
感光性保護基を含むカップリング剤及び、固体支持体の官能化のためのような、その使用
【課題】本発明は、感光性保護基を含む化合物、及び固体支持体の官能化のためのカップリング剤としてのそれらの使用に関する。本発明はまた、前記化合物により官能化された固体支持体、及び核酸分子のような関心のある生物分子の固定のための同じものの使用に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、感光性保護基を含むカップリング剤及び固体支持体の官能化のためのその使用に関する。本発明はまたこれらのカップリング剤で官能化された固体支持体にも関し、並びに関心のある生物分子、特に核酸分子の固定化のためのその使用にも関する。
【背景技術】
【0002】
関心のある生物分子の、特にオリゴヌクレオチド(ON)のような核酸の固体支持体への共有結合的なグラフティングのための様々な方法が既に提案されている。ONsの結合という特定の関係において、これらの方法は2つの主なカテゴリーに分けられ得る:
1)In situ合成、これはホスホルアミダイト経路を介する合成原理を利用して固体支持体にON分子を段階的に構築することからなる。特に国際出願WO 97/39151に記載されるこの方法は、高密度のONsを備えた表面の製造を可能にする。さらに、ブランキングプレート(blanking plate)と感光性保護基のセットの使用により、それは該支持体上での様々なONsの標的化を可能にする。しかしながら、この方法は、幾つかの大きな欠点を有する。特に、該支持体に結合されるONsを特徴付けることができず、また、修飾を伴うONs(ONs carrying modifications)を取り込むことが非常に困難である。
【0003】
2)デポジションによる、またはex situ合成、これは第一に従来のようにONを製造すること、そして第二に好適な化学反応を利用して固体支持体にそれを結合させることからなる。この第二の方法は特に、固体支持体に結合される前に特徴付けられ得るON分子を使用するという利点を有する。
【0004】
これ以降に記載する本発明は、より詳細には付着による合成方法のコンテクスト内にある。
【0005】
様々なタイプの付着による合成方法が先行技術において、特にONのグラフティングを配置させる様々な技術に関して既に記載されている。
【0006】
第一に、電気化学経路を使用する方法が挙げられ、これは、ピロール残基の電解重合を介して、例えばピロール基のような導電性基で予め官能化されたONを取り付けることからなる。このタイプの電気化学的ON堆積方法は、特に特許FR2 703 359に記載されている。これにもかかわらず、これは導電性支持体の使用を必要とする欠点を有し、これは最終成分を複雑化する。さらに、「Lab on a Chip」タイプのコンポーネントにおいて、様々な二次加工工程が電極の質を、ゆえにそれらの機能を損ない得る。
【0007】
第二に、光化学経路が挙げられ、これは表面及び使用される化学溶液の管理が外面化される(externalized)ため、より有望なようである。2つのアプローチが開発された:
i)表面と標的分子の間の直接的フォトグラフティング。この場合、該フォトグラフティングはあまり選択性のないフリーラジカル反応を伴うという欠点を有する;
ii)支持体上での感光性保護基で保護された官能基の遊離、これは標的ONとの引き続く反応を可能にして、それらの結合を生じる。この場合、使用される支持体は一般的には、その一端が該支持体の表面と結合するための官能基と、他端が相補的な化学官能基を有する標的分子と反応する官能基を有する二官能性化合物であるカップリング剤で官能化されており、該カップリング剤の前記反応性官能基は感光性基で保護される。しかし、競合するために、後者のアプローチは幾つかの基準を満たさなければならない。これらの基準は以下である:
−活性化された支持体と標的分子との間の反応が迅速でなければならない;この反応は、カップリング剤の反応性官能基がそれらの保護基から遊離される時にのみ起こり得る(活性化反応)。
【0008】
−この反応による収率は高くなければならない;
−カップリング剤を製造するための合成方法は、できる限り少ない工程を含み、実施されるように簡単でなければならない;
−穏やかな反応条件の使用(例えば、水);
−カップリング剤と標的分子との間で生じる反応は、このように官能化された支持体の非常にフレキシブルな使用を可能にするために、様々な温度及びpH条件下で安定でなければならない。
【0009】
現在、今日までに提案された様々な方法が、特に使用される様々なカップリング剤の特性のために、全てのこれらの基準を完全に十分に満たしていないことは明らかである。
【発明の開示】
【発明が解決しようとする課題】
【0010】
この理由のために、本発明者は、少ない工程を含む方法に従い容易に合成され、そして穏やかな条件下で実施できる、感光性基で保護された反応性官能基を有する新規の二官能性カップリング剤を提供するという目的を自分自身に与えており、これらのカップリング剤は、そこに標的生物分子を続いて結合することを目的とする固体支持体を官能化するために有利に使用できる。
【課題を解決するための手段】
【0011】
このように、本発明の第一の主題は、以下の式(I):
A−X−B−Proc (I):
(ここで:
− Aは固体支持体に結合するための官能基を表し、前記官能基は、アミン、ホスホルアミダイト、シラン及び活性エステル型の官能基から選択され;
− Xはスペーサーアームを表し;
− Bは、光脱保護の後、オキシアミン(−ONH2)型及び誘導体、或いはヒドラジド(−NH−NH2)及び誘導体の官能基を生じる基から選択される反応性官能基であり、前記官能基BはProc基で保護されている;
−Procは以下の式(II):
【0012】
【化1】

【0013】
(ここで:
− R1及びR2は同じでも異なってもよく、水素原子またはC1−C4アルキル基をで表す)
の感光性保護基である)
のカップリング剤である。
【0014】
本発明に従うと、官能基Aは、式(I)のカップリング剤が結合することを目的とする支持体の性質に従って選択される。活性エステル型の官能基は、好ましくN−ヒドロキシスクシンイミドで、または当業者に知られた任意の他の好適な活性化試薬、例えばペンタフルオロベンゼンでエステル化されたカルボン酸官能基である。
【0015】
スペーサーアームXは、所望の最終特性に応じて、長さ及び極性が変化し得る。例えば、それは無電荷疎水性鎖(飽和炭化水素ベース鎖など)またはグリセロールエーテルのような無電荷親水性鎖であり得る。
【0016】
本発明の特に好ましい実施形態に従うと、スペーサーアームXは、1〜13炭素原子を含む飽和炭化水素ベース鎖、ならびにトリエチレングリコール(TEG)及びヘキサメチレングリコール(HEG)のような炭素ベース鎖が4〜12炭素原子を含むグリコールエーテルから選択される。
【0017】
反応性官能基Bは、光脱保護の後に、少なくとも一つの相補的な化学官能基を備えた標的分子の共有結合を可能にする官能基である。これらの反応性官能基Bは、アミン官能基、アルコール、チオール、カルボキシルまたはカルボニルのような前記標的分子により保有される相補的な官能基と相互作用できる。相補的な官能基のペアのさらに網羅的なリストは、任意の有機化学モノグラフに容易に見られ得る。
【0018】
上記の式(II)の保護基について、そして、それらがアルキル基を表すとき、R1及びR2は互いに独立して、好ましくはメチル及びエチル基から選択され;該メチル基が最も特に好ましい。
【0019】
上記の式(I)のカップリング剤において、好ましくは特に以下のものが挙げられる:
i)− Aはホスホルアミダイト官能基を表し、
− Xは6炭素原子を有する飽和炭化水素ベース鎖を表し、
− Bはオキシアミン官能基(−ONH−)を表し、及び
− R1及びR2は水素原子またはメチル基を表す;
ii)− Aはトリ(C1−C4)アルコキシシランを表し、
− Xは2〜12炭素原子を有する飽和炭化水素ベース鎖を表し、
− Bはオキシアミン官能基(−ONH−)を表し、及び
− R1及びR2は水素原子またはメチル基を表す;
iii)− AはN−ヒドロキシスクシンイミドまたはペンタフルオロベンゼンでエステル化されたカルボキシリック官能基を表し、
− Xは−CH2−を表し、
− Bはオキシアミン官能基(−ONH−)を表し、及び
− R1及びR2は水素原子またはメチル基を表す。
【0020】
上記のポイントiii)で定義された化合物のうち、好ましくは、特に官能基Aがトリエトキシシランであり、そしてXが3または11炭素原子を有する飽和炭化水素ベース鎖である化合物である。上記の式(I)のカップリング剤は当業者によく知られた有機合成の原理に従い、また官能基A及びBの性質に依存して、容易に製造され得る。
【0021】
本発明の第一の実施形態に従い、そしてカップリング剤が式(I)の化合物(ここで結合官能基Aが例えばN−ヒドロキシスクシンイミドエステルの形態で活性化されたカルボン酸型であり、そして官能基Bがオキシアミン官能基である)であるとき、以下のスキーム1に対応する製造方法が好ましく使用される:
【0022】
【化2】
【0023】
このスキームにおいて、Procは上記で定義された式(II)の感光性保護基であり、そしてXは上記で示したものと同じ意味を有し得るスペーサーアームである。
【0024】
このスキームに従い、カルボキシリック官能基及びオキシアミン官能基を有する式(III)の化合物は、第一工程において、選択された式(II)のProc感光性保護基で保護され、式(IV)の化合物を生成し、次いで、第二工程において、式(IV)の化合物のカルボン酸官能基はN−ヒドロキシスクシンイミドで活性化され、式(I)の予期した化合物を生成する。
【0025】
本発明の第二の実施形態に従うと、カップリング剤が式(I)の化合物(ここで結合官能基Aはホスホルアミダイト官能基であり、そして官能基Bはオキシアミン官能基である)であるとき、以下のスキーム2に応じた製造方法が好ましく使用される:
【0026】
【化3】
【0027】
このスキームにおいて、X及びProcは上記で示したものと同じ意味を有し、Bはオキシアミン官能基を表し、そしてAは以下の基:
【0028】
【化4】
【0029】
(ここで、R及びR’は同じでも異なってもよく、1〜13炭素原子を有するアルキル基を表す)
を示す。
【0030】
このスキームに従うと、第一工程において、式(V)のハロゲン化アルコール(ここでHaloは臭素、塩素、ヨウ素またはフッ素のようなハロゲン原子を表す)はN−ヒドロキシフタルイミドと反応して(ハロゲン原子の求核置換)式(VI)の化合物を得、これは、第二工程で、ヒドラジン分解を受け、選択された式(II)の感光性Proc保護基で続いて保護されるオキシアミン官能基を遊離して式(VII)の化合物を生成し、これは、第三工程において、ホスフィチル化を受け、予期される式(I)の化合物が生成される。
【0031】
本発明の第三の実施形態に従うと、カップリング剤が式(I)の化合物(ここで結合官能基Aがシラン型であり、そして官能基Bがオキシアミン官能基である)であるとき、以下のスキーム3に応じた製造方法が好ましく使用される:
【0032】
【化5】
【0033】
この方法に従うと、式(IX)のアルケン化合物のオキシアミン官能基(ここで、Xは式(I)のカップリング剤に関して上記で示したものと同じ意味を有し得るスペーサーアームを表す)が保護されて、式(X)の化合物を得、これは続いてヒドロシリル化を受け、式(I)の化合物が生成される(ここで結合官能基Aはトリ(C1−C4)アルコキシシラン官能基である)。
【0034】
本発明に従う式(I)のカップリング剤は固体支持体の官能化のために使用され得る。ゆえに、本発明の主題は、固体支持体の官能化のための上記の定義した式(I)の少なくとも一つのカップリング剤の使用である。
【0035】
式(I)のカップリング剤の使用は、有利には光脱保護により容易に進み得る反応性官能基を備えた安定層で固体支持体の表面をすばやく修飾することを可能にする。
【0036】
このように、本発明の主題はまた、官能化された固体支持体の製造方法であり、これは、少なくとも一つの固体支持体の表面を、有機溶媒中の少なくとも一つの式(I)のカップリング剤の溶液と接触させる少なくとも一つの工程を含むことを特徴とする。
【0037】
該有機溶媒は、好ましくは、例えばトリクロロエチレン、ジメチルホルムアミド(DMF)及びシクロヘキサンのような非極性溶媒から選択される。
【0038】
式(I)のカップリング剤の溶液と固体支持体との接触は、好ましくは約4℃と80℃の間の温度で、約1〜48時間行われる。
【0039】
該支持体(substrate)は続いて1以上の溶媒、好ましくは連続して反応溶媒、無水エタノール及び/またはクロロホルムで洗浄し、そして好ましくは窒素で乾燥される。
【0040】
この方法は容易に実施できるという利点を有し、そして良好な密度の層を得ることを可能にする。特に、式(I)の化合物の自己組織化単層(SAM)で官能化された少なくとも一つの表面を有する固体支持体を得ることを可能にする。SAMsは、分子が組織化された分子の集合体であると定義され、ここで組織化は該分子の鎖の間の相互作用のためであり、安定した、単分子の、そして規則正しい異方性フィルムを生じる(A. ULMAN, Chem. Rev., 1996, 96, 1533-1554)。特に固体支持体の表面が、好ましくはトリクロロエチレンのような溶媒中で約10mMの溶液の、本発明に従う式(I)のカップリング剤(ここで結合官能基Aはシラン型)で官能化されるとき、そして接触が約4℃の温度で約24時間実施されるとき、このようなSAMsが得られ得る。
【0041】
さらに、本発明に従う方法を実施することにより得た表面は、固体支持体の表面に存在するカップリング剤の反応性官能基Bに対して相補的である化学官能基を含む関心のある生物分子を共有結合で固定化するための時間及び空間的に、選択的に容易に除去される感光性保護基で保護された多数の反応性官能基を直接提示する。
【0042】
本発明に従う式(I)のカップリング剤で官能化され得る固体支持体は、好ましくはガラス、セラミック(酸化物型)、シリコンまたはプラスチックから作製される支持体から選択され、前記支持体は少なくとも一つの水和された、ヒドロキシル化された、シラン化されたまたはアミン化表面、あるいは活性エステル型の表面を有する。このような表面は、よく知られた先行技術に従い容易に調製され得る。例えば、表面のヒドロキシル官能基を遊離するための酸加水分解を続いて受けるシランエポキシドによるシラン化によりヒドロキシル化された表面を含む固体支持体を製造することが可能である。
【0043】
これらの固体支持体は、少なくとも一つの平面または平面ではない、平滑なまたは構造化された表面を有し、例えばガラススライド、平面のプラスチックプレートまたはウェルを備えたプラスチックプレート、キャピラリーチューブあるいはポーラスまたは非ポーラスビーズの形態であり得る。
【0044】
上記のように、式(I)のカップリング剤の結合官能基Aは、前記カップリング剤が結合されることを意図する支持体の表面の性質に応じて選択される。
【0045】
このように、これらが少なくとも一つの活性エステル型の表面を有する支持体であるとき、官能基Aが好ましくはアミン官能基であり;そして、これらが少なくとも一つのヒドロキシル型の表面を有する支持体であるとき、官能基Aは好ましくはホスホルアミダイトまたはシラン官能基であり;これらが少なくとも一つのヒドリド型の表面を有する支持体であるとき、官能基Aは好ましくはシラン官能基であり、そして、これらがアミノ化表面を有する支持体であるとき、官能基Aは好ましくは活性エステル官能基である。
【0046】
ゆえに、本発明の主題はまた、上記で定義した1以上の式(I)のカップリング剤で官能化された少なくとも一つの表面を有する固体支持体である。
【0047】
このような支持体は、それらが脱保護された後の前記固体支持体の表面に存在する式(I)のカップリング剤の反応性官能基Bに対して相補的である、化学官能基を含む関心のある生物分子の固定化のために、そして特にDNA及びオリゴヌクレオチドのような核酸分子の共有結合的な固定化のために、有利に使用され得る。
【0048】
このように、本発明の主題はまた、前記固体支持体の表面に存在する式(I)のカップリング剤の反応性官能基Bに対して相補的である化学官能基を含む関心ある生物分子の、そして特にアミド結合(ペプチド)の形成を介した核酸(DNA、オリゴヌクレオチド)の、共有結合的な固定のための、上記の固体支持体の使用である。
【0049】
本発明の主題はまた、上記の固体支持体への、関心ある生物分子、特に核酸分子の固定化方法であり、これは該固体支持体の表面の少なくとも一部分の露光による式(I)の化合物の反応性官能基Bの光−脱保護、続いて、このように活性化された固体支持体を関心のある生物分子、特に核酸分子の溶液と接触させる第1工程を少なくとも含み、その結果、前記分子に備えられた、そして本発明に従う前記式(I)の化合物の反応性官能基Bに対して反応性を有する、少なくとも一つの化学官能基との間の共有結合の形成を介する前記分子の前記支持体への固定化を生じさせ、そしてこれらの2つの工程を必要に応じて繰り返すことを特徴とする。
【0050】
特に、光−脱保護工程は、該固体支持体の官能化された表面の一部のみに局在され得、例えばマスクを介して実施され得る。
【0051】
露光後、式(I)のカップリング剤の反応性官能基Bは脱保護(活性化)状態にあり、これは、該固体支持体の表面に存在する前記活性化反応性官能基Bに対して反応性を有する化学官能基を含む溶液中の任意の分子を捕獲し得るフリーな反応性官能基を有する領域を生み出す。
【0052】
特に、固定化工程は、式(I)の化合物のオキシアミン官能基Bと、関心ある生物分子、特に核酸分子のカルボニル化官能基との間のオキシム結合の形成を通して実施される。この反応は、穏やかな条件下、特に4と7の間のpHで実施できるという利点を有する。さらに、形成された結合(オキシム)は非常に良い安定性を示し、そして、更なる還元工程により、それを安定化させる必要がない。
【0053】
露光波長は好ましくは300〜400nm間にある。
【0054】
関心のある生物分子の溶液は、単純な水溶液または緩衝作用のある水溶液中のこれらの分子の溶液であり得る。
【0055】
接触を実施する期間は、好ましくは約5〜60分間であり、また好ましくは約4〜60℃間の温度で実施される。
【0056】
最後に、本発明の主題はまた、上記した、また本発明に従う固定化方法を実施することにより得られる、すなわち本発明に従う式(I)のカップリング剤の反応性官能基Bで形成された共有結合により関心のある前記生物分子、特に核酸分子が固定化された少なくとも一つの表面を有する、固体支持体(特に核酸チップ)である。
【0057】
上記アレンジメントの他に、本発明はまた、以下の記載から明らかになる他のアレンジメントも含有し、これは、式(I)のカップリング剤の製造の例、本発明に従う式(I)のカップリング剤によるキャピラリーチューブ型のガラス固体支持体または平面の固体支持体の官能化の例、オリゴヌクレオチドの固定化のためのこれらの官能化支持体の使用、ならびに添付の図1及び2
(ここで:
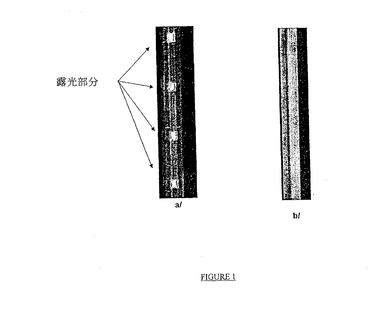
− 図1は、非官能化キャピラリーチューブ(タイプb/のキャピラリーチューブ)と比較した、フルオロフォアで標識された相補的なオリゴヌクレオチドによるハイブリダイゼーション後の、本発明に従う方法に従うキャピラリーチューブ型(タイプa/のキャピラリーチューブ)の固体支持体に結合したオリゴヌクレオチドにより放出された蛍光の写真を表す;
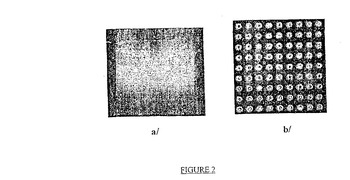
−図2は、非官能化支持体(タイプb/の支持体)と比較した、フルオロフォアで標識された相補的なオリゴヌクレオチドによるハイブリダイゼーション後の、本発明に従う方法に従う平面の固体支持体(タイプa/の支持体)に結合したオリゴヌクレオチドにより放出された蛍光の写真を表す
に関する。
【0058】
実施例1:式(I)に従うホスホルアミダイト誘導体の製造
【0059】
【化6】
【0060】
1)第一工程:化合物(1)の製造:オキシアミンの導入
【0061】
【化7】
【0062】
150mlのジメチルホルムアミド(DMF)中のN−ヒドロキシフタルイミド(2.70g;16.6mmol)と炭酸カリウム(4.6g;23.6mmol)の溶液を攪拌しながらアルゴン下、1時間50℃の温度に加熱した。続いて6−ブロモヘキサノール(3g;16.6mmol)を添加し、次いで反応混合液を再び50℃の温度で一晩攪拌した。ろ過後、該溶媒を減圧下で蒸発させた。続いて50mlの酢酸エチルを、このようにして得られた残渣に添加し、次いで有機相を0.1N 水酸化ナトリウムで、次いで塩化ナトリウムの飽和水溶液で洗浄した。続いて該有機相を無水硫酸ナトリウム上で乾燥させ、次いで蒸発させた。生成物(1)は85℃と88℃の間の融点を有する白色粉末の形態で得た(3.28g;12.5mmol;収率75%)。
【0063】
CDCl3中、周波数200MHzで実施したプロトン核磁気共鳴(1H-NMR)分析は以下の通りである:
δ ppm = 1.30-1.90 (8H, m, 4CH2), 3.60 (2H, t, J = 13 Hz, CH2O), 4.20 (2H, t, J = 13 Hz, CH2ON), 7.70-7.90 (4H, m, Ar-H).
MS (DCI, positive mode): Mcalc- 263 (C14H17NO4), m/z = 281 [M+NH4]+.
2)第二工程:化合物(2)を製造するための化合物(1)のトリチル化
【0064】
【化8】
【0065】
塩化トリチル(4.2g;15mmol)を、75mlの無水ピリジン中の上記の前記工程で得た化合物(1)(3.2g;12.5mmol)の溶液に添加した。該溶液をアルゴン下、一晩、周囲温度で攪拌した。続いて5mlのメタノールをゆっくり添加し、次いで該溶媒を減圧下で蒸発させた。続いて得られた残渣を100mlの酢酸エチルに溶解し、次いで有機相を水で、次いで塩化ナトリウムの飽和水溶液で洗浄した。続いて該有機相を無水硫酸ナトリウム下で乾燥させ、次いで蒸発させた。粗生成物をシリカゲルクロマトグラフィー(溶離剤 ジクロロメタン/シクロヘキサン:75/25、v/v)で精製した。生成物(2)は106−107℃で融解する白色粉末の形態で得た(6.27g;12.4mmol;収率99%)。
【0066】
1H-NMR分析(200 MHz, CDCl3)は以下の通りである:
δ ppm = 1.30-1.90 (8H, m, 4 CH2), 3.20 (2H, t, J = 13 Hz, CH-2O), 4.20 (2H, t, J = 13 Hz, CH2ON), 7.70-7.90 (4HAr, m, Ar-H-フタルイミド), 7.20-7.40 (15H, m, Ar-H-トリチル).
MS (ESI, positive mode): MCalc = 505 (C33H-31NO4), m/z = 528 [M+Na]+, m/z = 544 [M+K]+.
3)第三工程:化合物(3)を得るための化合物(2)のヒドラジン分解
【0067】
【化9】
【0068】
上記の第二工程において得られた化合物(2)(6.27g;12.4mmol)を、50mlのジクロロメタンに溶解し、次いで780mgのヒドラジン(24.8mmol)を添加した。得られた溶液を1時間半還流させ、次いでろ過し、減圧下で蒸発させた。粗生成物を、溶離剤がジクロロメタン、次いでジクロロメタンとメタノールが95/5(v/v)の混合液である、シリカゲルクロマトグラフィーで精製した。化合物(3)は収率90%でオイルの形態で得た(4.20g;112.2mmol)。
【0069】
1H-NMR(200 MHz, CDCl3):δ ppm = 1.30-1.80 (8H, m, 4 CH2), 3.10 (2H, t, J = 13 Hz, CH2O-Tri), 3.60 (2H, t, J = 13 Hz, CH2ON), 7.70-7.90 (15H, m, Ar-H--トリチル).
MS (ESI, positive mode): Mcalc= 375 (C25H29NO2), m/z = 376 [M+H]+, m/z = 398 [M+Na]+, m/z = 414 [M+K]+.
4)第四工程:化合物(4)を得るための化合物(3)のオキシアミンの保護
【0070】
【化10】
【0071】
上記の第三工程で製造した化合物(3)(4.20g;11.2mmol)を100mlのピリジンに溶解した。続いて5.40g(22.4mmol)の2−(2−ニトロフェニル)プロピルクロロホルメート(Cl−NPPOC)を含有する50mlのジクロロメタン溶液を滴下した。該反応混合液を暗所で1時間攪拌し、次いで減圧下で蒸発させた。続いて得られた残渣をジクロロメタンに溶解し、次いで有機相を塩化ナトリウムの飽和水溶液で洗浄した。該有機相を無水硫酸ナトリウム下で乾燥させ、次いで蒸発させた。粗生成物を溶離剤としてジクロロメタンを使用したシリカゲルクロマトグラフィーにより精製した。生成物(4)は収率89%でオレンジ色のオイル(5.80g;9.9mmol)の形態で得た。
1H-NMR (200 MHz, CDCl3): δ ppm = 1.30 (3H, d, J = 16 Hz, CH3), 1.50-1.80 (8H, m, 4 CH2), 3.0 (2H, t, J = 13 Hz, CH2O-Tri), 3.70 (2H, m, CH2ON, 1H, m CH), 4.20 (2H, t, J = 13 Hz, CH2OCO), 7.10-7.50 (15H, m, Ar-H-トリチル), 7.50-7.80 (4H, m, Ar-H- NPPOC).
MS (ESI, positive mode): Mcalc = 582 (C35H38N2O6), m/z = 605 [M+Na]+, m/z = 621 [M+K]+.
MS (ESI, negative mode): m/z = 581 [M-H]-.
5)化合物(5)を得るための化合物(4)の脱トリチル化
【0072】
【化11】
【0073】
上記の前記工程で得た5.80g(9.90mmol)の化合物(4)を、ジクロロメタン中、20%のトリフルオロ酢酸溶液50mlに溶解した。該混合液を3〜4時間周囲温度で攪拌し、次いで減圧下で蒸発させた。得られた残渣を100mlのジクロロメタンに溶解し、次いで有機相をNaClの飽和水溶液で洗浄した。該有機相を無水硫酸ナトリウム上で乾燥させ、次いで蒸発させた。粗生成物を、溶離剤としてジクロロメタン、次いで95/5(v/v)のジクロロメタン/メタノールを使用したシリカゲルクロマトグラフィーにより精製した。化合物(5)は収率90%でオレンジ色のオイル(3.0g;8.9mmol)の形態で得た。
1H-NMR (200 MHz, CDCl3): δ ppm = 1.30 (3H, d, J = 16 Hz, CH3), 1.50-1.80 (8H, m, 4 CH2), 3.70 (2H, m, CH2ON, 1H, m, CH), 4.30 (2H, t, CH2OCO, 2H, t, CH2OH), 7.50-7.80 (4H, m, Ar-H NPPOC).
MS(DVI, positive mode): Mcalc = 340 (C16H24N2O6), m/z = 454 [M+TFA]+.
6)第六工程:化合物(6)を製造するための化合物(5)のホスフィチル化
【0074】
【化12】
【0075】
上記の前記工程で得た化合物(5)(0.90g;2.7mmol)をアルゴン気流下、15mlのジクロロメタンに溶解し、次いでジイソプロピルエチルアミン(DIEA)(0.56ml,3.2mmol)を添加し、そして0.72mlの2−シアノエチルジイソプロピルクロロホスホルアミダイト(3.2mmol)を添加した。反応混合液を、周囲温度で4時間、出発生成物がなくなるまで攪拌した。40mlのジクロロメタンを次いで添加し、そして有機相を炭酸水素ナトリウム飽和水溶液で、次いでNaCl飽和水溶液で洗浄した。該有機相を無水硫酸ナトリウム下で乾燥させ、次いで蒸発させた。粗生成物を、溶離剤としてジクロロメタンを、次いで97/3(v/v)のジクロロメタン/メタノール混合液を使用してシリカゲルクロマトグラフィーにより精製した。化合物(6)は収率38%で黄色オイル(0.50g;1.0mmol)の形態で得た。
1H-NMR (200 MHz, CDCl3): δ ppm = 1.20-1.50 (15H, m, 4 CH3), 1.50-1.85 (8H, m, 4 CH2), 2.65 (2H, t, CH2CN), 3.80 (2H, m, CH2OH, 1H, m, CH, 4 H, m, 2 CH2OP), 4.30 (2H, m, CH2OCO, 2H, m, 2 CHN, 7.10-7.70 (4H Ar-H NPPOC).
31P-NMR (200 MHz, CDCl3): δ ppm = 120 (2 diastereoisomers).
実施例2:式(I)に従う活性エステル誘導体(化合物(8))の製造
【0076】
【化13】
【0077】
1)第一工程:前駆体酸(precursor acid)の製造
【0078】
【化14】
【0079】
カルボキシメトキシルアミンハイドロクロライド(1g;4.57mmol)を25mlの10%炭酸ナトリウム水溶液に溶解した。該溶液を0℃に冷却し、次いで、2.20g(9.1mmol)の2−(2−ニトロフェニル)プロピルクロロホルメート(Cl−NPPOC)を含有する20mlのジオキサン溶液を攪拌しながら滴下した。攪拌は、周囲温度で2〜3時間続けた。反応メディウムを蒸発させて、乾燥させた。250mlの水をこのようにして得た残渣に添加し、次いで水相を200mlのジエチルエーテルで洗浄した。次いで、該水相を1Nの塩酸溶液で酸性化してpH3とし、次いで250mlのジクロロメタンで3回抽出した。有機相を合わせ、そして無水硫酸ナトリウム上で乾燥させた。粗生成物を溶離剤としてジクロロメタン、次いで97/3(v/v)のジクロロメタン/メタノール混合液を使用してシリカゲルクロマトグラフィーにより精製した。化合物(7)は収率90%で88〜92℃間で融解する白色粉末の形態で得た(1.20g;4.0mmol)。
1H-NMR (200 MHz, CDCl3): δ ppm = 1.40 (3H, d, J = 16 Hz, CH3), 3.70 (1H, m, CH), 4.30 (2H, m, CH2O) 4.40 (s, 2H, COCH2O), 7.30-7.60 (4H, m, Ar-H NPPOC).
13C-NMR (300 MHz, CDCl3): δ ppm = 18 (CH2), 33 (CH2), 67 (CH3), 70 (CH3), 75 (CH3), 123 (quat), 137 (quat), 138 (quat), 133 (quat), 136 (CH), 151 (CH), 159 (CH), 172 (CH), 173 (CH).
MS (ESI, positive mode): Mcalc = 298 (C12H14N2O7), m/z = 321 [M+Na]+, m/z = 337 [M+K]+.
MS (ESI, negative mode): m/z = 297 [M-H]-, m/z = 595 [2M-H]-.
2)第二工程:N−ヒドロキシスクシンイミド活性エステル(8)の製造
【0080】
【化15】
【0081】
上記の前記工程で得た酸(7)(1.20g;4.0mmol)を15mlの無水ジクロロメタンに溶解し、次いでジシクロカルボジイミド(DCC)(0.82g;4.4mmol)を、続いてN−ヒドロキシスクシンイミド(0.457g;4.4mmol)を添加した。反応メディウムを一晩攪拌した。ろ過後、該溶媒を減圧下で蒸発させた。粗生成物を溶離剤として酢酸エチルを使用して速いシリカゲルクロマトグラフィーにより精製した。化合物(8)は収率67%で黄色粉末の形態で得た(1.0g;2.68mmol)。
実施例3:ペンタフルオロフェノール(9)により活性化された式(I)のエステル誘導体の製造
【0082】
【化16】
【0083】
上記の実施例2の第一工程で得た化合物(7)500mg(1.6mmol)を5mlのジクロロメタンに溶解し、次いで372mg(1.9mmol)のペンタフルオロフェノールを添加し、そしてジクロロメタン中、1mlのDCC(360mg,1.9mmol)溶液を滴下した。混合液を4時間攪拌し、次いで形成したジシクロウレア(dicyclourea、DCU)沈殿物をろ過により除いた。溶媒蒸発後、化合物(9)は収率100%でオイルの形態で得た(740mg,1.9mmol)。
1H-NMR (200 MHz, CDCl3): δ ppm = 1.30 (3H, d, J = 16 Hz, CH3, 3.70 (1H, m, CH), 4.30 (2 H, m, CH2O), 4.70 (s, 2H, COCH2O), 7.30-7.70, (4H, m, Ar-H NPPOC).
19F-NMR (200 MHz, CDCl3): δ ppm = -166.0 (1F, t), -164.0 (1F, d), -162.0 (1F, t), -157.0 (1F, t), -152.50 (1F, d).
実施例4:式(I)のトリエトキシシラン誘導体の製造
【0084】
【化17】
【0085】
1)第一工程:N−ニトロフェニルプロピルオキシカルボニルアリルオキシアミン(10)の製造
【0086】
【化18】
【0087】
0℃の温度に冷却された25mlの無水ピリジン中、3.250g(29.6mmol;1.1当量)の市販のアリルオキシアミン溶液に、6g(27mmol)の2−(2−ニトロフェニル)プロピルクロロホルメート(Cl−NPPOC)を含有するジクロロメタン溶液15mlを滴下した。30分攪拌後、該溶媒を蒸発させ、次いでトルエンとともに蒸発させた。反応粗生成物を酢酸エチルに溶解した。有機相を洗浄し、無水硫酸ナトリウム上で乾燥させ、次いで減圧下で蒸発させた。溶離剤として酢酸エチル/ヘキサン:1/4(v/v)の混合液を使用したシリカゲルクロマトグラフィーによる精製後、化合物(10)を収率72%(5.76g)で得た。
1H-NMR (200 MHz, CDCl3): δ ppm = 1.40 (d, 6H, Si-CH2-CH3), 3.70 (q, 1H, Ar-CH-CH3), 4.30 (m, 4H, CH2O, OCH2), 5.30 (dd, 2H, CH2), 5.90 (m, 1H, CH), 7.20 (s, 1H, NH), 7.30-7.80 (m, 4H, H-Ar).
2)第二工程:化合物(11)を製造するためのヒドロシリル化
8mlのトリエトキシシラン中、上記の前工程で得た1.67g(5.6mmol)の化合物(10)の溶液に、20μlのカールステッド触媒を滴下した。60℃で一晩攪拌後、溶媒を真空下、蒸留により蒸発させた。反応粗生成物を溶離剤として1/4(v/v)酢酸エチル/ヘキサン混合液を使用してシリカゲルクロマトグラフィーにより精製し、収率60%で予期した化合物(11)を得た(1.33g)。
1H-NMR (200 MHz, CDCl3): δ ppm = 0.65 (m, 2H, CH2Si), 1.20 (t, 3H, CH3-CH2-O), 1.40 (d, 3H, CH3-CH), 1.70 (m, 3H, CH2, CH-CH3), 3.80 (m, 4H, O-CH2-CH3, CH2O), 4.30 (m, 2H, CH2-O-CO), 7.30-7.80 (m, 5H, NH, H-Ar).
MS (エレクトロスプレー, positive mode): Mcalc = 444, m/e = 44.7 [M+H]+.
以下の固体支持体の製造例は、キャピラリーチューブの形態(長さ3cm及び直径100μmのガラスキャピラリーチューブ)において本発明に従う式(I)のカップリング剤を使用して実施した。しかしながら、これらの例は他のタイプの支持体の平面の形態に置き換えられるということを明確に理解すべきである。
【0088】
実施例5:シラン型の結合官能基Aを含む式(I)のカップリング剤で官能化された固体支持体の製造
この例において、上記の実施例4で製造した化合物(11)を式(I)のカップリング剤として使用した。
【0089】
化合物(11)により官能化された固体支持体は、以下の反応スキーム:
【0090】
【化19】
【0091】
に従う無機固体支持体(mineral solid support)(ガラスキャピラリーチューブ)の表面のシラン化に関する工程による場合、単一工程で得られる。
【0092】
これを行うために、キャピラリーチューブは1/1 水/エタノール混合液中、6M水酸化ナトリウム溶液で満たされ、そして溶液を周囲温度で2時間該キャピラリーチューブに残す。
【0093】
続いて、該キャピラリーチューブを水で徹底的に洗浄し、次いで窒素で乾燥させる。
【0094】
続いて、該キャピラリーチューブをトリクロロエチレン中、10mMの化合物(11)の溶液で満たし、そして周囲温度で一晩作用させ続けた。
【0095】
続いて、該キャピラリーチューブをトリクロロエチレン、無水エタノール、次いでクロロホルムで徹底的に洗浄し、最終的に窒素で乾燥させる。
【0096】
そして、その内表面が化合物(11)の相で官能化された、すなわち光感受性オキシアミン表面のガラスキャピラリーチューブを得た。
【0097】
実施例6:活性エステル型の結合官能基Aを含む式(I)のカップリング剤で官能化された固体支持体の製造
この実施例において、NHS−活性エステル型の結合官能基Aを含む、上記の実施例2で製造した化合物(8)を式(I)のカップリング剤として使用した。
【0098】
NHS−活性エステルカップリング剤により官能化された固体支持体は以下の反応スキーム:
【0099】
【化20】
【0100】
に従う、2つの工程で得る。
【0101】
これに従うと、第一工程では、固体支持体の無機表面がアミン官能基を含む表面に変換され、そして第二工程では、前記表面アミン官能基が化合物(8)と反応する。
【0102】
1)第一工程:アミン官能基を含む表面の製造
ガラスキャピラリーチューブを1/1 水/エタノール混合液中、6M 水酸化ナトリウム溶液で満たし、次いで該溶液を周囲温度で2時間該キャピラリーチューブに残した。
【0103】
続いて、該キャピラリーチューブを水で徹底的に洗浄し、次いで窒素で乾燥させた。
【0104】
続いて、該キャピラリーチューブを、95°エタノール中、10%のトリエトキシアミノプロピルシラン溶液で満たし、該溶液を周囲温度で一晩、該キャピラリーチューブに残した。続いて、該キャピラリーチューブをエタノールで洗浄し、そして窒素で乾燥させた。
【0105】
その内表面がアミン官能基で被覆されたキャピラリーチューブを得た。
【0106】
2)第二工程:化合物(8)による該支持体の官能化
上記の第一工程で得たキャピラリーチューブを、ジメチルホルムアミド(DMF)中、10mMの化合物(8)の溶液で満たし、そして該溶液を周囲温度で一晩、該キャピラリーチューブに残した。
【0107】
続いて、該キャピラリーチューブをDMFで、次いでエタノールで洗浄し、そして最終的に窒素で乾燥させた。
【0108】
その内表面が化合物(8)の層で官能化された、すなわち光感受性オキシアミン表面のガラスキャピラリーチューブをこのようにして得た。
【0109】
実施例7:関心ある生物分子の固定化のための官能化された支持体の使用
この実施例において、上記実施例5で得た固体支持体をオリゴヌクレオチド(ONs)の固定化のために使用した。しかしながら、これらの支持体の他の関心ある生物分子の固定化のための使用もまた妥当であると明確に理解すべきである。
【0110】
オリゴヌクレオチドの場合、該固体支持体の表面でのこれらの分子のグラフティング及びこれらの存在の確認は、4つの工程を含む:
(i)露光:これは光作用下で、該固体支持体の表面上の反応性官能基Bを部分的にフリーにすることを可能にする工程である。この官能基Bは生物分子のグラフティングを可能にする。
【0111】
(ii)固定化:これは露光工程後、ONにより保持されるアルデヒド官能基と固体支持体の表面で光脱保護された官能基Bとの間での反応による、ON、それ自身の、グラフディングの工程である。
【0112】
(iii)ハイブリダイゼーション:これは該固体支持体の表面上にグラフトされたONの、フルオロフォアを備えたその相補的な配列による認識による、所望のONの起こり得るグラフティングの確認工程である。
【0113】
(iv)スキャナーによる蛍光の検出:これは、フルオロフォアにより放たれる任意のシグナルの加工(processing)工程である。
【0114】
I)手順:
1)露光工程
上記実施例5で製造した化合物(11)で官能化されたキャピラリーチューブを1/1 ピリジン/水混合液で満たした。
【0115】
続いて、キャピラリーチューブの露光を、波長365nm、スロットサイズ160μmに調整した、強度4 mW/mm2の紫外線A(UVA)放射露光デバイスを使用して5秒間、前記チューブの様々な選択された部分で実施し、前記露光デバイスは100Wランプを備えた。
【0116】
続いて、該キャピラリーチューブは水で洗浄し、次いで窒素で乾燥させた。
【0117】
2)ON固定化工程
このように第一工程に従い処理されたキャピラリーチューブを、水中、10μMの、アルデヒド官能基を有するONs溶液とインキュベートし、次いで全体を30分間作用させ続けた。
【0118】
続いて、該キャピラリーチューブを水で、次いで0.2%のラウリル硫酸ナトリウム溶液で、そして再び水で洗浄し、次いで最終的に窒素で乾燥させた。
【0119】
3)ハイブリダイゼーション工程
上記の工程2)に従いONsが固定化されたキャピラリーチューブを、CY3フルオロフォアを有する相補的なONs溶液で満たし、次いで全体を40℃の温度で1時間作用させ続けた。
【0120】
続いて、該キャピラリーチューブを0.2%クエン酸ナトリウム溶液で洗浄した。
【0121】
4)リーディング工程
フルオロフォアで標識されたONsが上記の工程3)に従いハイブリダイズしたキャピラリーチューブをガラススライド上に置き、次いでジェノミクスソリューション社(the company Genomic’s Solution)から販売されている蛍光リーダーに導入した。
【0122】
同じ測定はまた、式(I)のカップリング剤による官能化工程を除いては、テストキャピラリーチューブ(type a/)のための上記のものと同じ処理を行ったコントロールキャピラリーチューブ(type b/)について実施した。
【0123】
II/結果
得られた蛍光画像を添付の図1に表し、ここで露光された部分のみ蛍光していることに気付き得る。さらに、本発明に従う式(I)のカップリング剤で官能化されたキャピラリーチューブ(type a/)のみが、所望のONsの光−局所的固定化に対応する蛍光セグメントを含み、非官能化キャピラリーチューブ(type b/)は完全に非蛍光であることが分かる。
【0124】
この実施例は、本発明に従う固体支持体が、特定の及び局所的な様式における関心ある分子の固定化のために有利に使用され得ることを実証する。
【0125】
実施例8:式(I)のトリエトキシシラン誘導体の製造(15)
【0126】
【化21】
【0127】
1)第一工程:化合物(12)の製造:オキシアミンの導入
【0128】
【化22】
【0129】
250mlのジメチルホルムアミド(DMF)中、N−ヒドロキシフタルイミド(3.50g;21mmol)と炭酸カリウム(8g;41mmol)の溶液を攪拌しながら1時間アルゴン下、50℃の温度で加熱した。続いて、11−ブロモウンデセン(5.0g;21mmol)を添加し、次いで反応混合液を再び3時間、50℃の温度で攪拌した。ろ過後、該溶媒を減圧下で蒸発させた。続いて、100mlのジクロロメタンをこのようにして得た残渣に添加し、次いで有機相を0.1N水酸化ナトリウムで、次いで塩化ナトリウムの飽和水溶液で洗浄した。続いて該有機相を無水硫酸ナトリウム上で乾燥させ、次いで蒸発させた。生成物(12)を38〜40℃間の融点を有する白色粉末の形態で得た(6.45g;20.4mmol;収率97%)。
【0130】
CDCl3中、周波数200MHzで実施したプロトン核磁気共鳴(1H-NMR)分析は以下の通りである:
δ ppm = 1.20-1.50 (12 H, m, 6 CH2); 1.78 (2 H, m, J = 7 Hz, C2H3CH2CH2CH2); 2.02 (2 H, m, C2H3CH2CH2); 4.18 (2 H, t, J = 6 Hz, CH2ON); 4.95 (2 H, m, CH2CHCH2); 5.79
(1 H, m, CH2CHCH2); 7.70-7.85 (4 H, m, Ar-H).
MS (ESI, positive mode): Mcalc - 315 (C19H25NO3), m/z = 338 [M+Na]+.
2)第二工程:化合物(13)を製造するための化合物(12)のヒドラジン分解
【0131】
【化23】
【0132】
上記の第一工程で得た化合物(12)(3.0g;9.64mmol)を30mlジクロロメタンに溶解し、次いで966mgのヒドラジン(19,3mmol)を添加した。得られた溶液を3時間還流し、次いでろ過し、次いで減圧下で蒸発させた。粗生成物を溶離剤として酢酸エチルとシクロヘキサン(v/v:1/2)の混合液を使用して、シリカゲルクロマトグラフィーで精製した。化合物(13)を収率93%で半透明オイルの形態で得た(1.65g;8.94mmol)。
1H-NMR (200 MHz, CDCl3): δ ppm = 1.20-1.50 (12 H, m, 4 CH2), 1.56 (2 H, m, C2H3CH2CH2CH2), 2.03 (2 H, q, C2H3CH2CH2), 3.65 (2 H, t, J = 6 Hz, CH2ON), 4.96 (2 H, m, CH2CHCH2), 5.80 (1 H, m, CH2CHCH2).
MS (ESI, positive mode): Mcalc = 185 (C11H23NO), m/z = 186 [M+H]+.
3)第三工程:化合物(14)を得るための化合物(13)のオキシアミンの保護
【0133】
【化24】
【0134】
上記の第二工程で製造した化合物(13)(470mg;2.54mmol)を2mlのピリジンに溶解させた。続いて、620mg(5.08mmol)の2−(2−ニトロフェニル)プロピルクロロホルメート(Cl−NPPOC)を含む3mlのジクロロメタン溶液を滴下した。反応混合液を暗所で2時間攪拌し、次いでろ過し、次いで減圧下で蒸発させた。続いて得られた残渣をジクロロメタンに溶解し、次いで有機相を10%(w/v)の炭酸ナトリウム水溶液で、次いで塩酸水溶液(1N)で、最終的に塩化ナトリウムの飽和水溶液で洗浄した。有機相を無水硫酸ナトリウム上で乾燥させ、次いで蒸発させた。粗生成物を、溶離剤として酢酸エチルとシクロヘキサン(v/v:1/4)の混合液を使用して、シリカゲルクロマトグラフィーで精製した。化合物(14)を収率88%のオレンジ色オイルの形態で得た(873mg;2.22mmol)。
1H-NMR (200 MHz, CDCl3):δ ppm = 1.20-1.40 (12 H, m, 6 CH2), 1.35 (3 H, d, J = 7 Hz, CH3), 1.56 (2 H, m C3H3CH2CH2CH2), 2.03 (2 H, m, C2H3CH2CH2), 3.65 (2 H, m, CH2ON, 1H, m, CH), 4.28 (2H, m, CH2OCO), 4.96 (2 H, m, CH2CHCH2), 5.80 (1 H, m, CH2CHCH2), 7.20-7.80 (4 H, m, Ar-H NPPOC).
MS (ESI, positive mode): Mcalc= 392 (C21H32N2O5), m/z = 415 [M+Na]+, m/z = 410 [M+NH4]+.
4)第四工程:化合物(15)を得るための化合物(14)のヒドロシリル化
【0135】
【化25】
【0136】
上記の第三工程に記載の方法に従い得た化合物(14)1.28g(3.2mmol)を、5mlのトリエトキシシラン及び14μl(0.25当量)カールステッド触媒を含む溶液に添加した。反応混合液を60℃の温度で2時間攪拌し、次いで溶媒を蒸発させ、減圧下でDMFと共蒸発させた(5〜6回)。予測した化合物(15)を収率83%のオイルの形態で得た(1.49g; 2.68mmol)。
1H-NMR (200 MHz, CDCl3): δ ppm = 0.6 (2 H, m, CH2Si), 1.20-1.40 (16 H, m, 6 CH2, 9H 3 CH3CH2OSi), 1.35 (3 H, d, J = 7 Hz, CH3), 3.65 (2 H, m, CH2ON, 1H, m, CH), 4.28 (2 H, m, CH2OCO), 7.20-7.80 (4 H, m, Ar-H NPPOC).
MS (ESI, positive mode): Mcalc = 556 (C27H48N2O8Si), m/z = 579 [M+Na]+.
実施例9:シラン型の結合官能基Aを含む式(I)のカップリング剤で官能化された平面の固体支持体の製造
この実施例において、上記の実施例8で製造した化合物(15)を式(I)のカップリング剤として使用した。
【0137】
1)第一工程:表面の再水和(活性化)
不活化SiO2官能基を有する固体の平面のケイ素支持体を、4mlの水と3mlのエタノールを含み、さらに水酸化ナトリウム(1g NaOH)を含む溶液に1時間浸漬させた。続いて、該支持体を超純粋で、次いで0.2Nの塩酸溶液で、最終的に水で洗浄して、活性化SiOH官能基を含む表面を生成した。次いで、該支持体をアルゴンで乾燥させた。
【0138】
2)第二工程:化合物(15)を有する支持体のシラン化
このように活性化された支持体をトルエン/トリエチルアミン(v/v:97/3)中、5mMの化合物(15)(7ml)溶液に、一晩、80℃で浸した。続いて、該支持体をトルエンで、次いでエタノールで洗浄し、最終的にアルゴン下で乾燥させた。続いて、このように化合物(15)で官能化された支持体を110℃で3時間、インキュベーターに置いた。
【0139】
実施例10:関心のある生物分子の固定化のための式(I)の化合物で官能化された固体平面支持体の使用
上記の実施例7でまさに述べたように、固体支持体の表面でのONグラフティング及びそれらの存在の確認は4つの工程を含む:露光、固定化、ハイブリダイゼーション及び最終的にスキャナーによる蛍光の検出。
【0140】
I)手順:
1)第一工程:支持体の露光
上記実施例9に記載された方法に従う化合物(15)で官能化され、その上に光の透過を可能にする100μm×100μmの透明なスポットを含むプラスチックマスクが適用されている固体平面支持体を、1/1(v/v)のピリジン/水混合液の液滴で被覆した。該支持体の表面全体の、従ってマスクを通した露光を、波長365nm、強度4mW/mm2である紫外線A(UVA)放射露光デバイスを使用して15秒間実施し、前記露光デバイスは100Wランプを備えていた。
【0141】
次いで、該固体平面支持体を水で洗浄し、窒素下で乾燥させた。
【0142】
2)第二工程:ON固定化
このように第一工程に従い処理された平面支持体を、水中、10μMの、アルデヒド官能基を有するON溶液とインキュベートし、全体を30分間作用し続けた。
【0143】
次いで、該支持体を水で、次いで0.2%ラウリル硫酸ナトリウム溶液で、次いで再び水で洗浄し、最終的に窒素で乾燥させた。
【0144】
3)第三工程:ハイブリダイゼーション
ONsが上記の工程2)に従い固定化された固体支持体を、CY3フルオロフォアを有する相補的なONs溶液で満たし、次いで全体を40℃の温度で1時間作用し続けた。続いて、該支持体を0.2%クエン酸ナトリウム溶液で洗浄した。
【0145】
4)第四工程:リーディング
フルオロフォアで標識されたONsが上記の工程3)に従いハイブリダイズした支持体をガラススライド上に置き、次いでジェノミクスソリューション社(the company Genomic’s Solution)から販売されている蛍光リーダーに導入した。
【0146】
同じ測定はまた、化合物(15)による官能化工程を除いては、テストキャピラリーチューブ(type b/)について上記のものと同じ処理を行った固体平面支持体(type b/)について実施した。
【0147】
II/結果
得られた蛍光画像を添付の図2に表し、ここで露光された部分のみ蛍光があることに気付き得る。
【0148】
さらに、本発明に従うカップリング剤(15)で官能化された固体平面支持体(type a/の支持体))のみが、所望のONsの光−局所的固定化に対応する蛍光スポットを含み、非官能化支持体(type b/の支持体))は完全に非蛍光であることが分かる。
【0149】
この実施例は、本発明に従う固体支持体が、特定の及び局所的な様式における関心ある分子の固定化のために有利に使用され得ることを実証する。
【図面の簡単な説明】
【0150】
【図1】図1は、非官能化キャピラリーチューブ(タイプb/のキャピラリーチューブ)と比較した、フルオロフォアで標識された相補的なオリゴヌクレオチドによるハイブリダイゼーション後の、本発明に従う方法に従うキャピラリーチューブ型(タイプa/のキャピラリーチューブ)の固体支持体に結合したオリゴヌクレオチドにより放出された蛍光の写真を表す。
【図2】図2は、非官能化支持体(タイプb/の支持体)と比較した、フルオロフォアで標識された相補的なオリゴヌクレオチドによるハイブリダイゼーション後の、本発明に従う方法に従う平面の固体支持体(タイプa/の支持体)に結合したオリゴヌクレオチドにより放出された蛍光の写真を表す。
【技術分野】
【0001】
本発明は、感光性保護基を含むカップリング剤及び固体支持体の官能化のためのその使用に関する。本発明はまたこれらのカップリング剤で官能化された固体支持体にも関し、並びに関心のある生物分子、特に核酸分子の固定化のためのその使用にも関する。
【背景技術】
【0002】
関心のある生物分子の、特にオリゴヌクレオチド(ON)のような核酸の固体支持体への共有結合的なグラフティングのための様々な方法が既に提案されている。ONsの結合という特定の関係において、これらの方法は2つの主なカテゴリーに分けられ得る:
1)In situ合成、これはホスホルアミダイト経路を介する合成原理を利用して固体支持体にON分子を段階的に構築することからなる。特に国際出願WO 97/39151に記載されるこの方法は、高密度のONsを備えた表面の製造を可能にする。さらに、ブランキングプレート(blanking plate)と感光性保護基のセットの使用により、それは該支持体上での様々なONsの標的化を可能にする。しかしながら、この方法は、幾つかの大きな欠点を有する。特に、該支持体に結合されるONsを特徴付けることができず、また、修飾を伴うONs(ONs carrying modifications)を取り込むことが非常に困難である。
【0003】
2)デポジションによる、またはex situ合成、これは第一に従来のようにONを製造すること、そして第二に好適な化学反応を利用して固体支持体にそれを結合させることからなる。この第二の方法は特に、固体支持体に結合される前に特徴付けられ得るON分子を使用するという利点を有する。
【0004】
これ以降に記載する本発明は、より詳細には付着による合成方法のコンテクスト内にある。
【0005】
様々なタイプの付着による合成方法が先行技術において、特にONのグラフティングを配置させる様々な技術に関して既に記載されている。
【0006】
第一に、電気化学経路を使用する方法が挙げられ、これは、ピロール残基の電解重合を介して、例えばピロール基のような導電性基で予め官能化されたONを取り付けることからなる。このタイプの電気化学的ON堆積方法は、特に特許FR2 703 359に記載されている。これにもかかわらず、これは導電性支持体の使用を必要とする欠点を有し、これは最終成分を複雑化する。さらに、「Lab on a Chip」タイプのコンポーネントにおいて、様々な二次加工工程が電極の質を、ゆえにそれらの機能を損ない得る。
【0007】
第二に、光化学経路が挙げられ、これは表面及び使用される化学溶液の管理が外面化される(externalized)ため、より有望なようである。2つのアプローチが開発された:
i)表面と標的分子の間の直接的フォトグラフティング。この場合、該フォトグラフティングはあまり選択性のないフリーラジカル反応を伴うという欠点を有する;
ii)支持体上での感光性保護基で保護された官能基の遊離、これは標的ONとの引き続く反応を可能にして、それらの結合を生じる。この場合、使用される支持体は一般的には、その一端が該支持体の表面と結合するための官能基と、他端が相補的な化学官能基を有する標的分子と反応する官能基を有する二官能性化合物であるカップリング剤で官能化されており、該カップリング剤の前記反応性官能基は感光性基で保護される。しかし、競合するために、後者のアプローチは幾つかの基準を満たさなければならない。これらの基準は以下である:
−活性化された支持体と標的分子との間の反応が迅速でなければならない;この反応は、カップリング剤の反応性官能基がそれらの保護基から遊離される時にのみ起こり得る(活性化反応)。
【0008】
−この反応による収率は高くなければならない;
−カップリング剤を製造するための合成方法は、できる限り少ない工程を含み、実施されるように簡単でなければならない;
−穏やかな反応条件の使用(例えば、水);
−カップリング剤と標的分子との間で生じる反応は、このように官能化された支持体の非常にフレキシブルな使用を可能にするために、様々な温度及びpH条件下で安定でなければならない。
【0009】
現在、今日までに提案された様々な方法が、特に使用される様々なカップリング剤の特性のために、全てのこれらの基準を完全に十分に満たしていないことは明らかである。
【発明の開示】
【発明が解決しようとする課題】
【0010】
この理由のために、本発明者は、少ない工程を含む方法に従い容易に合成され、そして穏やかな条件下で実施できる、感光性基で保護された反応性官能基を有する新規の二官能性カップリング剤を提供するという目的を自分自身に与えており、これらのカップリング剤は、そこに標的生物分子を続いて結合することを目的とする固体支持体を官能化するために有利に使用できる。
【課題を解決するための手段】
【0011】
このように、本発明の第一の主題は、以下の式(I):
A−X−B−Proc (I):
(ここで:
− Aは固体支持体に結合するための官能基を表し、前記官能基は、アミン、ホスホルアミダイト、シラン及び活性エステル型の官能基から選択され;
− Xはスペーサーアームを表し;
− Bは、光脱保護の後、オキシアミン(−ONH2)型及び誘導体、或いはヒドラジド(−NH−NH2)及び誘導体の官能基を生じる基から選択される反応性官能基であり、前記官能基BはProc基で保護されている;
−Procは以下の式(II):
【0012】
【化1】
【0013】
(ここで:
− R1及びR2は同じでも異なってもよく、水素原子またはC1−C4アルキル基をで表す)
の感光性保護基である)
のカップリング剤である。
【0014】
本発明に従うと、官能基Aは、式(I)のカップリング剤が結合することを目的とする支持体の性質に従って選択される。活性エステル型の官能基は、好ましくN−ヒドロキシスクシンイミドで、または当業者に知られた任意の他の好適な活性化試薬、例えばペンタフルオロベンゼンでエステル化されたカルボン酸官能基である。
【0015】
スペーサーアームXは、所望の最終特性に応じて、長さ及び極性が変化し得る。例えば、それは無電荷疎水性鎖(飽和炭化水素ベース鎖など)またはグリセロールエーテルのような無電荷親水性鎖であり得る。
【0016】
本発明の特に好ましい実施形態に従うと、スペーサーアームXは、1〜13炭素原子を含む飽和炭化水素ベース鎖、ならびにトリエチレングリコール(TEG)及びヘキサメチレングリコール(HEG)のような炭素ベース鎖が4〜12炭素原子を含むグリコールエーテルから選択される。
【0017】
反応性官能基Bは、光脱保護の後に、少なくとも一つの相補的な化学官能基を備えた標的分子の共有結合を可能にする官能基である。これらの反応性官能基Bは、アミン官能基、アルコール、チオール、カルボキシルまたはカルボニルのような前記標的分子により保有される相補的な官能基と相互作用できる。相補的な官能基のペアのさらに網羅的なリストは、任意の有機化学モノグラフに容易に見られ得る。
【0018】
上記の式(II)の保護基について、そして、それらがアルキル基を表すとき、R1及びR2は互いに独立して、好ましくはメチル及びエチル基から選択され;該メチル基が最も特に好ましい。
【0019】
上記の式(I)のカップリング剤において、好ましくは特に以下のものが挙げられる:
i)− Aはホスホルアミダイト官能基を表し、
− Xは6炭素原子を有する飽和炭化水素ベース鎖を表し、
− Bはオキシアミン官能基(−ONH−)を表し、及び
− R1及びR2は水素原子またはメチル基を表す;
ii)− Aはトリ(C1−C4)アルコキシシランを表し、
− Xは2〜12炭素原子を有する飽和炭化水素ベース鎖を表し、
− Bはオキシアミン官能基(−ONH−)を表し、及び
− R1及びR2は水素原子またはメチル基を表す;
iii)− AはN−ヒドロキシスクシンイミドまたはペンタフルオロベンゼンでエステル化されたカルボキシリック官能基を表し、
− Xは−CH2−を表し、
− Bはオキシアミン官能基(−ONH−)を表し、及び
− R1及びR2は水素原子またはメチル基を表す。
【0020】
上記のポイントiii)で定義された化合物のうち、好ましくは、特に官能基Aがトリエトキシシランであり、そしてXが3または11炭素原子を有する飽和炭化水素ベース鎖である化合物である。上記の式(I)のカップリング剤は当業者によく知られた有機合成の原理に従い、また官能基A及びBの性質に依存して、容易に製造され得る。
【0021】
本発明の第一の実施形態に従い、そしてカップリング剤が式(I)の化合物(ここで結合官能基Aが例えばN−ヒドロキシスクシンイミドエステルの形態で活性化されたカルボン酸型であり、そして官能基Bがオキシアミン官能基である)であるとき、以下のスキーム1に対応する製造方法が好ましく使用される:
【0022】
【化2】
【0023】
このスキームにおいて、Procは上記で定義された式(II)の感光性保護基であり、そしてXは上記で示したものと同じ意味を有し得るスペーサーアームである。
【0024】
このスキームに従い、カルボキシリック官能基及びオキシアミン官能基を有する式(III)の化合物は、第一工程において、選択された式(II)のProc感光性保護基で保護され、式(IV)の化合物を生成し、次いで、第二工程において、式(IV)の化合物のカルボン酸官能基はN−ヒドロキシスクシンイミドで活性化され、式(I)の予期した化合物を生成する。
【0025】
本発明の第二の実施形態に従うと、カップリング剤が式(I)の化合物(ここで結合官能基Aはホスホルアミダイト官能基であり、そして官能基Bはオキシアミン官能基である)であるとき、以下のスキーム2に応じた製造方法が好ましく使用される:
【0026】
【化3】
【0027】
このスキームにおいて、X及びProcは上記で示したものと同じ意味を有し、Bはオキシアミン官能基を表し、そしてAは以下の基:
【0028】
【化4】
【0029】
(ここで、R及びR’は同じでも異なってもよく、1〜13炭素原子を有するアルキル基を表す)
を示す。
【0030】
このスキームに従うと、第一工程において、式(V)のハロゲン化アルコール(ここでHaloは臭素、塩素、ヨウ素またはフッ素のようなハロゲン原子を表す)はN−ヒドロキシフタルイミドと反応して(ハロゲン原子の求核置換)式(VI)の化合物を得、これは、第二工程で、ヒドラジン分解を受け、選択された式(II)の感光性Proc保護基で続いて保護されるオキシアミン官能基を遊離して式(VII)の化合物を生成し、これは、第三工程において、ホスフィチル化を受け、予期される式(I)の化合物が生成される。
【0031】
本発明の第三の実施形態に従うと、カップリング剤が式(I)の化合物(ここで結合官能基Aがシラン型であり、そして官能基Bがオキシアミン官能基である)であるとき、以下のスキーム3に応じた製造方法が好ましく使用される:
【0032】
【化5】
【0033】
この方法に従うと、式(IX)のアルケン化合物のオキシアミン官能基(ここで、Xは式(I)のカップリング剤に関して上記で示したものと同じ意味を有し得るスペーサーアームを表す)が保護されて、式(X)の化合物を得、これは続いてヒドロシリル化を受け、式(I)の化合物が生成される(ここで結合官能基Aはトリ(C1−C4)アルコキシシラン官能基である)。
【0034】
本発明に従う式(I)のカップリング剤は固体支持体の官能化のために使用され得る。ゆえに、本発明の主題は、固体支持体の官能化のための上記の定義した式(I)の少なくとも一つのカップリング剤の使用である。
【0035】
式(I)のカップリング剤の使用は、有利には光脱保護により容易に進み得る反応性官能基を備えた安定層で固体支持体の表面をすばやく修飾することを可能にする。
【0036】
このように、本発明の主題はまた、官能化された固体支持体の製造方法であり、これは、少なくとも一つの固体支持体の表面を、有機溶媒中の少なくとも一つの式(I)のカップリング剤の溶液と接触させる少なくとも一つの工程を含むことを特徴とする。
【0037】
該有機溶媒は、好ましくは、例えばトリクロロエチレン、ジメチルホルムアミド(DMF)及びシクロヘキサンのような非極性溶媒から選択される。
【0038】
式(I)のカップリング剤の溶液と固体支持体との接触は、好ましくは約4℃と80℃の間の温度で、約1〜48時間行われる。
【0039】
該支持体(substrate)は続いて1以上の溶媒、好ましくは連続して反応溶媒、無水エタノール及び/またはクロロホルムで洗浄し、そして好ましくは窒素で乾燥される。
【0040】
この方法は容易に実施できるという利点を有し、そして良好な密度の層を得ることを可能にする。特に、式(I)の化合物の自己組織化単層(SAM)で官能化された少なくとも一つの表面を有する固体支持体を得ることを可能にする。SAMsは、分子が組織化された分子の集合体であると定義され、ここで組織化は該分子の鎖の間の相互作用のためであり、安定した、単分子の、そして規則正しい異方性フィルムを生じる(A. ULMAN, Chem. Rev., 1996, 96, 1533-1554)。特に固体支持体の表面が、好ましくはトリクロロエチレンのような溶媒中で約10mMの溶液の、本発明に従う式(I)のカップリング剤(ここで結合官能基Aはシラン型)で官能化されるとき、そして接触が約4℃の温度で約24時間実施されるとき、このようなSAMsが得られ得る。
【0041】
さらに、本発明に従う方法を実施することにより得た表面は、固体支持体の表面に存在するカップリング剤の反応性官能基Bに対して相補的である化学官能基を含む関心のある生物分子を共有結合で固定化するための時間及び空間的に、選択的に容易に除去される感光性保護基で保護された多数の反応性官能基を直接提示する。
【0042】
本発明に従う式(I)のカップリング剤で官能化され得る固体支持体は、好ましくはガラス、セラミック(酸化物型)、シリコンまたはプラスチックから作製される支持体から選択され、前記支持体は少なくとも一つの水和された、ヒドロキシル化された、シラン化されたまたはアミン化表面、あるいは活性エステル型の表面を有する。このような表面は、よく知られた先行技術に従い容易に調製され得る。例えば、表面のヒドロキシル官能基を遊離するための酸加水分解を続いて受けるシランエポキシドによるシラン化によりヒドロキシル化された表面を含む固体支持体を製造することが可能である。
【0043】
これらの固体支持体は、少なくとも一つの平面または平面ではない、平滑なまたは構造化された表面を有し、例えばガラススライド、平面のプラスチックプレートまたはウェルを備えたプラスチックプレート、キャピラリーチューブあるいはポーラスまたは非ポーラスビーズの形態であり得る。
【0044】
上記のように、式(I)のカップリング剤の結合官能基Aは、前記カップリング剤が結合されることを意図する支持体の表面の性質に応じて選択される。
【0045】
このように、これらが少なくとも一つの活性エステル型の表面を有する支持体であるとき、官能基Aが好ましくはアミン官能基であり;そして、これらが少なくとも一つのヒドロキシル型の表面を有する支持体であるとき、官能基Aは好ましくはホスホルアミダイトまたはシラン官能基であり;これらが少なくとも一つのヒドリド型の表面を有する支持体であるとき、官能基Aは好ましくはシラン官能基であり、そして、これらがアミノ化表面を有する支持体であるとき、官能基Aは好ましくは活性エステル官能基である。
【0046】
ゆえに、本発明の主題はまた、上記で定義した1以上の式(I)のカップリング剤で官能化された少なくとも一つの表面を有する固体支持体である。
【0047】
このような支持体は、それらが脱保護された後の前記固体支持体の表面に存在する式(I)のカップリング剤の反応性官能基Bに対して相補的である、化学官能基を含む関心のある生物分子の固定化のために、そして特にDNA及びオリゴヌクレオチドのような核酸分子の共有結合的な固定化のために、有利に使用され得る。
【0048】
このように、本発明の主題はまた、前記固体支持体の表面に存在する式(I)のカップリング剤の反応性官能基Bに対して相補的である化学官能基を含む関心ある生物分子の、そして特にアミド結合(ペプチド)の形成を介した核酸(DNA、オリゴヌクレオチド)の、共有結合的な固定のための、上記の固体支持体の使用である。
【0049】
本発明の主題はまた、上記の固体支持体への、関心ある生物分子、特に核酸分子の固定化方法であり、これは該固体支持体の表面の少なくとも一部分の露光による式(I)の化合物の反応性官能基Bの光−脱保護、続いて、このように活性化された固体支持体を関心のある生物分子、特に核酸分子の溶液と接触させる第1工程を少なくとも含み、その結果、前記分子に備えられた、そして本発明に従う前記式(I)の化合物の反応性官能基Bに対して反応性を有する、少なくとも一つの化学官能基との間の共有結合の形成を介する前記分子の前記支持体への固定化を生じさせ、そしてこれらの2つの工程を必要に応じて繰り返すことを特徴とする。
【0050】
特に、光−脱保護工程は、該固体支持体の官能化された表面の一部のみに局在され得、例えばマスクを介して実施され得る。
【0051】
露光後、式(I)のカップリング剤の反応性官能基Bは脱保護(活性化)状態にあり、これは、該固体支持体の表面に存在する前記活性化反応性官能基Bに対して反応性を有する化学官能基を含む溶液中の任意の分子を捕獲し得るフリーな反応性官能基を有する領域を生み出す。
【0052】
特に、固定化工程は、式(I)の化合物のオキシアミン官能基Bと、関心ある生物分子、特に核酸分子のカルボニル化官能基との間のオキシム結合の形成を通して実施される。この反応は、穏やかな条件下、特に4と7の間のpHで実施できるという利点を有する。さらに、形成された結合(オキシム)は非常に良い安定性を示し、そして、更なる還元工程により、それを安定化させる必要がない。
【0053】
露光波長は好ましくは300〜400nm間にある。
【0054】
関心のある生物分子の溶液は、単純な水溶液または緩衝作用のある水溶液中のこれらの分子の溶液であり得る。
【0055】
接触を実施する期間は、好ましくは約5〜60分間であり、また好ましくは約4〜60℃間の温度で実施される。
【0056】
最後に、本発明の主題はまた、上記した、また本発明に従う固定化方法を実施することにより得られる、すなわち本発明に従う式(I)のカップリング剤の反応性官能基Bで形成された共有結合により関心のある前記生物分子、特に核酸分子が固定化された少なくとも一つの表面を有する、固体支持体(特に核酸チップ)である。
【0057】
上記アレンジメントの他に、本発明はまた、以下の記載から明らかになる他のアレンジメントも含有し、これは、式(I)のカップリング剤の製造の例、本発明に従う式(I)のカップリング剤によるキャピラリーチューブ型のガラス固体支持体または平面の固体支持体の官能化の例、オリゴヌクレオチドの固定化のためのこれらの官能化支持体の使用、ならびに添付の図1及び2
(ここで:
− 図1は、非官能化キャピラリーチューブ(タイプb/のキャピラリーチューブ)と比較した、フルオロフォアで標識された相補的なオリゴヌクレオチドによるハイブリダイゼーション後の、本発明に従う方法に従うキャピラリーチューブ型(タイプa/のキャピラリーチューブ)の固体支持体に結合したオリゴヌクレオチドにより放出された蛍光の写真を表す;
−図2は、非官能化支持体(タイプb/の支持体)と比較した、フルオロフォアで標識された相補的なオリゴヌクレオチドによるハイブリダイゼーション後の、本発明に従う方法に従う平面の固体支持体(タイプa/の支持体)に結合したオリゴヌクレオチドにより放出された蛍光の写真を表す
に関する。
【0058】
実施例1:式(I)に従うホスホルアミダイト誘導体の製造
【0059】
【化6】
【0060】
1)第一工程:化合物(1)の製造:オキシアミンの導入
【0061】
【化7】
【0062】
150mlのジメチルホルムアミド(DMF)中のN−ヒドロキシフタルイミド(2.70g;16.6mmol)と炭酸カリウム(4.6g;23.6mmol)の溶液を攪拌しながらアルゴン下、1時間50℃の温度に加熱した。続いて6−ブロモヘキサノール(3g;16.6mmol)を添加し、次いで反応混合液を再び50℃の温度で一晩攪拌した。ろ過後、該溶媒を減圧下で蒸発させた。続いて50mlの酢酸エチルを、このようにして得られた残渣に添加し、次いで有機相を0.1N 水酸化ナトリウムで、次いで塩化ナトリウムの飽和水溶液で洗浄した。続いて該有機相を無水硫酸ナトリウム上で乾燥させ、次いで蒸発させた。生成物(1)は85℃と88℃の間の融点を有する白色粉末の形態で得た(3.28g;12.5mmol;収率75%)。
【0063】
CDCl3中、周波数200MHzで実施したプロトン核磁気共鳴(1H-NMR)分析は以下の通りである:
δ ppm = 1.30-1.90 (8H, m, 4CH2), 3.60 (2H, t, J = 13 Hz, CH2O), 4.20 (2H, t, J = 13 Hz, CH2ON), 7.70-7.90 (4H, m, Ar-H).
MS (DCI, positive mode): Mcalc- 263 (C14H17NO4), m/z = 281 [M+NH4]+.
2)第二工程:化合物(2)を製造するための化合物(1)のトリチル化
【0064】
【化8】
【0065】
塩化トリチル(4.2g;15mmol)を、75mlの無水ピリジン中の上記の前記工程で得た化合物(1)(3.2g;12.5mmol)の溶液に添加した。該溶液をアルゴン下、一晩、周囲温度で攪拌した。続いて5mlのメタノールをゆっくり添加し、次いで該溶媒を減圧下で蒸発させた。続いて得られた残渣を100mlの酢酸エチルに溶解し、次いで有機相を水で、次いで塩化ナトリウムの飽和水溶液で洗浄した。続いて該有機相を無水硫酸ナトリウム下で乾燥させ、次いで蒸発させた。粗生成物をシリカゲルクロマトグラフィー(溶離剤 ジクロロメタン/シクロヘキサン:75/25、v/v)で精製した。生成物(2)は106−107℃で融解する白色粉末の形態で得た(6.27g;12.4mmol;収率99%)。
【0066】
1H-NMR分析(200 MHz, CDCl3)は以下の通りである:
δ ppm = 1.30-1.90 (8H, m, 4 CH2), 3.20 (2H, t, J = 13 Hz, CH-2O), 4.20 (2H, t, J = 13 Hz, CH2ON), 7.70-7.90 (4HAr, m, Ar-H-フタルイミド), 7.20-7.40 (15H, m, Ar-H-トリチル).
MS (ESI, positive mode): MCalc = 505 (C33H-31NO4), m/z = 528 [M+Na]+, m/z = 544 [M+K]+.
3)第三工程:化合物(3)を得るための化合物(2)のヒドラジン分解
【0067】
【化9】
【0068】
上記の第二工程において得られた化合物(2)(6.27g;12.4mmol)を、50mlのジクロロメタンに溶解し、次いで780mgのヒドラジン(24.8mmol)を添加した。得られた溶液を1時間半還流させ、次いでろ過し、減圧下で蒸発させた。粗生成物を、溶離剤がジクロロメタン、次いでジクロロメタンとメタノールが95/5(v/v)の混合液である、シリカゲルクロマトグラフィーで精製した。化合物(3)は収率90%でオイルの形態で得た(4.20g;112.2mmol)。
【0069】
1H-NMR(200 MHz, CDCl3):δ ppm = 1.30-1.80 (8H, m, 4 CH2), 3.10 (2H, t, J = 13 Hz, CH2O-Tri), 3.60 (2H, t, J = 13 Hz, CH2ON), 7.70-7.90 (15H, m, Ar-H--トリチル).
MS (ESI, positive mode): Mcalc= 375 (C25H29NO2), m/z = 376 [M+H]+, m/z = 398 [M+Na]+, m/z = 414 [M+K]+.
4)第四工程:化合物(4)を得るための化合物(3)のオキシアミンの保護
【0070】
【化10】
【0071】
上記の第三工程で製造した化合物(3)(4.20g;11.2mmol)を100mlのピリジンに溶解した。続いて5.40g(22.4mmol)の2−(2−ニトロフェニル)プロピルクロロホルメート(Cl−NPPOC)を含有する50mlのジクロロメタン溶液を滴下した。該反応混合液を暗所で1時間攪拌し、次いで減圧下で蒸発させた。続いて得られた残渣をジクロロメタンに溶解し、次いで有機相を塩化ナトリウムの飽和水溶液で洗浄した。該有機相を無水硫酸ナトリウム下で乾燥させ、次いで蒸発させた。粗生成物を溶離剤としてジクロロメタンを使用したシリカゲルクロマトグラフィーにより精製した。生成物(4)は収率89%でオレンジ色のオイル(5.80g;9.9mmol)の形態で得た。
1H-NMR (200 MHz, CDCl3): δ ppm = 1.30 (3H, d, J = 16 Hz, CH3), 1.50-1.80 (8H, m, 4 CH2), 3.0 (2H, t, J = 13 Hz, CH2O-Tri), 3.70 (2H, m, CH2ON, 1H, m CH), 4.20 (2H, t, J = 13 Hz, CH2OCO), 7.10-7.50 (15H, m, Ar-H-トリチル), 7.50-7.80 (4H, m, Ar-H- NPPOC).
MS (ESI, positive mode): Mcalc = 582 (C35H38N2O6), m/z = 605 [M+Na]+, m/z = 621 [M+K]+.
MS (ESI, negative mode): m/z = 581 [M-H]-.
5)化合物(5)を得るための化合物(4)の脱トリチル化
【0072】
【化11】
【0073】
上記の前記工程で得た5.80g(9.90mmol)の化合物(4)を、ジクロロメタン中、20%のトリフルオロ酢酸溶液50mlに溶解した。該混合液を3〜4時間周囲温度で攪拌し、次いで減圧下で蒸発させた。得られた残渣を100mlのジクロロメタンに溶解し、次いで有機相をNaClの飽和水溶液で洗浄した。該有機相を無水硫酸ナトリウム上で乾燥させ、次いで蒸発させた。粗生成物を、溶離剤としてジクロロメタン、次いで95/5(v/v)のジクロロメタン/メタノールを使用したシリカゲルクロマトグラフィーにより精製した。化合物(5)は収率90%でオレンジ色のオイル(3.0g;8.9mmol)の形態で得た。
1H-NMR (200 MHz, CDCl3): δ ppm = 1.30 (3H, d, J = 16 Hz, CH3), 1.50-1.80 (8H, m, 4 CH2), 3.70 (2H, m, CH2ON, 1H, m, CH), 4.30 (2H, t, CH2OCO, 2H, t, CH2OH), 7.50-7.80 (4H, m, Ar-H NPPOC).
MS(DVI, positive mode): Mcalc = 340 (C16H24N2O6), m/z = 454 [M+TFA]+.
6)第六工程:化合物(6)を製造するための化合物(5)のホスフィチル化
【0074】
【化12】
【0075】
上記の前記工程で得た化合物(5)(0.90g;2.7mmol)をアルゴン気流下、15mlのジクロロメタンに溶解し、次いでジイソプロピルエチルアミン(DIEA)(0.56ml,3.2mmol)を添加し、そして0.72mlの2−シアノエチルジイソプロピルクロロホスホルアミダイト(3.2mmol)を添加した。反応混合液を、周囲温度で4時間、出発生成物がなくなるまで攪拌した。40mlのジクロロメタンを次いで添加し、そして有機相を炭酸水素ナトリウム飽和水溶液で、次いでNaCl飽和水溶液で洗浄した。該有機相を無水硫酸ナトリウム下で乾燥させ、次いで蒸発させた。粗生成物を、溶離剤としてジクロロメタンを、次いで97/3(v/v)のジクロロメタン/メタノール混合液を使用してシリカゲルクロマトグラフィーにより精製した。化合物(6)は収率38%で黄色オイル(0.50g;1.0mmol)の形態で得た。
1H-NMR (200 MHz, CDCl3): δ ppm = 1.20-1.50 (15H, m, 4 CH3), 1.50-1.85 (8H, m, 4 CH2), 2.65 (2H, t, CH2CN), 3.80 (2H, m, CH2OH, 1H, m, CH, 4 H, m, 2 CH2OP), 4.30 (2H, m, CH2OCO, 2H, m, 2 CHN, 7.10-7.70 (4H Ar-H NPPOC).
31P-NMR (200 MHz, CDCl3): δ ppm = 120 (2 diastereoisomers).
実施例2:式(I)に従う活性エステル誘導体(化合物(8))の製造
【0076】
【化13】
【0077】
1)第一工程:前駆体酸(precursor acid)の製造
【0078】
【化14】
【0079】
カルボキシメトキシルアミンハイドロクロライド(1g;4.57mmol)を25mlの10%炭酸ナトリウム水溶液に溶解した。該溶液を0℃に冷却し、次いで、2.20g(9.1mmol)の2−(2−ニトロフェニル)プロピルクロロホルメート(Cl−NPPOC)を含有する20mlのジオキサン溶液を攪拌しながら滴下した。攪拌は、周囲温度で2〜3時間続けた。反応メディウムを蒸発させて、乾燥させた。250mlの水をこのようにして得た残渣に添加し、次いで水相を200mlのジエチルエーテルで洗浄した。次いで、該水相を1Nの塩酸溶液で酸性化してpH3とし、次いで250mlのジクロロメタンで3回抽出した。有機相を合わせ、そして無水硫酸ナトリウム上で乾燥させた。粗生成物を溶離剤としてジクロロメタン、次いで97/3(v/v)のジクロロメタン/メタノール混合液を使用してシリカゲルクロマトグラフィーにより精製した。化合物(7)は収率90%で88〜92℃間で融解する白色粉末の形態で得た(1.20g;4.0mmol)。
1H-NMR (200 MHz, CDCl3): δ ppm = 1.40 (3H, d, J = 16 Hz, CH3), 3.70 (1H, m, CH), 4.30 (2H, m, CH2O) 4.40 (s, 2H, COCH2O), 7.30-7.60 (4H, m, Ar-H NPPOC).
13C-NMR (300 MHz, CDCl3): δ ppm = 18 (CH2), 33 (CH2), 67 (CH3), 70 (CH3), 75 (CH3), 123 (quat), 137 (quat), 138 (quat), 133 (quat), 136 (CH), 151 (CH), 159 (CH), 172 (CH), 173 (CH).
MS (ESI, positive mode): Mcalc = 298 (C12H14N2O7), m/z = 321 [M+Na]+, m/z = 337 [M+K]+.
MS (ESI, negative mode): m/z = 297 [M-H]-, m/z = 595 [2M-H]-.
2)第二工程:N−ヒドロキシスクシンイミド活性エステル(8)の製造
【0080】
【化15】
【0081】
上記の前記工程で得た酸(7)(1.20g;4.0mmol)を15mlの無水ジクロロメタンに溶解し、次いでジシクロカルボジイミド(DCC)(0.82g;4.4mmol)を、続いてN−ヒドロキシスクシンイミド(0.457g;4.4mmol)を添加した。反応メディウムを一晩攪拌した。ろ過後、該溶媒を減圧下で蒸発させた。粗生成物を溶離剤として酢酸エチルを使用して速いシリカゲルクロマトグラフィーにより精製した。化合物(8)は収率67%で黄色粉末の形態で得た(1.0g;2.68mmol)。
実施例3:ペンタフルオロフェノール(9)により活性化された式(I)のエステル誘導体の製造
【0082】
【化16】
【0083】
上記の実施例2の第一工程で得た化合物(7)500mg(1.6mmol)を5mlのジクロロメタンに溶解し、次いで372mg(1.9mmol)のペンタフルオロフェノールを添加し、そしてジクロロメタン中、1mlのDCC(360mg,1.9mmol)溶液を滴下した。混合液を4時間攪拌し、次いで形成したジシクロウレア(dicyclourea、DCU)沈殿物をろ過により除いた。溶媒蒸発後、化合物(9)は収率100%でオイルの形態で得た(740mg,1.9mmol)。
1H-NMR (200 MHz, CDCl3): δ ppm = 1.30 (3H, d, J = 16 Hz, CH3, 3.70 (1H, m, CH), 4.30 (2 H, m, CH2O), 4.70 (s, 2H, COCH2O), 7.30-7.70, (4H, m, Ar-H NPPOC).
19F-NMR (200 MHz, CDCl3): δ ppm = -166.0 (1F, t), -164.0 (1F, d), -162.0 (1F, t), -157.0 (1F, t), -152.50 (1F, d).
実施例4:式(I)のトリエトキシシラン誘導体の製造
【0084】
【化17】
【0085】
1)第一工程:N−ニトロフェニルプロピルオキシカルボニルアリルオキシアミン(10)の製造
【0086】
【化18】
【0087】
0℃の温度に冷却された25mlの無水ピリジン中、3.250g(29.6mmol;1.1当量)の市販のアリルオキシアミン溶液に、6g(27mmol)の2−(2−ニトロフェニル)プロピルクロロホルメート(Cl−NPPOC)を含有するジクロロメタン溶液15mlを滴下した。30分攪拌後、該溶媒を蒸発させ、次いでトルエンとともに蒸発させた。反応粗生成物を酢酸エチルに溶解した。有機相を洗浄し、無水硫酸ナトリウム上で乾燥させ、次いで減圧下で蒸発させた。溶離剤として酢酸エチル/ヘキサン:1/4(v/v)の混合液を使用したシリカゲルクロマトグラフィーによる精製後、化合物(10)を収率72%(5.76g)で得た。
1H-NMR (200 MHz, CDCl3): δ ppm = 1.40 (d, 6H, Si-CH2-CH3), 3.70 (q, 1H, Ar-CH-CH3), 4.30 (m, 4H, CH2O, OCH2), 5.30 (dd, 2H, CH2), 5.90 (m, 1H, CH), 7.20 (s, 1H, NH), 7.30-7.80 (m, 4H, H-Ar).
2)第二工程:化合物(11)を製造するためのヒドロシリル化
8mlのトリエトキシシラン中、上記の前工程で得た1.67g(5.6mmol)の化合物(10)の溶液に、20μlのカールステッド触媒を滴下した。60℃で一晩攪拌後、溶媒を真空下、蒸留により蒸発させた。反応粗生成物を溶離剤として1/4(v/v)酢酸エチル/ヘキサン混合液を使用してシリカゲルクロマトグラフィーにより精製し、収率60%で予期した化合物(11)を得た(1.33g)。
1H-NMR (200 MHz, CDCl3): δ ppm = 0.65 (m, 2H, CH2Si), 1.20 (t, 3H, CH3-CH2-O), 1.40 (d, 3H, CH3-CH), 1.70 (m, 3H, CH2, CH-CH3), 3.80 (m, 4H, O-CH2-CH3, CH2O), 4.30 (m, 2H, CH2-O-CO), 7.30-7.80 (m, 5H, NH, H-Ar).
MS (エレクトロスプレー, positive mode): Mcalc = 444, m/e = 44.7 [M+H]+.
以下の固体支持体の製造例は、キャピラリーチューブの形態(長さ3cm及び直径100μmのガラスキャピラリーチューブ)において本発明に従う式(I)のカップリング剤を使用して実施した。しかしながら、これらの例は他のタイプの支持体の平面の形態に置き換えられるということを明確に理解すべきである。
【0088】
実施例5:シラン型の結合官能基Aを含む式(I)のカップリング剤で官能化された固体支持体の製造
この例において、上記の実施例4で製造した化合物(11)を式(I)のカップリング剤として使用した。
【0089】
化合物(11)により官能化された固体支持体は、以下の反応スキーム:
【0090】
【化19】
【0091】
に従う無機固体支持体(mineral solid support)(ガラスキャピラリーチューブ)の表面のシラン化に関する工程による場合、単一工程で得られる。
【0092】
これを行うために、キャピラリーチューブは1/1 水/エタノール混合液中、6M水酸化ナトリウム溶液で満たされ、そして溶液を周囲温度で2時間該キャピラリーチューブに残す。
【0093】
続いて、該キャピラリーチューブを水で徹底的に洗浄し、次いで窒素で乾燥させる。
【0094】
続いて、該キャピラリーチューブをトリクロロエチレン中、10mMの化合物(11)の溶液で満たし、そして周囲温度で一晩作用させ続けた。
【0095】
続いて、該キャピラリーチューブをトリクロロエチレン、無水エタノール、次いでクロロホルムで徹底的に洗浄し、最終的に窒素で乾燥させる。
【0096】
そして、その内表面が化合物(11)の相で官能化された、すなわち光感受性オキシアミン表面のガラスキャピラリーチューブを得た。
【0097】
実施例6:活性エステル型の結合官能基Aを含む式(I)のカップリング剤で官能化された固体支持体の製造
この実施例において、NHS−活性エステル型の結合官能基Aを含む、上記の実施例2で製造した化合物(8)を式(I)のカップリング剤として使用した。
【0098】
NHS−活性エステルカップリング剤により官能化された固体支持体は以下の反応スキーム:
【0099】
【化20】
【0100】
に従う、2つの工程で得る。
【0101】
これに従うと、第一工程では、固体支持体の無機表面がアミン官能基を含む表面に変換され、そして第二工程では、前記表面アミン官能基が化合物(8)と反応する。
【0102】
1)第一工程:アミン官能基を含む表面の製造
ガラスキャピラリーチューブを1/1 水/エタノール混合液中、6M 水酸化ナトリウム溶液で満たし、次いで該溶液を周囲温度で2時間該キャピラリーチューブに残した。
【0103】
続いて、該キャピラリーチューブを水で徹底的に洗浄し、次いで窒素で乾燥させた。
【0104】
続いて、該キャピラリーチューブを、95°エタノール中、10%のトリエトキシアミノプロピルシラン溶液で満たし、該溶液を周囲温度で一晩、該キャピラリーチューブに残した。続いて、該キャピラリーチューブをエタノールで洗浄し、そして窒素で乾燥させた。
【0105】
その内表面がアミン官能基で被覆されたキャピラリーチューブを得た。
【0106】
2)第二工程:化合物(8)による該支持体の官能化
上記の第一工程で得たキャピラリーチューブを、ジメチルホルムアミド(DMF)中、10mMの化合物(8)の溶液で満たし、そして該溶液を周囲温度で一晩、該キャピラリーチューブに残した。
【0107】
続いて、該キャピラリーチューブをDMFで、次いでエタノールで洗浄し、そして最終的に窒素で乾燥させた。
【0108】
その内表面が化合物(8)の層で官能化された、すなわち光感受性オキシアミン表面のガラスキャピラリーチューブをこのようにして得た。
【0109】
実施例7:関心ある生物分子の固定化のための官能化された支持体の使用
この実施例において、上記実施例5で得た固体支持体をオリゴヌクレオチド(ONs)の固定化のために使用した。しかしながら、これらの支持体の他の関心ある生物分子の固定化のための使用もまた妥当であると明確に理解すべきである。
【0110】
オリゴヌクレオチドの場合、該固体支持体の表面でのこれらの分子のグラフティング及びこれらの存在の確認は、4つの工程を含む:
(i)露光:これは光作用下で、該固体支持体の表面上の反応性官能基Bを部分的にフリーにすることを可能にする工程である。この官能基Bは生物分子のグラフティングを可能にする。
【0111】
(ii)固定化:これは露光工程後、ONにより保持されるアルデヒド官能基と固体支持体の表面で光脱保護された官能基Bとの間での反応による、ON、それ自身の、グラフディングの工程である。
【0112】
(iii)ハイブリダイゼーション:これは該固体支持体の表面上にグラフトされたONの、フルオロフォアを備えたその相補的な配列による認識による、所望のONの起こり得るグラフティングの確認工程である。
【0113】
(iv)スキャナーによる蛍光の検出:これは、フルオロフォアにより放たれる任意のシグナルの加工(processing)工程である。
【0114】
I)手順:
1)露光工程
上記実施例5で製造した化合物(11)で官能化されたキャピラリーチューブを1/1 ピリジン/水混合液で満たした。
【0115】
続いて、キャピラリーチューブの露光を、波長365nm、スロットサイズ160μmに調整した、強度4 mW/mm2の紫外線A(UVA)放射露光デバイスを使用して5秒間、前記チューブの様々な選択された部分で実施し、前記露光デバイスは100Wランプを備えた。
【0116】
続いて、該キャピラリーチューブは水で洗浄し、次いで窒素で乾燥させた。
【0117】
2)ON固定化工程
このように第一工程に従い処理されたキャピラリーチューブを、水中、10μMの、アルデヒド官能基を有するONs溶液とインキュベートし、次いで全体を30分間作用させ続けた。
【0118】
続いて、該キャピラリーチューブを水で、次いで0.2%のラウリル硫酸ナトリウム溶液で、そして再び水で洗浄し、次いで最終的に窒素で乾燥させた。
【0119】
3)ハイブリダイゼーション工程
上記の工程2)に従いONsが固定化されたキャピラリーチューブを、CY3フルオロフォアを有する相補的なONs溶液で満たし、次いで全体を40℃の温度で1時間作用させ続けた。
【0120】
続いて、該キャピラリーチューブを0.2%クエン酸ナトリウム溶液で洗浄した。
【0121】
4)リーディング工程
フルオロフォアで標識されたONsが上記の工程3)に従いハイブリダイズしたキャピラリーチューブをガラススライド上に置き、次いでジェノミクスソリューション社(the company Genomic’s Solution)から販売されている蛍光リーダーに導入した。
【0122】
同じ測定はまた、式(I)のカップリング剤による官能化工程を除いては、テストキャピラリーチューブ(type a/)のための上記のものと同じ処理を行ったコントロールキャピラリーチューブ(type b/)について実施した。
【0123】
II/結果
得られた蛍光画像を添付の図1に表し、ここで露光された部分のみ蛍光していることに気付き得る。さらに、本発明に従う式(I)のカップリング剤で官能化されたキャピラリーチューブ(type a/)のみが、所望のONsの光−局所的固定化に対応する蛍光セグメントを含み、非官能化キャピラリーチューブ(type b/)は完全に非蛍光であることが分かる。
【0124】
この実施例は、本発明に従う固体支持体が、特定の及び局所的な様式における関心ある分子の固定化のために有利に使用され得ることを実証する。
【0125】
実施例8:式(I)のトリエトキシシラン誘導体の製造(15)
【0126】
【化21】
【0127】
1)第一工程:化合物(12)の製造:オキシアミンの導入
【0128】
【化22】
【0129】
250mlのジメチルホルムアミド(DMF)中、N−ヒドロキシフタルイミド(3.50g;21mmol)と炭酸カリウム(8g;41mmol)の溶液を攪拌しながら1時間アルゴン下、50℃の温度で加熱した。続いて、11−ブロモウンデセン(5.0g;21mmol)を添加し、次いで反応混合液を再び3時間、50℃の温度で攪拌した。ろ過後、該溶媒を減圧下で蒸発させた。続いて、100mlのジクロロメタンをこのようにして得た残渣に添加し、次いで有機相を0.1N水酸化ナトリウムで、次いで塩化ナトリウムの飽和水溶液で洗浄した。続いて該有機相を無水硫酸ナトリウム上で乾燥させ、次いで蒸発させた。生成物(12)を38〜40℃間の融点を有する白色粉末の形態で得た(6.45g;20.4mmol;収率97%)。
【0130】
CDCl3中、周波数200MHzで実施したプロトン核磁気共鳴(1H-NMR)分析は以下の通りである:
δ ppm = 1.20-1.50 (12 H, m, 6 CH2); 1.78 (2 H, m, J = 7 Hz, C2H3CH2CH2CH2); 2.02 (2 H, m, C2H3CH2CH2); 4.18 (2 H, t, J = 6 Hz, CH2ON); 4.95 (2 H, m, CH2CHCH2); 5.79
(1 H, m, CH2CHCH2); 7.70-7.85 (4 H, m, Ar-H).
MS (ESI, positive mode): Mcalc - 315 (C19H25NO3), m/z = 338 [M+Na]+.
2)第二工程:化合物(13)を製造するための化合物(12)のヒドラジン分解
【0131】
【化23】
【0132】
上記の第一工程で得た化合物(12)(3.0g;9.64mmol)を30mlジクロロメタンに溶解し、次いで966mgのヒドラジン(19,3mmol)を添加した。得られた溶液を3時間還流し、次いでろ過し、次いで減圧下で蒸発させた。粗生成物を溶離剤として酢酸エチルとシクロヘキサン(v/v:1/2)の混合液を使用して、シリカゲルクロマトグラフィーで精製した。化合物(13)を収率93%で半透明オイルの形態で得た(1.65g;8.94mmol)。
1H-NMR (200 MHz, CDCl3): δ ppm = 1.20-1.50 (12 H, m, 4 CH2), 1.56 (2 H, m, C2H3CH2CH2CH2), 2.03 (2 H, q, C2H3CH2CH2), 3.65 (2 H, t, J = 6 Hz, CH2ON), 4.96 (2 H, m, CH2CHCH2), 5.80 (1 H, m, CH2CHCH2).
MS (ESI, positive mode): Mcalc = 185 (C11H23NO), m/z = 186 [M+H]+.
3)第三工程:化合物(14)を得るための化合物(13)のオキシアミンの保護
【0133】
【化24】
【0134】
上記の第二工程で製造した化合物(13)(470mg;2.54mmol)を2mlのピリジンに溶解させた。続いて、620mg(5.08mmol)の2−(2−ニトロフェニル)プロピルクロロホルメート(Cl−NPPOC)を含む3mlのジクロロメタン溶液を滴下した。反応混合液を暗所で2時間攪拌し、次いでろ過し、次いで減圧下で蒸発させた。続いて得られた残渣をジクロロメタンに溶解し、次いで有機相を10%(w/v)の炭酸ナトリウム水溶液で、次いで塩酸水溶液(1N)で、最終的に塩化ナトリウムの飽和水溶液で洗浄した。有機相を無水硫酸ナトリウム上で乾燥させ、次いで蒸発させた。粗生成物を、溶離剤として酢酸エチルとシクロヘキサン(v/v:1/4)の混合液を使用して、シリカゲルクロマトグラフィーで精製した。化合物(14)を収率88%のオレンジ色オイルの形態で得た(873mg;2.22mmol)。
1H-NMR (200 MHz, CDCl3):δ ppm = 1.20-1.40 (12 H, m, 6 CH2), 1.35 (3 H, d, J = 7 Hz, CH3), 1.56 (2 H, m C3H3CH2CH2CH2), 2.03 (2 H, m, C2H3CH2CH2), 3.65 (2 H, m, CH2ON, 1H, m, CH), 4.28 (2H, m, CH2OCO), 4.96 (2 H, m, CH2CHCH2), 5.80 (1 H, m, CH2CHCH2), 7.20-7.80 (4 H, m, Ar-H NPPOC).
MS (ESI, positive mode): Mcalc= 392 (C21H32N2O5), m/z = 415 [M+Na]+, m/z = 410 [M+NH4]+.
4)第四工程:化合物(15)を得るための化合物(14)のヒドロシリル化
【0135】
【化25】
【0136】
上記の第三工程に記載の方法に従い得た化合物(14)1.28g(3.2mmol)を、5mlのトリエトキシシラン及び14μl(0.25当量)カールステッド触媒を含む溶液に添加した。反応混合液を60℃の温度で2時間攪拌し、次いで溶媒を蒸発させ、減圧下でDMFと共蒸発させた(5〜6回)。予測した化合物(15)を収率83%のオイルの形態で得た(1.49g; 2.68mmol)。
1H-NMR (200 MHz, CDCl3): δ ppm = 0.6 (2 H, m, CH2Si), 1.20-1.40 (16 H, m, 6 CH2, 9H 3 CH3CH2OSi), 1.35 (3 H, d, J = 7 Hz, CH3), 3.65 (2 H, m, CH2ON, 1H, m, CH), 4.28 (2 H, m, CH2OCO), 7.20-7.80 (4 H, m, Ar-H NPPOC).
MS (ESI, positive mode): Mcalc = 556 (C27H48N2O8Si), m/z = 579 [M+Na]+.
実施例9:シラン型の結合官能基Aを含む式(I)のカップリング剤で官能化された平面の固体支持体の製造
この実施例において、上記の実施例8で製造した化合物(15)を式(I)のカップリング剤として使用した。
【0137】
1)第一工程:表面の再水和(活性化)
不活化SiO2官能基を有する固体の平面のケイ素支持体を、4mlの水と3mlのエタノールを含み、さらに水酸化ナトリウム(1g NaOH)を含む溶液に1時間浸漬させた。続いて、該支持体を超純粋で、次いで0.2Nの塩酸溶液で、最終的に水で洗浄して、活性化SiOH官能基を含む表面を生成した。次いで、該支持体をアルゴンで乾燥させた。
【0138】
2)第二工程:化合物(15)を有する支持体のシラン化
このように活性化された支持体をトルエン/トリエチルアミン(v/v:97/3)中、5mMの化合物(15)(7ml)溶液に、一晩、80℃で浸した。続いて、該支持体をトルエンで、次いでエタノールで洗浄し、最終的にアルゴン下で乾燥させた。続いて、このように化合物(15)で官能化された支持体を110℃で3時間、インキュベーターに置いた。
【0139】
実施例10:関心のある生物分子の固定化のための式(I)の化合物で官能化された固体平面支持体の使用
上記の実施例7でまさに述べたように、固体支持体の表面でのONグラフティング及びそれらの存在の確認は4つの工程を含む:露光、固定化、ハイブリダイゼーション及び最終的にスキャナーによる蛍光の検出。
【0140】
I)手順:
1)第一工程:支持体の露光
上記実施例9に記載された方法に従う化合物(15)で官能化され、その上に光の透過を可能にする100μm×100μmの透明なスポットを含むプラスチックマスクが適用されている固体平面支持体を、1/1(v/v)のピリジン/水混合液の液滴で被覆した。該支持体の表面全体の、従ってマスクを通した露光を、波長365nm、強度4mW/mm2である紫外線A(UVA)放射露光デバイスを使用して15秒間実施し、前記露光デバイスは100Wランプを備えていた。
【0141】
次いで、該固体平面支持体を水で洗浄し、窒素下で乾燥させた。
【0142】
2)第二工程:ON固定化
このように第一工程に従い処理された平面支持体を、水中、10μMの、アルデヒド官能基を有するON溶液とインキュベートし、全体を30分間作用し続けた。
【0143】
次いで、該支持体を水で、次いで0.2%ラウリル硫酸ナトリウム溶液で、次いで再び水で洗浄し、最終的に窒素で乾燥させた。
【0144】
3)第三工程:ハイブリダイゼーション
ONsが上記の工程2)に従い固定化された固体支持体を、CY3フルオロフォアを有する相補的なONs溶液で満たし、次いで全体を40℃の温度で1時間作用し続けた。続いて、該支持体を0.2%クエン酸ナトリウム溶液で洗浄した。
【0145】
4)第四工程:リーディング
フルオロフォアで標識されたONsが上記の工程3)に従いハイブリダイズした支持体をガラススライド上に置き、次いでジェノミクスソリューション社(the company Genomic’s Solution)から販売されている蛍光リーダーに導入した。
【0146】
同じ測定はまた、化合物(15)による官能化工程を除いては、テストキャピラリーチューブ(type b/)について上記のものと同じ処理を行った固体平面支持体(type b/)について実施した。
【0147】
II/結果
得られた蛍光画像を添付の図2に表し、ここで露光された部分のみ蛍光があることに気付き得る。
【0148】
さらに、本発明に従うカップリング剤(15)で官能化された固体平面支持体(type a/の支持体))のみが、所望のONsの光−局所的固定化に対応する蛍光スポットを含み、非官能化支持体(type b/の支持体))は完全に非蛍光であることが分かる。
【0149】
この実施例は、本発明に従う固体支持体が、特定の及び局所的な様式における関心ある分子の固定化のために有利に使用され得ることを実証する。
【図面の簡単な説明】
【0150】
【図1】図1は、非官能化キャピラリーチューブ(タイプb/のキャピラリーチューブ)と比較した、フルオロフォアで標識された相補的なオリゴヌクレオチドによるハイブリダイゼーション後の、本発明に従う方法に従うキャピラリーチューブ型(タイプa/のキャピラリーチューブ)の固体支持体に結合したオリゴヌクレオチドにより放出された蛍光の写真を表す。
【図2】図2は、非官能化支持体(タイプb/の支持体)と比較した、フルオロフォアで標識された相補的なオリゴヌクレオチドによるハイブリダイゼーション後の、本発明に従う方法に従う平面の固体支持体(タイプa/の支持体)に結合したオリゴヌクレオチドにより放出された蛍光の写真を表す。
【特許請求の範囲】
【請求項1】
以下の式(I):
A−X−B−Proc (I):
(ここで:
− Aは固体支持体に付着するための官能基を表し、前記官能基は、アミン、ホスホルアミダイト、シラン及び活性エステル型の官能基から選択され;
− Xはスペーサーアームを表し;
− Bは、光脱保護の後、オキシアミン(−ONH2)型及び誘導体、或いはヒドラジド(−NH−NH2)及び誘導体の官能基を生じる基から選択される反応性官能基であり、前記官能基BはProc基で保護されている;
−Procは以下の式(II):
【化1】
(ここで:
− R1及びR2は同じでも異なってもよく、水素原子またはC1−C4アルキル基を表す)
の感光性保護基である)
のカップリング剤。
【請求項2】
該スペーサーアームXが、無電荷疎水性鎖または無電荷親水性鎖であることを特徴とする、請求項1に記載のカップリング剤。
【請求項3】
該スペーサーアームXが、1〜13炭素原子を含む飽和炭化水素ベース鎖、ならびに炭素ベース鎖が4〜12炭素原子を含むグリコールエーテルから選択されることを特徴とする、請求項2に記載のカップリング剤。
【請求項4】
R1及びR2が互いに独立して、メチル及びエチル基から選択されるアルキル基を表すことを特徴とする、前記請求項のいずれかに記載のカップリング剤。
【請求項5】
i)− Aはホスホルアミダイト官能基を表し、
− Xは6炭素原子を有する飽和炭化水素ベース鎖を表し、
− Bはオキシアミン官能基(−ONH−)を表し、
及び
− R1及びR2は水素原子またはメチル基を表す;
ii)− Aはトリ(C1−C4)アルコキシシランを表し、
− Xは2〜12炭素原子を有する飽和炭化水素ベース鎖を表し、
− Bはオキシアミン官能基(−ONH−)を表し、及び
− R1及びR2は水素原子またはメチル基を表す;
iii)− AはN−ヒドロキシスクシンイミドまたはペンタフルオロベンゼンでエステル化されたカルボキシリック官能基を表し、
− Xは−CH2−を表し、
− Bはオキシアミン官能基(−ONH−)を表し、及び
− R1及びR2は水素原子またはメチル基を表す、
ものから選択されることを特徴とする、前記請求項のいずれかに記載のカップリング剤。
【請求項6】
該化合物iii)が、該官能基Aがトリエトキシシランであり、そしてXが3または11炭素原子を有する飽和炭化水素ベース鎖である化合物から選択されることを特徴とする、請求項5に記載のカップリング剤。
【請求項7】
固体支持体の官能化のための、請求項1〜6のいずれかに定義された少なくとも一つの式(I)のカップリング剤の使用。
【請求項8】
少なくとも一つの固体支持体の表面を、請求項1〜6のいずれかに記載の少なくとも一つの式(I)のカップリング剤の溶液と、有機溶媒中で、接触させる少なくとも一つの工程を含むことを特徴とする、官能化された固体支持体の製造方法。
【請求項9】
式(I)のカップリング剤の溶液と該固体支持体との接触が、4〜80℃間の温度で1〜48時間実施されることを特徴とする、請求項8に記載の方法。
【請求項10】
該固体支持体が、ガラス、セラミック、シリコンまたはプラスチックから作製される支持体から選択され、前記支持体が、少なくとも一つの水和された、ヒドロキシル化された、シラン化されたまたはアミノ化された表面、あるいは活性エステル型の表面を有することを特徴とする、請求項8または9に記載の方法。
【請求項11】
該支持体が、少なくとも一つの平面または平面ではない、平滑または構造化された表面を有し、ガラススライド、平面のプラスチックプレートまたはウェルを備えたプラスチックプレート、キャピラリーチューブあるいはポーラスまたは非ポーラスビーズの形態にあることを特徴とする、請求項8〜10のいずれかに記載の方法。
【請求項12】
請求項1〜6のいずれかに定義された1以上の式(I)のカップリング剤で官能化された少なくとも一つの表面を有することを特徴とする固体支持体。
【請求項13】
前記固体支持体の表面に存在する式(I)のカップリング剤の反応性官能基Bに対して相補的である化学官能基を含む関心のある生物分子を共有結合で固定化するための、請求項12に記載の少なくとも一つの固体支持体の使用。
【請求項14】
関心のある生物分子が核酸分子であることを特徴とする、請求項13に記載の使用。
【請求項15】
固体支持体の表面の少なくとも一部の露光による式(I)の化合物の反応性官能基Bの光−脱保護、続いて、このように活性化された固体支持体を関心のある生物分子の溶液と接触させる第1工程を少なくとも含み、その結果、前記固体支持体上で前記分子に保有され、そして前記式(I)の化合物の反応性官能基Bに対して反応性を有する、少なくとも一つの化学官能基間の共有結合の形成を介する前記分子の固定化を生じさせ、そしてこれらの2つの工程を必要に応じて繰り返すことを特徴とする、請求項14に記載の固体支持体への関心のある生物分子の固定化方法。
【請求項16】
該光−脱保護工程が、該固体支持体の官能化された表面の一部のみに局在されることを特徴とする、請求項15に記載の方法。
【請求項17】
該固定化工程が、式(I)の化合物のオキシアミン官能基Bと、関心ある生物分子のカルボニル化官能基との間のオキシム結合の形成を通して実施されることを特徴とする、請求項15または16に記載の方法。
【請求項18】
該固定化工程が4と7の間のpHで実施されることを特徴とする、請求項17に記載の方法。
【請求項19】
露光波長が300〜400nm間であることを特徴とする、請求項16〜18のいずれかに記載の方法。
【請求項20】
請求項15〜19のいずれかに記載の方法に従い得られ、及び請求項1〜6のいずれかに記載の式(I)のカップリング剤の反応性官能基Bと形成された共有結合により関心のある前記生物分子が固定された少なくとも一つの表面を有することを特徴とする固体支持体。
【請求項1】
以下の式(I):
A−X−B−Proc (I):
(ここで:
− Aは固体支持体に付着するための官能基を表し、前記官能基は、アミン、ホスホルアミダイト、シラン及び活性エステル型の官能基から選択され;
− Xはスペーサーアームを表し;
− Bは、光脱保護の後、オキシアミン(−ONH2)型及び誘導体、或いはヒドラジド(−NH−NH2)及び誘導体の官能基を生じる基から選択される反応性官能基であり、前記官能基BはProc基で保護されている;
−Procは以下の式(II):
【化1】
(ここで:
− R1及びR2は同じでも異なってもよく、水素原子またはC1−C4アルキル基を表す)
の感光性保護基である)
のカップリング剤。
【請求項2】
該スペーサーアームXが、無電荷疎水性鎖または無電荷親水性鎖であることを特徴とする、請求項1に記載のカップリング剤。
【請求項3】
該スペーサーアームXが、1〜13炭素原子を含む飽和炭化水素ベース鎖、ならびに炭素ベース鎖が4〜12炭素原子を含むグリコールエーテルから選択されることを特徴とする、請求項2に記載のカップリング剤。
【請求項4】
R1及びR2が互いに独立して、メチル及びエチル基から選択されるアルキル基を表すことを特徴とする、前記請求項のいずれかに記載のカップリング剤。
【請求項5】
i)− Aはホスホルアミダイト官能基を表し、
− Xは6炭素原子を有する飽和炭化水素ベース鎖を表し、
− Bはオキシアミン官能基(−ONH−)を表し、
及び
− R1及びR2は水素原子またはメチル基を表す;
ii)− Aはトリ(C1−C4)アルコキシシランを表し、
− Xは2〜12炭素原子を有する飽和炭化水素ベース鎖を表し、
− Bはオキシアミン官能基(−ONH−)を表し、及び
− R1及びR2は水素原子またはメチル基を表す;
iii)− AはN−ヒドロキシスクシンイミドまたはペンタフルオロベンゼンでエステル化されたカルボキシリック官能基を表し、
− Xは−CH2−を表し、
− Bはオキシアミン官能基(−ONH−)を表し、及び
− R1及びR2は水素原子またはメチル基を表す、
ものから選択されることを特徴とする、前記請求項のいずれかに記載のカップリング剤。
【請求項6】
該化合物iii)が、該官能基Aがトリエトキシシランであり、そしてXが3または11炭素原子を有する飽和炭化水素ベース鎖である化合物から選択されることを特徴とする、請求項5に記載のカップリング剤。
【請求項7】
固体支持体の官能化のための、請求項1〜6のいずれかに定義された少なくとも一つの式(I)のカップリング剤の使用。
【請求項8】
少なくとも一つの固体支持体の表面を、請求項1〜6のいずれかに記載の少なくとも一つの式(I)のカップリング剤の溶液と、有機溶媒中で、接触させる少なくとも一つの工程を含むことを特徴とする、官能化された固体支持体の製造方法。
【請求項9】
式(I)のカップリング剤の溶液と該固体支持体との接触が、4〜80℃間の温度で1〜48時間実施されることを特徴とする、請求項8に記載の方法。
【請求項10】
該固体支持体が、ガラス、セラミック、シリコンまたはプラスチックから作製される支持体から選択され、前記支持体が、少なくとも一つの水和された、ヒドロキシル化された、シラン化されたまたはアミノ化された表面、あるいは活性エステル型の表面を有することを特徴とする、請求項8または9に記載の方法。
【請求項11】
該支持体が、少なくとも一つの平面または平面ではない、平滑または構造化された表面を有し、ガラススライド、平面のプラスチックプレートまたはウェルを備えたプラスチックプレート、キャピラリーチューブあるいはポーラスまたは非ポーラスビーズの形態にあることを特徴とする、請求項8〜10のいずれかに記載の方法。
【請求項12】
請求項1〜6のいずれかに定義された1以上の式(I)のカップリング剤で官能化された少なくとも一つの表面を有することを特徴とする固体支持体。
【請求項13】
前記固体支持体の表面に存在する式(I)のカップリング剤の反応性官能基Bに対して相補的である化学官能基を含む関心のある生物分子を共有結合で固定化するための、請求項12に記載の少なくとも一つの固体支持体の使用。
【請求項14】
関心のある生物分子が核酸分子であることを特徴とする、請求項13に記載の使用。
【請求項15】
固体支持体の表面の少なくとも一部の露光による式(I)の化合物の反応性官能基Bの光−脱保護、続いて、このように活性化された固体支持体を関心のある生物分子の溶液と接触させる第1工程を少なくとも含み、その結果、前記固体支持体上で前記分子に保有され、そして前記式(I)の化合物の反応性官能基Bに対して反応性を有する、少なくとも一つの化学官能基間の共有結合の形成を介する前記分子の固定化を生じさせ、そしてこれらの2つの工程を必要に応じて繰り返すことを特徴とする、請求項14に記載の固体支持体への関心のある生物分子の固定化方法。
【請求項16】
該光−脱保護工程が、該固体支持体の官能化された表面の一部のみに局在されることを特徴とする、請求項15に記載の方法。
【請求項17】
該固定化工程が、式(I)の化合物のオキシアミン官能基Bと、関心ある生物分子のカルボニル化官能基との間のオキシム結合の形成を通して実施されることを特徴とする、請求項15または16に記載の方法。
【請求項18】
該固定化工程が4と7の間のpHで実施されることを特徴とする、請求項17に記載の方法。
【請求項19】
露光波長が300〜400nm間であることを特徴とする、請求項16〜18のいずれかに記載の方法。
【請求項20】
請求項15〜19のいずれかに記載の方法に従い得られ、及び請求項1〜6のいずれかに記載の式(I)のカップリング剤の反応性官能基Bと形成された共有結合により関心のある前記生物分子が固定された少なくとも一つの表面を有することを特徴とする固体支持体。
【図1】

【図2】
【図2】
【公表番号】特表2008−508239(P2008−508239A)
【公表日】平成20年3月21日(2008.3.21)
【国際特許分類】
【出願番号】特願2007−523103(P2007−523103)
【出願日】平成17年7月11日(2005.7.11)
【国際出願番号】PCT/FR2005/001786
【国際公開番号】WO2006/024722
【国際公開日】平成18年3月9日(2006.3.9)
【出願人】(506423291)コミサリア ア レネルジィ アトミーク (85)
【出願人】(505181011)ユニベルシテ ジョセフ フーリエ (3)
【氏名又は名称原語表記】UNIVERSITE JOSEPH FOURIER
【住所又は居所原語表記】Domaine Universitaire de Saint−Martin d’Heres,F−38041 GRENOBLE,FRANCE
【Fターム(参考)】
【公表日】平成20年3月21日(2008.3.21)
【国際特許分類】
【出願日】平成17年7月11日(2005.7.11)
【国際出願番号】PCT/FR2005/001786
【国際公開番号】WO2006/024722
【国際公開日】平成18年3月9日(2006.3.9)
【出願人】(506423291)コミサリア ア レネルジィ アトミーク (85)
【出願人】(505181011)ユニベルシテ ジョセフ フーリエ (3)
【氏名又は名称原語表記】UNIVERSITE JOSEPH FOURIER
【住所又は居所原語表記】Domaine Universitaire de Saint−Martin d’Heres,F−38041 GRENOBLE,FRANCE
【Fターム(参考)】
[ Back to top ]