
抗体の生成方法
本発明は一般的に、癌原遺伝子を発現している動物において抗体生産細胞及び抗体を生産するための方法に関する。本発明はまた、自己寛容などの免疫束縛を通常受けている抗原に特異的な抗体の効率的な生産のための方法に関する。本発明はさらに、従来行われている抗体生産B細胞の骨髄腫融合パートナーとの融合を必要としない、抗体生産細胞及び抗体の生産に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は一般的に、癌原遺伝子を発現している動物において抗体生産細胞及び抗体を生産するための方法に関する。本発明はまた、自己寛容などの免疫束縛を通常受けている抗原に特異的な抗体の効率的な生産のための方法に関する。本発明はさらに、従来行われている抗体生産B細胞の骨髄腫融合パートナーとの融合を必要としない、抗体生産細胞及び抗体の生産に関する。
【0002】
(関連出願の相互参照)
本願は、2007年3月13日に出願せる米国仮出願第60/894654号、及び2007年5月18日に出願せる米国仮出願第60/939042号の米国特許法第119条(e)を相互参照する。各出願の開示全体を、引用することによりあらゆる目的で本明細書に援用する。
【背景技術】
【0003】
抗体、特にモノクローナル抗体の使用は、ルーチンの診断法のみならず基礎及び臨床研究の様々な分野に変革をもたらしている。モノクローナル抗体の臨床応用は、癌治療用の薬物の主たる新たな供給源として浮上してきた。例えば、CD20と称される表面蛋白質に対する抗体は、NHL患者や、さらには抗体が媒介する自己免疫疾患の患者の予後を劇的に改善している。
【0004】
高力価の中和抗体の発生は、多くのウイルス感染を患者が撃退する能力に関連している。最近の東南アジアでの鳥インフルエンザ(フル)の伝播は、感染しても生き存えた個体が、有効なB細胞抗体依存性の中和応答を惹起できる人々でもあることを示した。同様の知見が、その前年にSARS生存者の場合で示された。インフルエンザが原因となる米国での年間死亡者数は、年間30,000〜50,000名にのぼると見られる。インフルエンザによる全世界の死亡者数は、米国だけでの数字の20〜30倍高いと見られる。特に脆弱な2集団は、幼児と高齢者である。受動的に供給でき可能性のある中和抗体の開発は、インフルエンザ又はHIVなどの他のウイルスが原因となる死亡率を劇的に減少させるはずである。
【0005】
2005年5月の時点で、米国市場に18種の治療用モノクローナル抗体製品があった。世界的には、ほぼすべての消耗性疾患を処置するために200社より多くが開発中の、推定で500種のモノクローナル抗体製品があった。これらのモノクローナル抗体製品のうちおよそ80種が、臨床試験中である。モノクローナル抗体に対する世界市場は、2008年に167億ドルに増加すると見積もられる。
【発明の概要】
【発明が解決しようとする課題】
【0006】
モノクローナル抗体を生成するための従来からのアプローチは、マウス、又は最適な他の動物の超免疫化に基づいている。脾臓由来の抗体生産細胞はその後、収集して骨髄腫細胞融合パートナーに融合される。それらの抗体生産遺伝子を保持する細胞の選択は、順式及び逆式選択(forward and reverse selection)手順により成し遂げられる。これは、非常に強力な技術であることがわかってはいるものの、基礎生物学により課される制限のため、得ることができたはずの可能な特異性全体の90%より多くを逸する結果を招きやすい。それらの制限のいくつかに、「自己寛容」と称される機構や、抗体生産細胞と骨髄腫融合パートナーとの間の融合を成功裡に達成するのに必要な特定の要件が包含される。従って、上に挙げた制限に関連する問題を解決し、且つ目的の抗体を同定するのに必要な合計時間を短縮する、改善された抗体の生成方法が、当該技術分野で必要とされている。
【課題を解決するための手段】
【0007】
本発明の一態様は、MYCを誘導性に過剰発現する動物を、その動物においてMYCが過剰発現されない条件下で抗原に導き、その動物からB細胞を回収し、そしてMYCが過剰発現される条件下でそのB細胞を培養することによって、抗体生産細胞を生産するための方法に関する。
【0008】
いくつかの実施形態で、方法はさらに、動物を抗原に導く工程の後に、その動物においてMYCが過剰発現される条件下に動物を維持する工程を含む。
【0009】
いくつかの実施形態で、動物は、主にその動物のB細胞においてMYCを誘導性に過剰発現する。
【0010】
いくつかの実施形態で、動物は、MMTV−rtTA/TRE−MYCマウスである。いくつかの実施形態で、動物を抗原に導く工程は、抗原をコード化しているDNAを、その動物に遺伝的に移入することを含む。いくつかの実施形態で、抗原は自己抗原を含む。いくつかの実施形態で、動物はさらに、抗原を発現する。いくつかの実施形態で、抗原はgp120又はgp41などのHIV蛋白質を含む。
【0011】
いくつかの実施形態で、抗原は血球凝集素などのインフルエンザウイルス由来の抗原を含む。
【0012】
いくつかの実施形態で、マウスは、その動物を抗原に導く工程の間、MYC発現を抑止する抗生物質上に維持され、B細胞を培養する工程は、抗生物質非存在下に行なって抗体生産細胞を生産する。
【0013】
いくつかの実施形態で、方法はさらに、マウスを抗原に導く工程の後に、抗生物質への曝露からその動物を外す工程を含む。いくつかの実施形態で、抗生物質はドキシサイクリンである。
【0014】
本発明の別の態様は、MYCを誘導性に過剰発現する動物を、その動物においてMYCが過剰発現されない条件下で抗原に導き、その動物からB細胞を回収し、MYCが過剰発現される条件下でそのB細胞を培養し、そしてB細胞培養物から抗体を回収することによって抗体を生産するための方法に関する。
【0015】
いくつかの実施形態で、方法はさらに、動物を抗原に導く工程の後に、その動物においてMYCが過剰発現される条件下に動物を維持する工程を含む。
【0016】
いくつかの実施形態で、動物は主にその動物のB細胞においてMYCを誘導性に過剰発現する。
【0017】
いくつかの実施形態で、動物はMMTV−rtTA/TRE−MYCマウスである。いくつかの実施形態で、動物を抗原に導く工程は、抗原をコード化しているDNAを、その動物に遺伝的に移入することを含む。
【0018】
いくつかの実施形態で、抗原は自己抗原を含む。いくつかの実施形態で、動物はさらに、抗原を発現する。
【0019】
いくつかの実施形態で、抗原はgp120又はgp41などのHIV蛋白質を含む。
【0020】
いくつかの実施形態で、抗原は血球凝集素などのインフルエンザウイルス由来の抗原を含む。
【0021】
いくつかの実施形態で、抗体はヒト化抗体である。
【0022】
本発明の別の態様は、B細胞の重鎖又は軽鎖遺伝子のVDJ連結領域をコード化している核酸分子を、MYCを誘導性に過剰発現し且つB細胞の生産を防止する遺伝的修飾を含む動物からの骨髄由来の幹細胞に導入し、その骨髄由来の幹細胞をレシピエント動物に移入し、そのレシピエント動物からB細胞を回収し、MYCが過剰発現される条件下でそのB細胞を培養し、そしてB細胞培養物から抗体を回収することによって抗体を生産するための方法に関する。
【0023】
いくつかの実施形態で、方法はさらに、MYCがレシピエント動物において過剰発現される条件下にレシピエント動物を維持する工程を、その骨髄由来の幹細胞をレシピエント動物に移入する工程の後に含む。
【0024】
いくつかの実施形態で、方法はさらに、MYCがレシピエント動物において過剰発現されない条件下にレシピエント動物を維持する工程を、その骨髄由来の幹細胞をレシピエント動物に移入する工程の後に含む。
【0025】
いくつかの実施形態で、第1工程からのB細胞はヒトB細胞である。
【0026】
いくつかの実施形態で、B細胞はヒトから単離される。
【0027】
いくつかの実施形態で、ヒトは、抗体が媒介する自己免疫疾患に罹患している。
【0028】
いくつかの実施形態で、第1工程は、骨髄由来の幹細胞に、VDJ連結領域をコード化している核酸分子をレトロウイルスにより形質導入することを含む。
【0029】
いくつかの実施形態で、VDJ連結領域をコード化している核酸分子は、目的の抗原に選択的に結合する単離ヒトB細胞からクローニングされる。
【0030】
いくつかの実施形態で、VDJ連結領域をコード化している核酸分子は、ヒトドナーから得たB細胞に認められるIgH及びIgL配列より再配置したVDJ領域のPCR増幅断片である。
【0031】
いくつかの実施形態で、ヒトドナーは、健常ドナー、及び抗体が媒介する自己免疫疾患の患者からなる群より選択される。
【0032】
いくつかの実施形態で、骨髄由来の幹細胞はさらに、ヒトIgHをコード化している核酸分子、及びヒトIgLをコード化している核酸分子で形質導入される。
【0033】
いくつかの実施形態で、ヒトIgHをコード化している核酸分子とヒトIgLをコード化している核酸分子は、同じ核酸分子である。
【0034】
いくつかの実施形態で、ヒトIgHとヒトIgLをコード化している核酸分子の一方又は両方は、VDJ領域をコード化する同じ核酸分子である。
【0035】
いくつかの実施形態で、骨髄由来の幹細胞は、ヒト又はマウスからのものである。
【0036】
いくつかの実施形態で、マウスは、Rag−2−/−、SCID、DNA−PK−/−、Ku70−/−、Ku80−/−、XRCC4−/−及びμMT−/−からなるリストより選択される遺伝的修飾を含む、MMTV−rtTA/TRE−MYCマウスである。
【0037】
いくつかの実施形態で、レシピエント動物は致死的に照射されたマウスである。
【0038】
いくつかの実施形態で、レシピエント動物はSCIDマウスである。
【0039】
いくつかの実施形態で、動物は、VDJ領域に選択的に結合する抗原にインビボで導かれる。
【0040】
いくつかの実施形態で、抗体イソタイプは、IgA又はIgGである。
【0041】
いくつかの実施形態で、抗体のFc領域を遺伝的に修飾して、抗体が自己免疫反応及び関連免疫複合体沈着の問題を誘発する能力を最小にしている。
【0042】
本発明の別の態様は、細胞の生存及び増殖を促進する癌原遺伝子を誘導性に過剰発現する動物を、その癌原遺伝子がその動物において過剰発現されない条件下で抗原に導き、その動物からB細胞を回収し、そして癌原遺伝子が過剰発現される条件下でそのB細胞を培養することを含む、抗体生産細胞を生産するための方法に関する。
【0043】
本発明の別の態様は、細胞の生存及び増殖を促進する癌原遺伝子を誘導性に過剰発現する動物を、その癌原遺伝子がその動物において過剰発現されない条件下で抗原に導き、その動物からB細胞を回収し、癌原遺伝子が過剰発現される条件下でそのB細胞を培養し、そしてそのB細胞培養物から抗体を回収することによる、抗体を生産するための方法に関する。
【0044】
本発明の別の態様は、抗原をコード化している核酸分子を、細胞の生存及び増殖を促進する癌原遺伝子を誘導性に過剰発現する動物からの骨髄由来の幹細胞に導入し、その骨髄由来の幹細胞をレシピエント動物に移入し、癌原遺伝子がその動物において過剰発現される条件下にレシピエント動物を維持し、そのレシピエント動物からB細胞を回収し、癌原遺伝子が過剰発現される条件下でそのB細胞を培養し、そしてB細胞培養物から抗体を回収することによる、抗体を生産するための方法に関する。
【0045】
本発明の別の態様は、細胞の生存及び増殖を促進する癌原遺伝子をコード化している核酸分子を、動物からの骨髄由来の幹細胞に導入し、その骨髄由来の幹細胞をレシピエント動物に移入し、そのレシピエント動物からB細胞を回収し、抗原をコード化している核酸分子をそのB細胞に導入し、癌原遺伝子が過剰発現される条件下でそのB細胞を培養し、そしてB細胞培養物から抗体を回収することによる、抗体を生産するための方法に関する。
【0046】
本発明の別の態様は、細胞の生存及び増殖を促進する癌原遺伝子をコード化している核酸分子を、動物からの骨髄由来の幹細胞に導入し、その骨髄由来の幹細胞を第1レシピエント動物に移入して、その第1レシピエント動物からB細胞を回収し、抗原をコード化している核酸分子をそのB細胞に導入し、そのB細胞を第2レシピエント動物に移入し、その動物において癌原遺伝子が過剰発現される条件下に第2レシピエント動物を維持し、その第2レシピエント動物からB細胞を回収し、癌原遺伝子が過剰発現される条件下でそのB細胞を培養し、そしてB細胞培養物から抗体を回収することによる、抗体を生産するための方法に関する。
【0047】
いくつかの実施形態で、癌原遺伝子はMYCである。
【0048】
いくつかの実施形態で、骨髄由来の幹細胞は、抗アポトーシス蛋白質をコード化している核酸分子で形質導入されている。
【0049】
いくつかの実施形態で、抗アポトーシス蛋白質は、Bcl−2である。
【0050】
いくつかの実施形態で、核酸分子は、レトロウイルスにより導入される。
【0051】
いくつかの実施形態で、骨髄由来の幹細胞は、ヒトからのものである。
【0052】
いくつかの実施形態で、骨髄由来の幹細胞は、条件付きで不死化された長寿命の造血幹細胞である。
【0053】
いくつかの実施形態で、レシピエント動物は、亜致死的に照射されたNOD/SCIDマウスである。
【0054】
いくつかの実施形態で、動物は、ヒトIg座位をコード化している核酸分子に対して遺伝子導入されている。
【図面の簡単な説明】
【0055】
【図1】図1は、TBLK6及びTBLK7細胞系の表面上での、CD138(Y軸)及びCD40(X軸)の発現を(上部パネル)、そしてTBLK6及びTBLK7細胞系からのIgM分泌の分析を(下部パネル)示す。
【図2】図2は、Eμ−MYC/BCRHEL/sHEL遺伝子導入マウスで生じる腫瘍及び細胞系の表面表現型を(上部パネル)、そしてEμ−MYC/BCRHEL/sHELマウスで生じる腫瘍及び細胞系における免疫グロブリン生産(A)及びHEL特異的力価(B)を(下部パネル)示す。
【図3】図3は、MYCの急激な過剰発現後の活性化B細胞の外観を(上部パネル)、そしてMYCの連続的な過剰発現の間での活性化B細胞の蓄積を(下部パネル)示す。
【図4】図4は、MYCの過剰発現後の血清中の自己抗体の蓄積を(上部パネル)、そしてMYCの過剰発現後の腎臓における自己抗体及び免疫複合体の蓄積を(下部パネル)示す。
【図5】図5は、HELを発現するPRV変異体の致死的攻撃からの、新規HEL特異抗体でのマウスの保護を示す。
【図6】図6は、レトロウイルスによるキメラマウスで発生するB細胞腫瘍の表面表現型を示す。
【図7】図7は、レトロウイルスによるキメラマウスから得た血清と、HAを発現している細胞溶解液との反応性を実証するウエスタンブロット分析を示す。
【図8】図8は、レトロウイルスによるキメラマウスから得た、マウス血清の血球凝集阻害分析を示す。
【図9】図9は、MYCを過剰発現しているマウスの使用により得られる抗体特異性をヒト化する、レトロウイルスのベクターによる2種のアプローチを示す。
【発明を実施するための形態】
【0056】
本発明は、従来の抗体生産に伴う問題の多くを克服する、抗体生産細胞及び抗体を生産するための新規方法を提供する。一般的に、本発明は、癌原遺伝子を発現している動物、例えば、MYC又はAkt(特に好ましくはMYC)を過剰発現する動物において、抗体生産細胞及び抗体を迅速に生産するための方法に関する。本明細書に開示する方法によって、抗体生産B細胞を骨髄腫融合パートナーと融合させる必要なしに、モノクローナル抗体を生産することが可能になり、抗体を生産するのに必要な時間が短縮される。本明細書に開示する方法はさらに、免疫化されたマウスからのB細胞が骨髄腫パートナーと首尾良く融合するために、従来の抗体生産技術に必要とされるような細胞周期の特定段階にあることの必要性を排除する。本発明はまた、自己寛容などの免疫束縛を通常受けている抗原に特異的な抗体の効率的な生産も可能にする。例えば、本方法は、自己抗原に対するモノクローナル抗体を生産するために使用してもよい。
【0057】
本発明者らは、MYCの過多が、可溶性抗原に対するB細胞寛容を破壊できることを実証している。例えば、MYCを過剰発現している、BCRHEL遺伝子導入B細胞は、sHELに対する活発な応答を惹起し、悪性化が始まる前にポリクローナル自己免疫性リンパ球増殖性疾患を発症させる(図3及び4参照)。自己反応性B細胞におけるMYCの過剰発現は、MYCが増殖及び生存シグナルをもたらす能力によって、B細胞をT細胞の援助に対して非依存性とすることができる。MYCを過剰発現している、自己反応性B細胞の増殖した集団は、その同族抗原への連続曝露と、MYCの過剰発現との双方に伴い、依存性のままであるB細胞リンパ腫を発生する。腫瘍担持マウスからのリンパ節、脾臓及び骨髄より収集したB細胞を用いて、BCRHEL導入遺伝子を発現し、且つ抗HEL IgMを分泌する多くの細胞系を、初代細胞の骨髄腫融合パートナーへの融合を行うことなく樹立できる(図2参照)。本プロトコルは、以下に記載の通り、事実上任意の抗原に容易に適用できる。
【0058】
同様の結果が、2つのさらなる動物でのプロトコルを用いて得られている。一例は、Ars.AlマウスとΕμ−MYC株との間の交雑種に由来するマウスである。それらのマウスでは、バーキット様リンパ腫の発生が平均して36日齢に起こる。この腫瘍は成熟した活性化B細胞で構成されている。それらの細胞は、表面にIgMを発現する。このように、本発明者らは、MYC過剰発現が、低親和性の抗DNA抗体という面において自己反応性B細胞に対する寛容を破壊できることを実証している。第2の例では、食餌からのドキシサイクリン排除後に、B細胞特異的で一時的に調節されるMYCの過剰発現を可能とする、MMTV−rtTA/TRE−MYCマウスを利用する。ドキシサイクリン含有食餌を4月齢でマウスから離すと、マウスは活性化された末梢B細胞、抗核抗体を血清に、免疫複合体沈着物を腎臓に蓄積し、また6週以内(平均的な例では、42日)にB細胞リンパ腫を発生した。重要なのは、骨髄腫パートナー細胞への融合を行わずに、腫瘍から細胞系を樹立できていることである。
【0059】
本発明は、少なくとも三態様のシステムの生物学に基づき、現今のアプローチよりも効力が有意に高められた、既知の特異性を有する抗体を生産する新規のアプローチを提供する。第一は、自己寛容の機構により課されるバリアの存在で、このシステムにより免疫束縛なしに抗体を生成できるようになる。このアプローチで、標準的な(当該技術分野の水準で)免疫化手順によって達成できない抗体特異性の生成が可能になる。さらに、標準的免疫化プロトコルで自己寛容を破壊する簡単な手段はない。第二は、目的の同族抗原が、B細胞異常増殖をインビボで推進し、リンパ腫を発生しうる抗体生産細胞と、得られるモノクローナル抗体生産細胞系の選択を可能とする。これにより、得られるモノクローナル抗体のスクリーニングを直接的に行うことができるため、開発のための時間が加速される。第三は、これらのマウスで生じる腫瘍から抗体生産細胞系を生成してクローン増殖する能力により、抗体生産脾臓B細胞を骨髄腫融合パートナーに融合させる必要性が排除される。従来の融合プロセスは、かなり非効率的で、免疫化後に増殖するB細胞の画分の不死化が可能になるにすぎず、このため、モノクローナル抗体の形態で誘導しうる特異性の数が限定されてしまう。この非効率性の理由は多くあり、例えば、細胞周期のS期にあるB細胞の必要性、細胞に保持される必要のあるB細胞と骨髄腫融合パートナーとに由来する染色体の適切な数及び組み合わせ、HAT及び6−TG選択で生存する必要性、等である。まとめると、モノクローナル抗体の生成のための本発明の新規なアプローチは、従来のアプローチよりも随分短い時間枠にて、より多くの抗体を提供する。現在利用できるツールを用いた、そのアプローチへのいくつかの変更があり、選択されるその実施形態を以下にまとめる。
【0060】
本発明の一実施形態で、抗体を生産するために使用される好適なヒト以外の動物には、抗体を生産でき、且つ細胞の生存及び増殖を促進する少なくとも1種の癌原遺伝子を(例えば、遺伝的修飾、又は天然、若しくは定方向突然変異の結果として)過剰発現する、ヒト以外の任意の動物が挙げられる。本発明の方法で用いられるヒト以外の動物によって過剰発現されるべき好ましい癌原遺伝子としては、限定しないが、MYC又はAkt(ミリスチル化物)が挙げられ、MYCが特に好ましい。一例として、MYCを過剰発現しているマウスを本明細書で詳説するが、他の好適な癌原遺伝子及び他の動物の使用を含む本プロトコルの改変や、プロトコルを実施する際の技術的詳細の改変は、本明細書の開示に基づいて企図されるはずであり、それらが本発明に包含されることは理解されるべきである。
【0061】
MYCを過剰発現している任意の動物を、本発明に使用してもよい。このような動物には、好ましくはヒト以外の哺乳動物が含まれ、より好ましくは齧歯類、さらにより好ましくはマウスが挙げられる。特に有用な動物は、MYCを主にB細胞集団に過剰発現しており、その例としてΕμ−MYCマウス株などが挙げられる。好ましい動物にはまた、誘導可能な態様でMYCを過剰発現するものも含まれる。これらの動物では、MYC過剰発現は、一時的な態様で調節でき、例えば、抗体の生産が開始するまで抑制できる。例えば、MMTV−rtTA/TRE−MYCマウス株に、ドキシサイクリン又はテトラサイクリンを誕生から目的の抗体を生産するための使用時までの間、投与してもよく、これにより自然発症リンパ増殖性疾患の形成が最小化される。MYC過剰発現は、マウスの食餌からドキシサイクリン又はテトラサイクリンを除去することによって開始させることができる。本発明での使用に好適な、多くのさらなる誘導可能な遺伝子発現/抑制システムが、当該技術分野で知られている。好適なマウスは、レトロウイルスの形質導入及び養子移入技術によって作り出してもよい。例えば、TRE−MYCマウスからの骨髄細胞又は精製B細胞を、レトロウイルスによりrtTAで形質導入し、レシピエントマウスに移入するか、又はインビトロで培養してもよい。得られる細胞は、移入又は培養の前に、さらに(例えば、目的の抗原で)形質導入してもよい。
【0062】
本発明の特定の実施形態で、MYCを過剰発現している細胞を保有している動物を、インビボで抗原に導き(例えば、暴露し)、抗原に対する免疫応答を惹起させてもよい。例えば、一態様で、当該技術分野で周知の、動物に外来抗原を導入する従来の経路によって(例えば、免疫化と類似の技術を用いて)、目的の抗原に動物を暴露してもよい。
【0063】
或いは、好ましい実施形態で、MYCを誘導性に発現する細胞を保有する動物を、当該技術分野で一般的な技術により目的の抗原を発現するように設計してもよく、又は目的の抗原を発現している細胞を動物に導入してもよい。例として、骨髄造血幹細胞の、レトロウイルスが媒介する形質導入;遺伝子導入動物の作製、若しくはMYCを過剰発現している動物と、抗原を発現する既存の遺伝子導入動物との交雑;又は動物への遺伝子送達のための他の任意の方法が挙げられる。いくつかの実施形態で、抗体の生産が望まれるまでMYCが過剰発現されない条件下に動物を維持する。動物は次いで、MYCが過剰発現されて目的の抗原に対して特異的な抗体を生産するB細胞を生成する条件下に維持してもよい。本発明者らは、抗原に対して特異的なB細胞は、MYC過剰発現がなければ抗原に対して寛容化されることになると考えている。いったんMYCの過剰発現が誘導されれば、B細胞は新自己抗原に対する寛容を破壊でき、抗原に対して特異的な抗体を生成することができる。
【0064】
正常AN−I細胞集団(B細胞発生時にT3としても知られる集団、自己抗原結合に際して骨髄において反応不顕性となったB細胞と考えられるもの)由来の細胞系を、複数のアプローチによって作製してもよく、そのうちのいくつかを以下に記載及び例証する。一実施形態で、AN−I集団は、受胎以降ドキシサイクリンにて維持されたMMTV−rtTA/TRE−MYCマウス由来ものである。本発明の方法によれば、それらのマウスは、目的の特異抗原も発現すべきである。前記のとおり、これは骨髄造血幹細胞の、レトロウイルスが媒介する形質導入、標準的遺伝子導入のアプローチ、又はマウスへの遺伝子送達のためのその他の任意の方法によって容易に成し遂げることができる。細胞は、およそ6週齡のマウスや、或いは野生型マウス、又は導入遺伝子の1つを保因するのみのマウスのいずれかの脾臓から単離できる。細胞は、インビトロで、好適なリンパ球培地(例えば、RPMI1640、10%ウシ胎児血清、pen/strep、L−グルタミン、2−βメルカプトエタノール、ピルビン酸ナトリウム、Hepes及び非必須アミノ酸)において、ドキシサイクリン(50nM)の存在下又は非存在下に蒔くことができる。細胞は例えば、24穴プレートにおいて、およそ2x106細胞/mlの密度で蒔かれるが、これらの工程は当業者によって容易に至適化又は改変できる。培地は通常、週1回交換する。このアプローチは、細胞の供給源及び状態(すなわち腫瘍性又は正常細胞、臓器等)によって、10〜70%の効率で細胞系を提供できる。細胞はクローン性の増殖について視覚的に調べてもよい。約14〜28日に増殖し始めるいずれのクローンでも、6穴プレートにおいて、最終的に組織培養フラスコにまで、ゆっくりと増殖させることができる。全てのマルチクローン性細胞系を、以前報告され、当該技術分野で公知の通りに、分析の前に限界希釈法によって単一細胞クローニングに付してもよい。
【0065】
別の実施形態で、目的の抗体を生産できるB細胞系の形質転換は、インビボで起こさせてもよい。例としては、受胎以降、ドキシサイクリン含有食餌にて維持されていたMMTV−rtTA/TRE−MYCマウスのコホート(cDNAをコード化しているプラスミドから目的抗原も発現するもの)を、6週齢で正常マウス固形飼料に切り替える。このマウスは、B細胞リンパ腫の発生に関わる臨床徴候(毛皮汚損、外見的に明白なリンパ節腫脹、脱水、不活発、後肢麻痺−上行性等)について毎日検査できる。さらに、マウスは定期的に供血でき、目的の抗原への反応性について調べることができる。一旦マウスが腫瘍を発生すれば、そのリンパ節、脾臓、骨髄、及び血清を採取する。こうして得られる細胞懸濁液は、FACS分析に使用してもよく、細胞を蒔いて前記のように細胞系を生成し、そして生存可能な初代腫瘍組織が継続的に得られるようにするため、残余の細胞を10%DMSO中で凍結する。血清は、目的の抗原を発現する細胞を染色するのに使用して、また特異的抗原に対するものであるウエスタンブロット及びELISAアッセイ用に使用してもよい。コントロールマウスには通常、野生型、及び単独に遺伝子導入したマウスが含まれる。
【0066】
別の実施形態で、MMTV−rtTA/TRE−MYCマウス(前記同様、受胎以降ドキシサイクリン含有食餌にて維持されているもの)からの精製B細胞を、選択抗原を発現するようにインビトロで形質導入できる。形質導入した細胞はその後、ドキシサイクリン(移入後のB細胞においてMYCの過剰発現を誘導するもの)にて維持されていないレシピエントマウス(例えば、野生型マウス)に養子移入する。マウスは次いで、前記のように生成された細胞系、及びB細胞リンパ腫の発生に伴う臨床徴候について毎日試験することができる。
【0067】
モノクローナル抗体生産細胞の樹立を成し遂げるためのさらなるアプローチは、細胞の生存及び増殖を促進する(そして好ましくは、調節可能(誘導可能、制御可能)である)癌原遺伝子と、アポトーシスを阻害する蛋白質をコード化している核酸分子との選択された組み合わせを発現するマウスからの、骨髄レトロウイルスのキメラの使用を含む。本実施形態で、組み合わせの例として、限定しないが、MYC−ER及びBcl−2が挙げられる。この後者のアプローチは、発現されるMYCを活性にするために4−ヒドロキシタモキシフェン(4OHT)を添加することを含む。本発明者らは、長寿命の造血幹細胞の条件付き不死化に関する実験においてMYC機能を首尾良く調節するような構築体を使用している。この場合、骨髄キメラマウスのコホートは、以下のように5FU富化骨髄由来幹細胞を用いて生成してもよい。骨髄由来造血幹細胞の場合、長寿命のHSCを富化するために、そしてそれらのインビボ増殖を誘導するために、5mg/マウスの5−フルオロウラシル(5FU)を静脈内に投与する。骨髄細胞は、5日後に大腿骨及び脛側から収集する。低張溶解バッファーを用いて、赤血球細胞を溶解する。残る細胞を、培地で2回洗浄して、15%熱不活性化ウシ胎児血清、ペニシリン/ストレプトマイシン、L−グルタミン、非必須アミノ酸、組換えヒトIL−3、IL−6及び幹細胞因子(SCF)を補充したDMEM培地を含む24穴プレートに2x106細胞/mlの濃度で蒔く。前記の通り、骨髄細胞は選択抗原を発現するように、インビトロで形質導入してもよい。細胞を第1スピン感染の前に24時間培養して、24時間毎に3回、この手順に付す。
【0068】
最後のスピン感染の翌日に、細胞をフローサイトメトリーによって分析する。例として、レンチウイルスにより形質導入した骨髄に由来するHSCを用いて、致死的に照射されたマウスを再構成してもよく、12週後のリンパ系器官におけるレポーター遺伝子GFPの発現を用いて細胞を追跡できる。この場合、マウスは正常末梢リンパ系区画(骨髄移植後8〜12週)を再構成してもよい。脾臓、GFP+AN−1/T3細胞は次いで、それらマウスから単離し、前記のようにインビトロ不死化プロトコルに用いてもよい。重要な相違は、システムからドキシサイクリンを排除する代わりに、4OHTを培地に添加することである。或いは、lmg/マウスの4OHTで週1回腹腔内に処置した野生型レシピエントマウスのコホートに、ソートされたGFP+AN−1/T3細胞を養子移入できる。マウスは、B細胞リンパ腫又は白血病の発生に伴う臨床徴候の出現につき毎日モニターする。得られる腫瘍を収集し、MYC機能のレギュレーターとしてドキシサイクリンの代わりに4OHTを用い、本節に既に記載したようにB細胞系を生成するために使用できる。
【0069】
別の実施形態で、条件付きで不死化した骨髄由来の幹細胞系を、前記方法における5FU富化骨髄由来幹細胞の代わりに使用してもよい。いくつかの実施形態で、細胞の生存及び増殖を促進する(そして好ましくは調節可能(誘導可能、制御可能)である)癌原遺伝子と、アポトーシスを阻害する蛋白質をコード化している核酸分子との前記組み合わせ(MYC−ERとBcl−2との組み合わせが例示される)を発現する細胞系を使用してもよい。条件付きで不死化した骨髄由来の幹細胞系及びそれらの作製の詳細な説明及び例は、国際公開第WO2007/047583号パンフレットに記載されており、その内容を引用することにより本明細書に援用する。WO2007/047583号パンフレットは、癌原遺伝子と抗アポトーシス遺伝子及び本発明で使用してもよいそれらの誘導体とのさらなる組み合わせの詳細な説明を提供する。特定の実施形態で、細胞系は、選択抗原をそれらが発現するように、インビトロで形質導入してもよい。
【0070】
目的の抗原に対して特異的な抗体のポリクローナル集団や、さらにはそれらの抗体を生産する細胞は、動物の組織(脾臓、リンパ節等)又は血清から直接、単離してもよい。別の実施形態で、抗体生産細胞のモノクローナル集団を、標準的手順により単離してもよく、またモノクローナル抗体を培養培地から回収してもよい。
【0071】
本発明の方法はまた、ヒト抗体を生産する細胞系を作製するのに使用してもよい。本実施形態を成し遂げるための2種の例示的技術として、(1)ヒト免疫グロブリン座位(IgH及びIgL)をコード化している遺伝子導入BAC構築体を保因するマウス株の、Eμ−MYC株若しくはMMTV−rtTA/TRE−MYC導入遺伝子を含むマウスのいずれかへの交雑(繁殖による)、又は(2)MYC−ER(又は別の好適な誘導可能癌原遺伝子)に対するcDNAを含む、レトロウイルスの骨髄キメラの使用が挙げられる。この後者のアプローチで、目的の同族抗原は、レトロウイルスによりコード化されるcDNAから新自己抗原として、従来の導入遺伝子から、又は遺伝子及び蛋白質送達の他の手段によって発現される。前記のものに比較した場合、本実施形態における重要な相違は、本設定にて生産される抗体がヒト蛋白質になるはずであるということである。
【0072】
ヒト抗体を生産する細胞系を作製する第2の方法は、所定の目的蛋白質に対する血清抗体価を有するヒトから得られる末梢血B細胞の単離に基づいている。B細胞は、標準的アプローチを用いて精製する。精製B細胞は、目的の特異性を備えたようなB細胞に対して富化するために、目的の蛋白質を被覆したプラスチックプレート上にパニングすることができる。或いは、目的の蛋白質は、磁気ビーズに接合させ、次いで目的の特異性を備えたB細胞を単離するのに使用できる。それらの細胞は、その後、単一細胞RT−PCR用のTerasakiプレートに単一細胞ソートすることができる。単離したB細胞の各々に対するIgH及びIgL遺伝子に対するVDJ連結領域につき、cDNAを生成する。それらのcDNA断片は次いで、ヒトIgH及び/又はIgLをコード化し、且つ多重クローニング部位をそれらの分子が普通はそれら自体のVDJ配列を有するはずである位置に含む、レトロウイルスベクター(LTR−IgH−IRES−IgL−LTRをコード化するレトロウイルスのベクター、又はIgLに対するもの、IgHのいずれか1つ)へとクローニングできる。得られるレトロウイルスを配列決定して、MYCを過剰発現し、且つB細胞の生産を防止する遺伝的修飾を含む、マウス由来の5FU富化骨髄由来HSCを形質導入するのに使用できる。例として、MMTV−rtTA/TRE−MYC/Rag−1−/−マウス、Eμ−MYC/Rag−1−/−マウス、又はIgH/IgLレトロウイルス及びMYC−ERに対するもので同時感染したRag−1−/−骨髄が挙げられる。さらなる例として、Rag−2−/−、SCID、DNA−PK−/−、Ku70−/−、Ku80−/−、XRCC4−/−、μMT−/−等の座位の遺伝的除去を含むマウスと交雑した、前記のMYCを過剰発現しているマウスの、任意のものが挙げられる。それらの細胞は次いで、前記のプロトコル(レトロウイルスの骨髄キメラマウス用)を用いて致死的に照射されたマウスを再構成するのに使用できる。特異的抗原をコード化している核酸をマウスに形質導入するのに、任意の手段(遺伝子銃、裸(naked)DNA免疫化、遺伝子導入マウス、レンチウイルスでの「遺伝子導入」マウス、組換え蛋白質の直接注射、その遺伝子に対するcDNAをコード化するワクシニアウイルス、組換えアデノウイルス、組換え酵母、又は哺乳動物細胞、tat−融合蛋白質等)を使用できる。
【0073】
本アプローチより得られる細胞系は、目的の蛋白質に特異的なヒト免疫グロブリン配列をコード化することになる。これは、ウイルス感染、腫瘍、細菌及び真菌等を含む様々な病原及び疾患に対する阻害抗体のカクテルの生成のための強力な新規アプローチである。本発明はまた、MMTV−tTA/TRE−MYCマウスを使用することで得られる抗体特異性をヒト化する方法も含んでいる。この場合、IgL座位にあるマウスIgH及びVJ領域より由来する、再配置したVDJ連結配列をPCR増幅する。それらの配列は、MMTV−tTA/TRE−MYCマウスより発生し、目的の抗原に対する抗体を生産することが既に示されているクローン細胞系から得ることができる。PCR増幅断片は、その中にPCR断片がクローニングされる多重クローニング部位を含むヒトIgH及びIgL配列をコード化するレトロウイルスのプラスミドへとクローニングできる(図9参照)。IgH及びIgL配列は、両方のcDNAが同じウイルスベクターから発現されるように、IRESエレメントによって離間されている。異なるウイルスベクターの使用により、所定の特異性を備えた、異なる抗体イソタイプ(IgA、IgG、IgM、IgE及びその異なるサブグループ)の生成が可能になる。さらに、Fc領域をコード化している配列をさらに修飾して、得られる抗体が自己免疫反応及び関連免疫複合体沈着の問題を誘発する能力を最小にしてもよい。得られるレトロウイルスは、Rag−1−/−/MMTV−tTA/TRE−MYCマウスから得られる骨髄由来造血幹細胞を形質導入するのに使用される。形質導入した細胞は、致死的に照射されたC57/BL6野生型マウスのコホートに移植できる。この結果得られるマウスは、一抗原に対して単一特異性であり、ヒト化抗体の追加された特徴のすべてを備えたモノクローナル抗体生産細胞を生成できる。
【0074】
別の実施形態で、目的の抗原に対するモノクローナルヒト抗体は、以下の方法によって生産できる。所定の細胞系に見られるIgH及びIgLからの特異的VDJ連結配列を増幅する代わりに、抗体が媒介する自己免疫疾患(例えば、シェーグレン症候群、ハシモト甲状腺炎、全身性エリテマトーデス、ワルデンストロームマクログロブリン血症等)に罹患している患者や、非ホジキンリンパ腫(バーキットリンパ腫、濾胞様リンパ腫、びまん性大細胞型B細胞性リンパ腫、MGUS及び複数の骨髄腫)に罹患している患者、又は健常ドナーのいずれかから得たB細胞に見られるIgH及びIgL配列から再配置したVDJに対するPCR増幅断片を単離できる。PCR断片は、前記レトロウイルス構築体にクローニングされる(これらのライブラリーの生成を円滑にするよう、2つの異なるレトロウイルスの構築体としても調製できる)。次いで、レトロウイルスのライブラリーを使用して、前記のように骨髄キメラマウスを生成するために、Rag−−/−/MMTV−tTA/TRE−MYCマウスから得られた骨髄由来造血幹細胞(HSC)を形質導入できる。骨髄キメラマウスは、ヒト免疫グロブリンを発現するB細胞のみを作り、免疫化の準備が整うまで(MYC過剰発現を抑制するために)ドキシサイクリン含有食餌にて維持できる。これらのマウスは、(それらのB細胞でのMYC過剰発現を果たすため)ドキシサイクリンの非存在下に免疫化できる。反応性の抗原特異的B細胞は、特異的抗原に対するパニングによってインビトロで単離及び濃縮できる。この細胞は、それらを不死化して、ヒト抗体を生産するモノクローナル細胞系を作るために、MYC過剰発現下に成長させることができる。
【0075】
前記の通り、このアプローチを使用して、例えば、特異的抗原に対するヒトIgA抗体を含め、異なるイソタイプを有するヒト抗体を特異的に単離できる。IgA抗体は、予防用に後で強く求められるが、IgA生産を意図的に誘導するために従来の抗体生産方法を用いてどのように動物を免疫化するかは不明なままである。本発明を用いて、この問題が解決される。さらに、本明細書で定義するいずれの特異性も、前記の通り別のFc骨格に容易に移入できる。
【0076】
得られる細胞系は、多量の抗体を至適に生産しない可能性がある。これは、IgH及びIgLをコード化するcDNAをPCRに基づき増幅することによって解消できる。それらcDNAの発現ベクターへのクローニングは、次いで骨髄腫細胞系(又は他のB細胞リンパ腫細胞系)を形質導入して抗体生産を増大させるのに使用できる。
【0077】
第3のアプローチは、レトロウイルスにより目的の抗原でヒトctlt−HSC細胞系を形質導入して、亜致死的に照射されたNOD/SCIDマウスに、それらの形質導入ctlt−HSC細胞系を用いて移植することである。マウスに4OHTを注射し、得られる白血病/リンパ腫を培養してヒトモノクローナル抗体生産細胞系を作製できる。その利点は、モノクローナル抗体全体がヒト遺伝子によりコード化され、インビボで成熟できることである。第4の可能性は、亜致死的に照射され、且つヒトctlt−HSC細胞系で再構成されたNOD/SCIDマウスに由来する脾臓成熟ヒトB細胞を単離することである。MYC過剰発現なしに発生した成熟B細胞を、目的の抗原でレトロウイルスにより形質導入できる。細胞は、亜致死的に照射されたNOD/SCIDマウスに戻し移植してそれらのマウスに4OHTを注射することによりインビボで形質転換させるか、又は4OHTの存在下にインビトロ培養で維持することができる。これにより、目的の抗原の核酸配列のみで開始して2〜3週で、新規ヒトモノクローナル抗体の発生が可能になりうる。MYCが自己抗原に対するB細胞寛容を破壊する能力は、T細胞免疫優性エピトープに関わらず多くの特異性に対する抗体の発生をもたらすかもしれない。MYCがB細胞寛容を破壊する機構は、自己反応性B細胞をT細胞の援助に非依存的なものとすることに関わり、よってそれらを蛋白質配列の特定部分に応答する束縛から開放することもできる。本アプローチは、自己蛋白質を模倣し、免疫系によって通常は無視され、従って寛容機構が概して、それらの残基に対する良好な応答を妨げる、ウイルスによりコード化されたエピトープを明らかにすることができるかもしれない。本発明は従って、それらの共通ドメインを良好なワクチン候補とする新規の方法を提供する。
【0078】
本明細書に開示の技術は、癌及び自己免疫などの感染症及び慢性疾病を明らかにする、検出及び処置用の診断及び治療用抗体の迅速な開発のための新しいストラテジーを提供する。抗体生産のための既存の手順に比べて、本発明の速度及び効率のために、新しい感染作用物質が現れた後すぐに、これらの抗体を生成することができる。例えば、疾患の突然の激増(例えば、パンデミックインフルエンザ(フル))の原因になるウイルスに特異的な抗体を、現行法により生成されるものよりもずっと短期間で治療剤としての使用に備えるものとしうる。本発明の方法を用いて生産される抗体は、あらゆる型及びクラスの抗体を初めとして、抗体結合断片及び抗体の誘導体を含んでもよい。さらに詳細には、本発明の方法により生産される抗体は、かかる抗体を含有する血清、又は様々な程度に精製されている抗体を含むことができる。本発明の全抗体は、ポリクローナル又はモノクローナルでありうる。或いは、全抗体の機能性等価物、例えば、1種以上の抗体ドメインが切欠されたか又は存在しない抗原結合断片(例えば、Fv、Fab、Fab’,又はF(ab)2断片)、さらには、単鎖抗体、ヒト抗体、ヒト化抗体(前記)、1種より多くのエピトープに結合できる抗体(例えば、二重特異性抗体)、若しくは1種以上の異なる抗原に結合できる抗体(例えば、二重又は多重特異性抗体)を含む遺伝的に操作した抗体又はその抗原結合断片なども、本発明の方法によって生産されうる。
【0079】
多種多様な抗原に選択的に結合することができる抗体を、本発明の方法によって生産できる。一般的に、動物に導入すると免疫応答を誘導できる任意の抗原が、本発明での使用に好適である。さらに、自己抗原などの、正常動物では通常自己寛容機構を受けているはずの抗原も、本発明で使用してもよい。例えば、抗原としては、限定しないが、ウイルス抗原、真菌抗原、細菌抗原、蠕虫抗原、寄生虫抗原、外寄生生物抗原、原生動物抗原、又は他の任意の感染作用物質からの抗原を含め、病原に関わる任意の抗原を挙げることができる。抗原としてはまた、病原性又は細胞性のいずれの出所からのものであれ、特定の疾患又は状態に関連する任意の抗原も挙げることができ、これらには限定しないが、癌(腫瘍)抗原、自己免疫疾患に関わる抗原(例えば、糖尿病抗原)、アレルギー抗原(アレルゲン)、1種以上の変異アミノ酸を内包している哺乳動物細胞分子、哺乳動物細胞によって通常は出生前又は新生児期に発現される蛋白質、疫学的作用物質(例えばウイルス)の挿入により発現が誘導される蛋白質、遺伝子転座により発現が誘導される蛋白質、及び調節配列の突然変異により発現が誘導される蛋白質が含まれる。これらの抗原は、天然抗原又は遺伝操作された抗原で、なんらかの方法によって修飾されているもの(例えば、配列変化又は融合蛋白質の生成)であることができる。
【0080】
一態様で、抗原は、アデノウイルス、アレナウイルス、ブニアウイルス、コロナウイルス、コクサッキーウイルス、サイトメガロウイルス、エプスタインバーウイルス、フラビウイルス、ヘパドナウイルス、肝炎ウイルス、ヘルペスウイルス、インフルエンザウイルス、レンチウイルス、麻疹ウイルス、ムンプスウイルス、ミクソウイルス、発癌ウイルス、オルソミクソウイルス、パピローマウイルス、パポバウイルス、パラインフルエンザウイルス、パラミクソウイルス、パルボウイルス、ピコルナウイルス、ポックスウイルス、狂犬病ウイルス、呼吸器合胞体ウイルス、レオウイルス、ラブドウイルス、風疹ウイルス、トガウイルス、及び水痘ウイルスなどのウイルス由来のものであるが、これらに限定されることはない。他のウイルスとして、T−リンパ増殖性ウイルス、例えば、ヒトT細胞リンパ増殖性ウイルス(HTLV−I及びHTLV−IIなどのHTLV)、ウシ白血病ウイルス(BLVS)及びネコ白血病ウイルス(FLV)が挙げられる。レンチウイルスとしては、限定しないが、ヒト(HIV−I又はHIV−2を含むHIV)、サル(SIV)、ネコ(FIV)及びイヌ(CIV)免疫不全ウイルスが挙げられよう。
【0081】
本発明の新規の手順によるモノクローナル抗体の生成により、HIV変異体のクレード全体を中和する潜在的能力のある新規特異性の発生をもたらすことができる。これは、例えば、ウイルスのエンベロープ蛋白質の構造上の必須構成要素の標的化、又は代わりに、宿主コレセプター蛋白質のいずれかによって達成できよう。
【0082】
好ましい一実施形態で、本発明の方法は、血球凝集素(HA)蛋白質又はノイラミニダーゼ(NA)蛋白質などのインフルエンザウイルス蛋白質に特異的な抗体を生成するのに使用してもよい。いくつかの実施形態で、HA蛋白質は、Hl、H2、H3、H4、H5、H6、H7、H8、H9、H10、H11、H12、H13、H14、H15及びH16から選択される。一態様で、HA蛋白質はH5である。一態様で、NA蛋白質は、Nl、N2、N3、N4、N5、N6、N7、N8及びN9から選択される。一態様で、NA蛋白質はN5である。特定の実施形態で、抗体はHA蛋白質のサブユニットに対して特異的である。
【0083】
別の態様で、抗原は、Aspergillus、Bordatella、Brugia、Candida、Ch1amydia、Coccidia、Cryptococcus、Dirofilaria、Escherichia、Francisella、Gonococcus、Histoplasma、Leishmania、Mycobacterium、Mycoplasma、Paramecium、Pertussis、Plasmodium、Pneumococcus、Pneumocystis、Rickettsia、Salmonella、Shigella、Staphylococcus、Streptococcus、Toxoplasma、Vibriocholerae Yersiniaからなる属より選択される感染作用物質由来のものである。一態様で、感染作用物質は、Plasmodium falciparum(熱帯熱マラリア原虫) ox Plasmodium vivax(三日熱マラリア原虫)から選択される。
【0084】
一態様で、抗原は、Enterobacteriaceae、Micrococcaceae、Vibrionaceae、Pasteurellaceae、Mycoplasmataceae、及びRickettsiaceaeより選択されるファミリー由来の細菌からのものである。一態様で、細菌は、Pseudomonas、Bordetella、Mycobacterium、Vibrio、Bacillus、Salmonella、Francisella、Staphylococcus、Streptococcus、Escherichia、Enterococcus、Pasteurella、及びYersiniaから選択される属のものである。一態様で、細菌は、Pseudomonas aeruginosa、Pseudomonas mallei、Pseudomonas pseudomallei、Bordetella pertussis、Mycobacterium tuberculosis、Mycobacterium leprae、Francisella tularensis、Vibrio cholerae、Bacillus anthracis、Salmonella enteric、Yersinia pestis、Escherichia coli及びBordetella bronchisepticaから選択される種由来である。
【0085】
好適な抗原として、ウイルスが結合する細胞性レセプター(例えば、CD4等)も挙げられる。
【0086】
本発明によれば、「〜に選択的に結合する」の用語は、抗体又はその抗原結合断片が、特定の蛋白質又は他の抗原に優先的に結合する能力を意味する。さらに具体的には、「選択的に結合する」の用語は、ある蛋白質と別のものとの特異的結合(例えば、抗原に対する、抗体又はその抗原結合断片)であって、結合のレベルが、任意の標準的アッセイ(例えば、イムノアッセイ)によって測定して、そのアッセイに対するバックグラウンコントロールよりも統計的に有意に高いことを意味する。例えば、イムノアッセイを実施する場合、コントロールは通常、抗体又は抗原結合断片を単独で含む(すなわち、抗原の非存在下の)反応ウェル/チューブであり、抗原非存在下での抗体又はその抗原結合断片による反応性の量(例えば、ウェルへの非特異的結合)が、バックグラウンドと考えられる。結合は、酵素イムノアッセイ(例えば、ELISA)、イムノブロットアッセイ等を含む、当該技術分野の水準にある様々な方法を用いて測定できる。
【0087】
本発明によれば、本発明での使用に好適な抗原として、同じ抗原由来の2種以上の免疫原性ドメイン若しくはエピトープ;同じ細胞、組織若しくは生物由来の2種以上の抗原、免疫原性ドメイン、若しくはエピトープ;及び/又は異なる細胞、組織若しくは生物由来の2種以上の異なる抗原、免疫原性ドメイン、若しくはエピトープを挙げることができる。実際、本発明は、1度の免疫化後に多重特異性のモノクローナル抗体を生成するための方法を提供する。例えば、本明細書に記載のマウス(例えば、MMTV−tTA/TRE−MYCマウス)を、細菌又は酵母にて組換え蛋白質として生成され、GST−融合蛋白質として作製された抗原などの2種以上の免疫原性ドメイン、エピトープ、又は他の抗原で、標準的な免疫化技術を用いて免疫化してもよい。免疫化されたマウスからのB細胞は次いで、例えば、精製蛋白質抗原で被覆されたプレート上にパニングすることによって単離してもよい。単離された抗原特異的B細胞は、MYC過剰発現を活性化し、継続的抗原の存在下にB細胞をインビトロで形質転換するために、抗生物質(例えば、ドキシサイクリン)の非存在下に培養してもよい。得られる細胞系はその後、抗体生産及び特異性についてスクリーニングしてもよい。このアプローチで、一度に200もの抗原に特異的なモノクローナル抗体を迅速に生産できるようになり、多くの蛋白質に対するモノクローナル抗体の供給を豊富にすることによって、プロテオミクスの分野での主な障壁を解消しうる。
【0088】
前記の方法は、1つの抗原又は複数の抗原の、多くの又はすべてのエピトープを認識する中和抗体組成物を生成するのに使用してもよい。例えば、1つのウイルスのいくつかの抗原で、又はいくつかの関連ウイルス株からの特定の抗原の複数の変異体でマウスを免疫化してもよい。単離された抗原特異的B細胞は、次いで抗原の特定のエピトープに対して特異的な抗体を生産する細胞についてパニングしてもよい。これらはその後、合わせて、ウイルス、又はウイルス株のパネルからの特異的抗原の全ての変異体の、殆ど又は全てのエピトープに結合する抗体組成物を生産してもよい。同様に、類似のエピトープとの交差反応性なしに、1つの特定の抗原又はエピトープへ結合することにつき、同様の方法によって抗体を選択してもよい。
【0089】
本発明によって生産される、予防又は治療剤として有用な抗体は、通常は組成物(製剤)の形態で提供される。本発明の一実施形態で、医薬組成物又は製剤は、有効な量の抗体又はその抗原結合断片、及び薬理学的に許容できる担体から調製される。薬理学的に許容できる担体は、当業者に周知である。本発明によれば、「薬理学的に許容できる担体」には、薬理学的に許容できる賦形剤及び/又は薬理学的に許容できる送達媒体で、好適なインビボの部位に製剤又は組成物を投与するための使用に好適なものが含まれる。薬理学的に許容できる担体は、抗体が患者の標的部位に到達すれば、その抗体が作用でき、好ましくはその患者に治療的恩恵をもたらすことのできる形態の製剤において、使用される抗体を維持できるものでもよい。従って、本発明はさらに、本発明によって生産される抗体又は誘導体の1種以上を用いて個体を処置する方法も包含する。本発明は、一実施形態で、病原による感染(例えば、コロナウイルス感染)若しくはそれに起因する疾患を含む、状態若しくは疾患を有するか、又はそれを発生する危険のある動物を処置する(その状態又は疾患の予防及び/又は治療的処置を含む)方法を包含する。この方法は、その状態若しくは疾患を有するか、又はそれを発生する危険のある動物に、本明細書に記載の通り本発明によって生産される1種以上の抗体又はその機能性誘導体を投与して、その疾患又は状態に起因する少なくとも1つの症状をその動物において予防又は低減することを含め、疾患又は状態を低減又は予防する工程を含む。本発明はマウスを用いて例証されているが、当該技術分野で知られている抗体生産用の任意の実験動物を使用してもよいことは当業者に理解されるであろう。本発明の方法は、細胞又はその組織を含め、抗体生産に好適な任意の生物で使用できる。好ましい動物として、脊椎動物綱の任意の動物、制限なく霊長類、齧歯類、家畜及び家庭用ペットを含む哺乳類(すなわち、哺乳動物)が挙げられる。一般的に、抗体の生産において、例えば、限定しないが、ウサギ、ヒツジ、ハムスター、モルモット、マウス、ラット又はニワトリなどの好適な実験動物が、それに対する抗体を所望される抗原に暴露される。好ましい実施形態で、動物は「ヒト化」動物、例えば、ヒト抗体を生じるヒトctlt−HSC細胞系及びヒトモノクローナル生産細胞系で再構成されたNOD/SCIDマウスである。
【0090】
(一般定義)
本発明の実施には、特に断らない限り、当業者に周知の、分子生物学(組換え技術を含む)、微生物学、細胞生物学、生化学、核酸化学、及び免疫学の常用の技術を利用しうる。そのような技術は、Molecular Cloning:A Laboratory Manual,第2版(Sambrookら、1989)およびMolecular Cloning:A Laboratory Manual,第3版(Sambrook及びRussel,2001),(本明細書では併せて「Sambrook」として引用する);Current Protocols in Molecular Biology(F.M.Ausubelら編、1987,2001年までの追補も含む);PCR:The Polymerase Chain Reaction,(Mullisら、編、1994);Harlow及びLane(1988)Antibodies,A Laboratory Manual,Cold Spring Harbor Publications,New York;Harlow及びLane(1999)Using Antibodies:A Laboratory Manual Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY(本明細書では併せて「Harlow及びLane」として引用する),Beaucageら、編、Current Protocols in Nucleic Acid Chemistry John Wiley & Sons,Inc.,New York,2000);並びにVaccines,S.Plotkin及びW.Orenstein,編、第3版(1999)などの文献において充分に説明されている。
【0091】
本発明によれば、本明細書で一般的に用いられる用語「抗原」は、自然発生的に、若しくは合成により得られる蛋白質(ペプチド、部分蛋白質、全長蛋白質)のあらゆる部分、細胞性組成物(全細胞、細胞溶解液又は破壊細胞)、生物(生物全体、溶解液、若しくは破壊細胞)、又は炭水化物(癌細胞で発現されているものなど)や他の分子若しくはその一部を意味する。抗原は、その抗原が投与される個体の細胞及び組織内で遭遇する同一若しくは類似の抗原に対する、抗原特異的な免疫応答(例えば、体液性免疫応答及び/又は細胞媒介性免疫応答)を誘発する。或いは、抗原は免疫寛容原として作用できる。免疫応答の刺激を言う場合、「抗原」の用語は、「免疫原」の用語と互換的に用ることができる。抗原は、単一エピトープほどに小さくてもよいし、又はより大きくてもよく、また複数のエピトープを含むことができる。従って、抗原のサイズは、約5〜12アミノ酸(例えば、ペプチド)と小さくてよいし、全長蛋白質ほどの大きさでもよく、これには多量体、及び融合蛋白質、キメラ蛋白質、全細胞、微生物全体、又はその一部(例えば、全細胞の溶解液や、微生物の抽出物)が含まれる。さらに、抗原には炭水化物が含まれうる。
【0092】
所定抗原の「免疫原性ドメイン」は、動物に投与された場合に免疫原として作用する少なくとも1つのエピトープを含む、抗原の任意の部分、断片又はエピトープ(例えば、ペプチド断片、サブユニット、抗体エピトープ又は他のコンホメーションに基づくエピトープ)でありうる。例えば、単一蛋白質が、複数の異なる免疫原性ドメインを含みうる。免疫原性ドメインは、体液性免疫応答の場合のように蛋白質内で線状の配列である必要はない。
【0093】
エピトープは、本明細書で、所定抗原内の、免疫応答を誘発するのに充分な単一免疫原性部位、又は所定抗原内の、免疫応答を抑制、削除若しくは不活性化させるのに充分な、単一免疫寛容原性部位と定義する。当業者は、T細胞エピトープのサイズ及び組成がB細胞エピトープと異なること、並びにクラスI MHC経路を通じて提示されるエピトープは、クラスII MHC経路を通じて提示されるエピトープと異なることを認識するであろう。エピトープは、線状配列であっても、又はコンホメーションに基づくエピトープ(保存結合領域)であってもよい。「ワクチン接種」又は「免疫化」は、単独又はアジュバントと共に抗原を投与することの結果としての、抗原又はその部分に対する免疫応答の誘発(誘導)を意味する。免疫化の概念は、当該技術分野で周知である。抗原の投与により誘発される免疫応答は、抗原を投与しないときと比較した場合の、免疫応答の任意の様相(例えば、細胞媒介性応答、体液性応答、サイトカイン生産)の検出可能な任意の変化でありうる。
【0094】
本発明によれば、抗体は免疫グロブリンドメインを含むことを特徴とし、よって、それらは蛋白質の免疫グロブリンスーパーファミリーのメンバーである。一般的には、抗体分子は2種の鎖を含んでいる。鎖の1方の型は、重鎖又はH鎖と称され、もう一方の型は軽鎖又はL鎖と称される。2つの鎖は、等モル比で存在し、各抗体分子は通常、2つのH鎖と2つのL鎖とを有している。2つのH鎖はジスルフィド結合によって連結され、H鎖の各々はジスルフィド結合によってL鎖に連結されている。L鎖は、ラムダ(λ)及びカッパ(κ)鎖と称される2種のみである。これに対し、H鎖の主要クラスは5つあり、イソタイプと称される。その5つのクラスには、免疫グロブリンM(IgM又はμ)、免疫グロブリンD(IgD又はδ)、免疫グロブリンG(IgG又はλ)、免疫グロブリンA(IgA又はα)、及び免疫グロブリンE(IgE又はε)が含まれる。このようなイソタイプ間の弁別的特性は、免疫グロブリンの定常ドメインによって定義付けられ、それらを以下に詳説する。ヒト免疫グロブリン分子は、9つのイソタイプ、IgM、IgD、IgE、IgG1(γl)、IgG2(γ2)、IgG3(γ3)及びIgG4(γ4)を含むIgGの4つのサブクラス、並びにIgAl(αl)及びIgA2(α2)を含むIgAの2つのサブクラスを含む。ヒトにおいて、IgGサブクラス3及びIgMは、最も強力な補体活性化因子(古典的補体系)であるが、IgGサブクラス1と、ずっと程度は低いがIgGサブクラス2は、古典的補体系の中度から低度の活性化因子である。IgG4サブクラスは補体系(古典的又は副補体系)を活性化しない。副補体系を活性化することが知られている唯一のヒト免疫グロブリンイソタイプは、IgAである。マウスでは、IgGサブクラスはIgG1、IgG2a、IgG2b及びIgG3である。マウスIgG1は補体を活性化しないが、IgG2a、IgG2b及びIgG3は補体活性化因子である。
【0095】
免疫グロブリン分子の各々のH又はL鎖は、L鎖可変ドメイン(VLドメイン)及びL鎖定常ドメイン(CLドメイン)、並びにH鎖可変ドメイン(VHドメイン)及びH鎖定常ドメイン(CHドメイン)と称される2つの領域を含む。完全なCHドメインは、3つのサブドメイン(CH1、CH2、CH3)、及びヒンジ領域を含む。合わせて、1つのH鎖と1つのL鎖が、免疫グロブリン可変領域を有する免疫グロブリン分子の腕を形成できる。完全な免疫グロブリン分子は、2つの会合した(例えば、ジスルフィド結合した)腕を含んでいる。よって、免疫グロブリン全体の各々の腕は、VH+L領域、及びCH+L領域を含んでいる。本願明細書で用いる「可変領域」又は「V領域」という用語は、VH+L領域(Fv断片としても知られている)、VL領域又はVH領域を意味する。本願明細書で用いる「定常領域」又は「C領域」という用語もやはり、CH+L領域、CL領域又はCH領域を意味する。
【0096】
プロテアーゼを用いた免疫グロブリンの限定消化で、2つの断片を生産してもよい。抗原結合断片は、Fab、Fab’、又はF(ab’)2断片と称される。抗原への結合能力を欠く断片は、Fc断片と称される。Fab断片は、VH領域及びCH領域の一部(CH1ドメイン)と対合するL鎖(VL+CLドメイン)を有する、免疫グロブリン分子の一方の腕を含んでいる。Fab’断片は、Fab断片に対応しており、ヒンジ領域の一部がCH1ドメインに付着している。F(ab’)2断片は、典型的にはヒンジ領域において、ジスルフィド結合を介して通常互いに共有結合している2つのFab’断片に対応する。
【0097】
CHドメインは免疫グロブリンのイソタイプを決定付け、そのイソタイプに応じて異なる機能的特性が与えられる。例えば、μ定常領域は、IgM分子の五量体の凝集体形成を可能とし、α定常領域は二量体の形成を可能とする。
【0098】
免疫グロブリン分子の抗原特異性は、可変、すなわちV領域のアミノ酸配列によって与えられる。このように、異なる免疫グロブリン分子のV領域は、それらの抗原特異性に応じて有意に変動しうる。V領域の特定の部分は、他の部分よりも保存されており、フレームワーク領域(FW領域)と称される。これに対し、V領域の特定の部分は、可変性が高く、超可変領域と命名されている。VLとVHドメインが免疫グロブリン分子内で対合すると、各ドメイン由来の超可変領域が会合して、抗原結合部位を形成する超可変ループを創出する。よって、超可変ループが免疫グロブリンの特異性を決定し、それらの表面が抗原に対して相補的であるので、相補性決定領域(CDR)と命名されている。
【0099】
V領域のさらなる可変性は、免疫グロブリンV領域をコード化する遺伝子セグメントの組み合わせの可変性によって与えられる。免疫グロブリン遺伝子は、体細胞性に再配置して免疫グロブリン分子をコード化する再配置免疫グロブリン遺伝子を形成する、複数の生殖系遺伝子セグメントを含む。VL領域は、L鎖V遺伝子セグメント及びJ遺伝子セグメント(連結セグメント)によってコード化される。VH領域は、H鎖V遺伝子セグメント、D遺伝子セグメント(多様性セグメント)及びJ遺伝子セグメント(連結セグメント)によってコード化される。
【0100】
L鎖及びH鎖V遺伝子セグメントの両者とも、実質的なアミノ酸配列可変性の3つの領域を有している。かかる領域は、それぞれL鎖CDR1、CDR2及びCDR3、並びにH鎖CDR1、CDR2及びCDR3と称される。L鎖CDR1の長さは、異なるVL領域間で実質的に変動しうる。例えば、CDR1の長さは、約7アミノ酸から約17アミノ酸までで変動しうる。これに対し、L鎖CDR2及びCDR3の長さは、通常は異なるVL領域間で変動しない。H鎖CDR3の長さは、異なるVH領域間で実質的に変動しうる。例えば、CDR3の長さは、約1アミノ酸から約20アミノ酸までで変動しうる。各H及びL鎖CDR領域には、FW領域が隣接している。
【0101】
免疫グロブリン分子の他の機能面として、免疫グロブリン分子の価数、免疫グロブリン分子の親和性、及び免疫グロブリン分子の結合力が挙げられる。本願明細書で用いる「親和性」は、免疫グロブリン分子上の単一部位で免疫グロブリン分子が抗原に結合する(すなわち、一価抗原に結合する一価Fab断片結合)強度を意味する。親和性は、免疫グロブリンが抗原に結合する強度の合計を意味する結合力とは異なっている。免疫グロブリン結合親和性は、当該技術分野で標準的な技術を使用して測定でき、この例として競合的結合技術、平衡透析法又はBIAコア法が挙げられる。本願明細書で用いる「価数」とは、免疫グロブリン分子あたりの異なる抗原結合部位の数(すなわち、抗原結合断片の抗体分子1つあたりの抗原結合部位の数)を意味する。例えば、一価免疫グロブリン分子は、一度に1つの抗原にしか結合できないが、二価免疫グロブリン分子は、一度に2種以上の抗原に結合できる、等々である。
【0102】
「個体」は、脊椎動物、好ましくは哺乳動物、より好ましくはヒトである。哺乳動物としては、限定しないが、家畜、競技用動物、ペット、霊長類、マウス及びラットが挙げられる。「個体」の用語は、「動物」、「被検者」又は「患者」の用語と互換的に使用できる。
【0103】
本発明の様々な態様は、以下の実験に記載されている。これらの実験結果は、例示目的のみで、本発明の範囲を限定することは意図しない。
【実施例】
【0104】
(実施例1)
以下の実施例は、MMTV−tTA/TRE−MYCマウスに由来するB細胞系の生産を示す。
【0105】
B細胞特異的に誘導性にMYCを過剰発現できるマウスのAN1/T3集団からB細胞系を生成するために、生後8週間、MMTV−tTA/TRE−MYCマウスをドキシサイクリンにて維持し、その後正常食餌に切り替えた。そのマウスで、外見的に顕著なリンパ節腫脹及び脾腫大が発生し、リンパ系異常増殖に伴い一貫して認められる多くの臨床徴候(毛皮汚損、弯曲姿勢、努力性呼吸、貧血、臓器肥大等)を表した。マウスを安楽死させて、それらのリンパ節及び脾臓を分析用に集めた。リンパ節のいくつか及び脾臓の一部から、単一細胞懸濁液を生成した。それらの細胞をフローサイトメトリー分析に使用した。腫瘍を初期に特徴付けすることで、活性化B細胞の高い有病率が実証された。同じ細胞のいくつかを用いて、培養物を播種し、B細胞系を生成させた。これらの細胞は、リンパ球培地(RPMI 1640、10%ウシ胎児血清、ペニシリン/ストレプトマイシン、L−グルタミン、HEPES、非必須アミノ酸、ピルビン酸ナトリウム及び2−β−メルカプトエタノール)で培養した。およそ14〜21日後に、ウェルのいくつかが細胞系のクローン増殖を呈し始めた。細胞が大きなフラスコでの成長に適応し、細胞の一部は凍結保存するまで、注意深く増殖させた。
【0106】
最初に2種の細胞系を選び、TBLK6及びTBLK7と命名した。両細胞系の試料をCD138(Y軸)及びCD40(X軸)に特異的な抗体で染色し、フローサイトメトリーによって分析した。図1(上部パネル)に示す通り、2種の細胞系は、異なるレベルのCD138発現を示し、CD40発現はほとんど乃至は全く示さなかった。播種後の組織培養培地への免疫グロブリン分泌のレベルも測定した。各細胞系由来の細胞の規定数(105細胞)を、1mlの成長培地単独、又はIL−4、IL−6若しくは両者のいずれかを補充した培地を含む24穴プレートのウェルに播種した。培養開始後、1、3又は4日に上清の試料を採取した。その後、抗IgM捕獲ELISA用に上清を使用した。図1(下部パネル)に表す結果より、両細胞系は自発的に免疫グロブリンをそれらの生育培地に分泌するが、最初の接種物にIL−4及びIL−6を添加することによって分泌のレベルを増加できることが示される。次いで、TBLK6及びTBLK7両者により分泌される免疫グロブリンはIgMであることを実証した。真のモノクローナル集団を生成し続けるために、それらの細胞系の両方を単一細胞にクローン化できる。さらに、セルソーティングによってAN1/T3集団を単離するために使用できるMMTV−tTA/TRE−MYCバイジェニックマウスのコホートを、現在生成している。
【0107】
(実施例2)
以下の実施例は、Eμ−MYC/BCRHEL/sHEL遺伝子導入マウスで生じる腫瘍及び細胞系の表面表現型とHEL特異的抗体生産を実証する。
【0108】
野生型マウス(塗りつぶしたヒストグラム)、BCRHEL遺伝子導入マウス(明灰色実線)、BCRHEL/sHELマウス(網点灰色線)、及びEμ−MYC/BCRHEL/sHEL三重遺伝子導入マウス(黒実線)から得た細胞を、表示の表面マーカーに対して特異的な抗体で染色し、フローサイトメトリーによって分析した。図2(上部パネル)に示すデータは、B220+脾臓細胞上の表示マーカーの発現を表す。
【0109】
細胞系TBL−I、TBL−8、TBL−14及びTTLN9(全て、Eμ−MYC/BCRHEL/sHELマウスで生じる腫瘍由来)の105細胞を、24穴プレートに、サイトカインを全く追加しない1mlの生育培地中へ播種した。4日後に上清の試料を採取して、総IgMの濃度(A)、さらにHEL特異的IgMの力価(B)についてアッセイした。様々なコントロールマウスからの血清も含めて、細胞系における抗体生産を比較する手段を提供する。これらのマウスには、野生型C57/BL6マウス(WT)、BCRHEL遺伝子導入マウス(BCR−tg)、sHEL遺伝子導入マウス(Ag−tg)、BCRHEL/sHEL二重遺伝子導入マウス(BCR/Ag−tg)、Eμ−MYCマウス及び腫瘍担持Eμ−MYC/BCRHEL/sHEL三重遺伝子導入マウス(BL)が含まれた。図2(下部パネル)に示す結果は、Eμ−MYC/BCRHEL/sHELマウスで生じる腫瘍及び細胞系におけるHEL特異的力価を示す。
【0110】
(実施例3)
以下の実施例は、MYCがB細胞寛容を破壊して、抗原誘発、MYC−依存性B細胞リンパ腫を起こすことができることを実証する。
【0111】
寛容を破壊することによって、MYCはB及びT細胞を自己抗原による持続的刺激に曝して、細胞増殖、及び確実になるゲノムの障害を助長できる力をもたらすのかもしれない。MYCを過剰発現しているB細胞の新生物発生前状態の検討は、B細胞寛容の調節におけるMYCの新規な役割の解明につながる。
【0112】
フローサイトメトリー分析を、野生型マウス(塗りつぶしたヒストグラム)、ずっとドキシサイクリンにて維持しておいたMMTV−rtTA/TRE−M7C/BCRHEL/sHELマウス(明灰色線)、及び安楽死の1週間前にドキシサイクリンから離脱させておいたMMTV−rtTA/TRE−MYC/BCRHEL/sHELマウス(暗灰色線)から得たリンパ節細胞について実施した。図3(上部パネル)は、B細胞、CD69(A)、及びB7−2(CD86)(B)の抗原依存性活性化後に上方調節される2種の分子に対する抗体で細胞を染色した場合の結果を示す。CD69及びB7−2の示唆レベルは、細胞のB220+画分で表れ、フローサイトメトリーによりチトクロムC染色細胞上でゲート開閉することによって確認される。結果は、MYCの急激な過剰発現後に活性化B細胞が出現することを示唆している。
【0113】
図3(下部パネル)は、活性化B細胞の蓄積に、MYCの連続的な過剰発現が必要であることを示す。グラフの各データポイントは、個々のマウスのリンパ節で検出された活性化B細胞の数を表す。4匹のマウスのコホートを、各時間のポイントに用いた。この図は、誘導されたMMTV−rtTA/TRE−M7C/BCRHEL/sHELマウスでの活性化B細胞の蓄積の惹起及び維持におけるMYCの必要性を示す。
【0114】
野生型マウス(1)、BCRHELマウス(2)、BCRHEL/sHELマウス(3)、顕性腫瘍の発生前のEμ−MYC/BCRHEL/sHELマウス(4)、ずっとドキシサイクリンにて維持しておいたMMTV−rtTA/TRE−M7C/BCRHEL/sHELマウス(5)、及び血清採取前28日にドキシサイクリンから離脱しておいたMMTV−rtTA/TRE−M7C/BCRHEL/sHELマウス(6)から血清を得て、HEL(A)、又は総血清免疫グロブリン(B)に対するELISAによって三重でアッセイした。図4(上部パネル)に表す結果は、MYCの過剰発現後の血清中の自己抗体の蓄積を示す。
【0115】
MYCの過剰発現後の腎臓における自己抗体及び免疫複合体の蓄積を調べるために、野生型マウス(A)又はEμ−MYC/BCRHEL/sHELマウス(B)から組織学的検討用の腎臓を得た。組織を切片としてヘマトキシリン及びエオシンで染色し、顕微鏡画像を得た。倍率は100倍であった。免疫蛍光のために、野生型マウス(C)又はEμ−MYC/BCRHEL/sHELマウス(D)から腎臓を得た。凍結組織を切片化し、IgMに対するローダミン接合抗体で染色した。倍率は5倍であった。図4(下部パネル)に表す結果は、MYCの過剰発現後の腎臓における自己抗体及び免疫複合体の蓄積を示す。
【0116】
そうでなければ遺伝子導入自己抗原に対して免疫寛容のマウスが、B細胞系統中でMYCが単独で発現されれると、抗原に対する免疫応答を開始することを観察している。応答性B細胞は、活性化表現型に変換し、腎臓の免疫複合体病を引き起こす自己抗体を生産した。寛容の断絶の確立及び維持の双方に、MYCは必要であった。これらのマウスは、リンパ腫も発生し、それはBCR及びMYCに由来するシグナル間の協働の結果である可能性が最も高かった。これらの効果は、MYCがサイトカインの代替として働く能力に起因するかもしれない。MYCがB細胞増殖及び生存の双方に対するサイトカインの効果を模倣でき、実際にそれらの効果が必要とされることを見出した。我々のデータは、MYC過剰発現が、免疫学的寛容の断絶の確立及び維持に必要であることを示唆している。抗原依存的にMYCを過剰発現する自己反応性B細胞を不死化する能力によって、寛容B細胞の短い寿命によって限定されることは最早なく、また反応不顕性B細胞が骨髄腫細胞系と生産的に融合する周期に入ることを最早必要としないことから、ハイブリドーマ細胞系の生成のための従来からのアプローチよりもさらに多くの特異性の安定化が可能になる。
【0117】
(実施例4)
以下の実施例は、B細胞特異的な、Doxで調節されるMYCを過剰発現しているマウスを使用した、自己反応性バックグラウンドからのB細胞系の生成を実証している
【0118】
前記の結果は、MYCの過多が、可溶性自己抗原に対するB細胞寛容を破壊できることを示している。それらの研究では、MYCを過剰発現している、BCRHEL遺伝子導入B細胞は、HELに対する活発な応答を惹起し、悪性化が始まる前にポリクローナル自己免疫性リンパ球増殖性疾患を発症させることができた(図3及び4)。自己反応性B細胞におけるMYCの過剰発現は、MYCの増殖及び生存シグナルを提供する能力によって、B細胞をT細胞の援助に対して非依存性とすることができることが、我々の研究により示唆された。MYCを過剰発現している、自己反応性B細胞の増殖した集団は、その同族抗原への連続曝露と、MYCの過剰発現との双方に伴い、依存性のままのB細胞リンパ腫を生成し始めた。腫瘍担持マウスからのリンパ節、脾臓及び骨髄よりB細胞を収集でき、そしてBCRHEL導入遺伝子を発現し、且つ抗HEL IgMを分泌する多くの細胞系を、初代細胞の骨髄腫融合パートナーへの融合を行うことなく樹立できた。
【0119】
2つのさらなる環境を使用して、同様の結果を得ることができている。一例では、Ars/AlマウスをΕμ−MYC株に交雑した。それらのマウス(n=9マウス)は、平均して第36日齢でバーキット様リンパ腫を発生した。腫瘍は、成熟した活性化B細胞で構成されていた。それらの細胞は、表面にIgMを発現していた。それらの結果は、MYC過剰発現が、低親和性の抗DNA抗体という面において、自己反応性B細胞に対する寛容を破壊する能力を示している。第2例では、MMTV−rtTA/TRE−MYCマウスを用いた。それらのマウスは、食餌からのドキシサイクリンの排除後に、B細胞特異的で一時的に調節されたMYCの過剰発現を可能とする。ドキシサイクリン含有食餌を4月齢でマウスから離すと、活性化された末梢B細胞、抗核抗体を血清に、免疫複合体沈着物を腎臓に蓄積し、また6週以内にB細胞リンパ腫を発生した(平均的な例は、42日)。骨髄腫パートナー細胞への融合を行わずに、腫瘍から細胞系を樹立できている。これらの結果は、自己反応性B細胞レセプターを発現するB細胞系を生成する、新規アプローチとしての本システムの使用を示唆している。
【0120】
(実施例5)
以下の実施例は、MYCを過剰発現しているマウスで生産される抗体が、インビボで機能することを実証する。
【0121】
MMTV−tTA/TRE−MYCマウスにて生成したHEL特異抗体がインビボで機能しうるかどうかを調べるために、致死的なウイルス感染のモデルを使用することにした。ブタ狂犬病ウイルス(PRV)は、静脈内投与後にマウスで致死的となることがこれまでに示されている、アルファ−ヘルペスウイルスのメンバーである。PRVの変異体を2種構築した。一例では、US−9と称される遺伝子に対する配列の1つをGFPに融合させた。この構築体により、異なる設定にてウイルスで感染した細胞、及びウイルスの追跡が可能になる。重要なこととして、US−9蛋白質は、なお生産されており、その機能を保持している。ウイルスはその付加的な遺伝的カーゴにもかかわらず完全に病原性がある。HELに対する読み取り枠をコード化するPRVの変異体も生成した。感染細胞のウエスタンブロット分析によって、本ウイルス変異体がHELを発現することが示された(データは示さず)。
【0122】
GFPを発現しているウイルス又はHELを発現しているウイルス接種液(109PFUの力価を有するウイルス上清200μl)を、1:500に希釈したHEL特異抗体と氷上で1時間インキュベートした。混合液は次いで、4匹のマウスのコホートに静脈注射した。その後マウスは、ウイルス及び抗体混合液の投与後4日間モニターし、PRV感染に関わる重篤な神経学的臨床徴候を呈したら安楽死させた。図5に示す通り、GFPを発現しているウイルス(実線)は、HEL特異抗体の存在による影響を受けなかった。そのコホートでの死亡の動態は、野生型PRV株を用いて以前に発明者が得たものに類似していた。これに対し、HELを発現しているウイルス(破線)を与えたマウスでの死亡の動態は、有意に遅延し、そのマウスのコホートは、GFPを発現しているウイルスを注射したマウスのほぼ2倍も生き延びた。これらの実験に使用したウイルスは、US−9融合蛋白質を一過性に発現するのみであった。細胞への侵入後、それらは野生型PRVを生産した。死亡の遅延は、抗体がウイルス感染を阻害する能力を示唆している。
【0123】
(実施例6)
以下の実施例は、表現型としてH5N1を用いて、感染作用物質に対する新規抗体の発生を実証する。
【0124】
新自己抗原としてシステムに導入することによりMMTV−tTA/TRE−MYCマウスが目的の抗原に対する新規抗体を生成する能力を試験するために、レトロウイルスの骨髄キメラマウスの生成に依存するアプローチを用いた。pMISCV−H5−IRES−GFP(pMIG−H5)を生成するために、A/Ty/Ont/7732/66(H5N9)単離物からのヘマグルチニン(HA)のコーディング領域(cDNAのORFをコード化している部分−クローンに非翻訳部分が存在していた)を、pMIGにサブクローニングした。5−FUで処置したTRE−MYCマウスからの骨髄を、pMIG−H5及びpMIG−tTAで形質導入した。GFP発現のフローサイトメトリー分析により定量すると、およそ60%の細胞が形質導入されていた。3日後に、致死的に照射された(800/400R)マウス(Bl/6レシピエント)のコホートに細胞を移植した。骨髄キメラマウスをSEPTRA上に維持し、血液学的悪性疾患の外見的に顕著な臨床徴候を毎日観察した。再構成後6週で、数匹のマウスが、弯曲姿勢、毛皮汚損、外的に触知可能な腫瘍(脾腫大及びリンパ節腫脹)及び呼吸促迫を含む腫瘍発生の臨床徴候を示し始めた。再構成後7〜8週の間で、100%のマウスが腫瘍発生の徴候を呈した。マウスを屠殺し、GFP発現の証左のための蛍光顕微鏡によって分析した。屠殺されたマウスの100%に脾腫大、並びにリンパ節、骨髄及び脾臓にGFP発現があった。
【0125】
リンパ節及び脾臓を採取して、24穴プレートに蒔く単一細胞懸濁液を生成するのに使用した。残存細胞を培養して細胞系を生成させ始めるか、又はその後の分析用に凍結保存した。細胞をB細胞マーカーB220及びIgMに対して特異的な抗体を用いて染色し、その発現につきフローサイトメトリーによって分析した。図6は、GFP陽性細胞は、B細胞マーカーB220及びIgMの双方を発現することを示している。腫瘍は、成熟した活性化B細胞で構成される、MYC誘発性の抗原依存性腫瘍を生じる、成熟した活性化(ブラスティング)B細胞(バーキットリンパ腫のマウスモデルで観察されたものと同様)で構成されていた。これらの細胞を培養系に入れて、クローン増殖させた集団を、最初の接種後8日に開始して継代した。これは、モノクローナル抗体生産への現今のアプローチで通常成し遂げられるものより有意に速いタイムラインである。さらに、この新規アプローチで血球凝集素に対する新規抗体を生成できるようになる加速された時間フレームが、このアプローチを、新しく新生の感染症及び他の生物学的脅威に対する新規の中和抗体の発生のための迅速な応答プラットフォームとするはずである。骨髄細胞は、さらに特異性を導き、又は追加の抗原を加えるべく、今後マウスの大きなコホートを再構成するためにも凍結できる。マウスからその器官を収集した時間に血清を採取して、−20℃で保管した。腫瘍がH5 HA蛋白質に対する抗体を生成しているかどうかを確かめるために、血清を1:5000の希釈にてウエスタンブロット分析で使用した。pMIG−HAをトランスフェクトした293細胞由来の蛋白質溶解液(TritonX−100系溶解バッファー)、A/Ty/Ont/7732/66単離物由来の、HAを発現しているプラスミド、又は陽性コントロールプラスミドpCDNA3−H5を、SDS−PAGE(12%ゲル)によって分離し、PVDF膜に転写して、その膜を希釈血清でプローブ探査し、その後HRPを接合した二次抗体とインキュベーションした。図7は、腫瘍担持マウス由来の血清は、pMIG−HA(中央のレーン)又は陽性コントロールプラスミドpCDNA3−H5(右のレーン)のいずれかを発現している293細胞由来のHAと反応するが、トランスフェクトしていない293細胞由来の溶解液又はpMIGベクターのみをトランスフェクトした細胞由来の溶解液(左のレーン)とは反応しなかったことを示している。
【0126】
HA蛋白質は、およそ38〜40kDのバンドとして現れる。低分子量のバンド(19Kd)は、非特異的であるらしく、良好なローディングコントロールとして働く。展開されたバンド形成パターンは、H5、及び細胞におけるその正常な成熟及び処理の間に発生する切断産物と一致している。成熟HAは、2つのサブユニット(HA1及びHA2)で構成される。HA1サブユニット(〜40kDa)は、分子の球状頭部を形成し、表面糖蛋白質及び糖脂質上の宿主細胞シアル酸レセプターへの結合を担う。HA2サブユニット(〜20kDa)サブユニットは、侵入の間、宿主細胞のエンドソーム膜とのウイルスエンベロープの融合を担う。プロテアーゼ切断部位は、これらの2つのサブユニットを分かち、宿主プロテアーゼによる切断が、宿主細胞への侵入に必要である。ウイルスを中和するエピトープの大部分がHA1サブユニットに認められ、ウイルス−レセプター相互作用を阻害する。
【0127】
血清がウイルス−レセプター相互作用を遮断する能力を試験するために、H5N2、H1N1、H7N2、H3N2、及びH6N8サブタイプを含む多様なインフルエンザA単離物を用いた血球凝集阻害及びウイルス中和アッセイを使用した(図8)。3匹のマウス(1〜3)から、H5/tTA BM形質導入後6〜8週に単離した血清、又はリン酸緩衝性生理食塩水(PBS)(C1)を、マイクロタイタープレートの二重のウェルに沿って連続希釈した。連続希釈後に、インフルエンザAウイルスA/Mal/WI/944/82(H5N2)、A/NY/1469/02(H1N1)、又はPBS(ウイルスなし)の4種の凝集単位を各ウェルに添加して、30分間インキュベートした。次に、シチメンチョウ赤血球細胞を添加して30分間インキュベートし、血球凝集活性を検出した。第1カラムは、1:20の最終血清濃度を含有する。
【0128】
これらのデータは、H5N1に対して生成した新規抗体は、異なるクレード(H1N1)からのウイルスも中和できたことを示している。これは、MYCの過剰発現を用いる新しいアプローチが、ウイルスによりコード化されるエピトープで、自己蛋白質をおそらく模倣し、寛容機構がそれらの残基に対する良好な応答を妨げるために通常は無視されるものを明らかにできることを示唆している。これにより、特定のタイプ(インフルエンザ、又は他のタイプ)の異なるウイルス間の共通の残基を同定して、治療用に開発することができるようになるかもしれない。
【0129】
リンパ節及び脾臓細胞由来の細胞系を作製でき、その上清をHAとの反応性について試験して、HA蛋白質に対する抗体を生産する細胞系を生成できることを立証することができる。これらの上清及び/又は血清は、H5を含むインフルエンザウイルスを中和する能力についても試験できる。中和アッセイには、ある抗体が、インフルエンザ(フル)ウイルス及びヒツジ赤血球細胞を用いる凝集アッセイや、上皮性細胞のインビトロ、又はマウスのインビボの有効な感染を阻害できるかどうかの判定を含みうる。本発明のさらなる開示及び実施形態を、添付の原稿に見出すことができ、これを本願に引用してその全体を援用する。
【0130】
本発明の様々な実施形態を本明細書に詳細に記載しているが、それらの実施形態の改変及び適応が当業者に想起されるであろうことは明らかである。しかし、このような改変及び適応は添付の特許請求の範囲に記載の本発明の範囲に含まれることは特に理解されるべきである。
【技術分野】
【0001】
本発明は一般的に、癌原遺伝子を発現している動物において抗体生産細胞及び抗体を生産するための方法に関する。本発明はまた、自己寛容などの免疫束縛を通常受けている抗原に特異的な抗体の効率的な生産のための方法に関する。本発明はさらに、従来行われている抗体生産B細胞の骨髄腫融合パートナーとの融合を必要としない、抗体生産細胞及び抗体の生産に関する。
【0002】
(関連出願の相互参照)
本願は、2007年3月13日に出願せる米国仮出願第60/894654号、及び2007年5月18日に出願せる米国仮出願第60/939042号の米国特許法第119条(e)を相互参照する。各出願の開示全体を、引用することによりあらゆる目的で本明細書に援用する。
【背景技術】
【0003】
抗体、特にモノクローナル抗体の使用は、ルーチンの診断法のみならず基礎及び臨床研究の様々な分野に変革をもたらしている。モノクローナル抗体の臨床応用は、癌治療用の薬物の主たる新たな供給源として浮上してきた。例えば、CD20と称される表面蛋白質に対する抗体は、NHL患者や、さらには抗体が媒介する自己免疫疾患の患者の予後を劇的に改善している。
【0004】
高力価の中和抗体の発生は、多くのウイルス感染を患者が撃退する能力に関連している。最近の東南アジアでの鳥インフルエンザ(フル)の伝播は、感染しても生き存えた個体が、有効なB細胞抗体依存性の中和応答を惹起できる人々でもあることを示した。同様の知見が、その前年にSARS生存者の場合で示された。インフルエンザが原因となる米国での年間死亡者数は、年間30,000〜50,000名にのぼると見られる。インフルエンザによる全世界の死亡者数は、米国だけでの数字の20〜30倍高いと見られる。特に脆弱な2集団は、幼児と高齢者である。受動的に供給でき可能性のある中和抗体の開発は、インフルエンザ又はHIVなどの他のウイルスが原因となる死亡率を劇的に減少させるはずである。
【0005】
2005年5月の時点で、米国市場に18種の治療用モノクローナル抗体製品があった。世界的には、ほぼすべての消耗性疾患を処置するために200社より多くが開発中の、推定で500種のモノクローナル抗体製品があった。これらのモノクローナル抗体製品のうちおよそ80種が、臨床試験中である。モノクローナル抗体に対する世界市場は、2008年に167億ドルに増加すると見積もられる。
【発明の概要】
【発明が解決しようとする課題】
【0006】
モノクローナル抗体を生成するための従来からのアプローチは、マウス、又は最適な他の動物の超免疫化に基づいている。脾臓由来の抗体生産細胞はその後、収集して骨髄腫細胞融合パートナーに融合される。それらの抗体生産遺伝子を保持する細胞の選択は、順式及び逆式選択(forward and reverse selection)手順により成し遂げられる。これは、非常に強力な技術であることがわかってはいるものの、基礎生物学により課される制限のため、得ることができたはずの可能な特異性全体の90%より多くを逸する結果を招きやすい。それらの制限のいくつかに、「自己寛容」と称される機構や、抗体生産細胞と骨髄腫融合パートナーとの間の融合を成功裡に達成するのに必要な特定の要件が包含される。従って、上に挙げた制限に関連する問題を解決し、且つ目的の抗体を同定するのに必要な合計時間を短縮する、改善された抗体の生成方法が、当該技術分野で必要とされている。
【課題を解決するための手段】
【0007】
本発明の一態様は、MYCを誘導性に過剰発現する動物を、その動物においてMYCが過剰発現されない条件下で抗原に導き、その動物からB細胞を回収し、そしてMYCが過剰発現される条件下でそのB細胞を培養することによって、抗体生産細胞を生産するための方法に関する。
【0008】
いくつかの実施形態で、方法はさらに、動物を抗原に導く工程の後に、その動物においてMYCが過剰発現される条件下に動物を維持する工程を含む。
【0009】
いくつかの実施形態で、動物は、主にその動物のB細胞においてMYCを誘導性に過剰発現する。
【0010】
いくつかの実施形態で、動物は、MMTV−rtTA/TRE−MYCマウスである。いくつかの実施形態で、動物を抗原に導く工程は、抗原をコード化しているDNAを、その動物に遺伝的に移入することを含む。いくつかの実施形態で、抗原は自己抗原を含む。いくつかの実施形態で、動物はさらに、抗原を発現する。いくつかの実施形態で、抗原はgp120又はgp41などのHIV蛋白質を含む。
【0011】
いくつかの実施形態で、抗原は血球凝集素などのインフルエンザウイルス由来の抗原を含む。
【0012】
いくつかの実施形態で、マウスは、その動物を抗原に導く工程の間、MYC発現を抑止する抗生物質上に維持され、B細胞を培養する工程は、抗生物質非存在下に行なって抗体生産細胞を生産する。
【0013】
いくつかの実施形態で、方法はさらに、マウスを抗原に導く工程の後に、抗生物質への曝露からその動物を外す工程を含む。いくつかの実施形態で、抗生物質はドキシサイクリンである。
【0014】
本発明の別の態様は、MYCを誘導性に過剰発現する動物を、その動物においてMYCが過剰発現されない条件下で抗原に導き、その動物からB細胞を回収し、MYCが過剰発現される条件下でそのB細胞を培養し、そしてB細胞培養物から抗体を回収することによって抗体を生産するための方法に関する。
【0015】
いくつかの実施形態で、方法はさらに、動物を抗原に導く工程の後に、その動物においてMYCが過剰発現される条件下に動物を維持する工程を含む。
【0016】
いくつかの実施形態で、動物は主にその動物のB細胞においてMYCを誘導性に過剰発現する。
【0017】
いくつかの実施形態で、動物はMMTV−rtTA/TRE−MYCマウスである。いくつかの実施形態で、動物を抗原に導く工程は、抗原をコード化しているDNAを、その動物に遺伝的に移入することを含む。
【0018】
いくつかの実施形態で、抗原は自己抗原を含む。いくつかの実施形態で、動物はさらに、抗原を発現する。
【0019】
いくつかの実施形態で、抗原はgp120又はgp41などのHIV蛋白質を含む。
【0020】
いくつかの実施形態で、抗原は血球凝集素などのインフルエンザウイルス由来の抗原を含む。
【0021】
いくつかの実施形態で、抗体はヒト化抗体である。
【0022】
本発明の別の態様は、B細胞の重鎖又は軽鎖遺伝子のVDJ連結領域をコード化している核酸分子を、MYCを誘導性に過剰発現し且つB細胞の生産を防止する遺伝的修飾を含む動物からの骨髄由来の幹細胞に導入し、その骨髄由来の幹細胞をレシピエント動物に移入し、そのレシピエント動物からB細胞を回収し、MYCが過剰発現される条件下でそのB細胞を培養し、そしてB細胞培養物から抗体を回収することによって抗体を生産するための方法に関する。
【0023】
いくつかの実施形態で、方法はさらに、MYCがレシピエント動物において過剰発現される条件下にレシピエント動物を維持する工程を、その骨髄由来の幹細胞をレシピエント動物に移入する工程の後に含む。
【0024】
いくつかの実施形態で、方法はさらに、MYCがレシピエント動物において過剰発現されない条件下にレシピエント動物を維持する工程を、その骨髄由来の幹細胞をレシピエント動物に移入する工程の後に含む。
【0025】
いくつかの実施形態で、第1工程からのB細胞はヒトB細胞である。
【0026】
いくつかの実施形態で、B細胞はヒトから単離される。
【0027】
いくつかの実施形態で、ヒトは、抗体が媒介する自己免疫疾患に罹患している。
【0028】
いくつかの実施形態で、第1工程は、骨髄由来の幹細胞に、VDJ連結領域をコード化している核酸分子をレトロウイルスにより形質導入することを含む。
【0029】
いくつかの実施形態で、VDJ連結領域をコード化している核酸分子は、目的の抗原に選択的に結合する単離ヒトB細胞からクローニングされる。
【0030】
いくつかの実施形態で、VDJ連結領域をコード化している核酸分子は、ヒトドナーから得たB細胞に認められるIgH及びIgL配列より再配置したVDJ領域のPCR増幅断片である。
【0031】
いくつかの実施形態で、ヒトドナーは、健常ドナー、及び抗体が媒介する自己免疫疾患の患者からなる群より選択される。
【0032】
いくつかの実施形態で、骨髄由来の幹細胞はさらに、ヒトIgHをコード化している核酸分子、及びヒトIgLをコード化している核酸分子で形質導入される。
【0033】
いくつかの実施形態で、ヒトIgHをコード化している核酸分子とヒトIgLをコード化している核酸分子は、同じ核酸分子である。
【0034】
いくつかの実施形態で、ヒトIgHとヒトIgLをコード化している核酸分子の一方又は両方は、VDJ領域をコード化する同じ核酸分子である。
【0035】
いくつかの実施形態で、骨髄由来の幹細胞は、ヒト又はマウスからのものである。
【0036】
いくつかの実施形態で、マウスは、Rag−2−/−、SCID、DNA−PK−/−、Ku70−/−、Ku80−/−、XRCC4−/−及びμMT−/−からなるリストより選択される遺伝的修飾を含む、MMTV−rtTA/TRE−MYCマウスである。
【0037】
いくつかの実施形態で、レシピエント動物は致死的に照射されたマウスである。
【0038】
いくつかの実施形態で、レシピエント動物はSCIDマウスである。
【0039】
いくつかの実施形態で、動物は、VDJ領域に選択的に結合する抗原にインビボで導かれる。
【0040】
いくつかの実施形態で、抗体イソタイプは、IgA又はIgGである。
【0041】
いくつかの実施形態で、抗体のFc領域を遺伝的に修飾して、抗体が自己免疫反応及び関連免疫複合体沈着の問題を誘発する能力を最小にしている。
【0042】
本発明の別の態様は、細胞の生存及び増殖を促進する癌原遺伝子を誘導性に過剰発現する動物を、その癌原遺伝子がその動物において過剰発現されない条件下で抗原に導き、その動物からB細胞を回収し、そして癌原遺伝子が過剰発現される条件下でそのB細胞を培養することを含む、抗体生産細胞を生産するための方法に関する。
【0043】
本発明の別の態様は、細胞の生存及び増殖を促進する癌原遺伝子を誘導性に過剰発現する動物を、その癌原遺伝子がその動物において過剰発現されない条件下で抗原に導き、その動物からB細胞を回収し、癌原遺伝子が過剰発現される条件下でそのB細胞を培養し、そしてそのB細胞培養物から抗体を回収することによる、抗体を生産するための方法に関する。
【0044】
本発明の別の態様は、抗原をコード化している核酸分子を、細胞の生存及び増殖を促進する癌原遺伝子を誘導性に過剰発現する動物からの骨髄由来の幹細胞に導入し、その骨髄由来の幹細胞をレシピエント動物に移入し、癌原遺伝子がその動物において過剰発現される条件下にレシピエント動物を維持し、そのレシピエント動物からB細胞を回収し、癌原遺伝子が過剰発現される条件下でそのB細胞を培養し、そしてB細胞培養物から抗体を回収することによる、抗体を生産するための方法に関する。
【0045】
本発明の別の態様は、細胞の生存及び増殖を促進する癌原遺伝子をコード化している核酸分子を、動物からの骨髄由来の幹細胞に導入し、その骨髄由来の幹細胞をレシピエント動物に移入し、そのレシピエント動物からB細胞を回収し、抗原をコード化している核酸分子をそのB細胞に導入し、癌原遺伝子が過剰発現される条件下でそのB細胞を培養し、そしてB細胞培養物から抗体を回収することによる、抗体を生産するための方法に関する。
【0046】
本発明の別の態様は、細胞の生存及び増殖を促進する癌原遺伝子をコード化している核酸分子を、動物からの骨髄由来の幹細胞に導入し、その骨髄由来の幹細胞を第1レシピエント動物に移入して、その第1レシピエント動物からB細胞を回収し、抗原をコード化している核酸分子をそのB細胞に導入し、そのB細胞を第2レシピエント動物に移入し、その動物において癌原遺伝子が過剰発現される条件下に第2レシピエント動物を維持し、その第2レシピエント動物からB細胞を回収し、癌原遺伝子が過剰発現される条件下でそのB細胞を培養し、そしてB細胞培養物から抗体を回収することによる、抗体を生産するための方法に関する。
【0047】
いくつかの実施形態で、癌原遺伝子はMYCである。
【0048】
いくつかの実施形態で、骨髄由来の幹細胞は、抗アポトーシス蛋白質をコード化している核酸分子で形質導入されている。
【0049】
いくつかの実施形態で、抗アポトーシス蛋白質は、Bcl−2である。
【0050】
いくつかの実施形態で、核酸分子は、レトロウイルスにより導入される。
【0051】
いくつかの実施形態で、骨髄由来の幹細胞は、ヒトからのものである。
【0052】
いくつかの実施形態で、骨髄由来の幹細胞は、条件付きで不死化された長寿命の造血幹細胞である。
【0053】
いくつかの実施形態で、レシピエント動物は、亜致死的に照射されたNOD/SCIDマウスである。
【0054】
いくつかの実施形態で、動物は、ヒトIg座位をコード化している核酸分子に対して遺伝子導入されている。
【図面の簡単な説明】
【0055】
【図1】図1は、TBLK6及びTBLK7細胞系の表面上での、CD138(Y軸)及びCD40(X軸)の発現を(上部パネル)、そしてTBLK6及びTBLK7細胞系からのIgM分泌の分析を(下部パネル)示す。
【図2】図2は、Eμ−MYC/BCRHEL/sHEL遺伝子導入マウスで生じる腫瘍及び細胞系の表面表現型を(上部パネル)、そしてEμ−MYC/BCRHEL/sHELマウスで生じる腫瘍及び細胞系における免疫グロブリン生産(A)及びHEL特異的力価(B)を(下部パネル)示す。
【図3】図3は、MYCの急激な過剰発現後の活性化B細胞の外観を(上部パネル)、そしてMYCの連続的な過剰発現の間での活性化B細胞の蓄積を(下部パネル)示す。
【図4】図4は、MYCの過剰発現後の血清中の自己抗体の蓄積を(上部パネル)、そしてMYCの過剰発現後の腎臓における自己抗体及び免疫複合体の蓄積を(下部パネル)示す。
【図5】図5は、HELを発現するPRV変異体の致死的攻撃からの、新規HEL特異抗体でのマウスの保護を示す。
【図6】図6は、レトロウイルスによるキメラマウスで発生するB細胞腫瘍の表面表現型を示す。
【図7】図7は、レトロウイルスによるキメラマウスから得た血清と、HAを発現している細胞溶解液との反応性を実証するウエスタンブロット分析を示す。
【図8】図8は、レトロウイルスによるキメラマウスから得た、マウス血清の血球凝集阻害分析を示す。
【図9】図9は、MYCを過剰発現しているマウスの使用により得られる抗体特異性をヒト化する、レトロウイルスのベクターによる2種のアプローチを示す。
【発明を実施するための形態】
【0056】
本発明は、従来の抗体生産に伴う問題の多くを克服する、抗体生産細胞及び抗体を生産するための新規方法を提供する。一般的に、本発明は、癌原遺伝子を発現している動物、例えば、MYC又はAkt(特に好ましくはMYC)を過剰発現する動物において、抗体生産細胞及び抗体を迅速に生産するための方法に関する。本明細書に開示する方法によって、抗体生産B細胞を骨髄腫融合パートナーと融合させる必要なしに、モノクローナル抗体を生産することが可能になり、抗体を生産するのに必要な時間が短縮される。本明細書に開示する方法はさらに、免疫化されたマウスからのB細胞が骨髄腫パートナーと首尾良く融合するために、従来の抗体生産技術に必要とされるような細胞周期の特定段階にあることの必要性を排除する。本発明はまた、自己寛容などの免疫束縛を通常受けている抗原に特異的な抗体の効率的な生産も可能にする。例えば、本方法は、自己抗原に対するモノクローナル抗体を生産するために使用してもよい。
【0057】
本発明者らは、MYCの過多が、可溶性抗原に対するB細胞寛容を破壊できることを実証している。例えば、MYCを過剰発現している、BCRHEL遺伝子導入B細胞は、sHELに対する活発な応答を惹起し、悪性化が始まる前にポリクローナル自己免疫性リンパ球増殖性疾患を発症させる(図3及び4参照)。自己反応性B細胞におけるMYCの過剰発現は、MYCが増殖及び生存シグナルをもたらす能力によって、B細胞をT細胞の援助に対して非依存性とすることができる。MYCを過剰発現している、自己反応性B細胞の増殖した集団は、その同族抗原への連続曝露と、MYCの過剰発現との双方に伴い、依存性のままであるB細胞リンパ腫を発生する。腫瘍担持マウスからのリンパ節、脾臓及び骨髄より収集したB細胞を用いて、BCRHEL導入遺伝子を発現し、且つ抗HEL IgMを分泌する多くの細胞系を、初代細胞の骨髄腫融合パートナーへの融合を行うことなく樹立できる(図2参照)。本プロトコルは、以下に記載の通り、事実上任意の抗原に容易に適用できる。
【0058】
同様の結果が、2つのさらなる動物でのプロトコルを用いて得られている。一例は、Ars.AlマウスとΕμ−MYC株との間の交雑種に由来するマウスである。それらのマウスでは、バーキット様リンパ腫の発生が平均して36日齢に起こる。この腫瘍は成熟した活性化B細胞で構成されている。それらの細胞は、表面にIgMを発現する。このように、本発明者らは、MYC過剰発現が、低親和性の抗DNA抗体という面において自己反応性B細胞に対する寛容を破壊できることを実証している。第2の例では、食餌からのドキシサイクリン排除後に、B細胞特異的で一時的に調節されるMYCの過剰発現を可能とする、MMTV−rtTA/TRE−MYCマウスを利用する。ドキシサイクリン含有食餌を4月齢でマウスから離すと、マウスは活性化された末梢B細胞、抗核抗体を血清に、免疫複合体沈着物を腎臓に蓄積し、また6週以内(平均的な例では、42日)にB細胞リンパ腫を発生した。重要なのは、骨髄腫パートナー細胞への融合を行わずに、腫瘍から細胞系を樹立できていることである。
【0059】
本発明は、少なくとも三態様のシステムの生物学に基づき、現今のアプローチよりも効力が有意に高められた、既知の特異性を有する抗体を生産する新規のアプローチを提供する。第一は、自己寛容の機構により課されるバリアの存在で、このシステムにより免疫束縛なしに抗体を生成できるようになる。このアプローチで、標準的な(当該技術分野の水準で)免疫化手順によって達成できない抗体特異性の生成が可能になる。さらに、標準的免疫化プロトコルで自己寛容を破壊する簡単な手段はない。第二は、目的の同族抗原が、B細胞異常増殖をインビボで推進し、リンパ腫を発生しうる抗体生産細胞と、得られるモノクローナル抗体生産細胞系の選択を可能とする。これにより、得られるモノクローナル抗体のスクリーニングを直接的に行うことができるため、開発のための時間が加速される。第三は、これらのマウスで生じる腫瘍から抗体生産細胞系を生成してクローン増殖する能力により、抗体生産脾臓B細胞を骨髄腫融合パートナーに融合させる必要性が排除される。従来の融合プロセスは、かなり非効率的で、免疫化後に増殖するB細胞の画分の不死化が可能になるにすぎず、このため、モノクローナル抗体の形態で誘導しうる特異性の数が限定されてしまう。この非効率性の理由は多くあり、例えば、細胞周期のS期にあるB細胞の必要性、細胞に保持される必要のあるB細胞と骨髄腫融合パートナーとに由来する染色体の適切な数及び組み合わせ、HAT及び6−TG選択で生存する必要性、等である。まとめると、モノクローナル抗体の生成のための本発明の新規なアプローチは、従来のアプローチよりも随分短い時間枠にて、より多くの抗体を提供する。現在利用できるツールを用いた、そのアプローチへのいくつかの変更があり、選択されるその実施形態を以下にまとめる。
【0060】
本発明の一実施形態で、抗体を生産するために使用される好適なヒト以外の動物には、抗体を生産でき、且つ細胞の生存及び増殖を促進する少なくとも1種の癌原遺伝子を(例えば、遺伝的修飾、又は天然、若しくは定方向突然変異の結果として)過剰発現する、ヒト以外の任意の動物が挙げられる。本発明の方法で用いられるヒト以外の動物によって過剰発現されるべき好ましい癌原遺伝子としては、限定しないが、MYC又はAkt(ミリスチル化物)が挙げられ、MYCが特に好ましい。一例として、MYCを過剰発現しているマウスを本明細書で詳説するが、他の好適な癌原遺伝子及び他の動物の使用を含む本プロトコルの改変や、プロトコルを実施する際の技術的詳細の改変は、本明細書の開示に基づいて企図されるはずであり、それらが本発明に包含されることは理解されるべきである。
【0061】
MYCを過剰発現している任意の動物を、本発明に使用してもよい。このような動物には、好ましくはヒト以外の哺乳動物が含まれ、より好ましくは齧歯類、さらにより好ましくはマウスが挙げられる。特に有用な動物は、MYCを主にB細胞集団に過剰発現しており、その例としてΕμ−MYCマウス株などが挙げられる。好ましい動物にはまた、誘導可能な態様でMYCを過剰発現するものも含まれる。これらの動物では、MYC過剰発現は、一時的な態様で調節でき、例えば、抗体の生産が開始するまで抑制できる。例えば、MMTV−rtTA/TRE−MYCマウス株に、ドキシサイクリン又はテトラサイクリンを誕生から目的の抗体を生産するための使用時までの間、投与してもよく、これにより自然発症リンパ増殖性疾患の形成が最小化される。MYC過剰発現は、マウスの食餌からドキシサイクリン又はテトラサイクリンを除去することによって開始させることができる。本発明での使用に好適な、多くのさらなる誘導可能な遺伝子発現/抑制システムが、当該技術分野で知られている。好適なマウスは、レトロウイルスの形質導入及び養子移入技術によって作り出してもよい。例えば、TRE−MYCマウスからの骨髄細胞又は精製B細胞を、レトロウイルスによりrtTAで形質導入し、レシピエントマウスに移入するか、又はインビトロで培養してもよい。得られる細胞は、移入又は培養の前に、さらに(例えば、目的の抗原で)形質導入してもよい。
【0062】
本発明の特定の実施形態で、MYCを過剰発現している細胞を保有している動物を、インビボで抗原に導き(例えば、暴露し)、抗原に対する免疫応答を惹起させてもよい。例えば、一態様で、当該技術分野で周知の、動物に外来抗原を導入する従来の経路によって(例えば、免疫化と類似の技術を用いて)、目的の抗原に動物を暴露してもよい。
【0063】
或いは、好ましい実施形態で、MYCを誘導性に発現する細胞を保有する動物を、当該技術分野で一般的な技術により目的の抗原を発現するように設計してもよく、又は目的の抗原を発現している細胞を動物に導入してもよい。例として、骨髄造血幹細胞の、レトロウイルスが媒介する形質導入;遺伝子導入動物の作製、若しくはMYCを過剰発現している動物と、抗原を発現する既存の遺伝子導入動物との交雑;又は動物への遺伝子送達のための他の任意の方法が挙げられる。いくつかの実施形態で、抗体の生産が望まれるまでMYCが過剰発現されない条件下に動物を維持する。動物は次いで、MYCが過剰発現されて目的の抗原に対して特異的な抗体を生産するB細胞を生成する条件下に維持してもよい。本発明者らは、抗原に対して特異的なB細胞は、MYC過剰発現がなければ抗原に対して寛容化されることになると考えている。いったんMYCの過剰発現が誘導されれば、B細胞は新自己抗原に対する寛容を破壊でき、抗原に対して特異的な抗体を生成することができる。
【0064】
正常AN−I細胞集団(B細胞発生時にT3としても知られる集団、自己抗原結合に際して骨髄において反応不顕性となったB細胞と考えられるもの)由来の細胞系を、複数のアプローチによって作製してもよく、そのうちのいくつかを以下に記載及び例証する。一実施形態で、AN−I集団は、受胎以降ドキシサイクリンにて維持されたMMTV−rtTA/TRE−MYCマウス由来ものである。本発明の方法によれば、それらのマウスは、目的の特異抗原も発現すべきである。前記のとおり、これは骨髄造血幹細胞の、レトロウイルスが媒介する形質導入、標準的遺伝子導入のアプローチ、又はマウスへの遺伝子送達のためのその他の任意の方法によって容易に成し遂げることができる。細胞は、およそ6週齡のマウスや、或いは野生型マウス、又は導入遺伝子の1つを保因するのみのマウスのいずれかの脾臓から単離できる。細胞は、インビトロで、好適なリンパ球培地(例えば、RPMI1640、10%ウシ胎児血清、pen/strep、L−グルタミン、2−βメルカプトエタノール、ピルビン酸ナトリウム、Hepes及び非必須アミノ酸)において、ドキシサイクリン(50nM)の存在下又は非存在下に蒔くことができる。細胞は例えば、24穴プレートにおいて、およそ2x106細胞/mlの密度で蒔かれるが、これらの工程は当業者によって容易に至適化又は改変できる。培地は通常、週1回交換する。このアプローチは、細胞の供給源及び状態(すなわち腫瘍性又は正常細胞、臓器等)によって、10〜70%の効率で細胞系を提供できる。細胞はクローン性の増殖について視覚的に調べてもよい。約14〜28日に増殖し始めるいずれのクローンでも、6穴プレートにおいて、最終的に組織培養フラスコにまで、ゆっくりと増殖させることができる。全てのマルチクローン性細胞系を、以前報告され、当該技術分野で公知の通りに、分析の前に限界希釈法によって単一細胞クローニングに付してもよい。
【0065】
別の実施形態で、目的の抗体を生産できるB細胞系の形質転換は、インビボで起こさせてもよい。例としては、受胎以降、ドキシサイクリン含有食餌にて維持されていたMMTV−rtTA/TRE−MYCマウスのコホート(cDNAをコード化しているプラスミドから目的抗原も発現するもの)を、6週齢で正常マウス固形飼料に切り替える。このマウスは、B細胞リンパ腫の発生に関わる臨床徴候(毛皮汚損、外見的に明白なリンパ節腫脹、脱水、不活発、後肢麻痺−上行性等)について毎日検査できる。さらに、マウスは定期的に供血でき、目的の抗原への反応性について調べることができる。一旦マウスが腫瘍を発生すれば、そのリンパ節、脾臓、骨髄、及び血清を採取する。こうして得られる細胞懸濁液は、FACS分析に使用してもよく、細胞を蒔いて前記のように細胞系を生成し、そして生存可能な初代腫瘍組織が継続的に得られるようにするため、残余の細胞を10%DMSO中で凍結する。血清は、目的の抗原を発現する細胞を染色するのに使用して、また特異的抗原に対するものであるウエスタンブロット及びELISAアッセイ用に使用してもよい。コントロールマウスには通常、野生型、及び単独に遺伝子導入したマウスが含まれる。
【0066】
別の実施形態で、MMTV−rtTA/TRE−MYCマウス(前記同様、受胎以降ドキシサイクリン含有食餌にて維持されているもの)からの精製B細胞を、選択抗原を発現するようにインビトロで形質導入できる。形質導入した細胞はその後、ドキシサイクリン(移入後のB細胞においてMYCの過剰発現を誘導するもの)にて維持されていないレシピエントマウス(例えば、野生型マウス)に養子移入する。マウスは次いで、前記のように生成された細胞系、及びB細胞リンパ腫の発生に伴う臨床徴候について毎日試験することができる。
【0067】
モノクローナル抗体生産細胞の樹立を成し遂げるためのさらなるアプローチは、細胞の生存及び増殖を促進する(そして好ましくは、調節可能(誘導可能、制御可能)である)癌原遺伝子と、アポトーシスを阻害する蛋白質をコード化している核酸分子との選択された組み合わせを発現するマウスからの、骨髄レトロウイルスのキメラの使用を含む。本実施形態で、組み合わせの例として、限定しないが、MYC−ER及びBcl−2が挙げられる。この後者のアプローチは、発現されるMYCを活性にするために4−ヒドロキシタモキシフェン(4OHT)を添加することを含む。本発明者らは、長寿命の造血幹細胞の条件付き不死化に関する実験においてMYC機能を首尾良く調節するような構築体を使用している。この場合、骨髄キメラマウスのコホートは、以下のように5FU富化骨髄由来幹細胞を用いて生成してもよい。骨髄由来造血幹細胞の場合、長寿命のHSCを富化するために、そしてそれらのインビボ増殖を誘導するために、5mg/マウスの5−フルオロウラシル(5FU)を静脈内に投与する。骨髄細胞は、5日後に大腿骨及び脛側から収集する。低張溶解バッファーを用いて、赤血球細胞を溶解する。残る細胞を、培地で2回洗浄して、15%熱不活性化ウシ胎児血清、ペニシリン/ストレプトマイシン、L−グルタミン、非必須アミノ酸、組換えヒトIL−3、IL−6及び幹細胞因子(SCF)を補充したDMEM培地を含む24穴プレートに2x106細胞/mlの濃度で蒔く。前記の通り、骨髄細胞は選択抗原を発現するように、インビトロで形質導入してもよい。細胞を第1スピン感染の前に24時間培養して、24時間毎に3回、この手順に付す。
【0068】
最後のスピン感染の翌日に、細胞をフローサイトメトリーによって分析する。例として、レンチウイルスにより形質導入した骨髄に由来するHSCを用いて、致死的に照射されたマウスを再構成してもよく、12週後のリンパ系器官におけるレポーター遺伝子GFPの発現を用いて細胞を追跡できる。この場合、マウスは正常末梢リンパ系区画(骨髄移植後8〜12週)を再構成してもよい。脾臓、GFP+AN−1/T3細胞は次いで、それらマウスから単離し、前記のようにインビトロ不死化プロトコルに用いてもよい。重要な相違は、システムからドキシサイクリンを排除する代わりに、4OHTを培地に添加することである。或いは、lmg/マウスの4OHTで週1回腹腔内に処置した野生型レシピエントマウスのコホートに、ソートされたGFP+AN−1/T3細胞を養子移入できる。マウスは、B細胞リンパ腫又は白血病の発生に伴う臨床徴候の出現につき毎日モニターする。得られる腫瘍を収集し、MYC機能のレギュレーターとしてドキシサイクリンの代わりに4OHTを用い、本節に既に記載したようにB細胞系を生成するために使用できる。
【0069】
別の実施形態で、条件付きで不死化した骨髄由来の幹細胞系を、前記方法における5FU富化骨髄由来幹細胞の代わりに使用してもよい。いくつかの実施形態で、細胞の生存及び増殖を促進する(そして好ましくは調節可能(誘導可能、制御可能)である)癌原遺伝子と、アポトーシスを阻害する蛋白質をコード化している核酸分子との前記組み合わせ(MYC−ERとBcl−2との組み合わせが例示される)を発現する細胞系を使用してもよい。条件付きで不死化した骨髄由来の幹細胞系及びそれらの作製の詳細な説明及び例は、国際公開第WO2007/047583号パンフレットに記載されており、その内容を引用することにより本明細書に援用する。WO2007/047583号パンフレットは、癌原遺伝子と抗アポトーシス遺伝子及び本発明で使用してもよいそれらの誘導体とのさらなる組み合わせの詳細な説明を提供する。特定の実施形態で、細胞系は、選択抗原をそれらが発現するように、インビトロで形質導入してもよい。
【0070】
目的の抗原に対して特異的な抗体のポリクローナル集団や、さらにはそれらの抗体を生産する細胞は、動物の組織(脾臓、リンパ節等)又は血清から直接、単離してもよい。別の実施形態で、抗体生産細胞のモノクローナル集団を、標準的手順により単離してもよく、またモノクローナル抗体を培養培地から回収してもよい。
【0071】
本発明の方法はまた、ヒト抗体を生産する細胞系を作製するのに使用してもよい。本実施形態を成し遂げるための2種の例示的技術として、(1)ヒト免疫グロブリン座位(IgH及びIgL)をコード化している遺伝子導入BAC構築体を保因するマウス株の、Eμ−MYC株若しくはMMTV−rtTA/TRE−MYC導入遺伝子を含むマウスのいずれかへの交雑(繁殖による)、又は(2)MYC−ER(又は別の好適な誘導可能癌原遺伝子)に対するcDNAを含む、レトロウイルスの骨髄キメラの使用が挙げられる。この後者のアプローチで、目的の同族抗原は、レトロウイルスによりコード化されるcDNAから新自己抗原として、従来の導入遺伝子から、又は遺伝子及び蛋白質送達の他の手段によって発現される。前記のものに比較した場合、本実施形態における重要な相違は、本設定にて生産される抗体がヒト蛋白質になるはずであるということである。
【0072】
ヒト抗体を生産する細胞系を作製する第2の方法は、所定の目的蛋白質に対する血清抗体価を有するヒトから得られる末梢血B細胞の単離に基づいている。B細胞は、標準的アプローチを用いて精製する。精製B細胞は、目的の特異性を備えたようなB細胞に対して富化するために、目的の蛋白質を被覆したプラスチックプレート上にパニングすることができる。或いは、目的の蛋白質は、磁気ビーズに接合させ、次いで目的の特異性を備えたB細胞を単離するのに使用できる。それらの細胞は、その後、単一細胞RT−PCR用のTerasakiプレートに単一細胞ソートすることができる。単離したB細胞の各々に対するIgH及びIgL遺伝子に対するVDJ連結領域につき、cDNAを生成する。それらのcDNA断片は次いで、ヒトIgH及び/又はIgLをコード化し、且つ多重クローニング部位をそれらの分子が普通はそれら自体のVDJ配列を有するはずである位置に含む、レトロウイルスベクター(LTR−IgH−IRES−IgL−LTRをコード化するレトロウイルスのベクター、又はIgLに対するもの、IgHのいずれか1つ)へとクローニングできる。得られるレトロウイルスを配列決定して、MYCを過剰発現し、且つB細胞の生産を防止する遺伝的修飾を含む、マウス由来の5FU富化骨髄由来HSCを形質導入するのに使用できる。例として、MMTV−rtTA/TRE−MYC/Rag−1−/−マウス、Eμ−MYC/Rag−1−/−マウス、又はIgH/IgLレトロウイルス及びMYC−ERに対するもので同時感染したRag−1−/−骨髄が挙げられる。さらなる例として、Rag−2−/−、SCID、DNA−PK−/−、Ku70−/−、Ku80−/−、XRCC4−/−、μMT−/−等の座位の遺伝的除去を含むマウスと交雑した、前記のMYCを過剰発現しているマウスの、任意のものが挙げられる。それらの細胞は次いで、前記のプロトコル(レトロウイルスの骨髄キメラマウス用)を用いて致死的に照射されたマウスを再構成するのに使用できる。特異的抗原をコード化している核酸をマウスに形質導入するのに、任意の手段(遺伝子銃、裸(naked)DNA免疫化、遺伝子導入マウス、レンチウイルスでの「遺伝子導入」マウス、組換え蛋白質の直接注射、その遺伝子に対するcDNAをコード化するワクシニアウイルス、組換えアデノウイルス、組換え酵母、又は哺乳動物細胞、tat−融合蛋白質等)を使用できる。
【0073】
本アプローチより得られる細胞系は、目的の蛋白質に特異的なヒト免疫グロブリン配列をコード化することになる。これは、ウイルス感染、腫瘍、細菌及び真菌等を含む様々な病原及び疾患に対する阻害抗体のカクテルの生成のための強力な新規アプローチである。本発明はまた、MMTV−tTA/TRE−MYCマウスを使用することで得られる抗体特異性をヒト化する方法も含んでいる。この場合、IgL座位にあるマウスIgH及びVJ領域より由来する、再配置したVDJ連結配列をPCR増幅する。それらの配列は、MMTV−tTA/TRE−MYCマウスより発生し、目的の抗原に対する抗体を生産することが既に示されているクローン細胞系から得ることができる。PCR増幅断片は、その中にPCR断片がクローニングされる多重クローニング部位を含むヒトIgH及びIgL配列をコード化するレトロウイルスのプラスミドへとクローニングできる(図9参照)。IgH及びIgL配列は、両方のcDNAが同じウイルスベクターから発現されるように、IRESエレメントによって離間されている。異なるウイルスベクターの使用により、所定の特異性を備えた、異なる抗体イソタイプ(IgA、IgG、IgM、IgE及びその異なるサブグループ)の生成が可能になる。さらに、Fc領域をコード化している配列をさらに修飾して、得られる抗体が自己免疫反応及び関連免疫複合体沈着の問題を誘発する能力を最小にしてもよい。得られるレトロウイルスは、Rag−1−/−/MMTV−tTA/TRE−MYCマウスから得られる骨髄由来造血幹細胞を形質導入するのに使用される。形質導入した細胞は、致死的に照射されたC57/BL6野生型マウスのコホートに移植できる。この結果得られるマウスは、一抗原に対して単一特異性であり、ヒト化抗体の追加された特徴のすべてを備えたモノクローナル抗体生産細胞を生成できる。
【0074】
別の実施形態で、目的の抗原に対するモノクローナルヒト抗体は、以下の方法によって生産できる。所定の細胞系に見られるIgH及びIgLからの特異的VDJ連結配列を増幅する代わりに、抗体が媒介する自己免疫疾患(例えば、シェーグレン症候群、ハシモト甲状腺炎、全身性エリテマトーデス、ワルデンストロームマクログロブリン血症等)に罹患している患者や、非ホジキンリンパ腫(バーキットリンパ腫、濾胞様リンパ腫、びまん性大細胞型B細胞性リンパ腫、MGUS及び複数の骨髄腫)に罹患している患者、又は健常ドナーのいずれかから得たB細胞に見られるIgH及びIgL配列から再配置したVDJに対するPCR増幅断片を単離できる。PCR断片は、前記レトロウイルス構築体にクローニングされる(これらのライブラリーの生成を円滑にするよう、2つの異なるレトロウイルスの構築体としても調製できる)。次いで、レトロウイルスのライブラリーを使用して、前記のように骨髄キメラマウスを生成するために、Rag−−/−/MMTV−tTA/TRE−MYCマウスから得られた骨髄由来造血幹細胞(HSC)を形質導入できる。骨髄キメラマウスは、ヒト免疫グロブリンを発現するB細胞のみを作り、免疫化の準備が整うまで(MYC過剰発現を抑制するために)ドキシサイクリン含有食餌にて維持できる。これらのマウスは、(それらのB細胞でのMYC過剰発現を果たすため)ドキシサイクリンの非存在下に免疫化できる。反応性の抗原特異的B細胞は、特異的抗原に対するパニングによってインビトロで単離及び濃縮できる。この細胞は、それらを不死化して、ヒト抗体を生産するモノクローナル細胞系を作るために、MYC過剰発現下に成長させることができる。
【0075】
前記の通り、このアプローチを使用して、例えば、特異的抗原に対するヒトIgA抗体を含め、異なるイソタイプを有するヒト抗体を特異的に単離できる。IgA抗体は、予防用に後で強く求められるが、IgA生産を意図的に誘導するために従来の抗体生産方法を用いてどのように動物を免疫化するかは不明なままである。本発明を用いて、この問題が解決される。さらに、本明細書で定義するいずれの特異性も、前記の通り別のFc骨格に容易に移入できる。
【0076】
得られる細胞系は、多量の抗体を至適に生産しない可能性がある。これは、IgH及びIgLをコード化するcDNAをPCRに基づき増幅することによって解消できる。それらcDNAの発現ベクターへのクローニングは、次いで骨髄腫細胞系(又は他のB細胞リンパ腫細胞系)を形質導入して抗体生産を増大させるのに使用できる。
【0077】
第3のアプローチは、レトロウイルスにより目的の抗原でヒトctlt−HSC細胞系を形質導入して、亜致死的に照射されたNOD/SCIDマウスに、それらの形質導入ctlt−HSC細胞系を用いて移植することである。マウスに4OHTを注射し、得られる白血病/リンパ腫を培養してヒトモノクローナル抗体生産細胞系を作製できる。その利点は、モノクローナル抗体全体がヒト遺伝子によりコード化され、インビボで成熟できることである。第4の可能性は、亜致死的に照射され、且つヒトctlt−HSC細胞系で再構成されたNOD/SCIDマウスに由来する脾臓成熟ヒトB細胞を単離することである。MYC過剰発現なしに発生した成熟B細胞を、目的の抗原でレトロウイルスにより形質導入できる。細胞は、亜致死的に照射されたNOD/SCIDマウスに戻し移植してそれらのマウスに4OHTを注射することによりインビボで形質転換させるか、又は4OHTの存在下にインビトロ培養で維持することができる。これにより、目的の抗原の核酸配列のみで開始して2〜3週で、新規ヒトモノクローナル抗体の発生が可能になりうる。MYCが自己抗原に対するB細胞寛容を破壊する能力は、T細胞免疫優性エピトープに関わらず多くの特異性に対する抗体の発生をもたらすかもしれない。MYCがB細胞寛容を破壊する機構は、自己反応性B細胞をT細胞の援助に非依存的なものとすることに関わり、よってそれらを蛋白質配列の特定部分に応答する束縛から開放することもできる。本アプローチは、自己蛋白質を模倣し、免疫系によって通常は無視され、従って寛容機構が概して、それらの残基に対する良好な応答を妨げる、ウイルスによりコード化されたエピトープを明らかにすることができるかもしれない。本発明は従って、それらの共通ドメインを良好なワクチン候補とする新規の方法を提供する。
【0078】
本明細書に開示の技術は、癌及び自己免疫などの感染症及び慢性疾病を明らかにする、検出及び処置用の診断及び治療用抗体の迅速な開発のための新しいストラテジーを提供する。抗体生産のための既存の手順に比べて、本発明の速度及び効率のために、新しい感染作用物質が現れた後すぐに、これらの抗体を生成することができる。例えば、疾患の突然の激増(例えば、パンデミックインフルエンザ(フル))の原因になるウイルスに特異的な抗体を、現行法により生成されるものよりもずっと短期間で治療剤としての使用に備えるものとしうる。本発明の方法を用いて生産される抗体は、あらゆる型及びクラスの抗体を初めとして、抗体結合断片及び抗体の誘導体を含んでもよい。さらに詳細には、本発明の方法により生産される抗体は、かかる抗体を含有する血清、又は様々な程度に精製されている抗体を含むことができる。本発明の全抗体は、ポリクローナル又はモノクローナルでありうる。或いは、全抗体の機能性等価物、例えば、1種以上の抗体ドメインが切欠されたか又は存在しない抗原結合断片(例えば、Fv、Fab、Fab’,又はF(ab)2断片)、さらには、単鎖抗体、ヒト抗体、ヒト化抗体(前記)、1種より多くのエピトープに結合できる抗体(例えば、二重特異性抗体)、若しくは1種以上の異なる抗原に結合できる抗体(例えば、二重又は多重特異性抗体)を含む遺伝的に操作した抗体又はその抗原結合断片なども、本発明の方法によって生産されうる。
【0079】
多種多様な抗原に選択的に結合することができる抗体を、本発明の方法によって生産できる。一般的に、動物に導入すると免疫応答を誘導できる任意の抗原が、本発明での使用に好適である。さらに、自己抗原などの、正常動物では通常自己寛容機構を受けているはずの抗原も、本発明で使用してもよい。例えば、抗原としては、限定しないが、ウイルス抗原、真菌抗原、細菌抗原、蠕虫抗原、寄生虫抗原、外寄生生物抗原、原生動物抗原、又は他の任意の感染作用物質からの抗原を含め、病原に関わる任意の抗原を挙げることができる。抗原としてはまた、病原性又は細胞性のいずれの出所からのものであれ、特定の疾患又は状態に関連する任意の抗原も挙げることができ、これらには限定しないが、癌(腫瘍)抗原、自己免疫疾患に関わる抗原(例えば、糖尿病抗原)、アレルギー抗原(アレルゲン)、1種以上の変異アミノ酸を内包している哺乳動物細胞分子、哺乳動物細胞によって通常は出生前又は新生児期に発現される蛋白質、疫学的作用物質(例えばウイルス)の挿入により発現が誘導される蛋白質、遺伝子転座により発現が誘導される蛋白質、及び調節配列の突然変異により発現が誘導される蛋白質が含まれる。これらの抗原は、天然抗原又は遺伝操作された抗原で、なんらかの方法によって修飾されているもの(例えば、配列変化又は融合蛋白質の生成)であることができる。
【0080】
一態様で、抗原は、アデノウイルス、アレナウイルス、ブニアウイルス、コロナウイルス、コクサッキーウイルス、サイトメガロウイルス、エプスタインバーウイルス、フラビウイルス、ヘパドナウイルス、肝炎ウイルス、ヘルペスウイルス、インフルエンザウイルス、レンチウイルス、麻疹ウイルス、ムンプスウイルス、ミクソウイルス、発癌ウイルス、オルソミクソウイルス、パピローマウイルス、パポバウイルス、パラインフルエンザウイルス、パラミクソウイルス、パルボウイルス、ピコルナウイルス、ポックスウイルス、狂犬病ウイルス、呼吸器合胞体ウイルス、レオウイルス、ラブドウイルス、風疹ウイルス、トガウイルス、及び水痘ウイルスなどのウイルス由来のものであるが、これらに限定されることはない。他のウイルスとして、T−リンパ増殖性ウイルス、例えば、ヒトT細胞リンパ増殖性ウイルス(HTLV−I及びHTLV−IIなどのHTLV)、ウシ白血病ウイルス(BLVS)及びネコ白血病ウイルス(FLV)が挙げられる。レンチウイルスとしては、限定しないが、ヒト(HIV−I又はHIV−2を含むHIV)、サル(SIV)、ネコ(FIV)及びイヌ(CIV)免疫不全ウイルスが挙げられよう。
【0081】
本発明の新規の手順によるモノクローナル抗体の生成により、HIV変異体のクレード全体を中和する潜在的能力のある新規特異性の発生をもたらすことができる。これは、例えば、ウイルスのエンベロープ蛋白質の構造上の必須構成要素の標的化、又は代わりに、宿主コレセプター蛋白質のいずれかによって達成できよう。
【0082】
好ましい一実施形態で、本発明の方法は、血球凝集素(HA)蛋白質又はノイラミニダーゼ(NA)蛋白質などのインフルエンザウイルス蛋白質に特異的な抗体を生成するのに使用してもよい。いくつかの実施形態で、HA蛋白質は、Hl、H2、H3、H4、H5、H6、H7、H8、H9、H10、H11、H12、H13、H14、H15及びH16から選択される。一態様で、HA蛋白質はH5である。一態様で、NA蛋白質は、Nl、N2、N3、N4、N5、N6、N7、N8及びN9から選択される。一態様で、NA蛋白質はN5である。特定の実施形態で、抗体はHA蛋白質のサブユニットに対して特異的である。
【0083】
別の態様で、抗原は、Aspergillus、Bordatella、Brugia、Candida、Ch1amydia、Coccidia、Cryptococcus、Dirofilaria、Escherichia、Francisella、Gonococcus、Histoplasma、Leishmania、Mycobacterium、Mycoplasma、Paramecium、Pertussis、Plasmodium、Pneumococcus、Pneumocystis、Rickettsia、Salmonella、Shigella、Staphylococcus、Streptococcus、Toxoplasma、Vibriocholerae Yersiniaからなる属より選択される感染作用物質由来のものである。一態様で、感染作用物質は、Plasmodium falciparum(熱帯熱マラリア原虫) ox Plasmodium vivax(三日熱マラリア原虫)から選択される。
【0084】
一態様で、抗原は、Enterobacteriaceae、Micrococcaceae、Vibrionaceae、Pasteurellaceae、Mycoplasmataceae、及びRickettsiaceaeより選択されるファミリー由来の細菌からのものである。一態様で、細菌は、Pseudomonas、Bordetella、Mycobacterium、Vibrio、Bacillus、Salmonella、Francisella、Staphylococcus、Streptococcus、Escherichia、Enterococcus、Pasteurella、及びYersiniaから選択される属のものである。一態様で、細菌は、Pseudomonas aeruginosa、Pseudomonas mallei、Pseudomonas pseudomallei、Bordetella pertussis、Mycobacterium tuberculosis、Mycobacterium leprae、Francisella tularensis、Vibrio cholerae、Bacillus anthracis、Salmonella enteric、Yersinia pestis、Escherichia coli及びBordetella bronchisepticaから選択される種由来である。
【0085】
好適な抗原として、ウイルスが結合する細胞性レセプター(例えば、CD4等)も挙げられる。
【0086】
本発明によれば、「〜に選択的に結合する」の用語は、抗体又はその抗原結合断片が、特定の蛋白質又は他の抗原に優先的に結合する能力を意味する。さらに具体的には、「選択的に結合する」の用語は、ある蛋白質と別のものとの特異的結合(例えば、抗原に対する、抗体又はその抗原結合断片)であって、結合のレベルが、任意の標準的アッセイ(例えば、イムノアッセイ)によって測定して、そのアッセイに対するバックグラウンコントロールよりも統計的に有意に高いことを意味する。例えば、イムノアッセイを実施する場合、コントロールは通常、抗体又は抗原結合断片を単独で含む(すなわち、抗原の非存在下の)反応ウェル/チューブであり、抗原非存在下での抗体又はその抗原結合断片による反応性の量(例えば、ウェルへの非特異的結合)が、バックグラウンドと考えられる。結合は、酵素イムノアッセイ(例えば、ELISA)、イムノブロットアッセイ等を含む、当該技術分野の水準にある様々な方法を用いて測定できる。
【0087】
本発明によれば、本発明での使用に好適な抗原として、同じ抗原由来の2種以上の免疫原性ドメイン若しくはエピトープ;同じ細胞、組織若しくは生物由来の2種以上の抗原、免疫原性ドメイン、若しくはエピトープ;及び/又は異なる細胞、組織若しくは生物由来の2種以上の異なる抗原、免疫原性ドメイン、若しくはエピトープを挙げることができる。実際、本発明は、1度の免疫化後に多重特異性のモノクローナル抗体を生成するための方法を提供する。例えば、本明細書に記載のマウス(例えば、MMTV−tTA/TRE−MYCマウス)を、細菌又は酵母にて組換え蛋白質として生成され、GST−融合蛋白質として作製された抗原などの2種以上の免疫原性ドメイン、エピトープ、又は他の抗原で、標準的な免疫化技術を用いて免疫化してもよい。免疫化されたマウスからのB細胞は次いで、例えば、精製蛋白質抗原で被覆されたプレート上にパニングすることによって単離してもよい。単離された抗原特異的B細胞は、MYC過剰発現を活性化し、継続的抗原の存在下にB細胞をインビトロで形質転換するために、抗生物質(例えば、ドキシサイクリン)の非存在下に培養してもよい。得られる細胞系はその後、抗体生産及び特異性についてスクリーニングしてもよい。このアプローチで、一度に200もの抗原に特異的なモノクローナル抗体を迅速に生産できるようになり、多くの蛋白質に対するモノクローナル抗体の供給を豊富にすることによって、プロテオミクスの分野での主な障壁を解消しうる。
【0088】
前記の方法は、1つの抗原又は複数の抗原の、多くの又はすべてのエピトープを認識する中和抗体組成物を生成するのに使用してもよい。例えば、1つのウイルスのいくつかの抗原で、又はいくつかの関連ウイルス株からの特定の抗原の複数の変異体でマウスを免疫化してもよい。単離された抗原特異的B細胞は、次いで抗原の特定のエピトープに対して特異的な抗体を生産する細胞についてパニングしてもよい。これらはその後、合わせて、ウイルス、又はウイルス株のパネルからの特異的抗原の全ての変異体の、殆ど又は全てのエピトープに結合する抗体組成物を生産してもよい。同様に、類似のエピトープとの交差反応性なしに、1つの特定の抗原又はエピトープへ結合することにつき、同様の方法によって抗体を選択してもよい。
【0089】
本発明によって生産される、予防又は治療剤として有用な抗体は、通常は組成物(製剤)の形態で提供される。本発明の一実施形態で、医薬組成物又は製剤は、有効な量の抗体又はその抗原結合断片、及び薬理学的に許容できる担体から調製される。薬理学的に許容できる担体は、当業者に周知である。本発明によれば、「薬理学的に許容できる担体」には、薬理学的に許容できる賦形剤及び/又は薬理学的に許容できる送達媒体で、好適なインビボの部位に製剤又は組成物を投与するための使用に好適なものが含まれる。薬理学的に許容できる担体は、抗体が患者の標的部位に到達すれば、その抗体が作用でき、好ましくはその患者に治療的恩恵をもたらすことのできる形態の製剤において、使用される抗体を維持できるものでもよい。従って、本発明はさらに、本発明によって生産される抗体又は誘導体の1種以上を用いて個体を処置する方法も包含する。本発明は、一実施形態で、病原による感染(例えば、コロナウイルス感染)若しくはそれに起因する疾患を含む、状態若しくは疾患を有するか、又はそれを発生する危険のある動物を処置する(その状態又は疾患の予防及び/又は治療的処置を含む)方法を包含する。この方法は、その状態若しくは疾患を有するか、又はそれを発生する危険のある動物に、本明細書に記載の通り本発明によって生産される1種以上の抗体又はその機能性誘導体を投与して、その疾患又は状態に起因する少なくとも1つの症状をその動物において予防又は低減することを含め、疾患又は状態を低減又は予防する工程を含む。本発明はマウスを用いて例証されているが、当該技術分野で知られている抗体生産用の任意の実験動物を使用してもよいことは当業者に理解されるであろう。本発明の方法は、細胞又はその組織を含め、抗体生産に好適な任意の生物で使用できる。好ましい動物として、脊椎動物綱の任意の動物、制限なく霊長類、齧歯類、家畜及び家庭用ペットを含む哺乳類(すなわち、哺乳動物)が挙げられる。一般的に、抗体の生産において、例えば、限定しないが、ウサギ、ヒツジ、ハムスター、モルモット、マウス、ラット又はニワトリなどの好適な実験動物が、それに対する抗体を所望される抗原に暴露される。好ましい実施形態で、動物は「ヒト化」動物、例えば、ヒト抗体を生じるヒトctlt−HSC細胞系及びヒトモノクローナル生産細胞系で再構成されたNOD/SCIDマウスである。
【0090】
(一般定義)
本発明の実施には、特に断らない限り、当業者に周知の、分子生物学(組換え技術を含む)、微生物学、細胞生物学、生化学、核酸化学、及び免疫学の常用の技術を利用しうる。そのような技術は、Molecular Cloning:A Laboratory Manual,第2版(Sambrookら、1989)およびMolecular Cloning:A Laboratory Manual,第3版(Sambrook及びRussel,2001),(本明細書では併せて「Sambrook」として引用する);Current Protocols in Molecular Biology(F.M.Ausubelら編、1987,2001年までの追補も含む);PCR:The Polymerase Chain Reaction,(Mullisら、編、1994);Harlow及びLane(1988)Antibodies,A Laboratory Manual,Cold Spring Harbor Publications,New York;Harlow及びLane(1999)Using Antibodies:A Laboratory Manual Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY(本明細書では併せて「Harlow及びLane」として引用する),Beaucageら、編、Current Protocols in Nucleic Acid Chemistry John Wiley & Sons,Inc.,New York,2000);並びにVaccines,S.Plotkin及びW.Orenstein,編、第3版(1999)などの文献において充分に説明されている。
【0091】
本発明によれば、本明細書で一般的に用いられる用語「抗原」は、自然発生的に、若しくは合成により得られる蛋白質(ペプチド、部分蛋白質、全長蛋白質)のあらゆる部分、細胞性組成物(全細胞、細胞溶解液又は破壊細胞)、生物(生物全体、溶解液、若しくは破壊細胞)、又は炭水化物(癌細胞で発現されているものなど)や他の分子若しくはその一部を意味する。抗原は、その抗原が投与される個体の細胞及び組織内で遭遇する同一若しくは類似の抗原に対する、抗原特異的な免疫応答(例えば、体液性免疫応答及び/又は細胞媒介性免疫応答)を誘発する。或いは、抗原は免疫寛容原として作用できる。免疫応答の刺激を言う場合、「抗原」の用語は、「免疫原」の用語と互換的に用ることができる。抗原は、単一エピトープほどに小さくてもよいし、又はより大きくてもよく、また複数のエピトープを含むことができる。従って、抗原のサイズは、約5〜12アミノ酸(例えば、ペプチド)と小さくてよいし、全長蛋白質ほどの大きさでもよく、これには多量体、及び融合蛋白質、キメラ蛋白質、全細胞、微生物全体、又はその一部(例えば、全細胞の溶解液や、微生物の抽出物)が含まれる。さらに、抗原には炭水化物が含まれうる。
【0092】
所定抗原の「免疫原性ドメイン」は、動物に投与された場合に免疫原として作用する少なくとも1つのエピトープを含む、抗原の任意の部分、断片又はエピトープ(例えば、ペプチド断片、サブユニット、抗体エピトープ又は他のコンホメーションに基づくエピトープ)でありうる。例えば、単一蛋白質が、複数の異なる免疫原性ドメインを含みうる。免疫原性ドメインは、体液性免疫応答の場合のように蛋白質内で線状の配列である必要はない。
【0093】
エピトープは、本明細書で、所定抗原内の、免疫応答を誘発するのに充分な単一免疫原性部位、又は所定抗原内の、免疫応答を抑制、削除若しくは不活性化させるのに充分な、単一免疫寛容原性部位と定義する。当業者は、T細胞エピトープのサイズ及び組成がB細胞エピトープと異なること、並びにクラスI MHC経路を通じて提示されるエピトープは、クラスII MHC経路を通じて提示されるエピトープと異なることを認識するであろう。エピトープは、線状配列であっても、又はコンホメーションに基づくエピトープ(保存結合領域)であってもよい。「ワクチン接種」又は「免疫化」は、単独又はアジュバントと共に抗原を投与することの結果としての、抗原又はその部分に対する免疫応答の誘発(誘導)を意味する。免疫化の概念は、当該技術分野で周知である。抗原の投与により誘発される免疫応答は、抗原を投与しないときと比較した場合の、免疫応答の任意の様相(例えば、細胞媒介性応答、体液性応答、サイトカイン生産)の検出可能な任意の変化でありうる。
【0094】
本発明によれば、抗体は免疫グロブリンドメインを含むことを特徴とし、よって、それらは蛋白質の免疫グロブリンスーパーファミリーのメンバーである。一般的には、抗体分子は2種の鎖を含んでいる。鎖の1方の型は、重鎖又はH鎖と称され、もう一方の型は軽鎖又はL鎖と称される。2つの鎖は、等モル比で存在し、各抗体分子は通常、2つのH鎖と2つのL鎖とを有している。2つのH鎖はジスルフィド結合によって連結され、H鎖の各々はジスルフィド結合によってL鎖に連結されている。L鎖は、ラムダ(λ)及びカッパ(κ)鎖と称される2種のみである。これに対し、H鎖の主要クラスは5つあり、イソタイプと称される。その5つのクラスには、免疫グロブリンM(IgM又はμ)、免疫グロブリンD(IgD又はδ)、免疫グロブリンG(IgG又はλ)、免疫グロブリンA(IgA又はα)、及び免疫グロブリンE(IgE又はε)が含まれる。このようなイソタイプ間の弁別的特性は、免疫グロブリンの定常ドメインによって定義付けられ、それらを以下に詳説する。ヒト免疫グロブリン分子は、9つのイソタイプ、IgM、IgD、IgE、IgG1(γl)、IgG2(γ2)、IgG3(γ3)及びIgG4(γ4)を含むIgGの4つのサブクラス、並びにIgAl(αl)及びIgA2(α2)を含むIgAの2つのサブクラスを含む。ヒトにおいて、IgGサブクラス3及びIgMは、最も強力な補体活性化因子(古典的補体系)であるが、IgGサブクラス1と、ずっと程度は低いがIgGサブクラス2は、古典的補体系の中度から低度の活性化因子である。IgG4サブクラスは補体系(古典的又は副補体系)を活性化しない。副補体系を活性化することが知られている唯一のヒト免疫グロブリンイソタイプは、IgAである。マウスでは、IgGサブクラスはIgG1、IgG2a、IgG2b及びIgG3である。マウスIgG1は補体を活性化しないが、IgG2a、IgG2b及びIgG3は補体活性化因子である。
【0095】
免疫グロブリン分子の各々のH又はL鎖は、L鎖可変ドメイン(VLドメイン)及びL鎖定常ドメイン(CLドメイン)、並びにH鎖可変ドメイン(VHドメイン)及びH鎖定常ドメイン(CHドメイン)と称される2つの領域を含む。完全なCHドメインは、3つのサブドメイン(CH1、CH2、CH3)、及びヒンジ領域を含む。合わせて、1つのH鎖と1つのL鎖が、免疫グロブリン可変領域を有する免疫グロブリン分子の腕を形成できる。完全な免疫グロブリン分子は、2つの会合した(例えば、ジスルフィド結合した)腕を含んでいる。よって、免疫グロブリン全体の各々の腕は、VH+L領域、及びCH+L領域を含んでいる。本願明細書で用いる「可変領域」又は「V領域」という用語は、VH+L領域(Fv断片としても知られている)、VL領域又はVH領域を意味する。本願明細書で用いる「定常領域」又は「C領域」という用語もやはり、CH+L領域、CL領域又はCH領域を意味する。
【0096】
プロテアーゼを用いた免疫グロブリンの限定消化で、2つの断片を生産してもよい。抗原結合断片は、Fab、Fab’、又はF(ab’)2断片と称される。抗原への結合能力を欠く断片は、Fc断片と称される。Fab断片は、VH領域及びCH領域の一部(CH1ドメイン)と対合するL鎖(VL+CLドメイン)を有する、免疫グロブリン分子の一方の腕を含んでいる。Fab’断片は、Fab断片に対応しており、ヒンジ領域の一部がCH1ドメインに付着している。F(ab’)2断片は、典型的にはヒンジ領域において、ジスルフィド結合を介して通常互いに共有結合している2つのFab’断片に対応する。
【0097】
CHドメインは免疫グロブリンのイソタイプを決定付け、そのイソタイプに応じて異なる機能的特性が与えられる。例えば、μ定常領域は、IgM分子の五量体の凝集体形成を可能とし、α定常領域は二量体の形成を可能とする。
【0098】
免疫グロブリン分子の抗原特異性は、可変、すなわちV領域のアミノ酸配列によって与えられる。このように、異なる免疫グロブリン分子のV領域は、それらの抗原特異性に応じて有意に変動しうる。V領域の特定の部分は、他の部分よりも保存されており、フレームワーク領域(FW領域)と称される。これに対し、V領域の特定の部分は、可変性が高く、超可変領域と命名されている。VLとVHドメインが免疫グロブリン分子内で対合すると、各ドメイン由来の超可変領域が会合して、抗原結合部位を形成する超可変ループを創出する。よって、超可変ループが免疫グロブリンの特異性を決定し、それらの表面が抗原に対して相補的であるので、相補性決定領域(CDR)と命名されている。
【0099】
V領域のさらなる可変性は、免疫グロブリンV領域をコード化する遺伝子セグメントの組み合わせの可変性によって与えられる。免疫グロブリン遺伝子は、体細胞性に再配置して免疫グロブリン分子をコード化する再配置免疫グロブリン遺伝子を形成する、複数の生殖系遺伝子セグメントを含む。VL領域は、L鎖V遺伝子セグメント及びJ遺伝子セグメント(連結セグメント)によってコード化される。VH領域は、H鎖V遺伝子セグメント、D遺伝子セグメント(多様性セグメント)及びJ遺伝子セグメント(連結セグメント)によってコード化される。
【0100】
L鎖及びH鎖V遺伝子セグメントの両者とも、実質的なアミノ酸配列可変性の3つの領域を有している。かかる領域は、それぞれL鎖CDR1、CDR2及びCDR3、並びにH鎖CDR1、CDR2及びCDR3と称される。L鎖CDR1の長さは、異なるVL領域間で実質的に変動しうる。例えば、CDR1の長さは、約7アミノ酸から約17アミノ酸までで変動しうる。これに対し、L鎖CDR2及びCDR3の長さは、通常は異なるVL領域間で変動しない。H鎖CDR3の長さは、異なるVH領域間で実質的に変動しうる。例えば、CDR3の長さは、約1アミノ酸から約20アミノ酸までで変動しうる。各H及びL鎖CDR領域には、FW領域が隣接している。
【0101】
免疫グロブリン分子の他の機能面として、免疫グロブリン分子の価数、免疫グロブリン分子の親和性、及び免疫グロブリン分子の結合力が挙げられる。本願明細書で用いる「親和性」は、免疫グロブリン分子上の単一部位で免疫グロブリン分子が抗原に結合する(すなわち、一価抗原に結合する一価Fab断片結合)強度を意味する。親和性は、免疫グロブリンが抗原に結合する強度の合計を意味する結合力とは異なっている。免疫グロブリン結合親和性は、当該技術分野で標準的な技術を使用して測定でき、この例として競合的結合技術、平衡透析法又はBIAコア法が挙げられる。本願明細書で用いる「価数」とは、免疫グロブリン分子あたりの異なる抗原結合部位の数(すなわち、抗原結合断片の抗体分子1つあたりの抗原結合部位の数)を意味する。例えば、一価免疫グロブリン分子は、一度に1つの抗原にしか結合できないが、二価免疫グロブリン分子は、一度に2種以上の抗原に結合できる、等々である。
【0102】
「個体」は、脊椎動物、好ましくは哺乳動物、より好ましくはヒトである。哺乳動物としては、限定しないが、家畜、競技用動物、ペット、霊長類、マウス及びラットが挙げられる。「個体」の用語は、「動物」、「被検者」又は「患者」の用語と互換的に使用できる。
【0103】
本発明の様々な態様は、以下の実験に記載されている。これらの実験結果は、例示目的のみで、本発明の範囲を限定することは意図しない。
【実施例】
【0104】
(実施例1)
以下の実施例は、MMTV−tTA/TRE−MYCマウスに由来するB細胞系の生産を示す。
【0105】
B細胞特異的に誘導性にMYCを過剰発現できるマウスのAN1/T3集団からB細胞系を生成するために、生後8週間、MMTV−tTA/TRE−MYCマウスをドキシサイクリンにて維持し、その後正常食餌に切り替えた。そのマウスで、外見的に顕著なリンパ節腫脹及び脾腫大が発生し、リンパ系異常増殖に伴い一貫して認められる多くの臨床徴候(毛皮汚損、弯曲姿勢、努力性呼吸、貧血、臓器肥大等)を表した。マウスを安楽死させて、それらのリンパ節及び脾臓を分析用に集めた。リンパ節のいくつか及び脾臓の一部から、単一細胞懸濁液を生成した。それらの細胞をフローサイトメトリー分析に使用した。腫瘍を初期に特徴付けすることで、活性化B細胞の高い有病率が実証された。同じ細胞のいくつかを用いて、培養物を播種し、B細胞系を生成させた。これらの細胞は、リンパ球培地(RPMI 1640、10%ウシ胎児血清、ペニシリン/ストレプトマイシン、L−グルタミン、HEPES、非必須アミノ酸、ピルビン酸ナトリウム及び2−β−メルカプトエタノール)で培養した。およそ14〜21日後に、ウェルのいくつかが細胞系のクローン増殖を呈し始めた。細胞が大きなフラスコでの成長に適応し、細胞の一部は凍結保存するまで、注意深く増殖させた。
【0106】
最初に2種の細胞系を選び、TBLK6及びTBLK7と命名した。両細胞系の試料をCD138(Y軸)及びCD40(X軸)に特異的な抗体で染色し、フローサイトメトリーによって分析した。図1(上部パネル)に示す通り、2種の細胞系は、異なるレベルのCD138発現を示し、CD40発現はほとんど乃至は全く示さなかった。播種後の組織培養培地への免疫グロブリン分泌のレベルも測定した。各細胞系由来の細胞の規定数(105細胞)を、1mlの成長培地単独、又はIL−4、IL−6若しくは両者のいずれかを補充した培地を含む24穴プレートのウェルに播種した。培養開始後、1、3又は4日に上清の試料を採取した。その後、抗IgM捕獲ELISA用に上清を使用した。図1(下部パネル)に表す結果より、両細胞系は自発的に免疫グロブリンをそれらの生育培地に分泌するが、最初の接種物にIL−4及びIL−6を添加することによって分泌のレベルを増加できることが示される。次いで、TBLK6及びTBLK7両者により分泌される免疫グロブリンはIgMであることを実証した。真のモノクローナル集団を生成し続けるために、それらの細胞系の両方を単一細胞にクローン化できる。さらに、セルソーティングによってAN1/T3集団を単離するために使用できるMMTV−tTA/TRE−MYCバイジェニックマウスのコホートを、現在生成している。
【0107】
(実施例2)
以下の実施例は、Eμ−MYC/BCRHEL/sHEL遺伝子導入マウスで生じる腫瘍及び細胞系の表面表現型とHEL特異的抗体生産を実証する。
【0108】
野生型マウス(塗りつぶしたヒストグラム)、BCRHEL遺伝子導入マウス(明灰色実線)、BCRHEL/sHELマウス(網点灰色線)、及びEμ−MYC/BCRHEL/sHEL三重遺伝子導入マウス(黒実線)から得た細胞を、表示の表面マーカーに対して特異的な抗体で染色し、フローサイトメトリーによって分析した。図2(上部パネル)に示すデータは、B220+脾臓細胞上の表示マーカーの発現を表す。
【0109】
細胞系TBL−I、TBL−8、TBL−14及びTTLN9(全て、Eμ−MYC/BCRHEL/sHELマウスで生じる腫瘍由来)の105細胞を、24穴プレートに、サイトカインを全く追加しない1mlの生育培地中へ播種した。4日後に上清の試料を採取して、総IgMの濃度(A)、さらにHEL特異的IgMの力価(B)についてアッセイした。様々なコントロールマウスからの血清も含めて、細胞系における抗体生産を比較する手段を提供する。これらのマウスには、野生型C57/BL6マウス(WT)、BCRHEL遺伝子導入マウス(BCR−tg)、sHEL遺伝子導入マウス(Ag−tg)、BCRHEL/sHEL二重遺伝子導入マウス(BCR/Ag−tg)、Eμ−MYCマウス及び腫瘍担持Eμ−MYC/BCRHEL/sHEL三重遺伝子導入マウス(BL)が含まれた。図2(下部パネル)に示す結果は、Eμ−MYC/BCRHEL/sHELマウスで生じる腫瘍及び細胞系におけるHEL特異的力価を示す。
【0110】
(実施例3)
以下の実施例は、MYCがB細胞寛容を破壊して、抗原誘発、MYC−依存性B細胞リンパ腫を起こすことができることを実証する。
【0111】
寛容を破壊することによって、MYCはB及びT細胞を自己抗原による持続的刺激に曝して、細胞増殖、及び確実になるゲノムの障害を助長できる力をもたらすのかもしれない。MYCを過剰発現しているB細胞の新生物発生前状態の検討は、B細胞寛容の調節におけるMYCの新規な役割の解明につながる。
【0112】
フローサイトメトリー分析を、野生型マウス(塗りつぶしたヒストグラム)、ずっとドキシサイクリンにて維持しておいたMMTV−rtTA/TRE−M7C/BCRHEL/sHELマウス(明灰色線)、及び安楽死の1週間前にドキシサイクリンから離脱させておいたMMTV−rtTA/TRE−MYC/BCRHEL/sHELマウス(暗灰色線)から得たリンパ節細胞について実施した。図3(上部パネル)は、B細胞、CD69(A)、及びB7−2(CD86)(B)の抗原依存性活性化後に上方調節される2種の分子に対する抗体で細胞を染色した場合の結果を示す。CD69及びB7−2の示唆レベルは、細胞のB220+画分で表れ、フローサイトメトリーによりチトクロムC染色細胞上でゲート開閉することによって確認される。結果は、MYCの急激な過剰発現後に活性化B細胞が出現することを示唆している。
【0113】
図3(下部パネル)は、活性化B細胞の蓄積に、MYCの連続的な過剰発現が必要であることを示す。グラフの各データポイントは、個々のマウスのリンパ節で検出された活性化B細胞の数を表す。4匹のマウスのコホートを、各時間のポイントに用いた。この図は、誘導されたMMTV−rtTA/TRE−M7C/BCRHEL/sHELマウスでの活性化B細胞の蓄積の惹起及び維持におけるMYCの必要性を示す。
【0114】
野生型マウス(1)、BCRHELマウス(2)、BCRHEL/sHELマウス(3)、顕性腫瘍の発生前のEμ−MYC/BCRHEL/sHELマウス(4)、ずっとドキシサイクリンにて維持しておいたMMTV−rtTA/TRE−M7C/BCRHEL/sHELマウス(5)、及び血清採取前28日にドキシサイクリンから離脱しておいたMMTV−rtTA/TRE−M7C/BCRHEL/sHELマウス(6)から血清を得て、HEL(A)、又は総血清免疫グロブリン(B)に対するELISAによって三重でアッセイした。図4(上部パネル)に表す結果は、MYCの過剰発現後の血清中の自己抗体の蓄積を示す。
【0115】
MYCの過剰発現後の腎臓における自己抗体及び免疫複合体の蓄積を調べるために、野生型マウス(A)又はEμ−MYC/BCRHEL/sHELマウス(B)から組織学的検討用の腎臓を得た。組織を切片としてヘマトキシリン及びエオシンで染色し、顕微鏡画像を得た。倍率は100倍であった。免疫蛍光のために、野生型マウス(C)又はEμ−MYC/BCRHEL/sHELマウス(D)から腎臓を得た。凍結組織を切片化し、IgMに対するローダミン接合抗体で染色した。倍率は5倍であった。図4(下部パネル)に表す結果は、MYCの過剰発現後の腎臓における自己抗体及び免疫複合体の蓄積を示す。
【0116】
そうでなければ遺伝子導入自己抗原に対して免疫寛容のマウスが、B細胞系統中でMYCが単独で発現されれると、抗原に対する免疫応答を開始することを観察している。応答性B細胞は、活性化表現型に変換し、腎臓の免疫複合体病を引き起こす自己抗体を生産した。寛容の断絶の確立及び維持の双方に、MYCは必要であった。これらのマウスは、リンパ腫も発生し、それはBCR及びMYCに由来するシグナル間の協働の結果である可能性が最も高かった。これらの効果は、MYCがサイトカインの代替として働く能力に起因するかもしれない。MYCがB細胞増殖及び生存の双方に対するサイトカインの効果を模倣でき、実際にそれらの効果が必要とされることを見出した。我々のデータは、MYC過剰発現が、免疫学的寛容の断絶の確立及び維持に必要であることを示唆している。抗原依存的にMYCを過剰発現する自己反応性B細胞を不死化する能力によって、寛容B細胞の短い寿命によって限定されることは最早なく、また反応不顕性B細胞が骨髄腫細胞系と生産的に融合する周期に入ることを最早必要としないことから、ハイブリドーマ細胞系の生成のための従来からのアプローチよりもさらに多くの特異性の安定化が可能になる。
【0117】
(実施例4)
以下の実施例は、B細胞特異的な、Doxで調節されるMYCを過剰発現しているマウスを使用した、自己反応性バックグラウンドからのB細胞系の生成を実証している
【0118】
前記の結果は、MYCの過多が、可溶性自己抗原に対するB細胞寛容を破壊できることを示している。それらの研究では、MYCを過剰発現している、BCRHEL遺伝子導入B細胞は、HELに対する活発な応答を惹起し、悪性化が始まる前にポリクローナル自己免疫性リンパ球増殖性疾患を発症させることができた(図3及び4)。自己反応性B細胞におけるMYCの過剰発現は、MYCの増殖及び生存シグナルを提供する能力によって、B細胞をT細胞の援助に対して非依存性とすることができることが、我々の研究により示唆された。MYCを過剰発現している、自己反応性B細胞の増殖した集団は、その同族抗原への連続曝露と、MYCの過剰発現との双方に伴い、依存性のままのB細胞リンパ腫を生成し始めた。腫瘍担持マウスからのリンパ節、脾臓及び骨髄よりB細胞を収集でき、そしてBCRHEL導入遺伝子を発現し、且つ抗HEL IgMを分泌する多くの細胞系を、初代細胞の骨髄腫融合パートナーへの融合を行うことなく樹立できた。
【0119】
2つのさらなる環境を使用して、同様の結果を得ることができている。一例では、Ars/AlマウスをΕμ−MYC株に交雑した。それらのマウス(n=9マウス)は、平均して第36日齢でバーキット様リンパ腫を発生した。腫瘍は、成熟した活性化B細胞で構成されていた。それらの細胞は、表面にIgMを発現していた。それらの結果は、MYC過剰発現が、低親和性の抗DNA抗体という面において、自己反応性B細胞に対する寛容を破壊する能力を示している。第2例では、MMTV−rtTA/TRE−MYCマウスを用いた。それらのマウスは、食餌からのドキシサイクリンの排除後に、B細胞特異的で一時的に調節されたMYCの過剰発現を可能とする。ドキシサイクリン含有食餌を4月齢でマウスから離すと、活性化された末梢B細胞、抗核抗体を血清に、免疫複合体沈着物を腎臓に蓄積し、また6週以内にB細胞リンパ腫を発生した(平均的な例は、42日)。骨髄腫パートナー細胞への融合を行わずに、腫瘍から細胞系を樹立できている。これらの結果は、自己反応性B細胞レセプターを発現するB細胞系を生成する、新規アプローチとしての本システムの使用を示唆している。
【0120】
(実施例5)
以下の実施例は、MYCを過剰発現しているマウスで生産される抗体が、インビボで機能することを実証する。
【0121】
MMTV−tTA/TRE−MYCマウスにて生成したHEL特異抗体がインビボで機能しうるかどうかを調べるために、致死的なウイルス感染のモデルを使用することにした。ブタ狂犬病ウイルス(PRV)は、静脈内投与後にマウスで致死的となることがこれまでに示されている、アルファ−ヘルペスウイルスのメンバーである。PRVの変異体を2種構築した。一例では、US−9と称される遺伝子に対する配列の1つをGFPに融合させた。この構築体により、異なる設定にてウイルスで感染した細胞、及びウイルスの追跡が可能になる。重要なこととして、US−9蛋白質は、なお生産されており、その機能を保持している。ウイルスはその付加的な遺伝的カーゴにもかかわらず完全に病原性がある。HELに対する読み取り枠をコード化するPRVの変異体も生成した。感染細胞のウエスタンブロット分析によって、本ウイルス変異体がHELを発現することが示された(データは示さず)。
【0122】
GFPを発現しているウイルス又はHELを発現しているウイルス接種液(109PFUの力価を有するウイルス上清200μl)を、1:500に希釈したHEL特異抗体と氷上で1時間インキュベートした。混合液は次いで、4匹のマウスのコホートに静脈注射した。その後マウスは、ウイルス及び抗体混合液の投与後4日間モニターし、PRV感染に関わる重篤な神経学的臨床徴候を呈したら安楽死させた。図5に示す通り、GFPを発現しているウイルス(実線)は、HEL特異抗体の存在による影響を受けなかった。そのコホートでの死亡の動態は、野生型PRV株を用いて以前に発明者が得たものに類似していた。これに対し、HELを発現しているウイルス(破線)を与えたマウスでの死亡の動態は、有意に遅延し、そのマウスのコホートは、GFPを発現しているウイルスを注射したマウスのほぼ2倍も生き延びた。これらの実験に使用したウイルスは、US−9融合蛋白質を一過性に発現するのみであった。細胞への侵入後、それらは野生型PRVを生産した。死亡の遅延は、抗体がウイルス感染を阻害する能力を示唆している。
【0123】
(実施例6)
以下の実施例は、表現型としてH5N1を用いて、感染作用物質に対する新規抗体の発生を実証する。
【0124】
新自己抗原としてシステムに導入することによりMMTV−tTA/TRE−MYCマウスが目的の抗原に対する新規抗体を生成する能力を試験するために、レトロウイルスの骨髄キメラマウスの生成に依存するアプローチを用いた。pMISCV−H5−IRES−GFP(pMIG−H5)を生成するために、A/Ty/Ont/7732/66(H5N9)単離物からのヘマグルチニン(HA)のコーディング領域(cDNAのORFをコード化している部分−クローンに非翻訳部分が存在していた)を、pMIGにサブクローニングした。5−FUで処置したTRE−MYCマウスからの骨髄を、pMIG−H5及びpMIG−tTAで形質導入した。GFP発現のフローサイトメトリー分析により定量すると、およそ60%の細胞が形質導入されていた。3日後に、致死的に照射された(800/400R)マウス(Bl/6レシピエント)のコホートに細胞を移植した。骨髄キメラマウスをSEPTRA上に維持し、血液学的悪性疾患の外見的に顕著な臨床徴候を毎日観察した。再構成後6週で、数匹のマウスが、弯曲姿勢、毛皮汚損、外的に触知可能な腫瘍(脾腫大及びリンパ節腫脹)及び呼吸促迫を含む腫瘍発生の臨床徴候を示し始めた。再構成後7〜8週の間で、100%のマウスが腫瘍発生の徴候を呈した。マウスを屠殺し、GFP発現の証左のための蛍光顕微鏡によって分析した。屠殺されたマウスの100%に脾腫大、並びにリンパ節、骨髄及び脾臓にGFP発現があった。
【0125】
リンパ節及び脾臓を採取して、24穴プレートに蒔く単一細胞懸濁液を生成するのに使用した。残存細胞を培養して細胞系を生成させ始めるか、又はその後の分析用に凍結保存した。細胞をB細胞マーカーB220及びIgMに対して特異的な抗体を用いて染色し、その発現につきフローサイトメトリーによって分析した。図6は、GFP陽性細胞は、B細胞マーカーB220及びIgMの双方を発現することを示している。腫瘍は、成熟した活性化B細胞で構成される、MYC誘発性の抗原依存性腫瘍を生じる、成熟した活性化(ブラスティング)B細胞(バーキットリンパ腫のマウスモデルで観察されたものと同様)で構成されていた。これらの細胞を培養系に入れて、クローン増殖させた集団を、最初の接種後8日に開始して継代した。これは、モノクローナル抗体生産への現今のアプローチで通常成し遂げられるものより有意に速いタイムラインである。さらに、この新規アプローチで血球凝集素に対する新規抗体を生成できるようになる加速された時間フレームが、このアプローチを、新しく新生の感染症及び他の生物学的脅威に対する新規の中和抗体の発生のための迅速な応答プラットフォームとするはずである。骨髄細胞は、さらに特異性を導き、又は追加の抗原を加えるべく、今後マウスの大きなコホートを再構成するためにも凍結できる。マウスからその器官を収集した時間に血清を採取して、−20℃で保管した。腫瘍がH5 HA蛋白質に対する抗体を生成しているかどうかを確かめるために、血清を1:5000の希釈にてウエスタンブロット分析で使用した。pMIG−HAをトランスフェクトした293細胞由来の蛋白質溶解液(TritonX−100系溶解バッファー)、A/Ty/Ont/7732/66単離物由来の、HAを発現しているプラスミド、又は陽性コントロールプラスミドpCDNA3−H5を、SDS−PAGE(12%ゲル)によって分離し、PVDF膜に転写して、その膜を希釈血清でプローブ探査し、その後HRPを接合した二次抗体とインキュベーションした。図7は、腫瘍担持マウス由来の血清は、pMIG−HA(中央のレーン)又は陽性コントロールプラスミドpCDNA3−H5(右のレーン)のいずれかを発現している293細胞由来のHAと反応するが、トランスフェクトしていない293細胞由来の溶解液又はpMIGベクターのみをトランスフェクトした細胞由来の溶解液(左のレーン)とは反応しなかったことを示している。
【0126】
HA蛋白質は、およそ38〜40kDのバンドとして現れる。低分子量のバンド(19Kd)は、非特異的であるらしく、良好なローディングコントロールとして働く。展開されたバンド形成パターンは、H5、及び細胞におけるその正常な成熟及び処理の間に発生する切断産物と一致している。成熟HAは、2つのサブユニット(HA1及びHA2)で構成される。HA1サブユニット(〜40kDa)は、分子の球状頭部を形成し、表面糖蛋白質及び糖脂質上の宿主細胞シアル酸レセプターへの結合を担う。HA2サブユニット(〜20kDa)サブユニットは、侵入の間、宿主細胞のエンドソーム膜とのウイルスエンベロープの融合を担う。プロテアーゼ切断部位は、これらの2つのサブユニットを分かち、宿主プロテアーゼによる切断が、宿主細胞への侵入に必要である。ウイルスを中和するエピトープの大部分がHA1サブユニットに認められ、ウイルス−レセプター相互作用を阻害する。
【0127】
血清がウイルス−レセプター相互作用を遮断する能力を試験するために、H5N2、H1N1、H7N2、H3N2、及びH6N8サブタイプを含む多様なインフルエンザA単離物を用いた血球凝集阻害及びウイルス中和アッセイを使用した(図8)。3匹のマウス(1〜3)から、H5/tTA BM形質導入後6〜8週に単離した血清、又はリン酸緩衝性生理食塩水(PBS)(C1)を、マイクロタイタープレートの二重のウェルに沿って連続希釈した。連続希釈後に、インフルエンザAウイルスA/Mal/WI/944/82(H5N2)、A/NY/1469/02(H1N1)、又はPBS(ウイルスなし)の4種の凝集単位を各ウェルに添加して、30分間インキュベートした。次に、シチメンチョウ赤血球細胞を添加して30分間インキュベートし、血球凝集活性を検出した。第1カラムは、1:20の最終血清濃度を含有する。
【0128】
これらのデータは、H5N1に対して生成した新規抗体は、異なるクレード(H1N1)からのウイルスも中和できたことを示している。これは、MYCの過剰発現を用いる新しいアプローチが、ウイルスによりコード化されるエピトープで、自己蛋白質をおそらく模倣し、寛容機構がそれらの残基に対する良好な応答を妨げるために通常は無視されるものを明らかにできることを示唆している。これにより、特定のタイプ(インフルエンザ、又は他のタイプ)の異なるウイルス間の共通の残基を同定して、治療用に開発することができるようになるかもしれない。
【0129】
リンパ節及び脾臓細胞由来の細胞系を作製でき、その上清をHAとの反応性について試験して、HA蛋白質に対する抗体を生産する細胞系を生成できることを立証することができる。これらの上清及び/又は血清は、H5を含むインフルエンザウイルスを中和する能力についても試験できる。中和アッセイには、ある抗体が、インフルエンザ(フル)ウイルス及びヒツジ赤血球細胞を用いる凝集アッセイや、上皮性細胞のインビトロ、又はマウスのインビボの有効な感染を阻害できるかどうかの判定を含みうる。本発明のさらなる開示及び実施形態を、添付の原稿に見出すことができ、これを本願に引用してその全体を援用する。
【0130】
本発明の様々な実施形態を本明細書に詳細に記載しているが、それらの実施形態の改変及び適応が当業者に想起されるであろうことは明らかである。しかし、このような改変及び適応は添付の特許請求の範囲に記載の本発明の範囲に含まれることは特に理解されるべきである。
【特許請求の範囲】
【請求項1】
a)MYCを誘導性に過剰発現する動物を、前記動物においてMYCが過剰発現されない条件下で抗原に導き;b)前記動物からB細胞を回収し;及びc)MYCが過剰発現される条件下で前記B細胞を培養する工程を含む、抗体生産細胞を生産するための方法。
【請求項2】
前記動物を前記抗原に導く工程の後に、前記動物においてMYCが過剰発現される条件下に前記動物を維持する工程をさらに含む、請求項1記載の方法。
【請求項3】
前記動物は、主に前記動物の前記B細胞においてMYCを誘導性に過剰発現する、請求項に記載の方法。
【請求項4】
前記動物は、MMTV−rtTA/TRE−MYCマウスである、請求項1記載の方法。
【請求項5】
a)前記マウスは、前記動物を前記抗原に導く工程の間、MYC発現を抑止する抗生物質上に維持され;及びb)前記B細胞を培養する工程は、前記抗生物質の非存在下で行って抗体生産細胞を生産する、請求項4記載の方法。
【請求項6】
前記マウスを前記抗原に導く工程の後に、前記抗生物質への曝露から前記動物を外す工程をさらに含む、請求項5記載の方法。
【請求項7】
前記抗生物質はドキシサイクリンである、請求項5記載の方法。
【請求項8】
前記動物を前記抗原に導く工程は、前記抗原をコード化しているDNAを、前記動物に遺伝的に移入する工程を含む、請求項1記載の方法。
【請求項9】
前記抗原は自己抗原を含む、請求項1記載の方法。
【請求項10】
前記動物はさらに前記抗原を発現する、請求項1記載の方法。
【請求項11】
前記抗原はHIV蛋白質を含む、請求項1記載の方法。
【請求項12】
前記抗原はgp120又はgp41を含む、請求項1記載の方法。
【請求項13】
前記抗原はインフルエンザウイルス由来の抗原を含む、請求項1記載の方法。
【請求項14】
前記抗原は血球凝集素である、請求項13記載の方法。
【請求項15】
a)MYCを誘導性に過剰発現する動物を、前記動物においてMYCが過剰発現されない条件下で抗原に導き;b)前記動物からB細胞を回収し;c)MYCが過剰発現される条件下で前記B細胞を培養し;及びd)前記B細胞培養物から抗体を回収する工程を含む、抗体を生産するための方法。
【請求項16】
前記動物を前記抗原に導く工程の後に、前記動物においてMYCが過剰発現される条件下に前記動物を維持する工程をさらに含む、請求項15記載の方法。
【請求項17】
前記動物は主に前記動物の前記B細胞においてMYCを誘導性に過剰発現する、請求項15記載の方法。
【請求項18】
前記動物はMMTV−rtTA/TRE−MYCマウスである、請求項15記載の方法。
【請求項19】
前記動物を前記抗原に導く工程は、前記抗原をコード化しているDNAを、前記動物に遺伝的に移入する工程を含む、請求項15記載の方法。
【請求項20】
前記抗原は自己抗原を含む、請求項15記載の方法。
【請求項21】
前記動物はさらに前記抗原を発現する、請求項15記載の方法。
【請求項22】
前記抗原はHIV蛋白質を含む、請求項15記載の方法。
【請求項23】
前記抗原はgp120又はgp41を含む、請求項15記載の方法。
【請求項24】
前記抗原はインフルエンザウイルス由来の抗原を含む、請求項15記載の方法。
【請求項25】
前記抗原は血球凝集素である、請求項24記載の方法。
【請求項26】
前記抗体はヒト化抗体である、請求項15記載の方法。
【請求項27】
a)B細胞の重鎖又は軽鎖遺伝子のVDJ連結領域をコード化している核酸分子を、MYCを誘導性に過剰発現し且つ前記B細胞の生産を防止する遺伝的修飾を含む動物からの骨髄由来の幹細胞に導入し;b)前記骨髄由来の幹細胞をレシピエント動物に移入し;c)前記レシピエント動物からB細胞を回収し;d)MYCが過剰発現される条件下で前記B細胞を培養し、及びe)前記B細胞培養物から抗体を回収する工程を含む、抗体を生産するための方法。
【請求項28】
MYCが前記レシピエント動物において過剰発現される条件下に前記レシピエント動物を維持する工程を、前記骨髄由来の幹細胞を前記レシピエント動物に移入する工程の後にさらに含む、請求項27記載の方法。
【請求項29】
MYCが前記レシピエント動物において過剰発現されない条件下に前記レシピエント動物を維持する工程を、前記骨髄由来の幹細胞を前記レシピエント動物に移入する工程の後にさらに含む、請求項27記載の方法。
【請求項30】
前記工程(a)からの前記B細胞はヒトB細胞である、請求項27記載の方法。
【請求項31】
前記B細胞はヒトから単離される、請求項30記載の方法。
【請求項32】
前記ヒトは、抗体が媒介する自己免疫疾患に罹患している、請求項31記載の方法。
【請求項33】
前記工程(a)は、前記骨髄由来の幹細胞に、VDJ連結領域をコード化している前記核酸分子をレトロウイルスにより形質導入する工程を含む、請求項27記載の方法。
【請求項34】
前記VDJ連結領域をコード化している前記核酸分子は、目的の抗原に選択的に結合する単離ヒトB細胞からクローニングされる、請求項33記載の方法。
【請求項35】
前記VDJ連結領域をコード化している前記核酸分子は、ヒトドナーから得たB細胞に認められるIgH及びIgL配列より再配置したVDJ領域のPCR増幅断片である、請求項33記載の方法。
【請求項36】
前記ヒトドナーは、健常ドナー、及び抗体が媒介する自己免疫疾患の患者からなる群より選択される、請求項35記載の方法。
【請求項37】
前記骨髄由来の幹細胞はさらに、ヒトIgHをコード化している核酸分子、及びヒトIgLをコード化している核酸分子で形質導入される、請求項33記載の方法。
【請求項38】
前記ヒトIgHをコード化している前記核酸分子と前記ヒトIgLをコード化している前記核酸分子は、同じ核酸分子である、請求項37記載の方法。
【請求項39】
前記ヒトIgHと前記ヒトIgLをコード化している核酸分子の一方又は両方は、前記VDJ領域をコード化する同じ核酸分子である、請求項37記載の方法。
【請求項40】
前記骨髄由来の幹細胞はヒトからのものである、請求項27記載の方法。
【請求項41】
前記骨髄由来の幹細胞はマウスからのものである、請求項27記載の方法。
【請求項42】
前記マウスは、Rag−2−/−、SCID、DNA−PK−/−、Ku70−/−、Ku80−/−、XRCC4−/−及びμMT−/−からなるリストより選択される遺伝的修飾を含む、MMTV−rtTA/TRE−MYCマウスである、請求項41記載の方法。
【請求項43】
前記レシピエント動物は致死的に照射されたマウスである、請求項27記載の方法。
【請求項44】
前記レシピエント動物はSCIDマウスである、請求項27記載の方法。
【請求項45】
前記動物は、前記VDJ領域に選択的に結合する抗原にインビボで導かれる、請求項27記載の方法。
【請求項46】
前記抗体のイソタイプはIgAである、請求項27記載の方法。
【請求項47】
前記抗体のイソタイプはIgGである、請求項27記載の方法。
【請求項48】
前記抗体のFc領域を遺伝的に修飾して、前記抗体が自己免疫反応及び関連免疫複合体沈着問題を誘発する能力を最小にしている、請求項27記載の方法。
【請求項49】
a)細胞の生存及び増殖を促進する癌原遺伝子を誘導性に過剰発現する動物を、前記癌原遺伝子が前記動物において過剰発現されない条件下で抗原に導き;b)前記動物からB細胞を回収し;及びc)前記癌原遺伝子が過剰発現される条件下で前記B細胞を培養する工程を含む、抗体生産細胞を生産するための方法。
【請求項50】
a)細胞の生存及び増殖を促進する癌原遺伝子を誘導性に過剰発現する動物を、前記癌原遺伝子が前記動物において過剰発現されない条件下で抗原に導き;b)前記動物からB細胞を回収し、;c)前記癌原遺伝子が過剰発現される条件下で前記B細胞を培養し;及びd)前記B細胞培養物から抗体を回収する工程を含む、抗体を生産するための方法。
【請求項51】
a)抗原をコード化している核酸分子を、細胞の生存及び増殖を促進する癌原遺伝子を誘導性に過剰発現する動物からの骨髄由来の幹細胞に導入し;b)前記骨髄由来の幹細胞をレシピエント動物に移入し;c)前記癌原遺伝子が前記動物において過剰発現される条件下に前記レシピエント動物を維持し;d)前記レシピエント動物からB細胞を回収し;e)前記癌原遺伝子が過剰発現される条件下で前記B細胞を培養し;及びf)前記B細胞培養物から抗体を回収する工程を含む、抗体を生産するための方法。
【請求項52】
a)細胞の生存及び増殖を促進する癌原遺伝子をコード化している核酸分子を、動物からの骨髄由来の幹細胞に導入し;b)前記骨髄由来の幹細胞をレシピエント動物に移入し;c)前記レシピエント動物からB細胞を回収し;d)抗原をコード化している核酸分子を前記B細胞に導入し;e)前記癌原遺伝子が過剰発現される条件下で前記B細胞を培養し;及びf)前記B細胞培養物から抗体を回収する工程を含む、抗体を生産するための方法。
【請求項53】
a)細胞の生存及び増殖を促進する癌原遺伝子をコード化している核酸分子を、動物からの骨髄由来の幹細胞に導入し;b)前記骨髄由来の幹細胞を第1レシピエント動物に移入し;c)前記第1レシピエント動物からB細胞を回収し;d)抗原をコード化している核酸分子を前記B細胞に導入し;e)前記B細胞を第2レシピエント動物に移入し;f)前記動物において前記癌原遺伝子が過剰発現される条件下に前記第2レシピエント動物を維持し;g)前記第2レシピエント動物からB細胞を回収し;h)前記癌原遺伝子が過剰発現される条件下で前記B細胞を培養し;及びi)前記B細胞培養物から抗体を回収する工程を含む、抗体を生産するための方法。
【請求項54】
前記癌原遺伝子はMYCである、請求項51記載の方法。
【請求項55】
前記骨髄由来の幹細胞は、抗アポトーシス蛋白質をコード化している核酸分子で形質導入されている、請求項52記載の方法。
【請求項56】
前記抗アポトーシス蛋白質はBcl−2である、請求項55記載の方法。
【請求項57】
前記核酸分子は、レトロウイルスにより導入される、請求項52記載の方法。
【請求項58】
前記骨髄由来の幹細胞はヒトからのものである、請求項52記載の方法。
【請求項59】
前記骨髄由来の幹細胞は、条件付きで不死化された長寿命の造血幹細胞である、請求項52記載の方法。
【請求項60】
前記レシピエント動物は、亜致死的に照射されたNOD/SCIDマウスである、請求項51のいずれか1つに記載の方法。
【請求項61】
前記動物は、ヒトIg座位をコード化している核酸分子に対して遺伝子導入されている、請求項15のいずれか1つに記載の方法。
【請求項1】
a)MYCを誘導性に過剰発現する動物を、前記動物においてMYCが過剰発現されない条件下で抗原に導き;b)前記動物からB細胞を回収し;及びc)MYCが過剰発現される条件下で前記B細胞を培養する工程を含む、抗体生産細胞を生産するための方法。
【請求項2】
前記動物を前記抗原に導く工程の後に、前記動物においてMYCが過剰発現される条件下に前記動物を維持する工程をさらに含む、請求項1記載の方法。
【請求項3】
前記動物は、主に前記動物の前記B細胞においてMYCを誘導性に過剰発現する、請求項に記載の方法。
【請求項4】
前記動物は、MMTV−rtTA/TRE−MYCマウスである、請求項1記載の方法。
【請求項5】
a)前記マウスは、前記動物を前記抗原に導く工程の間、MYC発現を抑止する抗生物質上に維持され;及びb)前記B細胞を培養する工程は、前記抗生物質の非存在下で行って抗体生産細胞を生産する、請求項4記載の方法。
【請求項6】
前記マウスを前記抗原に導く工程の後に、前記抗生物質への曝露から前記動物を外す工程をさらに含む、請求項5記載の方法。
【請求項7】
前記抗生物質はドキシサイクリンである、請求項5記載の方法。
【請求項8】
前記動物を前記抗原に導く工程は、前記抗原をコード化しているDNAを、前記動物に遺伝的に移入する工程を含む、請求項1記載の方法。
【請求項9】
前記抗原は自己抗原を含む、請求項1記載の方法。
【請求項10】
前記動物はさらに前記抗原を発現する、請求項1記載の方法。
【請求項11】
前記抗原はHIV蛋白質を含む、請求項1記載の方法。
【請求項12】
前記抗原はgp120又はgp41を含む、請求項1記載の方法。
【請求項13】
前記抗原はインフルエンザウイルス由来の抗原を含む、請求項1記載の方法。
【請求項14】
前記抗原は血球凝集素である、請求項13記載の方法。
【請求項15】
a)MYCを誘導性に過剰発現する動物を、前記動物においてMYCが過剰発現されない条件下で抗原に導き;b)前記動物からB細胞を回収し;c)MYCが過剰発現される条件下で前記B細胞を培養し;及びd)前記B細胞培養物から抗体を回収する工程を含む、抗体を生産するための方法。
【請求項16】
前記動物を前記抗原に導く工程の後に、前記動物においてMYCが過剰発現される条件下に前記動物を維持する工程をさらに含む、請求項15記載の方法。
【請求項17】
前記動物は主に前記動物の前記B細胞においてMYCを誘導性に過剰発現する、請求項15記載の方法。
【請求項18】
前記動物はMMTV−rtTA/TRE−MYCマウスである、請求項15記載の方法。
【請求項19】
前記動物を前記抗原に導く工程は、前記抗原をコード化しているDNAを、前記動物に遺伝的に移入する工程を含む、請求項15記載の方法。
【請求項20】
前記抗原は自己抗原を含む、請求項15記載の方法。
【請求項21】
前記動物はさらに前記抗原を発現する、請求項15記載の方法。
【請求項22】
前記抗原はHIV蛋白質を含む、請求項15記載の方法。
【請求項23】
前記抗原はgp120又はgp41を含む、請求項15記載の方法。
【請求項24】
前記抗原はインフルエンザウイルス由来の抗原を含む、請求項15記載の方法。
【請求項25】
前記抗原は血球凝集素である、請求項24記載の方法。
【請求項26】
前記抗体はヒト化抗体である、請求項15記載の方法。
【請求項27】
a)B細胞の重鎖又は軽鎖遺伝子のVDJ連結領域をコード化している核酸分子を、MYCを誘導性に過剰発現し且つ前記B細胞の生産を防止する遺伝的修飾を含む動物からの骨髄由来の幹細胞に導入し;b)前記骨髄由来の幹細胞をレシピエント動物に移入し;c)前記レシピエント動物からB細胞を回収し;d)MYCが過剰発現される条件下で前記B細胞を培養し、及びe)前記B細胞培養物から抗体を回収する工程を含む、抗体を生産するための方法。
【請求項28】
MYCが前記レシピエント動物において過剰発現される条件下に前記レシピエント動物を維持する工程を、前記骨髄由来の幹細胞を前記レシピエント動物に移入する工程の後にさらに含む、請求項27記載の方法。
【請求項29】
MYCが前記レシピエント動物において過剰発現されない条件下に前記レシピエント動物を維持する工程を、前記骨髄由来の幹細胞を前記レシピエント動物に移入する工程の後にさらに含む、請求項27記載の方法。
【請求項30】
前記工程(a)からの前記B細胞はヒトB細胞である、請求項27記載の方法。
【請求項31】
前記B細胞はヒトから単離される、請求項30記載の方法。
【請求項32】
前記ヒトは、抗体が媒介する自己免疫疾患に罹患している、請求項31記載の方法。
【請求項33】
前記工程(a)は、前記骨髄由来の幹細胞に、VDJ連結領域をコード化している前記核酸分子をレトロウイルスにより形質導入する工程を含む、請求項27記載の方法。
【請求項34】
前記VDJ連結領域をコード化している前記核酸分子は、目的の抗原に選択的に結合する単離ヒトB細胞からクローニングされる、請求項33記載の方法。
【請求項35】
前記VDJ連結領域をコード化している前記核酸分子は、ヒトドナーから得たB細胞に認められるIgH及びIgL配列より再配置したVDJ領域のPCR増幅断片である、請求項33記載の方法。
【請求項36】
前記ヒトドナーは、健常ドナー、及び抗体が媒介する自己免疫疾患の患者からなる群より選択される、請求項35記載の方法。
【請求項37】
前記骨髄由来の幹細胞はさらに、ヒトIgHをコード化している核酸分子、及びヒトIgLをコード化している核酸分子で形質導入される、請求項33記載の方法。
【請求項38】
前記ヒトIgHをコード化している前記核酸分子と前記ヒトIgLをコード化している前記核酸分子は、同じ核酸分子である、請求項37記載の方法。
【請求項39】
前記ヒトIgHと前記ヒトIgLをコード化している核酸分子の一方又は両方は、前記VDJ領域をコード化する同じ核酸分子である、請求項37記載の方法。
【請求項40】
前記骨髄由来の幹細胞はヒトからのものである、請求項27記載の方法。
【請求項41】
前記骨髄由来の幹細胞はマウスからのものである、請求項27記載の方法。
【請求項42】
前記マウスは、Rag−2−/−、SCID、DNA−PK−/−、Ku70−/−、Ku80−/−、XRCC4−/−及びμMT−/−からなるリストより選択される遺伝的修飾を含む、MMTV−rtTA/TRE−MYCマウスである、請求項41記載の方法。
【請求項43】
前記レシピエント動物は致死的に照射されたマウスである、請求項27記載の方法。
【請求項44】
前記レシピエント動物はSCIDマウスである、請求項27記載の方法。
【請求項45】
前記動物は、前記VDJ領域に選択的に結合する抗原にインビボで導かれる、請求項27記載の方法。
【請求項46】
前記抗体のイソタイプはIgAである、請求項27記載の方法。
【請求項47】
前記抗体のイソタイプはIgGである、請求項27記載の方法。
【請求項48】
前記抗体のFc領域を遺伝的に修飾して、前記抗体が自己免疫反応及び関連免疫複合体沈着問題を誘発する能力を最小にしている、請求項27記載の方法。
【請求項49】
a)細胞の生存及び増殖を促進する癌原遺伝子を誘導性に過剰発現する動物を、前記癌原遺伝子が前記動物において過剰発現されない条件下で抗原に導き;b)前記動物からB細胞を回収し;及びc)前記癌原遺伝子が過剰発現される条件下で前記B細胞を培養する工程を含む、抗体生産細胞を生産するための方法。
【請求項50】
a)細胞の生存及び増殖を促進する癌原遺伝子を誘導性に過剰発現する動物を、前記癌原遺伝子が前記動物において過剰発現されない条件下で抗原に導き;b)前記動物からB細胞を回収し、;c)前記癌原遺伝子が過剰発現される条件下で前記B細胞を培養し;及びd)前記B細胞培養物から抗体を回収する工程を含む、抗体を生産するための方法。
【請求項51】
a)抗原をコード化している核酸分子を、細胞の生存及び増殖を促進する癌原遺伝子を誘導性に過剰発現する動物からの骨髄由来の幹細胞に導入し;b)前記骨髄由来の幹細胞をレシピエント動物に移入し;c)前記癌原遺伝子が前記動物において過剰発現される条件下に前記レシピエント動物を維持し;d)前記レシピエント動物からB細胞を回収し;e)前記癌原遺伝子が過剰発現される条件下で前記B細胞を培養し;及びf)前記B細胞培養物から抗体を回収する工程を含む、抗体を生産するための方法。
【請求項52】
a)細胞の生存及び増殖を促進する癌原遺伝子をコード化している核酸分子を、動物からの骨髄由来の幹細胞に導入し;b)前記骨髄由来の幹細胞をレシピエント動物に移入し;c)前記レシピエント動物からB細胞を回収し;d)抗原をコード化している核酸分子を前記B細胞に導入し;e)前記癌原遺伝子が過剰発現される条件下で前記B細胞を培養し;及びf)前記B細胞培養物から抗体を回収する工程を含む、抗体を生産するための方法。
【請求項53】
a)細胞の生存及び増殖を促進する癌原遺伝子をコード化している核酸分子を、動物からの骨髄由来の幹細胞に導入し;b)前記骨髄由来の幹細胞を第1レシピエント動物に移入し;c)前記第1レシピエント動物からB細胞を回収し;d)抗原をコード化している核酸分子を前記B細胞に導入し;e)前記B細胞を第2レシピエント動物に移入し;f)前記動物において前記癌原遺伝子が過剰発現される条件下に前記第2レシピエント動物を維持し;g)前記第2レシピエント動物からB細胞を回収し;h)前記癌原遺伝子が過剰発現される条件下で前記B細胞を培養し;及びi)前記B細胞培養物から抗体を回収する工程を含む、抗体を生産するための方法。
【請求項54】
前記癌原遺伝子はMYCである、請求項51記載の方法。
【請求項55】
前記骨髄由来の幹細胞は、抗アポトーシス蛋白質をコード化している核酸分子で形質導入されている、請求項52記載の方法。
【請求項56】
前記抗アポトーシス蛋白質はBcl−2である、請求項55記載の方法。
【請求項57】
前記核酸分子は、レトロウイルスにより導入される、請求項52記載の方法。
【請求項58】
前記骨髄由来の幹細胞はヒトからのものである、請求項52記載の方法。
【請求項59】
前記骨髄由来の幹細胞は、条件付きで不死化された長寿命の造血幹細胞である、請求項52記載の方法。
【請求項60】
前記レシピエント動物は、亜致死的に照射されたNOD/SCIDマウスである、請求項51のいずれか1つに記載の方法。
【請求項61】
前記動物は、ヒトIg座位をコード化している核酸分子に対して遺伝子導入されている、請求項15のいずれか1つに記載の方法。
【図1】


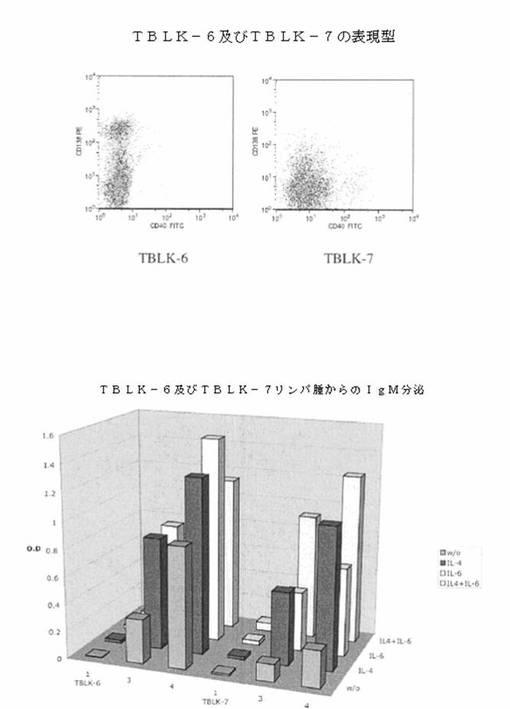
【図2】

【図3】
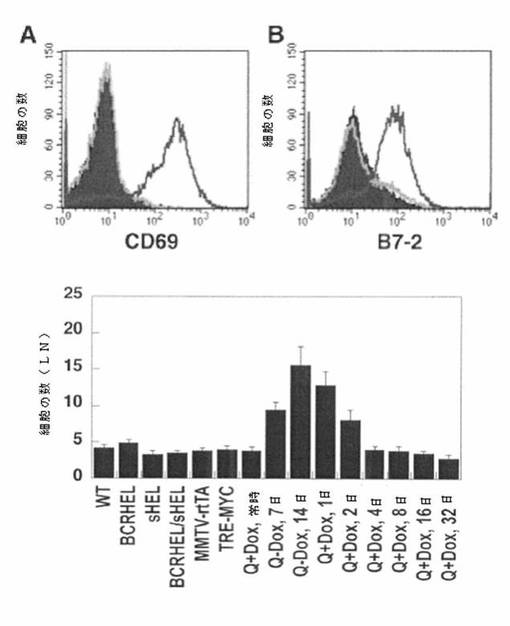
【図4】

【図5】
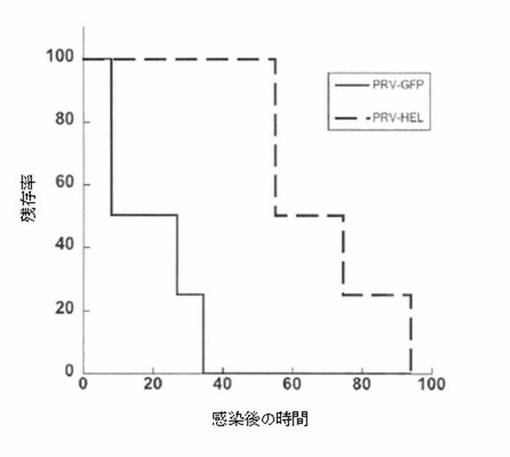
【図6】

【図7】

【図8】

【図9】
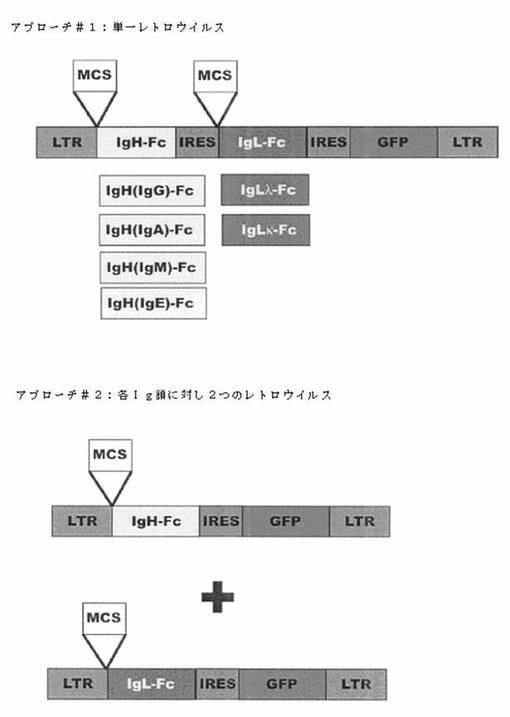
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公表番号】特表2010−521170(P2010−521170A)
【公表日】平成22年6月24日(2010.6.24)
【国際特許分類】
【出願番号】特願2009−553785(P2009−553785)
【出願日】平成20年3月13日(2008.3.13)
【国際出願番号】PCT/US2008/056896
【国際公開番号】WO2008/112922
【国際公開日】平成20年9月18日(2008.9.18)
【出願人】(500343614)ナショナル ジューイッシュ ヘルス (6)
【氏名又は名称原語表記】National Jewish Health)
【Fターム(参考)】
【公表日】平成22年6月24日(2010.6.24)
【国際特許分類】
【出願日】平成20年3月13日(2008.3.13)
【国際出願番号】PCT/US2008/056896
【国際公開番号】WO2008/112922
【国際公開日】平成20年9月18日(2008.9.18)
【出願人】(500343614)ナショナル ジューイッシュ ヘルス (6)
【氏名又は名称原語表記】National Jewish Health)
【Fターム(参考)】
[ Back to top ]