
感光性樹脂組成物、およびこれを用いた物品、及びネガ型パターン形成方法
【課題】高感度で、ポリイミド前駆体の種類を問わず溶解性コントラストを得られ、結果的に十分なプロセスマージンを保ちつつ、形状が良好なパターンを得ることができる感光性樹脂組成物を提供する。
【解決手段】一般式(1)で表される光塩基発生剤、及びポリイミド前駆体を含有する、感光性樹脂組成物。

(Arは、特定のアントラセニル基、特定のアントラキノリル基、又は特定のピレニル基のいずれかであり、他の各符号は、明細書中で定義したとおりである。)
【解決手段】一般式(1)で表される光塩基発生剤、及びポリイミド前駆体を含有する、感光性樹脂組成物。
(Arは、特定のアントラセニル基、特定のアントラキノリル基、又は特定のピレニル基のいずれかであり、他の各符号は、明細書中で定義したとおりである。)
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、解像性に優れ、低コストで、ポリイミド前駆体の構造上適用可能な選択肢の範囲が広い感光性樹脂組成物に関し、特に、電磁波によるパターニング工程を経て形成される製品又は部材の材料(例えば、電子部品、光学製品、光学部品の成形材料、層形成材料又は接着剤など)として好適に利用することが出来る感光性樹脂組成物、及び、当該樹脂組成物を用いて作製した物品、並びに当該樹脂組成物を用いたネガ型パターン形成方法に関するものである。
【背景技術】
【0002】
従来から、半導体素子の表面保護膜や層間絶縁膜、電子部品の絶縁材料として、耐熱性、電気特性、機械特性に優れたポリイミド樹脂が使用されてきた(非特許文献1)。
半導体集積回路やプリント基板上の回路パターン形成は、素材表面へのレジスト剤の造膜、所定箇所への露光、エッチング等による不要箇所の除去、基板表面の洗浄作業等の煩雑で多岐に亘る工程を経て行われることから、回路パターンの製造工程を簡略化するために、露光、現像によるパターン形成後も必要な部分のレジストを絶縁材料としてそのまま残して用いることができる耐熱感光性材料が望まれている。これらの材料として、ポリイミドをベースポリマーとした耐熱感光性材料が提案されている。
【0003】
このような感光性ポリイミドとしては、例えば、特許文献1において、ポリイミド前駆体と重クロム酸塩からなる系が最初に提案された。しかしながら、この材料は、実用的な光感度を有するとともに膜形成能が高いなどの長所を有する反面、保存安定性に欠け、またポリイミド中にクロムイオンが残存することなどの欠点があり、実用には至らなかった。さらに、特許文献2には、ポリイミド前駆体であるポリアミック酸に感光性基をエステル結合で導入した化合物が、特許文献3には、ポリイミド前駆体にメタクリロイル基を持つアミン化合物をポリアミック酸に添加し、アミノ基とカルボキシル基をイオン結合させた化合物が紹介されている。しかしながら、エステル結合に代表される共有結合型感光性ポリイミドは、合成プロセスが煩雑であり、コストが嵩む点が問題点として挙げられる。また、イオン結合型感光性ポリイミドは、ポリイミド骨格と感光性基の結合力が小さく、露光部も溶解されることから残膜率が低下し、厚膜化が困難である点が問題点として挙げられる(非特許文献2)。また、これらの化合物の多くは、有機溶剤現像性のものであり、コスト面および環境負荷面を鑑みると、アルカリ水溶液により現像可能な化合物の方が望ましい。
【0004】
このようなポリイミド前駆体は、耐熱性、機械特性に優れるように芳香族系モノマーを基本骨格に用いている。一般的に、芳香族環を基本骨格に有するポリイミド前駆体は、可視領域から紫外領域にかけて透過率が下がっていく傾向があり、特にi線(波長:365nm)未満、特に350nm未満の波長領域に強い吸収を有していることから、紫外−可視光照射時において透光性が低い。そのため、感光性ポリイミドは、露光部において光化学反応が十分に進行せず、低感度であったり、パターンの形状が悪化するという問題があった。耐熱感光性材料の適用範囲が広がるにつれ、材料要求は多種多様のものになってきており、感光性ポリイミドに厚膜形成能が求められている。形成パターンが厚膜の場合においては、光透過性が低い問題はさらに深刻になる。そのため、膜物性および感度の面において共に優れた耐熱性感光性樹脂を実現するためには、i線(波長:365nm)、h線(波長:405nm)領域に、光反応活性を有する感光性システムの構築が望ましい。
【0005】
近年、新しいパターン形成材料の1つとして、光塩基発生剤が注目されている(例えば特許文献4)。しかしながら、既存の光塩基発生剤をポリイミド前駆体の系に適用するには吸収波長の点に問題があった。すなわち、既存の光塩基発生剤は350nm以下に吸収波長を持つものが多く、光反応によるイミド化促進剤としてポリイミド前駆体に添加すると、ポリイミド前駆体と光塩基発生剤の吸収波長が重なることから感度面で問題が生じる。そのため、耐熱性、機械特性および、感度の面において、共に優れた耐熱性感光性樹脂を実現するために、350nm以上、望ましくは、400nm以上の波長領域、例えばi線(波長:365nm)、h線(波長:405nm)領域に、光反応活性を有する塩基発生剤が求められている。
【0006】
【特許文献1】特公昭49−17374号公報
【特許文献2】特公昭55−30207号公報
【特許文献3】特開昭54−145794号公報
【特許文献4】特開2006−189591号公報
【非特許文献1】「最新ポリイミド〜基礎と応用」,株式会社エヌー・ティー・エス,2002年,p.327〜338
【非特許文献2】「電子部品用高分子材料の最新動向III」,株式会社住ベテクノリサーチ, 2004年,p.36〜39
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明は上記実情を鑑みて成し遂げられたものであり、その目的は、高感度で、ポリイミド前駆体の種類を問わず大きな溶解性コントラストを得られ、結果的に十分なプロセスマージンを保ちつつ、形状が良好なパターンを得ることができる感光性樹脂組成物を提供することにある。
【課題を解決するための手段】
【0008】
本発明に係る感光性樹脂組成物は、一般式(1)で表される光塩基発生剤、及びポリイミド前駆体を含有する。
【0009】
【化1】
(式(1)中、Arは、下記一般式(I)で示されるアントラセニル基、下記一般式(II)で示されるアントラキノリル基、及び下記一般式(III)で示されるピレニル基からなる群より選ばれる何れかの基を表し、
R1及びR2は夫々独立して、水素原子、炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基を表し、R1及びR2の少なくとも1つは水素原子ではなく、R1及びR2はこれらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成していても良く、
R3及びR4は夫々独立して、水素原子又は炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基を表す。)
【0010】
【化2】
(式(I)中、R5〜R13は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【0011】
【化3】
(式(II)中、R14〜R20は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【0012】
【化4】
(式(III)中、R21〜R29は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【0013】
本発明者らは、塩基発生部位としてカルバミン酸エステル結合と、上記特定のアントラセニル基、上記特定のアントラキノリル基、又は上記特定のピレニル基とを含む上記式(1)で表される化合物が、350nm以上の波長領域において光反応活性を有する光塩基発生剤として機能し、ポリイミド前駆体と組み合わせることにより高感度の感光性ポリイミドを達成し得ることを見出し、本発明に至った。
【0014】
上記光塩基発生剤は、上記特定の構造を有することにより、350nm以上の波長領域に光反応活性を有し得るため、ポリイミド前駆体と組み合わせても、感度良く、露光部のみ塩基の作用によってイミド化を促進させることが可能である。上記光塩基発生剤は導入する置換基によっては400nm以上の波長領域に吸収帯を持たせることも可能である。このため、i線(波長:365nm)未満に広い吸収帯を有している芳香族環を基本骨格に有するポリイミド前駆体と吸収波長が重なることなく、高感度の光塩基発生剤として機能するので、感光性樹脂組成物の塗膜又は成形体上の電磁波照射部位と非照射部位の間での溶解性差を大きくでき、結果的に、十分なプロセスマージンを保ちつつ、形状が良好なパターンを得ることができる。
【0015】
本発明の感光性樹脂組成物においては、前記一般式(1)で表される光塩基発生剤におけるR1及びR2が、炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基であることが、発生する塩基性物質の触媒効果が大きい点から好ましい。
【0016】
本発明の感光性樹脂組成物においては、前記一般式(1)で表される光塩基発生剤におけるR1及びR2が、これらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成しているものであることが、発生する塩基性物質の触媒効果が大きい点から好ましい。
【0017】
本発明の感光性樹脂組成物に用いられるポリイミド前駆体としては、それ自体が塩基性物質の作用によって最終生成物への反応が促進される化合物、中でも、それ自体が塩基性物質の作用によって最終生成物への反応が促進され、且つ加熱により溶解性が変化する化合物として、ポリアミック酸のようなポリイミド前駆体を用いることが好ましい。このようなポリイミド前駆体を用いると、耐熱性及び機械特性に優れた感光性ポリイミド樹脂組成物を得ることができる。
本発明によれば、従来、露光部と未露光部の間で溶解性のコントラストを取りにくかったポリイミド前駆体についても、溶解阻害剤、溶解抑制剤の適用なしで良好なパターン形状を得ることができる。
【0018】
本発明の一実施形態においては、感光性樹脂組成物に増感剤を添加することにより、照射感度を向上させることができる。
【0019】
本発明の感光性樹脂組成物においては、前記光塩基発生剤の5%重量減少温度が170℃以上であることが、当該感光性樹脂組成物の塗膜に対して露光後現像前に行うイミド化の温度において光塩基発生剤が分解し難くなる点から好ましい。
【0020】
また、上記本発明の感光性樹脂組成物は、広範な構造のポリイミド前駆体を選択できる為、それによって得られる硬化物は、耐熱性、寸法安定性、絶縁性等のポリイミドが特徴的に有する機能を付与することが可能であることから、ポリイミドが適用されている公知の全ての部材用のフィルム、塗膜又は3次元構造物として好適である。
特に、本発明に係る感光性組成物は、主にパターン形成材料(レジスト)として用いられ、それによって形成されたパターンは、永久膜として耐熱性や絶縁性を付与する成分として機能し、例えば、カラーフィルター、フレキシブルディスプレー用フィルム、半導体装置、電子部品、層間絶縁膜、配線被覆膜、光回路、光回路部品、反射防止膜、その他の光学部材、又は建築材料を形成するのに適している。
【0021】
さらに本発明は、前記本発明に係る感光性樹脂組成物又はその硬化物により少なくとも一部分が形成されている、印刷物、カラーフィルター、フレキシブルディスプレー用フィルム、半導体装置、電子部品、層間絶縁膜、配線被覆膜、光回路、光回路部品、反射防止膜、ホログラム、光学部材又は建築材料いずれかの物品を提供する。
【0022】
さらに本発明は、上記感光性樹脂組成物を用いるネガ型パターン形成方法を提供するものでもある。本発明に係るネガ型パターン形成方法は、上記感光性樹脂組成物からなる塗膜又は成形体の表面に、所定のパターン状に電磁波を照射し、必要に応じて後処理(通常は、加熱処理)を行って前記塗膜又は成形体の電磁波照射部位の溶解性を選択的に低下させた後、現像することを特徴とする。
上記ネガ型パターン形成方法においては、ポリイミド前駆体と、光塩基発生剤として上記式(1)で表されるような光塩基発生剤を組み合わせて用いることにより、感光性樹脂組成物からなる塗膜又は成形体の表面を現像液から保護するためのレジスト膜を用いずに、現像を行うネガ型パターン形成が可能である。
【発明の効果】
【0023】
以上に述べたように、本発明によれば、350nm以上の波長領域、例えばi線(波長:365nm)領域に、光反応活性を有する新規な光塩基発生剤を用いて、ポリイミド前駆体に添加剤を混合するという簡便な手法で、感光性ポリイミド樹脂組成物を調製し用いることができる。上記式(1)で表される光塩基発生剤は、電磁波照射による光分解反応により、塩基性物質であるアミンを発生させるので、塩基が触媒として作用する反応を有する種々の構造のポリイミド前駆体に適用することができる。したがって、本発明に係る感光性樹脂組成物は、パターン形成プロセスに制限を受けることなく、最終的なポリイミドの構造を広範囲から選択することができ、耐熱性、機械特性に優れる感光性ポリイミド樹脂組成物として利用できる。
本発明によれば、従来、露光部と未露光部の間で溶解性のコントラストを取りにくかったポリイミド前駆体について、溶解阻害剤、溶解抑制剤の適用なしで良好なパターン形状を得ることができる。
【発明を実施するための最良の形態】
【0024】
本発明は、光塩基発生剤を用いた感光性樹脂組成物、及び当該感光性樹脂組成物を用いた物品、及びネガ型パターン形成方法を含むものである。以下、感光性樹脂組成物から順に説明する。
なお、本発明において、上記式(1)で表されるような光塩基発生剤の結合を開裂させる光、電磁波とは、光分解反応を引き起こすことが可能なものであればよく、可視及び非可視領域の波長の電磁波だけでなく、電子線のような粒子線、及び、電磁波と粒子線を総称する放射線又は電離放射線が含まれる。
【0025】
本発明の感光性樹脂組成物は、一般式(1)で表される光塩基発生剤、及びポリイミド前駆体を含有する。
【0026】
【化5】
(式(1)中、Arは、下記一般式(I)で示されるアントラセニル基、下記一般式(II)で示されるアントラキノリル基、及び下記一般式(III)で示されるピレニル基からなる群より選ばれる何れかの基を表し、
R1及びR2は夫々独立して、水素原子、炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基を表し、R1及びR2の少なくとも1つは水素原子ではなく、R1及びR2はこれらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成していても良く、
R3及びR4は夫々独立して、水素原子又は炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基を表す。)
【0027】
【化6】
(式(I)中、R5〜R13は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【0028】
【化7】
(式(II)中、R14〜R20は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【0029】
【化8】
(式(III)中、R21〜R29は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【0030】
本発明に用いられる上記式(1)で表される光塩基発生剤は、電磁波照射による光分解反応により、塩基性物質であるアミンを発生させる。一方、ポリイミド前駆体は、例えば、塩基性物質の触媒作用によって、イミド化反応が開始される温度を下げることができる。つまり、ポリイミド前駆体を光塩基発生剤と共存させておき、電磁波の照射により塩基を発生させることで、電磁波の照射された部位は、ポリイミド前駆体の最終生成物への反応が促進され、より低温でイミド化を進行させることができる。本発明の感光性樹脂組成物を用いて、パターンを得るには、例えば、パターンを残したい場所に電磁波を照射した後、塩基性物質が存在する場所ではイミド化が進行し、塩基性物質の存在していない場所ではイミド化が進行しない温度で、加熱を行う。その結果、塩基性物質が存在する場所、すなわち、電磁波を照射した場所のみイミド化が進行し溶解性が低下する為、所定の現像液(有機溶媒や、アルカリ水溶液等)で現像することで、パターンを得ることができる。その後、目的に応じて、更に加熱を行って、ポリイミドパターンとすることができる。
【0031】
本発明に用いられる上記式(1)で表される光塩基発生剤は、特に、上記特定のアントラセニル基、上記特定のアントラキノリル基、又は上記特定のピレニル基が導入されていることにより、化合物が吸収する光の波長が長くなっている。また特定のアントラセニル基、上記特定のアントラキノリル基、又は上記特定のピレニル基に適宜置換基を導入することにより、より長波長側の光を吸収することが可能になる。更に、特定のアントラセニル基、上記特定のアントラキノリル基、又は上記特定のピレニル基が結合した炭素に、適宜置換基を導入することにより、長波長の光に対する感度を向上させることができる。このようにして、本発明に用いられる光塩基発生剤は、上記特定の構造を有することにより、350nm以上の波長領域に光反応活性を有し得るため、ポリイミド前駆体と組み合わせても、感度良く、露光部のみ塩基の作用によってイミド化を促進させることが可能である。上記光塩基発生剤は導入する置換基によっては400nm以上の波長領域に光反応活性を持たせることも可能である。本発明に用いられる光塩基発生剤は、高感度の光塩基発生剤として機能するので、感光性樹脂組成物の塗膜又は成形体上の電磁波照射部位と非照射部位の間での溶解性差を大きくでき、結果的に、十分なプロセスマージンを保ちつつ、形状が良好なパターンを得ることができる。
【0032】
まず、上記一般式(1)で表される光塩基発生剤について説明する。光塩基発生剤とは、光照射によりその化学構造が分解し、塩基性物質を発生するものをいう。
上記一般式(1)において、R1及びR2は夫々独立して、水素原子、炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基を表すか、或いは、R1及びR2は連結して、これらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成するものを表す。R1及びR2の少なくとも1つは水素原子ではないが、これは、両方とも水素原子であると、化合物の安定性が悪く、発生するアミンもアンモニアとなってしまうため塩基発生剤として有用ではないからである。
【0033】
R1及びR2の位置に導入する水素原子の数ならびに置換基の種類を変更することにより、アミンの塩基性度や熱物性、溶解度などの物性を変化させることが可能である。光塩基発生剤から発生させたいアミンが合成又は市販で入手可能であれば、発生させたいアミンに合わせて、R1及びR2は特に制限されず、適宜設計することが可能である。
より塩基性度の高いアミンの方が、例えば後述するポリイミド前駆体のイミド化における脱水縮合反応等に対する触媒作用が強く、より少量の添加で、より低い温度での脱水縮合反応等における触媒効果の発現が可能となる。つまりは、上記式(1)で表される化合物自身の電磁波に対する感度が低い場合でも、発生した塩基性物質の触媒効果が大きい為、感光性樹脂組成物としての見た目の感度は向上する。
上記のような触媒効果等の、発生した塩基性物質が与える効果が大きい点から、上記式(1)で表される化合物の電磁波の吸収に伴う解裂反応によって発生する塩基性物質は、脂肪族アミンが好ましい。そのような点から、上記式(1)で表される化合物のR1及びR2は、夫々独立して、水素原子、炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基、或いは、R1及びR2は連結して、これらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成するものであって、R1及びR2の少なくとも1つは水素原子ではないものが選択されている。
【0034】
その中でも、塩基性の観点から、発生する塩基性物質は、2級の脂肪族アミンが好ましい。しかしながら脂肪族1級アミンを用いた場合でも、芳香族アミンを用いた場合に比べて、十分な触媒効果を得ることができる。そのため、脂肪族アミンの中でも、更に、5%重量減少温度や50%重量減少温度、熱分解温度といった熱物性や、溶解性といった他の物性面や、合成の簡便性やコストといった観点から、アミンを適宜選択することが望ましい。
【0035】
2級の脂肪族アミンを発生させ、高感度を達成し、さらには、露光部未露光部の溶解性コントラストを大きくする観点から、R1及びR2が、炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基であることが好ましい。或いは、R1及びR2が、連結して、これらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成しているものであることが好ましい。
【0036】
R1及びR2で示される炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基は、直鎖状アルキル基、分岐状アルキル基、及び、環状アルキル基、又はこれらの組み合わせからなるアルキル基が挙げられる。当該アルキル基は、芳香族基等の置換基を有していても良い。例えば、炭素数1〜20の直鎖又は分岐の飽和又は不飽和アルキル基、炭素数4〜20のシクロアルキル基、炭素数7〜20のフェノキシアルキル基、炭素数7〜20のアリールアルキル基、炭素数1〜20のヒドロキシアルキル基などが挙げられる。
【0037】
炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基としては、具体的には、例えばメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、シクロブチル基、n−ペンチル基、イソペンチル基、sec−ペンチル基、tert−ペンチル基、ネオペンチル基、2−メチルブチル基、1,2−ジメチルプロピル基、1−エチルプロピル基、シクロペンチル基、n−ヘキシル基、イソヘキシル基、sec−ヘキシル基、tert−ヘキシル基、ネオヘキシル基、2−メチルペンチル基、1,2−ジメチルブチル基、2,3−ジメチルブチル基、1−エチルブチル基、シクロヘキシル基、n−ヘプチル基、イソヘプチル基、sec−ヘプチル基、tert−ヘプチル基、ネオヘプチル基、シクロヘプチル基、n−オクチル基、イソオクチル基、sec−オクチル基、tert−オクチル基、ネオオクチル基、2−エチルヘキシル基、シクロオクチル基、n−ノニル基、イソノニル基、sec−ノニル基、tert−ノニル基、ネオノニル基、シクロノニル基、n−デシル基、イソデシル基、sec−デシル基、tert−デシル基、ネオデシル基、シクロデシル基、n−ウンデシル基、n−ドデシル基、n−トリデシル基、n−テトラデシル基、n−ペンタデシル基、n−ヘキサデシル基、n−ヘプタデシル基、n−オクタデシル基、イソボルニル基、ノルボルニル基、アダマンチル基、メチルアダマンチル基、ベンジル基、ビニル基、1−プロペニル基、2−プロペニル基、イソプロペニル基、ブテニル基、ペンテニル基、ヘキセニル基等が挙げられるが、これらに限定されるものではない。中でも単位重量あたりの塩基発生量および製造の容易さの点から置換基を有してもよい炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基が好ましく、更に置換基を有してもよい炭素数1〜6の直鎖状、分枝状若しくは環状の飽和アルキル基が好ましく、具体的には、エチル基、n−プロピル基、n−ブチル基、n−ペンチル基、n−ヘキシル基が好ましい。
また置換基としては、ハロゲン原子、ヒドロキシル基、メルカプト基、シアノ基、シリル基、シラノール基、アルコキシ基、ニトロ基、アリール基、アセチル基、アセトキシ基、不飽和アルキルエーテル基、アリールエーテル基、不飽和アルキルチオエーテル基、アリールチオエーテル基等が挙げられる。なお、本発明において、「置換基を有してもよい炭素数1〜20のアルキル基」の炭素数は、アルキル基部分の炭素数であり、置換基中の炭素数は含めないものとする。
【0038】
一般式(I)において、R1及びR2が、連結して、これらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成しても良い。このような場合も、発生する塩基性物質の触媒効果が大きい点から好ましい。
この場合においては、R1とR2とで、2つの結合手が共に窒素原子と結合する炭素数3〜8のアルキレン基を形成し、含窒素脂肪族環を形成する。当該含窒素脂肪族環は、置換基を有していても良く、更にR1とR2が結合している窒素原子以外のヘテロ原子を上記炭素数3〜8のアルキレン基鎖中に有していてもよい。ヘテロ原子を鎖中に有する場合の結合としては、例えば、エーテル結合、チオエーテル結合、カルボニル結合、エステル結合、アミド結合、ウレタン結合、カーボネート結合などが挙げられる。
【0039】
R1及びR2の2つが連結して環状になって、R1及びR2が結合している窒素原子を含む含窒素脂肪族環を形成している場合の含窒素脂肪族環としては、例えば、アゼチジン(4員環)、ピロリジン(5員環)、ピペリジン(6員環)、ヘキサメチレンイミン環(7員環)、ヘプタメチレンイミン環(8員環)、オクタメチレンイミン環(9員環)等が挙げられる。更に、含窒素脂肪族環には、R1及びR2が結合している窒素原子の他にヘテロ原子を含んでいても良く、このような窒素原子以外のヘテロ原子を更に含む、含窒素脂肪族環としては、モルホリン、チオモルホリン、オキサゾリジン、チアゾリジンなどが挙げられる。含窒素脂肪族環には上述したような置換基を有していても良い。置換基としては、直鎖または分岐アルキル基が好ましい。なお、本発明において、「置換基を有してもよい炭素数3〜8の含窒素脂肪族環」の炭素数は、含窒素脂肪族環部分の炭素数であり、置換基中の炭素数は含めないものとする。
【0040】
上述のような、置換基を有していても良い炭素数3〜8の含窒素脂肪族環としては、ピロリジン環、ピペリジン環、ヘキサメチレンイミン環、ヘプタメチレンイミン環、2,5−ジメチルピロリジン環、2,6−ジメチルピペリジン環等の(窒素原子以外のヘテロ原子不含である)メチル基等のアルキル基が置換していてもよい炭素数4〜7の含窒素脂肪族環が好ましく、中でもピロリジン環、ピペリジン環、ヘキサメチレンイミン環、ヘプタメチレンイミン環等の(窒素原子以外のヘテロ原子を不含かつ無置換である)炭素数4〜7の含窒素脂肪族環がより好ましく、その中でも(窒素原子以外のヘテロ原子不含かつ無置換である)炭素数5の含窒素脂肪族環であるピペリジン環がさらに好ましい。これら好ましい具体例の炭素数4〜7の含窒素脂肪族環を有する一般式(1)で示される化合物は、発生する塩基性物質の触媒効果が大きく、且つ、安価かつ容易に製造できるという点で有用である。
【0041】
アルキル置換体の具体例としては、メチルアゼチジンなどのモノアルキルアゼチジン、ジメチルアゼチジンなどのジアルキルアゼチジン、トリメチルアゼチジンなどのトリアルキルアゼチジン、メチルピロリジンなどのモノアルキルピロリジン、ジメチルピロリジンなどのジアルキルピロリジン、トリメチルピロリジンなどのトリアルキルピロリジン、テトラメチルピロリジンなどのテトラアルキルピロリジン、メチルピペリジンなどのモノアルキルピペリジン、ジメチルピペリジンなどのジアルキルピペリジン、トリメチルピペリジンなどのトリアルキルピペリジン、テトラメチルピペリジンなどのテトラアルキルピペリジン、ペンタメチルピペリジンなどのペンタアルキルピペリジン等が挙げられる。
【0042】
一般式(1)におけるR3及びR4は夫々独立して、水素原子、炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基を表す。R3及びR4は水素原子でも良いが、R3及びR4に適宜、上記アルキル基を導入することにより、h線等の長波長に対する感度を向上させることができる。
【0043】
R3及びR4において、炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基としては、具体的には、例えばメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、シクロブチル基、n−ペンチル基、イソペンチル基、sec−ペンチル基、tert−ペンチル基、ネオペンチル基、2−メチルブチル基、1,2−ジメチルプロピル基、1−エチルプロピル基、シクロペンチル基、n−ヘキシル基、イソヘキシル基、sec−ヘキシル基、tert−ヘキシル基、ネオヘキシル基、2−メチルペンチル基、1,2−ジメチルブチル基、2,3−ジメチルブチル基、1−エチルブチル基、シクロヘキシル基、n−ヘプチル基、イソヘプチル基、sec−ヘプチル基、tert−ヘプチル基、ネオヘプチル基、シクロヘプチル基、n−オクチル基、イソオクチル基、sec−オクチル基、tert−オクチル基、ネオオクチル基、2−エチルヘキシル基、シクロオクチル基、n−ノニル基、イソノニル基、sec−ノニル基、tert−ノニル基、ネオノニル基、シクロノニル基、n−デシル基、イソデシル基、sec−デシル基、tert−デシル基、ネオデシル基、シクロデシル基、ノルボルニル基、アダマンチル基等が挙げられ、中でも炭素数1〜6の直鎖状飽和アルキル基が好ましく、更に炭素数1〜3の直鎖状飽和アルキル基が好ましく、具体的には、メチル基、エチル基、n−プロピル基が好ましく、その中でも炭素数1のアルキル基であるメチル基がより好ましい。
【0044】
一般式(1)におけるR3及びR4としては、光分解反応における感度の面から、1つが炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基であることが好ましい。
また、光分解反応における感度の面からは、R3及びR4が、ともに炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基であることがさらに好ましい。
【0045】
上記一般式(1)のArに該当する上記一般式(I)、(II)、及び(III)において、R5〜R29は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。
R5〜R29は全て水素原子であっても良いが、R5〜R29に置換基を導入させることにより、溶剤溶解性を向上したり、感度を向上したり、吸収波長をより長波長側にシフトさせることができる。この吸収波長をシフトする程度(シフト値)は、置換基の種類によって相違する。このシフト値については、「有機化学のスペクトルによる同定法 第5版(R.M.Silverstein著、281頁、1993年東京化学同人発行)」に記載の表が参考となる。
【0046】
ハロゲン原子としては、具体的には、例えば、フッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられる。
炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基としては、具体的には、例えばメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、シクロブチル基、n−ペンチル基、イソペンチル基、sec−ペンチル基、tert−ペンチル基、ネオペンチル基、2−メチルブチル基、1,2−ジメチルプロピル基、1−エチルプロピル基、シクロペンチル基、n−ヘキシル基、イソヘキシル基、sec−ヘキシル基、tert−ヘキシル基、ネオヘキシル基、2−メチルペンチル基、1,2−ジメチルブチル基、2,3−ジメチルブチル基、1−エチルブチル基、シクロヘキシル基、n−ヘプチル基、イソヘプチル基、sec−ヘプチル基、tert−ヘプチル基、ネオヘプチル基、シクロヘプチル基、n−オクチル基、イソオクチル基、sec−オクチル基、tert−オクチル基、ネオオクチル基、2−エチルヘキシル基、シクロオクチル基、n−ノニル基、イソノニル基、sec−ノニル基、tert−ノニル基、ネオノニル基、シクロノニル基、n−デシル基、イソデシル基、sec−デシル基、tert−デシル基、ネオデシル基、シクロデシル基、ノルボルニル基、アダマンチル基等が挙げられ、中でも炭素数1〜6の直鎖状飽和アルキル基が好ましく、更に炭素数1〜3の直鎖状の飽和アルキル基が好ましく、具体的には、メチル基、エチル基、n−プロピル基が好ましい。
【0047】
本発明の上記一般式(1)で示される光塩基発生剤のうち、より具体的な光塩基発生剤としては、一般式(1)におけるR1及びR2が、共に炭素数2〜10の直鎖状の飽和アルキル基であって、かつ下記一般式(2)で示される光塩基発生剤、並びに、一般式(1)におけるR1及びR2が、連結して、置換基を有していても良い炭素数4〜8の含窒素脂肪族環を形成するものであって、かつ下記一般式(3)で示される光塩基発生剤が挙げられる。これらの化合物は、他の一般式(1)で示される化合物と比較して、安価かつ容易に製造でき、従来の光塩基発生剤が感光する光と比べてより長波長の光の照射によっても、より効率的に塩基を発生できる光塩基発生剤となり得るという点において、好ましい化合物である。
【0048】
【化9】
(式(2)中、p及びqは夫々独立して、1〜9の整数を表し、R3、R4、及びArは、式(1)と同じである。)
【0049】
【化10】
(式(3)中、rは1〜5の整数を表し、R3、R4、及びArは、式(1)と同じである。)
【0050】
上記一般式(II)におけるp及びqとしては、1〜7がより好ましい。
上記一般式(III)におけるrとしては、1〜3がより好ましい。
【0051】
また、上記一般式(2)で示される光塩基発生剤の好ましい具体例としては、一般式(2)におけるR3及びR4が共に水素原子であり、Arは、上記一般式(I)においてR5〜R13がすべて水素原子であるアントラセニル基であって、かつp及びqが共に1、すなわちR1及びR2が、共に炭素数2のアルキル基であるエチル基であるものが挙げられ、より具体的には、式(4)で示される光塩基発生剤が、好ましいものとして挙げられる。
【0052】
【化11】
【0053】
さらにまた、上記一般式(3)で示される光塩基発生剤の好ましい具体例としては、一般式(3)におけるR3及びR4が共に水素原子であり、Arは、上記一般式(I)においてR5〜R13がすべて水素原子であるアントラセニル基、上記一般式(II)においてR14〜R20がすべて水素原子であるアントラキノリル基、及び上記一般式(II)においてR21〜R29がすべて水素原子であるピレニル基からなるより選ばれるいずれかの基であって、かつrが2、すなわちR1及びR2が結合している窒素原子と共に(窒素原子以外のヘテロ原子不含かつ無置換である)炭素数5の含窒素脂肪族環であるピペリジン環を形成するものである化合物が挙げられ、より具体的には、下記式(5)で示される光塩基発生剤、下記式(6)で示される光塩基発生剤、及び下記式(7)で示される光塩基発生剤が、好ましいものとして挙げられる。
【0054】
【化12】
【0055】
【化13】
【0056】
【化14】
【0057】
そのほか、一般式(1)で表される光塩基発生剤としては、たとえば、以下の化学式で表されるものが例示できるが、これらに限定されるものではない。
【0058】
【化15】
(式中、−Meとは、メチル基を表す。)
【0059】
【化16】
(式中、−Meとは、メチル基を表す。)
【0060】
【化17】
(式中、−Meとは、メチル基を表す。)
【0061】
本発明の一般式(1)で表される光塩基発生剤の製造方法としては、例えば、下記一般式(8)で示されるアルコールとハロゲン化ギ酸エステルとを、要すれば有機溶媒の存在下で反応させた後、下記一般式(9)で示されるアミンを反応させればよい。
【0062】
【化18】
(式中、Ar、R3及びR4、R1及びR2は、式(1)と同じである。)
【0063】
より具体的な製造方法としては、例えば上記一般式(8)で示されるアルコールと、当該アルコールに対して、通常0.8〜10当量、好ましくは0.8〜3当量の例えばクロロギ酸−4−ニトロフェニル、ブロモギ酸−2−ニトロフェニル等のハロゲン化ギ酸エステルとを、上記アルコールに対して、通常0.8〜20当量、好ましくは0.8〜7当量の例えばトリエチルアミン等の3級アミンの存在下、要すれば脱水ジメチルアセトアミド(脱水DMAc)等の脱水有機溶媒中で、通常−20℃〜100℃、好ましくは0℃〜60℃で反応させることにより、相当する炭酸エステル(カルボナート)を得る(第一工程)。
【0064】
上記第一工程で使用される一般式(8)で示されるアルコールとしては、例えば9−アントラセンメタノール、2−ヒドロキシメチルアントラキノン、1−ピレンメタノールが挙げられる。これらのアルコールは、市販のものを用いるか、常法により合成したものを、設計に合わせて適宜用いればよい。
【0065】
次いで、得られた炭酸エステル(カルボナート)と当該炭酸エステル(カルボナート)に対して、通常0.8〜10当量、好ましくは0.8〜3当量の上記一般式(9)で示されるアミンとを、要すればジクロロメタン等の有機溶媒中で、通常−20℃〜100℃、好ましくは0℃〜60℃で、反応させることにより(第二工程)、本発明の一般式(1)で示される化合物を得ることができる。
上記第二工程で使用される一般式(9)で示されるアミンは、市販のものを用いるか、常法により合成したものを適宜用いればよく、特に限定されない。上述したように、光を照射して発生させたいアミンを設計に合わせて用いるようにする。
具体的には、例えば、ジエチルアミン、ジ−n−プロピルアミン、ジイソプロピルアミン、ジ−n−ブチルアミン、ジイソブチルアミン、ジ−sec−ブチルアミン、ジ−tert−ブチルアミン、ジシクロブチルアミン、ジ−n−ペンチルアミン、ジイソペンチルアミン、ジ−sec−ペンチルアミン、ジ−tert−ペンチルアミン、ジネオペンチルアミン、ジ−2−メチルブチルアミン、ジ−1,2−ジメチルプロピルアミン、ジ−1−エチルプロピルアミン、ジシクロペンチルアミン、ジ−n−ヘキシルアミン、ジイソヘキシルアミン、ジ−sec−ヘキシルアミン、ジ−tert−ヘキシルアミン、ジネオヘキシルアミン、ジ−2−メチルペンチルアミン、ジ−1,2−ジメチルブチルアミン、ジ−2,3−ジメチルブチルアミン、ジ−1−エチルブチルアミン、ジシクロヘキシルアミン、ジ−n−ヘプチルアミン、ジイソヘプチルアミン、ジ−sec−ヘプチルアミン、ジ−tert−ヘプチルアミン、ジネオヘプチルアミン、ジシクロヘプチルアミン、ジ−n−オクチルアミン、ジイソオクチルアミン、ジ−sec−オクチルアミン、ジ−tert−オクチルアミン、ジネオオクチルアミン、ジ−2−エチルヘキシルアミン、ジシクロオクチルアミン、ジ−n−ノニルアミン、ジイソノニルアミン、ジ−sec−ノニルアミン、ジ−tert−ノニルアミン、ジネオノニルアミン、ジシクロノニルアミン、ジ−n−デシルアミン、ジイソデシルアミン、ジ−sec−デシルアミン、ジ−tert−デシルアミン、ジネオデシルアミン、ジシクロデシルアミン、ジノルボルニルアミン、ジアダマンチルアミン、エチルメチルアミン、メチル−n−プロピルアミン、メチルイソプロピルアミン、エチル−n−プロピルアミン、エチルイソプロピルアミン、n−プロピルイソプロピルアミン等の炭素数1〜10の直鎖状、分枝状若しくは環状のジアルキルアミン、アゼチジン、ピロリジン、2,5−ジメチルピロリジン、ピペリジン、2,6−ジメチルピペリジン、ヘキサメチレンイミン、ヘプタメチレンイミン、オクタメチレンイミン、オキサゾリジン、チアゾリジン、モルホリン、チオモルホリン等が挙げられるが、これらに限定されるものではない。
このようにして、光塩基発生剤から発生させたいアミンが合成又は市販で入手可能であれば、発生させたいアミンに合わせて、R1及びR2は特に制限されず、適宜設計し合成することが可能である。
【0066】
なお、本発明に用いられる一般式(1)で表される光塩基発生剤を製造する方法は上記に限られない。別法としては、例えば上記一般式(8)で示されるアルコールと、当該アルコールに対して、通常0.8〜10当量、好ましくは0.8〜3当量の下記一般式(10)で示される化合物とを、要すれば上記アルコールに対して、通常0.8〜5当量、好ましくは0.8〜3当量の例えば水素化ナトリウム等の塩基の存在下、要すれば脱水テトラヒドロフラン(脱水THF)等の有機溶媒中で、通常0℃〜120℃、好ましくは20℃〜100℃で反応させることにより、本発明に用いられる一般式(1)で表される光塩基発生剤を得ることができる。当該法では、目的とする化合物を1工程で合成することができる。
【0067】
【化19】
(式中、Xはハロゲン原子を表し、R1及びR2は式(1)と同じである。)
【0068】
一般式(10)におけるXで示されるハロゲン原子としては、具体的には、例えば塩素原子、臭素原子、ヨウ素原子等が挙げられ、中でも塩素原子、臭素原子が好ましく、その中でも塩素原子がより好ましい。
上記反応で使用される一般式(10)で示される化合物は、市販のものを用いるか、常法により合成したものを適宜用いればよい。
【0069】
以上のようにして得られる、式(1)で表される光塩基発生剤、および上記式(1)で表される光塩基発生剤の光分解反応により生じる塩基性物質は、本発明の光塩基発生剤を含有する感光性樹脂組成物の塗膜に対して露光後現像前に行う、加熱の温度(パターン形成用の部分的なイミド化の温度)において分解しないことが好ましい。具体的には、上記式(1)で表される光塩基発生剤や、光分解反応により生じる塩基性物質を加熱して初期の重量から5%重量が減少したときの温度(5%重量減少温度)は170℃、更に好ましくは200℃以上であることが望ましい。
【0070】
更には、本発明の光塩基発生剤を含有する感光性樹脂組成物が、製品として用いられる場合、感光性樹脂組成物中に塩基性物質が残存しないことが好ましいので、現像後に行う加熱のプロセス(完全イミド化のプロセス)で分解、または揮発してしまう塩基性物質であることが好ましい。具体的には、光分解反応により生じる塩基性物質を加熱して初期の重量から50%重量が減少したときの温度(50%重量減少温度)が400℃以下であることが好ましい。
【0071】
一般的な露光光源である高圧水銀灯の代表的な発光波長は、436nm、405nm、365nmであるため、本発明の前記式(1)で表される光塩基発生剤は、350nm以上の波長の電磁波を吸収することが望ましく、400nm以上の波長の電磁波の吸収を有することがさらに好ましい。芳香族環を基本骨格に有するポリイミド前駆体は、350nm未満に広い吸収帯を有している場合が多い為、光塩基発生剤が365nm以上、更に400nm以上の波長の電磁波の吸収を有する場合には、ポリイミド前駆体と光塩基発生剤の吸収波長が重なることなく、感度を向上することが可能になるからである。一般的な露光光源である高圧水銀灯の代表的な発光波長を用いる点からは、中でも前記光塩基発生剤は405nmに吸収を有することが好ましい。
【0072】
また、本発明の前記式(1)で表される光塩基発生剤は、350nm以上、更に350nm〜500nmの波長の電磁波に対して光分解性を有することが好ましい。より好ましくは、400nm以上、更に400nm〜500nmの波長の電磁波に対して光分解性を有することが好ましい。
中でも前記光塩基発生剤は、436nm、及び405nmの波長の電磁波のうち少なくとも1つの波長に吸収を有するだけでなく、405nmの波長の電磁波に対して光分解性を有することが好ましい。436nm、及び405nmの波長の電磁波のうち少なくとも1つの波長に吸収を有していてもこのような波長の電磁波に対して光分解性を有しない場合もある。
405nmの波長の電磁波に対して光分解性を有するかどうかは、例えば、i線(波長:365nm)を全く通さないフィルターを介して高圧水銀灯を用いて光塩基発生剤に照射して、光塩基発生剤が分解するか否か、或いは塩基性物質を発生させるか否かを観測することによって判断できる。
【0073】
次に、ポリイミド前駆体について説明する。
本発明に用いるポリイミド前駆体は、なんらかの溶媒(有機溶剤、又は水溶液)に可溶なものであることが好ましい。溶媒(有機溶剤、又は水溶液)に可溶なものであると、ポリイミド前駆体の当該溶媒に対する溶解性を変化させることにより、その可溶な溶媒を現像液として用いて、適宜、有機溶剤、塩基性水溶液、酸性水溶液、又は中性水溶液による現像をすることが可能になる。
ここで、ある溶媒に可溶とは、具体的には、基板上に形成された塗膜の25℃における当該溶媒に対する溶解速度が、100Å/sec以上を目安とする。当該溶解速度は1000Å/sec以上であることがさらに好ましい。
例えば、塩基性水溶液に可溶なものは、具体的には、基板上に形成された塗膜の25℃における0.1wt%のテトラメチルアンモニウムヒドロキシド(TMAH)水溶液に対する溶解速度が、100Å/sec以上である。当該溶解速度は1000Å/sec以上であることがさらに好ましい。さらには、より一般的に用いられる現像液である2.38重量%のテトラメチルアンモニウムヒドロキシド水溶液に対する溶解速度が、100Å/sec以上であることが好ましく、1000Å/sec以上であることがさらに好ましい。上記定義による溶解速度が100Å/secより小さい場合、現像時間が遅くなり作業性、生産性が悪くなると共に、露光部、未露光部間の溶解性コントラストが得にくくなる。
したがって、本発明の感光性樹脂組成物のある溶媒に対しての溶解速度は、25℃における当該溶媒に対する溶解速度が、100Å/sec以上であることが好ましく、1000Å/sec以上であることがさらに好ましい。
【0074】
上記溶解速度を測定する具体的手順としては、無アルカリガラス等の基板上に形成されたポリイミド前駆体の塗膜を、25℃に調温され、撹拌された現像液(0.1重量%TMAH水溶液または、2.38重量%TMAH水溶液等の塩基性水溶液、有機溶剤等)に一定時間、浸漬し、蒸留水でリンス後、乾燥させた後で測定した膜厚と、初期膜厚との差を、膜減り量とし、その膜減り量を、現像液に浸漬した時間で割ったものが、25℃における単位時間当たりの溶解速度ということになる。
【0075】
また、露光部と未露光部の間に十分な溶解性コントラストを得るために、感光性樹脂組成物を実際に所定の感光パターン形成プロセスにおいて用いた時に、パターン状露光、及び、必要に応じて後工程(通常は加熱工程)を行って得られる、現像工程前における未露光部位と露光部位の現像液に対する溶解性の比(未露光部位の現像液に対する単位時間当たりの溶解速度/露光部位の現像液に対する単位時間当たりの溶解速度)が、10以上であることが好ましい。
単位時間当たりの溶解速度は、上記の方法と同様にして求められ、感光性樹脂組成物の塗膜にパターン露光を行い、露光後の加熱を行った後に、露光部、未露光部の溶解速度を、それぞれ求める。
【0076】
本発明においては、塩基性物質の作用によって最終生成物への反応が促進されるポリイミド前駆体が用いられる。ここで、ポリイミド前駆体が、塩基性物質の作用によって最終生成物への反応が促進される態様には、ポリイミド前駆体が塩基性物質の作用のみによって最終生成物に変化する態様のみならず、塩基性物質の作用によってポリイミド前駆体の最終生成物への反応温度が、塩基性物質の作用がない場合に比べて低下するような態様が含まれる。
このような塩基性物質の存在の有無により反応温度差が出来る場合には、反応温度差を利用して、塩基性物質と共存するポリイミド前駆体のみが最終生成物へと反応する適切な温度で加熱することにより、塩基性物質と共存するポリイミド前駆体のみが最終生成物へと反応しある溶媒への溶解性が変化する。従って、塩基性物質の存在の有無によって、ポリイミド前駆体のある溶媒への溶解性を変化させることが可能となり、ひいては当該溶媒を現像液として用いて現像によるパターニングが可能になる。よって、本発明に用いられるポリイミド前駆体としては、塩基性物質の作用によって最終生成物への反応が促進され、且つ、加熱により溶解性が、加熱前に比べて低く変化するポリイミド前駆体が好適に用いられる。
【0077】
ここで、ポリイミド前駆体としては、下記式(11)で表されるようなポリアミック酸が好適に用いられる。
【0078】
【化20】
(式(11)中、R31は4価の有機基である。R32は2価の有機基である。)
【0079】
なお、R31の4価は酸と結合するための価数のみを示しているが、他に更なる置換基を有していても良い。同様に、R32の2価はアミンと結合するための価数のみを示しているが、他に更なる置換基を有していても良い。
ポリアミック酸は、酸2無水物とジアミンを溶液中で混合するのみで得られるので、1段階の反応で合成することができ、合成が容易で低コストで入手できるので好ましい。
【0080】
ポリアミック酸のように、塩基の触媒作用によって熱硬化温度が低下するポリイミド前駆体を用いる場合には、先ず、そのようなポリアミック酸と、前記光塩基発生剤を組み合わせた感光性樹脂組成物の塗膜又は成形体上のパターンを残したい部分に電磁波を照射する。すると、照射部には、塩基性物質が発生し、その部分のイミド化温度が選択的に低下する。次に、照射部はイミド化反応が起こるが、非照射部はイミド化反応が起こらない処理温度で加熱し、照射部のみ少なくとも現像液に溶解しない程度に部分的にイミド化させる。次に、所定の現像液(有機溶媒や塩基性水溶液等)で非照射部を溶解して熱硬化物からなるパターンを形成する。このパターンを、更に必要に応じ更に加熱してイミド化を完結させる。以上の工程によって、所望の2次元樹脂パターン(一般的な平面パターン)又は3次元樹脂パターン(立体的に成形された形状)が得られる。
【0081】
本発明においては、上記式(1)で表されるような化合物が、高感度の光塩基発生剤として機能し、感光性樹脂組成物の塗膜又は成形体上の電磁波照射部位と非照射部位の間での溶解性差を大きくできるので、有機溶媒ではなく、塩基性水溶液を用いる場合でも優れた現像性が得られる。
副次的な効果として、用いるポリイミド前駆体がポリアミック酸である場合、塩基性物質の触媒効果によりイミド化に要する温度が低くても充分な為、最終キュア温度を300℃未満、更に好ましくは250℃以下まで下げることが可能である。従来のポリアミック酸はイミド化するために最終キュア温度を300℃以上とする必要があった為、用途が制限されていたが、最終キュア温度を下げることが可能になったことによって、より広範囲の用途に適用が可能である。
【0082】
また、ポリイミド前駆体に関して、最終的に得られるポリイミドの耐熱性及び寸法安定性の要求が厳しい用途に対しては、酸二無水物由来の部分が芳香族構造を有し、さらにジアミン由来の部分も芳香族構造を含む全芳香族ポリイミド前駆体であることが好ましい。それゆえジアミン成分由来の構造も芳香族ジアミンから誘導される構造であることが好ましい。
ここで、全芳香族ポリイミド前駆体とは、芳香族酸成分と芳香族アミン成分の共重合、又は、芳香族酸/アミノ成分の重合により得られるポリイミド前駆体及びその誘導体である。また、芳香族酸成分とは、ポリイミド骨格を形成する4つの酸基が全て芳香族環上に置換している化合物であり、芳香族アミン成分とは、ポリイミド骨格を形成する2つのアミノ基が両方とも芳香族環上に置換している化合物であり、芳香族酸/アミノ成分とはポリイミド骨格を形成する酸基とアミノ基がいずれも芳香族環上に置換している化合物である。ただし、後述する原料の具体例から明らかなように、全ての酸基又はアミノ基が同じ芳香環上に存在する必要はない。
【0083】
本発明のポリイミド前駆体を製造する方法としては、従来公知の手法を適用することができる。例えば、(1)酸二無水物とジアミンから前駆体であるポリアミック酸を合成する手法。(2)酸二無水物に1価のアルコールやアミノ化合物、エポキシ化合物等を反応させ合成した、エステル酸やアミド酸モノマーのカルボン酸に、ジアミノ化合物やその誘導体を反応させてポリイミド前駆体を合成する手法などが挙げられるがこれに限定されない。
【0084】
本発明のポリイミド前駆体に適用可能な酸二無水物としては、例えば、エチレンテトラカルボン酸二無水物、ブタンテトラカルボン酸二無水物、シクロブタンテトラカルボン酸二無水物、メチルシクロブタンテトラカルボン酸二無水物、シクロペンタンテトラカルボン酸二無水物などの脂肪族テトラカルボン酸二無水物;ピロメリット酸二無水物、3,3’,4,4’−ベンゾフェノンテトラカルボン酸二無水物、2,2’,3,3’−ベンゾフェノンテトラカルボン酸二無水物、2,3’,3,4’−ベンゾフェノンテトラカルボン酸二無水物、3,3’,4,4’−ビフェニルテトラカルボン酸二無水物、2,2’,3,3’−ビフェニルテトラカルボン酸二無水物、2,3’,3,4’−ビフェニルテトラカルボン酸二無水物、2,2’,6,6’−ビフェニルテトラカルボン酸二無水物、2,2−ビス(3,4−ジカルボキシフェニル)プロパン二無水物、2,2−ビス(2,3−ジカルボキシフェニル)プロパン二無水物、ビス(3,4−ジカルボキシフェニル)エーテル二無水物、ビス(3,4−ジカルボキシフェニル)スルホン二無水物、1,1−ビス(2,3−ジカルボキシフェニル)エタン二無水物、ビス(2,3−ジカルボキシフェニル)メタン二無水物、ビス(3,4−ジカルボキシフェニル)メタン二無水物、2,2−ビス(3,4−ジカルボキシフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物、2,2−ビス(2,3−ジカルボキシフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物、1,3−ビス〔(3,4−ジカルボキシ)ベンゾイル〕ベンゼン二無水物、1,4−ビス〔(3,4−ジカルボキシ)ベンゾイル〕ベンゼン二無水物、2,2−ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}プロパン二無水物、
【0085】
2,2−ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}プロパン二無水物、ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}ケトン二無水物、ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}ケトン二無水物、4,4’−ビス〔4−(1,2−ジカルボキシ)フェノキシ〕ビフェニル二無水物、4,4’−ビス〔3−(1,2−ジカルボキシ)フェノキシ〕ビフェニル二無水物、ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}ケトン二無水物、ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}ケトン二無水物、ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}スルホン二無水物、ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}スルホン二無水物、ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}スルフィド二無水物、ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}スルフィド二無水物、2,2−ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物、2,2−ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物、2,3,6,7−ナフタレンテトラカルボン酸二無水物、1,1,1,3,3,3−ヘキサフルオロ−2,2−ビス(2,3−又は3,4−ジカルボキシフェニル)プロパン二無水物、1,4,5,8−ナフタレンテトラカルボン酸二無水物、1,2,5,6−ナフタレンテトラカルボン酸二無水物、1,2,3,4−ベンゼンテトラカルボン酸二無水物、3,4,9,10−ぺリレンテトラカルボン酸二無水物、2,3,6,7−アントラセンテトラカルボン酸二無水物、1,2,7,8−フェナントレンテトラカルボン酸二無水物、ピリジンテトラカルボン酸二無水物、スルホニルジフタル酸無水物、m−ターフェニル−3,3’,4,4’−テトラカルボン酸二無水物、p−ターフェニル−3,3’,4,4’−テトラカルボン酸二無水物などの芳香族テトラカルボン酸二無水物等が挙げられる。これらは単独あるいは2種以上混合して用いられる。そして、特に好ましく用いられるテトラカルボン酸二無水物としてピロメリット酸二無水物、3,3’,4,4’−ベンゾフェノンテトラカルボン酸二無水物、3,3’,4,4’−ビフェニルテトラカルボン酸二無水物、2,2’,6,6’−ビフェニルテトラカルボン酸二無水物、ビス(3,4−ジカルボキシフェニル)エーテル二無水物、2,2−ビス(3,4−ジカルボキシフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物が挙げられる。
【0086】
併用する酸二無水物としてフッ素が導入された酸二無水物や、脂環骨格を有する酸二無水物を用いると、透明性をそれほど損なわずに溶解性や熱膨張率等の物性を調整することが可能である。また、ピロメリット酸無水物、3,3’,4,4’−ビフェニルテトラカルボン酸二無水物、1,4,5,8−ナフタレンテトラカルボン酸二無水物などの剛直な酸二無水物を用いると、最終的に得られるポリイミドの線熱膨張係数が小さくなるが、透明性の向上を阻害する傾向があるので、共重合割合に注意しながら併用してもよい。
【0087】
一方、アミン成分も、1種類のジアミン単独で、または2種類以上のジアミンを併用して用いることができる。用いられるジアミン成分は限定されるわけではないが、p−フェニレンジアミン、m−フェニレンジアミン、o−フェニレンジアミン、3,3’−ジアミノジフェニルエーテル、3,4’−ジアミノジフェニルエーテル、4,4’−ジアミノジフェニルエーテル、3,3’−ジアミノジフェニルスルフィド、3,4’−ジアミノジフェニルスルフィド、4,4’−ジアミノジフェニルスルフィド、3,3’−ジアミノジフェニルスルホン、3,4’−ジアミノジフェニルスルホン、4,4’−ジアミノジフェニルスルホン、3,3’−ジアミノベンゾフェノン、4,4’−ジアミノベンゾフェノン、3,4’−ジアミノベンゾフェノン、3,3’−ジアミノジフェニルメタン、4,4’−ジアミノジフェニルメタン、3,4’−ジアミノジフェニルメタン、2,2−ジ(3−アミノフェニル)プロパン、2,2−ジ(4−アミノフェニル)プロパン、2−(3−アミノフェニル)−2−(4−アミノフェニル)プロパン、2,2−ジ(3−アミノフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン、2,2−ジ(4−アミノフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン、2−(3−アミノフェニル)−2−(4−アミノフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン、1,1−ジ(3−アミノフェニル)−1−フェニルエタン、1,1−ジ(4−アミノフェニル)−1−フェニルエタン、1−(3−アミノフェニル)−1−(4−アミノフェニル)−1−フェニルエタン、1,3−ビス(3−アミノフェノキシ)ベンゼン、1,3−ビス(4−アミノフェノキシ)ベンゼン、1,4−ビス(3−アミノフェノキシ)ベンゼン、1,4−ビス(4−アミノフェノキシ)ベンゼン、1,3−ビス(3−アミノベンゾイル)ベンゼン、1,3−ビス(4−アミノベンゾイル)ベンゼン、1,4−ビス(3−アミノベンゾイル)ベンゼン、1,4−ビス(4−アミノベンゾイル)ベンゼン、1,3−ビス(3−アミノ−α,α−ジメチルベンジル)ベンゼン、1,3−ビス(4−アミノ−α,α−ジメチルベンジル)ベンゼン、1,4−ビス(3−アミノ−α,α−ジメチルベンジル)ベンゼン、1,4−ビス(4−アミノ−α,α−ジメチルベンジル)ベンゼン、1,3−ビス(3−アミノ−α,α−ジトリフルオロメチルベンジル)ベンゼン、1,3−ビス(4−アミノ−α,α−ジトリフルオロメチルベンジル)ベンゼン、1,4−ビス(3−アミノ−α,α−ジトリフルオロメチルベンジル)ベンゼン、1,4−ビス(4−アミノ−α,α−ジトリフルオロメチルベンジル)ベンゼン、2,6−ビス(3−アミノフェノキシ)ベンゾニトリル、2,6−ビス(3−アミノフェノキシ)ピリジン、4,4’−ビス(3−アミノフェノキシ)ビフェニル、4,4’−ビス(4−アミノフェノキシ)ビフェニル、ビス[4−(3−アミノフェノキシ)フェニル]ケトン、ビス[4−(4−アミノフェノキシ)フェニル]ケトン、ビス[4−(3−アミノフェノキシ)フェニル]スルフィド、ビス[4−(4−アミノフェノキシ)フェニル]スルフィド、
【0088】
ビス[4−(3−アミノフェノキシ)フェニル]スルホン、ビス[4−(4−アミノフェノキシ)フェニル]スルホン、ビス[4−(3−アミノフェノキシ)フェニル]エーテル、ビス[4−(4−アミノフェノキシ)フェニル]エーテル、2,2−ビス[4−(3−アミノフェノキシ)フェニル]プロパン、2,2−ビス[4−(4−アミノフェノキシ)フェニル]プロパン、2,2−ビス[3−(3−アミノフェノキシ)フェニル]−1,1,1,3,3,3−ヘキサフルオロプロパン、2,2−ビス[4−(4−アミノフェノキシ)フェニル]−1,1,1,3,3,3−ヘキサフルオロプロパン、1,3−ビス[4−(3−アミノフェノキシ)ベンゾイル]ベンゼン、1,3−ビス[4−(4−アミノフェノキシ)ベンゾイル]ベンゼン、1,4−ビス[4−(3−アミノフェノキシ)ベンゾイル]ベンゼン、1,4−ビス[4−(4−アミノフェノキシ)ベンゾイル]ベンゼン、1,3−ビス[4−(3−アミノフェノキシ)−α,α−ジメチルベンジル]ベンゼン、1,3−ビス[4−(4−アミノフェノキシ)−α,α−ジメチルベンジル]ベンゼン、1,4−ビス[4−(3−アミノフェノキシ)−α,α−ジメチルベンジル]ベンゼン、1,4−ビス[4−(4−アミノフェノキシ)−α,α−ジメチルベンジル]ベンゼン、4,4’−ビス[4−(4−アミノフェノキシ)ベンゾイル]ジフェニルエーテル、4,4’−ビス[4−(4−アミノ−α,α−ジメチルベンジル)フェノキシ]ベンゾフェノン、4,4’−ビス[4−(4−アミノ−α,α−ジメチルベンジル)フェノキシ]ジフェニルスルホン、4,4’−ビス[4−(4−アミノフェノキシ)フェノキシ]ジフェニルスルホン、3,3’−ジアミノ−4,4’−ジフェノキシベンゾフェノン、3,3’−ジアミノ−4,4’−ジビフェノキシベンゾフェノン、3,3’−ジアミノ−4−フェノキシベンゾフェノン、3,3’−ジアミノ−4−ビフェノキシベンゾフェノン、6,6’−ビス(3−アミノフェノキシ)−3,3,3’,3’−テトラメチル−1,1’−スピロビインダン、6,6’−ビス(4−アミノフェノキシ)−3,3,3’,3’−テトラメチル−1,1’−スピロビインダンのような芳香族アミン;
【0089】
1,3−ビス(3−アミノプロピル)テトラメチルジシロキサン、1,3−ビス(4−アミノブチル)テトラメチルジシロキサン、α,ω−ビス(3−アミノプロピル)ポリジメチルシロキサン、α,ω−ビス(3−アミノブチル)ポリジメチルシロキサン、ビス(アミノメチル)エーテル、ビス(2−アミノエチル)エーテル、ビス(3−アミノプロピル)エーテル、ビス(2−アミノメトキシ)エチル]エーテル、ビス[2−(2−アミノエトキシ)エチル]エーテル、ビス[2−(3−アミノプロトキシ)エチル]エーテル、1,2−ビス(アミノメトキシ)エタン、1,2−ビス(2−アミノエトキシ)エタン、1,2−ビス[2−(アミノメトキシ)エトキシ]エタン、1,2−ビス[2−(2−アミノエトキシ)エトキシ]エタン、エチレングリコールビス(3−アミノプロピル)エーテル、ジエチレングリコールビス(3−アミノプロピル)エーテル、トリエチレングリコールビス(3−アミノプロピル)エーテル、エチレンジアミン、1,3−ジアミノプロパン、1,4−ジアミノブタン、1,5−ジアミノペンタン、1,6−ジアミノヘキサン、1,7−ジアミノヘプタン、1,8−ジアミノオクタン、1,9−ジアミノノナン、1,10−ジアミノデカン、1,11−ジアミノウンデカン、1,12−ジアミノドデカンのような脂肪族アミン;
【0090】
1,2−ジアミノシクロヘキサン、1,3−ジアミノシクロヘキサン、1,4−ジアミノシクロヘキサン、1,2−ジ(2−アミノエチル)シクロヘキサン、1,3−ジ(2−アミノエチル)シクロヘキサン、1,4−ジ(2−アミノエチル)シクロヘキサン、ビス(4−アミノシクロへキシル)メタン、2,6−ビス(アミノメチル)ビシクロ[2.2.1]ヘプタン、2,5−ビス(アミノメチル)ビシクロ[2.2.1]ヘプタンのような脂環式ジアミンなどが挙げられる。グアナミン類としては、アセトグアナミン、ベンゾグアナミンなどを挙げることができ、また、上記ジアミンの芳香環上水素原子の一部若しくは全てをフルオロ基、メチル基、メトキシ基、トリフルオロメチル基、又はトリフルオロメトキシ基から選ばれた置換基で置換したジアミンも使用することができる。
さらに目的に応じ、架橋点となるエチニル基、ベンゾシクロブテン−4’−イル基、ビニル基、アリル基、シアノ基、イソシアネート基、及びイソプロペニル基のいずれか1種又は2種以上を、上記ジアミンの芳香環上水素原子の一部若しくは全てに置換基として導入しても使用することができる。
【0091】
ジアミンは、目的の物性によって選択することができ、p−フェニレンジアミンなどの剛直なジアミンを用いれば、最終的に得られるポリイミドは低膨張率となる。剛直なジアミンとしては、同一の芳香環に2つアミノ基が結合しているジアミンとして、p−フェニレンジアミン、m−フェニレンジアミン、1,4−ジアミノナフタレン、1,5−ジアミノナフタレン、2、6−ジアミノナフタレン、2,7−ジアミノナフタレン、1,4−ジアミノアントラセンなどが挙げられる。
さらに、2つ以上の芳香族環が単結合により結合し、2つ以上のアミノ基がそれぞれ別々の芳香族環上に直接又は置換基の一部として結合しているジアミンが挙げられ、例えば、下記式(12)により表されるものがある。具体例としては、ベンジジン等が挙げられる。
【0092】
【化21】
(aは1以上の自然数、アミノ基はベンゼン環同士の結合に対して、メタ位または、パラ位に結合する。)
【0093】
さらに、上記式(12)において、他のベンゼン環との結合に関与せず、ベンゼン環上のアミノ基が置換していない位置に置換基を有するジアミンも用いることができる。これら置換基は、1価の有機基であるがそれらは互いに結合していてもよい。
具体例としては、2,2’−ジメチル−4,4’−ジアミノビフェニル、2,2’−ジトリフルオロメチル−4,4’−ジアミノビフェニル、3,3’−ジクロロ−4,4’−ジアミノビフェニル、3,3’−ジメトキシ−4,4’−ジアミノビフェニル、3,3’−ジメチル−4,4’−ジアミノビフェニル等が挙げられる。
また、最終的に得られるポリイミドを光導波路、光回路部品として用いる場合には、芳香環の置換基としてフッ素を導入すると1μm以上の波長の電磁波に対しての透過率を向上させることができる。
【0094】
一方、ジアミンとして、1,3−ビス(3−アミノプロピル)テトラメチルジシロキサンなどのシロキサン骨格を有するジアミンを用いると、最終的に得られるポリイミドの弾性率が低下し、ガラス転移温度を低下させることができる。
ここで、選択されるジアミンは耐熱性の観点より芳香族ジアミンが好ましいが、目的の物性に応じてジアミンの全体の60モル%、好ましくは40モル%を超えない範囲で、脂肪族ジアミンやシロキサン系ジアミン等の芳香族以外のジアミンを用いても良い。
【0095】
一方、ポリイミド前駆体を合成するには、例えば、アミン成分として4,4’−ジアミノジフェニルエーテルをN−メチルピロリドンなどの有機極性溶媒に溶解させた溶液を冷却しながら、そこへ等モルの3,3’,4,4’−ビフェニルテトラカルボン酸二無水物を徐々に加え撹拌し、ポリイミド前駆体溶液を得ることができる。
このようにして合成されるポリイミド前駆体は、最終的に得られるポリイミドに耐熱性及び寸法安定性を求める場合には、芳香族酸成分及び/又は芳香族アミン成分の共重合割合ができるだけ大きいことが好ましい。具体的には、イミド構造の繰り返し単位を構成する酸成分に占める芳香族酸成分の割合が50モル%以上、特に70モル%以上であることが好ましく、イミド構造の繰り返し単位を構成するアミン成分に占める芳香族アミン成分の割合が40モル%以上、特に60モル%以上であることが好ましく、全芳香族ポリイミドであることが特に好ましい。
【0096】
ポリイミド前駆体は、感光性樹脂組成物とした際の感度を高め、マスクパターンを正確に再現するパターン形状を得るために、5μmの膜厚のときに、露光波長に対して少なくとも5%以上の透過率を示すことが好ましく、15%以上の透過率を示すことが更に好ましい。
露光波長に対してポリイミド前駆体の透過率が高いということは、それだけ、光のロスが少ないということであり、高感度の感光性樹脂組成物を得ることができる。
【0097】
また、一般的な露光光源である高圧水銀灯を用いて露光を行う場合には、少なくとも436nm、405nm、365nmの波長の電磁波のうち1つの波長の電磁波に対する透過率が、厚み5μmのフィルムに成膜した時で好ましくは5%以上、更に好ましくは15%、より更に好ましくは50%以上である。
【0098】
ポリイミド前駆体の重量平均分子量は、その用途にもよるが、3,000〜1,000,000の範囲であることが好ましく、5,000〜500,000の範囲であることがさらに好ましく、10,000〜500,000の範囲であることがさらに好ましい。重量平均分子量が3,000未満であると、塗膜又はフィルムとした場合に十分な強度が得られにくい。また、加熱処理等を施しポリイミドなどの高分子とした際の膜の強度も低くなる。一方、重量平均分子量が1,000,000を超えると粘度が上昇し、溶解性も落ちてくるため、表面が平滑で膜厚が均一な塗膜又はフィルムが得られにくい。
ここで用いている分子量とは、ゲル浸透クロマトグラフィー(GPC)によるポリスチレン換算の値のことをいい、ポリイミド前駆体そのものの分子量でも良いし、無水酢酸等で化学的イミド化処理を行った後のものでも良い。
【0099】
なお、ポリイミド前駆体合成時における溶媒は、極性溶媒が望ましく、代表的なものとして、N−メチル−2−ピロリドン、N−アセチル−2−ピロリドン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N,N−ジエチルホルムアミド、N,N−ジエチルアセトアミド、N,N−ジメチルメトキシアセトアミド、ジメチルスルホキシド、ヘキサメチルフォスホアミド、ピリジン、ジメチルスルホン、テトラメチレンスルホン、ジメチルテトラメチレンスルホン、ジエチレングリコールジメチルエーテル、シクロペンタノン、γ−ブチロラクトン、α−アセチル−γ−ブチロラクトン等があり、これらの溶媒は単独であるいは2種類以上を組み合わせて用いられる。この他にも溶媒として組合せて用いられるものとしてベンゼン、ベンゾニトリル、1,4−ジオキサン、テトラヒドロフラン、ブチロラクトン、キシレン、トルエン、シクロヘキサノン等の非極性溶媒が挙げられ、これらの溶媒は、原料の分散媒、反応調節剤、あるいは生成物からの溶媒の揮散調節剤、皮膜平滑剤などとして使用される。
【0100】
本発明に係る感光性樹脂組成物は、前記光塩基発生剤と、前記ポリイミド前駆体と、溶媒だけの単純な混合物であってもよいが、さらに、増感剤、光又は熱硬化性成分、ポリイミド前駆体以外の非重合性バインダー樹脂、その他の成分を配合して、感光性樹脂組成物を調製してもよい。
感光性樹脂組成物を溶解、分散又は希釈する溶剤としては各種の汎用溶剤を用いることが出来る。また、ポリイミド前駆体としてポリアミック酸を用いる場合には、ポリアミック酸の合成反応により得られた溶液をそのまま用い、そこに必要に応じて他の成分を混合しても良い。
【0101】
前記光塩基発生剤の吸収波長がポリイミド前駆体の吸収波長と重なる部分があり、十分な感度が得られない場合において、感度向上の手段として、増感剤の添加が効果を発揮する場合がある。また、ポリイミド前駆体を透過する電磁波の波長帯に前記光塩基発生剤が吸収波長を有する場合においても、感度向上の手段として、増感剤を添加することができる。ただし、増感剤の添加によるポリイミド前駆体の含有率の減少に伴う、得られるパターンの膜物性、特に膜強度や耐熱性の低下に関して考慮に入れる必要がある。
増感剤と呼ばれる化合物の具体例としては、チオキサントン及び、ジエチルチオキサントンなどのその誘導体、シアニン及び、その誘導体、メロシアニン及び、その誘導体、クマリン系及び、その誘導体、ケトクマリン及び、その誘導体、ケトビスクマリン、及びその誘導体、シクロペンタノン及び、その誘導体、シクロヘキサノン及び、その誘導体、チオピリリウム塩及び、その誘導体、キノリン系及び、その誘導体、スチリルキノリン系及び、その誘導体、チオキサンテン系、キサンテン系及び、その誘導体、オキソノール系及び、その誘導体、ローダミン系及び、その誘導体、ピリリウム塩及び、その誘導体等が挙げられる。
【0102】
シアニン、メロシアニン及び、その誘導体の具体例としては、3,3’−ジカルボキシエチル−2,2’チオシアニンブロミド、1−カルボキシメチル−1’−カルボキシエチル−2,2’−キノシアニンブロミド、1,3’−ジエチル−2,2’−キノチアシアニンヨ−ジド、3−エチル−5−[(3−エチル−2(3H)−ベンゾチアゾリデン)エチリデン]−2−チオキソ−4−オキサゾリジン等が挙げられる。
クマリン、ケトクマリン及び、その誘導体の具体例としては、3−(2’−ベンゾイミダゾール)−7−ジエチルアミノクマリン、3,3’−カルボニルビス(7−ジエチルアミノクマリン)、3,3’−カルボニルビスクマリン、3,3’−カルボニルビス(5,7−ジメトキシクマリン)、3,3’−カルボニルビス(7−アセトキシクマリン)等が挙げられる。
チオキサントン及び、その誘導体の具体例としては、ジエチルチオキサントン、イソプロピルチオキサントンなどが挙げられる。
【0103】
さらに他にはベンゾフェノン、アセトフェノン、アントロン、p,p’−テトラメチルジアミノベンゾフェノン(ミヒラーケトン)、フェナントレン、2−ニトロフルオレン、5−ニトロアセナフテン、ベンゾキノン、N−アセチル−p−ニトロアニリン、p−ニトロアニリン、2−エチルアントラキノン、2−ターシャリーブチルアントラキノン、N−アセチル−4−ニトロ−1−ナフチルアミン、ピクラミド、1,2−ベンズアンスラキノン、3−メチル−1,3−ジアザ−1,9−ベンズアンスロン、p,p’−テトラエチルジアミノベンゾフェノン、2−クロロ−4−ニトロアニリン、ジベンザルアセトン、1,2−ナフトキノン、2,5−ビス−(4’−ジエチルアミノベンザル)−シクロペンタン、2,6−ビス−(4’−ジエチルアミノベンザル)−シクロヘキサノン、2,6−ビス−(4’−ジメチルアミノベンザル)−4−メチル−シクロヘキサノン、2,6−ビス−(4’−ジエチルアミノベンザル)−4−メチル−シクロヘキサノン、4,4’−ビス−(ジメチルアミノ)−カルコン、4,4’−ビス−(ジエチルアミノ)−カルコン、p−ジメチルアミノベンジリデンインダノン、1,3−ビス−(4’−ジメチルアミノベンザル)−アセトン、1,3−ビス−(4’−ジエチルアミノベンザル)−アセトン、N−フェニル−ジエタノールアミン、N−p−トリル−ジエチルアミン、などが挙げられる。
本発明ではこれらの増感剤を1種または2種以上使用することができる。
【0104】
また、組成物に使用される溶剤としては、例えば、ジエチルエーテル、テトラヒドロフラン、ジオキサン、エチレングリコールジメチルエーテル、エチレングリコールジエチルエーテル、プロピレングリコールジメチルエーテル、プロピレングリコールジエチルエーテル等のエーテル類;エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、プロピレングリコールモノメチルエーテル、プロピレングリコールモノエチルエーテル、ジエチレングリコールモノメチルエーテル、ジエチレングリコールモノエチルエーテル等のグリコールモノエーテル類(いわゆるセロソルブ類);メチルエチルケトン、アセトン、メチルイソブチルケトン、シクロペンタノン、シクロヘキサノンなどのケトン類;酢酸エチル、酢酸ブチル、酢酸n−プロピル、酢酸i−プロピル、酢酸n−ブチル、酢酸i−ブチル、前記グリコールモノエーテル類の酢酸エステル(例えば、メチルセロソルブアセテート、エチルセロソルブアセテート)、メトキシプロピルアセテート、エトキシプロピルアセテート、蓚酸ジメチル、乳酸メチル、乳酸エチル等のエステル類;エタノール、プロパノール、ブタノール、ヘキサノール、シクロヘキサノール、エチレングリコール、ジエチレングリコール、グリセリン等のアルコール類;塩化メチレン、1,1−ジクロロエタン、1,2−ジクロロエチレン、1−クロロプロパン、1−クロロブタン、1−クロロペンタン、クロロベンゼン、ブロムベンゼン、o−ジクロロベンゼン、m−ジクロロベンゼン等のハロゲン化炭化水素類;N,N−ジメチルホルムアミド、N,N−ジエチルホルムアミド、N,N−ジメチルアセトアミド、N,N−ジエチルアセトアミド等のアミド類;N−メチルピロリドンなどのピロリドン類;γ−ブチロラクトン等のラクトン類;ジメチルスルホキシドなどのスルホキシド類、その他の有機極性溶媒類等が挙げられ、更には、ベンゼン、トルエン、キシレン等の芳香族炭化水素類、及び、その他の有機非極性溶媒類等も挙げられる。これらの溶媒は単独若しくは組み合わせて用いられる。
中でも、N−メチル−2−ピロリドン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N,N−ジエチルホルムアミド、N,N−ジエチルアセトアミド、N,N−ジメチルメトキシアセトアミド、ジメチルスルホキシド、ヘキサメチルフォスホアミド、N−アセチル−2−ピロリドン、ピリジン、ジメチルスルホン、テトラメチレンスルホン、ジメチルテトラメチレンスルホン、ジエチレングリコールジメチルエーテル、シクロペンタノン、γ−ブチロラクトン、α−アセチル−γ−ブチロラクトン等の極性溶媒が好適なものとして挙げられる。
【0105】
光硬化性成分としては、エチレン性不飽和結合を1つ又は2つ以上有する化合物を用いることができ、例えば、アミド系モノマー、(メタ)アクリレートモノマー、ウレタン(メタ)アクリレートオリゴマー、ポリエステル(メタ)アクリレートオリゴマー、エポキシ(メタ)アクリレート、及びヒドロキシル基含有(メタ)アクリレート、スチレン等の芳香族ビニル化合物を挙げることができる。また、ポリイミド前駆体が、ポリアミック酸等のカルボン酸成分を構造内に有する場合には、3級アミノ基を有するエチレン性不飽和結合含有化合物を用いると、ポリイミド前駆体のカルボン酸とイオン結合を形成し、感光性樹脂組成物としたときの露光部、未露光部の溶解速度のコントラストが大きくなる。
【0106】
このようなエチレン性不飽和結合を有する光硬化性化合物を用いる場合には、さらに光ラジカル発生剤を添加してもよい。光ラジカル発生剤としては、例えば、ベンゾイン、ベンゾインメチルエーテル、ベンゾインエチルエーテル及びベンゾインイソプロピルエーテル等のベンゾインとそのアルキルエーテル;アセトフェノン、2,2−ジメトキシ−2−フェニルアセトフェノン、2,2−ジエトキシ−2−フェニルアセトフェノン、1,1−ジクロロアセトフェノン、1−ヒドロキシアセトフェノン、1−ヒドロキシシクロヘキシルフェニルケトン及び2−メチル−1−[4−(メチルチオ)フェニル]−2−モルフォリノ−プロパン−1−オン等のアセトフェノン;2−メチルアントラキノン、2−エチルアントラキノン、2−ターシャリ-ブチルアントラキノン、1−クロロアントラキノン及び2−アミルアントラキノン等のアントラキノン;2,4−ジメチルチオキサントン、2,4−ジエチルチオキサントン、2−クロロチオキサントン及び2,4−ジイソピルチオキサントン等のチオキサントン;アセトフェノンジメチルケタール及びベンジルジメチルケタール等のケタール;2,4,6−トリメチルベンゾイルジフェニルホスフィンオキシド等のモノアシルホスフィンオキシドあるいはビスアシルホスフィンオキシド類;ベンゾフェノン等のベンゾフェノン類;並びにキサントン類等が挙げられる。
【0107】
本発明に係る樹脂組成物に加工特性や各種機能性を付与するために、その他に様々な有機又は無機の低分子又は高分子化合物を配合してもよい。例えば、染料、界面活性剤、レベリング剤、可塑剤、微粒子等を用いることができる。微粒子には、ポリスチレン、ポリテトラフルオロエチレン等の有機微粒子、コロイダルシリカ、カーボン、層状珪酸塩等の無機微粒子等が含まれ、それらは多孔質や中空構造であってもよい。また、その機能又は形態としては顔料、フィラー、繊維等がある。
【0108】
本発明に係る感光性樹脂組成物において、前記ポリイミド前駆体(固形分)は、得られるパターンの膜物性、特に膜強度や耐熱性の点から、感光性樹脂組成物の固形分全体に対し、30重量%以上、50重量%以上含有することが好ましい。また、前記式(1)で表される光塩基発生剤は、感光性樹脂組成物に含まれるポリイミド前駆体の固形分100重量部に対し、通常、0.01〜50重量部、好ましくは0.1〜30重量部の範囲内で含有させることが好ましい。0.01重量部未満であると環化反応促進効果が不十分となる傾向があり、50重量部を超えると最終的に得られる樹脂硬化物に求められる諸物性を満たしにくくなる。
また、上記増感剤の配合量はポリイミド前駆体の固形分100重量部に対して50重量部未満とすることが好ましく、30重量部未満とすることがより好ましい。また、最終的に得られる樹脂硬化物に求められる諸物性の低下を防ぐため、前記本発明に係る式(1)で表される光塩基発生剤と増感剤の合計がポリイミド前駆体100重量部に対して50重量部以下であることが望ましい。
また、その他の任意成分の配合割合は、感光性樹脂組成物の固形分全体に対し、0.1重量%〜20重量%の範囲が好ましい。0.1重量%未満だと、添加物を添加した効果が発揮されにくく、20重量%を超えると、最終的に得られる樹脂硬化物の特性が最終生成物に反映されにくい。なお、感光性樹脂組成物の固形分とは溶剤以外の全成分であり、液状のモノマー成分も固形分に含まれる。
【0109】
本発明に係る感光性樹脂組成物は、さまざまなコーティングプロセスや成形プロセスに用いられて、フィルムや3次元的形状の成形物を作製することができる。
【0110】
本発明の感光性樹脂組成物より得られるポリイミドは、耐熱性、寸法安定性、絶縁性等の本来の特性も損なわれておらず、良好である。
例えば、本発明の感光性樹脂組成物から得られるポリイミドの窒素中で測定した5%重量減少温度は、250℃以上が好ましく、300℃以上がさらに好ましい。特に、はんだリフローの工程を通るような電子部品等の用途に用いる場合は、5%重量減少温度が300℃以下であると、はんだリフローの工程で発生した分解ガスにより気泡等の不具合が発生する恐れがある。
ここで、5%重量減少温度とは、熱重量分析装置を用いて重量減少を測定した時に、サンプルの重量が初期重量から5%減少した時点(換言すればサンプル重量が初期の95%となった時点)の温度である。同様に10%重量減少温度とはサンプル重量が初期重量から10%減少した時点の温度である。
【0111】
本発明の感光性樹脂組成物から得られるポリイミドのガラス転移温度は、耐熱性の観点からは高ければ高いほど良いが、光導波路のように熱成形プロセスが考えられる用途においては、120℃〜450℃程度のガラス転移温度を示すことが好ましく、200℃〜400℃程度のガラス転移温度を示すことがさらに好ましい。ここで本発明におけるガラス転移温度は、感光性樹脂組成物から得られるポリイミドをフィルム形状にすることが出来る場合には、動的粘弾性測定によって、tanδ(tanδ=損失弾性率(E’’)/貯蔵弾性率(E’))のピーク温度から求められる。動的粘弾性測定としては、例えば、粘弾性測定装置Solid Analyzer RSA II(Rheometric Scientific社製)によって、周波数3Hz、昇温速度5℃/minにより行うことができる。感光性樹脂組成物から得られるポリイミドをフィルム形状にできない場合には、示差熱分析装置(DSC)のベースラインの変曲点の温度で判断する。
【0112】
本発明の感光性樹脂組成物から得られるポリイミドの寸法安定性の観点から、線熱膨張係数は60ppm以下が好ましく、40ppm以下がさらに好ましい。半導体素子等の製造プロセスにおいてシリコンウェハ上に膜を形成する場合には、密着性、基板のそりの観点から20ppm以下がさらに好ましい。ここで、本発明における線熱膨張係数とは、本発明で得られる感光性樹脂組成物から得られるポリイミドのフィルムの熱機械的分析装置(TMA)によって求めることができる。熱機械的分析装置(例えばThermo Plus TMA8310(リガク社製))によって、昇温速度を10℃/min、評価サンプルの断面積当たりの加重が同じになるように引張り加重を1g/25000μm2として得られる。
【0113】
以上に述べたように、本発明に係る感光性ポリイミド樹脂組成物は、上記式(1)で表される化合物が、高感度の光塩基発生剤として機能することで、多種多様なポリイミド前駆体を適用することができ、最終的に得られるポリイミドの構造を広範囲から選択することができる。
また、本発明によれば、ポリイミド前駆体に前記本発明に係る式(1)で表される光塩基発生剤を混合するだけという簡便な手法で感光性ポリイミド樹脂組成物を得ることができることから、コストパフォーマンスにも優れる。
さらには、電磁波の照射により発生したアミンの触媒効果により、イミド化等の最終生成物への反応に要する処理温度を低減できる為、プロセスへの不可や製品への熱によるダメージを低減することが可能である。
【0114】
本発明に係る感光性樹脂組成物は、印刷インキ、接着剤、充填剤、電子材料、光回路部品、成形材料、レジスト材料、建築材料、3次元造形、光学部材等、樹脂材料が用いられる公知の全ての分野・製品に利用できる。
【0115】
本発明に係る感光性樹脂組成物は、耐熱性、寸法安定性、絶縁性等の特性が有効とされる広範な分野・製品、例えば、塗料又は印刷インキ、或いは、カラーフィルター、フレキシブルディスプレー用フィルム、半導体装置、電子部品、層間絶縁膜、配線被覆膜、光回路、光回路部品、反射防止膜、ホログラム、光学部材又は建築材料の形成材料として好適に用いられる。例えば具体的には、半導体装置用バッファーコート膜、多層配線板の層間絶縁膜等が挙げられる。
【0116】
特に、本発明の感光性樹脂組成物は、主にパターン形成材料(レジスト)として用いられ、それによって形成されたパターンはポリイミドからなる永久膜として耐熱性や絶縁性を付与する成分として機能するため、例えば、カラーフィルター、フレキシブルディスプレー用フィルム、電子部品、半導体装置、層間絶縁膜、配線被覆膜、光回路、光回路部品、反射防止膜、その他の光学部材又は電子部材を形成するのに適している。
【0117】
また、本発明においては、本発明に係る感光性樹脂組成物又はその熱硬化物により少なくとも一部分が形成されている、印刷物、カラーフィルター、フレキシブルディスプレー用フィルム、半導体装置、電子部品、層間絶縁膜、配線被覆膜、光回路、光回路部品、反射防止膜、ホログラム、光学部材又は建築材料いずれかの物品が提供される。
【0118】
次に、本発明に係るネガ型パターン形成方法を説明する。
本発明に係るネガ型パターン形成方法は、前記本発明に係る感光性樹脂組成物からなる塗膜又は成形体の表面に、所定のパターン状に電磁波を照射し、必要に応じて熱処理等の後処理を行って、前記塗膜又は成形体の電磁波照射部位の溶解性を選択的に低下させた後、現像することを特徴とする。
本発明に係る感光性樹脂組成物を何らかの支持体上に塗布し、所定のパターン状に電磁波を照射すると、露光部においてのみ、前記光塩基性物質が分解して塩基性物質を生成する。塩基性物質は、露光部のポリイミド前駆体の最終生成物への反応を促進する触媒として作用する。
【0119】
塩基性物質により、露光部のポリイミド前駆体が直接的に最終生成物へ反応して、露光部のポリイミド前駆体のみ、ある溶媒に対する溶解性が選択的に低下される場合には、露光後に特に後処理なく、当該露光部の溶解性が低下した溶媒を現像液として用いて、溶解性が低下していない未露光部のみを溶解することにより、現像することが可能になる。
【0120】
本発明の感光性樹脂組成物は、N−メチル−2−ピロリドン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N,N−ジエチルホルムアミド、N,N−ジエチルアセトアミド、N,N−ジメチルメトキシアセトアミド、ジメチルスルホキシド、ヘキサメチルフォスホアミド、N−アセチル−2−ピロリドン、ピリジン、ジメチルスルホン、テトラメチレンスルホン、ジメチルテトラメチレンスルホン、ジエチレングリコールジメチルエーテル、シクロペンタノン、γ−ブチロラクトン、α−アセチル−γ−ブチロラクトン等の極性溶媒に溶解後、浸漬法、スプレー法、スクリーン印刷法、スピンコート法などによって、シリコンウエハ、金属基板、セラミック基板などの基材表面に塗布し、加熱して溶剤の大部分を除くことにより、基材表面に粘着性のない塗膜を与えることができる。塗膜の厚みには特に制限はないが、0.5〜50μmであることが好ましく、感度および現像速度面から1.0〜20μmであることがより望ましい。塗布した塗膜の乾燥条件としては、例えば、80〜100℃、1分〜20分が挙げられる。
【0121】
この塗膜に、所定のパターンを有するマスクを通して、電磁波を照射しパターン状に露光後を行い、加熱後、膜の未露光部分を、適切な現像液で現像して除去することにより、所望のパターン化された膜を得ることができる。
【0122】
露光工程に用いられる露光方法や露光装置は特に限定されることなく、密着露光でも間接露光でも良くg線ステッパ、i線ステッパ、超高圧水銀灯を用いるコンタクト/プロキシミティ露光機、ミラープロジェクション露光機、又はその他の紫外線、可視光線、X線、電子線などを照射可能な投影機や線源を使用することができる。
【0123】
本発明に係るネガ型パターン形成方法においては、露光工程と現像工程の間に、必要に応じて熱処理などの後処理を行っても良い。ここでの後処理は、前記塗膜又は成形体の電磁波照射部位の、ある溶媒に対する溶解性を選択的に低下させるための処理である。
熱処理等の後処理は、例えば、塩基性物質と共存する露光部のポリイミド前駆体に対してのみ、最終生成物へ反応させる処理とする。従って、熱処理をする場合には、例えば、塩基性物質が存在する露光部と、塩基性物質が存在しない未露光部とで、ポリイミド前駆体の環化率が異なるようになる温度で行うことが好ましい。
【0124】
たとえばポリアミック酸をイミド化する場合、この段階での熱処理の好ましい温度範囲は、通常60℃〜200℃程度である。熱処理温度が60℃より低いと、イミド化の効率が悪く、現実的なプロセス条件で露光部、未露光部のイミド化率の差を創出することが難しくなる。一方、熱処理温度が200℃以上であると、電磁波の吸収に伴う分子内解裂反応により、塩基性物質を生成する中性の化合物が熱分解したり、アミンが存在していない未露光部でもイミド化が進行したりして、露光部と未露光部の溶解性の差が出にくい。
具体的には、例えば、120〜200℃で、1分〜20分加熱を行う。
この熱処理は、公知の方法であればどの方法でもよく、具体的に例示すると、空気、又は窒素雰囲気下の循環オーブン、ホットプレートによる加熱などが挙げられるが、特に限定されない。
【0125】
現像工程に用いられる現像液としては、特に限定されず、塩基性水溶液、有機溶剤など、用いられるポリイミド前駆体に合わせて適宜選択することが可能である。
塩基性水溶液としては、特に限定されないが、例えば、濃度が、0.01重量%〜10重量%、好ましくは、0.05重量%〜5重量%のテトラメチルアンモニウムヒドロキシド(TMAH)水溶液の他、ジエタノールアミン、ジエチルアミノエタノール、水酸化ナトリウム、水酸化カリウム、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、炭酸水素カリウム、トリエチルアミン、ジエチルアミン、メチルアミン、ジメチルアミン、酢酸ジメチルアミノエチル、ジメチルアミノエタノール、ジメチルアミノエチルメタクリレート、シクロヘキシルアミン、エチレンジアミン、ヘキサメチレンジアミン、テトラメチルアンモニウムなどの水溶液等が挙げられる。
溶質は、1種類でも2種類以上でも良く、全体の重量の50%以上、さらに好ましくは70%以上、水が含まれていれば有機溶媒等を含んでいても良い。
また、有機溶剤としては、特に限定されないが、N−メチル−2−ピロリドン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、ジメチルスルホキシド、γ−ブチロラクロン、ジメチルアクリルアミドなどの極性溶媒、メタノール、エタノール、イソプロパノールなどのアルコール類、酢酸エチル、プロピレングリコールモノメチルエーテルアセテートなどのエステル類、シクロペンタノン、シクロヘキサノン、イソブチルケトン、メチルイソブチルケトンなどのケトン類などを、単独であるいは2種類以上を組み合わせて添加してもよい。現像後は水にて洗浄を行う。この場合においてもエタノール、イソプロピルアルコールなどのアルコール類、乳酸エチル、プロピレングリコールモノメチルエーテルアセテートなどのエステル類などを水に加えても良い。
【0126】
現像後は必要に応じて水または貧溶媒でリンスを行い、80〜100℃で乾燥しパターンを安定なものとする。このレリーフパターンを、耐熱性のあるものとするために180〜500℃、好ましくは200〜350℃の温度で数十分から数時間加熱することにより、完全にイミド化を進行させ、パターン化された高耐熱性樹脂層が形成される。
【実施例】
【0127】
[合成例1:光塩基発生剤1の合成]
(1)アントラセン−9−イル−メチル−4’−ニトロフェニルカルボナートの合成(第一工程)
9−アントラセンメタノール5.0g(24mmol;和光純薬工業(株)製)を脱水ジメチルアセトアミド(脱水DMAc)250mLに溶解させた溶液に、トリエチルアミン7.3g(72mmol)を加えた。その溶液に、クロロギ酸−4−ニトロフェニル4.9g(24mmol;和光純薬工業(株)製)を添加した後、室温で24時間攪拌して反応させた。反応終了後、反応液に氷水を投入し、この混合液をジクロロメタンで抽出し、さらに抽出後の有機層を水で洗浄後、当該有機層を濃縮した。次いで、濃縮残渣に水を投入し、そこで生じた結晶を濾取後、得られた結晶を乾燥することにより、黄色結晶のアントラセン−9−イル−メチル−4’−ニトロフェニルカルボナート4.8g(収率:53%)を得た。以下に1H−NMRの測定結果を示す。
1H−NMR(400MHz,CDCl3)δ(ppm):6.39(2H,s,OCH2),7.36(2H,d,J=9.3Hz,ArH),7.51−7.54(2H,m,ArH),7.60−7.65(2H,m,ArH),8.06(2H,d,J=8.7Hz,ArH),8.24(2H,d,J=9.3Hz,ArH),8.41(2H,d,J=8.7Hz,ArH),8.57(1H,s,ArH)
【0128】
(2)アントラセン−9−イル−メトキシカルボニルピペリジンの合成(第二工程)
上記(1)で得られたアントラセン−9−イル−メチル−4’−ニトロフェニルカルボナート4.8g(13mmol)をジクロロメタン100mLに溶解させた溶液に、ピペリジン1.4g(16mmol)を加えた後、室温で1時間攪拌して反応させた。反応終了後、反応液を水で洗浄し、洗浄後の有機層を濃縮した。得られた濃縮残渣をカラムクロマトグラフィー(充填剤:シリカゲル(ワコーゲルC−200:和光純薬工業(株)製)、展開溶媒:ジクロロメタン)で精製することにより、下記式(5)で表されるアントラセン−9−イル−メトキシカルボニルピペリジン1.3g(収率:32%、淡黄色結晶)を得た。以下に1H−NMR及び融点の測定結果を示す。
1H−NMR(400MHz,CDCl3)δ(ppm):1.39(2H,br,CH2),1.53(4H,br,2×CH2),3.28(2H,br,NCH2),3.46(4H,br,NCH2),6.15(2H,s,OCH2),7.49−7.59(4H,m,ArH),8.03(2H,d,J=8.8Hz,ArH),8.41(2H,d,J=8.8Hz,ArH),8.50(1H,s,ArH)
融点:130−132℃
【0129】
【化22】
【0130】
[合成例2:光塩基発生剤2の合成]
(1)アントラキノン−2−イル−メチル−4’−ニトロフェニルカルボナートの合成(第一工程)
2−ヒドロキシメチルアントラキノン3.0g(12mmol;東京化成工業(株)製)を脱水ジメチルアセトアミド(脱水DMAc)100mLに溶解させた溶液に、トリエチルアミン3.8g(37mmol)を加えた。その溶液に、クロロギ酸−4−ニトロフェニル2.5g(12mmol;和光純薬工業(株)製)を添加した後、室温で24時間攪拌して反応させた。反応終了後、反応液に氷水を投入し、そこで生じた結晶を濾取後、得られた結晶を乾燥することにより、淡黄色結晶のアントラキノン−2−イル−メチル−4’−ニトロフェニルカルボナート2.4g(収率:47%)を得た。以下に1H−NMRの測定結果を示す。
1H−NMR(400MHz,CDCl3)δ(ppm):5.47(2H,s,OCH2),7.42(2H,d,J=9.3Hz,ArH),7.82−7.85(3H,m,ArH),8.29−8.39(4H,m,ArH),8.30(2H,d,J=9.3Hz,ArH)
【0131】
(2)アントラキノン−2−イル−メトキシカルボニルピペリジンの合成(第二工程)
上記(1)で得られたアントラキノン−2−イル−メチル−4’−ニトロフェニルカルボナート2.4g(5.8mmol)を脱水ジメチルアセトアミド(脱水DMAc)450mLに溶解させた溶液に、ピペリジン0.60g(7.0mmol)を加えた後、室温で1時間攪拌して反応させた。反応終了後、反応液に氷水を投入し、そこで生じた結晶を濾取後、得られた結晶を乾燥することにより、下記式(6)で表されるアントラキノン−2−イル−メトキシカルボニルピペリジン1.1g(収率:55%、淡黄色結晶)を得た。以下に1H−NMR及び融点の測定結果を示す。
1H−NMR(400MHz,CDCl3)δ(ppm):1.55−1.63(6H,m,3×CH2),3.49(4H,br,2×NCH2),5.29(2H,s,OCH2),7.76−7.83(3H,m,ArH),8.27−8.34(4H,m,ArH)
融点:146−147℃
【0132】
【化23】
【0133】
[合成例3:光塩基発生剤3の合成]
(1)ピレン−1−イル−メチル−4’−ニトロフェニルカルボナートの合成(第一工程)
1−ピレンメタノール5.0g(22mmol;東京化成工業(株)製)を脱水ジメチルアセトアミド(脱水DMAc)200mLに溶解させた溶液に、トリエチルアミン6.5g(65mmol)を加えた。その溶液に、クロロギ酸−4−ニトロフェニル4.3g(22mmol;和光純薬工業(株)製)を添加した後、室温で24時間攪拌して反応させた。反応終了後、反応液に氷水を投入し、この混合液をジクロロメタンで抽出し、さらに抽出後の有機層を水で洗浄後、当該有機層を濃縮した。次いで、濃縮残渣にトルエンを投入し、そこで生じた結晶を濾取後、得られた結晶を乾燥することにより、黄色結晶のピレン−1−イル−メチル−4’−ニトロフェニルカルボナート5.9g(収率:68%)を得た。以下に1H−NMRの測定結果を示す。
1H−NMR(400MHz,CDCl3)δ(ppm):6.02(2H,s,OCH2),7.25(2H,d,J=9.3Hz,ArH),7.98−8.20(9H,m,ArH),8.27(2H,d,J=9.3Hz,ArH)
【0134】
(2)ピレン−1−イル−メトキシカルボニルピペリジンの合成(第二工程)
上記(1)で得られたピレン−1−イル−メチル−4’−ニトロフェニルカルボナート5.9g(15mmol)を脱水ジメチルアセトアミド(脱水DMAc)50mLに溶解させた溶液に、ピペリジン1.4g(16mmol)を加えた後、室温で2時間攪拌して反応させた。反応終了後、反応液に氷水を投入し、この混合液をジクロロメタンで抽出し、さらに抽出後の有機層を水で洗浄後、当該有機層を濃縮した。得られた濃縮残渣をカラムクロマトグラフィー(充填剤:シリカゲル(ワコーゲルC−200:和光純薬工業(株)製)、展開溶媒:ジクロロメタン)で精製することにより、下記式(7)で表されるピレン−1−イル−メトキシカルボニルピペリジン1.6g(収率:32%、橙色結晶)を得た。以下に1H−NMR及び融点の測定結果を示す。
1H−NMR(400MHz,CDCl3)δ(ppm):1.54(6H,br,3×CH2),3.43(4H,br,2×NCH2),5.83(2H,s,OCH2),7.99−8.34(9H,m,ArH)
融点:118−120℃
【0135】
【化24】
【0136】
[合成例4:光塩基発生剤4の合成]
50%水素化ナトリウム1.6g(33mmol)及び脱水テトラヒドロフラン(脱水THF)4mLを仕込んだ溶液に、9−アントラセンメタノール6.3g(30mmol;和光純薬工業(株)製)を脱水テトラヒドロフラン(脱水THF)26mLに溶解させた溶液を滴下した。次いで、その溶液に、N,N−ジエチルカルバモイルクロリド4.5g(33mmol;シグマアルドリッチジャパン(株)製)の脱水テトラヒドロフラン(脱水THF)溶液4mLを添加した後、60℃で2時間攪拌して反応させた。反応終了後、反応液を冷却し、冷却した溶液にn−ヘキサンを加え、さらにこの溶液を水で洗浄し、洗浄後の有機層を濃縮した。得られた濃縮残渣をカラムクロマトグラフィー(充填剤:シリカゲル(ワコーゲルC−200:和光純薬工業(株)製)、展開溶媒:n−ヘプタン)で精製することにより、下記式(4)で表されるアントラセン−9−イル−メトキシカルボニル−N,N−ジエチルアミン5.7g(収率:61%、黄色結晶)を得た。以下に1H−NMR及び融点の測定結果を示す。
1H−NMR(400MHz,CDCl3)δ(ppm):0.91(3H,br,CH3),1.14(3H,br,CH3),3.14(2H,br,NCH2),3.35(2H,br,NCH2),6.14(2H,s,OCH2),7.48(2H,dd,J=8.0,6.8Hz,ArH),7.57(2H,dd,J=8.4,6.8Hz,ArH),8.04(2H,d,J=8.0Hz,ArH),8.46(2H,d,J=8.4Hz,ArH),8.52(1H,s,ArH)
融点:72−74℃
【0137】
【化25】
【0138】
[合成例5:ポリイミド前駆体1の合成]
窒素置換した500mL4つ口セパラブルフラスコに、4,4’−ジアミノジフェニルエーテル20.0g(100mmol)および脱水N−メチルピロリドン200mLを入れ、氷浴下で撹拌して溶解させた。この溶液に3,3’,4,4’−ビフェニルテトラカルボン酸二無水物29.4g(100mmol)を加え、氷浴下で2時間攪拌した。反応溶液をアセトンにより再沈殿し、濾取して得られた沈殿物を室温で8時間減圧乾燥することにより、ポリアミド酸(ポリイミド前駆体1)を白色固体として定量的に得た。
【0139】
<試験>
(1)モル吸光係数の測定
合成例1〜4で得られた光塩基発生剤1〜4をそれぞれ、電子天秤を用いて秤量し、メスフラスコを用いることにより、濃度5×10−5mol/Lのアセトニトリル溶液を調製した。この溶液を石英セル((株)東新理興製、TOS−UV−10(1cm×1cm×4cm))に入れ、分光光度計(島津製作所社製UV−2550)により190〜800nmの波長範囲での紫外−可視吸収スペクトルを測定した。スペクトルで得られた吸光度から、下式によりモル吸光係数ε(365、405、436nm)を測定した。結果を表1に示す。
モル吸光係数(ε)=(吸光度)/モル濃度(mol/L)/光路長(cm)
【0140】
【表1】
【0141】
(2)光分解能の測定
光塩基発生剤について、石英製NMRチューブ中に電子天秤を用いて1.0mg秤量し、重アセトニトリル0.5mLを加え溶解させた。このサンプルに、350nm以下の波長を透過しないフィルター1を介して高圧水銀灯(ウシオ電機社製SPOT CURE SP−III 250UA、ランプ型番:USH−255BY)の全波長をフィルター通過前100J/cm2(i線換算:紫外線照度計:ウシオ電機社製UIT−150、受光器:UVD−S365)、フィルター通過後18.2J/cm2(i線換算:紫外線照度計:ウシオ電機社製UIT−150、受光器:UVD−S365)により光を照射し、照射前後のNMRスペクトルの比較を行うことにより、i(365nm)線以上の波長領域における光分解性の評価を行った。図1にフィルター1の透過率曲線を示す。光分解性の評価結果を表2に示す。
【0142】
【表2】
【0143】
(3)熱安定性の測定
光塩基発生剤について、DTG−60(島津製作所製)を用いて30℃から600℃まで昇温速度10℃/minでTG−DTA測定を行った。5%重量減少温度を算出し、耐熱性の評価を行った。耐熱性の評価結果を表3に示す。
【0144】
【表3】
【0145】
(実施例1)
上記光塩基発生剤1を0.2g、上記ポリイミド前駆体1を1g、N−メチルピロリドン9gに溶解させ、本発明の感光性樹脂組成物(感光性樹脂組成物1)を得た。
【0146】
(実施例2〜4)
実施例1における光塩基発生剤1を、それぞれ光塩基発生剤2〜4に代えた以外は、実施例1と同様にして、感光性樹脂組成物(感光性樹脂組成物2〜4)を得た。
【0147】
[評価:パターン形成]
感光性樹脂組成物1を、ガラス板上に乾燥後膜厚2μmになるようにスピンコートし、100℃のホットプレート上で15分間乾燥させた。そこへ、手動露光装置(大日本科研製、MA−1100)でi線換算で、2000mJ/cm2の紫外−可視光線照射を行い、その後、155℃のホットプレート上で10分間加熱したのち、テトラメチルアンモニウムハイドロオキサイド 2.38%溶液にイソプロパノールを10wt%添加した溶液に浸漬した。その結果、露光部が現像液に溶解せず残存したパターンを得ることができた。さらに、それらのサンプルを300℃で1時間加熱しイミド化を行った。
この結果から、本発明の感光性樹脂組成物は、良好なパターンを形成することできることが明らかとなった。
【図面の簡単な説明】
【0148】
【図1】図1は、フィルター1の透過率曲線を示した図である。
【技術分野】
【0001】
本発明は、解像性に優れ、低コストで、ポリイミド前駆体の構造上適用可能な選択肢の範囲が広い感光性樹脂組成物に関し、特に、電磁波によるパターニング工程を経て形成される製品又は部材の材料(例えば、電子部品、光学製品、光学部品の成形材料、層形成材料又は接着剤など)として好適に利用することが出来る感光性樹脂組成物、及び、当該樹脂組成物を用いて作製した物品、並びに当該樹脂組成物を用いたネガ型パターン形成方法に関するものである。
【背景技術】
【0002】
従来から、半導体素子の表面保護膜や層間絶縁膜、電子部品の絶縁材料として、耐熱性、電気特性、機械特性に優れたポリイミド樹脂が使用されてきた(非特許文献1)。
半導体集積回路やプリント基板上の回路パターン形成は、素材表面へのレジスト剤の造膜、所定箇所への露光、エッチング等による不要箇所の除去、基板表面の洗浄作業等の煩雑で多岐に亘る工程を経て行われることから、回路パターンの製造工程を簡略化するために、露光、現像によるパターン形成後も必要な部分のレジストを絶縁材料としてそのまま残して用いることができる耐熱感光性材料が望まれている。これらの材料として、ポリイミドをベースポリマーとした耐熱感光性材料が提案されている。
【0003】
このような感光性ポリイミドとしては、例えば、特許文献1において、ポリイミド前駆体と重クロム酸塩からなる系が最初に提案された。しかしながら、この材料は、実用的な光感度を有するとともに膜形成能が高いなどの長所を有する反面、保存安定性に欠け、またポリイミド中にクロムイオンが残存することなどの欠点があり、実用には至らなかった。さらに、特許文献2には、ポリイミド前駆体であるポリアミック酸に感光性基をエステル結合で導入した化合物が、特許文献3には、ポリイミド前駆体にメタクリロイル基を持つアミン化合物をポリアミック酸に添加し、アミノ基とカルボキシル基をイオン結合させた化合物が紹介されている。しかしながら、エステル結合に代表される共有結合型感光性ポリイミドは、合成プロセスが煩雑であり、コストが嵩む点が問題点として挙げられる。また、イオン結合型感光性ポリイミドは、ポリイミド骨格と感光性基の結合力が小さく、露光部も溶解されることから残膜率が低下し、厚膜化が困難である点が問題点として挙げられる(非特許文献2)。また、これらの化合物の多くは、有機溶剤現像性のものであり、コスト面および環境負荷面を鑑みると、アルカリ水溶液により現像可能な化合物の方が望ましい。
【0004】
このようなポリイミド前駆体は、耐熱性、機械特性に優れるように芳香族系モノマーを基本骨格に用いている。一般的に、芳香族環を基本骨格に有するポリイミド前駆体は、可視領域から紫外領域にかけて透過率が下がっていく傾向があり、特にi線(波長:365nm)未満、特に350nm未満の波長領域に強い吸収を有していることから、紫外−可視光照射時において透光性が低い。そのため、感光性ポリイミドは、露光部において光化学反応が十分に進行せず、低感度であったり、パターンの形状が悪化するという問題があった。耐熱感光性材料の適用範囲が広がるにつれ、材料要求は多種多様のものになってきており、感光性ポリイミドに厚膜形成能が求められている。形成パターンが厚膜の場合においては、光透過性が低い問題はさらに深刻になる。そのため、膜物性および感度の面において共に優れた耐熱性感光性樹脂を実現するためには、i線(波長:365nm)、h線(波長:405nm)領域に、光反応活性を有する感光性システムの構築が望ましい。
【0005】
近年、新しいパターン形成材料の1つとして、光塩基発生剤が注目されている(例えば特許文献4)。しかしながら、既存の光塩基発生剤をポリイミド前駆体の系に適用するには吸収波長の点に問題があった。すなわち、既存の光塩基発生剤は350nm以下に吸収波長を持つものが多く、光反応によるイミド化促進剤としてポリイミド前駆体に添加すると、ポリイミド前駆体と光塩基発生剤の吸収波長が重なることから感度面で問題が生じる。そのため、耐熱性、機械特性および、感度の面において、共に優れた耐熱性感光性樹脂を実現するために、350nm以上、望ましくは、400nm以上の波長領域、例えばi線(波長:365nm)、h線(波長:405nm)領域に、光反応活性を有する塩基発生剤が求められている。
【0006】
【特許文献1】特公昭49−17374号公報
【特許文献2】特公昭55−30207号公報
【特許文献3】特開昭54−145794号公報
【特許文献4】特開2006−189591号公報
【非特許文献1】「最新ポリイミド〜基礎と応用」,株式会社エヌー・ティー・エス,2002年,p.327〜338
【非特許文献2】「電子部品用高分子材料の最新動向III」,株式会社住ベテクノリサーチ, 2004年,p.36〜39
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明は上記実情を鑑みて成し遂げられたものであり、その目的は、高感度で、ポリイミド前駆体の種類を問わず大きな溶解性コントラストを得られ、結果的に十分なプロセスマージンを保ちつつ、形状が良好なパターンを得ることができる感光性樹脂組成物を提供することにある。
【課題を解決するための手段】
【0008】
本発明に係る感光性樹脂組成物は、一般式(1)で表される光塩基発生剤、及びポリイミド前駆体を含有する。
【0009】
【化1】
(式(1)中、Arは、下記一般式(I)で示されるアントラセニル基、下記一般式(II)で示されるアントラキノリル基、及び下記一般式(III)で示されるピレニル基からなる群より選ばれる何れかの基を表し、
R1及びR2は夫々独立して、水素原子、炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基を表し、R1及びR2の少なくとも1つは水素原子ではなく、R1及びR2はこれらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成していても良く、
R3及びR4は夫々独立して、水素原子又は炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基を表す。)
【0010】
【化2】
(式(I)中、R5〜R13は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【0011】
【化3】
(式(II)中、R14〜R20は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【0012】
【化4】
(式(III)中、R21〜R29は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【0013】
本発明者らは、塩基発生部位としてカルバミン酸エステル結合と、上記特定のアントラセニル基、上記特定のアントラキノリル基、又は上記特定のピレニル基とを含む上記式(1)で表される化合物が、350nm以上の波長領域において光反応活性を有する光塩基発生剤として機能し、ポリイミド前駆体と組み合わせることにより高感度の感光性ポリイミドを達成し得ることを見出し、本発明に至った。
【0014】
上記光塩基発生剤は、上記特定の構造を有することにより、350nm以上の波長領域に光反応活性を有し得るため、ポリイミド前駆体と組み合わせても、感度良く、露光部のみ塩基の作用によってイミド化を促進させることが可能である。上記光塩基発生剤は導入する置換基によっては400nm以上の波長領域に吸収帯を持たせることも可能である。このため、i線(波長:365nm)未満に広い吸収帯を有している芳香族環を基本骨格に有するポリイミド前駆体と吸収波長が重なることなく、高感度の光塩基発生剤として機能するので、感光性樹脂組成物の塗膜又は成形体上の電磁波照射部位と非照射部位の間での溶解性差を大きくでき、結果的に、十分なプロセスマージンを保ちつつ、形状が良好なパターンを得ることができる。
【0015】
本発明の感光性樹脂組成物においては、前記一般式(1)で表される光塩基発生剤におけるR1及びR2が、炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基であることが、発生する塩基性物質の触媒効果が大きい点から好ましい。
【0016】
本発明の感光性樹脂組成物においては、前記一般式(1)で表される光塩基発生剤におけるR1及びR2が、これらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成しているものであることが、発生する塩基性物質の触媒効果が大きい点から好ましい。
【0017】
本発明の感光性樹脂組成物に用いられるポリイミド前駆体としては、それ自体が塩基性物質の作用によって最終生成物への反応が促進される化合物、中でも、それ自体が塩基性物質の作用によって最終生成物への反応が促進され、且つ加熱により溶解性が変化する化合物として、ポリアミック酸のようなポリイミド前駆体を用いることが好ましい。このようなポリイミド前駆体を用いると、耐熱性及び機械特性に優れた感光性ポリイミド樹脂組成物を得ることができる。
本発明によれば、従来、露光部と未露光部の間で溶解性のコントラストを取りにくかったポリイミド前駆体についても、溶解阻害剤、溶解抑制剤の適用なしで良好なパターン形状を得ることができる。
【0018】
本発明の一実施形態においては、感光性樹脂組成物に増感剤を添加することにより、照射感度を向上させることができる。
【0019】
本発明の感光性樹脂組成物においては、前記光塩基発生剤の5%重量減少温度が170℃以上であることが、当該感光性樹脂組成物の塗膜に対して露光後現像前に行うイミド化の温度において光塩基発生剤が分解し難くなる点から好ましい。
【0020】
また、上記本発明の感光性樹脂組成物は、広範な構造のポリイミド前駆体を選択できる為、それによって得られる硬化物は、耐熱性、寸法安定性、絶縁性等のポリイミドが特徴的に有する機能を付与することが可能であることから、ポリイミドが適用されている公知の全ての部材用のフィルム、塗膜又は3次元構造物として好適である。
特に、本発明に係る感光性組成物は、主にパターン形成材料(レジスト)として用いられ、それによって形成されたパターンは、永久膜として耐熱性や絶縁性を付与する成分として機能し、例えば、カラーフィルター、フレキシブルディスプレー用フィルム、半導体装置、電子部品、層間絶縁膜、配線被覆膜、光回路、光回路部品、反射防止膜、その他の光学部材、又は建築材料を形成するのに適している。
【0021】
さらに本発明は、前記本発明に係る感光性樹脂組成物又はその硬化物により少なくとも一部分が形成されている、印刷物、カラーフィルター、フレキシブルディスプレー用フィルム、半導体装置、電子部品、層間絶縁膜、配線被覆膜、光回路、光回路部品、反射防止膜、ホログラム、光学部材又は建築材料いずれかの物品を提供する。
【0022】
さらに本発明は、上記感光性樹脂組成物を用いるネガ型パターン形成方法を提供するものでもある。本発明に係るネガ型パターン形成方法は、上記感光性樹脂組成物からなる塗膜又は成形体の表面に、所定のパターン状に電磁波を照射し、必要に応じて後処理(通常は、加熱処理)を行って前記塗膜又は成形体の電磁波照射部位の溶解性を選択的に低下させた後、現像することを特徴とする。
上記ネガ型パターン形成方法においては、ポリイミド前駆体と、光塩基発生剤として上記式(1)で表されるような光塩基発生剤を組み合わせて用いることにより、感光性樹脂組成物からなる塗膜又は成形体の表面を現像液から保護するためのレジスト膜を用いずに、現像を行うネガ型パターン形成が可能である。
【発明の効果】
【0023】
以上に述べたように、本発明によれば、350nm以上の波長領域、例えばi線(波長:365nm)領域に、光反応活性を有する新規な光塩基発生剤を用いて、ポリイミド前駆体に添加剤を混合するという簡便な手法で、感光性ポリイミド樹脂組成物を調製し用いることができる。上記式(1)で表される光塩基発生剤は、電磁波照射による光分解反応により、塩基性物質であるアミンを発生させるので、塩基が触媒として作用する反応を有する種々の構造のポリイミド前駆体に適用することができる。したがって、本発明に係る感光性樹脂組成物は、パターン形成プロセスに制限を受けることなく、最終的なポリイミドの構造を広範囲から選択することができ、耐熱性、機械特性に優れる感光性ポリイミド樹脂組成物として利用できる。
本発明によれば、従来、露光部と未露光部の間で溶解性のコントラストを取りにくかったポリイミド前駆体について、溶解阻害剤、溶解抑制剤の適用なしで良好なパターン形状を得ることができる。
【発明を実施するための最良の形態】
【0024】
本発明は、光塩基発生剤を用いた感光性樹脂組成物、及び当該感光性樹脂組成物を用いた物品、及びネガ型パターン形成方法を含むものである。以下、感光性樹脂組成物から順に説明する。
なお、本発明において、上記式(1)で表されるような光塩基発生剤の結合を開裂させる光、電磁波とは、光分解反応を引き起こすことが可能なものであればよく、可視及び非可視領域の波長の電磁波だけでなく、電子線のような粒子線、及び、電磁波と粒子線を総称する放射線又は電離放射線が含まれる。
【0025】
本発明の感光性樹脂組成物は、一般式(1)で表される光塩基発生剤、及びポリイミド前駆体を含有する。
【0026】
【化5】
(式(1)中、Arは、下記一般式(I)で示されるアントラセニル基、下記一般式(II)で示されるアントラキノリル基、及び下記一般式(III)で示されるピレニル基からなる群より選ばれる何れかの基を表し、
R1及びR2は夫々独立して、水素原子、炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基を表し、R1及びR2の少なくとも1つは水素原子ではなく、R1及びR2はこれらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成していても良く、
R3及びR4は夫々独立して、水素原子又は炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基を表す。)
【0027】
【化6】
(式(I)中、R5〜R13は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【0028】
【化7】
(式(II)中、R14〜R20は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【0029】
【化8】
(式(III)中、R21〜R29は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【0030】
本発明に用いられる上記式(1)で表される光塩基発生剤は、電磁波照射による光分解反応により、塩基性物質であるアミンを発生させる。一方、ポリイミド前駆体は、例えば、塩基性物質の触媒作用によって、イミド化反応が開始される温度を下げることができる。つまり、ポリイミド前駆体を光塩基発生剤と共存させておき、電磁波の照射により塩基を発生させることで、電磁波の照射された部位は、ポリイミド前駆体の最終生成物への反応が促進され、より低温でイミド化を進行させることができる。本発明の感光性樹脂組成物を用いて、パターンを得るには、例えば、パターンを残したい場所に電磁波を照射した後、塩基性物質が存在する場所ではイミド化が進行し、塩基性物質の存在していない場所ではイミド化が進行しない温度で、加熱を行う。その結果、塩基性物質が存在する場所、すなわち、電磁波を照射した場所のみイミド化が進行し溶解性が低下する為、所定の現像液(有機溶媒や、アルカリ水溶液等)で現像することで、パターンを得ることができる。その後、目的に応じて、更に加熱を行って、ポリイミドパターンとすることができる。
【0031】
本発明に用いられる上記式(1)で表される光塩基発生剤は、特に、上記特定のアントラセニル基、上記特定のアントラキノリル基、又は上記特定のピレニル基が導入されていることにより、化合物が吸収する光の波長が長くなっている。また特定のアントラセニル基、上記特定のアントラキノリル基、又は上記特定のピレニル基に適宜置換基を導入することにより、より長波長側の光を吸収することが可能になる。更に、特定のアントラセニル基、上記特定のアントラキノリル基、又は上記特定のピレニル基が結合した炭素に、適宜置換基を導入することにより、長波長の光に対する感度を向上させることができる。このようにして、本発明に用いられる光塩基発生剤は、上記特定の構造を有することにより、350nm以上の波長領域に光反応活性を有し得るため、ポリイミド前駆体と組み合わせても、感度良く、露光部のみ塩基の作用によってイミド化を促進させることが可能である。上記光塩基発生剤は導入する置換基によっては400nm以上の波長領域に光反応活性を持たせることも可能である。本発明に用いられる光塩基発生剤は、高感度の光塩基発生剤として機能するので、感光性樹脂組成物の塗膜又は成形体上の電磁波照射部位と非照射部位の間での溶解性差を大きくでき、結果的に、十分なプロセスマージンを保ちつつ、形状が良好なパターンを得ることができる。
【0032】
まず、上記一般式(1)で表される光塩基発生剤について説明する。光塩基発生剤とは、光照射によりその化学構造が分解し、塩基性物質を発生するものをいう。
上記一般式(1)において、R1及びR2は夫々独立して、水素原子、炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基を表すか、或いは、R1及びR2は連結して、これらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成するものを表す。R1及びR2の少なくとも1つは水素原子ではないが、これは、両方とも水素原子であると、化合物の安定性が悪く、発生するアミンもアンモニアとなってしまうため塩基発生剤として有用ではないからである。
【0033】
R1及びR2の位置に導入する水素原子の数ならびに置換基の種類を変更することにより、アミンの塩基性度や熱物性、溶解度などの物性を変化させることが可能である。光塩基発生剤から発生させたいアミンが合成又は市販で入手可能であれば、発生させたいアミンに合わせて、R1及びR2は特に制限されず、適宜設計することが可能である。
より塩基性度の高いアミンの方が、例えば後述するポリイミド前駆体のイミド化における脱水縮合反応等に対する触媒作用が強く、より少量の添加で、より低い温度での脱水縮合反応等における触媒効果の発現が可能となる。つまりは、上記式(1)で表される化合物自身の電磁波に対する感度が低い場合でも、発生した塩基性物質の触媒効果が大きい為、感光性樹脂組成物としての見た目の感度は向上する。
上記のような触媒効果等の、発生した塩基性物質が与える効果が大きい点から、上記式(1)で表される化合物の電磁波の吸収に伴う解裂反応によって発生する塩基性物質は、脂肪族アミンが好ましい。そのような点から、上記式(1)で表される化合物のR1及びR2は、夫々独立して、水素原子、炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基、或いは、R1及びR2は連結して、これらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成するものであって、R1及びR2の少なくとも1つは水素原子ではないものが選択されている。
【0034】
その中でも、塩基性の観点から、発生する塩基性物質は、2級の脂肪族アミンが好ましい。しかしながら脂肪族1級アミンを用いた場合でも、芳香族アミンを用いた場合に比べて、十分な触媒効果を得ることができる。そのため、脂肪族アミンの中でも、更に、5%重量減少温度や50%重量減少温度、熱分解温度といった熱物性や、溶解性といった他の物性面や、合成の簡便性やコストといった観点から、アミンを適宜選択することが望ましい。
【0035】
2級の脂肪族アミンを発生させ、高感度を達成し、さらには、露光部未露光部の溶解性コントラストを大きくする観点から、R1及びR2が、炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基であることが好ましい。或いは、R1及びR2が、連結して、これらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成しているものであることが好ましい。
【0036】
R1及びR2で示される炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基は、直鎖状アルキル基、分岐状アルキル基、及び、環状アルキル基、又はこれらの組み合わせからなるアルキル基が挙げられる。当該アルキル基は、芳香族基等の置換基を有していても良い。例えば、炭素数1〜20の直鎖又は分岐の飽和又は不飽和アルキル基、炭素数4〜20のシクロアルキル基、炭素数7〜20のフェノキシアルキル基、炭素数7〜20のアリールアルキル基、炭素数1〜20のヒドロキシアルキル基などが挙げられる。
【0037】
炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基としては、具体的には、例えばメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、シクロブチル基、n−ペンチル基、イソペンチル基、sec−ペンチル基、tert−ペンチル基、ネオペンチル基、2−メチルブチル基、1,2−ジメチルプロピル基、1−エチルプロピル基、シクロペンチル基、n−ヘキシル基、イソヘキシル基、sec−ヘキシル基、tert−ヘキシル基、ネオヘキシル基、2−メチルペンチル基、1,2−ジメチルブチル基、2,3−ジメチルブチル基、1−エチルブチル基、シクロヘキシル基、n−ヘプチル基、イソヘプチル基、sec−ヘプチル基、tert−ヘプチル基、ネオヘプチル基、シクロヘプチル基、n−オクチル基、イソオクチル基、sec−オクチル基、tert−オクチル基、ネオオクチル基、2−エチルヘキシル基、シクロオクチル基、n−ノニル基、イソノニル基、sec−ノニル基、tert−ノニル基、ネオノニル基、シクロノニル基、n−デシル基、イソデシル基、sec−デシル基、tert−デシル基、ネオデシル基、シクロデシル基、n−ウンデシル基、n−ドデシル基、n−トリデシル基、n−テトラデシル基、n−ペンタデシル基、n−ヘキサデシル基、n−ヘプタデシル基、n−オクタデシル基、イソボルニル基、ノルボルニル基、アダマンチル基、メチルアダマンチル基、ベンジル基、ビニル基、1−プロペニル基、2−プロペニル基、イソプロペニル基、ブテニル基、ペンテニル基、ヘキセニル基等が挙げられるが、これらに限定されるものではない。中でも単位重量あたりの塩基発生量および製造の容易さの点から置換基を有してもよい炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基が好ましく、更に置換基を有してもよい炭素数1〜6の直鎖状、分枝状若しくは環状の飽和アルキル基が好ましく、具体的には、エチル基、n−プロピル基、n−ブチル基、n−ペンチル基、n−ヘキシル基が好ましい。
また置換基としては、ハロゲン原子、ヒドロキシル基、メルカプト基、シアノ基、シリル基、シラノール基、アルコキシ基、ニトロ基、アリール基、アセチル基、アセトキシ基、不飽和アルキルエーテル基、アリールエーテル基、不飽和アルキルチオエーテル基、アリールチオエーテル基等が挙げられる。なお、本発明において、「置換基を有してもよい炭素数1〜20のアルキル基」の炭素数は、アルキル基部分の炭素数であり、置換基中の炭素数は含めないものとする。
【0038】
一般式(I)において、R1及びR2が、連結して、これらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成しても良い。このような場合も、発生する塩基性物質の触媒効果が大きい点から好ましい。
この場合においては、R1とR2とで、2つの結合手が共に窒素原子と結合する炭素数3〜8のアルキレン基を形成し、含窒素脂肪族環を形成する。当該含窒素脂肪族環は、置換基を有していても良く、更にR1とR2が結合している窒素原子以外のヘテロ原子を上記炭素数3〜8のアルキレン基鎖中に有していてもよい。ヘテロ原子を鎖中に有する場合の結合としては、例えば、エーテル結合、チオエーテル結合、カルボニル結合、エステル結合、アミド結合、ウレタン結合、カーボネート結合などが挙げられる。
【0039】
R1及びR2の2つが連結して環状になって、R1及びR2が結合している窒素原子を含む含窒素脂肪族環を形成している場合の含窒素脂肪族環としては、例えば、アゼチジン(4員環)、ピロリジン(5員環)、ピペリジン(6員環)、ヘキサメチレンイミン環(7員環)、ヘプタメチレンイミン環(8員環)、オクタメチレンイミン環(9員環)等が挙げられる。更に、含窒素脂肪族環には、R1及びR2が結合している窒素原子の他にヘテロ原子を含んでいても良く、このような窒素原子以外のヘテロ原子を更に含む、含窒素脂肪族環としては、モルホリン、チオモルホリン、オキサゾリジン、チアゾリジンなどが挙げられる。含窒素脂肪族環には上述したような置換基を有していても良い。置換基としては、直鎖または分岐アルキル基が好ましい。なお、本発明において、「置換基を有してもよい炭素数3〜8の含窒素脂肪族環」の炭素数は、含窒素脂肪族環部分の炭素数であり、置換基中の炭素数は含めないものとする。
【0040】
上述のような、置換基を有していても良い炭素数3〜8の含窒素脂肪族環としては、ピロリジン環、ピペリジン環、ヘキサメチレンイミン環、ヘプタメチレンイミン環、2,5−ジメチルピロリジン環、2,6−ジメチルピペリジン環等の(窒素原子以外のヘテロ原子不含である)メチル基等のアルキル基が置換していてもよい炭素数4〜7の含窒素脂肪族環が好ましく、中でもピロリジン環、ピペリジン環、ヘキサメチレンイミン環、ヘプタメチレンイミン環等の(窒素原子以外のヘテロ原子を不含かつ無置換である)炭素数4〜7の含窒素脂肪族環がより好ましく、その中でも(窒素原子以外のヘテロ原子不含かつ無置換である)炭素数5の含窒素脂肪族環であるピペリジン環がさらに好ましい。これら好ましい具体例の炭素数4〜7の含窒素脂肪族環を有する一般式(1)で示される化合物は、発生する塩基性物質の触媒効果が大きく、且つ、安価かつ容易に製造できるという点で有用である。
【0041】
アルキル置換体の具体例としては、メチルアゼチジンなどのモノアルキルアゼチジン、ジメチルアゼチジンなどのジアルキルアゼチジン、トリメチルアゼチジンなどのトリアルキルアゼチジン、メチルピロリジンなどのモノアルキルピロリジン、ジメチルピロリジンなどのジアルキルピロリジン、トリメチルピロリジンなどのトリアルキルピロリジン、テトラメチルピロリジンなどのテトラアルキルピロリジン、メチルピペリジンなどのモノアルキルピペリジン、ジメチルピペリジンなどのジアルキルピペリジン、トリメチルピペリジンなどのトリアルキルピペリジン、テトラメチルピペリジンなどのテトラアルキルピペリジン、ペンタメチルピペリジンなどのペンタアルキルピペリジン等が挙げられる。
【0042】
一般式(1)におけるR3及びR4は夫々独立して、水素原子、炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基を表す。R3及びR4は水素原子でも良いが、R3及びR4に適宜、上記アルキル基を導入することにより、h線等の長波長に対する感度を向上させることができる。
【0043】
R3及びR4において、炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基としては、具体的には、例えばメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、シクロブチル基、n−ペンチル基、イソペンチル基、sec−ペンチル基、tert−ペンチル基、ネオペンチル基、2−メチルブチル基、1,2−ジメチルプロピル基、1−エチルプロピル基、シクロペンチル基、n−ヘキシル基、イソヘキシル基、sec−ヘキシル基、tert−ヘキシル基、ネオヘキシル基、2−メチルペンチル基、1,2−ジメチルブチル基、2,3−ジメチルブチル基、1−エチルブチル基、シクロヘキシル基、n−ヘプチル基、イソヘプチル基、sec−ヘプチル基、tert−ヘプチル基、ネオヘプチル基、シクロヘプチル基、n−オクチル基、イソオクチル基、sec−オクチル基、tert−オクチル基、ネオオクチル基、2−エチルヘキシル基、シクロオクチル基、n−ノニル基、イソノニル基、sec−ノニル基、tert−ノニル基、ネオノニル基、シクロノニル基、n−デシル基、イソデシル基、sec−デシル基、tert−デシル基、ネオデシル基、シクロデシル基、ノルボルニル基、アダマンチル基等が挙げられ、中でも炭素数1〜6の直鎖状飽和アルキル基が好ましく、更に炭素数1〜3の直鎖状飽和アルキル基が好ましく、具体的には、メチル基、エチル基、n−プロピル基が好ましく、その中でも炭素数1のアルキル基であるメチル基がより好ましい。
【0044】
一般式(1)におけるR3及びR4としては、光分解反応における感度の面から、1つが炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基であることが好ましい。
また、光分解反応における感度の面からは、R3及びR4が、ともに炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基であることがさらに好ましい。
【0045】
上記一般式(1)のArに該当する上記一般式(I)、(II)、及び(III)において、R5〜R29は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。
R5〜R29は全て水素原子であっても良いが、R5〜R29に置換基を導入させることにより、溶剤溶解性を向上したり、感度を向上したり、吸収波長をより長波長側にシフトさせることができる。この吸収波長をシフトする程度(シフト値)は、置換基の種類によって相違する。このシフト値については、「有機化学のスペクトルによる同定法 第5版(R.M.Silverstein著、281頁、1993年東京化学同人発行)」に記載の表が参考となる。
【0046】
ハロゲン原子としては、具体的には、例えば、フッ素原子、塩素原子、臭素原子、ヨウ素原子等が挙げられる。
炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基としては、具体的には、例えばメチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、シクロブチル基、n−ペンチル基、イソペンチル基、sec−ペンチル基、tert−ペンチル基、ネオペンチル基、2−メチルブチル基、1,2−ジメチルプロピル基、1−エチルプロピル基、シクロペンチル基、n−ヘキシル基、イソヘキシル基、sec−ヘキシル基、tert−ヘキシル基、ネオヘキシル基、2−メチルペンチル基、1,2−ジメチルブチル基、2,3−ジメチルブチル基、1−エチルブチル基、シクロヘキシル基、n−ヘプチル基、イソヘプチル基、sec−ヘプチル基、tert−ヘプチル基、ネオヘプチル基、シクロヘプチル基、n−オクチル基、イソオクチル基、sec−オクチル基、tert−オクチル基、ネオオクチル基、2−エチルヘキシル基、シクロオクチル基、n−ノニル基、イソノニル基、sec−ノニル基、tert−ノニル基、ネオノニル基、シクロノニル基、n−デシル基、イソデシル基、sec−デシル基、tert−デシル基、ネオデシル基、シクロデシル基、ノルボルニル基、アダマンチル基等が挙げられ、中でも炭素数1〜6の直鎖状飽和アルキル基が好ましく、更に炭素数1〜3の直鎖状の飽和アルキル基が好ましく、具体的には、メチル基、エチル基、n−プロピル基が好ましい。
【0047】
本発明の上記一般式(1)で示される光塩基発生剤のうち、より具体的な光塩基発生剤としては、一般式(1)におけるR1及びR2が、共に炭素数2〜10の直鎖状の飽和アルキル基であって、かつ下記一般式(2)で示される光塩基発生剤、並びに、一般式(1)におけるR1及びR2が、連結して、置換基を有していても良い炭素数4〜8の含窒素脂肪族環を形成するものであって、かつ下記一般式(3)で示される光塩基発生剤が挙げられる。これらの化合物は、他の一般式(1)で示される化合物と比較して、安価かつ容易に製造でき、従来の光塩基発生剤が感光する光と比べてより長波長の光の照射によっても、より効率的に塩基を発生できる光塩基発生剤となり得るという点において、好ましい化合物である。
【0048】
【化9】
(式(2)中、p及びqは夫々独立して、1〜9の整数を表し、R3、R4、及びArは、式(1)と同じである。)
【0049】
【化10】
(式(3)中、rは1〜5の整数を表し、R3、R4、及びArは、式(1)と同じである。)
【0050】
上記一般式(II)におけるp及びqとしては、1〜7がより好ましい。
上記一般式(III)におけるrとしては、1〜3がより好ましい。
【0051】
また、上記一般式(2)で示される光塩基発生剤の好ましい具体例としては、一般式(2)におけるR3及びR4が共に水素原子であり、Arは、上記一般式(I)においてR5〜R13がすべて水素原子であるアントラセニル基であって、かつp及びqが共に1、すなわちR1及びR2が、共に炭素数2のアルキル基であるエチル基であるものが挙げられ、より具体的には、式(4)で示される光塩基発生剤が、好ましいものとして挙げられる。
【0052】
【化11】
【0053】
さらにまた、上記一般式(3)で示される光塩基発生剤の好ましい具体例としては、一般式(3)におけるR3及びR4が共に水素原子であり、Arは、上記一般式(I)においてR5〜R13がすべて水素原子であるアントラセニル基、上記一般式(II)においてR14〜R20がすべて水素原子であるアントラキノリル基、及び上記一般式(II)においてR21〜R29がすべて水素原子であるピレニル基からなるより選ばれるいずれかの基であって、かつrが2、すなわちR1及びR2が結合している窒素原子と共に(窒素原子以外のヘテロ原子不含かつ無置換である)炭素数5の含窒素脂肪族環であるピペリジン環を形成するものである化合物が挙げられ、より具体的には、下記式(5)で示される光塩基発生剤、下記式(6)で示される光塩基発生剤、及び下記式(7)で示される光塩基発生剤が、好ましいものとして挙げられる。
【0054】
【化12】
【0055】
【化13】
【0056】
【化14】
【0057】
そのほか、一般式(1)で表される光塩基発生剤としては、たとえば、以下の化学式で表されるものが例示できるが、これらに限定されるものではない。
【0058】
【化15】
(式中、−Meとは、メチル基を表す。)
【0059】
【化16】
(式中、−Meとは、メチル基を表す。)
【0060】
【化17】
(式中、−Meとは、メチル基を表す。)
【0061】
本発明の一般式(1)で表される光塩基発生剤の製造方法としては、例えば、下記一般式(8)で示されるアルコールとハロゲン化ギ酸エステルとを、要すれば有機溶媒の存在下で反応させた後、下記一般式(9)で示されるアミンを反応させればよい。
【0062】
【化18】
(式中、Ar、R3及びR4、R1及びR2は、式(1)と同じである。)
【0063】
より具体的な製造方法としては、例えば上記一般式(8)で示されるアルコールと、当該アルコールに対して、通常0.8〜10当量、好ましくは0.8〜3当量の例えばクロロギ酸−4−ニトロフェニル、ブロモギ酸−2−ニトロフェニル等のハロゲン化ギ酸エステルとを、上記アルコールに対して、通常0.8〜20当量、好ましくは0.8〜7当量の例えばトリエチルアミン等の3級アミンの存在下、要すれば脱水ジメチルアセトアミド(脱水DMAc)等の脱水有機溶媒中で、通常−20℃〜100℃、好ましくは0℃〜60℃で反応させることにより、相当する炭酸エステル(カルボナート)を得る(第一工程)。
【0064】
上記第一工程で使用される一般式(8)で示されるアルコールとしては、例えば9−アントラセンメタノール、2−ヒドロキシメチルアントラキノン、1−ピレンメタノールが挙げられる。これらのアルコールは、市販のものを用いるか、常法により合成したものを、設計に合わせて適宜用いればよい。
【0065】
次いで、得られた炭酸エステル(カルボナート)と当該炭酸エステル(カルボナート)に対して、通常0.8〜10当量、好ましくは0.8〜3当量の上記一般式(9)で示されるアミンとを、要すればジクロロメタン等の有機溶媒中で、通常−20℃〜100℃、好ましくは0℃〜60℃で、反応させることにより(第二工程)、本発明の一般式(1)で示される化合物を得ることができる。
上記第二工程で使用される一般式(9)で示されるアミンは、市販のものを用いるか、常法により合成したものを適宜用いればよく、特に限定されない。上述したように、光を照射して発生させたいアミンを設計に合わせて用いるようにする。
具体的には、例えば、ジエチルアミン、ジ−n−プロピルアミン、ジイソプロピルアミン、ジ−n−ブチルアミン、ジイソブチルアミン、ジ−sec−ブチルアミン、ジ−tert−ブチルアミン、ジシクロブチルアミン、ジ−n−ペンチルアミン、ジイソペンチルアミン、ジ−sec−ペンチルアミン、ジ−tert−ペンチルアミン、ジネオペンチルアミン、ジ−2−メチルブチルアミン、ジ−1,2−ジメチルプロピルアミン、ジ−1−エチルプロピルアミン、ジシクロペンチルアミン、ジ−n−ヘキシルアミン、ジイソヘキシルアミン、ジ−sec−ヘキシルアミン、ジ−tert−ヘキシルアミン、ジネオヘキシルアミン、ジ−2−メチルペンチルアミン、ジ−1,2−ジメチルブチルアミン、ジ−2,3−ジメチルブチルアミン、ジ−1−エチルブチルアミン、ジシクロヘキシルアミン、ジ−n−ヘプチルアミン、ジイソヘプチルアミン、ジ−sec−ヘプチルアミン、ジ−tert−ヘプチルアミン、ジネオヘプチルアミン、ジシクロヘプチルアミン、ジ−n−オクチルアミン、ジイソオクチルアミン、ジ−sec−オクチルアミン、ジ−tert−オクチルアミン、ジネオオクチルアミン、ジ−2−エチルヘキシルアミン、ジシクロオクチルアミン、ジ−n−ノニルアミン、ジイソノニルアミン、ジ−sec−ノニルアミン、ジ−tert−ノニルアミン、ジネオノニルアミン、ジシクロノニルアミン、ジ−n−デシルアミン、ジイソデシルアミン、ジ−sec−デシルアミン、ジ−tert−デシルアミン、ジネオデシルアミン、ジシクロデシルアミン、ジノルボルニルアミン、ジアダマンチルアミン、エチルメチルアミン、メチル−n−プロピルアミン、メチルイソプロピルアミン、エチル−n−プロピルアミン、エチルイソプロピルアミン、n−プロピルイソプロピルアミン等の炭素数1〜10の直鎖状、分枝状若しくは環状のジアルキルアミン、アゼチジン、ピロリジン、2,5−ジメチルピロリジン、ピペリジン、2,6−ジメチルピペリジン、ヘキサメチレンイミン、ヘプタメチレンイミン、オクタメチレンイミン、オキサゾリジン、チアゾリジン、モルホリン、チオモルホリン等が挙げられるが、これらに限定されるものではない。
このようにして、光塩基発生剤から発生させたいアミンが合成又は市販で入手可能であれば、発生させたいアミンに合わせて、R1及びR2は特に制限されず、適宜設計し合成することが可能である。
【0066】
なお、本発明に用いられる一般式(1)で表される光塩基発生剤を製造する方法は上記に限られない。別法としては、例えば上記一般式(8)で示されるアルコールと、当該アルコールに対して、通常0.8〜10当量、好ましくは0.8〜3当量の下記一般式(10)で示される化合物とを、要すれば上記アルコールに対して、通常0.8〜5当量、好ましくは0.8〜3当量の例えば水素化ナトリウム等の塩基の存在下、要すれば脱水テトラヒドロフラン(脱水THF)等の有機溶媒中で、通常0℃〜120℃、好ましくは20℃〜100℃で反応させることにより、本発明に用いられる一般式(1)で表される光塩基発生剤を得ることができる。当該法では、目的とする化合物を1工程で合成することができる。
【0067】
【化19】
(式中、Xはハロゲン原子を表し、R1及びR2は式(1)と同じである。)
【0068】
一般式(10)におけるXで示されるハロゲン原子としては、具体的には、例えば塩素原子、臭素原子、ヨウ素原子等が挙げられ、中でも塩素原子、臭素原子が好ましく、その中でも塩素原子がより好ましい。
上記反応で使用される一般式(10)で示される化合物は、市販のものを用いるか、常法により合成したものを適宜用いればよい。
【0069】
以上のようにして得られる、式(1)で表される光塩基発生剤、および上記式(1)で表される光塩基発生剤の光分解反応により生じる塩基性物質は、本発明の光塩基発生剤を含有する感光性樹脂組成物の塗膜に対して露光後現像前に行う、加熱の温度(パターン形成用の部分的なイミド化の温度)において分解しないことが好ましい。具体的には、上記式(1)で表される光塩基発生剤や、光分解反応により生じる塩基性物質を加熱して初期の重量から5%重量が減少したときの温度(5%重量減少温度)は170℃、更に好ましくは200℃以上であることが望ましい。
【0070】
更には、本発明の光塩基発生剤を含有する感光性樹脂組成物が、製品として用いられる場合、感光性樹脂組成物中に塩基性物質が残存しないことが好ましいので、現像後に行う加熱のプロセス(完全イミド化のプロセス)で分解、または揮発してしまう塩基性物質であることが好ましい。具体的には、光分解反応により生じる塩基性物質を加熱して初期の重量から50%重量が減少したときの温度(50%重量減少温度)が400℃以下であることが好ましい。
【0071】
一般的な露光光源である高圧水銀灯の代表的な発光波長は、436nm、405nm、365nmであるため、本発明の前記式(1)で表される光塩基発生剤は、350nm以上の波長の電磁波を吸収することが望ましく、400nm以上の波長の電磁波の吸収を有することがさらに好ましい。芳香族環を基本骨格に有するポリイミド前駆体は、350nm未満に広い吸収帯を有している場合が多い為、光塩基発生剤が365nm以上、更に400nm以上の波長の電磁波の吸収を有する場合には、ポリイミド前駆体と光塩基発生剤の吸収波長が重なることなく、感度を向上することが可能になるからである。一般的な露光光源である高圧水銀灯の代表的な発光波長を用いる点からは、中でも前記光塩基発生剤は405nmに吸収を有することが好ましい。
【0072】
また、本発明の前記式(1)で表される光塩基発生剤は、350nm以上、更に350nm〜500nmの波長の電磁波に対して光分解性を有することが好ましい。より好ましくは、400nm以上、更に400nm〜500nmの波長の電磁波に対して光分解性を有することが好ましい。
中でも前記光塩基発生剤は、436nm、及び405nmの波長の電磁波のうち少なくとも1つの波長に吸収を有するだけでなく、405nmの波長の電磁波に対して光分解性を有することが好ましい。436nm、及び405nmの波長の電磁波のうち少なくとも1つの波長に吸収を有していてもこのような波長の電磁波に対して光分解性を有しない場合もある。
405nmの波長の電磁波に対して光分解性を有するかどうかは、例えば、i線(波長:365nm)を全く通さないフィルターを介して高圧水銀灯を用いて光塩基発生剤に照射して、光塩基発生剤が分解するか否か、或いは塩基性物質を発生させるか否かを観測することによって判断できる。
【0073】
次に、ポリイミド前駆体について説明する。
本発明に用いるポリイミド前駆体は、なんらかの溶媒(有機溶剤、又は水溶液)に可溶なものであることが好ましい。溶媒(有機溶剤、又は水溶液)に可溶なものであると、ポリイミド前駆体の当該溶媒に対する溶解性を変化させることにより、その可溶な溶媒を現像液として用いて、適宜、有機溶剤、塩基性水溶液、酸性水溶液、又は中性水溶液による現像をすることが可能になる。
ここで、ある溶媒に可溶とは、具体的には、基板上に形成された塗膜の25℃における当該溶媒に対する溶解速度が、100Å/sec以上を目安とする。当該溶解速度は1000Å/sec以上であることがさらに好ましい。
例えば、塩基性水溶液に可溶なものは、具体的には、基板上に形成された塗膜の25℃における0.1wt%のテトラメチルアンモニウムヒドロキシド(TMAH)水溶液に対する溶解速度が、100Å/sec以上である。当該溶解速度は1000Å/sec以上であることがさらに好ましい。さらには、より一般的に用いられる現像液である2.38重量%のテトラメチルアンモニウムヒドロキシド水溶液に対する溶解速度が、100Å/sec以上であることが好ましく、1000Å/sec以上であることがさらに好ましい。上記定義による溶解速度が100Å/secより小さい場合、現像時間が遅くなり作業性、生産性が悪くなると共に、露光部、未露光部間の溶解性コントラストが得にくくなる。
したがって、本発明の感光性樹脂組成物のある溶媒に対しての溶解速度は、25℃における当該溶媒に対する溶解速度が、100Å/sec以上であることが好ましく、1000Å/sec以上であることがさらに好ましい。
【0074】
上記溶解速度を測定する具体的手順としては、無アルカリガラス等の基板上に形成されたポリイミド前駆体の塗膜を、25℃に調温され、撹拌された現像液(0.1重量%TMAH水溶液または、2.38重量%TMAH水溶液等の塩基性水溶液、有機溶剤等)に一定時間、浸漬し、蒸留水でリンス後、乾燥させた後で測定した膜厚と、初期膜厚との差を、膜減り量とし、その膜減り量を、現像液に浸漬した時間で割ったものが、25℃における単位時間当たりの溶解速度ということになる。
【0075】
また、露光部と未露光部の間に十分な溶解性コントラストを得るために、感光性樹脂組成物を実際に所定の感光パターン形成プロセスにおいて用いた時に、パターン状露光、及び、必要に応じて後工程(通常は加熱工程)を行って得られる、現像工程前における未露光部位と露光部位の現像液に対する溶解性の比(未露光部位の現像液に対する単位時間当たりの溶解速度/露光部位の現像液に対する単位時間当たりの溶解速度)が、10以上であることが好ましい。
単位時間当たりの溶解速度は、上記の方法と同様にして求められ、感光性樹脂組成物の塗膜にパターン露光を行い、露光後の加熱を行った後に、露光部、未露光部の溶解速度を、それぞれ求める。
【0076】
本発明においては、塩基性物質の作用によって最終生成物への反応が促進されるポリイミド前駆体が用いられる。ここで、ポリイミド前駆体が、塩基性物質の作用によって最終生成物への反応が促進される態様には、ポリイミド前駆体が塩基性物質の作用のみによって最終生成物に変化する態様のみならず、塩基性物質の作用によってポリイミド前駆体の最終生成物への反応温度が、塩基性物質の作用がない場合に比べて低下するような態様が含まれる。
このような塩基性物質の存在の有無により反応温度差が出来る場合には、反応温度差を利用して、塩基性物質と共存するポリイミド前駆体のみが最終生成物へと反応する適切な温度で加熱することにより、塩基性物質と共存するポリイミド前駆体のみが最終生成物へと反応しある溶媒への溶解性が変化する。従って、塩基性物質の存在の有無によって、ポリイミド前駆体のある溶媒への溶解性を変化させることが可能となり、ひいては当該溶媒を現像液として用いて現像によるパターニングが可能になる。よって、本発明に用いられるポリイミド前駆体としては、塩基性物質の作用によって最終生成物への反応が促進され、且つ、加熱により溶解性が、加熱前に比べて低く変化するポリイミド前駆体が好適に用いられる。
【0077】
ここで、ポリイミド前駆体としては、下記式(11)で表されるようなポリアミック酸が好適に用いられる。
【0078】
【化20】
(式(11)中、R31は4価の有機基である。R32は2価の有機基である。)
【0079】
なお、R31の4価は酸と結合するための価数のみを示しているが、他に更なる置換基を有していても良い。同様に、R32の2価はアミンと結合するための価数のみを示しているが、他に更なる置換基を有していても良い。
ポリアミック酸は、酸2無水物とジアミンを溶液中で混合するのみで得られるので、1段階の反応で合成することができ、合成が容易で低コストで入手できるので好ましい。
【0080】
ポリアミック酸のように、塩基の触媒作用によって熱硬化温度が低下するポリイミド前駆体を用いる場合には、先ず、そのようなポリアミック酸と、前記光塩基発生剤を組み合わせた感光性樹脂組成物の塗膜又は成形体上のパターンを残したい部分に電磁波を照射する。すると、照射部には、塩基性物質が発生し、その部分のイミド化温度が選択的に低下する。次に、照射部はイミド化反応が起こるが、非照射部はイミド化反応が起こらない処理温度で加熱し、照射部のみ少なくとも現像液に溶解しない程度に部分的にイミド化させる。次に、所定の現像液(有機溶媒や塩基性水溶液等)で非照射部を溶解して熱硬化物からなるパターンを形成する。このパターンを、更に必要に応じ更に加熱してイミド化を完結させる。以上の工程によって、所望の2次元樹脂パターン(一般的な平面パターン)又は3次元樹脂パターン(立体的に成形された形状)が得られる。
【0081】
本発明においては、上記式(1)で表されるような化合物が、高感度の光塩基発生剤として機能し、感光性樹脂組成物の塗膜又は成形体上の電磁波照射部位と非照射部位の間での溶解性差を大きくできるので、有機溶媒ではなく、塩基性水溶液を用いる場合でも優れた現像性が得られる。
副次的な効果として、用いるポリイミド前駆体がポリアミック酸である場合、塩基性物質の触媒効果によりイミド化に要する温度が低くても充分な為、最終キュア温度を300℃未満、更に好ましくは250℃以下まで下げることが可能である。従来のポリアミック酸はイミド化するために最終キュア温度を300℃以上とする必要があった為、用途が制限されていたが、最終キュア温度を下げることが可能になったことによって、より広範囲の用途に適用が可能である。
【0082】
また、ポリイミド前駆体に関して、最終的に得られるポリイミドの耐熱性及び寸法安定性の要求が厳しい用途に対しては、酸二無水物由来の部分が芳香族構造を有し、さらにジアミン由来の部分も芳香族構造を含む全芳香族ポリイミド前駆体であることが好ましい。それゆえジアミン成分由来の構造も芳香族ジアミンから誘導される構造であることが好ましい。
ここで、全芳香族ポリイミド前駆体とは、芳香族酸成分と芳香族アミン成分の共重合、又は、芳香族酸/アミノ成分の重合により得られるポリイミド前駆体及びその誘導体である。また、芳香族酸成分とは、ポリイミド骨格を形成する4つの酸基が全て芳香族環上に置換している化合物であり、芳香族アミン成分とは、ポリイミド骨格を形成する2つのアミノ基が両方とも芳香族環上に置換している化合物であり、芳香族酸/アミノ成分とはポリイミド骨格を形成する酸基とアミノ基がいずれも芳香族環上に置換している化合物である。ただし、後述する原料の具体例から明らかなように、全ての酸基又はアミノ基が同じ芳香環上に存在する必要はない。
【0083】
本発明のポリイミド前駆体を製造する方法としては、従来公知の手法を適用することができる。例えば、(1)酸二無水物とジアミンから前駆体であるポリアミック酸を合成する手法。(2)酸二無水物に1価のアルコールやアミノ化合物、エポキシ化合物等を反応させ合成した、エステル酸やアミド酸モノマーのカルボン酸に、ジアミノ化合物やその誘導体を反応させてポリイミド前駆体を合成する手法などが挙げられるがこれに限定されない。
【0084】
本発明のポリイミド前駆体に適用可能な酸二無水物としては、例えば、エチレンテトラカルボン酸二無水物、ブタンテトラカルボン酸二無水物、シクロブタンテトラカルボン酸二無水物、メチルシクロブタンテトラカルボン酸二無水物、シクロペンタンテトラカルボン酸二無水物などの脂肪族テトラカルボン酸二無水物;ピロメリット酸二無水物、3,3’,4,4’−ベンゾフェノンテトラカルボン酸二無水物、2,2’,3,3’−ベンゾフェノンテトラカルボン酸二無水物、2,3’,3,4’−ベンゾフェノンテトラカルボン酸二無水物、3,3’,4,4’−ビフェニルテトラカルボン酸二無水物、2,2’,3,3’−ビフェニルテトラカルボン酸二無水物、2,3’,3,4’−ビフェニルテトラカルボン酸二無水物、2,2’,6,6’−ビフェニルテトラカルボン酸二無水物、2,2−ビス(3,4−ジカルボキシフェニル)プロパン二無水物、2,2−ビス(2,3−ジカルボキシフェニル)プロパン二無水物、ビス(3,4−ジカルボキシフェニル)エーテル二無水物、ビス(3,4−ジカルボキシフェニル)スルホン二無水物、1,1−ビス(2,3−ジカルボキシフェニル)エタン二無水物、ビス(2,3−ジカルボキシフェニル)メタン二無水物、ビス(3,4−ジカルボキシフェニル)メタン二無水物、2,2−ビス(3,4−ジカルボキシフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物、2,2−ビス(2,3−ジカルボキシフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物、1,3−ビス〔(3,4−ジカルボキシ)ベンゾイル〕ベンゼン二無水物、1,4−ビス〔(3,4−ジカルボキシ)ベンゾイル〕ベンゼン二無水物、2,2−ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}プロパン二無水物、
【0085】
2,2−ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}プロパン二無水物、ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}ケトン二無水物、ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}ケトン二無水物、4,4’−ビス〔4−(1,2−ジカルボキシ)フェノキシ〕ビフェニル二無水物、4,4’−ビス〔3−(1,2−ジカルボキシ)フェノキシ〕ビフェニル二無水物、ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}ケトン二無水物、ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}ケトン二無水物、ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}スルホン二無水物、ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}スルホン二無水物、ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}スルフィド二無水物、ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}スルフィド二無水物、2,2−ビス{4−〔4−(1,2−ジカルボキシ)フェノキシ〕フェニル}−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物、2,2−ビス{4−〔3−(1,2−ジカルボキシ)フェノキシ〕フェニル}−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物、2,3,6,7−ナフタレンテトラカルボン酸二無水物、1,1,1,3,3,3−ヘキサフルオロ−2,2−ビス(2,3−又は3,4−ジカルボキシフェニル)プロパン二無水物、1,4,5,8−ナフタレンテトラカルボン酸二無水物、1,2,5,6−ナフタレンテトラカルボン酸二無水物、1,2,3,4−ベンゼンテトラカルボン酸二無水物、3,4,9,10−ぺリレンテトラカルボン酸二無水物、2,3,6,7−アントラセンテトラカルボン酸二無水物、1,2,7,8−フェナントレンテトラカルボン酸二無水物、ピリジンテトラカルボン酸二無水物、スルホニルジフタル酸無水物、m−ターフェニル−3,3’,4,4’−テトラカルボン酸二無水物、p−ターフェニル−3,3’,4,4’−テトラカルボン酸二無水物などの芳香族テトラカルボン酸二無水物等が挙げられる。これらは単独あるいは2種以上混合して用いられる。そして、特に好ましく用いられるテトラカルボン酸二無水物としてピロメリット酸二無水物、3,3’,4,4’−ベンゾフェノンテトラカルボン酸二無水物、3,3’,4,4’−ビフェニルテトラカルボン酸二無水物、2,2’,6,6’−ビフェニルテトラカルボン酸二無水物、ビス(3,4−ジカルボキシフェニル)エーテル二無水物、2,2−ビス(3,4−ジカルボキシフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン二無水物が挙げられる。
【0086】
併用する酸二無水物としてフッ素が導入された酸二無水物や、脂環骨格を有する酸二無水物を用いると、透明性をそれほど損なわずに溶解性や熱膨張率等の物性を調整することが可能である。また、ピロメリット酸無水物、3,3’,4,4’−ビフェニルテトラカルボン酸二無水物、1,4,5,8−ナフタレンテトラカルボン酸二無水物などの剛直な酸二無水物を用いると、最終的に得られるポリイミドの線熱膨張係数が小さくなるが、透明性の向上を阻害する傾向があるので、共重合割合に注意しながら併用してもよい。
【0087】
一方、アミン成分も、1種類のジアミン単独で、または2種類以上のジアミンを併用して用いることができる。用いられるジアミン成分は限定されるわけではないが、p−フェニレンジアミン、m−フェニレンジアミン、o−フェニレンジアミン、3,3’−ジアミノジフェニルエーテル、3,4’−ジアミノジフェニルエーテル、4,4’−ジアミノジフェニルエーテル、3,3’−ジアミノジフェニルスルフィド、3,4’−ジアミノジフェニルスルフィド、4,4’−ジアミノジフェニルスルフィド、3,3’−ジアミノジフェニルスルホン、3,4’−ジアミノジフェニルスルホン、4,4’−ジアミノジフェニルスルホン、3,3’−ジアミノベンゾフェノン、4,4’−ジアミノベンゾフェノン、3,4’−ジアミノベンゾフェノン、3,3’−ジアミノジフェニルメタン、4,4’−ジアミノジフェニルメタン、3,4’−ジアミノジフェニルメタン、2,2−ジ(3−アミノフェニル)プロパン、2,2−ジ(4−アミノフェニル)プロパン、2−(3−アミノフェニル)−2−(4−アミノフェニル)プロパン、2,2−ジ(3−アミノフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン、2,2−ジ(4−アミノフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン、2−(3−アミノフェニル)−2−(4−アミノフェニル)−1,1,1,3,3,3−ヘキサフルオロプロパン、1,1−ジ(3−アミノフェニル)−1−フェニルエタン、1,1−ジ(4−アミノフェニル)−1−フェニルエタン、1−(3−アミノフェニル)−1−(4−アミノフェニル)−1−フェニルエタン、1,3−ビス(3−アミノフェノキシ)ベンゼン、1,3−ビス(4−アミノフェノキシ)ベンゼン、1,4−ビス(3−アミノフェノキシ)ベンゼン、1,4−ビス(4−アミノフェノキシ)ベンゼン、1,3−ビス(3−アミノベンゾイル)ベンゼン、1,3−ビス(4−アミノベンゾイル)ベンゼン、1,4−ビス(3−アミノベンゾイル)ベンゼン、1,4−ビス(4−アミノベンゾイル)ベンゼン、1,3−ビス(3−アミノ−α,α−ジメチルベンジル)ベンゼン、1,3−ビス(4−アミノ−α,α−ジメチルベンジル)ベンゼン、1,4−ビス(3−アミノ−α,α−ジメチルベンジル)ベンゼン、1,4−ビス(4−アミノ−α,α−ジメチルベンジル)ベンゼン、1,3−ビス(3−アミノ−α,α−ジトリフルオロメチルベンジル)ベンゼン、1,3−ビス(4−アミノ−α,α−ジトリフルオロメチルベンジル)ベンゼン、1,4−ビス(3−アミノ−α,α−ジトリフルオロメチルベンジル)ベンゼン、1,4−ビス(4−アミノ−α,α−ジトリフルオロメチルベンジル)ベンゼン、2,6−ビス(3−アミノフェノキシ)ベンゾニトリル、2,6−ビス(3−アミノフェノキシ)ピリジン、4,4’−ビス(3−アミノフェノキシ)ビフェニル、4,4’−ビス(4−アミノフェノキシ)ビフェニル、ビス[4−(3−アミノフェノキシ)フェニル]ケトン、ビス[4−(4−アミノフェノキシ)フェニル]ケトン、ビス[4−(3−アミノフェノキシ)フェニル]スルフィド、ビス[4−(4−アミノフェノキシ)フェニル]スルフィド、
【0088】
ビス[4−(3−アミノフェノキシ)フェニル]スルホン、ビス[4−(4−アミノフェノキシ)フェニル]スルホン、ビス[4−(3−アミノフェノキシ)フェニル]エーテル、ビス[4−(4−アミノフェノキシ)フェニル]エーテル、2,2−ビス[4−(3−アミノフェノキシ)フェニル]プロパン、2,2−ビス[4−(4−アミノフェノキシ)フェニル]プロパン、2,2−ビス[3−(3−アミノフェノキシ)フェニル]−1,1,1,3,3,3−ヘキサフルオロプロパン、2,2−ビス[4−(4−アミノフェノキシ)フェニル]−1,1,1,3,3,3−ヘキサフルオロプロパン、1,3−ビス[4−(3−アミノフェノキシ)ベンゾイル]ベンゼン、1,3−ビス[4−(4−アミノフェノキシ)ベンゾイル]ベンゼン、1,4−ビス[4−(3−アミノフェノキシ)ベンゾイル]ベンゼン、1,4−ビス[4−(4−アミノフェノキシ)ベンゾイル]ベンゼン、1,3−ビス[4−(3−アミノフェノキシ)−α,α−ジメチルベンジル]ベンゼン、1,3−ビス[4−(4−アミノフェノキシ)−α,α−ジメチルベンジル]ベンゼン、1,4−ビス[4−(3−アミノフェノキシ)−α,α−ジメチルベンジル]ベンゼン、1,4−ビス[4−(4−アミノフェノキシ)−α,α−ジメチルベンジル]ベンゼン、4,4’−ビス[4−(4−アミノフェノキシ)ベンゾイル]ジフェニルエーテル、4,4’−ビス[4−(4−アミノ−α,α−ジメチルベンジル)フェノキシ]ベンゾフェノン、4,4’−ビス[4−(4−アミノ−α,α−ジメチルベンジル)フェノキシ]ジフェニルスルホン、4,4’−ビス[4−(4−アミノフェノキシ)フェノキシ]ジフェニルスルホン、3,3’−ジアミノ−4,4’−ジフェノキシベンゾフェノン、3,3’−ジアミノ−4,4’−ジビフェノキシベンゾフェノン、3,3’−ジアミノ−4−フェノキシベンゾフェノン、3,3’−ジアミノ−4−ビフェノキシベンゾフェノン、6,6’−ビス(3−アミノフェノキシ)−3,3,3’,3’−テトラメチル−1,1’−スピロビインダン、6,6’−ビス(4−アミノフェノキシ)−3,3,3’,3’−テトラメチル−1,1’−スピロビインダンのような芳香族アミン;
【0089】
1,3−ビス(3−アミノプロピル)テトラメチルジシロキサン、1,3−ビス(4−アミノブチル)テトラメチルジシロキサン、α,ω−ビス(3−アミノプロピル)ポリジメチルシロキサン、α,ω−ビス(3−アミノブチル)ポリジメチルシロキサン、ビス(アミノメチル)エーテル、ビス(2−アミノエチル)エーテル、ビス(3−アミノプロピル)エーテル、ビス(2−アミノメトキシ)エチル]エーテル、ビス[2−(2−アミノエトキシ)エチル]エーテル、ビス[2−(3−アミノプロトキシ)エチル]エーテル、1,2−ビス(アミノメトキシ)エタン、1,2−ビス(2−アミノエトキシ)エタン、1,2−ビス[2−(アミノメトキシ)エトキシ]エタン、1,2−ビス[2−(2−アミノエトキシ)エトキシ]エタン、エチレングリコールビス(3−アミノプロピル)エーテル、ジエチレングリコールビス(3−アミノプロピル)エーテル、トリエチレングリコールビス(3−アミノプロピル)エーテル、エチレンジアミン、1,3−ジアミノプロパン、1,4−ジアミノブタン、1,5−ジアミノペンタン、1,6−ジアミノヘキサン、1,7−ジアミノヘプタン、1,8−ジアミノオクタン、1,9−ジアミノノナン、1,10−ジアミノデカン、1,11−ジアミノウンデカン、1,12−ジアミノドデカンのような脂肪族アミン;
【0090】
1,2−ジアミノシクロヘキサン、1,3−ジアミノシクロヘキサン、1,4−ジアミノシクロヘキサン、1,2−ジ(2−アミノエチル)シクロヘキサン、1,3−ジ(2−アミノエチル)シクロヘキサン、1,4−ジ(2−アミノエチル)シクロヘキサン、ビス(4−アミノシクロへキシル)メタン、2,6−ビス(アミノメチル)ビシクロ[2.2.1]ヘプタン、2,5−ビス(アミノメチル)ビシクロ[2.2.1]ヘプタンのような脂環式ジアミンなどが挙げられる。グアナミン類としては、アセトグアナミン、ベンゾグアナミンなどを挙げることができ、また、上記ジアミンの芳香環上水素原子の一部若しくは全てをフルオロ基、メチル基、メトキシ基、トリフルオロメチル基、又はトリフルオロメトキシ基から選ばれた置換基で置換したジアミンも使用することができる。
さらに目的に応じ、架橋点となるエチニル基、ベンゾシクロブテン−4’−イル基、ビニル基、アリル基、シアノ基、イソシアネート基、及びイソプロペニル基のいずれか1種又は2種以上を、上記ジアミンの芳香環上水素原子の一部若しくは全てに置換基として導入しても使用することができる。
【0091】
ジアミンは、目的の物性によって選択することができ、p−フェニレンジアミンなどの剛直なジアミンを用いれば、最終的に得られるポリイミドは低膨張率となる。剛直なジアミンとしては、同一の芳香環に2つアミノ基が結合しているジアミンとして、p−フェニレンジアミン、m−フェニレンジアミン、1,4−ジアミノナフタレン、1,5−ジアミノナフタレン、2、6−ジアミノナフタレン、2,7−ジアミノナフタレン、1,4−ジアミノアントラセンなどが挙げられる。
さらに、2つ以上の芳香族環が単結合により結合し、2つ以上のアミノ基がそれぞれ別々の芳香族環上に直接又は置換基の一部として結合しているジアミンが挙げられ、例えば、下記式(12)により表されるものがある。具体例としては、ベンジジン等が挙げられる。
【0092】
【化21】
(aは1以上の自然数、アミノ基はベンゼン環同士の結合に対して、メタ位または、パラ位に結合する。)
【0093】
さらに、上記式(12)において、他のベンゼン環との結合に関与せず、ベンゼン環上のアミノ基が置換していない位置に置換基を有するジアミンも用いることができる。これら置換基は、1価の有機基であるがそれらは互いに結合していてもよい。
具体例としては、2,2’−ジメチル−4,4’−ジアミノビフェニル、2,2’−ジトリフルオロメチル−4,4’−ジアミノビフェニル、3,3’−ジクロロ−4,4’−ジアミノビフェニル、3,3’−ジメトキシ−4,4’−ジアミノビフェニル、3,3’−ジメチル−4,4’−ジアミノビフェニル等が挙げられる。
また、最終的に得られるポリイミドを光導波路、光回路部品として用いる場合には、芳香環の置換基としてフッ素を導入すると1μm以上の波長の電磁波に対しての透過率を向上させることができる。
【0094】
一方、ジアミンとして、1,3−ビス(3−アミノプロピル)テトラメチルジシロキサンなどのシロキサン骨格を有するジアミンを用いると、最終的に得られるポリイミドの弾性率が低下し、ガラス転移温度を低下させることができる。
ここで、選択されるジアミンは耐熱性の観点より芳香族ジアミンが好ましいが、目的の物性に応じてジアミンの全体の60モル%、好ましくは40モル%を超えない範囲で、脂肪族ジアミンやシロキサン系ジアミン等の芳香族以外のジアミンを用いても良い。
【0095】
一方、ポリイミド前駆体を合成するには、例えば、アミン成分として4,4’−ジアミノジフェニルエーテルをN−メチルピロリドンなどの有機極性溶媒に溶解させた溶液を冷却しながら、そこへ等モルの3,3’,4,4’−ビフェニルテトラカルボン酸二無水物を徐々に加え撹拌し、ポリイミド前駆体溶液を得ることができる。
このようにして合成されるポリイミド前駆体は、最終的に得られるポリイミドに耐熱性及び寸法安定性を求める場合には、芳香族酸成分及び/又は芳香族アミン成分の共重合割合ができるだけ大きいことが好ましい。具体的には、イミド構造の繰り返し単位を構成する酸成分に占める芳香族酸成分の割合が50モル%以上、特に70モル%以上であることが好ましく、イミド構造の繰り返し単位を構成するアミン成分に占める芳香族アミン成分の割合が40モル%以上、特に60モル%以上であることが好ましく、全芳香族ポリイミドであることが特に好ましい。
【0096】
ポリイミド前駆体は、感光性樹脂組成物とした際の感度を高め、マスクパターンを正確に再現するパターン形状を得るために、5μmの膜厚のときに、露光波長に対して少なくとも5%以上の透過率を示すことが好ましく、15%以上の透過率を示すことが更に好ましい。
露光波長に対してポリイミド前駆体の透過率が高いということは、それだけ、光のロスが少ないということであり、高感度の感光性樹脂組成物を得ることができる。
【0097】
また、一般的な露光光源である高圧水銀灯を用いて露光を行う場合には、少なくとも436nm、405nm、365nmの波長の電磁波のうち1つの波長の電磁波に対する透過率が、厚み5μmのフィルムに成膜した時で好ましくは5%以上、更に好ましくは15%、より更に好ましくは50%以上である。
【0098】
ポリイミド前駆体の重量平均分子量は、その用途にもよるが、3,000〜1,000,000の範囲であることが好ましく、5,000〜500,000の範囲であることがさらに好ましく、10,000〜500,000の範囲であることがさらに好ましい。重量平均分子量が3,000未満であると、塗膜又はフィルムとした場合に十分な強度が得られにくい。また、加熱処理等を施しポリイミドなどの高分子とした際の膜の強度も低くなる。一方、重量平均分子量が1,000,000を超えると粘度が上昇し、溶解性も落ちてくるため、表面が平滑で膜厚が均一な塗膜又はフィルムが得られにくい。
ここで用いている分子量とは、ゲル浸透クロマトグラフィー(GPC)によるポリスチレン換算の値のことをいい、ポリイミド前駆体そのものの分子量でも良いし、無水酢酸等で化学的イミド化処理を行った後のものでも良い。
【0099】
なお、ポリイミド前駆体合成時における溶媒は、極性溶媒が望ましく、代表的なものとして、N−メチル−2−ピロリドン、N−アセチル−2−ピロリドン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N,N−ジエチルホルムアミド、N,N−ジエチルアセトアミド、N,N−ジメチルメトキシアセトアミド、ジメチルスルホキシド、ヘキサメチルフォスホアミド、ピリジン、ジメチルスルホン、テトラメチレンスルホン、ジメチルテトラメチレンスルホン、ジエチレングリコールジメチルエーテル、シクロペンタノン、γ−ブチロラクトン、α−アセチル−γ−ブチロラクトン等があり、これらの溶媒は単独であるいは2種類以上を組み合わせて用いられる。この他にも溶媒として組合せて用いられるものとしてベンゼン、ベンゾニトリル、1,4−ジオキサン、テトラヒドロフラン、ブチロラクトン、キシレン、トルエン、シクロヘキサノン等の非極性溶媒が挙げられ、これらの溶媒は、原料の分散媒、反応調節剤、あるいは生成物からの溶媒の揮散調節剤、皮膜平滑剤などとして使用される。
【0100】
本発明に係る感光性樹脂組成物は、前記光塩基発生剤と、前記ポリイミド前駆体と、溶媒だけの単純な混合物であってもよいが、さらに、増感剤、光又は熱硬化性成分、ポリイミド前駆体以外の非重合性バインダー樹脂、その他の成分を配合して、感光性樹脂組成物を調製してもよい。
感光性樹脂組成物を溶解、分散又は希釈する溶剤としては各種の汎用溶剤を用いることが出来る。また、ポリイミド前駆体としてポリアミック酸を用いる場合には、ポリアミック酸の合成反応により得られた溶液をそのまま用い、そこに必要に応じて他の成分を混合しても良い。
【0101】
前記光塩基発生剤の吸収波長がポリイミド前駆体の吸収波長と重なる部分があり、十分な感度が得られない場合において、感度向上の手段として、増感剤の添加が効果を発揮する場合がある。また、ポリイミド前駆体を透過する電磁波の波長帯に前記光塩基発生剤が吸収波長を有する場合においても、感度向上の手段として、増感剤を添加することができる。ただし、増感剤の添加によるポリイミド前駆体の含有率の減少に伴う、得られるパターンの膜物性、特に膜強度や耐熱性の低下に関して考慮に入れる必要がある。
増感剤と呼ばれる化合物の具体例としては、チオキサントン及び、ジエチルチオキサントンなどのその誘導体、シアニン及び、その誘導体、メロシアニン及び、その誘導体、クマリン系及び、その誘導体、ケトクマリン及び、その誘導体、ケトビスクマリン、及びその誘導体、シクロペンタノン及び、その誘導体、シクロヘキサノン及び、その誘導体、チオピリリウム塩及び、その誘導体、キノリン系及び、その誘導体、スチリルキノリン系及び、その誘導体、チオキサンテン系、キサンテン系及び、その誘導体、オキソノール系及び、その誘導体、ローダミン系及び、その誘導体、ピリリウム塩及び、その誘導体等が挙げられる。
【0102】
シアニン、メロシアニン及び、その誘導体の具体例としては、3,3’−ジカルボキシエチル−2,2’チオシアニンブロミド、1−カルボキシメチル−1’−カルボキシエチル−2,2’−キノシアニンブロミド、1,3’−ジエチル−2,2’−キノチアシアニンヨ−ジド、3−エチル−5−[(3−エチル−2(3H)−ベンゾチアゾリデン)エチリデン]−2−チオキソ−4−オキサゾリジン等が挙げられる。
クマリン、ケトクマリン及び、その誘導体の具体例としては、3−(2’−ベンゾイミダゾール)−7−ジエチルアミノクマリン、3,3’−カルボニルビス(7−ジエチルアミノクマリン)、3,3’−カルボニルビスクマリン、3,3’−カルボニルビス(5,7−ジメトキシクマリン)、3,3’−カルボニルビス(7−アセトキシクマリン)等が挙げられる。
チオキサントン及び、その誘導体の具体例としては、ジエチルチオキサントン、イソプロピルチオキサントンなどが挙げられる。
【0103】
さらに他にはベンゾフェノン、アセトフェノン、アントロン、p,p’−テトラメチルジアミノベンゾフェノン(ミヒラーケトン)、フェナントレン、2−ニトロフルオレン、5−ニトロアセナフテン、ベンゾキノン、N−アセチル−p−ニトロアニリン、p−ニトロアニリン、2−エチルアントラキノン、2−ターシャリーブチルアントラキノン、N−アセチル−4−ニトロ−1−ナフチルアミン、ピクラミド、1,2−ベンズアンスラキノン、3−メチル−1,3−ジアザ−1,9−ベンズアンスロン、p,p’−テトラエチルジアミノベンゾフェノン、2−クロロ−4−ニトロアニリン、ジベンザルアセトン、1,2−ナフトキノン、2,5−ビス−(4’−ジエチルアミノベンザル)−シクロペンタン、2,6−ビス−(4’−ジエチルアミノベンザル)−シクロヘキサノン、2,6−ビス−(4’−ジメチルアミノベンザル)−4−メチル−シクロヘキサノン、2,6−ビス−(4’−ジエチルアミノベンザル)−4−メチル−シクロヘキサノン、4,4’−ビス−(ジメチルアミノ)−カルコン、4,4’−ビス−(ジエチルアミノ)−カルコン、p−ジメチルアミノベンジリデンインダノン、1,3−ビス−(4’−ジメチルアミノベンザル)−アセトン、1,3−ビス−(4’−ジエチルアミノベンザル)−アセトン、N−フェニル−ジエタノールアミン、N−p−トリル−ジエチルアミン、などが挙げられる。
本発明ではこれらの増感剤を1種または2種以上使用することができる。
【0104】
また、組成物に使用される溶剤としては、例えば、ジエチルエーテル、テトラヒドロフラン、ジオキサン、エチレングリコールジメチルエーテル、エチレングリコールジエチルエーテル、プロピレングリコールジメチルエーテル、プロピレングリコールジエチルエーテル等のエーテル類;エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、プロピレングリコールモノメチルエーテル、プロピレングリコールモノエチルエーテル、ジエチレングリコールモノメチルエーテル、ジエチレングリコールモノエチルエーテル等のグリコールモノエーテル類(いわゆるセロソルブ類);メチルエチルケトン、アセトン、メチルイソブチルケトン、シクロペンタノン、シクロヘキサノンなどのケトン類;酢酸エチル、酢酸ブチル、酢酸n−プロピル、酢酸i−プロピル、酢酸n−ブチル、酢酸i−ブチル、前記グリコールモノエーテル類の酢酸エステル(例えば、メチルセロソルブアセテート、エチルセロソルブアセテート)、メトキシプロピルアセテート、エトキシプロピルアセテート、蓚酸ジメチル、乳酸メチル、乳酸エチル等のエステル類;エタノール、プロパノール、ブタノール、ヘキサノール、シクロヘキサノール、エチレングリコール、ジエチレングリコール、グリセリン等のアルコール類;塩化メチレン、1,1−ジクロロエタン、1,2−ジクロロエチレン、1−クロロプロパン、1−クロロブタン、1−クロロペンタン、クロロベンゼン、ブロムベンゼン、o−ジクロロベンゼン、m−ジクロロベンゼン等のハロゲン化炭化水素類;N,N−ジメチルホルムアミド、N,N−ジエチルホルムアミド、N,N−ジメチルアセトアミド、N,N−ジエチルアセトアミド等のアミド類;N−メチルピロリドンなどのピロリドン類;γ−ブチロラクトン等のラクトン類;ジメチルスルホキシドなどのスルホキシド類、その他の有機極性溶媒類等が挙げられ、更には、ベンゼン、トルエン、キシレン等の芳香族炭化水素類、及び、その他の有機非極性溶媒類等も挙げられる。これらの溶媒は単独若しくは組み合わせて用いられる。
中でも、N−メチル−2−ピロリドン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N,N−ジエチルホルムアミド、N,N−ジエチルアセトアミド、N,N−ジメチルメトキシアセトアミド、ジメチルスルホキシド、ヘキサメチルフォスホアミド、N−アセチル−2−ピロリドン、ピリジン、ジメチルスルホン、テトラメチレンスルホン、ジメチルテトラメチレンスルホン、ジエチレングリコールジメチルエーテル、シクロペンタノン、γ−ブチロラクトン、α−アセチル−γ−ブチロラクトン等の極性溶媒が好適なものとして挙げられる。
【0105】
光硬化性成分としては、エチレン性不飽和結合を1つ又は2つ以上有する化合物を用いることができ、例えば、アミド系モノマー、(メタ)アクリレートモノマー、ウレタン(メタ)アクリレートオリゴマー、ポリエステル(メタ)アクリレートオリゴマー、エポキシ(メタ)アクリレート、及びヒドロキシル基含有(メタ)アクリレート、スチレン等の芳香族ビニル化合物を挙げることができる。また、ポリイミド前駆体が、ポリアミック酸等のカルボン酸成分を構造内に有する場合には、3級アミノ基を有するエチレン性不飽和結合含有化合物を用いると、ポリイミド前駆体のカルボン酸とイオン結合を形成し、感光性樹脂組成物としたときの露光部、未露光部の溶解速度のコントラストが大きくなる。
【0106】
このようなエチレン性不飽和結合を有する光硬化性化合物を用いる場合には、さらに光ラジカル発生剤を添加してもよい。光ラジカル発生剤としては、例えば、ベンゾイン、ベンゾインメチルエーテル、ベンゾインエチルエーテル及びベンゾインイソプロピルエーテル等のベンゾインとそのアルキルエーテル;アセトフェノン、2,2−ジメトキシ−2−フェニルアセトフェノン、2,2−ジエトキシ−2−フェニルアセトフェノン、1,1−ジクロロアセトフェノン、1−ヒドロキシアセトフェノン、1−ヒドロキシシクロヘキシルフェニルケトン及び2−メチル−1−[4−(メチルチオ)フェニル]−2−モルフォリノ−プロパン−1−オン等のアセトフェノン;2−メチルアントラキノン、2−エチルアントラキノン、2−ターシャリ-ブチルアントラキノン、1−クロロアントラキノン及び2−アミルアントラキノン等のアントラキノン;2,4−ジメチルチオキサントン、2,4−ジエチルチオキサントン、2−クロロチオキサントン及び2,4−ジイソピルチオキサントン等のチオキサントン;アセトフェノンジメチルケタール及びベンジルジメチルケタール等のケタール;2,4,6−トリメチルベンゾイルジフェニルホスフィンオキシド等のモノアシルホスフィンオキシドあるいはビスアシルホスフィンオキシド類;ベンゾフェノン等のベンゾフェノン類;並びにキサントン類等が挙げられる。
【0107】
本発明に係る樹脂組成物に加工特性や各種機能性を付与するために、その他に様々な有機又は無機の低分子又は高分子化合物を配合してもよい。例えば、染料、界面活性剤、レベリング剤、可塑剤、微粒子等を用いることができる。微粒子には、ポリスチレン、ポリテトラフルオロエチレン等の有機微粒子、コロイダルシリカ、カーボン、層状珪酸塩等の無機微粒子等が含まれ、それらは多孔質や中空構造であってもよい。また、その機能又は形態としては顔料、フィラー、繊維等がある。
【0108】
本発明に係る感光性樹脂組成物において、前記ポリイミド前駆体(固形分)は、得られるパターンの膜物性、特に膜強度や耐熱性の点から、感光性樹脂組成物の固形分全体に対し、30重量%以上、50重量%以上含有することが好ましい。また、前記式(1)で表される光塩基発生剤は、感光性樹脂組成物に含まれるポリイミド前駆体の固形分100重量部に対し、通常、0.01〜50重量部、好ましくは0.1〜30重量部の範囲内で含有させることが好ましい。0.01重量部未満であると環化反応促進効果が不十分となる傾向があり、50重量部を超えると最終的に得られる樹脂硬化物に求められる諸物性を満たしにくくなる。
また、上記増感剤の配合量はポリイミド前駆体の固形分100重量部に対して50重量部未満とすることが好ましく、30重量部未満とすることがより好ましい。また、最終的に得られる樹脂硬化物に求められる諸物性の低下を防ぐため、前記本発明に係る式(1)で表される光塩基発生剤と増感剤の合計がポリイミド前駆体100重量部に対して50重量部以下であることが望ましい。
また、その他の任意成分の配合割合は、感光性樹脂組成物の固形分全体に対し、0.1重量%〜20重量%の範囲が好ましい。0.1重量%未満だと、添加物を添加した効果が発揮されにくく、20重量%を超えると、最終的に得られる樹脂硬化物の特性が最終生成物に反映されにくい。なお、感光性樹脂組成物の固形分とは溶剤以外の全成分であり、液状のモノマー成分も固形分に含まれる。
【0109】
本発明に係る感光性樹脂組成物は、さまざまなコーティングプロセスや成形プロセスに用いられて、フィルムや3次元的形状の成形物を作製することができる。
【0110】
本発明の感光性樹脂組成物より得られるポリイミドは、耐熱性、寸法安定性、絶縁性等の本来の特性も損なわれておらず、良好である。
例えば、本発明の感光性樹脂組成物から得られるポリイミドの窒素中で測定した5%重量減少温度は、250℃以上が好ましく、300℃以上がさらに好ましい。特に、はんだリフローの工程を通るような電子部品等の用途に用いる場合は、5%重量減少温度が300℃以下であると、はんだリフローの工程で発生した分解ガスにより気泡等の不具合が発生する恐れがある。
ここで、5%重量減少温度とは、熱重量分析装置を用いて重量減少を測定した時に、サンプルの重量が初期重量から5%減少した時点(換言すればサンプル重量が初期の95%となった時点)の温度である。同様に10%重量減少温度とはサンプル重量が初期重量から10%減少した時点の温度である。
【0111】
本発明の感光性樹脂組成物から得られるポリイミドのガラス転移温度は、耐熱性の観点からは高ければ高いほど良いが、光導波路のように熱成形プロセスが考えられる用途においては、120℃〜450℃程度のガラス転移温度を示すことが好ましく、200℃〜400℃程度のガラス転移温度を示すことがさらに好ましい。ここで本発明におけるガラス転移温度は、感光性樹脂組成物から得られるポリイミドをフィルム形状にすることが出来る場合には、動的粘弾性測定によって、tanδ(tanδ=損失弾性率(E’’)/貯蔵弾性率(E’))のピーク温度から求められる。動的粘弾性測定としては、例えば、粘弾性測定装置Solid Analyzer RSA II(Rheometric Scientific社製)によって、周波数3Hz、昇温速度5℃/minにより行うことができる。感光性樹脂組成物から得られるポリイミドをフィルム形状にできない場合には、示差熱分析装置(DSC)のベースラインの変曲点の温度で判断する。
【0112】
本発明の感光性樹脂組成物から得られるポリイミドの寸法安定性の観点から、線熱膨張係数は60ppm以下が好ましく、40ppm以下がさらに好ましい。半導体素子等の製造プロセスにおいてシリコンウェハ上に膜を形成する場合には、密着性、基板のそりの観点から20ppm以下がさらに好ましい。ここで、本発明における線熱膨張係数とは、本発明で得られる感光性樹脂組成物から得られるポリイミドのフィルムの熱機械的分析装置(TMA)によって求めることができる。熱機械的分析装置(例えばThermo Plus TMA8310(リガク社製))によって、昇温速度を10℃/min、評価サンプルの断面積当たりの加重が同じになるように引張り加重を1g/25000μm2として得られる。
【0113】
以上に述べたように、本発明に係る感光性ポリイミド樹脂組成物は、上記式(1)で表される化合物が、高感度の光塩基発生剤として機能することで、多種多様なポリイミド前駆体を適用することができ、最終的に得られるポリイミドの構造を広範囲から選択することができる。
また、本発明によれば、ポリイミド前駆体に前記本発明に係る式(1)で表される光塩基発生剤を混合するだけという簡便な手法で感光性ポリイミド樹脂組成物を得ることができることから、コストパフォーマンスにも優れる。
さらには、電磁波の照射により発生したアミンの触媒効果により、イミド化等の最終生成物への反応に要する処理温度を低減できる為、プロセスへの不可や製品への熱によるダメージを低減することが可能である。
【0114】
本発明に係る感光性樹脂組成物は、印刷インキ、接着剤、充填剤、電子材料、光回路部品、成形材料、レジスト材料、建築材料、3次元造形、光学部材等、樹脂材料が用いられる公知の全ての分野・製品に利用できる。
【0115】
本発明に係る感光性樹脂組成物は、耐熱性、寸法安定性、絶縁性等の特性が有効とされる広範な分野・製品、例えば、塗料又は印刷インキ、或いは、カラーフィルター、フレキシブルディスプレー用フィルム、半導体装置、電子部品、層間絶縁膜、配線被覆膜、光回路、光回路部品、反射防止膜、ホログラム、光学部材又は建築材料の形成材料として好適に用いられる。例えば具体的には、半導体装置用バッファーコート膜、多層配線板の層間絶縁膜等が挙げられる。
【0116】
特に、本発明の感光性樹脂組成物は、主にパターン形成材料(レジスト)として用いられ、それによって形成されたパターンはポリイミドからなる永久膜として耐熱性や絶縁性を付与する成分として機能するため、例えば、カラーフィルター、フレキシブルディスプレー用フィルム、電子部品、半導体装置、層間絶縁膜、配線被覆膜、光回路、光回路部品、反射防止膜、その他の光学部材又は電子部材を形成するのに適している。
【0117】
また、本発明においては、本発明に係る感光性樹脂組成物又はその熱硬化物により少なくとも一部分が形成されている、印刷物、カラーフィルター、フレキシブルディスプレー用フィルム、半導体装置、電子部品、層間絶縁膜、配線被覆膜、光回路、光回路部品、反射防止膜、ホログラム、光学部材又は建築材料いずれかの物品が提供される。
【0118】
次に、本発明に係るネガ型パターン形成方法を説明する。
本発明に係るネガ型パターン形成方法は、前記本発明に係る感光性樹脂組成物からなる塗膜又は成形体の表面に、所定のパターン状に電磁波を照射し、必要に応じて熱処理等の後処理を行って、前記塗膜又は成形体の電磁波照射部位の溶解性を選択的に低下させた後、現像することを特徴とする。
本発明に係る感光性樹脂組成物を何らかの支持体上に塗布し、所定のパターン状に電磁波を照射すると、露光部においてのみ、前記光塩基性物質が分解して塩基性物質を生成する。塩基性物質は、露光部のポリイミド前駆体の最終生成物への反応を促進する触媒として作用する。
【0119】
塩基性物質により、露光部のポリイミド前駆体が直接的に最終生成物へ反応して、露光部のポリイミド前駆体のみ、ある溶媒に対する溶解性が選択的に低下される場合には、露光後に特に後処理なく、当該露光部の溶解性が低下した溶媒を現像液として用いて、溶解性が低下していない未露光部のみを溶解することにより、現像することが可能になる。
【0120】
本発明の感光性樹脂組成物は、N−メチル−2−ピロリドン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N,N−ジエチルホルムアミド、N,N−ジエチルアセトアミド、N,N−ジメチルメトキシアセトアミド、ジメチルスルホキシド、ヘキサメチルフォスホアミド、N−アセチル−2−ピロリドン、ピリジン、ジメチルスルホン、テトラメチレンスルホン、ジメチルテトラメチレンスルホン、ジエチレングリコールジメチルエーテル、シクロペンタノン、γ−ブチロラクトン、α−アセチル−γ−ブチロラクトン等の極性溶媒に溶解後、浸漬法、スプレー法、スクリーン印刷法、スピンコート法などによって、シリコンウエハ、金属基板、セラミック基板などの基材表面に塗布し、加熱して溶剤の大部分を除くことにより、基材表面に粘着性のない塗膜を与えることができる。塗膜の厚みには特に制限はないが、0.5〜50μmであることが好ましく、感度および現像速度面から1.0〜20μmであることがより望ましい。塗布した塗膜の乾燥条件としては、例えば、80〜100℃、1分〜20分が挙げられる。
【0121】
この塗膜に、所定のパターンを有するマスクを通して、電磁波を照射しパターン状に露光後を行い、加熱後、膜の未露光部分を、適切な現像液で現像して除去することにより、所望のパターン化された膜を得ることができる。
【0122】
露光工程に用いられる露光方法や露光装置は特に限定されることなく、密着露光でも間接露光でも良くg線ステッパ、i線ステッパ、超高圧水銀灯を用いるコンタクト/プロキシミティ露光機、ミラープロジェクション露光機、又はその他の紫外線、可視光線、X線、電子線などを照射可能な投影機や線源を使用することができる。
【0123】
本発明に係るネガ型パターン形成方法においては、露光工程と現像工程の間に、必要に応じて熱処理などの後処理を行っても良い。ここでの後処理は、前記塗膜又は成形体の電磁波照射部位の、ある溶媒に対する溶解性を選択的に低下させるための処理である。
熱処理等の後処理は、例えば、塩基性物質と共存する露光部のポリイミド前駆体に対してのみ、最終生成物へ反応させる処理とする。従って、熱処理をする場合には、例えば、塩基性物質が存在する露光部と、塩基性物質が存在しない未露光部とで、ポリイミド前駆体の環化率が異なるようになる温度で行うことが好ましい。
【0124】
たとえばポリアミック酸をイミド化する場合、この段階での熱処理の好ましい温度範囲は、通常60℃〜200℃程度である。熱処理温度が60℃より低いと、イミド化の効率が悪く、現実的なプロセス条件で露光部、未露光部のイミド化率の差を創出することが難しくなる。一方、熱処理温度が200℃以上であると、電磁波の吸収に伴う分子内解裂反応により、塩基性物質を生成する中性の化合物が熱分解したり、アミンが存在していない未露光部でもイミド化が進行したりして、露光部と未露光部の溶解性の差が出にくい。
具体的には、例えば、120〜200℃で、1分〜20分加熱を行う。
この熱処理は、公知の方法であればどの方法でもよく、具体的に例示すると、空気、又は窒素雰囲気下の循環オーブン、ホットプレートによる加熱などが挙げられるが、特に限定されない。
【0125】
現像工程に用いられる現像液としては、特に限定されず、塩基性水溶液、有機溶剤など、用いられるポリイミド前駆体に合わせて適宜選択することが可能である。
塩基性水溶液としては、特に限定されないが、例えば、濃度が、0.01重量%〜10重量%、好ましくは、0.05重量%〜5重量%のテトラメチルアンモニウムヒドロキシド(TMAH)水溶液の他、ジエタノールアミン、ジエチルアミノエタノール、水酸化ナトリウム、水酸化カリウム、炭酸ナトリウム、炭酸カリウム、炭酸水素ナトリウム、炭酸水素カリウム、トリエチルアミン、ジエチルアミン、メチルアミン、ジメチルアミン、酢酸ジメチルアミノエチル、ジメチルアミノエタノール、ジメチルアミノエチルメタクリレート、シクロヘキシルアミン、エチレンジアミン、ヘキサメチレンジアミン、テトラメチルアンモニウムなどの水溶液等が挙げられる。
溶質は、1種類でも2種類以上でも良く、全体の重量の50%以上、さらに好ましくは70%以上、水が含まれていれば有機溶媒等を含んでいても良い。
また、有機溶剤としては、特に限定されないが、N−メチル−2−ピロリドン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、ジメチルスルホキシド、γ−ブチロラクロン、ジメチルアクリルアミドなどの極性溶媒、メタノール、エタノール、イソプロパノールなどのアルコール類、酢酸エチル、プロピレングリコールモノメチルエーテルアセテートなどのエステル類、シクロペンタノン、シクロヘキサノン、イソブチルケトン、メチルイソブチルケトンなどのケトン類などを、単独であるいは2種類以上を組み合わせて添加してもよい。現像後は水にて洗浄を行う。この場合においてもエタノール、イソプロピルアルコールなどのアルコール類、乳酸エチル、プロピレングリコールモノメチルエーテルアセテートなどのエステル類などを水に加えても良い。
【0126】
現像後は必要に応じて水または貧溶媒でリンスを行い、80〜100℃で乾燥しパターンを安定なものとする。このレリーフパターンを、耐熱性のあるものとするために180〜500℃、好ましくは200〜350℃の温度で数十分から数時間加熱することにより、完全にイミド化を進行させ、パターン化された高耐熱性樹脂層が形成される。
【実施例】
【0127】
[合成例1:光塩基発生剤1の合成]
(1)アントラセン−9−イル−メチル−4’−ニトロフェニルカルボナートの合成(第一工程)
9−アントラセンメタノール5.0g(24mmol;和光純薬工業(株)製)を脱水ジメチルアセトアミド(脱水DMAc)250mLに溶解させた溶液に、トリエチルアミン7.3g(72mmol)を加えた。その溶液に、クロロギ酸−4−ニトロフェニル4.9g(24mmol;和光純薬工業(株)製)を添加した後、室温で24時間攪拌して反応させた。反応終了後、反応液に氷水を投入し、この混合液をジクロロメタンで抽出し、さらに抽出後の有機層を水で洗浄後、当該有機層を濃縮した。次いで、濃縮残渣に水を投入し、そこで生じた結晶を濾取後、得られた結晶を乾燥することにより、黄色結晶のアントラセン−9−イル−メチル−4’−ニトロフェニルカルボナート4.8g(収率:53%)を得た。以下に1H−NMRの測定結果を示す。
1H−NMR(400MHz,CDCl3)δ(ppm):6.39(2H,s,OCH2),7.36(2H,d,J=9.3Hz,ArH),7.51−7.54(2H,m,ArH),7.60−7.65(2H,m,ArH),8.06(2H,d,J=8.7Hz,ArH),8.24(2H,d,J=9.3Hz,ArH),8.41(2H,d,J=8.7Hz,ArH),8.57(1H,s,ArH)
【0128】
(2)アントラセン−9−イル−メトキシカルボニルピペリジンの合成(第二工程)
上記(1)で得られたアントラセン−9−イル−メチル−4’−ニトロフェニルカルボナート4.8g(13mmol)をジクロロメタン100mLに溶解させた溶液に、ピペリジン1.4g(16mmol)を加えた後、室温で1時間攪拌して反応させた。反応終了後、反応液を水で洗浄し、洗浄後の有機層を濃縮した。得られた濃縮残渣をカラムクロマトグラフィー(充填剤:シリカゲル(ワコーゲルC−200:和光純薬工業(株)製)、展開溶媒:ジクロロメタン)で精製することにより、下記式(5)で表されるアントラセン−9−イル−メトキシカルボニルピペリジン1.3g(収率:32%、淡黄色結晶)を得た。以下に1H−NMR及び融点の測定結果を示す。
1H−NMR(400MHz,CDCl3)δ(ppm):1.39(2H,br,CH2),1.53(4H,br,2×CH2),3.28(2H,br,NCH2),3.46(4H,br,NCH2),6.15(2H,s,OCH2),7.49−7.59(4H,m,ArH),8.03(2H,d,J=8.8Hz,ArH),8.41(2H,d,J=8.8Hz,ArH),8.50(1H,s,ArH)
融点:130−132℃
【0129】
【化22】
【0130】
[合成例2:光塩基発生剤2の合成]
(1)アントラキノン−2−イル−メチル−4’−ニトロフェニルカルボナートの合成(第一工程)
2−ヒドロキシメチルアントラキノン3.0g(12mmol;東京化成工業(株)製)を脱水ジメチルアセトアミド(脱水DMAc)100mLに溶解させた溶液に、トリエチルアミン3.8g(37mmol)を加えた。その溶液に、クロロギ酸−4−ニトロフェニル2.5g(12mmol;和光純薬工業(株)製)を添加した後、室温で24時間攪拌して反応させた。反応終了後、反応液に氷水を投入し、そこで生じた結晶を濾取後、得られた結晶を乾燥することにより、淡黄色結晶のアントラキノン−2−イル−メチル−4’−ニトロフェニルカルボナート2.4g(収率:47%)を得た。以下に1H−NMRの測定結果を示す。
1H−NMR(400MHz,CDCl3)δ(ppm):5.47(2H,s,OCH2),7.42(2H,d,J=9.3Hz,ArH),7.82−7.85(3H,m,ArH),8.29−8.39(4H,m,ArH),8.30(2H,d,J=9.3Hz,ArH)
【0131】
(2)アントラキノン−2−イル−メトキシカルボニルピペリジンの合成(第二工程)
上記(1)で得られたアントラキノン−2−イル−メチル−4’−ニトロフェニルカルボナート2.4g(5.8mmol)を脱水ジメチルアセトアミド(脱水DMAc)450mLに溶解させた溶液に、ピペリジン0.60g(7.0mmol)を加えた後、室温で1時間攪拌して反応させた。反応終了後、反応液に氷水を投入し、そこで生じた結晶を濾取後、得られた結晶を乾燥することにより、下記式(6)で表されるアントラキノン−2−イル−メトキシカルボニルピペリジン1.1g(収率:55%、淡黄色結晶)を得た。以下に1H−NMR及び融点の測定結果を示す。
1H−NMR(400MHz,CDCl3)δ(ppm):1.55−1.63(6H,m,3×CH2),3.49(4H,br,2×NCH2),5.29(2H,s,OCH2),7.76−7.83(3H,m,ArH),8.27−8.34(4H,m,ArH)
融点:146−147℃
【0132】
【化23】
【0133】
[合成例3:光塩基発生剤3の合成]
(1)ピレン−1−イル−メチル−4’−ニトロフェニルカルボナートの合成(第一工程)
1−ピレンメタノール5.0g(22mmol;東京化成工業(株)製)を脱水ジメチルアセトアミド(脱水DMAc)200mLに溶解させた溶液に、トリエチルアミン6.5g(65mmol)を加えた。その溶液に、クロロギ酸−4−ニトロフェニル4.3g(22mmol;和光純薬工業(株)製)を添加した後、室温で24時間攪拌して反応させた。反応終了後、反応液に氷水を投入し、この混合液をジクロロメタンで抽出し、さらに抽出後の有機層を水で洗浄後、当該有機層を濃縮した。次いで、濃縮残渣にトルエンを投入し、そこで生じた結晶を濾取後、得られた結晶を乾燥することにより、黄色結晶のピレン−1−イル−メチル−4’−ニトロフェニルカルボナート5.9g(収率:68%)を得た。以下に1H−NMRの測定結果を示す。
1H−NMR(400MHz,CDCl3)δ(ppm):6.02(2H,s,OCH2),7.25(2H,d,J=9.3Hz,ArH),7.98−8.20(9H,m,ArH),8.27(2H,d,J=9.3Hz,ArH)
【0134】
(2)ピレン−1−イル−メトキシカルボニルピペリジンの合成(第二工程)
上記(1)で得られたピレン−1−イル−メチル−4’−ニトロフェニルカルボナート5.9g(15mmol)を脱水ジメチルアセトアミド(脱水DMAc)50mLに溶解させた溶液に、ピペリジン1.4g(16mmol)を加えた後、室温で2時間攪拌して反応させた。反応終了後、反応液に氷水を投入し、この混合液をジクロロメタンで抽出し、さらに抽出後の有機層を水で洗浄後、当該有機層を濃縮した。得られた濃縮残渣をカラムクロマトグラフィー(充填剤:シリカゲル(ワコーゲルC−200:和光純薬工業(株)製)、展開溶媒:ジクロロメタン)で精製することにより、下記式(7)で表されるピレン−1−イル−メトキシカルボニルピペリジン1.6g(収率:32%、橙色結晶)を得た。以下に1H−NMR及び融点の測定結果を示す。
1H−NMR(400MHz,CDCl3)δ(ppm):1.54(6H,br,3×CH2),3.43(4H,br,2×NCH2),5.83(2H,s,OCH2),7.99−8.34(9H,m,ArH)
融点:118−120℃
【0135】
【化24】
【0136】
[合成例4:光塩基発生剤4の合成]
50%水素化ナトリウム1.6g(33mmol)及び脱水テトラヒドロフラン(脱水THF)4mLを仕込んだ溶液に、9−アントラセンメタノール6.3g(30mmol;和光純薬工業(株)製)を脱水テトラヒドロフラン(脱水THF)26mLに溶解させた溶液を滴下した。次いで、その溶液に、N,N−ジエチルカルバモイルクロリド4.5g(33mmol;シグマアルドリッチジャパン(株)製)の脱水テトラヒドロフラン(脱水THF)溶液4mLを添加した後、60℃で2時間攪拌して反応させた。反応終了後、反応液を冷却し、冷却した溶液にn−ヘキサンを加え、さらにこの溶液を水で洗浄し、洗浄後の有機層を濃縮した。得られた濃縮残渣をカラムクロマトグラフィー(充填剤:シリカゲル(ワコーゲルC−200:和光純薬工業(株)製)、展開溶媒:n−ヘプタン)で精製することにより、下記式(4)で表されるアントラセン−9−イル−メトキシカルボニル−N,N−ジエチルアミン5.7g(収率:61%、黄色結晶)を得た。以下に1H−NMR及び融点の測定結果を示す。
1H−NMR(400MHz,CDCl3)δ(ppm):0.91(3H,br,CH3),1.14(3H,br,CH3),3.14(2H,br,NCH2),3.35(2H,br,NCH2),6.14(2H,s,OCH2),7.48(2H,dd,J=8.0,6.8Hz,ArH),7.57(2H,dd,J=8.4,6.8Hz,ArH),8.04(2H,d,J=8.0Hz,ArH),8.46(2H,d,J=8.4Hz,ArH),8.52(1H,s,ArH)
融点:72−74℃
【0137】
【化25】
【0138】
[合成例5:ポリイミド前駆体1の合成]
窒素置換した500mL4つ口セパラブルフラスコに、4,4’−ジアミノジフェニルエーテル20.0g(100mmol)および脱水N−メチルピロリドン200mLを入れ、氷浴下で撹拌して溶解させた。この溶液に3,3’,4,4’−ビフェニルテトラカルボン酸二無水物29.4g(100mmol)を加え、氷浴下で2時間攪拌した。反応溶液をアセトンにより再沈殿し、濾取して得られた沈殿物を室温で8時間減圧乾燥することにより、ポリアミド酸(ポリイミド前駆体1)を白色固体として定量的に得た。
【0139】
<試験>
(1)モル吸光係数の測定
合成例1〜4で得られた光塩基発生剤1〜4をそれぞれ、電子天秤を用いて秤量し、メスフラスコを用いることにより、濃度5×10−5mol/Lのアセトニトリル溶液を調製した。この溶液を石英セル((株)東新理興製、TOS−UV−10(1cm×1cm×4cm))に入れ、分光光度計(島津製作所社製UV−2550)により190〜800nmの波長範囲での紫外−可視吸収スペクトルを測定した。スペクトルで得られた吸光度から、下式によりモル吸光係数ε(365、405、436nm)を測定した。結果を表1に示す。
モル吸光係数(ε)=(吸光度)/モル濃度(mol/L)/光路長(cm)
【0140】
【表1】
【0141】
(2)光分解能の測定
光塩基発生剤について、石英製NMRチューブ中に電子天秤を用いて1.0mg秤量し、重アセトニトリル0.5mLを加え溶解させた。このサンプルに、350nm以下の波長を透過しないフィルター1を介して高圧水銀灯(ウシオ電機社製SPOT CURE SP−III 250UA、ランプ型番:USH−255BY)の全波長をフィルター通過前100J/cm2(i線換算:紫外線照度計:ウシオ電機社製UIT−150、受光器:UVD−S365)、フィルター通過後18.2J/cm2(i線換算:紫外線照度計:ウシオ電機社製UIT−150、受光器:UVD−S365)により光を照射し、照射前後のNMRスペクトルの比較を行うことにより、i(365nm)線以上の波長領域における光分解性の評価を行った。図1にフィルター1の透過率曲線を示す。光分解性の評価結果を表2に示す。
【0142】
【表2】
【0143】
(3)熱安定性の測定
光塩基発生剤について、DTG−60(島津製作所製)を用いて30℃から600℃まで昇温速度10℃/minでTG−DTA測定を行った。5%重量減少温度を算出し、耐熱性の評価を行った。耐熱性の評価結果を表3に示す。
【0144】
【表3】
【0145】
(実施例1)
上記光塩基発生剤1を0.2g、上記ポリイミド前駆体1を1g、N−メチルピロリドン9gに溶解させ、本発明の感光性樹脂組成物(感光性樹脂組成物1)を得た。
【0146】
(実施例2〜4)
実施例1における光塩基発生剤1を、それぞれ光塩基発生剤2〜4に代えた以外は、実施例1と同様にして、感光性樹脂組成物(感光性樹脂組成物2〜4)を得た。
【0147】
[評価:パターン形成]
感光性樹脂組成物1を、ガラス板上に乾燥後膜厚2μmになるようにスピンコートし、100℃のホットプレート上で15分間乾燥させた。そこへ、手動露光装置(大日本科研製、MA−1100)でi線換算で、2000mJ/cm2の紫外−可視光線照射を行い、その後、155℃のホットプレート上で10分間加熱したのち、テトラメチルアンモニウムハイドロオキサイド 2.38%溶液にイソプロパノールを10wt%添加した溶液に浸漬した。その結果、露光部が現像液に溶解せず残存したパターンを得ることができた。さらに、それらのサンプルを300℃で1時間加熱しイミド化を行った。
この結果から、本発明の感光性樹脂組成物は、良好なパターンを形成することできることが明らかとなった。
【図面の簡単な説明】
【0148】
【図1】図1は、フィルター1の透過率曲線を示した図である。
【特許請求の範囲】
【請求項1】
一般式(1)で表される光塩基発生剤、及びポリイミド前駆体を含有する、感光性樹脂組成物。
【化1】
(式(1)中、Arは、下記一般式(I)で示されるアントラセニル基、下記一般式(II)で示されるアントラキノリル基、及び下記一般式(III)で示されるピレニル基からなる群より選ばれる何れかの基を表し、
R1及びR2は夫々独立して、水素原子、炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基を表し、R1及びR2の少なくとも1つは水素原子ではなく、R1及びR2はこれらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成していても良く、
R3及びR4は夫々独立して、水素原子又は炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基を表す。)
【化2】
(式(I)中、R5〜R13は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【化3】
(式(II)中、R14〜R20は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【化4】
(式(III)中、R21〜R29は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【請求項2】
前記一般式(1)で表される光塩基発生剤におけるR1及びR2が、炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基である、請求項1に記載の感光性樹脂組成物。
【請求項3】
前記一般式(1)で表される光塩基発生剤におけるR1及びR2が、これらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成しているものである、請求項1に記載の感光性樹脂組成物。
【請求項4】
前記ポリイミド前駆体は、それ自体が塩基性物質の作用によって最終生成物への反応が促進されるものである、請求項1乃至3のいずれかに記載の感光性樹脂組成物。
【請求項5】
前記ポリイミド前駆体は、それ自体が塩基性物質の作用によって最終生成物への反応が促進され、且つ、加熱により溶解性が変化するものである、請求項1乃至4のいずれかに記載の感光性樹脂組成物。
【請求項6】
更に、増感剤を含有する請求項1乃至5のいずれかに記載の感光性樹脂組成物。
【請求項7】
前記ポリイミド前駆体がポリアミック酸である、請求項1乃至6のいずれかに記載の感光性樹脂組成物。
【請求項8】
前記光塩基発生剤が365nm以上の波長の電磁波に対して光分解性を有する、請求項1乃至7のいずれかに記載の感光性樹脂組成物。
【請求項9】
前記光塩基発生剤の5%重量減少温度が170℃以上である、請求項1乃至8のいずれかに記載の感光性樹脂組成物。
【請求項10】
塗料又は印刷インキ、或いは、カラーフィルター、フレキシブルディスプレー用フィルム、半導体装置、電子部品、層間絶縁膜、配線被覆膜、光回路、光回路部品、反射防止膜、ホログラム、光学部材又は建築材料の形成材料として用いられる、請求項1乃至9のいずれかに記載の感光性樹脂組成物。
【請求項11】
前記請求項1乃至10のいずれかに記載の感光性樹脂組成物又はその硬化物により少なくとも一部分が形成されている、印刷物、カラーフィルター、フレキシブルディスプレー用フィルム、半導体装置、電子部品、層間絶縁膜、配線被覆膜、光回路、光回路部品、反射防止膜、ホログラム、光学部材又は建築材料いずれかの物品。
【請求項12】
前記請求項1乃至10のいずれかに記載の感光性樹脂組成物からなる塗膜又は成形体の表面に、所定のパターン状に電磁波を照射し、前記塗膜又は成形体の電磁波照射部位の溶解性を選択的に低下させた後、現像する、ネガ型パターン形成方法。
【請求項13】
電磁波を照射後、加熱処理を行って、前記塗膜又は成形体の電磁波照射部位の溶解性を選択的に低下させる、請求項12に記載のネガ型パターン形成方法。
【請求項1】
一般式(1)で表される光塩基発生剤、及びポリイミド前駆体を含有する、感光性樹脂組成物。
【化1】
(式(1)中、Arは、下記一般式(I)で示されるアントラセニル基、下記一般式(II)で示されるアントラキノリル基、及び下記一般式(III)で示されるピレニル基からなる群より選ばれる何れかの基を表し、
R1及びR2は夫々独立して、水素原子、炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基を表し、R1及びR2の少なくとも1つは水素原子ではなく、R1及びR2はこれらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成していても良く、
R3及びR4は夫々独立して、水素原子又は炭素数1〜10の直鎖状、分枝状若しくは環状の飽和アルキル基を表す。)
【化2】
(式(I)中、R5〜R13は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【化3】
(式(II)中、R14〜R20は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【化4】
(式(III)中、R21〜R29は夫々独立して、水素原子、ハロゲン原子又は炭素数1〜10の直鎖状若しくは分枝状の飽和アルキル基を表す。)
【請求項2】
前記一般式(1)で表される光塩基発生剤におけるR1及びR2が、炭素数1〜20の置換基を有していても良い飽和又は不飽和アルキル基である、請求項1に記載の感光性樹脂組成物。
【請求項3】
前記一般式(1)で表される光塩基発生剤におけるR1及びR2が、これらが結合している窒素原子と共に、置換基を有していても良い炭素数3〜8の含窒素脂肪族環を形成しているものである、請求項1に記載の感光性樹脂組成物。
【請求項4】
前記ポリイミド前駆体は、それ自体が塩基性物質の作用によって最終生成物への反応が促進されるものである、請求項1乃至3のいずれかに記載の感光性樹脂組成物。
【請求項5】
前記ポリイミド前駆体は、それ自体が塩基性物質の作用によって最終生成物への反応が促進され、且つ、加熱により溶解性が変化するものである、請求項1乃至4のいずれかに記載の感光性樹脂組成物。
【請求項6】
更に、増感剤を含有する請求項1乃至5のいずれかに記載の感光性樹脂組成物。
【請求項7】
前記ポリイミド前駆体がポリアミック酸である、請求項1乃至6のいずれかに記載の感光性樹脂組成物。
【請求項8】
前記光塩基発生剤が365nm以上の波長の電磁波に対して光分解性を有する、請求項1乃至7のいずれかに記載の感光性樹脂組成物。
【請求項9】
前記光塩基発生剤の5%重量減少温度が170℃以上である、請求項1乃至8のいずれかに記載の感光性樹脂組成物。
【請求項10】
塗料又は印刷インキ、或いは、カラーフィルター、フレキシブルディスプレー用フィルム、半導体装置、電子部品、層間絶縁膜、配線被覆膜、光回路、光回路部品、反射防止膜、ホログラム、光学部材又は建築材料の形成材料として用いられる、請求項1乃至9のいずれかに記載の感光性樹脂組成物。
【請求項11】
前記請求項1乃至10のいずれかに記載の感光性樹脂組成物又はその硬化物により少なくとも一部分が形成されている、印刷物、カラーフィルター、フレキシブルディスプレー用フィルム、半導体装置、電子部品、層間絶縁膜、配線被覆膜、光回路、光回路部品、反射防止膜、ホログラム、光学部材又は建築材料いずれかの物品。
【請求項12】
前記請求項1乃至10のいずれかに記載の感光性樹脂組成物からなる塗膜又は成形体の表面に、所定のパターン状に電磁波を照射し、前記塗膜又は成形体の電磁波照射部位の溶解性を選択的に低下させた後、現像する、ネガ型パターン形成方法。
【請求項13】
電磁波を照射後、加熱処理を行って、前記塗膜又は成形体の電磁波照射部位の溶解性を選択的に低下させる、請求項12に記載のネガ型パターン形成方法。
【図1】

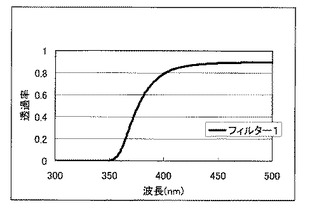
【公開番号】特開2010−133996(P2010−133996A)
【公開日】平成22年6月17日(2010.6.17)
【国際特許分類】
【出願番号】特願2008−306996(P2008−306996)
【出願日】平成20年12月2日(2008.12.2)
【出願人】(000002897)大日本印刷株式会社 (14,506)
【Fターム(参考)】
【公開日】平成22年6月17日(2010.6.17)
【国際特許分類】
【出願日】平成20年12月2日(2008.12.2)
【出願人】(000002897)大日本印刷株式会社 (14,506)
【Fターム(参考)】
[ Back to top ]