
抗ウイルス剤
抗ウイルス剤
本発明は,キナーゼを調節することにより,特にウイルス感染の媒介に関与する宿主細胞キナーゼを阻害することにより,ウイルス,特にポックスウイルスによる感染を阻害または治療する方法を提供する。ウイルス感染を阻害しうる薬剤を選択するために,感染の間にウイルスが必要とする宿主細胞の細胞蛋白質を同定し,評価し,および分類する方法が開示される。システムバイオロジーの方法を用いることにより,到来したウイルスの細胞表面への最初の接着から,侵入,転写,複製,生合成,および子孫粒子のアセンブリまで,ウイルス/宿主細胞相互作用を調べる。この方法は,siRNAスクリーニングプラットフォームを利用し,遺伝子サイレンシングを用いて,「ウイルスインフェクトーム」,すなわち,ウイルスが感染を確立し,および感染サイクルを進行させるのに必要とする細胞蛋白質の集合をマッピングする。インフェクトームをチャート分析することにより,ウイルス感染に関与する宿主細胞蛋白質の同定によりウイルスの生物学に関する情報が提供され,ウイルスが細胞において増殖感染を確立することを防止する新規な抗ウイルス剤の開発が可能となる。

本発明は,キナーゼを調節することにより,特にウイルス感染の媒介に関与する宿主細胞キナーゼを阻害することにより,ウイルス,特にポックスウイルスによる感染を阻害または治療する方法を提供する。ウイルス感染を阻害しうる薬剤を選択するために,感染の間にウイルスが必要とする宿主細胞の細胞蛋白質を同定し,評価し,および分類する方法が開示される。システムバイオロジーの方法を用いることにより,到来したウイルスの細胞表面への最初の接着から,侵入,転写,複製,生合成,および子孫粒子のアセンブリまで,ウイルス/宿主細胞相互作用を調べる。この方法は,siRNAスクリーニングプラットフォームを利用し,遺伝子サイレンシングを用いて,「ウイルスインフェクトーム」,すなわち,ウイルスが感染を確立し,および感染サイクルを進行させるのに必要とする細胞蛋白質の集合をマッピングする。インフェクトームをチャート分析することにより,ウイルス感染に関与する宿主細胞蛋白質の同定によりウイルスの生物学に関する情報が提供され,ウイルスが細胞において増殖感染を確立することを防止する新規な抗ウイルス剤の開発が可能となる。
【発明の詳細な説明】
【技術分野】
【0001】
クロスリファレンス
本出願は,米国特許仮出願60/945,740(2007年6月22日出願,その全体は参照として本明細書に組み込まれる)に基づく優先権を主張する。
【背景技術】
【0002】
発明の背景
抗ウイルス剤は,ウイルス感染の治療に用いられる一群の医薬品である。抗ウイルス剤は,抗微生物剤の1種であり,その大部分は抗生物質,抗真菌剤および抗寄生虫剤を含む。広範囲の病原菌をカバーしうる抗菌剤とは異なり,抗ウイルス剤はスペクトルが狭く,限定された効力しか持たない傾向にある。
【0003】
抗ウイルス剤の出現は,生物の遺伝子および分子機能に関する知識が拡大して,生物医学の研究者がウイルスの構造および機能を理解できるようになったこと,新規薬剤を発見する技術が進歩したこと,および致死性のある後天性免疫不全症候群(AIDS)の流行の原因であるヒト免疫不全ウイルス(HIV)を扱うように医療専門家に圧力がかけられたことの産物である。
【0004】
季節性ヒトインフルエンザについての継続する問題および将来のパンデミックの脅威のため,インフルエンザに対する抗ウイルス剤の開発は高い優先度を有する(Fauci 2006)。ワクチン接種はなお予防の基礎であるが,抗ウイルス剤は流行およびパンデミックの可能性に対する世界的規模の戦いにおける要素を構成する(Pleshka et al.2006)。抗ウイルス剤の有用性はウイルス中の抗原の変化により影響を受けないため,抗ウイルス剤はワクチンに比べて利点を有する。このことは,出現した種に対してワクチンが入手可能となる前にこれらを用いることができることを意味する。また,これらは成立した病気に対しても有効である。予防においては,これらは感染に対して防御し,ウイルスの広がりを減少させ,免疫感作に対して有用な補完として作用する。これらの利点のため,インフルエンザパンデミックのリスクに関するいくつかの米国政府系委員会は,このウイルスに対する新規な抗ウイルス剤を開発する研究を推奨してきた。
【0005】
インフルエンザに対する現在入手可能な抗ウイルス剤は,ウイルスM2−チャネル(アマンチジン,リマンチジン)またはノイラミニダーゼ(NA)(オセルタミビルおよびザナミビル)のいずれかを阻害するものである(Pleshka et al.2006)。前者はヒトインフルエンザA株に対する予防および治療用に安全かつ有用であるが,ほとんど処方されない。NA阻害剤は最も一般的に使用されている。これらはトリH5N1を含むインフルエンザA株に対して活性があり,インフルエンザBに対しても作用する。オセルタミビルは予防および治療に用いられているが,ザナミビル(吸入用)は予防用としてはまだ認可されていない。
【0006】
現在の抗ウイルス薬剤(および一般にウイルス蛋白質を標的とする薬剤)にともなう主要な問題点は,ウイルス遺伝子中の点突然変異による耐性の出現である。M2チャネルブロッカーについては,完全耐性の出現は急速である。NA阻害剤の場合には,耐性株は出現するが,幸運なことにこれまでのところこれらは野生型と比較して適応度および病原性が低い。しかし,耐性株が発生する可能性は問題として残っている。
【0007】
ウイルスそれ自体および標的としてのその蛋白質に焦点が当てられているが,ウイルス感染に関与する宿主細胞蛋白質に干渉する新世代の抗ウイルス剤を開発することは有用である。これらの蛋白質の活性を阻害することにより,子孫ウイルスの複製および産生を阻害することが可能となるであろう。
【発明の概要】
【課題を解決するための手段】
【0008】
発明の概要
本発明は,ウイルス感染において役割を果たす宿主細胞蛋白質を同定する方法を提供する。これらの宿主細胞標的蛋白質を同定することにより,これらを標的とするウイルス感染の治療的介入のための薬剤の同定が可能となる。また,これらの宿主細胞蛋白質を調節してウイルス感染を治療および/または予防する薬剤および方法も提供される。本発明は,宿主細胞蛋白質を調節することにより,宿主細胞においてウイルス感染を阻害または低減させる薬剤を提供する。さらに,本発明は,ウイルス感染の治療に用いることができるキットを提供する。
【0009】
1つの観点においては,本発明は,ポックスウイルス感染を治療する方法を提供し,この方法は,治療を必要とする被検動物に有効量のキナーゼ調節剤を投与することを含む。1つの態様においては,被検動物はヒトである。1つの態様においては,前記マクロピノサイトーシス経路の前記阻害剤は,PAK1;DYRK3;PTK9;およびGPRK2Lからなる群より選択されるキナーゼの阻害剤である。1つの態様においては,前記キナーゼ調節剤は宿主細胞キナーゼ調節剤である。1つの態様においては,キナーゼ調節剤は,キナーゼを標的とするドミナント・ネガティブ分子,siRNA,shRNA,抗体または小分子である。1つの態様においては,キナーゼ調節剤はsiRNAである。1つの態様においては,キナーゼ調節剤はCEP−1347である。1つの態様においては,前記宿主細胞キナーゼ調節剤は宿主細胞キナーゼ阻害剤である。1つの態様においては,前記宿主細胞キナーゼ阻害剤は,PAK1;DYRK3;PTK9;およびGPRK2Lからなる群より選択されるキナーゼの阻害剤である。1つの態様においては,ポックスウイルスは天然痘ウイルスである。1つの態様においては,ポックスウイルスはワクシニアウイルスである。1つの態様においては,感染は呼吸器感染である。
【0010】
別の観点においては,本発明は,ウイルス感染を治療する方法を提供し,この方法は,治療を必要とする被検動物に有効量のマクロピノサイトーシス経路の調節剤を投与することを含む。1つの態様においては,被検動物はヒトである。1つの態様においては,マクロピノサイトーシス経路の前記調節剤は前記マクロピノサイトーシス経路の阻害剤である。1つの態様においては,前記阻害剤はキナーゼ阻害剤である。1つの態様においては,前記阻害剤は宿主細胞キナーゼ阻害剤である。1つの態様においては,前記宿主細胞キナーゼ阻害剤は,PAK1;DYRK3;PTK9;およびGPRK2Lからなる群より選択されるキナーゼの阻害剤である。1つの態様においては,阻害剤はCEP−1347である。1つの態様においては,前記ウイルスはポックスウイルスである。1つの態様においては,前記ウイルスは天然痘ウイルスである。1つの態様においては,前記ウイルスはワクシニアウイルスである。
【0011】
別の観点においては,本発明は,細胞をキナーゼ阻害剤およびウイルスと接触させ,キナーゼ阻害剤がウイルスによる細胞の感染を阻害するか否かを判定することを含む方法を提供する。1つの態様においてはキナーゼ阻害剤は,PAK1;DYRK3;PTK9;およびGPRK2Lからなる群より選択されるキナーゼを阻害する。別の態様においては,キナーゼ阻害剤は,キナーゼを標的とするドミナント・ネガティブ分子,siRNA,shRNA,抗体または小分子からなる群より選択される。別の態様においては,ウイルスはインフルエンザウイルスまたはポックスウイルス,例えばワクシニアまたは天然痘ウイルスである。別の態様においては,接触はインビトロで行われる。
【0012】
参照
本明細書において言及されるすべての刊行物,特許および特許出願は,個々の刊行物,特許および特許出願が特定的にかつ個別に本明細書に参照として取り込まれるように,本明細書に参照として取り込まれる。
【0013】
本発明の新規な特徴は,特に添付の特許請求の範囲に記載される。本発明の原理を用いる例示的態様を記載する下記の詳細な説明および添付の図面を参照することにより,本発明の特徴および利点をより理解することができる。
【図面の簡単な説明】
【0014】
【図1】図1は,成熟ビリオン侵入の間のサーフィンおよび膜摂動を示す。
【図2】図2は,p21活性化キナーゼ−1(PAK1)がMV侵入に必要であることを示す。
【図3】図3は,ワクシニアMVが細胞に侵入するためにマクロピノサイトーシスを利用することを示す。
【図4】図4は,ワクシニアMVが内在化にPSを必要とすることを示す。
【図5】図5は,PAK1の活性化はAd3エンドサイトーシスおよび感染に必要であるが,Ad5には必要ではないことを示す。
【図6】図6は,マクロピノサイトーシスを用いるAd3感染の経路を示す。
【図7】図7は,MVをHeLa細胞に加えた後のEGFR活性化を示す。
【図8】図8は,EGFR阻害剤324674(Calbiochem)がMV侵入を遮断し,これは低pH融合によりバイパスされることを示す。
【発明を実施するための形態】
【0015】
発明の詳細な説明
本発明の多くの態様が本明細書に示され記載されるが,当業者にはそのような態様は例示のためにのみ提供されていることが明らかであろう。当業者は本発明から逸脱することなく多くの改変,変更および置き換えをなすことができる。本発明の実施に際しては,本明細書に記載される本明細書の態様の種々の代替物を用いることができることが理解される。下記の特許請求の範囲は本発明の範囲を規定し,これらの特許請求の範囲の範囲内の方法および構造およびそれらの均等物もカバーすることが意図される。
【0016】
本発明の方法は,ウイルスが感染,複製および/または伝播に利用する宿主細胞遺伝子を同定することを含む。また,同定された宿主細胞遺伝子によりコードされる特異的宿主細胞蛋白質を標的とする薬剤を同定する方法も開示される。さらに,本発明は,同定された宿主細胞標的を調節するための薬剤および方法を含む。そのような薬剤および方法は,ウイルス感染の治療に適している。そのような宿主細胞標的の調節は,宿主細胞標的の活性化または阻害のいずれかを含むことができる。したがって,非ウイルス蛋白質,例えばキナーゼ等の宿主細胞蛋白質の活性を調節,例えば阻害する化合物を,抗ウイルス医薬製剤として用いる。
【0017】
1つの態様においては,本発明の方法は,被検動物,例えば多数のウイルスのいずれかによるヒトの感染を阻害する抗ウイルス剤の開発に用いることができる。1つの態様においては,本発明の方法は,呼吸器ウイルスによる宿主の感染を阻害する抗ウイルス剤の開発に用いられる。呼吸器ウイルスは,最も一般的には,浮遊する飛沫または鼻汁により伝染し,広範な種類の病気につながりうる。呼吸器ウイルスとしては,呼吸器合胞体ウイルス(RSV),インフルエンザウイルス,コロナウイルス,例えばSARS,アデノウイルス,パラインフルエンザウイルスおよびライノウイルスが挙げられる。
【0018】
I.ウイルス
1つの態様においてはウイルス,例えば,ポックスウイルス,アデノウイルスまたは本明細書に記載される任意のウイルスが感染または複製に必要とする宿主細胞蛋白質が同定される。アデノウイルスは,最も一般的には呼吸器疾患を引き起こす。アデノウイルス感染により引き起こされる呼吸器疾患の症状は,一般的な風邪症候群から肺炎,クループ,および気管支炎まで様々である。免疫系に障害をもつ患者は特にアデノウイルス感染の重篤な合併症を起こしやすい。急性呼吸器疾患(ARD)は,第二次世界大戦の間に軍隊入隊者の間で最初に認められ,混雑およびストレスの状態の中でアデノウイルス感染により引き起こされうる。アデノウイルスは,中程度のサイズ(90−100nm)の,二本鎖DNAを含むエンベロープを有さない正二十面体ウイルスである。ヒト感染を引き起こしうる49種の免疫学的に区別しうるタイプ(6種の亜属:A−F)がある。アデノウイルスは,化学的または物理的作用および不利なpH条件に対して非常に安定であり,身体の外で長期間生存することが可能である。ある種のアデノウイルス,例えばAD2およびAd5(C種)は,感染性侵入にクラスリン媒介性エンドサイトーシスおよびマクロピノサイトーシスを利用する。他のアデノウイルス,例えばAd3(B種)は,感染性侵入にダイナミン依存性エンドサイトーシスおよびマクロピノサイトーシスを利用する。
【0019】
1つの態様においては,ポックスウイルスが感染または複製に必要とする宿主細胞蛋白質が同定される。ポックスウイルスは,一般にエンベロープを有する。このウイルスは,約200nmx300nmのサイズを有する。DNAは直鎖状の二本鎖である。ウイルスファミリーPoxviridaeは,オルトポックスウイルス属を含み,これは天然痘の原因であるVariolavera種を含む。このウイルスには2つの形態,すなわち大痘瘡と小痘瘡がある。天然痘は,典型的には,通常は感染したヒトの呼吸器系から浮遊した天然痘ウイルスの吸入によりヒトからヒトに伝染する。したがって,これらのウイルスの阻害はバイオテロに対する防御として有用である。ワクシニアもまた感染性ポックスウイルスである。
【0020】
1つの態様においては,呼吸器合胞体ウイルス(RSV)が感染または複製に必要とする宿主細胞蛋白質が同定される。RSVは新生児および1歳未満の小児における細気管支炎および肺炎の最も一般的な原因である。最も頻繁には,病気は発熱,鼻水,咳,および時には喘鳴から始まる。最初のRSV感染の間,25%から40%の新生児および年少の小児は細気管支炎または肺炎の兆候または症状を有しており,0.5%から2%は入院を必要とする。ほとんどの小児は8から15日間で病気から回復する。RSV感染で入院する小児の大部分は,生後6か月未満である。RSVはまた一生の間に反復感染を引き起こし,通常は中程度から重症の風邪様症状と伴う。しかし,いずれの年齢の間でも,特に老齢者および心臓,肺または免疫系に障害を有する者の間で,重症の下部呼吸管疾患を引き起こしうる。RSVはネガティブセンスのエンベロープRNAウイルスである。ビリオンは様々な形状およびサイズであり(平均直径は120−300nm),環境中で不安定であり(環境表面上では数時間しか生存しない),石けん水および消毒剤で容易に不活性化する。
【0021】
1つの態様においては,ヒトパラインフルエンザウイルス(HPIV)が感染または複製に必要とする宿主細胞蛋白質が同定される。HPIVは,呼吸器合胞体ウイルス(RSV)についで2番目に一般的な幼い小児における下部呼吸管疾患の原因である。RSVと同様に,HPIVは,一生の間に反復感染を引き起こし,通常は上部呼吸管疾患(例えば,風邪および/または咽喉炎)として現れる。HPIVはまた,特に老齢者および免疫系の障害をもつ患者の間で,反復感染により重篤な下部呼吸管疾患(例えば,肺炎,気管支炎,および細気管支炎)を引き起こしうる。4種のHPIVのそれぞれは,異なる臨床的および疫学的特徴を有する。HPIV−1およびHPIV−2の最も独特の臨床的特徴はクループ(すなわち,喉頭気管気管支炎)である。HPIV−1は小児のクループの主な原因であり,一方HPIV−2はあまり頻繁に検出されない。HPIV−1および−2の両方とも他の上部および下部呼吸管疾患を引き起こしうる。HPIV−3はしばしば細気管支炎および肺炎に関連している。HPIV−4はあまり頻繁に検出されず,これはおそらく重篤な疾患を引き起こさないためであろう。HPIVのインキュベーション期間は一般に1−7日間である。HPIVは,ネガティブセンスの一本鎖RNAウイルスであり,その表面にF蛋白質およびヘマグルチニン−ノイラミニダーゼ糖蛋白質の"スパイク"を持つ。HPIVには4つの血清型(1から4)および2つのサブタイプ(4aおよび4b)がある。ビリオンは様々なサイズ(平均直径は150から300nm)および形状であり,環境中で不安定であり(環境表面上で数時間の生存),石けん水により容易に不活性化される。
【0022】
1つの態様においては,コロナウイルスが感染または複製に必要とする宿主細胞蛋白質が同定される。コロナウイルスはCoronaviridaeファミリーに属する動物ウイルスの属である。コロナウイルスは,エンベロープを持つウイルスであり,ポジティブセンスの一本鎖RNAゲノムおよびらせん対称を有する。コロナウイルスのゲノムサイズは約16から31キロベースの範囲であり,RNAウイルスとしては異常に大きい。"コロナウイルス"との名称は,ラテン語のcorona(王冠を意味する)に由来する。これは,ウイルスエンベロープが電子顕微鏡下で小さな球状構造の特徴的な環の王冠を持つように見えるためである。この形態学は,実際にウイルスのスパイクペプロマーにより形成させており,これはウイルスの表面に存在して宿主指向性を決定する蛋白質である。コロナウイルスはニドウイルス目に分類され,この目のすべてのウイルスは感染の間に3’末端を共有する入れ子集合のサブゲノムmRNAを生成するため,巣を意味するラテン語のnidusから名付けられている。すべてのコロナウイルスの全体構造に寄与する蛋白質は,スパイク,エンベロープ,膜およびヌクレオカプシドである。SARSの特定の場合においては,S上の規定されたレセプター結合ドメインがウイルスのその細胞性レセプターであるアンジオテンシン変換酵素2への結合を媒介する。
【0023】
1つの態様においては,ライノウイルスが感染または複製に必要とする宿主細胞蛋白質が同定される。ライノウイルス("鼻"を意味するギリシャ語のrhin−に由来)はPicornaviridaeファミリーのウイルスの属である。ライノウイルスはヒトに感染性をもつ最も一般的なウイルスであり,一般的な風邪の原因ウイルスである。風邪の症状を引き起こすウイルス血清型は105を越えており,ライノウイルスは全症例の約50%の原因である。ライノウイルスは7.2から8.5kbの長さの一本鎖のポジティブセンスRNAゲノムを有する。ゲノムの5’末端にはウイルスによりコードされる蛋白質があり,哺乳動物のmRNAと同様に,3’ポリAテールが存在する。構造蛋白質はゲノムの5’領域にコードされており,末端は非構造性である。これはすべてのピコルナウイルスについて同じである。ウイルス粒子それ自体は,エンベロープをもたず,二十面体の構造を有する。
【0024】
1つの態様においては,インフルエンザウイルスが感染または複製に必要とする宿主細胞蛋白質が同定される。インフルエンザウイルスはウイルスのオルトミクソウイルスファミリーに属する。このファミリーには,トゴトウイルスおよびドーリウイルスも含まれる。ヒトおよび他の種に感染するインフルエンザウイルスとしては,いくつかの型およびサブタイプが知られている。A型インフルエンザ型インフルエンザウイルスは,ヒト,トリ,ブタ,ウマ,アザラシおよび他の動物に感染するが,野生の鳥類がこれらのウイルスの天然の宿主である。A型インフルエンザウイルスはいくつかのサブタイプに分類され,ウイルスの表面上の2つの蛋白質,すなわちヘマグルチニン(HA)およびノイラミニダーゼ(NA)に基づいて命名されている。例えば,“H7N2ウイルス”はHA7蛋白質およびNA2蛋白質を有するインフルエンザAサブタイプを表す。同様に,“H5N1”ウイルスはHA5蛋白質およびNA1蛋白質を有する。16種のHAサブタイプおよび9種のNAサブタイプが知られている。HAとNA蛋白質との多くの異なる組み合わせが可能である。現在,いくつかのインフルエンザAサブタイプ(すなわち,H1N1,H1N2,およびH3N2)のみがヒトの間で一般に流行している。他のサブタイプは主として他の動物種において見いだされる。例えば,H7N7およびH3N8ウイルスはウマに病気を引き起こし,またH3N8はイヌに病気を引き起こすことが最近示された(http://www.cdc.gov/flu/avian/gen-info/flu-viruses.htm)。
【0025】
インフルエンザ感染に関与する宿主細胞蛋白質を標的とする抗ウイルス剤を用いて,高リスク群(病院組織,高齢者ケア施設,免疫が抑制されている個体)をケースバイケースで保護することができる。抗ウイルス剤の可能性のある用途は,トリH5N1により引き起こされるかインフルエンザウイルスの他の株により引き起こされるかにかかわらず,将来のパンデミックの拡大および重篤性を制限することである。サブタイプH5およびH7のトリA型インフルエンザウイルス,例えばH5N1,H7N7,およびH7N3ウイルスは,高い病原性と関連づけられており,これらのウイルスによるヒトの感染は,軽症(H7N3,H7N7)から重症かつ致命的な疾患(H7N7,H5N1)まで様々である。病原性の低いウイルスの感染によるヒトの病気は,非常に軽い症状(例えば結膜炎)からインフルエンザ様の病気まで確認されている。ヒトに感染したことがある病原性の低いウイルスの例には,H7N7,H9N2,およびH7N2が含まれる。(http://www.cdc.gov/flu/avian/gen-info/flu-viruses.htm)。
【0026】
B型インフルエンザウイルスは通常はヒトに見いだされるが,アザラシにも感染しうる。A型インフルエンザウイルスとは異なり,これらのウイルスはサブタイプにしたがって分類されていない。B型インフルエンザウイルスはヒトの間で罹患および死亡を引き起こしうるが,一般にA型インフルエンザウイルスと比べて重大な流行を伴わない。B型インフルエンザウイルスはヒトでの流行を引き起こしうるが,パンデミクスを引き起こしたことはない(http://www.cdc.gov/flu/avian/gen-info/flu-viruses.htm)。
【0027】
C型インフルエンザウイルスウイルスはヒトに軽い病気を引き起こし,流行またはパンデミックを引き起こさない。これらのウイルスはイヌおよびブタにも感染することができる。これらのウイルスはサブタイプにしたがって分類されていない(http://www.cdc.gov/flu/avian/gen-info/flu-viruses.htm)。
【0028】
インフルエンザウイルスは,細胞表面レセプターの特異性および細胞指向性に関して互いに異なるが,共通する侵入経路を利用する。これらの経路を図表化し,ウイルスインフルエンザの伝染,侵入,複製,生合成,アセンブリ,または退出に関与する宿主細胞蛋白質を同定することにより,現存する,および出現するインフルエンザ種に対する一般的薬剤の開発が可能となる。薬剤はまた,同様の経路を利用する非関連ウイルスに対しても有用性を示すであろう。例えば,薬剤は,インフルエンザウイルスに加えて多数の異なるウイルスに対して気道上皮細胞を保護することができる。
【0029】
本明細書に記載される方法は,任意のウイルス,例えば,アベルソン白血病ウイルス,アベルソンネズミ白血病ウイルス,アベルソンウイルス,急性喉頭気管気管支炎ウイルス,アデレード川ウイルス,アデノ随伴ウイルス群,アデノウイルス,アフリカウマ病ウイルス,アフリカブタ熱ウイルス,AIDSウイルス,アリューシャンミンク病パルボウイルス,アルファレトロウイルス,アルファウイルス,ALV関連ウイルス,アマパリウイルス,アフタウイルス,アクアレオウイルス,アルボウイルス,アルボウイルスC,アルボウイルス群A,アルボウイルス群B,アレナウイルス群,アルゼンチン出血熱ウイルス,アルゼンチン出血熱ウイルス,アーテリウイルス,アストロウイルス,アテリネヘルペスウイルス群,オジェッキー病ウイルス,オーラウイルス,オースダック病ウイルス,オーストラリアコウモリリッサウイルス,トリアデノウイルス,トリ赤芽球症ウイルス,トリ感染性気管支炎ウイルス,トリ白血病ウイルス,トリ白血病ウイルス,トリリンパ腫症ウイルス,トリ骨髄芽球症ウイルス,トリパラミクソウイルス,トリ気脳炎ウイルス,トリ細網内皮症ウイルス,トリ肉腫ウイルス,トリC型レトロウイルス群,トリヘパドナウイルス,トリ痘ウイルス,Bウイルス,B19ウイルス,ババンキウイルス,ヒヒヘルペスウイルス,バキュロウイルス,バーマー森林ウイルス,ベラルウイルス,ベリマーウイルス,ベータレトロウイルス,バーナウイルス,ビトナーウイルス,BKウイルス,ブラッククリーク運河ウイルス,ブルータングウイルス,ボリビア出血熱ウイルス,ボマ病ウイルス,ヒツジボーダー病ウイルス,ボルナウイルス,ウシアルファヘルペスウイルス1,ウシアルファヘルペスウイルス2,ウシコロナウイルス,ウシ一過性熱ウイルス,ウシ免疫不全ウイルス,ウシ白血病ウイルス,ウシ白血病ウイルス,ウシ乳頭炎ウイルス,ウシパピローマウイルス,ウシ丘疹口内炎ウイルス,ウシパルボウイルス,ウシ合胞体ウイルス,ウシC型オンコウイルス,ウシウイルス下痢ウイルス,バギークリークウイルス,弾丸形ウイルス群,ブニヤンウエラウイルス超群,ブニヤウイルス,バーキットリンパ腫ウイルス,ブワンバ熱,CAウイルス,カリシウイルス,カリフォルニア脳炎ウイルス,ラクダ痘ウイルス,カナリ痘ウイルス,イヌヘルペスウイルス,イヌコロナウイルス,イヌジステンパーウイルス,イヌヘルペスウイルス,イヌ微小ウイルス,イヌパルボウイルス,カノデルガディトウイルス,ヤギ関節炎ウイルス,ヤギ脳炎ウイルス,ヤギヘルペスウイルス,ヤギ痘ウイルス,カルジオウイルス,テンジクネズミヘルペスウイルス1,オナガザルヘルペスウイルス1,オナガザルヘルペスウイルス1,オナガザルヘルペスウイルス2,チャンジプラウイルス,チャンギノラウイルス,ブチナマズウイルス,シャルルビルウイルス,ニワトリ痘ウイルス,チクングンヤウイルス,チンパンジーヘルペスウイルス,チャブレオウイルス,シロザケウイルス,コカルウイルス,ギンザケレオウイルス,媾疹ウイルス,コロラドダニ熱ウイルス,コルチウイルス,コロンビアSKウイルス,風邪ウイルス,感染性膿瘡ウイルス,感染性膿疱性皮膚炎ウイルス,コロナウイルス,コリパルタウイルス,鼻風邪ウイルス,牛痘ウイルス,コクサッキーウイルス,CPV(細胞質多角体病ウイルス),コオロギ麻痺ウイルス,クリミア−コンゴ出血熱ウイルス,クループ関連ウイルス,クリプトウイルス,サイポウイルス,サイトメガロウイルス,サイトメガロウイルス群,細胞質多角体病ウイルス,シカパピローマウイルス,デルタレトロウイルス,デングウイルス,デンソウイルス,デペンドウイルス,ドーリウイルス,ディプロルナウイルス,ショウジョウバエCウイルス,カモB型肝炎ウイルス,カモ肝炎ウイルス1,カモ肝炎ウイルス2,デュオウイルス,デュベンヘージウイルス,変形ウイングウイルスDWV,東部ウマ脳炎ウイルス,東部ウマ脳脊髄炎ウイルス,EBウイルス,エボラウイルス,エボラ様ウイルス,エコーウイルス,エコーウイルス,エコーウイルス10,エコーウイルス28,エコーウイルス9,欠肢症ウイルス,EEEウイルス,EIAウイルス,EIAウイルス,脳炎ウイルス,脳心筋炎群ウイルス,脳心筋炎ウイルス,エンテロウイルス,酵素上昇ウイルス,酵素上昇ウイルス(LDH),流行性出血熱ウイルス,流行性出血病ウイルス,エプスタインバーウイルス,ウマ科アルファヘルペスウイルス1,ウマ科アルファヘルペスウイルス4,ウマ科ヘルペスウイルス2,ウマ流産ウイルス,ウマ動脈炎ウイルス,ウマ脳症ウイルス,ウマ感染性貧血ウイルス,ウマ麻疹ウイルス,ウマ鼻肺炎ウイルス,ウマライノウイルス,ユーベナングウイルス,ヨーロッパエルクパピローマウイルス,ヨーロッパブタ熱ウイルス,エバーグレイズウイルス,ヤッチウイルス,ネコヘルペスウイルス1,ネコカリシウイルス,ネコ線維肉腫ウイルス,ネコヘルペスウイルス,ネコ免疫不全ウイルス,ネコ感染性腹膜炎ウイルス,ネコ白血病/肉腫ウイルス,ネコ白血病ウイルス,ネコ汎白血球減少症ウイルス,ネコパルボウイルス,ネコ肉腫ウイルス,ネコ合胞体ウイルス,フィロウイルス,フランダーズウイルス,フラビウイルス,口蹄疫ウイルス,フォートモーガンウイルス,フォーコーナーズハンタウイルス,家禽アデノウイルス1,家禽痘ウイルス,フレンドウイルス,ガンマレトロウイルス,GB肝炎ウイルス,GBウイルス,ドイツ麻疹ウイルス,ゲタウイルス,テナガザル白血病ウイルス,腺熱ウイルス,ヤギ痘ウイルス,ゴールデンシンナーウイルス,ゴノメタウイルス,ガチョウパルボウイルス,顆粒病ウイルス,グロスウイルス,ジリスB型肝炎ウイルス,群Aアルボウイルス,グアナリトウイルス,テンジクネズミサイトメガロウイルス,テンジクネズミC型ウイルス,ハンターンウイルス,ハンタウイルス,ホンビノスガイレオウイルス,野ウサギ線維腫ウイルス,HCMV(ヒトサイトメガロウイルス),赤血球吸着ウイルス2,日本の赤血球凝集ウイルス,出血熱ウイルス,ヘンドラウイルス,ヘニパウイルス,肝炎ウイルス,A型肝炎ウイルス,B型肝炎ウイルス群,C型肝炎ウイルス,D型肝炎ウイルス,デルタ肝炎ウイルス,E型肝炎ウイルス,F型肝炎ウイルス,G型肝炎ウイルス,非A非B肝炎ウイルス,肝炎ウイルス,肝炎ウイルス(非ヒト),肝脳脊髄炎レオウイルス3,肝ウイルス,ヘロンB型肝炎ウイルス,ヘルペスBウイルス,ヘルペス単純ウイルス,ヘルペス単純ウイルス1,ヘルペス単純ウイルス2,ヘルペスウイルス,ヘルペスウイルス7,ヘルペスウイルスクモザル,ヘルペスウイルスヒト,ヘルペスウイルス感染,ヘルペスウイルスリスザル,ヘルペスウイルスブタ,ヘルペスウイルス水痘,ハイランドJウイルス,ヒラメラブドウイルス,ブタコレラウイルス,ヒトアデノウイルス2,ヒトアルファヘルペスウイルス1,ヒトアルファヘルペスウイルス2,ヒトアルファヘルペスウイルス3,ヒトBリンパ球指向性ウイルス,ヒトベータヘルペスウイルス5,ヒトコロナウイルス,ヒトサイトメガロウイルス群,ヒト泡沫状ウイルス,ヒトガンマヘルペスウイルス4,ヒトガンマヘルペスウイルス6,ヒトA型肝炎ウイルス,ヒトヘルペスウイルス1群,ヒトヘルペスウイルス2群,ヒトヘルペスウイルス3群,ヒトヘルペスウイルス4群,ヒトヘルペスウイルス6,ヒトヘルペスウイルス8,ヒト免疫不全ウイルス,ヒト免疫不全ウイルス1,ヒト免疫不全ウイルス2,ヒトパピローマウイルス,ヒトT細胞白血病ウイルス,ヒトT細胞白血病ウイルスI,ヒトT細胞白血病ウイルスII,ヒトT細胞白血病ウイルスIII,ヒトT細胞リンパ腫ウイルスI,ヒトT細胞リンパ腫ウイルスII,1型ヒトT細胞リンパ球指向性ウイルス,2型ヒトT細胞リンパ球指向性ウイルス,ヒトTリンパ球指向性ウイルスI,ヒトTリンパ球指向性ウイルスII,ヒトTリンパ球指向性ウイルスIII,生痕ウイルス,新生児胃腸炎ウイルス,感染性ウシ鼻気管炎ウイルス,感染性造血壊死ウイルス,感染性膵臓壊死ウイルス,インフルエンザウイルスA,インフルエンザウイルスB,インフルエンザウイルスC,インフルエンザウイルスD,インフルエンザウイルスpr8,昆虫虹色ウイルス,昆虫ウイルス,イリドウイルス,日本Bウイルス,日本脳炎ウイルス,JCウイルス,フニンウイルス,カポジ肉腫関連ヘルペスウイルス,ケメロボウイルス,キラムラットウイルス,クラマスウイルス,コロンゴウイルス,韓国出血熱ウイルス,クンバウイルス,キサヌール森林病ウイルス,キジラガーシュウイルス,ラクロスウイルス,乳酸デヒドロゲナーゼ上昇ウイルス,乳酸デヒドロゲナーゼウイルス,ラゴスコウモリウイルス,ラングールウイルス,ウサギパルボウイルス,ラッサ熱ウイルス,ラッサウイルス,潜在的ラットウイルス,LCMウイルス,リーキーウイルス,レンチウイルス,ウサギ痘ウイルス,白血病ウイルス,白血病ウイルス,ランピースキン病ウイルス,リンパ節症関連ウイルス,リンパ球クリプトウイルス,リンパ球性脈絡髄膜炎ウイルス,リンパ増殖ウイルス群,マシュポウイルス,マッドイッチウイルス,哺乳動物B型オンコウイルス群,哺乳動物B型レトロウイルス,哺乳動物C型レトロウイルス群,哺乳動物D型レトロウイルス,乳癌ウイルス,マプエラウイルス,マールブルグウイルス,マールブルグ様ウイルス,メイソンファイザーサルウイルス,マストアデノウイルス,マヤロウイルス,MEウイルス,麻疹ウイルス,メナングルウイルス,メンゴウイルス,メンゴウイルス,ミデルブルグウイルス,乳牛結節ウイルス,ミンク腸炎ウイルス,マウスの微小ウイルス,MLV関連ウイルス,MMウイルス,モコラウイルス,軟体動物痘ウイルス,伝染性軟属腫ウイルス,サルBウイルス,サル痘ウイルス,モノネガウイルス,麻疹ウイルス,マウントエルゴンコウモリウイルス,マウスサイトメガロウイルス,マウス脳脊髄炎ウイルス,マウス肝炎ウイルス,マウスKウイルス,マウス白血病ウイルス,マウス乳癌ウイルス,マウス微小ウイルス,マウス肺炎ウイルス,マウスポリオウイルス,マウスポリオーマウイルス,マウス肉腫ウイルス,マウス痘ウイルス,モザンビークウイルス,ムカンボウイルス,粘膜病ウイルス,ムンプスウイルス,ネズミベータヘルペスウイルス1,ネズミサイトメガロウイルス2,ネズミサイトメガロウイルス群,ネズミ脳脊髄炎ウイルス,ネズミ肝炎ウイルス,ネズミ白血病ウイルス,ネズミ結節誘発ウイルス,ネズミポリオーマウイルス,ネズミ肉腫ウイルス,ムーロメガロウイルス,マリーバレー脳炎ウイルス,粘液腫ウイルス,ミクソウイルス,多形性ミクソウイルス,ミクソウイルスパロチチディス,ナイロビヒツジ病ウイルス,ナイロウイルス,ナニルナウイルス,ナリバウイルス,デュモウイルス,ニースリングウイルス,ネルソン湾ウイルス,神経指向性ウイルス,新世界アレナウイルス,新生児肺炎ウイルス,ニューカッスル病ウイルス,ニパウイルス,非細胞変性ウイルス,ノーウォークウイルス,核多角体病ウイルス(NPV),ニップルネックウイルス,オニョンニョンウイルス,オッケルボウイルス,発癌性ウイルス,発癌性ウイルス様粒子,オンコルナウイルス,オルビウイルス,伝染性膿疱性皮膚炎ウイルス,オロポーシェウイルス,オルトヘパディナウイルス,オルトミクソウイルス,オルトポックスウイルス,オルトレオウイルス,オロンゴ,ヒツジパピローマウイルス,ウシカタル熱ウイルス,ヨザルヘルペスウイルス,パリアンウイルス,パピローマウイルス,パピローマウイルスシルビラギ,パポバウイルス,パラインフルエンザウイルス,パラインフルエンザウイルス1型,パラインフルエンザウイルス2型,パラインフルエンザウイルス3型,パラインフルエンザウイルス4型,パラミクソウイルス,パラポックスウイルス,パラワクシニアウイルス,パルボウイルス,パルボウイルスB19,パルボウイルス群,ペスチウイルス,静脈ウイルス,アザラシジステンパーウイルス,ピコディナウイルス,ピコルナウイルス,ブタサイトメガロウイルス,ハト痘ウイルス,ピリーウイルス,ピクサナウイルス,
マウスの肺炎ウイルス,肺炎ウイルス,ポリオウイルス,ポリオウイルス,ポリディナウイルス,多角体病ウイルス,ポリオーマウイルス,ポリオーマウイルス,ウシポリオーマウイルス,オナガザルポリオーマウイルス,ヒトポリオーマウイルス2,ポリオーマウイルスマッカカエ1,ネズミポリオーマウイルス1,ネズミポリオーマウイルス2,ヒヒポリオーマウイルス1,ヒヒポリオーマウイルス2,ポリオーマウイルスシルビラギ,ポンギンヘルペスウイルス1,ブタ流行性下痢ウイルス,ブタ赤血球凝集性脳脊髄炎ウイルス,ブタパルボウイルス,ブタ伝染性胃腸炎ウイルス,ブタC型ウイルス,ポックスウイルス,ポックスウイルス,天然痘ポックスウイルス,プロスペクトヒルウイルス,プロウイルス,偽牛痘ウイルス,仮性狂犬病ウイルス,オウム痘ウイルス,ウズラ痘ウイルス,ウサギ線維腫ウイルス,ウサギ腎臓空胞化ウイルス,ウサギパピローマウイルス,狂犬病ウイルス,アライグマパルボウイルス,アライグマ痘ウイルス,ラニケートウイルス,ラットサイトメガロウイルス,ラットパルボウイルス,ラットウイルス,ラウシャーウイルス,組換えワクシニアウイルス,組換えウイルス,レオウイルス,レオウイルス1,レオウイルス2,レオウイルス3,爬虫類C型ウイルス,呼吸器感染ウイルス,呼吸器合胞体ウイルス,呼吸器ウイルス,細網内皮症ウイルス,ラブドウイルス,ラブドウイルスカルピア,ラジノウイルス,ライノウイルス,リジディオウイルス,リフトバレー熱ウイルス,ライリーウイルス,牛疫ウイルス,RNA腫瘍ウイルス,ロス川ウイルス,ロタウイルス,ルージョールウイルス,ラウス肉腫ウイルス,風疹ウイルス,麻疹ウイルス,ルビウイルス,ロシア秋脳炎ウイルス,SA11サルウイルス,SA2ウイルス,サビアウイルス,サギヤマウイルス,サイミリンヘルペスウイルス1,唾液腺ウイルス,サシチョウバエ熱ウイルス群,サンジンバウイルス,SARSウイルス,SDAV(唾液腺涙腺炎ウイルス),アザラシ痘ウイルス,セムリキ森林熱ウイルス,ソウルウイルス,ヒツジ痘ウイルス,ショープ線維腫ウイルス,ショープパピローマウイルス,サル泡沫状ウイルス,サルA型肝炎ウイルス,サルヒト免疫不全ウイルス,サル免疫不全ウイルス,サルパラインフルエンザウイルス,サルT細胞リンパ球指向性ウイルス,サルウイルス,サルウイルス40,シンプレックスウイルス,シンモンブレウイルス,シンドビスウイルス,天然痘ウイルス,南アメリカ出血熱ウイルス,スズメ痘ウイルス,スプマウイルス,リス線維腫ウイルス,リスザルレトロウイルス,SSV1ウイルス群,STLV(サルTリンパ球指向性ウイルス)I型,STLV(サルTリンパ球指向性ウイルス)II型,STLV(サルTリンパ球指向性ウイルス)III型,口内炎丘疹ウイルス,下顎ウイルス,イノシシアルファヘルペスウイルス1,ブタヘルペスウイルス2,スイポックスウイルス,沼地熱ウイルス,豚痘ウイルス,スイスマウス白血病ウイルス,TACウイルス,タカリベウイルス群,タカリベウイルス,タナポックスウイルス,タテラポックスウイルス,テンチレオウイルス,タイラー脳脊髄炎ウイルス,タイラーウイルス,トゴトウイルス,トッタパレイヤンウイルス,ダニ媒介性脳炎ウイルス,チオマンウイルス,トガウイルス,トロウイルス,腫瘍ウイルス,ツパイアウイルス,シチメンチョウ鼻気管炎ウイルス,シチメンチョウ痘ウイルス,C型レトロウイルス,D型オンコウイルス,D型レトロウイルス群,潰瘍性疾患ラブドウイルス,ウナウイルス,ウクニエミウイルス群,ワクシニアウイルス,空胞化ウイルス,水痘帯状疱疹ウイルス,水痘ウイルス,バリコラウイルス,大痘瘡ウイルス,天然痘ウイルス,バシンギシュー病ウイルス,VEEウイルス,ベネズエラウマ脳炎ウイルス,ベネズエラウマ脳脊髄炎ウイルス,ベネズエラ出血熱ウイルス,水疱性口内炎ウイルス,小胞性ウイルス,ビリュイスクウイルス,毒ヘビレトロウイルス,ウイルス出血性敗血症ウイルス,ビスナマエディウイルス,ビスナウイルス,ハタネズミ痘ウイルス,VSV(水疱性口内炎ウイルス),ウォーラルウイルス,ウォリゴウイルス,いぼウイルス,WEEウイルス,西ナイルウイルス,西部ウマ脳炎ウイルス,西部ウマ脳脊髄炎ウイルス,ワタロアウイルス,冬季嘔吐症ウイルス,ウッドチャックB型肝炎ウイルス,ウーリーモンキー肉腫ウイルス,創傷腫瘍ウイルス,WRSVウイルス,ヤバサル腫瘍ウイルス,ヤバウイルス,ヤタポックスウイルス,黄熱ウイルス,およびヤグボダノバックウイルスにより引き起こされる感染の治療用の薬剤の開発および/または同定に有用である。1つの態様においては,各ウイルスについて,ウイルス感染の特定の段階,例えば細胞侵入または複製サイクルの間にウイルス感染に関与する宿主細胞遺伝子の一覧を含むインフェクトームが作成される。
【0030】
II.ウイルス感染経路
本明細書に開示される宿主細胞標的は,好ましくはウイルス複製および/または感染経路において役割を果たす。そのような宿主細胞標的を標的とすることにより,ウイルスの複製および/または感染経路を調節することができる。好ましい態様においては,同定された宿主細胞標的は,適当な薬剤を用いて直接的にまたは間接的に調節される。そのような適当な薬剤としては,小分子治療剤,蛋白質治療剤,または核酸治療剤が挙げられる。そのような宿主細胞標的の調節はまた,宿主細胞標的の上流または下流のシグナリング経路の物質を標的とすることにより行うこともできる。
【0031】
他のウイルスと同様に,インフルエンザウイルスの複製には6つの相が含まれる;伝染,侵入,複製,生合成,アセンブリ,および退出。侵入は,エンドサイトーシスにより起こり,複製およびvRNPアセンブリは核で生じ,ウイルスは細胞膜から発芽する。感染した患者においては,ウイルスは気道上皮細胞を標的とする。好ましくは,本明細書に記載される方法においては,そのような経路に関与する少なくとも1つの宿主細胞標的が調節される。
【0032】
あるウイルスについては,宿主細胞の感染の間に関与する段階の解明において大きな進歩があった。例えば,1980年代初期に開始された実験では,インフルエンザウイルスは,他のウイルス,例えばアルファ−およびラブドウイルスと共有する要素を用いる段階的なエンドサイトーシスの侵入プログラムにしたたうことが示された(Marsh and Helenius 1989;Whittaker 2006)。段階は次のとおりである:1)細胞表面上のシアル酸含有複合糖質レセプターへの最初の付着;2)ウイルス粒子により誘導されるシグナリング;3)クラスリン依存性およびクラスリン非依存性の細胞メカニズムによるエンドサイトーシス;4)後期エンドソームからの酸誘導性,ヘマグルチニン(HA)媒介性浸透;5)酸により活性化されるM2およびマトリクス蛋白質(M1)依存性のカプシドの被覆の除去;および,6)vRNPのサイトゾル間輸送および核内輸送。これらの工程は,宿主細胞から,ソーティングレセプター,ベシクル形成機構,キナーゼ媒介性制御,オルガネラの酸性化,およびおそらくは細胞骨格の活性の形での支援に依存する。
【0033】
インフルエンザの細胞表面への付着は,HA1サブユニットが末端シアル酸残基をもつオリゴ糖成分を有する細胞表面の糖蛋白質および糖脂質に結合することにより生ずる(Skehel and Wiley 2000)。シアル酸と次の糖を連結する結合は種特異性に寄与する。H5N1等のトリの種は,a−(2,3)−結合を好み,ヒト種はa−(2,6)−結合を好む(Matrosovich 2006)。上皮細胞においては,結合は頂端表面上の微絨毛に優先的に生じ,エンドサイトーシスはこれらの拡大の基部において生ずる(Matlin 1982)。レセプターの結合が細胞を侵襲用に用意するシグナルを誘導するか否かはまだ明らかではないが,おそらくそうであろう。これは,蛋白質キナーゼCの活性化およびホスファチジルイノシトール−3−リン酸(PI3P)の合成が有効な侵入に必要であるからである(Sieczkarski et al.2003;Whittaker 2006)。
【0034】
エンドサイトーシスの内在化は,結合から数分以内に生ずる(Matlin 1982;Yoshimura and Ohnishi 1984)。組織培養細胞においては,インフルエンザウイルスは,3つの異なるタイプの細胞プロセスを利用する:1)予め存在するクラスリンにより被覆されたくぼみ,2)ウイルスにより誘導されるクラスリンにより被覆されたくぼみ,および3)見える被覆をもたないベシクル中のエンドサイトーシス(Matlin 1982;Sieczkarski and Whittaker 2002;Rust et al.2004),結果を参照)。蛍光ウイルスを用いるビデオ顕微鏡は,ウイルス粒子が細胞周辺でアクチン媒介性の急速な動きを示した後,微小管により媒介されるマイナス末端に向けて細胞の核周辺部へと輸送されることを示した。生きた細胞のイメージングは,ウイルス粒子が最初に移動性の周辺初期エンドソームの小集団に入り,これがウイルス粒子を細胞質中のより深い位置に運んだ後,浸透が生ずることを示した(Lakadamyali et al.2003;Rust et al.2004)。エンドサイトーシスのプロセスは,蛋白質キナーゼおよび脂質キナーゼ,プロテアソーム,ならびにRabsおよびユビキチン依存性輸送因子により制御されている(Khor et al.2003;Whittaker 2006)。
【0035】
膜浸透段階は,三量体の準安定性HAの低pH−媒介性活性化,およびこのI型ウイルスF蛋白質の膜融合可能コンフォメーションへの変換により媒介される(Maeda et al.1981;White et al.1982)。これは内在化の約16分後に起こり,pH閾値は株によって様々であり,5.0−5.6の範囲である。標的膜は中期または後期エンドソームの限定膜である。融合のメカニズムはよく研究されている(Kielian and Rey 2006)。さらに,融合それ自体は,脂質二重膜以外および機能的酸性化システム以外には宿主細胞成分を必要としないことが観察された(Maeda et al.1981;White et al.1982)。浸透段階は,リソソーム指向性弱塩基,カルボン酸イオノフォア,およびプロトンポンプ阻害剤等の薬剤により阻害される(Matlin 1982;Whittaker 2006)。
【0036】
侵入するvRNPの核内輸送を可能とするためには,カプシドが解体される必要がある。この段階は,アマンタジン感受性のM2チャネルによるウイルス内部の酸性化を含み,vRNPからのMlの解離を引き起こす(Bukrinskaya et al.1982;Martin and Helenius 1991;Pinto et al.1992)。個々のvRNPの核膜孔複合体への輸送および核内への移行は,細胞性核輸送レセプターに依存する(O’Neill et al.1995;Cros et al.2005)。ウイルスRNA(正鎖および負鎖の合成)の複製および転写は,核内のクロマチンと密に会合した複合体中で生ずる。これらの段階の多くはウイルスポリメラーゼにより触媒されるが,細胞性因子,例えばRNAポリメラーゼ活性化因子,シャペロンHSP90,hCLE,およびヒトスプライシング因子UAP56が関与していることが明らかである。ウイルス遺伝子発現は,細胞性キナーゼに依存する制御系である,転写レベルにおける複雑な細胞制御を受けている(Whittaker 2006)。
【0037】
インフルエンザ粒子の最終的なアセンブリは,細胞膜における発芽プロセスの間に行われる。上皮細胞においては,発芽は頂端膜ドメインにおいてのみ生ずる(Rodriguez−Boulan 1983)。まず,子孫vRNPが核質の中の核エンベロープに,次に核から細胞質に輸送され,最後に細胞周辺に蓄積する。核からの退出はウイルス蛋白質NEPおよびM1,および種々の細胞蛋白質,例えばCRM1(核外輸送レセプター),カスパーゼ,およびおそらくはある種の核蛋白質シャペロンに依存する。リン酸化は,M1およびNEP合成を制御することにより,およびMAPK/ERK系を介して,核外輸送において役割を果たす(Bui et al.1996;Ludwig 2006)。
【0038】
ウイルスの3つの膜蛋白質は,ER中で合成され,フォールディングされ,アセンブリされてオリゴマーとなる(Doms et al.1993)。これらはゴルジ複合体を通過し,その炭水化物成分の修飾および蛋白質分解酵素による切断により成熟する。これらは細胞膜に到達した後,発芽プロセスにおいてM1およびvRNPと会合し,その結果,8つすべてのvRNPを取り込み,脂質を除くほとんどの宿主細胞成分を排出する。
【0039】
インフルエンザ感染は,いくつかのシグナリングカスケード,例えば,MAPK経路(ERK,JNK,p38およびBMK−1/ERK5),IkB/NF−kBシグナリングモジュール,Raf/MEK/ERKカスケード,およびプログラムされた細胞死の活性化を伴う(Ludwig 2006)。その結果,感染の進行を制限する種々の影響,例えば,IFNbの転写的活性化,アポトーシス性細胞死,および後期エンドソームからのウイルスの脱出の妨害が生ずる(Ludwig 2006)。
【0040】
ウイルスと細胞との相互作用の最も先の研究は,組織培養または卵に適合したウイルス株を用いる組織培養において行われた。これらの例におけるウイルスは,レセプター結合および指向性に影響を与える変化が誘導されるよう適合された(Matrosovich 2006)。野生型の病原性の株の感染はウイルスと宿主蛋白質との相互作用のより自然の状況を提供する。ヒト気道においては,インフルエンザAおよびBは主として上部呼吸管のNeuSAca−(2,6)−Galを有する非繊毛上皮細胞に感染し,一方,トリ種は気道のより深くでa−(2,3)−結合シアル酸を有する繊毛上皮細胞に感染することが知られている(Matrosovich et al.2004a)。
【0041】
III.マクロピノサイトーシスによるウイルスの細胞内への侵入
本発明の1つの観点は,マクロピノサイトーシスによるウイルス侵入経路に関与する蛋白質を標的とする抗ウイルス療法である。好ましくは,標的蛋白質は宿主細胞蛋白質である。好ましい標的は,キナーゼおよびキナーゼ経路の蛋白質である。好ましい標的としては,PAK1;DYRK3;PTK9;GPRK2L;Cdc42;および/またはRac1が挙げられる。好ましくは,マクロピノサイトーシス経路をポックスウイルス感染の治療の標的とする。好ましいポックスウイルスは,天然痘の原因ウイルスである天然痘ウイルスである。
【0042】
単一の作用メカニズムに限定されることを意図するものではないが,ポックスウイルス,例えば,ワクシニアウイルスは,宿主細胞に侵入するためにマクロピノサイトーシスおよびアポトーシス擬態を利用することが見いだされた(図6)。マクロピノサイトーシスは,大量の液体が封入され内在化されるプロセスである。この経路には,細胞膜再編成,エンドサイトーシスベシクルの形成,および膜の波打ち部位における葉状仮足の閉鎖が関与し,マクロピノソームが形成される(Lanzavecchia,A(1996)Curr Opin Immunol 8348−354;Sieczkarski and Whittaker(2002)J Gen Virol 83:1535−1545)。Rho GTPase(West et al.(2000)Curr Biol 10:839−848),ARF6(Radhakrishna et al.(1996)J Cell Biol 134:935−947),および1型ホスファチジルイノシトール−3キナーゼ(PI3−K)(Hooshmand−Rad et al.(1997)Exp Cell Res234:434−441)が,マクロピノサイトーシスに関与している。さらに,2つのRab,GTPaseであるRab5およびRab34/Rahは,マクロピノソームの形成における関与が示唆されている(Li et al.(1997)J Biol Chem,272:10337−10340;Sun et al.(2003)J Biol Chem 278:4063−4071)。Rab34/Rahはアクチンとともに膜の波打ちおよび新生マクロピノソームに局在することができ,その過剰発現はマクロピノサイトーシスを促進しうる(Sun et al.(2003)J Biol Chem 278:4063−4071)。Rab5はRab34/Rahとともにマクロピノソームに局在することができる(Sun et al.(2003)J Biol Chem278:4063−4071)。
【0043】
本発明者らは,原型ポックスウイルスであるワクシニアウイルスがその宿主細胞に侵入する際に用いるメカニズムを分析した。ウイルスは組織培養細胞の糸状偽足に結合し,これに沿って細胞体に向かって移動し,アクチンおよびアクチン付随蛋白質を含む大きな,過渡的な膜ブレブの突出を誘導することが認められた。ブレブが引っ込むにつれて,ウイルスは細胞内小胞中に内在化された。ブレブ形成を阻害する阻害剤,例えばブレビスタチン,EIPA,またはPAK1のドミナント・ネガティブコンストラクトは,感染もブロックした。阻害プロファイルから,MVは,アポトーシス後の細胞残存物の排除に関与するエンドサイトーシスプロセスであるマクロピノサイトーシスを誘導することが明らかになった。マクロピノサイトーシスによるアポトーシス体の内在化の決定因子はホスファチジルセリン(PS)の暴露であることから,ならびにMVの膜はこのリン脂質が非常に豊富であることから,異なる脂質組成を有するウイルスを調製した。PSの存在はブレブ形成マクロピノサイトーシス,および感染能において役割を果たす。この結果は,ワクシニアウイルスは,ウイルス誘導性マクロピノサイトーシスおよびアポトーシス擬態を用いることを示す。
【0044】
別の態様においては,アデノウイルス,好ましくは流行性結膜炎,ぜんそく状態の増悪,罹患率および死亡率に関連するB種ヒトアデノウイルス血清型3(Ad3)の治療のために,マクロピノサイトーシス経路を標的とする。好ましくは,アデノウイルスに対する治療薬は,アクチン,蛋白質キナーゼC,ナトリウム−プロトン交換体,Rac1,PAK1,およびC末端アデノウイルスE1A結合蛋白質−1(CtBP1)を標的とする。
【0045】
単一の作用のメカニズムに限定されることを意図するものではないが,Ad3は上皮および造血細胞に侵入するためにダイナミン非依存性マクロピノサイトーシスを利用することが認められた。感染性のAd3マクロピノサイトーシスは,アクチン,蛋白質キナーゼC,ナトリウム−プロトン交換体,およびRac1を標的とする阻害剤に対して感受性であるが,Cdc42に対してはそうではない。これは,p21活性化キナーゼ1(PAK1),および先天的な免疫応答を含む膜通過および転写抑制に関与する二機能性蛋白質であるC末端アデノウイルスE1A結合蛋白質−1(CtBP1)のウイルス活性化を必要とする。CtBP1はPAK1によりリン酸化され,細胞膜およびマクロピノソームにリクルートされ,同時に転写抑制解除が生ずる。これらを合わせて,Ad3は,抗原提示用に設計された,転写的抗ウイルス宿主遺伝子活性化によりウイルス宿主範囲を広げる,先天的なエンドサイトーシスの免疫経路を妨害する。
【0046】
IV.ウイルス感染において役割を果たす宿主細胞蛋白質を同定するための方法および装置
ウイルスによる細胞侵入および増殖感染には,段階的なプログラムが関与しており,ここでは,わずかの事象がウイルス蛋白質および酵素により媒介され,残りの事象は細胞性機能に依存する。関与する細胞性蛋白質の完全な目録を得るためには,システムバイオロジーのアプローチを利用する態様が非常に有用である。組織培養細胞におけるインフルエンザ感染に関与する必須遺伝子の系統的同定を含む態様は,発見の有益な手段を提供する。siRNAのゲノムワイドなライブラリおよびハイスループット装置プラットフォームを含むシステムバイオロジーのアプローチは,ウイルス感染に関与する宿主細胞蛋白質を大量の候補物質蛋白質から迅速かつ効率的に同定することができる。
【0047】
1つの態様においては,自動化ハイスループットsiRNAスクリーニング技術をゲノムデータベース情報と組み合わせて用いて,宿主細胞蛋白質の系統的同定を行う。ここでは,ゲノムデータベースは,そのゲノム配列が知られている任意の種,例えば,ヒト,マウス,またはトリ種に由来するものであることができる。ある態様においては,高度なロボティクスおよびスクリーニング技術,例えば,the RNAi Image−based Screening Center(RISC)をもつスクリーニングプラットフォームを用いることができる。siRNAスクリーニングは,任意の適当な宿主細胞または細胞株,例えば,マウスまたはヒト宿主細胞,例えば,気道上皮細胞,またはHeLa MZ細胞,HeLa Kyoto細胞,またはA549細胞等の宿主細胞株を用いて行うことができる。他の適当な細胞株としては,16HBEと称される気管支細胞株,THEと称される気管細胞株,ならびに空気−液体界面(いわゆるALI培養)でよく分化した培養多列粘液線毛上皮を形成する市販のヒト気道上皮細胞培養物(HBEpC,Promocell,Heidelberg Germanyから購入)が挙げられる。そのような細胞はインフルエンザ感染のモデルとして用いることができることが知られている(Matrosovich et al.2004)。ある態様においては,関連するかまたは必要なウイルス侵入レセプター(例えば,HIV−1についてはCD4およびCXCR4)を発現するようトランスフォームされている安定な宿主細胞株を作製することができる。宿主細胞は,機能的効力について予め確認されているsiRNAのゲノムライブラリを用いてスクリーニングすることができる。ある態様においては,siRNAのゲノムライブラリは,商業的供給元,例えばQiagenから入手することができる。
【0048】
1つの態様においてはHeLa細胞を宿主細胞として用いる。HeLa細胞は,siRNAトランスフェクションによる有効なサイレンシングが可能である。インフルエンザウイルスの試験を含む態様は,単一のインフルエンザウイルスが細胞膜の被覆されたまたは被覆されていないくぼみの両方に結合することを示す。10分後,ウイルスは被覆されたおよび被覆されていない小さいベシクル中に存在し,30分後にはエンドソームと一致する見かけのより大きいベシクル中で多くが検出される。すなわち,ウイルス侵入の形態学は,内在化がゆっくりであることを除き,MDCK細胞において観察されたものと似ている。さらに,インフルエンザウイルスがエンドソーム構造に入って出る軌跡を,初期エンドソームを緑に標識するRab5−GFPおよび後期エンドソームを赤に標識するRab7−RFPを発現するHela細胞を用いて追跡した。さらに,HeLa細胞を用いて,感染,転写,およびウイルス蛋白質合成の初期段階を研究し,または後期段階のいくつか,例えばvRNPの核外輸送の障害についてスクリーニングすることができる。
【0049】
別の態様においてはA549細胞を宿主細胞として用いる。A549細胞は,呼吸器ウイルス,例えばインフルエンザウイルス感染の研究を含む態様において特に有用である。A549細胞は気管支起源の上皮細胞株であり,インフルエンザ感染研究において広く用いられている(Ehrhardt et al.2006)。A549細胞は,インフルエンザ疾患の間にインサイチューで感染した宿主細胞に非常に類似した系を提供する。さらに,A549細胞は,子孫ウイルスの放出および二次感染を含む全複製サイクルを分析する可能性を提供する。インフルエンザ研究およびアッセイにおいてしばしば用いられるMDCK細胞とは異なり,A549細胞はヒト起源であり,siRNAにより容易にトランスフェクションすることができる(Graeser 2004)。さらに別の態様においては,次の2つのインフルエンザウイルスを試験して,培養A549細胞におけるウイルスの伝播および二次感染を自動化ハイスループットフォーマットで分析する:1)トリH7N7ウイルス,ほとんどの細胞株においてそのHAはセクレターゼ切断により活性化される(Wurzer et al.2003);および2)ヒトインフルエンザ株,例えばX31/Aichi/68,および96,384,768,1152,1440,1536,3072ウエルプレート,または他のマルチウエルプレートのフォーマットと適合するトリプシン重層方式。
【0050】
さらに,本発明の態様は自動化スクリーニングプラットフォームを用いて実施しうることが企図される。ここでは,スクリーニングプラットフォームは,液体を操作するロボット,例えばTecanおよび2つの自動化顕微鏡,例えばCell Worx(Applied Precision Instruments)を含むことができる。自動化スクリーニングプラットフォームを用いてハイスループット実験手順を実施しうることが理解される。さらに,コンピュータおよび実験的努力を平行に組み合わせて,siRNAアッセイを最適に適用し,完全に自動化されたデータトラッキング,画像分析,定量,および統計学的分析のためのソフトウエアを設定することができる。
【0051】
大規模の態様においては,宿主細胞株の全ゲノムをカバーするsiRNAを用いてスクリーニングを行う。別の態様においては,宿主細胞株のゲノムのサブセット(例えば,少なくとも600,100,2000,3000,4000,5000,6000,7000,8000,9000,10000,15000,20000,25000,または30000の遺伝子)をカバーするsiRNAを用いてスクリーニングを行う。例えば,1つの可能な態様においては,ヒトゲノムの少なくとも7,000の遺伝子をカバーするsiRNAを用いてスクリーニングを行う。RISCプラットフォームにより,調べるべき各ウイルス株について2つの異なる細胞株を用いて7,000遺伝子のスクリーニングを2−4週間以内に完了することが可能である。次に,ユーザ仕様のMatLabプラグインを用いて,データセットを詳しく分析して管理する。MatLabプラグインは,生成された画像中でデータを自動的に定量することができ,低品質の画像を自動的に廃棄し,データ分析の構造安定性および再現性を判定する品質管理アルゴリズムを含むことができる。分析が完了すれば,結果からウイルス侵入に関与する宿主蛋白質を同定することができる。ウイルスインフェクトームライブラリは,元々はcDNAマイクロアレイの分析用に作成されたがRNAiデータセットとともに用いるように大幅に改変されたバイオインフォマティクスのツール上に構築する。大きなデータセットの強固な統計学は,ほとんどの重みが非常に顕著な表現型に与えられることを確実にする。特定の表現型は,試験する各遺伝子について少なくとも3つのsiRNAを用い,ある遺伝子に対する3つのsiRNAのうち2つが同様の効果を示すことを必要とすることにより重み付けされる。
【0052】
ある態様では,画像に基づくアッセイを利用することができる。これはプレートリーダーより感度が高く,したがってウイルス感染の背後にある細胞生物学に関する追加の情報が得られる。これらの態様においては,平静対照では平均で10−20%の細胞しか感染しないため,高い感度が望ましい。低い「ベースライン」は,より効率的なsiRNAサイレンシングと関連しており,感染における増加と減少との間を区別することができる。この判定は,感染経路に関する最適な情報を提供する。
【0053】
大きなフォーマットのsiRNAスクリーニング(例えば宿主細胞ゲノムのサブセットまたは全体をカバーする大きな遺伝子セット)を含むある態様においては,自動化液体操作ロボット,例えば,96,384,768,1152,1440,1536,3072ウエルプレート,または他のマルチウエルプレートを操作しうるTecanを用いる。生成したデータ(ウエルあたり9個の画像;スクリーンあたり1,430,784個の画像,約3.8TBに対応)をNASサーバに自動的に動かすアルゴリズムを用いる。さらに別の態様においては,高いバッファー容量,例えば1,2,3,4,5,6,7,8,9,または10TBは,一次的なネットワーク障害により分析プロセスが遅れないことを保証する。さらに別の態様においては,これらの大きな画像のセットの中から分析されていない画像を連続的に探すアルゴリズムは,自動的に画像を分析待ち行列に配置する。これらの態様のいくつかにおいては,MatLab画像分析プラグインを用いる。さらに,スクリーニングからの「生の」データについてバイオインフォマティクス評価を行って擬陽性を排除することにより,複雑なプロセスに関与する細胞システムを再構築することができる。このことにより,各侵入経路および他の感染関連プロセスに特異的な分子機構の鍵となる標的宿主細胞蛋白質の定義が可能となる。ある態様においては,用いられる基準には,強いRNAi表現型および広い細胞タイプ依存性が含まれる。
【0054】
上述の方法は,siRNAについて実施されてきた。しかし,他の適当な分子種,例えば,有機または無機化合物,抗体等の蛋白質,またはアンチセンスRNA等の核酸種も用いることができる。
【0055】
本発明の核酸療法は,天然の核酸であっても,修飾された核酸であっても,核酸の類似体であってもよい。核酸類似体としては,例えば,ペプチド核酸(PNA),ロック核酸(LNA),トレオース核酸(TNA),拡大塩基DNA(xDNAまたはyDNA)が挙げられる。同様に,ホスホロチオエートまたはホスホネート骨格修飾核酸も包含される。
【0056】
V.キナーゼ
ある態様においては,ウイルス感染を調節するとして同定される宿主細胞蛋白質はキナーゼである。さらに別の態様においては,宿主細胞蛋白質は,PAK1,Cdc42,Rac1,DYRK3,PTK9,およびGPRK2Lである。数百種のヒトキナーゼが知られており,これらはいくつかの異なるファミリーに分類され,種々の疾患状態において役割を果たすことが知られている(Manning et al.2002)。
【0057】
キナーゼの阻害剤としては,例えば,ドミナント・ネガティブ分子,siRNA,shRNA,抗体および小分子が挙げられる。ドミナント・ネガティブ分子には,例えば,分子内または分子間の蛋白質−蛋白質相互作用のインターフェースをブロックすることにより,インビトロまたはインビボで蛋白質の機能を妨害する分子が含まれる。ドミナント・ネガティブ分子としては,例えば,蛋白質標的のフラグメント(変異体フラグメントを含む)および標的蛋白質の非機能的変異体が挙げられる。抗体としては,例えば,完全イムノグロブリン,一本鎖抗体およびイムノグロブリンの特定の結合部位が挙げられる。小分子としては,例えば,約5000Daまで,約2000Daまで,または約1000Daまでの質量を有する有機または無機の非ポリマー性分子が挙げられる。
【0058】
A.PAK1
PAK1機能の分子メカニズムについては多くの見識が存在する(概説として,Parrini,MC et al.(2005)Biochem Soc Trans 33:646−648)。PAK1は自己阻害ホモダイマーとして存在することができる(Lei,M et al.(2000)Cell 102,387−397)。1つのPAK1分子のN末端制御ドメインは別のPAK1分子のC末端の触媒ドメインに結合してこれを阻害することができる。PAK1はGTP結合形のCdc42およびRac1に結合することにより活性化されることができる。これらの分子への結合は,制御ドメインのフォールディングを変化させることができ,これはPAK1ホモダイマーの解離につながる(Lei,M et al.(2000)上掲;Parrini,MC et al.(2002)Mol Cell.9,73−83.)。PAK1はまた,GTPaseであるRac2,Rac3,TC10,CHP,およびWrch−1に結合し,活性化されることができる(概説として,Zhao and Manser(2005)Biochem J.386,201−214)。これらのGTPaseによる結合および活性化は,N末端制御ドメインないしPBD(p21結合ドメイン)中の残基が媒介することができる。Cdc42およびRacはCdc42およびPAK1のRac相互作用結合ドメイン(CRIB)(アミノ酸75−90)(Burbelo et al.(1995)J Biol Chem.8,29071−29074)にわずかに結合することができ,キナーゼ阻害ドメイン(KI)を挟む配列は結合親和性に寄与しうる(Knaus and Bokoch(1998)Int J Biochem Cell Biol.30,857−862;Sells,MA and Chernoff,J(1997)Trends Cell Biol.7,162−167;Lei,M et al.(2000)上掲)。CRIBドメインのN末端の短いリジンリッチセグメント(PAK1アミノ酸66−68)は,Rac GTPaseの結合を媒介することができる(Knaus,UG and BokochGM(1998)上掲)。
【0059】
KIドメインは,約90nMのKiで触媒ドメインを阻害することができる(Zhao et al.(1998)Mol.Cell.Biol.18:2153−2163)。PAK1のKIドメインの残基Leu−107,ならびにKIドメインの他のアミノ酸が,この阻害の接点として寄与しうる(Lei et al.(2000)上掲)。PAK1のKI領域は,活性部位の2つの構造成分(ヘリックスCおよび活性化ループ)を安定化することができる。KIセグメントのリジンは,触媒に役割を果たす2つのアスパラギン酸残基とともに塩架橋を形成することにより,活性部位をブロックすることができる。このKIポリペプチドは,PAK活性化をブロックすることができる(Zhao et al.(1998)上掲)。PAK1のPBDを含むペプチドのCdc42への結合における結合定数は10−50nMの範囲であることが報告されている(Thompson et al.(1998)Biochemistry 37:7885−7891)。
【0060】
PAK1のN末端制御ドメインも,PAK相互作用交換因子(PIX)に結合することができる2つの保存されたPXXP SH3(Srcホモロジー3)結合モチーフおよび保存SH3結合部位を含む(Manser et al.(1998)Mol.Cell 1:183−192)。1番目の保存SH3結合部位は,アダプター蛋白質であるNckに結合することができ(Bokoch et al.(1996)J.Biol.Chem.271:25746−25749),2番目の部位はGrb2に結合することができる(Puto et al.(2003)J.Biol.Chem.278:9388−9393)。
【0061】
GTPaseの結合は,KIドメインの触媒ドメインとの相互作用を破壊するコンフォメーションの変化を引き起こすことができ,キナーゼ活性に寄与しうる自己リン酸化を可能とすることが示唆されている(Lei,M et al.(2000)上掲)。自己リン酸化は,PAK1を活性状態へとスイッチすることができる。PAK1の触媒ドメインの活性化ループ中のThr−423の自己リン酸化は,自己阻害からの解除および外因性基質に対する触媒機能を維持することができる(Yu et al.(1998)Biochem.J.334:121−131;Gatti et al.(1999)J.Biol.Chem.274:8022−8028;Zenke et al.(1999)J.Biol.Chem.274:32565−32573)。PAK1はPDK1(3−ホスホイノシチド依存性キナーゼ1)により修飾されることができる(King et al.(2000)J.Biol.Chem.275:41201−41209)。Ser−144(KIドメイン中の保存残基)におけるαPAKの自己リン酸化はキナーゼ活性化に寄与することができるが(Chong et al.(2001)J.Biol.Chem.276:17347−17353),一方,PAK1の自己リン酸化部位Ser−198/203はPIX−PAK相互作用を下方制御することができる。
【0062】
PAK1は,RacおよびCdc42 GTPaseとは独立して活性化されることができる。限定されたプロテアーゼ媒介性切断は,PAKキナーゼの自己リン酸化および活性を刺激することができる(Brenner et al.(1995)J.Biol.Chem.270:21121−21128;Roig et al.(1998)Vitam.Horm.62,167−198)。SH3含有NckおよびGrb2アダプター蛋白質を介するPAK1の膜リクルートメントは,キナーゼ活性を刺激することができる(Lu et al.(1997)Curr.Biol.785−94;Daniels et al.(1998)EMBO J.274:6047−6050)。この活性化は,重要なThr−423残基におけるPDK1によるリン酸化(King et al.(2000)J.Biol.Chem.275:41201−41209)またはスフィンゴシン等の脂質との相互作用を含むことができ,これはGTPase非依存性様式でキナーゼを活性化することができる(Bokoch et al.(1998)J.Biol.Chem.273:8137−8144)。PIXを介してPAKと間接的に関連しうるGIT1(G−蛋白質共役レセプターキナーゼ−相互作用標的1)もまた,Rho GTPaseを必要としないメカニズムによりPAKを活性化することができる(Loo et al.(2004)Mol.Cell.Biol.24:3849−3859)。
【0063】
PAK1はまた,焦点接着関連蛋白質PIX(Coolとも称される)と複合体を形成することができる。2つの異なる遺伝子(αPIXおよびβPIX)に由来する複数のPIX蛋白質は,そのSH3ドメインを介してPAKに結合することができる(Manser et al.(1998)Mol.Cell1:183−192;Bagrodia et al.(1999)J.Biol.Chem.274:22393−22400)。αPIXを欠失した細胞の分析から,走化性白血球のCdc42媒介性方向センシングにおけるPIX−PAK複合体の役割が示唆されている(Li et al.(2003)Cell 114:215−227)。PIXのGIT1(PKL/CAT1としても知られる(Turner et al.(1999)J.Cell Biol.145:851−863;Bagrodia et al.(1999)上掲)との会合は,パキシリンに結合することにより焦点接着を標的とすることができる(Turner et al.(1999)上掲)。GIT1の過剰発現により,焦点接着の分解およびパキシリンの喪失が生じうる(Loo et al.(2004)Mol.Cell.Biol.24:3849−3859)。すなわち,GIT1およびPIXは両方とも運動性細胞および細胞−細胞接合の前縁の焦点接着に局在してPAKを活性化することができる(Zegers et al.(2003)EMBO J.22:4155−4165;Zhao et al.(2000)Mol.Cell.Biol.20:6354−6363;Manabe et al.(2002)J.CellSci.115:1497−1510)。
【0064】
2つの関連するヒト蛋白質ホスファターゼは,PAK1を例えばThr−423で脱リン酸化することができる(Koh et al.(2002)Curr.Biol.12:317−321)。これらのホスファターゼは,POPX1(PIX1のパートナー)およびPOPX2であり,これらは様々な形のPIXに結合して,PAKを含む多量体複合体を形成するすることができる。活性なPAK1の細胞における効果は,これらのホスファターゼのいずれかの過剰発現により拮抗されうる(Manabe et al.(2002)上掲)。
【0065】
他の蛋白質キナーゼはPAK機能を下方制御するかもしれない。AktはPAK1をSer−21でリン酸化することができ,この修飾は,NckのPAK1N−末端への結合を低下させることができるが,キナーゼ活性は増加する(Zhao et al.(2000)上掲;Tang et al.(2000)J.Biol.Chem.275:9106−9109)。
【0066】
PAK1は,マクロピノサイトーシスの制御に関与している。活性化されたPAK1変異体(T423E)は,ストレス線維および焦点接着複合体の崩壊,および葉状仮足の形成(Sells et al.(1997)Curr Biol.7:202−210;Manser et al.(1997)Mol Cell Biol 17:1129−1143),およびアクチン骨格の再編成を誘発しうる。PAK1が関与するキナーゼ活性および蛋白質−蛋白質相互作用は,アクチン細胞骨格に影響を及ぼしうる(Sells et al.(1997)Curr Biol.7:202−210;Turner et al.(1999)J Cell Biol.145:851−863)。
【0067】
本発明の方法および組成物において用いることができるPAK1の阻害剤としては,例えば,内皮細胞移動を調節することができるPAK1の残基1−74(MSNNGLDIQDKPPAPPMRNTSTMIGAGSKDAGTLNHGSKPLPPNPEEKKKKDRFYRSILPGDKTNKKKEKERPE;(配列番号1)を含むドミナント・ネガティブPAK1(Kiosses et al.(1999)J.Cell Biol.147:831−843);PAK1の最初のプロリンリッチドメインの13アミノ酸(KPPAPPMRNTSTM;(配列番号2));HIV tat蛋白質のポリ塩基性配列に融合させたこれらの残基(YGRKKRRQRRRGKPPAPPMRNTSTM;(配列番号3))(Kiosses et al.(2002)Circ.Res.90:697−702);PAK1自己阻害ドメインを含み,マクロピノサイトーシスをブロックすることができるPAK1のアミノ酸83−149のフラグメント,HTIHVGFDAVTGEFTGMPEQWARLLQTSNITKSEQKKNPQAVLDVLEFYNSKKTSNSQKYMSFTDKS(配列番号4)(Dharmawardhane et al.(2000)Mol.Biol.Cell11:3341−3352;Barradeau et al.米国特許7,364,887);PAK1との結合に影響をおよぼしうる一酸化窒素シンターゼ(DLC1/PIN)のダイニン軽鎖−1/蛋白質阻害剤のペプチド(Kumar et al.米国特許7,067,633);ドミナント・ネガティブPAK1(PAK1(K299R))(Naor,米国特許公開20050090474)が挙げられる。
【0068】
本発明の方法および組成物において用いることができるPak1の間接的阻害剤としては,例えば,PAK1キナーゼ活性を低下させることができるヒストン脱アセチル化阻害剤FK228(Hirokawa et al.(2005)Can.Biol.Ther.4:956−960);それぞれSrcファミリーキナーゼおよびETKを阻害することによりPAK1の活性化を低下させることができるチロシン−キナーゼ阻害剤PP1およびAG879(He et al.(2004)Can.Biol.Ther.3:96−101;He et al.米国特許公開20030153009);およびPAK1活性を低下させるPP1とAG879,GL−2003の水溶性誘導体との組み合わせ(Hirokawa et al.(2006)Cancer Letters 245:242−251)が挙げられる。
【0069】
本発明の方法および組成物において用いることができるPAK1の別の阻害剤には,インビトロおよびインビボでのPAK1の直接阻害剤であるCEP−1347(Nheu,TV et al.(2002)Cancer J.8,328−336)が含まれる。
【0070】
本発明の方法および組成物において用いることができるPAK1のさらに別の阻害剤には,VanEykら(米国特許6,248,549)に開示されるものが含まれる。
【0071】
本発明の方法および組成物において用いることができるさらに別の阻害剤としては,PAK1に対するsiRNAが挙げられる:siPAK1−0AGAGCTGCTACAGCATCAA(配列番号6),siPAK1−1GACAUCCAACAGCCAGAAA(配列番号7),siPAK1−2GAGAAAGAGCGGCCAGAGA(配列番号8),hPAK1−6UACCAGCACUAUGAUUGGA(配列番号9),siPAK1−7UCUGUAUACACACGGUCUG(配列番号10)(Nasoff et al.2007米国特許公開US20070128204(2006年12月1日出願),およびQiagenから入手可能な3つのsiRNAオリゴ(PAK1_p1,PAK1_p2,およびPAK1_p3)(表1)。これらのsiRNAは,OiagenによりRT−PCRを用いて評価され,約70%以上の標的遺伝子mRNAのノックダウンを与えることが示されている。これらのsiRNAは対称の2bpの3’オーバーハングをもつ21bpデュープレックスである。
【0072】
【表1】
【0073】
B.DYRK3
哺乳動物DYRK3(REDK,hYAK3)は,Ser/Thr部位を標的としうるMAPK関連蛋白質キナーゼである。DYRK3は,コンセンサスキナーゼサブドメインVIIとVIIIとの間の保存YXYモチーフ(ないしループ)におけるチロシン(自己)リン酸化により活性化されることができる。DYRK3は赤血球リニアージの造血細胞で選択的に高レベルで発現されることができる(Geiger,JN et al.(2001)Blood 97:901−910;Lord,KA et al.(2000)Blood 95:2838−2846)。初代ネズミおよびヒト造血系前駆細胞においてDYRK3をアンチセンスオリゴヌクレオチドで阻害すると,コロニー形成単位−赤血球(赤芽球の最後から2番目の前駆細胞)の産生に影響を及ぼしうる。DYRK3活性は,その予想(自己)リン酸化ループ中のTyr333の存在に依存することができ,ループの酸性化は活性化でありうる(Li,K. et al.(2002)J Biol Chem 49,47052−47060)。DYRK3はキナーゼドメインならびにユニークなC末端ドメイン依存性が関与するメカニズムにより作用して,PKAに依存する経路によりCREBおよびCRE応答経路を制御することができる(Li,K.et al.(2002)上掲)。FDC造血系前駆細胞におけるDYRK3の発現はアポトーシスを制御しうる(Li,K.et al.(2002)上掲)。
【0074】
本発明の方法および組成物において用いることができるDYRK3の阻害剤としては,DYRK3/hYAK3のキノリン阻害剤(米国特許7,087,758),YAK3/DYRK3の阻害剤であるGSK626616AC,(http://clinicaltrials.gov/show/NCT00443170),3−カルボキシキノリン誘導体DYKR3/YAK3(Burgess et al.米国特許公開20060106058),およびQiagenから入手しうる3つのsiRNAオリゴ(DYRK3_p1,DYRK3_p2,およびDYRK3_p3)(表2)が挙げられる。これらのsiRNAは,OiagenによりRT−PCRを用いて評価され,約70%以上の標的遺伝子mRNAのノックダウンを与えることが示されている。これらのsiRNAは対称の2bpの3’オーバーハングをもつ21bpデュープレックスである。
【0075】
【表2】
【0076】
C.PTK9(TWF1)
ツインフィリン1は,短いリンカー領域により連結される2つのADF/コフィリン様(ADF−H)ドメインおよびそれに続く20残基のC末端テールから構成される。2つのADF−Hドメインは互いに約20%のホモロジーを有する(Lappalainen et al.,(1998)Mol.Biol.Cell 9:1951−1959.)。
【0077】
ヒトツインフィリンは,元々はチロシンキナーゼとして同定されたが(Beeler et al.,(1994)Mol.Cell.Biol.14:982−988),研究により,これはキナーゼ活性を持たず(Vartiainen et al.,(2000)Mol.Cell.Biol.20:1772−1783;Rohwer et al.,(1999)Eur.J.Biochem.263:518−525),ツインフィリンは既知の蛋白質キナーゼに対して配列ホモロジーを持たないことが示された。むしろ,ツインフィリンは,アクチンモノマーに結合することができる(Goode et al.,(1998)J.CellBiol.142:723−733;Vartiainen et al.,(2000)上掲;Wahlstrom et al.,(2001)J.CellBiol.155:787−796)。ツインフィリンの機能に関する情報は,部分的には,酵母およびDrosophilaのホモログの研究から得られた。ツインフィリンはアクチンモノマーと1:1の複合体を形成するようである。ツインフィリンはアクチンモノマーを有効に隔離することができる(Goode et al.,(1998)上掲;Vartiainen et al.,(2000)上掲;Wahlstrom et al.,(2001)上掲)。ツインフィリンは,ADP−アクチンモノマーと相互作用して,そのヌクレオチド交換およびフィラメントのアセンブリを阻害することができる(Palmgren et al.,(2001)J.CellBiol.155:251−260)。ツインフィリンは新たに脱重合したアセンブリ不能ADP−アクチンモノマーと相互作用するかもしれない。
【0078】
ツインフィリンは点状の細胞質染色パターンを有し,培養哺乳動物細胞においてアクチンモノマーおよびフィラメントを含む細胞性プロセスに局在することができる(Vartiainen et al.,(2000)上掲)。ツインフィリンとキャッピング蛋白質との間の直接相互作用は,ツインフィリンの急速なアクチンフィラメントアセンブリの部位への局在化を媒介することができる(Palmgren et al.,(2001)上掲)。
【0079】
本発明の方法および組成物において用いることができるTWF1/PTK9の阻害剤としては,shRNA,例えば,SigmaTRC(The RNAi Consortium)#:TRCN 0000011013;クローンID:NM_002822.3−907s1c1;受託番号:NM_198974.1,NM_002822.3;CCGGGCCTGGATACACATGCAGTATCTCGAGATACTGCATGTGTATCCAGGCTTTTT(配列番号30);Sigma TRC#TRCN 0000006364;クローンID:NM_002822.3−2014s1c1;受託番号:NM_198974.1,NM_002822.3;CCGGCCGAGCAAATACTCAGATTTACTCGAGTAAATCTGAGTATTTGCTCGGTTTTT(配列番号31);Sigma TRC#TRCN 0000006365;クローンID:NM_002822.3−364s1c1;受託番号:NM_198974.1,NM_002822.3;CCGGCCAGGGATATGAATGGATATTCTCGAGAATATCCATTCATATCCCTGGTTTTT(配列番号32);Sigma TRC#TRCN 0000006366;クローンID:NM_002822.3−474s1c1;受託番号:NM_198974.1,NM_002822.3;CCGGGCCACATTAAAGATGAAGTATCTCGAGATACTTCATCTTTAATGTGGCTTTTT(配列番号33);Sigma TRC#:TRCN 0000006367;クローンID:NM_002822.3−962s1c1;受託番号:NM_198974.1,NM_002822.3;CCGGCGTCTGCTAGAAATTGTAGAACTCGAGTTCTACAATTTCTAGCAGACGTTTTT(配列番号34)が挙げられる。
【0080】
また,本発明の方法および組成物において用いることができるPTK9/TWF1の阻害剤には,PTK9既設計キメラRNAi(Cat.#H00005756−R04,Abnova)およびPTK9評価済みStealth(商標)Duo Pak(Cat.#12938068,Invitrogen)が含まれる。
【0081】
D.GPRK2L(GRK4)
G−蛋白質共役レセプター(GPCR)キナーゼ(GRK)は,3つのファミリーに体系づけられるセリン/トレオニンキナーゼである(Penela et al.,(2003)Cell Signal 15:973−981)。1つのファミリーはGRK4ファミリーであり,これはGRK4,GRK5,およびGRK6から構成される。GRK4サブファミリーの特徴としては,以下のものが挙げられる:a)C末端システイン残基のパルミトイル化(GRK4/6について)または負に荷電した膜リン脂質とC末端近くの正に荷電したドメインとの相互作用(GRK5について)による膜局在化,b)ホスファチジルイノシトールビスホスフェート結合(N末端ドメインへの)による活性化,およびc)カルシウムセンサー蛋白質,例えばカルモジュリンによる阻害(Pronin et al.,(1997)J.Biol.Chem.272:18273−18280;Pitcher et al.,(1998)Annu.Rev.Biochem.67:653−692;Kohout and Lefkowitz,(2003)Mol Pharmacol.63:9−18;Willets et al.,(2003)Trends Pharmacol.Sci.24:626−633)。
【0082】
GRK4には4つのRNAスプライシング変種が同定されている:α,β,γおよびδ(Premont et al.,(1996)J.Biol.Chem.271:6403−6410)。GRK4αは全長型である。GRK4βは,エクソン2によりコードされる配列を欠失しており,このためN末端近くのホスファチジルイノシトールビスホスフェート結合ドメインを含む32アミノ酸が欠失している。GRK4γはエクソン15によりコードされる配列を欠失しており,このため,C末端近くの46アミノ酸が欠失している。最も短い型のGRK4δは,選択的にスプライシングされたエクソンの両方によりコードされる配列を欠失している。
【0083】
GRKはGCPR脱感作において役割を果たしている。GPCRはアゴニストによる活性化によって脱感作を生じうる;このプロセスにより,連続的アゴニスト刺激下でのレセプター応答が減少しうる(Ferguson et al.,(1996)Can.J.Physiol.Pharmacol.74:1095−1110;Gainetdinov et al.,(2004)Annu.Rev.Neurosci.27:107−144)。GRK媒介性リン酸化は,レセプター/G蛋白質相互作用を低下させ,アレスチン結合を開始させることができる。アレスチンとの会合はさらにG蛋白質カップリングを減少させ,レセプターのエンドサイトーシスを増強することができる。内在化されたGPCRは,さらに別のシグナリング経路に関与することができ,細胞膜へのリサイクル用に選別され,または分解の標的となる(Ferguson et al.,(1996)Can.J.Physiol.Pharmacol.74:1095−1110;Penela et al.,(2003)Cell Signal 15:973−981;Gainetdinov et al.,(2004)Annu.Rev.Neurosci.27:107−144)。
【0084】
GRK4はまた,GPCRのアゴニスト非依存性リン酸化を刺激することができる。例えば,GRK4をD1レセプターと共発現させると,アゴニストを加えてもわずかにしか増加しなかったレセプターがリン酸化される(Rankin et al.(2006)Mol.Pharmacol.69:759−769)。アゴニスト結合の非存在下におけるGRK4αによるD1レセプターのリン酸化により,アゴニスト誘導性cAMP蓄積の減少,基底レセプター内在化の増加,および全レセプターの数の減少が生じうる。
【0085】
本発明の方法および組成物において用いることができるGRK4の阻害剤としては,例えば,アンチセンス−オリゴヌクレオチド(As−Odn),209 5’−CATGAAGTTCTCCAGTTCCAT−3’ 189(配列番号19)(Sanada et al.(2006)Hypertension 47:1131−1139),カルモジュリン(Iacovelli et al.(1999)FASEB J.13:1−8),ヘパリン(GRK4αの阻害剤;Sallese et al.(1997)J.Biol.Chem.272:10188−10198),およびQiagenから入手しうる3つのsiRNAオリゴ(GPRK2L_p1,GPRK2L_p2,およびGPRK2L_p3)(表3)が挙げられる。これらのsiRNAは,OiagenによりRT−PCRを用いて評価され,約70%以上の標的遺伝子mRNAのノックダウンを与えることが示されている。これらのsiRNAは対称の2bpの3’オーバーハングをもつ21bpデュープレックスである。
【0086】
【表3】
【0087】
E.Rac1
Rac1は小さいシグナリングG蛋白質であり,GTPaseのRhoファミリーのメンバーである。Rac1はPAK1の標的である。本発明の方法および組成物において用いることができるRac1の阻害剤としては,例えば,Rac1阻害剤W56(MVDGKPVNLGLWDTAG;(配列番号44);Cat.No.2221;Tocris bioscience),Rac1阻害剤(Cat.No.553502;Calbiochem),Rac1阻害剤NSC23766,N6−[2−[[4−(ジエチルアミノ)−1−メチルブチル]アミノ]−6−メチル−4−ピリミジニル]−2−メチル−4,6−キノリンジアミン三塩酸塩(Cat.No.2161;Tocris bioscience)が挙げられる。
【0088】
F.Cdc42
Cdc42はRho−サブファミリーの小さいGTPaseであり,細胞形態,移動,エンドサイトーシスおよび細胞サイクル進行を管理するシグナリング経路を制御することができる。本発明の方法および組成物において用いることができるCdc42の阻害剤としては,例えば,セクラミンB(Pelish et al.(2006)Biochem.Pharmacol.71:1720−1726);セクラミンA(Xu et al.(2006)Org Biomol Chem4:4149−4157);およびACK42(Nur−E−Kamal et al.(1999)Oncogene 18:7787−7793)が挙げられる。
【0089】
VI.トランスジェニック細胞および非ヒト哺乳動物
トランスジェニック動物モデル,例えば組換えおよびノックアウト動物は,本明細書に記載される宿主核酸から生成することができる。トランスジェニック非ヒト哺乳動物の例としては,限定されないが,マウス,ラット,トリ,ウシおよびブタが挙げられる。ある例においては,トランスジェニック非ヒト哺乳動物はキナーゼに関連する標的配列の1またはそれ以上がノックアウトされており,ウイルス感受性,例えばインフルエンザまたはポックスウイルスによる感染の感受性が低下している。そのようなノックアウト動物は,ウイルス感染の段階の研究に,および動物からヒトへのウイルスの伝染の低下に有用である。さらに,本明細書に記載されるものと同じ標的を利用する動物ウイルスを動物において分析することができる。
【0090】
所望の遺伝子をノックアウトまたは機能的に欠失させるために用いられる配列の発現は,適切なプロモーター配列を選択することにより制御することができる。例えば,構成的プロモーターは機能的に欠失した遺伝子が動物により決して発現されないことを確実にするために用いることができる。これに対し,誘導可能なプロモーターは,トランスジェニック動物が目的とする遺伝子を発現するかまたは発現しない時を制御するために用いることができる。誘導可能なプロモーターの例には,組織特異的プロモーターおよび特定の刺激(例えば,光,酸素,化学物質濃度)に応答性または非応答性のプロモーターが含まれ,例えば,テトラサイクリン/ドキシサイクリン制御プロモーター(TET−off,TET−on),エクダイソン誘導性プロモーター,およびCre/loxPリコンビナーゼ系が挙げられる。
【0091】
1つの態様においては,ヒトキナーゼ遺伝子を持つかまたは内因性キナーゼ遺伝子が破壊されているトランスジェニックマウスを,インフルエンザまたはポックスウイルス等の種々の哺乳動物ウイルスに暴露した後に試験することができる。比較データは,そのウイルスおよび関連するウイルスのライフサイクルについての知見を提供することができる。さらに,さもなくば感染(例えばインフルエンザ)に対して感受性であるノックアウト動物(例えばブタ)を作製して,遺伝子の破壊によって付与される感染に対する耐性を判定することができる。
【0092】
別の態様においては,ヒトキナーゼ遺伝子を持つかまたは内因性キナーゼ遺伝子が破壊されているトランスジェニックブタを作製し,これを動物モデルとして用いて,インフルエンザまたはポックスウイルス感染等のウイルス感染に対する感受性を判定することができる。トランスジェニック動物,ならびにトランスジェニック動物を製造し使用する方法は,種々の特許および公開特許,例えば,WO01/43540;WO02/19811;米国特許公開2001−0044937および2002−0066117;および米国特許5,859,308;6,281,408;および6,376,743(これらは参照として本明細書に組み込まれる)に記載されている。
【0093】
VII.キナーゼ阻害剤の合理的設計
本発明の1つの観点は,蛋白質キナーゼ,例えば宿主細胞のウイルス感染に関与する蛋白質キナーゼを調節する薬剤に関する。ある態様においては,薬剤は,抗体,無機化合物,有機化合物,蛋白質/ペプチド薬剤または小分子,例えばsiRNAであることができる。好ましくは,薬剤は,PAK1,Cdc42,Rac1,DYRK3,PTK9,およびGPRK2Lを阻害することができる。ある態様においては,そのような薬剤は,インビトロでおよびインビボで抗ウイルス効果を発揮する。
【0094】
本発明のさらに別の観点は,宿主キナーゼを阻害する組成物を得るおよび/または作製する方法に関し,この方法は,阻害剤を設計し;その薬剤が宿主キナーゼを阻害するか否かを試験し;これを用いて宿主キナーゼを阻害する組成物を作製することを含む。ある態様においては,本発明は,キナーゼ調節剤であり,2以上の宿主キナーゼを阻害することができる薬剤を設計し,試験する方法に関する。
【0095】
本発明の合理的設計方法は,PAK1,Cdc42,Rac1,DYRK3,PTK9,およびGPRK2Lの構造についての現在の理解の助けを受けている。好ましくはキナーゼのX線構造を用いて,試験阻害剤のキナーゼへの結合を調べる。典型的には,候補物質の酵素への結合の「強さ」と,その薬剤のインビトロでの細胞活性との間には直接の相関がある。
【0096】
薬剤が無機または有機化合物である態様においては,前記化合物は,完全にコンピュータによる方法を用いて設計し試験することができ,または,そのような設計および試験の一部をコンピュータによって行い,残りをウエットラボ技術を用いて行うことができる。
【0097】
宿主細胞のウイルス感染に関与する蛋白質キナーゼを阻害するリード化合物は,種々の方法を用いて同定することができる。1つの態様においては,コンピュータを使用した“インシリコ”方法論により,標的宿主細胞キナーゼを阻害するようリード化合物を設計する。ケモゲノミクスのツール,例えばKinase Tool kit(商標)を用いて,バイオインフォマティクス,医薬化学およびコンピュータによる知識源の組み合わせを用いて,ATP部位特異的キナーゼ阻害剤を設計することができる。モデリングおよびディスプレイ手法を用いて,X線結晶構造の重ね合わせとサブポケット類似性分析により,この情報を増強することができる。既知のキナーゼ阻害剤の大部分はATP拮抗性であり,触媒ドメイン中の結合部位を標的とする。しかし,有用な阻害剤はまた,結合部位中の結合したATPによって占められていない領域を占めるものであってもよい。利用可能な結晶構造について蛋白質データバンクを調査すると,結合した阻害剤のまわりのATP部位の活性化されたまたは部分的に活性化されたコンフォメーションが,広くすべてのキナーゼについて同じであることが認められた(Birault 2006)。さらに,一致した限定された数の一次残基が小分子の結合に関与していることが明らかである。系統発生学的に異なるキナーゼ,例えば,p38αおよびFGF−1は,ATP結合部位において三次元構造上の共通性を示す。この構造的類似性は,特定の個々のキナーゼ,ならびにより大きな分類の後続的に関連するキナーゼを阻害する新規なキナーゼ阻害剤を同定するのに有用な基盤を提供する。
【0098】
別の態様においては,リード化合物は,コンピュータのフィルタを用いて,既知の化合物のデータベースからリード化合物を同定することにより発見される。これらのデータベースのいくつかは,数百万種類の化合物を含みうる。フィルタは,適当なADMET(吸収,分布,代謝,排泄,毒性)特性を組み込むよう設計される。これらの医薬化学的取り扱い用のフィルタは,所望の化学的特性のリストに基づく。ADMETモデリングを化合物の最適化の間に用いて,所望の特性を持つと思われる化合物を含む許容可能な特性空間を定義する。ある態様においては,2以上のコンピュータフィルタを適用して既知の化合物を分析する。適用しうるフィルタとしては,限定されないが,the Lipinski filter(5のルール),the Veber(2のルール)filter,Chem GPS,MDDR filter,Shoichet’s Aggregators,Martinfilter,Ghosefilter,Eganfilter,Med Chemtractibility filter,Leadlikeness,Caco−2 permeation filterおよびthe Muegge filterが挙げられる。これらのフィルタを,所望の特性,例えば,水溶性,分子量,SlogP,およびH結合ドナーまたはアクセプターの数を有する任意の化合物をスクリーニングするよう設定することができる。
【0099】
別の態様においては,特定のファミリーのキナーゼを阻害することが知られているかまたは予測される薬剤,例えば無機または有機化合物のライブラリを,ウイルス感染を調節する宿主細胞蛋白質を同定するために用いたものと同じ系を用いて,ウイルス感染を阻害する能力について試験する。ある態様においては,スクリーニングは,siRNAのライブラリを化合物のライブラリで置き換えて,同様にして行う。それぞれのウイルスについて化学物質のスクリーニングの結果をsiRNAスクリーニングの結果と比較して,ランク付けした化合物のリストを作成する。さらに別の態様においては,化合物スクリーニングにおいてウイルス感染を阻害する能力を示した上位のヒット化合物,例えば上位5,10,15,20または25種の化合物についてインビトロ酵素アッセイを行う。ここでは,上位の化合物が宿主細胞標的キナーゼ,またはキナーゼシグナリング経路の上流または下流のキナーゼを阻害する能力についてプロファイリングする。さらに別の態様においては,ウイルス感染の阻害において,および/または宿主細胞キナーゼ標的化の特異性において最も高い有効性を示すトップ化合物を,ウイルス感染の動物モデルを用いて毒性およびインビボ効力について試験する。
【0100】
ある態様においては,特定のキナーゼ,例えば,PAK1,DYRK3,PTK9,およびGPRK2L,またはキナーゼシグナリング経路においてPAK1,DYRK3,PTK9,およびGPRK2Lの上流または下流にあるキナーゼまたは他の物質を標的とする薬剤が同定され,または開発される。
【0101】
試験は,設計された薬剤を宿主細胞キナーゼに対する阻害活性について評価することを含む。ある態様においては,設計された薬剤の集合をコンピュータによる方法により評価して,物理的に薬剤を合成することなく,宿主細胞キナーゼを阻害するその活性を推定する。そのようなコンピュータによる方法は,薬剤の他の特性,例えば,溶解性,膜浸透性,代謝および毒性を推定するためにも用いることができる。
【0102】
ある態様においては,試験は,設計した薬剤を合成し,ウエットラボ手法の1またはそれ以上の生物学的アッセイにおいて,宿主細胞キナーゼを阻害するか,および/またはウイルス感染を阻害するその活性について評価することを含む。
【0103】
次に,合成された薬剤の活性を,宿主細胞キナーゼの阻害,および/またはウイルス感染の阻害を直接的にまたは間接的に反映する生物学的アッセイにより評価することができる。代表的な生物学的アッセイとしては,限定されないが,1)キナーゼ阻害の無細胞実験;2)ウイルス阻害の無細胞実験3)ウイルス感染の阻害の全細胞実験(例えば,ウイルスの伝染,侵入,複製,生合成,アセンブリ,または退出);および4)ウイルス感染に対する効力のインビボ動物モデル,例えば,特定のウイルスに感染したマウス,トリ,霊長類またはブタモデルが挙げられる。
【0104】
インビトロでアッセイに関しては,候補薬剤が宿主細胞キナーゼを阻害する能力は,薬剤を宿主細胞キナーゼの活性測定用のアッセイ混合物と接触させ,薬剤の存在下および非存在下における酵素の活性を測定することにより評価することができる。宿主細胞キナーゼの活性が薬剤の存在下で非存在下におけるより低いことは宿主細胞キナーゼ阻害剤であることを示す。
【0105】
無細胞宿主細胞キナーゼアッセイの一例は,Clerk and Sugden,FEBS Letters,426:93−96(1998)(本明細書に参照として取り込まれる)に記載されるものである。別の例示的システムは,キナーゼプロファイリング技術であるAMBITプラットフォーム(Kinomescan)である。プラットフォームを用いて,分子相互作用を同定し,連結されていない小分子の複数のキナーゼのATP部位への結合の定量的測定特異性を決定することができる。例えば,プラットフォームを用いて阻害剤を分析し,薬剤が他の「オフターゲット」キナーゼと比較していかに強くその目的とするキナーゼ標的に結合するかを明らかにする。この「オフターゲット」結合を用いて,阻害剤の副作用を同定することができ,またはある種の阻害剤を他のウイルスについて評価することを正当化することができる。
【0106】
ウイルス感染に対する応答を反映するために用いられる動物モデルを用いて,インビボでの宿主細胞キナーゼ阻害活性を評価することができる。動物モデルの例としては,限定されないが,マウス,ラット,フェレット,テンジクネズミ,ブタ(Susscrofa),ウマ,霊長類,およびウマが挙げられる。
【0107】
ある態様においては,薬剤の活性ないし効力は,全細胞および/またはIC50またはED50値のインビボアッセイにより測定して,複数の宿主キナーゼに対して同様であり,これは下記に詳細に説明する。ある態様においては,複数の宿主細胞キナーゼに対する単一の薬剤の効力の相違は,約1000倍以下である。さらに別の態様においては,効力の相違は約100倍以下である。さらに別の特定の態様においては,効力の相違は約10倍以下である。
【0108】
VIII.治療方法
本発明の1つの態様は,キナーゼを阻害してウイルス感染を阻害するかまたは低減させる薬剤を含む医薬組成物およびキットを用いる方法に関する。本発明の別の態様は,被検動物を治療するための方法,医薬組成物およびキットを提供する。本明細書において用いる場合,"被検動物"との用語は,ヒトならびに他の哺乳動物を含む。本明細書において用いる場合,"治療する"との用語は治療上の有益性および/または予防上の有益性を達成することを含む。治療上の有益性とは,原因となるウイルス感染の根絶または軽減を意味する。また,治療上の有益性は,被検動物がなお原因となるウイルスに罹患しているにもかかわらず被検動物において改善が観察されるように,原因となるウイルス感染に伴う生理学的症状の1またはそれ以上を根絶または軽減することによっても達成される。
【0109】
予防上の有益性が望ましい態様においては,本発明の医薬組成物を,ウイルス感染,例えば,インフルエンザ,またはHIV感染を発病するリスクを有する患者,またはまだ病気の診断がされていなくてもウイルス感染の生理学的症状の1またはそれ以上を訴える患者に投与することができる。投与によりウイルス感染が発症することを予防することができ,または,発症するウイルス感染を低下させ,減少させ,短期にし,および/またはさもなくば軽減することができる。医薬組成物は,標的キナーゼ活性を調節することができる。ここでは,調節するとの用語は,標的キナーゼの阻害,あるいは標的キナーゼの活性化を含む。
【0110】
蛋白質キナーゼの活性を低下させることはまた,キナーゼを"阻害する"とも表される。"阻害する"との用語,およびその文法的活用形,例えば"阻害的"とは,完全な阻害は必要ではないが,キナーゼ活性の低下を表す。ある態様においては,そのような低下は,阻害効果がない場合,例えば阻害剤の非存在下における酵素の活性の少なくとも50%,少なくとも75%,少なくとも90%,および少なくとも95%であることができる。逆に,"阻害しない"との語句およびその文法的活用形は,薬剤の存在下で酵素活性の20%未満,10%未満,および5%未満の低下がある状況を表す。さらに,"実質的に阻害しない"との語句およびその文法的活用形は,薬剤の存在下で酵素活性の30%未満,20%未満,およびある態様においては10%未満の低下がある状況を表す。
【0111】
蛋白質キナーゼの活性を増加させることは,キナーゼを"活性化する"とも称される。"活性化される"との用語,およびその文法的活用形,例えば,"活性化する"とは,完全に活性化される必要はないが,キナーゼ活性の増加を表す。ある態様においては,そのような増加は,活性化効果がない場合に,例えば,アクチベータの非存在下で,酵素の活性の少なくとも50%,少なくとも75%,少なくとも90%,および95%であることができる。逆に,"活性化しない"との語句およびその文法的活用形は,薬剤の存在下において酵素活性の20%未満,10%未満,および5%未満の増加がある状況を表す。さらに,"実質的に活性化しない"との語句およびその文法的活用形は,薬剤の存在下において酵素活性の30%未満,20%未満,およびある態様においては10%未満の増加がある状況を表す。
【0112】
酵素活性を低下させる能力は,薬剤または薬剤の組み合わせの酵素に向けたまたはこれに対する効力または活性の尺度である。効力は,無細胞,全細胞および/またはインビボアッセイにより,IC50,Kiおよび/またはED50値として測定することができる。IC50値は,所定の条件下で酵素活性を半分(50%)阻害するのに必要な薬剤の濃度を表す。Ki価は阻害剤の酵素への結合の平衡アフィニティー定数を表す。ED50値は,生物学的アッセイにおいて最大の半分の応答を引き起こすのに必要な薬剤の用量を表す。当業者はこれらの尺度の詳細を理解することができ,生化学,酵素学等の標準的な教科書に見いだすことができる。
【0113】
本発明はまた,ウイルス感染を治療するために用いることができるキットを含む。これらのキットは,キナーゼを阻害する薬剤または薬剤の組み合わせを含み,ある態様においては,本明細書に記載される種々の方法およびアプローチにしたがってキットを使用する方法を教示する指示を含む。そのようなキットはまた,薬剤の活性および/または利点を示す情報,例えば,科学参考文献,パッケージ挿入材料,臨床的結果,および/またはこれらの概要を含んでいてもよい。そのような情報は,種々の実験,例えば,インビボモデルを含む実験動物を用いた実験およびヒトの臨床試験に基づく実験の結果に基づくものでありうる。本明細書に記載されるキットは,健康管理者,例えば,医師,看護師,薬剤師,処方薬剤職員等に提供し,販売し,および/または販売促進することができる。
【0114】
A.siRNA療法
二本鎖オリゴヌクレオチドは,一方の鎖のオリゴヌクレオチド配列が第2の鎖のオリゴヌクレオチド配列に相補的である2つの別々のオリゴヌクレオチド配列のアセンブリによって形成される。そのような二本鎖オリゴヌクレオチドは一般に,2つの別々のオリゴヌクレオチド(例えばsiRNA)から,またはそれ自体でフォールディングして二本鎖構造を形成する単一の分子(例えば,shRNAないし短ヘアピンRNA)から組み立てられる。当該技術分野において知られるこれらの二本鎖オリゴヌクレオチドはすべて,デュープレックスの各鎖は異なるヌクレオチド配列を有し,一方のヌクレオチド配列領域(ガイド配列またはアンチセンス配列)のみが標的核酸配列に相補的であり,他方の鎖(センス配列)は標的核酸配列に相同のヌクレオチド配列を含むという,共通の特徴を有する。
【0115】
二本鎖RNAにより誘導される遺伝子サイレンシングは,少なくとも3つの異なるレベルで生じうる:(i)転写不活性化,すなわちRNAによりガイドされるDNAまたはヒストンメチル化;(ii)siRNAにより誘導されるmRNA分解;および(iii)mRNAにより誘導される転写減衰。RNA哺乳動物細胞における誘導性サイレンシング(RNA干渉ないしRNAi)の主要なメカニズムはmRNA分解であると一般に考えられている。RNA干渉(RNAi)は,遺伝子発現を翻訳の段階で阻害するか,または特定の遺伝子の転写を立体的に妨害するメカニズムである。特定のRNAi経路蛋白質は,dsRNAにより誘導されて標的メッセンジャーRNA(mRNA)に向かい,ここで標的を"切断"する,すなわち,これをもはや蛋白質に翻訳されない小さい部分に分解する。RNAiを哺乳動物細胞において用いる最初の試みは,dsRNAの長鎖の使用に焦点が当てられていた。しかし,RNAiを誘導しようとするこれらの試みはほとんど成功しなかった。これは,部分的には,インターフェロン応答が誘導され,標的特異的ではなく一般的な蛋白質合成の阻害が生じたためである。すなわち,長いdsRNAは哺乳動物系においては実行可能なRNAiの選択肢ではない。別の結果は,遺伝子が転写される程度に影響を及ぼす遺伝子のエピジェネティックな変化,すなわち,ヒストン修飾およびDNAメチル化の変化である。
【0116】
最近,短い(18−30bp)RNAデュープレックスを哺乳動物培養細胞に導入すると,インターフェロン応答を誘導することなく標的mRNAの配列特異的阻害が実現可能であることが示された。これらの短いdsRNAのある種のものは,小阻害RNA("siRNA")と称され,モル濃度以下で触媒的に作用して,細胞中で標的mRNAの95%以上を切断することができる。siRNA活性のメカニズムの説明,ならびにその用途のいくつかは,下記に記載されている:Provost et al.,Ribonuclease Activity and RNA Binding of Recombinant Human Dicer,E.M.B.O.J.,2002 Nov.1;21(21):5864−5874;Tabara et al.,The dsRNA Binding Protein RDE−4 Interacts with RDE−1,DCR−1 and a DexH−box Helicaseto Direct RNAi in C.elegans,Cell 2002,June 28;109(7):861−71;Ketting et al.,Dicer Functions in RNA Interference and in Synthesis of Small RNA Involved in Developmental Timing in C.elegans;Martinez et al.,Single−Stranded Antisense siRNA Guide Target RNA Cleavage in RNAi,Cell 2002,September.6;110(5):563;Hutvagner&Zamore,Amicro RNA in a multiple−turnover RNAi enzyme complex,Science 2002,297:2056。
【0117】
機構的な観点からは,長い二本鎖RNAを植物および無脊椎動物細胞に導入すると,Dicerとして知られるIII型エンドヌクレアーゼにより分解されてsiRNAとなる(Sharp,RNAinterference−−2001,Genes Dev.2001,15:485.Dicer,a ribonuclease−III−like enzyme,processes the dsRNA into 19−23 base pair short interfering RNAs with characteristic two base 3’ overhangs;Bernstein,Caudy,Hammond,&Hannon,Role for a bidentate ribonuclease in the initiation step of RNA interference,Nature 2001,409:363)。次にsiRNAはRNA誘導性サイレンシング複合体(RISC)中に取り込まれ,ここで1またはそれ以上のヘリカーゼがsiRNAデュープレックスをほどき,相補的なアンチセンス鎖が標的認識をガイドできるようにする(Nykanen,Haley,&Zamore,ATP requirements and small interfering RNA structure in the RNA interference pathway,Cell 2001,107:309)。適当な標的mRNAに結合すると,RISC中の1またはそれ以上のエンドヌクレアーゼは標的を切断して,サイレンシングを誘導する(Elbashir,Lendeckel,&Tuschl,RNA interference is mediated by 21−and 22−nucleotide RNAs,Genes Dev 2001,15:188,FIG.1.)。
【0118】
一般に,アンチセンス配列は活性なRISC複合体中に保持され,配列特異的RNA干渉を媒介するためにアンチセンス配列と標的配列との相補的塩基対形成によりRISCを標的ヌクレオチド配列へとガイドする。当該技術分野においては,細胞培養系によってはある種の未修飾siRNAが"オフターゲット"効果を示す場合があることが知られている。このオフターゲット効果にはRISC複合体中のsiRNAのアンチセンス配列ではなくセンス配列が関与しているとの仮説がある(例えば,Schwarz et al.,2003,Cell,115,199−208を参照)。この場合,センス配列はRISC複合体を意図した標的配列とは異なる配列(オフターゲット配列)に向かわせ,その結果,オフターゲット配列が阻害されると考えられている。これらの二本鎖核酸分子においては,各鎖は異なる標的核酸配列に相補的である。しかし,これらのdsRNAにより影響を受けるオフターゲットは完全に予測可能なわけではなく,非特異的である。
【0119】
"siRNA"との用語は,RNA干渉(RNAi)経路を誘導する小さい阻害性RNAデュープレックスを表す。これらの分子は,種々の長さであることができ(一般に18−30塩基対),アンチセンス鎖中の標的mRNAに対して種々の程度の相補性を含む。すべてではないがある種のsiRNAは,センス鎖および/またはアンチセンス鎖の5’または3’末端に塩基対形成しないオーバーハング塩基を有する。"siRNA"との用語は,2つの別々の鎖のデュープレックス,ならびにデュープレックス領域を含むヘアピン構造を形成しうる一本鎖を含む。小干渉RNA(siRNA)は,短干渉RNAまたはサイレンシングRNAとしても知られており,生物学において種々の役割を果たす20−25ヌクレオチドの長さの一群の二本鎖RNA分子である。
【0120】
2つのRNA鎖は完全に相補的である必要はないが,鎖はハイブリダイズしてデュープレックス構造を形成するのに十分に相補的でなければならない。ある場合には,相補的RNA鎖は,30ヌクレオチド未満,好ましくは25ヌクレオチド未満の長さであり,より好ましくは19−24ヌクレオチドの長さ,より好ましくは20−23ヌクレオチドの長さ,さらにより好ましくは22ヌクレオチドの長さである。本発明のdsRNAはさらに,少なくとも1つの一本鎖ヌクレオチドオーバーハングを含んでいてもよい。本発明のdsRNAはさらに,置換されるかまたは化学的に修飾されたヌクレオチドを含んでいてもよい。下記に詳細に説明するように,dsRNAは当該技術分野において知られる標準的な方法により合成することができる。
【0121】
siRNAは,これらが培養細胞株において誘導するサイレンシングのレベルまたは程度に基づいて,5つの群に分類することができる(非機能的,準機能的,機能的,高機能的,および超機能的)。本明細書において用いる場合,これらの定義は,siRNAが100nMの濃度で前記細胞株にトランスフェクトされ,トランスフェクションのおよそ24時間後であって,トランスフェクション後72時間を越えない時点でサイレンシングのレベルが試験される一連の状態に基づく。この文脈において,"非機能的siRNA"とは,50%未満(<50%)の標的サイレンシングを誘導するsiRNAとして定義される。"準機能的siRNA"は50−79%の標的サイレンシングを誘導する。"機能的siRNA"は80−95%の遺伝子サイレンシングを誘導する分子である。"高機能的siRNA"は,95%より高い遺伝子サイレンシングを誘導する分子である。"超機能的siRNA"は,特定の群の分子である。この書面の目的のために,超機能的siRNAは,以下の分子として定義される:(1)ナノモル以下の濃度(例えば,1ナノモル未満)でトランスフェクションしたときに特定の標的の95%より高いサイレンシングを誘導する;および/または(2)96時間を越えて機能的(またはより高い)レベルのサイレンシングを誘導する。これらの相対的機能性(絶対的であることを意図するものではないが)を用いて,機能ゲノミクス,標的同定および治療などの用途のために,siRNAを特定の標的と比較することができる。
【0122】
A.マイクロRNA療法
マイクロRNA(miRNA)は約21−23ヌクレオチドの長さの一本鎖RNA分子であり,遺伝子発現を制御する。miRNAは,遺伝子によりコードされ,DNAから転写されるが,蛋白質には翻訳されない(非コーディングRNA)。その代わりに,これはpri−miRNAとして知られる一次転写産物からプロセシングされて,pre−miRNAと称される短いステム−ループ構造となり,最後に機能的miRNAとなる。成熟miRNA分子は,1またはそれ以上のメッセンジャーRNA(mRNA)分子に部分的に相補的であり,その主な機能は遺伝子発現を下方制御することである。
【0123】
IX.製剤,投与の経路,および有効量
本発明のさらに別の観点は,本発明の薬剤またはその組み合わせを含む医薬組成物の製剤,投与経路および有効投与量に関する。そのような医薬組成物は,上述のように,ウイルス感染の治療に用いることができる。
【0124】
薬剤またはその薬学的に許容しうる塩は,単独で,または1またはそれ以上の他の薬剤との組み合わせで,または1またはそれ以上の他の形態との組み合わせで提供することができる。例えば,製剤は,各薬剤の相対的効力および意図される適応症に応じて,1またはそれ以上の薬剤を特定の比率で含んでいてもよい。例えば,2つの異なる宿主標的を標的とする組成物において,効力が類似する場合,約1:1比の薬剤を用いることができる。2つの形態は,同じ単位投与形態中で,例えば1つのクリーム,座剤,錠剤,カプセル,エアロゾルスプレイ,または飲料に溶解すべき粉体包中に一緒に製剤することができる。あるいは,それぞれの形態を,例えば2つのクリーム,2つの座剤,2つの錠剤,2つのカプセル,錠剤と錠剤を溶解するための液体,2つのエアロゾルスプレイ,または粉体包と粉体を溶解するための液体等の,別々の単位として製剤してもよい。
【0125】
“薬学的に許容しうる塩”との用語は,本発明において用いられる薬剤の生物学的有効性および特性を維持し,生物学的にまたは他の点で望ましくないものではない塩を意味する。例えば,薬学的に許容しうる塩は,本発明の薬剤がキナーゼ,例えば,PAK1,DYRK3,PTK9,およびGPRK2Lからなる群より選択されるキナーゼを阻害する有益な効果を妨害しない。
【0126】
典型的な塩は,無機イオン,例えば,ナトリウム,カリウム,カルシウム,マグネシウムイオン等の塩である。そのような塩には,無機または有機酸,例えば,塩酸,臭化水素酸,リン酸,硝酸,硫酸,メタンスルホン酸,p−トルエンスルホン酸,酢酸,フマル酸,コハク酸,乳酸,マンデル酸,リンゴ酸,クエン酸,酒石酸またはマレイン酸との塩が含まれる。さらに,薬剤がカルボキシ基または他の酸性の基を持つ場合,これを無機または有機塩基との薬学的に許容しうる付加塩に変換することができる。適当な塩基の例としては,水酸化ナトリウム,水酸化カリウム,アンモニア,シクロヘキシルアミン,ジシクロヘキシルアミン,エタノールアミン,ジエタノールアミン,トリエタノールアミン等が挙げられる。
【0127】
薬学的に許容しうるエステルまたはアミドとは,本発明において用いられる薬剤の生物学的有効性および特性を維持し,生物学的にまたは他の点で望ましくないものではないエステルまたはアミドを表す。例えば,エステルまたはアミドは,本発明の薬剤がキナーゼ,例えば,PAK1,DYRK3,PTK9,およびGPRK2Lからなる群より選択されるキナーゼを阻害する有益な効果を妨害しない。典型的なエステルとしては,エチル,メチル,イソブチル,エチレングリコール等が挙げられる。典型的なアミドとしては,未置換アミド,アルキルアミド,ジアルキルアミド等が挙げられる。
【0128】
ある態様においては,薬剤は,1またはそれ以上の他の化合物,形態,および/または薬剤,例えば上述したものと組み合わせて投与することができる。キナーゼ阻害剤と1またはそれ以上の他の活性薬剤との組み合わせを含む医薬組成物は,ある一定のモル比を含むよう製剤することができる。例えば,約99:1から約1:99のモル比のキナーゼ阻害剤:他の活性薬剤を用いることができる。この態様の一部においては,キナーゼ阻害剤:他の活性薬剤のモル比の範囲は,約80:20−約20:80;約75:25−約25:75,約70:30−約30:70,約66:33−約33:66,約60:40−約40:60;約50:50;および約90:10−約10:90から選択される。キナーゼ阻害剤:他の活性薬剤のモル比は,約1:9であってもよく,ある態様においては約1:1であってもよい。2つの薬剤,形態および/または化合物は,同じ投与単位として,例えば,1つのクリーム,座剤,錠剤,カプセル,または飲料に溶解すべき粉体パケット中で一緒に製剤してもよく,またはそれぞれの薬剤,形態,および/または化合物を別々の単位として,例えば,2つのクリーム,座剤,錠剤,2つのカプセル,錠剤と錠剤を溶解するための液体,エアロゾルスプレイ,粉体のパックおよび粉体を溶解するための液体等で製剤してもよい。
【0129】
必要であるかまたは望ましい場合には,薬剤および/または薬剤の組み合わせは,さらに別の薬剤とともに投与してもよい。本発明の薬剤および/または薬剤の組み合わせとともに投与することができる薬剤の選択は,少なくとも部分的には,治療すべき状態によって異なる。本発明の製剤において有用な特定の薬剤としては,例えば,ウイルス感染に治療効果を有する任意の薬剤,例えば,炎症性状態を治療するために用いられる薬剤が挙げられる。例えば,インフルエンザの治療用には,ある態様においては,本発明の組成物はさらに,1またはそれ以上のNSAID等の抗炎症剤,例えば,イブプロフェン,ナプロキセン,アセトミノフェン,ケトプロフェン,またはアスピリンを含んでいてもよい。インフルエンザの治療用の別の態様においては,本発明の製剤はさらに,1またはそれ以上の慣用の抗インフルエンザウイルス薬,例えば,アマンタジン,リマンタジン,ザナミビル,およびオセルタミビルを含んでいてもよ。HIV等のレトロウイルス感染の治療においては,本発明の製剤はさらに,1またはそれ以上の慣用の抗ウイルス剤,例えば,プロテアーゼ阻害剤(ロピナビル/リトナビル{Kaletra},インジナビル{Crixivan},リトナビル{Norvir},ネルフィナビル{Viracept},サキナビルハードゲルカプセル{Invirase},アタザナビル{Reyataz},アンプレナビル{Agenerase},フォサンプレナビル{Telzir},チプラナビル{Aptivus}),リバーストランスクリプターゼ阻害剤,例えば,非ヌクレオシドおよびヌクレオシド/ヌクレオチド阻害剤(AZT{ジドブジン,Retrovir},ddI{ジダノシン,Videx},3TC{ラミブジン,Epivir},d4T{スタブジン,Zerit},アバカビル{Ziagen},FTC{エントリシタビン,Emtriva},テノフォビル{Viread},エファビレンツ{Sustiva}およびネビラピン{Viramune}),融合阻害剤T20{エンフビルチド,Fuzeon},インテグラーゼ阻害剤(MK−0518およびGS−9137),および成熟阻害剤(PA−457{Bevirimat})を含んでいてもよい。別の例としては,製剤はさらに1またはそれ以上のサプリメント,例えばビタミンC,Eまたは他の抗酸化剤を含んでいてもよい。
【0130】
薬剤(またはその薬学的に許容しうる塩,エステルまたはアミド)は,それ自体で投与してもよく,活性薬剤が1またはそれ以上の薬学的に許容しうる担体との混合剤ないし混合物である医薬組成物の形で投与してもよい。
【0131】
本明細書において用いる場合,医薬組成物とは,被験者に投与すべく調製された任意の組成物でありうる。本発明にしたがって使用するための医薬組成物は,1またはそれ以上の生理学的に許容しうる担体を用いて,例えば,賦形剤,希釈剤,および/または,例えば活性な薬剤を投与可能な調製物に加工することを容易にする助剤を含むよう,慣用の手法で製剤することができる。適当な製剤は,少なくとも部分的には,選択される投与経路によって異なるであろう。本発明において有用な薬剤,またはその薬学的に許容しうる塩,エステルまたはアミドは,投与の多くの経路ないしモードを用いて,例えば,経口,頬,局所,経直腸,経皮,経粘膜,皮下,静脈内,および筋肉内適用,ならびに吸入により,患者にデリバリーすることができる。
【0132】
経口投与用には,薬剤は,活性薬剤を当該技術分野においてよく知られる薬学的に許容しうる担体と混合することにより容易に製剤することができる。そのような担体は,本発明の薬剤を,錠剤として,例えば,チュアブル錠,丸薬,糖衣錠,カプセル,トローチ,硬質キャンディー,液体,ゲル,シロップ,スラリー,粉体,懸濁液,エリキシル,ウエハス等として,治療すべき患者が経口で摂取できるように製剤することを可能とする。そのような製剤は,薬学的に許容しうる担体,例えば,固形希釈剤または増量剤,滅菌水性媒体および種々の無毒性有機溶媒を含むことができる。一般に,本発明の薬剤は,経口投与用形態の全組成の約0.5%,約5%,約10%,約20%,または約30%から約50%,約60%,約70%,約80%または約90重量%の範囲の濃度レベルで,所望の単位投与形態を与えるのに十分な量で含まれる。
【0133】
経口で使用するための水性懸濁液は,本発明の薬剤を,薬学的に許容しうる賦形剤,例えば,懸濁剤(例えば,メチルセルロース),湿潤剤(例えば,レシチン,リソレシチンおよび/または長鎖脂肪族アルコール),ならびに着色剤,保存剤,甘味料等とともに含むことができる。
【0134】
ある態様においては,例えば大きな親油性成分の存在のため,薬剤を溶液とするために油または非水性溶媒が必要であるかもしれない。あるいは,乳濁液,懸濁液,または他の調製物,例えば,リポソーム調製物を用いてもよい。リポソーム製剤に関しては,病気の治療用のリポソームを製造するための既知の任意の方法を用いることができる。例えば,Bangham et al.,J.Mol.Biol.23:238−252(1965)およびSzoka et al.,Proc.Natl Acad.Sci.USA 75:4194−4198(1978)(本明細書に参照として取り込まれる)を参照。リポソームにリガンドを結合させて,これらの組成物が特定の作用部位に向かうようにしてもよい。本発明の薬剤はまた食品,例えばクリームチーズ,バター,サラダドレッシング,またはアイスクリーム中に組み込んで,可溶化,投与,および/またはある患者集団におけるコンプライアンスを容易にすることができる。
【0135】
経口使用用の医薬製剤は,固体賦形剤として得ることができ,得られた混合物を任意にすりつぶし,顆粒の混合物を加工し,所望の場合には適当な助剤を加えた後,錠剤または糖衣錠コアを得ることができる。適当な賦形剤は,特に,糖等の増量剤,例えばラクトース,ショ糖,マンニトール,またはソルビトール;香味要素,セルロース調製物,例えば,トウモロコシデンプン,小麦デンプン,米デンプン,ジャガイモデンプン,ゼラチン,トラガガントガム,メチルセルロース,ヒドロキシプロピルメチルセルロース,カルボキシメチルセルロースナトリウム,および/またはポリビニルピロリドン(PVP)である。所望の場合には,崩壊剤,例えば,架橋ポリビニルピロリドン,寒天,またはアルギン酸またはその塩,例えばアルギン酸ナトリウムを加えてもよい。薬剤はまた,持続放出製剤として製剤してもよい。
【0136】
糖衣錠のコアは,適当なコーティングとともに提供することができる。この目的のために,濃縮糖溶液を用いることができ,これは任意にアラビアゴム,タルク,ポリビニルピロリドン,カルボポールゲル,ポリエチレングリコール,および/または二酸化チタン,ラッカー溶液,および適当な有機溶媒または溶媒混合物を含むことができる。活性薬剤の異なる組み合わせの識別または特徴付けのために,染料または顔料を錠剤または糖衣錠のコーティングに加えてもよい。
【0137】
経口で使用することができる医薬製剤としては,ゼラチンから形成されるプッシュフィットカプセル,ならびにゼラチンと可塑剤,例えばグリセロールまたはソルビトールから形成される柔らかい密封カプセルが挙げられる。プッシュフィットカプセルは,活性成分を,ラクトース等の増量剤,スターチ等の結合剤,および/またはタルクまたはステアリン酸マグネシウム等の潤滑剤,および任意に,安定剤等との混合物として含むことができる。軟カプセルにおいては,活性薬剤は適当な液体,例えば油脂,液体パラフィン,または液体ポリエチレングリコールに溶解または懸濁することができる。さらに,安定剤を加えてもよい。経口投与用のすべての製剤は,投与に適した用量でなければならない。
【0138】
注射用には,本発明の薬剤を水性溶液,例えば,限定されないが,生理学的に適合性のバッファ,例えば,ハンクス溶液,リンゲル,または生理学的食塩水バッファ中で製剤することができる。そのような組成物はまた,1またはそれ以上の賦形剤,例えば,保存剤,溶解剤,増量剤,潤滑剤,安定剤,アルブミン等を含むことができる。製剤の方法は当該技術分野において知られており,例えば,Remington’s Pharmaceutical Sciences,最新版,Mack Publishing Co.,Easton Pに開示されている。
【0139】
上述した製剤に加えて,薬剤はまた,デポ調製物として製剤してもよい。そのような長期作用性製剤は,移植または経皮デリバリー(例えば皮下または筋肉内),筋肉内注射または経皮パッチの使用により投与することができる。すなわち,例えば,薬剤は,適当なポリマー性または疎水性材料(例えば,許容しうる油中の乳濁液として)またはイオン交換樹脂とともに,または溶けにくい誘導体として,例えば溶けにくい塩として製剤することができる。
【0140】
ある態様においては,本発明の1またはそれ以上の薬剤を含む医薬組成物は,局所的に投与したとき,または感染の特定の部位またはその近くに注射したとき,局所的および領域的効果を発揮する。例えば,粘稠液体,ゲル,ゼリー,クリーム,ローション,軟膏,座剤,泡状物,またはエアロゾルスプレイの直接局所投与を局所投与用に用いて,例えば,局所的および/または領域的効果を生じさせてもよい。そのような製剤用の薬学的に適したベヒクルとしては,例えば,低級脂肪族アルコール,ポリグリコール(例えば,グリセロールまたはポリエチレングリコール),脂肪酸のエステル,油,脂肪,シリコーン等が挙げられる。そのような製剤はまた,保存剤(例えば,p−ヒドロキシ安息香酸エステル)および/または抗酸化剤(例えば,アスコルビン酸およびトコフェロール)を含んでいてもよい。Dermatological Formulations:Percutaneous absorption,Barry(Ed.),Marcel Dekker Incl,1983も参照。ある態様においては,キナーゼ阻害剤を含む局部/局所製剤を用いて,表皮または粘膜のウイルス感染を治療する。
【0141】
本発明の医薬組成物は,化粧品用のまたは皮膚科学的に許容しうる担体を含んでいてもよ。そのような担体は,皮膚,爪,粘膜,組織および/または頭髪と適合性であり,これらの要件を満たす慣用的に用いられている任意の化粧品用のまたは皮膚科学的担体を含むことができる。そのような担体は,当業者が容易に選択することができる。皮膚用軟膏の製剤に際しては,本発明の薬剤または薬剤の組み合わせを油性炭化水素基剤,無水吸収基剤,油中水吸収基剤,水中油の水除去可能基剤および/または水溶性基剤中で製剤することができる。
【0142】
本発明にしたがう組成物は,局所適用に適した任意の形態,例えば,水性,水性−アルコール性または油性溶液,ローションまたは血清分散物,水性,無水または油性ゲル,油脂相を水性相中に分散させることにより得られる乳濁液(O/Wないし水中油)またはその逆(W/Oないし油中水),マイクロ乳濁液,あるいはマイクロカプセル,マイクロ粒子または脂質ベシクルのイオン性および/または非イオン性分散物であることができる。これらの組成物は,一般的な方法にしたがって製造することができる。本発明の薬剤の他,本発明にしたがう組成物の種々の構成成分の量は,当該技術分野において慣用的に用いられている量である。特に,これらの組成物は,保護,治療またはケア用の,または皮膚洗浄用の顔面,手,身体および/または粘膜用のクリーム,ミルク,ローション,ゲルまたは泡状物を構成する。組成物はまた,石けんまたはクレジングバーを構成する固体調製物からなるものであってもよい。
【0143】
本発明の組成物はまた,化粧品および皮膚科学の分野で一般的な助剤,例えば,親水性または親油性ゲル化剤,親水性または親油性の活性薬剤,保存剤,抗酸化剤,溶媒,香料,増量剤,サンスクリーン,臭い吸収剤および染料を含んでいてもよい。これらの種々の助剤の量は,該当する分野において一般的に用いられている量であり,例えば,組成物の総重量の約0.01%−約20%である。これらの助剤は,その性質によって,油脂相に,水性相に,および/または脂質ベシクル中に導入することができる。
【0144】
ある態様においては,眼のウイルス感染は,本発明の薬剤または薬剤の組み合わせを含む眼科溶液,懸濁液,軟膏または挿入物を用いて有効に治療することができる。
【0145】
ある態様においては,耳のウイルス感染は,本発明の薬剤または薬剤の組み合わせを含む耳用溶液,懸濁液,軟膏または挿入物を用いて有効に治療することができる。
【0146】
ある態様においては,本発明の薬剤は,懸濁液の形ではなく,可溶化形でデリバリーし,このことにより,作用部位へのより速く定量的な吸収が可能となる。一般に,ゼリー,クリーム,ローション,座剤および軟膏等本発明の薬剤により長く暴露される領域を提供することができるが,一方,溶液中の製剤,例えば,スプレイは,より迅速な短時間の暴露を提供する。
【0147】
局部/局所適用に関連するある態様においては,医薬組成物は,1またはそれ以上の浸透促進剤を含むことができる。例えば,製剤は,本発明の薬剤または薬剤の組み合わせが透過バリア,例えば皮膚を越える浸透を増加させるかまたはそのデリバリーを助ける適当な個体またはゲル相の担体または賦形剤を含んでいてもよい。これらの浸透促進化合物の多くは局所用製剤の分野で知られており,例えば,水,アルコール類(例えば,メタノール,エタノール,2−プロパノール等のテルペン),スルホキシド類(例えば,ジメチルスルホキシド,デシルメチルスルホキシド,テトラデシルメチルスルホキシド),ピロリドン類(例えば,2−ピロリドン,N−メチル−2−ピロリドン,N−(2−ヒドロキシエチル)ピロリドン),ラウロカプラム,アセトン,ジメチルアセトアミド,ジメチルホルムアミド,テトラヒドロフルフリルアルコール,L−α−アミノ酸,アニオン性,カチオン性,両イオン性または非イオン性界面活性剤(例えば,ミリスチン酸イソプロピルおよびラウリル硫酸ナトリウム),脂肪酸,脂肪族アルコール(例えばオレイン酸),アミン,アミド,クロフィブリン酸アミド,ヘキサメチレンラウラミド,蛋白質分解酵素,α−ビスアボロール,d−リモネン,尿素およびN,N−ジエチル−m−トルアミド等が含まれる。さらに別の例としては,保湿剤(例えば尿素),グリコール(例えば,プロピレングリコールおよびポリエチレングリコール),グリセロールモノラウレート,アルカン,アルカノール,ORGELASE,炭酸カルシウム,リン酸カルシウム,種々の糖類,デンプン,セルロース誘導体,ゼラチン,および/または他のポリマーが挙げられる。ある態様においては,医薬組成物は,1またはそれ以上のそのような浸透促進剤を含むことができる。
【0148】
ある態様においては,局部/局所に適用するための医薬組成物は,1またはそれ以上の抗微生物性保存剤,例えば,四級アンモニウム化合物,有機水銀剤,p−ヒドロキシ安息香酸,芳香族アルコール,クロロブタノール等を含むことができる。
【0149】
胃腸のウイルス感染は,経口または経直腸でデリバリーされる,本発明の薬剤または薬剤の組み合わせを含む溶液,懸濁液,軟膏,浣腸剤および/または座剤を用いて有効に治療することができる。
【0150】
呼吸器ウイルス感染は,本発明の薬剤または薬剤の組み合わせを含むエアロゾル溶液,懸濁液または乾燥粉体を用いて有効に治療することができる。吸入による投与は,肺のウイルス感染,例えばインフルエンザの治療に特に有用である。エアロゾルは,呼吸器系または鼻腔経路を通して投与することができる。例えば,当業者は,本発明の組成物を適当な担体,例えば,薬学的に許容しうる噴射剤に懸濁するかまたは溶解し,経鼻スプレイまたは吸入を用いて肺に直接投与しうることを理解するであろう。例えば,キナーゼ阻害剤を含むエアロゾル製剤は,例えば鼻スプレイまたは吸入剤として投与するための噴射剤または溶媒と噴射剤との混合物に溶解し,懸濁し,または乳化することができる。エアロゾル製剤は,当該技術分野において一般に用いられているように,任意の許容可能な噴射剤,例えば化粧品用のまたは皮膚科学的にまたは薬学的に許容しうる噴射剤を加圧下に含むことができる。
【0151】
鼻腔投与用のエアロゾル製剤は,一般に液滴またはスプレイの形で鼻腔に投与するよう設計された水性溶液である。鼻腔溶液は一般に等張であり約5.5から約6.5のpHを維持するようわずかに緩衝されている点において鼻分泌物と類似しうるが,この範囲外のpH値を用いることもできる。抗微生物剤または保存剤を製剤中に含めてもよい。
【0152】
吸入用のエアロゾル製剤および吸入抗原は本発明の薬剤または薬剤の組み合わせが鼻腔または口腔呼吸器経路で投与されたときに被験者の呼吸器樹中に運ばれるよう設計することができる。吸入溶液は,例えば,ネブライザにより投与することができる。細かく粉砕したまたは液体の薬剤を含む吸入剤または送気剤は,薬剤または薬剤の組み合わせの溶液または懸濁液が,例えば吹き出しを助けるために噴射剤に含まれる医薬エアロゾルとして呼吸器系にデリバリーすることができる。噴射剤は,液化ガス,例えば,ハロカーボン,例えば,フッ化炭素,例えばフッ化塩化炭化水素,ヒドロクロロフルオロカーボン,およびヒドロクロロカーボン,ならびに炭化水素および炭化水素エーテルであることができる。
【0153】
本発明において有用なハロカーボン噴射剤としては,すべての水素がフッ素で置き換えられているフッ化炭素噴射剤,すべての水素が塩素および少なくとも1つのフッ素で置き換えられている塩化フッ化炭素噴射剤,水素含有フッ化炭素噴射剤,および水素含有塩化フッ化炭素噴射剤が挙げられる。ハロカーボン噴射剤は,Johnson,米国特許5,376,359(1994年12月27日発行);Byronら,米国特許5,190,029(1993年5月2日発行);およびPurewalら,米国特許5,776,434(1998年7月7日発行)に記載されている。本発明において有用な炭化水素噴射剤としては,例えば,プロパン,イソブタン,n−ブタン,ペンタン,イソペンタンおよびネオペンタンが挙げられる。炭化水素のブレンドもまた噴射剤として用いることができる。エーテル噴射剤は,例えば,ジメチルエーテルおよびエーテルが挙げられる。本発明のエアロゾル製剤はまた,2以上の噴射剤を含んでいてもよい。例えば,エアロゾル製剤は,同じクラスからの2種以上の噴射剤,例えば,2種またはそれ以上のフッ化炭素を含んでいてもよく;または異なるクラスからの2種以上,3種以上,または4種以上の噴射剤,例えば,フッ化炭化水素および炭化水素を含んでいてもよい。本発明の医薬組成物はまた,圧縮ガス,例えば,二酸化炭素,亜酸化窒素または窒素等の不活性ガスとともに調剤してもよい。
【0154】
エアロゾル製剤はまた,他の成分,例えば,エタノール,イソプロパノール,プロピレングリコール,ならびに界面活性剤または他の成分,例えば,油および界面活性剤を含んでいてもよい。これらの成分は,製剤を安定化させ,および/または,バルブ部材を潤滑化させるよう作用することができる。
【0155】
エアロゾル製剤は,加圧下で充填して,溶液,懸濁液,乳濁液,粉体および半固体調製物を用いてエアロゾルとして製剤することができる。例えば,溶液エアロゾル製剤は,本発明の薬剤,例えば,キナーゼ阻害剤の溶液を,(実質的に)純粋な噴射剤中に,または噴射剤と溶媒との混合物として含むことができる。溶媒は,薬剤を溶解するために,および/または噴射剤の蒸発を遅延させるために用いることができる。本発明において有用な溶媒としは,例えば,水,エタノールおよびグリコールが挙げられる。適当な溶媒の任意の組み合わせを用いることができ,任意にこれを保存剤,抗酸化剤,および/または他のエアロゾル成分と組み合わせてもよい。
【0156】
エアロゾル製剤はまた,分散液または懸濁液であってもよい。懸濁液エアロゾル製剤は,本発明の薬剤,例えばキナーゼ阻害剤の懸濁液または本発明の薬剤の組み合わせ,および分散剤を含むことができる。本発明において有用な分散剤としては,例えば,ソルビタントリオレエート,オレイルアルコール,オレイン酸,レシチンおよびコーン油が挙げられる。懸濁エアロゾル製剤はまた,潤滑剤,保存剤,抗酸化剤,および/または他のエアロゾル成分を含んでいてもよい。
【0157】
エアロゾル製剤は,乳濁液と同様にして製剤することができる。乳濁液エアロゾル製剤は,例えば,エタノール等のアルコール,界面活性剤,水および噴射剤,ならびに本発明の薬剤,例えばキナーゼ阻害剤または薬剤の組み合わせを含むことができる。用いることができる界面活性剤は,非イオン性,アニオン性またはカチオン性であってもよい。乳濁液エアロゾル製剤の1つの例は,例えば,エタノール,界面活性剤,水および噴射剤を含む。乳濁液エアロゾル製剤の別の例は,例えば,植物油,モノステアリン酸グリセリルおよびプロパンを含むことができる。
【0158】
本発明において用いるのに適した医薬組成物には,活性成分が有効量で,すなわち,少なくとも1つのウイルス感染を有する宿主において治療的および/または予防的有益性を達成するのに有効な量で存在する組成物が含まれる。特定の用途に有効な実際の量は,治療している状態,被験者の状態,製剤,および投与の経路,ならびに当業者に知られる他の因子によって異なるであろう。有効量のキナーゼ阻害剤の決定は,本明細書の開示に鑑みて十分に当業者の能力の範囲内であり,日常的な最適化手法を用いて決定される。
【0159】
ヒトにおいて用いるための有効量は,動物モデルから決定することができる。例えば,動物において有効であることが見いだされた循環,肝臓,局所,および/または胃腸濃度を達成するよう,ヒト用の投与量を製剤することができる。当業者は,特に本明細書に記載される動物モデル実験データに鑑みて,ヒトにおいて用いるための有効量を決定することができる。当業者は,動物データ,および他のタイプの同様のデータに基づいて,ヒトに適した本発明の組成物の有効量を決定することができる。
【0160】
本発明の薬剤または薬剤の組み合わせについて述べる場合,有効量とは一般に,医療または医薬品の分野における種々の規制機関ないし諮問機関(例えば,FDA,AMA)のいずれかにより,または製造元または供給元により推奨または認可された投与量範囲,投与のモード,処方物等を表す。
【0161】
さらに,キナーゼ阻害剤の適切な投与量は,インビトロの実験結果に基づいて決定することができる。例えば,ある薬剤がキナーゼ,例えばPAK1,DYRK3,PTK9,およびGPRK2Lを阻害するインビトロの効力は,同様の生物学的効果を達成するのに有効なインビボ投与量の開発に有用な情報を提供する。
【0162】
ある態様においては,本発明の薬剤の投与は間欠的であってもよく,例えば,2日に1回,3日に1回,5日に1回,1週間に1回,1か月に1または2回等の投与であってもよい。ある態様においては,量,形態,および/または種々の形態の量は,投与の異なる時点で様々であってもよい。
【0163】
当業者は,患者において特定の薬剤の投与の効果をモニタリングすることができる。例えば,HIVウイルスの負荷レベルは,当該技術分野において標準的な手法,例えば,CD4細胞のカウントの測定,および/またはPCRによるウイルスレベルの検出により測定することができる。他の手法も当業者には明らかである。
【0164】
バイオテロリズム
本発明は,バイオテロリストの攻撃により引き起こされるウイルス感染の治療に用いることができる。バイオテロリストの攻撃に用いることができるウイルスとしては,例えば,天然痘を引き起こす大痘瘡ウイルス;脳炎ウイルス,例えば西部ウマ脳炎ウイルス,東部ウマ脳炎ウイルス,ベネズエラウマ脳炎ウイルス,およびアレナウイルス(Lassa,Machupo),ブンヤウイルス,フィロウイルス(Ebola,Marburg),および出血熱を引き起こすフラビウイルスが挙げられる。本発明は,バイオテロリストの攻撃により引き起こされるウイルス感染の治療において用いて,対応医療を強化するために備蓄することができる。
【実施例】
【0165】
実施例1
ワクシニアウイルスの侵入メカニズムの評価
ワクシニアウイルスの細胞内への侵入について研究するため,まず,コア蛋白質の1つであるA5のN末端に単量体黄色蛍光蛋白質(mYFP−MV)のタグを付けた組換え成熟ウイルス粒子(MV)を作製した。これをEGFPタグ付きアクチンを発現するHeLa細胞に加えると,明るく蛍光を発する多くのウイルスが糸状仮足に結合し,糸状偽足に沿って細胞体に向かって移動する(「サーフィンする」)ことが見いだされた(図1A)。この移動は,一般に滑らかであり,中断されず,アクチンの逆行の流れとほぼ同じ速度であった(約1μm/分;図1B)。MVが細胞体に到達すると,劇的な変化が起こった。すなわち,細胞膜のウイルスと接触した位置から大きなブレブが突出した。この拡張は30+/−4秒間続き,ブレブは10+/−2秒間拡張したままでであり,その後ウイルスとともに引っ込められた(図1C)。ブレブ形成のピークはウイルス添加後30分間続き,細胞の40%が1またはそれ以上のブレブを示した(図1D)。
【0166】
間接免疫蛍光法は,ブレブは,アクチン−GFPに加えて,種々のアクチン付随蛋白質,例えば,Rac1,RhoA,エズリンおよびコルタクチン(図1E)を含むことを示した。ミオシンII阻害剤であるブレビスタチン(Limouze et al.,2004)は,ブレブの形成を妨害し,ウイルス感染を62%阻害した。このことは,ブレブ形成が増殖性侵入において役割を果たすことを示唆する(図1F)。複数のHeLaおよびBSC40ミドリザル腎臓細胞株において同様の結果が見られた。
【0167】
ウイルスの一部は糸状偽足を用いて細胞体に到達するのみならず,これらはシグナリングカスケードを誘発し,その結果,過渡的な膜ブレブが形成された。ブレブは,細胞運動,細胞質分裂,およびアポトーシスの間に観察されるものと似ていた(Charras et al.,2006;Fishkind et al.,1991;Mills et al.,1998)。
【0168】
図1は,成熟ビリオン侵入の間のサーフィンおよび膜摂動を示す。図1Aは,糸状仮足に沿ったMVのサーフィンを示す。組換えA5−YFP MVを過渡的にトランスフェクトしたGFP−アクチンを発現するHeLa細胞に加えた。画像は,37℃で1Hzで2.5分間取得した。時点はビリオンのリアルタイム画像に対応する。矢印は個々のビリオンを示す。図1Bは,MVのサーフィンの速度を示す。36個の個別のビリオンの速度を,時間あたり移動した距離の相違により測定した(μm/分間)。図1Cは膜ブレブ形成の誘導を示す。組換えA5−YFP MVをGFP−アクチンを発現するHeLa細胞に加え,37℃で1Hzで2.5分間画像を取得した。矢印はブレブが崩壊した部位におけるアクチンパッチ形成を示す。図1Dは,MVにより誘導される細胞のブレブ形成の経時変化を示す。MVをHeLa細胞に4℃で1時間結合させた。細胞を洗浄し,示された時間37℃にシフトした後,4%FAで固定した。各時点につき50個の細胞をブレブ形成について評点し,細胞のブレブ形成の未感染対照に対するパーセントとして示す。実験は三重に行い,結果を平均した。図1Eは,ブレブに局在化する細胞性因子の決定を示す。示される蛍光タグ付き蛋白質で過渡的にトランスフェクトしたHeLa細胞にMVを感染させるか,または未処理のままにした。細胞を感染後30分間固定し(mpi),共焦点顕微鏡により分析した。細胞1個あたり10個のZステーク(Z−stakes)を回収し,Z投影で示した。図1Fはブレブ形成および感染能を示す。HeLa細胞を種々の濃度のブレビスタチンで前処理した後,初期/後期ウイルスプロモーターからEGFPを発現する組換えMV(EGFP−MV)に感染させた。感染した細胞のパーセンテージをFACS分析により測定した。感染した細胞の対照感染に対するパーセンテージを示す。実験は三重に行い,結果を平均した。
【0169】
次に,関与する特定の細胞性キナーゼを同定するために,HeLa細胞において干渉(si)RNAを用いて50種類のキナーゼをサイレンシングすることによりスクリーニングを行った。単一のsiRNAでトランスフェクションした後,早期/後期プロモーターからEGFPを発現する組換えMV(EGFP−MV)を加え,12時間後にEGFP発現について細胞を分析した。各遺伝子につき3つの異なるsiRNAを用い,模擬感染細胞または対照siRNAでトランスフェクションした細胞と比較して3倍のEGFPシグナルの抑制を有意とした。50種類のキナーゼのうち,PAK1がEGFP発現を阻害することが見いだされ,これは,ウイルスの結合,侵入,転写,または初期遺伝子の翻訳が抑制されたことを意味する。
【0170】
感染におけるPAK1の必要性を検証するために,まず,2つの追加のsiRNAを用い,イムノブロッティングにより判定して,これらがPAK1の82%および76%のノックダウンを引き起こすことを見いだした(図2A;上)。蛍光活性化細胞ソーティング(FACS)に基づく感染アッセイは,感染した細胞の数がそれぞれ74%および68%減少したことを示した(図2A;下)。先の報告は,残基83−149(AID,自己阻害ドメイン)を含むPAK1ドメインの過剰発現はマクロピノサイトーシスを阻害することを示している(Dharmawardhane et al.,2000)。HeLa細胞においては,AIDの発現はwtPAK1を過剰発現する細胞と比較して感染を80%阻害することが見いだされた。変異型のAID(AIDL107F)の発現は影響を与えなかった(図2B)。
【0171】
PAK1のトレオニン残基423のリン酸化は,マクロピノサイトーシスの活性化に役割を果たしている(Dharmawardhane et al.,2000)。MVを細胞に加えると,10分以内にリン酸化PAK1が検出され,PAK1は60分間にわたって活性化されたままであった(図2C)。最大応答は,30mpiに見られた。この結果は,ビリオン取り込みおよびウイルスコアのエンドソーム放出の経時変化と一致していた(Townsley et al.,2006)。これらを合わせると,この結果は,PAK1活性がワクシニアMVの細胞内への増殖性侵入において役割を果たすことを示す。明らかに,ウイルスはRac1の活性化を誘発し,その結果,PAK1およびアクチン動力学に関与する下流の他の因子が活性化される。
【0172】
図2は,p21活性化キナーゼ−1(PAK1)がMV侵入に必要であることを示す。図2Aは,PAK1のsiRNAノックダウンがMV感染に及ぼす影響を示す。HeLa細胞をPAK1に対する2つの別々に確認したsiRNA(Qiagen;A:TCCACTGATTGCTGCAGCTAA(配列番号12);B:TTGAAGAGAACTGCAACTGAA(配列番号13)でトランスフェクションした。処理の36時間後,細胞にEGFP−MVをMOI=1で感染させ,2hpiで回収して分析した。感染した細胞のパーセンテージをFACS分析により求めた。実験は三重に行い,結果を平均した。PAK1に対するイムノブロット分析(a−PAK1;Santa Cruz)を行って,PAK1蛋白質レベルの低下を確認した(下パネル)。1レーンあたり合計50μgの細胞溶解物を負荷し,モック処理細胞(−−)に対する残存PAK1蛋白質の%を求めた。アクチンに対するイムノブロットを負荷対照として用いた。図2Bは,MVの感染能に及ぼすドミナント・ネガティブPAK1の影響を示す。HeLa細胞を蛍光タグ付きの野生型PAK1(WT),PAK1自己阻害ドメイン(AID),または変異型AID(AIDL107F)で過渡的にトランスフェクションした。細胞をEGFP−MVでMOI=1で感染させた。4hpiにおいて細胞を固定し,アクチンを染色した。細胞をトランスフェクションした蛋白質(赤),ウイルス感染(緑)およびアクチン(青)について共焦点顕微鏡により分析した。実験は三重に行い,1回の実験あたり100個のトランスフェクトした細胞を感染について評点した。結果は,トランスフェクトした/感染細胞の平均パーセンテージとして示す。図2Cは,MV感染の間のPAK1の活性化を示す。MVを4℃で1時間HeLa細胞に結合させた。細胞を冷PBSで2回洗浄した。予熱した培地を加えて,感染物を37℃にシフトした後,示された時点で回収した。PAK1およびリン酸化PAK1(a−PAK1−Thr423;Cell Signaling)についてイムノブロット分析を行った。
【0173】
PAK1はマクロピノサイトーシス,すなわち,多くの異なる細胞タイプにおいて大量の液体および細胞膜の内在化につながる,リガンドにより誘導されるエンドサイトーシスのプロセスに関与することが知られている(Falcone et al.,2006)。マクロピノサイトーシスは動的なアクチン再編成に依存し,これはコレステロール,ならびに小さいGTPaseであるRac1,CDC42,およびArf6を必要とする(Kirkham and Parton,2005)。さらに,PAK1に加えて,Na+/H+交換体,チロシンキナーゼ,および少なくとも1つのセリントレオニンキナーゼ,PKCも関与する(Dharmawardhane et al.,2000;Grimmer et al.,2002;Hewlett et al.,1994;Veithen et al.,1996)。ダイナミンは必要ではない(Damke et al.,1994)。
【0174】
MV侵入にマクロピノサイトーシスが関与しているか否かを試験するために,種々の広範な範囲のキナーゼ阻害剤を試験した。スタウロスポリン(セリン/トレオニンキナーゼ阻害剤)は感染を65%まで,ゲニステイン(チロシンキナーゼ阻害剤)は82%まで,ワートマニン(PI3キナーゼ阻害剤)は63%まで阻害した(図3A)。次に,アクチンアセンブリを防止する(サイトカラシンD),アクチンを脱重合させる(ラトルンクリンA),またはアクチンフィラメントを安定化させる(ジャスパキノリドA)薬剤も,感染の用量依存的阻害を40%から90%の範囲で引き起こすことを見いだした(図3B)。顕微鏡により,MVにより誘導されるブレブの形成がこれらのアクチン阻害剤により阻害されることが示された。Na+/H+交換体およびマクロピノサイトーシスの阻害剤であるEIPAも感染を90%阻害した(図3C)。構成的に活性なArf6の発現は,wtArf6を過剰発現する細胞と比較して感染を78%阻害したが(図3D),ドミナント・ネガティブダイナミン(K44A)の過剰発現は影響を与えなかった。MVによる感染はコレステロール依存性であり,Rac1を必要とすることは,すでに他の研究者により示されている(Chung et al.,2005;Locker et al.,2000)。まとめると,阻害はマクロピノサイトーシスプロセスと完全に一致した。
【0175】
エンドサイトーシスプロセスのマクロピノサイトーシス的性質は,同時に生ずる液相マーカーの内在化とも一致した。蛍光MVを含むエンドサイトーシスの小胞は,液相マーカーである568−デキストランについて陽性であったが,クラスリン媒介性エンドサイトーシスにより内在化されるリガンドである568−トランスフェリンについて陰性であった(図3E)。100μMのEIPAを含めると,MVのFITC−デキストラン陽性小胞への内在化が防止された(図3E)。
【0176】
図3は,ワクシニアMVが細胞に侵入するのにマクロピノサイトーシスを利用することを示す。図3Aおよび3Bは,MV感染に必要な一般的キナーゼおよびアクチン骨格を示す。一連のキナーゼ阻害剤およびアクチン作用性薬剤がMVの感染能に及ぼす影響をFACS分析により調べた。アッセイは,下記の材料および方法の節に概説するようにして行った。結果は,3回の実験の平均であり,未処理対照に対する感染細胞のパーセンテージで示す。図3Cは,EIPAによるMVの感染能の阻害を示す。細胞をNa+/H+交換およびマクロピノサイトーシスの阻害剤であるEIPA(5−N−エチル−N−イソプロピルアミロリド;Sigma Aldrich)で前処理し,次にEGFP−MVを感染させ,下記の材料および方法の節に記載されるようにしてFACSにより分析した。実験は三重に行い,結果は薬剤非存在下における対照感染に対する感染細胞のパーセンテージで表す。図3DはMVの感染能に及ぼすドミナント・ネガティブArf6の影響を示す。HeLa細胞を蛍光タグ付き野生型Arf6(WT)または構成的活性化型Arf6(C/A)で過渡的にトランスフェクションした。細胞はEGFP−MVでMOI=1で感染させた。4hpiにおいて,細胞を固定し,アクチンを染色した。細胞を,トランスフェクションした蛋白質(赤),ウイルス感染(緑)およびアクチン(青)について共焦点顕微鏡により分析した。実験は三重に行い,1回の実験に100個のトランスフェクションした細胞を用いて,感染について評点した。結果は,トランスフェクション/感染した細胞の平均パーセンテージとして示す。図3EはMVのエンドサイトーシスの小胞中への内在化を示す。細胞を100μMのEIPAで処理するかまたは未処理とした。細胞は,mYFP−MVとMOI=1で4℃で1時間結合させるか,または未感染とした。細胞を冷PBSで2回洗浄し,37℃に15分間シフトした。次に細胞を液相マーカーである10kDaの568−デキストラン(0.5mg/ml)または568−トランスフェリン(Tfn)(200ng/ml)で,EIPAの存在下または非存在下で10分間パルスした。低pHで軽く洗浄することにより表面に結合したデキストランおよびTfnを除去した後,固定した。サンプルは共焦点顕微鏡で分析した。1つの画像あたり10個のZsテークを集め,Z投影として表示した。
【0177】
ホスファチジルセリン(PS)の役割をさらに詳細に分析するため,単離したMV粒子を0.5%NP40を用いて抽出し,次に界面活性剤および抽出された脂質を除去した。これにより,感染能は劇的に低下したが(図4A),ウイルス構造蛋白質の損失はなかった(データ示さず)。蛍光MVを用いて,抽出したウイルスは細胞に結合するが,ブレブ形成およびエンドサイトーシスを誘導することができなかったことを示した。未処理のMVが細胞の内部にクラスター化している8時間後でさえも,抽出したビリオンはなお細胞表面に留まっており,このことは,エンドサイトーシスに欠陥があることを示唆する(図4B)。PSの役割を試験するために,脱脂質化したウイルスを異なる脂質組成をもつリポソームの存在下でインキュベーションすることにより,脂質を戻し入れた(addback)。いずれの脂質を用いるかにかかわらず,再脂質化したウイルスは細胞に結合することができた(図4C)。しかし,PSを有するもののみ,ブレブ形成およびエンドサイトーシスを誘導することができた。さらに,PSを再び加えると,元の感染能の大部分(90%)およびプラーク形成がレスキューされた(図4DおよびE)。
【0178】
PS結合蛋白質であるアネキシン−A5(ANX5)をEGFPタグ付きの形で用いて,ウイルスのPSが実際にウイルス膜の外葉に暴露されていることを示した。ウイルスは,この試薬に暴露したときにわずかに染色され(図4F),ウイルスPSをANX5でマスキングすると,MVの細胞への結合には影響を及ぼさずに感染が95%阻害されることが見いだされた(図4G)。ウイルスを加える前に細胞をANX5で処理すると,ウイルスの結合または感染能に及ぼす影響は観察されなかった。これらの結果は,マクロピノサイトーシスプロセスの開始にウイルス粒子中のPSが必要であることを示す。
【0179】
最後に,ANX5を用いて,感染細胞からMVを放出する感染細胞の溶解が,アポトーシスまたは壊死のいずれにより引き起こされるかを判定した。細胞アポトーシスの特徴である表面暴露PSの存在について,ウイルスプラークを分析した(Martin et al.,1995)。ウイルスプラークを囲む細胞は大量の表面暴露PSを含むが,非感染細胞またはプラーク間の細胞はこれを含まないことが見いだされた(図4H)。このことは,感染の後期において,ワクシニアウイルスはアポトーシスを誘導し,これがMVが退出する手段であることを示す。すなわち,ウイルスは,感染細胞からのアポトーシス体と一緒に,隣り合う非感染細胞に暴露される。
【0180】
図4は,ワクシニアMVは内在化にPSを必要とすることを示す。図4Aは,MVの感染能にはウイルス脂質が必要であることを示す。ビリオンから種々の濃度のNP40(0.1−1.0%)を用いて脂質を抽出した。補正後,BSC40細胞におけるタイター測定(pfu/ml)によりビリオンの感染能を測定した。図4Bは,脂質を抽出したMVが細胞に結合しうるが細胞に侵入できないことを示す。0.5%NP40で処理したかまたは未処理のmYFP−MVをMOI=1で細胞に加えた。細胞は,30mpiまたは8hpiで固定し,アクチンについて染色し,共焦点顕微鏡によりウイルスの結合および感染について可視化した。図4Cは,MVの結合はビリオン膜の脂質構成成分には依存しないことを示す。1x109個のmYFP−MVを,未処理で,または脂質を抽出して,または続いて異なる脂質(MおよびM’)とともに戻した。戻した後,ビリオンをHeLa細胞に4℃で1時間結合させ,冷PBSで2回洗浄し,下記の材料および方法の節にしたがって,FACS分析により分析した。結果は3回の独立した実験として示す。図4Dは,MVの感染能はビリオン膜中のPSに依存することを示す。1x109個のEGFP−MVを脂質抽出するかまたは未処理で異なる脂質に戻した(下記の材料および方法の節を参照)。戻した後,ビリオンをHeLa細胞に4℃で1時間結合させた。PBSで2回洗浄し,37℃で2時間感染を進行させた後に,FACS分析を行った。3回の独立した実験の結果を示す。図4Eは,MVによる増殖感染はビリオン膜中のPSに依存することを示す。1x109個のWR MVを脂質抽出するかまたは未処理で異なる脂質に戻した。戻した後,ビリオンの感染能をBSC40細胞におけるタイター評定(pfu/ml)により測定した。結果は3回の独立した実験で示す。図4Fは,ウイルス膜PSがMVの表面への暴露されることを示す。ウイルス膜上のPSの存在は,組換え488アネキシンV(ANX5)を用いて示した。ビリオンは,Vybrant(登録商標)アポトーシスアッセイキット#2(Molecular Probes)に提供されるプロトコルを適合させて分析した。簡単には,ビリオンを25μlの1Xアネキシン結合バッファ中で,2μlの488ANX5とともに室温で15分間インキュベーションした。ビリオンをペレット化し,結合バッファで1回洗浄し,カバースリップに結合させた後,共焦点顕微鏡により可視化した。mYFP−MVを可視化して陽性対照とした。図4Gは,MV膜PSのマスキングは感染を妨害することを示す。結合:HeLa細胞またはmYFP−MVをANX5で,製造元の指針にしたがって(細胞)または上述の条件で(MV)前処理した。処理後,ANX5と結合した細胞を未処理のビリオンとともに,またはANX5処理mYFPビリオンを未処理細胞とともにインキュベーションした。mYFP−MVのHeLa細胞への結合を陽性対照として用いた。実験は三重に行い,結果は対照に対するビリオン結合細胞のパーセントとして示した。感染:実験は,EGFP−MVを用いる結合と同様にして行った。図4Hは,アポトーシス性細胞についてウイルスプラークが濃縮されていることを示す。BSC40細胞単層をwt−MVに感染させ,感染は24時間進行させた。次に細胞をPBSで2回洗浄し,製造元のプロトコルにしたがって,生存,アポトーシスおよび壊死細胞について分析した(Vybrant(登録商標)アポトーシスアッセイキット#2;Molecular Probes)。488ANX5は緑で,ヨウ化プロピジウムは赤で示される。未感染細胞を染色対照として用いた。
【0181】
材料および方法:
細胞株およびウイルス
BSC40霊長類およびHeLa ATCC細胞の単層を10%ウシ胎児血清(FCS)を含むダルベッコ改変イーグル培地(DMEM;Gibco BRL)中で37℃で維持した。野生型(wt)ワクシニアウイルス(WR株),WRE/LEGFP(MV−EGFP)(Paula Traktman,Medical College of Wisconsinより供与),およびコア蛋白質の蛍光タイプを含むWRであるA5(mYFP−MV)を示されるようにして用いた。すべてのウイルスのストックは,ブレフェルジンAの存在下で調製した。ウイルスは,細胞質溶解物から超遠心分離により25−40%のショ糖勾配中36%ショ糖のバンドとして精製した。
【0182】
蛍光活性化セルソーティング(FACS)
結合アッセイ:mYFP−MVをHeLa細胞(wtまたは処理済)に,無血清DMEM中で4℃で1時間,感染多重度(MOI)=1で結合させた。ビリオン結合細胞を4℃にシフトし,PBSで2回洗浄し,プレートからトリプシン処理し,ホルムアルデヒド(FA)で氷上で30分間固定した。固定した細胞を遠心分離により回収し,PBSで1回洗浄し,再び回収し,PBSに懸濁してFACS分析を行った。各サンプルから合計で10,000事象を分析し,未結合およびmYFP−MV結合対照に対する相対的mYFP発現について評点した。感染アッセイ:EGFP−MVを無血清DMEM中で薬剤の存在下で4℃で1時間HeLa細胞に結合させた。すべのアッセイはMOI=1で行った。結合後,細胞を冷PBSで2回洗浄し,予熱した培地を加えた。細胞を37℃にシフトし,感染を2時間進行させた後,固定し,上述のようにしてFACS用に調製した。薬剤スクリーニング:HeLa細胞を感染前に種々の濃度の示された薬剤で15分間前処理した。細胞を,無血清DMEM中で薬剤の存在下で,4℃で1時間EGFP−MVに結合させた。すべてのアッセイはMOI=1で行った。結合後,細胞を冷PBSで2回洗浄し,薬剤を含む予熱した培地を加えた。次に細胞を37℃にシフトし,感染を2時間進行させた。細胞を固定し,上述のようにしてFACS分析用に調製した。すべてのFACS分析は,薬剤なしの対照感染に対する感染細胞のパーセンテージとして表した。
【0183】
ビリオンの界面活性剤抽出
合計で1.0x109のワクシニアウイルスwt,EGFP−MV,またはmYFP−MVビリオン(上述のようにして精製)を,100mM Tris(pH9.0)および種々の濃度のNP−40:すなわち,0,0.1,0.5,および1.0%(vol/vol)を含む反応混合物中で37℃で60分間インキュベーションした。抽出後,可溶性画分および粒子画分(それぞれビリオンの膜およびコア成分を表す)を沈降(16,000xg,30分間,室温)により分離した。次にサンプルをBSC40細胞上でタイターを測定して,ウイルスタイター(PFU数/ミリリットル)を決定した。すべてのタイター測定は三重に行い,結果を平均した。
【0184】
ビリオン脂質の戻し入れ
種々の脂質組成をもつリポソームを調製し,Oie(Oie,1985#2008)の方法にしたがって,脂質を抽出したビリオンを再構築した。簡単には,脂質を抽出したビリオンを,種々の濃度のPS(20μg/mlまたは200μg/ml)またはGM1(20μg/ml)を組み込んだPC系リポソーム(200μg/ml)とともに37℃で2時間インキュベーションした。ビリオンを遠心分離により回収し,次に洗浄し,バッファーに再懸濁した。再構築ビリオンをFACSおよび顕微鏡による結合および侵入アッセイに供し,ならびにウイルス収量のタイターを測定した。
【0185】
実施例2
アデノウイルス血清型3ウイルスの侵入メカニズムの評価
材料および方法
細胞およびウイルス
細胞は,記載されるようにして,10%FCS(GIBCO−BRL)を含むDME(GIBCO−BRL)で低い継代数で成長させた(Suomalainen et al.,1999)。ヒト黒色腫M21 litter(表面発現αvインテグリン陰性)およびM21L細胞(細胞表面αvインテグリン陽性)はDr.D.Cheresh(Scripps Research Institute,La Jolla,CA,Felding−Habermann et al.,1992)から供与された。K562慢性骨髄性白血病細胞は記載されるようにして成長させた(Nagel et al.,2003)。CD46またはCARを安定して発現するBHK細胞は,CD46またはCARのBC1アイソフォームのいずれかをコードするプラスミドを安定的にトランスフェクトすることにより調製した(Sirena et al.,2005)。Ad3およびAd2ts1は,記載されるようにして成長させ単離した(Greber et al.,1996)。Ad3のテキサスレッドによる標識は公表されているようにして行った(Nakano and Greber,2000)。(3H)−チミジン標識Ad3は,公表されているようにして製造した(Greber et al.,1993)。
【0186】
cDNA,蛋白質および化学物質
CtBP1−S/BARSをコードするcDNAはDr.A.Colanzi(Dep.Of Cell Biology and Oncology,S.MariaImbaro,Italy)から入手した。pCMV−myc CtBP1−Swtは,PCR増幅CtBP1−Swt(それぞれSalIおよびNotIで切断)をpCMV骨格ベクター(Stratagene)中にライゲーションすることにより生成した。Myc−CtBP3 D355Aは,Quik Change R部位特異的変異導入キット(Stratagene)を用い,下記のプライマー5’−CTGGGCCAGCATGGCCCCTGCTGTGGTG−3’(配列番号21)および5’−CACCACAGCAGGGGCCATGCTGGCCCAG−3’(配列番号22)を用いて生成した(Bonazzi et al.,2005)。得られたcDNAをシークエンシングにより確認した。K44A−dyn2およびdyn2wt発現プラスミドはDr.C.Lamaze(Pasteur Institute)より入手した。Pak1wtおよび阻害的ドメイン発現ベクターは,J.Chernoff(Fox Chase Cancer Center,Philadelphia,PA)より入手した。Toxin B(0.5mg/ml)はDrs.F.HofmannおよびK.Aktories(University of Freiburg,Freiburg,Germany)より入手した。PKC阻害剤Go6976(1μM)およびGo6983(1μM)はCalbiochem(Juro Supply)より購入し,Na+/H+交換阻害剤EIPA(100μM)はAlexis Corporationより購入し,サイトカラシンD(5μM)およびジャスパラキノリド(500nM)はCalbiochemより購入した。メチル−ベータ−シクロデキストリン(50mM)によるコレステロール飢餓は,先に公表されているようにして行った(Imelli et al.,2004)。Ad3可溶性ファイバーノブ(最終濃度5μg/mlで使用)はP.Fender(Grenoble,France)から入手した。ダイナソールはDr.J.S.Siegel(Organic Chemistry Institute,University of Zurich)の合成したものを用いた。CtBP1−L/Sに対する抗体は,BD Transduction laboratoriesから,PAK1抗体(C−19)はSanta Cruz Biotechnology(Santa Cruz,CA)から,リン酸化PAK1(T423)に対する抗体はCell Signalling Technologyから入手した。
【0187】
エンドサイトーシスおよびAd−eGFP形質導入
細胞を冷却下で,RPMI−0.2%BSA中で(3H)チミジン標識Ad3(1μg/ml;3X109粒子,35mmディッシュ上)とともに60分間インキュベーションし,冷RPMI−BSAで洗浄し,RPMI−BSAとともに示された時間暖めた。細胞を冷RPMI−BSAおよび冷PBSで2回洗浄した。細胞外ウイルス粒子を細胞から除去するために,細胞を冷2%トリプシン−EDTA(GIBCO)とともに冷却下で60分間インキュベーションし,PBS−2%FCSを加え,細胞を500xgで遠心分離した。洗浄工程を2回繰り返した。熱SDS(0.4%)中に細胞溶解物を調製し,20G医療シリンジで剪断し,Beckman Coulter Scintillation System LS3801を用いて,液体シンチレーションカウント(Ready Safe;Beckman Coulter)により放射活性を測定した。トリプシンなしの対照細胞の計数を100%対照として用いた。薬剤実験のためには,細胞をRPMI−BSA中の薬剤とともに37℃で30分間プレインキュベーションした。形質導入実験のためには,細胞を温RPMI−BSAで洗浄し,1μg/mlのウイルスとともに60分間インキュベーションし,RPMI−BSAで数回洗浄し,水浴中で4時間(A549細胞),5時間(HeLa細胞),8時間(M21およびM21L細胞)および16時間(K562細胞)インキュベーションした。細胞を冷PBSで洗浄し,冷却下で2%トリプシンで,次に2%PBS−FCSで処理し,フローサイトメトリ(Beckman FC500 cytometer)で分析した。サンプル1つあたり少なくとも10000個の生存細胞を計数した。薬剤実験のためには,細胞をRPMI−BSA中の阻害剤で37℃で30分間前処理した後,薬剤の存在下で60分間温感染させ,次に薬剤を含まない培地で洗浄し,さらにインキュベーションした。
【0188】
デキストラン取り込み
細胞を,先に記載されるようにして(Meier et al.,2002),5μg/mlの冷Ad3とともにプレインキュベーションし,温RPMI−BSAで洗浄し,0.5mg/mlのデキストラン−FITC(lysine−fixable 10kDa,Molecular Probes)を含むRPMI−BSA中で37℃で10分間暖めた。デキストラン取り込みは,細胞を冷RPMI−BSAおよびPBSで洗浄(3回繰り返し)することにより停止させた。表面に結合したデキストランは,冷0.1M酢酸ナトリウム(pH5.5),0.05M NaCl中で10分間酸処理することにより除去した。FACS分析用には,細胞をPBS中2%トリプシン(GIBCO−BRL)中で氷上で25分間剥離させ,2mlの7%FCS/PBSを含む6mlのポリプロピレンチューブ(no2063;Falcon,Becton Dickinson)に移し,290xgでペレット化し,2%FCS/PBSに再懸濁した。サンプル1個あたり少なくとも10000個の生存細胞をフローサイトメータで計数した(Beckman FC500サイトメータ)。
【0189】
透過型電子顕微鏡,およびBSA−金の取り込み
冷Ad3またはAd2ts1(50μg/ml,感染多重度[MOI]5000)で60分間結合させた後,適宜洗浄および内在化を行い,細胞を0.1Mカコジル酸ナトリウムバッファ,pH7.4(CaCo)中2%ホルムアルデヒド−1.5%グルタルアルデヒドで一晩固定し,CaCoで数回洗浄し,二回蒸留水中の1%OsO4(Electron Microscopa Sciences)および1.5%フェリシアン化カリウム(FeK3N6)で4℃で60分間固定後処理を行った(Simionescu and Siminonescuの方法を改変)。標本を0.1Mカコジル酸ナトリウムですすぎ,0.05Mカコジル酸ナトリウム中1%タンニン酸で室温で45分間対比染色し,1%硫酸ナトリウムで洗浄し,H2Oですすぎ,H2O中2%酢酸ウラニルで一晩染色し,先に記載されるようにしてEpon中に包埋した(Nakano et al.,2000)。ウイルス粒子は,細胞膜,エンドソームおよびサイトゾルの超薄切片を倍率50000xで定量し,透過電子顕微鏡で加速電圧80,000Vで観察した(Zeiss EM902A)。15nmのコロイド金は,HAuCl4のクエン酸還元により調製した(Frens,1973;Horisberger and Rosset,1977)。20mlのコロイド金溶液(pHを5.9に調節)に,50μlの10mg/mlBSA(Sigma,無脂肪酸)溶液を加えた(DeRoe et al.,1987)。BSA金複合体を安定化させるために,1mlの1%PEG−20000(Roth,Switzerland)を加え,サンプルを28,000gで60分間遠心分離し,ペレットを2mlの金バッファ(0.2%PEG−20000を含む滅菌濾過済みPBS)に溶解し,4℃で保存した。BSA金内在化は,Ad3またはAd2−ts1の冷結合後に,BSA金のRPMI−BSA(約0.1mg/mlのBSA)中1:1希釈を用いて,37℃で10分間行った。
【0190】
蛍光顕微鏡および免疫蛍光
細胞を種々のDNAコンストラクトで30時間トランスフェクションした後,Fugene6(Roche,製造元の指針にしたがう)を用いた実験を行った。細胞を,Ad3−eGFPまたはAd5−eGFPで37℃で60分間感染させ,洗浄し,37℃で15時間インキュベーションした。細胞を固定し,DAKOを用いてマウントした。デキストランおよびトランスフェリン取り込み用には,細胞を5μg/mlのAd3で冷却下でシンクロナイズさせ,温洗浄し,RPMI−BSA中の0.5mg/mlデキストラン−TRおよび20μg/mlのトランスフェリン−Alexa647の混合物で37℃で30分間パルスし(水浴),次に5分間チェイスし,固定し,DAKOを用いてマウントした。共焦点レーザー走査顕微鏡(CLSM)は,Ar−ArKrレーザー,He−Ne543−594レーザー,He−Ne633レーザー,405nmのダイオードレーザーおよび63x油浸対物レンズ(N.A.1.4PLAPO)を備えたLeica−DM SP2 RXA2−TCS−AOBS顕微鏡(Leica Microsystems,Wetzlar,Germany)により行った。ピンホール値1.0,エアリー1,得られる光学切片約0.48μm,ボクセル0.233x0.233x0.48μm。ズーム倍率は2であった。画像のプロセシングはLeicaand Photoshopソフトウエア(Adobe)を用いて行い,細胞の合計投影の蛍光強度はImage J(webpage:http://rsb.info.nih.gov/ij/)を用いて決定した。デキストラン陽性エンドソームを用いるCtBP1−L/S共局在用には,細胞をAd3−TR(2μg/ml)で氷上で60分間冷シンクロナイズさせ,温RPMI−BSAで洗浄し,0.5mg/mlのデキストラン−FITCで37℃で10分間パルスした。細胞をRPMI−BSAおよびPBSでよく洗浄し,固定し,CtBP1−L/Sマウスモノクローナル抗体(BD Transduction laboratories)および二次抗体としてAlexa647−コンジュゲート化ヤギ抗マウス抗体を用いて,免疫蛍光により分析した。
【0191】
siRNAトランスフェクション
K562細胞を,クラスリン重鎖(AACCUGCGGUCUGGAGUCAAC(配列番号23);Qiagen(Hinrichsen et al.,2003))およびCtBP1/CtBP3(CCGUCAAGCAGAUGAGACAUU(配列番号24);GGAUAGAGACCACGCCAGUUU(配列番号25);Dharmacon(Bonazzi et al.,2005))に対するsiRNAで,Nucleofector I(Amaxa;programT−03)を製造元の指針にしたがって用いて,トランスフェクトした。非標的化siRNA配列(Qiagen,またはDharmacon)のトランスフェクションを対照として用いた。トランスフェクションは第0日および第2日に行い,ウエスタンブロッティングおよび実験用の細胞溶解物は第4日に回収した。HeLa細胞を,クラスリン重鎖,CtBP1/CtBP3,またはPAK1(確認済siRNACat.SI00605703およびSI00605696;Qiagen)に対するsiRNAで,リポフェクタミン2000(Invitrogen)を製造元の指針にしたがって用いてトランスフェクトした。トランスフェクションは第0日および第2日に2回行い,ウエスタンブロッティングおよび実験用の細胞溶解物は第4日に回収した。A549細胞を,クラスリン重鎖,CtBP1/CtBP3,PAK1またはダイナミン2{GACAUGAUCCUGCAGUUCA(配列番号26),Qiagen,Huang,2004#15509}に対するsiRNAで,リポフェクタミン2000を上述のようにして用いてトランスフェクトした。
【0192】
ウエスタンブロッティング
細胞を35mmディッシュで成長させ,リン酸緩衝化食塩水(PBS)で洗浄し,500μlの2%熱SDS中で溶解した。溶解物を20ゲージの針に数回通過させ,95℃で30秒間加熱した。16,000×gで10分間遠心分離した後,150μlの上清を50μlのサンプルバッファー(200mM Tris/HCl,pH6.8,8%SDS,0.4%ブロモフェノールブルー,40%グリセロール,167mMジチオスレイトール)と混合し,95℃で10分間加熱した。抽出物を10%SDS−PAGEで分離し,Hybond−ECLニトロセルロース膜(Amersham Biosciences,Zurich,Switzerland)に移し,50mMTris/100mM塩化ナトリウム/0.1%Tween,pH7.4(TNT)中の5%ドライミルクでブロッキングした。免疫学的プロービング(PAK1ブロットについては3%ミルク,CtBP1ブロットについては0.2%BSA)の後,ECLPlus試薬(Amersham Biosciences)でHRPコンジュゲート化抗体を検出した。フィルタを,100mMβ−メルカプトエタノール,2%SDS,62.5mMTris/HCl,pH6.7で50℃で30分間ストリッピングした後,TNTでよく洗浄し,5%ドライミルクでブロッキングし,3%ミルクを含むTNT中の抗カルネキシン抗体(Dr.A.Helenius,Zurichより寄付)で再プローブした。
【0193】
図5は,Ad3のエンドサイトーシスおよび感染にはPAK1の活性化が必要であるが,Ad5には必要ではないことを示す。図5Aは,Ad3およびAd5がPAK1を活性化することを示す。HeLa細胞を0.5μg/mlのAd3,Ad5またはts1(約5X105細胞)とともに,冷却中で60分間インキュベーションし,洗浄し,異なる時間暖めた。細胞をホスファターゼおよびプロテアーゼ阻害剤を含む冷PBSで洗浄し,ディッシュからはがし,阻害剤を含む100μlのPBSに再懸濁し,ジチオスレイトールを含むSDSサンプルバッファと混合し,加熱し,SDS−PAGE(1レーンあたり105個の細胞に相当)およびそれぞれPAK1およびリン酸化PAK1(T423)に対するウエスタンブロッティングにより,ECLを検出法として用いて分析した。図5B:野生型またはドミナント・ネガティブPAK1(阻害ドメインID)を発現するHeLa細胞にAd3−eGFPまたはAd5−eGFPを形質導入するか,または,Ad3感染の際のデキストラン−FITCまたはAlexa647で標識したトランスフェリンの取り込みについて評価した。図5C:HeLa細胞を,PAK1に対するsiRNAであるP2およびP8またはns siRNAで72時間トランスフェクトし(ダブルトランスフェクション,20pmoles/ml siRNA),Ad3−eGFPまたはAd5−eGFPを6時間感染させ,フローサイトメトリでeGFP発現について分析した。1x105個のトランスフェクトした細胞をウエスタンブロッティング(WB)によりPAK1含有量について分析した。図5D:Ad3のエンドソームエスケープを,それぞれ抗PAK1 siRNA P2およびns siRNAでトランスフェクトしたHeLa細胞における薄片EMにより測定した。結果は,細胞膜,エンドソーム中およびサイトゾル中にビリオンを示した。
【0194】
実施例3
ウイルス感染の間のEGFRの役割
Rac1エフェクターであるPAK1は,ワクシニア成熟ビリオン(MV)の侵入に必要であり,RhoAファミリーのGTPaseであるRac1およびRhoAは侵入プロセスの間に活性化される(Mercer and Helenius(2008)Science320:531−535)。次に,これらのGTPaseは,種々の細胞表面レセプターにより活性化されることができる(Schiller(2006)Cell Signal 18:1834−1843)。その1つは表皮成長因子レセプター(EGFR,Erb1)である。これまでワクシニア侵入におけるEGFRの関与については意見が分かれている(Marsh and Eppstein(1987)J Cell Biochem.34:239−245;Eppstein et al.(1985)Nature 318:663−665;Hugin and Hauser(1994)J.Virol.68:8409−8412)。
【0195】
MV感染の経時変化を用いて,ワクシニア感染の間のEGFRの活性化状態を評価した。EGFR活性化は,チロシン1173でリン酸化されたEGFRを認識する抗体を用いてモニターした。EGFRはウイルスを加えてから5分以内に強く活性化され,そのピークは感染後15分であった(図7)。
【0196】
EGFR阻害剤である324674(Calbiochem)は,MV侵入を有効に妨害し,低pH融合により容易にバイパスされた(図8)。
【0197】
これらの結果は,ワクシニアMVは感染の間にEGFRを活性化することができ,この活性化が侵入に必要であることを示す。さらに,MVがEGFRの下流に存在するPAK1の活性化を誘導したことが示唆される。これらのデータは,MV侵入および感染に必要なシグナリング経路についてのさらなる洞察を提供し,他の潜在的細胞性抗ウイルス標的およびポックスウイルス感染に対する治療法を示唆する。
【0198】
材料および方法
蛍光活性化細胞ソーティング(FACS)感染アッセイ
薬剤スクリーニング:HeLa細胞は,感染前に,示される薬剤で種々の濃度で15分間前処理した。EGFP発現MVを,4℃で無血清DMEM中で薬剤の存在下で1時間結合させた。すべてのアッセイはMOI=1で実施した。結合後,細胞を冷PBSで2回洗浄し,次に予温した薬剤含有培地を加えた。細胞を37℃にシフトさせ,2時間感染させた。細胞をPBSで2回洗浄し,プレートをトリプシン処理し,4%ホルムアミド(FA)中で氷上30分間固定した。固定した細胞を遠心分離により回収し,PBSで洗浄し,再び回収し,PBS中に懸濁して,FACS Calibur System(BD Biosciences)を用いるFACS分析を行った。すべてのFACS分析は,三重に行い,薬剤なしの対照感染に対する感染細胞の平均パーセンテージで表した。エラーバーは,実験間の標準偏差を表す。
【0199】
参考文献
Bangham, A.D., M.M. Standish, and J.C. Watkins. 1965. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 13(1): 238-52.
Birault, V., C.J.Harris, J. Le, M. Lipkin, R. Nerella, and A. Stevens. 2006. Bringing kinases into focus: efficient drug design through the use of chemogenomic toolkits.Curr. Med. Chem. 13(15): 1735-48,
Bui M, Whittaker G, Helenius A (1996) The protein kinase inhibitor, H7, directly blocks M1-ediated nuclear export of infleunza virus RNPs. submitted.
Bukrinskaya AG, Vorkunova NK, Kornilayeva GV, Naramanbetova RA, Vorkunova GK (1982) Influenza virus uncoating in infected cells and effect of rimantadine. J Gen Virol 60: 49-59.
Cros JF, Garcia-Sastre A, Palese P (2005) An unconventional NLS is critical for the nuclear import of the influenza A virus nucleoprotein and ribonucleoprotein. Traffic 6(3): 205- 213.
Doms RW, Lamb RA, Rose JK, Helenius A (1993) Folding and assembly of viral membrane proteins. Virology 193(2): 545-562.
Ehrhardt C, Marjuki H, Wolff T, Nurnberg B, Planz O et al. (2006) Bivalent role of the phosphatidylinositol-3-kinase (PI3K) during influenza virus infection and host cell defence. Cell Microbiol 8(8): 1336-1348.
Graeser RU, Sarah; Brosig, Michaela; Kubbutat, Michael H.G. (2004) Highly efficient gene silencing using 2-for-Silencing siRNA Duplexes targeting protein kinase genes. QIAGEN News 2004 e32.
Huang YT, Turchek BM (2000) Mink lung cells and mixed mink lung and A549 cells for rapid detection of influenza virus and other respiratory viruses. J Clin Microbiol 38(1): 422- 423.
Khor R, McElroy LJ, Whittaker GR (2003) The ubiquitin-vacuolar protein sorting system is selectively required during entry of influenza virus into host cells. Traffic 4(12): 857- 868.
Kielian M, Rey FA (2006) Virus membrane-fusion proteins: more than one way to make a hairpin. Nat Rev Microbiol 4(1): 67-76.
Ludwig S (2006) Signaling and apoptosis in influenza virus-infected cells. In: Kawaoka Y, editor. Influenza Virology: Current Topics by Yoshihiro Kawaoka: Caister Academic Press, pp. 37-64.
Maeda T, Kawasaki K, Ohnishi S (1981) Interaction of influenza virus hemagglutinin with target membrane lipids is a key step in virus-induced hemolysis and fusion a pH 5.2. Proc Natl Acad Sci USA 78: 4133-4137.
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. (2002) The protein kinase complement of the human genome. Science 298(5600):1912-34.
Marsh M, Helenius A (1989) Virus entry into animal cells. Adv Virus Res 36: 107-151.
Martin K, Helenius A (1991) Nuclear transport of influenza virus ribonucleoproteins: the viral matrix protein (M1) promotes export and inhibits import. Cell 67: 117-130.
Matlin KS, H. Reggio, A. Helenius, and K. Simons (1982) Infectious entry pathway of influenza virus in a canine kidney cell line. J Cell Biol 91: 601-613.
Matrosovich M, Matrosovich T, Garten W, Klenk HD (2006) New low-viscosity overlay medium for viral plaque assays. Virol J 3(1): 63.
Matrosovich M, N.; Klenk, Hans-Dieter; Kawaoka, Yoshihiro (2006) Receptor specificity, host range, and pathogenicity of influenza viruses. In: Kawaoka Y, editor. Influenza Virology: Current Topics by Yoshihiro Kawaoka: Caister Academic Press, pp. 37-64.
Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk HD (2004a) Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc Natl Acad Sci U S A 101(13): 4620-4624.
Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk HD (2004b) Neuraminidase is important for the initiation of influenza virus infection in human airway epithelium. J Virol 78(22): 12665-12667.
O'Neill RE, Jaskunas R, Blobel G, Palese P, Moroianu J (1995) Nuclear import of influenza virus RNA can be mediated by viral nucleoprotein and transport factors required for protein import. J Biol Chem 270: 22701-22704.
Pleshka S, Ludwig S, Wolff T, Planz (2006) Anti-viral approaches against infleunza viruses. In: Bogner E, Holzenburg A, editors. Newconceptys of antiviral therapy. Dordrecht: Springer, pp. 115-188.
Rodriguez-Boulan E (1983) Polarized assembly of enveloped viruses from cultured epithelial cells. Methods in Enzymology 98: 487-501.
Rust MJ, Lakadamyali M, Zhang F, Zhuang X (2004) Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat Struct Mol Biol 11(6): 567- 573.
Sieczkarski SB, Whittaker GR (2002) Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J Virol 76(20): 10455-10464.
Sieczkarski SB, Brown HA, Whittaker GR (2003) Role of protein kinase C betall in influenza virus entry via late endosomes. J Virol 77(1): 460-469.
Skehel J, Wiley D (2000) Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69: 531 - 569.
Szoka, F. Jr., and D. Papahadjopoulos. 1978. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc. Natl. Acad. Sci. USA. 75(9): 4194-8.
White J, Kartenbeck J, Helenius A (1982) Membrane fusion activity of Influenza virus. EMBO J 1:217-222.
Whittaker GRD, P. (2006) Entry and intracellular transport of influenza virus. In: Kawaoka Y, editor. Influenza Virology: Current Topics by Yoshihiro Kawaoka: Caister Academic Press, pp. 37-64.
Wurzer WJ, Planz O, Ehrhardt C, Giner M, Silberzahn T et al. (2003) Caspase 3 activation is essential for efficient influenza virus propagation. Embo J 22(11): 2717-2728.
Yoshimura A, Ohnishi S (1984) Uncoating of influenza virus in endosomes. Journal of Virology 51 (2): 497-504.
Bonazzi, M., S. Spano, G. Turacchio, C. Cericola, C. Valente, A. Colanzi, H.S. Kweon, V.W. Hsu, E.V. Polishchuck, R.S. Polishchuck, M. Sallese, T. Pulvirenti, D. Corda, and A. Luini. 2005. CtBP3/BARS drives membrane fission in dynamin-independent transport pathways. Nat Cell Biol. 7:570-80.
Charras, G.T., C.K. Hu, M. Coughlin, and T.J. Mitchison. 2006. Reassembly of contractile actin cortex in cell blebs. J Cell Biol. 175:477-90.
Chung, C.S., C.Y. Huang, and W. Chang. 2005. Vaccinia virus penetration requires cholesterol and results in specific viral envelope proteins associated with lipid rafts. J Virol. 79:1623-34.
Damke, H., T. Baba, D.E. Warnock, and S.L. Schmid. 1994. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J Cell Biol. 127:915-34.
De Roe, C., P.J. Courtoy, and P. Baudhuin. 1987. A model of protein-colloidal gold interactions. J Histochem Cytochem. 35:1191-8.
Dharmawardhane, S., A. Schurmann, M.A. Sells, J. Chernoff, S.L. Schmid, and G.M. Bokoch. 2000. Regulation of macropinocytosis by p21-activated kinase-1. Mol Biol Cell. 11:3341-52.
Falcone, S., E. Cocucci, P. Podini, T. Kirchhausen, E. Clementi, and J. Meldolesi. 2006. Macropinocytosis: regulated coordination of endocytic and exocytic membrane traffic events. J Cell Sci. 119:4758-69.
Felding-Habermann, B., B.M. Mueller, C.A. Romerdahl, and D.A. Cheresh. 1992. Involvement of integrin alpha V gene expression in human melanoma tumorigenicity. J Clin Invest. 89:2018-22.
Fishkind, D.J., L.G. Cao, and Y.L. Wang. 1991. Microinjection of the catalytic fragment of myosin light chain kinase into dividing cells: effects on mitosis and cytokinesis. J Cell Biol. 114:967-75.
Frens, G. 1973. Controlled nucleation for the regulation of particle size in monodisperse gold suspensions. Nature Phys Sci. 241:20-12.
Greber, U.F., P. Webster, J. Weber, and A. Helenius. 1996. The role of the adenovirus protease in virus entry into cells. EMBO J. 15:1766-1777.
Greber, U.F., M. Willetts, P. Webster, and A. Helenius. 1993. Stepwise dismantling of adenovirus 2 during entry into cells. Cell. 75:477-486.
Grimmer, S., B. van Deurs, and K. Sandvig. 2002. Membrane ruffling and macropinocytosis in A431 cells require cholesterol. J Cell Sci. 115:2953-62.
Hewlett, L.J., A.R. Prescott, and C. Watts. 1994. The coated pit and macropinocytic pathways serve distinct endosome populations. J Cell Biol. 124:689-703.
Hinrichsen, L., J. Harborth, L. Andrees, K. Weber, and E.J. Ungewickell. 2003. Effect of clathrin heavy chain- and alpha-adaptin-specific small inhibitory RNAs on endocytic accessory proteins and receptor trafficking in HeLa cells. J Biol Chem. 278:45160-70.
Horisberger, M., and J. Rosset. 1977. Colloidal gold, a useful marker for transmission and scanning electron microscopy. J Histochem Cytochem. 25:295-305.
Imelli, N., O. Meier, K. Boucke, S. Hemmi, and U.F. Greber. 2004. Cholesterol is required for endocytosis and endosomal escape of adenovirus type 2. J Virol. 78:3089-98.
Kirkham, M., and R.G. Parton. 2005. Clathrin-independent endocytosis: new insights into caveolae and non-caveolar lipid raft carriers. Biochim Biophys Acta. 1746:349-63.
Limouze, J., A.F. Straight, T. Mitchison, and J.R. Sellers. 2004. Specificity of blebbistatin, an inhibitor of myosin II. J Muscle Res Cell Motil. 25:337-41.
Locker, J.K., A. Kuehn, S. Schleich, G. Rutter, H. Hohenberg, R. Wepf, and G. Griffiths. 2000. Entry of the two infectious forms of vaccinia virus at the plasma membane is signaling-dependent for the IMV but not the EEV. Mol.Biol.Cell. 11:2497-2511.
Martin, S.J., C.P. Reutelingsperger, A.J. McGahon, J.A. Rader, R.C. van Schie, D.M. LaFace, and D.R. Green. 1995. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 182:1545-56.
Meier, O., K. Boucke, S. Vig, S. Keller, R.P. Stidwill, S. Hemmi, and U.F. Greber. 2002. Adenovirus triggers macropinocytosis and endosomal leakage together with its clathrin mediated uptake. J. Cell Biol. 158:1119-1131.
Mills, J.C., N.L. Stone, J. Erhardt, and R.N. Pittman. 1998. Apoptotic membrane blebbing is regulated by myosin light chain phosphorylation. J Cell Biol. 140:627-36.
Nagel, H., S. Maag, A. Tassis, F.O. Nestle, U.F. Greber, and S. Hemmi. 2003. The alphavbeta5 integrin of hematopoietic and nonhematopoietic cells is a transduction receptor of RGD-4C fiber-modified adenoviruses. Gene Ther. 10:1643-53.
Nakano, M.Y., K. Boucke, M. Suomalainen, R.P. Stidwill, and U.F. Greber. 2000. The first step of adenovirus type 2 disassembly occurs at the cell surface, independently of endocytosis and escape to the cytosol. J Virol. 74:7085-95.
Nakano, M.Y., and U.F. Greber. 2000. Quantitative microscopy of fluorescent adenovirus entry. J. Struct. Biol. 129:57-68.
Sirena, D., Z. Ruzsics, W. Schaffner, U.F. Greber, and S. Hemmi. 2005. The nucleotide sequence and a first generation gene transfer vector of species B human adenovirus serotype 3. Virology. 343:283-98.
Suomalainen, M., M.Y. Nakano, K. Boucke, S. Keller, R.P. Stidwill, and U.F. Greber. 1999. Microtubule-dependent minus and plus end-directed motilities are competing processes for nuclear targeting of adenovirus. J. Cell Biol. 144:657-672.
Townsley, A.C., A.S. Weisberg, T.R. Wagenaar, and B. Moss. 2006. Vaccinia virus entry into cells via a low-pH-dependent endosomal pathway. J Virol. 80:8899-908.
Veithen, A., P. Cupers, P. Baudhuin, and P.J. Courtoy. 1996. v-Src induces constitutive macropinocytosis in rat fibroblasts. J Cell Sci. 109 ( Pt 8):2005-12.
【技術分野】
【0001】
クロスリファレンス
本出願は,米国特許仮出願60/945,740(2007年6月22日出願,その全体は参照として本明細書に組み込まれる)に基づく優先権を主張する。
【背景技術】
【0002】
発明の背景
抗ウイルス剤は,ウイルス感染の治療に用いられる一群の医薬品である。抗ウイルス剤は,抗微生物剤の1種であり,その大部分は抗生物質,抗真菌剤および抗寄生虫剤を含む。広範囲の病原菌をカバーしうる抗菌剤とは異なり,抗ウイルス剤はスペクトルが狭く,限定された効力しか持たない傾向にある。
【0003】
抗ウイルス剤の出現は,生物の遺伝子および分子機能に関する知識が拡大して,生物医学の研究者がウイルスの構造および機能を理解できるようになったこと,新規薬剤を発見する技術が進歩したこと,および致死性のある後天性免疫不全症候群(AIDS)の流行の原因であるヒト免疫不全ウイルス(HIV)を扱うように医療専門家に圧力がかけられたことの産物である。
【0004】
季節性ヒトインフルエンザについての継続する問題および将来のパンデミックの脅威のため,インフルエンザに対する抗ウイルス剤の開発は高い優先度を有する(Fauci 2006)。ワクチン接種はなお予防の基礎であるが,抗ウイルス剤は流行およびパンデミックの可能性に対する世界的規模の戦いにおける要素を構成する(Pleshka et al.2006)。抗ウイルス剤の有用性はウイルス中の抗原の変化により影響を受けないため,抗ウイルス剤はワクチンに比べて利点を有する。このことは,出現した種に対してワクチンが入手可能となる前にこれらを用いることができることを意味する。また,これらは成立した病気に対しても有効である。予防においては,これらは感染に対して防御し,ウイルスの広がりを減少させ,免疫感作に対して有用な補完として作用する。これらの利点のため,インフルエンザパンデミックのリスクに関するいくつかの米国政府系委員会は,このウイルスに対する新規な抗ウイルス剤を開発する研究を推奨してきた。
【0005】
インフルエンザに対する現在入手可能な抗ウイルス剤は,ウイルスM2−チャネル(アマンチジン,リマンチジン)またはノイラミニダーゼ(NA)(オセルタミビルおよびザナミビル)のいずれかを阻害するものである(Pleshka et al.2006)。前者はヒトインフルエンザA株に対する予防および治療用に安全かつ有用であるが,ほとんど処方されない。NA阻害剤は最も一般的に使用されている。これらはトリH5N1を含むインフルエンザA株に対して活性があり,インフルエンザBに対しても作用する。オセルタミビルは予防および治療に用いられているが,ザナミビル(吸入用)は予防用としてはまだ認可されていない。
【0006】
現在の抗ウイルス薬剤(および一般にウイルス蛋白質を標的とする薬剤)にともなう主要な問題点は,ウイルス遺伝子中の点突然変異による耐性の出現である。M2チャネルブロッカーについては,完全耐性の出現は急速である。NA阻害剤の場合には,耐性株は出現するが,幸運なことにこれまでのところこれらは野生型と比較して適応度および病原性が低い。しかし,耐性株が発生する可能性は問題として残っている。
【0007】
ウイルスそれ自体および標的としてのその蛋白質に焦点が当てられているが,ウイルス感染に関与する宿主細胞蛋白質に干渉する新世代の抗ウイルス剤を開発することは有用である。これらの蛋白質の活性を阻害することにより,子孫ウイルスの複製および産生を阻害することが可能となるであろう。
【発明の概要】
【課題を解決するための手段】
【0008】
発明の概要
本発明は,ウイルス感染において役割を果たす宿主細胞蛋白質を同定する方法を提供する。これらの宿主細胞標的蛋白質を同定することにより,これらを標的とするウイルス感染の治療的介入のための薬剤の同定が可能となる。また,これらの宿主細胞蛋白質を調節してウイルス感染を治療および/または予防する薬剤および方法も提供される。本発明は,宿主細胞蛋白質を調節することにより,宿主細胞においてウイルス感染を阻害または低減させる薬剤を提供する。さらに,本発明は,ウイルス感染の治療に用いることができるキットを提供する。
【0009】
1つの観点においては,本発明は,ポックスウイルス感染を治療する方法を提供し,この方法は,治療を必要とする被検動物に有効量のキナーゼ調節剤を投与することを含む。1つの態様においては,被検動物はヒトである。1つの態様においては,前記マクロピノサイトーシス経路の前記阻害剤は,PAK1;DYRK3;PTK9;およびGPRK2Lからなる群より選択されるキナーゼの阻害剤である。1つの態様においては,前記キナーゼ調節剤は宿主細胞キナーゼ調節剤である。1つの態様においては,キナーゼ調節剤は,キナーゼを標的とするドミナント・ネガティブ分子,siRNA,shRNA,抗体または小分子である。1つの態様においては,キナーゼ調節剤はsiRNAである。1つの態様においては,キナーゼ調節剤はCEP−1347である。1つの態様においては,前記宿主細胞キナーゼ調節剤は宿主細胞キナーゼ阻害剤である。1つの態様においては,前記宿主細胞キナーゼ阻害剤は,PAK1;DYRK3;PTK9;およびGPRK2Lからなる群より選択されるキナーゼの阻害剤である。1つの態様においては,ポックスウイルスは天然痘ウイルスである。1つの態様においては,ポックスウイルスはワクシニアウイルスである。1つの態様においては,感染は呼吸器感染である。
【0010】
別の観点においては,本発明は,ウイルス感染を治療する方法を提供し,この方法は,治療を必要とする被検動物に有効量のマクロピノサイトーシス経路の調節剤を投与することを含む。1つの態様においては,被検動物はヒトである。1つの態様においては,マクロピノサイトーシス経路の前記調節剤は前記マクロピノサイトーシス経路の阻害剤である。1つの態様においては,前記阻害剤はキナーゼ阻害剤である。1つの態様においては,前記阻害剤は宿主細胞キナーゼ阻害剤である。1つの態様においては,前記宿主細胞キナーゼ阻害剤は,PAK1;DYRK3;PTK9;およびGPRK2Lからなる群より選択されるキナーゼの阻害剤である。1つの態様においては,阻害剤はCEP−1347である。1つの態様においては,前記ウイルスはポックスウイルスである。1つの態様においては,前記ウイルスは天然痘ウイルスである。1つの態様においては,前記ウイルスはワクシニアウイルスである。
【0011】
別の観点においては,本発明は,細胞をキナーゼ阻害剤およびウイルスと接触させ,キナーゼ阻害剤がウイルスによる細胞の感染を阻害するか否かを判定することを含む方法を提供する。1つの態様においてはキナーゼ阻害剤は,PAK1;DYRK3;PTK9;およびGPRK2Lからなる群より選択されるキナーゼを阻害する。別の態様においては,キナーゼ阻害剤は,キナーゼを標的とするドミナント・ネガティブ分子,siRNA,shRNA,抗体または小分子からなる群より選択される。別の態様においては,ウイルスはインフルエンザウイルスまたはポックスウイルス,例えばワクシニアまたは天然痘ウイルスである。別の態様においては,接触はインビトロで行われる。
【0012】
参照
本明細書において言及されるすべての刊行物,特許および特許出願は,個々の刊行物,特許および特許出願が特定的にかつ個別に本明細書に参照として取り込まれるように,本明細書に参照として取り込まれる。
【0013】
本発明の新規な特徴は,特に添付の特許請求の範囲に記載される。本発明の原理を用いる例示的態様を記載する下記の詳細な説明および添付の図面を参照することにより,本発明の特徴および利点をより理解することができる。
【図面の簡単な説明】
【0014】
【図1】図1は,成熟ビリオン侵入の間のサーフィンおよび膜摂動を示す。
【図2】図2は,p21活性化キナーゼ−1(PAK1)がMV侵入に必要であることを示す。
【図3】図3は,ワクシニアMVが細胞に侵入するためにマクロピノサイトーシスを利用することを示す。
【図4】図4は,ワクシニアMVが内在化にPSを必要とすることを示す。
【図5】図5は,PAK1の活性化はAd3エンドサイトーシスおよび感染に必要であるが,Ad5には必要ではないことを示す。
【図6】図6は,マクロピノサイトーシスを用いるAd3感染の経路を示す。
【図7】図7は,MVをHeLa細胞に加えた後のEGFR活性化を示す。
【図8】図8は,EGFR阻害剤324674(Calbiochem)がMV侵入を遮断し,これは低pH融合によりバイパスされることを示す。
【発明を実施するための形態】
【0015】
発明の詳細な説明
本発明の多くの態様が本明細書に示され記載されるが,当業者にはそのような態様は例示のためにのみ提供されていることが明らかであろう。当業者は本発明から逸脱することなく多くの改変,変更および置き換えをなすことができる。本発明の実施に際しては,本明細書に記載される本明細書の態様の種々の代替物を用いることができることが理解される。下記の特許請求の範囲は本発明の範囲を規定し,これらの特許請求の範囲の範囲内の方法および構造およびそれらの均等物もカバーすることが意図される。
【0016】
本発明の方法は,ウイルスが感染,複製および/または伝播に利用する宿主細胞遺伝子を同定することを含む。また,同定された宿主細胞遺伝子によりコードされる特異的宿主細胞蛋白質を標的とする薬剤を同定する方法も開示される。さらに,本発明は,同定された宿主細胞標的を調節するための薬剤および方法を含む。そのような薬剤および方法は,ウイルス感染の治療に適している。そのような宿主細胞標的の調節は,宿主細胞標的の活性化または阻害のいずれかを含むことができる。したがって,非ウイルス蛋白質,例えばキナーゼ等の宿主細胞蛋白質の活性を調節,例えば阻害する化合物を,抗ウイルス医薬製剤として用いる。
【0017】
1つの態様においては,本発明の方法は,被検動物,例えば多数のウイルスのいずれかによるヒトの感染を阻害する抗ウイルス剤の開発に用いることができる。1つの態様においては,本発明の方法は,呼吸器ウイルスによる宿主の感染を阻害する抗ウイルス剤の開発に用いられる。呼吸器ウイルスは,最も一般的には,浮遊する飛沫または鼻汁により伝染し,広範な種類の病気につながりうる。呼吸器ウイルスとしては,呼吸器合胞体ウイルス(RSV),インフルエンザウイルス,コロナウイルス,例えばSARS,アデノウイルス,パラインフルエンザウイルスおよびライノウイルスが挙げられる。
【0018】
I.ウイルス
1つの態様においてはウイルス,例えば,ポックスウイルス,アデノウイルスまたは本明細書に記載される任意のウイルスが感染または複製に必要とする宿主細胞蛋白質が同定される。アデノウイルスは,最も一般的には呼吸器疾患を引き起こす。アデノウイルス感染により引き起こされる呼吸器疾患の症状は,一般的な風邪症候群から肺炎,クループ,および気管支炎まで様々である。免疫系に障害をもつ患者は特にアデノウイルス感染の重篤な合併症を起こしやすい。急性呼吸器疾患(ARD)は,第二次世界大戦の間に軍隊入隊者の間で最初に認められ,混雑およびストレスの状態の中でアデノウイルス感染により引き起こされうる。アデノウイルスは,中程度のサイズ(90−100nm)の,二本鎖DNAを含むエンベロープを有さない正二十面体ウイルスである。ヒト感染を引き起こしうる49種の免疫学的に区別しうるタイプ(6種の亜属:A−F)がある。アデノウイルスは,化学的または物理的作用および不利なpH条件に対して非常に安定であり,身体の外で長期間生存することが可能である。ある種のアデノウイルス,例えばAD2およびAd5(C種)は,感染性侵入にクラスリン媒介性エンドサイトーシスおよびマクロピノサイトーシスを利用する。他のアデノウイルス,例えばAd3(B種)は,感染性侵入にダイナミン依存性エンドサイトーシスおよびマクロピノサイトーシスを利用する。
【0019】
1つの態様においては,ポックスウイルスが感染または複製に必要とする宿主細胞蛋白質が同定される。ポックスウイルスは,一般にエンベロープを有する。このウイルスは,約200nmx300nmのサイズを有する。DNAは直鎖状の二本鎖である。ウイルスファミリーPoxviridaeは,オルトポックスウイルス属を含み,これは天然痘の原因であるVariolavera種を含む。このウイルスには2つの形態,すなわち大痘瘡と小痘瘡がある。天然痘は,典型的には,通常は感染したヒトの呼吸器系から浮遊した天然痘ウイルスの吸入によりヒトからヒトに伝染する。したがって,これらのウイルスの阻害はバイオテロに対する防御として有用である。ワクシニアもまた感染性ポックスウイルスである。
【0020】
1つの態様においては,呼吸器合胞体ウイルス(RSV)が感染または複製に必要とする宿主細胞蛋白質が同定される。RSVは新生児および1歳未満の小児における細気管支炎および肺炎の最も一般的な原因である。最も頻繁には,病気は発熱,鼻水,咳,および時には喘鳴から始まる。最初のRSV感染の間,25%から40%の新生児および年少の小児は細気管支炎または肺炎の兆候または症状を有しており,0.5%から2%は入院を必要とする。ほとんどの小児は8から15日間で病気から回復する。RSV感染で入院する小児の大部分は,生後6か月未満である。RSVはまた一生の間に反復感染を引き起こし,通常は中程度から重症の風邪様症状と伴う。しかし,いずれの年齢の間でも,特に老齢者および心臓,肺または免疫系に障害を有する者の間で,重症の下部呼吸管疾患を引き起こしうる。RSVはネガティブセンスのエンベロープRNAウイルスである。ビリオンは様々な形状およびサイズであり(平均直径は120−300nm),環境中で不安定であり(環境表面上では数時間しか生存しない),石けん水および消毒剤で容易に不活性化する。
【0021】
1つの態様においては,ヒトパラインフルエンザウイルス(HPIV)が感染または複製に必要とする宿主細胞蛋白質が同定される。HPIVは,呼吸器合胞体ウイルス(RSV)についで2番目に一般的な幼い小児における下部呼吸管疾患の原因である。RSVと同様に,HPIVは,一生の間に反復感染を引き起こし,通常は上部呼吸管疾患(例えば,風邪および/または咽喉炎)として現れる。HPIVはまた,特に老齢者および免疫系の障害をもつ患者の間で,反復感染により重篤な下部呼吸管疾患(例えば,肺炎,気管支炎,および細気管支炎)を引き起こしうる。4種のHPIVのそれぞれは,異なる臨床的および疫学的特徴を有する。HPIV−1およびHPIV−2の最も独特の臨床的特徴はクループ(すなわち,喉頭気管気管支炎)である。HPIV−1は小児のクループの主な原因であり,一方HPIV−2はあまり頻繁に検出されない。HPIV−1および−2の両方とも他の上部および下部呼吸管疾患を引き起こしうる。HPIV−3はしばしば細気管支炎および肺炎に関連している。HPIV−4はあまり頻繁に検出されず,これはおそらく重篤な疾患を引き起こさないためであろう。HPIVのインキュベーション期間は一般に1−7日間である。HPIVは,ネガティブセンスの一本鎖RNAウイルスであり,その表面にF蛋白質およびヘマグルチニン−ノイラミニダーゼ糖蛋白質の"スパイク"を持つ。HPIVには4つの血清型(1から4)および2つのサブタイプ(4aおよび4b)がある。ビリオンは様々なサイズ(平均直径は150から300nm)および形状であり,環境中で不安定であり(環境表面上で数時間の生存),石けん水により容易に不活性化される。
【0022】
1つの態様においては,コロナウイルスが感染または複製に必要とする宿主細胞蛋白質が同定される。コロナウイルスはCoronaviridaeファミリーに属する動物ウイルスの属である。コロナウイルスは,エンベロープを持つウイルスであり,ポジティブセンスの一本鎖RNAゲノムおよびらせん対称を有する。コロナウイルスのゲノムサイズは約16から31キロベースの範囲であり,RNAウイルスとしては異常に大きい。"コロナウイルス"との名称は,ラテン語のcorona(王冠を意味する)に由来する。これは,ウイルスエンベロープが電子顕微鏡下で小さな球状構造の特徴的な環の王冠を持つように見えるためである。この形態学は,実際にウイルスのスパイクペプロマーにより形成させており,これはウイルスの表面に存在して宿主指向性を決定する蛋白質である。コロナウイルスはニドウイルス目に分類され,この目のすべてのウイルスは感染の間に3’末端を共有する入れ子集合のサブゲノムmRNAを生成するため,巣を意味するラテン語のnidusから名付けられている。すべてのコロナウイルスの全体構造に寄与する蛋白質は,スパイク,エンベロープ,膜およびヌクレオカプシドである。SARSの特定の場合においては,S上の規定されたレセプター結合ドメインがウイルスのその細胞性レセプターであるアンジオテンシン変換酵素2への結合を媒介する。
【0023】
1つの態様においては,ライノウイルスが感染または複製に必要とする宿主細胞蛋白質が同定される。ライノウイルス("鼻"を意味するギリシャ語のrhin−に由来)はPicornaviridaeファミリーのウイルスの属である。ライノウイルスはヒトに感染性をもつ最も一般的なウイルスであり,一般的な風邪の原因ウイルスである。風邪の症状を引き起こすウイルス血清型は105を越えており,ライノウイルスは全症例の約50%の原因である。ライノウイルスは7.2から8.5kbの長さの一本鎖のポジティブセンスRNAゲノムを有する。ゲノムの5’末端にはウイルスによりコードされる蛋白質があり,哺乳動物のmRNAと同様に,3’ポリAテールが存在する。構造蛋白質はゲノムの5’領域にコードされており,末端は非構造性である。これはすべてのピコルナウイルスについて同じである。ウイルス粒子それ自体は,エンベロープをもたず,二十面体の構造を有する。
【0024】
1つの態様においては,インフルエンザウイルスが感染または複製に必要とする宿主細胞蛋白質が同定される。インフルエンザウイルスはウイルスのオルトミクソウイルスファミリーに属する。このファミリーには,トゴトウイルスおよびドーリウイルスも含まれる。ヒトおよび他の種に感染するインフルエンザウイルスとしては,いくつかの型およびサブタイプが知られている。A型インフルエンザ型インフルエンザウイルスは,ヒト,トリ,ブタ,ウマ,アザラシおよび他の動物に感染するが,野生の鳥類がこれらのウイルスの天然の宿主である。A型インフルエンザウイルスはいくつかのサブタイプに分類され,ウイルスの表面上の2つの蛋白質,すなわちヘマグルチニン(HA)およびノイラミニダーゼ(NA)に基づいて命名されている。例えば,“H7N2ウイルス”はHA7蛋白質およびNA2蛋白質を有するインフルエンザAサブタイプを表す。同様に,“H5N1”ウイルスはHA5蛋白質およびNA1蛋白質を有する。16種のHAサブタイプおよび9種のNAサブタイプが知られている。HAとNA蛋白質との多くの異なる組み合わせが可能である。現在,いくつかのインフルエンザAサブタイプ(すなわち,H1N1,H1N2,およびH3N2)のみがヒトの間で一般に流行している。他のサブタイプは主として他の動物種において見いだされる。例えば,H7N7およびH3N8ウイルスはウマに病気を引き起こし,またH3N8はイヌに病気を引き起こすことが最近示された(http://www.cdc.gov/flu/avian/gen-info/flu-viruses.htm)。
【0025】
インフルエンザ感染に関与する宿主細胞蛋白質を標的とする抗ウイルス剤を用いて,高リスク群(病院組織,高齢者ケア施設,免疫が抑制されている個体)をケースバイケースで保護することができる。抗ウイルス剤の可能性のある用途は,トリH5N1により引き起こされるかインフルエンザウイルスの他の株により引き起こされるかにかかわらず,将来のパンデミックの拡大および重篤性を制限することである。サブタイプH5およびH7のトリA型インフルエンザウイルス,例えばH5N1,H7N7,およびH7N3ウイルスは,高い病原性と関連づけられており,これらのウイルスによるヒトの感染は,軽症(H7N3,H7N7)から重症かつ致命的な疾患(H7N7,H5N1)まで様々である。病原性の低いウイルスの感染によるヒトの病気は,非常に軽い症状(例えば結膜炎)からインフルエンザ様の病気まで確認されている。ヒトに感染したことがある病原性の低いウイルスの例には,H7N7,H9N2,およびH7N2が含まれる。(http://www.cdc.gov/flu/avian/gen-info/flu-viruses.htm)。
【0026】
B型インフルエンザウイルスは通常はヒトに見いだされるが,アザラシにも感染しうる。A型インフルエンザウイルスとは異なり,これらのウイルスはサブタイプにしたがって分類されていない。B型インフルエンザウイルスはヒトの間で罹患および死亡を引き起こしうるが,一般にA型インフルエンザウイルスと比べて重大な流行を伴わない。B型インフルエンザウイルスはヒトでの流行を引き起こしうるが,パンデミクスを引き起こしたことはない(http://www.cdc.gov/flu/avian/gen-info/flu-viruses.htm)。
【0027】
C型インフルエンザウイルスウイルスはヒトに軽い病気を引き起こし,流行またはパンデミックを引き起こさない。これらのウイルスはイヌおよびブタにも感染することができる。これらのウイルスはサブタイプにしたがって分類されていない(http://www.cdc.gov/flu/avian/gen-info/flu-viruses.htm)。
【0028】
インフルエンザウイルスは,細胞表面レセプターの特異性および細胞指向性に関して互いに異なるが,共通する侵入経路を利用する。これらの経路を図表化し,ウイルスインフルエンザの伝染,侵入,複製,生合成,アセンブリ,または退出に関与する宿主細胞蛋白質を同定することにより,現存する,および出現するインフルエンザ種に対する一般的薬剤の開発が可能となる。薬剤はまた,同様の経路を利用する非関連ウイルスに対しても有用性を示すであろう。例えば,薬剤は,インフルエンザウイルスに加えて多数の異なるウイルスに対して気道上皮細胞を保護することができる。
【0029】
本明細書に記載される方法は,任意のウイルス,例えば,アベルソン白血病ウイルス,アベルソンネズミ白血病ウイルス,アベルソンウイルス,急性喉頭気管気管支炎ウイルス,アデレード川ウイルス,アデノ随伴ウイルス群,アデノウイルス,アフリカウマ病ウイルス,アフリカブタ熱ウイルス,AIDSウイルス,アリューシャンミンク病パルボウイルス,アルファレトロウイルス,アルファウイルス,ALV関連ウイルス,アマパリウイルス,アフタウイルス,アクアレオウイルス,アルボウイルス,アルボウイルスC,アルボウイルス群A,アルボウイルス群B,アレナウイルス群,アルゼンチン出血熱ウイルス,アルゼンチン出血熱ウイルス,アーテリウイルス,アストロウイルス,アテリネヘルペスウイルス群,オジェッキー病ウイルス,オーラウイルス,オースダック病ウイルス,オーストラリアコウモリリッサウイルス,トリアデノウイルス,トリ赤芽球症ウイルス,トリ感染性気管支炎ウイルス,トリ白血病ウイルス,トリ白血病ウイルス,トリリンパ腫症ウイルス,トリ骨髄芽球症ウイルス,トリパラミクソウイルス,トリ気脳炎ウイルス,トリ細網内皮症ウイルス,トリ肉腫ウイルス,トリC型レトロウイルス群,トリヘパドナウイルス,トリ痘ウイルス,Bウイルス,B19ウイルス,ババンキウイルス,ヒヒヘルペスウイルス,バキュロウイルス,バーマー森林ウイルス,ベラルウイルス,ベリマーウイルス,ベータレトロウイルス,バーナウイルス,ビトナーウイルス,BKウイルス,ブラッククリーク運河ウイルス,ブルータングウイルス,ボリビア出血熱ウイルス,ボマ病ウイルス,ヒツジボーダー病ウイルス,ボルナウイルス,ウシアルファヘルペスウイルス1,ウシアルファヘルペスウイルス2,ウシコロナウイルス,ウシ一過性熱ウイルス,ウシ免疫不全ウイルス,ウシ白血病ウイルス,ウシ白血病ウイルス,ウシ乳頭炎ウイルス,ウシパピローマウイルス,ウシ丘疹口内炎ウイルス,ウシパルボウイルス,ウシ合胞体ウイルス,ウシC型オンコウイルス,ウシウイルス下痢ウイルス,バギークリークウイルス,弾丸形ウイルス群,ブニヤンウエラウイルス超群,ブニヤウイルス,バーキットリンパ腫ウイルス,ブワンバ熱,CAウイルス,カリシウイルス,カリフォルニア脳炎ウイルス,ラクダ痘ウイルス,カナリ痘ウイルス,イヌヘルペスウイルス,イヌコロナウイルス,イヌジステンパーウイルス,イヌヘルペスウイルス,イヌ微小ウイルス,イヌパルボウイルス,カノデルガディトウイルス,ヤギ関節炎ウイルス,ヤギ脳炎ウイルス,ヤギヘルペスウイルス,ヤギ痘ウイルス,カルジオウイルス,テンジクネズミヘルペスウイルス1,オナガザルヘルペスウイルス1,オナガザルヘルペスウイルス1,オナガザルヘルペスウイルス2,チャンジプラウイルス,チャンギノラウイルス,ブチナマズウイルス,シャルルビルウイルス,ニワトリ痘ウイルス,チクングンヤウイルス,チンパンジーヘルペスウイルス,チャブレオウイルス,シロザケウイルス,コカルウイルス,ギンザケレオウイルス,媾疹ウイルス,コロラドダニ熱ウイルス,コルチウイルス,コロンビアSKウイルス,風邪ウイルス,感染性膿瘡ウイルス,感染性膿疱性皮膚炎ウイルス,コロナウイルス,コリパルタウイルス,鼻風邪ウイルス,牛痘ウイルス,コクサッキーウイルス,CPV(細胞質多角体病ウイルス),コオロギ麻痺ウイルス,クリミア−コンゴ出血熱ウイルス,クループ関連ウイルス,クリプトウイルス,サイポウイルス,サイトメガロウイルス,サイトメガロウイルス群,細胞質多角体病ウイルス,シカパピローマウイルス,デルタレトロウイルス,デングウイルス,デンソウイルス,デペンドウイルス,ドーリウイルス,ディプロルナウイルス,ショウジョウバエCウイルス,カモB型肝炎ウイルス,カモ肝炎ウイルス1,カモ肝炎ウイルス2,デュオウイルス,デュベンヘージウイルス,変形ウイングウイルスDWV,東部ウマ脳炎ウイルス,東部ウマ脳脊髄炎ウイルス,EBウイルス,エボラウイルス,エボラ様ウイルス,エコーウイルス,エコーウイルス,エコーウイルス10,エコーウイルス28,エコーウイルス9,欠肢症ウイルス,EEEウイルス,EIAウイルス,EIAウイルス,脳炎ウイルス,脳心筋炎群ウイルス,脳心筋炎ウイルス,エンテロウイルス,酵素上昇ウイルス,酵素上昇ウイルス(LDH),流行性出血熱ウイルス,流行性出血病ウイルス,エプスタインバーウイルス,ウマ科アルファヘルペスウイルス1,ウマ科アルファヘルペスウイルス4,ウマ科ヘルペスウイルス2,ウマ流産ウイルス,ウマ動脈炎ウイルス,ウマ脳症ウイルス,ウマ感染性貧血ウイルス,ウマ麻疹ウイルス,ウマ鼻肺炎ウイルス,ウマライノウイルス,ユーベナングウイルス,ヨーロッパエルクパピローマウイルス,ヨーロッパブタ熱ウイルス,エバーグレイズウイルス,ヤッチウイルス,ネコヘルペスウイルス1,ネコカリシウイルス,ネコ線維肉腫ウイルス,ネコヘルペスウイルス,ネコ免疫不全ウイルス,ネコ感染性腹膜炎ウイルス,ネコ白血病/肉腫ウイルス,ネコ白血病ウイルス,ネコ汎白血球減少症ウイルス,ネコパルボウイルス,ネコ肉腫ウイルス,ネコ合胞体ウイルス,フィロウイルス,フランダーズウイルス,フラビウイルス,口蹄疫ウイルス,フォートモーガンウイルス,フォーコーナーズハンタウイルス,家禽アデノウイルス1,家禽痘ウイルス,フレンドウイルス,ガンマレトロウイルス,GB肝炎ウイルス,GBウイルス,ドイツ麻疹ウイルス,ゲタウイルス,テナガザル白血病ウイルス,腺熱ウイルス,ヤギ痘ウイルス,ゴールデンシンナーウイルス,ゴノメタウイルス,ガチョウパルボウイルス,顆粒病ウイルス,グロスウイルス,ジリスB型肝炎ウイルス,群Aアルボウイルス,グアナリトウイルス,テンジクネズミサイトメガロウイルス,テンジクネズミC型ウイルス,ハンターンウイルス,ハンタウイルス,ホンビノスガイレオウイルス,野ウサギ線維腫ウイルス,HCMV(ヒトサイトメガロウイルス),赤血球吸着ウイルス2,日本の赤血球凝集ウイルス,出血熱ウイルス,ヘンドラウイルス,ヘニパウイルス,肝炎ウイルス,A型肝炎ウイルス,B型肝炎ウイルス群,C型肝炎ウイルス,D型肝炎ウイルス,デルタ肝炎ウイルス,E型肝炎ウイルス,F型肝炎ウイルス,G型肝炎ウイルス,非A非B肝炎ウイルス,肝炎ウイルス,肝炎ウイルス(非ヒト),肝脳脊髄炎レオウイルス3,肝ウイルス,ヘロンB型肝炎ウイルス,ヘルペスBウイルス,ヘルペス単純ウイルス,ヘルペス単純ウイルス1,ヘルペス単純ウイルス2,ヘルペスウイルス,ヘルペスウイルス7,ヘルペスウイルスクモザル,ヘルペスウイルスヒト,ヘルペスウイルス感染,ヘルペスウイルスリスザル,ヘルペスウイルスブタ,ヘルペスウイルス水痘,ハイランドJウイルス,ヒラメラブドウイルス,ブタコレラウイルス,ヒトアデノウイルス2,ヒトアルファヘルペスウイルス1,ヒトアルファヘルペスウイルス2,ヒトアルファヘルペスウイルス3,ヒトBリンパ球指向性ウイルス,ヒトベータヘルペスウイルス5,ヒトコロナウイルス,ヒトサイトメガロウイルス群,ヒト泡沫状ウイルス,ヒトガンマヘルペスウイルス4,ヒトガンマヘルペスウイルス6,ヒトA型肝炎ウイルス,ヒトヘルペスウイルス1群,ヒトヘルペスウイルス2群,ヒトヘルペスウイルス3群,ヒトヘルペスウイルス4群,ヒトヘルペスウイルス6,ヒトヘルペスウイルス8,ヒト免疫不全ウイルス,ヒト免疫不全ウイルス1,ヒト免疫不全ウイルス2,ヒトパピローマウイルス,ヒトT細胞白血病ウイルス,ヒトT細胞白血病ウイルスI,ヒトT細胞白血病ウイルスII,ヒトT細胞白血病ウイルスIII,ヒトT細胞リンパ腫ウイルスI,ヒトT細胞リンパ腫ウイルスII,1型ヒトT細胞リンパ球指向性ウイルス,2型ヒトT細胞リンパ球指向性ウイルス,ヒトTリンパ球指向性ウイルスI,ヒトTリンパ球指向性ウイルスII,ヒトTリンパ球指向性ウイルスIII,生痕ウイルス,新生児胃腸炎ウイルス,感染性ウシ鼻気管炎ウイルス,感染性造血壊死ウイルス,感染性膵臓壊死ウイルス,インフルエンザウイルスA,インフルエンザウイルスB,インフルエンザウイルスC,インフルエンザウイルスD,インフルエンザウイルスpr8,昆虫虹色ウイルス,昆虫ウイルス,イリドウイルス,日本Bウイルス,日本脳炎ウイルス,JCウイルス,フニンウイルス,カポジ肉腫関連ヘルペスウイルス,ケメロボウイルス,キラムラットウイルス,クラマスウイルス,コロンゴウイルス,韓国出血熱ウイルス,クンバウイルス,キサヌール森林病ウイルス,キジラガーシュウイルス,ラクロスウイルス,乳酸デヒドロゲナーゼ上昇ウイルス,乳酸デヒドロゲナーゼウイルス,ラゴスコウモリウイルス,ラングールウイルス,ウサギパルボウイルス,ラッサ熱ウイルス,ラッサウイルス,潜在的ラットウイルス,LCMウイルス,リーキーウイルス,レンチウイルス,ウサギ痘ウイルス,白血病ウイルス,白血病ウイルス,ランピースキン病ウイルス,リンパ節症関連ウイルス,リンパ球クリプトウイルス,リンパ球性脈絡髄膜炎ウイルス,リンパ増殖ウイルス群,マシュポウイルス,マッドイッチウイルス,哺乳動物B型オンコウイルス群,哺乳動物B型レトロウイルス,哺乳動物C型レトロウイルス群,哺乳動物D型レトロウイルス,乳癌ウイルス,マプエラウイルス,マールブルグウイルス,マールブルグ様ウイルス,メイソンファイザーサルウイルス,マストアデノウイルス,マヤロウイルス,MEウイルス,麻疹ウイルス,メナングルウイルス,メンゴウイルス,メンゴウイルス,ミデルブルグウイルス,乳牛結節ウイルス,ミンク腸炎ウイルス,マウスの微小ウイルス,MLV関連ウイルス,MMウイルス,モコラウイルス,軟体動物痘ウイルス,伝染性軟属腫ウイルス,サルBウイルス,サル痘ウイルス,モノネガウイルス,麻疹ウイルス,マウントエルゴンコウモリウイルス,マウスサイトメガロウイルス,マウス脳脊髄炎ウイルス,マウス肝炎ウイルス,マウスKウイルス,マウス白血病ウイルス,マウス乳癌ウイルス,マウス微小ウイルス,マウス肺炎ウイルス,マウスポリオウイルス,マウスポリオーマウイルス,マウス肉腫ウイルス,マウス痘ウイルス,モザンビークウイルス,ムカンボウイルス,粘膜病ウイルス,ムンプスウイルス,ネズミベータヘルペスウイルス1,ネズミサイトメガロウイルス2,ネズミサイトメガロウイルス群,ネズミ脳脊髄炎ウイルス,ネズミ肝炎ウイルス,ネズミ白血病ウイルス,ネズミ結節誘発ウイルス,ネズミポリオーマウイルス,ネズミ肉腫ウイルス,ムーロメガロウイルス,マリーバレー脳炎ウイルス,粘液腫ウイルス,ミクソウイルス,多形性ミクソウイルス,ミクソウイルスパロチチディス,ナイロビヒツジ病ウイルス,ナイロウイルス,ナニルナウイルス,ナリバウイルス,デュモウイルス,ニースリングウイルス,ネルソン湾ウイルス,神経指向性ウイルス,新世界アレナウイルス,新生児肺炎ウイルス,ニューカッスル病ウイルス,ニパウイルス,非細胞変性ウイルス,ノーウォークウイルス,核多角体病ウイルス(NPV),ニップルネックウイルス,オニョンニョンウイルス,オッケルボウイルス,発癌性ウイルス,発癌性ウイルス様粒子,オンコルナウイルス,オルビウイルス,伝染性膿疱性皮膚炎ウイルス,オロポーシェウイルス,オルトヘパディナウイルス,オルトミクソウイルス,オルトポックスウイルス,オルトレオウイルス,オロンゴ,ヒツジパピローマウイルス,ウシカタル熱ウイルス,ヨザルヘルペスウイルス,パリアンウイルス,パピローマウイルス,パピローマウイルスシルビラギ,パポバウイルス,パラインフルエンザウイルス,パラインフルエンザウイルス1型,パラインフルエンザウイルス2型,パラインフルエンザウイルス3型,パラインフルエンザウイルス4型,パラミクソウイルス,パラポックスウイルス,パラワクシニアウイルス,パルボウイルス,パルボウイルスB19,パルボウイルス群,ペスチウイルス,静脈ウイルス,アザラシジステンパーウイルス,ピコディナウイルス,ピコルナウイルス,ブタサイトメガロウイルス,ハト痘ウイルス,ピリーウイルス,ピクサナウイルス,
マウスの肺炎ウイルス,肺炎ウイルス,ポリオウイルス,ポリオウイルス,ポリディナウイルス,多角体病ウイルス,ポリオーマウイルス,ポリオーマウイルス,ウシポリオーマウイルス,オナガザルポリオーマウイルス,ヒトポリオーマウイルス2,ポリオーマウイルスマッカカエ1,ネズミポリオーマウイルス1,ネズミポリオーマウイルス2,ヒヒポリオーマウイルス1,ヒヒポリオーマウイルス2,ポリオーマウイルスシルビラギ,ポンギンヘルペスウイルス1,ブタ流行性下痢ウイルス,ブタ赤血球凝集性脳脊髄炎ウイルス,ブタパルボウイルス,ブタ伝染性胃腸炎ウイルス,ブタC型ウイルス,ポックスウイルス,ポックスウイルス,天然痘ポックスウイルス,プロスペクトヒルウイルス,プロウイルス,偽牛痘ウイルス,仮性狂犬病ウイルス,オウム痘ウイルス,ウズラ痘ウイルス,ウサギ線維腫ウイルス,ウサギ腎臓空胞化ウイルス,ウサギパピローマウイルス,狂犬病ウイルス,アライグマパルボウイルス,アライグマ痘ウイルス,ラニケートウイルス,ラットサイトメガロウイルス,ラットパルボウイルス,ラットウイルス,ラウシャーウイルス,組換えワクシニアウイルス,組換えウイルス,レオウイルス,レオウイルス1,レオウイルス2,レオウイルス3,爬虫類C型ウイルス,呼吸器感染ウイルス,呼吸器合胞体ウイルス,呼吸器ウイルス,細網内皮症ウイルス,ラブドウイルス,ラブドウイルスカルピア,ラジノウイルス,ライノウイルス,リジディオウイルス,リフトバレー熱ウイルス,ライリーウイルス,牛疫ウイルス,RNA腫瘍ウイルス,ロス川ウイルス,ロタウイルス,ルージョールウイルス,ラウス肉腫ウイルス,風疹ウイルス,麻疹ウイルス,ルビウイルス,ロシア秋脳炎ウイルス,SA11サルウイルス,SA2ウイルス,サビアウイルス,サギヤマウイルス,サイミリンヘルペスウイルス1,唾液腺ウイルス,サシチョウバエ熱ウイルス群,サンジンバウイルス,SARSウイルス,SDAV(唾液腺涙腺炎ウイルス),アザラシ痘ウイルス,セムリキ森林熱ウイルス,ソウルウイルス,ヒツジ痘ウイルス,ショープ線維腫ウイルス,ショープパピローマウイルス,サル泡沫状ウイルス,サルA型肝炎ウイルス,サルヒト免疫不全ウイルス,サル免疫不全ウイルス,サルパラインフルエンザウイルス,サルT細胞リンパ球指向性ウイルス,サルウイルス,サルウイルス40,シンプレックスウイルス,シンモンブレウイルス,シンドビスウイルス,天然痘ウイルス,南アメリカ出血熱ウイルス,スズメ痘ウイルス,スプマウイルス,リス線維腫ウイルス,リスザルレトロウイルス,SSV1ウイルス群,STLV(サルTリンパ球指向性ウイルス)I型,STLV(サルTリンパ球指向性ウイルス)II型,STLV(サルTリンパ球指向性ウイルス)III型,口内炎丘疹ウイルス,下顎ウイルス,イノシシアルファヘルペスウイルス1,ブタヘルペスウイルス2,スイポックスウイルス,沼地熱ウイルス,豚痘ウイルス,スイスマウス白血病ウイルス,TACウイルス,タカリベウイルス群,タカリベウイルス,タナポックスウイルス,タテラポックスウイルス,テンチレオウイルス,タイラー脳脊髄炎ウイルス,タイラーウイルス,トゴトウイルス,トッタパレイヤンウイルス,ダニ媒介性脳炎ウイルス,チオマンウイルス,トガウイルス,トロウイルス,腫瘍ウイルス,ツパイアウイルス,シチメンチョウ鼻気管炎ウイルス,シチメンチョウ痘ウイルス,C型レトロウイルス,D型オンコウイルス,D型レトロウイルス群,潰瘍性疾患ラブドウイルス,ウナウイルス,ウクニエミウイルス群,ワクシニアウイルス,空胞化ウイルス,水痘帯状疱疹ウイルス,水痘ウイルス,バリコラウイルス,大痘瘡ウイルス,天然痘ウイルス,バシンギシュー病ウイルス,VEEウイルス,ベネズエラウマ脳炎ウイルス,ベネズエラウマ脳脊髄炎ウイルス,ベネズエラ出血熱ウイルス,水疱性口内炎ウイルス,小胞性ウイルス,ビリュイスクウイルス,毒ヘビレトロウイルス,ウイルス出血性敗血症ウイルス,ビスナマエディウイルス,ビスナウイルス,ハタネズミ痘ウイルス,VSV(水疱性口内炎ウイルス),ウォーラルウイルス,ウォリゴウイルス,いぼウイルス,WEEウイルス,西ナイルウイルス,西部ウマ脳炎ウイルス,西部ウマ脳脊髄炎ウイルス,ワタロアウイルス,冬季嘔吐症ウイルス,ウッドチャックB型肝炎ウイルス,ウーリーモンキー肉腫ウイルス,創傷腫瘍ウイルス,WRSVウイルス,ヤバサル腫瘍ウイルス,ヤバウイルス,ヤタポックスウイルス,黄熱ウイルス,およびヤグボダノバックウイルスにより引き起こされる感染の治療用の薬剤の開発および/または同定に有用である。1つの態様においては,各ウイルスについて,ウイルス感染の特定の段階,例えば細胞侵入または複製サイクルの間にウイルス感染に関与する宿主細胞遺伝子の一覧を含むインフェクトームが作成される。
【0030】
II.ウイルス感染経路
本明細書に開示される宿主細胞標的は,好ましくはウイルス複製および/または感染経路において役割を果たす。そのような宿主細胞標的を標的とすることにより,ウイルスの複製および/または感染経路を調節することができる。好ましい態様においては,同定された宿主細胞標的は,適当な薬剤を用いて直接的にまたは間接的に調節される。そのような適当な薬剤としては,小分子治療剤,蛋白質治療剤,または核酸治療剤が挙げられる。そのような宿主細胞標的の調節はまた,宿主細胞標的の上流または下流のシグナリング経路の物質を標的とすることにより行うこともできる。
【0031】
他のウイルスと同様に,インフルエンザウイルスの複製には6つの相が含まれる;伝染,侵入,複製,生合成,アセンブリ,および退出。侵入は,エンドサイトーシスにより起こり,複製およびvRNPアセンブリは核で生じ,ウイルスは細胞膜から発芽する。感染した患者においては,ウイルスは気道上皮細胞を標的とする。好ましくは,本明細書に記載される方法においては,そのような経路に関与する少なくとも1つの宿主細胞標的が調節される。
【0032】
あるウイルスについては,宿主細胞の感染の間に関与する段階の解明において大きな進歩があった。例えば,1980年代初期に開始された実験では,インフルエンザウイルスは,他のウイルス,例えばアルファ−およびラブドウイルスと共有する要素を用いる段階的なエンドサイトーシスの侵入プログラムにしたたうことが示された(Marsh and Helenius 1989;Whittaker 2006)。段階は次のとおりである:1)細胞表面上のシアル酸含有複合糖質レセプターへの最初の付着;2)ウイルス粒子により誘導されるシグナリング;3)クラスリン依存性およびクラスリン非依存性の細胞メカニズムによるエンドサイトーシス;4)後期エンドソームからの酸誘導性,ヘマグルチニン(HA)媒介性浸透;5)酸により活性化されるM2およびマトリクス蛋白質(M1)依存性のカプシドの被覆の除去;および,6)vRNPのサイトゾル間輸送および核内輸送。これらの工程は,宿主細胞から,ソーティングレセプター,ベシクル形成機構,キナーゼ媒介性制御,オルガネラの酸性化,およびおそらくは細胞骨格の活性の形での支援に依存する。
【0033】
インフルエンザの細胞表面への付着は,HA1サブユニットが末端シアル酸残基をもつオリゴ糖成分を有する細胞表面の糖蛋白質および糖脂質に結合することにより生ずる(Skehel and Wiley 2000)。シアル酸と次の糖を連結する結合は種特異性に寄与する。H5N1等のトリの種は,a−(2,3)−結合を好み,ヒト種はa−(2,6)−結合を好む(Matrosovich 2006)。上皮細胞においては,結合は頂端表面上の微絨毛に優先的に生じ,エンドサイトーシスはこれらの拡大の基部において生ずる(Matlin 1982)。レセプターの結合が細胞を侵襲用に用意するシグナルを誘導するか否かはまだ明らかではないが,おそらくそうであろう。これは,蛋白質キナーゼCの活性化およびホスファチジルイノシトール−3−リン酸(PI3P)の合成が有効な侵入に必要であるからである(Sieczkarski et al.2003;Whittaker 2006)。
【0034】
エンドサイトーシスの内在化は,結合から数分以内に生ずる(Matlin 1982;Yoshimura and Ohnishi 1984)。組織培養細胞においては,インフルエンザウイルスは,3つの異なるタイプの細胞プロセスを利用する:1)予め存在するクラスリンにより被覆されたくぼみ,2)ウイルスにより誘導されるクラスリンにより被覆されたくぼみ,および3)見える被覆をもたないベシクル中のエンドサイトーシス(Matlin 1982;Sieczkarski and Whittaker 2002;Rust et al.2004),結果を参照)。蛍光ウイルスを用いるビデオ顕微鏡は,ウイルス粒子が細胞周辺でアクチン媒介性の急速な動きを示した後,微小管により媒介されるマイナス末端に向けて細胞の核周辺部へと輸送されることを示した。生きた細胞のイメージングは,ウイルス粒子が最初に移動性の周辺初期エンドソームの小集団に入り,これがウイルス粒子を細胞質中のより深い位置に運んだ後,浸透が生ずることを示した(Lakadamyali et al.2003;Rust et al.2004)。エンドサイトーシスのプロセスは,蛋白質キナーゼおよび脂質キナーゼ,プロテアソーム,ならびにRabsおよびユビキチン依存性輸送因子により制御されている(Khor et al.2003;Whittaker 2006)。
【0035】
膜浸透段階は,三量体の準安定性HAの低pH−媒介性活性化,およびこのI型ウイルスF蛋白質の膜融合可能コンフォメーションへの変換により媒介される(Maeda et al.1981;White et al.1982)。これは内在化の約16分後に起こり,pH閾値は株によって様々であり,5.0−5.6の範囲である。標的膜は中期または後期エンドソームの限定膜である。融合のメカニズムはよく研究されている(Kielian and Rey 2006)。さらに,融合それ自体は,脂質二重膜以外および機能的酸性化システム以外には宿主細胞成分を必要としないことが観察された(Maeda et al.1981;White et al.1982)。浸透段階は,リソソーム指向性弱塩基,カルボン酸イオノフォア,およびプロトンポンプ阻害剤等の薬剤により阻害される(Matlin 1982;Whittaker 2006)。
【0036】
侵入するvRNPの核内輸送を可能とするためには,カプシドが解体される必要がある。この段階は,アマンタジン感受性のM2チャネルによるウイルス内部の酸性化を含み,vRNPからのMlの解離を引き起こす(Bukrinskaya et al.1982;Martin and Helenius 1991;Pinto et al.1992)。個々のvRNPの核膜孔複合体への輸送および核内への移行は,細胞性核輸送レセプターに依存する(O’Neill et al.1995;Cros et al.2005)。ウイルスRNA(正鎖および負鎖の合成)の複製および転写は,核内のクロマチンと密に会合した複合体中で生ずる。これらの段階の多くはウイルスポリメラーゼにより触媒されるが,細胞性因子,例えばRNAポリメラーゼ活性化因子,シャペロンHSP90,hCLE,およびヒトスプライシング因子UAP56が関与していることが明らかである。ウイルス遺伝子発現は,細胞性キナーゼに依存する制御系である,転写レベルにおける複雑な細胞制御を受けている(Whittaker 2006)。
【0037】
インフルエンザ粒子の最終的なアセンブリは,細胞膜における発芽プロセスの間に行われる。上皮細胞においては,発芽は頂端膜ドメインにおいてのみ生ずる(Rodriguez−Boulan 1983)。まず,子孫vRNPが核質の中の核エンベロープに,次に核から細胞質に輸送され,最後に細胞周辺に蓄積する。核からの退出はウイルス蛋白質NEPおよびM1,および種々の細胞蛋白質,例えばCRM1(核外輸送レセプター),カスパーゼ,およびおそらくはある種の核蛋白質シャペロンに依存する。リン酸化は,M1およびNEP合成を制御することにより,およびMAPK/ERK系を介して,核外輸送において役割を果たす(Bui et al.1996;Ludwig 2006)。
【0038】
ウイルスの3つの膜蛋白質は,ER中で合成され,フォールディングされ,アセンブリされてオリゴマーとなる(Doms et al.1993)。これらはゴルジ複合体を通過し,その炭水化物成分の修飾および蛋白質分解酵素による切断により成熟する。これらは細胞膜に到達した後,発芽プロセスにおいてM1およびvRNPと会合し,その結果,8つすべてのvRNPを取り込み,脂質を除くほとんどの宿主細胞成分を排出する。
【0039】
インフルエンザ感染は,いくつかのシグナリングカスケード,例えば,MAPK経路(ERK,JNK,p38およびBMK−1/ERK5),IkB/NF−kBシグナリングモジュール,Raf/MEK/ERKカスケード,およびプログラムされた細胞死の活性化を伴う(Ludwig 2006)。その結果,感染の進行を制限する種々の影響,例えば,IFNbの転写的活性化,アポトーシス性細胞死,および後期エンドソームからのウイルスの脱出の妨害が生ずる(Ludwig 2006)。
【0040】
ウイルスと細胞との相互作用の最も先の研究は,組織培養または卵に適合したウイルス株を用いる組織培養において行われた。これらの例におけるウイルスは,レセプター結合および指向性に影響を与える変化が誘導されるよう適合された(Matrosovich 2006)。野生型の病原性の株の感染はウイルスと宿主蛋白質との相互作用のより自然の状況を提供する。ヒト気道においては,インフルエンザAおよびBは主として上部呼吸管のNeuSAca−(2,6)−Galを有する非繊毛上皮細胞に感染し,一方,トリ種は気道のより深くでa−(2,3)−結合シアル酸を有する繊毛上皮細胞に感染することが知られている(Matrosovich et al.2004a)。
【0041】
III.マクロピノサイトーシスによるウイルスの細胞内への侵入
本発明の1つの観点は,マクロピノサイトーシスによるウイルス侵入経路に関与する蛋白質を標的とする抗ウイルス療法である。好ましくは,標的蛋白質は宿主細胞蛋白質である。好ましい標的は,キナーゼおよびキナーゼ経路の蛋白質である。好ましい標的としては,PAK1;DYRK3;PTK9;GPRK2L;Cdc42;および/またはRac1が挙げられる。好ましくは,マクロピノサイトーシス経路をポックスウイルス感染の治療の標的とする。好ましいポックスウイルスは,天然痘の原因ウイルスである天然痘ウイルスである。
【0042】
単一の作用メカニズムに限定されることを意図するものではないが,ポックスウイルス,例えば,ワクシニアウイルスは,宿主細胞に侵入するためにマクロピノサイトーシスおよびアポトーシス擬態を利用することが見いだされた(図6)。マクロピノサイトーシスは,大量の液体が封入され内在化されるプロセスである。この経路には,細胞膜再編成,エンドサイトーシスベシクルの形成,および膜の波打ち部位における葉状仮足の閉鎖が関与し,マクロピノソームが形成される(Lanzavecchia,A(1996)Curr Opin Immunol 8348−354;Sieczkarski and Whittaker(2002)J Gen Virol 83:1535−1545)。Rho GTPase(West et al.(2000)Curr Biol 10:839−848),ARF6(Radhakrishna et al.(1996)J Cell Biol 134:935−947),および1型ホスファチジルイノシトール−3キナーゼ(PI3−K)(Hooshmand−Rad et al.(1997)Exp Cell Res234:434−441)が,マクロピノサイトーシスに関与している。さらに,2つのRab,GTPaseであるRab5およびRab34/Rahは,マクロピノソームの形成における関与が示唆されている(Li et al.(1997)J Biol Chem,272:10337−10340;Sun et al.(2003)J Biol Chem 278:4063−4071)。Rab34/Rahはアクチンとともに膜の波打ちおよび新生マクロピノソームに局在することができ,その過剰発現はマクロピノサイトーシスを促進しうる(Sun et al.(2003)J Biol Chem 278:4063−4071)。Rab5はRab34/Rahとともにマクロピノソームに局在することができる(Sun et al.(2003)J Biol Chem278:4063−4071)。
【0043】
本発明者らは,原型ポックスウイルスであるワクシニアウイルスがその宿主細胞に侵入する際に用いるメカニズムを分析した。ウイルスは組織培養細胞の糸状偽足に結合し,これに沿って細胞体に向かって移動し,アクチンおよびアクチン付随蛋白質を含む大きな,過渡的な膜ブレブの突出を誘導することが認められた。ブレブが引っ込むにつれて,ウイルスは細胞内小胞中に内在化された。ブレブ形成を阻害する阻害剤,例えばブレビスタチン,EIPA,またはPAK1のドミナント・ネガティブコンストラクトは,感染もブロックした。阻害プロファイルから,MVは,アポトーシス後の細胞残存物の排除に関与するエンドサイトーシスプロセスであるマクロピノサイトーシスを誘導することが明らかになった。マクロピノサイトーシスによるアポトーシス体の内在化の決定因子はホスファチジルセリン(PS)の暴露であることから,ならびにMVの膜はこのリン脂質が非常に豊富であることから,異なる脂質組成を有するウイルスを調製した。PSの存在はブレブ形成マクロピノサイトーシス,および感染能において役割を果たす。この結果は,ワクシニアウイルスは,ウイルス誘導性マクロピノサイトーシスおよびアポトーシス擬態を用いることを示す。
【0044】
別の態様においては,アデノウイルス,好ましくは流行性結膜炎,ぜんそく状態の増悪,罹患率および死亡率に関連するB種ヒトアデノウイルス血清型3(Ad3)の治療のために,マクロピノサイトーシス経路を標的とする。好ましくは,アデノウイルスに対する治療薬は,アクチン,蛋白質キナーゼC,ナトリウム−プロトン交換体,Rac1,PAK1,およびC末端アデノウイルスE1A結合蛋白質−1(CtBP1)を標的とする。
【0045】
単一の作用のメカニズムに限定されることを意図するものではないが,Ad3は上皮および造血細胞に侵入するためにダイナミン非依存性マクロピノサイトーシスを利用することが認められた。感染性のAd3マクロピノサイトーシスは,アクチン,蛋白質キナーゼC,ナトリウム−プロトン交換体,およびRac1を標的とする阻害剤に対して感受性であるが,Cdc42に対してはそうではない。これは,p21活性化キナーゼ1(PAK1),および先天的な免疫応答を含む膜通過および転写抑制に関与する二機能性蛋白質であるC末端アデノウイルスE1A結合蛋白質−1(CtBP1)のウイルス活性化を必要とする。CtBP1はPAK1によりリン酸化され,細胞膜およびマクロピノソームにリクルートされ,同時に転写抑制解除が生ずる。これらを合わせて,Ad3は,抗原提示用に設計された,転写的抗ウイルス宿主遺伝子活性化によりウイルス宿主範囲を広げる,先天的なエンドサイトーシスの免疫経路を妨害する。
【0046】
IV.ウイルス感染において役割を果たす宿主細胞蛋白質を同定するための方法および装置
ウイルスによる細胞侵入および増殖感染には,段階的なプログラムが関与しており,ここでは,わずかの事象がウイルス蛋白質および酵素により媒介され,残りの事象は細胞性機能に依存する。関与する細胞性蛋白質の完全な目録を得るためには,システムバイオロジーのアプローチを利用する態様が非常に有用である。組織培養細胞におけるインフルエンザ感染に関与する必須遺伝子の系統的同定を含む態様は,発見の有益な手段を提供する。siRNAのゲノムワイドなライブラリおよびハイスループット装置プラットフォームを含むシステムバイオロジーのアプローチは,ウイルス感染に関与する宿主細胞蛋白質を大量の候補物質蛋白質から迅速かつ効率的に同定することができる。
【0047】
1つの態様においては,自動化ハイスループットsiRNAスクリーニング技術をゲノムデータベース情報と組み合わせて用いて,宿主細胞蛋白質の系統的同定を行う。ここでは,ゲノムデータベースは,そのゲノム配列が知られている任意の種,例えば,ヒト,マウス,またはトリ種に由来するものであることができる。ある態様においては,高度なロボティクスおよびスクリーニング技術,例えば,the RNAi Image−based Screening Center(RISC)をもつスクリーニングプラットフォームを用いることができる。siRNAスクリーニングは,任意の適当な宿主細胞または細胞株,例えば,マウスまたはヒト宿主細胞,例えば,気道上皮細胞,またはHeLa MZ細胞,HeLa Kyoto細胞,またはA549細胞等の宿主細胞株を用いて行うことができる。他の適当な細胞株としては,16HBEと称される気管支細胞株,THEと称される気管細胞株,ならびに空気−液体界面(いわゆるALI培養)でよく分化した培養多列粘液線毛上皮を形成する市販のヒト気道上皮細胞培養物(HBEpC,Promocell,Heidelberg Germanyから購入)が挙げられる。そのような細胞はインフルエンザ感染のモデルとして用いることができることが知られている(Matrosovich et al.2004)。ある態様においては,関連するかまたは必要なウイルス侵入レセプター(例えば,HIV−1についてはCD4およびCXCR4)を発現するようトランスフォームされている安定な宿主細胞株を作製することができる。宿主細胞は,機能的効力について予め確認されているsiRNAのゲノムライブラリを用いてスクリーニングすることができる。ある態様においては,siRNAのゲノムライブラリは,商業的供給元,例えばQiagenから入手することができる。
【0048】
1つの態様においてはHeLa細胞を宿主細胞として用いる。HeLa細胞は,siRNAトランスフェクションによる有効なサイレンシングが可能である。インフルエンザウイルスの試験を含む態様は,単一のインフルエンザウイルスが細胞膜の被覆されたまたは被覆されていないくぼみの両方に結合することを示す。10分後,ウイルスは被覆されたおよび被覆されていない小さいベシクル中に存在し,30分後にはエンドソームと一致する見かけのより大きいベシクル中で多くが検出される。すなわち,ウイルス侵入の形態学は,内在化がゆっくりであることを除き,MDCK細胞において観察されたものと似ている。さらに,インフルエンザウイルスがエンドソーム構造に入って出る軌跡を,初期エンドソームを緑に標識するRab5−GFPおよび後期エンドソームを赤に標識するRab7−RFPを発現するHela細胞を用いて追跡した。さらに,HeLa細胞を用いて,感染,転写,およびウイルス蛋白質合成の初期段階を研究し,または後期段階のいくつか,例えばvRNPの核外輸送の障害についてスクリーニングすることができる。
【0049】
別の態様においてはA549細胞を宿主細胞として用いる。A549細胞は,呼吸器ウイルス,例えばインフルエンザウイルス感染の研究を含む態様において特に有用である。A549細胞は気管支起源の上皮細胞株であり,インフルエンザ感染研究において広く用いられている(Ehrhardt et al.2006)。A549細胞は,インフルエンザ疾患の間にインサイチューで感染した宿主細胞に非常に類似した系を提供する。さらに,A549細胞は,子孫ウイルスの放出および二次感染を含む全複製サイクルを分析する可能性を提供する。インフルエンザ研究およびアッセイにおいてしばしば用いられるMDCK細胞とは異なり,A549細胞はヒト起源であり,siRNAにより容易にトランスフェクションすることができる(Graeser 2004)。さらに別の態様においては,次の2つのインフルエンザウイルスを試験して,培養A549細胞におけるウイルスの伝播および二次感染を自動化ハイスループットフォーマットで分析する:1)トリH7N7ウイルス,ほとんどの細胞株においてそのHAはセクレターゼ切断により活性化される(Wurzer et al.2003);および2)ヒトインフルエンザ株,例えばX31/Aichi/68,および96,384,768,1152,1440,1536,3072ウエルプレート,または他のマルチウエルプレートのフォーマットと適合するトリプシン重層方式。
【0050】
さらに,本発明の態様は自動化スクリーニングプラットフォームを用いて実施しうることが企図される。ここでは,スクリーニングプラットフォームは,液体を操作するロボット,例えばTecanおよび2つの自動化顕微鏡,例えばCell Worx(Applied Precision Instruments)を含むことができる。自動化スクリーニングプラットフォームを用いてハイスループット実験手順を実施しうることが理解される。さらに,コンピュータおよび実験的努力を平行に組み合わせて,siRNAアッセイを最適に適用し,完全に自動化されたデータトラッキング,画像分析,定量,および統計学的分析のためのソフトウエアを設定することができる。
【0051】
大規模の態様においては,宿主細胞株の全ゲノムをカバーするsiRNAを用いてスクリーニングを行う。別の態様においては,宿主細胞株のゲノムのサブセット(例えば,少なくとも600,100,2000,3000,4000,5000,6000,7000,8000,9000,10000,15000,20000,25000,または30000の遺伝子)をカバーするsiRNAを用いてスクリーニングを行う。例えば,1つの可能な態様においては,ヒトゲノムの少なくとも7,000の遺伝子をカバーするsiRNAを用いてスクリーニングを行う。RISCプラットフォームにより,調べるべき各ウイルス株について2つの異なる細胞株を用いて7,000遺伝子のスクリーニングを2−4週間以内に完了することが可能である。次に,ユーザ仕様のMatLabプラグインを用いて,データセットを詳しく分析して管理する。MatLabプラグインは,生成された画像中でデータを自動的に定量することができ,低品質の画像を自動的に廃棄し,データ分析の構造安定性および再現性を判定する品質管理アルゴリズムを含むことができる。分析が完了すれば,結果からウイルス侵入に関与する宿主蛋白質を同定することができる。ウイルスインフェクトームライブラリは,元々はcDNAマイクロアレイの分析用に作成されたがRNAiデータセットとともに用いるように大幅に改変されたバイオインフォマティクスのツール上に構築する。大きなデータセットの強固な統計学は,ほとんどの重みが非常に顕著な表現型に与えられることを確実にする。特定の表現型は,試験する各遺伝子について少なくとも3つのsiRNAを用い,ある遺伝子に対する3つのsiRNAのうち2つが同様の効果を示すことを必要とすることにより重み付けされる。
【0052】
ある態様では,画像に基づくアッセイを利用することができる。これはプレートリーダーより感度が高く,したがってウイルス感染の背後にある細胞生物学に関する追加の情報が得られる。これらの態様においては,平静対照では平均で10−20%の細胞しか感染しないため,高い感度が望ましい。低い「ベースライン」は,より効率的なsiRNAサイレンシングと関連しており,感染における増加と減少との間を区別することができる。この判定は,感染経路に関する最適な情報を提供する。
【0053】
大きなフォーマットのsiRNAスクリーニング(例えば宿主細胞ゲノムのサブセットまたは全体をカバーする大きな遺伝子セット)を含むある態様においては,自動化液体操作ロボット,例えば,96,384,768,1152,1440,1536,3072ウエルプレート,または他のマルチウエルプレートを操作しうるTecanを用いる。生成したデータ(ウエルあたり9個の画像;スクリーンあたり1,430,784個の画像,約3.8TBに対応)をNASサーバに自動的に動かすアルゴリズムを用いる。さらに別の態様においては,高いバッファー容量,例えば1,2,3,4,5,6,7,8,9,または10TBは,一次的なネットワーク障害により分析プロセスが遅れないことを保証する。さらに別の態様においては,これらの大きな画像のセットの中から分析されていない画像を連続的に探すアルゴリズムは,自動的に画像を分析待ち行列に配置する。これらの態様のいくつかにおいては,MatLab画像分析プラグインを用いる。さらに,スクリーニングからの「生の」データについてバイオインフォマティクス評価を行って擬陽性を排除することにより,複雑なプロセスに関与する細胞システムを再構築することができる。このことにより,各侵入経路および他の感染関連プロセスに特異的な分子機構の鍵となる標的宿主細胞蛋白質の定義が可能となる。ある態様においては,用いられる基準には,強いRNAi表現型および広い細胞タイプ依存性が含まれる。
【0054】
上述の方法は,siRNAについて実施されてきた。しかし,他の適当な分子種,例えば,有機または無機化合物,抗体等の蛋白質,またはアンチセンスRNA等の核酸種も用いることができる。
【0055】
本発明の核酸療法は,天然の核酸であっても,修飾された核酸であっても,核酸の類似体であってもよい。核酸類似体としては,例えば,ペプチド核酸(PNA),ロック核酸(LNA),トレオース核酸(TNA),拡大塩基DNA(xDNAまたはyDNA)が挙げられる。同様に,ホスホロチオエートまたはホスホネート骨格修飾核酸も包含される。
【0056】
V.キナーゼ
ある態様においては,ウイルス感染を調節するとして同定される宿主細胞蛋白質はキナーゼである。さらに別の態様においては,宿主細胞蛋白質は,PAK1,Cdc42,Rac1,DYRK3,PTK9,およびGPRK2Lである。数百種のヒトキナーゼが知られており,これらはいくつかの異なるファミリーに分類され,種々の疾患状態において役割を果たすことが知られている(Manning et al.2002)。
【0057】
キナーゼの阻害剤としては,例えば,ドミナント・ネガティブ分子,siRNA,shRNA,抗体および小分子が挙げられる。ドミナント・ネガティブ分子には,例えば,分子内または分子間の蛋白質−蛋白質相互作用のインターフェースをブロックすることにより,インビトロまたはインビボで蛋白質の機能を妨害する分子が含まれる。ドミナント・ネガティブ分子としては,例えば,蛋白質標的のフラグメント(変異体フラグメントを含む)および標的蛋白質の非機能的変異体が挙げられる。抗体としては,例えば,完全イムノグロブリン,一本鎖抗体およびイムノグロブリンの特定の結合部位が挙げられる。小分子としては,例えば,約5000Daまで,約2000Daまで,または約1000Daまでの質量を有する有機または無機の非ポリマー性分子が挙げられる。
【0058】
A.PAK1
PAK1機能の分子メカニズムについては多くの見識が存在する(概説として,Parrini,MC et al.(2005)Biochem Soc Trans 33:646−648)。PAK1は自己阻害ホモダイマーとして存在することができる(Lei,M et al.(2000)Cell 102,387−397)。1つのPAK1分子のN末端制御ドメインは別のPAK1分子のC末端の触媒ドメインに結合してこれを阻害することができる。PAK1はGTP結合形のCdc42およびRac1に結合することにより活性化されることができる。これらの分子への結合は,制御ドメインのフォールディングを変化させることができ,これはPAK1ホモダイマーの解離につながる(Lei,M et al.(2000)上掲;Parrini,MC et al.(2002)Mol Cell.9,73−83.)。PAK1はまた,GTPaseであるRac2,Rac3,TC10,CHP,およびWrch−1に結合し,活性化されることができる(概説として,Zhao and Manser(2005)Biochem J.386,201−214)。これらのGTPaseによる結合および活性化は,N末端制御ドメインないしPBD(p21結合ドメイン)中の残基が媒介することができる。Cdc42およびRacはCdc42およびPAK1のRac相互作用結合ドメイン(CRIB)(アミノ酸75−90)(Burbelo et al.(1995)J Biol Chem.8,29071−29074)にわずかに結合することができ,キナーゼ阻害ドメイン(KI)を挟む配列は結合親和性に寄与しうる(Knaus and Bokoch(1998)Int J Biochem Cell Biol.30,857−862;Sells,MA and Chernoff,J(1997)Trends Cell Biol.7,162−167;Lei,M et al.(2000)上掲)。CRIBドメインのN末端の短いリジンリッチセグメント(PAK1アミノ酸66−68)は,Rac GTPaseの結合を媒介することができる(Knaus,UG and BokochGM(1998)上掲)。
【0059】
KIドメインは,約90nMのKiで触媒ドメインを阻害することができる(Zhao et al.(1998)Mol.Cell.Biol.18:2153−2163)。PAK1のKIドメインの残基Leu−107,ならびにKIドメインの他のアミノ酸が,この阻害の接点として寄与しうる(Lei et al.(2000)上掲)。PAK1のKI領域は,活性部位の2つの構造成分(ヘリックスCおよび活性化ループ)を安定化することができる。KIセグメントのリジンは,触媒に役割を果たす2つのアスパラギン酸残基とともに塩架橋を形成することにより,活性部位をブロックすることができる。このKIポリペプチドは,PAK活性化をブロックすることができる(Zhao et al.(1998)上掲)。PAK1のPBDを含むペプチドのCdc42への結合における結合定数は10−50nMの範囲であることが報告されている(Thompson et al.(1998)Biochemistry 37:7885−7891)。
【0060】
PAK1のN末端制御ドメインも,PAK相互作用交換因子(PIX)に結合することができる2つの保存されたPXXP SH3(Srcホモロジー3)結合モチーフおよび保存SH3結合部位を含む(Manser et al.(1998)Mol.Cell 1:183−192)。1番目の保存SH3結合部位は,アダプター蛋白質であるNckに結合することができ(Bokoch et al.(1996)J.Biol.Chem.271:25746−25749),2番目の部位はGrb2に結合することができる(Puto et al.(2003)J.Biol.Chem.278:9388−9393)。
【0061】
GTPaseの結合は,KIドメインの触媒ドメインとの相互作用を破壊するコンフォメーションの変化を引き起こすことができ,キナーゼ活性に寄与しうる自己リン酸化を可能とすることが示唆されている(Lei,M et al.(2000)上掲)。自己リン酸化は,PAK1を活性状態へとスイッチすることができる。PAK1の触媒ドメインの活性化ループ中のThr−423の自己リン酸化は,自己阻害からの解除および外因性基質に対する触媒機能を維持することができる(Yu et al.(1998)Biochem.J.334:121−131;Gatti et al.(1999)J.Biol.Chem.274:8022−8028;Zenke et al.(1999)J.Biol.Chem.274:32565−32573)。PAK1はPDK1(3−ホスホイノシチド依存性キナーゼ1)により修飾されることができる(King et al.(2000)J.Biol.Chem.275:41201−41209)。Ser−144(KIドメイン中の保存残基)におけるαPAKの自己リン酸化はキナーゼ活性化に寄与することができるが(Chong et al.(2001)J.Biol.Chem.276:17347−17353),一方,PAK1の自己リン酸化部位Ser−198/203はPIX−PAK相互作用を下方制御することができる。
【0062】
PAK1は,RacおよびCdc42 GTPaseとは独立して活性化されることができる。限定されたプロテアーゼ媒介性切断は,PAKキナーゼの自己リン酸化および活性を刺激することができる(Brenner et al.(1995)J.Biol.Chem.270:21121−21128;Roig et al.(1998)Vitam.Horm.62,167−198)。SH3含有NckおよびGrb2アダプター蛋白質を介するPAK1の膜リクルートメントは,キナーゼ活性を刺激することができる(Lu et al.(1997)Curr.Biol.785−94;Daniels et al.(1998)EMBO J.274:6047−6050)。この活性化は,重要なThr−423残基におけるPDK1によるリン酸化(King et al.(2000)J.Biol.Chem.275:41201−41209)またはスフィンゴシン等の脂質との相互作用を含むことができ,これはGTPase非依存性様式でキナーゼを活性化することができる(Bokoch et al.(1998)J.Biol.Chem.273:8137−8144)。PIXを介してPAKと間接的に関連しうるGIT1(G−蛋白質共役レセプターキナーゼ−相互作用標的1)もまた,Rho GTPaseを必要としないメカニズムによりPAKを活性化することができる(Loo et al.(2004)Mol.Cell.Biol.24:3849−3859)。
【0063】
PAK1はまた,焦点接着関連蛋白質PIX(Coolとも称される)と複合体を形成することができる。2つの異なる遺伝子(αPIXおよびβPIX)に由来する複数のPIX蛋白質は,そのSH3ドメインを介してPAKに結合することができる(Manser et al.(1998)Mol.Cell1:183−192;Bagrodia et al.(1999)J.Biol.Chem.274:22393−22400)。αPIXを欠失した細胞の分析から,走化性白血球のCdc42媒介性方向センシングにおけるPIX−PAK複合体の役割が示唆されている(Li et al.(2003)Cell 114:215−227)。PIXのGIT1(PKL/CAT1としても知られる(Turner et al.(1999)J.Cell Biol.145:851−863;Bagrodia et al.(1999)上掲)との会合は,パキシリンに結合することにより焦点接着を標的とすることができる(Turner et al.(1999)上掲)。GIT1の過剰発現により,焦点接着の分解およびパキシリンの喪失が生じうる(Loo et al.(2004)Mol.Cell.Biol.24:3849−3859)。すなわち,GIT1およびPIXは両方とも運動性細胞および細胞−細胞接合の前縁の焦点接着に局在してPAKを活性化することができる(Zegers et al.(2003)EMBO J.22:4155−4165;Zhao et al.(2000)Mol.Cell.Biol.20:6354−6363;Manabe et al.(2002)J.CellSci.115:1497−1510)。
【0064】
2つの関連するヒト蛋白質ホスファターゼは,PAK1を例えばThr−423で脱リン酸化することができる(Koh et al.(2002)Curr.Biol.12:317−321)。これらのホスファターゼは,POPX1(PIX1のパートナー)およびPOPX2であり,これらは様々な形のPIXに結合して,PAKを含む多量体複合体を形成するすることができる。活性なPAK1の細胞における効果は,これらのホスファターゼのいずれかの過剰発現により拮抗されうる(Manabe et al.(2002)上掲)。
【0065】
他の蛋白質キナーゼはPAK機能を下方制御するかもしれない。AktはPAK1をSer−21でリン酸化することができ,この修飾は,NckのPAK1N−末端への結合を低下させることができるが,キナーゼ活性は増加する(Zhao et al.(2000)上掲;Tang et al.(2000)J.Biol.Chem.275:9106−9109)。
【0066】
PAK1は,マクロピノサイトーシスの制御に関与している。活性化されたPAK1変異体(T423E)は,ストレス線維および焦点接着複合体の崩壊,および葉状仮足の形成(Sells et al.(1997)Curr Biol.7:202−210;Manser et al.(1997)Mol Cell Biol 17:1129−1143),およびアクチン骨格の再編成を誘発しうる。PAK1が関与するキナーゼ活性および蛋白質−蛋白質相互作用は,アクチン細胞骨格に影響を及ぼしうる(Sells et al.(1997)Curr Biol.7:202−210;Turner et al.(1999)J Cell Biol.145:851−863)。
【0067】
本発明の方法および組成物において用いることができるPAK1の阻害剤としては,例えば,内皮細胞移動を調節することができるPAK1の残基1−74(MSNNGLDIQDKPPAPPMRNTSTMIGAGSKDAGTLNHGSKPLPPNPEEKKKKDRFYRSILPGDKTNKKKEKERPE;(配列番号1)を含むドミナント・ネガティブPAK1(Kiosses et al.(1999)J.Cell Biol.147:831−843);PAK1の最初のプロリンリッチドメインの13アミノ酸(KPPAPPMRNTSTM;(配列番号2));HIV tat蛋白質のポリ塩基性配列に融合させたこれらの残基(YGRKKRRQRRRGKPPAPPMRNTSTM;(配列番号3))(Kiosses et al.(2002)Circ.Res.90:697−702);PAK1自己阻害ドメインを含み,マクロピノサイトーシスをブロックすることができるPAK1のアミノ酸83−149のフラグメント,HTIHVGFDAVTGEFTGMPEQWARLLQTSNITKSEQKKNPQAVLDVLEFYNSKKTSNSQKYMSFTDKS(配列番号4)(Dharmawardhane et al.(2000)Mol.Biol.Cell11:3341−3352;Barradeau et al.米国特許7,364,887);PAK1との結合に影響をおよぼしうる一酸化窒素シンターゼ(DLC1/PIN)のダイニン軽鎖−1/蛋白質阻害剤のペプチド(Kumar et al.米国特許7,067,633);ドミナント・ネガティブPAK1(PAK1(K299R))(Naor,米国特許公開20050090474)が挙げられる。
【0068】
本発明の方法および組成物において用いることができるPak1の間接的阻害剤としては,例えば,PAK1キナーゼ活性を低下させることができるヒストン脱アセチル化阻害剤FK228(Hirokawa et al.(2005)Can.Biol.Ther.4:956−960);それぞれSrcファミリーキナーゼおよびETKを阻害することによりPAK1の活性化を低下させることができるチロシン−キナーゼ阻害剤PP1およびAG879(He et al.(2004)Can.Biol.Ther.3:96−101;He et al.米国特許公開20030153009);およびPAK1活性を低下させるPP1とAG879,GL−2003の水溶性誘導体との組み合わせ(Hirokawa et al.(2006)Cancer Letters 245:242−251)が挙げられる。
【0069】
本発明の方法および組成物において用いることができるPAK1の別の阻害剤には,インビトロおよびインビボでのPAK1の直接阻害剤であるCEP−1347(Nheu,TV et al.(2002)Cancer J.8,328−336)が含まれる。
【0070】
本発明の方法および組成物において用いることができるPAK1のさらに別の阻害剤には,VanEykら(米国特許6,248,549)に開示されるものが含まれる。
【0071】
本発明の方法および組成物において用いることができるさらに別の阻害剤としては,PAK1に対するsiRNAが挙げられる:siPAK1−0AGAGCTGCTACAGCATCAA(配列番号6),siPAK1−1GACAUCCAACAGCCAGAAA(配列番号7),siPAK1−2GAGAAAGAGCGGCCAGAGA(配列番号8),hPAK1−6UACCAGCACUAUGAUUGGA(配列番号9),siPAK1−7UCUGUAUACACACGGUCUG(配列番号10)(Nasoff et al.2007米国特許公開US20070128204(2006年12月1日出願),およびQiagenから入手可能な3つのsiRNAオリゴ(PAK1_p1,PAK1_p2,およびPAK1_p3)(表1)。これらのsiRNAは,OiagenによりRT−PCRを用いて評価され,約70%以上の標的遺伝子mRNAのノックダウンを与えることが示されている。これらのsiRNAは対称の2bpの3’オーバーハングをもつ21bpデュープレックスである。
【0072】
【表1】
【0073】
B.DYRK3
哺乳動物DYRK3(REDK,hYAK3)は,Ser/Thr部位を標的としうるMAPK関連蛋白質キナーゼである。DYRK3は,コンセンサスキナーゼサブドメインVIIとVIIIとの間の保存YXYモチーフ(ないしループ)におけるチロシン(自己)リン酸化により活性化されることができる。DYRK3は赤血球リニアージの造血細胞で選択的に高レベルで発現されることができる(Geiger,JN et al.(2001)Blood 97:901−910;Lord,KA et al.(2000)Blood 95:2838−2846)。初代ネズミおよびヒト造血系前駆細胞においてDYRK3をアンチセンスオリゴヌクレオチドで阻害すると,コロニー形成単位−赤血球(赤芽球の最後から2番目の前駆細胞)の産生に影響を及ぼしうる。DYRK3活性は,その予想(自己)リン酸化ループ中のTyr333の存在に依存することができ,ループの酸性化は活性化でありうる(Li,K. et al.(2002)J Biol Chem 49,47052−47060)。DYRK3はキナーゼドメインならびにユニークなC末端ドメイン依存性が関与するメカニズムにより作用して,PKAに依存する経路によりCREBおよびCRE応答経路を制御することができる(Li,K.et al.(2002)上掲)。FDC造血系前駆細胞におけるDYRK3の発現はアポトーシスを制御しうる(Li,K.et al.(2002)上掲)。
【0074】
本発明の方法および組成物において用いることができるDYRK3の阻害剤としては,DYRK3/hYAK3のキノリン阻害剤(米国特許7,087,758),YAK3/DYRK3の阻害剤であるGSK626616AC,(http://clinicaltrials.gov/show/NCT00443170),3−カルボキシキノリン誘導体DYKR3/YAK3(Burgess et al.米国特許公開20060106058),およびQiagenから入手しうる3つのsiRNAオリゴ(DYRK3_p1,DYRK3_p2,およびDYRK3_p3)(表2)が挙げられる。これらのsiRNAは,OiagenによりRT−PCRを用いて評価され,約70%以上の標的遺伝子mRNAのノックダウンを与えることが示されている。これらのsiRNAは対称の2bpの3’オーバーハングをもつ21bpデュープレックスである。
【0075】
【表2】
【0076】
C.PTK9(TWF1)
ツインフィリン1は,短いリンカー領域により連結される2つのADF/コフィリン様(ADF−H)ドメインおよびそれに続く20残基のC末端テールから構成される。2つのADF−Hドメインは互いに約20%のホモロジーを有する(Lappalainen et al.,(1998)Mol.Biol.Cell 9:1951−1959.)。
【0077】
ヒトツインフィリンは,元々はチロシンキナーゼとして同定されたが(Beeler et al.,(1994)Mol.Cell.Biol.14:982−988),研究により,これはキナーゼ活性を持たず(Vartiainen et al.,(2000)Mol.Cell.Biol.20:1772−1783;Rohwer et al.,(1999)Eur.J.Biochem.263:518−525),ツインフィリンは既知の蛋白質キナーゼに対して配列ホモロジーを持たないことが示された。むしろ,ツインフィリンは,アクチンモノマーに結合することができる(Goode et al.,(1998)J.CellBiol.142:723−733;Vartiainen et al.,(2000)上掲;Wahlstrom et al.,(2001)J.CellBiol.155:787−796)。ツインフィリンの機能に関する情報は,部分的には,酵母およびDrosophilaのホモログの研究から得られた。ツインフィリンはアクチンモノマーと1:1の複合体を形成するようである。ツインフィリンはアクチンモノマーを有効に隔離することができる(Goode et al.,(1998)上掲;Vartiainen et al.,(2000)上掲;Wahlstrom et al.,(2001)上掲)。ツインフィリンは,ADP−アクチンモノマーと相互作用して,そのヌクレオチド交換およびフィラメントのアセンブリを阻害することができる(Palmgren et al.,(2001)J.CellBiol.155:251−260)。ツインフィリンは新たに脱重合したアセンブリ不能ADP−アクチンモノマーと相互作用するかもしれない。
【0078】
ツインフィリンは点状の細胞質染色パターンを有し,培養哺乳動物細胞においてアクチンモノマーおよびフィラメントを含む細胞性プロセスに局在することができる(Vartiainen et al.,(2000)上掲)。ツインフィリンとキャッピング蛋白質との間の直接相互作用は,ツインフィリンの急速なアクチンフィラメントアセンブリの部位への局在化を媒介することができる(Palmgren et al.,(2001)上掲)。
【0079】
本発明の方法および組成物において用いることができるTWF1/PTK9の阻害剤としては,shRNA,例えば,SigmaTRC(The RNAi Consortium)#:TRCN 0000011013;クローンID:NM_002822.3−907s1c1;受託番号:NM_198974.1,NM_002822.3;CCGGGCCTGGATACACATGCAGTATCTCGAGATACTGCATGTGTATCCAGGCTTTTT(配列番号30);Sigma TRC#TRCN 0000006364;クローンID:NM_002822.3−2014s1c1;受託番号:NM_198974.1,NM_002822.3;CCGGCCGAGCAAATACTCAGATTTACTCGAGTAAATCTGAGTATTTGCTCGGTTTTT(配列番号31);Sigma TRC#TRCN 0000006365;クローンID:NM_002822.3−364s1c1;受託番号:NM_198974.1,NM_002822.3;CCGGCCAGGGATATGAATGGATATTCTCGAGAATATCCATTCATATCCCTGGTTTTT(配列番号32);Sigma TRC#TRCN 0000006366;クローンID:NM_002822.3−474s1c1;受託番号:NM_198974.1,NM_002822.3;CCGGGCCACATTAAAGATGAAGTATCTCGAGATACTTCATCTTTAATGTGGCTTTTT(配列番号33);Sigma TRC#:TRCN 0000006367;クローンID:NM_002822.3−962s1c1;受託番号:NM_198974.1,NM_002822.3;CCGGCGTCTGCTAGAAATTGTAGAACTCGAGTTCTACAATTTCTAGCAGACGTTTTT(配列番号34)が挙げられる。
【0080】
また,本発明の方法および組成物において用いることができるPTK9/TWF1の阻害剤には,PTK9既設計キメラRNAi(Cat.#H00005756−R04,Abnova)およびPTK9評価済みStealth(商標)Duo Pak(Cat.#12938068,Invitrogen)が含まれる。
【0081】
D.GPRK2L(GRK4)
G−蛋白質共役レセプター(GPCR)キナーゼ(GRK)は,3つのファミリーに体系づけられるセリン/トレオニンキナーゼである(Penela et al.,(2003)Cell Signal 15:973−981)。1つのファミリーはGRK4ファミリーであり,これはGRK4,GRK5,およびGRK6から構成される。GRK4サブファミリーの特徴としては,以下のものが挙げられる:a)C末端システイン残基のパルミトイル化(GRK4/6について)または負に荷電した膜リン脂質とC末端近くの正に荷電したドメインとの相互作用(GRK5について)による膜局在化,b)ホスファチジルイノシトールビスホスフェート結合(N末端ドメインへの)による活性化,およびc)カルシウムセンサー蛋白質,例えばカルモジュリンによる阻害(Pronin et al.,(1997)J.Biol.Chem.272:18273−18280;Pitcher et al.,(1998)Annu.Rev.Biochem.67:653−692;Kohout and Lefkowitz,(2003)Mol Pharmacol.63:9−18;Willets et al.,(2003)Trends Pharmacol.Sci.24:626−633)。
【0082】
GRK4には4つのRNAスプライシング変種が同定されている:α,β,γおよびδ(Premont et al.,(1996)J.Biol.Chem.271:6403−6410)。GRK4αは全長型である。GRK4βは,エクソン2によりコードされる配列を欠失しており,このためN末端近くのホスファチジルイノシトールビスホスフェート結合ドメインを含む32アミノ酸が欠失している。GRK4γはエクソン15によりコードされる配列を欠失しており,このため,C末端近くの46アミノ酸が欠失している。最も短い型のGRK4δは,選択的にスプライシングされたエクソンの両方によりコードされる配列を欠失している。
【0083】
GRKはGCPR脱感作において役割を果たしている。GPCRはアゴニストによる活性化によって脱感作を生じうる;このプロセスにより,連続的アゴニスト刺激下でのレセプター応答が減少しうる(Ferguson et al.,(1996)Can.J.Physiol.Pharmacol.74:1095−1110;Gainetdinov et al.,(2004)Annu.Rev.Neurosci.27:107−144)。GRK媒介性リン酸化は,レセプター/G蛋白質相互作用を低下させ,アレスチン結合を開始させることができる。アレスチンとの会合はさらにG蛋白質カップリングを減少させ,レセプターのエンドサイトーシスを増強することができる。内在化されたGPCRは,さらに別のシグナリング経路に関与することができ,細胞膜へのリサイクル用に選別され,または分解の標的となる(Ferguson et al.,(1996)Can.J.Physiol.Pharmacol.74:1095−1110;Penela et al.,(2003)Cell Signal 15:973−981;Gainetdinov et al.,(2004)Annu.Rev.Neurosci.27:107−144)。
【0084】
GRK4はまた,GPCRのアゴニスト非依存性リン酸化を刺激することができる。例えば,GRK4をD1レセプターと共発現させると,アゴニストを加えてもわずかにしか増加しなかったレセプターがリン酸化される(Rankin et al.(2006)Mol.Pharmacol.69:759−769)。アゴニスト結合の非存在下におけるGRK4αによるD1レセプターのリン酸化により,アゴニスト誘導性cAMP蓄積の減少,基底レセプター内在化の増加,および全レセプターの数の減少が生じうる。
【0085】
本発明の方法および組成物において用いることができるGRK4の阻害剤としては,例えば,アンチセンス−オリゴヌクレオチド(As−Odn),209 5’−CATGAAGTTCTCCAGTTCCAT−3’ 189(配列番号19)(Sanada et al.(2006)Hypertension 47:1131−1139),カルモジュリン(Iacovelli et al.(1999)FASEB J.13:1−8),ヘパリン(GRK4αの阻害剤;Sallese et al.(1997)J.Biol.Chem.272:10188−10198),およびQiagenから入手しうる3つのsiRNAオリゴ(GPRK2L_p1,GPRK2L_p2,およびGPRK2L_p3)(表3)が挙げられる。これらのsiRNAは,OiagenによりRT−PCRを用いて評価され,約70%以上の標的遺伝子mRNAのノックダウンを与えることが示されている。これらのsiRNAは対称の2bpの3’オーバーハングをもつ21bpデュープレックスである。
【0086】
【表3】
【0087】
E.Rac1
Rac1は小さいシグナリングG蛋白質であり,GTPaseのRhoファミリーのメンバーである。Rac1はPAK1の標的である。本発明の方法および組成物において用いることができるRac1の阻害剤としては,例えば,Rac1阻害剤W56(MVDGKPVNLGLWDTAG;(配列番号44);Cat.No.2221;Tocris bioscience),Rac1阻害剤(Cat.No.553502;Calbiochem),Rac1阻害剤NSC23766,N6−[2−[[4−(ジエチルアミノ)−1−メチルブチル]アミノ]−6−メチル−4−ピリミジニル]−2−メチル−4,6−キノリンジアミン三塩酸塩(Cat.No.2161;Tocris bioscience)が挙げられる。
【0088】
F.Cdc42
Cdc42はRho−サブファミリーの小さいGTPaseであり,細胞形態,移動,エンドサイトーシスおよび細胞サイクル進行を管理するシグナリング経路を制御することができる。本発明の方法および組成物において用いることができるCdc42の阻害剤としては,例えば,セクラミンB(Pelish et al.(2006)Biochem.Pharmacol.71:1720−1726);セクラミンA(Xu et al.(2006)Org Biomol Chem4:4149−4157);およびACK42(Nur−E−Kamal et al.(1999)Oncogene 18:7787−7793)が挙げられる。
【0089】
VI.トランスジェニック細胞および非ヒト哺乳動物
トランスジェニック動物モデル,例えば組換えおよびノックアウト動物は,本明細書に記載される宿主核酸から生成することができる。トランスジェニック非ヒト哺乳動物の例としては,限定されないが,マウス,ラット,トリ,ウシおよびブタが挙げられる。ある例においては,トランスジェニック非ヒト哺乳動物はキナーゼに関連する標的配列の1またはそれ以上がノックアウトされており,ウイルス感受性,例えばインフルエンザまたはポックスウイルスによる感染の感受性が低下している。そのようなノックアウト動物は,ウイルス感染の段階の研究に,および動物からヒトへのウイルスの伝染の低下に有用である。さらに,本明細書に記載されるものと同じ標的を利用する動物ウイルスを動物において分析することができる。
【0090】
所望の遺伝子をノックアウトまたは機能的に欠失させるために用いられる配列の発現は,適切なプロモーター配列を選択することにより制御することができる。例えば,構成的プロモーターは機能的に欠失した遺伝子が動物により決して発現されないことを確実にするために用いることができる。これに対し,誘導可能なプロモーターは,トランスジェニック動物が目的とする遺伝子を発現するかまたは発現しない時を制御するために用いることができる。誘導可能なプロモーターの例には,組織特異的プロモーターおよび特定の刺激(例えば,光,酸素,化学物質濃度)に応答性または非応答性のプロモーターが含まれ,例えば,テトラサイクリン/ドキシサイクリン制御プロモーター(TET−off,TET−on),エクダイソン誘導性プロモーター,およびCre/loxPリコンビナーゼ系が挙げられる。
【0091】
1つの態様においては,ヒトキナーゼ遺伝子を持つかまたは内因性キナーゼ遺伝子が破壊されているトランスジェニックマウスを,インフルエンザまたはポックスウイルス等の種々の哺乳動物ウイルスに暴露した後に試験することができる。比較データは,そのウイルスおよび関連するウイルスのライフサイクルについての知見を提供することができる。さらに,さもなくば感染(例えばインフルエンザ)に対して感受性であるノックアウト動物(例えばブタ)を作製して,遺伝子の破壊によって付与される感染に対する耐性を判定することができる。
【0092】
別の態様においては,ヒトキナーゼ遺伝子を持つかまたは内因性キナーゼ遺伝子が破壊されているトランスジェニックブタを作製し,これを動物モデルとして用いて,インフルエンザまたはポックスウイルス感染等のウイルス感染に対する感受性を判定することができる。トランスジェニック動物,ならびにトランスジェニック動物を製造し使用する方法は,種々の特許および公開特許,例えば,WO01/43540;WO02/19811;米国特許公開2001−0044937および2002−0066117;および米国特許5,859,308;6,281,408;および6,376,743(これらは参照として本明細書に組み込まれる)に記載されている。
【0093】
VII.キナーゼ阻害剤の合理的設計
本発明の1つの観点は,蛋白質キナーゼ,例えば宿主細胞のウイルス感染に関与する蛋白質キナーゼを調節する薬剤に関する。ある態様においては,薬剤は,抗体,無機化合物,有機化合物,蛋白質/ペプチド薬剤または小分子,例えばsiRNAであることができる。好ましくは,薬剤は,PAK1,Cdc42,Rac1,DYRK3,PTK9,およびGPRK2Lを阻害することができる。ある態様においては,そのような薬剤は,インビトロでおよびインビボで抗ウイルス効果を発揮する。
【0094】
本発明のさらに別の観点は,宿主キナーゼを阻害する組成物を得るおよび/または作製する方法に関し,この方法は,阻害剤を設計し;その薬剤が宿主キナーゼを阻害するか否かを試験し;これを用いて宿主キナーゼを阻害する組成物を作製することを含む。ある態様においては,本発明は,キナーゼ調節剤であり,2以上の宿主キナーゼを阻害することができる薬剤を設計し,試験する方法に関する。
【0095】
本発明の合理的設計方法は,PAK1,Cdc42,Rac1,DYRK3,PTK9,およびGPRK2Lの構造についての現在の理解の助けを受けている。好ましくはキナーゼのX線構造を用いて,試験阻害剤のキナーゼへの結合を調べる。典型的には,候補物質の酵素への結合の「強さ」と,その薬剤のインビトロでの細胞活性との間には直接の相関がある。
【0096】
薬剤が無機または有機化合物である態様においては,前記化合物は,完全にコンピュータによる方法を用いて設計し試験することができ,または,そのような設計および試験の一部をコンピュータによって行い,残りをウエットラボ技術を用いて行うことができる。
【0097】
宿主細胞のウイルス感染に関与する蛋白質キナーゼを阻害するリード化合物は,種々の方法を用いて同定することができる。1つの態様においては,コンピュータを使用した“インシリコ”方法論により,標的宿主細胞キナーゼを阻害するようリード化合物を設計する。ケモゲノミクスのツール,例えばKinase Tool kit(商標)を用いて,バイオインフォマティクス,医薬化学およびコンピュータによる知識源の組み合わせを用いて,ATP部位特異的キナーゼ阻害剤を設計することができる。モデリングおよびディスプレイ手法を用いて,X線結晶構造の重ね合わせとサブポケット類似性分析により,この情報を増強することができる。既知のキナーゼ阻害剤の大部分はATP拮抗性であり,触媒ドメイン中の結合部位を標的とする。しかし,有用な阻害剤はまた,結合部位中の結合したATPによって占められていない領域を占めるものであってもよい。利用可能な結晶構造について蛋白質データバンクを調査すると,結合した阻害剤のまわりのATP部位の活性化されたまたは部分的に活性化されたコンフォメーションが,広くすべてのキナーゼについて同じであることが認められた(Birault 2006)。さらに,一致した限定された数の一次残基が小分子の結合に関与していることが明らかである。系統発生学的に異なるキナーゼ,例えば,p38αおよびFGF−1は,ATP結合部位において三次元構造上の共通性を示す。この構造的類似性は,特定の個々のキナーゼ,ならびにより大きな分類の後続的に関連するキナーゼを阻害する新規なキナーゼ阻害剤を同定するのに有用な基盤を提供する。
【0098】
別の態様においては,リード化合物は,コンピュータのフィルタを用いて,既知の化合物のデータベースからリード化合物を同定することにより発見される。これらのデータベースのいくつかは,数百万種類の化合物を含みうる。フィルタは,適当なADMET(吸収,分布,代謝,排泄,毒性)特性を組み込むよう設計される。これらの医薬化学的取り扱い用のフィルタは,所望の化学的特性のリストに基づく。ADMETモデリングを化合物の最適化の間に用いて,所望の特性を持つと思われる化合物を含む許容可能な特性空間を定義する。ある態様においては,2以上のコンピュータフィルタを適用して既知の化合物を分析する。適用しうるフィルタとしては,限定されないが,the Lipinski filter(5のルール),the Veber(2のルール)filter,Chem GPS,MDDR filter,Shoichet’s Aggregators,Martinfilter,Ghosefilter,Eganfilter,Med Chemtractibility filter,Leadlikeness,Caco−2 permeation filterおよびthe Muegge filterが挙げられる。これらのフィルタを,所望の特性,例えば,水溶性,分子量,SlogP,およびH結合ドナーまたはアクセプターの数を有する任意の化合物をスクリーニングするよう設定することができる。
【0099】
別の態様においては,特定のファミリーのキナーゼを阻害することが知られているかまたは予測される薬剤,例えば無機または有機化合物のライブラリを,ウイルス感染を調節する宿主細胞蛋白質を同定するために用いたものと同じ系を用いて,ウイルス感染を阻害する能力について試験する。ある態様においては,スクリーニングは,siRNAのライブラリを化合物のライブラリで置き換えて,同様にして行う。それぞれのウイルスについて化学物質のスクリーニングの結果をsiRNAスクリーニングの結果と比較して,ランク付けした化合物のリストを作成する。さらに別の態様においては,化合物スクリーニングにおいてウイルス感染を阻害する能力を示した上位のヒット化合物,例えば上位5,10,15,20または25種の化合物についてインビトロ酵素アッセイを行う。ここでは,上位の化合物が宿主細胞標的キナーゼ,またはキナーゼシグナリング経路の上流または下流のキナーゼを阻害する能力についてプロファイリングする。さらに別の態様においては,ウイルス感染の阻害において,および/または宿主細胞キナーゼ標的化の特異性において最も高い有効性を示すトップ化合物を,ウイルス感染の動物モデルを用いて毒性およびインビボ効力について試験する。
【0100】
ある態様においては,特定のキナーゼ,例えば,PAK1,DYRK3,PTK9,およびGPRK2L,またはキナーゼシグナリング経路においてPAK1,DYRK3,PTK9,およびGPRK2Lの上流または下流にあるキナーゼまたは他の物質を標的とする薬剤が同定され,または開発される。
【0101】
試験は,設計された薬剤を宿主細胞キナーゼに対する阻害活性について評価することを含む。ある態様においては,設計された薬剤の集合をコンピュータによる方法により評価して,物理的に薬剤を合成することなく,宿主細胞キナーゼを阻害するその活性を推定する。そのようなコンピュータによる方法は,薬剤の他の特性,例えば,溶解性,膜浸透性,代謝および毒性を推定するためにも用いることができる。
【0102】
ある態様においては,試験は,設計した薬剤を合成し,ウエットラボ手法の1またはそれ以上の生物学的アッセイにおいて,宿主細胞キナーゼを阻害するか,および/またはウイルス感染を阻害するその活性について評価することを含む。
【0103】
次に,合成された薬剤の活性を,宿主細胞キナーゼの阻害,および/またはウイルス感染の阻害を直接的にまたは間接的に反映する生物学的アッセイにより評価することができる。代表的な生物学的アッセイとしては,限定されないが,1)キナーゼ阻害の無細胞実験;2)ウイルス阻害の無細胞実験3)ウイルス感染の阻害の全細胞実験(例えば,ウイルスの伝染,侵入,複製,生合成,アセンブリ,または退出);および4)ウイルス感染に対する効力のインビボ動物モデル,例えば,特定のウイルスに感染したマウス,トリ,霊長類またはブタモデルが挙げられる。
【0104】
インビトロでアッセイに関しては,候補薬剤が宿主細胞キナーゼを阻害する能力は,薬剤を宿主細胞キナーゼの活性測定用のアッセイ混合物と接触させ,薬剤の存在下および非存在下における酵素の活性を測定することにより評価することができる。宿主細胞キナーゼの活性が薬剤の存在下で非存在下におけるより低いことは宿主細胞キナーゼ阻害剤であることを示す。
【0105】
無細胞宿主細胞キナーゼアッセイの一例は,Clerk and Sugden,FEBS Letters,426:93−96(1998)(本明細書に参照として取り込まれる)に記載されるものである。別の例示的システムは,キナーゼプロファイリング技術であるAMBITプラットフォーム(Kinomescan)である。プラットフォームを用いて,分子相互作用を同定し,連結されていない小分子の複数のキナーゼのATP部位への結合の定量的測定特異性を決定することができる。例えば,プラットフォームを用いて阻害剤を分析し,薬剤が他の「オフターゲット」キナーゼと比較していかに強くその目的とするキナーゼ標的に結合するかを明らかにする。この「オフターゲット」結合を用いて,阻害剤の副作用を同定することができ,またはある種の阻害剤を他のウイルスについて評価することを正当化することができる。
【0106】
ウイルス感染に対する応答を反映するために用いられる動物モデルを用いて,インビボでの宿主細胞キナーゼ阻害活性を評価することができる。動物モデルの例としては,限定されないが,マウス,ラット,フェレット,テンジクネズミ,ブタ(Susscrofa),ウマ,霊長類,およびウマが挙げられる。
【0107】
ある態様においては,薬剤の活性ないし効力は,全細胞および/またはIC50またはED50値のインビボアッセイにより測定して,複数の宿主キナーゼに対して同様であり,これは下記に詳細に説明する。ある態様においては,複数の宿主細胞キナーゼに対する単一の薬剤の効力の相違は,約1000倍以下である。さらに別の態様においては,効力の相違は約100倍以下である。さらに別の特定の態様においては,効力の相違は約10倍以下である。
【0108】
VIII.治療方法
本発明の1つの態様は,キナーゼを阻害してウイルス感染を阻害するかまたは低減させる薬剤を含む医薬組成物およびキットを用いる方法に関する。本発明の別の態様は,被検動物を治療するための方法,医薬組成物およびキットを提供する。本明細書において用いる場合,"被検動物"との用語は,ヒトならびに他の哺乳動物を含む。本明細書において用いる場合,"治療する"との用語は治療上の有益性および/または予防上の有益性を達成することを含む。治療上の有益性とは,原因となるウイルス感染の根絶または軽減を意味する。また,治療上の有益性は,被検動物がなお原因となるウイルスに罹患しているにもかかわらず被検動物において改善が観察されるように,原因となるウイルス感染に伴う生理学的症状の1またはそれ以上を根絶または軽減することによっても達成される。
【0109】
予防上の有益性が望ましい態様においては,本発明の医薬組成物を,ウイルス感染,例えば,インフルエンザ,またはHIV感染を発病するリスクを有する患者,またはまだ病気の診断がされていなくてもウイルス感染の生理学的症状の1またはそれ以上を訴える患者に投与することができる。投与によりウイルス感染が発症することを予防することができ,または,発症するウイルス感染を低下させ,減少させ,短期にし,および/またはさもなくば軽減することができる。医薬組成物は,標的キナーゼ活性を調節することができる。ここでは,調節するとの用語は,標的キナーゼの阻害,あるいは標的キナーゼの活性化を含む。
【0110】
蛋白質キナーゼの活性を低下させることはまた,キナーゼを"阻害する"とも表される。"阻害する"との用語,およびその文法的活用形,例えば"阻害的"とは,完全な阻害は必要ではないが,キナーゼ活性の低下を表す。ある態様においては,そのような低下は,阻害効果がない場合,例えば阻害剤の非存在下における酵素の活性の少なくとも50%,少なくとも75%,少なくとも90%,および少なくとも95%であることができる。逆に,"阻害しない"との語句およびその文法的活用形は,薬剤の存在下で酵素活性の20%未満,10%未満,および5%未満の低下がある状況を表す。さらに,"実質的に阻害しない"との語句およびその文法的活用形は,薬剤の存在下で酵素活性の30%未満,20%未満,およびある態様においては10%未満の低下がある状況を表す。
【0111】
蛋白質キナーゼの活性を増加させることは,キナーゼを"活性化する"とも称される。"活性化される"との用語,およびその文法的活用形,例えば,"活性化する"とは,完全に活性化される必要はないが,キナーゼ活性の増加を表す。ある態様においては,そのような増加は,活性化効果がない場合に,例えば,アクチベータの非存在下で,酵素の活性の少なくとも50%,少なくとも75%,少なくとも90%,および95%であることができる。逆に,"活性化しない"との語句およびその文法的活用形は,薬剤の存在下において酵素活性の20%未満,10%未満,および5%未満の増加がある状況を表す。さらに,"実質的に活性化しない"との語句およびその文法的活用形は,薬剤の存在下において酵素活性の30%未満,20%未満,およびある態様においては10%未満の増加がある状況を表す。
【0112】
酵素活性を低下させる能力は,薬剤または薬剤の組み合わせの酵素に向けたまたはこれに対する効力または活性の尺度である。効力は,無細胞,全細胞および/またはインビボアッセイにより,IC50,Kiおよび/またはED50値として測定することができる。IC50値は,所定の条件下で酵素活性を半分(50%)阻害するのに必要な薬剤の濃度を表す。Ki価は阻害剤の酵素への結合の平衡アフィニティー定数を表す。ED50値は,生物学的アッセイにおいて最大の半分の応答を引き起こすのに必要な薬剤の用量を表す。当業者はこれらの尺度の詳細を理解することができ,生化学,酵素学等の標準的な教科書に見いだすことができる。
【0113】
本発明はまた,ウイルス感染を治療するために用いることができるキットを含む。これらのキットは,キナーゼを阻害する薬剤または薬剤の組み合わせを含み,ある態様においては,本明細書に記載される種々の方法およびアプローチにしたがってキットを使用する方法を教示する指示を含む。そのようなキットはまた,薬剤の活性および/または利点を示す情報,例えば,科学参考文献,パッケージ挿入材料,臨床的結果,および/またはこれらの概要を含んでいてもよい。そのような情報は,種々の実験,例えば,インビボモデルを含む実験動物を用いた実験およびヒトの臨床試験に基づく実験の結果に基づくものでありうる。本明細書に記載されるキットは,健康管理者,例えば,医師,看護師,薬剤師,処方薬剤職員等に提供し,販売し,および/または販売促進することができる。
【0114】
A.siRNA療法
二本鎖オリゴヌクレオチドは,一方の鎖のオリゴヌクレオチド配列が第2の鎖のオリゴヌクレオチド配列に相補的である2つの別々のオリゴヌクレオチド配列のアセンブリによって形成される。そのような二本鎖オリゴヌクレオチドは一般に,2つの別々のオリゴヌクレオチド(例えばsiRNA)から,またはそれ自体でフォールディングして二本鎖構造を形成する単一の分子(例えば,shRNAないし短ヘアピンRNA)から組み立てられる。当該技術分野において知られるこれらの二本鎖オリゴヌクレオチドはすべて,デュープレックスの各鎖は異なるヌクレオチド配列を有し,一方のヌクレオチド配列領域(ガイド配列またはアンチセンス配列)のみが標的核酸配列に相補的であり,他方の鎖(センス配列)は標的核酸配列に相同のヌクレオチド配列を含むという,共通の特徴を有する。
【0115】
二本鎖RNAにより誘導される遺伝子サイレンシングは,少なくとも3つの異なるレベルで生じうる:(i)転写不活性化,すなわちRNAによりガイドされるDNAまたはヒストンメチル化;(ii)siRNAにより誘導されるmRNA分解;および(iii)mRNAにより誘導される転写減衰。RNA哺乳動物細胞における誘導性サイレンシング(RNA干渉ないしRNAi)の主要なメカニズムはmRNA分解であると一般に考えられている。RNA干渉(RNAi)は,遺伝子発現を翻訳の段階で阻害するか,または特定の遺伝子の転写を立体的に妨害するメカニズムである。特定のRNAi経路蛋白質は,dsRNAにより誘導されて標的メッセンジャーRNA(mRNA)に向かい,ここで標的を"切断"する,すなわち,これをもはや蛋白質に翻訳されない小さい部分に分解する。RNAiを哺乳動物細胞において用いる最初の試みは,dsRNAの長鎖の使用に焦点が当てられていた。しかし,RNAiを誘導しようとするこれらの試みはほとんど成功しなかった。これは,部分的には,インターフェロン応答が誘導され,標的特異的ではなく一般的な蛋白質合成の阻害が生じたためである。すなわち,長いdsRNAは哺乳動物系においては実行可能なRNAiの選択肢ではない。別の結果は,遺伝子が転写される程度に影響を及ぼす遺伝子のエピジェネティックな変化,すなわち,ヒストン修飾およびDNAメチル化の変化である。
【0116】
最近,短い(18−30bp)RNAデュープレックスを哺乳動物培養細胞に導入すると,インターフェロン応答を誘導することなく標的mRNAの配列特異的阻害が実現可能であることが示された。これらの短いdsRNAのある種のものは,小阻害RNA("siRNA")と称され,モル濃度以下で触媒的に作用して,細胞中で標的mRNAの95%以上を切断することができる。siRNA活性のメカニズムの説明,ならびにその用途のいくつかは,下記に記載されている:Provost et al.,Ribonuclease Activity and RNA Binding of Recombinant Human Dicer,E.M.B.O.J.,2002 Nov.1;21(21):5864−5874;Tabara et al.,The dsRNA Binding Protein RDE−4 Interacts with RDE−1,DCR−1 and a DexH−box Helicaseto Direct RNAi in C.elegans,Cell 2002,June 28;109(7):861−71;Ketting et al.,Dicer Functions in RNA Interference and in Synthesis of Small RNA Involved in Developmental Timing in C.elegans;Martinez et al.,Single−Stranded Antisense siRNA Guide Target RNA Cleavage in RNAi,Cell 2002,September.6;110(5):563;Hutvagner&Zamore,Amicro RNA in a multiple−turnover RNAi enzyme complex,Science 2002,297:2056。
【0117】
機構的な観点からは,長い二本鎖RNAを植物および無脊椎動物細胞に導入すると,Dicerとして知られるIII型エンドヌクレアーゼにより分解されてsiRNAとなる(Sharp,RNAinterference−−2001,Genes Dev.2001,15:485.Dicer,a ribonuclease−III−like enzyme,processes the dsRNA into 19−23 base pair short interfering RNAs with characteristic two base 3’ overhangs;Bernstein,Caudy,Hammond,&Hannon,Role for a bidentate ribonuclease in the initiation step of RNA interference,Nature 2001,409:363)。次にsiRNAはRNA誘導性サイレンシング複合体(RISC)中に取り込まれ,ここで1またはそれ以上のヘリカーゼがsiRNAデュープレックスをほどき,相補的なアンチセンス鎖が標的認識をガイドできるようにする(Nykanen,Haley,&Zamore,ATP requirements and small interfering RNA structure in the RNA interference pathway,Cell 2001,107:309)。適当な標的mRNAに結合すると,RISC中の1またはそれ以上のエンドヌクレアーゼは標的を切断して,サイレンシングを誘導する(Elbashir,Lendeckel,&Tuschl,RNA interference is mediated by 21−and 22−nucleotide RNAs,Genes Dev 2001,15:188,FIG.1.)。
【0118】
一般に,アンチセンス配列は活性なRISC複合体中に保持され,配列特異的RNA干渉を媒介するためにアンチセンス配列と標的配列との相補的塩基対形成によりRISCを標的ヌクレオチド配列へとガイドする。当該技術分野においては,細胞培養系によってはある種の未修飾siRNAが"オフターゲット"効果を示す場合があることが知られている。このオフターゲット効果にはRISC複合体中のsiRNAのアンチセンス配列ではなくセンス配列が関与しているとの仮説がある(例えば,Schwarz et al.,2003,Cell,115,199−208を参照)。この場合,センス配列はRISC複合体を意図した標的配列とは異なる配列(オフターゲット配列)に向かわせ,その結果,オフターゲット配列が阻害されると考えられている。これらの二本鎖核酸分子においては,各鎖は異なる標的核酸配列に相補的である。しかし,これらのdsRNAにより影響を受けるオフターゲットは完全に予測可能なわけではなく,非特異的である。
【0119】
"siRNA"との用語は,RNA干渉(RNAi)経路を誘導する小さい阻害性RNAデュープレックスを表す。これらの分子は,種々の長さであることができ(一般に18−30塩基対),アンチセンス鎖中の標的mRNAに対して種々の程度の相補性を含む。すべてではないがある種のsiRNAは,センス鎖および/またはアンチセンス鎖の5’または3’末端に塩基対形成しないオーバーハング塩基を有する。"siRNA"との用語は,2つの別々の鎖のデュープレックス,ならびにデュープレックス領域を含むヘアピン構造を形成しうる一本鎖を含む。小干渉RNA(siRNA)は,短干渉RNAまたはサイレンシングRNAとしても知られており,生物学において種々の役割を果たす20−25ヌクレオチドの長さの一群の二本鎖RNA分子である。
【0120】
2つのRNA鎖は完全に相補的である必要はないが,鎖はハイブリダイズしてデュープレックス構造を形成するのに十分に相補的でなければならない。ある場合には,相補的RNA鎖は,30ヌクレオチド未満,好ましくは25ヌクレオチド未満の長さであり,より好ましくは19−24ヌクレオチドの長さ,より好ましくは20−23ヌクレオチドの長さ,さらにより好ましくは22ヌクレオチドの長さである。本発明のdsRNAはさらに,少なくとも1つの一本鎖ヌクレオチドオーバーハングを含んでいてもよい。本発明のdsRNAはさらに,置換されるかまたは化学的に修飾されたヌクレオチドを含んでいてもよい。下記に詳細に説明するように,dsRNAは当該技術分野において知られる標準的な方法により合成することができる。
【0121】
siRNAは,これらが培養細胞株において誘導するサイレンシングのレベルまたは程度に基づいて,5つの群に分類することができる(非機能的,準機能的,機能的,高機能的,および超機能的)。本明細書において用いる場合,これらの定義は,siRNAが100nMの濃度で前記細胞株にトランスフェクトされ,トランスフェクションのおよそ24時間後であって,トランスフェクション後72時間を越えない時点でサイレンシングのレベルが試験される一連の状態に基づく。この文脈において,"非機能的siRNA"とは,50%未満(<50%)の標的サイレンシングを誘導するsiRNAとして定義される。"準機能的siRNA"は50−79%の標的サイレンシングを誘導する。"機能的siRNA"は80−95%の遺伝子サイレンシングを誘導する分子である。"高機能的siRNA"は,95%より高い遺伝子サイレンシングを誘導する分子である。"超機能的siRNA"は,特定の群の分子である。この書面の目的のために,超機能的siRNAは,以下の分子として定義される:(1)ナノモル以下の濃度(例えば,1ナノモル未満)でトランスフェクションしたときに特定の標的の95%より高いサイレンシングを誘導する;および/または(2)96時間を越えて機能的(またはより高い)レベルのサイレンシングを誘導する。これらの相対的機能性(絶対的であることを意図するものではないが)を用いて,機能ゲノミクス,標的同定および治療などの用途のために,siRNAを特定の標的と比較することができる。
【0122】
A.マイクロRNA療法
マイクロRNA(miRNA)は約21−23ヌクレオチドの長さの一本鎖RNA分子であり,遺伝子発現を制御する。miRNAは,遺伝子によりコードされ,DNAから転写されるが,蛋白質には翻訳されない(非コーディングRNA)。その代わりに,これはpri−miRNAとして知られる一次転写産物からプロセシングされて,pre−miRNAと称される短いステム−ループ構造となり,最後に機能的miRNAとなる。成熟miRNA分子は,1またはそれ以上のメッセンジャーRNA(mRNA)分子に部分的に相補的であり,その主な機能は遺伝子発現を下方制御することである。
【0123】
IX.製剤,投与の経路,および有効量
本発明のさらに別の観点は,本発明の薬剤またはその組み合わせを含む医薬組成物の製剤,投与経路および有効投与量に関する。そのような医薬組成物は,上述のように,ウイルス感染の治療に用いることができる。
【0124】
薬剤またはその薬学的に許容しうる塩は,単独で,または1またはそれ以上の他の薬剤との組み合わせで,または1またはそれ以上の他の形態との組み合わせで提供することができる。例えば,製剤は,各薬剤の相対的効力および意図される適応症に応じて,1またはそれ以上の薬剤を特定の比率で含んでいてもよい。例えば,2つの異なる宿主標的を標的とする組成物において,効力が類似する場合,約1:1比の薬剤を用いることができる。2つの形態は,同じ単位投与形態中で,例えば1つのクリーム,座剤,錠剤,カプセル,エアロゾルスプレイ,または飲料に溶解すべき粉体包中に一緒に製剤することができる。あるいは,それぞれの形態を,例えば2つのクリーム,2つの座剤,2つの錠剤,2つのカプセル,錠剤と錠剤を溶解するための液体,2つのエアロゾルスプレイ,または粉体包と粉体を溶解するための液体等の,別々の単位として製剤してもよい。
【0125】
“薬学的に許容しうる塩”との用語は,本発明において用いられる薬剤の生物学的有効性および特性を維持し,生物学的にまたは他の点で望ましくないものではない塩を意味する。例えば,薬学的に許容しうる塩は,本発明の薬剤がキナーゼ,例えば,PAK1,DYRK3,PTK9,およびGPRK2Lからなる群より選択されるキナーゼを阻害する有益な効果を妨害しない。
【0126】
典型的な塩は,無機イオン,例えば,ナトリウム,カリウム,カルシウム,マグネシウムイオン等の塩である。そのような塩には,無機または有機酸,例えば,塩酸,臭化水素酸,リン酸,硝酸,硫酸,メタンスルホン酸,p−トルエンスルホン酸,酢酸,フマル酸,コハク酸,乳酸,マンデル酸,リンゴ酸,クエン酸,酒石酸またはマレイン酸との塩が含まれる。さらに,薬剤がカルボキシ基または他の酸性の基を持つ場合,これを無機または有機塩基との薬学的に許容しうる付加塩に変換することができる。適当な塩基の例としては,水酸化ナトリウム,水酸化カリウム,アンモニア,シクロヘキシルアミン,ジシクロヘキシルアミン,エタノールアミン,ジエタノールアミン,トリエタノールアミン等が挙げられる。
【0127】
薬学的に許容しうるエステルまたはアミドとは,本発明において用いられる薬剤の生物学的有効性および特性を維持し,生物学的にまたは他の点で望ましくないものではないエステルまたはアミドを表す。例えば,エステルまたはアミドは,本発明の薬剤がキナーゼ,例えば,PAK1,DYRK3,PTK9,およびGPRK2Lからなる群より選択されるキナーゼを阻害する有益な効果を妨害しない。典型的なエステルとしては,エチル,メチル,イソブチル,エチレングリコール等が挙げられる。典型的なアミドとしては,未置換アミド,アルキルアミド,ジアルキルアミド等が挙げられる。
【0128】
ある態様においては,薬剤は,1またはそれ以上の他の化合物,形態,および/または薬剤,例えば上述したものと組み合わせて投与することができる。キナーゼ阻害剤と1またはそれ以上の他の活性薬剤との組み合わせを含む医薬組成物は,ある一定のモル比を含むよう製剤することができる。例えば,約99:1から約1:99のモル比のキナーゼ阻害剤:他の活性薬剤を用いることができる。この態様の一部においては,キナーゼ阻害剤:他の活性薬剤のモル比の範囲は,約80:20−約20:80;約75:25−約25:75,約70:30−約30:70,約66:33−約33:66,約60:40−約40:60;約50:50;および約90:10−約10:90から選択される。キナーゼ阻害剤:他の活性薬剤のモル比は,約1:9であってもよく,ある態様においては約1:1であってもよい。2つの薬剤,形態および/または化合物は,同じ投与単位として,例えば,1つのクリーム,座剤,錠剤,カプセル,または飲料に溶解すべき粉体パケット中で一緒に製剤してもよく,またはそれぞれの薬剤,形態,および/または化合物を別々の単位として,例えば,2つのクリーム,座剤,錠剤,2つのカプセル,錠剤と錠剤を溶解するための液体,エアロゾルスプレイ,粉体のパックおよび粉体を溶解するための液体等で製剤してもよい。
【0129】
必要であるかまたは望ましい場合には,薬剤および/または薬剤の組み合わせは,さらに別の薬剤とともに投与してもよい。本発明の薬剤および/または薬剤の組み合わせとともに投与することができる薬剤の選択は,少なくとも部分的には,治療すべき状態によって異なる。本発明の製剤において有用な特定の薬剤としては,例えば,ウイルス感染に治療効果を有する任意の薬剤,例えば,炎症性状態を治療するために用いられる薬剤が挙げられる。例えば,インフルエンザの治療用には,ある態様においては,本発明の組成物はさらに,1またはそれ以上のNSAID等の抗炎症剤,例えば,イブプロフェン,ナプロキセン,アセトミノフェン,ケトプロフェン,またはアスピリンを含んでいてもよい。インフルエンザの治療用の別の態様においては,本発明の製剤はさらに,1またはそれ以上の慣用の抗インフルエンザウイルス薬,例えば,アマンタジン,リマンタジン,ザナミビル,およびオセルタミビルを含んでいてもよ。HIV等のレトロウイルス感染の治療においては,本発明の製剤はさらに,1またはそれ以上の慣用の抗ウイルス剤,例えば,プロテアーゼ阻害剤(ロピナビル/リトナビル{Kaletra},インジナビル{Crixivan},リトナビル{Norvir},ネルフィナビル{Viracept},サキナビルハードゲルカプセル{Invirase},アタザナビル{Reyataz},アンプレナビル{Agenerase},フォサンプレナビル{Telzir},チプラナビル{Aptivus}),リバーストランスクリプターゼ阻害剤,例えば,非ヌクレオシドおよびヌクレオシド/ヌクレオチド阻害剤(AZT{ジドブジン,Retrovir},ddI{ジダノシン,Videx},3TC{ラミブジン,Epivir},d4T{スタブジン,Zerit},アバカビル{Ziagen},FTC{エントリシタビン,Emtriva},テノフォビル{Viread},エファビレンツ{Sustiva}およびネビラピン{Viramune}),融合阻害剤T20{エンフビルチド,Fuzeon},インテグラーゼ阻害剤(MK−0518およびGS−9137),および成熟阻害剤(PA−457{Bevirimat})を含んでいてもよい。別の例としては,製剤はさらに1またはそれ以上のサプリメント,例えばビタミンC,Eまたは他の抗酸化剤を含んでいてもよい。
【0130】
薬剤(またはその薬学的に許容しうる塩,エステルまたはアミド)は,それ自体で投与してもよく,活性薬剤が1またはそれ以上の薬学的に許容しうる担体との混合剤ないし混合物である医薬組成物の形で投与してもよい。
【0131】
本明細書において用いる場合,医薬組成物とは,被験者に投与すべく調製された任意の組成物でありうる。本発明にしたがって使用するための医薬組成物は,1またはそれ以上の生理学的に許容しうる担体を用いて,例えば,賦形剤,希釈剤,および/または,例えば活性な薬剤を投与可能な調製物に加工することを容易にする助剤を含むよう,慣用の手法で製剤することができる。適当な製剤は,少なくとも部分的には,選択される投与経路によって異なるであろう。本発明において有用な薬剤,またはその薬学的に許容しうる塩,エステルまたはアミドは,投与の多くの経路ないしモードを用いて,例えば,経口,頬,局所,経直腸,経皮,経粘膜,皮下,静脈内,および筋肉内適用,ならびに吸入により,患者にデリバリーすることができる。
【0132】
経口投与用には,薬剤は,活性薬剤を当該技術分野においてよく知られる薬学的に許容しうる担体と混合することにより容易に製剤することができる。そのような担体は,本発明の薬剤を,錠剤として,例えば,チュアブル錠,丸薬,糖衣錠,カプセル,トローチ,硬質キャンディー,液体,ゲル,シロップ,スラリー,粉体,懸濁液,エリキシル,ウエハス等として,治療すべき患者が経口で摂取できるように製剤することを可能とする。そのような製剤は,薬学的に許容しうる担体,例えば,固形希釈剤または増量剤,滅菌水性媒体および種々の無毒性有機溶媒を含むことができる。一般に,本発明の薬剤は,経口投与用形態の全組成の約0.5%,約5%,約10%,約20%,または約30%から約50%,約60%,約70%,約80%または約90重量%の範囲の濃度レベルで,所望の単位投与形態を与えるのに十分な量で含まれる。
【0133】
経口で使用するための水性懸濁液は,本発明の薬剤を,薬学的に許容しうる賦形剤,例えば,懸濁剤(例えば,メチルセルロース),湿潤剤(例えば,レシチン,リソレシチンおよび/または長鎖脂肪族アルコール),ならびに着色剤,保存剤,甘味料等とともに含むことができる。
【0134】
ある態様においては,例えば大きな親油性成分の存在のため,薬剤を溶液とするために油または非水性溶媒が必要であるかもしれない。あるいは,乳濁液,懸濁液,または他の調製物,例えば,リポソーム調製物を用いてもよい。リポソーム製剤に関しては,病気の治療用のリポソームを製造するための既知の任意の方法を用いることができる。例えば,Bangham et al.,J.Mol.Biol.23:238−252(1965)およびSzoka et al.,Proc.Natl Acad.Sci.USA 75:4194−4198(1978)(本明細書に参照として取り込まれる)を参照。リポソームにリガンドを結合させて,これらの組成物が特定の作用部位に向かうようにしてもよい。本発明の薬剤はまた食品,例えばクリームチーズ,バター,サラダドレッシング,またはアイスクリーム中に組み込んで,可溶化,投与,および/またはある患者集団におけるコンプライアンスを容易にすることができる。
【0135】
経口使用用の医薬製剤は,固体賦形剤として得ることができ,得られた混合物を任意にすりつぶし,顆粒の混合物を加工し,所望の場合には適当な助剤を加えた後,錠剤または糖衣錠コアを得ることができる。適当な賦形剤は,特に,糖等の増量剤,例えばラクトース,ショ糖,マンニトール,またはソルビトール;香味要素,セルロース調製物,例えば,トウモロコシデンプン,小麦デンプン,米デンプン,ジャガイモデンプン,ゼラチン,トラガガントガム,メチルセルロース,ヒドロキシプロピルメチルセルロース,カルボキシメチルセルロースナトリウム,および/またはポリビニルピロリドン(PVP)である。所望の場合には,崩壊剤,例えば,架橋ポリビニルピロリドン,寒天,またはアルギン酸またはその塩,例えばアルギン酸ナトリウムを加えてもよい。薬剤はまた,持続放出製剤として製剤してもよい。
【0136】
糖衣錠のコアは,適当なコーティングとともに提供することができる。この目的のために,濃縮糖溶液を用いることができ,これは任意にアラビアゴム,タルク,ポリビニルピロリドン,カルボポールゲル,ポリエチレングリコール,および/または二酸化チタン,ラッカー溶液,および適当な有機溶媒または溶媒混合物を含むことができる。活性薬剤の異なる組み合わせの識別または特徴付けのために,染料または顔料を錠剤または糖衣錠のコーティングに加えてもよい。
【0137】
経口で使用することができる医薬製剤としては,ゼラチンから形成されるプッシュフィットカプセル,ならびにゼラチンと可塑剤,例えばグリセロールまたはソルビトールから形成される柔らかい密封カプセルが挙げられる。プッシュフィットカプセルは,活性成分を,ラクトース等の増量剤,スターチ等の結合剤,および/またはタルクまたはステアリン酸マグネシウム等の潤滑剤,および任意に,安定剤等との混合物として含むことができる。軟カプセルにおいては,活性薬剤は適当な液体,例えば油脂,液体パラフィン,または液体ポリエチレングリコールに溶解または懸濁することができる。さらに,安定剤を加えてもよい。経口投与用のすべての製剤は,投与に適した用量でなければならない。
【0138】
注射用には,本発明の薬剤を水性溶液,例えば,限定されないが,生理学的に適合性のバッファ,例えば,ハンクス溶液,リンゲル,または生理学的食塩水バッファ中で製剤することができる。そのような組成物はまた,1またはそれ以上の賦形剤,例えば,保存剤,溶解剤,増量剤,潤滑剤,安定剤,アルブミン等を含むことができる。製剤の方法は当該技術分野において知られており,例えば,Remington’s Pharmaceutical Sciences,最新版,Mack Publishing Co.,Easton Pに開示されている。
【0139】
上述した製剤に加えて,薬剤はまた,デポ調製物として製剤してもよい。そのような長期作用性製剤は,移植または経皮デリバリー(例えば皮下または筋肉内),筋肉内注射または経皮パッチの使用により投与することができる。すなわち,例えば,薬剤は,適当なポリマー性または疎水性材料(例えば,許容しうる油中の乳濁液として)またはイオン交換樹脂とともに,または溶けにくい誘導体として,例えば溶けにくい塩として製剤することができる。
【0140】
ある態様においては,本発明の1またはそれ以上の薬剤を含む医薬組成物は,局所的に投与したとき,または感染の特定の部位またはその近くに注射したとき,局所的および領域的効果を発揮する。例えば,粘稠液体,ゲル,ゼリー,クリーム,ローション,軟膏,座剤,泡状物,またはエアロゾルスプレイの直接局所投与を局所投与用に用いて,例えば,局所的および/または領域的効果を生じさせてもよい。そのような製剤用の薬学的に適したベヒクルとしては,例えば,低級脂肪族アルコール,ポリグリコール(例えば,グリセロールまたはポリエチレングリコール),脂肪酸のエステル,油,脂肪,シリコーン等が挙げられる。そのような製剤はまた,保存剤(例えば,p−ヒドロキシ安息香酸エステル)および/または抗酸化剤(例えば,アスコルビン酸およびトコフェロール)を含んでいてもよい。Dermatological Formulations:Percutaneous absorption,Barry(Ed.),Marcel Dekker Incl,1983も参照。ある態様においては,キナーゼ阻害剤を含む局部/局所製剤を用いて,表皮または粘膜のウイルス感染を治療する。
【0141】
本発明の医薬組成物は,化粧品用のまたは皮膚科学的に許容しうる担体を含んでいてもよ。そのような担体は,皮膚,爪,粘膜,組織および/または頭髪と適合性であり,これらの要件を満たす慣用的に用いられている任意の化粧品用のまたは皮膚科学的担体を含むことができる。そのような担体は,当業者が容易に選択することができる。皮膚用軟膏の製剤に際しては,本発明の薬剤または薬剤の組み合わせを油性炭化水素基剤,無水吸収基剤,油中水吸収基剤,水中油の水除去可能基剤および/または水溶性基剤中で製剤することができる。
【0142】
本発明にしたがう組成物は,局所適用に適した任意の形態,例えば,水性,水性−アルコール性または油性溶液,ローションまたは血清分散物,水性,無水または油性ゲル,油脂相を水性相中に分散させることにより得られる乳濁液(O/Wないし水中油)またはその逆(W/Oないし油中水),マイクロ乳濁液,あるいはマイクロカプセル,マイクロ粒子または脂質ベシクルのイオン性および/または非イオン性分散物であることができる。これらの組成物は,一般的な方法にしたがって製造することができる。本発明の薬剤の他,本発明にしたがう組成物の種々の構成成分の量は,当該技術分野において慣用的に用いられている量である。特に,これらの組成物は,保護,治療またはケア用の,または皮膚洗浄用の顔面,手,身体および/または粘膜用のクリーム,ミルク,ローション,ゲルまたは泡状物を構成する。組成物はまた,石けんまたはクレジングバーを構成する固体調製物からなるものであってもよい。
【0143】
本発明の組成物はまた,化粧品および皮膚科学の分野で一般的な助剤,例えば,親水性または親油性ゲル化剤,親水性または親油性の活性薬剤,保存剤,抗酸化剤,溶媒,香料,増量剤,サンスクリーン,臭い吸収剤および染料を含んでいてもよい。これらの種々の助剤の量は,該当する分野において一般的に用いられている量であり,例えば,組成物の総重量の約0.01%−約20%である。これらの助剤は,その性質によって,油脂相に,水性相に,および/または脂質ベシクル中に導入することができる。
【0144】
ある態様においては,眼のウイルス感染は,本発明の薬剤または薬剤の組み合わせを含む眼科溶液,懸濁液,軟膏または挿入物を用いて有効に治療することができる。
【0145】
ある態様においては,耳のウイルス感染は,本発明の薬剤または薬剤の組み合わせを含む耳用溶液,懸濁液,軟膏または挿入物を用いて有効に治療することができる。
【0146】
ある態様においては,本発明の薬剤は,懸濁液の形ではなく,可溶化形でデリバリーし,このことにより,作用部位へのより速く定量的な吸収が可能となる。一般に,ゼリー,クリーム,ローション,座剤および軟膏等本発明の薬剤により長く暴露される領域を提供することができるが,一方,溶液中の製剤,例えば,スプレイは,より迅速な短時間の暴露を提供する。
【0147】
局部/局所適用に関連するある態様においては,医薬組成物は,1またはそれ以上の浸透促進剤を含むことができる。例えば,製剤は,本発明の薬剤または薬剤の組み合わせが透過バリア,例えば皮膚を越える浸透を増加させるかまたはそのデリバリーを助ける適当な個体またはゲル相の担体または賦形剤を含んでいてもよい。これらの浸透促進化合物の多くは局所用製剤の分野で知られており,例えば,水,アルコール類(例えば,メタノール,エタノール,2−プロパノール等のテルペン),スルホキシド類(例えば,ジメチルスルホキシド,デシルメチルスルホキシド,テトラデシルメチルスルホキシド),ピロリドン類(例えば,2−ピロリドン,N−メチル−2−ピロリドン,N−(2−ヒドロキシエチル)ピロリドン),ラウロカプラム,アセトン,ジメチルアセトアミド,ジメチルホルムアミド,テトラヒドロフルフリルアルコール,L−α−アミノ酸,アニオン性,カチオン性,両イオン性または非イオン性界面活性剤(例えば,ミリスチン酸イソプロピルおよびラウリル硫酸ナトリウム),脂肪酸,脂肪族アルコール(例えばオレイン酸),アミン,アミド,クロフィブリン酸アミド,ヘキサメチレンラウラミド,蛋白質分解酵素,α−ビスアボロール,d−リモネン,尿素およびN,N−ジエチル−m−トルアミド等が含まれる。さらに別の例としては,保湿剤(例えば尿素),グリコール(例えば,プロピレングリコールおよびポリエチレングリコール),グリセロールモノラウレート,アルカン,アルカノール,ORGELASE,炭酸カルシウム,リン酸カルシウム,種々の糖類,デンプン,セルロース誘導体,ゼラチン,および/または他のポリマーが挙げられる。ある態様においては,医薬組成物は,1またはそれ以上のそのような浸透促進剤を含むことができる。
【0148】
ある態様においては,局部/局所に適用するための医薬組成物は,1またはそれ以上の抗微生物性保存剤,例えば,四級アンモニウム化合物,有機水銀剤,p−ヒドロキシ安息香酸,芳香族アルコール,クロロブタノール等を含むことができる。
【0149】
胃腸のウイルス感染は,経口または経直腸でデリバリーされる,本発明の薬剤または薬剤の組み合わせを含む溶液,懸濁液,軟膏,浣腸剤および/または座剤を用いて有効に治療することができる。
【0150】
呼吸器ウイルス感染は,本発明の薬剤または薬剤の組み合わせを含むエアロゾル溶液,懸濁液または乾燥粉体を用いて有効に治療することができる。吸入による投与は,肺のウイルス感染,例えばインフルエンザの治療に特に有用である。エアロゾルは,呼吸器系または鼻腔経路を通して投与することができる。例えば,当業者は,本発明の組成物を適当な担体,例えば,薬学的に許容しうる噴射剤に懸濁するかまたは溶解し,経鼻スプレイまたは吸入を用いて肺に直接投与しうることを理解するであろう。例えば,キナーゼ阻害剤を含むエアロゾル製剤は,例えば鼻スプレイまたは吸入剤として投与するための噴射剤または溶媒と噴射剤との混合物に溶解し,懸濁し,または乳化することができる。エアロゾル製剤は,当該技術分野において一般に用いられているように,任意の許容可能な噴射剤,例えば化粧品用のまたは皮膚科学的にまたは薬学的に許容しうる噴射剤を加圧下に含むことができる。
【0151】
鼻腔投与用のエアロゾル製剤は,一般に液滴またはスプレイの形で鼻腔に投与するよう設計された水性溶液である。鼻腔溶液は一般に等張であり約5.5から約6.5のpHを維持するようわずかに緩衝されている点において鼻分泌物と類似しうるが,この範囲外のpH値を用いることもできる。抗微生物剤または保存剤を製剤中に含めてもよい。
【0152】
吸入用のエアロゾル製剤および吸入抗原は本発明の薬剤または薬剤の組み合わせが鼻腔または口腔呼吸器経路で投与されたときに被験者の呼吸器樹中に運ばれるよう設計することができる。吸入溶液は,例えば,ネブライザにより投与することができる。細かく粉砕したまたは液体の薬剤を含む吸入剤または送気剤は,薬剤または薬剤の組み合わせの溶液または懸濁液が,例えば吹き出しを助けるために噴射剤に含まれる医薬エアロゾルとして呼吸器系にデリバリーすることができる。噴射剤は,液化ガス,例えば,ハロカーボン,例えば,フッ化炭素,例えばフッ化塩化炭化水素,ヒドロクロロフルオロカーボン,およびヒドロクロロカーボン,ならびに炭化水素および炭化水素エーテルであることができる。
【0153】
本発明において有用なハロカーボン噴射剤としては,すべての水素がフッ素で置き換えられているフッ化炭素噴射剤,すべての水素が塩素および少なくとも1つのフッ素で置き換えられている塩化フッ化炭素噴射剤,水素含有フッ化炭素噴射剤,および水素含有塩化フッ化炭素噴射剤が挙げられる。ハロカーボン噴射剤は,Johnson,米国特許5,376,359(1994年12月27日発行);Byronら,米国特許5,190,029(1993年5月2日発行);およびPurewalら,米国特許5,776,434(1998年7月7日発行)に記載されている。本発明において有用な炭化水素噴射剤としては,例えば,プロパン,イソブタン,n−ブタン,ペンタン,イソペンタンおよびネオペンタンが挙げられる。炭化水素のブレンドもまた噴射剤として用いることができる。エーテル噴射剤は,例えば,ジメチルエーテルおよびエーテルが挙げられる。本発明のエアロゾル製剤はまた,2以上の噴射剤を含んでいてもよい。例えば,エアロゾル製剤は,同じクラスからの2種以上の噴射剤,例えば,2種またはそれ以上のフッ化炭素を含んでいてもよく;または異なるクラスからの2種以上,3種以上,または4種以上の噴射剤,例えば,フッ化炭化水素および炭化水素を含んでいてもよい。本発明の医薬組成物はまた,圧縮ガス,例えば,二酸化炭素,亜酸化窒素または窒素等の不活性ガスとともに調剤してもよい。
【0154】
エアロゾル製剤はまた,他の成分,例えば,エタノール,イソプロパノール,プロピレングリコール,ならびに界面活性剤または他の成分,例えば,油および界面活性剤を含んでいてもよい。これらの成分は,製剤を安定化させ,および/または,バルブ部材を潤滑化させるよう作用することができる。
【0155】
エアロゾル製剤は,加圧下で充填して,溶液,懸濁液,乳濁液,粉体および半固体調製物を用いてエアロゾルとして製剤することができる。例えば,溶液エアロゾル製剤は,本発明の薬剤,例えば,キナーゼ阻害剤の溶液を,(実質的に)純粋な噴射剤中に,または噴射剤と溶媒との混合物として含むことができる。溶媒は,薬剤を溶解するために,および/または噴射剤の蒸発を遅延させるために用いることができる。本発明において有用な溶媒としは,例えば,水,エタノールおよびグリコールが挙げられる。適当な溶媒の任意の組み合わせを用いることができ,任意にこれを保存剤,抗酸化剤,および/または他のエアロゾル成分と組み合わせてもよい。
【0156】
エアロゾル製剤はまた,分散液または懸濁液であってもよい。懸濁液エアロゾル製剤は,本発明の薬剤,例えばキナーゼ阻害剤の懸濁液または本発明の薬剤の組み合わせ,および分散剤を含むことができる。本発明において有用な分散剤としては,例えば,ソルビタントリオレエート,オレイルアルコール,オレイン酸,レシチンおよびコーン油が挙げられる。懸濁エアロゾル製剤はまた,潤滑剤,保存剤,抗酸化剤,および/または他のエアロゾル成分を含んでいてもよい。
【0157】
エアロゾル製剤は,乳濁液と同様にして製剤することができる。乳濁液エアロゾル製剤は,例えば,エタノール等のアルコール,界面活性剤,水および噴射剤,ならびに本発明の薬剤,例えばキナーゼ阻害剤または薬剤の組み合わせを含むことができる。用いることができる界面活性剤は,非イオン性,アニオン性またはカチオン性であってもよい。乳濁液エアロゾル製剤の1つの例は,例えば,エタノール,界面活性剤,水および噴射剤を含む。乳濁液エアロゾル製剤の別の例は,例えば,植物油,モノステアリン酸グリセリルおよびプロパンを含むことができる。
【0158】
本発明において用いるのに適した医薬組成物には,活性成分が有効量で,すなわち,少なくとも1つのウイルス感染を有する宿主において治療的および/または予防的有益性を達成するのに有効な量で存在する組成物が含まれる。特定の用途に有効な実際の量は,治療している状態,被験者の状態,製剤,および投与の経路,ならびに当業者に知られる他の因子によって異なるであろう。有効量のキナーゼ阻害剤の決定は,本明細書の開示に鑑みて十分に当業者の能力の範囲内であり,日常的な最適化手法を用いて決定される。
【0159】
ヒトにおいて用いるための有効量は,動物モデルから決定することができる。例えば,動物において有効であることが見いだされた循環,肝臓,局所,および/または胃腸濃度を達成するよう,ヒト用の投与量を製剤することができる。当業者は,特に本明細書に記載される動物モデル実験データに鑑みて,ヒトにおいて用いるための有効量を決定することができる。当業者は,動物データ,および他のタイプの同様のデータに基づいて,ヒトに適した本発明の組成物の有効量を決定することができる。
【0160】
本発明の薬剤または薬剤の組み合わせについて述べる場合,有効量とは一般に,医療または医薬品の分野における種々の規制機関ないし諮問機関(例えば,FDA,AMA)のいずれかにより,または製造元または供給元により推奨または認可された投与量範囲,投与のモード,処方物等を表す。
【0161】
さらに,キナーゼ阻害剤の適切な投与量は,インビトロの実験結果に基づいて決定することができる。例えば,ある薬剤がキナーゼ,例えばPAK1,DYRK3,PTK9,およびGPRK2Lを阻害するインビトロの効力は,同様の生物学的効果を達成するのに有効なインビボ投与量の開発に有用な情報を提供する。
【0162】
ある態様においては,本発明の薬剤の投与は間欠的であってもよく,例えば,2日に1回,3日に1回,5日に1回,1週間に1回,1か月に1または2回等の投与であってもよい。ある態様においては,量,形態,および/または種々の形態の量は,投与の異なる時点で様々であってもよい。
【0163】
当業者は,患者において特定の薬剤の投与の効果をモニタリングすることができる。例えば,HIVウイルスの負荷レベルは,当該技術分野において標準的な手法,例えば,CD4細胞のカウントの測定,および/またはPCRによるウイルスレベルの検出により測定することができる。他の手法も当業者には明らかである。
【0164】
バイオテロリズム
本発明は,バイオテロリストの攻撃により引き起こされるウイルス感染の治療に用いることができる。バイオテロリストの攻撃に用いることができるウイルスとしては,例えば,天然痘を引き起こす大痘瘡ウイルス;脳炎ウイルス,例えば西部ウマ脳炎ウイルス,東部ウマ脳炎ウイルス,ベネズエラウマ脳炎ウイルス,およびアレナウイルス(Lassa,Machupo),ブンヤウイルス,フィロウイルス(Ebola,Marburg),および出血熱を引き起こすフラビウイルスが挙げられる。本発明は,バイオテロリストの攻撃により引き起こされるウイルス感染の治療において用いて,対応医療を強化するために備蓄することができる。
【実施例】
【0165】
実施例1
ワクシニアウイルスの侵入メカニズムの評価
ワクシニアウイルスの細胞内への侵入について研究するため,まず,コア蛋白質の1つであるA5のN末端に単量体黄色蛍光蛋白質(mYFP−MV)のタグを付けた組換え成熟ウイルス粒子(MV)を作製した。これをEGFPタグ付きアクチンを発現するHeLa細胞に加えると,明るく蛍光を発する多くのウイルスが糸状仮足に結合し,糸状偽足に沿って細胞体に向かって移動する(「サーフィンする」)ことが見いだされた(図1A)。この移動は,一般に滑らかであり,中断されず,アクチンの逆行の流れとほぼ同じ速度であった(約1μm/分;図1B)。MVが細胞体に到達すると,劇的な変化が起こった。すなわち,細胞膜のウイルスと接触した位置から大きなブレブが突出した。この拡張は30+/−4秒間続き,ブレブは10+/−2秒間拡張したままでであり,その後ウイルスとともに引っ込められた(図1C)。ブレブ形成のピークはウイルス添加後30分間続き,細胞の40%が1またはそれ以上のブレブを示した(図1D)。
【0166】
間接免疫蛍光法は,ブレブは,アクチン−GFPに加えて,種々のアクチン付随蛋白質,例えば,Rac1,RhoA,エズリンおよびコルタクチン(図1E)を含むことを示した。ミオシンII阻害剤であるブレビスタチン(Limouze et al.,2004)は,ブレブの形成を妨害し,ウイルス感染を62%阻害した。このことは,ブレブ形成が増殖性侵入において役割を果たすことを示唆する(図1F)。複数のHeLaおよびBSC40ミドリザル腎臓細胞株において同様の結果が見られた。
【0167】
ウイルスの一部は糸状偽足を用いて細胞体に到達するのみならず,これらはシグナリングカスケードを誘発し,その結果,過渡的な膜ブレブが形成された。ブレブは,細胞運動,細胞質分裂,およびアポトーシスの間に観察されるものと似ていた(Charras et al.,2006;Fishkind et al.,1991;Mills et al.,1998)。
【0168】
図1は,成熟ビリオン侵入の間のサーフィンおよび膜摂動を示す。図1Aは,糸状仮足に沿ったMVのサーフィンを示す。組換えA5−YFP MVを過渡的にトランスフェクトしたGFP−アクチンを発現するHeLa細胞に加えた。画像は,37℃で1Hzで2.5分間取得した。時点はビリオンのリアルタイム画像に対応する。矢印は個々のビリオンを示す。図1Bは,MVのサーフィンの速度を示す。36個の個別のビリオンの速度を,時間あたり移動した距離の相違により測定した(μm/分間)。図1Cは膜ブレブ形成の誘導を示す。組換えA5−YFP MVをGFP−アクチンを発現するHeLa細胞に加え,37℃で1Hzで2.5分間画像を取得した。矢印はブレブが崩壊した部位におけるアクチンパッチ形成を示す。図1Dは,MVにより誘導される細胞のブレブ形成の経時変化を示す。MVをHeLa細胞に4℃で1時間結合させた。細胞を洗浄し,示された時間37℃にシフトした後,4%FAで固定した。各時点につき50個の細胞をブレブ形成について評点し,細胞のブレブ形成の未感染対照に対するパーセントとして示す。実験は三重に行い,結果を平均した。図1Eは,ブレブに局在化する細胞性因子の決定を示す。示される蛍光タグ付き蛋白質で過渡的にトランスフェクトしたHeLa細胞にMVを感染させるか,または未処理のままにした。細胞を感染後30分間固定し(mpi),共焦点顕微鏡により分析した。細胞1個あたり10個のZステーク(Z−stakes)を回収し,Z投影で示した。図1Fはブレブ形成および感染能を示す。HeLa細胞を種々の濃度のブレビスタチンで前処理した後,初期/後期ウイルスプロモーターからEGFPを発現する組換えMV(EGFP−MV)に感染させた。感染した細胞のパーセンテージをFACS分析により測定した。感染した細胞の対照感染に対するパーセンテージを示す。実験は三重に行い,結果を平均した。
【0169】
次に,関与する特定の細胞性キナーゼを同定するために,HeLa細胞において干渉(si)RNAを用いて50種類のキナーゼをサイレンシングすることによりスクリーニングを行った。単一のsiRNAでトランスフェクションした後,早期/後期プロモーターからEGFPを発現する組換えMV(EGFP−MV)を加え,12時間後にEGFP発現について細胞を分析した。各遺伝子につき3つの異なるsiRNAを用い,模擬感染細胞または対照siRNAでトランスフェクションした細胞と比較して3倍のEGFPシグナルの抑制を有意とした。50種類のキナーゼのうち,PAK1がEGFP発現を阻害することが見いだされ,これは,ウイルスの結合,侵入,転写,または初期遺伝子の翻訳が抑制されたことを意味する。
【0170】
感染におけるPAK1の必要性を検証するために,まず,2つの追加のsiRNAを用い,イムノブロッティングにより判定して,これらがPAK1の82%および76%のノックダウンを引き起こすことを見いだした(図2A;上)。蛍光活性化細胞ソーティング(FACS)に基づく感染アッセイは,感染した細胞の数がそれぞれ74%および68%減少したことを示した(図2A;下)。先の報告は,残基83−149(AID,自己阻害ドメイン)を含むPAK1ドメインの過剰発現はマクロピノサイトーシスを阻害することを示している(Dharmawardhane et al.,2000)。HeLa細胞においては,AIDの発現はwtPAK1を過剰発現する細胞と比較して感染を80%阻害することが見いだされた。変異型のAID(AIDL107F)の発現は影響を与えなかった(図2B)。
【0171】
PAK1のトレオニン残基423のリン酸化は,マクロピノサイトーシスの活性化に役割を果たしている(Dharmawardhane et al.,2000)。MVを細胞に加えると,10分以内にリン酸化PAK1が検出され,PAK1は60分間にわたって活性化されたままであった(図2C)。最大応答は,30mpiに見られた。この結果は,ビリオン取り込みおよびウイルスコアのエンドソーム放出の経時変化と一致していた(Townsley et al.,2006)。これらを合わせると,この結果は,PAK1活性がワクシニアMVの細胞内への増殖性侵入において役割を果たすことを示す。明らかに,ウイルスはRac1の活性化を誘発し,その結果,PAK1およびアクチン動力学に関与する下流の他の因子が活性化される。
【0172】
図2は,p21活性化キナーゼ−1(PAK1)がMV侵入に必要であることを示す。図2Aは,PAK1のsiRNAノックダウンがMV感染に及ぼす影響を示す。HeLa細胞をPAK1に対する2つの別々に確認したsiRNA(Qiagen;A:TCCACTGATTGCTGCAGCTAA(配列番号12);B:TTGAAGAGAACTGCAACTGAA(配列番号13)でトランスフェクションした。処理の36時間後,細胞にEGFP−MVをMOI=1で感染させ,2hpiで回収して分析した。感染した細胞のパーセンテージをFACS分析により求めた。実験は三重に行い,結果を平均した。PAK1に対するイムノブロット分析(a−PAK1;Santa Cruz)を行って,PAK1蛋白質レベルの低下を確認した(下パネル)。1レーンあたり合計50μgの細胞溶解物を負荷し,モック処理細胞(−−)に対する残存PAK1蛋白質の%を求めた。アクチンに対するイムノブロットを負荷対照として用いた。図2Bは,MVの感染能に及ぼすドミナント・ネガティブPAK1の影響を示す。HeLa細胞を蛍光タグ付きの野生型PAK1(WT),PAK1自己阻害ドメイン(AID),または変異型AID(AIDL107F)で過渡的にトランスフェクションした。細胞をEGFP−MVでMOI=1で感染させた。4hpiにおいて細胞を固定し,アクチンを染色した。細胞をトランスフェクションした蛋白質(赤),ウイルス感染(緑)およびアクチン(青)について共焦点顕微鏡により分析した。実験は三重に行い,1回の実験あたり100個のトランスフェクトした細胞を感染について評点した。結果は,トランスフェクトした/感染細胞の平均パーセンテージとして示す。図2Cは,MV感染の間のPAK1の活性化を示す。MVを4℃で1時間HeLa細胞に結合させた。細胞を冷PBSで2回洗浄した。予熱した培地を加えて,感染物を37℃にシフトした後,示された時点で回収した。PAK1およびリン酸化PAK1(a−PAK1−Thr423;Cell Signaling)についてイムノブロット分析を行った。
【0173】
PAK1はマクロピノサイトーシス,すなわち,多くの異なる細胞タイプにおいて大量の液体および細胞膜の内在化につながる,リガンドにより誘導されるエンドサイトーシスのプロセスに関与することが知られている(Falcone et al.,2006)。マクロピノサイトーシスは動的なアクチン再編成に依存し,これはコレステロール,ならびに小さいGTPaseであるRac1,CDC42,およびArf6を必要とする(Kirkham and Parton,2005)。さらに,PAK1に加えて,Na+/H+交換体,チロシンキナーゼ,および少なくとも1つのセリントレオニンキナーゼ,PKCも関与する(Dharmawardhane et al.,2000;Grimmer et al.,2002;Hewlett et al.,1994;Veithen et al.,1996)。ダイナミンは必要ではない(Damke et al.,1994)。
【0174】
MV侵入にマクロピノサイトーシスが関与しているか否かを試験するために,種々の広範な範囲のキナーゼ阻害剤を試験した。スタウロスポリン(セリン/トレオニンキナーゼ阻害剤)は感染を65%まで,ゲニステイン(チロシンキナーゼ阻害剤)は82%まで,ワートマニン(PI3キナーゼ阻害剤)は63%まで阻害した(図3A)。次に,アクチンアセンブリを防止する(サイトカラシンD),アクチンを脱重合させる(ラトルンクリンA),またはアクチンフィラメントを安定化させる(ジャスパキノリドA)薬剤も,感染の用量依存的阻害を40%から90%の範囲で引き起こすことを見いだした(図3B)。顕微鏡により,MVにより誘導されるブレブの形成がこれらのアクチン阻害剤により阻害されることが示された。Na+/H+交換体およびマクロピノサイトーシスの阻害剤であるEIPAも感染を90%阻害した(図3C)。構成的に活性なArf6の発現は,wtArf6を過剰発現する細胞と比較して感染を78%阻害したが(図3D),ドミナント・ネガティブダイナミン(K44A)の過剰発現は影響を与えなかった。MVによる感染はコレステロール依存性であり,Rac1を必要とすることは,すでに他の研究者により示されている(Chung et al.,2005;Locker et al.,2000)。まとめると,阻害はマクロピノサイトーシスプロセスと完全に一致した。
【0175】
エンドサイトーシスプロセスのマクロピノサイトーシス的性質は,同時に生ずる液相マーカーの内在化とも一致した。蛍光MVを含むエンドサイトーシスの小胞は,液相マーカーである568−デキストランについて陽性であったが,クラスリン媒介性エンドサイトーシスにより内在化されるリガンドである568−トランスフェリンについて陰性であった(図3E)。100μMのEIPAを含めると,MVのFITC−デキストラン陽性小胞への内在化が防止された(図3E)。
【0176】
図3は,ワクシニアMVが細胞に侵入するのにマクロピノサイトーシスを利用することを示す。図3Aおよび3Bは,MV感染に必要な一般的キナーゼおよびアクチン骨格を示す。一連のキナーゼ阻害剤およびアクチン作用性薬剤がMVの感染能に及ぼす影響をFACS分析により調べた。アッセイは,下記の材料および方法の節に概説するようにして行った。結果は,3回の実験の平均であり,未処理対照に対する感染細胞のパーセンテージで示す。図3Cは,EIPAによるMVの感染能の阻害を示す。細胞をNa+/H+交換およびマクロピノサイトーシスの阻害剤であるEIPA(5−N−エチル−N−イソプロピルアミロリド;Sigma Aldrich)で前処理し,次にEGFP−MVを感染させ,下記の材料および方法の節に記載されるようにしてFACSにより分析した。実験は三重に行い,結果は薬剤非存在下における対照感染に対する感染細胞のパーセンテージで表す。図3DはMVの感染能に及ぼすドミナント・ネガティブArf6の影響を示す。HeLa細胞を蛍光タグ付き野生型Arf6(WT)または構成的活性化型Arf6(C/A)で過渡的にトランスフェクションした。細胞はEGFP−MVでMOI=1で感染させた。4hpiにおいて,細胞を固定し,アクチンを染色した。細胞を,トランスフェクションした蛋白質(赤),ウイルス感染(緑)およびアクチン(青)について共焦点顕微鏡により分析した。実験は三重に行い,1回の実験に100個のトランスフェクションした細胞を用いて,感染について評点した。結果は,トランスフェクション/感染した細胞の平均パーセンテージとして示す。図3EはMVのエンドサイトーシスの小胞中への内在化を示す。細胞を100μMのEIPAで処理するかまたは未処理とした。細胞は,mYFP−MVとMOI=1で4℃で1時間結合させるか,または未感染とした。細胞を冷PBSで2回洗浄し,37℃に15分間シフトした。次に細胞を液相マーカーである10kDaの568−デキストラン(0.5mg/ml)または568−トランスフェリン(Tfn)(200ng/ml)で,EIPAの存在下または非存在下で10分間パルスした。低pHで軽く洗浄することにより表面に結合したデキストランおよびTfnを除去した後,固定した。サンプルは共焦点顕微鏡で分析した。1つの画像あたり10個のZsテークを集め,Z投影として表示した。
【0177】
ホスファチジルセリン(PS)の役割をさらに詳細に分析するため,単離したMV粒子を0.5%NP40を用いて抽出し,次に界面活性剤および抽出された脂質を除去した。これにより,感染能は劇的に低下したが(図4A),ウイルス構造蛋白質の損失はなかった(データ示さず)。蛍光MVを用いて,抽出したウイルスは細胞に結合するが,ブレブ形成およびエンドサイトーシスを誘導することができなかったことを示した。未処理のMVが細胞の内部にクラスター化している8時間後でさえも,抽出したビリオンはなお細胞表面に留まっており,このことは,エンドサイトーシスに欠陥があることを示唆する(図4B)。PSの役割を試験するために,脱脂質化したウイルスを異なる脂質組成をもつリポソームの存在下でインキュベーションすることにより,脂質を戻し入れた(addback)。いずれの脂質を用いるかにかかわらず,再脂質化したウイルスは細胞に結合することができた(図4C)。しかし,PSを有するもののみ,ブレブ形成およびエンドサイトーシスを誘導することができた。さらに,PSを再び加えると,元の感染能の大部分(90%)およびプラーク形成がレスキューされた(図4DおよびE)。
【0178】
PS結合蛋白質であるアネキシン−A5(ANX5)をEGFPタグ付きの形で用いて,ウイルスのPSが実際にウイルス膜の外葉に暴露されていることを示した。ウイルスは,この試薬に暴露したときにわずかに染色され(図4F),ウイルスPSをANX5でマスキングすると,MVの細胞への結合には影響を及ぼさずに感染が95%阻害されることが見いだされた(図4G)。ウイルスを加える前に細胞をANX5で処理すると,ウイルスの結合または感染能に及ぼす影響は観察されなかった。これらの結果は,マクロピノサイトーシスプロセスの開始にウイルス粒子中のPSが必要であることを示す。
【0179】
最後に,ANX5を用いて,感染細胞からMVを放出する感染細胞の溶解が,アポトーシスまたは壊死のいずれにより引き起こされるかを判定した。細胞アポトーシスの特徴である表面暴露PSの存在について,ウイルスプラークを分析した(Martin et al.,1995)。ウイルスプラークを囲む細胞は大量の表面暴露PSを含むが,非感染細胞またはプラーク間の細胞はこれを含まないことが見いだされた(図4H)。このことは,感染の後期において,ワクシニアウイルスはアポトーシスを誘導し,これがMVが退出する手段であることを示す。すなわち,ウイルスは,感染細胞からのアポトーシス体と一緒に,隣り合う非感染細胞に暴露される。
【0180】
図4は,ワクシニアMVは内在化にPSを必要とすることを示す。図4Aは,MVの感染能にはウイルス脂質が必要であることを示す。ビリオンから種々の濃度のNP40(0.1−1.0%)を用いて脂質を抽出した。補正後,BSC40細胞におけるタイター測定(pfu/ml)によりビリオンの感染能を測定した。図4Bは,脂質を抽出したMVが細胞に結合しうるが細胞に侵入できないことを示す。0.5%NP40で処理したかまたは未処理のmYFP−MVをMOI=1で細胞に加えた。細胞は,30mpiまたは8hpiで固定し,アクチンについて染色し,共焦点顕微鏡によりウイルスの結合および感染について可視化した。図4Cは,MVの結合はビリオン膜の脂質構成成分には依存しないことを示す。1x109個のmYFP−MVを,未処理で,または脂質を抽出して,または続いて異なる脂質(MおよびM’)とともに戻した。戻した後,ビリオンをHeLa細胞に4℃で1時間結合させ,冷PBSで2回洗浄し,下記の材料および方法の節にしたがって,FACS分析により分析した。結果は3回の独立した実験として示す。図4Dは,MVの感染能はビリオン膜中のPSに依存することを示す。1x109個のEGFP−MVを脂質抽出するかまたは未処理で異なる脂質に戻した(下記の材料および方法の節を参照)。戻した後,ビリオンをHeLa細胞に4℃で1時間結合させた。PBSで2回洗浄し,37℃で2時間感染を進行させた後に,FACS分析を行った。3回の独立した実験の結果を示す。図4Eは,MVによる増殖感染はビリオン膜中のPSに依存することを示す。1x109個のWR MVを脂質抽出するかまたは未処理で異なる脂質に戻した。戻した後,ビリオンの感染能をBSC40細胞におけるタイター評定(pfu/ml)により測定した。結果は3回の独立した実験で示す。図4Fは,ウイルス膜PSがMVの表面への暴露されることを示す。ウイルス膜上のPSの存在は,組換え488アネキシンV(ANX5)を用いて示した。ビリオンは,Vybrant(登録商標)アポトーシスアッセイキット#2(Molecular Probes)に提供されるプロトコルを適合させて分析した。簡単には,ビリオンを25μlの1Xアネキシン結合バッファ中で,2μlの488ANX5とともに室温で15分間インキュベーションした。ビリオンをペレット化し,結合バッファで1回洗浄し,カバースリップに結合させた後,共焦点顕微鏡により可視化した。mYFP−MVを可視化して陽性対照とした。図4Gは,MV膜PSのマスキングは感染を妨害することを示す。結合:HeLa細胞またはmYFP−MVをANX5で,製造元の指針にしたがって(細胞)または上述の条件で(MV)前処理した。処理後,ANX5と結合した細胞を未処理のビリオンとともに,またはANX5処理mYFPビリオンを未処理細胞とともにインキュベーションした。mYFP−MVのHeLa細胞への結合を陽性対照として用いた。実験は三重に行い,結果は対照に対するビリオン結合細胞のパーセントとして示した。感染:実験は,EGFP−MVを用いる結合と同様にして行った。図4Hは,アポトーシス性細胞についてウイルスプラークが濃縮されていることを示す。BSC40細胞単層をwt−MVに感染させ,感染は24時間進行させた。次に細胞をPBSで2回洗浄し,製造元のプロトコルにしたがって,生存,アポトーシスおよび壊死細胞について分析した(Vybrant(登録商標)アポトーシスアッセイキット#2;Molecular Probes)。488ANX5は緑で,ヨウ化プロピジウムは赤で示される。未感染細胞を染色対照として用いた。
【0181】
材料および方法:
細胞株およびウイルス
BSC40霊長類およびHeLa ATCC細胞の単層を10%ウシ胎児血清(FCS)を含むダルベッコ改変イーグル培地(DMEM;Gibco BRL)中で37℃で維持した。野生型(wt)ワクシニアウイルス(WR株),WRE/LEGFP(MV−EGFP)(Paula Traktman,Medical College of Wisconsinより供与),およびコア蛋白質の蛍光タイプを含むWRであるA5(mYFP−MV)を示されるようにして用いた。すべてのウイルスのストックは,ブレフェルジンAの存在下で調製した。ウイルスは,細胞質溶解物から超遠心分離により25−40%のショ糖勾配中36%ショ糖のバンドとして精製した。
【0182】
蛍光活性化セルソーティング(FACS)
結合アッセイ:mYFP−MVをHeLa細胞(wtまたは処理済)に,無血清DMEM中で4℃で1時間,感染多重度(MOI)=1で結合させた。ビリオン結合細胞を4℃にシフトし,PBSで2回洗浄し,プレートからトリプシン処理し,ホルムアルデヒド(FA)で氷上で30分間固定した。固定した細胞を遠心分離により回収し,PBSで1回洗浄し,再び回収し,PBSに懸濁してFACS分析を行った。各サンプルから合計で10,000事象を分析し,未結合およびmYFP−MV結合対照に対する相対的mYFP発現について評点した。感染アッセイ:EGFP−MVを無血清DMEM中で薬剤の存在下で4℃で1時間HeLa細胞に結合させた。すべのアッセイはMOI=1で行った。結合後,細胞を冷PBSで2回洗浄し,予熱した培地を加えた。細胞を37℃にシフトし,感染を2時間進行させた後,固定し,上述のようにしてFACS用に調製した。薬剤スクリーニング:HeLa細胞を感染前に種々の濃度の示された薬剤で15分間前処理した。細胞を,無血清DMEM中で薬剤の存在下で,4℃で1時間EGFP−MVに結合させた。すべてのアッセイはMOI=1で行った。結合後,細胞を冷PBSで2回洗浄し,薬剤を含む予熱した培地を加えた。次に細胞を37℃にシフトし,感染を2時間進行させた。細胞を固定し,上述のようにしてFACS分析用に調製した。すべてのFACS分析は,薬剤なしの対照感染に対する感染細胞のパーセンテージとして表した。
【0183】
ビリオンの界面活性剤抽出
合計で1.0x109のワクシニアウイルスwt,EGFP−MV,またはmYFP−MVビリオン(上述のようにして精製)を,100mM Tris(pH9.0)および種々の濃度のNP−40:すなわち,0,0.1,0.5,および1.0%(vol/vol)を含む反応混合物中で37℃で60分間インキュベーションした。抽出後,可溶性画分および粒子画分(それぞれビリオンの膜およびコア成分を表す)を沈降(16,000xg,30分間,室温)により分離した。次にサンプルをBSC40細胞上でタイターを測定して,ウイルスタイター(PFU数/ミリリットル)を決定した。すべてのタイター測定は三重に行い,結果を平均した。
【0184】
ビリオン脂質の戻し入れ
種々の脂質組成をもつリポソームを調製し,Oie(Oie,1985#2008)の方法にしたがって,脂質を抽出したビリオンを再構築した。簡単には,脂質を抽出したビリオンを,種々の濃度のPS(20μg/mlまたは200μg/ml)またはGM1(20μg/ml)を組み込んだPC系リポソーム(200μg/ml)とともに37℃で2時間インキュベーションした。ビリオンを遠心分離により回収し,次に洗浄し,バッファーに再懸濁した。再構築ビリオンをFACSおよび顕微鏡による結合および侵入アッセイに供し,ならびにウイルス収量のタイターを測定した。
【0185】
実施例2
アデノウイルス血清型3ウイルスの侵入メカニズムの評価
材料および方法
細胞およびウイルス
細胞は,記載されるようにして,10%FCS(GIBCO−BRL)を含むDME(GIBCO−BRL)で低い継代数で成長させた(Suomalainen et al.,1999)。ヒト黒色腫M21 litter(表面発現αvインテグリン陰性)およびM21L細胞(細胞表面αvインテグリン陽性)はDr.D.Cheresh(Scripps Research Institute,La Jolla,CA,Felding−Habermann et al.,1992)から供与された。K562慢性骨髄性白血病細胞は記載されるようにして成長させた(Nagel et al.,2003)。CD46またはCARを安定して発現するBHK細胞は,CD46またはCARのBC1アイソフォームのいずれかをコードするプラスミドを安定的にトランスフェクトすることにより調製した(Sirena et al.,2005)。Ad3およびAd2ts1は,記載されるようにして成長させ単離した(Greber et al.,1996)。Ad3のテキサスレッドによる標識は公表されているようにして行った(Nakano and Greber,2000)。(3H)−チミジン標識Ad3は,公表されているようにして製造した(Greber et al.,1993)。
【0186】
cDNA,蛋白質および化学物質
CtBP1−S/BARSをコードするcDNAはDr.A.Colanzi(Dep.Of Cell Biology and Oncology,S.MariaImbaro,Italy)から入手した。pCMV−myc CtBP1−Swtは,PCR増幅CtBP1−Swt(それぞれSalIおよびNotIで切断)をpCMV骨格ベクター(Stratagene)中にライゲーションすることにより生成した。Myc−CtBP3 D355Aは,Quik Change R部位特異的変異導入キット(Stratagene)を用い,下記のプライマー5’−CTGGGCCAGCATGGCCCCTGCTGTGGTG−3’(配列番号21)および5’−CACCACAGCAGGGGCCATGCTGGCCCAG−3’(配列番号22)を用いて生成した(Bonazzi et al.,2005)。得られたcDNAをシークエンシングにより確認した。K44A−dyn2およびdyn2wt発現プラスミドはDr.C.Lamaze(Pasteur Institute)より入手した。Pak1wtおよび阻害的ドメイン発現ベクターは,J.Chernoff(Fox Chase Cancer Center,Philadelphia,PA)より入手した。Toxin B(0.5mg/ml)はDrs.F.HofmannおよびK.Aktories(University of Freiburg,Freiburg,Germany)より入手した。PKC阻害剤Go6976(1μM)およびGo6983(1μM)はCalbiochem(Juro Supply)より購入し,Na+/H+交換阻害剤EIPA(100μM)はAlexis Corporationより購入し,サイトカラシンD(5μM)およびジャスパラキノリド(500nM)はCalbiochemより購入した。メチル−ベータ−シクロデキストリン(50mM)によるコレステロール飢餓は,先に公表されているようにして行った(Imelli et al.,2004)。Ad3可溶性ファイバーノブ(最終濃度5μg/mlで使用)はP.Fender(Grenoble,France)から入手した。ダイナソールはDr.J.S.Siegel(Organic Chemistry Institute,University of Zurich)の合成したものを用いた。CtBP1−L/Sに対する抗体は,BD Transduction laboratoriesから,PAK1抗体(C−19)はSanta Cruz Biotechnology(Santa Cruz,CA)から,リン酸化PAK1(T423)に対する抗体はCell Signalling Technologyから入手した。
【0187】
エンドサイトーシスおよびAd−eGFP形質導入
細胞を冷却下で,RPMI−0.2%BSA中で(3H)チミジン標識Ad3(1μg/ml;3X109粒子,35mmディッシュ上)とともに60分間インキュベーションし,冷RPMI−BSAで洗浄し,RPMI−BSAとともに示された時間暖めた。細胞を冷RPMI−BSAおよび冷PBSで2回洗浄した。細胞外ウイルス粒子を細胞から除去するために,細胞を冷2%トリプシン−EDTA(GIBCO)とともに冷却下で60分間インキュベーションし,PBS−2%FCSを加え,細胞を500xgで遠心分離した。洗浄工程を2回繰り返した。熱SDS(0.4%)中に細胞溶解物を調製し,20G医療シリンジで剪断し,Beckman Coulter Scintillation System LS3801を用いて,液体シンチレーションカウント(Ready Safe;Beckman Coulter)により放射活性を測定した。トリプシンなしの対照細胞の計数を100%対照として用いた。薬剤実験のためには,細胞をRPMI−BSA中の薬剤とともに37℃で30分間プレインキュベーションした。形質導入実験のためには,細胞を温RPMI−BSAで洗浄し,1μg/mlのウイルスとともに60分間インキュベーションし,RPMI−BSAで数回洗浄し,水浴中で4時間(A549細胞),5時間(HeLa細胞),8時間(M21およびM21L細胞)および16時間(K562細胞)インキュベーションした。細胞を冷PBSで洗浄し,冷却下で2%トリプシンで,次に2%PBS−FCSで処理し,フローサイトメトリ(Beckman FC500 cytometer)で分析した。サンプル1つあたり少なくとも10000個の生存細胞を計数した。薬剤実験のためには,細胞をRPMI−BSA中の阻害剤で37℃で30分間前処理した後,薬剤の存在下で60分間温感染させ,次に薬剤を含まない培地で洗浄し,さらにインキュベーションした。
【0188】
デキストラン取り込み
細胞を,先に記載されるようにして(Meier et al.,2002),5μg/mlの冷Ad3とともにプレインキュベーションし,温RPMI−BSAで洗浄し,0.5mg/mlのデキストラン−FITC(lysine−fixable 10kDa,Molecular Probes)を含むRPMI−BSA中で37℃で10分間暖めた。デキストラン取り込みは,細胞を冷RPMI−BSAおよびPBSで洗浄(3回繰り返し)することにより停止させた。表面に結合したデキストランは,冷0.1M酢酸ナトリウム(pH5.5),0.05M NaCl中で10分間酸処理することにより除去した。FACS分析用には,細胞をPBS中2%トリプシン(GIBCO−BRL)中で氷上で25分間剥離させ,2mlの7%FCS/PBSを含む6mlのポリプロピレンチューブ(no2063;Falcon,Becton Dickinson)に移し,290xgでペレット化し,2%FCS/PBSに再懸濁した。サンプル1個あたり少なくとも10000個の生存細胞をフローサイトメータで計数した(Beckman FC500サイトメータ)。
【0189】
透過型電子顕微鏡,およびBSA−金の取り込み
冷Ad3またはAd2ts1(50μg/ml,感染多重度[MOI]5000)で60分間結合させた後,適宜洗浄および内在化を行い,細胞を0.1Mカコジル酸ナトリウムバッファ,pH7.4(CaCo)中2%ホルムアルデヒド−1.5%グルタルアルデヒドで一晩固定し,CaCoで数回洗浄し,二回蒸留水中の1%OsO4(Electron Microscopa Sciences)および1.5%フェリシアン化カリウム(FeK3N6)で4℃で60分間固定後処理を行った(Simionescu and Siminonescuの方法を改変)。標本を0.1Mカコジル酸ナトリウムですすぎ,0.05Mカコジル酸ナトリウム中1%タンニン酸で室温で45分間対比染色し,1%硫酸ナトリウムで洗浄し,H2Oですすぎ,H2O中2%酢酸ウラニルで一晩染色し,先に記載されるようにしてEpon中に包埋した(Nakano et al.,2000)。ウイルス粒子は,細胞膜,エンドソームおよびサイトゾルの超薄切片を倍率50000xで定量し,透過電子顕微鏡で加速電圧80,000Vで観察した(Zeiss EM902A)。15nmのコロイド金は,HAuCl4のクエン酸還元により調製した(Frens,1973;Horisberger and Rosset,1977)。20mlのコロイド金溶液(pHを5.9に調節)に,50μlの10mg/mlBSA(Sigma,無脂肪酸)溶液を加えた(DeRoe et al.,1987)。BSA金複合体を安定化させるために,1mlの1%PEG−20000(Roth,Switzerland)を加え,サンプルを28,000gで60分間遠心分離し,ペレットを2mlの金バッファ(0.2%PEG−20000を含む滅菌濾過済みPBS)に溶解し,4℃で保存した。BSA金内在化は,Ad3またはAd2−ts1の冷結合後に,BSA金のRPMI−BSA(約0.1mg/mlのBSA)中1:1希釈を用いて,37℃で10分間行った。
【0190】
蛍光顕微鏡および免疫蛍光
細胞を種々のDNAコンストラクトで30時間トランスフェクションした後,Fugene6(Roche,製造元の指針にしたがう)を用いた実験を行った。細胞を,Ad3−eGFPまたはAd5−eGFPで37℃で60分間感染させ,洗浄し,37℃で15時間インキュベーションした。細胞を固定し,DAKOを用いてマウントした。デキストランおよびトランスフェリン取り込み用には,細胞を5μg/mlのAd3で冷却下でシンクロナイズさせ,温洗浄し,RPMI−BSA中の0.5mg/mlデキストラン−TRおよび20μg/mlのトランスフェリン−Alexa647の混合物で37℃で30分間パルスし(水浴),次に5分間チェイスし,固定し,DAKOを用いてマウントした。共焦点レーザー走査顕微鏡(CLSM)は,Ar−ArKrレーザー,He−Ne543−594レーザー,He−Ne633レーザー,405nmのダイオードレーザーおよび63x油浸対物レンズ(N.A.1.4PLAPO)を備えたLeica−DM SP2 RXA2−TCS−AOBS顕微鏡(Leica Microsystems,Wetzlar,Germany)により行った。ピンホール値1.0,エアリー1,得られる光学切片約0.48μm,ボクセル0.233x0.233x0.48μm。ズーム倍率は2であった。画像のプロセシングはLeicaand Photoshopソフトウエア(Adobe)を用いて行い,細胞の合計投影の蛍光強度はImage J(webpage:http://rsb.info.nih.gov/ij/)を用いて決定した。デキストラン陽性エンドソームを用いるCtBP1−L/S共局在用には,細胞をAd3−TR(2μg/ml)で氷上で60分間冷シンクロナイズさせ,温RPMI−BSAで洗浄し,0.5mg/mlのデキストラン−FITCで37℃で10分間パルスした。細胞をRPMI−BSAおよびPBSでよく洗浄し,固定し,CtBP1−L/Sマウスモノクローナル抗体(BD Transduction laboratories)および二次抗体としてAlexa647−コンジュゲート化ヤギ抗マウス抗体を用いて,免疫蛍光により分析した。
【0191】
siRNAトランスフェクション
K562細胞を,クラスリン重鎖(AACCUGCGGUCUGGAGUCAAC(配列番号23);Qiagen(Hinrichsen et al.,2003))およびCtBP1/CtBP3(CCGUCAAGCAGAUGAGACAUU(配列番号24);GGAUAGAGACCACGCCAGUUU(配列番号25);Dharmacon(Bonazzi et al.,2005))に対するsiRNAで,Nucleofector I(Amaxa;programT−03)を製造元の指針にしたがって用いて,トランスフェクトした。非標的化siRNA配列(Qiagen,またはDharmacon)のトランスフェクションを対照として用いた。トランスフェクションは第0日および第2日に行い,ウエスタンブロッティングおよび実験用の細胞溶解物は第4日に回収した。HeLa細胞を,クラスリン重鎖,CtBP1/CtBP3,またはPAK1(確認済siRNACat.SI00605703およびSI00605696;Qiagen)に対するsiRNAで,リポフェクタミン2000(Invitrogen)を製造元の指針にしたがって用いてトランスフェクトした。トランスフェクションは第0日および第2日に2回行い,ウエスタンブロッティングおよび実験用の細胞溶解物は第4日に回収した。A549細胞を,クラスリン重鎖,CtBP1/CtBP3,PAK1またはダイナミン2{GACAUGAUCCUGCAGUUCA(配列番号26),Qiagen,Huang,2004#15509}に対するsiRNAで,リポフェクタミン2000を上述のようにして用いてトランスフェクトした。
【0192】
ウエスタンブロッティング
細胞を35mmディッシュで成長させ,リン酸緩衝化食塩水(PBS)で洗浄し,500μlの2%熱SDS中で溶解した。溶解物を20ゲージの針に数回通過させ,95℃で30秒間加熱した。16,000×gで10分間遠心分離した後,150μlの上清を50μlのサンプルバッファー(200mM Tris/HCl,pH6.8,8%SDS,0.4%ブロモフェノールブルー,40%グリセロール,167mMジチオスレイトール)と混合し,95℃で10分間加熱した。抽出物を10%SDS−PAGEで分離し,Hybond−ECLニトロセルロース膜(Amersham Biosciences,Zurich,Switzerland)に移し,50mMTris/100mM塩化ナトリウム/0.1%Tween,pH7.4(TNT)中の5%ドライミルクでブロッキングした。免疫学的プロービング(PAK1ブロットについては3%ミルク,CtBP1ブロットについては0.2%BSA)の後,ECLPlus試薬(Amersham Biosciences)でHRPコンジュゲート化抗体を検出した。フィルタを,100mMβ−メルカプトエタノール,2%SDS,62.5mMTris/HCl,pH6.7で50℃で30分間ストリッピングした後,TNTでよく洗浄し,5%ドライミルクでブロッキングし,3%ミルクを含むTNT中の抗カルネキシン抗体(Dr.A.Helenius,Zurichより寄付)で再プローブした。
【0193】
図5は,Ad3のエンドサイトーシスおよび感染にはPAK1の活性化が必要であるが,Ad5には必要ではないことを示す。図5Aは,Ad3およびAd5がPAK1を活性化することを示す。HeLa細胞を0.5μg/mlのAd3,Ad5またはts1(約5X105細胞)とともに,冷却中で60分間インキュベーションし,洗浄し,異なる時間暖めた。細胞をホスファターゼおよびプロテアーゼ阻害剤を含む冷PBSで洗浄し,ディッシュからはがし,阻害剤を含む100μlのPBSに再懸濁し,ジチオスレイトールを含むSDSサンプルバッファと混合し,加熱し,SDS−PAGE(1レーンあたり105個の細胞に相当)およびそれぞれPAK1およびリン酸化PAK1(T423)に対するウエスタンブロッティングにより,ECLを検出法として用いて分析した。図5B:野生型またはドミナント・ネガティブPAK1(阻害ドメインID)を発現するHeLa細胞にAd3−eGFPまたはAd5−eGFPを形質導入するか,または,Ad3感染の際のデキストラン−FITCまたはAlexa647で標識したトランスフェリンの取り込みについて評価した。図5C:HeLa細胞を,PAK1に対するsiRNAであるP2およびP8またはns siRNAで72時間トランスフェクトし(ダブルトランスフェクション,20pmoles/ml siRNA),Ad3−eGFPまたはAd5−eGFPを6時間感染させ,フローサイトメトリでeGFP発現について分析した。1x105個のトランスフェクトした細胞をウエスタンブロッティング(WB)によりPAK1含有量について分析した。図5D:Ad3のエンドソームエスケープを,それぞれ抗PAK1 siRNA P2およびns siRNAでトランスフェクトしたHeLa細胞における薄片EMにより測定した。結果は,細胞膜,エンドソーム中およびサイトゾル中にビリオンを示した。
【0194】
実施例3
ウイルス感染の間のEGFRの役割
Rac1エフェクターであるPAK1は,ワクシニア成熟ビリオン(MV)の侵入に必要であり,RhoAファミリーのGTPaseであるRac1およびRhoAは侵入プロセスの間に活性化される(Mercer and Helenius(2008)Science320:531−535)。次に,これらのGTPaseは,種々の細胞表面レセプターにより活性化されることができる(Schiller(2006)Cell Signal 18:1834−1843)。その1つは表皮成長因子レセプター(EGFR,Erb1)である。これまでワクシニア侵入におけるEGFRの関与については意見が分かれている(Marsh and Eppstein(1987)J Cell Biochem.34:239−245;Eppstein et al.(1985)Nature 318:663−665;Hugin and Hauser(1994)J.Virol.68:8409−8412)。
【0195】
MV感染の経時変化を用いて,ワクシニア感染の間のEGFRの活性化状態を評価した。EGFR活性化は,チロシン1173でリン酸化されたEGFRを認識する抗体を用いてモニターした。EGFRはウイルスを加えてから5分以内に強く活性化され,そのピークは感染後15分であった(図7)。
【0196】
EGFR阻害剤である324674(Calbiochem)は,MV侵入を有効に妨害し,低pH融合により容易にバイパスされた(図8)。
【0197】
これらの結果は,ワクシニアMVは感染の間にEGFRを活性化することができ,この活性化が侵入に必要であることを示す。さらに,MVがEGFRの下流に存在するPAK1の活性化を誘導したことが示唆される。これらのデータは,MV侵入および感染に必要なシグナリング経路についてのさらなる洞察を提供し,他の潜在的細胞性抗ウイルス標的およびポックスウイルス感染に対する治療法を示唆する。
【0198】
材料および方法
蛍光活性化細胞ソーティング(FACS)感染アッセイ
薬剤スクリーニング:HeLa細胞は,感染前に,示される薬剤で種々の濃度で15分間前処理した。EGFP発現MVを,4℃で無血清DMEM中で薬剤の存在下で1時間結合させた。すべてのアッセイはMOI=1で実施した。結合後,細胞を冷PBSで2回洗浄し,次に予温した薬剤含有培地を加えた。細胞を37℃にシフトさせ,2時間感染させた。細胞をPBSで2回洗浄し,プレートをトリプシン処理し,4%ホルムアミド(FA)中で氷上30分間固定した。固定した細胞を遠心分離により回収し,PBSで洗浄し,再び回収し,PBS中に懸濁して,FACS Calibur System(BD Biosciences)を用いるFACS分析を行った。すべてのFACS分析は,三重に行い,薬剤なしの対照感染に対する感染細胞の平均パーセンテージで表した。エラーバーは,実験間の標準偏差を表す。
【0199】
参考文献
Bangham, A.D., M.M. Standish, and J.C. Watkins. 1965. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 13(1): 238-52.
Birault, V., C.J.Harris, J. Le, M. Lipkin, R. Nerella, and A. Stevens. 2006. Bringing kinases into focus: efficient drug design through the use of chemogenomic toolkits.Curr. Med. Chem. 13(15): 1735-48,
Bui M, Whittaker G, Helenius A (1996) The protein kinase inhibitor, H7, directly blocks M1-ediated nuclear export of infleunza virus RNPs. submitted.
Bukrinskaya AG, Vorkunova NK, Kornilayeva GV, Naramanbetova RA, Vorkunova GK (1982) Influenza virus uncoating in infected cells and effect of rimantadine. J Gen Virol 60: 49-59.
Cros JF, Garcia-Sastre A, Palese P (2005) An unconventional NLS is critical for the nuclear import of the influenza A virus nucleoprotein and ribonucleoprotein. Traffic 6(3): 205- 213.
Doms RW, Lamb RA, Rose JK, Helenius A (1993) Folding and assembly of viral membrane proteins. Virology 193(2): 545-562.
Ehrhardt C, Marjuki H, Wolff T, Nurnberg B, Planz O et al. (2006) Bivalent role of the phosphatidylinositol-3-kinase (PI3K) during influenza virus infection and host cell defence. Cell Microbiol 8(8): 1336-1348.
Graeser RU, Sarah; Brosig, Michaela; Kubbutat, Michael H.G. (2004) Highly efficient gene silencing using 2-for-Silencing siRNA Duplexes targeting protein kinase genes. QIAGEN News 2004 e32.
Huang YT, Turchek BM (2000) Mink lung cells and mixed mink lung and A549 cells for rapid detection of influenza virus and other respiratory viruses. J Clin Microbiol 38(1): 422- 423.
Khor R, McElroy LJ, Whittaker GR (2003) The ubiquitin-vacuolar protein sorting system is selectively required during entry of influenza virus into host cells. Traffic 4(12): 857- 868.
Kielian M, Rey FA (2006) Virus membrane-fusion proteins: more than one way to make a hairpin. Nat Rev Microbiol 4(1): 67-76.
Ludwig S (2006) Signaling and apoptosis in influenza virus-infected cells. In: Kawaoka Y, editor. Influenza Virology: Current Topics by Yoshihiro Kawaoka: Caister Academic Press, pp. 37-64.
Maeda T, Kawasaki K, Ohnishi S (1981) Interaction of influenza virus hemagglutinin with target membrane lipids is a key step in virus-induced hemolysis and fusion a pH 5.2. Proc Natl Acad Sci USA 78: 4133-4137.
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. (2002) The protein kinase complement of the human genome. Science 298(5600):1912-34.
Marsh M, Helenius A (1989) Virus entry into animal cells. Adv Virus Res 36: 107-151.
Martin K, Helenius A (1991) Nuclear transport of influenza virus ribonucleoproteins: the viral matrix protein (M1) promotes export and inhibits import. Cell 67: 117-130.
Matlin KS, H. Reggio, A. Helenius, and K. Simons (1982) Infectious entry pathway of influenza virus in a canine kidney cell line. J Cell Biol 91: 601-613.
Matrosovich M, Matrosovich T, Garten W, Klenk HD (2006) New low-viscosity overlay medium for viral plaque assays. Virol J 3(1): 63.
Matrosovich M, N.; Klenk, Hans-Dieter; Kawaoka, Yoshihiro (2006) Receptor specificity, host range, and pathogenicity of influenza viruses. In: Kawaoka Y, editor. Influenza Virology: Current Topics by Yoshihiro Kawaoka: Caister Academic Press, pp. 37-64.
Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk HD (2004a) Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc Natl Acad Sci U S A 101(13): 4620-4624.
Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk HD (2004b) Neuraminidase is important for the initiation of influenza virus infection in human airway epithelium. J Virol 78(22): 12665-12667.
O'Neill RE, Jaskunas R, Blobel G, Palese P, Moroianu J (1995) Nuclear import of influenza virus RNA can be mediated by viral nucleoprotein and transport factors required for protein import. J Biol Chem 270: 22701-22704.
Pleshka S, Ludwig S, Wolff T, Planz (2006) Anti-viral approaches against infleunza viruses. In: Bogner E, Holzenburg A, editors. Newconceptys of antiviral therapy. Dordrecht: Springer, pp. 115-188.
Rodriguez-Boulan E (1983) Polarized assembly of enveloped viruses from cultured epithelial cells. Methods in Enzymology 98: 487-501.
Rust MJ, Lakadamyali M, Zhang F, Zhuang X (2004) Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat Struct Mol Biol 11(6): 567- 573.
Sieczkarski SB, Whittaker GR (2002) Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J Virol 76(20): 10455-10464.
Sieczkarski SB, Brown HA, Whittaker GR (2003) Role of protein kinase C betall in influenza virus entry via late endosomes. J Virol 77(1): 460-469.
Skehel J, Wiley D (2000) Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69: 531 - 569.
Szoka, F. Jr., and D. Papahadjopoulos. 1978. Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation. Proc. Natl. Acad. Sci. USA. 75(9): 4194-8.
White J, Kartenbeck J, Helenius A (1982) Membrane fusion activity of Influenza virus. EMBO J 1:217-222.
Whittaker GRD, P. (2006) Entry and intracellular transport of influenza virus. In: Kawaoka Y, editor. Influenza Virology: Current Topics by Yoshihiro Kawaoka: Caister Academic Press, pp. 37-64.
Wurzer WJ, Planz O, Ehrhardt C, Giner M, Silberzahn T et al. (2003) Caspase 3 activation is essential for efficient influenza virus propagation. Embo J 22(11): 2717-2728.
Yoshimura A, Ohnishi S (1984) Uncoating of influenza virus in endosomes. Journal of Virology 51 (2): 497-504.
Bonazzi, M., S. Spano, G. Turacchio, C. Cericola, C. Valente, A. Colanzi, H.S. Kweon, V.W. Hsu, E.V. Polishchuck, R.S. Polishchuck, M. Sallese, T. Pulvirenti, D. Corda, and A. Luini. 2005. CtBP3/BARS drives membrane fission in dynamin-independent transport pathways. Nat Cell Biol. 7:570-80.
Charras, G.T., C.K. Hu, M. Coughlin, and T.J. Mitchison. 2006. Reassembly of contractile actin cortex in cell blebs. J Cell Biol. 175:477-90.
Chung, C.S., C.Y. Huang, and W. Chang. 2005. Vaccinia virus penetration requires cholesterol and results in specific viral envelope proteins associated with lipid rafts. J Virol. 79:1623-34.
Damke, H., T. Baba, D.E. Warnock, and S.L. Schmid. 1994. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J Cell Biol. 127:915-34.
De Roe, C., P.J. Courtoy, and P. Baudhuin. 1987. A model of protein-colloidal gold interactions. J Histochem Cytochem. 35:1191-8.
Dharmawardhane, S., A. Schurmann, M.A. Sells, J. Chernoff, S.L. Schmid, and G.M. Bokoch. 2000. Regulation of macropinocytosis by p21-activated kinase-1. Mol Biol Cell. 11:3341-52.
Falcone, S., E. Cocucci, P. Podini, T. Kirchhausen, E. Clementi, and J. Meldolesi. 2006. Macropinocytosis: regulated coordination of endocytic and exocytic membrane traffic events. J Cell Sci. 119:4758-69.
Felding-Habermann, B., B.M. Mueller, C.A. Romerdahl, and D.A. Cheresh. 1992. Involvement of integrin alpha V gene expression in human melanoma tumorigenicity. J Clin Invest. 89:2018-22.
Fishkind, D.J., L.G. Cao, and Y.L. Wang. 1991. Microinjection of the catalytic fragment of myosin light chain kinase into dividing cells: effects on mitosis and cytokinesis. J Cell Biol. 114:967-75.
Frens, G. 1973. Controlled nucleation for the regulation of particle size in monodisperse gold suspensions. Nature Phys Sci. 241:20-12.
Greber, U.F., P. Webster, J. Weber, and A. Helenius. 1996. The role of the adenovirus protease in virus entry into cells. EMBO J. 15:1766-1777.
Greber, U.F., M. Willetts, P. Webster, and A. Helenius. 1993. Stepwise dismantling of adenovirus 2 during entry into cells. Cell. 75:477-486.
Grimmer, S., B. van Deurs, and K. Sandvig. 2002. Membrane ruffling and macropinocytosis in A431 cells require cholesterol. J Cell Sci. 115:2953-62.
Hewlett, L.J., A.R. Prescott, and C. Watts. 1994. The coated pit and macropinocytic pathways serve distinct endosome populations. J Cell Biol. 124:689-703.
Hinrichsen, L., J. Harborth, L. Andrees, K. Weber, and E.J. Ungewickell. 2003. Effect of clathrin heavy chain- and alpha-adaptin-specific small inhibitory RNAs on endocytic accessory proteins and receptor trafficking in HeLa cells. J Biol Chem. 278:45160-70.
Horisberger, M., and J. Rosset. 1977. Colloidal gold, a useful marker for transmission and scanning electron microscopy. J Histochem Cytochem. 25:295-305.
Imelli, N., O. Meier, K. Boucke, S. Hemmi, and U.F. Greber. 2004. Cholesterol is required for endocytosis and endosomal escape of adenovirus type 2. J Virol. 78:3089-98.
Kirkham, M., and R.G. Parton. 2005. Clathrin-independent endocytosis: new insights into caveolae and non-caveolar lipid raft carriers. Biochim Biophys Acta. 1746:349-63.
Limouze, J., A.F. Straight, T. Mitchison, and J.R. Sellers. 2004. Specificity of blebbistatin, an inhibitor of myosin II. J Muscle Res Cell Motil. 25:337-41.
Locker, J.K., A. Kuehn, S. Schleich, G. Rutter, H. Hohenberg, R. Wepf, and G. Griffiths. 2000. Entry of the two infectious forms of vaccinia virus at the plasma membane is signaling-dependent for the IMV but not the EEV. Mol.Biol.Cell. 11:2497-2511.
Martin, S.J., C.P. Reutelingsperger, A.J. McGahon, J.A. Rader, R.C. van Schie, D.M. LaFace, and D.R. Green. 1995. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 182:1545-56.
Meier, O., K. Boucke, S. Vig, S. Keller, R.P. Stidwill, S. Hemmi, and U.F. Greber. 2002. Adenovirus triggers macropinocytosis and endosomal leakage together with its clathrin mediated uptake. J. Cell Biol. 158:1119-1131.
Mills, J.C., N.L. Stone, J. Erhardt, and R.N. Pittman. 1998. Apoptotic membrane blebbing is regulated by myosin light chain phosphorylation. J Cell Biol. 140:627-36.
Nagel, H., S. Maag, A. Tassis, F.O. Nestle, U.F. Greber, and S. Hemmi. 2003. The alphavbeta5 integrin of hematopoietic and nonhematopoietic cells is a transduction receptor of RGD-4C fiber-modified adenoviruses. Gene Ther. 10:1643-53.
Nakano, M.Y., K. Boucke, M. Suomalainen, R.P. Stidwill, and U.F. Greber. 2000. The first step of adenovirus type 2 disassembly occurs at the cell surface, independently of endocytosis and escape to the cytosol. J Virol. 74:7085-95.
Nakano, M.Y., and U.F. Greber. 2000. Quantitative microscopy of fluorescent adenovirus entry. J. Struct. Biol. 129:57-68.
Sirena, D., Z. Ruzsics, W. Schaffner, U.F. Greber, and S. Hemmi. 2005. The nucleotide sequence and a first generation gene transfer vector of species B human adenovirus serotype 3. Virology. 343:283-98.
Suomalainen, M., M.Y. Nakano, K. Boucke, S. Keller, R.P. Stidwill, and U.F. Greber. 1999. Microtubule-dependent minus and plus end-directed motilities are competing processes for nuclear targeting of adenovirus. J. Cell Biol. 144:657-672.
Townsley, A.C., A.S. Weisberg, T.R. Wagenaar, and B. Moss. 2006. Vaccinia virus entry into cells via a low-pH-dependent endosomal pathway. J Virol. 80:8899-908.
Veithen, A., P. Cupers, P. Baudhuin, and P.J. Courtoy. 1996. v-Src induces constitutive macropinocytosis in rat fibroblasts. J Cell Sci. 109 ( Pt 8):2005-12.
【特許請求の範囲】
【請求項1】
ポックスウイルス感染を治療する方法であって,治療を必要とする被検動物に,有効量のキナーゼ調節剤を投与することを含む方法。
【請求項2】
被検動物はヒトである,請求項1記載の方法。
【請求項3】
キナーゼ調節剤はPAK1;DYRK3;PTK9;およびGPRK2Lからなる群より選択されるキナーゼを調節する,請求項2記載の方法。
【請求項4】
前記キナーゼ調節剤は宿主細胞キナーゼ調節剤である,請求項1記載の方法。
【請求項5】
キナーゼ調節剤は,キナーゼを標的とするドミナント・ネガティブ分子,siRNA,shRNA,抗体または小分子である,請求項1記載の方法。
【請求項6】
キナーゼ調節剤はsiRNAである,請求項1記載の方法。
【請求項7】
前記宿主細胞キナーゼ調節剤は宿主細胞キナーゼ阻害剤である,請求項4記載の方法。
【請求項8】
前記宿主細胞キナーゼ阻害剤は,PAK1;DYRK3;PTK9;およびGPRK2Lからなる群より選択されるキナーゼの阻害剤である,請求項7記載の方法。
【請求項9】
キナーゼ調節剤はCEP−1347である,請求項1記載の方法。
【請求項10】
ポックスウイルスは天然痘ウイルスである,請求項1記載の方法。
【請求項11】
ポックスウイルスはワクシニアウイルスである,請求項1記載の方法。
【請求項12】
感染は呼吸器感染である,請求項1記載の方法。
【請求項13】
ウイルス感染を治療する方法であって,治療を必要とする被検動物に有効量のマクロピノサイトーシス経路の調節剤を投与することを含む方法。
【請求項14】
被検動物はヒトである,請求項13記載の方法。
【請求項15】
マクロピノサイトーシス経路の前記調節剤は,前記マクロピノサイトーシス経路の阻害剤である,請求項13記載の方法。
【請求項16】
前記阻害剤はキナーゼ阻害剤である,請求項15記載の方法。
【請求項17】
前記阻害剤は宿主細胞キナーゼ阻害剤である,請求項15記載の方法。
【請求項18】
前記宿主細胞キナーゼ阻害剤は,PAK1;DYRK3;PTK9;およびGPRK2Lからなる群より選択されるキナーゼの阻害剤である,請求項17記載の方法。
【請求項19】
阻害剤はCEP−1347である,請求項18記載の方法。
【請求項20】
前記ウイルスはポックスウイルスである,請求項13記載の方法。
【請求項21】
前記ウイルスは天然痘ウイルスである,請求項13記載の方法。
【請求項22】
前記ウイルスはワクシニアウイルスである,請求項13記載の方法。
【請求項23】
a) 細胞をキナーゼ阻害剤およびウイルスと接触させ,そして
b) キナーゼ阻害剤がウイルスによる細胞の感染を阻害するか否かを判定する,
ことを含む方法。
【請求項24】
ウイルスはポックスウイルスである,請求項23記載の方法。
【請求項25】
ウイルスはインフルエンザウイルスである,請求項23記載の方法。
【請求項26】
ウイルスはワクシニアまたは天然痘である,請求項23記載の方法。
【請求項27】
キナーゼ阻害剤は,PAK1;DYRK3;PTK9;およびGPRK2Lからなる群より選択されるキナーゼを阻害する,請求項23記載の方法。
【請求項28】
キナーゼ阻害剤は,キナーゼを標的とするドミナント・ネガティブ分子,siRNA,shRNA,抗体または小分子からなる群より選択される,請求項23記載の方法。
【請求項29】
接触はインビトロで行われる,請求項23記載の方法。
【請求項1】
ポックスウイルス感染を治療する方法であって,治療を必要とする被検動物に,有効量のキナーゼ調節剤を投与することを含む方法。
【請求項2】
被検動物はヒトである,請求項1記載の方法。
【請求項3】
キナーゼ調節剤はPAK1;DYRK3;PTK9;およびGPRK2Lからなる群より選択されるキナーゼを調節する,請求項2記載の方法。
【請求項4】
前記キナーゼ調節剤は宿主細胞キナーゼ調節剤である,請求項1記載の方法。
【請求項5】
キナーゼ調節剤は,キナーゼを標的とするドミナント・ネガティブ分子,siRNA,shRNA,抗体または小分子である,請求項1記載の方法。
【請求項6】
キナーゼ調節剤はsiRNAである,請求項1記載の方法。
【請求項7】
前記宿主細胞キナーゼ調節剤は宿主細胞キナーゼ阻害剤である,請求項4記載の方法。
【請求項8】
前記宿主細胞キナーゼ阻害剤は,PAK1;DYRK3;PTK9;およびGPRK2Lからなる群より選択されるキナーゼの阻害剤である,請求項7記載の方法。
【請求項9】
キナーゼ調節剤はCEP−1347である,請求項1記載の方法。
【請求項10】
ポックスウイルスは天然痘ウイルスである,請求項1記載の方法。
【請求項11】
ポックスウイルスはワクシニアウイルスである,請求項1記載の方法。
【請求項12】
感染は呼吸器感染である,請求項1記載の方法。
【請求項13】
ウイルス感染を治療する方法であって,治療を必要とする被検動物に有効量のマクロピノサイトーシス経路の調節剤を投与することを含む方法。
【請求項14】
被検動物はヒトである,請求項13記載の方法。
【請求項15】
マクロピノサイトーシス経路の前記調節剤は,前記マクロピノサイトーシス経路の阻害剤である,請求項13記載の方法。
【請求項16】
前記阻害剤はキナーゼ阻害剤である,請求項15記載の方法。
【請求項17】
前記阻害剤は宿主細胞キナーゼ阻害剤である,請求項15記載の方法。
【請求項18】
前記宿主細胞キナーゼ阻害剤は,PAK1;DYRK3;PTK9;およびGPRK2Lからなる群より選択されるキナーゼの阻害剤である,請求項17記載の方法。
【請求項19】
阻害剤はCEP−1347である,請求項18記載の方法。
【請求項20】
前記ウイルスはポックスウイルスである,請求項13記載の方法。
【請求項21】
前記ウイルスは天然痘ウイルスである,請求項13記載の方法。
【請求項22】
前記ウイルスはワクシニアウイルスである,請求項13記載の方法。
【請求項23】
a) 細胞をキナーゼ阻害剤およびウイルスと接触させ,そして
b) キナーゼ阻害剤がウイルスによる細胞の感染を阻害するか否かを判定する,
ことを含む方法。
【請求項24】
ウイルスはポックスウイルスである,請求項23記載の方法。
【請求項25】
ウイルスはインフルエンザウイルスである,請求項23記載の方法。
【請求項26】
ウイルスはワクシニアまたは天然痘である,請求項23記載の方法。
【請求項27】
キナーゼ阻害剤は,PAK1;DYRK3;PTK9;およびGPRK2Lからなる群より選択されるキナーゼを阻害する,請求項23記載の方法。
【請求項28】
キナーゼ阻害剤は,キナーゼを標的とするドミナント・ネガティブ分子,siRNA,shRNA,抗体または小分子からなる群より選択される,請求項23記載の方法。
【請求項29】
接触はインビトロで行われる,請求項23記載の方法。
【図1】

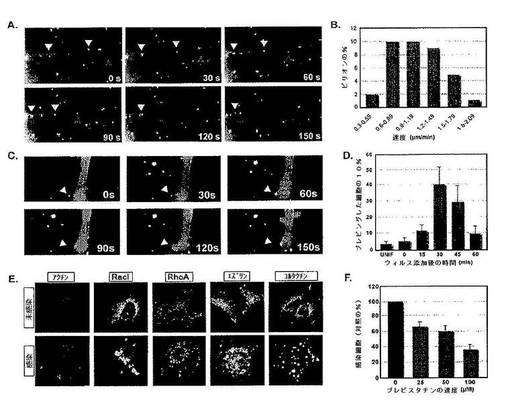
【図2】
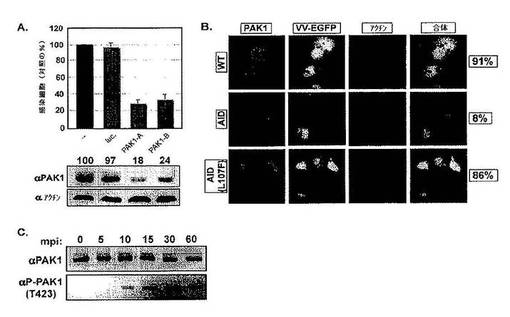
【図3】
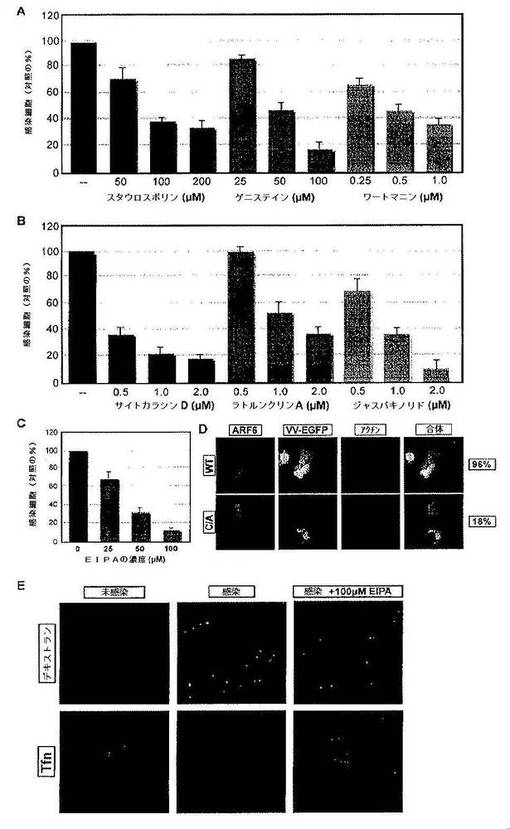
【図4】
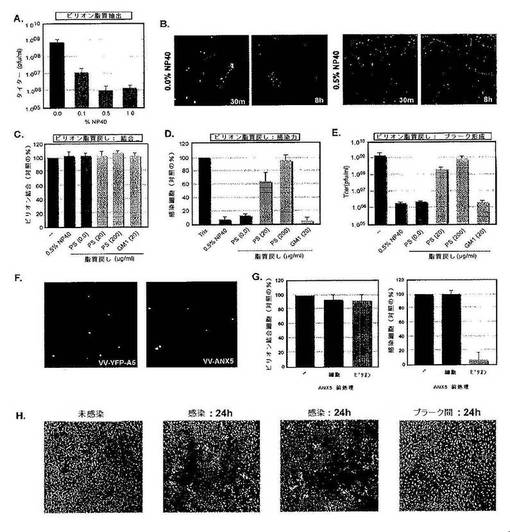
【図5】
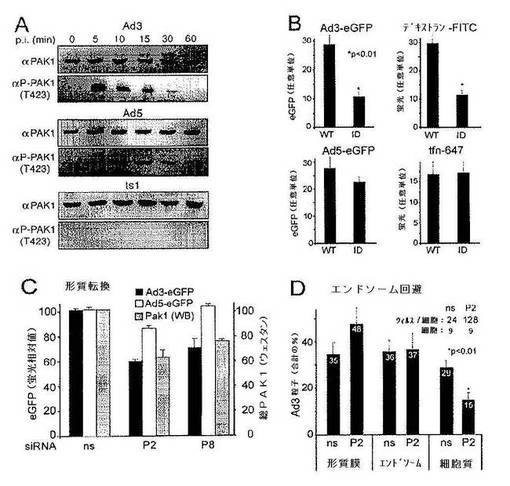
【図6】

【図7】
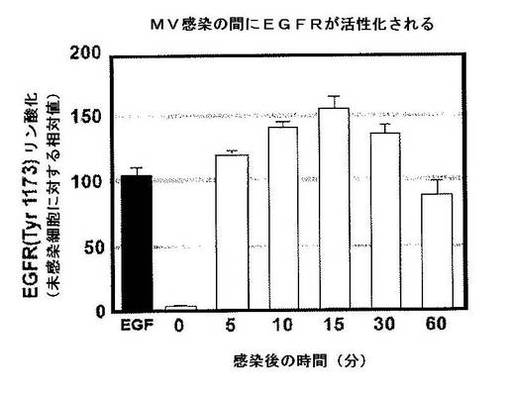
【図8】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2010−530870(P2010−530870A)
【公表日】平成22年9月16日(2010.9.16)
【国際特許分類】
【出願番号】特願2010−512811(P2010−512811)
【出願日】平成20年6月20日(2008.6.20)
【国際出願番号】PCT/IB2008/002644
【国際公開番号】WO2009/001224
【国際公開日】平成20年12月31日(2008.12.31)
【出願人】(509351203)
【Fターム(参考)】
【公表日】平成22年9月16日(2010.9.16)
【国際特許分類】
【出願日】平成20年6月20日(2008.6.20)
【国際出願番号】PCT/IB2008/002644
【国際公開番号】WO2009/001224
【国際公開日】平成20年12月31日(2008.12.31)
【出願人】(509351203)
【Fターム(参考)】
[ Back to top ]