
微細結晶
【課題】本発明の目的は、例えば溶解性、安定性、バイオアベイラビリティ、医薬製剤中の分散性等が良好な化合物1の結晶およびその結晶を含む固体医薬製剤を提供することにある。
【解決手段】溶解性、安定性、バイオアベイラビリティ、医薬製剤中の分散性等が良好な50μm未満の平均粒径を有する(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの微細結晶、50μm未満の平均粒径を有し、かつ結晶化度が20%以上である(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの微細結晶等を提供する。また、これらを含むことを特徴とする固体医薬製剤を提供する。
【解決手段】溶解性、安定性、バイオアベイラビリティ、医薬製剤中の分散性等が良好な50μm未満の平均粒径を有する(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの微細結晶、50μm未満の平均粒径を有し、かつ結晶化度が20%以上である(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの微細結晶等を提供する。また、これらを含むことを特徴とする固体医薬製剤を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオン(以下、化合物1という)の結晶およびその結晶を含む固体医薬製剤に関する。
【背景技術】
【0002】
化合物1はアデノシンA2受容体拮抗作用を示し、アデノシンA2受容体亢進作用に基づく各種疾患、例えばパーキンソン病、老人性痴呆症、うつ病、喘息、骨粗しょう症等の治療等に有用である(特許文献1、特許文献2)。化合物1を含むキサンチン誘導体が、吸入投与を目的として微粉化した粉末の状態で用いられることが知られている(特許文献1)。また、化合物1の結晶が知られている(特許文献2)。上記引用文献に示された方法で合成された化合物1の結晶は、(1)水に対する溶解性が低い、(2)結晶の形状が短径数μm×長径数100μm以上の針状結晶であるという特徴を有するため、製剤化の工程操作中に化合物1の結晶が凝集するという課題を有している。水に対する溶解性が低い薬物は、消化管内での溶解性の低さと溶解速度の遅さから、一般的にバイオアベイラビリティが低いと言われている。化合物1に関しても溶解性、溶解速度等の向上、バイオアベイラビリティの改善等が望まれている。一方、製剤化の工程操作中に生じる化合物1の結晶の凝集は、化合物1の結晶および添加剤の流動性に影響を与え、製剤化工程における化合物1の結晶の取り扱いや固体製剤中の化合物1の分散性の面で問題になっている。また、化合物1は特に光に不安定で、構造中の二重結合部分(ビニレン部分)が異性化しやすいことが知られており(非特許文献1)、化合物1を含有する医薬製剤を調製する際の取り扱いに留意が必要である。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】欧州特許第0590919号明細書
【特許文献2】特開平9-040652号公報
【非特許文献】
【0004】
【非特許文献1】バイオオーガニック・メディシナル・ケミストリー・レターズ(Bioorg. Med. Chem. Lett.)、7巻、2349-2352ページ(1997年)
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明の目的は、例えば溶解性、安定性、バイオアベイラビリティ、医薬製剤中の分散性等が良好な化合物1の結晶およびその結晶を含む固体医薬製剤を提供することにある。
【課題を解決するための手段】
【0006】
本発明は、以下の(1)〜(13)に関する。
(1) 50μm未満の平均粒径を有する
【化1】

で表される(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの微細結晶。
(2) (E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの微細結晶の平均粒径が0.5〜20μmである上記(1)記載の微細結晶。
(3) 結晶化度が20%以上である上記(1)または(2)記載の(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの微細結晶。
(4) 結晶化度が30%以上である上記(1)または(2)記載の(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの微細結晶。
(5) 結晶化度が40%以上である上記(1)または(2)記載の(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの微細結晶。
(6) 上記(1)〜(5)のいずれかに記載の微細結晶を含むことを特徴とする固体医薬製剤。
(7) 結晶化度が20%以上である(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶と分散剤を含有する固体分散体。
(8) (E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶化度が30%以上である上記(7)記載の固体分散体。
(9) (E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶化度が40%以上である上記(7)記載の固体分散体。
(10) 上記(7)〜(9)のいずれかに記載の固体分散体を含むことを特徴とする固体医薬製剤。
(11) 結晶化度が20%以上である(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンを含むことを特徴とする固体医薬製剤。
(12) (E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶化度が30%以上である上記(11)記載の固体医薬製剤。
(13) (E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶化度が40%以上である上記(11)記載の固体医薬製剤。
【発明の効果】
【0007】
本発明により、例えば溶解性、安定性、吸収性、医薬製剤中の分散性等が良好な化合物1の結晶およびその結晶を含む固体医薬製剤が提供される。
【図面の簡単な説明】
【0008】
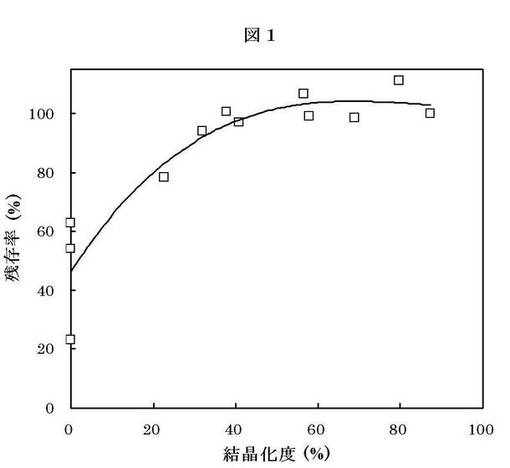
【図1】試験例1での化合物1の結晶化度と化合物1の光安定性の相関を示したものである。縦軸は化合物1の残存率(%)を表し、横軸は化合物1の結晶化度(%)を表す。
【0009】
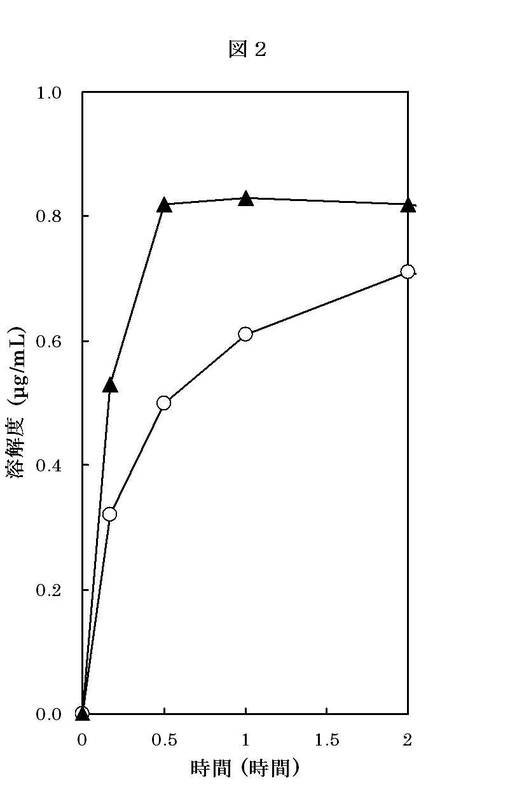
【図2】化合物1の結晶の平均粒径と化合物1の溶解度との関係を示したものである。縦軸は化合物1の溶解度(μg/mL)を表し、横軸は経過時間(時間)を表す。グラフ上の各プロットの意味は、以下の通りである。-○-:結晶Aの溶解度(μg/mL)-▲-:微細結晶Aの溶解度(μg/mL)
【発明を実施するための形態】
【0010】
本明細書において、「(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオン」(「化合物1」)という記載は、非晶性の化合物1、結晶性の化合物1またはそれらの混合物を意味し、原料として使用される「化合物1」においては、その結晶化度、平均粒径等も限定されない。
【0011】
本発明の「50μm未満の平均粒径を有する(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの微細結晶」(「化合物1の微細結晶」)は、平均粒径が50μm未満の結晶性の化合物1であれば特に限定されないが、中でも0.5〜20μmの平均粒径を有する微細結晶が好ましい。さらに結晶化度が20%以上である「化合物1の微細結晶」が好ましく、中でも結晶化度が30%以上である「化合物1の微細結晶」がより好ましく、さらに結晶化度が40%以上である「化合物1の微細結晶」が最も好ましい。なお、これらの平均粒径は、例えばレーザー回折・錯乱式粒度分布測定装置(例えばMASTERSIZER 2000 Ver.2.00J;MALVERN社製等)や画像解析装置(例えばLUZEX(R) AP;ニコレ社製等)等を用いて測定され、粒度分布の平均値として算出される。また、これらの結晶化度は粉末X線回折装置(例えばJDX8030;日本電子製等)を用いて特定の回折角2qにおける回折ピークの積分強度を測定することにより算出される。
【0012】
本発明の「化合物1の微細結晶」の調製法は特に限定されないが、例えば欧州特許第0590919号明細書、特開平9-040652号公報等に記載の方法またはそれらに準じた方法により得られる「50μm以上の平均粒径を有する(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶」(「化合物1の結晶」)を粉砕および/または篩い分けすることにより調製され、粉砕および/または篩い分けは適宜組み合わせて数回行ってもよい。粉砕は一般的に使用される粉砕機で行うことができ、該粉砕機として、例えば乳鉢、メカノミル、ジェットミル等を用い、例えば粉砕機の回転速度、「化合物1の結晶」の供給速度、粉砕の時間等を適宜調整することにより所望の平均粒径および/または結晶化度を有する「化合物1の微細結晶」を得ることができる。中でもジェットミルによる粉砕が好ましく、例えば「化合物1の結晶」の供給速度10〜1000g/分、粉砕圧力0.01〜1.0MPaで、「化合物1の結晶」を粉砕することができる。
【0013】
本発明の「化合物1の微細結晶」を含む固体医薬製剤は、上記の「化合物1の微細結晶」を含む固体医薬製剤であればいずれでもよく、例えば
(a)上記の方法で得られる「化合物1の微細結晶」と添加剤とを混合し製剤化したもの、
(b)上記の方法で得られる「化合物1の結晶」と添加剤とを混合し、上記「化合物1の微細結晶」の調製法と同様にして得られる混合物を粉砕および/または篩い分けした後、製剤化したもの、
(c)「化合物1」と分散剤から固体分散体を調製した後、該固体分散体と添加剤とを混合し製剤化したもの
等があげられる。なお本発明の固体医薬製剤中の「化合物1の微細結晶」の含有量は、好ましくは0.001%〜80%、さらに好ましくは0.1%〜50%である。
【0014】
該固体分散体は、「化合物1」または「化合物1の結晶」とこれを分散することができる分散剤から調製される固体分散体であり、該固体分散体中の化合物1における結晶部分が上記「化合物1の微細結晶」の平均粒径または平均粒径および結晶化度を有するものであれば特に限定されない。分散剤としては、例えばヒドロキシプロピルメチルセルロース(HPMC)、ポリビニルピロリドン(PVP)、ヒドロキシプロピルセルロース(HPC)等の高分子が好ましい。また「化合物1」または「化合物1の結晶」と分散剤の配合比は、1:0.1〜1:5(重量比)であることが好ましく、中でも1:0.1〜1:3(重量比)であることが好ましい。また該固体分散体の製造法も特に限定されないが、例えば欧州特許第0590919号明細書、特開平9-040652号公報等に記載の方法またはそれらに準じた方法により得られる「化合物1」または「化合物1の結晶」と分散剤から、例えば混合粉砕法、溶媒法等の通常の方法で調製することにより得ることができる。
【0015】
混合粉砕法としては、例えば「化合物1の結晶」を分散剤とともに混合機器等を用いて混合したものを、一般的に使用される粉砕機例えば乳鉢、メカノミル、ジェットミル等を用いて粉砕する方法等があげられ、例えば粉砕機の回転速度、「化合物1の結晶」の供給速度、粉砕の時間等を適宜調整することにより所望の平均粒径または平均粒径および結晶化度を有する「化合物1の微細結晶」を含む固体分散体を得ることができる。中でもジェットミルによる粉砕が好ましい。
【0016】
溶媒法としては、例えば「化合物1」または「化合物1の結晶」を分散剤とともに有機溶媒に溶解または分散し、ついで有機溶媒を常法により減圧下または常圧下で除去する方法等があげられる。具体的には、例えば流動層造粒装置、攪拌造粒装置、噴霧造粒装置、噴霧乾燥造粒装置、真空乾燥造粒装置等が用いられ、所望により一般的に使用される粉砕機、例えば乳鉢、メカノミル、ジェットミル等を用いて粉砕する方法を組み合わせてもよい。有機溶媒としては、「化合物1」または「化合物1の結晶」を溶解するものであれば特に限定されないが、例えばジクロロメタン、ジクロロエタン、クロロホルム等のハロゲン化炭化水素類、アセトン、メチルエチルケトン等のケトン類、メタノール、エタノール等のアルコール類、テトラヒドロフラン等のエーテル類、酢酸エチル等のエステル類、ジメチルホルムアミド、ジメチルアセトアミド等のアミド類等があげられる。
【0017】
添加剤としては、例えば賦形剤、結合剤、崩壊剤、滑沢剤、可塑剤、界面活性剤、コーティング剤、着色剤、矯味剤、酸味剤等があげられ、これらは製剤の種類に応じて適宜用いることができる。
賦形剤としては、例えば白糖、ブドウ糖、蔗糖、マンニトール、ラクトース等の糖類、トウモロコシ澱粉、バレイショ澱粉等の澱粉、結晶セルロース、微結晶セルロース等のセルロース類等があげられる。
結合剤としては、例えばポリビニルアルコール、ヒドロキシプロピルセルロース、ゼラチン、メチルセルロース、エチルセルロース、ポリビニルピロリドン等があげられる。
崩壊剤としては、例えばトウモロコシ澱粉、バレイショ澱粉等の澱粉、寒天、ゼラチン末、結晶セルロース、アルギン酸ナトリウム、クロスポビドン等があげられる。
滑沢剤としては、例えばステアリン酸マグネシウム、タルク等があげられる。
可塑剤としては、例えば植物油、グリセリン等があげられる。
界面活性剤としては、例えばラウリル硫酸ナトリウム、ポリソルベート80、脂肪酸エステル等があげられる。
コーティング剤としては、例えば白糖、ヒドロキシプロピルセルロース等の糖衣、ゼラチン、グリセリン、ソルビトール等の膠衣等があげられる。
着色剤としては、例えば食用色素等があげられ、矯味剤としては、例えば、サッカリンナトリウム、アスパルテーム、ステビア等があげられ、酸味剤としては、例えばクエン酸、リンゴ酸、酒石酸等があげられる。
【0018】
「結晶化度が20%以上である(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶」(「結晶化度が20%以上である化合物1の結晶」)は、結晶化度が20%以上である結晶性の化合物1であれば特に限定されないが、中でも結晶化度が30%以上である結晶性の化合物1が好ましく、さらに結晶化度が40%以上である結晶性の化合物1がより好ましい。またこれらの製造法も特に限定されないが、例えば欧州特許第0590919号明細書、特開平9-040652号公報等に記載の方法またはそれらに準じた方法により得ることができる。
【0019】
また、「結晶化度が20%以上である化合物1の結晶」を含む固体医薬製剤は、上記の「結晶化度が20%以上である化合物1の結晶」を含む固体医薬製剤であればいずれでもよく、該固体医薬製剤の製造法も特に限定されないが、例えば上記「化合物1の微細結晶」を含む固体医薬製剤の製造法と同様の方法をあげることができる。
【0020】
また、「結晶化度が20%以上である化合物1の結晶」と分散剤を含有する固体分散体は、「化合物1」または「化合物1の結晶」とこれを分散することができる分散剤から調製される固体分散体であり、該固体分散体中の化合物1における結晶部分が20%以上の結晶化度を有するものであれば平均粒径等は特に限定されず、該固体分散体の製造法も特に限定されないが、例えば上記「化合物1の微細結晶」と分散剤を含有する固体分散体の製造法と同様の方法をあげることができる。分散剤としては、例えばHPMC、PVP、HPC等が好ましい。また「化合物1」または「化合物1の結晶」と分散剤の配合比は、1:0.1〜1:5(重量比)であることが好ましく、中でも1:0.1〜1:3(重量比)であることが好ましい。
【0021】
本発明の固体医薬製剤の製剤形態としては、例えば糖衣錠等の錠剤、散剤、顆粒剤、カプセル剤、丸剤、トローチ剤、懸濁内用液剤等があげられ、これら製剤は製剤学の技術分野においてよく知られている混合工程、粉砕工程、篩い分け工程、造粒加工工程、整粒加工工程、打錠工程、乾燥工程、カプセル充填工程、コーティング工程等の製剤化工程を組み合わせることにより製造できる。
【0022】
以下に試験例により本発明の効果を具体的に説明する。
試験例1:化合物1の結晶化度と光安定性
<試料調製方法>
以下、「化合物1の結晶」とHPMCから固体分散体を調製した。
「化合物1の結晶」としては、特開平9-040652号公報に記載の方法により得られた未粉砕の「化合物1の結晶」(結晶化度87.2%)を使用した。
試料Aは、未粉砕の「化合物1の結晶」(10g)とHPMCとを、表1に示した配合比でジクロロメタンに溶解し、溶媒を留去した後、得られた固体を錠剤粉砕機(YM-100、湯山製作所製)で、粉砕翼の回転数10000rpmで1分間粉砕することにより得られた。
試料Bは、未粉砕の「化合物1の結晶」(100g)とHPMCとを、表1に示した配合比で混合した後、表1に示した粉砕機を用いて粉砕することにより得られた。
検量線作成用の物理混合物(標品)は、未粉砕の「化合物1の結晶」とHPMCとを種々の割合でそれぞれとり、200×150mmのビニール袋中でよく振りまぜて調製した。
【0023】
<相対結晶化度の測定方法>
各試料における化合物1の結晶化度(以下、結晶化度ということもある)は、粉末X線回折装置により回折角2qを0°から40°まで変化させて各試料の回折ピークを測定し、下記の方法により算出した。
相対結晶化度の算出に用いる検量線は、種々の割合で調製した物理混合物を用いて、回折角2q=約16°における回折ピークの積分強度を測定し、それぞれの物理混合物中の「結晶性の化合物1」の量と回折ピークの積分強度の比率をプロットして作成した。なお、結晶化度87.2%である未粉砕の「化合物1の結晶」を相対結晶化度100%(「結晶性の化合物1」の含有率100%)である標準試料、HPMCを相対結晶化度0%(「結晶性の化合物1」の含有率0%)である試料とした。
試料の回折角2q=約16°における回折ピークの積分強度を測定し、上記の検量線に基づいて、測定した積分強度から各試料中の「結晶性の化合物1」の量を算出し、各試料の相対結晶化度(%)を「化合物1」の量(「結晶性の化合物1」と「非晶性の化合物1」)に対する「結晶性の化合物1」量の比率として下記の式により求めた。結晶化度(%)は、ここで求めた相対結晶化度(%)と未粉砕の「化合物1の結晶」の結晶化度87.2%から比例計算することにより決定した。
相対結晶化度(%)=(「結晶性化合物1の量」/「化合物1」の量)×100
【0024】
<光安定性の測定法>
各試料の光安定性は、下記の方法により試料中の「化合物1」の残存率(%)を測定することで追跡した。
試料を秤量してつめた透明ガラスバイアルを蛍光灯下約5000 lxで保存し、各試料とも照射開始の8時間後にサンプリングした。サンプリングした試料を水とアセトニトリルの混合溶媒(水:アセトニトリル=60:40)に溶解した後、高速液体クロマトグラフィー(HPLC)により試料中の「化合物1」の量を定量した。照射前の「化合物1」の量を100%とし、これに対する照射後の「化合物1」の量を残存率(%)として求めた。
試料AおよびBの結晶化度(%)および「化合物1」の残存率(%)を第1表に、結晶化度と安定性の相関を図1に示す。
なお、HPLCの測定条件は以下の通りである。
分析機器:LC-6シリーズ(島津製作所製)
カラム:Inertsil ODS-2(φ6×150mm)
カラム温度:25℃
移動相:50 mmol/L KH2PO4(pH 6.1, KOH)/アセトニトリル=60/40
流速:1.2 mL/分
検出条件:UV 248nm
【0025】
【表1】
【0026】
以上の結果、化合物1の結晶化度と光安定性には正の相関があり、「化合物1の結晶」の結晶化度が20%以上、好ましくは30%以上で光照射下での分解が減少することが判明した。つまり、一連の粉砕、分散化等の製剤化工程において「化合物1の結晶」の結晶化度を一定値以上に保持できれば、または一定値以上の結晶化度を有する「化合物1の結晶」を製剤化工程で用いれば、光照射下でも分解物の増加はみられず製剤化工程での化合物1の安定性を維持できると考えられる。
【0027】
試験例2:化合物1の結晶の平均粒径と溶解度
実施例2で得られた結晶A(結晶の平均粒径;167μm、2mg)および微細結晶A(微細結晶の粒径D100=8.7μm、2mg)を用いて、それぞれの水(200 mL)に対する室温での溶解度を測定した。
経過時間に対する結晶Aおよび微細結晶Aの溶解度(μg/mL)を図2に示す。
【0028】
以上の結果、平均粒径の小さい微細結晶Aは、結晶Aと比較して、溶解速度が早く、良好な化合物1の溶解性を有することが判明した。
【0029】
また、微細結晶Aでは、製剤化の工程操作中に化合物1の結晶が凝集するという現象はみられず、微細結晶Aは結晶Aと比較して分散性に優れることも判明した。
【0030】
試験例3:経口吸収性の比較
実施例3で得られた結晶B(結晶の平均粒径;181μm)および微細結晶B(結晶の平均粒径;11μm)を、それぞれ0.5重量/容量%メチルセルロース水溶液に懸濁させ、投与薬液(0.3mg/mL)を調製した。得られた投与薬液をSD系雄性ラット(体重 209〜233g;日本チャールス・リバー)に10mL/kgの容量で経口投与した。投与から0.25、0.5、1、2、4、6、8、12および24時間後の時点で、それぞれヘパリン処理されたキャピラリーチューブを用いて、経時的にラットの尾静脈より血液(各回約0.3mL)を採取した。得られた血液を遠心分離(1950×g、10分間、4℃)し、血漿を分離した。得られた血漿中の「化合物1」の濃度をHPLCにより測定し、ラット3例での平均値を算出した。
結晶Bおよび微細結晶Bをそれぞれラットに経口投与した場合の最高血漿中濃度(Cmax)、投与時点から最後に定量できた時点までの血漿中濃度-時間曲線下面積(AUC0-t)および投与時点から無限大時間までの血漿中濃度-時間曲線下面積(AUC0-∞)を第2表に示す。
なお、HPLCの測定条件は以下の通りである。
分析機器:L-7000シリーズ(日立製作所製)
カラム:ULTRON VX-ODS(φ4.6×150mm)
カラム温度:30℃
移動相:10 mmol/L 酢酸緩衝液(pH 5.7)/アセトニトリル= 53/47
流速:1.0 mL/分
検出条件:UV 360 nm
【0031】
【表2】
【0032】
以上の結果、平均粒径の小さい微細結晶Bを経口投与した場合、結晶Bを経口投与した場合と比較して、高いCmax、AUC0-tおよびAUC0-∞が得られ、平均粒径の小さい微細結晶Bの方が結晶Bよりも優れた経口吸収性を有していることが判明した。
【0033】
以下に実施例により本発明を具体的に説明するが、これらは本発明の一態様を具体的に説明的に例示するものであって本発明を限定するものではない。
【実施例1】
【0034】
「化合物1の結晶」(1kg)をジェットミル(PJM I-1.5;日本ニューマチック社製)に投入し、供給速度50g/分、粉砕圧力0.4MPaで粉砕することにより、平均粒径24μmである「化合物1の微細結晶」(950g)を得た。なお、平均粒径は画像解析装置(Image Command 5098;オリンパス工学工業社製、湿式法)により測定した。
【実施例2】
【0035】
特開平9-040652号公報に記載の方法により、未粉砕の「化合物1の結晶」(結晶A、結晶の平均粒径;167μm)を得た。上記結晶Aをジェットミル(PJM I-1.5;日本ニューマチック社製)で、供給速度50g/分、粉砕圧力0.4MPaで粉砕することにより「化合物1の微細結晶」(微細結晶A、微細結晶の粒径D100=8.7μm;粒子の100%が8.7μm以下であることを表す、結晶化度84.6%)を得た。なお、平均粒径は画像解析装置(Image Command 5098;オリンパス工学工業社製、湿式法)により測定した。
【実施例3】
【0036】
特開平9-040652号公報に記載の方法により、未粉砕の「化合物1の結晶」(結晶B、結晶の平均粒径;181μm、結晶化度71.6%)を得た。上記結晶Bをジェットミル(PJM-100SP;日本ニューマチック社製)で、供給速度50g/分、粉砕圧力0.25MPaで粉砕することにより「化合物1の微細結晶」(微細結晶B、結晶の平均粒径;11μm、結晶化度67.3%)を得た。なお、平均粒径は画像解析装置(LUZEX(R) AP、ニコレ社製)により測定した。
【実施例4】
【0037】
錠剤(1)
実施例1で得られた化合物1の微細結晶 40 mg
ラクトース 110 mg
結晶セルロース 44 mg
ポリビニルピロリドン 4 mg
ステアリン酸マグネシウム 2 mg
上記であげた物質を混合し、通常の方法で圧縮する。
【実施例5】
【0038】
カプセル剤
実施例1で得られた化合物1の微細結晶 10 mg
ラクトース 60 mg
トウモロコシ澱粉 27 mg
ポリビニルピロリドン 2 mg
ステアリン酸マグネシウム 1 mg
上記であげた物質を混合し、通常の方法で造粒し、硬質ゼラチンカプセルに充填する。
【産業上の利用可能性】
【0039】
本発明により、例えば溶解性、安定性、吸収性、医薬製剤中の分散性等が良好な化合物1の結晶およびその結晶を含む固体医薬製剤が提供される。
【技術分野】
【0001】
本発明は、(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオン(以下、化合物1という)の結晶およびその結晶を含む固体医薬製剤に関する。
【背景技術】
【0002】
化合物1はアデノシンA2受容体拮抗作用を示し、アデノシンA2受容体亢進作用に基づく各種疾患、例えばパーキンソン病、老人性痴呆症、うつ病、喘息、骨粗しょう症等の治療等に有用である(特許文献1、特許文献2)。化合物1を含むキサンチン誘導体が、吸入投与を目的として微粉化した粉末の状態で用いられることが知られている(特許文献1)。また、化合物1の結晶が知られている(特許文献2)。上記引用文献に示された方法で合成された化合物1の結晶は、(1)水に対する溶解性が低い、(2)結晶の形状が短径数μm×長径数100μm以上の針状結晶であるという特徴を有するため、製剤化の工程操作中に化合物1の結晶が凝集するという課題を有している。水に対する溶解性が低い薬物は、消化管内での溶解性の低さと溶解速度の遅さから、一般的にバイオアベイラビリティが低いと言われている。化合物1に関しても溶解性、溶解速度等の向上、バイオアベイラビリティの改善等が望まれている。一方、製剤化の工程操作中に生じる化合物1の結晶の凝集は、化合物1の結晶および添加剤の流動性に影響を与え、製剤化工程における化合物1の結晶の取り扱いや固体製剤中の化合物1の分散性の面で問題になっている。また、化合物1は特に光に不安定で、構造中の二重結合部分(ビニレン部分)が異性化しやすいことが知られており(非特許文献1)、化合物1を含有する医薬製剤を調製する際の取り扱いに留意が必要である。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】欧州特許第0590919号明細書
【特許文献2】特開平9-040652号公報
【非特許文献】
【0004】
【非特許文献1】バイオオーガニック・メディシナル・ケミストリー・レターズ(Bioorg. Med. Chem. Lett.)、7巻、2349-2352ページ(1997年)
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明の目的は、例えば溶解性、安定性、バイオアベイラビリティ、医薬製剤中の分散性等が良好な化合物1の結晶およびその結晶を含む固体医薬製剤を提供することにある。
【課題を解決するための手段】
【0006】
本発明は、以下の(1)〜(13)に関する。
(1) 50μm未満の平均粒径を有する
【化1】
で表される(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの微細結晶。
(2) (E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの微細結晶の平均粒径が0.5〜20μmである上記(1)記載の微細結晶。
(3) 結晶化度が20%以上である上記(1)または(2)記載の(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの微細結晶。
(4) 結晶化度が30%以上である上記(1)または(2)記載の(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの微細結晶。
(5) 結晶化度が40%以上である上記(1)または(2)記載の(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの微細結晶。
(6) 上記(1)〜(5)のいずれかに記載の微細結晶を含むことを特徴とする固体医薬製剤。
(7) 結晶化度が20%以上である(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶と分散剤を含有する固体分散体。
(8) (E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶化度が30%以上である上記(7)記載の固体分散体。
(9) (E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶化度が40%以上である上記(7)記載の固体分散体。
(10) 上記(7)〜(9)のいずれかに記載の固体分散体を含むことを特徴とする固体医薬製剤。
(11) 結晶化度が20%以上である(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンを含むことを特徴とする固体医薬製剤。
(12) (E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶化度が30%以上である上記(11)記載の固体医薬製剤。
(13) (E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶化度が40%以上である上記(11)記載の固体医薬製剤。
【発明の効果】
【0007】
本発明により、例えば溶解性、安定性、吸収性、医薬製剤中の分散性等が良好な化合物1の結晶およびその結晶を含む固体医薬製剤が提供される。
【図面の簡単な説明】
【0008】
【図1】試験例1での化合物1の結晶化度と化合物1の光安定性の相関を示したものである。縦軸は化合物1の残存率(%)を表し、横軸は化合物1の結晶化度(%)を表す。
【0009】
【図2】化合物1の結晶の平均粒径と化合物1の溶解度との関係を示したものである。縦軸は化合物1の溶解度(μg/mL)を表し、横軸は経過時間(時間)を表す。グラフ上の各プロットの意味は、以下の通りである。-○-:結晶Aの溶解度(μg/mL)-▲-:微細結晶Aの溶解度(μg/mL)
【発明を実施するための形態】
【0010】
本明細書において、「(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオン」(「化合物1」)という記載は、非晶性の化合物1、結晶性の化合物1またはそれらの混合物を意味し、原料として使用される「化合物1」においては、その結晶化度、平均粒径等も限定されない。
【0011】
本発明の「50μm未満の平均粒径を有する(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの微細結晶」(「化合物1の微細結晶」)は、平均粒径が50μm未満の結晶性の化合物1であれば特に限定されないが、中でも0.5〜20μmの平均粒径を有する微細結晶が好ましい。さらに結晶化度が20%以上である「化合物1の微細結晶」が好ましく、中でも結晶化度が30%以上である「化合物1の微細結晶」がより好ましく、さらに結晶化度が40%以上である「化合物1の微細結晶」が最も好ましい。なお、これらの平均粒径は、例えばレーザー回折・錯乱式粒度分布測定装置(例えばMASTERSIZER 2000 Ver.2.00J;MALVERN社製等)や画像解析装置(例えばLUZEX(R) AP;ニコレ社製等)等を用いて測定され、粒度分布の平均値として算出される。また、これらの結晶化度は粉末X線回折装置(例えばJDX8030;日本電子製等)を用いて特定の回折角2qにおける回折ピークの積分強度を測定することにより算出される。
【0012】
本発明の「化合物1の微細結晶」の調製法は特に限定されないが、例えば欧州特許第0590919号明細書、特開平9-040652号公報等に記載の方法またはそれらに準じた方法により得られる「50μm以上の平均粒径を有する(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶」(「化合物1の結晶」)を粉砕および/または篩い分けすることにより調製され、粉砕および/または篩い分けは適宜組み合わせて数回行ってもよい。粉砕は一般的に使用される粉砕機で行うことができ、該粉砕機として、例えば乳鉢、メカノミル、ジェットミル等を用い、例えば粉砕機の回転速度、「化合物1の結晶」の供給速度、粉砕の時間等を適宜調整することにより所望の平均粒径および/または結晶化度を有する「化合物1の微細結晶」を得ることができる。中でもジェットミルによる粉砕が好ましく、例えば「化合物1の結晶」の供給速度10〜1000g/分、粉砕圧力0.01〜1.0MPaで、「化合物1の結晶」を粉砕することができる。
【0013】
本発明の「化合物1の微細結晶」を含む固体医薬製剤は、上記の「化合物1の微細結晶」を含む固体医薬製剤であればいずれでもよく、例えば
(a)上記の方法で得られる「化合物1の微細結晶」と添加剤とを混合し製剤化したもの、
(b)上記の方法で得られる「化合物1の結晶」と添加剤とを混合し、上記「化合物1の微細結晶」の調製法と同様にして得られる混合物を粉砕および/または篩い分けした後、製剤化したもの、
(c)「化合物1」と分散剤から固体分散体を調製した後、該固体分散体と添加剤とを混合し製剤化したもの
等があげられる。なお本発明の固体医薬製剤中の「化合物1の微細結晶」の含有量は、好ましくは0.001%〜80%、さらに好ましくは0.1%〜50%である。
【0014】
該固体分散体は、「化合物1」または「化合物1の結晶」とこれを分散することができる分散剤から調製される固体分散体であり、該固体分散体中の化合物1における結晶部分が上記「化合物1の微細結晶」の平均粒径または平均粒径および結晶化度を有するものであれば特に限定されない。分散剤としては、例えばヒドロキシプロピルメチルセルロース(HPMC)、ポリビニルピロリドン(PVP)、ヒドロキシプロピルセルロース(HPC)等の高分子が好ましい。また「化合物1」または「化合物1の結晶」と分散剤の配合比は、1:0.1〜1:5(重量比)であることが好ましく、中でも1:0.1〜1:3(重量比)であることが好ましい。また該固体分散体の製造法も特に限定されないが、例えば欧州特許第0590919号明細書、特開平9-040652号公報等に記載の方法またはそれらに準じた方法により得られる「化合物1」または「化合物1の結晶」と分散剤から、例えば混合粉砕法、溶媒法等の通常の方法で調製することにより得ることができる。
【0015】
混合粉砕法としては、例えば「化合物1の結晶」を分散剤とともに混合機器等を用いて混合したものを、一般的に使用される粉砕機例えば乳鉢、メカノミル、ジェットミル等を用いて粉砕する方法等があげられ、例えば粉砕機の回転速度、「化合物1の結晶」の供給速度、粉砕の時間等を適宜調整することにより所望の平均粒径または平均粒径および結晶化度を有する「化合物1の微細結晶」を含む固体分散体を得ることができる。中でもジェットミルによる粉砕が好ましい。
【0016】
溶媒法としては、例えば「化合物1」または「化合物1の結晶」を分散剤とともに有機溶媒に溶解または分散し、ついで有機溶媒を常法により減圧下または常圧下で除去する方法等があげられる。具体的には、例えば流動層造粒装置、攪拌造粒装置、噴霧造粒装置、噴霧乾燥造粒装置、真空乾燥造粒装置等が用いられ、所望により一般的に使用される粉砕機、例えば乳鉢、メカノミル、ジェットミル等を用いて粉砕する方法を組み合わせてもよい。有機溶媒としては、「化合物1」または「化合物1の結晶」を溶解するものであれば特に限定されないが、例えばジクロロメタン、ジクロロエタン、クロロホルム等のハロゲン化炭化水素類、アセトン、メチルエチルケトン等のケトン類、メタノール、エタノール等のアルコール類、テトラヒドロフラン等のエーテル類、酢酸エチル等のエステル類、ジメチルホルムアミド、ジメチルアセトアミド等のアミド類等があげられる。
【0017】
添加剤としては、例えば賦形剤、結合剤、崩壊剤、滑沢剤、可塑剤、界面活性剤、コーティング剤、着色剤、矯味剤、酸味剤等があげられ、これらは製剤の種類に応じて適宜用いることができる。
賦形剤としては、例えば白糖、ブドウ糖、蔗糖、マンニトール、ラクトース等の糖類、トウモロコシ澱粉、バレイショ澱粉等の澱粉、結晶セルロース、微結晶セルロース等のセルロース類等があげられる。
結合剤としては、例えばポリビニルアルコール、ヒドロキシプロピルセルロース、ゼラチン、メチルセルロース、エチルセルロース、ポリビニルピロリドン等があげられる。
崩壊剤としては、例えばトウモロコシ澱粉、バレイショ澱粉等の澱粉、寒天、ゼラチン末、結晶セルロース、アルギン酸ナトリウム、クロスポビドン等があげられる。
滑沢剤としては、例えばステアリン酸マグネシウム、タルク等があげられる。
可塑剤としては、例えば植物油、グリセリン等があげられる。
界面活性剤としては、例えばラウリル硫酸ナトリウム、ポリソルベート80、脂肪酸エステル等があげられる。
コーティング剤としては、例えば白糖、ヒドロキシプロピルセルロース等の糖衣、ゼラチン、グリセリン、ソルビトール等の膠衣等があげられる。
着色剤としては、例えば食用色素等があげられ、矯味剤としては、例えば、サッカリンナトリウム、アスパルテーム、ステビア等があげられ、酸味剤としては、例えばクエン酸、リンゴ酸、酒石酸等があげられる。
【0018】
「結晶化度が20%以上である(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶」(「結晶化度が20%以上である化合物1の結晶」)は、結晶化度が20%以上である結晶性の化合物1であれば特に限定されないが、中でも結晶化度が30%以上である結晶性の化合物1が好ましく、さらに結晶化度が40%以上である結晶性の化合物1がより好ましい。またこれらの製造法も特に限定されないが、例えば欧州特許第0590919号明細書、特開平9-040652号公報等に記載の方法またはそれらに準じた方法により得ることができる。
【0019】
また、「結晶化度が20%以上である化合物1の結晶」を含む固体医薬製剤は、上記の「結晶化度が20%以上である化合物1の結晶」を含む固体医薬製剤であればいずれでもよく、該固体医薬製剤の製造法も特に限定されないが、例えば上記「化合物1の微細結晶」を含む固体医薬製剤の製造法と同様の方法をあげることができる。
【0020】
また、「結晶化度が20%以上である化合物1の結晶」と分散剤を含有する固体分散体は、「化合物1」または「化合物1の結晶」とこれを分散することができる分散剤から調製される固体分散体であり、該固体分散体中の化合物1における結晶部分が20%以上の結晶化度を有するものであれば平均粒径等は特に限定されず、該固体分散体の製造法も特に限定されないが、例えば上記「化合物1の微細結晶」と分散剤を含有する固体分散体の製造法と同様の方法をあげることができる。分散剤としては、例えばHPMC、PVP、HPC等が好ましい。また「化合物1」または「化合物1の結晶」と分散剤の配合比は、1:0.1〜1:5(重量比)であることが好ましく、中でも1:0.1〜1:3(重量比)であることが好ましい。
【0021】
本発明の固体医薬製剤の製剤形態としては、例えば糖衣錠等の錠剤、散剤、顆粒剤、カプセル剤、丸剤、トローチ剤、懸濁内用液剤等があげられ、これら製剤は製剤学の技術分野においてよく知られている混合工程、粉砕工程、篩い分け工程、造粒加工工程、整粒加工工程、打錠工程、乾燥工程、カプセル充填工程、コーティング工程等の製剤化工程を組み合わせることにより製造できる。
【0022】
以下に試験例により本発明の効果を具体的に説明する。
試験例1:化合物1の結晶化度と光安定性
<試料調製方法>
以下、「化合物1の結晶」とHPMCから固体分散体を調製した。
「化合物1の結晶」としては、特開平9-040652号公報に記載の方法により得られた未粉砕の「化合物1の結晶」(結晶化度87.2%)を使用した。
試料Aは、未粉砕の「化合物1の結晶」(10g)とHPMCとを、表1に示した配合比でジクロロメタンに溶解し、溶媒を留去した後、得られた固体を錠剤粉砕機(YM-100、湯山製作所製)で、粉砕翼の回転数10000rpmで1分間粉砕することにより得られた。
試料Bは、未粉砕の「化合物1の結晶」(100g)とHPMCとを、表1に示した配合比で混合した後、表1に示した粉砕機を用いて粉砕することにより得られた。
検量線作成用の物理混合物(標品)は、未粉砕の「化合物1の結晶」とHPMCとを種々の割合でそれぞれとり、200×150mmのビニール袋中でよく振りまぜて調製した。
【0023】
<相対結晶化度の測定方法>
各試料における化合物1の結晶化度(以下、結晶化度ということもある)は、粉末X線回折装置により回折角2qを0°から40°まで変化させて各試料の回折ピークを測定し、下記の方法により算出した。
相対結晶化度の算出に用いる検量線は、種々の割合で調製した物理混合物を用いて、回折角2q=約16°における回折ピークの積分強度を測定し、それぞれの物理混合物中の「結晶性の化合物1」の量と回折ピークの積分強度の比率をプロットして作成した。なお、結晶化度87.2%である未粉砕の「化合物1の結晶」を相対結晶化度100%(「結晶性の化合物1」の含有率100%)である標準試料、HPMCを相対結晶化度0%(「結晶性の化合物1」の含有率0%)である試料とした。
試料の回折角2q=約16°における回折ピークの積分強度を測定し、上記の検量線に基づいて、測定した積分強度から各試料中の「結晶性の化合物1」の量を算出し、各試料の相対結晶化度(%)を「化合物1」の量(「結晶性の化合物1」と「非晶性の化合物1」)に対する「結晶性の化合物1」量の比率として下記の式により求めた。結晶化度(%)は、ここで求めた相対結晶化度(%)と未粉砕の「化合物1の結晶」の結晶化度87.2%から比例計算することにより決定した。
相対結晶化度(%)=(「結晶性化合物1の量」/「化合物1」の量)×100
【0024】
<光安定性の測定法>
各試料の光安定性は、下記の方法により試料中の「化合物1」の残存率(%)を測定することで追跡した。
試料を秤量してつめた透明ガラスバイアルを蛍光灯下約5000 lxで保存し、各試料とも照射開始の8時間後にサンプリングした。サンプリングした試料を水とアセトニトリルの混合溶媒(水:アセトニトリル=60:40)に溶解した後、高速液体クロマトグラフィー(HPLC)により試料中の「化合物1」の量を定量した。照射前の「化合物1」の量を100%とし、これに対する照射後の「化合物1」の量を残存率(%)として求めた。
試料AおよびBの結晶化度(%)および「化合物1」の残存率(%)を第1表に、結晶化度と安定性の相関を図1に示す。
なお、HPLCの測定条件は以下の通りである。
分析機器:LC-6シリーズ(島津製作所製)
カラム:Inertsil ODS-2(φ6×150mm)
カラム温度:25℃
移動相:50 mmol/L KH2PO4(pH 6.1, KOH)/アセトニトリル=60/40
流速:1.2 mL/分
検出条件:UV 248nm
【0025】
【表1】
【0026】
以上の結果、化合物1の結晶化度と光安定性には正の相関があり、「化合物1の結晶」の結晶化度が20%以上、好ましくは30%以上で光照射下での分解が減少することが判明した。つまり、一連の粉砕、分散化等の製剤化工程において「化合物1の結晶」の結晶化度を一定値以上に保持できれば、または一定値以上の結晶化度を有する「化合物1の結晶」を製剤化工程で用いれば、光照射下でも分解物の増加はみられず製剤化工程での化合物1の安定性を維持できると考えられる。
【0027】
試験例2:化合物1の結晶の平均粒径と溶解度
実施例2で得られた結晶A(結晶の平均粒径;167μm、2mg)および微細結晶A(微細結晶の粒径D100=8.7μm、2mg)を用いて、それぞれの水(200 mL)に対する室温での溶解度を測定した。
経過時間に対する結晶Aおよび微細結晶Aの溶解度(μg/mL)を図2に示す。
【0028】
以上の結果、平均粒径の小さい微細結晶Aは、結晶Aと比較して、溶解速度が早く、良好な化合物1の溶解性を有することが判明した。
【0029】
また、微細結晶Aでは、製剤化の工程操作中に化合物1の結晶が凝集するという現象はみられず、微細結晶Aは結晶Aと比較して分散性に優れることも判明した。
【0030】
試験例3:経口吸収性の比較
実施例3で得られた結晶B(結晶の平均粒径;181μm)および微細結晶B(結晶の平均粒径;11μm)を、それぞれ0.5重量/容量%メチルセルロース水溶液に懸濁させ、投与薬液(0.3mg/mL)を調製した。得られた投与薬液をSD系雄性ラット(体重 209〜233g;日本チャールス・リバー)に10mL/kgの容量で経口投与した。投与から0.25、0.5、1、2、4、6、8、12および24時間後の時点で、それぞれヘパリン処理されたキャピラリーチューブを用いて、経時的にラットの尾静脈より血液(各回約0.3mL)を採取した。得られた血液を遠心分離(1950×g、10分間、4℃)し、血漿を分離した。得られた血漿中の「化合物1」の濃度をHPLCにより測定し、ラット3例での平均値を算出した。
結晶Bおよび微細結晶Bをそれぞれラットに経口投与した場合の最高血漿中濃度(Cmax)、投与時点から最後に定量できた時点までの血漿中濃度-時間曲線下面積(AUC0-t)および投与時点から無限大時間までの血漿中濃度-時間曲線下面積(AUC0-∞)を第2表に示す。
なお、HPLCの測定条件は以下の通りである。
分析機器:L-7000シリーズ(日立製作所製)
カラム:ULTRON VX-ODS(φ4.6×150mm)
カラム温度:30℃
移動相:10 mmol/L 酢酸緩衝液(pH 5.7)/アセトニトリル= 53/47
流速:1.0 mL/分
検出条件:UV 360 nm
【0031】
【表2】
【0032】
以上の結果、平均粒径の小さい微細結晶Bを経口投与した場合、結晶Bを経口投与した場合と比較して、高いCmax、AUC0-tおよびAUC0-∞が得られ、平均粒径の小さい微細結晶Bの方が結晶Bよりも優れた経口吸収性を有していることが判明した。
【0033】
以下に実施例により本発明を具体的に説明するが、これらは本発明の一態様を具体的に説明的に例示するものであって本発明を限定するものではない。
【実施例1】
【0034】
「化合物1の結晶」(1kg)をジェットミル(PJM I-1.5;日本ニューマチック社製)に投入し、供給速度50g/分、粉砕圧力0.4MPaで粉砕することにより、平均粒径24μmである「化合物1の微細結晶」(950g)を得た。なお、平均粒径は画像解析装置(Image Command 5098;オリンパス工学工業社製、湿式法)により測定した。
【実施例2】
【0035】
特開平9-040652号公報に記載の方法により、未粉砕の「化合物1の結晶」(結晶A、結晶の平均粒径;167μm)を得た。上記結晶Aをジェットミル(PJM I-1.5;日本ニューマチック社製)で、供給速度50g/分、粉砕圧力0.4MPaで粉砕することにより「化合物1の微細結晶」(微細結晶A、微細結晶の粒径D100=8.7μm;粒子の100%が8.7μm以下であることを表す、結晶化度84.6%)を得た。なお、平均粒径は画像解析装置(Image Command 5098;オリンパス工学工業社製、湿式法)により測定した。
【実施例3】
【0036】
特開平9-040652号公報に記載の方法により、未粉砕の「化合物1の結晶」(結晶B、結晶の平均粒径;181μm、結晶化度71.6%)を得た。上記結晶Bをジェットミル(PJM-100SP;日本ニューマチック社製)で、供給速度50g/分、粉砕圧力0.25MPaで粉砕することにより「化合物1の微細結晶」(微細結晶B、結晶の平均粒径;11μm、結晶化度67.3%)を得た。なお、平均粒径は画像解析装置(LUZEX(R) AP、ニコレ社製)により測定した。
【実施例4】
【0037】
錠剤(1)
実施例1で得られた化合物1の微細結晶 40 mg
ラクトース 110 mg
結晶セルロース 44 mg
ポリビニルピロリドン 4 mg
ステアリン酸マグネシウム 2 mg
上記であげた物質を混合し、通常の方法で圧縮する。
【実施例5】
【0038】
カプセル剤
実施例1で得られた化合物1の微細結晶 10 mg
ラクトース 60 mg
トウモロコシ澱粉 27 mg
ポリビニルピロリドン 2 mg
ステアリン酸マグネシウム 1 mg
上記であげた物質を混合し、通常の方法で造粒し、硬質ゼラチンカプセルに充填する。
【産業上の利用可能性】
【0039】
本発明により、例えば溶解性、安定性、吸収性、医薬製剤中の分散性等が良好な化合物1の結晶およびその結晶を含む固体医薬製剤が提供される。
【特許請求の範囲】
【請求項1】
【化2】
で表される(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶を、ジェットミルを用いて粉砕して、該結晶の平均粒径を0.5μm以上50μm未満にする工程および
得られた微細結晶と添加剤を混合し、製剤化する工程を含むことを特徴とする、(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンを含有する固体医薬製剤の製造方法。
【請求項2】
(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶を、ジェットミルを用いて粉砕して、該結晶の平均粒径を0.5〜20μmにする工程および
得られた微細結晶と添加剤を混合し、製剤化する工程を含むことを特徴とする、(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンを含有する固体医薬製剤の製造方法。
【請求項3】
(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶と添加剤を混合する工程、
得られた混合物を、ジェットミルを用いて粉砕して、該結晶の平均粒径を0.5μm以上50μm未満にする工程および
得られた粉砕後の混合物を製剤化する工程を含むことを特徴とする、(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンを含有する固体医薬製剤の製造方法。
【請求項4】
(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶と添加剤を混合する工程、
得られた混合物を、ジェットミルを用いて粉砕して、該結晶の平均粒径を0.5〜20μmにする工程および
得られた粉砕後の混合物を製剤化する工程を含むことを特徴とする、(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンを含有する固体医薬製剤の製造方法。
【請求項1】
【化2】
で表される(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶を、ジェットミルを用いて粉砕して、該結晶の平均粒径を0.5μm以上50μm未満にする工程および
得られた微細結晶と添加剤を混合し、製剤化する工程を含むことを特徴とする、(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンを含有する固体医薬製剤の製造方法。
【請求項2】
(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶を、ジェットミルを用いて粉砕して、該結晶の平均粒径を0.5〜20μmにする工程および
得られた微細結晶と添加剤を混合し、製剤化する工程を含むことを特徴とする、(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンを含有する固体医薬製剤の製造方法。
【請求項3】
(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶と添加剤を混合する工程、
得られた混合物を、ジェットミルを用いて粉砕して、該結晶の平均粒径を0.5μm以上50μm未満にする工程および
得られた粉砕後の混合物を製剤化する工程を含むことを特徴とする、(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンを含有する固体医薬製剤の製造方法。
【請求項4】
(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンの結晶と添加剤を混合する工程、
得られた混合物を、ジェットミルを用いて粉砕して、該結晶の平均粒径を0.5〜20μmにする工程および
得られた粉砕後の混合物を製剤化する工程を含むことを特徴とする、(E)-8-(3,4-ジメトキシスチリル)-1,3-ジエチル-7-メチル-3,7-ジヒドロ-1H-プリン-2,6-ジオンを含有する固体医薬製剤の製造方法。
【図1】

【図2】
【図2】
【公開番号】特開2010−195836(P2010−195836A)
【公開日】平成22年9月9日(2010.9.9)
【国際特許分類】
【出願番号】特願2010−138014(P2010−138014)
【出願日】平成22年6月17日(2010.6.17)
【分割の表示】特願2005−506044(P2005−506044)の分割
【原出願日】平成16年5月7日(2004.5.7)
【出願人】(000001029)協和発酵キリン株式会社 (276)
【Fターム(参考)】
【公開日】平成22年9月9日(2010.9.9)
【国際特許分類】
【出願日】平成22年6月17日(2010.6.17)
【分割の表示】特願2005−506044(P2005−506044)の分割
【原出願日】平成16年5月7日(2004.5.7)
【出願人】(000001029)協和発酵キリン株式会社 (276)
【Fターム(参考)】
[ Back to top ]