
微細粉砕及び微細な種での結晶化により有機結晶微細粒子組成物を製造する方法及び装置、並びにそれらの使用
本発明は、活性有機化合物の結晶粒子を製造する方法に関する。この方法は、湿式粉砕プロセスによって微細な種を生成する工程と、微細な種を結晶化プロセスに付す工程とを含む。得られる結晶粒子は約100μm未満の平均粒径を有する。本発明はまた、本発明の方法によって製造される結晶粒子及び薬学的に許容可能な担体を含む医薬組成物も提供する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、活性有機化合物の結晶粒子を製造する方法及び装置、並びにそれらの使用に関する。
【背景技術】
【0002】
例えば医薬品有効成分(「API」)のような活性有機化合物の製造の際、固形物の形成は、溶液相中での結晶化、その後に続く分離及び乾燥によって達成される場合が最も多い。多くの場合、乾燥した活性有機化合物は、最終生成物の適正な配合を確実にするのに必要な粒径プロファイルに達するようにさらに処理されねばならない。得られる粒径は実質的に様々であり得るが、大半の場合、微細な活性医薬成分の粉体は300μm未満の平均サイズを有する。しかしながら、低水溶性及び/又は低透過性の医薬品ターゲットにより40μm未満の粒径の結晶に対する強い必要性が存在する。配合物中の粒子が小さいことにより、体内への輸送のためにより広い表面積が与えられる。
【0003】
許容可能な粒径プロファイルに達するために、エアジェット分級粉砕、ピン粉砕(pin milling)又はハンマー粉砕等の乾式粉砕工程を行うことが一般的である。医薬品処理に通常用いられる乾式粉砕装置のいくつかの例としては、Hosakawa Micron(www.hosokawamicron.com)製の乾式粉砕装置(例えばピンミル:Alpine(登録商標)UPZ Fine Impact Mills、例えば流動エアジェットミル:Alpine(登録商標)AFG Fluidized Bed Opposed Jet Mills)、Fluid Energy製の乾式粉砕装置、Quadro Engineering製の乾式粉砕装置及び非特許文献1に記載の乾式粉砕装置が挙げられる。乾式粉砕工程を用いて、粒子の凝集物を破砕してその元のサイズにし、及び/又は元の粒子を破砕してより小さな片にすることができる。
【0004】
プロセス工学の観点から、乾式粉砕は多くの動作上の問題をもたらし、費用がかかる。一つの大きな問題は、活性化合物に対する作業者曝露の抑制である。非常に強力な化合物の場合、乾式粉砕は、ダスティングを低く保つのに高価な工学的制御を必要とするであろう。さらに、工学的制御はダスト爆発を最小限にとどめるのに必要とされるであろう。乾式粉砕の他の動作上の問題としては、高温での溶融又はミルの内部部品への付着による、乾式ミルの内部での物質の蓄積が挙げられる。ピン粉砕では、このような不十分な粉砕性能はそれぞれ「メルトバック」又は「フラッギング」と一般に呼ばれ、さらには非晶質物質の生成、ミルの目詰まり、及び物質が処理される際にミルを出る粒径の変化をもたらす可能性がある。化合物によっては処理中にミルを腐食し、API生成物において許容不可能な高レベルの汚染をもたらす。したがって、結晶化により直接、目的の粒径分布(PSD)の結晶を形成し、粒子仕上げ工程として乾式粉砕を回避することが望ましい。
【0005】
不都合なことに、溶液結晶化により直接又は湿式粉砕技法により直接、製造する方法はない。一つの進展として、固体スラリーのロータステータ粉砕があり、その後に分離が続く。ロータステータ粉砕は通常、20μmを上回る平均サイズの粒子を製造する。不都合なことに、大半の場合では、摩砕(attrition)がこの粉砕プロセスに見られることが多い。非常に小さな粒子がバイモーダル(bimodal:双峰性)粒径を残したまま元の粒子から削り取られる場合に摩砕が生じる(非特許文献2)。多くの場合、ロータステータ粉砕の結果、これらの微細粒子の存在により濾過工程が著しく低速となる。さらに、直接圧縮又はローラ圧着技法を用いるバイモーダルな生成物の配合が問題である。小さなAPI粒子のモノモーダルな生成物の形成は、仕上げ工程として乾式粉砕を用いない場合に有益であろう。
【0006】
結晶化による、液体中に溶解している溶質からの新たな固体相の形成は、以下の2つの経路:(1)新たな粒子の核形成によって、又は(2)既存の粒子上での溶質の堆積による成長によって生じることが一般的に認められている。核形成は、結晶化装置内の異物質上で生じるか又は溶液から均一に生じ得る。特許文献1及び特許文献2は、析出の際の溶質の多くの新たな粒子の大量な核形成によって生成される小さな粒子、さらにはナノ粒子を記載している。これらのプロセスでは、系の特性を溶媒組成、温度又は反応を用いて変えることで、溶質に高い過飽和をもたらし、この過飽和により、高速な核形成及び結晶化に至る。核形成による多くの粒子の発生により、結晶化工程の終了時点に小さな粒径分布がもたらされるため、乾式粉砕の必要がなくなる。
【0007】
上記の核形成プロセスの重大な弱点は、高過飽和下では、オストワルドの段階則(Ostwald’s rule)(非特許文献3)によって説明されるように、望ましくない固体状形態(結晶形態/結晶内の分子パッキング)が生じる可能性があることである。炭酸カルシウムについて、様々な結晶形態の生成がKabasci他によって立証されている(非特許文献4)。医薬化合物が同じAPIについていくつかの異なる結晶形態を示すことは一般的であり、したがって、これらの核形成を引き出す技術の使用は特定の用途と見なされる。さらに、高過飽和及びそれに伴う核形成を含むプロセスは、溶媒分子又は不純物を内包した結晶を生じる可能性がある。一般に、医薬品のために選択される精製及び分離プロセスは、高化学純度及び適正な固体状形態の生成物をもたらすべきであり、核形成事象が著しく影響を及ぼすプロセスは望ましくない。
【0008】
最終生成物の形態学的特性を制御しようとする際、微粒子工学では生成物の種粒子を用いて結晶化中に結晶成長のテンプレートを与える傾向が高い。播種は、過飽和度を制限することによって粒径、結晶形態及び化学純度を制御するのに役立ち得る。種々の粉砕法が種原料(seed stock)を生成するのに用いられてきた。結晶化種子用に小さな粒子を生成して適度なサイズの粒子を得るのに乾式粉砕が通常用いられてきた。この手法は、先に説明した乾式粉砕に伴う工学的問題及び安全面の問題をなくすが、種生成に関して湿式粉砕法ほど望ましくない。
【0009】
ロータステータ湿式粉砕は、20μmよりも大きいという実際の制限がある比較的大きな活性有機粒子を生成するのに用いることができることが示されている。その一方、20μmよりも大きくするように粉砕することは、小さな片がバイモーダル粒径分布をもたらす摩砕方式では粉砕時間を延ばす必要がある(非特許文献2)。ロータステータ湿式粉砕された生成物を種として用いる結晶化によって大きな粒子が生じ、多くの場合、バイモーダル粒径分布が生じることが分かっている。所望の小さなサイズの結晶又はモノモーダル物質を形成するには、その後に乾式粉砕工程が必要とされる。このような種の生成方法は理想的ではない。
【0010】
超音波破砕(sonication)は、結晶化のために大きな種を生成するのに用いられる別の技法である。例えば、100μmよりも大きい生成物をもたらす超音波破砕が示されている(特許文献3)。近年、400mm未満の粒子を有する医薬品の直接配合のための最終生成物流を形成するのに媒体粉砕(媒体ミル粉砕)が用いられている(特許文献4)が、その次の結晶化において湿式粉砕された微細な種を用いることはこれまで示されていない。媒体粉砕及びその有用性の概説が特許文献5に記載されている。
【0011】
上記特許は、媒体ミル及び媒体ミルビーズの構成について可能な材料を記載している。これらの材料は特許文献6に挙げられており、この特許では媒体は砂、ビーズ、シリンダ、ペレット、セラミック又はプラスチックから構成されることができると記載されている。この特許は、ミルを金属、鋼合金又はセラミックから構成することができること、及び、ミルにセラミックで内張りされてもよいことを開示している。ポリスチレンを含むプラスチック樹脂が特に有用であるとして述べられている。特許文献7は、安定剤の存在下で有機色素を1μm未満に粉砕するための酸化ジルコニウムビーズの使用を開示している。ミル構築物のいくつかの組合せを本発明の実施に用いてもよい。一実施の形態では、セラミックビーズ及びセラミックミルが用いられる。さらなる実施の形態では、セラミックビーズ及びクロムで内張りされたミルが用いられる。
【特許文献1】米国特許第5,314,506号
【特許文献2】米国特許出願公開第2004/0091546号
【特許文献3】米国特許第3,892,539号
【特許文献4】米国特許第5,145,684号
【特許文献5】米国特許第6,634,576号
【特許文献6】米国特許第3,804,653号
【特許文献7】米国特許第4,950,586号
【非特許文献1】Perry’s Chemical Engineer's Handbook(Sixth edition ed. Robert H. Perry and Don Green),Section8
【非特許文献2】American Pharmaceutical Review Vol 7,Issue 5,pp 120-123 -“Rotor Stator Milling of API's...”
【非特許文献3】Threlfall−vol 7 no 6 2003 Organic Process Research and Development
【非特許文献4】Trans IChemE,vol 74,Part A,October 1996
【非特許文献5】Chemical Engineering Progress,September 1997,P34“Take some Solid Steps to improve Crystallization”
【非特許文献6】Johnson and Prud'homme(Australian Journal of Chemistry 56(10): 1021−1024(2003)
【非特許文献7】Marcant and David(AIChE Journal Nov 1991 vol 37,No 11)
【非特許文献8】Chemical Engineering Transactions,“Proceedings of the 15 th International Symposium on Industrial Crystallization 2002”(Volume 1 2002, p 65, published by ADIC−Associazione Italiana Di Engegneria Chemi
【非特許文献9】Pohl and Schubert Partec 2004“dispersion and deagglomeration of nanoparticles in aqueous solutions”
【非特許文献10】McCausland et.Al.Chemical Engineering Progress July 2001 P.56−61
【非特許文献11】The Handbook of Industrial Mixing(Ed. Paul,et al,2004,Wiley Interscience)
【非特許文献12】The Handbook of Pharmaceutical Excipients(3rd edition,editor Arthur H.Kibbe, 2000,American Pharmaceutical Association,London)
【非特許文献13】Physicians Desk Reference(51 edition, 20O1,Medical Economics Co.,Montvale, NJ)
【発明の開示】
【発明が解決しようとする課題】
【0012】
概して、配合要求を満たすように乾式粉砕を省くために十分に制御されたサイズ又は表面積の活性有機物、特に医薬生成物を製造することができる結晶化プロセスが依然として必要とされている。医薬産業では、そのバイオアベイラビリティの増大及び/又は溶解率を理由に、より小さな粒子が一貫して求められている。同様に、必要な結晶形態及び十分に制御された結晶純度を有する化学化合物をもたらすことも重要である。本発明では、約0.1μm〜約20μmの範囲の平均粒径を有する湿式粉砕した微細な種が、制御された粒径分布、結晶形態及び純度を有することから、微細な活性有機固体粒子の生成に対し、特に活性医薬品成分の結晶化に対し驚くほど有効であることが示されている。本発明のさらなる利点として、下流粉砕の必要性がなくなるため、これらのプロセスに伴うことが多い衛生面及び安全面の危険性がなくなることが挙げられる。
【課題を解決するための手段】
【0013】
本発明は、活性有機化合物の結晶粒子を製造する方法を提供する。本方法は、湿式粉砕プロセスによって微細な種(micro-seed)を生成する工程と、微細な種を結晶化プロセスに付す工程とを含む。湿式粉砕(wet milling)プロセスによって生成される微細な種は、約0.1μm〜約20μmの平均粒径を有する。得られる結晶粒子は、100μm未満の平均粒径を有する。
【0014】
結晶化工程に関して、本発明は2つのプロセスを含む。第1の結晶化プロセスは、3つの工程プロセス:媒体粉砕を用いて微細な種のスラリーを生成すること、微細な種の一部を溶解すること、及び微細な種で活性有機化合物を結晶化することである。
【0015】
第2の結晶化プロセスもまた、微細な種のスラリーを生成すること、結晶化される生成物の溶液を生成すること、及びこの溶液とスラリーを合わせることを含む3つの工程プロセスである。この第2の結晶化プロセスの一実施の形態では、微細な種のスラリー及び生成物の溶液は合わさると高速微細混合する。
【0016】
第2の結晶化プロセスを達成するために、3つの処理構成のうちの1つを個別に用いるか又は組合せて用いてもよい。一つの構成はバッチ式処理構成であり、別の構成は半連続式処理構成であり、3つ目は連続式処理構成である。
【0017】
また、再循環ループを第2の結晶化プロセスと共に用いてもよい。第2の結晶化プロセスの一実施の形態では、再循環ループをバッチ式処理構成の一部として用いる。第2の結晶化プロセスの別の実施の形態では、再循環ループを半連続式処理構成の一部として用いる。第2の結晶化プロセスのさらに別の実施の形態では、再循環ループを連続式処理構成の一部として用いる。
【0018】
第2の結晶化プロセスは2つのタイプの溶媒流を用いる。一実施の形態では、溶媒系は水系溶媒流であり、別の実施の形態では、溶媒系は有機溶媒系であり、さらに別の実施の形態では、溶媒系は混合溶媒流である。
【0019】
さらに、補助エネルギー装置を第2の結晶化プロセスと共に用いてもよい。第1の実施の形態ではこの補助エネルギー装置は混合用T継手であり、第2の実施の形態では混合用エルボであり、第3の実施の形態ではスタティックミキサーであり、第4の実施の形態では超音波破砕機であり、第5の実施の形態ではロータステータホモジナイザーである。
【0020】
さらに、本発明の活性有機化合物は、鎮痛薬、抗炎症剤、駆虫薬、抗不整脈薬、気管支喘息治療薬、抗菌剤、抗凝血薬、抗うつ剤、抗糖尿病薬、抗てんかん薬、抗ヒスタミン剤、血圧降下薬、抗ムスカリン薬、抗ミコバクテリア薬、抗悪性腫瘍薬、免疫抑制剤、抗甲状腺薬、抗ウイルス薬、抗不安剤、鎮静剤、収れん薬、βアドレナリン作動性受容体遮断薬、造影剤、コルチコステロイド、咳止め薬、診断薬、画像診断薬、ドーパミン作動薬、止血剤、免疫学的薬剤、脂質調節剤、筋弛緩剤、副交感神経作働薬、副甲状腺カルシトニン、プロスタグランジン、放射性医薬品、性ホルモン、抗アレルギー薬、刺激剤、交感神経作働薬、甲状腺薬、血管拡張剤、及びキサンチンを含む群から選択される医薬品であってもよい。
【0021】
さらに、本発明は、本明細書に記載の方法によって製造される結晶粒子及び薬学的に許容可能な担体を含む医薬組成物をさらに提供する。
【発明を実施するための最良の形態】
【0022】
本発明の微細粉砕及び結晶化プロセス(「MMC」)は、微細な種粒子での、約100μm未満、例えば約60μm未満、さらには約40μm未満等の平均体積粒径への成長を含む。たいていの場合、生成物は、結晶化のために添加される種の量に応じて約3μm〜約40μmの範囲である。微細な種は、平均体積分析により、約0.1μm〜約20μm、例えば約1μm〜約10μmの範囲とすることができる。種は、複数の湿式粉砕装置、例えば媒体粉砕等によって生成することができる。平均1μm未満の粒子を用いてもよい。しかしながら、このサイズ範囲は、成長結晶中に粒子が分散したままである場合に得られるAPI粒径が約0.5%〜約15%の通常の種レベルを用いる従来の分離技法に望ましい粒径よりも小さいため、微細な種より魅力的ではない。
【0023】
本発明の方法(MMC)は、微細な種のスラリーを生成すること、及び結晶化される生成物を含有する溶液を生成することを含む。これら2つの流れを組合せて生成物の結晶化を行う。大半の場合では、結晶化は、結晶化を推進するように生成物の溶解性変化及び濃度変化を操作することによって続行される。これらの操作により、種上に溶質を堆積させるための推進力を与える過飽和系がもたらされる。播種を行っている間の過飽和レベル、及びその後の結晶化が、核形成に対する成長条件を高めるレベルで制御される。本発明では、本方法は、新たな粒子の発生を制御しつつ、微細な種での成長を促し易くするように設計される。結晶化方法の概説(成長及び核形成プロセス条件の説明を含む)をPriceが行っている(非特許文献5)。
【0024】
本発明のMMC法の微細な種粒子及び生成物粒子は、複数の特定の利点を有する。微細な種粒子は表面積対体積の比が高く、したがって、成長速度が所定の過飽和度で大きな種粒子に比して著しく高まる。高密集の種粒子では、異物質上での核形成が回避されるため、結晶化は低い過飽和度での既存の種粒子上における成長の1つとされる。そのため、API粒子のサイズ及び形態が種粒子の特性によって制御される。
【0025】
一般に、所望の結晶形態が最も安定している反応器条件での動作、及び所望の結晶形態での播種が好適である。粒子−粒子の衝突が著しく重量の少ない物体間で生じるため、小さな粒子はせん断による粒子摩砕に対する感度が低いことが見出されている。モノモーダルな種で開始する場合、本発明の方法は、光学顕微鏡写真及びレーザ散乱法によって確認されるようなモノモーダル粒径分布をもたらす。得られる生成物の単分散粒径により、複合プロセスを微細粒子仕上げに魅力的な方法にする下流濾過及び配合に適応可能である。
【0026】
本発明は、いずれもの析出した活性有機粒子又は結晶化した活性有機粒子、例えば、医薬品、バイオ医薬品、ニュートラシューティカルズ、診断剤、農業用化学物質、殺虫剤、除草剤、色素、食品成分、食品配合物、飲料物、ファインケミカルズ及び化粧品の製造に用いることができるが、説明を容易にするため、主として医薬品を特に対象とする。他の産業部門で使用される有機化合物のための結晶粒子/析出粒子は、本明細書に記載の同じ一般的技法を用いて製造することができる。
【0027】
微細な種の存在下での成長を促すように過飽和を生じさせるいずれの方法も本発明に適応可能である。結晶化を操作する一般的な方法は、溶媒組成変化、温度変化、化学反応の使用、又は蒸留の使用を含む。反応結晶化には1つ又は複数の試薬からの最終APIの配合が必要とされるが、生成されたAPIは過飽和となり、生成物の過飽和が結晶化の源となる。過飽和を生じさせる結晶化法及び核形成と成長との相互作用の概説をPriceが行っている(非特許文献5)。この文献はその全体が参照により本願に援用される。
【0028】
溶質への微細な種の添加又は微細な種への溶質の添加は、いくつかの方法、例えばバッチ式結晶化、セミバッチ式結晶化又は半連続式結晶化で達成することができる。これらの技法は当業者には一般的であり、他の結晶化装置構成への拡張も期待される。さらに、これらの方法の組合せも利用することができる。
【0029】
バッチ式結晶化は通常、温度を変えるか又は溶媒を蒸留によって除去して過飽和を生じさせる結晶化を含む。セミバッチ式結晶化は通常、溶質槽又は溶質用の反応前駆体への溶媒又は試薬の連続添加を含む。バッチ式結晶化及びセミバッチ式結晶化では、種は通常、溶質槽に添加され、これにより、種の添加時点で又は種の添加の結果として過飽和する。図6及び図7を参照されたい。
【0030】
半連続式結晶化は、結晶化全体を通して反応器内の液体相の含量をほぼ一定に保つように設計される。非溶媒(貧溶媒(anti-solvent)とも呼ぶ)による半連続式結晶化では、種の流れが反応器に添加され、その後、溶液中に溶解した溶質を含有する流れ及び非溶媒流の双方の同時添加が行われる。ここで、結晶化は成分が添加される速度と同様の速度で行われる。図8を参照されたい。反応結晶化の例示的な図式を図9に挙げる。
【0031】
MMC法に選択される流れの化学組成は、結晶化される化合物に応じて決まる。したがって、水性流、有機流又は水性流と有機流の混合流を用いることができる。
【0032】
本発明のプロセスでは、微細な種のサイズへの湿式粉砕には、下流の生成プロセスでの乾式粉砕の必要性を制限することが必要である。もっぱら選別機が約1μm〜約10μmの範囲の平均最適サイズの粒子を提供することができる。高エネルギーの流体力学キャビテーション又は高強度の超音波破砕、高エネルギーのボールミル粉砕又は媒体粉砕、及び高圧均質化等の粉砕法が、約1μm〜約10μmの範囲の平均最適サイズを有する微細な種を製造するのに用いることができる技術の代表的なものである。
【0033】
本発明の一実施形態では、媒体粉砕は目的のサイズに対して種の粒径を縮小するのに効果的な湿式粉砕法である。さらに、媒体粉砕は粉砕プロセス時にAPIの結晶度を維持することが分かっている。用いられる媒体ビーズのサイズは、例えば約0.5mm〜約4mmの範囲である。
【0034】
本発明の湿式粉砕プロセスの際に変更することができる追加パラメータとしては、所望の微細な種のサイズを得るための生成物濃度、粉砕温度及びミル速度が挙げられる。
【0035】
API生成物流に対して媒体粉砕作業を行って、コロイド安定剤の存在下で0.5mm以下の特殊ビーズを用いて平均サイズが1ミクロン未満の粒子を生成した。表面活性剤が、1ミクロン未満で活性であるコロイド粒子間の結合力を克服し、配合のために分散粒子流を供給する。この供給流を微細な種として本発明に用いることができる。本発明による結晶化は、結晶化に十分な分散速度が用いられる場合に最も予測可能である。種として粒子の凝集を用いることは、凝集物の数及びサイズが様々である可能性があるため望ましくない。したがって、0.1μm〜0.5μmの種結晶を本発明において用いることができ、その場合、有機化合物が分散粒子として自己安定化しない限り、コロイド安定剤を用いることが望ましい。
【0036】
本発明のプロセスは主として、既存の種粒子での成長のプロセスであるため、微細な種の量及びサイズがAPI粒径の主要な決定因子である。可変量の種を添加して結晶化後に所望の粒径分布(PSD)を得ることができる。典型的な種の量(種のスラリーの溶媒相中に溶解していない物質)は、結晶化される活性成分の量に対して約0.1重量%〜20重量%の範囲である。成長結晶では、少ない種の導入により、より大きな粒子が生じる。例えば、少量の種により生成物の粒径が増大して60μmを上回ることができるが、結晶化は潜在的に、核形成を回避すると共にこれらの種での成長を促すように非常に低速である可能性がある。約0.5%〜15%の種レベルが1μm〜10μmの微細な種で開始する場合の妥当な充填である。
【0037】
別の実施形態では、MMC法は、
(1)約0.1μm〜20μmの平均サイズを有する微細な種を生成する、湿式粉砕プロセスを用いること、及び
(2)100μm未満の平均サイズを有する結晶粒子をもたらす、微細な種で活性有機化合物を結晶化すること、
を含む。
【0038】
さらなる実施形態では、MMC法は、
(1)約0.1μm〜20μmの平均サイズを有する微細な種を生成する、湿式粉砕プロセスを用いること、
(2)微細な種の一部を溶解すること、及び
(3)100μm未満の平均サイズを有する結晶粒子をもたらす、微細な種で活性有機化合物を結晶化すること、
を含む。
【0039】
溶解プロセスは、加熱、pH変化、溶媒組成等の変化を含み得る。これにより、得られる粒径分布が種よりもわずかに大きいにすぎない分布となる。いくつかの例では、微細な種の粒径が適度に大きくなるだけで生成物には十分であり、したがって、50%以上の種レベルを用いることができる。
【0040】
一実施形態では、微細な種は分離し、乾燥生成物として充填され得る。
【0041】
本発明のMMC法は大幅にスケール拡大可能である。スケールごとの適正な機器設計により、全てのスケールでロバスト性能が可能となり得る。確実なスケールアップに用いられ得る2つの特徴は以下の通りである:1)活性結晶化系への物質の添加の際の高速微細混合、及び2)所望でない凝集の粒子分散のためのエネルギー装置の包含。これらの特徴を含む結晶化装置設計は、本発明のスケールアップに適応可能である。
【0042】
高速微細混合は、生成物の結晶化の特徴的な誘導時間に対して分子レベルでの2つの流れの高速混合時間を意味する。これらの概念は、非特許文献6及び非特許文献7によって詳細に説明されている。これらの著者らのグループは双方とも、微細混合時間が結晶化又は析出の成果に影響を及ぼし得ることを強調している。したがって、著者らは、微細混合時間が短いことが有利であることを強調している。溶媒、濃縮液又は試薬添加剤について、この高速微細混合により、核形成事象につながる可能性がある濃度勾配が低減又は排除される。
【0043】
本発明の一実施形態では、過飽和度は微細な種での成長を促すように低く保たれる。いくつかの例では、結晶化の動態は高速であり、核形成を実質的に回避することができない。適した高速ミキサーは、これらの例では、試薬流を高速混合すると共に試薬の局部的な高濃度を回避することによって核形成を制限するように選択されるものとする。微細な種が溶質を有する結晶化装置に添加される場合、高速微細混合による種の分散は、結晶化が行われる際に微細な種の凝集を制限することが必要である。
【0044】
さらに、Hunslowの論文(非特許文献8)は、粒子の凝集が局部的な過飽和レベルに直接関連することを教示している。したがって、高速微細混合はまた、この状況の場合に凝集を最小限度にとどめる上でも役立つ。高速ミキサーの選択は、ミキサーの選択によって粒子摩砕のレベルに対してバランスがとれていなければならない。粒子−粒子間による粒子の発生、又は種粒子の存在下での粒子−結晶化装置間の表面相互作用による粒子の発生を引き起こすメカニズムは一般的に、第2の核形成と呼ばれ、大半の結晶化において或る程度生じることが予測される。機器の選択により、この作用の程度を変えることができる。
【0045】
小さなサイズの活性有機化合物は凝集し、次いで、結晶化の際に凝集物上に塊を堆積させることによって凝集する。粒子が凝集すると、API粒径は、成長が個々の種粒子にのみ生じ、且つ凝集物が存在しない場合よりも大きくなるであろう。医薬用途によっては、凝集は望ましくなく、その理由は、凝集した粒子を含んだプロセスをスケールアップすることがより困難となる可能性があるからである。これらの状況では、凝集に首尾よく対処する、微細な種を使用する方法を発展させることが望ましい。
【0046】
一般に、粒子が受けるエネルギー密度は解凝集を得るのに十分でなければならず、結晶化の際に、分散系を維持するのに十分な周波数で粒子をエネルギー密度に晒さなければならない。補助エネルギー装置は粒子を分散することによって凝集を最小限にとどめるのに役立つ。エネルギー装置の機能は、軽く凝縮した物質を分解する粒子衝突を生じさせるか又は凝集物をよじって(torque)破砕するせん断場を生じさせることである。このエネルギー装置は、適正に設計されたタンク攪拌機又は流体を圧送するのに用いる再循環パイプと同じくらい単純なものとすることができる。流体ポンプは高エネルギー装置であり、結晶化プロセスに影響を及ぼし得る。これらの装置は、凝集物が強固でないか又は生成物が装置に頻繁に晒される場合では十分である。強力なせん断環境を与えるにはロータステータ湿式ミルが有用であり、これらは粒子自体が摩砕しない場合に最も有用である。結晶化装置に適用される超音波破砕エネルギーが、容易に凝集してより強固な凝集物を形成する化合物の凝集を制限することが分かっている。結晶化の終了時点での超音波破砕又はエネルギー装置の適用も凝集物を破砕するのに有用であり得るが、凝集物は結晶化時間の終了まで著しく強固である可能性があるため、結晶化の最中よりも望ましくない。超音波破砕ホーン(sonication horn)もまた、主要な粒子を破砕することなく凝集物を軽く破砕することに関与し得る音波を与える。
【0047】
針状結晶が微細有機物の処理に好機を示す。特に、微細有機物の濾過速度が一般に低速である。本発明の一態様は、結晶化中の超音波破砕の使用である。超音波破砕は、幅方向への針状結晶の成長を促すことができ、濾過に対しより堅固な生成物をもたらす。針状結晶のために微細な種を生成するのに超音波破砕を使用することもまた、特に有利である。針状により、長手方向軸を中心に破砕する傾向があり、同様の幅の結晶がもたらされるが、長さはより短い。
【0048】
超音波破砕(超音波は典型的には10kHz〜60kHz)の基本技術は非常に複雑であり、上首尾の解凝集の基本的なメカニズムは不明確であるが、超音波破砕が解凝集(deaggregation or deagglomeration)時に効果的であることがよく知られている(非特許文献9)。機構プロセスの非限定的な説明として、超音波破砕は凝集物の破砕のために高出力密度ひいては高強度の超音波を与える。キャビテーション気泡が音波の負圧期間中に形成され、これらの気泡が高速に潰れることにより、解凝集に有用な衝撃波、高い温度及び圧力がもたらされる。本発明では、種粒子及び成長した粒子はたいていの場合は著しく破砕されず、したがって、超音波破砕の高エネルギー事象が、粒子を摩砕することなく分散粒子での成長を促すのに特に効果的であることが分かっている。
【0049】
近年、化学薬品の超音波破砕での作業は結晶化に入り込んでいる。核形成の誘導時間を減らすか又は適度な過飽和度で簡単な核形成を与えるのに超音波を使用することに焦点が置かれてきた。このことは、先駆的な固体(solids apriori)の非存在下で、又は固体種をバッチ濃縮液に添加する必要なく、種床生成の再現性を高めるのに有用である(非特許文献10)。この手法は、微細な種の存在が最終生成物特性、特に結晶形態を示す本教示とは対照的である。
【0050】
MMC法におけるように分散した微細な種の粒子での制御された成長のために医薬品の結晶化に超音波破砕を適用することは独特である。さらに、本発明に示されるように首尾よい解凝集に必要とされる超音波破砕力は比較的小さく、結晶化の終了時点で総バッチの1リットル当たり10ワット未満、好ましくは、結晶化の終了時点で総バッチの1リットル当たり1ワット未満である。超音波破砕のための機器の設計及び技術のリサーチが実際のリサーチ領域である。本発明に適用可能なフローセルのいくつかの例は、エネルギー装置として再循環ループで使用するいくつかの製造業者(例えばBranson WF3-16、及び、例えばTelsonics SRR46 series)によって市販されている。
【0051】
微細混合のための方法及び補助エネルギー装置を組み入れるための方法を提供するために再循環ループを使用することは、スケールアップに特に有利であることが示されている。主要な概念は、従来の結晶化装置(通常は攪拌タンク)からの微細混合及びエネルギー入力の要求を軽減し、特定化した機能性ゾーンを形成することである。攪拌タンク結晶化装置は、攪拌タンクの外部で独立して制御されるシステムに対して微細混合及び補助エネルギー入力を用いるブレンド装置として働くことができる。この手法は、大スケールの製造のためのスケーラブルな結晶化システムの一例である。このシステムの実際のエミュレーションを図3に挙げる。微細混合は、高エネルギー散逸領域又は高乱流領域に流れを加えることによって最もよく達成される。この流れをパイプの中央の再循環ループ内の乱流領域に加えることが一実施形態である。この例では、少なくとも1m/秒の速度が従来のパイプ流に推奨されているが、提供される微細混合は必ずしも高速である必要はない。この例は、試薬を追加する位置を限定せず、試薬を添加する方法が適正な微細混合を達成するのに重要である。パイプライン及び攪拌容器中での混合の概念は、非特許文献11に記載されている。
【0052】
結晶化装置の再循環速度は、再循環ループ全体を通して結晶化の終了時点でバッチの一体積の相当量を移動する時間又は結晶化の終了時点の循環時間(turnover time)まで定量することができる。容器の循環時間は個別に様々であり、バッチが補助エネルギー装置に晒されて生成物の凝集を制限することができる周波数の関数である。大スケールの製造のための典型的な循環時間は約5分〜約30分の範囲であるが、これに限定されない。生成物結晶の凝集物は通常、結晶化による塊の堆積を伴うため、結晶化の速度は、解凝集を可能にするのに必要とされる循環時間を延ばすように低速となり得る。
【0053】
得られる生成物の粒子のサイズ及び表面積は、種又は結晶化バッチに補助添加剤を添加することによって増大し得る。一実施形態では、添加剤は結晶化装置内での種及び結晶の分散に役立ち、この分散により粒子凝集を制限する。補助添加剤の添加は他の目的、例えば、生成物の酸化の還元等のために又は容器の側面に固着する化合物を制限するために用いることができる。補助添加剤は、純粋な活性成分をもたらす分離工程によって実質的に取り除かれてもよい。界面活性剤特性を有する物質は、MMC法の粉砕工程、播種工程及び結晶化工程のスラリー特性を高めるのに有用である。
【0054】
補助添加剤としては、限定するものではないが、不活性希釈剤、両親媒性コポリマー、可溶化剤、乳化剤、懸濁剤、アジュバント、湿潤剤、甘味料、風味料、及び香料、等張剤、コロイド分散剤、及び界面活性剤、例えば限定するものではないが、ジミリストイルホスファチジルグリセロール等の荷電リン脂質;アルギン酸、アルギン酸塩、アカシア、アカシアガム、1,3ブチレングリコール、塩化ベンザルコニウム、コロイダル二酸化ケイ素、セトステアリルアルコール、セトマクロゴル乳化ワックス、カゼイン、ステアリン酸カルシウム、塩化セチルピリジニウム、セチルアルコール、コレステロール、炭酸カルシウム、ステアリン酸スクロースとジステアリン酸スクロースとの混合物であるCrodestas F−110(登録商標)(Croda Inc.)、クレイ、カオリン及びベントナイト、セルロースの誘導体、並びにヒドロキシプロピルメチルセルロース(HPMC)等のそれらの塩、カルボキシメチルセルロースナトリウム、カルボキシメチルセルロース、及びその塩、ヒドロキシプロピルセルロース、メチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロースフタレート、非結晶性セルロース、リン酸二カルシウム、ドデシルトリメチルアンモニウムブロミド、デキストラン、スルホコハク酸ナトリウムのジアルキルエステル(例えば、American CyanamidのAerosol OT(登録商標)、ゼラチン、グリセロール、グリセロールモノステアレート、グルコース、Olin 10−G(登録商標)又は界面活性剤10−G(登録商標)(Olin Chemicals(Stamford, Conn.))としても知られるp−イソノニルフェノキシポリ−(グリシドール);オクタノイル−N−メチルグルカミド、デカノイル−N−メチルグルカミド等のグルカミド;ヘプタノイル−N−メチルグルカミド、ラクトース、レシチン(ホスファチド)、n−ドデシルβ−D−マルトシド等のマルトシド;マンニトール、ステアリン酸マグネシウム、ケイ酸アルミニウムマグネシウム、綿実油、トウモロコシ胚芽油、オリーブ油、ヒマシ油、及びゴマ油等の油;パラフィン、ジャガイモでんぷん、ポリエチレングリコール(例えば、Union CarbideのCarbowax 3350(登録商標)及びCarbowax 1450(登録商標)、並びにCarbopol 934(登録商標))ポリオキシエチレンアルキルエーテル(例えば、セトマクロゴール1000等のマクロゴールエーテル)、ポリオキシエチレンソルビタン脂肪酸エステル(例えば、ICI specialty chemicalsの市販のTween(登録商標))、ポリオキシエチレンヒマシ油誘導体、ポリオキシエチレンステアレート、ポリビニルアルコール(PVA)、ポリビニルピロリドン(PVP)、リン酸塩、酸化エチレン及びホルムアルデヒドを含む4−(1,1,3,3−テトラメチルブチル)フェノールポリマー(チロキサポール、スペリオン(superione)、及びトリトンとしても知られる)、全てのポロキサマー及びポラキサミン(例えば、BASF Corporation(Mount Olive, NJ)から入手可能な、Pluronics F68LF(登録商標)、F87(登録商標)、F108(登録商標)、及びtetronic 908(登録商標))、ピラノシド、例えば、n−ヘキシルβ−D−グルコピラノシド、n−ヘプチルβ−D−グルコピラノシド、n−オクチルβ−D−グルコピラノシド、n−デシルβ−D−グルコピラノシド、n−デシルβ−D−マルトピラノシド、n−ドデシルβ−D−グルコピラノシド;第4級アンモニウム化合物、ケイ酸、クエン酸ナトリウム、でんぷん、ソルビタンエステル、炭酸ナトリウム、固体ポリエチレングリコール、ドデシル硫酸ナトリウム、ラウリル硫酸ナトリウム(例えば、DuPont corporationのDUPONOL P(登録商標))ステアリン酸、スクロース、タピオカでんぷん、タルク、n−ヘプチルβ−D−チオグルコシド等のチオグルコシド、トラガカント、トリエタノールアミン、及びアルキルアリールポリエーテルスルホネートであるTriton X−200(登録商標)((Rhom and Haas)等が挙げられる。不活性希釈剤、可溶化剤、乳化剤、アジュバント、湿潤剤、等張剤、コロイド分散剤及び界面活性剤は、市販されているか、又は当該技術分野で既知の技法で調製することができる。
【0055】
同様に、プロセス性能を適応させるための結晶成長調節剤等の市販されていない所望な化学構造物を合成することが可能である。混合前又は混合後のプロセス溶媒流への添加に適したこれらの又は他の薬学的賦形剤のうち多くの特性は、非特許文献12に挙げられており、この開示はその全体が参照により本明細書に援用される。
【0056】
本発明のMMC法では、微細粒子は最終混合液中に形成される。微細粒子を含有する最終溶媒濃縮は、透析、蒸留、ワイパー式薄膜蒸発(wiped film evaporation)、遠心分離、凍結乾燥、濾過、滅菌濾過、抽出、超臨界流体抽出及び噴霧乾燥が挙げられるがこれらに限定されない複数の後処理プロセスによって変更され得る。これらのプロセスは通常、微細粒子の形成後に行われるが、形成プロセス中に行われてもよい。
【0057】
溶液相中の生成物の高溶解性は、乾燥の際に粒子の液相中の残存溶質の堆積をもたらし、これが、結晶化の際に形成される元の粒子の軽い凝集につながる。配合後の薬剤粒子の溶解は多くの場合、凝集物に対する元の粒径の表面積に影響されやすい。軽い凝集物は配合処理中に破砕されて、許容可能なバイオアベラビリティを有する生成物をもたらすことができる。
【0058】
粒径を測定する際、適正な測定具を選択するように注意を払わねばならない。例えば、粒径を測定するのに用いられる典型的なレーザ光走査技法は、凝集物を元の粒子になるように破砕することができない可能性があるため、誤った示度をもたらし得る。したがって、生成物の粒径分析は元の粒径ではなく大きな凝集物を示し得る。光走査技法に対する表面積測定は、以下の例に記載のように好適な測定技法である。しかしながら、平均粒径は従来のレーザ光走査装置を用いて測定されてもよい。具体的には、乾燥生成物の分析は1気圧〜3気圧のSympatec Helos社製分析機と同様の機械において好適である。一般に、生成物の表面積及び粒径が対象の粒子の形状に応じて直接関わる。
【0059】
粒径分析について問題となることが多い粒子の一形態は、長さ対幅のアスペクト比が6を超える針形状である。このタイプの粒子は、顕微鏡写真が小さなサイズの一貫した生成物が製造されていることを示している場合、バイモダール粒径分布を示すことができる。本発明では、アスペクト比が6未満である場合、粒径は、乾式分析における光走査によって、Sympatec Helos社製分析機で測定される。アスペクト比が6以上である場合、光学顕微鏡を用いて結晶の最長寸法で粒径を測定する。
【0060】
医薬配合物を製造するための、MMC法の生成物の続く後処理は典型的に、製品の性能又は市販品としての製品の承認を向上させるのに有利である。限定するものではないが、ローラー圧着、湿式造粒、直接圧縮、又は直接カプセル充填等のプロセスが全て可能である。特に、当該産業の需要を満たすような、MMC法の生成物を含む医薬組成物を製造することができ、これらの配合物は、上述のような種々のタイプの補助添加剤を含む。MMC法及び続く配合用の化合物の、見込まれるが、限定するものではない種類としては、鎮痛薬、抗炎症剤、駆虫薬、抗不整脈薬、気管支喘息治療薬、抗菌剤、抗凝血薬、抗うつ剤、抗糖尿病薬、抗てんかん薬、抗ヒスタミン剤、血圧降下薬、抗ムスカリン薬、抗ミコバクテリア薬、抗悪性腫瘍薬、免疫抑制剤、抗甲状腺薬、抗ウイルス薬、抗不安剤、鎮静剤、収れん薬、βアドレナリン作動性受容体遮断薬、造影剤、コルチコステロイド、咳止め薬、診断薬、画像診断薬、ドーパミン作動薬、止血剤、免疫学的薬剤、脂質調節剤、筋弛緩剤、副交感神経作働薬、副甲状腺カルシトニン、プロスタグランジン、放射性医薬品、性ホルモン、抗アレルギー薬、刺激剤、交感神経作働薬、甲状腺薬、血管拡張剤、及びキサンチンが挙げられる。薬剤物質としては、経皮パッチ等の他の方法を使用することも考えられるが、経口投与及び静脈内投与及び吸入投与を目的とするものが挙げられる。この薬剤物質は、任意の医薬有機活性化合物及び前駆体化合物から選択することができる。これらの種類の薬剤の記載、及び各種類における化学種のリストは、非特許文献13に見ることができ、この開示はその全体が参照により本明細書に援用される。薬剤物質は、市販されており、及び/又は当該技術分野で既知の技法で調製することができる。
【0061】
本明細書中で使用される場合、用語「結晶化」及び/又は「析出」は、流体から粒子を製造する任意の手法を含み、このような手法としては、古典的な溶媒/貧溶媒結晶化/析出、温度依存結晶化/析出、「塩析」結晶化/析出、pH依存反応、「冷却誘起(cooling driven)」結晶化/析出、化学反応及び/又は物理反応に基づく結晶化/析出等が挙げられるが、これらに限定されない。
【0062】
本明細書中で使用される場合、用語「バイオ医薬品」は、生物源に由来するか、又は生物源(例えば、タンパク質、ペプチド、ワクチン、核酸、免疫グロブリン、多糖、細胞産物、植物抽出物、動物抽出物、組換タンパク質、酵素、又はそれらの組合せ)からの産物に相当するように化学合成される任意の治療用化合物を含む。
【0063】
本明細書中で使用される場合、用語「溶媒」及び「貧溶媒」はそれぞれ、物質が実質的に溶解される流体、及び所望の物質を結晶化/析出させるか、又は溶液から溶離させる流体を意味する。
【0064】
本発明の方法及び装置は、広範な医薬物質を結晶化するのに利用することができる。本発明に従って結晶化し得る水溶性医薬物質及び水不溶性医薬物質としては、限定するものではないが、アナボリックステロイド、滋養強壮薬、鎮痛薬、麻酔薬、制酸剤、抗不整脈薬、気管支喘息治療薬、抗菌剤、抗齲蝕剤、抗凝血薬、抗コリン作動薬(anticolonergics)、抗痙攣薬(anticonvulsants)、抗うつ剤、抗糖尿病薬、止瀉薬、制吐薬、抗てんかん薬、抗真菌剤(antifungals)、駆虫薬、痔疾薬、抗ヒスタミン剤、抗ホルモン、血圧降下薬、抗低血圧薬、抗炎症剤、抗ムスカリン薬、抗ミコバクテリア薬(antimycotics)、抗悪性腫瘍薬、抗肥満薬、歯石防止薬、抗原虫薬、抗精神病薬、防腐剤、鎮痙薬(anti-spasmotics)、抗血栓薬(anti-thrombics)、鎮咳薬、抗ウイルス薬、抗不安薬、収斂剤(astringensts)、βアドレナリン作動性受容体遮断薬、胆汁酸、ブレスフレッシュナー、気管支鎮痙薬(bronchospasmolytic drugs)、気管支拡張剤(bronchodilators)、カルシウムチャンネル遮断薬、強心配糖体、避妊薬、コルチコステロイド、鬱血除去薬、診断薬、消化薬、利尿薬、ドーパミン作動薬、電解質、催吐薬、去痰薬、止血剤、ホルモン、ホルモン置換療法薬、催眠剤、血糖降下薬、免疫抑制剤、性的不全治療薬、緩下剤、脂質調節剤、粘液溶解薬、筋弛緩剤、非ステロイド系抗炎症剤、栄養補助剤、鎮痛剤(pain relievers)、副交感神経抑制剤(parasympathicolytics)、副交感神経作働薬、プロスタグランジン(prostagladins)、精神刺激剤、向精神薬、鎮静剤、性ステロイド、鎮痙薬、ステロイド、刺激剤、スルホンアミド、交感神経遮断(sympathicolytics)、交感神経様作用薬(sumpathicomimetics)、交感神経作働薬、甲状腺作働薬(thyreomimetics)、抗甲状腺薬(thyreostatic drugs)、血管拡張剤、ビタミン、キサンチン、並びにそれらの混合物が挙げられる。
【0065】
本発明による医薬組成物は、本明細書に記載の粒子及び薬学的に許容可能な担体を含む。好適な薬学的に許容可能な担体は、当業者にとって既知のものである。これらとしては、非経口注射用、固体形態又は液体形態の経口投与用、及び直腸投与用等の無毒性の生理学的に許容可能な担体、アジュバント又は賦形剤が挙げられる。本発明の医薬組成物は、限定するものではないが静脈内投与を含む、経口投与及び非経口投与の用途に有用である。
【0066】
以下の実施例は、本発明のMMC法を実施する方法の限定的ではない記載を挙げている。
【0067】
以下の実施例において、微細な種の粒子は、600mlディスクミル及びDYNO(登録商標)ミルから成るKDLモデルの2つのミルのうちの1つによって作製した。このミルチャンバにはクロム処理を行い、攪拌ディスクはイットリウム安定化酸化ジルコニウムのものとした。ミルには、約1900グラムの均一直径のイットリウム安定化酸化ジルコニウムラウンドビーズを充填した。160ml攪拌Mini−Cerミルはセラミックチャンバ及びセラミック攪拌機を備えており、Netzsch Incによって製造されたものとした。ミルには、約500グラムの様々なサイズの均一直径のイットリウム安定化酸化ジルコニウムビーズを充填した。これらのミルのビーズは、Norstone(登録商標)Inc.(Wyncote, Pennsylvania)によって供給されたものとした。それらは、高度に研磨されており、且つTOSOH USA, Inc.によってオリジナルで製造されている。
【0068】
粒子の表面積は、特に指定のない限り、GEMINI 2360(Micromeritics(登録商標) Instrument Corporation Inc.(Norcross, Georgia)製)によるBET多点解析を用いて分析した。
【0069】
光学顕微鏡によって粒子の顕微鏡写真を撮影した。顕微鏡写真は、特に断りのない限り結晶化の終了時点での結晶化スラリーのものである。
【0070】
乾燥ケーキの粒径分布は、特に断りのない限り、HELOS OASIS(SYMPATEC Gbh(http://www.sympatec.com/))機内でレーザ光回折を用いて分析した。この同一の機械には、粉砕材料のスラリー又は結晶化による生成物スラリーを分析することができるスラリーセルも備えられていた。Isopar G(登録商標)担体流体へのレシチンの添加及び超音波破砕の適用を含む、分析のための標準的な技法を用いた。
【実施例】
【0071】
[実施例1]
化合物A=Cox II阻害剤
この一連のセミバッチ式結晶化は、媒体粉砕によって表面積が広い微細な種を形成することができること、及び結晶化の際に導入される微細な種の量を変えて可変の表面積及び粒径の最終生成物を製造する作用を示す。最終生成物の表面積はジェット粉砕した物質に匹敵し得る。また、粉砕後であるが結晶化プロセスの前の、微細な種への補助添加剤の添加により、得られる生成物の表面積を増大させることができることを示す実験も示す。貧溶媒を添加して結晶化させた。
【0072】
化合物Aのジェット粉砕
Hosakawa Micron,Inc.の100AFGジェットミルの場合に、1mm〜1.9mm範囲の典型的な条件のノズル、43psig〜45psigのジェット圧及び7000rpm〜21000rpmを用いて、化合物Aをジェット粉砕した。物質の得られた表面積は2.5m2/gであった。
【0073】
実施例1A〜実施例1Eの微細な種の粉砕
0日目、1mmのイットリウム安定化酸化ジルコニウムビーズを収容しているディスクミルを50%のn−ヘプタン及び50%のトルエンで洗浄し、ミルの内容物を容積式ポンプを介した空気による処理のために移した。ミルに接続されている容器に、60グラムの化合物A及び1066グラムの50:50のトルエン:ヘプタン(重量に基づく)を充填した。混合物を、ミル収容タンク内で25℃の温度で攪拌した。次いで、混合物を900ml/分の速度で60分間ミル内を再循環させた。この時間中、ミルは6.8m/秒の先端速度で作動した。タンク内スラリーを20分、40分及び60分の時点で採取して顕微鏡によって粉砕プロセスを確認した。60分後、スラリーを、表1及び2の結晶化の実験での後の使用のためにガラスジャーに詰めた。ジャーの微細な種のスラリーを焼結ガラス漏斗で濾過し、フィルタケーキを真空オーブン内で60℃で乾燥することによって、溶液中に溶解していない微細な種の濃度を求めた。この値は、種の充填に基づいて記録した。乾燥後のフィルタケーキの表面積を標準BET等温線によって測定したところ、3.4m2/gであることが分かった。
【0074】
結晶化1A及び結晶化1B
一連のバッチ式貧溶媒結晶化を以下の:
1)化合物Aをトルエン及びヘプタン中に室温で溶解して、表1に概説するように視覚的に透明な溶液を得ること(「初期」充填)、
2)微細な種の存在により結晶化を開始した粉砕工程からの指定量の微細な種のスラリー、及び微細な種のスラリーを添加した追加の貧溶媒を添加すること、
3)この貧溶媒を用いてn−ヘプタンを一分量ずつ添加して結晶化すること(なお、充填は添加と添加との間に少なくとも30分、間をおいて4時間〜12時間かけて行われた)、及び
4)得られたスラリーを濾過して限定量のヘプタン(約2ケーキ体積〜10ケーキ体積)で洗浄してから60℃で乾燥させて、表面積の分析(後処理)に適した乾燥ケーキを得ること、
によって行った。
【0075】
手順及び生成量は表1に記載する。
【0076】
【表1】
【0077】
実施例1Bに対応する顕微鏡写真を示す図10を参照されたい。スケールバーは10μmを示す。
【0078】
結晶化1C、結晶化1D及び結晶化1E
実施例1A及び実施例1Bの基本手順に続いて第2の一連のバッチを導入し、ここでは貧溶媒を12時間にわたって連続添加した(実施例1C〜実施例1E)。実施例1Dでは、イオン性界面活性剤レシチンオイル(食品グレード)を、バッチへの添加前に媒体ミルからの微細な種のスラリーに添加した。実施例1Eでは、非イオン性界面活性剤Triton X−100(登録商標)(Sigma Aldrich)を、バッチへの添加前に媒体ミルからの微細な種のスラリーに添加した。非イオン性又はイオン性の界面活性剤の添加により、表2に記載のそれらの結晶化から得られた生成物の表面積が増大した。
【0079】
【表2】
【0080】
[実施例2]
化合物A=Cox II阻害剤
この一連の実施例は、非イオン性又はイオン性の界面活性剤等の補助添加剤を微細な種の湿式粉砕プロセスに添加するとスラリーの物理的な取扱い性を高めることができることを示す。結晶化プロセスで使用するために粉砕後に補助添加剤を微細な種のスラリーに添加した結果、上記の実施例1D及び実施例1Eに示すのと同様に生成物表面積が増大した。さらに、異なる表面積の結晶化後に物質を得る必要に応じて粉砕時間を変えることができることを示すために、スラリーの試料を15分及び60分の時点で採取した。ここでも同様に、表面積はジェット粉砕した物質の表面積に匹敵し、しかも本発明のプロセスによって直接生成された。
【0081】
実施例2A及び実施例2Bの微細な種の粉砕
0日目、1mmのイットリウム安定化酸化ジルコニウムビーズを収容しているディスクミルを50%のn−ヘプタン及び50%のトルエンで洗浄し、ミルの内容物を容積式ポンプからの空気による処理のために移した。60グラムの化合物A及び1083グラムの50:50のトルエン:ヘプタン(重量に基づく)を、ミルに接続されている容器に充填した。総計10グラムのTriton X−100も添加した。混合物を、ミル収容タンク内で21℃の温度で攪拌し、次いで、混合物を900ml/分の速度で60分間ミル内を再循環させた。この時間中、ミルは6.8m/秒の先端速度で作動した。タンク内スラリーのうちの少量を15分、30分及び45分の時点で採取して顕微鏡によって粉砕プロセスを確認した。粉砕から60分後、スラリーを後の使用のためにガラスジャーに詰めた。ジャーの微細な種のスラリーの一部を0.2μmフィルタ漏斗で濾過して、溶液中に溶解していない微細な種の濃度を求めた。フィルタケーキを限定量の貧溶媒ヘプタンで洗浄し、真空オーブン内で60℃で乾燥させた。固体としての微細な種のスラリーの濃度は4.1重量%であった。この濃度は、粉砕プロセスの際に非イオン性界面活性剤を用いなかった実施例1の対応する微細な種のスラリーよりも約30%高かった。この違いは、粉砕システムにおける物理的な損失の低減に貢献し得る。乾燥後のフィルタケーキの表面積を標準BET等温線によって測定したところ、3.9m2/gであることが分かった。
【0082】
実施例2C及び実施例2Dの微細な種の粉砕
0日目、1mmのイットリウム安定化酸化ジルコニウムビーズを収容しているディスクミルを50%のn−ヘプタン及び50%のトルエンで洗浄し、ミルの内容物を容積式ポンプからの空気による処理のために移した。60グラムの化合物A及び1074グラムの50:50のトルエン:ヘプタン(重量に基づく)を、ミルに接続されている容器に充填した。総計125グラムのレシチンオイルも添加した。混合物を、ミル収容タンク内で20℃の温度で攪拌した。次いで、混合物を900ml/分の速度で60分間ミル内を再循環させた。ミルの出口温度は21℃であった。この時間中、ミルは6.8m/秒の先端速度で作動した。タンク内スラリーのうちの少量を15分、30分及び45分の時点で採取して顕微鏡によって粉砕プロセスを確認した。粉砕から60分後、スラリーを後の使用のためにガラスジャーに詰めた。ジャーの微細な種のスラリーの一部を0.2μmフィルタ漏斗で濾過し、溶液中に溶解していない微細な種の濃度を求めた。フィルタケーキを限定量の貧溶媒ヘプタンで洗浄し、真空オーブン内で60℃で乾燥させた。固体としての微細な種のスラリーの濃度は4.8重量%であった。この濃度は、粉砕プロセスの際にイオン性界面活性剤を用いなかった実施例1の対応する微細な種のスラリーよりも約50%高かった。この違いは、粉砕システムにおける物理的な損失の低減に貢献し得る。乾燥後のフィルタケーキの表面積を標準BET等温線によって測定したところ、5.3m2/gであることが分かった。
【0083】
結晶化2A、結晶化2B、結晶化2C及び結晶化2D
一連のバッチ式貧溶媒結晶化を以下:
1)化合物Aをトルエン及びヘプタン中に溶解すること(これにより、視覚的に透明な溶液が得られた(表3中の「初期」充填))、
2)さらに多くの非イオン性又はイオン性の界面活性剤を微細な種に添加した後で、表3に示すように指定量の微細な種のスラリーを添加すること、
3)n−ヘプタンを連続的に添加して結晶化すること、及び
4)得られたスラリーを濾過して2ケーキ体積〜10ケーキ体積のヘプタンで洗浄してから60℃で乾燥させて、表面積の分析(後処理)のために乾燥ケーキを得ること、
によって行った。
【0084】
手順及び生成量は表3に記載する。
【0085】
【表3】
【0086】
[実施例3]
化合物B=Cox II阻害剤
この一連の実施例は、「メルトバック」を示すことが分かっている化合物に対するピン粉砕の代わりとなることができることを示す。結晶の形態は、化合物Bの4つの他の可能な結晶形態が知られていてもプロセス全体を通して制御される。結晶化は高温で行われた。この例は、表面積が異なるレベルの微細な種の添加によって制御されることができることを示す。
【0087】
化合物Bのピン粉砕
化合物Bを、Alpine(登録商標)UPZ160ミル(Hosakawa)の一般的な条件及び高プロセス窒素流を用いて薬学的使用のためにピン粉砕した。この化合物はその低融点に起因して粉砕することが困難である。処理温度をこの化合物の融点未満に保つように、処理の間、0℃の低温窒素を40SCFM(標準立方フィート毎分)でミルのピンリンス(pin-rinse)として施した。粉砕は特別な工程なしには可能ではなかった。得られた物質の表面積は0.9m2/gであった。
【0088】
実施例3A及び実施例3Bの微細な種の粉砕
0日目、1mmのイットリウム安定化酸化ジルコニウムビーズを収容しているディスクミルを50%のn−ヘプタン及び50%のトルエンで洗浄し、ミルの内容物を容積式ポンプからの空気による処理のために移した。60グラムの化合物B及び1066グラムの50:50のトルエン:ヘプタン(重量に基づく)を、ミルに接続されている容器に充填した。混合物をミル収容タンク内で25℃の温度で攪拌し、次いで、混合物を900ml/分の速度で60分間ミル内を再循環させた。この時間中、ミルは6.8m/秒の先端速度で作動した。ミルの出口温度は25℃であった。タンク内スラリーのうちの少量を15分、30分及び45分の時点で採取して顕微鏡によって粉砕プロセスを確認した。粉砕から総計60分後、スラリーを後の使用のためにガラスジャーに詰めた。微細な種のスラリーの1つのジャーから、122.8gをフィルタ漏斗で濾過し、フィルタケーキを限定量の貧溶媒ヘプタンで洗浄した。総計9.7グラムの湿潤ケーキを回収した。次いで、これを真空オーブン内で60℃で乾燥させた。乾燥後のフィルタケーキの表面積を標準BET等温線によって測定したところ、5.7m2/gであることが分かった。
【0089】
結晶化3A及び結晶化3B
一連のバッチ式貧溶媒結晶化を以下:
1)化合物Bを50ml攪拌容器内でトルエン及びヘプタン中に50℃で溶解し、表4に「初期」充填として示すように視覚的に透明な溶液が得られること、
2)粉砕工程からの指定量の微細な種のスラリーを添加すること(微細な種及び微細な種のスラリーを添加した追加の貧溶媒の存在による結晶化を開始した)、
3)n−ヘプタンを連続的に添加して結晶化すること、及び
4)得られたスラリーを室温で濾過し、2ケーキ体積〜10ケーキ体積のヘプタンで洗浄してから60℃で乾燥させて、表面積の分析のために乾燥ケーキを得ること、
によって行った。
【0090】
手順及び生成量は表4に記載する。
【0091】
【表4】
【0092】
図11は、再循環粉砕から0.5分後の実施例3Bの微細粉砕スラリーの顕微鏡写真である。図12は、再循環粉砕から15分後の実施例3Bの微細粉砕スラリーの顕微鏡写真である。図13は、再循環粉砕から60分後の実施例3Bの微細粉砕スラリーの顕微鏡写真である。図14は、実施例3Bの結晶化後の最終生成物に相当する顕微鏡写真を示す。スケールバーは10μmを表す。
【0093】
[実施例4]
化合物C=BK1拮抗剤
一連の実施例は、本発明の方法を用いて複数の薬剤類に対応することができることを示す。また、最終生成物の表面積は異なるサイズの微細な種を用いることによって制御されることができることも示す。微細な種のサイズを異なる粉砕時間量を用いて変えることができる。この実施例での粉砕工程によって生成される種粒子はサイズが1μmを超える。化合物Cは低融点を有するため、乾燥粉砕の際に「メルトバック」を回避するのにMMC法が有用である。低温窒素をピンミルのピンリンスとして施して有意な量の物質を粉砕することができる。
【0094】
実施例4A及び実施例4Bの微細な種の粉砕
0日目、1mmのイットリウム安定化酸化ジルコニウムビーズを収容しているディスクミルを50重量%のn−ヘプタン及び50重量%のトルエンで洗浄し、ミルの内容物を容積式ポンプからの空気による処理のために移した。60グラムの化合物C及び1066グラムの50:50のトルエン:ヘプタン(重量に基づく)を、ミルに接続されている容器に充填した。混合物をミル収容タンク内で19℃の温度で攪拌し、次いで、混合物を900ml/分の速度で60分間ミル内を再循環させた。この時間中、ミルは6.8m/秒の先端速度で作動した。ミルの出口温度は20℃であった。タンク内スラリーのうちの少量を0分、15分、30分及び45分の時点で採取して顕微鏡によって粉砕プロセスを確認した。粉砕から総計60分後、スラリーを後の使用のためにガラスジャーに詰めた。スラリーの試料を、レシチン、及びISOPAR G(登録商標)での120秒の超音波破砕を用いて、SYMPATEC(登録商標)光拡散湿式細胞分析機で分析した。図24及び図25は、微細な種の粒径分布を示す。15分間粉砕した微細な種の場合、平均体積粒径は3.9μmであり、粒子の95体積%は9.8μm未満であった。60分間粉砕した微細な種の場合、平均体積粒径は2.35μmであり、粒子の95体積%は5.2μm未満であり、より長く粉砕した微細な種を用いるとより変化の急な粒径分布を示す。上記のいくつかの実施例におけるように、粉砕後15分〜60分の微細な種のスラリーの一部を濾過し、ヘプタンで洗浄し、60℃で乾燥させた。乾燥後、フィルタケーキの表面積を標準BET等温線によって測定したところ、15分の粉砕の場合は4.6m2/gであり、60分の粉砕の場合は6.6m2/gであることが分かった。このデータは、微細な種のサイズ及び表面積がプロセスパラメータによって制御されることができることを示す。
【0095】
結晶化4A及び結晶化4B
2つのバッチ式貧溶媒結晶化を以下の:
1)化合物Cを、オーバヘッド攪拌機によって攪拌した75ml容器内でトルエン及びヘプタン中に43℃で溶解すること(これにより、視覚的に透明な溶液が得られた(「初期」充填))、
2)in−situでの光後方錯乱によって視認される固体を形成することなく、スラリーを40℃に冷却して過飽和溶液を生成すること、
3)粉砕工程からの指定量の微細な種のスラリーを添加すること、
4)n−ヘプタンを連続的に添加して結晶化すること、及び
5)得られたスラリーを室温で濾過し、2ケーキ体積〜10ケーキ体積のヘプタンで洗浄してから60℃で乾燥させて、表面積の分析のために乾燥ケーキを得ること、
によって行った。
【0096】
手順及び生成量は表5に記載する。
【0097】
【表5】
【0098】
図15は実施例4Bの最終生成物の顕微鏡写真を示す。
【0099】
[実施例5]
化合物D=ビスホスホネート
本実施例は、従来の結晶化とその後に続く乾燥ケーキのピン粉砕によって得られる粒径をMMC法によって繰り返すことができることを示す。本実施例はまた、温度クールダウン結晶化(temperature cooldown crystallization)及び別の薬剤類を示す。異なるサイズの媒体ビーズを用い、プロセスは水系とした。
【0100】
従来の手法
化合物Dを60℃で100g/lの水中に溶解した。この化合物を0℃に冷却し、同時に200g/lに蒸留して結晶化生成物を得た。物質を濾過し、乾燥させ、一般的なピン粉砕条件を用いてピン粉砕した。この生成物のピン粉砕は特に困難である。ミルの機能は、40kgの物質を処理する毎に、ミルを停止してピンを洗浄した場合にのみ維持した。このプロセスにより、顕微鏡写真によって視覚的に分析されたように5μm〜40μmの生成物が生じた。
【0101】
実施例5の微細な種の粉砕
0日目、ディスクミルに1890gの1.5mmのイットリウム安定化酸化ジルコニウムビーズを充填し、脱イオン水で洗浄した。ミルの内容物を容積式ポンプからの空気による処理のために移した。34グラムの化合物D及び207グラムの脱イオン水(水重量に基づく)を、ミルに接続されている容器に充填した。混合物を、ミル収容タンク内で攪拌し、その上、630ml/分の速度で10分間ミル内を再循環させた。この時間中、ミルは6.8m/秒の先端速度で作動した。ミルの出口温度は20℃であった。タンク内スラリーのうちの少量を0分及び5分の時点で採取して顕微鏡によって粉砕プロセスを確認した。粉砕から10分後、スラリーを後の使用のためにガラスジャーに詰めた。微細な種の顕微鏡写真は1.5mmビーズの場合では1.0mmビーズの場合での実験よりも大きなサイズを示した。
【0102】
結晶化5
0日目、温度クールダウン結晶化を、オーバヘッド攪拌機によって攪拌した75ml容器内の95gの水中に14.0gの化合物Dを溶解することによって行い、これにより、視覚的に透明な溶液を得た。容器を囲んでいるジャケットの温度をこの溶解のために66℃に保持した。スラリーを、ジャケットを64℃にすることによって冷却して、固形物を形成することなく過飽和溶液を生成した。過飽和は視認され、in−situでの光後方錯乱によって確認した。粉砕工程から総計4.0グラムのスラリーの微細な種を添加し、ジャケット温度を61℃に変えた。次いで、ジャケットを61℃〜48℃に4時間かけて冷却し、48℃から20℃に7時間かけて冷却した。視覚的な粒径分析のために微細な種のスラリーの顕微鏡写真を分析した。平均長は17μmであり、平均幅は8μmであった。このサイズは、医薬用途に必要とされるサイズに近似している。図16は、実施例5の最終生成物の顕微鏡写真である。
【0103】
[実施例6]
化合物F=セロトニン拮抗薬
この一連の実施例は、MMC法が、イヌの血漿レベルによって測定された場合に、AFGジェットミルによって生成された生成物のバイオアベイラビリティに対処することができることを示す。この一連の実施例は、より小さな粒径(より広い表面積)を有する生成物を促すために、結晶化容器内に設置される補助エネルギー装置(この場合では超音波破砕機)の利用をさらに示す。実施例6は、粉砕プロセスにおけるビーズがより小さいことにより、微細な種の同じ充填を用いた場合により広い表面積の微細な種及びより広い表面積の生成物がもたらされることを示す。この実施例は、より高いレベル、ここでは20%の種の使用により、生成物の表面積を増大させることができることを示す。本実施例は、混合した水性有機溶媒を用いた半連続式プロセスである。化合物Fはいくつかの多形体を有することが知られており、本発明によるプロセスは所望の多形体を生成した。このことは、医薬品処理に対するMMC法の実現可能性を示す。
【0104】
AFG粉砕
物質を1mmノズル、50psigジェット圧、9000rpm〜18000rpmで100AFGにより粉砕したところ、表面積が0.6m2/gであった。
【0105】
実施例6の微細な種#1の粉砕
0日目、1890グラムの1.5mmのイットリウム安定化酸化ジルコニウムビーズを収容しているディスクミルを、60体積%のイソプロパノール(IPA)及び40体積%の脱イオン水で洗浄した。ミルの内容物を容積式ポンプからの空気による処理のために移した。ミルに接続された容器に、18.5グラムの化合物F及び220グラムの60/40のIPA/水を充填した。この混合物を、ミル収容タンク内で攪拌し、その上、600ml/分〜900ml/分の速度で15分間ミル内を再循環させた。この時間中、ミルは6.8m/秒の先端速度で作動し、ミルの出口温度は30℃未満であった。タンク内スラリーのうちの少量を0分、5分及び10分の時点で採取して顕微鏡によって粉砕プロセスを確認した。粉砕から15分後、スラリーを後の使用のためにガラスジャーに詰めた。
【0106】
実施例6の微細な種#2の粉砕
上記の粉砕#1の手順を繰り返した。ただし媒体として1894グラムの1.0mmのイットリウム安定化酸化ジルコニウムビーズを使用した。
【0107】
半連続式結晶化
半連続式結晶化を、指定の充填時間、微細な種のスラリー濃縮液及び貧溶媒の同時添加によって達成した。濃縮液の添加の間、溶媒比を維持した。充填を、容器の対向する両側で攪拌機付近の液体−ガス表面の下で22ゲージ針を介して行った。攪拌のために75ml容器にはオーバヘッド攪拌機を用い、及び8mm超音波破砕プローブを液体−ガス面の下に設置した。表6に示すように、結晶化の間、超音波破砕プローブを約10ワットの電力で作動させた。媒体粉砕した種#2を用いる実験の場合、濃縮液を充填したときと同じ速度でバッチ式の濃縮液添加の終了時点に追加の水を添加して、溶媒比を4:3から1:2のIPA:水に変えた。このことは、母液損失を下げることによって収率を約5%高めるために行われ、粒径に著しく影響を及ぼさなかった。後処理は、減圧による室温でのスラリーの濾過、及び空気による乾燥又は40℃の真空オーブン内での乾燥を含んだ。
【0108】
表6の実施例6Cの収率を85%になるように定量化した。この実験をX線回折によって示したところ、所望のヘミ水和物の形態がもたらされた。
【0109】
【表6】
【0110】
後配合(Post Formulation)及び使用
実施例6Cの固形生成物とAFG粉砕試料を並行して(side by side study)配合し、従来の医薬品成分を用いて直接充填カプセルとした。MMCの実施例6Cのイヌについての曲線下面積(AUC(24時間))を、AFGで粉砕した物質と比較したところ、等しいバイオパフォーマンスを得たことを示した。その結果を図26に挙げる。
【0111】
[実施例7]
化合物G=DP IV阻害剤
本実施例は、大きな粒子(50μmよりも大きい)を本発明のMMC法によって一貫して作製することができることを示す。粒径は種々の異なる種の充填(loads)を用いて目的に合わせることができる。
【0112】
媒体粉砕
0日目、KDL媒体ミルを80/20のIPA/水で洗浄し、空気を送って乾燥させた。80/20(重量に基づく)のIPA/水中、100mg/gでの化合物Gのスラリーを再循環モードで、300ml/分の速度で120分間ミル内に給送した。得られた微細な種の粒径は、光回析によって測定された場合に4.7μmの平均サイズを有した。
【0113】
結晶化
一連の結晶化を、実施例7の媒体粉砕した微細な種を用いて行った。これらの結晶化において、種の量は様々であった。70/30(重量に基づく)のIPA/水中、220mg/gでの化合物Gのバッチを70℃より高く加熱して固形物を溶解した。視覚的に透明な液体が得られた。バッチを65℃〜67℃に冷却して過飽和を生じさせた。バッチを、表7に示すような微細な種のレベルで播種した(種のスラリーに添加した乾燥生成物のグラム対バッチ内での種のスラリーのグラム)。バッチを3時間エージングし、5時間かけて室温に冷却した。イソプロピルアルコール貧溶媒を15分〜30分の期間をかけて充填して、80/20(重量に基づく)のIPA/水に達した。バッチを1時間エージングし、減圧濾過し、45℃のオーブン内で減圧乾燥させた。粒径を、湿潤状態で約30ワットで30秒超音波破砕を用いて粒度分布測定(Microtrac)粒径光回析を介して分析した。以下の結果を得た。
【0114】
【表7】
【0115】
[実施例8]
化合物D=ビスホスホネート
本実施例は、MMC法のスケールアップと、スケールアップ時の容器の混合特性を高める再循環ループの利用とを示す。本実施例は、再循環ループ内に設置されるより高い強度のエネルギー装置(ここではスタティックミキサー)により、最終生成物の表面積を増大させることができることをさらに示す。この一連の例は、ピン粉砕した生成物に匹敵するプロファイルを示す。
【0116】
ピン粉砕
化合物Dを結晶化した。この生成物をピン粉砕し、得られた粒径を光回析によって測定したところ、18.7μmであり、95%が50μm未満であった。表面積は0.53m2/gであった。
【0117】
実施例8の微細な種の粉砕
一連の媒体粉砕の実験を行って結晶化のために微細な種を供給した。0日目、ディスクミルに1.5mmのイットリウム安定化酸化ジルコニウムビーズを充填し、次いで脱イオン水で洗浄した。ミルの内容物を容積式ポンプからの空気による処理のために移した。1リットル当たり100グラムの脱イオン水濃度に等しいスラリーを、ミルに接続された容器に充填した。この混合物を、ミル収容タンク内で攪拌し、その上、900ml/分の速度でミル内を再循環させた。この時間中、ミルは6.8m/秒の先端速度で作動し、ミルの出口温度は25℃であった。粉砕後、スラリーを次の使用のためにガラスジャーに詰めた。
【0118】
結晶化8
一連の温度クールダウン結晶化を、オーバヘッド攪拌機を用いて攪拌した容器内の2500gの脱イオン水中で化合物Dを250g溶解することによって行った。容器を囲んでいるジャケットの温度を上げ、バッチ温度を60℃〜62℃に上げて、バッチを視覚的に透明な溶液に溶解した。スラリーを52℃に冷却して、視認されるほどの固形物を形成することなく過飽和溶液を生成した。総計115ミリリットルの微細な種のスラリーを、反応器の上部を介して容器に添加し、52℃〜53℃で30分間エージングした。バッチを5℃に冷却し、少なくとも1時間エージングし、次いで、真空フィルタを用いて濾過冷却し、45℃で減圧乾燥させた。
【0119】
最終溶媒組成にある母液中の生成物の濃度に基づき、少なくとも80%の収率がこの一連の実施例について予測される。粒子の表面積をBET等温線及び光回析によって分析した。実験8Aの粒子は強固に凝集し、光回析機の測定能力の限界を上回った。図4に示すような再循環ループの追加により生成物の表面積が増大した。再循環ループ内のより高エネルギーな装置であるスタティックミキサーの追加により、乾燥生成物をピン粉砕することによってもたらされる表面積に匹敵する、より広い表面積がもたらされた。
【0120】
【表8】
【0121】
実施例8Aの結果は、MMC法をスケールアップするのに選択される装置が生成物の結果を変えることができることを示す。混合に役立たせるために再循環ループを容器に追加することは、本発明の一実施形態である。さらに、実施例8Cは、補助エネルギー装置を追加することにより、再循環ループ内により高いエネルギーをもたらし、それによって表面積が増大した生成物をもたらすことができることを示す。実施例8Cの表面積は、ピン粉砕によってもたらされる表面積に合致する。再循環ループ又は補助エネルギー装置なしでもたらされた結晶化により、図17及び図18に示すように比較的低い表面積及び大きな粒径の目に見える程度に凝集した物質がもたらされる。
【0122】
[実施例9]
化合物E=脂質低下化合物
本実施例は、貧溶媒を用いる半連続式結晶化を示し、ここでは貧溶媒及び濃縮液の複数の充填回数に対処することができる。生成物の表面積を増大させるのに超音波破砕が有用であることが示されている。ここでは、より小さな0.8mmのビーズを用いて、或る範囲のビーズサイズを本発明の方法に従って用いることができることを示した。
【0123】
従来の乾燥粉砕手法
化合物Eをジェット粉砕した。得られた表面積の詳細は、生成物当たり1.4m2/g〜2.9m2/gであった。
【0124】
実施例9の微細な種の粉砕
0日目、ディスクミルに乾燥状態の0.8mmのイットリウム安定化酸化ジルコニウムビーズを充填した。ミルに接続された容器に、1000mlの60/40のMeOH/水を充填し、次いで、生成物の性能のための補助添加剤として0.2グラムのブチル化ヒドロキシアニソール(butylated hydroxy anisole)(BHA)を充填した。混合物を、ミル収容タンク内で攪拌し、その上、900ml/分の速度で30分間ミル内を再循環させた。この時間中、ミルは6.8m/秒の先端速度で作動し、ミルの出口温度は21℃であった。タンク内スラリーのうちの少量を0分及び30分の時点で採取して、顕微鏡によって粉砕プロセスを確認した。粉砕から総計30分後、スラリーを後の使用のためにガラスジャーに詰めた。微細な種の平均サイズは約2μmであることが示された。
【0125】
結晶化9A、結晶化9B、結晶化9C及び結晶化9D
半連続式貧溶媒結晶化を以下の:
1)1リットルのメタノール中に60gの化合物Eを溶解することによって濃縮液を生成すること(なお、生成物の酸化を防止するために、この流れに総計0.2gのブチルヒドロキシアニソールを添加した)、
2)粉砕からの5mlの微細な種のスラリーを充填すると共に5mlの60/40のメタノール/水(体積に基づく)を添加することによって、微細な種床を形成すること(なお、22mm径ブレードを用いて600rpmで100ml攪拌容器に充填を行った)、
3)56ミリリットルの濃縮液及び36ミリリットルの脱イオン水貧溶媒を、別個のシリンジポンプを介して容器に同時充填すること、
4)バッチを室温で1時間エージングすること(なお、約10ワットの電力での超音波破砕を、8mmプローブ(Telesonics社製のDG30)を使用して濃縮液添加の際に且つ1時間のエージング期間の際に結晶化装置に直接適用した)、及び
5)得られたスラリーを室温で濾過してから45℃で減圧乾燥して、表面積の分析のために乾燥ケーキを得ること(なお、粒径を乾燥固形物の光回析によって測定した)、
によって行った。
【0126】
最終溶媒組成にある母液中の生成物の濃度に基づき、この実施例のセットについて少なくとも80%の収率が予測される。実験は同一の反応器システムを使用して行った。
【0127】
手順及び生成量は表9に記載する。
【0128】
【表9】
【0129】
実施例9A及び実施例9Bの生成物の顕微鏡写真をそれぞれ図19及び図20に示す。生成物は個々の結晶の長さ以外は同様である。図19は図21と比較することができ、ここでは、いかなる核形成も抑制するためにより低い超音波破砕出力(sonication power)及びより長い添加時間を用いてプロセスをスケールアップした。
【0130】
[実施例10]
化合物E=脂質低下化合物
本実施例は、本発明の方法が特殊な化学製品用の市販製品の体積レベルに対するスケールアップに適応可能であったことを示す。ここでは、15kgスケールの生成物が半連続式バッチ法を用いて1つのバッチで生成される。首尾よいスケールアップをもたらした再循環ループのより大きなスケールのエミュレーションが記載されている。再循環速度は18分のバッチの循環時間に対応し、実際の速度は大スケールな製造プロセスに対応する。超音波破砕出力密度は1つのバッチ当たり約0.7W/kgであり、実際のレベルは大スケールな製造プロセスに対応する。結晶化生成物を従来の製造機器を用いて後処理した。多くの医薬品の場合のように、生成物は酸素感応性であり、全ての流れは窒素流又は減圧印加を用いてガスを抜いた。補助添加剤であるブチルヒドロキシアニソール(BHA)を生成物の安定剤として使用した。
【0131】
実施例10の微細な種の粉砕
総計1.49kgの未粉砕のままの化合物E、9.3kgの脱イオン水、14kgのメタノール及び8.14gのBHAを、攪拌機を備えたジャケット付き30リットルガラス容器に充填して、容器の内容物をブレンドした。このスラリーに窒素を充填して溶液のガスを抜き、窒素スイープを粉砕プロセス全体を通して用いて、系を不活性に保持した。大量の固形物を充填すると、物質は湿潤中に凝集(clumping)を示した。物質を解凝集(declump)するために、3/8インチID(内径)再循環ラインを、ロータステータミル(粗歯付きのIKA(登録商標)Works T−50)を収容している容器に接続した。バッチを湿式ミル内を30分間再循環させて、大きな塊の固形物を破砕した。IKA Worksミルをポンプとして用いて、この工程中少なくとも2倍のバッチ量を再循環させた。この再循環工程により生成物の粒径が著しく縮小した。
【0132】
バッチを粉砕して微細な種にするために、第2の再循環ラインを図1におけるように構成した。ポンプは蠕動性のMasterflexとし、ミルはNetzsch媒体ミル(型番号「Minicer」)とした。ミルに135mlの1mmのイットリウム安定化酸化ジルコニウムビーズ(約500グラム)を充填した。次いで、Masterflex(登録商標)容積式ポンプを用いてバッチスラリーを300ml/分の速度でMinicerミル内を再循環させた。ミルは2202rpmで作動し、これは6.8m/秒の先端速度に対応する。ミル及びバッチ容器はグリコール浴によって冷却して、粉砕プロセス全体を通してバッチスラリー温度を25℃未満の温度に維持した。バッチスラリーを総計41時間粉砕した。粉砕したスラリーを室温で一晩エージングし、次いで、次の3時間以内の使用のために媒体ミルからポリドラムに排出した。粉砕したスラリーは微細な種の流れであった。スラリーの一部を0.2μmフィルタで濾過し、40℃の真空オーブン内で乾燥させた後に分析した。スラリーの排出時点で、粉砕した固形物の表面積は4.05m2/gであり、平均体積粒径は2.1μmであり、粒子の95体積%は4.8μm未満であった。Helos分析機を使用した。
【0133】
実施例10の結晶化
再循環ループの設定:より大きなスケールの機器は図3の設定と同様であるが、ただし、インラインレーザ後方錯乱プローブ(inline laser backscattering probe)を用いて、スラリー中の粒子のコード長(chord length)をリアルタイムで測定し、第1の混合装置の前に種を充填した。100ガロン攪拌タンクの底部からの再循環ループは以下の:
1)ダイアフラムポンプと、
2)コード長監視のための集光反射測定プローブ(focused beam reflectance measurement probe)と、
3)必要に応じて種のスラリーを採取及び充填するための3/8インチ弁ポートと、
4)ドラムからの脱イオン水貧溶媒の添加のためにポンプに接続された高速混合装置と、
5)2リットルのフロースルーセルにおいて2インチ径及び22インチ長のラジアル型超音波破砕ホーンから構成されるエネルギー装置(なお、超音波破砕機はTelsonics社製であり、2000Wの発電機によって給電された)と、
6)ドラムからのバッチ式濃縮液添加のためにポンプに接続された高速混合装置と、
7)スラリーの再循環速度を測定する質量計と、
8)主要な結晶化装置に戻るパイプ(内径は13/16インチ)と、
から構成した。
【0134】
貧溶媒流:先に清浄し脱イオン水で洗浄した容器に、総計250kgの脱イオン水を充填した。脱イオン水はいくつかの真空パージ及び窒素圧パージを用いてガスを抜いた。水を50ガロンドラム内に入れ、使用するまで閉じたままにした。この流れは貧溶媒流であった。
【0135】
バッチ流:メタノールで洗浄した容器に、総計14kgの化合物Eの活性医薬品成分(API)、144kgのメタノール(先にガスを抜いてある)及び80gのBHA阻害剤を充填した。化合物Eの濃縮液を50ガロンドラム内に入れ、使用するまで閉じたままにした。これはバッチ流であった。
【0136】
微細な種のスラリーの生成:先に生成した総計36kgの60/40(体積/体積)のメタノール/水の溶液を100ガロン結晶化装置に充填した。溶液を、再循環ループを用いて約25kg/分で再循環した。超音波破砕機のラジアル型プローブを350W電力で設定し、Lasentec(登録商標)FBRMプローブを測定のために作動した。上記の実施例に記載の微細な種のスラリーを、3/8インチ種充填ポート用のT継手を介して再循環ループに充填し、種の床を20℃〜25℃で超音波破砕により15分間再循環させた。これはバッチ用の微細な種であった。
【0137】
結晶化のための充填:容器攪拌機は22インチ径であり、結晶化のために3m/秒で回転した。総計129kgの脱イオン水を、メタノールバッチ濃縮液中の168kgの化合物Eと共に微細な種に10時間かけて同時に一定の充填速度で充填した。結晶化全体を通して、350Wでの連続超音波破砕を加えながらバッチを20℃〜25℃に保持した。試料を添加から1時間後、3時間後、6時間後及び10時間後に採取して、結晶化の進み具合を確認した。同時添加を完了した後、84kgの脱イオン水を20℃〜25℃で超音波破砕により2時間かけて一定の充填速度で充填した。余分な水の貧溶媒の追加を行い、生成物の溶解度を下げることによって収率を高めた。充填は低速で行い、核形成に対する結晶の成長を促した。
【0138】
脱イオン水の充填後、バッチを20℃〜25℃で超音波破砕により1時間エージングして、結晶の完全な成長を確実にした。図21に示すように光学顕微鏡を用いて結晶スラリーの写真を収集した。図21は、制御されていない核形成が存在することに起因する小さな粒子が全くない状態で粒子が単分散していることを示す。再循環ループをオフにして、バッチを20℃〜25℃で一晩エージングした。濾過による後処理及びバッチの乾燥を続いて行った。
【0139】
実施例10の後処理
濾過及び乾燥:容器内での一晩のエージング後、バッチを室温で濾過した。1mg/g未満の化合物Eの濃度を有する総計385kgの母液を回収した。容器の壁からバッチフィルタにかけて洗浄すると共にフィルタ内の生成物を洗浄するために、先に生成した総計20kgの50/50(v/v)のメタノール/水を、スプレーボールを介して結晶化装置に充填した。総計40kgの洗浄水及び残存母液を回収した。濾過及びケーキへの少なくとも1時間の窒素圧の印加後、湿潤ケーキを全てフィルタから取り出し、トレイに載せ、減圧下で40℃で48時間、大型トレイ内で乾燥させた。この時点で、ケーキにおける残存水及びメタノールは0.5重量%しかなかった。総計14.5kgの乾燥ケーキをトレイドライヤーから取り出したところ、特に物理的な損失を考慮すると93.5%の高収率が得られることが示された。平均体積粒径は8.8μmであり、粒子の95体積%は20.3μm未満であった。この表面積は、BET窒素吸着によって測定した場合に1.7m2であった。これらの結果は、プロセスのスケールアップを示す実施例10の実験材料に匹敵した。
【0140】
図21は図19と比較することができる。結晶は同様のサイズ及び形態を有した。ここでは、単位体積当たりの超音波破砕出力は、実験室において1リットル当たり100Wから1リットル当たり1ワット未満まで低下したが、性能は許容範囲であった。したがって、超音波破砕力の実用レベルがすべてのスケールで支障なく用いられることができることを示す。
【0141】
[実施例11]
化合物D=ビスホスホネート
本実施例はクールダウンバッチ結晶化のスケールアップを示す。スケールアップの場合、乱流流速(1m/秒の平均線速度)での再循環ループ及び二重T継手のエネルギー装置を用いて結晶化の際の微細な種及び生成物の分散に役立たせることによって結晶の凝集を防止することができることも示す。本実施例は、超音波破砕することなく凝集が形成されないようにすることが可能であることをさらに示す。
【0142】
実施例11の微細な種の粉砕
手順は実施例10の手順と同様である。ただし、異なる生成物供給流と共に用いられるDYNO(登録商標)−Mill Type KDLA媒体ミルを使用した。DYNO(登録商標)−Millに495mlの1.5mmのイットリウム安定化酸化ジルコニウムビーズを充填し、脱イオン水をミル内に再循環させてビーズを湿潤した。次いで余分な水を捨てた。総計1.0kgの化合物Dを30リットル容器内の10リットルの脱イオン水に充填した。この充填は、水中の部分溶解を考慮した後の主要なバッチに対する溶液からの3重量%に相当する。スラリーをロータ/ステータミル内に15分間再循環させ、次いで一晩エージングした。次いで、スラリーを0.9l/分の速度でMasterflexポンプを介して媒体ミル内を再循環させた。ミルの先端速度は6.8m/秒に設定した。粉砕を5時間行った。スラリーをミルからドラムに排出した。スラリーの試料を0.2μmフィルタで濾過し、アセトン(約0.1g/l未満の溶解度)で洗浄して試料を乾燥しやすくした。試料を真空オーブン内で乾燥させて分析した。体積平均粒径は3.19μmであり、粒子の95%は7.8μm未満であった。プロファイルはユニモーダル(uimodal:単峰性)であった。表面積は窒素吸着により1.7m2/gであった。
【0143】
実施例11の結晶化
メカニカル設定:結晶化装置のために、上記の実施例10と同じ機器設定を用いた。エネルギー装置は図5に示したような「二重T継手」から構成した。ラインは急に直角に曲がった(sharp-right-angle-turn)3/4インチIDスチールパイプから成っていた。流れは出口で合流する。
【0144】
バッチ結晶化:総計22kgの化合物Dを220リットルの脱イオン水に充填して60℃で溶解した。100ガロンタンク内の溶解液を攪拌し、60℃に維持し、29kg/分の流速で再循環ループに再循環させた。バッチを51℃〜52℃に冷却して種の充填のために過飽和を生じさせた。再循環ライン内の平均線速度(体積流量/横断面積)はラインの大部分では1.4m/秒〜1.7m/秒であり、バッチの循環時間は9分であった。本実施例では、再循環ラインは乱流再循環ループと共にエネルギー装置として二重T継手を含んでいた。容器は4m/秒の先端速度で攪拌した。
【0145】
微細な種のスラリーを、ダイアフラムポンプ及び3/8インチ種充填ポートを介して一定の速度で4分かけて再循環ループに充填した。充填を再循環ループ内へ直接施して、種のスラリーの分散を促した。バッチを種の充填によって50℃〜52℃に冷却し、その温度で30分間エージングし、次いで、制御された線形的なクールダウンを介して10時間かけて1℃〜3℃に冷却した。スラリーの光学顕微鏡写真を図22に示すように撮影した。図22に示すように、粒子は、制御されていない核形成があることに起因する小さな粒子が全くない状態で単分散した。
【0146】
実施例11の後処理
濾過及び乾燥:クールダウン後、バッチを1℃〜3℃で一晩エージングし、次いで、ポリフィルタクロス(poly filter cloth)(Shaffer社から入手可能なKAVON(商標)ブランド909ウイーブ)を設けた、予め冷却して(1℃〜3℃)攪拌したフィルタドライヤー(Cogeim 0.25m2)内で濾過した。湿潤ケーキを3連続の65kgアセトンスラリー洗浄(溶媒充填、内容物の数分間の攪拌、その後の濾過から成る)により洗浄した。これらの洗浄を用いて、乾燥の際に固形物の凝集をもたらすのに十分高い生成物濃度の残存母液を除去した。アセトンにより洗浄した固形物を、フィルタジャケットの25℃の流体によって同じフィルタ内で十分な減圧下で乾燥させて詰めた。顕微鏡写真は、ケーキの凝集が全くなく、乾燥ケーキの体積平均粒径は20.6μmであることを示した。粒子の95体積%は、Helos乾式粒子分析機を用いて41mm未満であった。表面積はBET窒素吸着により0.40m2/gであった。これらの結果は、実施例8B及び実施例8Cのラボスケール実験に匹敵する。これは、粒子の不十分な分散を結晶化の際に利用した実施例8Aの結果とは対照的である。
[実施例12]
【0147】
化合物D=ビスホスホネート
本実施例は、動作条件の選択及び所与の生成物でのMMC用のエネルギー装置の選択における融通性を示す。本実施例は製造スケールでの動作の第3の実施例でもある。本実施例は、実施例11と同じメカニカル設定及び手順を用いているが、クールダウン時間を10時間から3時間に短縮し、循環時間を9分から18分に延ばしたことを強調しておく。これらの作用の結果、核形成の可能性がより高くなり、再循環ループ及びエネルギー装置に対する暴露の頻度がより少なくなることで、結晶化装置内に形成されるいかなる凝集物も破砕されて分散粒子となる。エネルギー装置内でのより高速の固形物堆積速度及びより低速の再循環速度は、二重T継手の代わりにより高い強度のエネルギー装置である12インチ長及び2インチ幅のTelsonic rapid probe(1lのフロースルーセル内で800Wの電力で動作する)を用いることによって相殺した。種の充填も10重量%まで高めて実施例11よりも著しく小さい生成物を得るようにした。
【0148】
種の生成:本手順は、生成物及びミル準備について実施例11の手順に従った。ここでは、3.48kgの化合物Dの純水及び33kgの脱イオン水を30l容器に充填して、0.45l/分〜0.9l/分の流速で16時間、DYNO(登録商標)−Mill Type KDLAに再循環させた。得られた生成物の粒径は2.8μmの平均体積であり、粒子の95%が6.4μm未満であった。表面積は2.0m2/gであった。
【0149】
バッチ結晶化:本手順は実施例11の手順と同等であるが、ただし、100ガロンタンク内の水中に溶解した22kgの化合物Dを15kg/分程度の流速でバッチ全体を通して再循環ループに再循環させた。バッチを約53℃〜54℃に冷却して種の充填のために過飽和を生じさせた。
【0150】
微細な種のスラリーを、ダイアフラムポンプ及び3/8インチ充填ポートを介して一定速度で8分かけて再循環ループに充填した。充填を再循環ループ内に直接施して、種のスラリーの分散を促した。バッチを種の充填によって約50℃〜52℃に冷却し、この温度で30分間エージングし、次いで、制御された線形的なクールダウンを介して約1℃〜3℃に3時間かけて冷却した。スラリーの光学顕微鏡写真を図23におけるように撮影した。図23は、制御されていない核形成があることに起因する小さな粒子が全くない状態で粒子が単分散することを示す。物質を実施例11におけるように濾過によって後処理し、洗浄し、乾燥させた。結晶化の状態及び結果を以下に示す。
【0151】
【表10】
【0152】
本出願は、2006年3月14日に出願された米国仮特許出願第60/782,169号(その全体が参照により本明細書に援用される)の優先的な利益を主張する。
【図面の簡単な説明】
【0153】
【図1】再循環モードにある媒体粉砕に必要な典型的な構成部品を示し、当該構成部品はブレンド容器と、流体ポンプと、媒体ミルと、ブレンド容器に戻る再循環ラインとを含む。一回通し式粉砕(single pass milling)は再循環せず、生成物をミルから回収レシーバ(collection receiver)に単に給送するだけである。一回通しモードでは、ポンプの代わりに蒸留缶からの圧力伝達を用いることができる。一回通しを複数回行うことで同様の生成物プロファイルを再循環モードとして達成することができる。
【図2】実施例1〜実施例7、及び実施例9用に設定される結晶化容器を示す。実施例1では、針付きシリンジを用いて、貧溶媒を10秒未満で1分量ずつ高速充填した。任意選択的に、超音波破砕機プローブ及び/又は光拡散プローブを加えることができる。
【図3】実施例10、実施例11及び実施例12におけるように、微細粉砕及び結晶化プロセスのスケールアップに適応可能に示されている設定の一例を示す。結晶化容器と再循環ループの構成部品とが示されている。
【図4】実施例8に記載のプロセスを示し、ここでは、外部の再循環ループが補助エネルギー装置の用途に用いられている。このエネルギー装置は静止しており、ここでは、ミキサー内を通る流体の流れにより、圧力降下及び乱流の動きによって系内にエネルギーの導入が行われる。2つのストリームとスタティックミキサーとのインピンジメントを促す、図示のように配置された2つのT継手から成る二重T継手は、Koflo Corp製の「kenics helical style」の継手とした。
【図5】実施例11に用いられる二重T継手の補助エネルギー装置を示す。ラインは急に直角に曲がった3/4インチIDスチールパイプから作製される。2つの流れが出口で合流する。
【図6】可能な結晶化プロセスの概要であって、当該プロセスは、微細な種のスラリーを生成すること、結晶化される生成物の濃縮溶液を生成すること、及び結晶化を開始するようにスラリーを濃縮液と合わせることを含む、結晶化プロセスの概要である。過飽和を生じさせる複数の方法によってさらなる結晶化が可能であり、そのうちのいくつかを挙げている。
【図7】バッチ式結晶化法の一例である。
【図8】半連続式結晶化法の一例である。
【図9】バッチ式反応結晶化法の一例である。試薬A及びBを反応させて、結晶化される生成物を生成する反応シナリオが示されている。
【図10】実施例1Bの生成物の顕微鏡写真である。
【図11】再循環微細粉砕から0.5分後の、実施例3Bの微細粉砕プロセスの生成物の顕微鏡写真である。
【図12】再循環微細粉砕から15分後の、実施例3Bの微細粉砕プロセスの生成物の顕微鏡写真である。
【図13】再循環微細粉砕から60分後の、実施例3Bの微細粉砕プロセスの生成物の顕微鏡写真である。
【図14】実施例3Bの結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図15】実施例4Bの結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図16】実施例5の結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図17】実施例8Aの結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図18】実施例8Bの結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図19】実施例9Aの結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図20】実施例9Bの結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図21】実施例10の結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図22】実施例11の結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図23】実施例12の結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図24】再循環微細粉砕から15分後の、実施例3Bの微細粉砕プロセスの生成物についての粒径分布記録である。
【図25】再循環微細粉砕から60分後の、実施例3Bの微細粉砕プロセスの生成物についての粒径分布記録である。
【図26】実施例6におけるような微細粉砕及び結晶化プロセス又は乾式粉砕プロセスについて、直接充填カプセルの挿入から初めの24時間の血流中の化合物Fの血漿レベルを比較して3匹のイヌについて収集された薬物動態学的データに関する記録である。
【技術分野】
【0001】
本発明は、活性有機化合物の結晶粒子を製造する方法及び装置、並びにそれらの使用に関する。
【背景技術】
【0002】
例えば医薬品有効成分(「API」)のような活性有機化合物の製造の際、固形物の形成は、溶液相中での結晶化、その後に続く分離及び乾燥によって達成される場合が最も多い。多くの場合、乾燥した活性有機化合物は、最終生成物の適正な配合を確実にするのに必要な粒径プロファイルに達するようにさらに処理されねばならない。得られる粒径は実質的に様々であり得るが、大半の場合、微細な活性医薬成分の粉体は300μm未満の平均サイズを有する。しかしながら、低水溶性及び/又は低透過性の医薬品ターゲットにより40μm未満の粒径の結晶に対する強い必要性が存在する。配合物中の粒子が小さいことにより、体内への輸送のためにより広い表面積が与えられる。
【0003】
許容可能な粒径プロファイルに達するために、エアジェット分級粉砕、ピン粉砕(pin milling)又はハンマー粉砕等の乾式粉砕工程を行うことが一般的である。医薬品処理に通常用いられる乾式粉砕装置のいくつかの例としては、Hosakawa Micron(www.hosokawamicron.com)製の乾式粉砕装置(例えばピンミル:Alpine(登録商標)UPZ Fine Impact Mills、例えば流動エアジェットミル:Alpine(登録商標)AFG Fluidized Bed Opposed Jet Mills)、Fluid Energy製の乾式粉砕装置、Quadro Engineering製の乾式粉砕装置及び非特許文献1に記載の乾式粉砕装置が挙げられる。乾式粉砕工程を用いて、粒子の凝集物を破砕してその元のサイズにし、及び/又は元の粒子を破砕してより小さな片にすることができる。
【0004】
プロセス工学の観点から、乾式粉砕は多くの動作上の問題をもたらし、費用がかかる。一つの大きな問題は、活性化合物に対する作業者曝露の抑制である。非常に強力な化合物の場合、乾式粉砕は、ダスティングを低く保つのに高価な工学的制御を必要とするであろう。さらに、工学的制御はダスト爆発を最小限にとどめるのに必要とされるであろう。乾式粉砕の他の動作上の問題としては、高温での溶融又はミルの内部部品への付着による、乾式ミルの内部での物質の蓄積が挙げられる。ピン粉砕では、このような不十分な粉砕性能はそれぞれ「メルトバック」又は「フラッギング」と一般に呼ばれ、さらには非晶質物質の生成、ミルの目詰まり、及び物質が処理される際にミルを出る粒径の変化をもたらす可能性がある。化合物によっては処理中にミルを腐食し、API生成物において許容不可能な高レベルの汚染をもたらす。したがって、結晶化により直接、目的の粒径分布(PSD)の結晶を形成し、粒子仕上げ工程として乾式粉砕を回避することが望ましい。
【0005】
不都合なことに、溶液結晶化により直接又は湿式粉砕技法により直接、製造する方法はない。一つの進展として、固体スラリーのロータステータ粉砕があり、その後に分離が続く。ロータステータ粉砕は通常、20μmを上回る平均サイズの粒子を製造する。不都合なことに、大半の場合では、摩砕(attrition)がこの粉砕プロセスに見られることが多い。非常に小さな粒子がバイモーダル(bimodal:双峰性)粒径を残したまま元の粒子から削り取られる場合に摩砕が生じる(非特許文献2)。多くの場合、ロータステータ粉砕の結果、これらの微細粒子の存在により濾過工程が著しく低速となる。さらに、直接圧縮又はローラ圧着技法を用いるバイモーダルな生成物の配合が問題である。小さなAPI粒子のモノモーダルな生成物の形成は、仕上げ工程として乾式粉砕を用いない場合に有益であろう。
【0006】
結晶化による、液体中に溶解している溶質からの新たな固体相の形成は、以下の2つの経路:(1)新たな粒子の核形成によって、又は(2)既存の粒子上での溶質の堆積による成長によって生じることが一般的に認められている。核形成は、結晶化装置内の異物質上で生じるか又は溶液から均一に生じ得る。特許文献1及び特許文献2は、析出の際の溶質の多くの新たな粒子の大量な核形成によって生成される小さな粒子、さらにはナノ粒子を記載している。これらのプロセスでは、系の特性を溶媒組成、温度又は反応を用いて変えることで、溶質に高い過飽和をもたらし、この過飽和により、高速な核形成及び結晶化に至る。核形成による多くの粒子の発生により、結晶化工程の終了時点に小さな粒径分布がもたらされるため、乾式粉砕の必要がなくなる。
【0007】
上記の核形成プロセスの重大な弱点は、高過飽和下では、オストワルドの段階則(Ostwald’s rule)(非特許文献3)によって説明されるように、望ましくない固体状形態(結晶形態/結晶内の分子パッキング)が生じる可能性があることである。炭酸カルシウムについて、様々な結晶形態の生成がKabasci他によって立証されている(非特許文献4)。医薬化合物が同じAPIについていくつかの異なる結晶形態を示すことは一般的であり、したがって、これらの核形成を引き出す技術の使用は特定の用途と見なされる。さらに、高過飽和及びそれに伴う核形成を含むプロセスは、溶媒分子又は不純物を内包した結晶を生じる可能性がある。一般に、医薬品のために選択される精製及び分離プロセスは、高化学純度及び適正な固体状形態の生成物をもたらすべきであり、核形成事象が著しく影響を及ぼすプロセスは望ましくない。
【0008】
最終生成物の形態学的特性を制御しようとする際、微粒子工学では生成物の種粒子を用いて結晶化中に結晶成長のテンプレートを与える傾向が高い。播種は、過飽和度を制限することによって粒径、結晶形態及び化学純度を制御するのに役立ち得る。種々の粉砕法が種原料(seed stock)を生成するのに用いられてきた。結晶化種子用に小さな粒子を生成して適度なサイズの粒子を得るのに乾式粉砕が通常用いられてきた。この手法は、先に説明した乾式粉砕に伴う工学的問題及び安全面の問題をなくすが、種生成に関して湿式粉砕法ほど望ましくない。
【0009】
ロータステータ湿式粉砕は、20μmよりも大きいという実際の制限がある比較的大きな活性有機粒子を生成するのに用いることができることが示されている。その一方、20μmよりも大きくするように粉砕することは、小さな片がバイモーダル粒径分布をもたらす摩砕方式では粉砕時間を延ばす必要がある(非特許文献2)。ロータステータ湿式粉砕された生成物を種として用いる結晶化によって大きな粒子が生じ、多くの場合、バイモーダル粒径分布が生じることが分かっている。所望の小さなサイズの結晶又はモノモーダル物質を形成するには、その後に乾式粉砕工程が必要とされる。このような種の生成方法は理想的ではない。
【0010】
超音波破砕(sonication)は、結晶化のために大きな種を生成するのに用いられる別の技法である。例えば、100μmよりも大きい生成物をもたらす超音波破砕が示されている(特許文献3)。近年、400mm未満の粒子を有する医薬品の直接配合のための最終生成物流を形成するのに媒体粉砕(媒体ミル粉砕)が用いられている(特許文献4)が、その次の結晶化において湿式粉砕された微細な種を用いることはこれまで示されていない。媒体粉砕及びその有用性の概説が特許文献5に記載されている。
【0011】
上記特許は、媒体ミル及び媒体ミルビーズの構成について可能な材料を記載している。これらの材料は特許文献6に挙げられており、この特許では媒体は砂、ビーズ、シリンダ、ペレット、セラミック又はプラスチックから構成されることができると記載されている。この特許は、ミルを金属、鋼合金又はセラミックから構成することができること、及び、ミルにセラミックで内張りされてもよいことを開示している。ポリスチレンを含むプラスチック樹脂が特に有用であるとして述べられている。特許文献7は、安定剤の存在下で有機色素を1μm未満に粉砕するための酸化ジルコニウムビーズの使用を開示している。ミル構築物のいくつかの組合せを本発明の実施に用いてもよい。一実施の形態では、セラミックビーズ及びセラミックミルが用いられる。さらなる実施の形態では、セラミックビーズ及びクロムで内張りされたミルが用いられる。
【特許文献1】米国特許第5,314,506号
【特許文献2】米国特許出願公開第2004/0091546号
【特許文献3】米国特許第3,892,539号
【特許文献4】米国特許第5,145,684号
【特許文献5】米国特許第6,634,576号
【特許文献6】米国特許第3,804,653号
【特許文献7】米国特許第4,950,586号
【非特許文献1】Perry’s Chemical Engineer's Handbook(Sixth edition ed. Robert H. Perry and Don Green),Section8
【非特許文献2】American Pharmaceutical Review Vol 7,Issue 5,pp 120-123 -“Rotor Stator Milling of API's...”
【非特許文献3】Threlfall−vol 7 no 6 2003 Organic Process Research and Development
【非特許文献4】Trans IChemE,vol 74,Part A,October 1996
【非特許文献5】Chemical Engineering Progress,September 1997,P34“Take some Solid Steps to improve Crystallization”
【非特許文献6】Johnson and Prud'homme(Australian Journal of Chemistry 56(10): 1021−1024(2003)
【非特許文献7】Marcant and David(AIChE Journal Nov 1991 vol 37,No 11)
【非特許文献8】Chemical Engineering Transactions,“Proceedings of the 15 th International Symposium on Industrial Crystallization 2002”(Volume 1 2002, p 65, published by ADIC−Associazione Italiana Di Engegneria Chemi
【非特許文献9】Pohl and Schubert Partec 2004“dispersion and deagglomeration of nanoparticles in aqueous solutions”
【非特許文献10】McCausland et.Al.Chemical Engineering Progress July 2001 P.56−61
【非特許文献11】The Handbook of Industrial Mixing(Ed. Paul,et al,2004,Wiley Interscience)
【非特許文献12】The Handbook of Pharmaceutical Excipients(3rd edition,editor Arthur H.Kibbe, 2000,American Pharmaceutical Association,London)
【非特許文献13】Physicians Desk Reference(51 edition, 20O1,Medical Economics Co.,Montvale, NJ)
【発明の開示】
【発明が解決しようとする課題】
【0012】
概して、配合要求を満たすように乾式粉砕を省くために十分に制御されたサイズ又は表面積の活性有機物、特に医薬生成物を製造することができる結晶化プロセスが依然として必要とされている。医薬産業では、そのバイオアベイラビリティの増大及び/又は溶解率を理由に、より小さな粒子が一貫して求められている。同様に、必要な結晶形態及び十分に制御された結晶純度を有する化学化合物をもたらすことも重要である。本発明では、約0.1μm〜約20μmの範囲の平均粒径を有する湿式粉砕した微細な種が、制御された粒径分布、結晶形態及び純度を有することから、微細な活性有機固体粒子の生成に対し、特に活性医薬品成分の結晶化に対し驚くほど有効であることが示されている。本発明のさらなる利点として、下流粉砕の必要性がなくなるため、これらのプロセスに伴うことが多い衛生面及び安全面の危険性がなくなることが挙げられる。
【課題を解決するための手段】
【0013】
本発明は、活性有機化合物の結晶粒子を製造する方法を提供する。本方法は、湿式粉砕プロセスによって微細な種(micro-seed)を生成する工程と、微細な種を結晶化プロセスに付す工程とを含む。湿式粉砕(wet milling)プロセスによって生成される微細な種は、約0.1μm〜約20μmの平均粒径を有する。得られる結晶粒子は、100μm未満の平均粒径を有する。
【0014】
結晶化工程に関して、本発明は2つのプロセスを含む。第1の結晶化プロセスは、3つの工程プロセス:媒体粉砕を用いて微細な種のスラリーを生成すること、微細な種の一部を溶解すること、及び微細な種で活性有機化合物を結晶化することである。
【0015】
第2の結晶化プロセスもまた、微細な種のスラリーを生成すること、結晶化される生成物の溶液を生成すること、及びこの溶液とスラリーを合わせることを含む3つの工程プロセスである。この第2の結晶化プロセスの一実施の形態では、微細な種のスラリー及び生成物の溶液は合わさると高速微細混合する。
【0016】
第2の結晶化プロセスを達成するために、3つの処理構成のうちの1つを個別に用いるか又は組合せて用いてもよい。一つの構成はバッチ式処理構成であり、別の構成は半連続式処理構成であり、3つ目は連続式処理構成である。
【0017】
また、再循環ループを第2の結晶化プロセスと共に用いてもよい。第2の結晶化プロセスの一実施の形態では、再循環ループをバッチ式処理構成の一部として用いる。第2の結晶化プロセスの別の実施の形態では、再循環ループを半連続式処理構成の一部として用いる。第2の結晶化プロセスのさらに別の実施の形態では、再循環ループを連続式処理構成の一部として用いる。
【0018】
第2の結晶化プロセスは2つのタイプの溶媒流を用いる。一実施の形態では、溶媒系は水系溶媒流であり、別の実施の形態では、溶媒系は有機溶媒系であり、さらに別の実施の形態では、溶媒系は混合溶媒流である。
【0019】
さらに、補助エネルギー装置を第2の結晶化プロセスと共に用いてもよい。第1の実施の形態ではこの補助エネルギー装置は混合用T継手であり、第2の実施の形態では混合用エルボであり、第3の実施の形態ではスタティックミキサーであり、第4の実施の形態では超音波破砕機であり、第5の実施の形態ではロータステータホモジナイザーである。
【0020】
さらに、本発明の活性有機化合物は、鎮痛薬、抗炎症剤、駆虫薬、抗不整脈薬、気管支喘息治療薬、抗菌剤、抗凝血薬、抗うつ剤、抗糖尿病薬、抗てんかん薬、抗ヒスタミン剤、血圧降下薬、抗ムスカリン薬、抗ミコバクテリア薬、抗悪性腫瘍薬、免疫抑制剤、抗甲状腺薬、抗ウイルス薬、抗不安剤、鎮静剤、収れん薬、βアドレナリン作動性受容体遮断薬、造影剤、コルチコステロイド、咳止め薬、診断薬、画像診断薬、ドーパミン作動薬、止血剤、免疫学的薬剤、脂質調節剤、筋弛緩剤、副交感神経作働薬、副甲状腺カルシトニン、プロスタグランジン、放射性医薬品、性ホルモン、抗アレルギー薬、刺激剤、交感神経作働薬、甲状腺薬、血管拡張剤、及びキサンチンを含む群から選択される医薬品であってもよい。
【0021】
さらに、本発明は、本明細書に記載の方法によって製造される結晶粒子及び薬学的に許容可能な担体を含む医薬組成物をさらに提供する。
【発明を実施するための最良の形態】
【0022】
本発明の微細粉砕及び結晶化プロセス(「MMC」)は、微細な種粒子での、約100μm未満、例えば約60μm未満、さらには約40μm未満等の平均体積粒径への成長を含む。たいていの場合、生成物は、結晶化のために添加される種の量に応じて約3μm〜約40μmの範囲である。微細な種は、平均体積分析により、約0.1μm〜約20μm、例えば約1μm〜約10μmの範囲とすることができる。種は、複数の湿式粉砕装置、例えば媒体粉砕等によって生成することができる。平均1μm未満の粒子を用いてもよい。しかしながら、このサイズ範囲は、成長結晶中に粒子が分散したままである場合に得られるAPI粒径が約0.5%〜約15%の通常の種レベルを用いる従来の分離技法に望ましい粒径よりも小さいため、微細な種より魅力的ではない。
【0023】
本発明の方法(MMC)は、微細な種のスラリーを生成すること、及び結晶化される生成物を含有する溶液を生成することを含む。これら2つの流れを組合せて生成物の結晶化を行う。大半の場合では、結晶化は、結晶化を推進するように生成物の溶解性変化及び濃度変化を操作することによって続行される。これらの操作により、種上に溶質を堆積させるための推進力を与える過飽和系がもたらされる。播種を行っている間の過飽和レベル、及びその後の結晶化が、核形成に対する成長条件を高めるレベルで制御される。本発明では、本方法は、新たな粒子の発生を制御しつつ、微細な種での成長を促し易くするように設計される。結晶化方法の概説(成長及び核形成プロセス条件の説明を含む)をPriceが行っている(非特許文献5)。
【0024】
本発明のMMC法の微細な種粒子及び生成物粒子は、複数の特定の利点を有する。微細な種粒子は表面積対体積の比が高く、したがって、成長速度が所定の過飽和度で大きな種粒子に比して著しく高まる。高密集の種粒子では、異物質上での核形成が回避されるため、結晶化は低い過飽和度での既存の種粒子上における成長の1つとされる。そのため、API粒子のサイズ及び形態が種粒子の特性によって制御される。
【0025】
一般に、所望の結晶形態が最も安定している反応器条件での動作、及び所望の結晶形態での播種が好適である。粒子−粒子の衝突が著しく重量の少ない物体間で生じるため、小さな粒子はせん断による粒子摩砕に対する感度が低いことが見出されている。モノモーダルな種で開始する場合、本発明の方法は、光学顕微鏡写真及びレーザ散乱法によって確認されるようなモノモーダル粒径分布をもたらす。得られる生成物の単分散粒径により、複合プロセスを微細粒子仕上げに魅力的な方法にする下流濾過及び配合に適応可能である。
【0026】
本発明は、いずれもの析出した活性有機粒子又は結晶化した活性有機粒子、例えば、医薬品、バイオ医薬品、ニュートラシューティカルズ、診断剤、農業用化学物質、殺虫剤、除草剤、色素、食品成分、食品配合物、飲料物、ファインケミカルズ及び化粧品の製造に用いることができるが、説明を容易にするため、主として医薬品を特に対象とする。他の産業部門で使用される有機化合物のための結晶粒子/析出粒子は、本明細書に記載の同じ一般的技法を用いて製造することができる。
【0027】
微細な種の存在下での成長を促すように過飽和を生じさせるいずれの方法も本発明に適応可能である。結晶化を操作する一般的な方法は、溶媒組成変化、温度変化、化学反応の使用、又は蒸留の使用を含む。反応結晶化には1つ又は複数の試薬からの最終APIの配合が必要とされるが、生成されたAPIは過飽和となり、生成物の過飽和が結晶化の源となる。過飽和を生じさせる結晶化法及び核形成と成長との相互作用の概説をPriceが行っている(非特許文献5)。この文献はその全体が参照により本願に援用される。
【0028】
溶質への微細な種の添加又は微細な種への溶質の添加は、いくつかの方法、例えばバッチ式結晶化、セミバッチ式結晶化又は半連続式結晶化で達成することができる。これらの技法は当業者には一般的であり、他の結晶化装置構成への拡張も期待される。さらに、これらの方法の組合せも利用することができる。
【0029】
バッチ式結晶化は通常、温度を変えるか又は溶媒を蒸留によって除去して過飽和を生じさせる結晶化を含む。セミバッチ式結晶化は通常、溶質槽又は溶質用の反応前駆体への溶媒又は試薬の連続添加を含む。バッチ式結晶化及びセミバッチ式結晶化では、種は通常、溶質槽に添加され、これにより、種の添加時点で又は種の添加の結果として過飽和する。図6及び図7を参照されたい。
【0030】
半連続式結晶化は、結晶化全体を通して反応器内の液体相の含量をほぼ一定に保つように設計される。非溶媒(貧溶媒(anti-solvent)とも呼ぶ)による半連続式結晶化では、種の流れが反応器に添加され、その後、溶液中に溶解した溶質を含有する流れ及び非溶媒流の双方の同時添加が行われる。ここで、結晶化は成分が添加される速度と同様の速度で行われる。図8を参照されたい。反応結晶化の例示的な図式を図9に挙げる。
【0031】
MMC法に選択される流れの化学組成は、結晶化される化合物に応じて決まる。したがって、水性流、有機流又は水性流と有機流の混合流を用いることができる。
【0032】
本発明のプロセスでは、微細な種のサイズへの湿式粉砕には、下流の生成プロセスでの乾式粉砕の必要性を制限することが必要である。もっぱら選別機が約1μm〜約10μmの範囲の平均最適サイズの粒子を提供することができる。高エネルギーの流体力学キャビテーション又は高強度の超音波破砕、高エネルギーのボールミル粉砕又は媒体粉砕、及び高圧均質化等の粉砕法が、約1μm〜約10μmの範囲の平均最適サイズを有する微細な種を製造するのに用いることができる技術の代表的なものである。
【0033】
本発明の一実施形態では、媒体粉砕は目的のサイズに対して種の粒径を縮小するのに効果的な湿式粉砕法である。さらに、媒体粉砕は粉砕プロセス時にAPIの結晶度を維持することが分かっている。用いられる媒体ビーズのサイズは、例えば約0.5mm〜約4mmの範囲である。
【0034】
本発明の湿式粉砕プロセスの際に変更することができる追加パラメータとしては、所望の微細な種のサイズを得るための生成物濃度、粉砕温度及びミル速度が挙げられる。
【0035】
API生成物流に対して媒体粉砕作業を行って、コロイド安定剤の存在下で0.5mm以下の特殊ビーズを用いて平均サイズが1ミクロン未満の粒子を生成した。表面活性剤が、1ミクロン未満で活性であるコロイド粒子間の結合力を克服し、配合のために分散粒子流を供給する。この供給流を微細な種として本発明に用いることができる。本発明による結晶化は、結晶化に十分な分散速度が用いられる場合に最も予測可能である。種として粒子の凝集を用いることは、凝集物の数及びサイズが様々である可能性があるため望ましくない。したがって、0.1μm〜0.5μmの種結晶を本発明において用いることができ、その場合、有機化合物が分散粒子として自己安定化しない限り、コロイド安定剤を用いることが望ましい。
【0036】
本発明のプロセスは主として、既存の種粒子での成長のプロセスであるため、微細な種の量及びサイズがAPI粒径の主要な決定因子である。可変量の種を添加して結晶化後に所望の粒径分布(PSD)を得ることができる。典型的な種の量(種のスラリーの溶媒相中に溶解していない物質)は、結晶化される活性成分の量に対して約0.1重量%〜20重量%の範囲である。成長結晶では、少ない種の導入により、より大きな粒子が生じる。例えば、少量の種により生成物の粒径が増大して60μmを上回ることができるが、結晶化は潜在的に、核形成を回避すると共にこれらの種での成長を促すように非常に低速である可能性がある。約0.5%〜15%の種レベルが1μm〜10μmの微細な種で開始する場合の妥当な充填である。
【0037】
別の実施形態では、MMC法は、
(1)約0.1μm〜20μmの平均サイズを有する微細な種を生成する、湿式粉砕プロセスを用いること、及び
(2)100μm未満の平均サイズを有する結晶粒子をもたらす、微細な種で活性有機化合物を結晶化すること、
を含む。
【0038】
さらなる実施形態では、MMC法は、
(1)約0.1μm〜20μmの平均サイズを有する微細な種を生成する、湿式粉砕プロセスを用いること、
(2)微細な種の一部を溶解すること、及び
(3)100μm未満の平均サイズを有する結晶粒子をもたらす、微細な種で活性有機化合物を結晶化すること、
を含む。
【0039】
溶解プロセスは、加熱、pH変化、溶媒組成等の変化を含み得る。これにより、得られる粒径分布が種よりもわずかに大きいにすぎない分布となる。いくつかの例では、微細な種の粒径が適度に大きくなるだけで生成物には十分であり、したがって、50%以上の種レベルを用いることができる。
【0040】
一実施形態では、微細な種は分離し、乾燥生成物として充填され得る。
【0041】
本発明のMMC法は大幅にスケール拡大可能である。スケールごとの適正な機器設計により、全てのスケールでロバスト性能が可能となり得る。確実なスケールアップに用いられ得る2つの特徴は以下の通りである:1)活性結晶化系への物質の添加の際の高速微細混合、及び2)所望でない凝集の粒子分散のためのエネルギー装置の包含。これらの特徴を含む結晶化装置設計は、本発明のスケールアップに適応可能である。
【0042】
高速微細混合は、生成物の結晶化の特徴的な誘導時間に対して分子レベルでの2つの流れの高速混合時間を意味する。これらの概念は、非特許文献6及び非特許文献7によって詳細に説明されている。これらの著者らのグループは双方とも、微細混合時間が結晶化又は析出の成果に影響を及ぼし得ることを強調している。したがって、著者らは、微細混合時間が短いことが有利であることを強調している。溶媒、濃縮液又は試薬添加剤について、この高速微細混合により、核形成事象につながる可能性がある濃度勾配が低減又は排除される。
【0043】
本発明の一実施形態では、過飽和度は微細な種での成長を促すように低く保たれる。いくつかの例では、結晶化の動態は高速であり、核形成を実質的に回避することができない。適した高速ミキサーは、これらの例では、試薬流を高速混合すると共に試薬の局部的な高濃度を回避することによって核形成を制限するように選択されるものとする。微細な種が溶質を有する結晶化装置に添加される場合、高速微細混合による種の分散は、結晶化が行われる際に微細な種の凝集を制限することが必要である。
【0044】
さらに、Hunslowの論文(非特許文献8)は、粒子の凝集が局部的な過飽和レベルに直接関連することを教示している。したがって、高速微細混合はまた、この状況の場合に凝集を最小限度にとどめる上でも役立つ。高速ミキサーの選択は、ミキサーの選択によって粒子摩砕のレベルに対してバランスがとれていなければならない。粒子−粒子間による粒子の発生、又は種粒子の存在下での粒子−結晶化装置間の表面相互作用による粒子の発生を引き起こすメカニズムは一般的に、第2の核形成と呼ばれ、大半の結晶化において或る程度生じることが予測される。機器の選択により、この作用の程度を変えることができる。
【0045】
小さなサイズの活性有機化合物は凝集し、次いで、結晶化の際に凝集物上に塊を堆積させることによって凝集する。粒子が凝集すると、API粒径は、成長が個々の種粒子にのみ生じ、且つ凝集物が存在しない場合よりも大きくなるであろう。医薬用途によっては、凝集は望ましくなく、その理由は、凝集した粒子を含んだプロセスをスケールアップすることがより困難となる可能性があるからである。これらの状況では、凝集に首尾よく対処する、微細な種を使用する方法を発展させることが望ましい。
【0046】
一般に、粒子が受けるエネルギー密度は解凝集を得るのに十分でなければならず、結晶化の際に、分散系を維持するのに十分な周波数で粒子をエネルギー密度に晒さなければならない。補助エネルギー装置は粒子を分散することによって凝集を最小限にとどめるのに役立つ。エネルギー装置の機能は、軽く凝縮した物質を分解する粒子衝突を生じさせるか又は凝集物をよじって(torque)破砕するせん断場を生じさせることである。このエネルギー装置は、適正に設計されたタンク攪拌機又は流体を圧送するのに用いる再循環パイプと同じくらい単純なものとすることができる。流体ポンプは高エネルギー装置であり、結晶化プロセスに影響を及ぼし得る。これらの装置は、凝集物が強固でないか又は生成物が装置に頻繁に晒される場合では十分である。強力なせん断環境を与えるにはロータステータ湿式ミルが有用であり、これらは粒子自体が摩砕しない場合に最も有用である。結晶化装置に適用される超音波破砕エネルギーが、容易に凝集してより強固な凝集物を形成する化合物の凝集を制限することが分かっている。結晶化の終了時点での超音波破砕又はエネルギー装置の適用も凝集物を破砕するのに有用であり得るが、凝集物は結晶化時間の終了まで著しく強固である可能性があるため、結晶化の最中よりも望ましくない。超音波破砕ホーン(sonication horn)もまた、主要な粒子を破砕することなく凝集物を軽く破砕することに関与し得る音波を与える。
【0047】
針状結晶が微細有機物の処理に好機を示す。特に、微細有機物の濾過速度が一般に低速である。本発明の一態様は、結晶化中の超音波破砕の使用である。超音波破砕は、幅方向への針状結晶の成長を促すことができ、濾過に対しより堅固な生成物をもたらす。針状結晶のために微細な種を生成するのに超音波破砕を使用することもまた、特に有利である。針状により、長手方向軸を中心に破砕する傾向があり、同様の幅の結晶がもたらされるが、長さはより短い。
【0048】
超音波破砕(超音波は典型的には10kHz〜60kHz)の基本技術は非常に複雑であり、上首尾の解凝集の基本的なメカニズムは不明確であるが、超音波破砕が解凝集(deaggregation or deagglomeration)時に効果的であることがよく知られている(非特許文献9)。機構プロセスの非限定的な説明として、超音波破砕は凝集物の破砕のために高出力密度ひいては高強度の超音波を与える。キャビテーション気泡が音波の負圧期間中に形成され、これらの気泡が高速に潰れることにより、解凝集に有用な衝撃波、高い温度及び圧力がもたらされる。本発明では、種粒子及び成長した粒子はたいていの場合は著しく破砕されず、したがって、超音波破砕の高エネルギー事象が、粒子を摩砕することなく分散粒子での成長を促すのに特に効果的であることが分かっている。
【0049】
近年、化学薬品の超音波破砕での作業は結晶化に入り込んでいる。核形成の誘導時間を減らすか又は適度な過飽和度で簡単な核形成を与えるのに超音波を使用することに焦点が置かれてきた。このことは、先駆的な固体(solids apriori)の非存在下で、又は固体種をバッチ濃縮液に添加する必要なく、種床生成の再現性を高めるのに有用である(非特許文献10)。この手法は、微細な種の存在が最終生成物特性、特に結晶形態を示す本教示とは対照的である。
【0050】
MMC法におけるように分散した微細な種の粒子での制御された成長のために医薬品の結晶化に超音波破砕を適用することは独特である。さらに、本発明に示されるように首尾よい解凝集に必要とされる超音波破砕力は比較的小さく、結晶化の終了時点で総バッチの1リットル当たり10ワット未満、好ましくは、結晶化の終了時点で総バッチの1リットル当たり1ワット未満である。超音波破砕のための機器の設計及び技術のリサーチが実際のリサーチ領域である。本発明に適用可能なフローセルのいくつかの例は、エネルギー装置として再循環ループで使用するいくつかの製造業者(例えばBranson WF3-16、及び、例えばTelsonics SRR46 series)によって市販されている。
【0051】
微細混合のための方法及び補助エネルギー装置を組み入れるための方法を提供するために再循環ループを使用することは、スケールアップに特に有利であることが示されている。主要な概念は、従来の結晶化装置(通常は攪拌タンク)からの微細混合及びエネルギー入力の要求を軽減し、特定化した機能性ゾーンを形成することである。攪拌タンク結晶化装置は、攪拌タンクの外部で独立して制御されるシステムに対して微細混合及び補助エネルギー入力を用いるブレンド装置として働くことができる。この手法は、大スケールの製造のためのスケーラブルな結晶化システムの一例である。このシステムの実際のエミュレーションを図3に挙げる。微細混合は、高エネルギー散逸領域又は高乱流領域に流れを加えることによって最もよく達成される。この流れをパイプの中央の再循環ループ内の乱流領域に加えることが一実施形態である。この例では、少なくとも1m/秒の速度が従来のパイプ流に推奨されているが、提供される微細混合は必ずしも高速である必要はない。この例は、試薬を追加する位置を限定せず、試薬を添加する方法が適正な微細混合を達成するのに重要である。パイプライン及び攪拌容器中での混合の概念は、非特許文献11に記載されている。
【0052】
結晶化装置の再循環速度は、再循環ループ全体を通して結晶化の終了時点でバッチの一体積の相当量を移動する時間又は結晶化の終了時点の循環時間(turnover time)まで定量することができる。容器の循環時間は個別に様々であり、バッチが補助エネルギー装置に晒されて生成物の凝集を制限することができる周波数の関数である。大スケールの製造のための典型的な循環時間は約5分〜約30分の範囲であるが、これに限定されない。生成物結晶の凝集物は通常、結晶化による塊の堆積を伴うため、結晶化の速度は、解凝集を可能にするのに必要とされる循環時間を延ばすように低速となり得る。
【0053】
得られる生成物の粒子のサイズ及び表面積は、種又は結晶化バッチに補助添加剤を添加することによって増大し得る。一実施形態では、添加剤は結晶化装置内での種及び結晶の分散に役立ち、この分散により粒子凝集を制限する。補助添加剤の添加は他の目的、例えば、生成物の酸化の還元等のために又は容器の側面に固着する化合物を制限するために用いることができる。補助添加剤は、純粋な活性成分をもたらす分離工程によって実質的に取り除かれてもよい。界面活性剤特性を有する物質は、MMC法の粉砕工程、播種工程及び結晶化工程のスラリー特性を高めるのに有用である。
【0054】
補助添加剤としては、限定するものではないが、不活性希釈剤、両親媒性コポリマー、可溶化剤、乳化剤、懸濁剤、アジュバント、湿潤剤、甘味料、風味料、及び香料、等張剤、コロイド分散剤、及び界面活性剤、例えば限定するものではないが、ジミリストイルホスファチジルグリセロール等の荷電リン脂質;アルギン酸、アルギン酸塩、アカシア、アカシアガム、1,3ブチレングリコール、塩化ベンザルコニウム、コロイダル二酸化ケイ素、セトステアリルアルコール、セトマクロゴル乳化ワックス、カゼイン、ステアリン酸カルシウム、塩化セチルピリジニウム、セチルアルコール、コレステロール、炭酸カルシウム、ステアリン酸スクロースとジステアリン酸スクロースとの混合物であるCrodestas F−110(登録商標)(Croda Inc.)、クレイ、カオリン及びベントナイト、セルロースの誘導体、並びにヒドロキシプロピルメチルセルロース(HPMC)等のそれらの塩、カルボキシメチルセルロースナトリウム、カルボキシメチルセルロース、及びその塩、ヒドロキシプロピルセルロース、メチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロースフタレート、非結晶性セルロース、リン酸二カルシウム、ドデシルトリメチルアンモニウムブロミド、デキストラン、スルホコハク酸ナトリウムのジアルキルエステル(例えば、American CyanamidのAerosol OT(登録商標)、ゼラチン、グリセロール、グリセロールモノステアレート、グルコース、Olin 10−G(登録商標)又は界面活性剤10−G(登録商標)(Olin Chemicals(Stamford, Conn.))としても知られるp−イソノニルフェノキシポリ−(グリシドール);オクタノイル−N−メチルグルカミド、デカノイル−N−メチルグルカミド等のグルカミド;ヘプタノイル−N−メチルグルカミド、ラクトース、レシチン(ホスファチド)、n−ドデシルβ−D−マルトシド等のマルトシド;マンニトール、ステアリン酸マグネシウム、ケイ酸アルミニウムマグネシウム、綿実油、トウモロコシ胚芽油、オリーブ油、ヒマシ油、及びゴマ油等の油;パラフィン、ジャガイモでんぷん、ポリエチレングリコール(例えば、Union CarbideのCarbowax 3350(登録商標)及びCarbowax 1450(登録商標)、並びにCarbopol 934(登録商標))ポリオキシエチレンアルキルエーテル(例えば、セトマクロゴール1000等のマクロゴールエーテル)、ポリオキシエチレンソルビタン脂肪酸エステル(例えば、ICI specialty chemicalsの市販のTween(登録商標))、ポリオキシエチレンヒマシ油誘導体、ポリオキシエチレンステアレート、ポリビニルアルコール(PVA)、ポリビニルピロリドン(PVP)、リン酸塩、酸化エチレン及びホルムアルデヒドを含む4−(1,1,3,3−テトラメチルブチル)フェノールポリマー(チロキサポール、スペリオン(superione)、及びトリトンとしても知られる)、全てのポロキサマー及びポラキサミン(例えば、BASF Corporation(Mount Olive, NJ)から入手可能な、Pluronics F68LF(登録商標)、F87(登録商標)、F108(登録商標)、及びtetronic 908(登録商標))、ピラノシド、例えば、n−ヘキシルβ−D−グルコピラノシド、n−ヘプチルβ−D−グルコピラノシド、n−オクチルβ−D−グルコピラノシド、n−デシルβ−D−グルコピラノシド、n−デシルβ−D−マルトピラノシド、n−ドデシルβ−D−グルコピラノシド;第4級アンモニウム化合物、ケイ酸、クエン酸ナトリウム、でんぷん、ソルビタンエステル、炭酸ナトリウム、固体ポリエチレングリコール、ドデシル硫酸ナトリウム、ラウリル硫酸ナトリウム(例えば、DuPont corporationのDUPONOL P(登録商標))ステアリン酸、スクロース、タピオカでんぷん、タルク、n−ヘプチルβ−D−チオグルコシド等のチオグルコシド、トラガカント、トリエタノールアミン、及びアルキルアリールポリエーテルスルホネートであるTriton X−200(登録商標)((Rhom and Haas)等が挙げられる。不活性希釈剤、可溶化剤、乳化剤、アジュバント、湿潤剤、等張剤、コロイド分散剤及び界面活性剤は、市販されているか、又は当該技術分野で既知の技法で調製することができる。
【0055】
同様に、プロセス性能を適応させるための結晶成長調節剤等の市販されていない所望な化学構造物を合成することが可能である。混合前又は混合後のプロセス溶媒流への添加に適したこれらの又は他の薬学的賦形剤のうち多くの特性は、非特許文献12に挙げられており、この開示はその全体が参照により本明細書に援用される。
【0056】
本発明のMMC法では、微細粒子は最終混合液中に形成される。微細粒子を含有する最終溶媒濃縮は、透析、蒸留、ワイパー式薄膜蒸発(wiped film evaporation)、遠心分離、凍結乾燥、濾過、滅菌濾過、抽出、超臨界流体抽出及び噴霧乾燥が挙げられるがこれらに限定されない複数の後処理プロセスによって変更され得る。これらのプロセスは通常、微細粒子の形成後に行われるが、形成プロセス中に行われてもよい。
【0057】
溶液相中の生成物の高溶解性は、乾燥の際に粒子の液相中の残存溶質の堆積をもたらし、これが、結晶化の際に形成される元の粒子の軽い凝集につながる。配合後の薬剤粒子の溶解は多くの場合、凝集物に対する元の粒径の表面積に影響されやすい。軽い凝集物は配合処理中に破砕されて、許容可能なバイオアベラビリティを有する生成物をもたらすことができる。
【0058】
粒径を測定する際、適正な測定具を選択するように注意を払わねばならない。例えば、粒径を測定するのに用いられる典型的なレーザ光走査技法は、凝集物を元の粒子になるように破砕することができない可能性があるため、誤った示度をもたらし得る。したがって、生成物の粒径分析は元の粒径ではなく大きな凝集物を示し得る。光走査技法に対する表面積測定は、以下の例に記載のように好適な測定技法である。しかしながら、平均粒径は従来のレーザ光走査装置を用いて測定されてもよい。具体的には、乾燥生成物の分析は1気圧〜3気圧のSympatec Helos社製分析機と同様の機械において好適である。一般に、生成物の表面積及び粒径が対象の粒子の形状に応じて直接関わる。
【0059】
粒径分析について問題となることが多い粒子の一形態は、長さ対幅のアスペクト比が6を超える針形状である。このタイプの粒子は、顕微鏡写真が小さなサイズの一貫した生成物が製造されていることを示している場合、バイモダール粒径分布を示すことができる。本発明では、アスペクト比が6未満である場合、粒径は、乾式分析における光走査によって、Sympatec Helos社製分析機で測定される。アスペクト比が6以上である場合、光学顕微鏡を用いて結晶の最長寸法で粒径を測定する。
【0060】
医薬配合物を製造するための、MMC法の生成物の続く後処理は典型的に、製品の性能又は市販品としての製品の承認を向上させるのに有利である。限定するものではないが、ローラー圧着、湿式造粒、直接圧縮、又は直接カプセル充填等のプロセスが全て可能である。特に、当該産業の需要を満たすような、MMC法の生成物を含む医薬組成物を製造することができ、これらの配合物は、上述のような種々のタイプの補助添加剤を含む。MMC法及び続く配合用の化合物の、見込まれるが、限定するものではない種類としては、鎮痛薬、抗炎症剤、駆虫薬、抗不整脈薬、気管支喘息治療薬、抗菌剤、抗凝血薬、抗うつ剤、抗糖尿病薬、抗てんかん薬、抗ヒスタミン剤、血圧降下薬、抗ムスカリン薬、抗ミコバクテリア薬、抗悪性腫瘍薬、免疫抑制剤、抗甲状腺薬、抗ウイルス薬、抗不安剤、鎮静剤、収れん薬、βアドレナリン作動性受容体遮断薬、造影剤、コルチコステロイド、咳止め薬、診断薬、画像診断薬、ドーパミン作動薬、止血剤、免疫学的薬剤、脂質調節剤、筋弛緩剤、副交感神経作働薬、副甲状腺カルシトニン、プロスタグランジン、放射性医薬品、性ホルモン、抗アレルギー薬、刺激剤、交感神経作働薬、甲状腺薬、血管拡張剤、及びキサンチンが挙げられる。薬剤物質としては、経皮パッチ等の他の方法を使用することも考えられるが、経口投与及び静脈内投与及び吸入投与を目的とするものが挙げられる。この薬剤物質は、任意の医薬有機活性化合物及び前駆体化合物から選択することができる。これらの種類の薬剤の記載、及び各種類における化学種のリストは、非特許文献13に見ることができ、この開示はその全体が参照により本明細書に援用される。薬剤物質は、市販されており、及び/又は当該技術分野で既知の技法で調製することができる。
【0061】
本明細書中で使用される場合、用語「結晶化」及び/又は「析出」は、流体から粒子を製造する任意の手法を含み、このような手法としては、古典的な溶媒/貧溶媒結晶化/析出、温度依存結晶化/析出、「塩析」結晶化/析出、pH依存反応、「冷却誘起(cooling driven)」結晶化/析出、化学反応及び/又は物理反応に基づく結晶化/析出等が挙げられるが、これらに限定されない。
【0062】
本明細書中で使用される場合、用語「バイオ医薬品」は、生物源に由来するか、又は生物源(例えば、タンパク質、ペプチド、ワクチン、核酸、免疫グロブリン、多糖、細胞産物、植物抽出物、動物抽出物、組換タンパク質、酵素、又はそれらの組合せ)からの産物に相当するように化学合成される任意の治療用化合物を含む。
【0063】
本明細書中で使用される場合、用語「溶媒」及び「貧溶媒」はそれぞれ、物質が実質的に溶解される流体、及び所望の物質を結晶化/析出させるか、又は溶液から溶離させる流体を意味する。
【0064】
本発明の方法及び装置は、広範な医薬物質を結晶化するのに利用することができる。本発明に従って結晶化し得る水溶性医薬物質及び水不溶性医薬物質としては、限定するものではないが、アナボリックステロイド、滋養強壮薬、鎮痛薬、麻酔薬、制酸剤、抗不整脈薬、気管支喘息治療薬、抗菌剤、抗齲蝕剤、抗凝血薬、抗コリン作動薬(anticolonergics)、抗痙攣薬(anticonvulsants)、抗うつ剤、抗糖尿病薬、止瀉薬、制吐薬、抗てんかん薬、抗真菌剤(antifungals)、駆虫薬、痔疾薬、抗ヒスタミン剤、抗ホルモン、血圧降下薬、抗低血圧薬、抗炎症剤、抗ムスカリン薬、抗ミコバクテリア薬(antimycotics)、抗悪性腫瘍薬、抗肥満薬、歯石防止薬、抗原虫薬、抗精神病薬、防腐剤、鎮痙薬(anti-spasmotics)、抗血栓薬(anti-thrombics)、鎮咳薬、抗ウイルス薬、抗不安薬、収斂剤(astringensts)、βアドレナリン作動性受容体遮断薬、胆汁酸、ブレスフレッシュナー、気管支鎮痙薬(bronchospasmolytic drugs)、気管支拡張剤(bronchodilators)、カルシウムチャンネル遮断薬、強心配糖体、避妊薬、コルチコステロイド、鬱血除去薬、診断薬、消化薬、利尿薬、ドーパミン作動薬、電解質、催吐薬、去痰薬、止血剤、ホルモン、ホルモン置換療法薬、催眠剤、血糖降下薬、免疫抑制剤、性的不全治療薬、緩下剤、脂質調節剤、粘液溶解薬、筋弛緩剤、非ステロイド系抗炎症剤、栄養補助剤、鎮痛剤(pain relievers)、副交感神経抑制剤(parasympathicolytics)、副交感神経作働薬、プロスタグランジン(prostagladins)、精神刺激剤、向精神薬、鎮静剤、性ステロイド、鎮痙薬、ステロイド、刺激剤、スルホンアミド、交感神経遮断(sympathicolytics)、交感神経様作用薬(sumpathicomimetics)、交感神経作働薬、甲状腺作働薬(thyreomimetics)、抗甲状腺薬(thyreostatic drugs)、血管拡張剤、ビタミン、キサンチン、並びにそれらの混合物が挙げられる。
【0065】
本発明による医薬組成物は、本明細書に記載の粒子及び薬学的に許容可能な担体を含む。好適な薬学的に許容可能な担体は、当業者にとって既知のものである。これらとしては、非経口注射用、固体形態又は液体形態の経口投与用、及び直腸投与用等の無毒性の生理学的に許容可能な担体、アジュバント又は賦形剤が挙げられる。本発明の医薬組成物は、限定するものではないが静脈内投与を含む、経口投与及び非経口投与の用途に有用である。
【0066】
以下の実施例は、本発明のMMC法を実施する方法の限定的ではない記載を挙げている。
【0067】
以下の実施例において、微細な種の粒子は、600mlディスクミル及びDYNO(登録商標)ミルから成るKDLモデルの2つのミルのうちの1つによって作製した。このミルチャンバにはクロム処理を行い、攪拌ディスクはイットリウム安定化酸化ジルコニウムのものとした。ミルには、約1900グラムの均一直径のイットリウム安定化酸化ジルコニウムラウンドビーズを充填した。160ml攪拌Mini−Cerミルはセラミックチャンバ及びセラミック攪拌機を備えており、Netzsch Incによって製造されたものとした。ミルには、約500グラムの様々なサイズの均一直径のイットリウム安定化酸化ジルコニウムビーズを充填した。これらのミルのビーズは、Norstone(登録商標)Inc.(Wyncote, Pennsylvania)によって供給されたものとした。それらは、高度に研磨されており、且つTOSOH USA, Inc.によってオリジナルで製造されている。
【0068】
粒子の表面積は、特に指定のない限り、GEMINI 2360(Micromeritics(登録商標) Instrument Corporation Inc.(Norcross, Georgia)製)によるBET多点解析を用いて分析した。
【0069】
光学顕微鏡によって粒子の顕微鏡写真を撮影した。顕微鏡写真は、特に断りのない限り結晶化の終了時点での結晶化スラリーのものである。
【0070】
乾燥ケーキの粒径分布は、特に断りのない限り、HELOS OASIS(SYMPATEC Gbh(http://www.sympatec.com/))機内でレーザ光回折を用いて分析した。この同一の機械には、粉砕材料のスラリー又は結晶化による生成物スラリーを分析することができるスラリーセルも備えられていた。Isopar G(登録商標)担体流体へのレシチンの添加及び超音波破砕の適用を含む、分析のための標準的な技法を用いた。
【実施例】
【0071】
[実施例1]
化合物A=Cox II阻害剤
この一連のセミバッチ式結晶化は、媒体粉砕によって表面積が広い微細な種を形成することができること、及び結晶化の際に導入される微細な種の量を変えて可変の表面積及び粒径の最終生成物を製造する作用を示す。最終生成物の表面積はジェット粉砕した物質に匹敵し得る。また、粉砕後であるが結晶化プロセスの前の、微細な種への補助添加剤の添加により、得られる生成物の表面積を増大させることができることを示す実験も示す。貧溶媒を添加して結晶化させた。
【0072】
化合物Aのジェット粉砕
Hosakawa Micron,Inc.の100AFGジェットミルの場合に、1mm〜1.9mm範囲の典型的な条件のノズル、43psig〜45psigのジェット圧及び7000rpm〜21000rpmを用いて、化合物Aをジェット粉砕した。物質の得られた表面積は2.5m2/gであった。
【0073】
実施例1A〜実施例1Eの微細な種の粉砕
0日目、1mmのイットリウム安定化酸化ジルコニウムビーズを収容しているディスクミルを50%のn−ヘプタン及び50%のトルエンで洗浄し、ミルの内容物を容積式ポンプを介した空気による処理のために移した。ミルに接続されている容器に、60グラムの化合物A及び1066グラムの50:50のトルエン:ヘプタン(重量に基づく)を充填した。混合物を、ミル収容タンク内で25℃の温度で攪拌した。次いで、混合物を900ml/分の速度で60分間ミル内を再循環させた。この時間中、ミルは6.8m/秒の先端速度で作動した。タンク内スラリーを20分、40分及び60分の時点で採取して顕微鏡によって粉砕プロセスを確認した。60分後、スラリーを、表1及び2の結晶化の実験での後の使用のためにガラスジャーに詰めた。ジャーの微細な種のスラリーを焼結ガラス漏斗で濾過し、フィルタケーキを真空オーブン内で60℃で乾燥することによって、溶液中に溶解していない微細な種の濃度を求めた。この値は、種の充填に基づいて記録した。乾燥後のフィルタケーキの表面積を標準BET等温線によって測定したところ、3.4m2/gであることが分かった。
【0074】
結晶化1A及び結晶化1B
一連のバッチ式貧溶媒結晶化を以下の:
1)化合物Aをトルエン及びヘプタン中に室温で溶解して、表1に概説するように視覚的に透明な溶液を得ること(「初期」充填)、
2)微細な種の存在により結晶化を開始した粉砕工程からの指定量の微細な種のスラリー、及び微細な種のスラリーを添加した追加の貧溶媒を添加すること、
3)この貧溶媒を用いてn−ヘプタンを一分量ずつ添加して結晶化すること(なお、充填は添加と添加との間に少なくとも30分、間をおいて4時間〜12時間かけて行われた)、及び
4)得られたスラリーを濾過して限定量のヘプタン(約2ケーキ体積〜10ケーキ体積)で洗浄してから60℃で乾燥させて、表面積の分析(後処理)に適した乾燥ケーキを得ること、
によって行った。
【0075】
手順及び生成量は表1に記載する。
【0076】
【表1】
【0077】
実施例1Bに対応する顕微鏡写真を示す図10を参照されたい。スケールバーは10μmを示す。
【0078】
結晶化1C、結晶化1D及び結晶化1E
実施例1A及び実施例1Bの基本手順に続いて第2の一連のバッチを導入し、ここでは貧溶媒を12時間にわたって連続添加した(実施例1C〜実施例1E)。実施例1Dでは、イオン性界面活性剤レシチンオイル(食品グレード)を、バッチへの添加前に媒体ミルからの微細な種のスラリーに添加した。実施例1Eでは、非イオン性界面活性剤Triton X−100(登録商標)(Sigma Aldrich)を、バッチへの添加前に媒体ミルからの微細な種のスラリーに添加した。非イオン性又はイオン性の界面活性剤の添加により、表2に記載のそれらの結晶化から得られた生成物の表面積が増大した。
【0079】
【表2】
【0080】
[実施例2]
化合物A=Cox II阻害剤
この一連の実施例は、非イオン性又はイオン性の界面活性剤等の補助添加剤を微細な種の湿式粉砕プロセスに添加するとスラリーの物理的な取扱い性を高めることができることを示す。結晶化プロセスで使用するために粉砕後に補助添加剤を微細な種のスラリーに添加した結果、上記の実施例1D及び実施例1Eに示すのと同様に生成物表面積が増大した。さらに、異なる表面積の結晶化後に物質を得る必要に応じて粉砕時間を変えることができることを示すために、スラリーの試料を15分及び60分の時点で採取した。ここでも同様に、表面積はジェット粉砕した物質の表面積に匹敵し、しかも本発明のプロセスによって直接生成された。
【0081】
実施例2A及び実施例2Bの微細な種の粉砕
0日目、1mmのイットリウム安定化酸化ジルコニウムビーズを収容しているディスクミルを50%のn−ヘプタン及び50%のトルエンで洗浄し、ミルの内容物を容積式ポンプからの空気による処理のために移した。60グラムの化合物A及び1083グラムの50:50のトルエン:ヘプタン(重量に基づく)を、ミルに接続されている容器に充填した。総計10グラムのTriton X−100も添加した。混合物を、ミル収容タンク内で21℃の温度で攪拌し、次いで、混合物を900ml/分の速度で60分間ミル内を再循環させた。この時間中、ミルは6.8m/秒の先端速度で作動した。タンク内スラリーのうちの少量を15分、30分及び45分の時点で採取して顕微鏡によって粉砕プロセスを確認した。粉砕から60分後、スラリーを後の使用のためにガラスジャーに詰めた。ジャーの微細な種のスラリーの一部を0.2μmフィルタ漏斗で濾過して、溶液中に溶解していない微細な種の濃度を求めた。フィルタケーキを限定量の貧溶媒ヘプタンで洗浄し、真空オーブン内で60℃で乾燥させた。固体としての微細な種のスラリーの濃度は4.1重量%であった。この濃度は、粉砕プロセスの際に非イオン性界面活性剤を用いなかった実施例1の対応する微細な種のスラリーよりも約30%高かった。この違いは、粉砕システムにおける物理的な損失の低減に貢献し得る。乾燥後のフィルタケーキの表面積を標準BET等温線によって測定したところ、3.9m2/gであることが分かった。
【0082】
実施例2C及び実施例2Dの微細な種の粉砕
0日目、1mmのイットリウム安定化酸化ジルコニウムビーズを収容しているディスクミルを50%のn−ヘプタン及び50%のトルエンで洗浄し、ミルの内容物を容積式ポンプからの空気による処理のために移した。60グラムの化合物A及び1074グラムの50:50のトルエン:ヘプタン(重量に基づく)を、ミルに接続されている容器に充填した。総計125グラムのレシチンオイルも添加した。混合物を、ミル収容タンク内で20℃の温度で攪拌した。次いで、混合物を900ml/分の速度で60分間ミル内を再循環させた。ミルの出口温度は21℃であった。この時間中、ミルは6.8m/秒の先端速度で作動した。タンク内スラリーのうちの少量を15分、30分及び45分の時点で採取して顕微鏡によって粉砕プロセスを確認した。粉砕から60分後、スラリーを後の使用のためにガラスジャーに詰めた。ジャーの微細な種のスラリーの一部を0.2μmフィルタ漏斗で濾過し、溶液中に溶解していない微細な種の濃度を求めた。フィルタケーキを限定量の貧溶媒ヘプタンで洗浄し、真空オーブン内で60℃で乾燥させた。固体としての微細な種のスラリーの濃度は4.8重量%であった。この濃度は、粉砕プロセスの際にイオン性界面活性剤を用いなかった実施例1の対応する微細な種のスラリーよりも約50%高かった。この違いは、粉砕システムにおける物理的な損失の低減に貢献し得る。乾燥後のフィルタケーキの表面積を標準BET等温線によって測定したところ、5.3m2/gであることが分かった。
【0083】
結晶化2A、結晶化2B、結晶化2C及び結晶化2D
一連のバッチ式貧溶媒結晶化を以下:
1)化合物Aをトルエン及びヘプタン中に溶解すること(これにより、視覚的に透明な溶液が得られた(表3中の「初期」充填))、
2)さらに多くの非イオン性又はイオン性の界面活性剤を微細な種に添加した後で、表3に示すように指定量の微細な種のスラリーを添加すること、
3)n−ヘプタンを連続的に添加して結晶化すること、及び
4)得られたスラリーを濾過して2ケーキ体積〜10ケーキ体積のヘプタンで洗浄してから60℃で乾燥させて、表面積の分析(後処理)のために乾燥ケーキを得ること、
によって行った。
【0084】
手順及び生成量は表3に記載する。
【0085】
【表3】
【0086】
[実施例3]
化合物B=Cox II阻害剤
この一連の実施例は、「メルトバック」を示すことが分かっている化合物に対するピン粉砕の代わりとなることができることを示す。結晶の形態は、化合物Bの4つの他の可能な結晶形態が知られていてもプロセス全体を通して制御される。結晶化は高温で行われた。この例は、表面積が異なるレベルの微細な種の添加によって制御されることができることを示す。
【0087】
化合物Bのピン粉砕
化合物Bを、Alpine(登録商標)UPZ160ミル(Hosakawa)の一般的な条件及び高プロセス窒素流を用いて薬学的使用のためにピン粉砕した。この化合物はその低融点に起因して粉砕することが困難である。処理温度をこの化合物の融点未満に保つように、処理の間、0℃の低温窒素を40SCFM(標準立方フィート毎分)でミルのピンリンス(pin-rinse)として施した。粉砕は特別な工程なしには可能ではなかった。得られた物質の表面積は0.9m2/gであった。
【0088】
実施例3A及び実施例3Bの微細な種の粉砕
0日目、1mmのイットリウム安定化酸化ジルコニウムビーズを収容しているディスクミルを50%のn−ヘプタン及び50%のトルエンで洗浄し、ミルの内容物を容積式ポンプからの空気による処理のために移した。60グラムの化合物B及び1066グラムの50:50のトルエン:ヘプタン(重量に基づく)を、ミルに接続されている容器に充填した。混合物をミル収容タンク内で25℃の温度で攪拌し、次いで、混合物を900ml/分の速度で60分間ミル内を再循環させた。この時間中、ミルは6.8m/秒の先端速度で作動した。ミルの出口温度は25℃であった。タンク内スラリーのうちの少量を15分、30分及び45分の時点で採取して顕微鏡によって粉砕プロセスを確認した。粉砕から総計60分後、スラリーを後の使用のためにガラスジャーに詰めた。微細な種のスラリーの1つのジャーから、122.8gをフィルタ漏斗で濾過し、フィルタケーキを限定量の貧溶媒ヘプタンで洗浄した。総計9.7グラムの湿潤ケーキを回収した。次いで、これを真空オーブン内で60℃で乾燥させた。乾燥後のフィルタケーキの表面積を標準BET等温線によって測定したところ、5.7m2/gであることが分かった。
【0089】
結晶化3A及び結晶化3B
一連のバッチ式貧溶媒結晶化を以下:
1)化合物Bを50ml攪拌容器内でトルエン及びヘプタン中に50℃で溶解し、表4に「初期」充填として示すように視覚的に透明な溶液が得られること、
2)粉砕工程からの指定量の微細な種のスラリーを添加すること(微細な種及び微細な種のスラリーを添加した追加の貧溶媒の存在による結晶化を開始した)、
3)n−ヘプタンを連続的に添加して結晶化すること、及び
4)得られたスラリーを室温で濾過し、2ケーキ体積〜10ケーキ体積のヘプタンで洗浄してから60℃で乾燥させて、表面積の分析のために乾燥ケーキを得ること、
によって行った。
【0090】
手順及び生成量は表4に記載する。
【0091】
【表4】
【0092】
図11は、再循環粉砕から0.5分後の実施例3Bの微細粉砕スラリーの顕微鏡写真である。図12は、再循環粉砕から15分後の実施例3Bの微細粉砕スラリーの顕微鏡写真である。図13は、再循環粉砕から60分後の実施例3Bの微細粉砕スラリーの顕微鏡写真である。図14は、実施例3Bの結晶化後の最終生成物に相当する顕微鏡写真を示す。スケールバーは10μmを表す。
【0093】
[実施例4]
化合物C=BK1拮抗剤
一連の実施例は、本発明の方法を用いて複数の薬剤類に対応することができることを示す。また、最終生成物の表面積は異なるサイズの微細な種を用いることによって制御されることができることも示す。微細な種のサイズを異なる粉砕時間量を用いて変えることができる。この実施例での粉砕工程によって生成される種粒子はサイズが1μmを超える。化合物Cは低融点を有するため、乾燥粉砕の際に「メルトバック」を回避するのにMMC法が有用である。低温窒素をピンミルのピンリンスとして施して有意な量の物質を粉砕することができる。
【0094】
実施例4A及び実施例4Bの微細な種の粉砕
0日目、1mmのイットリウム安定化酸化ジルコニウムビーズを収容しているディスクミルを50重量%のn−ヘプタン及び50重量%のトルエンで洗浄し、ミルの内容物を容積式ポンプからの空気による処理のために移した。60グラムの化合物C及び1066グラムの50:50のトルエン:ヘプタン(重量に基づく)を、ミルに接続されている容器に充填した。混合物をミル収容タンク内で19℃の温度で攪拌し、次いで、混合物を900ml/分の速度で60分間ミル内を再循環させた。この時間中、ミルは6.8m/秒の先端速度で作動した。ミルの出口温度は20℃であった。タンク内スラリーのうちの少量を0分、15分、30分及び45分の時点で採取して顕微鏡によって粉砕プロセスを確認した。粉砕から総計60分後、スラリーを後の使用のためにガラスジャーに詰めた。スラリーの試料を、レシチン、及びISOPAR G(登録商標)での120秒の超音波破砕を用いて、SYMPATEC(登録商標)光拡散湿式細胞分析機で分析した。図24及び図25は、微細な種の粒径分布を示す。15分間粉砕した微細な種の場合、平均体積粒径は3.9μmであり、粒子の95体積%は9.8μm未満であった。60分間粉砕した微細な種の場合、平均体積粒径は2.35μmであり、粒子の95体積%は5.2μm未満であり、より長く粉砕した微細な種を用いるとより変化の急な粒径分布を示す。上記のいくつかの実施例におけるように、粉砕後15分〜60分の微細な種のスラリーの一部を濾過し、ヘプタンで洗浄し、60℃で乾燥させた。乾燥後、フィルタケーキの表面積を標準BET等温線によって測定したところ、15分の粉砕の場合は4.6m2/gであり、60分の粉砕の場合は6.6m2/gであることが分かった。このデータは、微細な種のサイズ及び表面積がプロセスパラメータによって制御されることができることを示す。
【0095】
結晶化4A及び結晶化4B
2つのバッチ式貧溶媒結晶化を以下の:
1)化合物Cを、オーバヘッド攪拌機によって攪拌した75ml容器内でトルエン及びヘプタン中に43℃で溶解すること(これにより、視覚的に透明な溶液が得られた(「初期」充填))、
2)in−situでの光後方錯乱によって視認される固体を形成することなく、スラリーを40℃に冷却して過飽和溶液を生成すること、
3)粉砕工程からの指定量の微細な種のスラリーを添加すること、
4)n−ヘプタンを連続的に添加して結晶化すること、及び
5)得られたスラリーを室温で濾過し、2ケーキ体積〜10ケーキ体積のヘプタンで洗浄してから60℃で乾燥させて、表面積の分析のために乾燥ケーキを得ること、
によって行った。
【0096】
手順及び生成量は表5に記載する。
【0097】
【表5】
【0098】
図15は実施例4Bの最終生成物の顕微鏡写真を示す。
【0099】
[実施例5]
化合物D=ビスホスホネート
本実施例は、従来の結晶化とその後に続く乾燥ケーキのピン粉砕によって得られる粒径をMMC法によって繰り返すことができることを示す。本実施例はまた、温度クールダウン結晶化(temperature cooldown crystallization)及び別の薬剤類を示す。異なるサイズの媒体ビーズを用い、プロセスは水系とした。
【0100】
従来の手法
化合物Dを60℃で100g/lの水中に溶解した。この化合物を0℃に冷却し、同時に200g/lに蒸留して結晶化生成物を得た。物質を濾過し、乾燥させ、一般的なピン粉砕条件を用いてピン粉砕した。この生成物のピン粉砕は特に困難である。ミルの機能は、40kgの物質を処理する毎に、ミルを停止してピンを洗浄した場合にのみ維持した。このプロセスにより、顕微鏡写真によって視覚的に分析されたように5μm〜40μmの生成物が生じた。
【0101】
実施例5の微細な種の粉砕
0日目、ディスクミルに1890gの1.5mmのイットリウム安定化酸化ジルコニウムビーズを充填し、脱イオン水で洗浄した。ミルの内容物を容積式ポンプからの空気による処理のために移した。34グラムの化合物D及び207グラムの脱イオン水(水重量に基づく)を、ミルに接続されている容器に充填した。混合物を、ミル収容タンク内で攪拌し、その上、630ml/分の速度で10分間ミル内を再循環させた。この時間中、ミルは6.8m/秒の先端速度で作動した。ミルの出口温度は20℃であった。タンク内スラリーのうちの少量を0分及び5分の時点で採取して顕微鏡によって粉砕プロセスを確認した。粉砕から10分後、スラリーを後の使用のためにガラスジャーに詰めた。微細な種の顕微鏡写真は1.5mmビーズの場合では1.0mmビーズの場合での実験よりも大きなサイズを示した。
【0102】
結晶化5
0日目、温度クールダウン結晶化を、オーバヘッド攪拌機によって攪拌した75ml容器内の95gの水中に14.0gの化合物Dを溶解することによって行い、これにより、視覚的に透明な溶液を得た。容器を囲んでいるジャケットの温度をこの溶解のために66℃に保持した。スラリーを、ジャケットを64℃にすることによって冷却して、固形物を形成することなく過飽和溶液を生成した。過飽和は視認され、in−situでの光後方錯乱によって確認した。粉砕工程から総計4.0グラムのスラリーの微細な種を添加し、ジャケット温度を61℃に変えた。次いで、ジャケットを61℃〜48℃に4時間かけて冷却し、48℃から20℃に7時間かけて冷却した。視覚的な粒径分析のために微細な種のスラリーの顕微鏡写真を分析した。平均長は17μmであり、平均幅は8μmであった。このサイズは、医薬用途に必要とされるサイズに近似している。図16は、実施例5の最終生成物の顕微鏡写真である。
【0103】
[実施例6]
化合物F=セロトニン拮抗薬
この一連の実施例は、MMC法が、イヌの血漿レベルによって測定された場合に、AFGジェットミルによって生成された生成物のバイオアベイラビリティに対処することができることを示す。この一連の実施例は、より小さな粒径(より広い表面積)を有する生成物を促すために、結晶化容器内に設置される補助エネルギー装置(この場合では超音波破砕機)の利用をさらに示す。実施例6は、粉砕プロセスにおけるビーズがより小さいことにより、微細な種の同じ充填を用いた場合により広い表面積の微細な種及びより広い表面積の生成物がもたらされることを示す。この実施例は、より高いレベル、ここでは20%の種の使用により、生成物の表面積を増大させることができることを示す。本実施例は、混合した水性有機溶媒を用いた半連続式プロセスである。化合物Fはいくつかの多形体を有することが知られており、本発明によるプロセスは所望の多形体を生成した。このことは、医薬品処理に対するMMC法の実現可能性を示す。
【0104】
AFG粉砕
物質を1mmノズル、50psigジェット圧、9000rpm〜18000rpmで100AFGにより粉砕したところ、表面積が0.6m2/gであった。
【0105】
実施例6の微細な種#1の粉砕
0日目、1890グラムの1.5mmのイットリウム安定化酸化ジルコニウムビーズを収容しているディスクミルを、60体積%のイソプロパノール(IPA)及び40体積%の脱イオン水で洗浄した。ミルの内容物を容積式ポンプからの空気による処理のために移した。ミルに接続された容器に、18.5グラムの化合物F及び220グラムの60/40のIPA/水を充填した。この混合物を、ミル収容タンク内で攪拌し、その上、600ml/分〜900ml/分の速度で15分間ミル内を再循環させた。この時間中、ミルは6.8m/秒の先端速度で作動し、ミルの出口温度は30℃未満であった。タンク内スラリーのうちの少量を0分、5分及び10分の時点で採取して顕微鏡によって粉砕プロセスを確認した。粉砕から15分後、スラリーを後の使用のためにガラスジャーに詰めた。
【0106】
実施例6の微細な種#2の粉砕
上記の粉砕#1の手順を繰り返した。ただし媒体として1894グラムの1.0mmのイットリウム安定化酸化ジルコニウムビーズを使用した。
【0107】
半連続式結晶化
半連続式結晶化を、指定の充填時間、微細な種のスラリー濃縮液及び貧溶媒の同時添加によって達成した。濃縮液の添加の間、溶媒比を維持した。充填を、容器の対向する両側で攪拌機付近の液体−ガス表面の下で22ゲージ針を介して行った。攪拌のために75ml容器にはオーバヘッド攪拌機を用い、及び8mm超音波破砕プローブを液体−ガス面の下に設置した。表6に示すように、結晶化の間、超音波破砕プローブを約10ワットの電力で作動させた。媒体粉砕した種#2を用いる実験の場合、濃縮液を充填したときと同じ速度でバッチ式の濃縮液添加の終了時点に追加の水を添加して、溶媒比を4:3から1:2のIPA:水に変えた。このことは、母液損失を下げることによって収率を約5%高めるために行われ、粒径に著しく影響を及ぼさなかった。後処理は、減圧による室温でのスラリーの濾過、及び空気による乾燥又は40℃の真空オーブン内での乾燥を含んだ。
【0108】
表6の実施例6Cの収率を85%になるように定量化した。この実験をX線回折によって示したところ、所望のヘミ水和物の形態がもたらされた。
【0109】
【表6】
【0110】
後配合(Post Formulation)及び使用
実施例6Cの固形生成物とAFG粉砕試料を並行して(side by side study)配合し、従来の医薬品成分を用いて直接充填カプセルとした。MMCの実施例6Cのイヌについての曲線下面積(AUC(24時間))を、AFGで粉砕した物質と比較したところ、等しいバイオパフォーマンスを得たことを示した。その結果を図26に挙げる。
【0111】
[実施例7]
化合物G=DP IV阻害剤
本実施例は、大きな粒子(50μmよりも大きい)を本発明のMMC法によって一貫して作製することができることを示す。粒径は種々の異なる種の充填(loads)を用いて目的に合わせることができる。
【0112】
媒体粉砕
0日目、KDL媒体ミルを80/20のIPA/水で洗浄し、空気を送って乾燥させた。80/20(重量に基づく)のIPA/水中、100mg/gでの化合物Gのスラリーを再循環モードで、300ml/分の速度で120分間ミル内に給送した。得られた微細な種の粒径は、光回析によって測定された場合に4.7μmの平均サイズを有した。
【0113】
結晶化
一連の結晶化を、実施例7の媒体粉砕した微細な種を用いて行った。これらの結晶化において、種の量は様々であった。70/30(重量に基づく)のIPA/水中、220mg/gでの化合物Gのバッチを70℃より高く加熱して固形物を溶解した。視覚的に透明な液体が得られた。バッチを65℃〜67℃に冷却して過飽和を生じさせた。バッチを、表7に示すような微細な種のレベルで播種した(種のスラリーに添加した乾燥生成物のグラム対バッチ内での種のスラリーのグラム)。バッチを3時間エージングし、5時間かけて室温に冷却した。イソプロピルアルコール貧溶媒を15分〜30分の期間をかけて充填して、80/20(重量に基づく)のIPA/水に達した。バッチを1時間エージングし、減圧濾過し、45℃のオーブン内で減圧乾燥させた。粒径を、湿潤状態で約30ワットで30秒超音波破砕を用いて粒度分布測定(Microtrac)粒径光回析を介して分析した。以下の結果を得た。
【0114】
【表7】
【0115】
[実施例8]
化合物D=ビスホスホネート
本実施例は、MMC法のスケールアップと、スケールアップ時の容器の混合特性を高める再循環ループの利用とを示す。本実施例は、再循環ループ内に設置されるより高い強度のエネルギー装置(ここではスタティックミキサー)により、最終生成物の表面積を増大させることができることをさらに示す。この一連の例は、ピン粉砕した生成物に匹敵するプロファイルを示す。
【0116】
ピン粉砕
化合物Dを結晶化した。この生成物をピン粉砕し、得られた粒径を光回析によって測定したところ、18.7μmであり、95%が50μm未満であった。表面積は0.53m2/gであった。
【0117】
実施例8の微細な種の粉砕
一連の媒体粉砕の実験を行って結晶化のために微細な種を供給した。0日目、ディスクミルに1.5mmのイットリウム安定化酸化ジルコニウムビーズを充填し、次いで脱イオン水で洗浄した。ミルの内容物を容積式ポンプからの空気による処理のために移した。1リットル当たり100グラムの脱イオン水濃度に等しいスラリーを、ミルに接続された容器に充填した。この混合物を、ミル収容タンク内で攪拌し、その上、900ml/分の速度でミル内を再循環させた。この時間中、ミルは6.8m/秒の先端速度で作動し、ミルの出口温度は25℃であった。粉砕後、スラリーを次の使用のためにガラスジャーに詰めた。
【0118】
結晶化8
一連の温度クールダウン結晶化を、オーバヘッド攪拌機を用いて攪拌した容器内の2500gの脱イオン水中で化合物Dを250g溶解することによって行った。容器を囲んでいるジャケットの温度を上げ、バッチ温度を60℃〜62℃に上げて、バッチを視覚的に透明な溶液に溶解した。スラリーを52℃に冷却して、視認されるほどの固形物を形成することなく過飽和溶液を生成した。総計115ミリリットルの微細な種のスラリーを、反応器の上部を介して容器に添加し、52℃〜53℃で30分間エージングした。バッチを5℃に冷却し、少なくとも1時間エージングし、次いで、真空フィルタを用いて濾過冷却し、45℃で減圧乾燥させた。
【0119】
最終溶媒組成にある母液中の生成物の濃度に基づき、少なくとも80%の収率がこの一連の実施例について予測される。粒子の表面積をBET等温線及び光回析によって分析した。実験8Aの粒子は強固に凝集し、光回析機の測定能力の限界を上回った。図4に示すような再循環ループの追加により生成物の表面積が増大した。再循環ループ内のより高エネルギーな装置であるスタティックミキサーの追加により、乾燥生成物をピン粉砕することによってもたらされる表面積に匹敵する、より広い表面積がもたらされた。
【0120】
【表8】
【0121】
実施例8Aの結果は、MMC法をスケールアップするのに選択される装置が生成物の結果を変えることができることを示す。混合に役立たせるために再循環ループを容器に追加することは、本発明の一実施形態である。さらに、実施例8Cは、補助エネルギー装置を追加することにより、再循環ループ内により高いエネルギーをもたらし、それによって表面積が増大した生成物をもたらすことができることを示す。実施例8Cの表面積は、ピン粉砕によってもたらされる表面積に合致する。再循環ループ又は補助エネルギー装置なしでもたらされた結晶化により、図17及び図18に示すように比較的低い表面積及び大きな粒径の目に見える程度に凝集した物質がもたらされる。
【0122】
[実施例9]
化合物E=脂質低下化合物
本実施例は、貧溶媒を用いる半連続式結晶化を示し、ここでは貧溶媒及び濃縮液の複数の充填回数に対処することができる。生成物の表面積を増大させるのに超音波破砕が有用であることが示されている。ここでは、より小さな0.8mmのビーズを用いて、或る範囲のビーズサイズを本発明の方法に従って用いることができることを示した。
【0123】
従来の乾燥粉砕手法
化合物Eをジェット粉砕した。得られた表面積の詳細は、生成物当たり1.4m2/g〜2.9m2/gであった。
【0124】
実施例9の微細な種の粉砕
0日目、ディスクミルに乾燥状態の0.8mmのイットリウム安定化酸化ジルコニウムビーズを充填した。ミルに接続された容器に、1000mlの60/40のMeOH/水を充填し、次いで、生成物の性能のための補助添加剤として0.2グラムのブチル化ヒドロキシアニソール(butylated hydroxy anisole)(BHA)を充填した。混合物を、ミル収容タンク内で攪拌し、その上、900ml/分の速度で30分間ミル内を再循環させた。この時間中、ミルは6.8m/秒の先端速度で作動し、ミルの出口温度は21℃であった。タンク内スラリーのうちの少量を0分及び30分の時点で採取して、顕微鏡によって粉砕プロセスを確認した。粉砕から総計30分後、スラリーを後の使用のためにガラスジャーに詰めた。微細な種の平均サイズは約2μmであることが示された。
【0125】
結晶化9A、結晶化9B、結晶化9C及び結晶化9D
半連続式貧溶媒結晶化を以下の:
1)1リットルのメタノール中に60gの化合物Eを溶解することによって濃縮液を生成すること(なお、生成物の酸化を防止するために、この流れに総計0.2gのブチルヒドロキシアニソールを添加した)、
2)粉砕からの5mlの微細な種のスラリーを充填すると共に5mlの60/40のメタノール/水(体積に基づく)を添加することによって、微細な種床を形成すること(なお、22mm径ブレードを用いて600rpmで100ml攪拌容器に充填を行った)、
3)56ミリリットルの濃縮液及び36ミリリットルの脱イオン水貧溶媒を、別個のシリンジポンプを介して容器に同時充填すること、
4)バッチを室温で1時間エージングすること(なお、約10ワットの電力での超音波破砕を、8mmプローブ(Telesonics社製のDG30)を使用して濃縮液添加の際に且つ1時間のエージング期間の際に結晶化装置に直接適用した)、及び
5)得られたスラリーを室温で濾過してから45℃で減圧乾燥して、表面積の分析のために乾燥ケーキを得ること(なお、粒径を乾燥固形物の光回析によって測定した)、
によって行った。
【0126】
最終溶媒組成にある母液中の生成物の濃度に基づき、この実施例のセットについて少なくとも80%の収率が予測される。実験は同一の反応器システムを使用して行った。
【0127】
手順及び生成量は表9に記載する。
【0128】
【表9】
【0129】
実施例9A及び実施例9Bの生成物の顕微鏡写真をそれぞれ図19及び図20に示す。生成物は個々の結晶の長さ以外は同様である。図19は図21と比較することができ、ここでは、いかなる核形成も抑制するためにより低い超音波破砕出力(sonication power)及びより長い添加時間を用いてプロセスをスケールアップした。
【0130】
[実施例10]
化合物E=脂質低下化合物
本実施例は、本発明の方法が特殊な化学製品用の市販製品の体積レベルに対するスケールアップに適応可能であったことを示す。ここでは、15kgスケールの生成物が半連続式バッチ法を用いて1つのバッチで生成される。首尾よいスケールアップをもたらした再循環ループのより大きなスケールのエミュレーションが記載されている。再循環速度は18分のバッチの循環時間に対応し、実際の速度は大スケールな製造プロセスに対応する。超音波破砕出力密度は1つのバッチ当たり約0.7W/kgであり、実際のレベルは大スケールな製造プロセスに対応する。結晶化生成物を従来の製造機器を用いて後処理した。多くの医薬品の場合のように、生成物は酸素感応性であり、全ての流れは窒素流又は減圧印加を用いてガスを抜いた。補助添加剤であるブチルヒドロキシアニソール(BHA)を生成物の安定剤として使用した。
【0131】
実施例10の微細な種の粉砕
総計1.49kgの未粉砕のままの化合物E、9.3kgの脱イオン水、14kgのメタノール及び8.14gのBHAを、攪拌機を備えたジャケット付き30リットルガラス容器に充填して、容器の内容物をブレンドした。このスラリーに窒素を充填して溶液のガスを抜き、窒素スイープを粉砕プロセス全体を通して用いて、系を不活性に保持した。大量の固形物を充填すると、物質は湿潤中に凝集(clumping)を示した。物質を解凝集(declump)するために、3/8インチID(内径)再循環ラインを、ロータステータミル(粗歯付きのIKA(登録商標)Works T−50)を収容している容器に接続した。バッチを湿式ミル内を30分間再循環させて、大きな塊の固形物を破砕した。IKA Worksミルをポンプとして用いて、この工程中少なくとも2倍のバッチ量を再循環させた。この再循環工程により生成物の粒径が著しく縮小した。
【0132】
バッチを粉砕して微細な種にするために、第2の再循環ラインを図1におけるように構成した。ポンプは蠕動性のMasterflexとし、ミルはNetzsch媒体ミル(型番号「Minicer」)とした。ミルに135mlの1mmのイットリウム安定化酸化ジルコニウムビーズ(約500グラム)を充填した。次いで、Masterflex(登録商標)容積式ポンプを用いてバッチスラリーを300ml/分の速度でMinicerミル内を再循環させた。ミルは2202rpmで作動し、これは6.8m/秒の先端速度に対応する。ミル及びバッチ容器はグリコール浴によって冷却して、粉砕プロセス全体を通してバッチスラリー温度を25℃未満の温度に維持した。バッチスラリーを総計41時間粉砕した。粉砕したスラリーを室温で一晩エージングし、次いで、次の3時間以内の使用のために媒体ミルからポリドラムに排出した。粉砕したスラリーは微細な種の流れであった。スラリーの一部を0.2μmフィルタで濾過し、40℃の真空オーブン内で乾燥させた後に分析した。スラリーの排出時点で、粉砕した固形物の表面積は4.05m2/gであり、平均体積粒径は2.1μmであり、粒子の95体積%は4.8μm未満であった。Helos分析機を使用した。
【0133】
実施例10の結晶化
再循環ループの設定:より大きなスケールの機器は図3の設定と同様であるが、ただし、インラインレーザ後方錯乱プローブ(inline laser backscattering probe)を用いて、スラリー中の粒子のコード長(chord length)をリアルタイムで測定し、第1の混合装置の前に種を充填した。100ガロン攪拌タンクの底部からの再循環ループは以下の:
1)ダイアフラムポンプと、
2)コード長監視のための集光反射測定プローブ(focused beam reflectance measurement probe)と、
3)必要に応じて種のスラリーを採取及び充填するための3/8インチ弁ポートと、
4)ドラムからの脱イオン水貧溶媒の添加のためにポンプに接続された高速混合装置と、
5)2リットルのフロースルーセルにおいて2インチ径及び22インチ長のラジアル型超音波破砕ホーンから構成されるエネルギー装置(なお、超音波破砕機はTelsonics社製であり、2000Wの発電機によって給電された)と、
6)ドラムからのバッチ式濃縮液添加のためにポンプに接続された高速混合装置と、
7)スラリーの再循環速度を測定する質量計と、
8)主要な結晶化装置に戻るパイプ(内径は13/16インチ)と、
から構成した。
【0134】
貧溶媒流:先に清浄し脱イオン水で洗浄した容器に、総計250kgの脱イオン水を充填した。脱イオン水はいくつかの真空パージ及び窒素圧パージを用いてガスを抜いた。水を50ガロンドラム内に入れ、使用するまで閉じたままにした。この流れは貧溶媒流であった。
【0135】
バッチ流:メタノールで洗浄した容器に、総計14kgの化合物Eの活性医薬品成分(API)、144kgのメタノール(先にガスを抜いてある)及び80gのBHA阻害剤を充填した。化合物Eの濃縮液を50ガロンドラム内に入れ、使用するまで閉じたままにした。これはバッチ流であった。
【0136】
微細な種のスラリーの生成:先に生成した総計36kgの60/40(体積/体積)のメタノール/水の溶液を100ガロン結晶化装置に充填した。溶液を、再循環ループを用いて約25kg/分で再循環した。超音波破砕機のラジアル型プローブを350W電力で設定し、Lasentec(登録商標)FBRMプローブを測定のために作動した。上記の実施例に記載の微細な種のスラリーを、3/8インチ種充填ポート用のT継手を介して再循環ループに充填し、種の床を20℃〜25℃で超音波破砕により15分間再循環させた。これはバッチ用の微細な種であった。
【0137】
結晶化のための充填:容器攪拌機は22インチ径であり、結晶化のために3m/秒で回転した。総計129kgの脱イオン水を、メタノールバッチ濃縮液中の168kgの化合物Eと共に微細な種に10時間かけて同時に一定の充填速度で充填した。結晶化全体を通して、350Wでの連続超音波破砕を加えながらバッチを20℃〜25℃に保持した。試料を添加から1時間後、3時間後、6時間後及び10時間後に採取して、結晶化の進み具合を確認した。同時添加を完了した後、84kgの脱イオン水を20℃〜25℃で超音波破砕により2時間かけて一定の充填速度で充填した。余分な水の貧溶媒の追加を行い、生成物の溶解度を下げることによって収率を高めた。充填は低速で行い、核形成に対する結晶の成長を促した。
【0138】
脱イオン水の充填後、バッチを20℃〜25℃で超音波破砕により1時間エージングして、結晶の完全な成長を確実にした。図21に示すように光学顕微鏡を用いて結晶スラリーの写真を収集した。図21は、制御されていない核形成が存在することに起因する小さな粒子が全くない状態で粒子が単分散していることを示す。再循環ループをオフにして、バッチを20℃〜25℃で一晩エージングした。濾過による後処理及びバッチの乾燥を続いて行った。
【0139】
実施例10の後処理
濾過及び乾燥:容器内での一晩のエージング後、バッチを室温で濾過した。1mg/g未満の化合物Eの濃度を有する総計385kgの母液を回収した。容器の壁からバッチフィルタにかけて洗浄すると共にフィルタ内の生成物を洗浄するために、先に生成した総計20kgの50/50(v/v)のメタノール/水を、スプレーボールを介して結晶化装置に充填した。総計40kgの洗浄水及び残存母液を回収した。濾過及びケーキへの少なくとも1時間の窒素圧の印加後、湿潤ケーキを全てフィルタから取り出し、トレイに載せ、減圧下で40℃で48時間、大型トレイ内で乾燥させた。この時点で、ケーキにおける残存水及びメタノールは0.5重量%しかなかった。総計14.5kgの乾燥ケーキをトレイドライヤーから取り出したところ、特に物理的な損失を考慮すると93.5%の高収率が得られることが示された。平均体積粒径は8.8μmであり、粒子の95体積%は20.3μm未満であった。この表面積は、BET窒素吸着によって測定した場合に1.7m2であった。これらの結果は、プロセスのスケールアップを示す実施例10の実験材料に匹敵した。
【0140】
図21は図19と比較することができる。結晶は同様のサイズ及び形態を有した。ここでは、単位体積当たりの超音波破砕出力は、実験室において1リットル当たり100Wから1リットル当たり1ワット未満まで低下したが、性能は許容範囲であった。したがって、超音波破砕力の実用レベルがすべてのスケールで支障なく用いられることができることを示す。
【0141】
[実施例11]
化合物D=ビスホスホネート
本実施例はクールダウンバッチ結晶化のスケールアップを示す。スケールアップの場合、乱流流速(1m/秒の平均線速度)での再循環ループ及び二重T継手のエネルギー装置を用いて結晶化の際の微細な種及び生成物の分散に役立たせることによって結晶の凝集を防止することができることも示す。本実施例は、超音波破砕することなく凝集が形成されないようにすることが可能であることをさらに示す。
【0142】
実施例11の微細な種の粉砕
手順は実施例10の手順と同様である。ただし、異なる生成物供給流と共に用いられるDYNO(登録商標)−Mill Type KDLA媒体ミルを使用した。DYNO(登録商標)−Millに495mlの1.5mmのイットリウム安定化酸化ジルコニウムビーズを充填し、脱イオン水をミル内に再循環させてビーズを湿潤した。次いで余分な水を捨てた。総計1.0kgの化合物Dを30リットル容器内の10リットルの脱イオン水に充填した。この充填は、水中の部分溶解を考慮した後の主要なバッチに対する溶液からの3重量%に相当する。スラリーをロータ/ステータミル内に15分間再循環させ、次いで一晩エージングした。次いで、スラリーを0.9l/分の速度でMasterflexポンプを介して媒体ミル内を再循環させた。ミルの先端速度は6.8m/秒に設定した。粉砕を5時間行った。スラリーをミルからドラムに排出した。スラリーの試料を0.2μmフィルタで濾過し、アセトン(約0.1g/l未満の溶解度)で洗浄して試料を乾燥しやすくした。試料を真空オーブン内で乾燥させて分析した。体積平均粒径は3.19μmであり、粒子の95%は7.8μm未満であった。プロファイルはユニモーダル(uimodal:単峰性)であった。表面積は窒素吸着により1.7m2/gであった。
【0143】
実施例11の結晶化
メカニカル設定:結晶化装置のために、上記の実施例10と同じ機器設定を用いた。エネルギー装置は図5に示したような「二重T継手」から構成した。ラインは急に直角に曲がった(sharp-right-angle-turn)3/4インチIDスチールパイプから成っていた。流れは出口で合流する。
【0144】
バッチ結晶化:総計22kgの化合物Dを220リットルの脱イオン水に充填して60℃で溶解した。100ガロンタンク内の溶解液を攪拌し、60℃に維持し、29kg/分の流速で再循環ループに再循環させた。バッチを51℃〜52℃に冷却して種の充填のために過飽和を生じさせた。再循環ライン内の平均線速度(体積流量/横断面積)はラインの大部分では1.4m/秒〜1.7m/秒であり、バッチの循環時間は9分であった。本実施例では、再循環ラインは乱流再循環ループと共にエネルギー装置として二重T継手を含んでいた。容器は4m/秒の先端速度で攪拌した。
【0145】
微細な種のスラリーを、ダイアフラムポンプ及び3/8インチ種充填ポートを介して一定の速度で4分かけて再循環ループに充填した。充填を再循環ループ内へ直接施して、種のスラリーの分散を促した。バッチを種の充填によって50℃〜52℃に冷却し、その温度で30分間エージングし、次いで、制御された線形的なクールダウンを介して10時間かけて1℃〜3℃に冷却した。スラリーの光学顕微鏡写真を図22に示すように撮影した。図22に示すように、粒子は、制御されていない核形成があることに起因する小さな粒子が全くない状態で単分散した。
【0146】
実施例11の後処理
濾過及び乾燥:クールダウン後、バッチを1℃〜3℃で一晩エージングし、次いで、ポリフィルタクロス(poly filter cloth)(Shaffer社から入手可能なKAVON(商標)ブランド909ウイーブ)を設けた、予め冷却して(1℃〜3℃)攪拌したフィルタドライヤー(Cogeim 0.25m2)内で濾過した。湿潤ケーキを3連続の65kgアセトンスラリー洗浄(溶媒充填、内容物の数分間の攪拌、その後の濾過から成る)により洗浄した。これらの洗浄を用いて、乾燥の際に固形物の凝集をもたらすのに十分高い生成物濃度の残存母液を除去した。アセトンにより洗浄した固形物を、フィルタジャケットの25℃の流体によって同じフィルタ内で十分な減圧下で乾燥させて詰めた。顕微鏡写真は、ケーキの凝集が全くなく、乾燥ケーキの体積平均粒径は20.6μmであることを示した。粒子の95体積%は、Helos乾式粒子分析機を用いて41mm未満であった。表面積はBET窒素吸着により0.40m2/gであった。これらの結果は、実施例8B及び実施例8Cのラボスケール実験に匹敵する。これは、粒子の不十分な分散を結晶化の際に利用した実施例8Aの結果とは対照的である。
[実施例12]
【0147】
化合物D=ビスホスホネート
本実施例は、動作条件の選択及び所与の生成物でのMMC用のエネルギー装置の選択における融通性を示す。本実施例は製造スケールでの動作の第3の実施例でもある。本実施例は、実施例11と同じメカニカル設定及び手順を用いているが、クールダウン時間を10時間から3時間に短縮し、循環時間を9分から18分に延ばしたことを強調しておく。これらの作用の結果、核形成の可能性がより高くなり、再循環ループ及びエネルギー装置に対する暴露の頻度がより少なくなることで、結晶化装置内に形成されるいかなる凝集物も破砕されて分散粒子となる。エネルギー装置内でのより高速の固形物堆積速度及びより低速の再循環速度は、二重T継手の代わりにより高い強度のエネルギー装置である12インチ長及び2インチ幅のTelsonic rapid probe(1lのフロースルーセル内で800Wの電力で動作する)を用いることによって相殺した。種の充填も10重量%まで高めて実施例11よりも著しく小さい生成物を得るようにした。
【0148】
種の生成:本手順は、生成物及びミル準備について実施例11の手順に従った。ここでは、3.48kgの化合物Dの純水及び33kgの脱イオン水を30l容器に充填して、0.45l/分〜0.9l/分の流速で16時間、DYNO(登録商標)−Mill Type KDLAに再循環させた。得られた生成物の粒径は2.8μmの平均体積であり、粒子の95%が6.4μm未満であった。表面積は2.0m2/gであった。
【0149】
バッチ結晶化:本手順は実施例11の手順と同等であるが、ただし、100ガロンタンク内の水中に溶解した22kgの化合物Dを15kg/分程度の流速でバッチ全体を通して再循環ループに再循環させた。バッチを約53℃〜54℃に冷却して種の充填のために過飽和を生じさせた。
【0150】
微細な種のスラリーを、ダイアフラムポンプ及び3/8インチ充填ポートを介して一定速度で8分かけて再循環ループに充填した。充填を再循環ループ内に直接施して、種のスラリーの分散を促した。バッチを種の充填によって約50℃〜52℃に冷却し、この温度で30分間エージングし、次いで、制御された線形的なクールダウンを介して約1℃〜3℃に3時間かけて冷却した。スラリーの光学顕微鏡写真を図23におけるように撮影した。図23は、制御されていない核形成があることに起因する小さな粒子が全くない状態で粒子が単分散することを示す。物質を実施例11におけるように濾過によって後処理し、洗浄し、乾燥させた。結晶化の状態及び結果を以下に示す。
【0151】
【表10】
【0152】
本出願は、2006年3月14日に出願された米国仮特許出願第60/782,169号(その全体が参照により本明細書に援用される)の優先的な利益を主張する。
【図面の簡単な説明】
【0153】
【図1】再循環モードにある媒体粉砕に必要な典型的な構成部品を示し、当該構成部品はブレンド容器と、流体ポンプと、媒体ミルと、ブレンド容器に戻る再循環ラインとを含む。一回通し式粉砕(single pass milling)は再循環せず、生成物をミルから回収レシーバ(collection receiver)に単に給送するだけである。一回通しモードでは、ポンプの代わりに蒸留缶からの圧力伝達を用いることができる。一回通しを複数回行うことで同様の生成物プロファイルを再循環モードとして達成することができる。
【図2】実施例1〜実施例7、及び実施例9用に設定される結晶化容器を示す。実施例1では、針付きシリンジを用いて、貧溶媒を10秒未満で1分量ずつ高速充填した。任意選択的に、超音波破砕機プローブ及び/又は光拡散プローブを加えることができる。
【図3】実施例10、実施例11及び実施例12におけるように、微細粉砕及び結晶化プロセスのスケールアップに適応可能に示されている設定の一例を示す。結晶化容器と再循環ループの構成部品とが示されている。
【図4】実施例8に記載のプロセスを示し、ここでは、外部の再循環ループが補助エネルギー装置の用途に用いられている。このエネルギー装置は静止しており、ここでは、ミキサー内を通る流体の流れにより、圧力降下及び乱流の動きによって系内にエネルギーの導入が行われる。2つのストリームとスタティックミキサーとのインピンジメントを促す、図示のように配置された2つのT継手から成る二重T継手は、Koflo Corp製の「kenics helical style」の継手とした。
【図5】実施例11に用いられる二重T継手の補助エネルギー装置を示す。ラインは急に直角に曲がった3/4インチIDスチールパイプから作製される。2つの流れが出口で合流する。
【図6】可能な結晶化プロセスの概要であって、当該プロセスは、微細な種のスラリーを生成すること、結晶化される生成物の濃縮溶液を生成すること、及び結晶化を開始するようにスラリーを濃縮液と合わせることを含む、結晶化プロセスの概要である。過飽和を生じさせる複数の方法によってさらなる結晶化が可能であり、そのうちのいくつかを挙げている。
【図7】バッチ式結晶化法の一例である。
【図8】半連続式結晶化法の一例である。
【図9】バッチ式反応結晶化法の一例である。試薬A及びBを反応させて、結晶化される生成物を生成する反応シナリオが示されている。
【図10】実施例1Bの生成物の顕微鏡写真である。
【図11】再循環微細粉砕から0.5分後の、実施例3Bの微細粉砕プロセスの生成物の顕微鏡写真である。
【図12】再循環微細粉砕から15分後の、実施例3Bの微細粉砕プロセスの生成物の顕微鏡写真である。
【図13】再循環微細粉砕から60分後の、実施例3Bの微細粉砕プロセスの生成物の顕微鏡写真である。
【図14】実施例3Bの結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図15】実施例4Bの結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図16】実施例5の結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図17】実施例8Aの結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図18】実施例8Bの結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図19】実施例9Aの結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図20】実施例9Bの結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図21】実施例10の結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図22】実施例11の結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図23】実施例12の結晶化の終了時点での生成物のスラリーの顕微鏡写真である。
【図24】再循環微細粉砕から15分後の、実施例3Bの微細粉砕プロセスの生成物についての粒径分布記録である。
【図25】再循環微細粉砕から60分後の、実施例3Bの微細粉砕プロセスの生成物についての粒径分布記録である。
【図26】実施例6におけるような微細粉砕及び結晶化プロセス又は乾式粉砕プロセスについて、直接充填カプセルの挿入から初めの24時間の血流中の化合物Fの血漿レベルを比較して3匹のイヌについて収集された薬物動態学的データに関する記録である。
【特許請求の範囲】
【請求項1】
活性有機化合物の結晶粒子を製造する方法であって、
微細な種を結晶化プロセスに付すことからなり、
前記微細な種が湿式粉砕プロセスによって生成され、約0.1μm〜約20μmの平均粒径を有し、得られる結晶粒子が100μm未満の平均粒径を有する方法。
【請求項2】
得られる結晶粒子の前記平均粒径が60μm未満である、請求項1に記載の方法。
【請求項3】
前記微細な種の前記平均粒径が約0.5μm〜20μmである、請求項1に記載の方法。
【請求項4】
前記微細な種の前記平均粒径が約1μm〜10μmである、請求項1に記載の方法。
【請求項5】
前記湿式粉砕プロセスの際、キャビテーションミル、ボールミル、媒体ミル又は超音波破砕が用いられる、請求項1に記載の方法。
【請求項6】
前記媒体ミル又は前記ボール媒体が0.5mm〜4mmのビーズを用いる、請求項5に記載の方法。
【請求項7】
セラミックミル及びセラミックビーズが用いられるか、又はクロムで内張りされたミル及びセラミックビーズが用いられる、請求項6に記載の方法。
【請求項8】
前記活性有機化合物が医薬品である、請求項1に記載の方法。
【請求項9】
前記医薬品が、鎮痛薬、抗炎症剤、駆虫薬、抗不整脈薬、気管支喘息治療薬、抗菌剤、抗凝血薬、抗うつ剤、抗糖尿病薬、抗てんかん薬、抗ヒスタミン剤、血圧降下薬、抗ムスカリン薬、抗ミコバクテリア薬、抗悪性腫瘍薬、免疫抑制剤、抗甲状腺薬、抗ウイルス薬、抗不安剤、鎮静剤、収れん薬、βアドレナリン作動性受容体遮断薬、造影剤、コルチコステロイド、咳止め薬、診断薬、画像診断薬、ドーパミン作動薬、止血剤、免疫学的薬剤、脂質調節剤、筋弛緩剤、副交感神経作働薬、副甲状腺カルシトニン、プロスタグランジン、放射性医薬品、性ホルモン、抗アレルギー薬、刺激剤、交感神経作働薬、甲状腺薬、血管拡張剤、及びキサンチンから成る群から選択される、請求項8に記載の方法。
【請求項10】
請求項1に記載の方法において製造される結晶粒子及び薬学的に許容可能な担体からなる、医薬組成物。
【請求項11】
前記結晶化プロセスが、
(1)前記微細な種のスラリーを生成する工程、
(2)結晶化される前記生成物の溶液を生成する工程、及び(combining)工程、
を含む、請求項1に記載の方法。
【請求項12】
前記結晶化プロセスが、バッチ式、半連続式又は連続式の処理構成を用いることを含む、請求項11に記載の方法。
【請求項13】
前記結晶化プロセスの際、再循環ループが用いられる、請求項12に記載の方法。
【請求項14】
前記結晶化プロセスの前記溶媒系が主として水系溶媒流、主として有機溶媒流又は混合溶媒流を含む、請求項11に記載の方法。
【請求項15】
前記結晶化プロセスの際、補助エネルギー装置が用いられる、請求項11に記載の方法。
【請求項16】
前記補助エネルギー装置が、混合用T継手、混合用エルボ、スタティックミキサー、超音波破砕機又はロータステータホモジナイザーである、請求項15に記載の方法。
【請求項17】
前記補助エネルギー装置が前記結晶化プロセスの終了時点に用いられる、請求項15に記載の方法。
【請求項18】
前記補助エネルギー装置が再循環ループ内に設置される、請求項15に記載の方法。
【請求項19】
前記結晶化プロセスが、微細な種、バッチ溶液、試薬溶液又は貧溶媒を再循環ループ又は高い混合強度の領域に添加することをさらに含む、請求項11に記載の方法。
【請求項20】
前記結晶化プロセスが、1つ又は複数の補助添加剤をさらに含む、請求項11に記載の方法。
【請求項21】
前記微細な種のスラリー及び前記生成物の溶液が合わさると高速微細混合する、請求項11に記載の方法。
【請求項22】
前記結晶化プロセスが、
(1)媒体粉砕を用いて前記微細な種のスラリーを生成する工程、
(2)前記微細な種の一部を溶解する工程、及び
(3)前記活性有機化合物を前記微細な種で結晶化する工程、
を含む、請求項1に記載の方法。
【請求項23】
得られる結晶粒子が、前記微細な種の形態に対応する結晶形態を有する、請求項1に記載の方法。
【請求項1】
活性有機化合物の結晶粒子を製造する方法であって、
微細な種を結晶化プロセスに付すことからなり、
前記微細な種が湿式粉砕プロセスによって生成され、約0.1μm〜約20μmの平均粒径を有し、得られる結晶粒子が100μm未満の平均粒径を有する方法。
【請求項2】
得られる結晶粒子の前記平均粒径が60μm未満である、請求項1に記載の方法。
【請求項3】
前記微細な種の前記平均粒径が約0.5μm〜20μmである、請求項1に記載の方法。
【請求項4】
前記微細な種の前記平均粒径が約1μm〜10μmである、請求項1に記載の方法。
【請求項5】
前記湿式粉砕プロセスの際、キャビテーションミル、ボールミル、媒体ミル又は超音波破砕が用いられる、請求項1に記載の方法。
【請求項6】
前記媒体ミル又は前記ボール媒体が0.5mm〜4mmのビーズを用いる、請求項5に記載の方法。
【請求項7】
セラミックミル及びセラミックビーズが用いられるか、又はクロムで内張りされたミル及びセラミックビーズが用いられる、請求項6に記載の方法。
【請求項8】
前記活性有機化合物が医薬品である、請求項1に記載の方法。
【請求項9】
前記医薬品が、鎮痛薬、抗炎症剤、駆虫薬、抗不整脈薬、気管支喘息治療薬、抗菌剤、抗凝血薬、抗うつ剤、抗糖尿病薬、抗てんかん薬、抗ヒスタミン剤、血圧降下薬、抗ムスカリン薬、抗ミコバクテリア薬、抗悪性腫瘍薬、免疫抑制剤、抗甲状腺薬、抗ウイルス薬、抗不安剤、鎮静剤、収れん薬、βアドレナリン作動性受容体遮断薬、造影剤、コルチコステロイド、咳止め薬、診断薬、画像診断薬、ドーパミン作動薬、止血剤、免疫学的薬剤、脂質調節剤、筋弛緩剤、副交感神経作働薬、副甲状腺カルシトニン、プロスタグランジン、放射性医薬品、性ホルモン、抗アレルギー薬、刺激剤、交感神経作働薬、甲状腺薬、血管拡張剤、及びキサンチンから成る群から選択される、請求項8に記載の方法。
【請求項10】
請求項1に記載の方法において製造される結晶粒子及び薬学的に許容可能な担体からなる、医薬組成物。
【請求項11】
前記結晶化プロセスが、
(1)前記微細な種のスラリーを生成する工程、
(2)結晶化される前記生成物の溶液を生成する工程、及び(combining)工程、
を含む、請求項1に記載の方法。
【請求項12】
前記結晶化プロセスが、バッチ式、半連続式又は連続式の処理構成を用いることを含む、請求項11に記載の方法。
【請求項13】
前記結晶化プロセスの際、再循環ループが用いられる、請求項12に記載の方法。
【請求項14】
前記結晶化プロセスの前記溶媒系が主として水系溶媒流、主として有機溶媒流又は混合溶媒流を含む、請求項11に記載の方法。
【請求項15】
前記結晶化プロセスの際、補助エネルギー装置が用いられる、請求項11に記載の方法。
【請求項16】
前記補助エネルギー装置が、混合用T継手、混合用エルボ、スタティックミキサー、超音波破砕機又はロータステータホモジナイザーである、請求項15に記載の方法。
【請求項17】
前記補助エネルギー装置が前記結晶化プロセスの終了時点に用いられる、請求項15に記載の方法。
【請求項18】
前記補助エネルギー装置が再循環ループ内に設置される、請求項15に記載の方法。
【請求項19】
前記結晶化プロセスが、微細な種、バッチ溶液、試薬溶液又は貧溶媒を再循環ループ又は高い混合強度の領域に添加することをさらに含む、請求項11に記載の方法。
【請求項20】
前記結晶化プロセスが、1つ又は複数の補助添加剤をさらに含む、請求項11に記載の方法。
【請求項21】
前記微細な種のスラリー及び前記生成物の溶液が合わさると高速微細混合する、請求項11に記載の方法。
【請求項22】
前記結晶化プロセスが、
(1)媒体粉砕を用いて前記微細な種のスラリーを生成する工程、
(2)前記微細な種の一部を溶解する工程、及び
(3)前記活性有機化合物を前記微細な種で結晶化する工程、
を含む、請求項1に記載の方法。
【請求項23】
得られる結晶粒子が、前記微細な種の形態に対応する結晶形態を有する、請求項1に記載の方法。
【図1】

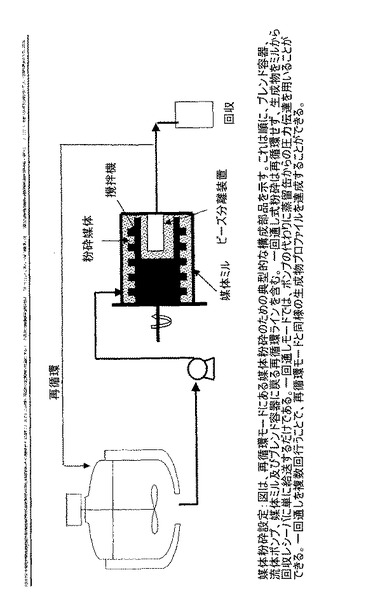
【図2】
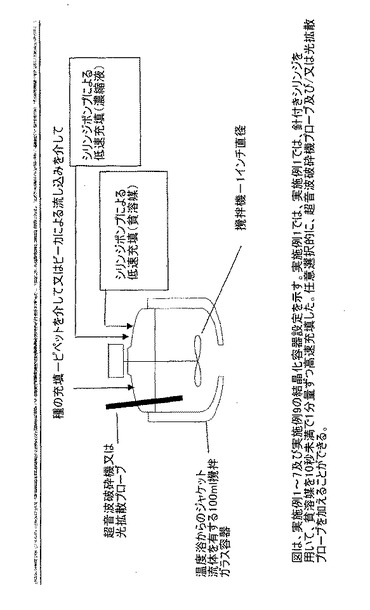
【図3】
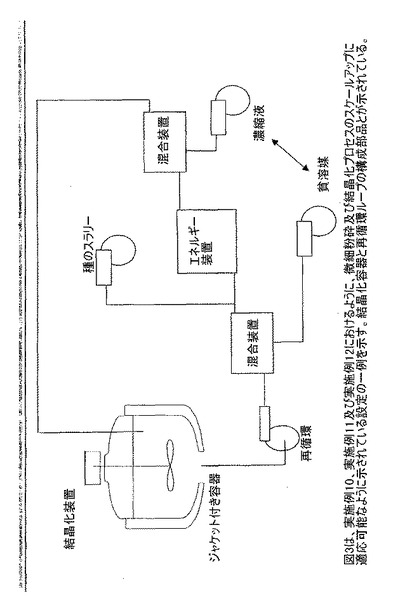
【図4】
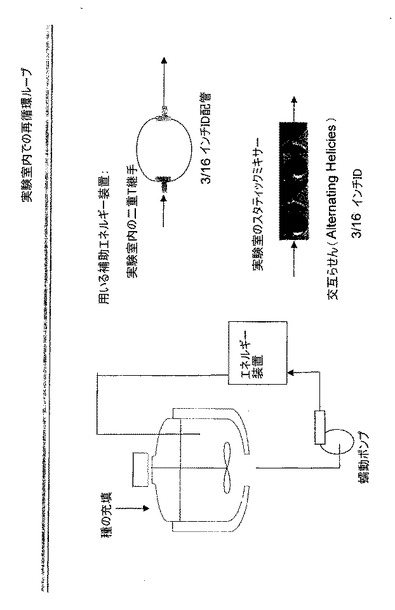
【図5】
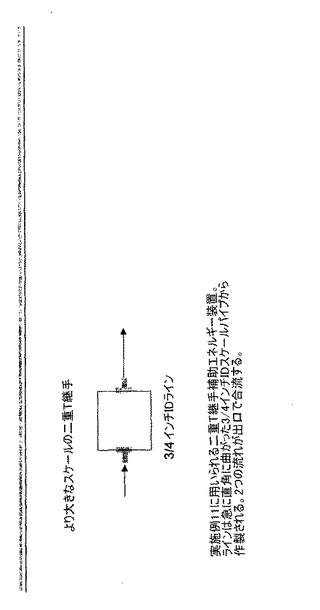
【図6】
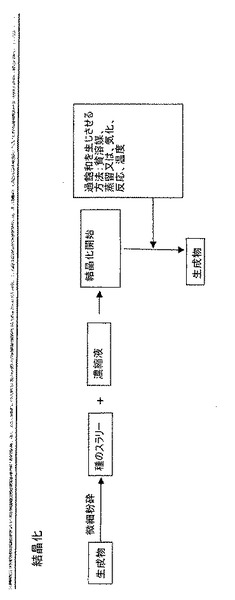
【図7】
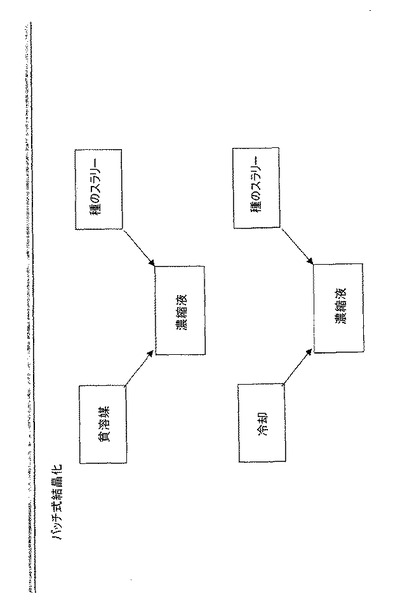
【図8】
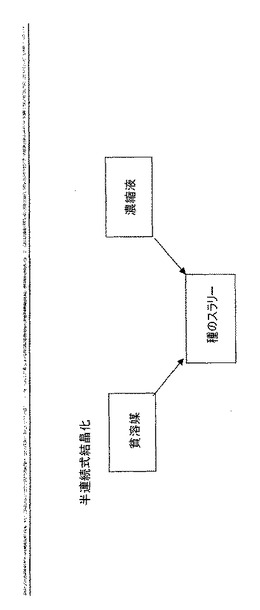
【図9】
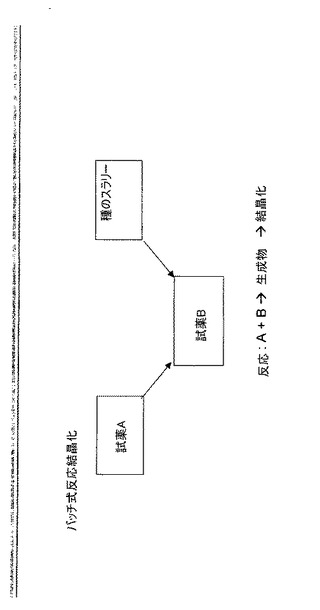
【図24】
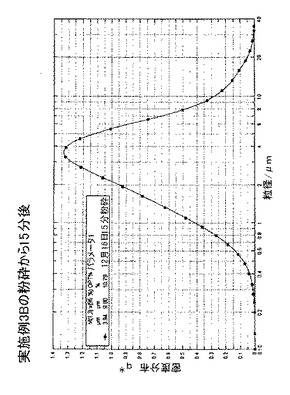
【図25】
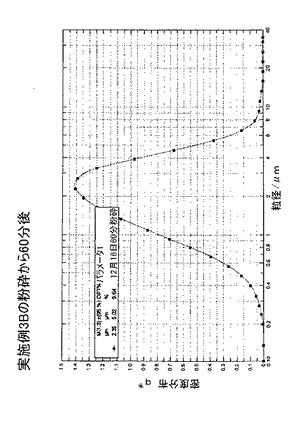
【図26】
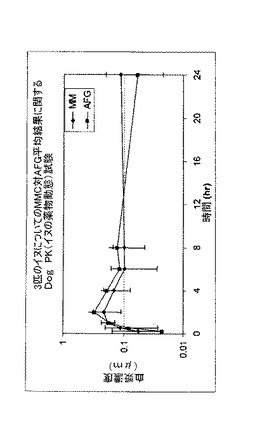
【図10】
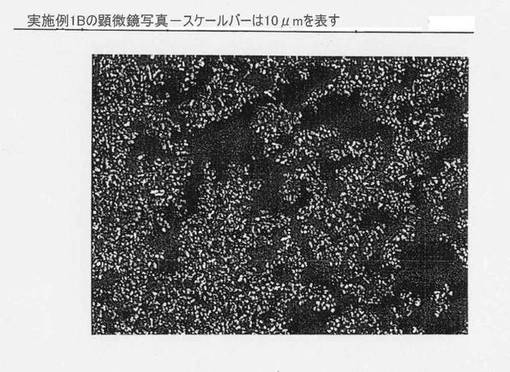
【図11】
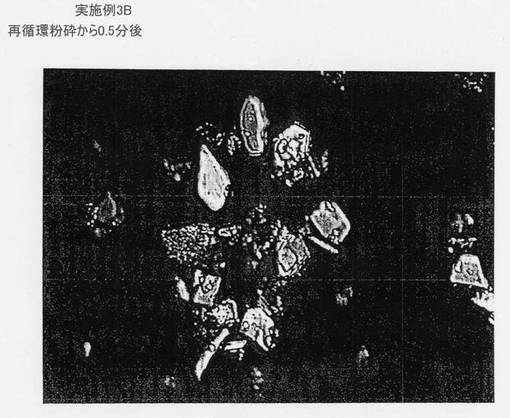
【図12】

【図13】

【図14】

【図15】

【図16】

【図17】
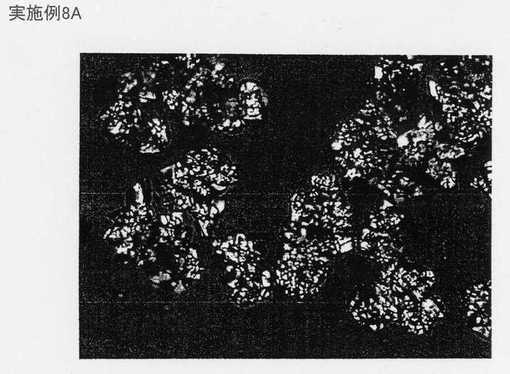
【図18】

【図19】
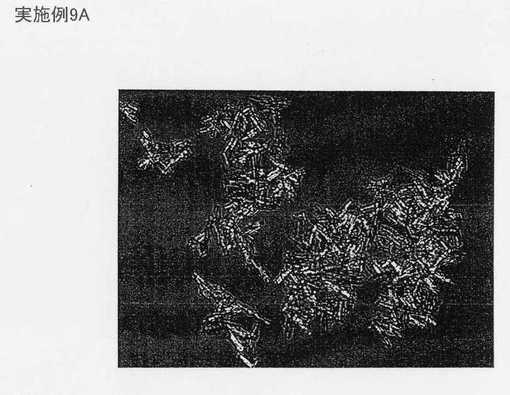
【図20】

【図21】

【図22】

【図23】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図24】
【図25】
【図26】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【公表番号】特表2009−529982(P2009−529982A)
【公表日】平成21年8月27日(2009.8.27)
【国際特許分類】
【出願番号】特願2009−500573(P2009−500573)
【出願日】平成19年3月12日(2007.3.12)
【国際出願番号】PCT/US2007/063785
【国際公開番号】WO2007/106768
【国際公開日】平成19年9月20日(2007.9.20)
【出願人】(390023526)メルク エンド カムパニー インコーポレーテッド (924)
【氏名又は名称原語表記】MERCK & COMPANY INCOPORATED
【Fターム(参考)】
【公表日】平成21年8月27日(2009.8.27)
【国際特許分類】
【出願日】平成19年3月12日(2007.3.12)
【国際出願番号】PCT/US2007/063785
【国際公開番号】WO2007/106768
【国際公開日】平成19年9月20日(2007.9.20)
【出願人】(390023526)メルク エンド カムパニー インコーポレーテッド (924)
【氏名又は名称原語表記】MERCK & COMPANY INCOPORATED
【Fターム(参考)】
[ Back to top ]