
抗ウイルスペプチドのシステイン酸誘導体
本発明は、ウイルス感染の阻害剤であり、および/または抗膜融合特性を示す、改善された水溶性を有しているC34ペプチド誘導体に関する。具体的には、本発明は、ヒト免疫不全ウイルス(HIV)、RSウイルス(RSV)、ヒトパラインフルエンザウイルス(HPV)、麻疹ウイルス(MeV)、およびサル免疫不全ウイルス(SIV)のそれぞれのウイルス感染を処置するための長期作用を持つ、これらのウイルスに対する阻害活性を有するC34誘導体に関する。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願への相互参照
この出願は、共に2007年5月16日に出願されたU.S.第60/938,380号およびU.S.第60/938,394号への優先権を主張する。上記の出願の内容は、それらの全体が参考として本明細書に援用される。
【背景技術】
【0002】
発明の背景
非感染細胞への1型ヒト免疫不全ウイルス(HIV−1)の進入には主に3つの工程が含まれる:(i)gp120のCD4受容体に対する結合、(ii)それに続く共受容体CXCR4またはCCR5に対する結合、そして(iii)HIV−1膜貫通糖タンパク質gp41の細胞外ドメインの一連の立体構造の変化(これは、最終的に感染を起こさせる膜融合事象を誘発するために重要である)。RSウイルス(RSV)、ヒト3型パラインフルエンザウイルス(HPIV−3)、麻疹ウイルス、およびサル免疫不全ウイルス(SIV)のようなウイルスは、gp41様タンパク質を含むHIVと高度な構造的類似性および機能的類似性を示す。
【0003】
いくつかの低分子薬物候補(CD4またはCCR5共受容体に対する結合を阻害するものを含む)は、ヒトの臨床試験の段階にあるか、または販売の認可が近いかのいずれかにある(非特許文献1;非特許文献2)。いくつかの合成のペプチドは、膜融合に関連する事象(非感染細胞へのレトロウイルスの感染の阻害を含む)を阻害するか、あるいは壊すことが知られている。例えば、合成ペプチドC34、T1249、DP−107、およびT−20(DP−178)(これらは、gp41中の異なるドメインに由来する)はHIV−1感染およびHIV−1に誘導される細胞と細胞の融合の強力な阻害剤である。
【0004】
T−20(DP−178、エンフュービルタイド、Fuzeon(登録商標)、Trimeris/Roche Applied Sciences)は、HIV−1 gp41のCHR配列をベースとする合成ペプチドであり、gp41の立体構造の再編成を標的化すると考えられている。T−20の阻害は、6ヘリックスの束状構造の形成の阻害を生じるgp41のNHR領域の疎水性溝に結合するその能力に起因すると広く考えられている(非特許文献3)。この考えとは対照的に、最近の研究では、T−20がgp41およびgp120中の複数の部位を標的化できることが示唆された(非特許文献4)。例えば、T−20は膜の表面に結合してオリゴマーを形成し、それによって感染細胞の血漿膜でのgp41の動員とオリゴマー形成を阻害する(非特許文献5;非特許文献6)。さらに、ペプチド配列666WASLWNWF673を有している膜貫通ドメインにすぐ隣接している領域内にあるgp41の細胞外ドメインが、gp41のNHRよりもT−20に対してより親和性の高い部位を構成することもまた示されている(非特許文献5;非特許文献6)。
【0005】
T−20と重複しているがgp41のコイルドコイルキャビティ結合残基(colied−coil cavity binding residues)628WMEW631を含むペプチド配列からなる別のC−ペプチドC34は、NHR領域の疎水性溝についてgp41のCHRと競合することが公知である(非特許文献4)。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】Meanwell NA,Kadow JF、Curr Opinion Drug Disc & Develop(2003)6:451−461
【非特許文献2】Olson WC,Maddon P J、Curr Drug Targets−Infectious Disord(2003)3:283−294
【非特許文献3】Kliger Y,Shai Y、J Mol Biol(2000)295:163−168
【非特許文献4】Liu Sら、J Biol Chem(2005)280:11259−11273
【非特許文献5】Munoz−Barroso Iら、J Cell Biol(1998)140:315−23
【非特許文献6】Kliger Yら、J Biol Chem(2001)276:1391−1397
【発明の概要】
【発明が解決しようとする課題】
【0007】
当該分野で記載されている抗ウイルスペプチドまたは抗膜融合性ペプチドの多くは、強力な抗ウイルス活性および/または抗膜融合活性を示すが、これらのペプチドは生理学的pHの水性処方物中での溶解度は低く、さらにインビボでの血漿半減期も短い。したがって、既存の抗ウイルスペプチドおよび/または抗膜融合性ペプチドの溶解度を増大させ、そして半減期を延長させ、それにより水溶性であり、インビボでの作用期間の長い抗ウイルスペプチドおよび/または抗膜融合性ペプチドを提供するための方法が必要である。
【課題を解決するための手段】
【0008】
本発明は、改変前のペプチドと比較して生理学的pHの水溶液中で高い溶解度を有する改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドに、少なくとも一部関係する。1つの実施形態においては、本発明のペプチドは、1つ以上の極性基または極性部分(例えば、1つ以上のシステイン酸)を含むように改変され、それによって水溶液中でのそれらの溶解度が増大する。改変されたペプチドにはさらに、改変されたペプチドが血液成分または担体タンパク質(例えば、アルブミン(例えば、ヒト血清アルブミンまたは組み換え体アルブミン))上の利用できる官能基と反応できるように化学反応性部分を含めることができ、それによって改変されたペプチドのインビボでの安定性が増大する。複数の実施形態においては、改変されたペプチドは、血液成分または担体タンパク質(例えば、アルブミン(例えば、ヒト血清アルブミン、組み換え体アルブミン、または他の担体タンパク質))に結合させられる。これらの改変されたペプチドまたはそれらの結合体は、それによって、例えば、より頻繁に、またはさらには持続的にペプチドを投与する必要性を低減する。本発明の改変されたペプチドは、多数のウイルス(ヒト免疫不全ウイルス(HIV)、ヒトRSウイルス(RSV)、ヒトパラインフルエンザウイルス(HPIV)、麻疹ウイルス(MeV)およびサル免疫不全ウイルス(SIV)を含む)の感染を緩和するために、例えば、予防的および/または治療的に使用することができる。ウイルストランスフェクション(例えば、肝炎ウイルス、エプスタイン−バーウイルスおよび他の関連するウイルス)に関与する他のペプチドの改変もまた、本発明の範囲に含まれる。
【0009】
したがって、本発明の1つの態様においては、改変前のペプチドと比較して、約5から8までの範囲のpH(例えば、生理学的pH)で水溶液または水中で高い溶解度を有する、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドを特徴とする。1つの実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドは、濃縮された水溶液(例えば、水溶液(例えば、等張性または高塩濃度の水溶液)中で約10mg/mlから500mg/ml、約10mg/mlから400mg/ml、約10mg/mlから300mg/ml、約10mg/mlから200mg/ml、約10mg/mlから180mg/ml、約40mg/mlから180mg/ml、約60mg/mlから180mg/ml、または約90mg/mlから100mg/mlの範囲の濃度)中で実質的に溶解したままである(例えば、約5から8までの範囲のpH(例えば、生理学的pH)では、約40%未満、30%、20%、10%が水または水溶液中に沈殿する)。複数の実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドは、改変前のペプチドよりも少なくとも約1.3倍、1.5倍、1.8倍、2倍、2.3倍、2.5倍、2.8倍、3倍、または3.5倍大きい溶解限度(すなわち、透明な溶液を維持する最大濃度)を示す。複数の実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドは、約5から8までの範囲のpHで、等張性水溶液中で少なくとも約20mg/ml、25mg/ml、30mg/ml、35mg/ml、または40mg/mlの溶解限度を有する。「水溶液」としては、本明細書中で使用される場合は、水、生理食塩溶液(例えば、等張性の溶液)、水の中に作られた緩衝液(例えば、リン酸ナトリウム緩衝液)、水性ゲル、および対象(例えば、ヒト対象)への投与(例えば、皮下投与、静脈内投与、肺投与、筋肉内投与、もしくは腹腔内投与)に適しているpHの水性処方物;あるいは製造プロセスに適しているpHの処方物が挙げられるが、これらに限定されない。
【0010】
複数の実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドには、1つ以上の極性部分が含まれる。1つの実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドには1つ以上の極性部分が含まれ、これは、生理学的pHで電荷を持つか、または生理学的pHでは電荷を持たないかのいずれかである。いくつかの実施形態においては、側鎖は中性であり得、例えば、水素結合または他の非共有相互作用によって水溶液中での改変されたペプチドの全体的な溶解度を増大させることができる。例えば、特定の例においては、酸素基または窒素基を持つ中性の側鎖はバルク溶媒に水素結合することができ、ペプチドの全体的な溶解度を増大させるために使用することができる。いくつかの実施形態においては、側鎖は任意の自然界には存在しない極性の側鎖または中性の側鎖(例えば、20種類の自然界に存在しているアミノ酸の中には見られない側鎖)であり得る。
【0011】
複数の実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドの極性部分には以下の構造が含まれる:
【0012】
【化1】

例えば、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドには、1つ以上のシステイン酸を含めることができる。複数の実施形態においては、システイン酸は以下の構造を有する:
【0013】
【化2】
本明細書中に開示されるペプチドの溶解度を増大させることができるさらなる適切な側鎖は、本発明の開示の利点を前提として、当業者によって容易に選択されるであろう。
【0014】
他の実施形態においては、1つ以上の極性部分(例えば、システイン酸)が抗ウイルスペプチドおよび/または抗膜融合性ペプチドのN末端あるいはC末端に付加される。他の実施形態においては、1つ以上の極性部分が、抗ウイルスペプチドおよび/または抗膜融合性ペプチドの内部配列に付加される。
【0015】
1つの実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドには、gp41のコイルドコイルキャビティ結合残基の少なくとも一部が含まれる。例えば、ペプチドには、残基628WMEW631(配列番号1)、またはそれらに対する1つまでのアミノ酸置換(例えば、保存的置換または非保存的置換)あるいは付加を有しているアミノ酸配列が含まれ得る。他の実施形態においては、抗ウイルスペプチドおよび/または抗膜融合性ペプチドには、アミノ酸配列628WMEWDREINNYTSLIHSLIEESQNQQEKNEQELL661(アミノ酸C1からC34に相当する)(配列番号2)、あるいはそれらに対して5個まで、4個、3個、2個、または1個のアミノ酸置換(例えば、保存的置換もしくは非保存的置換)、欠失、または付加を持つアミノ酸配列に由来する、天然のC34のアミノ酸配列全体あるいは一部が含まれる。
【0016】
他の実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドには、DP107ペプチドおよびDP178ペプチド、ならびにそれらのアナログのアミノ酸配列が含まれる。これには、DP107およびDP178が由来するHIVのgp41領域に相当し、抗ウイルス活性および/または抗膜融合活性を示す他の(非HIV)ウイルスに由来するアミノ酸配列からなるペプチドが含まれる。さらに具体的には、これらのペプチドは、特に、ヒトRSウイルス(RSV)、ヒトパラインフルエンザウイルス(HPV)、麻疹ウイルス(MeV)、およびサル免疫不全ウイルス(SIV)に対して抗ウイルス活性を示すことができる。本発明はまた、引用により本明細書中に具体的に組み入れられるUS05/0070475の配列番号1から配列番号86の改変されたペプチドにも関する。
【0017】
複数の実施形態においては、本発明の改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドにはさらに、改変されたペプチドが血液成分または担体タンパク質上の利用できる官能基と反応して安定な共有結合を形成できるように1つ以上の化学反応性部分または基が含まれ得、それによって結合したペプチド形態が生じる。1つの実施形態においては、改変されたペプチドには、1つ以上の血液成分(例えば、アルブミン)上の1つ以上のアミノ基、ヒドロキシル基、またはチオール基と反応して安定な共有結合を形成する1つ以上の反応性基が含まれる。例えば、ペプチド反応性基アルブミン結合体は、約1:1のアルブミンに対するペプチドのモル比であり得る。通常、結合は、反応性基とアルブミン(例えば、ヒトアルブミン)のアミノ酸34(Cys34)との間での共有結合によって起こる。
【0018】
別の実施形態においては、反応性基はマレイミド含有基(例えば、MPA(マレイミドプロピオン酸)またはGMBA(γ−マレイミド−ブチララミド))(これは、血液タンパク質(移動性の血液タンパク質(例えば、アルブミン)を含む)上のチオール基と反応する)であり得る。反応性の改変または反応性基にはさらに、1つ以上のリンカーを含めることができる。複数の実施形態においては、リンカーは、(2−アミノ)エトキシ酢酸(AEA)、[2−(2−アミノ)エトキシ)]エトキシ酢酸(AEEA)、エチレンジアミン(EDA)のうちの1つ以上;アルキル鎖(C1〜C10)、例えば、8−アミノオクタン酸(AOA)、8−アミノプロパン酸(APA)、または4−アミノ安息香酸(APhA)の1つ以上から選択される。リンカーを持つ反応性基、またはリンカーを持たない反応性基を、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドのN末端あるいはC末端に、通常は、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドのC末端に付加することができる。他の実施形態においては、反応性基は、改変されたペプチドの内部残基に結合させられる(例えば、内部リジン残基のεNH2基;内部セリン残基(例えば、C34のセリン13)のヒドロキシル基に結合させられる)。C34の改変されたペプチドの限定ではない例は、WO02/096935(その全体が引用により本明細書中に組み入れられる)に開示されている。
【0019】
通常は、1つ以上の極性部分(例えば、システイン酸)が改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドの一方の末端(例えば、N末端)に付加されれば、反応性基は反対側の末端(例えば、C末端)に付加される。例えば、改変されたペプチドは以下の立体配置のうちの1つを有し得る:
[極性部分(例えば、システイン酸)−改変されたペプチド−リンカーn−反応性基](VI);または
[反応性基−リンカーn−改変されたペプチド−極性部分(例えば、システイン酸)](VII)。
式中、反応性基は、例えば、リンカーを持つマレイミド含有基またはリンカーを持たないマレイミド含有基であり得、例えば、nは、0、1、2、3、4、またはそれ以上のリンカーであり得る。1つ以上のリンカーが存在する場合は、リンカーは同じである(例えば、AEEA−AEEA)場合も、また異なる(例えば、AEEA−EDAもしくはAEA−AEEA)場合もある。
【0020】
特定の複数の実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドに含められるさらなる基は以下の式(I)を有する化合物であり得る。
【0021】
(VIII)(R1)m−X−(R2)n
式(VIII)中では、mとnの和は少なくとも1であり、mとnはそれぞれが0以上の整数である。例えば、mが0である場合は、nは1以上であり、nが0である場合は、mは1以上である。Xは、抗ウイルスペプチドおよび/または抗膜融合性ペプチド、例えば、C34、T20、T1249、あるいは、それらのアナログまたは誘導体(例えば、それらのマレイミド誘導体を含む)である。R1が存在し、R2が存在しない場合は、R1はX基のN末端に存在する。R1が存在せず、R2が存在する場合は、R2はX基のC末端に存在する。
【0022】
特定の例においては、R1とR2はそれぞれ独立して、式(IX)を有する化合物から選択され得る。
【0023】
【化3】
式(IX)のコア構造はアミノ酸のコア構造と類似しており、これにはアミノ基、α炭素、およびカルボキシル基が含まれる。ペプチド誘導体中のR1基とR2基の正確な位置に応じて、複数の基を式(IX)中の様々な原子を介してペプチドに結合させることができる。例えば、R1が式(IX)を有する化合物である場合には、R1を式(IX)のカルボキシル基を介してペプチドに結合させて、R1のカルボキシル基とペプチドのアミノ基との間にペプチド結合をもたらすことができる。R2が式(IX)を有する化合物である場合には、R2を式(IX)のアミノ基を介してペプチドに結合させて、R2のアミノ基とペプチドのカルボキシ基との間にペプチド結合をもたらすことができる。
【0024】
いくつかの実施形態においては、式(IX)のR3基は、20種類の自然界に存在しているアミノ酸の中に一般的に見られる極性の電荷を持たない基以外の、任意の極性の電荷を持たない基であり得る。例えば、R3基は、スルホニル基(HS=(O)2)、スルホキシド基(HS=O)、スルホン酸基(HO−S=(O)2)、ハロアルキル基、二級アミン、三級アミン、ヒドロキシル基、または極性であるかもしくは全くの中性であり、水溶液中でのペプチド誘導体の全体的な可溶性を増大させることができる他の側鎖基であり得るか、あるいは、それらを含み得る。例えば、水素結合することができる基を持つ側鎖は、ペプチドの全体的な溶解度を増大させるために使用され得る。特定の例においては、側鎖は、リンカーまたは他の種との望ましくない副反応が実質的な程度には起こらないように、非反応性であることが好ましい。いくつかの例においては、R3についての上記基はα炭素から間隔をあけて、例えば、1〜3個の炭素原子の間隔をあけて存在し得る。特定の例においては、R3は、式(X)−(XV)を有する化合物が提供されるように選択され得る。
【0025】
【化4】
本明細書中に開示されるペプチドの溶解度を増大させることができるさらなる適切な側鎖は、本開示の利点を前提として、当業者によって容易に選択されるであろう。
【0026】
特定の複数の実施形態においては、R1基とR2基は、ペプチド結合体の二次構造全体に、または特定の場合には三次構造には実質的には影響を及ぼさない。ペプチド結合体の二次構造に実質的には影響を及ぼさないことにより、ペプチド結合体の全体的な活性は、誘導体化されていないペプチドの全体的な活性よりも感知できるほどに下回ることはないはずである。
【0027】
他の実施形態においては、ペプチド誘導体は、式(XVI)に示されるような組成物の形態をとり得る。
【0028】
(XVI) X1−(R1)m−X2−(R2)n
式(XVI)においては、X1とX2は、互いに連結させられると、例えば、C34、T20、もしくはT1249、またはそれらの変異体を提供するであろうペプチドの部分を提示する。式(XVI)においては、R1とR2は、式(IX)に関して上記で議論した基のうちの任意のものであり得、mとnの和は1以上の整数であり、mまたはnのいずれかが0であり得る可能性がある。式(XVI)においては、ペプチド鎖の中央に基が挿入されている。そのような挿入は、ペプチドの酵素消化、それに続くR1基もしくはR2基またはそれらの両方の挿入、次いでペプチド断片を互いに結合させることを含む多くの様々な方法を使用して行われ得る。
【0029】
特定の複数の実施形態においては、本明細書中に開示される化合物は、ペプチドのN末端、C末端に、またはペプチドの1つ以上のアミノ酸の側鎖を介して、1つ以上のさらなる基に連結させられ得る。例えば、式(XVII)−(XX)に模式的に示される組成物が作成され得る。
【0030】
【化5】
式(XVII)−(XX)においては、Lは、例えば、(2−アミノ)エトキシ酢酸(AEA)、エチレンジアミン(EDA)、2−[2−(2−アミノ)エトキシ]エトキシ酢酸(AEAA)、アルキル鎖モチーフ(C1〜C10)(例えば、グリシン)、3−アミノプロピオン酸(APA)、8−アミノオクタン酸(AOA)、4−アミノ安息香酸(APhA)などのようなリンカーであり、R1とR2は、本明細書中で議論された基のうちの任意のものであり得る。リンカーは、ペプチドの任意のアミノ酸を介して、例えば、リジンのアミノ基、チオール基、ペプチドの1つ以上のアミノ酸側鎖残基の中のヒドロキシル基を介してペプチドに対して結合させられ得るか、あるいはペプチドのN末端またはC末端に結合させられ得る。X基、X1基、およびX2基は、ペプチド(X)またはペプチド断片(X1およびX2)である。式(XIX)および式(XX)中に示されるP基は、リンカーLを介して誘導されたペプチドに結合させることができるタンパク質を示す。例示的なタンパク質としては、血液タンパク質または担体タンパク質(例えば、ヒト血清アルブミン、組み換え体アルブミン、免疫グロブリンもしくはその断片、トランスフェリン、または他の適切なタンパク質)が挙げられる。
【0031】
タンパク質結合体(式(XIX)および式(XX))は、エキソビボまたはインビボで生産され得る。インビボでの生産が行われる場合には、化合物(例えば、式(XVII)および式(XVIII)に示されるもの)が対象に導入され得、インビボタンパク質(例えば、アルブミン)と反応させられ得る。
【0032】
本発明の抗ウイルスペプチドおよび/または抗膜融合性ペプチドは、1つ以上のアミノ酸の置換または付加を有し得る。例えば、ペプチドは、1つ以上の保存的置換もしくは非保存的置換を有し得る。特定の複数の実施形態においては、改変されたペプチドにはさらに、1つ以上のアミノ酸残基を含めることができる。例えば、C34の改変されたペプチドは、任意に、28位の本来のリジン(Lys28)のアルギニンでの置換を有することができ、そして/あるいは、Lys残基(またはC末端に本明細書中に記載される反応性基(例えば、AEEA−MPA)に対して直接または間接的に共有結合させられるそのε−窒素原子で修飾されたリジン残基)を付加することができる。基Lys(ε−AEEA−MPA)においては、AEEA−MPAは、リジンのεNH2基に結合させられることが理解されるはずである。
【0033】
本発明のC34の改変された抗ウイルスペプチドおよび/または改変された抗膜融合性ペプチドの限定ではない例としては、以下の配列が挙げられる:
CA化合物I:(C34に直接連結させられたシステイン酸(CA);本明細書中ではCA−C34とも呼ばれる(配列番号3))。
【0034】
【化6】
CA化合物II:(28位に本来のリジン(Lys28)のアルギニンでの置換を有しているC34に直接連結させられたシステイン酸(CA);本明細書中ではCA−C34(Arg28)とも呼ばれる(配列番号4))。
【0035】
【化7】
CA化合物III:(35位にさらなるリジン残基(Lys35)を有しているC34に直接連結させられたシステイン酸(CA)、ここでは、リジンのεNH2基はリンカー(AEEA−MPA)を介して反応性基に結合させられる;本明細書中ではCA−C34−Lys35(ε−AEEA−MPA)とも呼ばれる(配列番号5))。
【0036】
【化8】
および
CA化合物IV:(28位に本来のリジン(Lys28)のアルギニンでの置換;35位にさらなるリジン残基(Lys35)を有しているC34に直接連結させられたシステイン酸(CA)、ここでは、リジンのεNH2基はリンカー(AEEA−MPA)を介して反応性基に結合させられる;本明細書中ではCA−C34(Arg28)−Lys35(ε−AEEA−MPA)とも呼ばれる(配列番号6))。
【0037】
【化9】
なお別の態様においては、本発明は、1つ以上の血液成分上の利用できる官能基に結合させられた1つ以上の化学反応性の改変を有している、本明細書中に記載される改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドの結合体を特徴とする。本発明の1つの実施形態においては、改変されたペプチドには、血液成分上のアミノ基、ヒドロキシル基、またはチオール基に結合させられて安定な共有結合を形成する反応性基が含まれる。マレイミド基は改変されたペプチドに直接結合させることができ、また、例えばリンカー(例えば、本明細書中に記載されるようなリンカー)を介して間接的に結合させることもできる。本発明の別の実施形態においては、反応性基は血液タンパク質(アルブミンのような移動性の血液タンパク質を含む)上のチオール基と反応するマレイミドであり得る。ペプチド−反応性基アルブミン結合体は、約1:1のアルブミンに対するペプチドのモル比であり得る。通常、結合は、反応性基とヒトアルブミンのアミノ酸34(Cys34)との間での共有結合によって起こる。
【0038】
改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドには、結合体を形成するように1つ以上の血液成分(例えば、血清アルブミン)に共有結合する能力を有している反応性部分(例えば、マレイミド含有基)が含まれ得る。結合工程は、インビボで、例えば、対象に対する改変されたペプチドの投与後に起こり得る。あるいは、結合工程は、エキソビボまたはインビトロで、例えば、反応性基を含む改変されたペプチドが血液成分(例えば、アルブミン)と接触させられることによって起こり得る。C34、DP107、DP178などの結合体の調製と使用は、それらの全体が引用により本明細書中に組み入れられるWO02/096935およびUS05/0070475に開示されている。インビボまたはエキソビボで形成させられた結合体は、対象(例えば、ヒト対象)においてHIV、RSV、HPV、MeV、またはSIVのようなウイルスのウイルス活性および/または膜融合性活性を阻害することにおいて有用である。
【0039】
別の態様においては、本発明は、本明細書中に記載される抗ウイルスペプチドおよび/または抗膜融合性ペプチドあるいは改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドと薬学的に許容される担体を含む組成物(例えば、薬学的組成物)を特徴とする。複数の実施形態においては、組成物は、注射(例えば、皮下注射または血管内注射)、ならびに肺送達、筋肉内送達、および/または腹腔内送達に適している。他の実施形態においては、組成物は製造プロセスに適している。
【0040】
他の実施形態においては、組成物は濃縮され、例えば、約5から8までの範囲のpHの水溶液(例えば、等張性水溶液または高塩濃度の水溶液)中で、約10mg/mlから500mg/ml、約10mg/mlから400mg/ml、約10mg/mlから300mg/ml、約10mg/mlから200mg/ml、約10mg/mlから180mg/ml、約40mg/mlから150mg/ml、約60mg/mlから125mg/ml、または約90mg/mlから100mg/mlの濃度である。
【0041】
別の態様においては、本発明は、本明細書中に記載される改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドあるいはそれらの結合体を含む、ウイルス感染の予防および/または処置に使用される方法と組成物を特徴とする。この方法には、ウイルス感染に伴う1つ以上の症状を軽減するために、本明細書中に記載される改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチド、あるいはそれらの結合体の有効量(例えば、予防量または治療量)を処置が必要な対象(例えば、ヒト対象)に投与する工程が含まれる。処置または予防することができる例示的なウイルス感染としては、AIDS、ヒトRSウイルス(RSV)、ヒトパラインフルエンザウイルス(HPV)、麻疹ウイルス(MeV)、およびサル免疫不全ウイルス(SIV)が挙げられる。したがって、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチド、あるいはそれらの結合体を使用してウイルスが関係している疾患または障害の1つ以上の症状を軽減する、または阻害する、あるいはその発症を防ぐ、または遅らせるための方法が開示される。予防的(例えば、疾患もしくは障害の1つ以上の症状の発症または再発を防ぐ、軽減する、あるいは遅らせるための)使用の場合には、対象は、疾患または障害の1つ以上の症状を有している場合も、また有していない場合もある。例えば、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドあるいはそれらの結合体は、何らかの検出可能な症状の発現の前に、または全てではなく少なくとも一部の症状が検出された後に投与することができる。治療的使用の場合には、処置によって、対象の疾患または障害が改善する、治癒する、維持される、またはその期間が短くなり得る。治療的使用においては、対象は、症状の一部が発現している場合も、また症状が完全に発現している場合もある。典型的な場合には、処置によって、医師が検出可能な程度に対象の疾患または障害が改善するか、あるいは、疾患または障害の悪化が防がれる。
【0042】
対象(例えば、ヒト対象)においてHIV、RSV、HPV、MeV、またはSIVの1つ以上の活性を阻害するための方法と組成物が開示される。この方法には、本明細書中に記載される改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチド、あるいはそれらの結合体の有効量(例えば、予防量または治療量)を処置が必要な対象に投与する工程が含まれる。
【0043】
本発明の改変されたペプチドはまた、精製と製造プロセスを容易にすることにおいても有用である。なぜなら、改変されたペプチドの高い溶解度によってより濃縮された反応溶液が可能となり、ひいては大規模な製造プロセスが容易となるからである。したがって、本発明はまた、抗ウイルスペプチドおよび/または抗膜融合性ペプチドの溶解度を増大させるための方法も特徴とする。この方法には、1つ以上の極性部分(例えば、1つ以上のシステイン酸)を含む改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチド(例えば、本明細書中に記載される改変されたペプチド)を提供する工程;および改変されたペプチドの溶液(例えば、本明細書中に記載される薬学的組成物、または製造調製物(manufacturing preparation))を調製する工程が含まれる。この方法には、任意に、溶液中の改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドの溶解度を決定する工程(例えば、溶液中の改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドの試料を得、そして試料の濁度および/または乳白光(opalescence)を評価することによる)が含まれ得る。
【0044】
別の態様においては、本発明は、抗ウイルスペプチドおよび/または抗膜融合性ペプチドの調製(例えば、結合(例えば、大規模な結合))を促進するための方法を特徴とする。この方法には、1つ以上の極性部分(例えば、1つ以上のシステイン酸)を含む改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチド(例えば、本明細書中に記載される改変されたペプチド)を提供する工程;および高濃度(例えば、本明細書中に記載されるような高濃度)の改変されたペプチドを有している改変されたペプチドの溶液を調製する工程が含まれる。
【0045】
本明細書中で使用される場合は、冠詞「a」と「an」は、1つまたは2つ以上(例えば、少なくとも1つ)のその冠詞の文法上の対象物をいう。
【0046】
用語「または」は、特段明記されない限りは、用語「および/または」を意味するように本明細書中で使用され、そして「および/または」と互換的に使用される。
【0047】
用語「タンパク質」と「ポリペプチド」は本明細書中では互換的に使用される。
【0048】
「約」と「およそ」は、一般的には、測定の性質または精度を前提とした、測定された量についての許容される誤差の程度を意味するものとする。例示的な誤差の程度は、提供される値または値の範囲の20パーセント(%)以内であり、通常は10%以内、より典型的には5%以内である。
【0049】
本出願全体を通じて引用される全ての刊行物、係属中の特許出願、公開された特許出願(WO02/096935およびUS05/0070475を含む)、および公開された特許の内容は、それらの全体が引用により本明細書中に組み入れられる。
【0050】
本発明の他の特徴、目的、および利点は、明細書と図面から、そして特許請求の範囲から明らかであろう。
【図面の簡単な説明】
【0051】
【図1】図1は、天然のC34(白四角)と比較した、対照の存在下の末梢血単核細胞(PBMC)中でのHIV−1IIIB複製の阻害(黒菱形)を示している線グラフである。
【図2】図2は、ヒト血清アルブミンに結合させたC34−Lys35(ε−AEEA−MPA)(C34−Lys35(ε−AEEA−MPA):HSA)(白四角)と比較した、対照の存在下でのPBMC中のHIV−1IIIB複製の阻害(黒菱形)を示している線グラフである。
【図3】図3は、N末端にシステイン酸を、そしてC末端に付加されたリジンのεNH2に結合させられたAEEA−MPA有しているC34のアルブミン結合体(ヒト血清アルブミンに結合させられたCA−C34−Lys35(ε−AEEA−MPA)(CA−C34−Lys35(ε−AEEA−MPA):HSA))(白四角)と比較した、対照の存在下でのPBMC中のHIV−1IIIB複製の阻害(黒菱形)を示している線グラフである。
【図4】図4は、MPA−AEEAリンカーによってC34のトリプトファンのN末端αアミノ基に結合させられたアルブミンの結合体(本明細書中ではPC−1505とも呼ばれる;MPA−(AEEA)−C34)(白四角)と比較した、対照の存在下でのPBMC中のHIV−1IIIB複製の阻害(黒菱形)を示している線グラフである。
【図5】図5Aは、Sprague−Dawleyラットへの静脈内投与または皮下投与のいずれかの後の、C34ペプチドと化合物VIII(本明細書中ではPC−1505;MPA−(AEEA)−C34;およびAC−CpdVIIIとも呼ばれる)の薬物動態曲線を示す。図5Bは、Sprague−Dawley ラットへの静脈内投与または皮下投与のいずれかの後のrHAの薬物動態曲線と比較した、化合物VIIIの薬物動態曲線を示す。曲線を重ねあわすことにより、ヒト血清アルブミンのシステイン34に対する化学結合連結マレイミド−化合物VIIIの安定性、さらには、腎臓でのクリアランスとペプチダーゼ消化に対する化合物VIIIの安定性についての明白なサポートする証拠が提供される。
【図6】図6は、HIVIIIbを使用したPBMC中でのいくつかの改変された抗膜融合性ペプチドの活性の結果をまとめている表である。
【発明を実施するための形態】
【0052】
(発明の詳細な説明)
改変前のペプチドと比較して、生理学的pHの水溶液中で高い溶解度を有している改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドが開示される。1つの実施形態においては、本発明のペプチドは、1つ以上の極性部分(例えば、1つ以上のシステイン酸)を含むように改変され、それにより水溶液中でのそれらの溶解度が増大する。改変されたペプチドにはさらに、改変されたペプチドが血液成分または担体タンパク質(例えば、アルブミン)上の利用できる官能基と反応できるように化学反応性部分を含めることができ、これにより改変されたペプチドのインビボでの安定性が増大する。本発明の改変されたペプチドは、多数のウイルス(ヒト免疫不全ウイルス(HIV)、ヒトRSウイルス(RSV)、ヒトパラインフルエンザウイルス(HPV)、麻疹ウイルス(MeV)、およびサル免疫不全ウイルス(SIV)を含む)の感染を緩和するために、例えば、予防的および/または治療的に使用することができる。
【0053】
特定の用語は以下のように定義される:
抗ウイルスペプチド:本明細書中で使用される場合は、「抗ウイルスペプチド」は、例えば、細胞−細胞融合または遊離ウイルス感染を阻害することにより、細胞のウイルス感染を阻害するペプチドをいうものとする。感染の経路には、膜融合(外被ウイルスの場合に生じる場合)またはウイルス構造および細胞構造に関与するいくつかの他の融合事象が含まれ得る。特定のウイルスによるウイルス感染を阻害するペプチドは、特定のウイルス(例えば、特に抗HIVペプチド、抗RSVペプチドなど)に関して言及され得る。
【0054】
抗膜融合性ペプチド:「抗膜融合性ペプチド」は、ペプチドが存在しない場合に起こる膜融合のレベルと比較して、2つ以上の要素の間(例えば、ウイルス−細胞または細胞−細胞)での膜融合事象のレベルを阻害するかまたは減少させる能力を示すペプチドである。
【0055】
HIVおよび抗HIVペプチド:ヒト免疫不全ウイルス(HIV)(これは、後天性免疫不全症候群(AIDS)の原因となる)は、レトロウイルスのレンチウイルファミリーのメンバーである。HIVには2つの一般的なタイプ:HIV−1とHIV−2があり、それぞれについて様々な株が同定されている。HIVは、CD−4+細胞を標的化し、そしてウイルスの進入は、HIVタンパク質gp41のCD−4+細胞表面受容体に対する結合に依存する。抗HIVペプチドは、HIVに対する抗ウイルス活性(遊離ウイルスによるCD−4+細胞感染の阻害、および/または感染したCD−4+細胞と非感染CD−4+細胞との間でのHIVに誘導される合胞体形成の阻害を含む)を示すペプチドをいう。
【0056】
SIVおよび抗SIVペプチド:サル免疫不全ウイルス(SIV)は、感受性のあるサルにおいて後天性免疫不全症候群(AIDS)様の疾病を引き起こすレンチウイルスである。抗SIVペプチドは、SIVに対する抗ウイルス活性(SIVウイルスによる細胞の感染の阻害、および感染した細胞と非感染細胞との間での合胞体形成の阻害を含む)を示すペプチドである。
【0057】
RSVおよび抗RSVペプチド:RSウイルス(RSV)は呼吸器病原体であり、このウイルスが細気管支炎(小通気道の炎症)および肺炎を引き起こす可能性がある乳児および小児においては特に危険である。RSVは逆鎖(negative sense)の一本鎖RNAウイルスであり、ウイルスのParamyxoviridaeファミリーのメンバーである。RSVの感染の経路は、通常は、気道(すなわち、鼻、喉頭、気管および気管支ならびに細気管支)により粘膜を介する。抗RSVペプチドは、RSVに対する抗ウイルス活性(遊離RSVウイルスによる粘膜細胞感染および感染した細胞と非感染細胞との間での合胞体形成の阻害を含む)を示すペプチドである。
【0058】
HPVおよび抗HPVペプチド:RSVと同様に、ヒトパラインフルエンザウイルス(HPIVまたはHPV)は呼吸器疾患の別の主原因であり、RSVと同様に、ウイルスのParamyxoviridaeファミリーのメンバーである、逆鎖の一本鎖RNAウイルスである。HPIVには4種類の認識されている血清型(HPIV−1、HPIV−2、HPIV−3、およびHPIV−4)がある。HPIV−1は、小児のクループの主原因であり、HPIV−1およびHPIV−2の両方は、呼吸器上部および呼吸器下気の疾病を引き起こす。HPIV−3は、より頻繁に細気管支炎および肺炎に関与する。抗HPVペプチドは、HPVに対する抗ウイルス活性(遊離HPVウイルスによる感染および感染した細胞と非感染細胞との間での合胞体形成の阻害を含む)を示すペプチドである。
【0059】
Mevおよび抗Mevペプチド:麻疹ウイルス(VMまたはMeV)は、ウイルスのParamyxoviridaeファミリーに属する、外被逆鎖一本鎖RNAウイルスである。RSVおよびHPVと同様に、MeVは呼吸器疾患を引き起こし、そしてまた、さらなる日和見感染の原因となる免疫抑制も生じる。場合によっては、MeVは、脳の感染を確立し、重篤な神経性合併症を導く可能性がある。抗MeVペプチドは、Mevに対する抗ウイルス活性(遊離Mevウイルスによる感染および感染した細胞と非感染細胞との間での合胞体形成の阻害を含む)を示すペプチドである。
【0060】
C34およびC34アナログ:用語「C34」は、gp41コイルドコイルキャビティ結合残基の部分をいう。例えば、このペプチドにはgp41の残基628WMEW631(配列番号1)、またはgp41の残基628WMEWDREINNYTSLIHSLIEESQNQQEKNEQELL661(配列番号2)が含まれ得る。
【0061】
C34のアナログには、それらの短縮型、欠失型、挿入型、および/またはアミノ酸置換型(例えば、保存的置換もしくは非保存的置換)が含まれ得る。欠失は、C34ペプチドの1つ以上のアミノ酸残基の除去からなり得、これには、ペプチド配列の1つの連続する部分、または複数の部分の除去が含まれる場合もある。挿入には、1つのアミノ酸残基またはひとつづきの残基が含まれ得、挿入は、C34ペプチドのカルボキシ末端もしくはアミノ末端、またはペプチドの内部位置に作製され得る。
【0062】
DP−178およびDP178アナログ:特段明記されるか、または文脈により明らかにされない限りは、DP−178は、HIV−1単離物LAI(HIVLAI)のgp41糖タンパク質のアミノ酸残基638-673に相当し、配列:YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF(配列番号7)を有する36アミノ酸のDP−178ペプチドを意味する。
【0063】
DP178のアナログには、それらの短縮型、欠失型、挿入型、および/またはアミノ酸置換型(例えば、保存的置換もしくは非保存的置換)が含まれ得る。ペプチドの短縮には、3〜36の間のアミノ酸のペプチドが含まれ得る。欠失は、DP178ペプチドからの1つ以上のアミノ酸残基の除去からなり得、これには、ペプチド配列の1つの連続する部分または複数の部分の除去が含まれる場合もある。挿入には、1つのアミノ酸残基またはひとつづきの残基が含まれ得、挿入は、DP178ペプチドのカルボキシ末端もしくはアミノ末端、またはペプチドの内部位置に作製され得る。
【0064】
DP178ペプチドアナログは、そのアミノ酸配列がDP178が由来するgp41領域に対応するHIV−1LAI以外のウイルスのペプチド領域、ならびにそれらの短縮型、欠失型、または挿入型のアミノ酸配列からなるペプチドである。このような他のウイルスとしては、他のHIV単離物(例えば、HIV−2NIHZ、RSウイルス(RSV)、ヒトパラインフルエンザウイルス(HPV)、サル免疫不全ウイルス(SIV)および麻疹ウイルス(MeV))が挙げられるがこれらに限定されない。DP178アナログはまた、ALLMOTI5、107×178×4およびPLZIP検索モチーフ(米国特許第6,013,263号、同第6,017,536号および同第6,020,459号に記載されており、そして本明細書中に引用される)により同定されるかまたは認識されるこれらのペプチド配列をいい、これらは、DP178に対して構造的類似性および/またはアミノ酸モチーフ類似性を有する。DP178アナログはさらに、この用語が米国特許第6,013,263号、同第6,017,536号、および同第6,020,459号において定義されているように、「DP178様」と記載されるペプチドをいう。
【0065】
DP107およびDP107アナログ:特段明記されないか、または文脈によって明らかにされない限りは、DP107は、HIV−1単離物LAI(HIVLAI)のgp41タンパク質のアミノ酸残基558〜595に相当し、配列:NNLLRAIEAQQHLLQLTVWQIKQLQARILAVERYLKDQ(配列番号8)を有する、38アミノ酸のDP107ペプチドを意味する。
【0066】
DP107のアナログには、その短縮型、欠失型、挿入型、および/またはアミノ酸置換型(例えば、保存的置換もしくは非保存的置換)が含まれ得る。ペプチドの短縮には、3〜38の間のアミノ酸のペプチドが含まれ得る。欠失は、DP107ペプチドからの1つ以上のアミノ酸残基の除去からなり得、これには、ペプチド配列の1つの連続する部分または複数の部分の除去が含まれる場合もある。挿入には、1つのアミノ酸残基またはひとつづきの残基が含まれ得、挿入は、DP107ペプチドのカルボキシ末端もしくはアミノ末端、またはペプチドの内部位置に作製され得る。
【0067】
DP107ペプチドアナログは、これらのアミノ酸配列が、DP107が由来するgp41領域に対応するHIV−1LA1、ならびにそれらの短縮型、欠失型、および/または挿入型以外のウイルスのペプチド領域のアミノ酸配列からなるペプチドである。このような他のウイルスとしては、他のHIV単離物(例えば、HIV−2NIHZ、RSウイルス(RSV)、ヒトパラインフルエンザウイルス(HPV)、サル免疫不全ウイルス(SIV)、および麻疹ウイルス(MeV))を挙げることができるが、これらに限定されない。DP107アナログはまた、米国特許第6,013,263号、同第6,017,536号および6,020,459号に記載されており、そして本明細書中に引用されるALLMOTI5、107×178×4およびPLZIP検索モチーフによって同定されたかまたは認識されたこれらのペプチド配列をいい、これらはDP107に対して構造的類似性および/またはアミノ酸モチーフ類似性を有する。DP107アナログは、この用語が、米国特許第6,013,263号、同第6,017,536号および6,020,459号に定義されているように、さらに、「DP107様」と記載されるペプチドもいう。
【0068】
反応性基:反応性基は、共有結合を形成することができる化学基である。このような反応性基は、C34、DP−107、DP−178、もしくはT−1249ペプチドまたはそれらのアナログ、あるいは目的の他の抗ウイルスペプチドまたは抗膜融合性ペプチドに連結させられるか、または結合させられる。反応性基は、一般的には、水性環境中で安定であり、そして通常は、カルボキシ基、ホスホリル基、または従来のアシル基であり、エステルまたは混合無水物のいずれか、あるいはイミデートとして存在し、それによって、移動性の血液成分上の標的部位の、アミノ基、ヒドロキシまたはチオールのような官能基と共有結合を形成することができる。大部分については、これらのエステルは、フェノール性化合物を含むか、またはチオールエステル、アルキルエステル、リン酸エステルなどである。
【0069】
官能基:官能基は、改変された抗ウイルスペプチド上の反応性基が反応して共有結合を形成する、血液成分上の基である。官能基としては、通常、エステル反応性部分に結合するためのヒドロキシル基、マレイミド、イミデート、およびチオエステル基に結合するためのチオール基;カルボキシ基、ホスホリル基、またはアシル基に結合するためのアミノ基、およびアミノ基に結合するためのカルボキシル基が挙げられる。
【0070】
血液成分または担体タンパク質:血液成分は、固定されている場合も、また移動性である場合もある。固定された血液成分は非移動性の血液成分であり、これには、組織、膜受容体、間質タンパク質、フィブリンタンパク質、コラーゲン、血小板、内皮細胞、上皮細胞、およびそれらが結合した膜および膜受容体、身体の細胞(somatic body cell)、骨格細胞および平滑筋細胞、神経成分、骨細胞および破骨細胞、ならびに全ての身体組織(特に、循環系およびリンパ系に関連するもの)が含まれる。移動性の血液成分は、任意の長い期間にわたって(一般的には5分を超えない、より通常は1分間)固定された状態にはない血液成分である。これらの血液成分は、膜結合しておらず、そして長期間にわたって血液中に存在し、そして少なくとも0.1μg/mlの最小濃度で存在する。移動性の血液成分には担体タンパク質が含まれる。移動性の血液成分としては、血清アルブミン、トランスフェリン、フェリチン、および免疫グロブリン(例えば、IgMおよびIgG)が挙げられる。移動性の血液成分の半減期は、少なくとも約12時間である。血液成分のさらなる例としては、フェリチン、ステロイド結合タンパク質、トランスフェリン、チロキシン結合タンパク質、およびα−2−マクログロブリンが挙げられる。通常は、血清アルブミンおよびIgGがより好ましく、血清アルブミン(例えば、ヒト血清アルブミン)が最も好ましい。アルブミンはまた、組み換えによって、またはゲノム供給源(例えば、酵母、細菌(例えば、E.coli)、哺乳動物細胞(例えば、チャイニーズハムスター卵巣(CHO)細胞)、トランスジェニック植物、トランスジェニック動物)に由来する場合もある。したがって、用語「血液成分」には、対象から生化学的に精製されたタンパク質、ならびに組み換えによって作製されたタンパク質が含まれる。
【0071】
保護基:保護基は、ペプチド誘導体をそれ自体と反応しないよう保護するために利用される化学部分である。様々な保護基が、本明細書中に開示され、そして米国特許第5,493,007号(これは、引用により本明細書中に組み入れられる)にも開示されている。このような保護基としては、アセチル、フルオレニルメチルオキシカルボニル(Fmoc)、t−ブチルオキシカルボニル(Boc)、ベンジルオキシカルボニル(CBZ)などが挙げられる。特定の保護されたアミノ酸が表1に示される。
【0072】
【表1】
連結基:連結(スペーサー)基は、反応性部分を抗ウイルスペプチドまたは抗膜融合性ペプチドに連結させるかまたは接続する、化学部分である。連結基には、1つ以上のアルキル部分、アルコキシ部分、アルケニル部分、アルキル部分で置換されたアルキニル部分またはアミノ部分、シクロアルキル部分、多環式部分、アリール部分、ポリアリール部分、置換されたアリール部分、複素環式部分、および置換された複素環式部分が含まれ得る。連結基には、(2−アミノ)エトキシ酢酸(AEA)、[2−(2−アミノ)エトキシ)]エトキシ酢酸(AEEA)、エチレンジアミン(EDA);1つ以上のアルキル鎖(C1〜C10)(例えば、8−アミノオクタン酸(AOA)、8−アミノプロパン酸(APA)、または4−アミノ安息香酸(APhA))が含まれ得る。
【0073】
感受性官能基:感受性官能基は、抗ウイルスペプチドおよび/または抗膜融合性ペプチド上に可能性のある反応部位を提示する原子の群である。存在する場合には、感受性官能基は、リンカー−反応性基改変のための付着点として選択され得る。感受性官能基としては、カルボキシル基、アミノ基、チオール基、およびヒドロキシル基が挙げられるが、これらに限定されない。
【0074】
改変されたペプチド:改変されたペプチドは、反応性基の付着によって改変された抗ウイルスおよび/または抗膜融合性ペプチドである。反応性基は、連結基を介して、または任意に連結基を使用することなくのいずれかで、ペプチドに付着させられ得る。1つ以上のさらなるアミノ酸がペプチドに付加されて、反応性部分の付着が容易にされ得ることもまた想定される。改変されたペプチドは、血液成分との結合がインビボで起こるように、インビボで投与され得るか、またはこれらは、最初に血液成分または担体タンパク質に対してインビトロで結合させられ(例えば、組み換えによって生産されたタンパク質(例えば、組み換え体アルブミン、免疫グロブリン、またはトランスフェリン)を使用して)、そして得られた結合ペプチド(以下に定義される)が、インビボで投与され得る。
【0075】
結合ペプチド:結合ペプチドは、改変されたペプチドの反応性基と血液成分の官能基との間で形成された(連結基を伴うか、または伴わないで)共有結合を介して血液成分に結合させられた、改変されたペプチドである。本出願全体を通じて使用される場合には、用語「結合ペプチド」は、特定の結合ペプチド(例えば、「結合C34」または「結合DP107」)をいうように、より特化され得る。
【0076】
複数の実施形態においては、本発明の改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドには、結合体を形成するように血液成分(より具体的には、血清アルブミン)に共有結合する能力を有しているマレイミド含有基が含まれる。対象への抗ウイルスペプチドおよび/または抗膜融合性ペプチドのマレイミド誘導体の投与によって、血液成分(例えば、血清アルブミン)に対するペプチドのインビボでの結合が生じ得る。改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドを血液成分または担体タンパク質(例えば、アルブミン)と接触させることによって、エキソビボ(またはインビボ)で結合体を調製することもまた本発明に含まれる。この場合は、アルブミンは、様々な供給源から(例えば、血液試料中、精製されたアルブミン、組み換え体アルブミン(アルブミンの改変された形態(例えば、アミノ酸置換、挿入、および/または欠失を有する形態)を含む)など)提供することができる。C34とアルブミンの結合体の調製と使用は、WO02/096935に十分に開示されており、同様の調製と使用が本発明の結合体に適用される。対象の中でインビボで形成させられた結合体とエキソビボで調製された結合体はいずれも、対象に投与される場合には、対応する融合ペプチド阻害剤(fusion peptide inhibitor)の抗膜融合活性を示すために、したがって、対象においてHIV、RSV、HPV、MeV、またはSIVの活性を阻害するために有用である。
【0077】
これらの定義を考慮すると、本発明は、存在する抗ウイルスペプチドおよび抗膜融合性ペプチドの特性を利用する。ペプチドによって阻害され得るウイルスとしては、例えば、米国特許第6,013,263号、同第6,017,536号および同第6,020,459号において表V〜VIIおよびIX〜XIVに列挙されたウイルスのすべての種が挙げられるが、これらに限定されない。これらのウイルスとしては、例えば、ヒトレトロウイルス(HIV−1、HIV−2、およびヒトTリンパ球ウイルス(HTLV−IおよびHTLV−II)を含む)、および非ヒトレトロウイルス(ウシ白血症ウイルス、ネコ肉腫ウイルス、ネコ白血症ウイルス、サル免疫不全ウイルス(SIV)、サル肉腫ウイルス、サル白血病、およびヒツジ進行性肺炎ウイルスを含む)が挙げられる。非レトロウイルスウイルスはまた、本発明のペプチドによって阻害され得、これには以下が含まれる:ヒトRSウイルス(RSV)、イヌジステンパーウイルス、ニューカッスル病ウイルス、ヒトパラインフルエンザウイルス(HPIV)、インフルエンザウイルス、麻疹ウイルス(MeV)、エプスタイン−バーウイルス、B型肝炎ウイルス、およびサルマソン−ファイザーウイルス。非外被ウイルスはまた本発明のペプチドによって阻害され得、そして非外被ウイルスとしては、ピコルナウイルス(例えば、ポリオウイルス)、A型肝炎ウイルス、エンテロウイルス、エコーウイルス、コクサッキーウイルス、パポバウイルス(例えば、パピローマウイルス)、パルボウイルス、アデノウイルス、およびレオウイルスが挙げられるがこれらに限定されない。
【0078】
一例として、本出願の背景のセクションで議論されたように、HIV融合タンパク質の作用機構が記載されており、そしてペプチドの抗ウイルス特性および抗膜融合特性は十分に確立されている。カルボキシ末端の外部ドメイン配列に対応する合成ペプチド(例えば、LAI株のHIV−1クラスBのアミノ酸残基643〜678、または類似する株に由来する残基638〜673、ならびに残基558〜595)は、低濃度でウイルスに媒介される細胞と細胞の融合を完全に阻害することが示されている。本発明のペプチドは、天然のウイルスgp41のロイシンジッパー領域と競合し、それによりウイルスの細胞内への融合/感染の干渉が生じる。
【0079】
本発明によってはさらに、DAC(商標)(Drug Activity Complex)技術を用いて、選択された抗ウイルスペプチドおよび/または抗膜融合性ペプチドを改変し、タンパク質担体上へのペプチドの選択的結合を通じて、このペプチドに対して生体利用性、半減期の延長、およびより優れた分布を付与するために使用される、ペプチドの抗ウイルス特性を改変することのない方法と試薬が提供される。本発明のために選択されたる担体(しかし、これに限定されない)は、マレイミド部分で改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドによるその遊離のチオールを通じて結合させられたアルブミンである。
【0080】
抗ウイルス阻害剤および/または抗膜融合性阻害剤
いくつかのペプチド配列は、HIV−1融合/感染の予防について極めて強力であると文献に記載されている。例として、ペプチドC34、DP107、DP178は、融合に関係があるgp41の立体構造に結合する。従って、本発明の1つの実施形態においては、C34様、DP178様、およびDP178様ペプチドが改変される。同様に、本発明の他の実施形態には、HIVに対して使用されるC34様、DP107様、およびDP107様ペプチド、ならびにRSV、HPV、MeVおよびSIVウイルス中に見られるDP107およびDP178のペプチドアナログの改変が含まれる。
【0081】
改変されたC34ペプチドまたはアナログ
特定の複数の実施形態においては、本発明の改変されたC34ペプチドには、以下の式(I)を有する化合物であり得る、ペプチドに含められるさらなる基が含まれる。
【0082】
(VIII)(R1)m−X−(R2)n
式(VIII)においては、mとnの和は少なくとも1であり、mとnはそれぞれが0以上の整数である。例えば、mが0である場合は、nは1以上であり、nが0である場合は、mは1以上である。Xは、ペプチド、ペプチド断片、またはタンパク質、例えば、C34、T20、T1249、あるいは、それらの誘導体(例えば、それらのマレイミド誘導体を含む)である。R1が存在し、R2が存在しない場合は、R1はX基のN末端に存在する。R1が存在せず、R2が存在する場合は、R2はX基のC末端に存在する。
【0083】
特定の例においては、R1とR2はそれぞれ独立して、式(IX)を有する化合物から選択され得る。
【0084】
【化10】
式(IX)のコア構造はアミノ酸のコア構造と類似しており、これにはアミノ基、α炭素、およびカルボキシル基が含まれる。ペプチド誘導体中のR1基とR2基の正確な位置に応じて、複数の基を式(IX)中の様々な原子を介してペプチドに結合させることができる。例えば、R1が式(IX)を有する化合物である場合には、R1を式(IX)のカルボキシル基を介してペプチドに結合させて、R1のカルボキシル基とペプチドのアミノ基との間にペプチド結合をもたらすことができる。R2が式(IX)を有する化合物である場合には、R2を式(IX)のアミノ基を介してペプチドに結合させて、R2のアミノ基とペプチドのカルボキシ基との間にペプチド結合をもたらすことができる。
【0085】
いくつかの例においては、式(IX)のR3基は、20種類の自然界に存在しているアミノ酸の中に一般的に見られる極性の電荷を持たない基以外の、任意の極性の電荷を持たない基であり得る。例えば、R3基は、スルホニル基(HS=(O)2)、スルホキシド基(HS=O)、スルホン酸基(HO−S=(O)2)、ハロアルキル基、二級アミン、三級アミン、ヒドロキシル基、または極性であるかもしくは全くの中性であり、水溶液中でのペプチド誘導体の全体的な可溶性を増大させることができる他の側鎖基であり得るか、あるいは、それらを含み得る。例えば、水素結合することができる基を持つ側鎖は、ペプチドの全体的な溶解度を増大させるために使用され得る。特定の例においては、側鎖は、リンカーまたは他の種との望ましくない副反応が実質的な程度には起こらないように、非反応性であることが好ましい。いくつかの例においては、R3についての上記基はα炭素から間隔をあけて、例えば、1〜3個の炭素原子の間隔をあけて存在し得る。
【0086】
特定の例においては、R3は、式(X)−(XV)を有する化合物が提供されるように選択され得る。
【0087】
【化11】
本明細書中に開示されるペプチドの溶解度を増大させることができるさらなる適切な側鎖は、本開示の利点を前提として、当業者によって容易に選択されるであろう。
【0088】
特定の複数の実施形態においては、R1基とR2基は、ペプチド結合体の二次構造全体に、または特定の場合には三次構造には実質的には影響を及ぼさない。ペプチド結合体の二次構造に実質的には影響を及ぼさないことにより、ペプチド結合体の全体的な活性は、誘導体化されていないペプチドの全体的な活性よりも感知できるほどに下回ることはないはずである。
【0089】
他の実施形態においては、ペプチド誘導体は、式(XVI)に示されるような組成物の形態をとり得る。
【0090】
(XVI) X1−(R1)m−X2−(R2)n
式(XVI)においては、X1とX2は、互いに連結させられると、例えば、C34、T20、もしくはT1249を提供するであろうペプチドの部分を提示する。式(XVI)においては、R1とR2は、式(IX)に関して上記で議論された基のうちの任意のものであり得、mとnの和は1以上の整数であり、mまたはnのいずれかが0であり得る可能性がある。式(XVI)においては、ペプチド鎖の中央に基が挿入されている。そのような挿入は、ペプチドの酵素消化、それに続くR1基もしくはR2基またはそれらの両方の挿入、次いでペプチド断片を互いに結合させることを含む多くの様々な方法を使用して行われ得る。
【0091】
本明細書中に記載されるC34のシステイン酸誘導体の合成は、ペプチドの作製の間の手作業による介入を伴うSymphony Peptide Synthesizerの自動化された固相手順を使用して行われる(本明細書中の実施例1〜5を参照のこと)。合成は、Fmoc保護されたRamageアミドリンカー樹脂上で、Fmoc保護されたアミノ酸を使用して行った。カップリングは、O−ベンゾトリアゾール−1−イル−N,N,N’,N’−テトラメチル−ウロニウムヘキサフルオロホスフェート(HBTU)とジイソプロピルエチルアミン(DIEA)を活性化因子の混合物としてN,N−ジメチルホルムアミド(DMF)溶液中で使用することによって行った。Fmoc保護基を20%のピペリジン/DMFを使用して除去した。ペプチドが樹脂から切り離されると遊離のNα末端が生じるように、Boc保護されたアミノ酸をN末端に使用した。シグマコーティッドガラス反応容器を合成の際に使用した。
【0092】
特定の複数の実施形態においては、ペプチドの部分は、例えば、Merrifield,1986.Solid phase synthesis.Science.232:341−347によって記載されているように従来の固相合成技術を使用して合成され得る。簡単に説明すると、ブロッキング基がアミノ酸のN末端に付加され、アミノ酸のカルボキシル基はジシクロヘキシルカルボジイミド(DCCD)との反応によって活性化され得る。活性化されたアミノ酸は、遊離のN末端と樹脂またはビーズに結合させられたC末端を有しているアミノ酸と反応させられ得る。アミノブロックされたジペプチジル化合物の形成の後、酸処理によってイソブチレン、二酸化炭素、および樹脂もしくはビーズに結合させられたジペプチドの生産が生じる。さらなるアミノ酸が、これらの工程を繰り返すことによってジペプチドビーズに対して付加され得る。加えて、本明細書中に開示されるアミノ酸誘導体はまた、類似する反応を使用して鎖に沿った任意の点でペプチド鎖に付加される場合もある。したがって、式(IX)を有する化合物を提供するために所望されるペプチド中のどの位置にでもR1基またはR2基を挿入することが可能である。
【0093】
特定の複数の実施形態においては、本明細書中に開示される化合物は、ペプチドのN末端、C末端に、またはペプチドの1つ以上のアミノ酸の側鎖を介して、1つ以上のさらなる基に連結させられ得る。例えば、式(XVII)−(XX)に模式的に示される組成物が作成され得る。
【0094】
【化12】
式(XVII)−(XX)においては、Lは、例えば、(2−アミノ)エトキシ酢酸(AEA)、エチレンジアミン(EDA)、2−[2−(2−アミノ)エトキシ]エトキシ酢酸(AEAA)、アルキル鎖モチーフ(C1〜C10)(例えば、グリシン)、3−アミノプロピオン酸(APA)、8−アミノオクタン酸(AOA)、4−アミノ安息香酸(APhA)などのようなリンカーであり、R1とR2は、本明細書中で議論された基のうちの任意のものであり得る。リンカーは、ペプチドの任意のアミノ酸を介して、例えば、ペプチド中のリジンのεアミノ基を介して、ペプチドのN末端またはC末端に結合させられ得る。X基、X1基、およびX2基は、ペプチド(X)またはペプチド断片(X1およびX2)である。式(XIX)および式(XX)中に示されるP基は、リンカーLを介して誘導されたペプチドに結合させることができるタンパク質を示す。例示的なタンパク質としては、血液タンパク質、ヒト血清アルブミン、組み換え体アルブミン、または他の適切なタンパク質が挙げられる。
【0095】
タンパク質結合体(式(XIX)および式(XX))は、エキソビボまたはインビボで生産され得る。インビボでの生産が行われる場合には、化合物(例えば、式(XVII)および式(XVIII)に示されるもの)が対象に導入され得、インビボタンパク質(例えば、アルブミン)と反応させられ得る。
【0096】
本発明のC34の改変された抗ウイルスペプチドおよび/または改変された抗膜融合性ペプチドの限定ではない例としては、以下の配列が挙げられる:
CA化合物I:(C34に直接連結させられたシステイン酸(CA);本明細書中ではCA−C34とも呼ばれる(配列番号3))。
【0097】
【化13】
CA化合物II:(28位に本来のリジン(Lys28)のアルギニンでの置換を有しているC34に直接連結させられたシステイン酸(CA);本明細書中ではCA−C34(Arg28)とも呼ばれる(配列番号4))。
【0098】
【化14】
CA化合物III:(35位にさらなるリジン残基(Lys35)を有しているC34に直接連結させられたシステイン酸(CA)、ここでは、リジンのεNH2基はリンカー(AEEA−MPA)を介して反応性基に結合させられる;本明細書中ではCA−C34−Lys35(ε−AEEA−MPA)とも呼ばれる(配列番号5))。
【0099】
【化15】
CA化合物IV:(28位に本来のリジン(Lys28)のアルギニンでの置換;35位にさらなるリジン残基(Lys35)を有しているC34に直接連結させられたシステイン酸(CA)、ここでは、リジンのεNH2基はリンカー(AEEA−MPA)を介して反応性基に結合させられる;本明細書中ではCA−C34(Arg28)−Lys35(ε−AEEA−MPA)とも呼ばれる(配列番号6))。
【0100】
【化16】
本出願の教示に従って改変することができる改変されたC34ペプチドのさらなる例にはまた、以下のアミノ酸配列も含まれる:
【0101】
【化17】
【0102】
【化18】
改変されたC34ペプチドの限定ではない例は、以下に示される式I〜式VIIIの化合物であり、これらはインビボまたはエキソビボのいずれかで血液成分上のチオール基と反応させて安定な共有結合を形成させることができる。これらの化合物の合成はWO02/096935(その内容は引用により本明細書中に具体的に組み入れられる)に記載されている。
【0103】
【化19】
【0104】
【化20】
DP178およびDP107
DP178ペプチド
DP178ペプチドは、HIV−1LAI単離物に由来する膜貫通タンパク質gp41のアミノ酸残基638から673に相当し、36アミノ酸の配列を有する(アミノ末端からカルボキシ末端に向かって読まれる):
NH2−YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF−COOH(配列番号7)
全長のDP178(36マー)に加えて、本発明のペプチドには3個から36個の間のアミノ酸残基のペプチドを含むDP178ペプチドの短縮型(すなわち、トリペプチドから36マーのポリペプチドまでの範囲のペプチド)が含まれる。これらの短縮型ペプチドは表2および表3に示される。
【0105】
加えて、DP178ペプチドのアミノ酸置換型もまた本発明の範囲内にある。HIV−1とHIV−2外被タンパク質は構造的には異なるが、HIV−1およびHIV−2のDP178に対応する領域内には著しいアミノ酸保存が存在する。アミノ酸の保存は、周期性の性質であり、これは構造および/または機能のいくらかの保存を示唆している。従って、アミノ酸置換の1つの可能なクラスには、本発明のDP178ペプチドの構造を安定化させると推定さられるこれらのアミノ酸の変化が含まれる。本明細書中に記載されるDP178およびDP178アナログの配列を利用して、当業者は容易にDP178コンセンサス配列を作製し、そして好ましいアミノ酸置換を提示する保存されたアミノ酸残基を、これらから確定することができる。
【0106】
アミノ酸置換は保存された性質の置換である場合も、また保存されていない性質の置換である場合もある。保存されたアミノ酸置換は、類似する電荷、大きさ、および/または疎水性特性のアミノ酸での、DP178ペプチド配列の1以上のアミノ酸の置換(例えば、グルタミン酸(E)のアスパラギン酸(D)へのアミノ酸置換)からなる。非保存的置換は、類似していない電荷、大きさ、および/または疎水性特性を有しているアミノ酸での、DP178ペプチド配列の1以上のアミノ酸の置換(例えば、グルタミン酸(E)のバリン(V)への置換)からなる。
【0107】
DP178のアミノ酸の挿入は、1つのアミノ酸残基または残基のひとつづきの残基からなり得る。挿入は、DP178またはDP178短縮ポリペプチドのカルボキシ末端またはアミノ末端に、ならびにペプチドに対する内部位置に作製され得る。
【0108】
このような挿入は、一般的には2〜15アミノ酸の長さの範囲であろう。目的のペプチドのカルボキシ末端またはアミノ末端のいずれかに作製された挿入はより広い大きさの範囲の挿入であり得、約2〜約50アミノ酸の挿入が好ましいことが意図される。1つ以上のそのような挿入は、そのような挿入が上記の107×178×4、ALLMOTI5またはPLZIP検索モチーフによってなおも認識されされ得るペプチドを生じる限りは、DP178またはDP178短縮型に導入され得る。
【0109】
好ましいアミノ末端への挿入またはカルボキシ末端への挿入は、約2〜約50のアミノ酸残基の長さの範囲のペプチドであり、これらは、実際のDP178 gp41アミノ酸配列に対して、それぞれアミノまたはカルボキシのいずれかのgp41タンパク質領域に対応する。従って、好ましいアミノ末端またはカルボキシ末端へのアミノ酸の挿入には、gp41タンパク質のDP178領域に対してすぐアミノ側またはカルボキシ側に見られるgp41アミノ酸配列が含まれるであろう。
【0110】
DP178またはDP178短縮型の欠失もまた本発明の範囲に含まれる。このような欠失は、DP178またはDP178様ペプチド配列からの1以上のアミノ酸の除去からなり、得られるペプチド配列の長さの下限は、4〜6アミノ酸である。
【0111】
このような欠失には、ペプチド配列の1つの連続する部分または1つより多くの不連続な部分が含まれ得る。1以上のこのような欠失は、このような欠失が上記の107×178×4、ALLMOTI5またはPLZIP検索モチーフによって、なおも認識されされ得るペプチドを生じる限りは、DP178またはDP178短縮型に導入され得る。
【0112】
DP107ペプチド
DP107は38アミノ酸のペプチドであり、これは強力な抗ウイルス活性を示し、そしここに示されるHIV−1LA1単離体膜貫通(TM)gp41糖タンパク質の残基558〜595に対応する:
NH2−NNLLRAIEAQQHLLQLTVWQIKQLQARILAVERYLKDQ−COOH(配列番号17)。
【0113】
全長のDP107の38マーに加えて、DP107ペプチドには、3個から38個の間のアミノ酸残基のペプチドを含むDP107ペプチドの短縮(すなわち、トリペプチドから38マーのポリペプチドまでの大きさの範囲のペプチド)が含まれる。これらのペプチドは、US2005/0070475の表4および表5に示される。
【0114】
加えて、DP178ペプチドのアミノ酸置換体もまた本発明の範囲に含まれる。DP178と同様に、これもまた周期性の性質のものである、HIV−1およびHIV−2のDP107に対応する領域内にもまた高度なアミノ酸保存が存在し、これは、構造および/または機能の保存を示唆している。従って、アミノ酸置換の1つの可能なクラスには、本発明のDP107ペプチドの構造を安定化させると推定されるこれらのアミノ酸の変化が含まれる。本明細書中に記載されるDP107およびDP107アナログ配列を利用して、当業者は、容易にDP107コンセンサス配列を作製し、そして好ましいアミノ酸置換を提示するであろう保存されたアミノ酸残基を、これらから確定することができる。
【0115】
アミノ酸置換は保存された性質の置換である場合も、また保存されていない性質の置換である場合もある。保存的アミノ酸置換は、類似する電荷、大きさ、および/または疎水性特性のアミノ酸での、DP107ペプチド配列の1以上のアミノ酸の置換(例えば、グルタミン酸(E)のアスパラギン酸(D)へのアミノ酸置換)からなる。非保存的置換は、類似していない電荷、大きさ、および/または疎水性特性を有しているアミノ酸での、DP107ペプチド配列の1以上のアミノ酸の置換(例えば、グルタミン酸(E)のバリン(V)への置換)からなる。
【0116】
アミノ酸の挿入は、単一のアミノ酸残基またはひとつづきの残基からなり得る。挿入は、DP107またはDP107短縮型ペプチドのカルボキシ末端またはアミノ末端に、ならびにペプチドに対して内部位置に作製され得る。
【0117】
このような挿入は、一般的には、2〜15アミノ酸の長さの範囲であろう。目的のペプチドのカルボキシ末端またはアミノ末端のいずれかに作製された挿入はより広い大きさの範囲の挿入であり得、約2〜約50アミノ酸が好ましいことが意図される。1以上のこのような挿入は、このような挿入が上記の107×178×4、ALLMOTI5またはPLZIP検索モチーフによってなおも認識されされ得るペプチドを生じる限りは、DP107またはDP107短縮型に導入され得る。
【0118】
好ましいアミノ末端への挿入またはカルボキシ末端への挿入は、約2〜約50までのアミノ酸残基の長さの範囲のペプチドであり、実際のDP107 gp41アミノ酸配列に対して、それぞれアミノまたはカルボキシのいずれかでgp41タンパク質領域に対応する。従って、好ましいアミノ末端またはカルボキシ末端へのアミノ酸の挿入には、gp41タンパク質のDP107領域に対してすぐアミノ側またはカルボキシ側に見られるgp41アミノ酸配列が含まれる。
【0119】
DP107またはDP107短縮型の欠失もまた本発明の範囲に含まれる。このような欠失は、DP107またはDP107様ペプチド配列からの1以上のアミノ酸の除去からなり、得られるペプチド配列の長さの下限は、4〜6アミノ酸である。
【0120】
このような欠失には、ペプチド配列の1つの連続する部分または1つより多くの不連続な部分が含まれ得る。1以上のこのような欠失は、このような欠失が上記の107×178×4、ALLMOTI5またはPLZIP検索モチーフによってなおも認識されされ得るペプチドを生じる限りは、DP107またはDP107短縮型に導入され得る。
【0121】
DP107およびDP107短縮型は、米国特許第5,656,480号にさらに十分に記載されている。
【0122】
DP107およびDP178アナログ
上記の本発明のDP178、DP178短縮型、DP107およびDP107短縮型配列のアナログに対応するペプチドは、他のウイルス(例えば、非HIV−1外被ウイルス、非外被ウイルスおよび他の非ウイルス性生物を含む)中に見ることができる。
【0123】
このようなDP178アナログおよびDP107アナログは、例えば、外被化されたウイルスの膜貫通(「TM」)タンパク質中に存在するペプチド配列に対応し得、そして非外被生物および非ウイルス性生物中に存在するペプチド配列に対応し得る。このようなペプチドは、抗膜融合性活性、抗ウイルス活性、最も具体的には天然の配列が見られるウイルスに特異的である抗ウイルス活性を示す場合があり、また、コイルドコイルペプチド構造が関与している細胞間プロセスを調節する能力を示す場合もある。
【0124】
DP178アナログ
DP178アナログは、そのアミノ酸配列が、例えば、DP178が誘導されたgp41ペプチド領域に対応する他の(すなわち、HIV−1以外の)ウイルスのペプチド領域のアミノ酸配列からなるペプチドである。そのようなウイルスとしては、他のHIV−1単離物およびHIV−2単離物を挙げることができるが、これらに限定されない。
【0125】
他の(すなわち、非HIV−1LAI)HIV−1単離体の対応するgp41ペプチド領域由来のDP178アナログとしては、例えば、以下に示されるペプチド配列を挙げることができる。
【0126】
【化21】
配列番号18、配列番号19、および配列番号20のペプチドは、それぞれ、HIV−lSF2、HIV−1RF、およびHIV−1MNに由来する。他のDP178アナログとしてはHIV−2から誘導されたアナログが挙げられ、これには、US2005/0070475の配列番号6および配列番号7のペプチドが含まれる(これらはそれぞれ、HIV−2RODおよびHIV−2NIHZに由来する)。さらに別の有用なアナログとしては、US2005/0070475の配列番号8および配列番号9のペプチドが挙げられ、これらは、抗ウイルス活性を示すことが明らかにされている。
【0127】
本発明においては、DP178アナログは、そのアミノ酸配列がgp41タンパク質のDP178領域に対応するペプチドを提示することが好ましく、本明細書中に開示されるペプチドにはさらに、約2〜約50までのアミノ酸残基の長さの範囲であり、実際のDP178アミノ酸配列に対するアミノまたはカルボキシのいずれかのgp41タンパク質領域に対応するアミノ酸配列が含まれ得ることもまた意図される。
【0128】
US2005/0070475の表6および表7には、HIV−2NIHZDP178アナログのいくつかの可能な短縮が示されており、これには、3個から36個の間のアミノ酸残基のペプチド(すなわち、トリペプチドから36マーのポリペプチドまでの大きさの範囲のペプチド)が含まれ得る。ペプチド配列は、これらの表中ではアミノ末端(左)からカルボキシ末端(右)方向で列挙されている。
【0129】
さらなるDP178アナログおよびDP107アナログ
DP178およびDP107アナログは、例えば、上記の107×178×4、ALLMOTI5またはPLZIPのコンピューター補助の検索ストラテジーのうちの1つ以上を利用することによって、認識されるかまたは同定される。この検索ストラテジーは、DP107および/またはDP178の構造的特徴および/またはアミノ酸配列特徴と類似した、構造的特徴および/またはアミノ酸配列特徴を有すると予想されるさらなるペプチド領域を同定する。
【0130】
検索ストラテジーは、米国特許第6,013,263号、同第6,017,536号および同第6,020,459号の第9節に示される例に完全に記載されている。この検索ストラテジーは、一部が、DP107およびDP178から推定される一次アミノ酸モチーフに基づくが、一次アミノ酸配列の相同性の検索だけに基づくものではない。なぜなら、そのようなタンパク質配列の相同性は存在するが、主要なウイルス群の間ではないからである。例えば、一次アミノ酸配列の相同性は、HIV−1の異なる株のTMタンパク質内、またはサル免疫不全ウイルス(SIV)の異なる単離物のTMタンパク質内で高い。
【0131】
米国特許第6,013,263号、同第6,017,536号、および同第6,020,459号に開示されるコンピューター検索ストラテジーでは、DP107またはDP178に類似するタンパク質の領域の同定に成功した。この検索ストラテジーは、市販の配列データベースパッケージ(好ましくはPC/Gene)を用いて使用されるように設計されている。
【0132】
米国特許第6,013,263号、同第6,017,536号、および同第6,020,459号においては、一連の検索モチーフ(107×178×4、ALLMOTI5、およびPLZIPモチーフ)は、厳密な範囲から幅広い範囲までストリンジェンシーの範囲が設計され、操作されたが、107×178×4が好ましい。配列は、米国特許第6,013,263号、同第6,017,536号、および同第6,020,459号のそのような検索モチーフ(例えば、表V〜表XIVに列挙されたようなモチーフ)を介して同定され、抗膜融合性(例えば、抗ウイルス)活性を強く示し、抗膜融合性(例えば、抗ウイルス)化合物の同定においてさらに有用であり得る。
【0133】
他の抗ウイルスペプチド
抗RSVペプチド
抗RSVペプチドには、RSVによるウイルス感染を阻害することがさらに同定されている、RSV中の対応するペプチド配列から同定されたDP178および/またはDP107アナログが含まれる。目的のそのようなペプチドには、US2005/0070475の表16のペプチドおよび配列番号10〜配列番号30のペプチドが含まれる。これらのペプチドを合成するための詳細なプロトコールはUS2005/0070475に開示されており、その内容は引用により本明細書中に具体的に組み入れられる。特に興味深いのは、以下のペプチドである:
【0134】
【化22】
US2005/0070475の配列番号10のペプチドは、RSVのF2領域に由来し、そして、DP107およびDP178ペプチドに対応していると記載されている検索モチーフ(すなわち、「DP107/178様」)を使用して、米国特許第6,103,236号、および同第6,020,459号の中で同定された。配列番号21〜配列番号23のペプチドはそれぞれ、US2005/0070475の配列番号10のペプチドの中に含まれるアミノ酸配列を有し、そして各々が、抗RSV活性を示し、特に、50μg/ml未満の濃度でRSV感染Hep−2細胞と非感染Hep−2細胞との間での融合および合胞体形成を阻害することが示されている。
【0135】
US2005/0070475の配列番号11のペプチドは、RSVのF1領域に由来し、そして、DP107に対応していると記載されている検索モチーフ(すなわち、「DP107様」)を使用して、米国特許第6,103,236号、および同第6,020,459号の中で同定された。配列番号24のペプチドには、US2005/0070475の配列番号10のペプチドの中に含まれるアミノ酸配列が含まれ、同様に、抗RSV活性を示し、特に、50μg/ml未満の濃度でRSV感染Hep−2細胞と非感染Hep−2細胞との間での融合および合胞体形成を阻害することが示されている。
【0136】
抗HPIVペプチド
抗HPIVペプチドには、HPIV中の対応するペプチド配列から同定されたDP178および/またはDP107アナログが含まれ、そしてHPIVによるウイルス感染を阻害することがさらに同定されている。目的のそのようなペプチドには、US2005/0070475の表17および配列番号31〜配列番号62のペプチドが含まれる。これらのペプチドを合成するための詳細なプロトコールはUS2005/0070475に開示されており、その内容は引用により本明細書中に具体的に組み入れられる。特に興味深いのは、以下のペプチドである:
【0137】
【化23】
US2005/0070475の配列番号31のペプチドは、HPIV−3のF1領域に由来し、そして、DP107に対応していると記載されている検索モチーフ(すなわち、「DP107様」)を用いて、米国特許第6,103,236号、および同第6,020,459号の中で同定された。配列番号25および配列番号26のペプチドはそれぞれ、US2005/0070475の配列番号30のペプチドの中に含まれるアミノ酸配列を有し、そしてそれぞれが、抗HPIV−3活性を示し、特に、1μg/ml未満の濃度でHPIV−3感染Hep2細胞と非感染CV−1W細胞との間での融合および合胞体形成を阻害することが示されている。
【0138】
US2005/0070475の配列番号32のペプチドもまた、HPIV−3のF1領域に由来し、そして、DP178に対応していると記載されている検索モチーフ(すなわち、「DP178様」)を用いて、米国特許第6,103,236号、および同第6,020,459号の中で同定された。配列番号27および配列番号28〜配列番号32のペプチドはそれぞれ、US2005/0070475の配列番号32のペプチドの中に含まれるアミノ酸配列を有し、そしてそれぞれがまた、抗HPIV−3活性を示し、特に、1μg/ml未満の濃度でHPIV−3感染Hep2細胞と非感染CV−1W細胞のと間での融合および合胞体形成を阻害することが示されている。
【0139】
抗MeVペプチド
抗MeVペプチドは、麻疹ウイルス(MeV)によるウイルス感染を阻害することがさらに同定されている、麻疹ウイルス中の対応するペプチド配列から同定されたDP178および/またはDP107アナログである。特定の目的のそのようなペプチドには、US2005/0070475の表19のペプチドおよび配列番号74〜配列番号86のペプチドが含まれる。これらのペプチドを合成するための詳細なプロトコールはUS2005/0070475に開示されており、その内容は引用により本明細書中に具体的に組み入れられる。特に興味深いのは、以下に列挙されるペプチドである:
【0140】
【化24】
麻疹ウイルス由来の配列は、DP178に対応していると記載されている検索モチーフ(すなわち、「DP178様」)を使用して、米国特許第6,103,236号、および同第6,020,459号の中で同定された。配列番号33、配列番号34、配列番号35および配列番号36のペプチドはそれぞれ、そのように同定されたアミノ酸配列を有し、そしてそれぞれが、抗MeV活性を示し、特に、1μg/ml未満の濃度でMeV感染Hep2と非感染Vero細胞との間での融合および合胞体形成を阻害することが示されている。
【0141】
抗SIVペプチド
抗SIVペプチドは、SIVによるウイルス感染を阻害することがさらに同定されている、SIV中の対応するペプチド配列から同定されたDP178および/またはDP107アナログである。目的のそのようなペプチドには、US2005/0070475の表18のペプチドおよび配列番号63〜配列番号73までのペプチドが含まれる。これらのペプチドを合成するための詳細なプロトコールはUS2005/0070475に開示されており、その内容は引用により本明細書中に具体的に組み入れられる。特に興味深いのは、以下のペプチドである:
【0142】
【化25】
【0143】
【化26】
SIV膜貫通融合タンパク質由来の配列は、DP178に対応していると記載されている検索モチーフ(すなわち、「DP178様」)を使用して、米国特許第6,103,236号、および同第6,020,459号の中で同定された。配列番号37〜配列番号46のペプチドはそれぞれ、そのように同定されたアミノ酸配列を有し、そしてそれぞれが、粗ペプチドとして強力な抗SIV活性を示すことが示されている。
【0144】
さらなるウイルス阻害剤(viral inhibitor)および融合阻害剤(fusion inhibotor)
表現「ウイルス阻害剤誘導体」は、抗膜融合性化合物または侵入阻害剤(または非抗膜融合性)化合物から選択されたウイルス阻害剤の任意の改変体あるいは誘導体を意味するように意図される。
【0145】
抗膜融合性化合物としては以下が挙げられるが、これらに限定されない:エンフュービルタイド;C34;T−1249;TRI−899;TRI−999;5−ヘリックス;N36 Mut(e.g);NCCG−gp41;DP−107;M41−P;N36;M87o;FM−006;ADS−J1;C14リンクミド(C14 linkmid);C34コイル;ヘモリジンA;IQN17;IQN23;SC34EK;SPI−30,014;SPI−70,038;T−1249−HSA;T−649;T−651;TRI−1144;C14;MBP−107;scC34;SJ−2176;T−1249−トランスフェリン;p26;p38;ADS−J2;C52L;クローン3抗体;D5 IgG;D5 scFc;F240 scFv;シフュービルタイド(sifuvirtide);IZN−36;T−1249ミメティボディ(mimetibody);N−36−E;NB−2;NB−64;S−29−I;テアフラビン−3,3’−ジガラート;VIRIP;シアマイシンI;シアマイシンII。
【0146】
侵入阻害剤(または非抗膜融合性)化合物としては以下が挙げられるが、これらに限定されない:AMD−070;SPC−3;KRH−2731;AMD−8664;FC−131;HIV−1 Tatアナログ;KRH−1120;KRH−1636;POL−2438;T−134;T−140;ストローマ細胞由来因子1;ALX40−4C;AMD−3100;T−22;TJN−151;AM−1401;EradicAideウイルスマクロファージ炎症タンパク質II;AMD−3451;コノクルボン;マラビロック;ビクリビロック;INCB−9471;INCB−15,050;DAPTA;PRO−140;HGS−004;SCH−C;TAK−652;TAK−220;ニフェビロック(nifeviroc);AMD−887;抗CD63 MAb;AOP−RANTES;CPMD−167;E−913;FLSC R/T−IgG1;HGS−101;NIBR−1282;ノナカイン(nonakine);PSC−RANTES;sCD4−17b;SCH−350,634;MIP−1α;MIP−1β;RANTES;アプラビロック;ペプチドT;TAK−779;pCLXSNベクター;UCB−35,625;J−113,863;CLIV;I−309;EGCG;エピガロカテキンガレート(Epigallocathechin gallate);HB−19;λ−カラギーナン;PC−515;カードラン硫酸塩;OKU−40;OKU−41;VGV−1;ジンテルビル(Zintevir);AR−177;T−30,177;コハク酸化アルブミン;NSC0−658,586;ISIS−5320;RP−400c;SA−1042;C31G;サヴィ(Savvy);PRO−542;rCD4−IgG2;BMS−488,043;BMS−378,806;DES−6;12p1;アクチノヒビン;BlockAide/VP;CD4M33;CT−319;CT−326;シアノビリン−N;DCM−205;DES−10;グリフィスシン;HNG−105;NBD−556;NBD−557;PEG−シアノビリン−N;シトビリン(scytovirin);sCD4;デキストリン−2−硫酸塩;F−105;FP−21,399;TNX−355;B4 MAb;R−15−K;sCD38(51−75)MBP;PRO−2000;NSC−13,778;SB−673,461M;SB−673,462M;rsCD4;Ac(Ala10,11)RANTES(2−14);IC−9564;RPR−103,611;Immudel−gp120;スリゴビル;IQP−0410;アセチル化トリヨードサイロニン;SP−01A;DEB−025;CSA−54;HGS−H/A27;SP−10;VIR−5103;BMS−433,771;TMC−353,121;NSC−650,898;ミケラミンB;NSC−692,906;TG−102;VIR−576;MEDI−488;CovX−Body;CNI−H0294。
【0147】
抗ウイルスおよび抗膜融合性ペプチドの改変
本発明では、抗ウイルス活性および/または抗膜融合性活性を示すペプチドを改変することが意図され、DP−107およびDP−178ならびにそれらのアナログのそのような改変が含まれる。そのような改変されたペプチドは、共有結合を介して血液成分上の利用可能な反応性官能基と反応することができる。本発明はまた、そのような改変、血液成分とのそのような組合せ、およびそれらの使用のための方法に関する。これらの方法には、非結合ペプチドの患者への投与と比較して、結合させられた抗ウイルスペプチド誘導体の有効な治療薬の寿命を延長させる工程が含まれる。改変されたペプチドは、DAC(商標)(薬物親和性複合体)として設計されたタイプのペプチドであり、これには、抗ウイルスペプチド分子と、移動性の血液タンパク質の反応性官能基と反応することができる化学反応基を伴う連結基が含まれる。血液成分またはタンパク質との反応によって、改変されたペプチド、すなわちDACを、血液を介して適切な部位または受容体に送達することができる。
【0148】
タンパク質上の官能基と共有結合を形成させるためには、反応性基として多種多様な活性なカルボキシル基、特にエステルを使用することができ、この場合、ヒドロキシル部分は、ペプチドを改変するために必要とされるレベルで生理学的に許容される。多数の異なるヒドロキシル基がこれらの反応性基に使用され得るが、最も便利なものは、N−ヒドロキシスクシンイミド(すなわち、NHS)、N−ヒドロキシ−スルホスクシンイミド(sulfo−NHS)であろう。本発明の好ましい実施形態においては、タンパク質上の官能基はチオール基であり、そして反応性基はマレイミド含有基(例えば、γ−マレイミド−ブチラールアミド(GMBA)またはマレイミドプロピオン酸(MPA)であろう。
【0149】
第一級アミンは、NHSエステルについての重要な標的である。タンパク質のN末端上に存在する接近することができるα−アミン基はNHSエステルと反応する。しかし、タンパク質のα−アミノ基は、NHSカップリングには望ましくもない場合があり、また、利用可能できない場合もある。5種類のアミノ酸はそれらの側鎖中に窒素を有するが、リジンのε−アミンだけが有意にNHSエステルと反応する。以下の反応スキームに示されるように、NHSエステルの結合反応が第一級アミンと反応してN−ヒドロキシスクシンイミドを放出する場合には、アミド結合が形成される。
【0150】
【化27】
本発明の好ましい実施形態において、このタンパク質上の官能基はチオール基であり、化学反応性基はマレイミド含有基(例えば、MPAまたはGMBA(γ−マレイミド−ブチラールアミド)であろう。マレイミド基は、反応混合物のpHが6.5と7.4の間に維持される場合には、ペプチド上のスルフヒドリル基について最も選択的である。pH7.0では、マレイミド基のスルフヒドリルとの反応の速度は、アミンとの反応の速度よりも1000倍速い。以下の反応スキームに示されるように、マレイミド基とスルフヒドリルとの間で安定なチオエーテル結合が形成され、これは、生理学的な条件下では切断され得ない。
【0151】
【化28−1】
特異的標識
好ましくは、本発明の改変されたペプチドは、移動性の血液タンパク質上のチオール基と特異的に反応するように設計される。このような反応は、好ましくは、移動性血液タンパク質(例えば、血清アルブミンまたはIgG)上のチオール基へのマレイミドの連結(例えば、GMBS、MPAまたは他のマレイミドから調製される)で改変されたこのペプチドの共有結合によって確立される。
【0152】
特定の状況では、マレイミドでの特異的標識は、NHSおよびsulfo−NHSのような基での移動性タンパク質の非特異的な標識化を上回るいくつかの利点をもたらす。チオール基は、インビボではアミノ基よりも少ない量で存在する。従って、本発明のマレイミドで改変されたペプチド(すなわち、マレイミドペプチド)は、より少ないタンパク質に共有結合するであろう。例えば、アルブミン(最も豊富な血液タンパク質)中には、チオール基が1つだけ存在する。従って、ペプチド−マレイミド−アルブミン結合体は、約1:1のモル比でペプチド対アルブミンを含む傾向があるだろう。アルブミンに加えて、IgG分子(クラスII)もまた遊離のチオールを有する。IgG分子と血清アルブミンが、血液中の可溶タンパク質の大部分を構成するので、これらはまた、マレイミド改変ペプチドに共有結合させるために利用できる血液中の遊離のチオール基の大部分を構成する。
【0153】
さらに、遊離のチオール含有血液タンパク質(IgGを含む)の間でさえ、アルブミン自体の特有の特徴が原因で、マレイミドでの特異的標識はペプチド−マレイミド−アルブミン結合体の優先的な形成を導く。アルブミンの単一の遊離のチオール基は種間で高度に保存されており、アミノ酸残基34(Cys34)に位置する。最近、アルブミンのCys34が、他の遊離チオール含有タンパク質上の遊離のチオールと比べて、高い反応性を有することが明らかにされていた。これは、アルブミンのCys34に対する非常に低いpK値(5.5)に一部原因がある。これは、一般的にシステイン残基についての典型的なpK値(典型的には約8)よりもはるかに低い。この低いpKが原因で、正常な生理学的条件下では、アルブミンのCys34は主にイオン化された形態であり、これはその反応性を劇的に高める。Cys34の低いpK値に加えて、Cys34の反応性を高める別の因子はその位置であり、この位置は、アルブミンの領域Vの一つのループの表面に近い間隙の中にある。この位置は、Cys34を全ての種類のリガンドに対して極めて利用しやすくし、そしてこの位置は、遊離ラジカルトラップおよび遊離チオールスカベンジャーとしてのCys34の生物学的役割において重要な因子である。これらの特性は、Cys34をマレイミド−ペプチドに非常に反応性にし、反応速度の加速は、他の遊離のチオール含有タンパク質とのマレイミド−ペプチドの反応速度に比べて、1000倍であり得る。
【0154】
ペプチド−マレイミド−アルブミン結合体の別の利点は、詳細にはCys34において、ペプチド対アルブミンを1:1で含むことに関連する再現性である。他の技術(例えば、グルタルアルデヒド、DCC、EDCおよび例えば、遊離のアミンの他の化学活性化)には、この選択性が欠けている。例えば、アルブミンには52個のリジン残基が含まれ、このうちの25〜30個は、アルブミンの表面に位置し、したがって結合のために接近することができる。これらのリジン残基の活性化、あるいはこれらのリジン残基を介するカップリングのためのペプチドの別の改変によっては、結合体の不均一な集団が生じる。ペプチド対アルブミンの1:1のモル比が使用される場合でもなお、生成物は、複数の結合体産物からなり、そのいくつかには、アルブミン1分子当たり0、1、2以上のペプチドが含まれており、それぞれが、25〜30個の利用可能なリジン部位のうちの任意1つ以上に無作為に結合させられたペプチドを有する。多数の可能な組み合わせが提供される場合には、正確な組成および個々の結合体のバッチの特性決定は困難になり、バッチ間の再現性はほとんど不可能であり、このような結合体の治療薬としての望ましさを低下させてしまう。加えて、アルブミンのリジン残基を介する結合は、アルブミン1分子当たりにより多くの治療薬を送達するという利点を少なくとも有するようであるが、複数の研究によって、治療薬対アルブミンの1:1の比が好ましいことが示されている。Stehleらによる論文「The Loading Rate Determines Tumor Targeting properties of Methotrexate−Albumin Conjugates in Rats」、Anti−Cancer Drugs,第8巻,677−685頁(1988)では、著者らは、グルタルアルデヒドを介して結合させられたアルブミンに対する抗癌剤メトトレキサートの1:1の比が最も有望な結果を生じたことを報告した。これらの結合体は腫瘍細胞によって優先的に取り込まれたが、5:1〜20:1のメトトレキサート分子を保有している結合体は、変化したHPLCプロフィールを有し、そしてインビボで肝臓に迅速に取り込まれた。これらの高い比では、アルブミンに対する立体構造の変化は、治療用担体としてのその有効性を低下させると推論された。
【0155】
マレイミド−ペプチドのインビボでの制御された投与を通じて、アルブミンとIgGのインビボでの特異的標識を制御することができる。典型的な投与においては、投与されたマレイミド−ペプチドのうちの80〜90%がアルブミンを標識し、5%未満はIgGを標識するであろう。グルタチオンのような遊離のチオールの微量標識もまた起こるであろう。このような特異的標識は、インビボでの使用に好ましい。なぜなら、これにより、投与された薬剤の概算された半減期の正確な計算が可能となるからである。
【0156】
制御された特異的なインビボ標識化を提供することに加えて、マレイミド−ペプチドは、血清アルブミンとIgGのエキソビボでの特異的標識を提供することができる。このようなエキソビボでの標識には、血清アルブミンおよび/またはIgGを含む、血液、血清または生理食塩溶液へのマレイミド−ペプチドの添加が含まれる。一旦、マレイミド−ペプチドとの結合がエキソビボで起こると、血液、血清、または生理食塩溶液は、インビボ処置のために患者の血液に再投与することができる。
【0157】
NHS−ペプチドとは対照的に、マレイミド−ペプチドは、一般的には、水溶液の存在下および遊離のアミンの存在下では極めて安定である。マレイミド−ペプチドが遊離のチオールとだけ反応するので、保護基は、一般的には、マレイミド−ペプチドがそれ自体と反応することを防ぐ必要はない。加えて、改変されたペプチドの高い安定性によって、インビボでの使用に適している高度に精製された産物を調製するためのHPLCのようなさらなる精製工程の使用が可能となる。最後に、高い化学的安定性によって、製品の有効期間をより長くすることができる。
【0158】
非特異的標識
本発明の抗ウイルスペプチドはまた、血液成分の非特異的標識のために改変される場合もある。アミノ基に対する結合、特に非特異的標識のためのアミド結合の形成を伴う結合もまた使用されるであろう。そのような結合を形成させるためには、化学反応基として多種多様な活性カルボキシル基、特にエステルが使用され得、この場合、ヒドロキシル部分は、必要とされるレベルで生理学的に許容される。多数の異なるヒドロキシル基がこれらの結合剤において使用され得るが、最も便利なものはN−ヒドロキシスクシンイミド(NHS)およびN−ヒドロキシ−スルホスクシンイミド(sulfo−NHS)であろう。
【0159】
利用され得る他の結合剤は、米国特許第5,612,034号に記載されている。
【0160】
改変されたペプチドの化学反応基がインビボで反応し得る様々な部位としては、細胞、特に赤血球(成熟赤血球)および血小板、ならびにタンパク質(例えば、免疫グロブリン(IgGおよびIgMを含む)、血清アルブミン、フェリチン、ステロイド結合タンパク質、トランスフェリン、サイロキシン結合タンパク、α−2−マクログロブリンなど)が挙げられる。改変されたペプチドが反応するそれらの受容体(これは、長期間生存しない)は、一般的に、約3日以内にヒト宿主から排除されるであろう。上記に示されるタンパク質(細胞のタンパク質を含む)は、血中濃度に基づいて、特に半減期に関しては、少なくとも3日間残存し、そして5日間以上残存する場合もあるであろう(通常は、60日間を超えず、より通常は30日間を超えない)。
【0161】
ほとんどの部分について、反応は、血液中の移動性の成分、具体的には血液タンパク質および細胞、さらに具体的には血液タンパク質および成熟赤血球との反応であろう。「移動性」によっては、その成分が、任意の延長された期間(一般的には5分間を超えない、より通常は1分間)固定された状況にはないことが意図されるが、血液成分のうちのいくつかは、延長された期間比較的固定された状態で存在し得る。最初は、機能化されたタンパク質および細胞の比較的不均質な集団が存在するであろう。しかし、ほとんどの部分については、数日以内の集団は、血流中の機能化されたタンパク質の半減期に依存して、最初の集団から実質的に変化するであろう。従って、通常は約3日以内またはそれ以後には、IgGは、血流において優勢な機能化されたタンパク質となるであろう。
【0162】
通常は、投与後5日までに、IgG、血清アルブミンおよび成熟赤血球は、血液中の結合成分の少なくとも約60モル%、通常は少なくとも約75モル%を占め、IgG、IgM(実質的により少ない程度まで)および血清アルブミンは、非細胞性結合成分の少なくとも約50モル%、通常は少なくとも約75モル%、より通常は約80モル%となるであろう。
【0163】
血液成分に対する非特異的な改変されたペプチドの所望される結合体は、患者への改変されたペプチドの投与によってインビボで調製され得る。患者はヒトまたは他の哺乳動物であり得る。その投与は、ボーラスの形態で行われる場合も、また、流れを計器で調節して注入することなどによって、ゆっくりと経時的に導入される場合もある。
【0164】
所望される場合、目的の結合体はまた、血液を本発明の改変されたペプチドと組み合わせ、これにより血液成分上の反応性官能基への改変されたペプチドの共有結合が可能となること、次いで、宿主に結合させられた血液を戻すかまたは投与することによってエキソビボで調製され得る。さらに、これはまた、初めに、個々の血液成分または限られた数の成分(例えば、赤血球、免疫グロブリン、血清アルブミンなど)を精製し、そしてその成分または複数の成分をエキソビボで化学的に反応性の改変されたペプチドと組み合わせることによって達成され得る。次いで、機能化された血液または血液成分は、目的の治療的に有効な結合体をインビボで提供するために宿主に戻され得る。血液はまた、エキソビボでの操作の際の凝固を防ぐために処理され得る。
【0165】
改変された抗ウイルスペプチドおよび抗膜融合性ペプチドの合成
A.ペプチド合成
本発明の抗ウイルスペプチドおよび/または抗膜融合性ペプチドは、当業者に公知の固相ペプチド化学の標準的な方法によって合成され得る。例えば、ペプチドは、Applied Biosystem synthesizerを使用して、Steward and Young(Steward,J.M.and Young,J.D.,Solid
Phase Peptide Synthesis,第2版.,Pierce Chemical Company,Rockford,III.,(1984))によって記載された手順に従って固相化学技術によって合成され得る。次いで、同様に、複数のペプチド断片が合成され、その後、互いに連結させられることによって、より大きなペプチドが形成させられる。これらの合成ペプチドはまた、特定の位置にアミノ酸置換を有して作製することもできる。
【0166】
固相ペプチド合成について、多くの技術の概要が、J.M.Stewart and J.D.Young,Solid Phase Peptide Synthesis,W.H.Freeman Co.(San Francisco),1963、およびJ.Meienhofer,Hormonal Proteins and Peptides,第2巻 46頁、Academic Press(New York),1973に見ることができる。伝統的な溶液合成については、G.Schroder and K.Lupke,The Peptides,第1巻、Acacemic Press(New York)を参照のこと。一般的には、これらの方法には、1つ以上のアミノ酸または適切に保護されたアミノ酸の、成長しつつあるペプチド鎖への連続的付加が含まれる。通常、第1のアミノ酸のアミノ基またはカルボキシル基のいずれかが、適切な保護基によって保護される。次いで、保護されたアミノ酸または誘導されたアミノ酸は、不活性な固体支持体に付着させられるか、あるいは、アミド結合を形成させるために適している条件下で、適切に保護された相補性(アミノもしくはカルボキシル)基を有している配列中に次のアミノ酸を付加することによって溶液中で利用されるかのいずれかである。次いで、保護基がこの新たに付加されたアミノ酸残基から除去され、そして(適切に保護された)次のアミノ酸が付加されるなどが行われる。
【0167】
全ての所望されるアミノ酸が、適切な配列内に連結させられた後、任意の残っている保護基(および任意の固体支持体)が連続的に、または同時に除去されて、最終的なポリペプチドにされる。この一般的手順の単純な改変によって、2つ以上のアミノ酸を一度に成長しつつある鎖に対して、例えば、保護されたトリペプチドを適切に保護されたジペプチドと(キラル中心をラセミ化させない条件下で)カップリングさせることによって付加させることが可能であり、脱保護の後に、ペンタペプチドが形成させられる。
【0168】
本発明の化合物を調製する特に好ましい方法には固相ペプチド合成が含まれる。この場合、アミノ酸α−N−末端は、酸感受性基または塩基感受性基によって保護される。そのような保護基は、ペプチド結合形成の条件に対して安定であるという特性を有するべきであり、一方、成長しつつあるペプチド鎖の破壊またはその中に含まれる任意のキラル中心のラセミ化を伴わずに容易に除去できる。適切な保護基は、9−フルオレニルメチルオキシカルボニル(Fmoc)、t−ブチルオキシカルボニル(Boc)、ベンジルオキシカルボニル(Cbz)、ビフェニルイソプロピルオキシカルボニル、t−アミルオキシカルボニル、イソボルニルオキシカルボニル、α,α−ジメチル−3,5−ジメトキシベンジルオキシカルボニル、o−ニトロフェニルスルフェニル、2−シアノ−t−ブチルオキシカルボニルなどである。9−フルオレニルメチルオキシカルボニル(Fmoc)保護基は、本発明のペプチドの合成に特に好ましい。他の好ましい側鎖保護基は、リジンおよびアルギニンのような側鎖アミノ基については、2,2,5,7,8−ペンタメチルクロマン−6−スルホニル(pmc)基、ニトロ基、p−トルエンスルホニル基、4−メトキシベンゼン−スルホニル基、Cbz基、Boc基、およびアダマンチルオキシカルボニル基であり;チロシンについては、ベンジル基、o−ブロモベンジルオキシカルボニル基、2,6−ジクロロベンジル基、イソプロピル基、t−ブチル(t−Bu)基、シクロヘキシル基、シクロペニル基およびアセチル(Ac)基であり;セリンについては、t−ブチル基、ベンジル基およびテトラヒドロピラニル基であり;ヒスチジンについては、トリチル基、ベンジル基、Cbz基、p−トルエンスルホニル基および2,4−ジニトロフェニル基であり;トリプトファンについては、ホルミル基であり;アスパラギン酸およびグルタミン酸については、ベンジル基およびt−ブチル基であり、さらに、システインについては、トリフェニルメチル(トリチル)基である。
【0169】
固相ペプチド合成方法において、α−C−末端アミノ酸は、適切な固体支持体または樹脂に付着させらる。上記合成に有用な適切な固体支持体は、段階的な縮合−脱保護反応の試薬および反応条件に対して不活性であり、さらに、使用される媒体に不溶性である材料である。α−C−末端カルボキシペプチドの合成のための好ましい固体支持体は、4−ヒドロキシメチルフェノキシメチル−コポリ(スチレン−1%ジビニルベンゼン)である。α−C−末端アミドペプチドに好ましい固相支持体は、Applied Biosystems(Foster City,Calif.)から入手することができる4−(2’,4’−ジメトキシフェニル−Fmoc−アミノメチル)フェノキシアセトアミドエチル樹脂である。α−C−末端アミノ酸は、4−ジメチルアミノピリジン(DMAP)、1−ヒドロキシベンゾトリアゾール(HOBT)、ベンゾトリアゾール−1−イルオキシ−トリス(ジメチルアミノ)ホスホニウム−ヘキサフルオロホスフェート(BOP)またはビス(2−オキソ−3−オキサゾリジニル)ホスフィンクロリド(BOPCl)を伴ってまたは伴わずに、N,N’−ジシクロヘキシルカルボジイミド(DCC)、N,N’−ジイソプロピルカルボジイミド(DIC)またはO−ベンゾトリアゾル−1−イル−N,N,N’,N’,−テトラメチルウロニウム−ヘキサフロオロホスフェート(HBTU)によって樹脂に結合させられ、結合は、溶媒(例えば、ジクロロメタンまたはDMF)中で10℃から50℃の間の温度で約1時間〜24時間の間媒介される。
【0170】
固体支持体が、4−(2’,4’−ジメトキシフェニル−Fmoc−アミノメチル−)フェノキシ−アセトアミドエチル樹脂である場合、Fmoc基は、上記のα−C−末端アミノ酸とのカップリングの前に、第二級アミン、好ましくはピペリジンで切断される。脱保護された4−(2’,4’−ジメトキシフェニル−Fmoc−アミノメチル−)フェノキシ−アセトアミドエチル樹脂へのカップリングのための好ましい方法は、DMF中のO−ベンゾトリアゾール−1−イル−N,N,N’,N’,−テトラメチルウロニウムヘキサフロオロ−ホスフェート(HBTU、1当量)および1−ヒドロキシベンゾトリアゾール(HOBT、1当量)である。保護されたアミノ酸の連続的なカップリングは、当該分野で周知であるように、自動ポリペプチド合成装置の中で行わせることができる。好ましい実施形態においては、成長しつつあるペプチド鎖のα−N−末端アミノ酸はFmocで保護される。成長しつつあるペプチドのα−N−末端側からのFmoc保護基の除去は、第二級アミン、好ましくはピペリジンでの処理によって達成される。次いで、個々の保護されたアミノ酸は、約3倍のモル過剰量で導入され、そして好ましくは、カップリングはDMF中で行われる。カップリング剤は、通常、O−ベンゾトリアゾール−1−イル−N,N,N’,N’,−テトラメチルウロニウム−ヘキサフロオロホスフェート(HBTU、1当量)および1−ヒドロキシベンゾトリアゾール(HOBT、1当量)である。
【0171】
固相合成の終点において、このポリペプチドは、連続的または1回の操作のいずれかで、樹脂から取り出され、脱保護される。ポリペプチドの取り出しと脱保護は、チオアニソール、水、エタンジチオールおよびトリフルオロ酢酸を含む切断試薬(cleavage
reagent)で樹脂が結合したポリペプチドを処理することによって1回の操作で達成され得る。ポリペプチドのα−C−末端がアルキルアミドである場合は、樹脂は、アルキルアミンを用いてアミノ分解によって切断される。あるいは、ペプチドは、例えば、メタノールを用いるエステル交換反応、それに続くアミノ分解、または直接的なアミド基交換によって取り出され得る。保護されたペプチドは、この時点で精製され得るか、または次の工程に直接使用され得る。側鎖保護基の除去は、上記の切断カクテル(cleavage cocktail)を使用して達成される。完全に脱保護されたペプチドは、任意または全ての以下の型:弱塩基樹脂上のイオン交換(酢酸塩の形態);非誘導体化ポリスチレン−ジビニルベンゼン上の疎水性吸着クロマトグラフィー(例えば、Amberlite XAD);シリカゲル吸着クロマトグラフィー;カルボキシメチルセルロース上でのイオン交換クロマトグラフィー;分配クロマトグラフィー(例えば、Sephadex G−25、LH−20)または向流分配;高速液体クロマトグラフィー(HPLC)(特に、オクチル−もしくはオクタデシルシリル−シリカ結合相充填剤上での逆相HPLC)を利用する一連のクロマトグラフィー工程によって精製される。これらのITPの分子量は、Fast Atom Bombardment(FAB)Mass Spectroscopyを使用して決定される。
【0172】
N−末端保護基
上記で議論されたように、用語「N−保護基」とは、アミノ酸もしくはペプチドのα−N−末端を保護すること、あるいはアミノ酸もしくはペプチドのアミノ基を、合成手順の間の望ましくない反応から保護することが意図される基をいう。一般的に、使用されるN−保護基は、Greene、「Protective Groups In Organic Synthesis」(John Wiley&Sons、New York(1981))(これは、引用により本明細書中に組み入れられる)に開示されている。加えて、保護基を、例えば、酵素的加水分解によってインビボで容易に切断されて生物学的に活性なペアレント(parent)を放出するるプロドラッグとして使用することができる。α−N−保護基には、低級アルカノイル基(例えば、ホルミル基、アセチル基(「Ac」)、プロピオニル基、ピバロイル基、t−ブチルアセチル基など;他のアシル基(2−クロロアセチル基、2−ブロモアセチル基、トリフルオロアセチル基、トリクロロアセチル基、フタリル基、o−ニトロフェノキシアセチル基、クロロブチリル基、ベンゾイル基、4−クロロベンゾイル基、4−ブロモベンゾイル基、4−ニトロベンゾイル基などを含む);スルホニル基(例えば、ベンゼンスルホニル基、p−トルエンスルホニル基など);カルバメート形成基(例えば、ベンジルオキシカルボニル基、p−クロロベンジルオキシカルボニル基、p−メトキシベンジルオキシカルボニル基、p−ニトロベンジルオキシカルボニル基、2−ニトロベンジルオキシカルボニル基、p−ブロモベンジルオキシカルボニル基、3,4−ジメトキシベンジルオキシカルボニル基、3,5−ジメトキシベンジルオキシカルボニル基、2,4−ジメトキシベンジルオキシカルボニル基、4−エトキシベンジルオキシカルボニル基、2−ニトロ−4,5−ジメトキシベンジルオキシカルボニル基、3,4,5−トリメトキシベンジルオキシカルボニル基、1−(p−ビフェニルイル)−1−メチルエトキシカルボニル基、α,α−ジメチル−3,5−ジ−メトキシベンジルオキシカルボニル基、ベンズヒドリルオキシカルボニル基、t−ブチルオキシカルボニル基(Boc)、ジイソプロピルメトキシカルボニル基、イソプロピルオキシカルボニル基、エトキシカルボニル基、メトキシカルボニル基、アリルオキシカルボニル基、2,2,2−トリクロロエトキシカルボニル基、フェノキシカルボニル基、4−ニトロフェノキシカルボニル基、フルオレニル−9−メトキシカルボニル基、シクロペンチルオキシカルボニル基、アダマンチルオキシカルボニル基、シクロヘキシルオキシカルボニル基、フェニルチオカルボニル基など);アリールアルキル基(例えば、ベンジル基、トリフェニルメチル基、ベンジルオキシメチル基、9−フルオレニルメチルオキシカルボニル基(Fmoc)など)、ならびにシリル基(例えば、トリメチルシリルなど)が含まれる。
【0173】
カルボキシ保護基
上記で議論されたように、用語「カルボキシ保護基」とは、化合物の他の機能性部位に関する反応が実施されながらカルボン酸官能基をブロックもしくは保護するために使用されるカルボン酸保護エステル基またはアミド基をいう。カルボキシ保護基は、Greene、「Protective Groups in Organic Synthesis」152〜186頁(1981)(これは引用により本明細書中に組み入れられる)に開示されている。加えて、カルボキシ保護基はプロドラッグとして使用することができ、それによってカルボキシ保護基は、例えば、酵素的加水分解によって容易にインビボで切断されて、生物学的に活性なペアレントを放出することができる。そのようなカルボキシ保護基は当業者に周知であり、米国特許第3,840,556号および同第3,719,667号にて記載されているように、ペニシリンおよびセファロスポリンの分野におけるカルボキシル基の保護において広く使用されている。これらの開示は引用により本明細書中に組み入れられる。代表的なカルボキシ保護基は、C1〜C8低級アルキル基(例えば、メチル基、エチル基またはt−ブチル基など);アリールアルキル基(例えば、フェネチル基またはベンジル基、ならびに、アルコキシベンジル基またはニトロベンジル基などのようなそれらの置換誘導体);アリールアルケニル基(例えば、フェニルエテニル基など);アリールおよびその置換された誘導体(例えば、5−インダニル基など);ジアルキルアミノアルキル基(例えば、ジメチルアミノエチル基など);アルカノイルオキシアルキル基(例えば、アセトキシメチル基、ブチリルオキシメチル基、バレリルオキシメチル基、イソブチリルオキシメチル基、イソバレリルオキシメチル基、1−(プロピオニルオキシ)−1−エチル基、1−(ピバロイルオキシ)−1−エチル基、1−メチル−1−(プロピオニルオキシ)−1−エチル基、ピバロイルオキシメチル基、プロピオニルオキシメチル基など);シクロアルカノイルオキシアルキル基(例えば、シクロプロピルカルボニルオキシメチル基、シクロブチルカルボニルオキシメチル基、シクロペンチルカルボニルオキシメチル基、シクロヘキシルカルボニルオキシメチル基など);アロイルオキシアルキル基(例えば、ベンゾイルオキシメチル基、ベンゾイルオキシエチル基など);アリールアルキルカルボニルオキシアルキル基(例えば、ベンジルカルボニルオキシメチル基、2−ベンジルカルボニルオキシエチル基など);アルコキシカルボニルアルキル基またはシクロアルキルオキシカルボニルアルキル基(例えば、メトキシカルボニルメチル基、シクロヘキシルオキシカルボニルメチル基、1−メトキシカルボニル−1−エチル基など);アルコキシカルボニルオキシアルキル基またはシクロアルキルオキシカルボニルオキシアルキル基(例えば、メトキシカルボニルオキシメチル基、t−ブチルオキシカルボニルオキシメチル基、1−エトキシカルボニルオキシ−1−エチル基、1−シクロへキシルオキシカルボニルオキシ−1−エチル基など);アリールオキシカルボニルオキシアルキル基(例えば、2−(フェノキシカルボニルオキシ)エチル基、2−(5−インダニルオキシカルボニルオキシ)エチル基など);アルコキシアルキルカルボニルオキシアルキル基(例えば、2−(1−メトキシ−2−メチルプロパン−2−オイルオキシ)エチル基など);アリールアルキルオキシカルボニルオキシアルキル基(例えば、2−(ベンジルオキシカルボニルオキシ)エチル基など);アリールアルケニルオキシカルボニルオキシアルキル基(例えば、2−(3−フェニルプロペン−2−イルオキシカルボニルオキシ)エチル基など);アルコキシカルボニルアミノアルキル基(例えば、t−ブチルオキシカルボニルアミノメチル基など);アルキルアミノカルボニルアミノアルキル基(例えば、メチルアミノカルボニルアミノメチル基など);アルカノイルアミノアルキル基(例えば、アセチルアミノメチル基など);複素環式カルボニルオキシアルキル基(例えば、4−メチルピペラジニルカルボニルオキシメチル基など);ジアルキルアミノカルボニルアルキル基(例えば、ジメチルアミノカルボニルメチル基、ジエチルアミノカルボニルメチル基など);(5−(低級アルキル)−2−オキソ−1,3−ジオキソレン−4−イル)アルキル基(例えば、(5−t−ブチル−2−オキソ−1,3−ジオキソレン−4−イル)メチル基など);ならびに、(5−フェニル−2−オキソ−1,3−ジオキソレン−4−イル)アルキル基(例えば、(5−フェニル−2−オキソ−1,3−ジオキソレン−4−イル)メチル基など)である。
【0174】
代表的なアミドカルボキシ保護基は、アミノカルボニル基および低級アルキルアミノカルボニル基である。
【0175】
本発明の好ましいカルボキシ保護された化合物には複数の化合物があり、ここでは、その保護されたカルボキシ基は、低級アルキルエステル、シクロアルキルエステルもしくはアリールアルキルエステル(例えば、メチルエステル、エチルエステル、プロピルエステル、イソプロピルエステル、ブチルエステル、sec−ブチルエステル、イソブチルエステル、アミルエステル、イソアミルエステル、オクチルエステル、シクロヘキシルエステル、フェニルエチルエステルなど)、またはアルカノイルオキシアルキルエステル、シクロアルカノイルオキシアルキルエステル、アロイルオキシアルキルエステル、もしくはアリールアルキルカルボニルオキシアルキルエステルである。好ましいアミドカルボキシ保護基は、低級アルキルアミノカルボニル基である。例えば、アスパラギン酸は、酸不安定基(例えば、t−ブチル基)によってα−C−末端で保護され得、そして水素化不安定基(例えば、ベンジル基)によってβ−C−末端で保護され得、次いで、合成の間に選択的に脱保護され得る。
【0176】
ペプチドの改変
本発明の改変されたペプチドの生産様式は、そのペプチドを含む種々の要素の性質に応じて広く変化するであろう。合成手順は、単純であるように、高収率を提供するように、そして高度に精製された安定な生成物を可能とするように選択されるであろう。通常は、化学反応基は、合成の最終段階で、例えば、カルボキシル基を用いて作製され、エステル化されて活性エステルが形成させられるであろう。本発明の改変されたペプチドの生産のための特定の方法が以下に記載される。
【0177】
具体的には、選択されたペプチドは、抗ウイルス活性について最初にアッセイされ、次いでペプチドのN末端、C末端または内部のいずれかのみで、連結基を用いて改変される。その後、この改変されたペプチド連結基の抗ウイルス活性がアッセイされる。抗ウイルス活性が劇的に低下(すなわち、10分の1未満に低下)しない場合は、その後、改変されたペプチド連結基の安定性がそのインビボ寿命によって測定される。この安定性が所望されるレベルにまで改善されていなければ、次いでペプチドは、別の部位で改変され、そして所望されるレベルの抗ウイルスおよび安定性が得られるまでこの手順が繰り返される。
【0178】
さらに具体的には、リンカーおよび反応基での改変を受けさせるために選択された個々のペプチドが、以下の基準に従って改変されるであろう:末端のカルボン酸基がペプチド上で利用可能であり、そしてこの末端のカルボン酸基が抗ウイルス活性の保持に重要ではなく、他の感受性官能基がペプチド上に存在しない場合は、このカルボン酸が、リンカー反応基改変のための結合点として選択されるであろう。末端のカルボン酸基が抗ウイルス活性に関与している場合、またはカルボン酸が利用できない場合は、抗ウイルス活性の保持に重要ではない任意の他の感受性官能基が、リンカー反応要素の改変のための結合点として選択されるであろう。いくつかの感受性官能基をペプチド上で利用できる場合は、リンカー/反応要素の付加および保護された全ての感受性官能基の脱保護後に、抗ウイルス活性の保持がなおも得られるような様式で、保護基の組み合わせが使用されるであろう。利用できる感受性官能基がペプチド上にない場合、またはより単純な改変経路が所望される場合は、合成による試みにより、抗ウイルス活性の保持が維持されるような様式で、もともとのペプチドの改変が可能となるであろう。この場合、改変はペプチドの反対側の末端で起こるであろう。
【0179】
NHS誘導体は、ペプチド中に他の感受性官能基が存在しない場合にカルボン酸から合成され得る。特に、このようなペプチドは、無水CH2Cl2およびEDC中でN−ヒドロキシスクシンイミドと反応させられ、そしてこの生成物は、クロマトグラフィーによって精製されるか、またはNHS誘導体を生じさせるための適切な溶媒系から再結晶化させられる。
【0180】
あるいは、NHS誘導体は、アミノ基および/またはチオール基と、カルボン酸を含むペプチドから合成され得る。分子中に遊離のアミノ基またはチオール基が存在する場合は、NHS誘導体の付加が行われる前に、これらの感受性官能基が保護されることが好ましい。例えば、分子に遊離のアミノ基が含まれる場合は、Fmoc保護されたアミンまたは好ましくはtBoc保護されたアミンへのアミンの変換が、上記化学反応が行われる前に必要とされる。アミン官能基は、NHS誘導体の調製後も脱保護されないであろう。従って、この方法は、所望される抗ウイルス効果を誘導するために、そのアミン基が遊離状態であることが必要ではない化合物に対してのみ適用される。アミノ基が分子のもともとの特性を保持するために遊離状態であることが必要とされる場合は、以下に記載される別のタイプの化学反応が行われなければならない。
【0181】
加えて、NHS誘導体は、アミノ基またはチオール基が含まれ、そしてカルボン酸が含まれていないペプチドから合成され得る。選択された分子にカルボン酸が含まれていない場合は、一連の二官能性リンカーを使用して、その分子を反応性NHS誘導体へと変換させることができる。例えば、DMF中に溶解させられ、そして遊離アミノ含有分子に付加させられたエチレングリコール−ビス(スクシンイミジルスクシネート)(EGS)およびトリエチルアミン(EGSの方が有利なように10:1の比で)は、モノNHS誘導体を生じるであろう。チオールで誘導された分子からNHS誘導体を生じさせるためには、DMF中のN−[−マレイミドブチリルオキシ]スクシンイミドエステル(GMBS)とトリエチルアミンを使用することができる。マレイミド基は遊離のチオールと反応し、そしてNHS誘導体は、シリカ上でのクロマトグラフィーまたはHPLCによって反応混合物から精製されるであろう。
【0182】
NHS誘導体はまた、複数の感受性官能基を含むペプチドからも合成され得る。それぞれの場合について、異なる様式で分析と解明がなされなければならないであろう。しかし、市販されている多量の保護基および二官能性リンカーのお陰で、本発明は、ペプチドを改変するために、好ましくはわずか1つの化学的工程で(上記に記載されるように)、または2工程(上記に記載されたように、感受性基の事前の保護を含む)もしくは3工程(保護、活性化、および脱保護)で任意のペプチドに適用することができる。例外的な状況下でのみ、ペプチドを活性なNHSまたはマレイミド誘導体に変換させるために、複数の(3工程を超える)合成工程が必要であろう。
【0183】
マレイミド誘導体はまた、遊離のアミノ基と遊離のカルボン酸を含むペプチドからも合成され得る。アミノ誘導された分子からマレイミド誘導体を生じさせるためには、DMF中のN−[γ−マレイミドブチリルオキシ]スクシンイミドエステル(GMBS)とトリエチルアミンを使用することができる。スクシンイミドエステル基は遊離のアミノと反応し、そしてマレイミド誘導体は、結晶化によって、またはシリカ上でのクロマトグラフィーによって、またはHPLCによって、反応混合物から精製されるであろう。
【0184】
最後に、マレイミド誘導体は、複数の他の感受性官能基を含み、そして遊離のカルボン酸を含まないペプチドから合成され得る。選択された分子にカルボン酸が含まれていない場合は、一連の二官能性架橋試薬を使用して、分子を反応性NHS誘導体へと変換させることができる。例えば、DMF中でのHBTU/HOBt/DIEA活性化を使用して、マレイミドプロピオン酸(MPA)を遊離のアミンにカップリングさせて、遊離のアミンとMPAのカルボン酸基との反応を通してマレイミド誘導体を生じさせることができる。
【0185】
多くの他の市販されているヘテロ二官能性架橋試薬を、必要とされる場合には代替的に使用することができる。多数の二官能性化合物を要素への架橋のために利用することができる。例示的な試薬としては、以下が挙げられる:アジドベンゾイルヒドラジド、N−[4−(p−アジドサリチルアミノ)ブチル]−3’−[2’−ピリジルジチオ)プロピオンアミド)、ビス−スルホスクシンイミジルスベレート、ジメチルアジピイミデート、ジスクシンイミジル酒石酸塩、N−γ−マレイミドブチリルオキシスクシンイミドエステル、N−ヒドロキシスルホスクシンイミジル−4−アジドベンゾエート、N−スクシンイミジル[4−アジドフェニル]−1,3’−ジチオプロピオネート、N−スクシンイミジル[4−ヨードアセチル]アミノベンゾエート、グルタルアルデヒド、およびスクシンイミジル4−[N−マレイミドメチル]シクロヘキサン−1−カルボキシレート。
【0186】
改変された抗ウイルスペプチドの使用
本発明の改変された抗ウイルスペプチドは、ウイルス感染に罹患している患者の処置において治療薬として使用され得、そして以下に記載される方法および当該分野で公知の他の方法に従って患者に投与することができる。改変されたペプチドの有効な治療的投薬量は、当業者に周知の手順によって決定され得、そしてこのペプチドの潜在的な毒性に関する任意の懸念が考慮されるであろう。
【0187】
改変されたペプチドはまた、これまでには感染していない個体に対して予防的に投与することもできる。このことは、個体がウイルス伝染の危険性が高い感染した個体と接触した場合に起こり得るように、個体がウイルスに曝される高い危険性に供されている場合に有利であり得る。このことは、ウイルス(例えば、HIVウイルス)に対する公知の治療法がある場合には特に有利であり得る。一例として、改変された抗HIVペプチドの予防的な投与は、医療施設の労働者がHIV感染個体に由来する血液に曝されている状況において、または個体をHIVウイルスに曝す可能性があるリスクの高い活動にその個体が従事している他の状況において有利であろう。
【0188】
改変された抗ウイルスペプチドおよび抗膜融合性ペプチドの投与
一般的に、改変されたペプチドは、生理学的に許容される媒体(例えば、脱イオン水、リン酸緩衝化生理食塩水(PBS)、生理食塩水、水性エタノールまたは他のアルコール、血漿、タンパク様溶液、マンニトール、水性グルコース、アルコール、植物油など)の中で投与される。含まれ得る他の添加物としては、緩衝液(この場合、媒体は、約5〜10の範囲のpHで一般的に緩衝化され、緩衝液は一般的には約50〜250mMの濃度の範囲であろう)、塩(塩濃度は、一般的には約5〜500mMの範囲であろう)、生理的に許容される安定剤などが挙げられる。組成物は、保存および輸送に便利なように凍結乾燥させられる場合がある。
【0189】
本発明の改変されたペプチドは、大部分について、非経口的に(例えば、静脈内に(IV)、動脈内に(IA)、筋肉内に(IM)、皮下に(SC)など)投与される。投与は、適切な状況では、輸注により得る。官能基の反応が比較的遅いいくつかの場合には、投与は、経口的、経鼻的、直腸的、経皮的またはエーロゾルであり得、この場合、結合体の性質により脈管系への移動が可能となる。通常は、一回の注入が使用されるが、所望される場合には、2回以上の注入が使用される場合がある。改変されたペプチドは、任意の都合のよい手段(注射器、トロカール、カテーテルなどを含む)によって投与され得る。
【0190】
特定の実施形態においては、改変されたペプチドは、当該分野で公知の方法によって肺手段によって投与されるであろう。ペプチドまたはタンパク質のエアゾール乾燥粉末形態の肺深部への送達のための技術は、Pattonら(1997)Chemtech 27(12):34−38に開示されている。ペプチドの肺投与を開示しているさらなる参考文献としては、Senior,K.ら(2000)PSTT,第3巻:281−282;Gumbleton,M.(2006)Advanced Drug Delivery Reviews 58:993−995;「Drug Delivery:Pulmonary Delivery」と題されたNewhouse,M.T.(2006)Encyclopedia of Pharmaceutical Technology;およびLabiris,N.R.(2003)J.Clin.Pharmacology 56:600−612が挙げられる。これらの参考文献の全ての内容が引用により本明細書中に組み入れられる。
【0191】
投与の特定の様式は、投与される量、1回のボーラスであるかまたは継続的な投与であるかなどに依存して変化するであろう。好ましくは、投与は静脈内であり、この場合、導入の部位は、本発明にとって重要ではなく、好ましくは急速な血流が存在する部位(例えば、静脈内、末梢静脈または中心静脈)である。他の経路は、投与が、遅延性放出技術または保護マトリックスと結びつけられる使用を見出し得る。この意図は、改変されたペプチドが血液中で効率的に分布されることであり、その結果、血液成分と反応することができる。結合体の濃度は、一般的には、約1pg/ml〜50mg/mlまでの範囲で広範に変化するであろう。静脈内に投与される総量は、一般的には、約0.1mg/ml〜約50mg/ml、約5mg/ml〜40mg/ml、約10mg/ml〜30mg/ml、約10mg/ml〜20mg/ml、または約5mg/ml〜15mg/ml、約1mg/ml〜約10mg/ml、あるいは、約1mg/ml〜5mg/mlの範囲であろう。
【0192】
血液の長期間にわたり生存する成分(例えば、免疫グロブリン、血清アルブミン、赤血球および血小板)に結合させることにより、多数の利点が結果として生じる。ペプチドの活性は、数日〜数週間延長される。この期間に投与は1回のみ必要である。活性化合物は、大きい分子に主に結合されるので(ここでは、他の生理学的プロセスを妨げるように細胞内に取り込まれる可能性は低い)、より優れた特異性を得ることができる。
【0193】
改変されたペプチドの存在のモニタリング
哺乳動物宿主の血液は、1回以上、改変されたペプチド化合物の存在についてモニターされ得る。宿主の血液の一部または試料を採取することにより、ペプチドが治療的に活性であるために十分な量で長期間生存する血液成分に結合されているかどうかを決定することができ、そしてその後、血液中のペプチド化合物のレベルを決定することができる。所望される場合は、ペプチドがどの血液成分に結合されるかもまた決定することができる。これは、非特異的改変ペプチドが使用される場合に特に重要である。特定のマレイミドで改変されたペプチドに関して、血清アルブミンおよびIgGの半減期を算出することは、はるかに簡単である。
【0194】
免疫アッセイ
本発明の別の態様は、抗ウイルスペプチドおよび/もしくはアナログ、またはそれらの誘導体および結合体の、生物学的試料(例えば、血液)における濃度を、これらのペプチド、ペプチドアナログ、またはそれらの誘導体および結合体に対して特異的な抗体を用いて決定するための方法、ならびにこのようなペプチド、アナログおよび/またはこれらの誘導体もしくは結合体に強く関係している毒性についての処置としてのこのような抗体の使用に関する。患者におけるインビボでのこのペプチドの高い安定性と寿命は、処置の際に新しい問題(毒性に関する高い可能性を含む)を導く可能性があるので、このことは有利である。
【0195】
特定のペプチド、そのペプチドアナログまたは誘導体に対して特異性を有している抗治療薬剤抗体(モノクローナル抗体またはポリクローナル抗体のいずれか)の使用は、任意のこのような問題を媒介する手助けをし得る。抗体は、特定のペプチド、そのアナログもしくは誘導体を用いて、またはその因子の免疫原性断片、あるいはこの因子の抗原決定基に対応する合成された免疫原を用いて免疫された宿主から作製され得るか、またはそれらから導かれ得る。好ましい抗体は、ペプチド、ペプチドアナログ、もしくは誘導体の天然の形態、改変された形態、および結合させられた形態に対して、高い特異性と親和性を有するであろう。このような抗体はまた、酵素、蛍光色素、または放射性標識で標識することもできる。
【0196】
改変されたペプチドに特異的な抗体は、ペプチド特異的抗体の誘導のために精製されたペプチドを使用するすることにより作製され得る。抗体の誘導によっては、動物中への注射による免疫応答の刺激だけではなく、合成抗体または他の特異的結合分子の作製における類似の工程(例えば、組換え免疫グロブリンライブラリーのスクリーニング)も意図される。モノクローナル抗体およびポリクローナル抗体の両方が、当該分野で周知の手順により作成され得る。
【0197】
抗ペプチド抗体が、改変されたペプチド、そのアナログまたは誘導体の投与によって誘導された毒性を処置するために使用され得、そしてエキソビボでも、またインビボでも使用され得る。エキソビボでの方法には、固体支持体に固定された抗治療薬剤抗体を使用する、毒性に対する免疫透析処置が含まれるであろう。インビボでの方法には、抗体−薬剤複合体のクリアランスを誘導するために有効な量の抗治療薬剤抗体の投与が含まれる。
【0198】
抗体は、無菌条件下で抗体と血液を接触させることによって、エキソビボで患者の血液から、改変されたペプチド、そのアナログまたは誘導体、およびその結合体を除去するために使用され得る。例えば、抗体は、カラムマトリックス上に固着、あるいは固定することができ、そして患者の血液を患者から取り出して、マトリックス上を通過させることができる。改変されたペプチド、ペプチドアナログ、誘導体または結合体は抗体に結合し、そして低濃度のペプチド、アナログ、誘導体または結合体を含む血液が、その後、患者の循環系に戻され得る。除去されるペプチド化合物の量は、圧力および流速を調整することによって制御することができる。
【0199】
患者の血液の血漿成分からのペプチド、アナログ、誘導体および結合体の優先的な除去は、例えば、半透膜の使用によって、またはそうでなければ、抗治療薬抗体を含むマトリックス上に血漿成分を通過させる前に、当該分野で公知の方法により細胞成分から血漿成分をまず分離することによって行うことができる。あるいは、ペプチドが結合させられた血球(赤血球細胞を含む)の優先的な除去は、患者の血液中の血球を回収し、濃縮し、そして患者の血液の血清成分の排除のためにそれらの細胞を固着させられた抗治療薬抗体と接触させることによって行うことができる。
【0200】
抗治療薬抗体は、処置のためにペプチド、アナログ、誘導体または結合体が投与された患者に、インビボで非経口的に投与することができる。抗体は、ペプチド化合物および結合体に結合するであろう。一旦ペプチドに結合されると、活性は、完全にはブロックされないとしても、妨害され、それによって患者の血流中のペプチド化合物の生物学的有効濃度が低下し、そして有害な副作用が最少となるであろう。加えて、結合させられた抗体−ペプチド複合体は、患者の血流からのペプチド化合物と結合体のクリアランスを容易にするであろう。
【0201】
十分に記載されてきた本発明は、以下の非限定的な実施例を参照することにより、さらに認識され、そして理解することができる。
【実施例】
【0202】
(実施例1〜5)
C34のシステイン酸誘導体の合成と精製
C34のシステイン酸誘導体の合成は、ペプチドの作製の間に手作業による介入を伴うSymphony Peptide Synthesizer上での自動化された固相手順を使用して行った。合成は、Fmoc保護されたRamageアミドリンカー樹脂上で、Fmoc保護されたアミノ酸を使用して行った。カップリングは、O−ベンゾトリアゾール−1−イル−N,N,N’,N’−テトラメチル−ウロニウムヘキサフルオロホスフェート(HBTU)とジイソプロピルエチルアミン(DIEA)を活性化因子の混合物としてN,N−ジメチルホルムアミド(DMF)溶液中で使用することによって行った。Fmoc保護基を20%のピペリジン/DMFを使用して除去した。ペプチドが樹脂から切り離されると遊離のNα末端が生じるように、Boc保護されたアミノ酸をN末端に使用した。シグマコーティッドガラス反応容器を合成の際に使用した。
【0203】
(実施例2)
CA−C34の合成
CA−C34は以下のアミノ酸配列を有する:
【0204】
【化28−2】
CA−C34の改変されたペプチドを以下のように合成した:
工程1:100μモルスケールでのCA−C34の固相ペプチド合成を、手作業による固相合成、自動化された固相合成、Symphony Peptide SynthesizerおよびRamage樹脂を使用して行った。以下の保護されたアミノ酸を順番に樹脂に付加させた:
【0205】
【化28−3】
これらをN,N−ジメチルホルムアミド(DMF)に溶解させ、そして配列に従って、O−ベンゾトリアゾール−1−イル−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)およびジイソプロピルエチルアミン(DIEA)を使用して活性化させた。Fmoc保護基の除去は、N,N−ジメチルホルムアミド(DMF)中の20%(V/V)ピペリジンの溶液を使用して20分間行った(工程1)。
【0206】
工程2:ペプチドを、85%のTFA/5%のTIS/5%のチオアニソールおよび5%のフェノールを使用して樹脂から切断し、続いてドライアイス冷(0〜4℃)Et2Oによって沈殿させ、回収した。
【0207】
(実施例3)
CA−C34(Arg28)の合成
CA−C34(Arg28)は以下のアミノ酸配列を有する:
【0208】
【化28−4】
工程1:100μモルスケールでのCA−C34(Arg28)の固相ペプチド合成を、手作業による固相合成、自動化された固相合成、Symphony Peptide SynthesizerおよびRamage樹脂を使用して行った。以下の保護されたアミノ酸を順番に樹脂に付加させた:
【0209】
【化28−5】
これらをN,N−ジメチルホルムアミド(DMF)に溶解させ、そして配列に従って、O−ベンゾトリアゾール−1−イル−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)およびジイソプロピルエチルアミン(DIEA)を使用して活性化させた。Fmoc保護基の除去は、N,N−ジメチルホルムアミド(DMF)中の20%(V/V)ピペリジンの溶液を使用して20分間行った(工程1)。
【0210】
工程2:ペプチドを、85%のTFA/5%のTIS/5%のチオアニソールおよび5%のフェノールを使用して樹脂から切断し、続いてドライアイス冷(0〜4℃)Et2Oによって沈殿させ、回収した。
【0211】
(実施例4)
CA−C34−Lys35(ε−AEEA−MPA)の合成
CA−C34−Lys35(ε−AEEA−MPA)は以下のアミノ酸配列を有する:
【0212】
【化28−6】
工程1:100μモルスケールでのCA−C34−Lys35(ε−AEEA−MPA)の固相ペプチド合成を、手作業による固相合成、自動化された固相合成、Symphony Peptide SynthesizerおよびRamage樹脂を使用して行った。以下の保護されたアミノ酸を順番に樹脂に付加させた:
【0213】
【化28−7】
これらをN,N−ジメチルホルムアミド(DMF)に溶解させ、そして配列に従って、O−ベンゾトリアゾール−1−イル−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)およびジイソプロピルエチルアミン(DIEA)を使用して活性化させた。Fmoc保護基の除去は、N,N−ジメチルホルムアミド(DMF)中の20%(V/V)ピペリジンの溶液を使用して20分間行った(工程1)。
【0214】
工程2:Lys(Aloc)基の選択的脱保護を手作業で、そして5mLのC6H6:CHCl3(1:1):2.5%のNMM(v:v):5%のAcOH(v:v)中に溶解させた3等量のPd(PPh3)4の溶液で樹脂を2時間処理することによって行った(工程2)。その後、樹脂をCHCl3(6×5mL)、DCM中の20%のAcOH(6×5mL)、DCM(6×5mL)、およびDMF(6×5mL)で洗浄した。
【0215】
工程3:その後、合成をFmoc−AEEA−OHと3−マレイミドプロピオン酸の付加のために再度自動化した(工程3)。AEEA上の保護基(Fmoc)を先に記載したように除去し、それぞれのカップリングの間に、樹脂をN,N−ジメチルホルムアミド(DMF)で3回、そしてイソプロパノールで3回洗浄した。
【0216】
工程4:ペプチドを85%のTFA/5%のTIS/5%のチオアニソールおよび5%のフェノールを使用して樹脂から切断し、続いてドライアイス冷(0〜4℃)Et2Oによって沈殿させ(工程4)、回収した。
【0217】
(実施例5)
CA−C34(Arg28)−Lys35(ε−AEEA−MPA)
CA−C34(Arg28)−Lys35(ε−AEEA−MPA)は以下の配列を有する:
【0218】
【化28−8】
工程1:100μモルスケールでのCA−C34(Arg28)−Lys35(ε−AEEA−MPA)の固相ペプチド合成を、手作業による固相合成、自動化された固相合成、Symphony Peptide SynthesizerおよびRamage樹脂を使用して行った。以下の保護されたアミノ酸を順番に樹脂に付加させた:
【0219】
【化28−9】
これらをN,N−ジメチルホルムアミド(DMF)に溶解させ、そして配列に従って、O−ベンゾトリアゾール−1−イル−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)およびジイソプロピルエチルアミン(DIEA)を使用して活性化させた。Fmoc保護基の除去は、N,N−ジメチルホルムアミド(DMF)中の20%(V/V)ピペリジンの溶液を使用して20分間行った(工程1)。
【0220】
工程2:Lys(Aloc)基の選択的脱保護を手作業で、そして5mLのC6H6:CHCl3(1:1):2.5%のNMM(v:v):5%のAcOH(v:v)中に溶解させた3等量のPd(PPh3)4の溶液で樹脂を2時間処理することによって行った(工程2)。その後、樹脂をCHCl3(6×5mL)、DCM中の20%のAcOH(6×5mL)、DCM(6×5mL)、およびDMF(6×5mL)で洗浄した。
【0221】
工程3:その後、合成をFmoc−AEEA−OHと3−マレイミドプロピオン酸の付加のために再度自動化した(工程3)。AEEA上の保護基(Fmoc)を先に記載したように除去し、それぞれのカップリングの間に、樹脂をN,N−ジメチルホルムアミド(DMF)で3回、そしてイソプロパノールで3回洗浄した。
【0222】
工程4:ペプチドを85%のTFA/5%のTIS/5%のチオアニソールおよび5%のフェノールを使用して樹脂から切断し、続いてドライアイス冷(0〜4℃)Et2Oによって沈殿させ(工程4)、回収した。
【0223】
精製手順:
C34の改変されたペプチドをそれぞれ、Varian(Dynamax)分取バイナリーHPLCシステムを使用して、分取逆相HPLCによって精製した。
【0224】
例示的な誘導体の精製は、水/TFA混合物(H2O中の0.1%のTFA;溶媒A)とアセトニトリル/TFA(CH3CN中の0.1%のTFA;溶媒B)で平衡化させたPhenomenex Luna 10μフェニル−ヘキシル、50mm×250mmカラム(粒子10μ)を使用して行った。溶離は、28〜38%のB勾配を180分間にわたって50mL/分で流すことによって行った。ペプチドを含む画分を、214nmと254nmのUV吸光度(Varian Dynamax UVD II)によって検出した。
【0225】
画分を25mLのアリコートの中に回収した。所望される生成物を含む画分を、LC/MS上への直接の注入後の質量の検出によって同定した。選択した画分をその後、分析用HPLC(20分間にわたる20〜60%のB;Phenomenex Luna 5μフェニル−ヘキシル、10mm×250mmカラム、0.5mL/分)によって分析して、≧90%の純度を有している画分をプールするために同定した。プールを液体窒素を使用して凍結乾燥させ、その後、少なくとも2日間凍結乾燥させて白色粉末を得た。
【0226】
IV−フローチャート:
同じ合成反応スキームを全ての誘導体に使用した。CA−C34とCA−C34−Lys35(ε−AEEA−MPA)についての反応スキームを以下のフローチャートにおいて説明する。もちろん、AEEAおよびMPAの付加に伴うAloc除去工程はCA−C34については省略した。
【0227】
【化29】
(実施例6)
C34のシステイン酸誘導体の溶解度アッセイ
a.溶解度アッセイは、100mMのリン酸ナトリウム(開始時pH8)水溶液中で行った。高い緩衝液濃度がHPLCによる精製の後にC34誘導体とともに残っている過剰量のトリフルオロ酢酸(TFA)を中和するために有用であることが明らかにされている。
【0228】
5.8〜7.0の最終的なpHが様々なC34誘導体の溶解度を維持するためには適している。したがって、開始緩衝液をpH8.0で調製し;そしてC34誘導体の可溶化によりおよそ6.3の最終的なpHが生じた。表2は、N末端にシステイン酸(CA)を持つかまたは持たない、そしてC末端にLys35(ε−AEEA−MPA)を持つかまたは持たないC34の溶解限度を示す。さらに、表1に示す最終的なオスモル濃度は、これらの最終的な溶液が等張性であることを明らかにしている。
【0229】
【表2】
天然のC34は15.75mg/mlでは溶解性であることが明らかであり、得られる溶液は数分以内にゲルを形成する。C34−Lys35(ε−AEEA−MPA)は、これが溶液に投入されると直ちにゲルを形成し、さらに緩衝液を加えても決して化合物を可溶化させることはできない。表1から注目され得るように、これらの化合物の両方のN末端でのシステイン酸の付加は、C34に対して有意に高い溶解度(すなわち、それぞれ29.3mg/mlおよび33.8mg/ml)を付与する。
【0230】
b.500mMのリン酸ナトリウム緩衝液(pH8.0)中で30mg/ml〜35mg/mlのC34に連結させられたN末端が改変されたAEEA−MPA(W1(AEEA−MPA)−C34)、およびAEEAリンカーを介してMPAに連結させられた35位にリジン付加を有しているN末端システイン酸が改変されたC34(CA K35(AEEA−MPA−C34))の溶解度
1Mのリン酸ナトリウム緩衝液(pH8.0)
A−2Mのリン酸二水素ナトリウム、無水物、UQAM、OM−27、S1835 1M=141.96g/1L、2M=13.3568g/40mlのnanopure H2O。
B−2Mのリン酸一ナトリウム、一水和物、UQAM、OM−27、S1820 1M=137.99g/1L、2M=11.0392g/40mlのnanopure H2O
C−Mix 30ml nanopure H2O+およそ28mlのリン酸二水素ナトリウム+1.5mlのリン酸一ナトリウム。pHが8.0であるかどうかを確認する。容量をリン酸二水素ナトリウムで調整する
D−最終pHを8に(実際には7.98)に調整する。
【0231】
500mMのリン酸ナトリウム緩衝液(pH8.0)
1Mのリン酸ナトリウム緩衝液(pH8.0)を等量のnanopure H2O(濾過していない)と混合する。最終的なpHは8.0である。
【0232】
c.500mMのリン酸ナトリウム(pH8.0)中で100mg/mlでのW1(AEEA−MPA)−C34および(CA K35(AEEA−MPA−C34))の溶解度
化合物:
W1(AEEA−MPA)−C34 Batch B,ロット番号JC−205−12,Inventory no 1139.
塩を含む1M=4769.1=12.64mg重量
塩を含まない1M=4541.1=12.0357mg
純度=90.8%=100mg/mlについて109.3ulの500mMのリン酸ナトリウム(pH8.0)中の10.928mg。
【0233】
注釈:ガラスバイアル中の粉末に対して緩衝液を加えた。1分間のボルテックス(中速)後は可溶性であった。3つの粒子が残った。最終的なpHは6.82であった(Chemistry DepartmentによるpHメーターで測定した、500mMのNaP pH8緩衝液は8.1であった)。
【0234】
(CA K35(AEEA−MPA−C34))Batch B,ロット番号PB−262−01,Inventory no 1352.
塩を含む1M=5165.2=12.11mg重量
塩を含まない1M=4820.2=11.30mg
純度=92.8%=100mg/mlについて104.9ulの500mMのリン酸ナトリウム(pH8.0)中10.487mg。
【0235】
注釈:ガラスバイアルの中の粉末に対して緩衝液を加えた。1分30秒間のボルテックス(中速)後はほとんど可溶性であった。2つの小さいペレットがガラスバイアルの底にあった。3分後、これらの2つの小さいペレットは溶解した。最終的なpHは6.75であった(Chemistry DepartmentによるpHメーターで測定した、500mMのNaP pH8緩衝液は8.1であった)。
【0236】
W1(AEEA−MPA)−C34と(CA K35(AEEA−MPA−C34))は、一度溶解すれば黄色である。W1(AEEA−MPA)−C34はさらに濃い色である。
【0237】
結論:
W1(AEEA−MPA)−C34化合物と(CA K35(AEEA−MPA−C34))化合物は、500mMのリン酸ナトリウム緩衝液(pH8.0)中で100mg/mlでは可溶性である。それらの最終的なpHは許容される限度、すなわち6.8を上回る。これらの化合物についてのpHの許容限度は6.2である。
【0238】
d.500mMのリン酸ナトリウム(pH8.0)中の150mg/mlでのW1(AEEA−MPA)−C34および(CA K35(AEEA−MPA−C34))の溶解度
W1(AEEA−MPA)−C34 Batch B,ロット番号JC−205−12,Inventory no 1139.
塩を含む1M=4769.1=11.97mg重量
塩を含まない1M=4541.1=11.398mg
純度=90.8%=150mg/mlについて69ulの500mMのリン酸ナトリウム(pH8.0)中の10.35mg。
【0239】
注釈:緩衝液をガラスバイアル中の粉末に対して加えた。1分間のボルテックス(中速〜高速)後は可溶性であった。最終的なpHは6.60であった(Chemistry DepartmentによるpHメーターで測定した、500mMのNaP pH8緩衝液は8.23であった)。
【0240】
(CA K35(AEEA−MPA−C34))Batch B,ロット番号PB−262−01,Inventory no 1352.
塩を含む1M=5165.2=12.48mg重量
塩を含まない1M=4820.2=11.64mg
純度=92.8%=150mg/mlについて72.05ulの500mMのリン酸ナトリウム(pH8.0)中の10.808mg。
【0241】
注釈:ガラスバイアルの中の粉末に対して緩衝液を加えた。1分間のボルテックス(中速〜高速)後はほとんど可溶性であった。1つの小さいペレットがガラスバイアルの底にあった。10分後、この小さいペレットは溶解した。最終的なpHは6.57であった(Chemistry DepartmentによるpHメーターで測定した、500mMのNaP pH8緩衝液は8.23であった)。
【0242】
W1(AEEA−MPA)−C34と(CA K35(AEEA−MPA−C34))は一度溶解すると黄色である。W1(AEEA−MPA)−C34はさらに濃い色である。
【0243】
結論:W1(AEEA−MPA)−C34と(CA K35(AEEA−MPA−C34))はいずれも、500mMのリン酸ナトリウム緩衝液(pH8.0)中では150mg/mlで可溶性である。
【0244】
(実施例7)
C34のシステイン酸誘導体の活性アッセイ
第1の活性アッセイ:抗HIV−1に誘導される細胞と細胞の融合のアッセイ
様々な抗膜融合性化合物とそれぞれの予め形成させたアルブミン結合体の効力を、New York Blood Center,310 East 67th Street,New York,N.Y.10021においてShibo Jiang博士と共同研究者らによって設計されたHIV−1に誘導される細胞と細胞の融合のアッセイを使用して評価した。HIVに誘導される細胞と細胞の融合に対するC34の阻害活性は以前に記載されているように検出した(Jiang,S.L.ら(2000)A Convenient Cell Fusion Assay for Rapid Screening for HIV Entry Inhibitors,Proc.SPIE 3926:212−219)。
【0245】
簡単に説明すると、化合物を最初に、ストック溶液としてリン酸−クエン酸緩衝液(pH7.0)中に100μMに稀釈し、その後、1000nM、500nM、250nM、100nM、50nM、25nM、5nM、1nMに培養培地中にさらに稀釈した。50μlのこの化合物溶液をカルセイン−AM(Molecular Probes,Inc.,Eugene,OR)で標識した50μlのHIV−1IIIB感染H9細胞(H9/HIV−1IIIB)と、2×105細胞/mlで混合した。37℃で2時間の共培養の後、カルセインで標識されたH9/HIV−1IIIB細胞を、MT−2細胞と融合したものまたは融合していないものを両方、それぞれ、485nMおよび535nmのフィルター励起波長を使用して、接眼ミクロメーターディスク(10×10mm sq.)と20×の対物レンズを備えた倒立蛍光顕微鏡(Zeiss,Gennany)下でカウントした。融合した細胞は融合していない細胞よりもはるかに大きく(少なくとも2倍)、したがって、融合した細胞中での蛍光の強度は、1つの細胞から2つ以上の細胞へのカルセインの拡散が原因で、融合していない細胞中での蛍光強度よりも弱い。1ウェルあたり4つの領域を試験し、細胞融合の割合を以下の式によって計算した:融合した細胞/(融合した細胞+融合していない細胞)×100%。
【0246】
ポジティブ対照のウェルに、50μlのカルセインで標識したHIV感染細胞を添加した。ネガティブ対照のウェルには、培養培地と、カルセインで標識した非感染H9細胞を添加した。細胞融合の阻害率(%)を以下の式を使用して計算した:[1−(E−N)/(P−N)]×100%、式中、「E」は実験グループにおける細胞融合の割合(%)を示し、「P」は、試験化合物を添加しなかったポジティブ対照グループにおける融合の割合(%)を示し、「N」は、カルセインで標識したH9/HIV−1IIIB細胞をカルセインで標識したH9細胞で置換したネガティブ対照グループにおける融合の割合(%)を意味する。抗ウイルス化合物による細胞融合の50%の阻害のための濃度(IC50)を、T.C.Chou博士から懇意によって提供されたコンピュータープログラム(Chou,T.C.and Hayball,M.P.,CalcuSyn:Windows(登録商標) software for dose effect analysis(1991)Ferguson,MO63135,USA,BIOSOFT)を使用して計算した。
【0247】
表3は、N末端にシステイン酸を持つかまたは持たない、そして基Lys(ε−AEEA−MPA)を介してヒト血清アルブミン(HSA)に結合させられているかまたは結合させられていないC34の抗膜融合活性を示す。
【0248】
【表3】
表3に示すように、C34の抗膜融合活性とCA−C34の抗膜融合活性との間では有意な差は観察されなかった。したがって、C34のN末端でのシステイン酸の付加は、C34の抗膜融合活性に対してネガティブな影響は与えない。
【0249】
表2もまた、それらのC末端へのC34およびCA−C34に対するLys(ε−AEEA−MPA)のカップリング、それに続くHSAに対するそれらの結合が、それらの抗膜融合活性に対して有意なネガティブな影響を持たないことを示している。
【0250】
(実施例8)
第2の活性のアッセイ:ヒトPBMC中でのHIVIIIbの複製の阻害
化合物の抗HIV効力と細胞傷害性を、HIV−1株IIIBを使用したPBMCをベースとするアッセイにおいて急性感染後に評価した。これらの実験は、以下に記載するプロトコールにしたがってSouthern Research Institute,Infectious Disease Research Department,431 Aviation Way,Frederick,MD.で行った。
【0251】
a.PBMCのHIV−1感染
HIVおよびHBVについて血清反応陰性である新鮮なヒトPBMCを、スクリーニングしたドナーから単離し、そしてBiological Specialty Corporation Colmar,PAからも商業的に提供された。細胞を低速での遠心分離とPBS中への再懸濁によって2〜3回ペレット化/洗浄して、混入している血小板を取り除いた。その後、白血球保有血をダルベッコのリン酸緩衝化生理食塩水(Dulbecco’s Phosphate Buffered Saline)(DPBS)で稀釈し、50mLの遠心管の中でLymphocyte Separation Medium(LSM;Mediatech,Inc.によるCellgro(登録商標);密度1.078+/−0.002g/ml;カタログ番号85−072−CL)の上に積層させ、次いで遠心分離した。バフィーコート層を、得られた界面から穏やかに吸引し、その後、低速遠心分離によってPBSで洗浄した。3回目の洗浄の後、細胞をウシ胎児血清(FBS)、L−グルタミン、ペニシリン、ストレプトマイシン、およびフィトヘマグルチニン(PHA−P;Sigma,St−Louis,MO)を補充したRPMI 1640中に再懸濁させた。細胞を37℃でインキュベートした。2日間のインキュベーションの後、PBMCを遠心分離し、FBS、L−グルタミン、ペニシリン、ストレプトマイシン、および組み換え体ヒトIL−2(R&D Systems,Inc.,Minneapolis,MN)を含むRPMI 1640中に再懸濁させた。IL−2を、PHA分裂刺激によって開始させた細胞分裂を維持するために培養培地中に含めた。細胞を最長で2週間培養物中で維持し、単球を組織培養フラスコへの接着の結果として培養物から枯渇させた。
【0252】
標準的なPBMCアッセイについては、少なくとも2人の正常なドナーに由来するPHA−Pで刺激した細胞をプールし、新鮮な培地中に稀釈し、Infectious Disease Research department of Southern Research Instituteによって開発された標準的な形式で96ウェル丸底マイクロプレートの内部ウェルにプレートした。2人以上のドナーに由来するPBMCのプールを、HIV感染の量的および質的相違と、初代リンパ球集団のPHAとIL−2に対する全体的な応答によって生じる個々のドナーの間で観察されるばらつきを最小にするために使用した。それぞれのプレートには、ウイルス/細胞対照ウェル(細胞+ウイルス)、実験用ウェル(化合物+細胞+ウイルス)、および化合物対照ウェル(細胞傷害性のMTSモニタリングに必要な、化合物+培地、細胞なし)を含めた。試験化合物の希釈物をマイクロタイターチューブの中に調製し、それぞれの濃度を標準的な形式を使用して適切なウェルの中に置いた。次いで、PBMCに対する化合物の稀釈物の添加後、予め決定した希釈率のウイルスストック溶液を、それぞれの試験ウェルの中に入れた(最終的なMOI約0.1)。ウイルスストック溶液は、NIAID AIDS Research and Reference Reagent Programから入手した継代数の低い臨床的な単離物HIV−1IIIBから調製した。−80℃で保存したHIV−1IIIBの予め滴定したアリコートを、使用の直前に生物学的安全キャビネットの中で室温になるように迅速に融解させた。HIV−1はPBMCに対して細胞変性性ではないので、同じアッセイプレートを抗ウイルス効力と細胞傷害性の測定の両方に使用することができる。PBMC培養物を、37℃、5%のCO2で感染後7日間維持した。
【0253】
b.逆転写活性アッセイ
マイクロタイタープレートをベースとする逆転写酵素(RT)反応を利用した(Buckheitら、AIDS Research and Human Retroviruses 7:295−302,1991)。滴定したチミジン三リン酸(3H−TTP、80Ci/mmol,NEN)を、1mCi/mlで1:1のdH2O:エタノール中に入れた。ポリrA:オリゴdT鋳型:プライマー(Pharmacia)を、150μlのポリrA(20mg/ml)を0.5mlのオリゴdT(20単位/ml)および5.35mlの滅菌dH2Oと一緒にし、続いてアリコートする(1.0ml)ことによってストック溶液として調製し、−20℃で保存した。RT反応緩衝液は毎日新しく調製し、これは、125μlの1.0MのEGTA、125μlのdH2O、125μlの20%のTriton X100、50μlの1.0MのTris(pH7.4)、50μlの1.0MのDTT、および40μlの1.0MのMgCl2からなる。最終的な反応混合物は、1部の3H−TTP、4部のdH2O、2.5部のポリrA:オリゴdTストック、および2.5部の反応緩衝液を合わせることによって調製した。10μlのこの反応混合物を丸底マイクロタイタープレートに入れ、15μlのウイルスを含む上清を添加し、混合した。プレートを37℃で60分間インキュベートした。インキュベーション後、反応容量をDE81フィルターマット(Wallac)上にスポットし、5%のリン酸ナトリウム緩衝液または2×SSC(Life Technologies)の中でそれぞれ5分間を5回、蒸留水中でそれぞれ1分間を2回、70%のエタノール中でそれぞれ1分間を2回洗浄し、そしてその後乾燥させた。取り込まれた放射活性(カウント毎分(counts per minute)、CPM)を、標準的な液体シンチレーション技術を使用して定量した。
【0254】
c.細胞傷害性を測定するためのPBMCの生存性についてのMTS染色
アッセイの最後に、アッセイプレートを可溶性のテトラゾリウムをベースとする色素MTS(CellTiter Reagent,Promega)で染色して、細胞の生存性を決定し、化合物の細胞傷害性を定量した。MTSは、代謝活性のある細胞のミトコンドリア酵素によって代謝されて、可溶性のホルマザン産物を生じ、それにより、細胞の生存性と化合物の細胞傷害性の迅速な定量的分析が可能となる。MTSは、使用の前に調製の必要のない安定な溶液である。アッセイの最後に、1ウェルあたり20μlのMTS試薬を添加した。HIV PBMCアッセイのために、ウェルを37℃で4時間インキュベートした。インキュベーションの間隔は、それぞれの細胞のタイプの中での最適な色素の還元について経験的に決定された時間に基づいて選択した。接着性のプレートシーラーをリッドの代わりに使用し、密閉したプレートを可溶性のホルマザン産物を混合するために数回反転させ、プレートをMolecular Devices Vmaxプレートリーダーを用いて490/650nmで分光光度測定によって読み取った。
【0255】
d.データ分析
社内コンピュータープログラム(in−house computer program)を使用して、IC50(ウイルスの複製の50%の阻害)、IC90(ウイルスの複製の90%の阻害)、TC50(50%の細胞傷害性)、TC90(90%の細胞傷害性)、および治療指数(TI=TC50/IC50)を計算した。データのグラフ表示を伴う抗ウイルス活性と細胞傷害性の両方についての生データは、個々の化合物の活性をまとめている印刷物に提供される。AZTを抗HIVアッセイにおいて関連するポジティブ対照化合物と平行して評価した。
【0256】
e.結果
図1は、天然のC34によるPBMC中でのHIV−1IIIB複製の阻害を示す(曲線◆を参照のこと)。この化合物は、以下に示すように、PBMCに対して有意な細胞傷害性の影響を全く示さなかった(曲線□を参照のこと)。
【0257】
図2は、C末端に付加されたリジンのεNH2上にAEEA−MPAを有しているC34のアルブミン結合体(すなわち、C34−Lys35(ε−AEEA−MPA):HSA)によるPBMC中でのHIV−1IIIB複製の阻害を示す(曲線◆を参照のこと)。この化合物は、以下に示すように、PBMCに対して細胞傷害性の影響を全く示さなかった(曲線□を参照のこと)。
【0258】
図3は、N末端にシステイン酸を有しており、C末端に付加されたリジンのεNH2上にAEEA−MPAを有しているC34のアルブミン結合体(すなわち、CA−C34−Lys35(ε−AEEA−MPA):HSA)によるPBMC中でのHIV−1IIIB複製の阻害を示す(曲線◆を参照のこと)。この化合物は、以下に示すように、PBMCに対して細胞傷害性の影響を全く示さなかった(曲線□を参照のこと)。
【0259】
図1、2、および3に説明するデータに基づいて、両方のアルブミン結合体のIC50値を、天然のC34と比較して表4に提供する。
【0260】
【表4】
表4は、天然のC34、C34のアルブミン結合体、およびCA−C34のアルブミン結合体についての類似する抗HIV活性を示す。結論として、N末端でのシステイン酸の付加と、その後のLys35(ε−AEEA−MPA)を介するアルブミンへの結合は、このアッセイにおいてC34の活性にネガティブな影響を与えることはない。
【0261】
(実施例8)
さらなる抗膜融合性ペプチド誘導体
実験手順
以下の手順を以下に詳細に議論するような結果を得るために行った実験全体を通じて使用した。
【0262】
CHRペプチドアナログの合成は、ペプチドの作成の間に手作業による介入を伴うSymphony Peptide Synthesizer上での自動化された固相手順を使用して行った。合成は、Fmoc保護されたRamageアミドリンカー樹脂上で、Fmoc保護されたアミノ酸を使用して行った。カップリングは、O−ベンゾトリアゾール−1−イル−N,N,N’,N’−テトラメチル−ウロニウムヘキサフルオロホスフェート(HBTU)とジイソプロピルエチルアミン(DIEA)を活性化因子の混合物としてN,N−ジメチルホルムアミド(DMF)溶液中で使用することによって行った。Fmoc保護基を20%のピペリジン/DMFを使用して除去した。ペプチドが樹脂から切り離された後の遊離のα−N末端を作成するために、Boc保護されたアミノ酸をN末端に使用した。シグマコーティッドガラス反応容器を合成の際に使用した。
【0263】
マレイミドを分子のC末端部分に配置する場合(表5、アルブミン結合化合物VII、およびアセチル化結合化合物X)は、ペプチドの固相合成は、Fmoc−Lys(Aloc)の付加によって開始した。Alocは酸性培地に対して安定な特異的な直交する保護基である。その後、ペプチド鎖を、それらの側鎖が酸性培地に対して不安定な基で保護されているアミノ酸の連続する付加によって固体支持体上で伸張させた。ペプチド鎖が完了したら、C末端リジン上のAloc保護基を、テトラキストリフェニルホスフィンパラジウムを使用して選択的に除去した。その後、Fmoc−アミノエトキシエトキシ酢酸(AEEA)リンカーを保護されていないリジンに対して化学的にカップリングさせた。伝統的なFmocの脱保護プロトコールにしたがって、マレイミドプロピオン酸(MPA)をAEEAスペーサーに化学的にカップリングさせた。最後に、酸不安定性の保護基をペプチドから切り離し、その後ペプチドを固体支持体から強酸の混合物を使用して切断した。マレイミドを分子(表5、マレイミド−化合物VIII、アルブミン結合化合物VIII)、およびアルブミン結合MPA−AEEA−化合物VIIIのN末端部分に配置する場合は、ペプチドの固相合成は融合ペプチド阻害剤の天然のアミノ酸配列によって開始した。
【0264】
【表5−1】
【0265】
【表5−2】
ペプチドの精製
生成物をそれぞれ、Varian(Dynamax)分取バイナリーHPLCシステムを使用して、分取逆相HPLCによって精製した。全てのDACペプチドの精製は、水/TFA混合物(H2O中の0.1%のTFA;溶媒A)とアセトニトリル/TFA(CH3CN中の0.1%のTFA;溶媒B)で平衡化させたPhenomenex Lunaフェニル−ヘキシル(10ミクロン、50mm×250mm)カラムを使用して行った。溶離は、様々な勾配の溶媒Bを180分間かけて50mL/分で流すことによって行った。ペプチドを含む画分を、214nmと254nmのUV吸光度(Varian Dynamax UVD II)によって検出した。
【0266】
画分を25mLのアリコートの中に回収した。所望される生成物を含む画分を、LC/MS上への直接の注入後の質量の検出によって同定した。選択した画分をその後、分析用HPLC(20分間にわたる20〜60%のB;Phenomenex Luna 5 micronフェニル−ヘキシル、10mm×250mmカラム、0.5mL/分)によって分析して、≧90%の純度を有している画分をプールするために同定した。その後、このプールを液体窒素を使用して凍結乾燥させ、続いて、少なくとも2日間凍結乾燥させて白色粉末を得た。
【0267】
アルブミン結合体の調製
HSAのシステイン−34に対するマレイミド−C34およびマレイミド−T−20誘導体の結合と、それに続く疎水性相互作用クロマトグラフィーを使用した精製は、最近、有効なプロセスとなった。結合工程には、それぞれのマレイミド−ペプチドをHSAの25%溶液(Cortex−Biochem,San Leandro,CA)と混合する工程、および37℃で30分間インキュベートする工程が含まれる。AKTA精製装置(GE Healthcare)を使用して、得られた混合物を、5mMのオクタン酸ナトリウムと750mMの(NH4)2SO4からなる20mMのリン酸ナトリウム緩衝液(pH7)中に平衡化させたブチルセファロース4ファストフロー樹脂(GE Healthcare)を充填した50mlのカラム上に直接、2.5ml/分の流速でロードした。これらの条件下では、C34−HSA結合体は疎水性樹脂上に吸着したが、原則として全ての結合しなかったHSAはカラムの空隙容量の中に溶離した。それぞれの結合体を、4倍を超えるカラム容量の徐々に低下する(NH4)2SO4濃度(750nM〜0mM)の直線的勾配をかけることによって遊離の(未反応の)マレイミド−C34誘導体の全てからさらに精製した。それぞれの精製した結合体を、その後、脱塩し、10kDaの超遠心分離フィルターデバイス(Amicon;Millipore,Bedford,MA)を使用して水の中で濃縮した。最後に、それぞれの結合体の溶液を、pH7の等張性の緩衝溶液中に再度処方した。それぞれの精製した試料の質量スペクトルによって、最も豊富なタンパク質産物がそれぞれのマレイミド誘導体を持つHSAの1:1の共有複合体に相当することを確認し、それぞれの精製した試料の逆相HPLC分析によって、原則として全ての結合しなかった(遊離の)マレイミド誘導体の除去を確認した。個々のアルブミン結合体を滅菌した0.9%のNaClを使用して処方し、T−20(San Francisco General Hospital pharmacyから入手した)を注射用の滅菌水中に溶解させ、HClでpH7に調整した。
【0268】
新しいヒトPBMC中での抗HIV効力の評価
HIV−1 IIIBは、Robert C.Gallo博士の懇意によりAIDS Research and Reference Reagent Program,Division of AIDS,NIAID,NIHを通じて入手した(Popovic ME,Read−Connole E,Gallo RC(1984)T4 positive human neoplastic cell lines susceptible to and permissive for HTLV−III.Lancet ii:1472−1473;Popovic M,Sarngadharan MG,Read E,Gallo RC(1984)Detection,isolation,and continuous production of cytopathic retroviruses(HTLV−III)from patients with AIDS and pre−AIDS.Science 224:497−500;Ratner Lら(1985)Complete nucleotide sequence of the AIDS virus,HTLV−III.Nature 313:277−283).HIVおよびHBVについて血清反応陰性である新しいヒト末梢血単核細胞(PBMC)をスクリーニングしたドナーの血液(Biological Specialty Corporation;Colmar,PA)から、製造業者の説明書にしたがって、Lymphocyte Separation Medium(LSM;Mediatech,Inc.によるCellgro(登録商標);密度1.078+/−0.002 g/ml)単離した。細胞を4μg/mLのフィトヘマグルチニン(PHA;Sigma)中での48〜72時間のインキュベーションによって刺激した。分裂刺激を、20U/mLの組み換え体ヒトIL−2(R&D sysてms、Inc)の培養培地への添加によって維持した。少なくとも2人のドナーに由来するPHAで刺激したPBMCをプールし、新鮮な培地中に稀釈し、96ウェルプレートに対して5×104細胞/ウェルで添加した。細胞を9種類の異なる試験化合物濃度(3連のウェル/濃度)の存在下で感染させ(最終的なMOI約0.1)、7日間インキュベートした。ウイルス阻害のレベルを決定するために、細胞を含まない上清試料を逆転写酵素活性の分析のために回収した(Buckheit RW,Swanstrom R(1991)Characterization of an HIV−1 isolate displaying an apparent absence of virion−associated reverse transcriptase activity.AIDS Res Hum Retrovir 7:295−302)。上清試料の除去後、化合物の細胞傷害性を、製造業者の説明書にしたがって3−(4,5−ジメチルチアゾール−2−イル)−5−(3−カルボキシメトキシフェニル)−2−(4−スルホフェニル)−2H−テトラゾリウム(MTS;CellTiter 96 Reagent,Promega)の添加によって測定した。社内コンピュータープログラムを使用して、IC50(ウイルスの複製の50%の阻害)、IC90(ウイルスの複製の90%の阻害)、TC50(細胞生存性の50%の低下)、および選択性指数(IC50/TC50)を決定した。AZT(ヌクレオシド逆転写酵素阻害剤)をアッセイ対照化合物として用いた。
【0269】
ウイルス
以下の試薬は、AIDS Research and Reference Reagent Program,Division of AIDS,NIAID,NIHを通じて入手し:pNL4−3はMalcolm Martinから入手した(Adachi Aら(1986)Production of acquired immunodeficiency syndrome−associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone.J Virol 59:284−291.)。
【0270】
AIDS Reagent ProgramによるNL4−3には、gp41中に予想しなかった変異形DIV(G36D)変異が含まれており、これは、インビトロでT−20に対して8倍の耐性を付与する。T−20−感受性NL4−3(NL4−3G)を、gp41のアミノ酸位置36でコンセンサス配列と一致するように部位特異的突然変異誘発によって変更させた(アスパラギン酸をグリシンで置き換えた)。NL4−3GおよびNL4−3D(もともとのクローン)のストックを、293T細胞のトランスフェクションと3日目の上清の回収によって調製した。ウイルスストックは、ELISAによるp24検出を用いて、PHAで活性化させたPBMC中での50%終点アッセイによって滴定した。
【0271】
結果
PBMCをベースとするアッセイを使用したインビトロでの抗ウイルス活性
それぞれのアルブミン結合体の抗ウイルス活性を、HIV−1 IIIBに対するPBMCをベースとするアッセイを使用してインビトロでもともとのペプチド阻害剤に対して比較した(Popovic ME,Read−Connole E,Gallo RC(1984)T4 positive human neoplastic cell lines susceptible to and permissive for HTLV−III.Lancet ii:1472−1473;Popovic M,Sarngadharan MG,Read E,Gallo RC(1984)Detection,isolation,and continuous production of cytopathic retroviruses(HTLV−III)from patients with AIDS and pre−AIDS.Science 224:497−500;Ratner Lら(1985)Complete nucleotide sequence of the AIDS virus,HTLV−III.Nature 313:277−283;Buckheit RW,Swanstrom R(1991)Characterization of an HIV−1 isolate displaying an apparent absence of virion−associated reverse transcriptase activity.AIDS Res Hum Retrovir 7:295−302)。興味深いことに、PC−1505(アルブミン結合化合物VIII)、化合物C(アルブミン結合化合物VII)、および化合物D(アルブミン結合化合物VI)の抗ウイルス活性は全て、インビトロではC34ペプチドおよびT−20に対して本質的に等しい効力があった。すなわち、C34ペプチドのN末端(PC−1505(アルブミン結合化合物VIII)および化合物C(アルブミン結合化合物VII)またはC末端(化合物D(アルブミン結合化合物VI))のいずれかでの反応性マレイミド基の置換、それに続くアルブミンの結合は、融合阻害剤の抗ウイルス活性を変化させることはなかった(表7)。T−20へのアルブミンの結合の後は、反応性ペプチドが、結合がペプチドのN末端で起こるように設計されている場合(化合物E アルブミン結合化合物III)には、抗ウイルス活性は極めて良好に保存されていたが、ペプチドのC末端でアルブミンに対する結合が起こる場合には、このペプチドの抗ウイルス活性の有意な低下が観察された(化合物F 化合物X−does cpd Fは、Eriksonにおいて同等であった(化合物I−VIII))。
【0272】
【表6】
C34ペプチド、化合物VIII、およびrHAのラットでの薬物動態プロフィール
この研究で観察された抗ウイルス活性が、遊離のペプチド、あるいはマレイミドとシステイン−34との間での共有結合の可逆性ではなく、アルブミン結合体の作用が原因であることを確認するために、全てのアルブミン結合体を試験前に精製してあらゆる結合していないペプチドを除去し、化合物VIIIの薬物動態プロフィールを、ラットにおいてC34ペプチド(図5A)とrHA(図5B)に対して比較した。明らかに、C34ペプチドの暴露はアルブミン結合の後に激的に改善され、予め形成させた結合体−化合物VIIIの薬物動態プロフィールがrHAの薬物動態プロフィールと重なり合うという事実によって、C34ペプチドがアルブミンの半減期に近い半減期をとることを確認した。C34ペプチドとHSAについて測定した薬物動態曲線の重ね合わせはまた、予め形成させた結合体−化合物VIIIの静脈内または皮下投与のいずれかの後、少なくとも30時間、Balb/cマウスを使用しても観察された(データは示さない、ラットでのアルブミンのT1/2よりもマウスでのアルブミンのT1/2は短い)。逆に、結合体からのC34ペプチドのゆっくりとした持続的な放出は、予め形成させた結合体−化合物VIIIの全暴露がrHAのものよりも劣るので、2つの薬物動態プロフィールはもはや重なり合わななくなるであろう。さらに、結合体から放出されたC34ペプチドは、インビボでは極めて短い半減期に曝され、長期持続型の予め形成させた結合体−化合物VIIIに対して比較すると、抗ウイルス有効性は限定される。したがって、システイン−34に対してマレイミドを連結させる結合はインビボで極めて安定であり、C34ペプチドは迅速な腎臓でのクリアランスとペプチダーゼによる分解に対してより安定となる。まとめると、インビトロおよびインビボでの全てのアルブミン結合体についての抗ウイルス活性は、マレイミド−システイン−34結合の可逆性ではなく、化学的に安定な結合体の作用だけによると結論付けることができる。
【0273】
考察
HIV gp41のN末端ヘリックス領域(NHR)とC末端ヘリックス領域(CHR)の配列をベースとする合成のペプチドは、多段階の融合プロセスの間に露出させられたgp41結合部位について競合することによってHIVの進入を阻害することが示されている(Chan DC,Fass D,Berger J M,Kim P S(1997)Core structure of gp41 from the HIV envelope glycoprotein.Cell 89:263−273;Chan DC,Chutkowski CT,Kim PS(1998)Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target.Proc Natl Acad Sci USA 95:15613−15617)。臨床では、これらのペプチドのほとんどの成功は、gp41のCHRに由来するT−20(Trimeris/Roche Applied SciencesによるFuzeon(登録商標))である。低分子と比較すると、ペプチドの商業的有用性は、多くの場合はそれらのコスト、ならびにそれらの短い半減期とインビトロでの低い分布によって限定される。本発明者らは、他のクラスのペプチドについてすでに行われているように、ヒトアルブミンのシステイン−34に共有結合するようにCHRペプチド(C34およびT−20)を操作することによって、これらの欠点に取り組もうと考えた(Holmes DLら(2000)Site specific 1:1 opioid:albumin conjugate with in vitro activity and long in vivo duration.Bioconj Chem 11:439−444;Leger Rら(2003)Synthesis and in vitro analysis of atrial natriuretic peptide−albumin conjugates.Bioorg & Med Chem Lett 13:3571−3575;Leger Rら(2004)Kringle 5 peptide−albumin conjugates with anti−migratory activity.Bioorg & Med Chem Lett 14:841−845;Leger Rら(2004)Identification of CJC−1131−albumin bioconjugate as a stable and bioactive GLP−1(7−36)analog.Bioorg & Med Chem Lett 14:4395−4398;Jette Lら(2005)Human growth hormone−releasing factor(hGRF)1−29 albumin bioconjugates activate the GRF receptor on the anterior pituitary in rats:Identification of CJC−1295 as a long−lasting GRF analog.Endocrinology 146:3052−3058;Thibaudeau Kら(2005)Synthesis and evaluation of insulin−human serum albumin conjugates.Bioconj Chem 16:1000−1008)。すなわち、本発明者らは、CHR−ペプチド−HSA結合体がもともとの融合阻害剤のはるかに短い半減期とは反対に、アルブミンの半減期に近い体内での半減期となるであろうと仮定した。
【0274】
本明細書中に示した結果は、gp41のNHRが、もともと考えられていたよりも利用しやすいことを示唆している。例えば、予め形成させた結合体−化合物VIIIを使用して示されるようにそのような競合阻害を利用できるようにするためには、gp41は、標的細胞が存在しない条件で(すなわち、細胞を含まないウイルスまたは非感染細胞の状況下で)NHR領域を露出させる配座平衡に関与し得るか、あるいは、「エントリークロウ(entry claw)」の中に形成されるプレヘアピン中間体(Sougrat Rら(2007)Electron tomography of the contact between T cells and SIV/HIV−1:Implications for viral entry.PLoS Pathogens 3:0571−0581)が、6ヘリックスの束状構造の形成、その後の脂質の混合および膜貫通工程の前に十分に溶媒に露出させられる。すなわち、予め形成させた結合体−化合物VIIIについて約71kDaのMwを用いた場合には、本発明者らの結果は、gp41のNHR−三量体を評価するための分子量カットオフが、これまでに報告されている(すなわち、<25kDa)よりもはるかに大きいことを示唆している(11)。第2に、C34ペプチドのN末端セグメント628WMEW631は、HIV−1の侵入を阻害するC34ペプチドの能力に不可欠であると仮定されるgp41コイルドコイルキャビティ結合残基を提示する(18,19)。したがって、予め形成させた結合体−化合物VIII(AEEAリンカーからなる)または化合物C予め形成させた結合体−化合物VII(AEEAリンカーが存在しない)のいずれかの場合においては、どのようにすれば、C34ペプチドの628WMEW631セグメントをgp41のNHRに到達させることが可能であるか、そして同時に、アルブミンの表面に対して永久的に結合し、近位に接近して配置されるか?予め形成させた結合体−化合物VIIIおよび化合物C予め形成させた結合体−化合物VIIの抗ウイルス活性の保持についての1つの可能性のある説明は、血清アルブミンがいくつかの立体構造状態をとるように誘導することができる高度に可撓性のタンパク質であるという事実である(Peters T,Jr(1996)All about albumin−biochemistry,genetics,and medical applications,Copyright by Academic Press,Inc)。例えば、C34ペプチドはアルブミンのシステイン−34に永久的に結合させられるので、アルブミンの拘束されていないN末端ドメイン(すなわち、ジスルフィド結合が存在しない)内での局所立体構造転位は、gp41のNHR領域上への融合阻害剤の正確な挿入を容易にするための部分的な巻き戻しを生じさせることができる。したがって、システイン−34上以外のアルブミン分子内のほかの場所にC34ペプチドを配置することにより、この融合阻害剤についての抗ウイルス活性の同様の保存が導かれるかどうか(例えば、リジン残基、N末端、またはC末端)、あるいは、抗ウイルス活性の同様の保存が、他の豊富に存在する高分子量の血清タンパク質(例えば、トランスフェリンまたはIgG)に対するC34ペプチドの永久的な結合の後に観察されるであろうかどうかは明らかにはなっていない。したがって、アルブミン分子が、単にタンパク質貨物(protein cargo)としての役割を担うだけではなく、活発な参加型の役割を担っている可能性もある。例えば、マレイル化(maleylated)−、アコニチニル化−、およびスクシニル化されたアルブミンは、インビトロで強力なHIV−1侵入阻害剤として機能する(35〜38)。
【0275】
加えて、C34ペプチドの中に見られる34個のアミノ酸残基のうちの24個が、T−20の中で見られるアミノ酸残基と重複すると仮定すると、T−20は、このペプチドC末端での修飾後にアルブミン結合体についてより少ない候補となることが可能であるかどうか、一方では、抗ウイルス活性の保持の改善が、T−20がそのN末端で改変された場合に観察されるかどうか?この知見についての1つの可能性のある説明は、T−20が原因であるHIV−1の阻害機構がC34ペプチドのものとは異なることを示唆している最近の証拠である(Liu Sら(2005)J Biol Chem 280:11259−11273;Munoz−Barroso I,ら(1998)J Cell Biol 140:315−23;Kliger Yら(2001)J Biol Chem 276:1391−1397)。例えば、T−20はまた、血漿膜へのgp41の動員、その後の脂質混合工程後のオリゴマー形成を阻害することが示されているが、C34ペプチドは6ヘリックスの束状構造の形成後はその阻害効果を発揮することができないことが明らかにされている(Liu Sら(2005)J Biol Chem 280:11259−11273)。すなわち、T−20は、血漿膜へのその挿入後にそのような阻害機能を行うこと、そしてT20の疎水性C末端セグメント666WASLWNWF673は、これらの疎水性相互作用の達成に重要と考えられることが提案されている(Munoz−Barroso I,ら(1998)J Cell Biol 140:315−23;Kliger Yら(2001)J Biol Chem 276:1391−1397)。さらに具体的には、T−20は、血漿膜の近位に配置されるgp41中の対応する配列に結合することによって、gp41の動員とオリゴマー形成を阻害する(Munoz−Barroso I,Durell S,Sakaguchi K,Appella E,Blumenthal R(1998)Dilation of the human immunodeficiency virus−1 envelope glycoprotein fusion pore revealed by the inhibitory action of a synthetic peptide from gp41.J Cell Biol 140:315−23;Kliger Yら(2001)J Biol Chem 276:1391−1397)。したがって、666WASLWNWF673配列がアルブミン分子にすぐ隣接して配置されている化合物F化合物Xについて観察された抗ウイルス活性の大幅な低下は、結合していない(遊離の)T−20ペプチドと同程度に有効に、脂質混合工程後にこのペプチドが機能できないことに寄与している可能性がある。逆に、666WASLWNWF673配列は、化合物E(化合物IX)の構成においては立体構造上の拘束が少ない。まとめると、本発明者らの結果は、T−20とC34ペプチドがHIV−1融合の同じ工程では機能していないことを示唆した報告を明白にサポートする証拠を提供する。
【0276】
本明細書中に示される結果は、膜融合とウイルスの侵入について類似する機構を採用するHIVウイルスと他のウイルスの阻害のための、この新規のクラスのアルブミン−ペプチド結合体についての原理の証明を確立する。結合していない(遊離の)ペプチド阻害剤と比較すると、アルブミン結合体は、全HIV感染細胞のおよそ98%の解剖学上のホームを示すリンパ系に対する有意に改善された暴露を導き得る(Stebbing J,Gazzard B,Douek DC(2004)Where does HIV live? N Engl J Med 350:1872−1880)。この改善は、血清アルブミンについて観察された血漿濃度比に対する有意な定常状態リンパに(Bent−Hansen L(1991)Whole body capillary exchange of albumin.Acta Physiol Scand Suppl 603:5−10(Review);Porter CJH,Charman SA(2000)Lymphatic transport of proteins after subcutaneous administration.J Pharm Sci 89:297−310)、および16〜20kDaより大きい皮下注射されたタンパク質について観察された効率的なリンパの取り込み、輸送、および透過性に(Porter CJH,Charman SA(2000)Lymphatic transport of proteins after subcutaneous administration.J Pharm Sci 89:297−310)主に原因があると予想され得る。
【0277】
最後に、C34ペプチドおよび多くの他の抗膜融合性ペプチド中に見られる疎水性残基の高い含有量が原因で、アルブミン結合体はまた、これらが皮下送達に適している単純な水性処方物中に入れられる場合には、ペプチドのこのファミリーについて一般的に観察される低い溶解限度の矯正をも助け得る。例えば、C34ペプチドの溶解限度は、水性緩衝液中では1mg/mlを超えないことが明らかであるが、PC−1505の溶解限度は、アルブミンの溶解限度と類似することが明らかにされており、これはC34ペプチドのおよそ16mg/mlに相当する(すなわち、25%(w/v)の溶液=PC−1505の250mg/ml、およそC34ペプチドの16mg/ml)。
【0278】
まとめると、アルブミンのシステイン−34を介する抗膜融合性ペプチドの結合によって、gp41のNHRトリマーに一般的に関係している立体的なブロックが克服され、それにより、強力であり、強い中和活性を示す新規のさらに分子量の大きい分子の発見の期待がもたらされる。アルブミン結合C34ペプチドHIV−1融合阻害剤の一例であるPC−1505は、T−20よりも少ない投与頻度しか必要とせず、そしておそらく、ヒトにおいては、T−20耐性HIV−1に対する有効な薬剤である。
【0279】
(実施例9)
さらなる抗膜融合性ペプチド誘導体
図6は、記載した抗膜融合性のいくつかの結合体(PC(予め形成させた複合体)と示す)のインビトロでの抗HIV活性を示している表を示す。アッセイは、本明細書中の実施例に記載したように行った。
【0280】
本発明の特定の実施形態が記載され、説明されてきたが、当業者は、本発明はこれらの実施形態の任意の詳細に限定されるように意図されるのではなく、むしろ添付の特許請求の範囲によって定義されることを理解するであろう。
【技術分野】
【0001】
関連出願への相互参照
この出願は、共に2007年5月16日に出願されたU.S.第60/938,380号およびU.S.第60/938,394号への優先権を主張する。上記の出願の内容は、それらの全体が参考として本明細書に援用される。
【背景技術】
【0002】
発明の背景
非感染細胞への1型ヒト免疫不全ウイルス(HIV−1)の進入には主に3つの工程が含まれる:(i)gp120のCD4受容体に対する結合、(ii)それに続く共受容体CXCR4またはCCR5に対する結合、そして(iii)HIV−1膜貫通糖タンパク質gp41の細胞外ドメインの一連の立体構造の変化(これは、最終的に感染を起こさせる膜融合事象を誘発するために重要である)。RSウイルス(RSV)、ヒト3型パラインフルエンザウイルス(HPIV−3)、麻疹ウイルス、およびサル免疫不全ウイルス(SIV)のようなウイルスは、gp41様タンパク質を含むHIVと高度な構造的類似性および機能的類似性を示す。
【0003】
いくつかの低分子薬物候補(CD4またはCCR5共受容体に対する結合を阻害するものを含む)は、ヒトの臨床試験の段階にあるか、または販売の認可が近いかのいずれかにある(非特許文献1;非特許文献2)。いくつかの合成のペプチドは、膜融合に関連する事象(非感染細胞へのレトロウイルスの感染の阻害を含む)を阻害するか、あるいは壊すことが知られている。例えば、合成ペプチドC34、T1249、DP−107、およびT−20(DP−178)(これらは、gp41中の異なるドメインに由来する)はHIV−1感染およびHIV−1に誘導される細胞と細胞の融合の強力な阻害剤である。
【0004】
T−20(DP−178、エンフュービルタイド、Fuzeon(登録商標)、Trimeris/Roche Applied Sciences)は、HIV−1 gp41のCHR配列をベースとする合成ペプチドであり、gp41の立体構造の再編成を標的化すると考えられている。T−20の阻害は、6ヘリックスの束状構造の形成の阻害を生じるgp41のNHR領域の疎水性溝に結合するその能力に起因すると広く考えられている(非特許文献3)。この考えとは対照的に、最近の研究では、T−20がgp41およびgp120中の複数の部位を標的化できることが示唆された(非特許文献4)。例えば、T−20は膜の表面に結合してオリゴマーを形成し、それによって感染細胞の血漿膜でのgp41の動員とオリゴマー形成を阻害する(非特許文献5;非特許文献6)。さらに、ペプチド配列666WASLWNWF673を有している膜貫通ドメインにすぐ隣接している領域内にあるgp41の細胞外ドメインが、gp41のNHRよりもT−20に対してより親和性の高い部位を構成することもまた示されている(非特許文献5;非特許文献6)。
【0005】
T−20と重複しているがgp41のコイルドコイルキャビティ結合残基(colied−coil cavity binding residues)628WMEW631を含むペプチド配列からなる別のC−ペプチドC34は、NHR領域の疎水性溝についてgp41のCHRと競合することが公知である(非特許文献4)。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】Meanwell NA,Kadow JF、Curr Opinion Drug Disc & Develop(2003)6:451−461
【非特許文献2】Olson WC,Maddon P J、Curr Drug Targets−Infectious Disord(2003)3:283−294
【非特許文献3】Kliger Y,Shai Y、J Mol Biol(2000)295:163−168
【非特許文献4】Liu Sら、J Biol Chem(2005)280:11259−11273
【非特許文献5】Munoz−Barroso Iら、J Cell Biol(1998)140:315−23
【非特許文献6】Kliger Yら、J Biol Chem(2001)276:1391−1397
【発明の概要】
【発明が解決しようとする課題】
【0007】
当該分野で記載されている抗ウイルスペプチドまたは抗膜融合性ペプチドの多くは、強力な抗ウイルス活性および/または抗膜融合活性を示すが、これらのペプチドは生理学的pHの水性処方物中での溶解度は低く、さらにインビボでの血漿半減期も短い。したがって、既存の抗ウイルスペプチドおよび/または抗膜融合性ペプチドの溶解度を増大させ、そして半減期を延長させ、それにより水溶性であり、インビボでの作用期間の長い抗ウイルスペプチドおよび/または抗膜融合性ペプチドを提供するための方法が必要である。
【課題を解決するための手段】
【0008】
本発明は、改変前のペプチドと比較して生理学的pHの水溶液中で高い溶解度を有する改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドに、少なくとも一部関係する。1つの実施形態においては、本発明のペプチドは、1つ以上の極性基または極性部分(例えば、1つ以上のシステイン酸)を含むように改変され、それによって水溶液中でのそれらの溶解度が増大する。改変されたペプチドにはさらに、改変されたペプチドが血液成分または担体タンパク質(例えば、アルブミン(例えば、ヒト血清アルブミンまたは組み換え体アルブミン))上の利用できる官能基と反応できるように化学反応性部分を含めることができ、それによって改変されたペプチドのインビボでの安定性が増大する。複数の実施形態においては、改変されたペプチドは、血液成分または担体タンパク質(例えば、アルブミン(例えば、ヒト血清アルブミン、組み換え体アルブミン、または他の担体タンパク質))に結合させられる。これらの改変されたペプチドまたはそれらの結合体は、それによって、例えば、より頻繁に、またはさらには持続的にペプチドを投与する必要性を低減する。本発明の改変されたペプチドは、多数のウイルス(ヒト免疫不全ウイルス(HIV)、ヒトRSウイルス(RSV)、ヒトパラインフルエンザウイルス(HPIV)、麻疹ウイルス(MeV)およびサル免疫不全ウイルス(SIV)を含む)の感染を緩和するために、例えば、予防的および/または治療的に使用することができる。ウイルストランスフェクション(例えば、肝炎ウイルス、エプスタイン−バーウイルスおよび他の関連するウイルス)に関与する他のペプチドの改変もまた、本発明の範囲に含まれる。
【0009】
したがって、本発明の1つの態様においては、改変前のペプチドと比較して、約5から8までの範囲のpH(例えば、生理学的pH)で水溶液または水中で高い溶解度を有する、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドを特徴とする。1つの実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドは、濃縮された水溶液(例えば、水溶液(例えば、等張性または高塩濃度の水溶液)中で約10mg/mlから500mg/ml、約10mg/mlから400mg/ml、約10mg/mlから300mg/ml、約10mg/mlから200mg/ml、約10mg/mlから180mg/ml、約40mg/mlから180mg/ml、約60mg/mlから180mg/ml、または約90mg/mlから100mg/mlの範囲の濃度)中で実質的に溶解したままである(例えば、約5から8までの範囲のpH(例えば、生理学的pH)では、約40%未満、30%、20%、10%が水または水溶液中に沈殿する)。複数の実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドは、改変前のペプチドよりも少なくとも約1.3倍、1.5倍、1.8倍、2倍、2.3倍、2.5倍、2.8倍、3倍、または3.5倍大きい溶解限度(すなわち、透明な溶液を維持する最大濃度)を示す。複数の実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドは、約5から8までの範囲のpHで、等張性水溶液中で少なくとも約20mg/ml、25mg/ml、30mg/ml、35mg/ml、または40mg/mlの溶解限度を有する。「水溶液」としては、本明細書中で使用される場合は、水、生理食塩溶液(例えば、等張性の溶液)、水の中に作られた緩衝液(例えば、リン酸ナトリウム緩衝液)、水性ゲル、および対象(例えば、ヒト対象)への投与(例えば、皮下投与、静脈内投与、肺投与、筋肉内投与、もしくは腹腔内投与)に適しているpHの水性処方物;あるいは製造プロセスに適しているpHの処方物が挙げられるが、これらに限定されない。
【0010】
複数の実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドには、1つ以上の極性部分が含まれる。1つの実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドには1つ以上の極性部分が含まれ、これは、生理学的pHで電荷を持つか、または生理学的pHでは電荷を持たないかのいずれかである。いくつかの実施形態においては、側鎖は中性であり得、例えば、水素結合または他の非共有相互作用によって水溶液中での改変されたペプチドの全体的な溶解度を増大させることができる。例えば、特定の例においては、酸素基または窒素基を持つ中性の側鎖はバルク溶媒に水素結合することができ、ペプチドの全体的な溶解度を増大させるために使用することができる。いくつかの実施形態においては、側鎖は任意の自然界には存在しない極性の側鎖または中性の側鎖(例えば、20種類の自然界に存在しているアミノ酸の中には見られない側鎖)であり得る。
【0011】
複数の実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドの極性部分には以下の構造が含まれる:
【0012】
【化1】
例えば、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドには、1つ以上のシステイン酸を含めることができる。複数の実施形態においては、システイン酸は以下の構造を有する:
【0013】
【化2】
本明細書中に開示されるペプチドの溶解度を増大させることができるさらなる適切な側鎖は、本発明の開示の利点を前提として、当業者によって容易に選択されるであろう。
【0014】
他の実施形態においては、1つ以上の極性部分(例えば、システイン酸)が抗ウイルスペプチドおよび/または抗膜融合性ペプチドのN末端あるいはC末端に付加される。他の実施形態においては、1つ以上の極性部分が、抗ウイルスペプチドおよび/または抗膜融合性ペプチドの内部配列に付加される。
【0015】
1つの実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドには、gp41のコイルドコイルキャビティ結合残基の少なくとも一部が含まれる。例えば、ペプチドには、残基628WMEW631(配列番号1)、またはそれらに対する1つまでのアミノ酸置換(例えば、保存的置換または非保存的置換)あるいは付加を有しているアミノ酸配列が含まれ得る。他の実施形態においては、抗ウイルスペプチドおよび/または抗膜融合性ペプチドには、アミノ酸配列628WMEWDREINNYTSLIHSLIEESQNQQEKNEQELL661(アミノ酸C1からC34に相当する)(配列番号2)、あるいはそれらに対して5個まで、4個、3個、2個、または1個のアミノ酸置換(例えば、保存的置換もしくは非保存的置換)、欠失、または付加を持つアミノ酸配列に由来する、天然のC34のアミノ酸配列全体あるいは一部が含まれる。
【0016】
他の実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドには、DP107ペプチドおよびDP178ペプチド、ならびにそれらのアナログのアミノ酸配列が含まれる。これには、DP107およびDP178が由来するHIVのgp41領域に相当し、抗ウイルス活性および/または抗膜融合活性を示す他の(非HIV)ウイルスに由来するアミノ酸配列からなるペプチドが含まれる。さらに具体的には、これらのペプチドは、特に、ヒトRSウイルス(RSV)、ヒトパラインフルエンザウイルス(HPV)、麻疹ウイルス(MeV)、およびサル免疫不全ウイルス(SIV)に対して抗ウイルス活性を示すことができる。本発明はまた、引用により本明細書中に具体的に組み入れられるUS05/0070475の配列番号1から配列番号86の改変されたペプチドにも関する。
【0017】
複数の実施形態においては、本発明の改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドにはさらに、改変されたペプチドが血液成分または担体タンパク質上の利用できる官能基と反応して安定な共有結合を形成できるように1つ以上の化学反応性部分または基が含まれ得、それによって結合したペプチド形態が生じる。1つの実施形態においては、改変されたペプチドには、1つ以上の血液成分(例えば、アルブミン)上の1つ以上のアミノ基、ヒドロキシル基、またはチオール基と反応して安定な共有結合を形成する1つ以上の反応性基が含まれる。例えば、ペプチド反応性基アルブミン結合体は、約1:1のアルブミンに対するペプチドのモル比であり得る。通常、結合は、反応性基とアルブミン(例えば、ヒトアルブミン)のアミノ酸34(Cys34)との間での共有結合によって起こる。
【0018】
別の実施形態においては、反応性基はマレイミド含有基(例えば、MPA(マレイミドプロピオン酸)またはGMBA(γ−マレイミド−ブチララミド))(これは、血液タンパク質(移動性の血液タンパク質(例えば、アルブミン)を含む)上のチオール基と反応する)であり得る。反応性の改変または反応性基にはさらに、1つ以上のリンカーを含めることができる。複数の実施形態においては、リンカーは、(2−アミノ)エトキシ酢酸(AEA)、[2−(2−アミノ)エトキシ)]エトキシ酢酸(AEEA)、エチレンジアミン(EDA)のうちの1つ以上;アルキル鎖(C1〜C10)、例えば、8−アミノオクタン酸(AOA)、8−アミノプロパン酸(APA)、または4−アミノ安息香酸(APhA)の1つ以上から選択される。リンカーを持つ反応性基、またはリンカーを持たない反応性基を、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドのN末端あるいはC末端に、通常は、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドのC末端に付加することができる。他の実施形態においては、反応性基は、改変されたペプチドの内部残基に結合させられる(例えば、内部リジン残基のεNH2基;内部セリン残基(例えば、C34のセリン13)のヒドロキシル基に結合させられる)。C34の改変されたペプチドの限定ではない例は、WO02/096935(その全体が引用により本明細書中に組み入れられる)に開示されている。
【0019】
通常は、1つ以上の極性部分(例えば、システイン酸)が改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドの一方の末端(例えば、N末端)に付加されれば、反応性基は反対側の末端(例えば、C末端)に付加される。例えば、改変されたペプチドは以下の立体配置のうちの1つを有し得る:
[極性部分(例えば、システイン酸)−改変されたペプチド−リンカーn−反応性基](VI);または
[反応性基−リンカーn−改変されたペプチド−極性部分(例えば、システイン酸)](VII)。
式中、反応性基は、例えば、リンカーを持つマレイミド含有基またはリンカーを持たないマレイミド含有基であり得、例えば、nは、0、1、2、3、4、またはそれ以上のリンカーであり得る。1つ以上のリンカーが存在する場合は、リンカーは同じである(例えば、AEEA−AEEA)場合も、また異なる(例えば、AEEA−EDAもしくはAEA−AEEA)場合もある。
【0020】
特定の複数の実施形態においては、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドに含められるさらなる基は以下の式(I)を有する化合物であり得る。
【0021】
(VIII)(R1)m−X−(R2)n
式(VIII)中では、mとnの和は少なくとも1であり、mとnはそれぞれが0以上の整数である。例えば、mが0である場合は、nは1以上であり、nが0である場合は、mは1以上である。Xは、抗ウイルスペプチドおよび/または抗膜融合性ペプチド、例えば、C34、T20、T1249、あるいは、それらのアナログまたは誘導体(例えば、それらのマレイミド誘導体を含む)である。R1が存在し、R2が存在しない場合は、R1はX基のN末端に存在する。R1が存在せず、R2が存在する場合は、R2はX基のC末端に存在する。
【0022】
特定の例においては、R1とR2はそれぞれ独立して、式(IX)を有する化合物から選択され得る。
【0023】
【化3】
式(IX)のコア構造はアミノ酸のコア構造と類似しており、これにはアミノ基、α炭素、およびカルボキシル基が含まれる。ペプチド誘導体中のR1基とR2基の正確な位置に応じて、複数の基を式(IX)中の様々な原子を介してペプチドに結合させることができる。例えば、R1が式(IX)を有する化合物である場合には、R1を式(IX)のカルボキシル基を介してペプチドに結合させて、R1のカルボキシル基とペプチドのアミノ基との間にペプチド結合をもたらすことができる。R2が式(IX)を有する化合物である場合には、R2を式(IX)のアミノ基を介してペプチドに結合させて、R2のアミノ基とペプチドのカルボキシ基との間にペプチド結合をもたらすことができる。
【0024】
いくつかの実施形態においては、式(IX)のR3基は、20種類の自然界に存在しているアミノ酸の中に一般的に見られる極性の電荷を持たない基以外の、任意の極性の電荷を持たない基であり得る。例えば、R3基は、スルホニル基(HS=(O)2)、スルホキシド基(HS=O)、スルホン酸基(HO−S=(O)2)、ハロアルキル基、二級アミン、三級アミン、ヒドロキシル基、または極性であるかもしくは全くの中性であり、水溶液中でのペプチド誘導体の全体的な可溶性を増大させることができる他の側鎖基であり得るか、あるいは、それらを含み得る。例えば、水素結合することができる基を持つ側鎖は、ペプチドの全体的な溶解度を増大させるために使用され得る。特定の例においては、側鎖は、リンカーまたは他の種との望ましくない副反応が実質的な程度には起こらないように、非反応性であることが好ましい。いくつかの例においては、R3についての上記基はα炭素から間隔をあけて、例えば、1〜3個の炭素原子の間隔をあけて存在し得る。特定の例においては、R3は、式(X)−(XV)を有する化合物が提供されるように選択され得る。
【0025】
【化4】
本明細書中に開示されるペプチドの溶解度を増大させることができるさらなる適切な側鎖は、本開示の利点を前提として、当業者によって容易に選択されるであろう。
【0026】
特定の複数の実施形態においては、R1基とR2基は、ペプチド結合体の二次構造全体に、または特定の場合には三次構造には実質的には影響を及ぼさない。ペプチド結合体の二次構造に実質的には影響を及ぼさないことにより、ペプチド結合体の全体的な活性は、誘導体化されていないペプチドの全体的な活性よりも感知できるほどに下回ることはないはずである。
【0027】
他の実施形態においては、ペプチド誘導体は、式(XVI)に示されるような組成物の形態をとり得る。
【0028】
(XVI) X1−(R1)m−X2−(R2)n
式(XVI)においては、X1とX2は、互いに連結させられると、例えば、C34、T20、もしくはT1249、またはそれらの変異体を提供するであろうペプチドの部分を提示する。式(XVI)においては、R1とR2は、式(IX)に関して上記で議論した基のうちの任意のものであり得、mとnの和は1以上の整数であり、mまたはnのいずれかが0であり得る可能性がある。式(XVI)においては、ペプチド鎖の中央に基が挿入されている。そのような挿入は、ペプチドの酵素消化、それに続くR1基もしくはR2基またはそれらの両方の挿入、次いでペプチド断片を互いに結合させることを含む多くの様々な方法を使用して行われ得る。
【0029】
特定の複数の実施形態においては、本明細書中に開示される化合物は、ペプチドのN末端、C末端に、またはペプチドの1つ以上のアミノ酸の側鎖を介して、1つ以上のさらなる基に連結させられ得る。例えば、式(XVII)−(XX)に模式的に示される組成物が作成され得る。
【0030】
【化5】
式(XVII)−(XX)においては、Lは、例えば、(2−アミノ)エトキシ酢酸(AEA)、エチレンジアミン(EDA)、2−[2−(2−アミノ)エトキシ]エトキシ酢酸(AEAA)、アルキル鎖モチーフ(C1〜C10)(例えば、グリシン)、3−アミノプロピオン酸(APA)、8−アミノオクタン酸(AOA)、4−アミノ安息香酸(APhA)などのようなリンカーであり、R1とR2は、本明細書中で議論された基のうちの任意のものであり得る。リンカーは、ペプチドの任意のアミノ酸を介して、例えば、リジンのアミノ基、チオール基、ペプチドの1つ以上のアミノ酸側鎖残基の中のヒドロキシル基を介してペプチドに対して結合させられ得るか、あるいはペプチドのN末端またはC末端に結合させられ得る。X基、X1基、およびX2基は、ペプチド(X)またはペプチド断片(X1およびX2)である。式(XIX)および式(XX)中に示されるP基は、リンカーLを介して誘導されたペプチドに結合させることができるタンパク質を示す。例示的なタンパク質としては、血液タンパク質または担体タンパク質(例えば、ヒト血清アルブミン、組み換え体アルブミン、免疫グロブリンもしくはその断片、トランスフェリン、または他の適切なタンパク質)が挙げられる。
【0031】
タンパク質結合体(式(XIX)および式(XX))は、エキソビボまたはインビボで生産され得る。インビボでの生産が行われる場合には、化合物(例えば、式(XVII)および式(XVIII)に示されるもの)が対象に導入され得、インビボタンパク質(例えば、アルブミン)と反応させられ得る。
【0032】
本発明の抗ウイルスペプチドおよび/または抗膜融合性ペプチドは、1つ以上のアミノ酸の置換または付加を有し得る。例えば、ペプチドは、1つ以上の保存的置換もしくは非保存的置換を有し得る。特定の複数の実施形態においては、改変されたペプチドにはさらに、1つ以上のアミノ酸残基を含めることができる。例えば、C34の改変されたペプチドは、任意に、28位の本来のリジン(Lys28)のアルギニンでの置換を有することができ、そして/あるいは、Lys残基(またはC末端に本明細書中に記載される反応性基(例えば、AEEA−MPA)に対して直接または間接的に共有結合させられるそのε−窒素原子で修飾されたリジン残基)を付加することができる。基Lys(ε−AEEA−MPA)においては、AEEA−MPAは、リジンのεNH2基に結合させられることが理解されるはずである。
【0033】
本発明のC34の改変された抗ウイルスペプチドおよび/または改変された抗膜融合性ペプチドの限定ではない例としては、以下の配列が挙げられる:
CA化合物I:(C34に直接連結させられたシステイン酸(CA);本明細書中ではCA−C34とも呼ばれる(配列番号3))。
【0034】
【化6】
CA化合物II:(28位に本来のリジン(Lys28)のアルギニンでの置換を有しているC34に直接連結させられたシステイン酸(CA);本明細書中ではCA−C34(Arg28)とも呼ばれる(配列番号4))。
【0035】
【化7】
CA化合物III:(35位にさらなるリジン残基(Lys35)を有しているC34に直接連結させられたシステイン酸(CA)、ここでは、リジンのεNH2基はリンカー(AEEA−MPA)を介して反応性基に結合させられる;本明細書中ではCA−C34−Lys35(ε−AEEA−MPA)とも呼ばれる(配列番号5))。
【0036】
【化8】
および
CA化合物IV:(28位に本来のリジン(Lys28)のアルギニンでの置換;35位にさらなるリジン残基(Lys35)を有しているC34に直接連結させられたシステイン酸(CA)、ここでは、リジンのεNH2基はリンカー(AEEA−MPA)を介して反応性基に結合させられる;本明細書中ではCA−C34(Arg28)−Lys35(ε−AEEA−MPA)とも呼ばれる(配列番号6))。
【0037】
【化9】
なお別の態様においては、本発明は、1つ以上の血液成分上の利用できる官能基に結合させられた1つ以上の化学反応性の改変を有している、本明細書中に記載される改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドの結合体を特徴とする。本発明の1つの実施形態においては、改変されたペプチドには、血液成分上のアミノ基、ヒドロキシル基、またはチオール基に結合させられて安定な共有結合を形成する反応性基が含まれる。マレイミド基は改変されたペプチドに直接結合させることができ、また、例えばリンカー(例えば、本明細書中に記載されるようなリンカー)を介して間接的に結合させることもできる。本発明の別の実施形態においては、反応性基は血液タンパク質(アルブミンのような移動性の血液タンパク質を含む)上のチオール基と反応するマレイミドであり得る。ペプチド−反応性基アルブミン結合体は、約1:1のアルブミンに対するペプチドのモル比であり得る。通常、結合は、反応性基とヒトアルブミンのアミノ酸34(Cys34)との間での共有結合によって起こる。
【0038】
改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドには、結合体を形成するように1つ以上の血液成分(例えば、血清アルブミン)に共有結合する能力を有している反応性部分(例えば、マレイミド含有基)が含まれ得る。結合工程は、インビボで、例えば、対象に対する改変されたペプチドの投与後に起こり得る。あるいは、結合工程は、エキソビボまたはインビトロで、例えば、反応性基を含む改変されたペプチドが血液成分(例えば、アルブミン)と接触させられることによって起こり得る。C34、DP107、DP178などの結合体の調製と使用は、それらの全体が引用により本明細書中に組み入れられるWO02/096935およびUS05/0070475に開示されている。インビボまたはエキソビボで形成させられた結合体は、対象(例えば、ヒト対象)においてHIV、RSV、HPV、MeV、またはSIVのようなウイルスのウイルス活性および/または膜融合性活性を阻害することにおいて有用である。
【0039】
別の態様においては、本発明は、本明細書中に記載される抗ウイルスペプチドおよび/または抗膜融合性ペプチドあるいは改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドと薬学的に許容される担体を含む組成物(例えば、薬学的組成物)を特徴とする。複数の実施形態においては、組成物は、注射(例えば、皮下注射または血管内注射)、ならびに肺送達、筋肉内送達、および/または腹腔内送達に適している。他の実施形態においては、組成物は製造プロセスに適している。
【0040】
他の実施形態においては、組成物は濃縮され、例えば、約5から8までの範囲のpHの水溶液(例えば、等張性水溶液または高塩濃度の水溶液)中で、約10mg/mlから500mg/ml、約10mg/mlから400mg/ml、約10mg/mlから300mg/ml、約10mg/mlから200mg/ml、約10mg/mlから180mg/ml、約40mg/mlから150mg/ml、約60mg/mlから125mg/ml、または約90mg/mlから100mg/mlの濃度である。
【0041】
別の態様においては、本発明は、本明細書中に記載される改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドあるいはそれらの結合体を含む、ウイルス感染の予防および/または処置に使用される方法と組成物を特徴とする。この方法には、ウイルス感染に伴う1つ以上の症状を軽減するために、本明細書中に記載される改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチド、あるいはそれらの結合体の有効量(例えば、予防量または治療量)を処置が必要な対象(例えば、ヒト対象)に投与する工程が含まれる。処置または予防することができる例示的なウイルス感染としては、AIDS、ヒトRSウイルス(RSV)、ヒトパラインフルエンザウイルス(HPV)、麻疹ウイルス(MeV)、およびサル免疫不全ウイルス(SIV)が挙げられる。したがって、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチド、あるいはそれらの結合体を使用してウイルスが関係している疾患または障害の1つ以上の症状を軽減する、または阻害する、あるいはその発症を防ぐ、または遅らせるための方法が開示される。予防的(例えば、疾患もしくは障害の1つ以上の症状の発症または再発を防ぐ、軽減する、あるいは遅らせるための)使用の場合には、対象は、疾患または障害の1つ以上の症状を有している場合も、また有していない場合もある。例えば、改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドあるいはそれらの結合体は、何らかの検出可能な症状の発現の前に、または全てではなく少なくとも一部の症状が検出された後に投与することができる。治療的使用の場合には、処置によって、対象の疾患または障害が改善する、治癒する、維持される、またはその期間が短くなり得る。治療的使用においては、対象は、症状の一部が発現している場合も、また症状が完全に発現している場合もある。典型的な場合には、処置によって、医師が検出可能な程度に対象の疾患または障害が改善するか、あるいは、疾患または障害の悪化が防がれる。
【0042】
対象(例えば、ヒト対象)においてHIV、RSV、HPV、MeV、またはSIVの1つ以上の活性を阻害するための方法と組成物が開示される。この方法には、本明細書中に記載される改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチド、あるいはそれらの結合体の有効量(例えば、予防量または治療量)を処置が必要な対象に投与する工程が含まれる。
【0043】
本発明の改変されたペプチドはまた、精製と製造プロセスを容易にすることにおいても有用である。なぜなら、改変されたペプチドの高い溶解度によってより濃縮された反応溶液が可能となり、ひいては大規模な製造プロセスが容易となるからである。したがって、本発明はまた、抗ウイルスペプチドおよび/または抗膜融合性ペプチドの溶解度を増大させるための方法も特徴とする。この方法には、1つ以上の極性部分(例えば、1つ以上のシステイン酸)を含む改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチド(例えば、本明細書中に記載される改変されたペプチド)を提供する工程;および改変されたペプチドの溶液(例えば、本明細書中に記載される薬学的組成物、または製造調製物(manufacturing preparation))を調製する工程が含まれる。この方法には、任意に、溶液中の改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドの溶解度を決定する工程(例えば、溶液中の改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドの試料を得、そして試料の濁度および/または乳白光(opalescence)を評価することによる)が含まれ得る。
【0044】
別の態様においては、本発明は、抗ウイルスペプチドおよび/または抗膜融合性ペプチドの調製(例えば、結合(例えば、大規模な結合))を促進するための方法を特徴とする。この方法には、1つ以上の極性部分(例えば、1つ以上のシステイン酸)を含む改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチド(例えば、本明細書中に記載される改変されたペプチド)を提供する工程;および高濃度(例えば、本明細書中に記載されるような高濃度)の改変されたペプチドを有している改変されたペプチドの溶液を調製する工程が含まれる。
【0045】
本明細書中で使用される場合は、冠詞「a」と「an」は、1つまたは2つ以上(例えば、少なくとも1つ)のその冠詞の文法上の対象物をいう。
【0046】
用語「または」は、特段明記されない限りは、用語「および/または」を意味するように本明細書中で使用され、そして「および/または」と互換的に使用される。
【0047】
用語「タンパク質」と「ポリペプチド」は本明細書中では互換的に使用される。
【0048】
「約」と「およそ」は、一般的には、測定の性質または精度を前提とした、測定された量についての許容される誤差の程度を意味するものとする。例示的な誤差の程度は、提供される値または値の範囲の20パーセント(%)以内であり、通常は10%以内、より典型的には5%以内である。
【0049】
本出願全体を通じて引用される全ての刊行物、係属中の特許出願、公開された特許出願(WO02/096935およびUS05/0070475を含む)、および公開された特許の内容は、それらの全体が引用により本明細書中に組み入れられる。
【0050】
本発明の他の特徴、目的、および利点は、明細書と図面から、そして特許請求の範囲から明らかであろう。
【図面の簡単な説明】
【0051】
【図1】図1は、天然のC34(白四角)と比較した、対照の存在下の末梢血単核細胞(PBMC)中でのHIV−1IIIB複製の阻害(黒菱形)を示している線グラフである。
【図2】図2は、ヒト血清アルブミンに結合させたC34−Lys35(ε−AEEA−MPA)(C34−Lys35(ε−AEEA−MPA):HSA)(白四角)と比較した、対照の存在下でのPBMC中のHIV−1IIIB複製の阻害(黒菱形)を示している線グラフである。
【図3】図3は、N末端にシステイン酸を、そしてC末端に付加されたリジンのεNH2に結合させられたAEEA−MPA有しているC34のアルブミン結合体(ヒト血清アルブミンに結合させられたCA−C34−Lys35(ε−AEEA−MPA)(CA−C34−Lys35(ε−AEEA−MPA):HSA))(白四角)と比較した、対照の存在下でのPBMC中のHIV−1IIIB複製の阻害(黒菱形)を示している線グラフである。
【図4】図4は、MPA−AEEAリンカーによってC34のトリプトファンのN末端αアミノ基に結合させられたアルブミンの結合体(本明細書中ではPC−1505とも呼ばれる;MPA−(AEEA)−C34)(白四角)と比較した、対照の存在下でのPBMC中のHIV−1IIIB複製の阻害(黒菱形)を示している線グラフである。
【図5】図5Aは、Sprague−Dawleyラットへの静脈内投与または皮下投与のいずれかの後の、C34ペプチドと化合物VIII(本明細書中ではPC−1505;MPA−(AEEA)−C34;およびAC−CpdVIIIとも呼ばれる)の薬物動態曲線を示す。図5Bは、Sprague−Dawley ラットへの静脈内投与または皮下投与のいずれかの後のrHAの薬物動態曲線と比較した、化合物VIIIの薬物動態曲線を示す。曲線を重ねあわすことにより、ヒト血清アルブミンのシステイン34に対する化学結合連結マレイミド−化合物VIIIの安定性、さらには、腎臓でのクリアランスとペプチダーゼ消化に対する化合物VIIIの安定性についての明白なサポートする証拠が提供される。
【図6】図6は、HIVIIIbを使用したPBMC中でのいくつかの改変された抗膜融合性ペプチドの活性の結果をまとめている表である。
【発明を実施するための形態】
【0052】
(発明の詳細な説明)
改変前のペプチドと比較して、生理学的pHの水溶液中で高い溶解度を有している改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドが開示される。1つの実施形態においては、本発明のペプチドは、1つ以上の極性部分(例えば、1つ以上のシステイン酸)を含むように改変され、それにより水溶液中でのそれらの溶解度が増大する。改変されたペプチドにはさらに、改変されたペプチドが血液成分または担体タンパク質(例えば、アルブミン)上の利用できる官能基と反応できるように化学反応性部分を含めることができ、これにより改変されたペプチドのインビボでの安定性が増大する。本発明の改変されたペプチドは、多数のウイルス(ヒト免疫不全ウイルス(HIV)、ヒトRSウイルス(RSV)、ヒトパラインフルエンザウイルス(HPV)、麻疹ウイルス(MeV)、およびサル免疫不全ウイルス(SIV)を含む)の感染を緩和するために、例えば、予防的および/または治療的に使用することができる。
【0053】
特定の用語は以下のように定義される:
抗ウイルスペプチド:本明細書中で使用される場合は、「抗ウイルスペプチド」は、例えば、細胞−細胞融合または遊離ウイルス感染を阻害することにより、細胞のウイルス感染を阻害するペプチドをいうものとする。感染の経路には、膜融合(外被ウイルスの場合に生じる場合)またはウイルス構造および細胞構造に関与するいくつかの他の融合事象が含まれ得る。特定のウイルスによるウイルス感染を阻害するペプチドは、特定のウイルス(例えば、特に抗HIVペプチド、抗RSVペプチドなど)に関して言及され得る。
【0054】
抗膜融合性ペプチド:「抗膜融合性ペプチド」は、ペプチドが存在しない場合に起こる膜融合のレベルと比較して、2つ以上の要素の間(例えば、ウイルス−細胞または細胞−細胞)での膜融合事象のレベルを阻害するかまたは減少させる能力を示すペプチドである。
【0055】
HIVおよび抗HIVペプチド:ヒト免疫不全ウイルス(HIV)(これは、後天性免疫不全症候群(AIDS)の原因となる)は、レトロウイルスのレンチウイルファミリーのメンバーである。HIVには2つの一般的なタイプ:HIV−1とHIV−2があり、それぞれについて様々な株が同定されている。HIVは、CD−4+細胞を標的化し、そしてウイルスの進入は、HIVタンパク質gp41のCD−4+細胞表面受容体に対する結合に依存する。抗HIVペプチドは、HIVに対する抗ウイルス活性(遊離ウイルスによるCD−4+細胞感染の阻害、および/または感染したCD−4+細胞と非感染CD−4+細胞との間でのHIVに誘導される合胞体形成の阻害を含む)を示すペプチドをいう。
【0056】
SIVおよび抗SIVペプチド:サル免疫不全ウイルス(SIV)は、感受性のあるサルにおいて後天性免疫不全症候群(AIDS)様の疾病を引き起こすレンチウイルスである。抗SIVペプチドは、SIVに対する抗ウイルス活性(SIVウイルスによる細胞の感染の阻害、および感染した細胞と非感染細胞との間での合胞体形成の阻害を含む)を示すペプチドである。
【0057】
RSVおよび抗RSVペプチド:RSウイルス(RSV)は呼吸器病原体であり、このウイルスが細気管支炎(小通気道の炎症)および肺炎を引き起こす可能性がある乳児および小児においては特に危険である。RSVは逆鎖(negative sense)の一本鎖RNAウイルスであり、ウイルスのParamyxoviridaeファミリーのメンバーである。RSVの感染の経路は、通常は、気道(すなわち、鼻、喉頭、気管および気管支ならびに細気管支)により粘膜を介する。抗RSVペプチドは、RSVに対する抗ウイルス活性(遊離RSVウイルスによる粘膜細胞感染および感染した細胞と非感染細胞との間での合胞体形成の阻害を含む)を示すペプチドである。
【0058】
HPVおよび抗HPVペプチド:RSVと同様に、ヒトパラインフルエンザウイルス(HPIVまたはHPV)は呼吸器疾患の別の主原因であり、RSVと同様に、ウイルスのParamyxoviridaeファミリーのメンバーである、逆鎖の一本鎖RNAウイルスである。HPIVには4種類の認識されている血清型(HPIV−1、HPIV−2、HPIV−3、およびHPIV−4)がある。HPIV−1は、小児のクループの主原因であり、HPIV−1およびHPIV−2の両方は、呼吸器上部および呼吸器下気の疾病を引き起こす。HPIV−3は、より頻繁に細気管支炎および肺炎に関与する。抗HPVペプチドは、HPVに対する抗ウイルス活性(遊離HPVウイルスによる感染および感染した細胞と非感染細胞との間での合胞体形成の阻害を含む)を示すペプチドである。
【0059】
Mevおよび抗Mevペプチド:麻疹ウイルス(VMまたはMeV)は、ウイルスのParamyxoviridaeファミリーに属する、外被逆鎖一本鎖RNAウイルスである。RSVおよびHPVと同様に、MeVは呼吸器疾患を引き起こし、そしてまた、さらなる日和見感染の原因となる免疫抑制も生じる。場合によっては、MeVは、脳の感染を確立し、重篤な神経性合併症を導く可能性がある。抗MeVペプチドは、Mevに対する抗ウイルス活性(遊離Mevウイルスによる感染および感染した細胞と非感染細胞との間での合胞体形成の阻害を含む)を示すペプチドである。
【0060】
C34およびC34アナログ:用語「C34」は、gp41コイルドコイルキャビティ結合残基の部分をいう。例えば、このペプチドにはgp41の残基628WMEW631(配列番号1)、またはgp41の残基628WMEWDREINNYTSLIHSLIEESQNQQEKNEQELL661(配列番号2)が含まれ得る。
【0061】
C34のアナログには、それらの短縮型、欠失型、挿入型、および/またはアミノ酸置換型(例えば、保存的置換もしくは非保存的置換)が含まれ得る。欠失は、C34ペプチドの1つ以上のアミノ酸残基の除去からなり得、これには、ペプチド配列の1つの連続する部分、または複数の部分の除去が含まれる場合もある。挿入には、1つのアミノ酸残基またはひとつづきの残基が含まれ得、挿入は、C34ペプチドのカルボキシ末端もしくはアミノ末端、またはペプチドの内部位置に作製され得る。
【0062】
DP−178およびDP178アナログ:特段明記されるか、または文脈により明らかにされない限りは、DP−178は、HIV−1単離物LAI(HIVLAI)のgp41糖タンパク質のアミノ酸残基638-673に相当し、配列:YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF(配列番号7)を有する36アミノ酸のDP−178ペプチドを意味する。
【0063】
DP178のアナログには、それらの短縮型、欠失型、挿入型、および/またはアミノ酸置換型(例えば、保存的置換もしくは非保存的置換)が含まれ得る。ペプチドの短縮には、3〜36の間のアミノ酸のペプチドが含まれ得る。欠失は、DP178ペプチドからの1つ以上のアミノ酸残基の除去からなり得、これには、ペプチド配列の1つの連続する部分または複数の部分の除去が含まれる場合もある。挿入には、1つのアミノ酸残基またはひとつづきの残基が含まれ得、挿入は、DP178ペプチドのカルボキシ末端もしくはアミノ末端、またはペプチドの内部位置に作製され得る。
【0064】
DP178ペプチドアナログは、そのアミノ酸配列がDP178が由来するgp41領域に対応するHIV−1LAI以外のウイルスのペプチド領域、ならびにそれらの短縮型、欠失型、または挿入型のアミノ酸配列からなるペプチドである。このような他のウイルスとしては、他のHIV単離物(例えば、HIV−2NIHZ、RSウイルス(RSV)、ヒトパラインフルエンザウイルス(HPV)、サル免疫不全ウイルス(SIV)および麻疹ウイルス(MeV))が挙げられるがこれらに限定されない。DP178アナログはまた、ALLMOTI5、107×178×4およびPLZIP検索モチーフ(米国特許第6,013,263号、同第6,017,536号および同第6,020,459号に記載されており、そして本明細書中に引用される)により同定されるかまたは認識されるこれらのペプチド配列をいい、これらは、DP178に対して構造的類似性および/またはアミノ酸モチーフ類似性を有する。DP178アナログはさらに、この用語が米国特許第6,013,263号、同第6,017,536号、および同第6,020,459号において定義されているように、「DP178様」と記載されるペプチドをいう。
【0065】
DP107およびDP107アナログ:特段明記されないか、または文脈によって明らかにされない限りは、DP107は、HIV−1単離物LAI(HIVLAI)のgp41タンパク質のアミノ酸残基558〜595に相当し、配列:NNLLRAIEAQQHLLQLTVWQIKQLQARILAVERYLKDQ(配列番号8)を有する、38アミノ酸のDP107ペプチドを意味する。
【0066】
DP107のアナログには、その短縮型、欠失型、挿入型、および/またはアミノ酸置換型(例えば、保存的置換もしくは非保存的置換)が含まれ得る。ペプチドの短縮には、3〜38の間のアミノ酸のペプチドが含まれ得る。欠失は、DP107ペプチドからの1つ以上のアミノ酸残基の除去からなり得、これには、ペプチド配列の1つの連続する部分または複数の部分の除去が含まれる場合もある。挿入には、1つのアミノ酸残基またはひとつづきの残基が含まれ得、挿入は、DP107ペプチドのカルボキシ末端もしくはアミノ末端、またはペプチドの内部位置に作製され得る。
【0067】
DP107ペプチドアナログは、これらのアミノ酸配列が、DP107が由来するgp41領域に対応するHIV−1LA1、ならびにそれらの短縮型、欠失型、および/または挿入型以外のウイルスのペプチド領域のアミノ酸配列からなるペプチドである。このような他のウイルスとしては、他のHIV単離物(例えば、HIV−2NIHZ、RSウイルス(RSV)、ヒトパラインフルエンザウイルス(HPV)、サル免疫不全ウイルス(SIV)、および麻疹ウイルス(MeV))を挙げることができるが、これらに限定されない。DP107アナログはまた、米国特許第6,013,263号、同第6,017,536号および6,020,459号に記載されており、そして本明細書中に引用されるALLMOTI5、107×178×4およびPLZIP検索モチーフによって同定されたかまたは認識されたこれらのペプチド配列をいい、これらはDP107に対して構造的類似性および/またはアミノ酸モチーフ類似性を有する。DP107アナログは、この用語が、米国特許第6,013,263号、同第6,017,536号および6,020,459号に定義されているように、さらに、「DP107様」と記載されるペプチドもいう。
【0068】
反応性基:反応性基は、共有結合を形成することができる化学基である。このような反応性基は、C34、DP−107、DP−178、もしくはT−1249ペプチドまたはそれらのアナログ、あるいは目的の他の抗ウイルスペプチドまたは抗膜融合性ペプチドに連結させられるか、または結合させられる。反応性基は、一般的には、水性環境中で安定であり、そして通常は、カルボキシ基、ホスホリル基、または従来のアシル基であり、エステルまたは混合無水物のいずれか、あるいはイミデートとして存在し、それによって、移動性の血液成分上の標的部位の、アミノ基、ヒドロキシまたはチオールのような官能基と共有結合を形成することができる。大部分については、これらのエステルは、フェノール性化合物を含むか、またはチオールエステル、アルキルエステル、リン酸エステルなどである。
【0069】
官能基:官能基は、改変された抗ウイルスペプチド上の反応性基が反応して共有結合を形成する、血液成分上の基である。官能基としては、通常、エステル反応性部分に結合するためのヒドロキシル基、マレイミド、イミデート、およびチオエステル基に結合するためのチオール基;カルボキシ基、ホスホリル基、またはアシル基に結合するためのアミノ基、およびアミノ基に結合するためのカルボキシル基が挙げられる。
【0070】
血液成分または担体タンパク質:血液成分は、固定されている場合も、また移動性である場合もある。固定された血液成分は非移動性の血液成分であり、これには、組織、膜受容体、間質タンパク質、フィブリンタンパク質、コラーゲン、血小板、内皮細胞、上皮細胞、およびそれらが結合した膜および膜受容体、身体の細胞(somatic body cell)、骨格細胞および平滑筋細胞、神経成分、骨細胞および破骨細胞、ならびに全ての身体組織(特に、循環系およびリンパ系に関連するもの)が含まれる。移動性の血液成分は、任意の長い期間にわたって(一般的には5分を超えない、より通常は1分間)固定された状態にはない血液成分である。これらの血液成分は、膜結合しておらず、そして長期間にわたって血液中に存在し、そして少なくとも0.1μg/mlの最小濃度で存在する。移動性の血液成分には担体タンパク質が含まれる。移動性の血液成分としては、血清アルブミン、トランスフェリン、フェリチン、および免疫グロブリン(例えば、IgMおよびIgG)が挙げられる。移動性の血液成分の半減期は、少なくとも約12時間である。血液成分のさらなる例としては、フェリチン、ステロイド結合タンパク質、トランスフェリン、チロキシン結合タンパク質、およびα−2−マクログロブリンが挙げられる。通常は、血清アルブミンおよびIgGがより好ましく、血清アルブミン(例えば、ヒト血清アルブミン)が最も好ましい。アルブミンはまた、組み換えによって、またはゲノム供給源(例えば、酵母、細菌(例えば、E.coli)、哺乳動物細胞(例えば、チャイニーズハムスター卵巣(CHO)細胞)、トランスジェニック植物、トランスジェニック動物)に由来する場合もある。したがって、用語「血液成分」には、対象から生化学的に精製されたタンパク質、ならびに組み換えによって作製されたタンパク質が含まれる。
【0071】
保護基:保護基は、ペプチド誘導体をそれ自体と反応しないよう保護するために利用される化学部分である。様々な保護基が、本明細書中に開示され、そして米国特許第5,493,007号(これは、引用により本明細書中に組み入れられる)にも開示されている。このような保護基としては、アセチル、フルオレニルメチルオキシカルボニル(Fmoc)、t−ブチルオキシカルボニル(Boc)、ベンジルオキシカルボニル(CBZ)などが挙げられる。特定の保護されたアミノ酸が表1に示される。
【0072】
【表1】
連結基:連結(スペーサー)基は、反応性部分を抗ウイルスペプチドまたは抗膜融合性ペプチドに連結させるかまたは接続する、化学部分である。連結基には、1つ以上のアルキル部分、アルコキシ部分、アルケニル部分、アルキル部分で置換されたアルキニル部分またはアミノ部分、シクロアルキル部分、多環式部分、アリール部分、ポリアリール部分、置換されたアリール部分、複素環式部分、および置換された複素環式部分が含まれ得る。連結基には、(2−アミノ)エトキシ酢酸(AEA)、[2−(2−アミノ)エトキシ)]エトキシ酢酸(AEEA)、エチレンジアミン(EDA);1つ以上のアルキル鎖(C1〜C10)(例えば、8−アミノオクタン酸(AOA)、8−アミノプロパン酸(APA)、または4−アミノ安息香酸(APhA))が含まれ得る。
【0073】
感受性官能基:感受性官能基は、抗ウイルスペプチドおよび/または抗膜融合性ペプチド上に可能性のある反応部位を提示する原子の群である。存在する場合には、感受性官能基は、リンカー−反応性基改変のための付着点として選択され得る。感受性官能基としては、カルボキシル基、アミノ基、チオール基、およびヒドロキシル基が挙げられるが、これらに限定されない。
【0074】
改変されたペプチド:改変されたペプチドは、反応性基の付着によって改変された抗ウイルスおよび/または抗膜融合性ペプチドである。反応性基は、連結基を介して、または任意に連結基を使用することなくのいずれかで、ペプチドに付着させられ得る。1つ以上のさらなるアミノ酸がペプチドに付加されて、反応性部分の付着が容易にされ得ることもまた想定される。改変されたペプチドは、血液成分との結合がインビボで起こるように、インビボで投与され得るか、またはこれらは、最初に血液成分または担体タンパク質に対してインビトロで結合させられ(例えば、組み換えによって生産されたタンパク質(例えば、組み換え体アルブミン、免疫グロブリン、またはトランスフェリン)を使用して)、そして得られた結合ペプチド(以下に定義される)が、インビボで投与され得る。
【0075】
結合ペプチド:結合ペプチドは、改変されたペプチドの反応性基と血液成分の官能基との間で形成された(連結基を伴うか、または伴わないで)共有結合を介して血液成分に結合させられた、改変されたペプチドである。本出願全体を通じて使用される場合には、用語「結合ペプチド」は、特定の結合ペプチド(例えば、「結合C34」または「結合DP107」)をいうように、より特化され得る。
【0076】
複数の実施形態においては、本発明の改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドには、結合体を形成するように血液成分(より具体的には、血清アルブミン)に共有結合する能力を有しているマレイミド含有基が含まれる。対象への抗ウイルスペプチドおよび/または抗膜融合性ペプチドのマレイミド誘導体の投与によって、血液成分(例えば、血清アルブミン)に対するペプチドのインビボでの結合が生じ得る。改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドを血液成分または担体タンパク質(例えば、アルブミン)と接触させることによって、エキソビボ(またはインビボ)で結合体を調製することもまた本発明に含まれる。この場合は、アルブミンは、様々な供給源から(例えば、血液試料中、精製されたアルブミン、組み換え体アルブミン(アルブミンの改変された形態(例えば、アミノ酸置換、挿入、および/または欠失を有する形態)を含む)など)提供することができる。C34とアルブミンの結合体の調製と使用は、WO02/096935に十分に開示されており、同様の調製と使用が本発明の結合体に適用される。対象の中でインビボで形成させられた結合体とエキソビボで調製された結合体はいずれも、対象に投与される場合には、対応する融合ペプチド阻害剤(fusion peptide inhibitor)の抗膜融合活性を示すために、したがって、対象においてHIV、RSV、HPV、MeV、またはSIVの活性を阻害するために有用である。
【0077】
これらの定義を考慮すると、本発明は、存在する抗ウイルスペプチドおよび抗膜融合性ペプチドの特性を利用する。ペプチドによって阻害され得るウイルスとしては、例えば、米国特許第6,013,263号、同第6,017,536号および同第6,020,459号において表V〜VIIおよびIX〜XIVに列挙されたウイルスのすべての種が挙げられるが、これらに限定されない。これらのウイルスとしては、例えば、ヒトレトロウイルス(HIV−1、HIV−2、およびヒトTリンパ球ウイルス(HTLV−IおよびHTLV−II)を含む)、および非ヒトレトロウイルス(ウシ白血症ウイルス、ネコ肉腫ウイルス、ネコ白血症ウイルス、サル免疫不全ウイルス(SIV)、サル肉腫ウイルス、サル白血病、およびヒツジ進行性肺炎ウイルスを含む)が挙げられる。非レトロウイルスウイルスはまた、本発明のペプチドによって阻害され得、これには以下が含まれる:ヒトRSウイルス(RSV)、イヌジステンパーウイルス、ニューカッスル病ウイルス、ヒトパラインフルエンザウイルス(HPIV)、インフルエンザウイルス、麻疹ウイルス(MeV)、エプスタイン−バーウイルス、B型肝炎ウイルス、およびサルマソン−ファイザーウイルス。非外被ウイルスはまた本発明のペプチドによって阻害され得、そして非外被ウイルスとしては、ピコルナウイルス(例えば、ポリオウイルス)、A型肝炎ウイルス、エンテロウイルス、エコーウイルス、コクサッキーウイルス、パポバウイルス(例えば、パピローマウイルス)、パルボウイルス、アデノウイルス、およびレオウイルスが挙げられるがこれらに限定されない。
【0078】
一例として、本出願の背景のセクションで議論されたように、HIV融合タンパク質の作用機構が記載されており、そしてペプチドの抗ウイルス特性および抗膜融合特性は十分に確立されている。カルボキシ末端の外部ドメイン配列に対応する合成ペプチド(例えば、LAI株のHIV−1クラスBのアミノ酸残基643〜678、または類似する株に由来する残基638〜673、ならびに残基558〜595)は、低濃度でウイルスに媒介される細胞と細胞の融合を完全に阻害することが示されている。本発明のペプチドは、天然のウイルスgp41のロイシンジッパー領域と競合し、それによりウイルスの細胞内への融合/感染の干渉が生じる。
【0079】
本発明によってはさらに、DAC(商標)(Drug Activity Complex)技術を用いて、選択された抗ウイルスペプチドおよび/または抗膜融合性ペプチドを改変し、タンパク質担体上へのペプチドの選択的結合を通じて、このペプチドに対して生体利用性、半減期の延長、およびより優れた分布を付与するために使用される、ペプチドの抗ウイルス特性を改変することのない方法と試薬が提供される。本発明のために選択されたる担体(しかし、これに限定されない)は、マレイミド部分で改変された抗ウイルスペプチドおよび/または抗膜融合性ペプチドによるその遊離のチオールを通じて結合させられたアルブミンである。
【0080】
抗ウイルス阻害剤および/または抗膜融合性阻害剤
いくつかのペプチド配列は、HIV−1融合/感染の予防について極めて強力であると文献に記載されている。例として、ペプチドC34、DP107、DP178は、融合に関係があるgp41の立体構造に結合する。従って、本発明の1つの実施形態においては、C34様、DP178様、およびDP178様ペプチドが改変される。同様に、本発明の他の実施形態には、HIVに対して使用されるC34様、DP107様、およびDP107様ペプチド、ならびにRSV、HPV、MeVおよびSIVウイルス中に見られるDP107およびDP178のペプチドアナログの改変が含まれる。
【0081】
改変されたC34ペプチドまたはアナログ
特定の複数の実施形態においては、本発明の改変されたC34ペプチドには、以下の式(I)を有する化合物であり得る、ペプチドに含められるさらなる基が含まれる。
【0082】
(VIII)(R1)m−X−(R2)n
式(VIII)においては、mとnの和は少なくとも1であり、mとnはそれぞれが0以上の整数である。例えば、mが0である場合は、nは1以上であり、nが0である場合は、mは1以上である。Xは、ペプチド、ペプチド断片、またはタンパク質、例えば、C34、T20、T1249、あるいは、それらの誘導体(例えば、それらのマレイミド誘導体を含む)である。R1が存在し、R2が存在しない場合は、R1はX基のN末端に存在する。R1が存在せず、R2が存在する場合は、R2はX基のC末端に存在する。
【0083】
特定の例においては、R1とR2はそれぞれ独立して、式(IX)を有する化合物から選択され得る。
【0084】
【化10】
式(IX)のコア構造はアミノ酸のコア構造と類似しており、これにはアミノ基、α炭素、およびカルボキシル基が含まれる。ペプチド誘導体中のR1基とR2基の正確な位置に応じて、複数の基を式(IX)中の様々な原子を介してペプチドに結合させることができる。例えば、R1が式(IX)を有する化合物である場合には、R1を式(IX)のカルボキシル基を介してペプチドに結合させて、R1のカルボキシル基とペプチドのアミノ基との間にペプチド結合をもたらすことができる。R2が式(IX)を有する化合物である場合には、R2を式(IX)のアミノ基を介してペプチドに結合させて、R2のアミノ基とペプチドのカルボキシ基との間にペプチド結合をもたらすことができる。
【0085】
いくつかの例においては、式(IX)のR3基は、20種類の自然界に存在しているアミノ酸の中に一般的に見られる極性の電荷を持たない基以外の、任意の極性の電荷を持たない基であり得る。例えば、R3基は、スルホニル基(HS=(O)2)、スルホキシド基(HS=O)、スルホン酸基(HO−S=(O)2)、ハロアルキル基、二級アミン、三級アミン、ヒドロキシル基、または極性であるかもしくは全くの中性であり、水溶液中でのペプチド誘導体の全体的な可溶性を増大させることができる他の側鎖基であり得るか、あるいは、それらを含み得る。例えば、水素結合することができる基を持つ側鎖は、ペプチドの全体的な溶解度を増大させるために使用され得る。特定の例においては、側鎖は、リンカーまたは他の種との望ましくない副反応が実質的な程度には起こらないように、非反応性であることが好ましい。いくつかの例においては、R3についての上記基はα炭素から間隔をあけて、例えば、1〜3個の炭素原子の間隔をあけて存在し得る。
【0086】
特定の例においては、R3は、式(X)−(XV)を有する化合物が提供されるように選択され得る。
【0087】
【化11】
本明細書中に開示されるペプチドの溶解度を増大させることができるさらなる適切な側鎖は、本開示の利点を前提として、当業者によって容易に選択されるであろう。
【0088】
特定の複数の実施形態においては、R1基とR2基は、ペプチド結合体の二次構造全体に、または特定の場合には三次構造には実質的には影響を及ぼさない。ペプチド結合体の二次構造に実質的には影響を及ぼさないことにより、ペプチド結合体の全体的な活性は、誘導体化されていないペプチドの全体的な活性よりも感知できるほどに下回ることはないはずである。
【0089】
他の実施形態においては、ペプチド誘導体は、式(XVI)に示されるような組成物の形態をとり得る。
【0090】
(XVI) X1−(R1)m−X2−(R2)n
式(XVI)においては、X1とX2は、互いに連結させられると、例えば、C34、T20、もしくはT1249を提供するであろうペプチドの部分を提示する。式(XVI)においては、R1とR2は、式(IX)に関して上記で議論された基のうちの任意のものであり得、mとnの和は1以上の整数であり、mまたはnのいずれかが0であり得る可能性がある。式(XVI)においては、ペプチド鎖の中央に基が挿入されている。そのような挿入は、ペプチドの酵素消化、それに続くR1基もしくはR2基またはそれらの両方の挿入、次いでペプチド断片を互いに結合させることを含む多くの様々な方法を使用して行われ得る。
【0091】
本明細書中に記載されるC34のシステイン酸誘導体の合成は、ペプチドの作製の間の手作業による介入を伴うSymphony Peptide Synthesizerの自動化された固相手順を使用して行われる(本明細書中の実施例1〜5を参照のこと)。合成は、Fmoc保護されたRamageアミドリンカー樹脂上で、Fmoc保護されたアミノ酸を使用して行った。カップリングは、O−ベンゾトリアゾール−1−イル−N,N,N’,N’−テトラメチル−ウロニウムヘキサフルオロホスフェート(HBTU)とジイソプロピルエチルアミン(DIEA)を活性化因子の混合物としてN,N−ジメチルホルムアミド(DMF)溶液中で使用することによって行った。Fmoc保護基を20%のピペリジン/DMFを使用して除去した。ペプチドが樹脂から切り離されると遊離のNα末端が生じるように、Boc保護されたアミノ酸をN末端に使用した。シグマコーティッドガラス反応容器を合成の際に使用した。
【0092】
特定の複数の実施形態においては、ペプチドの部分は、例えば、Merrifield,1986.Solid phase synthesis.Science.232:341−347によって記載されているように従来の固相合成技術を使用して合成され得る。簡単に説明すると、ブロッキング基がアミノ酸のN末端に付加され、アミノ酸のカルボキシル基はジシクロヘキシルカルボジイミド(DCCD)との反応によって活性化され得る。活性化されたアミノ酸は、遊離のN末端と樹脂またはビーズに結合させられたC末端を有しているアミノ酸と反応させられ得る。アミノブロックされたジペプチジル化合物の形成の後、酸処理によってイソブチレン、二酸化炭素、および樹脂もしくはビーズに結合させられたジペプチドの生産が生じる。さらなるアミノ酸が、これらの工程を繰り返すことによってジペプチドビーズに対して付加され得る。加えて、本明細書中に開示されるアミノ酸誘導体はまた、類似する反応を使用して鎖に沿った任意の点でペプチド鎖に付加される場合もある。したがって、式(IX)を有する化合物を提供するために所望されるペプチド中のどの位置にでもR1基またはR2基を挿入することが可能である。
【0093】
特定の複数の実施形態においては、本明細書中に開示される化合物は、ペプチドのN末端、C末端に、またはペプチドの1つ以上のアミノ酸の側鎖を介して、1つ以上のさらなる基に連結させられ得る。例えば、式(XVII)−(XX)に模式的に示される組成物が作成され得る。
【0094】
【化12】
式(XVII)−(XX)においては、Lは、例えば、(2−アミノ)エトキシ酢酸(AEA)、エチレンジアミン(EDA)、2−[2−(2−アミノ)エトキシ]エトキシ酢酸(AEAA)、アルキル鎖モチーフ(C1〜C10)(例えば、グリシン)、3−アミノプロピオン酸(APA)、8−アミノオクタン酸(AOA)、4−アミノ安息香酸(APhA)などのようなリンカーであり、R1とR2は、本明細書中で議論された基のうちの任意のものであり得る。リンカーは、ペプチドの任意のアミノ酸を介して、例えば、ペプチド中のリジンのεアミノ基を介して、ペプチドのN末端またはC末端に結合させられ得る。X基、X1基、およびX2基は、ペプチド(X)またはペプチド断片(X1およびX2)である。式(XIX)および式(XX)中に示されるP基は、リンカーLを介して誘導されたペプチドに結合させることができるタンパク質を示す。例示的なタンパク質としては、血液タンパク質、ヒト血清アルブミン、組み換え体アルブミン、または他の適切なタンパク質が挙げられる。
【0095】
タンパク質結合体(式(XIX)および式(XX))は、エキソビボまたはインビボで生産され得る。インビボでの生産が行われる場合には、化合物(例えば、式(XVII)および式(XVIII)に示されるもの)が対象に導入され得、インビボタンパク質(例えば、アルブミン)と反応させられ得る。
【0096】
本発明のC34の改変された抗ウイルスペプチドおよび/または改変された抗膜融合性ペプチドの限定ではない例としては、以下の配列が挙げられる:
CA化合物I:(C34に直接連結させられたシステイン酸(CA);本明細書中ではCA−C34とも呼ばれる(配列番号3))。
【0097】
【化13】
CA化合物II:(28位に本来のリジン(Lys28)のアルギニンでの置換を有しているC34に直接連結させられたシステイン酸(CA);本明細書中ではCA−C34(Arg28)とも呼ばれる(配列番号4))。
【0098】
【化14】
CA化合物III:(35位にさらなるリジン残基(Lys35)を有しているC34に直接連結させられたシステイン酸(CA)、ここでは、リジンのεNH2基はリンカー(AEEA−MPA)を介して反応性基に結合させられる;本明細書中ではCA−C34−Lys35(ε−AEEA−MPA)とも呼ばれる(配列番号5))。
【0099】
【化15】
CA化合物IV:(28位に本来のリジン(Lys28)のアルギニンでの置換;35位にさらなるリジン残基(Lys35)を有しているC34に直接連結させられたシステイン酸(CA)、ここでは、リジンのεNH2基はリンカー(AEEA−MPA)を介して反応性基に結合させられる;本明細書中ではCA−C34(Arg28)−Lys35(ε−AEEA−MPA)とも呼ばれる(配列番号6))。
【0100】
【化16】
本出願の教示に従って改変することができる改変されたC34ペプチドのさらなる例にはまた、以下のアミノ酸配列も含まれる:
【0101】
【化17】
【0102】
【化18】
改変されたC34ペプチドの限定ではない例は、以下に示される式I〜式VIIIの化合物であり、これらはインビボまたはエキソビボのいずれかで血液成分上のチオール基と反応させて安定な共有結合を形成させることができる。これらの化合物の合成はWO02/096935(その内容は引用により本明細書中に具体的に組み入れられる)に記載されている。
【0103】
【化19】
【0104】
【化20】
DP178およびDP107
DP178ペプチド
DP178ペプチドは、HIV−1LAI単離物に由来する膜貫通タンパク質gp41のアミノ酸残基638から673に相当し、36アミノ酸の配列を有する(アミノ末端からカルボキシ末端に向かって読まれる):
NH2−YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF−COOH(配列番号7)
全長のDP178(36マー)に加えて、本発明のペプチドには3個から36個の間のアミノ酸残基のペプチドを含むDP178ペプチドの短縮型(すなわち、トリペプチドから36マーのポリペプチドまでの範囲のペプチド)が含まれる。これらの短縮型ペプチドは表2および表3に示される。
【0105】
加えて、DP178ペプチドのアミノ酸置換型もまた本発明の範囲内にある。HIV−1とHIV−2外被タンパク質は構造的には異なるが、HIV−1およびHIV−2のDP178に対応する領域内には著しいアミノ酸保存が存在する。アミノ酸の保存は、周期性の性質であり、これは構造および/または機能のいくらかの保存を示唆している。従って、アミノ酸置換の1つの可能なクラスには、本発明のDP178ペプチドの構造を安定化させると推定さられるこれらのアミノ酸の変化が含まれる。本明細書中に記載されるDP178およびDP178アナログの配列を利用して、当業者は容易にDP178コンセンサス配列を作製し、そして好ましいアミノ酸置換を提示する保存されたアミノ酸残基を、これらから確定することができる。
【0106】
アミノ酸置換は保存された性質の置換である場合も、また保存されていない性質の置換である場合もある。保存されたアミノ酸置換は、類似する電荷、大きさ、および/または疎水性特性のアミノ酸での、DP178ペプチド配列の1以上のアミノ酸の置換(例えば、グルタミン酸(E)のアスパラギン酸(D)へのアミノ酸置換)からなる。非保存的置換は、類似していない電荷、大きさ、および/または疎水性特性を有しているアミノ酸での、DP178ペプチド配列の1以上のアミノ酸の置換(例えば、グルタミン酸(E)のバリン(V)への置換)からなる。
【0107】
DP178のアミノ酸の挿入は、1つのアミノ酸残基または残基のひとつづきの残基からなり得る。挿入は、DP178またはDP178短縮ポリペプチドのカルボキシ末端またはアミノ末端に、ならびにペプチドに対する内部位置に作製され得る。
【0108】
このような挿入は、一般的には2〜15アミノ酸の長さの範囲であろう。目的のペプチドのカルボキシ末端またはアミノ末端のいずれかに作製された挿入はより広い大きさの範囲の挿入であり得、約2〜約50アミノ酸の挿入が好ましいことが意図される。1つ以上のそのような挿入は、そのような挿入が上記の107×178×4、ALLMOTI5またはPLZIP検索モチーフによってなおも認識されされ得るペプチドを生じる限りは、DP178またはDP178短縮型に導入され得る。
【0109】
好ましいアミノ末端への挿入またはカルボキシ末端への挿入は、約2〜約50のアミノ酸残基の長さの範囲のペプチドであり、これらは、実際のDP178 gp41アミノ酸配列に対して、それぞれアミノまたはカルボキシのいずれかのgp41タンパク質領域に対応する。従って、好ましいアミノ末端またはカルボキシ末端へのアミノ酸の挿入には、gp41タンパク質のDP178領域に対してすぐアミノ側またはカルボキシ側に見られるgp41アミノ酸配列が含まれるであろう。
【0110】
DP178またはDP178短縮型の欠失もまた本発明の範囲に含まれる。このような欠失は、DP178またはDP178様ペプチド配列からの1以上のアミノ酸の除去からなり、得られるペプチド配列の長さの下限は、4〜6アミノ酸である。
【0111】
このような欠失には、ペプチド配列の1つの連続する部分または1つより多くの不連続な部分が含まれ得る。1以上のこのような欠失は、このような欠失が上記の107×178×4、ALLMOTI5またはPLZIP検索モチーフによって、なおも認識されされ得るペプチドを生じる限りは、DP178またはDP178短縮型に導入され得る。
【0112】
DP107ペプチド
DP107は38アミノ酸のペプチドであり、これは強力な抗ウイルス活性を示し、そしここに示されるHIV−1LA1単離体膜貫通(TM)gp41糖タンパク質の残基558〜595に対応する:
NH2−NNLLRAIEAQQHLLQLTVWQIKQLQARILAVERYLKDQ−COOH(配列番号17)。
【0113】
全長のDP107の38マーに加えて、DP107ペプチドには、3個から38個の間のアミノ酸残基のペプチドを含むDP107ペプチドの短縮(すなわち、トリペプチドから38マーのポリペプチドまでの大きさの範囲のペプチド)が含まれる。これらのペプチドは、US2005/0070475の表4および表5に示される。
【0114】
加えて、DP178ペプチドのアミノ酸置換体もまた本発明の範囲に含まれる。DP178と同様に、これもまた周期性の性質のものである、HIV−1およびHIV−2のDP107に対応する領域内にもまた高度なアミノ酸保存が存在し、これは、構造および/または機能の保存を示唆している。従って、アミノ酸置換の1つの可能なクラスには、本発明のDP107ペプチドの構造を安定化させると推定されるこれらのアミノ酸の変化が含まれる。本明細書中に記載されるDP107およびDP107アナログ配列を利用して、当業者は、容易にDP107コンセンサス配列を作製し、そして好ましいアミノ酸置換を提示するであろう保存されたアミノ酸残基を、これらから確定することができる。
【0115】
アミノ酸置換は保存された性質の置換である場合も、また保存されていない性質の置換である場合もある。保存的アミノ酸置換は、類似する電荷、大きさ、および/または疎水性特性のアミノ酸での、DP107ペプチド配列の1以上のアミノ酸の置換(例えば、グルタミン酸(E)のアスパラギン酸(D)へのアミノ酸置換)からなる。非保存的置換は、類似していない電荷、大きさ、および/または疎水性特性を有しているアミノ酸での、DP107ペプチド配列の1以上のアミノ酸の置換(例えば、グルタミン酸(E)のバリン(V)への置換)からなる。
【0116】
アミノ酸の挿入は、単一のアミノ酸残基またはひとつづきの残基からなり得る。挿入は、DP107またはDP107短縮型ペプチドのカルボキシ末端またはアミノ末端に、ならびにペプチドに対して内部位置に作製され得る。
【0117】
このような挿入は、一般的には、2〜15アミノ酸の長さの範囲であろう。目的のペプチドのカルボキシ末端またはアミノ末端のいずれかに作製された挿入はより広い大きさの範囲の挿入であり得、約2〜約50アミノ酸が好ましいことが意図される。1以上のこのような挿入は、このような挿入が上記の107×178×4、ALLMOTI5またはPLZIP検索モチーフによってなおも認識されされ得るペプチドを生じる限りは、DP107またはDP107短縮型に導入され得る。
【0118】
好ましいアミノ末端への挿入またはカルボキシ末端への挿入は、約2〜約50までのアミノ酸残基の長さの範囲のペプチドであり、実際のDP107 gp41アミノ酸配列に対して、それぞれアミノまたはカルボキシのいずれかでgp41タンパク質領域に対応する。従って、好ましいアミノ末端またはカルボキシ末端へのアミノ酸の挿入には、gp41タンパク質のDP107領域に対してすぐアミノ側またはカルボキシ側に見られるgp41アミノ酸配列が含まれる。
【0119】
DP107またはDP107短縮型の欠失もまた本発明の範囲に含まれる。このような欠失は、DP107またはDP107様ペプチド配列からの1以上のアミノ酸の除去からなり、得られるペプチド配列の長さの下限は、4〜6アミノ酸である。
【0120】
このような欠失には、ペプチド配列の1つの連続する部分または1つより多くの不連続な部分が含まれ得る。1以上のこのような欠失は、このような欠失が上記の107×178×4、ALLMOTI5またはPLZIP検索モチーフによってなおも認識されされ得るペプチドを生じる限りは、DP107またはDP107短縮型に導入され得る。
【0121】
DP107およびDP107短縮型は、米国特許第5,656,480号にさらに十分に記載されている。
【0122】
DP107およびDP178アナログ
上記の本発明のDP178、DP178短縮型、DP107およびDP107短縮型配列のアナログに対応するペプチドは、他のウイルス(例えば、非HIV−1外被ウイルス、非外被ウイルスおよび他の非ウイルス性生物を含む)中に見ることができる。
【0123】
このようなDP178アナログおよびDP107アナログは、例えば、外被化されたウイルスの膜貫通(「TM」)タンパク質中に存在するペプチド配列に対応し得、そして非外被生物および非ウイルス性生物中に存在するペプチド配列に対応し得る。このようなペプチドは、抗膜融合性活性、抗ウイルス活性、最も具体的には天然の配列が見られるウイルスに特異的である抗ウイルス活性を示す場合があり、また、コイルドコイルペプチド構造が関与している細胞間プロセスを調節する能力を示す場合もある。
【0124】
DP178アナログ
DP178アナログは、そのアミノ酸配列が、例えば、DP178が誘導されたgp41ペプチド領域に対応する他の(すなわち、HIV−1以外の)ウイルスのペプチド領域のアミノ酸配列からなるペプチドである。そのようなウイルスとしては、他のHIV−1単離物およびHIV−2単離物を挙げることができるが、これらに限定されない。
【0125】
他の(すなわち、非HIV−1LAI)HIV−1単離体の対応するgp41ペプチド領域由来のDP178アナログとしては、例えば、以下に示されるペプチド配列を挙げることができる。
【0126】
【化21】
配列番号18、配列番号19、および配列番号20のペプチドは、それぞれ、HIV−lSF2、HIV−1RF、およびHIV−1MNに由来する。他のDP178アナログとしてはHIV−2から誘導されたアナログが挙げられ、これには、US2005/0070475の配列番号6および配列番号7のペプチドが含まれる(これらはそれぞれ、HIV−2RODおよびHIV−2NIHZに由来する)。さらに別の有用なアナログとしては、US2005/0070475の配列番号8および配列番号9のペプチドが挙げられ、これらは、抗ウイルス活性を示すことが明らかにされている。
【0127】
本発明においては、DP178アナログは、そのアミノ酸配列がgp41タンパク質のDP178領域に対応するペプチドを提示することが好ましく、本明細書中に開示されるペプチドにはさらに、約2〜約50までのアミノ酸残基の長さの範囲であり、実際のDP178アミノ酸配列に対するアミノまたはカルボキシのいずれかのgp41タンパク質領域に対応するアミノ酸配列が含まれ得ることもまた意図される。
【0128】
US2005/0070475の表6および表7には、HIV−2NIHZDP178アナログのいくつかの可能な短縮が示されており、これには、3個から36個の間のアミノ酸残基のペプチド(すなわち、トリペプチドから36マーのポリペプチドまでの大きさの範囲のペプチド)が含まれ得る。ペプチド配列は、これらの表中ではアミノ末端(左)からカルボキシ末端(右)方向で列挙されている。
【0129】
さらなるDP178アナログおよびDP107アナログ
DP178およびDP107アナログは、例えば、上記の107×178×4、ALLMOTI5またはPLZIPのコンピューター補助の検索ストラテジーのうちの1つ以上を利用することによって、認識されるかまたは同定される。この検索ストラテジーは、DP107および/またはDP178の構造的特徴および/またはアミノ酸配列特徴と類似した、構造的特徴および/またはアミノ酸配列特徴を有すると予想されるさらなるペプチド領域を同定する。
【0130】
検索ストラテジーは、米国特許第6,013,263号、同第6,017,536号および同第6,020,459号の第9節に示される例に完全に記載されている。この検索ストラテジーは、一部が、DP107およびDP178から推定される一次アミノ酸モチーフに基づくが、一次アミノ酸配列の相同性の検索だけに基づくものではない。なぜなら、そのようなタンパク質配列の相同性は存在するが、主要なウイルス群の間ではないからである。例えば、一次アミノ酸配列の相同性は、HIV−1の異なる株のTMタンパク質内、またはサル免疫不全ウイルス(SIV)の異なる単離物のTMタンパク質内で高い。
【0131】
米国特許第6,013,263号、同第6,017,536号、および同第6,020,459号に開示されるコンピューター検索ストラテジーでは、DP107またはDP178に類似するタンパク質の領域の同定に成功した。この検索ストラテジーは、市販の配列データベースパッケージ(好ましくはPC/Gene)を用いて使用されるように設計されている。
【0132】
米国特許第6,013,263号、同第6,017,536号、および同第6,020,459号においては、一連の検索モチーフ(107×178×4、ALLMOTI5、およびPLZIPモチーフ)は、厳密な範囲から幅広い範囲までストリンジェンシーの範囲が設計され、操作されたが、107×178×4が好ましい。配列は、米国特許第6,013,263号、同第6,017,536号、および同第6,020,459号のそのような検索モチーフ(例えば、表V〜表XIVに列挙されたようなモチーフ)を介して同定され、抗膜融合性(例えば、抗ウイルス)活性を強く示し、抗膜融合性(例えば、抗ウイルス)化合物の同定においてさらに有用であり得る。
【0133】
他の抗ウイルスペプチド
抗RSVペプチド
抗RSVペプチドには、RSVによるウイルス感染を阻害することがさらに同定されている、RSV中の対応するペプチド配列から同定されたDP178および/またはDP107アナログが含まれる。目的のそのようなペプチドには、US2005/0070475の表16のペプチドおよび配列番号10〜配列番号30のペプチドが含まれる。これらのペプチドを合成するための詳細なプロトコールはUS2005/0070475に開示されており、その内容は引用により本明細書中に具体的に組み入れられる。特に興味深いのは、以下のペプチドである:
【0134】
【化22】
US2005/0070475の配列番号10のペプチドは、RSVのF2領域に由来し、そして、DP107およびDP178ペプチドに対応していると記載されている検索モチーフ(すなわち、「DP107/178様」)を使用して、米国特許第6,103,236号、および同第6,020,459号の中で同定された。配列番号21〜配列番号23のペプチドはそれぞれ、US2005/0070475の配列番号10のペプチドの中に含まれるアミノ酸配列を有し、そして各々が、抗RSV活性を示し、特に、50μg/ml未満の濃度でRSV感染Hep−2細胞と非感染Hep−2細胞との間での融合および合胞体形成を阻害することが示されている。
【0135】
US2005/0070475の配列番号11のペプチドは、RSVのF1領域に由来し、そして、DP107に対応していると記載されている検索モチーフ(すなわち、「DP107様」)を使用して、米国特許第6,103,236号、および同第6,020,459号の中で同定された。配列番号24のペプチドには、US2005/0070475の配列番号10のペプチドの中に含まれるアミノ酸配列が含まれ、同様に、抗RSV活性を示し、特に、50μg/ml未満の濃度でRSV感染Hep−2細胞と非感染Hep−2細胞との間での融合および合胞体形成を阻害することが示されている。
【0136】
抗HPIVペプチド
抗HPIVペプチドには、HPIV中の対応するペプチド配列から同定されたDP178および/またはDP107アナログが含まれ、そしてHPIVによるウイルス感染を阻害することがさらに同定されている。目的のそのようなペプチドには、US2005/0070475の表17および配列番号31〜配列番号62のペプチドが含まれる。これらのペプチドを合成するための詳細なプロトコールはUS2005/0070475に開示されており、その内容は引用により本明細書中に具体的に組み入れられる。特に興味深いのは、以下のペプチドである:
【0137】
【化23】
US2005/0070475の配列番号31のペプチドは、HPIV−3のF1領域に由来し、そして、DP107に対応していると記載されている検索モチーフ(すなわち、「DP107様」)を用いて、米国特許第6,103,236号、および同第6,020,459号の中で同定された。配列番号25および配列番号26のペプチドはそれぞれ、US2005/0070475の配列番号30のペプチドの中に含まれるアミノ酸配列を有し、そしてそれぞれが、抗HPIV−3活性を示し、特に、1μg/ml未満の濃度でHPIV−3感染Hep2細胞と非感染CV−1W細胞との間での融合および合胞体形成を阻害することが示されている。
【0138】
US2005/0070475の配列番号32のペプチドもまた、HPIV−3のF1領域に由来し、そして、DP178に対応していると記載されている検索モチーフ(すなわち、「DP178様」)を用いて、米国特許第6,103,236号、および同第6,020,459号の中で同定された。配列番号27および配列番号28〜配列番号32のペプチドはそれぞれ、US2005/0070475の配列番号32のペプチドの中に含まれるアミノ酸配列を有し、そしてそれぞれがまた、抗HPIV−3活性を示し、特に、1μg/ml未満の濃度でHPIV−3感染Hep2細胞と非感染CV−1W細胞のと間での融合および合胞体形成を阻害することが示されている。
【0139】
抗MeVペプチド
抗MeVペプチドは、麻疹ウイルス(MeV)によるウイルス感染を阻害することがさらに同定されている、麻疹ウイルス中の対応するペプチド配列から同定されたDP178および/またはDP107アナログである。特定の目的のそのようなペプチドには、US2005/0070475の表19のペプチドおよび配列番号74〜配列番号86のペプチドが含まれる。これらのペプチドを合成するための詳細なプロトコールはUS2005/0070475に開示されており、その内容は引用により本明細書中に具体的に組み入れられる。特に興味深いのは、以下に列挙されるペプチドである:
【0140】
【化24】
麻疹ウイルス由来の配列は、DP178に対応していると記載されている検索モチーフ(すなわち、「DP178様」)を使用して、米国特許第6,103,236号、および同第6,020,459号の中で同定された。配列番号33、配列番号34、配列番号35および配列番号36のペプチドはそれぞれ、そのように同定されたアミノ酸配列を有し、そしてそれぞれが、抗MeV活性を示し、特に、1μg/ml未満の濃度でMeV感染Hep2と非感染Vero細胞との間での融合および合胞体形成を阻害することが示されている。
【0141】
抗SIVペプチド
抗SIVペプチドは、SIVによるウイルス感染を阻害することがさらに同定されている、SIV中の対応するペプチド配列から同定されたDP178および/またはDP107アナログである。目的のそのようなペプチドには、US2005/0070475の表18のペプチドおよび配列番号63〜配列番号73までのペプチドが含まれる。これらのペプチドを合成するための詳細なプロトコールはUS2005/0070475に開示されており、その内容は引用により本明細書中に具体的に組み入れられる。特に興味深いのは、以下のペプチドである:
【0142】
【化25】
【0143】
【化26】
SIV膜貫通融合タンパク質由来の配列は、DP178に対応していると記載されている検索モチーフ(すなわち、「DP178様」)を使用して、米国特許第6,103,236号、および同第6,020,459号の中で同定された。配列番号37〜配列番号46のペプチドはそれぞれ、そのように同定されたアミノ酸配列を有し、そしてそれぞれが、粗ペプチドとして強力な抗SIV活性を示すことが示されている。
【0144】
さらなるウイルス阻害剤(viral inhibitor)および融合阻害剤(fusion inhibotor)
表現「ウイルス阻害剤誘導体」は、抗膜融合性化合物または侵入阻害剤(または非抗膜融合性)化合物から選択されたウイルス阻害剤の任意の改変体あるいは誘導体を意味するように意図される。
【0145】
抗膜融合性化合物としては以下が挙げられるが、これらに限定されない:エンフュービルタイド;C34;T−1249;TRI−899;TRI−999;5−ヘリックス;N36 Mut(e.g);NCCG−gp41;DP−107;M41−P;N36;M87o;FM−006;ADS−J1;C14リンクミド(C14 linkmid);C34コイル;ヘモリジンA;IQN17;IQN23;SC34EK;SPI−30,014;SPI−70,038;T−1249−HSA;T−649;T−651;TRI−1144;C14;MBP−107;scC34;SJ−2176;T−1249−トランスフェリン;p26;p38;ADS−J2;C52L;クローン3抗体;D5 IgG;D5 scFc;F240 scFv;シフュービルタイド(sifuvirtide);IZN−36;T−1249ミメティボディ(mimetibody);N−36−E;NB−2;NB−64;S−29−I;テアフラビン−3,3’−ジガラート;VIRIP;シアマイシンI;シアマイシンII。
【0146】
侵入阻害剤(または非抗膜融合性)化合物としては以下が挙げられるが、これらに限定されない:AMD−070;SPC−3;KRH−2731;AMD−8664;FC−131;HIV−1 Tatアナログ;KRH−1120;KRH−1636;POL−2438;T−134;T−140;ストローマ細胞由来因子1;ALX40−4C;AMD−3100;T−22;TJN−151;AM−1401;EradicAideウイルスマクロファージ炎症タンパク質II;AMD−3451;コノクルボン;マラビロック;ビクリビロック;INCB−9471;INCB−15,050;DAPTA;PRO−140;HGS−004;SCH−C;TAK−652;TAK−220;ニフェビロック(nifeviroc);AMD−887;抗CD63 MAb;AOP−RANTES;CPMD−167;E−913;FLSC R/T−IgG1;HGS−101;NIBR−1282;ノナカイン(nonakine);PSC−RANTES;sCD4−17b;SCH−350,634;MIP−1α;MIP−1β;RANTES;アプラビロック;ペプチドT;TAK−779;pCLXSNベクター;UCB−35,625;J−113,863;CLIV;I−309;EGCG;エピガロカテキンガレート(Epigallocathechin gallate);HB−19;λ−カラギーナン;PC−515;カードラン硫酸塩;OKU−40;OKU−41;VGV−1;ジンテルビル(Zintevir);AR−177;T−30,177;コハク酸化アルブミン;NSC0−658,586;ISIS−5320;RP−400c;SA−1042;C31G;サヴィ(Savvy);PRO−542;rCD4−IgG2;BMS−488,043;BMS−378,806;DES−6;12p1;アクチノヒビン;BlockAide/VP;CD4M33;CT−319;CT−326;シアノビリン−N;DCM−205;DES−10;グリフィスシン;HNG−105;NBD−556;NBD−557;PEG−シアノビリン−N;シトビリン(scytovirin);sCD4;デキストリン−2−硫酸塩;F−105;FP−21,399;TNX−355;B4 MAb;R−15−K;sCD38(51−75)MBP;PRO−2000;NSC−13,778;SB−673,461M;SB−673,462M;rsCD4;Ac(Ala10,11)RANTES(2−14);IC−9564;RPR−103,611;Immudel−gp120;スリゴビル;IQP−0410;アセチル化トリヨードサイロニン;SP−01A;DEB−025;CSA−54;HGS−H/A27;SP−10;VIR−5103;BMS−433,771;TMC−353,121;NSC−650,898;ミケラミンB;NSC−692,906;TG−102;VIR−576;MEDI−488;CovX−Body;CNI−H0294。
【0147】
抗ウイルスおよび抗膜融合性ペプチドの改変
本発明では、抗ウイルス活性および/または抗膜融合性活性を示すペプチドを改変することが意図され、DP−107およびDP−178ならびにそれらのアナログのそのような改変が含まれる。そのような改変されたペプチドは、共有結合を介して血液成分上の利用可能な反応性官能基と反応することができる。本発明はまた、そのような改変、血液成分とのそのような組合せ、およびそれらの使用のための方法に関する。これらの方法には、非結合ペプチドの患者への投与と比較して、結合させられた抗ウイルスペプチド誘導体の有効な治療薬の寿命を延長させる工程が含まれる。改変されたペプチドは、DAC(商標)(薬物親和性複合体)として設計されたタイプのペプチドであり、これには、抗ウイルスペプチド分子と、移動性の血液タンパク質の反応性官能基と反応することができる化学反応基を伴う連結基が含まれる。血液成分またはタンパク質との反応によって、改変されたペプチド、すなわちDACを、血液を介して適切な部位または受容体に送達することができる。
【0148】
タンパク質上の官能基と共有結合を形成させるためには、反応性基として多種多様な活性なカルボキシル基、特にエステルを使用することができ、この場合、ヒドロキシル部分は、ペプチドを改変するために必要とされるレベルで生理学的に許容される。多数の異なるヒドロキシル基がこれらの反応性基に使用され得るが、最も便利なものは、N−ヒドロキシスクシンイミド(すなわち、NHS)、N−ヒドロキシ−スルホスクシンイミド(sulfo−NHS)であろう。本発明の好ましい実施形態においては、タンパク質上の官能基はチオール基であり、そして反応性基はマレイミド含有基(例えば、γ−マレイミド−ブチラールアミド(GMBA)またはマレイミドプロピオン酸(MPA)であろう。
【0149】
第一級アミンは、NHSエステルについての重要な標的である。タンパク質のN末端上に存在する接近することができるα−アミン基はNHSエステルと反応する。しかし、タンパク質のα−アミノ基は、NHSカップリングには望ましくもない場合があり、また、利用可能できない場合もある。5種類のアミノ酸はそれらの側鎖中に窒素を有するが、リジンのε−アミンだけが有意にNHSエステルと反応する。以下の反応スキームに示されるように、NHSエステルの結合反応が第一級アミンと反応してN−ヒドロキシスクシンイミドを放出する場合には、アミド結合が形成される。
【0150】
【化27】
本発明の好ましい実施形態において、このタンパク質上の官能基はチオール基であり、化学反応性基はマレイミド含有基(例えば、MPAまたはGMBA(γ−マレイミド−ブチラールアミド)であろう。マレイミド基は、反応混合物のpHが6.5と7.4の間に維持される場合には、ペプチド上のスルフヒドリル基について最も選択的である。pH7.0では、マレイミド基のスルフヒドリルとの反応の速度は、アミンとの反応の速度よりも1000倍速い。以下の反応スキームに示されるように、マレイミド基とスルフヒドリルとの間で安定なチオエーテル結合が形成され、これは、生理学的な条件下では切断され得ない。
【0151】
【化28−1】
特異的標識
好ましくは、本発明の改変されたペプチドは、移動性の血液タンパク質上のチオール基と特異的に反応するように設計される。このような反応は、好ましくは、移動性血液タンパク質(例えば、血清アルブミンまたはIgG)上のチオール基へのマレイミドの連結(例えば、GMBS、MPAまたは他のマレイミドから調製される)で改変されたこのペプチドの共有結合によって確立される。
【0152】
特定の状況では、マレイミドでの特異的標識は、NHSおよびsulfo−NHSのような基での移動性タンパク質の非特異的な標識化を上回るいくつかの利点をもたらす。チオール基は、インビボではアミノ基よりも少ない量で存在する。従って、本発明のマレイミドで改変されたペプチド(すなわち、マレイミドペプチド)は、より少ないタンパク質に共有結合するであろう。例えば、アルブミン(最も豊富な血液タンパク質)中には、チオール基が1つだけ存在する。従って、ペプチド−マレイミド−アルブミン結合体は、約1:1のモル比でペプチド対アルブミンを含む傾向があるだろう。アルブミンに加えて、IgG分子(クラスII)もまた遊離のチオールを有する。IgG分子と血清アルブミンが、血液中の可溶タンパク質の大部分を構成するので、これらはまた、マレイミド改変ペプチドに共有結合させるために利用できる血液中の遊離のチオール基の大部分を構成する。
【0153】
さらに、遊離のチオール含有血液タンパク質(IgGを含む)の間でさえ、アルブミン自体の特有の特徴が原因で、マレイミドでの特異的標識はペプチド−マレイミド−アルブミン結合体の優先的な形成を導く。アルブミンの単一の遊離のチオール基は種間で高度に保存されており、アミノ酸残基34(Cys34)に位置する。最近、アルブミンのCys34が、他の遊離チオール含有タンパク質上の遊離のチオールと比べて、高い反応性を有することが明らかにされていた。これは、アルブミンのCys34に対する非常に低いpK値(5.5)に一部原因がある。これは、一般的にシステイン残基についての典型的なpK値(典型的には約8)よりもはるかに低い。この低いpKが原因で、正常な生理学的条件下では、アルブミンのCys34は主にイオン化された形態であり、これはその反応性を劇的に高める。Cys34の低いpK値に加えて、Cys34の反応性を高める別の因子はその位置であり、この位置は、アルブミンの領域Vの一つのループの表面に近い間隙の中にある。この位置は、Cys34を全ての種類のリガンドに対して極めて利用しやすくし、そしてこの位置は、遊離ラジカルトラップおよび遊離チオールスカベンジャーとしてのCys34の生物学的役割において重要な因子である。これらの特性は、Cys34をマレイミド−ペプチドに非常に反応性にし、反応速度の加速は、他の遊離のチオール含有タンパク質とのマレイミド−ペプチドの反応速度に比べて、1000倍であり得る。
【0154】
ペプチド−マレイミド−アルブミン結合体の別の利点は、詳細にはCys34において、ペプチド対アルブミンを1:1で含むことに関連する再現性である。他の技術(例えば、グルタルアルデヒド、DCC、EDCおよび例えば、遊離のアミンの他の化学活性化)には、この選択性が欠けている。例えば、アルブミンには52個のリジン残基が含まれ、このうちの25〜30個は、アルブミンの表面に位置し、したがって結合のために接近することができる。これらのリジン残基の活性化、あるいはこれらのリジン残基を介するカップリングのためのペプチドの別の改変によっては、結合体の不均一な集団が生じる。ペプチド対アルブミンの1:1のモル比が使用される場合でもなお、生成物は、複数の結合体産物からなり、そのいくつかには、アルブミン1分子当たり0、1、2以上のペプチドが含まれており、それぞれが、25〜30個の利用可能なリジン部位のうちの任意1つ以上に無作為に結合させられたペプチドを有する。多数の可能な組み合わせが提供される場合には、正確な組成および個々の結合体のバッチの特性決定は困難になり、バッチ間の再現性はほとんど不可能であり、このような結合体の治療薬としての望ましさを低下させてしまう。加えて、アルブミンのリジン残基を介する結合は、アルブミン1分子当たりにより多くの治療薬を送達するという利点を少なくとも有するようであるが、複数の研究によって、治療薬対アルブミンの1:1の比が好ましいことが示されている。Stehleらによる論文「The Loading Rate Determines Tumor Targeting properties of Methotrexate−Albumin Conjugates in Rats」、Anti−Cancer Drugs,第8巻,677−685頁(1988)では、著者らは、グルタルアルデヒドを介して結合させられたアルブミンに対する抗癌剤メトトレキサートの1:1の比が最も有望な結果を生じたことを報告した。これらの結合体は腫瘍細胞によって優先的に取り込まれたが、5:1〜20:1のメトトレキサート分子を保有している結合体は、変化したHPLCプロフィールを有し、そしてインビボで肝臓に迅速に取り込まれた。これらの高い比では、アルブミンに対する立体構造の変化は、治療用担体としてのその有効性を低下させると推論された。
【0155】
マレイミド−ペプチドのインビボでの制御された投与を通じて、アルブミンとIgGのインビボでの特異的標識を制御することができる。典型的な投与においては、投与されたマレイミド−ペプチドのうちの80〜90%がアルブミンを標識し、5%未満はIgGを標識するであろう。グルタチオンのような遊離のチオールの微量標識もまた起こるであろう。このような特異的標識は、インビボでの使用に好ましい。なぜなら、これにより、投与された薬剤の概算された半減期の正確な計算が可能となるからである。
【0156】
制御された特異的なインビボ標識化を提供することに加えて、マレイミド−ペプチドは、血清アルブミンとIgGのエキソビボでの特異的標識を提供することができる。このようなエキソビボでの標識には、血清アルブミンおよび/またはIgGを含む、血液、血清または生理食塩溶液へのマレイミド−ペプチドの添加が含まれる。一旦、マレイミド−ペプチドとの結合がエキソビボで起こると、血液、血清、または生理食塩溶液は、インビボ処置のために患者の血液に再投与することができる。
【0157】
NHS−ペプチドとは対照的に、マレイミド−ペプチドは、一般的には、水溶液の存在下および遊離のアミンの存在下では極めて安定である。マレイミド−ペプチドが遊離のチオールとだけ反応するので、保護基は、一般的には、マレイミド−ペプチドがそれ自体と反応することを防ぐ必要はない。加えて、改変されたペプチドの高い安定性によって、インビボでの使用に適している高度に精製された産物を調製するためのHPLCのようなさらなる精製工程の使用が可能となる。最後に、高い化学的安定性によって、製品の有効期間をより長くすることができる。
【0158】
非特異的標識
本発明の抗ウイルスペプチドはまた、血液成分の非特異的標識のために改変される場合もある。アミノ基に対する結合、特に非特異的標識のためのアミド結合の形成を伴う結合もまた使用されるであろう。そのような結合を形成させるためには、化学反応基として多種多様な活性カルボキシル基、特にエステルが使用され得、この場合、ヒドロキシル部分は、必要とされるレベルで生理学的に許容される。多数の異なるヒドロキシル基がこれらの結合剤において使用され得るが、最も便利なものはN−ヒドロキシスクシンイミド(NHS)およびN−ヒドロキシ−スルホスクシンイミド(sulfo−NHS)であろう。
【0159】
利用され得る他の結合剤は、米国特許第5,612,034号に記載されている。
【0160】
改変されたペプチドの化学反応基がインビボで反応し得る様々な部位としては、細胞、特に赤血球(成熟赤血球)および血小板、ならびにタンパク質(例えば、免疫グロブリン(IgGおよびIgMを含む)、血清アルブミン、フェリチン、ステロイド結合タンパク質、トランスフェリン、サイロキシン結合タンパク、α−2−マクログロブリンなど)が挙げられる。改変されたペプチドが反応するそれらの受容体(これは、長期間生存しない)は、一般的に、約3日以内にヒト宿主から排除されるであろう。上記に示されるタンパク質(細胞のタンパク質を含む)は、血中濃度に基づいて、特に半減期に関しては、少なくとも3日間残存し、そして5日間以上残存する場合もあるであろう(通常は、60日間を超えず、より通常は30日間を超えない)。
【0161】
ほとんどの部分について、反応は、血液中の移動性の成分、具体的には血液タンパク質および細胞、さらに具体的には血液タンパク質および成熟赤血球との反応であろう。「移動性」によっては、その成分が、任意の延長された期間(一般的には5分間を超えない、より通常は1分間)固定された状況にはないことが意図されるが、血液成分のうちのいくつかは、延長された期間比較的固定された状態で存在し得る。最初は、機能化されたタンパク質および細胞の比較的不均質な集団が存在するであろう。しかし、ほとんどの部分については、数日以内の集団は、血流中の機能化されたタンパク質の半減期に依存して、最初の集団から実質的に変化するであろう。従って、通常は約3日以内またはそれ以後には、IgGは、血流において優勢な機能化されたタンパク質となるであろう。
【0162】
通常は、投与後5日までに、IgG、血清アルブミンおよび成熟赤血球は、血液中の結合成分の少なくとも約60モル%、通常は少なくとも約75モル%を占め、IgG、IgM(実質的により少ない程度まで)および血清アルブミンは、非細胞性結合成分の少なくとも約50モル%、通常は少なくとも約75モル%、より通常は約80モル%となるであろう。
【0163】
血液成分に対する非特異的な改変されたペプチドの所望される結合体は、患者への改変されたペプチドの投与によってインビボで調製され得る。患者はヒトまたは他の哺乳動物であり得る。その投与は、ボーラスの形態で行われる場合も、また、流れを計器で調節して注入することなどによって、ゆっくりと経時的に導入される場合もある。
【0164】
所望される場合、目的の結合体はまた、血液を本発明の改変されたペプチドと組み合わせ、これにより血液成分上の反応性官能基への改変されたペプチドの共有結合が可能となること、次いで、宿主に結合させられた血液を戻すかまたは投与することによってエキソビボで調製され得る。さらに、これはまた、初めに、個々の血液成分または限られた数の成分(例えば、赤血球、免疫グロブリン、血清アルブミンなど)を精製し、そしてその成分または複数の成分をエキソビボで化学的に反応性の改変されたペプチドと組み合わせることによって達成され得る。次いで、機能化された血液または血液成分は、目的の治療的に有効な結合体をインビボで提供するために宿主に戻され得る。血液はまた、エキソビボでの操作の際の凝固を防ぐために処理され得る。
【0165】
改変された抗ウイルスペプチドおよび抗膜融合性ペプチドの合成
A.ペプチド合成
本発明の抗ウイルスペプチドおよび/または抗膜融合性ペプチドは、当業者に公知の固相ペプチド化学の標準的な方法によって合成され得る。例えば、ペプチドは、Applied Biosystem synthesizerを使用して、Steward and Young(Steward,J.M.and Young,J.D.,Solid
Phase Peptide Synthesis,第2版.,Pierce Chemical Company,Rockford,III.,(1984))によって記載された手順に従って固相化学技術によって合成され得る。次いで、同様に、複数のペプチド断片が合成され、その後、互いに連結させられることによって、より大きなペプチドが形成させられる。これらの合成ペプチドはまた、特定の位置にアミノ酸置換を有して作製することもできる。
【0166】
固相ペプチド合成について、多くの技術の概要が、J.M.Stewart and J.D.Young,Solid Phase Peptide Synthesis,W.H.Freeman Co.(San Francisco),1963、およびJ.Meienhofer,Hormonal Proteins and Peptides,第2巻 46頁、Academic Press(New York),1973に見ることができる。伝統的な溶液合成については、G.Schroder and K.Lupke,The Peptides,第1巻、Acacemic Press(New York)を参照のこと。一般的には、これらの方法には、1つ以上のアミノ酸または適切に保護されたアミノ酸の、成長しつつあるペプチド鎖への連続的付加が含まれる。通常、第1のアミノ酸のアミノ基またはカルボキシル基のいずれかが、適切な保護基によって保護される。次いで、保護されたアミノ酸または誘導されたアミノ酸は、不活性な固体支持体に付着させられるか、あるいは、アミド結合を形成させるために適している条件下で、適切に保護された相補性(アミノもしくはカルボキシル)基を有している配列中に次のアミノ酸を付加することによって溶液中で利用されるかのいずれかである。次いで、保護基がこの新たに付加されたアミノ酸残基から除去され、そして(適切に保護された)次のアミノ酸が付加されるなどが行われる。
【0167】
全ての所望されるアミノ酸が、適切な配列内に連結させられた後、任意の残っている保護基(および任意の固体支持体)が連続的に、または同時に除去されて、最終的なポリペプチドにされる。この一般的手順の単純な改変によって、2つ以上のアミノ酸を一度に成長しつつある鎖に対して、例えば、保護されたトリペプチドを適切に保護されたジペプチドと(キラル中心をラセミ化させない条件下で)カップリングさせることによって付加させることが可能であり、脱保護の後に、ペンタペプチドが形成させられる。
【0168】
本発明の化合物を調製する特に好ましい方法には固相ペプチド合成が含まれる。この場合、アミノ酸α−N−末端は、酸感受性基または塩基感受性基によって保護される。そのような保護基は、ペプチド結合形成の条件に対して安定であるという特性を有するべきであり、一方、成長しつつあるペプチド鎖の破壊またはその中に含まれる任意のキラル中心のラセミ化を伴わずに容易に除去できる。適切な保護基は、9−フルオレニルメチルオキシカルボニル(Fmoc)、t−ブチルオキシカルボニル(Boc)、ベンジルオキシカルボニル(Cbz)、ビフェニルイソプロピルオキシカルボニル、t−アミルオキシカルボニル、イソボルニルオキシカルボニル、α,α−ジメチル−3,5−ジメトキシベンジルオキシカルボニル、o−ニトロフェニルスルフェニル、2−シアノ−t−ブチルオキシカルボニルなどである。9−フルオレニルメチルオキシカルボニル(Fmoc)保護基は、本発明のペプチドの合成に特に好ましい。他の好ましい側鎖保護基は、リジンおよびアルギニンのような側鎖アミノ基については、2,2,5,7,8−ペンタメチルクロマン−6−スルホニル(pmc)基、ニトロ基、p−トルエンスルホニル基、4−メトキシベンゼン−スルホニル基、Cbz基、Boc基、およびアダマンチルオキシカルボニル基であり;チロシンについては、ベンジル基、o−ブロモベンジルオキシカルボニル基、2,6−ジクロロベンジル基、イソプロピル基、t−ブチル(t−Bu)基、シクロヘキシル基、シクロペニル基およびアセチル(Ac)基であり;セリンについては、t−ブチル基、ベンジル基およびテトラヒドロピラニル基であり;ヒスチジンについては、トリチル基、ベンジル基、Cbz基、p−トルエンスルホニル基および2,4−ジニトロフェニル基であり;トリプトファンについては、ホルミル基であり;アスパラギン酸およびグルタミン酸については、ベンジル基およびt−ブチル基であり、さらに、システインについては、トリフェニルメチル(トリチル)基である。
【0169】
固相ペプチド合成方法において、α−C−末端アミノ酸は、適切な固体支持体または樹脂に付着させらる。上記合成に有用な適切な固体支持体は、段階的な縮合−脱保護反応の試薬および反応条件に対して不活性であり、さらに、使用される媒体に不溶性である材料である。α−C−末端カルボキシペプチドの合成のための好ましい固体支持体は、4−ヒドロキシメチルフェノキシメチル−コポリ(スチレン−1%ジビニルベンゼン)である。α−C−末端アミドペプチドに好ましい固相支持体は、Applied Biosystems(Foster City,Calif.)から入手することができる4−(2’,4’−ジメトキシフェニル−Fmoc−アミノメチル)フェノキシアセトアミドエチル樹脂である。α−C−末端アミノ酸は、4−ジメチルアミノピリジン(DMAP)、1−ヒドロキシベンゾトリアゾール(HOBT)、ベンゾトリアゾール−1−イルオキシ−トリス(ジメチルアミノ)ホスホニウム−ヘキサフルオロホスフェート(BOP)またはビス(2−オキソ−3−オキサゾリジニル)ホスフィンクロリド(BOPCl)を伴ってまたは伴わずに、N,N’−ジシクロヘキシルカルボジイミド(DCC)、N,N’−ジイソプロピルカルボジイミド(DIC)またはO−ベンゾトリアゾル−1−イル−N,N,N’,N’,−テトラメチルウロニウム−ヘキサフロオロホスフェート(HBTU)によって樹脂に結合させられ、結合は、溶媒(例えば、ジクロロメタンまたはDMF)中で10℃から50℃の間の温度で約1時間〜24時間の間媒介される。
【0170】
固体支持体が、4−(2’,4’−ジメトキシフェニル−Fmoc−アミノメチル−)フェノキシ−アセトアミドエチル樹脂である場合、Fmoc基は、上記のα−C−末端アミノ酸とのカップリングの前に、第二級アミン、好ましくはピペリジンで切断される。脱保護された4−(2’,4’−ジメトキシフェニル−Fmoc−アミノメチル−)フェノキシ−アセトアミドエチル樹脂へのカップリングのための好ましい方法は、DMF中のO−ベンゾトリアゾール−1−イル−N,N,N’,N’,−テトラメチルウロニウムヘキサフロオロ−ホスフェート(HBTU、1当量)および1−ヒドロキシベンゾトリアゾール(HOBT、1当量)である。保護されたアミノ酸の連続的なカップリングは、当該分野で周知であるように、自動ポリペプチド合成装置の中で行わせることができる。好ましい実施形態においては、成長しつつあるペプチド鎖のα−N−末端アミノ酸はFmocで保護される。成長しつつあるペプチドのα−N−末端側からのFmoc保護基の除去は、第二級アミン、好ましくはピペリジンでの処理によって達成される。次いで、個々の保護されたアミノ酸は、約3倍のモル過剰量で導入され、そして好ましくは、カップリングはDMF中で行われる。カップリング剤は、通常、O−ベンゾトリアゾール−1−イル−N,N,N’,N’,−テトラメチルウロニウム−ヘキサフロオロホスフェート(HBTU、1当量)および1−ヒドロキシベンゾトリアゾール(HOBT、1当量)である。
【0171】
固相合成の終点において、このポリペプチドは、連続的または1回の操作のいずれかで、樹脂から取り出され、脱保護される。ポリペプチドの取り出しと脱保護は、チオアニソール、水、エタンジチオールおよびトリフルオロ酢酸を含む切断試薬(cleavage
reagent)で樹脂が結合したポリペプチドを処理することによって1回の操作で達成され得る。ポリペプチドのα−C−末端がアルキルアミドである場合は、樹脂は、アルキルアミンを用いてアミノ分解によって切断される。あるいは、ペプチドは、例えば、メタノールを用いるエステル交換反応、それに続くアミノ分解、または直接的なアミド基交換によって取り出され得る。保護されたペプチドは、この時点で精製され得るか、または次の工程に直接使用され得る。側鎖保護基の除去は、上記の切断カクテル(cleavage cocktail)を使用して達成される。完全に脱保護されたペプチドは、任意または全ての以下の型:弱塩基樹脂上のイオン交換(酢酸塩の形態);非誘導体化ポリスチレン−ジビニルベンゼン上の疎水性吸着クロマトグラフィー(例えば、Amberlite XAD);シリカゲル吸着クロマトグラフィー;カルボキシメチルセルロース上でのイオン交換クロマトグラフィー;分配クロマトグラフィー(例えば、Sephadex G−25、LH−20)または向流分配;高速液体クロマトグラフィー(HPLC)(特に、オクチル−もしくはオクタデシルシリル−シリカ結合相充填剤上での逆相HPLC)を利用する一連のクロマトグラフィー工程によって精製される。これらのITPの分子量は、Fast Atom Bombardment(FAB)Mass Spectroscopyを使用して決定される。
【0172】
N−末端保護基
上記で議論されたように、用語「N−保護基」とは、アミノ酸もしくはペプチドのα−N−末端を保護すること、あるいはアミノ酸もしくはペプチドのアミノ基を、合成手順の間の望ましくない反応から保護することが意図される基をいう。一般的に、使用されるN−保護基は、Greene、「Protective Groups In Organic Synthesis」(John Wiley&Sons、New York(1981))(これは、引用により本明細書中に組み入れられる)に開示されている。加えて、保護基を、例えば、酵素的加水分解によってインビボで容易に切断されて生物学的に活性なペアレント(parent)を放出するるプロドラッグとして使用することができる。α−N−保護基には、低級アルカノイル基(例えば、ホルミル基、アセチル基(「Ac」)、プロピオニル基、ピバロイル基、t−ブチルアセチル基など;他のアシル基(2−クロロアセチル基、2−ブロモアセチル基、トリフルオロアセチル基、トリクロロアセチル基、フタリル基、o−ニトロフェノキシアセチル基、クロロブチリル基、ベンゾイル基、4−クロロベンゾイル基、4−ブロモベンゾイル基、4−ニトロベンゾイル基などを含む);スルホニル基(例えば、ベンゼンスルホニル基、p−トルエンスルホニル基など);カルバメート形成基(例えば、ベンジルオキシカルボニル基、p−クロロベンジルオキシカルボニル基、p−メトキシベンジルオキシカルボニル基、p−ニトロベンジルオキシカルボニル基、2−ニトロベンジルオキシカルボニル基、p−ブロモベンジルオキシカルボニル基、3,4−ジメトキシベンジルオキシカルボニル基、3,5−ジメトキシベンジルオキシカルボニル基、2,4−ジメトキシベンジルオキシカルボニル基、4−エトキシベンジルオキシカルボニル基、2−ニトロ−4,5−ジメトキシベンジルオキシカルボニル基、3,4,5−トリメトキシベンジルオキシカルボニル基、1−(p−ビフェニルイル)−1−メチルエトキシカルボニル基、α,α−ジメチル−3,5−ジ−メトキシベンジルオキシカルボニル基、ベンズヒドリルオキシカルボニル基、t−ブチルオキシカルボニル基(Boc)、ジイソプロピルメトキシカルボニル基、イソプロピルオキシカルボニル基、エトキシカルボニル基、メトキシカルボニル基、アリルオキシカルボニル基、2,2,2−トリクロロエトキシカルボニル基、フェノキシカルボニル基、4−ニトロフェノキシカルボニル基、フルオレニル−9−メトキシカルボニル基、シクロペンチルオキシカルボニル基、アダマンチルオキシカルボニル基、シクロヘキシルオキシカルボニル基、フェニルチオカルボニル基など);アリールアルキル基(例えば、ベンジル基、トリフェニルメチル基、ベンジルオキシメチル基、9−フルオレニルメチルオキシカルボニル基(Fmoc)など)、ならびにシリル基(例えば、トリメチルシリルなど)が含まれる。
【0173】
カルボキシ保護基
上記で議論されたように、用語「カルボキシ保護基」とは、化合物の他の機能性部位に関する反応が実施されながらカルボン酸官能基をブロックもしくは保護するために使用されるカルボン酸保護エステル基またはアミド基をいう。カルボキシ保護基は、Greene、「Protective Groups in Organic Synthesis」152〜186頁(1981)(これは引用により本明細書中に組み入れられる)に開示されている。加えて、カルボキシ保護基はプロドラッグとして使用することができ、それによってカルボキシ保護基は、例えば、酵素的加水分解によって容易にインビボで切断されて、生物学的に活性なペアレントを放出することができる。そのようなカルボキシ保護基は当業者に周知であり、米国特許第3,840,556号および同第3,719,667号にて記載されているように、ペニシリンおよびセファロスポリンの分野におけるカルボキシル基の保護において広く使用されている。これらの開示は引用により本明細書中に組み入れられる。代表的なカルボキシ保護基は、C1〜C8低級アルキル基(例えば、メチル基、エチル基またはt−ブチル基など);アリールアルキル基(例えば、フェネチル基またはベンジル基、ならびに、アルコキシベンジル基またはニトロベンジル基などのようなそれらの置換誘導体);アリールアルケニル基(例えば、フェニルエテニル基など);アリールおよびその置換された誘導体(例えば、5−インダニル基など);ジアルキルアミノアルキル基(例えば、ジメチルアミノエチル基など);アルカノイルオキシアルキル基(例えば、アセトキシメチル基、ブチリルオキシメチル基、バレリルオキシメチル基、イソブチリルオキシメチル基、イソバレリルオキシメチル基、1−(プロピオニルオキシ)−1−エチル基、1−(ピバロイルオキシ)−1−エチル基、1−メチル−1−(プロピオニルオキシ)−1−エチル基、ピバロイルオキシメチル基、プロピオニルオキシメチル基など);シクロアルカノイルオキシアルキル基(例えば、シクロプロピルカルボニルオキシメチル基、シクロブチルカルボニルオキシメチル基、シクロペンチルカルボニルオキシメチル基、シクロヘキシルカルボニルオキシメチル基など);アロイルオキシアルキル基(例えば、ベンゾイルオキシメチル基、ベンゾイルオキシエチル基など);アリールアルキルカルボニルオキシアルキル基(例えば、ベンジルカルボニルオキシメチル基、2−ベンジルカルボニルオキシエチル基など);アルコキシカルボニルアルキル基またはシクロアルキルオキシカルボニルアルキル基(例えば、メトキシカルボニルメチル基、シクロヘキシルオキシカルボニルメチル基、1−メトキシカルボニル−1−エチル基など);アルコキシカルボニルオキシアルキル基またはシクロアルキルオキシカルボニルオキシアルキル基(例えば、メトキシカルボニルオキシメチル基、t−ブチルオキシカルボニルオキシメチル基、1−エトキシカルボニルオキシ−1−エチル基、1−シクロへキシルオキシカルボニルオキシ−1−エチル基など);アリールオキシカルボニルオキシアルキル基(例えば、2−(フェノキシカルボニルオキシ)エチル基、2−(5−インダニルオキシカルボニルオキシ)エチル基など);アルコキシアルキルカルボニルオキシアルキル基(例えば、2−(1−メトキシ−2−メチルプロパン−2−オイルオキシ)エチル基など);アリールアルキルオキシカルボニルオキシアルキル基(例えば、2−(ベンジルオキシカルボニルオキシ)エチル基など);アリールアルケニルオキシカルボニルオキシアルキル基(例えば、2−(3−フェニルプロペン−2−イルオキシカルボニルオキシ)エチル基など);アルコキシカルボニルアミノアルキル基(例えば、t−ブチルオキシカルボニルアミノメチル基など);アルキルアミノカルボニルアミノアルキル基(例えば、メチルアミノカルボニルアミノメチル基など);アルカノイルアミノアルキル基(例えば、アセチルアミノメチル基など);複素環式カルボニルオキシアルキル基(例えば、4−メチルピペラジニルカルボニルオキシメチル基など);ジアルキルアミノカルボニルアルキル基(例えば、ジメチルアミノカルボニルメチル基、ジエチルアミノカルボニルメチル基など);(5−(低級アルキル)−2−オキソ−1,3−ジオキソレン−4−イル)アルキル基(例えば、(5−t−ブチル−2−オキソ−1,3−ジオキソレン−4−イル)メチル基など);ならびに、(5−フェニル−2−オキソ−1,3−ジオキソレン−4−イル)アルキル基(例えば、(5−フェニル−2−オキソ−1,3−ジオキソレン−4−イル)メチル基など)である。
【0174】
代表的なアミドカルボキシ保護基は、アミノカルボニル基および低級アルキルアミノカルボニル基である。
【0175】
本発明の好ましいカルボキシ保護された化合物には複数の化合物があり、ここでは、その保護されたカルボキシ基は、低級アルキルエステル、シクロアルキルエステルもしくはアリールアルキルエステル(例えば、メチルエステル、エチルエステル、プロピルエステル、イソプロピルエステル、ブチルエステル、sec−ブチルエステル、イソブチルエステル、アミルエステル、イソアミルエステル、オクチルエステル、シクロヘキシルエステル、フェニルエチルエステルなど)、またはアルカノイルオキシアルキルエステル、シクロアルカノイルオキシアルキルエステル、アロイルオキシアルキルエステル、もしくはアリールアルキルカルボニルオキシアルキルエステルである。好ましいアミドカルボキシ保護基は、低級アルキルアミノカルボニル基である。例えば、アスパラギン酸は、酸不安定基(例えば、t−ブチル基)によってα−C−末端で保護され得、そして水素化不安定基(例えば、ベンジル基)によってβ−C−末端で保護され得、次いで、合成の間に選択的に脱保護され得る。
【0176】
ペプチドの改変
本発明の改変されたペプチドの生産様式は、そのペプチドを含む種々の要素の性質に応じて広く変化するであろう。合成手順は、単純であるように、高収率を提供するように、そして高度に精製された安定な生成物を可能とするように選択されるであろう。通常は、化学反応基は、合成の最終段階で、例えば、カルボキシル基を用いて作製され、エステル化されて活性エステルが形成させられるであろう。本発明の改変されたペプチドの生産のための特定の方法が以下に記載される。
【0177】
具体的には、選択されたペプチドは、抗ウイルス活性について最初にアッセイされ、次いでペプチドのN末端、C末端または内部のいずれかのみで、連結基を用いて改変される。その後、この改変されたペプチド連結基の抗ウイルス活性がアッセイされる。抗ウイルス活性が劇的に低下(すなわち、10分の1未満に低下)しない場合は、その後、改変されたペプチド連結基の安定性がそのインビボ寿命によって測定される。この安定性が所望されるレベルにまで改善されていなければ、次いでペプチドは、別の部位で改変され、そして所望されるレベルの抗ウイルスおよび安定性が得られるまでこの手順が繰り返される。
【0178】
さらに具体的には、リンカーおよび反応基での改変を受けさせるために選択された個々のペプチドが、以下の基準に従って改変されるであろう:末端のカルボン酸基がペプチド上で利用可能であり、そしてこの末端のカルボン酸基が抗ウイルス活性の保持に重要ではなく、他の感受性官能基がペプチド上に存在しない場合は、このカルボン酸が、リンカー反応基改変のための結合点として選択されるであろう。末端のカルボン酸基が抗ウイルス活性に関与している場合、またはカルボン酸が利用できない場合は、抗ウイルス活性の保持に重要ではない任意の他の感受性官能基が、リンカー反応要素の改変のための結合点として選択されるであろう。いくつかの感受性官能基をペプチド上で利用できる場合は、リンカー/反応要素の付加および保護された全ての感受性官能基の脱保護後に、抗ウイルス活性の保持がなおも得られるような様式で、保護基の組み合わせが使用されるであろう。利用できる感受性官能基がペプチド上にない場合、またはより単純な改変経路が所望される場合は、合成による試みにより、抗ウイルス活性の保持が維持されるような様式で、もともとのペプチドの改変が可能となるであろう。この場合、改変はペプチドの反対側の末端で起こるであろう。
【0179】
NHS誘導体は、ペプチド中に他の感受性官能基が存在しない場合にカルボン酸から合成され得る。特に、このようなペプチドは、無水CH2Cl2およびEDC中でN−ヒドロキシスクシンイミドと反応させられ、そしてこの生成物は、クロマトグラフィーによって精製されるか、またはNHS誘導体を生じさせるための適切な溶媒系から再結晶化させられる。
【0180】
あるいは、NHS誘導体は、アミノ基および/またはチオール基と、カルボン酸を含むペプチドから合成され得る。分子中に遊離のアミノ基またはチオール基が存在する場合は、NHS誘導体の付加が行われる前に、これらの感受性官能基が保護されることが好ましい。例えば、分子に遊離のアミノ基が含まれる場合は、Fmoc保護されたアミンまたは好ましくはtBoc保護されたアミンへのアミンの変換が、上記化学反応が行われる前に必要とされる。アミン官能基は、NHS誘導体の調製後も脱保護されないであろう。従って、この方法は、所望される抗ウイルス効果を誘導するために、そのアミン基が遊離状態であることが必要ではない化合物に対してのみ適用される。アミノ基が分子のもともとの特性を保持するために遊離状態であることが必要とされる場合は、以下に記載される別のタイプの化学反応が行われなければならない。
【0181】
加えて、NHS誘導体は、アミノ基またはチオール基が含まれ、そしてカルボン酸が含まれていないペプチドから合成され得る。選択された分子にカルボン酸が含まれていない場合は、一連の二官能性リンカーを使用して、その分子を反応性NHS誘導体へと変換させることができる。例えば、DMF中に溶解させられ、そして遊離アミノ含有分子に付加させられたエチレングリコール−ビス(スクシンイミジルスクシネート)(EGS)およびトリエチルアミン(EGSの方が有利なように10:1の比で)は、モノNHS誘導体を生じるであろう。チオールで誘導された分子からNHS誘導体を生じさせるためには、DMF中のN−[−マレイミドブチリルオキシ]スクシンイミドエステル(GMBS)とトリエチルアミンを使用することができる。マレイミド基は遊離のチオールと反応し、そしてNHS誘導体は、シリカ上でのクロマトグラフィーまたはHPLCによって反応混合物から精製されるであろう。
【0182】
NHS誘導体はまた、複数の感受性官能基を含むペプチドからも合成され得る。それぞれの場合について、異なる様式で分析と解明がなされなければならないであろう。しかし、市販されている多量の保護基および二官能性リンカーのお陰で、本発明は、ペプチドを改変するために、好ましくはわずか1つの化学的工程で(上記に記載されるように)、または2工程(上記に記載されたように、感受性基の事前の保護を含む)もしくは3工程(保護、活性化、および脱保護)で任意のペプチドに適用することができる。例外的な状況下でのみ、ペプチドを活性なNHSまたはマレイミド誘導体に変換させるために、複数の(3工程を超える)合成工程が必要であろう。
【0183】
マレイミド誘導体はまた、遊離のアミノ基と遊離のカルボン酸を含むペプチドからも合成され得る。アミノ誘導された分子からマレイミド誘導体を生じさせるためには、DMF中のN−[γ−マレイミドブチリルオキシ]スクシンイミドエステル(GMBS)とトリエチルアミンを使用することができる。スクシンイミドエステル基は遊離のアミノと反応し、そしてマレイミド誘導体は、結晶化によって、またはシリカ上でのクロマトグラフィーによって、またはHPLCによって、反応混合物から精製されるであろう。
【0184】
最後に、マレイミド誘導体は、複数の他の感受性官能基を含み、そして遊離のカルボン酸を含まないペプチドから合成され得る。選択された分子にカルボン酸が含まれていない場合は、一連の二官能性架橋試薬を使用して、分子を反応性NHS誘導体へと変換させることができる。例えば、DMF中でのHBTU/HOBt/DIEA活性化を使用して、マレイミドプロピオン酸(MPA)を遊離のアミンにカップリングさせて、遊離のアミンとMPAのカルボン酸基との反応を通してマレイミド誘導体を生じさせることができる。
【0185】
多くの他の市販されているヘテロ二官能性架橋試薬を、必要とされる場合には代替的に使用することができる。多数の二官能性化合物を要素への架橋のために利用することができる。例示的な試薬としては、以下が挙げられる:アジドベンゾイルヒドラジド、N−[4−(p−アジドサリチルアミノ)ブチル]−3’−[2’−ピリジルジチオ)プロピオンアミド)、ビス−スルホスクシンイミジルスベレート、ジメチルアジピイミデート、ジスクシンイミジル酒石酸塩、N−γ−マレイミドブチリルオキシスクシンイミドエステル、N−ヒドロキシスルホスクシンイミジル−4−アジドベンゾエート、N−スクシンイミジル[4−アジドフェニル]−1,3’−ジチオプロピオネート、N−スクシンイミジル[4−ヨードアセチル]アミノベンゾエート、グルタルアルデヒド、およびスクシンイミジル4−[N−マレイミドメチル]シクロヘキサン−1−カルボキシレート。
【0186】
改変された抗ウイルスペプチドの使用
本発明の改変された抗ウイルスペプチドは、ウイルス感染に罹患している患者の処置において治療薬として使用され得、そして以下に記載される方法および当該分野で公知の他の方法に従って患者に投与することができる。改変されたペプチドの有効な治療的投薬量は、当業者に周知の手順によって決定され得、そしてこのペプチドの潜在的な毒性に関する任意の懸念が考慮されるであろう。
【0187】
改変されたペプチドはまた、これまでには感染していない個体に対して予防的に投与することもできる。このことは、個体がウイルス伝染の危険性が高い感染した個体と接触した場合に起こり得るように、個体がウイルスに曝される高い危険性に供されている場合に有利であり得る。このことは、ウイルス(例えば、HIVウイルス)に対する公知の治療法がある場合には特に有利であり得る。一例として、改変された抗HIVペプチドの予防的な投与は、医療施設の労働者がHIV感染個体に由来する血液に曝されている状況において、または個体をHIVウイルスに曝す可能性があるリスクの高い活動にその個体が従事している他の状況において有利であろう。
【0188】
改変された抗ウイルスペプチドおよび抗膜融合性ペプチドの投与
一般的に、改変されたペプチドは、生理学的に許容される媒体(例えば、脱イオン水、リン酸緩衝化生理食塩水(PBS)、生理食塩水、水性エタノールまたは他のアルコール、血漿、タンパク様溶液、マンニトール、水性グルコース、アルコール、植物油など)の中で投与される。含まれ得る他の添加物としては、緩衝液(この場合、媒体は、約5〜10の範囲のpHで一般的に緩衝化され、緩衝液は一般的には約50〜250mMの濃度の範囲であろう)、塩(塩濃度は、一般的には約5〜500mMの範囲であろう)、生理的に許容される安定剤などが挙げられる。組成物は、保存および輸送に便利なように凍結乾燥させられる場合がある。
【0189】
本発明の改変されたペプチドは、大部分について、非経口的に(例えば、静脈内に(IV)、動脈内に(IA)、筋肉内に(IM)、皮下に(SC)など)投与される。投与は、適切な状況では、輸注により得る。官能基の反応が比較的遅いいくつかの場合には、投与は、経口的、経鼻的、直腸的、経皮的またはエーロゾルであり得、この場合、結合体の性質により脈管系への移動が可能となる。通常は、一回の注入が使用されるが、所望される場合には、2回以上の注入が使用される場合がある。改変されたペプチドは、任意の都合のよい手段(注射器、トロカール、カテーテルなどを含む)によって投与され得る。
【0190】
特定の実施形態においては、改変されたペプチドは、当該分野で公知の方法によって肺手段によって投与されるであろう。ペプチドまたはタンパク質のエアゾール乾燥粉末形態の肺深部への送達のための技術は、Pattonら(1997)Chemtech 27(12):34−38に開示されている。ペプチドの肺投与を開示しているさらなる参考文献としては、Senior,K.ら(2000)PSTT,第3巻:281−282;Gumbleton,M.(2006)Advanced Drug Delivery Reviews 58:993−995;「Drug Delivery:Pulmonary Delivery」と題されたNewhouse,M.T.(2006)Encyclopedia of Pharmaceutical Technology;およびLabiris,N.R.(2003)J.Clin.Pharmacology 56:600−612が挙げられる。これらの参考文献の全ての内容が引用により本明細書中に組み入れられる。
【0191】
投与の特定の様式は、投与される量、1回のボーラスであるかまたは継続的な投与であるかなどに依存して変化するであろう。好ましくは、投与は静脈内であり、この場合、導入の部位は、本発明にとって重要ではなく、好ましくは急速な血流が存在する部位(例えば、静脈内、末梢静脈または中心静脈)である。他の経路は、投与が、遅延性放出技術または保護マトリックスと結びつけられる使用を見出し得る。この意図は、改変されたペプチドが血液中で効率的に分布されることであり、その結果、血液成分と反応することができる。結合体の濃度は、一般的には、約1pg/ml〜50mg/mlまでの範囲で広範に変化するであろう。静脈内に投与される総量は、一般的には、約0.1mg/ml〜約50mg/ml、約5mg/ml〜40mg/ml、約10mg/ml〜30mg/ml、約10mg/ml〜20mg/ml、または約5mg/ml〜15mg/ml、約1mg/ml〜約10mg/ml、あるいは、約1mg/ml〜5mg/mlの範囲であろう。
【0192】
血液の長期間にわたり生存する成分(例えば、免疫グロブリン、血清アルブミン、赤血球および血小板)に結合させることにより、多数の利点が結果として生じる。ペプチドの活性は、数日〜数週間延長される。この期間に投与は1回のみ必要である。活性化合物は、大きい分子に主に結合されるので(ここでは、他の生理学的プロセスを妨げるように細胞内に取り込まれる可能性は低い)、より優れた特異性を得ることができる。
【0193】
改変されたペプチドの存在のモニタリング
哺乳動物宿主の血液は、1回以上、改変されたペプチド化合物の存在についてモニターされ得る。宿主の血液の一部または試料を採取することにより、ペプチドが治療的に活性であるために十分な量で長期間生存する血液成分に結合されているかどうかを決定することができ、そしてその後、血液中のペプチド化合物のレベルを決定することができる。所望される場合は、ペプチドがどの血液成分に結合されるかもまた決定することができる。これは、非特異的改変ペプチドが使用される場合に特に重要である。特定のマレイミドで改変されたペプチドに関して、血清アルブミンおよびIgGの半減期を算出することは、はるかに簡単である。
【0194】
免疫アッセイ
本発明の別の態様は、抗ウイルスペプチドおよび/もしくはアナログ、またはそれらの誘導体および結合体の、生物学的試料(例えば、血液)における濃度を、これらのペプチド、ペプチドアナログ、またはそれらの誘導体および結合体に対して特異的な抗体を用いて決定するための方法、ならびにこのようなペプチド、アナログおよび/またはこれらの誘導体もしくは結合体に強く関係している毒性についての処置としてのこのような抗体の使用に関する。患者におけるインビボでのこのペプチドの高い安定性と寿命は、処置の際に新しい問題(毒性に関する高い可能性を含む)を導く可能性があるので、このことは有利である。
【0195】
特定のペプチド、そのペプチドアナログまたは誘導体に対して特異性を有している抗治療薬剤抗体(モノクローナル抗体またはポリクローナル抗体のいずれか)の使用は、任意のこのような問題を媒介する手助けをし得る。抗体は、特定のペプチド、そのアナログもしくは誘導体を用いて、またはその因子の免疫原性断片、あるいはこの因子の抗原決定基に対応する合成された免疫原を用いて免疫された宿主から作製され得るか、またはそれらから導かれ得る。好ましい抗体は、ペプチド、ペプチドアナログ、もしくは誘導体の天然の形態、改変された形態、および結合させられた形態に対して、高い特異性と親和性を有するであろう。このような抗体はまた、酵素、蛍光色素、または放射性標識で標識することもできる。
【0196】
改変されたペプチドに特異的な抗体は、ペプチド特異的抗体の誘導のために精製されたペプチドを使用するすることにより作製され得る。抗体の誘導によっては、動物中への注射による免疫応答の刺激だけではなく、合成抗体または他の特異的結合分子の作製における類似の工程(例えば、組換え免疫グロブリンライブラリーのスクリーニング)も意図される。モノクローナル抗体およびポリクローナル抗体の両方が、当該分野で周知の手順により作成され得る。
【0197】
抗ペプチド抗体が、改変されたペプチド、そのアナログまたは誘導体の投与によって誘導された毒性を処置するために使用され得、そしてエキソビボでも、またインビボでも使用され得る。エキソビボでの方法には、固体支持体に固定された抗治療薬剤抗体を使用する、毒性に対する免疫透析処置が含まれるであろう。インビボでの方法には、抗体−薬剤複合体のクリアランスを誘導するために有効な量の抗治療薬剤抗体の投与が含まれる。
【0198】
抗体は、無菌条件下で抗体と血液を接触させることによって、エキソビボで患者の血液から、改変されたペプチド、そのアナログまたは誘導体、およびその結合体を除去するために使用され得る。例えば、抗体は、カラムマトリックス上に固着、あるいは固定することができ、そして患者の血液を患者から取り出して、マトリックス上を通過させることができる。改変されたペプチド、ペプチドアナログ、誘導体または結合体は抗体に結合し、そして低濃度のペプチド、アナログ、誘導体または結合体を含む血液が、その後、患者の循環系に戻され得る。除去されるペプチド化合物の量は、圧力および流速を調整することによって制御することができる。
【0199】
患者の血液の血漿成分からのペプチド、アナログ、誘導体および結合体の優先的な除去は、例えば、半透膜の使用によって、またはそうでなければ、抗治療薬抗体を含むマトリックス上に血漿成分を通過させる前に、当該分野で公知の方法により細胞成分から血漿成分をまず分離することによって行うことができる。あるいは、ペプチドが結合させられた血球(赤血球細胞を含む)の優先的な除去は、患者の血液中の血球を回収し、濃縮し、そして患者の血液の血清成分の排除のためにそれらの細胞を固着させられた抗治療薬抗体と接触させることによって行うことができる。
【0200】
抗治療薬抗体は、処置のためにペプチド、アナログ、誘導体または結合体が投与された患者に、インビボで非経口的に投与することができる。抗体は、ペプチド化合物および結合体に結合するであろう。一旦ペプチドに結合されると、活性は、完全にはブロックされないとしても、妨害され、それによって患者の血流中のペプチド化合物の生物学的有効濃度が低下し、そして有害な副作用が最少となるであろう。加えて、結合させられた抗体−ペプチド複合体は、患者の血流からのペプチド化合物と結合体のクリアランスを容易にするであろう。
【0201】
十分に記載されてきた本発明は、以下の非限定的な実施例を参照することにより、さらに認識され、そして理解することができる。
【実施例】
【0202】
(実施例1〜5)
C34のシステイン酸誘導体の合成と精製
C34のシステイン酸誘導体の合成は、ペプチドの作製の間に手作業による介入を伴うSymphony Peptide Synthesizer上での自動化された固相手順を使用して行った。合成は、Fmoc保護されたRamageアミドリンカー樹脂上で、Fmoc保護されたアミノ酸を使用して行った。カップリングは、O−ベンゾトリアゾール−1−イル−N,N,N’,N’−テトラメチル−ウロニウムヘキサフルオロホスフェート(HBTU)とジイソプロピルエチルアミン(DIEA)を活性化因子の混合物としてN,N−ジメチルホルムアミド(DMF)溶液中で使用することによって行った。Fmoc保護基を20%のピペリジン/DMFを使用して除去した。ペプチドが樹脂から切り離されると遊離のNα末端が生じるように、Boc保護されたアミノ酸をN末端に使用した。シグマコーティッドガラス反応容器を合成の際に使用した。
【0203】
(実施例2)
CA−C34の合成
CA−C34は以下のアミノ酸配列を有する:
【0204】
【化28−2】
CA−C34の改変されたペプチドを以下のように合成した:
工程1:100μモルスケールでのCA−C34の固相ペプチド合成を、手作業による固相合成、自動化された固相合成、Symphony Peptide SynthesizerおよびRamage樹脂を使用して行った。以下の保護されたアミノ酸を順番に樹脂に付加させた:
【0205】
【化28−3】
これらをN,N−ジメチルホルムアミド(DMF)に溶解させ、そして配列に従って、O−ベンゾトリアゾール−1−イル−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)およびジイソプロピルエチルアミン(DIEA)を使用して活性化させた。Fmoc保護基の除去は、N,N−ジメチルホルムアミド(DMF)中の20%(V/V)ピペリジンの溶液を使用して20分間行った(工程1)。
【0206】
工程2:ペプチドを、85%のTFA/5%のTIS/5%のチオアニソールおよび5%のフェノールを使用して樹脂から切断し、続いてドライアイス冷(0〜4℃)Et2Oによって沈殿させ、回収した。
【0207】
(実施例3)
CA−C34(Arg28)の合成
CA−C34(Arg28)は以下のアミノ酸配列を有する:
【0208】
【化28−4】
工程1:100μモルスケールでのCA−C34(Arg28)の固相ペプチド合成を、手作業による固相合成、自動化された固相合成、Symphony Peptide SynthesizerおよびRamage樹脂を使用して行った。以下の保護されたアミノ酸を順番に樹脂に付加させた:
【0209】
【化28−5】
これらをN,N−ジメチルホルムアミド(DMF)に溶解させ、そして配列に従って、O−ベンゾトリアゾール−1−イル−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)およびジイソプロピルエチルアミン(DIEA)を使用して活性化させた。Fmoc保護基の除去は、N,N−ジメチルホルムアミド(DMF)中の20%(V/V)ピペリジンの溶液を使用して20分間行った(工程1)。
【0210】
工程2:ペプチドを、85%のTFA/5%のTIS/5%のチオアニソールおよび5%のフェノールを使用して樹脂から切断し、続いてドライアイス冷(0〜4℃)Et2Oによって沈殿させ、回収した。
【0211】
(実施例4)
CA−C34−Lys35(ε−AEEA−MPA)の合成
CA−C34−Lys35(ε−AEEA−MPA)は以下のアミノ酸配列を有する:
【0212】
【化28−6】
工程1:100μモルスケールでのCA−C34−Lys35(ε−AEEA−MPA)の固相ペプチド合成を、手作業による固相合成、自動化された固相合成、Symphony Peptide SynthesizerおよびRamage樹脂を使用して行った。以下の保護されたアミノ酸を順番に樹脂に付加させた:
【0213】
【化28−7】
これらをN,N−ジメチルホルムアミド(DMF)に溶解させ、そして配列に従って、O−ベンゾトリアゾール−1−イル−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)およびジイソプロピルエチルアミン(DIEA)を使用して活性化させた。Fmoc保護基の除去は、N,N−ジメチルホルムアミド(DMF)中の20%(V/V)ピペリジンの溶液を使用して20分間行った(工程1)。
【0214】
工程2:Lys(Aloc)基の選択的脱保護を手作業で、そして5mLのC6H6:CHCl3(1:1):2.5%のNMM(v:v):5%のAcOH(v:v)中に溶解させた3等量のPd(PPh3)4の溶液で樹脂を2時間処理することによって行った(工程2)。その後、樹脂をCHCl3(6×5mL)、DCM中の20%のAcOH(6×5mL)、DCM(6×5mL)、およびDMF(6×5mL)で洗浄した。
【0215】
工程3:その後、合成をFmoc−AEEA−OHと3−マレイミドプロピオン酸の付加のために再度自動化した(工程3)。AEEA上の保護基(Fmoc)を先に記載したように除去し、それぞれのカップリングの間に、樹脂をN,N−ジメチルホルムアミド(DMF)で3回、そしてイソプロパノールで3回洗浄した。
【0216】
工程4:ペプチドを85%のTFA/5%のTIS/5%のチオアニソールおよび5%のフェノールを使用して樹脂から切断し、続いてドライアイス冷(0〜4℃)Et2Oによって沈殿させ(工程4)、回収した。
【0217】
(実施例5)
CA−C34(Arg28)−Lys35(ε−AEEA−MPA)
CA−C34(Arg28)−Lys35(ε−AEEA−MPA)は以下の配列を有する:
【0218】
【化28−8】
工程1:100μモルスケールでのCA−C34(Arg28)−Lys35(ε−AEEA−MPA)の固相ペプチド合成を、手作業による固相合成、自動化された固相合成、Symphony Peptide SynthesizerおよびRamage樹脂を使用して行った。以下の保護されたアミノ酸を順番に樹脂に付加させた:
【0219】
【化28−9】
これらをN,N−ジメチルホルムアミド(DMF)に溶解させ、そして配列に従って、O−ベンゾトリアゾール−1−イル−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)およびジイソプロピルエチルアミン(DIEA)を使用して活性化させた。Fmoc保護基の除去は、N,N−ジメチルホルムアミド(DMF)中の20%(V/V)ピペリジンの溶液を使用して20分間行った(工程1)。
【0220】
工程2:Lys(Aloc)基の選択的脱保護を手作業で、そして5mLのC6H6:CHCl3(1:1):2.5%のNMM(v:v):5%のAcOH(v:v)中に溶解させた3等量のPd(PPh3)4の溶液で樹脂を2時間処理することによって行った(工程2)。その後、樹脂をCHCl3(6×5mL)、DCM中の20%のAcOH(6×5mL)、DCM(6×5mL)、およびDMF(6×5mL)で洗浄した。
【0221】
工程3:その後、合成をFmoc−AEEA−OHと3−マレイミドプロピオン酸の付加のために再度自動化した(工程3)。AEEA上の保護基(Fmoc)を先に記載したように除去し、それぞれのカップリングの間に、樹脂をN,N−ジメチルホルムアミド(DMF)で3回、そしてイソプロパノールで3回洗浄した。
【0222】
工程4:ペプチドを85%のTFA/5%のTIS/5%のチオアニソールおよび5%のフェノールを使用して樹脂から切断し、続いてドライアイス冷(0〜4℃)Et2Oによって沈殿させ(工程4)、回収した。
【0223】
精製手順:
C34の改変されたペプチドをそれぞれ、Varian(Dynamax)分取バイナリーHPLCシステムを使用して、分取逆相HPLCによって精製した。
【0224】
例示的な誘導体の精製は、水/TFA混合物(H2O中の0.1%のTFA;溶媒A)とアセトニトリル/TFA(CH3CN中の0.1%のTFA;溶媒B)で平衡化させたPhenomenex Luna 10μフェニル−ヘキシル、50mm×250mmカラム(粒子10μ)を使用して行った。溶離は、28〜38%のB勾配を180分間にわたって50mL/分で流すことによって行った。ペプチドを含む画分を、214nmと254nmのUV吸光度(Varian Dynamax UVD II)によって検出した。
【0225】
画分を25mLのアリコートの中に回収した。所望される生成物を含む画分を、LC/MS上への直接の注入後の質量の検出によって同定した。選択した画分をその後、分析用HPLC(20分間にわたる20〜60%のB;Phenomenex Luna 5μフェニル−ヘキシル、10mm×250mmカラム、0.5mL/分)によって分析して、≧90%の純度を有している画分をプールするために同定した。プールを液体窒素を使用して凍結乾燥させ、その後、少なくとも2日間凍結乾燥させて白色粉末を得た。
【0226】
IV−フローチャート:
同じ合成反応スキームを全ての誘導体に使用した。CA−C34とCA−C34−Lys35(ε−AEEA−MPA)についての反応スキームを以下のフローチャートにおいて説明する。もちろん、AEEAおよびMPAの付加に伴うAloc除去工程はCA−C34については省略した。
【0227】
【化29】
(実施例6)
C34のシステイン酸誘導体の溶解度アッセイ
a.溶解度アッセイは、100mMのリン酸ナトリウム(開始時pH8)水溶液中で行った。高い緩衝液濃度がHPLCによる精製の後にC34誘導体とともに残っている過剰量のトリフルオロ酢酸(TFA)を中和するために有用であることが明らかにされている。
【0228】
5.8〜7.0の最終的なpHが様々なC34誘導体の溶解度を維持するためには適している。したがって、開始緩衝液をpH8.0で調製し;そしてC34誘導体の可溶化によりおよそ6.3の最終的なpHが生じた。表2は、N末端にシステイン酸(CA)を持つかまたは持たない、そしてC末端にLys35(ε−AEEA−MPA)を持つかまたは持たないC34の溶解限度を示す。さらに、表1に示す最終的なオスモル濃度は、これらの最終的な溶液が等張性であることを明らかにしている。
【0229】
【表2】
天然のC34は15.75mg/mlでは溶解性であることが明らかであり、得られる溶液は数分以内にゲルを形成する。C34−Lys35(ε−AEEA−MPA)は、これが溶液に投入されると直ちにゲルを形成し、さらに緩衝液を加えても決して化合物を可溶化させることはできない。表1から注目され得るように、これらの化合物の両方のN末端でのシステイン酸の付加は、C34に対して有意に高い溶解度(すなわち、それぞれ29.3mg/mlおよび33.8mg/ml)を付与する。
【0230】
b.500mMのリン酸ナトリウム緩衝液(pH8.0)中で30mg/ml〜35mg/mlのC34に連結させられたN末端が改変されたAEEA−MPA(W1(AEEA−MPA)−C34)、およびAEEAリンカーを介してMPAに連結させられた35位にリジン付加を有しているN末端システイン酸が改変されたC34(CA K35(AEEA−MPA−C34))の溶解度
1Mのリン酸ナトリウム緩衝液(pH8.0)
A−2Mのリン酸二水素ナトリウム、無水物、UQAM、OM−27、S1835 1M=141.96g/1L、2M=13.3568g/40mlのnanopure H2O。
B−2Mのリン酸一ナトリウム、一水和物、UQAM、OM−27、S1820 1M=137.99g/1L、2M=11.0392g/40mlのnanopure H2O
C−Mix 30ml nanopure H2O+およそ28mlのリン酸二水素ナトリウム+1.5mlのリン酸一ナトリウム。pHが8.0であるかどうかを確認する。容量をリン酸二水素ナトリウムで調整する
D−最終pHを8に(実際には7.98)に調整する。
【0231】
500mMのリン酸ナトリウム緩衝液(pH8.0)
1Mのリン酸ナトリウム緩衝液(pH8.0)を等量のnanopure H2O(濾過していない)と混合する。最終的なpHは8.0である。
【0232】
c.500mMのリン酸ナトリウム(pH8.0)中で100mg/mlでのW1(AEEA−MPA)−C34および(CA K35(AEEA−MPA−C34))の溶解度
化合物:
W1(AEEA−MPA)−C34 Batch B,ロット番号JC−205−12,Inventory no 1139.
塩を含む1M=4769.1=12.64mg重量
塩を含まない1M=4541.1=12.0357mg
純度=90.8%=100mg/mlについて109.3ulの500mMのリン酸ナトリウム(pH8.0)中の10.928mg。
【0233】
注釈:ガラスバイアル中の粉末に対して緩衝液を加えた。1分間のボルテックス(中速)後は可溶性であった。3つの粒子が残った。最終的なpHは6.82であった(Chemistry DepartmentによるpHメーターで測定した、500mMのNaP pH8緩衝液は8.1であった)。
【0234】
(CA K35(AEEA−MPA−C34))Batch B,ロット番号PB−262−01,Inventory no 1352.
塩を含む1M=5165.2=12.11mg重量
塩を含まない1M=4820.2=11.30mg
純度=92.8%=100mg/mlについて104.9ulの500mMのリン酸ナトリウム(pH8.0)中10.487mg。
【0235】
注釈:ガラスバイアルの中の粉末に対して緩衝液を加えた。1分30秒間のボルテックス(中速)後はほとんど可溶性であった。2つの小さいペレットがガラスバイアルの底にあった。3分後、これらの2つの小さいペレットは溶解した。最終的なpHは6.75であった(Chemistry DepartmentによるpHメーターで測定した、500mMのNaP pH8緩衝液は8.1であった)。
【0236】
W1(AEEA−MPA)−C34と(CA K35(AEEA−MPA−C34))は、一度溶解すれば黄色である。W1(AEEA−MPA)−C34はさらに濃い色である。
【0237】
結論:
W1(AEEA−MPA)−C34化合物と(CA K35(AEEA−MPA−C34))化合物は、500mMのリン酸ナトリウム緩衝液(pH8.0)中で100mg/mlでは可溶性である。それらの最終的なpHは許容される限度、すなわち6.8を上回る。これらの化合物についてのpHの許容限度は6.2である。
【0238】
d.500mMのリン酸ナトリウム(pH8.0)中の150mg/mlでのW1(AEEA−MPA)−C34および(CA K35(AEEA−MPA−C34))の溶解度
W1(AEEA−MPA)−C34 Batch B,ロット番号JC−205−12,Inventory no 1139.
塩を含む1M=4769.1=11.97mg重量
塩を含まない1M=4541.1=11.398mg
純度=90.8%=150mg/mlについて69ulの500mMのリン酸ナトリウム(pH8.0)中の10.35mg。
【0239】
注釈:緩衝液をガラスバイアル中の粉末に対して加えた。1分間のボルテックス(中速〜高速)後は可溶性であった。最終的なpHは6.60であった(Chemistry DepartmentによるpHメーターで測定した、500mMのNaP pH8緩衝液は8.23であった)。
【0240】
(CA K35(AEEA−MPA−C34))Batch B,ロット番号PB−262−01,Inventory no 1352.
塩を含む1M=5165.2=12.48mg重量
塩を含まない1M=4820.2=11.64mg
純度=92.8%=150mg/mlについて72.05ulの500mMのリン酸ナトリウム(pH8.0)中の10.808mg。
【0241】
注釈:ガラスバイアルの中の粉末に対して緩衝液を加えた。1分間のボルテックス(中速〜高速)後はほとんど可溶性であった。1つの小さいペレットがガラスバイアルの底にあった。10分後、この小さいペレットは溶解した。最終的なpHは6.57であった(Chemistry DepartmentによるpHメーターで測定した、500mMのNaP pH8緩衝液は8.23であった)。
【0242】
W1(AEEA−MPA)−C34と(CA K35(AEEA−MPA−C34))は一度溶解すると黄色である。W1(AEEA−MPA)−C34はさらに濃い色である。
【0243】
結論:W1(AEEA−MPA)−C34と(CA K35(AEEA−MPA−C34))はいずれも、500mMのリン酸ナトリウム緩衝液(pH8.0)中では150mg/mlで可溶性である。
【0244】
(実施例7)
C34のシステイン酸誘導体の活性アッセイ
第1の活性アッセイ:抗HIV−1に誘導される細胞と細胞の融合のアッセイ
様々な抗膜融合性化合物とそれぞれの予め形成させたアルブミン結合体の効力を、New York Blood Center,310 East 67th Street,New York,N.Y.10021においてShibo Jiang博士と共同研究者らによって設計されたHIV−1に誘導される細胞と細胞の融合のアッセイを使用して評価した。HIVに誘導される細胞と細胞の融合に対するC34の阻害活性は以前に記載されているように検出した(Jiang,S.L.ら(2000)A Convenient Cell Fusion Assay for Rapid Screening for HIV Entry Inhibitors,Proc.SPIE 3926:212−219)。
【0245】
簡単に説明すると、化合物を最初に、ストック溶液としてリン酸−クエン酸緩衝液(pH7.0)中に100μMに稀釈し、その後、1000nM、500nM、250nM、100nM、50nM、25nM、5nM、1nMに培養培地中にさらに稀釈した。50μlのこの化合物溶液をカルセイン−AM(Molecular Probes,Inc.,Eugene,OR)で標識した50μlのHIV−1IIIB感染H9細胞(H9/HIV−1IIIB)と、2×105細胞/mlで混合した。37℃で2時間の共培養の後、カルセインで標識されたH9/HIV−1IIIB細胞を、MT−2細胞と融合したものまたは融合していないものを両方、それぞれ、485nMおよび535nmのフィルター励起波長を使用して、接眼ミクロメーターディスク(10×10mm sq.)と20×の対物レンズを備えた倒立蛍光顕微鏡(Zeiss,Gennany)下でカウントした。融合した細胞は融合していない細胞よりもはるかに大きく(少なくとも2倍)、したがって、融合した細胞中での蛍光の強度は、1つの細胞から2つ以上の細胞へのカルセインの拡散が原因で、融合していない細胞中での蛍光強度よりも弱い。1ウェルあたり4つの領域を試験し、細胞融合の割合を以下の式によって計算した:融合した細胞/(融合した細胞+融合していない細胞)×100%。
【0246】
ポジティブ対照のウェルに、50μlのカルセインで標識したHIV感染細胞を添加した。ネガティブ対照のウェルには、培養培地と、カルセインで標識した非感染H9細胞を添加した。細胞融合の阻害率(%)を以下の式を使用して計算した:[1−(E−N)/(P−N)]×100%、式中、「E」は実験グループにおける細胞融合の割合(%)を示し、「P」は、試験化合物を添加しなかったポジティブ対照グループにおける融合の割合(%)を示し、「N」は、カルセインで標識したH9/HIV−1IIIB細胞をカルセインで標識したH9細胞で置換したネガティブ対照グループにおける融合の割合(%)を意味する。抗ウイルス化合物による細胞融合の50%の阻害のための濃度(IC50)を、T.C.Chou博士から懇意によって提供されたコンピュータープログラム(Chou,T.C.and Hayball,M.P.,CalcuSyn:Windows(登録商標) software for dose effect analysis(1991)Ferguson,MO63135,USA,BIOSOFT)を使用して計算した。
【0247】
表3は、N末端にシステイン酸を持つかまたは持たない、そして基Lys(ε−AEEA−MPA)を介してヒト血清アルブミン(HSA)に結合させられているかまたは結合させられていないC34の抗膜融合活性を示す。
【0248】
【表3】
表3に示すように、C34の抗膜融合活性とCA−C34の抗膜融合活性との間では有意な差は観察されなかった。したがって、C34のN末端でのシステイン酸の付加は、C34の抗膜融合活性に対してネガティブな影響は与えない。
【0249】
表2もまた、それらのC末端へのC34およびCA−C34に対するLys(ε−AEEA−MPA)のカップリング、それに続くHSAに対するそれらの結合が、それらの抗膜融合活性に対して有意なネガティブな影響を持たないことを示している。
【0250】
(実施例8)
第2の活性のアッセイ:ヒトPBMC中でのHIVIIIbの複製の阻害
化合物の抗HIV効力と細胞傷害性を、HIV−1株IIIBを使用したPBMCをベースとするアッセイにおいて急性感染後に評価した。これらの実験は、以下に記載するプロトコールにしたがってSouthern Research Institute,Infectious Disease Research Department,431 Aviation Way,Frederick,MD.で行った。
【0251】
a.PBMCのHIV−1感染
HIVおよびHBVについて血清反応陰性である新鮮なヒトPBMCを、スクリーニングしたドナーから単離し、そしてBiological Specialty Corporation Colmar,PAからも商業的に提供された。細胞を低速での遠心分離とPBS中への再懸濁によって2〜3回ペレット化/洗浄して、混入している血小板を取り除いた。その後、白血球保有血をダルベッコのリン酸緩衝化生理食塩水(Dulbecco’s Phosphate Buffered Saline)(DPBS)で稀釈し、50mLの遠心管の中でLymphocyte Separation Medium(LSM;Mediatech,Inc.によるCellgro(登録商標);密度1.078+/−0.002g/ml;カタログ番号85−072−CL)の上に積層させ、次いで遠心分離した。バフィーコート層を、得られた界面から穏やかに吸引し、その後、低速遠心分離によってPBSで洗浄した。3回目の洗浄の後、細胞をウシ胎児血清(FBS)、L−グルタミン、ペニシリン、ストレプトマイシン、およびフィトヘマグルチニン(PHA−P;Sigma,St−Louis,MO)を補充したRPMI 1640中に再懸濁させた。細胞を37℃でインキュベートした。2日間のインキュベーションの後、PBMCを遠心分離し、FBS、L−グルタミン、ペニシリン、ストレプトマイシン、および組み換え体ヒトIL−2(R&D Systems,Inc.,Minneapolis,MN)を含むRPMI 1640中に再懸濁させた。IL−2を、PHA分裂刺激によって開始させた細胞分裂を維持するために培養培地中に含めた。細胞を最長で2週間培養物中で維持し、単球を組織培養フラスコへの接着の結果として培養物から枯渇させた。
【0252】
標準的なPBMCアッセイについては、少なくとも2人の正常なドナーに由来するPHA−Pで刺激した細胞をプールし、新鮮な培地中に稀釈し、Infectious Disease Research department of Southern Research Instituteによって開発された標準的な形式で96ウェル丸底マイクロプレートの内部ウェルにプレートした。2人以上のドナーに由来するPBMCのプールを、HIV感染の量的および質的相違と、初代リンパ球集団のPHAとIL−2に対する全体的な応答によって生じる個々のドナーの間で観察されるばらつきを最小にするために使用した。それぞれのプレートには、ウイルス/細胞対照ウェル(細胞+ウイルス)、実験用ウェル(化合物+細胞+ウイルス)、および化合物対照ウェル(細胞傷害性のMTSモニタリングに必要な、化合物+培地、細胞なし)を含めた。試験化合物の希釈物をマイクロタイターチューブの中に調製し、それぞれの濃度を標準的な形式を使用して適切なウェルの中に置いた。次いで、PBMCに対する化合物の稀釈物の添加後、予め決定した希釈率のウイルスストック溶液を、それぞれの試験ウェルの中に入れた(最終的なMOI約0.1)。ウイルスストック溶液は、NIAID AIDS Research and Reference Reagent Programから入手した継代数の低い臨床的な単離物HIV−1IIIBから調製した。−80℃で保存したHIV−1IIIBの予め滴定したアリコートを、使用の直前に生物学的安全キャビネットの中で室温になるように迅速に融解させた。HIV−1はPBMCに対して細胞変性性ではないので、同じアッセイプレートを抗ウイルス効力と細胞傷害性の測定の両方に使用することができる。PBMC培養物を、37℃、5%のCO2で感染後7日間維持した。
【0253】
b.逆転写活性アッセイ
マイクロタイタープレートをベースとする逆転写酵素(RT)反応を利用した(Buckheitら、AIDS Research and Human Retroviruses 7:295−302,1991)。滴定したチミジン三リン酸(3H−TTP、80Ci/mmol,NEN)を、1mCi/mlで1:1のdH2O:エタノール中に入れた。ポリrA:オリゴdT鋳型:プライマー(Pharmacia)を、150μlのポリrA(20mg/ml)を0.5mlのオリゴdT(20単位/ml)および5.35mlの滅菌dH2Oと一緒にし、続いてアリコートする(1.0ml)ことによってストック溶液として調製し、−20℃で保存した。RT反応緩衝液は毎日新しく調製し、これは、125μlの1.0MのEGTA、125μlのdH2O、125μlの20%のTriton X100、50μlの1.0MのTris(pH7.4)、50μlの1.0MのDTT、および40μlの1.0MのMgCl2からなる。最終的な反応混合物は、1部の3H−TTP、4部のdH2O、2.5部のポリrA:オリゴdTストック、および2.5部の反応緩衝液を合わせることによって調製した。10μlのこの反応混合物を丸底マイクロタイタープレートに入れ、15μlのウイルスを含む上清を添加し、混合した。プレートを37℃で60分間インキュベートした。インキュベーション後、反応容量をDE81フィルターマット(Wallac)上にスポットし、5%のリン酸ナトリウム緩衝液または2×SSC(Life Technologies)の中でそれぞれ5分間を5回、蒸留水中でそれぞれ1分間を2回、70%のエタノール中でそれぞれ1分間を2回洗浄し、そしてその後乾燥させた。取り込まれた放射活性(カウント毎分(counts per minute)、CPM)を、標準的な液体シンチレーション技術を使用して定量した。
【0254】
c.細胞傷害性を測定するためのPBMCの生存性についてのMTS染色
アッセイの最後に、アッセイプレートを可溶性のテトラゾリウムをベースとする色素MTS(CellTiter Reagent,Promega)で染色して、細胞の生存性を決定し、化合物の細胞傷害性を定量した。MTSは、代謝活性のある細胞のミトコンドリア酵素によって代謝されて、可溶性のホルマザン産物を生じ、それにより、細胞の生存性と化合物の細胞傷害性の迅速な定量的分析が可能となる。MTSは、使用の前に調製の必要のない安定な溶液である。アッセイの最後に、1ウェルあたり20μlのMTS試薬を添加した。HIV PBMCアッセイのために、ウェルを37℃で4時間インキュベートした。インキュベーションの間隔は、それぞれの細胞のタイプの中での最適な色素の還元について経験的に決定された時間に基づいて選択した。接着性のプレートシーラーをリッドの代わりに使用し、密閉したプレートを可溶性のホルマザン産物を混合するために数回反転させ、プレートをMolecular Devices Vmaxプレートリーダーを用いて490/650nmで分光光度測定によって読み取った。
【0255】
d.データ分析
社内コンピュータープログラム(in−house computer program)を使用して、IC50(ウイルスの複製の50%の阻害)、IC90(ウイルスの複製の90%の阻害)、TC50(50%の細胞傷害性)、TC90(90%の細胞傷害性)、および治療指数(TI=TC50/IC50)を計算した。データのグラフ表示を伴う抗ウイルス活性と細胞傷害性の両方についての生データは、個々の化合物の活性をまとめている印刷物に提供される。AZTを抗HIVアッセイにおいて関連するポジティブ対照化合物と平行して評価した。
【0256】
e.結果
図1は、天然のC34によるPBMC中でのHIV−1IIIB複製の阻害を示す(曲線◆を参照のこと)。この化合物は、以下に示すように、PBMCに対して有意な細胞傷害性の影響を全く示さなかった(曲線□を参照のこと)。
【0257】
図2は、C末端に付加されたリジンのεNH2上にAEEA−MPAを有しているC34のアルブミン結合体(すなわち、C34−Lys35(ε−AEEA−MPA):HSA)によるPBMC中でのHIV−1IIIB複製の阻害を示す(曲線◆を参照のこと)。この化合物は、以下に示すように、PBMCに対して細胞傷害性の影響を全く示さなかった(曲線□を参照のこと)。
【0258】
図3は、N末端にシステイン酸を有しており、C末端に付加されたリジンのεNH2上にAEEA−MPAを有しているC34のアルブミン結合体(すなわち、CA−C34−Lys35(ε−AEEA−MPA):HSA)によるPBMC中でのHIV−1IIIB複製の阻害を示す(曲線◆を参照のこと)。この化合物は、以下に示すように、PBMCに対して細胞傷害性の影響を全く示さなかった(曲線□を参照のこと)。
【0259】
図1、2、および3に説明するデータに基づいて、両方のアルブミン結合体のIC50値を、天然のC34と比較して表4に提供する。
【0260】
【表4】
表4は、天然のC34、C34のアルブミン結合体、およびCA−C34のアルブミン結合体についての類似する抗HIV活性を示す。結論として、N末端でのシステイン酸の付加と、その後のLys35(ε−AEEA−MPA)を介するアルブミンへの結合は、このアッセイにおいてC34の活性にネガティブな影響を与えることはない。
【0261】
(実施例8)
さらなる抗膜融合性ペプチド誘導体
実験手順
以下の手順を以下に詳細に議論するような結果を得るために行った実験全体を通じて使用した。
【0262】
CHRペプチドアナログの合成は、ペプチドの作成の間に手作業による介入を伴うSymphony Peptide Synthesizer上での自動化された固相手順を使用して行った。合成は、Fmoc保護されたRamageアミドリンカー樹脂上で、Fmoc保護されたアミノ酸を使用して行った。カップリングは、O−ベンゾトリアゾール−1−イル−N,N,N’,N’−テトラメチル−ウロニウムヘキサフルオロホスフェート(HBTU)とジイソプロピルエチルアミン(DIEA)を活性化因子の混合物としてN,N−ジメチルホルムアミド(DMF)溶液中で使用することによって行った。Fmoc保護基を20%のピペリジン/DMFを使用して除去した。ペプチドが樹脂から切り離された後の遊離のα−N末端を作成するために、Boc保護されたアミノ酸をN末端に使用した。シグマコーティッドガラス反応容器を合成の際に使用した。
【0263】
マレイミドを分子のC末端部分に配置する場合(表5、アルブミン結合化合物VII、およびアセチル化結合化合物X)は、ペプチドの固相合成は、Fmoc−Lys(Aloc)の付加によって開始した。Alocは酸性培地に対して安定な特異的な直交する保護基である。その後、ペプチド鎖を、それらの側鎖が酸性培地に対して不安定な基で保護されているアミノ酸の連続する付加によって固体支持体上で伸張させた。ペプチド鎖が完了したら、C末端リジン上のAloc保護基を、テトラキストリフェニルホスフィンパラジウムを使用して選択的に除去した。その後、Fmoc−アミノエトキシエトキシ酢酸(AEEA)リンカーを保護されていないリジンに対して化学的にカップリングさせた。伝統的なFmocの脱保護プロトコールにしたがって、マレイミドプロピオン酸(MPA)をAEEAスペーサーに化学的にカップリングさせた。最後に、酸不安定性の保護基をペプチドから切り離し、その後ペプチドを固体支持体から強酸の混合物を使用して切断した。マレイミドを分子(表5、マレイミド−化合物VIII、アルブミン結合化合物VIII)、およびアルブミン結合MPA−AEEA−化合物VIIIのN末端部分に配置する場合は、ペプチドの固相合成は融合ペプチド阻害剤の天然のアミノ酸配列によって開始した。
【0264】
【表5−1】
【0265】
【表5−2】
ペプチドの精製
生成物をそれぞれ、Varian(Dynamax)分取バイナリーHPLCシステムを使用して、分取逆相HPLCによって精製した。全てのDACペプチドの精製は、水/TFA混合物(H2O中の0.1%のTFA;溶媒A)とアセトニトリル/TFA(CH3CN中の0.1%のTFA;溶媒B)で平衡化させたPhenomenex Lunaフェニル−ヘキシル(10ミクロン、50mm×250mm)カラムを使用して行った。溶離は、様々な勾配の溶媒Bを180分間かけて50mL/分で流すことによって行った。ペプチドを含む画分を、214nmと254nmのUV吸光度(Varian Dynamax UVD II)によって検出した。
【0266】
画分を25mLのアリコートの中に回収した。所望される生成物を含む画分を、LC/MS上への直接の注入後の質量の検出によって同定した。選択した画分をその後、分析用HPLC(20分間にわたる20〜60%のB;Phenomenex Luna 5 micronフェニル−ヘキシル、10mm×250mmカラム、0.5mL/分)によって分析して、≧90%の純度を有している画分をプールするために同定した。その後、このプールを液体窒素を使用して凍結乾燥させ、続いて、少なくとも2日間凍結乾燥させて白色粉末を得た。
【0267】
アルブミン結合体の調製
HSAのシステイン−34に対するマレイミド−C34およびマレイミド−T−20誘導体の結合と、それに続く疎水性相互作用クロマトグラフィーを使用した精製は、最近、有効なプロセスとなった。結合工程には、それぞれのマレイミド−ペプチドをHSAの25%溶液(Cortex−Biochem,San Leandro,CA)と混合する工程、および37℃で30分間インキュベートする工程が含まれる。AKTA精製装置(GE Healthcare)を使用して、得られた混合物を、5mMのオクタン酸ナトリウムと750mMの(NH4)2SO4からなる20mMのリン酸ナトリウム緩衝液(pH7)中に平衡化させたブチルセファロース4ファストフロー樹脂(GE Healthcare)を充填した50mlのカラム上に直接、2.5ml/分の流速でロードした。これらの条件下では、C34−HSA結合体は疎水性樹脂上に吸着したが、原則として全ての結合しなかったHSAはカラムの空隙容量の中に溶離した。それぞれの結合体を、4倍を超えるカラム容量の徐々に低下する(NH4)2SO4濃度(750nM〜0mM)の直線的勾配をかけることによって遊離の(未反応の)マレイミド−C34誘導体の全てからさらに精製した。それぞれの精製した結合体を、その後、脱塩し、10kDaの超遠心分離フィルターデバイス(Amicon;Millipore,Bedford,MA)を使用して水の中で濃縮した。最後に、それぞれの結合体の溶液を、pH7の等張性の緩衝溶液中に再度処方した。それぞれの精製した試料の質量スペクトルによって、最も豊富なタンパク質産物がそれぞれのマレイミド誘導体を持つHSAの1:1の共有複合体に相当することを確認し、それぞれの精製した試料の逆相HPLC分析によって、原則として全ての結合しなかった(遊離の)マレイミド誘導体の除去を確認した。個々のアルブミン結合体を滅菌した0.9%のNaClを使用して処方し、T−20(San Francisco General Hospital pharmacyから入手した)を注射用の滅菌水中に溶解させ、HClでpH7に調整した。
【0268】
新しいヒトPBMC中での抗HIV効力の評価
HIV−1 IIIBは、Robert C.Gallo博士の懇意によりAIDS Research and Reference Reagent Program,Division of AIDS,NIAID,NIHを通じて入手した(Popovic ME,Read−Connole E,Gallo RC(1984)T4 positive human neoplastic cell lines susceptible to and permissive for HTLV−III.Lancet ii:1472−1473;Popovic M,Sarngadharan MG,Read E,Gallo RC(1984)Detection,isolation,and continuous production of cytopathic retroviruses(HTLV−III)from patients with AIDS and pre−AIDS.Science 224:497−500;Ratner Lら(1985)Complete nucleotide sequence of the AIDS virus,HTLV−III.Nature 313:277−283).HIVおよびHBVについて血清反応陰性である新しいヒト末梢血単核細胞(PBMC)をスクリーニングしたドナーの血液(Biological Specialty Corporation;Colmar,PA)から、製造業者の説明書にしたがって、Lymphocyte Separation Medium(LSM;Mediatech,Inc.によるCellgro(登録商標);密度1.078+/−0.002 g/ml)単離した。細胞を4μg/mLのフィトヘマグルチニン(PHA;Sigma)中での48〜72時間のインキュベーションによって刺激した。分裂刺激を、20U/mLの組み換え体ヒトIL−2(R&D sysてms、Inc)の培養培地への添加によって維持した。少なくとも2人のドナーに由来するPHAで刺激したPBMCをプールし、新鮮な培地中に稀釈し、96ウェルプレートに対して5×104細胞/ウェルで添加した。細胞を9種類の異なる試験化合物濃度(3連のウェル/濃度)の存在下で感染させ(最終的なMOI約0.1)、7日間インキュベートした。ウイルス阻害のレベルを決定するために、細胞を含まない上清試料を逆転写酵素活性の分析のために回収した(Buckheit RW,Swanstrom R(1991)Characterization of an HIV−1 isolate displaying an apparent absence of virion−associated reverse transcriptase activity.AIDS Res Hum Retrovir 7:295−302)。上清試料の除去後、化合物の細胞傷害性を、製造業者の説明書にしたがって3−(4,5−ジメチルチアゾール−2−イル)−5−(3−カルボキシメトキシフェニル)−2−(4−スルホフェニル)−2H−テトラゾリウム(MTS;CellTiter 96 Reagent,Promega)の添加によって測定した。社内コンピュータープログラムを使用して、IC50(ウイルスの複製の50%の阻害)、IC90(ウイルスの複製の90%の阻害)、TC50(細胞生存性の50%の低下)、および選択性指数(IC50/TC50)を決定した。AZT(ヌクレオシド逆転写酵素阻害剤)をアッセイ対照化合物として用いた。
【0269】
ウイルス
以下の試薬は、AIDS Research and Reference Reagent Program,Division of AIDS,NIAID,NIHを通じて入手し:pNL4−3はMalcolm Martinから入手した(Adachi Aら(1986)Production of acquired immunodeficiency syndrome−associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone.J Virol 59:284−291.)。
【0270】
AIDS Reagent ProgramによるNL4−3には、gp41中に予想しなかった変異形DIV(G36D)変異が含まれており、これは、インビトロでT−20に対して8倍の耐性を付与する。T−20−感受性NL4−3(NL4−3G)を、gp41のアミノ酸位置36でコンセンサス配列と一致するように部位特異的突然変異誘発によって変更させた(アスパラギン酸をグリシンで置き換えた)。NL4−3GおよびNL4−3D(もともとのクローン)のストックを、293T細胞のトランスフェクションと3日目の上清の回収によって調製した。ウイルスストックは、ELISAによるp24検出を用いて、PHAで活性化させたPBMC中での50%終点アッセイによって滴定した。
【0271】
結果
PBMCをベースとするアッセイを使用したインビトロでの抗ウイルス活性
それぞれのアルブミン結合体の抗ウイルス活性を、HIV−1 IIIBに対するPBMCをベースとするアッセイを使用してインビトロでもともとのペプチド阻害剤に対して比較した(Popovic ME,Read−Connole E,Gallo RC(1984)T4 positive human neoplastic cell lines susceptible to and permissive for HTLV−III.Lancet ii:1472−1473;Popovic M,Sarngadharan MG,Read E,Gallo RC(1984)Detection,isolation,and continuous production of cytopathic retroviruses(HTLV−III)from patients with AIDS and pre−AIDS.Science 224:497−500;Ratner Lら(1985)Complete nucleotide sequence of the AIDS virus,HTLV−III.Nature 313:277−283;Buckheit RW,Swanstrom R(1991)Characterization of an HIV−1 isolate displaying an apparent absence of virion−associated reverse transcriptase activity.AIDS Res Hum Retrovir 7:295−302)。興味深いことに、PC−1505(アルブミン結合化合物VIII)、化合物C(アルブミン結合化合物VII)、および化合物D(アルブミン結合化合物VI)の抗ウイルス活性は全て、インビトロではC34ペプチドおよびT−20に対して本質的に等しい効力があった。すなわち、C34ペプチドのN末端(PC−1505(アルブミン結合化合物VIII)および化合物C(アルブミン結合化合物VII)またはC末端(化合物D(アルブミン結合化合物VI))のいずれかでの反応性マレイミド基の置換、それに続くアルブミンの結合は、融合阻害剤の抗ウイルス活性を変化させることはなかった(表7)。T−20へのアルブミンの結合の後は、反応性ペプチドが、結合がペプチドのN末端で起こるように設計されている場合(化合物E アルブミン結合化合物III)には、抗ウイルス活性は極めて良好に保存されていたが、ペプチドのC末端でアルブミンに対する結合が起こる場合には、このペプチドの抗ウイルス活性の有意な低下が観察された(化合物F 化合物X−does cpd Fは、Eriksonにおいて同等であった(化合物I−VIII))。
【0272】
【表6】
C34ペプチド、化合物VIII、およびrHAのラットでの薬物動態プロフィール
この研究で観察された抗ウイルス活性が、遊離のペプチド、あるいはマレイミドとシステイン−34との間での共有結合の可逆性ではなく、アルブミン結合体の作用が原因であることを確認するために、全てのアルブミン結合体を試験前に精製してあらゆる結合していないペプチドを除去し、化合物VIIIの薬物動態プロフィールを、ラットにおいてC34ペプチド(図5A)とrHA(図5B)に対して比較した。明らかに、C34ペプチドの暴露はアルブミン結合の後に激的に改善され、予め形成させた結合体−化合物VIIIの薬物動態プロフィールがrHAの薬物動態プロフィールと重なり合うという事実によって、C34ペプチドがアルブミンの半減期に近い半減期をとることを確認した。C34ペプチドとHSAについて測定した薬物動態曲線の重ね合わせはまた、予め形成させた結合体−化合物VIIIの静脈内または皮下投与のいずれかの後、少なくとも30時間、Balb/cマウスを使用しても観察された(データは示さない、ラットでのアルブミンのT1/2よりもマウスでのアルブミンのT1/2は短い)。逆に、結合体からのC34ペプチドのゆっくりとした持続的な放出は、予め形成させた結合体−化合物VIIIの全暴露がrHAのものよりも劣るので、2つの薬物動態プロフィールはもはや重なり合わななくなるであろう。さらに、結合体から放出されたC34ペプチドは、インビボでは極めて短い半減期に曝され、長期持続型の予め形成させた結合体−化合物VIIIに対して比較すると、抗ウイルス有効性は限定される。したがって、システイン−34に対してマレイミドを連結させる結合はインビボで極めて安定であり、C34ペプチドは迅速な腎臓でのクリアランスとペプチダーゼによる分解に対してより安定となる。まとめると、インビトロおよびインビボでの全てのアルブミン結合体についての抗ウイルス活性は、マレイミド−システイン−34結合の可逆性ではなく、化学的に安定な結合体の作用だけによると結論付けることができる。
【0273】
考察
HIV gp41のN末端ヘリックス領域(NHR)とC末端ヘリックス領域(CHR)の配列をベースとする合成のペプチドは、多段階の融合プロセスの間に露出させられたgp41結合部位について競合することによってHIVの進入を阻害することが示されている(Chan DC,Fass D,Berger J M,Kim P S(1997)Core structure of gp41 from the HIV envelope glycoprotein.Cell 89:263−273;Chan DC,Chutkowski CT,Kim PS(1998)Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target.Proc Natl Acad Sci USA 95:15613−15617)。臨床では、これらのペプチドのほとんどの成功は、gp41のCHRに由来するT−20(Trimeris/Roche Applied SciencesによるFuzeon(登録商標))である。低分子と比較すると、ペプチドの商業的有用性は、多くの場合はそれらのコスト、ならびにそれらの短い半減期とインビトロでの低い分布によって限定される。本発明者らは、他のクラスのペプチドについてすでに行われているように、ヒトアルブミンのシステイン−34に共有結合するようにCHRペプチド(C34およびT−20)を操作することによって、これらの欠点に取り組もうと考えた(Holmes DLら(2000)Site specific 1:1 opioid:albumin conjugate with in vitro activity and long in vivo duration.Bioconj Chem 11:439−444;Leger Rら(2003)Synthesis and in vitro analysis of atrial natriuretic peptide−albumin conjugates.Bioorg & Med Chem Lett 13:3571−3575;Leger Rら(2004)Kringle 5 peptide−albumin conjugates with anti−migratory activity.Bioorg & Med Chem Lett 14:841−845;Leger Rら(2004)Identification of CJC−1131−albumin bioconjugate as a stable and bioactive GLP−1(7−36)analog.Bioorg & Med Chem Lett 14:4395−4398;Jette Lら(2005)Human growth hormone−releasing factor(hGRF)1−29 albumin bioconjugates activate the GRF receptor on the anterior pituitary in rats:Identification of CJC−1295 as a long−lasting GRF analog.Endocrinology 146:3052−3058;Thibaudeau Kら(2005)Synthesis and evaluation of insulin−human serum albumin conjugates.Bioconj Chem 16:1000−1008)。すなわち、本発明者らは、CHR−ペプチド−HSA結合体がもともとの融合阻害剤のはるかに短い半減期とは反対に、アルブミンの半減期に近い体内での半減期となるであろうと仮定した。
【0274】
本明細書中に示した結果は、gp41のNHRが、もともと考えられていたよりも利用しやすいことを示唆している。例えば、予め形成させた結合体−化合物VIIIを使用して示されるようにそのような競合阻害を利用できるようにするためには、gp41は、標的細胞が存在しない条件で(すなわち、細胞を含まないウイルスまたは非感染細胞の状況下で)NHR領域を露出させる配座平衡に関与し得るか、あるいは、「エントリークロウ(entry claw)」の中に形成されるプレヘアピン中間体(Sougrat Rら(2007)Electron tomography of the contact between T cells and SIV/HIV−1:Implications for viral entry.PLoS Pathogens 3:0571−0581)が、6ヘリックスの束状構造の形成、その後の脂質の混合および膜貫通工程の前に十分に溶媒に露出させられる。すなわち、予め形成させた結合体−化合物VIIIについて約71kDaのMwを用いた場合には、本発明者らの結果は、gp41のNHR−三量体を評価するための分子量カットオフが、これまでに報告されている(すなわち、<25kDa)よりもはるかに大きいことを示唆している(11)。第2に、C34ペプチドのN末端セグメント628WMEW631は、HIV−1の侵入を阻害するC34ペプチドの能力に不可欠であると仮定されるgp41コイルドコイルキャビティ結合残基を提示する(18,19)。したがって、予め形成させた結合体−化合物VIII(AEEAリンカーからなる)または化合物C予め形成させた結合体−化合物VII(AEEAリンカーが存在しない)のいずれかの場合においては、どのようにすれば、C34ペプチドの628WMEW631セグメントをgp41のNHRに到達させることが可能であるか、そして同時に、アルブミンの表面に対して永久的に結合し、近位に接近して配置されるか?予め形成させた結合体−化合物VIIIおよび化合物C予め形成させた結合体−化合物VIIの抗ウイルス活性の保持についての1つの可能性のある説明は、血清アルブミンがいくつかの立体構造状態をとるように誘導することができる高度に可撓性のタンパク質であるという事実である(Peters T,Jr(1996)All about albumin−biochemistry,genetics,and medical applications,Copyright by Academic Press,Inc)。例えば、C34ペプチドはアルブミンのシステイン−34に永久的に結合させられるので、アルブミンの拘束されていないN末端ドメイン(すなわち、ジスルフィド結合が存在しない)内での局所立体構造転位は、gp41のNHR領域上への融合阻害剤の正確な挿入を容易にするための部分的な巻き戻しを生じさせることができる。したがって、システイン−34上以外のアルブミン分子内のほかの場所にC34ペプチドを配置することにより、この融合阻害剤についての抗ウイルス活性の同様の保存が導かれるかどうか(例えば、リジン残基、N末端、またはC末端)、あるいは、抗ウイルス活性の同様の保存が、他の豊富に存在する高分子量の血清タンパク質(例えば、トランスフェリンまたはIgG)に対するC34ペプチドの永久的な結合の後に観察されるであろうかどうかは明らかにはなっていない。したがって、アルブミン分子が、単にタンパク質貨物(protein cargo)としての役割を担うだけではなく、活発な参加型の役割を担っている可能性もある。例えば、マレイル化(maleylated)−、アコニチニル化−、およびスクシニル化されたアルブミンは、インビトロで強力なHIV−1侵入阻害剤として機能する(35〜38)。
【0275】
加えて、C34ペプチドの中に見られる34個のアミノ酸残基のうちの24個が、T−20の中で見られるアミノ酸残基と重複すると仮定すると、T−20は、このペプチドC末端での修飾後にアルブミン結合体についてより少ない候補となることが可能であるかどうか、一方では、抗ウイルス活性の保持の改善が、T−20がそのN末端で改変された場合に観察されるかどうか?この知見についての1つの可能性のある説明は、T−20が原因であるHIV−1の阻害機構がC34ペプチドのものとは異なることを示唆している最近の証拠である(Liu Sら(2005)J Biol Chem 280:11259−11273;Munoz−Barroso I,ら(1998)J Cell Biol 140:315−23;Kliger Yら(2001)J Biol Chem 276:1391−1397)。例えば、T−20はまた、血漿膜へのgp41の動員、その後の脂質混合工程後のオリゴマー形成を阻害することが示されているが、C34ペプチドは6ヘリックスの束状構造の形成後はその阻害効果を発揮することができないことが明らかにされている(Liu Sら(2005)J Biol Chem 280:11259−11273)。すなわち、T−20は、血漿膜へのその挿入後にそのような阻害機能を行うこと、そしてT20の疎水性C末端セグメント666WASLWNWF673は、これらの疎水性相互作用の達成に重要と考えられることが提案されている(Munoz−Barroso I,ら(1998)J Cell Biol 140:315−23;Kliger Yら(2001)J Biol Chem 276:1391−1397)。さらに具体的には、T−20は、血漿膜の近位に配置されるgp41中の対応する配列に結合することによって、gp41の動員とオリゴマー形成を阻害する(Munoz−Barroso I,Durell S,Sakaguchi K,Appella E,Blumenthal R(1998)Dilation of the human immunodeficiency virus−1 envelope glycoprotein fusion pore revealed by the inhibitory action of a synthetic peptide from gp41.J Cell Biol 140:315−23;Kliger Yら(2001)J Biol Chem 276:1391−1397)。したがって、666WASLWNWF673配列がアルブミン分子にすぐ隣接して配置されている化合物F化合物Xについて観察された抗ウイルス活性の大幅な低下は、結合していない(遊離の)T−20ペプチドと同程度に有効に、脂質混合工程後にこのペプチドが機能できないことに寄与している可能性がある。逆に、666WASLWNWF673配列は、化合物E(化合物IX)の構成においては立体構造上の拘束が少ない。まとめると、本発明者らの結果は、T−20とC34ペプチドがHIV−1融合の同じ工程では機能していないことを示唆した報告を明白にサポートする証拠を提供する。
【0276】
本明細書中に示される結果は、膜融合とウイルスの侵入について類似する機構を採用するHIVウイルスと他のウイルスの阻害のための、この新規のクラスのアルブミン−ペプチド結合体についての原理の証明を確立する。結合していない(遊離の)ペプチド阻害剤と比較すると、アルブミン結合体は、全HIV感染細胞のおよそ98%の解剖学上のホームを示すリンパ系に対する有意に改善された暴露を導き得る(Stebbing J,Gazzard B,Douek DC(2004)Where does HIV live? N Engl J Med 350:1872−1880)。この改善は、血清アルブミンについて観察された血漿濃度比に対する有意な定常状態リンパに(Bent−Hansen L(1991)Whole body capillary exchange of albumin.Acta Physiol Scand Suppl 603:5−10(Review);Porter CJH,Charman SA(2000)Lymphatic transport of proteins after subcutaneous administration.J Pharm Sci 89:297−310)、および16〜20kDaより大きい皮下注射されたタンパク質について観察された効率的なリンパの取り込み、輸送、および透過性に(Porter CJH,Charman SA(2000)Lymphatic transport of proteins after subcutaneous administration.J Pharm Sci 89:297−310)主に原因があると予想され得る。
【0277】
最後に、C34ペプチドおよび多くの他の抗膜融合性ペプチド中に見られる疎水性残基の高い含有量が原因で、アルブミン結合体はまた、これらが皮下送達に適している単純な水性処方物中に入れられる場合には、ペプチドのこのファミリーについて一般的に観察される低い溶解限度の矯正をも助け得る。例えば、C34ペプチドの溶解限度は、水性緩衝液中では1mg/mlを超えないことが明らかであるが、PC−1505の溶解限度は、アルブミンの溶解限度と類似することが明らかにされており、これはC34ペプチドのおよそ16mg/mlに相当する(すなわち、25%(w/v)の溶液=PC−1505の250mg/ml、およそC34ペプチドの16mg/ml)。
【0278】
まとめると、アルブミンのシステイン−34を介する抗膜融合性ペプチドの結合によって、gp41のNHRトリマーに一般的に関係している立体的なブロックが克服され、それにより、強力であり、強い中和活性を示す新規のさらに分子量の大きい分子の発見の期待がもたらされる。アルブミン結合C34ペプチドHIV−1融合阻害剤の一例であるPC−1505は、T−20よりも少ない投与頻度しか必要とせず、そしておそらく、ヒトにおいては、T−20耐性HIV−1に対する有効な薬剤である。
【0279】
(実施例9)
さらなる抗膜融合性ペプチド誘導体
図6は、記載した抗膜融合性のいくつかの結合体(PC(予め形成させた複合体)と示す)のインビトロでの抗HIV活性を示している表を示す。アッセイは、本明細書中の実施例に記載したように行った。
【0280】
本発明の特定の実施形態が記載され、説明されてきたが、当業者は、本発明はこれらの実施形態の任意の詳細に限定されるように意図されるのではなく、むしろ添付の特許請求の範囲によって定義されることを理解するであろう。
【特許請求の範囲】
【請求項1】
改変された抗膜融合性ペプチドまたはその結合体であって、該ペプチドは、改変前の該ペプチドと比較して、約5から8までの範囲のpHの水溶液中で高い溶解度を有するように改変され、該改変されたペプチドは以下:
a)約10mg/ml〜180mg/mlの範囲の濃度で、該水溶液において約10%未満の沈殿を示す;
b)改変前の該ペプチドよりも少なくとも約2.5倍高い溶解限度を有する;および
c)該水溶液中で少なくとも約20mg/mlの溶解限度を有する、
の特性を有する、改変された抗膜融合性ペプチドまたはその結合体。
【請求項2】
前記改変されたペプチドに、生理学的pHで電荷を持つかまたは電荷を持たないかのいずれかである1つ以上の極性部分が含まれている、請求項1に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項3】
前記改変されたペプチドの1つ以上の極性部分に、20種類の天然に存在するアミノ酸の中には見られない1つ以上の極性側鎖または中性側鎖が含まれている、請求項2に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項4】
前記改変されたペプチドの1つ以上の極性部分に1つ以上のシステイン酸が含まれている、請求項3に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項5】
前記改変されたペプチドの1つ以上のシステイン酸が、前記改変された抗膜融合性ペプチドのN末端またはC末端に付加されている、請求項1または請求項4に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項6】
前記改変されたペプチドの1つ以上の極性部分が、該ペプチドの二次構造または三次構造に実質的に影響を与えない、請求項3に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項7】
前記改変されたペプチドにgp41コイルドコイルキャビティ結合残基の少なくとも一部分が含まれている、請求項1または請求項4に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項8】
前記改変されたペプチドにアミノ酸628WMEWDREINNYTSLIHSLIEESQNQQEKNEQELL661(配列番号2)に由来するC34のアミノ酸配列、またはそれに対する2個までのアミノ酸置換、挿入、もしくは欠失が含まれている、請求項7に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項9】
前記改変されたペプチドに、DP107およびDP178のアミノ酸配列、またはそれに対する2個までのアミノ酸置換、挿入、もしくは欠失が含まれている、請求項7に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項10】
前記改変されたペプチドが血液成分または担体タンパク質上の利用可能な官能基と反応して前記結合体の安定な共有結合を形成することができるように、1つ以上の化学反応性部分がさらに含まれている、請求項7に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項11】
前記反応性部分がマレイミド含有基である、請求項10に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項12】
以下:(2−アミノ)エトキシ酢酸(AEA)、[2−(2−アミノ)エトキシ)]エトキシ酢酸(AEEA)、エチレンジアミン(EDA);8−アミノオクタン酸(AOA)、8−アミノプロパン酸(APA)、および4−アミノ安息香酸(APhA)のような1つ以上のアルキル鎖(C1−C10)、
からなる群より選択される1つ以上のリンカーがさらに含まれている、請求項10に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項13】
リンカーを持つかまたはリンカーを持たない前記反応性部分が、前記改変されたペプチドのC末端に付加されており、前記1つ以上の極性部分が、該改変された抗膜融合性ペプチドのN末端に付加されている、請求項10に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項14】
リンカーを持つかまたはリンカーを持たない前記反応性部分が、前記改変されたペプチドのN末端に付加されており、前記1つ以上の極性部分が、該改変された抗膜融合性ペプチドのC末端に付加されている、請求項10に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項15】
前記血液成分または担体タンパク質がアルブミンである、請求項10に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項16】
前記アルブミンが組み換え体である、請求項15に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項17】
前記アルブミンが共有結合させられている、請求項16に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項18】
以下のような構造:
[(システイン酸)−改変されたペプチド−リンカーn−反応性基];または、[反応性基−リンカーn−改変されたペプチド−(システイン酸)]
を有している、改変された抗膜融合性ペプチドまたはその結合体であって、式中、
該反応性基は、例えば、リンカーを持つかまたはリンカーを持たない、ヒト血清アルブミンに共有結合させられたマレイミド含有基であり;
nは、(2−アミノ)エトキシ酢酸(AEA)、[2−(2−アミノ)エトキシ)]エトキシ酢酸(AEEA)、エチレンジアミン(EDA);8−アミノオクタン酸(AOA)、8−アミノプロパン酸(APA)、および4−アミノ安息香酸(APhA)のような1つ以上のアルキル鎖(C1−C10)からなる群より選択される0、1、2、3、4、またはそれ以上のリンカーであり得;そして
前記改変されたペプチドには、アミノ酸628WMEWDREINNYTSLIHSLIEESQNQQEKNEQELL661(配列番号2)に由来するC34のアミノ酸配列、またはそれに対する2個までのアミノ酸置換もしくは付加が含まれる、
改変された抗膜融合性ペプチドまたはその結合体。
【請求項19】
以下の式:
(I) (R1)m−X−(R2)n
を有している改変された抗膜融合性ペプチドまたはその結合体であって、
式(I)において、mとnの和は少なくとも1であり、mとnはそれぞれが0以上の整数であり;
Xには、C34、DP107、DP178、またはそれらのアナログのアミノ酸配列が含まれ;
R1が存在し、R2が存在しない場合は、R1はX基のN末端に存在し、R1が存在せず、R2が存在する場合は、R2はX基のC末端に存在する、
改変された抗膜融合性ペプチドまたはその結合体。
【請求項20】
請求項19に記載の改変された抗膜融合性ペプチドまたはその結合体であって、R1とR2はそれぞれ独立して、式(II):
【化30】
を有する化合物から選択され、ここで、
式(II)のコア構造はアミノ酸のコア構造と類似しており、これにはアミノ基、α炭素、およびカルボキシル基が含まれ;そして
ここでは、式(II)のR3基には、スルホニル基(HS=(O)2)、スルホキシド基(HS=O)、スルホン酸基(HO−S=(O)2)、ハロアルキル基、二級アミン、三級アミン、ヒドロキシル基、または極性であるかもしくは全くの中性であり、水溶液中でのペプチド誘導体の全体的な可溶性を増大させることができる他の側鎖基が含まれる、
改変された抗膜融合性ペプチドまたはその結合体。
【請求項21】
前記R1基とR2基が、前記ペプチドの二次構造または三次構造全体には実質的には影響を及ぼさない請求項20に記載の改変された抗膜融合性ペプチドまたはその結合体であって、該ペプチド結合体の二次構造に実質的には影響を及ぼさないことにより、ペプチド結合体の全体的な活性が、誘導体化されていないペプチドの全体的な活性よりも感知できるほどに下回ることはないはずである、改変された抗膜融合性ペプチドまたはその結合体。
【請求項22】
以下:
CA化合物I:(C34に直接連結させられたシステイン酸(CA);本明細書中ではCA−C34とも呼ばれる(配列番号3)):
【化31】
CA化合物II:(28位に本来のリジン(Lys28)のアルギニンでの置換を有しているC34に直接連結させられたシステイン酸(CA);本明細書中ではCA−C34(Arg28)とも呼ばれる)(配列番号4):
【化32】
CA化合物III:(35位にさらなるリジン残基(Lys35)を有しているC34に直接連結させられたシステイン酸(CA)であって、ここでは、リジンのεNH2基はリンカー(AEEA−MPA)を介して反応性基に結合させられる;本明細書中ではCA−C34−Lys35(ε−AEEA−MPA)とも呼ばれる)(配列番号5):
【化33】
および
CA化合物IV:(28位に本来のリジン(Lys28)のアルギニンでの置換;35位にさらなるリジン残基(Lys35)を有しているC34に直接連結させられたシステイン酸(CA)であって、ここでは、リジンのεNH2基はリンカー(AEEA−MPA)を介して反応性基に結合させられる;本明細書中ではCA−C34(Arg28)−Lys35(ε−AEEA−MPA)とも呼ばれる)(配列番号6)
【化34】
からなる群より選択される構造を有する、改変された抗膜融合性ペプチド。
【請求項23】
請求項1、4、18、19、または22のいずれかに記載の改変された抗膜融合性ペプチドを含む結合体。
【請求項24】
安定な共有結合を形成させるためにアルブミン上の1つ以上のアミノ基、ヒドロキシル基、またはチオール基に対して、リンカーを用いてまたはリンカーを用いることなく結合させられた、請求項8に記載の改変された抗膜融合性ペプチドを含む結合体。
【請求項25】
安定な共有結合を形成させるためにアルブミン上の1つ以上のアミノ基、ヒドロキシル基、またはチオール基に対して、リンカーを用いてまたはリンカーを用いることなく結合させられた、請求項10に記載の改変された抗膜融合性ペプチドを含む結合体。
【請求項26】
安定な共有結合を形成させるためにアルブミン上の1つ以上のアミノ基、ヒドロキシル基、またはチオール基に対して、リンカーを用いてまたはリンカーを用いることなく結合させられた、請求項11に記載の改変された抗膜融合性ペプチドを含む結合体。
【請求項27】
安定な共有結合を形成させるためにアルブミン上の1つ以上のアミノ基、ヒドロキシル基、またはチオール基に対して、リンカーを用いてまたはリンカーを用いることなく結合させられた、請求項12に記載の改変された抗膜融合性ペプチドを含む結合体。
【請求項28】
安定な共有結合を形成させるためにアルブミン上の1つ以上のアミノ基、ヒドロキシル基、またはチオール基に対して、リンカーを用いてまたはリンカーを用いることなく結合させられた、請求項13に記載の改変された抗膜融合性ペプチドを含む結合体。
【請求項29】
安定な共有結合を形成させるためにアルブミン上の1つ以上のアミノ基、ヒドロキシル基、またはチオール基に対して、リンカーを用いてまたはリンカーを用いることなく結合させられた、請求項15に記載の改変された抗膜融合性ペプチドを含む結合体。
【請求項30】
安定な共有結合を形成させるためにアルブミン上の1つ以上のアミノ基、ヒドロキシル基、またはチオール基に対して、リンカーを用いてまたはリンカーを用いることなく結合させられた、請求項22に記載の改変された抗膜融合性ペプチドを含む結合体。
【請求項31】
皮下投与、静脈内投与、または肺投与に適している、請求項8に記載の改変された抗膜融合性ペプチドまたはその結合体と薬学的に許容される担体とを含む薬学的組成物。
【請求項32】
皮下投与、静脈内投与、または肺投与に適している、請求項22に記載の改変された抗膜融合性ペプチドまたはその結合体と薬学的に許容される担体とを含む薬学的組成物。
【請求項33】
皮下投与、静脈内投与、または肺投与に適している、請求項23に記載の結合体と薬学的に許容される担体とを含む薬学的組成物。
【請求項34】
皮下投与、静脈内投与、または肺投与に適している、請求項24に記載の結合体と薬学的に許容される担体とを含む薬学的組成物。
【請求項35】
対象におけるヒト免疫不全ウイルス(HIV)感染、RSウイルス(RSV)、ヒト3型パラインフルエンザウイルス(HPIV−3)、麻疹ウイルス(MeV)、およびサル免疫不全ウイルス(SIV)からなる群より選択されるウイルスの処置または予防方法であって、
請求項8に記載の改変された抗膜融合性ペプチドまたはその結合体を、該ウイルスを有しているかまたは該ウイルスを有するリスクがある該対象に対して投与し、それによって感染を処置または予防する工程を含む、方法。
【請求項36】
対象におけるヒト免疫不全ウイルス(HIV)感染、RSウイルス(RSV)、ヒト3型パラインフルエンザウイルス(HPIV−3)、麻疹ウイルス(MeV)、およびサル免疫不全ウイルス(SIV)からなる群より選択されるウイルスの処置または予防方法であって、
請求項22に記載の改変された抗膜融合性ペプチドまたはその結合体を、該ウイルスを有しているかまたは該ウイルスを有するリスクがある該対象に対して投与し、それによって感染を処置または予防する工程を含む、方法。
【請求項37】
対象におけるヒト免疫不全ウイルス(HIV)感染、RSウイルス(RSV)、ヒト3型パラインフルエンザウイルス(HPIV−3)、麻疹ウイルス(MeV)、およびサル免疫不全ウイルス(SIV)からなる群より選択されるウイルスの処置または予防方法であって、
請求項23に記載の結合体を、該ウイルスを有しているかまたは該ウイルスを有するリスクがある該対象に対して投与し、それによって感染を処置または予防する工程を含む、方法。
【請求項38】
対象におけるヒト免疫不全ウイルス(HIV)感染、RSウイルス(RSV)、ヒト3型パラインフルエンザウイルス(HPIV−3)、麻疹ウイルス(MeV)、およびサル免疫不全ウイルス(SIV)からなる群より選択されるウイルスの処置または予防方法であって、
請求項24に記載の結合体を、該ウイルスを有しているかまたは該ウイルスを有するリスクがある対象に対して投与し、それによって感染を処置または予防する工程を含む、方法。
【請求項39】
対象においてヒト免疫不全ウイルス(HIV)感染、RSウイルス(RSV)、ヒト3型パラインフルエンザウイルス(HPIV−3)、麻疹ウイルス(MeV)、およびサル免疫不全ウイルス(SIV)のうちの1つ以上の活性を阻害するための方法であって、処置が必要な該対象に対して有効量の請求項8に記載の改変された抗膜融合性ペプチドまたはその結合体を投与する工程を含む、方法
【請求項40】
対象においてヒト免疫不全ウイルス(HIV)感染、RSウイルス(RSV)、ヒト3型パラインフルエンザウイルス(HPIV−3)、麻疹ウイルス、およびサル免疫不全ウイルス(SIV)のうちの1つ以上の活性を阻害するための方法であって、処置が必要な該対象に対して有効量の請求項22に記載の改変された抗膜融合性ペプチドまたはその結合体を投与する工程を含む、方法。
【請求項41】
抗膜融合性ペプチドの大規模な調製を増進するための方法であって、該方法は:
請求項8に記載の改変された抗膜融合性ペプチドを提供する工程;および
少なくとも100mg/mlの該改変されたペプチドの濃度を有する該改変されたペプチドの溶液を調製する工程、
を含む、方法。
【請求項1】
改変された抗膜融合性ペプチドまたはその結合体であって、該ペプチドは、改変前の該ペプチドと比較して、約5から8までの範囲のpHの水溶液中で高い溶解度を有するように改変され、該改変されたペプチドは以下:
a)約10mg/ml〜180mg/mlの範囲の濃度で、該水溶液において約10%未満の沈殿を示す;
b)改変前の該ペプチドよりも少なくとも約2.5倍高い溶解限度を有する;および
c)該水溶液中で少なくとも約20mg/mlの溶解限度を有する、
の特性を有する、改変された抗膜融合性ペプチドまたはその結合体。
【請求項2】
前記改変されたペプチドに、生理学的pHで電荷を持つかまたは電荷を持たないかのいずれかである1つ以上の極性部分が含まれている、請求項1に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項3】
前記改変されたペプチドの1つ以上の極性部分に、20種類の天然に存在するアミノ酸の中には見られない1つ以上の極性側鎖または中性側鎖が含まれている、請求項2に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項4】
前記改変されたペプチドの1つ以上の極性部分に1つ以上のシステイン酸が含まれている、請求項3に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項5】
前記改変されたペプチドの1つ以上のシステイン酸が、前記改変された抗膜融合性ペプチドのN末端またはC末端に付加されている、請求項1または請求項4に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項6】
前記改変されたペプチドの1つ以上の極性部分が、該ペプチドの二次構造または三次構造に実質的に影響を与えない、請求項3に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項7】
前記改変されたペプチドにgp41コイルドコイルキャビティ結合残基の少なくとも一部分が含まれている、請求項1または請求項4に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項8】
前記改変されたペプチドにアミノ酸628WMEWDREINNYTSLIHSLIEESQNQQEKNEQELL661(配列番号2)に由来するC34のアミノ酸配列、またはそれに対する2個までのアミノ酸置換、挿入、もしくは欠失が含まれている、請求項7に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項9】
前記改変されたペプチドに、DP107およびDP178のアミノ酸配列、またはそれに対する2個までのアミノ酸置換、挿入、もしくは欠失が含まれている、請求項7に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項10】
前記改変されたペプチドが血液成分または担体タンパク質上の利用可能な官能基と反応して前記結合体の安定な共有結合を形成することができるように、1つ以上の化学反応性部分がさらに含まれている、請求項7に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項11】
前記反応性部分がマレイミド含有基である、請求項10に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項12】
以下:(2−アミノ)エトキシ酢酸(AEA)、[2−(2−アミノ)エトキシ)]エトキシ酢酸(AEEA)、エチレンジアミン(EDA);8−アミノオクタン酸(AOA)、8−アミノプロパン酸(APA)、および4−アミノ安息香酸(APhA)のような1つ以上のアルキル鎖(C1−C10)、
からなる群より選択される1つ以上のリンカーがさらに含まれている、請求項10に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項13】
リンカーを持つかまたはリンカーを持たない前記反応性部分が、前記改変されたペプチドのC末端に付加されており、前記1つ以上の極性部分が、該改変された抗膜融合性ペプチドのN末端に付加されている、請求項10に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項14】
リンカーを持つかまたはリンカーを持たない前記反応性部分が、前記改変されたペプチドのN末端に付加されており、前記1つ以上の極性部分が、該改変された抗膜融合性ペプチドのC末端に付加されている、請求項10に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項15】
前記血液成分または担体タンパク質がアルブミンである、請求項10に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項16】
前記アルブミンが組み換え体である、請求項15に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項17】
前記アルブミンが共有結合させられている、請求項16に記載の改変された抗膜融合性ペプチドまたはその結合体。
【請求項18】
以下のような構造:
[(システイン酸)−改変されたペプチド−リンカーn−反応性基];または、[反応性基−リンカーn−改変されたペプチド−(システイン酸)]
を有している、改変された抗膜融合性ペプチドまたはその結合体であって、式中、
該反応性基は、例えば、リンカーを持つかまたはリンカーを持たない、ヒト血清アルブミンに共有結合させられたマレイミド含有基であり;
nは、(2−アミノ)エトキシ酢酸(AEA)、[2−(2−アミノ)エトキシ)]エトキシ酢酸(AEEA)、エチレンジアミン(EDA);8−アミノオクタン酸(AOA)、8−アミノプロパン酸(APA)、および4−アミノ安息香酸(APhA)のような1つ以上のアルキル鎖(C1−C10)からなる群より選択される0、1、2、3、4、またはそれ以上のリンカーであり得;そして
前記改変されたペプチドには、アミノ酸628WMEWDREINNYTSLIHSLIEESQNQQEKNEQELL661(配列番号2)に由来するC34のアミノ酸配列、またはそれに対する2個までのアミノ酸置換もしくは付加が含まれる、
改変された抗膜融合性ペプチドまたはその結合体。
【請求項19】
以下の式:
(I) (R1)m−X−(R2)n
を有している改変された抗膜融合性ペプチドまたはその結合体であって、
式(I)において、mとnの和は少なくとも1であり、mとnはそれぞれが0以上の整数であり;
Xには、C34、DP107、DP178、またはそれらのアナログのアミノ酸配列が含まれ;
R1が存在し、R2が存在しない場合は、R1はX基のN末端に存在し、R1が存在せず、R2が存在する場合は、R2はX基のC末端に存在する、
改変された抗膜融合性ペプチドまたはその結合体。
【請求項20】
請求項19に記載の改変された抗膜融合性ペプチドまたはその結合体であって、R1とR2はそれぞれ独立して、式(II):
【化30】
を有する化合物から選択され、ここで、
式(II)のコア構造はアミノ酸のコア構造と類似しており、これにはアミノ基、α炭素、およびカルボキシル基が含まれ;そして
ここでは、式(II)のR3基には、スルホニル基(HS=(O)2)、スルホキシド基(HS=O)、スルホン酸基(HO−S=(O)2)、ハロアルキル基、二級アミン、三級アミン、ヒドロキシル基、または極性であるかもしくは全くの中性であり、水溶液中でのペプチド誘導体の全体的な可溶性を増大させることができる他の側鎖基が含まれる、
改変された抗膜融合性ペプチドまたはその結合体。
【請求項21】
前記R1基とR2基が、前記ペプチドの二次構造または三次構造全体には実質的には影響を及ぼさない請求項20に記載の改変された抗膜融合性ペプチドまたはその結合体であって、該ペプチド結合体の二次構造に実質的には影響を及ぼさないことにより、ペプチド結合体の全体的な活性が、誘導体化されていないペプチドの全体的な活性よりも感知できるほどに下回ることはないはずである、改変された抗膜融合性ペプチドまたはその結合体。
【請求項22】
以下:
CA化合物I:(C34に直接連結させられたシステイン酸(CA);本明細書中ではCA−C34とも呼ばれる(配列番号3)):
【化31】
CA化合物II:(28位に本来のリジン(Lys28)のアルギニンでの置換を有しているC34に直接連結させられたシステイン酸(CA);本明細書中ではCA−C34(Arg28)とも呼ばれる)(配列番号4):
【化32】
CA化合物III:(35位にさらなるリジン残基(Lys35)を有しているC34に直接連結させられたシステイン酸(CA)であって、ここでは、リジンのεNH2基はリンカー(AEEA−MPA)を介して反応性基に結合させられる;本明細書中ではCA−C34−Lys35(ε−AEEA−MPA)とも呼ばれる)(配列番号5):
【化33】
および
CA化合物IV:(28位に本来のリジン(Lys28)のアルギニンでの置換;35位にさらなるリジン残基(Lys35)を有しているC34に直接連結させられたシステイン酸(CA)であって、ここでは、リジンのεNH2基はリンカー(AEEA−MPA)を介して反応性基に結合させられる;本明細書中ではCA−C34(Arg28)−Lys35(ε−AEEA−MPA)とも呼ばれる)(配列番号6)
【化34】
からなる群より選択される構造を有する、改変された抗膜融合性ペプチド。
【請求項23】
請求項1、4、18、19、または22のいずれかに記載の改変された抗膜融合性ペプチドを含む結合体。
【請求項24】
安定な共有結合を形成させるためにアルブミン上の1つ以上のアミノ基、ヒドロキシル基、またはチオール基に対して、リンカーを用いてまたはリンカーを用いることなく結合させられた、請求項8に記載の改変された抗膜融合性ペプチドを含む結合体。
【請求項25】
安定な共有結合を形成させるためにアルブミン上の1つ以上のアミノ基、ヒドロキシル基、またはチオール基に対して、リンカーを用いてまたはリンカーを用いることなく結合させられた、請求項10に記載の改変された抗膜融合性ペプチドを含む結合体。
【請求項26】
安定な共有結合を形成させるためにアルブミン上の1つ以上のアミノ基、ヒドロキシル基、またはチオール基に対して、リンカーを用いてまたはリンカーを用いることなく結合させられた、請求項11に記載の改変された抗膜融合性ペプチドを含む結合体。
【請求項27】
安定な共有結合を形成させるためにアルブミン上の1つ以上のアミノ基、ヒドロキシル基、またはチオール基に対して、リンカーを用いてまたはリンカーを用いることなく結合させられた、請求項12に記載の改変された抗膜融合性ペプチドを含む結合体。
【請求項28】
安定な共有結合を形成させるためにアルブミン上の1つ以上のアミノ基、ヒドロキシル基、またはチオール基に対して、リンカーを用いてまたはリンカーを用いることなく結合させられた、請求項13に記載の改変された抗膜融合性ペプチドを含む結合体。
【請求項29】
安定な共有結合を形成させるためにアルブミン上の1つ以上のアミノ基、ヒドロキシル基、またはチオール基に対して、リンカーを用いてまたはリンカーを用いることなく結合させられた、請求項15に記載の改変された抗膜融合性ペプチドを含む結合体。
【請求項30】
安定な共有結合を形成させるためにアルブミン上の1つ以上のアミノ基、ヒドロキシル基、またはチオール基に対して、リンカーを用いてまたはリンカーを用いることなく結合させられた、請求項22に記載の改変された抗膜融合性ペプチドを含む結合体。
【請求項31】
皮下投与、静脈内投与、または肺投与に適している、請求項8に記載の改変された抗膜融合性ペプチドまたはその結合体と薬学的に許容される担体とを含む薬学的組成物。
【請求項32】
皮下投与、静脈内投与、または肺投与に適している、請求項22に記載の改変された抗膜融合性ペプチドまたはその結合体と薬学的に許容される担体とを含む薬学的組成物。
【請求項33】
皮下投与、静脈内投与、または肺投与に適している、請求項23に記載の結合体と薬学的に許容される担体とを含む薬学的組成物。
【請求項34】
皮下投与、静脈内投与、または肺投与に適している、請求項24に記載の結合体と薬学的に許容される担体とを含む薬学的組成物。
【請求項35】
対象におけるヒト免疫不全ウイルス(HIV)感染、RSウイルス(RSV)、ヒト3型パラインフルエンザウイルス(HPIV−3)、麻疹ウイルス(MeV)、およびサル免疫不全ウイルス(SIV)からなる群より選択されるウイルスの処置または予防方法であって、
請求項8に記載の改変された抗膜融合性ペプチドまたはその結合体を、該ウイルスを有しているかまたは該ウイルスを有するリスクがある該対象に対して投与し、それによって感染を処置または予防する工程を含む、方法。
【請求項36】
対象におけるヒト免疫不全ウイルス(HIV)感染、RSウイルス(RSV)、ヒト3型パラインフルエンザウイルス(HPIV−3)、麻疹ウイルス(MeV)、およびサル免疫不全ウイルス(SIV)からなる群より選択されるウイルスの処置または予防方法であって、
請求項22に記載の改変された抗膜融合性ペプチドまたはその結合体を、該ウイルスを有しているかまたは該ウイルスを有するリスクがある該対象に対して投与し、それによって感染を処置または予防する工程を含む、方法。
【請求項37】
対象におけるヒト免疫不全ウイルス(HIV)感染、RSウイルス(RSV)、ヒト3型パラインフルエンザウイルス(HPIV−3)、麻疹ウイルス(MeV)、およびサル免疫不全ウイルス(SIV)からなる群より選択されるウイルスの処置または予防方法であって、
請求項23に記載の結合体を、該ウイルスを有しているかまたは該ウイルスを有するリスクがある該対象に対して投与し、それによって感染を処置または予防する工程を含む、方法。
【請求項38】
対象におけるヒト免疫不全ウイルス(HIV)感染、RSウイルス(RSV)、ヒト3型パラインフルエンザウイルス(HPIV−3)、麻疹ウイルス(MeV)、およびサル免疫不全ウイルス(SIV)からなる群より選択されるウイルスの処置または予防方法であって、
請求項24に記載の結合体を、該ウイルスを有しているかまたは該ウイルスを有するリスクがある対象に対して投与し、それによって感染を処置または予防する工程を含む、方法。
【請求項39】
対象においてヒト免疫不全ウイルス(HIV)感染、RSウイルス(RSV)、ヒト3型パラインフルエンザウイルス(HPIV−3)、麻疹ウイルス(MeV)、およびサル免疫不全ウイルス(SIV)のうちの1つ以上の活性を阻害するための方法であって、処置が必要な該対象に対して有効量の請求項8に記載の改変された抗膜融合性ペプチドまたはその結合体を投与する工程を含む、方法
【請求項40】
対象においてヒト免疫不全ウイルス(HIV)感染、RSウイルス(RSV)、ヒト3型パラインフルエンザウイルス(HPIV−3)、麻疹ウイルス、およびサル免疫不全ウイルス(SIV)のうちの1つ以上の活性を阻害するための方法であって、処置が必要な該対象に対して有効量の請求項22に記載の改変された抗膜融合性ペプチドまたはその結合体を投与する工程を含む、方法。
【請求項41】
抗膜融合性ペプチドの大規模な調製を増進するための方法であって、該方法は:
請求項8に記載の改変された抗膜融合性ペプチドを提供する工程;および
少なくとも100mg/mlの該改変されたペプチドの濃度を有する該改変されたペプチドの溶液を調製する工程、
を含む、方法。
【図1】

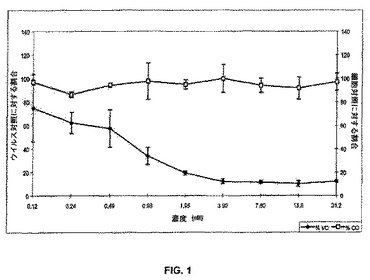
【図2】
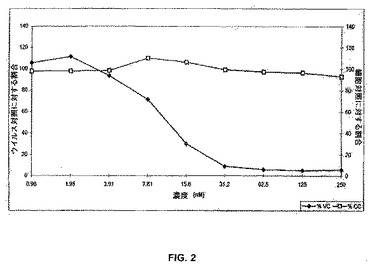
【図3】

【図4】
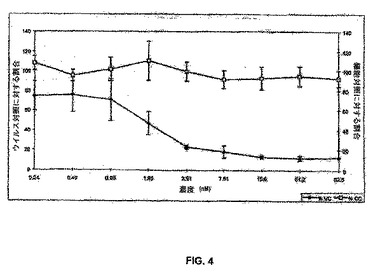
【図5】

【図6】
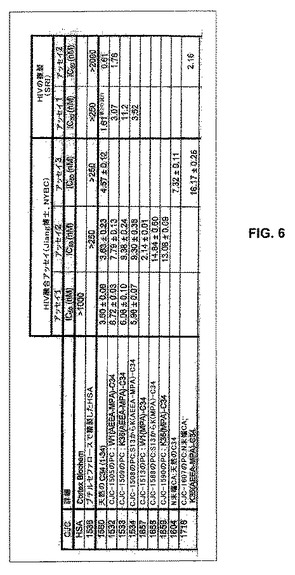
【図2】
【図3】
【図4】
【図5】
【図6】
【公表番号】特表2010−527376(P2010−527376A)
【公表日】平成22年8月12日(2010.8.12)
【国際特許分類】
【出願番号】特願2010−508621(P2010−508621)
【出願日】平成20年5月16日(2008.5.16)
【国際出願番号】PCT/US2008/064010
【国際公開番号】WO2008/144584
【国際公開日】平成20年11月27日(2008.11.27)
【出願人】(507340636)コンジュケム バイオテクノロジーズ インコーポレイテッド (18)
【Fターム(参考)】
【公表日】平成22年8月12日(2010.8.12)
【国際特許分類】
【出願日】平成20年5月16日(2008.5.16)
【国際出願番号】PCT/US2008/064010
【国際公開番号】WO2008/144584
【国際公開日】平成20年11月27日(2008.11.27)
【出願人】(507340636)コンジュケム バイオテクノロジーズ インコーポレイテッド (18)
【Fターム(参考)】
[ Back to top ]