
抗がん剤
【課題】安全性の高い新規抗がん剤を提供する。
【解決手段】下記の一般式(1)で表されるパルミチン酸誘導体又は薬理的に許容されるその塩を有効成分として含有することを特徴とする抗がん剤。

(式中、R、R’は水素原子又は低級アルキル基を表す)上記の発明において、下記の一般式(2)で表されるパルミチン酸誘導体又は薬理的に許容されるその塩を有効成分として含有することを特徴とする抗がん剤。
【解決手段】下記の一般式(1)で表されるパルミチン酸誘導体又は薬理的に許容されるその塩を有効成分として含有することを特徴とする抗がん剤。
(式中、R、R’は水素原子又は低級アルキル基を表す)上記の発明において、下記の一般式(2)で表されるパルミチン酸誘導体又は薬理的に許容されるその塩を有効成分として含有することを特徴とする抗がん剤。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は抗がん剤に関し、詳細には、パルミチン酸誘導体を有効成分とする抗がん剤に関する。
【背景技術】
【0002】
がんは、今日最も死亡率の高い疾患で、治療法には三大療法として化学療法、放射線療法、外科療法がある。化学療法は、マイトマイシンC、アクチノマイシンDなどの抗腫瘍性抗生物質、5−フルオロウラシル、メトトレキサートなどの代謝拮抗剤、シクロホスファミド、ニムスチンなどのアルキル化剤、アナストロゾール、エチニルエストラジオールなどのホルモン剤、シスプラチン、オキサリプラチンなどのプラチナ製剤、イリノテカン、エトポシドなどの植物アルカロイドのような抗がん剤が用いられている。しかし、抗がん剤は、がん細胞のみに選択的に作用するのではなく、正常細胞にも作用するため、抗がん剤の投与を受けた患者は嘔吐、食欲不振、悪心、脱毛、骨髄抑制、肝機能障害、腎機能障害、心機能障害など種々の副作用によりQOL(Quality of Life、生活の質)を著しく低下させられるという大きな問題があり、抗がん活性に優れ正常細胞に対する毒性が低く安全性の高い抗がん剤が常々望まれている。
【0003】
従来、上記のように多種多様の抗がん剤があるが、安全性の高い抗がん剤として不飽和脂肪酸を有効成分とする抗腫瘍剤の提案がある(特許文献1、特許文献2参照)。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開2003−171272号公報
【特許文献2】特開2005−247754号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明は、生体内脂質由来の低分子化合物で飽和脂肪酸であるパルミチン酸の誘導体を有効成分とする、抗がん活性に優れ、安全性の高い新規な抗がん剤を提供することを課題とする。
【課題を解決するための手段】
【0006】
上記の課題を解決する本発明は、下記の一般式(1)で表されるパルミチン酸誘導体又は薬理的に許容されるその塩を有効成分として含有することを特徴とする抗がん剤を要旨とする。
【0007】
【化1】
(式中、R、R’は水素原子又は低級アルキル基を表す)
【0008】
本発明は、上記の発明において、下記の一般式(2)で表されるパルミチン酸誘導体又は薬理的に許容されるその塩を有効成分として含有することを特徴とする抗がん剤を要旨とする。
【0009】
【化2】
【0010】
本発明は、上記の発明において、下記の一般式(3)で表されるパルミチン酸誘導体の塩酸塩を有効成分として含有することを特徴とする抗がん剤を要旨とする。
【0011】
【化3】
【発明の効果】
【0012】
本発明の抗がん剤は、抗がん活性に優れ安全性が高いので、投与を受けた患者のQOLの低下を防止し、がんを効果的に治療できる医薬、またがんの予防、再発を抑制できる医薬として有用である。
【図面の簡単な説明】
【0013】
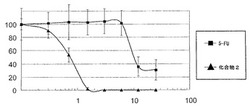
【図1】一般式(3)で表されるパルミチン酸誘導体及び5-フルオロウラシルをヒト大腸がん細胞株HT29に暴露し、がん細胞生存率を測定した結果に基づき作成された増殖曲線(用量反応曲線)である。
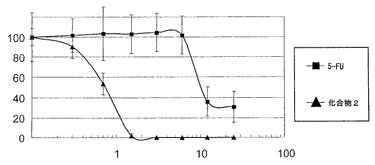
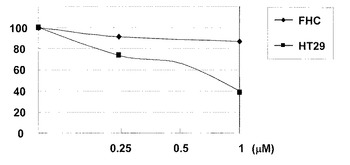
【図2】一般式(3)で表されるパルミチン酸誘導体をヒト大腸正常細胞株FHC及びヒト大腸がん細胞株HT29に暴露し、各細胞生存率を測定した結果に基づき作成された増殖曲線(用量反応曲線)である。
【発明を実施するための形態】
【0014】
上記の一般式(1)で表されるパルミチン酸誘導体において、R、R’が低級アルキル基である場合、任意の位置に置換できる低級アルキル基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert-ブチル基、ペンチル基、ヘキシル基等の炭素数1〜6の直鎖又は分枝鎖状のアルキル基を例示できる。
【0015】
上記の一般式(1)で表されるパルミチン酸誘導体において、R、R’が水素原子である場合、パルミチン酸誘導体は上記の一般式(2)で表され、1-パルミトイル-4-ピペリジノピペリジン(1-Palmitoyl-4-piperidinopiperidine)(以下、「化合物1」ということがある)である。また、当該パルミチン酸誘導体が塩酸塩である場合、パルミチン酸誘導体は一般式(3)で表され、1-パルミトイル-4-ピペリジノピペリジン塩酸塩(1-Palmitoyl-4-piperidinopiperidine hydrochloride)(以下、「化合物2」ということがある)である。
【0016】
上記の一般式(1)で表されるパルミチン酸誘導体は、公知のアミドの合成法により製造できる。例えば、パルミチン酸とアミン成分の4-ピペリジノピペリジン又は低級アルキル基で置換された4-ピペリジノピペリジンをカルボジイミドなどの脱水縮合剤の存在下に反応させることにより製造できる。また、パルミチン酸ハロゲン化物、パルミチン酸無水物又はパルミチン酸エステルとアミン成分の4-ピペリジノピペリジン又は低級アルキル基で置換された4-ピペリジノピペリジンと反応させることにより製造できる。反応終了後に抽出、分配、カラムクロマトグラフィー等の公知の分離、精製手段を用いて単離することができる。
【0017】
上記の一般式(3)で表される化合物2は、パルミチン酸とジメチルホルムアミドとアミン成分の4-ピペリジノピペリジンを脱水縮合剤の存在下に反応させることにより製造できる。反応終了後に抽出、分配、カラムクロマトグラフィー等の公知の分離、精製手段を用いて単離することができる。また、上記の一般式(1)で表されるパルミチン酸誘導体において、R、R’が低級アルキル基であり、パルミチン酸誘導体が塩酸塩である場合、4-ピペリジノピペリジンに代え、低級アルキル基で置換された4-ピペリジノピペリジンを用いる。
【0018】
上記の一般式(1)で表されるパルミチン酸誘導体は、遊離のものでも、また薬理的に許容される塩、プロドラッグとすることもできる。塩は、酸付加塩でも塩基付加塩でも良い。酸付加塩を形成する酸は、リン酸、塩酸、硫酸、硝酸等の無機酸、メタンスルホン酸、クエン酸、フマル酸、コハク酸、酒石酸、シュウ酸、マレイン酸、酢酸、リンゴ酸、乳酸、アスコルビン酸等の有機酸を例示できる。塩基付加塩を形成する塩基は、水酸化アンモニウム、アルカリ金属水酸化物、アルカリ土類金属水酸化物、炭酸塩、炭酸水素塩等の無機塩基、エタノールアミン、トリエチルアミン、トリス(ヒドロキシメチル)アミノメタン等の有機塩基を例示できる。上記の一般式(1)で表されるパルミチン酸誘導体は、溶媒和物とすることができる。このような溶媒和物としては、水和物、アルコール和物などを例示できる。
【0019】
上記の一般式(1)で表されるパルミチン酸誘導体は、大腸がん、胃がん、食道がん、結腸がん、肝臓がん、膵臓がん、乳がん、肺がん、胆嚢がん、胆管がん、胆道がん、直腸がん、卵巣がん、子宮がん、腎がん、膀胱がん、前立腺がん、骨肉腫、脳腫瘍、白血病、筋肉腫、皮膚がん、悪性黒色腫、悪性リンパ腫、舌がん、骨髄腫、甲状腺がん、皮膚転移がん、皮膚黒色腫などの治療に用いることができるがこれらに限定されず、また前がん病変の治療に用いることもできる。
【0020】
上記の一般式(1)で表されるパルミチン酸誘導体の抗がん剤としての投与形態は、特に限定されず、経口又は非経口のいずれの投与形態でもよい。また、投与形態に応じて適当な剤形とすることができ、例えば注射剤、カプセル剤、錠剤、顆粒剤、散剤、丸剤、細粒剤などの経口剤、直腸投与剤、油脂性坐剤、水性坐剤などの各種製剤に調製することができる。
【0021】
上記の各種製剤は、薬理的に許容され通常用いられる添加剤、例えば賦形剤、結合剤、滑沢剤、崩壊剤、界面活性剤、流動性促進剤などを適宜添加して調製できる。賦形剤として、例えば、乳糖、果糖、ブドウ糖、コーンスターチ、ソルビット、結晶セルロースなどが、結合剤として、例えば、メチルセルロース、エチルセルロース、アラビアゴム、ゼラチン、ヒドロキシプロピルセルロース、ポリビニルピロリドンなどが、滑沢剤として、例えば、タルク、ステアリン酸マグネシウム、ポリエチレングリコール、硬化植物油などが、崩壊剤として、例えば、澱粉、アルギン酸ナトリウム、ゼラチン、炭酸カルシウム、クエン酸カルシウム、デキストリン、炭酸マグネシウム、合成ケイ酸マグネシウムなどが、界面活性剤として、例えば、ラウリル硫酸ナトリウム、大豆レシチン、ショ糖脂肪酸エステル、ポリソルベート80などが、流動性促進剤として、例えば、軽質無水ケイ酸、乾燥水酸化アルミニウムゲル、合成ケイ酸アルミニウム、ケイ酸マグネシウムなどが、その他の添加剤としては、シロップ、ワセリン、グリセリン、エタノール、プロピレングリコール、クエン酸、塩化ナトリウム、亜硝酸ソーダ、リン酸ナトリウムなどが挙げられる。
【0022】
上記の一般式(1)で表されるパルミチン酸誘導体の投与量は、用法、患者の年齢、性別、症状の程度などを考慮して適宜増減できるが、通常成人1日当り200〜400mg好ましくは100〜200mgで、これを1日1回又は数回に分けて投与できる。
【実施例】
【0023】
次いで、本発明を実施例を挙げて説明するが、本発明は以下の実施例に限定されるものではない。
【0024】
〔実施例1〕(化合物1の合成)
パルミチン酸 (2.6 g, 10 mmol、和光純薬工業社) を塩化チオニル (5mL、和光純薬工業社)と2時間還流した後、過剰の塩化チオニルを留去し(減圧下)、この中に、4-ピペリジノピペリジン (1.7 g, 10 mmol、和光純薬工業社) を含むテトラヒドロフラン溶液を加え、3時間還流した。反応液は炭酸水素ナトリウム溶液に滴下し、酢酸エチルで分液した。酢酸エチル層は濃縮後、シリカゲルカラムクロマトグラフィー(展開溶媒:クロロホルム−メタノール 10:1)で精製した。化合物1は、淡褐色の結晶となって固化した(融点:低すぎて測定不能)。
【0025】
C26H50N2O MW 406; EIMS m/z (rel. int.): 406 [M]+ (22), 167 (44), 124 (100), 84 (29); 1H-NMR (500 MHz, CDCl3) δ:0.86 (3H, t, J = 5.7 Hz), 1.23 (22H, br s), 1.35-1.47 (4H, m), 1.55-1.63(7H, m), 1.84 (1H, d, J= 12.6 Hz), 2.56-2.63 (6H, m), 2.97 (1H, t, J = 12.6 Hz), 3.89 (1H, d, J= 13.2 Hz), 4.69 (1H, d, J = 13.2 Hz).
【0026】
〔実施例2〕(化合物2の合成)
パルミチン酸 (2.6 g, 10 mmol、和光純薬工業社) と1-ヒドロキシベンゾトリアゾール (1.4 g, 11 mmol、和光純薬工業社) をジメチルホルムアミド40 mlに溶解し、4-ピペリジノピペリジン (1.7 g, 11 mmol、和光純薬工業社) を加え、氷浴上で15分間撹拌した(A液)。別に縮合剤である1−エチル−3−(3−ジメチルアミノプロピル)−カルボジイミド(和光純薬工業社)を20 mlのジメチルホルムアミドに溶解した (B液)。A液を氷浴上で撹拌しながらB液を滴下した。氷浴上で2時間、次いで室温にて12時間撹拌を続け、下記の処理を行った。
(処理法)
反応生成物にクロロホルム50 mlを加え、次の (1)および(2) の溶液と分液操作を行った。
(1) 1.0%塩酸溶液(2回)、(2) 飽和食塩水(2回)
得られたクロロホルム層は濃縮後、シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル混合液)にて精製し、化合物2を得た。
【0027】
mp. 189℃; EIMS m/z (rel. int.): 406 [M]+ (42), 377 (12), 322 (11), 167 (46), 124 (100), 110 (20), 84 (30); 1H-NMR (400 MHz, CDCl3) δ: 0.88 (3H, t, J = 6.8 Hz), 1.25 (26H, br s), 1.60-1.69 (5H, m), 2.17 (2H, m), 2.30 (2H, dd, J = 8.8, 7.8 Hz), 2.53 (2H, m), 3.08 (2H, m), 3.26 (2H, m), 4.01 (1H, m), 4.86 (1H, m), 2.00-3.40 (br s).
【0028】
〔実施例3〕(化合物2の抗がん活性評価)
1.試薬の調製
化合物2は、実施例2で製造したものを蒸留水に溶かし、抗がん活性の測定に使用した。ギムザ染色液(Sigma社: Cat. No. S128-4L)の25 mLを蒸留水475 mLにて希釈して使用した。大腸がん治療に使用される代表的な抗がん剤の5-フルオロウラシル(5-fluorouracil)(以下、「5-FU」ということがある)を抗がん活性の比較対象とする薬剤とした。5-FUは、ジメチルスルホキシド(DMSO)(Sigma 社: Cat. No. D8418-250ML)に溶かして使用した。
2.ヒトがん細胞株の培養
ヒト大腸がん細胞株HT29(American Type Culture Collection(ATCC))とヒト大腸正常細胞株FHC(American Type Culture Collection(ATCC))をそれぞれ10 %ウシ胎児血清(FBS) (BioWest社: Cat. No. S1500)添加DMEM培地 (和光純薬工業社: Cat. No. 041-29775)中で37℃、5%CO2条件下にて培養した。培養装置は、Napco 5400 CO2インキュベーター(Napco社)を使用した。
【0029】
3.(化合物2のヒト大腸がん細胞株HT29に対する抗がん活性評価)
ヒト大腸がん細胞株HT29を6ウエル・プレート(日本ベクトン・デイッキンソン社、BD Falcon(TM): Cat. No. 353046)に500細胞/ウエルの濃度で蒔いて一晩培養した後、化合物2と5-FUをそれぞれ0、0.3、0.7、1.5、3.0、6.0、12.0、25.0μMの濃度でがん細胞に暴露し、5日間培養した(0μMの培地には、化合物2は蒸留水を、5-FUはDMSOを0.1%になるようにそれぞれ添加した)。培養後、各ウエルをリン酸緩衝生理食塩水(phosphate buffered saline 、以下「PBS」と略す)で2回洗浄し、各ウエル当たり2mLの100%メタノールで10分間、がん細胞を固定した。風乾し、各ウエル当たり2mLのギムザ染色液で30分間染色した後水道水で洗浄した。風乾後、染色されたがん細胞のコロニー数を目視にて計測した。
【0030】
上記のヒト大腸がん細胞株HT29を用いた細胞増殖アッセイの結果に基づき図1に示す増殖曲線(用量反応曲線)を作成した。化合物2のIC50値(50%増殖抑制率)を測定し、これを抗がん活性の指標とした。図1のグラフの縦軸は、がん細胞生存率(%)とし、横軸は化合物2の濃度(μM)とした。化合物2の濃度が0μMのときの細胞生存率を100%として、化合物2の各濃度での細胞生存率を換算した。その結果、化合物2の大腸がん細胞株HT29株に対するIC50値は、0.74μMであった。一方、5-FUのIC50値は、9μMであった。図1より化合物2の濃度が3μMのとき、がん細胞を100%死滅させているが、同じ濃度の5-FUでは全てのがん細胞が生存している。化合物2のIC50値は、5-FUと比較して12倍小さい値である。これは、化合物2が5-FUに比べてより低濃度で抗がん活性を発揮することを示す。このことにより、化合物2は、大腸がんの化学療法に使用される第1選択の抗がん剤である5-FUより強い抗がん活性を有することが示唆された。
【0031】
4.(化合物2のヒト大腸正常細胞株FHCとヒト大腸がん細胞株HT29に対する毒性の比較)
化合物2のヒト大腸正常細胞株FHCとヒト大腸がん細胞株HT29に対する毒性を検討した。各細胞株をそれぞれ6cmディッシュ (Nalge Nunc International社: Cat. No. 150288)に2x104細胞/ディッシュの濃度で蒔き一晩培養した後、各細胞株に対して化合物2を0、0.25、1.0μMの濃度で暴露し、3日間培養した(0μMの培地には、蒸留水を0.1%になるように添加した)。培養後、培地を除去し0.25%トリプシン-EDTA溶液(Sigma 社:Cat. No. T4049)1 mLを加え10分間37℃でインキュベーションした後、細胞をディッシュから剥がした。剥がした細胞に3mLの1xPBSを添加し細胞をよくほぐした後Coulter Counter Z1 (Becton Dickinson社)にて細胞数を測定した。
【0032】
上記の細胞増殖アッセイの結果に基づき図2に示す増殖曲線を作成した。化合物2の曝露下でのヒト大腸がん細胞株HT29及びヒト大腸正常細胞株FHCに対する生存率を測定し、これを正常細胞に対する毒性評価の指針とした。図2のグラフの縦軸は、各細胞生存率
(%)とし、横軸は化合物2の濃度(μM)とした。図2のグラフは、化合物2の濃度が0μMのときの細胞生存率を100%として、化合物2の各濃度の細胞生存率を換算した。その結果、ヒト大腸がん細胞株HT29の細胞が60%死滅した時の化合物2の濃度は1μMであり、この濃度における大腸正常細胞株FHCの細胞生存率は85%であった。このことにより、化合物2は、1μMまでの抗がん活性を呈する濃度において正常細胞に対する毒性は極めて少ないことが示唆された。
【産業上の利用可能性】
【0033】
本発明は、強力な抗がん活性を発揮し、毒性の少ない新規な抗がん剤とし医薬品工業上の有用性が期待される。
【技術分野】
【0001】
本発明は抗がん剤に関し、詳細には、パルミチン酸誘導体を有効成分とする抗がん剤に関する。
【背景技術】
【0002】
がんは、今日最も死亡率の高い疾患で、治療法には三大療法として化学療法、放射線療法、外科療法がある。化学療法は、マイトマイシンC、アクチノマイシンDなどの抗腫瘍性抗生物質、5−フルオロウラシル、メトトレキサートなどの代謝拮抗剤、シクロホスファミド、ニムスチンなどのアルキル化剤、アナストロゾール、エチニルエストラジオールなどのホルモン剤、シスプラチン、オキサリプラチンなどのプラチナ製剤、イリノテカン、エトポシドなどの植物アルカロイドのような抗がん剤が用いられている。しかし、抗がん剤は、がん細胞のみに選択的に作用するのではなく、正常細胞にも作用するため、抗がん剤の投与を受けた患者は嘔吐、食欲不振、悪心、脱毛、骨髄抑制、肝機能障害、腎機能障害、心機能障害など種々の副作用によりQOL(Quality of Life、生活の質)を著しく低下させられるという大きな問題があり、抗がん活性に優れ正常細胞に対する毒性が低く安全性の高い抗がん剤が常々望まれている。
【0003】
従来、上記のように多種多様の抗がん剤があるが、安全性の高い抗がん剤として不飽和脂肪酸を有効成分とする抗腫瘍剤の提案がある(特許文献1、特許文献2参照)。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開2003−171272号公報
【特許文献2】特開2005−247754号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明は、生体内脂質由来の低分子化合物で飽和脂肪酸であるパルミチン酸の誘導体を有効成分とする、抗がん活性に優れ、安全性の高い新規な抗がん剤を提供することを課題とする。
【課題を解決するための手段】
【0006】
上記の課題を解決する本発明は、下記の一般式(1)で表されるパルミチン酸誘導体又は薬理的に許容されるその塩を有効成分として含有することを特徴とする抗がん剤を要旨とする。
【0007】
【化1】
(式中、R、R’は水素原子又は低級アルキル基を表す)
【0008】
本発明は、上記の発明において、下記の一般式(2)で表されるパルミチン酸誘導体又は薬理的に許容されるその塩を有効成分として含有することを特徴とする抗がん剤を要旨とする。
【0009】
【化2】
【0010】
本発明は、上記の発明において、下記の一般式(3)で表されるパルミチン酸誘導体の塩酸塩を有効成分として含有することを特徴とする抗がん剤を要旨とする。
【0011】
【化3】
【発明の効果】
【0012】
本発明の抗がん剤は、抗がん活性に優れ安全性が高いので、投与を受けた患者のQOLの低下を防止し、がんを効果的に治療できる医薬、またがんの予防、再発を抑制できる医薬として有用である。
【図面の簡単な説明】
【0013】
【図1】一般式(3)で表されるパルミチン酸誘導体及び5-フルオロウラシルをヒト大腸がん細胞株HT29に暴露し、がん細胞生存率を測定した結果に基づき作成された増殖曲線(用量反応曲線)である。
【図2】一般式(3)で表されるパルミチン酸誘導体をヒト大腸正常細胞株FHC及びヒト大腸がん細胞株HT29に暴露し、各細胞生存率を測定した結果に基づき作成された増殖曲線(用量反応曲線)である。
【発明を実施するための形態】
【0014】
上記の一般式(1)で表されるパルミチン酸誘導体において、R、R’が低級アルキル基である場合、任意の位置に置換できる低級アルキル基としては、メチル基、エチル基、プロピル基、イソプロピル基、ブチル基、tert-ブチル基、ペンチル基、ヘキシル基等の炭素数1〜6の直鎖又は分枝鎖状のアルキル基を例示できる。
【0015】
上記の一般式(1)で表されるパルミチン酸誘導体において、R、R’が水素原子である場合、パルミチン酸誘導体は上記の一般式(2)で表され、1-パルミトイル-4-ピペリジノピペリジン(1-Palmitoyl-4-piperidinopiperidine)(以下、「化合物1」ということがある)である。また、当該パルミチン酸誘導体が塩酸塩である場合、パルミチン酸誘導体は一般式(3)で表され、1-パルミトイル-4-ピペリジノピペリジン塩酸塩(1-Palmitoyl-4-piperidinopiperidine hydrochloride)(以下、「化合物2」ということがある)である。
【0016】
上記の一般式(1)で表されるパルミチン酸誘導体は、公知のアミドの合成法により製造できる。例えば、パルミチン酸とアミン成分の4-ピペリジノピペリジン又は低級アルキル基で置換された4-ピペリジノピペリジンをカルボジイミドなどの脱水縮合剤の存在下に反応させることにより製造できる。また、パルミチン酸ハロゲン化物、パルミチン酸無水物又はパルミチン酸エステルとアミン成分の4-ピペリジノピペリジン又は低級アルキル基で置換された4-ピペリジノピペリジンと反応させることにより製造できる。反応終了後に抽出、分配、カラムクロマトグラフィー等の公知の分離、精製手段を用いて単離することができる。
【0017】
上記の一般式(3)で表される化合物2は、パルミチン酸とジメチルホルムアミドとアミン成分の4-ピペリジノピペリジンを脱水縮合剤の存在下に反応させることにより製造できる。反応終了後に抽出、分配、カラムクロマトグラフィー等の公知の分離、精製手段を用いて単離することができる。また、上記の一般式(1)で表されるパルミチン酸誘導体において、R、R’が低級アルキル基であり、パルミチン酸誘導体が塩酸塩である場合、4-ピペリジノピペリジンに代え、低級アルキル基で置換された4-ピペリジノピペリジンを用いる。
【0018】
上記の一般式(1)で表されるパルミチン酸誘導体は、遊離のものでも、また薬理的に許容される塩、プロドラッグとすることもできる。塩は、酸付加塩でも塩基付加塩でも良い。酸付加塩を形成する酸は、リン酸、塩酸、硫酸、硝酸等の無機酸、メタンスルホン酸、クエン酸、フマル酸、コハク酸、酒石酸、シュウ酸、マレイン酸、酢酸、リンゴ酸、乳酸、アスコルビン酸等の有機酸を例示できる。塩基付加塩を形成する塩基は、水酸化アンモニウム、アルカリ金属水酸化物、アルカリ土類金属水酸化物、炭酸塩、炭酸水素塩等の無機塩基、エタノールアミン、トリエチルアミン、トリス(ヒドロキシメチル)アミノメタン等の有機塩基を例示できる。上記の一般式(1)で表されるパルミチン酸誘導体は、溶媒和物とすることができる。このような溶媒和物としては、水和物、アルコール和物などを例示できる。
【0019】
上記の一般式(1)で表されるパルミチン酸誘導体は、大腸がん、胃がん、食道がん、結腸がん、肝臓がん、膵臓がん、乳がん、肺がん、胆嚢がん、胆管がん、胆道がん、直腸がん、卵巣がん、子宮がん、腎がん、膀胱がん、前立腺がん、骨肉腫、脳腫瘍、白血病、筋肉腫、皮膚がん、悪性黒色腫、悪性リンパ腫、舌がん、骨髄腫、甲状腺がん、皮膚転移がん、皮膚黒色腫などの治療に用いることができるがこれらに限定されず、また前がん病変の治療に用いることもできる。
【0020】
上記の一般式(1)で表されるパルミチン酸誘導体の抗がん剤としての投与形態は、特に限定されず、経口又は非経口のいずれの投与形態でもよい。また、投与形態に応じて適当な剤形とすることができ、例えば注射剤、カプセル剤、錠剤、顆粒剤、散剤、丸剤、細粒剤などの経口剤、直腸投与剤、油脂性坐剤、水性坐剤などの各種製剤に調製することができる。
【0021】
上記の各種製剤は、薬理的に許容され通常用いられる添加剤、例えば賦形剤、結合剤、滑沢剤、崩壊剤、界面活性剤、流動性促進剤などを適宜添加して調製できる。賦形剤として、例えば、乳糖、果糖、ブドウ糖、コーンスターチ、ソルビット、結晶セルロースなどが、結合剤として、例えば、メチルセルロース、エチルセルロース、アラビアゴム、ゼラチン、ヒドロキシプロピルセルロース、ポリビニルピロリドンなどが、滑沢剤として、例えば、タルク、ステアリン酸マグネシウム、ポリエチレングリコール、硬化植物油などが、崩壊剤として、例えば、澱粉、アルギン酸ナトリウム、ゼラチン、炭酸カルシウム、クエン酸カルシウム、デキストリン、炭酸マグネシウム、合成ケイ酸マグネシウムなどが、界面活性剤として、例えば、ラウリル硫酸ナトリウム、大豆レシチン、ショ糖脂肪酸エステル、ポリソルベート80などが、流動性促進剤として、例えば、軽質無水ケイ酸、乾燥水酸化アルミニウムゲル、合成ケイ酸アルミニウム、ケイ酸マグネシウムなどが、その他の添加剤としては、シロップ、ワセリン、グリセリン、エタノール、プロピレングリコール、クエン酸、塩化ナトリウム、亜硝酸ソーダ、リン酸ナトリウムなどが挙げられる。
【0022】
上記の一般式(1)で表されるパルミチン酸誘導体の投与量は、用法、患者の年齢、性別、症状の程度などを考慮して適宜増減できるが、通常成人1日当り200〜400mg好ましくは100〜200mgで、これを1日1回又は数回に分けて投与できる。
【実施例】
【0023】
次いで、本発明を実施例を挙げて説明するが、本発明は以下の実施例に限定されるものではない。
【0024】
〔実施例1〕(化合物1の合成)
パルミチン酸 (2.6 g, 10 mmol、和光純薬工業社) を塩化チオニル (5mL、和光純薬工業社)と2時間還流した後、過剰の塩化チオニルを留去し(減圧下)、この中に、4-ピペリジノピペリジン (1.7 g, 10 mmol、和光純薬工業社) を含むテトラヒドロフラン溶液を加え、3時間還流した。反応液は炭酸水素ナトリウム溶液に滴下し、酢酸エチルで分液した。酢酸エチル層は濃縮後、シリカゲルカラムクロマトグラフィー(展開溶媒:クロロホルム−メタノール 10:1)で精製した。化合物1は、淡褐色の結晶となって固化した(融点:低すぎて測定不能)。
【0025】
C26H50N2O MW 406; EIMS m/z (rel. int.): 406 [M]+ (22), 167 (44), 124 (100), 84 (29); 1H-NMR (500 MHz, CDCl3) δ:0.86 (3H, t, J = 5.7 Hz), 1.23 (22H, br s), 1.35-1.47 (4H, m), 1.55-1.63(7H, m), 1.84 (1H, d, J= 12.6 Hz), 2.56-2.63 (6H, m), 2.97 (1H, t, J = 12.6 Hz), 3.89 (1H, d, J= 13.2 Hz), 4.69 (1H, d, J = 13.2 Hz).
【0026】
〔実施例2〕(化合物2の合成)
パルミチン酸 (2.6 g, 10 mmol、和光純薬工業社) と1-ヒドロキシベンゾトリアゾール (1.4 g, 11 mmol、和光純薬工業社) をジメチルホルムアミド40 mlに溶解し、4-ピペリジノピペリジン (1.7 g, 11 mmol、和光純薬工業社) を加え、氷浴上で15分間撹拌した(A液)。別に縮合剤である1−エチル−3−(3−ジメチルアミノプロピル)−カルボジイミド(和光純薬工業社)を20 mlのジメチルホルムアミドに溶解した (B液)。A液を氷浴上で撹拌しながらB液を滴下した。氷浴上で2時間、次いで室温にて12時間撹拌を続け、下記の処理を行った。
(処理法)
反応生成物にクロロホルム50 mlを加え、次の (1)および(2) の溶液と分液操作を行った。
(1) 1.0%塩酸溶液(2回)、(2) 飽和食塩水(2回)
得られたクロロホルム層は濃縮後、シリカゲルカラムクロマトグラフィー(展開溶媒:ヘキサン−酢酸エチル混合液)にて精製し、化合物2を得た。
【0027】
mp. 189℃; EIMS m/z (rel. int.): 406 [M]+ (42), 377 (12), 322 (11), 167 (46), 124 (100), 110 (20), 84 (30); 1H-NMR (400 MHz, CDCl3) δ: 0.88 (3H, t, J = 6.8 Hz), 1.25 (26H, br s), 1.60-1.69 (5H, m), 2.17 (2H, m), 2.30 (2H, dd, J = 8.8, 7.8 Hz), 2.53 (2H, m), 3.08 (2H, m), 3.26 (2H, m), 4.01 (1H, m), 4.86 (1H, m), 2.00-3.40 (br s).
【0028】
〔実施例3〕(化合物2の抗がん活性評価)
1.試薬の調製
化合物2は、実施例2で製造したものを蒸留水に溶かし、抗がん活性の測定に使用した。ギムザ染色液(Sigma社: Cat. No. S128-4L)の25 mLを蒸留水475 mLにて希釈して使用した。大腸がん治療に使用される代表的な抗がん剤の5-フルオロウラシル(5-fluorouracil)(以下、「5-FU」ということがある)を抗がん活性の比較対象とする薬剤とした。5-FUは、ジメチルスルホキシド(DMSO)(Sigma 社: Cat. No. D8418-250ML)に溶かして使用した。
2.ヒトがん細胞株の培養
ヒト大腸がん細胞株HT29(American Type Culture Collection(ATCC))とヒト大腸正常細胞株FHC(American Type Culture Collection(ATCC))をそれぞれ10 %ウシ胎児血清(FBS) (BioWest社: Cat. No. S1500)添加DMEM培地 (和光純薬工業社: Cat. No. 041-29775)中で37℃、5%CO2条件下にて培養した。培養装置は、Napco 5400 CO2インキュベーター(Napco社)を使用した。
【0029】
3.(化合物2のヒト大腸がん細胞株HT29に対する抗がん活性評価)
ヒト大腸がん細胞株HT29を6ウエル・プレート(日本ベクトン・デイッキンソン社、BD Falcon(TM): Cat. No. 353046)に500細胞/ウエルの濃度で蒔いて一晩培養した後、化合物2と5-FUをそれぞれ0、0.3、0.7、1.5、3.0、6.0、12.0、25.0μMの濃度でがん細胞に暴露し、5日間培養した(0μMの培地には、化合物2は蒸留水を、5-FUはDMSOを0.1%になるようにそれぞれ添加した)。培養後、各ウエルをリン酸緩衝生理食塩水(phosphate buffered saline 、以下「PBS」と略す)で2回洗浄し、各ウエル当たり2mLの100%メタノールで10分間、がん細胞を固定した。風乾し、各ウエル当たり2mLのギムザ染色液で30分間染色した後水道水で洗浄した。風乾後、染色されたがん細胞のコロニー数を目視にて計測した。
【0030】
上記のヒト大腸がん細胞株HT29を用いた細胞増殖アッセイの結果に基づき図1に示す増殖曲線(用量反応曲線)を作成した。化合物2のIC50値(50%増殖抑制率)を測定し、これを抗がん活性の指標とした。図1のグラフの縦軸は、がん細胞生存率(%)とし、横軸は化合物2の濃度(μM)とした。化合物2の濃度が0μMのときの細胞生存率を100%として、化合物2の各濃度での細胞生存率を換算した。その結果、化合物2の大腸がん細胞株HT29株に対するIC50値は、0.74μMであった。一方、5-FUのIC50値は、9μMであった。図1より化合物2の濃度が3μMのとき、がん細胞を100%死滅させているが、同じ濃度の5-FUでは全てのがん細胞が生存している。化合物2のIC50値は、5-FUと比較して12倍小さい値である。これは、化合物2が5-FUに比べてより低濃度で抗がん活性を発揮することを示す。このことにより、化合物2は、大腸がんの化学療法に使用される第1選択の抗がん剤である5-FUより強い抗がん活性を有することが示唆された。
【0031】
4.(化合物2のヒト大腸正常細胞株FHCとヒト大腸がん細胞株HT29に対する毒性の比較)
化合物2のヒト大腸正常細胞株FHCとヒト大腸がん細胞株HT29に対する毒性を検討した。各細胞株をそれぞれ6cmディッシュ (Nalge Nunc International社: Cat. No. 150288)に2x104細胞/ディッシュの濃度で蒔き一晩培養した後、各細胞株に対して化合物2を0、0.25、1.0μMの濃度で暴露し、3日間培養した(0μMの培地には、蒸留水を0.1%になるように添加した)。培養後、培地を除去し0.25%トリプシン-EDTA溶液(Sigma 社:Cat. No. T4049)1 mLを加え10分間37℃でインキュベーションした後、細胞をディッシュから剥がした。剥がした細胞に3mLの1xPBSを添加し細胞をよくほぐした後Coulter Counter Z1 (Becton Dickinson社)にて細胞数を測定した。
【0032】
上記の細胞増殖アッセイの結果に基づき図2に示す増殖曲線を作成した。化合物2の曝露下でのヒト大腸がん細胞株HT29及びヒト大腸正常細胞株FHCに対する生存率を測定し、これを正常細胞に対する毒性評価の指針とした。図2のグラフの縦軸は、各細胞生存率
(%)とし、横軸は化合物2の濃度(μM)とした。図2のグラフは、化合物2の濃度が0μMのときの細胞生存率を100%として、化合物2の各濃度の細胞生存率を換算した。その結果、ヒト大腸がん細胞株HT29の細胞が60%死滅した時の化合物2の濃度は1μMであり、この濃度における大腸正常細胞株FHCの細胞生存率は85%であった。このことにより、化合物2は、1μMまでの抗がん活性を呈する濃度において正常細胞に対する毒性は極めて少ないことが示唆された。
【産業上の利用可能性】
【0033】
本発明は、強力な抗がん活性を発揮し、毒性の少ない新規な抗がん剤とし医薬品工業上の有用性が期待される。
【特許請求の範囲】
【請求項1】
下記の一般式(1)で表されるパルミチン酸誘導体又は薬理的に許容されるその塩を有効成分として含有することを特徴とする抗がん剤。
【化1】
(式中、R、R’は水素原子又は低級アルキル基を表す)
【請求項2】
下記の一般式(2)で表されるパルミチン酸誘導体又は薬理的に許容されるその塩を有効成分として含有することを特徴とする請求項1に記載の抗がん剤。
【化2】
【請求項3】
下記の一般式(3)で表されるパルミチン酸誘導体の塩酸塩を有効成分として含有することを特徴とする請求項2に記載の抗がん剤。
【化3】
【請求項1】
下記の一般式(1)で表されるパルミチン酸誘導体又は薬理的に許容されるその塩を有効成分として含有することを特徴とする抗がん剤。
【化1】
(式中、R、R’は水素原子又は低級アルキル基を表す)
【請求項2】
下記の一般式(2)で表されるパルミチン酸誘導体又は薬理的に許容されるその塩を有効成分として含有することを特徴とする請求項1に記載の抗がん剤。
【化2】
【請求項3】
下記の一般式(3)で表されるパルミチン酸誘導体の塩酸塩を有効成分として含有することを特徴とする請求項2に記載の抗がん剤。
【化3】
【図1】

【図2】
【図2】
【公開番号】特開2011−207855(P2011−207855A)
【公開日】平成23年10月20日(2011.10.20)
【国際特許分類】
【出願番号】特願2010−79755(P2010−79755)
【出願日】平成22年3月30日(2010.3.30)
【出願人】(310022501)
【出願人】(597018325)
【Fターム(参考)】
【公開日】平成23年10月20日(2011.10.20)
【国際特許分類】
【出願日】平成22年3月30日(2010.3.30)
【出願人】(310022501)
【出願人】(597018325)
【Fターム(参考)】
[ Back to top ]