
悪性腫瘍治療薬
【課題】ステビオシド由来の悪性腫瘍治療薬として有用な新たな化合物を提供する。
【解決手段】一般式(1)

(式中、R1はトリフルオロメチルビニル基、フェニル基又はメチルフェニルビニル基を示す)
で表されるステビオール誘導体、及びこれを有効成分とする医薬。
【解決手段】一般式(1)
(式中、R1はトリフルオロメチルビニル基、フェニル基又はメチルフェニルビニル基を示す)
で表されるステビオール誘導体、及びこれを有効成分とする医薬。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ステビア由来のジテルペノイドの新規な化学修飾物及びこれを有効成分とする悪性腫瘍治療薬に関する。
【背景技術】
【0002】
植物は古来より医薬品資源として注目されており、これまでにも様々な医薬品あるいは医薬品素材が植物中に見出されている。植物由来の悪性腫瘍治療薬としては、イチイ科の植物からの抽出物であるパクリタキセル(タキソール);クロタキカズラ科クサミズキ、タマミズキ科カンレンボクに含まれるアルカロイドであるカンプトテシン;及びキョウチクトウ科ニチニチソウに含まれるインドールアルカロイドであるビンクリスチン、ビンブラスチン等が知られている。
【0003】
ステビオシドは、ステビア(Stevia rebaudiana Bertoni:キク科)の葉に豊富に存在するジテルペノイド配糖体であり、甘味料として広く知られており、また抗炎症作用及び発がん抑制作用を有することが知られている(非特許文献1)。また、本発明者らは、ステビオシドから製造したイソステビオールの微生物による構造修飾物の抗発がんプロモーター作用(非特許文献2)、DNAポリイソメラーゼ及びメポイソメラーゼ阻害活性(非特許文献3)について報告した。さらに、ステビオシドの加水分解生成物であるイソステビオール及びステビオールを化学修飾し、それらの腫瘍細胞傷害活性についても報告されている(非特許文献4〜6)。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Biol.Pharm.Bull.,25,1488−1490(2002)
【非特許文献2】J.Nat.Prod.,67,407−410(2004)
【非特許文献3】Life Sci.,77,2127−2140(2005)
【非特許文献4】Chem.Pharm.Bull.,2004,52,1117
【非特許文献5】Bioorg.Med.Chem.Lett.2009,19,1818
【非特許文献6】第52回香料・テルペンおよび精油化学に関する討論会講演要旨集2PIII−4(平成20年10月)
【発明の概要】
【発明が解決しようとする課題】
【0005】
しかしながら、これらステビオシド誘導体のがんに対する作用は十分でなく、さらに強力な抗悪性腫瘍活性を有する化合物の創出が望まれる。
従って、本発明の課題は、ステビオシド由来の悪性腫瘍治療薬として有用な新たな化合物を提供することにある。
【課題を解決するための手段】
【0006】
そこで、本発明者は、ステビオシドの加水分解生成物であるステビオールの化学修飾を行い、5位のカルボキシル基の変換に着目した。まず、5位のカルボキシル基を還元して、5−ヒドロキシメチル体とし、当該ヒドロキシメチル基をトリフルオロメチルアクリロイル化、ベンゾイル化又はメチルシンナミル化した化合物を合成し、それらの腫瘍細胞傷害活性を検討したところ、これらの化合物が胃癌、肺癌及び乳癌細胞に対する強力な傷害活性を有すること、さらにはアポトーシス誘導作用も有することを見出し、本発明を完成した。
【0007】
すなわち、本発明は、一般式(1)
【0008】
【化1】
【0009】
(式中、R1はトリフルオロメチルビニル基、フェニル基又はメチルフェニルビニル基を示す)
で表されるステビオール誘導体を提供するものである。
【0010】
また、本発明は、一般式(1)で表される化合物を有効成分とする医薬を提供するものである。
さらに、本発明は、一般式(1)で表される化合物を有効成分とするアポトーシス誘導薬を提供するものである。
【発明の効果】
【0011】
本発明によれば、胃癌、肺癌及び乳癌等の患者数が多く、死亡率も高い悪性腫瘍に対して有用な悪性腫瘍治療薬が提供できる。本発明化合物は、アポトーシス誘導作用により悪性腫瘍細胞傷害活性を示すと考えられる。
【図面の簡単な説明】
【0012】
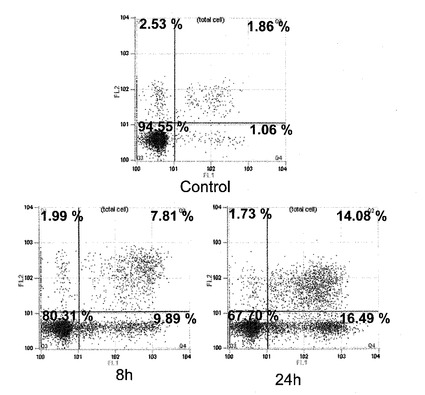
【図1】化合物(1a)処理A549細胞のフローサイトメトリーの結果(8hr、24hr)を示す図である。
【図2】化合物(1b)処理A549細胞のフローサイトメトリーの結果(8hr、24hr)を示す図である。
【図3】化合物(1c)処理A549細胞のフローサイトメトリーの結果(8hr、24hr)を示す図である。
【図4】化合物(1d)処理A549細胞のフローサイトメトリーの結果(8hr、24hr)を示す図である。
【図5】化合物(1e)処理A549細胞のフローサイトメトリーの結果(24hr)を示す図である。
【図6】化合物(1a)〜(1d)処理A549細胞のHoechst染色結果を示す図である。
【図7】化合物(1e)の処理A549細胞のHoechst染色結果を示す図である。
【発明を実施するための形態】
【0013】
一般式(1)中、R1はトリフルオロメチルビニル基、フェニル基又はメチルフェニルビニル基を示す。ここでメチルフェニルビニル基には、2−メチルフェニルビニル基、3−メチルフェニルビニル基、4−メチルフェニルビニル基が挙げられる。
【0014】
本発明化合物を列挙すれば、次のとおりである。
【0015】
【化2】
【0016】
本発明化合物(1)は、例えばステビオール(A)をメチルエステル化して得られるステビオールメチルエステル(B)を還元して得られる化合物(C)に、安息香酸、メチルケイヒ酸、トリフルオロメチルアクリル酸を用いてアシル化することにより製造することができる。
【0017】
【化3】
【0018】
(式中、R1は前記と同じ)
【0019】
ステビオール(A)は、ステビアから抽出することにより得られるステビオシド(式(A)のステビオースの13−O−β−D−Glc−(1→2)−β−Glc,19−D−β−D−Glc配糖体)を加水分解することにより得ることができる(非特許文献4)。ステビオール(A)のメチルエステル化反応は、ステビオール(A)にジアゾメタン等のメチル化剤を反応させることにより行うことができる。
【0020】
化合物(B)の還元は、例えば水素化アルミニウムリチウム(LiAlH4)等の還元剤を用いて行われる。反応は、例えばジエチルエーテル、ジオキサン等のエーテル系溶媒等の存在下に、LiAlH4を添加して、室温〜還流温度で0.5〜10時間行えばよい。
【0021】
化合物(C)とR1COOHとの反応は、ジ−2−ピリジルチオノカーボネート等の縮合剤及び4−ジメチルアミノピリジン等の有機塩基の存在下に室温から還流温度で0.5〜50時間行えばよい。
【0022】
得られた本発明化合物(1)は、洗浄、再結晶、各種クロマトグラフィー等の手段により精製することができる。
【0023】
後記実施例から明らかなように、本発明化合物(1)は、胃癌細胞、肺癌細胞、乳癌細胞等に対して優れた細胞傷害活性を有する。また、その細胞傷害活性はアポトーシス誘導活性に基づくものである。従って、本発明化合物は、ヒトを含む哺乳動物の悪性腫瘍治療薬として有用である。
本発明の悪性腫瘍治療薬の対象となる悪性腫瘍としては、固形腫瘍が好ましい。固形腫瘍としては、肺癌、大腸癌、胃癌、乳癌、前立腺癌、甲状腺癌などの上皮細胞癌;及び平滑筋肉腫、骨肉腫などの肉腫が挙げられるが、特に胃癌、肺癌、乳癌が好ましい。
【0024】
本発明の医薬は本発明化合物(1)に賦形剤、結合剤、滑沢剤、崩壊剤、被覆剤、乳化剤、懸濁化剤、溶剤、安定化剤、吸収助剤、軟膏基剤等の1以上の薬学的に許容される担体を適宜添加し、常法により経口投与用、注射投与用、直腸内投与用、外用などの剤形に製剤化することによって得られる。
経口投与用の製剤としては、顆粒、錠剤、糖衣錠、カプセル剤、丸剤、液剤、乳剤、懸濁剤等が;注射投与用の製剤としては、静脈内注射、筋肉内注射、皮下注射、点滴注射用の製剤などが;直腸内投与用の製剤としては、坐薬軟カプセル等が好ましい。
本発明の医薬は上記の如き製剤として、ヒトを含む哺乳動物に投与することができる。
本発明の医薬は、本発明化合物として、1日当り約1〜500mg/kgを1〜4回投与するのが好ましい。
【実施例】
【0025】
次に実施例を挙げて本発明をさらに詳細に説明する。
【0026】
参考例1
ent−kaur−16−ene−13,19−diol(エント−カウル−16−エン−13,19−ジオール)(C)の調製
ステビオールメチルエステル(195mg)をジエチルエーテル(43mL)に溶解し、水素化アルミニウムリチウム(430mg)を加え、5時間加熱還流を行った。水、希塩酸で処理後、ジエチルエーテルで抽出した。ジエチルエーテル層は、水、飽和炭酸水素ナトリウム水溶液にて洗浄後、溶媒を除去した。反応生成物はシリカゲル薄層クロマトグラフィー(展開液:ヘキサン−酢酸エチル=7:3)にて精製し,ent−kaur−16−ene−13,19−diol(98mg)を得た。
【0027】
実施例1
ent−kaur−16−ene−13,19−diol 19−O−benzoate(エント−カウル−16−エン−13,19−ジオール 19−O−ベンゾエート;化合物(1a)の調製
ent−kaur−16−ene−13,19−diol(20mg)をトルエン(0.4mL)に溶解し、安息香酸(38mg)、4−ジメチルアミノピリジン(4−DMAP;16mg)、及びdi−2−pyridyl thionocarbonate(DPTC;30mg)を加え室温で24時間攪拌した。水で希釈後、ジエチルエーテルで抽出した。ジエチルエーテル層は水洗浄し、無水硫酸ナトリウムで脱水後、溶媒を除去した。反応生成物はシリカゲル薄層クロマトグラフィー(展開液:ヘキサン−酢酸エチル=7:3)にて精製し、化合物(1a)(8.4mg)を得た。
【0028】
1H-NMR(CDCl3, 400MHz) δ: 1.08 (s; 3H), 1.09 (s; 3H), 4.12 (d, J = 11.0Hz; 1H), 4.49 (d, J = 11.0Hz; 1H), 4.82 (brs; 1H), 4.99 (brs; 1H), 7.45 (t, J = 7.8Hz; 2H), 7.56 (t, J = 7.4Hz; 1H), 8.04 (brd, J = 7.1Hz; 2H); HR-ESI-MS (positive ion mode) : m/z 431.2554 (C27H36O3Na計算値:431.2562).
【0029】
実施例2
ent−kaur−16−ene−13,19−diol 19−O−4−methylcinnamate(エント−カウル−16−エン−13,19−ジオール 19−O−4―メチルシンナメート;化合物(1b)の調製
ent−kaur−16−ene−13,19−diol(31mg)をトルエン(1.2mL)に溶解し、4−メチルケイ皮酸(32mg)、4−ジメチルアミノピリジン(4−DMAP;21mg)、及びdi−2−pyridyl thionocarbonate(DPTC;45mg)を加え室温で24時間攪拌した。水で希釈後、ジエチルエーテルで抽出した。ジエチルエーテル層は水洗浄し、無水硫酸ナトリウムで脱水後、溶媒を除去した。反応生成物はシリカゲル薄層クロマトグラフィー(展開液:ヘキサン−酢酸エチル=7:3)にて精製し、化合物(1b)(17mg)を得た。
【0030】
1H-NMR(CDCl3, 400MHz) δ: 1.01 (s; 3H), 1.07 (s; 3H), 2.37 (s; 3H), 4.00 (d, J = 11.0Hz; 1H), 4.36 (d, J = 11.0Hz; 1H), 4.81 (brs; 1H), 4.98 (brs; 1H),6.38 (d, J = 16.0Hz; 1H), 7.18 (d, J = 8.2Hz; 2H), 7.42 (d, J = 8.2Hz; 2H), 7.63 (d, J = 16.0Hz; 1H); HR-ESI-MS (positive ion mode) : m/z 471.2881 (C30H40O3Na計算値:471.2875).
【0031】
実施例3
ent−kaur−16−ene−13,19−diol 19−O−3−methylcinnamate(エント−カウル−16−エン−13,19−ジオール 19−O−3―メチルシンナメート;化合物(1c)の調製
ent−kaur−16−ene−13,19−diol(40mg)をトルエン(1.6mL)に溶解し、3−メチルケイ皮酸(46mg)、4−ジメチルアミノピリジン(4−DMAP;28mg)、及びdi−2−pyridyl thionocarbonate(DPTC;63mg)を加え室温で24時間攪拌した。水で希釈後、ジエチルエーテルで抽出した。ジエチルエーテル層は水洗浄し、無水硫酸ナトリウムで脱水後、溶媒を除去した。反応生成物はシリカゲル薄層クロマトグラフィー(展開液:ヘキサン−酢酸エチル=7:3)にて精製し、化合物(1c)(35mg)を得た。
【0032】
1H-NMR(CDCl3, 400MHz) δ: 1.01 (s; 3H), 1.07 (s; 3H), 2.37 (s; 3H), 4.01 (d, J = 11.0Hz; 1H), 4.36 (d, J = 11.0Hz; 1H), 4.82 (brs; 1H), 4.98 (brs; 1H),6.42 (d, J = 16.0Hz; 1H), 7.19 (d, J = 7.3Hz; 1H),7.27 (t, J = 7.8Hz; 1H), 7.33 (d, J = 6.9Hz; 1H), 7.34 (s; 1H), 7.63 (d, J = 16.0Hz; 1H); HR-ESI-MS (positive ion mode): m/z 471.2885 (C30H40O3Na計算値:471.2875).
【0033】
実施例4
ent−kaur−16−ene−13,19−diol 19−O−2−methylcinnamate(エント−カウル−16−エン−13,19−ジオール 19−O−2―メチルシンナメート;化合物(1d)の調製
ent−kaur−16−ene−13,19−diol(30mg)をトルエン(1.2mL)に溶解し、2−メチルケイ皮酸(35mg)、4−ジメチルアミノピリジン(4−DMAP;23mg)、及びdi−2−pyridyl thionocarbonate(DPTC;46mg)を加え室温で24時間攪拌した。水で希釈後、ジエチルエーテルで抽出した。ジエチルエーテル層は水洗浄し、無水硫酸ナトリウムで脱水後、溶媒を除去した。反応生成物はシリカゲル薄層クロマトグラフィー(展開液:ヘキサン−酢酸エチル=7:3)にて精製し、化合物(1d)(28mg)を得た。
【0034】
1H-NMR(CDCl3, 400MHz) δ: 1.02 (s; 3H), 1.08 (s; 3H), 2.43 (s; 3H), 4.04 (d, J = 11.0Hz; 1H), 4.35 (d, J = 11.0Hz; 1H), 4.82 (brs; 1H), 4.98 (brs; 1H), 6.35 (d, J = 16.0Hz; 1H), 7.19 (d, J = 7.3Hz; 1H), 7.20 (t, J = 7.3Hz; 1H), 7.27 (dt, J = 1.4, 7.3Hz; 1H), 7.56 (d, J = 7.8Hz; 1H), 7.97 (d, J = 16.0Hz; 1H); HR-ESI-MS (positive ion mode) : m/z 471.2867 (C30H40O3Na計算値:471.2875).
【0035】
実施例5
ent−kaur−16−ene−13,19−diol 19−O−β−trifluoromethyl acrylate(エント−カウル−16−エン−13,19−ジオール 19−O−トリフルオロメチルアクリレート;化合物(1e)の調製
ent−kaur−16−ene−13,19−diol(30mg)をトルエン(1.2mL)に溶解し、β−トリフルオロメチルアクリル酸(44mg)、4−ジメチルアミノピリジン(4−DMAP;23mg)、及びdi−2−pyridyl thionocarbonate(DPTC;46mg)を加え室温で24時間攪拌した。水で希釈後、ジエチルエーテルで抽出した。ジエチルエーテル層は水洗浄し、無水硫酸ナトリウムで脱水後、溶媒を除去した。反応生成物はシリカゲル薄層クロマトグラフィー(展開液:ヘキサン−酢酸エチル=7:3)にて精製し、化合物(1e)(16mg)を得た。
【0036】
1H-NMR(CDCl3, 400MHz) δ: 0.98 (s; 3H), 1.04 (s; 3H), 4.01 (d, J = 11.0Hz; 1H), 4.39 (d, J = 11.0Hz; 1H), 4.82 (brs; 1H), 4.98 (brs; 1H), 6.49 (dd, J = 2.1, 15.8Hz; 1H), 6.75 (dq, J = 6.6, 15.8Hz; 1H); HR-APCI-MS (positive ion mode): m/z 409.2346 (C24H32O3F3計算値:409.2354).
【0037】
実施例6(腫瘍細胞傷害試験及びその傷害機構の解析)
1.細胞傷害活性試験
3種類の腫瘍細胞を用いて細胞傷害活性を評価した:ヒト乳癌由来細胞株SKBR3、ヒト肺癌由来細胞株A549及びヒト胃癌由来細細胞株AZ521を用いた。
各細胞を96wellマイクロプレートに播種し、24時間培養した。本発明化合物を添加し、48時間作用後、0.5%MTT溶液を加え、3時間培養した。その後、0.04N HCl/isopropanolを加えTop:570nm、Bottom:630nmにて吸光度を測定し、コントロールとの比較により細胞生存率を算出した。EC50はコントロールと比較して細胞を50%死滅させる効果濃度のことであり、GraphPad Prism computer program(Ver.4.0)を用いて求めた。
【0038】
細胞傷害活性試験に供したSKBR3、A549及びAZ521の細胞数は何れも3×103cells/wellである。
【0039】
2.フローサイトメトリー(Annexin V−Propidium Iodide(PI)二重染色法)
本試験はAnnexin V−FITC kit[Bender MedSystems,BMS306FI]を用いて行った。A549を6wellプレートに播種し(1×105cells/well)、24時間培養した。本発明化合物(40μM又は30μM)を作用させた後、細胞を回収し、遠心分離(1000×g,5min)を行った。遠心分離後、上清を除去し、Binding buffer 195μL及びAnnexin V5μLを加えピペッティングを行い、15分間暗所で染色した。その後、遠心分離(1000×g,5min)を行い、上清を除去し、Binding buffer 490μL及びPI 10μLを加えピペッティングし、フローサイトメーターにて解析を行った。
【0040】
3.結果
3−1.細胞傷害活性試験
本発明化合物の細胞傷害活性の結果を表1に示した。胃癌細胞(AZ521)、肺癌細胞(A549)及び乳癌細胞(SKBR3)に対して、参照化合物であるシスプラチンをはるかに上回る細胞傷害活性を示した。
【0041】
【表1】
【0042】
3−2.フローサイトメトリー(Annexin V−Propidium Iodide(PI)二重染色法)
本発明化合物の細胞傷害性がアポトーシスによることを確認するために本発明化合物20μM〜40μM(化合物(1a)は40μM、化合物(1b)〜(1d)は30μM、化合物(1e)は20μM)を作用させた肺癌細胞A549についてAnnexin V−PI二重染色を行い、フローサイトメトリーを行った(図1〜5)。時間依存的にAnnexin V 陽性/PI陰性細胞群の増加が確認され(8hr、24hr)、本発明化合物がアポトーシスを誘導することを確認した。
【0043】
3−3.Hoechst染色
20μM〜30μMの化合物(1a)〜(1e)を24時間作用させた肺癌細胞A549についてHoechst 33342染色を行った。その結果、図6及び7に示すように、化合物(1a)〜(1e)のいずれの化合物処理によっても細胞核の凝集・断片化が観察されたことから、本発明化合物がアポトーシスを誘導することが判明した。
【0044】
また、化合物(1b)を作用させたA549細胞におけるカスパーゼ遺伝子解析をしたところ、いずれのカスパーゼも活性化されていることから、本発明化合物によるアポトーシス誘導は、カスパーゼ3、8、9を介したシグナル伝達によるものと考えられた。
【0045】
本発明化合物がヒト胃癌由来細胞株AZ521、ヒト肺癌由来細胞株A549及びヒト乳癌細胞株SKBR3に対して参照化合物のシスプラチンと比較してはるかに強い細胞傷害活性を有することが明らかになった。また、本発明化合物はA549細胞に対しアポトーシス誘導により細胞傷害活性を示すことが確認された。
【技術分野】
【0001】
本発明は、ステビア由来のジテルペノイドの新規な化学修飾物及びこれを有効成分とする悪性腫瘍治療薬に関する。
【背景技術】
【0002】
植物は古来より医薬品資源として注目されており、これまでにも様々な医薬品あるいは医薬品素材が植物中に見出されている。植物由来の悪性腫瘍治療薬としては、イチイ科の植物からの抽出物であるパクリタキセル(タキソール);クロタキカズラ科クサミズキ、タマミズキ科カンレンボクに含まれるアルカロイドであるカンプトテシン;及びキョウチクトウ科ニチニチソウに含まれるインドールアルカロイドであるビンクリスチン、ビンブラスチン等が知られている。
【0003】
ステビオシドは、ステビア(Stevia rebaudiana Bertoni:キク科)の葉に豊富に存在するジテルペノイド配糖体であり、甘味料として広く知られており、また抗炎症作用及び発がん抑制作用を有することが知られている(非特許文献1)。また、本発明者らは、ステビオシドから製造したイソステビオールの微生物による構造修飾物の抗発がんプロモーター作用(非特許文献2)、DNAポリイソメラーゼ及びメポイソメラーゼ阻害活性(非特許文献3)について報告した。さらに、ステビオシドの加水分解生成物であるイソステビオール及びステビオールを化学修飾し、それらの腫瘍細胞傷害活性についても報告されている(非特許文献4〜6)。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Biol.Pharm.Bull.,25,1488−1490(2002)
【非特許文献2】J.Nat.Prod.,67,407−410(2004)
【非特許文献3】Life Sci.,77,2127−2140(2005)
【非特許文献4】Chem.Pharm.Bull.,2004,52,1117
【非特許文献5】Bioorg.Med.Chem.Lett.2009,19,1818
【非特許文献6】第52回香料・テルペンおよび精油化学に関する討論会講演要旨集2PIII−4(平成20年10月)
【発明の概要】
【発明が解決しようとする課題】
【0005】
しかしながら、これらステビオシド誘導体のがんに対する作用は十分でなく、さらに強力な抗悪性腫瘍活性を有する化合物の創出が望まれる。
従って、本発明の課題は、ステビオシド由来の悪性腫瘍治療薬として有用な新たな化合物を提供することにある。
【課題を解決するための手段】
【0006】
そこで、本発明者は、ステビオシドの加水分解生成物であるステビオールの化学修飾を行い、5位のカルボキシル基の変換に着目した。まず、5位のカルボキシル基を還元して、5−ヒドロキシメチル体とし、当該ヒドロキシメチル基をトリフルオロメチルアクリロイル化、ベンゾイル化又はメチルシンナミル化した化合物を合成し、それらの腫瘍細胞傷害活性を検討したところ、これらの化合物が胃癌、肺癌及び乳癌細胞に対する強力な傷害活性を有すること、さらにはアポトーシス誘導作用も有することを見出し、本発明を完成した。
【0007】
すなわち、本発明は、一般式(1)
【0008】
【化1】
【0009】
(式中、R1はトリフルオロメチルビニル基、フェニル基又はメチルフェニルビニル基を示す)
で表されるステビオール誘導体を提供するものである。
【0010】
また、本発明は、一般式(1)で表される化合物を有効成分とする医薬を提供するものである。
さらに、本発明は、一般式(1)で表される化合物を有効成分とするアポトーシス誘導薬を提供するものである。
【発明の効果】
【0011】
本発明によれば、胃癌、肺癌及び乳癌等の患者数が多く、死亡率も高い悪性腫瘍に対して有用な悪性腫瘍治療薬が提供できる。本発明化合物は、アポトーシス誘導作用により悪性腫瘍細胞傷害活性を示すと考えられる。
【図面の簡単な説明】
【0012】
【図1】化合物(1a)処理A549細胞のフローサイトメトリーの結果(8hr、24hr)を示す図である。
【図2】化合物(1b)処理A549細胞のフローサイトメトリーの結果(8hr、24hr)を示す図である。
【図3】化合物(1c)処理A549細胞のフローサイトメトリーの結果(8hr、24hr)を示す図である。
【図4】化合物(1d)処理A549細胞のフローサイトメトリーの結果(8hr、24hr)を示す図である。
【図5】化合物(1e)処理A549細胞のフローサイトメトリーの結果(24hr)を示す図である。
【図6】化合物(1a)〜(1d)処理A549細胞のHoechst染色結果を示す図である。
【図7】化合物(1e)の処理A549細胞のHoechst染色結果を示す図である。
【発明を実施するための形態】
【0013】
一般式(1)中、R1はトリフルオロメチルビニル基、フェニル基又はメチルフェニルビニル基を示す。ここでメチルフェニルビニル基には、2−メチルフェニルビニル基、3−メチルフェニルビニル基、4−メチルフェニルビニル基が挙げられる。
【0014】
本発明化合物を列挙すれば、次のとおりである。
【0015】
【化2】
【0016】
本発明化合物(1)は、例えばステビオール(A)をメチルエステル化して得られるステビオールメチルエステル(B)を還元して得られる化合物(C)に、安息香酸、メチルケイヒ酸、トリフルオロメチルアクリル酸を用いてアシル化することにより製造することができる。
【0017】
【化3】
【0018】
(式中、R1は前記と同じ)
【0019】
ステビオール(A)は、ステビアから抽出することにより得られるステビオシド(式(A)のステビオースの13−O−β−D−Glc−(1→2)−β−Glc,19−D−β−D−Glc配糖体)を加水分解することにより得ることができる(非特許文献4)。ステビオール(A)のメチルエステル化反応は、ステビオール(A)にジアゾメタン等のメチル化剤を反応させることにより行うことができる。
【0020】
化合物(B)の還元は、例えば水素化アルミニウムリチウム(LiAlH4)等の還元剤を用いて行われる。反応は、例えばジエチルエーテル、ジオキサン等のエーテル系溶媒等の存在下に、LiAlH4を添加して、室温〜還流温度で0.5〜10時間行えばよい。
【0021】
化合物(C)とR1COOHとの反応は、ジ−2−ピリジルチオノカーボネート等の縮合剤及び4−ジメチルアミノピリジン等の有機塩基の存在下に室温から還流温度で0.5〜50時間行えばよい。
【0022】
得られた本発明化合物(1)は、洗浄、再結晶、各種クロマトグラフィー等の手段により精製することができる。
【0023】
後記実施例から明らかなように、本発明化合物(1)は、胃癌細胞、肺癌細胞、乳癌細胞等に対して優れた細胞傷害活性を有する。また、その細胞傷害活性はアポトーシス誘導活性に基づくものである。従って、本発明化合物は、ヒトを含む哺乳動物の悪性腫瘍治療薬として有用である。
本発明の悪性腫瘍治療薬の対象となる悪性腫瘍としては、固形腫瘍が好ましい。固形腫瘍としては、肺癌、大腸癌、胃癌、乳癌、前立腺癌、甲状腺癌などの上皮細胞癌;及び平滑筋肉腫、骨肉腫などの肉腫が挙げられるが、特に胃癌、肺癌、乳癌が好ましい。
【0024】
本発明の医薬は本発明化合物(1)に賦形剤、結合剤、滑沢剤、崩壊剤、被覆剤、乳化剤、懸濁化剤、溶剤、安定化剤、吸収助剤、軟膏基剤等の1以上の薬学的に許容される担体を適宜添加し、常法により経口投与用、注射投与用、直腸内投与用、外用などの剤形に製剤化することによって得られる。
経口投与用の製剤としては、顆粒、錠剤、糖衣錠、カプセル剤、丸剤、液剤、乳剤、懸濁剤等が;注射投与用の製剤としては、静脈内注射、筋肉内注射、皮下注射、点滴注射用の製剤などが;直腸内投与用の製剤としては、坐薬軟カプセル等が好ましい。
本発明の医薬は上記の如き製剤として、ヒトを含む哺乳動物に投与することができる。
本発明の医薬は、本発明化合物として、1日当り約1〜500mg/kgを1〜4回投与するのが好ましい。
【実施例】
【0025】
次に実施例を挙げて本発明をさらに詳細に説明する。
【0026】
参考例1
ent−kaur−16−ene−13,19−diol(エント−カウル−16−エン−13,19−ジオール)(C)の調製
ステビオールメチルエステル(195mg)をジエチルエーテル(43mL)に溶解し、水素化アルミニウムリチウム(430mg)を加え、5時間加熱還流を行った。水、希塩酸で処理後、ジエチルエーテルで抽出した。ジエチルエーテル層は、水、飽和炭酸水素ナトリウム水溶液にて洗浄後、溶媒を除去した。反応生成物はシリカゲル薄層クロマトグラフィー(展開液:ヘキサン−酢酸エチル=7:3)にて精製し,ent−kaur−16−ene−13,19−diol(98mg)を得た。
【0027】
実施例1
ent−kaur−16−ene−13,19−diol 19−O−benzoate(エント−カウル−16−エン−13,19−ジオール 19−O−ベンゾエート;化合物(1a)の調製
ent−kaur−16−ene−13,19−diol(20mg)をトルエン(0.4mL)に溶解し、安息香酸(38mg)、4−ジメチルアミノピリジン(4−DMAP;16mg)、及びdi−2−pyridyl thionocarbonate(DPTC;30mg)を加え室温で24時間攪拌した。水で希釈後、ジエチルエーテルで抽出した。ジエチルエーテル層は水洗浄し、無水硫酸ナトリウムで脱水後、溶媒を除去した。反応生成物はシリカゲル薄層クロマトグラフィー(展開液:ヘキサン−酢酸エチル=7:3)にて精製し、化合物(1a)(8.4mg)を得た。
【0028】
1H-NMR(CDCl3, 400MHz) δ: 1.08 (s; 3H), 1.09 (s; 3H), 4.12 (d, J = 11.0Hz; 1H), 4.49 (d, J = 11.0Hz; 1H), 4.82 (brs; 1H), 4.99 (brs; 1H), 7.45 (t, J = 7.8Hz; 2H), 7.56 (t, J = 7.4Hz; 1H), 8.04 (brd, J = 7.1Hz; 2H); HR-ESI-MS (positive ion mode) : m/z 431.2554 (C27H36O3Na計算値:431.2562).
【0029】
実施例2
ent−kaur−16−ene−13,19−diol 19−O−4−methylcinnamate(エント−カウル−16−エン−13,19−ジオール 19−O−4―メチルシンナメート;化合物(1b)の調製
ent−kaur−16−ene−13,19−diol(31mg)をトルエン(1.2mL)に溶解し、4−メチルケイ皮酸(32mg)、4−ジメチルアミノピリジン(4−DMAP;21mg)、及びdi−2−pyridyl thionocarbonate(DPTC;45mg)を加え室温で24時間攪拌した。水で希釈後、ジエチルエーテルで抽出した。ジエチルエーテル層は水洗浄し、無水硫酸ナトリウムで脱水後、溶媒を除去した。反応生成物はシリカゲル薄層クロマトグラフィー(展開液:ヘキサン−酢酸エチル=7:3)にて精製し、化合物(1b)(17mg)を得た。
【0030】
1H-NMR(CDCl3, 400MHz) δ: 1.01 (s; 3H), 1.07 (s; 3H), 2.37 (s; 3H), 4.00 (d, J = 11.0Hz; 1H), 4.36 (d, J = 11.0Hz; 1H), 4.81 (brs; 1H), 4.98 (brs; 1H),6.38 (d, J = 16.0Hz; 1H), 7.18 (d, J = 8.2Hz; 2H), 7.42 (d, J = 8.2Hz; 2H), 7.63 (d, J = 16.0Hz; 1H); HR-ESI-MS (positive ion mode) : m/z 471.2881 (C30H40O3Na計算値:471.2875).
【0031】
実施例3
ent−kaur−16−ene−13,19−diol 19−O−3−methylcinnamate(エント−カウル−16−エン−13,19−ジオール 19−O−3―メチルシンナメート;化合物(1c)の調製
ent−kaur−16−ene−13,19−diol(40mg)をトルエン(1.6mL)に溶解し、3−メチルケイ皮酸(46mg)、4−ジメチルアミノピリジン(4−DMAP;28mg)、及びdi−2−pyridyl thionocarbonate(DPTC;63mg)を加え室温で24時間攪拌した。水で希釈後、ジエチルエーテルで抽出した。ジエチルエーテル層は水洗浄し、無水硫酸ナトリウムで脱水後、溶媒を除去した。反応生成物はシリカゲル薄層クロマトグラフィー(展開液:ヘキサン−酢酸エチル=7:3)にて精製し、化合物(1c)(35mg)を得た。
【0032】
1H-NMR(CDCl3, 400MHz) δ: 1.01 (s; 3H), 1.07 (s; 3H), 2.37 (s; 3H), 4.01 (d, J = 11.0Hz; 1H), 4.36 (d, J = 11.0Hz; 1H), 4.82 (brs; 1H), 4.98 (brs; 1H),6.42 (d, J = 16.0Hz; 1H), 7.19 (d, J = 7.3Hz; 1H),7.27 (t, J = 7.8Hz; 1H), 7.33 (d, J = 6.9Hz; 1H), 7.34 (s; 1H), 7.63 (d, J = 16.0Hz; 1H); HR-ESI-MS (positive ion mode): m/z 471.2885 (C30H40O3Na計算値:471.2875).
【0033】
実施例4
ent−kaur−16−ene−13,19−diol 19−O−2−methylcinnamate(エント−カウル−16−エン−13,19−ジオール 19−O−2―メチルシンナメート;化合物(1d)の調製
ent−kaur−16−ene−13,19−diol(30mg)をトルエン(1.2mL)に溶解し、2−メチルケイ皮酸(35mg)、4−ジメチルアミノピリジン(4−DMAP;23mg)、及びdi−2−pyridyl thionocarbonate(DPTC;46mg)を加え室温で24時間攪拌した。水で希釈後、ジエチルエーテルで抽出した。ジエチルエーテル層は水洗浄し、無水硫酸ナトリウムで脱水後、溶媒を除去した。反応生成物はシリカゲル薄層クロマトグラフィー(展開液:ヘキサン−酢酸エチル=7:3)にて精製し、化合物(1d)(28mg)を得た。
【0034】
1H-NMR(CDCl3, 400MHz) δ: 1.02 (s; 3H), 1.08 (s; 3H), 2.43 (s; 3H), 4.04 (d, J = 11.0Hz; 1H), 4.35 (d, J = 11.0Hz; 1H), 4.82 (brs; 1H), 4.98 (brs; 1H), 6.35 (d, J = 16.0Hz; 1H), 7.19 (d, J = 7.3Hz; 1H), 7.20 (t, J = 7.3Hz; 1H), 7.27 (dt, J = 1.4, 7.3Hz; 1H), 7.56 (d, J = 7.8Hz; 1H), 7.97 (d, J = 16.0Hz; 1H); HR-ESI-MS (positive ion mode) : m/z 471.2867 (C30H40O3Na計算値:471.2875).
【0035】
実施例5
ent−kaur−16−ene−13,19−diol 19−O−β−trifluoromethyl acrylate(エント−カウル−16−エン−13,19−ジオール 19−O−トリフルオロメチルアクリレート;化合物(1e)の調製
ent−kaur−16−ene−13,19−diol(30mg)をトルエン(1.2mL)に溶解し、β−トリフルオロメチルアクリル酸(44mg)、4−ジメチルアミノピリジン(4−DMAP;23mg)、及びdi−2−pyridyl thionocarbonate(DPTC;46mg)を加え室温で24時間攪拌した。水で希釈後、ジエチルエーテルで抽出した。ジエチルエーテル層は水洗浄し、無水硫酸ナトリウムで脱水後、溶媒を除去した。反応生成物はシリカゲル薄層クロマトグラフィー(展開液:ヘキサン−酢酸エチル=7:3)にて精製し、化合物(1e)(16mg)を得た。
【0036】
1H-NMR(CDCl3, 400MHz) δ: 0.98 (s; 3H), 1.04 (s; 3H), 4.01 (d, J = 11.0Hz; 1H), 4.39 (d, J = 11.0Hz; 1H), 4.82 (brs; 1H), 4.98 (brs; 1H), 6.49 (dd, J = 2.1, 15.8Hz; 1H), 6.75 (dq, J = 6.6, 15.8Hz; 1H); HR-APCI-MS (positive ion mode): m/z 409.2346 (C24H32O3F3計算値:409.2354).
【0037】
実施例6(腫瘍細胞傷害試験及びその傷害機構の解析)
1.細胞傷害活性試験
3種類の腫瘍細胞を用いて細胞傷害活性を評価した:ヒト乳癌由来細胞株SKBR3、ヒト肺癌由来細胞株A549及びヒト胃癌由来細細胞株AZ521を用いた。
各細胞を96wellマイクロプレートに播種し、24時間培養した。本発明化合物を添加し、48時間作用後、0.5%MTT溶液を加え、3時間培養した。その後、0.04N HCl/isopropanolを加えTop:570nm、Bottom:630nmにて吸光度を測定し、コントロールとの比較により細胞生存率を算出した。EC50はコントロールと比較して細胞を50%死滅させる効果濃度のことであり、GraphPad Prism computer program(Ver.4.0)を用いて求めた。
【0038】
細胞傷害活性試験に供したSKBR3、A549及びAZ521の細胞数は何れも3×103cells/wellである。
【0039】
2.フローサイトメトリー(Annexin V−Propidium Iodide(PI)二重染色法)
本試験はAnnexin V−FITC kit[Bender MedSystems,BMS306FI]を用いて行った。A549を6wellプレートに播種し(1×105cells/well)、24時間培養した。本発明化合物(40μM又は30μM)を作用させた後、細胞を回収し、遠心分離(1000×g,5min)を行った。遠心分離後、上清を除去し、Binding buffer 195μL及びAnnexin V5μLを加えピペッティングを行い、15分間暗所で染色した。その後、遠心分離(1000×g,5min)を行い、上清を除去し、Binding buffer 490μL及びPI 10μLを加えピペッティングし、フローサイトメーターにて解析を行った。
【0040】
3.結果
3−1.細胞傷害活性試験
本発明化合物の細胞傷害活性の結果を表1に示した。胃癌細胞(AZ521)、肺癌細胞(A549)及び乳癌細胞(SKBR3)に対して、参照化合物であるシスプラチンをはるかに上回る細胞傷害活性を示した。
【0041】
【表1】
【0042】
3−2.フローサイトメトリー(Annexin V−Propidium Iodide(PI)二重染色法)
本発明化合物の細胞傷害性がアポトーシスによることを確認するために本発明化合物20μM〜40μM(化合物(1a)は40μM、化合物(1b)〜(1d)は30μM、化合物(1e)は20μM)を作用させた肺癌細胞A549についてAnnexin V−PI二重染色を行い、フローサイトメトリーを行った(図1〜5)。時間依存的にAnnexin V 陽性/PI陰性細胞群の増加が確認され(8hr、24hr)、本発明化合物がアポトーシスを誘導することを確認した。
【0043】
3−3.Hoechst染色
20μM〜30μMの化合物(1a)〜(1e)を24時間作用させた肺癌細胞A549についてHoechst 33342染色を行った。その結果、図6及び7に示すように、化合物(1a)〜(1e)のいずれの化合物処理によっても細胞核の凝集・断片化が観察されたことから、本発明化合物がアポトーシスを誘導することが判明した。
【0044】
また、化合物(1b)を作用させたA549細胞におけるカスパーゼ遺伝子解析をしたところ、いずれのカスパーゼも活性化されていることから、本発明化合物によるアポトーシス誘導は、カスパーゼ3、8、9を介したシグナル伝達によるものと考えられた。
【0045】
本発明化合物がヒト胃癌由来細胞株AZ521、ヒト肺癌由来細胞株A549及びヒト乳癌細胞株SKBR3に対して参照化合物のシスプラチンと比較してはるかに強い細胞傷害活性を有することが明らかになった。また、本発明化合物はA549細胞に対しアポトーシス誘導により細胞傷害活性を示すことが確認された。
【特許請求の範囲】
【請求項1】
一般式(1)
【化1】
(式中、R1はトリフルオロメチルビニル基、フェニル基又はメチルフェニルビニル基を示す)
で表されるステビオール誘導体。
【請求項2】
請求項1記載の化合物を有効成分とする医薬。
【請求項3】
悪性腫瘍治療薬である請求項2記載の医薬。
【請求項4】
胃癌、肺癌又は乳癌の治療薬である請求項2又は3記載の医薬。
【請求項5】
請求項1記載の化合物を有効成分とするアポトーシス誘導薬。
【請求項1】
一般式(1)
【化1】
(式中、R1はトリフルオロメチルビニル基、フェニル基又はメチルフェニルビニル基を示す)
で表されるステビオール誘導体。
【請求項2】
請求項1記載の化合物を有効成分とする医薬。
【請求項3】
悪性腫瘍治療薬である請求項2記載の医薬。
【請求項4】
胃癌、肺癌又は乳癌の治療薬である請求項2又は3記載の医薬。
【請求項5】
請求項1記載の化合物を有効成分とするアポトーシス誘導薬。
【図1】


【図2】

【図3】
【図4】

【図5】

【図6】

【図7】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2011−74065(P2011−74065A)
【公開日】平成23年4月14日(2011.4.14)
【国際特許分類】
【出願番号】特願2010−179201(P2010−179201)
【出願日】平成22年8月10日(2010.8.10)
【出願人】(899000057)学校法人日本大学 (650)
【Fターム(参考)】
【公開日】平成23年4月14日(2011.4.14)
【国際特許分類】
【出願日】平成22年8月10日(2010.8.10)
【出願人】(899000057)学校法人日本大学 (650)
【Fターム(参考)】
[ Back to top ]