
微細粒状体の製造方法
【解決課題】分散媒中において難溶性ないし非溶性の各種薬剤の高度かつ均一な微細粒状体を形成する方法を提供する。
【手段】分散媒に対して難溶性ないし非溶性のゲスト分子を、ホスト分子により包接するあるいは界面活性剤により乳化することによって、分散媒中にゲスト分子の微細粒状体を製造する方法であって、圧力容器中に、ゲスト分子、およびホスト分子ないし界面活性剤を含む分散媒を仕込み、この圧力容器に二酸化炭素を導入して、加圧し、所定時間撹拌を行なって包接あるいは乳化処理した後、圧力容器内を減圧して二酸化炭素を排出することにより、分散媒中のゲスト分子の微細粒状体を製造することを特徴とするものである。撹拌は静止型混合機により行われるものであることが望ましい。
【手段】分散媒に対して難溶性ないし非溶性のゲスト分子を、ホスト分子により包接するあるいは界面活性剤により乳化することによって、分散媒中にゲスト分子の微細粒状体を製造する方法であって、圧力容器中に、ゲスト分子、およびホスト分子ないし界面活性剤を含む分散媒を仕込み、この圧力容器に二酸化炭素を導入して、加圧し、所定時間撹拌を行なって包接あるいは乳化処理した後、圧力容器内を減圧して二酸化炭素を排出することにより、分散媒中のゲスト分子の微細粒状体を製造することを特徴とするものである。撹拌は静止型混合機により行われるものであることが望ましい。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、微細粒状体の製造方法に関するものである。詳しく述べると本発明は、高圧二酸化炭素により加圧することにより、分散媒中において各種薬剤の微細粒状体を形成する方法に関するものである。
【背景技術】
【0002】
現在、医療・医薬分野で製造されている薬物の多くは、水に対する溶解性が低く、そのため薬物のバイオアベイラビリティ(生物学的利用能)が大幅に低下するという問題が生じている。このような難溶性薬物の溶解性改善の方法の一つに、シクロデキストリン(CD)及びその誘導体と薬物の包接化がある。しかし、従来の方法においては、難溶性薬物とCD溶液の包接化は、薬物を有機溶媒等に溶解させた状態でCD溶液と包接化するという問題があり、医療・医薬分野において使用する薬剤の処理方法として適当なものであるとはいい難いものである。
【0003】
また、例えば、樹脂組成物や塗料組成物等の中に、可塑剤、各種安定化剤、結晶核剤等の各種添加剤を添加しようとする場合、これらの添加剤を組成物中に均一かつ微分散させることが樹脂組成物や塗料組成物の特性向上の上で大きな鍵となる。
【0004】
ところで、従来、超臨界状態の二酸化炭素を用いて各種物質の微細粒状体を形成することが、広く研究されている。
【0005】
例えば、特許文献1には、金属ナノ粒子の製造方法として、亜臨界ないし超臨界の二酸化炭素を反応媒体とし、ビス(2−エチルヘキシル)スルホコハク酸ナトリウムおよび2,3,3,3,4,4,5,5−オクタフルオロ−1−ペンタノールを活性剤として使用し、水/二酸化炭素からなる逆ミクロエマルジョンを形成し、ミセル中に含まれる金属イオンを、ナトリウムトリアセトキシボロハイドライト等の還元剤により還元して金属ナノ粒子を得る方法が開示されている。
【0006】
また、特許文献2には、圧力容器内で40〜65℃、10〜30MPaで、超臨界ニ酸化炭素、リン脂質等のリポソーム膜構成成分、医薬化合物および製薬助剤を混合して懸濁液を得、次いで、系内を減圧して二酸化炭素を排出することにより、該医薬化合物が内包されたリポソームの水性分散液を調製し、その後前記リン脂質の転移温度〜(転移温度+10)℃で、所定時間インキュベートしてリポソーム含有製剤を製造することが開示されている。
【0007】
特許文献3には、界面活性物質が樹脂粒子中または表面に存在せず、かつ粒度分布がシャープな樹脂粒子を得る方法として、樹脂(b)の溶融液または樹脂(b)の溶剤溶液、あるいは樹脂(b)の前駆体(b0)またはその溶剤溶液を、無機ないし有機の各種微粒子(A)が分散している液状又は超臨界状態の二酸化炭素(X)からなる分散媒体(X0)に分散して得られた微粒子(C1)の分散体(X1)を、(必要に応じて(b0)を重合反応させた後)、圧力を減圧することにより分散体(X1)から(X)を除くことにより樹脂粒子(C)を得ることを特徴とする、微粒子(A)が樹脂(b)からなる樹脂粒子(B)の表面に付着されてなる樹脂粒子(C)の製造方法が、また、特許文献4には、親水性成分が樹脂粒子中または表面に存在せず、かつ粒度分布がシャープな樹脂粒子を得る方法として、樹脂(b)もしくは樹脂(b)の溶剤溶液、または樹脂(b)の前駆体(b0)またはその溶剤溶液を、液状又は超臨界状態の二酸化炭素(X)中に分散させ、圧力を減圧することにより液状又は超臨界状態の二酸化炭素(X)を除去することにより、樹脂粒子を製造する方法において、ジメチルシロキサン基及びフッ素を含有する少なくとも一方の基を有する分散安定剤(D)と無機ないし有機の各種微粒子(A)が樹脂(b)からなる樹脂粒子(B)の表面に付着されてなる樹脂粒子(C)の製造方法が開示されている。
【0008】
また、特許文献5には、超臨界状態の二酸化炭素流体を用い常温常圧下で微粒子を塗布して被塗布物の表面上に超微粒子膜を形成する方法であって、真空状態ないし真空状態に近い状態とした高圧容器に数十μm以下の微粒子である微粒子を注入し、超臨界状態の二酸化炭素流体を注入し、それらを撹拌して超臨界状態の二酸化炭素を分散媒体とする微粒子の高分散状態になった混合物とし、該混合物をノズルから被塗物に噴射することで膜を形成する微粒子膜形成方法を開示している。
【0009】
このように、従来、各種薬剤の微細粒状体を形成する上で、超臨界状態の二酸化炭素を分散媒として使用することが行われているが、上記したような包接化において超臨界状態の二酸化炭素を用いることは行われていなかった。
【0010】
また、例えば、樹脂組成物の調製方法として、上記したように界面活性剤等を用いることなく、樹脂成分と添加剤とを超臨界状態の二酸化炭素中で直接分散させようとする試みは行われていたものの、添加剤を微細な粒状体として分散配合することが十分になされず、添加剤による改質効果の向上に限度があるものであった。
【特許文献1】特開2005−290481号公報
【特許文献2】特開2006−298844号公報
【特許文献3】特開2006−321830号公報
【特許文献4】特開2007−277551号公報
【特許文献5】特開2008−12448号公報
【発明の開示】
【発明が解決しようとする課題】
【0011】
従って本発明は、上記したような従来技術における問題点を鑑み、分散媒中において難溶性ないし非溶性の各種薬剤の高度かつ均一な微細粒状体を形成する方法を提供することを課題とするものである。
【0012】
本発明はさらに、有機溶媒を用いることなく、水に難溶性ないし非溶性の各種薬剤を包接化により効率よく、微細粒状体に形成する方法を提供することを課題とするものである。
【0013】
本発明はまた、樹脂組成物や塗料組成物等の各種組成物中に添加する各種添加剤を安定な微細粒状体の形態とし、これを組成物中に配合することで組成物の高い改質効果を得ることのできる微細粒状体の形成方法を提供することを課題とするものである。
【課題を解決するための手段】
【0014】
上記課題を解決する本発明は、分散媒に対して難溶性ないし非溶性のゲスト分子を、ホスト分子により包接するあるいは界面活性剤により乳化することによって、分散媒中にゲスト分子の微細粒状体を製造する方法であって、圧力容器中に、ゲスト分子、およびホスト分子ないし界面活性剤を含む分散媒を仕込み、この圧力容器に二酸化炭素を導入して、1〜100MPaの操作圧まで加圧し、所定時間撹拌を行なって包接あるいは乳化処理した後、圧力容器内を減圧して二酸化炭素を排出することにより、分散媒中のゲスト分子の微細粒状体を製造することを特徴とするものである。
【0015】
本発明はまた、撹拌が静止型混合機により行われるものである微細粒状体の製造方法を示すものである。
【0016】
本発明はまた、分散媒が水ないし水系溶液である上記微細粒状体の製造方法を示すものである。
【0017】
本発明はさらに、ゲスト分子が標準状態(SATP、25℃、105Pa)において、固体状のものである微細粒状体の製造方法を示すものである。
【0018】
本発明はさらに、ゲスト分子が、S−(+)−イブプロフェン、プロゲステロン、ケトプロフェンおよびグリセオフルビンからなる群から選ばれてなるいずれか1つの化合物である微細粒状体の製造方法を示すものである。
【0019】
本発明はさらに、ホスト分子がシクロデキストリンおよびその誘導体からなる群から選ばれてなるものである微細粒状体の製造方法を示すものである。
【0020】
本発明はまた、ゲスト分子が樹脂ないしゴム成分を含有する組成物用の添加剤である微粒粒状体の製造方法を示すものである。
【0021】
本発明はさらに、ゲスト分子がシリコーンオイルである微粒粒状体の製造方法を示すものである。
【0022】
上記課題を解決する本発明はまた、上記の製造方法により得られた分散媒中のゲスト分子の微細粒状体を樹脂組成物中に添加することを特徴とする樹脂組成物の製造方法である。
【0023】
本発明はまた、樹脂組成物がポリアミド系樹脂組成物であり、シリコーンオイルであるゲスト分子の微細粒状体を添加混合し、これを成形加工することを特徴とする、透明度の改善されたポリアミド系樹脂製品の製造方法を示すものである。
【0024】
本発明はさらに、当該製造方法により得られたことを特徴とする透明度の改善されたポリアミド系樹脂製品を示すものである。
【0025】
本発明はまた、前記製造方法により得られた、分散媒中のゲスト分子の微細粒状体を、さらに他の液状体に添加吸収させることを特徴とする微細粒状体分散体の製造方法を示すものである。
【0026】
本発明はさらに、前記製造方法により得られた、分散媒中のゲスト分子の微細粒状体を、被塗物表面へと吹きつけることを特徴とする微細粒状体被膜の形成方法を示すものである。
【発明の効果】
【0027】
本発明によれば、分散媒中において難溶性ないし非溶性の各種薬剤の高度かつ均一な微細粒状体を形成することができ、例えば、水に対して難溶性ないし非溶性のゲスト分子を、有機溶媒を用いることなく、ホスト分子により包接化して微細粒状化することができ、医療・医薬分野で製造されている薬物の溶解性を改善し、薬物のバイオアベイラビリティを大きく向上させることができる。
【0028】
また、本発明によれば、樹脂組成物や塗料組成物等の各種組成物中に添加する各種添加剤を安定な微細粒状体の形態とし、これを組成物中に配合することで組成物の高い改質効果を得ることのできる。具体的には例えば、ナイロン等のポリアミド系樹脂組成物に、撥水性、帯電防止性、柔軟性あるいは潤滑性等を付与することを目的とする改質剤としてシリコーンオイルを添加しようとする場合に、当該シリコーンオイルを本発明の製造方法により微細粒状化して、樹脂組成物に配合することで、上記した所期の特性の改質効果に加え、樹脂組成物製品の透明性を大きく改善できる。
【発明を実施するための最良の形態】
【0029】
以下、本発明を実施形態に基づき、より詳細に説明する。
【0030】
使用原料等
本発明において用いられる分散媒としては、特に限定されるものではないが、医療・医薬分野で用いられるゲスト分子を取り扱う場合、また、環境保全性の観点からは、水、あるいは、水にメタノール、エタノール、イソプロパノール等の低級アルコール、酸、アルカリ、塩等の水溶性ないし水混和性化合物を添加してなる水系溶媒を用いることが望ましく、特に水が好ましい。
【0031】
また、分散媒に対して難溶性ないし非溶性のゲスト分子としても特に限定されるものではなく、各種の薬剤を用いることができる。なお、本明細書において、「分散媒に対して難溶性ないし非溶性」とは、一般に難溶性ないし非溶性と言われるものであれば包含され得、厳密に規定されるものではないが、例えば、標準状態(SATP、25℃、105Pa)の分散媒(特に、水)に対する溶解度が1μg/mL程度以下のものが少なくとも含まれるものである。
【0032】
このような、分散媒に対して難溶性ないし非溶性のゲスト分子として、具体的には、例えば、医療・医薬分野における解熱剤、抗炎症剤、鎮痛剤、精神安定剤、鎮静剤、抗腫瘍剤、抗菌剤、抗生物質、抗高脂血症剤、鎮咳去たん剤、筋弛緩剤、抗てんかん剤、抗潰瘍剤、抗うつ剤、抗アレルギー剤、強心剤、不整脈治療剤、血管拡張剤、降圧利尿剤、糖尿治療剤、抗結核剤、抗リウマチ剤、ステロイド剤、麻薬拮抗剤、ホルモン剤、脂溶性ビタミン剤、抗凝血剤、虚血性疾患治療薬、免疫疾患治療薬、アルツハイマー病治療薬、脳血管攣縮治療薬、脳血栓治療薬、緑内障治療薬、高眼圧症治療薬、関節炎治療薬、抗セプシス薬、抗セプティックショック薬、抗喘息薬、頻尿・尿失禁治療薬、アトピー性皮膚炎治療薬、アレルギー性鼻炎治療薬、動物薬組成物等、また、各種樹脂組成物、塗料組成物、インキ組成物等に添加される、可塑剤、各種安定化剤、結晶核剤、ワックス成分、消泡剤、難燃化剤、離型剤、有機着色剤等の各種添加剤、その他、化粧料組成物、農薬組成物、殺虫剤、殺菌剤、除草剤、飲食用組成物等を挙げることができるが、これらに何ら限定されるものではない。
【0033】
また、分散媒に対して難溶性ないし非溶性のゲスト分子としては、例えば、標準状態(SATP、25℃、105Pa)において、固体状のものであり、加圧により融点降下を生じ、本発明の製造方法における製造条件下において融液状態となり得る物質が、好ましい例として挙げることができる。このような物質は、従来の方法においては、微小粒状体とすることが困難であり、本発明の製造方法を用いることが、特に有用なものであるからである。
【0034】
このような標準状態(SATP、25℃、105Pa)において、固体状の難溶性ないし非溶性のゲスト分子としては、具体的には、例えば、S−(+)−イブプロフェン、プロゲステロン、ケトプロフェン、グリセオフルビン等の薬剤を例示することができるが、何らこれらに限定されるものではない。
【0035】
また、ゲスト分子が樹脂用添加剤である場合、好ましい一例としては、ジメチルシリコーンオイル、メチルフェニルシリコーンオイル、各種変性シリコーンオイル等のシリコーンオイルを挙げることができるが、もちろん何らこれらに限定されるものではない。
ルことが知られている。
【0036】
一方、このようなゲスト分子を包接するホスト分子としては、特に限定されるものではないが、例えば、各種天然型シクロデキストリンおよび各種化学修飾型シクロデキストリン、各種クラウンエーテル、各種シクロファン、各種カリックスアレーン等を挙げることができ、なかでも好ましいものとして、α―シクロデキストリン、β―シクロデキストリン、γ―シクロデキストリン、δ―シクロデキストリンおよびこれらを化学修飾したもの等のシクロデキストリンおよびその誘導体を挙げることができる。ここで言う化学修飾とは、各種シクロデキストリン中の水酸基をアルキル化、ヒドロキシアルキル化したもの等を指す。この時のアルキル基としては、直鎖状、分枝状のいずれでも良く、その構造中に不飽和結合を含むものでも良いが、好ましいものとしてメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、t−ブチル基、ヒドロキシメチル、ヒドロキシエチル、ヒドロキシプロピル等を挙げることができ、このような各種化学修飾型シクロデキストリンのなかでも、ヒドロキシプロピル−β−シクロデキストリンを、特に好ましいものとして挙げることができる。
【0037】
なお、ゲスト分子をホスト分子で包接する態様において、ゲスト分子とホスト分子との配合比としては、使用するゲスト分子およびホスト分子の種類等によっても異なるものであり、特に限定されるものではないが、例えば、ゲスト分子とホスト分子の仕込みモル比で、ゲスト分子:ホスト分子=1:0.5〜1:3.5程度、より好ましくは、1:0.5〜1:2.5程度とすることが望ましい。
【0038】
また、ゲスト分子を乳化する界面活性剤としても、特に限定されるものではなく、また使用するゲスト分子および分散媒の種類によっても左右されるものであるが、例えば、ポリオキシエチレンポリオキシプロピレングリコール、ポリオキシエチレン硬化ヒマシ油、ポリオキシエチレンラウリルエーテル等のポリオキシエチレンアルキルエーテル、ポリオキシエチレンソルビタン脂肪酸エステル(ポリソルベート)、モノステアリン酸ソルビタン等のソルビタン脂肪酸エステル等の非イオン性界面活性剤、塩化ベンザルコニウム、塩化ベンゼトニウム、塩化セチルピリジニウム等のアニオン性界面活性剤、ステアリン酸カルシウム、ステアリン酸マグネシウム、ラウリル硫酸ナトリウム等のカチオン性界面活性剤等が挙げられる。また、アンモニウムカルボキシレートパーフルオロエーテル等のフッ素基を有する界面活性剤等が挙げられる。
【0039】
製造条件等
本発明の微細粒状体の製造方法においては、上記したような分散媒に対して難溶性ないし非溶性のゲスト分子の微細粒状体を製造するにおいて、平衡セル等の圧力容器中に、当該ゲスト分子、並びにこれを包接するホスト分子またはこれを乳化する界面活性剤および分散媒を仕込み、この圧力容器に高圧の二酸化炭素、好ましくは亜臨界ないし超臨界状態の二酸化炭素を導入して加圧し、所定時間撹拌下に保持することで、ゲスト分子の包接化ないしは乳化により、分散媒中にゲスト分子の微細粒状体を形成した後、圧力容器内を減圧して二酸化炭素を排出することにより、ゲスト分子の微細粒状体の分散液を得るものである。
【0040】
ここで、ゲスト分子が、標準状態(SATP、25℃、105Pa)において、固体状のものである場合には、これを包接するホスト分子またはこれを乳化する界面活性剤および分散媒と混合するに先立ち、予め、二酸化炭素により加圧した後、さらに、二酸化炭素の加圧下で、ホスト分子又はこれを乳化する界面活性剤および分散媒と撹拌下に混合しても良い。すなわち、このような標準状態で固体状のゲスト分子、例えば、S-(+)-イブプロフェンは、二酸化炭素による加圧で、融点降下を生じることが知られており(J.Chem and Eng Data.,2005)、予め、二酸化炭素により加圧することで融解し融液状態とした後、包接化ないし乳化するほうが、効率良く、微小粒状体化することができるためである。
【0041】
本発明の製造方法において、微小粒状体化を行う際の二酸化炭素による加圧条件としては、用いられるゲスト分子の種類、操作時の温度条件等によっても左右されるが、例えば、1〜100MPa、より好ましくは1〜50MPa、最も好ましくは1〜40MPaであることが望ましい。圧力が1MPa未満であると、良好な微小粒状体を形成できないためであり、一方、圧力が100MPaを超えるものであると、設備上および運転操作上で不利となるためである。
【0042】
なお、二酸化炭素の超臨界状態とは、二酸化炭素の臨界点(31.1℃、7.38MPa)を超える温度・圧力条件下にある二酸化炭素を指し、亜臨界状態の二酸化炭素とは、圧力または温度のいずれかのみが臨界点を越えた状態にある二酸化炭素(本発明に関しては主として圧力のみが臨界点を超えた状態である。)を指し、また液状の二酸化炭素とは、二酸化炭素の三重点(−56.6℃、0.518MPa)と二酸化炭素の臨界点を通る気液境界線、臨界温度の等温線、及び固液境界線に囲まれた部分の温度・圧力条件下にある二酸化炭素を指すものである。
【0043】
また、本発明の製造方法において、二酸化炭素による加圧下で微小粒状体化を行う際の温度条件としては、用いられるゲスト分子の種類、操作時の二酸化炭素の圧力条件等にも左右され、特に限定されるものではないが、例えば、0〜200℃、より好ましくは0〜180℃、さらに好ましくは0〜150℃とすることが望ましい。
【0044】
また、本発明においては、このように二酸化炭素による加圧下において、ゲスト分子を、ホスト分子又はこれを乳化する界面活性剤および分散媒と撹拌下に混合するが、この撹拌装置としては、例えば、マグネティックスターラーや各種撹拌子を備え機械的に回転駆動される撹拌型混合機や、管内に混合エレメントを有してなる静止型混合機(スタティックミキサー)等を用いることができるが、このうち、静止型混合機を用いることが望ましい。
【0045】
ここで、本明細書において述べる静止型混合機(スタティックミキサー)とは、「流体通路の構成によって、レイノルズ数の広範囲にわたって流体混合ができるとともに、機械的可動部を持たない流体通路の構造体」であり、分割、転換、反転の3つの混合原理により撹拌を行う混合機である。
【0046】
静止型混合機としては、例えば、日本フローコントロール株式会社製のスパイラルタイプスタティックミキサー、特に、スタティックミキサー 85シリーズ等を好ましい例として挙げることができるが、これらに何ら限定されるものではない。
【0047】
本発明の製造方法、殊に、ゲスト分子をホスト分子により包接する態様においては、この撹拌が十分なものでないと、加圧条件下においても、ゲスト分子の流体が分散媒中であまり広がらず、包切度が低下して、良好な微細粒状体が得られない虞れがあるために、十分な撹拌効果が得られる静止型混合機を用いることが特に望ましい。
【0048】
なお、撹拌装置として、マグネティックスターラーや機械的に回転駆動される撹拌型混合機を用いる場合においても、十分な撹拌がなされるように、撹拌子の形状や撹拌速度を適切なものとすることが望ましく、例えば、マグネティックスターラーの場合、撹拌子として星型等の複雑形状のものを用いたり、圧力容器の容積に比して通常より大きい撹拌子を用いる、あるいは回転数を高速、具体的には例えば、500rpm以上、好ましくは800rpm〜1000rpmのものとするなどとして、撹拌時の分散媒液面形状が大きく変化するないし渦流が形成される状態とすることが望ましい。
【0049】
このように二酸化炭素による加圧条件下で、ゲスト分子を、ホスト分子又はこれを乳化する界面活性剤および分散媒と撹拌下に所定時間、例えば、1〜60分間、より好ましくは1〜30分間混合することによって、ゲスト分子のホスト分子による包接化、あるいはゲスト分子の界面活性剤による乳化が促進され、ゲスト分子の均一な微細粒状体、具体的には例えば、平均粒子径1〜105nm、より好ましくは5〜5000nmを分散媒中に形成することができる。
【0050】
その後、圧力容器内を減圧して二酸化炭素を排出することにより、分散媒中のゲスト分子の微細粒状体の分散液を取り出す。
【0051】
このようにして取り出されたゲスト粒子の微細粒状体の分散液は、そのまま分散液として回収する、あるいは必要に応じて、他の液状体、例えば、エチレングリコール、グリセリン、流動パラフィン、水等に添加吸収させたり、そのまま、被塗物表面へと吹きつけることにより微細粒状体被膜を形成する等の処理にかけられることができる。
【0052】
得られたゲスト粒子の微細粒状体の分散液は、ゲスト分子が医療・医薬分野における薬剤である場合、そのまま、あるいは生理食塩水、リン酸緩衝溶液等の水系溶媒中で、他の成分と容易に調合されることで、所定の医薬組成物へと調製でき、その後、必要に応じて、乾燥、賦形化等の処理を施される。
【0053】
また、ゲスト分子が各種樹脂組成物、ゴム組成物、塗料組成物、インキ組成物等の樹脂ないしゴム成分を含有する組成物用の添加剤である場合にも、これら組成物中に、そのまま、あるいは上記したように、他の液状体に添加吸収させた後、溶融混練、溶液ないし分散媒中で混合する等により、配合されることで、組成物中に均一かつ微分散化させることができ、優れた改質効果を発揮することができる。
【0054】
なお、この場合の樹脂ないしゴム成分を含有する組成物としても、特に限定されるものではなく、例えば、ポリオレフィン樹脂系、ポリスチレン樹脂系、ポリ塩化ビニル樹脂系、ポリ塩化ビニリデン樹脂系、ポリアクリロニトリル樹脂系、ポリアミド樹脂系、ポリエーテルイミド系、ポリアミドイミド系、ポリエステル樹脂系、ポリカーボネート樹脂系、ポリアセタール樹脂系、酢酸ビニル樹脂系、ポリビニルアセタール系(例えば、ボリビニルブチラール樹脂)、熱可塑性ポリウレタンエラストマー系、アクリル樹脂系、ポリフェニレン樹脂系、フッ素樹脂系、ポリビニルアルコール系、ポリビニルピロリドン、セルロース樹脂系、変性セルロース樹脂系、フェノール樹脂系、尿素樹脂系、メラミン樹脂系、フラン樹脂系、アルキド樹脂系、不飽和ポリエステル樹脂系、ジアリルフタレート樹脂系、エポキシ樹脂系、シリコーン樹脂系、ポリイミド樹脂系、ポリウレタン樹脂系、グアナミン樹脂系及びフッ素樹脂系、天然ゴム系、ポリブタジエンゴム系、 スチレンブタジエンゴム系、ポリイソプレンゴム系、クロロプレンゴム系、アクリロニトリルブタジエンゴム系、ブチルゴム系、エチレンプロピレンゴム系、アクリルゴム系、ウレタンゴム系、エピクロヒドリンゴム系、フッ素ゴム系、シリコーンゴム系等、あるいはこれらの二種以上の混合系などの公知の各種の組成物が含まれる。
【0055】
なお、本発明に係る製造方法により得られたゲスト粒子の微細粒状体の分散液は、減圧して二酸化炭素を排出した後においても、比較的安定してその微細粒状体を維持することができ、例えば、密閉容器中で1週間保存後において、その平均粒子径の変動率が10%以下程度、1月保存後においても、20%以下程度となり得るものである。
【0056】
具体的態様1
次に、本発明に係る微細粒状体の製造方法につき、好ましい具体的態様に基づき、さらに詳細に説明する。
【0057】
本発明の微細粒状体の製造方法を適用する好ましい一例として、ゲスト分子であるS−(+)−イブプロフェンのホスト分子であるシクロデキストリンないしその誘導体による包接化を挙げることができる。なお、この場合の分散媒としては、水を用いることが好ましい。
【0058】
S−(+)−イブプロフェンを二酸化炭素で加圧した場合、S−(+)−イブプロフェンの融解温度と圧力との間には、次のような関係が見られる。
【0059】
【表1】

S−(+)−イブプロフェンを、シクロデキストリンないしその誘導体との共存下においた場合においても、S−(+)−イブプロフェンのみの場合とほぼ同様な融解温度低下が見られ、包接化処理の処理温度条件下で、少なくともS−(+)−イブプロフェンの融解が生じる圧力以上の圧力まで加圧することによって、包接化が迅速に進行し、水に対するS−(+)−イブプロフェンの溶解量を高めることができる。好ましくは、処理温度が0〜50℃、加圧条件が1〜5MPaであり、この場合、撹拌時間としては、撹拌条件にもよるが、1〜60分程度、より好ましくは5〜15分程度とすることが望ましい。すなわち、5分程度で包接が十分に平衡に達し、それ以上の時間撹拌処理を行っても、水に対するS−(+)−イブプロフェンの溶解量はほとんど向上しないかあるいは逆に若干低下してしまうためである。
【0060】
なお、S−(+)−イブプロフェンを、シクロデキストリンないしその誘導体の水溶液と混合するに先立ち、予め、S−(+)−イブプロフェンを上記したような所定条件下にて二酸化炭素により加圧して、融解させた後、同じく所定条件下の二酸化炭素により加圧しながらシクロデキストリンないしその誘導体の水溶液へと撹拌下に混合しても良く、この場合、より迅速かつ良好な包接化を進行させることができる。
【0061】
なお、撹拌条件としては、上記したように静止型混合機を用いるか、あるいはマグネティックスターラーを用いる場合、上記したように、撹拌子として星型等の複雑形状のものを用いたり、圧力容器の容積に比して通常より大きい撹拌子を用いる、あるいはその回転数を500rpm以上、好ましくは800rpm〜1000rpmのものとすることが望ましく、このようにしてS−(+)−イブプロフェンとシクロデキストリンとの接触効率を高めることで、S−(+)−イブプロフェンの微細な液滴が生成し包接効率が上昇することによって、水に対するS−(+)−イブプロフェンの溶解量を高めることができる。
【0062】
また、包接化におけるゲスト分子としてのS−(+)−イブプロフェンとホスト分子としてのシクロデキストリンないしその誘導体との配合割合としては、特に限定されないが、仕込みモル比で、ゲスト分子:ホスト分子=1:0.5〜1:3.5程度、より好ましくは、1:0.8〜1:1.2程度とすることが望ましい。
【0063】
また、シクロデキストリンないしその誘導体の水溶液中における濃度としては、特に限定されるものではないが、1g/1ml程度が適当であり、さらに、水に対するS−(+)−イブプロフェンの仕込み量としても、特に限定されるものではないが、水100mlに対し、S−(+)−イブプロフェン500mg程度が適当である。
【0064】
以上示したような条件下で、S−(+)−イブプロフェンをシクロデキストリンないしその誘導体を包接化処理することにより、減圧後においてもS−(+)−イブプロフェンの固体の析出を見ることなく、S−(+)−イブプロフェンの仕込み量の約80%ないしそれ以上の量を包接化により水に溶解させることが可能である。
【0065】
具体的態様2
次に、本発明の微細粒状体の製造方法を適用する好ましい別の一例として、シリコーンオイルの界面活性剤による乳化、およびこのようにして得られたシリコーンオイルエマルジョンを配合してなるポリアミド系樹脂組成物を挙げることができる。なお、この場合の分散媒としては、特に限定されるものではないが、水を用いることが好ましい。
【0066】
シリコーンオイルと、界面活性剤および分散媒である水を、本発明の製造方法に基づき、圧力容器内で、酸化炭素を導入して加圧し、所定時間撹拌を行って乳化処理することで、シリコーンオイルのO/Wエマルジョンを調製することができる。用いる界面活性剤としては、陰イオン界面活性剤、陽イオン界面活性剤、両性イオン界面活性剤、非イオン界面活性剤のいずれのタイプのものを用いることも可能である。特に限定されるものではないが、陰イオン界面活性剤としては、例えば、各種脂肪酸ナトリウム、各種脂肪酸カリウム、アルファスルホ脂肪酸エステルナトリウム等の脂肪酸系界面活性剤、各種直鎖アルキルベンゼンスルホン酸ナトリウム等の直鎖アルキルベンゼン系陰イオン界面活性剤、各種アルキル硫酸エステルナトリウム、各種アルキルエーテル硫酸エステルナトリウム等の高級アルコール系陰イオン界面活性剤、各種アルファオレフィンスルホン酸ナトリウム等のアルファオレフィン系陰イオン界面活性剤、各種アルキルスルホン酸ナトリウム等のノルマルパラフィン系陰イオン界面活性剤などが例示でき、また陽イオン界面活性剤としては、例えば、各種アルキルトリメチルアンモニウム塩、各種ジアルキルジメチルアンモニウム塩等の第四級アンモニウム塩系陽イオン界面活性剤などが例示でき、また、両性イオン界面活性剤としては、例えば、各種アルキルアミノ脂肪酸ナトリウム等のアミノ酸系両性イオン界面活性剤、各種アルキルベタイン等のベタイン系両性イオン界面活性剤、各種アルキルアミンオキシド等のアミンオキシド系両性イオン界面活性剤などが例示でき、非イオン界面活性剤としては、例えば、各種ショ糖脂肪酸エステル、各種ソルビタン脂肪酸、各種ポリオキシエチレンソルビタン脂肪酸エステル等のエステル型非イオン界面活性剤、ポリオキシエチレンデシルエーテル、ポリオキシエチレンドデシルエーテルなどといった各種ポリオキシエチレンアルキルエーテル、各種ポリオキシエチレンアルキルフェニルエーテル等のエーテル型非イオン界面活性剤、さらに、各種エステル・エーテル型非イオン界面活性剤、脂肪酸アルカノールアミドなどといったその他の非イオン界面活性剤などが例示できるが、これらに何ら限定されるものではない。
【0067】
また、特に限定されるものではないが、例えば、処理温度が0〜100℃、加圧条件が1〜100MPaであり、この場合、撹拌時間としては、撹拌条件にもよるが、1〜60分程度、より好ましくは1〜30分程度とすることが望ましい。
【0068】
なお、撹拌条件としては、乳化される分子が標準状態(SATP)においても液状であるシリコーンオイルであるため、上記したS−(+)−イブプロフェンのように標準状態(SATP)において固体である場合と比較して、より穏和なもので十分であり、例えば、マグネティックスターラーを用いる場合、回転数を200rpm以上、好ましくは300〜500rpm程度のものとすることができる。なお、撹拌条件としては、何らこれに限定されるものではない。
【0069】
また、シリコーンオイルと界面活性剤との配合割合としては、特に限定されないが、仕込みモル比で、シリコーンオイル:界面活性剤=1:10-5〜1:10-2程度、より好ましくは、1:10-4〜1:10-2程度とすることが望ましい。
【0070】
また、界面活性剤の水溶液中における濃度としては、特に限定されるものではないが、0.01〜10質量%程度が適当であり、さらに、シリコーンオイルに対する水の仕込み量としても、特に限定されるものではないが、シリコーンオイル1mgに対し、水5〜10ml程度が適当である。
【0071】
以上示したような条件下で、シリコーンオイルを界面活性剤で乳化処理することにより、所定粒子径、例えば、平均粒子径1〜5000nm、代表的には200nm前後のシリコーンオイル液滴が均一に分散してなるO/Wエマルジョンが調製でき、減圧後においても長期間、液滴粒径の変動の少ない安定したエマルジョンとなる。
【0072】
ここで、シリコーンオイルのO/Wエマルジョンを、二酸化炭素による加圧条件下で処理すると、シリコーンオイル液滴の粒径は、加圧処理前のものに比べ、加圧処理終了直後(すなわち、所定時間加圧処理した後減圧した直後)のものの方が、大きくなっている傾向がある。この現象の詳細な作用機序は、明らかではないが、加圧処理終了のシリコーンオイルのO/Wエマルジョンに窒素をバブリングしてやると、バブリング経過時間が長くなるにつれ、シリコーンオイル液滴の粒径の大きさは低下し、一方、曝気することなく密閉状態で放置した場合、長期間経っても、窒素バブリングした場合と比較して、シリコーンオイル液滴の粒径の大きさは明らかに大きく、粒径の低下傾向が小さいことから、エマルジョン中のシリコーンオイル液滴は、二酸化炭素によって膨らんでいるものと思われる。また、シリコーンオイルのO/Wエマルジョンは、二酸化炭素での加圧下(観察のため、撹拌は行っていない)では、オイルリッチな上相と、水が大部分を占める下相とに二相分離し、その後、減圧すると、再び、均一なエマルジョンへと戻っていることが判った。撹拌下においても、微小領域中ではこのような相分離および再均質化が生じているものと思われ、上記したようなシリコーンオイル液滴の粒径変化は、このような相分離および再均質化を経ることによる、再分散の効果も含むものであると思われる。なお、このような現象は、二酸化炭素での加圧処理に特有のものであり、例えば、窒素を用いて同様に加圧処理を行っても、加圧下でシリコーンオイルエマルジョンの相分離現象は生じない。
【0073】
このように二酸化炭素による加圧下に調製されたシリコーンオイルのO/Wエマルジョンは、各種樹脂組成物の撥水性、帯電防止性、柔軟性あるいは潤滑性等を付与することを目的とする改質剤として好適に用いることができるが、上記したような特性の改質に加え、その樹脂組成物の透明度を改善させることが可能であり、特に、ナイロン6、ナイロン66、ナイロン610、ナイロン612、ナイロン11、ナイロン12等のナイロンに代表されるポリアミド樹脂系組成物中への改質剤として好適に使用することが可能である。なお、このような樹脂組成物の透明度の改善効果も、二酸化炭素での加圧処理に特有のものであり、例えば、窒素を用いて同様に加圧処理を行ったものを用いても、樹脂組成物の透明度の改善効果は見られないものである。
【0074】
なお、ポリアミド系樹脂組成物等の樹脂組成物に当該シリコーンオイルのO/Wエマルジョンを添加する場合には、樹脂組成物にシリコーンオイルのO/Wエマルジョンを溶融混練すれば良い。なお、シリコーンオイルのO/Wエマルジョンの添加量としては、透明度を改善しようとする場合、比較的少量であることが好ましく、ポリアミド系樹脂組成物100質量部あたり0.001〜10質量部、代表的には約0.1質量部程度が望ましい。
【実施例】
【0075】
以下、本発明を実施例に基づきより具体的に説明する。
【0076】
<実施例1および比較例1> S−(+)−イブプロフェンの包接化
(予備実験)
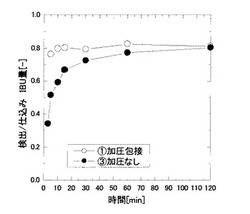
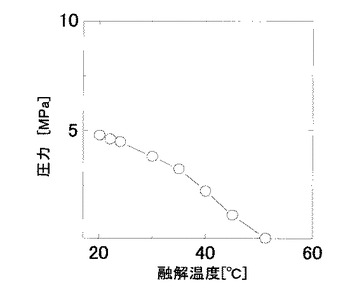
S−(+)−イブプロフェン(以下、IBUと略記する場合もある。)のCO2加圧条件でのp−T線図を図1に示す。図1からCO2加圧条件条件下ではIBUの融点は降下することが確認できた。また、ヒドロキシプロピル−β―シクロデキストリン溶液(以下、HPCD溶液と略記する場合もある。)中においても同様の融点降下が確認された。この図から、IBUがHPCD溶液中で融液として存在する領域を調べ、操作条件として設定した。
【0077】
(実験操作)
HPCD濃度29.6mMのHPCD水溶液10mlと粉末状のIBUを、HPCD:IBU=1:1の仕込みモル比で、ステンレス鋼製の耐圧の反応器に仕込み、系を35℃に保温した。その反応器にポンプでCO2を導入、操作圧である8MPaまで加圧し、マグネチックスターラー(500rpm)を用いて攪拌し包接化を行った(実施例1)。また攪拌時間を反応時間とした。減圧後得られた溶液は、時間を置かず紫外可視分光光度計を用い溶液中のIBU量を測定した。
【0078】
一方、比較のために、二酸化炭素による加圧を行わない系(大気圧:0.1MPa)で同様にIBUの包接化を行った(比較例1)。なお、比較例1では、圧力以外は実施例1と同様の条件で実験を行った。
【0079】
(結果及び考察)
吸光度分析において、水に溶解したIBUと包接されたIBUのスペクトルのピークが同位置に存在するため、検出薬物量は、水への溶解量を含んだ値となるが、水への溶解量は全体の検出IBU量に対して1〜3%とほとんど無視できる値である、そのため、薬物が包接された割合を示す包接度を(包接度=検出IBU量/仕込みIBU量)と定義することにした。
【0080】
図2に、攪拌時間を変化させたときの包接度の経時変化を示す。図2に示す結果から包接平衡に達する時間が大幅に短縮されたことから包接反応が大幅に加速されたことがわかった。
【0081】
<実施例2〜4、比較例2および参考例1〜2> S−(+)−イブプロフェンの包接化
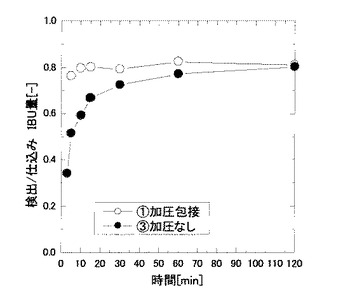
操作圧である二酸化炭素の圧力を、0.1MPa(比較例2)、2MPa(参考例1)、4MPa(実施例2)、6MPa(実施例3)、8MPa(実施例4)にそれぞれ設定し、撹拌時間を5分間に設定する以外は、実施例1と同様にして包接化処理を行い、圧力条件の変化による包接度の変化を調べた。その結果を図3に示す。
【0082】
実験時の温度(35℃)におけるIBUの融解圧力は、3.25MPaであり、図3より、融解圧力前後において包接度の上昇が確認された。このことから、加圧によりIBUが融解し、融液の状態でHPCD溶液と接触したことにより、包接効率が上昇したものと考えられた。
<実施例5および比較例3> S−(+)−イブプロフェンの包接化
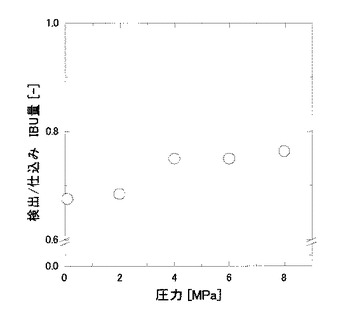
撹拌速度の包接化への影響を調べるため、撹拌時間を5分間に設定し、マグネチックスターラーの回転数を種々変動させる以外は、実施例1および比較例1と同様にして、撹拌速度の変化による包接度の変化を調べた。その結果を図4に示す。
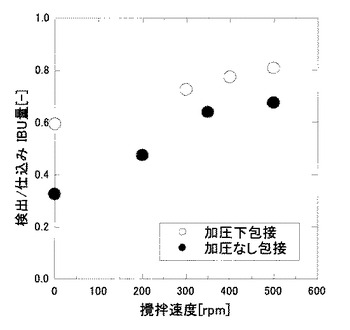
【0083】
図4に示すように、攪拌速度の上昇とともに包接度の値も上昇した。このことから、IBUとHPCDの接触効率の上昇によって包接効率も上昇することが確認された。また、全ての攪拌速度において、実施例である加圧条件下における包接度の方が、比較例である非加圧条件下におけるものよりも高い値を示した。このことは、融液を攪拌することによって微小な液滴が生成され、それによって界面積が増え、包接効率が上昇したためであると考えられた。
【0084】
<実施例6> S−(+)−イブプロフェンの包接化
撹拌装置として、マグネチックスターラーに代えて、スタティックミキサー(日本フローコントロール社製、型番85−212)を用いた以外は、実施例1と同様の条件下において、包接化処理を行った。
【0085】
その結果、スタティックミキサーを用いた実施例6においては、マグネチックスターラーを用いた実施例1と比較して、より微小な液滴が形成され界面積が増大したことが確認され、スタティックミキサーを用いることでより効率的かつ良好な包接化がなされ得ること明らかとなった。
【0086】
<実施例7〜10> シリコーンオイルのエマルジョン
まず、市販のジメチルシリコーンオイルの非イオン性界面活性剤によるエマルジョン(松本シリコーンソフナー#302、松本油脂株式会社製)におけるオイル液滴の平均粒子径を、ダイナミック光散乱高度計(NICOMP 380ZLS Zeta potential/Particle sizer、パーティクル・サイジング・システムズ社製)(以下、DLSと略記することもある。)を用いて測定した。その結果、このエマルジョンにおける、オイル液滴の平均粒子径は182.1nmであった。
【0087】
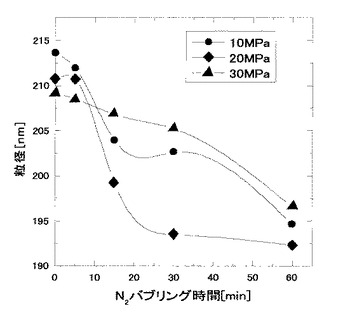
次いで、このジメチルシリコーンオイルエマルジョンを、ステンレス鋼製の耐圧の反応器に仕込み、系を50℃に保温した。マグネチックスターラー(150rpm)を用いて攪拌しながら、その反応器にポンプでCO2を導入、操作圧である10、20、30MPaまでそれぞれ加圧し、15分間撹拌処理を行った。減圧後得られたエマルジョンにおけるオイル液滴の平均粒子径を直ちに、上記と同様にDLSにより測定したところ、図5に示すように、いずれの加圧条件でも、オイル液滴の平均粒子径は210nmに近いものであり、加圧処理前のものよりもエマルジョンにおけるオイル液滴の平均粒子径が増大していた。
【0088】
さらに、このオイル液滴の平均粒子径の加圧処理前後における変動の要因を調べるため、加圧処理により得られたエマルジョンをN2でバブリング処理し、バブリング時間が5分、10分、15分、30分、および60分となった時点で、エマルジョンにおけるオイル液滴の平均粒子径をDLSにより測定した。一方で、加圧処理により得られたエマルジョンを、少量(3ml)サンプル瓶に採取し、曝気することなく密閉して1週間放置した後、エマルジョンにおけるオイル液滴の平均粒子径を同様にDLSにより測定した。
【0089】
その結果、図5に示すように、いずれの圧力条件により処理したものも、N2でバブリング処理した場合、時間が経過するに従って、マルジョンにおけるオイル液滴の平均粒子径は低下した。一方で、密閉放置後のものにおいては、操作圧10、20、30MPaで加圧処理したものそれぞれにつき、208.2nm、201.1nm、202.7nmといずれも平均粒子径として200nm以上の値を保っており、N2でバブリング処理した場合と比べ、明らかに、平均粒子径の低下傾向が小さいものであった。このことから、二酸化炭素による加圧処理を行った場合、シリコーンオイルのエマルジョンにおけるオイル液滴が二酸化炭素により膨らんでいるものと思われた。
【0090】
<参考例3> 二酸化炭素加圧下におけるシリコーンオイルエマルジョンの挙動
二酸化炭素による加圧下におけるシリコーンオイルエマルジョンの挙動を調べるために、光学窓付平衡セル内に、市販のジメチルシリコーンオイルの非イオン性界面活性剤によるエマルジョン(松本シリコーンソフナー#302、松本油脂株式会社製)を入れ、50℃まで昇温させた後、二酸化炭素によって10mPaまで加圧し、その状態を30分間維持した後、20mPaまで昇圧し、その状態でさらに30分間維持した後、減圧し、各時間におけるエマルジョンの状態を観察した。その結果を、図6に示す。図6に示されるように、加圧状態においては単一相であったエマルジョンが二相に分離し、減圧することによって再び単一相のエマルジョンとなっていた。
【0091】
なお、別途、エマルジョンに赤色水性インキを添加して着色し、同様の実験を行ったところ、加圧状態における相分離エマルジョンの上相は着色しておらす、一方下相は加圧とともに白濁が薄くなり赤色透明に近くなったことから、上相はシリコーンオイルが大部分を占め、下相は水が大部分を占めるエマルジョンであると判断できた。
【0092】
得られた結果から、上記したようなシリコーンオイル液滴の粒径変化は、このような相分離および再均質化を経ることによる、再分散の効果も含むものであると思われた。
【0093】
<比較例4> 窒素加圧下におけるシリコーンオイルエマルジョンの挙動
比較のために、二酸化炭素に変えて窒素により加圧する以外は、参考例3と同様にして、シリコーンオイルエマルジョンを加圧処理し、窒素加圧下におけるシリコーンオイルエマルジョンの挙動を調べた。その結果を図7に示す。
【0094】
図7に示すように、窒素による加圧においては、参考例3におけるようなシリコーンオイルエマルジョンの相分離は生起せず、エマルジョンの相分離は二酸化炭素加圧に特有なものであることが判明した。
【0095】
<実施例11〜12および比較例5> 二酸化炭素加圧処理後におけるシリコーンオイルエマルジョンの特性
二酸化炭素加圧処理前後のシリコーンエマルジョンの粘度および濡れ性を調べた。
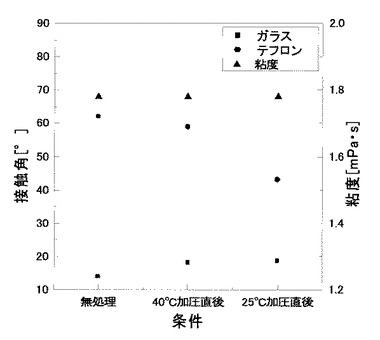
まず、市販のジメチルシリコーンオイルの非イオン性界面活性剤によるエマルジョン(松本シリコーンソフナー#302、松本油脂株式会社製)を、ステンレス鋼製の耐圧の反応器に仕込み、系を25℃(実施例11)、40℃(実施例12)にそれぞれに保持し、マグネチックスターラー(800rpm)を用いて攪拌しながら、その反応器にポンプでCO2を導入、操作圧である12MPaまでそれぞれ加圧し、15分間撹拌処理を行ったのち、減圧した。このようにしてそれぞれ得られた二酸化炭素加圧処理直後のシリコーンオイルエマルジョンと、未処理の原料であるシリコーンオイルエマルジョン(比較例5)に関して、音叉型振動式粘度計(VIBRO VISCOMETER SV-10、株式会社エー・アンド・デイ製)を用いて粘度測定を行った。また、θ/2法を用いて、ガラスおよびテフロン(登録商標)表面における接触角を測定した。得られた結果を、図8に示す。
【0096】
図8に示すように、本発明の実施例に係るシリコーンオイルエマルジョンは、二酸化炭素加圧処理を施さなかった比較例のシリコーンオイルエマルジョンと比較して、粘度特性およびガラスに対する濡れ性はあまり変わらないものの、テフロン(登録商標)に対する濡れ性が大きく向上しているのが判った。
【0097】
<実施例13〜14および比較例5> ポリアミド系樹脂製品
上記実施例11〜12におけるものと同様の条件下で二酸化炭素加圧処理したシリコーンオイルエマルジョンを、原料ナイロン(ナイロン6:ナイロン66=4:1の質量比のブレンド物)と溶融混練した後、押出し成形を行い直径3mmの紐状に成形した(実施例13〜14)。なお、シリコーンオイルエマルジョンの添加量は、原料ナイロン1kgに対し、それぞれ1g、3gおよび5gとし、また押出条件としては押出回転速度270rpm、押出温度230℃とした。
【0098】
一方比較のために、加圧未処理のジメチルシリコーンオイルエマルジョン(松本シリコーンソフナー#302、松本油脂株式会社製)を上記と同様の条件下で原料ナイロンと溶融混練した後、押出し成形を行い紐状に成形した(比較例5)。
【0099】
得られた紐状成形品に関し、その透明度および結晶化度を測定した。
【0100】
なお、透明度は、各紐状成形品より長さ18.6mmの試料を切り出し、この試料の一端部に光源を配置し、他端部に光量子センサを配置すると共に、試料の側周面を黒色テープで覆うことにより構成してなる透明度定量化装置を用い、試料を通過した光の強さ(I)[mA]と、同距離でのブランク(中空)にした場合に計測される光の強さ(I0)[mA]とを測定し、以下の式により透過度(%)を算出した。
【0101】
透過度(%)=(I/I0)×100
また、結晶化度Xcについては、示差走査熱量測定法(DSC法)に基づき、以下の式より算出した。
【0102】
結晶化度Xc=(Hc/Hc0)×100
(Hc:DSC曲線の融解熱[J/g」、Hc0:結晶化度100%の場合の融解熱[J/g」)
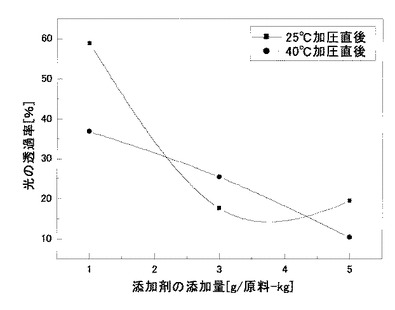
その結果、比較例5のもの(透過率0〜3%)に比べ、実施例13〜14のものは、いずれも透過率の向上が見られ、特に図9に示すように、シリコーンオイルエマルジョンの添加量が1gであった場合には、非常に良好な透明性の改善効果が見られた。
【0103】
なお、結晶化度については、比較例5および実施例13〜14のものいずれにおいても、約20%前後であり、その有意差は見られず、透明性の改善効果は、二酸化炭素加圧処理のジメチルシリコーンオイルエマルジョンの添加によるものであるが、結晶化度の変化によるものではないことが考えられた。
【図面の簡単な説明】
【0104】
【図1】は、S−(+)−イブプロフェンのCO2加圧条件での融点変動を示すp−T線図である。
【図2】は、攪拌時間を変化させたときの、S−(+)−イブプロフェンのヒドロキシプロピル−β―シクロデキストリンによる包接度の経時変化を示すグラフである。
【図3】は、二酸化炭素による加圧条件を変化させたときの、S−(+)−イブプロフェンのヒドロキシプロピル−β―シクロデキストリンによる包接度の変化を示すグラフである。
【図4】は、撹拌速度を変化させたときの、S−(+)−イブプロフェンのヒドロキシプロピル−β―シクロデキストリンによる包接度の変化を示すグラフである。
【図5】は、二酸化炭素加圧処理を行ったシリコーンオイルエマルジョンに対し、窒素バブリングを行った場合における、エマルジョン中のオイル液滴の平均粒子径の経時変化を示すグラフである。
【図6】は、二酸化炭素加圧下におけるシリコーンオイルエマルジョンの状態変化を示す写真図である。
【図7】は、窒素加圧下におけるシリコーンオイルエマルジョンの状態変化を示す写真図である。
【図8】は、二酸化炭素加圧処理前後のシリコーンエマルジョンの粘度および濡れ性の変化を示すグラフである。
【図9】は、二酸化炭素加圧処理されたシリコーンオイルエマルジョンを添加したナイロン樹脂製品の光透過率とエマルジョンの添加量との関係を示すグラフである。
【技術分野】
【0001】
本発明は、微細粒状体の製造方法に関するものである。詳しく述べると本発明は、高圧二酸化炭素により加圧することにより、分散媒中において各種薬剤の微細粒状体を形成する方法に関するものである。
【背景技術】
【0002】
現在、医療・医薬分野で製造されている薬物の多くは、水に対する溶解性が低く、そのため薬物のバイオアベイラビリティ(生物学的利用能)が大幅に低下するという問題が生じている。このような難溶性薬物の溶解性改善の方法の一つに、シクロデキストリン(CD)及びその誘導体と薬物の包接化がある。しかし、従来の方法においては、難溶性薬物とCD溶液の包接化は、薬物を有機溶媒等に溶解させた状態でCD溶液と包接化するという問題があり、医療・医薬分野において使用する薬剤の処理方法として適当なものであるとはいい難いものである。
【0003】
また、例えば、樹脂組成物や塗料組成物等の中に、可塑剤、各種安定化剤、結晶核剤等の各種添加剤を添加しようとする場合、これらの添加剤を組成物中に均一かつ微分散させることが樹脂組成物や塗料組成物の特性向上の上で大きな鍵となる。
【0004】
ところで、従来、超臨界状態の二酸化炭素を用いて各種物質の微細粒状体を形成することが、広く研究されている。
【0005】
例えば、特許文献1には、金属ナノ粒子の製造方法として、亜臨界ないし超臨界の二酸化炭素を反応媒体とし、ビス(2−エチルヘキシル)スルホコハク酸ナトリウムおよび2,3,3,3,4,4,5,5−オクタフルオロ−1−ペンタノールを活性剤として使用し、水/二酸化炭素からなる逆ミクロエマルジョンを形成し、ミセル中に含まれる金属イオンを、ナトリウムトリアセトキシボロハイドライト等の還元剤により還元して金属ナノ粒子を得る方法が開示されている。
【0006】
また、特許文献2には、圧力容器内で40〜65℃、10〜30MPaで、超臨界ニ酸化炭素、リン脂質等のリポソーム膜構成成分、医薬化合物および製薬助剤を混合して懸濁液を得、次いで、系内を減圧して二酸化炭素を排出することにより、該医薬化合物が内包されたリポソームの水性分散液を調製し、その後前記リン脂質の転移温度〜(転移温度+10)℃で、所定時間インキュベートしてリポソーム含有製剤を製造することが開示されている。
【0007】
特許文献3には、界面活性物質が樹脂粒子中または表面に存在せず、かつ粒度分布がシャープな樹脂粒子を得る方法として、樹脂(b)の溶融液または樹脂(b)の溶剤溶液、あるいは樹脂(b)の前駆体(b0)またはその溶剤溶液を、無機ないし有機の各種微粒子(A)が分散している液状又は超臨界状態の二酸化炭素(X)からなる分散媒体(X0)に分散して得られた微粒子(C1)の分散体(X1)を、(必要に応じて(b0)を重合反応させた後)、圧力を減圧することにより分散体(X1)から(X)を除くことにより樹脂粒子(C)を得ることを特徴とする、微粒子(A)が樹脂(b)からなる樹脂粒子(B)の表面に付着されてなる樹脂粒子(C)の製造方法が、また、特許文献4には、親水性成分が樹脂粒子中または表面に存在せず、かつ粒度分布がシャープな樹脂粒子を得る方法として、樹脂(b)もしくは樹脂(b)の溶剤溶液、または樹脂(b)の前駆体(b0)またはその溶剤溶液を、液状又は超臨界状態の二酸化炭素(X)中に分散させ、圧力を減圧することにより液状又は超臨界状態の二酸化炭素(X)を除去することにより、樹脂粒子を製造する方法において、ジメチルシロキサン基及びフッ素を含有する少なくとも一方の基を有する分散安定剤(D)と無機ないし有機の各種微粒子(A)が樹脂(b)からなる樹脂粒子(B)の表面に付着されてなる樹脂粒子(C)の製造方法が開示されている。
【0008】
また、特許文献5には、超臨界状態の二酸化炭素流体を用い常温常圧下で微粒子を塗布して被塗布物の表面上に超微粒子膜を形成する方法であって、真空状態ないし真空状態に近い状態とした高圧容器に数十μm以下の微粒子である微粒子を注入し、超臨界状態の二酸化炭素流体を注入し、それらを撹拌して超臨界状態の二酸化炭素を分散媒体とする微粒子の高分散状態になった混合物とし、該混合物をノズルから被塗物に噴射することで膜を形成する微粒子膜形成方法を開示している。
【0009】
このように、従来、各種薬剤の微細粒状体を形成する上で、超臨界状態の二酸化炭素を分散媒として使用することが行われているが、上記したような包接化において超臨界状態の二酸化炭素を用いることは行われていなかった。
【0010】
また、例えば、樹脂組成物の調製方法として、上記したように界面活性剤等を用いることなく、樹脂成分と添加剤とを超臨界状態の二酸化炭素中で直接分散させようとする試みは行われていたものの、添加剤を微細な粒状体として分散配合することが十分になされず、添加剤による改質効果の向上に限度があるものであった。
【特許文献1】特開2005−290481号公報
【特許文献2】特開2006−298844号公報
【特許文献3】特開2006−321830号公報
【特許文献4】特開2007−277551号公報
【特許文献5】特開2008−12448号公報
【発明の開示】
【発明が解決しようとする課題】
【0011】
従って本発明は、上記したような従来技術における問題点を鑑み、分散媒中において難溶性ないし非溶性の各種薬剤の高度かつ均一な微細粒状体を形成する方法を提供することを課題とするものである。
【0012】
本発明はさらに、有機溶媒を用いることなく、水に難溶性ないし非溶性の各種薬剤を包接化により効率よく、微細粒状体に形成する方法を提供することを課題とするものである。
【0013】
本発明はまた、樹脂組成物や塗料組成物等の各種組成物中に添加する各種添加剤を安定な微細粒状体の形態とし、これを組成物中に配合することで組成物の高い改質効果を得ることのできる微細粒状体の形成方法を提供することを課題とするものである。
【課題を解決するための手段】
【0014】
上記課題を解決する本発明は、分散媒に対して難溶性ないし非溶性のゲスト分子を、ホスト分子により包接するあるいは界面活性剤により乳化することによって、分散媒中にゲスト分子の微細粒状体を製造する方法であって、圧力容器中に、ゲスト分子、およびホスト分子ないし界面活性剤を含む分散媒を仕込み、この圧力容器に二酸化炭素を導入して、1〜100MPaの操作圧まで加圧し、所定時間撹拌を行なって包接あるいは乳化処理した後、圧力容器内を減圧して二酸化炭素を排出することにより、分散媒中のゲスト分子の微細粒状体を製造することを特徴とするものである。
【0015】
本発明はまた、撹拌が静止型混合機により行われるものである微細粒状体の製造方法を示すものである。
【0016】
本発明はまた、分散媒が水ないし水系溶液である上記微細粒状体の製造方法を示すものである。
【0017】
本発明はさらに、ゲスト分子が標準状態(SATP、25℃、105Pa)において、固体状のものである微細粒状体の製造方法を示すものである。
【0018】
本発明はさらに、ゲスト分子が、S−(+)−イブプロフェン、プロゲステロン、ケトプロフェンおよびグリセオフルビンからなる群から選ばれてなるいずれか1つの化合物である微細粒状体の製造方法を示すものである。
【0019】
本発明はさらに、ホスト分子がシクロデキストリンおよびその誘導体からなる群から選ばれてなるものである微細粒状体の製造方法を示すものである。
【0020】
本発明はまた、ゲスト分子が樹脂ないしゴム成分を含有する組成物用の添加剤である微粒粒状体の製造方法を示すものである。
【0021】
本発明はさらに、ゲスト分子がシリコーンオイルである微粒粒状体の製造方法を示すものである。
【0022】
上記課題を解決する本発明はまた、上記の製造方法により得られた分散媒中のゲスト分子の微細粒状体を樹脂組成物中に添加することを特徴とする樹脂組成物の製造方法である。
【0023】
本発明はまた、樹脂組成物がポリアミド系樹脂組成物であり、シリコーンオイルであるゲスト分子の微細粒状体を添加混合し、これを成形加工することを特徴とする、透明度の改善されたポリアミド系樹脂製品の製造方法を示すものである。
【0024】
本発明はさらに、当該製造方法により得られたことを特徴とする透明度の改善されたポリアミド系樹脂製品を示すものである。
【0025】
本発明はまた、前記製造方法により得られた、分散媒中のゲスト分子の微細粒状体を、さらに他の液状体に添加吸収させることを特徴とする微細粒状体分散体の製造方法を示すものである。
【0026】
本発明はさらに、前記製造方法により得られた、分散媒中のゲスト分子の微細粒状体を、被塗物表面へと吹きつけることを特徴とする微細粒状体被膜の形成方法を示すものである。
【発明の効果】
【0027】
本発明によれば、分散媒中において難溶性ないし非溶性の各種薬剤の高度かつ均一な微細粒状体を形成することができ、例えば、水に対して難溶性ないし非溶性のゲスト分子を、有機溶媒を用いることなく、ホスト分子により包接化して微細粒状化することができ、医療・医薬分野で製造されている薬物の溶解性を改善し、薬物のバイオアベイラビリティを大きく向上させることができる。
【0028】
また、本発明によれば、樹脂組成物や塗料組成物等の各種組成物中に添加する各種添加剤を安定な微細粒状体の形態とし、これを組成物中に配合することで組成物の高い改質効果を得ることのできる。具体的には例えば、ナイロン等のポリアミド系樹脂組成物に、撥水性、帯電防止性、柔軟性あるいは潤滑性等を付与することを目的とする改質剤としてシリコーンオイルを添加しようとする場合に、当該シリコーンオイルを本発明の製造方法により微細粒状化して、樹脂組成物に配合することで、上記した所期の特性の改質効果に加え、樹脂組成物製品の透明性を大きく改善できる。
【発明を実施するための最良の形態】
【0029】
以下、本発明を実施形態に基づき、より詳細に説明する。
【0030】
使用原料等
本発明において用いられる分散媒としては、特に限定されるものではないが、医療・医薬分野で用いられるゲスト分子を取り扱う場合、また、環境保全性の観点からは、水、あるいは、水にメタノール、エタノール、イソプロパノール等の低級アルコール、酸、アルカリ、塩等の水溶性ないし水混和性化合物を添加してなる水系溶媒を用いることが望ましく、特に水が好ましい。
【0031】
また、分散媒に対して難溶性ないし非溶性のゲスト分子としても特に限定されるものではなく、各種の薬剤を用いることができる。なお、本明細書において、「分散媒に対して難溶性ないし非溶性」とは、一般に難溶性ないし非溶性と言われるものであれば包含され得、厳密に規定されるものではないが、例えば、標準状態(SATP、25℃、105Pa)の分散媒(特に、水)に対する溶解度が1μg/mL程度以下のものが少なくとも含まれるものである。
【0032】
このような、分散媒に対して難溶性ないし非溶性のゲスト分子として、具体的には、例えば、医療・医薬分野における解熱剤、抗炎症剤、鎮痛剤、精神安定剤、鎮静剤、抗腫瘍剤、抗菌剤、抗生物質、抗高脂血症剤、鎮咳去たん剤、筋弛緩剤、抗てんかん剤、抗潰瘍剤、抗うつ剤、抗アレルギー剤、強心剤、不整脈治療剤、血管拡張剤、降圧利尿剤、糖尿治療剤、抗結核剤、抗リウマチ剤、ステロイド剤、麻薬拮抗剤、ホルモン剤、脂溶性ビタミン剤、抗凝血剤、虚血性疾患治療薬、免疫疾患治療薬、アルツハイマー病治療薬、脳血管攣縮治療薬、脳血栓治療薬、緑内障治療薬、高眼圧症治療薬、関節炎治療薬、抗セプシス薬、抗セプティックショック薬、抗喘息薬、頻尿・尿失禁治療薬、アトピー性皮膚炎治療薬、アレルギー性鼻炎治療薬、動物薬組成物等、また、各種樹脂組成物、塗料組成物、インキ組成物等に添加される、可塑剤、各種安定化剤、結晶核剤、ワックス成分、消泡剤、難燃化剤、離型剤、有機着色剤等の各種添加剤、その他、化粧料組成物、農薬組成物、殺虫剤、殺菌剤、除草剤、飲食用組成物等を挙げることができるが、これらに何ら限定されるものではない。
【0033】
また、分散媒に対して難溶性ないし非溶性のゲスト分子としては、例えば、標準状態(SATP、25℃、105Pa)において、固体状のものであり、加圧により融点降下を生じ、本発明の製造方法における製造条件下において融液状態となり得る物質が、好ましい例として挙げることができる。このような物質は、従来の方法においては、微小粒状体とすることが困難であり、本発明の製造方法を用いることが、特に有用なものであるからである。
【0034】
このような標準状態(SATP、25℃、105Pa)において、固体状の難溶性ないし非溶性のゲスト分子としては、具体的には、例えば、S−(+)−イブプロフェン、プロゲステロン、ケトプロフェン、グリセオフルビン等の薬剤を例示することができるが、何らこれらに限定されるものではない。
【0035】
また、ゲスト分子が樹脂用添加剤である場合、好ましい一例としては、ジメチルシリコーンオイル、メチルフェニルシリコーンオイル、各種変性シリコーンオイル等のシリコーンオイルを挙げることができるが、もちろん何らこれらに限定されるものではない。
ルことが知られている。
【0036】
一方、このようなゲスト分子を包接するホスト分子としては、特に限定されるものではないが、例えば、各種天然型シクロデキストリンおよび各種化学修飾型シクロデキストリン、各種クラウンエーテル、各種シクロファン、各種カリックスアレーン等を挙げることができ、なかでも好ましいものとして、α―シクロデキストリン、β―シクロデキストリン、γ―シクロデキストリン、δ―シクロデキストリンおよびこれらを化学修飾したもの等のシクロデキストリンおよびその誘導体を挙げることができる。ここで言う化学修飾とは、各種シクロデキストリン中の水酸基をアルキル化、ヒドロキシアルキル化したもの等を指す。この時のアルキル基としては、直鎖状、分枝状のいずれでも良く、その構造中に不飽和結合を含むものでも良いが、好ましいものとしてメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、t−ブチル基、ヒドロキシメチル、ヒドロキシエチル、ヒドロキシプロピル等を挙げることができ、このような各種化学修飾型シクロデキストリンのなかでも、ヒドロキシプロピル−β−シクロデキストリンを、特に好ましいものとして挙げることができる。
【0037】
なお、ゲスト分子をホスト分子で包接する態様において、ゲスト分子とホスト分子との配合比としては、使用するゲスト分子およびホスト分子の種類等によっても異なるものであり、特に限定されるものではないが、例えば、ゲスト分子とホスト分子の仕込みモル比で、ゲスト分子:ホスト分子=1:0.5〜1:3.5程度、より好ましくは、1:0.5〜1:2.5程度とすることが望ましい。
【0038】
また、ゲスト分子を乳化する界面活性剤としても、特に限定されるものではなく、また使用するゲスト分子および分散媒の種類によっても左右されるものであるが、例えば、ポリオキシエチレンポリオキシプロピレングリコール、ポリオキシエチレン硬化ヒマシ油、ポリオキシエチレンラウリルエーテル等のポリオキシエチレンアルキルエーテル、ポリオキシエチレンソルビタン脂肪酸エステル(ポリソルベート)、モノステアリン酸ソルビタン等のソルビタン脂肪酸エステル等の非イオン性界面活性剤、塩化ベンザルコニウム、塩化ベンゼトニウム、塩化セチルピリジニウム等のアニオン性界面活性剤、ステアリン酸カルシウム、ステアリン酸マグネシウム、ラウリル硫酸ナトリウム等のカチオン性界面活性剤等が挙げられる。また、アンモニウムカルボキシレートパーフルオロエーテル等のフッ素基を有する界面活性剤等が挙げられる。
【0039】
製造条件等
本発明の微細粒状体の製造方法においては、上記したような分散媒に対して難溶性ないし非溶性のゲスト分子の微細粒状体を製造するにおいて、平衡セル等の圧力容器中に、当該ゲスト分子、並びにこれを包接するホスト分子またはこれを乳化する界面活性剤および分散媒を仕込み、この圧力容器に高圧の二酸化炭素、好ましくは亜臨界ないし超臨界状態の二酸化炭素を導入して加圧し、所定時間撹拌下に保持することで、ゲスト分子の包接化ないしは乳化により、分散媒中にゲスト分子の微細粒状体を形成した後、圧力容器内を減圧して二酸化炭素を排出することにより、ゲスト分子の微細粒状体の分散液を得るものである。
【0040】
ここで、ゲスト分子が、標準状態(SATP、25℃、105Pa)において、固体状のものである場合には、これを包接するホスト分子またはこれを乳化する界面活性剤および分散媒と混合するに先立ち、予め、二酸化炭素により加圧した後、さらに、二酸化炭素の加圧下で、ホスト分子又はこれを乳化する界面活性剤および分散媒と撹拌下に混合しても良い。すなわち、このような標準状態で固体状のゲスト分子、例えば、S-(+)-イブプロフェンは、二酸化炭素による加圧で、融点降下を生じることが知られており(J.Chem and Eng Data.,2005)、予め、二酸化炭素により加圧することで融解し融液状態とした後、包接化ないし乳化するほうが、効率良く、微小粒状体化することができるためである。
【0041】
本発明の製造方法において、微小粒状体化を行う際の二酸化炭素による加圧条件としては、用いられるゲスト分子の種類、操作時の温度条件等によっても左右されるが、例えば、1〜100MPa、より好ましくは1〜50MPa、最も好ましくは1〜40MPaであることが望ましい。圧力が1MPa未満であると、良好な微小粒状体を形成できないためであり、一方、圧力が100MPaを超えるものであると、設備上および運転操作上で不利となるためである。
【0042】
なお、二酸化炭素の超臨界状態とは、二酸化炭素の臨界点(31.1℃、7.38MPa)を超える温度・圧力条件下にある二酸化炭素を指し、亜臨界状態の二酸化炭素とは、圧力または温度のいずれかのみが臨界点を越えた状態にある二酸化炭素(本発明に関しては主として圧力のみが臨界点を超えた状態である。)を指し、また液状の二酸化炭素とは、二酸化炭素の三重点(−56.6℃、0.518MPa)と二酸化炭素の臨界点を通る気液境界線、臨界温度の等温線、及び固液境界線に囲まれた部分の温度・圧力条件下にある二酸化炭素を指すものである。
【0043】
また、本発明の製造方法において、二酸化炭素による加圧下で微小粒状体化を行う際の温度条件としては、用いられるゲスト分子の種類、操作時の二酸化炭素の圧力条件等にも左右され、特に限定されるものではないが、例えば、0〜200℃、より好ましくは0〜180℃、さらに好ましくは0〜150℃とすることが望ましい。
【0044】
また、本発明においては、このように二酸化炭素による加圧下において、ゲスト分子を、ホスト分子又はこれを乳化する界面活性剤および分散媒と撹拌下に混合するが、この撹拌装置としては、例えば、マグネティックスターラーや各種撹拌子を備え機械的に回転駆動される撹拌型混合機や、管内に混合エレメントを有してなる静止型混合機(スタティックミキサー)等を用いることができるが、このうち、静止型混合機を用いることが望ましい。
【0045】
ここで、本明細書において述べる静止型混合機(スタティックミキサー)とは、「流体通路の構成によって、レイノルズ数の広範囲にわたって流体混合ができるとともに、機械的可動部を持たない流体通路の構造体」であり、分割、転換、反転の3つの混合原理により撹拌を行う混合機である。
【0046】
静止型混合機としては、例えば、日本フローコントロール株式会社製のスパイラルタイプスタティックミキサー、特に、スタティックミキサー 85シリーズ等を好ましい例として挙げることができるが、これらに何ら限定されるものではない。
【0047】
本発明の製造方法、殊に、ゲスト分子をホスト分子により包接する態様においては、この撹拌が十分なものでないと、加圧条件下においても、ゲスト分子の流体が分散媒中であまり広がらず、包切度が低下して、良好な微細粒状体が得られない虞れがあるために、十分な撹拌効果が得られる静止型混合機を用いることが特に望ましい。
【0048】
なお、撹拌装置として、マグネティックスターラーや機械的に回転駆動される撹拌型混合機を用いる場合においても、十分な撹拌がなされるように、撹拌子の形状や撹拌速度を適切なものとすることが望ましく、例えば、マグネティックスターラーの場合、撹拌子として星型等の複雑形状のものを用いたり、圧力容器の容積に比して通常より大きい撹拌子を用いる、あるいは回転数を高速、具体的には例えば、500rpm以上、好ましくは800rpm〜1000rpmのものとするなどとして、撹拌時の分散媒液面形状が大きく変化するないし渦流が形成される状態とすることが望ましい。
【0049】
このように二酸化炭素による加圧条件下で、ゲスト分子を、ホスト分子又はこれを乳化する界面活性剤および分散媒と撹拌下に所定時間、例えば、1〜60分間、より好ましくは1〜30分間混合することによって、ゲスト分子のホスト分子による包接化、あるいはゲスト分子の界面活性剤による乳化が促進され、ゲスト分子の均一な微細粒状体、具体的には例えば、平均粒子径1〜105nm、より好ましくは5〜5000nmを分散媒中に形成することができる。
【0050】
その後、圧力容器内を減圧して二酸化炭素を排出することにより、分散媒中のゲスト分子の微細粒状体の分散液を取り出す。
【0051】
このようにして取り出されたゲスト粒子の微細粒状体の分散液は、そのまま分散液として回収する、あるいは必要に応じて、他の液状体、例えば、エチレングリコール、グリセリン、流動パラフィン、水等に添加吸収させたり、そのまま、被塗物表面へと吹きつけることにより微細粒状体被膜を形成する等の処理にかけられることができる。
【0052】
得られたゲスト粒子の微細粒状体の分散液は、ゲスト分子が医療・医薬分野における薬剤である場合、そのまま、あるいは生理食塩水、リン酸緩衝溶液等の水系溶媒中で、他の成分と容易に調合されることで、所定の医薬組成物へと調製でき、その後、必要に応じて、乾燥、賦形化等の処理を施される。
【0053】
また、ゲスト分子が各種樹脂組成物、ゴム組成物、塗料組成物、インキ組成物等の樹脂ないしゴム成分を含有する組成物用の添加剤である場合にも、これら組成物中に、そのまま、あるいは上記したように、他の液状体に添加吸収させた後、溶融混練、溶液ないし分散媒中で混合する等により、配合されることで、組成物中に均一かつ微分散化させることができ、優れた改質効果を発揮することができる。
【0054】
なお、この場合の樹脂ないしゴム成分を含有する組成物としても、特に限定されるものではなく、例えば、ポリオレフィン樹脂系、ポリスチレン樹脂系、ポリ塩化ビニル樹脂系、ポリ塩化ビニリデン樹脂系、ポリアクリロニトリル樹脂系、ポリアミド樹脂系、ポリエーテルイミド系、ポリアミドイミド系、ポリエステル樹脂系、ポリカーボネート樹脂系、ポリアセタール樹脂系、酢酸ビニル樹脂系、ポリビニルアセタール系(例えば、ボリビニルブチラール樹脂)、熱可塑性ポリウレタンエラストマー系、アクリル樹脂系、ポリフェニレン樹脂系、フッ素樹脂系、ポリビニルアルコール系、ポリビニルピロリドン、セルロース樹脂系、変性セルロース樹脂系、フェノール樹脂系、尿素樹脂系、メラミン樹脂系、フラン樹脂系、アルキド樹脂系、不飽和ポリエステル樹脂系、ジアリルフタレート樹脂系、エポキシ樹脂系、シリコーン樹脂系、ポリイミド樹脂系、ポリウレタン樹脂系、グアナミン樹脂系及びフッ素樹脂系、天然ゴム系、ポリブタジエンゴム系、 スチレンブタジエンゴム系、ポリイソプレンゴム系、クロロプレンゴム系、アクリロニトリルブタジエンゴム系、ブチルゴム系、エチレンプロピレンゴム系、アクリルゴム系、ウレタンゴム系、エピクロヒドリンゴム系、フッ素ゴム系、シリコーンゴム系等、あるいはこれらの二種以上の混合系などの公知の各種の組成物が含まれる。
【0055】
なお、本発明に係る製造方法により得られたゲスト粒子の微細粒状体の分散液は、減圧して二酸化炭素を排出した後においても、比較的安定してその微細粒状体を維持することができ、例えば、密閉容器中で1週間保存後において、その平均粒子径の変動率が10%以下程度、1月保存後においても、20%以下程度となり得るものである。
【0056】
具体的態様1
次に、本発明に係る微細粒状体の製造方法につき、好ましい具体的態様に基づき、さらに詳細に説明する。
【0057】
本発明の微細粒状体の製造方法を適用する好ましい一例として、ゲスト分子であるS−(+)−イブプロフェンのホスト分子であるシクロデキストリンないしその誘導体による包接化を挙げることができる。なお、この場合の分散媒としては、水を用いることが好ましい。
【0058】
S−(+)−イブプロフェンを二酸化炭素で加圧した場合、S−(+)−イブプロフェンの融解温度と圧力との間には、次のような関係が見られる。
【0059】
【表1】
S−(+)−イブプロフェンを、シクロデキストリンないしその誘導体との共存下においた場合においても、S−(+)−イブプロフェンのみの場合とほぼ同様な融解温度低下が見られ、包接化処理の処理温度条件下で、少なくともS−(+)−イブプロフェンの融解が生じる圧力以上の圧力まで加圧することによって、包接化が迅速に進行し、水に対するS−(+)−イブプロフェンの溶解量を高めることができる。好ましくは、処理温度が0〜50℃、加圧条件が1〜5MPaであり、この場合、撹拌時間としては、撹拌条件にもよるが、1〜60分程度、より好ましくは5〜15分程度とすることが望ましい。すなわち、5分程度で包接が十分に平衡に達し、それ以上の時間撹拌処理を行っても、水に対するS−(+)−イブプロフェンの溶解量はほとんど向上しないかあるいは逆に若干低下してしまうためである。
【0060】
なお、S−(+)−イブプロフェンを、シクロデキストリンないしその誘導体の水溶液と混合するに先立ち、予め、S−(+)−イブプロフェンを上記したような所定条件下にて二酸化炭素により加圧して、融解させた後、同じく所定条件下の二酸化炭素により加圧しながらシクロデキストリンないしその誘導体の水溶液へと撹拌下に混合しても良く、この場合、より迅速かつ良好な包接化を進行させることができる。
【0061】
なお、撹拌条件としては、上記したように静止型混合機を用いるか、あるいはマグネティックスターラーを用いる場合、上記したように、撹拌子として星型等の複雑形状のものを用いたり、圧力容器の容積に比して通常より大きい撹拌子を用いる、あるいはその回転数を500rpm以上、好ましくは800rpm〜1000rpmのものとすることが望ましく、このようにしてS−(+)−イブプロフェンとシクロデキストリンとの接触効率を高めることで、S−(+)−イブプロフェンの微細な液滴が生成し包接効率が上昇することによって、水に対するS−(+)−イブプロフェンの溶解量を高めることができる。
【0062】
また、包接化におけるゲスト分子としてのS−(+)−イブプロフェンとホスト分子としてのシクロデキストリンないしその誘導体との配合割合としては、特に限定されないが、仕込みモル比で、ゲスト分子:ホスト分子=1:0.5〜1:3.5程度、より好ましくは、1:0.8〜1:1.2程度とすることが望ましい。
【0063】
また、シクロデキストリンないしその誘導体の水溶液中における濃度としては、特に限定されるものではないが、1g/1ml程度が適当であり、さらに、水に対するS−(+)−イブプロフェンの仕込み量としても、特に限定されるものではないが、水100mlに対し、S−(+)−イブプロフェン500mg程度が適当である。
【0064】
以上示したような条件下で、S−(+)−イブプロフェンをシクロデキストリンないしその誘導体を包接化処理することにより、減圧後においてもS−(+)−イブプロフェンの固体の析出を見ることなく、S−(+)−イブプロフェンの仕込み量の約80%ないしそれ以上の量を包接化により水に溶解させることが可能である。
【0065】
具体的態様2
次に、本発明の微細粒状体の製造方法を適用する好ましい別の一例として、シリコーンオイルの界面活性剤による乳化、およびこのようにして得られたシリコーンオイルエマルジョンを配合してなるポリアミド系樹脂組成物を挙げることができる。なお、この場合の分散媒としては、特に限定されるものではないが、水を用いることが好ましい。
【0066】
シリコーンオイルと、界面活性剤および分散媒である水を、本発明の製造方法に基づき、圧力容器内で、酸化炭素を導入して加圧し、所定時間撹拌を行って乳化処理することで、シリコーンオイルのO/Wエマルジョンを調製することができる。用いる界面活性剤としては、陰イオン界面活性剤、陽イオン界面活性剤、両性イオン界面活性剤、非イオン界面活性剤のいずれのタイプのものを用いることも可能である。特に限定されるものではないが、陰イオン界面活性剤としては、例えば、各種脂肪酸ナトリウム、各種脂肪酸カリウム、アルファスルホ脂肪酸エステルナトリウム等の脂肪酸系界面活性剤、各種直鎖アルキルベンゼンスルホン酸ナトリウム等の直鎖アルキルベンゼン系陰イオン界面活性剤、各種アルキル硫酸エステルナトリウム、各種アルキルエーテル硫酸エステルナトリウム等の高級アルコール系陰イオン界面活性剤、各種アルファオレフィンスルホン酸ナトリウム等のアルファオレフィン系陰イオン界面活性剤、各種アルキルスルホン酸ナトリウム等のノルマルパラフィン系陰イオン界面活性剤などが例示でき、また陽イオン界面活性剤としては、例えば、各種アルキルトリメチルアンモニウム塩、各種ジアルキルジメチルアンモニウム塩等の第四級アンモニウム塩系陽イオン界面活性剤などが例示でき、また、両性イオン界面活性剤としては、例えば、各種アルキルアミノ脂肪酸ナトリウム等のアミノ酸系両性イオン界面活性剤、各種アルキルベタイン等のベタイン系両性イオン界面活性剤、各種アルキルアミンオキシド等のアミンオキシド系両性イオン界面活性剤などが例示でき、非イオン界面活性剤としては、例えば、各種ショ糖脂肪酸エステル、各種ソルビタン脂肪酸、各種ポリオキシエチレンソルビタン脂肪酸エステル等のエステル型非イオン界面活性剤、ポリオキシエチレンデシルエーテル、ポリオキシエチレンドデシルエーテルなどといった各種ポリオキシエチレンアルキルエーテル、各種ポリオキシエチレンアルキルフェニルエーテル等のエーテル型非イオン界面活性剤、さらに、各種エステル・エーテル型非イオン界面活性剤、脂肪酸アルカノールアミドなどといったその他の非イオン界面活性剤などが例示できるが、これらに何ら限定されるものではない。
【0067】
また、特に限定されるものではないが、例えば、処理温度が0〜100℃、加圧条件が1〜100MPaであり、この場合、撹拌時間としては、撹拌条件にもよるが、1〜60分程度、より好ましくは1〜30分程度とすることが望ましい。
【0068】
なお、撹拌条件としては、乳化される分子が標準状態(SATP)においても液状であるシリコーンオイルであるため、上記したS−(+)−イブプロフェンのように標準状態(SATP)において固体である場合と比較して、より穏和なもので十分であり、例えば、マグネティックスターラーを用いる場合、回転数を200rpm以上、好ましくは300〜500rpm程度のものとすることができる。なお、撹拌条件としては、何らこれに限定されるものではない。
【0069】
また、シリコーンオイルと界面活性剤との配合割合としては、特に限定されないが、仕込みモル比で、シリコーンオイル:界面活性剤=1:10-5〜1:10-2程度、より好ましくは、1:10-4〜1:10-2程度とすることが望ましい。
【0070】
また、界面活性剤の水溶液中における濃度としては、特に限定されるものではないが、0.01〜10質量%程度が適当であり、さらに、シリコーンオイルに対する水の仕込み量としても、特に限定されるものではないが、シリコーンオイル1mgに対し、水5〜10ml程度が適当である。
【0071】
以上示したような条件下で、シリコーンオイルを界面活性剤で乳化処理することにより、所定粒子径、例えば、平均粒子径1〜5000nm、代表的には200nm前後のシリコーンオイル液滴が均一に分散してなるO/Wエマルジョンが調製でき、減圧後においても長期間、液滴粒径の変動の少ない安定したエマルジョンとなる。
【0072】
ここで、シリコーンオイルのO/Wエマルジョンを、二酸化炭素による加圧条件下で処理すると、シリコーンオイル液滴の粒径は、加圧処理前のものに比べ、加圧処理終了直後(すなわち、所定時間加圧処理した後減圧した直後)のものの方が、大きくなっている傾向がある。この現象の詳細な作用機序は、明らかではないが、加圧処理終了のシリコーンオイルのO/Wエマルジョンに窒素をバブリングしてやると、バブリング経過時間が長くなるにつれ、シリコーンオイル液滴の粒径の大きさは低下し、一方、曝気することなく密閉状態で放置した場合、長期間経っても、窒素バブリングした場合と比較して、シリコーンオイル液滴の粒径の大きさは明らかに大きく、粒径の低下傾向が小さいことから、エマルジョン中のシリコーンオイル液滴は、二酸化炭素によって膨らんでいるものと思われる。また、シリコーンオイルのO/Wエマルジョンは、二酸化炭素での加圧下(観察のため、撹拌は行っていない)では、オイルリッチな上相と、水が大部分を占める下相とに二相分離し、その後、減圧すると、再び、均一なエマルジョンへと戻っていることが判った。撹拌下においても、微小領域中ではこのような相分離および再均質化が生じているものと思われ、上記したようなシリコーンオイル液滴の粒径変化は、このような相分離および再均質化を経ることによる、再分散の効果も含むものであると思われる。なお、このような現象は、二酸化炭素での加圧処理に特有のものであり、例えば、窒素を用いて同様に加圧処理を行っても、加圧下でシリコーンオイルエマルジョンの相分離現象は生じない。
【0073】
このように二酸化炭素による加圧下に調製されたシリコーンオイルのO/Wエマルジョンは、各種樹脂組成物の撥水性、帯電防止性、柔軟性あるいは潤滑性等を付与することを目的とする改質剤として好適に用いることができるが、上記したような特性の改質に加え、その樹脂組成物の透明度を改善させることが可能であり、特に、ナイロン6、ナイロン66、ナイロン610、ナイロン612、ナイロン11、ナイロン12等のナイロンに代表されるポリアミド樹脂系組成物中への改質剤として好適に使用することが可能である。なお、このような樹脂組成物の透明度の改善効果も、二酸化炭素での加圧処理に特有のものであり、例えば、窒素を用いて同様に加圧処理を行ったものを用いても、樹脂組成物の透明度の改善効果は見られないものである。
【0074】
なお、ポリアミド系樹脂組成物等の樹脂組成物に当該シリコーンオイルのO/Wエマルジョンを添加する場合には、樹脂組成物にシリコーンオイルのO/Wエマルジョンを溶融混練すれば良い。なお、シリコーンオイルのO/Wエマルジョンの添加量としては、透明度を改善しようとする場合、比較的少量であることが好ましく、ポリアミド系樹脂組成物100質量部あたり0.001〜10質量部、代表的には約0.1質量部程度が望ましい。
【実施例】
【0075】
以下、本発明を実施例に基づきより具体的に説明する。
【0076】
<実施例1および比較例1> S−(+)−イブプロフェンの包接化
(予備実験)
S−(+)−イブプロフェン(以下、IBUと略記する場合もある。)のCO2加圧条件でのp−T線図を図1に示す。図1からCO2加圧条件条件下ではIBUの融点は降下することが確認できた。また、ヒドロキシプロピル−β―シクロデキストリン溶液(以下、HPCD溶液と略記する場合もある。)中においても同様の融点降下が確認された。この図から、IBUがHPCD溶液中で融液として存在する領域を調べ、操作条件として設定した。
【0077】
(実験操作)
HPCD濃度29.6mMのHPCD水溶液10mlと粉末状のIBUを、HPCD:IBU=1:1の仕込みモル比で、ステンレス鋼製の耐圧の反応器に仕込み、系を35℃に保温した。その反応器にポンプでCO2を導入、操作圧である8MPaまで加圧し、マグネチックスターラー(500rpm)を用いて攪拌し包接化を行った(実施例1)。また攪拌時間を反応時間とした。減圧後得られた溶液は、時間を置かず紫外可視分光光度計を用い溶液中のIBU量を測定した。
【0078】
一方、比較のために、二酸化炭素による加圧を行わない系(大気圧:0.1MPa)で同様にIBUの包接化を行った(比較例1)。なお、比較例1では、圧力以外は実施例1と同様の条件で実験を行った。
【0079】
(結果及び考察)
吸光度分析において、水に溶解したIBUと包接されたIBUのスペクトルのピークが同位置に存在するため、検出薬物量は、水への溶解量を含んだ値となるが、水への溶解量は全体の検出IBU量に対して1〜3%とほとんど無視できる値である、そのため、薬物が包接された割合を示す包接度を(包接度=検出IBU量/仕込みIBU量)と定義することにした。
【0080】
図2に、攪拌時間を変化させたときの包接度の経時変化を示す。図2に示す結果から包接平衡に達する時間が大幅に短縮されたことから包接反応が大幅に加速されたことがわかった。
【0081】
<実施例2〜4、比較例2および参考例1〜2> S−(+)−イブプロフェンの包接化
操作圧である二酸化炭素の圧力を、0.1MPa(比較例2)、2MPa(参考例1)、4MPa(実施例2)、6MPa(実施例3)、8MPa(実施例4)にそれぞれ設定し、撹拌時間を5分間に設定する以外は、実施例1と同様にして包接化処理を行い、圧力条件の変化による包接度の変化を調べた。その結果を図3に示す。
【0082】
実験時の温度(35℃)におけるIBUの融解圧力は、3.25MPaであり、図3より、融解圧力前後において包接度の上昇が確認された。このことから、加圧によりIBUが融解し、融液の状態でHPCD溶液と接触したことにより、包接効率が上昇したものと考えられた。
<実施例5および比較例3> S−(+)−イブプロフェンの包接化
撹拌速度の包接化への影響を調べるため、撹拌時間を5分間に設定し、マグネチックスターラーの回転数を種々変動させる以外は、実施例1および比較例1と同様にして、撹拌速度の変化による包接度の変化を調べた。その結果を図4に示す。
【0083】
図4に示すように、攪拌速度の上昇とともに包接度の値も上昇した。このことから、IBUとHPCDの接触効率の上昇によって包接効率も上昇することが確認された。また、全ての攪拌速度において、実施例である加圧条件下における包接度の方が、比較例である非加圧条件下におけるものよりも高い値を示した。このことは、融液を攪拌することによって微小な液滴が生成され、それによって界面積が増え、包接効率が上昇したためであると考えられた。
【0084】
<実施例6> S−(+)−イブプロフェンの包接化
撹拌装置として、マグネチックスターラーに代えて、スタティックミキサー(日本フローコントロール社製、型番85−212)を用いた以外は、実施例1と同様の条件下において、包接化処理を行った。
【0085】
その結果、スタティックミキサーを用いた実施例6においては、マグネチックスターラーを用いた実施例1と比較して、より微小な液滴が形成され界面積が増大したことが確認され、スタティックミキサーを用いることでより効率的かつ良好な包接化がなされ得ること明らかとなった。
【0086】
<実施例7〜10> シリコーンオイルのエマルジョン
まず、市販のジメチルシリコーンオイルの非イオン性界面活性剤によるエマルジョン(松本シリコーンソフナー#302、松本油脂株式会社製)におけるオイル液滴の平均粒子径を、ダイナミック光散乱高度計(NICOMP 380ZLS Zeta potential/Particle sizer、パーティクル・サイジング・システムズ社製)(以下、DLSと略記することもある。)を用いて測定した。その結果、このエマルジョンにおける、オイル液滴の平均粒子径は182.1nmであった。
【0087】
次いで、このジメチルシリコーンオイルエマルジョンを、ステンレス鋼製の耐圧の反応器に仕込み、系を50℃に保温した。マグネチックスターラー(150rpm)を用いて攪拌しながら、その反応器にポンプでCO2を導入、操作圧である10、20、30MPaまでそれぞれ加圧し、15分間撹拌処理を行った。減圧後得られたエマルジョンにおけるオイル液滴の平均粒子径を直ちに、上記と同様にDLSにより測定したところ、図5に示すように、いずれの加圧条件でも、オイル液滴の平均粒子径は210nmに近いものであり、加圧処理前のものよりもエマルジョンにおけるオイル液滴の平均粒子径が増大していた。
【0088】
さらに、このオイル液滴の平均粒子径の加圧処理前後における変動の要因を調べるため、加圧処理により得られたエマルジョンをN2でバブリング処理し、バブリング時間が5分、10分、15分、30分、および60分となった時点で、エマルジョンにおけるオイル液滴の平均粒子径をDLSにより測定した。一方で、加圧処理により得られたエマルジョンを、少量(3ml)サンプル瓶に採取し、曝気することなく密閉して1週間放置した後、エマルジョンにおけるオイル液滴の平均粒子径を同様にDLSにより測定した。
【0089】
その結果、図5に示すように、いずれの圧力条件により処理したものも、N2でバブリング処理した場合、時間が経過するに従って、マルジョンにおけるオイル液滴の平均粒子径は低下した。一方で、密閉放置後のものにおいては、操作圧10、20、30MPaで加圧処理したものそれぞれにつき、208.2nm、201.1nm、202.7nmといずれも平均粒子径として200nm以上の値を保っており、N2でバブリング処理した場合と比べ、明らかに、平均粒子径の低下傾向が小さいものであった。このことから、二酸化炭素による加圧処理を行った場合、シリコーンオイルのエマルジョンにおけるオイル液滴が二酸化炭素により膨らんでいるものと思われた。
【0090】
<参考例3> 二酸化炭素加圧下におけるシリコーンオイルエマルジョンの挙動
二酸化炭素による加圧下におけるシリコーンオイルエマルジョンの挙動を調べるために、光学窓付平衡セル内に、市販のジメチルシリコーンオイルの非イオン性界面活性剤によるエマルジョン(松本シリコーンソフナー#302、松本油脂株式会社製)を入れ、50℃まで昇温させた後、二酸化炭素によって10mPaまで加圧し、その状態を30分間維持した後、20mPaまで昇圧し、その状態でさらに30分間維持した後、減圧し、各時間におけるエマルジョンの状態を観察した。その結果を、図6に示す。図6に示されるように、加圧状態においては単一相であったエマルジョンが二相に分離し、減圧することによって再び単一相のエマルジョンとなっていた。
【0091】
なお、別途、エマルジョンに赤色水性インキを添加して着色し、同様の実験を行ったところ、加圧状態における相分離エマルジョンの上相は着色しておらす、一方下相は加圧とともに白濁が薄くなり赤色透明に近くなったことから、上相はシリコーンオイルが大部分を占め、下相は水が大部分を占めるエマルジョンであると判断できた。
【0092】
得られた結果から、上記したようなシリコーンオイル液滴の粒径変化は、このような相分離および再均質化を経ることによる、再分散の効果も含むものであると思われた。
【0093】
<比較例4> 窒素加圧下におけるシリコーンオイルエマルジョンの挙動
比較のために、二酸化炭素に変えて窒素により加圧する以外は、参考例3と同様にして、シリコーンオイルエマルジョンを加圧処理し、窒素加圧下におけるシリコーンオイルエマルジョンの挙動を調べた。その結果を図7に示す。
【0094】
図7に示すように、窒素による加圧においては、参考例3におけるようなシリコーンオイルエマルジョンの相分離は生起せず、エマルジョンの相分離は二酸化炭素加圧に特有なものであることが判明した。
【0095】
<実施例11〜12および比較例5> 二酸化炭素加圧処理後におけるシリコーンオイルエマルジョンの特性
二酸化炭素加圧処理前後のシリコーンエマルジョンの粘度および濡れ性を調べた。
まず、市販のジメチルシリコーンオイルの非イオン性界面活性剤によるエマルジョン(松本シリコーンソフナー#302、松本油脂株式会社製)を、ステンレス鋼製の耐圧の反応器に仕込み、系を25℃(実施例11)、40℃(実施例12)にそれぞれに保持し、マグネチックスターラー(800rpm)を用いて攪拌しながら、その反応器にポンプでCO2を導入、操作圧である12MPaまでそれぞれ加圧し、15分間撹拌処理を行ったのち、減圧した。このようにしてそれぞれ得られた二酸化炭素加圧処理直後のシリコーンオイルエマルジョンと、未処理の原料であるシリコーンオイルエマルジョン(比較例5)に関して、音叉型振動式粘度計(VIBRO VISCOMETER SV-10、株式会社エー・アンド・デイ製)を用いて粘度測定を行った。また、θ/2法を用いて、ガラスおよびテフロン(登録商標)表面における接触角を測定した。得られた結果を、図8に示す。
【0096】
図8に示すように、本発明の実施例に係るシリコーンオイルエマルジョンは、二酸化炭素加圧処理を施さなかった比較例のシリコーンオイルエマルジョンと比較して、粘度特性およびガラスに対する濡れ性はあまり変わらないものの、テフロン(登録商標)に対する濡れ性が大きく向上しているのが判った。
【0097】
<実施例13〜14および比較例5> ポリアミド系樹脂製品
上記実施例11〜12におけるものと同様の条件下で二酸化炭素加圧処理したシリコーンオイルエマルジョンを、原料ナイロン(ナイロン6:ナイロン66=4:1の質量比のブレンド物)と溶融混練した後、押出し成形を行い直径3mmの紐状に成形した(実施例13〜14)。なお、シリコーンオイルエマルジョンの添加量は、原料ナイロン1kgに対し、それぞれ1g、3gおよび5gとし、また押出条件としては押出回転速度270rpm、押出温度230℃とした。
【0098】
一方比較のために、加圧未処理のジメチルシリコーンオイルエマルジョン(松本シリコーンソフナー#302、松本油脂株式会社製)を上記と同様の条件下で原料ナイロンと溶融混練した後、押出し成形を行い紐状に成形した(比較例5)。
【0099】
得られた紐状成形品に関し、その透明度および結晶化度を測定した。
【0100】
なお、透明度は、各紐状成形品より長さ18.6mmの試料を切り出し、この試料の一端部に光源を配置し、他端部に光量子センサを配置すると共に、試料の側周面を黒色テープで覆うことにより構成してなる透明度定量化装置を用い、試料を通過した光の強さ(I)[mA]と、同距離でのブランク(中空)にした場合に計測される光の強さ(I0)[mA]とを測定し、以下の式により透過度(%)を算出した。
【0101】
透過度(%)=(I/I0)×100
また、結晶化度Xcについては、示差走査熱量測定法(DSC法)に基づき、以下の式より算出した。
【0102】
結晶化度Xc=(Hc/Hc0)×100
(Hc:DSC曲線の融解熱[J/g」、Hc0:結晶化度100%の場合の融解熱[J/g」)
その結果、比較例5のもの(透過率0〜3%)に比べ、実施例13〜14のものは、いずれも透過率の向上が見られ、特に図9に示すように、シリコーンオイルエマルジョンの添加量が1gであった場合には、非常に良好な透明性の改善効果が見られた。
【0103】
なお、結晶化度については、比較例5および実施例13〜14のものいずれにおいても、約20%前後であり、その有意差は見られず、透明性の改善効果は、二酸化炭素加圧処理のジメチルシリコーンオイルエマルジョンの添加によるものであるが、結晶化度の変化によるものではないことが考えられた。
【図面の簡単な説明】
【0104】
【図1】は、S−(+)−イブプロフェンのCO2加圧条件での融点変動を示すp−T線図である。
【図2】は、攪拌時間を変化させたときの、S−(+)−イブプロフェンのヒドロキシプロピル−β―シクロデキストリンによる包接度の経時変化を示すグラフである。
【図3】は、二酸化炭素による加圧条件を変化させたときの、S−(+)−イブプロフェンのヒドロキシプロピル−β―シクロデキストリンによる包接度の変化を示すグラフである。
【図4】は、撹拌速度を変化させたときの、S−(+)−イブプロフェンのヒドロキシプロピル−β―シクロデキストリンによる包接度の変化を示すグラフである。
【図5】は、二酸化炭素加圧処理を行ったシリコーンオイルエマルジョンに対し、窒素バブリングを行った場合における、エマルジョン中のオイル液滴の平均粒子径の経時変化を示すグラフである。
【図6】は、二酸化炭素加圧下におけるシリコーンオイルエマルジョンの状態変化を示す写真図である。
【図7】は、窒素加圧下におけるシリコーンオイルエマルジョンの状態変化を示す写真図である。
【図8】は、二酸化炭素加圧処理前後のシリコーンエマルジョンの粘度および濡れ性の変化を示すグラフである。
【図9】は、二酸化炭素加圧処理されたシリコーンオイルエマルジョンを添加したナイロン樹脂製品の光透過率とエマルジョンの添加量との関係を示すグラフである。
【特許請求の範囲】
【請求項1】
分散媒に対して難溶性ないし非溶性のゲスト分子を、ホスト分子により包接するあるいは界面活性剤により乳化することによって、分散媒中にゲスト分子の微細粒状体を製造する方法であって、圧力容器中に、ゲスト分子、およびホスト分子ないし界面活性剤を含む分散媒を仕込み、この圧力容器に二酸化炭素を導入して、1〜100MPaの操作圧まで加圧し、所定時間撹拌を行なって包接あるいは乳化処理した後、圧力容器内を減圧して二酸化炭素を排出することにより、分散媒中のゲスト分子の微細粒状体を製造する方法。
【請求項2】
撹拌が静止型混合機により行われるものである請求項1に記載の微細粒状体の製造方法。
【請求項3】
分散媒が水ないし水系溶液である請求項1または2に記載の微細粒状体の製造方法。
【請求項4】
ゲスト分子が標準状態(SATP、25℃、105Pa)において、固体状のものである請求項1〜3のいずれかに記載の微細粒状体の製造方法。
【請求項5】
ゲスト分子が、S−(+)−イブプロフェン、プロゲステロン、ケトプロフェンおよびグリセオフルビンからなる群から選ばれてなるいずれか1つの化合物である請求項1〜4のいずれかに記載の微細粒状体の製造方法。
【請求項6】
ホスト分子がシクロデキストリンおよびその誘導体からなる群から選ばれてなるものである請求項1〜5のいずれかに記載の微細粒状体の製造方法。
【請求項7】
ゲスト分子が、樹脂ないしゴム成分を含有する組成物用の添加剤である請求項1〜3のいずれかに記載の微粒粒状体の製造方法。
【請求項8】
ゲスト分子がシリコーンオイルである請求項7に記載の微粒粒状体の製造方法。
【請求項9】
請求項7または8に記載の製造方法により得られた分散媒中のゲスト分子の微細粒状体を樹脂組成物中に添加することを特徴とする樹脂組成物の製造方法。
【請求項10】
請求項9に記載の樹脂組成物がポリアミド系樹脂組成物であり、シリコーンオイルであるゲスト分子の微細粒状体を添加混合し、これを成形加工することを特徴とする、透明度の改善されたポリアミド系樹脂製品の製造方法。
【請求項11】
請求項10に記載の製造方法により得られたことを特徴とする透明度の改善されたポリアミド系樹脂製品。
【請求項12】
請求項1〜3のいずれかに記載の製造方法により得られた、分散媒中のゲスト分子の微細粒状体を、さらに他の液状体に添加吸収させることを特徴とする微細粒状体分散体の製造方法。
【請求項13】
請求項1〜3のいずれかに記載の製造方法により得られた、分散媒中のゲスト分子の微細粒状体を、被塗物表面へと吹きつけることを特徴とする微細粒状体被膜の形成方法。
【請求項1】
分散媒に対して難溶性ないし非溶性のゲスト分子を、ホスト分子により包接するあるいは界面活性剤により乳化することによって、分散媒中にゲスト分子の微細粒状体を製造する方法であって、圧力容器中に、ゲスト分子、およびホスト分子ないし界面活性剤を含む分散媒を仕込み、この圧力容器に二酸化炭素を導入して、1〜100MPaの操作圧まで加圧し、所定時間撹拌を行なって包接あるいは乳化処理した後、圧力容器内を減圧して二酸化炭素を排出することにより、分散媒中のゲスト分子の微細粒状体を製造する方法。
【請求項2】
撹拌が静止型混合機により行われるものである請求項1に記載の微細粒状体の製造方法。
【請求項3】
分散媒が水ないし水系溶液である請求項1または2に記載の微細粒状体の製造方法。
【請求項4】
ゲスト分子が標準状態(SATP、25℃、105Pa)において、固体状のものである請求項1〜3のいずれかに記載の微細粒状体の製造方法。
【請求項5】
ゲスト分子が、S−(+)−イブプロフェン、プロゲステロン、ケトプロフェンおよびグリセオフルビンからなる群から選ばれてなるいずれか1つの化合物である請求項1〜4のいずれかに記載の微細粒状体の製造方法。
【請求項6】
ホスト分子がシクロデキストリンおよびその誘導体からなる群から選ばれてなるものである請求項1〜5のいずれかに記載の微細粒状体の製造方法。
【請求項7】
ゲスト分子が、樹脂ないしゴム成分を含有する組成物用の添加剤である請求項1〜3のいずれかに記載の微粒粒状体の製造方法。
【請求項8】
ゲスト分子がシリコーンオイルである請求項7に記載の微粒粒状体の製造方法。
【請求項9】
請求項7または8に記載の製造方法により得られた分散媒中のゲスト分子の微細粒状体を樹脂組成物中に添加することを特徴とする樹脂組成物の製造方法。
【請求項10】
請求項9に記載の樹脂組成物がポリアミド系樹脂組成物であり、シリコーンオイルであるゲスト分子の微細粒状体を添加混合し、これを成形加工することを特徴とする、透明度の改善されたポリアミド系樹脂製品の製造方法。
【請求項11】
請求項10に記載の製造方法により得られたことを特徴とする透明度の改善されたポリアミド系樹脂製品。
【請求項12】
請求項1〜3のいずれかに記載の製造方法により得られた、分散媒中のゲスト分子の微細粒状体を、さらに他の液状体に添加吸収させることを特徴とする微細粒状体分散体の製造方法。
【請求項13】
請求項1〜3のいずれかに記載の製造方法により得られた、分散媒中のゲスト分子の微細粒状体を、被塗物表面へと吹きつけることを特徴とする微細粒状体被膜の形成方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図8】
【図9】
【図6】

【図7】

【図2】
【図3】
【図4】
【図5】
【図8】
【図9】
【図6】
【図7】
【公開番号】特開2009−208035(P2009−208035A)
【公開日】平成21年9月17日(2009.9.17)
【国際特許分類】
【出願番号】特願2008−55933(P2008−55933)
【出願日】平成20年3月6日(2008.3.6)
【出願人】(508069567)エコラボ株式会社 (1)
【出願人】(508069578)
【Fターム(参考)】
【公開日】平成21年9月17日(2009.9.17)
【国際特許分類】
【出願日】平成20年3月6日(2008.3.6)
【出願人】(508069567)エコラボ株式会社 (1)
【出願人】(508069578)
【Fターム(参考)】
[ Back to top ]