
微細自己集合体
【課題】 糖鎖にオリゴ糖を用いて微細自己凝集体を作成することにより、独特の高次構造を有する微細自己凝集体を得た。
【解決手段】 本発明は、下記一般式
【化1】

(式中、Gは2〜5の単糖が結合したオリゴ糖残基(二糖類を除く)を表し、Rは炭素数6〜25の炭化水素基を表す。)で表わされる構造を有するO−グリコシド型糖脂質を水に分散し、この分散液から凝集させることにより得られた微細自己集合体。この微細自己集合体は、マイクロダブルヘリカルファイバー状凝集体、マイクロヘリカルディスク状凝集体、微細チューブ状凝集体、及びマイクロファイバー状凝集体などの形態をとる。
【解決手段】 本発明は、下記一般式
【化1】
(式中、Gは2〜5の単糖が結合したオリゴ糖残基(二糖類を除く)を表し、Rは炭素数6〜25の炭化水素基を表す。)で表わされる構造を有するO−グリコシド型糖脂質を水に分散し、この分散液から凝集させることにより得られた微細自己集合体。この微細自己集合体は、マイクロダブルヘリカルファイバー状凝集体、マイクロヘリカルディスク状凝集体、微細チューブ状凝集体、及びマイクロファイバー状凝集体などの形態をとる。
【発明の詳細な説明】
【技術分野】
【0001】
この発明は、O−グリコシド型糖脂質から成る微細自己集合体に関し、より詳細には、糖鎖にオリゴ糖を用いたO−グリコシド型糖脂質から成る微細自己集合体に関する。
【背景技術】
【0002】
従来の天然植物資源から分離精製したカルダノールを出発原料として合成されたグルコース置換長鎖アルキルフェノール誘導体であるカルダニルグルコシドは、水中において加熱溶解し徐冷することで糖鎖の水素結合によりナノチューブ状凝集体を形成することが知られている(非特許文献1)。
【0003】
【非特許文献1】G. John, M. Masuda, Y. Okada, K. Yase, and T. Shimizu, Adv. Mat., 13, 715 (2001)
【発明の開示】
【発明が解決しようとする課題】
【0004】
糖部分がグルコースの場合には糖鎖置換長鎖アルキルフェノール誘導体の微細自己凝集体が製造されるが、このグルコースの代わりにガラクトースを用いて、同様に糖鎖置換長鎖アルキルフェノール誘導体であるカルダニルガラクトシドを作製すると、これは水中において加熱溶解し徐冷すると結晶になる。
このような知見に基づき、本発明では、糖鎖にオリゴ糖を用いて糖鎖置換長鎖炭化水素フェノール誘導体を作製し、その凝集物の形態についての検討を行った。
【課題を解決するための手段】
【0005】
本発明は、糖鎖にオリゴ糖を用いることにより、オリゴ糖由来の特性を微細自己凝集体に付加させ、独特の高次構造を有する微細自己凝集体を作成することに成功した。
【0006】
即ち、本発明は、下記一般式
【化1】
(式中、Gは2〜5の単糖が結合したオリゴ糖残基を表し、Rは炭素数6〜25の炭化水素基を表す。)で表わされる構造を有するO−グリコシド型糖脂質から成る微細自己集合体である。
【発明の効果】
【0007】
本発明の微細自己集合体は、ナノファイバー状凝集体、マイクロファイバー状凝集体、マイクロヘリカルファイバー状凝集体、ナノチューブ状凝集体、及びマイクロヘリカルディスク状凝集体などの様々な高次構造を有するO−グリコシド型糖脂質の微細自己凝集体であり、また用いたオリゴ糖の特性が付加された微細自己凝集体である。この微細自己集合体は、医療用の判定剤、吸着剤として用いることができる他、食品化学工業、農林業、繊維加工業、電子情報などの分野において乳化剤、安定剤、分散剤、湿潤剤、及びナノ若しくはマイクロ部品等として利用できる。
【発明を実施するための最良の形態】
【0008】
本発明で用いる界面活性有機化合物は、下記一般式
【化1】
で表わされるO−グリコシド型糖脂質である。
本発明においては、炭化水素基(R)は−O−G基に対してo位、m位又はp位のいずれにあってもよいが、メタ(m)位にあることが好ましい。
前記一般式(化1)におけるGは、糖残基数2〜5、好ましくは2のオリゴ糖鎖の還元末端水酸基を除いた残基、すなわち還元末端の炭素原子がO−グリコシド結合に関与しているオリゴ糖残基である。このようなものとしては、例えばラクトース、メリビオース、セロビオース、及びガラクトシル−α(1→4)ガラクトースなど市販で入手可能な化合物、又は化学合成あるいは酵素合成で得られるオリゴ糖などが挙げられる。還元末端のアノマー位のグリコシド結合はα−アノマー及びβ−アノマー及びそれらの混合物のいずれであってもよい。
【0009】
一方、前記一般式(化1)におけるRは、炭素数が6〜25、好ましくは14〜16、より好ましくは15の炭化水素基であり、好ましくは飽和又は二重結合を1〜5、好ましくは1〜3含む不飽和の脂肪族炭化水素から成る脂肪族炭化水素である。この炭化水素は好ましくは直鎖である。このような炭化水素基としては、例えば、ドデシル基、トリデシル基、テトラデシル基、ペンタデシル基、ヘキサデシル基、ヘプタデシル基、オクタデシル基や、これらに不飽和結合としてモノエン、ジエン、トリエンなどを含むものが挙げられるが、原料の入手が容易であるという点で、8−ペンタデセニル基、8,10−ペンタデカジエニル基、8,10、12−ペンタデカトリエニル基が好ましい。
【0010】
前記一般式(化1)で表わされるオリゴ糖置換長鎖炭化水素フェノール誘導体は例えば次に示す方法により製造することができる。
水酸基がすべてアセチル基で保護されたオリゴ糖を一般式
【化2】
(式中、Rは前記と同様である。)で表わされる長鎖炭化水素フェノールに添加し、水酸基部分にグリコシド結合させ、次に糖残基のアセチル保護基を除去することにより、オリゴ糖置換長鎖炭化水素フェノール誘導体を得ることができる。
【0011】
水酸基がすべてアセチル基で保護されたオリゴ糖として、ラクトース オクタアセテート、セロビオース オクタアセテートなどの市販の化合物を用いるか、メリビオース、4-O-α-D-ガラクトピラノシル-D-ガラクトピラノースなどの市販のオリゴ糖や化学合成あるいは酵素合成して得られたオリゴ糖をアセチル化したものを用いることができる。
アセチル化の手順として下記に一例を示す。オリゴ糖をドライピリジンに溶解させ、ジメチルアミノピリジンを加え、室温〜40℃で1時間〜1晩磁気撹拌する。この際、無水酢酸を反応系に加えておくと反応が速く進行する。トルエンとエタノール の混合溶媒で5〜7回共沸させピリジンを除き、残渣をクロロホルムに溶かし、飽和炭酸水素ナトリウム水溶液及び水で数回洗う。濃縮しシリカゲルカラムで精製し、濃縮して乾燥させる。
【0012】
次に本発明のオリゴ糖置換長鎖炭化水素フェノール誘導体を製造するための望ましい態様について説明する。
前記一般式(化2)で表わされる長鎖炭化水素フェノールの水酸基部分に水酸基がすべてアセチル基で保護された糖残基数2〜30のオリゴ糖をグリコシド結合させる。この反応においてオリゴ糖の還元末端のアノマー位を活性化させるために活性化剤としてトリメチルシリル トリフルオロメタンスルフォネートやボロン トリフルオライド−エチル エーテル 錯体を加えるが、オリゴ糖:長鎖アルキルフェノール:活性化剤=1 : 1〜1.5 : 1〜1.2のモル比で混合、反応させることが望ましい。用いるオリゴ糖や長鎖アルキルフェノールの種類に合わせて活性剤の種類や反応温度を変えると収率が高くなる。例えば、反応しにくいオリゴ糖や長鎖アルキルフェノールの場合、活性化剤としてボロン トリフルオライド−エチル エーテル 錯体を反応温度は室温にするなどやや厳しい条件にした方がよい。ただし、あまり厳しい条件に設定するとオリゴ糖鎖が分解するので収率が低くなる。比較的反応しやすいオリゴ糖や長鎖アルキルフェノールの場合は活性化剤としてトリメチルシリル トリフルオロメタンスルフォネートを反応温度は0℃付近という温和な条件が望ましい。得られた化合物を脱アセチル化するためにメタノールに溶解し、室温でナトリウムメトキシド/メタノール溶液を作用させる。強酸性イオン交換樹脂を加えることで中和させ、シリカゲルカラムクロマトグラフィーで精製することで、前記一般式(化1)のオリゴ糖置換長鎖炭化水素フェノール誘導体を得ることができる。
【0013】
本発明の微細自己集合体の製法に制限はないが、上記O−グリコシド型糖脂質を水に分散後、マントルヒーターを用いて加熱、約20分沸騰し、室温まで自然冷却、ナノチューブが出来るまで室温に放置することにより得ることができる(特願2000−271192、特願2001−363762等)。更に詳細には、得られた前記一般式(化1)のオリゴ糖置換長鎖炭化水素フェノール誘導体1 mgを水中40 mLに60℃で超音波により分散させ、120℃で加熱溶解させる。徐冷し放置すると水分散液として微細自己凝集体を得る。得られた凝集体の形態、大きさなどは溶液を乾かさずに、あるいは溶液を基板に滴下乾燥させるか、凍結乾燥させてから基板に置き、光学顕微鏡及び走査型電子顕微鏡で観察することにより確認できる。
この微細自己集合体を製造する際に、上記化1で表されるO−グリコシド型糖脂質の一種類(単一品)を用いてもよいし、2種以上の混合物(例えば、実施例1で用いた天然カルダノール(4種類の混合物)から成る脂質から成るO−グリコシド型糖脂質の混合物等)を用いてもよい。
【実施例】
【0014】
以下、実施例にて本発明を例証するが、本発明を限定することを意図するものではない。
製造例1
カシューナッツオイルを約400Paで2回真空蒸留し、220℃から235℃の沸点をもつ成分を集めてカルダノールを得た。100 mLナス型フラスコにβ-ラクトース オクタアセテート(SIGMA社製)2.00 g、カルダノール0.89 g、モレキュラーシーブ4A 3 g、及びドライトルエン 30 mLを加えて窒素雰囲気下室温で1時間磁気撹拌した。反応系を氷浴により0℃に冷却してから、窒素雰囲気下でシリンジに測り取ったトリメチルシリル トリフルオロメタンスルフォネート0.53 mLを滴下ロートにより反応系に滴下した。2時間磁気撹拌した後、反応系にトリエチルアミン4.1 mLを加えてしばらくの間磁気撹拌することにより反応を停止させた。酢酸エチルで洗いながらセライトで吸引ろ過し、濃縮した。シリカゲルカラム(カラムの直径:5 cm;カラムの長さ:30 cm;シリカゲル粉末体積:550 mL;展開溶媒:トルエン : 酢酸エチル = 2 : 1;ドレイン:250 mL)に添加し、フラクションコレクターにより約20 mLずつ分画した。29番目から53番目のフラクションを集めて濃縮してクーゲル(60℃、30分間)で乾燥させ、カルダニル-2,3,4, 6,2',3',6'-ヘプタ-O-アセチル-β-D-ラクトシドを得た。収量:0.39 g。Rf = 0.45(展開溶媒:トルエン : 酢酸エチル = 2 : 1)。
【0015】
次に、50 mLナス型フラスコに上記で得たカルダニル-2,3,4, 6,2',3',6'-ヘプタ-O-アセチル-β-D-ラクトシド300 mgを採り、メタノール 3 mL及びテトラヒドロフラン 3 mLを加えて溶解させた。28 %ナトリウムメトキシド/メタノール溶液3滴を加えて溶液のpHを調べてみるとpH = 9であった。室温で2時間磁気撹拌した。反応系にDowex 50を加えて反応系を中和することにより反応を停止させた。テトラヒドロフランで洗いながらセライトにより吸引ろ過してDowex 50の粒を除いてから濃縮した。シリカゲルカラム(カラムの直径:5 cm;カラムの長さ:30 cm;シリカゲル粉末体積:450 mL;展開溶媒:クロロホルム : メタノール = 10 : 3;ドレイン:230 mL)に添加し、フラクションコレクターにより約10 mLずつ分画した。41番目から200番目のフラクションを集めて濃縮してクーゲル(60℃、30分間)で乾燥させ、カルダニル(混合型)-β-D-ラクトシドを得た。収量:184 mg。Rf = 0.61(展開溶媒:クロロホルム : メタノール = 10 : 3)。
1H-NMR (400 MHz; CD3OD; r.t.): δ7.19-6.82 (m, フェニル基), 5.36 (m, -CH2CHCHCH2-), 4.93 (d, J1,2 = 7.2 Hz, H-1), 4.39 (d, J1,2 = 7.6 Hz, H-1'), 3.90 - 3.13 (m, 糖鎖), 2.79 (m, -CHCHCH2CHCH-), 2.57 (t, J = 7.6 Hz, -C6H4CH2-), 2.04 (m, -CH2CH2CHCHCH2 CH2-), 1.60 - 1.29 (m, -CH2-), 0.90 ppm (m, -CH3).
【0016】
実施例1
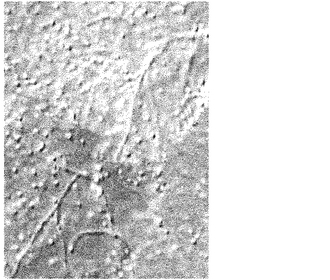
500 mLナス型フラスコに製造例1で得たカルダニル-β-D-ラクトシド5 mgを採り、水200 mLを加えた。マントルヒーターを用いて4時間磁気撹拌しながら還流させた。数日間室温で放置して置くと、白い沈殿物が生じた。この沈殿物の形状を光学顕微鏡及び透過型電子顕微鏡で確認したところ、図1及び2に示すナノファイバー状凝集体が観察された。このナノファイバー状凝集体は、直径が約800 nmの枝別れしたファイバーであった。
【0017】
製造例2
カシューナッツオイルを蒸留することで分離した飽和型、モノエン型、ジエン型、及びトリエン型のカルダノールの混合物約5mLをシリカゲルカラム(カラムの直径:5 cm;カラムの長さ:30 cm;シリカゲル粉末体積:450 mL;展開溶媒:トルエン : 酢酸エチル = 10 : 1;ドレイン:250 mL)に添加し、フラクションコレクターにより約20 mLずつ分画した。11番目から23番目のフラクションを集めて濃縮してクーゲル(60℃、30分間)で乾燥させた。
得られたカルダノール 584 mgをシリカゲルカラム(カラムの直径:3 cm;カラムの長さ:30 cm;シリカゲル粉末体積:200 mL;展開溶媒:ヘキサン : 酢酸エチル = 9 : 1;ドレイン:80 mL)に添加し、フラクションコレクターにより約7 mLずつ分画した。40番目から46番目のフラクションを集めて濃縮してクーゲル(60℃、30分間)で乾燥させた。得られたカルダノールはモノエン型のみのカルダノールであった。収量:143 mg。Rf = 0.63(展開溶媒:ヘキサン : 酢酸エチル = 9 : 1)。
1H-NMR (400 MHz; CDCl3; r.t.): δ7.16-6.63 (m, フェニル基), 5.34 (m, -CH2CHCHCH2-), 4.67 (d, J = 4.4 Hz, -OH), 2.55 (t, J = 7.8 Hz, -C6H4CH2-), 2.02 (m, -CH2CHCHCH2-), 1.61 - 1.26 (m, -CH2-), 0.88 ppm (t, J = 6.8 Hz, -CH3).
【0018】
製造例3
50 mLナス型フラスコにβ-ラクトース オクタアセテート271 mg、製造例3で得たカルダノール(モノエン型)121 mg、モレキュラーシーブ 4A 0.4 g、及びドライトルエン 4 mLを加えてアルゴン雰囲気下室温で1時間磁気撹拌した。反応系を氷浴により0℃に冷却してから、アルゴン雰囲気下でシリンジに測り取ったトリメチルシリル トリフルオロメタンスルフォネート72 μLをシリンジにより反応系に滴下した。2時間磁気撹拌した後、反応系にトリエチルアミン0.6 mLを加えてしばらくの間磁気撹拌することにより反応を停止させた。酢酸エチルで洗いながらセライトで吸引ろ過し、濃縮した。シリカゲルカラム(カラムの直径:2 cm;カラムの長さ:30 cm;シリカゲル粉末体積:80 mL;展開溶媒:トルエン : 酢酸エチル = 2 : 1;ドレイン:40 mL)に添加し、フラクションコレクターにより約6 mLずつ分画した。16番目から22番目のフラクションを集めて濃縮してクーゲル(60℃、30分間)で乾燥させ、カルダニル(モノエン型)-2,3,4, 6,2',3',6'-ヘプタ-O-アセチル-β-D-ラクトシドを得た。収量:54 mg。Rf = 0.39(展開溶媒:トルエン : 酢酸エチル = 2 : 1)。
【0019】
30 mLナス型フラスコに上記で得たカルダニル(モノエン型)-2,3,4, 6,2',3',6'-ヘプタ-O-アセチル-β-D-ラクトシド54 mgを採り、メタノール 1 mL及びテトラヒドロフラン 1 mLを加えて溶解させた。28 %ナトリウムメトキシド/メタノール溶液1滴を加えて溶液のpHを調べてみるとpH = 9であった。室温で1時間磁気撹拌した。反応系にDowex 50を加えて反応系を中和することにより反応を停止させた。テトラヒドロフランで洗いながらセライトにより吸引ろ過してDowex 50の粒を除いてから濃縮した。シリカゲルカラム(カラムの直径:1 cm;カラムの長さ:30 cm;シリカゲル粉末体積:50 mL;展開溶媒:クロロホルム : メタノール = 5 : 1;ドレイン:0 mL)に添加し、フラクションコレクターにより約10 mLずつ分画した。20番目から45番目のフラクションを集めて濃縮してクーゲル(60℃、30分間)で乾燥させ、カルダニル(モノエン型)-β-D-ラクトシドを得た。収量:28 mg。Rf = 0.29(展開溶媒:クロロホルム : メタノール = 10 : 3)。
元素分析:C33H54O11; calc.: C: 63.24%, H: 8.68%; found: C: 63.23%, H: 8.86%. 1H-NMR (400 MHz; CD3OD; r.t.): δ 7.19-6.82 (m, フェニル基), 5.34 (m, -CH2CHCHCH2-), 4.93 (d, J1,2 = 7.6 Hz, H-1), 4.39 (d, J1,2 = 7.6 Hz, H-1'), 3.93 - 3.13 (m, 糖鎖), 2.57 (t, J = 7.8 Hz, -C6H4CH2-), 2.02 (m, -CH2CHCHCH2-), 1.60 - 1.28 (m, -CH2-), 0.90 ppm (t, J = 6.8 Hz, -CH3).
【0020】
実施例2
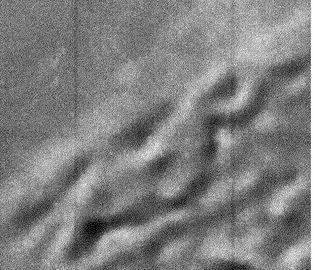
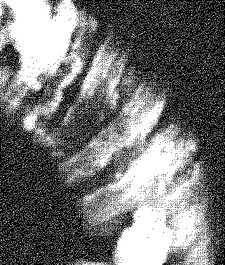
200 mL三角フラスコに製造例3で得たカルダニル(モノエン型)-β-D-ラクトシド1 mgを採り、水40 mLを加えた。60℃の水浴中で超音波に20分間曝してからオートクレーブで120℃20分間加熱した。そのままオートクレーブの蓋を開けずに一晩オートクレーブ中に放置しておいてから室温に取り出すと、白い沈殿物が生じていた。この沈殿物の形状を光学顕微鏡及び走査型電子顕微鏡で確認したところ、マイクロダブルヘリカルファイバー状凝集体(図3及び4)、マイクロヘリカルディスク状凝集体(図5及び6)、微細チューブ状凝集体、及びマイクロファイバー状凝集体などの混合物が観察された。
図3及び4に示すマイクロダブルヘリカルファイバー状凝集体は、幅が約3μmの2本のヘリカルリボン状凝集体が緩やかな角度で交互に絡み合って直径が約8μmの網目を連続して形成した凝集体であった。
図5及び6に示すマイクロヘリカルディスク状凝集体は、幅が約1μmのヘリカルリボン状凝集体が急な角度で巻いてディスク(円盤)状に近い形になったものがさらにお互いに中心軸方向に凝集して直径が約4〜5μmの円柱体を形成した凝集体であった。
【図面の簡単な説明】
【0021】
【図1】実施例1で得たナノファイバー状凝集体の光学顕微鏡像を示す図である。写真のサイズは縦175μm×横127μmである。
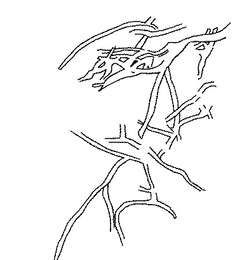
【図2】図1をトレースした図である。
【図3】実施例2で得られたマイクロダブルヘリカルファイバー状凝集体の光学顕微鏡像を示す図である。写真のサイズは縦28μm×横36μmである。
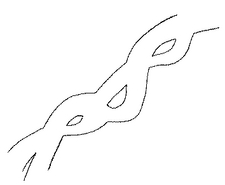
【図4】図3をトレースした図である。
【図5】実施例2で得られたマイクロヘリカルディスク状凝集体の走査型電子顕微鏡像を示す図である。写真のサイズは縦8.3μm×横7.1μmである。
【図6】図5をトレースした図である。
【技術分野】
【0001】
この発明は、O−グリコシド型糖脂質から成る微細自己集合体に関し、より詳細には、糖鎖にオリゴ糖を用いたO−グリコシド型糖脂質から成る微細自己集合体に関する。
【背景技術】
【0002】
従来の天然植物資源から分離精製したカルダノールを出発原料として合成されたグルコース置換長鎖アルキルフェノール誘導体であるカルダニルグルコシドは、水中において加熱溶解し徐冷することで糖鎖の水素結合によりナノチューブ状凝集体を形成することが知られている(非特許文献1)。
【0003】
【非特許文献1】G. John, M. Masuda, Y. Okada, K. Yase, and T. Shimizu, Adv. Mat., 13, 715 (2001)
【発明の開示】
【発明が解決しようとする課題】
【0004】
糖部分がグルコースの場合には糖鎖置換長鎖アルキルフェノール誘導体の微細自己凝集体が製造されるが、このグルコースの代わりにガラクトースを用いて、同様に糖鎖置換長鎖アルキルフェノール誘導体であるカルダニルガラクトシドを作製すると、これは水中において加熱溶解し徐冷すると結晶になる。
このような知見に基づき、本発明では、糖鎖にオリゴ糖を用いて糖鎖置換長鎖炭化水素フェノール誘導体を作製し、その凝集物の形態についての検討を行った。
【課題を解決するための手段】
【0005】
本発明は、糖鎖にオリゴ糖を用いることにより、オリゴ糖由来の特性を微細自己凝集体に付加させ、独特の高次構造を有する微細自己凝集体を作成することに成功した。
【0006】
即ち、本発明は、下記一般式
【化1】
(式中、Gは2〜5の単糖が結合したオリゴ糖残基を表し、Rは炭素数6〜25の炭化水素基を表す。)で表わされる構造を有するO−グリコシド型糖脂質から成る微細自己集合体である。
【発明の効果】
【0007】
本発明の微細自己集合体は、ナノファイバー状凝集体、マイクロファイバー状凝集体、マイクロヘリカルファイバー状凝集体、ナノチューブ状凝集体、及びマイクロヘリカルディスク状凝集体などの様々な高次構造を有するO−グリコシド型糖脂質の微細自己凝集体であり、また用いたオリゴ糖の特性が付加された微細自己凝集体である。この微細自己集合体は、医療用の判定剤、吸着剤として用いることができる他、食品化学工業、農林業、繊維加工業、電子情報などの分野において乳化剤、安定剤、分散剤、湿潤剤、及びナノ若しくはマイクロ部品等として利用できる。
【発明を実施するための最良の形態】
【0008】
本発明で用いる界面活性有機化合物は、下記一般式
【化1】
で表わされるO−グリコシド型糖脂質である。
本発明においては、炭化水素基(R)は−O−G基に対してo位、m位又はp位のいずれにあってもよいが、メタ(m)位にあることが好ましい。
前記一般式(化1)におけるGは、糖残基数2〜5、好ましくは2のオリゴ糖鎖の還元末端水酸基を除いた残基、すなわち還元末端の炭素原子がO−グリコシド結合に関与しているオリゴ糖残基である。このようなものとしては、例えばラクトース、メリビオース、セロビオース、及びガラクトシル−α(1→4)ガラクトースなど市販で入手可能な化合物、又は化学合成あるいは酵素合成で得られるオリゴ糖などが挙げられる。還元末端のアノマー位のグリコシド結合はα−アノマー及びβ−アノマー及びそれらの混合物のいずれであってもよい。
【0009】
一方、前記一般式(化1)におけるRは、炭素数が6〜25、好ましくは14〜16、より好ましくは15の炭化水素基であり、好ましくは飽和又は二重結合を1〜5、好ましくは1〜3含む不飽和の脂肪族炭化水素から成る脂肪族炭化水素である。この炭化水素は好ましくは直鎖である。このような炭化水素基としては、例えば、ドデシル基、トリデシル基、テトラデシル基、ペンタデシル基、ヘキサデシル基、ヘプタデシル基、オクタデシル基や、これらに不飽和結合としてモノエン、ジエン、トリエンなどを含むものが挙げられるが、原料の入手が容易であるという点で、8−ペンタデセニル基、8,10−ペンタデカジエニル基、8,10、12−ペンタデカトリエニル基が好ましい。
【0010】
前記一般式(化1)で表わされるオリゴ糖置換長鎖炭化水素フェノール誘導体は例えば次に示す方法により製造することができる。
水酸基がすべてアセチル基で保護されたオリゴ糖を一般式
【化2】
(式中、Rは前記と同様である。)で表わされる長鎖炭化水素フェノールに添加し、水酸基部分にグリコシド結合させ、次に糖残基のアセチル保護基を除去することにより、オリゴ糖置換長鎖炭化水素フェノール誘導体を得ることができる。
【0011】
水酸基がすべてアセチル基で保護されたオリゴ糖として、ラクトース オクタアセテート、セロビオース オクタアセテートなどの市販の化合物を用いるか、メリビオース、4-O-α-D-ガラクトピラノシル-D-ガラクトピラノースなどの市販のオリゴ糖や化学合成あるいは酵素合成して得られたオリゴ糖をアセチル化したものを用いることができる。
アセチル化の手順として下記に一例を示す。オリゴ糖をドライピリジンに溶解させ、ジメチルアミノピリジンを加え、室温〜40℃で1時間〜1晩磁気撹拌する。この際、無水酢酸を反応系に加えておくと反応が速く進行する。トルエンとエタノール の混合溶媒で5〜7回共沸させピリジンを除き、残渣をクロロホルムに溶かし、飽和炭酸水素ナトリウム水溶液及び水で数回洗う。濃縮しシリカゲルカラムで精製し、濃縮して乾燥させる。
【0012】
次に本発明のオリゴ糖置換長鎖炭化水素フェノール誘導体を製造するための望ましい態様について説明する。
前記一般式(化2)で表わされる長鎖炭化水素フェノールの水酸基部分に水酸基がすべてアセチル基で保護された糖残基数2〜30のオリゴ糖をグリコシド結合させる。この反応においてオリゴ糖の還元末端のアノマー位を活性化させるために活性化剤としてトリメチルシリル トリフルオロメタンスルフォネートやボロン トリフルオライド−エチル エーテル 錯体を加えるが、オリゴ糖:長鎖アルキルフェノール:活性化剤=1 : 1〜1.5 : 1〜1.2のモル比で混合、反応させることが望ましい。用いるオリゴ糖や長鎖アルキルフェノールの種類に合わせて活性剤の種類や反応温度を変えると収率が高くなる。例えば、反応しにくいオリゴ糖や長鎖アルキルフェノールの場合、活性化剤としてボロン トリフルオライド−エチル エーテル 錯体を反応温度は室温にするなどやや厳しい条件にした方がよい。ただし、あまり厳しい条件に設定するとオリゴ糖鎖が分解するので収率が低くなる。比較的反応しやすいオリゴ糖や長鎖アルキルフェノールの場合は活性化剤としてトリメチルシリル トリフルオロメタンスルフォネートを反応温度は0℃付近という温和な条件が望ましい。得られた化合物を脱アセチル化するためにメタノールに溶解し、室温でナトリウムメトキシド/メタノール溶液を作用させる。強酸性イオン交換樹脂を加えることで中和させ、シリカゲルカラムクロマトグラフィーで精製することで、前記一般式(化1)のオリゴ糖置換長鎖炭化水素フェノール誘導体を得ることができる。
【0013】
本発明の微細自己集合体の製法に制限はないが、上記O−グリコシド型糖脂質を水に分散後、マントルヒーターを用いて加熱、約20分沸騰し、室温まで自然冷却、ナノチューブが出来るまで室温に放置することにより得ることができる(特願2000−271192、特願2001−363762等)。更に詳細には、得られた前記一般式(化1)のオリゴ糖置換長鎖炭化水素フェノール誘導体1 mgを水中40 mLに60℃で超音波により分散させ、120℃で加熱溶解させる。徐冷し放置すると水分散液として微細自己凝集体を得る。得られた凝集体の形態、大きさなどは溶液を乾かさずに、あるいは溶液を基板に滴下乾燥させるか、凍結乾燥させてから基板に置き、光学顕微鏡及び走査型電子顕微鏡で観察することにより確認できる。
この微細自己集合体を製造する際に、上記化1で表されるO−グリコシド型糖脂質の一種類(単一品)を用いてもよいし、2種以上の混合物(例えば、実施例1で用いた天然カルダノール(4種類の混合物)から成る脂質から成るO−グリコシド型糖脂質の混合物等)を用いてもよい。
【実施例】
【0014】
以下、実施例にて本発明を例証するが、本発明を限定することを意図するものではない。
製造例1
カシューナッツオイルを約400Paで2回真空蒸留し、220℃から235℃の沸点をもつ成分を集めてカルダノールを得た。100 mLナス型フラスコにβ-ラクトース オクタアセテート(SIGMA社製)2.00 g、カルダノール0.89 g、モレキュラーシーブ4A 3 g、及びドライトルエン 30 mLを加えて窒素雰囲気下室温で1時間磁気撹拌した。反応系を氷浴により0℃に冷却してから、窒素雰囲気下でシリンジに測り取ったトリメチルシリル トリフルオロメタンスルフォネート0.53 mLを滴下ロートにより反応系に滴下した。2時間磁気撹拌した後、反応系にトリエチルアミン4.1 mLを加えてしばらくの間磁気撹拌することにより反応を停止させた。酢酸エチルで洗いながらセライトで吸引ろ過し、濃縮した。シリカゲルカラム(カラムの直径:5 cm;カラムの長さ:30 cm;シリカゲル粉末体積:550 mL;展開溶媒:トルエン : 酢酸エチル = 2 : 1;ドレイン:250 mL)に添加し、フラクションコレクターにより約20 mLずつ分画した。29番目から53番目のフラクションを集めて濃縮してクーゲル(60℃、30分間)で乾燥させ、カルダニル-2,3,4, 6,2',3',6'-ヘプタ-O-アセチル-β-D-ラクトシドを得た。収量:0.39 g。Rf = 0.45(展開溶媒:トルエン : 酢酸エチル = 2 : 1)。
【0015】
次に、50 mLナス型フラスコに上記で得たカルダニル-2,3,4, 6,2',3',6'-ヘプタ-O-アセチル-β-D-ラクトシド300 mgを採り、メタノール 3 mL及びテトラヒドロフラン 3 mLを加えて溶解させた。28 %ナトリウムメトキシド/メタノール溶液3滴を加えて溶液のpHを調べてみるとpH = 9であった。室温で2時間磁気撹拌した。反応系にDowex 50を加えて反応系を中和することにより反応を停止させた。テトラヒドロフランで洗いながらセライトにより吸引ろ過してDowex 50の粒を除いてから濃縮した。シリカゲルカラム(カラムの直径:5 cm;カラムの長さ:30 cm;シリカゲル粉末体積:450 mL;展開溶媒:クロロホルム : メタノール = 10 : 3;ドレイン:230 mL)に添加し、フラクションコレクターにより約10 mLずつ分画した。41番目から200番目のフラクションを集めて濃縮してクーゲル(60℃、30分間)で乾燥させ、カルダニル(混合型)-β-D-ラクトシドを得た。収量:184 mg。Rf = 0.61(展開溶媒:クロロホルム : メタノール = 10 : 3)。
1H-NMR (400 MHz; CD3OD; r.t.): δ7.19-6.82 (m, フェニル基), 5.36 (m, -CH2CHCHCH2-), 4.93 (d, J1,2 = 7.2 Hz, H-1), 4.39 (d, J1,2 = 7.6 Hz, H-1'), 3.90 - 3.13 (m, 糖鎖), 2.79 (m, -CHCHCH2CHCH-), 2.57 (t, J = 7.6 Hz, -C6H4CH2-), 2.04 (m, -CH2CH2CHCHCH2 CH2-), 1.60 - 1.29 (m, -CH2-), 0.90 ppm (m, -CH3).
【0016】
実施例1
500 mLナス型フラスコに製造例1で得たカルダニル-β-D-ラクトシド5 mgを採り、水200 mLを加えた。マントルヒーターを用いて4時間磁気撹拌しながら還流させた。数日間室温で放置して置くと、白い沈殿物が生じた。この沈殿物の形状を光学顕微鏡及び透過型電子顕微鏡で確認したところ、図1及び2に示すナノファイバー状凝集体が観察された。このナノファイバー状凝集体は、直径が約800 nmの枝別れしたファイバーであった。
【0017】
製造例2
カシューナッツオイルを蒸留することで分離した飽和型、モノエン型、ジエン型、及びトリエン型のカルダノールの混合物約5mLをシリカゲルカラム(カラムの直径:5 cm;カラムの長さ:30 cm;シリカゲル粉末体積:450 mL;展開溶媒:トルエン : 酢酸エチル = 10 : 1;ドレイン:250 mL)に添加し、フラクションコレクターにより約20 mLずつ分画した。11番目から23番目のフラクションを集めて濃縮してクーゲル(60℃、30分間)で乾燥させた。
得られたカルダノール 584 mgをシリカゲルカラム(カラムの直径:3 cm;カラムの長さ:30 cm;シリカゲル粉末体積:200 mL;展開溶媒:ヘキサン : 酢酸エチル = 9 : 1;ドレイン:80 mL)に添加し、フラクションコレクターにより約7 mLずつ分画した。40番目から46番目のフラクションを集めて濃縮してクーゲル(60℃、30分間)で乾燥させた。得られたカルダノールはモノエン型のみのカルダノールであった。収量:143 mg。Rf = 0.63(展開溶媒:ヘキサン : 酢酸エチル = 9 : 1)。
1H-NMR (400 MHz; CDCl3; r.t.): δ7.16-6.63 (m, フェニル基), 5.34 (m, -CH2CHCHCH2-), 4.67 (d, J = 4.4 Hz, -OH), 2.55 (t, J = 7.8 Hz, -C6H4CH2-), 2.02 (m, -CH2CHCHCH2-), 1.61 - 1.26 (m, -CH2-), 0.88 ppm (t, J = 6.8 Hz, -CH3).
【0018】
製造例3
50 mLナス型フラスコにβ-ラクトース オクタアセテート271 mg、製造例3で得たカルダノール(モノエン型)121 mg、モレキュラーシーブ 4A 0.4 g、及びドライトルエン 4 mLを加えてアルゴン雰囲気下室温で1時間磁気撹拌した。反応系を氷浴により0℃に冷却してから、アルゴン雰囲気下でシリンジに測り取ったトリメチルシリル トリフルオロメタンスルフォネート72 μLをシリンジにより反応系に滴下した。2時間磁気撹拌した後、反応系にトリエチルアミン0.6 mLを加えてしばらくの間磁気撹拌することにより反応を停止させた。酢酸エチルで洗いながらセライトで吸引ろ過し、濃縮した。シリカゲルカラム(カラムの直径:2 cm;カラムの長さ:30 cm;シリカゲル粉末体積:80 mL;展開溶媒:トルエン : 酢酸エチル = 2 : 1;ドレイン:40 mL)に添加し、フラクションコレクターにより約6 mLずつ分画した。16番目から22番目のフラクションを集めて濃縮してクーゲル(60℃、30分間)で乾燥させ、カルダニル(モノエン型)-2,3,4, 6,2',3',6'-ヘプタ-O-アセチル-β-D-ラクトシドを得た。収量:54 mg。Rf = 0.39(展開溶媒:トルエン : 酢酸エチル = 2 : 1)。
【0019】
30 mLナス型フラスコに上記で得たカルダニル(モノエン型)-2,3,4, 6,2',3',6'-ヘプタ-O-アセチル-β-D-ラクトシド54 mgを採り、メタノール 1 mL及びテトラヒドロフラン 1 mLを加えて溶解させた。28 %ナトリウムメトキシド/メタノール溶液1滴を加えて溶液のpHを調べてみるとpH = 9であった。室温で1時間磁気撹拌した。反応系にDowex 50を加えて反応系を中和することにより反応を停止させた。テトラヒドロフランで洗いながらセライトにより吸引ろ過してDowex 50の粒を除いてから濃縮した。シリカゲルカラム(カラムの直径:1 cm;カラムの長さ:30 cm;シリカゲル粉末体積:50 mL;展開溶媒:クロロホルム : メタノール = 5 : 1;ドレイン:0 mL)に添加し、フラクションコレクターにより約10 mLずつ分画した。20番目から45番目のフラクションを集めて濃縮してクーゲル(60℃、30分間)で乾燥させ、カルダニル(モノエン型)-β-D-ラクトシドを得た。収量:28 mg。Rf = 0.29(展開溶媒:クロロホルム : メタノール = 10 : 3)。
元素分析:C33H54O11; calc.: C: 63.24%, H: 8.68%; found: C: 63.23%, H: 8.86%. 1H-NMR (400 MHz; CD3OD; r.t.): δ 7.19-6.82 (m, フェニル基), 5.34 (m, -CH2CHCHCH2-), 4.93 (d, J1,2 = 7.6 Hz, H-1), 4.39 (d, J1,2 = 7.6 Hz, H-1'), 3.93 - 3.13 (m, 糖鎖), 2.57 (t, J = 7.8 Hz, -C6H4CH2-), 2.02 (m, -CH2CHCHCH2-), 1.60 - 1.28 (m, -CH2-), 0.90 ppm (t, J = 6.8 Hz, -CH3).
【0020】
実施例2
200 mL三角フラスコに製造例3で得たカルダニル(モノエン型)-β-D-ラクトシド1 mgを採り、水40 mLを加えた。60℃の水浴中で超音波に20分間曝してからオートクレーブで120℃20分間加熱した。そのままオートクレーブの蓋を開けずに一晩オートクレーブ中に放置しておいてから室温に取り出すと、白い沈殿物が生じていた。この沈殿物の形状を光学顕微鏡及び走査型電子顕微鏡で確認したところ、マイクロダブルヘリカルファイバー状凝集体(図3及び4)、マイクロヘリカルディスク状凝集体(図5及び6)、微細チューブ状凝集体、及びマイクロファイバー状凝集体などの混合物が観察された。
図3及び4に示すマイクロダブルヘリカルファイバー状凝集体は、幅が約3μmの2本のヘリカルリボン状凝集体が緩やかな角度で交互に絡み合って直径が約8μmの網目を連続して形成した凝集体であった。
図5及び6に示すマイクロヘリカルディスク状凝集体は、幅が約1μmのヘリカルリボン状凝集体が急な角度で巻いてディスク(円盤)状に近い形になったものがさらにお互いに中心軸方向に凝集して直径が約4〜5μmの円柱体を形成した凝集体であった。
【図面の簡単な説明】
【0021】
【図1】実施例1で得たナノファイバー状凝集体の光学顕微鏡像を示す図である。写真のサイズは縦175μm×横127μmである。
【図2】図1をトレースした図である。
【図3】実施例2で得られたマイクロダブルヘリカルファイバー状凝集体の光学顕微鏡像を示す図である。写真のサイズは縦28μm×横36μmである。
【図4】図3をトレースした図である。
【図5】実施例2で得られたマイクロヘリカルディスク状凝集体の走査型電子顕微鏡像を示す図である。写真のサイズは縦8.3μm×横7.1μmである。
【図6】図5をトレースした図である。
【特許請求の範囲】
【請求項1】
下記一般式
【化1】
(式中、Gは2〜5の単糖が結合したオリゴ糖残基(二糖類を除く)を表し、Rは炭素数6〜25の炭化水素基を表す。)で表わされる構造を有するO−グリコシド型糖脂質から成る微細自己集合体。
【請求項2】
下記一般式
【化1】
(式中、Gは2〜5の単糖が結合したオリゴ糖残基を表し、Rは炭素数6〜25の炭化水素基を表す。)で表わされる構造を有するO−グリコシド型糖脂質の2種以上の混合物から成る微細自己集合体。
【請求項3】
下記一般式
【化1】
(式中、Gは2〜5の単糖が結合したオリゴ糖残基(二糖類を除く)を表し、Rは炭素数6〜25の炭化水素基を表す。)で表わされる構造を有するO−グリコシド型糖脂質を水に分散し、この分散液から凝集させることにより得られた微細自己集合体。
【請求項4】
下記一般式
【化1】
(式中、Gは2〜5の単糖が結合したオリゴ糖残基を表し、Rは炭素数6〜25の炭化水素基を表す。)で表わされる構造を有するO−グリコシド型糖脂質の2種以上の混合物を水に分散し、この分散液から凝集させることにより得られた微細自己集合体。
【請求項5】
前記一般式(化1)において、炭化水素基(R)が−O−G基に対してメタ位にある請求項1〜4のいずれか一項に記載の微細自己集合体。
【請求項1】
下記一般式
【化1】
(式中、Gは2〜5の単糖が結合したオリゴ糖残基(二糖類を除く)を表し、Rは炭素数6〜25の炭化水素基を表す。)で表わされる構造を有するO−グリコシド型糖脂質から成る微細自己集合体。
【請求項2】
下記一般式
【化1】
(式中、Gは2〜5の単糖が結合したオリゴ糖残基を表し、Rは炭素数6〜25の炭化水素基を表す。)で表わされる構造を有するO−グリコシド型糖脂質の2種以上の混合物から成る微細自己集合体。
【請求項3】
下記一般式
【化1】
(式中、Gは2〜5の単糖が結合したオリゴ糖残基(二糖類を除く)を表し、Rは炭素数6〜25の炭化水素基を表す。)で表わされる構造を有するO−グリコシド型糖脂質を水に分散し、この分散液から凝集させることにより得られた微細自己集合体。
【請求項4】
下記一般式
【化1】
(式中、Gは2〜5の単糖が結合したオリゴ糖残基を表し、Rは炭素数6〜25の炭化水素基を表す。)で表わされる構造を有するO−グリコシド型糖脂質の2種以上の混合物を水に分散し、この分散液から凝集させることにより得られた微細自己集合体。
【請求項5】
前記一般式(化1)において、炭化水素基(R)が−O−G基に対してメタ位にある請求項1〜4のいずれか一項に記載の微細自己集合体。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2006−143723(P2006−143723A)
【公開日】平成18年6月8日(2006.6.8)
【国際特許分類】
【出願番号】特願2005−331525(P2005−331525)
【出願日】平成17年11月16日(2005.11.16)
【分割の表示】特願2002−49238(P2002−49238)の分割
【原出願日】平成14年2月26日(2002.2.26)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【Fターム(参考)】
【公開日】平成18年6月8日(2006.6.8)
【国際特許分類】
【出願日】平成17年11月16日(2005.11.16)
【分割の表示】特願2002−49238(P2002−49238)の分割
【原出願日】平成14年2月26日(2002.2.26)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【Fターム(参考)】
[ Back to top ]