
慢性リンパ性白血病細胞に由来するポリペプチドおよび抗体、ならびにそれらの使用
【課題】慢性リンパ性白血病細胞に由来するポリペプチドおよび抗体、ならびにそれらの使用の提供。
【解決手段】本発明は、癌を処置するための方法であって、癌に罹患している被験体に対して、i)CD200とCD200レセプターとの間の相互作用を妨害して、それによりCD200の免疫抑制効果を阻害し、かつii)OX−2/CD200またはOX−2/CD200レセプターに結合する部分を含むポリペプチド融合分子を使用して癌細胞を殺す治療組成物を投与する工程を包含する方法を提供する。
【解決手段】本発明は、癌を処置するための方法であって、癌に罹患している被験体に対して、i)CD200とCD200レセプターとの間の相互作用を妨害して、それによりCD200の免疫抑制効果を阻害し、かつii)OX−2/CD200またはOX−2/CD200レセプターに結合する部分を含むポリペプチド融合分子を使用して癌細胞を殺す治療組成物を投与する工程を包含する方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願)
本出願は、2005年6月30日に提出された米国特許出願第11/171,567号に対する優先権を主張する。この米国特許出願第11/171,567号は、2004年11月23日に提出された米国特許出願第10/996,316号の一部継続である。米国特許出願第10/996,316号は、2004年7月20日に提出された米国特許出願第10/894,672号の一部継続である。米国特許出願第10/894,672号は、2003年12月15日に提出された米国特許出願第10/736,188号の一部継続である。米国特許出願第10/736,188号は、次に、2001年12月10日に提出されたPCT/US01/47931の一部継続である。PCT/US01/47931は、2000年12月8日に提出された米国仮出願第60/254,113号に対する優先権を主張する国際出願である。米国仮出願第60/254,113号は、2003年3月4日に提出された米国特許出願第10/379,151号の一部継続である。上述の米国出願、国際出願および仮出願の開示全体は、参考として本明細書中に援用される。
【0002】
(技術分野)
2つの機構の組合せを提供する療法を使用する癌処置を開示する。さらに詳細には、本開示は:1)免疫抑制を遮断して、それにより癌細胞の根絶を促進するために、CD200とそのレセプターとの間の相互作用を妨害する;2)a)補体媒介性または抗体依存性細胞傷害、あるいはb)CD200標的部分を含む融合分子を使用して細胞を標的化することのどちらかによって、癌細胞を直接殺す療法を使用して、癌を処置することに関する。
【背景技術】
【0003】
(背景)
慢性リンパ球性白血病(CLL)は、白血球の疾患であり、西半球における白血病の最も一般的な形である。CLLは、ゆっくり増殖するが、長い寿命を持つ悪性リンパ球の増殖に関連する疾患の別の群を示す。CLLは、たとえば古典型および混合型のB細胞慢性リンパ球性白血病(B−CLL)、B細胞およびT細胞前リンパ球性白血病、ヘアリー細胞白血病、および大顆粒リンパ球性白血病を含む各種のカテゴリーに分類される。
CLLのすべての異なる種類のうち、B−CLLは全白血病の約30パーセントを占める。50歳を超えた個人でより頻繁に発生するが、より若年者にも次第に見られるようになっている。B−CLLは、形態的に正常であるが、生物学的に未成熟であり、機能損失につながるBリンパ球の蓄積を特徴とする。リンパ球は正常には、感染と戦うように機能する。しかしながらB−CLLでは、リンパ球が血液および骨髄に蓄積して、リンパ節の腫れを引き起こす。正常な骨髄および血液細胞の生成が減少して、患者は重篤な貧血はもちろんのこと、血小板数の減少もしばしば経験する。このことは、生命を脅かす出血の危険および白血液細胞数の減少による深刻な感染の発現をもたらす可能性がある。
【0004】
白血病などの疾患をさらに理解するために、その病因論、病原および生物学に関する研究のためのツールとして使用できる適切な細胞株を有することが重要である。悪性ヒトBリンパ細胞株の例は、プレB急性リンパ芽球性白血病(Reh)、びまん性大細胞リンパ腫(WSU−DLCL2)、およびワルデンシュトレームマクログロブリン血症(WSU−WM)を含む。あいにく既存の細胞株の多くは、白血病およびリンパ腫の臨床的に最も一般的な種類を示さない。
【0005】
インビトロでのエプスタイン−バールウイルス(EBV)感染の使用は、悪性細胞を代表する一部のCLL由来細胞株、特にB−CLL細胞株をもたらした。これらの細胞株の表現型は、生体内腫瘍の表現型とは異なり、代わりにB−CLL系の特徴は、リンパ芽球様細胞株の特徴と類似する傾向がある。EBV感染を用いてB−CLL細胞を不死化する試みは、あまり成功しなかった。この理由ははっきりしないが、EBVレセプター発現、結合または摂取の不足によらないことは既知である。Wellsらは、B−CLL細胞が細胞周期のG1/S期で停止されることと、変換関連EBV DNAが発現されなかったことを見出した。EBVとB−CLL細胞との間の相互作用が、正常なB細胞との間の相互作用と異なることを示唆する。EBV変換CLL細胞株はさらに分化して、EBVによって不死化されたリンパ芽球様細胞株(LCL)にさらに類似した形態を示すと思われる。
【0006】
EBV陰性CLL細胞株のWSU−CLLが以前に確立されている(非特許文献1)。しかしながら、他のそのような細胞株は知られていない。
【0007】
各種の機構が、癌およびCLLを含む疾患状態に対する生体反応で役割を果たす。たとえばCD4+Tヘルパー細胞は、刺激因子をエフェクター細胞に供給することによって、各種の悪性腫瘍に対して有効な免疫反応で不可欠な役割を果たす。細胞傷害性T細胞は、癌細胞を除去するのに最も有効な細胞であると考えられ、Tヘルパー細胞は、IL−2およびIFN−γなどのTh1サイトカインを分泌することによって細胞傷害性T細胞を刺激する。各種の悪性腫瘍において、Tヘルパー細胞は、健常者に見られる細胞と比べて変化した表現型を有することが示されている。顕著に変化した特徴の1つは、Th1サイトカイン生成の低下およびTh2サイトカイン生成への変化である(たとえば非特許文献2;非特許文献3;非特許文献4;非特許文献5;非特許文献6;非特許文献7;非特許文献8;非特許文献9;非特許文献10を参照)。Th1プロフィールへのそのサイトカイン変化を逆転することは、T細胞の抗腫瘍効果を増強することが証明されている(非特許文献11;非特許文献12を参照)。
【0008】
腫瘍細胞がTh1からTh2へのTヘルパー細胞のサイトカイン発現を駆動させる能力の基礎を成す機構は、IL−10またはTGF−βなどのサイトカインの分泌はもちろんのこと、免疫系の細胞と相互作用する表面分子の発現も含む。免疫グロブリン遺伝子ファミリの分子に高度の相同性を持つ樹状細胞の表面に発現された細胞である、OX−2/CD200は、免疫抑制に関与しており(非特許文献13)、OX−2/CD200発現細胞をTh1サイトカイン生成の刺激を阻害できる証拠が提供されている。Gorczynskiらはマウスモデルにおいて、OX−2/CD200 Fcの輸液が白血病腫瘍細胞を使用した動物モデルにおける腫瘍細胞の拒絶を抑制することを示した(非特許文献14)。
【0009】
特にT細胞の活性を増強できる範囲で、癌またはCLLの罹患者を処置する改良方法が望ましい。
【先行技術文献】
【非特許文献】
【0010】
【非特許文献1】Mohammad et al.,,Leukemia(1996)10(1):130−7
【非特許文献2】Kiani,et al.,Haematologica(2003)88:754−761
【非特許文献3】Maggio,et al.,Ann Oncol(2002)13 Suppl 1:52−56
【非特許文献4】Ito, et al.,Cancer(1999)85:2359−2367
【非特許文献5】Podhorecka, et al.,Leuk Res(2002)26:657−660
【非特許文献6】Tatsumi,et al.,J Exp Med(2002)196:619−628
【非特許文献7】Agarwal,et al.,Immunol Invest(2003)32:17−30
【非特許文献8】Smyth,et al.,Ann Surg Oncol(2003)10:455− 462
【非特許文献9】Contasta, et al.,Cancer Biother Radiopharm(2003)18:549−557
【非特許文献10】Lauerova, et al.,Neoplasma(2002)49:159−166
【非特許文献11】Winter,et al.,Immunology(2003)108:409−419
【非特許文献12】Inagawa,et al.,Anticancer Res(1998)18:3957−3964
【非特許文献13】Gorczynski et al.,Transplantation(1998)65:1106−1114
【非特許文献14】Clin Exp Immunol(2001)126:220−229
【発明の概要】
【課題を解決するための手段】
【0011】
(概要)
1つの実施形態において、EBVによる不死化によって確立されていない悪性起源のCLL細胞株が提供される。細胞株は一次CLL細胞に由来し、ATCCアクセション番号PTA−3920で寄託されている。好ましい実施形態において、細胞株はCLL−AATである。CLL−AATはB−CLL一次細胞に由来するB−CLL細胞株である。
【0012】
さらなる局面において、CLL−AAT細胞株を使用して、CLLの診断および/または処置に有用なモノクローナル抗体を産生させる。抗体は、本明細書で免疫原として開示された細胞を使用することによって産生でき、それゆえモノクローナル抗体を単離できる動物での免疫反応を上昇させる。そのような抗体の配列が決定でき、抗体またはその変異体は組換え技法によって生成できる。この局面において、「変異体」は、モノクローナル抗体の配列に基づくキメラ、CDRグラフト、ヒト化および完全ヒト抗体を含む。
【0013】
さらに組換えライブラリーに由来する抗体(「ファージ抗体」)は、本明細書で述べた細胞、またはそれに由来するポリペプチドを、標的特異性に基づいて抗体を単離するためのベイトとして用いて選択できる。
なおさらなる局面において、抗体は、一次CLL細胞、またはそれに由来する抗原を用いて抗体ライブラリーをパニングすることによって産生でき、そしてCLL細胞株、たとえば本明細書で述べるCLL細胞株を使用してさらにスクリーニングおよび/または特徴付けできる。したがって、CLL細胞株への抗体の結合を評価することを含む、CLLに特異性の抗体を特徴付ける方法が提供される。
【0014】
さらなる局面において、当業者に公知の方法によって、たとえば免疫沈降とそれに続く質量分光分析によって、CLL−AAT細胞株を利用してCLL細胞内で特異的に発現されるタンパク質を同定するための方法が提供される。そのようなタンパク質は、CLL−AAT細胞株において、またはCLL患者に由来する一次細胞において特異的に発現できる。
【0015】
低分子ライブラリー(多くが市販されている)は、細胞の増殖特徴を調節できる因子を同定するための細胞ベースアッセイにおいて、CLL−AAT細胞株を使用してスクリーニングできる。たとえばCLL−AAT細胞株でアポトーシスを調節する、またはその増殖および/または増殖を阻害する因子を同定できる。そのような因子は、治療化合物の開発のための候補である。
【0016】
CLL−AAT細胞株から単離された核酸は、CLL特異性遺伝子を同定するサブトラクティブハイブリダイゼーション実験で、またはマイクロアレイ分析(たとえば遺伝子チップ実験)で使用できる。転写がCLL細胞において調節される遺伝子を同定できる。この方法で同定されたポリペプチドまたは核酸遺伝子生成物は、CLLの抗体または小型細胞療法の開発のための手がかりとして有用である。
【0017】
好ましい局面において、CLL−AAT細胞株は、細胞によってインターナライズされる細胞表面成分に結合する、インターナライズする抗体を同定するために使用できる。そのような抗体は、治療に使用するための候補である。特に、細胞質内で安定なままであり、細胞内結合活性を維持する単鎖抗体は、この方法でスクリーニングできる。
【0018】
なお別の局面において、患者が悪性癌細胞によってアップレギュレートされたポリペプチドについてスクリーニングされ、アップレギュレートされたポリペプチドの代謝経路を妨害する抗体が患者に投与される治療処置が説明されている。
【0019】
本開示はさらに、OX−2/CD200が被験体においてアップレギュレートされるかどうかに関する判定が行われ、もしそうならば被験体に免疫反応を増強する療法投与するための方法に関する。OX2/CD200のアップレギュレーションは、OX2/CD200レベルを測定することによって直接、またはOX2/CD200と相関する任意のマーカーのレベルをモニターすることによって判定できる。適切な免疫調節療法は、T細胞または抗原提示細胞の負の調節を遮断する因子の投与、T細胞、癌ワクチン、免疫系を刺激する一般的なアジュバントの正の副刺激を増強する因子の投与あるいはIL−2、GM−CSFおよびIFN−γなどのサイトカインによる処置を含む。特に有用な実施形態において、免疫反応を増強する療法は、1つ以上の他の免疫調節療法と場合により組合せた、OX−2/CD200に結合するポリペプチドの投与を含む。別の実施形態において、ポリペプチドはOX−2/CD200レセプターに結合する。
【0020】
別の局面において、本開示による方法を使用して、OX−2/CD200が被験体にてアップレギュレートされる疾患状態を、OX−2/CD200に結合するポリペプチドまたはOX−2/CD200レセプターを該疾患状態に罹患した被験体に投与することによって処置する。1つの実施形態において、これらの方法によって処置される疾患状態は、癌、特に他の実施形態においてCLLを含む。
【0021】
特に有用な実施形態において、本開示による癌療法は、i)免疫抑制を遮断して、それにより癌細胞の根絶を促進するために、CD200とそのレセプターとの間の相互作用を妨害する抗体を投与することと;ii)癌細胞を直接殺すために、CD200標的部分を含む融合分子を投与することとを含む。あるいは抗体は、補体媒介性または抗体依存性細胞傷害によって癌細胞を直接殺す。
【0022】
本開示による別の実施形態において、治療処置の進行をモニターするための方法が提供される。該方法は、免疫調節療法を投与する工程と、療法の有効性を判定するために、被験体におけるOX−2/CD200レベルを少なくとも2回判定する工程とを含む。
【0023】
別の局面において、本開示は、癌細胞によって発現された分子の免疫調節効果を評価するための方法を提供する。これらの方法では、癌細胞によって発現またはアップレギュレートされる分子を同定する(たとえば実験的に、またはデータベースから)。癌細胞、リンパ球および癌細胞によって発現またはアップレギュレートされる分子を被験体に投与して、癌細胞の増殖の速度をモニターする。投与されるリンパ球の数は、a)癌細胞の増殖を減速するのに十分であるように、またはb)癌細胞の増殖を減速するのに不十分であるようにのいずれかに規定される。癌細胞によって発現またはアップレギュレートされる分子は、分子または分子自体の活性部分として、あるいは分子またはその部分を自然に生成または発現する細胞を投与することによって、あるいは分子またはその部分を生成または発現するように操作された細胞を投与することによって投与できる。腫瘍細胞およびドナーリンパ球が単独で投与された場合の増殖速度と比較して、癌細胞の増殖速度の何らかの変化が観察される場合、該分子は免疫調節効果を有すると考えられる。たとえば投与されるリンパ球の数が癌細胞の増殖を減速するのに十分であり、観察された腫瘍細胞の増殖速度が腫瘍細胞およびドナーリンパ球が単独で投与されたときの増殖速度と比較して高い場合、癌細胞によって発現またはアップレギュレートされる分子は免疫抑制性であると見なされる。別の例として、投与されるリンパ球の数が癌細胞の増殖を減速するのに不十分であり、観察された腫瘍細胞の増殖速度が腫瘍細胞およびドナーリンパ球が単独で投与されたときの増殖速度と比較して低い場合、該化合物は免疫増強性であるとみなされる。いったん分子の免疫調節効果が確立されると、本明細書で述べた実施形態に従って、分子の活性を増強または阻害する化合物が同定できる。増強または阻害効果は、癌細胞によって発現またはアップレギュレートされた分子との直接作用の結果でありうるか、あるいは癌細胞によって発現またはアップレギュレートされた化合物の代謝経路における他の分子との間の相互作用の結果でありうる。
【0024】
別の局面において、本開示は化合物の免疫調節効果を評価するための方法を提供する。これらの方法では、癌細胞、リンパ球および評価される化合物を被験体に投与して、癌細胞の増殖の速度をモニターする。投与されるリンパ球の数は、a)癌細胞の増殖を減速するのに十分であるように、またはb)癌細胞の増殖を減速するのに不十分であるように、のいずれかに規定される。評価される化合物は、化合物自体として、または該化合物を自然に生成する細胞を投与することによって、または該化合物を生成するように操作された細胞を投与することによって投与できる。腫瘍細胞およびドナーリンパ球が単独で投与された場合の増殖速度と比較して、癌細胞の増殖速度の何らかの変化が観察される場合、該分子は免疫調節効果を有すると考えられる。たとえば投与されるリンパ球の数が癌細胞の増殖を減速するのに十分であり、観察された腫瘍細胞の増殖速度が腫瘍細胞およびドナーリンパ球が単独で投与されたときの増殖速度と比較して高い場合、癌細胞によって発現またはアップレギュレートされる分子は免疫抑制性であると見なされる。別の例として、投与されるリンパ球の数が癌細胞の増殖を減速するのに不十分であり、観察された腫瘍細胞の増殖速度が腫瘍細胞およびドナーリンパ球が単独で投与されたときの増殖速度と比較して低い場合、該化合物は免疫増強性であるとみなされる。
例えば、本願発明は以下の項目を提供する。
(項目1)
癌を処置するための方法であって、
癌に罹患している被験体に対して、
i)CD200とCD200レセプターとの間の相互作用を妨害して、それによりCD200の免疫抑制効果を阻害し、かつ
ii)OX−2/CD200またはOX−2/CD200レセプターに結合する部分を含むポリペプチド融合分子を使用して癌細胞を殺す
治療組成物を投与する工程;
を包含する、方法。
(項目2)
前記治療組成物を投与する工程が、前記被験体に対して、OX−2/CD200に結合する抗体を投与することを包含する、項目1に記載の方法。
(項目3)
前記治療組成物を投与する工程が、前記被験体に対して、OX−2/CD200に結合するモノクローナル抗体を投与することを包含する、項目1に記載の方法。
(項目4)
前記治療組成物を投与する工程が、前記被験体に対して、OX−2/CD200レセプターに結合する抗体を投与することを包含する、項目1に記載の方法。
(項目5)
前記治療組成物を投与する工程が、前記被験体に対して、OX−2/CD200レセプターに結合するモノクローナル抗体を投与することを包含する、項目1に記載の方法。
(項目6)
前記癌に罹患している被験体が、CLL患者である、項目1に記載の方法。
(項目7)
前記融合分子が、毒素を含む、項目1に記載の方法。
(項目8)
前記融合分子が、高エネルギー放射線エミッターを含む、項目1に記載の方法。
(項目9)
前記融合分子が、細胞傷害性T細胞活性またはNK細胞活性を増強するサイトカインもしくはケモカインを含む、項目1に記載の方法。
(項目10)
前記融合分子が、T細胞を誘引するケモカインを含む、項目1に記載の方法。
(項目11)
癌を処置するための方法であって、
CD200に結合する抗体を投与する工程;
を包含し、
a)CD200とそのレセプターとの間の相互作用が遮断され、
b)CD200を発現する癌細胞が殺される、
方法。
(項目12)
癌を処置するための方法であって、
CD200に結合する抗体を投与する工程;
を包含し、
a)CD200とそのレセプターとの間の相互作用が遮断され、
b)該癌に対する細胞傷害性T細胞活性またはNK細胞活性が増強される、
方法。
(項目13)
前記細胞傷害性T細胞活性またはNK細胞活性の増強が、IL−2、IL−12、IL−18、IL−13、およびIL−5からなる群より選択されるサイトカインと融合したCD200に結合する抗体によって達成される、項目12に記載の癌を処置するための方法。
(項目14)
癌を処置するための方法であって、
CD200に結合する抗体を投与する工程;
を包含し、
a)CD200とそのレセプターとの間の相互作用が遮断され、
b)T細胞が腫瘍細胞に誘引される、
方法。
(項目15)
T細胞誘引が、MIG、IP−10およびI−TACからなる群より選択されるケモカインと融合したCD200に結合する抗体によって達成される、項目14に記載の癌を処置するための方法。
(項目16)
OX−2/CD200抗原を保持する細胞を標的とする第一の部分と;
細胞の死を促進する第二の部分と;
を含む、融合分子。
(項目17)
前記第一の部分が、OX−2/CD200に結合する抗体を含む、項目16に記載の融合分子。
(項目18)
前記第一の部分が、OX−2/CD200に結合するモノクローナル抗体を含む、項目16に記載の融合分子。
(項目19)
癌を処置するための方法であって、
癌患者に対して、
(i)CD200とCD200レセプターとの間の相互作用を妨害して、それによりCD200の免疫抑制効果を阻害し、かつ
(ii)補体媒介性細胞傷害または抗体依存性細胞傷害によって癌細胞を殺す、
抗体を投与する工程;
を包含する、
方法。
(項目20)
(i)CD200とCD200レセプターとの間の相互作用を妨害して、それによりCD200の免疫抑制効果を阻害し、かつ(ii)補体媒介性細胞傷害または抗体依存性細胞傷害によって癌細胞を殺す、抗体。
(項目21)
項目20に記載の抗体と、薬学的に受容可能なキャリアとを含む、組成物。
(項目22)
前記治療組成物を投与する工程が、配列番号5、12および13からなる群より選択される配列を有する軽鎖CDR1領域を含む抗体を投与することを包含する、項目1に記載の方法。
(項目23)
前記治療組成物を投与する工程が、配列番号21および23からなる群より選択される配列を有する軽鎖CDR2領域を含む抗体を投与することを包含する、項目1に記載の方法。
(項目24)
前記治療組成物を投与する工程が、配列番号29、37および38からなる群より選択される配列を有する軽鎖CDR3領域を含む抗体を投与することを包含する、項目1に記載の方法。
(項目25)
前記治療組成物を投与する工程が、配列番号50、55および56からなる群より選択される配列を有する重鎖CDR1領域を含む抗体を投与することを包含する、項目1に記載の方法。
(項目26)
前記治療組成物を投与する工程が、配列番号69、74および75からなる群より選択される配列を有する重鎖CDR2領域を含む抗体を投与することを包含する、項目1に記載の方法。
(項目27)
前記治療組成物を投与する工程が、配列番号88、93および94からなる群より選択される配列を有する重鎖CDR3領域を含む抗体を投与することを包含する、項目1に記載の方法。
(項目28)
前記第一の部分が、配列番号5、12、13、21、23、29、37、38、50、55、56、69、74、75、88、93および94からなる群より選択される1つ以上のアミノ酸配列を含むポリペプチドを含む、項目16に記載の融合分子。
(項目29)
前記治療組成物が、ヒト化抗体を含む、項目2〜5のいずれかに記載の方法。
(項目30)
前記治療組成物が、Fv、scFv、Fab’およびF(ab’)2を含む、項目2〜5のいずれかに記載の方法。
(項目31)
前記抗体が、ヒト化抗体を含む、項目11〜15のいずれかに記載の方法。
(項目32)
前記抗体が、Fv、scFv、Fab’およびF(ab’)2を含む、項目11〜15のいずれかに記載の方法。
(項目33)
前記抗体が、ヒト化抗体を含む、項目17〜18のいずれかに記載の融合分子。
(項目34)
前記抗体が、Fv、scFv、Fab’およびF(ab’)2を含む、項目17〜18のいずれかに記載の融合分子。
(項目35)
前記治療組成物が、ヒト化抗体を含む、項目22〜27のいずれかに記載の方法。
(項目36)
前記治療組成物が、Fv、scFv、Fab’およびF(ab’)2を含む、項目22〜27のいずれかに記載の方法。
(項目37)
前記抗体が、ヒト化抗体を含む、項目21に記載の組成物。
(項目38)
前記抗体が、Fv、scFv、Fab’およびF(ab’)2を含む、項目21に記載の組成物。
(項目39)
前記ポリペプチドが、ヒト化抗体を含む、項目28に記載の融合分子。
(項目40)
前記ポリペプチドが、Fv、scFv、Fab’およびF(ab’)2を含む、項目28に記載の融合分子。
(項目41)
配列番号111〜199からなる群より選択されるポリペプチド配列を含む、CD200を発現する細胞に結合する抗体。
(項目42)
配列番号111、114、118、133、136、140、141、149、155、162、163、165、174、181、187、188、189、195および199からなる群より選択されるポリペプチド配列を含む、項目41に記載の抗体。
(項目43)
配列番号200〜211からなる群より選択されるポリペプチド配列を含む、項目41に記載の抗体。
(項目44)
前記抗体が、ヒト化抗体を含む、項目41に記載の抗体。
(項目45)
前記抗体が、Fv、scFv、Fab’およびF(ab’)2を含む、項目41に記載の抗体。
(項目46)
癌を処置するための方法であって、
癌に罹患している被験体に対して、CD200を発現する細胞に結合する抗体を含有する治療組成物を投与する工程;
を包含し、該抗体は、1つ以上のTh1サイトカインの生成を促進するのに十分な量で存在する、方法。
(項目47)
前記抗体が、配列番号111〜199からなる群より選択されるポリペプチド配列を含む、項目46に記載の方法。
(項目48)
前記抗体が、配列番号111、114、118、133、136、140、141、149、155、162、163、165、174、181、187、188、189、195および199からなる群より選択されるポリペプチド配列を含む、項目46に記載の方法。
(項目49)
前記抗体が、配列番号200〜211からなる群より選択されるポリペプチド配列を含む、項目46に記載の方法。
(項目50)
前記抗体が、ヒト化抗体を含む、項目46に記載の方法。
(項目51)
前記抗体が、Fv、scFv、Fab’およびF(ab’)2を含む、項目46に記載の方法。
(項目52)
前記癌が、CLLである、項目46に記載の方法。
(項目53)
腫瘍細胞を殺すための方法であって、
癌に罹患している被験体に対して、CD200を発現する細胞に結合する抗体を含む治療組成物を投与する工程;
を包含し、該抗体は、抗体依存性細胞媒介性細胞傷害を引き起こすのに十分な量で存在する、方法。
(項目54)
前記抗体が、配列番号111〜199からなる群より選択されるポリペプチド配列を含む、項目53に記載の方法。
(項目55)
前記抗体が、配列番号111、114、118、133、136、140、141、149、155、162、163、165、174、181、187、188、189、195および199からなる群より選択されるポリペプチド配列を含む、項目53に記載の方法。
(項目56)
前記抗体が、配列番号200−211からなる群より選択されるポリペプチド配列を含む、項目53に記載の方法。
(項目57)
前記抗体が、ヒト化抗体を含む、項目53に記載の方法。
(項目58)
前記抗体が、Fv、scFv、Fab’およびF(ab’)2を含む、項目53に記載の方法。
(項目59)
前記癌が、CLLである、項目53に記載の方法。
(項目60)
OX−2/CD200抗原を保持する細胞を標的とする第一の部分であって、配列番号111〜199からなる群より選択されるポリペプチド配列を含む第一の部分と;
細胞の死を促進する第二の部分と;
を含む、融合細胞。
(項目61)
前記第一の部分が、配列番号111、114、118、133、136、140、141、149、155、162、163、165、174、181、187、188、189、195および199からなる群より選択されるポリペプチド配列を含む、項目60に記載の融合分子。
(項目62)
前記第一の部分が、配列番号200〜211からなる群より選択されるポリペプチド配列を含む、項目61に記載の融合分子。
(項目63)
配列番号111〜199からなる群より選択されるポリペプチド配列を含む抗体と、薬学的に受容可能なキャリアとを含む、組成物。
(項目64)
前記抗体が、ヒト化抗体を含む、項目63に記載の組成物。
(項目65)
前記抗体が、Fv、scFv、Fab’およびF(ab’)2を含む、項目63に記載の組成物。
(項目66)
癌を処置するための方法であって、
癌に罹患している被験体に対して、CD200を発現する細胞に結合する抗体を含む治療組成物を投与する工程;
を包含し、該抗体は、1つ以上のTh1サイトカインの生成を促進するのに十分な量、かつ補体媒介性細胞傷害または抗体依存性細胞傷害を引き起こすのに十分な量で存在する、方法。
(項目67)
配列番号200〜211からなる群より選択されるポリペプチド配列を含む、CD200を発現する細胞に結合するキメラ抗体。
(項目68)
OX−2/CD200が被験体においてアップレギュレートされるかどうかを判定する工程;および
該被験体に対して、免疫反応を向上させる療法を投与する工程;
を包含する、方法。
(項目69)
前記療法が、OX−2/CD200に結合するかまたはOX−2/CD200レセプターに結合するポリペプチドを投与することを包含し、該ポリペプチドは、OX−2/CD200の免疫抑制効果を阻害するのに十分な量で投与される、項目68に記載の方法。
(項目70)
前記被験体が、癌患者である、項目68に記載の方法。
(項目71)
前記被験体が、CLL患者である、項目68に記載の方法。
(項目72)
前記ポリペプチドが、抗体を含む、項目69に記載の方法。
(項目73)
前記ポリペプチドが、ヒト化抗体を含む、項目69に記載の方法。
(項目74)
前記ポリペプチドが、Fv、scFv、Fab’およびF(ab’)2を含む、項目69に記載の方法。
(項目75)
前記OX−2/CD200が被験体においてアップレギュレートされるかどうかを判定する工程が、
癌患者から組織を取得すること;
CD200レベルが、対応する正常組織で見出されたレベルの少なくとも2倍であるかどうかを評価すること;
を包含する、項目68に記載の方法。
(項目76)
血液が、前記癌患者から取得される組織である、項目75に記載の方法。
(項目77)
組織が生検を行うことによって癌患者から取得される、項目75に記載の方法。
(項目78)
CD200レベルが、抗CD200抗体を癌細胞マーカーと組合せて使用するFACS分析を実施することによって評価される、項目75に記載の方法。
(項目79)
前記癌細胞マーカーが、CD38またはCD19である、項目75に記載の方法。
(項目80)
前記患者が、造血癌に罹患している、項目75に記載の方法。
(項目81)
前記患者が、CLLに罹患している、項目75に記載の方法。
(項目82)
前記療法が、OX−2/CD200に結合するかまたはOX−2/CD200レセプターに結合するポリペプチドを投与する前に既存の調節T細胞を排除することを包含し、該ポリペプチドは、OX−2/CD200免疫抑制効果を阻害するのに十分な量で投与される、項目69に記載の方法。
(項目83)
前記療法が、抗CD25抗体およびシクロホスファミドからなる群より選択される試薬の調節T細胞排除量を投与することを包含する、項目82に記載の方法。
(項目84)
前記療法が、骨髄切除療法を投与することをさらに包含する、項目69に記載の方法。
(項目85)
前記療法が、骨髄移植をさらに包含する、項目84に記載の方法。
(項目86)
前記療法が、CLL反応性T細胞の移動をさらに包含する、項目84に記載の方法。
(項目87)
前記療法が、癌ワクチンを投与することをさらに包含する、項目69に記載の方法。
(項目88)
前記癌ワクチンが、CLL細胞を負荷した樹状細胞;CLL細胞を負荷した樹状細胞に由来するタンパク質、ペプチドまたはRNA;患者由来熱ショックタンパク質タンパク質;腫瘍ペプチド;および腫瘍タンパク質;からなる群より選択される、項目87に記載の方法。
(項目89)
前記療法が、免疫賦活化合物の有効免疫刺激量を投与することをさらに包含する、項目69に記載の方法。
(項目90)
前記免疫賦活化合物が、CpG、toll様レセプターアゴニストおよび抗CTLA−4抗体からなる群より選択される、項目89に記載の方法。
(項目91)
前記療法が、抗PDL1抗体、抗PDL1抗体、抗IL−10抗体および抗IL−6抗体からなる群より選択される化合物の有効免疫抑制性遮断量を投与することをさらに包含する、項目69に記載の方法。
(項目92)
前記療法が、T細胞または抗原提示細胞の負の調節を遮断可能な因子の有効量を投与することを包含する、項目69に記載の方法。
(項目93)
前記因子が、抗CTLA4抗体、抗PD−L1抗体、抗PDL−2抗体および抗PD−1抗体からなる群より選択される、項目92に記載の方法。
(項目94)
前記療法が、T細胞の正の副刺激を増強可能な因子の有効量を投与することを包含する、項目69に記載の方法。
(項目95)
前記因子が、抗CD40抗体および抗4−1BB抗体からなる群より選択される、項目94に記載の方法。
(項目96)
前記療法が、癌ワクチンを投与することを包含する、項目69に記載の方法。
(項目97)
前記癌ワクチンが、CLL細胞を負荷した樹状細胞;CLL細胞を負荷した樹状細胞に由来するタンパク質、ペプチドまたはRNA;患者由来熱ショックタンパク質タンパク質;腫瘍ペプチド;および腫瘍タンパク質;からなる群より選択される、項目96に記載の方法。
(項目98)
前記療法が、先天免疫系のレセプターを活性化可能な因子の有効量を投与することを包含する、項目69に記載の方法。
(項目99)
前記因子が、CpG、Luivac、Biostim、Ribominyl、Imudon、Bronchovaxomからなる群より選択される、項目98に記載の方法。
(項目100)
前記療法が、サイトカインの有効な免疫増強量を投与することを包含する、項目69に記載の方法。
(項目101)
前記サイトカインが、IL−2、GM−CSFおよびIFN−γからなる群より選択される、項目100に記載の方法。
(項目102)
治療処置の進行をモニターするための方法であって、
被験体に対して、免疫調節療法を投与する工程;および
該療法の有効性を判定するために、被験体において少なくとも2倍のOX−2/CD200レベルを判定する工程;
を包含する、方法。
(項目103)
前記被験体におけるOX−2/CD200レベルが、前記免疫調節療法の投与前に判定され、次に、該被験体におけるOX−2/CD200レベルが、該療法の少なくとも1回の後に判定される、項目102に記載の方法。
(項目104)
前記免疫調節療法の投薬量または頻度を調整する工程をさらに包含する、項目102に記載の方法。
(項目105)
OX−2/CD200レベルが、OX−2/CD200と相関するマーカーを検出することによって判定される、項目102に記載の方法。
(項目106)
癌細胞によって発現される分子の免疫調節効果を評価するための方法であって、
被験体に対して、癌細胞によって自然に発現された分子、癌細胞およびリンパ球を投与する工程;および
該癌細胞の増殖の速度をモニターする工程;
を包含し、
該癌細胞およびドナーリンパ球のみが投与された場合の増殖速度と比較して、該癌細胞の増殖速度に何らかの変化が観察された場合に、該分子は免疫調節効果を有すると見なされる、方法。
(項目107)
前記癌細胞およびドナーリンパ球のみが投与された場合の増殖速度と比較して該癌細胞の増殖の速度の低下が観察される場合に、前記分子は、免疫増強性であると見なされる、項目106に記載の方法。
(項目108)
前記癌細胞およびドナーリンパ球のみが投与された場合の増殖速度と比較して該癌細胞のより高い増殖速度が観察される場合に、前記分子は、免疫抑制性であると見なされる、項目106に記載の方法。
(項目109)
前記癌細胞によって自然に発現される分子が、該化合物を自然に発現する細胞を注射することによって投与される、項目106に記載の方法。
(項目110)
前記癌細胞によって自然に発現される分子が、該分子を発現するように操作された細胞を注射することによって投与される、項目106に記載の方法。
(項目111)
前記癌細胞によって発現される分子をデータベースから同定する工程をさらに包含する、項目106に記載の方法。
(項目112)
前記癌細胞によって発現される分子を実験的に同定する工程をさらに包含する、項目106に記載の方法。
(項目113)
前記リンパ球が、ヒトである、項目106に記載の方法。
(項目114)
前記ヒトリンパ球が、末梢血リンパ球(PBL)、T細胞、B細胞、および樹状細胞からなる群より選択される、項目113に記載の方法。
(項目115)
前記リンパ球が、ヒト末梢血リンパ球(PBL)である、項目106に記載の方法。
(項目116)
前記投与されるリンパ球の数が、癌細胞の増殖速度を低下させるのには不十分である、項目106に記載の方法。
(項目117)
前記投与されるリンパ球の数が、投与される癌細胞の数よりも少ない、項目106に記載の方法。
(項目118)
前記投与されるリンパ球の数が、約100万細胞〜約200万細胞である、項目106に記載の方法。
(項目119)
前記投与されるリンパ球の数が、癌細胞の増殖速度を低下させるのに十分である、項目106に記載の方法。
(項目120)
前記投与されるリンパ球の数が、投与される癌細胞の数以上である、項目106に記載の方法。
(項目121)
前記投与されるリンパ球の数が、約500万細胞〜約1,000万細胞である、項目106に記載の方法。
(項目122)
前記癌細胞の投与が、RAJIおよびNamalwaからなる群より選択される少なくとも1つのリンパ腫細胞株を前記被験体に対して注射することを包含する、項目106に記載の方法。
(項目123)
前記被験体が、免疫不全マウスである、項目106に記載の方法。
(項目124)
前記投与されるリンパ球の数が、前記癌細胞の増殖を減速するのに十分であり、前記癌細胞により発現される分子は、前記癌細胞およびリンパ球のみが投与された場合の増殖速度と比較して観察される癌細胞の増殖速度が高い場合に、免疫抑制性であると見なされる、項目106に記載の方法。
(項目125)
前記投与されるリンパ球の数が、癌細胞の増殖を減速するのに不十分であり、前記癌細胞によって発現される分子は、前記癌細胞およびリンパ球のみが投与された場合の増殖速度と比較して観察される癌細胞の増殖速度が低い場合に、免疫増強性であると見なされる、項目106に記載の方法。
(項目126)
リンパ球および癌細胞のみを対照被験体に投与することによって、前記癌細胞の増殖速度を判定する工程;
をさらに包含する、項目106に記載の方法。
(項目127)
前記癌細胞によって発現される分子を投与する前に、リンパ球および癌細胞のみを前記被験体に対して投与することによって、前記癌細胞の増殖速度を判定する工程;
をさらに包含する、項目106に記載の方法。
(項目128)
化合物の免疫調節効果を評価するための方法であって、
被験体に対して、評価される化合物、癌細胞およびリンパ球を投与する工程;および
前記癌細胞の増殖の速度をモニターする工程;
を包含し、
該癌細胞およびドナーリンパ球のみが投与された場合の増殖速度と比較して、該癌細胞の増殖速度に何らかの変化が観察された場合に、該化合物は、免疫調節効果を有すると見なされる、方法。
(項目129)
前記癌細胞およびドナーリンパ球のみが投与された場合の増殖速度と比較して前記癌細胞の増殖の速度の低下が観察される場合に、前記化合物は、免疫増強性であると見なされる、項目128に記載の方法。
(項目130)
前記化合物の存在下での前記癌細胞の増殖速度が、前記癌細胞およびドナーリンパ球のみが投与される場合の増殖速度よりも高い場合に、該化合物は、免疫抑制性であると見なされる、項目128に記載の方法。
(項目131)
前記評価される化合物が、抗体である、項目128に記載の方法。
(項目132)
前記癌細胞の投与が、前記被験体に対して、RAJIおよびNamalwaからなる群より選択される少なくとも1つのリンパ腫細胞株を注射することを包含する、項目128に記載の方法。
(項目133)
前記投与される癌細胞が、免疫抑制分子を発現する、項目128に記載の方法。
(項目134)
前記投与される癌細胞が、CD200を発現する、項目128に記載の方法。
(項目135)
CD200の発現が、前記投与される癌細胞によってアップレギュレートされる、項目128に記載の方法。
(項目136)
前記リンパ球が、ヒトである、項目128に記載の方法。
(項目137)
前記ヒトリンパ球が、末梢血リンパ球(PBL)、T細胞、B細胞、および樹状細胞からなる群より選択される、項目136に記載の方法。
(項目138)
前記リンパ球が、ヒト末梢血リンパ球(PBL)である、項目128に記載の方法。
(項目139)
前記投与されるヒトPBLの数が、投与される癌細胞の数よりも少ない、項目138に記載の方法。
(項目140)
前記投与されるヒトPBLの数が、約100万細胞〜約200万細胞である、項目138に記載の方法。
(項目141)
前記投与されるヒトPBLの数が、投与される癌細胞の数以上である、項目138に記載の方法。
(項目142)
前記投与されるヒトPBLの数が、約500万細胞〜約1,000万細胞である、項目138に記載の方法。
(項目143)
前記被験体が、免疫不全マウスである、項目128に記載の方法。
(項目144)
前記投与されるリンパ球の数が、前記癌細胞のみと投与された場合に該癌細胞の増殖速度を減速するのに十分であり、前記化合物は、該化合物の存在下での該癌細胞の増殖速度が、前記癌細胞およびドナーリンパ球のみが投与された場合の増殖速度よりも大きい場合に、免疫抑制性であると見なされる、項目128に記載の方法。
(項目145)
前記投与されるリンパ球の数が、前記癌細胞のみと投与された場合に該癌細胞の増殖速度を減速するのには不十分であり、前記化合物は、前記癌細胞およびドナーリンパ球のみが投与された場合の増殖速度と比較して該癌細胞の増殖の速度の低下が観察される場合に、免疫増強性であると見なされる、項目128に記載の方法。
(項目146)
化合物の免疫調節効果を評価するための方法であって、
評価される化合物、癌細胞および約2×106個未満のリンパ球を、被験体に対して投与する工程;および
該癌細胞の増殖の速度をモニターする工程;
を包含し、
該癌細胞およびドナーリンパ球のみが投与された場合の増殖速度と比較して該癌細胞の増殖の速度の低下が観察される場合に、該化合物は、免疫増強性であると見なされる、方法。
(項目147)
前記投与される癌細胞が、免疫抑制分子を発現する、項目146に記載の方法。
(項目148)
前記投与される癌細胞が、CD200を発現する、項目146に記載の方法。
(項目149)
CD200の発現が、前記投与される癌細胞によってアップレギュレートされる、項目146に記載の方法。
(項目150)
化合物の免疫調節効果を評価するための方法であって、
評価される化合物、癌細胞および約5×106個のリンパ球を被験体に対して投与する工程;および
該癌細胞の増殖の速度をモニターする工程;
を包含し、
該化合物の存在下での該癌細胞の増殖の速度が、該癌細胞およびドナーリンパ球のみが投与される場合の増殖速度よりも高い場合に、該化合物は、免疫抑制性であると見なされる、方法。
(項目151)
前記投与される癌細胞が、免疫抑制分子を発現する、項目150に記載の方法。
(項目152)
前記投与される癌細胞が、CD200を発現する、項目150に記載の方法。
(項目153)
CD200の発現が、前記投与される癌細胞によってアップレギュレートされる、項目150に記載の方法。
【図面の簡単な説明】
【0025】
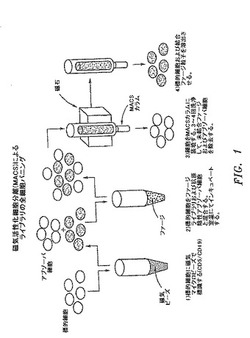
【図1】磁気活性化細胞分離(MACS)による抗体ライブラリーの細胞表面パニングに関与する代表的な工程を図式的に示す。
【図2】選択したscFvクローンの一次B−CLL細胞への結合および正常ヒトPBMCへの結合の非存在を示す、全細胞ELISAの結果を示すグラフである。この図および他の図の呼称2°+3°は、マウス抗HAによって染色され、抗マウス抗体のみを検出する負の対照ウェルを指す。この図および他の図の呼称であるRSC−Sライブラリーは、元のウサギscFv非パニングライブラリーから調製した溶解性抗体を指す。この図および他の図の呼称であるR3/RSC−Sプールは、3回目のパニングによるscFv抗体のプール全体から調製した溶解性抗体を指す。抗CD5抗体を正の対照として使用して、同数のB−CLLおよびPBMC細胞が各ウェルにて平板培養されることを検証した。
【図3a】選択したscFv抗体の一次B−CLL細胞および正常一次ヒトB細胞への結合を比較する、全細胞ELISAの結果を示す。抗CD19抗体を正の対照として使用して、同数のB−CLLおよび正常B細胞が各ウェルにて平板培養されることを検証した。他の対照は図2の凡例に示した通りであった。
【図3b】選択したscFv抗体の一次B−CLL細胞および正常一次ヒトB細胞への結合を比較する、全細胞ELISAの結果を示す。抗CD19抗体を正の対照として使用して、同数のB−CLLおよび正常B細胞が各ウェルにて平板培養されることを検証した。他の対照は図2の凡例に示した通りであった。
【図4a】scFvクローンが患者特異性(すなわちイディオタイプ)または血液型特異性(すなわちHLA)抗原に結合するかどうかを判定するために使用された全細胞ELISAの結果を示す。各クローンを3名の別個のB−CLL患者から単離したPBMCへの結合について試験した。1つの患者サンプルのみに結合したクローンを患者または血液型特異性と見なした。
【図4b】scFvクローンが患者特異性(すなわちイディオタイプ)または血液型特異性(すなわちHLA)抗原に結合するかどうかを判定するために使用された全細胞ELISAの結果を示す。各クローンを3名の別個のB−CLL患者から単離したPBMCへの結合について試験した。1つの患者サンプルのみに結合したクローンを患者または血液型特異性と見なした。
【図5a】scFvクローンの一次B−CLL細胞および3つのヒト白血病細胞株への結合を比較する全細胞ELISAの結果を示す。Ramosは、バーキットリンパ腫に由来する成熟B細胞株である。RLは、非ホジキンリンパ腫に由来する成熟B細胞株である。TF−1は、赤白血病に由来する赤芽球細胞株である。
【図5b】scFvクローンの一次B−CLL細胞および3つのヒト白血病細胞株への結合を比較する全細胞ELISAの結果を示す。Ramosは、バーキットリンパ腫に由来する成熟B細胞株である。RLは、非ホジキンリンパ腫に由来する成熟B細胞株である。TF−1は、赤白血病に由来する赤芽球細胞株である。
【図6a】scFvクローンの一次B−CLL細胞およびB−CLL患者に由来する細胞株であるCLL−AATへの結合を比較する全細胞ELISAの結果を示す。TF−1細胞は負の対照として含まれている。
【図6b】scFvクローンの一次B−CLL細胞およびB−CLL患者に由来する細胞株であるCLL−AATへの結合を比較する全細胞ELISAの結果を示す。TF−1細胞は負の対照として含まれている。
【図6c】scFvクローンの一次B−CLL細胞およびB−CLL患者に由来する細胞株であるCLL−AATへの結合を比較する全細胞ELISAの結果を示す。TF−1細胞は負の対照として含まれている。
【図7】3色フローサイトメトリーによって分析した、本開示によるscFv抗体の結合特異性を示す。正常な末梢血単核細胞において、scFv−9によって認識された抗原は、Bリンパ球で中程度に発現され、Tリンパ球のサブ個体群で弱く発現される。正常ドナーからのPBMCは、抗CD5−FITC、抗CD19−PerCP、およびscFv−9/抗HA−ビオチン/ストレプトアビジン−PEを使用して、3色フローサイトメトリーによって分析した。
【図8a】本開示によるscFv抗体によって認識された抗原の発現レベルを示す。scFv−3およびscFv−9によって認識された抗原は、CLL−AAT細胞株が由来する一次CLL腫瘍で過剰発現される。CLL−AAT細胞株を確立するために使用したCLL患者からの一次PBMCまたは正常ドナーからのPBMCをscFv抗体で染色して、フローサイトメトリーによって分析した。scFv−3およびscFv−9は、平均蛍光強度によって測定されたように、CLL細胞を正常PBMCよりも明るく染色する。
【図8b】本開示によるscFv抗体によって認識された抗原の発現レベルを示す。scFv−3およびscFv−9によって認識された抗原は、CLL−AAT細胞株が由来する一次CLL腫瘍で過剰発現される。CLL−AAT細胞株を確立するために使用したCLL患者からの一次PBMCまたは正常ドナーからのPBMCをscFv抗体で染色して、フローサイトメトリーによって分析した。scFv−3およびscFv−9は、平均蛍光強度によって測定されたように、CLL細胞を正常PBMCよりも明るく染色する。
【図8c】本開示によるscFv抗体によって認識された抗原の発現レベルを示す。scFv−3およびscFv−9によって認識された抗原は、CLL−AAT細胞株が由来する一次CLL腫瘍で過剰発現される。CLL−AAT細胞株を確立するために使用したCLL患者からの一次PBMCまたは正常ドナーからのPBMCをscFv抗体で染色して、フローサイトメトリーによって分析した。scFv−3およびscFv−9は、平均蛍光強度によって測定されたように、CLL細胞を正常PBMCよりも明るく染色する。
【図9a−1】選択したscFv抗体のCDR配列および結合特異性の概要を提供する。
【図9a−2】選択したscFv抗体のCDR配列および結合特異性の概要を提供する。
【図9B−1】選択したscFv抗体のCDR配列および結合特異性の概要を提供する。
【図9B−2】選択したscFv抗体のCDR配列および結合特異性の概要を提供する。
【図9C】選択したscFv抗体のCDR配列および結合特異性の概要を提供する。
【図10】図8a〜8cで説明した一次CLL細胞対正常PBMCでのscFv抗原の発現レベルを比較する、フローサイトメトリー結果の概要を示す表2である。
【図11】scFv−9抗原の発現レベルを、CLL患者から単離した末梢血単核細胞中のCD38+細胞のパーセンテージと比較する、フローサイトメトリー結果の概要を示す表である。
【図12】免疫沈降および質量分光分析によるscFv抗原の同定を示す。CLL−AAT細胞を0.5mg/mlスルホ−NHS−LC−ビオチン(Pierce)のPBSによる溶液、pH8.0によって30分間標識した。未反応ビオチンを除去するためのPBSによる徹底的な洗浄の後、細胞を窒素キャビテーションによって破壊して、ミクロソーム画分を分画遠心法によって単離した。ミクロソーム画分をNP40溶解緩衝液中に再懸濁させて、正常ウサギ血清およびタンパク質Aセファロースによって徹底的に前処理した。抗原をラット抗HAアガロースビーズ(Roche)に結合したHA−tagged scFv抗体によって免疫沈降させた。免疫沈降の後、抗原をSDS−PAGEによって分離して、ストレプトアビジン−アルカリホスファターゼ(AP)を使用したウェスタンブロットによって、またはCoomassie G−250染色によって検出した。CLL−AAT細胞に結合しない抗体であるscFv−7を負の対照として使用した。抗原バンドをCoomassie染色ゲルから切除して、質量分光分析(MS)によって同定した。The Scripps Research InstituteのProteomics Core Facility(ラホーヤ、カリフォルニア州)にてMALDI−MSを実施した。μLC/MS/MSはHarvard Microchemistry Facility(ケンブリッジ、マサチューセッツ州)にて実施した。
【図13】3つのscFv抗体がヒトOX−2/CD200 cDNAクローンによって一時的にトランスフェクションされた293−EBNA細胞に特異的に結合することを示す。OX−2/CD200 cDNAをRT−PCRによってCLL細胞からクローニングして、哺乳類発現ベクターpCEP4(Invitrogen)内に挿入する。PCEP4−CD200プラスミドまたは対応する空のベクターpCEP4は、Polyfect試薬(QIAGEN)を用いて、293−EBNA細胞内にトランスフェクションした。トランスフェクションの2日後、細胞を、scFv抗体への結合についてフローサイトメトリーによって分析した。
【図14】OX−2/CD200トランスフェクション細胞の存在が、IL−2およびIFN−γなどのTh1サイトカインのダウンレギュレーションを引き起こしたことを示す。抗OX−2/CD200抗体の30μg/mlでの添加は、Th1反応を完全に回復した。
【図15】混合リンパ球反応におけるCLL細胞の存在が、IL−2に対するTh1反応のダウンレギュレーションを引き起こしたことを示す。
【図16】混合リンパ球反応におけるCLL細胞の存在が、IFN−γに対するTh1反応のダウンレギュレーションを引き起こしたことを示す。
【図17A】ヒトPBL細胞の存在下または非存在下のどちらかでのRAJI細胞4×106個を皮下注射されたNOD/SCIDマウスのすべての群の腫瘍体積の平均+/−SDを示す。
【図17B】ヒトPBL細胞の存在下または非存在下のどちらかでのRAJI細胞4×106個を皮下注射されたNOD/SCIDマウスのすべての群の腫瘍体積の平均+/−SDを示す。
【図18】2つのパラメトリック検定(Studentのt検定およびWelchの検定)および1つのノンパラメトリック検定(Wilcox検定)を使用して実施した統計解析の結果を示す。
【図19A】ヤギ抗マウスIgG Fc抗体に捕獲されたCD200−Fcに対する3回目のパニングの後の、代表的なIgG1カッパクローンのELISAの結果を示す。
【図19B】ヤギ抗マウスIgG Fc抗体に捕獲されたCD200−Fcに対する3回目のパニングの後の、代表的なIgG2aカッパクローンのELISAの結果を示す。
【図19C】マイクロタイターウェルに直接コーティングしたCD200−Fcに対する3回目のパニングの後の、代表的なIgG1カッパクローンのELISAの結果を示す。
【図19D】マイクロタイターウェルに直接コーティングしたCD200−Fcに対する3回目のパニングの後の、代表的なIgG2aカッパクローンのELISAの結果を示す。
【図20A】ヤギ抗マウスIgG Fcに捕獲されたCD200−Fcで選択された代表的なIgG1クローンのフローサイトメトリーの結果を示す。
【図20B】ヤギ抗マウスIgG Fcに捕獲されたCD200−Fcで選択された代表的なIgG2aクローンのフローサイトメトリーの結果を示す。
【図20C】直接コーティングされたCD200−Fcで選択された代表的なIgG1クローンのフローサイトメトリーの結果を示す。
【図20D】直接コーティングされたCD200−Fcで選択された代表的なIgG2aクローンのフローサイトメトリーの結果を示す。
【図21a−1】CD200特異性クローンの重鎖相補性領域の推定アミノ酸配列を示す。
【図21a−2】CD200特異性クローンの重鎖相補性領域の推定アミノ酸配列を示す。
【図21b−1】CD200特異性クローンの重鎖相補性領域の推定アミノ酸配列を示す。
【図21b−2】CD200特異性クローンの重鎖相補性領域の推定アミノ酸配列を示す。
【図22】蛍光ビーズアッセイにおける、選択したクローンがCD200のそのレセプター(CD200R)との間の相互作用を遮断する能力を示す。
【図23】キメラ化のための選択されたCD200 Fabの推定アミノ酸配列を示す。
【図24】小規模一過性トランスフェクションの培養上澄みから得たキメラIgGのELISA結果を示す。
【図25】CD200に対して作られたすべての抗体がレセプターリガンド相互作用を非常によく遮断することを示す、精製IgGのビーズ阻害アッセイの結果を示す。
【図26A】CLL細胞の存在が混合リンパ球反応で観察されたIFN−γおよびIL−2生成の大半を完全に排除したが、抗体のいずれかの存在がこれらのTh1サイトカインの生成を許容したことを示す。
【図26B】CLL細胞の存在が混合リンパ球反応で観察されたIFN−γおよびIL−2生成の大半を完全に排除したが、抗体のいずれかの存在がこれらのTh1サイトカインの生成を許容したことを示す。
【図26C】抗体の存在下でIL−10生成がダウンレギュレートされたことを示す。
【図27】抗体依存性細胞媒介性細胞傷害性アッセイ(ADCC)での腫瘍細胞を発現するCD200を殺す能力を示す。マウスキメラCD200抗体すべてが、CD200陽性細胞と共に培養されたときに、同様の溶解レベルを生じた。
【図28】対応のない両側Student t検定によって解析され、腫瘍細胞のみを投与された群と比較された、各種のPBLドナーを使用する10回の実験の代表的な例を示す。有意差は500万または1000万のPBLを投与された群で観察されたが、第32日以降100万のPBLを投与された群では観察されなかった。
【図29】対応のない両側Student t検定によって解析され、腫瘍細胞のみを投与された群と比較された、各種のPBLドナーを使用する10回の実験の代表的な例を示す。有意差は両方のドナーで1000万のPBLを投与された群で観察されたが、第8日以降200万のPBLを投与された群では観察されなかった。
【図30a】レンチウイルスベクターから形質導入された腫瘍細胞を発現するCD200に感染したRAJI細胞が、その親RAJI細胞よりもいくらか低速に増殖するように見えたことを示す。形質導入細胞および親細胞との増殖差は、統計的有意性には達しなかった。
【図30b】PBL 5または10×106を注射したときに、PBLの存在が逆CD200(非機能性CD200)によって形質導入されたRAJI細胞における腫瘍増殖を最大84%減少させたことを示し、この特定のドナーがRAJI腫瘍細胞を非常に強く拒絶することを示唆している。
【図30c】腫瘍細胞でのCD200発現が免疫系に腫瘍増殖の減速を実際に防止させることを示唆する結果を示す。またこの試験は、免疫抑制性化合物または分子を評価するためのRAJI/PBLモデルの有用性を証明する。
【図31a】RAJI/PBLモデルで見られる効果が、他の腫瘍細胞モデルでも観察できるかどうかを示す。Namalwa腫瘍細胞が、形質導入細胞と親細胞との間での有意差なしに、迅速な腫瘍増殖を生じたことを示す。
【図31b】RAJI/PBLモデルで見られる効果が、他の腫瘍細胞モデルでも観察できるかどうかを示す。PBLの存在が腫瘍増殖を約50%減速させたことを示す。両側Student t検定の結果は、PBL処置群と、腫瘍細胞のみを投与された群との差が統計的に有意であることを示した。
【図31c】RAJI/PBLモデルで見られる効果が、他の腫瘍細胞モデルでも観察できるかどうかを示す。Namalwa細胞を発現するCD200およびPBLを投与された群の腫瘍増殖が、Namalwa細胞を投与された群の腫瘍増殖と同様であったことを示す。これらのデータにより、腫瘍細胞でのCD200発現がヒト免疫系による腫瘍増殖の減速を防止することが確認される。
【図31d】RAJI/PBLモデルで見られる効果が、他の腫瘍細胞モデルでも観察できるかどうかを示す。Namalwa細胞を発現するCD200およびPBLを投与された群の腫瘍増殖が、Namalwa細胞を投与された群の腫瘍増殖と同様であったことを示す。これらのデータにより、腫瘍細胞でのCD200発現がヒト免疫系による腫瘍増殖の減速を防止することが確認される。
【図32】RAJI PBLモデルが免疫増強化合物の有効性を評価する効率的な方法であることを証明する結果を示す。
【図33】腫瘍細胞におけるCD200発現が、免疫細胞による腫瘍増殖の抑制を防止したことを示す。
【図34】腫瘍細胞におけるCD200発現が、免疫細胞による腫瘍増殖の抑制を防止したことを示す。
【発明を実施するための形態】
【0026】
(好ましい実施形態の詳細な説明)
本開示により、OX−2/CD200が被験体においてアップレギュレートされるかどうかを判定して、そうならば被験体に免疫反応を増強する療法を投与するための方法を提供する。適切な免疫調節療法の説明例は、T細胞または抗原提示細胞の負の調節を遮断する因子(たとえば抗CTLA4抗体、抗PD−L1抗体、抗PDL−2抗体、抗PD−1抗体など)の投与、あるいはT細胞の正の副刺激を増強する因子(たとえば抗CD40抗体または抗4−1BB抗体)の投与、あるいNK細胞数またはT細胞活性を増加させる因子(たとえば単独の、あるいはIMiD、サリドマイド、またはサリドマイド類似物質などのインヒビタと組合された、抗CD200抗体)の投与を含む。さらに免疫調節療法は、癌ワクチン、たとえば腫瘍細胞、腫瘍RNAまたは腫瘍DNAを負荷(load)した樹状細胞、腫瘍タンパク質または腫瘍ペプチド、患者由来熱ショックタンパク質(hsp)あるいはCpG、Luivac、Biostim、Ribominyl、Imudon、Bronchovaxomなどの免疫系を各種のレベルで刺激する一般的なアジュバントあるいは先天免疫系のレセプター(たとえばtoll様レセプター)を活性化する他の任意の化合物でありうる。また免疫調節療法は、IL−2、GM−CSFおよびIFN−γなどのサイトカインによる処置を含むことができる。
【0027】
特に有用な実施形態において、免疫反応を増強する療法は、単独での、または上述した免疫調節療法の1つと組合せた、OX−2/CD200に結合するポリペプチドの投与である。一般に、本開示で利用するポリペプチドは、当業者に公知である各種の技法を使用して構成できる。1つの実施形態において、ポリペプチドは化学合成によって得られる。他の実施形態において、ポリペプチドは抗体であるか、あるいは1つ以上の抗体の1つの断片または複数の断片から構成される。
【0028】
好ましくは、本開示の方法で利用するポリペプチドは、CLL細胞株から得られる。「CLL」は本明細書で使用するように、これに限定されるわけではないが、B細胞CLL(「B−CLL」)を含むB細胞およびT細胞の、これに限定されるわけではないが、各種の発生ステージを含む、任意のリンパ球を伴う慢性リンパ球性白血病を指す。B−CLLは本明細書で使用するように、CD5+、CD23+、CD20dim+、sIgdim+であり、細胞周期のG0/G1にて停止する成熟B細胞表現型による白血病を指す。さらなる局面において、CLL細胞株は、癌およびCLLを含む、OX−2/CD200がアップレギュレートされる疾患状態の診断および/または処置で有用な、抗体を含むポリペプチドを産生するために使用される。
【0029】
本明細書で使用するように、「抗体」という用語は、選択した標的に結合できる完全抗体または抗体断片を指す。Fab、Fv、scFv、Fab’およびF(ab’)2、モノクローナルおよびポリクローナル抗体、操作された抗体(キメラ、CDRグラフトおよびヒト化、完全ヒト抗体、および人工的に選択された抗体を含む)、ならびにファージ提示または代わりの技法を使用して生成された合成または半合成抗体が含まれる。小型断片、たとえばFvおよびscFvは、その小さなサイズおよび結果として生じる優れた組織分布によって、診断および治療用途のために好都合な特性を所有する。
【0030】
抗体は、本明細書で免疫原として開示された細胞を使用することによって産生でき、それによりモノクローナル抗体を単離できる動物における免疫反応を上昇させる。そのような抗体の配列を決定でき、組換え技法によって抗体またはその変異体を生成できる。本局面において、「変異体」は、モノクローナル抗体の配列をベースとするキメラ、CDRグラフト、ヒト化および完全ヒト抗体はもちろんのこと、OX−2/CD200に結合できるポリペプチドも含む。
【0031】
さらに組換えライブラリー(「ファージ抗体」)に由来する抗体は、標的特異性に基づいて抗体またはポリペプチドを単離するためのベイトとして、本明細書で述べた細胞、またはそれに由来するポリペプチドを使用して選択できる。
【0032】
なおさらなる局面において、抗体またはポリペプチドは、一次CLL細胞、またはそれに由来する抗原を使用して抗体ライブラリーをパニングすることによって、そしてさらにCLL細胞株、たとえば本明細書で述べるCLL細胞株を使用してスクリーニングおよび/または特徴付けることによって産生できる。したがってCLLに対して特異性の抗体またはポリペプチドを特徴付ける方法が提供され、該方法は抗体またはポリペプチドのCLL細胞株への結合を評価する工程を含む。
【0033】
(細胞株の調製)
細胞株は、当業者に公知の確立された方法に従って生成できる。一般に細胞株は、患者に由来する一次細胞を、培養物中に不死化細胞が自発的に産生されるまで培養することによって生成される。これらの細胞を次に単離して、さらに培養してアポトーシスに対して耐性を示すクローン細胞個体群または細胞を生成する。
【0034】
たとえばCLL細胞は、CLLに罹患している患者から採取した末梢血から単離できる。細胞を洗浄して、場合により存在する細胞の種類を決定するために免疫分類できる。次に細胞を培地、たとえばIL−4を含有する培地中で培養できる。好都合には、培養プロセスの間に培地の全部または一部を1回以上交換する。それによって細胞株を単離でき、培養物中の増殖向上によって同定できるであろう。
【0035】
1つの実施形態において、EBVによる不死化によって確立されない悪性起源のCLL細胞株が提供される。「悪性起源」は、たとえばEBVによって形質転換される非増殖性細胞とは対照的に、悪性CLL一次細胞からの細胞株の誘導を指す。本開示による有用な細胞株は、表現型においてそれ自体悪性であっても、なくてもよい。「悪性」表現型を有するCLL細胞は、細胞増殖の反復サイクルを特徴とする、基材培地とは結合していない細胞増殖を示し、アポトーシスに対する耐性を示す。一次CLL細胞に由来した細胞株は、ATCCアクセション番号PTA−3920で寄託されている。好ましい実施形態において、細胞株はCLL−AATである。CLL−AATは、B−CLL一次細胞に由来するB−CLL細胞株である。
【0036】
1つの実施形態において、CLL細胞で特異的に発現されたタンパク質は、当業者に公知の方法、たとえば免疫沈降とそれに続く質量分光分析によって、CLL−AAT細胞株を利用して同定される。そのようなタンパク質は、CLL−AAT細胞株において、またはCLL患者に由来する一次細胞において特異的に発現できる。
【0037】
小型細胞ライブラリー(多くは市販されている)は、細胞の増殖特徴を調節できる因子を同定するために、細胞ベースアッセイでCLL−AAT細胞株を使用してスクリーニングできる。たとえばCLL−AAT細胞株においてアポトーシスを調節する、またはその増殖および/または増殖を阻害する因子を同定できる。そのような因子は、治療化合物の開発のための候補である。
【0038】
CLL−AAT細胞株から単離された核酸は、CLL特異性遺伝子を同定するサブトラクティブハイブリダイゼーション実験またはマイクロアレイ分析(たとえば遺伝子チップ実験)で使用できる。CLL細胞において調節される遺伝子を同定できる。この方法で同定されるポリペプチドまたは核酸遺伝子生成物は、CLLの抗体または低分子療法の開発のための手がかりとして有用である。
【0039】
1つの実施形態において、CLL−AAT細胞株は、細胞表面成分に結合して、次に細胞によってインターナライズされる、インターナライズする抗体を同定するために使用できる。そのような抗体は、治療用途の候補である。特に細胞質中で安定のままであり、細胞内結合活性を保持する単鎖抗体は、この方法でスクリーニングできる。
【0040】
(モノクローナル抗体の調製)
組換えDNA技術は、本開示によって生成された抗体を改良するために使用できる。それゆえキメラ抗体は、診断または治療用途においてその免疫原性を低下させるために構成できる。さらに免疫原性は、CDRグラフト、そして場合によりフレームワーク修飾によって抗体をヒト化することによって最小限にできる。その内容が参考として本明細書中に援用されるU.S.Patent No.5,225,539を参照。
【0041】
抗体は動物血清から得られるか、あるいはモノクローナル抗体またはその断片の場合は細胞培養で生成できる。組換えDNA技術を使用して、細菌または好ましくは哺乳類細胞培養物中で確立された手順に従って抗体を生成できる。選択した細胞培養系は好ましくは抗体生成物を分泌する。
【0042】
別の実施形態において、本明細書で開示した抗体の生成のためのプロセスは、ハイブリッドベクターによって形質転換された宿主、たとえばE.coliまたは哺乳類細胞を培養することを含む。該ベクターは、適正な読み枠内にて抗体タンパク質をコードする第2のDNA配列に連結されたシグナルペプチドをコードする第1のDNA配列に作動的に連結されたプロモータを含有する1つ以上の発現カセットを含む。該抗体タンパク質は次に収集および単離される。場合により、発現カセットは、適正な読み枠内でシグナルペプチドのそれぞれ個別に作動的に連結された抗体タンパク質をコードする多シストロン性、たとえば2シストロン性DNA配列に作動的に連結されたプロモータを含むことができる。
【0043】
ハイブリドーマ細胞または哺乳類細胞のインビトロでの増殖は、哺乳類血清(たとえばウシ胎仔血清)、または微量元素および増殖維持サプリメント(たとえばフィーダー細胞、たとえば正常マウス腹腔滲出細胞、脾臓細胞、骨髄マクロファージ、2−アミノエタノール、インスリン、トランスフェリン、低密度リポタンパク質、オレイン酸など)を場合により補充した、慣例の標準培地(たとえばダルベッコ変法イーグル培地(DMEM)またはRPMI1640培地)を含む適切な培地中で実施される。細菌細胞または酵母細胞である宿主細胞の増殖は、当分野で公知の適切な培地中で同様に実施される。たとえばバクテリアでは適切な培地は、培地LE、NZCYM、NZYM、NZM、Terrific Broth、SOB、SOC、2xYT、またはM9最小培地を含む。酵母では、適切な培地は、培地YPD、YEPD、最小培地、またはComplete Minimal Dropout Mediumを含む。
【0044】
インビトロ生成は、比較的純粋な抗体調製物を提供し、より多くの量の所望の抗体を与えるためのスケールアップを可能にする。細菌細胞、酵母、植物、または哺乳類細胞培養のための技法は当分野で公知であり、均質懸濁培養(たとえばエアリフトリアクタ内または連続スターラーリアクタ内で)、そして固定化またはエントラップ細胞培養(たとえば中空繊維、マイクロカプセル内、アガロースマイクロビーズまたはセラミックカートリッジ上で)を含む。
【0045】
大量の所望の抗体は、哺乳類細胞を生体内で増殖することによっても得られる。この目的のために、所望の抗体を生成するハイブリドーマ細胞を組織適合哺乳類に注射して、抗体生成腫瘍の増殖を引き起こす。場合により、該動物は炭化水素、特にプリスタン(テトラメチル−ペンタデカン)などの鉱油によって刺激できる。1〜3週間後に、抗体をこれらの哺乳類の体液から単離する。たとえば適切なミエローマ細胞とBalb/cマウスからの抗体生成脾臓細胞との、または所望の抗体を生成するハイブリドーマ細胞株Sp2/0に由来するトランスフェクション細胞との融合によって得たハイブリドーマ細胞を、場合によってプリスチンによって前処置したBalb/cマウスに腹腔内注射する。1〜2週間後に腹水を該動物から採取する。
【0046】
上述の、または他の技法は、たとえばその開示が参照により本明細書にすべて組み入れられているKohler and Milstein,(1975)Nature 256:495−497;U.S.Patent No.4,376,110;Harlow
and Lane,Antibodies:a Laboratory Manual,(1988)Cold Spring Harborで議論されている。組換え抗体分子の調製技法は、上の参考文献に、またたとえばWO97/08320;U.S.Patent No.5,427,908;U.S.Patent No.5,508,717;Smith,1985,Science,Vol.225,pp 1315−1317;Parmley and Smith,1988,Gene 73,pp 305−318;De La Cruz et al.,1988,Journal of Biological Chemistry,263 pp 4318−4322;U.S.Patent No.5,403,484;U.S.Patent No.5223409;WO88/06630;WO92/15679;U.S.Patent No.5780279;U.S.Patent No.5571698;U.S.Patent No.6040136;Davis et al.,1999,Cancer Metastasis Rev.,18(4):421−5;Taylor,et al.,Nucleic Acids Research 20(1992):6287−6295;Tomizuka et al.,Proc.Natl.Academy of Sciences USA 97(2)(2000):722−727にも述べられている。これらのすべての参考文献の内容は、参考として本明細書中に援用される。
【0047】
細胞培養物上澄みを所望の抗体について、CLL細胞の免疫蛍光染色によって、免疫ブロッティングによって、酵素イムノアッセイによって、たとえばサンドイッチアッセイまたはドットアッセイ、あるいはラジオイムノアッセイによって選択的にスクリーニングする。
【0048】
抗体の単離では、培養物上澄み中または腹水中の免疫グロブリンを、たとえば硫酸アンモニウムによる沈降、ポリエチレングリコールなどの吸湿材による透析、選択膜による濾過などによって濃縮できる。必要および/または所望ならば、慣習的なクロマトグラフィー法、たとえばゲル濾過、イオン交換クロマトグラフィー、DEAE−セルロースでのクロマトグラフィーおよび/または(免疫)アフィニティクロマトグラフィー、たとえば本開示によるCLL細胞株に由来する1つ以上の表面ポリペプチドによって、あるいはタンパク質−AまたはGを用いたアフィニティクロマトグラフィーによって精製される。
【0049】
別の実施形態は、適切な哺乳類、たとえばウサギがプールされたCLL患者サンプルによって免疫化されることを特徴とする、細胞株に対して作られた抗体を分泌する細菌細胞株の調製のためのプロセスを提供する。免疫化ウサギから生成されたファージ提示ライブラリーが当分野で公知の方法(たとえば参考として本明細書中に援用される、各種の参考文献で開示された方法)に従って、構成され、所望の抗体についてパニングされる。
【0050】
モノクローナル抗体を分泌するハイブリドーマ細胞も検討される。好ましいハイブリドーマ細胞は遺伝的に安定であり、所望の特異性の本明細書で述べたモノクローナル抗体を分泌して、解凍および再クローニングによって深温冷凍培養物から活性化できる。
【0051】
別の実施形態において、本明細書で述べるCLL細胞株に対して作られたモノクローナル抗体を分泌するハイブリドーマ細胞株の調製のためのプロセスが提供される。そのプロセスでは、適切な哺乳類、たとえばBalb/cマウスを、本開示で述べた細胞に由来する1つ以上のポリペプチドまたはその抗原断片、細胞株自体、あるいは上述したような精製ポリペプチドを含有する抗原キャリアによって免疫化する。免疫化哺乳類の抗体生成細胞を培養物中で短期間増殖させるか、適切なミエローマ細胞株の細胞と融合させる。融合で得られたハイブリッド細胞をクローニングして、所望の抗体を分泌する細胞クローンを選択する。たとえば本細胞株によって免疫化したBalb/cマウスの脾臓細胞をミエローマ細胞株PAIまたはミエローマ細胞株Sp2/0−Ag 14の細胞と融合させて、得られたハイブリッド細胞を所望の抗体の分泌についてスクリーニングして、陽性ハイブリドーマ細胞をクローニングする。
【0052】
好ましいのは、本開示に従って複数回、たとえば4〜6回、数ヶ月に渡って、たとえば2〜4ヶ月の間、細胞株106〜107細胞を皮下および/または腹腔内注射することによって、Balb/cマウスを免疫化することを特徴とする、ハイブリドーマ細胞株の調製のプロセスである。免疫化マウスからの脾臓細胞は、最後の注射から2〜4日後に採取して、融合プロモータ、好ましくはポリエチレングリコールの存在下でミエローマ細胞株PAIの細胞と融合させる。好ましくは、約30%〜約50%のポリエチレングリコール(分子量約4000)を含有する溶液中で、ミエローマ細胞を3〜20倍過剰の免疫化マウスからの脾臓細胞と融合させる。融合後、細胞を、選択培地、たとえばHAT培地を添加した上述したような適切な培地中にて、正常ミエローマ細胞が所望のハイブリドーマ細胞を過剰増殖させるのを防止するために一定間隔で膨張させる。
【0053】
さらなる実施形態において、上述した細胞株に対して作られた抗体の重鎖可変ドメインおよび/または軽鎖可変ドメインをコードするインサートを含む組換えDNAが生成される。DNAという用語は、コード単鎖DNA、前記コードDNAおよびその相補性DNAより成る2本鎖DNA、またはこれらの相補性(単鎖)DNA自体を含む。
【0054】
さらに本明細書で開示された細胞株に対して作られた抗体の重鎖可変ドメインおよび/または軽鎖可変ドメインをコードするDNAは、重鎖可変ドメインおよび/または軽鎖可変ドメインをコードする真正DNA配列を有する酵素または化学合成DNA、あるいはその変異体でありうる。真正DNAの変異体は、1つ以上のアミノ酸が除去または1つ以上の他のアミノ酸と交換された、上述の抗体の重鎖可変ドメインおよび/または軽鎖可変ドメインをコードするDNAである。好ましくは前記修飾は、ヒト化および発現最適化用途における抗体の重鎖可変ドメインおよび/または軽鎖可変ドメインのCDRの外側である。変異体DNAという用語は、1つ以上のヌクレオチドが同じアミノ酸をコードする新しいコドンを持つ他のヌクレオチドと交換される、サイレント変異体も含む。変異体配列という用語は、縮重配列も含む。縮重配列は、最初にコードされたアミノ酸配列の変化を生じることなく、制限数のヌクレオチドが他のヌクレオチドによって置換されるという点で、遺伝子コードの意味の範囲内で縮重される。そのような縮重配列は、重鎖マウス可変ドメインおよび/または軽鎖マウス可変ドメインの最適発現を得るために、特異性宿主、特にE.coliによって好まれる、その各種の制限部位および/または特定のコドンの頻度のために有用なことがある。
【0055】
変異体という用語は、当分野で周知の方法に従って真正DNAのインビトロ変異誘発によって得られたDNA変異体を含むものとする。
【0056】
完全テトラマー免疫グロブリン分子の構築およびキメラ抗体の発現では、重鎖および軽鎖可変ドメインをコードする組換えDNAインサートを重鎖および軽鎖定常ドメインをコードする対応するDNAと融合させて、たとえばハイブリッドベクター内への包含後に、適切な宿主細胞内へ移動させる。
【0057】
ヒト定常ドメインgに融合された本明細書で開示された細胞株に対して作られた抗体の重鎖マウス可変ドメインをコードするインサートを含む組換えDNA、たとえばγ1、γ2、γ3またはγ4、好ましくはγ1またはγ4も提供される。ヒト定常ドメインκまたはλ、好ましくはκに縮合された本明細書で開示された細胞株に対して作られた抗体の軽鎖マウス可変ドメインをコードするインサートを含む組換えDNAも提供される。
【0058】
別の実施形態は、重鎖可変ドメインおよび軽鎖可変ドメインが、宿主細胞における抗体の処理を促進するシグナル配列を場合により含むスペーサー基によって連結される組換えポリペプチドをコードする組換えDNAおよび/または抗体および/または開裂部位および/またはペプチドスペーサーおよび/またはエフェクター分子の精製を促進するペプチドをコードするDNAに関する。
【0059】
エフェクター分子をコードするDNAは、診断または治療用途で有用なエフェクター分子をコードするDNAであるものとする。それゆえ毒素または酵素、特にプロドラッグの活性化を触媒できる酵素であるエフェクター分子が特に示される。そのようなエフェクター分子をコードするDNAは、天然発生型酵素または毒素コードDNA、またはその変異体の配列を有し、当分野で周知の方法によって調製できる。
本抗体/ポリペプチドの使用
本明細書で利用するポリペプチドおよび/または抗体は、特に診断および治療用途のために示されている。
【0060】
本抗体は、治療剤として癌患者、特にこれに限定されるわけではないがCLL患者に投与できる。一部の実施形態において、抗体はCD200とそのレセプターとの間の相互作用を妨害できる。この妨害は、CD200の免疫抑制効果を遮断できる。この方法で免疫反応を改善することによって、そのような抗体は癌細胞の根絶を促進できる。
【0061】
抗CD200抗体は、他の免疫調節化合物、ワクチンまたは化学療法と組合せて投与することもできる。たとえば抗CD25またはシクロホスファミドなどの試薬による既存の調節T細胞の除去は、抗CD200処置を開始する前に、1つの特定の有用な実施形態で実現される。また骨髄切除療法と、それに続く骨髄移植またはCLL細胞と反応するT細胞の養子移入の治療有効性は、抗CD200療法によって向上する。さらに抗CD200処置は、癌ワクチン、たとえばCLL細胞を負荷(load)した樹状細胞、そのような細胞に由来するタンパク質、ペプチドまたはRNA、患者由来熱ショックタンパク質(hsp)、腫瘍ペプチドまたは腫瘍タンパク質の有効性を実質的に向上させることができる。他の実施形態において、抗CD200抗体は免疫賦活化合物、たとえばCpG、toll様レセプターアゴニストまたは他の任意のアジュバント、抗CTLA−4抗体などと組合せて使用される。なお他の実施形態において、抗CD200処置の有効性は、免疫抑制性機構、たとえば抗PDL1および/または2抗体、抗IL−10抗体、抗IL−6抗体などを遮断することによって改善される。なお他の実施形態において、抗CD200処置の有効性は、NK細胞数またはT細胞を増加させる因子、たとえば低分子インヒビタIMiD、サリドマイド、またはサリドマイド類似物質の投与によって改善される)。
【0062】
本開示による抗CD200抗体は、診断ツールとして使用できる。たとえば造血性癌の患者から採取した血液を使用して、CD200の発現は、CLL細胞上のたとえばCD38およびCD19などの適切な癌細胞マーカーと組合せて抗CD200抗体を使用するFACS分析によって、癌細胞上で評価できる。抗CD200抗体による処置には、正常B細胞で見出されるレベルよりも少なくとも1.4倍高いCD200レベルの患者を選択できる。
【0063】
本抗CD200抗体を診断ツールとして使用する別の例において、悪性腫瘍を持つ患者からの生検を採取して、抗CD200抗体を使用するFACS分析によってCD200の発現を判定する。腫瘍細胞がCD200を、対応する正常組織と比較して少なくとも1.4倍高いレベルで発現する場合、免疫調節療法のために癌患者を選択する。免疫調節療法は抗CD200療法でありうるが、患者の免疫系に影響を及ぼす他の任意の療法でもよい。適切な免疫調節療法の例は、T細胞または抗原提示細胞の負の調節を遮断する因子(たとえば抗CTLA4、抗PD−L1、抗PDL−2、抗PD−1)の投与またはT細胞の正の副刺激を増強する因子(たとえば抗CD40または抗4−1BB)の投与を含む。さらに免疫調節療法は、NK細胞数またはT細胞活性を増加させる因子(たとえば単独の、あるいはIMiD、サリドマイド、またはサリドマイド類似物質などのインヒビタと組合された、抗CD200抗体)の投与あるいは調節T細胞を消耗する因子(たとえば単独の、またはONTAKと組合せた抗CD200抗体)の投与でもよい。さらに免疫調節療法は、癌ワクチン、たとえば腫瘍細胞、腫瘍RNAまたは腫瘍DNAを負荷(load)した樹状細胞、腫瘍タンパク質または腫瘍ペプチド、患者由来熱ショックタンパク質(hsp)あるいはCpG、Luivac、Biostim、Ribominyl、Imudon、Bronchovaxomなどの免疫系を各種のレベルで刺激する一般的なアジュバントあるいは先天免疫系のレセプター(たとえばtoll様レセプター)を活性化する他の任意の化合物でありうる。または療法は、IL−2、GM−CSFおよびIFN−γなどのサイトカインによる処置を含む。
【0064】
本開示による別の実施形態において、治療処置の進行および/または有効性をモニターするための方法が提供される。該方法は、療法の有効性を決定するために、被験体におけるOX−2/CD200レベルを少なくとも2回決定する工程とを含む。たとえばOX−2/CD200の前処置レベルが確認でき、療法の少なくとも1回の投与の後に、OX−2/CD200のレベルを再度決定できる。OX−2/CD200レベルの低下は、有効な処置を示す。OX−2/CD200レベルの測定は、開業医によって療法の投薬量または頻度を増加するための指針として使用できる。OX−2/CD200レベルを直接モニターできることを、あるいはOX−2/CD200に相関する任意のマーカーをモニターできることをもちろん理解すべきである。
【0065】
本抗体はまた、生体内で癌性細胞を検出するために利用できる。これは抗体を標識することと、標識した抗体を被験体に投与することと、次に被験体を撮像することとによって実現される。本開示による診断撮像に有用な標識の例は、131I、111In、123I、99mTc、32P、125I、3H、14C、および188Rhなどの放射性標識、フルオレセインおよびローダミンなどの蛍光標識、核磁気共鳴活性標識、陽電子放出断層撮影(「PET」)スキャナによって検出できる陽電子放出同位体、ルシフェリンなどの化学発光剤、ならびにペルオキシダーゼまたはホスファターゼなどの酵素マーカーである。短距離検出器プローブ、たとえば経直腸プローブによって検出可能な短距離放射線エミッター、たとえば同位体も利用できる。抗体は、そのような試薬によって、当分野で公知の技法を使用して標識できる。たとえば抗体の放射性標識に関する技法については、参考として本明細書中に援用されるWensel and Meares,Radioimmunoimaging and Radioimmunotherapy,Elsevier,N.Y.(1983)を参照。参考として本明細書中に援用されるD.Colcher et al.,“Use of Monoclonal Antibodies
as Radiopharmaceuticals for the Localization of Human Carcinoma Xenografts in Athymic Mice”,Meth.Enzymol.121:802−816(1986)も参照。
【0066】
本開示による放射性標識抗体は、インビトロ診断試験に使用できる。抗体、その結合部分、プローブ、またはリガンドの特異性活性は、半減期、放射性標識の同位体純度、および生物因子に標識が包含される方法によって変わる。イムノアッセイ試験では、一般に特異性活性が高くなればなるほど、感度がより良好になる。放射性同位体によって抗体を標識するための手順は、当分野で一般に公知である。
【0067】
放射性標識抗体は患者に投与することができ、患者内で抗体が反応する抗原を持つ癌細胞に局在化して、たとえばγカメラまたは放出断層撮影を使用する放射核スキャンなどの公知の技法を用いて、生体内で検出または「撮像」される。たとえば参考として本明細書中に援用されるA.R.Bradwell et al.,“Developments
in Antibody Imaging”,Monoclonal Antibodies for Cancer Detection and Therapy,R.W.Baldwin et al.,(eds.),pp.65−85(Academic
Press 1985)を参照。あるいは、Brookhaven National
Laboratoryに配置されているPet VIと呼ばれるような陽電子放出横断断層撮影スキャナを放射性標識が陽電子(たとえば11C、18F、15O、および13N)を放出するところで使用できる。
【0068】
蛍光団および発色団標識生物因子は、当分野で周知の標準部分から調製できる。抗体および他のタンパク質は約310nmまでの波長を有する光を吸収するため、蛍光部分は、310nmを超える、好ましくは400nmを超える波長にて実質的な吸収を有するように選択すべきである。各種の適切な蛍光団および発色団は、参考として本明細書中に援用されるStryer,Science,162:526(1968)およびBrand,L.et al.,Annual Review of Biochemistry,41:843−868(1972)によって述べられている。抗体は、参考として本明細書中に援用されるU.S.Patent Nos.3,940,475、4,289,747、および4,376,110に開示された手順などの従来手順によって、蛍光発色団基によって標識できる。
【0069】
他の実施形態において、二重特異性抗体が検討される。二重特異性抗体は、少なくとも2つの異なる抗原に結合特異性を有するモノクローナル、好ましくはヒトまたはヒト化抗体である。この場合では、結合特異性の一方が癌細胞のCD200抗原に対してであり、他方が他の任意の抗原に対して、好ましくは細胞表面タンパク質またはレセプターまたはレセプターサブユニットに対してである。
【0070】
二重特異性抗体を作成するための方法は、当業者の範囲内である。伝統的には、二重特異性抗体の組換え生成は、2つの重鎖が異なる特異性を有する、2つの免疫グロブリン重鎖/軽鎖対の同時発現に基づいている(Milstein and Cuello,Nature,305:537−539(1983))。所望の結合特異性を持つ抗体可変ドメイン(抗体−抗原複合部位)を免疫グロブリン定常ドメイン配列に融合することができる。融合は好ましくは、ヒンジ、CH2、およびCH3領域の少なくとも一部を含む、免疫グロブリン重鎖定常ドメインによってである。免疫グロブリン重鎖融合、そして所望ならば免疫グロブリン軽鎖をコードするDNAは、独立した発現ベクターに挿入されて、適切な宿主生物内へ同時トランスフェクションされる。二重特異性抗体を産生する説明的な現在周知のさらなる詳細事項については、たとえばSuresh et al.,Methods in Enzymology,121:210(1986);WO 96/27011;Brennan et al.,Science 229:81(1985);Shalaby et al.,J.Exp.Med.175:217−225(1992);Kostelny et al.,J.Immunol.148(5):1547−1553(1992);Hollinger et al.,Proc.Natl.Acad.Sci.USA 90:6444−6448(1993);およびGruber et al.,J.Immunol.152:5368(1994);およびTutt et al.,J.Immunol.147:60(1991)を参照。
【0071】
本抗体は、癌性細胞を生体内で直接殺すために、または切除するためにも使用できる。これは、(場合により細胞傷害薬に融合できる)抗体を、そのような処置を必要とする被験体に投与することを含む。抗体は癌細胞のCD200を認識するため、抗体が結合するそのような任意の細胞が破壊される。
【0072】
癌細胞を殺すために、または切除するために抗体を単独で使用するとき、そのような致死または切除は、内因性宿主免疫機能、たとえば補体媒介性または抗体依存性細胞傷害を開始することによって実施できる。細胞をこの方法で殺すかどうかを判断するためのアッセイは、当業者の範囲内である。
【0073】
本開示の抗体は、各種の細胞傷害性化合物を送達するために使用できる。任意の細胞傷害性化合物を本抗体に融合できる。融合は化学的または遺伝子的に実現できる(たとえば発現を介して、単一の融合分子として)。細胞傷害性化合物は生物製剤、たとえばポリペプチド、または低分子でありうる。当業者が認識するように、低分子には化学融合が使用されるが、これに対して生物化合物では、化学または遺伝子融合のどちらかを使用できる。
【0074】
細胞傷害性化合物の非制限的な例は、治療薬は治療薬、放射線を放出する化合物、植物、真菌、または細菌起源の分子、生物タンパク質、およびその混合物を含む。細胞傷害性薬は、細胞内作用細胞傷害性薬、たとえば短距離、高エネルギーα−エミッターを含む、たとえば短距離放射線エミッターでありうる。酵素活性毒素およびその断片はたとえば、ジフテリア毒素A断片、ジフテリア毒素の非結合活性断片、(Pseudomonas aeruginosaからの)外毒素A、リシンA鎖、アブリンA鎖、モデクシンA鎖、アルファ−サクリン、あるAleurites fordiiタンパク質、あるDianthinタンパク質、Phytolacca americanaタンパク質(PAP、PAPIIおよびPAP−S)、Morodica charantiaインヒビタ、クルシン、クロチン、Saponaria officinalisインヒビタ、ゲロニン、マイトジリン、レストリクトシン、フェノマイシン、およびエノマイシンによって例示される。免疫毒素の酵素活性ポリペプチドを調製する手順は、参考として本明細書中に援用されるWO84/03508およびWO85/03508に述べられている。ある細胞傷害性部分は、たとえばアドリアマイシン、クロラムブシル、ダウノマイシン、メトトレキサート、ネオカルチノスタチン、および白金に由来する。
【0075】
抗体を細胞傷害性剤と結合する手順は上述されており、当業者の範囲内である。
【0076】
あるいは抗体は高エネルギー放射線エミッター、たとえば放射性同位体、たとえば131I、腫瘍部位に局在化するときに複数の細胞直径の致死を引き起こすγ−エミッターに結合できる。たとえば参考として本明細書中に援用される、S.E.Order,“Analysis,Results,and Future Prospective of
the Therapeutic Use of Radiolabeled Antibody in Cancer Therapy”,Monoclonal Antibodies for Cancer Detection and Therapy,R.W.Baldwin et al.(eds.),pp 303−316(Academic Press 1985)を参照。他の適切な放射性同位体は、α−エミッター、たとえば212Bi、213Bi、および211At、ならびにβ−エミッター、たとえば186Reおよび90Yを含む。
【0077】
本抗体(純抗体、標識抗体、毒素に融合された抗体などにかかわらず)の抗体投与の経路は、公知の方法、たとえば静脈内、腹腔内、脳内、筋肉内、皮下、眼内、動脈内、髄腔内、吸入または病巣内経路による注射または輸液、あるいは持続放出システムによる。該抗体は好ましくは、輸液によって、またはボーラス注射によって連続的に投与される。抗体を局所的または全身的方法で投与できる。
【0078】
本抗体は、薬学的に受容可能なキャリアとの混合物として調製できる。本出願の化合物の処方および投与のための技法は、“Remington’s Pharmaceutical Sciences,”Mack Publishing Co.,Easton,PA,最新版に見出すことができる。本治療組成物は静脈内に、あるいは鼻または肺を通じて、好ましくは液体または粉末エアゾール(凍結乾燥)として投与できる。該組成物は、所望通りに非経口的に、または皮下的にも投与できる。全身投与する場合、該治療組成物は、滅菌されており、発熱物質を含まず、pH、等張性、および安定性に十分に注意を払った非経口的に許容される溶液とすべきである。これらの条件は当業者に公知である。
【0079】
使用するのに適した製薬組成物は、本抗体の1つ以上がその本来の目的を達成するために有効な量で含有される組成物を含む。さらに詳細には、治療的有効量は、疾患の症状を防止、軽減または改善するために、あるいは処置される被験体の生存を延長するために有効な抗体の量を意味する。治療的有効量の決定は、特に本明細書で提供する詳細な開示に照らして、十分に当業者の能力の範囲内である。治療的有効投薬量は、インビトロおよび生体内方法を使用して決定できる。
【0080】
一部の実施形態において、本CD200結合抗体は、CD200によって白血病細胞を直接標的化することによって、CLLにおける免疫抑制を遮断する利益を提供する。詳細には、免疫系の刺激によって、脾臓およびリンパ節からのCLL細胞の根絶を可能にできる。出願人は、これらの微環境からのCLL細胞の根絶が、単にB細胞を標的とする因子(たとえばアレムツズマブ)によって成功したことを一切認識していない。これに対して、CLL反応性T細胞は、抗体よりもこれらの臓器へより良好なアクセスを有することができる。他の実施形態において、直接細胞致死は、CLL細胞に抗CD200Abをタグ付けすることによって実現される。
【0081】
特に有用な実施形態において、直接細胞致死およびTh1プロフィールへの免疫反応の推進の組合せは、癌処置への特に強力な手法を提供する。それゆえ1つの実施形態において、CD200に結合して、a)CD200とそのレセプターとの間の相互作用を遮断して、同時にb)CD200を発現する癌細胞を直接殺す抗体または抗体断片を癌患者に投与する癌処置が提供される。癌細胞が殺す機構は、これに限定されるわけではないが、抗体依存性細胞傷害(ADCC)または補体依存性細胞傷害(CDC);毒素との融合;放射性標識との融合;細胞致死に関与する生物因子、たとえばグランザイムBまたはパーフォリンとの融合;細胞傷害性ウイルスとの融合;TNF−αまたはIFN−αなどのサイトカインとの融合を含むことができる。代わりの実施形態において、癌処置は、a)CD200とそのレセプターとの間の相互作用を遮断して、同時にb)腫瘍に対する細胞傷害性T細胞またはNK細胞活性を増強する抗体を投与することを含む。細胞傷害性T細胞またはNK細胞活性のそのような増強はたとえば、たとえばIL−2、IL−12、IL−18、IL−13、およびIL−5などのサイトカインと融合させることにより組合せられる。加えてそのような増強は、IMiD、サリドマイド、またはサリドマイド類似物質などのインヒビタと組合せた抗CD200抗体の投与によって実現できる。
【0082】
なお別の実施形態において、癌処置は、a)CD200とそのレセプターとの間の相互作用を遮断して、同時にb)T細胞を腫瘍細胞に誘引する抗体を投与することを含む。T細胞誘引は、AbをMIG、IP−10、1−TAC、CCL21、CCL5またはLIGHTなどのケモカインと融合することによって実現できる。免疫抑制の遮断および腫瘍細胞の抗体標的化による直接致死の複合作用は、有効性の向上をもたらす独自の手法である。
【0083】
上の開示は抗体に関してであったが、一部の実施形態では、そのような抗体に由来するポリペプチドを本開示に従って利用できる。
【0084】
(CLL細胞株の使用)
CLL細胞株は、CLL、癌、およびOX−2/CD200のアップレギュレートされたレベルを特徴とする他の疾患状態、たとえばメラノーマの診断および処置を開発するための重要なツールを提供するため、その開発には多くの利点がある。
【0085】
本開示による細胞株は、CLLおよびOX−2/CD200のアップレギュレートされたレベルを特徴とする他の疾患状態に関する病因論、病原および生物学についてのインビトロ試験に使用できる。これはこれらの疾患の療法に有用である適切な因子の同定を補助する。
【0086】
細胞株は、上で言及したようなCLL、癌、およびOX−2/CD200のアップレギュレートされたレベルを特徴とする他の疾患状態(たとえばメラノーマ)のインビトロおよび生体内診断のための、そして他の方法によって、たとえば抗体ライブラリーをCLL患者に由来する一次細胞および/または抗原によってパニングすることによって生成された抗体のスクリーニングおよび/または特徴付けのための、ポリペプチドおよび/またはモノクローナル抗体を生成するためにも使用できる。
【0087】
細胞株はそのようなものとして使用できるか、またはそれから抗原を誘導できる。好都合なことに、そのような抗原は、CLLに対して特異性の細胞表面抗原である。それらは本開示による細胞株から直接単離できる。あるいは本明細書で述べた細胞株から作成したcDNA発現ライブラリーは、抗CLL抗体の選択および特徴付けならびに新規CLL特異性抗原の同定に有用である、CLL特異性抗原を発現するために使用できる。
【0088】
モノクローナル抗体療法を使用するCLLの処置は、当分野で提案されている。近年、Hainsworth(Oncologist 5(5)(2000)376−384)は、モノクローナル抗体に由来する最新の療法について述べている。リンパ球性白血病は特に、リンパ球腫瘍での複数のリンパ球特異性抗原の存在によるこの治療手法のための良好な候補であると考えられる。
【0089】
既存の抗体療法(たとえば、Bリンパ球の表現で発現される、CD20抗原に対して作られたリツキシマブTM)は、あるリンパ球性疾患に対してうまく使用されてきた。しかしながら低密度CD20抗原は、CLLにおけるBリンパ球の表面で発現される(Almasri et al.,Am.J.Hematol.,40(4)(1992)259−263)。
【0090】
それゆえ本明細書で述べるCLL細胞株は、本CLL細胞株の1つ以上の抗原決定基に対して特異性を有する新規の抗CLL抗体およびポリペプチドの開発、そしてCLL、癌、およびOX−2/CD200のアップレギュレートされたレベルを特徴とする他の疾患状態の療法および診断でのその使用を可能にする。
【0091】
抗体またはポリペプチドは、OX−2/CD200が通常相互作用するレセプターに結合して、それによりOX−2/CD200が該レセプターに結合するのを防止または阻害できる。なお別の代案として、抗体はOX−2/CD200の発現を調節する抗原に結合して、それによりOX−2/CD200の正常な、または上昇した発現を防止または阻害できる。OX−2/CD200の存在が免疫反応の低下と関連しているため、OX−2/CD200の代謝経路を妨害することが望ましいので、患者の免疫系は疾患状態、たとえば癌またはCLLから、さらに効果的に防御できる。
【0092】
特に有用な実施形態において、ポリペプチドはOX−2/CD200に結合する。1つの実施形態において、ポリペプチドは、OX−2/CD200に結合して、OX−2/CD200が他の分子またはレセプターと相互作用するのを防止または阻害する抗体でありうる。OX−2/CD200を過剰発現するCLL細胞および他の細胞がTh1サイトカインの生成を大幅に減少させるので、OX−2/CD200に結合する抗CD200抗体またはポリペプチドの、OX−2/CD200のアップレギュレートされたレベルを有する被験体への投与は、Th1サイトカインプロフィールを回復する。それゆえこれらのポリペプチドおよび/または抗体は、CLLおよび他の癌またはOX−2/CD200を過剰発現する疾患の処置における有用な治療剤でありうる。
【0093】
それゆえ別の実施形態において、本開示の方法は、被験体をOX−2/CD200の存在についてスクリーニングする工程と、OX−2/CD200に結合するポリペプチドを投与する工程とを含む。もちろん、OX−2/CD200の存在を直接モニターできること、あるいはOX−2/CD200と相関する任意のマーカーを検知できることを理解すべきである。特に有用な実施形態において、CLL患者をOX−2/CD200の過剰発現についてスクリーニングして、OX−2/CD200に結合する抗体を患者に投与する。1つのそのような抗体は、Serotec Inc.(3200 Atlantic Ave,Suite 105,Raleigh,NC 27604)から市販されている抗CD200抗体である。以下で詳細に述べるように、別のそのような抗体は、OX−2/CD200に結合するscFv−9である(図9B)。
【0094】
別の局面において、本開示は、癌細胞によって発現された分子の免疫調節効果を評価するための方法を提供する。これらの方法では、癌細胞によって発現またはアップレギュレートされる分子を最初に同定する。該分子は、データベースから、または実験的に同定できる。癌細胞によって発現またはアップレギュレートされる分子を同定するデータベースが公知であり、たとえばNCI60癌マイクロアレイプロジェクト(http://genome−www.stanford.edu/nci60/)(Ross et al.,Nature Genetics 24:227−34,2000)、癌腫分類(http://www.gnf.org/cancer/epican/)(Andrew
I.Su et al.,“Molecular Classification of Human Carcinomas by Use of Gene Expression Signatures.”Cancer Research 61:7388−7393,2001)、およびリンパ腫/白血病分子プロファイリングプロジェクト(http://llmpp.nih.gov/lymphoma/)(Alizadeh
et al.,Nature 403:503−11,2000)を含む。
【0095】
癌細胞によって発現またはアップレギュレートされる分子を同定するための実験方法も公知であり、たとえばマイクロアレイ実験、定量的PCR、FACS、およびノーザン分析を含む。
【0096】
癌細胞、リンパ球および癌細胞によって発現される以前に同定された分子を被験体に投与して、癌細胞の増殖の速度をモニターする。本方法では、任意の種類の癌細胞を利用できる。一部の実施形態において、癌細胞は免疫抑制性化合物を発現する。特に有用な実施形態において、癌細胞はCD200を発現、または過剰発現さえする。適切な癌細胞は、これに限定されるわけではないが、RAJIまたはNamalwa細胞株などのリンパ腫細胞株を含む。投与される癌細胞の量は、約1×106〜約20×106個の範囲である。
【0097】
本プロセスでは、任意の種類のリンパ球を利用できる。適切なリンパ球はたとえばPBL、T細胞、細胞傷害性T細胞、樹状細胞またはNK細胞を含む。特に有用な実施形態において、リンパ球はヒトリンパ球、詳細にはヒトPBLである。投与されるリンパ球の数は、a)癌細胞の増殖を減速するのに十分であるように、またはb)癌細胞の増殖を減速するのに不十分であるようにのいずれかで規定される。投与されるリンパ球の量は、投与されるリンパ球の数が癌細胞の増殖を減速するのに十分であるようにするときには、投与された癌細胞の数と同じか、それ以上でよい。実施形態において、投与されるリンパ球の数が癌細胞の増殖を減速するのに十分であるようにするときには、投与されるリンパ球の数は約5×106〜約10×106個でよい。投与されるリンパ球の量は、投与されるリンパ球の数が癌細胞の増殖を減速するのに不十分であるようにするときは、投与された癌細胞の数よりも少なくてよい。実施形態において、投与されるリンパ球の数が癌細胞の増殖を減速するのに不十分であるようにするときは、投与されるリンパ球の量は約1×106〜約4×106個の範囲でありうる。上述の量は例示的であり、他の適切な量を実験的に決定して本方法で使用できる。
【0098】
癌細胞によって発現またはアップレギュレートされる分子は、(単離されたか、または組換え産生されたかのいずれかの)分子またはその活性部分自体として、あるいは分子を自然に生成する細胞を投与することによって、あるいは分子またはその部分を生成するために操作された細胞を投与することによって、投与できる。所望の分子を発現させるために細胞を操作するための方法は、当業者に公知である。投与される分子の量は、健常者に見出される量を超える任意の量でよい。たとえば投与される分子の量は、健常者の同じ種類の細胞で見出される量の1.4倍超〜健常者の同じ種類の細胞で見出される量の約10,000倍超でよい。評価される分子がある程度の免疫調節活性を有することが既知であること、または既知でないことを理解すべきである。それゆえ本方法は、分子の免疫調節効果を確認するためにはもちろんのこと、最初からそのような活性を判定するためにも使用される。
【0099】
癌細胞、リンパ球および分子を投与する被験体として、任意の小動物を選択できる。被験体は好都合に免疫不全にすることができる。適切な小動物はたとえば免疫不全マウス、照射ラット、照射モルモットなどを含む。
【0100】
癌細胞の増殖速度は、旧来の技法を使用してモニターできる。たとえば腫瘍増殖は、長さおよび幅をカリパスで測定することによってモニターできる。腫瘍体積はたとえば、腫瘍の長さに腫瘍の幅を掛けて、次に腫瘍の幅の半分を掛けることに基づいて算出できる。癌細胞増殖の速度は、たとえば週に3回など、定期的に測定できる。癌細胞およびリンパ球のみが投与されたときの癌細胞の増殖速度は、癌細胞およびリンパ球のみを対照被験体に投与して、腫瘍サイズを定期的に測定することによって決定できる。あるいは癌細胞およびリンパ球のみを最初に投与することができ、いったんベースライン増殖速度が確立されたら、癌細胞によって発現またはアップレギュレートされる分子を次に被験体に投与でき、第2の投与後の癌細胞の増殖の速度を測定できる。あるいは全身モデルによって、癌細胞増殖の速度をFACS、生存または他の旧来の技法を使用してモニターできる。
【0101】
腫瘍細胞およびドナーリンパ球のみが投与されたときの増殖速度と比較して、癌細胞の増殖速度の何らかの変化が観察される場合、分子は免疫調節効果を有すると考えられる。投与されるリンパ球の数が癌細胞の増殖を減速させるのに十分であり、観察された腫瘍細胞の増殖速度が、腫瘍細胞およびドナーリンパ球のみが投与されたときの増殖速度と比較して高い場合、癌細胞によって発現またはアップレギュレートされる分子は免疫抑制性であると見なされる。投与されるリンパ球の数が癌細胞の増殖を減速するのに不十分であり、観察された腫瘍細胞の増殖速度が、腫瘍細胞およびドナーリンパ球のみが投与されたときの増殖速度と比較して低い場合、分子は免疫増強性であると見なされる。通例、観察された癌細胞の増殖速度は、腫瘍細胞およびドナーリンパ球のみが投与されたときの増殖速度よりも約20〜約1,000%高く、免疫抑制または免疫増強効果が統計的に有意となる。
【0102】
分子の免疫調節効果がいったん確立されると、本明細書に述べた実施形態によって、分子の活性の増強または阻害のどちらかを行う化合物を同定できる。免疫調節効果を有することが以前に見出された、分子の活性の増強または阻害のどちらかを行う化合物は、免疫調節効果を提供するタンパク質/タンパク質相互作用を変化させる任意の化合物でありうる。増強または阻害効果は、癌細胞によって発現またはアップレギュレートされる化合物との直接相互作用の結果でありうるか、あるいは癌細胞によって発現またはアップレギュレートされる化合物の代謝経路において他の化合物との間の相互作用の結果でありうる。
【0103】
たとえば、免疫調節効果を有することが以前に見出された分子、分子が相互作用するレセプター、または免疫調節効果に関与する代謝経路におけるある他の分子と相互作用する、抗体または機能性抗体断片を同定できる。抗体(抗体ライブラリーを含む)を作成するための、そしてそれらを阻害または増強効果についてスクリーニングするための技法は、当業者に明らかとなるであろう。別の例として、低分子を阻害または増強効果についてスクリーニングできる。低分子ライブラリーを阻害または増強効果についてスクリーニングする技法は、当業者に明らかとなるであろう。
【0104】
別の局面において、化合物の免疫調節効果を評価するための方法が本開示によって検討される。ヒト免疫系に作用する化合物または分子の免疫調節特性を証明することは、小動物モデルでは非常に困難である。しばしば該化合物は小動物の免疫系に作用せず、マウスにおけるヒト免疫系の再構成を必要とする。再構成は、各種の胎生免疫臓器をマウスにグラフトすることによって、またはヒトリンパ球の注射によって実現できるが、今日までに説明されたモデルのいずれも、化合物または分子の免疫調節特性の証明に有用であることが判明していない。免疫系は癌細胞の根絶に重要な役割を果たすと考えられている。癌細胞は、免疫抑制性レセプターのアップレギュレーションによって免疫系を回避するための方法を見出している。
【0105】
化合物の免疫調節効果を評価する本方法は、ドナーリンパ球(たとえばPBL)を輸液される白血病患者で観察されたグラフト対白血病効果を模倣するモデルによって実現され、患者の80%で寛解を引き起こす。該方法は、癌細胞、リンパ球および評価される化合物を被験体に投与することを含み、癌細胞の増殖の速度をモニターする。
【0106】
本方法では任意の種類の癌細胞を利用できる。一部の実施形態では、癌細胞は免疫抑制性化合物を発現する。特に有用な実施形態において、癌細胞はCD200を発現、または過剰発現さえする。適切な癌細胞は、これに限定されるわけではないが、RAJIまたはNamalwa細胞株などのリンパ腫細胞株を含む。投与される癌細胞の量は、約1×106〜約20×106個の範囲である。
【0107】
本プロセスでは、任意の種類のリンパ球を利用できる。適切なリンパ球はたとえば、PBL、樹状細胞、T細胞、細胞傷害性T細胞、NK細胞を含む。特に有用な実施形態において、リンパ球はヒトリンパ球、詳細にはヒトPBLである。投与されるリンパ球の数は、a)癌細胞の増殖を減速するのに十分であるように、またはb)癌細胞の増殖を減速するのに不十分であるように、のいずれかで規定される。投与されるリンパ球の量は、投与されるリンパ球の数が癌細胞の増殖を減速するのに十分であるようにするときには、投与された癌細胞の数と同じか、それ以上でよい。実施形態において、投与されるリンパ球の数が癌細胞の増殖を減速するのに十分であるようにするときには、投与されるリンパ球の数は約5×106〜約10×106個でよい。投与されるリンパ球の量は、投与されるリンパ球の数が癌細胞の増殖を減速するのに不十分であるようにするときは、投与された癌細胞の数よりも少なくてよい。実施形態において、投与されるリンパ球の数が癌細胞の増殖を減速するのに不十分であるようにするときは、投与されるリンパ球の量は約1×106〜約4×106個の範囲でありうる。上述の量は例示的であり、他の適切な量を実験的に決定して本方法で使用できる。
【0108】
評価される化合物は、免疫調節効果が決定されることを求めている任意の化合物でよい。化合物の説明例は、本明細書で上述した抗体およびペプチドを含む。投与される、評価される化合物の量は、約1mg/kg〜約200mg/kgの範囲でありうる。評価される化合物は、化合物自体として、あるいは化合物を自然に生成する細胞を投与することによって、あるいは化合物を生成するために操作された細胞を投与することによって、投与できる。評価される化合物がある程度の免疫調節活性を有することが既知であること、または既知でないことを理解すべきである。それゆえ本方法は、分子の免疫調節効果を確認するためにはもちろんのこと、最初からそのような活性を判定するためにも使用される。
【0109】
癌細胞、リンパ球および分子を投与する被験体として、任意の小動物を選択できる。被験体は好都合に免疫不全にすることができる。適切な小動物はたとえば免疫不全マウス、照射ラット、照射モルモットなどを含む。
【0110】
癌細胞の増殖速度は、旧来の技法を使用してモニターできる。たとえば腫瘍増殖は、長さおよび幅をカリパスで測定することによってモニターできる。腫瘍体積はたとえば、腫瘍の長さに腫瘍の幅を掛けて、次に腫瘍の幅の半分を掛けることに基づいて算出できる。癌細胞増殖の速度は、たとえば週に3回など、定期的に測定できる。癌細胞およびリンパ球のみが投与されたときの癌細胞の増殖速度は、癌細胞およびリンパ球のみを対照被験体に投与して、腫瘍サイズを定期的に測定することによって決定できる。あるいは癌細胞およびリンパ球のみを最初に投与することができ、いったんベースライン増殖速度が確立されたら、評価される化合物を次に被験体に投与でき、第2の投与後の癌細胞の増殖の速度を測定できる。
【0111】
リンパ腫細胞株RAJIまたはNamalwaなどの癌細胞を免疫不全マウスに注射するとき、500万〜1000万個のPBLの投与は、腫瘍増殖の著しい減速を引き起こす。これに対して、少ない数のPBL(ドナーによって1〜200万個)は、腫瘍増殖を減速しない。
【0112】
腫瘍細胞およびドナーリンパ球のみが投与されたときの増殖速度と比較して、癌細胞の増殖速度の何らかの変化が観察される場合、分子は免疫調節効果を有すると考えられる。投与されるリンパ球の数が癌細胞の増殖を減速させるのに十分であり、観察された腫瘍細胞の増殖速度が、腫瘍細胞およびドナーリンパ球のみが投与されたときの増殖速度と比較して高い場合、化合物は免疫抑制性であると見なされる。投与されるリンパ球の数が癌細胞の増殖を減速するのに不十分であり、観察された腫瘍細胞の増殖速度が、腫瘍細胞およびドナーリンパ球のみが投与されたときの増殖速度と比較して低い場合、化合物は免疫増強性であると見なされる。
【0113】
当業者が本明細書で述べた組成物および方法をより良好に実施できるようにするために、以下の実施例を例示目的で与える。
【実施例】
【0114】
(実施例1)
(細胞株CLL−AATの単離)
(細胞株の確立)
CLLと診断された患者から末梢血を採取した。WBCカウントは1.6×108/mlであった。単核細胞をHistopaque−1077密度勾配遠心分離(Sigma
Diagnostics,セントルイス、ミズーリ州)によって単離した。細胞を10%熱不活性化ウシ胎仔血清(FBS)を添加したイスコーブ変法ダルベッコ培地(IMDM)で2回洗浄して、氷冷IMDM/10% FBS 5ml中で再懸濁させた。トリパンブルーで染色することによって生細胞をカウントした。細胞を同じ体積の85% FBS/15% DMSOと混合して、貯蔵するために液体窒素中で1mlの分割量として凍結させた。
【0115】
免疫表現型検査は、CD45+リンパ球個体群の>90%がIgD、カッパ軽鎖、CD5、CD19、およびCD23を発現することを示した。この個体群は、低レベルのIgMおよびCD20も発現した。細胞の約50%が高レベルのCD38を発現した。細胞はラムダ軽鎖、CD10およびCD138では陰性であった。
【0116】
細胞の分割量を解凍、洗浄、そして20%熱不活性化FBS、2mM L−グルタミン、100単位/mlペニシリン、100μg/mlストレプトマイシン、50μM 2−メルカプトエタノール、および5ng/ml組換えヒトIL−4(R & D Systems、ミネアポリス、ミネソタ州)を添加したIMDM中で107個/mLの密度にて再懸濁させた。細胞を37℃にて加湿5% CO2雰囲気中で培養した。一定の増殖が観察されるまで、培地を4日ごとに一部交換した。5週間後、培養物中の細胞の数は、約4日ごとに倍増しはじめた。この細胞株をCLL−AATと名付けた。
【0117】
(細胞株の特徴付け)
フローサイトメトリーによる細胞株の免疫表現型検査は、IgM、カッパ軽鎖、CD23、CD38、およびCD138の高い発現、CD19およびCD20の中程度の発現、そしてIgDおよびCD5の弱い発現を示した。細胞株はラムダ軽鎖、CD4、CD8、およびCD10に対して陰性であった。
細胞株の免疫表現型検査も、全細胞ELISAによって、一次B−CLL細胞への特異性結合のために選択されたウサギscFv抗体のパネルを使用して実施した。これらのCLL特異性scFv抗体はすべて、CLL−AAT細胞株も認識した。これに対して、scFvの大多数は、B細胞リンパ腫に由来する2つの細胞株:バーキットリンパ腫細胞株のRamos、および非ホジキンリンパ腫細胞株のRLに結合しなかった。
【0118】
(実施例2)
(抗体ファージ提示および細胞表面パニングを使用する、B−CLL特異性細胞表面抗原に対するscFv抗体の選択)
(免疫化およびscFv抗体ライブラリー構成)
末梢血単核細胞(PBMC)をScripps Clinic(ラホーヤ、カリフォルニア州)にてCLL患者から採取した血液から単離した。ウサギ2匹をCLLの異なるドナー10名からプールしたPBMC 2×107個によって免疫化した。3回の免疫化、すなわち2回の皮下注射と、それに続く1回の静脈内注射を3週間の間隔で実施した。血清力価は、フローサイトメトリーを使用して血清IgGの一次CLL細胞への結合を測定することによって確認した。最後の免疫化の5日後に、該動物から脾臓、骨髄、およびPBMCを採取した。Tri−Reagent(Molecular Research Center,Inc)を使用して、全RNAをこれらの組織から単離した。単鎖Fv(scFv)抗体ファージ提示ライブラリーを前に述べたように構成した(Barbas et al.,(2001)Phage Display:A Laboratory Manual,Cold Spring Harbor Laboratory Press,コールドスプリングハーバー、ニューヨーク州)。細胞表面パニングでは、再増幅ライブラリーからのファージミド粒子をポリエチレングリコー(PEG)によって沈降させ、1%ウシ血清アルブミン(BSA)を含有するリン酸緩衝食塩水(PBS)で再懸濁させて、PBSで一晩透析した。
【0119】
(細胞表面パニングによる抗体選択)
ライブラリーは、Siegelら(1997,J.Immunol.Methods 206:73−85)によって述べられているように、磁気活性化細胞分離装置(MACS)を用いた正負の選択によりCLL細胞表面特異性抗体について濃縮した。簡潔には、scFv抗体ライブラリーからのファージミド粒子をMPBS(2%脱脂粉乳、PBS中0.02%アジ化ナトリウムPBS、pH7.4)中で1時間、25℃にてプレインキュベートして、非特異性結合部位を遮断した。一次CLL細胞約107個を常磁性マイクロビーズ(Miltenyi Biotec,サニーベール、カリフォルニア州)に結合したマウス抗CD5 IgGおよびマウス抗CD19 IgGで標識した。未結合マイクロビーズを洗浄によって除去した。標識CLL細胞(「標的細胞」)を過剰な「抗原−負のアブゾーバ細胞」と混合して、ペレット化して、ファージ粒子50μl(cfu 1010〜1011cfu)中へ再懸濁させた。アブゾーバ細胞は、細胞表面に非特異的に付着するファージはもちろんのこと、標的およびアブゾーバ細胞の両方に存在する「共通」抗原に対して特異性のファージも吸収するように作用する。使用したアブゾーバ細胞は、免疫磁気的負の選択(StemSep system,StemCell Technologies,バンクーバー、カナダ)によって末梢血から単離されたTF−1細胞(ヒト赤白血病細胞株)または正常ヒトB細胞のどちらかであった。アブゾーバ細胞の標的細胞に対する比は体積で約10倍であった。25℃での30分間のインキュベーションの後、細胞/ファージ混合物をMiniMACS MS+分離カラムに移した。カラムをMPBS 0.5mlで2回、そしてPBSで1回洗浄して、未結合ファージおよびアブゾーバ細胞を除去した。標的細胞をPBS 1ml中でカラムから溶出させて、最高速度の微量遠心機で15秒間に渡ってペレット化した。捕獲されたファージ粒子は、酸溶出緩衝液(グリシンによってpHを2.2に調整した0.1N HCl、および1mg/ml BSA)200μl中で標的細胞を再懸濁させることによって溶出させた。25℃での10分間のインキュベーションの後、緩衝液を2M Tris塩基、pH10.5 12μLによって中和して、溶出したファージを次回のパニングの間に、E.coli中で増幅した。各回のパニングでは、インプットおよびアウトプットファージ力価を決定した。インプット力価は、標的細胞/アブゾーバ細胞混合物に添加した再増幅ファージ粒子の数であり、アウトプット力価は、標的細胞から溶出した捕獲ファージの数である。濃縮因子(E)は、式E=(Rnアウトプット/Rnインプット)/(R1アウトプット/R1インプット)、式中、R1=1回およびRn=2、3、または4回を使用して計算する。大半の場合、102〜103倍の濃縮因子は、3回目または4回目に達成されるはずである。
【0120】
(パニング後の濃縮抗体プールの分析)
3〜5回のパニングの後、捕獲ファージのプールをCLL細胞への結合について、フローサイトメトリーおよび/または全細胞ELISAによってアッセイした:
1.HA−tagged溶解性抗体の形で全体のプールを生成するために、非サプレッサ系のE.coli(たとえばTOP10F’)2mlにファージミド粒子1μl(cfu 109〜1010)を感染させた。元のパニングされていないライブラリーを負の対照として使用した。カルベニシリンを最終濃度10μMまで添加して、培養物を37℃にて250rpmで振とうしながら1時間インキュベートした。50μg/mlカルベニシリンを含有するSB培地8mlを添加して、培養物を〜0.8のOD 600まで培養した。IPTGを最終濃度1mMまで添加して、LacプロモータからscFv発現を誘発して、37℃での振とうを4時間継続した。培養物を3000xgにて15分間遠心分離にかけた。溶解性抗体を含有する上澄みを濾過して、−20℃にて1mlの分割量で貯蔵した。
【0121】
2.scFv抗体プールの標的細胞対アブゾーバ細胞への結合を、フローサイトメトリーによって、高親和性ラット抗HA(クローン3F10、Roche Molecular Biochemicals)を二次抗体として、PE結合ロバ抗ラットを三次抗体として使用して決定した。
【0122】
3.抗体プールの標的細胞対アブゾーバ細胞への結合も、全細胞ELISAによって以下に述べるように決定した。
【0123】
(パニング後の個別scFvクローンのスクリーニング)
パニング後に個別scFvクローンをスクリーニングするために、TOP10F’細胞にファージプールを上述のように感染させて、カルベニシリンおよびテトラサイクリンを含有するLBプレートに散布して、37℃にて一晩インキュベートした。個別のコロニーを、ウェル当たりSB−カルベニシリン培地を0.6〜1.0ml含有する深96ウェルプレートへ接種した。培養物をHiGro振とうインキュベータ(GeneMachines,サンカルロス、カリフォルニア州)で520rpmおよび37℃にて6〜8時間培養した。この時点で、各ウェルから90μlの分割量をDMSO 10μlを含有する深96ウェルプレートに移した。この複製プレートを−80℃にて貯蔵した。元のプレートにIPTGを最終濃度1mMまで添加して、振とうを3時間継続した。プレートを3000xgにて15分間遠心分離にかけた。溶解性scFv抗体を含有する上澄みを別の深96ウェルプレートに移して、−20℃にて貯蔵した。
【0124】
HA−tagged scFv抗体をスクリーニングするための感受性全細胞ELISA方法を開発した:
1.ELISAプレートをコンカナバリンA(0.1M NaHCO3、pH8.6、0.1mM CaCl2中10mg/ml)でコーティングする。
【0125】
2.プレートをPBSで洗浄した後、PBS 50μl中の標的細胞またはアブゾーバ細胞0.5〜1×105個を各ウェルに添加して、プレートを250xgにて10分間遠心分離にかける。
【0126】
3.PBS中の0.02%グルタルアルデヒド50μlを添加して、細胞を40℃にて一晩固定する。
【0127】
4.PBSで洗浄した後、非特異性結合部位を、4%脱脂粉乳を含有するPBSによって室温にて3時間遮断する。
【0128】
5.細胞を溶解性、HA−tagged scFvまたはFab抗体(TOP10F’上澄み)50μlによって室温で2時間インキュベートして、次にPBSで6回洗浄する。
【0129】
6.結合した抗体を、マウス抗HA二次抗体(クローン12CA5)およびアルカリホスファターゼ(AP)結合抗マウスIgG三次抗体を使用して検出する。シグナルの約10倍の増幅は、AMDEX AP結合ヒツジ抗マウスIgGを三次抗体(Amersham Pharmacia Biotech)として使用することによって得られる。AMDEX抗体はデキストラン主鎖を介して複数のAP分子に結合される。アルカリホスファターゼ基質PNPPによって呈色され、マイクロプレートリーダーを使用して405nmにて測定する。
【0130】
scFvクローンの一次スクリーニングは、一次CLL細胞対正常ヒトPBMCに対するELISAによって行った。CLL細胞で陽性であり、正常PBMCで陰性であるクローンは、正常ヒトB細胞、ヒトB細胞株、TF−1細胞、およびCLL−AAT細胞株に対するELISAによって再スクリーニングした。クローンは別個の患者3名から単離したCLL細胞に対するELISAによっても再スクリーニングして、患者特異性または血液型特異性抗原を認識するクローンを除去した。代表的なELISAによる結果を図2〜6に示し、図9A〜9Cにまとめる。
【0131】
得られたユニークなscFv抗体クローンの数をDNAフィンガープリンティングおよび配列決定によって決定した。scFv DNAインサートをプラスミドからPCRによって増幅して、制限酵素BstNIによって消化した。得られた断片を4%アガロースゲル上で分離して、臭化エチジウムによって染色した。異なる制限断片パターンを有するクローンは、異なるアミノ酸配列を持つ必要がある。同一パターンを持つクローンはおそらく、同様または同一の配列を有する。ユニークなBstNIフィンガープリントを有するクローンは、DNA配列決定によってさらに分析した。25個の異なる配列が見出され、密接に関連する相補性決定領域を持つ16グループの抗体にクラスター化できた(図9A〜9C)。
【0132】
(scFv抗体のフローサイトメトリーによる特徴付け)
複数のscFv抗体の結合特異性を3色フローサイトメトリーによって分析した(図7)。正常ドナーから単離したPBMCをFITC結合抗CD5およびPerCP結合抗CD19によって染色した。scFv抗体による染色は、ビオチン結合抗HAを二次抗体として、そしてPE結合ストレプトアビジンを使用して行った。3つの抗体、すなわちscFv−2、scFv−3、およびscFv−6は、CD19+Bリンパ球個体群を特異的に認識することが見出された(データは示さず)。第4の抗体scFv−9は、2つの別個の細胞個体群:CD19+Bリンパ球およびCD5+Tリンパ球のサブセットを認識した(図7)。T細胞サブセットのさらなる特徴付けは、それがCD4+CD8−TH細胞のサブ個体群であることを示した(データは示さず)。
【0133】
scFv抗体によって認識された抗原が一次CLL細胞で過剰発現されるかどうかを判定するために、CLL患者5名および正常ドナー5名からのPMBCをscFvで染色して、フローサイトメトリーによって比較した(図8および表2)。陽性細胞個体群の平均蛍光強度を比較することによって、CLL細胞対正常細胞での抗原の相対発現レベルを決定できる。1つの抗体、すなわちscFv−2は正常PBMCよりも低い強度でCLL細胞を常に染色したのに対して、scFv−3およびscFv−6はどちらも、正常PBMCよりも鮮やかにCLL細胞を常に染色した。第4の抗体scFv−9は、CLLサンプル5つのうち2つを正常PBMCよりもはるかに強く染色したが、残り3つのCLLサンプルには中程度に鮮やかな染色のみを与えた(図8および表2)。これはscFv−3およびscFv−6の抗原が、すべてではないにしても大半のCLL腫瘍にて約1.4倍過剰発現されるのに対して、scFv−9がCLL腫瘍のサブセットにて3〜6倍過剰発現されることを示す。
【0134】
CLL患者は2つのほぼ等しい群に分割できる:不十分な予後の患者(中間生存期間8年)および好ましい予後の患者(中間生存期間26年)。CLLについて複数の好ましくない予後の指標が確認されており、最も顕著には体細胞変異のないVH遺伝子の存在および高いパーセンテージのCD38+B細胞の存在である。CLL患者のサブセットのみで過剰発現された抗原を認識するため、scFv−9抗原過剰発現がCLL患者10名からの血液サンプル中のパーセンテージと相関したかどうかを判定することが求められた(図11)。結果は、scFv−9抗原過剰発現およびCD38+細胞のパーセントが相互に完全に独立していることを示す。
【0135】
(scFv抗体によって認識された抗原の、免疫沈降(IP)および質量分光分析(MS)による同定)
これらの抗体に対する抗原を同定するために、scFvを使用して、細胞表面ビオチン化CLL−AAT細胞のミクロソーム画分から調製した溶解物から抗原を免疫沈降させた(図12)。免疫沈降された抗原をSDS−PAGEによって精製して、マトリックス支援レーザーイオン化質量分光分析(MALDI−MS)または毛細管逆相HPLCナノ電子スプレータンデム質量分光分析(μLC/MS/MS)によって同定した(データは示さず)。scFv−2は、110kd抗原をRLおよびCLL−AAT細胞の両方から免疫沈降させた(図12)。この抗原をMALDI−MSによってB細胞特異性マーカーCD19として同定した。scFv−3およびscFv−6はどちらも、45kd抗原をCLL−AAT細胞から免疫沈降させた(示さず)。この抗原はMALDI−MSによって、CLLおよび活性化B細胞の公知のマーカーであるCD23として同定された。scFv−9は、50kd抗原をCLL−AAT細胞から免疫沈降させた(図12)。この抗原はμLC/MS/MSによって、B細胞、活性化CD4+T細胞、および胸腺細胞の公知のマーカーであるOX−2/CD200として同定された。OX−2/CD200は、一部の非リンパ球細胞、たとえばニューロンおよび内皮細胞でも発現される。
【0136】
(実施例3)
OX−2/CD200を発現する細胞が、サイトカイン反応をTh1反応(IL−2、IFN−γ)からTh2反応(IL−4、IL−10)に変化させる能力を、ドナー1名からの単球由来マクロファージ/樹状細胞および別のドナーからの血液由来T細胞を用いた混合リンパ球反応にて評価した。OX−2/CD200発現細胞源として、以下で述べるOX−2/CD200トランスフェクションEBNA細胞またはCLL患者サンプルのどちらかを使用した。
【0137】
(293−EBNA細胞のトランスフェクション)
293−EBNA細胞(Invitrogen)を100mm皿に2.5×106個にて播種した。24時間後、Polyfect試薬(QIAGEN)をメーカーの説明書に従って使用して、細胞を一過性トランスフェクションした。細胞を、ベクターpCEP4(Invitrogen)中のOX−2/CD200 cDNA 7.2μgおよびpAdVAntageベクター(Promega)0.8μgと同時トランスフェクションした。負の対照として、空のpCEP4ベクターおよびpAdVAntageと同時トランスフェクションした。トランスフェクションの48時間後、scFv−9抗体を用いたフローサイトメトリーで決定されたように、細胞の約90%がOX−2/CD200をその表面で発現した。
【0138】
(血液単球からの樹状細胞/マクロファージの成熟)
軟膜をSan Diego Blood Bankから取得し、Ficollを使用して一次血液リンパ球(PBL)を単離した。2%ヒト血清を含有するイーグル最小必須培地(EMEM)中で細胞を1時間に渡って付着させ、続いてPBSによって激しく洗浄した。細胞をGM−CSF、IL−4およびIFN−γまたはM−CSFのいずれかの存在下で、3日後にリポポリサッカライド(LPS)を添加して、または添加せずに5日間培養した。成熟した細胞を収集して、γ−照射器(Shepherd Mark I Model 30照射器(Cs137))を使用して2000RADを照射した。
【0139】
(混合リンパ球反応)
混合リンパ球反応は、24ウェルプレートに樹状細胞/マクロファージ500,000およびレスポンダ細胞1×106個を使用して構成した。レスポンダ細胞は、Ficollを使用して末梢血から精製したT細胞濃縮リンパ球であった。T細胞は、細胞を組織培養フラスコ内で1時間インキュベートして、非付着性細胞画分を取ることによって濃縮した。OX−2/CD200トランスフェクションEBNA細胞またはCLL細胞500,000個を抗CD200抗体(完全IgGに変換されたscFv−9)30μg/mlの存在下または非存在下で、リンパ球添加の2〜4時間前にマクロファージ/樹状細胞に添加した。上澄みを48時間および68時間後に採取して、サイトカインの存在について分析した。
【0140】
(scFv−9の完全IgGへの変換)
scFv−9の軽鎖および重鎖V遺伝子をオーバーラップPCRによって、各遺伝子の可変領域をヒトラムダ軽鎖定常領域遺伝子、およびヒトIgG1重鎖定常領域CH1遺伝子とそれぞれ連結するプライマーを用いて増幅した。scFv−9の軽鎖遺伝子および重鎖遺伝子の可変領域は、特異性プライマーによって増幅して、ヒトラムダ軽鎖定常領域遺伝子およびIgG1重鎖定常領域CH1遺伝子は以下のような特異性プライマーによって個別に増幅した:
【0141】
【化1】

増幅生成物を精製して、オーバーラップPCRを実施した。
【0142】
最終生成物は、Xba I/Sac I(軽鎖)およびXho I/Pin AI(重鎖)によって消化して、ヒトFab発現ベクターのPAX243hGL内へクローニングした(その開示が参照によって本明細書に組み入れられている、公開されたInternational Application WO 2004/078937を参照)。DNAクローンをPCRエラーについてDNA配列決定によって分析した。hCMV IEプロモータ遺伝子をNot I/Xho I部位(重鎖の前)に挿入した。ベクターをXba I/Pin AI/EcoR I/Nhe Iにならびに軽鎖およびhCMV IEプロモータを含有する3472bp断片よって消化して、重鎖遺伝子をIgG1発現ベクターのXba I/Pin AI部位に移動した。
【0143】
(サイトカイン分析)
完全IgGに変換されたscFv−9の、混合リンパ球反応におけるサイトカインプロフィールに対する効果を判定した。
【0144】
組織培養物上澄み中に見出されたIL−2、IFN−γ、IL−4、IL−10およびIL−6などのサイトカインを、ELISAを使用して定量した。各サイトカインの一致した捕獲および検出抗体の対をR+D Systems(ミネアポリス、ミネソタ州)から取得して、組換えヒトサイトカインを使用して各サイトカインの標準曲線を作成した。抗サイトカイン捕獲抗体をPBS中のプレートに最適濃度にてコーティングした。一晩のインキュベーションの後、プレートを洗浄して、1% BSAおよび5%スクロースを含有するPBSによって1時間遮断した。0.05% Tweenを含有するPBSによる3回の洗浄の後、1% BSAを含有するPBS中に上澄みを2倍または10倍希釈として添加した。捕獲サイトカインを適切なビオチン化抗サイトカイン抗体によって検出して、アルカリホスファターゼ結合ストレプトアビジンおよびSigmaS基質の添加を続けた。呈色は、ELISAプレートリーダー(Molecular Devices)によって評価した。
【0145】
図14に示すように、OX−2/CD200トランスフェクションされたが、未トランスフェクションではない細胞の存在は、IL−2およびIFN−γなどのTh1サイトカインのダウンレギュレーションを生じた。30μg/mlでの抗CD200抗体の添加はTh1反応を完全に回復し、抗体がOX−2/CD200とそのレセプターとの間の相互作用を遮断したことを示す。
【0146】
図15および16に示すように、混合リンパ球反応におけるCLL細胞の存在は、Th1反応のダウンレギュレーションを生じた(図15はIL−2の結果を示す;図16はIFN−γの結果を示す)。このことは、OX−2/CD200を過剰発現する細胞(IB、EM、HS、BH)だけでなく、OX−2/CD200を過剰発現しなかったCLL細胞(JR、JGおよびGB)(これらの細胞の発現レベルを図11に示す)にも当てはまる。しかしながら抗CD200抗体はOX−2/CD200を過剰発現する細胞においてTh1反応を回復するだけであり、OX−2/CD200を発現する患者では、マクロファージでのOX−2/CD200のそのレセプターとの間の相互作用を排除することがTh1反応を回復するのに十分であることを示した。OX−2/CD200を過剰発現しなかった患者では、Th1反応のダウンレギュレーションに他の機構が関与しているように思われた。
抗CD200の腫瘍拒絶に対する効果を試験するための動物モデル
RAJIリンパ腫の腫瘍増殖がPBLの同時注射によって防止されるモデルを確立した。NOD/SCIDマウスにRAJI細胞4×106個、異なるドナーからのヒトPBL
1×106、5×106または10×106細胞の存在下または非存在下のどちらかで皮下注射した。腫瘍の長さおよび幅はもちろんのこと、体重も週に3回測定した。すべての群の腫瘍体積の平均+/−SDを図17AおよびBに示す。2つのパラメトリック検定(Student t検定およびWelchの検定)および1つのノンパラメトリック検定(Wilcox検定)を使用して、統計分析を実施した。統計分析の結果は図18に見出される。RAJI細胞は許容される変動の皮下腫瘍を形成する。拒絶は、特定のドナーおよびPBL細胞数に依存する。PBL 1×106個は腫瘍増殖を防止するには不十分であった。第22〜43日がPBL 5×106個のドナー2および第36日から開始するPBL 5×106または1×107個のドナー3は、腫瘍増殖を著しく低減した。ドナー4は、第48日以降はほとんど有意である。
【0147】
抗CD200の効果を試験するために、RAJI細胞にCD200を安定にトランスフェクションする。動物には、前の段落で述べたように注射する。CD200トランスフェクション細胞の存在下では、腫瘍はヒトPBLが存在しても増殖する。このモデルでの腫瘍拒絶を評価するために、抗CD200抗体を投与する。
【0148】
また液状腫瘍モデルを確立する。RAJI細胞をNOD/SCIDマウスに腹腔内注射する。細胞は骨髄、脾臓、リンパ節および他の臓器に分散して、麻痺を引き起こす。ヒトPBLの同時注射は、腫瘍増殖を防止または減速する。マウスを運動障害および麻痺の徴候について評価することによって、腫瘍増殖をモニターする。いったんこのような徴候が観察されたら、マウスを殺処分して、腫瘍細胞の数を骨髄、脾臓、リンパ節および血液を含む各種の臓器において、FACS分析およびPCRによって評価する。
【0149】
皮下モデルと同様に、CD200トランスフェクション細胞を腹腔内注射する。それらはヒトPBLが存在しても増殖する。抗CD200による処置は、腫瘍拒絶または腫瘍増殖の減速を生じる。
【0150】
(実施例4)
(ライブラリー構成)
マウスは、マウスIgG Fcに融合されたバキュロウイルス発現組換えCD200細胞外ドメイン(CD200−Fc)(Orbigen Inc.,サンディエゴ、カリフォルニア州)および全長CD200を含有するベクターを一過性トランスフェクションされた293−EBNA細胞によって交互に免疫化した。全RNAは、マウス脾臓からTRI試薬(Molecular Research Center,Inc.,シンシナティ、オハイオ州)をメーカーのプロトコルに従って使用して調製した。メッセンジャRNA(mRNA)は、Oligotex(QIAGEN Inc.,バレンシア、カリフォルニア州)をメーカーのマニュアルに従って使用して精製した。第1鎖cDNAはSuperScript II RTase(Invitrogen Life Technologies,カールスバッド、カリフォルニア州)をメーカーのプロトコルに従って使用して合成した。第1鎖cDNAを制限エンドヌクレアーゼによって消化して、第2鎖cDNAを、2003年3月27日に公開された、公開PCT application WO03/025202A2で十分に説明されている方法に従って合成した。第2鎖cDNAをPCR精製キット(QIAGEN)によって浄化して、シングルプライマー増幅を2003年3月27日に公開された、公開PCT application WO03/025202A2に述べられている方法に従って実施した。増幅生成物をプールして、PCR精製キットによって精製した。カッパ軽鎖をXba IおよびBspE Iで消化して、IgG1およびIgG2a重鎖をXho IおよびBin Iによって消化した。消化した断片はゲル抽出キット(QIAGEN)を用いてアガロースゲルから精製して、2004年9月16日に公開された公開PCT application WO/04078937A2で述べられているように、PAX313m/hGベクター内へクローニングした。
【0151】
(ライブラリーのパニング)
ライブラリー(IgG1カッパおよびIgG2aカッパ)は、マイクロタイター(Costar Group,ベテスダ、メリーランド州)に直接コーティングされた、またはヤギ抗マウスIgG Fc特異性抗体(Sigma−Aldrich Corp.,セントルイス、ミズーリ州)によって捕獲されたいずれかのCD200−Fcにパニングした。ライブラリーファージの調製では、エレクトロコンピテントXL1−Blue細胞(Stratagene、ラホーヤ、カリフォルニア州)をライブラリーDNAによって電気穿孔して、SOC培地中で1時間、そしてSB培地中で2時間、カルベニシリンを用いて培養した。ファージ生成は、VCS M13ヘルパーファージ(Amersham Biosciences Corp.,ピスカタウェイ、ニュージャージー州)および1mM
IPTGの添加により、30℃にて一晩に渡って誘発させた。培養物を遠沈させて、ファージを4%ポリエチレングリコールおよび3% NaClによって沈降させた。ファージを遠沈させて、関係のない抗原である、マウスIgG Fcに融合されたバキュロウイルス発現細胞外ドメイン(FLJ32028−Fc)であるFLJ32028(Orbigen,サンディエゴ)を溶解性競合物として含有する、1% BSA/PBSに再懸濁させた。直接コーティングしたCD200−Fcに対するパニングでは、4つのウェルをCD200−Fc(0.1M NaHCO3 pH8.6の5μg/ml)100μlによって4℃にて一晩コーティングした。ウェルをリン酸塩緩衝食塩水(PBS)、pH7.0で5回洗浄して、1%ウシ血清アルブミン(BSA)/PBSによって37℃にて1時間に渡って遮断した。ヤギ抗マウスIgG Fcに捕獲されたCD200−Fcに対するパニングでは、4つのマイクロタイターウェルをヤギ抗マウスIgG Fc 100μl(PBS中20μg/ml)によって4℃にて一晩コーティングした。ウェルをPBSによって5回洗浄して、CD200−Fc(PBS中の20μg/ml)100μlによって37℃にて1時間に渡ってインキュベートした。ウェルをPBSによって5回洗浄して、1% BSA/PBSによって37℃にて1時間に渡って遮断した。パニングの直接コーティングおよび捕獲方法では、ブロッカーは別のライブラリー(その開示全体が参考として本明細書中に援用される、2004年6月2日に提出されたPCT application serial No.PCT/US04/17118(未公開)の実施例3に述べたライブラリー)のFLJ32028に対するパニングから得られた溶解性Fabの混合物によって置換されて、マウスIgG Fcのエピトープをマスクして、ウェルを37℃にて30分間インキュベートした。これらのマスキングFabは、CD200−Fcにも結合することが示されている。ライブラリーファージをマスキングFabの上部に添加して、ウェルを37℃にて約1.5時間インキュベートした。未結合ファージをPBSによって厳密性を増強しながら洗浄して(1回目は3回、2回目は5回、3回目および4回目は10回)、各洗浄のたびに5分間のインキュベーションならびにピペットによる出し入れ5回を行った。結合ファージは、1mg/ml BSAを含む0.1M HCl
100μl、pH2.2によって2回溶出させ、2M Tris塩基、pH11.5で中和した。新しく培養したER2738細胞を溶出させたファージによって感染させて、カルベニシリンおよびグルコースを含有するLBアガロースプレート上へ滴定した。次回のパニングのために、残りのファージをVCS M13ヘルパーファージおよび1mM IPTGの添加によって30〜37℃にて一晩増殖させる。
【0152】
(ライブラリーのスクリーニング)
3および4回目の滴定プレートからの95コロニーを12.5μg/mlテトラサイクリンおよび50μg/mlカルベニシリンを含有するSB 1ml中で37℃にて約6時間培養した。VCS M13ヘルパーファージを添加して、培養物を37℃にて2時間インキュベートした。1mM IPTGおよび70μg/mlカナマイシンを添加して、Fab−ファージ生成を30℃にて一晩誘発した。マイクロタイターウェルをウサギ抗マウスIgG F(ab’)2(PBS中4μg/ml)50μl、CD200−Fc(0.1M NaHCO3中4μg/ml、pH8.6)、またはFLJ32028−Fc(0.1M NaHCO3中4μg/ml、pH8.6)によって4℃にて一晩コーティングした。ウェルをPBSによって3回洗浄して、1% BSA/PBS 100μlによって37℃にて1時間遮断した。培養物を遠沈させた。Fab−ファージを含有する培養物上澄みとブロッカーを交換して、ウェルを37℃にて1.5〜2時間インキュベートした。残りのFab−ファージは、フローサイトメトリーのために−80℃にて貯蔵した。プレートをPBSで3回洗浄して、アルカリホスファターゼ(AP)結合ヤギ抗マウスIgGF(ab’)2抗体50μl(Pierce)(1% BSA/PBS中1:500)によって37℃にて1時間に渡って検出した。プレートをPBSによって3回洗浄して、AP基質(Sigma−Aldrich)によってpNPP緩衝液中で展開した。3回目からのクローンのほぼすべてがすでに、CD200に対して特異的に陽性である(図19A〜D)。クローンを高スループットフローサイトメトリー分析によってもスクリーニングした。CD200(1×105細胞)を含む293細胞一過性トランスフェクション100マイクロリットルを96ウェルプレート(Costar)に分割した。Fab−ファージ50マイクロリットルを細胞に添加して、ピペット操作によって混合して、氷上で30分間インキュベートした。細胞を0.01% NaN3を含有する1% BSA/PBSによって2回洗浄した。細胞を0.01% NaN3を含有する1% BSA/PBS中のPE結合ヤギ抗マウスIgG抗体100μl(Sigma−Aldrich)中に再懸濁させて、氷上で30分間インキュベートした。細胞を0.01% NaN3を含有する1% BSA/PBSによって2回洗浄して、PBS中1%パラホルムアルデヒド200μlに再懸濁させた。CD200発現細胞への陽性結合を示す代表的なクローンを図20A〜Dに示す。
【0153】
DNA配列を分析して、重鎖の推定アミノ酸配列を相補性決定領域3(CDR3)に従ってグループ化した(図21A、B)。それらを17の群に分けた。
【0154】
(蛍光ビーズアッセイ)
さらなる分析のために23個のクローンを選択した。それらはcG2aR3B5、dG1R3A5、cG2aR3A2、dG2aR3B2、dG1R3A1、cG2aR3Al、cG2aR3B、dG1R3B、cG1R3A、cG1R3A、cG1R3A1、dG1R3B、dG1R3B、cG1R3C、dG2aR3C、dG2aR3A1、cG2aR3B、cG2aR3B、dG1R3B、cG2aR3B、cG2aR3C、dG1R3H、およびdG2aR3A6であった。選択したFabのDNAをSpe I/Nhe Iによって、遺伝子III除去および溶解性Fab発現および精製のために消化した。精製したFabは、CD200とそのレセプター(CD200R)との間の相互作用を遮断するその能力について蛍光ビーズアッセイで評価した。TransFluoSpheresカルボキシラート修飾ミクロスフェア(488/645)(Molecular Probes Invitrogen Detection Technologies,ユージーン、オレゴン州)にストレプトアビジンを、続いてビオチン標識抗ヒトFc抗体および暴露ウイルス生成CD200−Fcタンパク質ををコーティングした。293個の細胞にCD200Rを一過性トランスフェクションした。細胞表面発現をFACS分析によって確認した。CD200コーティングビーズ100万個を各種の量の抗CD200FabまたはキメラIgGによって10分間プレインキュベートしてから、CD200Rトランスフェクション細胞50,000個を添加した。37℃でのインキュベーションの30分後、1% BSAを含有するTris緩衝液によって細胞を洗浄して、FACS Caliburを使用して分析した。Fabのc1A10、c2aB7、およびd1A5は、Fab 6.7μg/mlにてCD200およびCD200Rの相互作用の最善の遮断を示した(図22)。これらのクローンは、図21Aおよび/またはBにおいてそれぞれcG1R3A10、cG2aR3B7およびdG1R3A5と呼ばれる。
【0155】
(キメラ化およびIgG変換)
キメラ化およびIgG変換のために6つの抗体を選択した(図23を参照)。それらはc1A10(cG1R3A10)、c2aA10(cG2aR3A10)、c2aB7(cG2aR3B7)、d1A5(dG1R3A5)、d1B5(dG1R3B5)、およびd1B10(dG1R3B10)であった。キメラ化のために、オーバーラップPCRを実施して、マウスカッパ鎖可変領域およびヒトカッパ鎖定常領域を連結した。マウス重鎖可変領域をクローニングのために、部分ヒトIgG1定常領域およびApa I部位を含有する3’プライマーによって増幅した。増幅されたカッパ鎖断片および重鎖断片を、カッパ軽鎖ではXba I/Not Iに、重鎖断片ではXho I/Apa IにヒトIgG1定常領域を含有するPAX243hGKベクター内へクローニングした(公開されたInternational Application WO 2004/078937を参照)。キメラFabのCD200への結合は、ELISAおよびフローサイトメトリーによって確認した。Not I/Xho Iでの重鎖発現、次に軽鎖および重鎖の、Xba I/Pin AI部位におけるヒトIgG1発現ベクター内への移動のために、これらのキメラFabは、ヒトサイトメガロウイルス前初期プロモータ(hCMV IE Pro)配列の挿入によって、IgGへ変換される。このベクターは、哺乳類細胞での軽鎖発現のために、追加のhCMV IE Pro配列上流Xba I部位を含む。DNA配列を確認して、哺乳類細胞トランスフェクションのためにHiSpeed Maxi prepカラム(QIAGEN)を使用してmaxi prep DNAを調製した。293−EBNA細胞での一過性トランスフェクションをEffectene(QIAGEN)をメーカーのプロトコルに従って使用し、pAdVAntageベクター(Promega US,マジソン、ウィスコンシン州)を添加して実施した。NS0細胞への安定な細胞株トランスフェクションは、Effecteneをメーカーのプロトコルに従って使用して実施した。小規模一過性トランスフェクションの後、各抗体の培養物上澄みをELISAによって試験した(図24)。大規模一過性トランスフェクションの後、各IgGを培養物上澄みから、FPLCを用いた抗ヒトIgG F(ab’)2アフィニティカラム(Amersham Biosciences)によって精製した。
【0156】
精製したIgGは、Fabについて述べたようにビーズ阻害アッセイで試験した。CD200に対して作られた抗体はすべて、以下の図25に示すように、レセプターリガンド相互作用を非常に良好に遮断した。
【0157】
(混合リンパ球反応)
CD200のそのレセプターとの間の相互作用の遮断も、CD200の存在下での混合リンパ球反応で観察されたTh1からTh2へのサイトカイン移動を防止するかどうかを評価した。San Diego Blood Bankから軟膜を取得して、Histopaque(Sigma− Aldrich)を使用して血液リンパ球(PBL)を単離した。2%ヒト血清を含有するEMEM中で細胞を1時間に渡って付着させ、続いてPBSによって激しく洗浄した。細胞をGM−CSF、IL−4およびIFN−γの存在下で5日間培養した。成熟した細胞を収集して、γ−照射器(University of California San Diego)を使用して2000RADを照射した。混合リンパ球反応は、24ウェルプレートに樹状細胞/マクロファージ500,000個およびレスポンダ細胞1×106個を使用して構成した。レスポンダ細胞は、Histopaqueを使用して末梢血から精製したT細胞濃縮リンパ球であった。T細胞は、細胞を組織培養フラスコ内で1時間インキュベートして、非付着性細胞画分を取ることによって濃縮した。CD200発現一次照射CLL細胞500000個を、各種の量の抗CD200抗体の存在下または非存在下で樹状細胞に、リンパ球添加の2〜4時間前に添加した。上澄みを48時間および68時間後に採取して、IL−2、IFN−γ、IL−4、IL−10およびIL−6などのサイトカインをELISAを使用して定量した。各サイトカインの一致した捕獲および検出抗体の対をR+D Systems(ミネアポリス、ミネソタ州)から取得して、組換えヒトサイトカインを使用して各サイトカインの標準曲線を作成した。抗サイトカイン捕獲抗体をPBS中のプレートに最適濃度にてコーティングした。一晩のインキュベーションの後、プレートを洗浄して、1% BSAおよび5%スクロースを含有するPBSによって1時間遮断した。0.05% Tweenを含有するPBSによる3回の洗浄の後、1% BSAを含有するPBS中の指示された希釈物に上澄みを添加した。捕獲サイトカインを適切なビオチン化抗サイトカイン抗体によって検出して、アルカリホスファターゼ結合ストレプトアビジンおよびSigmaS基質の添加を続けた。呈色は、ELISAプレートリーダー(Molecular Devices Corp.,サニーベール、カリフォルニア州)によって評価した。図26AおよびBに示すように、CLL細胞の存在は、混合リンパ球反応で観察されたIFN−γおよびIL−2生成の大半を完全に排除した。いずれかの抗体の存在は、Th1サイトカインの生成を許容した(図26AおよびB)。これに対して、IL−10の生成は、抗体の存在下でダウンレギュレートされた(図26Cを参照)。
【0158】
(抗体依存性細胞媒介性細胞傷害性アッセイ)
さらに6つのキメラマウス抗CD200抗体を、抗体依存性細胞媒介性細胞傷害性アッセイ(ADCC)でCD200発現腫瘍細胞を殺すその能力について評価した。CD200をトランスフェクションされた293−EBNA細胞に培地0.5ml中、37℃にて、100μCi/100万細胞で標識した。3回の洗浄後、細胞をカウントして、培地(10% ヒトAB血清を添加したRPMI)中で20万/mlにて再懸濁させて、50μl(10,000細胞/ウェル)を3つずつ、96ウェル丸底プレートに分配した。最終濃度20μg/mlを達成するために、抗CD200抗体20μlを各ウェルに分配した。末梢血単核細胞(エフェクター細胞)をFicoll勾配で単離して、赤血球を塩化アンモニウムによって溶解させ、洗浄して、培地に再懸濁させて、細胞50μlを各ウェルに分配した。アッセイプレートを回転させて(1,500rpm/5分/弱ブレーキ)、細胞培養インキュベータに移した。4時間後、アッセイプレートを前と同様に回転させた。上澄み36μlをピコプレートに移して、microscint−20カクテル250μlと混合して、オービタルシェーカーに2分間置いて、Top countで読み取った。図27に示すように、CD200陽性細胞と培養したときに、マウスキメラCD200抗体すべてが同様の溶解レベルを生じた。CD200陰性細胞では溶解は観察されなかった。加えて溶解の程度は、アイソタイプ対照抗体d2A6(抗FLJ32028抗体)と比較したときに、統計的に有意であった(p<0.05)。
【0159】
(実施例5)
(Raji/PBLモデル)
NOD.CB17−Prkdc<scid>マウス(Jackson Laboratory)に、RAJI細胞4×106個(ATCC)を含有するRPMI 200μlをPBL 0、100万、500万または1000万個と共に皮下注射した。各群にマウス9または10匹を入れた。PBLを全塩化アンモニウム250mlから単離した。腫瘍増殖は、カリパスで長さおよび幅を測定することによって、週3回モニターした。腫瘍増殖は、長さx幅x幅/2に基づいて計算した。
【0160】
腫瘍細胞のみを与えた群と比較したPBLを注射した群間の相違を、対応のない両側Student t検定によって分析した。有意差は、PBL 500万または1000万個を与えた群では見られたが、第32日からPBL 100万個を与えた群では見られなかった。図28に示したデータは、各種のPBLドナーを使用した実験10回の代表的な例である。
【0161】
(Namalwa PBLモデル)
NOD.CB17−Prkdc<scid>マウス(Jackson Laboratory、バーハーバー、メイン州)に、Namalwa細胞4×106個(ATCC)を含有するRPMI 200μlを、PBL 0、200万または1000万個と共に皮下注射した。各群にマウス9または10匹を入れた。PBLを全血250mlからhistopaque勾配で単離して、0.9%塩化アンモニウムを使用して赤血球溶解を続けた。腫瘍増殖は、カリパスで長さおよび幅を測定することによって、週3回モニターした。腫瘍増殖は、長さx幅x幅/2に基づいて計算した。
【0162】
図29は、対応のない両側Student t検定によって分析した、腫瘍細胞のみを与えた群と比較したPBLを注射した群間の相違を示す。有意差は、両方のドナーでPBL 1000万個を与えた群で見られたが、第8日からPBL 200万個を与えた群では見られなかった。
【0163】
(安定なCD200発現細胞株の生成)
安定なCD200発現RajiおよびNamalwa細胞株は、Virapower Lentiviral Expression System(Invitrogen,カールスバッド、カリフォルニア州)を使用して産生した。CD200 cDNAを一次CLL細胞からRT−PCRによって、フォワードプライマー5’−GACAAGCTTGCAAGGATGGAGAGGCTGGTGA−3’(配列番号212)およびリバースプライマー5’−GACGGATCCGCCCCTTTTCCTCCTGCTTTTCTC−3’(配列番号213)を使用して単離した。PCR生成物をGatewayエントリベクターpCR8/GW/TOPO−TAへクローニングして、個々のクローンを配列決定した。正しい配列のクローンをセンスおよびアンチセンス方向の両方でレンチウイルスベクターpLenti6/V5/DESTおよびpLenti6/UbC/V5/DEST内へ、Gateway技術(Invitrogen,カールスバッド、カリフォルニア州)を用いて組込んだ。この2つのベクターの主な相違は、CD200発現を引き起こすために使用されたプロモータである:pLenti6/V5/DESTがヒトCMV前初期プロモータを含有するのに対して、pLenti6/UbC/V5/DESTはヒトユビキチンCプロモータを含有する。
【0164】
高力価VSV−Gシュードタイプレンチウイルスストックを293−FT細胞の一過性同時トランスフェクションによって、製造者が推奨するように生成した。RajiまたはNamalwa細胞は、12μg/mlポリブレンを含有する増殖培地1ml中に細胞106個を再懸濁させて、レンチウイルスストック1mlを添加することによって形質導入した。37℃にて一晩インキュベートした後、ウイルスを含有する培地を除去して、新しい培地4mlと交換した。2日後、感染した細胞をCD200発現についてフローサイトメトリーによって分析した。すべての実験で、細胞の≧70%がCD200+であったのに対して、親細胞株および負の対照(アンチセンスCD200)ウイルスを形質導入された細胞ではCD200は検出できなかった。
【0165】
CD200を過剰発現するクローン細胞株を単離するために、感染した細胞をブラストサイジンによって13日間に渡って選択した。使用したブラストサイジンの濃度はRaji細胞では6μg/ml、Namalwa細胞では2μg/mlであった。次に安定なクローンを、ブラストサイジン耐性細胞の希釈を制限することによって96ウェルプレート内へ単離した。クローンは、96ウェル形式でフローサイトメトリーによって、PE結合マウス抗ヒトCD200(クローンMRC OX104、Serotec)および高スループットサンプラーを装備したBD FACSCaliburを使用してスクリーニングした。2000 Rajiおよび2000 Namalwaクローン全体をスクリーニングした後、CD200発現が最高であるこれらのクローンをさらなる特徴付けの後に旧来の技法を用いて膨張させた。
【0166】
(RAJI/PBLモデルにおけるCD200の免疫抑制効果)
開示した方法に従って、CD200をCLL細胞でアップレギュレートされることを証明した。この分子のアップレギュレーションは、潜在的に免疫抑制性でありうる。CD200を発現する癌細胞が免疫系に癌細胞を根絶させるかどうかを試験するために、通常はCD200を発現しないRAJI細胞に、CD200をコードするレンチウイルスベクター系を上述のように感染させた。CD200を安定して発現するRAJIクローンを選択した。ベクター感染の効果を確実なくすための対照として、CD200の逆転非機能形(CD200rev)を発現するクローンも選択した。CD200を発現するRAJI細胞、CD200REVまたは親RAJI細胞をNOD.CB17−Prkdc<scid>マウスに皮下注射した。試験には以下の群を含めた:
群1:RAJI 4×106個、皮下;マウス9匹
この群は、レンチウイルス形質導入細胞が親細胞と同様の増殖を示すようにする必要があった。
【0167】
群2:RAJICD200 4×106個、皮下;マウス9匹
この群は、CD200形質導入細胞が親細胞と同様の増殖を示すようにする必要があった。またこの群は、最大の腫瘍増殖を与えるであろう。群3および群4をこの群と比較した。
【0168】
群3:RAJICD200 4×106個+PBL 5×106個、皮下;マウス9匹
このPBL数は、以前の実験で一部のマウスの腫瘍増殖を低減することが示されている。拒絶は、細胞1000万個ほど強力ではないが、CD200がある数の細胞のみに影響を及ぼしうるかどうか判定するためであり、5×106個は、拒絶を得るために使用できるPBLの最小量である;拒絶は、CD200の存在によって防止されるはずである。
【0169】
群4:RAJICD200 4×106個+PBL 10×106個、皮下;マウス8匹
これはRAJI/PBLモデルで拒絶を確認するための、PBLの最適数である。目的は、CD200発現がこの拒絶を防止するということである。
【0170】
群5:RAJICD200rev 4×106個、皮下;マウス9匹
この群は、レンチウイルス形質導入細胞が親細胞と同様の増殖を示すようにする必要があった。
【0171】
群6:RAJICD200rev 4×106個+PBL 2×106個、皮下;マウス9匹
この数のPBLは、強力な拒絶または腫瘍増殖の低減を生じるはずがない。これは群3および群4(最大予想腫瘍増殖)の正の対照である。この群で拒絶がない場合、ドナーPBLは過剰に活性化されて、CD200による効果の不足を説明できるほうから開始する。
【0172】
群7:RAJICD200rev 4×106個+PBL 5×106個、皮下;マウス9匹
CD200群3で観察されたすべての効果がCD200に実際に関連しており、レンチウイルス形質導入には関連していないという対照。
【0173】
群8:RAJICD200rev 4×106個+PBL 10×106個、皮下;マウス8匹
CD200群4で観察されたすべての効果がCD200に実際に関連しており、レンチウイルス形質導入には関連していないという対照。
【0174】
許容限界を超えたサイズに達した腫瘍に基づいて、第38日に動物を殺処分した。動物4匹/群から腫瘍を除去した。腫瘍2個/群をOCTで凍結させて、他の2個は細胞を単離して、FACSによってCD200発現について分析するために使用した。図30(a〜c)は、本試験の結果を示す。
【0175】
RAJICD200細胞は多少より低速に増殖するように思われるが、形質導入細胞と親細胞との増殖差は、図30(a)に示すように統計的有意には達しなかった。
【0176】
PBL 5または10×106個を注射したときに、PBLの存在は腫瘍増殖を84%まで減速したが、一般にPBL 10×106個は、PBL 5×106個と比較して、時間に関してより強力な減速を引き起こした。親腫瘍細胞と比較した増殖の減速は第20日から著しかった。PBL 2×106個は第22日〜第29日に著しい腫瘍増殖の減速を引き起こしたが、その減速は後の時点で克服された(図30(b))。本試験は、この特定のドナーがRAJI腫瘍細胞を非常に強力に拒絶することを示した。
【0177】
CD200発現RAJI細胞およびPBLを投与した群の腫瘍増殖は、RAJI細胞のみを投与した群の腫瘍増殖とは著しく異なっていなかったが、PBL 10×106個を投与されたマウスは腫瘍増殖減速の傾向を示し、ただし差は、腫瘍が100m3に達した後のどの時点でも統計的有意には達しなかった。RAJI細胞およびPBL 5×106個を投与した群の各マウスは第2の腫瘍を発症して、一部のマウスは第7日という早期に発症したが、これは任意の他の群では観察されなかった。分析のために、第2の腫瘍を第1の腫瘍に加えて、合せたサイズを図30(c)に示す。
【0178】
このような結果は、腫瘍細胞でのCD200発現が実際に、免疫系による腫瘍増殖の減速を防止することを示す。また本試験は、免疫抑制性化合物または分子を評価するための、RAJI/PBLモデルの有用性を証明する。
【0179】
(Namalwa/PBLモデルでのCD200の免疫抑制効果)
RAJI/PBLモデルで見られる効果が他の腫瘍モデルでも見られるかどうかを評価するために、Namalwa腫瘍細胞もレンチウイルスCD200系によって感染させて、安定なクローンを選択した。ベクター感染の効果を確実なくすための対照として、CD200の逆転非機能形(CD200rev)を発現するクローンも選択した。NOD.CB17−Prkdc<scid>マウスに、以下のスキームに従って図31(a)〜(d)に示すように注射した:
群1:Namalwa 4×106個、皮下;マウス9匹
この群は、レンチウイルス形質導入細胞が親細胞と同様の増殖を示すようにする必要があった。
【0180】
群2:Namalwa CD200(1D12Ub)4×106個、皮下;マウス9匹
この群は、CD200形質導入細胞が親細胞と同様の増殖を示すようにする必要があった。またこの群は、最大の腫瘍増殖を与えるであろう。群3および群4をこの群と比較した。
【0181】
群3:Namalwa CD200(1D12Ub)4×106個+PBL 5×106個、皮下;マウス9匹
このPBL数は、以前の実験で一部のマウスの腫瘍増殖を低減することが示されている。拒絶は、細胞1000万個ほど強力ではないが、CD200がある数の細胞のみに影響を及ぼすことが可能で、これが最小PBLである場合、拒絶を得るために使用できる;拒絶はCD200の存在によって防止されるであろう。
【0182】
群4:Namalwa CD200(1D12Ub)4×106個+PBL 10×106個、皮下;マウス8匹
これはNamalwa/PBLモデルで拒絶を確認するための、PBLの最適数である。これはCD200発現がこの拒絶を防止することを示すために実施した。
【0183】
群5:Namalwa CD200rev(C5Ubrev)4×106個、皮下;マウス9匹
この群は、レンチウイルス形質導入細胞が親細胞と同様の増殖を示すようにする必要があった。
【0184】
群6:Namalwa CD200rev 4×106個+PBL 2×106個、皮下;マウス9
この数のPBLは、強力な拒絶または腫瘍増殖の低減を生じるはずがない。これは群3および群4(最大予想腫瘍増殖)の正の対照である。この群で拒絶がない場合、ドナーPBLは過剰に活性化されて、CD200による効果の不足を説明できるほうから開始する。
【0185】
群7:Namalwa CD200rev 4×106個+PBL 5×106個、皮下;マウス9匹
この群は、CD200に関連するが、レンチウイルス形質導入には関連しないCD200群3の任意の効果を検出するための対照として必要であった。
【0186】
群8:Namalwa CD200rev 4×106個+PBL 10×106個、皮下;マウス8匹
この群は、CD200に関連するが、レンチウイルス形質導入には関連しないCD200群4の任意の効果を検出するための対照として必要であった。
【0187】
腫瘍の長さおよび幅を週3回評価した。
【0188】
すべての腫瘍細胞が迅速な腫瘍増殖を引き起こした。形質導入細胞と親細胞との間に著しい増殖差はなかった。腫瘍は図31(a)に示すように、以前に観察されたよりも侵襲的に増殖する。
【0189】
図31(b)は、PBLの存在が腫瘍増殖を約50%減速することを示す。この傾向は第2日から観察された。PBL処置群対腫瘍細胞のみを投与した群の差は、PBLを200万個または1000万個注射したときに、第17日および第19日に統計的に有意である(両側Student t検定によって決定されたように)。PBL 500万個の注射は、腫瘍増殖の減速を引き起こしたが、有意には達しなかった。
【0190】
CD200発現Namalwa細胞およびPBLを投与した群の腫瘍増殖は、Namalwa細胞のみを投与した群の腫瘍増殖と同様であった(図31(c))。
【0191】
これらのデータは、腫瘍細胞でのCD200発現がヒト免疫系による腫瘍増殖の減速を防止することを確認している。
【0192】
(抗CD200抗体による、RAJI/PBLモデルにおけるCD200の免疫抑制効果の遮断)
抗CD200抗体が腫瘍細胞で発現されたCD200の免疫抑制効果を遮断できるかどうかを評価するために、CD200が形質導入されたRAJI細胞をNOD.CB17−Prkdc<scid>マウスに皮下注射して、キメラ抗CD200抗体d1B5およびc2aB7ならびに腫瘍細胞を結合しない抗体(alxn4100)の存在下または非存在下でPBLが腫瘍増殖を低減する能力を評価した。抗体を最初に腫瘍細胞と共に10mg/kgまた2.5mg/kgで、次に以下に示す濃度で週2回静脈内投与した。以下の群を構成した:
1.RAJICD200 4×106個;マウス10匹
2.RAJICD200 4×106個+PBL 5×106個;マウス10匹
3.RAJICD200 4×106個+PBL 5×106個+100mg/kg d1B5;マウス12匹
4.RAJICD200 4×106個+PBL 5×106個+20mg/kg d1B5;マウス9匹
5.RAJICD200 4×106個+PBL 5×106個+100mg/kg c2aB7;マウス11匹
6.RAJICD200 4×106個+PBL 5×106個+20mg/kg c2aB7;マウス9匹
7.RAJICD200 4×106個+PBL 5×106個+100mg/kg alxn4100;マウス9匹。
【0193】
腫瘍の長さおよび幅を週に3回測定して、腫瘍体積を腫瘍の長さ*幅*幅/2によって計算した。図33は、予想通り、腫瘍細胞でのCD200発現が免疫細胞による腫瘍増殖の低減を防止することを示している。しかしながら抗CD200抗体の添加は、腫瘍体積を50〜75%縮小した。抗体による増殖の低減は、第18日から試験の終りまでのStudent t検定およびMann Whitney検定によって決定されたように、統計的に有意であった。これに対して、対照抗体による処置は、腫瘍増殖を低減しなかった。これらのデータは、抗癌処置での抗CD200の有用性を証明する。また本試験は、免疫調節治療を評価するための、RAJI/PBLモデルの有用性を証明する。
【0194】
(抗CD200抗体による、Namalwa/PBLモデルにおけるCD200の免疫抑制効果の遮断)
RAJI/PBLモデルにおいて抗CD200抗体によって見られた効果が他の腫瘍モデルでも観察できるかどうかを評価するために、CD200が形質導入されたRAJI細胞をNOD.CB17−Prkdc<scid>マウスに皮下注射して、キメラ抗CD200抗体d1B5およびc2aB7ならびに腫瘍細胞を結合しない抗体(alxn4100)の存在下または非存在下でPBLが腫瘍増殖を低減する能力を評価した。以下に示す濃度の抗体を最初に腫瘍細胞と共に、次に腫瘍0.5cm以内では週2回、皮下投与した。以下の群を構成した(別途記載しない限り、マウス10匹/群):
1.NamalwaCD200 4×106個
2.NamalwaCD200 4×106個+PBL 5×106個;マウス12匹
3.NamalwaCD200 4×106個+PBL 5×106個+10mg/kg d1B5;マウス12匹
4.NamalwaCD200 4×106個+PBL 5×106個+2.5mg/kg d1B5
5.NamalwaCD200 4×106個+PBL 5×106個+10mg/kg c2aB7;マウス12匹
6.NamalwaCD200 4×106個+PBL 5×106個+2.5mg/kg c2aB7
7.NamalwaCD200 4×106個+PBL 5×106個+10mg/kg alxn4100。
【0195】
腫瘍の長さおよび幅を週に3回測定して、腫瘍体積を腫瘍の長さ*幅*幅/2によって計算した。図34は、予想通り、腫瘍細胞でのCD200発現が免疫細胞による腫瘍増殖の低減を防止することを示している。しかしながら抗CD200抗体の添加は、腫瘍体積を最大97%縮小した。抗体による増殖の低減は、第12日から試験の終りまでのStudent t検定およびMann Whitney検定によって決定されたように、統計的に有意であった。これに対して、対照抗体による処置は、腫瘍増殖を低減しなかった。これらのデータによって、抗癌処置での抗CD200の有用性、および免疫調節療法での腫瘍/PBLモデルの使用が確認される。
【0196】
(RAJI/PBLモデルにおける化合物の潜在的な免疫増強効果の検出)
CD3カラム(Miltenyi)を使用してPBLから単離したT細胞を、AZND1(抗DC−SIGN抗体)の腹水調製物または対照抗体(BB5)の腹水調製物と共にインビトロでインキュベートした。NOD.CB17−Prkdc<scid>マウスに次のスキームに従って注射した:
群6:マウス8匹、RAJI 4×106個+AZND1増強T細胞0.8×106+PBL 2×106皮下
群5:マウス10匹、RAJI 4×106個、皮下
群4:マウス8匹、RAJI 4×106個+PBL 8×106個、皮下
群3:マウス8匹、RAJI 4×106個+新しいT細胞0.8×106個、皮下
群2:マウス8匹、RAJI 4×106個+AZND1増強T細胞0.8×106個、皮下
群1:マウス8匹、RAJI 4×106個+BB5.1増強T細胞0.8×106個、皮下
腫瘍の長さおよび幅を3回/週で測定した。
【0197】
T細胞は負の対照BB5でインキュベートしたT細胞は腫瘍増殖を低減しなかったが、AZND1処置T細胞は腫瘍増殖を著しく低減しなかった(図32を参照)。
【0198】
これらの結果は、免疫増強化合物の有効性を評価するためにRAJ/PBLモデルを使用できることを証明する。
【0199】
(実施例6)
(CLL患者でのCD200アップレギュレーションの判定)
CLL患者15名からのリンパ球をFITC結合抗CD5(e−bioscience)、APC結合抗CD19(e−bioscience)およびPE結合抗CD200(Serotec)によって染色した。健常ドナーからのリンパ球をそれに従って染色した。CD5+CD19+細胞でのCD200発現を判定した。以下の表に示すように、CD200発現のレベルはCLL患者サンプル間で変化したが、すべてのCLLサンプルが、正常B細胞でのCD200発現と比較して、上昇したレベル(1.6〜4倍の範囲)のより高いCD200発現を示した。上昇したレベルのCD200発現を示すCCL患者を、本明細書で述べる方法によるCD200処置のために選択した。
【0200】
(正常B細胞と比較したB−CLL細胞でのCD200発現のFACS分析)
【0201】
【化2】
(参考文献)
以下の参考文献は、本発明が関係する最新技術をより十分に説明するために、参考として本明細書中に援用される。以下のこのような刊行物または上で参照により組み入れられたものと、本開示との不一致は、本開示を支持して解決されるものとする。
【0202】
【化3】
本明細書で開示された実施形態に対して各種の改良を行えることが理解されるであろう。たとえば当業者が認識するように、本明細書で述べた特異性配列は、OX−2/CD200を結合するのに使用されるポリペプチド、抗体または抗体断片の機能に必ずしも悪影響を及ぼすことなく、わずかに変更できる。たとえば抗体配列内の1個または複数のアミノ酸の置換は、抗体または断片の機能を破壊することなく頻繁に行うことができる。それゆえ本明細書で述べた特異性抗体に対して70%を超える同一性の程度を有するポリペプチドまたは抗体が本開示の範囲内であることを理解すべきである。特に有用な実施形態において、本明細書で述べた特異性抗体に対して約80%を超える同一性を有する抗体が検討される。他の有用な実施形態において、本明細書で述べた特異性抗体に対して約90%を超える同一性を有する抗体が検討される。したがって上の記述は、制限的としてではなく、単に好ましい実施形態の例示として解釈すべきである。当業者は、本開示の範囲および精神から逸脱せずに他の改良を構想するであろう。
【技術分野】
【0001】
(関連出願)
本出願は、2005年6月30日に提出された米国特許出願第11/171,567号に対する優先権を主張する。この米国特許出願第11/171,567号は、2004年11月23日に提出された米国特許出願第10/996,316号の一部継続である。米国特許出願第10/996,316号は、2004年7月20日に提出された米国特許出願第10/894,672号の一部継続である。米国特許出願第10/894,672号は、2003年12月15日に提出された米国特許出願第10/736,188号の一部継続である。米国特許出願第10/736,188号は、次に、2001年12月10日に提出されたPCT/US01/47931の一部継続である。PCT/US01/47931は、2000年12月8日に提出された米国仮出願第60/254,113号に対する優先権を主張する国際出願である。米国仮出願第60/254,113号は、2003年3月4日に提出された米国特許出願第10/379,151号の一部継続である。上述の米国出願、国際出願および仮出願の開示全体は、参考として本明細書中に援用される。
【0002】
(技術分野)
2つの機構の組合せを提供する療法を使用する癌処置を開示する。さらに詳細には、本開示は:1)免疫抑制を遮断して、それにより癌細胞の根絶を促進するために、CD200とそのレセプターとの間の相互作用を妨害する;2)a)補体媒介性または抗体依存性細胞傷害、あるいはb)CD200標的部分を含む融合分子を使用して細胞を標的化することのどちらかによって、癌細胞を直接殺す療法を使用して、癌を処置することに関する。
【背景技術】
【0003】
(背景)
慢性リンパ球性白血病(CLL)は、白血球の疾患であり、西半球における白血病の最も一般的な形である。CLLは、ゆっくり増殖するが、長い寿命を持つ悪性リンパ球の増殖に関連する疾患の別の群を示す。CLLは、たとえば古典型および混合型のB細胞慢性リンパ球性白血病(B−CLL)、B細胞およびT細胞前リンパ球性白血病、ヘアリー細胞白血病、および大顆粒リンパ球性白血病を含む各種のカテゴリーに分類される。
CLLのすべての異なる種類のうち、B−CLLは全白血病の約30パーセントを占める。50歳を超えた個人でより頻繁に発生するが、より若年者にも次第に見られるようになっている。B−CLLは、形態的に正常であるが、生物学的に未成熟であり、機能損失につながるBリンパ球の蓄積を特徴とする。リンパ球は正常には、感染と戦うように機能する。しかしながらB−CLLでは、リンパ球が血液および骨髄に蓄積して、リンパ節の腫れを引き起こす。正常な骨髄および血液細胞の生成が減少して、患者は重篤な貧血はもちろんのこと、血小板数の減少もしばしば経験する。このことは、生命を脅かす出血の危険および白血液細胞数の減少による深刻な感染の発現をもたらす可能性がある。
【0004】
白血病などの疾患をさらに理解するために、その病因論、病原および生物学に関する研究のためのツールとして使用できる適切な細胞株を有することが重要である。悪性ヒトBリンパ細胞株の例は、プレB急性リンパ芽球性白血病(Reh)、びまん性大細胞リンパ腫(WSU−DLCL2)、およびワルデンシュトレームマクログロブリン血症(WSU−WM)を含む。あいにく既存の細胞株の多くは、白血病およびリンパ腫の臨床的に最も一般的な種類を示さない。
【0005】
インビトロでのエプスタイン−バールウイルス(EBV)感染の使用は、悪性細胞を代表する一部のCLL由来細胞株、特にB−CLL細胞株をもたらした。これらの細胞株の表現型は、生体内腫瘍の表現型とは異なり、代わりにB−CLL系の特徴は、リンパ芽球様細胞株の特徴と類似する傾向がある。EBV感染を用いてB−CLL細胞を不死化する試みは、あまり成功しなかった。この理由ははっきりしないが、EBVレセプター発現、結合または摂取の不足によらないことは既知である。Wellsらは、B−CLL細胞が細胞周期のG1/S期で停止されることと、変換関連EBV DNAが発現されなかったことを見出した。EBVとB−CLL細胞との間の相互作用が、正常なB細胞との間の相互作用と異なることを示唆する。EBV変換CLL細胞株はさらに分化して、EBVによって不死化されたリンパ芽球様細胞株(LCL)にさらに類似した形態を示すと思われる。
【0006】
EBV陰性CLL細胞株のWSU−CLLが以前に確立されている(非特許文献1)。しかしながら、他のそのような細胞株は知られていない。
【0007】
各種の機構が、癌およびCLLを含む疾患状態に対する生体反応で役割を果たす。たとえばCD4+Tヘルパー細胞は、刺激因子をエフェクター細胞に供給することによって、各種の悪性腫瘍に対して有効な免疫反応で不可欠な役割を果たす。細胞傷害性T細胞は、癌細胞を除去するのに最も有効な細胞であると考えられ、Tヘルパー細胞は、IL−2およびIFN−γなどのTh1サイトカインを分泌することによって細胞傷害性T細胞を刺激する。各種の悪性腫瘍において、Tヘルパー細胞は、健常者に見られる細胞と比べて変化した表現型を有することが示されている。顕著に変化した特徴の1つは、Th1サイトカイン生成の低下およびTh2サイトカイン生成への変化である(たとえば非特許文献2;非特許文献3;非特許文献4;非特許文献5;非特許文献6;非特許文献7;非特許文献8;非特許文献9;非特許文献10を参照)。Th1プロフィールへのそのサイトカイン変化を逆転することは、T細胞の抗腫瘍効果を増強することが証明されている(非特許文献11;非特許文献12を参照)。
【0008】
腫瘍細胞がTh1からTh2へのTヘルパー細胞のサイトカイン発現を駆動させる能力の基礎を成す機構は、IL−10またはTGF−βなどのサイトカインの分泌はもちろんのこと、免疫系の細胞と相互作用する表面分子の発現も含む。免疫グロブリン遺伝子ファミリの分子に高度の相同性を持つ樹状細胞の表面に発現された細胞である、OX−2/CD200は、免疫抑制に関与しており(非特許文献13)、OX−2/CD200発現細胞をTh1サイトカイン生成の刺激を阻害できる証拠が提供されている。Gorczynskiらはマウスモデルにおいて、OX−2/CD200 Fcの輸液が白血病腫瘍細胞を使用した動物モデルにおける腫瘍細胞の拒絶を抑制することを示した(非特許文献14)。
【0009】
特にT細胞の活性を増強できる範囲で、癌またはCLLの罹患者を処置する改良方法が望ましい。
【先行技術文献】
【非特許文献】
【0010】
【非特許文献1】Mohammad et al.,,Leukemia(1996)10(1):130−7
【非特許文献2】Kiani,et al.,Haematologica(2003)88:754−761
【非特許文献3】Maggio,et al.,Ann Oncol(2002)13 Suppl 1:52−56
【非特許文献4】Ito, et al.,Cancer(1999)85:2359−2367
【非特許文献5】Podhorecka, et al.,Leuk Res(2002)26:657−660
【非特許文献6】Tatsumi,et al.,J Exp Med(2002)196:619−628
【非特許文献7】Agarwal,et al.,Immunol Invest(2003)32:17−30
【非特許文献8】Smyth,et al.,Ann Surg Oncol(2003)10:455− 462
【非特許文献9】Contasta, et al.,Cancer Biother Radiopharm(2003)18:549−557
【非特許文献10】Lauerova, et al.,Neoplasma(2002)49:159−166
【非特許文献11】Winter,et al.,Immunology(2003)108:409−419
【非特許文献12】Inagawa,et al.,Anticancer Res(1998)18:3957−3964
【非特許文献13】Gorczynski et al.,Transplantation(1998)65:1106−1114
【非特許文献14】Clin Exp Immunol(2001)126:220−229
【発明の概要】
【課題を解決するための手段】
【0011】
(概要)
1つの実施形態において、EBVによる不死化によって確立されていない悪性起源のCLL細胞株が提供される。細胞株は一次CLL細胞に由来し、ATCCアクセション番号PTA−3920で寄託されている。好ましい実施形態において、細胞株はCLL−AATである。CLL−AATはB−CLL一次細胞に由来するB−CLL細胞株である。
【0012】
さらなる局面において、CLL−AAT細胞株を使用して、CLLの診断および/または処置に有用なモノクローナル抗体を産生させる。抗体は、本明細書で免疫原として開示された細胞を使用することによって産生でき、それゆえモノクローナル抗体を単離できる動物での免疫反応を上昇させる。そのような抗体の配列が決定でき、抗体またはその変異体は組換え技法によって生成できる。この局面において、「変異体」は、モノクローナル抗体の配列に基づくキメラ、CDRグラフト、ヒト化および完全ヒト抗体を含む。
【0013】
さらに組換えライブラリーに由来する抗体(「ファージ抗体」)は、本明細書で述べた細胞、またはそれに由来するポリペプチドを、標的特異性に基づいて抗体を単離するためのベイトとして用いて選択できる。
なおさらなる局面において、抗体は、一次CLL細胞、またはそれに由来する抗原を用いて抗体ライブラリーをパニングすることによって産生でき、そしてCLL細胞株、たとえば本明細書で述べるCLL細胞株を使用してさらにスクリーニングおよび/または特徴付けできる。したがって、CLL細胞株への抗体の結合を評価することを含む、CLLに特異性の抗体を特徴付ける方法が提供される。
【0014】
さらなる局面において、当業者に公知の方法によって、たとえば免疫沈降とそれに続く質量分光分析によって、CLL−AAT細胞株を利用してCLL細胞内で特異的に発現されるタンパク質を同定するための方法が提供される。そのようなタンパク質は、CLL−AAT細胞株において、またはCLL患者に由来する一次細胞において特異的に発現できる。
【0015】
低分子ライブラリー(多くが市販されている)は、細胞の増殖特徴を調節できる因子を同定するための細胞ベースアッセイにおいて、CLL−AAT細胞株を使用してスクリーニングできる。たとえばCLL−AAT細胞株でアポトーシスを調節する、またはその増殖および/または増殖を阻害する因子を同定できる。そのような因子は、治療化合物の開発のための候補である。
【0016】
CLL−AAT細胞株から単離された核酸は、CLL特異性遺伝子を同定するサブトラクティブハイブリダイゼーション実験で、またはマイクロアレイ分析(たとえば遺伝子チップ実験)で使用できる。転写がCLL細胞において調節される遺伝子を同定できる。この方法で同定されたポリペプチドまたは核酸遺伝子生成物は、CLLの抗体または小型細胞療法の開発のための手がかりとして有用である。
【0017】
好ましい局面において、CLL−AAT細胞株は、細胞によってインターナライズされる細胞表面成分に結合する、インターナライズする抗体を同定するために使用できる。そのような抗体は、治療に使用するための候補である。特に、細胞質内で安定なままであり、細胞内結合活性を維持する単鎖抗体は、この方法でスクリーニングできる。
【0018】
なお別の局面において、患者が悪性癌細胞によってアップレギュレートされたポリペプチドについてスクリーニングされ、アップレギュレートされたポリペプチドの代謝経路を妨害する抗体が患者に投与される治療処置が説明されている。
【0019】
本開示はさらに、OX−2/CD200が被験体においてアップレギュレートされるかどうかに関する判定が行われ、もしそうならば被験体に免疫反応を増強する療法投与するための方法に関する。OX2/CD200のアップレギュレーションは、OX2/CD200レベルを測定することによって直接、またはOX2/CD200と相関する任意のマーカーのレベルをモニターすることによって判定できる。適切な免疫調節療法は、T細胞または抗原提示細胞の負の調節を遮断する因子の投与、T細胞、癌ワクチン、免疫系を刺激する一般的なアジュバントの正の副刺激を増強する因子の投与あるいはIL−2、GM−CSFおよびIFN−γなどのサイトカインによる処置を含む。特に有用な実施形態において、免疫反応を増強する療法は、1つ以上の他の免疫調節療法と場合により組合せた、OX−2/CD200に結合するポリペプチドの投与を含む。別の実施形態において、ポリペプチドはOX−2/CD200レセプターに結合する。
【0020】
別の局面において、本開示による方法を使用して、OX−2/CD200が被験体にてアップレギュレートされる疾患状態を、OX−2/CD200に結合するポリペプチドまたはOX−2/CD200レセプターを該疾患状態に罹患した被験体に投与することによって処置する。1つの実施形態において、これらの方法によって処置される疾患状態は、癌、特に他の実施形態においてCLLを含む。
【0021】
特に有用な実施形態において、本開示による癌療法は、i)免疫抑制を遮断して、それにより癌細胞の根絶を促進するために、CD200とそのレセプターとの間の相互作用を妨害する抗体を投与することと;ii)癌細胞を直接殺すために、CD200標的部分を含む融合分子を投与することとを含む。あるいは抗体は、補体媒介性または抗体依存性細胞傷害によって癌細胞を直接殺す。
【0022】
本開示による別の実施形態において、治療処置の進行をモニターするための方法が提供される。該方法は、免疫調節療法を投与する工程と、療法の有効性を判定するために、被験体におけるOX−2/CD200レベルを少なくとも2回判定する工程とを含む。
【0023】
別の局面において、本開示は、癌細胞によって発現された分子の免疫調節効果を評価するための方法を提供する。これらの方法では、癌細胞によって発現またはアップレギュレートされる分子を同定する(たとえば実験的に、またはデータベースから)。癌細胞、リンパ球および癌細胞によって発現またはアップレギュレートされる分子を被験体に投与して、癌細胞の増殖の速度をモニターする。投与されるリンパ球の数は、a)癌細胞の増殖を減速するのに十分であるように、またはb)癌細胞の増殖を減速するのに不十分であるようにのいずれかに規定される。癌細胞によって発現またはアップレギュレートされる分子は、分子または分子自体の活性部分として、あるいは分子またはその部分を自然に生成または発現する細胞を投与することによって、あるいは分子またはその部分を生成または発現するように操作された細胞を投与することによって投与できる。腫瘍細胞およびドナーリンパ球が単独で投与された場合の増殖速度と比較して、癌細胞の増殖速度の何らかの変化が観察される場合、該分子は免疫調節効果を有すると考えられる。たとえば投与されるリンパ球の数が癌細胞の増殖を減速するのに十分であり、観察された腫瘍細胞の増殖速度が腫瘍細胞およびドナーリンパ球が単独で投与されたときの増殖速度と比較して高い場合、癌細胞によって発現またはアップレギュレートされる分子は免疫抑制性であると見なされる。別の例として、投与されるリンパ球の数が癌細胞の増殖を減速するのに不十分であり、観察された腫瘍細胞の増殖速度が腫瘍細胞およびドナーリンパ球が単独で投与されたときの増殖速度と比較して低い場合、該化合物は免疫増強性であるとみなされる。いったん分子の免疫調節効果が確立されると、本明細書で述べた実施形態に従って、分子の活性を増強または阻害する化合物が同定できる。増強または阻害効果は、癌細胞によって発現またはアップレギュレートされた分子との直接作用の結果でありうるか、あるいは癌細胞によって発現またはアップレギュレートされた化合物の代謝経路における他の分子との間の相互作用の結果でありうる。
【0024】
別の局面において、本開示は化合物の免疫調節効果を評価するための方法を提供する。これらの方法では、癌細胞、リンパ球および評価される化合物を被験体に投与して、癌細胞の増殖の速度をモニターする。投与されるリンパ球の数は、a)癌細胞の増殖を減速するのに十分であるように、またはb)癌細胞の増殖を減速するのに不十分であるように、のいずれかに規定される。評価される化合物は、化合物自体として、または該化合物を自然に生成する細胞を投与することによって、または該化合物を生成するように操作された細胞を投与することによって投与できる。腫瘍細胞およびドナーリンパ球が単独で投与された場合の増殖速度と比較して、癌細胞の増殖速度の何らかの変化が観察される場合、該分子は免疫調節効果を有すると考えられる。たとえば投与されるリンパ球の数が癌細胞の増殖を減速するのに十分であり、観察された腫瘍細胞の増殖速度が腫瘍細胞およびドナーリンパ球が単独で投与されたときの増殖速度と比較して高い場合、癌細胞によって発現またはアップレギュレートされる分子は免疫抑制性であると見なされる。別の例として、投与されるリンパ球の数が癌細胞の増殖を減速するのに不十分であり、観察された腫瘍細胞の増殖速度が腫瘍細胞およびドナーリンパ球が単独で投与されたときの増殖速度と比較して低い場合、該化合物は免疫増強性であるとみなされる。
例えば、本願発明は以下の項目を提供する。
(項目1)
癌を処置するための方法であって、
癌に罹患している被験体に対して、
i)CD200とCD200レセプターとの間の相互作用を妨害して、それによりCD200の免疫抑制効果を阻害し、かつ
ii)OX−2/CD200またはOX−2/CD200レセプターに結合する部分を含むポリペプチド融合分子を使用して癌細胞を殺す
治療組成物を投与する工程;
を包含する、方法。
(項目2)
前記治療組成物を投与する工程が、前記被験体に対して、OX−2/CD200に結合する抗体を投与することを包含する、項目1に記載の方法。
(項目3)
前記治療組成物を投与する工程が、前記被験体に対して、OX−2/CD200に結合するモノクローナル抗体を投与することを包含する、項目1に記載の方法。
(項目4)
前記治療組成物を投与する工程が、前記被験体に対して、OX−2/CD200レセプターに結合する抗体を投与することを包含する、項目1に記載の方法。
(項目5)
前記治療組成物を投与する工程が、前記被験体に対して、OX−2/CD200レセプターに結合するモノクローナル抗体を投与することを包含する、項目1に記載の方法。
(項目6)
前記癌に罹患している被験体が、CLL患者である、項目1に記載の方法。
(項目7)
前記融合分子が、毒素を含む、項目1に記載の方法。
(項目8)
前記融合分子が、高エネルギー放射線エミッターを含む、項目1に記載の方法。
(項目9)
前記融合分子が、細胞傷害性T細胞活性またはNK細胞活性を増強するサイトカインもしくはケモカインを含む、項目1に記載の方法。
(項目10)
前記融合分子が、T細胞を誘引するケモカインを含む、項目1に記載の方法。
(項目11)
癌を処置するための方法であって、
CD200に結合する抗体を投与する工程;
を包含し、
a)CD200とそのレセプターとの間の相互作用が遮断され、
b)CD200を発現する癌細胞が殺される、
方法。
(項目12)
癌を処置するための方法であって、
CD200に結合する抗体を投与する工程;
を包含し、
a)CD200とそのレセプターとの間の相互作用が遮断され、
b)該癌に対する細胞傷害性T細胞活性またはNK細胞活性が増強される、
方法。
(項目13)
前記細胞傷害性T細胞活性またはNK細胞活性の増強が、IL−2、IL−12、IL−18、IL−13、およびIL−5からなる群より選択されるサイトカインと融合したCD200に結合する抗体によって達成される、項目12に記載の癌を処置するための方法。
(項目14)
癌を処置するための方法であって、
CD200に結合する抗体を投与する工程;
を包含し、
a)CD200とそのレセプターとの間の相互作用が遮断され、
b)T細胞が腫瘍細胞に誘引される、
方法。
(項目15)
T細胞誘引が、MIG、IP−10およびI−TACからなる群より選択されるケモカインと融合したCD200に結合する抗体によって達成される、項目14に記載の癌を処置するための方法。
(項目16)
OX−2/CD200抗原を保持する細胞を標的とする第一の部分と;
細胞の死を促進する第二の部分と;
を含む、融合分子。
(項目17)
前記第一の部分が、OX−2/CD200に結合する抗体を含む、項目16に記載の融合分子。
(項目18)
前記第一の部分が、OX−2/CD200に結合するモノクローナル抗体を含む、項目16に記載の融合分子。
(項目19)
癌を処置するための方法であって、
癌患者に対して、
(i)CD200とCD200レセプターとの間の相互作用を妨害して、それによりCD200の免疫抑制効果を阻害し、かつ
(ii)補体媒介性細胞傷害または抗体依存性細胞傷害によって癌細胞を殺す、
抗体を投与する工程;
を包含する、
方法。
(項目20)
(i)CD200とCD200レセプターとの間の相互作用を妨害して、それによりCD200の免疫抑制効果を阻害し、かつ(ii)補体媒介性細胞傷害または抗体依存性細胞傷害によって癌細胞を殺す、抗体。
(項目21)
項目20に記載の抗体と、薬学的に受容可能なキャリアとを含む、組成物。
(項目22)
前記治療組成物を投与する工程が、配列番号5、12および13からなる群より選択される配列を有する軽鎖CDR1領域を含む抗体を投与することを包含する、項目1に記載の方法。
(項目23)
前記治療組成物を投与する工程が、配列番号21および23からなる群より選択される配列を有する軽鎖CDR2領域を含む抗体を投与することを包含する、項目1に記載の方法。
(項目24)
前記治療組成物を投与する工程が、配列番号29、37および38からなる群より選択される配列を有する軽鎖CDR3領域を含む抗体を投与することを包含する、項目1に記載の方法。
(項目25)
前記治療組成物を投与する工程が、配列番号50、55および56からなる群より選択される配列を有する重鎖CDR1領域を含む抗体を投与することを包含する、項目1に記載の方法。
(項目26)
前記治療組成物を投与する工程が、配列番号69、74および75からなる群より選択される配列を有する重鎖CDR2領域を含む抗体を投与することを包含する、項目1に記載の方法。
(項目27)
前記治療組成物を投与する工程が、配列番号88、93および94からなる群より選択される配列を有する重鎖CDR3領域を含む抗体を投与することを包含する、項目1に記載の方法。
(項目28)
前記第一の部分が、配列番号5、12、13、21、23、29、37、38、50、55、56、69、74、75、88、93および94からなる群より選択される1つ以上のアミノ酸配列を含むポリペプチドを含む、項目16に記載の融合分子。
(項目29)
前記治療組成物が、ヒト化抗体を含む、項目2〜5のいずれかに記載の方法。
(項目30)
前記治療組成物が、Fv、scFv、Fab’およびF(ab’)2を含む、項目2〜5のいずれかに記載の方法。
(項目31)
前記抗体が、ヒト化抗体を含む、項目11〜15のいずれかに記載の方法。
(項目32)
前記抗体が、Fv、scFv、Fab’およびF(ab’)2を含む、項目11〜15のいずれかに記載の方法。
(項目33)
前記抗体が、ヒト化抗体を含む、項目17〜18のいずれかに記載の融合分子。
(項目34)
前記抗体が、Fv、scFv、Fab’およびF(ab’)2を含む、項目17〜18のいずれかに記載の融合分子。
(項目35)
前記治療組成物が、ヒト化抗体を含む、項目22〜27のいずれかに記載の方法。
(項目36)
前記治療組成物が、Fv、scFv、Fab’およびF(ab’)2を含む、項目22〜27のいずれかに記載の方法。
(項目37)
前記抗体が、ヒト化抗体を含む、項目21に記載の組成物。
(項目38)
前記抗体が、Fv、scFv、Fab’およびF(ab’)2を含む、項目21に記載の組成物。
(項目39)
前記ポリペプチドが、ヒト化抗体を含む、項目28に記載の融合分子。
(項目40)
前記ポリペプチドが、Fv、scFv、Fab’およびF(ab’)2を含む、項目28に記載の融合分子。
(項目41)
配列番号111〜199からなる群より選択されるポリペプチド配列を含む、CD200を発現する細胞に結合する抗体。
(項目42)
配列番号111、114、118、133、136、140、141、149、155、162、163、165、174、181、187、188、189、195および199からなる群より選択されるポリペプチド配列を含む、項目41に記載の抗体。
(項目43)
配列番号200〜211からなる群より選択されるポリペプチド配列を含む、項目41に記載の抗体。
(項目44)
前記抗体が、ヒト化抗体を含む、項目41に記載の抗体。
(項目45)
前記抗体が、Fv、scFv、Fab’およびF(ab’)2を含む、項目41に記載の抗体。
(項目46)
癌を処置するための方法であって、
癌に罹患している被験体に対して、CD200を発現する細胞に結合する抗体を含有する治療組成物を投与する工程;
を包含し、該抗体は、1つ以上のTh1サイトカインの生成を促進するのに十分な量で存在する、方法。
(項目47)
前記抗体が、配列番号111〜199からなる群より選択されるポリペプチド配列を含む、項目46に記載の方法。
(項目48)
前記抗体が、配列番号111、114、118、133、136、140、141、149、155、162、163、165、174、181、187、188、189、195および199からなる群より選択されるポリペプチド配列を含む、項目46に記載の方法。
(項目49)
前記抗体が、配列番号200〜211からなる群より選択されるポリペプチド配列を含む、項目46に記載の方法。
(項目50)
前記抗体が、ヒト化抗体を含む、項目46に記載の方法。
(項目51)
前記抗体が、Fv、scFv、Fab’およびF(ab’)2を含む、項目46に記載の方法。
(項目52)
前記癌が、CLLである、項目46に記載の方法。
(項目53)
腫瘍細胞を殺すための方法であって、
癌に罹患している被験体に対して、CD200を発現する細胞に結合する抗体を含む治療組成物を投与する工程;
を包含し、該抗体は、抗体依存性細胞媒介性細胞傷害を引き起こすのに十分な量で存在する、方法。
(項目54)
前記抗体が、配列番号111〜199からなる群より選択されるポリペプチド配列を含む、項目53に記載の方法。
(項目55)
前記抗体が、配列番号111、114、118、133、136、140、141、149、155、162、163、165、174、181、187、188、189、195および199からなる群より選択されるポリペプチド配列を含む、項目53に記載の方法。
(項目56)
前記抗体が、配列番号200−211からなる群より選択されるポリペプチド配列を含む、項目53に記載の方法。
(項目57)
前記抗体が、ヒト化抗体を含む、項目53に記載の方法。
(項目58)
前記抗体が、Fv、scFv、Fab’およびF(ab’)2を含む、項目53に記載の方法。
(項目59)
前記癌が、CLLである、項目53に記載の方法。
(項目60)
OX−2/CD200抗原を保持する細胞を標的とする第一の部分であって、配列番号111〜199からなる群より選択されるポリペプチド配列を含む第一の部分と;
細胞の死を促進する第二の部分と;
を含む、融合細胞。
(項目61)
前記第一の部分が、配列番号111、114、118、133、136、140、141、149、155、162、163、165、174、181、187、188、189、195および199からなる群より選択されるポリペプチド配列を含む、項目60に記載の融合分子。
(項目62)
前記第一の部分が、配列番号200〜211からなる群より選択されるポリペプチド配列を含む、項目61に記載の融合分子。
(項目63)
配列番号111〜199からなる群より選択されるポリペプチド配列を含む抗体と、薬学的に受容可能なキャリアとを含む、組成物。
(項目64)
前記抗体が、ヒト化抗体を含む、項目63に記載の組成物。
(項目65)
前記抗体が、Fv、scFv、Fab’およびF(ab’)2を含む、項目63に記載の組成物。
(項目66)
癌を処置するための方法であって、
癌に罹患している被験体に対して、CD200を発現する細胞に結合する抗体を含む治療組成物を投与する工程;
を包含し、該抗体は、1つ以上のTh1サイトカインの生成を促進するのに十分な量、かつ補体媒介性細胞傷害または抗体依存性細胞傷害を引き起こすのに十分な量で存在する、方法。
(項目67)
配列番号200〜211からなる群より選択されるポリペプチド配列を含む、CD200を発現する細胞に結合するキメラ抗体。
(項目68)
OX−2/CD200が被験体においてアップレギュレートされるかどうかを判定する工程;および
該被験体に対して、免疫反応を向上させる療法を投与する工程;
を包含する、方法。
(項目69)
前記療法が、OX−2/CD200に結合するかまたはOX−2/CD200レセプターに結合するポリペプチドを投与することを包含し、該ポリペプチドは、OX−2/CD200の免疫抑制効果を阻害するのに十分な量で投与される、項目68に記載の方法。
(項目70)
前記被験体が、癌患者である、項目68に記載の方法。
(項目71)
前記被験体が、CLL患者である、項目68に記載の方法。
(項目72)
前記ポリペプチドが、抗体を含む、項目69に記載の方法。
(項目73)
前記ポリペプチドが、ヒト化抗体を含む、項目69に記載の方法。
(項目74)
前記ポリペプチドが、Fv、scFv、Fab’およびF(ab’)2を含む、項目69に記載の方法。
(項目75)
前記OX−2/CD200が被験体においてアップレギュレートされるかどうかを判定する工程が、
癌患者から組織を取得すること;
CD200レベルが、対応する正常組織で見出されたレベルの少なくとも2倍であるかどうかを評価すること;
を包含する、項目68に記載の方法。
(項目76)
血液が、前記癌患者から取得される組織である、項目75に記載の方法。
(項目77)
組織が生検を行うことによって癌患者から取得される、項目75に記載の方法。
(項目78)
CD200レベルが、抗CD200抗体を癌細胞マーカーと組合せて使用するFACS分析を実施することによって評価される、項目75に記載の方法。
(項目79)
前記癌細胞マーカーが、CD38またはCD19である、項目75に記載の方法。
(項目80)
前記患者が、造血癌に罹患している、項目75に記載の方法。
(項目81)
前記患者が、CLLに罹患している、項目75に記載の方法。
(項目82)
前記療法が、OX−2/CD200に結合するかまたはOX−2/CD200レセプターに結合するポリペプチドを投与する前に既存の調節T細胞を排除することを包含し、該ポリペプチドは、OX−2/CD200免疫抑制効果を阻害するのに十分な量で投与される、項目69に記載の方法。
(項目83)
前記療法が、抗CD25抗体およびシクロホスファミドからなる群より選択される試薬の調節T細胞排除量を投与することを包含する、項目82に記載の方法。
(項目84)
前記療法が、骨髄切除療法を投与することをさらに包含する、項目69に記載の方法。
(項目85)
前記療法が、骨髄移植をさらに包含する、項目84に記載の方法。
(項目86)
前記療法が、CLL反応性T細胞の移動をさらに包含する、項目84に記載の方法。
(項目87)
前記療法が、癌ワクチンを投与することをさらに包含する、項目69に記載の方法。
(項目88)
前記癌ワクチンが、CLL細胞を負荷した樹状細胞;CLL細胞を負荷した樹状細胞に由来するタンパク質、ペプチドまたはRNA;患者由来熱ショックタンパク質タンパク質;腫瘍ペプチド;および腫瘍タンパク質;からなる群より選択される、項目87に記載の方法。
(項目89)
前記療法が、免疫賦活化合物の有効免疫刺激量を投与することをさらに包含する、項目69に記載の方法。
(項目90)
前記免疫賦活化合物が、CpG、toll様レセプターアゴニストおよび抗CTLA−4抗体からなる群より選択される、項目89に記載の方法。
(項目91)
前記療法が、抗PDL1抗体、抗PDL1抗体、抗IL−10抗体および抗IL−6抗体からなる群より選択される化合物の有効免疫抑制性遮断量を投与することをさらに包含する、項目69に記載の方法。
(項目92)
前記療法が、T細胞または抗原提示細胞の負の調節を遮断可能な因子の有効量を投与することを包含する、項目69に記載の方法。
(項目93)
前記因子が、抗CTLA4抗体、抗PD−L1抗体、抗PDL−2抗体および抗PD−1抗体からなる群より選択される、項目92に記載の方法。
(項目94)
前記療法が、T細胞の正の副刺激を増強可能な因子の有効量を投与することを包含する、項目69に記載の方法。
(項目95)
前記因子が、抗CD40抗体および抗4−1BB抗体からなる群より選択される、項目94に記載の方法。
(項目96)
前記療法が、癌ワクチンを投与することを包含する、項目69に記載の方法。
(項目97)
前記癌ワクチンが、CLL細胞を負荷した樹状細胞;CLL細胞を負荷した樹状細胞に由来するタンパク質、ペプチドまたはRNA;患者由来熱ショックタンパク質タンパク質;腫瘍ペプチド;および腫瘍タンパク質;からなる群より選択される、項目96に記載の方法。
(項目98)
前記療法が、先天免疫系のレセプターを活性化可能な因子の有効量を投与することを包含する、項目69に記載の方法。
(項目99)
前記因子が、CpG、Luivac、Biostim、Ribominyl、Imudon、Bronchovaxomからなる群より選択される、項目98に記載の方法。
(項目100)
前記療法が、サイトカインの有効な免疫増強量を投与することを包含する、項目69に記載の方法。
(項目101)
前記サイトカインが、IL−2、GM−CSFおよびIFN−γからなる群より選択される、項目100に記載の方法。
(項目102)
治療処置の進行をモニターするための方法であって、
被験体に対して、免疫調節療法を投与する工程;および
該療法の有効性を判定するために、被験体において少なくとも2倍のOX−2/CD200レベルを判定する工程;
を包含する、方法。
(項目103)
前記被験体におけるOX−2/CD200レベルが、前記免疫調節療法の投与前に判定され、次に、該被験体におけるOX−2/CD200レベルが、該療法の少なくとも1回の後に判定される、項目102に記載の方法。
(項目104)
前記免疫調節療法の投薬量または頻度を調整する工程をさらに包含する、項目102に記載の方法。
(項目105)
OX−2/CD200レベルが、OX−2/CD200と相関するマーカーを検出することによって判定される、項目102に記載の方法。
(項目106)
癌細胞によって発現される分子の免疫調節効果を評価するための方法であって、
被験体に対して、癌細胞によって自然に発現された分子、癌細胞およびリンパ球を投与する工程;および
該癌細胞の増殖の速度をモニターする工程;
を包含し、
該癌細胞およびドナーリンパ球のみが投与された場合の増殖速度と比較して、該癌細胞の増殖速度に何らかの変化が観察された場合に、該分子は免疫調節効果を有すると見なされる、方法。
(項目107)
前記癌細胞およびドナーリンパ球のみが投与された場合の増殖速度と比較して該癌細胞の増殖の速度の低下が観察される場合に、前記分子は、免疫増強性であると見なされる、項目106に記載の方法。
(項目108)
前記癌細胞およびドナーリンパ球のみが投与された場合の増殖速度と比較して該癌細胞のより高い増殖速度が観察される場合に、前記分子は、免疫抑制性であると見なされる、項目106に記載の方法。
(項目109)
前記癌細胞によって自然に発現される分子が、該化合物を自然に発現する細胞を注射することによって投与される、項目106に記載の方法。
(項目110)
前記癌細胞によって自然に発現される分子が、該分子を発現するように操作された細胞を注射することによって投与される、項目106に記載の方法。
(項目111)
前記癌細胞によって発現される分子をデータベースから同定する工程をさらに包含する、項目106に記載の方法。
(項目112)
前記癌細胞によって発現される分子を実験的に同定する工程をさらに包含する、項目106に記載の方法。
(項目113)
前記リンパ球が、ヒトである、項目106に記載の方法。
(項目114)
前記ヒトリンパ球が、末梢血リンパ球(PBL)、T細胞、B細胞、および樹状細胞からなる群より選択される、項目113に記載の方法。
(項目115)
前記リンパ球が、ヒト末梢血リンパ球(PBL)である、項目106に記載の方法。
(項目116)
前記投与されるリンパ球の数が、癌細胞の増殖速度を低下させるのには不十分である、項目106に記載の方法。
(項目117)
前記投与されるリンパ球の数が、投与される癌細胞の数よりも少ない、項目106に記載の方法。
(項目118)
前記投与されるリンパ球の数が、約100万細胞〜約200万細胞である、項目106に記載の方法。
(項目119)
前記投与されるリンパ球の数が、癌細胞の増殖速度を低下させるのに十分である、項目106に記載の方法。
(項目120)
前記投与されるリンパ球の数が、投与される癌細胞の数以上である、項目106に記載の方法。
(項目121)
前記投与されるリンパ球の数が、約500万細胞〜約1,000万細胞である、項目106に記載の方法。
(項目122)
前記癌細胞の投与が、RAJIおよびNamalwaからなる群より選択される少なくとも1つのリンパ腫細胞株を前記被験体に対して注射することを包含する、項目106に記載の方法。
(項目123)
前記被験体が、免疫不全マウスである、項目106に記載の方法。
(項目124)
前記投与されるリンパ球の数が、前記癌細胞の増殖を減速するのに十分であり、前記癌細胞により発現される分子は、前記癌細胞およびリンパ球のみが投与された場合の増殖速度と比較して観察される癌細胞の増殖速度が高い場合に、免疫抑制性であると見なされる、項目106に記載の方法。
(項目125)
前記投与されるリンパ球の数が、癌細胞の増殖を減速するのに不十分であり、前記癌細胞によって発現される分子は、前記癌細胞およびリンパ球のみが投与された場合の増殖速度と比較して観察される癌細胞の増殖速度が低い場合に、免疫増強性であると見なされる、項目106に記載の方法。
(項目126)
リンパ球および癌細胞のみを対照被験体に投与することによって、前記癌細胞の増殖速度を判定する工程;
をさらに包含する、項目106に記載の方法。
(項目127)
前記癌細胞によって発現される分子を投与する前に、リンパ球および癌細胞のみを前記被験体に対して投与することによって、前記癌細胞の増殖速度を判定する工程;
をさらに包含する、項目106に記載の方法。
(項目128)
化合物の免疫調節効果を評価するための方法であって、
被験体に対して、評価される化合物、癌細胞およびリンパ球を投与する工程;および
前記癌細胞の増殖の速度をモニターする工程;
を包含し、
該癌細胞およびドナーリンパ球のみが投与された場合の増殖速度と比較して、該癌細胞の増殖速度に何らかの変化が観察された場合に、該化合物は、免疫調節効果を有すると見なされる、方法。
(項目129)
前記癌細胞およびドナーリンパ球のみが投与された場合の増殖速度と比較して前記癌細胞の増殖の速度の低下が観察される場合に、前記化合物は、免疫増強性であると見なされる、項目128に記載の方法。
(項目130)
前記化合物の存在下での前記癌細胞の増殖速度が、前記癌細胞およびドナーリンパ球のみが投与される場合の増殖速度よりも高い場合に、該化合物は、免疫抑制性であると見なされる、項目128に記載の方法。
(項目131)
前記評価される化合物が、抗体である、項目128に記載の方法。
(項目132)
前記癌細胞の投与が、前記被験体に対して、RAJIおよびNamalwaからなる群より選択される少なくとも1つのリンパ腫細胞株を注射することを包含する、項目128に記載の方法。
(項目133)
前記投与される癌細胞が、免疫抑制分子を発現する、項目128に記載の方法。
(項目134)
前記投与される癌細胞が、CD200を発現する、項目128に記載の方法。
(項目135)
CD200の発現が、前記投与される癌細胞によってアップレギュレートされる、項目128に記載の方法。
(項目136)
前記リンパ球が、ヒトである、項目128に記載の方法。
(項目137)
前記ヒトリンパ球が、末梢血リンパ球(PBL)、T細胞、B細胞、および樹状細胞からなる群より選択される、項目136に記載の方法。
(項目138)
前記リンパ球が、ヒト末梢血リンパ球(PBL)である、項目128に記載の方法。
(項目139)
前記投与されるヒトPBLの数が、投与される癌細胞の数よりも少ない、項目138に記載の方法。
(項目140)
前記投与されるヒトPBLの数が、約100万細胞〜約200万細胞である、項目138に記載の方法。
(項目141)
前記投与されるヒトPBLの数が、投与される癌細胞の数以上である、項目138に記載の方法。
(項目142)
前記投与されるヒトPBLの数が、約500万細胞〜約1,000万細胞である、項目138に記載の方法。
(項目143)
前記被験体が、免疫不全マウスである、項目128に記載の方法。
(項目144)
前記投与されるリンパ球の数が、前記癌細胞のみと投与された場合に該癌細胞の増殖速度を減速するのに十分であり、前記化合物は、該化合物の存在下での該癌細胞の増殖速度が、前記癌細胞およびドナーリンパ球のみが投与された場合の増殖速度よりも大きい場合に、免疫抑制性であると見なされる、項目128に記載の方法。
(項目145)
前記投与されるリンパ球の数が、前記癌細胞のみと投与された場合に該癌細胞の増殖速度を減速するのには不十分であり、前記化合物は、前記癌細胞およびドナーリンパ球のみが投与された場合の増殖速度と比較して該癌細胞の増殖の速度の低下が観察される場合に、免疫増強性であると見なされる、項目128に記載の方法。
(項目146)
化合物の免疫調節効果を評価するための方法であって、
評価される化合物、癌細胞および約2×106個未満のリンパ球を、被験体に対して投与する工程;および
該癌細胞の増殖の速度をモニターする工程;
を包含し、
該癌細胞およびドナーリンパ球のみが投与された場合の増殖速度と比較して該癌細胞の増殖の速度の低下が観察される場合に、該化合物は、免疫増強性であると見なされる、方法。
(項目147)
前記投与される癌細胞が、免疫抑制分子を発現する、項目146に記載の方法。
(項目148)
前記投与される癌細胞が、CD200を発現する、項目146に記載の方法。
(項目149)
CD200の発現が、前記投与される癌細胞によってアップレギュレートされる、項目146に記載の方法。
(項目150)
化合物の免疫調節効果を評価するための方法であって、
評価される化合物、癌細胞および約5×106個のリンパ球を被験体に対して投与する工程;および
該癌細胞の増殖の速度をモニターする工程;
を包含し、
該化合物の存在下での該癌細胞の増殖の速度が、該癌細胞およびドナーリンパ球のみが投与される場合の増殖速度よりも高い場合に、該化合物は、免疫抑制性であると見なされる、方法。
(項目151)
前記投与される癌細胞が、免疫抑制分子を発現する、項目150に記載の方法。
(項目152)
前記投与される癌細胞が、CD200を発現する、項目150に記載の方法。
(項目153)
CD200の発現が、前記投与される癌細胞によってアップレギュレートされる、項目150に記載の方法。
【図面の簡単な説明】
【0025】
【図1】磁気活性化細胞分離(MACS)による抗体ライブラリーの細胞表面パニングに関与する代表的な工程を図式的に示す。
【図2】選択したscFvクローンの一次B−CLL細胞への結合および正常ヒトPBMCへの結合の非存在を示す、全細胞ELISAの結果を示すグラフである。この図および他の図の呼称2°+3°は、マウス抗HAによって染色され、抗マウス抗体のみを検出する負の対照ウェルを指す。この図および他の図の呼称であるRSC−Sライブラリーは、元のウサギscFv非パニングライブラリーから調製した溶解性抗体を指す。この図および他の図の呼称であるR3/RSC−Sプールは、3回目のパニングによるscFv抗体のプール全体から調製した溶解性抗体を指す。抗CD5抗体を正の対照として使用して、同数のB−CLLおよびPBMC細胞が各ウェルにて平板培養されることを検証した。
【図3a】選択したscFv抗体の一次B−CLL細胞および正常一次ヒトB細胞への結合を比較する、全細胞ELISAの結果を示す。抗CD19抗体を正の対照として使用して、同数のB−CLLおよび正常B細胞が各ウェルにて平板培養されることを検証した。他の対照は図2の凡例に示した通りであった。
【図3b】選択したscFv抗体の一次B−CLL細胞および正常一次ヒトB細胞への結合を比較する、全細胞ELISAの結果を示す。抗CD19抗体を正の対照として使用して、同数のB−CLLおよび正常B細胞が各ウェルにて平板培養されることを検証した。他の対照は図2の凡例に示した通りであった。
【図4a】scFvクローンが患者特異性(すなわちイディオタイプ)または血液型特異性(すなわちHLA)抗原に結合するかどうかを判定するために使用された全細胞ELISAの結果を示す。各クローンを3名の別個のB−CLL患者から単離したPBMCへの結合について試験した。1つの患者サンプルのみに結合したクローンを患者または血液型特異性と見なした。
【図4b】scFvクローンが患者特異性(すなわちイディオタイプ)または血液型特異性(すなわちHLA)抗原に結合するかどうかを判定するために使用された全細胞ELISAの結果を示す。各クローンを3名の別個のB−CLL患者から単離したPBMCへの結合について試験した。1つの患者サンプルのみに結合したクローンを患者または血液型特異性と見なした。
【図5a】scFvクローンの一次B−CLL細胞および3つのヒト白血病細胞株への結合を比較する全細胞ELISAの結果を示す。Ramosは、バーキットリンパ腫に由来する成熟B細胞株である。RLは、非ホジキンリンパ腫に由来する成熟B細胞株である。TF−1は、赤白血病に由来する赤芽球細胞株である。
【図5b】scFvクローンの一次B−CLL細胞および3つのヒト白血病細胞株への結合を比較する全細胞ELISAの結果を示す。Ramosは、バーキットリンパ腫に由来する成熟B細胞株である。RLは、非ホジキンリンパ腫に由来する成熟B細胞株である。TF−1は、赤白血病に由来する赤芽球細胞株である。
【図6a】scFvクローンの一次B−CLL細胞およびB−CLL患者に由来する細胞株であるCLL−AATへの結合を比較する全細胞ELISAの結果を示す。TF−1細胞は負の対照として含まれている。
【図6b】scFvクローンの一次B−CLL細胞およびB−CLL患者に由来する細胞株であるCLL−AATへの結合を比較する全細胞ELISAの結果を示す。TF−1細胞は負の対照として含まれている。
【図6c】scFvクローンの一次B−CLL細胞およびB−CLL患者に由来する細胞株であるCLL−AATへの結合を比較する全細胞ELISAの結果を示す。TF−1細胞は負の対照として含まれている。
【図7】3色フローサイトメトリーによって分析した、本開示によるscFv抗体の結合特異性を示す。正常な末梢血単核細胞において、scFv−9によって認識された抗原は、Bリンパ球で中程度に発現され、Tリンパ球のサブ個体群で弱く発現される。正常ドナーからのPBMCは、抗CD5−FITC、抗CD19−PerCP、およびscFv−9/抗HA−ビオチン/ストレプトアビジン−PEを使用して、3色フローサイトメトリーによって分析した。
【図8a】本開示によるscFv抗体によって認識された抗原の発現レベルを示す。scFv−3およびscFv−9によって認識された抗原は、CLL−AAT細胞株が由来する一次CLL腫瘍で過剰発現される。CLL−AAT細胞株を確立するために使用したCLL患者からの一次PBMCまたは正常ドナーからのPBMCをscFv抗体で染色して、フローサイトメトリーによって分析した。scFv−3およびscFv−9は、平均蛍光強度によって測定されたように、CLL細胞を正常PBMCよりも明るく染色する。
【図8b】本開示によるscFv抗体によって認識された抗原の発現レベルを示す。scFv−3およびscFv−9によって認識された抗原は、CLL−AAT細胞株が由来する一次CLL腫瘍で過剰発現される。CLL−AAT細胞株を確立するために使用したCLL患者からの一次PBMCまたは正常ドナーからのPBMCをscFv抗体で染色して、フローサイトメトリーによって分析した。scFv−3およびscFv−9は、平均蛍光強度によって測定されたように、CLL細胞を正常PBMCよりも明るく染色する。
【図8c】本開示によるscFv抗体によって認識された抗原の発現レベルを示す。scFv−3およびscFv−9によって認識された抗原は、CLL−AAT細胞株が由来する一次CLL腫瘍で過剰発現される。CLL−AAT細胞株を確立するために使用したCLL患者からの一次PBMCまたは正常ドナーからのPBMCをscFv抗体で染色して、フローサイトメトリーによって分析した。scFv−3およびscFv−9は、平均蛍光強度によって測定されたように、CLL細胞を正常PBMCよりも明るく染色する。
【図9a−1】選択したscFv抗体のCDR配列および結合特異性の概要を提供する。
【図9a−2】選択したscFv抗体のCDR配列および結合特異性の概要を提供する。
【図9B−1】選択したscFv抗体のCDR配列および結合特異性の概要を提供する。
【図9B−2】選択したscFv抗体のCDR配列および結合特異性の概要を提供する。
【図9C】選択したscFv抗体のCDR配列および結合特異性の概要を提供する。
【図10】図8a〜8cで説明した一次CLL細胞対正常PBMCでのscFv抗原の発現レベルを比較する、フローサイトメトリー結果の概要を示す表2である。
【図11】scFv−9抗原の発現レベルを、CLL患者から単離した末梢血単核細胞中のCD38+細胞のパーセンテージと比較する、フローサイトメトリー結果の概要を示す表である。
【図12】免疫沈降および質量分光分析によるscFv抗原の同定を示す。CLL−AAT細胞を0.5mg/mlスルホ−NHS−LC−ビオチン(Pierce)のPBSによる溶液、pH8.0によって30分間標識した。未反応ビオチンを除去するためのPBSによる徹底的な洗浄の後、細胞を窒素キャビテーションによって破壊して、ミクロソーム画分を分画遠心法によって単離した。ミクロソーム画分をNP40溶解緩衝液中に再懸濁させて、正常ウサギ血清およびタンパク質Aセファロースによって徹底的に前処理した。抗原をラット抗HAアガロースビーズ(Roche)に結合したHA−tagged scFv抗体によって免疫沈降させた。免疫沈降の後、抗原をSDS−PAGEによって分離して、ストレプトアビジン−アルカリホスファターゼ(AP)を使用したウェスタンブロットによって、またはCoomassie G−250染色によって検出した。CLL−AAT細胞に結合しない抗体であるscFv−7を負の対照として使用した。抗原バンドをCoomassie染色ゲルから切除して、質量分光分析(MS)によって同定した。The Scripps Research InstituteのProteomics Core Facility(ラホーヤ、カリフォルニア州)にてMALDI−MSを実施した。μLC/MS/MSはHarvard Microchemistry Facility(ケンブリッジ、マサチューセッツ州)にて実施した。
【図13】3つのscFv抗体がヒトOX−2/CD200 cDNAクローンによって一時的にトランスフェクションされた293−EBNA細胞に特異的に結合することを示す。OX−2/CD200 cDNAをRT−PCRによってCLL細胞からクローニングして、哺乳類発現ベクターpCEP4(Invitrogen)内に挿入する。PCEP4−CD200プラスミドまたは対応する空のベクターpCEP4は、Polyfect試薬(QIAGEN)を用いて、293−EBNA細胞内にトランスフェクションした。トランスフェクションの2日後、細胞を、scFv抗体への結合についてフローサイトメトリーによって分析した。
【図14】OX−2/CD200トランスフェクション細胞の存在が、IL−2およびIFN−γなどのTh1サイトカインのダウンレギュレーションを引き起こしたことを示す。抗OX−2/CD200抗体の30μg/mlでの添加は、Th1反応を完全に回復した。
【図15】混合リンパ球反応におけるCLL細胞の存在が、IL−2に対するTh1反応のダウンレギュレーションを引き起こしたことを示す。
【図16】混合リンパ球反応におけるCLL細胞の存在が、IFN−γに対するTh1反応のダウンレギュレーションを引き起こしたことを示す。
【図17A】ヒトPBL細胞の存在下または非存在下のどちらかでのRAJI細胞4×106個を皮下注射されたNOD/SCIDマウスのすべての群の腫瘍体積の平均+/−SDを示す。
【図17B】ヒトPBL細胞の存在下または非存在下のどちらかでのRAJI細胞4×106個を皮下注射されたNOD/SCIDマウスのすべての群の腫瘍体積の平均+/−SDを示す。
【図18】2つのパラメトリック検定(Studentのt検定およびWelchの検定)および1つのノンパラメトリック検定(Wilcox検定)を使用して実施した統計解析の結果を示す。
【図19A】ヤギ抗マウスIgG Fc抗体に捕獲されたCD200−Fcに対する3回目のパニングの後の、代表的なIgG1カッパクローンのELISAの結果を示す。
【図19B】ヤギ抗マウスIgG Fc抗体に捕獲されたCD200−Fcに対する3回目のパニングの後の、代表的なIgG2aカッパクローンのELISAの結果を示す。
【図19C】マイクロタイターウェルに直接コーティングしたCD200−Fcに対する3回目のパニングの後の、代表的なIgG1カッパクローンのELISAの結果を示す。
【図19D】マイクロタイターウェルに直接コーティングしたCD200−Fcに対する3回目のパニングの後の、代表的なIgG2aカッパクローンのELISAの結果を示す。
【図20A】ヤギ抗マウスIgG Fcに捕獲されたCD200−Fcで選択された代表的なIgG1クローンのフローサイトメトリーの結果を示す。
【図20B】ヤギ抗マウスIgG Fcに捕獲されたCD200−Fcで選択された代表的なIgG2aクローンのフローサイトメトリーの結果を示す。
【図20C】直接コーティングされたCD200−Fcで選択された代表的なIgG1クローンのフローサイトメトリーの結果を示す。
【図20D】直接コーティングされたCD200−Fcで選択された代表的なIgG2aクローンのフローサイトメトリーの結果を示す。
【図21a−1】CD200特異性クローンの重鎖相補性領域の推定アミノ酸配列を示す。
【図21a−2】CD200特異性クローンの重鎖相補性領域の推定アミノ酸配列を示す。
【図21b−1】CD200特異性クローンの重鎖相補性領域の推定アミノ酸配列を示す。
【図21b−2】CD200特異性クローンの重鎖相補性領域の推定アミノ酸配列を示す。
【図22】蛍光ビーズアッセイにおける、選択したクローンがCD200のそのレセプター(CD200R)との間の相互作用を遮断する能力を示す。
【図23】キメラ化のための選択されたCD200 Fabの推定アミノ酸配列を示す。
【図24】小規模一過性トランスフェクションの培養上澄みから得たキメラIgGのELISA結果を示す。
【図25】CD200に対して作られたすべての抗体がレセプターリガンド相互作用を非常によく遮断することを示す、精製IgGのビーズ阻害アッセイの結果を示す。
【図26A】CLL細胞の存在が混合リンパ球反応で観察されたIFN−γおよびIL−2生成の大半を完全に排除したが、抗体のいずれかの存在がこれらのTh1サイトカインの生成を許容したことを示す。
【図26B】CLL細胞の存在が混合リンパ球反応で観察されたIFN−γおよびIL−2生成の大半を完全に排除したが、抗体のいずれかの存在がこれらのTh1サイトカインの生成を許容したことを示す。
【図26C】抗体の存在下でIL−10生成がダウンレギュレートされたことを示す。
【図27】抗体依存性細胞媒介性細胞傷害性アッセイ(ADCC)での腫瘍細胞を発現するCD200を殺す能力を示す。マウスキメラCD200抗体すべてが、CD200陽性細胞と共に培養されたときに、同様の溶解レベルを生じた。
【図28】対応のない両側Student t検定によって解析され、腫瘍細胞のみを投与された群と比較された、各種のPBLドナーを使用する10回の実験の代表的な例を示す。有意差は500万または1000万のPBLを投与された群で観察されたが、第32日以降100万のPBLを投与された群では観察されなかった。
【図29】対応のない両側Student t検定によって解析され、腫瘍細胞のみを投与された群と比較された、各種のPBLドナーを使用する10回の実験の代表的な例を示す。有意差は両方のドナーで1000万のPBLを投与された群で観察されたが、第8日以降200万のPBLを投与された群では観察されなかった。
【図30a】レンチウイルスベクターから形質導入された腫瘍細胞を発現するCD200に感染したRAJI細胞が、その親RAJI細胞よりもいくらか低速に増殖するように見えたことを示す。形質導入細胞および親細胞との増殖差は、統計的有意性には達しなかった。
【図30b】PBL 5または10×106を注射したときに、PBLの存在が逆CD200(非機能性CD200)によって形質導入されたRAJI細胞における腫瘍増殖を最大84%減少させたことを示し、この特定のドナーがRAJI腫瘍細胞を非常に強く拒絶することを示唆している。
【図30c】腫瘍細胞でのCD200発現が免疫系に腫瘍増殖の減速を実際に防止させることを示唆する結果を示す。またこの試験は、免疫抑制性化合物または分子を評価するためのRAJI/PBLモデルの有用性を証明する。
【図31a】RAJI/PBLモデルで見られる効果が、他の腫瘍細胞モデルでも観察できるかどうかを示す。Namalwa腫瘍細胞が、形質導入細胞と親細胞との間での有意差なしに、迅速な腫瘍増殖を生じたことを示す。
【図31b】RAJI/PBLモデルで見られる効果が、他の腫瘍細胞モデルでも観察できるかどうかを示す。PBLの存在が腫瘍増殖を約50%減速させたことを示す。両側Student t検定の結果は、PBL処置群と、腫瘍細胞のみを投与された群との差が統計的に有意であることを示した。
【図31c】RAJI/PBLモデルで見られる効果が、他の腫瘍細胞モデルでも観察できるかどうかを示す。Namalwa細胞を発現するCD200およびPBLを投与された群の腫瘍増殖が、Namalwa細胞を投与された群の腫瘍増殖と同様であったことを示す。これらのデータにより、腫瘍細胞でのCD200発現がヒト免疫系による腫瘍増殖の減速を防止することが確認される。
【図31d】RAJI/PBLモデルで見られる効果が、他の腫瘍細胞モデルでも観察できるかどうかを示す。Namalwa細胞を発現するCD200およびPBLを投与された群の腫瘍増殖が、Namalwa細胞を投与された群の腫瘍増殖と同様であったことを示す。これらのデータにより、腫瘍細胞でのCD200発現がヒト免疫系による腫瘍増殖の減速を防止することが確認される。
【図32】RAJI PBLモデルが免疫増強化合物の有効性を評価する効率的な方法であることを証明する結果を示す。
【図33】腫瘍細胞におけるCD200発現が、免疫細胞による腫瘍増殖の抑制を防止したことを示す。
【図34】腫瘍細胞におけるCD200発現が、免疫細胞による腫瘍増殖の抑制を防止したことを示す。
【発明を実施するための形態】
【0026】
(好ましい実施形態の詳細な説明)
本開示により、OX−2/CD200が被験体においてアップレギュレートされるかどうかを判定して、そうならば被験体に免疫反応を増強する療法を投与するための方法を提供する。適切な免疫調節療法の説明例は、T細胞または抗原提示細胞の負の調節を遮断する因子(たとえば抗CTLA4抗体、抗PD−L1抗体、抗PDL−2抗体、抗PD−1抗体など)の投与、あるいはT細胞の正の副刺激を増強する因子(たとえば抗CD40抗体または抗4−1BB抗体)の投与、あるいNK細胞数またはT細胞活性を増加させる因子(たとえば単独の、あるいはIMiD、サリドマイド、またはサリドマイド類似物質などのインヒビタと組合された、抗CD200抗体)の投与を含む。さらに免疫調節療法は、癌ワクチン、たとえば腫瘍細胞、腫瘍RNAまたは腫瘍DNAを負荷(load)した樹状細胞、腫瘍タンパク質または腫瘍ペプチド、患者由来熱ショックタンパク質(hsp)あるいはCpG、Luivac、Biostim、Ribominyl、Imudon、Bronchovaxomなどの免疫系を各種のレベルで刺激する一般的なアジュバントあるいは先天免疫系のレセプター(たとえばtoll様レセプター)を活性化する他の任意の化合物でありうる。また免疫調節療法は、IL−2、GM−CSFおよびIFN−γなどのサイトカインによる処置を含むことができる。
【0027】
特に有用な実施形態において、免疫反応を増強する療法は、単独での、または上述した免疫調節療法の1つと組合せた、OX−2/CD200に結合するポリペプチドの投与である。一般に、本開示で利用するポリペプチドは、当業者に公知である各種の技法を使用して構成できる。1つの実施形態において、ポリペプチドは化学合成によって得られる。他の実施形態において、ポリペプチドは抗体であるか、あるいは1つ以上の抗体の1つの断片または複数の断片から構成される。
【0028】
好ましくは、本開示の方法で利用するポリペプチドは、CLL細胞株から得られる。「CLL」は本明細書で使用するように、これに限定されるわけではないが、B細胞CLL(「B−CLL」)を含むB細胞およびT細胞の、これに限定されるわけではないが、各種の発生ステージを含む、任意のリンパ球を伴う慢性リンパ球性白血病を指す。B−CLLは本明細書で使用するように、CD5+、CD23+、CD20dim+、sIgdim+であり、細胞周期のG0/G1にて停止する成熟B細胞表現型による白血病を指す。さらなる局面において、CLL細胞株は、癌およびCLLを含む、OX−2/CD200がアップレギュレートされる疾患状態の診断および/または処置で有用な、抗体を含むポリペプチドを産生するために使用される。
【0029】
本明細書で使用するように、「抗体」という用語は、選択した標的に結合できる完全抗体または抗体断片を指す。Fab、Fv、scFv、Fab’およびF(ab’)2、モノクローナルおよびポリクローナル抗体、操作された抗体(キメラ、CDRグラフトおよびヒト化、完全ヒト抗体、および人工的に選択された抗体を含む)、ならびにファージ提示または代わりの技法を使用して生成された合成または半合成抗体が含まれる。小型断片、たとえばFvおよびscFvは、その小さなサイズおよび結果として生じる優れた組織分布によって、診断および治療用途のために好都合な特性を所有する。
【0030】
抗体は、本明細書で免疫原として開示された細胞を使用することによって産生でき、それによりモノクローナル抗体を単離できる動物における免疫反応を上昇させる。そのような抗体の配列を決定でき、組換え技法によって抗体またはその変異体を生成できる。本局面において、「変異体」は、モノクローナル抗体の配列をベースとするキメラ、CDRグラフト、ヒト化および完全ヒト抗体はもちろんのこと、OX−2/CD200に結合できるポリペプチドも含む。
【0031】
さらに組換えライブラリー(「ファージ抗体」)に由来する抗体は、標的特異性に基づいて抗体またはポリペプチドを単離するためのベイトとして、本明細書で述べた細胞、またはそれに由来するポリペプチドを使用して選択できる。
【0032】
なおさらなる局面において、抗体またはポリペプチドは、一次CLL細胞、またはそれに由来する抗原を使用して抗体ライブラリーをパニングすることによって、そしてさらにCLL細胞株、たとえば本明細書で述べるCLL細胞株を使用してスクリーニングおよび/または特徴付けることによって産生できる。したがってCLLに対して特異性の抗体またはポリペプチドを特徴付ける方法が提供され、該方法は抗体またはポリペプチドのCLL細胞株への結合を評価する工程を含む。
【0033】
(細胞株の調製)
細胞株は、当業者に公知の確立された方法に従って生成できる。一般に細胞株は、患者に由来する一次細胞を、培養物中に不死化細胞が自発的に産生されるまで培養することによって生成される。これらの細胞を次に単離して、さらに培養してアポトーシスに対して耐性を示すクローン細胞個体群または細胞を生成する。
【0034】
たとえばCLL細胞は、CLLに罹患している患者から採取した末梢血から単離できる。細胞を洗浄して、場合により存在する細胞の種類を決定するために免疫分類できる。次に細胞を培地、たとえばIL−4を含有する培地中で培養できる。好都合には、培養プロセスの間に培地の全部または一部を1回以上交換する。それによって細胞株を単離でき、培養物中の増殖向上によって同定できるであろう。
【0035】
1つの実施形態において、EBVによる不死化によって確立されない悪性起源のCLL細胞株が提供される。「悪性起源」は、たとえばEBVによって形質転換される非増殖性細胞とは対照的に、悪性CLL一次細胞からの細胞株の誘導を指す。本開示による有用な細胞株は、表現型においてそれ自体悪性であっても、なくてもよい。「悪性」表現型を有するCLL細胞は、細胞増殖の反復サイクルを特徴とする、基材培地とは結合していない細胞増殖を示し、アポトーシスに対する耐性を示す。一次CLL細胞に由来した細胞株は、ATCCアクセション番号PTA−3920で寄託されている。好ましい実施形態において、細胞株はCLL−AATである。CLL−AATは、B−CLL一次細胞に由来するB−CLL細胞株である。
【0036】
1つの実施形態において、CLL細胞で特異的に発現されたタンパク質は、当業者に公知の方法、たとえば免疫沈降とそれに続く質量分光分析によって、CLL−AAT細胞株を利用して同定される。そのようなタンパク質は、CLL−AAT細胞株において、またはCLL患者に由来する一次細胞において特異的に発現できる。
【0037】
小型細胞ライブラリー(多くは市販されている)は、細胞の増殖特徴を調節できる因子を同定するために、細胞ベースアッセイでCLL−AAT細胞株を使用してスクリーニングできる。たとえばCLL−AAT細胞株においてアポトーシスを調節する、またはその増殖および/または増殖を阻害する因子を同定できる。そのような因子は、治療化合物の開発のための候補である。
【0038】
CLL−AAT細胞株から単離された核酸は、CLL特異性遺伝子を同定するサブトラクティブハイブリダイゼーション実験またはマイクロアレイ分析(たとえば遺伝子チップ実験)で使用できる。CLL細胞において調節される遺伝子を同定できる。この方法で同定されるポリペプチドまたは核酸遺伝子生成物は、CLLの抗体または低分子療法の開発のための手がかりとして有用である。
【0039】
1つの実施形態において、CLL−AAT細胞株は、細胞表面成分に結合して、次に細胞によってインターナライズされる、インターナライズする抗体を同定するために使用できる。そのような抗体は、治療用途の候補である。特に細胞質中で安定のままであり、細胞内結合活性を保持する単鎖抗体は、この方法でスクリーニングできる。
【0040】
(モノクローナル抗体の調製)
組換えDNA技術は、本開示によって生成された抗体を改良するために使用できる。それゆえキメラ抗体は、診断または治療用途においてその免疫原性を低下させるために構成できる。さらに免疫原性は、CDRグラフト、そして場合によりフレームワーク修飾によって抗体をヒト化することによって最小限にできる。その内容が参考として本明細書中に援用されるU.S.Patent No.5,225,539を参照。
【0041】
抗体は動物血清から得られるか、あるいはモノクローナル抗体またはその断片の場合は細胞培養で生成できる。組換えDNA技術を使用して、細菌または好ましくは哺乳類細胞培養物中で確立された手順に従って抗体を生成できる。選択した細胞培養系は好ましくは抗体生成物を分泌する。
【0042】
別の実施形態において、本明細書で開示した抗体の生成のためのプロセスは、ハイブリッドベクターによって形質転換された宿主、たとえばE.coliまたは哺乳類細胞を培養することを含む。該ベクターは、適正な読み枠内にて抗体タンパク質をコードする第2のDNA配列に連結されたシグナルペプチドをコードする第1のDNA配列に作動的に連結されたプロモータを含有する1つ以上の発現カセットを含む。該抗体タンパク質は次に収集および単離される。場合により、発現カセットは、適正な読み枠内でシグナルペプチドのそれぞれ個別に作動的に連結された抗体タンパク質をコードする多シストロン性、たとえば2シストロン性DNA配列に作動的に連結されたプロモータを含むことができる。
【0043】
ハイブリドーマ細胞または哺乳類細胞のインビトロでの増殖は、哺乳類血清(たとえばウシ胎仔血清)、または微量元素および増殖維持サプリメント(たとえばフィーダー細胞、たとえば正常マウス腹腔滲出細胞、脾臓細胞、骨髄マクロファージ、2−アミノエタノール、インスリン、トランスフェリン、低密度リポタンパク質、オレイン酸など)を場合により補充した、慣例の標準培地(たとえばダルベッコ変法イーグル培地(DMEM)またはRPMI1640培地)を含む適切な培地中で実施される。細菌細胞または酵母細胞である宿主細胞の増殖は、当分野で公知の適切な培地中で同様に実施される。たとえばバクテリアでは適切な培地は、培地LE、NZCYM、NZYM、NZM、Terrific Broth、SOB、SOC、2xYT、またはM9最小培地を含む。酵母では、適切な培地は、培地YPD、YEPD、最小培地、またはComplete Minimal Dropout Mediumを含む。
【0044】
インビトロ生成は、比較的純粋な抗体調製物を提供し、より多くの量の所望の抗体を与えるためのスケールアップを可能にする。細菌細胞、酵母、植物、または哺乳類細胞培養のための技法は当分野で公知であり、均質懸濁培養(たとえばエアリフトリアクタ内または連続スターラーリアクタ内で)、そして固定化またはエントラップ細胞培養(たとえば中空繊維、マイクロカプセル内、アガロースマイクロビーズまたはセラミックカートリッジ上で)を含む。
【0045】
大量の所望の抗体は、哺乳類細胞を生体内で増殖することによっても得られる。この目的のために、所望の抗体を生成するハイブリドーマ細胞を組織適合哺乳類に注射して、抗体生成腫瘍の増殖を引き起こす。場合により、該動物は炭化水素、特にプリスタン(テトラメチル−ペンタデカン)などの鉱油によって刺激できる。1〜3週間後に、抗体をこれらの哺乳類の体液から単離する。たとえば適切なミエローマ細胞とBalb/cマウスからの抗体生成脾臓細胞との、または所望の抗体を生成するハイブリドーマ細胞株Sp2/0に由来するトランスフェクション細胞との融合によって得たハイブリドーマ細胞を、場合によってプリスチンによって前処置したBalb/cマウスに腹腔内注射する。1〜2週間後に腹水を該動物から採取する。
【0046】
上述の、または他の技法は、たとえばその開示が参照により本明細書にすべて組み入れられているKohler and Milstein,(1975)Nature 256:495−497;U.S.Patent No.4,376,110;Harlow
and Lane,Antibodies:a Laboratory Manual,(1988)Cold Spring Harborで議論されている。組換え抗体分子の調製技法は、上の参考文献に、またたとえばWO97/08320;U.S.Patent No.5,427,908;U.S.Patent No.5,508,717;Smith,1985,Science,Vol.225,pp 1315−1317;Parmley and Smith,1988,Gene 73,pp 305−318;De La Cruz et al.,1988,Journal of Biological Chemistry,263 pp 4318−4322;U.S.Patent No.5,403,484;U.S.Patent No.5223409;WO88/06630;WO92/15679;U.S.Patent No.5780279;U.S.Patent No.5571698;U.S.Patent No.6040136;Davis et al.,1999,Cancer Metastasis Rev.,18(4):421−5;Taylor,et al.,Nucleic Acids Research 20(1992):6287−6295;Tomizuka et al.,Proc.Natl.Academy of Sciences USA 97(2)(2000):722−727にも述べられている。これらのすべての参考文献の内容は、参考として本明細書中に援用される。
【0047】
細胞培養物上澄みを所望の抗体について、CLL細胞の免疫蛍光染色によって、免疫ブロッティングによって、酵素イムノアッセイによって、たとえばサンドイッチアッセイまたはドットアッセイ、あるいはラジオイムノアッセイによって選択的にスクリーニングする。
【0048】
抗体の単離では、培養物上澄み中または腹水中の免疫グロブリンを、たとえば硫酸アンモニウムによる沈降、ポリエチレングリコールなどの吸湿材による透析、選択膜による濾過などによって濃縮できる。必要および/または所望ならば、慣習的なクロマトグラフィー法、たとえばゲル濾過、イオン交換クロマトグラフィー、DEAE−セルロースでのクロマトグラフィーおよび/または(免疫)アフィニティクロマトグラフィー、たとえば本開示によるCLL細胞株に由来する1つ以上の表面ポリペプチドによって、あるいはタンパク質−AまたはGを用いたアフィニティクロマトグラフィーによって精製される。
【0049】
別の実施形態は、適切な哺乳類、たとえばウサギがプールされたCLL患者サンプルによって免疫化されることを特徴とする、細胞株に対して作られた抗体を分泌する細菌細胞株の調製のためのプロセスを提供する。免疫化ウサギから生成されたファージ提示ライブラリーが当分野で公知の方法(たとえば参考として本明細書中に援用される、各種の参考文献で開示された方法)に従って、構成され、所望の抗体についてパニングされる。
【0050】
モノクローナル抗体を分泌するハイブリドーマ細胞も検討される。好ましいハイブリドーマ細胞は遺伝的に安定であり、所望の特異性の本明細書で述べたモノクローナル抗体を分泌して、解凍および再クローニングによって深温冷凍培養物から活性化できる。
【0051】
別の実施形態において、本明細書で述べるCLL細胞株に対して作られたモノクローナル抗体を分泌するハイブリドーマ細胞株の調製のためのプロセスが提供される。そのプロセスでは、適切な哺乳類、たとえばBalb/cマウスを、本開示で述べた細胞に由来する1つ以上のポリペプチドまたはその抗原断片、細胞株自体、あるいは上述したような精製ポリペプチドを含有する抗原キャリアによって免疫化する。免疫化哺乳類の抗体生成細胞を培養物中で短期間増殖させるか、適切なミエローマ細胞株の細胞と融合させる。融合で得られたハイブリッド細胞をクローニングして、所望の抗体を分泌する細胞クローンを選択する。たとえば本細胞株によって免疫化したBalb/cマウスの脾臓細胞をミエローマ細胞株PAIまたはミエローマ細胞株Sp2/0−Ag 14の細胞と融合させて、得られたハイブリッド細胞を所望の抗体の分泌についてスクリーニングして、陽性ハイブリドーマ細胞をクローニングする。
【0052】
好ましいのは、本開示に従って複数回、たとえば4〜6回、数ヶ月に渡って、たとえば2〜4ヶ月の間、細胞株106〜107細胞を皮下および/または腹腔内注射することによって、Balb/cマウスを免疫化することを特徴とする、ハイブリドーマ細胞株の調製のプロセスである。免疫化マウスからの脾臓細胞は、最後の注射から2〜4日後に採取して、融合プロモータ、好ましくはポリエチレングリコールの存在下でミエローマ細胞株PAIの細胞と融合させる。好ましくは、約30%〜約50%のポリエチレングリコール(分子量約4000)を含有する溶液中で、ミエローマ細胞を3〜20倍過剰の免疫化マウスからの脾臓細胞と融合させる。融合後、細胞を、選択培地、たとえばHAT培地を添加した上述したような適切な培地中にて、正常ミエローマ細胞が所望のハイブリドーマ細胞を過剰増殖させるのを防止するために一定間隔で膨張させる。
【0053】
さらなる実施形態において、上述した細胞株に対して作られた抗体の重鎖可変ドメインおよび/または軽鎖可変ドメインをコードするインサートを含む組換えDNAが生成される。DNAという用語は、コード単鎖DNA、前記コードDNAおよびその相補性DNAより成る2本鎖DNA、またはこれらの相補性(単鎖)DNA自体を含む。
【0054】
さらに本明細書で開示された細胞株に対して作られた抗体の重鎖可変ドメインおよび/または軽鎖可変ドメインをコードするDNAは、重鎖可変ドメインおよび/または軽鎖可変ドメインをコードする真正DNA配列を有する酵素または化学合成DNA、あるいはその変異体でありうる。真正DNAの変異体は、1つ以上のアミノ酸が除去または1つ以上の他のアミノ酸と交換された、上述の抗体の重鎖可変ドメインおよび/または軽鎖可変ドメインをコードするDNAである。好ましくは前記修飾は、ヒト化および発現最適化用途における抗体の重鎖可変ドメインおよび/または軽鎖可変ドメインのCDRの外側である。変異体DNAという用語は、1つ以上のヌクレオチドが同じアミノ酸をコードする新しいコドンを持つ他のヌクレオチドと交換される、サイレント変異体も含む。変異体配列という用語は、縮重配列も含む。縮重配列は、最初にコードされたアミノ酸配列の変化を生じることなく、制限数のヌクレオチドが他のヌクレオチドによって置換されるという点で、遺伝子コードの意味の範囲内で縮重される。そのような縮重配列は、重鎖マウス可変ドメインおよび/または軽鎖マウス可変ドメインの最適発現を得るために、特異性宿主、特にE.coliによって好まれる、その各種の制限部位および/または特定のコドンの頻度のために有用なことがある。
【0055】
変異体という用語は、当分野で周知の方法に従って真正DNAのインビトロ変異誘発によって得られたDNA変異体を含むものとする。
【0056】
完全テトラマー免疫グロブリン分子の構築およびキメラ抗体の発現では、重鎖および軽鎖可変ドメインをコードする組換えDNAインサートを重鎖および軽鎖定常ドメインをコードする対応するDNAと融合させて、たとえばハイブリッドベクター内への包含後に、適切な宿主細胞内へ移動させる。
【0057】
ヒト定常ドメインgに融合された本明細書で開示された細胞株に対して作られた抗体の重鎖マウス可変ドメインをコードするインサートを含む組換えDNA、たとえばγ1、γ2、γ3またはγ4、好ましくはγ1またはγ4も提供される。ヒト定常ドメインκまたはλ、好ましくはκに縮合された本明細書で開示された細胞株に対して作られた抗体の軽鎖マウス可変ドメインをコードするインサートを含む組換えDNAも提供される。
【0058】
別の実施形態は、重鎖可変ドメインおよび軽鎖可変ドメインが、宿主細胞における抗体の処理を促進するシグナル配列を場合により含むスペーサー基によって連結される組換えポリペプチドをコードする組換えDNAおよび/または抗体および/または開裂部位および/またはペプチドスペーサーおよび/またはエフェクター分子の精製を促進するペプチドをコードするDNAに関する。
【0059】
エフェクター分子をコードするDNAは、診断または治療用途で有用なエフェクター分子をコードするDNAであるものとする。それゆえ毒素または酵素、特にプロドラッグの活性化を触媒できる酵素であるエフェクター分子が特に示される。そのようなエフェクター分子をコードするDNAは、天然発生型酵素または毒素コードDNA、またはその変異体の配列を有し、当分野で周知の方法によって調製できる。
本抗体/ポリペプチドの使用
本明細書で利用するポリペプチドおよび/または抗体は、特に診断および治療用途のために示されている。
【0060】
本抗体は、治療剤として癌患者、特にこれに限定されるわけではないがCLL患者に投与できる。一部の実施形態において、抗体はCD200とそのレセプターとの間の相互作用を妨害できる。この妨害は、CD200の免疫抑制効果を遮断できる。この方法で免疫反応を改善することによって、そのような抗体は癌細胞の根絶を促進できる。
【0061】
抗CD200抗体は、他の免疫調節化合物、ワクチンまたは化学療法と組合せて投与することもできる。たとえば抗CD25またはシクロホスファミドなどの試薬による既存の調節T細胞の除去は、抗CD200処置を開始する前に、1つの特定の有用な実施形態で実現される。また骨髄切除療法と、それに続く骨髄移植またはCLL細胞と反応するT細胞の養子移入の治療有効性は、抗CD200療法によって向上する。さらに抗CD200処置は、癌ワクチン、たとえばCLL細胞を負荷(load)した樹状細胞、そのような細胞に由来するタンパク質、ペプチドまたはRNA、患者由来熱ショックタンパク質(hsp)、腫瘍ペプチドまたは腫瘍タンパク質の有効性を実質的に向上させることができる。他の実施形態において、抗CD200抗体は免疫賦活化合物、たとえばCpG、toll様レセプターアゴニストまたは他の任意のアジュバント、抗CTLA−4抗体などと組合せて使用される。なお他の実施形態において、抗CD200処置の有効性は、免疫抑制性機構、たとえば抗PDL1および/または2抗体、抗IL−10抗体、抗IL−6抗体などを遮断することによって改善される。なお他の実施形態において、抗CD200処置の有効性は、NK細胞数またはT細胞を増加させる因子、たとえば低分子インヒビタIMiD、サリドマイド、またはサリドマイド類似物質の投与によって改善される)。
【0062】
本開示による抗CD200抗体は、診断ツールとして使用できる。たとえば造血性癌の患者から採取した血液を使用して、CD200の発現は、CLL細胞上のたとえばCD38およびCD19などの適切な癌細胞マーカーと組合せて抗CD200抗体を使用するFACS分析によって、癌細胞上で評価できる。抗CD200抗体による処置には、正常B細胞で見出されるレベルよりも少なくとも1.4倍高いCD200レベルの患者を選択できる。
【0063】
本抗CD200抗体を診断ツールとして使用する別の例において、悪性腫瘍を持つ患者からの生検を採取して、抗CD200抗体を使用するFACS分析によってCD200の発現を判定する。腫瘍細胞がCD200を、対応する正常組織と比較して少なくとも1.4倍高いレベルで発現する場合、免疫調節療法のために癌患者を選択する。免疫調節療法は抗CD200療法でありうるが、患者の免疫系に影響を及ぼす他の任意の療法でもよい。適切な免疫調節療法の例は、T細胞または抗原提示細胞の負の調節を遮断する因子(たとえば抗CTLA4、抗PD−L1、抗PDL−2、抗PD−1)の投与またはT細胞の正の副刺激を増強する因子(たとえば抗CD40または抗4−1BB)の投与を含む。さらに免疫調節療法は、NK細胞数またはT細胞活性を増加させる因子(たとえば単独の、あるいはIMiD、サリドマイド、またはサリドマイド類似物質などのインヒビタと組合された、抗CD200抗体)の投与あるいは調節T細胞を消耗する因子(たとえば単独の、またはONTAKと組合せた抗CD200抗体)の投与でもよい。さらに免疫調節療法は、癌ワクチン、たとえば腫瘍細胞、腫瘍RNAまたは腫瘍DNAを負荷(load)した樹状細胞、腫瘍タンパク質または腫瘍ペプチド、患者由来熱ショックタンパク質(hsp)あるいはCpG、Luivac、Biostim、Ribominyl、Imudon、Bronchovaxomなどの免疫系を各種のレベルで刺激する一般的なアジュバントあるいは先天免疫系のレセプター(たとえばtoll様レセプター)を活性化する他の任意の化合物でありうる。または療法は、IL−2、GM−CSFおよびIFN−γなどのサイトカインによる処置を含む。
【0064】
本開示による別の実施形態において、治療処置の進行および/または有効性をモニターするための方法が提供される。該方法は、療法の有効性を決定するために、被験体におけるOX−2/CD200レベルを少なくとも2回決定する工程とを含む。たとえばOX−2/CD200の前処置レベルが確認でき、療法の少なくとも1回の投与の後に、OX−2/CD200のレベルを再度決定できる。OX−2/CD200レベルの低下は、有効な処置を示す。OX−2/CD200レベルの測定は、開業医によって療法の投薬量または頻度を増加するための指針として使用できる。OX−2/CD200レベルを直接モニターできることを、あるいはOX−2/CD200に相関する任意のマーカーをモニターできることをもちろん理解すべきである。
【0065】
本抗体はまた、生体内で癌性細胞を検出するために利用できる。これは抗体を標識することと、標識した抗体を被験体に投与することと、次に被験体を撮像することとによって実現される。本開示による診断撮像に有用な標識の例は、131I、111In、123I、99mTc、32P、125I、3H、14C、および188Rhなどの放射性標識、フルオレセインおよびローダミンなどの蛍光標識、核磁気共鳴活性標識、陽電子放出断層撮影(「PET」)スキャナによって検出できる陽電子放出同位体、ルシフェリンなどの化学発光剤、ならびにペルオキシダーゼまたはホスファターゼなどの酵素マーカーである。短距離検出器プローブ、たとえば経直腸プローブによって検出可能な短距離放射線エミッター、たとえば同位体も利用できる。抗体は、そのような試薬によって、当分野で公知の技法を使用して標識できる。たとえば抗体の放射性標識に関する技法については、参考として本明細書中に援用されるWensel and Meares,Radioimmunoimaging and Radioimmunotherapy,Elsevier,N.Y.(1983)を参照。参考として本明細書中に援用されるD.Colcher et al.,“Use of Monoclonal Antibodies
as Radiopharmaceuticals for the Localization of Human Carcinoma Xenografts in Athymic Mice”,Meth.Enzymol.121:802−816(1986)も参照。
【0066】
本開示による放射性標識抗体は、インビトロ診断試験に使用できる。抗体、その結合部分、プローブ、またはリガンドの特異性活性は、半減期、放射性標識の同位体純度、および生物因子に標識が包含される方法によって変わる。イムノアッセイ試験では、一般に特異性活性が高くなればなるほど、感度がより良好になる。放射性同位体によって抗体を標識するための手順は、当分野で一般に公知である。
【0067】
放射性標識抗体は患者に投与することができ、患者内で抗体が反応する抗原を持つ癌細胞に局在化して、たとえばγカメラまたは放出断層撮影を使用する放射核スキャンなどの公知の技法を用いて、生体内で検出または「撮像」される。たとえば参考として本明細書中に援用されるA.R.Bradwell et al.,“Developments
in Antibody Imaging”,Monoclonal Antibodies for Cancer Detection and Therapy,R.W.Baldwin et al.,(eds.),pp.65−85(Academic
Press 1985)を参照。あるいは、Brookhaven National
Laboratoryに配置されているPet VIと呼ばれるような陽電子放出横断断層撮影スキャナを放射性標識が陽電子(たとえば11C、18F、15O、および13N)を放出するところで使用できる。
【0068】
蛍光団および発色団標識生物因子は、当分野で周知の標準部分から調製できる。抗体および他のタンパク質は約310nmまでの波長を有する光を吸収するため、蛍光部分は、310nmを超える、好ましくは400nmを超える波長にて実質的な吸収を有するように選択すべきである。各種の適切な蛍光団および発色団は、参考として本明細書中に援用されるStryer,Science,162:526(1968)およびBrand,L.et al.,Annual Review of Biochemistry,41:843−868(1972)によって述べられている。抗体は、参考として本明細書中に援用されるU.S.Patent Nos.3,940,475、4,289,747、および4,376,110に開示された手順などの従来手順によって、蛍光発色団基によって標識できる。
【0069】
他の実施形態において、二重特異性抗体が検討される。二重特異性抗体は、少なくとも2つの異なる抗原に結合特異性を有するモノクローナル、好ましくはヒトまたはヒト化抗体である。この場合では、結合特異性の一方が癌細胞のCD200抗原に対してであり、他方が他の任意の抗原に対して、好ましくは細胞表面タンパク質またはレセプターまたはレセプターサブユニットに対してである。
【0070】
二重特異性抗体を作成するための方法は、当業者の範囲内である。伝統的には、二重特異性抗体の組換え生成は、2つの重鎖が異なる特異性を有する、2つの免疫グロブリン重鎖/軽鎖対の同時発現に基づいている(Milstein and Cuello,Nature,305:537−539(1983))。所望の結合特異性を持つ抗体可変ドメイン(抗体−抗原複合部位)を免疫グロブリン定常ドメイン配列に融合することができる。融合は好ましくは、ヒンジ、CH2、およびCH3領域の少なくとも一部を含む、免疫グロブリン重鎖定常ドメインによってである。免疫グロブリン重鎖融合、そして所望ならば免疫グロブリン軽鎖をコードするDNAは、独立した発現ベクターに挿入されて、適切な宿主生物内へ同時トランスフェクションされる。二重特異性抗体を産生する説明的な現在周知のさらなる詳細事項については、たとえばSuresh et al.,Methods in Enzymology,121:210(1986);WO 96/27011;Brennan et al.,Science 229:81(1985);Shalaby et al.,J.Exp.Med.175:217−225(1992);Kostelny et al.,J.Immunol.148(5):1547−1553(1992);Hollinger et al.,Proc.Natl.Acad.Sci.USA 90:6444−6448(1993);およびGruber et al.,J.Immunol.152:5368(1994);およびTutt et al.,J.Immunol.147:60(1991)を参照。
【0071】
本抗体は、癌性細胞を生体内で直接殺すために、または切除するためにも使用できる。これは、(場合により細胞傷害薬に融合できる)抗体を、そのような処置を必要とする被験体に投与することを含む。抗体は癌細胞のCD200を認識するため、抗体が結合するそのような任意の細胞が破壊される。
【0072】
癌細胞を殺すために、または切除するために抗体を単独で使用するとき、そのような致死または切除は、内因性宿主免疫機能、たとえば補体媒介性または抗体依存性細胞傷害を開始することによって実施できる。細胞をこの方法で殺すかどうかを判断するためのアッセイは、当業者の範囲内である。
【0073】
本開示の抗体は、各種の細胞傷害性化合物を送達するために使用できる。任意の細胞傷害性化合物を本抗体に融合できる。融合は化学的または遺伝子的に実現できる(たとえば発現を介して、単一の融合分子として)。細胞傷害性化合物は生物製剤、たとえばポリペプチド、または低分子でありうる。当業者が認識するように、低分子には化学融合が使用されるが、これに対して生物化合物では、化学または遺伝子融合のどちらかを使用できる。
【0074】
細胞傷害性化合物の非制限的な例は、治療薬は治療薬、放射線を放出する化合物、植物、真菌、または細菌起源の分子、生物タンパク質、およびその混合物を含む。細胞傷害性薬は、細胞内作用細胞傷害性薬、たとえば短距離、高エネルギーα−エミッターを含む、たとえば短距離放射線エミッターでありうる。酵素活性毒素およびその断片はたとえば、ジフテリア毒素A断片、ジフテリア毒素の非結合活性断片、(Pseudomonas aeruginosaからの)外毒素A、リシンA鎖、アブリンA鎖、モデクシンA鎖、アルファ−サクリン、あるAleurites fordiiタンパク質、あるDianthinタンパク質、Phytolacca americanaタンパク質(PAP、PAPIIおよびPAP−S)、Morodica charantiaインヒビタ、クルシン、クロチン、Saponaria officinalisインヒビタ、ゲロニン、マイトジリン、レストリクトシン、フェノマイシン、およびエノマイシンによって例示される。免疫毒素の酵素活性ポリペプチドを調製する手順は、参考として本明細書中に援用されるWO84/03508およびWO85/03508に述べられている。ある細胞傷害性部分は、たとえばアドリアマイシン、クロラムブシル、ダウノマイシン、メトトレキサート、ネオカルチノスタチン、および白金に由来する。
【0075】
抗体を細胞傷害性剤と結合する手順は上述されており、当業者の範囲内である。
【0076】
あるいは抗体は高エネルギー放射線エミッター、たとえば放射性同位体、たとえば131I、腫瘍部位に局在化するときに複数の細胞直径の致死を引き起こすγ−エミッターに結合できる。たとえば参考として本明細書中に援用される、S.E.Order,“Analysis,Results,and Future Prospective of
the Therapeutic Use of Radiolabeled Antibody in Cancer Therapy”,Monoclonal Antibodies for Cancer Detection and Therapy,R.W.Baldwin et al.(eds.),pp 303−316(Academic Press 1985)を参照。他の適切な放射性同位体は、α−エミッター、たとえば212Bi、213Bi、および211At、ならびにβ−エミッター、たとえば186Reおよび90Yを含む。
【0077】
本抗体(純抗体、標識抗体、毒素に融合された抗体などにかかわらず)の抗体投与の経路は、公知の方法、たとえば静脈内、腹腔内、脳内、筋肉内、皮下、眼内、動脈内、髄腔内、吸入または病巣内経路による注射または輸液、あるいは持続放出システムによる。該抗体は好ましくは、輸液によって、またはボーラス注射によって連続的に投与される。抗体を局所的または全身的方法で投与できる。
【0078】
本抗体は、薬学的に受容可能なキャリアとの混合物として調製できる。本出願の化合物の処方および投与のための技法は、“Remington’s Pharmaceutical Sciences,”Mack Publishing Co.,Easton,PA,最新版に見出すことができる。本治療組成物は静脈内に、あるいは鼻または肺を通じて、好ましくは液体または粉末エアゾール(凍結乾燥)として投与できる。該組成物は、所望通りに非経口的に、または皮下的にも投与できる。全身投与する場合、該治療組成物は、滅菌されており、発熱物質を含まず、pH、等張性、および安定性に十分に注意を払った非経口的に許容される溶液とすべきである。これらの条件は当業者に公知である。
【0079】
使用するのに適した製薬組成物は、本抗体の1つ以上がその本来の目的を達成するために有効な量で含有される組成物を含む。さらに詳細には、治療的有効量は、疾患の症状を防止、軽減または改善するために、あるいは処置される被験体の生存を延長するために有効な抗体の量を意味する。治療的有効量の決定は、特に本明細書で提供する詳細な開示に照らして、十分に当業者の能力の範囲内である。治療的有効投薬量は、インビトロおよび生体内方法を使用して決定できる。
【0080】
一部の実施形態において、本CD200結合抗体は、CD200によって白血病細胞を直接標的化することによって、CLLにおける免疫抑制を遮断する利益を提供する。詳細には、免疫系の刺激によって、脾臓およびリンパ節からのCLL細胞の根絶を可能にできる。出願人は、これらの微環境からのCLL細胞の根絶が、単にB細胞を標的とする因子(たとえばアレムツズマブ)によって成功したことを一切認識していない。これに対して、CLL反応性T細胞は、抗体よりもこれらの臓器へより良好なアクセスを有することができる。他の実施形態において、直接細胞致死は、CLL細胞に抗CD200Abをタグ付けすることによって実現される。
【0081】
特に有用な実施形態において、直接細胞致死およびTh1プロフィールへの免疫反応の推進の組合せは、癌処置への特に強力な手法を提供する。それゆえ1つの実施形態において、CD200に結合して、a)CD200とそのレセプターとの間の相互作用を遮断して、同時にb)CD200を発現する癌細胞を直接殺す抗体または抗体断片を癌患者に投与する癌処置が提供される。癌細胞が殺す機構は、これに限定されるわけではないが、抗体依存性細胞傷害(ADCC)または補体依存性細胞傷害(CDC);毒素との融合;放射性標識との融合;細胞致死に関与する生物因子、たとえばグランザイムBまたはパーフォリンとの融合;細胞傷害性ウイルスとの融合;TNF−αまたはIFN−αなどのサイトカインとの融合を含むことができる。代わりの実施形態において、癌処置は、a)CD200とそのレセプターとの間の相互作用を遮断して、同時にb)腫瘍に対する細胞傷害性T細胞またはNK細胞活性を増強する抗体を投与することを含む。細胞傷害性T細胞またはNK細胞活性のそのような増強はたとえば、たとえばIL−2、IL−12、IL−18、IL−13、およびIL−5などのサイトカインと融合させることにより組合せられる。加えてそのような増強は、IMiD、サリドマイド、またはサリドマイド類似物質などのインヒビタと組合せた抗CD200抗体の投与によって実現できる。
【0082】
なお別の実施形態において、癌処置は、a)CD200とそのレセプターとの間の相互作用を遮断して、同時にb)T細胞を腫瘍細胞に誘引する抗体を投与することを含む。T細胞誘引は、AbをMIG、IP−10、1−TAC、CCL21、CCL5またはLIGHTなどのケモカインと融合することによって実現できる。免疫抑制の遮断および腫瘍細胞の抗体標的化による直接致死の複合作用は、有効性の向上をもたらす独自の手法である。
【0083】
上の開示は抗体に関してであったが、一部の実施形態では、そのような抗体に由来するポリペプチドを本開示に従って利用できる。
【0084】
(CLL細胞株の使用)
CLL細胞株は、CLL、癌、およびOX−2/CD200のアップレギュレートされたレベルを特徴とする他の疾患状態、たとえばメラノーマの診断および処置を開発するための重要なツールを提供するため、その開発には多くの利点がある。
【0085】
本開示による細胞株は、CLLおよびOX−2/CD200のアップレギュレートされたレベルを特徴とする他の疾患状態に関する病因論、病原および生物学についてのインビトロ試験に使用できる。これはこれらの疾患の療法に有用である適切な因子の同定を補助する。
【0086】
細胞株は、上で言及したようなCLL、癌、およびOX−2/CD200のアップレギュレートされたレベルを特徴とする他の疾患状態(たとえばメラノーマ)のインビトロおよび生体内診断のための、そして他の方法によって、たとえば抗体ライブラリーをCLL患者に由来する一次細胞および/または抗原によってパニングすることによって生成された抗体のスクリーニングおよび/または特徴付けのための、ポリペプチドおよび/またはモノクローナル抗体を生成するためにも使用できる。
【0087】
細胞株はそのようなものとして使用できるか、またはそれから抗原を誘導できる。好都合なことに、そのような抗原は、CLLに対して特異性の細胞表面抗原である。それらは本開示による細胞株から直接単離できる。あるいは本明細書で述べた細胞株から作成したcDNA発現ライブラリーは、抗CLL抗体の選択および特徴付けならびに新規CLL特異性抗原の同定に有用である、CLL特異性抗原を発現するために使用できる。
【0088】
モノクローナル抗体療法を使用するCLLの処置は、当分野で提案されている。近年、Hainsworth(Oncologist 5(5)(2000)376−384)は、モノクローナル抗体に由来する最新の療法について述べている。リンパ球性白血病は特に、リンパ球腫瘍での複数のリンパ球特異性抗原の存在によるこの治療手法のための良好な候補であると考えられる。
【0089】
既存の抗体療法(たとえば、Bリンパ球の表現で発現される、CD20抗原に対して作られたリツキシマブTM)は、あるリンパ球性疾患に対してうまく使用されてきた。しかしながら低密度CD20抗原は、CLLにおけるBリンパ球の表面で発現される(Almasri et al.,Am.J.Hematol.,40(4)(1992)259−263)。
【0090】
それゆえ本明細書で述べるCLL細胞株は、本CLL細胞株の1つ以上の抗原決定基に対して特異性を有する新規の抗CLL抗体およびポリペプチドの開発、そしてCLL、癌、およびOX−2/CD200のアップレギュレートされたレベルを特徴とする他の疾患状態の療法および診断でのその使用を可能にする。
【0091】
抗体またはポリペプチドは、OX−2/CD200が通常相互作用するレセプターに結合して、それによりOX−2/CD200が該レセプターに結合するのを防止または阻害できる。なお別の代案として、抗体はOX−2/CD200の発現を調節する抗原に結合して、それによりOX−2/CD200の正常な、または上昇した発現を防止または阻害できる。OX−2/CD200の存在が免疫反応の低下と関連しているため、OX−2/CD200の代謝経路を妨害することが望ましいので、患者の免疫系は疾患状態、たとえば癌またはCLLから、さらに効果的に防御できる。
【0092】
特に有用な実施形態において、ポリペプチドはOX−2/CD200に結合する。1つの実施形態において、ポリペプチドは、OX−2/CD200に結合して、OX−2/CD200が他の分子またはレセプターと相互作用するのを防止または阻害する抗体でありうる。OX−2/CD200を過剰発現するCLL細胞および他の細胞がTh1サイトカインの生成を大幅に減少させるので、OX−2/CD200に結合する抗CD200抗体またはポリペプチドの、OX−2/CD200のアップレギュレートされたレベルを有する被験体への投与は、Th1サイトカインプロフィールを回復する。それゆえこれらのポリペプチドおよび/または抗体は、CLLおよび他の癌またはOX−2/CD200を過剰発現する疾患の処置における有用な治療剤でありうる。
【0093】
それゆえ別の実施形態において、本開示の方法は、被験体をOX−2/CD200の存在についてスクリーニングする工程と、OX−2/CD200に結合するポリペプチドを投与する工程とを含む。もちろん、OX−2/CD200の存在を直接モニターできること、あるいはOX−2/CD200と相関する任意のマーカーを検知できることを理解すべきである。特に有用な実施形態において、CLL患者をOX−2/CD200の過剰発現についてスクリーニングして、OX−2/CD200に結合する抗体を患者に投与する。1つのそのような抗体は、Serotec Inc.(3200 Atlantic Ave,Suite 105,Raleigh,NC 27604)から市販されている抗CD200抗体である。以下で詳細に述べるように、別のそのような抗体は、OX−2/CD200に結合するscFv−9である(図9B)。
【0094】
別の局面において、本開示は、癌細胞によって発現された分子の免疫調節効果を評価するための方法を提供する。これらの方法では、癌細胞によって発現またはアップレギュレートされる分子を最初に同定する。該分子は、データベースから、または実験的に同定できる。癌細胞によって発現またはアップレギュレートされる分子を同定するデータベースが公知であり、たとえばNCI60癌マイクロアレイプロジェクト(http://genome−www.stanford.edu/nci60/)(Ross et al.,Nature Genetics 24:227−34,2000)、癌腫分類(http://www.gnf.org/cancer/epican/)(Andrew
I.Su et al.,“Molecular Classification of Human Carcinomas by Use of Gene Expression Signatures.”Cancer Research 61:7388−7393,2001)、およびリンパ腫/白血病分子プロファイリングプロジェクト(http://llmpp.nih.gov/lymphoma/)(Alizadeh
et al.,Nature 403:503−11,2000)を含む。
【0095】
癌細胞によって発現またはアップレギュレートされる分子を同定するための実験方法も公知であり、たとえばマイクロアレイ実験、定量的PCR、FACS、およびノーザン分析を含む。
【0096】
癌細胞、リンパ球および癌細胞によって発現される以前に同定された分子を被験体に投与して、癌細胞の増殖の速度をモニターする。本方法では、任意の種類の癌細胞を利用できる。一部の実施形態において、癌細胞は免疫抑制性化合物を発現する。特に有用な実施形態において、癌細胞はCD200を発現、または過剰発現さえする。適切な癌細胞は、これに限定されるわけではないが、RAJIまたはNamalwa細胞株などのリンパ腫細胞株を含む。投与される癌細胞の量は、約1×106〜約20×106個の範囲である。
【0097】
本プロセスでは、任意の種類のリンパ球を利用できる。適切なリンパ球はたとえばPBL、T細胞、細胞傷害性T細胞、樹状細胞またはNK細胞を含む。特に有用な実施形態において、リンパ球はヒトリンパ球、詳細にはヒトPBLである。投与されるリンパ球の数は、a)癌細胞の増殖を減速するのに十分であるように、またはb)癌細胞の増殖を減速するのに不十分であるようにのいずれかで規定される。投与されるリンパ球の量は、投与されるリンパ球の数が癌細胞の増殖を減速するのに十分であるようにするときには、投与された癌細胞の数と同じか、それ以上でよい。実施形態において、投与されるリンパ球の数が癌細胞の増殖を減速するのに十分であるようにするときには、投与されるリンパ球の数は約5×106〜約10×106個でよい。投与されるリンパ球の量は、投与されるリンパ球の数が癌細胞の増殖を減速するのに不十分であるようにするときは、投与された癌細胞の数よりも少なくてよい。実施形態において、投与されるリンパ球の数が癌細胞の増殖を減速するのに不十分であるようにするときは、投与されるリンパ球の量は約1×106〜約4×106個の範囲でありうる。上述の量は例示的であり、他の適切な量を実験的に決定して本方法で使用できる。
【0098】
癌細胞によって発現またはアップレギュレートされる分子は、(単離されたか、または組換え産生されたかのいずれかの)分子またはその活性部分自体として、あるいは分子を自然に生成する細胞を投与することによって、あるいは分子またはその部分を生成するために操作された細胞を投与することによって、投与できる。所望の分子を発現させるために細胞を操作するための方法は、当業者に公知である。投与される分子の量は、健常者に見出される量を超える任意の量でよい。たとえば投与される分子の量は、健常者の同じ種類の細胞で見出される量の1.4倍超〜健常者の同じ種類の細胞で見出される量の約10,000倍超でよい。評価される分子がある程度の免疫調節活性を有することが既知であること、または既知でないことを理解すべきである。それゆえ本方法は、分子の免疫調節効果を確認するためにはもちろんのこと、最初からそのような活性を判定するためにも使用される。
【0099】
癌細胞、リンパ球および分子を投与する被験体として、任意の小動物を選択できる。被験体は好都合に免疫不全にすることができる。適切な小動物はたとえば免疫不全マウス、照射ラット、照射モルモットなどを含む。
【0100】
癌細胞の増殖速度は、旧来の技法を使用してモニターできる。たとえば腫瘍増殖は、長さおよび幅をカリパスで測定することによってモニターできる。腫瘍体積はたとえば、腫瘍の長さに腫瘍の幅を掛けて、次に腫瘍の幅の半分を掛けることに基づいて算出できる。癌細胞増殖の速度は、たとえば週に3回など、定期的に測定できる。癌細胞およびリンパ球のみが投与されたときの癌細胞の増殖速度は、癌細胞およびリンパ球のみを対照被験体に投与して、腫瘍サイズを定期的に測定することによって決定できる。あるいは癌細胞およびリンパ球のみを最初に投与することができ、いったんベースライン増殖速度が確立されたら、癌細胞によって発現またはアップレギュレートされる分子を次に被験体に投与でき、第2の投与後の癌細胞の増殖の速度を測定できる。あるいは全身モデルによって、癌細胞増殖の速度をFACS、生存または他の旧来の技法を使用してモニターできる。
【0101】
腫瘍細胞およびドナーリンパ球のみが投与されたときの増殖速度と比較して、癌細胞の増殖速度の何らかの変化が観察される場合、分子は免疫調節効果を有すると考えられる。投与されるリンパ球の数が癌細胞の増殖を減速させるのに十分であり、観察された腫瘍細胞の増殖速度が、腫瘍細胞およびドナーリンパ球のみが投与されたときの増殖速度と比較して高い場合、癌細胞によって発現またはアップレギュレートされる分子は免疫抑制性であると見なされる。投与されるリンパ球の数が癌細胞の増殖を減速するのに不十分であり、観察された腫瘍細胞の増殖速度が、腫瘍細胞およびドナーリンパ球のみが投与されたときの増殖速度と比較して低い場合、分子は免疫増強性であると見なされる。通例、観察された癌細胞の増殖速度は、腫瘍細胞およびドナーリンパ球のみが投与されたときの増殖速度よりも約20〜約1,000%高く、免疫抑制または免疫増強効果が統計的に有意となる。
【0102】
分子の免疫調節効果がいったん確立されると、本明細書に述べた実施形態によって、分子の活性の増強または阻害のどちらかを行う化合物を同定できる。免疫調節効果を有することが以前に見出された、分子の活性の増強または阻害のどちらかを行う化合物は、免疫調節効果を提供するタンパク質/タンパク質相互作用を変化させる任意の化合物でありうる。増強または阻害効果は、癌細胞によって発現またはアップレギュレートされる化合物との直接相互作用の結果でありうるか、あるいは癌細胞によって発現またはアップレギュレートされる化合物の代謝経路において他の化合物との間の相互作用の結果でありうる。
【0103】
たとえば、免疫調節効果を有することが以前に見出された分子、分子が相互作用するレセプター、または免疫調節効果に関与する代謝経路におけるある他の分子と相互作用する、抗体または機能性抗体断片を同定できる。抗体(抗体ライブラリーを含む)を作成するための、そしてそれらを阻害または増強効果についてスクリーニングするための技法は、当業者に明らかとなるであろう。別の例として、低分子を阻害または増強効果についてスクリーニングできる。低分子ライブラリーを阻害または増強効果についてスクリーニングする技法は、当業者に明らかとなるであろう。
【0104】
別の局面において、化合物の免疫調節効果を評価するための方法が本開示によって検討される。ヒト免疫系に作用する化合物または分子の免疫調節特性を証明することは、小動物モデルでは非常に困難である。しばしば該化合物は小動物の免疫系に作用せず、マウスにおけるヒト免疫系の再構成を必要とする。再構成は、各種の胎生免疫臓器をマウスにグラフトすることによって、またはヒトリンパ球の注射によって実現できるが、今日までに説明されたモデルのいずれも、化合物または分子の免疫調節特性の証明に有用であることが判明していない。免疫系は癌細胞の根絶に重要な役割を果たすと考えられている。癌細胞は、免疫抑制性レセプターのアップレギュレーションによって免疫系を回避するための方法を見出している。
【0105】
化合物の免疫調節効果を評価する本方法は、ドナーリンパ球(たとえばPBL)を輸液される白血病患者で観察されたグラフト対白血病効果を模倣するモデルによって実現され、患者の80%で寛解を引き起こす。該方法は、癌細胞、リンパ球および評価される化合物を被験体に投与することを含み、癌細胞の増殖の速度をモニターする。
【0106】
本方法では任意の種類の癌細胞を利用できる。一部の実施形態では、癌細胞は免疫抑制性化合物を発現する。特に有用な実施形態において、癌細胞はCD200を発現、または過剰発現さえする。適切な癌細胞は、これに限定されるわけではないが、RAJIまたはNamalwa細胞株などのリンパ腫細胞株を含む。投与される癌細胞の量は、約1×106〜約20×106個の範囲である。
【0107】
本プロセスでは、任意の種類のリンパ球を利用できる。適切なリンパ球はたとえば、PBL、樹状細胞、T細胞、細胞傷害性T細胞、NK細胞を含む。特に有用な実施形態において、リンパ球はヒトリンパ球、詳細にはヒトPBLである。投与されるリンパ球の数は、a)癌細胞の増殖を減速するのに十分であるように、またはb)癌細胞の増殖を減速するのに不十分であるように、のいずれかで規定される。投与されるリンパ球の量は、投与されるリンパ球の数が癌細胞の増殖を減速するのに十分であるようにするときには、投与された癌細胞の数と同じか、それ以上でよい。実施形態において、投与されるリンパ球の数が癌細胞の増殖を減速するのに十分であるようにするときには、投与されるリンパ球の数は約5×106〜約10×106個でよい。投与されるリンパ球の量は、投与されるリンパ球の数が癌細胞の増殖を減速するのに不十分であるようにするときは、投与された癌細胞の数よりも少なくてよい。実施形態において、投与されるリンパ球の数が癌細胞の増殖を減速するのに不十分であるようにするときは、投与されるリンパ球の量は約1×106〜約4×106個の範囲でありうる。上述の量は例示的であり、他の適切な量を実験的に決定して本方法で使用できる。
【0108】
評価される化合物は、免疫調節効果が決定されることを求めている任意の化合物でよい。化合物の説明例は、本明細書で上述した抗体およびペプチドを含む。投与される、評価される化合物の量は、約1mg/kg〜約200mg/kgの範囲でありうる。評価される化合物は、化合物自体として、あるいは化合物を自然に生成する細胞を投与することによって、あるいは化合物を生成するために操作された細胞を投与することによって、投与できる。評価される化合物がある程度の免疫調節活性を有することが既知であること、または既知でないことを理解すべきである。それゆえ本方法は、分子の免疫調節効果を確認するためにはもちろんのこと、最初からそのような活性を判定するためにも使用される。
【0109】
癌細胞、リンパ球および分子を投与する被験体として、任意の小動物を選択できる。被験体は好都合に免疫不全にすることができる。適切な小動物はたとえば免疫不全マウス、照射ラット、照射モルモットなどを含む。
【0110】
癌細胞の増殖速度は、旧来の技法を使用してモニターできる。たとえば腫瘍増殖は、長さおよび幅をカリパスで測定することによってモニターできる。腫瘍体積はたとえば、腫瘍の長さに腫瘍の幅を掛けて、次に腫瘍の幅の半分を掛けることに基づいて算出できる。癌細胞増殖の速度は、たとえば週に3回など、定期的に測定できる。癌細胞およびリンパ球のみが投与されたときの癌細胞の増殖速度は、癌細胞およびリンパ球のみを対照被験体に投与して、腫瘍サイズを定期的に測定することによって決定できる。あるいは癌細胞およびリンパ球のみを最初に投与することができ、いったんベースライン増殖速度が確立されたら、評価される化合物を次に被験体に投与でき、第2の投与後の癌細胞の増殖の速度を測定できる。
【0111】
リンパ腫細胞株RAJIまたはNamalwaなどの癌細胞を免疫不全マウスに注射するとき、500万〜1000万個のPBLの投与は、腫瘍増殖の著しい減速を引き起こす。これに対して、少ない数のPBL(ドナーによって1〜200万個)は、腫瘍増殖を減速しない。
【0112】
腫瘍細胞およびドナーリンパ球のみが投与されたときの増殖速度と比較して、癌細胞の増殖速度の何らかの変化が観察される場合、分子は免疫調節効果を有すると考えられる。投与されるリンパ球の数が癌細胞の増殖を減速させるのに十分であり、観察された腫瘍細胞の増殖速度が、腫瘍細胞およびドナーリンパ球のみが投与されたときの増殖速度と比較して高い場合、化合物は免疫抑制性であると見なされる。投与されるリンパ球の数が癌細胞の増殖を減速するのに不十分であり、観察された腫瘍細胞の増殖速度が、腫瘍細胞およびドナーリンパ球のみが投与されたときの増殖速度と比較して低い場合、化合物は免疫増強性であると見なされる。
【0113】
当業者が本明細書で述べた組成物および方法をより良好に実施できるようにするために、以下の実施例を例示目的で与える。
【実施例】
【0114】
(実施例1)
(細胞株CLL−AATの単離)
(細胞株の確立)
CLLと診断された患者から末梢血を採取した。WBCカウントは1.6×108/mlであった。単核細胞をHistopaque−1077密度勾配遠心分離(Sigma
Diagnostics,セントルイス、ミズーリ州)によって単離した。細胞を10%熱不活性化ウシ胎仔血清(FBS)を添加したイスコーブ変法ダルベッコ培地(IMDM)で2回洗浄して、氷冷IMDM/10% FBS 5ml中で再懸濁させた。トリパンブルーで染色することによって生細胞をカウントした。細胞を同じ体積の85% FBS/15% DMSOと混合して、貯蔵するために液体窒素中で1mlの分割量として凍結させた。
【0115】
免疫表現型検査は、CD45+リンパ球個体群の>90%がIgD、カッパ軽鎖、CD5、CD19、およびCD23を発現することを示した。この個体群は、低レベルのIgMおよびCD20も発現した。細胞の約50%が高レベルのCD38を発現した。細胞はラムダ軽鎖、CD10およびCD138では陰性であった。
【0116】
細胞の分割量を解凍、洗浄、そして20%熱不活性化FBS、2mM L−グルタミン、100単位/mlペニシリン、100μg/mlストレプトマイシン、50μM 2−メルカプトエタノール、および5ng/ml組換えヒトIL−4(R & D Systems、ミネアポリス、ミネソタ州)を添加したIMDM中で107個/mLの密度にて再懸濁させた。細胞を37℃にて加湿5% CO2雰囲気中で培養した。一定の増殖が観察されるまで、培地を4日ごとに一部交換した。5週間後、培養物中の細胞の数は、約4日ごとに倍増しはじめた。この細胞株をCLL−AATと名付けた。
【0117】
(細胞株の特徴付け)
フローサイトメトリーによる細胞株の免疫表現型検査は、IgM、カッパ軽鎖、CD23、CD38、およびCD138の高い発現、CD19およびCD20の中程度の発現、そしてIgDおよびCD5の弱い発現を示した。細胞株はラムダ軽鎖、CD4、CD8、およびCD10に対して陰性であった。
細胞株の免疫表現型検査も、全細胞ELISAによって、一次B−CLL細胞への特異性結合のために選択されたウサギscFv抗体のパネルを使用して実施した。これらのCLL特異性scFv抗体はすべて、CLL−AAT細胞株も認識した。これに対して、scFvの大多数は、B細胞リンパ腫に由来する2つの細胞株:バーキットリンパ腫細胞株のRamos、および非ホジキンリンパ腫細胞株のRLに結合しなかった。
【0118】
(実施例2)
(抗体ファージ提示および細胞表面パニングを使用する、B−CLL特異性細胞表面抗原に対するscFv抗体の選択)
(免疫化およびscFv抗体ライブラリー構成)
末梢血単核細胞(PBMC)をScripps Clinic(ラホーヤ、カリフォルニア州)にてCLL患者から採取した血液から単離した。ウサギ2匹をCLLの異なるドナー10名からプールしたPBMC 2×107個によって免疫化した。3回の免疫化、すなわち2回の皮下注射と、それに続く1回の静脈内注射を3週間の間隔で実施した。血清力価は、フローサイトメトリーを使用して血清IgGの一次CLL細胞への結合を測定することによって確認した。最後の免疫化の5日後に、該動物から脾臓、骨髄、およびPBMCを採取した。Tri−Reagent(Molecular Research Center,Inc)を使用して、全RNAをこれらの組織から単離した。単鎖Fv(scFv)抗体ファージ提示ライブラリーを前に述べたように構成した(Barbas et al.,(2001)Phage Display:A Laboratory Manual,Cold Spring Harbor Laboratory Press,コールドスプリングハーバー、ニューヨーク州)。細胞表面パニングでは、再増幅ライブラリーからのファージミド粒子をポリエチレングリコー(PEG)によって沈降させ、1%ウシ血清アルブミン(BSA)を含有するリン酸緩衝食塩水(PBS)で再懸濁させて、PBSで一晩透析した。
【0119】
(細胞表面パニングによる抗体選択)
ライブラリーは、Siegelら(1997,J.Immunol.Methods 206:73−85)によって述べられているように、磁気活性化細胞分離装置(MACS)を用いた正負の選択によりCLL細胞表面特異性抗体について濃縮した。簡潔には、scFv抗体ライブラリーからのファージミド粒子をMPBS(2%脱脂粉乳、PBS中0.02%アジ化ナトリウムPBS、pH7.4)中で1時間、25℃にてプレインキュベートして、非特異性結合部位を遮断した。一次CLL細胞約107個を常磁性マイクロビーズ(Miltenyi Biotec,サニーベール、カリフォルニア州)に結合したマウス抗CD5 IgGおよびマウス抗CD19 IgGで標識した。未結合マイクロビーズを洗浄によって除去した。標識CLL細胞(「標的細胞」)を過剰な「抗原−負のアブゾーバ細胞」と混合して、ペレット化して、ファージ粒子50μl(cfu 1010〜1011cfu)中へ再懸濁させた。アブゾーバ細胞は、細胞表面に非特異的に付着するファージはもちろんのこと、標的およびアブゾーバ細胞の両方に存在する「共通」抗原に対して特異性のファージも吸収するように作用する。使用したアブゾーバ細胞は、免疫磁気的負の選択(StemSep system,StemCell Technologies,バンクーバー、カナダ)によって末梢血から単離されたTF−1細胞(ヒト赤白血病細胞株)または正常ヒトB細胞のどちらかであった。アブゾーバ細胞の標的細胞に対する比は体積で約10倍であった。25℃での30分間のインキュベーションの後、細胞/ファージ混合物をMiniMACS MS+分離カラムに移した。カラムをMPBS 0.5mlで2回、そしてPBSで1回洗浄して、未結合ファージおよびアブゾーバ細胞を除去した。標的細胞をPBS 1ml中でカラムから溶出させて、最高速度の微量遠心機で15秒間に渡ってペレット化した。捕獲されたファージ粒子は、酸溶出緩衝液(グリシンによってpHを2.2に調整した0.1N HCl、および1mg/ml BSA)200μl中で標的細胞を再懸濁させることによって溶出させた。25℃での10分間のインキュベーションの後、緩衝液を2M Tris塩基、pH10.5 12μLによって中和して、溶出したファージを次回のパニングの間に、E.coli中で増幅した。各回のパニングでは、インプットおよびアウトプットファージ力価を決定した。インプット力価は、標的細胞/アブゾーバ細胞混合物に添加した再増幅ファージ粒子の数であり、アウトプット力価は、標的細胞から溶出した捕獲ファージの数である。濃縮因子(E)は、式E=(Rnアウトプット/Rnインプット)/(R1アウトプット/R1インプット)、式中、R1=1回およびRn=2、3、または4回を使用して計算する。大半の場合、102〜103倍の濃縮因子は、3回目または4回目に達成されるはずである。
【0120】
(パニング後の濃縮抗体プールの分析)
3〜5回のパニングの後、捕獲ファージのプールをCLL細胞への結合について、フローサイトメトリーおよび/または全細胞ELISAによってアッセイした:
1.HA−tagged溶解性抗体の形で全体のプールを生成するために、非サプレッサ系のE.coli(たとえばTOP10F’)2mlにファージミド粒子1μl(cfu 109〜1010)を感染させた。元のパニングされていないライブラリーを負の対照として使用した。カルベニシリンを最終濃度10μMまで添加して、培養物を37℃にて250rpmで振とうしながら1時間インキュベートした。50μg/mlカルベニシリンを含有するSB培地8mlを添加して、培養物を〜0.8のOD 600まで培養した。IPTGを最終濃度1mMまで添加して、LacプロモータからscFv発現を誘発して、37℃での振とうを4時間継続した。培養物を3000xgにて15分間遠心分離にかけた。溶解性抗体を含有する上澄みを濾過して、−20℃にて1mlの分割量で貯蔵した。
【0121】
2.scFv抗体プールの標的細胞対アブゾーバ細胞への結合を、フローサイトメトリーによって、高親和性ラット抗HA(クローン3F10、Roche Molecular Biochemicals)を二次抗体として、PE結合ロバ抗ラットを三次抗体として使用して決定した。
【0122】
3.抗体プールの標的細胞対アブゾーバ細胞への結合も、全細胞ELISAによって以下に述べるように決定した。
【0123】
(パニング後の個別scFvクローンのスクリーニング)
パニング後に個別scFvクローンをスクリーニングするために、TOP10F’細胞にファージプールを上述のように感染させて、カルベニシリンおよびテトラサイクリンを含有するLBプレートに散布して、37℃にて一晩インキュベートした。個別のコロニーを、ウェル当たりSB−カルベニシリン培地を0.6〜1.0ml含有する深96ウェルプレートへ接種した。培養物をHiGro振とうインキュベータ(GeneMachines,サンカルロス、カリフォルニア州)で520rpmおよび37℃にて6〜8時間培養した。この時点で、各ウェルから90μlの分割量をDMSO 10μlを含有する深96ウェルプレートに移した。この複製プレートを−80℃にて貯蔵した。元のプレートにIPTGを最終濃度1mMまで添加して、振とうを3時間継続した。プレートを3000xgにて15分間遠心分離にかけた。溶解性scFv抗体を含有する上澄みを別の深96ウェルプレートに移して、−20℃にて貯蔵した。
【0124】
HA−tagged scFv抗体をスクリーニングするための感受性全細胞ELISA方法を開発した:
1.ELISAプレートをコンカナバリンA(0.1M NaHCO3、pH8.6、0.1mM CaCl2中10mg/ml)でコーティングする。
【0125】
2.プレートをPBSで洗浄した後、PBS 50μl中の標的細胞またはアブゾーバ細胞0.5〜1×105個を各ウェルに添加して、プレートを250xgにて10分間遠心分離にかける。
【0126】
3.PBS中の0.02%グルタルアルデヒド50μlを添加して、細胞を40℃にて一晩固定する。
【0127】
4.PBSで洗浄した後、非特異性結合部位を、4%脱脂粉乳を含有するPBSによって室温にて3時間遮断する。
【0128】
5.細胞を溶解性、HA−tagged scFvまたはFab抗体(TOP10F’上澄み)50μlによって室温で2時間インキュベートして、次にPBSで6回洗浄する。
【0129】
6.結合した抗体を、マウス抗HA二次抗体(クローン12CA5)およびアルカリホスファターゼ(AP)結合抗マウスIgG三次抗体を使用して検出する。シグナルの約10倍の増幅は、AMDEX AP結合ヒツジ抗マウスIgGを三次抗体(Amersham Pharmacia Biotech)として使用することによって得られる。AMDEX抗体はデキストラン主鎖を介して複数のAP分子に結合される。アルカリホスファターゼ基質PNPPによって呈色され、マイクロプレートリーダーを使用して405nmにて測定する。
【0130】
scFvクローンの一次スクリーニングは、一次CLL細胞対正常ヒトPBMCに対するELISAによって行った。CLL細胞で陽性であり、正常PBMCで陰性であるクローンは、正常ヒトB細胞、ヒトB細胞株、TF−1細胞、およびCLL−AAT細胞株に対するELISAによって再スクリーニングした。クローンは別個の患者3名から単離したCLL細胞に対するELISAによっても再スクリーニングして、患者特異性または血液型特異性抗原を認識するクローンを除去した。代表的なELISAによる結果を図2〜6に示し、図9A〜9Cにまとめる。
【0131】
得られたユニークなscFv抗体クローンの数をDNAフィンガープリンティングおよび配列決定によって決定した。scFv DNAインサートをプラスミドからPCRによって増幅して、制限酵素BstNIによって消化した。得られた断片を4%アガロースゲル上で分離して、臭化エチジウムによって染色した。異なる制限断片パターンを有するクローンは、異なるアミノ酸配列を持つ必要がある。同一パターンを持つクローンはおそらく、同様または同一の配列を有する。ユニークなBstNIフィンガープリントを有するクローンは、DNA配列決定によってさらに分析した。25個の異なる配列が見出され、密接に関連する相補性決定領域を持つ16グループの抗体にクラスター化できた(図9A〜9C)。
【0132】
(scFv抗体のフローサイトメトリーによる特徴付け)
複数のscFv抗体の結合特異性を3色フローサイトメトリーによって分析した(図7)。正常ドナーから単離したPBMCをFITC結合抗CD5およびPerCP結合抗CD19によって染色した。scFv抗体による染色は、ビオチン結合抗HAを二次抗体として、そしてPE結合ストレプトアビジンを使用して行った。3つの抗体、すなわちscFv−2、scFv−3、およびscFv−6は、CD19+Bリンパ球個体群を特異的に認識することが見出された(データは示さず)。第4の抗体scFv−9は、2つの別個の細胞個体群:CD19+Bリンパ球およびCD5+Tリンパ球のサブセットを認識した(図7)。T細胞サブセットのさらなる特徴付けは、それがCD4+CD8−TH細胞のサブ個体群であることを示した(データは示さず)。
【0133】
scFv抗体によって認識された抗原が一次CLL細胞で過剰発現されるかどうかを判定するために、CLL患者5名および正常ドナー5名からのPMBCをscFvで染色して、フローサイトメトリーによって比較した(図8および表2)。陽性細胞個体群の平均蛍光強度を比較することによって、CLL細胞対正常細胞での抗原の相対発現レベルを決定できる。1つの抗体、すなわちscFv−2は正常PBMCよりも低い強度でCLL細胞を常に染色したのに対して、scFv−3およびscFv−6はどちらも、正常PBMCよりも鮮やかにCLL細胞を常に染色した。第4の抗体scFv−9は、CLLサンプル5つのうち2つを正常PBMCよりもはるかに強く染色したが、残り3つのCLLサンプルには中程度に鮮やかな染色のみを与えた(図8および表2)。これはscFv−3およびscFv−6の抗原が、すべてではないにしても大半のCLL腫瘍にて約1.4倍過剰発現されるのに対して、scFv−9がCLL腫瘍のサブセットにて3〜6倍過剰発現されることを示す。
【0134】
CLL患者は2つのほぼ等しい群に分割できる:不十分な予後の患者(中間生存期間8年)および好ましい予後の患者(中間生存期間26年)。CLLについて複数の好ましくない予後の指標が確認されており、最も顕著には体細胞変異のないVH遺伝子の存在および高いパーセンテージのCD38+B細胞の存在である。CLL患者のサブセットのみで過剰発現された抗原を認識するため、scFv−9抗原過剰発現がCLL患者10名からの血液サンプル中のパーセンテージと相関したかどうかを判定することが求められた(図11)。結果は、scFv−9抗原過剰発現およびCD38+細胞のパーセントが相互に完全に独立していることを示す。
【0135】
(scFv抗体によって認識された抗原の、免疫沈降(IP)および質量分光分析(MS)による同定)
これらの抗体に対する抗原を同定するために、scFvを使用して、細胞表面ビオチン化CLL−AAT細胞のミクロソーム画分から調製した溶解物から抗原を免疫沈降させた(図12)。免疫沈降された抗原をSDS−PAGEによって精製して、マトリックス支援レーザーイオン化質量分光分析(MALDI−MS)または毛細管逆相HPLCナノ電子スプレータンデム質量分光分析(μLC/MS/MS)によって同定した(データは示さず)。scFv−2は、110kd抗原をRLおよびCLL−AAT細胞の両方から免疫沈降させた(図12)。この抗原をMALDI−MSによってB細胞特異性マーカーCD19として同定した。scFv−3およびscFv−6はどちらも、45kd抗原をCLL−AAT細胞から免疫沈降させた(示さず)。この抗原はMALDI−MSによって、CLLおよび活性化B細胞の公知のマーカーであるCD23として同定された。scFv−9は、50kd抗原をCLL−AAT細胞から免疫沈降させた(図12)。この抗原はμLC/MS/MSによって、B細胞、活性化CD4+T細胞、および胸腺細胞の公知のマーカーであるOX−2/CD200として同定された。OX−2/CD200は、一部の非リンパ球細胞、たとえばニューロンおよび内皮細胞でも発現される。
【0136】
(実施例3)
OX−2/CD200を発現する細胞が、サイトカイン反応をTh1反応(IL−2、IFN−γ)からTh2反応(IL−4、IL−10)に変化させる能力を、ドナー1名からの単球由来マクロファージ/樹状細胞および別のドナーからの血液由来T細胞を用いた混合リンパ球反応にて評価した。OX−2/CD200発現細胞源として、以下で述べるOX−2/CD200トランスフェクションEBNA細胞またはCLL患者サンプルのどちらかを使用した。
【0137】
(293−EBNA細胞のトランスフェクション)
293−EBNA細胞(Invitrogen)を100mm皿に2.5×106個にて播種した。24時間後、Polyfect試薬(QIAGEN)をメーカーの説明書に従って使用して、細胞を一過性トランスフェクションした。細胞を、ベクターpCEP4(Invitrogen)中のOX−2/CD200 cDNA 7.2μgおよびpAdVAntageベクター(Promega)0.8μgと同時トランスフェクションした。負の対照として、空のpCEP4ベクターおよびpAdVAntageと同時トランスフェクションした。トランスフェクションの48時間後、scFv−9抗体を用いたフローサイトメトリーで決定されたように、細胞の約90%がOX−2/CD200をその表面で発現した。
【0138】
(血液単球からの樹状細胞/マクロファージの成熟)
軟膜をSan Diego Blood Bankから取得し、Ficollを使用して一次血液リンパ球(PBL)を単離した。2%ヒト血清を含有するイーグル最小必須培地(EMEM)中で細胞を1時間に渡って付着させ、続いてPBSによって激しく洗浄した。細胞をGM−CSF、IL−4およびIFN−γまたはM−CSFのいずれかの存在下で、3日後にリポポリサッカライド(LPS)を添加して、または添加せずに5日間培養した。成熟した細胞を収集して、γ−照射器(Shepherd Mark I Model 30照射器(Cs137))を使用して2000RADを照射した。
【0139】
(混合リンパ球反応)
混合リンパ球反応は、24ウェルプレートに樹状細胞/マクロファージ500,000およびレスポンダ細胞1×106個を使用して構成した。レスポンダ細胞は、Ficollを使用して末梢血から精製したT細胞濃縮リンパ球であった。T細胞は、細胞を組織培養フラスコ内で1時間インキュベートして、非付着性細胞画分を取ることによって濃縮した。OX−2/CD200トランスフェクションEBNA細胞またはCLL細胞500,000個を抗CD200抗体(完全IgGに変換されたscFv−9)30μg/mlの存在下または非存在下で、リンパ球添加の2〜4時間前にマクロファージ/樹状細胞に添加した。上澄みを48時間および68時間後に採取して、サイトカインの存在について分析した。
【0140】
(scFv−9の完全IgGへの変換)
scFv−9の軽鎖および重鎖V遺伝子をオーバーラップPCRによって、各遺伝子の可変領域をヒトラムダ軽鎖定常領域遺伝子、およびヒトIgG1重鎖定常領域CH1遺伝子とそれぞれ連結するプライマーを用いて増幅した。scFv−9の軽鎖遺伝子および重鎖遺伝子の可変領域は、特異性プライマーによって増幅して、ヒトラムダ軽鎖定常領域遺伝子およびIgG1重鎖定常領域CH1遺伝子は以下のような特異性プライマーによって個別に増幅した:
【0141】
【化1】
増幅生成物を精製して、オーバーラップPCRを実施した。
【0142】
最終生成物は、Xba I/Sac I(軽鎖)およびXho I/Pin AI(重鎖)によって消化して、ヒトFab発現ベクターのPAX243hGL内へクローニングした(その開示が参照によって本明細書に組み入れられている、公開されたInternational Application WO 2004/078937を参照)。DNAクローンをPCRエラーについてDNA配列決定によって分析した。hCMV IEプロモータ遺伝子をNot I/Xho I部位(重鎖の前)に挿入した。ベクターをXba I/Pin AI/EcoR I/Nhe Iにならびに軽鎖およびhCMV IEプロモータを含有する3472bp断片よって消化して、重鎖遺伝子をIgG1発現ベクターのXba I/Pin AI部位に移動した。
【0143】
(サイトカイン分析)
完全IgGに変換されたscFv−9の、混合リンパ球反応におけるサイトカインプロフィールに対する効果を判定した。
【0144】
組織培養物上澄み中に見出されたIL−2、IFN−γ、IL−4、IL−10およびIL−6などのサイトカインを、ELISAを使用して定量した。各サイトカインの一致した捕獲および検出抗体の対をR+D Systems(ミネアポリス、ミネソタ州)から取得して、組換えヒトサイトカインを使用して各サイトカインの標準曲線を作成した。抗サイトカイン捕獲抗体をPBS中のプレートに最適濃度にてコーティングした。一晩のインキュベーションの後、プレートを洗浄して、1% BSAおよび5%スクロースを含有するPBSによって1時間遮断した。0.05% Tweenを含有するPBSによる3回の洗浄の後、1% BSAを含有するPBS中に上澄みを2倍または10倍希釈として添加した。捕獲サイトカインを適切なビオチン化抗サイトカイン抗体によって検出して、アルカリホスファターゼ結合ストレプトアビジンおよびSigmaS基質の添加を続けた。呈色は、ELISAプレートリーダー(Molecular Devices)によって評価した。
【0145】
図14に示すように、OX−2/CD200トランスフェクションされたが、未トランスフェクションではない細胞の存在は、IL−2およびIFN−γなどのTh1サイトカインのダウンレギュレーションを生じた。30μg/mlでの抗CD200抗体の添加はTh1反応を完全に回復し、抗体がOX−2/CD200とそのレセプターとの間の相互作用を遮断したことを示す。
【0146】
図15および16に示すように、混合リンパ球反応におけるCLL細胞の存在は、Th1反応のダウンレギュレーションを生じた(図15はIL−2の結果を示す;図16はIFN−γの結果を示す)。このことは、OX−2/CD200を過剰発現する細胞(IB、EM、HS、BH)だけでなく、OX−2/CD200を過剰発現しなかったCLL細胞(JR、JGおよびGB)(これらの細胞の発現レベルを図11に示す)にも当てはまる。しかしながら抗CD200抗体はOX−2/CD200を過剰発現する細胞においてTh1反応を回復するだけであり、OX−2/CD200を発現する患者では、マクロファージでのOX−2/CD200のそのレセプターとの間の相互作用を排除することがTh1反応を回復するのに十分であることを示した。OX−2/CD200を過剰発現しなかった患者では、Th1反応のダウンレギュレーションに他の機構が関与しているように思われた。
抗CD200の腫瘍拒絶に対する効果を試験するための動物モデル
RAJIリンパ腫の腫瘍増殖がPBLの同時注射によって防止されるモデルを確立した。NOD/SCIDマウスにRAJI細胞4×106個、異なるドナーからのヒトPBL
1×106、5×106または10×106細胞の存在下または非存在下のどちらかで皮下注射した。腫瘍の長さおよび幅はもちろんのこと、体重も週に3回測定した。すべての群の腫瘍体積の平均+/−SDを図17AおよびBに示す。2つのパラメトリック検定(Student t検定およびWelchの検定)および1つのノンパラメトリック検定(Wilcox検定)を使用して、統計分析を実施した。統計分析の結果は図18に見出される。RAJI細胞は許容される変動の皮下腫瘍を形成する。拒絶は、特定のドナーおよびPBL細胞数に依存する。PBL 1×106個は腫瘍増殖を防止するには不十分であった。第22〜43日がPBL 5×106個のドナー2および第36日から開始するPBL 5×106または1×107個のドナー3は、腫瘍増殖を著しく低減した。ドナー4は、第48日以降はほとんど有意である。
【0147】
抗CD200の効果を試験するために、RAJI細胞にCD200を安定にトランスフェクションする。動物には、前の段落で述べたように注射する。CD200トランスフェクション細胞の存在下では、腫瘍はヒトPBLが存在しても増殖する。このモデルでの腫瘍拒絶を評価するために、抗CD200抗体を投与する。
【0148】
また液状腫瘍モデルを確立する。RAJI細胞をNOD/SCIDマウスに腹腔内注射する。細胞は骨髄、脾臓、リンパ節および他の臓器に分散して、麻痺を引き起こす。ヒトPBLの同時注射は、腫瘍増殖を防止または減速する。マウスを運動障害および麻痺の徴候について評価することによって、腫瘍増殖をモニターする。いったんこのような徴候が観察されたら、マウスを殺処分して、腫瘍細胞の数を骨髄、脾臓、リンパ節および血液を含む各種の臓器において、FACS分析およびPCRによって評価する。
【0149】
皮下モデルと同様に、CD200トランスフェクション細胞を腹腔内注射する。それらはヒトPBLが存在しても増殖する。抗CD200による処置は、腫瘍拒絶または腫瘍増殖の減速を生じる。
【0150】
(実施例4)
(ライブラリー構成)
マウスは、マウスIgG Fcに融合されたバキュロウイルス発現組換えCD200細胞外ドメイン(CD200−Fc)(Orbigen Inc.,サンディエゴ、カリフォルニア州)および全長CD200を含有するベクターを一過性トランスフェクションされた293−EBNA細胞によって交互に免疫化した。全RNAは、マウス脾臓からTRI試薬(Molecular Research Center,Inc.,シンシナティ、オハイオ州)をメーカーのプロトコルに従って使用して調製した。メッセンジャRNA(mRNA)は、Oligotex(QIAGEN Inc.,バレンシア、カリフォルニア州)をメーカーのマニュアルに従って使用して精製した。第1鎖cDNAはSuperScript II RTase(Invitrogen Life Technologies,カールスバッド、カリフォルニア州)をメーカーのプロトコルに従って使用して合成した。第1鎖cDNAを制限エンドヌクレアーゼによって消化して、第2鎖cDNAを、2003年3月27日に公開された、公開PCT application WO03/025202A2で十分に説明されている方法に従って合成した。第2鎖cDNAをPCR精製キット(QIAGEN)によって浄化して、シングルプライマー増幅を2003年3月27日に公開された、公開PCT application WO03/025202A2に述べられている方法に従って実施した。増幅生成物をプールして、PCR精製キットによって精製した。カッパ軽鎖をXba IおよびBspE Iで消化して、IgG1およびIgG2a重鎖をXho IおよびBin Iによって消化した。消化した断片はゲル抽出キット(QIAGEN)を用いてアガロースゲルから精製して、2004年9月16日に公開された公開PCT application WO/04078937A2で述べられているように、PAX313m/hGベクター内へクローニングした。
【0151】
(ライブラリーのパニング)
ライブラリー(IgG1カッパおよびIgG2aカッパ)は、マイクロタイター(Costar Group,ベテスダ、メリーランド州)に直接コーティングされた、またはヤギ抗マウスIgG Fc特異性抗体(Sigma−Aldrich Corp.,セントルイス、ミズーリ州)によって捕獲されたいずれかのCD200−Fcにパニングした。ライブラリーファージの調製では、エレクトロコンピテントXL1−Blue細胞(Stratagene、ラホーヤ、カリフォルニア州)をライブラリーDNAによって電気穿孔して、SOC培地中で1時間、そしてSB培地中で2時間、カルベニシリンを用いて培養した。ファージ生成は、VCS M13ヘルパーファージ(Amersham Biosciences Corp.,ピスカタウェイ、ニュージャージー州)および1mM
IPTGの添加により、30℃にて一晩に渡って誘発させた。培養物を遠沈させて、ファージを4%ポリエチレングリコールおよび3% NaClによって沈降させた。ファージを遠沈させて、関係のない抗原である、マウスIgG Fcに融合されたバキュロウイルス発現細胞外ドメイン(FLJ32028−Fc)であるFLJ32028(Orbigen,サンディエゴ)を溶解性競合物として含有する、1% BSA/PBSに再懸濁させた。直接コーティングしたCD200−Fcに対するパニングでは、4つのウェルをCD200−Fc(0.1M NaHCO3 pH8.6の5μg/ml)100μlによって4℃にて一晩コーティングした。ウェルをリン酸塩緩衝食塩水(PBS)、pH7.0で5回洗浄して、1%ウシ血清アルブミン(BSA)/PBSによって37℃にて1時間に渡って遮断した。ヤギ抗マウスIgG Fcに捕獲されたCD200−Fcに対するパニングでは、4つのマイクロタイターウェルをヤギ抗マウスIgG Fc 100μl(PBS中20μg/ml)によって4℃にて一晩コーティングした。ウェルをPBSによって5回洗浄して、CD200−Fc(PBS中の20μg/ml)100μlによって37℃にて1時間に渡ってインキュベートした。ウェルをPBSによって5回洗浄して、1% BSA/PBSによって37℃にて1時間に渡って遮断した。パニングの直接コーティングおよび捕獲方法では、ブロッカーは別のライブラリー(その開示全体が参考として本明細書中に援用される、2004年6月2日に提出されたPCT application serial No.PCT/US04/17118(未公開)の実施例3に述べたライブラリー)のFLJ32028に対するパニングから得られた溶解性Fabの混合物によって置換されて、マウスIgG Fcのエピトープをマスクして、ウェルを37℃にて30分間インキュベートした。これらのマスキングFabは、CD200−Fcにも結合することが示されている。ライブラリーファージをマスキングFabの上部に添加して、ウェルを37℃にて約1.5時間インキュベートした。未結合ファージをPBSによって厳密性を増強しながら洗浄して(1回目は3回、2回目は5回、3回目および4回目は10回)、各洗浄のたびに5分間のインキュベーションならびにピペットによる出し入れ5回を行った。結合ファージは、1mg/ml BSAを含む0.1M HCl
100μl、pH2.2によって2回溶出させ、2M Tris塩基、pH11.5で中和した。新しく培養したER2738細胞を溶出させたファージによって感染させて、カルベニシリンおよびグルコースを含有するLBアガロースプレート上へ滴定した。次回のパニングのために、残りのファージをVCS M13ヘルパーファージおよび1mM IPTGの添加によって30〜37℃にて一晩増殖させる。
【0152】
(ライブラリーのスクリーニング)
3および4回目の滴定プレートからの95コロニーを12.5μg/mlテトラサイクリンおよび50μg/mlカルベニシリンを含有するSB 1ml中で37℃にて約6時間培養した。VCS M13ヘルパーファージを添加して、培養物を37℃にて2時間インキュベートした。1mM IPTGおよび70μg/mlカナマイシンを添加して、Fab−ファージ生成を30℃にて一晩誘発した。マイクロタイターウェルをウサギ抗マウスIgG F(ab’)2(PBS中4μg/ml)50μl、CD200−Fc(0.1M NaHCO3中4μg/ml、pH8.6)、またはFLJ32028−Fc(0.1M NaHCO3中4μg/ml、pH8.6)によって4℃にて一晩コーティングした。ウェルをPBSによって3回洗浄して、1% BSA/PBS 100μlによって37℃にて1時間遮断した。培養物を遠沈させた。Fab−ファージを含有する培養物上澄みとブロッカーを交換して、ウェルを37℃にて1.5〜2時間インキュベートした。残りのFab−ファージは、フローサイトメトリーのために−80℃にて貯蔵した。プレートをPBSで3回洗浄して、アルカリホスファターゼ(AP)結合ヤギ抗マウスIgGF(ab’)2抗体50μl(Pierce)(1% BSA/PBS中1:500)によって37℃にて1時間に渡って検出した。プレートをPBSによって3回洗浄して、AP基質(Sigma−Aldrich)によってpNPP緩衝液中で展開した。3回目からのクローンのほぼすべてがすでに、CD200に対して特異的に陽性である(図19A〜D)。クローンを高スループットフローサイトメトリー分析によってもスクリーニングした。CD200(1×105細胞)を含む293細胞一過性トランスフェクション100マイクロリットルを96ウェルプレート(Costar)に分割した。Fab−ファージ50マイクロリットルを細胞に添加して、ピペット操作によって混合して、氷上で30分間インキュベートした。細胞を0.01% NaN3を含有する1% BSA/PBSによって2回洗浄した。細胞を0.01% NaN3を含有する1% BSA/PBS中のPE結合ヤギ抗マウスIgG抗体100μl(Sigma−Aldrich)中に再懸濁させて、氷上で30分間インキュベートした。細胞を0.01% NaN3を含有する1% BSA/PBSによって2回洗浄して、PBS中1%パラホルムアルデヒド200μlに再懸濁させた。CD200発現細胞への陽性結合を示す代表的なクローンを図20A〜Dに示す。
【0153】
DNA配列を分析して、重鎖の推定アミノ酸配列を相補性決定領域3(CDR3)に従ってグループ化した(図21A、B)。それらを17の群に分けた。
【0154】
(蛍光ビーズアッセイ)
さらなる分析のために23個のクローンを選択した。それらはcG2aR3B5、dG1R3A5、cG2aR3A2、dG2aR3B2、dG1R3A1、cG2aR3Al、cG2aR3B、dG1R3B、cG1R3A、cG1R3A、cG1R3A1、dG1R3B、dG1R3B、cG1R3C、dG2aR3C、dG2aR3A1、cG2aR3B、cG2aR3B、dG1R3B、cG2aR3B、cG2aR3C、dG1R3H、およびdG2aR3A6であった。選択したFabのDNAをSpe I/Nhe Iによって、遺伝子III除去および溶解性Fab発現および精製のために消化した。精製したFabは、CD200とそのレセプター(CD200R)との間の相互作用を遮断するその能力について蛍光ビーズアッセイで評価した。TransFluoSpheresカルボキシラート修飾ミクロスフェア(488/645)(Molecular Probes Invitrogen Detection Technologies,ユージーン、オレゴン州)にストレプトアビジンを、続いてビオチン標識抗ヒトFc抗体および暴露ウイルス生成CD200−Fcタンパク質ををコーティングした。293個の細胞にCD200Rを一過性トランスフェクションした。細胞表面発現をFACS分析によって確認した。CD200コーティングビーズ100万個を各種の量の抗CD200FabまたはキメラIgGによって10分間プレインキュベートしてから、CD200Rトランスフェクション細胞50,000個を添加した。37℃でのインキュベーションの30分後、1% BSAを含有するTris緩衝液によって細胞を洗浄して、FACS Caliburを使用して分析した。Fabのc1A10、c2aB7、およびd1A5は、Fab 6.7μg/mlにてCD200およびCD200Rの相互作用の最善の遮断を示した(図22)。これらのクローンは、図21Aおよび/またはBにおいてそれぞれcG1R3A10、cG2aR3B7およびdG1R3A5と呼ばれる。
【0155】
(キメラ化およびIgG変換)
キメラ化およびIgG変換のために6つの抗体を選択した(図23を参照)。それらはc1A10(cG1R3A10)、c2aA10(cG2aR3A10)、c2aB7(cG2aR3B7)、d1A5(dG1R3A5)、d1B5(dG1R3B5)、およびd1B10(dG1R3B10)であった。キメラ化のために、オーバーラップPCRを実施して、マウスカッパ鎖可変領域およびヒトカッパ鎖定常領域を連結した。マウス重鎖可変領域をクローニングのために、部分ヒトIgG1定常領域およびApa I部位を含有する3’プライマーによって増幅した。増幅されたカッパ鎖断片および重鎖断片を、カッパ軽鎖ではXba I/Not Iに、重鎖断片ではXho I/Apa IにヒトIgG1定常領域を含有するPAX243hGKベクター内へクローニングした(公開されたInternational Application WO 2004/078937を参照)。キメラFabのCD200への結合は、ELISAおよびフローサイトメトリーによって確認した。Not I/Xho Iでの重鎖発現、次に軽鎖および重鎖の、Xba I/Pin AI部位におけるヒトIgG1発現ベクター内への移動のために、これらのキメラFabは、ヒトサイトメガロウイルス前初期プロモータ(hCMV IE Pro)配列の挿入によって、IgGへ変換される。このベクターは、哺乳類細胞での軽鎖発現のために、追加のhCMV IE Pro配列上流Xba I部位を含む。DNA配列を確認して、哺乳類細胞トランスフェクションのためにHiSpeed Maxi prepカラム(QIAGEN)を使用してmaxi prep DNAを調製した。293−EBNA細胞での一過性トランスフェクションをEffectene(QIAGEN)をメーカーのプロトコルに従って使用し、pAdVAntageベクター(Promega US,マジソン、ウィスコンシン州)を添加して実施した。NS0細胞への安定な細胞株トランスフェクションは、Effecteneをメーカーのプロトコルに従って使用して実施した。小規模一過性トランスフェクションの後、各抗体の培養物上澄みをELISAによって試験した(図24)。大規模一過性トランスフェクションの後、各IgGを培養物上澄みから、FPLCを用いた抗ヒトIgG F(ab’)2アフィニティカラム(Amersham Biosciences)によって精製した。
【0156】
精製したIgGは、Fabについて述べたようにビーズ阻害アッセイで試験した。CD200に対して作られた抗体はすべて、以下の図25に示すように、レセプターリガンド相互作用を非常に良好に遮断した。
【0157】
(混合リンパ球反応)
CD200のそのレセプターとの間の相互作用の遮断も、CD200の存在下での混合リンパ球反応で観察されたTh1からTh2へのサイトカイン移動を防止するかどうかを評価した。San Diego Blood Bankから軟膜を取得して、Histopaque(Sigma− Aldrich)を使用して血液リンパ球(PBL)を単離した。2%ヒト血清を含有するEMEM中で細胞を1時間に渡って付着させ、続いてPBSによって激しく洗浄した。細胞をGM−CSF、IL−4およびIFN−γの存在下で5日間培養した。成熟した細胞を収集して、γ−照射器(University of California San Diego)を使用して2000RADを照射した。混合リンパ球反応は、24ウェルプレートに樹状細胞/マクロファージ500,000個およびレスポンダ細胞1×106個を使用して構成した。レスポンダ細胞は、Histopaqueを使用して末梢血から精製したT細胞濃縮リンパ球であった。T細胞は、細胞を組織培養フラスコ内で1時間インキュベートして、非付着性細胞画分を取ることによって濃縮した。CD200発現一次照射CLL細胞500000個を、各種の量の抗CD200抗体の存在下または非存在下で樹状細胞に、リンパ球添加の2〜4時間前に添加した。上澄みを48時間および68時間後に採取して、IL−2、IFN−γ、IL−4、IL−10およびIL−6などのサイトカインをELISAを使用して定量した。各サイトカインの一致した捕獲および検出抗体の対をR+D Systems(ミネアポリス、ミネソタ州)から取得して、組換えヒトサイトカインを使用して各サイトカインの標準曲線を作成した。抗サイトカイン捕獲抗体をPBS中のプレートに最適濃度にてコーティングした。一晩のインキュベーションの後、プレートを洗浄して、1% BSAおよび5%スクロースを含有するPBSによって1時間遮断した。0.05% Tweenを含有するPBSによる3回の洗浄の後、1% BSAを含有するPBS中の指示された希釈物に上澄みを添加した。捕獲サイトカインを適切なビオチン化抗サイトカイン抗体によって検出して、アルカリホスファターゼ結合ストレプトアビジンおよびSigmaS基質の添加を続けた。呈色は、ELISAプレートリーダー(Molecular Devices Corp.,サニーベール、カリフォルニア州)によって評価した。図26AおよびBに示すように、CLL細胞の存在は、混合リンパ球反応で観察されたIFN−γおよびIL−2生成の大半を完全に排除した。いずれかの抗体の存在は、Th1サイトカインの生成を許容した(図26AおよびB)。これに対して、IL−10の生成は、抗体の存在下でダウンレギュレートされた(図26Cを参照)。
【0158】
(抗体依存性細胞媒介性細胞傷害性アッセイ)
さらに6つのキメラマウス抗CD200抗体を、抗体依存性細胞媒介性細胞傷害性アッセイ(ADCC)でCD200発現腫瘍細胞を殺すその能力について評価した。CD200をトランスフェクションされた293−EBNA細胞に培地0.5ml中、37℃にて、100μCi/100万細胞で標識した。3回の洗浄後、細胞をカウントして、培地(10% ヒトAB血清を添加したRPMI)中で20万/mlにて再懸濁させて、50μl(10,000細胞/ウェル)を3つずつ、96ウェル丸底プレートに分配した。最終濃度20μg/mlを達成するために、抗CD200抗体20μlを各ウェルに分配した。末梢血単核細胞(エフェクター細胞)をFicoll勾配で単離して、赤血球を塩化アンモニウムによって溶解させ、洗浄して、培地に再懸濁させて、細胞50μlを各ウェルに分配した。アッセイプレートを回転させて(1,500rpm/5分/弱ブレーキ)、細胞培養インキュベータに移した。4時間後、アッセイプレートを前と同様に回転させた。上澄み36μlをピコプレートに移して、microscint−20カクテル250μlと混合して、オービタルシェーカーに2分間置いて、Top countで読み取った。図27に示すように、CD200陽性細胞と培養したときに、マウスキメラCD200抗体すべてが同様の溶解レベルを生じた。CD200陰性細胞では溶解は観察されなかった。加えて溶解の程度は、アイソタイプ対照抗体d2A6(抗FLJ32028抗体)と比較したときに、統計的に有意であった(p<0.05)。
【0159】
(実施例5)
(Raji/PBLモデル)
NOD.CB17−Prkdc<scid>マウス(Jackson Laboratory)に、RAJI細胞4×106個(ATCC)を含有するRPMI 200μlをPBL 0、100万、500万または1000万個と共に皮下注射した。各群にマウス9または10匹を入れた。PBLを全塩化アンモニウム250mlから単離した。腫瘍増殖は、カリパスで長さおよび幅を測定することによって、週3回モニターした。腫瘍増殖は、長さx幅x幅/2に基づいて計算した。
【0160】
腫瘍細胞のみを与えた群と比較したPBLを注射した群間の相違を、対応のない両側Student t検定によって分析した。有意差は、PBL 500万または1000万個を与えた群では見られたが、第32日からPBL 100万個を与えた群では見られなかった。図28に示したデータは、各種のPBLドナーを使用した実験10回の代表的な例である。
【0161】
(Namalwa PBLモデル)
NOD.CB17−Prkdc<scid>マウス(Jackson Laboratory、バーハーバー、メイン州)に、Namalwa細胞4×106個(ATCC)を含有するRPMI 200μlを、PBL 0、200万または1000万個と共に皮下注射した。各群にマウス9または10匹を入れた。PBLを全血250mlからhistopaque勾配で単離して、0.9%塩化アンモニウムを使用して赤血球溶解を続けた。腫瘍増殖は、カリパスで長さおよび幅を測定することによって、週3回モニターした。腫瘍増殖は、長さx幅x幅/2に基づいて計算した。
【0162】
図29は、対応のない両側Student t検定によって分析した、腫瘍細胞のみを与えた群と比較したPBLを注射した群間の相違を示す。有意差は、両方のドナーでPBL 1000万個を与えた群で見られたが、第8日からPBL 200万個を与えた群では見られなかった。
【0163】
(安定なCD200発現細胞株の生成)
安定なCD200発現RajiおよびNamalwa細胞株は、Virapower Lentiviral Expression System(Invitrogen,カールスバッド、カリフォルニア州)を使用して産生した。CD200 cDNAを一次CLL細胞からRT−PCRによって、フォワードプライマー5’−GACAAGCTTGCAAGGATGGAGAGGCTGGTGA−3’(配列番号212)およびリバースプライマー5’−GACGGATCCGCCCCTTTTCCTCCTGCTTTTCTC−3’(配列番号213)を使用して単離した。PCR生成物をGatewayエントリベクターpCR8/GW/TOPO−TAへクローニングして、個々のクローンを配列決定した。正しい配列のクローンをセンスおよびアンチセンス方向の両方でレンチウイルスベクターpLenti6/V5/DESTおよびpLenti6/UbC/V5/DEST内へ、Gateway技術(Invitrogen,カールスバッド、カリフォルニア州)を用いて組込んだ。この2つのベクターの主な相違は、CD200発現を引き起こすために使用されたプロモータである:pLenti6/V5/DESTがヒトCMV前初期プロモータを含有するのに対して、pLenti6/UbC/V5/DESTはヒトユビキチンCプロモータを含有する。
【0164】
高力価VSV−Gシュードタイプレンチウイルスストックを293−FT細胞の一過性同時トランスフェクションによって、製造者が推奨するように生成した。RajiまたはNamalwa細胞は、12μg/mlポリブレンを含有する増殖培地1ml中に細胞106個を再懸濁させて、レンチウイルスストック1mlを添加することによって形質導入した。37℃にて一晩インキュベートした後、ウイルスを含有する培地を除去して、新しい培地4mlと交換した。2日後、感染した細胞をCD200発現についてフローサイトメトリーによって分析した。すべての実験で、細胞の≧70%がCD200+であったのに対して、親細胞株および負の対照(アンチセンスCD200)ウイルスを形質導入された細胞ではCD200は検出できなかった。
【0165】
CD200を過剰発現するクローン細胞株を単離するために、感染した細胞をブラストサイジンによって13日間に渡って選択した。使用したブラストサイジンの濃度はRaji細胞では6μg/ml、Namalwa細胞では2μg/mlであった。次に安定なクローンを、ブラストサイジン耐性細胞の希釈を制限することによって96ウェルプレート内へ単離した。クローンは、96ウェル形式でフローサイトメトリーによって、PE結合マウス抗ヒトCD200(クローンMRC OX104、Serotec)および高スループットサンプラーを装備したBD FACSCaliburを使用してスクリーニングした。2000 Rajiおよび2000 Namalwaクローン全体をスクリーニングした後、CD200発現が最高であるこれらのクローンをさらなる特徴付けの後に旧来の技法を用いて膨張させた。
【0166】
(RAJI/PBLモデルにおけるCD200の免疫抑制効果)
開示した方法に従って、CD200をCLL細胞でアップレギュレートされることを証明した。この分子のアップレギュレーションは、潜在的に免疫抑制性でありうる。CD200を発現する癌細胞が免疫系に癌細胞を根絶させるかどうかを試験するために、通常はCD200を発現しないRAJI細胞に、CD200をコードするレンチウイルスベクター系を上述のように感染させた。CD200を安定して発現するRAJIクローンを選択した。ベクター感染の効果を確実なくすための対照として、CD200の逆転非機能形(CD200rev)を発現するクローンも選択した。CD200を発現するRAJI細胞、CD200REVまたは親RAJI細胞をNOD.CB17−Prkdc<scid>マウスに皮下注射した。試験には以下の群を含めた:
群1:RAJI 4×106個、皮下;マウス9匹
この群は、レンチウイルス形質導入細胞が親細胞と同様の増殖を示すようにする必要があった。
【0167】
群2:RAJICD200 4×106個、皮下;マウス9匹
この群は、CD200形質導入細胞が親細胞と同様の増殖を示すようにする必要があった。またこの群は、最大の腫瘍増殖を与えるであろう。群3および群4をこの群と比較した。
【0168】
群3:RAJICD200 4×106個+PBL 5×106個、皮下;マウス9匹
このPBL数は、以前の実験で一部のマウスの腫瘍増殖を低減することが示されている。拒絶は、細胞1000万個ほど強力ではないが、CD200がある数の細胞のみに影響を及ぼしうるかどうか判定するためであり、5×106個は、拒絶を得るために使用できるPBLの最小量である;拒絶は、CD200の存在によって防止されるはずである。
【0169】
群4:RAJICD200 4×106個+PBL 10×106個、皮下;マウス8匹
これはRAJI/PBLモデルで拒絶を確認するための、PBLの最適数である。目的は、CD200発現がこの拒絶を防止するということである。
【0170】
群5:RAJICD200rev 4×106個、皮下;マウス9匹
この群は、レンチウイルス形質導入細胞が親細胞と同様の増殖を示すようにする必要があった。
【0171】
群6:RAJICD200rev 4×106個+PBL 2×106個、皮下;マウス9匹
この数のPBLは、強力な拒絶または腫瘍増殖の低減を生じるはずがない。これは群3および群4(最大予想腫瘍増殖)の正の対照である。この群で拒絶がない場合、ドナーPBLは過剰に活性化されて、CD200による効果の不足を説明できるほうから開始する。
【0172】
群7:RAJICD200rev 4×106個+PBL 5×106個、皮下;マウス9匹
CD200群3で観察されたすべての効果がCD200に実際に関連しており、レンチウイルス形質導入には関連していないという対照。
【0173】
群8:RAJICD200rev 4×106個+PBL 10×106個、皮下;マウス8匹
CD200群4で観察されたすべての効果がCD200に実際に関連しており、レンチウイルス形質導入には関連していないという対照。
【0174】
許容限界を超えたサイズに達した腫瘍に基づいて、第38日に動物を殺処分した。動物4匹/群から腫瘍を除去した。腫瘍2個/群をOCTで凍結させて、他の2個は細胞を単離して、FACSによってCD200発現について分析するために使用した。図30(a〜c)は、本試験の結果を示す。
【0175】
RAJICD200細胞は多少より低速に増殖するように思われるが、形質導入細胞と親細胞との増殖差は、図30(a)に示すように統計的有意には達しなかった。
【0176】
PBL 5または10×106個を注射したときに、PBLの存在は腫瘍増殖を84%まで減速したが、一般にPBL 10×106個は、PBL 5×106個と比較して、時間に関してより強力な減速を引き起こした。親腫瘍細胞と比較した増殖の減速は第20日から著しかった。PBL 2×106個は第22日〜第29日に著しい腫瘍増殖の減速を引き起こしたが、その減速は後の時点で克服された(図30(b))。本試験は、この特定のドナーがRAJI腫瘍細胞を非常に強力に拒絶することを示した。
【0177】
CD200発現RAJI細胞およびPBLを投与した群の腫瘍増殖は、RAJI細胞のみを投与した群の腫瘍増殖とは著しく異なっていなかったが、PBL 10×106個を投与されたマウスは腫瘍増殖減速の傾向を示し、ただし差は、腫瘍が100m3に達した後のどの時点でも統計的有意には達しなかった。RAJI細胞およびPBL 5×106個を投与した群の各マウスは第2の腫瘍を発症して、一部のマウスは第7日という早期に発症したが、これは任意の他の群では観察されなかった。分析のために、第2の腫瘍を第1の腫瘍に加えて、合せたサイズを図30(c)に示す。
【0178】
このような結果は、腫瘍細胞でのCD200発現が実際に、免疫系による腫瘍増殖の減速を防止することを示す。また本試験は、免疫抑制性化合物または分子を評価するための、RAJI/PBLモデルの有用性を証明する。
【0179】
(Namalwa/PBLモデルでのCD200の免疫抑制効果)
RAJI/PBLモデルで見られる効果が他の腫瘍モデルでも見られるかどうかを評価するために、Namalwa腫瘍細胞もレンチウイルスCD200系によって感染させて、安定なクローンを選択した。ベクター感染の効果を確実なくすための対照として、CD200の逆転非機能形(CD200rev)を発現するクローンも選択した。NOD.CB17−Prkdc<scid>マウスに、以下のスキームに従って図31(a)〜(d)に示すように注射した:
群1:Namalwa 4×106個、皮下;マウス9匹
この群は、レンチウイルス形質導入細胞が親細胞と同様の増殖を示すようにする必要があった。
【0180】
群2:Namalwa CD200(1D12Ub)4×106個、皮下;マウス9匹
この群は、CD200形質導入細胞が親細胞と同様の増殖を示すようにする必要があった。またこの群は、最大の腫瘍増殖を与えるであろう。群3および群4をこの群と比較した。
【0181】
群3:Namalwa CD200(1D12Ub)4×106個+PBL 5×106個、皮下;マウス9匹
このPBL数は、以前の実験で一部のマウスの腫瘍増殖を低減することが示されている。拒絶は、細胞1000万個ほど強力ではないが、CD200がある数の細胞のみに影響を及ぼすことが可能で、これが最小PBLである場合、拒絶を得るために使用できる;拒絶はCD200の存在によって防止されるであろう。
【0182】
群4:Namalwa CD200(1D12Ub)4×106個+PBL 10×106個、皮下;マウス8匹
これはNamalwa/PBLモデルで拒絶を確認するための、PBLの最適数である。これはCD200発現がこの拒絶を防止することを示すために実施した。
【0183】
群5:Namalwa CD200rev(C5Ubrev)4×106個、皮下;マウス9匹
この群は、レンチウイルス形質導入細胞が親細胞と同様の増殖を示すようにする必要があった。
【0184】
群6:Namalwa CD200rev 4×106個+PBL 2×106個、皮下;マウス9
この数のPBLは、強力な拒絶または腫瘍増殖の低減を生じるはずがない。これは群3および群4(最大予想腫瘍増殖)の正の対照である。この群で拒絶がない場合、ドナーPBLは過剰に活性化されて、CD200による効果の不足を説明できるほうから開始する。
【0185】
群7:Namalwa CD200rev 4×106個+PBL 5×106個、皮下;マウス9匹
この群は、CD200に関連するが、レンチウイルス形質導入には関連しないCD200群3の任意の効果を検出するための対照として必要であった。
【0186】
群8:Namalwa CD200rev 4×106個+PBL 10×106個、皮下;マウス8匹
この群は、CD200に関連するが、レンチウイルス形質導入には関連しないCD200群4の任意の効果を検出するための対照として必要であった。
【0187】
腫瘍の長さおよび幅を週3回評価した。
【0188】
すべての腫瘍細胞が迅速な腫瘍増殖を引き起こした。形質導入細胞と親細胞との間に著しい増殖差はなかった。腫瘍は図31(a)に示すように、以前に観察されたよりも侵襲的に増殖する。
【0189】
図31(b)は、PBLの存在が腫瘍増殖を約50%減速することを示す。この傾向は第2日から観察された。PBL処置群対腫瘍細胞のみを投与した群の差は、PBLを200万個または1000万個注射したときに、第17日および第19日に統計的に有意である(両側Student t検定によって決定されたように)。PBL 500万個の注射は、腫瘍増殖の減速を引き起こしたが、有意には達しなかった。
【0190】
CD200発現Namalwa細胞およびPBLを投与した群の腫瘍増殖は、Namalwa細胞のみを投与した群の腫瘍増殖と同様であった(図31(c))。
【0191】
これらのデータは、腫瘍細胞でのCD200発現がヒト免疫系による腫瘍増殖の減速を防止することを確認している。
【0192】
(抗CD200抗体による、RAJI/PBLモデルにおけるCD200の免疫抑制効果の遮断)
抗CD200抗体が腫瘍細胞で発現されたCD200の免疫抑制効果を遮断できるかどうかを評価するために、CD200が形質導入されたRAJI細胞をNOD.CB17−Prkdc<scid>マウスに皮下注射して、キメラ抗CD200抗体d1B5およびc2aB7ならびに腫瘍細胞を結合しない抗体(alxn4100)の存在下または非存在下でPBLが腫瘍増殖を低減する能力を評価した。抗体を最初に腫瘍細胞と共に10mg/kgまた2.5mg/kgで、次に以下に示す濃度で週2回静脈内投与した。以下の群を構成した:
1.RAJICD200 4×106個;マウス10匹
2.RAJICD200 4×106個+PBL 5×106個;マウス10匹
3.RAJICD200 4×106個+PBL 5×106個+100mg/kg d1B5;マウス12匹
4.RAJICD200 4×106個+PBL 5×106個+20mg/kg d1B5;マウス9匹
5.RAJICD200 4×106個+PBL 5×106個+100mg/kg c2aB7;マウス11匹
6.RAJICD200 4×106個+PBL 5×106個+20mg/kg c2aB7;マウス9匹
7.RAJICD200 4×106個+PBL 5×106個+100mg/kg alxn4100;マウス9匹。
【0193】
腫瘍の長さおよび幅を週に3回測定して、腫瘍体積を腫瘍の長さ*幅*幅/2によって計算した。図33は、予想通り、腫瘍細胞でのCD200発現が免疫細胞による腫瘍増殖の低減を防止することを示している。しかしながら抗CD200抗体の添加は、腫瘍体積を50〜75%縮小した。抗体による増殖の低減は、第18日から試験の終りまでのStudent t検定およびMann Whitney検定によって決定されたように、統計的に有意であった。これに対して、対照抗体による処置は、腫瘍増殖を低減しなかった。これらのデータは、抗癌処置での抗CD200の有用性を証明する。また本試験は、免疫調節治療を評価するための、RAJI/PBLモデルの有用性を証明する。
【0194】
(抗CD200抗体による、Namalwa/PBLモデルにおけるCD200の免疫抑制効果の遮断)
RAJI/PBLモデルにおいて抗CD200抗体によって見られた効果が他の腫瘍モデルでも観察できるかどうかを評価するために、CD200が形質導入されたRAJI細胞をNOD.CB17−Prkdc<scid>マウスに皮下注射して、キメラ抗CD200抗体d1B5およびc2aB7ならびに腫瘍細胞を結合しない抗体(alxn4100)の存在下または非存在下でPBLが腫瘍増殖を低減する能力を評価した。以下に示す濃度の抗体を最初に腫瘍細胞と共に、次に腫瘍0.5cm以内では週2回、皮下投与した。以下の群を構成した(別途記載しない限り、マウス10匹/群):
1.NamalwaCD200 4×106個
2.NamalwaCD200 4×106個+PBL 5×106個;マウス12匹
3.NamalwaCD200 4×106個+PBL 5×106個+10mg/kg d1B5;マウス12匹
4.NamalwaCD200 4×106個+PBL 5×106個+2.5mg/kg d1B5
5.NamalwaCD200 4×106個+PBL 5×106個+10mg/kg c2aB7;マウス12匹
6.NamalwaCD200 4×106個+PBL 5×106個+2.5mg/kg c2aB7
7.NamalwaCD200 4×106個+PBL 5×106個+10mg/kg alxn4100。
【0195】
腫瘍の長さおよび幅を週に3回測定して、腫瘍体積を腫瘍の長さ*幅*幅/2によって計算した。図34は、予想通り、腫瘍細胞でのCD200発現が免疫細胞による腫瘍増殖の低減を防止することを示している。しかしながら抗CD200抗体の添加は、腫瘍体積を最大97%縮小した。抗体による増殖の低減は、第12日から試験の終りまでのStudent t検定およびMann Whitney検定によって決定されたように、統計的に有意であった。これに対して、対照抗体による処置は、腫瘍増殖を低減しなかった。これらのデータによって、抗癌処置での抗CD200の有用性、および免疫調節療法での腫瘍/PBLモデルの使用が確認される。
【0196】
(RAJI/PBLモデルにおける化合物の潜在的な免疫増強効果の検出)
CD3カラム(Miltenyi)を使用してPBLから単離したT細胞を、AZND1(抗DC−SIGN抗体)の腹水調製物または対照抗体(BB5)の腹水調製物と共にインビトロでインキュベートした。NOD.CB17−Prkdc<scid>マウスに次のスキームに従って注射した:
群6:マウス8匹、RAJI 4×106個+AZND1増強T細胞0.8×106+PBL 2×106皮下
群5:マウス10匹、RAJI 4×106個、皮下
群4:マウス8匹、RAJI 4×106個+PBL 8×106個、皮下
群3:マウス8匹、RAJI 4×106個+新しいT細胞0.8×106個、皮下
群2:マウス8匹、RAJI 4×106個+AZND1増強T細胞0.8×106個、皮下
群1:マウス8匹、RAJI 4×106個+BB5.1増強T細胞0.8×106個、皮下
腫瘍の長さおよび幅を3回/週で測定した。
【0197】
T細胞は負の対照BB5でインキュベートしたT細胞は腫瘍増殖を低減しなかったが、AZND1処置T細胞は腫瘍増殖を著しく低減しなかった(図32を参照)。
【0198】
これらの結果は、免疫増強化合物の有効性を評価するためにRAJ/PBLモデルを使用できることを証明する。
【0199】
(実施例6)
(CLL患者でのCD200アップレギュレーションの判定)
CLL患者15名からのリンパ球をFITC結合抗CD5(e−bioscience)、APC結合抗CD19(e−bioscience)およびPE結合抗CD200(Serotec)によって染色した。健常ドナーからのリンパ球をそれに従って染色した。CD5+CD19+細胞でのCD200発現を判定した。以下の表に示すように、CD200発現のレベルはCLL患者サンプル間で変化したが、すべてのCLLサンプルが、正常B細胞でのCD200発現と比較して、上昇したレベル(1.6〜4倍の範囲)のより高いCD200発現を示した。上昇したレベルのCD200発現を示すCCL患者を、本明細書で述べる方法によるCD200処置のために選択した。
【0200】
(正常B細胞と比較したB−CLL細胞でのCD200発現のFACS分析)
【0201】
【化2】
(参考文献)
以下の参考文献は、本発明が関係する最新技術をより十分に説明するために、参考として本明細書中に援用される。以下のこのような刊行物または上で参照により組み入れられたものと、本開示との不一致は、本開示を支持して解決されるものとする。
【0202】
【化3】
本明細書で開示された実施形態に対して各種の改良を行えることが理解されるであろう。たとえば当業者が認識するように、本明細書で述べた特異性配列は、OX−2/CD200を結合するのに使用されるポリペプチド、抗体または抗体断片の機能に必ずしも悪影響を及ぼすことなく、わずかに変更できる。たとえば抗体配列内の1個または複数のアミノ酸の置換は、抗体または断片の機能を破壊することなく頻繁に行うことができる。それゆえ本明細書で述べた特異性抗体に対して70%を超える同一性の程度を有するポリペプチドまたは抗体が本開示の範囲内であることを理解すべきである。特に有用な実施形態において、本明細書で述べた特異性抗体に対して約80%を超える同一性を有する抗体が検討される。他の有用な実施形態において、本明細書で述べた特異性抗体に対して約90%を超える同一性を有する抗体が検討される。したがって上の記述は、制限的としてではなく、単に好ましい実施形態の例示として解釈すべきである。当業者は、本開示の範囲および精神から逸脱せずに他の改良を構想するであろう。
【特許請求の範囲】
【請求項1】
本願明細書に記載された発明。
【請求項1】
本願明細書に記載された発明。
【図1】

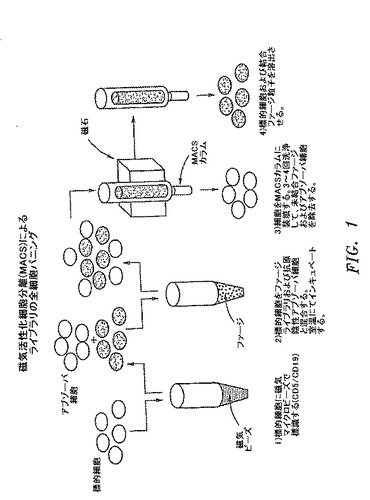
【図2】
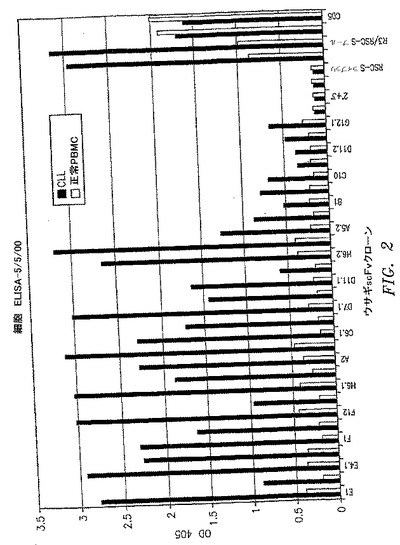
【図3a】
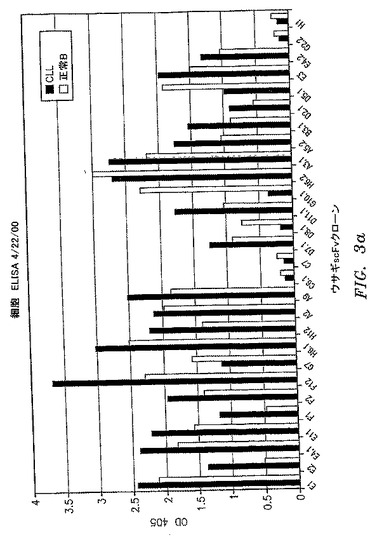
【図3b】
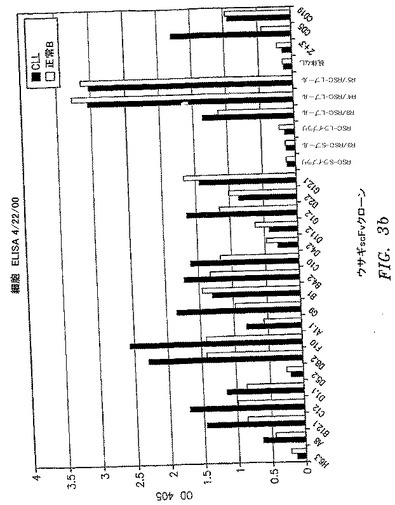
【図4a】
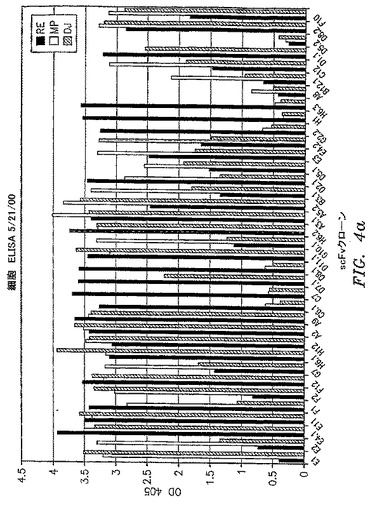
【図4b】
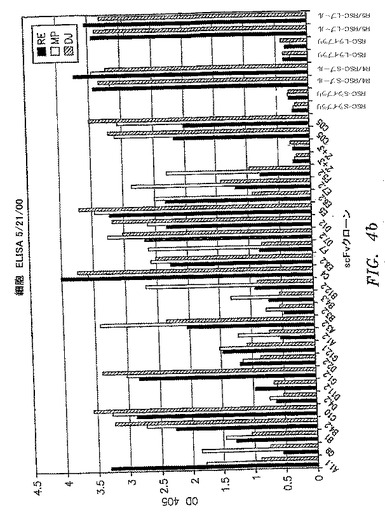
【図5a】
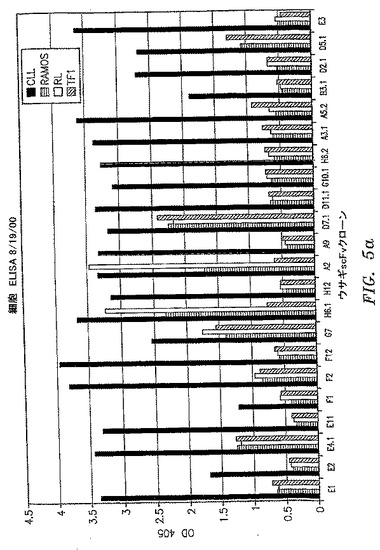
【図5b】

【図6a】
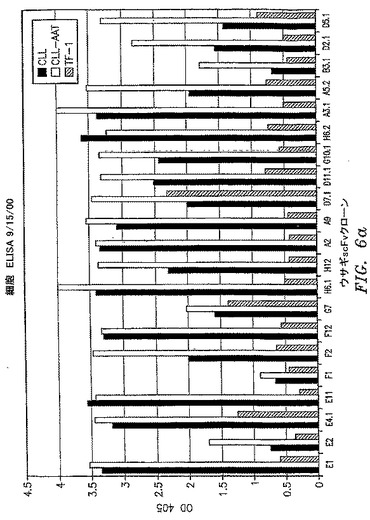
【図6b】
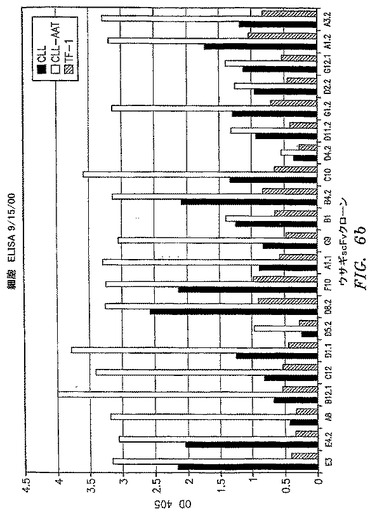
【図6c】
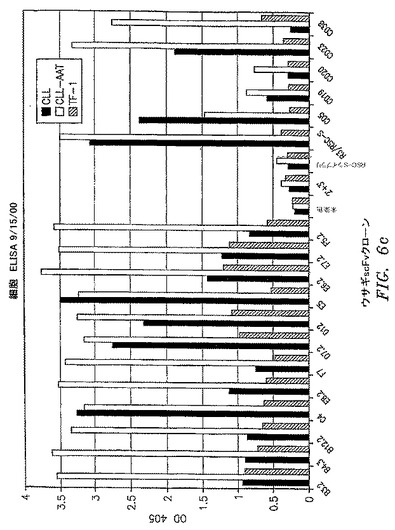
【図7】
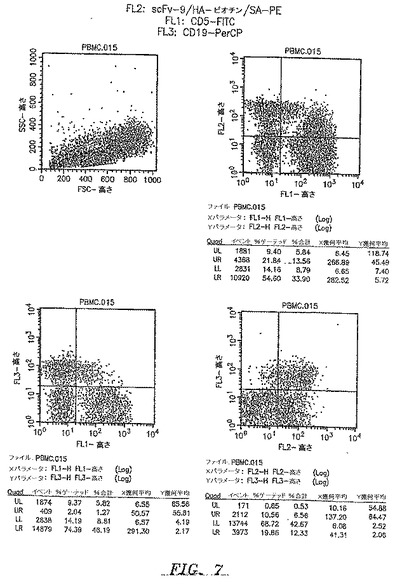
【図8a】
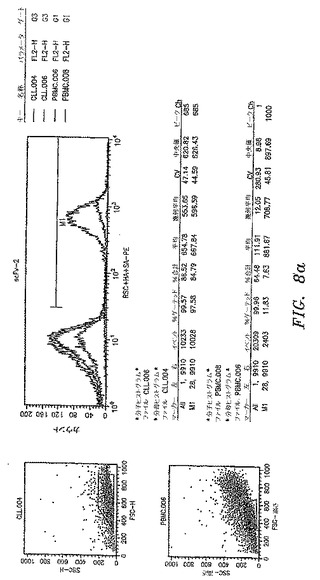
【図8b】
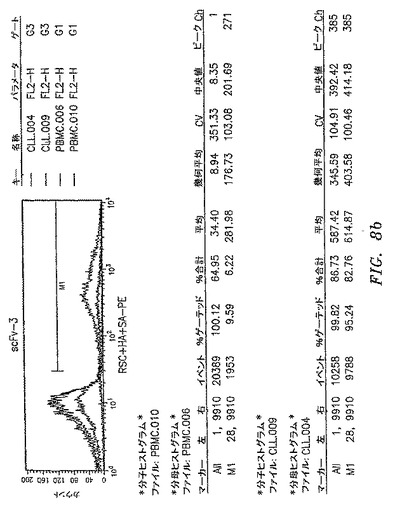
【図8c】
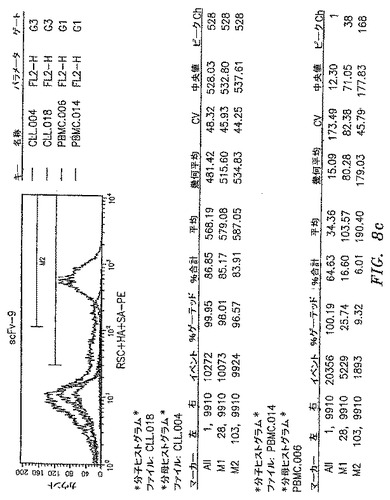
【図9a−1】

【図9a−2】
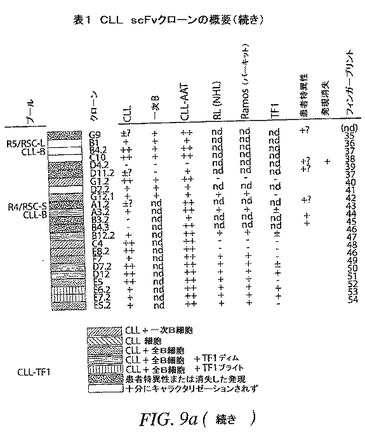
【図9B−1】

【図9B−2】

【図9C】
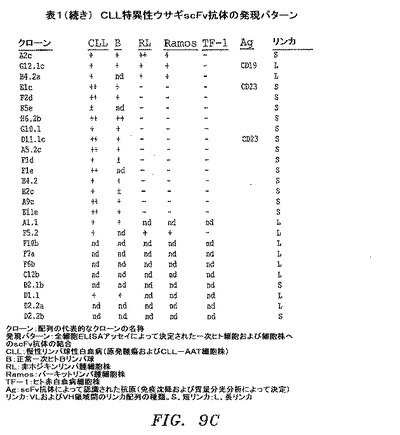
【図10】
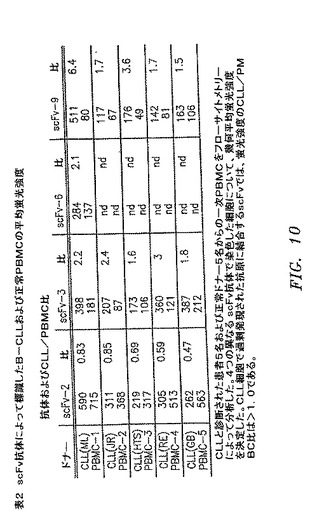
【図11】

【図12】
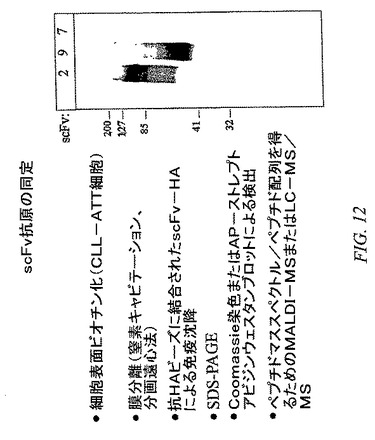
【図13】
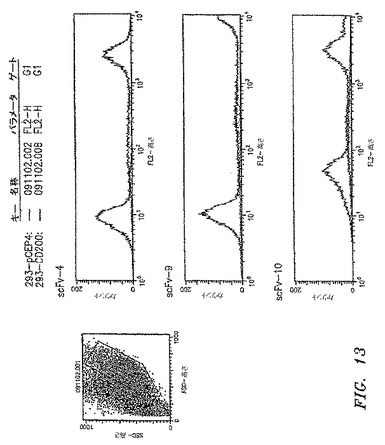
【図14】
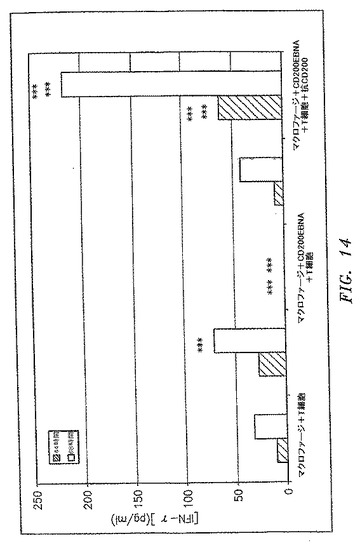
【図15】
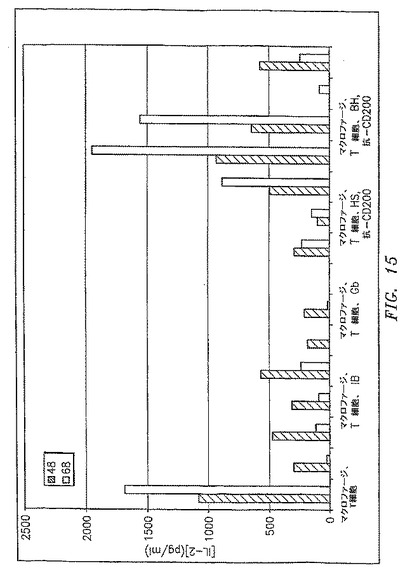
【図16】
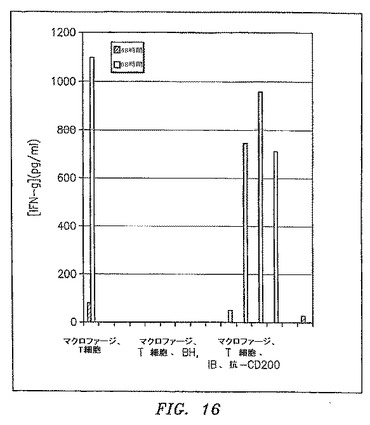
【図17A】
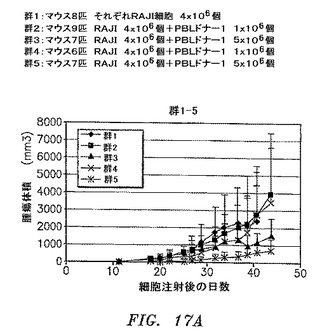
【図17B】
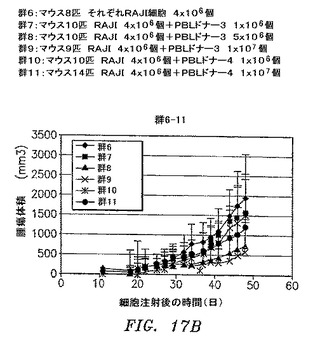
【図18】
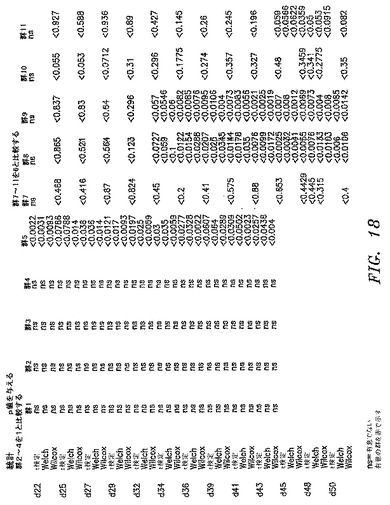
【図19A】
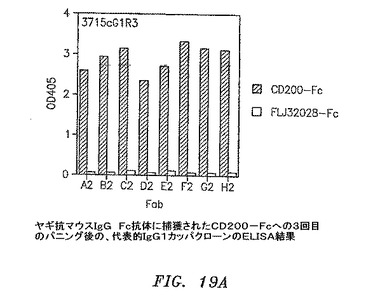
【図19B】
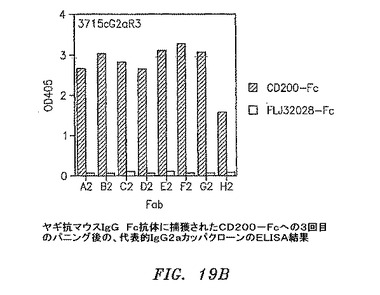
【図19C】
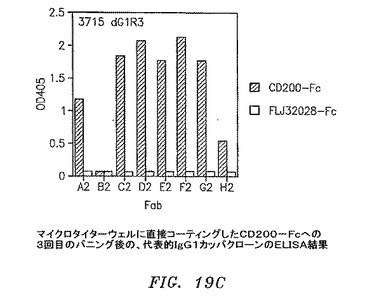
【図19D】
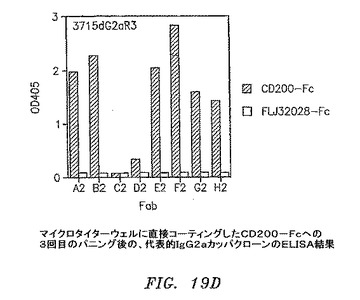
【図20A】
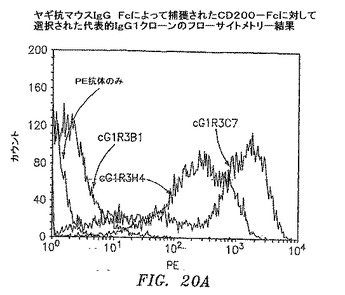
【図20B】
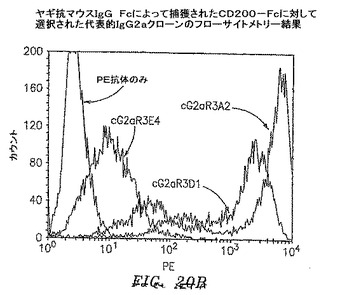
【図20C】
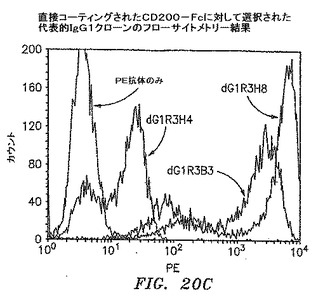
【図20D】
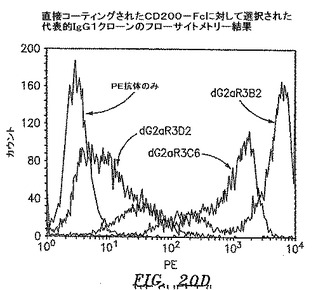
【図21a−1】
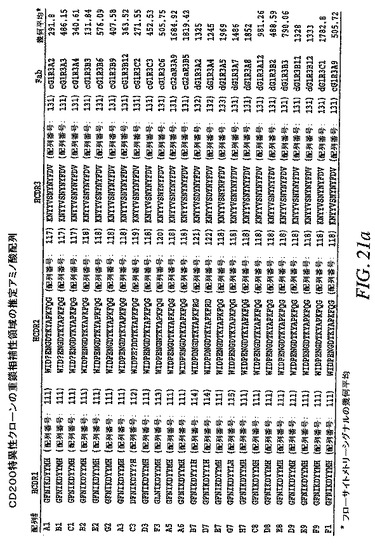
【図21a−2】
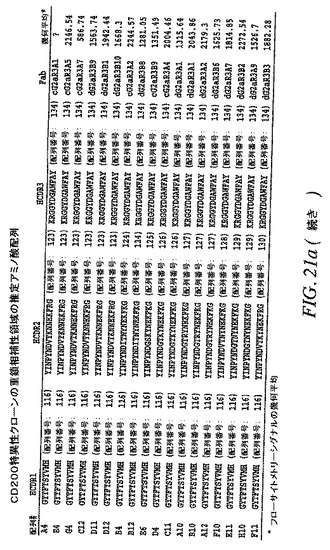
【図21b−1】
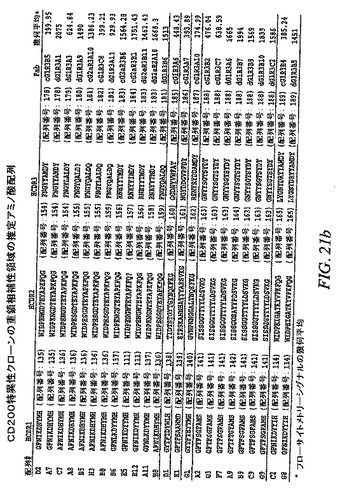
【図21b−2】

【図22】
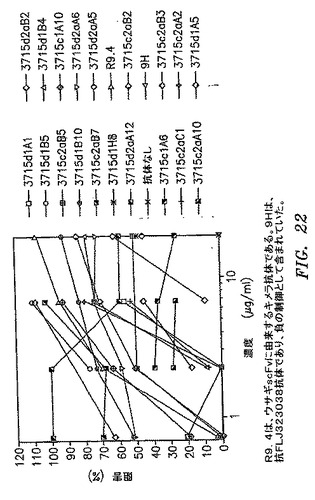
【図23】

【図24】
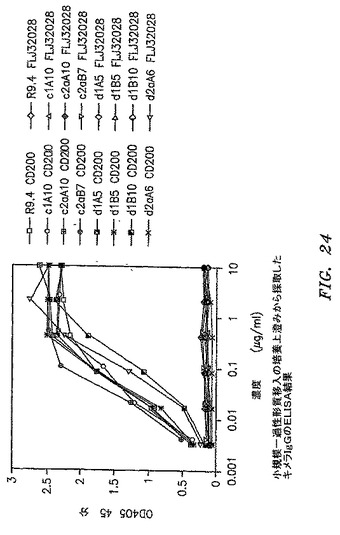
【図25】
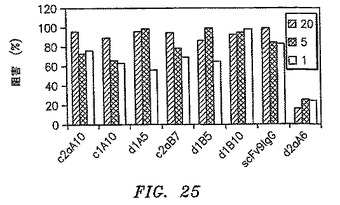
【図26A】
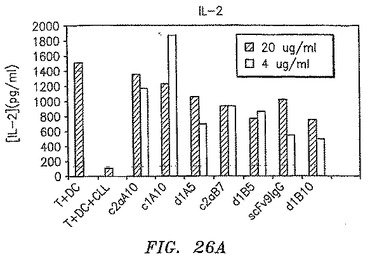
【図26B】
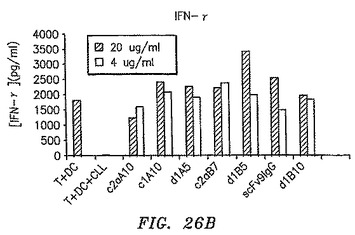
【図26C】
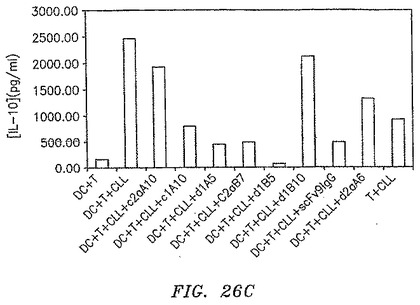
【図27】
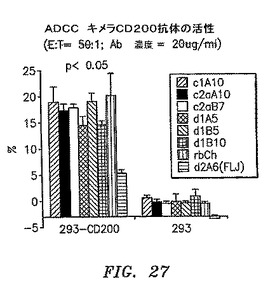
【図28】
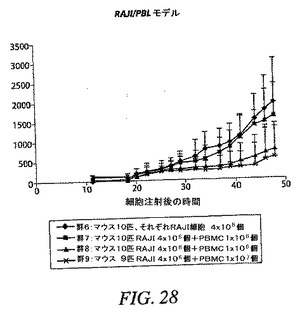
【図29】
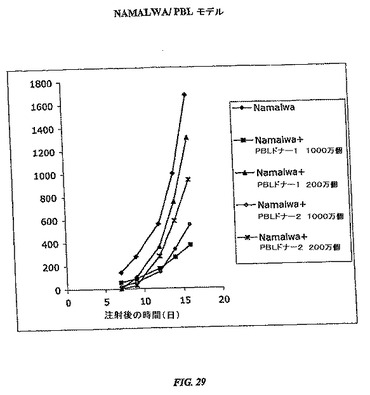
【図30a】
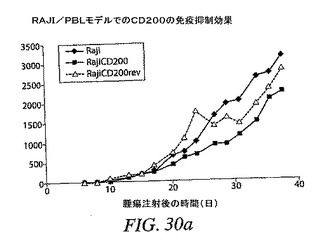
【図30b】
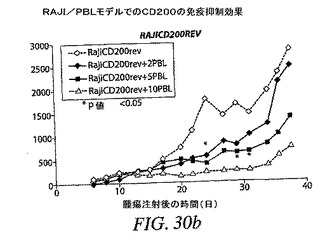
【図30c】
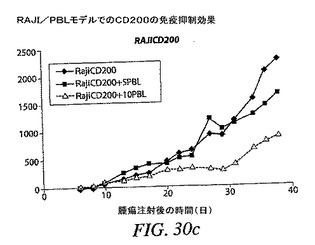
【図31a】
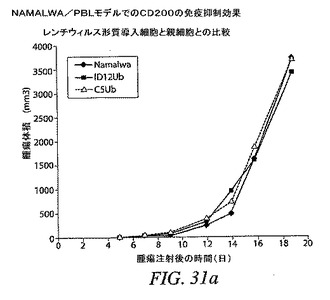
【図31b】
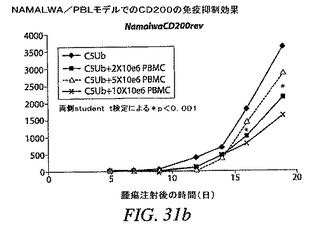
【図31c】
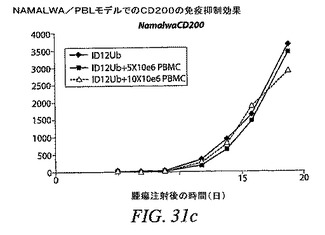
【図31d】
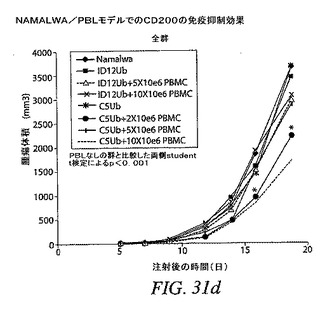
【図32】
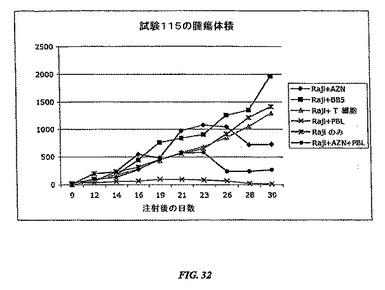
【図33】
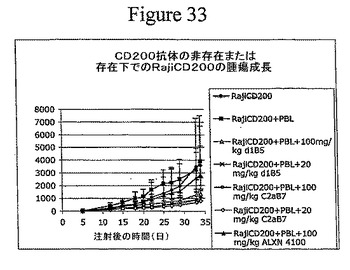
【図34】
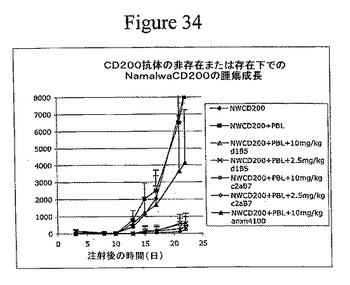
【図2】
【図3a】
【図3b】
【図4a】
【図4b】
【図5a】
【図5b】
【図6a】
【図6b】
【図6c】
【図7】
【図8a】
【図8b】
【図8c】
【図9a−1】
【図9a−2】
【図9B−1】
【図9B−2】
【図9C】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17A】
【図17B】
【図18】
【図19A】
【図19B】
【図19C】
【図19D】
【図20A】
【図20B】
【図20C】
【図20D】
【図21a−1】
【図21a−2】
【図21b−1】
【図21b−2】
【図22】
【図23】
【図24】
【図25】
【図26A】
【図26B】
【図26C】
【図27】
【図28】
【図29】
【図30a】
【図30b】
【図30c】
【図31a】
【図31b】
【図31c】
【図31d】
【図32】
【図33】
【図34】
【公開番号】特開2012−111780(P2012−111780A)
【公開日】平成24年6月14日(2012.6.14)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−53212(P2012−53212)
【出願日】平成24年3月9日(2012.3.9)
【分割の表示】特願2007−522632(P2007−522632)の分割
【原出願日】平成17年7月19日(2005.7.19)
【出願人】(503102674)アレクシオン ファーマシューティカルズ, インコーポレイテッド (51)
【Fターム(参考)】
【公開日】平成24年6月14日(2012.6.14)
【国際特許分類】
【出願番号】特願2012−53212(P2012−53212)
【出願日】平成24年3月9日(2012.3.9)
【分割の表示】特願2007−522632(P2007−522632)の分割
【原出願日】平成17年7月19日(2005.7.19)
【出願人】(503102674)アレクシオン ファーマシューティカルズ, インコーポレイテッド (51)
【Fターム(参考)】
[ Back to top ]