に非常に近い種であるM. tuberculosis に由来する。これら2種類の細菌はそれぞれ人間および家畜において結核の原因となる。M. tuberculosis 由来の蛋白質は M. bovis 内で発現され、シグナルペプチドを有する細胞によって培地中に分泌される。
【0023】
M. bovisは人間のワクチン処理に関して上記の利点を有するとともに、配列 No.2および配列 No.3に対応する蛋白質は人間において強い免疫応答を誘導するので、M. tuberculosis に由来する蛋白質の一部を含むハイブリッド蛋白質をM. bovisで生産するのが特に有利である。
【0024】
ワクチン処理の対象となる病原性微生物抗原は特別な方法で与えられない限り、人体内で非常に微弱な応答しか誘導できないことは広く知られている。
【0025】
本発明は下記2つの方法でこの問題を解決する:
(1) ハイブリッド蛋白質をM. bovis BCGの表面に存在させ、および/または細菌によって排出させ、
(2) 強力な免疫応答を誘導することが分かっている抗原決定基すなわち配列 No.2または配列 No.3を有する蛋白質の一方の抗原決定基を、単独で注入した場合には弱い免疫応答しか誘導しない抗原決定基と組み合わせる。
【0026】
配列 No.2または配列 No.3蛋白質の一方の抗原決定基を組み合わせることによって、ハイブリッド蛋白質の第2の抗原決定基に対する免疫応答が増幅される。この現象はおそらく、ハプテン担体効果と関連させることができる。
【0027】
このような操作が、M. leprae 由来の蛋白質、例えばWieles達による論文(1994, 上記参照)に記載の蛋白質等では不可能であることは明らかである。なぜなら、M. tuberculosis とM.lepraeとの間にははるかに大きな違いがあるため、そのような蛋白質は適切に発現されない可能性があり、このM. leprae 蛋白によって誘導される免疫誘導はあまりよく知られていないからである。さらに、ワクチン処理の目的で病原性種から蛋白質を導入することで人体の健康を損なう危険性があり、製薬業界はそのようなリスクを進んで負うものではない。
【0028】
以上の点から、蛋白質配列 No.2および配列 No.3とWieles達の論文(1994, 上記参照)に記載のM. leprae 蛋白質とは、それらの配列の明らかな相同性(図17参照)にもかかわらず、互いに区別される。
【0029】
本発明はさらに、上記定義の蛋白質または微生物を少なくとも一つ含むワクチンまたは薬剤に関するものである。
非グラフト化蛋白質を含むワクチンは、結核に対して個人を免疫するために使用することができ、M. bovis以外の生物原因物質から得られたエピトープを含むグラフト化蛋白質は他の病気に対する免疫処理で使用することができる。
【0030】
指標として、一人に対する1回投与量当たり1〜500 μgの蛋白質あるいは一人あたり103〜107組み換えバクテリアを皮内投与することができる。
本発明のさらに他の対象は、薬学上許容される希釈剤またはアジュバントと組み合わせた上記蛋白質または微生物を少なくとも薬学的に有効な量含む医薬組成物にある。
本発明のさらに他の対象は、特定の結核抗体の存在を調べる生物学的液体を上記蛋白質と接触させる結核に特異的な抗体の検出方法にある。
【0031】
上記蛋白質は担体に固定されているのが好ましい。この検出法はウェスタンブロット(イムノインプリント)法、酵素イムノアッセイ(ELISA) 法またはラジオイムノアッセイ(RIA) 法で蛋白質と、免疫反応を可能にするバッファー溶液と、必要に応じて用いられる生成した抗原−抗体複合体の検出物質とを含む試験用キットを用いて行うことができる。
【0032】
以下、図面を参照して本発明を具体的に説明するが、本発明が下記の説明に限定されるものではない。
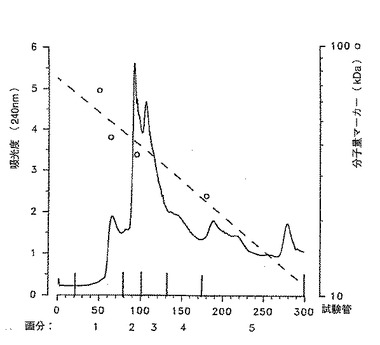
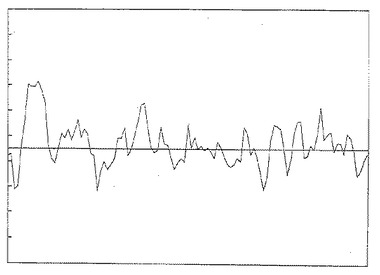
[図1]は後述の条件下でイオン交換カラムに保持されない M. tuberculosis画分を分子濾過 (si 300) した場合の240nm における吸光度 (OD) 曲線。
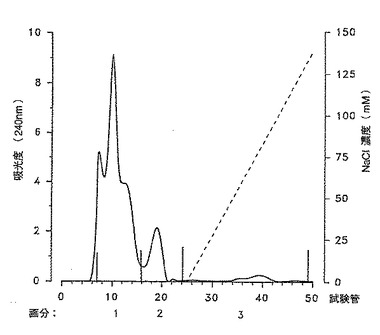
[図2]は上記分子濾過によって得られた画分1に由来する分子を高速イオン交換カラム (DEAE) で分離した場合の220nm における吸光度曲線。
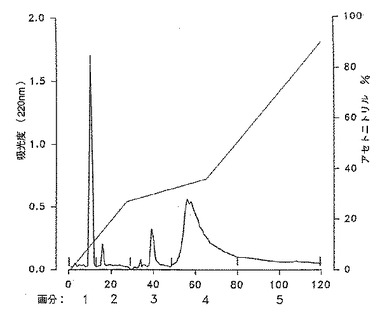
[図3]は上記イオン交換クロマトグラフィーで得られた画分1を逆相カラムクロマトグラフィーにかけた時の220nm における吸光度曲線。
【0033】
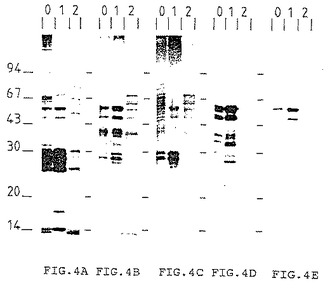
[図4A]〜[図4E]はそれぞれ下記で得られたPVDF膜 (メンブレン) の写真:
(1) PVDF膜上に移した分子(4A)の着色剤
Aurodyne着色(Amersyam)、
(2) 生菌(4B)または死菌(4C)によって免疫処理したモルモット
由来の血清混合物
(3) BCG 由来の精製抗原で免疫処理したウサギの血清(4D)
(Infection and Immunity (1993) 61 742-750)
(4) 参照番号I-1081のモノクロナル抗体(4E)
これらPVDF膜には、事前に、低圧イオン交換カラムで分離した画分をアクリルアミドゲル電気泳動で分離した分子をブロットした。レーン0は開始材料に対応し、レーン1は保持されなかった画分に、レーン2は保持された画分に対応する。
【0034】
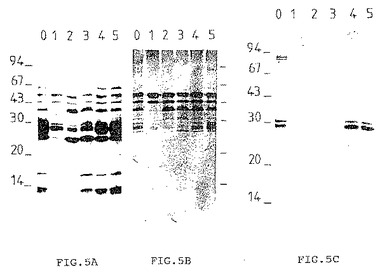
[図5A]〜[図5E]はSi 300ゲル濾過カラムで得られた5つの画分(1〜5)および低圧DEAEカラムで保持されなかった画分(0)をマイグレートさせて得られるゲルに相当するPVDF膜を示している。同一のゲルを複数のPVDFに移した後、蛋白質着色剤[Aurodye, Amersham (5A)]、生菌(5B)または死菌(5C)で免疫処理したモルモット由来の血清あるいは兎血清(5D)またはモノクロナル抗体(5E)を用いてそれぞれの膜を検出した。
【0035】
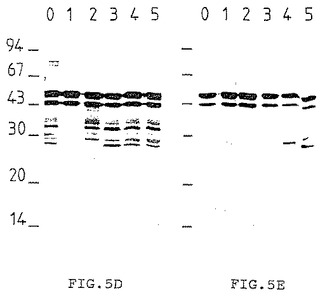
[図6A]〜[図6E]は高速イオン交換カラムで得られた画分(1〜3)と分子篩を用いた濾過で得られた画分1(ウェル0)をマイグレートさせて得られるゲルに対応するPVDF膜を示す。上記膜はそれぞれ下記を用いて検出した:
蛋白質着色剤(6A)、
生菌(6B)および死菌(6C)を用いて免疫処理したモルモットの血清に由来する抗体、
兎血清(6D)、
モノクロナル抗体(6E)
【0036】
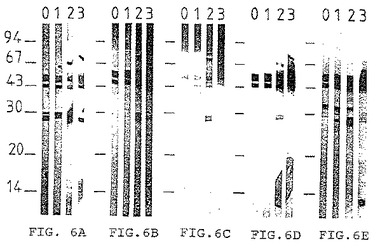
[図7A]〜[図7D]はイオン交換カラムで得られた画分1(0)および逆相クロマトグライフィーで得られた画分(1〜5)の移動に相当するゲルをブロットした膜を同じコードを有する図6A〜6B、6D〜6Eと同一の試薬で検出した結果を示す。
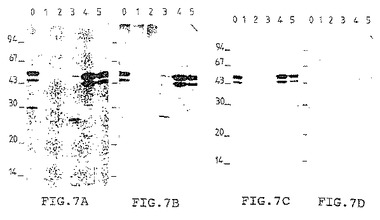
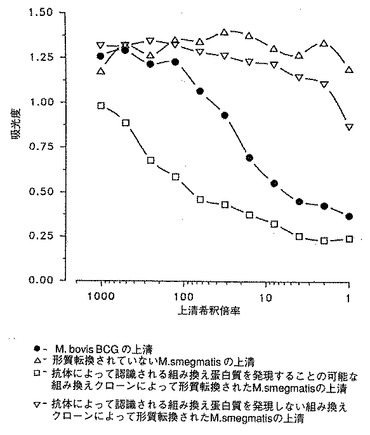
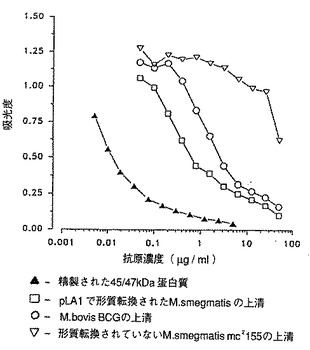
[図8]は、M. smegmatisにおいてM. tuberculosis H37Rv を発現させるための遺伝子ライブラリーのスクリーニングを示す。M. bovis BCG、形質転換されていないM. smegmatisおよび抗体によって認識される組み換え蛋白質を発現するまたはしない組み換えクローンによって形質転換されたM. smegmatisの上清を各種希釈率でテストした。
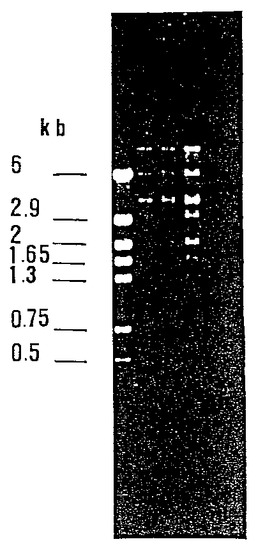
[図9]はライブラリーより選択され、E. coli で電気穿孔(エレクトロポレ、electropore))し、アルカリ溶解で抽出した3つのコスミドのアガロールゲル上での移動を示す。
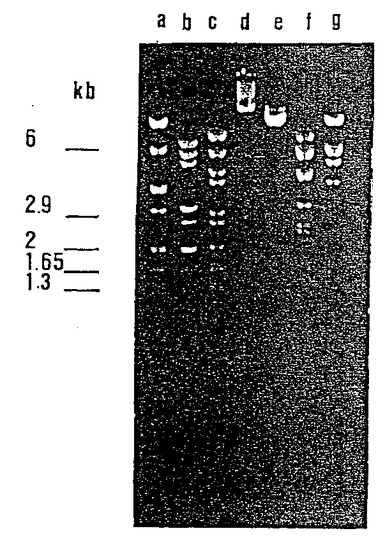
[図10]はBamHI(a)、SmaI(b) 、HpaI(c) 、NotI(d) 、SspI(e),EcoRI(f)およびHindIII(g)で処理したE. coli NM554 から抽出された pLA1 のコスミドDNAのゲル上での移動を示す。
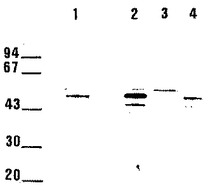
【0037】
[図11]は 45/47kDa 蛋白質のマイコバクテリアにおける発現を示し、培養7日目の細菌培養液の上清を洗浄し、Amicon PM10
膜上で濃縮、凍結乾燥し、免疫インプリントで分析したもの。1/500 に希釈した兎血清のポリクロナル抗体によって蛋白質を検出した。
ウェルはそれぞれ下記 (1)〜(4) を含む:
(1) M. bovis BCG由来の精製した45/47 kDa蛋白質0.25μg
(2) pLA1によって形質転換されたM.smegmatis mc2155の上清5μg
(3) 形質転換されていないM. smegmatis mc2155 の上清5μg
(4) M. bovis BCG上清5μg。
【0038】
[図12]はマイコバクテリアにおける45/47kDa 蛋白質の発現を示し、細菌培養液の上清を洗浄し、Amicon PM10 膜上で濃縮した後、凍結乾燥し、競合ELISA 法で分析した。凍結乾燥させた各種濃度の上清を8000倍に希釈した兎のポリクロナル血清で検出し、混合物を予め精製蛋白質を固定したウェルに写した。
[図13A]、[図13B]は pUC18の pLA1 コスミドをBamHI で処理して得られる断片をリゲーションして得られた各種 pUC18: :M.tuberculosis H37Rv組み換えクローンのプラスミドプロフィール(13A)と、BamHI 制限プロフィール(13B)である。この図は検討した36個のクローンのうち21個を示す。ウェル『P』は基準ベクターであるpUC18 に相当し、ウェル『m』はpKN プラスミドをPvuII によって開裂させた断片であるサイズマーカーに相当する。
【0039】
[図14]は45/47kDa 蛋白質のE.coliにおける発現を可能にする挿入断片の制限地図である。両方向にクローニングされた3kbの挿入断片を含むpLA34 およびpLA4プラスミドから欠失によって一群のクローンを得た。矢印はこれらクローンから『直接』および『逆』プライマーを用いて配列決定する時の方向を示す。
B , Bam HI S , Sma I E , EcoR I K , Kpn I
H , Hind III Sa, Sal I Sp, Sph I
【0040】
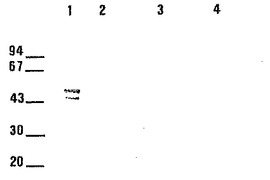
[図15]はE. coli における45/47kD蛋白質の発現を示し、細菌培養液の溶解物を免疫インプリントで分析した。DEAE上で精製後、臭化シアンによって活性化したSepharose-4Bカラム上に固定したE. coli の溶解物上に吸着させた兎のポリクロナル抗体によって蛋白質を検出した。
ウェルはそれぞれ下記 (1)〜(4) を含む:
(1) 精製された45/47kDa 蛋白質0.2 μg
(2) pLA34-2 で形質転換したE. coli XL-Blue の溶解物25μg
(3) pLA34 で形質転換したE. coli XL-Blue の溶解物25μg
(4) 形質転換していないE. coli XL-Blue の溶解物25μg。
【0041】
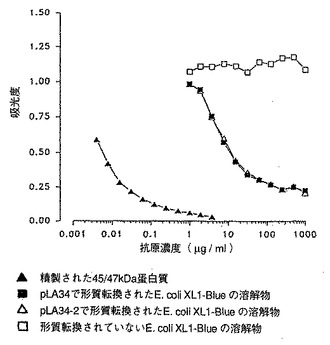
[図16]は E. coliにおける45/47kDa 蛋白質の発現を示し、競合ElISA で分析した細菌培養液の溶解物は未精製の状態で使用した。
[図17]は本発明による配列 No.2とM. leprae (mln 431) 由来の蛋白質の配列とを比較したものである。
[図18]は配列 No.2の蛋白質の疎水性度プロフィールである。
【実施例】
【0042】
実施例1
M.tuberculosis抗原の精製法
1) 抗原の入手
ソートン(Sauton's)の合成培地130 mlを入れたフラスコを用い、BCG の培養に関する通常の方法 (Gheorghiu et al.,Bull.Institut Pasteur 1983, 81:281-288)で、M. tuberculosis (H37Rv株) の培養物を作った。37℃で20日間培養後、培地を回収し、室温で沈降分離および濾過(0.22 μm) した。これらの操作は安全上の理由からグローブボックス内で実施した。回収および濾過した培地は安全フード内で再び0.22μmフィルターを用いて濾過した後、下記の操作に使用した。
窒素雰囲気下、2bar 、4℃でAmicon(PM10)膜に取り、4%のブタノールを含む逆浸透水(retro-osmosed water) を用いて強く洗浄し、元の量の10〜20倍に濃縮した。Amicon PM10 膜で除外されなかった分子を含む濃縮培地を凍結乾燥し、重量測定後、−20℃で粉末状態で保存した。以下の精製操作に使用した12gの開始材料は70リットルの培地から得られたものである。
精製操作
【0043】
2) 低速イオン交換カラム
約240 mlの Triacyl Mゲル(SEPRACOR)の高さが300 mm、直径32 mm の調製用低速イオン交換カラムを準備した。4%のブタノールを含む緩衝生理食塩水(10mM Na2HPO4 /NaH2PO4, pH=7 および 10mM NaCl)を用いてカラムを平衡化した。
上記段階で濃縮後に凍結乾燥して調製した材料を、上記緩衝化生理食塩水に溶解し、40,000Gで12分間超遠心分離した。遠心後の溶液の上部4/5だけを回収し、蠕動ポンプを用いてイオン交換カラムに添加した。カラムに保持されない第1主要画分を回収した。緩衝生理食塩水(10mM Na2HPO4 /NaH2PO4,pH=7.5および1M NaCl)を用いて溶出し、第2画分を得た。2バールの圧力下で Amicon (PM10)膜に取り、4%ブタノールを含む逆浸透水を用いてそれぞれの画分を強く洗浄し、約15倍に濃縮した。カラムに保持されない画分は2.9 gの材料および大部分の分子を含んでおり、これを次の段階で精製した。カラムに保持され、塩溶液で溶出した画分には約1.01gの材料が含まれていた。
【0044】
3) ゲル濾過
50×750 mm(SERVA) の調製用高速カラム(Si300 カラム 3μm)を4%ブタノールを含む緩衝生理食塩水(50mM Na2HPO4,KH2HPO4 を用いてpH=7.5に調整したもの)を用いて平衡化した。
この溶液は予め膜(0.22 μm) を用いて濾過しておく。カラムの流速は1.25ml bar/分に調節し、45bar の設定最大圧には到達しなかった。
カラムに添加する材料は緩衝液/ブタノール溶液を用いて濃度50mg/mlに調製した。10mlのサンプルを調製し、−20℃で凍結した。解凍後に再び濾過してカラムに添加した10mlのサンプルには約500mg の未精製材料が含有されていた。典型的な分離法である240nm での吸光度プロフィールを図1に示す。このプロフィールに基づいて選択された5つの主要画分を4℃で濃縮し、Amicon PM10 膜上で4%のブタノールを含む逆浸透水を用いて強く洗浄した。濃縮された画分をそれぞれ凍結乾燥し、重量測定し、−20℃で保存した。この段階で得られた画分1は生菌を用いて免疫処理したモルモット由来の抗体または結核患者由来の抗体によって認識される主要な分子を含んでいた。この画分のみを用いて以下の段階を行う。
【0045】
4) イオン交換カラム
4%のブタノールを含む緩衝生理食塩水(10mM Na2HPO4 /NaH2PO4, pH=7.5 および10mM NaCl)を用いて、DEAE-TSK 5PW調製用カラム 21.5 ×150mm (LKB) を平衡化した。流量6ml/分で最大圧力は30bar 以下であった。溶出バッファーに関してはNaCl濃度のみを変化させた(1M)。合計100 mgの上記材料を含む4mlのサンプルを注入した後、図2に示す直線状濃度勾配を与えた。240nm での吸光度プロフィールに従って主要画分を回収した。これらの画分を濃縮し、Amicon PM10 膜上で4%ブタノールを含む逆浸透水を用いて洗浄し、凍結乾燥した。重量測定後、各画分を−20℃で保存した。この段階で得られた画分1のみが生きた細菌を用いて免疫処理したモルモット由来の抗体によって認識される分子の大部分を含んでいた。この画分を用いて次の分離段階を行った。
【0046】
5) 逆相カラム
4.6 ×250mm の RP300C8 10μm(Aquapore Brownlee Lab.)カラムを0.22μmのフィルターで濾過した酢酸アンモニウムバッファー(20mM NH4COOCH3) を用い、最大圧力115bar、流速2ml/分で平衡化した。10mgを含む1mlのサンプルを注入後、図3のプロフィールに従って90%のアセトニトリルを含む溶出バッファーを用いて溶出した。220nm における吸光度プロフィールから5つの主要な画分が分離され、それら画分を減圧下に40℃で蒸発し、濃縮し、その後凍結乾燥した。
【0047】
6) 抗原の免疫検出
laemmli の通常の方法(Nature, 1970, 277: 680-685)に従って、10%ポリアミドを含む0.1 %SDS変性ゲルを調製した。精製段階で得られた10〜2μgの材料を含むサンプルを5%メルカプトエタノール、3%のSDSおよび痕跡量のブルモフェノールブルーを含むバッファーに溶解し、ゲルのウエルに10μlずつ添加した。電気泳動してブロモフェノールブルーをゲルの端まで移動させた後、一夜、中程度の電場を加えてサンプル中に存在する分子をPVDFシート(Millipore) 上に移した。[Harlow and lane, Antibodies, A laboratory Manual, Cold Spring Harbor Laboratory (Publishers), 1988]。
PVDEシートをコマシーブルー溶液で1分以内で着色し、その後脱色して分子量マーカーを識別可能とし、その形状を鉛筆でなぞる。完全に脱色した後、PBS +Triton X100 3%を用いて室温で30分間シートを洗浄し、その後、PBS 単独で5分間ずつ3回洗浄した。次いで、5%の脱脂粉乳を含むPBS を用いて37℃で1時間シートを飽和し、PBS +Tween 20(0.2%)で3回洗浄した。
PBS +Tween 20バッファー(0.2%)+粉末ミルク(5%)で20倍に希釈した抗血清を用い、37℃で1時間30分、定期的に攪拌しながらインキュベートした。さらにPBS +Tween を用いて3回洗浄し、次いで、アルカリホスファターゼを用いて標識した抗イムノグロブリン抗体と一緒にインキュベートした。ホスファターゼ(Biosys) を用いて標識した人間とモルモットの抗イムノグロブリン抗体を PBS+Tween20 (0.2%)+ミルク(5%)で最終希釈倍率を2500倍にして使用した。37℃で1時間30分インキュベート後、PBS +Tween を用いてPVDFシートを3回洗浄し、BCIPおよびNBT (Harlow and Lane 上記)を含む検出バッファー中で室温で5〜10分間培養した。反応を停止させ、乾燥させた後シートの写真を撮影した。
【0048】
7) アミノ酸組成物
BeckmannのLS6300分析装置を使用して、Institut Pasteur
Organic Chemistry Departmentで各クロマトグラフィー画分について合計アミノ酸組成を分析した。
45-47kD 蛋白質のアミノ酸頻度(amino acid frequency)で表した合計組成は以下のとおり:
ASN/ASP:10.4%;THR:5.7 %;SER:5.6 %;GLN/GLU:6.3 %;GLY:7.1 %;ALA:19.3%;VAL:6.2 %;ILE:2.2 %;LEU:4.4
%;TYR:2.2 %;PHE:2.4 %;LYS:2.7 %;ARG:2.7 %;PRO:20.9%。
【0049】
実施例2
M. tuberculosis 蛋白および蛋白画分の免疫特異性の決定と、生菌を用いて免疫処理したモルモットの抗体で認識される抗原の単離
12〜15匹のモルモット群(Hartley のメス、試験開始時の重量=250〜300g)に生きたマイコバクテリア(2×107単位のBCG を0.1 ml生理食塩水に添加したものを2回皮内注射)または同じ株のマイコバクテリア0.2 mgを熱殺菌(120℃、30分)したものを完全フロイントアジュバントに生理食塩水を懸濁して(1/1) 成るエマルション溶液0.5 mlとして筋肉注射で投与した。免疫処理してから7〜12ヶ月後に各モルモット群から血清サンプルをとり、濾過(0.22 μm) 後、少量に分割し、−20℃で凍結保存した。いくつかの群の血清試験を行った(生菌で免疫処理したもの5群と死菌を用いて免疫処理したもの6群)。それぞれの免疫処理タイプを代表する血清群を用いて得られた結果を示す。同じ免疫処理方法ではグループ間の差は最小であった。
【0050】
1) 低速イオン交換カラムを用いた分離段階
培養培地(Amicon PM10 膜を用いて洗浄・濃縮後、凍結乾燥したもの)を超遠心分離し、その後低速イオン交換カラムに添加した。カラムに保持されなかった画分と、高モル濃度緩衝溶液で溶出した画分との2種類の画分が得られ、これらをAmiconPM10膜を用いて洗浄・濃縮し、凍結乾燥した。
各画分(10 μm) をSDS ゲルのトラックに添加し、電気泳動した後、PVDF膜に移し(ブロットし)、免疫検出法で各種血清と反応する分子の大部分を含む画分を識別した。
【0051】
[図4]は同一のゲルを、蛋白質用の着色剤(Aurodye-Amersham)(4A)、生菌(4B)または死菌(4C)で免疫処理したモルモットの血清を用いて検出した免疫インプリントを示す。免疫インプリント4Dおよび4FはBCG と同一の分子に対する兎血清 (Infection and Immunity, 1993, 61, 742 〜750)およびパスツール研究所のCNCM (Collection Nationale de Cultures de Micro-organismes) に寄託されたモノクロナル抗体を生産するI-1081ハイブリドーマの上清を用いて検出した。カラムに保持されたなかった画分のみが、生菌または死菌を用いて免疫処理されたモルモット由来の血清によって認識される45/47kDa分子または上記ハイブリドーマの上清によって認識される45/47kDa分子を含んでいた。
【0052】
2) Si300 を用いた分子濾過段階
上記段階でカラムに保持されなかった画分を、500 mgの材料を含む10mlのサンプルとしてSi300 カラムに添加した。図1に示したプロフィールに従って画分1〜5を分離し、一連の添加によって得られた精製物を合わせ、洗浄、濃縮、凍結乾燥した。
各画分(10μg)をSDSゲルのトラックに添加し、電気泳動を行ってPVDF膜に移し、免疫検出法で各種血清と反応する蛋白質の大部分を含む画分を同定した。
[図5]は同一のゲルを、蛋白質着色剤(Aurodye Amersham)、生菌(5B)または死菌(5C)で免疫処理したモルモットに由来する血清を用いて検出した免疫インプリントを示す。免疫インプリント5Dおよび5Eは、BCG から精製した分子に対する兎血清およびI-1081モノクロナル抗体を用いて検出した。
画分1に存在する45kDおよび47kDの2種類の抗原は主として生菌を用いて免疫処理した動物に由来する抗体、ポリクロナル兎血清またはモノクロナル抗体によって認識された。この画分を第2の精製段階用に選択した。
【0053】
3) イオン交換段階
上記画分のサンプル100 mgをDEAE−TSK調製カラムに添加し、NaCl濃度勾配を付けて溶出させた。溶出分子の220nmにおけるプロフィールから主要な3つの画分(図2)が画定された。一連の添加で得られた各画分を集め、洗浄、濃縮および凍結乾燥した。
上記画分を5μgずつSDSゲル電気泳動し、PVDFシート上の免疫インプリントを蛋白質着色剤(Aurodye)(図6A)、生菌(図6B)または死菌(図6C)を用いて免疫処理したモルモットの血清、兎血清(図6D)あるいはモノクロナル抗体(図6E)を用いて検出した。1−DEAE画分は、死菌を用いて免疫処理した動物の抗体によって認識される抗原をごくわずかしか含んでいなかった。一方この同じ1−DEAE画分は生菌を用いて免疫処理されたモルモット由来の抗体および兎血清およびモノクロナル抗体によって強く認識される45/47kD のダブレットを含んでいた。この画分1−DEAEを用いて次の精製段階を行った。
【0054】
4) 逆相カラム段階
10μm RP 300 カラムを、酢酸アンモニウムバッファー(20mM)で平衡化し、上記1−DEAE画分を最大5〜10mg含むサンプル1mlを添加した。図3に示す0〜90%のアセトニトリル濃度勾配を付けて溶出し、5つの主要画分を回収した。これらの画分から40℃で減圧蒸発してアセトニトリルの大部分を除去/濃縮し、凍結乾燥した。
Aurodye を用いた蛋白質の着色(図6)では、画分4(アセトニトリル濃度30〜50%)が生菌を用いて免疫処理した動物由来の抗体あるいは兎血清中に存在する抗体またはモノクロナル抗体によって認識される分子の大部分を含んでいた。
【0055】
実施例3
Mycobacterium tuberculosis由来の45/47kD蛋白質のMycobacterium smegmatis およびEscherichia coliへのクローニングと発現
1) 材料および方法
1.1 菌株とその成長条件、上清と菌抽出物の調製
ソートンの合成培地で M. bovis BCG (1173P2 株)を37℃で7日間培養し、上清液を0.22μmの膜で濾過した。この上清液を4%ブタノールの存在下で未精製状態で保存するかAmicon-PM 膜を用いて濃縮後に凍結乾燥した。
M. segmatis mc2155 (Snapper 達、Molecular Microbiol.4, 1911-1919, 1990)を、7H9 +OADC液体培地で37℃で7日間培養した。pYUB18:M. tuberculosisライブラリー由来のコスミドによって形質転換した各M. smegmatis mc2 155クローンを25mg/mlのカナマイシン存在下で培養した。その後培養液を5000rpm で15分間遠心分離し、培養液の上清を分離して、4%ブタノール存在下で4℃で保存した。培地組成によって妨害されないELISA 試験はこれらの調製物を用いて行った。クローン培養液の上清をSDS-PAGEゲル上で分析する際には、それらをソートン合成培地で37℃で7日間培養し、0.22μm膜を用いて培養物の上清を濾過し、Amicon-PM10 膜で濃縮し、凍結乾燥した。
【0056】
E. coli NM554 およびXL 1-Blue 株を固体または液体Luria-Bertani (LB)培地で37℃で培養した。pUC18 プラスミドで形質転換したE. coli XL 1-Blue クローンを25μm/lのアンピシリン存在下で培養した。
E. coli XL1-BlueとpUC18:M. tuberculosis 組み換えプラスミドで形質転換したクローンとを一夜(16h) 培養した後、培養液を一連の急激に凍結/溶解(−70℃/+60℃)することによって培養液溶解物を調製した。溶解物を遠心分離し、上清を分離し、−20℃で保存した。これら調製物からの蛋白質の分離はBCA 法で行った(Pierce)。
【0057】
1.2 クローニングベクター
使用したM. tuberculosis の遺伝子ライブラリー(Jacobs達1991、上記)は、M. segmatis mc2155への電気穿孔法により、Stewart Coleによって作製された。本出願人は400 個の組み換え体を使用した。
ライブラリーはコスミドのシャトルベクターpYU818内で作製した。このシャトルベクターは、pYUB12プラスミド (Snapper 達、Proc. Natl. Acad. Sci., USA, 1988, 85:6987-6991)に由来するもので、このプラスミドはλバクテリオファージのCos 配列が挿入されてファージ溶解物状態のライブラリーにおける組み換えコスミドの増幅と良好な保持が可能になっている。このライブラリーは以下の方法で作製した。
すなわち、M. tuberculosis H37Rv 株のゲノムDNAを、最大35kbから45kbの断片が得られるような条件で、酵素Sau 3aで部分的に処理し、これらの断片を精製後、pYUB18にリゲーションし、制限エンドヌクレアーゼBamHI で処理後、脱燐酸基化を行った。
【0058】
pUC18 プラスミドベクター(Yanisch-Perron 達、Gene, 1985, 33: 103-119)を用いて、E. coli XL-Blue へのサブクローニングを行った。この多コピープラスミドはβガラクトシダーゼの末端アミノ酸断片をコードしたE. coli のラクトースオペロンに由来するDNAを含む。この断片はイソプロピルβ−D−チオガラクトピラノシド(IPTG)によって誘導可能で、E. coli XL1-Blue宿主株によってコードされる不完全なβガラクトシダーゼフォームと供にアルファ相補性(alpha-complementation) を確立することができる。従って、外部DNA の挿入によって、アルファ相補性の全廃が誘導される。組み換えプラスミドは、宿主株に形質転換されるとコロニーが白色を呈することによって確認され、これに対して細菌がpUC18 プラスミドで形質転換されるとコロニーは青色を示す。このスクリーニングは、IPTGおよびX-Gal 酵素基質の存在下で行った。
【0059】
1.3 分子生物学的方法
1.3.1 M.Smegmatis mc2 155コスミドの抽出
いくつかの変更を加えてM. smegmatis用に適応させたアルカリ溶菌法(Jacobs 達、1991, 上記) を用いて組み換え pYUB18:M. tuberculosis コスミドの抽出を行った。培養5日目(指数増殖期終了後)に細菌を回収し、5000rpm で10分間遠心分離した。細菌残留物(3ml)を5mlの溶液A(50 mMグルコース、25mMトリスHCl pH8、10mM EDTA 、リゾチーム10mg/ml) に再懸濁し、37℃で20分間インキュベートした。2倍量(10ml)の溶液B(0.2N NaOH、1%SDS)を添加し、逆混合した。混合物を65℃で30分間インキュベートし、その後、4℃で15分間インキュベートした。最後に1.5 倍量(7.5ml) の溶液C(5mM 酢酸カリウム、酢酸11.5%)を添加し、逆混合した。混合物を4℃で30分間インキュベートした。続いて調製物を4℃で13000rpm、15分間の遠心分離し、上清を回収して、同量のフェノール/クロロホルム50/50で処理した。
抽出後、試験管を4000rpm で10分間の遠心分離した。水相を清潔な試験管に移し、−20℃で保存しておいた2倍量のエタノールを用いて2回処理した。反転後、これを−20℃で少なくとも1時間保存し、その後、12000rpmで20分間遠心分離した。最後に残留物を同量の70%エタノール(−20℃に保存しておいたもの)で洗浄し、Speed-Vac で5分間乾燥させた。乾燥残留物を500 μlの滅菌水に懸濁し、−20℃で保存した。
【0060】
1.3.2. E. coli プラスミドの抽出および精製
pYUB18コスミドおよびpUC18 組み換えプラスミドの迅速な抽出はアルカリ溶菌法(Birnboim 達、Nucleic Acids Res., 1979,
7: 1513)で行った。
アルカリ溶解操作後、関連するコスミドと組み換えプラスミドとを、臭化エチジウムの存在下で塩化セシウム濃度勾配超遠心分離で精製した(Maniatis 達、Cold Spring Harbor, New
York: Cold Spring Harbor Laboratory Press, 1982)。
【0061】
1.3.3 形質転換技術
塩化カルシウムを用いた化学的方法
この一般的方法は pUC18組み換えプラスミトでE. coli XL1-Blueを形質転換するのに使用した。まず最初にコンペテントバクテリアを調製した。20mlの2YT 培地に、一夜培養したプレカルチャーを1/100 の割合で植えつけた。細菌を、攪拌しながら37℃で2時間、OD=0.6 になるまで培養し、その後4℃、400
rpm で10分間、遠心分離した。残留物を8mlの100mM CaCl2 に懸濁し、氷浴中に15分間保持し、その後再び4℃で4000rpm 、10分間の遠心分離した。最後に残留物を1.6 mlの100mMCaCl2に懸濁し、氷浴中で30分間保持した。
こうして調製したコンピテントバクテリアはすぐに形質転換に利用するか、あるいは4℃で数日間保存してもよい。形質転換時には2μlのDNA に200 μlのコンピテントバクテリアを混合した。混合物を氷浴中で45分間保存し、その後42℃で2分間の温度ショックを加えた。800 μlの2YT 培地を添加し、その後攪拌しながら調製物を37℃で1時間インキュベートし、Mlアンピシリンプレートに50μl〜200 μl/プレートの割合で拡げた。翌日、コロニーをカウントし、形質転換効率を計算した。
【0062】
物理的電気穿孔法
この方法は大型ベクターを用いてE. coli を形質転換するために使用した。すなわち、E. coli のNM554 株を、50kb以上のサイズを有する組み換えpYUB18コスミドを用いて電気穿孔にかけた。コンピテントバクテリアは新規に調製した。200 mlの 2YT培地に、一夜培養したプレカルチャーを1/100 の希釈率で植えつけ、37℃で3時間細菌を培養した。その後、6000rpm で10分間遠心分離した。残留物を4℃の滅菌水10mlに取り、その後4℃の滅菌水190 mlに懸濁した。細菌を、再び6000rpm で10分間遠心分離し、4℃の滅菌水10mlを用いて再度洗浄した。最後に残留物を400 μlの10%グリセロールに懸濁した。
電気穿孔はBio-Rad Gene Pulser を用いて行った。4mmのセル内で100 μlの細菌を1〜4μlのDNAと混合した。混合物に電気ショックを与え(2500V、25μF)、その後、1mlの2YT 培地を素早くセルに添加した。全体を試験管に移し、攪拌しながら37℃で1時間インキュベートした。インキュベート後培養液をML−アンピシリンプレートに50μl〜200 μl/プレートの割合で拡げた。翌日コロニーをカウントし、形質転換効率を計算した。
【0063】
1.3.4 酵素処理断片のクローニング
クローニングすべきDNAをBamHI 制限エンドヌクレアーゼで処理した。pUC18 プラスミドを同様の方法で処理した。所望のpYUB18組み換えコスミドから得られた断片を、T4 DNAリガーゼ酵素(Amersham)の活性を利用してプラスミドベクターにリゲーションした。リゲーションは、20μlの容量で16℃で一夜行った。リゲーション混合物全体を用いてE. coli XL1-Blueの形質転換を行った。遺伝情報発現後、全ての細菌を25μg/ml、(TPTG 、X-Gal)の割合でMl- アンピシリンプレートに拡げた。アルファ相補性が確立されないクローンが、コロニーの色(白色)によって確認された。
クローニングによる精製後、組み換えクローンを検討した。アルカリ溶解によってプラスミドDNA を抽出し、その後、制限エンドヌクレアーゼBamH Iによる処理の前または後に0.8 %のアガロールゲルを用いて分析した。
【0064】
1.3.5 制限地図の作製
両方向にクローニングされた3kbのBamH I-BamH I 断片を含むpLA34 およびpLA4組み換えプラスミドを、pUC18 マルチサイトリンカー(ポリリンカー)内の部位を有する各種の制限エンドヌクレアーゼで処理した。制限エンドヌクレアーゼBamH I、Hind III、Sph I 、Xba I 、SalI、Kpn I 、Eco RIおよびSma
I を用いて単一または二重の処理を行い、0.8 %アガロールゲルを用いて分析した。臭化エチジウムを用いてDNAを染色した後、それらの移動距離をマーカー(インターナルラボラトリースタンダード, Pvu II で処理したpKN プラスミド)と比較して各断片のサイズを決定した。
【0065】
1.4 蛋白質の検出方法
1.4.1 ELISA 法
競合ELISA 法によってポリクロナル血清(Romain達、1993,
上記)を用いて、細菌培養物より得られる各種調製物中の45/47kDa 蛋白質濃度を測定した。
この兎のポリクロナル血清は、通常の免疫処理方法(フロインドの不完全アジュバントに懸濁した精製蛋白質50μgを注射し、1ヶ月後に25μgを注射する)に従って45/47蛋白質に対して作製されたものである。
第1のマイクロプレートのウェルを炭酸塩緩衝液を用いて1μg/mlに調製した精製蛋白質溶液か15日目のMycobacterium1 bovis BCG 上清液(10μg/ml)のいずれかで覆った。37℃で1時間かけて抗原を固定し、マイクロプレートをPBS で5回洗浄した。第2のインキュベーションではPBS と0.5 %ゼラチンと4%ブタノールとを含む溶液を用いて37℃で1時間、ウェルを飽和させた。その後、PBS-Tween 0.1 %を用いてマイクロプレートを5回洗浄した。
【0066】
試験は以下のように行った:
第2のマイクロプレート内でPBS-Tween 0.1 %、0.25%ゼラチン、4 %ブタノールを用いて各種濃度(純粋、1/2 、1/4 、1/8 等)に希釈した分析すべき上清50μlと、PBS-Tween 0.1
%、0.25%ゼラチン、4 %ブタノールを用いて希釈倍率1/4000に調製した兎血清50μlとを37℃で1時間インキュベートし、次に混合物を第1のマイクロプレートに移して37℃で1時間インキュベートした。0.1 %のPBS-Tween を用いてマイクロプレートを10回洗浄した。最後に、兎に対する抗IgG H +L 複合抗体(Byosys)をアルカリホスファターゼを用いて標識し、 PBS-Tween 0.1 %、0.25%ゼラチン、4 %ブタノールを用いて希釈率1/4000に調製したものを37℃で1時間インキュベートした。PBS-Tween 0.1 %を用いてマイクロプレートを10回洗浄した。
最後に、酵素基質であるパラ−ニトロフェニルホスフェート(pNPP)を、NaHCO3、MgCl2、pH9.6 緩衝液を用いて40mg/24ml濃度に調製した状態で1時間または一夜インキュベートした。Titerteck Twinreader上、414nm および690nm でODを読み取った。
【0067】
1.4.2 免疫インプリント法
変性SDS−PAGEゲル上で行う通常のゲル電気泳動方法を採用した(Laemmli, Nature, 1970, 277:680-685)。その後、PVDF膜に電気的に移した(Towbin 達、Proc. Natl. Acad. Sci. USA, 1979, 76:4350-4354; Pluskal 達、Biotechniques,1986, 4:272-283)。
ゲル上で分析したサンプルを定量し、M. smegmatis上清については凍結乾燥体の重量μg (5μgを添加) を、E. coli 溶解物については蛋白質の重量μg(25 μgを添加) を測定した。
精製したM. bovis BCG蛋白質を、トラックあたり0.25μg蛋白質の濃度でゲル上に添加した。
膜上に移された蛋白質は、マイコバクテリア中で発現される蛋白質に対する兎のポリクロナル血清(希釈率:500 分の1)を用いて検出した。
E. coli 内の組み換え蛋白質を検出するために、これらポリクロナル抗体を DEAE(TrisacrylD) カラムで精製し、その後得られた免疫グロブリンを、臭化シアノゲン(Pharmacia) によって活性化したSepharose-4Bカラム上に固定されたE. coli の溶解物に吸着させた(Maniatis 達、1982) 。保持されなかった抗体は4℃で保存し、100 分の1の希釈率で膜に移された蛋白質を検出するために使用した。
アルカリホスファターゼで標識された種特異性のある抗Ig H+L 複合抗体(Bio-Sys) を3000分の1の希釈率で上記抗体を検出するために使用した。最後に、アルカリホスファターゼの活性は、2種類の人工色素産生基質、テトラゾリウムブルーと5-ブロモ-4- クロロ-3- インドリルホスフェートを用いて検出した。
【0068】
1.5 DNA配列決定
ヌクレオチドの配列決定は、2種類のクローンpLA34 およびpLA4から各種欠失によって得られたクローン群を使用して行った。欠失は作製した制限酵素地図に準じて選択した。
配列決定は2本鎖プラスミドDNAマトリクスを用いて行った。T7シーケンシングキット(Pharmacia) および35SATPを用いてサンガーの方法を適用した。
各種の欠失クローンおよびpUC18 プラスミドのユニバーサルプライマー(Direct and Reverase Primer)、続いて合成オリゴヌクレオチドを用いて配列を決定した。
配列は、2種類の相補鎖について決定した。
M. tuberculosis のゲノムDNAのGCの割合が高い(65 %)
コンプレッション領域は、7-Deaza dGTPすなわちdGTPの化学的類似物を含むT7 Deaza G/A 配列決定キット(Pharmacia) を用いて配列決定した。
【0069】
1.6 配列解析
得られた隣接する配列の比較および組合わはSTADENプログラムを用いてUnix(登録商標)で行った。EMBLおよびGen-Bankデータバンクの配列から検索した配列の相同性は GCGのFASTA およびT-FSTAプログラムを用いて作製した。
【0070】
2) 結果
2.1 M.tuberculosisに由来する45/47kDa蛋白質のM.smegmatis へのクローニングおよび発現
2.1.1 M. smegmatisにおいてM. tuberculosis を発現させるための遺伝子ライブラリーのスクリーニング
使用した遺伝子ライブラリー(Jacobs 達、1991, 上記)は、制限エンドヌクレアーゼSau 3aを用いてゲノムを部分的に切断して得られる40kbの断片をpYUB18コスミドベクター内にクローニングすることによって作製した。パルスフィールドゲル電気泳動によって4200kbと推定されるゲノムのサイズは、約100 〜150 個のクローンに含まれる。
競合ELISA を用いて液体培地中の蛋白質を決定した (Romain達、1993, 上記)。それによってM. bovis BCGの7日目の培養物の上清に含まれる45/47kDa蛋白質の量の検出および定義が可能になった(図8)。
この試験は以下のような利点を有する:優れた感度すなわち8000分の1に希釈したポリクロナル血清を用いることによって液体培地中の蛋白質を1ng/mlのオーダーで検出でき (Romain達、1993, 上記)、さらに一連のサンプルを迅速にスクリーニングするため操作が容易である。
【0071】
電気穿孔法でM. smegmatisに導入された400 個のpYUB18::M.tuberculosis H37Rv組み換えクローンをスクリーニングした。 そのために、各種のクローンを7H9 +OADC培地で7日間培養した。この試験では、培養液を遠心分離して得られる上清を分析して組み換え蛋白質を検索した。
M. bovis BCG 45/47kDa 蛋白質に特異的なモノクロナル抗体によって認識される蛋白質を発現する3つのクローンが見出された(図8)。この第1のスクリーニングでは、マイクロタイタープレートのウェルを、45/47kDa 蛋白質が全質量の2%と測定されたM. bovis BCG培養物の上清で覆った。選択した3種類のクローンを、マイクロタイタープレートのウェルを精製した45/47kDa 蛋白質で覆う第2の試験で確認した。
【0072】
2.1.2 選択された組み換えプラスミドの遺伝子分析
選択した各種コスミドを検討するために、変形アルカリ溶解によってM. smegmatis DNAを抽出した後、電気穿孔法によってE. coli NM554 に導入した。マイコバクテリアの染色体外DNAは細胞壁の複雑さ(溶解が困難である)およびベクターコピー数の少なさ(細菌1個に付き平均3〜10個) によって採取が困難である。E. coli NM554 に形質転換された3種類のクローンをML−カナマイシンプレート上で単離し、アルカリ溶菌によって抽出したコスミドDNAを0.8 %アガロールゲル上で分析した。
3つのクローンはサイズ 50kb 以上のDNAを含んでいた。制限エンドヌクレアーゼBam HIによる処理を行い、選択された3つのコスミドのプロフィールを識別した。これらは同一であることが明らかになった(図9)。プロフィールは、pYUB18ベクターに対応する12kbのバンドを示し、さらにクローニングされたDNA断片(約40kb)に相当する一連の低分子量バンドを示した。得られたバンドの数およびゲル上のそれらバンドの位置を考慮すれば、単離されたコスミドは同一であるとみなすことができる。
G+Cを多く含むDNAに対して幾分多くの開裂部位を有する制限エンドヌクレアーゼを用いて、pLA1コスミド単独の各種処理を行い、45/47 kDa蛋白質の遺伝子(単数または複数)を含むのに十分な中程度の長さを有する断片を識別し、それらのサブクローニングを行った(図10)。
【0073】
2.1.3 M. tuberculosis に由来する45/47 kDa蛋白質の M. smegmatisにおける発現
約40kbの挿入部分を含むpLA1コスミドによってM. smegmatisにおける組み換え蛋白質の発現が行われ、培養物上清中でポリクロナル抗体によって検出された。
発現された蛋白質のおおよそのサイズを決定するために、培養7日目の上清の凍結乾燥物を免疫インプリントにより分析した。M.smegmatis において発現された組み換え蛋白質は2種類の分子量45/47kDa を有し、これは明らかにM. bovis BCGにおいて発現されるものと同一である(図11)。
別の試験ではこれら組み換え蛋白質の発現レベルはM. bovisBCG に匹敵するものであった。凍結乾燥した上清から得られた所定量の蛋白質を競合ELISA で定量した。各種濃度の凍結乾燥した上清を、8000分の1に希釈した兎のポリクロナル血清を用いて検出した。組み換えM. smegmatisは、M. bovis BCGと比較して5倍量の蛋白質を発現した(図12)。
この挿入部分のサブクローニング並びに異種宿主(E. coli)
における組み換え蛋白質の分析を行って、これらの蛋白質をコードする遺伝子の数を決定した。
【0074】
2.2 M. tuberculosis に由来する45/47 kDa蛋白質のE. coli へのクローニングおよび発現
2.2.1 45/47蛋白質のE. coli におけるサブクローニングおよび発現
pLA1を異種宿主であるE. coli NM554 に形質転換すると、細菌培養物または溶解物の上清からは組み換え蛋白質は検出されなかった。これら蛋白質の発現を促進するために、コスミドをBam HIで処理して得られる断片をpUC18 プラスミドにサブクローニングする操作を行った(Yanisch-Perron 達、Gene, 1985,
33: 103,119)。
E. coli XL1-Blueに形質転換されたpUC18::M. tuberculosis組み換えプラスミドを宿主細菌のβガラクトシダーゼ発現の欠損によって選択した。一連の36個のクローンのうち『白色』クローンのプラスミドDNAを、アルカリ溶解および制限エンドヌクレアーゼBam HIによる処理を行って調製した。
アガロールゲル上で観察される得られたプラスミドのサイズは、数種類のプロフィールを示し、組み換えプラスミドが異なっていることが示された(図13A)。
【0075】
アガロールゲル上で観察されるクローニングされた挿入部分のサイズも、異なる制限プロフィールを示した(図13B)。これらのプロフィールは全て、pUC18 ベクターに相当する2.8kb
の断片と、クローニングされた挿入部分に相当するサイズの異なる一連の断片とを示した。
サイズが大きいためにクローニングの困難な12kbの断片を除いて、全ての処理断片を単独、2個または3個ずつクローニングした。
選択した36個のクローンについて、E. coli XL1-Blueにおける組み換え蛋白質の発現を誘導する能力に関するスクリーニングを行った。この試験は上記と同じ競合ELISA テストによって行った。
細菌培養物の上清中に組み換え蛋白質は検出されなかった。一方、3kbの挿入断片を少なくとも1つ含むクローンの細菌溶解物中に組み換え蛋白質が検出された。
この試験で測定された蛋白質の発現レベルは、プラミドのサイズによって影響されるものと思われた。検討した36個のクローンのうち、発現を可能にする2つのクローン、それぞれ3kb
および7kbの挿入部分を含むpLA34 およびpLA35 が見出された。
発現レベルは、表1(下記参照)の結果に示すようにpLA34 の方が高かった。
【0076】
2.2.2 pLA34 およびpLA34-2 クローンの制限地図
pUC18 のマルチサイトリンカー(ポリリンカー)中に存在する一般的な制限エンドヌクレアーゼに対する各種開裂部位を同定することによってpLA34 プラスミドについての制限地図を決定した(図14)。単一の制限部位Eco RIが、3kbを2kbおよび1kb の2つの断片に分断していた。
2kbのBamHI-EcoRI 挿入部位を有するpLA34-2 クローンは、上記のクローンから欠失によって作製された。このクローンもまた細菌溶解物内に組み換え蛋白質を発現させた(図15)。
細菌溶解物の免疫インプリント分析の結果、45/47kDa の2種類の分子量を有する蛋白質がM. bovis BCG内で発現される天然の蛋白質と明らかに同一であることが示された(図16)。
【0077】
2.2.3 M.tuberculosis H37Rvの45/47kDa 蛋白質をコードするヌクレオチド配列の分析
45/47kDa 蛋白質をコードした遺伝子の完全なヌクレオチド配列、上流の配列およびアミノ酸から推定される配列を、配列No. 1および配列 No.2に示す。
蛋白質ダブレットの発現を可能にする単一の遺伝子はヌクレオチド配列の1082から2056までの位置に975 塩基対を有する。 リボソーム固定のコンセンサス配列(Shine Dalgarno)は遺伝子の上流で確認された。
この遺伝子はGC含有率がM. tuberculosis の65%に比べて69.4%と高い。
この遺伝子から推定される蛋白質はシグナルペプチダーゼのANA 開裂部位を含む典型的なシグナル配列を有する。
この遺伝子は39アミノ酸のシグナル配列を含む325 アミノ酸の蛋白質をコードしている。
M. bovis BCGおよびM. tuberculosis から精製した蛋白質のアミノ酸組成を生化学的分析して得られる結果と蛋白質配列から類推される結果とを比較すると、非常に優れた一致が見られる(表2)。
従って、Mycobacterium smegmatis およびE. coli において2つの分子量を有する蛋白質の発現を可能にする単一の遺伝子が存在するという結論が導かれる。
【0078】
2.2.4 蛋白質配列の分析および配列の比較
推定されるアミノ酸配列から計算される分子量は28.7kDa である。
等電点の計算値は4.36である。この結果も精製したM. bovisBCG 蛋白質について行った生化学的な等電点の測定と非常によく一致する。
推定されるアミノ酸配列からプロリンとアラニンの含有率が高いことが示される(21.8 %および19.1%) 。
完全な配列は最近発表されたMycobacterium leprae由来の蛋白質との間に相同性を示す。2つの配列の比較を図17に示す。2つの蛋白質間の相同性率は65.4%である。Mycobacteriumlepraeについて報告されたこの蛋白質も分泌蛋白に特異的なシグナル配列を有する。
M. tuberculosis から導かれる蛋白質(本発明の対象:配列No. 2)の疎水性プロフィールを求めた。これを図18に示す。
【0079】
【表1】
【表2】
【0080】
【表3】
【表4】
【表5】
【表6】
【表7】
【表8】
【表9】
【図面の簡単な説明】
【0081】
【図1】イオン交換カラムに保持されない M. tuberculosis画分を分子濾過 (si 300) した場合の240nm における吸光度 (OD) 曲線。
【図2】上記分子濾過によって得られた画分1に由来する分子を高速イオン交換カラム (DEAE) で分離した場合の220nm における吸光度曲線。
【図3】上記イオン交換クロマトグラフィーで得られた画分1を逆相カラムクロマトグラフィーにかけた時の220nm における吸光度曲線。
【図4】A〜EはPVDF膜 (メンブレン)の写真:
【図5】A〜EはSi 300ゲル濾過カラムで得られた5つの画分(1〜5)および低圧DEAEカラムで保持されなかった画分(0)をマイグレートさせて得られるゲルに相当するPVDF膜を示す図。
【図6】A〜Eは高速イオン交換カラムで得られた画分(1〜3)と分子篩を用いた濾過で得られた画分1(ウェル0)をマイグレートさせて得られるゲルに対応するPVDF膜を示す図。
【図7】A〜Dはイオン交換カラムで得られた画分1(0)および逆相クロマトグライフィーで得られた画分(1〜5)の移動に相当するゲルをブロットした膜を同じコードを有する図6A〜6B、6D〜6Eと同一の試薬で検出した結果を示す図。
【図8】M. smegmatisにおいてM. tuberculosis H37Rv を発現させるための遺伝子ライブラリーのスクリーニングを示す図。
【図9】ライブラリーより選択され、E. coli で電気穿孔(エレクトロポレ、electropore))し、アルカリ溶解で抽出した3つのコスミドのアガロールゲル上での移動を示す図。
【図10】BamHI(a)、SmaI(b) 、HpaI(c) 、NotI(d) 、SspI(e),EcoRI(f)およびHindIII(g)で処理したE. coli NM554 から抽出された pLA1 のコスミドDNAのゲル上での移動を示す図。
【図11】45/47kDa 蛋白質のマイコバクテリアにおける発現を示す図。
【図12】マイコバクテリアにおける45/47kDa 蛋白質の発現を示す図。
【図13】A、Bは pUC18の pLA1 コスミドをBamHI で処理して得られる断片をリゲーションして得られた各種 pUC18: :M.tuberculosis H37Rv組み換えクローンのプラスミドプロフィール(13A)と、BamHI 制限プロフィール(13B)の図。
【図14】45/47kDa 蛋白質のE.coliにおける発現を可能にする挿入断片の制限地図
【図15】E. coli における45/47kD蛋白質の発現を示す図。
【図16】E. coliにおける45/47kDa 蛋白質の発現を示す図。
【図17】配列 No.2とM. leprae (mln 431) 由来の蛋白質の配列との比較図。
【図18】配列No. 2の蛋白質の疎水性プロフィール。
【技術分野】
【0001】
本発明は微生物蛋白と、この蛋白を産生する微生物とに関するものである。
本発明はさらに、この蛋白のワクチンまたは結核検出での利用に関するものである。
【背景技術】
【0002】
結核は現在でも世界中で公衆衛生上の問題になっている。結核を直接の原因とする年間の死者数は約300 万人で、新たな結核の件数は約1500万人である。この結核による死者の数は先進国でも高く、例えばフランスでは年間1500人に達する。公式な数字と正確な分析結果との差に関するルジョーの評価方法を考慮すると、この数字は2分の1から3分の1に過少評価されている。最近の結核患者数の増加あるいは少なくともこの疾患の発生頻度が減少せず、横ばいである状況は感染性HIV/AIDS疾患の拡大との関連で考える必要がある。結論として、結核はフランスや先進国でも依然として頻度の高い主要な感染症であり、発展途上国では単一の病気による死亡の主要な原因になっている。
【0003】
現在、患者から採取したサンプル中の細菌の培養で判定する診断結果の確度は結核患者の半分以下である。結核患者の80〜90%を占める細菌の検出が最も容易な肺結核の場合でも、喀出物の検査で陽性が示されるのは患者の半数に満たない。
【0004】
PCR(ポリメラーゼチェーンリアクションによる増幅)のような感度の高い方法の開発で常に問題になることはサンプル採取が難しいことである。すなわち、女性や子供は一般に唾を吐くことがなく、幼児のサンプルを得るには特殊な医学的介入(例えば神経節の生検や腰部穿刺による頭脊柱液の採取など)を必要とすることが多い。
しかも、サンプルの起源を管理できないため、サンプルをPCR法で使用することができないといったPCR法そのものに起因する制限が存在する。
【0005】
さらに、顕微鏡検査と培養による伝統的な細菌学的診断法は感度が低い(サンプル中に104〜105オーダーの細菌が必要)ため、既にかなり細菌が繁殖している状態、すなわち、かなり病気が進行した状態でなければならない。
従って、細菌自体の検出が困難または不可能なこの病気の診断では、Mycobacterium tuberculosis(結核菌)に対して特異的な抗体を用いる検出方法が大きな助けになるものと思われる。
研究者達は結核の血清学的診断方法を確立すべく努力してきた。この分野で行われた研究の概論は下記文献に記載されている。
【特許文献1】国際特許PCT WO92/21758号公報
【0006】
大抵の従来法では、生化学的特性を利用して蛋白質を予め単離する。本発明者はこの蛋白を単離した後に初めてこの蛋白の肺結核患者検出能力をテストした。
上記特許文献1(国際特許PCT WO92/21758号公報)には、結核患者または生菌で免疫処理したモルモットに由来する血清を用いて結核感染症の代表的な抗原を確実に選択する方法が開示されている。この方法では従来の試験法とは異なり、分子量44.5〜47.5kDのM.bovis 蛋白質を単離することができる。
これら蛋白質の1つのN末端に位置する17個のアミノ酸は下記のように決定されている:
【化1】
【0007】
下記文献にはこの国際特許に記載された結果の内容が要約されている。
【非特許文献1】ローマン達、1993、Infection and Immunity, 61, 742-750
【0008】
この論文は特に上記45〜47kDの複合蛋白質に対してウサギを免疫処理して得られる兎のポリクロナル免疫血清を用いた競合ELISA 試験について述べたものである。
これと平行して、ヤコブ(JACOBS)達によって結核菌の遺伝子ライブラリーが作られた(下記非特許文献2参照)。このライブラリーは多数の異なるクローンを含む。
【非特許文献2】1991, Method Enzymol., 204, 537-557
【0009】
別のマイコバクテリア種である癩菌(M. leprae) 由来の蛋白質がワイエル(WIELES)達によって同定されている (下記非特許文献3参照)。
【非特許文献3】Infection and Immunity, 62, 252-258, 1994
【0010】
この蛋白は43L とよばれ、ヌクレオチド配列から推定される分子量は約25.5Daである。そのN末端は M. bovis BCG において同定された45〜47kDa の複合蛋白質のN末端と47%の相同性を有し、その17個のアミノ酸配列は上記のとおりである。
既に述べたように、ヒトの医学分野では診断上の観点および治療上の観点では、マイコバクテリア、特に結核菌によって産生される蛋白質の正確な同定が主たる興味の対象である。
すなわち、実際に直面している未解決の課題は多くの疾患に対するワクチンの製造である。
もう1つの課題は、マイコバクテリアによって引き起こされる病気、例えば結核の検出である。
【発明の開示】
【発明が解決しようとする課題】
【0011】
従って、本出願人は、免疫応答において主要な役割を担うと期待される結核菌の蛋白の配列決定を行った。
本出願人は、上記45〜47kDの複合体に相当する蛋白質群が同一の遺伝子によってコードされるということ、そしてプロリン含有率が高いために分子量の計算値がポリアクリルアミドゲル上で推定される分子量と一致しないということを見出した。
【課題を解決するための手段】
【0012】
従って、本発明の対象は下記配列 No.2または配列 No.3のいずれか一方の少なくとも一部を含む蛋白質にある。
【化2】
【化3】
【0013】
本発明はさらに、配列 No.2または配列 No.3の少なくとも一部を含むハイブリッド蛋白質と、人間または動物の体内で免疫応答を誘導可能なペプチドまたは蛋白質の配列に関するものである。
【発明を実施するための最良の形態】
【0014】
抗原決定基は体液性および/または細胞性の応答を誘導可能なものであるのが好ましい。そのような決定基は各種のものにすることができ、特に複数のエピトープに対する抗体の合成を誘導可能な免疫源性組成物を得るために使用される抗原性蛋白断片、好ましくは糖蛋白にすることができる。
【0015】
これらのハイブリッド分子は、配列 No.2または配列 No.3を含む分子と、ジフテリア毒素、破傷風毒素、HBVウィルスのHBS抗原、ポリオウィルスの抗原、その他任意のウィルス毒素あるいは抗原の一部、特にエピトープとを一緒にしたもので構成することができる。
ハイブリッド分子の合成方法は、必要な蛋白質またはペプチド配列をコードしたハイブリッドDNAを製造するために遺伝子工学で用いられている方法にすることができる。
【0016】
本発明はさらに、アミノ酸配列中に配列 No.2と配列 No.3とを含む蛋白質またはこれらの配列の少なくとも一部を含むハイブリッド蛋白質に比べて機能的な変性を与えない二次的な相違点または一定の変化を有する蛋白質に関するものである。
【0017】
本発明は、配列 No.3に相当する蛋白質について算出される分子量 28779Da と、SDSゲル電気泳動で得られる複合体の分子量 45 〜47kDとの間の非常に大きな差を明らかにした点に注意されたい。この差はポリペプチド鎖中のプロリン頻度の高さ(21.7%)によるものと思われる。
【0018】
本発明はさらに他の対象は、上記定義の蛋白質をコードしたオリゴヌクレオチド、RNAまたはDNAにある。このヌクレオチドは下記の配列 No.1の少なくとも一部を含むのが有利である:
【化4】
【0019】
本発明はさらに、上記蛋白質を産生する微生物、特に上記蛋白質を分泌する微生物に関するものである。
【0020】
この微生物は Mycobacterium bovis BCGのような細菌であるのが好ましい。このバクテリアは結核に対する免疫を獲得する目的で既に人間で使用されている。
本発明のハイブリッド蛋白質を M.bovis BCGで製造する利点は、M. bovis BCGがワクチン目的で広く用いられている株であり、人間に無害であることが知られている点にある。この株は人体に注射後、15日〜1ヵ月かけて緩やかに増殖し、生体からの免疫応答の対象となる優れた抗原を発現する。
【0021】
これに対して、人間の癩病の原因物質である Mycobacteriumlepraeについてはほとんど知られていない。この細菌は現在でも培地で培養することができず、M. bovisと比較して成長期間が非常に長い。また、この細菌をワクチン処理に利用しない理由としては、この細菌のもつ病原性がある。
【0022】
配列 No.2または配列 No.3を有する蛋白質は、結核患者の体内に存在する抗体によって認識されるという利点があり、従って、先天的に免疫原性の高い抗原を構成する。
これらの蛋白質は M. bovis に非常に近い種であるM. tuberculosis に由来する。これら2種類の細菌はそれぞれ人間および家畜において結核の原因となる。M. tuberculosis 由来の蛋白質は M. bovis 内で発現され、シグナルペプチドを有する細胞によって培地中に分泌される。
【0023】
M. bovisは人間のワクチン処理に関して上記の利点を有するとともに、配列 No.2および配列 No.3に対応する蛋白質は人間において強い免疫応答を誘導するので、M. tuberculosis に由来する蛋白質の一部を含むハイブリッド蛋白質をM. bovisで生産するのが特に有利である。
【0024】
ワクチン処理の対象となる病原性微生物抗原は特別な方法で与えられない限り、人体内で非常に微弱な応答しか誘導できないことは広く知られている。
【0025】
本発明は下記2つの方法でこの問題を解決する:
(1) ハイブリッド蛋白質をM. bovis BCGの表面に存在させ、および/または細菌によって排出させ、
(2) 強力な免疫応答を誘導することが分かっている抗原決定基すなわち配列 No.2または配列 No.3を有する蛋白質の一方の抗原決定基を、単独で注入した場合には弱い免疫応答しか誘導しない抗原決定基と組み合わせる。
【0026】
配列 No.2または配列 No.3蛋白質の一方の抗原決定基を組み合わせることによって、ハイブリッド蛋白質の第2の抗原決定基に対する免疫応答が増幅される。この現象はおそらく、ハプテン担体効果と関連させることができる。
【0027】
このような操作が、M. leprae 由来の蛋白質、例えばWieles達による論文(1994, 上記参照)に記載の蛋白質等では不可能であることは明らかである。なぜなら、M. tuberculosis とM.lepraeとの間にははるかに大きな違いがあるため、そのような蛋白質は適切に発現されない可能性があり、このM. leprae 蛋白によって誘導される免疫誘導はあまりよく知られていないからである。さらに、ワクチン処理の目的で病原性種から蛋白質を導入することで人体の健康を損なう危険性があり、製薬業界はそのようなリスクを進んで負うものではない。
【0028】
以上の点から、蛋白質配列 No.2および配列 No.3とWieles達の論文(1994, 上記参照)に記載のM. leprae 蛋白質とは、それらの配列の明らかな相同性(図17参照)にもかかわらず、互いに区別される。
【0029】
本発明はさらに、上記定義の蛋白質または微生物を少なくとも一つ含むワクチンまたは薬剤に関するものである。
非グラフト化蛋白質を含むワクチンは、結核に対して個人を免疫するために使用することができ、M. bovis以外の生物原因物質から得られたエピトープを含むグラフト化蛋白質は他の病気に対する免疫処理で使用することができる。
【0030】
指標として、一人に対する1回投与量当たり1〜500 μgの蛋白質あるいは一人あたり103〜107組み換えバクテリアを皮内投与することができる。
本発明のさらに他の対象は、薬学上許容される希釈剤またはアジュバントと組み合わせた上記蛋白質または微生物を少なくとも薬学的に有効な量含む医薬組成物にある。
本発明のさらに他の対象は、特定の結核抗体の存在を調べる生物学的液体を上記蛋白質と接触させる結核に特異的な抗体の検出方法にある。
【0031】
上記蛋白質は担体に固定されているのが好ましい。この検出法はウェスタンブロット(イムノインプリント)法、酵素イムノアッセイ(ELISA) 法またはラジオイムノアッセイ(RIA) 法で蛋白質と、免疫反応を可能にするバッファー溶液と、必要に応じて用いられる生成した抗原−抗体複合体の検出物質とを含む試験用キットを用いて行うことができる。
【0032】
以下、図面を参照して本発明を具体的に説明するが、本発明が下記の説明に限定されるものではない。
[図1]は後述の条件下でイオン交換カラムに保持されない M. tuberculosis画分を分子濾過 (si 300) した場合の240nm における吸光度 (OD) 曲線。
[図2]は上記分子濾過によって得られた画分1に由来する分子を高速イオン交換カラム (DEAE) で分離した場合の220nm における吸光度曲線。
[図3]は上記イオン交換クロマトグラフィーで得られた画分1を逆相カラムクロマトグラフィーにかけた時の220nm における吸光度曲線。
【0033】
[図4A]〜[図4E]はそれぞれ下記で得られたPVDF膜 (メンブレン) の写真:
(1) PVDF膜上に移した分子(4A)の着色剤
Aurodyne着色(Amersyam)、
(2) 生菌(4B)または死菌(4C)によって免疫処理したモルモット
由来の血清混合物
(3) BCG 由来の精製抗原で免疫処理したウサギの血清(4D)
(Infection and Immunity (1993) 61 742-750)
(4) 参照番号I-1081のモノクロナル抗体(4E)
これらPVDF膜には、事前に、低圧イオン交換カラムで分離した画分をアクリルアミドゲル電気泳動で分離した分子をブロットした。レーン0は開始材料に対応し、レーン1は保持されなかった画分に、レーン2は保持された画分に対応する。
【0034】
[図5A]〜[図5E]はSi 300ゲル濾過カラムで得られた5つの画分(1〜5)および低圧DEAEカラムで保持されなかった画分(0)をマイグレートさせて得られるゲルに相当するPVDF膜を示している。同一のゲルを複数のPVDFに移した後、蛋白質着色剤[Aurodye, Amersham (5A)]、生菌(5B)または死菌(5C)で免疫処理したモルモット由来の血清あるいは兎血清(5D)またはモノクロナル抗体(5E)を用いてそれぞれの膜を検出した。
【0035】
[図6A]〜[図6E]は高速イオン交換カラムで得られた画分(1〜3)と分子篩を用いた濾過で得られた画分1(ウェル0)をマイグレートさせて得られるゲルに対応するPVDF膜を示す。上記膜はそれぞれ下記を用いて検出した:
蛋白質着色剤(6A)、
生菌(6B)および死菌(6C)を用いて免疫処理したモルモットの血清に由来する抗体、
兎血清(6D)、
モノクロナル抗体(6E)
【0036】
[図7A]〜[図7D]はイオン交換カラムで得られた画分1(0)および逆相クロマトグライフィーで得られた画分(1〜5)の移動に相当するゲルをブロットした膜を同じコードを有する図6A〜6B、6D〜6Eと同一の試薬で検出した結果を示す。
[図8]は、M. smegmatisにおいてM. tuberculosis H37Rv を発現させるための遺伝子ライブラリーのスクリーニングを示す。M. bovis BCG、形質転換されていないM. smegmatisおよび抗体によって認識される組み換え蛋白質を発現するまたはしない組み換えクローンによって形質転換されたM. smegmatisの上清を各種希釈率でテストした。
[図9]はライブラリーより選択され、E. coli で電気穿孔(エレクトロポレ、electropore))し、アルカリ溶解で抽出した3つのコスミドのアガロールゲル上での移動を示す。
[図10]はBamHI(a)、SmaI(b) 、HpaI(c) 、NotI(d) 、SspI(e),EcoRI(f)およびHindIII(g)で処理したE. coli NM554 から抽出された pLA1 のコスミドDNAのゲル上での移動を示す。
【0037】
[図11]は 45/47kDa 蛋白質のマイコバクテリアにおける発現を示し、培養7日目の細菌培養液の上清を洗浄し、Amicon PM10
膜上で濃縮、凍結乾燥し、免疫インプリントで分析したもの。1/500 に希釈した兎血清のポリクロナル抗体によって蛋白質を検出した。
ウェルはそれぞれ下記 (1)〜(4) を含む:
(1) M. bovis BCG由来の精製した45/47 kDa蛋白質0.25μg
(2) pLA1によって形質転換されたM.smegmatis mc2155の上清5μg
(3) 形質転換されていないM. smegmatis mc2155 の上清5μg
(4) M. bovis BCG上清5μg。
【0038】
[図12]はマイコバクテリアにおける45/47kDa 蛋白質の発現を示し、細菌培養液の上清を洗浄し、Amicon PM10 膜上で濃縮した後、凍結乾燥し、競合ELISA 法で分析した。凍結乾燥させた各種濃度の上清を8000倍に希釈した兎のポリクロナル血清で検出し、混合物を予め精製蛋白質を固定したウェルに写した。
[図13A]、[図13B]は pUC18の pLA1 コスミドをBamHI で処理して得られる断片をリゲーションして得られた各種 pUC18: :M.tuberculosis H37Rv組み換えクローンのプラスミドプロフィール(13A)と、BamHI 制限プロフィール(13B)である。この図は検討した36個のクローンのうち21個を示す。ウェル『P』は基準ベクターであるpUC18 に相当し、ウェル『m』はpKN プラスミドをPvuII によって開裂させた断片であるサイズマーカーに相当する。
【0039】
[図14]は45/47kDa 蛋白質のE.coliにおける発現を可能にする挿入断片の制限地図である。両方向にクローニングされた3kbの挿入断片を含むpLA34 およびpLA4プラスミドから欠失によって一群のクローンを得た。矢印はこれらクローンから『直接』および『逆』プライマーを用いて配列決定する時の方向を示す。
B , Bam HI S , Sma I E , EcoR I K , Kpn I
H , Hind III Sa, Sal I Sp, Sph I
【0040】
[図15]はE. coli における45/47kD蛋白質の発現を示し、細菌培養液の溶解物を免疫インプリントで分析した。DEAE上で精製後、臭化シアンによって活性化したSepharose-4Bカラム上に固定したE. coli の溶解物上に吸着させた兎のポリクロナル抗体によって蛋白質を検出した。
ウェルはそれぞれ下記 (1)〜(4) を含む:
(1) 精製された45/47kDa 蛋白質0.2 μg
(2) pLA34-2 で形質転換したE. coli XL-Blue の溶解物25μg
(3) pLA34 で形質転換したE. coli XL-Blue の溶解物25μg
(4) 形質転換していないE. coli XL-Blue の溶解物25μg。
【0041】
[図16]は E. coliにおける45/47kDa 蛋白質の発現を示し、競合ElISA で分析した細菌培養液の溶解物は未精製の状態で使用した。
[図17]は本発明による配列 No.2とM. leprae (mln 431) 由来の蛋白質の配列とを比較したものである。
[図18]は配列 No.2の蛋白質の疎水性度プロフィールである。
【実施例】
【0042】
実施例1
M.tuberculosis抗原の精製法
1) 抗原の入手
ソートン(Sauton's)の合成培地130 mlを入れたフラスコを用い、BCG の培養に関する通常の方法 (Gheorghiu et al.,Bull.Institut Pasteur 1983, 81:281-288)で、M. tuberculosis (H37Rv株) の培養物を作った。37℃で20日間培養後、培地を回収し、室温で沈降分離および濾過(0.22 μm) した。これらの操作は安全上の理由からグローブボックス内で実施した。回収および濾過した培地は安全フード内で再び0.22μmフィルターを用いて濾過した後、下記の操作に使用した。
窒素雰囲気下、2bar 、4℃でAmicon(PM10)膜に取り、4%のブタノールを含む逆浸透水(retro-osmosed water) を用いて強く洗浄し、元の量の10〜20倍に濃縮した。Amicon PM10 膜で除外されなかった分子を含む濃縮培地を凍結乾燥し、重量測定後、−20℃で粉末状態で保存した。以下の精製操作に使用した12gの開始材料は70リットルの培地から得られたものである。
精製操作
【0043】
2) 低速イオン交換カラム
約240 mlの Triacyl Mゲル(SEPRACOR)の高さが300 mm、直径32 mm の調製用低速イオン交換カラムを準備した。4%のブタノールを含む緩衝生理食塩水(10mM Na2HPO4 /NaH2PO4, pH=7 および 10mM NaCl)を用いてカラムを平衡化した。
上記段階で濃縮後に凍結乾燥して調製した材料を、上記緩衝化生理食塩水に溶解し、40,000Gで12分間超遠心分離した。遠心後の溶液の上部4/5だけを回収し、蠕動ポンプを用いてイオン交換カラムに添加した。カラムに保持されない第1主要画分を回収した。緩衝生理食塩水(10mM Na2HPO4 /NaH2PO4,pH=7.5および1M NaCl)を用いて溶出し、第2画分を得た。2バールの圧力下で Amicon (PM10)膜に取り、4%ブタノールを含む逆浸透水を用いてそれぞれの画分を強く洗浄し、約15倍に濃縮した。カラムに保持されない画分は2.9 gの材料および大部分の分子を含んでおり、これを次の段階で精製した。カラムに保持され、塩溶液で溶出した画分には約1.01gの材料が含まれていた。
【0044】
3) ゲル濾過
50×750 mm(SERVA) の調製用高速カラム(Si300 カラム 3μm)を4%ブタノールを含む緩衝生理食塩水(50mM Na2HPO4,KH2HPO4 を用いてpH=7.5に調整したもの)を用いて平衡化した。
この溶液は予め膜(0.22 μm) を用いて濾過しておく。カラムの流速は1.25ml bar/分に調節し、45bar の設定最大圧には到達しなかった。
カラムに添加する材料は緩衝液/ブタノール溶液を用いて濃度50mg/mlに調製した。10mlのサンプルを調製し、−20℃で凍結した。解凍後に再び濾過してカラムに添加した10mlのサンプルには約500mg の未精製材料が含有されていた。典型的な分離法である240nm での吸光度プロフィールを図1に示す。このプロフィールに基づいて選択された5つの主要画分を4℃で濃縮し、Amicon PM10 膜上で4%のブタノールを含む逆浸透水を用いて強く洗浄した。濃縮された画分をそれぞれ凍結乾燥し、重量測定し、−20℃で保存した。この段階で得られた画分1は生菌を用いて免疫処理したモルモット由来の抗体または結核患者由来の抗体によって認識される主要な分子を含んでいた。この画分のみを用いて以下の段階を行う。
【0045】
4) イオン交換カラム
4%のブタノールを含む緩衝生理食塩水(10mM Na2HPO4 /NaH2PO4, pH=7.5 および10mM NaCl)を用いて、DEAE-TSK 5PW調製用カラム 21.5 ×150mm (LKB) を平衡化した。流量6ml/分で最大圧力は30bar 以下であった。溶出バッファーに関してはNaCl濃度のみを変化させた(1M)。合計100 mgの上記材料を含む4mlのサンプルを注入した後、図2に示す直線状濃度勾配を与えた。240nm での吸光度プロフィールに従って主要画分を回収した。これらの画分を濃縮し、Amicon PM10 膜上で4%ブタノールを含む逆浸透水を用いて洗浄し、凍結乾燥した。重量測定後、各画分を−20℃で保存した。この段階で得られた画分1のみが生きた細菌を用いて免疫処理したモルモット由来の抗体によって認識される分子の大部分を含んでいた。この画分を用いて次の分離段階を行った。
【0046】
5) 逆相カラム
4.6 ×250mm の RP300C8 10μm(Aquapore Brownlee Lab.)カラムを0.22μmのフィルターで濾過した酢酸アンモニウムバッファー(20mM NH4COOCH3) を用い、最大圧力115bar、流速2ml/分で平衡化した。10mgを含む1mlのサンプルを注入後、図3のプロフィールに従って90%のアセトニトリルを含む溶出バッファーを用いて溶出した。220nm における吸光度プロフィールから5つの主要な画分が分離され、それら画分を減圧下に40℃で蒸発し、濃縮し、その後凍結乾燥した。
【0047】
6) 抗原の免疫検出
laemmli の通常の方法(Nature, 1970, 277: 680-685)に従って、10%ポリアミドを含む0.1 %SDS変性ゲルを調製した。精製段階で得られた10〜2μgの材料を含むサンプルを5%メルカプトエタノール、3%のSDSおよび痕跡量のブルモフェノールブルーを含むバッファーに溶解し、ゲルのウエルに10μlずつ添加した。電気泳動してブロモフェノールブルーをゲルの端まで移動させた後、一夜、中程度の電場を加えてサンプル中に存在する分子をPVDFシート(Millipore) 上に移した。[Harlow and lane, Antibodies, A laboratory Manual, Cold Spring Harbor Laboratory (Publishers), 1988]。
PVDEシートをコマシーブルー溶液で1分以内で着色し、その後脱色して分子量マーカーを識別可能とし、その形状を鉛筆でなぞる。完全に脱色した後、PBS +Triton X100 3%を用いて室温で30分間シートを洗浄し、その後、PBS 単独で5分間ずつ3回洗浄した。次いで、5%の脱脂粉乳を含むPBS を用いて37℃で1時間シートを飽和し、PBS +Tween 20(0.2%)で3回洗浄した。
PBS +Tween 20バッファー(0.2%)+粉末ミルク(5%)で20倍に希釈した抗血清を用い、37℃で1時間30分、定期的に攪拌しながらインキュベートした。さらにPBS +Tween を用いて3回洗浄し、次いで、アルカリホスファターゼを用いて標識した抗イムノグロブリン抗体と一緒にインキュベートした。ホスファターゼ(Biosys) を用いて標識した人間とモルモットの抗イムノグロブリン抗体を PBS+Tween20 (0.2%)+ミルク(5%)で最終希釈倍率を2500倍にして使用した。37℃で1時間30分インキュベート後、PBS +Tween を用いてPVDFシートを3回洗浄し、BCIPおよびNBT (Harlow and Lane 上記)を含む検出バッファー中で室温で5〜10分間培養した。反応を停止させ、乾燥させた後シートの写真を撮影した。
【0048】
7) アミノ酸組成物
BeckmannのLS6300分析装置を使用して、Institut Pasteur
Organic Chemistry Departmentで各クロマトグラフィー画分について合計アミノ酸組成を分析した。
45-47kD 蛋白質のアミノ酸頻度(amino acid frequency)で表した合計組成は以下のとおり:
ASN/ASP:10.4%;THR:5.7 %;SER:5.6 %;GLN/GLU:6.3 %;GLY:7.1 %;ALA:19.3%;VAL:6.2 %;ILE:2.2 %;LEU:4.4
%;TYR:2.2 %;PHE:2.4 %;LYS:2.7 %;ARG:2.7 %;PRO:20.9%。
【0049】
実施例2
M. tuberculosis 蛋白および蛋白画分の免疫特異性の決定と、生菌を用いて免疫処理したモルモットの抗体で認識される抗原の単離
12〜15匹のモルモット群(Hartley のメス、試験開始時の重量=250〜300g)に生きたマイコバクテリア(2×107単位のBCG を0.1 ml生理食塩水に添加したものを2回皮内注射)または同じ株のマイコバクテリア0.2 mgを熱殺菌(120℃、30分)したものを完全フロイントアジュバントに生理食塩水を懸濁して(1/1) 成るエマルション溶液0.5 mlとして筋肉注射で投与した。免疫処理してから7〜12ヶ月後に各モルモット群から血清サンプルをとり、濾過(0.22 μm) 後、少量に分割し、−20℃で凍結保存した。いくつかの群の血清試験を行った(生菌で免疫処理したもの5群と死菌を用いて免疫処理したもの6群)。それぞれの免疫処理タイプを代表する血清群を用いて得られた結果を示す。同じ免疫処理方法ではグループ間の差は最小であった。
【0050】
1) 低速イオン交換カラムを用いた分離段階
培養培地(Amicon PM10 膜を用いて洗浄・濃縮後、凍結乾燥したもの)を超遠心分離し、その後低速イオン交換カラムに添加した。カラムに保持されなかった画分と、高モル濃度緩衝溶液で溶出した画分との2種類の画分が得られ、これらをAmiconPM10膜を用いて洗浄・濃縮し、凍結乾燥した。
各画分(10 μm) をSDS ゲルのトラックに添加し、電気泳動した後、PVDF膜に移し(ブロットし)、免疫検出法で各種血清と反応する分子の大部分を含む画分を識別した。
【0051】
[図4]は同一のゲルを、蛋白質用の着色剤(Aurodye-Amersham)(4A)、生菌(4B)または死菌(4C)で免疫処理したモルモットの血清を用いて検出した免疫インプリントを示す。免疫インプリント4Dおよび4FはBCG と同一の分子に対する兎血清 (Infection and Immunity, 1993, 61, 742 〜750)およびパスツール研究所のCNCM (Collection Nationale de Cultures de Micro-organismes) に寄託されたモノクロナル抗体を生産するI-1081ハイブリドーマの上清を用いて検出した。カラムに保持されたなかった画分のみが、生菌または死菌を用いて免疫処理されたモルモット由来の血清によって認識される45/47kDa分子または上記ハイブリドーマの上清によって認識される45/47kDa分子を含んでいた。
【0052】
2) Si300 を用いた分子濾過段階
上記段階でカラムに保持されなかった画分を、500 mgの材料を含む10mlのサンプルとしてSi300 カラムに添加した。図1に示したプロフィールに従って画分1〜5を分離し、一連の添加によって得られた精製物を合わせ、洗浄、濃縮、凍結乾燥した。
各画分(10μg)をSDSゲルのトラックに添加し、電気泳動を行ってPVDF膜に移し、免疫検出法で各種血清と反応する蛋白質の大部分を含む画分を同定した。
[図5]は同一のゲルを、蛋白質着色剤(Aurodye Amersham)、生菌(5B)または死菌(5C)で免疫処理したモルモットに由来する血清を用いて検出した免疫インプリントを示す。免疫インプリント5Dおよび5Eは、BCG から精製した分子に対する兎血清およびI-1081モノクロナル抗体を用いて検出した。
画分1に存在する45kDおよび47kDの2種類の抗原は主として生菌を用いて免疫処理した動物に由来する抗体、ポリクロナル兎血清またはモノクロナル抗体によって認識された。この画分を第2の精製段階用に選択した。
【0053】
3) イオン交換段階
上記画分のサンプル100 mgをDEAE−TSK調製カラムに添加し、NaCl濃度勾配を付けて溶出させた。溶出分子の220nmにおけるプロフィールから主要な3つの画分(図2)が画定された。一連の添加で得られた各画分を集め、洗浄、濃縮および凍結乾燥した。
上記画分を5μgずつSDSゲル電気泳動し、PVDFシート上の免疫インプリントを蛋白質着色剤(Aurodye)(図6A)、生菌(図6B)または死菌(図6C)を用いて免疫処理したモルモットの血清、兎血清(図6D)あるいはモノクロナル抗体(図6E)を用いて検出した。1−DEAE画分は、死菌を用いて免疫処理した動物の抗体によって認識される抗原をごくわずかしか含んでいなかった。一方この同じ1−DEAE画分は生菌を用いて免疫処理されたモルモット由来の抗体および兎血清およびモノクロナル抗体によって強く認識される45/47kD のダブレットを含んでいた。この画分1−DEAEを用いて次の精製段階を行った。
【0054】
4) 逆相カラム段階
10μm RP 300 カラムを、酢酸アンモニウムバッファー(20mM)で平衡化し、上記1−DEAE画分を最大5〜10mg含むサンプル1mlを添加した。図3に示す0〜90%のアセトニトリル濃度勾配を付けて溶出し、5つの主要画分を回収した。これらの画分から40℃で減圧蒸発してアセトニトリルの大部分を除去/濃縮し、凍結乾燥した。
Aurodye を用いた蛋白質の着色(図6)では、画分4(アセトニトリル濃度30〜50%)が生菌を用いて免疫処理した動物由来の抗体あるいは兎血清中に存在する抗体またはモノクロナル抗体によって認識される分子の大部分を含んでいた。
【0055】
実施例3
Mycobacterium tuberculosis由来の45/47kD蛋白質のMycobacterium smegmatis およびEscherichia coliへのクローニングと発現
1) 材料および方法
1.1 菌株とその成長条件、上清と菌抽出物の調製
ソートンの合成培地で M. bovis BCG (1173P2 株)を37℃で7日間培養し、上清液を0.22μmの膜で濾過した。この上清液を4%ブタノールの存在下で未精製状態で保存するかAmicon-PM 膜を用いて濃縮後に凍結乾燥した。
M. segmatis mc2155 (Snapper 達、Molecular Microbiol.4, 1911-1919, 1990)を、7H9 +OADC液体培地で37℃で7日間培養した。pYUB18:M. tuberculosisライブラリー由来のコスミドによって形質転換した各M. smegmatis mc2 155クローンを25mg/mlのカナマイシン存在下で培養した。その後培養液を5000rpm で15分間遠心分離し、培養液の上清を分離して、4%ブタノール存在下で4℃で保存した。培地組成によって妨害されないELISA 試験はこれらの調製物を用いて行った。クローン培養液の上清をSDS-PAGEゲル上で分析する際には、それらをソートン合成培地で37℃で7日間培養し、0.22μm膜を用いて培養物の上清を濾過し、Amicon-PM10 膜で濃縮し、凍結乾燥した。
【0056】
E. coli NM554 およびXL 1-Blue 株を固体または液体Luria-Bertani (LB)培地で37℃で培養した。pUC18 プラスミドで形質転換したE. coli XL 1-Blue クローンを25μm/lのアンピシリン存在下で培養した。
E. coli XL1-BlueとpUC18:M. tuberculosis 組み換えプラスミドで形質転換したクローンとを一夜(16h) 培養した後、培養液を一連の急激に凍結/溶解(−70℃/+60℃)することによって培養液溶解物を調製した。溶解物を遠心分離し、上清を分離し、−20℃で保存した。これら調製物からの蛋白質の分離はBCA 法で行った(Pierce)。
【0057】
1.2 クローニングベクター
使用したM. tuberculosis の遺伝子ライブラリー(Jacobs達1991、上記)は、M. segmatis mc2155への電気穿孔法により、Stewart Coleによって作製された。本出願人は400 個の組み換え体を使用した。
ライブラリーはコスミドのシャトルベクターpYU818内で作製した。このシャトルベクターは、pYUB12プラスミド (Snapper 達、Proc. Natl. Acad. Sci., USA, 1988, 85:6987-6991)に由来するもので、このプラスミドはλバクテリオファージのCos 配列が挿入されてファージ溶解物状態のライブラリーにおける組み換えコスミドの増幅と良好な保持が可能になっている。このライブラリーは以下の方法で作製した。
すなわち、M. tuberculosis H37Rv 株のゲノムDNAを、最大35kbから45kbの断片が得られるような条件で、酵素Sau 3aで部分的に処理し、これらの断片を精製後、pYUB18にリゲーションし、制限エンドヌクレアーゼBamHI で処理後、脱燐酸基化を行った。
【0058】
pUC18 プラスミドベクター(Yanisch-Perron 達、Gene, 1985, 33: 103-119)を用いて、E. coli XL-Blue へのサブクローニングを行った。この多コピープラスミドはβガラクトシダーゼの末端アミノ酸断片をコードしたE. coli のラクトースオペロンに由来するDNAを含む。この断片はイソプロピルβ−D−チオガラクトピラノシド(IPTG)によって誘導可能で、E. coli XL1-Blue宿主株によってコードされる不完全なβガラクトシダーゼフォームと供にアルファ相補性(alpha-complementation) を確立することができる。従って、外部DNA の挿入によって、アルファ相補性の全廃が誘導される。組み換えプラスミドは、宿主株に形質転換されるとコロニーが白色を呈することによって確認され、これに対して細菌がpUC18 プラスミドで形質転換されるとコロニーは青色を示す。このスクリーニングは、IPTGおよびX-Gal 酵素基質の存在下で行った。
【0059】
1.3 分子生物学的方法
1.3.1 M.Smegmatis mc2 155コスミドの抽出
いくつかの変更を加えてM. smegmatis用に適応させたアルカリ溶菌法(Jacobs 達、1991, 上記) を用いて組み換え pYUB18:M. tuberculosis コスミドの抽出を行った。培養5日目(指数増殖期終了後)に細菌を回収し、5000rpm で10分間遠心分離した。細菌残留物(3ml)を5mlの溶液A(50 mMグルコース、25mMトリスHCl pH8、10mM EDTA 、リゾチーム10mg/ml) に再懸濁し、37℃で20分間インキュベートした。2倍量(10ml)の溶液B(0.2N NaOH、1%SDS)を添加し、逆混合した。混合物を65℃で30分間インキュベートし、その後、4℃で15分間インキュベートした。最後に1.5 倍量(7.5ml) の溶液C(5mM 酢酸カリウム、酢酸11.5%)を添加し、逆混合した。混合物を4℃で30分間インキュベートした。続いて調製物を4℃で13000rpm、15分間の遠心分離し、上清を回収して、同量のフェノール/クロロホルム50/50で処理した。
抽出後、試験管を4000rpm で10分間の遠心分離した。水相を清潔な試験管に移し、−20℃で保存しておいた2倍量のエタノールを用いて2回処理した。反転後、これを−20℃で少なくとも1時間保存し、その後、12000rpmで20分間遠心分離した。最後に残留物を同量の70%エタノール(−20℃に保存しておいたもの)で洗浄し、Speed-Vac で5分間乾燥させた。乾燥残留物を500 μlの滅菌水に懸濁し、−20℃で保存した。
【0060】
1.3.2. E. coli プラスミドの抽出および精製
pYUB18コスミドおよびpUC18 組み換えプラスミドの迅速な抽出はアルカリ溶菌法(Birnboim 達、Nucleic Acids Res., 1979,
7: 1513)で行った。
アルカリ溶解操作後、関連するコスミドと組み換えプラスミドとを、臭化エチジウムの存在下で塩化セシウム濃度勾配超遠心分離で精製した(Maniatis 達、Cold Spring Harbor, New
York: Cold Spring Harbor Laboratory Press, 1982)。
【0061】
1.3.3 形質転換技術
塩化カルシウムを用いた化学的方法
この一般的方法は pUC18組み換えプラスミトでE. coli XL1-Blueを形質転換するのに使用した。まず最初にコンペテントバクテリアを調製した。20mlの2YT 培地に、一夜培養したプレカルチャーを1/100 の割合で植えつけた。細菌を、攪拌しながら37℃で2時間、OD=0.6 になるまで培養し、その後4℃、400
rpm で10分間、遠心分離した。残留物を8mlの100mM CaCl2 に懸濁し、氷浴中に15分間保持し、その後再び4℃で4000rpm 、10分間の遠心分離した。最後に残留物を1.6 mlの100mMCaCl2に懸濁し、氷浴中で30分間保持した。
こうして調製したコンピテントバクテリアはすぐに形質転換に利用するか、あるいは4℃で数日間保存してもよい。形質転換時には2μlのDNA に200 μlのコンピテントバクテリアを混合した。混合物を氷浴中で45分間保存し、その後42℃で2分間の温度ショックを加えた。800 μlの2YT 培地を添加し、その後攪拌しながら調製物を37℃で1時間インキュベートし、Mlアンピシリンプレートに50μl〜200 μl/プレートの割合で拡げた。翌日、コロニーをカウントし、形質転換効率を計算した。
【0062】
物理的電気穿孔法
この方法は大型ベクターを用いてE. coli を形質転換するために使用した。すなわち、E. coli のNM554 株を、50kb以上のサイズを有する組み換えpYUB18コスミドを用いて電気穿孔にかけた。コンピテントバクテリアは新規に調製した。200 mlの 2YT培地に、一夜培養したプレカルチャーを1/100 の希釈率で植えつけ、37℃で3時間細菌を培養した。その後、6000rpm で10分間遠心分離した。残留物を4℃の滅菌水10mlに取り、その後4℃の滅菌水190 mlに懸濁した。細菌を、再び6000rpm で10分間遠心分離し、4℃の滅菌水10mlを用いて再度洗浄した。最後に残留物を400 μlの10%グリセロールに懸濁した。
電気穿孔はBio-Rad Gene Pulser を用いて行った。4mmのセル内で100 μlの細菌を1〜4μlのDNAと混合した。混合物に電気ショックを与え(2500V、25μF)、その後、1mlの2YT 培地を素早くセルに添加した。全体を試験管に移し、攪拌しながら37℃で1時間インキュベートした。インキュベート後培養液をML−アンピシリンプレートに50μl〜200 μl/プレートの割合で拡げた。翌日コロニーをカウントし、形質転換効率を計算した。
【0063】
1.3.4 酵素処理断片のクローニング
クローニングすべきDNAをBamHI 制限エンドヌクレアーゼで処理した。pUC18 プラスミドを同様の方法で処理した。所望のpYUB18組み換えコスミドから得られた断片を、T4 DNAリガーゼ酵素(Amersham)の活性を利用してプラスミドベクターにリゲーションした。リゲーションは、20μlの容量で16℃で一夜行った。リゲーション混合物全体を用いてE. coli XL1-Blueの形質転換を行った。遺伝情報発現後、全ての細菌を25μg/ml、(TPTG 、X-Gal)の割合でMl- アンピシリンプレートに拡げた。アルファ相補性が確立されないクローンが、コロニーの色(白色)によって確認された。
クローニングによる精製後、組み換えクローンを検討した。アルカリ溶解によってプラスミドDNA を抽出し、その後、制限エンドヌクレアーゼBamH Iによる処理の前または後に0.8 %のアガロールゲルを用いて分析した。
【0064】
1.3.5 制限地図の作製
両方向にクローニングされた3kbのBamH I-BamH I 断片を含むpLA34 およびpLA4組み換えプラスミドを、pUC18 マルチサイトリンカー(ポリリンカー)内の部位を有する各種の制限エンドヌクレアーゼで処理した。制限エンドヌクレアーゼBamH I、Hind III、Sph I 、Xba I 、SalI、Kpn I 、Eco RIおよびSma
I を用いて単一または二重の処理を行い、0.8 %アガロールゲルを用いて分析した。臭化エチジウムを用いてDNAを染色した後、それらの移動距離をマーカー(インターナルラボラトリースタンダード, Pvu II で処理したpKN プラスミド)と比較して各断片のサイズを決定した。
【0065】
1.4 蛋白質の検出方法
1.4.1 ELISA 法
競合ELISA 法によってポリクロナル血清(Romain達、1993,
上記)を用いて、細菌培養物より得られる各種調製物中の45/47kDa 蛋白質濃度を測定した。
この兎のポリクロナル血清は、通常の免疫処理方法(フロインドの不完全アジュバントに懸濁した精製蛋白質50μgを注射し、1ヶ月後に25μgを注射する)に従って45/47蛋白質に対して作製されたものである。
第1のマイクロプレートのウェルを炭酸塩緩衝液を用いて1μg/mlに調製した精製蛋白質溶液か15日目のMycobacterium1 bovis BCG 上清液(10μg/ml)のいずれかで覆った。37℃で1時間かけて抗原を固定し、マイクロプレートをPBS で5回洗浄した。第2のインキュベーションではPBS と0.5 %ゼラチンと4%ブタノールとを含む溶液を用いて37℃で1時間、ウェルを飽和させた。その後、PBS-Tween 0.1 %を用いてマイクロプレートを5回洗浄した。
【0066】
試験は以下のように行った:
第2のマイクロプレート内でPBS-Tween 0.1 %、0.25%ゼラチン、4 %ブタノールを用いて各種濃度(純粋、1/2 、1/4 、1/8 等)に希釈した分析すべき上清50μlと、PBS-Tween 0.1
%、0.25%ゼラチン、4 %ブタノールを用いて希釈倍率1/4000に調製した兎血清50μlとを37℃で1時間インキュベートし、次に混合物を第1のマイクロプレートに移して37℃で1時間インキュベートした。0.1 %のPBS-Tween を用いてマイクロプレートを10回洗浄した。最後に、兎に対する抗IgG H +L 複合抗体(Byosys)をアルカリホスファターゼを用いて標識し、 PBS-Tween 0.1 %、0.25%ゼラチン、4 %ブタノールを用いて希釈率1/4000に調製したものを37℃で1時間インキュベートした。PBS-Tween 0.1 %を用いてマイクロプレートを10回洗浄した。
最後に、酵素基質であるパラ−ニトロフェニルホスフェート(pNPP)を、NaHCO3、MgCl2、pH9.6 緩衝液を用いて40mg/24ml濃度に調製した状態で1時間または一夜インキュベートした。Titerteck Twinreader上、414nm および690nm でODを読み取った。
【0067】
1.4.2 免疫インプリント法
変性SDS−PAGEゲル上で行う通常のゲル電気泳動方法を採用した(Laemmli, Nature, 1970, 277:680-685)。その後、PVDF膜に電気的に移した(Towbin 達、Proc. Natl. Acad. Sci. USA, 1979, 76:4350-4354; Pluskal 達、Biotechniques,1986, 4:272-283)。
ゲル上で分析したサンプルを定量し、M. smegmatis上清については凍結乾燥体の重量μg (5μgを添加) を、E. coli 溶解物については蛋白質の重量μg(25 μgを添加) を測定した。
精製したM. bovis BCG蛋白質を、トラックあたり0.25μg蛋白質の濃度でゲル上に添加した。
膜上に移された蛋白質は、マイコバクテリア中で発現される蛋白質に対する兎のポリクロナル血清(希釈率:500 分の1)を用いて検出した。
E. coli 内の組み換え蛋白質を検出するために、これらポリクロナル抗体を DEAE(TrisacrylD) カラムで精製し、その後得られた免疫グロブリンを、臭化シアノゲン(Pharmacia) によって活性化したSepharose-4Bカラム上に固定されたE. coli の溶解物に吸着させた(Maniatis 達、1982) 。保持されなかった抗体は4℃で保存し、100 分の1の希釈率で膜に移された蛋白質を検出するために使用した。
アルカリホスファターゼで標識された種特異性のある抗Ig H+L 複合抗体(Bio-Sys) を3000分の1の希釈率で上記抗体を検出するために使用した。最後に、アルカリホスファターゼの活性は、2種類の人工色素産生基質、テトラゾリウムブルーと5-ブロモ-4- クロロ-3- インドリルホスフェートを用いて検出した。
【0068】
1.5 DNA配列決定
ヌクレオチドの配列決定は、2種類のクローンpLA34 およびpLA4から各種欠失によって得られたクローン群を使用して行った。欠失は作製した制限酵素地図に準じて選択した。
配列決定は2本鎖プラスミドDNAマトリクスを用いて行った。T7シーケンシングキット(Pharmacia) および35SATPを用いてサンガーの方法を適用した。
各種の欠失クローンおよびpUC18 プラスミドのユニバーサルプライマー(Direct and Reverase Primer)、続いて合成オリゴヌクレオチドを用いて配列を決定した。
配列は、2種類の相補鎖について決定した。
M. tuberculosis のゲノムDNAのGCの割合が高い(65 %)
コンプレッション領域は、7-Deaza dGTPすなわちdGTPの化学的類似物を含むT7 Deaza G/A 配列決定キット(Pharmacia) を用いて配列決定した。
【0069】
1.6 配列解析
得られた隣接する配列の比較および組合わはSTADENプログラムを用いてUnix(登録商標)で行った。EMBLおよびGen-Bankデータバンクの配列から検索した配列の相同性は GCGのFASTA およびT-FSTAプログラムを用いて作製した。
【0070】
2) 結果
2.1 M.tuberculosisに由来する45/47kDa蛋白質のM.smegmatis へのクローニングおよび発現
2.1.1 M. smegmatisにおいてM. tuberculosis を発現させるための遺伝子ライブラリーのスクリーニング
使用した遺伝子ライブラリー(Jacobs 達、1991, 上記)は、制限エンドヌクレアーゼSau 3aを用いてゲノムを部分的に切断して得られる40kbの断片をpYUB18コスミドベクター内にクローニングすることによって作製した。パルスフィールドゲル電気泳動によって4200kbと推定されるゲノムのサイズは、約100 〜150 個のクローンに含まれる。
競合ELISA を用いて液体培地中の蛋白質を決定した (Romain達、1993, 上記)。それによってM. bovis BCGの7日目の培養物の上清に含まれる45/47kDa蛋白質の量の検出および定義が可能になった(図8)。
この試験は以下のような利点を有する:優れた感度すなわち8000分の1に希釈したポリクロナル血清を用いることによって液体培地中の蛋白質を1ng/mlのオーダーで検出でき (Romain達、1993, 上記)、さらに一連のサンプルを迅速にスクリーニングするため操作が容易である。
【0071】
電気穿孔法でM. smegmatisに導入された400 個のpYUB18::M.tuberculosis H37Rv組み換えクローンをスクリーニングした。 そのために、各種のクローンを7H9 +OADC培地で7日間培養した。この試験では、培養液を遠心分離して得られる上清を分析して組み換え蛋白質を検索した。
M. bovis BCG 45/47kDa 蛋白質に特異的なモノクロナル抗体によって認識される蛋白質を発現する3つのクローンが見出された(図8)。この第1のスクリーニングでは、マイクロタイタープレートのウェルを、45/47kDa 蛋白質が全質量の2%と測定されたM. bovis BCG培養物の上清で覆った。選択した3種類のクローンを、マイクロタイタープレートのウェルを精製した45/47kDa 蛋白質で覆う第2の試験で確認した。
【0072】
2.1.2 選択された組み換えプラスミドの遺伝子分析
選択した各種コスミドを検討するために、変形アルカリ溶解によってM. smegmatis DNAを抽出した後、電気穿孔法によってE. coli NM554 に導入した。マイコバクテリアの染色体外DNAは細胞壁の複雑さ(溶解が困難である)およびベクターコピー数の少なさ(細菌1個に付き平均3〜10個) によって採取が困難である。E. coli NM554 に形質転換された3種類のクローンをML−カナマイシンプレート上で単離し、アルカリ溶菌によって抽出したコスミドDNAを0.8 %アガロールゲル上で分析した。
3つのクローンはサイズ 50kb 以上のDNAを含んでいた。制限エンドヌクレアーゼBam HIによる処理を行い、選択された3つのコスミドのプロフィールを識別した。これらは同一であることが明らかになった(図9)。プロフィールは、pYUB18ベクターに対応する12kbのバンドを示し、さらにクローニングされたDNA断片(約40kb)に相当する一連の低分子量バンドを示した。得られたバンドの数およびゲル上のそれらバンドの位置を考慮すれば、単離されたコスミドは同一であるとみなすことができる。
G+Cを多く含むDNAに対して幾分多くの開裂部位を有する制限エンドヌクレアーゼを用いて、pLA1コスミド単独の各種処理を行い、45/47 kDa蛋白質の遺伝子(単数または複数)を含むのに十分な中程度の長さを有する断片を識別し、それらのサブクローニングを行った(図10)。
【0073】
2.1.3 M. tuberculosis に由来する45/47 kDa蛋白質の M. smegmatisにおける発現
約40kbの挿入部分を含むpLA1コスミドによってM. smegmatisにおける組み換え蛋白質の発現が行われ、培養物上清中でポリクロナル抗体によって検出された。
発現された蛋白質のおおよそのサイズを決定するために、培養7日目の上清の凍結乾燥物を免疫インプリントにより分析した。M.smegmatis において発現された組み換え蛋白質は2種類の分子量45/47kDa を有し、これは明らかにM. bovis BCGにおいて発現されるものと同一である(図11)。
別の試験ではこれら組み換え蛋白質の発現レベルはM. bovisBCG に匹敵するものであった。凍結乾燥した上清から得られた所定量の蛋白質を競合ELISA で定量した。各種濃度の凍結乾燥した上清を、8000分の1に希釈した兎のポリクロナル血清を用いて検出した。組み換えM. smegmatisは、M. bovis BCGと比較して5倍量の蛋白質を発現した(図12)。
この挿入部分のサブクローニング並びに異種宿主(E. coli)
における組み換え蛋白質の分析を行って、これらの蛋白質をコードする遺伝子の数を決定した。
【0074】
2.2 M. tuberculosis に由来する45/47 kDa蛋白質のE. coli へのクローニングおよび発現
2.2.1 45/47蛋白質のE. coli におけるサブクローニングおよび発現
pLA1を異種宿主であるE. coli NM554 に形質転換すると、細菌培養物または溶解物の上清からは組み換え蛋白質は検出されなかった。これら蛋白質の発現を促進するために、コスミドをBam HIで処理して得られる断片をpUC18 プラスミドにサブクローニングする操作を行った(Yanisch-Perron 達、Gene, 1985,
33: 103,119)。
E. coli XL1-Blueに形質転換されたpUC18::M. tuberculosis組み換えプラスミドを宿主細菌のβガラクトシダーゼ発現の欠損によって選択した。一連の36個のクローンのうち『白色』クローンのプラスミドDNAを、アルカリ溶解および制限エンドヌクレアーゼBam HIによる処理を行って調製した。
アガロールゲル上で観察される得られたプラスミドのサイズは、数種類のプロフィールを示し、組み換えプラスミドが異なっていることが示された(図13A)。
【0075】
アガロールゲル上で観察されるクローニングされた挿入部分のサイズも、異なる制限プロフィールを示した(図13B)。これらのプロフィールは全て、pUC18 ベクターに相当する2.8kb
の断片と、クローニングされた挿入部分に相当するサイズの異なる一連の断片とを示した。
サイズが大きいためにクローニングの困難な12kbの断片を除いて、全ての処理断片を単独、2個または3個ずつクローニングした。
選択した36個のクローンについて、E. coli XL1-Blueにおける組み換え蛋白質の発現を誘導する能力に関するスクリーニングを行った。この試験は上記と同じ競合ELISA テストによって行った。
細菌培養物の上清中に組み換え蛋白質は検出されなかった。一方、3kbの挿入断片を少なくとも1つ含むクローンの細菌溶解物中に組み換え蛋白質が検出された。
この試験で測定された蛋白質の発現レベルは、プラミドのサイズによって影響されるものと思われた。検討した36個のクローンのうち、発現を可能にする2つのクローン、それぞれ3kb
および7kbの挿入部分を含むpLA34 およびpLA35 が見出された。
発現レベルは、表1(下記参照)の結果に示すようにpLA34 の方が高かった。
【0076】
2.2.2 pLA34 およびpLA34-2 クローンの制限地図
pUC18 のマルチサイトリンカー(ポリリンカー)中に存在する一般的な制限エンドヌクレアーゼに対する各種開裂部位を同定することによってpLA34 プラスミドについての制限地図を決定した(図14)。単一の制限部位Eco RIが、3kbを2kbおよび1kb の2つの断片に分断していた。
2kbのBamHI-EcoRI 挿入部位を有するpLA34-2 クローンは、上記のクローンから欠失によって作製された。このクローンもまた細菌溶解物内に組み換え蛋白質を発現させた(図15)。
細菌溶解物の免疫インプリント分析の結果、45/47kDa の2種類の分子量を有する蛋白質がM. bovis BCG内で発現される天然の蛋白質と明らかに同一であることが示された(図16)。
【0077】
2.2.3 M.tuberculosis H37Rvの45/47kDa 蛋白質をコードするヌクレオチド配列の分析
45/47kDa 蛋白質をコードした遺伝子の完全なヌクレオチド配列、上流の配列およびアミノ酸から推定される配列を、配列No. 1および配列 No.2に示す。
蛋白質ダブレットの発現を可能にする単一の遺伝子はヌクレオチド配列の1082から2056までの位置に975 塩基対を有する。 リボソーム固定のコンセンサス配列(Shine Dalgarno)は遺伝子の上流で確認された。
この遺伝子はGC含有率がM. tuberculosis の65%に比べて69.4%と高い。
この遺伝子から推定される蛋白質はシグナルペプチダーゼのANA 開裂部位を含む典型的なシグナル配列を有する。
この遺伝子は39アミノ酸のシグナル配列を含む325 アミノ酸の蛋白質をコードしている。
M. bovis BCGおよびM. tuberculosis から精製した蛋白質のアミノ酸組成を生化学的分析して得られる結果と蛋白質配列から類推される結果とを比較すると、非常に優れた一致が見られる(表2)。
従って、Mycobacterium smegmatis およびE. coli において2つの分子量を有する蛋白質の発現を可能にする単一の遺伝子が存在するという結論が導かれる。
【0078】
2.2.4 蛋白質配列の分析および配列の比較
推定されるアミノ酸配列から計算される分子量は28.7kDa である。
等電点の計算値は4.36である。この結果も精製したM. bovisBCG 蛋白質について行った生化学的な等電点の測定と非常によく一致する。
推定されるアミノ酸配列からプロリンとアラニンの含有率が高いことが示される(21.8 %および19.1%) 。
完全な配列は最近発表されたMycobacterium leprae由来の蛋白質との間に相同性を示す。2つの配列の比較を図17に示す。2つの蛋白質間の相同性率は65.4%である。Mycobacteriumlepraeについて報告されたこの蛋白質も分泌蛋白に特異的なシグナル配列を有する。
M. tuberculosis から導かれる蛋白質(本発明の対象:配列No. 2)の疎水性プロフィールを求めた。これを図18に示す。
【0079】
【表1】
【表2】
【0080】
【表3】
【表4】
【表5】
【表6】
【表7】
【表8】
【表9】
【図面の簡単な説明】
【0081】
【図1】イオン交換カラムに保持されない M. tuberculosis画分を分子濾過 (si 300) した場合の240nm における吸光度 (OD) 曲線。
【図2】上記分子濾過によって得られた画分1に由来する分子を高速イオン交換カラム (DEAE) で分離した場合の220nm における吸光度曲線。
【図3】上記イオン交換クロマトグラフィーで得られた画分1を逆相カラムクロマトグラフィーにかけた時の220nm における吸光度曲線。
【図4】A〜EはPVDF膜 (メンブレン)の写真:
【図5】A〜EはSi 300ゲル濾過カラムで得られた5つの画分(1〜5)および低圧DEAEカラムで保持されなかった画分(0)をマイグレートさせて得られるゲルに相当するPVDF膜を示す図。
【図6】A〜Eは高速イオン交換カラムで得られた画分(1〜3)と分子篩を用いた濾過で得られた画分1(ウェル0)をマイグレートさせて得られるゲルに対応するPVDF膜を示す図。
【図7】A〜Dはイオン交換カラムで得られた画分1(0)および逆相クロマトグライフィーで得られた画分(1〜5)の移動に相当するゲルをブロットした膜を同じコードを有する図6A〜6B、6D〜6Eと同一の試薬で検出した結果を示す図。
【図8】M. smegmatisにおいてM. tuberculosis H37Rv を発現させるための遺伝子ライブラリーのスクリーニングを示す図。
【図9】ライブラリーより選択され、E. coli で電気穿孔(エレクトロポレ、electropore))し、アルカリ溶解で抽出した3つのコスミドのアガロールゲル上での移動を示す図。
【図10】BamHI(a)、SmaI(b) 、HpaI(c) 、NotI(d) 、SspI(e),EcoRI(f)およびHindIII(g)で処理したE. coli NM554 から抽出された pLA1 のコスミドDNAのゲル上での移動を示す図。
【図11】45/47kDa 蛋白質のマイコバクテリアにおける発現を示す図。
【図12】マイコバクテリアにおける45/47kDa 蛋白質の発現を示す図。
【図13】A、Bは pUC18の pLA1 コスミドをBamHI で処理して得られる断片をリゲーションして得られた各種 pUC18: :M.tuberculosis H37Rv組み換えクローンのプラスミドプロフィール(13A)と、BamHI 制限プロフィール(13B)の図。
【図14】45/47kDa 蛋白質のE.coliにおける発現を可能にする挿入断片の制限地図
【図15】E. coli における45/47kD蛋白質の発現を示す図。
【図16】E. coliにおける45/47kDa 蛋白質の発現を示す図。
【図17】配列 No.2とM. leprae (mln 431) 由来の蛋白質の配列との比較図。
【図18】配列No. 2の蛋白質の疎水性プロフィール。
【特許請求の範囲】
【請求項1】
少なくとも下記配列 No.3の一部を有する蛋白質:
配列 No.3
【化1】
【請求項2】
少なくとも下記配列 No.2の一部を有する請求項1に記載の蛋白質:
配列 No.2
【化2】
【請求項3】
請求項1または2に記載の配列 No.2または配列 No.3の少なくとも一部と、免疫応答を誘導可能なペプチド配列または蛋白質とを含むハイブリッド蛋白質。
【請求項4】
免疫応答が体液性応答および/または細胞性応答である請求項3に記載の蛋白質。
【請求項5】
ペプチドまたは蛋白質が、ジフテリア毒素、破傷風毒素、HBV ウィルスのHBS 抗原またはポリオウィルスのVP1 抗原もしくはその他任意の毒素または抗原の一部、特にエピトープである請求項3または4に記載の蛋白質。
【請求項6】
請求項1〜5のいずれか一項に記載の蛋白質をコードするオリゴヌクレオチド。
【請求項7】
下記配列 No.1の少なくとも一部を有する請求項6に記載のDNA:
配列 No.1
【化3】
【請求項8】
請求項1〜5のいずれか一項に記載の蛋白質を産生する微生物。
【請求項9】
上記蛋白質の少なくとも一部が表面に存在する請求項8に記載の微生物。
【請求項10】
細菌である請求項9に記載の微生物。
【請求項11】
細菌、特にM. bovis BCGであることを特徴とする請求項8〜10のいずれか一項に記載の微生物。
【請求項12】
請求項1〜5および8〜11のいずれか一項に記載の蛋白質または微生物の有効量を薬学上許容される希釈剤またはアジュバントと組み合わせて含有する医薬組成物。
【請求項13】
請求項1〜5および8〜11のいずれか一項に記載の蛋白質または微生物を含む医薬またはワクチン。
【請求項14】
結核抗体を含有すると思われる生物液体を請求項1〜5のいずれか一項に記載の蛋白質と接触させることを特徴とする結核特異的抗体の検出方法。
【請求項15】
蛋白質を担体に固定する請求項14に記載の方法。
【請求項16】
請求項1〜5のいずれか一項に記載の蛋白質調製物と、プロセスを実施するための緩衝液とからなる請求項14または15に記載のプロセスを実施するための試験キット。
【請求項17】
生成した抗体−蛋白質複合体を検出するための試薬を含む請求項16に記載のキット。
【請求項18】
請求項1〜5のいずれか一項に記載の蛋白質と特異的に反応する抗体。
【請求項1】
少なくとも下記配列 No.3の一部を有する蛋白質:
配列 No.3
【化1】
【請求項2】
少なくとも下記配列 No.2の一部を有する請求項1に記載の蛋白質:
配列 No.2
【化2】
【請求項3】
請求項1または2に記載の配列 No.2または配列 No.3の少なくとも一部と、免疫応答を誘導可能なペプチド配列または蛋白質とを含むハイブリッド蛋白質。
【請求項4】
免疫応答が体液性応答および/または細胞性応答である請求項3に記載の蛋白質。
【請求項5】
ペプチドまたは蛋白質が、ジフテリア毒素、破傷風毒素、HBV ウィルスのHBS 抗原またはポリオウィルスのVP1 抗原もしくはその他任意の毒素または抗原の一部、特にエピトープである請求項3または4に記載の蛋白質。
【請求項6】
請求項1〜5のいずれか一項に記載の蛋白質をコードするオリゴヌクレオチド。
【請求項7】
下記配列 No.1の少なくとも一部を有する請求項6に記載のDNA:
配列 No.1
【化3】
【請求項8】
請求項1〜5のいずれか一項に記載の蛋白質を産生する微生物。
【請求項9】
上記蛋白質の少なくとも一部が表面に存在する請求項8に記載の微生物。
【請求項10】
細菌である請求項9に記載の微生物。
【請求項11】
細菌、特にM. bovis BCGであることを特徴とする請求項8〜10のいずれか一項に記載の微生物。
【請求項12】
請求項1〜5および8〜11のいずれか一項に記載の蛋白質または微生物の有効量を薬学上許容される希釈剤またはアジュバントと組み合わせて含有する医薬組成物。
【請求項13】
請求項1〜5および8〜11のいずれか一項に記載の蛋白質または微生物を含む医薬またはワクチン。
【請求項14】
結核抗体を含有すると思われる生物液体を請求項1〜5のいずれか一項に記載の蛋白質と接触させることを特徴とする結核特異的抗体の検出方法。
【請求項15】
蛋白質を担体に固定する請求項14に記載の方法。
【請求項16】
請求項1〜5のいずれか一項に記載の蛋白質調製物と、プロセスを実施するための緩衝液とからなる請求項14または15に記載のプロセスを実施するための試験キット。
【請求項17】
生成した抗体−蛋白質複合体を検出するための試薬を含む請求項16に記載のキット。
【請求項18】
請求項1〜5のいずれか一項に記載の蛋白質と特異的に反応する抗体。
【図1】

【図2】
【図3】
【図4】
【図5A−C】
【図5D−E】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】

【図14】
【図15】
【図16】
【図17】

【図18】
【図2】
【図3】
【図4】
【図5A−C】
【図5D−E】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【公開番号】特開2008−212160(P2008−212160A)
【公開日】平成20年9月18日(2008.9.18)
【国際特許分類】
【出願番号】特願2008−121105(P2008−121105)
【出願日】平成20年5月7日(2008.5.7)
【分割の表示】特願2006−241160(P2006−241160)の分割
【原出願日】平成8年1月31日(1996.1.31)
【出願人】(506269116)
【Fターム(参考)】
【公開日】平成20年9月18日(2008.9.18)
【国際特許分類】
【出願日】平成20年5月7日(2008.5.7)
【分割の表示】特願2006−241160(P2006−241160)の分割
【原出願日】平成8年1月31日(1996.1.31)
【出願人】(506269116)
【Fターム(参考)】
[ Back to top ]