
抗ヒスタミン剤と鎮咳剤と充血緩和剤とを徐放性製剤中に含む組成物
本発明は、風邪及びアレルギー兆候の治療のための経口製剤を提供する。各製剤は、抗ヒスタミン剤、鎮咳剤及び/又は充血緩和剤を1つの徐放性組成物にまとめたものである。本発明は、更にこれらの製剤の製造及び使用方法を提供すると同時に、抗ヒスタミン剤、鎮咳剤及び/又は充血緩和剤のうちの2種以上を含む経口徐放性組成物に含まれる単一の薬剤の乱用又は抽出を防ぐ方法を提供する。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
(関連出願の相互参照)
本願は、2009年5月1日に出願された米国特許仮出願第61/174,891号の優先権を主張するものであり、引用によりその内容すべてを本願明細書に援用する。
【0002】
(発明の背景)
年間で米国人がひく風邪は推定10億件、大人が年間で2〜4回、子どもは年間6〜10回風邪をひいている(国立アレルギー感染症研究所(NIAID)のファクトシート「The Common Cold(普通の風邪について)」www.niaid.nih.gov/factsheets/cold.htmにて閲覧可能、2004年12月)。米国人10人のうち約9人が風邪又は同様な上気道感染症に毎年少なくとも1回かかることになる。風邪は子供達のなかでは最も流行する病気であり、いずれの複合的な疾患よりも多発し、学校での子供の欠席理由全体の50%も占める(Micromedex Healthcare Series、「The Common Cold Etiology and Treatment(一般の風邪の原因と治療)」www.thomsonhc. com/hcs/librarian/NDで閲覧可能、2008年6月18日アクセス)。風邪が最もはやるのは、8月又は9月初旬から3月又は4月に及ぶ気温が低い数ヵ月の間である。大気温度の低下に伴い湿度も低くなりがちであり、室内や狭い部屋の中での人と人との交流が増えることから、風邪の原因となるウィルスの増殖を増長するようである(国立アレルギー感染症研究所(NIAID)のファクトシート「普通の風邪について」www.niaid.nih.gov/factsheets/cold.htmで閲覧可能、2004年12月アクセス)
【0003】
普通の風邪をひいた患者は、鼻水、鼻ずまり、鼻腔膜の腫れ、くしゃみ、のどの痛み、咳及び頭痛といった症状や徴候を示すのが一般的である(Micromedex Healthcare Series、「一般の風邪の原因と治療」(同サイト))。患者の70〜90%が鼻漏又はくしゃみを患い、65%が鼻づまり又はうっ血を起こし、25%が咳又は声かれをおこしている(同サイトより)。ほとんどの風邪が7〜14日間続く。2週間を超えて徴候が続く患者はアレルギーになっている可能性もある(同サイト)。
米国肺協会(American Lung Association)によると、風邪で病院に訪れる割合は、他のいずれの疾病の場合よりも多い(米国肺協会「Cold and Flu Guidelines: The Common Cold(風邪及び流感のガイドライン:普通の風邪)」www.lungusa.org.で閲覧可能、2008年6月16日アクセス)。実際のところ、普通の風邪による影響についての研究では、2003年だけで風邪に関連して病院をおとずれた件数は1億件を超え、費用は77億ドルであったことが示されている。また、この研究の概算によると、子どもたちが学校を休んだ日数は毎年1億8900万日となり、風邪の子どもを看病するために親は1億2600万日仕事を休んでいる(WebMD. 「Cost of the Common Cold: $40 Billion(普通の風邪によるコスト:400億ドル)」 www.medmutual.comにて閲覧可能、2003年2月24日アクセス;Fendrick等「The Economic Burden of Non-Influenza-Related Viral Respiratory Tract Infection in the United States.(米国における非インフルエンザ系ウィルスによる気道感染の経済的負荷)」Arch Intern Med. 163(4): 487〜494頁(2003))。風邪をひいた従業員が就労を休んだ1億5000万日に加えると、風邪に起因した労働損失の経済的影響は全部で200億ドルを超える(Fendrick(同書);Garibaldi RA,「Epidemiology of community-acquired respiratory tract infections in adults. Incidence, etiology, and impact(コミュニティーを襲う大人の気道感染についての流行病学。発生、病因及び影響)」 Am.J. Med. 78 (6B): 32〜37頁(1985);「普通の風邪について」国立アレルギー感染症研究所、www3.niaid.nih.gov/healthscience/healthtopics/colds/ 2008年6月11日検索;米国勢調査局www.Quickfacts.census.gov/qfd/states/00000.html2004年の概算)。
【0004】
米国において、咳や風邪の緩和を目的とする薬局の薬(OTC)及び処方箋による薬にかかった費用は、毎年30億ドルを超えている(WebMD(同サイト)、Fendrick(同書))。米国だけでも、普通の風邪で医師にかかる件数は年間で7,500万〜1億件になり、費用は控えめに見積もっても毎年77億ドルになる。米国人は徴候の緩和のために薬局の薬に29億ドル、さらに処方箋による薬に4億ドルを使っている(Fendrick(同書)、 Garibaldi(同書))。医者にかかった患者の3分の1を超える患者が抗生物質の処方箋を受け取っているが、これは不要な出費になるだけでなく(米国内では概算で4100万件の抗生物質の処方箋に対し年間11億ドルが支払われている)、このような薬剤の乱用にが抗生物質耐性を意味することにもなる(Fendrick(同書))。IMS Health社によると(2007年8月)、咳、風邪及び流感のための処方箋の数は年間でおよそ42,858,000件であった。すべての風邪の兆候を唯一の有効医薬成分(API)では治せないことから、単一成分の医薬品を多数使用するよりも、調合医薬品の方が、兆候緩和をもたらすのにより便利で、時にはより安価に提供できることが多い。
【0005】
本発明は、即放性及び/又は現在使用できる風邪やアレルギーの調合薬に比べて独特な長所を提供する。例えば、本発明の1つの実施形態は、3種類のAPI、即ち、抗ヒスタミン剤(例えばクロルフェニラミン)と鎮咳剤(例えばヒドロコドン)と充血緩和剤(例えばプソイドエフェドリン)の長所が、1つの徐放性(ER、例えば12時間)組成物中に統合されている。今まで、これらのAPIを一緒に使用する場合は、それぞれの即放性(IR)の剤形で1日に4〜6回の投与が必要であった。なぜならば、これらの3つのAPIの効果を示す単一の徐放性調合医薬品が現在は手に入らないからである。
上記APIの1種又は2種を含む徐放性医薬品は既に市場に存在する。同様に、3種類すべての薬剤を含む即放性医薬品もいくつかが市場に存在する。例えば、クロルフェニラミン(CPM)、ヒドロコドン(HC)及び/又はプソイドエフェドリン(PSE)を含む、即放性(IR)又は徐放性(ER)剤形の市場に出ているOTC又は処方箋薬としては、以下のものが挙げられる。
【0006】
プソイドエフェドリンを含む医薬品
Afrinol(登録商標)
Cenafed(登録商標)
D-イソエフェドリン
D-プソイドエフェドリン
Decofed(登録商標)
Dimetapp(登録商標) 充血緩和剤
Dimetapp(登録商標) 充血緩和剤、小児科用点滴薬
Drixoral(登録商標) 鼻の充血緩和剤
Efidac 24(登録商標)プソイドエフェドリン HCl
Eltor 120(登録商標)
Genaphed(登録商標)
イソエフェドリン
【0007】
Maxenal(登録商標)
Myfedrine(登録商標)
Novafed(登録商標)
Pedia Care(登録商標)
Pseudo 60's(登録商標)
Pseudo-12(登録商標)
プソイドエフェドリン塩酸塩
Sudafed 12 Hour(登録商標)
Sudafed 24 Hour(登録商標)
Sudogest
【0008】
クロルフェニラミンを含む医薬品
Aller-Chlor (登録商標)
Antagonate(登録商標)
Chlo-Amine(登録商標)
Chlor-Trimeton(登録商標)
Chlor-Tripolon(登録商標)
デキスクロルフェニラミンマレイン酸塩
Efidac 24(登録商標) クロルフェニラミンマレイン酸塩
Gen-Allerate(登録商標)
Haynon(登録商標)
Histadur(登録商標)
【0009】
Kloromin(登録商標)
Mylaramine(登録商標)
Novo-Pheniram(登録商標)
Phenetron(登録商標)
Piriton(登録商標)
Polaramine(登録商標)
Pyridamal 100(登録商標)
Telachlor(登録商標)
Teldrin(登録商標)
【0010】
ヒドロコドンを含む医薬品
Hycodan(登録商標) (臭化メチルホマトロピンを含む)
Lortab(登録商標) (アセトアミノフェンを含む)
Maxidone(登録商標) (アセトアミノフェンを含む)
Norco(登録商標) (アセトアミノフェンを含む)
Vicodin(登録商標) (アセトアミノフェンを含む)
Zydone(登録商標) (アセトアミノフェンを含む)
プソイドエフェドリンとクロルフェニラミンとを含む医薬品
Allerest(登録商標)
Anamine(登録商標)
Biohist-LA(登録商標)
Brexin(登録商標)
【0011】
Chlordrine(登録商標) SR
Chlor-Phed(登録商標)
Chlor-Trimeton(登録商標) (4時間及び12時間)
Deconamine(登録商標)
De-congestine TR(登録商標)
Dynahist ER(登録商標)
Histalet(登録商標)Syrup
Kronofed-A(登録商標) Kronocaps
Kronofed-A-Jr.(登録商標) Kronocaps
【0012】
ND(登録商標)Clear
Pseudoephed/Chlorphen 100(登録商標)
Rescon(登録商標)
Rescon(登録商標)Jr.
Rescon(登録商標)ED
Ryna(登録商標)
Sudafed(登録商標)Cold and Allergy
Tanafed(登録商標)
クロルフェニラミンとヒドロコドンとを含む医薬品
Tussionex(登録商標)Pennkinetic(登録商標)徐放性懸濁液
TussiCaps(登録商標)徐放性カプセル剤
S-T Forte(登録商標)2
【0013】
プソイドエフェドリンとヒドロコドンとを含む医薬品
Detussin(登録商標)Liquid
Histussin(登録商標)D Liquid
Tyrodone(登録商標)Liquid
クロルフェニラミンとプソイドエフェドリンとヒドロコドンとを含む医薬品
A-G Tussin(登録商標)
Atuss(登録商標)HD
Cordron-HC(登録商標)
Hexatussin(登録商標)
Histinex(登録商標)PV
Hydrocof-HC(登録商標)
Hydron(登録商標)PCS
Hydrotuss(登録商標)HC
Hyphed(登録商標)
【0014】
KG-Tussin(登録商標)
M-End(登録商標)
Notuss(登録商標)
P-V-Tussin(登録商標)Syrup
Pediatex(登録商標)HC
Q-V Tussin(登録商標)
Tussin-V(登録商標)
上記の3つの効果を示す製剤(CPMとPSEとHCとを同一医薬品に含む)は、いずれも3種成分すべてについての徐放性医薬品ではない。また、3種類すべての有効成分を含む経口組成物を患者に単回投与することにより、3種薬剤の血清レベルが12時間にわたり、該有効成分を含むFDAに承認された即放性の関連リスト収載医薬品(reference listed drug:RLD)組成物を12時間にわたって適切な投与回数で投与した際に得られる血清レベルと生物学的等価となるようにしたグループはいまだに構築されていない。
【0015】
同様に、ヒドロコドンとプソイドエフェドリンとを含む前記製剤(CMP等の他の有効成分を有していてもいなくてもよい)はいずれも徐放性医薬品ではない。ヒドロコドンとプソイドエフェドリンとを含む経口組成物の単回投与で、2種の血清レベルを12時間にわたり、ヒドロコドン及び/又はプソイドエフェドリン含有のFDAに承認された即放性RLD組成物を12時間にわたって適切な投与回数で投与した際に得られる血清レベルと生物学的等価となるようにしたグループはいまだに構築されていない。実際のところ、ヒドロコドンとプソイドエフェドリンの両方を含み(他の有効成分を有していてもいなくてもよい)、即放性又は徐放性である医薬品は、現在のところFDAに承認されているものはない。前記に示した市販医薬品リストは、FDA未承認であるが市場に出ている医薬品が挙げられていることを記しておかなければならない。
【0016】
本発明の別の実施形態に関連するが、OTC医薬品及び処方箋医薬品に含まれる薬剤の転用や乱用が近年段階的に拡大している。例えば、OTCの風邪及びアレルギー用錠剤の多くはプソイドエフェドリン又はエフェドリンを含むが、これを使ってメタンフェタミンとして知られる乱用薬物が秘密裏に製造されている。この薬物は「メス」「スピード」「クランク」又は「アイス」としても知られているが、中枢神経系をおかす強力で中毒性の覚醒剤である。メタンフェタミンは、ふかしたり吸い込んだり、注射したり飲み込めるように丸剤、カプセル剤、散剤にして違法で販売されている。
にわか作りの秘密の違法薬物研究所(meth labs)では、OCTの風邪やアレルギー用錠剤からプソイドエフェドリン又はエフェドリンが分離するまで、水、アルコール又は他の溶媒の溶液を使って数時間でプソイドエフェドリン又はエフェドリンを単離させる。その後、容易に手に入る一般的な家庭用品や簡単に査定できる(assessable)装置、即ち、アルコール、コールマンのホワイトガソリン、アセトン、道路用発煙筒(road flare)、排水溝クリーナー、ヨウ素、塩酸、岩塩、(エンジンなどの)点火燃料、コーヒーフィルター及びマッチ等を使って、プソイドエフェドリン又はエフェドリンをメタンフェタミンに変換する。薬物の製造方法は、インターネットを介して容易に入手できる「レシピ」や情報交換から入手できる。
米国ではメタンフェタミンの非合法取引や乱用が高まりつつあるが、プソイドエフェドリンを盗んだり転換することを防ぐ方策は厄介で費用がかかる。
【0017】
オピオイド類等の薬物を含む他の多くのOCT医薬品及び処方箋医薬品も、非合法的薬物乱用に流用する絶好のものである。例えば、ヒドロコドンは、風邪薬の鎮咳剤(咳抑制剤)として、また、処方箋薬に含まれる中程度から僅かに重篤な痛みを治療するための鎮痛剤として合法的に使用されている。ヒドロコドンは米国では最も多く処方されるアヘン含有鎮静剤で、2003年にはヒドロコドン含有薬品の1億1000万件を超える処方箋が調剤された。一般的にヒドロコドンの製造は非合法ではないが、目下、非合法的な転用や、病的高揚状態を得て苦痛緩和の効果を得るための直接的な乱用も見られる。偽のクリニックからの電話による処方箋、改竄した処方箋、窃盗、インターネットからの不法な購入を介して広範な転用が起こっている。
このように、不法な使用を目的とした薬剤の乱用や転用の可能性を回避できるOTC医薬品及び処方箋薬の調製及び販売へのニーズがある。例えば、にわか作りの研究所ではプソイドエフェドリン、エフェドリン及び/又はヒドロコドン等の薬剤が容易に抽出、分離できないような、風邪/咳及びアレルギー製剤へのニーズがある。同様に、非合法的研究所で秘密裏に製造されるかどうかには拘わらず、不正な目的のために容易に転用できない風邪/咳、痛みの緩和、筋肉弛緩の医薬品へのニーズが存在する。
【0018】
(発明の概要)
本発明の実施形態により、以下の有効成分(1)抗ヒスタミン剤、(2)鎮咳剤及び(3)充血緩和剤の1種以上を含む即放性(IR)製剤及び/又は調合医薬品のこれまでに関わる問題点や不利な点が解決される。現在市販されている多くの医薬品に対して、本発明は、3種すべての有効成分を徐放(ER)(例えば12時間)できる製剤を提供する。一例として、本発明は、クロルフェニラミン(抗ヒスタミン剤)、ヒドロコドン(麻酔性鎮咳剤)、プソイドエフェドリン(充血緩和剤)のIR剤形とER剤形との新規混合物を一つの医薬品にした経口製剤を提供する。結果的にこの製剤は1日2回投与が可能なER型調合品となり、以前から入手可能であったIR型(個々の成分ごと又は調合品を介してのいずれかで販売及び投与されてきた)と同じ効力を有する。また、この製剤は現存する単一及び調合のER製剤よりも以下の点で優れている。(a)即時及び長時間の薬剤伝達のためにIR及びER両方の薬剤投薬量を提供する。(b)3種の関連リスト収載医薬品(RLD)と生物学的等価な投薬量を単回投与形態で提供できる。(c)成分薬物の乱用及び転用を阻止する。
【0019】
本発明の実施形態の1つでは、充血緩和剤、鎮咳剤及び/又は抗ヒスタミン剤という有効成分を含む経口医薬組成物を患者へ単回投与することにより、上記有効成分を含有するFDA承認済みの即放性(IR)RLDを12時間にわたって適切な投与回数で投薬する際に得られる血清レベルと生物学的等価である有効成分の血清レベルを12時間にわたり得られる。即放性RLDの適切な投与回数とは、即放性RLDの12時間にわたる投与に関してFDAが承認している表示1つ以上で推奨される投与回数に相当する。
別の実施形態では、24時間を超える投薬時間にわたり有効成分の安定した血清レベルを得るため、充血緩和剤、鎮咳剤及び/又は抗ヒスタミン剤という有効成分を含む徐放性経口組成物を十分な投与回数で患者へ投薬することにより、上記有効成分を含有するFDA承認済み即放性医薬組成物1種以上を上記と同じ時間にわたり適切な投薬回数で投与した際に得られる血清レベルと生物学的等価である有効成分の血清レベルが得られる。即放性医薬品の適切な投与回数とは、FDAが承認する即放性医薬品1種以上の同じ時間にわたる投与について、FDAが承認する表示1つ以上で推奨する投与回数に相当する。更に本発明の別の実施形態は、ヒドロコドンとプソイドエフェドリンとを含む経口製剤であって、抗ヒスタミン剤を含むか否かにかかわらず上記有効成分が人体内で徐放される経口製剤を提供する。
【0020】
実施形態の1つでは、本発明製剤の徐放性(ER)部位が、懸濁液中の被覆ビーズ、粒子又はペレットの形態で存在することができ、即放性(IR)部位が懸濁液中に存在することができる。或いは、徐放性部位はカプセル剤、錠剤又は他の経口固形物といった固形の剤形を有し、即放性部位が前記徐放性部位の外側の第2の層又は媒体とすることもできる。実施形態の1つでは、12時間ごとに投与するように製剤化する。他の実施形態は、8時間ごと、16時間ごと、24時間ごと等といった投与用に製剤化されたものを含む。
同様に、別の実施形態においては、一つの徐放性調合医薬品が特定のIR/ER比を示し、徐放(ER)成分は粒子、ペレット又はビーズ内に存在し、即放(IR)部位が前記粒子、ペレット又はビーズの外側に存在する(例えば、シロップ剤中に懸濁、カプセル剤、錠剤等に含まれる散剤)。上記比率により、単回投与でも安定状態のいずれの条件においても、関連リスト収載医薬品(RLD)に対して生物学的等価(BE)な血清レベルが得られる。他の実施形態では、本発明の製剤を用いたヒトは、ある特定の血清レベル範囲(AUC、Tmax、T1/2等で測定)を経時的に得るが、該血清レベルは、単回投与でも安定状態のいずれの条件においても関連リスト収載医薬品(RLD)と生物学的等価(BE)である。
本発明の実施形態の1つは、抗ヒスタミン剤と鎮咳剤とを含み、任意で充血緩和剤を含んでいてもよい有効成分を即放剤形で含有する第1部位と、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを有効成分として徐放剤形で含む粒子、ペレット又はビーズを有する第2部位とを含有する、経口徐放性医薬組成物の製造方法に関する。別の実施形態では、本発明の方法は、即放性部位が最初に抗ヒスタミン剤と鎮咳剤とを含むが、充血緩和剤を含まず、徐放性部位が抗ヒスタミン剤と鎮咳剤とプソイドエフェドリンとを含むようにして組成物を製造する工程を有する方法である。関連の実施形態では、プソイドエフェドリン等の徐放性成分の1種以上を即放性ビヒクルに浸出させて、1種以上の成分による即放剤形としてもよい。
【0021】
また別の実施形態では、本発明の製剤は、1個のビーズ内に三種類すべての薬剤を含有する粒子を提供する。このような実施形態は、各薬剤をそれぞれ別のビーズで提供する既存の徐放性製剤に比べ、用量有効性、安全性及び転用や乱用に対する阻止力というすべての点において有利性を示す。
本発明の別の実施形態では、プソイドエフェドリン(又はエフェドリン等の化学的関連のある充血緩和剤)及び/又は麻酔性鎮咳剤(ヒドロコドン等)を含む徐放性粒子、ペレット又はビーズが、これらの薬剤を1種以上含む他の市販のOTC医薬品や処方箋医薬品と比較して、該薬剤の誤用、乱用又は不法な転用が出来ないように又はやりにくくするようにして、製剤に含まれる。例えば、本発明の製剤中の1個1個の徐放性粒子又はペレット又はビーズに、クロルフェニラミンやヒドロコドンといった抗ヒスタミン剤や鎮咳剤をはじめとする化合物と共にプソイドエフェドリン(又は関連化合物)が含有されていれば、違法薬物(meth)の製造や麻薬の単離を目的としたPSE(又はエフェドリン)及び/又は麻薬性鎮咳剤の最終製剤からの分離又は抽出を阻止、低減する。
【0022】
PSE、エフェドリン及び/又は麻薬を本発明の製剤から(例えばビーズから)抽出することは技術的には可能であるかもしれないが、抽出には複雑で高価な装置と技術が必要になる。違法薬物を不正に製造する者たちのほとんどは、そうまでして抽出作業をする装置及び/又はリソースには近付かない。特に、これらの薬剤を含有する他のOTC医薬品を用いて簡単にできることに比べればなおさらである。このように、PSE(もしくは関連化合物)又は麻薬性鎮咳剤を含有する市販のOTC製剤の多くとは異なり、更に自由に入手できる本発明製剤のOTC用にした医薬品は小売業者が販売することができる。
そこで本発明の実施形態は、現在市場にでている即放性及び/又は調合医薬品と比較すると特別な利点をもたらす。その利点は下記が挙げられる。(1)4時間から6時間ごとではなく1日2回の投与となることで、患者のコンプライアンスを向上させ、正しい用量の薬剤が伝達される。(2)処方箋が必要な咳や風邪の薬にプソイドエフェドリンの使用を導入すれば、免許をもった医学専門家を必ず巻き込むため、プソイドエフェドリンの使用は、例えば他の薬と一緒に投与する場合など、より適切な使用につながる。(3)懸濁液は錠剤よりも投与の選択肢が一層融通がきくため、個別化した患者の治療を斟酌し、兆候をもとに治療のバランスを調整することができる。(4)製剤の徐放性部位における有効医薬成分(API)の処理方法により、ヒドロコドン及び/又はプソイドエフェドリンが乱用及び/又は転用される可能性を削減する。(5)使用単位(4オンス)又は単位服用量(5〜10ml)を用いることにより、追跡がより簡単になり、転用の可能性も減少させることができる。
【0023】
更に本発明は、1種以上の薬剤を含むペレット、ビーズ又は粒子を有する徐放性成分を含み、該ペレット、ビーズ又は粒子がシロップ剤に懸濁している、新規な経口懸濁液製剤を提供する。前記シロップ剤も1種以上の薬剤を含んでいてもよい。当該分野では、経口懸濁液は他の液体製剤よりも安定性に優れる。
本発明は、製造工程中、ビーズの中に可溶性の非電解質成分を含有させることを想定している。そのような成分は、水がビーズ内に吸収されると溶けて、リザーバーから拡散し浸透圧を下げるため、ビーズの膨潤を抑えビーズ被膜の状態が損なわれるのを抑える。したがって、本発明は、薬剤充填ビーズが水を吸収する際に大量移動が可能である賦形剤の使用を包含するもので、それにより、ビーズ内部の圧力を低下させ、ビーズ被膜の崩壊を防ぎ薬剤放出の制御を容易にする。
このように、本発明は、水和性が高く水と強力に結合することができる不活性成分を高濃度に添加し、分散相である薬剤複合体と水との結合を妨害することにより、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明組成物の分散媒中における水分活性(water activity)を低下させることも包含する。このように、分散媒中の水は薬剤充填ビーズにそれほど十分には引きつけられないため、投与の前に薬剤が溶解することを抑えられ、薬剤は懸濁液の分散相中に閉じ込められる。
【0024】
したがって、本発明の実施形態の1つでは、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の剤形として、患者への投与時に持続的薬物放出を行うことができる熱力学的に安定した液体剤形の薬剤懸濁液を包含する。このような液体製剤は12、24時間にわたって48時間まで持続的放出が可能である。このように、本発明は、1日1回又は2回の投与しか必要でない、投与を確実に容易にする液体剤形を包含する。本発明では、可溶性の非電解質成分で比較的低分子量の成分が、薬剤複合体に含まれていてもよい。実施形態の1つは、薬剤-イオン交換マトリックス-複合体が1個のビーズである。また、本発明では、多孔質又は非多孔質の高分子膜で分散相が被覆されていてもよいことが考えられる。有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の実施形態の1つでは、分散媒中の水としっかりと結合することができる高度に水和化された賦形剤を分散媒にも含有することにより、分散媒中の水分活性が抑えられ、水が薬剤充填ビーズに引きつけられるのを最小限に抑え、投薬の前に薬剤が溶けないようにする。このような実施形態では、患者に投与されてから薬剤放出が開始される。例えば胃液又は膓液中という、水が高濃度に存在し、薬剤と同じ電荷を帯びた小さなイオンが存在する環境に懸濁液が配置された時、その小さなイオンにより拡散した二重の層は浸水し、薬剤の放出が引き起こされる。特定の実施形態において、上記剤形を投与した後、分散媒が患者の胃液によって希釈され、膜の水和化により膜はより一層多孔になり、ビーズからの薬剤の溶解と分散が可能になる。
【0025】
分散相は3種の薬剤又は有効成分を含む。本発明による液状の放出制御組成物において、その分散相は有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる。また、これらの薬剤が1個1個の粒子、ペレット又はビーズの中にまとまっていることも想定される。実施形態の1つは、これらの薬物が、薬物と反対の表面電荷を有する医薬的に許容できるイオン交換マトリックスと結合している。実施形態の1つでは、これらの薬物が、薬物の電荷と反対の表面電荷を有する同一の医薬的に許容できる イオン交換マトリックスと結合している。
実施形態の1つでは、薬剤又は有効成分が塩の形態で分散相に含まれる。別の実施形態では、イオン交換マトリックスが塩の形態で分散相に含まれる。ある実施形態では、1種以上の薬剤及び/又はイオン交換マトリックスが医薬的に許容できる塩の形態で分散相に含まれる。
【0026】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物に関する実施形態の1つでは、患者に投与する前は、分散相中の1種以上の薬剤の分散媒への放出速度は非常に遅く、分散媒及び分散相に存在する薬剤の総モル量を基準として5%未満である。本発明は、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる液状の放出制御組成物であって、患者への投与前に分散相から分散媒に放出される薬剤の量が、分散媒及び分散相に存在する薬剤の総モル量を基準として30%未満、25%未満、20%未満、15%未満、10%未満、5%未満、0.5%未満又は0.05%未満である組成物を想定している。こうした実施形態では、分散媒が1年を超えて、約2年を超えて、約3年を超えて、又は、約4年を超えて物理的にも化学的にも安定している。有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物に関する実施形態の1つでは、遊離な薬剤は実質的に分散媒に含まれない。さらに本発明の別の実施形態では、患者への投薬前に、分散相中の1種以上の薬剤が分散媒に放出されるもので、分散媒及び分散相中の薬剤の総モル量を基準に約10%、15%、20%、25%、30%、35%、40%、45%又は50%の薬剤が放出される。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物は、薬剤が分散媒中に存在することで、即ち、イオン交換マトリックスに結合していない薬剤が存在することで、安定性が保たれる。さらに、本発明のこのような組成物は、分散媒中の遊離薬剤の存在下、分散相から薬剤が適切に放出され続ける。本発明のこの組成物の別の実施形態では、塩の形態の薬剤が分散媒中に含有される。
【0027】
本発明の組成物は室温条件下で少なくとも1年、約2年、及び/又は、3年、4年又は5年の貯蔵寿命を有し、その間、この組成物の安定性及び薬剤放出プロファイル特性が変わらないこともわかっている。
また、本発明は、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の放出制御型液状医薬組成物の製造方法も想定するもので、分散相の調製には、有効成分とイオン交換マトリックス粉末との混合が含まれる。実施形態の1つでは、イオン又は塩の形態の有効成分を塩形態のイオン交換マトリックスと混合したもので分散相が調製される。
特定の実施形態では、塩の形態であるクロルフェニラミン、プソイドエフェドリン及びヒドロコドンを、他の1種以上の成分と水との存在下で、アルギン酸ナトリウム粉末と混合させる。他の成分の例は限定されないが、例えば、薬剤充填ビーズを形成する微晶質セルロース等が挙げられる。別の実施形態では、該薬剤充填ビーズが更にラクトースを含有する。特定の実施形態によると、得られたビーズをクエン酸トリエチル及びタルクの存在下でEUDRAGIT(登録商標)(オイドラギット)で被覆し、炉内で硬化させる。この被覆ビーズを、塩の形態であるクロルフェニラミン及びヒドロコドンと、水とショ糖を含む分散媒に懸濁させる。特定の実施形態では分散媒がシロップNF(SyrupNF)を含む。また、分散媒が更に防腐剤や他の不活性添加剤を含んでもよい。このような実施形態において、得られる液状の放出持続型医薬品は瓶の中で物理的安定性を保持することができ、また、患者への投与の際には医薬品の放出制御が可能である。本発明の剤形は、一度に2種以上の薬剤投与が必要な患者や慢性的な投薬を必要とする患者にとって、とりわけ有用である。
【0028】
特定の実施形態は、下記有効成分クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる液状放出制御型薬剤組成物を用いた、風邪の兆候の治療を想定する。実施形態の1つによると、即放性(IR)製剤、及び/又は、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む調合医薬品に関連したこれまでの問題点及び不便な点が、本発明により克服される。現在入手できる即放性及び/又は調合型の風邪及びアレルギー薬と比較して、本発明は特別な利点を提供する。本発明によると、3種類の医薬有効成分(API)、即ち、クロルフェニラミン(抗ヒスタミン剤)、ヒドロコドン(鎮咳剤)及びプソイドエフェドリン(充血緩和剤)の利点が1つの徐放性(ER)組成物(例えば12時間)の中に統合される。これまでは、上記有効成分を一緒に用いる場合、それぞれの即放(IR)型剤形で1日4〜6回の投与が必要であった。つまり、3種が有効となる調合型徐放性医薬品で単一のものは現在ないからである。
現在市販されている多くの医薬品に対して、本発明は、3種の有効成分すべての徐放(ER)(例えば12時間)を可能にする製剤を提供するものである。本発明は、クロルフェニラミン(抗ヒスタミン剤)、ヒドロコドン(麻酔性鎮咳剤)及びプソイドエフェドリン(充血緩和剤)の即放剤形及び徐放剤形の新規な混合物を単一の製品として有する経口製剤を提供する。結果的に、この製剤は1日2回の投与でこれまで利用できた即放剤形(個別の販売及び投与、或いは、調合医薬品として販売及び投与が行われいる)と同じ効力を有する徐放性調合医薬品となる。また、この製剤は、以下の点において現存する単独及び調合型の徐放性製剤よりも優れている。(a)薬剤の即時的な伝達と長時間にわたる伝達のための即放性及び徐放性の投薬量が提供される。(b)三種類のRLDと生物学的等価な用量が単回投与剤形で提供される。(c)各薬剤成分の乱用や転用を防ぐ。
【図面の簡単な説明】
【0029】
【図1(A)】図1(A)は、製剤Xの放出持続型懸濁液からのプソイドエフェドリンの放出プロファイルを示す。
【図1(B)】図1(B)は、被覆ビーズからのプソイドエフェドリンの放出プロファイルを示す。
【図2(A)】図2(A)は、製剤Xの放出持続型懸濁液からのヒドロコドンの放出プロファイルを示す。
【図2(B)】図2(B)は、被覆ビーズからのヒドロコドンの放出プロファイルを示す。
【図3(A)】図3(A)は、製剤Xの放出持続型懸濁液からのクロルフェニラミンの放出プロファイルを示す。
【図3(B)】図3(B)は、被覆ビーズからのクロルフェニラミンの放出プロファイルを示す。
【図4(A)】図4(A)は、塩基製剤の懸濁液の3週間後におけるプソイドエフェドリンの放出プロファイルを示す(n=3、容器4、5及び6)。
【図4(B)】図4(B)は、塩形態である製剤の懸濁液の3週間後におけるプソイドエフェドリン (プソイドエフェドリン塩酸塩) の放出プロファイルを示す(n=3、容器1、2及び3)。
【図5(A)】図5(A)は、塩基製剤の懸濁液の3週間後におけるヒドロコドンの放出プロファイルを示す(n=3、容器4、5及び6)。
【0030】
【図5(B)】図5(B)は、塩形態である製剤の懸濁液の3週間後におけるヒドロコドン(ヒドロコドン重酒石酸塩)の放出プロファイルを示す(n=3、容器1、2及び3)。
【図6(A)】図6(A)は、塩基製剤の懸濁液の3週間後におけるクロルフェニラミンの放出プロファイルを示す(n=3、容器4、5及び6)。
【図6(B)】図6(B)は、塩形態である製剤の懸濁液の3週間後におけるクロルフェニラミン (クロルフェニラミンマレイン酸塩)の放出プロファイルを示す(n=3、容器1、2及び3)。
【図7】図7は、アルギン酸に結合した有効成分ヒドロコドン(10 mg/5mL)を含む放出持続型懸濁液からの(時間=0)におけるヒドロコドンの放出プロファイルを示す。
【発明を実施するための形態】
【0031】
本願明細書で使用のごとく、「患者」という用語は、鳥(例えば家禽)又は哺乳類等のいずれの動物に限定されるものではなく、ヒト、家畜や農場の動物、動物園、スポーツ及びペットの動物、例えばウシ、ヒツジ、フェレット、ブタ、ウマ、ウサギ、ヤギなどの家庭用ペットや他の家畜動物が挙げられるが、これも限定されるものではない。本願明細書で使用のごとく、「対象」及び「患者」という用語は互換性をもって使用される。実施形態の1つでは患者が哺乳類で、非霊長類(例えば、ウシ、ブタ、ウマ、ネコ、イヌ、ネズミ等)及び霊長類(例えばサルやヒト)である。
【0032】
本願明細書で使用のごとく、「治療する」「治療」という用語は、治癒効果のある治療と予防的手段の両方を表わし、その目的は望ましくない生理的症状、機能障害、疾病を予防又は軽減すること、又は、好適又は所望の臨床結果を得ることである。本発明の目的にとって、好適又は所望の臨床結果とは、兆候を軽減すること、症状、障害、疾病の程度を下げること、症状、障害、疾病を安定状態にすること(即ち、悪化させないこと)、症状、障害、疾病の進行を遅らせたりゆっくりにさせること、症状、障害、疾病の状態を改善すること、(部位的又は全体的のいずれであっても)それが検出可能か否かにかかわらず鎮静させること、症状、障害、疾病の改善又は向上が挙げられるが、これらに限定されない。治療には、臨床的に重要であり過度な副作用のない細胞反応を引き出すことが含まれる。また、治療には、治療を受けない場合の予想寿命よりも生存を延長することを包含する。
本願明細書で使用のごとく、「電解質薬剤」という句は、分離又はプロトン化によってイオン化が可能な薬剤の医薬的に許容できるイオン状態を指す。
本願明細書で使用のごとく、「薬剤」という用語は有効成分を意味し、例えば治療上の活性成分である。「薬剤」「有効成分」「有効な医薬成分」(又は「API」)という用語は互換性を持って使用される。
本願明細書で使用のごとく、「徐放相」又は「徐放性部位」という用語は、組成物が患者に投与された後、長時間にわたり持続的に放出される医薬組成物の相又は部位を指す。液状の放出制御型組成物においては、前記用語は、組成物の分散固相、即ち分散相を指す。
【0033】
本願明細書で使用のごとく、「即放相」又は「即放性部位」という用語は、組成物が患者に投与された後、すぐに放出される医薬組成物の相又は部位を指す。液状の放出制御型組成物においては、前記用語は、組成物の液相、即ち分散媒を指す。
本願明細書で使用のごとく、「拡散可能な対イオン」という用語は、電解質薬剤をイオン交換マトリックスから置換することができる医薬的に許容できるイオンを指す。拡散可能な対イオンが正電荷を有する場合、本願明細書では拡散可能な対カチオンと称される。拡散可能な対カチオンの非制限的な例としては、例えば、ナトリウム、カリウム、マグネシウム又はカルシウムが挙げられる。拡散可能な対イオンが負電荷を有する場合、本願明細書では拡散可能な対アニオンと称される。拡散可能な対アニオンの非制限的な例としては、例えば、塩化物、臭化物、ヨウ化物及びリン酸塩が挙げられる。
本願明細書で使用のごとく、電解質薬剤と関連して使用される場合の「水溶性」という用語は、生理学的に適切なpH値の水100mlに対し電解質薬剤がおよそ3gを超える溶解性を意味する。特定の実施形態においては、水溶性という用語は、生理学的に適切なpH値の水100mlに対し電解質薬剤が1gを超える溶解性を意味する。
本願明細書で使用のごとく、薬剤又はイオン交換マトリックスと関連して使用される場合の「アミンの塩基の形態」という句は、実質的にすべてのアミン窒素原子がプロトン化していない状態で電荷が中性であることを意味する。
【0034】
本願明細書で使用のごとく、薬剤又はイオン交換マトリックスに関連して使用される場合の「酸の形態」という句は、実質的にすべての酸基が、解離されず電荷を有していない酸の形態にあることを意味する。
本願明細書で使用のごとく、「高度に水和した」という句は、水の熱力学的活性を抑えるのに十分な水素結合を有する成分を意味する。
略語の定義
ACCP:米国胸部疾患学会
ADME:吸収、分布、代謝、排泄
API:有効医薬成分
AUC:0時間から無限時間までの血漿中濃度曲線下面積
AUCt:0時間から最終測定時間(Ct)までの血漿中濃度曲線下面積
BA:バイオアベイラビリティ−(生物学的利用能)
BE:生物学的等価
Cmax:最高血漿中濃度、投薬期間中に観察された最高濃度
Cmin:最低血漿中濃度、 投薬期間中に観察された最低濃度
CPM:クロルフェニラミンマレイン酸塩
CNS:中枢神経系統
ER:徐放
FDA:米国食品医薬品局
【0035】
GRASE:一般的に安全かつ有効な物質
HC:ヒドロコドン
IND:臨床試験用新医薬品
IR:即放
OTC:一般用医薬品
PSE:プソイドエフェドリン
RLD:関連リスト収載医薬品
PK:薬物動態
SAE:重篤有害事象
t1/2:消失半減期
tmax:Cmaxが観察された時間
実施形態の1つにおいて、本発明は、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなり、親水コロイドのイオン交換マトリックスと該マトリックスと逆の電荷を有する薬剤とを水の存在下で混合して製造した薬剤充填ビーズを含む液状の放出持続製剤に関する。
【0036】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の液状の放出持続型製剤は、約8時間、約10時間、約12時間、約16時間、約18時間、約24時間、最長約48時間までの範囲で放出制御が可能である。ある実施形態では、本発明の液状放出持続型製剤中の薬剤の全モル量の内、約15%、20%、25%、30%、35%、40%又は45%から、約50%、55%、60%、65%、70%、75%、80%、85%、90%、95%又は100%に至る量が、製剤を患者に投与してから12時間、16時間又は24時間にわたって放出される。別の実施形態では、本発明の液状放出持続型製剤中の薬剤の全モル量の約40〜約100%、40〜60%又は45〜55%が、製剤を患者に投与してから12時間にわたって放出される。別の実施形態では、本発明の液状放出持続型製剤の徐放相の薬剤全モル量の約40〜約100%、40〜60%又は45〜55%が、製剤を患者に投与してから12時間にわたって放出される。別の実施形態では、本発明の液状放出持続型製剤中の薬剤全モル量の約40〜約100%、70〜100%又は90〜100%が、製剤を患者に投与してから24時間にわたって放出される。有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物における実施形態において、本発明の液状放出持続型製剤の徐放相の薬剤全モル量の約40〜約100%、70〜100%又は90〜100%が、製剤を患者に投与してから24時間にわたって放出される。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の実施形態では、液状放出持続型薬剤伝達システムが、薬剤を含有し薬剤放出を制御する物質により被覆されたビーズを含む。実施形態の1つでは、前記被膜は遮蔽バリアであるが、バイオアベイラビリティーに到達するまでは、その被膜を通って薬剤が拡散しなければならない。
【0037】
他の実施形態では、薬剤が例えば医薬的に許容できる塩など、塩の形態をとっている。薬剤の医薬的に許容できる好適な塩としては、ナトリウム、カリウム、リチウム;カルシウム、マグネシウム;アルミニウム及び亜鉛又は他の同様な金属;アンモニア及び有機アミン類、例えば非置換又はヒドロキシ置換のモノ-、ジ-又はトリアルキルアミン類;ジシクロヘキシルアミン;トリブチルアミン;ピリジン;N-メチル-N-エチルアミン;ジエチルアミン;トリエチルアミン;モノ-、ビス-又はトリス-(2-ヒドロキシ-低級アルキルアミン)、例えばモノ-、ビス-又はトリス-(2-ヒドロキシエチル)アミン、2-ヒドロキシ-t-ブチルアミン又はトリス-(ヒドロキシメチル)メチルアミン、N,N-ジ-低級アルキル-N-(ヒドロキシ低級アルキル)-アミン類、例えばN,N-ジメチル-N-(2-ヒドロキシエチル)アミン又はトリ-(2-ヒドロキシエチル)アミン;N-メチル-D-グルカミン;及びアミノ酸、例えばアルギニン、リシン;及び硫酸塩、クエン酸塩、酢酸塩、シュウ酸塩、塩化物、臭化物、ヨウ化物、硝酸塩、硫酸水素塩、リン酸塩、酸性リン酸塩(acid phosphate)、イソニコチン酸塩、乳酸塩、サリチル酸塩、酸性クエン酸塩、酒石酸塩、オレイン酸塩、タンニン酸塩、パントテン酸塩、重酒石酸塩、アスコルビン酸塩、コハク酸塩、マレイン酸塩、ゲンチシン酸塩(gentisinate)、フマル酸塩、グルコン酸塩、グルクロン酸塩、糖酸塩、ギ酸塩、安息香酸塩、グルタミン酸塩、メタンスルホン酸塩、エタンスルホン酸塩、ベンゼンスルホン酸塩、p-トルエンスルホン酸塩、塩酸塩、パモ酸塩(即ち、1,1 '-メチレン-ビス-(2-ヒドロキシ-3-ナフトエート)、エンボナート(embonate)、エストラート及びトシラート等が挙げられるが、これらに限定されない。つまり、薬剤を塩の形態で使用することにより多数の利点がもたらされる。薬剤物質は、いくつかの次善の物理化学特性や生物薬剤学的特性を有することが多いが、イオン化した塩基性又は酸性の薬剤分子を対イオンと組み合わせて薬剤を塩形態にすることで解決できる。くわえて、医薬的に許容できる、ヒトでの使用に最適な医薬品は、一般的に塩基の形態よりもむしろ塩形態のほうが製造業者からの入手が容易である。どの塩を選択するかは、イオン化基の酸性度又は塩基度、対イオンの安全性、医薬品表示(drug indications) 及び予定の剤形によりおおかた決まる。薬剤の医薬的に許容できる塩の形態をどのようにして決めるかは、この分野の当業者であればわかる(例えば、Kumar, L.等「Salt Selection in Drug Development」Pharmaceutical Technology 3(32) (2008)参照のこと)。
【0038】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物についての別の実施形態では、水和度が高く水と結合可能な成分が分散媒に含まれる。このような成分が水を分散媒から引き出すので、ビーズの外側かつ分散媒の中の水分活性がビーズ内よりも小さくなる。このような実施形態では、分散媒の水分活性が十分に低いので、製剤が投与され分散媒が希釈されるまで、薬剤充填ビーズへの水の拡散を妨ぎ、内部浸透圧が起きるのを防ぐ。本発明のいくつかの実施形態において、前記成分とは、ショ糖、デキストロース、マルトース、マニトール、ソルビトール、グリセリン又は低分子量ポリエチレングリコール等の非電解質賦形剤であるが、これらに制限されるものではない。このような実施形態において、本発明が意図する薬剤伝達システムは、水(例えば、胃液中の水分)により促進される。こうした実施形態では、分散媒中のビーズ外側の水分活性がビーズ内部の水分活性よりも大きい場合、水が被膜を通り拡散し、ビーズの可溶性成分を溶かし、拡散可能な遊離状態の薬剤のリザーバーを形成する。この遊離な状態の薬剤は、患者への薬剤放出の制御を可能にする被膜を透過することができる。
実施形態の1つにおいて、本発明の組成物の貯蔵寿命は6ヵ月以上である。実施形態によっては、患者への投与に先立ち、本発明の組成物は安定性を6ヵ月以上維持する。
更に、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の別の実施形態では、分散媒が水和度の高い賦形剤を含有する。具体的には、このような実施形態の1つにおいて、水和度の高い賦形剤が質量基準で50〜70%分散媒に含まれる。更に別の実施形態では、この水和度の高い賦形剤がショ糖である。
【0039】
本発明の組成物の実施形態の1つは、甘味料、香味料、着色剤、さらにこれらの混合物からなる群から選択される賦形剤を更に含む。別の実施形態では、該組成物は安定化剤、分散助剤及びこれらの混合物からなる群から選択される分散添加剤を更に含む。
本発明の組成物の特定の実施形態は、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる。
本発明のこうした実施形態の1つでは、医薬的に許容できる イオン交換マトリックス及び該イオン交換マトリックスに結合した水溶性電解質薬剤とが、分散相に更に含まれ、イオン交換マトリックスの表面電荷が電解質薬剤の表面電荷と相対する。本発明の別の実施形態では、分散相が膜で被覆されている。本発明の特定の実施形態では、この膜がポリマーである。膜は多孔質又は非多孔質でもよい。本発明の実施形態の1つでは、この膜が薬剤の拡散を制御する。本発明の更に別の実施形態では、分散相には薬剤充填ビーズが含まれ、該ビーズには、水がビーズに吸収された際に溶けてビーズから拡散することができ、ビーズ内の浸透圧を低減することができる低分子量で非電解質の可溶性賦形剤が含まれる。本発明の実施形態の1つは、この低分子賦形剤がラクトースである。本発明の別の実施形態では、分散媒中の水をひきつける水和度の高い賦形剤が分散媒中に更に含まれる。本発明は、水和度の高い賦形剤が高濃度であることを想定するもので、水和度の高い賦形剤が、質量基準で例えば50〜70%、55〜70%、45〜70%、55〜65%又は60〜70%の質量比で分散媒に含まれることを想定する。本発明の実施形態の1つでは、分散媒中の水和度の高い賦形剤がショ糖であり、例えば、質量基準でショ糖が65%である。本発明のある実施形態では、分散相及び/又は分散媒における薬剤又は有効成分の1種以上は塩基の形態ではない。本発明の別の実施形態では、分散相及び/又は分散媒中の薬剤又は有効成分の1種以上が塩の形態である。本発明の更に別の実施形態では、分散相が薬剤とイオン交換マトリックス粉末との混合物を含み、薬剤とイオン交換マトリックスが塩の形態、例えば医薬的に許容できる塩の形態である。
【0040】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の更に別の実施形態では、分散相及び/又は分散媒中の薬剤又は有効成分の1種以上は塩の形態ではない。このような実施形態の1つでは、分散相及び/又は分散媒における薬剤又は有効成分の1種以上が塩基の形態である。
イオン交換マトリックスは高分子有機化合物であってもよく、例えば、オリゴマー、コオリゴマー、ポリマー又はコポリマー;例えばゼオライトのような多孔質網状組織の無機固体;及び/又はこれらの組合せ等、表面が帯電し、反対の電荷を有するイオンを保持するものである。本願明細書で使用のごとく、「カチオン交換マトリックス」という句は、カチオン状態の薬剤を保持することができるイオン交換マトリックスを指す。本願明細書で使用のごとく、「アニオン交換マトリックス」という句はアニオン状態の薬剤を保持することができるイオン交換マトリックスを指す。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の設計及び適切な成分の選択は、治療用の有効成分(薬剤)の電荷に基づく。本発明は、非荷電の塩基又は酸の薬剤、或いは、カチオン性又はアニオン性薬剤であって、強力な電解質であるだけでなく、そのpKa(アニオン)より高い弱酸性薬剤及びそのpKa(カチオン)より低い弱塩基性薬剤である薬剤の投与に好適である。薬剤がイオンの場合、イオン交換マトリックスは薬剤イオンと反対の電荷を有していなければならない。薬剤が非電荷塩基又は酸の場合、イオン交換マトリックスはそれぞれ酸又は塩基の形態である。電解質薬剤がカチオンの場合、負の表面官能価(negative surface functionality)を有するアニオン交換マトリックスをイオン交換マトリックスとして使用しなければならない。本発明の組成物は、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなり、これらは正帯電したカチオン系薬剤である。
【0041】
カチオン交換マトリックス及びアニオン交換マトリックスは当該分野では周知である。有用なカチオン交換マトリックスの非制限的例としてはカチオン交換樹脂が挙げられ、例えば、スチレン-ジビニルベンゼン共重合体を含む重合体主鎖とスルホネート基の側鎖とを有する、Rohm and Haas社(ペンシルバニア州、フィラデルフィア)からAMBERLITE(商標)IRP69という商品名で販売されている樹脂;Rohm and Haas社からAMBERLITE(商標)IRP64及びIRP88という商品名で販売されている、カルボキシレート官能基を有するメタクリル酸-ジビニルベンゼン共重合体;アルギン酸塩、カルボキシメチルセルロース、クロスカルメロース、微晶質セルロース、キサンタンガム、カルボマー94等のカルボキシビニルポリマー、ゼラチン等の親水コロイド;又はこれらの混合物が挙げられる。実施形態の1つは、カチオン交換マトリックスがアルギン酸塩、カルボキシメチルセルロース、微晶質セルロース、キサンタンガム、カルボキシビニルポリマー、ゼラチン、又はこれらの混合物である。
好適なアニオン交換マトリックスの非制限的例としては、例えば、スチレン-ジビニルベンゼン共重合体を有する重合体主鎖とアンモニウム又はテトラアルキルアンモニウム官能基の側鎖とを有する、Rohm and Haas社(ペンシルバニア州、フィラデルフィア)からDUOLITE(商標)AP143という商品名で販売されている樹脂等のアニオン交換樹脂;ならびにキトサン、ポリリシン又はゼラチン等(これらに限定されない)の親水コロイド;ならびにこれらの混合物が挙げられる。実施形態の1つは、アニオン交換マトリックスがキトサン、ポリリシン、ゼラチン又はこれらの混合物である。
【0042】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の実施形態では、イオン交換マトリックスが水に不溶である。本発明のこのような組成物の別の実施形態では、イオン交換マトリックスが水溶性である。このような実施形態では、イオン交換マトリックスは分散媒と溶媒和化することができ、親水コロイドの実施形態がある。本発明は、親水コロイドとして、デンプン、寒天、セルロース、アルギン酸、グアーゴム、キサンタンガム, ゼラチン、アカシア、アルブミンなど、これらに限定はされないが湿式結合剤といった単純なものからマイクロスフェアの成分という新規なものにわたる用途に使用されてきた天然物質を非制限的例として含むものである。非制限的な合成例としては、メチルセルロース、カルボキシメチルセルロース、ヒドロキシプロピルメチルセルロース、メチルアクリル酸、ポリ乳酸、ポリグリコール酸、ポリ酸無水物など、従来からの湿式造粒などの用途から機能性塗料や生物分解性移植組織といったより現代的な非制限的用途に広く展開されているものが含まれる。
他の実施形態によると、カチオン交換マトリックスは親水コロイドである。このような実施形態では、カチオン交換マトリックスがアルギン酸塩、カルボキシメチルセルロース、微晶質セルロース、キサンタンガム、カルボマー94等のカルボキシビニルポリマー類、又はこれらの混合物である。実施形態によっては、親水コロイドが膨潤を抑えるために架橋されている。ある実施形態は、イオン交換物質がアルギン酸カルシウムである。別の実施形態は、イオン交換マトリックスがアルギン酸ナトリウムである。
【0043】
別の実施形態では、アニオン交換マトリックスが親水コロイドである。このような実施形態のアニオン交換マトリックスは、キトサン、ポリリシン、ゼラチン又はこれらの混合物である。実施形態によっては、親水コロイドが膨潤を抑えるために架橋されている。
水溶性の電解質薬剤はイオン交換マトリックスと結合し、イオン交換マトリックス薬剤複合物を形成する。
実施形態によっては、イオン交換マトリックス薬剤複合物が粒子状又はビーズの形態である。この粒子又はビーズは、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物において、液剤で経口投与が可能な大きさである。本発明の実施形態の1つでは、この粒子又はビーズが懸濁液中に沈殿しないような大きさ及び/又は密度である。実施形態によっては、粒子又はビーズは望ましくない患者属性を有していない。実施形態によっては、粒子又はビーズの直径は約0.01〜約2000μmの範囲である。別の実施形態では、約0.1〜約1000μm、更に別の実施形態では約1〜約1000μmの範囲である。粒子又はビーズの直径が2000μmを超える実施形態、3000μmを超える実施形態、又は5000μmを超える実施形態もある。粒子又はビーズの直径が2000μm未満、1000μm未満、500μm未満、50μm未満、1μm未満の実施形態もある。実施形態の1つは、粒子、ペレット又はビーズの直径が約600μmである。粒子、ペレット又はビーズの直径が約200μm、約300μm、約400μm、約500μm、約600μm、約700μm、約800μm又は約900μmの実施形態がある。
【0044】
コア部には固形剤形を形成するのに有用な医薬的に許容できる加工助剤を更に含んでもよく、デンプン、酸化チタンやシリカ等の増量剤、防腐剤、酸化防止剤等の安定化剤、植物油等の潤滑剤等をはじめとするが、これらに限定されるものではない。
実施形態の1つでは、イオン交換マトリックス薬剤複合物は低分子量の可溶性非電解質賦形剤を更に含有する。このような賦形剤は、ビーズが水を吸収した際に水に溶解し、ビーズから拡散することができるので、ビーズ内部の浸透圧を低下させる。この賦形剤の分子量は、ビーズを被覆するどんな膜も透過できるように十分に小さくなければならない。様々な実施形態によると、ビーズに含有される賦形剤の量は薬剤放出速度に影響を及ぼす可能性がある。ビーズ中に賦形剤は約5〜約10%、約10〜約20%、約20〜約30%、約30〜約40%、約40〜約45%含まれる。実施形態の1つは、賦形剤がラクトースである。このような実施形態では、製造工程において分散相に導入されたラクトースが多いほど、投与後のビーズからの薬剤の放出は速くなる。様々な実施形態として、ビーズ中にラクトースは約5〜約10%、約10〜約20%、約20〜約30%、約30〜約40%、約40〜約45%含まれる。実施形態によっては、ラクトースがビーズ中に約20〜約30%含まれる。可溶性非電解質賦形剤の他の例として、デキストロース、マルトース、マニトール、ソルビトール、グリセリン又は低分子量ポリエチレングリコールが本発明では挙げられるが、これらに限定されるものではない。
【0045】
実施形態の1つは、イオン交換マトリックス薬剤複合物が拡散制御皮膜を更に含む。この皮膜は、イオン交換マトリックス内への対イオンの拡散とマトリックスからの薬剤拡散を更に制御するうえで有用である。このように拡散制御皮膜は、患者に投与した後、分散媒及び/又は消化管内に電解質薬剤が放出されるのを制御するうえで有用である。本発明は、拡散制御を行ういずれの皮膜の使用も想定するものである。コーティング材料は、多数にわたる天然又は合成の膜形成剤のいずれでもよく、これらを単独又は相互に使用してもよい。また、可塑剤、顔料又は他の物質等の成分と共に使用してもよい。実施形態によっては、皮膜の成分は水には不溶でかつ水を通す。皮膜の透過性を変更する際に、水溶性物質の導入が有用となる場合がある。当該分野で拡散制御皮膜は公知である。非制限的な例として、SURELEASE(登録商標)(Colorcon社、ペンシルバニア州ウェストポイント)等のエチルセルロース; EUDRAGIT(登録商標)(Rohm Pharma社、ドイツ、Weiterstat)等のメチルメタクリレートポリマー;セルロースエステル類、セルロースジエステル類、セルローストリエステル類、セルロースエーテル類、セルロースエステル-エーテル、セルロースアシレート、セルロースジアシレート、セルローストリアシレート、セルロースアセテート、セルロースジアセテート、セルローストリアセテート、酢酸プロピオン酸セルロース、酢酸酪酸セルロースが挙げられる。皮膜がメチルメタクリレートポリマーの実施形態がある。
【0046】
実施形態の1つは、拡散制御皮膜が、エチルセルロース、メチルメタクリレート、セルロースエステル類、セルロースジエステル類、セルローストリエステル類、セルロースエーテル類、セルロースエステル-エーテル、セルロースアシレート、セルロースジアシレート、セルローストリアシレート、セルロースアセテート、セルロースジアセテート、セルローストリアセテート、酢酸プロピオン酸セルロース、酢酸酪酸セルロース 、これらの混合物からなる群から選択される。実施形態の1つは、拡散制御皮膜がエチルセルロース、メチルメタクリレート又はそれらの混合物である。別の実施形態は、拡散制御皮膜コーティング材が、コーティング材とイオン交換マトリックス薬剤複合物の全質量を基準として約20〜約30質量%である。
実施形態の1つは、イオン交換マトリックス薬剤複合物が、イオン交換マトリックス薬剤複合物及び拡散制御皮膜の全質量を基準として、約1%から約75%までの質量の拡散制御皮膜でコーティングされている。別の実施形態では5〜約50%、別の実施形態では約10〜約30%、更に別の実施形態では約20〜約25%である。一般に、コーティング材が多いほど薬剤放出が遅くなる。
実施形態の1つは、薬剤充填アルギン酸塩ビーズがEUDRAGIT(登録商標)RS 30D(Rohm社製)で十分にコーティングされ、コーティング材及び薬剤充填アルギン酸塩ビーズの全質量を基準として約20〜約30質量%のコーティング材を有する被覆ビーズとなる。
【0047】
別の実施形態では、イオン交換マトリックス薬剤複合物の拡散制御皮膜コーティング材が更に可塑剤を含む。可塑剤はコーティング材の可塑性を増強させ脆性を低下させるのに有用である。また、可塑剤は薬剤放出速度にも影響する。可塑剤はコーティング材のガラス転移温度を低下させるので、凝集性フィルムの中に添加された液滴を容易に融合させ、コーティング材の透過性に影響を与える。可塑剤は当該分野では公知である。可塑剤の非制限的例としては、クエン酸トリエチル、セバシン酸ジエチル、フタル酸ジエチル、クエン酸トリブチル、アセチルクエン酸トリブチルが挙げられる。実施形態の1つは可塑剤がクエン酸トリエチルである。実施形態の1つは、イオン交換マトリックス薬剤複合物は、イオン交換マトリックス薬剤複合物及び拡散制御膜の全質量を基準として、約0.1%から30%まで、約0.5%から約20%まで、約1〜約20%、約2〜約10%までの可塑剤を含有する。
【0048】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物は、イオン性成分の存在下で安定している。実施形態の1つでは、本発明の組成物がイオン性成分の存在下、分散媒中で安定している。このような実施形態において、本発明の組成物は、拡散可能な対イオンの存在下、分散媒中で安定している。別の実施形態では、本発明の組成物は、電解質薬剤、即ち、クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンの存在下、分散媒中で安定している。有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物は、このような薬剤が遊離な状態で分散媒中に存在していると、即ち、薬剤がイオン交換マトリックスに結合していない状態でいると、安定性が保持される。本発明の組成物は、分散媒中の遊離状態の薬剤の存在下、分散相からの薬剤放出プロファイルが適切に持続性を保持する。本発明の別の実施形態の分散媒は、有効成分がクロルフェニラミンとヒドロコドンとからなり、さらにプソイドエフェドリンを含んでいてもよく、即放剤形となる。このような実施形態では、分散媒中の薬剤はイオン交換マトリックスと結合していない。このような実施形態の1つでは、分散媒が塩の形態の薬剤を含有する。
実施形態の1つでは、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる液状放出制御型組成物は、分散媒中の遊離状態の薬剤及び/又は拡散可能な対イオンの存在下で安定している。このような実施形態の1つは、本発明の組成物が徐放相と即放相とを有し、即放相が遊離状態の薬剤を特定量含み、患者に投与する前に徐放相から即放相に放出される薬剤量は、分散媒及び分散相中の薬剤の全モル量を基準として30%未満、25%未満、20%未満、10%未満、5%未満、0.5%未満又は0.05%未満である。
【0049】
本発明は、3つの異なる治療徴候のために用いる3種の薬剤、即ち、アレルギー及び鼻漏の治療用のクロルフェニラミン、咳の治療用のヒドロコドン、鼻づまり治療用のプソイドエフェドリンを含む液状の放出制御型組成物に関する。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の液状放出制御型組成物の別の実施形態では、患者への投与前に、分散相の中の薬剤1種以上が分散媒中に浸出する。このような実施形態では、分散媒及び分散相中の薬剤の全モル量を基準に、薬剤の約10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、70%で、最大75%までが、患者への投与の前に分散相から分散媒に放出される。分散媒及び分散相中の薬剤の全モル量を基準に、実施形態の1つでは約15〜約35%、別の実施形態では約25%の薬剤が患者への投与の前に分散相から分散媒に放出される。
実施形態によっては、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の液状の放出持続型組成物の、即放相に含まれる薬剤対徐放相に含まれる前記と同じ薬剤の質量比(IR/ER比)が、約0:100、約5:100、約10:100、約15:85、約20:80、約25:75、約30:70、約35:65、約40:60、約45:50又は約50:50である。実施形態の1つは、薬剤のIR/ER比が約25:75である。本発明の特定の実施形態では、本発明の経口組成物の即放性部位対徐放性部位のクロルフェニラミンの質量比は約25:75、即放性部位対徐放性部位のヒドロコドンの質量比は約25:75、即放性部位対徐放性部位のプソイドエフェドリンの質量比は約25:75〜約0:100である。
【0050】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の他の実施形態では、液状の放出制御型医薬組成物1回の投与量1mlあたり0.1〜0.5、0.5〜1mg、1〜5mg、5〜10mg、10〜15mg、15〜20mg、20〜25mg、25〜30mg、30〜40mg、40〜50mg、50〜60mg、60〜70mg、70〜80mg、80〜90mg、90〜100mg、100〜120mg、120〜140mg、140〜160mg、160〜180mg、180〜200mg、200〜220mg、220〜240mg、240〜260mg、260〜280mg、280〜300mg、300〜350mg、350〜400mg、400〜450mg、450〜500mg、最大600mg、700mg、800mg、900mg、1000mgまでの各薬剤又は各有効成分が医薬組成物に含まれる。本発明の更に別の実施形態では、このような医薬組成物には、液状の放出制御型医薬組成物1回の投与量5mlあたり0.1〜0.5、0.5〜1mg、1〜5mg、5〜10mg、10〜15mg、15〜20mg、20〜25mg、25〜30mg、30〜40mg、40〜50mg、50〜60mg、60〜70mg、70〜80mg、80〜90mg、90〜100mg、100〜120mg、120〜140mg、140〜160mg、160〜180mg、180〜200mg、200〜220mg、220〜240mg、240〜260mg、260〜280mg、280〜300mg、300〜350mg、350〜400mg、400〜450mg、450〜500mg、最大600mg、700mg、800mg、900mg、1000mgまでの各薬剤又は各有効成分が含まれる。本発明の特定の実施形態では、液状の放出制御型医薬組成物1回の投与量5mlあたり1〜5mg、5〜10mg、10〜15mg、15〜20mg、20〜25mg、25〜30mg、30〜40mg、40〜50mg、最大100〜120mg、120〜140mg、140〜160mg、160〜180mg、180〜200mg、200〜220mg、220〜240mg、240〜260mg、260〜280mg、280〜300mgまでの各薬剤又は各有効成分が医薬組成物に含まれる。
【0051】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の実施形態によっては、水和度が高く分散媒中で水との結合が可能な賦形剤が高濃度に分散媒中に更に含まれる。本発明の薬剤は水性分散媒における溶解度が高いが、分散媒中に水和度の高い成分が存在することで、分散媒中の水が引き寄せられる。これは薬剤-イオン交換マトリックス複合物からの薬剤の溶解を開始するの必要である。製剤が投与され、胃液(ほぼ水)が分散媒を希釈したときにはじめて薬剤が溶けて、利用可能な状態になり、皮膜の透過及び/又はビーズからの拡散が始まる。このような実施形態では、遊離状態の薬剤は実質的に分散媒からなくなり、例えば、薬剤は0.5%未満又は0.05%未満しか分散媒には存在しない。本発明の様々な実施形態では、水和度の高い成分が分散媒中に質量比で約10〜約20%、約20〜約30%、約30〜約40%、約40〜約50%、約50〜約60%で存在する。実施形態によっては、該成分が約60〜約65%、最大約70%まで含まれる。本発明の他の実施形態は分散媒がショ糖又は他の糖分子を含む。このような実施形態では、ショ糖が質量比で10%より多く、20%より多く、30%より多く、40%より多く又は50%より多く分散媒中に含有される。有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の実施形態では、分散媒にショ糖が約65%かつ約70%以下含まれる(即ちSyrup NF)。本発明が包含する賦形剤の他の例としては、デキストロース、マニトール、フルクトース、ポリエチレングリコール、グリコール類及びグリセリン類が挙げられるが、これらに限定されるものではない。本発明の実施形態では、分散媒が質量比で10%より多くのデキストロース、マニトール、フルクトース、ポリエチレングリコール、グリコール又はグリセリンを、又は20%より多くのデキストロース、マニトール、フルクトース、ポリエチレングリコール、グリコール又はグリセリンを、又は30%より多くのデキストロース、マニトール、フルクトース、ポリエチレングリコール、グリコール又はグリセリンを、又は40%より多くのデキストロース、マニトール、フルクトース、ポリエチレングリコール、グリコール又はグリセリンを、又は50%を超えてデキストロース、マニトール、フルクトース、ポリエチレングリコール、グリコール又はグリセリンを含む。本発明の実施形態の1つは、分散媒中にデキストロース、マニトール、フルクトース、ポリエチレングリコール、グリコール又はグリセリンが約65%、かつ、デキストロース、マニトール、フルクトース、ポリエチレングリコール、グリコール又はグリセリンが約70%未満で含まれる。この分野の当業者であれば、同等に機能するであろう水和度の高い他の賦形剤を容易に決定することができる。
【0052】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の液状の放出制御型薬剤組成物は、本発明の意図する作用に賦形剤が悪影響を及ぼさないのであれば、安定化剤、分散剤等からなる群から選択される分散助剤をさらに含んでもよい。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の液状の放出制御型薬剤組成物は、本発明の意図する作用に賦形剤が悪影響を及ぼさないのであれば、例えば甘味料、風味剤、着色剤、増粘剤等の液状経口製剤に有用な賦形剤を更に含んでもよい。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の剤形の利点は、イオントラップ、浸透の制御、熱力学的均衡メカニズムが大概適切であり本質的に安定していること、また、当該分野の当業者であれば利用可能な、広く承認された従来からの医薬賦形剤の使用を通して効果を実現できるという事実である。
本発明で有用なカチオン系有効成分はクロルフェニラミン、ヒドロコドン又はプソイドエフェドリンである。
実施形態によると、塩の形態の前記電解質薬剤が有用な薬剤として挙げられる。薬剤の塩の形態として、マレイン酸塩、塩酸塩又は重酒石酸塩が好ましい。
【0053】
また、実施形態によっては、イオン交換マトリックスとの結合又は反応によりイオンを形成する中性状態の前記電解質薬剤が有用な薬剤として挙げられる。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の様々な実施形態において、イオン交換マトリックス薬剤複合物には、薬剤をイオンの状態に変換するのに十分な量のイオン交換マトリックスが含まれる。イオン交換マトリックス薬剤複合物には、薬剤をイオンの状態に変換するのに十分な量以上のイオン交換マトリックスが含まれていてもよい。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の液状の放出制御型薬剤組成物の実施形態の一つでは、薬剤又は有効成分が1個1個のの粒子、ペレット又はビーズの中で結合している。このような実施形態では、薬剤の表面電荷と逆の表面電荷を有する医薬的に許容できるイオン交換マトリックスに、薬剤が結合する。本発明の実施形態の1つは、薬剤の表面電荷と逆の表面電荷を有する同じ医薬的に許容できるイオン交換マトリックスに、薬剤が結合する。同一のイオン交換マトリックスに薬剤が結合することにより、組成物中の各薬剤の放出制御は妨害されることなく、各薬剤の放出を適切な速度にする。クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンは、負の表面官能価を有するイオン交換マトリックスと結合することが想定されている。 本発明の別の実施形態では、分散相が、医薬的に許容できる塩の形態の薬剤又は有効成分を含む。本発明の更に別の実施形態では、分散相が医薬的に許容できる塩の形態のイオン交換マトリックスを含む。
【0054】
実施形態によっては、本発明の組成物は有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなり、これらが1個1個の粒子又はペレット又はビーズの中に含まれるが、このような粒子、ペレット又はビーズの中で薬剤同士の化学的又は物理的相互作用はなく、単一の薬剤放出速度制御皮膜により満足のいく安定特性と適切な薬剤伝達プロファイルが得られる。
本発明の各薬剤をそれぞれ別々のビーズに含ませ、ビーズの混合を行う場合、その混合物が完全に均一ではない可能性があるので、薬剤の服用量が正しくない結果となる。不均一になるのは、無作為な変動によるものか、或いは、混合物中の2個以上のビーズの物性の差異、例えば重量や密度の差異によるものである。しかしながら、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとが1個の粒子又はペレット又はビーズの中で結合しているという上述の技術的方法によって、都合のよいことに、薬剤混合物の均質性を確保し、調合製剤におけるそれぞれの薬剤の服用量が均一したものとなり、所定の患者は各薬剤を一定量で摂取できる。更に、これらの薬剤を同一のイオン交換マトリックスに結合させることにより、分散相の中には薬剤結合樹脂複合物が1種類しかできないという利点が生まれる。更には、クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンを1個1個のの粒子、ペレット又はビーズ内で混合させることで、調合医薬品全体に存在する粒子、ペレット又はビーズの表面積を低減することができ、製品及び有効成分の安定性を増強することができる。
【0055】
本発明における上記の技術的方法のもう一つの利点は、同一の樹脂に結合したすべての薬剤が同様な又は同じ放出プロファイルを示し、本発明の各薬剤に関する投与量又は投与回数を違える必要がない。もしも異なる樹脂が使用されるならば、各薬剤の放出プロファイルは患者の食餌又は生理機能の差に影響を受けることもあるので、所定の患者にとって、ある薬剤は多すぎ、また別の薬剤は少なすぎる摂取になるという可能性がある。それに対して有利なことに、本発明の上述の技術的方法によると、クロルフェニラミンとヒドロコドンとプソイドエフェドリンについて同時に生物学的等価性を得ることが可能になる。また、このような薬剤を単一の放出方法を有する1個1個のビーズに配置することで、薬剤放出プロファイルが患者集団全体の中でより一貫性を有する。
クロルフェニラミンとヒドロコドンとプソイドエフェドリンとが1個1個の粒子又はペレット又はビーズの中で結合しているという上述の技術的方法の更なる利点は、このような薬剤組成物から個々の有効成分の抽出又は析出を難しくすることである。具体的には、このような有効成分が同一のイオン交換マトリックスに結合していることにより、いずれの単一の有効成分の抽出又は分離も当業者でなければ極めて難しくなる。例えば、これらの薬剤が1つのイオン交換マトリックスに結合していれば、1つの薬剤/1つのイオン交換マトリックスによる複合物間の密度等といった物性の差をもとに、1種の薬剤を他の薬剤から部分的に単離することは不可能である。本発明の前記技術的方法を用いて製造した製品からは、いずれの有効成分も1種を単離することは難しいため、製品の中のいずれか1種の成分を乱用又は不法使用しようとする潜在的可能性は低下する。即ち、本発明の技術的方法により、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとを含む調合医薬品は、薬剤の乱用や転用の潜在的可能性を抑えるように製造できることになる。
【0056】
更に別の実施形態では、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとが、それぞれの薬剤の表面電荷と逆の表面電荷を有する医薬的に許容できる異なるイオン交換マトリックスと結合している。
本発明の組成物の実施形態では、長期間にわたり、即ち、少なくとも1ヵ月、少なくとも3ヵ月、少なくとも6ヵ月、1年間、2年間又は3、4、5年以上にわたり安定している。具体的には、本発明の組成物は、前記の期間、化学的、物理的、微生物学的な安定性を維持する。この分野の当業者であれば、医薬組成物の化学的、物理的、微生物学的な安定性の評価方法は理解できよう。医薬組成物の安定特性、例えば、物理的、化学的、微生物学的な安定性次第で、患者に投与ができる最終組成物の状態で薬剤又は有効成分が瓶の中でどのくらいの期間保存できるかが決まる。実施形態の1つは、本発明の組成物は室温で少なくとも1ヵ月間、少なくとも3ヵ月間、少なくとも6ヵ月間、1年間、2年間、3、4、5年間以上安定している。このような実施形態では、本発明の組成物は、少なくとも1ヵ月間、少なくとも3ヵ月間、少なくとも6ヵ月間、1年間、2年間、3、4、5年間以上、化学的、物理的、微生物学的な安定性を有する。
【0057】
化学的安定性は、時間経過後の組成物中の薬剤又は有効成分の構造的保全性により証明される。化学的安定性はクロマトグラフィー分析及び/又は有効性の測定を用いて評価することができる。こうした評価により、組成物中の薬剤の存在、製品の劣化の有無が検出される。時間Xにおいて、ここでXはゼロ(即ち、組成物が製造された時)より長いある期間を指すが、時間ゼロ時点で存在していた薬剤の90〜100%が存在し、十分な構造的保全性を示すか、或いは、時間ゼロの時点で予想した構造的特性と比較して、同じか同等な構造的特性又は著しい差がない構造的特性を示す場合、薬剤は、時間Xにおいて化学的に安定しているとみなされる。実施形態の1つは、本発明の組成物が、少なくとも1ヵ月間、少なくとも3ヵ月間、少なくとも6ヵ月間、1年間、2年間、3、4、5年間以上、化学的安定性を有する。
【0058】
本発明の組成物の物理的安定性は、分散相を被覆する拡散制御皮膜又は機能的コーティングの保全性及びその透過性により証明される。物理的安定性は溶出分析により評価することができる。溶出分析は、時間ゼロ(即ち、組成物が製造された時点)又は時間X(Xはゼロより長い時間で、例えば、1週、2週、3週、1ヵ月、3ヵ月、6ヵ月、1年、2年、3年、4年又は5年)時点で開始して分析する。溶出テストは、患者の胃腸管内の環境と同等な化学的環境に組成物をいったん配置した際に放出される薬剤の量を示す。溶出テストは、薬剤が患者に投与された際の薬剤の放出速度を示す。溶出又は放出プロファイル、即ち、各測定時点での(薬剤を相応の化学環境のもとに配置後又は患者に投与後)放出された薬剤の量及び薬剤の放出速度が、薬剤組成物の物理的安定性を表わす。ある時点Xにおいて物理的に安定した薬剤組成物であれば、溶出分析を用いて評価した溶出又は放出プロファイルが、時間ゼロ(即ち、薬剤の製造直後に評価を行った時)において測定した薬剤組成物から予想されるものと同じ、同等、又は少なくとも著しく異ならない溶出又は放出プロファイルを示すであろう。また、物理的に安定した薬剤組成物であれば、測定における第1時点(即ち、測定開始時)において、予想されたある特定量の薬剤が放出される。具体的に言うならば、物理的に安定した組成物とは、測定における第1時点において、即放性部位を有していないものは薬剤放出は示さないか、或いは、非常に低い薬剤放出レベル、例えば、10%未満、5%未満又は1%未満の薬剤が徐放相から放出される。更に、物理的に安定した薬剤組成物であれば、組成物を相応の化学環境のもとに配置又は患者に投与した後、それぞれの次の時点では、薬剤放出速度が予想されたある特定の速度であり、放出される薬剤がある特定の量となる。本発明の組成物の実施形態では、少なくとも1ヵ月間、少なくとも3ヵ月間、少なくとも6ヵ月間、1年間、2年間、3、4、5年間以上、物理的安定性を有する。このような実施形態では、本発明の薬剤組成物は、少なくとも1ヵ月間、少なくとも3ヵ月間、少なくとも6ヵ月間、1年間、2年間、3、4、5年間以上瓶で保存することができ、その間物理的安定性が維持される。
【0059】
いくつかの実施形態では、本発明の組成物は微生物学的安定性も維持する。薬剤組成物の微生物学的安定性は、微生物による組成物の汚染の有無を示す。実施形態の1つは、本発明の組成物が、少なくとも1ヵ月間、少なくとも3ヵ月間、少なくとも6ヵ月間、1年間、2年間、3、4、5年間以上微生物学的安定性を有する。
本発明の液状の放出制御型組成物の安定特性により、この組成物は長い貯蔵寿命、即ち、1年以上の貯蔵寿命を有する。本発明の組成物は、室温条件下で、少なくとも1ヵ月間、少なくとも3ヵ月間、少なくとも6ヵ月間、約1年間、約2年間、3、4、5年間以上の貯蔵寿命を有し、その間、製剤の安定性及び薬剤放出プロファイル性は保たれると考えられる。他の実施形態の放出持続型製剤は、1ヵ月を超え、3ヵ月を超え、6ヵ月を超え、1年を超え、約2年間を超え、約3年間を超え、4年間を超え、5年間を超えて物理的にも化学的にも安定している。
本発明の別の態様は、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなり、徐放性部位と即放性部位とを有する調合製剤であって、患者に投与した際に、上記薬剤を含有する即放性医薬品と生物学的等価となる調合製剤を提供する。本発明の製剤は、上記薬剤のうちの2つ以上について生物学的等価性が同時に得られるという利点がある。本発明の別の実施形態は、鎮咳剤と抗ヒスタミン剤と充血緩和剤とを含み、徐放性部位と即放性部位とを有する非液状調合製剤であって、患者に投与した際に上記薬剤を含有する即放性医薬品と生物学的等価性となる、非液状調合製剤を提供する。
【0060】
本発明の態様の1つは、第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物に関するものであって、
第1の部位が即放性の形態で、有効成分として抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよく、
第2の部位が徐放性の形態で、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを3種の有効成分として含む粒子、ペレット又はビーズを含有し、
前記非液状経口薬剤組成物を患者へ単回投与した際に、少なくとも8時間にわたって得られる上記3種の有効成分の血漿中濃度が、前記有効成分で構成されるFDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価であり、
前記適切な投与回数とは、IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みの1つ以上のラベルで推奨される投与回数に対応する。
【0061】
更に、このような実施形態の1つは鎮咳剤が麻酔性鎮咳剤である。別の実施形態は、第1の部位が充血緩和剤を含まない。このような実施形態では、前記粒子、ペレット又はビーズが更に皮膜を有する。更なる実施形態では、前記抗ヒスタミン剤と鎮咳剤と充血緩和剤とが1個1個の粒子又はペレット又はビーズのなかに結合している。更に別の実施形態では、この粒子、ペレット又はビーズが更に医薬的に許容できるイオン交換マトリックスを含み、抗ヒスタミン剤、鎮咳剤及び充血緩和剤が該イオン交換マトリックスと結合している。このような実施形態では、甘味剤、風味剤、着色剤及びこれらの混合物からなる群から選択される賦形剤を更に含有する。別の実施形態では、安定化剤、分散剤及びこれらの混合物からなる群から選択される添加剤を更に含有する。
更に別の態様として、本発明は、第1の部位と第2の部位とを含む、非液状の経口徐放性薬剤組成物に関するもので、
第1の部位が即放性の形態で、有効成分として抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよく、
第2の部位が粒子、ペレット又はビーズで、徐放性の形態で抗ヒスタミン剤と鎮咳剤と充血緩和剤とを有効成分として含み、
前記3種の有効成分が24時間を超えて安定した血漿中濃度であるように前記非液状薬剤組成物を十分な投与回数で患者に投与した際、前記有効成分で構成されるFDA承認済みの即放性薬剤組成物1種以上を上記と同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となり、
前記適切な投与回数とは、FDA承認済みの即放性薬剤組成物1種以上の同じ期間にわたる投与に関して、FDA承認済みの1つ以上のラベルで推奨される投与回数に対応する。
【0062】
本発明の態様は、ある哺乳動物の少なくとも8時間にわたる抗ヒスタミン剤、鎮咳剤及び充血緩和剤の血漿中濃度を、FDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり同じ哺乳動物に適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価である血漿中濃度とする方法に関するもので、該方法は、
(a)第1の部位と第2の部位とを有する非液状経口徐放薬剤組成物であって、第1の部位が、即放性の形態で有効成分として抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよく、第2の部位が、徐放性の形態で有効成分として抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む粒子、ペレット又はビーズを含有する、非液状経口徐放薬剤組成物を前記哺乳動物に投与することと、
(b)少なくとも8時間にわたる前記3種の有効成分の血漿中濃度が、前記有効成分を含有するFDA承認済みのIR-RLD組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価とすることとを含み、前記適切な投与回数とは、IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みの1つ以上のラベルのなかで推奨される投与回数に対応する。
本発明の別の態様は、哺乳動物に非液状の経口徐放性(ER)薬剤組成物の投与後、抗ヒスタミン剤、鎮咳剤及び充血緩和剤の安定した血漿中濃度を得るための方法であって、前記血漿中濃度とは、活性成分と不活性成分とを含み、前記活性成分が抗ヒスタミン剤(例えばクロルフェニラミン)と鎮咳剤(例えばヒドロコドン)とプソイドエフェドリン(例えばプソイドエフェドリン)とからなる即放性(IR)組成物1種以上を同じ哺乳動物に投与した際に得られる血漿中濃度と生物学的等価な濃度で、該方法は、
【0063】
第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物であって、第1の部位が即放性の形態で、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを有効成分として含み、第2の部位が徐放性の形態で、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを有効成分として含む粒子、ペレット又はビーズを含有する、非液状経口徐放性薬剤組成物を前記哺乳動物に投与することを含み、
前記3種の有効成分が24時間を超えて安定した血漿中濃度であるように前記非液状経口徐放性薬剤組成物を十分な投与回数で哺乳動物に投与した際、前記有効成分を含有するFDA承認済みの即放性(IR)薬剤組成物1種以上を上記と同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となり、
前記FDA承認済みのIR薬剤組成物1種以上の適切な投与回数とは、FDA承認済みのIR薬剤組成物1種以上の同じ期間にわたる投与に関して、FDA承認済みの1つ以上のラベルの中で推奨される投与回数に対応し、
前記FDA承認済みのIR薬剤組成物1種以上の適切な投与回数が、経口ER薬剤組成物の十分な投与回数を上回るものである。
本発明の態様の1つは、医薬的有効成分(API)として抗ヒスタミン剤、鎮咳剤及び充血緩和剤を含有し、薬剤の3種すべてが徐放性(ER)を示す、新規な製剤に関する。例えば、本発明は、クロルフェニラミン、プソイドエフェドリン及びヒドロコドンの、即放剤形と徐放剤形の新規な混合物を1つの製品中に含む製剤を提供する。本発明の新規な製剤としては、3種すべての薬剤を含む既存のIR製品又は上記薬剤のうちの1種又は2種のみを含むIR 及び/又はER製品の混合物と同じ効力を持つ、1日2回の投与が可能なIR/ER調合製品となる製剤が挙げられる。
【0064】
抗ヒスタミン剤は、例えば、抗ヒスタミン性拮抗薬又は逆作動薬として関係のある細胞受容体、例えばH1受容体において作用することにより、体内におけるヒスタミンの放出又は活動を阻害する。ヒスタミンは、アレルギー、風邪及び流感(インフルエンザ)に関連した鬱血、くしゃみ、鼻水、鼻づまり、目のかゆみ、涙目を引き起こす。抗ヒスタミン剤はヒスタミンが細胞に付着し上記のような症状を起こさないようにする。抗ヒスタミン剤の例としては、クロルフェニラミン、ブロムフェニラミン、ジメンヒドリナート、ジフェンヒドラミン、ロラタジン、メクリジン及びクエチアピン等が挙げられる。本発明の実施形態の1つは、抗ヒスタミン剤がクロルフェニラミンである。「クロルフェニラミン」という用語には、この薬剤のいずれの形態も含まれ、具体的な実施形態は、クロルフェニラミンがクロルフェニラミンマレイン酸塩(CPM)で、これは化学名2-ピリジンプロパンアミン -(4-クロロフェニル)-N,N-ジメチル-, (Z)-2-ブテンジオエート(1 :1)としても公知である。
また、充血緩和剤も、風邪や流感、副鼻腔炎又はアレルギーを原因とする鼻づまりや鬱血の軽減を助けることができる。鼻、洞、胸部内の鬱血は、鼻や通気道の細胞膜中の血管が膨張、拡張したことによる。これらの細胞膜には、拡張(膨張及び充血)できる大容量の血管がたくさん通っている。この血管はヒスタミンの刺激により拡張する。これとは対照的に、充血緩和剤はこれらの細胞膜中の血管を収縮させ引き締めるが、細胞膜の外へ大量の血液を押し出すので血管が収縮し、通気道が再度開く。一般に、充血緩和剤は、アドレナリンという一種の刺激剤でもある天然の充血緩和剤と化学的に関連づけられる。最も一般的な経口充血緩和剤はプソイドエフェドリンとフェニレフリンである。本発明の実施形態は、充血緩和剤がプソイドエフェドリン(PSE)である。「プソイドエフェドリン」という用語は、この薬剤のいずれの形態も含まれ、具体的な実施形態はプソイドエフェドリンがプソイドエフェドリン塩酸塩であり、これは化学名ベンゼンメタノール,-[l-(メチルアミノ)エチル]-,[S-(R*,R*)]-, 塩酸塩としても公知である。
【0065】
一般的に、風邪薬は鎮咳剤と去痰剤という2つのグループに分類される。鎮咳剤は、咳が出るのを抑えたり軽減するのに使用される薬で、非麻酔性と麻酔性鎮咳剤とがある。ベンゾナテタート、デキストロメトルファン、カルベタペンタンは非麻酔性鎮咳剤の例である。デキストロメトルファン(鎮咳剤)とグアイフェネシン(去痰剤)を一緒に組み合わせることがある。麻酔性鎮咳剤の一例がヒドロコドン(HC)で、これは鎮痛剤でもあり、天然アヘンのコデイン及びテバインの2つに誘導される半合成オピオイドである。「ヒドロコドン」という用語には、この薬剤のいずれの形態も含まれる。本発明の実施形態は、鎮咳剤がヒドロコドン重酒石酸塩であって、これは化学名モルフィナン-6-オン, 4,5-エポキシ-3-メトキシ-17-メチル-,(5)-, [R-(R* ,R*)]-2,3-ジヒドロキシブタン-ジオエート (1 :1), 水和物(2:5)としても公知である。
抗ヒスタミン剤と充血緩和剤との調合品は、現在のところ、Actifed(登録商標)、Allegra-D(登録商標)、Chlor-Trimeton D(登録商標)、Claritin D(登録商標)、Contac(登録商標)、Co-Pyronil 2(登録商標)、Deconamine(登録商標)、Demazin(登録商標)、Dimetapp(登録商標)、Drixoral(登録商標)、Isoclor(登録商標)、Nolamine(登録商標)、Novafed A(登録商標)、Ornade(登録商標)、 Sudafed Plus(登録商標)、Tavist D(登録商標)、Triaminic(登録商標)及びTrinalin(登録商標)等が市販されている。鎮咳剤も鎮痛剤又は抗ヒスタミン剤等の他の薬剤と組み合わせたものが購入できる。総合感冒薬としても知られているこのような調合製品は多くの症状を同時に治療するものである。
【0066】
しかしながら、FDAのウェブサイトに記載されているように、FDA承認済みの即放性ヒドロコドン鎮咳製剤は、Hycodan(登録商標)、Mycodone(登録商標)及びTussigon(登録商標)に見られるように、ヒドロコドン重酒石酸塩と臭化メチルホマトロピンしか含まれていない。ヒドロコドンとクロルフェニラミンとを含む徐放性鎮咳製剤については、Tussionex Pennkinetic(登録商標)(懸濁液)とTussiCaps(登録商標)(カプセル剤)の2種しか現在のところ承認されていない。www.fda.gov/CDER/ drug/unapproved_drugs/hydrocodone_qa.htm参照のこと(2008年4月11日アクセス)。注目すべきは、ヒドロコドンとホマトロピンとを、グアイフェネシンのような去痰剤又はフェニレフリン もしくはプソイドエフェドリンのような充血緩和剤等の他の薬剤と組み合わせた咳抑制剤は、いずれの剤形も現在のところ許可されていない。このように、ヒドロコドンとPSEのような充血緩和剤とを含むFDA認可済みの市販薬剤は使用できない。
したがって、前述のごとく本発明は既存の市販調合製品とは異なるのである。なぜならば、本願明細書に記載の新規な製剤は、3種類の有効成分である抗ヒスタミン剤、鎮咳剤及び充血緩和剤を含有し、単一の経口製品によって3種類すべての有効成分が体内で徐放性を示すからである。実施形態の1つは、新規製剤が12時間ごとに投与される。他の実施形態では8時間ごと、16時間ごと、24時間ごと等で投与される。
【0067】
実施形態の1つは、この製剤が、咳、風邪、アレルギー兆候の治療を目的とした、徐放性被覆ペレットをシロップ剤に含む分散液である。例えば、該製剤は、10又は15mgのヒドロコドン重酒石酸塩と120mgのプソイドエフェドリン塩酸塩と8mgのクロルフェニラミンマレイン酸塩とを、大人の1回の服用量(5ml)中に一緒に含むことができる。薬剤は塩の形態で使用できるが、塩基の形態をはじめとする他の形態も使用できる。これらの有効成分は、処方箋医薬品としてもOTCの咳や風邪薬としても、各成分単独又は組み合わせて広範な人体投与テストがされている。
本発明の実施形態の1つは、製剤の徐放性(ER)部位が、懸濁液中の被覆ビーズ、粒子又はペレットに相当し、即放性(IR)部位が懸濁液中に存在する。実施形態の1つは、本発明の製剤が、UPM Pharmaceuticals社所有の特許公開公報(U.S. Ser. No. 10/724,276、2003年11月26日出願;U.S. Ser. No. 11/150,572、2005年6月9日出願;U.S. Ser. No. 11/198,937、2005年8月4日出願参照)に記載の技術を用いて調製される。徐放性ビーズ含有懸濁液の特定のタイプの製造と使用に関連する部分は、引用により本願明細書の記載に含まれるものとする。
【0068】
或いは、本発明におけるER部位は、カプセル剤、錠剤又は他の固形経口剤のような固体の剤形で構成され、IR部位は、前記ER部位の外側にある第2の層又は媒体であってもよい。実施形態によると、固形経口製剤には液体成分が含まれない。即ち、固形経口製剤には液相又は分散媒が含まれない。このような実施形態においては、固形経口剤のIR部位は液相又は分散媒を含まない。被験者に投与する前に、この固形経口製剤は、例えば分散媒等の液相と混合しない。同様に、実施形態の1つでは、単一の徐放性調合品で、徐放成分が粒子、ペレット又はビーズの内部で、即放性部位が外側にある(例えば、シロップ剤に懸濁又はカプセル剤、錠剤等の中の粉末)は特定のIR/ER比を示す。この比により、単回投与及び安定状態の条件で、関連リスト収載医薬品(RLD)と生物学的に等価となる血漿中濃度が得られる。
本発明の態様は、非液状の徐放性薬剤組成物に関する。本願明細書で使用の「非液状」という用語は、U.S. Ser. No. 10/724,276(2003年11月26日出願)、U.S. Ser. No. 11/150,572 (2005年6月9日出願)及びU.S. Ser. No. 11/198,937(2005年8月4日出願)に記載されているように、イオン交換マトリックス薬剤複合物を含有する分散相と分散媒とを有する液状組成物ではない組成物を指す。
別の実施形態では、本発明の製剤の投与により、ヒトにおいてある時間にわたり特定の範囲の血漿中濃度が得られるが(AUC、Tmax、T1/2等で測定)、前記血漿中濃度とは、12時間の徐放中、安全で効力のある濃度であり、単回投与の条件と安定状態の条件の両方で即放性RLDと生物学的等価である血漿濃度である。
【0069】
実施形態の1つは、経口徐放性薬剤組成物が第1の部位と第2の部位とを含み、第1の部位が即放状態で、有効成分として抗ヒスタミン剤と鎮咳剤とを含み、更に充血緩和剤を含んでいてもよく、第2の部位が粒子、ペレット又はビーズを含み、各粒子、ペレット又はビーズには有効成分として徐放状態で同じ抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む。別の実施形態では、この薬剤組成物を患者に単回投与すると、少なくとも8時間、例えば、8、12、18又は24時間にわたって上記3種の有効成分の血漿中濃度が、前記有効成分を含むFDA承認済みの即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となる。この適切な投与回数とは、FDA承認済みのIR薬剤組成物の同じ期間にわたる投与に関して、FDAが承認するラベルで推奨される投与回数に相当する。
実施形態によっては、徐放性部位が粒子、ペレット又はビーズの形態である。この粒子、ペレット又はビーズのサイズは、液剤又は固形製剤で経口投与できるサイズである。実施形態の1つは、粒子、ペレット又はビーズが懸濁液のなかで沈殿しないような大きさ及び/又は密度である。実施形態によっては、粒子、ペレット又はビーズの直径は約0.01〜約2000μmの範囲である。別の実施形態では、約0.1〜約1000μm、更に別の実施形態では約1〜約1000μmの範囲である。実施形態の1つは、粒子、ペレット又はビーズの直径が約600μmである。
【0070】
実施形態によると、抗ヒスタミン剤、鎮咳剤及びプソイドエフェドリンを含む懸濁液製剤であって、上記3種すべての薬剤が即放性及び徐放性を示す製剤で、徐放性粒子又は徐放性ペレット又は徐放性ビーズからのプソイドエフェドリンの放出速度が、抗ヒスタミン剤(例えばクロルフェニラミン)又は鎮咳剤(例えばヒドロコドン)の放出速度よりも速いという事実を利用している。例えば、このような製剤は、抗ヒスタミン剤、鎮咳剤及びプソイドエフェドリンを徐放性の粒子、ペレット又はビーズに含有させることができ、一方、即放性液体/ビヒクル部位は抗ヒスタミン剤と鎮咳剤とを含み、プソイドエフェドリンを含まない。このような特定の製剤であっても、3種すべての薬剤の即放及び徐放は、相当するRLDを例えば12時間にわたってFDA承認済みラベル表示のように2回以上の投与をした際の薬剤の放出と生物学的等価となるような即放と徐放を示す。
実施形態によっては、本発明の製剤における有効成分の担体として働く不活性成分は、アンモニオメタクリレート共重合体、ラクトース一水和物、メチルパラベン、微晶質セルロース、プロピルパラベン、純水、アルギン酸ナトリウム、ショ糖、タルク、二酸化チタン、クエン酸トリエチルが挙げられる。これらをはじめとする不活性成分を使って、2つの異なる相、即ち、即放用の薬剤を含みシロップ剤に溶解された分散媒と、薬剤の徐放性部位を含む被覆粒子、ペレット又はビーズを含有する分散相を調製して、本発明の製剤とすることができる。
【0071】
生物学的等価性(BE)は薬物動態学上の用語で、ある薬剤による2種類の調剤の生体内での生物学的な等価性を示すのに使用される。生物学的等価性要件とは、指定された医薬品の生体外及び/又は生体内試験のFDAが課した要件を指し、米国連邦規則集(CFR)第21巻320条1(f)項のもとで市場にだす条件として満たさなければならない。米国食品医薬品局(FDA)は、生物学的等価性を次のように定義している。「適切に計画された試験において、医薬的同等物又は医薬的代替物が同様な条件のもと、同じモル服用量で投与された際に、そこの含まれる有効成分又は有効部位が薬剤の活性サイトにおいて有効になる速度とその程度に著しい差がない」(米国連邦規則集(CFR)第21巻320条1(e)項);2002年7月2日にFDAが発行した「Bioavailability and Bioequivalence Studies for Orally Administered Drug Products - General Considerations (経口投与薬剤についてのバイオアベイラビリティーとバイオエクイバレンスの研究 -一般的考察)も参照のこと。サイトはwww.fda.gov/OHRMS/DOCKETS/98fr/02d-0258-gdl0001.pdf、2003年3月19日正式採用(米国連邦官報(Federal Register)、第68巻13316、2003年3月19日)。
BEは、適切な薬物動態学的パラメータで2つの薬を比較し、それらが許容範囲内かどうかを求めることにより測定できる。ある有効成分の「生物学的等価な」血漿中濃度とは、該有効成分を含む第1の医薬品の投与後に患者の血漿で測定した際、該有効成分に関する単回投与試験で測定したAUCinfinityならびに安定状態試験で測定したCmax、Cmin及びAUCinfinityの対数変換平均値が、90%信頼区間において、該有効成分を含む第2の医薬品の投与後に血漿で測定した該有効成分のCmax、Cmin及びAUCinfinityの80〜125%の範囲にあることを意味する。
【0072】
FDAでは、統計的分析を行ってBEを求めるの前に、薬物動態学的パラメータの対数変換を推奨している。従来のFDAが推奨するBE限界は、対数変換されたPKパラメータが90%信頼限界の範囲で互いの80〜125%内でなければならないというものである。本願明細書の実施形態の1つは、新規製剤が12時間の放出制御型医薬品である。この医薬品は、12時間にわたり2〜3回投与されるRLDの2回又は3回投与量と、t=0とt=6時間、又は、t=0とt=4時間とt=8時間の時点で比較すればよい。
単回投与試験とは、患者に対象の徐放性製品は1回だけ投与し、それに相当する即放性の関連リスト収載医薬品(RLD)は、該即放性RLDについてFDAが承認済みのラベル表示で指示されているような、12時間に相応する服用量で投薬する試験である。安定性試験とは、対象の徐放性製品と即放性の関連リスト収載医薬品(RLD)とを、有効成分の血漿中濃度が安定した濃度に達するまで、試験期間中繰り返し投与する試験である。「Cmax」とは、患者に投与した後、安定状態に到達した後に検出された最高血漿濃度(例えばng/ml)を指す。「Cmin」は、薬剤が安定状態に到達した後に患者から検出された最低血漿濃度(例えばng/ml)を指す。「AUCinfinity」又は「AUC」は、ある医薬品をある時間(例えば、8、12、24、48時間等)にわたり1回以上投与した後、血漿中の有効成分濃度を0時間から無限時間までプロットした曲線の下面積(例えばng/ml x hr)を指す。有効成分の「Cmin」、「Cmax」及び「AUCinfinity」は周知方法により測定できる。
【0073】
「関連リスト収載医薬品」又は「RLD」とは、医薬品簡略承認申請(ANDA)の承認を求める際に出願人が根拠にできる医薬品としてFDAが認定したリスト収載薬品を指す。このように、単一の薬剤を含有するRLDはFDAにより明示されている。ジェネリック薬品又は505(b)(2)出願のもとで提出された薬品に関して、ある有効成分についての製造業者が2社以上の場合、RLDの役割を果たす製品の供給元をどの製造業者にするかはFDAが選択する。本発明の目的のためには、各有効成分を別々に含有する即放性医薬品がRLDとして使われる。本発明の製剤と比較するためには、複数のRLDを組み合わせて、たとえばRLDの投与説明書にしたがって順次投与すればよい。同様に、本発明の目的のためには、「関連リスト収載医薬品」又は「RLD」という用語は、単独薬剤RLDが2つ以上混ざった液体も指す。例えば、RLDが、抗ヒスタミン剤のRLDと鎮咳剤のRLDと充血緩和剤のRLDとを含む混合液体であってもよい。クロルフェニラミンに対してFDAが承認しているRLDは、Chlor-Trimetonシロップ剤である。PSEに対してFDAが承認しているRLDはSudafedシロップ剤である。ヒドロコドンに対してFDAが承認しているRLDはHycodanシロップ剤である。本発明の製剤を投与して得られる血漿中濃度を比較し、生物学的等価性の評価を行う場合、単独の薬剤を含有するRLDを3種類含む混合液は、上記RLDのいずれのFDA承認済みラベル表示に従ってヒトへの投与を行うとよい。
2001年1月、FDAは生物学的等価性試験を説明するガイドラインを発表した。概して、試験のやりかた、データの分析方法等で、「Guidance for Industry: Statistical Approaches to Establishing Bioequivalence」(「Statistical Approaches」)という表題の文書として発表された。この文書の中で、FDAは、医薬品が制定法上のBEの定義を得ているかどうかの決定に使用する基準を規定した。本願明細書中、対象の医薬品が関連リスト収載医薬品に生物学的等価であるかを言及する場合、これらの文書に記載の「average BE」の定義を適用する。
【0074】
「Statistical Approaches」2頁に記載のごとく、FDAは以下のことを推奨している。『標準的な生体内BEテストのデザインは、テスト(T)薬品と参照(R)薬品を単回投与又は反復投与のいずれかで健康な被験者に別々に投与することを基礎とし、医薬品投与の2通りの順番にランダムに割り付ける。(中略)曲線下面積(AUC)や最高濃度(Cmax)等の薬物動態学的測定値の統計分析は2つの片側検定の方法を基準とし、T薬品とR薬品の投与後に求めた薬物動態学的測定値の平均値が同等かどうかを決める。このアプローチは平均生物学的等価性と称され、T薬品とR薬品の測定値の平均値(母幾何平均値(population geometric means))の比に関して90%信頼区間の計算も行う。生物学的等価性(BE)を確立するには、医薬品平均値の比について計算した信頼区間がBE範囲内、通常は80〜125%の中に収まらなければならない。』「Statistical Approaches」第IIB章、2頁(原文では強調)
BEデータの統計分析は、バイオアベイラビリティーのデータ(例えばAUC、Cmax、Cmin等)の対数変換用の統計モデルを基準とする。「Statistical Approaches」の手引書では、BE測定値の対数変換(自然対数又は底が10の対数)が提唱されている。データ分析については、「Statistical Approaches」では対数変換したBE測定値にパラメトリック(正規理論)法を用いることが推奨されている。『方程式2又は3に記載の基準を用いた平均生物学的等価性に関しては(第IV.A章)、μT-μR量の90%信頼区間を構築し、この信頼区間が区間[-θA, θA]に含まれる場合は平均生物学的等価性という結論に到達する、というのが一般的なアプローチである。(中略)対数変換したデータの平均値の差についての90%信頼区間は、実験計画に合った方法を使って計算しなければならない。』「Statistical Approaches」第VI.B章、10頁
【0075】
本発明の実施形態は、一般的に安全かつ有効な(GRASE)3種の有効成分、即ち、抗ヒスタミン剤、鎮咳剤及び充血緩和剤の利点を1つの徐放性薬品に統合するものである。現在、これらの薬剤を一緒に使用する場合は、これらの薬剤をそれぞれの即放性製剤で一日4〜6回投与することになる。3つの効果を発揮する1つにまとまった徐放性調合薬品はOTC医薬品も処方箋による医薬品でも利用できないからである。そこで、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含むIR/ER製剤であって、1つの医薬品を投与することで3種類すべての薬剤が徐放性を示す製剤であれば、3種類すべての有効成分は含有していない現行の医薬品及び/又は単に即放性の現行の医薬品よりも有利である。例えば、本発明の製剤を使用すると、患者にとって必要な薬の量が、即放性薬品の1日50〜55mlに比べて、例えば5ml等の少ない量で、風邪、流感又はアレルギーの症状を軽減することになる。また、本発明の製剤は、投与単位4オンスの便利な容器に入れて販売することもできる。また、患者にとっては薬を摂取する回数が減り、1日を通して苦痛軽減状態でいるために必要な服用量をとりそこなう危険性が小さくなる。患者のコンプライアンスという問題は決してなくなることがない、十分に認識された問題であることを考慮すると、単回投与(例えば12時間)で3種類すべての薬剤について生物学的等価性のある服用量を提供する本製剤は、現在利用できる製剤にくらべて著しい利点をもたらすものである。
本発明の実施形態は、クロルフェニラミン、プソイドエフェドリン及びヒドロコドンの即放剤形と徐放剤形との新規な混合物を1つの医薬品内に含む製剤を提供するもので、その単一医薬品は患者への投与回数を減らす一方で、上記薬剤を含有する即放性医薬品を摂取した患者と生物学的等価性が得られる。
【0076】
また、本発明は薬剤調合製剤及びそのような製剤の製造方法に関するもので、製剤は懸濁液又は固形カプセル剤もしくは錠剤の安定した徐放性経口薬剤で、粒子、ペレット又はビーズを含み、個々の粒子、ペレット又はビーズの中に2種以上の有効成分が含まれている。このアプローチは即放性製剤だけでなく、徐放性製剤に勝る多数の利点がある。
本発明の他の実施形態は、1種以上の薬剤を含むペレット、ビーズ又は粒子を有する徐放性成分を含有する新規な経口懸濁液製剤であって、前記ペレット、ビーズ又は粒子がシロップ剤に懸濁した製剤を提供する。シロップ剤も1種以上の薬剤を含んでもよい。本願明細書では、このような経口懸濁液の一例を挙げる。この経口懸濁液製剤は従来よりも優れた特性がある。例えば、例示する製剤の徐放性成分はビーズを有し、ビーズはイオン交換マトリックスと結合した3種類の薬剤を含有し、このビーズがシロップ剤の中で懸濁しているので、他の液体製剤よりも非常に高い製品安定性が本製剤に付与される。通常、水性の環境において薬剤は崩壊を起こす。固形剤形とすることで崩壊を最小限にできるが、液体製剤にみられる投与の容易さや投与量の融通性は損なわれる。少なくとも1つの実施形態では、本発明の剤形は以下の2段階のアプローチにより水への暴露を最小限にしている。(1)ビーズは、懸濁させるのに使用している液相に容易に溶けるというよりも、むしろ徐放性を示す。(2)液相はシロップ剤であるが、シロップ剤中の糖類により液相の水分活性が下げられる。また、水分活性を低下させ浸透圧を上昇させることにより、シロップ剤は次の働きをする。(a)薬剤の徐放性ビーズからシロップ剤への浸出を最小限に抑える。(b)ビーズの崩壊を防ぐ。逆にいえば、ビーズは患者が摂取した後、例えば、腸内で膨潤し崩壊することで薬剤を放出することができ、ERからの薬剤放出を可能にする。
【0077】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の液状の放出制御型薬剤組成物中のイオン交換マトリックス樹脂-薬剤複合物の濃度は、例えば、特定の薬剤に応じて、また、イオン交換マトリックス樹脂-薬剤複合物内の薬剤の含有量や治療すべき症状又は兆候、患者の年齢などに応じて広範囲にわたり変わるうる。こうした組成物の実施形態では、液状の放出制御型薬剤組成物中のイオン交換マトリックス樹脂-薬剤複合物の濃度の範囲は、液状の放出制御型薬剤組成物の全質量を基準に約5〜約90質量%である。この組成物の別の実施形態では、イオン交換マトリックス樹脂-薬剤複合物の質量の範囲は、液状の放出制御型薬剤組成物の全質量を基準に約10〜約50質量%である。更に別の実施形態では、イオン交換マトリックス樹脂-薬剤複合物の質量の範囲は、液状の放出制御型薬剤組成物の全質量を基準に約20〜約40質量%である。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の薬剤組成物の実施形態において、分散相の調製方法は、粉末状の薬剤と粉末状のイオン交換マトリックスとを混合することを含む。このような実施形態では、薬剤とイオン交換マトリックスは塩の形態のものを使えばよい。本発明の分散相を調製する粉末混合方法は、従来の方法に比べて費用効率及び時間効率がよい。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の薬剤組成物の実施形態の1つでは、イオン交換マトリックスがアルギン酸ナトリウムである。非制限的実施形態であるが、クロルフェニラミンマレイン酸塩、プソイドエフェドリン塩酸塩、ヒドロコドン重酒石酸塩の粉末を1種又は一緒にして、アルギン酸ナトリウム粉末と混合すればよい。別の実施形態は、薬剤とイオン交換粉末との混合物に、ラクトース、微晶質セルロース及び/又は他の賦形剤を添加してもよい。
【0078】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の薬剤組成物の別の実施形態では、薬剤とイオン交換粉末とを混合した後、水を加えて湿式の塊を作成し、押出と球状化を行う。得られた薬剤とイオン交換マトリックスとを含むコアビーズ又はペレットは流動床乾燥機で乾燥させればよい。実施形態の1つでは、個々のビーズ又は各ペレットに、同一のイオン交換マトリックスに結合した数種の有効成分が含まれる。例えば、個々のビーズ又は各ペレットに、アルギン酸ナトリウム粉末に結合したクロルフェニラミンマレイン酸塩、プソイドエフェドリン塩酸塩、ヒドロコドン重酒石酸塩が含まれる。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の薬剤組成物の実施形態では、流動床プロセッサー中、クエン酸トリエチル及びタルクの存在下、得られたビーズにEUDRAGIT(登録商標)をコーティングする。その後、被覆ビーズをタルクと混合して炉内で硬化させる。実施形態によると、被覆ビーズは2時間、4時間、8時間、16時間、24時間又は48時間硬化させる。実施形態によっては、被覆ビーズを約16〜約24時間硬化させる。被覆ビーズの硬化時間は、被覆ビーズからの薬剤の放出速度に影響を及ぼすことがある。
【0079】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる被覆ビーズを、塩の形態の薬剤と水とショ糖とを含む分散媒に懸濁させる。本発明の組成物の非制限的実施形態の1つでは、分散媒がクロルフェニラミンマレイン酸塩とヒドロコドン重酒石酸塩とを含む。本発明の組成物の別の実施形態では、分散媒が更にプソイドエフェドリン塩酸塩も含む。また、分散媒は防腐剤、矯味剤、他の不活性添加剤を更に含んでもよい。このような実施形態で得られた液状の放出持続型医薬品は、物理的安定性と化学的安定性を瓶の中で維持することができ、かつ、患者に投与する際は薬剤の放出を制御することができる。
本発明の態様の1つは、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンとからなる液状の放出制御型薬剤組成物の製造方法に関するもので、該方法は、
(a)クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる有効成分が単一粒子又は単一ペレット又は単一ビーズ内で結合している、粒子、ペレット又はビーズを作製する工程を含む、分散相を調製する工程と、
(b)クロルフェニラミン、ヒドロコドン、そして任意であるがプソイドエフェドリンを含んでいてもよい有効成分を含有する分散媒を調製する工程と、
【0080】
(c)前記粒子、ペレット又はビーズを皮膜でコーティングする工程と、
(d)前記ビーズを分散媒に分散させる工程とを含む。
前記方法の1つの実施形態は、分散相調製工程が、クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンを医薬的に許容できるイオン交換マトリックスと結合させる工程を更に含む。別の実施形態は、分散相調製工程が、クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンならびに医薬的に許容できるイオン交換マトリックスとからなる粒子、ペレット又はビーズを調製する工程を更に含み、個々の粒子、ペレット又はビーズ内ではクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンがイオン交換マトリックスに結合している。別の実施形態は、分散相調製工程が、クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンならびにイオン交換マトリックス粉末を混合する工程を更に含む。更に別の実施形態は、分散相調製工程が、クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンならびにイオン交換マトリックス粉末の湿式造粒、押出、球状化工程を更に含む。実施形態の1つは、クロルフェニラミン、ヒドロコドン、プソイドエフェドリン及びイオン交換マトリックスが塩の形態のものを使用する。
【0081】
本発明の製剤は、2種以上の薬剤の生物学的等価性を同時に実現できるところが利点の1つである。比較用医薬品の場合、各薬剤が同じ放出プロファイルにならないこともあり、異なる服用量や投与回数が必要になることがある。更に、徐放の方法が異なれば(例えば、異なる樹脂を使用)、各薬剤の放出プロファイルは患者の食餌又は生理機能の差に影響をうける可能性があり、患者によっては、ある薬剤は摂取しすぎで、別の薬剤の摂取は少なすぎるということがおきる。3種類の薬剤が、例えば単一の放出方法を有するビーズ1個1個の中に配置されることによって、薬剤放出プロファイルは患者全体の中でより一貫性があるものになる。
3種類の薬剤をそれぞれ別のビーズに配置した後にビーズを混合した場合、特に少量の場合、完全に均一な混合物ではないことがある。このような不均一が起きると、結果、薬剤の相対的服用量が不正確になる。即ち、ある薬剤は過多となり、別の薬剤は過少となる。この不均一は、無作為な変動によるだけでなく、3種のビーズ混合物において3種のビーズの物性が異なることに起因して起こることもある。例えば、プソイドエフェドリンはすべての薬剤のなかで占める割合(質量比)が高い。3種類の薬剤が同じ大きさと量のビーズそれぞれに充填されると、賦形剤に対してプソイドエフェドリンは量が多くなるので、プソイドエフェドリンのビーズは、ヒドロコドンのビーズ又はクロルフェニラミンのビーズと密度が異なる結果になる。このような密度の違いは均質性の欠如につながる。所定の温度及び圧力においてビーズが同じ密度になるように調整したとしても、条件が変わればそのまま維持することはできない。
【0082】
また、密度の違いを利用すれば、薬剤の種類に応じてビーズを故意に分離し、プソイドエフェドリンを集めて(say)薬剤の不正使用に転用することができる。一方、一粒のビーズの中にすべての薬剤が存在する場合、ビーズの物性の違いを基に1つの薬剤を他の薬剤から部分的に単離することは不可能である。更に、このような医薬品の製造方法であれば、粒子、ペレット又はビーズ1個に含まれるいずれの有効成分も乱用や転用を思いとどまらせ、阻止することになる。粒子、ペレット又はビーズ1個の中に2種以上の薬剤を混合又は注入することにより、乱用を目的として個人が医薬品から個々の有効成分を抽出又は分離することは容易ではない。更に、粒子、ペレット又はビーズ1個の中に2種以上の有効成分を混合又は注入することで、調合薬品全体に存在する粒子、ペレット又はビーズの表面積が低減するので、医薬品及び有効成分の安定性が増大する。
薬剤調合医薬品の実施形態の1つは、粒子、ペレット又はビーズ1個につき2種以上の有効成分を有する粒子、ペレット又はビーズを含有することにより、1つの有効成分を分離又は単離する潜在的可能性が減少する。同様に、本発明は、製剤に含まれるいずれか1つの成分についての乱用の潜在的可能性を低減する、風邪及びアレルギー用調合薬剤製剤の製造方法を提供する。例えば、製品に含まれる各ビーズが、医薬的に許容できるイオン交換マトリックスと、該イオン交換マトリックスに結合した2種以上の医薬的に許容できる有効薬剤成分とを含んでいてもよく、これは、UPM Pharmaceuticals社所有の特許公開公報(U.S. Ser. No. 10/724,276、U.S. Ser. No. 11/150,572、U.S. Ser. No. 11/198,937、引用により本願明細書の記載に含まれるものとする)に記載の特定のタイプの徐放性ビーズを含む懸濁液の製造と使用に関する技術を用いて調製してもよい。にわか作りの違法薬物研究所では、このような薬剤調合医薬品からはいずれの単一有効薬剤成分も簡単に抽出、分離又は単離することはできない。
【0083】
このように本発明の製剤は、咳/風邪の市場で現在入手可能な他の製剤とは異なる。例えば、Tussionex(登録商標)徐放性懸濁液は、咳止め/抗ヒスタミン剤の調合品で、ヒドロコドンとクロルフェニラミンを含み風邪やアレルギーによる咳や上気道の兆候をやわらげるのに使用される。この懸濁液型医薬品は薬剤-イオン交換樹脂ビーズを含有するが、懸濁液中の個々のビーズにはヒドロコドン又はクロルフェニラミンのいずれかが充填されており、両方の薬剤が1つのビーズには含まれていない。
また、本発明の製剤が現在市販されている製剤と異なる点は、インターネットが示す乱用薬物の製造又は単離を行うための、一般的な家庭用品、簡単な製造装置又はいわゆる「レシピ」は本発明の製剤には容易に利用できない、という点である。むしろ、本発明の製剤から対象となる乱用可能な薬物を分離することは、通常は大学や企業の研究所でしか利用できないような、例えば、複合クロマトグラフィー等の非常に技術的に高度な装置や科学的な熟練及び技術が必要となる。
本発明の別の実施形態では、イオン交換マトリックス薬剤粒子、ペレット又はビーズを有することにより、薬剤調合医薬品は乱用及び/又は転用の潜在的可能性を低減する。各ビーズには、医薬的に許容できるイオン交換マトリックスとイオン交換マトリックスに結合した2種以上の医薬的に許容できる有効成分とが含まれていてもよい。
【0084】
また、本発明は、少なくとも1種の有効成分の乱用を予防又は低減する方法も包含し、該方法は、薬剤調合医薬品を調製する工程を含むが、該医薬品が、粒子、ペレット又はビーズを含み、個々の粒子、ペレット又はビーズが医薬的に許容できるイオン交換マトリックスとイオン交換マトリックスに結合した2種以上の医薬的に許容できる有効成分とを含む。
別の実施形態では、単一の有効成分の抽出、単離又は分離の可能性を阻止又は低減するための方法が薬剤調合医薬品を調製する工程を含むが、該医薬品が、粒子、ペレット又はビーズを含み、個々の粒子、ペレット又はビーズが医薬的に許容できるイオン交換マトリックスとイオン交換マトリックスに結合した2種以上の医薬的に許容できる有効成分とを含む。マトリックス粒子、ペレット又はビーズに2種以上の有効成分が含まれているという事実により、工業レベルでない、クロマトグラフィー装置等の高品質装置をもたないにわか作りの違法薬物研究所で素人がいずれかの薬剤を単独に抽出又は分離することは、きわめて難しくなる。
別の実施形態は、本発明の薬剤調合医薬品により、患者が、例えば3つの異なる有効成分を適切かつ正確に投薬できるようになる。多数の製剤(例えば、異なる3種の医薬品で、それぞれに即放性有効成分が単独に含まれている)を使って患者が医者にかからず自分で治療する場合、適切かつ正確な投薬は困難であることが多く、とりわけ、1種又はそれ以上の薬剤の過剰投与又は過少投与をしないようにすること、及び/又は、副作用を最小限に抑えてすべての薬剤の治療効果を最大限に引き出すという点に関して、適切かつ正確な投薬は困難であることが多い。本発明の調合医薬品は、すべての関連薬剤が一つの剤形の状態で、例えば、12時間徐放型剤形で提供、投与されるので、上記のような問題が回避される。
【0085】
薬剤調合医薬品の1つの実施形態は懸濁液組成物で、徐放性薬剤を含有する被覆イオン交換マトリックス薬剤粒子、ペレット又はビーズを含む分散相を含み、更に任意であるが、即放性薬剤を含む分散媒であって、シロップ剤に溶解した分散媒を含んでいてもよい。
実施形態の1つは、使用する放出制御型薬剤組成物の投与量が、患者において、例えば、ヒトにおいて治療効果を達成するのに十分な量である。この「治療効果」という用語は、風邪、流感又はアレルギーに対するいずれの効果も意味し、咳の重篤度及び/又は回数、咳の兆候、鼻漏、鬱血又はくしゃみの低減などの症状緩和をはじめとするが、これらに制限されない効果、及び/又は、自覚的な健康状態の改善につながる他の生物学的効果を意味する。
本発明の具体的態様の1つは、は第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物に関するもので、
第1の部位が即放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよく、
第2の部位が徐放性の剤形で3種の有効成分として抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む粒子、ペレット又はビーズを含有し、
前記非液状経口薬剤組成物を患者へ単回投与した際に、少なくとも8時間にわたって得られる上記3種の有効成分の血漿中濃度が、前記有効成分で構成されるFDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となり、
前記適切な投与回数とは、IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みの1つ以上のラベルのなかで推奨される投与回数に対応する。
【0086】
前記組成物の実施形態の1つは鎮咳剤が麻酔性鎮咳剤である。前記組成物の別の実施形態は、第1の部位が充血緩和剤を含まず、抗ヒスタミン剤と鎮咳剤しか含まない。具体的実施形態は、抗ヒスタミン剤がクロルフェニラミンである。別の具体的実施形態は、鎮咳剤がヒドロコドンである。更に別の具体的実施形態は、充血緩和剤がプソイドエフェドリンである。前記組成物の実施形態の1つにおいて、粒子又はペレット又はビーズが更に皮膜を有する。
前記組成物の実施形態の1つは、前記抗ヒスタミン剤と鎮咳剤と充血緩和剤とが1個1個の粒子又はペレット又はビーズ内で結合している。このような実施形態では、粒子、ペレット又はビーズが医薬的に許容できるイオン交換マトリックスを更に含み、抗ヒスタミン剤、鎮咳剤及び充血緩和剤がイオン交換マトリックスと結合している。
上記組成物の実施形態によっては、甘味剤、風味剤、着色剤及びこれらの混合物からなる群から選択される賦形剤を更に含有する。別の実施形態では、安定化剤、分散剤及びこれらの混合物からなる群から選択される添加剤を更に含有する。
本発明の態様の1つは、第1の部位と第2の部位とを含む、非液状の経口徐放性薬剤組成物に関するもので、
第1の部位が即放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよく、
第2の部位が、有効成分として抗ヒスタミン剤と鎮咳剤と充血緩和剤とを徐放剤形で含む、粒子、ペレット又はビーズで、
【0087】
前記3種の有効成分が24時間を超えて安定した血漿中濃度であるように前記非液状薬剤組成物を十分な投与回数で患者に投与した際、前記有効成分の血漿中濃度が、前記有効成分で構成されるFDA承認済みの即放性薬剤組成物1種以上を上記と同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となり、
前記適切な投与回数とは、FDA承認済みの即放性薬剤組成物1種以上の同じ期間にわたる投与に関して、FDA承認済みの1つ以上のラベルのなかで推奨される投与回数に対応する。
本発明の別の態様は、ヒト被験者の咳、風邪、流感又はアレルギー兆候の治療方法に関するもので、本願明細書に記載の非液状経口徐放性薬剤組成物を該被験者に投与する工程を含む。このような実施形態において、医薬組成物は、1日1回又は1日2回投与ができる二段階放出(dual release)製剤としてヒトに投与される。
本発明の更に別の態様は、風邪、流感又はアレルギーに関連した咳、咳の兆候、鼻漏、鬱血又はくしゃみを少なくとも8時間にわたり治療する方法に関するもので、このような治療を必要とするヒトに対し、風邪又はアレルギーに関連した咳、咳の兆候、鼻漏、鬱血又はくしゃみを少なくとも8時間にわたり治療するのに有効な本願明細書記載の非液状薬剤組成物を単回投与することを含む方法に関する。
本発明の更に別の態様は、本願明細書記載の非液状経口徐放性薬剤組成物の製造方法に関するもので、
有効成分として、抗ヒスタミン剤と鎮咳剤とを含み、更に充血緩和剤を含んでもよい即放性部位を調製する工程と、
【0088】
抗ヒスタミン剤と鎮咳剤と充血緩和剤とを有効成分として含有する粒子、ペレット又はビーズの作製を含む徐放性部位を調製する工程と、
前記粒子、ペレット又はビーズを皮膜でコーティングする工程と、
前記徐放性部位と前記即放性部位とを混合する工程とを含む方法に関する。
前記方法の実施形態は、1個1個の粒子、ペレット又はビーズの中に2種以上の有効成分が結合している粒子、ペレット又はビーズを作製する工程を更に含む。このような方法の具体的実施形態の1つは、前記2種以上の有効成分を医薬的に許容できるイオン交換マトリックスと結合させる工程を更に含む。具体的実施形態は、抗ヒスタミン剤がクロルフェニラミンである。別の具体的実施形態は、鎮咳剤がヒドロコドンである。更に別の具体的実施形態は、充血緩和剤がプソイドエフェドリンである。
本発明の別の態様は、少なくとも8時間にわたる抗ヒスタミン剤、鎮咳剤及び充血緩和剤のある哺乳動物における血漿中濃度を、FDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり同じ哺乳動物に適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価である血漿中濃度にする方法に関するもので、該方法は、
第1の部位と第2の部位とを有する非液状経口徐放薬剤組成物であって、第1の部位が即放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよく、第2の部位が徐放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む粒子、ペレット又はビーズを含有する、非液状経口徐放薬剤組成物を前記哺乳動物に投与することと、
【0089】
少なくとも8時間にわたる前記3種の有効成分の血漿中濃度が、前記有効成分を含有するFDA承認済みのIR-RLD組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価とすることとを含み、前記適切な投与回数とは、IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みの1つ以上のラベルのなかで推奨される投与回数に対応する。
本発明の更に別の態様は、非液状の経口徐放性(ER)薬剤組成物の投与後、抗ヒスタミン剤、鎮咳剤及び充血緩和剤の安定した血漿中濃度を哺乳動物において得るための方法であって、前記血漿中濃度とは、活性成分と不活性成分とを含み、前記活性成分がクロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる即放性(IR)組成物1種以上を前記哺乳動物に投与した際に得られる血漿中濃度と生物学的等価であり、該方法は、
第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物であって、第1の部位が即放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含み、第2の部位が徐放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む粒子、ペレット又はビーズを含有する、非液状経口徐放性薬剤組成物を前記哺乳動物に投与することを含み、
【0090】
前記3種の有効成分が24時間を超えて安定した血漿中濃度であるように前記非液状経口徐放性薬剤組成物を十分な投与回数で哺乳動物に投与することにより、前記有効成分を含有するFDA承認済みの即放性(IR)薬剤組成物1種以上を上記と同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価な有効成分の血漿中濃度となり、
前記FDA承認済みのIR薬剤組成物1種以上の適切な投与回数とは、FDA承認済みのIR薬剤組成物1種以上の同じ期間にわたる投与に関して、FDAが承認するの1つ以上ラベルのなかで推奨される投与回数に対応し、
前記FDA承認済みのIR薬剤組成物1種以上の適切な投与回数が、経口ER薬剤組成物の十分な投与回数を上回るものである。
本発明の態様の1つは、第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物に関し、
前記第1の部位は、クロルフェニラミンとヒドロコドンとを含み、さらにプソイドエフェドリンを含んでもよい有効成分を即放剤形で含有し、
前記第2の部位は、3種の有効成分としてクロルフェニラミンとヒドロコドンとプソイドエフェドリンとを徐放剤形として含む粒子、ペレット又はビーズを含有し、
前記非液状経口薬剤組成物を患者へ単回投与することにより、少なくとも8時間にわたって得られる上記3種の有効成分の血漿中濃度が、前記有効成分で構成されるFDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となり、
前記適切な投与回数とは、IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みの1つ以上のラベルのなかで推奨される投与回数に対応する。
【0091】
前記薬剤組成物の実施形態によっては、少なくとも8時間という期間が12時間である。別の実施形態では、少なくとも8時間という期間が24時間である。
前記薬剤組成物の他の実施形態では、第1の部位にプソイドエフェドリンが含有されていない。
薬剤組成物の実施形態の1つは、経口固形剤形である。別の実施形態では、薬剤組成物は経口カプセル剤である。
前記薬剤組成物の実施形態によっては、粒子、ペレット又はビーズが更に皮膜を有する。このような実施形態で、該粒子、ペレット又はビーズは医薬的に許容できるイオン交換マトリックスを更に含み、このイオン交換マトリックスにクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンが結合している。
前記薬剤組成物の実施形態では、甘味剤、風味剤、着色剤及びこれらの混合物からなる群から選択される賦形剤を更に含有する。別の実施形態では、薬剤組成物が、安定化剤、分散剤及びこれらの混合物からなる群から選択される添加剤を更に含有する。
前記薬剤組成物の実施形態によっては、少なくとも8時間という期間が12時間であり、かつ、薬剤組成物が、8〜12mgのクロルフェニラミンマレイン酸塩と、10〜15mgのヒドロコドン重酒石酸塩と、少なくとも120mgのプソイドエフェドリンとからなる有効成分を1回の服用量5ml中に含む。他の実施形態では、少なくとも8時間という期間が24時間であり、かつ、薬剤組成物が、16〜24mgのクロルフェニラミンマレイン酸塩と、20〜30mgのヒドロコドン重酒石酸塩と、少なくとも240mgのプソイドエフェドリン塩酸塩とからなる有効成分を1回の服用量5ml中に含む。
【0092】
本発明の別の態様は、風邪、流感又はアレルギーに関連した咳、咳の兆候、鼻漏、鬱血又はくしゃみを少なくとも8時間にわたり治療する方法に関するもので、このような治療を必要とするヒトに対し、風邪又はアレルギーに関連する咳、咳の兆候、鼻漏、鬱血又はくしゃみを少なくとも8時間にわたり治療するのに有効な、有効成分がクロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる薬剤組成物を単回投与することを含む方法に関する。
前記方法の実施形態によっては、少なくとも8時間という期間が12時間である。この方法の別の実施形態では、少なくとも8時間という期間が24時間である。
本発明の態様の1つは、有効成分としてクロルフェニラミンとヒドロコドンとプソイドエフェドリンとを含む非液状経口医薬製剤を包含し、前記製剤により有効成分は即放性(IR)及び徐放性(ER)を示し、
前記製剤が即放性部位と徐放性部位とを有し、
前記非液状経口製剤を患者へ単回投与することにより、少なくとも8時間にわたるクロルフェニラミンとヒドロコドンとプソイドエフェドリンの血漿中濃度が、クロルフェニラミン、ヒドロコドン及び/又はプソイドエフェドリンを含有する即放性組成物1種以上を同じ時間にわたって2回以上投与した際に得られる血漿中濃度と生物学的等価となる。
【0093】
前記製剤の実施形態の1つは、少なくとも8時間という期間が12時間である。別の実施形態では、少なくとも8時間という期間が24時間である。
本発明の態様は、前記非液状経口医薬製剤の製造方法に関するもので、クロルフェニラミンとヒドロコドンとを含み、プソイドエフェドリンを含まない即放性部位を調製する工程を含む。
本発明の別の実施形態は前記非液状経口医薬製剤の製造方法であって、徐放性部位の調製工程を有し、該徐放性部位の調製工程が、1個1個の粒子、ペレット又はビーズがクロルフェニラミンとヒドロコドンとプソイドエフェドリンとを含有する、粒子、ペレット又はビーズを調製することを含み、更に、前記徐放性部位を前記即放性部位と混合する工程を含む製造方法である。
実施形態によっては、前記方法が、粒子、ペレット又はビーズを皮膜でコーティングする工程を、前記徐放性部位と即放性部位との混合工程の前に更に含む。
本発明の態様は、第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物に関するもので、
第1の部位が、クロルフェニラミンとヒドロコドンを含み、更にプソイドエフェドリンを含んでもよい有効成分を即放剤形で含有し、
第2の部位が、有効成分としてクロルフェニラミンとヒドロコドンとプソイドエフェドリンとを徐放剤形で含む粒子、ペレット又はビーズで、
【0094】
前記3種の有効成分が24時間を超えて安定した血漿中濃度であるように前記非液状薬剤組成物を十分な投与回数で患者に投与することにより、前記有効成分の血漿中濃度が、前記有効成分を含むFDA承認済みの即放性薬剤組成物1種以上を上記と同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となり、
前記適切な投与回数とは、FDA承認済みの即放性薬剤組成物1種以上の同じ期間にわたる投与に関して、1つ以上のFDAが承認するのラベルのなかで推奨される投与回数に対応する。
本発明の別の態様は、第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物に関するもので、
第1の部位が、クロルフェニラミンとヒドロコドンを含み、更にプソイドエフェドリンを含んでもよい有効成分を即放剤形で含有し、
第2の部位が、有効成分としてクロルフェニラミンとヒドロコドンとプソイドエフェドリンとを徐放剤形で含有する、粒子、ペレット又はビーズを含み、
前記非液状薬剤組成物を患者へ単回投与することにより、少なくとも8時間にわたって得られる上記3種の有効成分の血漿中濃度が、前記3種すべての有効成分を含むFDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となり、
前記適切な投与回数とは、IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みのラベルで推奨される投与回数に対応する。
【0095】
本発明の更に別の態様は、(1)クロルフェニラミン とヒドロコドンとを有効成分として含有する即放性(IR)部位と、(2)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとを有効成分として含む徐放性(ER)部位とを有する非液状経口医薬組成物に関するもので、
経口組成物におけるIR部位のクロルフェニラミン対ER部位のクロルフェニラミンの質量比が約25:75、IR部位のヒドロコドン対ER部位のヒドロコドンの質量比が約25:75、IR部位のプソイドエフェドリン対ER部位のプソイドエフェドリンの質量比が約0:100であって、
前記非液状経口組成物をヒトへ単回投与することにより、前記経口組成物に存在するヒドロコドンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を2回以上投与した際に得られるAUCinfinityと同等のヒドロコドンのAUCinfinityが得られ、
前記非液状経口組成物をヒトへ単回投与することにより、前記経口組成物に存在するプソイドエフェドリンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を2回以上投与した際に得られるAUCinfinityと同等のプソイドエフェドリンのAUCinfinityが得られる。
前記非液状経口組成物の実施形態によっては、非液状経口組成物をヒトへ単回投与することにより、前記経口組成物に存在するクロルフェニラミンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を2回以上投与した際に得られるAUCinfinityと同等のクロルフェニラミンのAUCinfinityが得られる。
【0096】
本発明の更に別の態様は、(1)クロルフェニラミン とヒドロコドンとを有効成分として含有する即放性(IR)部位と、(2)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとを有効成分として含む徐放性(ER)部位とを有する非液状経口医薬組成物に関するもので、
経口組成物におけるIR部位のクロルフェニラミン対ER部位のクロルフェニラミンの質量比が約25:75、IR部位のヒドロコドン対ER部位のヒドロコドンの質量比が約25:75、IR部位のプソイドエフェドリン対ER部位のプソイドエフェドリンの質量比が約0:100であって、
前記非液状経口組成物ではヒトにおけるヒドロコドンのAUCinfinityが、この経口組成物に比べてヒドロコドンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等であることを示し、12時間にわたり前記経口組成物は1回投与し、前記IR-RLDは時間がゼロのときと、6時間後の2回投与するもので、
前記非液状経口組成物ではヒトにおけるプソイドエフェドリンのAUCinfinityが、この経口組成物に比べてプソイドエフェドリンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等であることを示し、12時間にわたり前記経口組成物は1回投与し、前記IR-RLDは時間がゼロのときと、6時間後の2回投与するものである。
【0097】
実施形態により、前記非液状経口組成物ではヒトにおけるクロルフェニラミンのAUCinfinityが、この経口組成物に比べてクロルフェニラミンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等であることを示し、12時間にわたり前記経口組成物は1回投与し、前記IR-RLDは時間がゼロのときと、6時間後の2回投与するものである。
本発明の実施形態の1つは、ヒトの咳、風邪、流感又はアレルギーの兆候を治療する方法に関し、本願明細書前記の非液状経口徐放性薬剤組成物の1つを被験者に投与することを含む。
前記方法の実施形態では、医薬組成物が、1日1回又は1日2回投与ができる二段階投与組成物としてヒトに投与される。
本発明の態様によると、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを有効成分として含む非液状経口徐放性薬剤組成物に関するもので、血漿分析によると、3種類すべての有効成分の十分なAUCinfinityが得られ、この組成物はヒトに単回投与した後、少なくとも8時間にわたり治療効果が得られる。
本発明の態様によると、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとを有効成分として含む非液状経口徐放性薬剤組成物に関するもので、血漿分析によると、3種類すべての有効成分の十分なAUCinfinityが得られ、この組成物はヒトに単回投与した後、少なくとも8時間にわたる治療効果が得られる。
【0098】
前記態様の文脈での「治療効果」という用語は、風邪、流感又はアレルギーに対する効果を意味し、咳の重篤度及び/又は咳の回数、咳の兆候、鼻漏、鬱血又はくしゃみの低減などの兆候緩和をはじめとする(が制限されない)効果、及び/又は、自覚的なよい状態の改善につながる他の生物学的効果を意味する。
前記非液状薬剤組成物の実施形態の1つでは、少なくとも8時間という期間が12時間である。前記非液状薬剤組成物の別の実施形態では、少なくとも8時間という期間が24時間である。
本発明の実施形態の1つは、非液状経口徐放性薬剤組成物中に存在するプソイドエフェドリン又はエフェドリンの抽出、単離又は分離の可能性を阻止又は低減する方法を包含し、該方法は、
第1の部位と第2の部位とを含むように非液状経口徐放性薬剤組成物を調製する工程であって、
第1の部位が、即放剤形として、抗ヒスタミン剤、鎮咳剤又はその両方を含み、更にプソイドエフェドリン又はエフェドリンを含んでいてもよく、
第2の部位が粒子、ペレット又はビーズを含み、各粒子、ペレット又はビーズが有効成分としてプソイドエフェドリン又はエフェドリン、及び、抗ヒスタミン剤、鎮咳剤又はその両方を徐放剤形として含み、
更に、経口徐放性薬剤組成物中に存在するプソイドエフェドリン又はエフェドリンの抽出、単離又は分離が行えないようする、又は行いにくくする工程を含む。
【0099】
前記方法の具体的実施形態の1つは、鎮咳剤がヒドロコドンで、かつ、該方法が、非液状経口徐放性薬剤組成物に存在するヒドロコドンの抽出、単離又は分離が行えないようする、又は行いにくくする工程を更に含む。
別の実施形態において、前記方法は、プソイドエフェドリン又はエフェドリンの非液状経口徐放性薬剤組成物からの抽出、単離又は分離を、プソイドエフェドリン又はエフェドリンを含む即放性組成物と比べ、よりむずかしくする製造工程を更に含む。
本発明の実施形態の1つは、非液状経口徐放性薬剤組成物中に存在するプソイドエフェドリン又はエフェドリンの乱用の潜在的可能性を低減する方法に関し、該方法が、
第1の部位と第2の部位とを含むように非液状経口徐放性薬剤組成物を調製する工程を含む方法で、
前記第1の部位が、即放剤形として、抗ヒスタミン剤、鎮咳剤又はその両方を含み、更にプソイドエフェドリン又はエフェドリンを含んでいてもよく、
前記第2の部位が、有効成分として抗ヒスタミン剤、鎮咳剤及びプソイドエフェドリン又はエフェドリンを徐放剤形として含む粒子、ペレット又はビーズを含有する。
本発明の他の実施形態は、非液状経口徐放性薬剤組成物中に存在する麻酔性鎮咳剤又はプソイドエフェドリンの乱用の潜在的可能性を低減する方法に関するもので、該方法は、麻酔性鎮咳剤及びプソイドエフェドリンを有効成分として徐放剤形として含む粒子、ペレット又はビーズを有するように非液状経口徐放性薬剤組成物を調製する工程を含む。
【0100】
本発明の態様の1つは、風邪、流感又はアレルギーの兆候の治療に使用する固形の経口徐放性調合医薬製剤の製造方法に関するもので、プソイドエフェドリン又はエフェドリンを含有する即放性(IR)製剤に比べて、製剤中に含まれるプソイドエフェドリン又はエフェドリンに関する乱用の潜在的可能性を低減するもので、前記製造方法は、
粒子、ペレット又はビーズを含むように固形の経口徐放性薬剤組成物を調製する工程を含み、個々の粒子、ペレット又はビーズには2種以上の有効成分が徐放剤形で含まれ、また、その有効成分の少なくとも1種がプソイドエフェドリン又はエフェドリンであり、その有効成分の少なくとも1種はプソイドエフェドリン又はエフェドリンではない。
本発明の別の態様は、固形の経口徐放性薬剤組成物中に存在するプソイドエフェドリン又はエフェドリンの抽出、単離又は分離の可能性を阻止又は低減する方法に関するもので、該方法は、
粒子、ペレット又はビーズを含む固形の経口徐放性薬剤組成物を調製する工程を含み、個々の粒子、ペレット又はビーズの中には2種以上の医薬的に許容できる有効成分が徐放剤形で含まれ、その有効成分の少なくとも1種がプソイドエフェドリン又はエフェドリンであり、その有効成分の少なくとも1種はプソイドエフェドリン又はエフェドリンではない。
【0101】
本発明の別の態様は、ある哺乳動物において、少なくとも8時間にわたって3種の有効成分の血漿中濃度が、FDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり同じ哺乳動物に適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となるようにする方法に関するもので、該方法は、
(A)第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物であって、第1の部位が即放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよく、第2の部位が、有効成分として抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む個々の粒子、ペレット又はビーズを徐放剤形として含有する、非液状経口徐放薬剤組成物を前記哺乳動物に投与することと、
(B)少なくとも8時間にわたる前記3種の有効成分の血漿中濃度が、前記有効成分を含有するFDA承認済みのIR-RLD組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価になるようにすることとを含み、前記適切な投与回数とは、IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みの1つ以上のラベルのなかで推奨される投与回数に対応する。
本発明の別の態様は、ある哺乳動物において、少なくとも8時間にわたってクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンの血漿中濃度が、クロルフェニラミン、ヒドロコドン及び/又はプソイドエフェドリンを含有する即放性(IR)組成物1種以上を同じ期間にわたり2回以上同じ哺乳動物に投与した際に得られる血漿中濃度と生物学的等価となるようにする方法に関するもので、該方法は、
【0102】
(A)有効成分がクロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる単一の経口医薬製剤であって、前記有効成分が即放性(IR)と徐放性(ER)を示し、即放性(IR)部位と徐放性(ER)部位とを有する製剤を前記哺乳動物に投与することと、
(B)少なくとも8時間にわたるクロルフェニラミンとヒドロコドンとプソイドエフェドリンの血漿中濃度が、クロルフェニラミン、ヒドロコドン及び/又はプソイドエフェドリンを含有する即放性(IR)組成物1種以上を同じ期間にわたり2回以上投与した際に得られる血漿中濃度と生物学的等価になるようにすることとを含む。
前記方法の実施形態の1つでは、少なくとも8時間という期間が12時間である。前記方法の別の実施形態では、少なくとも8時間という期間が24時間である。
本発明の更に別の態様は、非液状の経口徐放性(ER)薬剤組成物の投与後、抗ヒスタミン剤、鎮咳剤及び充血緩和剤の安定した血漿中濃度を哺乳動物において得るための方法であって、前記血漿中濃度とは、抗ヒスタミン剤、鎮咳剤及び/又は充血緩和剤を含む即放性(IR)組成物1種以上を同じ哺乳動物に投与した際に得られる血漿中濃度と生物学的等価で、該方法は、
第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物であって、第1の部位が、抗ヒスタミン剤と鎮咳剤とを含み、更に充血緩和剤を含んでもよい有効成分を即放剤形で含有し、第2の部位が、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを有効成分として徐放剤形で含む粒子、ペレット又はビーズである、非液状経口徐放性薬剤組成物を前記哺乳動物に投与することを含み、
【0103】
前記3種の有効成分が24時間を超えて安定した血漿中濃度であるように前記経口徐放性薬剤組成物を十分な投与回数で哺乳動物に投与することにより、前記有効成分を含有するFDA承認済みの即放性(IR)薬剤組成物1種以上を上記と同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価な有効成分の血漿中濃度となり、
前記FDA承認済みのIR薬剤組成物1種以上の適切な投与回数とは、FDA承認済みのIR薬剤組成物1種以上の同じ期間にわたる投与に関して、FDAが承認するの1つ以上ラベルのなかで推奨される投与回数に対応し、
前記FDA承認済みのIR薬剤組成物1種以上の適切な投与回数が、経口ER薬剤組成物の十分な投与回数を上回るものである。
本発明の別の態様は、ある哺乳動物において、少なくとも8時間にわたって抗ヒスタミン剤、鎮咳剤及び充血緩和剤の血漿中濃度が、前記3種すべての有効成分を含有するFDA承認済み即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり同じ哺乳動物に適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価になるようにする方法に関し、該方法は、
(A)第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物であって、
前記第1の部位が、抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよい有効成分を即放剤形で含有し、
前記第2の部位が、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを有効成分として徐放剤形で含む粒子、ペレット又はビーズである、非液状経口徐放性薬剤組成物を前記哺乳動物に単回投与することと、
【0104】
(B)少なくとも8時間にわたり前記3種すべての有効成分の血漿中濃度が、3種すべての有効成分を含有するFDA承認済みのIR-RLD組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価にすることとを含み、
前記適切な投与回数とは、前記IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みのラベルで推奨される投与回数に対応する。
本発明の他の態様は、被験哺乳動物におけるヒドロコドンとプソイドエフェドリンのAUCinfinityを得る方法に関するもので、該方法は、
(A)少なくとも8時間という期間の最初に、(1)有効成分がクロルフェニラミンとヒドロコドンとからなる即放性(IR)部位と、(2)有効成分がクロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる徐放性(ER)部位とを有する徐放性(ER)経口医薬組成物を単回投与する工程であって、
該経口組成物におけるIR部位のクロルフェニラミン対ER部位のクロルフェニラミンの質量比が約25:75、IR部位のヒドロコドン対ER部位のヒドロコドンの質量比が約25:75、IR部位のプソイドエフェドリン対ER部位のプソイドエフェドリンの質量比が約0:100である工程と、
(B)前記経口組成物に存在するヒドロコドンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を前記と同じ期間にわたり2回以上投与した際に得られるAUCinfinityと同等のヒドロコドンのAUCinfinityを被験哺乳動物内に獲得する工程と、
【0105】
(C)前記経口組成物に存在するプソイドエフェドリンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を前記と同じ期間にわたり2回以上投与した際に得られるAUCinfinityと同等のプソイドエフェドリンのAUCinfinityを被験哺乳動物内に獲得する工程とを含む。
前記方法の実施形態の1つは、前記経口組成物に存在するプソイドエフェドリンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を前記と同じ期間にわたり2回以上投与した際に得られるAUCinfinityと同等のクロルフェニラミンのAUCinfinityを被験哺乳動物内に獲得する工程を更に含む。
本発明の別の態様は、被験哺乳動物におけるヒドロコドンとプソイドエフェドリンのAUCinfinityを得る方法に関するもので、該方法は、
(A)(1)有効成分がクロルフェニラミンとヒドロコドンとからなる即放性(IR)部位と、(2)有効成分がクロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる徐放性(ER)部位とを有する徐放性(ER)経口医薬組成物を投与する工程であって、
該経口組成物におけるIR部位のクロルフェニラミン対ER部位のクロルフェニラミンの質量比が約25:75、IR部位のヒドロコドン対ER部位のヒドロコドンの質量比が約25:75、IR部位のプソイドエフェドリン対ER部位のプソイドエフェドリンの質量比が約0:100である工程と、
【0106】
(B)前記経口組成物に比べてヒドロコドンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等のヒドロコドンのAUCinfinityを被験哺乳動物内に獲得する工程であって、前記経口組成物は12時間にわたる期間のなかで1回投与し、前記IR-RLDは時間がゼロのときと6時間後の2回投与する、工程と、
(C)前記経口組成物に比べてプソイドエフェドリンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等のプソイドエフェドリンのAUCinfinityを被験哺乳動物内に獲得する工程であって、前記経口組成物は12時間にわたる期間のなかで1回投与し、前記IR-RLDは時間がゼロのときと6時間後の2回投与する、工程とを有する。
前記方法の実施形態の1つは、(D)前記経口組成物に比べてクロルフェニラミンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等のクロルフェニラミンのAUCinfinityを被験哺乳動物内に獲得する工程であって、前記経口組成物は12時間にわたる期間のなかで1回投与し、前記IR-RLDは時間がゼロのときと6時間後の2回投与する、工程を更に有する。
本発明の更に別な態様は、非液状単一薬剤組成物をヒトに単回投与した後、少なくとも8時間にわたり、治療効果を得るのに十分な抗ヒスタミン剤、鎮咳剤、充血緩和剤のAUCinfinityを提供する方法であって、該方法は、(A)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとを有効成分として含む単一の非液状経口徐放性薬剤組成物をヒトに単回投与する工程と、(B)血漿分析により、単回投与後少なくとも8時間にわたり、ヒト被験者において治療効果が検出されるのに十分な3種類すべての有効成分のAUCinfinityを獲得する工程とを含む。
【0107】
本発明の具体的実施形態は、単一薬剤組成物をヒトに単回投与した後、少なくとも8時間にわたりヒト被験者の治療効果を得るのに十分なクロルフェニラミンとヒドロコドンとプソイドエフェドリンのAUCinfinityを提供する方法であって、該方法は、(A)有効成分がクロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる単一の経口徐放性薬剤組成物をヒト被験者に単回投与する工程と、(B)血漿分析により、単回投与後少なくとも8時間にわたり、ヒト被験者において治療効果が検出されるのに十分な3種類すべての有効成分のAUCinfinityを獲得する工程とを含む。
前記方法の実施形態の1つは、少なくとも8時間という期間が12時間である。前記方法の別の実施形態は、少なくとも8時間という期間が24時間である。
本発明の実施形態は、第1の部位と第2の部位とを有する固形の経口徐放性薬剤組成物に関するもので、
第1の部位が即放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよく、
第2の部位が徐放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む粒子、ペレット又はビーズを含有し、
前記経口薬剤組成物を患者に単回投与することにより、少なくとも8時間にわたる前記3種の有効成分の血漿中濃度が、前記有効成分を含有するFDA承認済みのIR-RLD組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となるもので、
前記適切な投与回数とは、前記IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みラベル1つ以上のなかで推奨される投与回数に対応する。
【0108】
本発明の実施形態の1つは、風邪、流感又はアレルギーの兆候の治療に使用する固形の経口徐放性調合医薬製剤の製造方法を包含するもので、該製剤は製剤中のいずれの有効成分についても、その有効成分を含有する即放性(IR)製剤に比べて、乱用の潜在的可能性を低減させており、前記製造方法は、
粒子、ペレット又はビーズを含むように固形の経口徐放性薬剤組成物を調製する工程を含み、各粒子、ペレット又はビーズには2種以上の有効成分が徐放剤形で含まれている。
本発明の更に別の実施形態は、固形の経口徐放性薬剤組成物から1つの有効成分が抽出、単離又は分離できる可能性を阻止又は低減する方法に関するもので、該方法は、
粒子、ペレット又はビーズを含み、その個々の粒子、ペレット又はビーズに2種以上の医薬的に許容できる有効成分が徐放剤形で含まれる、固形の経口徐放性薬剤組成物を調製する工程を含む。
本発明の実施形態は、非液状の経口徐放性薬剤組成物から、含有されているプソイドエフェドリン又はエフェドリンが抽出、単離又は分離できる可能性を阻止又は低減する方法に関するもので、該方法は、粒子、ペレット又はビーズを含み、その個々の粒子、ペレット又はビーズにプソイドエフェドリン又はエフェドリン及び抗ヒスタミン剤又は鎮咳剤又はその両方が有効成分として徐放剤形で含まれる、非液状の経口徐放性薬剤組成物を調製する工程を含む。
【0109】
本発明の実施形態は、非液状の経口徐放性薬剤組成物に含まれるプソイドエフェドリン又はエフェドリンの乱用の潜在的可能性を低減する方法に関するもので、該方法は、粒子、ペレット又はビーズを含み、その個々の粒子、ペレット又はビーズに抗ヒスタミン剤及び鎮咳剤及びプソイドエフェドリン又はエフェドリンが有効成分として徐放剤形で含まれる、非液状の経口徐放性薬剤組成物を調製する工程を含む。
別の具体的な実施形態では、非液状の経口徐放性薬剤組成物は、抗ヒスタミン剤、鎮咳剤及び充血緩和剤からなる有効成分を含有する。このような実施形態において、非液状薬剤組成物の即放性部位は、抗ヒスタミン剤と鎮咳剤とからなり、更に充血緩和剤を含んでいてもよい有効成分を含有する。別の実施形態は、非液状の薬剤組成物の徐放性部位が、抗ヒスタミン剤、鎮咳剤及び充血緩和剤からなる有効成分を含む。
更なる具体的な実施形態では、経口徐放性薬剤組成物が、クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる有効成分を含む。このような実施形態の1つでは、薬剤組成物の即放性部位が、クロルフェニラミンとヒドロコドンからなり、さらにプソイドエフェドリンを含んでもよい有効成分を含有する。別の実施形態では、薬剤組成物の徐放性部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む。
【0110】
以下の実施例は、本発明を理解するうえでの助けとするために記載するもので、本願明細書で記載及びクレームする本発明を具体的に限定するものとみなすべきではない。現在公知であるか、又は、当該分野の当業者が理解しうる範囲で後に展開される、あらゆる同等物の置換をはじめとする本発明の変更や、製剤の変更又は実験デザインにおける小さな変更は本発明の範囲内である。
実施例1
実施例1は「製剤X」の製造方法を記載する。「製剤X」は、咳、風邪及びアレルギーの兆候の治療を意図した徐放性被覆ぺレット含有シロップ剤の分散液である。製剤Xは、成人1回服用量(5ml)につき、15mgのヒドロコドン重酒石酸塩(HC、中枢作用性鎮咳剤)と、120mgのプソイドエフェドリン塩酸塩(PSE、交感神経作用様鼻充血緩和剤)と、8mgのクロルフェニラミンマレイン酸塩(CPM、抗ヒスタミン剤)とを一緒に含む。この製剤においては、塩の形態の薬剤が使用された。この製剤は、製造後、(4オンス)の使用単位容器(unit-of-use containers)に分けられた。
表1は、徐放性ペレット含有シロップ剤のIR/ER分散液である製剤Xの定量的組成の一例をあらわす表で、1回の投与量5ml(6.55g)を質量基準で表わしたものである。
【0111】
【表1】

【0112】
EPペレットにおけるAPI濃度に対するIRシロップにおけるAPI濃度の比は、現在市場に出ている即放性薬剤と生物学的等価になるような血漿の薬剤バイオアベイラビリティーが12時間得られるように設定された。
EPペレットにおけるAPI濃度に対するIRシロップにおけるAPI濃度の比は、現在市場に出ている即放性薬剤と生物学的等価になるような血漿の薬剤バイオアベイラビリティーが12時間得られるように設定された。
製剤Xのコアペレットの製造
製剤XのER成分にはコアペレットが含まれる。コアペレットの調製方法を以下に記載する。
0.207kgのクロルフェニラミンマレイン酸塩、4.138kgのプソイドエフェドリン塩酸塩、0.3879kgのヒドロコドン重酒石酸塩、2.500kgのラクトース 一水和物、0.5000kgのアルギン酸ナトリウム、17.27kgの微晶質セルロースを一緒に高剪断ミキサーに投入した。ミキサーの羽根車を200RPM、チョッパーを1500RPMにして5分間運転した。この粉体混合の後、約10.00kgの純水をおよそ1L/分の速度でミキサーに引き入れた。この間、チョッパーは使わず、羽根車は200 RPMで運転した。水を全量添加した後、羽根車は200RPM、チョッパーは1500RPMにして湿式造粒を1分間続けた。ミキサーから出した後、水分を含んだ塊を少なくとも30分間、開いた袋の中に放置した。
その後、0.7mmのスクリーンを有するドーム型一軸スクリュー押出機を45RPMで運転し、得られた水分を含む塊の押出成形を行った。この水分を含む塊の全量を処理するまで押出を続けた。
【0113】
スフェロナイザー(spheronizer)を使用し、3x3mmの円錐の先端を切ったパターン(truncated pattern)を有するディスクを約1000rpmで運転して、反復バッチ処理を行ってペレットを形成した。約3.8kgのバッチを9つ使い、球体化(spheronization)の運転はバッチごとにおよそ90秒にわたった。
球体化した材料を流動床乾燥機に入れた。このビーズを、入り口開始空気温度を60℃、毎分の全空気量を1500立方フィート( 約42.3m3)にして乾燥させた。引き続き、入り口空気温度と空気量を工程中に調整して適切な流動化を維持し、製品温度を50〜60℃に保持した。ビーズの水分が2%以下になるまで乾燥を続けた。流動床乾燥機を冷風モードで十分な時間運転して製品を室温に戻した後、乾燥したビーズを排出した。
自動ふるい振盪機のNo.22とNo.34のスクリーンを1200RPMで運転して乾燥ビーズの分粒を行った。No.22のスクリーンを通過し、No.34のスクリーンに残留したコアペレットを合格品とみなした。
製剤X用ペレットのコーティング
製剤Xのコアペレットのコーティング方法を以下に説明する。
【0114】
18.65kgのアンモニオメタクリレート共重合体タイプB分散液(オイドラギットRS30D)を、No. 20のスクリーンを使ってステンレス鋼製容器に投入した。0.7774kgのアンモニオメタクリレート共重合体タイプA分散液(オイドラギットRL30D)をNo. 20のスクリーンを使って同じ容器に投入した。この分散液の混合物をプロペラミキサーを約1000rpmにして混合した。別の容器に0.6087kgのクエン酸トリエチルと10.92kgの純水とを混合した。この混合液も、プロペラミキサーを使って、液中に空気をいれずに渦を作るのに十分な回転数で混合した。混合を続けながら、1.812kgのタルクをクエン酸トリエチルと水との混合物に添加した。5分間混合した後、撹拌器を止めて取り出した。クエン酸トリエチル、タルク及び水の混合物を、メタクリレート共重合体分散液が入った容器に、撹拌を続けながら移した。このコーティング系は、コーティング作業を通して十分に撹拌を続けて均一な分散液を維持した。
コアペレットのコーティングは、250umスクリーン付きのDベースプレートを使って、12インチのワースター(Wurster)型容器を有する流動床装置Glatt GPCG 30で行った。 カラム高さは底面から約2.5インチに設定し、開口部が1mmのノズルを使った。コアビーズ21.75kgを流動床装置に投入し、初期パラメータは以下のように設定した。入り口空気温度の目標範囲:40〜50℃、製品温度の目標範囲:27〜32℃、噴霧気圧の目標範囲:1.5〜2.5bar、空気体積の目標範囲:250〜450cfm、空気体積の合計:1500cfm、フィルターバッグの振盪回数:240秒ごとに5秒間。工程中に調整を行い、処理条件が範囲内に保たれるようにした。コーティング用分散液の噴霧は最初は20g/分の速さで行った。処理を続けるなかで、噴霧速度は最終的に100g/分まで上げた。
【0115】
コーティング用分散液をすべて適用した後、流動床装置を冷風モードで運転し、被覆ビーズを室温に戻してから排出した。その後、被覆ビーズとタルク75.00gとを5分間混合して排出した。
被覆ペレットは熱対流炉で硬化した。被覆ペレットをトレイにまき散らし、炉に入れた。炉の温度は55℃に設定し、ビーズを炉内に約16時間放置した。約16時間後、炉の電源を切りペレットを室温まで放冷した。
被覆ビーズの分粒は、 自動ふるい振盪機のNo.16とNo.34のスクリーンを1200RPMで運転して行った。No.16のスクリーンを通過し、No.34のスクリーンに残留した被覆ペレットを合格品とした。
製剤Xのコアペレット用ビヒクルの調製
製剤XのIR成分は液体を含み、製剤Xの「ビヒクル」又は「ビヒクルシロップ剤」とも呼ばれる。ビヒクルシロップ剤の製造方法は以下の通りである。
60リットルを収容するのに十分な容量のジャケット付きステンレス鋼製容器に、純水25.09kgを投入した。循環水温度制御装置を使って、作業を通して液体の温度を制御し、プロペラミキサーを使って撹拌した。カバーで水の蒸発を抑えた。液体中に空気が入らないように渦ができる速度で混合しながら、水を約70℃に加熱した。No.30のスクリーンを通過したプロピルパラベン0.01080kgを水に加え、プロピルパラベンが完全に溶解するまで混合を続けた。プロピルパラベンの溶解後、温度制御装置を50℃に設定し、ショ糖48.00kgをゆっくり容器に添加した。容器に蓋をして、ショ糖がすべて溶けるまで混合を続けた。
【0116】
ショ糖が完全に溶解したところで、温度制御装置を装置の最低温度に設定して、溶液を25℃まで放冷した。0.02642kgのクロルフェニラミンマレイン酸塩、0.04954kgのヒドロコドン重酒石酸塩、0.2680kgの人工イチゴ風味剤(粉末)、0.2160gの人工苦味矯味剤(粉末)、0.0720kgのスクラロースをシロップ剤に添加した。すべての固形物が溶解するまで混合を続けた。更に、撹拌を続けながらFD&C Red No.40アルミニウムレーキ0.0720kgと二酸化チタン0.1800kgをNo.30のスクリーンを使って容器に加え、均一な分散液を得た。(注:このビヒクルの例は、混合を停止すると沈殿する不溶物質を含む。)
この混合物の最終的な正味質量を求め、必要に応じて蒸発損失を埋め合わせるために純水を加えた。
製剤Xのための被覆ペレットとビヒクルシロップ剤との混合
被覆ペレットとビヒクルシロップ剤を混合して最終製品の製剤Xを作成する方法の1つを以下に説明する。
渦が形成されるようにビヒクルシロップ剤を一定して撹拌した。この撹拌を少なくとも10分行った後、24.00gの被覆ビーズを130.8gのビヒクルに添加し、服用量24回分又は120mLの製品を得た。
【0117】
実施例2 ヒトでの有効性
実施例2では製剤Xのヒトにおける有効性を評価するために行った試験について記載する。具体的には、実施例2では、徐放性製剤Xの単回投与と、HC、PSE又はCPMを含む即放性RLDを組み合わせたものの2回投与とを比較するために、健康な被験者16名で行った試験について記載する。この試験の目的の一つは、製剤Xの2つの製剤と、対応するRLDとの生物的等価性を求めることであった。
吸収
ヒドロコドン
ヒドロコドンは経口によりよく吸収されるが、腸や肝臓での代謝を伴う著しい初回通過効果を受ける。既に公表されている試験によると、10mgのHCを即放性経口剤形で5人の男性被験者に1回投与したところ、平均ピーク血漿濃度は23.6±5.2ng/mLで、Tmaxはおよそ1.3±0.3時間であった。「Hycodan(登録商標)」(www.rxmed.comにて入手可能、2008年6月23日アクセス);Stout, P., Farrell, L. 「Opioids - Effects on Human Performance and Behavior(オピオイド-ヒトの行動、挙動への影響)」Forensic Science Review. 15(1): 29-59 (2003)。すべてのヒドロコドン代謝産物は活性であり、ヒドロモルホン、ノルコデイン及び6-αと6-βヒドロキシ代謝産物を含む。Micromedex Health Care Series、DrugDex Evaluations「ヒドロコドン重酒石酸塩/イブプロフェン」http://www.thomsonhc.com/hcsにて閲覧可能、2008年7月1日アクセス、Cone等「Comparative metabolism of hydrocodone in man, rat, guinea pig, rabbit, and dog(ヒト、ネズミ、モルモット、ウサギ及びイヌにおける比較によるヒドロコドンの代謝)」Drug Metab Dispos 6:488-493 (1978)引用。
【0118】
表2は、「製剤X」の単回投与に対してHCの関連リスト収載医薬品(RLD)2回投与を行った際の、患者のヒドロコドン(HC)血漿中濃度に関するパラメータ、AUCinfinity、Cmax及びT1/2等の比較表である。治療Aとは、15mgのHC、120mgのPSE及び8mgのCPMを含有する「製剤X」の単回投与に対応し、治療Cは、7.5mgのHC、60mgのPSE及び4mgのCPMを含む、3つの単体RLDの混合液を2回投与することに相当する。
本試験では(表2の治療A及びCで表示されているように)、15mgのHCと120mgのPSEと8mgのCPMとを含む製剤Xを16人の被験者に投与した後のHCの平均ピーク血漿濃度は(Cmax)は17.54±4.75ng/mLであった。これに対し、7.5mgのHCを含有するRLDを0時間と6時間で2回投与した場合は、平均ピーク血漿濃度が25.64±6.56ng/mLであった。製剤Xの場合のTmax中央値は4時間であり、それに対してRLDの2回投与の場合のTmax中央値は7時間であった。製剤XのTmaxが科学文献にみられるもの比べた時に異なるのは、製剤X中の徐放性ペレットが時間をかけてHCを放出し、12時間の投薬を実現している事実による。この試験ではRLDを2回投与したため(既に公表されている試験では1回投与である)、RLDの2回投与で得られたピーク血漿濃度(Cmax)は製剤Xの場合よりも遅い時間に到達されている。
【0119】
【表2】
治療A:テスト製剤No.1 - 製剤X1q:15mgのHC、120mgのPSE、8mgのCPM
治療B:テスト製剤No.2 - 製剤X1q:10mgのHC、120mgのPSE、8mgのCPM
治療C:参考製剤No.1(治療A用)-RLD2q:7.5mgのHC、60mgのPSE、4mgのCPM
治療D:参考製剤No.2(治療B用)-RLD2q:5mgのHC、60mgのPSE、4mgのCPM
AUC=0時間から無限時間まで外挿した血漿中濃度−時間曲線下面積;AUCLAST= 0時間から量化できる最終血漿濃度までの血漿中濃度−時間曲線下面積;CMAX= 実測された最高血漿中濃度;CPM=クロルフェニラミンマレイン酸塩;HC=ヒドロコドン重酒石酸塩;PSE=プソイドエフェドリン塩酸塩;TMAX=最高血漿中濃度到達時間、T1/2=消失半減期、a=対数変換値の分析による指数結果
【0120】
プソイドエフェドリン
プソイドエフェドリンは消化管から容易に、かつ、ほとんど完全に吸収され、初回通過代謝を証明するものはない。既に公表されている試験では、Tmaxは投与から1.5〜2.4時間で見られ、バイオアベイラビリティーはすべての製剤で同じであり、食物による影響を受けない。Micromedex Health Care Series、DrugDex Evaluations「プソイドエフェドリン」http://www.thomsonhc.com/hcsにて閲覧可能、2008年7月1日アクセス;「Common cold and influenza management(普通の風邪とインフルエンザの管理)」MedScape(www.medscape.com/viewarticle/466063_3で閲覧可能、2008年6月26日アクセス);Graves DA等「Influence of a standard meal on the absorption of a controlled release pseudoephedrine suspension(標準的食事が及ぼす放出制御型プソイドエフェドリン懸濁液の吸収への影響)」Biopharm Drug Dispos. 5月-6月; 9(3):267-72 (1988)。
表3は、「製剤X」の単回投与に対してPSEのRLDを2回投与した場合の患者のプソイドエフェドリン(PSE)血漿中濃度に関するパラメータ、AUCinfinity、Cmax及びT1/2等の比較表である。治療Aとは、15mgのHC、120mgのPSE及び8mgのCPMを含有する「製剤X」の単回投与に対応し、治療Cは、60mgのPSE 、7.5mgのHC及び4mgのCPMを含む、3つの単体RLDの混合液を2回投与することに相当する。
【0121】
本試験では(表3の治療A及びCで表示されているように)、15mgのHCと120mgのPSEと8mgのCPMとを含む製剤Xを16人のヒト被験者に投与した後、PSEの平均ピーク血漿濃度は(Cmax)は292.05±49.94ng/mLであった。これに対し、60mgのPSEをそれぞれ含有する即放性RLDを2回投与した後は、平均ピーク血漿濃度が345.47±78.46ng/mLであった。製剤X投与の場合のTmax中央値は5時間であり、それに対してRLD投与後のTmax中央値は7時間であった。上記のごとく、製剤X中の徐放性ペレットが時間をかけてPSEを放出することで12時間の投薬になる。結果、この試験におけるRLDのピーク血漿濃度は、より遅い時間に到達されている。
【0122】
【表3】
治療A:テスト製剤No.1 - 製剤X1q:15mgのHC、120mgのPSE、8mgのCPM
治療B:テスト製剤No.2 - 製剤X1q:10mgのHC、120mgのPSE、8mgのCPM
治療C:参考製剤No.1(治療A用)- RLD2q:7.5mgのHC、60mgのPSE、4mgのCPM
治療D:参考製剤No.2(治療B用)- RLD2q:5mgのHC、60mgのPSE、4mgのCPM
AUC=0時間から無限時間まで外挿した血漿中濃度−時間曲線下面積;AUCLAST= 0時間から量化できる最終血漿濃度までの血漿中濃度−時間曲線下面積;CMAX= 実測された最高血漿中濃度;CPM=クロルフェニラミンマレイン酸塩;HC=ヒドロコドン重酒石酸塩;PSE=プソイドエフェドリン塩酸塩;TMAX=最高血漿中濃度到達時間、T1/2=消失半減期、a=対数変換値の分析による指数結果
【0123】
クロルフェニラミン
クロルフェニラミンは、経口投与後、容易かつ完全に吸収される。既に公表されている試験では、この薬剤は30〜60分以内に全身循環に現れ、2時間後にCmaxに到達し、その後の46時間で血漿濃度が低下する。Peets, E等「Metabolism of chlorpheniramine maleate in man (クロルフェニラミンマレイン酸塩の人体における代謝)」J. Pharmacol. Exp. Ther. 180:464-474-(1972);Micromedex Health Care Series、DrugDex Evaluations「クロルフェニラミン」http://www.thomsonhc.com/hcsで閲覧可能、2008年7月1日アクセス。クロルフェニラミンは経口バイオアベイラビリティーが41±16%で、食物摂取によりその吸収は遅くなるが、バイオアベイラビリティーは遅くならない。Rumore, M. M「Clinical pharmacokinetics of chlorpheniramine (クロルフェニラミンの臨床薬物動態学)」Drug Intel. Clin. Pharm. 18:701-707 (1984)。しかしながら、クロルフェニラミンは吸収中に胃腸粘膜で、また、肝臓での初回通過時に実質的に代謝されると考えられる。限られたデータであるが、従来の錠剤状のCPMを経口単回投与した場合は約25〜45%、溶剤の場合は35〜60%が薬剤の変化をおこさずに全身循環に到達することが示されている。また、限られたデータではあるが、従来の錠剤又は経口用液剤と比較して、この薬剤の徐放性調剤のバイオアベイラビリティーは低減する可能性も示されている。Micromedex Health Care Series:クロルフェニラミン(同サイト);Therapeutic Goods Administration(オーストラリア)「Core sedating antihistamines product information(抗ヒスタミン剤投与情報)」www.tga.gov.au/npmeds/pi-sedatingantihistamine.rtfにて閲覧可能、2008年6月26日アクセス。
【0124】
表4は、「製剤X」の単回投与に対してCPMのRLDを2回投与した場合の患者のクロルフェニラミン(CPM)血漿中濃度に関するパラメータ、AUCinfinity、Cmax及びT1/2等の比較表である。治療Aとは、15mgのHC、120mgのPSE及び8mgのCPMを含有する「製剤X」の単回投与に対応し、治療Cは、4mgのCPM 、7.5mgのHC及び60mgのPSEを含む、3つの単体RLDの混合液を2回投与することに相当する。
本試験では(表4の治療A及びCで表示されているように)、15mgのHCと120mgのPSEと8mgのCPMとを含む製剤Xを16人のヒト被験者に投与した後のCPMの平均ピーク血漿濃度は(Cmax)は21.20±6.30ng/mLであった。これに対し、4mgのCPMをそれぞれ含有するRLDを2回投与した後は、平均ピーク血漿濃度が28.89±7.92ng/mLであった。製剤X投与後9時間でTmax中央値が得られたが、RLD投与の場合は、9.50時間後であった。
【0125】
【表4】
治療A:テスト製剤No.1 - 製剤X1q:15mgのHC、120mgのPSE、8mgのCPM
治療B:テスト製剤No.2 - 製剤X1q:10mgのHC、120mgのPSE、8mgのCPM
治療C:参考製剤No.1(治療A用)- RLD2q:7.5mgのHC、60mgのPSE、4mgのCPM
治療D:参考製剤No.2(治療B用)- RLD2q:5mgのHC、60mgのPSE、4mgのCPM
AUC=0時間から無限時間まで外挿した血漿中濃度−時間曲線下面積;AUCLAST= 0時間から量化できる最終血漿濃度までの血漿中濃度−時間曲線下面積;CMAX= 実測された最高血漿中濃度;CPM=クロルフェニラミンマレイン酸塩;HC=ヒドロコドン重酒石酸塩;PSE=プソイドエフェドリン塩酸塩;TMAX=最高血漿中濃度到達時間、T1/2=消失半減期、a=対数変換値の分析による指数結果
【0126】
排出及び排泄
ヒドロコドン
ヒドロコドン の親化合物の排出半減期は3.8〜4.5時間である。Micromedex Health Care Series: ヒドロコドン、経口単回投与から最初の24時間で全排出のおよそ70%(同サイト); Stout, P.; Farrell, L.「Opioids - Effects on Human Performance and Behavior (オピオイド-ヒトの行動、挙動への影響)」 Forensic Science Review, 15(1): 29-59. (2003)
本試験では(表2の治療A及びCで表示されているように)、HCについての製剤XのT2/1を測定したところ7.22±1.45時間であった。これに対してRLDの場合は4.38±0.83時間であった。この差は、製剤Xが徐放性薬剤であることに因ると考えられた。
プソイドエフェドリン
PSEとその代謝産物は尿に排出され、服用量の90%までが投与から24時間以内にそのまま排出される。PSEの半減期はおよそ9〜16時間で、半減期は尿のpH値に影響されることがあり、アルカリ(pH8)の場合は延び、酸性(pH5)の場合は短くなる。Wishart, D.等「DrugBank: a comprehensive resource for in silico drug discovery and exploration(コンピュータによる薬物の発見と調査のための包括的リソース」Nucleic Acids Res. 34: D668-72 (2006);Micromedex Health Care Series: プソイドエフェドリン(同サイト)。
【0127】
本試験では(表3の治療A及びCで表示されているように)、PSEについての製剤XのT2/1の測定結果は6.48±1.40時間であった。これに対してRLDの場合は5.06±0.90時間であった。この差は、製剤Xが徐放性薬剤であることに因ると考えられた。
クロルフェニラミン
クロルフェニラミンの体内からの排出は、主にモノデスメチル化合物やジデスメチル化合物に代謝され、26%までが尿に排出される。腎臓による排出が全排出のおよそ50%を占め、3〜18%は薬剤が変化していない。腎臓による排出は尿流量が増えるほど、また、pH値が低いほど増加する。Micromedex Health Care Series:クロルフェニラミン(同サイト)参照のこと。1%未満が大便に排出される。CPMの半減期は20±5時間で、測定クリアランスは1.7±0.1mL/分/kgである。Rumore(同サイト)
本試験では(表4の治療A及びCで表示されているように)、CPMについての製剤XのT2/1は41.71±43.33時間であった。これに対してRLDの場合は19.37±9.66時間であった。しかしながら、サンプリングプロトコルでは、製剤X投与後24時間しかCPMを計測しなかった。わずか24時間の計測では、製剤XにおけるCPMの排出半減期を正確に測定するには不十分だったことが、T1/2にみられる変動性の一因となったのかもしれない。生物学的等価性は検出されるものと考えられる。
【0128】
表2〜表4に示すデータは、TmaxとT1/2について、必ずしも既に公表されている前記の数値を反映していない。その理由は、製剤Xが徐放性製剤だからである。TmaxとT1/2の公表されている数値は、血液中へ即座に取り込まれた後に移動することができる薬剤の数値である。製剤Xはすべての薬剤を即時的に放出しないのでTmaxは遅く、投与後数時間は薬剤が一定して足されているためT1/2とTmaxは変動するようである。同様に、表2〜表4に示されているRLDの数値も必ずしも公知の数値を反映していない。これは、既に公表されている試験では単回投与で数値を求めていたのに対し、表中の数値は6時間間隔で2回投与しているからである。本試験におけるRLDの2回目の投与が薬剤濃度の2つ目の急激なピークの理由である。結果としては、RLDの2回目の投与後、確かな薬剤ピーク濃度が得られる。結果、表2〜表4のRLDについてのTmaxは明らかに遅くなっているが、これは単に投与プロトコルに起因しているにすぎない。
【0129】
クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンの生物学的等価性
FDAは、単回投与試験の場合、AUCが生物学的等価(BE)を示すための唯一の適切な薬物動態学的(PK)パラメータとしているが、FDAは他のPKパラメータについての情報を要求している。表2〜表4には、製剤Xの2つの異なる処方とそれぞれに対応するRLDについてのPKデータが記載されている。表2は、HCについて、治療A(HC15mgを含む製剤X)と治療C(HC7.5mgを含むRLDを2回投与)を比較し、製剤XのAUCについての点推定はRLDの93.28%で、90%信頼区間(CI)が84.82〜102.58%であることを示している。これはFDAのBE標準である90%信頼区間(CI)の80〜125%を満たす。そのため、この試験ではHCは生物学的等価性を達成した。表3は、PSEについて治療Aと治療Cを比較し、製剤XのAUCについての点推定がRLDの95.45%で、90%信頼区間(CI)が88.55〜102.88%であることを示すので、生物学的等価性を達成している。表4は、CPMについて治療Aと治療Cを比較し、製剤XのAUCについての点推定がRLDの129.24%で、90%信頼区間(CI)が97.33〜171.60%であることを示している。この数値はBEを立証していない。しかしながら、本試験においてCPMのBEが確立できなかったのは、製剤XがCPM(HC及びPSEに加え)のBEを立証できなかったからではなく、本試験では、CPMの長い半減期を血漿採取中に考慮していなかったことによるものである。具体的に言うならば、最後の血漿サンプルの回収時、正確なT1/2が求められるところまでCPM濃度はまだ減衰していなかった。結果、AUCを求めるために必要な外挿が信頼性を欠いた。これについては、CMPのRLDと製剤Xが当てはまる。今後の試験によって、試料採取時間を適切なものにすれば、CPMのデータから製剤XがCPMのRLDと生物学的等価であることを示すことができよう。
【0130】
つまり、製剤XにおけるAUCの90%信頼限界は、HC及びPSEについてはRLDの80〜125%の範囲であり、少なくともこの2種の薬剤の生物学的等価性を示している。本試験では、CPMについてはAUCが明確な生物学的等価性を立証しなかった。これは、製剤Xの90%信頼区分が、CPMのRLDと比較すると97.33〜171.60%の範囲だったからである。すべての製剤にみられる高い個体内変動性とAUCO-INFとAUCO-LASTとの大きな差により、CPMのAUC分析は複雑であった。変動性にむけたテストサンプルの増大化とサンプリング時間の延長により、CPMの長い排出半減期(ある資料によると20〜24時間)を斟酌すれば、CPM含有RLDに対する製剤XのCPMの生物学的等価性は立証されるであろう。「Drug information: クロルフェニラミン」www.accessmedicine.com/ drugs. aspx?index=Cにて閲覧可能、2007年5月15日アクセス。
【0131】
即放性(IR)シロップ剤、徐放性(ER)ビーズ、lR/ERビーズ含有懸濁液におけるAPI量比を定めるための分析用HPLC法
APIとして抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む製剤を調製する際、分析用の方法を用いてAPIの採用すべき量又は比率を定める。それにより、該製剤を患者に投与した際、製剤は、3種のAPIすべての放出について徐放性を示す。
当該分野の当業者が本発明を製造及び使用できるように、十分に詳しく本発明を説明し例示してきたが、本発明の精神及び範囲から逸脱せずに、様々な代替、変更及び改善できることは明白である。本願明細書に記載した例は特定の実施形態を代表するものであり、例示であり、本発明の範囲を制限することを意図するものではない。その範囲内の変更や他の使用も当該分野の当業者には思いつくであろう。それらの変更は本発明の精神の範囲内に含まれ、クレームの範囲により規定される。
当該分野の当業者にとっては、本発明の範囲及び精神から逸脱することなく本願明細書に開示の発明に代替や変更が行えることは容易にわかることである。本発明のいくつかの態様を説明することを目的とした実施例に開示の特定の実施形態によって、本発明はその範囲を制限されることはなく、機能的に均等である実施形態は本発明の範囲に入る。実際、本願明細書に示し、記載した変更にくわえて様々な変更がこの分野の当業者には明らかとなり、それらは添付のクレームの範囲に入るものである。
本明細書で言及した特許及び出版物のすべては、本発明が関連している分野の当業者のレベルを示している。すべての特許及び出版物は、各文献を個々に具体的に示して援用するのと同じ程度に、引用により本願明細書の記載に含まれるものとする。
【0132】
実施例3
実施例3は、製剤X中のプソイドエフェドリン、ヒドロコドン及びクロルフェニラミンの放出プロファイルを、個々のビーズにクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンを含有する被覆ビーズからの各薬剤の放出プロファイルと比較するために行った生体外テストの結果を説明する。
製剤Xは、実施例1で説明したようにして調製した。実施例1にしたがって、個々のビーズがクロルフェニラミンとヒドロコドンとプソイドエフェドリンとを含む、被覆ビーズを調製した。時間ゼロサンプル(t=0)は、調製終了後から1週間以内に分析を行ったが、他のサンプルは、調製終了後から、湿度60%のチャンバーにおいて25℃で保存し(25/60)、1ヵ月(1mo)、3ヵ月(3mo)、6ヵ月(6mo)、9ヵ月(9mo)及び12ヵ月(12mo)後にそれぞれ分析した。高速液体クロマトグラフィー(HPLC)により定量化を行った。米国薬局方対応装置2(USP Apparatus 2)のパドルを100RPMで運転して、放出テストを行った。最初に、pH1.2のバッファー750mLを各容器に投入した。1.5時間後、pH調整液を250mL加えてpH6.8、容量1000mLとした。オートサンプラーは、0.5、4、8、12時間の時点でおよそ5mlのサンプルを抜き取るようにプログラムした。
【0133】
図1(A)は、製剤Xの液状放出持続型懸濁液からのプソイドエフェドリンの放出プロファイルを示す。図1(B)は、被覆ビーズからのプソイドエフェドリンの放出プロファイルを示す。図2(A)は、製剤Xの液状放出持続型懸濁液からのヒドロコドンの放出プロファイルを示す。図2(B)は、被覆ビーズからのヒドロコドンの放出プロファイルを示す。図3(A)は、製剤Xの液状放出持続型懸濁液からのクロルフェニラミンの放出プロファイルを示す。図3(B)は、被覆ビーズからのクロルフェニラミンの放出プロファイルを示す。
図1〜図3によると、製剤Xにおける各薬剤の即放性部位の存在について結果を正規化させると、製剤Xのプソイドエフェドリン、ヒドロコドン及びクロルフェニラミンの徐放性部位の放出速度は、被覆ビーズからのプソイドエフェドリン、ヒドロコドン及びクロルフェニラミンの放出速度と同等であることがわかる。図1〜図3に示された実験結果から、即放相中のイオン成分の存在、即ち、それぞれに塩の形態をとる遊離な薬剤の存在が、製剤Xの徐放性部位から持続的に放出される各薬剤の適切な放出速度を妨げていないことがわかる。また、図1〜図3は、タイムゼロの時点でテストした製剤Xのプソイドエフェドリン、ヒドロコドン及びクロルフェニラミンの徐放性部位の放出速度が、製造後1ヵ月、3ヵ月、6ヵ月、9ヵ月及び12ヵ月の間保存した製剤Xのプソイドエフェドリン、ヒドロコドン及びクロルフェニラミンの徐放性部位の放出速度と同等であることを示している。このように、図1〜図3に示す実験結果から、製剤Xは、1ヵ月、3ヵ月、6ヵ月、9ヵ月及び12ヵ月間室温で保存した後も安定性を保ち、各薬剤の持続性放出の適切な速度が保たれていることがわかる。
【0134】
実施例4
実施例4では、薬剤を塩基の形態で分散相に含む液状放出制御型組成物の放出プロファイルを、薬剤を塩の形態で分散相に含む液状放出制御型組成物の放出プロファイルと比較するために行った生体外テストの結果を説明する。
テストは2種類の組成物を使って行った。それぞれ、5mlの液体剤形で、プソイドエフェドリン塩酸塩30mg(IRが7.5mg、ERが22.5mg)、ヒドロコドン重酒石酸塩7.5mg(IRが1.87、ERが5.63mg)及びクロルフェニラミンマレイン酸塩6mg(IRが1.5mg、ERが4.5mg)相当を放出するようにした。第1の組成物は、18.4mgのプソイドエフェドリン、3.41mgのヒドロコドン及び1.58mgのクロルフェニラミンを含み、これらの薬剤は塩基の形態で、アルギン酸樹脂に結合させた。薬剤充填アルギネートビーズを作り、コーティングを行い、65%w/wのショ糖を含む分散媒に分散させた。第2の組成物は、22.5mgのプソイドフェドリン塩酸塩、5.62mgのヒドロコドン重酒石酸塩及び4.5mgのクロルフェニラミンマレイン酸塩を含み、アルギン酸ナトリウムに結合させた。第2の組成物においては、コアビーズを製造し、本質的には実施例1記載のようにコーティングを行い、できた被覆ビーズを65%w/wのショ糖を含む分散媒に分散させた。両者とも、即放性とした設計した塩の形態の薬剤の一回分投与部位が分散媒に含まれた。
【0135】
分析に先立ち、サンプルを室温で3週間保存した。その後、HPLCで定量化した。米国薬局方対応装置2のパドルを100RPMで運転させ、放出テストを行った。最初に、pH1.2のバッファー750mLを各容器に投入した。1.5時間後、pH調整液を250mL加えてpH6.8、容量1000mLとした。オートサンプラーは、以下の時点、5分、30分、60分、90分、4時間、8時間、12時間、16時間、20時間、24時間におよそ1mlを抜き取るようにプログラムした。
図4(A)は、プソイドエフェドリンの塩基性製剤の放出プロファイルを示す。 図4(B)は、プソイドエフェドリンの塩製剤の放出プロファイルを示す。図5(A)は、ヒドロコドンの塩基性製剤の放出プロファイルを示す。 図5(B)は、ヒドロコドンの塩製剤の放出プロファイルを示す。図6(A)は、クロルフェニラミンの塩基性製剤の放出プロファイルを示す。 図6(B)は、クロルフェニラミンの塩製剤の放出プロファイルを示す。
図4〜図6によると、液状放出持続型組成物においては、塩の形態のプソイドエフェドリン、ヒドロコドン及びクロルフェニラミンの放出速度は、塩基の形態のプソイドエフェドリン、ヒドロコドン及びクロルフェニラミンの放出速度とそれぞれ同等であることがわかる。詳細には、実験の各時点において、これらの薬剤が塩の形態でも塩基の形態でもほぼ同じか著しく異なることのない放出特性を示す。図4〜図6に示す実験結果から、これらの薬剤の適切な持続性放出プロファイルを得るために、塩の形態の薬剤を本発明の液状放出制御組成物に使用できることがわかる。
【0136】
実施例5
実施例5は、ヒドロコドンを唯一の有効成分として分散相に含む液状放出持続型組成物からのヒドロコドンの放出プロファイルを示す。
10mgのヒドロコドン塩基がアルギン酸マトリックスに結合して含まれる液状放出制御組成物を使って実験を行った。アルギン酸及び20%ラクトース一水和物に10mgのヒドロコドンが結合して含まれるように、ヒドロコドンアルギネートビーズを調製した。表5は、ヒドロコドンアルギネートビーズ製剤の定量組成の概要を示す。
【0137】
【表5】
【0138】
更に、ヒドロコドンアルギネートビーズを、1.5%Opadry/31.5%Eudragit RS30D(質量基準による質量)でコーティングした。被覆ヒドロコドンアルギネートビーズを、10mgのヒドロコドンに対して5mLのシロップ剤になるよう分散した。表6にヒドロコドン含有徐放性ペレットの分散液の定量組成の概略を示す。
【0139】
【表6】
【0140】
製造終了後、すぐにサンプルの含量検定を行った。HPLCで定量化した。米国薬局方対応装置2のパドルを100RPMで運転させ、放出テストを行った。最初に、pH1.2のバッファー750mLを各容器に投入した。1.5時間後、pH調整液を250mL加えてpH6.8、容量1000mLとした。オートサンプラーは、以下の時点、30分、1時間、1.5時間、2時間、3時間、4時間、6時間、8時間、10時間後におよそ1mlを抜き取るようにプログラムした。
図7は、アルギン酸マトリックに結合したヒドロコドンを含む液状放出制御組成物からのヒドロコドンの放出プロファイルを示す。
図7から、唯一の有効成分であるヒドロコドンをイオン交換マトリックスに結合させて含むビーズがシロップ剤に分散された液状放出持続性組成物からのヒドロコドンの放出速度がわかる。
【図1】
【図2】
【図3】
【図4】
【図5】
【図6】
【発明の詳細な説明】
【0001】
(関連出願の相互参照)
本願は、2009年5月1日に出願された米国特許仮出願第61/174,891号の優先権を主張するものであり、引用によりその内容すべてを本願明細書に援用する。
【0002】
(発明の背景)
年間で米国人がひく風邪は推定10億件、大人が年間で2〜4回、子どもは年間6〜10回風邪をひいている(国立アレルギー感染症研究所(NIAID)のファクトシート「The Common Cold(普通の風邪について)」www.niaid.nih.gov/factsheets/cold.htmにて閲覧可能、2004年12月)。米国人10人のうち約9人が風邪又は同様な上気道感染症に毎年少なくとも1回かかることになる。風邪は子供達のなかでは最も流行する病気であり、いずれの複合的な疾患よりも多発し、学校での子供の欠席理由全体の50%も占める(Micromedex Healthcare Series、「The Common Cold Etiology and Treatment(一般の風邪の原因と治療)」www.thomsonhc. com/hcs/librarian/NDで閲覧可能、2008年6月18日アクセス)。風邪が最もはやるのは、8月又は9月初旬から3月又は4月に及ぶ気温が低い数ヵ月の間である。大気温度の低下に伴い湿度も低くなりがちであり、室内や狭い部屋の中での人と人との交流が増えることから、風邪の原因となるウィルスの増殖を増長するようである(国立アレルギー感染症研究所(NIAID)のファクトシート「普通の風邪について」www.niaid.nih.gov/factsheets/cold.htmで閲覧可能、2004年12月アクセス)
【0003】
普通の風邪をひいた患者は、鼻水、鼻ずまり、鼻腔膜の腫れ、くしゃみ、のどの痛み、咳及び頭痛といった症状や徴候を示すのが一般的である(Micromedex Healthcare Series、「一般の風邪の原因と治療」(同サイト))。患者の70〜90%が鼻漏又はくしゃみを患い、65%が鼻づまり又はうっ血を起こし、25%が咳又は声かれをおこしている(同サイトより)。ほとんどの風邪が7〜14日間続く。2週間を超えて徴候が続く患者はアレルギーになっている可能性もある(同サイト)。
米国肺協会(American Lung Association)によると、風邪で病院に訪れる割合は、他のいずれの疾病の場合よりも多い(米国肺協会「Cold and Flu Guidelines: The Common Cold(風邪及び流感のガイドライン:普通の風邪)」www.lungusa.org.で閲覧可能、2008年6月16日アクセス)。実際のところ、普通の風邪による影響についての研究では、2003年だけで風邪に関連して病院をおとずれた件数は1億件を超え、費用は77億ドルであったことが示されている。また、この研究の概算によると、子どもたちが学校を休んだ日数は毎年1億8900万日となり、風邪の子どもを看病するために親は1億2600万日仕事を休んでいる(WebMD. 「Cost of the Common Cold: $40 Billion(普通の風邪によるコスト:400億ドル)」 www.medmutual.comにて閲覧可能、2003年2月24日アクセス;Fendrick等「The Economic Burden of Non-Influenza-Related Viral Respiratory Tract Infection in the United States.(米国における非インフルエンザ系ウィルスによる気道感染の経済的負荷)」Arch Intern Med. 163(4): 487〜494頁(2003))。風邪をひいた従業員が就労を休んだ1億5000万日に加えると、風邪に起因した労働損失の経済的影響は全部で200億ドルを超える(Fendrick(同書);Garibaldi RA,「Epidemiology of community-acquired respiratory tract infections in adults. Incidence, etiology, and impact(コミュニティーを襲う大人の気道感染についての流行病学。発生、病因及び影響)」 Am.J. Med. 78 (6B): 32〜37頁(1985);「普通の風邪について」国立アレルギー感染症研究所、www3.niaid.nih.gov/healthscience/healthtopics/colds/ 2008年6月11日検索;米国勢調査局www.Quickfacts.census.gov/qfd/states/00000.html2004年の概算)。
【0004】
米国において、咳や風邪の緩和を目的とする薬局の薬(OTC)及び処方箋による薬にかかった費用は、毎年30億ドルを超えている(WebMD(同サイト)、Fendrick(同書))。米国だけでも、普通の風邪で医師にかかる件数は年間で7,500万〜1億件になり、費用は控えめに見積もっても毎年77億ドルになる。米国人は徴候の緩和のために薬局の薬に29億ドル、さらに処方箋による薬に4億ドルを使っている(Fendrick(同書)、 Garibaldi(同書))。医者にかかった患者の3分の1を超える患者が抗生物質の処方箋を受け取っているが、これは不要な出費になるだけでなく(米国内では概算で4100万件の抗生物質の処方箋に対し年間11億ドルが支払われている)、このような薬剤の乱用にが抗生物質耐性を意味することにもなる(Fendrick(同書))。IMS Health社によると(2007年8月)、咳、風邪及び流感のための処方箋の数は年間でおよそ42,858,000件であった。すべての風邪の兆候を唯一の有効医薬成分(API)では治せないことから、単一成分の医薬品を多数使用するよりも、調合医薬品の方が、兆候緩和をもたらすのにより便利で、時にはより安価に提供できることが多い。
【0005】
本発明は、即放性及び/又は現在使用できる風邪やアレルギーの調合薬に比べて独特な長所を提供する。例えば、本発明の1つの実施形態は、3種類のAPI、即ち、抗ヒスタミン剤(例えばクロルフェニラミン)と鎮咳剤(例えばヒドロコドン)と充血緩和剤(例えばプソイドエフェドリン)の長所が、1つの徐放性(ER、例えば12時間)組成物中に統合されている。今まで、これらのAPIを一緒に使用する場合は、それぞれの即放性(IR)の剤形で1日に4〜6回の投与が必要であった。なぜならば、これらの3つのAPIの効果を示す単一の徐放性調合医薬品が現在は手に入らないからである。
上記APIの1種又は2種を含む徐放性医薬品は既に市場に存在する。同様に、3種類すべての薬剤を含む即放性医薬品もいくつかが市場に存在する。例えば、クロルフェニラミン(CPM)、ヒドロコドン(HC)及び/又はプソイドエフェドリン(PSE)を含む、即放性(IR)又は徐放性(ER)剤形の市場に出ているOTC又は処方箋薬としては、以下のものが挙げられる。
【0006】
プソイドエフェドリンを含む医薬品
Afrinol(登録商標)
Cenafed(登録商標)
D-イソエフェドリン
D-プソイドエフェドリン
Decofed(登録商標)
Dimetapp(登録商標) 充血緩和剤
Dimetapp(登録商標) 充血緩和剤、小児科用点滴薬
Drixoral(登録商標) 鼻の充血緩和剤
Efidac 24(登録商標)プソイドエフェドリン HCl
Eltor 120(登録商標)
Genaphed(登録商標)
イソエフェドリン
【0007】
Maxenal(登録商標)
Myfedrine(登録商標)
Novafed(登録商標)
Pedia Care(登録商標)
Pseudo 60's(登録商標)
Pseudo-12(登録商標)
プソイドエフェドリン塩酸塩
Sudafed 12 Hour(登録商標)
Sudafed 24 Hour(登録商標)
Sudogest
【0008】
クロルフェニラミンを含む医薬品
Aller-Chlor (登録商標)
Antagonate(登録商標)
Chlo-Amine(登録商標)
Chlor-Trimeton(登録商標)
Chlor-Tripolon(登録商標)
デキスクロルフェニラミンマレイン酸塩
Efidac 24(登録商標) クロルフェニラミンマレイン酸塩
Gen-Allerate(登録商標)
Haynon(登録商標)
Histadur(登録商標)
【0009】
Kloromin(登録商標)
Mylaramine(登録商標)
Novo-Pheniram(登録商標)
Phenetron(登録商標)
Piriton(登録商標)
Polaramine(登録商標)
Pyridamal 100(登録商標)
Telachlor(登録商標)
Teldrin(登録商標)
【0010】
ヒドロコドンを含む医薬品
Hycodan(登録商標) (臭化メチルホマトロピンを含む)
Lortab(登録商標) (アセトアミノフェンを含む)
Maxidone(登録商標) (アセトアミノフェンを含む)
Norco(登録商標) (アセトアミノフェンを含む)
Vicodin(登録商標) (アセトアミノフェンを含む)
Zydone(登録商標) (アセトアミノフェンを含む)
プソイドエフェドリンとクロルフェニラミンとを含む医薬品
Allerest(登録商標)
Anamine(登録商標)
Biohist-LA(登録商標)
Brexin(登録商標)
【0011】
Chlordrine(登録商標) SR
Chlor-Phed(登録商標)
Chlor-Trimeton(登録商標) (4時間及び12時間)
Deconamine(登録商標)
De-congestine TR(登録商標)
Dynahist ER(登録商標)
Histalet(登録商標)Syrup
Kronofed-A(登録商標) Kronocaps
Kronofed-A-Jr.(登録商標) Kronocaps
【0012】
ND(登録商標)Clear
Pseudoephed/Chlorphen 100(登録商標)
Rescon(登録商標)
Rescon(登録商標)Jr.
Rescon(登録商標)ED
Ryna(登録商標)
Sudafed(登録商標)Cold and Allergy
Tanafed(登録商標)
クロルフェニラミンとヒドロコドンとを含む医薬品
Tussionex(登録商標)Pennkinetic(登録商標)徐放性懸濁液
TussiCaps(登録商標)徐放性カプセル剤
S-T Forte(登録商標)2
【0013】
プソイドエフェドリンとヒドロコドンとを含む医薬品
Detussin(登録商標)Liquid
Histussin(登録商標)D Liquid
Tyrodone(登録商標)Liquid
クロルフェニラミンとプソイドエフェドリンとヒドロコドンとを含む医薬品
A-G Tussin(登録商標)
Atuss(登録商標)HD
Cordron-HC(登録商標)
Hexatussin(登録商標)
Histinex(登録商標)PV
Hydrocof-HC(登録商標)
Hydron(登録商標)PCS
Hydrotuss(登録商標)HC
Hyphed(登録商標)
【0014】
KG-Tussin(登録商標)
M-End(登録商標)
Notuss(登録商標)
P-V-Tussin(登録商標)Syrup
Pediatex(登録商標)HC
Q-V Tussin(登録商標)
Tussin-V(登録商標)
上記の3つの効果を示す製剤(CPMとPSEとHCとを同一医薬品に含む)は、いずれも3種成分すべてについての徐放性医薬品ではない。また、3種類すべての有効成分を含む経口組成物を患者に単回投与することにより、3種薬剤の血清レベルが12時間にわたり、該有効成分を含むFDAに承認された即放性の関連リスト収載医薬品(reference listed drug:RLD)組成物を12時間にわたって適切な投与回数で投与した際に得られる血清レベルと生物学的等価となるようにしたグループはいまだに構築されていない。
【0015】
同様に、ヒドロコドンとプソイドエフェドリンとを含む前記製剤(CMP等の他の有効成分を有していてもいなくてもよい)はいずれも徐放性医薬品ではない。ヒドロコドンとプソイドエフェドリンとを含む経口組成物の単回投与で、2種の血清レベルを12時間にわたり、ヒドロコドン及び/又はプソイドエフェドリン含有のFDAに承認された即放性RLD組成物を12時間にわたって適切な投与回数で投与した際に得られる血清レベルと生物学的等価となるようにしたグループはいまだに構築されていない。実際のところ、ヒドロコドンとプソイドエフェドリンの両方を含み(他の有効成分を有していてもいなくてもよい)、即放性又は徐放性である医薬品は、現在のところFDAに承認されているものはない。前記に示した市販医薬品リストは、FDA未承認であるが市場に出ている医薬品が挙げられていることを記しておかなければならない。
【0016】
本発明の別の実施形態に関連するが、OTC医薬品及び処方箋医薬品に含まれる薬剤の転用や乱用が近年段階的に拡大している。例えば、OTCの風邪及びアレルギー用錠剤の多くはプソイドエフェドリン又はエフェドリンを含むが、これを使ってメタンフェタミンとして知られる乱用薬物が秘密裏に製造されている。この薬物は「メス」「スピード」「クランク」又は「アイス」としても知られているが、中枢神経系をおかす強力で中毒性の覚醒剤である。メタンフェタミンは、ふかしたり吸い込んだり、注射したり飲み込めるように丸剤、カプセル剤、散剤にして違法で販売されている。
にわか作りの秘密の違法薬物研究所(meth labs)では、OCTの風邪やアレルギー用錠剤からプソイドエフェドリン又はエフェドリンが分離するまで、水、アルコール又は他の溶媒の溶液を使って数時間でプソイドエフェドリン又はエフェドリンを単離させる。その後、容易に手に入る一般的な家庭用品や簡単に査定できる(assessable)装置、即ち、アルコール、コールマンのホワイトガソリン、アセトン、道路用発煙筒(road flare)、排水溝クリーナー、ヨウ素、塩酸、岩塩、(エンジンなどの)点火燃料、コーヒーフィルター及びマッチ等を使って、プソイドエフェドリン又はエフェドリンをメタンフェタミンに変換する。薬物の製造方法は、インターネットを介して容易に入手できる「レシピ」や情報交換から入手できる。
米国ではメタンフェタミンの非合法取引や乱用が高まりつつあるが、プソイドエフェドリンを盗んだり転換することを防ぐ方策は厄介で費用がかかる。
【0017】
オピオイド類等の薬物を含む他の多くのOCT医薬品及び処方箋医薬品も、非合法的薬物乱用に流用する絶好のものである。例えば、ヒドロコドンは、風邪薬の鎮咳剤(咳抑制剤)として、また、処方箋薬に含まれる中程度から僅かに重篤な痛みを治療するための鎮痛剤として合法的に使用されている。ヒドロコドンは米国では最も多く処方されるアヘン含有鎮静剤で、2003年にはヒドロコドン含有薬品の1億1000万件を超える処方箋が調剤された。一般的にヒドロコドンの製造は非合法ではないが、目下、非合法的な転用や、病的高揚状態を得て苦痛緩和の効果を得るための直接的な乱用も見られる。偽のクリニックからの電話による処方箋、改竄した処方箋、窃盗、インターネットからの不法な購入を介して広範な転用が起こっている。
このように、不法な使用を目的とした薬剤の乱用や転用の可能性を回避できるOTC医薬品及び処方箋薬の調製及び販売へのニーズがある。例えば、にわか作りの研究所ではプソイドエフェドリン、エフェドリン及び/又はヒドロコドン等の薬剤が容易に抽出、分離できないような、風邪/咳及びアレルギー製剤へのニーズがある。同様に、非合法的研究所で秘密裏に製造されるかどうかには拘わらず、不正な目的のために容易に転用できない風邪/咳、痛みの緩和、筋肉弛緩の医薬品へのニーズが存在する。
【0018】
(発明の概要)
本発明の実施形態により、以下の有効成分(1)抗ヒスタミン剤、(2)鎮咳剤及び(3)充血緩和剤の1種以上を含む即放性(IR)製剤及び/又は調合医薬品のこれまでに関わる問題点や不利な点が解決される。現在市販されている多くの医薬品に対して、本発明は、3種すべての有効成分を徐放(ER)(例えば12時間)できる製剤を提供する。一例として、本発明は、クロルフェニラミン(抗ヒスタミン剤)、ヒドロコドン(麻酔性鎮咳剤)、プソイドエフェドリン(充血緩和剤)のIR剤形とER剤形との新規混合物を一つの医薬品にした経口製剤を提供する。結果的にこの製剤は1日2回投与が可能なER型調合品となり、以前から入手可能であったIR型(個々の成分ごと又は調合品を介してのいずれかで販売及び投与されてきた)と同じ効力を有する。また、この製剤は現存する単一及び調合のER製剤よりも以下の点で優れている。(a)即時及び長時間の薬剤伝達のためにIR及びER両方の薬剤投薬量を提供する。(b)3種の関連リスト収載医薬品(RLD)と生物学的等価な投薬量を単回投与形態で提供できる。(c)成分薬物の乱用及び転用を阻止する。
【0019】
本発明の実施形態の1つでは、充血緩和剤、鎮咳剤及び/又は抗ヒスタミン剤という有効成分を含む経口医薬組成物を患者へ単回投与することにより、上記有効成分を含有するFDA承認済みの即放性(IR)RLDを12時間にわたって適切な投与回数で投薬する際に得られる血清レベルと生物学的等価である有効成分の血清レベルを12時間にわたり得られる。即放性RLDの適切な投与回数とは、即放性RLDの12時間にわたる投与に関してFDAが承認している表示1つ以上で推奨される投与回数に相当する。
別の実施形態では、24時間を超える投薬時間にわたり有効成分の安定した血清レベルを得るため、充血緩和剤、鎮咳剤及び/又は抗ヒスタミン剤という有効成分を含む徐放性経口組成物を十分な投与回数で患者へ投薬することにより、上記有効成分を含有するFDA承認済み即放性医薬組成物1種以上を上記と同じ時間にわたり適切な投薬回数で投与した際に得られる血清レベルと生物学的等価である有効成分の血清レベルが得られる。即放性医薬品の適切な投与回数とは、FDAが承認する即放性医薬品1種以上の同じ時間にわたる投与について、FDAが承認する表示1つ以上で推奨する投与回数に相当する。更に本発明の別の実施形態は、ヒドロコドンとプソイドエフェドリンとを含む経口製剤であって、抗ヒスタミン剤を含むか否かにかかわらず上記有効成分が人体内で徐放される経口製剤を提供する。
【0020】
実施形態の1つでは、本発明製剤の徐放性(ER)部位が、懸濁液中の被覆ビーズ、粒子又はペレットの形態で存在することができ、即放性(IR)部位が懸濁液中に存在することができる。或いは、徐放性部位はカプセル剤、錠剤又は他の経口固形物といった固形の剤形を有し、即放性部位が前記徐放性部位の外側の第2の層又は媒体とすることもできる。実施形態の1つでは、12時間ごとに投与するように製剤化する。他の実施形態は、8時間ごと、16時間ごと、24時間ごと等といった投与用に製剤化されたものを含む。
同様に、別の実施形態においては、一つの徐放性調合医薬品が特定のIR/ER比を示し、徐放(ER)成分は粒子、ペレット又はビーズ内に存在し、即放(IR)部位が前記粒子、ペレット又はビーズの外側に存在する(例えば、シロップ剤中に懸濁、カプセル剤、錠剤等に含まれる散剤)。上記比率により、単回投与でも安定状態のいずれの条件においても、関連リスト収載医薬品(RLD)に対して生物学的等価(BE)な血清レベルが得られる。他の実施形態では、本発明の製剤を用いたヒトは、ある特定の血清レベル範囲(AUC、Tmax、T1/2等で測定)を経時的に得るが、該血清レベルは、単回投与でも安定状態のいずれの条件においても関連リスト収載医薬品(RLD)と生物学的等価(BE)である。
本発明の実施形態の1つは、抗ヒスタミン剤と鎮咳剤とを含み、任意で充血緩和剤を含んでいてもよい有効成分を即放剤形で含有する第1部位と、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを有効成分として徐放剤形で含む粒子、ペレット又はビーズを有する第2部位とを含有する、経口徐放性医薬組成物の製造方法に関する。別の実施形態では、本発明の方法は、即放性部位が最初に抗ヒスタミン剤と鎮咳剤とを含むが、充血緩和剤を含まず、徐放性部位が抗ヒスタミン剤と鎮咳剤とプソイドエフェドリンとを含むようにして組成物を製造する工程を有する方法である。関連の実施形態では、プソイドエフェドリン等の徐放性成分の1種以上を即放性ビヒクルに浸出させて、1種以上の成分による即放剤形としてもよい。
【0021】
また別の実施形態では、本発明の製剤は、1個のビーズ内に三種類すべての薬剤を含有する粒子を提供する。このような実施形態は、各薬剤をそれぞれ別のビーズで提供する既存の徐放性製剤に比べ、用量有効性、安全性及び転用や乱用に対する阻止力というすべての点において有利性を示す。
本発明の別の実施形態では、プソイドエフェドリン(又はエフェドリン等の化学的関連のある充血緩和剤)及び/又は麻酔性鎮咳剤(ヒドロコドン等)を含む徐放性粒子、ペレット又はビーズが、これらの薬剤を1種以上含む他の市販のOTC医薬品や処方箋医薬品と比較して、該薬剤の誤用、乱用又は不法な転用が出来ないように又はやりにくくするようにして、製剤に含まれる。例えば、本発明の製剤中の1個1個の徐放性粒子又はペレット又はビーズに、クロルフェニラミンやヒドロコドンといった抗ヒスタミン剤や鎮咳剤をはじめとする化合物と共にプソイドエフェドリン(又は関連化合物)が含有されていれば、違法薬物(meth)の製造や麻薬の単離を目的としたPSE(又はエフェドリン)及び/又は麻薬性鎮咳剤の最終製剤からの分離又は抽出を阻止、低減する。
【0022】
PSE、エフェドリン及び/又は麻薬を本発明の製剤から(例えばビーズから)抽出することは技術的には可能であるかもしれないが、抽出には複雑で高価な装置と技術が必要になる。違法薬物を不正に製造する者たちのほとんどは、そうまでして抽出作業をする装置及び/又はリソースには近付かない。特に、これらの薬剤を含有する他のOTC医薬品を用いて簡単にできることに比べればなおさらである。このように、PSE(もしくは関連化合物)又は麻薬性鎮咳剤を含有する市販のOTC製剤の多くとは異なり、更に自由に入手できる本発明製剤のOTC用にした医薬品は小売業者が販売することができる。
そこで本発明の実施形態は、現在市場にでている即放性及び/又は調合医薬品と比較すると特別な利点をもたらす。その利点は下記が挙げられる。(1)4時間から6時間ごとではなく1日2回の投与となることで、患者のコンプライアンスを向上させ、正しい用量の薬剤が伝達される。(2)処方箋が必要な咳や風邪の薬にプソイドエフェドリンの使用を導入すれば、免許をもった医学専門家を必ず巻き込むため、プソイドエフェドリンの使用は、例えば他の薬と一緒に投与する場合など、より適切な使用につながる。(3)懸濁液は錠剤よりも投与の選択肢が一層融通がきくため、個別化した患者の治療を斟酌し、兆候をもとに治療のバランスを調整することができる。(4)製剤の徐放性部位における有効医薬成分(API)の処理方法により、ヒドロコドン及び/又はプソイドエフェドリンが乱用及び/又は転用される可能性を削減する。(5)使用単位(4オンス)又は単位服用量(5〜10ml)を用いることにより、追跡がより簡単になり、転用の可能性も減少させることができる。
【0023】
更に本発明は、1種以上の薬剤を含むペレット、ビーズ又は粒子を有する徐放性成分を含み、該ペレット、ビーズ又は粒子がシロップ剤に懸濁している、新規な経口懸濁液製剤を提供する。前記シロップ剤も1種以上の薬剤を含んでいてもよい。当該分野では、経口懸濁液は他の液体製剤よりも安定性に優れる。
本発明は、製造工程中、ビーズの中に可溶性の非電解質成分を含有させることを想定している。そのような成分は、水がビーズ内に吸収されると溶けて、リザーバーから拡散し浸透圧を下げるため、ビーズの膨潤を抑えビーズ被膜の状態が損なわれるのを抑える。したがって、本発明は、薬剤充填ビーズが水を吸収する際に大量移動が可能である賦形剤の使用を包含するもので、それにより、ビーズ内部の圧力を低下させ、ビーズ被膜の崩壊を防ぎ薬剤放出の制御を容易にする。
このように、本発明は、水和性が高く水と強力に結合することができる不活性成分を高濃度に添加し、分散相である薬剤複合体と水との結合を妨害することにより、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明組成物の分散媒中における水分活性(water activity)を低下させることも包含する。このように、分散媒中の水は薬剤充填ビーズにそれほど十分には引きつけられないため、投与の前に薬剤が溶解することを抑えられ、薬剤は懸濁液の分散相中に閉じ込められる。
【0024】
したがって、本発明の実施形態の1つでは、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の剤形として、患者への投与時に持続的薬物放出を行うことができる熱力学的に安定した液体剤形の薬剤懸濁液を包含する。このような液体製剤は12、24時間にわたって48時間まで持続的放出が可能である。このように、本発明は、1日1回又は2回の投与しか必要でない、投与を確実に容易にする液体剤形を包含する。本発明では、可溶性の非電解質成分で比較的低分子量の成分が、薬剤複合体に含まれていてもよい。実施形態の1つは、薬剤-イオン交換マトリックス-複合体が1個のビーズである。また、本発明では、多孔質又は非多孔質の高分子膜で分散相が被覆されていてもよいことが考えられる。有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の実施形態の1つでは、分散媒中の水としっかりと結合することができる高度に水和化された賦形剤を分散媒にも含有することにより、分散媒中の水分活性が抑えられ、水が薬剤充填ビーズに引きつけられるのを最小限に抑え、投薬の前に薬剤が溶けないようにする。このような実施形態では、患者に投与されてから薬剤放出が開始される。例えば胃液又は膓液中という、水が高濃度に存在し、薬剤と同じ電荷を帯びた小さなイオンが存在する環境に懸濁液が配置された時、その小さなイオンにより拡散した二重の層は浸水し、薬剤の放出が引き起こされる。特定の実施形態において、上記剤形を投与した後、分散媒が患者の胃液によって希釈され、膜の水和化により膜はより一層多孔になり、ビーズからの薬剤の溶解と分散が可能になる。
【0025】
分散相は3種の薬剤又は有効成分を含む。本発明による液状の放出制御組成物において、その分散相は有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる。また、これらの薬剤が1個1個の粒子、ペレット又はビーズの中にまとまっていることも想定される。実施形態の1つは、これらの薬物が、薬物と反対の表面電荷を有する医薬的に許容できるイオン交換マトリックスと結合している。実施形態の1つでは、これらの薬物が、薬物の電荷と反対の表面電荷を有する同一の医薬的に許容できる イオン交換マトリックスと結合している。
実施形態の1つでは、薬剤又は有効成分が塩の形態で分散相に含まれる。別の実施形態では、イオン交換マトリックスが塩の形態で分散相に含まれる。ある実施形態では、1種以上の薬剤及び/又はイオン交換マトリックスが医薬的に許容できる塩の形態で分散相に含まれる。
【0026】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物に関する実施形態の1つでは、患者に投与する前は、分散相中の1種以上の薬剤の分散媒への放出速度は非常に遅く、分散媒及び分散相に存在する薬剤の総モル量を基準として5%未満である。本発明は、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる液状の放出制御組成物であって、患者への投与前に分散相から分散媒に放出される薬剤の量が、分散媒及び分散相に存在する薬剤の総モル量を基準として30%未満、25%未満、20%未満、15%未満、10%未満、5%未満、0.5%未満又は0.05%未満である組成物を想定している。こうした実施形態では、分散媒が1年を超えて、約2年を超えて、約3年を超えて、又は、約4年を超えて物理的にも化学的にも安定している。有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物に関する実施形態の1つでは、遊離な薬剤は実質的に分散媒に含まれない。さらに本発明の別の実施形態では、患者への投薬前に、分散相中の1種以上の薬剤が分散媒に放出されるもので、分散媒及び分散相中の薬剤の総モル量を基準に約10%、15%、20%、25%、30%、35%、40%、45%又は50%の薬剤が放出される。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物は、薬剤が分散媒中に存在することで、即ち、イオン交換マトリックスに結合していない薬剤が存在することで、安定性が保たれる。さらに、本発明のこのような組成物は、分散媒中の遊離薬剤の存在下、分散相から薬剤が適切に放出され続ける。本発明のこの組成物の別の実施形態では、塩の形態の薬剤が分散媒中に含有される。
【0027】
本発明の組成物は室温条件下で少なくとも1年、約2年、及び/又は、3年、4年又は5年の貯蔵寿命を有し、その間、この組成物の安定性及び薬剤放出プロファイル特性が変わらないこともわかっている。
また、本発明は、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の放出制御型液状医薬組成物の製造方法も想定するもので、分散相の調製には、有効成分とイオン交換マトリックス粉末との混合が含まれる。実施形態の1つでは、イオン又は塩の形態の有効成分を塩形態のイオン交換マトリックスと混合したもので分散相が調製される。
特定の実施形態では、塩の形態であるクロルフェニラミン、プソイドエフェドリン及びヒドロコドンを、他の1種以上の成分と水との存在下で、アルギン酸ナトリウム粉末と混合させる。他の成分の例は限定されないが、例えば、薬剤充填ビーズを形成する微晶質セルロース等が挙げられる。別の実施形態では、該薬剤充填ビーズが更にラクトースを含有する。特定の実施形態によると、得られたビーズをクエン酸トリエチル及びタルクの存在下でEUDRAGIT(登録商標)(オイドラギット)で被覆し、炉内で硬化させる。この被覆ビーズを、塩の形態であるクロルフェニラミン及びヒドロコドンと、水とショ糖を含む分散媒に懸濁させる。特定の実施形態では分散媒がシロップNF(SyrupNF)を含む。また、分散媒が更に防腐剤や他の不活性添加剤を含んでもよい。このような実施形態において、得られる液状の放出持続型医薬品は瓶の中で物理的安定性を保持することができ、また、患者への投与の際には医薬品の放出制御が可能である。本発明の剤形は、一度に2種以上の薬剤投与が必要な患者や慢性的な投薬を必要とする患者にとって、とりわけ有用である。
【0028】
特定の実施形態は、下記有効成分クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる液状放出制御型薬剤組成物を用いた、風邪の兆候の治療を想定する。実施形態の1つによると、即放性(IR)製剤、及び/又は、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む調合医薬品に関連したこれまでの問題点及び不便な点が、本発明により克服される。現在入手できる即放性及び/又は調合型の風邪及びアレルギー薬と比較して、本発明は特別な利点を提供する。本発明によると、3種類の医薬有効成分(API)、即ち、クロルフェニラミン(抗ヒスタミン剤)、ヒドロコドン(鎮咳剤)及びプソイドエフェドリン(充血緩和剤)の利点が1つの徐放性(ER)組成物(例えば12時間)の中に統合される。これまでは、上記有効成分を一緒に用いる場合、それぞれの即放(IR)型剤形で1日4〜6回の投与が必要であった。つまり、3種が有効となる調合型徐放性医薬品で単一のものは現在ないからである。
現在市販されている多くの医薬品に対して、本発明は、3種の有効成分すべての徐放(ER)(例えば12時間)を可能にする製剤を提供するものである。本発明は、クロルフェニラミン(抗ヒスタミン剤)、ヒドロコドン(麻酔性鎮咳剤)及びプソイドエフェドリン(充血緩和剤)の即放剤形及び徐放剤形の新規な混合物を単一の製品として有する経口製剤を提供する。結果的に、この製剤は1日2回の投与でこれまで利用できた即放剤形(個別の販売及び投与、或いは、調合医薬品として販売及び投与が行われいる)と同じ効力を有する徐放性調合医薬品となる。また、この製剤は、以下の点において現存する単独及び調合型の徐放性製剤よりも優れている。(a)薬剤の即時的な伝達と長時間にわたる伝達のための即放性及び徐放性の投薬量が提供される。(b)三種類のRLDと生物学的等価な用量が単回投与剤形で提供される。(c)各薬剤成分の乱用や転用を防ぐ。
【図面の簡単な説明】
【0029】
【図1(A)】図1(A)は、製剤Xの放出持続型懸濁液からのプソイドエフェドリンの放出プロファイルを示す。
【図1(B)】図1(B)は、被覆ビーズからのプソイドエフェドリンの放出プロファイルを示す。
【図2(A)】図2(A)は、製剤Xの放出持続型懸濁液からのヒドロコドンの放出プロファイルを示す。
【図2(B)】図2(B)は、被覆ビーズからのヒドロコドンの放出プロファイルを示す。
【図3(A)】図3(A)は、製剤Xの放出持続型懸濁液からのクロルフェニラミンの放出プロファイルを示す。
【図3(B)】図3(B)は、被覆ビーズからのクロルフェニラミンの放出プロファイルを示す。
【図4(A)】図4(A)は、塩基製剤の懸濁液の3週間後におけるプソイドエフェドリンの放出プロファイルを示す(n=3、容器4、5及び6)。
【図4(B)】図4(B)は、塩形態である製剤の懸濁液の3週間後におけるプソイドエフェドリン (プソイドエフェドリン塩酸塩) の放出プロファイルを示す(n=3、容器1、2及び3)。
【図5(A)】図5(A)は、塩基製剤の懸濁液の3週間後におけるヒドロコドンの放出プロファイルを示す(n=3、容器4、5及び6)。
【0030】
【図5(B)】図5(B)は、塩形態である製剤の懸濁液の3週間後におけるヒドロコドン(ヒドロコドン重酒石酸塩)の放出プロファイルを示す(n=3、容器1、2及び3)。
【図6(A)】図6(A)は、塩基製剤の懸濁液の3週間後におけるクロルフェニラミンの放出プロファイルを示す(n=3、容器4、5及び6)。
【図6(B)】図6(B)は、塩形態である製剤の懸濁液の3週間後におけるクロルフェニラミン (クロルフェニラミンマレイン酸塩)の放出プロファイルを示す(n=3、容器1、2及び3)。
【図7】図7は、アルギン酸に結合した有効成分ヒドロコドン(10 mg/5mL)を含む放出持続型懸濁液からの(時間=0)におけるヒドロコドンの放出プロファイルを示す。
【発明を実施するための形態】
【0031】
本願明細書で使用のごとく、「患者」という用語は、鳥(例えば家禽)又は哺乳類等のいずれの動物に限定されるものではなく、ヒト、家畜や農場の動物、動物園、スポーツ及びペットの動物、例えばウシ、ヒツジ、フェレット、ブタ、ウマ、ウサギ、ヤギなどの家庭用ペットや他の家畜動物が挙げられるが、これも限定されるものではない。本願明細書で使用のごとく、「対象」及び「患者」という用語は互換性をもって使用される。実施形態の1つでは患者が哺乳類で、非霊長類(例えば、ウシ、ブタ、ウマ、ネコ、イヌ、ネズミ等)及び霊長類(例えばサルやヒト)である。
【0032】
本願明細書で使用のごとく、「治療する」「治療」という用語は、治癒効果のある治療と予防的手段の両方を表わし、その目的は望ましくない生理的症状、機能障害、疾病を予防又は軽減すること、又は、好適又は所望の臨床結果を得ることである。本発明の目的にとって、好適又は所望の臨床結果とは、兆候を軽減すること、症状、障害、疾病の程度を下げること、症状、障害、疾病を安定状態にすること(即ち、悪化させないこと)、症状、障害、疾病の進行を遅らせたりゆっくりにさせること、症状、障害、疾病の状態を改善すること、(部位的又は全体的のいずれであっても)それが検出可能か否かにかかわらず鎮静させること、症状、障害、疾病の改善又は向上が挙げられるが、これらに限定されない。治療には、臨床的に重要であり過度な副作用のない細胞反応を引き出すことが含まれる。また、治療には、治療を受けない場合の予想寿命よりも生存を延長することを包含する。
本願明細書で使用のごとく、「電解質薬剤」という句は、分離又はプロトン化によってイオン化が可能な薬剤の医薬的に許容できるイオン状態を指す。
本願明細書で使用のごとく、「薬剤」という用語は有効成分を意味し、例えば治療上の活性成分である。「薬剤」「有効成分」「有効な医薬成分」(又は「API」)という用語は互換性を持って使用される。
本願明細書で使用のごとく、「徐放相」又は「徐放性部位」という用語は、組成物が患者に投与された後、長時間にわたり持続的に放出される医薬組成物の相又は部位を指す。液状の放出制御型組成物においては、前記用語は、組成物の分散固相、即ち分散相を指す。
【0033】
本願明細書で使用のごとく、「即放相」又は「即放性部位」という用語は、組成物が患者に投与された後、すぐに放出される医薬組成物の相又は部位を指す。液状の放出制御型組成物においては、前記用語は、組成物の液相、即ち分散媒を指す。
本願明細書で使用のごとく、「拡散可能な対イオン」という用語は、電解質薬剤をイオン交換マトリックスから置換することができる医薬的に許容できるイオンを指す。拡散可能な対イオンが正電荷を有する場合、本願明細書では拡散可能な対カチオンと称される。拡散可能な対カチオンの非制限的な例としては、例えば、ナトリウム、カリウム、マグネシウム又はカルシウムが挙げられる。拡散可能な対イオンが負電荷を有する場合、本願明細書では拡散可能な対アニオンと称される。拡散可能な対アニオンの非制限的な例としては、例えば、塩化物、臭化物、ヨウ化物及びリン酸塩が挙げられる。
本願明細書で使用のごとく、電解質薬剤と関連して使用される場合の「水溶性」という用語は、生理学的に適切なpH値の水100mlに対し電解質薬剤がおよそ3gを超える溶解性を意味する。特定の実施形態においては、水溶性という用語は、生理学的に適切なpH値の水100mlに対し電解質薬剤が1gを超える溶解性を意味する。
本願明細書で使用のごとく、薬剤又はイオン交換マトリックスと関連して使用される場合の「アミンの塩基の形態」という句は、実質的にすべてのアミン窒素原子がプロトン化していない状態で電荷が中性であることを意味する。
【0034】
本願明細書で使用のごとく、薬剤又はイオン交換マトリックスに関連して使用される場合の「酸の形態」という句は、実質的にすべての酸基が、解離されず電荷を有していない酸の形態にあることを意味する。
本願明細書で使用のごとく、「高度に水和した」という句は、水の熱力学的活性を抑えるのに十分な水素結合を有する成分を意味する。
略語の定義
ACCP:米国胸部疾患学会
ADME:吸収、分布、代謝、排泄
API:有効医薬成分
AUC:0時間から無限時間までの血漿中濃度曲線下面積
AUCt:0時間から最終測定時間(Ct)までの血漿中濃度曲線下面積
BA:バイオアベイラビリティ−(生物学的利用能)
BE:生物学的等価
Cmax:最高血漿中濃度、投薬期間中に観察された最高濃度
Cmin:最低血漿中濃度、 投薬期間中に観察された最低濃度
CPM:クロルフェニラミンマレイン酸塩
CNS:中枢神経系統
ER:徐放
FDA:米国食品医薬品局
【0035】
GRASE:一般的に安全かつ有効な物質
HC:ヒドロコドン
IND:臨床試験用新医薬品
IR:即放
OTC:一般用医薬品
PSE:プソイドエフェドリン
RLD:関連リスト収載医薬品
PK:薬物動態
SAE:重篤有害事象
t1/2:消失半減期
tmax:Cmaxが観察された時間
実施形態の1つにおいて、本発明は、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなり、親水コロイドのイオン交換マトリックスと該マトリックスと逆の電荷を有する薬剤とを水の存在下で混合して製造した薬剤充填ビーズを含む液状の放出持続製剤に関する。
【0036】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の液状の放出持続型製剤は、約8時間、約10時間、約12時間、約16時間、約18時間、約24時間、最長約48時間までの範囲で放出制御が可能である。ある実施形態では、本発明の液状放出持続型製剤中の薬剤の全モル量の内、約15%、20%、25%、30%、35%、40%又は45%から、約50%、55%、60%、65%、70%、75%、80%、85%、90%、95%又は100%に至る量が、製剤を患者に投与してから12時間、16時間又は24時間にわたって放出される。別の実施形態では、本発明の液状放出持続型製剤中の薬剤の全モル量の約40〜約100%、40〜60%又は45〜55%が、製剤を患者に投与してから12時間にわたって放出される。別の実施形態では、本発明の液状放出持続型製剤の徐放相の薬剤全モル量の約40〜約100%、40〜60%又は45〜55%が、製剤を患者に投与してから12時間にわたって放出される。別の実施形態では、本発明の液状放出持続型製剤中の薬剤全モル量の約40〜約100%、70〜100%又は90〜100%が、製剤を患者に投与してから24時間にわたって放出される。有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物における実施形態において、本発明の液状放出持続型製剤の徐放相の薬剤全モル量の約40〜約100%、70〜100%又は90〜100%が、製剤を患者に投与してから24時間にわたって放出される。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の実施形態では、液状放出持続型薬剤伝達システムが、薬剤を含有し薬剤放出を制御する物質により被覆されたビーズを含む。実施形態の1つでは、前記被膜は遮蔽バリアであるが、バイオアベイラビリティーに到達するまでは、その被膜を通って薬剤が拡散しなければならない。
【0037】
他の実施形態では、薬剤が例えば医薬的に許容できる塩など、塩の形態をとっている。薬剤の医薬的に許容できる好適な塩としては、ナトリウム、カリウム、リチウム;カルシウム、マグネシウム;アルミニウム及び亜鉛又は他の同様な金属;アンモニア及び有機アミン類、例えば非置換又はヒドロキシ置換のモノ-、ジ-又はトリアルキルアミン類;ジシクロヘキシルアミン;トリブチルアミン;ピリジン;N-メチル-N-エチルアミン;ジエチルアミン;トリエチルアミン;モノ-、ビス-又はトリス-(2-ヒドロキシ-低級アルキルアミン)、例えばモノ-、ビス-又はトリス-(2-ヒドロキシエチル)アミン、2-ヒドロキシ-t-ブチルアミン又はトリス-(ヒドロキシメチル)メチルアミン、N,N-ジ-低級アルキル-N-(ヒドロキシ低級アルキル)-アミン類、例えばN,N-ジメチル-N-(2-ヒドロキシエチル)アミン又はトリ-(2-ヒドロキシエチル)アミン;N-メチル-D-グルカミン;及びアミノ酸、例えばアルギニン、リシン;及び硫酸塩、クエン酸塩、酢酸塩、シュウ酸塩、塩化物、臭化物、ヨウ化物、硝酸塩、硫酸水素塩、リン酸塩、酸性リン酸塩(acid phosphate)、イソニコチン酸塩、乳酸塩、サリチル酸塩、酸性クエン酸塩、酒石酸塩、オレイン酸塩、タンニン酸塩、パントテン酸塩、重酒石酸塩、アスコルビン酸塩、コハク酸塩、マレイン酸塩、ゲンチシン酸塩(gentisinate)、フマル酸塩、グルコン酸塩、グルクロン酸塩、糖酸塩、ギ酸塩、安息香酸塩、グルタミン酸塩、メタンスルホン酸塩、エタンスルホン酸塩、ベンゼンスルホン酸塩、p-トルエンスルホン酸塩、塩酸塩、パモ酸塩(即ち、1,1 '-メチレン-ビス-(2-ヒドロキシ-3-ナフトエート)、エンボナート(embonate)、エストラート及びトシラート等が挙げられるが、これらに限定されない。つまり、薬剤を塩の形態で使用することにより多数の利点がもたらされる。薬剤物質は、いくつかの次善の物理化学特性や生物薬剤学的特性を有することが多いが、イオン化した塩基性又は酸性の薬剤分子を対イオンと組み合わせて薬剤を塩形態にすることで解決できる。くわえて、医薬的に許容できる、ヒトでの使用に最適な医薬品は、一般的に塩基の形態よりもむしろ塩形態のほうが製造業者からの入手が容易である。どの塩を選択するかは、イオン化基の酸性度又は塩基度、対イオンの安全性、医薬品表示(drug indications) 及び予定の剤形によりおおかた決まる。薬剤の医薬的に許容できる塩の形態をどのようにして決めるかは、この分野の当業者であればわかる(例えば、Kumar, L.等「Salt Selection in Drug Development」Pharmaceutical Technology 3(32) (2008)参照のこと)。
【0038】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物についての別の実施形態では、水和度が高く水と結合可能な成分が分散媒に含まれる。このような成分が水を分散媒から引き出すので、ビーズの外側かつ分散媒の中の水分活性がビーズ内よりも小さくなる。このような実施形態では、分散媒の水分活性が十分に低いので、製剤が投与され分散媒が希釈されるまで、薬剤充填ビーズへの水の拡散を妨ぎ、内部浸透圧が起きるのを防ぐ。本発明のいくつかの実施形態において、前記成分とは、ショ糖、デキストロース、マルトース、マニトール、ソルビトール、グリセリン又は低分子量ポリエチレングリコール等の非電解質賦形剤であるが、これらに制限されるものではない。このような実施形態において、本発明が意図する薬剤伝達システムは、水(例えば、胃液中の水分)により促進される。こうした実施形態では、分散媒中のビーズ外側の水分活性がビーズ内部の水分活性よりも大きい場合、水が被膜を通り拡散し、ビーズの可溶性成分を溶かし、拡散可能な遊離状態の薬剤のリザーバーを形成する。この遊離な状態の薬剤は、患者への薬剤放出の制御を可能にする被膜を透過することができる。
実施形態の1つにおいて、本発明の組成物の貯蔵寿命は6ヵ月以上である。実施形態によっては、患者への投与に先立ち、本発明の組成物は安定性を6ヵ月以上維持する。
更に、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の別の実施形態では、分散媒が水和度の高い賦形剤を含有する。具体的には、このような実施形態の1つにおいて、水和度の高い賦形剤が質量基準で50〜70%分散媒に含まれる。更に別の実施形態では、この水和度の高い賦形剤がショ糖である。
【0039】
本発明の組成物の実施形態の1つは、甘味料、香味料、着色剤、さらにこれらの混合物からなる群から選択される賦形剤を更に含む。別の実施形態では、該組成物は安定化剤、分散助剤及びこれらの混合物からなる群から選択される分散添加剤を更に含む。
本発明の組成物の特定の実施形態は、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる。
本発明のこうした実施形態の1つでは、医薬的に許容できる イオン交換マトリックス及び該イオン交換マトリックスに結合した水溶性電解質薬剤とが、分散相に更に含まれ、イオン交換マトリックスの表面電荷が電解質薬剤の表面電荷と相対する。本発明の別の実施形態では、分散相が膜で被覆されている。本発明の特定の実施形態では、この膜がポリマーである。膜は多孔質又は非多孔質でもよい。本発明の実施形態の1つでは、この膜が薬剤の拡散を制御する。本発明の更に別の実施形態では、分散相には薬剤充填ビーズが含まれ、該ビーズには、水がビーズに吸収された際に溶けてビーズから拡散することができ、ビーズ内の浸透圧を低減することができる低分子量で非電解質の可溶性賦形剤が含まれる。本発明の実施形態の1つは、この低分子賦形剤がラクトースである。本発明の別の実施形態では、分散媒中の水をひきつける水和度の高い賦形剤が分散媒中に更に含まれる。本発明は、水和度の高い賦形剤が高濃度であることを想定するもので、水和度の高い賦形剤が、質量基準で例えば50〜70%、55〜70%、45〜70%、55〜65%又は60〜70%の質量比で分散媒に含まれることを想定する。本発明の実施形態の1つでは、分散媒中の水和度の高い賦形剤がショ糖であり、例えば、質量基準でショ糖が65%である。本発明のある実施形態では、分散相及び/又は分散媒における薬剤又は有効成分の1種以上は塩基の形態ではない。本発明の別の実施形態では、分散相及び/又は分散媒中の薬剤又は有効成分の1種以上が塩の形態である。本発明の更に別の実施形態では、分散相が薬剤とイオン交換マトリックス粉末との混合物を含み、薬剤とイオン交換マトリックスが塩の形態、例えば医薬的に許容できる塩の形態である。
【0040】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の更に別の実施形態では、分散相及び/又は分散媒中の薬剤又は有効成分の1種以上は塩の形態ではない。このような実施形態の1つでは、分散相及び/又は分散媒における薬剤又は有効成分の1種以上が塩基の形態である。
イオン交換マトリックスは高分子有機化合物であってもよく、例えば、オリゴマー、コオリゴマー、ポリマー又はコポリマー;例えばゼオライトのような多孔質網状組織の無機固体;及び/又はこれらの組合せ等、表面が帯電し、反対の電荷を有するイオンを保持するものである。本願明細書で使用のごとく、「カチオン交換マトリックス」という句は、カチオン状態の薬剤を保持することができるイオン交換マトリックスを指す。本願明細書で使用のごとく、「アニオン交換マトリックス」という句はアニオン状態の薬剤を保持することができるイオン交換マトリックスを指す。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の設計及び適切な成分の選択は、治療用の有効成分(薬剤)の電荷に基づく。本発明は、非荷電の塩基又は酸の薬剤、或いは、カチオン性又はアニオン性薬剤であって、強力な電解質であるだけでなく、そのpKa(アニオン)より高い弱酸性薬剤及びそのpKa(カチオン)より低い弱塩基性薬剤である薬剤の投与に好適である。薬剤がイオンの場合、イオン交換マトリックスは薬剤イオンと反対の電荷を有していなければならない。薬剤が非電荷塩基又は酸の場合、イオン交換マトリックスはそれぞれ酸又は塩基の形態である。電解質薬剤がカチオンの場合、負の表面官能価(negative surface functionality)を有するアニオン交換マトリックスをイオン交換マトリックスとして使用しなければならない。本発明の組成物は、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなり、これらは正帯電したカチオン系薬剤である。
【0041】
カチオン交換マトリックス及びアニオン交換マトリックスは当該分野では周知である。有用なカチオン交換マトリックスの非制限的例としてはカチオン交換樹脂が挙げられ、例えば、スチレン-ジビニルベンゼン共重合体を含む重合体主鎖とスルホネート基の側鎖とを有する、Rohm and Haas社(ペンシルバニア州、フィラデルフィア)からAMBERLITE(商標)IRP69という商品名で販売されている樹脂;Rohm and Haas社からAMBERLITE(商標)IRP64及びIRP88という商品名で販売されている、カルボキシレート官能基を有するメタクリル酸-ジビニルベンゼン共重合体;アルギン酸塩、カルボキシメチルセルロース、クロスカルメロース、微晶質セルロース、キサンタンガム、カルボマー94等のカルボキシビニルポリマー、ゼラチン等の親水コロイド;又はこれらの混合物が挙げられる。実施形態の1つは、カチオン交換マトリックスがアルギン酸塩、カルボキシメチルセルロース、微晶質セルロース、キサンタンガム、カルボキシビニルポリマー、ゼラチン、又はこれらの混合物である。
好適なアニオン交換マトリックスの非制限的例としては、例えば、スチレン-ジビニルベンゼン共重合体を有する重合体主鎖とアンモニウム又はテトラアルキルアンモニウム官能基の側鎖とを有する、Rohm and Haas社(ペンシルバニア州、フィラデルフィア)からDUOLITE(商標)AP143という商品名で販売されている樹脂等のアニオン交換樹脂;ならびにキトサン、ポリリシン又はゼラチン等(これらに限定されない)の親水コロイド;ならびにこれらの混合物が挙げられる。実施形態の1つは、アニオン交換マトリックスがキトサン、ポリリシン、ゼラチン又はこれらの混合物である。
【0042】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の実施形態では、イオン交換マトリックスが水に不溶である。本発明のこのような組成物の別の実施形態では、イオン交換マトリックスが水溶性である。このような実施形態では、イオン交換マトリックスは分散媒と溶媒和化することができ、親水コロイドの実施形態がある。本発明は、親水コロイドとして、デンプン、寒天、セルロース、アルギン酸、グアーゴム、キサンタンガム, ゼラチン、アカシア、アルブミンなど、これらに限定はされないが湿式結合剤といった単純なものからマイクロスフェアの成分という新規なものにわたる用途に使用されてきた天然物質を非制限的例として含むものである。非制限的な合成例としては、メチルセルロース、カルボキシメチルセルロース、ヒドロキシプロピルメチルセルロース、メチルアクリル酸、ポリ乳酸、ポリグリコール酸、ポリ酸無水物など、従来からの湿式造粒などの用途から機能性塗料や生物分解性移植組織といったより現代的な非制限的用途に広く展開されているものが含まれる。
他の実施形態によると、カチオン交換マトリックスは親水コロイドである。このような実施形態では、カチオン交換マトリックスがアルギン酸塩、カルボキシメチルセルロース、微晶質セルロース、キサンタンガム、カルボマー94等のカルボキシビニルポリマー類、又はこれらの混合物である。実施形態によっては、親水コロイドが膨潤を抑えるために架橋されている。ある実施形態は、イオン交換物質がアルギン酸カルシウムである。別の実施形態は、イオン交換マトリックスがアルギン酸ナトリウムである。
【0043】
別の実施形態では、アニオン交換マトリックスが親水コロイドである。このような実施形態のアニオン交換マトリックスは、キトサン、ポリリシン、ゼラチン又はこれらの混合物である。実施形態によっては、親水コロイドが膨潤を抑えるために架橋されている。
水溶性の電解質薬剤はイオン交換マトリックスと結合し、イオン交換マトリックス薬剤複合物を形成する。
実施形態によっては、イオン交換マトリックス薬剤複合物が粒子状又はビーズの形態である。この粒子又はビーズは、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物において、液剤で経口投与が可能な大きさである。本発明の実施形態の1つでは、この粒子又はビーズが懸濁液中に沈殿しないような大きさ及び/又は密度である。実施形態によっては、粒子又はビーズは望ましくない患者属性を有していない。実施形態によっては、粒子又はビーズの直径は約0.01〜約2000μmの範囲である。別の実施形態では、約0.1〜約1000μm、更に別の実施形態では約1〜約1000μmの範囲である。粒子又はビーズの直径が2000μmを超える実施形態、3000μmを超える実施形態、又は5000μmを超える実施形態もある。粒子又はビーズの直径が2000μm未満、1000μm未満、500μm未満、50μm未満、1μm未満の実施形態もある。実施形態の1つは、粒子、ペレット又はビーズの直径が約600μmである。粒子、ペレット又はビーズの直径が約200μm、約300μm、約400μm、約500μm、約600μm、約700μm、約800μm又は約900μmの実施形態がある。
【0044】
コア部には固形剤形を形成するのに有用な医薬的に許容できる加工助剤を更に含んでもよく、デンプン、酸化チタンやシリカ等の増量剤、防腐剤、酸化防止剤等の安定化剤、植物油等の潤滑剤等をはじめとするが、これらに限定されるものではない。
実施形態の1つでは、イオン交換マトリックス薬剤複合物は低分子量の可溶性非電解質賦形剤を更に含有する。このような賦形剤は、ビーズが水を吸収した際に水に溶解し、ビーズから拡散することができるので、ビーズ内部の浸透圧を低下させる。この賦形剤の分子量は、ビーズを被覆するどんな膜も透過できるように十分に小さくなければならない。様々な実施形態によると、ビーズに含有される賦形剤の量は薬剤放出速度に影響を及ぼす可能性がある。ビーズ中に賦形剤は約5〜約10%、約10〜約20%、約20〜約30%、約30〜約40%、約40〜約45%含まれる。実施形態の1つは、賦形剤がラクトースである。このような実施形態では、製造工程において分散相に導入されたラクトースが多いほど、投与後のビーズからの薬剤の放出は速くなる。様々な実施形態として、ビーズ中にラクトースは約5〜約10%、約10〜約20%、約20〜約30%、約30〜約40%、約40〜約45%含まれる。実施形態によっては、ラクトースがビーズ中に約20〜約30%含まれる。可溶性非電解質賦形剤の他の例として、デキストロース、マルトース、マニトール、ソルビトール、グリセリン又は低分子量ポリエチレングリコールが本発明では挙げられるが、これらに限定されるものではない。
【0045】
実施形態の1つは、イオン交換マトリックス薬剤複合物が拡散制御皮膜を更に含む。この皮膜は、イオン交換マトリックス内への対イオンの拡散とマトリックスからの薬剤拡散を更に制御するうえで有用である。このように拡散制御皮膜は、患者に投与した後、分散媒及び/又は消化管内に電解質薬剤が放出されるのを制御するうえで有用である。本発明は、拡散制御を行ういずれの皮膜の使用も想定するものである。コーティング材料は、多数にわたる天然又は合成の膜形成剤のいずれでもよく、これらを単独又は相互に使用してもよい。また、可塑剤、顔料又は他の物質等の成分と共に使用してもよい。実施形態によっては、皮膜の成分は水には不溶でかつ水を通す。皮膜の透過性を変更する際に、水溶性物質の導入が有用となる場合がある。当該分野で拡散制御皮膜は公知である。非制限的な例として、SURELEASE(登録商標)(Colorcon社、ペンシルバニア州ウェストポイント)等のエチルセルロース; EUDRAGIT(登録商標)(Rohm Pharma社、ドイツ、Weiterstat)等のメチルメタクリレートポリマー;セルロースエステル類、セルロースジエステル類、セルローストリエステル類、セルロースエーテル類、セルロースエステル-エーテル、セルロースアシレート、セルロースジアシレート、セルローストリアシレート、セルロースアセテート、セルロースジアセテート、セルローストリアセテート、酢酸プロピオン酸セルロース、酢酸酪酸セルロースが挙げられる。皮膜がメチルメタクリレートポリマーの実施形態がある。
【0046】
実施形態の1つは、拡散制御皮膜が、エチルセルロース、メチルメタクリレート、セルロースエステル類、セルロースジエステル類、セルローストリエステル類、セルロースエーテル類、セルロースエステル-エーテル、セルロースアシレート、セルロースジアシレート、セルローストリアシレート、セルロースアセテート、セルロースジアセテート、セルローストリアセテート、酢酸プロピオン酸セルロース、酢酸酪酸セルロース 、これらの混合物からなる群から選択される。実施形態の1つは、拡散制御皮膜がエチルセルロース、メチルメタクリレート又はそれらの混合物である。別の実施形態は、拡散制御皮膜コーティング材が、コーティング材とイオン交換マトリックス薬剤複合物の全質量を基準として約20〜約30質量%である。
実施形態の1つは、イオン交換マトリックス薬剤複合物が、イオン交換マトリックス薬剤複合物及び拡散制御皮膜の全質量を基準として、約1%から約75%までの質量の拡散制御皮膜でコーティングされている。別の実施形態では5〜約50%、別の実施形態では約10〜約30%、更に別の実施形態では約20〜約25%である。一般に、コーティング材が多いほど薬剤放出が遅くなる。
実施形態の1つは、薬剤充填アルギン酸塩ビーズがEUDRAGIT(登録商標)RS 30D(Rohm社製)で十分にコーティングされ、コーティング材及び薬剤充填アルギン酸塩ビーズの全質量を基準として約20〜約30質量%のコーティング材を有する被覆ビーズとなる。
【0047】
別の実施形態では、イオン交換マトリックス薬剤複合物の拡散制御皮膜コーティング材が更に可塑剤を含む。可塑剤はコーティング材の可塑性を増強させ脆性を低下させるのに有用である。また、可塑剤は薬剤放出速度にも影響する。可塑剤はコーティング材のガラス転移温度を低下させるので、凝集性フィルムの中に添加された液滴を容易に融合させ、コーティング材の透過性に影響を与える。可塑剤は当該分野では公知である。可塑剤の非制限的例としては、クエン酸トリエチル、セバシン酸ジエチル、フタル酸ジエチル、クエン酸トリブチル、アセチルクエン酸トリブチルが挙げられる。実施形態の1つは可塑剤がクエン酸トリエチルである。実施形態の1つは、イオン交換マトリックス薬剤複合物は、イオン交換マトリックス薬剤複合物及び拡散制御膜の全質量を基準として、約0.1%から30%まで、約0.5%から約20%まで、約1〜約20%、約2〜約10%までの可塑剤を含有する。
【0048】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物は、イオン性成分の存在下で安定している。実施形態の1つでは、本発明の組成物がイオン性成分の存在下、分散媒中で安定している。このような実施形態において、本発明の組成物は、拡散可能な対イオンの存在下、分散媒中で安定している。別の実施形態では、本発明の組成物は、電解質薬剤、即ち、クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンの存在下、分散媒中で安定している。有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物は、このような薬剤が遊離な状態で分散媒中に存在していると、即ち、薬剤がイオン交換マトリックスに結合していない状態でいると、安定性が保持される。本発明の組成物は、分散媒中の遊離状態の薬剤の存在下、分散相からの薬剤放出プロファイルが適切に持続性を保持する。本発明の別の実施形態の分散媒は、有効成分がクロルフェニラミンとヒドロコドンとからなり、さらにプソイドエフェドリンを含んでいてもよく、即放剤形となる。このような実施形態では、分散媒中の薬剤はイオン交換マトリックスと結合していない。このような実施形態の1つでは、分散媒が塩の形態の薬剤を含有する。
実施形態の1つでは、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる液状放出制御型組成物は、分散媒中の遊離状態の薬剤及び/又は拡散可能な対イオンの存在下で安定している。このような実施形態の1つは、本発明の組成物が徐放相と即放相とを有し、即放相が遊離状態の薬剤を特定量含み、患者に投与する前に徐放相から即放相に放出される薬剤量は、分散媒及び分散相中の薬剤の全モル量を基準として30%未満、25%未満、20%未満、10%未満、5%未満、0.5%未満又は0.05%未満である。
【0049】
本発明は、3つの異なる治療徴候のために用いる3種の薬剤、即ち、アレルギー及び鼻漏の治療用のクロルフェニラミン、咳の治療用のヒドロコドン、鼻づまり治療用のプソイドエフェドリンを含む液状の放出制御型組成物に関する。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の液状放出制御型組成物の別の実施形態では、患者への投与前に、分散相の中の薬剤1種以上が分散媒中に浸出する。このような実施形態では、分散媒及び分散相中の薬剤の全モル量を基準に、薬剤の約10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、70%で、最大75%までが、患者への投与の前に分散相から分散媒に放出される。分散媒及び分散相中の薬剤の全モル量を基準に、実施形態の1つでは約15〜約35%、別の実施形態では約25%の薬剤が患者への投与の前に分散相から分散媒に放出される。
実施形態によっては、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の液状の放出持続型組成物の、即放相に含まれる薬剤対徐放相に含まれる前記と同じ薬剤の質量比(IR/ER比)が、約0:100、約5:100、約10:100、約15:85、約20:80、約25:75、約30:70、約35:65、約40:60、約45:50又は約50:50である。実施形態の1つは、薬剤のIR/ER比が約25:75である。本発明の特定の実施形態では、本発明の経口組成物の即放性部位対徐放性部位のクロルフェニラミンの質量比は約25:75、即放性部位対徐放性部位のヒドロコドンの質量比は約25:75、即放性部位対徐放性部位のプソイドエフェドリンの質量比は約25:75〜約0:100である。
【0050】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の他の実施形態では、液状の放出制御型医薬組成物1回の投与量1mlあたり0.1〜0.5、0.5〜1mg、1〜5mg、5〜10mg、10〜15mg、15〜20mg、20〜25mg、25〜30mg、30〜40mg、40〜50mg、50〜60mg、60〜70mg、70〜80mg、80〜90mg、90〜100mg、100〜120mg、120〜140mg、140〜160mg、160〜180mg、180〜200mg、200〜220mg、220〜240mg、240〜260mg、260〜280mg、280〜300mg、300〜350mg、350〜400mg、400〜450mg、450〜500mg、最大600mg、700mg、800mg、900mg、1000mgまでの各薬剤又は各有効成分が医薬組成物に含まれる。本発明の更に別の実施形態では、このような医薬組成物には、液状の放出制御型医薬組成物1回の投与量5mlあたり0.1〜0.5、0.5〜1mg、1〜5mg、5〜10mg、10〜15mg、15〜20mg、20〜25mg、25〜30mg、30〜40mg、40〜50mg、50〜60mg、60〜70mg、70〜80mg、80〜90mg、90〜100mg、100〜120mg、120〜140mg、140〜160mg、160〜180mg、180〜200mg、200〜220mg、220〜240mg、240〜260mg、260〜280mg、280〜300mg、300〜350mg、350〜400mg、400〜450mg、450〜500mg、最大600mg、700mg、800mg、900mg、1000mgまでの各薬剤又は各有効成分が含まれる。本発明の特定の実施形態では、液状の放出制御型医薬組成物1回の投与量5mlあたり1〜5mg、5〜10mg、10〜15mg、15〜20mg、20〜25mg、25〜30mg、30〜40mg、40〜50mg、最大100〜120mg、120〜140mg、140〜160mg、160〜180mg、180〜200mg、200〜220mg、220〜240mg、240〜260mg、260〜280mg、280〜300mgまでの各薬剤又は各有効成分が医薬組成物に含まれる。
【0051】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の実施形態によっては、水和度が高く分散媒中で水との結合が可能な賦形剤が高濃度に分散媒中に更に含まれる。本発明の薬剤は水性分散媒における溶解度が高いが、分散媒中に水和度の高い成分が存在することで、分散媒中の水が引き寄せられる。これは薬剤-イオン交換マトリックス複合物からの薬剤の溶解を開始するの必要である。製剤が投与され、胃液(ほぼ水)が分散媒を希釈したときにはじめて薬剤が溶けて、利用可能な状態になり、皮膜の透過及び/又はビーズからの拡散が始まる。このような実施形態では、遊離状態の薬剤は実質的に分散媒からなくなり、例えば、薬剤は0.5%未満又は0.05%未満しか分散媒には存在しない。本発明の様々な実施形態では、水和度の高い成分が分散媒中に質量比で約10〜約20%、約20〜約30%、約30〜約40%、約40〜約50%、約50〜約60%で存在する。実施形態によっては、該成分が約60〜約65%、最大約70%まで含まれる。本発明の他の実施形態は分散媒がショ糖又は他の糖分子を含む。このような実施形態では、ショ糖が質量比で10%より多く、20%より多く、30%より多く、40%より多く又は50%より多く分散媒中に含有される。有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の実施形態では、分散媒にショ糖が約65%かつ約70%以下含まれる(即ちSyrup NF)。本発明が包含する賦形剤の他の例としては、デキストロース、マニトール、フルクトース、ポリエチレングリコール、グリコール類及びグリセリン類が挙げられるが、これらに限定されるものではない。本発明の実施形態では、分散媒が質量比で10%より多くのデキストロース、マニトール、フルクトース、ポリエチレングリコール、グリコール又はグリセリンを、又は20%より多くのデキストロース、マニトール、フルクトース、ポリエチレングリコール、グリコール又はグリセリンを、又は30%より多くのデキストロース、マニトール、フルクトース、ポリエチレングリコール、グリコール又はグリセリンを、又は40%より多くのデキストロース、マニトール、フルクトース、ポリエチレングリコール、グリコール又はグリセリンを、又は50%を超えてデキストロース、マニトール、フルクトース、ポリエチレングリコール、グリコール又はグリセリンを含む。本発明の実施形態の1つは、分散媒中にデキストロース、マニトール、フルクトース、ポリエチレングリコール、グリコール又はグリセリンが約65%、かつ、デキストロース、マニトール、フルクトース、ポリエチレングリコール、グリコール又はグリセリンが約70%未満で含まれる。この分野の当業者であれば、同等に機能するであろう水和度の高い他の賦形剤を容易に決定することができる。
【0052】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の液状の放出制御型薬剤組成物は、本発明の意図する作用に賦形剤が悪影響を及ぼさないのであれば、安定化剤、分散剤等からなる群から選択される分散助剤をさらに含んでもよい。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の液状の放出制御型薬剤組成物は、本発明の意図する作用に賦形剤が悪影響を及ぼさないのであれば、例えば甘味料、風味剤、着色剤、増粘剤等の液状経口製剤に有用な賦形剤を更に含んでもよい。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の剤形の利点は、イオントラップ、浸透の制御、熱力学的均衡メカニズムが大概適切であり本質的に安定していること、また、当該分野の当業者であれば利用可能な、広く承認された従来からの医薬賦形剤の使用を通して効果を実現できるという事実である。
本発明で有用なカチオン系有効成分はクロルフェニラミン、ヒドロコドン又はプソイドエフェドリンである。
実施形態によると、塩の形態の前記電解質薬剤が有用な薬剤として挙げられる。薬剤の塩の形態として、マレイン酸塩、塩酸塩又は重酒石酸塩が好ましい。
【0053】
また、実施形態によっては、イオン交換マトリックスとの結合又は反応によりイオンを形成する中性状態の前記電解質薬剤が有用な薬剤として挙げられる。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の組成物の様々な実施形態において、イオン交換マトリックス薬剤複合物には、薬剤をイオンの状態に変換するのに十分な量のイオン交換マトリックスが含まれる。イオン交換マトリックス薬剤複合物には、薬剤をイオンの状態に変換するのに十分な量以上のイオン交換マトリックスが含まれていてもよい。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の液状の放出制御型薬剤組成物の実施形態の一つでは、薬剤又は有効成分が1個1個のの粒子、ペレット又はビーズの中で結合している。このような実施形態では、薬剤の表面電荷と逆の表面電荷を有する医薬的に許容できるイオン交換マトリックスに、薬剤が結合する。本発明の実施形態の1つは、薬剤の表面電荷と逆の表面電荷を有する同じ医薬的に許容できるイオン交換マトリックスに、薬剤が結合する。同一のイオン交換マトリックスに薬剤が結合することにより、組成物中の各薬剤の放出制御は妨害されることなく、各薬剤の放出を適切な速度にする。クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンは、負の表面官能価を有するイオン交換マトリックスと結合することが想定されている。 本発明の別の実施形態では、分散相が、医薬的に許容できる塩の形態の薬剤又は有効成分を含む。本発明の更に別の実施形態では、分散相が医薬的に許容できる塩の形態のイオン交換マトリックスを含む。
【0054】
実施形態によっては、本発明の組成物は有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなり、これらが1個1個の粒子又はペレット又はビーズの中に含まれるが、このような粒子、ペレット又はビーズの中で薬剤同士の化学的又は物理的相互作用はなく、単一の薬剤放出速度制御皮膜により満足のいく安定特性と適切な薬剤伝達プロファイルが得られる。
本発明の各薬剤をそれぞれ別々のビーズに含ませ、ビーズの混合を行う場合、その混合物が完全に均一ではない可能性があるので、薬剤の服用量が正しくない結果となる。不均一になるのは、無作為な変動によるものか、或いは、混合物中の2個以上のビーズの物性の差異、例えば重量や密度の差異によるものである。しかしながら、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとが1個の粒子又はペレット又はビーズの中で結合しているという上述の技術的方法によって、都合のよいことに、薬剤混合物の均質性を確保し、調合製剤におけるそれぞれの薬剤の服用量が均一したものとなり、所定の患者は各薬剤を一定量で摂取できる。更に、これらの薬剤を同一のイオン交換マトリックスに結合させることにより、分散相の中には薬剤結合樹脂複合物が1種類しかできないという利点が生まれる。更には、クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンを1個1個のの粒子、ペレット又はビーズ内で混合させることで、調合医薬品全体に存在する粒子、ペレット又はビーズの表面積を低減することができ、製品及び有効成分の安定性を増強することができる。
【0055】
本発明における上記の技術的方法のもう一つの利点は、同一の樹脂に結合したすべての薬剤が同様な又は同じ放出プロファイルを示し、本発明の各薬剤に関する投与量又は投与回数を違える必要がない。もしも異なる樹脂が使用されるならば、各薬剤の放出プロファイルは患者の食餌又は生理機能の差に影響を受けることもあるので、所定の患者にとって、ある薬剤は多すぎ、また別の薬剤は少なすぎる摂取になるという可能性がある。それに対して有利なことに、本発明の上述の技術的方法によると、クロルフェニラミンとヒドロコドンとプソイドエフェドリンについて同時に生物学的等価性を得ることが可能になる。また、このような薬剤を単一の放出方法を有する1個1個のビーズに配置することで、薬剤放出プロファイルが患者集団全体の中でより一貫性を有する。
クロルフェニラミンとヒドロコドンとプソイドエフェドリンとが1個1個の粒子又はペレット又はビーズの中で結合しているという上述の技術的方法の更なる利点は、このような薬剤組成物から個々の有効成分の抽出又は析出を難しくすることである。具体的には、このような有効成分が同一のイオン交換マトリックスに結合していることにより、いずれの単一の有効成分の抽出又は分離も当業者でなければ極めて難しくなる。例えば、これらの薬剤が1つのイオン交換マトリックスに結合していれば、1つの薬剤/1つのイオン交換マトリックスによる複合物間の密度等といった物性の差をもとに、1種の薬剤を他の薬剤から部分的に単離することは不可能である。本発明の前記技術的方法を用いて製造した製品からは、いずれの有効成分も1種を単離することは難しいため、製品の中のいずれか1種の成分を乱用又は不法使用しようとする潜在的可能性は低下する。即ち、本発明の技術的方法により、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとを含む調合医薬品は、薬剤の乱用や転用の潜在的可能性を抑えるように製造できることになる。
【0056】
更に別の実施形態では、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとが、それぞれの薬剤の表面電荷と逆の表面電荷を有する医薬的に許容できる異なるイオン交換マトリックスと結合している。
本発明の組成物の実施形態では、長期間にわたり、即ち、少なくとも1ヵ月、少なくとも3ヵ月、少なくとも6ヵ月、1年間、2年間又は3、4、5年以上にわたり安定している。具体的には、本発明の組成物は、前記の期間、化学的、物理的、微生物学的な安定性を維持する。この分野の当業者であれば、医薬組成物の化学的、物理的、微生物学的な安定性の評価方法は理解できよう。医薬組成物の安定特性、例えば、物理的、化学的、微生物学的な安定性次第で、患者に投与ができる最終組成物の状態で薬剤又は有効成分が瓶の中でどのくらいの期間保存できるかが決まる。実施形態の1つは、本発明の組成物は室温で少なくとも1ヵ月間、少なくとも3ヵ月間、少なくとも6ヵ月間、1年間、2年間、3、4、5年間以上安定している。このような実施形態では、本発明の組成物は、少なくとも1ヵ月間、少なくとも3ヵ月間、少なくとも6ヵ月間、1年間、2年間、3、4、5年間以上、化学的、物理的、微生物学的な安定性を有する。
【0057】
化学的安定性は、時間経過後の組成物中の薬剤又は有効成分の構造的保全性により証明される。化学的安定性はクロマトグラフィー分析及び/又は有効性の測定を用いて評価することができる。こうした評価により、組成物中の薬剤の存在、製品の劣化の有無が検出される。時間Xにおいて、ここでXはゼロ(即ち、組成物が製造された時)より長いある期間を指すが、時間ゼロ時点で存在していた薬剤の90〜100%が存在し、十分な構造的保全性を示すか、或いは、時間ゼロの時点で予想した構造的特性と比較して、同じか同等な構造的特性又は著しい差がない構造的特性を示す場合、薬剤は、時間Xにおいて化学的に安定しているとみなされる。実施形態の1つは、本発明の組成物が、少なくとも1ヵ月間、少なくとも3ヵ月間、少なくとも6ヵ月間、1年間、2年間、3、4、5年間以上、化学的安定性を有する。
【0058】
本発明の組成物の物理的安定性は、分散相を被覆する拡散制御皮膜又は機能的コーティングの保全性及びその透過性により証明される。物理的安定性は溶出分析により評価することができる。溶出分析は、時間ゼロ(即ち、組成物が製造された時点)又は時間X(Xはゼロより長い時間で、例えば、1週、2週、3週、1ヵ月、3ヵ月、6ヵ月、1年、2年、3年、4年又は5年)時点で開始して分析する。溶出テストは、患者の胃腸管内の環境と同等な化学的環境に組成物をいったん配置した際に放出される薬剤の量を示す。溶出テストは、薬剤が患者に投与された際の薬剤の放出速度を示す。溶出又は放出プロファイル、即ち、各測定時点での(薬剤を相応の化学環境のもとに配置後又は患者に投与後)放出された薬剤の量及び薬剤の放出速度が、薬剤組成物の物理的安定性を表わす。ある時点Xにおいて物理的に安定した薬剤組成物であれば、溶出分析を用いて評価した溶出又は放出プロファイルが、時間ゼロ(即ち、薬剤の製造直後に評価を行った時)において測定した薬剤組成物から予想されるものと同じ、同等、又は少なくとも著しく異ならない溶出又は放出プロファイルを示すであろう。また、物理的に安定した薬剤組成物であれば、測定における第1時点(即ち、測定開始時)において、予想されたある特定量の薬剤が放出される。具体的に言うならば、物理的に安定した組成物とは、測定における第1時点において、即放性部位を有していないものは薬剤放出は示さないか、或いは、非常に低い薬剤放出レベル、例えば、10%未満、5%未満又は1%未満の薬剤が徐放相から放出される。更に、物理的に安定した薬剤組成物であれば、組成物を相応の化学環境のもとに配置又は患者に投与した後、それぞれの次の時点では、薬剤放出速度が予想されたある特定の速度であり、放出される薬剤がある特定の量となる。本発明の組成物の実施形態では、少なくとも1ヵ月間、少なくとも3ヵ月間、少なくとも6ヵ月間、1年間、2年間、3、4、5年間以上、物理的安定性を有する。このような実施形態では、本発明の薬剤組成物は、少なくとも1ヵ月間、少なくとも3ヵ月間、少なくとも6ヵ月間、1年間、2年間、3、4、5年間以上瓶で保存することができ、その間物理的安定性が維持される。
【0059】
いくつかの実施形態では、本発明の組成物は微生物学的安定性も維持する。薬剤組成物の微生物学的安定性は、微生物による組成物の汚染の有無を示す。実施形態の1つは、本発明の組成物が、少なくとも1ヵ月間、少なくとも3ヵ月間、少なくとも6ヵ月間、1年間、2年間、3、4、5年間以上微生物学的安定性を有する。
本発明の液状の放出制御型組成物の安定特性により、この組成物は長い貯蔵寿命、即ち、1年以上の貯蔵寿命を有する。本発明の組成物は、室温条件下で、少なくとも1ヵ月間、少なくとも3ヵ月間、少なくとも6ヵ月間、約1年間、約2年間、3、4、5年間以上の貯蔵寿命を有し、その間、製剤の安定性及び薬剤放出プロファイル性は保たれると考えられる。他の実施形態の放出持続型製剤は、1ヵ月を超え、3ヵ月を超え、6ヵ月を超え、1年を超え、約2年間を超え、約3年間を超え、4年間を超え、5年間を超えて物理的にも化学的にも安定している。
本発明の別の態様は、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなり、徐放性部位と即放性部位とを有する調合製剤であって、患者に投与した際に、上記薬剤を含有する即放性医薬品と生物学的等価となる調合製剤を提供する。本発明の製剤は、上記薬剤のうちの2つ以上について生物学的等価性が同時に得られるという利点がある。本発明の別の実施形態は、鎮咳剤と抗ヒスタミン剤と充血緩和剤とを含み、徐放性部位と即放性部位とを有する非液状調合製剤であって、患者に投与した際に上記薬剤を含有する即放性医薬品と生物学的等価性となる、非液状調合製剤を提供する。
【0060】
本発明の態様の1つは、第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物に関するものであって、
第1の部位が即放性の形態で、有効成分として抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよく、
第2の部位が徐放性の形態で、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを3種の有効成分として含む粒子、ペレット又はビーズを含有し、
前記非液状経口薬剤組成物を患者へ単回投与した際に、少なくとも8時間にわたって得られる上記3種の有効成分の血漿中濃度が、前記有効成分で構成されるFDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価であり、
前記適切な投与回数とは、IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みの1つ以上のラベルで推奨される投与回数に対応する。
【0061】
更に、このような実施形態の1つは鎮咳剤が麻酔性鎮咳剤である。別の実施形態は、第1の部位が充血緩和剤を含まない。このような実施形態では、前記粒子、ペレット又はビーズが更に皮膜を有する。更なる実施形態では、前記抗ヒスタミン剤と鎮咳剤と充血緩和剤とが1個1個の粒子又はペレット又はビーズのなかに結合している。更に別の実施形態では、この粒子、ペレット又はビーズが更に医薬的に許容できるイオン交換マトリックスを含み、抗ヒスタミン剤、鎮咳剤及び充血緩和剤が該イオン交換マトリックスと結合している。このような実施形態では、甘味剤、風味剤、着色剤及びこれらの混合物からなる群から選択される賦形剤を更に含有する。別の実施形態では、安定化剤、分散剤及びこれらの混合物からなる群から選択される添加剤を更に含有する。
更に別の態様として、本発明は、第1の部位と第2の部位とを含む、非液状の経口徐放性薬剤組成物に関するもので、
第1の部位が即放性の形態で、有効成分として抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよく、
第2の部位が粒子、ペレット又はビーズで、徐放性の形態で抗ヒスタミン剤と鎮咳剤と充血緩和剤とを有効成分として含み、
前記3種の有効成分が24時間を超えて安定した血漿中濃度であるように前記非液状薬剤組成物を十分な投与回数で患者に投与した際、前記有効成分で構成されるFDA承認済みの即放性薬剤組成物1種以上を上記と同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となり、
前記適切な投与回数とは、FDA承認済みの即放性薬剤組成物1種以上の同じ期間にわたる投与に関して、FDA承認済みの1つ以上のラベルで推奨される投与回数に対応する。
【0062】
本発明の態様は、ある哺乳動物の少なくとも8時間にわたる抗ヒスタミン剤、鎮咳剤及び充血緩和剤の血漿中濃度を、FDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり同じ哺乳動物に適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価である血漿中濃度とする方法に関するもので、該方法は、
(a)第1の部位と第2の部位とを有する非液状経口徐放薬剤組成物であって、第1の部位が、即放性の形態で有効成分として抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよく、第2の部位が、徐放性の形態で有効成分として抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む粒子、ペレット又はビーズを含有する、非液状経口徐放薬剤組成物を前記哺乳動物に投与することと、
(b)少なくとも8時間にわたる前記3種の有効成分の血漿中濃度が、前記有効成分を含有するFDA承認済みのIR-RLD組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価とすることとを含み、前記適切な投与回数とは、IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みの1つ以上のラベルのなかで推奨される投与回数に対応する。
本発明の別の態様は、哺乳動物に非液状の経口徐放性(ER)薬剤組成物の投与後、抗ヒスタミン剤、鎮咳剤及び充血緩和剤の安定した血漿中濃度を得るための方法であって、前記血漿中濃度とは、活性成分と不活性成分とを含み、前記活性成分が抗ヒスタミン剤(例えばクロルフェニラミン)と鎮咳剤(例えばヒドロコドン)とプソイドエフェドリン(例えばプソイドエフェドリン)とからなる即放性(IR)組成物1種以上を同じ哺乳動物に投与した際に得られる血漿中濃度と生物学的等価な濃度で、該方法は、
【0063】
第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物であって、第1の部位が即放性の形態で、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを有効成分として含み、第2の部位が徐放性の形態で、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを有効成分として含む粒子、ペレット又はビーズを含有する、非液状経口徐放性薬剤組成物を前記哺乳動物に投与することを含み、
前記3種の有効成分が24時間を超えて安定した血漿中濃度であるように前記非液状経口徐放性薬剤組成物を十分な投与回数で哺乳動物に投与した際、前記有効成分を含有するFDA承認済みの即放性(IR)薬剤組成物1種以上を上記と同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となり、
前記FDA承認済みのIR薬剤組成物1種以上の適切な投与回数とは、FDA承認済みのIR薬剤組成物1種以上の同じ期間にわたる投与に関して、FDA承認済みの1つ以上のラベルの中で推奨される投与回数に対応し、
前記FDA承認済みのIR薬剤組成物1種以上の適切な投与回数が、経口ER薬剤組成物の十分な投与回数を上回るものである。
本発明の態様の1つは、医薬的有効成分(API)として抗ヒスタミン剤、鎮咳剤及び充血緩和剤を含有し、薬剤の3種すべてが徐放性(ER)を示す、新規な製剤に関する。例えば、本発明は、クロルフェニラミン、プソイドエフェドリン及びヒドロコドンの、即放剤形と徐放剤形の新規な混合物を1つの製品中に含む製剤を提供する。本発明の新規な製剤としては、3種すべての薬剤を含む既存のIR製品又は上記薬剤のうちの1種又は2種のみを含むIR 及び/又はER製品の混合物と同じ効力を持つ、1日2回の投与が可能なIR/ER調合製品となる製剤が挙げられる。
【0064】
抗ヒスタミン剤は、例えば、抗ヒスタミン性拮抗薬又は逆作動薬として関係のある細胞受容体、例えばH1受容体において作用することにより、体内におけるヒスタミンの放出又は活動を阻害する。ヒスタミンは、アレルギー、風邪及び流感(インフルエンザ)に関連した鬱血、くしゃみ、鼻水、鼻づまり、目のかゆみ、涙目を引き起こす。抗ヒスタミン剤はヒスタミンが細胞に付着し上記のような症状を起こさないようにする。抗ヒスタミン剤の例としては、クロルフェニラミン、ブロムフェニラミン、ジメンヒドリナート、ジフェンヒドラミン、ロラタジン、メクリジン及びクエチアピン等が挙げられる。本発明の実施形態の1つは、抗ヒスタミン剤がクロルフェニラミンである。「クロルフェニラミン」という用語には、この薬剤のいずれの形態も含まれ、具体的な実施形態は、クロルフェニラミンがクロルフェニラミンマレイン酸塩(CPM)で、これは化学名2-ピリジンプロパンアミン -(4-クロロフェニル)-N,N-ジメチル-, (Z)-2-ブテンジオエート(1 :1)としても公知である。
また、充血緩和剤も、風邪や流感、副鼻腔炎又はアレルギーを原因とする鼻づまりや鬱血の軽減を助けることができる。鼻、洞、胸部内の鬱血は、鼻や通気道の細胞膜中の血管が膨張、拡張したことによる。これらの細胞膜には、拡張(膨張及び充血)できる大容量の血管がたくさん通っている。この血管はヒスタミンの刺激により拡張する。これとは対照的に、充血緩和剤はこれらの細胞膜中の血管を収縮させ引き締めるが、細胞膜の外へ大量の血液を押し出すので血管が収縮し、通気道が再度開く。一般に、充血緩和剤は、アドレナリンという一種の刺激剤でもある天然の充血緩和剤と化学的に関連づけられる。最も一般的な経口充血緩和剤はプソイドエフェドリンとフェニレフリンである。本発明の実施形態は、充血緩和剤がプソイドエフェドリン(PSE)である。「プソイドエフェドリン」という用語は、この薬剤のいずれの形態も含まれ、具体的な実施形態はプソイドエフェドリンがプソイドエフェドリン塩酸塩であり、これは化学名ベンゼンメタノール,-[l-(メチルアミノ)エチル]-,[S-(R*,R*)]-, 塩酸塩としても公知である。
【0065】
一般的に、風邪薬は鎮咳剤と去痰剤という2つのグループに分類される。鎮咳剤は、咳が出るのを抑えたり軽減するのに使用される薬で、非麻酔性と麻酔性鎮咳剤とがある。ベンゾナテタート、デキストロメトルファン、カルベタペンタンは非麻酔性鎮咳剤の例である。デキストロメトルファン(鎮咳剤)とグアイフェネシン(去痰剤)を一緒に組み合わせることがある。麻酔性鎮咳剤の一例がヒドロコドン(HC)で、これは鎮痛剤でもあり、天然アヘンのコデイン及びテバインの2つに誘導される半合成オピオイドである。「ヒドロコドン」という用語には、この薬剤のいずれの形態も含まれる。本発明の実施形態は、鎮咳剤がヒドロコドン重酒石酸塩であって、これは化学名モルフィナン-6-オン, 4,5-エポキシ-3-メトキシ-17-メチル-,(5)-, [R-(R* ,R*)]-2,3-ジヒドロキシブタン-ジオエート (1 :1), 水和物(2:5)としても公知である。
抗ヒスタミン剤と充血緩和剤との調合品は、現在のところ、Actifed(登録商標)、Allegra-D(登録商標)、Chlor-Trimeton D(登録商標)、Claritin D(登録商標)、Contac(登録商標)、Co-Pyronil 2(登録商標)、Deconamine(登録商標)、Demazin(登録商標)、Dimetapp(登録商標)、Drixoral(登録商標)、Isoclor(登録商標)、Nolamine(登録商標)、Novafed A(登録商標)、Ornade(登録商標)、 Sudafed Plus(登録商標)、Tavist D(登録商標)、Triaminic(登録商標)及びTrinalin(登録商標)等が市販されている。鎮咳剤も鎮痛剤又は抗ヒスタミン剤等の他の薬剤と組み合わせたものが購入できる。総合感冒薬としても知られているこのような調合製品は多くの症状を同時に治療するものである。
【0066】
しかしながら、FDAのウェブサイトに記載されているように、FDA承認済みの即放性ヒドロコドン鎮咳製剤は、Hycodan(登録商標)、Mycodone(登録商標)及びTussigon(登録商標)に見られるように、ヒドロコドン重酒石酸塩と臭化メチルホマトロピンしか含まれていない。ヒドロコドンとクロルフェニラミンとを含む徐放性鎮咳製剤については、Tussionex Pennkinetic(登録商標)(懸濁液)とTussiCaps(登録商標)(カプセル剤)の2種しか現在のところ承認されていない。www.fda.gov/CDER/ drug/unapproved_drugs/hydrocodone_qa.htm参照のこと(2008年4月11日アクセス)。注目すべきは、ヒドロコドンとホマトロピンとを、グアイフェネシンのような去痰剤又はフェニレフリン もしくはプソイドエフェドリンのような充血緩和剤等の他の薬剤と組み合わせた咳抑制剤は、いずれの剤形も現在のところ許可されていない。このように、ヒドロコドンとPSEのような充血緩和剤とを含むFDA認可済みの市販薬剤は使用できない。
したがって、前述のごとく本発明は既存の市販調合製品とは異なるのである。なぜならば、本願明細書に記載の新規な製剤は、3種類の有効成分である抗ヒスタミン剤、鎮咳剤及び充血緩和剤を含有し、単一の経口製品によって3種類すべての有効成分が体内で徐放性を示すからである。実施形態の1つは、新規製剤が12時間ごとに投与される。他の実施形態では8時間ごと、16時間ごと、24時間ごと等で投与される。
【0067】
実施形態の1つは、この製剤が、咳、風邪、アレルギー兆候の治療を目的とした、徐放性被覆ペレットをシロップ剤に含む分散液である。例えば、該製剤は、10又は15mgのヒドロコドン重酒石酸塩と120mgのプソイドエフェドリン塩酸塩と8mgのクロルフェニラミンマレイン酸塩とを、大人の1回の服用量(5ml)中に一緒に含むことができる。薬剤は塩の形態で使用できるが、塩基の形態をはじめとする他の形態も使用できる。これらの有効成分は、処方箋医薬品としてもOTCの咳や風邪薬としても、各成分単独又は組み合わせて広範な人体投与テストがされている。
本発明の実施形態の1つは、製剤の徐放性(ER)部位が、懸濁液中の被覆ビーズ、粒子又はペレットに相当し、即放性(IR)部位が懸濁液中に存在する。実施形態の1つは、本発明の製剤が、UPM Pharmaceuticals社所有の特許公開公報(U.S. Ser. No. 10/724,276、2003年11月26日出願;U.S. Ser. No. 11/150,572、2005年6月9日出願;U.S. Ser. No. 11/198,937、2005年8月4日出願参照)に記載の技術を用いて調製される。徐放性ビーズ含有懸濁液の特定のタイプの製造と使用に関連する部分は、引用により本願明細書の記載に含まれるものとする。
【0068】
或いは、本発明におけるER部位は、カプセル剤、錠剤又は他の固形経口剤のような固体の剤形で構成され、IR部位は、前記ER部位の外側にある第2の層又は媒体であってもよい。実施形態によると、固形経口製剤には液体成分が含まれない。即ち、固形経口製剤には液相又は分散媒が含まれない。このような実施形態においては、固形経口剤のIR部位は液相又は分散媒を含まない。被験者に投与する前に、この固形経口製剤は、例えば分散媒等の液相と混合しない。同様に、実施形態の1つでは、単一の徐放性調合品で、徐放成分が粒子、ペレット又はビーズの内部で、即放性部位が外側にある(例えば、シロップ剤に懸濁又はカプセル剤、錠剤等の中の粉末)は特定のIR/ER比を示す。この比により、単回投与及び安定状態の条件で、関連リスト収載医薬品(RLD)と生物学的に等価となる血漿中濃度が得られる。
本発明の態様は、非液状の徐放性薬剤組成物に関する。本願明細書で使用の「非液状」という用語は、U.S. Ser. No. 10/724,276(2003年11月26日出願)、U.S. Ser. No. 11/150,572 (2005年6月9日出願)及びU.S. Ser. No. 11/198,937(2005年8月4日出願)に記載されているように、イオン交換マトリックス薬剤複合物を含有する分散相と分散媒とを有する液状組成物ではない組成物を指す。
別の実施形態では、本発明の製剤の投与により、ヒトにおいてある時間にわたり特定の範囲の血漿中濃度が得られるが(AUC、Tmax、T1/2等で測定)、前記血漿中濃度とは、12時間の徐放中、安全で効力のある濃度であり、単回投与の条件と安定状態の条件の両方で即放性RLDと生物学的等価である血漿濃度である。
【0069】
実施形態の1つは、経口徐放性薬剤組成物が第1の部位と第2の部位とを含み、第1の部位が即放状態で、有効成分として抗ヒスタミン剤と鎮咳剤とを含み、更に充血緩和剤を含んでいてもよく、第2の部位が粒子、ペレット又はビーズを含み、各粒子、ペレット又はビーズには有効成分として徐放状態で同じ抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む。別の実施形態では、この薬剤組成物を患者に単回投与すると、少なくとも8時間、例えば、8、12、18又は24時間にわたって上記3種の有効成分の血漿中濃度が、前記有効成分を含むFDA承認済みの即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となる。この適切な投与回数とは、FDA承認済みのIR薬剤組成物の同じ期間にわたる投与に関して、FDAが承認するラベルで推奨される投与回数に相当する。
実施形態によっては、徐放性部位が粒子、ペレット又はビーズの形態である。この粒子、ペレット又はビーズのサイズは、液剤又は固形製剤で経口投与できるサイズである。実施形態の1つは、粒子、ペレット又はビーズが懸濁液のなかで沈殿しないような大きさ及び/又は密度である。実施形態によっては、粒子、ペレット又はビーズの直径は約0.01〜約2000μmの範囲である。別の実施形態では、約0.1〜約1000μm、更に別の実施形態では約1〜約1000μmの範囲である。実施形態の1つは、粒子、ペレット又はビーズの直径が約600μmである。
【0070】
実施形態によると、抗ヒスタミン剤、鎮咳剤及びプソイドエフェドリンを含む懸濁液製剤であって、上記3種すべての薬剤が即放性及び徐放性を示す製剤で、徐放性粒子又は徐放性ペレット又は徐放性ビーズからのプソイドエフェドリンの放出速度が、抗ヒスタミン剤(例えばクロルフェニラミン)又は鎮咳剤(例えばヒドロコドン)の放出速度よりも速いという事実を利用している。例えば、このような製剤は、抗ヒスタミン剤、鎮咳剤及びプソイドエフェドリンを徐放性の粒子、ペレット又はビーズに含有させることができ、一方、即放性液体/ビヒクル部位は抗ヒスタミン剤と鎮咳剤とを含み、プソイドエフェドリンを含まない。このような特定の製剤であっても、3種すべての薬剤の即放及び徐放は、相当するRLDを例えば12時間にわたってFDA承認済みラベル表示のように2回以上の投与をした際の薬剤の放出と生物学的等価となるような即放と徐放を示す。
実施形態によっては、本発明の製剤における有効成分の担体として働く不活性成分は、アンモニオメタクリレート共重合体、ラクトース一水和物、メチルパラベン、微晶質セルロース、プロピルパラベン、純水、アルギン酸ナトリウム、ショ糖、タルク、二酸化チタン、クエン酸トリエチルが挙げられる。これらをはじめとする不活性成分を使って、2つの異なる相、即ち、即放用の薬剤を含みシロップ剤に溶解された分散媒と、薬剤の徐放性部位を含む被覆粒子、ペレット又はビーズを含有する分散相を調製して、本発明の製剤とすることができる。
【0071】
生物学的等価性(BE)は薬物動態学上の用語で、ある薬剤による2種類の調剤の生体内での生物学的な等価性を示すのに使用される。生物学的等価性要件とは、指定された医薬品の生体外及び/又は生体内試験のFDAが課した要件を指し、米国連邦規則集(CFR)第21巻320条1(f)項のもとで市場にだす条件として満たさなければならない。米国食品医薬品局(FDA)は、生物学的等価性を次のように定義している。「適切に計画された試験において、医薬的同等物又は医薬的代替物が同様な条件のもと、同じモル服用量で投与された際に、そこの含まれる有効成分又は有効部位が薬剤の活性サイトにおいて有効になる速度とその程度に著しい差がない」(米国連邦規則集(CFR)第21巻320条1(e)項);2002年7月2日にFDAが発行した「Bioavailability and Bioequivalence Studies for Orally Administered Drug Products - General Considerations (経口投与薬剤についてのバイオアベイラビリティーとバイオエクイバレンスの研究 -一般的考察)も参照のこと。サイトはwww.fda.gov/OHRMS/DOCKETS/98fr/02d-0258-gdl0001.pdf、2003年3月19日正式採用(米国連邦官報(Federal Register)、第68巻13316、2003年3月19日)。
BEは、適切な薬物動態学的パラメータで2つの薬を比較し、それらが許容範囲内かどうかを求めることにより測定できる。ある有効成分の「生物学的等価な」血漿中濃度とは、該有効成分を含む第1の医薬品の投与後に患者の血漿で測定した際、該有効成分に関する単回投与試験で測定したAUCinfinityならびに安定状態試験で測定したCmax、Cmin及びAUCinfinityの対数変換平均値が、90%信頼区間において、該有効成分を含む第2の医薬品の投与後に血漿で測定した該有効成分のCmax、Cmin及びAUCinfinityの80〜125%の範囲にあることを意味する。
【0072】
FDAでは、統計的分析を行ってBEを求めるの前に、薬物動態学的パラメータの対数変換を推奨している。従来のFDAが推奨するBE限界は、対数変換されたPKパラメータが90%信頼限界の範囲で互いの80〜125%内でなければならないというものである。本願明細書の実施形態の1つは、新規製剤が12時間の放出制御型医薬品である。この医薬品は、12時間にわたり2〜3回投与されるRLDの2回又は3回投与量と、t=0とt=6時間、又は、t=0とt=4時間とt=8時間の時点で比較すればよい。
単回投与試験とは、患者に対象の徐放性製品は1回だけ投与し、それに相当する即放性の関連リスト収載医薬品(RLD)は、該即放性RLDについてFDAが承認済みのラベル表示で指示されているような、12時間に相応する服用量で投薬する試験である。安定性試験とは、対象の徐放性製品と即放性の関連リスト収載医薬品(RLD)とを、有効成分の血漿中濃度が安定した濃度に達するまで、試験期間中繰り返し投与する試験である。「Cmax」とは、患者に投与した後、安定状態に到達した後に検出された最高血漿濃度(例えばng/ml)を指す。「Cmin」は、薬剤が安定状態に到達した後に患者から検出された最低血漿濃度(例えばng/ml)を指す。「AUCinfinity」又は「AUC」は、ある医薬品をある時間(例えば、8、12、24、48時間等)にわたり1回以上投与した後、血漿中の有効成分濃度を0時間から無限時間までプロットした曲線の下面積(例えばng/ml x hr)を指す。有効成分の「Cmin」、「Cmax」及び「AUCinfinity」は周知方法により測定できる。
【0073】
「関連リスト収載医薬品」又は「RLD」とは、医薬品簡略承認申請(ANDA)の承認を求める際に出願人が根拠にできる医薬品としてFDAが認定したリスト収載薬品を指す。このように、単一の薬剤を含有するRLDはFDAにより明示されている。ジェネリック薬品又は505(b)(2)出願のもとで提出された薬品に関して、ある有効成分についての製造業者が2社以上の場合、RLDの役割を果たす製品の供給元をどの製造業者にするかはFDAが選択する。本発明の目的のためには、各有効成分を別々に含有する即放性医薬品がRLDとして使われる。本発明の製剤と比較するためには、複数のRLDを組み合わせて、たとえばRLDの投与説明書にしたがって順次投与すればよい。同様に、本発明の目的のためには、「関連リスト収載医薬品」又は「RLD」という用語は、単独薬剤RLDが2つ以上混ざった液体も指す。例えば、RLDが、抗ヒスタミン剤のRLDと鎮咳剤のRLDと充血緩和剤のRLDとを含む混合液体であってもよい。クロルフェニラミンに対してFDAが承認しているRLDは、Chlor-Trimetonシロップ剤である。PSEに対してFDAが承認しているRLDはSudafedシロップ剤である。ヒドロコドンに対してFDAが承認しているRLDはHycodanシロップ剤である。本発明の製剤を投与して得られる血漿中濃度を比較し、生物学的等価性の評価を行う場合、単独の薬剤を含有するRLDを3種類含む混合液は、上記RLDのいずれのFDA承認済みラベル表示に従ってヒトへの投与を行うとよい。
2001年1月、FDAは生物学的等価性試験を説明するガイドラインを発表した。概して、試験のやりかた、データの分析方法等で、「Guidance for Industry: Statistical Approaches to Establishing Bioequivalence」(「Statistical Approaches」)という表題の文書として発表された。この文書の中で、FDAは、医薬品が制定法上のBEの定義を得ているかどうかの決定に使用する基準を規定した。本願明細書中、対象の医薬品が関連リスト収載医薬品に生物学的等価であるかを言及する場合、これらの文書に記載の「average BE」の定義を適用する。
【0074】
「Statistical Approaches」2頁に記載のごとく、FDAは以下のことを推奨している。『標準的な生体内BEテストのデザインは、テスト(T)薬品と参照(R)薬品を単回投与又は反復投与のいずれかで健康な被験者に別々に投与することを基礎とし、医薬品投与の2通りの順番にランダムに割り付ける。(中略)曲線下面積(AUC)や最高濃度(Cmax)等の薬物動態学的測定値の統計分析は2つの片側検定の方法を基準とし、T薬品とR薬品の投与後に求めた薬物動態学的測定値の平均値が同等かどうかを決める。このアプローチは平均生物学的等価性と称され、T薬品とR薬品の測定値の平均値(母幾何平均値(population geometric means))の比に関して90%信頼区間の計算も行う。生物学的等価性(BE)を確立するには、医薬品平均値の比について計算した信頼区間がBE範囲内、通常は80〜125%の中に収まらなければならない。』「Statistical Approaches」第IIB章、2頁(原文では強調)
BEデータの統計分析は、バイオアベイラビリティーのデータ(例えばAUC、Cmax、Cmin等)の対数変換用の統計モデルを基準とする。「Statistical Approaches」の手引書では、BE測定値の対数変換(自然対数又は底が10の対数)が提唱されている。データ分析については、「Statistical Approaches」では対数変換したBE測定値にパラメトリック(正規理論)法を用いることが推奨されている。『方程式2又は3に記載の基準を用いた平均生物学的等価性に関しては(第IV.A章)、μT-μR量の90%信頼区間を構築し、この信頼区間が区間[-θA, θA]に含まれる場合は平均生物学的等価性という結論に到達する、というのが一般的なアプローチである。(中略)対数変換したデータの平均値の差についての90%信頼区間は、実験計画に合った方法を使って計算しなければならない。』「Statistical Approaches」第VI.B章、10頁
【0075】
本発明の実施形態は、一般的に安全かつ有効な(GRASE)3種の有効成分、即ち、抗ヒスタミン剤、鎮咳剤及び充血緩和剤の利点を1つの徐放性薬品に統合するものである。現在、これらの薬剤を一緒に使用する場合は、これらの薬剤をそれぞれの即放性製剤で一日4〜6回投与することになる。3つの効果を発揮する1つにまとまった徐放性調合薬品はOTC医薬品も処方箋による医薬品でも利用できないからである。そこで、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含むIR/ER製剤であって、1つの医薬品を投与することで3種類すべての薬剤が徐放性を示す製剤であれば、3種類すべての有効成分は含有していない現行の医薬品及び/又は単に即放性の現行の医薬品よりも有利である。例えば、本発明の製剤を使用すると、患者にとって必要な薬の量が、即放性薬品の1日50〜55mlに比べて、例えば5ml等の少ない量で、風邪、流感又はアレルギーの症状を軽減することになる。また、本発明の製剤は、投与単位4オンスの便利な容器に入れて販売することもできる。また、患者にとっては薬を摂取する回数が減り、1日を通して苦痛軽減状態でいるために必要な服用量をとりそこなう危険性が小さくなる。患者のコンプライアンスという問題は決してなくなることがない、十分に認識された問題であることを考慮すると、単回投与(例えば12時間)で3種類すべての薬剤について生物学的等価性のある服用量を提供する本製剤は、現在利用できる製剤にくらべて著しい利点をもたらすものである。
本発明の実施形態は、クロルフェニラミン、プソイドエフェドリン及びヒドロコドンの即放剤形と徐放剤形との新規な混合物を1つの医薬品内に含む製剤を提供するもので、その単一医薬品は患者への投与回数を減らす一方で、上記薬剤を含有する即放性医薬品を摂取した患者と生物学的等価性が得られる。
【0076】
また、本発明は薬剤調合製剤及びそのような製剤の製造方法に関するもので、製剤は懸濁液又は固形カプセル剤もしくは錠剤の安定した徐放性経口薬剤で、粒子、ペレット又はビーズを含み、個々の粒子、ペレット又はビーズの中に2種以上の有効成分が含まれている。このアプローチは即放性製剤だけでなく、徐放性製剤に勝る多数の利点がある。
本発明の他の実施形態は、1種以上の薬剤を含むペレット、ビーズ又は粒子を有する徐放性成分を含有する新規な経口懸濁液製剤であって、前記ペレット、ビーズ又は粒子がシロップ剤に懸濁した製剤を提供する。シロップ剤も1種以上の薬剤を含んでもよい。本願明細書では、このような経口懸濁液の一例を挙げる。この経口懸濁液製剤は従来よりも優れた特性がある。例えば、例示する製剤の徐放性成分はビーズを有し、ビーズはイオン交換マトリックスと結合した3種類の薬剤を含有し、このビーズがシロップ剤の中で懸濁しているので、他の液体製剤よりも非常に高い製品安定性が本製剤に付与される。通常、水性の環境において薬剤は崩壊を起こす。固形剤形とすることで崩壊を最小限にできるが、液体製剤にみられる投与の容易さや投与量の融通性は損なわれる。少なくとも1つの実施形態では、本発明の剤形は以下の2段階のアプローチにより水への暴露を最小限にしている。(1)ビーズは、懸濁させるのに使用している液相に容易に溶けるというよりも、むしろ徐放性を示す。(2)液相はシロップ剤であるが、シロップ剤中の糖類により液相の水分活性が下げられる。また、水分活性を低下させ浸透圧を上昇させることにより、シロップ剤は次の働きをする。(a)薬剤の徐放性ビーズからシロップ剤への浸出を最小限に抑える。(b)ビーズの崩壊を防ぐ。逆にいえば、ビーズは患者が摂取した後、例えば、腸内で膨潤し崩壊することで薬剤を放出することができ、ERからの薬剤放出を可能にする。
【0077】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の液状の放出制御型薬剤組成物中のイオン交換マトリックス樹脂-薬剤複合物の濃度は、例えば、特定の薬剤に応じて、また、イオン交換マトリックス樹脂-薬剤複合物内の薬剤の含有量や治療すべき症状又は兆候、患者の年齢などに応じて広範囲にわたり変わるうる。こうした組成物の実施形態では、液状の放出制御型薬剤組成物中のイオン交換マトリックス樹脂-薬剤複合物の濃度の範囲は、液状の放出制御型薬剤組成物の全質量を基準に約5〜約90質量%である。この組成物の別の実施形態では、イオン交換マトリックス樹脂-薬剤複合物の質量の範囲は、液状の放出制御型薬剤組成物の全質量を基準に約10〜約50質量%である。更に別の実施形態では、イオン交換マトリックス樹脂-薬剤複合物の質量の範囲は、液状の放出制御型薬剤組成物の全質量を基準に約20〜約40質量%である。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の薬剤組成物の実施形態において、分散相の調製方法は、粉末状の薬剤と粉末状のイオン交換マトリックスとを混合することを含む。このような実施形態では、薬剤とイオン交換マトリックスは塩の形態のものを使えばよい。本発明の分散相を調製する粉末混合方法は、従来の方法に比べて費用効率及び時間効率がよい。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の薬剤組成物の実施形態の1つでは、イオン交換マトリックスがアルギン酸ナトリウムである。非制限的実施形態であるが、クロルフェニラミンマレイン酸塩、プソイドエフェドリン塩酸塩、ヒドロコドン重酒石酸塩の粉末を1種又は一緒にして、アルギン酸ナトリウム粉末と混合すればよい。別の実施形態は、薬剤とイオン交換粉末との混合物に、ラクトース、微晶質セルロース及び/又は他の賦形剤を添加してもよい。
【0078】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の薬剤組成物の別の実施形態では、薬剤とイオン交換粉末とを混合した後、水を加えて湿式の塊を作成し、押出と球状化を行う。得られた薬剤とイオン交換マトリックスとを含むコアビーズ又はペレットは流動床乾燥機で乾燥させればよい。実施形態の1つでは、個々のビーズ又は各ペレットに、同一のイオン交換マトリックスに結合した数種の有効成分が含まれる。例えば、個々のビーズ又は各ペレットに、アルギン酸ナトリウム粉末に結合したクロルフェニラミンマレイン酸塩、プソイドエフェドリン塩酸塩、ヒドロコドン重酒石酸塩が含まれる。
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる本発明の薬剤組成物の実施形態では、流動床プロセッサー中、クエン酸トリエチル及びタルクの存在下、得られたビーズにEUDRAGIT(登録商標)をコーティングする。その後、被覆ビーズをタルクと混合して炉内で硬化させる。実施形態によると、被覆ビーズは2時間、4時間、8時間、16時間、24時間又は48時間硬化させる。実施形態によっては、被覆ビーズを約16〜約24時間硬化させる。被覆ビーズの硬化時間は、被覆ビーズからの薬剤の放出速度に影響を及ぼすことがある。
【0079】
有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる被覆ビーズを、塩の形態の薬剤と水とショ糖とを含む分散媒に懸濁させる。本発明の組成物の非制限的実施形態の1つでは、分散媒がクロルフェニラミンマレイン酸塩とヒドロコドン重酒石酸塩とを含む。本発明の組成物の別の実施形態では、分散媒が更にプソイドエフェドリン塩酸塩も含む。また、分散媒は防腐剤、矯味剤、他の不活性添加剤を更に含んでもよい。このような実施形態で得られた液状の放出持続型医薬品は、物理的安定性と化学的安定性を瓶の中で維持することができ、かつ、患者に投与する際は薬剤の放出を制御することができる。
本発明の態様の1つは、有効成分がクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンとからなる液状の放出制御型薬剤組成物の製造方法に関するもので、該方法は、
(a)クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる有効成分が単一粒子又は単一ペレット又は単一ビーズ内で結合している、粒子、ペレット又はビーズを作製する工程を含む、分散相を調製する工程と、
(b)クロルフェニラミン、ヒドロコドン、そして任意であるがプソイドエフェドリンを含んでいてもよい有効成分を含有する分散媒を調製する工程と、
【0080】
(c)前記粒子、ペレット又はビーズを皮膜でコーティングする工程と、
(d)前記ビーズを分散媒に分散させる工程とを含む。
前記方法の1つの実施形態は、分散相調製工程が、クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンを医薬的に許容できるイオン交換マトリックスと結合させる工程を更に含む。別の実施形態は、分散相調製工程が、クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンならびに医薬的に許容できるイオン交換マトリックスとからなる粒子、ペレット又はビーズを調製する工程を更に含み、個々の粒子、ペレット又はビーズ内ではクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンがイオン交換マトリックスに結合している。別の実施形態は、分散相調製工程が、クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンならびにイオン交換マトリックス粉末を混合する工程を更に含む。更に別の実施形態は、分散相調製工程が、クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンならびにイオン交換マトリックス粉末の湿式造粒、押出、球状化工程を更に含む。実施形態の1つは、クロルフェニラミン、ヒドロコドン、プソイドエフェドリン及びイオン交換マトリックスが塩の形態のものを使用する。
【0081】
本発明の製剤は、2種以上の薬剤の生物学的等価性を同時に実現できるところが利点の1つである。比較用医薬品の場合、各薬剤が同じ放出プロファイルにならないこともあり、異なる服用量や投与回数が必要になることがある。更に、徐放の方法が異なれば(例えば、異なる樹脂を使用)、各薬剤の放出プロファイルは患者の食餌又は生理機能の差に影響をうける可能性があり、患者によっては、ある薬剤は摂取しすぎで、別の薬剤の摂取は少なすぎるということがおきる。3種類の薬剤が、例えば単一の放出方法を有するビーズ1個1個の中に配置されることによって、薬剤放出プロファイルは患者全体の中でより一貫性があるものになる。
3種類の薬剤をそれぞれ別のビーズに配置した後にビーズを混合した場合、特に少量の場合、完全に均一な混合物ではないことがある。このような不均一が起きると、結果、薬剤の相対的服用量が不正確になる。即ち、ある薬剤は過多となり、別の薬剤は過少となる。この不均一は、無作為な変動によるだけでなく、3種のビーズ混合物において3種のビーズの物性が異なることに起因して起こることもある。例えば、プソイドエフェドリンはすべての薬剤のなかで占める割合(質量比)が高い。3種類の薬剤が同じ大きさと量のビーズそれぞれに充填されると、賦形剤に対してプソイドエフェドリンは量が多くなるので、プソイドエフェドリンのビーズは、ヒドロコドンのビーズ又はクロルフェニラミンのビーズと密度が異なる結果になる。このような密度の違いは均質性の欠如につながる。所定の温度及び圧力においてビーズが同じ密度になるように調整したとしても、条件が変わればそのまま維持することはできない。
【0082】
また、密度の違いを利用すれば、薬剤の種類に応じてビーズを故意に分離し、プソイドエフェドリンを集めて(say)薬剤の不正使用に転用することができる。一方、一粒のビーズの中にすべての薬剤が存在する場合、ビーズの物性の違いを基に1つの薬剤を他の薬剤から部分的に単離することは不可能である。更に、このような医薬品の製造方法であれば、粒子、ペレット又はビーズ1個に含まれるいずれの有効成分も乱用や転用を思いとどまらせ、阻止することになる。粒子、ペレット又はビーズ1個の中に2種以上の薬剤を混合又は注入することにより、乱用を目的として個人が医薬品から個々の有効成分を抽出又は分離することは容易ではない。更に、粒子、ペレット又はビーズ1個の中に2種以上の有効成分を混合又は注入することで、調合薬品全体に存在する粒子、ペレット又はビーズの表面積が低減するので、医薬品及び有効成分の安定性が増大する。
薬剤調合医薬品の実施形態の1つは、粒子、ペレット又はビーズ1個につき2種以上の有効成分を有する粒子、ペレット又はビーズを含有することにより、1つの有効成分を分離又は単離する潜在的可能性が減少する。同様に、本発明は、製剤に含まれるいずれか1つの成分についての乱用の潜在的可能性を低減する、風邪及びアレルギー用調合薬剤製剤の製造方法を提供する。例えば、製品に含まれる各ビーズが、医薬的に許容できるイオン交換マトリックスと、該イオン交換マトリックスに結合した2種以上の医薬的に許容できる有効薬剤成分とを含んでいてもよく、これは、UPM Pharmaceuticals社所有の特許公開公報(U.S. Ser. No. 10/724,276、U.S. Ser. No. 11/150,572、U.S. Ser. No. 11/198,937、引用により本願明細書の記載に含まれるものとする)に記載の特定のタイプの徐放性ビーズを含む懸濁液の製造と使用に関する技術を用いて調製してもよい。にわか作りの違法薬物研究所では、このような薬剤調合医薬品からはいずれの単一有効薬剤成分も簡単に抽出、分離又は単離することはできない。
【0083】
このように本発明の製剤は、咳/風邪の市場で現在入手可能な他の製剤とは異なる。例えば、Tussionex(登録商標)徐放性懸濁液は、咳止め/抗ヒスタミン剤の調合品で、ヒドロコドンとクロルフェニラミンを含み風邪やアレルギーによる咳や上気道の兆候をやわらげるのに使用される。この懸濁液型医薬品は薬剤-イオン交換樹脂ビーズを含有するが、懸濁液中の個々のビーズにはヒドロコドン又はクロルフェニラミンのいずれかが充填されており、両方の薬剤が1つのビーズには含まれていない。
また、本発明の製剤が現在市販されている製剤と異なる点は、インターネットが示す乱用薬物の製造又は単離を行うための、一般的な家庭用品、簡単な製造装置又はいわゆる「レシピ」は本発明の製剤には容易に利用できない、という点である。むしろ、本発明の製剤から対象となる乱用可能な薬物を分離することは、通常は大学や企業の研究所でしか利用できないような、例えば、複合クロマトグラフィー等の非常に技術的に高度な装置や科学的な熟練及び技術が必要となる。
本発明の別の実施形態では、イオン交換マトリックス薬剤粒子、ペレット又はビーズを有することにより、薬剤調合医薬品は乱用及び/又は転用の潜在的可能性を低減する。各ビーズには、医薬的に許容できるイオン交換マトリックスとイオン交換マトリックスに結合した2種以上の医薬的に許容できる有効成分とが含まれていてもよい。
【0084】
また、本発明は、少なくとも1種の有効成分の乱用を予防又は低減する方法も包含し、該方法は、薬剤調合医薬品を調製する工程を含むが、該医薬品が、粒子、ペレット又はビーズを含み、個々の粒子、ペレット又はビーズが医薬的に許容できるイオン交換マトリックスとイオン交換マトリックスに結合した2種以上の医薬的に許容できる有効成分とを含む。
別の実施形態では、単一の有効成分の抽出、単離又は分離の可能性を阻止又は低減するための方法が薬剤調合医薬品を調製する工程を含むが、該医薬品が、粒子、ペレット又はビーズを含み、個々の粒子、ペレット又はビーズが医薬的に許容できるイオン交換マトリックスとイオン交換マトリックスに結合した2種以上の医薬的に許容できる有効成分とを含む。マトリックス粒子、ペレット又はビーズに2種以上の有効成分が含まれているという事実により、工業レベルでない、クロマトグラフィー装置等の高品質装置をもたないにわか作りの違法薬物研究所で素人がいずれかの薬剤を単独に抽出又は分離することは、きわめて難しくなる。
別の実施形態は、本発明の薬剤調合医薬品により、患者が、例えば3つの異なる有効成分を適切かつ正確に投薬できるようになる。多数の製剤(例えば、異なる3種の医薬品で、それぞれに即放性有効成分が単独に含まれている)を使って患者が医者にかからず自分で治療する場合、適切かつ正確な投薬は困難であることが多く、とりわけ、1種又はそれ以上の薬剤の過剰投与又は過少投与をしないようにすること、及び/又は、副作用を最小限に抑えてすべての薬剤の治療効果を最大限に引き出すという点に関して、適切かつ正確な投薬は困難であることが多い。本発明の調合医薬品は、すべての関連薬剤が一つの剤形の状態で、例えば、12時間徐放型剤形で提供、投与されるので、上記のような問題が回避される。
【0085】
薬剤調合医薬品の1つの実施形態は懸濁液組成物で、徐放性薬剤を含有する被覆イオン交換マトリックス薬剤粒子、ペレット又はビーズを含む分散相を含み、更に任意であるが、即放性薬剤を含む分散媒であって、シロップ剤に溶解した分散媒を含んでいてもよい。
実施形態の1つは、使用する放出制御型薬剤組成物の投与量が、患者において、例えば、ヒトにおいて治療効果を達成するのに十分な量である。この「治療効果」という用語は、風邪、流感又はアレルギーに対するいずれの効果も意味し、咳の重篤度及び/又は回数、咳の兆候、鼻漏、鬱血又はくしゃみの低減などの症状緩和をはじめとするが、これらに制限されない効果、及び/又は、自覚的な健康状態の改善につながる他の生物学的効果を意味する。
本発明の具体的態様の1つは、は第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物に関するもので、
第1の部位が即放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよく、
第2の部位が徐放性の剤形で3種の有効成分として抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む粒子、ペレット又はビーズを含有し、
前記非液状経口薬剤組成物を患者へ単回投与した際に、少なくとも8時間にわたって得られる上記3種の有効成分の血漿中濃度が、前記有効成分で構成されるFDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となり、
前記適切な投与回数とは、IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みの1つ以上のラベルのなかで推奨される投与回数に対応する。
【0086】
前記組成物の実施形態の1つは鎮咳剤が麻酔性鎮咳剤である。前記組成物の別の実施形態は、第1の部位が充血緩和剤を含まず、抗ヒスタミン剤と鎮咳剤しか含まない。具体的実施形態は、抗ヒスタミン剤がクロルフェニラミンである。別の具体的実施形態は、鎮咳剤がヒドロコドンである。更に別の具体的実施形態は、充血緩和剤がプソイドエフェドリンである。前記組成物の実施形態の1つにおいて、粒子又はペレット又はビーズが更に皮膜を有する。
前記組成物の実施形態の1つは、前記抗ヒスタミン剤と鎮咳剤と充血緩和剤とが1個1個の粒子又はペレット又はビーズ内で結合している。このような実施形態では、粒子、ペレット又はビーズが医薬的に許容できるイオン交換マトリックスを更に含み、抗ヒスタミン剤、鎮咳剤及び充血緩和剤がイオン交換マトリックスと結合している。
上記組成物の実施形態によっては、甘味剤、風味剤、着色剤及びこれらの混合物からなる群から選択される賦形剤を更に含有する。別の実施形態では、安定化剤、分散剤及びこれらの混合物からなる群から選択される添加剤を更に含有する。
本発明の態様の1つは、第1の部位と第2の部位とを含む、非液状の経口徐放性薬剤組成物に関するもので、
第1の部位が即放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよく、
第2の部位が、有効成分として抗ヒスタミン剤と鎮咳剤と充血緩和剤とを徐放剤形で含む、粒子、ペレット又はビーズで、
【0087】
前記3種の有効成分が24時間を超えて安定した血漿中濃度であるように前記非液状薬剤組成物を十分な投与回数で患者に投与した際、前記有効成分の血漿中濃度が、前記有効成分で構成されるFDA承認済みの即放性薬剤組成物1種以上を上記と同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となり、
前記適切な投与回数とは、FDA承認済みの即放性薬剤組成物1種以上の同じ期間にわたる投与に関して、FDA承認済みの1つ以上のラベルのなかで推奨される投与回数に対応する。
本発明の別の態様は、ヒト被験者の咳、風邪、流感又はアレルギー兆候の治療方法に関するもので、本願明細書に記載の非液状経口徐放性薬剤組成物を該被験者に投与する工程を含む。このような実施形態において、医薬組成物は、1日1回又は1日2回投与ができる二段階放出(dual release)製剤としてヒトに投与される。
本発明の更に別の態様は、風邪、流感又はアレルギーに関連した咳、咳の兆候、鼻漏、鬱血又はくしゃみを少なくとも8時間にわたり治療する方法に関するもので、このような治療を必要とするヒトに対し、風邪又はアレルギーに関連した咳、咳の兆候、鼻漏、鬱血又はくしゃみを少なくとも8時間にわたり治療するのに有効な本願明細書記載の非液状薬剤組成物を単回投与することを含む方法に関する。
本発明の更に別の態様は、本願明細書記載の非液状経口徐放性薬剤組成物の製造方法に関するもので、
有効成分として、抗ヒスタミン剤と鎮咳剤とを含み、更に充血緩和剤を含んでもよい即放性部位を調製する工程と、
【0088】
抗ヒスタミン剤と鎮咳剤と充血緩和剤とを有効成分として含有する粒子、ペレット又はビーズの作製を含む徐放性部位を調製する工程と、
前記粒子、ペレット又はビーズを皮膜でコーティングする工程と、
前記徐放性部位と前記即放性部位とを混合する工程とを含む方法に関する。
前記方法の実施形態は、1個1個の粒子、ペレット又はビーズの中に2種以上の有効成分が結合している粒子、ペレット又はビーズを作製する工程を更に含む。このような方法の具体的実施形態の1つは、前記2種以上の有効成分を医薬的に許容できるイオン交換マトリックスと結合させる工程を更に含む。具体的実施形態は、抗ヒスタミン剤がクロルフェニラミンである。別の具体的実施形態は、鎮咳剤がヒドロコドンである。更に別の具体的実施形態は、充血緩和剤がプソイドエフェドリンである。
本発明の別の態様は、少なくとも8時間にわたる抗ヒスタミン剤、鎮咳剤及び充血緩和剤のある哺乳動物における血漿中濃度を、FDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり同じ哺乳動物に適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価である血漿中濃度にする方法に関するもので、該方法は、
第1の部位と第2の部位とを有する非液状経口徐放薬剤組成物であって、第1の部位が即放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよく、第2の部位が徐放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む粒子、ペレット又はビーズを含有する、非液状経口徐放薬剤組成物を前記哺乳動物に投与することと、
【0089】
少なくとも8時間にわたる前記3種の有効成分の血漿中濃度が、前記有効成分を含有するFDA承認済みのIR-RLD組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価とすることとを含み、前記適切な投与回数とは、IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みの1つ以上のラベルのなかで推奨される投与回数に対応する。
本発明の更に別の態様は、非液状の経口徐放性(ER)薬剤組成物の投与後、抗ヒスタミン剤、鎮咳剤及び充血緩和剤の安定した血漿中濃度を哺乳動物において得るための方法であって、前記血漿中濃度とは、活性成分と不活性成分とを含み、前記活性成分がクロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる即放性(IR)組成物1種以上を前記哺乳動物に投与した際に得られる血漿中濃度と生物学的等価であり、該方法は、
第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物であって、第1の部位が即放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含み、第2の部位が徐放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む粒子、ペレット又はビーズを含有する、非液状経口徐放性薬剤組成物を前記哺乳動物に投与することを含み、
【0090】
前記3種の有効成分が24時間を超えて安定した血漿中濃度であるように前記非液状経口徐放性薬剤組成物を十分な投与回数で哺乳動物に投与することにより、前記有効成分を含有するFDA承認済みの即放性(IR)薬剤組成物1種以上を上記と同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価な有効成分の血漿中濃度となり、
前記FDA承認済みのIR薬剤組成物1種以上の適切な投与回数とは、FDA承認済みのIR薬剤組成物1種以上の同じ期間にわたる投与に関して、FDAが承認するの1つ以上ラベルのなかで推奨される投与回数に対応し、
前記FDA承認済みのIR薬剤組成物1種以上の適切な投与回数が、経口ER薬剤組成物の十分な投与回数を上回るものである。
本発明の態様の1つは、第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物に関し、
前記第1の部位は、クロルフェニラミンとヒドロコドンとを含み、さらにプソイドエフェドリンを含んでもよい有効成分を即放剤形で含有し、
前記第2の部位は、3種の有効成分としてクロルフェニラミンとヒドロコドンとプソイドエフェドリンとを徐放剤形として含む粒子、ペレット又はビーズを含有し、
前記非液状経口薬剤組成物を患者へ単回投与することにより、少なくとも8時間にわたって得られる上記3種の有効成分の血漿中濃度が、前記有効成分で構成されるFDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となり、
前記適切な投与回数とは、IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みの1つ以上のラベルのなかで推奨される投与回数に対応する。
【0091】
前記薬剤組成物の実施形態によっては、少なくとも8時間という期間が12時間である。別の実施形態では、少なくとも8時間という期間が24時間である。
前記薬剤組成物の他の実施形態では、第1の部位にプソイドエフェドリンが含有されていない。
薬剤組成物の実施形態の1つは、経口固形剤形である。別の実施形態では、薬剤組成物は経口カプセル剤である。
前記薬剤組成物の実施形態によっては、粒子、ペレット又はビーズが更に皮膜を有する。このような実施形態で、該粒子、ペレット又はビーズは医薬的に許容できるイオン交換マトリックスを更に含み、このイオン交換マトリックスにクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンが結合している。
前記薬剤組成物の実施形態では、甘味剤、風味剤、着色剤及びこれらの混合物からなる群から選択される賦形剤を更に含有する。別の実施形態では、薬剤組成物が、安定化剤、分散剤及びこれらの混合物からなる群から選択される添加剤を更に含有する。
前記薬剤組成物の実施形態によっては、少なくとも8時間という期間が12時間であり、かつ、薬剤組成物が、8〜12mgのクロルフェニラミンマレイン酸塩と、10〜15mgのヒドロコドン重酒石酸塩と、少なくとも120mgのプソイドエフェドリンとからなる有効成分を1回の服用量5ml中に含む。他の実施形態では、少なくとも8時間という期間が24時間であり、かつ、薬剤組成物が、16〜24mgのクロルフェニラミンマレイン酸塩と、20〜30mgのヒドロコドン重酒石酸塩と、少なくとも240mgのプソイドエフェドリン塩酸塩とからなる有効成分を1回の服用量5ml中に含む。
【0092】
本発明の別の態様は、風邪、流感又はアレルギーに関連した咳、咳の兆候、鼻漏、鬱血又はくしゃみを少なくとも8時間にわたり治療する方法に関するもので、このような治療を必要とするヒトに対し、風邪又はアレルギーに関連する咳、咳の兆候、鼻漏、鬱血又はくしゃみを少なくとも8時間にわたり治療するのに有効な、有効成分がクロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる薬剤組成物を単回投与することを含む方法に関する。
前記方法の実施形態によっては、少なくとも8時間という期間が12時間である。この方法の別の実施形態では、少なくとも8時間という期間が24時間である。
本発明の態様の1つは、有効成分としてクロルフェニラミンとヒドロコドンとプソイドエフェドリンとを含む非液状経口医薬製剤を包含し、前記製剤により有効成分は即放性(IR)及び徐放性(ER)を示し、
前記製剤が即放性部位と徐放性部位とを有し、
前記非液状経口製剤を患者へ単回投与することにより、少なくとも8時間にわたるクロルフェニラミンとヒドロコドンとプソイドエフェドリンの血漿中濃度が、クロルフェニラミン、ヒドロコドン及び/又はプソイドエフェドリンを含有する即放性組成物1種以上を同じ時間にわたって2回以上投与した際に得られる血漿中濃度と生物学的等価となる。
【0093】
前記製剤の実施形態の1つは、少なくとも8時間という期間が12時間である。別の実施形態では、少なくとも8時間という期間が24時間である。
本発明の態様は、前記非液状経口医薬製剤の製造方法に関するもので、クロルフェニラミンとヒドロコドンとを含み、プソイドエフェドリンを含まない即放性部位を調製する工程を含む。
本発明の別の実施形態は前記非液状経口医薬製剤の製造方法であって、徐放性部位の調製工程を有し、該徐放性部位の調製工程が、1個1個の粒子、ペレット又はビーズがクロルフェニラミンとヒドロコドンとプソイドエフェドリンとを含有する、粒子、ペレット又はビーズを調製することを含み、更に、前記徐放性部位を前記即放性部位と混合する工程を含む製造方法である。
実施形態によっては、前記方法が、粒子、ペレット又はビーズを皮膜でコーティングする工程を、前記徐放性部位と即放性部位との混合工程の前に更に含む。
本発明の態様は、第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物に関するもので、
第1の部位が、クロルフェニラミンとヒドロコドンを含み、更にプソイドエフェドリンを含んでもよい有効成分を即放剤形で含有し、
第2の部位が、有効成分としてクロルフェニラミンとヒドロコドンとプソイドエフェドリンとを徐放剤形で含む粒子、ペレット又はビーズで、
【0094】
前記3種の有効成分が24時間を超えて安定した血漿中濃度であるように前記非液状薬剤組成物を十分な投与回数で患者に投与することにより、前記有効成分の血漿中濃度が、前記有効成分を含むFDA承認済みの即放性薬剤組成物1種以上を上記と同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となり、
前記適切な投与回数とは、FDA承認済みの即放性薬剤組成物1種以上の同じ期間にわたる投与に関して、1つ以上のFDAが承認するのラベルのなかで推奨される投与回数に対応する。
本発明の別の態様は、第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物に関するもので、
第1の部位が、クロルフェニラミンとヒドロコドンを含み、更にプソイドエフェドリンを含んでもよい有効成分を即放剤形で含有し、
第2の部位が、有効成分としてクロルフェニラミンとヒドロコドンとプソイドエフェドリンとを徐放剤形で含有する、粒子、ペレット又はビーズを含み、
前記非液状薬剤組成物を患者へ単回投与することにより、少なくとも8時間にわたって得られる上記3種の有効成分の血漿中濃度が、前記3種すべての有効成分を含むFDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となり、
前記適切な投与回数とは、IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みのラベルで推奨される投与回数に対応する。
【0095】
本発明の更に別の態様は、(1)クロルフェニラミン とヒドロコドンとを有効成分として含有する即放性(IR)部位と、(2)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとを有効成分として含む徐放性(ER)部位とを有する非液状経口医薬組成物に関するもので、
経口組成物におけるIR部位のクロルフェニラミン対ER部位のクロルフェニラミンの質量比が約25:75、IR部位のヒドロコドン対ER部位のヒドロコドンの質量比が約25:75、IR部位のプソイドエフェドリン対ER部位のプソイドエフェドリンの質量比が約0:100であって、
前記非液状経口組成物をヒトへ単回投与することにより、前記経口組成物に存在するヒドロコドンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を2回以上投与した際に得られるAUCinfinityと同等のヒドロコドンのAUCinfinityが得られ、
前記非液状経口組成物をヒトへ単回投与することにより、前記経口組成物に存在するプソイドエフェドリンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を2回以上投与した際に得られるAUCinfinityと同等のプソイドエフェドリンのAUCinfinityが得られる。
前記非液状経口組成物の実施形態によっては、非液状経口組成物をヒトへ単回投与することにより、前記経口組成物に存在するクロルフェニラミンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を2回以上投与した際に得られるAUCinfinityと同等のクロルフェニラミンのAUCinfinityが得られる。
【0096】
本発明の更に別の態様は、(1)クロルフェニラミン とヒドロコドンとを有効成分として含有する即放性(IR)部位と、(2)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとを有効成分として含む徐放性(ER)部位とを有する非液状経口医薬組成物に関するもので、
経口組成物におけるIR部位のクロルフェニラミン対ER部位のクロルフェニラミンの質量比が約25:75、IR部位のヒドロコドン対ER部位のヒドロコドンの質量比が約25:75、IR部位のプソイドエフェドリン対ER部位のプソイドエフェドリンの質量比が約0:100であって、
前記非液状経口組成物ではヒトにおけるヒドロコドンのAUCinfinityが、この経口組成物に比べてヒドロコドンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等であることを示し、12時間にわたり前記経口組成物は1回投与し、前記IR-RLDは時間がゼロのときと、6時間後の2回投与するもので、
前記非液状経口組成物ではヒトにおけるプソイドエフェドリンのAUCinfinityが、この経口組成物に比べてプソイドエフェドリンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等であることを示し、12時間にわたり前記経口組成物は1回投与し、前記IR-RLDは時間がゼロのときと、6時間後の2回投与するものである。
【0097】
実施形態により、前記非液状経口組成物ではヒトにおけるクロルフェニラミンのAUCinfinityが、この経口組成物に比べてクロルフェニラミンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等であることを示し、12時間にわたり前記経口組成物は1回投与し、前記IR-RLDは時間がゼロのときと、6時間後の2回投与するものである。
本発明の実施形態の1つは、ヒトの咳、風邪、流感又はアレルギーの兆候を治療する方法に関し、本願明細書前記の非液状経口徐放性薬剤組成物の1つを被験者に投与することを含む。
前記方法の実施形態では、医薬組成物が、1日1回又は1日2回投与ができる二段階投与組成物としてヒトに投与される。
本発明の態様によると、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを有効成分として含む非液状経口徐放性薬剤組成物に関するもので、血漿分析によると、3種類すべての有効成分の十分なAUCinfinityが得られ、この組成物はヒトに単回投与した後、少なくとも8時間にわたり治療効果が得られる。
本発明の態様によると、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとを有効成分として含む非液状経口徐放性薬剤組成物に関するもので、血漿分析によると、3種類すべての有効成分の十分なAUCinfinityが得られ、この組成物はヒトに単回投与した後、少なくとも8時間にわたる治療効果が得られる。
【0098】
前記態様の文脈での「治療効果」という用語は、風邪、流感又はアレルギーに対する効果を意味し、咳の重篤度及び/又は咳の回数、咳の兆候、鼻漏、鬱血又はくしゃみの低減などの兆候緩和をはじめとする(が制限されない)効果、及び/又は、自覚的なよい状態の改善につながる他の生物学的効果を意味する。
前記非液状薬剤組成物の実施形態の1つでは、少なくとも8時間という期間が12時間である。前記非液状薬剤組成物の別の実施形態では、少なくとも8時間という期間が24時間である。
本発明の実施形態の1つは、非液状経口徐放性薬剤組成物中に存在するプソイドエフェドリン又はエフェドリンの抽出、単離又は分離の可能性を阻止又は低減する方法を包含し、該方法は、
第1の部位と第2の部位とを含むように非液状経口徐放性薬剤組成物を調製する工程であって、
第1の部位が、即放剤形として、抗ヒスタミン剤、鎮咳剤又はその両方を含み、更にプソイドエフェドリン又はエフェドリンを含んでいてもよく、
第2の部位が粒子、ペレット又はビーズを含み、各粒子、ペレット又はビーズが有効成分としてプソイドエフェドリン又はエフェドリン、及び、抗ヒスタミン剤、鎮咳剤又はその両方を徐放剤形として含み、
更に、経口徐放性薬剤組成物中に存在するプソイドエフェドリン又はエフェドリンの抽出、単離又は分離が行えないようする、又は行いにくくする工程を含む。
【0099】
前記方法の具体的実施形態の1つは、鎮咳剤がヒドロコドンで、かつ、該方法が、非液状経口徐放性薬剤組成物に存在するヒドロコドンの抽出、単離又は分離が行えないようする、又は行いにくくする工程を更に含む。
別の実施形態において、前記方法は、プソイドエフェドリン又はエフェドリンの非液状経口徐放性薬剤組成物からの抽出、単離又は分離を、プソイドエフェドリン又はエフェドリンを含む即放性組成物と比べ、よりむずかしくする製造工程を更に含む。
本発明の実施形態の1つは、非液状経口徐放性薬剤組成物中に存在するプソイドエフェドリン又はエフェドリンの乱用の潜在的可能性を低減する方法に関し、該方法が、
第1の部位と第2の部位とを含むように非液状経口徐放性薬剤組成物を調製する工程を含む方法で、
前記第1の部位が、即放剤形として、抗ヒスタミン剤、鎮咳剤又はその両方を含み、更にプソイドエフェドリン又はエフェドリンを含んでいてもよく、
前記第2の部位が、有効成分として抗ヒスタミン剤、鎮咳剤及びプソイドエフェドリン又はエフェドリンを徐放剤形として含む粒子、ペレット又はビーズを含有する。
本発明の他の実施形態は、非液状経口徐放性薬剤組成物中に存在する麻酔性鎮咳剤又はプソイドエフェドリンの乱用の潜在的可能性を低減する方法に関するもので、該方法は、麻酔性鎮咳剤及びプソイドエフェドリンを有効成分として徐放剤形として含む粒子、ペレット又はビーズを有するように非液状経口徐放性薬剤組成物を調製する工程を含む。
【0100】
本発明の態様の1つは、風邪、流感又はアレルギーの兆候の治療に使用する固形の経口徐放性調合医薬製剤の製造方法に関するもので、プソイドエフェドリン又はエフェドリンを含有する即放性(IR)製剤に比べて、製剤中に含まれるプソイドエフェドリン又はエフェドリンに関する乱用の潜在的可能性を低減するもので、前記製造方法は、
粒子、ペレット又はビーズを含むように固形の経口徐放性薬剤組成物を調製する工程を含み、個々の粒子、ペレット又はビーズには2種以上の有効成分が徐放剤形で含まれ、また、その有効成分の少なくとも1種がプソイドエフェドリン又はエフェドリンであり、その有効成分の少なくとも1種はプソイドエフェドリン又はエフェドリンではない。
本発明の別の態様は、固形の経口徐放性薬剤組成物中に存在するプソイドエフェドリン又はエフェドリンの抽出、単離又は分離の可能性を阻止又は低減する方法に関するもので、該方法は、
粒子、ペレット又はビーズを含む固形の経口徐放性薬剤組成物を調製する工程を含み、個々の粒子、ペレット又はビーズの中には2種以上の医薬的に許容できる有効成分が徐放剤形で含まれ、その有効成分の少なくとも1種がプソイドエフェドリン又はエフェドリンであり、その有効成分の少なくとも1種はプソイドエフェドリン又はエフェドリンではない。
【0101】
本発明の別の態様は、ある哺乳動物において、少なくとも8時間にわたって3種の有効成分の血漿中濃度が、FDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり同じ哺乳動物に適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となるようにする方法に関するもので、該方法は、
(A)第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物であって、第1の部位が即放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよく、第2の部位が、有効成分として抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む個々の粒子、ペレット又はビーズを徐放剤形として含有する、非液状経口徐放薬剤組成物を前記哺乳動物に投与することと、
(B)少なくとも8時間にわたる前記3種の有効成分の血漿中濃度が、前記有効成分を含有するFDA承認済みのIR-RLD組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価になるようにすることとを含み、前記適切な投与回数とは、IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みの1つ以上のラベルのなかで推奨される投与回数に対応する。
本発明の別の態様は、ある哺乳動物において、少なくとも8時間にわたってクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンの血漿中濃度が、クロルフェニラミン、ヒドロコドン及び/又はプソイドエフェドリンを含有する即放性(IR)組成物1種以上を同じ期間にわたり2回以上同じ哺乳動物に投与した際に得られる血漿中濃度と生物学的等価となるようにする方法に関するもので、該方法は、
【0102】
(A)有効成分がクロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる単一の経口医薬製剤であって、前記有効成分が即放性(IR)と徐放性(ER)を示し、即放性(IR)部位と徐放性(ER)部位とを有する製剤を前記哺乳動物に投与することと、
(B)少なくとも8時間にわたるクロルフェニラミンとヒドロコドンとプソイドエフェドリンの血漿中濃度が、クロルフェニラミン、ヒドロコドン及び/又はプソイドエフェドリンを含有する即放性(IR)組成物1種以上を同じ期間にわたり2回以上投与した際に得られる血漿中濃度と生物学的等価になるようにすることとを含む。
前記方法の実施形態の1つでは、少なくとも8時間という期間が12時間である。前記方法の別の実施形態では、少なくとも8時間という期間が24時間である。
本発明の更に別の態様は、非液状の経口徐放性(ER)薬剤組成物の投与後、抗ヒスタミン剤、鎮咳剤及び充血緩和剤の安定した血漿中濃度を哺乳動物において得るための方法であって、前記血漿中濃度とは、抗ヒスタミン剤、鎮咳剤及び/又は充血緩和剤を含む即放性(IR)組成物1種以上を同じ哺乳動物に投与した際に得られる血漿中濃度と生物学的等価で、該方法は、
第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物であって、第1の部位が、抗ヒスタミン剤と鎮咳剤とを含み、更に充血緩和剤を含んでもよい有効成分を即放剤形で含有し、第2の部位が、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを有効成分として徐放剤形で含む粒子、ペレット又はビーズである、非液状経口徐放性薬剤組成物を前記哺乳動物に投与することを含み、
【0103】
前記3種の有効成分が24時間を超えて安定した血漿中濃度であるように前記経口徐放性薬剤組成物を十分な投与回数で哺乳動物に投与することにより、前記有効成分を含有するFDA承認済みの即放性(IR)薬剤組成物1種以上を上記と同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価な有効成分の血漿中濃度となり、
前記FDA承認済みのIR薬剤組成物1種以上の適切な投与回数とは、FDA承認済みのIR薬剤組成物1種以上の同じ期間にわたる投与に関して、FDAが承認するの1つ以上ラベルのなかで推奨される投与回数に対応し、
前記FDA承認済みのIR薬剤組成物1種以上の適切な投与回数が、経口ER薬剤組成物の十分な投与回数を上回るものである。
本発明の別の態様は、ある哺乳動物において、少なくとも8時間にわたって抗ヒスタミン剤、鎮咳剤及び充血緩和剤の血漿中濃度が、前記3種すべての有効成分を含有するFDA承認済み即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり同じ哺乳動物に適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価になるようにする方法に関し、該方法は、
(A)第1の部位と第2の部位とを有する非液状経口徐放性薬剤組成物であって、
前記第1の部位が、抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよい有効成分を即放剤形で含有し、
前記第2の部位が、抗ヒスタミン剤と鎮咳剤と充血緩和剤とを有効成分として徐放剤形で含む粒子、ペレット又はビーズである、非液状経口徐放性薬剤組成物を前記哺乳動物に単回投与することと、
【0104】
(B)少なくとも8時間にわたり前記3種すべての有効成分の血漿中濃度が、3種すべての有効成分を含有するFDA承認済みのIR-RLD組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価にすることとを含み、
前記適切な投与回数とは、前記IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みのラベルで推奨される投与回数に対応する。
本発明の他の態様は、被験哺乳動物におけるヒドロコドンとプソイドエフェドリンのAUCinfinityを得る方法に関するもので、該方法は、
(A)少なくとも8時間という期間の最初に、(1)有効成分がクロルフェニラミンとヒドロコドンとからなる即放性(IR)部位と、(2)有効成分がクロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる徐放性(ER)部位とを有する徐放性(ER)経口医薬組成物を単回投与する工程であって、
該経口組成物におけるIR部位のクロルフェニラミン対ER部位のクロルフェニラミンの質量比が約25:75、IR部位のヒドロコドン対ER部位のヒドロコドンの質量比が約25:75、IR部位のプソイドエフェドリン対ER部位のプソイドエフェドリンの質量比が約0:100である工程と、
(B)前記経口組成物に存在するヒドロコドンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を前記と同じ期間にわたり2回以上投与した際に得られるAUCinfinityと同等のヒドロコドンのAUCinfinityを被験哺乳動物内に獲得する工程と、
【0105】
(C)前記経口組成物に存在するプソイドエフェドリンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を前記と同じ期間にわたり2回以上投与した際に得られるAUCinfinityと同等のプソイドエフェドリンのAUCinfinityを被験哺乳動物内に獲得する工程とを含む。
前記方法の実施形態の1つは、前記経口組成物に存在するプソイドエフェドリンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を前記と同じ期間にわたり2回以上投与した際に得られるAUCinfinityと同等のクロルフェニラミンのAUCinfinityを被験哺乳動物内に獲得する工程を更に含む。
本発明の別の態様は、被験哺乳動物におけるヒドロコドンとプソイドエフェドリンのAUCinfinityを得る方法に関するもので、該方法は、
(A)(1)有効成分がクロルフェニラミンとヒドロコドンとからなる即放性(IR)部位と、(2)有効成分がクロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる徐放性(ER)部位とを有する徐放性(ER)経口医薬組成物を投与する工程であって、
該経口組成物におけるIR部位のクロルフェニラミン対ER部位のクロルフェニラミンの質量比が約25:75、IR部位のヒドロコドン対ER部位のヒドロコドンの質量比が約25:75、IR部位のプソイドエフェドリン対ER部位のプソイドエフェドリンの質量比が約0:100である工程と、
【0106】
(B)前記経口組成物に比べてヒドロコドンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等のヒドロコドンのAUCinfinityを被験哺乳動物内に獲得する工程であって、前記経口組成物は12時間にわたる期間のなかで1回投与し、前記IR-RLDは時間がゼロのときと6時間後の2回投与する、工程と、
(C)前記経口組成物に比べてプソイドエフェドリンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等のプソイドエフェドリンのAUCinfinityを被験哺乳動物内に獲得する工程であって、前記経口組成物は12時間にわたる期間のなかで1回投与し、前記IR-RLDは時間がゼロのときと6時間後の2回投与する、工程とを有する。
前記方法の実施形態の1つは、(D)前記経口組成物に比べてクロルフェニラミンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等のクロルフェニラミンのAUCinfinityを被験哺乳動物内に獲得する工程であって、前記経口組成物は12時間にわたる期間のなかで1回投与し、前記IR-RLDは時間がゼロのときと6時間後の2回投与する、工程を更に有する。
本発明の更に別な態様は、非液状単一薬剤組成物をヒトに単回投与した後、少なくとも8時間にわたり、治療効果を得るのに十分な抗ヒスタミン剤、鎮咳剤、充血緩和剤のAUCinfinityを提供する方法であって、該方法は、(A)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとを有効成分として含む単一の非液状経口徐放性薬剤組成物をヒトに単回投与する工程と、(B)血漿分析により、単回投与後少なくとも8時間にわたり、ヒト被験者において治療効果が検出されるのに十分な3種類すべての有効成分のAUCinfinityを獲得する工程とを含む。
【0107】
本発明の具体的実施形態は、単一薬剤組成物をヒトに単回投与した後、少なくとも8時間にわたりヒト被験者の治療効果を得るのに十分なクロルフェニラミンとヒドロコドンとプソイドエフェドリンのAUCinfinityを提供する方法であって、該方法は、(A)有効成分がクロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる単一の経口徐放性薬剤組成物をヒト被験者に単回投与する工程と、(B)血漿分析により、単回投与後少なくとも8時間にわたり、ヒト被験者において治療効果が検出されるのに十分な3種類すべての有効成分のAUCinfinityを獲得する工程とを含む。
前記方法の実施形態の1つは、少なくとも8時間という期間が12時間である。前記方法の別の実施形態は、少なくとも8時間という期間が24時間である。
本発明の実施形態は、第1の部位と第2の部位とを有する固形の経口徐放性薬剤組成物に関するもので、
第1の部位が即放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤を含み、更に充血緩和剤を含んでもよく、
第2の部位が徐放性の剤形で有効成分として抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む粒子、ペレット又はビーズを含有し、
前記経口薬剤組成物を患者に単回投与することにより、少なくとも8時間にわたる前記3種の有効成分の血漿中濃度が、前記有効成分を含有するFDA承認済みのIR-RLD組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となるもので、
前記適切な投与回数とは、前記IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みラベル1つ以上のなかで推奨される投与回数に対応する。
【0108】
本発明の実施形態の1つは、風邪、流感又はアレルギーの兆候の治療に使用する固形の経口徐放性調合医薬製剤の製造方法を包含するもので、該製剤は製剤中のいずれの有効成分についても、その有効成分を含有する即放性(IR)製剤に比べて、乱用の潜在的可能性を低減させており、前記製造方法は、
粒子、ペレット又はビーズを含むように固形の経口徐放性薬剤組成物を調製する工程を含み、各粒子、ペレット又はビーズには2種以上の有効成分が徐放剤形で含まれている。
本発明の更に別の実施形態は、固形の経口徐放性薬剤組成物から1つの有効成分が抽出、単離又は分離できる可能性を阻止又は低減する方法に関するもので、該方法は、
粒子、ペレット又はビーズを含み、その個々の粒子、ペレット又はビーズに2種以上の医薬的に許容できる有効成分が徐放剤形で含まれる、固形の経口徐放性薬剤組成物を調製する工程を含む。
本発明の実施形態は、非液状の経口徐放性薬剤組成物から、含有されているプソイドエフェドリン又はエフェドリンが抽出、単離又は分離できる可能性を阻止又は低減する方法に関するもので、該方法は、粒子、ペレット又はビーズを含み、その個々の粒子、ペレット又はビーズにプソイドエフェドリン又はエフェドリン及び抗ヒスタミン剤又は鎮咳剤又はその両方が有効成分として徐放剤形で含まれる、非液状の経口徐放性薬剤組成物を調製する工程を含む。
【0109】
本発明の実施形態は、非液状の経口徐放性薬剤組成物に含まれるプソイドエフェドリン又はエフェドリンの乱用の潜在的可能性を低減する方法に関するもので、該方法は、粒子、ペレット又はビーズを含み、その個々の粒子、ペレット又はビーズに抗ヒスタミン剤及び鎮咳剤及びプソイドエフェドリン又はエフェドリンが有効成分として徐放剤形で含まれる、非液状の経口徐放性薬剤組成物を調製する工程を含む。
別の具体的な実施形態では、非液状の経口徐放性薬剤組成物は、抗ヒスタミン剤、鎮咳剤及び充血緩和剤からなる有効成分を含有する。このような実施形態において、非液状薬剤組成物の即放性部位は、抗ヒスタミン剤と鎮咳剤とからなり、更に充血緩和剤を含んでいてもよい有効成分を含有する。別の実施形態は、非液状の薬剤組成物の徐放性部位が、抗ヒスタミン剤、鎮咳剤及び充血緩和剤からなる有効成分を含む。
更なる具体的な実施形態では、経口徐放性薬剤組成物が、クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンからなる有効成分を含む。このような実施形態の1つでは、薬剤組成物の即放性部位が、クロルフェニラミンとヒドロコドンからなり、さらにプソイドエフェドリンを含んでもよい有効成分を含有する。別の実施形態では、薬剤組成物の徐放性部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む。
【0110】
以下の実施例は、本発明を理解するうえでの助けとするために記載するもので、本願明細書で記載及びクレームする本発明を具体的に限定するものとみなすべきではない。現在公知であるか、又は、当該分野の当業者が理解しうる範囲で後に展開される、あらゆる同等物の置換をはじめとする本発明の変更や、製剤の変更又は実験デザインにおける小さな変更は本発明の範囲内である。
実施例1
実施例1は「製剤X」の製造方法を記載する。「製剤X」は、咳、風邪及びアレルギーの兆候の治療を意図した徐放性被覆ぺレット含有シロップ剤の分散液である。製剤Xは、成人1回服用量(5ml)につき、15mgのヒドロコドン重酒石酸塩(HC、中枢作用性鎮咳剤)と、120mgのプソイドエフェドリン塩酸塩(PSE、交感神経作用様鼻充血緩和剤)と、8mgのクロルフェニラミンマレイン酸塩(CPM、抗ヒスタミン剤)とを一緒に含む。この製剤においては、塩の形態の薬剤が使用された。この製剤は、製造後、(4オンス)の使用単位容器(unit-of-use containers)に分けられた。
表1は、徐放性ペレット含有シロップ剤のIR/ER分散液である製剤Xの定量的組成の一例をあらわす表で、1回の投与量5ml(6.55g)を質量基準で表わしたものである。
【0111】
【表1】
【0112】
EPペレットにおけるAPI濃度に対するIRシロップにおけるAPI濃度の比は、現在市場に出ている即放性薬剤と生物学的等価になるような血漿の薬剤バイオアベイラビリティーが12時間得られるように設定された。
EPペレットにおけるAPI濃度に対するIRシロップにおけるAPI濃度の比は、現在市場に出ている即放性薬剤と生物学的等価になるような血漿の薬剤バイオアベイラビリティーが12時間得られるように設定された。
製剤Xのコアペレットの製造
製剤XのER成分にはコアペレットが含まれる。コアペレットの調製方法を以下に記載する。
0.207kgのクロルフェニラミンマレイン酸塩、4.138kgのプソイドエフェドリン塩酸塩、0.3879kgのヒドロコドン重酒石酸塩、2.500kgのラクトース 一水和物、0.5000kgのアルギン酸ナトリウム、17.27kgの微晶質セルロースを一緒に高剪断ミキサーに投入した。ミキサーの羽根車を200RPM、チョッパーを1500RPMにして5分間運転した。この粉体混合の後、約10.00kgの純水をおよそ1L/分の速度でミキサーに引き入れた。この間、チョッパーは使わず、羽根車は200 RPMで運転した。水を全量添加した後、羽根車は200RPM、チョッパーは1500RPMにして湿式造粒を1分間続けた。ミキサーから出した後、水分を含んだ塊を少なくとも30分間、開いた袋の中に放置した。
その後、0.7mmのスクリーンを有するドーム型一軸スクリュー押出機を45RPMで運転し、得られた水分を含む塊の押出成形を行った。この水分を含む塊の全量を処理するまで押出を続けた。
【0113】
スフェロナイザー(spheronizer)を使用し、3x3mmの円錐の先端を切ったパターン(truncated pattern)を有するディスクを約1000rpmで運転して、反復バッチ処理を行ってペレットを形成した。約3.8kgのバッチを9つ使い、球体化(spheronization)の運転はバッチごとにおよそ90秒にわたった。
球体化した材料を流動床乾燥機に入れた。このビーズを、入り口開始空気温度を60℃、毎分の全空気量を1500立方フィート( 約42.3m3)にして乾燥させた。引き続き、入り口空気温度と空気量を工程中に調整して適切な流動化を維持し、製品温度を50〜60℃に保持した。ビーズの水分が2%以下になるまで乾燥を続けた。流動床乾燥機を冷風モードで十分な時間運転して製品を室温に戻した後、乾燥したビーズを排出した。
自動ふるい振盪機のNo.22とNo.34のスクリーンを1200RPMで運転して乾燥ビーズの分粒を行った。No.22のスクリーンを通過し、No.34のスクリーンに残留したコアペレットを合格品とみなした。
製剤X用ペレットのコーティング
製剤Xのコアペレットのコーティング方法を以下に説明する。
【0114】
18.65kgのアンモニオメタクリレート共重合体タイプB分散液(オイドラギットRS30D)を、No. 20のスクリーンを使ってステンレス鋼製容器に投入した。0.7774kgのアンモニオメタクリレート共重合体タイプA分散液(オイドラギットRL30D)をNo. 20のスクリーンを使って同じ容器に投入した。この分散液の混合物をプロペラミキサーを約1000rpmにして混合した。別の容器に0.6087kgのクエン酸トリエチルと10.92kgの純水とを混合した。この混合液も、プロペラミキサーを使って、液中に空気をいれずに渦を作るのに十分な回転数で混合した。混合を続けながら、1.812kgのタルクをクエン酸トリエチルと水との混合物に添加した。5分間混合した後、撹拌器を止めて取り出した。クエン酸トリエチル、タルク及び水の混合物を、メタクリレート共重合体分散液が入った容器に、撹拌を続けながら移した。このコーティング系は、コーティング作業を通して十分に撹拌を続けて均一な分散液を維持した。
コアペレットのコーティングは、250umスクリーン付きのDベースプレートを使って、12インチのワースター(Wurster)型容器を有する流動床装置Glatt GPCG 30で行った。 カラム高さは底面から約2.5インチに設定し、開口部が1mmのノズルを使った。コアビーズ21.75kgを流動床装置に投入し、初期パラメータは以下のように設定した。入り口空気温度の目標範囲:40〜50℃、製品温度の目標範囲:27〜32℃、噴霧気圧の目標範囲:1.5〜2.5bar、空気体積の目標範囲:250〜450cfm、空気体積の合計:1500cfm、フィルターバッグの振盪回数:240秒ごとに5秒間。工程中に調整を行い、処理条件が範囲内に保たれるようにした。コーティング用分散液の噴霧は最初は20g/分の速さで行った。処理を続けるなかで、噴霧速度は最終的に100g/分まで上げた。
【0115】
コーティング用分散液をすべて適用した後、流動床装置を冷風モードで運転し、被覆ビーズを室温に戻してから排出した。その後、被覆ビーズとタルク75.00gとを5分間混合して排出した。
被覆ペレットは熱対流炉で硬化した。被覆ペレットをトレイにまき散らし、炉に入れた。炉の温度は55℃に設定し、ビーズを炉内に約16時間放置した。約16時間後、炉の電源を切りペレットを室温まで放冷した。
被覆ビーズの分粒は、 自動ふるい振盪機のNo.16とNo.34のスクリーンを1200RPMで運転して行った。No.16のスクリーンを通過し、No.34のスクリーンに残留した被覆ペレットを合格品とした。
製剤Xのコアペレット用ビヒクルの調製
製剤XのIR成分は液体を含み、製剤Xの「ビヒクル」又は「ビヒクルシロップ剤」とも呼ばれる。ビヒクルシロップ剤の製造方法は以下の通りである。
60リットルを収容するのに十分な容量のジャケット付きステンレス鋼製容器に、純水25.09kgを投入した。循環水温度制御装置を使って、作業を通して液体の温度を制御し、プロペラミキサーを使って撹拌した。カバーで水の蒸発を抑えた。液体中に空気が入らないように渦ができる速度で混合しながら、水を約70℃に加熱した。No.30のスクリーンを通過したプロピルパラベン0.01080kgを水に加え、プロピルパラベンが完全に溶解するまで混合を続けた。プロピルパラベンの溶解後、温度制御装置を50℃に設定し、ショ糖48.00kgをゆっくり容器に添加した。容器に蓋をして、ショ糖がすべて溶けるまで混合を続けた。
【0116】
ショ糖が完全に溶解したところで、温度制御装置を装置の最低温度に設定して、溶液を25℃まで放冷した。0.02642kgのクロルフェニラミンマレイン酸塩、0.04954kgのヒドロコドン重酒石酸塩、0.2680kgの人工イチゴ風味剤(粉末)、0.2160gの人工苦味矯味剤(粉末)、0.0720kgのスクラロースをシロップ剤に添加した。すべての固形物が溶解するまで混合を続けた。更に、撹拌を続けながらFD&C Red No.40アルミニウムレーキ0.0720kgと二酸化チタン0.1800kgをNo.30のスクリーンを使って容器に加え、均一な分散液を得た。(注:このビヒクルの例は、混合を停止すると沈殿する不溶物質を含む。)
この混合物の最終的な正味質量を求め、必要に応じて蒸発損失を埋め合わせるために純水を加えた。
製剤Xのための被覆ペレットとビヒクルシロップ剤との混合
被覆ペレットとビヒクルシロップ剤を混合して最終製品の製剤Xを作成する方法の1つを以下に説明する。
渦が形成されるようにビヒクルシロップ剤を一定して撹拌した。この撹拌を少なくとも10分行った後、24.00gの被覆ビーズを130.8gのビヒクルに添加し、服用量24回分又は120mLの製品を得た。
【0117】
実施例2 ヒトでの有効性
実施例2では製剤Xのヒトにおける有効性を評価するために行った試験について記載する。具体的には、実施例2では、徐放性製剤Xの単回投与と、HC、PSE又はCPMを含む即放性RLDを組み合わせたものの2回投与とを比較するために、健康な被験者16名で行った試験について記載する。この試験の目的の一つは、製剤Xの2つの製剤と、対応するRLDとの生物的等価性を求めることであった。
吸収
ヒドロコドン
ヒドロコドンは経口によりよく吸収されるが、腸や肝臓での代謝を伴う著しい初回通過効果を受ける。既に公表されている試験によると、10mgのHCを即放性経口剤形で5人の男性被験者に1回投与したところ、平均ピーク血漿濃度は23.6±5.2ng/mLで、Tmaxはおよそ1.3±0.3時間であった。「Hycodan(登録商標)」(www.rxmed.comにて入手可能、2008年6月23日アクセス);Stout, P., Farrell, L. 「Opioids - Effects on Human Performance and Behavior(オピオイド-ヒトの行動、挙動への影響)」Forensic Science Review. 15(1): 29-59 (2003)。すべてのヒドロコドン代謝産物は活性であり、ヒドロモルホン、ノルコデイン及び6-αと6-βヒドロキシ代謝産物を含む。Micromedex Health Care Series、DrugDex Evaluations「ヒドロコドン重酒石酸塩/イブプロフェン」http://www.thomsonhc.com/hcsにて閲覧可能、2008年7月1日アクセス、Cone等「Comparative metabolism of hydrocodone in man, rat, guinea pig, rabbit, and dog(ヒト、ネズミ、モルモット、ウサギ及びイヌにおける比較によるヒドロコドンの代謝)」Drug Metab Dispos 6:488-493 (1978)引用。
【0118】
表2は、「製剤X」の単回投与に対してHCの関連リスト収載医薬品(RLD)2回投与を行った際の、患者のヒドロコドン(HC)血漿中濃度に関するパラメータ、AUCinfinity、Cmax及びT1/2等の比較表である。治療Aとは、15mgのHC、120mgのPSE及び8mgのCPMを含有する「製剤X」の単回投与に対応し、治療Cは、7.5mgのHC、60mgのPSE及び4mgのCPMを含む、3つの単体RLDの混合液を2回投与することに相当する。
本試験では(表2の治療A及びCで表示されているように)、15mgのHCと120mgのPSEと8mgのCPMとを含む製剤Xを16人の被験者に投与した後のHCの平均ピーク血漿濃度は(Cmax)は17.54±4.75ng/mLであった。これに対し、7.5mgのHCを含有するRLDを0時間と6時間で2回投与した場合は、平均ピーク血漿濃度が25.64±6.56ng/mLであった。製剤Xの場合のTmax中央値は4時間であり、それに対してRLDの2回投与の場合のTmax中央値は7時間であった。製剤XのTmaxが科学文献にみられるもの比べた時に異なるのは、製剤X中の徐放性ペレットが時間をかけてHCを放出し、12時間の投薬を実現している事実による。この試験ではRLDを2回投与したため(既に公表されている試験では1回投与である)、RLDの2回投与で得られたピーク血漿濃度(Cmax)は製剤Xの場合よりも遅い時間に到達されている。
【0119】
【表2】
治療A:テスト製剤No.1 - 製剤X1q:15mgのHC、120mgのPSE、8mgのCPM
治療B:テスト製剤No.2 - 製剤X1q:10mgのHC、120mgのPSE、8mgのCPM
治療C:参考製剤No.1(治療A用)-RLD2q:7.5mgのHC、60mgのPSE、4mgのCPM
治療D:参考製剤No.2(治療B用)-RLD2q:5mgのHC、60mgのPSE、4mgのCPM
AUC=0時間から無限時間まで外挿した血漿中濃度−時間曲線下面積;AUCLAST= 0時間から量化できる最終血漿濃度までの血漿中濃度−時間曲線下面積;CMAX= 実測された最高血漿中濃度;CPM=クロルフェニラミンマレイン酸塩;HC=ヒドロコドン重酒石酸塩;PSE=プソイドエフェドリン塩酸塩;TMAX=最高血漿中濃度到達時間、T1/2=消失半減期、a=対数変換値の分析による指数結果
【0120】
プソイドエフェドリン
プソイドエフェドリンは消化管から容易に、かつ、ほとんど完全に吸収され、初回通過代謝を証明するものはない。既に公表されている試験では、Tmaxは投与から1.5〜2.4時間で見られ、バイオアベイラビリティーはすべての製剤で同じであり、食物による影響を受けない。Micromedex Health Care Series、DrugDex Evaluations「プソイドエフェドリン」http://www.thomsonhc.com/hcsにて閲覧可能、2008年7月1日アクセス;「Common cold and influenza management(普通の風邪とインフルエンザの管理)」MedScape(www.medscape.com/viewarticle/466063_3で閲覧可能、2008年6月26日アクセス);Graves DA等「Influence of a standard meal on the absorption of a controlled release pseudoephedrine suspension(標準的食事が及ぼす放出制御型プソイドエフェドリン懸濁液の吸収への影響)」Biopharm Drug Dispos. 5月-6月; 9(3):267-72 (1988)。
表3は、「製剤X」の単回投与に対してPSEのRLDを2回投与した場合の患者のプソイドエフェドリン(PSE)血漿中濃度に関するパラメータ、AUCinfinity、Cmax及びT1/2等の比較表である。治療Aとは、15mgのHC、120mgのPSE及び8mgのCPMを含有する「製剤X」の単回投与に対応し、治療Cは、60mgのPSE 、7.5mgのHC及び4mgのCPMを含む、3つの単体RLDの混合液を2回投与することに相当する。
【0121】
本試験では(表3の治療A及びCで表示されているように)、15mgのHCと120mgのPSEと8mgのCPMとを含む製剤Xを16人のヒト被験者に投与した後、PSEの平均ピーク血漿濃度は(Cmax)は292.05±49.94ng/mLであった。これに対し、60mgのPSEをそれぞれ含有する即放性RLDを2回投与した後は、平均ピーク血漿濃度が345.47±78.46ng/mLであった。製剤X投与の場合のTmax中央値は5時間であり、それに対してRLD投与後のTmax中央値は7時間であった。上記のごとく、製剤X中の徐放性ペレットが時間をかけてPSEを放出することで12時間の投薬になる。結果、この試験におけるRLDのピーク血漿濃度は、より遅い時間に到達されている。
【0122】
【表3】
治療A:テスト製剤No.1 - 製剤X1q:15mgのHC、120mgのPSE、8mgのCPM
治療B:テスト製剤No.2 - 製剤X1q:10mgのHC、120mgのPSE、8mgのCPM
治療C:参考製剤No.1(治療A用)- RLD2q:7.5mgのHC、60mgのPSE、4mgのCPM
治療D:参考製剤No.2(治療B用)- RLD2q:5mgのHC、60mgのPSE、4mgのCPM
AUC=0時間から無限時間まで外挿した血漿中濃度−時間曲線下面積;AUCLAST= 0時間から量化できる最終血漿濃度までの血漿中濃度−時間曲線下面積;CMAX= 実測された最高血漿中濃度;CPM=クロルフェニラミンマレイン酸塩;HC=ヒドロコドン重酒石酸塩;PSE=プソイドエフェドリン塩酸塩;TMAX=最高血漿中濃度到達時間、T1/2=消失半減期、a=対数変換値の分析による指数結果
【0123】
クロルフェニラミン
クロルフェニラミンは、経口投与後、容易かつ完全に吸収される。既に公表されている試験では、この薬剤は30〜60分以内に全身循環に現れ、2時間後にCmaxに到達し、その後の46時間で血漿濃度が低下する。Peets, E等「Metabolism of chlorpheniramine maleate in man (クロルフェニラミンマレイン酸塩の人体における代謝)」J. Pharmacol. Exp. Ther. 180:464-474-(1972);Micromedex Health Care Series、DrugDex Evaluations「クロルフェニラミン」http://www.thomsonhc.com/hcsで閲覧可能、2008年7月1日アクセス。クロルフェニラミンは経口バイオアベイラビリティーが41±16%で、食物摂取によりその吸収は遅くなるが、バイオアベイラビリティーは遅くならない。Rumore, M. M「Clinical pharmacokinetics of chlorpheniramine (クロルフェニラミンの臨床薬物動態学)」Drug Intel. Clin. Pharm. 18:701-707 (1984)。しかしながら、クロルフェニラミンは吸収中に胃腸粘膜で、また、肝臓での初回通過時に実質的に代謝されると考えられる。限られたデータであるが、従来の錠剤状のCPMを経口単回投与した場合は約25〜45%、溶剤の場合は35〜60%が薬剤の変化をおこさずに全身循環に到達することが示されている。また、限られたデータではあるが、従来の錠剤又は経口用液剤と比較して、この薬剤の徐放性調剤のバイオアベイラビリティーは低減する可能性も示されている。Micromedex Health Care Series:クロルフェニラミン(同サイト);Therapeutic Goods Administration(オーストラリア)「Core sedating antihistamines product information(抗ヒスタミン剤投与情報)」www.tga.gov.au/npmeds/pi-sedatingantihistamine.rtfにて閲覧可能、2008年6月26日アクセス。
【0124】
表4は、「製剤X」の単回投与に対してCPMのRLDを2回投与した場合の患者のクロルフェニラミン(CPM)血漿中濃度に関するパラメータ、AUCinfinity、Cmax及びT1/2等の比較表である。治療Aとは、15mgのHC、120mgのPSE及び8mgのCPMを含有する「製剤X」の単回投与に対応し、治療Cは、4mgのCPM 、7.5mgのHC及び60mgのPSEを含む、3つの単体RLDの混合液を2回投与することに相当する。
本試験では(表4の治療A及びCで表示されているように)、15mgのHCと120mgのPSEと8mgのCPMとを含む製剤Xを16人のヒト被験者に投与した後のCPMの平均ピーク血漿濃度は(Cmax)は21.20±6.30ng/mLであった。これに対し、4mgのCPMをそれぞれ含有するRLDを2回投与した後は、平均ピーク血漿濃度が28.89±7.92ng/mLであった。製剤X投与後9時間でTmax中央値が得られたが、RLD投与の場合は、9.50時間後であった。
【0125】
【表4】
治療A:テスト製剤No.1 - 製剤X1q:15mgのHC、120mgのPSE、8mgのCPM
治療B:テスト製剤No.2 - 製剤X1q:10mgのHC、120mgのPSE、8mgのCPM
治療C:参考製剤No.1(治療A用)- RLD2q:7.5mgのHC、60mgのPSE、4mgのCPM
治療D:参考製剤No.2(治療B用)- RLD2q:5mgのHC、60mgのPSE、4mgのCPM
AUC=0時間から無限時間まで外挿した血漿中濃度−時間曲線下面積;AUCLAST= 0時間から量化できる最終血漿濃度までの血漿中濃度−時間曲線下面積;CMAX= 実測された最高血漿中濃度;CPM=クロルフェニラミンマレイン酸塩;HC=ヒドロコドン重酒石酸塩;PSE=プソイドエフェドリン塩酸塩;TMAX=最高血漿中濃度到達時間、T1/2=消失半減期、a=対数変換値の分析による指数結果
【0126】
排出及び排泄
ヒドロコドン
ヒドロコドン の親化合物の排出半減期は3.8〜4.5時間である。Micromedex Health Care Series: ヒドロコドン、経口単回投与から最初の24時間で全排出のおよそ70%(同サイト); Stout, P.; Farrell, L.「Opioids - Effects on Human Performance and Behavior (オピオイド-ヒトの行動、挙動への影響)」 Forensic Science Review, 15(1): 29-59. (2003)
本試験では(表2の治療A及びCで表示されているように)、HCについての製剤XのT2/1を測定したところ7.22±1.45時間であった。これに対してRLDの場合は4.38±0.83時間であった。この差は、製剤Xが徐放性薬剤であることに因ると考えられた。
プソイドエフェドリン
PSEとその代謝産物は尿に排出され、服用量の90%までが投与から24時間以内にそのまま排出される。PSEの半減期はおよそ9〜16時間で、半減期は尿のpH値に影響されることがあり、アルカリ(pH8)の場合は延び、酸性(pH5)の場合は短くなる。Wishart, D.等「DrugBank: a comprehensive resource for in silico drug discovery and exploration(コンピュータによる薬物の発見と調査のための包括的リソース」Nucleic Acids Res. 34: D668-72 (2006);Micromedex Health Care Series: プソイドエフェドリン(同サイト)。
【0127】
本試験では(表3の治療A及びCで表示されているように)、PSEについての製剤XのT2/1の測定結果は6.48±1.40時間であった。これに対してRLDの場合は5.06±0.90時間であった。この差は、製剤Xが徐放性薬剤であることに因ると考えられた。
クロルフェニラミン
クロルフェニラミンの体内からの排出は、主にモノデスメチル化合物やジデスメチル化合物に代謝され、26%までが尿に排出される。腎臓による排出が全排出のおよそ50%を占め、3〜18%は薬剤が変化していない。腎臓による排出は尿流量が増えるほど、また、pH値が低いほど増加する。Micromedex Health Care Series:クロルフェニラミン(同サイト)参照のこと。1%未満が大便に排出される。CPMの半減期は20±5時間で、測定クリアランスは1.7±0.1mL/分/kgである。Rumore(同サイト)
本試験では(表4の治療A及びCで表示されているように)、CPMについての製剤XのT2/1は41.71±43.33時間であった。これに対してRLDの場合は19.37±9.66時間であった。しかしながら、サンプリングプロトコルでは、製剤X投与後24時間しかCPMを計測しなかった。わずか24時間の計測では、製剤XにおけるCPMの排出半減期を正確に測定するには不十分だったことが、T1/2にみられる変動性の一因となったのかもしれない。生物学的等価性は検出されるものと考えられる。
【0128】
表2〜表4に示すデータは、TmaxとT1/2について、必ずしも既に公表されている前記の数値を反映していない。その理由は、製剤Xが徐放性製剤だからである。TmaxとT1/2の公表されている数値は、血液中へ即座に取り込まれた後に移動することができる薬剤の数値である。製剤Xはすべての薬剤を即時的に放出しないのでTmaxは遅く、投与後数時間は薬剤が一定して足されているためT1/2とTmaxは変動するようである。同様に、表2〜表4に示されているRLDの数値も必ずしも公知の数値を反映していない。これは、既に公表されている試験では単回投与で数値を求めていたのに対し、表中の数値は6時間間隔で2回投与しているからである。本試験におけるRLDの2回目の投与が薬剤濃度の2つ目の急激なピークの理由である。結果としては、RLDの2回目の投与後、確かな薬剤ピーク濃度が得られる。結果、表2〜表4のRLDについてのTmaxは明らかに遅くなっているが、これは単に投与プロトコルに起因しているにすぎない。
【0129】
クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンの生物学的等価性
FDAは、単回投与試験の場合、AUCが生物学的等価(BE)を示すための唯一の適切な薬物動態学的(PK)パラメータとしているが、FDAは他のPKパラメータについての情報を要求している。表2〜表4には、製剤Xの2つの異なる処方とそれぞれに対応するRLDについてのPKデータが記載されている。表2は、HCについて、治療A(HC15mgを含む製剤X)と治療C(HC7.5mgを含むRLDを2回投与)を比較し、製剤XのAUCについての点推定はRLDの93.28%で、90%信頼区間(CI)が84.82〜102.58%であることを示している。これはFDAのBE標準である90%信頼区間(CI)の80〜125%を満たす。そのため、この試験ではHCは生物学的等価性を達成した。表3は、PSEについて治療Aと治療Cを比較し、製剤XのAUCについての点推定がRLDの95.45%で、90%信頼区間(CI)が88.55〜102.88%であることを示すので、生物学的等価性を達成している。表4は、CPMについて治療Aと治療Cを比較し、製剤XのAUCについての点推定がRLDの129.24%で、90%信頼区間(CI)が97.33〜171.60%であることを示している。この数値はBEを立証していない。しかしながら、本試験においてCPMのBEが確立できなかったのは、製剤XがCPM(HC及びPSEに加え)のBEを立証できなかったからではなく、本試験では、CPMの長い半減期を血漿採取中に考慮していなかったことによるものである。具体的に言うならば、最後の血漿サンプルの回収時、正確なT1/2が求められるところまでCPM濃度はまだ減衰していなかった。結果、AUCを求めるために必要な外挿が信頼性を欠いた。これについては、CMPのRLDと製剤Xが当てはまる。今後の試験によって、試料採取時間を適切なものにすれば、CPMのデータから製剤XがCPMのRLDと生物学的等価であることを示すことができよう。
【0130】
つまり、製剤XにおけるAUCの90%信頼限界は、HC及びPSEについてはRLDの80〜125%の範囲であり、少なくともこの2種の薬剤の生物学的等価性を示している。本試験では、CPMについてはAUCが明確な生物学的等価性を立証しなかった。これは、製剤Xの90%信頼区分が、CPMのRLDと比較すると97.33〜171.60%の範囲だったからである。すべての製剤にみられる高い個体内変動性とAUCO-INFとAUCO-LASTとの大きな差により、CPMのAUC分析は複雑であった。変動性にむけたテストサンプルの増大化とサンプリング時間の延長により、CPMの長い排出半減期(ある資料によると20〜24時間)を斟酌すれば、CPM含有RLDに対する製剤XのCPMの生物学的等価性は立証されるであろう。「Drug information: クロルフェニラミン」www.accessmedicine.com/ drugs. aspx?index=Cにて閲覧可能、2007年5月15日アクセス。
【0131】
即放性(IR)シロップ剤、徐放性(ER)ビーズ、lR/ERビーズ含有懸濁液におけるAPI量比を定めるための分析用HPLC法
APIとして抗ヒスタミン剤と鎮咳剤と充血緩和剤とを含む製剤を調製する際、分析用の方法を用いてAPIの採用すべき量又は比率を定める。それにより、該製剤を患者に投与した際、製剤は、3種のAPIすべての放出について徐放性を示す。
当該分野の当業者が本発明を製造及び使用できるように、十分に詳しく本発明を説明し例示してきたが、本発明の精神及び範囲から逸脱せずに、様々な代替、変更及び改善できることは明白である。本願明細書に記載した例は特定の実施形態を代表するものであり、例示であり、本発明の範囲を制限することを意図するものではない。その範囲内の変更や他の使用も当該分野の当業者には思いつくであろう。それらの変更は本発明の精神の範囲内に含まれ、クレームの範囲により規定される。
当該分野の当業者にとっては、本発明の範囲及び精神から逸脱することなく本願明細書に開示の発明に代替や変更が行えることは容易にわかることである。本発明のいくつかの態様を説明することを目的とした実施例に開示の特定の実施形態によって、本発明はその範囲を制限されることはなく、機能的に均等である実施形態は本発明の範囲に入る。実際、本願明細書に示し、記載した変更にくわえて様々な変更がこの分野の当業者には明らかとなり、それらは添付のクレームの範囲に入るものである。
本明細書で言及した特許及び出版物のすべては、本発明が関連している分野の当業者のレベルを示している。すべての特許及び出版物は、各文献を個々に具体的に示して援用するのと同じ程度に、引用により本願明細書の記載に含まれるものとする。
【0132】
実施例3
実施例3は、製剤X中のプソイドエフェドリン、ヒドロコドン及びクロルフェニラミンの放出プロファイルを、個々のビーズにクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンを含有する被覆ビーズからの各薬剤の放出プロファイルと比較するために行った生体外テストの結果を説明する。
製剤Xは、実施例1で説明したようにして調製した。実施例1にしたがって、個々のビーズがクロルフェニラミンとヒドロコドンとプソイドエフェドリンとを含む、被覆ビーズを調製した。時間ゼロサンプル(t=0)は、調製終了後から1週間以内に分析を行ったが、他のサンプルは、調製終了後から、湿度60%のチャンバーにおいて25℃で保存し(25/60)、1ヵ月(1mo)、3ヵ月(3mo)、6ヵ月(6mo)、9ヵ月(9mo)及び12ヵ月(12mo)後にそれぞれ分析した。高速液体クロマトグラフィー(HPLC)により定量化を行った。米国薬局方対応装置2(USP Apparatus 2)のパドルを100RPMで運転して、放出テストを行った。最初に、pH1.2のバッファー750mLを各容器に投入した。1.5時間後、pH調整液を250mL加えてpH6.8、容量1000mLとした。オートサンプラーは、0.5、4、8、12時間の時点でおよそ5mlのサンプルを抜き取るようにプログラムした。
【0133】
図1(A)は、製剤Xの液状放出持続型懸濁液からのプソイドエフェドリンの放出プロファイルを示す。図1(B)は、被覆ビーズからのプソイドエフェドリンの放出プロファイルを示す。図2(A)は、製剤Xの液状放出持続型懸濁液からのヒドロコドンの放出プロファイルを示す。図2(B)は、被覆ビーズからのヒドロコドンの放出プロファイルを示す。図3(A)は、製剤Xの液状放出持続型懸濁液からのクロルフェニラミンの放出プロファイルを示す。図3(B)は、被覆ビーズからのクロルフェニラミンの放出プロファイルを示す。
図1〜図3によると、製剤Xにおける各薬剤の即放性部位の存在について結果を正規化させると、製剤Xのプソイドエフェドリン、ヒドロコドン及びクロルフェニラミンの徐放性部位の放出速度は、被覆ビーズからのプソイドエフェドリン、ヒドロコドン及びクロルフェニラミンの放出速度と同等であることがわかる。図1〜図3に示された実験結果から、即放相中のイオン成分の存在、即ち、それぞれに塩の形態をとる遊離な薬剤の存在が、製剤Xの徐放性部位から持続的に放出される各薬剤の適切な放出速度を妨げていないことがわかる。また、図1〜図3は、タイムゼロの時点でテストした製剤Xのプソイドエフェドリン、ヒドロコドン及びクロルフェニラミンの徐放性部位の放出速度が、製造後1ヵ月、3ヵ月、6ヵ月、9ヵ月及び12ヵ月の間保存した製剤Xのプソイドエフェドリン、ヒドロコドン及びクロルフェニラミンの徐放性部位の放出速度と同等であることを示している。このように、図1〜図3に示す実験結果から、製剤Xは、1ヵ月、3ヵ月、6ヵ月、9ヵ月及び12ヵ月間室温で保存した後も安定性を保ち、各薬剤の持続性放出の適切な速度が保たれていることがわかる。
【0134】
実施例4
実施例4では、薬剤を塩基の形態で分散相に含む液状放出制御型組成物の放出プロファイルを、薬剤を塩の形態で分散相に含む液状放出制御型組成物の放出プロファイルと比較するために行った生体外テストの結果を説明する。
テストは2種類の組成物を使って行った。それぞれ、5mlの液体剤形で、プソイドエフェドリン塩酸塩30mg(IRが7.5mg、ERが22.5mg)、ヒドロコドン重酒石酸塩7.5mg(IRが1.87、ERが5.63mg)及びクロルフェニラミンマレイン酸塩6mg(IRが1.5mg、ERが4.5mg)相当を放出するようにした。第1の組成物は、18.4mgのプソイドエフェドリン、3.41mgのヒドロコドン及び1.58mgのクロルフェニラミンを含み、これらの薬剤は塩基の形態で、アルギン酸樹脂に結合させた。薬剤充填アルギネートビーズを作り、コーティングを行い、65%w/wのショ糖を含む分散媒に分散させた。第2の組成物は、22.5mgのプソイドフェドリン塩酸塩、5.62mgのヒドロコドン重酒石酸塩及び4.5mgのクロルフェニラミンマレイン酸塩を含み、アルギン酸ナトリウムに結合させた。第2の組成物においては、コアビーズを製造し、本質的には実施例1記載のようにコーティングを行い、できた被覆ビーズを65%w/wのショ糖を含む分散媒に分散させた。両者とも、即放性とした設計した塩の形態の薬剤の一回分投与部位が分散媒に含まれた。
【0135】
分析に先立ち、サンプルを室温で3週間保存した。その後、HPLCで定量化した。米国薬局方対応装置2のパドルを100RPMで運転させ、放出テストを行った。最初に、pH1.2のバッファー750mLを各容器に投入した。1.5時間後、pH調整液を250mL加えてpH6.8、容量1000mLとした。オートサンプラーは、以下の時点、5分、30分、60分、90分、4時間、8時間、12時間、16時間、20時間、24時間におよそ1mlを抜き取るようにプログラムした。
図4(A)は、プソイドエフェドリンの塩基性製剤の放出プロファイルを示す。 図4(B)は、プソイドエフェドリンの塩製剤の放出プロファイルを示す。図5(A)は、ヒドロコドンの塩基性製剤の放出プロファイルを示す。 図5(B)は、ヒドロコドンの塩製剤の放出プロファイルを示す。図6(A)は、クロルフェニラミンの塩基性製剤の放出プロファイルを示す。 図6(B)は、クロルフェニラミンの塩製剤の放出プロファイルを示す。
図4〜図6によると、液状放出持続型組成物においては、塩の形態のプソイドエフェドリン、ヒドロコドン及びクロルフェニラミンの放出速度は、塩基の形態のプソイドエフェドリン、ヒドロコドン及びクロルフェニラミンの放出速度とそれぞれ同等であることがわかる。詳細には、実験の各時点において、これらの薬剤が塩の形態でも塩基の形態でもほぼ同じか著しく異なることのない放出特性を示す。図4〜図6に示す実験結果から、これらの薬剤の適切な持続性放出プロファイルを得るために、塩の形態の薬剤を本発明の液状放出制御組成物に使用できることがわかる。
【0136】
実施例5
実施例5は、ヒドロコドンを唯一の有効成分として分散相に含む液状放出持続型組成物からのヒドロコドンの放出プロファイルを示す。
10mgのヒドロコドン塩基がアルギン酸マトリックスに結合して含まれる液状放出制御組成物を使って実験を行った。アルギン酸及び20%ラクトース一水和物に10mgのヒドロコドンが結合して含まれるように、ヒドロコドンアルギネートビーズを調製した。表5は、ヒドロコドンアルギネートビーズ製剤の定量組成の概要を示す。
【0137】
【表5】
【0138】
更に、ヒドロコドンアルギネートビーズを、1.5%Opadry/31.5%Eudragit RS30D(質量基準による質量)でコーティングした。被覆ヒドロコドンアルギネートビーズを、10mgのヒドロコドンに対して5mLのシロップ剤になるよう分散した。表6にヒドロコドン含有徐放性ペレットの分散液の定量組成の概略を示す。
【0139】
【表6】
【0140】
製造終了後、すぐにサンプルの含量検定を行った。HPLCで定量化した。米国薬局方対応装置2のパドルを100RPMで運転させ、放出テストを行った。最初に、pH1.2のバッファー750mLを各容器に投入した。1.5時間後、pH調整液を250mL加えてpH6.8、容量1000mLとした。オートサンプラーは、以下の時点、30分、1時間、1.5時間、2時間、3時間、4時間、6時間、8時間、10時間後におよそ1mlを抜き取るようにプログラムした。
図7は、アルギン酸マトリックに結合したヒドロコドンを含む液状放出制御組成物からのヒドロコドンの放出プロファイルを示す。
図7から、唯一の有効成分であるヒドロコドンをイオン交換マトリックスに結合させて含むビーズがシロップ剤に分散された液状放出持続性組成物からのヒドロコドンの放出速度がわかる。
【図1】
【図2】
【図3】
【図4】
【図5】
【図6】
【特許請求の範囲】
【請求項1】
クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含有する経口徐放薬剤組成物であって、前記薬剤組成物が第1の部位と第2の部位とを有し、
前記第1の部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、
前記第2の部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズを含み、
前記薬剤組成物を患者に単回投与すると、少なくとも8時間にわたって前記3種の有効成分の血漿中濃度が、前記有効成分を含むFDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価になり、
前記適切な投与回数が、前記IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みの1つ以上のラベルで推奨されている投与回数に対応する、経口徐放薬剤組成物。
【請求項2】
前記少なくとも8時間にわたる期間が12時間である、請求項1記載の薬剤組成物。
【請求項3】
前記少なくとも8時間にわたる期間が24時間である、請求項1記載の薬剤組成物。
【請求項4】
前記第1の部位において、前記有効成分がクロルフェニラミンとヒドロコドンとからなる、請求項1記載の薬剤組成物。
【請求項5】
前記薬剤組成物が経口懸濁液剤である、請求項1記載の薬剤組成物。
【請求項6】
前記薬剤組成物が経口固形剤形である、請求項1記載の薬剤組成物。
【請求項7】
前記薬剤組成物が経口カプセル剤形である、請求項1記載の薬剤組成物。
【請求項8】
前記粒子、ペレット又はビーズが更に皮膜を有する、請求項1記載の薬剤組成物。
【請求項9】
前記粒子、ペレット又はビーズが更に医薬的に許容できるイオン交換マトリックスを含み、前記クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンが前記イオン交換マトリックスと結合している、請求項1記載の薬剤組成物。
【請求項10】
甘味料、香味料、着色剤及びこれらの混合物からなる群から選択される賦形剤を更に含有する、請求項1記載の薬剤組成物。
【請求項11】
安定化剤、分散助剤及びこれらの混合物からなる群から選択される添加剤を更に含有する、請求項1記載の薬剤組成物。
【請求項12】
前記少なくとも8時間にわたる期間が12時間であり、かつ、単回投与5mlに対し、前記有効成分が、8〜12mgのクロルフェニラミンマレイン酸塩と、10〜15mgのヒドロコドン重酒石酸塩と、少なくとも120mgのプソイドエフェドリンとからなる、請求項1記載の薬剤組成物。
【請求項13】
前記少なくとも8時間という期間が24時間であり、かつ、単回投与5mlに対し、前記有効成分が、16〜24mgのクロルフェニラミンマレイン酸塩と、20〜30mgのヒドロコドン重酒石酸塩と、少なくとも240mgのプソイドエフェドリン塩酸塩とからなる、請求項1記載の薬剤組成物。
【請求項14】
風邪、流感又はアレルギーに関連した咳、咳の兆候、鼻漏、鬱血又はくしゃみを少なくとも8時間にわたり治療する方法で、このような治療を必要とするヒトに対し、風邪又はアレルギーに関連した咳、咳の兆候、鼻漏、鬱血又はくしゃみを少なくとも8時間にわたり治療するのに有効な、請求項1記載の薬剤組成物を単回投与することを含む方法。
【請求項15】
前記少なくとも8時間にわたる期間が12時間である、請求項4記載の方法。
【請求項16】
前記少なくとも8時間にわたる期間が24時間である、請求項4記載の方法。
【請求項17】
クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む、経口医薬製剤であって、前記製剤の前記有効成分が即放性(IR)と徐放性(ER)を示し、
前記製剤が即放性部位と徐放性部位とを有し、
前記即放性部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、
前記徐放性部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズを含み、
前記経口製剤を患者に単回投与すると、少なくとも8時間にわたってクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンの血漿中濃度が、クロルフェニラミン、ヒドロコドン及び/又はプソイドエフェドリンを含む即放性(IR)組成物1種以上を同じ期間にわたり2回以上投与した際に得られる血漿中濃度と生物学的等価となる、経口医薬製剤。
【請求項18】
前記少なくとも8時間にわたる期間が12時間である、請求項17記載の経口製剤。
【請求項19】
前記少なくとも8時間にわたる期間が24時間である、請求項17記載の経口製剤。
【請求項20】
前記製剤が経口懸濁液剤である、請求項17記載の経口製剤。
【請求項21】
請求項17記載の経口製剤の製造方法であって、前記即放性部位の前記有効成分がクロルフェニラミンとヒドロコドンとからなる、前記即放性部位を調製する工程を含む製造方法。
【請求項22】
請求項17記載の経口製剤の製造方法であって、前記徐放性部位を調製する工程を含み、 前記徐放性部位調製工程が、個々の粒子、ペレット又はビーズの有効成分がクロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる、粒子、ペレット又はビーズを作製する工程を有し、
前記製造方法が、前記徐放性部位と前記即放性部位とを混合する工程を更に含む製造方法。
【請求項23】
前記粒子、ペレット又はビーズを皮膜でコーティングする工程を、前記徐放性部位と前記即放性部位との混合工程の前に含む、請求項22記載の方法。
【請求項24】
クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む経口徐放性薬剤組成物であって、前記薬剤組成物が第1の部位と第2の部位とを有し、
前記第1の部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、
前記第2の部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含む粒子、ペレット又はビーズであり、
前記3種の有効成分が24時間を超えて安定した血漿中濃度であるように前記薬剤組成物を十分な投与回数で患者に投与した場合、前記有効成分を含むFDA承認済みの即放性薬剤組成物1種以上を上記と同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となる前記有効成分の血漿中濃度が提供され、
前記適切な投与回数が、前記FDA承認済みの即放性薬剤組成物1種以上の同じ期間にわたる投与に関して、FDA承認済みの1つ以上のラベルで推奨されている投与回数に対応する、経口徐放性薬剤組成物。
【請求項25】
クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む経口徐放性薬剤組成物であって、前記薬剤組成物が第1の部位と第2の部位とを有し、
前記第1の部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、
前記第2の部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズを含み、
前記薬剤組成物を患者へ単回投与すると、少なくとも8時間にわたって前記3種の有効成分の血漿中濃度が、前記3種の有効成分すべてを含有するFDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価になり、前記適切な投与回数が、前記IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みのラベルで推奨されている投与回数に対応する、経口徐放性薬剤組成物。
【請求項26】
(1)クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を含有する即放性(IR)部位と、(2)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む徐放性(ER)部位とを有する経口医薬組成物であって、
前記経口組成物における前記IR部位のクロルフェニラミン対前記ER部位のクロルフェニラミンの質量比が約25:75、前記IR部位のヒドロコドン対前記ER部位のヒドロコドンの質量比が約25:75、前記IR部位のプソイドエフェドリン対前記ER部位のプソイドエフェドリンの質量比が約0:100であって、
前記経口組成物をヒトへ単回投与することにより、前記経口組成物に存在するヒドロコドンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を2回以上投与した際に得られるAUCinfinityと同等のヒドロコドンのAUCinfinityが得られ、
前記経口組成物をヒトへ単回投与することにより、前記経口組成物に存在するプソイドエフェドリンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を2回以上投与した際に得られるAUCinfinityと同等のプソイドエフェドリンのAUCinfinityが得られる、経口医薬組成物。
【請求項27】
前記経口組成物をヒトへ単回投与することにより、前記経口組成物に存在するクロルフェニラミンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を2回以上投与した際に得られるAUCinfinityと同等のクロルフェニラミンのAUCinfinityが得られる、請求項26記載の経口組成物。
【請求項28】
(1)クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を含有する即放性(IR)部位と、(2)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む徐放性(ER)部位とを有する経口医薬組成物であって、
前記経口組成物における前記IR部位のクロルフェニラミン対前記ER部位のクロルフェニラミンの質量比が約25:75、前記IR部位のヒドロコドン対前記ER部位のヒドロコドンの質量比が約25:75、前記IR部位のプソイドエフェドリン対前記ER部位のプソイドエフェドリンの質量比が約0:100であり、
ヒトにおけるヒドロコドンのAUCinfinityが、前記経口組成物に比べてヒドロコドンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと、12時間にわたるなかで前記経口組成物は1回投与、前記IR-RLDは時間がゼロのときと6時間後の2回投与で、同等であることを前記経口組成物が示し、
ヒトにおけるプソイドエフェドリンのAUCinfinityが、前記経口組成物に比べてプソイドエフェドリンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと、12時間にわたるなかで前記経口組成物は1回投与、前記IR-RLDは時間がゼロのときと6時間後の2回投与で同等であることを前記経口組成物が示す、経口医薬組成物。
【請求項29】
ヒトにおけるクロルフェニラミンのAUCinfinityが、前記経口組成物に比べてクロルフェニラミンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと、12時間にわたるなかで前記経口組成物は1回投与、前記IR-RLDは時間がゼロのときと6時間後の2回投与で、同等であることを示す、請求項28記載の経口医薬組成物。
【請求項30】
ヒトにおける咳、風邪、流感又はアレルギーの兆候を治療する方法で、請求項1記載の経口徐放性薬剤組成物をヒトに投与する工程を含む方法。
【請求項31】
前記医薬組成物が、1日1回又は1日2回投与を可能にする二段階放出(dual release)製剤としてヒトに投与される、請求項30に記載の方法。
【請求項32】
クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む経口徐放性薬剤組成物であって、前記薬剤組成物が第1の部位と第2の部位とを有し、
前記第1の部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、
前記第2の部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズを含み、
血漿分析によると、ヒトに前記薬剤組成物を単回投与した後、少なくとも8時間にわたる治療効果を得るのに十分な3種類すべての有効成分のAUCinfinityが提供される、経口徐放性薬剤組成物。
【請求項33】
前記少なくとも8時間にわたる期間が12時間である、請求項32記載の薬剤組成物。
【請求項34】
前記少なくとも8時間にわたる期間が24時間である、請求項32記載の薬剤組成物。
【請求項35】
哺乳動物において、少なくとも8時間にわたるクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンの血漿中濃度を、FDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で同じ哺乳動物に投与した際に得られる血漿中濃度と生物学的等価にする方法であって、前記方法が、
(A)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含有する経口徐放性薬剤組成物であって、第1の部位と第2の部位とを有し、前記第1の部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、前記第2の部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズを含む、経口徐放性薬剤組成物を前記哺乳動物に投与することと、
(B)少なくとも8時間にわたる前記3種の有効成分の血漿中濃度を、前記有効成分を含有するFDAに承認されたIR-RLD組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価にすることを含み、前記適切な投与回数が、前記IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みの1つ以上のラベルで推奨されている投与回数に対応する、前記方法。
【請求項36】
哺乳動物において、少なくとも8時間にわたるクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンの血漿中濃度が、クロルフェニラミン、ヒドロコドン及び/又はプソイドエフェドリンを含む即放性(IR)組成物1種以上を同じ期間にわたり同じ哺乳動物に2回以上投与した際に得られる血漿中濃度と生物学的等価とする方法であって、
(A)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含有する単一の経口医薬製剤であって、前記有効成分が即放性(IR)と徐放性(ER)とを示し、前記製剤がIR部位とER部位とを有し、
前記IR部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、
前記ER部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズを含む、経口医薬製剤を前記哺乳動物に投与することと、
(B)少なくとも8時間にわたるクロルフェニラミンとヒドロコドンとプソイドエフェドリンの血漿中濃度を、クロルフェニラミン、ヒドロコドン及び/又はプソイドエフェドリンを含有するIR組成物1種以上を同じ期間にわたり2回以上投与した際に得られる血漿中濃度と生物学的等価にすることを含む、前記方法。
【請求項37】
前記少なくとも8時間にわたる期間が12時間である、請求項36記載の方法。
【請求項38】
前記少なくとも8時間にわたる期間が24時間である、請求項36記載の方法。
【請求項39】
哺乳動物に経口徐放性(ER)薬剤組成物の投与後、クロルフェニラミンとヒドロコドンとプソイドエフェドリンの安定した血漿中濃度を得る方法であって、前記血漿中濃度とは、抗ヒスタミン剤、鎮咳剤及び/又は充血緩和剤戸を含む即放性 (IR)組成物1種以上を同じ哺乳動物に投与した際に得られる血漿中濃度と生物学的等価な濃度であり、前記方法が、
第1の部位と第2の部位とを有する経口徐放性薬剤組成物であって、前記第1の部位がクロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、前記第2の部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズである経口徐放性薬剤組成物を、前記哺乳動物に投与することを含む方法であり、
前記3種の有効成分が24時間を超えて安定した血漿中濃度になるように前記経口徐放性薬剤組成物を十分な投与回数で前記哺乳動物に投与した場合、前記有効成分を含有するFDA承認済みの即放性(IR)薬剤組成物1種以上を上記と同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となる、前記有効成分の血漿中濃度が提供され、
前記FDA承認済みのIR薬剤組成物1種以上の適切な投与回数が、前記FDA承認済みのIR薬剤組成物1種以上の同じ期間にわたる投与に関して、FDA承認済みの1つ以上のラベルの中で推奨されている投与回数に対応し、
前記FDA承認済みのIR薬剤組成物1種以上の適切な投与回数が、前記経口ER薬剤組成物の十分な投与回数を上回っている、方法。
【請求項40】
哺乳動物において、少なくとも8時間にわたる有効成分としてのクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンの血漿中濃度が、前記3種すべての有効成分を含むFDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で同じ哺乳動物に投与した際に得られる血漿中濃度と生物学的等価とする方法であって、前記方法が、
(A)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含有する経口徐放性(ER)薬剤組成物であって、第1の部位と第2の部位とを有し、
前記第1の部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、
前記第2の部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズである、経口徐放性(ER)薬剤組成物を前記哺乳動物に単回投与することと、
(B)少なくとも8時間にわたる前記3種すべての有効成分の血漿中濃度を、前記3種すべての有効成分を含むFDAに承認されたIR-RLD組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価にすることとを含む方法で、
前記適切な投与回数が、前記IR-RLD組成物の同じ期間にわたる投与に関して、FDA承認済みのラベルの中で推奨されている投与回数に対応する、方法。
【請求項41】
被験哺乳動物におけるヒドロコドンとプソイドエフェドリンのAUCinfinityの値を得る方法であって、前記方法が、
(A)(1)クロルフェニラミンとヒドロコドンとからなる有効成分を含む即放性(IR)部位と、(2)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む徐放性(ER)部位とを有する徐放性(ER)経口医薬組成物を、少なくとも8時間という期間の最初に単回投与する工程であって、
前記経口組成物における前記IR部位のクロルフェニラミン対前記ER部位のクロルフェニラミンの質量比が約25:75、前記IR部位のヒドロコドン対前記ER部位のヒドロコドンの質量比が約25:75、前記IR部位のプソイドエフェドリン対前記ER部位のプソイドエフェドリンの質量比が約0:100である、投与工程と、
(B)前記経口組成物に存在するヒドロコドンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を前記と同じ期間にわたり2回以上投与した際に得られるAUCinfinityと同等のヒドロコドンのAUCinfinityを、前記被験哺乳動物内に獲得する工程と、
(C)前記経口組成物に存在するプソイドエフェドリンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を前記と同じ期間にわたり2回以上投与した際に得られるAUCinfinityと同等のプソイドエフェドリンのAUCinfinityを、前記被験哺乳動物内に獲得する工程とを含む方法。
【請求項42】
前記方法が、(D)前記経口組成物に存在するプソイドエフェドリンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を前記と同じ期間にわたり2回以上投与した際に得られるAUCinfinityと同等のクロルフェニラミンのAUCinfinityを、前記被験哺乳動物内に獲得する工程を更に含む、請求項41記載の方法。
【請求項43】
被験哺乳動物におけるヒドロコドンとプソイドエフェドリンのAUCinfinityの値を得る方法であって、前記方法が、
(A)(1)クロルフェニラミンとヒドロコドンとからなる有効成分を含有する即放性(IR)部位と、(2)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含有する徐放性(ER)部位とを有する徐放性(ER)経口医薬組成物を投与する工程であって、
前記経口組成物における前記IR部位のクロルフェニラミン対前記ER部位のクロルフェニラミンの質量比が約25:75、前記IR部位のヒドロコドン対前記ER部位のヒドロコドンの質量比が約25:75、前記IR部位のプソイドエフェドリン対前記ER部位のプソイドエフェドリンの質量比が約0:100である投与工程と、
(B)前記経口組成物に比べてヒドロコドンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等のヒドロコドンのAUCinfinityを、12時間にわたるなかで前記経口組成物は1回投与、前記IR-RLDは時間がゼロのときと6時間後の2回投与で、被験哺乳動物内に獲得する工程と、
(C)前記経口組成物に比べてプソイドエフェドリンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等のプソイドエフェドリンのAUCinfinityを、12時間にわたるなかで前記経口組成物は1回投与、前記IR-RLDは時間がゼロのときと6時間後の2回投与で、被験哺乳動物内に獲得する工程とを有する方法。
【請求項44】
前記方法が、(D)前記経口組成物に比べてクロルフェニラミンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等のクロルフェニラミンのAUCinfinityを、12時間にわたるなかで前記経口組成物は1回投与、前記IR-RLDは時間がゼロのときと6時間後の2回投与で、被験哺乳動物内に獲得する工程を更に有する、請求項43記載の方法。
【請求項45】
ヒトへの単一の薬剤組成物の単回投与後、ヒトが治療効果を得るのに十分なクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンのAUCinfinityを少なくとも8時間にわたり提供する方法であって、前記方法が、
(A)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む単一の経口徐放性薬剤組成物であって、第1の部位と第2の部位とを有し、
前記第1の部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、
前記第2の部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズを含む前記薬剤組成物を、ヒトに単回投与する工程と、
(B)血漿分析により、単回投与後少なくとも8時間にわたり、ヒト被験者において治療効果が検出されるのに十分な3種類すべての有効成分のAUCinfinityを獲得する工程とを含む、前記方法。
【請求項46】
前記少なくとも8時間にわたる期間が12時間である、請求項45記載の方法。
【請求項47】
前記少なくとも8時間にわたる期間が24時間である、請求項45記載の方法。
【請求項1】
クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含有する経口徐放薬剤組成物であって、前記薬剤組成物が第1の部位と第2の部位とを有し、
前記第1の部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、
前記第2の部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズを含み、
前記薬剤組成物を患者に単回投与すると、少なくとも8時間にわたって前記3種の有効成分の血漿中濃度が、前記有効成分を含むFDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価になり、
前記適切な投与回数が、前記IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みの1つ以上のラベルで推奨されている投与回数に対応する、経口徐放薬剤組成物。
【請求項2】
前記少なくとも8時間にわたる期間が12時間である、請求項1記載の薬剤組成物。
【請求項3】
前記少なくとも8時間にわたる期間が24時間である、請求項1記載の薬剤組成物。
【請求項4】
前記第1の部位において、前記有効成分がクロルフェニラミンとヒドロコドンとからなる、請求項1記載の薬剤組成物。
【請求項5】
前記薬剤組成物が経口懸濁液剤である、請求項1記載の薬剤組成物。
【請求項6】
前記薬剤組成物が経口固形剤形である、請求項1記載の薬剤組成物。
【請求項7】
前記薬剤組成物が経口カプセル剤形である、請求項1記載の薬剤組成物。
【請求項8】
前記粒子、ペレット又はビーズが更に皮膜を有する、請求項1記載の薬剤組成物。
【請求項9】
前記粒子、ペレット又はビーズが更に医薬的に許容できるイオン交換マトリックスを含み、前記クロルフェニラミン、ヒドロコドン及びプソイドエフェドリンが前記イオン交換マトリックスと結合している、請求項1記載の薬剤組成物。
【請求項10】
甘味料、香味料、着色剤及びこれらの混合物からなる群から選択される賦形剤を更に含有する、請求項1記載の薬剤組成物。
【請求項11】
安定化剤、分散助剤及びこれらの混合物からなる群から選択される添加剤を更に含有する、請求項1記載の薬剤組成物。
【請求項12】
前記少なくとも8時間にわたる期間が12時間であり、かつ、単回投与5mlに対し、前記有効成分が、8〜12mgのクロルフェニラミンマレイン酸塩と、10〜15mgのヒドロコドン重酒石酸塩と、少なくとも120mgのプソイドエフェドリンとからなる、請求項1記載の薬剤組成物。
【請求項13】
前記少なくとも8時間という期間が24時間であり、かつ、単回投与5mlに対し、前記有効成分が、16〜24mgのクロルフェニラミンマレイン酸塩と、20〜30mgのヒドロコドン重酒石酸塩と、少なくとも240mgのプソイドエフェドリン塩酸塩とからなる、請求項1記載の薬剤組成物。
【請求項14】
風邪、流感又はアレルギーに関連した咳、咳の兆候、鼻漏、鬱血又はくしゃみを少なくとも8時間にわたり治療する方法で、このような治療を必要とするヒトに対し、風邪又はアレルギーに関連した咳、咳の兆候、鼻漏、鬱血又はくしゃみを少なくとも8時間にわたり治療するのに有効な、請求項1記載の薬剤組成物を単回投与することを含む方法。
【請求項15】
前記少なくとも8時間にわたる期間が12時間である、請求項4記載の方法。
【請求項16】
前記少なくとも8時間にわたる期間が24時間である、請求項4記載の方法。
【請求項17】
クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む、経口医薬製剤であって、前記製剤の前記有効成分が即放性(IR)と徐放性(ER)を示し、
前記製剤が即放性部位と徐放性部位とを有し、
前記即放性部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、
前記徐放性部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズを含み、
前記経口製剤を患者に単回投与すると、少なくとも8時間にわたってクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンの血漿中濃度が、クロルフェニラミン、ヒドロコドン及び/又はプソイドエフェドリンを含む即放性(IR)組成物1種以上を同じ期間にわたり2回以上投与した際に得られる血漿中濃度と生物学的等価となる、経口医薬製剤。
【請求項18】
前記少なくとも8時間にわたる期間が12時間である、請求項17記載の経口製剤。
【請求項19】
前記少なくとも8時間にわたる期間が24時間である、請求項17記載の経口製剤。
【請求項20】
前記製剤が経口懸濁液剤である、請求項17記載の経口製剤。
【請求項21】
請求項17記載の経口製剤の製造方法であって、前記即放性部位の前記有効成分がクロルフェニラミンとヒドロコドンとからなる、前記即放性部位を調製する工程を含む製造方法。
【請求項22】
請求項17記載の経口製剤の製造方法であって、前記徐放性部位を調製する工程を含み、 前記徐放性部位調製工程が、個々の粒子、ペレット又はビーズの有効成分がクロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる、粒子、ペレット又はビーズを作製する工程を有し、
前記製造方法が、前記徐放性部位と前記即放性部位とを混合する工程を更に含む製造方法。
【請求項23】
前記粒子、ペレット又はビーズを皮膜でコーティングする工程を、前記徐放性部位と前記即放性部位との混合工程の前に含む、請求項22記載の方法。
【請求項24】
クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む経口徐放性薬剤組成物であって、前記薬剤組成物が第1の部位と第2の部位とを有し、
前記第1の部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、
前記第2の部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含む粒子、ペレット又はビーズであり、
前記3種の有効成分が24時間を超えて安定した血漿中濃度であるように前記薬剤組成物を十分な投与回数で患者に投与した場合、前記有効成分を含むFDA承認済みの即放性薬剤組成物1種以上を上記と同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となる前記有効成分の血漿中濃度が提供され、
前記適切な投与回数が、前記FDA承認済みの即放性薬剤組成物1種以上の同じ期間にわたる投与に関して、FDA承認済みの1つ以上のラベルで推奨されている投与回数に対応する、経口徐放性薬剤組成物。
【請求項25】
クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む経口徐放性薬剤組成物であって、前記薬剤組成物が第1の部位と第2の部位とを有し、
前記第1の部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、
前記第2の部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズを含み、
前記薬剤組成物を患者へ単回投与すると、少なくとも8時間にわたって前記3種の有効成分の血漿中濃度が、前記3種の有効成分すべてを含有するFDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価になり、前記適切な投与回数が、前記IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みのラベルで推奨されている投与回数に対応する、経口徐放性薬剤組成物。
【請求項26】
(1)クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を含有する即放性(IR)部位と、(2)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む徐放性(ER)部位とを有する経口医薬組成物であって、
前記経口組成物における前記IR部位のクロルフェニラミン対前記ER部位のクロルフェニラミンの質量比が約25:75、前記IR部位のヒドロコドン対前記ER部位のヒドロコドンの質量比が約25:75、前記IR部位のプソイドエフェドリン対前記ER部位のプソイドエフェドリンの質量比が約0:100であって、
前記経口組成物をヒトへ単回投与することにより、前記経口組成物に存在するヒドロコドンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を2回以上投与した際に得られるAUCinfinityと同等のヒドロコドンのAUCinfinityが得られ、
前記経口組成物をヒトへ単回投与することにより、前記経口組成物に存在するプソイドエフェドリンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を2回以上投与した際に得られるAUCinfinityと同等のプソイドエフェドリンのAUCinfinityが得られる、経口医薬組成物。
【請求項27】
前記経口組成物をヒトへ単回投与することにより、前記経口組成物に存在するクロルフェニラミンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を2回以上投与した際に得られるAUCinfinityと同等のクロルフェニラミンのAUCinfinityが得られる、請求項26記載の経口組成物。
【請求項28】
(1)クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を含有する即放性(IR)部位と、(2)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む徐放性(ER)部位とを有する経口医薬組成物であって、
前記経口組成物における前記IR部位のクロルフェニラミン対前記ER部位のクロルフェニラミンの質量比が約25:75、前記IR部位のヒドロコドン対前記ER部位のヒドロコドンの質量比が約25:75、前記IR部位のプソイドエフェドリン対前記ER部位のプソイドエフェドリンの質量比が約0:100であり、
ヒトにおけるヒドロコドンのAUCinfinityが、前記経口組成物に比べてヒドロコドンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと、12時間にわたるなかで前記経口組成物は1回投与、前記IR-RLDは時間がゼロのときと6時間後の2回投与で、同等であることを前記経口組成物が示し、
ヒトにおけるプソイドエフェドリンのAUCinfinityが、前記経口組成物に比べてプソイドエフェドリンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと、12時間にわたるなかで前記経口組成物は1回投与、前記IR-RLDは時間がゼロのときと6時間後の2回投与で同等であることを前記経口組成物が示す、経口医薬組成物。
【請求項29】
ヒトにおけるクロルフェニラミンのAUCinfinityが、前記経口組成物に比べてクロルフェニラミンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと、12時間にわたるなかで前記経口組成物は1回投与、前記IR-RLDは時間がゼロのときと6時間後の2回投与で、同等であることを示す、請求項28記載の経口医薬組成物。
【請求項30】
ヒトにおける咳、風邪、流感又はアレルギーの兆候を治療する方法で、請求項1記載の経口徐放性薬剤組成物をヒトに投与する工程を含む方法。
【請求項31】
前記医薬組成物が、1日1回又は1日2回投与を可能にする二段階放出(dual release)製剤としてヒトに投与される、請求項30に記載の方法。
【請求項32】
クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む経口徐放性薬剤組成物であって、前記薬剤組成物が第1の部位と第2の部位とを有し、
前記第1の部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、
前記第2の部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズを含み、
血漿分析によると、ヒトに前記薬剤組成物を単回投与した後、少なくとも8時間にわたる治療効果を得るのに十分な3種類すべての有効成分のAUCinfinityが提供される、経口徐放性薬剤組成物。
【請求項33】
前記少なくとも8時間にわたる期間が12時間である、請求項32記載の薬剤組成物。
【請求項34】
前記少なくとも8時間にわたる期間が24時間である、請求項32記載の薬剤組成物。
【請求項35】
哺乳動物において、少なくとも8時間にわたるクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンの血漿中濃度を、FDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で同じ哺乳動物に投与した際に得られる血漿中濃度と生物学的等価にする方法であって、前記方法が、
(A)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含有する経口徐放性薬剤組成物であって、第1の部位と第2の部位とを有し、前記第1の部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、前記第2の部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズを含む、経口徐放性薬剤組成物を前記哺乳動物に投与することと、
(B)少なくとも8時間にわたる前記3種の有効成分の血漿中濃度を、前記有効成分を含有するFDAに承認されたIR-RLD組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価にすることを含み、前記適切な投与回数が、前記IR-RLD組成物の同じ期間にわたる投与に関し、FDA承認済みの1つ以上のラベルで推奨されている投与回数に対応する、前記方法。
【請求項36】
哺乳動物において、少なくとも8時間にわたるクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンの血漿中濃度が、クロルフェニラミン、ヒドロコドン及び/又はプソイドエフェドリンを含む即放性(IR)組成物1種以上を同じ期間にわたり同じ哺乳動物に2回以上投与した際に得られる血漿中濃度と生物学的等価とする方法であって、
(A)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含有する単一の経口医薬製剤であって、前記有効成分が即放性(IR)と徐放性(ER)とを示し、前記製剤がIR部位とER部位とを有し、
前記IR部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、
前記ER部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズを含む、経口医薬製剤を前記哺乳動物に投与することと、
(B)少なくとも8時間にわたるクロルフェニラミンとヒドロコドンとプソイドエフェドリンの血漿中濃度を、クロルフェニラミン、ヒドロコドン及び/又はプソイドエフェドリンを含有するIR組成物1種以上を同じ期間にわたり2回以上投与した際に得られる血漿中濃度と生物学的等価にすることを含む、前記方法。
【請求項37】
前記少なくとも8時間にわたる期間が12時間である、請求項36記載の方法。
【請求項38】
前記少なくとも8時間にわたる期間が24時間である、請求項36記載の方法。
【請求項39】
哺乳動物に経口徐放性(ER)薬剤組成物の投与後、クロルフェニラミンとヒドロコドンとプソイドエフェドリンの安定した血漿中濃度を得る方法であって、前記血漿中濃度とは、抗ヒスタミン剤、鎮咳剤及び/又は充血緩和剤戸を含む即放性 (IR)組成物1種以上を同じ哺乳動物に投与した際に得られる血漿中濃度と生物学的等価な濃度であり、前記方法が、
第1の部位と第2の部位とを有する経口徐放性薬剤組成物であって、前記第1の部位がクロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、前記第2の部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズである経口徐放性薬剤組成物を、前記哺乳動物に投与することを含む方法であり、
前記3種の有効成分が24時間を超えて安定した血漿中濃度になるように前記経口徐放性薬剤組成物を十分な投与回数で前記哺乳動物に投与した場合、前記有効成分を含有するFDA承認済みの即放性(IR)薬剤組成物1種以上を上記と同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価となる、前記有効成分の血漿中濃度が提供され、
前記FDA承認済みのIR薬剤組成物1種以上の適切な投与回数が、前記FDA承認済みのIR薬剤組成物1種以上の同じ期間にわたる投与に関して、FDA承認済みの1つ以上のラベルの中で推奨されている投与回数に対応し、
前記FDA承認済みのIR薬剤組成物1種以上の適切な投与回数が、前記経口ER薬剤組成物の十分な投与回数を上回っている、方法。
【請求項40】
哺乳動物において、少なくとも8時間にわたる有効成分としてのクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンの血漿中濃度が、前記3種すべての有効成分を含むFDAに承認された即放性の関連リスト収載医薬品(IR-RLD)組成物を同じ期間にわたり適切な投与回数で同じ哺乳動物に投与した際に得られる血漿中濃度と生物学的等価とする方法であって、前記方法が、
(A)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含有する経口徐放性(ER)薬剤組成物であって、第1の部位と第2の部位とを有し、
前記第1の部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、
前記第2の部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズである、経口徐放性(ER)薬剤組成物を前記哺乳動物に単回投与することと、
(B)少なくとも8時間にわたる前記3種すべての有効成分の血漿中濃度を、前記3種すべての有効成分を含むFDAに承認されたIR-RLD組成物を同じ期間にわたり適切な投与回数で投与した際に得られる血漿中濃度と生物学的等価にすることとを含む方法で、
前記適切な投与回数が、前記IR-RLD組成物の同じ期間にわたる投与に関して、FDA承認済みのラベルの中で推奨されている投与回数に対応する、方法。
【請求項41】
被験哺乳動物におけるヒドロコドンとプソイドエフェドリンのAUCinfinityの値を得る方法であって、前記方法が、
(A)(1)クロルフェニラミンとヒドロコドンとからなる有効成分を含む即放性(IR)部位と、(2)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む徐放性(ER)部位とを有する徐放性(ER)経口医薬組成物を、少なくとも8時間という期間の最初に単回投与する工程であって、
前記経口組成物における前記IR部位のクロルフェニラミン対前記ER部位のクロルフェニラミンの質量比が約25:75、前記IR部位のヒドロコドン対前記ER部位のヒドロコドンの質量比が約25:75、前記IR部位のプソイドエフェドリン対前記ER部位のプソイドエフェドリンの質量比が約0:100である、投与工程と、
(B)前記経口組成物に存在するヒドロコドンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を前記と同じ期間にわたり2回以上投与した際に得られるAUCinfinityと同等のヒドロコドンのAUCinfinityを、前記被験哺乳動物内に獲得する工程と、
(C)前記経口組成物に存在するプソイドエフェドリンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を前記と同じ期間にわたり2回以上投与した際に得られるAUCinfinityと同等のプソイドエフェドリンのAUCinfinityを、前記被験哺乳動物内に獲得する工程とを含む方法。
【請求項42】
前記方法が、(D)前記経口組成物に存在するプソイドエフェドリンの半分以下の量を有する即放性の関連リスト収載医薬品(IR-RLD)を前記と同じ期間にわたり2回以上投与した際に得られるAUCinfinityと同等のクロルフェニラミンのAUCinfinityを、前記被験哺乳動物内に獲得する工程を更に含む、請求項41記載の方法。
【請求項43】
被験哺乳動物におけるヒドロコドンとプソイドエフェドリンのAUCinfinityの値を得る方法であって、前記方法が、
(A)(1)クロルフェニラミンとヒドロコドンとからなる有効成分を含有する即放性(IR)部位と、(2)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含有する徐放性(ER)部位とを有する徐放性(ER)経口医薬組成物を投与する工程であって、
前記経口組成物における前記IR部位のクロルフェニラミン対前記ER部位のクロルフェニラミンの質量比が約25:75、前記IR部位のヒドロコドン対前記ER部位のヒドロコドンの質量比が約25:75、前記IR部位のプソイドエフェドリン対前記ER部位のプソイドエフェドリンの質量比が約0:100である投与工程と、
(B)前記経口組成物に比べてヒドロコドンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等のヒドロコドンのAUCinfinityを、12時間にわたるなかで前記経口組成物は1回投与、前記IR-RLDは時間がゼロのときと6時間後の2回投与で、被験哺乳動物内に獲得する工程と、
(C)前記経口組成物に比べてプソイドエフェドリンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等のプソイドエフェドリンのAUCinfinityを、12時間にわたるなかで前記経口組成物は1回投与、前記IR-RLDは時間がゼロのときと6時間後の2回投与で、被験哺乳動物内に獲得する工程とを有する方法。
【請求項44】
前記方法が、(D)前記経口組成物に比べてクロルフェニラミンが半量である即放性の関連リスト収載医薬品(IR-RLD)を2回投与した際に得られるAUCinfinityと同等のクロルフェニラミンのAUCinfinityを、12時間にわたるなかで前記経口組成物は1回投与、前記IR-RLDは時間がゼロのときと6時間後の2回投与で、被験哺乳動物内に獲得する工程を更に有する、請求項43記載の方法。
【請求項45】
ヒトへの単一の薬剤組成物の単回投与後、ヒトが治療効果を得るのに十分なクロルフェニラミン、ヒドロコドン及びプソイドエフェドリンのAUCinfinityを少なくとも8時間にわたり提供する方法であって、前記方法が、
(A)クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を含む単一の経口徐放性薬剤組成物であって、第1の部位と第2の部位とを有し、
前記第1の部位が、クロルフェニラミンとヒドロコドンとからなり、更にプソイドエフェドリンを含んでいてもよい有効成分を即放剤形で含有し、
前記第2の部位が、クロルフェニラミンとヒドロコドンとプソイドエフェドリンとからなる有効成分を徐放剤形で含有する粒子、ペレット又はビーズを含む前記薬剤組成物を、ヒトに単回投与する工程と、
(B)血漿分析により、単回投与後少なくとも8時間にわたり、ヒト被験者において治療効果が検出されるのに十分な3種類すべての有効成分のAUCinfinityを獲得する工程とを含む、前記方法。
【請求項46】
前記少なくとも8時間にわたる期間が12時間である、請求項45記載の方法。
【請求項47】
前記少なくとも8時間にわたる期間が24時間である、請求項45記載の方法。
【図7】

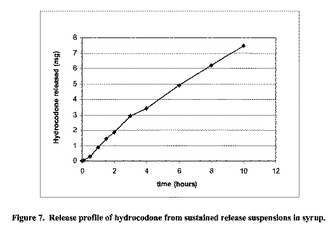
【公表番号】特表2012−525423(P2012−525423A)
【公表日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願番号】特願2012−508723(P2012−508723)
【出願日】平成22年4月29日(2010.4.29)
【国際出願番号】PCT/US2010/032949
【国際公開番号】WO2010/127100
【国際公開日】平成22年11月4日(2010.11.4)
【出願人】(511265729)アトリー ファーマシューティカルズ インコーポレイテッド (2)
【Fターム(参考)】
【公表日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願日】平成22年4月29日(2010.4.29)
【国際出願番号】PCT/US2010/032949
【国際公開番号】WO2010/127100
【国際公開日】平成22年11月4日(2010.11.4)
【出願人】(511265729)アトリー ファーマシューティカルズ インコーポレイテッド (2)
【Fターム(参考)】
[ Back to top ]