
抗体の製造方法
【課題】魚類を生かしたままの状態で、魚類に産生させた抗体を繰り返し得ることができる、抗体の製造方法の提供。
【解決手段】水泡を有する魚類を抗原投与の対象とすることにより、魚類を生かしたままの状態で、魚類に産生させた抗体を繰り返し得ることができる、抗体の製造方法。前記方法によって得られる抗体。
【解決手段】水泡を有する魚類を抗原投与の対象とすることにより、魚類を生かしたままの状態で、魚類に産生させた抗体を繰り返し得ることができる、抗体の製造方法。前記方法によって得られる抗体。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、抗体の製造方法に関する。さらに詳しくは、水泡を有する魚類に抗原を投与し、抗体を産生させる工程を含む、抗体の製造方法に関する。
【背景技術】
【0002】
現在、遺伝子組み換え技術により大腸菌、酵母等に有用なタンパク質を産生させ、また、これを抗原としてマウス等に投与することにより、抗体を産生させる技術が幅広く利用されている。
しかし、大腸菌などの原核生物に産生させたタンパク質は、多量体形成、糖鎖付加等の翻訳後修飾が正確に行われていない。また、酵母に産生させたタンパク質も完全な翻訳後修飾が行われていないため、本来の機能を有しない場合があるという問題があった。
【0003】
これらの問題を解決すべく、昆虫細胞や、マウス、ブタ、ヒト等の哺乳動物の細胞において有用なタンパク質を産生させる技術が利用されているが、細胞を培養するためのコストが高く、有用なタンパク質が得られるまでに時間がかかるという問題があった。
そこで、近年、バイオリアクターとして魚類を活用することに注目が集まっている。魚類由来のタンパク質は、哺乳動物と同様に多量体形成、糖鎖付加等の翻訳後修飾が行われるという利点がある。そこで特許文献1ではこの利点を生かし、トランスジェニック魚類に多量体糖タンパク質を産生させ、胚、仔魚、稚魚等の組織、血液中から回収する技術が開示されている。
【0004】
魚類は飼育が比較的容易であり、飼育コストも安価である。また、水温や給餌、日照時間、水質等によって産卵および採卵を制御できるなど、非常に扱いやすいという利点もある。
本発明者らは、抗原提示酵母を作成し、これをゼブラフィッシュ等の魚類に飼料として付与することで、魚類由来の特異的抗体を産生させる方法や、従来、困難とされてきたGPCR(G protein−coupled receptor;GPCR)等の膜タンパク質に対する抗体を魚類に産生させる(特願2009−83900)等、魚類による様々な技術を開発している(例えば、特許文献2、3、参照)。
しかし、これらの技術では、魚類全身から抗体を回収等するため、魚類を生かしたままの状態で、産生させた抗体を繰り返し得ることができなかった。
【0005】
本発明者らは、この問題を解決するため、本発明の開発において、水泡眼等の水泡を有する魚類に着目した。この種の金魚は観賞用として、国内外で幅広く飼育されている。
特許文献4において、水泡眼の水疱(水泡)内液が、魚類細胞の細胞保護的又は細胞増殖促進的に作用すること、魚類細胞の処理操作等において、未受精卵の卵質を維持した状態で効率的な操作を可能とすることが開示されている。
しかし、この特許文献では、水疱(水泡)内液に抗体等の有用なタンパク質を産生させること等については示唆すらされておらず、従来の技術においても、水泡を有する魚類を抗体の産生に利用することは試みられていなかった。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2001−86992号公報
【特許文献2】特開2007−255892号公報
【特許文献3】特開2009−73783号公報
【特許文献4】特開2009−183151号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明は、魚類を生かしたままの状態で、産生させた抗体を繰り返し得ることができる、抗体の製造方法の提供を課題とする。
【課題を解決するための手段】
【0008】
本発明者らは、上記課題を解決するために鋭意研究を行った結果、水泡眼等の水泡を有する魚類を抗原投与の対象とすることにより、魚類を生かしたままの状態で、これらの魚類に産生させた抗体を繰り返し得ることができる方法を見出し、本発明を完成するに至った。
【0009】
すなわち、本発明は次の(1)〜(8)の抗体の製造方法、該製造方法によって製造される抗体等に関する。
(1)水泡を有する魚類に抗原を投与し、抗体を産生させる工程を含む、抗体の製造方法。(2)抗原を投与する部位が水泡である上記(1)に記載の製造方法。
(3)抗原をオイルベースとともに、またはオイルベースと不活化大腸菌とともに投与する上記(1)または(2)に記載の製造方法。
(4)魚類に産生させた抗体を、魚類の水泡から採取する工程を含む、上記(1)〜(3)のいずれかに記載の製造方法。
(5)水泡を有する魚類が水泡眼またはらんちゅうである上記(1)〜(4)のいずれかに記載の製造方法。
(6)抗原がタンパク質または糖タンパク質である上記(1)〜(5)のいずれかに記載の製造方法。
(7)抗原がEGFP(enhanced green fluorescent protein;以下、EGFPと示す場合がある)、グルコアミラーゼまたはLGR3(leucine−rich repeat containing G protein−coupled receptor 3;以下、LGR3と示す場合がある)である上記(6)に記載の製造方法。
(8)上記(1)〜(7)のいずれかに記載の製造方法によって製造される抗体。
【発明の効果】
【0010】
本発明の製造方法により、魚類に抗体を産生させることにより、魚類を生かしたままの状態で大量の抗体を繰り返し製造することが可能となった。本発明の製造方法では、糖タンパク質等を抗原とする場合でも、有用な抗体を製造することができる、有用な抗体の製造方法である。
【図面の簡単な説明】
【0011】
【図1】ウェスタンブロット法の結果を示した図である(実施例1)。
【図2】ドットブロット法の結果を示した図である(実施例1)。
【図3】水泡眼(一例)の写真を示した図である(実施例1)。
【図4】タグの写真を示した図である(実施例1)。
【図5】抗原抗体反応の結果(EGFP−Hisタンパク質)を示した図である(実施例1)。
【図6】抗原抗体反応の結果(精製GA−His、培養上清GA−His)を示した図である(実施例1)。
【図7】pCold TF−hLGR3の配列を示した図である(実施例2)。
【図8】抗原抗体反応の結果を示した図である(実施例2)。
【図9】抗原抗体反応の結果を示した図である(実施例2)。
【発明を実施するための形態】
【0012】
本発明の「抗体の製造方法」とは、抗体を産生する生物を魚類とし、これに抗原を投与し、抗体を産生させる工程を含む方法のことをいう。
本発明の「抗体の製造方法」は、魚類が水泡を有する魚類であって、この水泡を有する魚類に抗原を投与し、抗体を産生させる工程を含む製造方法であればよく、抗体を製造するせるために、従来知られている他の工程や方法を含むものであってもよい。
【0013】
この方法において、「抗原を投与」は、魚類に抗体を産生させることが可能な投与方法であればどのような方法であってもよい。例えば、抗原となるタンパク質や抗原酵母等を飼料として経口的に投与したり、腹腔や水泡を投与部位として、抗原を直接、注射等によって投与したりすることが挙げられる。本発明においては特に、抗原の投与部位を水泡として、ここに直接抗原を投与することが好ましい。
【0014】
抗原は、そのまま投与してもよく、アジュバントと組み合わせて投与しても良い。アジュバントとしては、魚類の抗体産生を助けるものであれば、どのようなものであってもよいが、特にオイルベースをアジュバントとすることや、オイルベースと不活化大腸菌とを組み合わせたものをアジュバントとすることが好ましい。
【0015】
本発明の「抗体の製造方法」には、魚類に産生させた抗体を回収する工程も含むことができる。この抗体の回収は、魚類を生かしたまま回収できる方法で行うことが好ましく、特に、魚類の水泡から、抗体を含む水泡内液を回収する等の方法であることが好ましい。この場合、魚類の水泡に直接注射針等をさして、水泡内液を回収することにより、魚類を生かしたままで、魚類に産生させた抗体を回収することが可能となる。
【0016】
本発明における「水泡を有する魚類」としては、魚類の表皮に水泡を有する魚類であれば、いずれの魚種も含まれる。この「水泡を有する魚類」の水泡には、産生された抗体を含むリンパ液が含まれており、水泡内に貯留したこの液を注射器等で採取した場合、再貯留するという性質がある。このような「水泡を有する魚類」として、例えば、水泡眼、らんちゅうなどの金魚を挙げることができる。
【0017】
本発明の「抗原」とは、抗体を産生させるためのものであればどのようなものであってもよく、GPCR、LGR3などの膜貫通型タンパク質、EGFPなどの可溶性タンパク質や、α−ジストログリカン、グルコアミラーゼなどの糖タンパク質であってもよい。
魚類に投与する抗原の量は、魚類が抗体を産生できる量であれば特に問わないが、抗原となるこれらのタンパク質が100μg以上となるように、投与されることが特に好ましい。
【0018】
以下、実施例をあげて本発明をさらに詳細に説明するが、本発明はこれらに限定されるものではない。
【実施例1】
【0019】
I.抗原の調製
<抗原> 麹菌グルコアミラーゼ
1−1.プラスミドの構築
1)麹菌アスペルギルスオリザ由来の硝酸還元酵素遺伝子(以下、niaD遺伝子と示す場合がある)の大腸菌ベクターへの組み込み
目的遺伝子の起源生物(麹菌アスペルギルスオリザ)由来のゲノムDNAを鋳型として、プライマーA(配列表配列番号1)、プライマーB(配列表配列番号2)およびLA−Taqポリメラーゼ(宝ホールディングス)を混合し、以下の条件でPCRを行うことにより、niaD遺伝子(配列表配列番号3)を増幅した。
目的遺伝子の起源生物(麹菌アスペルギルスオリザ)由来のゲノムDNAは参考文献1に記載の定法に従い、調製した。
参考文献1:R.C.Garber and O.C.Yoder, Anal. Biochem.,135,416−422(1983)
<PCR条件>
96℃(5分間)を1サイクル
96℃(20秒間)、60℃(30秒間)、72℃(5分間)を30サイクル
72℃(7分間)を1サイクル
【0020】
得られたPCR増幅産物を制限酵素PstI−HindIIIで処理(37℃)した後、アガロースゲル電気泳動でniaD遺伝子のPstI−HindIII断片として切り出した。切り出しは、QIAquick Gel Extraction kit(QIAGEN)により行った。
切り出したniaD遺伝子のPstI−HindIII断片を大腸菌プラスミドpUC119(宝ホールディングス)のPstI−HindIII部位にDNA Ligation kit ver.1(宝ホールディングス)によりライゲーションし、大腸菌(E.coli)JM109株に形質転換することで、niaDマーカー遺伝子がサブクローニングされた大腸菌プラスミドpNIA2を得た。
大腸菌プラスミドpNIA2は、PstI、SalI部位の両方にunique siteとして遺伝子を導入することができる。
【0021】
2)麹菌アスペルギルスオリザ由来のグルコアミラーゼBターミネーター遺伝子(以下、glaBターミネーター遺伝子と示す)の大腸菌ベクターへの組み込み
麹菌ゲノムDNAを鋳型として、プライマーC(配列表配列番号4)とプライマーD(配列番号5)およびLA−Taqポリメラーゼ(宝ホールディングス)を混合し、以下の条件でPCRを行うことにより、glaBターミネーター遺伝子(配列表配列番号6)を増幅した。
<PCR条件>
96℃(5分間)を1サイクル
96℃(20秒間)、60℃(30秒間)、72℃(5分間)を30サイクル
72℃(7分間)を1サイクル
【0022】
得られたPCR増幅産物を制限酵素SalI−XhoIで処理(37℃)した後、アガロースゲル電気泳動でglaBターミネーター遺伝子のSalI−XhoI断片として切り出した。切り出しは、QIAquick Gel Extraction kit(QIAGEN)により行った。
切り出したglaBターミネーター遺伝子のSalI−XhoI断片を、上記1)で得た大腸菌プラスミドpNIA2のSalI部位にDNA Ligation kit ver.1(宝ホールディングス)によりライゲーションし、大腸菌(E.coli)JM109株に形質転換することで、glaBターミネーター遺伝子がサブクローニングされたプラスミドpNIATを得た。
大腸菌プラスミドpNIATは、PstI、SalI部位の両方に遺伝子をunique siteとして遺伝子を導入することができる。
【0023】
3)麹菌アスペルギルスオリザ由来のsodMプロモーター遺伝子(以下、sodMプロモーター遺伝子と示す)の組み込み
麹菌ゲノムDNAを鋳型として、プライマーE(配列表配列番号7)、プライマーF(配列表配列番号8)およびLA−Taqポリメラーゼ(宝ホールディングス)を混合し、以下の条件でPCRを行うことにより、麹菌由来sodMプロモーター遺伝子(配列表配列番号9)を増幅した。
<PCR条件>
96℃(5分間)を1サイクル
96℃(20秒間)、60℃(30秒間)、72℃(5分間)を30サイクル
72℃(7分間)を1サイクル
【0024】
得られたPCR増幅産物を制限酵素SalI−PstIで処理(37℃)した後、アガロースゲル電気泳動でsodMプロモーター遺伝子のSalI−PstI断片として切り出した。切り出しは、QIAquick Gel Extraction kit(QIAGEN)により行った。
切り出したsodMプロモーター遺伝子のSalI−PstI断片を、上記2)で得た大腸菌プラスミドpNIATのPstI−SalI部位にDNA Ligation kit ver.1(宝ホールディングス)によりライゲーションし、大腸菌(E.coli)JM109株に形質転換することで、sodMプロモーター遺伝子がサブクローニングされたプラスミドpNMBを得た。
大腸菌プラスミドpNMBは、SalI部位に麹菌で発現させたいタンパク質をコードする遺伝子をunique siteとして遺伝子を導入することができる。
【0025】
1−2.目的遺伝子の組み込み
1)ベクターの調製
上記1−1.で構築した大腸菌プラスミドpNMBを制限酵素SalIで処理(37℃)した後、dNTPを最終10mMとなるように添加してT4 DNAポリメラーゼ(宝ホールディングス)で1時間処理(37℃)した。さらに、バクテリア由来アルカリホスファターゼ(宝ホールディングス)で30分処理(50℃)した。処理後得られた物をPCRクリーンアップカラム(プロメガ社製)で溶出させ、ベクターとした。
【0026】
2)インサートの調製
麹菌で発現させたいタンパク質をコードする遺伝子を目的遺伝子として、上記ベクターにサブクローニングするインサートとして調製した。
目的遺伝子は、a)麹菌由来グルコアミラーゼB(glaB)遺伝子(配列表配列番号10(アミノ酸配列を配列表配列番号11に示した))、またはb)Hisタグ遺伝子をつなげた麹菌由来グルコアミラーゼB(glaB)遺伝子(配列表配列番号12(アミノ酸配列を配列表配列番号13に示した))であった。
上記1−1.1)で調製した目的遺伝子の起源生物(麹菌アスペルギルスオリザ)由来のゲノムDNAを鋳型として、目的遺伝子の開始コドンから下流30bpのセンス鎖のプライマー(5’末端をリン酸化)(配列表配列番号14)と、目的遺伝子の終止コドンから上流49bpのアンチセンス鎖のプライマー(5’末端をリン酸化)(配列表配列番号15)と、pfu Taqポリメラーゼ(東洋紡績株式会社)を混合し、以下の条件でPCRを行うことにより、目的遺伝子を増幅した。
得られたPCR増幅産物をPCRクリーンアップカラム(プロメガ社製)で処理溶出させ、インサートとした。
<PCR条件>
96℃(5分間)を1サイクル
96℃(20秒間)、60℃(30秒間)、72℃(5分間)を30サイクル
72℃(7分間)を1サイクル
【0027】
3)ベクターとインサートのライゲーション
上記1)で得たベクターおよび上記2)で得た各目的遺伝子のインサートを1:20(モル比)となるように添加して、DNA Ligation kit ver.1(宝ホールディングス)によりライゲーションを行い、大腸菌(E.coli)JM109株に形質転換した。
形質転換体から、各目的遺伝子の読み枠がsodMプロモーターと正方向にサブクローニングされているものを目的遺伝子発現プラスミドとした。
【0028】
1−3.目的遺伝子発現株の取得
PEG−カルシウム法により、上記1−2.で調製した目的遺伝子発現プラスミドにより、麹菌アスペルギルスオリザのniaD変異株(独立行政法人産業技術総合研究所特許生物寄託センターにFERM P−17707として寄託済み)を形質転換した。
【0029】
1)PEG−カルシウム法は、参考文献2に従い、以下の手順により行った。
即ち、麹菌胞子約108個を100ml DPY液体培地に接種し、28℃で18〜20時間静置培養を行った。こうして発芽させた後、ガラスビーズ(直径0.6cm)を培養フラスコに加え、30℃,130rpmの震とう培養でさらに24時間培養した。
菌糸を採集し、0.8M NaCl水溶液で菌糸を2〜3回洗い、洗った菌糸に10mlの新しく作ったプロトプラスト化溶液(15mg/ml Yatalase(タカラバイオ),10mg/ml Cellulase Onozuka R−10(Yakult Honsha),0.8M NaCl,10mM phosphate buffer(pH6.0),1mM DTT)を加え、30℃で3〜4時間ゆっくりと震とうし、細胞壁を消化してプロトプラストを調製した。
得られたプロトプラスト溶液をミラクロス(カルビオケム社)でろ過し、ろ液を更に4℃,2500g、5分間、遠心分離して回収した。
【0030】
プロトプラストを氷冷した溶液1(0.8M NaCl,10mM CaCl2,10mM Tris−HCl(pH8.0))で洗浄した後、2×108/mlになるように溶液1に懸濁して、その量に対して0.2容量になるように溶液2(40% PEG4000,50mM CaCl2,10mM Tris−HCl(pH8.0))を加え、静かに混合した。
形質転換は、0.1mlのプロトプラスト溶液を新しい15mlチューブに移し、15μl以下(3〜7μg)の目的DNAを加え、氷上で30分間放置した後、0.5ml溶液2を加えゆっくりと攪拌し、室温で20分間放置した。5ml溶液1を加えて、混合した後、4℃で2500gの速度で5分間遠心を行った。
プロトプラストを含んだ沈殿物を0.1mlの溶液1に懸濁し、0.8M NaClを含むCD再生培地に広げ、さらに、その上に、30℃まで冷却したCD軟寒天培地を添加した後、固化させ、28℃で4〜7日培養した。
参考文献2:Mol Gen Genet, 218,99−104,(1989)
【0031】
2)形質転換体のうち硝酸を単一窒素源とするツアペクドックス(Czapek−Dox)培地(2% グルコース、0.1% リン酸1水素2カリウム、0.05% 塩化カリウム、0.05% 硫酸マグネシウム、0.001% 硫酸鉄、0.3% 硝酸ナトリウム)で生育できる株を選択することにより、目的遺伝子発現プラスミドを保持する形質転換体を複数得た。
【0032】
1−4.目的遺伝子産物の調製
上記1−3.により得られた形質転換体をポテトデキストロース培地で培養することで胞子形成させ、滅菌水で胞子を回収した。
回収した胞子を、500ml容三角フラスコに入った100ml GPY液体培地(2% グルコース、1% ポリペプトン、0.5% イーストエキストラクト、0.1% リン酸1水素2カリウム、0.05% 塩化カリウム、0.05% 硫酸マグネシウム、0.001% 硫酸鉄、0.3% 硝酸ナトリウム)に最終胞子濃度1X106/mlとなるように植菌した。
これを30℃で3日間液体培養し、目的遺伝子産物が分泌発現した培養上清を回収した。
【0033】
1−5.培養上清の濃縮
上記1−4.で回収した培養上清の一部を、分画分子量10,000NMWLのセントリプレップ10(ミリポア社製)により遠心分離(3,000×g)し、濃縮した。
【0034】
1−6.培養上清に含まれるGA濃度の定量
上記1−4.で回収した培養上清に含まれるGA濃度は、SDS−PAGEによって培養上清からタンパク質を分離し、CBB染色像を画像解析ソフトのCS Analyzer 3.0(ATTO)により定量化することで推定した。
すなわち、次の1)〜5)の工程を行った。
1)下記1−7.で得た精製GAを3μgから11μgとなるようにPBSに溶解した(スタンダード)。
これと、1〜3μLの1x培養上清とにそれぞれ5xサンプルバッファー(2.5M Tris−HCl、10% グリセロール、0.05% Bromophenol Blue、10% SDS)を添加(終濃度1x)し、100℃で5分間煮沸することによってサンプルとした。
2)これらのサンプルから、SDS−PAGE(7.5% ポリアクリルアミドゲル、200V、20mA)によってタンパク質を分離した。
3)泳動後のゲルをCBB染色液(0.25% CBB−R250、50% メタノール、5% 酢酸)に浸漬し、室温で30分間振盪しながらタンパク質を染色した。
4)染色後のゲルを脱色液(25% メタノール、10% 酢酸)に浸漬し、室温で振盪しながらタンパク質に結合しなかったCBBを洗浄した。
5)脱色後のゲルをCCDカメラ(Light Capture II、ATTO)により撮影し、検出されたGAのバンドをCS Analyzer 3.0のゾーンデンシトメトリー解析によって定量化し、精製GAの積算値から検量線を作成した。
作成した検量線から推定された培養上清(濃縮なし)に含まれるGA濃度は約3.0μg/μLであった。
この結果より、上記1−5.にて5倍濃縮した培養上清を金魚用リンガー液{125mM NaCl、2.6mM KCl、10mM 2−[4−(2−Hydroxyethy l)−1−piperazinyl]ethanesulfonic acid(HEPES)(pH7.4)}によってGA濃度が4μg/μLとなるよう調製した(以下、これを培養上清GAまたは培養上清GA−Hisと示す場合がある)。
【0035】
1−7.目的遺伝子産物の精製
1)目的遺伝子産物
上記1−4.で回収した培養上清の一部を飽和濃度70%の硫酸アンモニウムを添加し、4℃で一晩撹拌しタンパク質を沈殿させた後、遠心分離により沈殿を取り除いた。
この培養上清をブチルトヨパール−650M(東ソー社製)樹脂を添加し、低温で一晩撹拌した後、樹脂を回収した。
回収した樹脂を1.5M 硫酸アンモニウム溶液で2回洗浄し、10mM 酢酸バッファー(pH5.0)で目的のGAタンパク質を溶出し、凍結乾燥したものを、精製GAタンパク質とした。
精製GAタンパク質は、金魚用リンガー液によりGA濃度が4μg/μLとなるよう調製した(以下、これを精製GAと示す場合がある)。
【0036】
2)Hisタグをもつ目的遺伝子産物
上記1−4.の目的遺伝子産物を含む培養上清をニッケルカラム(His GraviTrap column:GE Healthcare社)に添加した後、0.5M NaClおよび400mM イミダゾールを含む20mMリン酸ナトリウムバッファー(pH7.4)を添加してカラムに結合した組換えグルコアミラーゼを溶出した。
この溶出液を透析膜に移し、50mM酢酸ナトリウムバッファー(pH5.0)を含む1Lのバッファーに対し、4℃、3時間で透析を2回行い、Hisタグをもつ目的遺伝子産物を精製した。
精製したHisタグをもつ目的遺伝子産物は、金魚用リンガー液によりGA濃度が4μg/μLとなるよう調製した(以下、これを精製GA−Hisと示す場合がある)。
【0037】
<抗原> EGFP−Hisタンパク質
2−1.ベクターの作製(プラスミドの構築〜目的遺伝子の組み込み)
pCold TF DNA(タカラバイオ)を次のように改変することにより、csp Aプロモーターの下流に5’非翻訳領域、translation enhancing element(TEE)、EGFP、および3’末端に精製用のタグであるHisタグのDNA配列が連結したコンストラクトである、pColdTEE−EGFP−Hisベクターを作製した。
1)本発明者らがpXI(配列表配列番号16)の下流にEGFP(配列表配列番号17)を組み込んで作製したpXI−EGFPベクター(特願2009−107344号、参考文献3)を鋳型として、プライマーG(配列表配列番号18)、プライマーH(配列表配列番号19)およびTAKARA Ex Taq(タカラバイオ)を混合し、以下の条件でPCRを行うことにより、egfp遺伝子の開始コドンが制限酵素SmaIの認識配列に変換され、3’末端がHisタグのDNA配列に変換された断片を増幅した。
参考文献3:Johnson AD,Krieg PA.pXeX,a vector for efficient expression of cloned sequences in Xenopus embryos.Gene. 1994 Sep.
<PCR条件>
95℃(1分間)を1サイクル
95℃(10秒間)、55℃(30秒間)、72℃ (2分間)を30サイクル
72℃(5分間)を1サイクル
【0038】
2)上記1)の増幅産物(egfp遺伝子の開始コドンが制限酵素SmaIの認識配列に変換され、3’末端がHisタグのDNA配列に変換された断片)を鋳型として、プライマーG(配列表配列番号18)、プライマーI(配列表配列番号20)およびTAKARA Ex Taq(タカラバイオ)を混合し、上記1)と同様の条件でPCRを行うことにより、Hisタグの下流に制限酵素SfiIの認識配列が連結したegfp−his6遺伝子断片を増幅した。
【0039】
3)pCold−TF−DNA(タカラバイオ)を鋳型として、プライマーJ(配列表配列番号21)、プライマーK(配列表配列番号22)およびTAKARA Ex Taq(タカラバイオ)を混合し、以下の条件でPCRを行うことにより、TEEにSmaIの認識配列が連結し、pCold−TF−DNAのtranscription terminator配列の上流にSfiIの認識配列が連結するDNA断片を増幅した。
【0040】
上記3)で増幅したDNA断片と、SmaIおよびSfiIによって消化した上記2)のegfp−his6遺伝子断片を、DNA Ligation kit Mighty mix(タカラバイオ)によりライゲーションすることによってpColdTEE−EGFP−Hisを作製した。
【0041】
2−2.目的遺伝子産物の調製
上記2−1.で作製したpColdTEE−EGFP−Hisベクターにより大腸菌BL21を形質転換し、終濃度50μg/mLのアンピシリンナトリウムを含むLB平板培地に塗抹して、37℃で一晩保温しながら培養した。なお、特に記載のない場合、試薬は全て和光純薬工業株式会社の製品を使用した。
【0042】
1)生成したコロニーをLB−ampicillin液体培地に植菌し、37℃で12時間振盪しながら培養した。この培養上清を、あらかじめ作製した2xYT−Ampicillin液体培地{50μg/mL アンピシリンナトリウム、1.6% Bacto Tripton(BD)、1% 乾燥酵母エキス(ナカライテスク)、0.5% NaCl(pH 7.0)}に植菌し、波長600nmの吸光度が0.4〜0.5の範囲となるまで37℃で振盪しながら培養した。
その後、15℃で30分間静置し、0.1mM Isopropyl β−D−1−thiogalactopyranoside(IPTG)を培養上清の1/1000量を添加して、15℃で24時間振盪しながらEGFP−Hisタンパク質の発現を誘導した。
目視にてEGFP−Hisタンパク質の発現を確認した後、4℃で3000xg 15分間遠心分離して菌体を回収し、予冷した1xリン酸緩衝生理食塩水(PBS:137mM NaCl、2.7mM KCl、1.5mM KH2PO4、8.1mM Na2HPO4)により2回洗浄することで残留した培地成分を除去した。
【0043】
2)上記1)に続いて、40mLのNiカラム溶出緩衝液A{20mM リン酸ナトリウム、0.5M NaCl、20mM Imidazol(pH7.4)}を添加し、微量超音波細胞破砕機(MICROSON XL 2000、MISONIX)により菌体を破砕した。
次に、4℃で12000xg 30分間遠心分離して上清を回収し、0.20μmフィルター(ADVANTEC)によって夾雑物を除去して大腸菌発現タンパク質抽出液とした。
調製した大腸菌発現タンパク質抽出液をNi Sepharose担体(GEヘルスケア)を充填したNi カラム(GEヘルスケア)にペリスタポンプ(Bio Rad)によって添加することで、EGFP−Hisタンパク質を担体に結合させた。
【0044】
3)上記2)に続いて、Niカラム溶出緩衝液A{20mM リン酸ナトリウム、0.5M NaCl、20mM Imidazol(pH7.4)}およびNiカラム溶出緩衝液B{20mM リン酸ナトリウム、0.5M NaCl、500mM Imidazol(pH7.4)}による20mM〜500mM Imidazolの連続濃度勾配によってカラムからEGFP−Hisタンパク質を溶出した。
【0045】
4)次に、限外濾過膜(ミリポア)を用いて、EGFP−Hisタンパク質が溶出した画分のNi カラム溶出緩衝液をDEAEカラム溶出緩衝液A{20mM 2−amino−2−(hydroxymethyl)propane−1,3−diol(Tris)−HCl(pH8.0)}にバッファー置換したものをサンプルとし、あらかじめDEAE Sepharose Fast Flow担体(GEヘルスケア)を充填した陰イオン交換カラムに低圧クロマトグラフィーシステム(AKTAprime plus:GEヘルスケア)によって添加することで、EGFP−Hisタンパク質を担体に結合させ、DEAEカラム溶出緩衝液A{20mM Tris−HCl(pH8.0)}およびDEAEカラム溶出緩衝液B{20mM Tris−HCl、1M NaCl(pH8.0)}により、0M〜1M NaClの連続濃度勾配による溶出を行った。
【0046】
5)上記4)のカラムより溶出したEGFP−Hisタンパク質を含む画分の溶液を、2価イオンを含まない金魚用リンガー液{125mM NaCl、2.6mM KCl、10mM 2−[4−(2−Hydroxyethyl)−1−piperazinyl]ethanesulfonic acid(HEPES)(pH7.4)}に置換し、EGFP−Hisタンパク質濃度が4μg/μLになるように調製した。
【0047】
II.抗原抗体反応の確認
上記I.にて調製した抗原(精製GA、精製GA−HisおよびEGFP−Hisタンパク質)を抗体と作用させ、ウェスタンブロット法またはドットブロット法により、抗原抗体反応を確認した。
1.抗体
1)抗His抗体
Anti−His Antibody(Amersham Biosciences)2)抗GlaB抗体
グルコアミラーゼタンパク質全長のペプチド(配列表配列番号11)をデザインし、化学合成によりペプチド合成し、KLH(Keyhole Limpet Hemocyanin)をキャリアタンパク質としてコンジュゲーションしたペプチドを抗原としてウサギに免疫することにより抗体を得た(カスタム抗体作製;インビトロジェン社製)。
【0048】
2.ウェスタンブロット法
1)上記I.にて調製したEGFP−Hisタンパク質、精製GAおよび精製GA−Hisに5xサンプルバッファー(2.5M Tris,10% glycerol,0.05% Bromophenol Blue,10% SDS)をそれぞれ添加し(終濃度1x)、100℃で5分間煮沸してサンプルとした。
これらのサンプルを、10%ポリアクリルアミドゲルにより200V 20mAの設定で電気泳動し、タンパク質を分離した。
【0049】
2)次にセミドライブロッターによってタンパク質をPVDF膜(ATTO)に転写し、0.5% Tween20/PBS(0.5% PBST)で2回リンスして、5% スキムミルク/0.5%PBSTに浸漬して振盪しながら1時間ブロッキングした。
ブロッキング後のPVDF膜をPBSTで2回リンスし、0.5%PBSTで15分間x1回、5分間x2回洗浄して、余分なブロッキング液を除去した。
【0050】
3)続いて上記1.の抗His抗体および抗GlaB抗体をCan Get Signals(TOYOBO) Solution 1により、それぞれ1/3000および1/1000に希釈し、これにPVDF膜を浸漬して室温にて1時間振盪しながら反応させた。反応後のPVDF膜を0.5% PBSTで10分間x3回洗浄し、余分な抗体溶液を除去した。
4)次に、抗マウスIgG HRP標識二次抗体および抗ラビットIgG HRP標識二次抗体をCan Get Signals Solution 2によって、それぞれ1/25000および1/100000に希釈し、PVDF膜を浸漬して室温で1時間振盪しながら反応させた。
【0051】
5)反応後のPVDF膜をPBSTで10分間x3回洗浄し、発光基質液{Amersham ECL plus Western Blotting Detection Reagents(GE ヘルスケア)}に浸漬して室温で5分間反応させたのち、CCDカメラ[Light Capture(ATTO)]によって撮影し、CS Analyzer 3.0(ATTO)により発光を検出した。
【0052】
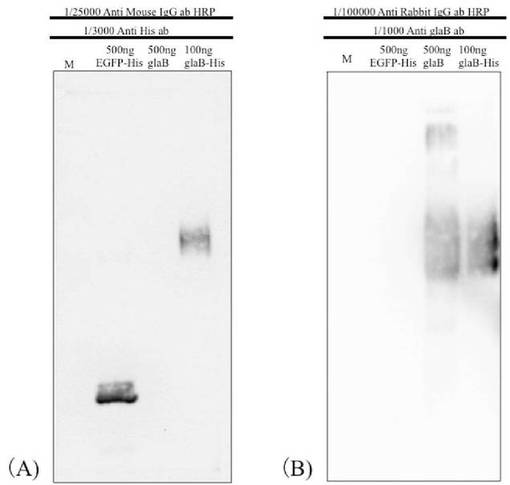
その結果、図1、A(抗His抗体と反応させたPVDF膜)に示されるように、抗His抗体によって、EGFP−Hisタンパク質、精製GAおよび精製GA−Hisをそれぞれ検出したところ、抗His抗体を反応させたPVDF膜ではEGFP−Hisタンパク質(図1、EGFP−His)および精製GA−His(図1、glaB−His)がそれぞれ検出された。
また、図1、B(抗GlaB抗体と反応させたPVDF膜)に示されるように、抗His抗体を反応させたPVDF膜では精製GA(図1、glaB)および精製GA−His(図1、glaB−His)がそれぞれ検出された。
従って、これらの結果から、精製GA−Hisを抗GlaB抗体および抗His抗体で検出できることが確認できた。
なお、図1では、各写真の上部に、各レーンに泳動したサンプル量および使用した一次抗体、二次抗体とその希釈倍率を示している(M:プレステインドSDS−PAGEスタンダード(Broad Range)(Bio Rad))。
【0053】
2.ドットブロット法
1)上記I.にて調製したEGFP−Hisタンパク質、精製GAおよび精製GA−Hisを、PBSにより、それぞれ250pg/μLから250ng/μLの範囲に希釈してサンプルとした。
2)PVDF膜をメタノールに浸漬して5分間振盪しながら置換し、その後超純水に浸漬して10分間x2回振盪しながら置換し、最後にPBSに浸漬して10分間以上振盪しながら置換することによって平衡化した。
3)上記1)のサンプル滴下直前にPVDF膜をプロワイプ(エリエール)に挟み込むことによって余分なPBSを除去し、パラフィルム上のPVDF膜へサンプルを2μLずつ滴下して風乾した。
【0054】
4)乾燥させたPVDF膜を{5% スキムミルク/0.05% Tween20 in TBS(20mM Tris−HCl,150mM NaCl pH7.5)(0.05% TBST)}によって平衡化およびブロッキングした。
5)続いて、PVDF膜を0.05% TBSTにより2回リンスし、10分間振盪しながら洗浄して5分間x2回振盪しながら洗浄し、余分なスキムミルク溶液を除去した。
6)続いて、Can Get Signals Solution 1により、上記1.の抗His抗体、抗GFP抗体(MBL)および抗GlaB抗体をそれぞれ1/3000に希釈し、PVDF膜を浸漬して室温で2時間振盪しながら反応させた。
反応後のPVDF膜は0.05% TBSTにより10分間x3回洗浄し、余分な抗体溶液を除去した。
【0055】
7)次に、Can Get Signals Solution 2によって、抗マウスIgG HRP標識二次抗体および抗ラビットIgG HRP標識二次抗体をそれぞれ1/25000および1/100000に希釈し、PVDF膜を浸漬して室温で1時間振盪しながら反応させた。
8)反応後のPVDF膜を0.05% TBSTにより10分間x3回洗浄し、ウェスタンブロット法と同様に発光基質液と反応させて発光を検出した。
【0056】
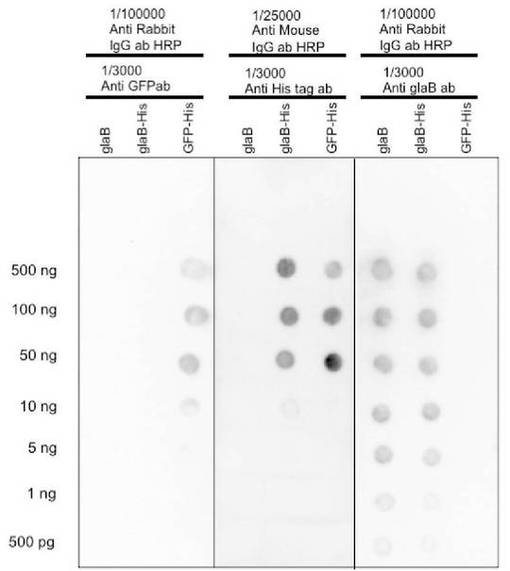
その結果、図2(左側)に示されるように、抗GFP抗体により、EGFP−Hisタンパク質、精製GAおよび精製GA−Hisの検出をそれぞれ行ったところ、10ngから500ngまでのEGFP−Hisタンパク質(図2、GFP−His)のスポットに反応が見られた。
また、図2(中央)に示されるように、抗His抗体により検出を行ったものは、10ngから500ngまでの精製GA−His(図2、glaB−His)のスポットおよび50ngから500ngまでのEGFP−Hisタンパク質(図2、GFP−His)のスポットに反応が見られた。
さらに、図2(右側)に示されるように、抗GlaB抗体により反応させたPVDF膜では500pgから500ngの精製GA(図2、glaB)および精製GA−His(図2、glaB−His)のスポットに反応が見られた。
以上の結果から、抗GlaB抗体および抗His抗体によって精製GA−Hisを検出できることが確認された。
【0057】
またこの条件では、1/1000に希釈した抗GlaB抗体は500pg以上の精製GAおよび精製GA−Hisを、1/3000に希釈した抗His抗体は10ng以上の精製GA−Hisおよび50ng以上のEGFP−Hisタンパク質を、1/3000に希釈した抗GFP抗体では10ng以上のEGFP−Hisタンパク質がそれぞれ検出できることも示された。
なお、図2でも、写真の上部に、各行に滴下したタンパク質の種類と、検出に使用した一次抗体および二次抗体とその希釈倍率をそれぞれ示した。写真の左側には、各列に滴下した各タンパク質量を示した。
【0058】
III.水泡を有する魚類による抗体産生
1.試料
1)水泡を有する魚類
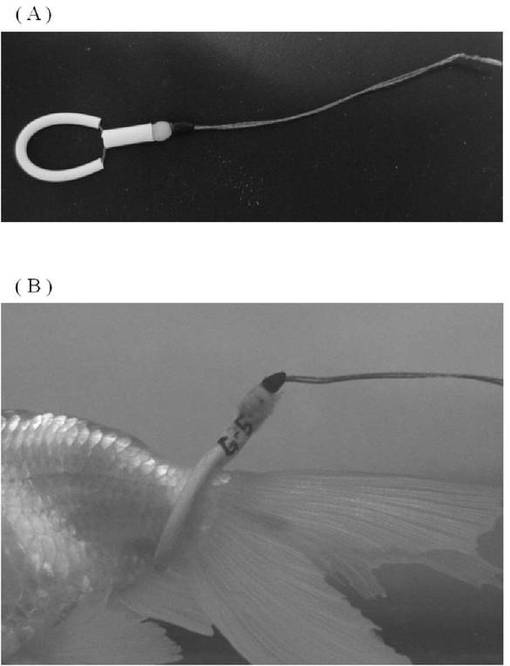
愛知県弥富市の丸照養魚場より入手した水泡眼を使用した。これらは、中和剤(金魚・メダカの中和剤、株式会社ニチドウ)を加えて残留塩素を中和した水道水により、水温20〜25℃で飼育した。本発明で使用した水泡眼(一例)を図3に写真で示した。
2)試薬
特に記載のない場合、試薬は全て和光純薬工業株式会社の製品を使用した。
3)抗原
上記I.で調製したEGFP−Hisタンパク質、精製GA−Hisまたは培養上清GA−Hisをそれぞれ抗原とした。
【0059】
4)アジュバント
次の(A)〜(D)を調製し、アジュバントとした。
(A)レシチンを含むオイルベース
(B)不活化結核菌を混合したオイルベース
(C)不活化麹菌を混合したオイルベース
(D)不活化大腸菌を混合したオイルベース
【0060】
(1)オイルベースの調製
オイルベースは、参考文献4を改変して、以下のように作製した。
すなわち、10gのグリセロールに0.1gの卵黄レシチンを添加し、60℃で保温しながらスターラーによって撹拌した。次に、10gの落花生油(ナカライテスク)を徐々に添加し、均一になるまで同様に撹拌してオイルベースとした。
参考文献4:J.A.REYNOLDS et al.1980(Adjuvant activity of a novel metabolizable lipid emulsion with inactivated viral vaccines. Reynolds JA, Harrington DG, Crabbs CL, Peters CJ, Di Luzio NR. Infect Immun. 1980 Jun;28(3):937−943.
【0061】
(2)不活化大腸菌、不活化麹菌の調製
大腸菌Escherichia coli DH5α株をLB培地に接種し、37℃で16時間振盪培養した。
麹菌Aspergillus oryzae OSI−1013株をDPY液体培地に接種し、28℃で18〜20時間静置培養した。
これらの菌をソニケーターで破砕し、不活化大腸菌および不活化麹菌を得た。
(3)不活化結核菌
結核菌H37 Ra, 乾燥(Difco Laboratories)を不活化結核菌として使用した。
(4)オイルベースと不活化大腸菌、不活化結核菌、不活化麹菌の混合
不活化大腸菌、不活化結核菌、不活化麹菌を、それぞれオイルベースに0.5mg/mLとなるよう添加した。
【0062】
(5)抗原とアジュバントの混合
(A)〜(D)のそれぞれのアジュバントと抗原溶液を1mL容シリンジ(テルモ)および18Gの試薬混合針(三商)により体積比1:1で混合し、油中水型エマルジョンを作製して免疫に使用した。菌体を混合したアジュバントは初回免疫時のみ使用し、2回目以降はオイルベースのみのアジュバントを使用した。
【0063】
2.抗体の産生
EGFP−Hisタンパク質を抗原とする試験区A〜E、精製GA−Hisを抗原とする試験区F〜J、培養上清GA−Hisを抗原とする試験区K,Lおよびコントロールとして試験区Mを作成し、各試験区における水泡眼は6匹とした。各試験区の抗原、アジュバントの組合せを表1に示した。
EGFP−Hisタンパク質を抗原とする試験区A〜E、およびコントロールの試験区Mの合計36匹は、6L水槽に一匹ずつ、同水系において飼育した。また、精製GA−Hisを抗原とする試験区F〜J、および培養上清GA−Hisを抗原とする試験区K,Lの合計36匹は個体識別のためのタグ(図4(A))を水泡眼の尾柄部分に装着し(図4(B))、60L水槽に1試験区(6匹)ごと飼育した。
【0064】
水泡眼への免疫は、試験区ごとに抗原のみ、またはアジュバントと混合してエマルジョン化した抗原を水泡眼の右側水泡に1匹当たり100μg注射することによって行った。免疫は14日に1回行い、70日間継続した。
免疫前および免疫と同日に水泡眼の左側水泡から1匹当たり50〜100μLの水泡液を採取した。免疫と同日に水泡液を採取する場合は、先に免疫した後、水泡液を採取した。
採取した水泡液は、4℃で1500xg 10分間遠心分離して上清を回収し、抗体価測定のためのサンプルとした。なお、調製したサンプルは抗原特異的な抗体の検出に使用するまで、−20℃で保管した。
【0065】
【表1】

【0066】
3.ドットブロット法による抗体検出
1)サンプル
上記2.において採取した水泡液を解凍した後、4℃で10000xg 10分間遠心分離して沈殿を除去してサンプルとした。
また、抗myc tag抗体(SANTA CRUZ)をネガティブコントロールとし、抗His抗体(Amersham Biosciences)をPBSで1/100〜1/5000の範囲に希釈したものをポジティブコントロールとした。
2)抗原
上記I.で調製したEGFP−Hisタンパク質および精製GA−HisをそれぞれPBSに置換したものを抗原とした。
【0067】
3)ドットブロット
抗原抗体反応の検出は、次の(1)〜(6)の手順により、サンドイッチ法によるドットブロット法によって行った。
(1)PVDF膜(ATTO)をメタノールに浸漬して5分間振盪しながら置換し、その後超純水に浸漬して10分間x2回振盪しながら置換した。最後に、PBSに浸漬して10分間以上振盪しながら置換することによって平衡化した。サンプル滴下直前にPVDF膜をプロワイプ(エリエール)に挟み込むことによって余分なPBSを除去し、パラフィルム上のPVDF膜へサンプルを2μLずつ滴下して風乾した。
(2)乾燥させたPVDF膜を5% スキムミルク/0.05% Tween 20 in TBS{20mM Tris−HCl、150mM NaCl(pH7.5)(0.05% TBST)}で平衡化およびブロッキングした。
続いて、PVDF膜を0.05% TBSTにより2回リンスし、10分間振盪しながら洗浄した。さらに、5分間x2回振盪しながら洗浄することによって、余分なスキムミルク溶液を除去した。
【0068】
(3)次に、Can Get Signals Solution 1(TOYOBO)によって抗原となるEGFP−Hisタンパク質あるいは精製GA−Hisをそれぞれ5ng/μLあるいは10ng/μLに希釈し、PVDF膜を浸漬して室温で2時間振盪しながら反応させた。反応後のPVDF膜は0.05% TBSTにより10分間x3回洗浄し、余分な抗原溶液を除去した。
(4)次に、Can Get Signals Solution 1によって抗GFP抗体(MBL)あるいは抗GA抗体(月桂冠より供与)をともに1/3,000に希釈し、PVDF膜を浸漬して室温で1時間振盪しながら反応させた。反応後のPVDF膜は0.05% TBSTにより10分間x3回洗浄し、余分な抗体溶液を除去した。
【0069】
(5)続いて、Can Get Signals Solution 2によって抗ラビットIgG HRP標識二次抗体を1/100,000に希釈し、PVDF膜を浸漬して室温で1時間振盪しながら反応させた。反応後のPVDF膜は0.05% TBSTにより10分間x3回洗浄し、余分な抗体溶液を除去した。
(6)反応後のPVDF膜をPBSTで10分間x3回洗浄し、発光基質液(Amersham ECL plus Western Blotting Detection Reagents、GE ヘルスケア)に浸漬して室温で5分間反応させた後、CCDカメラによって発光を検出した。
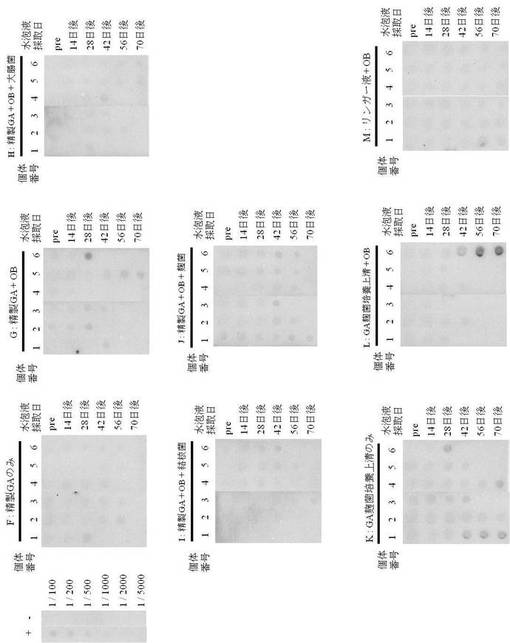
EGFP−Hisタンパク質を投与した試験区の結果を図5および表2に示し、精製GA−His、培養上清GA−Hisを投与した試験区の結果を図6および表3に示した。
【0070】
【表2】
【0071】
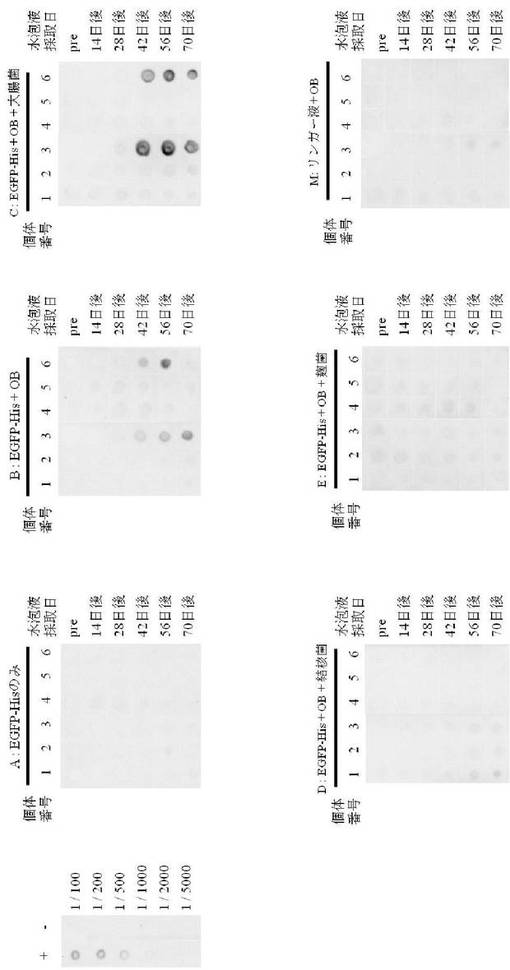
その結果、図5および表2に示されるように、抗原とともにアジュバントとしてオイルベースを投与した試験区Bの2匹と、オイルベースに不活化大腸菌を加えて免疫した試験区Cの2匹において、EGFP−Hisタンパク質(図5、EGFP−His、以下同様である)に対する抗体が検出され、さらに抗体価の増加が見られた。
しかしながら、抗原とともにオイルベースと結核菌を加えて免疫した試験区D、麹菌を加えて免疫した試験区EではEGFP−Hisタンパク質に対する抗体価の増加が認められなかった。また同様に、抗原のみを投与した試験区Aにおいても抗体価の増加は見られなかった。
【0072】
試験区BにおいてEGFP−Hisタンパク質に対する抗体価の増加が認められた2匹のうち、識別個体3では初回免疫から42日以降の水泡液において抗体価が増加し、特に56日後から70日後にかけて抗体価が徐々に増加していた。また、識別個体6では42日後に抗体価がわずかに増加し、56日後にさらに増加していた。
一方、試験区CにおいてEGFP−Hisタンパク質に対する抗体価の増加がみられた2匹のうち、識別個体3では42日後から抗体価が大きく増加し、70日後まで継続しており、また識別個体6でも42日後から抗体価が増加し、70日後まで継続していた。
【0073】
なお、図5では、左上に抗His抗体を反応させたポジティブコントロール(+)および抗myc tag抗体を反応させたネガティブコントロール(−)の結果を示している(希釈倍率:1/100〜1/1800)。
図5、各PVDF膜上部のアルファベットは各試験区、1〜6までの数字は水泡眼の個体識別番号を示し、アルファベットの右に各試験群の免疫条件(OB:オイルベース)を示している。また、右側には各列にはサンプル(水泡液)の採取日(pre:免疫前、その他は初回免疫から採取日までの日数)を示している。
【0074】
【表3】
【0075】
また、図6および表3に示されるように抗原とともにアジュバントとしてオイルベースを免疫した試験区Gの3匹において精製GA−Hisに対する抗体が検出され、抗体価も増加していた。
しかしながら、オイルベースに大腸菌を加えて免疫した試験区H、結核菌を加えて免疫した試験区I、麹菌を加えて免疫した試験区Jでは、精製GA−Hisに対する抗体価の増加は見られなかった。また同様に、抗原のみを投与した試験区Fにおいても抗体価の増加は検出されなかった。
【0076】
培養上清GA−His(図6、GA麹菌培養上清、以下同様である)を免疫した試験区KおよびLでは、抗原のみを免疫した試験区Kの3匹、抗原とともにオイルベースを免疫した試験区Lの1匹において培養上清GA−Hisに対する抗体価の増加が見られた。
試験区Kにおいて培養上清GA−Hisに対する抗体価の増加が検出された3匹のうち、識別個体1では初回免疫から42日後の水泡液から抗体価が増加し、70日後まで継続した。また、識別個体4では70日後の抗体価がわずかに増加し、識別個体6では28日後の抗体価が増加した。試験区Lについては、培養上清GA−Hisに対する抗体価の増加が検出された識別個体6では、初回免疫から42日後の水泡液における抗体価がわずかに増加し、56日後にさらに増加して70日後まで継続した。
コントロールとして金魚用リンガー液とともにオイルベースを免疫した試験区Mでは、EGFP−Hisタンパク質、精製GA−Hisおよび培養上清GA−Hisのいずれの抗原に対しても抗体価の増加は認められなかった。
【0077】
なお、図6では、左上に抗His抗体を反応させたポジティブコントロール(+)および抗myc tag抗体を反応させたネガティブコントロール(−)の結果を示している(希釈倍率:1/100〜1/5000)。
図6、各PVDF膜上部のアルファベットは各試験区、1〜6までの数字は水泡眼の個体識別番号を示し、アルファベットの右に各試験群の免疫条件(OB:オイルベース)を示している。また、右側には各列にはサンプル(水泡液)の採取日(pre:免疫前、その他は初回免疫から採取日までの日数)を示している。
【実施例2】
【0078】
I.抗原の調製
<抗原> LGR3
1−1.ベクターの作製(プラスミドの構築〜目的遺伝子の組み込み)
pCold TF DNA(タカラバイオ)を次のように改変することにより、発現ベクターpCold−TEE−His−hLGR3−LRRを作製した。
pCold TF DNA(タカラバイオ)上のトリガーファクター(TF)配列を除去するために、pCold TF DNAを鋳型とし、制限酵素SmaIおよびSfiI認識配列を持つプライマーによるPCRを行い、ベクターを増幅した。
同様に、pCR4−TOPO−hLGR3(インビトロジェン)を鋳型とし、制限酵素SmaIおよびSfiI認識配列を持つプライマーによるPCRを行い、hLGR3(human leucine−rich repeat containing G protein−coupled receptor 3;hLGR3)遺伝子のleucine−rich region(LRR)部分(配列表配列番23)(図7、hLGRマーカー部分(塩基:1695〜2173))を増幅した。
増幅産物を精製後、制限酵素処理に供し、DNA Ligation Kit(タカラバイオ)により、発現ベクターpCold−TEE−His−hLGR3−LRRを構築した。Escherichia coli HST08株によりコンストラクトを作製し、上記にて構築したpCold−TEE−His−hLGR3−LRRの塩基配列を確認したところ、構築前に予想した塩基配列と完全に一致した。従って、この結果より、コンストラクトが正しく構築されていることが確認できた。
【0079】
II.抗原抗体反応の確認
上記I.で作製したコンストラクト(発現ベクターpCold−TEE−His−hLGR3−LRR)を大腸菌origami株に形質転換した。
ダイレクトPCR法によって形質転換が確認された株を、5ml LB培地(アンピシリンを終濃度100μg/mlとなるように添加)に植菌し、37℃、180rpmで6時間前培養した。
前培養液1mlを250ml 2×YT培地に接種し、OD600=0.45−0.5になるまで37℃、130rpmで培養した後、速やかに15℃に急冷し30分間保冷した。30分後、終濃度が0.1mMになるようにIPTGを添加し、15℃、130rpm、24時間培養した。
菌体を集菌後、20mM リン酸バッファー(pH7.4、20mM イミダゾール、1.5M NaCl)に懸濁し、ソニケーション後、4℃、20,000×g、30分間遠心分離した。
上清を可溶性画分とし、また沈殿を6M 尿素を用いて可溶化不溶性画分とした。それぞれの画分をNi Sepharoseカラムを用いて精製し、Amicon Ultra(ミリポア)によって濃縮した後、SDS−PAGEで精製度を確認した。
【0080】
精製・濃縮した可溶性画分および可溶化不溶性画分をSDS−PAGEで分離後、抗His抗体(Anti−His Antibody(Amersham Biosciences))を用いて検出した結果を図8に示した。
その結果、可溶性画分および可溶化不溶性画分のいずれにおいても、目的分子量の位置にバンドが検出されたことから(図8、矢印部分)、目的タンパク質であるhLGR3タンパク質のLRR部分が発現していることが確認できた。なお、図8において、Mはマーカー、Sは可溶性画分、Pか可溶化不溶性画分のことを示している。
【0081】
III.水泡を有する魚類による抗体産生
1.試料
実施例1と同様の方法によって入手し2か月以上飼育した水泡眼(体長約8cmから10cmの健康な個体5匹)、実施例1と同様の試薬を試料とした。
また、上記II.で調製した可溶性画分(hLGR3タンパク質のLRR部分)を0.6μg/μlに調製したもの、または可溶化不溶性画分(hLGR3タンパク質のLRR部分)を0.1μg/μlに調製したものをそれぞれ抗原とし、表4の成分が均一になるまで混合したオイルベースをアジュバントとした。
【0082】
【表4】
【0083】
2.抗体の産生
次の(1)〜(5)の手順により、抗体の産生を行った。
(1)比較として免疫前の水泡眼の水泡液を100μl 注射器によりサンプリングした。
(2)上記(1)の後、免疫1回目として、上記II.で調製した抗原(0.6μg/μlの可溶性画分)90μlを等量のオイルベースと混合してエマルジョンとしたもの(合計180μl)を水泡眼の各個体の水泡内に注射した。
(3)上記(2)の免疫1回目から1週間後に、各個体から水泡液を100μl採取(サンプリング 1回目)した直後、免疫2回目として、上記II.で調製した抗原(0.1μg/μlの可溶化不溶性画分)を100μlを等量のオイルベースと混合してエマルジョンとしたもの(合計200μl)を水泡眼の各個体の水泡内に注射した。
(4)上記(3)の免疫2回目から1週間後に、各個体から水泡液を100μl採取(サンプリング 2回目)した。
(5)上記(4)のサンプリング 2回目から1週間後に、さらに、各個体から水泡液を100μl採取(サンプリング 3回目)した。
採取した水泡液はいずれも4℃で1,500×g 10分間遠心分離して上清を回収し、抗体価測定のためのサンプルとした。調製したサンプルは抗原特異的な抗体の検出に使用するまで、4℃で保存した。なお、この免疫期間中、死亡した個体はいなかった。
【0084】
3.ドットブロット法による抗体検出
1)サンプル
各個体において、上記1.において採取した水泡液を、1/2、1/10、1/20、1/100、1/200、1/1000、または1/2000にそれぞれ希釈したものをサンプルとした。
また、抗His抗体を検出用一次抗体とし、抗TF抗体(GenScript)をポジティブコントロールとし、抗AIF抗体(ラビット−ポリクローナル抗体)(PromoKine)をネガティブコントロールとした。
【0085】
2)抗原
上記I.II.と同様の手法によって、調製したHis−TF−hLGR3(図7、His部分(塩基:278〜295、Trigger Factor部分(塩基:296〜1657)、およびhLGRマーカー部分(塩基:1695〜2173)を含む)を終濃度5ng/μlとなるようにPBSに置換したものを抗原とした。
【0086】
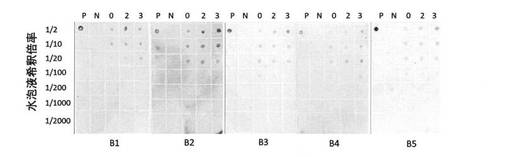
3)ドットブロット
抗原抗体反応の検出は、実施例1と同様の手順により、サンドイッチ法によるドットブロット法によって行った。
サンプルは、上記1)において調製したものを各2μl、図9に示したようにPVDF膜に滴下した。これに上記2)の抗原を反応させた後、上記1)の検出用一次抗体、ネガティブコントロール、もしくはポジティブコントロールを反応させた。その後、続いて、PBSによって抗マウスIgG HRP標識二次抗体(Cell Signaling)を1/25,000に希釈して反応させた。
反応後、洗浄等したPVDF膜を発光基質液(Amersham ECL plus Western Blotting Detection Reagents、GE ヘルスケア)に浸漬して室温で5分間反応させた後、CCDカメラによって発光を検出した。
【0087】
その結果、図9に示したように、抗His抗体を検出用一次抗体とした場合、水泡液を1/2希釈したサンプルにおいて、免疫前よりもサンプリング 2回目、さらにサンプリング 3回目のサンプルのシグナルが強くなったことが確認できた。従って、この結果より、水泡眼に抗原を投与したことにより、水泡液中に抗原に対する抗体が産生されていることが示唆された。
なお、図9において、下のB1〜B5は各サンプルの由来となる水泡眼の個体を示している。上の0は免疫前の水泡液、2はサンプリング 2回目の水泡液、3はサンプリング 3回目の水泡液を示しており、これらの水泡液の希釈倍率(1/2〜1/2000)を縦に示している。さらに、上のPはポジティブコントロール、Nはネガティブコントロールを示しており、これらのコントロールについては、上から1/100、1/200、1/500、1/1000、1/2000、1/5000に希釈したものを固相化している。
【0088】
以上の結果より、水泡を有する魚類によって抗体を製造することが可能であることが示された。水泡を有する魚類における抗体の製造では、水泡を有する魚類に対し、抗原を水泡内へ直接投与することができ、また、産生された抗体を水泡から直接回収できることも確認できた。
水泡から1回で直接回収できるリンパ液は1〜5mlであるため、1度の抗原の投与で産生された抗体を大量に回収することができる。また、抗体を含むリンパ液を採取後も、水泡眼が生きたままの状態であり、繰り返し水泡眼から抗体を含むリンパ液を継続的に採取することも可能であった。
【産業上の利用可能性】
【0089】
本発明により、大量かつ、魚類を生かしたままの状態で繰り返し、魚類に産生させた抗体を得ることが可能となった。本発明によって得られる抗体は、糖タンパク質等を抗原とするものも含まれ、哺乳動物等を対象とする抗体と同様に幅広く活用することができる。
【技術分野】
【0001】
本発明は、抗体の製造方法に関する。さらに詳しくは、水泡を有する魚類に抗原を投与し、抗体を産生させる工程を含む、抗体の製造方法に関する。
【背景技術】
【0002】
現在、遺伝子組み換え技術により大腸菌、酵母等に有用なタンパク質を産生させ、また、これを抗原としてマウス等に投与することにより、抗体を産生させる技術が幅広く利用されている。
しかし、大腸菌などの原核生物に産生させたタンパク質は、多量体形成、糖鎖付加等の翻訳後修飾が正確に行われていない。また、酵母に産生させたタンパク質も完全な翻訳後修飾が行われていないため、本来の機能を有しない場合があるという問題があった。
【0003】
これらの問題を解決すべく、昆虫細胞や、マウス、ブタ、ヒト等の哺乳動物の細胞において有用なタンパク質を産生させる技術が利用されているが、細胞を培養するためのコストが高く、有用なタンパク質が得られるまでに時間がかかるという問題があった。
そこで、近年、バイオリアクターとして魚類を活用することに注目が集まっている。魚類由来のタンパク質は、哺乳動物と同様に多量体形成、糖鎖付加等の翻訳後修飾が行われるという利点がある。そこで特許文献1ではこの利点を生かし、トランスジェニック魚類に多量体糖タンパク質を産生させ、胚、仔魚、稚魚等の組織、血液中から回収する技術が開示されている。
【0004】
魚類は飼育が比較的容易であり、飼育コストも安価である。また、水温や給餌、日照時間、水質等によって産卵および採卵を制御できるなど、非常に扱いやすいという利点もある。
本発明者らは、抗原提示酵母を作成し、これをゼブラフィッシュ等の魚類に飼料として付与することで、魚類由来の特異的抗体を産生させる方法や、従来、困難とされてきたGPCR(G protein−coupled receptor;GPCR)等の膜タンパク質に対する抗体を魚類に産生させる(特願2009−83900)等、魚類による様々な技術を開発している(例えば、特許文献2、3、参照)。
しかし、これらの技術では、魚類全身から抗体を回収等するため、魚類を生かしたままの状態で、産生させた抗体を繰り返し得ることができなかった。
【0005】
本発明者らは、この問題を解決するため、本発明の開発において、水泡眼等の水泡を有する魚類に着目した。この種の金魚は観賞用として、国内外で幅広く飼育されている。
特許文献4において、水泡眼の水疱(水泡)内液が、魚類細胞の細胞保護的又は細胞増殖促進的に作用すること、魚類細胞の処理操作等において、未受精卵の卵質を維持した状態で効率的な操作を可能とすることが開示されている。
しかし、この特許文献では、水疱(水泡)内液に抗体等の有用なタンパク質を産生させること等については示唆すらされておらず、従来の技術においても、水泡を有する魚類を抗体の産生に利用することは試みられていなかった。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2001−86992号公報
【特許文献2】特開2007−255892号公報
【特許文献3】特開2009−73783号公報
【特許文献4】特開2009−183151号公報
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明は、魚類を生かしたままの状態で、産生させた抗体を繰り返し得ることができる、抗体の製造方法の提供を課題とする。
【課題を解決するための手段】
【0008】
本発明者らは、上記課題を解決するために鋭意研究を行った結果、水泡眼等の水泡を有する魚類を抗原投与の対象とすることにより、魚類を生かしたままの状態で、これらの魚類に産生させた抗体を繰り返し得ることができる方法を見出し、本発明を完成するに至った。
【0009】
すなわち、本発明は次の(1)〜(8)の抗体の製造方法、該製造方法によって製造される抗体等に関する。
(1)水泡を有する魚類に抗原を投与し、抗体を産生させる工程を含む、抗体の製造方法。(2)抗原を投与する部位が水泡である上記(1)に記載の製造方法。
(3)抗原をオイルベースとともに、またはオイルベースと不活化大腸菌とともに投与する上記(1)または(2)に記載の製造方法。
(4)魚類に産生させた抗体を、魚類の水泡から採取する工程を含む、上記(1)〜(3)のいずれかに記載の製造方法。
(5)水泡を有する魚類が水泡眼またはらんちゅうである上記(1)〜(4)のいずれかに記載の製造方法。
(6)抗原がタンパク質または糖タンパク質である上記(1)〜(5)のいずれかに記載の製造方法。
(7)抗原がEGFP(enhanced green fluorescent protein;以下、EGFPと示す場合がある)、グルコアミラーゼまたはLGR3(leucine−rich repeat containing G protein−coupled receptor 3;以下、LGR3と示す場合がある)である上記(6)に記載の製造方法。
(8)上記(1)〜(7)のいずれかに記載の製造方法によって製造される抗体。
【発明の効果】
【0010】
本発明の製造方法により、魚類に抗体を産生させることにより、魚類を生かしたままの状態で大量の抗体を繰り返し製造することが可能となった。本発明の製造方法では、糖タンパク質等を抗原とする場合でも、有用な抗体を製造することができる、有用な抗体の製造方法である。
【図面の簡単な説明】
【0011】
【図1】ウェスタンブロット法の結果を示した図である(実施例1)。
【図2】ドットブロット法の結果を示した図である(実施例1)。
【図3】水泡眼(一例)の写真を示した図である(実施例1)。
【図4】タグの写真を示した図である(実施例1)。
【図5】抗原抗体反応の結果(EGFP−Hisタンパク質)を示した図である(実施例1)。
【図6】抗原抗体反応の結果(精製GA−His、培養上清GA−His)を示した図である(実施例1)。
【図7】pCold TF−hLGR3の配列を示した図である(実施例2)。
【図8】抗原抗体反応の結果を示した図である(実施例2)。
【図9】抗原抗体反応の結果を示した図である(実施例2)。
【発明を実施するための形態】
【0012】
本発明の「抗体の製造方法」とは、抗体を産生する生物を魚類とし、これに抗原を投与し、抗体を産生させる工程を含む方法のことをいう。
本発明の「抗体の製造方法」は、魚類が水泡を有する魚類であって、この水泡を有する魚類に抗原を投与し、抗体を産生させる工程を含む製造方法であればよく、抗体を製造するせるために、従来知られている他の工程や方法を含むものであってもよい。
【0013】
この方法において、「抗原を投与」は、魚類に抗体を産生させることが可能な投与方法であればどのような方法であってもよい。例えば、抗原となるタンパク質や抗原酵母等を飼料として経口的に投与したり、腹腔や水泡を投与部位として、抗原を直接、注射等によって投与したりすることが挙げられる。本発明においては特に、抗原の投与部位を水泡として、ここに直接抗原を投与することが好ましい。
【0014】
抗原は、そのまま投与してもよく、アジュバントと組み合わせて投与しても良い。アジュバントとしては、魚類の抗体産生を助けるものであれば、どのようなものであってもよいが、特にオイルベースをアジュバントとすることや、オイルベースと不活化大腸菌とを組み合わせたものをアジュバントとすることが好ましい。
【0015】
本発明の「抗体の製造方法」には、魚類に産生させた抗体を回収する工程も含むことができる。この抗体の回収は、魚類を生かしたまま回収できる方法で行うことが好ましく、特に、魚類の水泡から、抗体を含む水泡内液を回収する等の方法であることが好ましい。この場合、魚類の水泡に直接注射針等をさして、水泡内液を回収することにより、魚類を生かしたままで、魚類に産生させた抗体を回収することが可能となる。
【0016】
本発明における「水泡を有する魚類」としては、魚類の表皮に水泡を有する魚類であれば、いずれの魚種も含まれる。この「水泡を有する魚類」の水泡には、産生された抗体を含むリンパ液が含まれており、水泡内に貯留したこの液を注射器等で採取した場合、再貯留するという性質がある。このような「水泡を有する魚類」として、例えば、水泡眼、らんちゅうなどの金魚を挙げることができる。
【0017】
本発明の「抗原」とは、抗体を産生させるためのものであればどのようなものであってもよく、GPCR、LGR3などの膜貫通型タンパク質、EGFPなどの可溶性タンパク質や、α−ジストログリカン、グルコアミラーゼなどの糖タンパク質であってもよい。
魚類に投与する抗原の量は、魚類が抗体を産生できる量であれば特に問わないが、抗原となるこれらのタンパク質が100μg以上となるように、投与されることが特に好ましい。
【0018】
以下、実施例をあげて本発明をさらに詳細に説明するが、本発明はこれらに限定されるものではない。
【実施例1】
【0019】
I.抗原の調製
<抗原> 麹菌グルコアミラーゼ
1−1.プラスミドの構築
1)麹菌アスペルギルスオリザ由来の硝酸還元酵素遺伝子(以下、niaD遺伝子と示す場合がある)の大腸菌ベクターへの組み込み
目的遺伝子の起源生物(麹菌アスペルギルスオリザ)由来のゲノムDNAを鋳型として、プライマーA(配列表配列番号1)、プライマーB(配列表配列番号2)およびLA−Taqポリメラーゼ(宝ホールディングス)を混合し、以下の条件でPCRを行うことにより、niaD遺伝子(配列表配列番号3)を増幅した。
目的遺伝子の起源生物(麹菌アスペルギルスオリザ)由来のゲノムDNAは参考文献1に記載の定法に従い、調製した。
参考文献1:R.C.Garber and O.C.Yoder, Anal. Biochem.,135,416−422(1983)
<PCR条件>
96℃(5分間)を1サイクル
96℃(20秒間)、60℃(30秒間)、72℃(5分間)を30サイクル
72℃(7分間)を1サイクル
【0020】
得られたPCR増幅産物を制限酵素PstI−HindIIIで処理(37℃)した後、アガロースゲル電気泳動でniaD遺伝子のPstI−HindIII断片として切り出した。切り出しは、QIAquick Gel Extraction kit(QIAGEN)により行った。
切り出したniaD遺伝子のPstI−HindIII断片を大腸菌プラスミドpUC119(宝ホールディングス)のPstI−HindIII部位にDNA Ligation kit ver.1(宝ホールディングス)によりライゲーションし、大腸菌(E.coli)JM109株に形質転換することで、niaDマーカー遺伝子がサブクローニングされた大腸菌プラスミドpNIA2を得た。
大腸菌プラスミドpNIA2は、PstI、SalI部位の両方にunique siteとして遺伝子を導入することができる。
【0021】
2)麹菌アスペルギルスオリザ由来のグルコアミラーゼBターミネーター遺伝子(以下、glaBターミネーター遺伝子と示す)の大腸菌ベクターへの組み込み
麹菌ゲノムDNAを鋳型として、プライマーC(配列表配列番号4)とプライマーD(配列番号5)およびLA−Taqポリメラーゼ(宝ホールディングス)を混合し、以下の条件でPCRを行うことにより、glaBターミネーター遺伝子(配列表配列番号6)を増幅した。
<PCR条件>
96℃(5分間)を1サイクル
96℃(20秒間)、60℃(30秒間)、72℃(5分間)を30サイクル
72℃(7分間)を1サイクル
【0022】
得られたPCR増幅産物を制限酵素SalI−XhoIで処理(37℃)した後、アガロースゲル電気泳動でglaBターミネーター遺伝子のSalI−XhoI断片として切り出した。切り出しは、QIAquick Gel Extraction kit(QIAGEN)により行った。
切り出したglaBターミネーター遺伝子のSalI−XhoI断片を、上記1)で得た大腸菌プラスミドpNIA2のSalI部位にDNA Ligation kit ver.1(宝ホールディングス)によりライゲーションし、大腸菌(E.coli)JM109株に形質転換することで、glaBターミネーター遺伝子がサブクローニングされたプラスミドpNIATを得た。
大腸菌プラスミドpNIATは、PstI、SalI部位の両方に遺伝子をunique siteとして遺伝子を導入することができる。
【0023】
3)麹菌アスペルギルスオリザ由来のsodMプロモーター遺伝子(以下、sodMプロモーター遺伝子と示す)の組み込み
麹菌ゲノムDNAを鋳型として、プライマーE(配列表配列番号7)、プライマーF(配列表配列番号8)およびLA−Taqポリメラーゼ(宝ホールディングス)を混合し、以下の条件でPCRを行うことにより、麹菌由来sodMプロモーター遺伝子(配列表配列番号9)を増幅した。
<PCR条件>
96℃(5分間)を1サイクル
96℃(20秒間)、60℃(30秒間)、72℃(5分間)を30サイクル
72℃(7分間)を1サイクル
【0024】
得られたPCR増幅産物を制限酵素SalI−PstIで処理(37℃)した後、アガロースゲル電気泳動でsodMプロモーター遺伝子のSalI−PstI断片として切り出した。切り出しは、QIAquick Gel Extraction kit(QIAGEN)により行った。
切り出したsodMプロモーター遺伝子のSalI−PstI断片を、上記2)で得た大腸菌プラスミドpNIATのPstI−SalI部位にDNA Ligation kit ver.1(宝ホールディングス)によりライゲーションし、大腸菌(E.coli)JM109株に形質転換することで、sodMプロモーター遺伝子がサブクローニングされたプラスミドpNMBを得た。
大腸菌プラスミドpNMBは、SalI部位に麹菌で発現させたいタンパク質をコードする遺伝子をunique siteとして遺伝子を導入することができる。
【0025】
1−2.目的遺伝子の組み込み
1)ベクターの調製
上記1−1.で構築した大腸菌プラスミドpNMBを制限酵素SalIで処理(37℃)した後、dNTPを最終10mMとなるように添加してT4 DNAポリメラーゼ(宝ホールディングス)で1時間処理(37℃)した。さらに、バクテリア由来アルカリホスファターゼ(宝ホールディングス)で30分処理(50℃)した。処理後得られた物をPCRクリーンアップカラム(プロメガ社製)で溶出させ、ベクターとした。
【0026】
2)インサートの調製
麹菌で発現させたいタンパク質をコードする遺伝子を目的遺伝子として、上記ベクターにサブクローニングするインサートとして調製した。
目的遺伝子は、a)麹菌由来グルコアミラーゼB(glaB)遺伝子(配列表配列番号10(アミノ酸配列を配列表配列番号11に示した))、またはb)Hisタグ遺伝子をつなげた麹菌由来グルコアミラーゼB(glaB)遺伝子(配列表配列番号12(アミノ酸配列を配列表配列番号13に示した))であった。
上記1−1.1)で調製した目的遺伝子の起源生物(麹菌アスペルギルスオリザ)由来のゲノムDNAを鋳型として、目的遺伝子の開始コドンから下流30bpのセンス鎖のプライマー(5’末端をリン酸化)(配列表配列番号14)と、目的遺伝子の終止コドンから上流49bpのアンチセンス鎖のプライマー(5’末端をリン酸化)(配列表配列番号15)と、pfu Taqポリメラーゼ(東洋紡績株式会社)を混合し、以下の条件でPCRを行うことにより、目的遺伝子を増幅した。
得られたPCR増幅産物をPCRクリーンアップカラム(プロメガ社製)で処理溶出させ、インサートとした。
<PCR条件>
96℃(5分間)を1サイクル
96℃(20秒間)、60℃(30秒間)、72℃(5分間)を30サイクル
72℃(7分間)を1サイクル
【0027】
3)ベクターとインサートのライゲーション
上記1)で得たベクターおよび上記2)で得た各目的遺伝子のインサートを1:20(モル比)となるように添加して、DNA Ligation kit ver.1(宝ホールディングス)によりライゲーションを行い、大腸菌(E.coli)JM109株に形質転換した。
形質転換体から、各目的遺伝子の読み枠がsodMプロモーターと正方向にサブクローニングされているものを目的遺伝子発現プラスミドとした。
【0028】
1−3.目的遺伝子発現株の取得
PEG−カルシウム法により、上記1−2.で調製した目的遺伝子発現プラスミドにより、麹菌アスペルギルスオリザのniaD変異株(独立行政法人産業技術総合研究所特許生物寄託センターにFERM P−17707として寄託済み)を形質転換した。
【0029】
1)PEG−カルシウム法は、参考文献2に従い、以下の手順により行った。
即ち、麹菌胞子約108個を100ml DPY液体培地に接種し、28℃で18〜20時間静置培養を行った。こうして発芽させた後、ガラスビーズ(直径0.6cm)を培養フラスコに加え、30℃,130rpmの震とう培養でさらに24時間培養した。
菌糸を採集し、0.8M NaCl水溶液で菌糸を2〜3回洗い、洗った菌糸に10mlの新しく作ったプロトプラスト化溶液(15mg/ml Yatalase(タカラバイオ),10mg/ml Cellulase Onozuka R−10(Yakult Honsha),0.8M NaCl,10mM phosphate buffer(pH6.0),1mM DTT)を加え、30℃で3〜4時間ゆっくりと震とうし、細胞壁を消化してプロトプラストを調製した。
得られたプロトプラスト溶液をミラクロス(カルビオケム社)でろ過し、ろ液を更に4℃,2500g、5分間、遠心分離して回収した。
【0030】
プロトプラストを氷冷した溶液1(0.8M NaCl,10mM CaCl2,10mM Tris−HCl(pH8.0))で洗浄した後、2×108/mlになるように溶液1に懸濁して、その量に対して0.2容量になるように溶液2(40% PEG4000,50mM CaCl2,10mM Tris−HCl(pH8.0))を加え、静かに混合した。
形質転換は、0.1mlのプロトプラスト溶液を新しい15mlチューブに移し、15μl以下(3〜7μg)の目的DNAを加え、氷上で30分間放置した後、0.5ml溶液2を加えゆっくりと攪拌し、室温で20分間放置した。5ml溶液1を加えて、混合した後、4℃で2500gの速度で5分間遠心を行った。
プロトプラストを含んだ沈殿物を0.1mlの溶液1に懸濁し、0.8M NaClを含むCD再生培地に広げ、さらに、その上に、30℃まで冷却したCD軟寒天培地を添加した後、固化させ、28℃で4〜7日培養した。
参考文献2:Mol Gen Genet, 218,99−104,(1989)
【0031】
2)形質転換体のうち硝酸を単一窒素源とするツアペクドックス(Czapek−Dox)培地(2% グルコース、0.1% リン酸1水素2カリウム、0.05% 塩化カリウム、0.05% 硫酸マグネシウム、0.001% 硫酸鉄、0.3% 硝酸ナトリウム)で生育できる株を選択することにより、目的遺伝子発現プラスミドを保持する形質転換体を複数得た。
【0032】
1−4.目的遺伝子産物の調製
上記1−3.により得られた形質転換体をポテトデキストロース培地で培養することで胞子形成させ、滅菌水で胞子を回収した。
回収した胞子を、500ml容三角フラスコに入った100ml GPY液体培地(2% グルコース、1% ポリペプトン、0.5% イーストエキストラクト、0.1% リン酸1水素2カリウム、0.05% 塩化カリウム、0.05% 硫酸マグネシウム、0.001% 硫酸鉄、0.3% 硝酸ナトリウム)に最終胞子濃度1X106/mlとなるように植菌した。
これを30℃で3日間液体培養し、目的遺伝子産物が分泌発現した培養上清を回収した。
【0033】
1−5.培養上清の濃縮
上記1−4.で回収した培養上清の一部を、分画分子量10,000NMWLのセントリプレップ10(ミリポア社製)により遠心分離(3,000×g)し、濃縮した。
【0034】
1−6.培養上清に含まれるGA濃度の定量
上記1−4.で回収した培養上清に含まれるGA濃度は、SDS−PAGEによって培養上清からタンパク質を分離し、CBB染色像を画像解析ソフトのCS Analyzer 3.0(ATTO)により定量化することで推定した。
すなわち、次の1)〜5)の工程を行った。
1)下記1−7.で得た精製GAを3μgから11μgとなるようにPBSに溶解した(スタンダード)。
これと、1〜3μLの1x培養上清とにそれぞれ5xサンプルバッファー(2.5M Tris−HCl、10% グリセロール、0.05% Bromophenol Blue、10% SDS)を添加(終濃度1x)し、100℃で5分間煮沸することによってサンプルとした。
2)これらのサンプルから、SDS−PAGE(7.5% ポリアクリルアミドゲル、200V、20mA)によってタンパク質を分離した。
3)泳動後のゲルをCBB染色液(0.25% CBB−R250、50% メタノール、5% 酢酸)に浸漬し、室温で30分間振盪しながらタンパク質を染色した。
4)染色後のゲルを脱色液(25% メタノール、10% 酢酸)に浸漬し、室温で振盪しながらタンパク質に結合しなかったCBBを洗浄した。
5)脱色後のゲルをCCDカメラ(Light Capture II、ATTO)により撮影し、検出されたGAのバンドをCS Analyzer 3.0のゾーンデンシトメトリー解析によって定量化し、精製GAの積算値から検量線を作成した。
作成した検量線から推定された培養上清(濃縮なし)に含まれるGA濃度は約3.0μg/μLであった。
この結果より、上記1−5.にて5倍濃縮した培養上清を金魚用リンガー液{125mM NaCl、2.6mM KCl、10mM 2−[4−(2−Hydroxyethy l)−1−piperazinyl]ethanesulfonic acid(HEPES)(pH7.4)}によってGA濃度が4μg/μLとなるよう調製した(以下、これを培養上清GAまたは培養上清GA−Hisと示す場合がある)。
【0035】
1−7.目的遺伝子産物の精製
1)目的遺伝子産物
上記1−4.で回収した培養上清の一部を飽和濃度70%の硫酸アンモニウムを添加し、4℃で一晩撹拌しタンパク質を沈殿させた後、遠心分離により沈殿を取り除いた。
この培養上清をブチルトヨパール−650M(東ソー社製)樹脂を添加し、低温で一晩撹拌した後、樹脂を回収した。
回収した樹脂を1.5M 硫酸アンモニウム溶液で2回洗浄し、10mM 酢酸バッファー(pH5.0)で目的のGAタンパク質を溶出し、凍結乾燥したものを、精製GAタンパク質とした。
精製GAタンパク質は、金魚用リンガー液によりGA濃度が4μg/μLとなるよう調製した(以下、これを精製GAと示す場合がある)。
【0036】
2)Hisタグをもつ目的遺伝子産物
上記1−4.の目的遺伝子産物を含む培養上清をニッケルカラム(His GraviTrap column:GE Healthcare社)に添加した後、0.5M NaClおよび400mM イミダゾールを含む20mMリン酸ナトリウムバッファー(pH7.4)を添加してカラムに結合した組換えグルコアミラーゼを溶出した。
この溶出液を透析膜に移し、50mM酢酸ナトリウムバッファー(pH5.0)を含む1Lのバッファーに対し、4℃、3時間で透析を2回行い、Hisタグをもつ目的遺伝子産物を精製した。
精製したHisタグをもつ目的遺伝子産物は、金魚用リンガー液によりGA濃度が4μg/μLとなるよう調製した(以下、これを精製GA−Hisと示す場合がある)。
【0037】
<抗原> EGFP−Hisタンパク質
2−1.ベクターの作製(プラスミドの構築〜目的遺伝子の組み込み)
pCold TF DNA(タカラバイオ)を次のように改変することにより、csp Aプロモーターの下流に5’非翻訳領域、translation enhancing element(TEE)、EGFP、および3’末端に精製用のタグであるHisタグのDNA配列が連結したコンストラクトである、pColdTEE−EGFP−Hisベクターを作製した。
1)本発明者らがpXI(配列表配列番号16)の下流にEGFP(配列表配列番号17)を組み込んで作製したpXI−EGFPベクター(特願2009−107344号、参考文献3)を鋳型として、プライマーG(配列表配列番号18)、プライマーH(配列表配列番号19)およびTAKARA Ex Taq(タカラバイオ)を混合し、以下の条件でPCRを行うことにより、egfp遺伝子の開始コドンが制限酵素SmaIの認識配列に変換され、3’末端がHisタグのDNA配列に変換された断片を増幅した。
参考文献3:Johnson AD,Krieg PA.pXeX,a vector for efficient expression of cloned sequences in Xenopus embryos.Gene. 1994 Sep.
<PCR条件>
95℃(1分間)を1サイクル
95℃(10秒間)、55℃(30秒間)、72℃ (2分間)を30サイクル
72℃(5分間)を1サイクル
【0038】
2)上記1)の増幅産物(egfp遺伝子の開始コドンが制限酵素SmaIの認識配列に変換され、3’末端がHisタグのDNA配列に変換された断片)を鋳型として、プライマーG(配列表配列番号18)、プライマーI(配列表配列番号20)およびTAKARA Ex Taq(タカラバイオ)を混合し、上記1)と同様の条件でPCRを行うことにより、Hisタグの下流に制限酵素SfiIの認識配列が連結したegfp−his6遺伝子断片を増幅した。
【0039】
3)pCold−TF−DNA(タカラバイオ)を鋳型として、プライマーJ(配列表配列番号21)、プライマーK(配列表配列番号22)およびTAKARA Ex Taq(タカラバイオ)を混合し、以下の条件でPCRを行うことにより、TEEにSmaIの認識配列が連結し、pCold−TF−DNAのtranscription terminator配列の上流にSfiIの認識配列が連結するDNA断片を増幅した。
【0040】
上記3)で増幅したDNA断片と、SmaIおよびSfiIによって消化した上記2)のegfp−his6遺伝子断片を、DNA Ligation kit Mighty mix(タカラバイオ)によりライゲーションすることによってpColdTEE−EGFP−Hisを作製した。
【0041】
2−2.目的遺伝子産物の調製
上記2−1.で作製したpColdTEE−EGFP−Hisベクターにより大腸菌BL21を形質転換し、終濃度50μg/mLのアンピシリンナトリウムを含むLB平板培地に塗抹して、37℃で一晩保温しながら培養した。なお、特に記載のない場合、試薬は全て和光純薬工業株式会社の製品を使用した。
【0042】
1)生成したコロニーをLB−ampicillin液体培地に植菌し、37℃で12時間振盪しながら培養した。この培養上清を、あらかじめ作製した2xYT−Ampicillin液体培地{50μg/mL アンピシリンナトリウム、1.6% Bacto Tripton(BD)、1% 乾燥酵母エキス(ナカライテスク)、0.5% NaCl(pH 7.0)}に植菌し、波長600nmの吸光度が0.4〜0.5の範囲となるまで37℃で振盪しながら培養した。
その後、15℃で30分間静置し、0.1mM Isopropyl β−D−1−thiogalactopyranoside(IPTG)を培養上清の1/1000量を添加して、15℃で24時間振盪しながらEGFP−Hisタンパク質の発現を誘導した。
目視にてEGFP−Hisタンパク質の発現を確認した後、4℃で3000xg 15分間遠心分離して菌体を回収し、予冷した1xリン酸緩衝生理食塩水(PBS:137mM NaCl、2.7mM KCl、1.5mM KH2PO4、8.1mM Na2HPO4)により2回洗浄することで残留した培地成分を除去した。
【0043】
2)上記1)に続いて、40mLのNiカラム溶出緩衝液A{20mM リン酸ナトリウム、0.5M NaCl、20mM Imidazol(pH7.4)}を添加し、微量超音波細胞破砕機(MICROSON XL 2000、MISONIX)により菌体を破砕した。
次に、4℃で12000xg 30分間遠心分離して上清を回収し、0.20μmフィルター(ADVANTEC)によって夾雑物を除去して大腸菌発現タンパク質抽出液とした。
調製した大腸菌発現タンパク質抽出液をNi Sepharose担体(GEヘルスケア)を充填したNi カラム(GEヘルスケア)にペリスタポンプ(Bio Rad)によって添加することで、EGFP−Hisタンパク質を担体に結合させた。
【0044】
3)上記2)に続いて、Niカラム溶出緩衝液A{20mM リン酸ナトリウム、0.5M NaCl、20mM Imidazol(pH7.4)}およびNiカラム溶出緩衝液B{20mM リン酸ナトリウム、0.5M NaCl、500mM Imidazol(pH7.4)}による20mM〜500mM Imidazolの連続濃度勾配によってカラムからEGFP−Hisタンパク質を溶出した。
【0045】
4)次に、限外濾過膜(ミリポア)を用いて、EGFP−Hisタンパク質が溶出した画分のNi カラム溶出緩衝液をDEAEカラム溶出緩衝液A{20mM 2−amino−2−(hydroxymethyl)propane−1,3−diol(Tris)−HCl(pH8.0)}にバッファー置換したものをサンプルとし、あらかじめDEAE Sepharose Fast Flow担体(GEヘルスケア)を充填した陰イオン交換カラムに低圧クロマトグラフィーシステム(AKTAprime plus:GEヘルスケア)によって添加することで、EGFP−Hisタンパク質を担体に結合させ、DEAEカラム溶出緩衝液A{20mM Tris−HCl(pH8.0)}およびDEAEカラム溶出緩衝液B{20mM Tris−HCl、1M NaCl(pH8.0)}により、0M〜1M NaClの連続濃度勾配による溶出を行った。
【0046】
5)上記4)のカラムより溶出したEGFP−Hisタンパク質を含む画分の溶液を、2価イオンを含まない金魚用リンガー液{125mM NaCl、2.6mM KCl、10mM 2−[4−(2−Hydroxyethyl)−1−piperazinyl]ethanesulfonic acid(HEPES)(pH7.4)}に置換し、EGFP−Hisタンパク質濃度が4μg/μLになるように調製した。
【0047】
II.抗原抗体反応の確認
上記I.にて調製した抗原(精製GA、精製GA−HisおよびEGFP−Hisタンパク質)を抗体と作用させ、ウェスタンブロット法またはドットブロット法により、抗原抗体反応を確認した。
1.抗体
1)抗His抗体
Anti−His Antibody(Amersham Biosciences)2)抗GlaB抗体
グルコアミラーゼタンパク質全長のペプチド(配列表配列番号11)をデザインし、化学合成によりペプチド合成し、KLH(Keyhole Limpet Hemocyanin)をキャリアタンパク質としてコンジュゲーションしたペプチドを抗原としてウサギに免疫することにより抗体を得た(カスタム抗体作製;インビトロジェン社製)。
【0048】
2.ウェスタンブロット法
1)上記I.にて調製したEGFP−Hisタンパク質、精製GAおよび精製GA−Hisに5xサンプルバッファー(2.5M Tris,10% glycerol,0.05% Bromophenol Blue,10% SDS)をそれぞれ添加し(終濃度1x)、100℃で5分間煮沸してサンプルとした。
これらのサンプルを、10%ポリアクリルアミドゲルにより200V 20mAの設定で電気泳動し、タンパク質を分離した。
【0049】
2)次にセミドライブロッターによってタンパク質をPVDF膜(ATTO)に転写し、0.5% Tween20/PBS(0.5% PBST)で2回リンスして、5% スキムミルク/0.5%PBSTに浸漬して振盪しながら1時間ブロッキングした。
ブロッキング後のPVDF膜をPBSTで2回リンスし、0.5%PBSTで15分間x1回、5分間x2回洗浄して、余分なブロッキング液を除去した。
【0050】
3)続いて上記1.の抗His抗体および抗GlaB抗体をCan Get Signals(TOYOBO) Solution 1により、それぞれ1/3000および1/1000に希釈し、これにPVDF膜を浸漬して室温にて1時間振盪しながら反応させた。反応後のPVDF膜を0.5% PBSTで10分間x3回洗浄し、余分な抗体溶液を除去した。
4)次に、抗マウスIgG HRP標識二次抗体および抗ラビットIgG HRP標識二次抗体をCan Get Signals Solution 2によって、それぞれ1/25000および1/100000に希釈し、PVDF膜を浸漬して室温で1時間振盪しながら反応させた。
【0051】
5)反応後のPVDF膜をPBSTで10分間x3回洗浄し、発光基質液{Amersham ECL plus Western Blotting Detection Reagents(GE ヘルスケア)}に浸漬して室温で5分間反応させたのち、CCDカメラ[Light Capture(ATTO)]によって撮影し、CS Analyzer 3.0(ATTO)により発光を検出した。
【0052】
その結果、図1、A(抗His抗体と反応させたPVDF膜)に示されるように、抗His抗体によって、EGFP−Hisタンパク質、精製GAおよび精製GA−Hisをそれぞれ検出したところ、抗His抗体を反応させたPVDF膜ではEGFP−Hisタンパク質(図1、EGFP−His)および精製GA−His(図1、glaB−His)がそれぞれ検出された。
また、図1、B(抗GlaB抗体と反応させたPVDF膜)に示されるように、抗His抗体を反応させたPVDF膜では精製GA(図1、glaB)および精製GA−His(図1、glaB−His)がそれぞれ検出された。
従って、これらの結果から、精製GA−Hisを抗GlaB抗体および抗His抗体で検出できることが確認できた。
なお、図1では、各写真の上部に、各レーンに泳動したサンプル量および使用した一次抗体、二次抗体とその希釈倍率を示している(M:プレステインドSDS−PAGEスタンダード(Broad Range)(Bio Rad))。
【0053】
2.ドットブロット法
1)上記I.にて調製したEGFP−Hisタンパク質、精製GAおよび精製GA−Hisを、PBSにより、それぞれ250pg/μLから250ng/μLの範囲に希釈してサンプルとした。
2)PVDF膜をメタノールに浸漬して5分間振盪しながら置換し、その後超純水に浸漬して10分間x2回振盪しながら置換し、最後にPBSに浸漬して10分間以上振盪しながら置換することによって平衡化した。
3)上記1)のサンプル滴下直前にPVDF膜をプロワイプ(エリエール)に挟み込むことによって余分なPBSを除去し、パラフィルム上のPVDF膜へサンプルを2μLずつ滴下して風乾した。
【0054】
4)乾燥させたPVDF膜を{5% スキムミルク/0.05% Tween20 in TBS(20mM Tris−HCl,150mM NaCl pH7.5)(0.05% TBST)}によって平衡化およびブロッキングした。
5)続いて、PVDF膜を0.05% TBSTにより2回リンスし、10分間振盪しながら洗浄して5分間x2回振盪しながら洗浄し、余分なスキムミルク溶液を除去した。
6)続いて、Can Get Signals Solution 1により、上記1.の抗His抗体、抗GFP抗体(MBL)および抗GlaB抗体をそれぞれ1/3000に希釈し、PVDF膜を浸漬して室温で2時間振盪しながら反応させた。
反応後のPVDF膜は0.05% TBSTにより10分間x3回洗浄し、余分な抗体溶液を除去した。
【0055】
7)次に、Can Get Signals Solution 2によって、抗マウスIgG HRP標識二次抗体および抗ラビットIgG HRP標識二次抗体をそれぞれ1/25000および1/100000に希釈し、PVDF膜を浸漬して室温で1時間振盪しながら反応させた。
8)反応後のPVDF膜を0.05% TBSTにより10分間x3回洗浄し、ウェスタンブロット法と同様に発光基質液と反応させて発光を検出した。
【0056】
その結果、図2(左側)に示されるように、抗GFP抗体により、EGFP−Hisタンパク質、精製GAおよび精製GA−Hisの検出をそれぞれ行ったところ、10ngから500ngまでのEGFP−Hisタンパク質(図2、GFP−His)のスポットに反応が見られた。
また、図2(中央)に示されるように、抗His抗体により検出を行ったものは、10ngから500ngまでの精製GA−His(図2、glaB−His)のスポットおよび50ngから500ngまでのEGFP−Hisタンパク質(図2、GFP−His)のスポットに反応が見られた。
さらに、図2(右側)に示されるように、抗GlaB抗体により反応させたPVDF膜では500pgから500ngの精製GA(図2、glaB)および精製GA−His(図2、glaB−His)のスポットに反応が見られた。
以上の結果から、抗GlaB抗体および抗His抗体によって精製GA−Hisを検出できることが確認された。
【0057】
またこの条件では、1/1000に希釈した抗GlaB抗体は500pg以上の精製GAおよび精製GA−Hisを、1/3000に希釈した抗His抗体は10ng以上の精製GA−Hisおよび50ng以上のEGFP−Hisタンパク質を、1/3000に希釈した抗GFP抗体では10ng以上のEGFP−Hisタンパク質がそれぞれ検出できることも示された。
なお、図2でも、写真の上部に、各行に滴下したタンパク質の種類と、検出に使用した一次抗体および二次抗体とその希釈倍率をそれぞれ示した。写真の左側には、各列に滴下した各タンパク質量を示した。
【0058】
III.水泡を有する魚類による抗体産生
1.試料
1)水泡を有する魚類
愛知県弥富市の丸照養魚場より入手した水泡眼を使用した。これらは、中和剤(金魚・メダカの中和剤、株式会社ニチドウ)を加えて残留塩素を中和した水道水により、水温20〜25℃で飼育した。本発明で使用した水泡眼(一例)を図3に写真で示した。
2)試薬
特に記載のない場合、試薬は全て和光純薬工業株式会社の製品を使用した。
3)抗原
上記I.で調製したEGFP−Hisタンパク質、精製GA−Hisまたは培養上清GA−Hisをそれぞれ抗原とした。
【0059】
4)アジュバント
次の(A)〜(D)を調製し、アジュバントとした。
(A)レシチンを含むオイルベース
(B)不活化結核菌を混合したオイルベース
(C)不活化麹菌を混合したオイルベース
(D)不活化大腸菌を混合したオイルベース
【0060】
(1)オイルベースの調製
オイルベースは、参考文献4を改変して、以下のように作製した。
すなわち、10gのグリセロールに0.1gの卵黄レシチンを添加し、60℃で保温しながらスターラーによって撹拌した。次に、10gの落花生油(ナカライテスク)を徐々に添加し、均一になるまで同様に撹拌してオイルベースとした。
参考文献4:J.A.REYNOLDS et al.1980(Adjuvant activity of a novel metabolizable lipid emulsion with inactivated viral vaccines. Reynolds JA, Harrington DG, Crabbs CL, Peters CJ, Di Luzio NR. Infect Immun. 1980 Jun;28(3):937−943.
【0061】
(2)不活化大腸菌、不活化麹菌の調製
大腸菌Escherichia coli DH5α株をLB培地に接種し、37℃で16時間振盪培養した。
麹菌Aspergillus oryzae OSI−1013株をDPY液体培地に接種し、28℃で18〜20時間静置培養した。
これらの菌をソニケーターで破砕し、不活化大腸菌および不活化麹菌を得た。
(3)不活化結核菌
結核菌H37 Ra, 乾燥(Difco Laboratories)を不活化結核菌として使用した。
(4)オイルベースと不活化大腸菌、不活化結核菌、不活化麹菌の混合
不活化大腸菌、不活化結核菌、不活化麹菌を、それぞれオイルベースに0.5mg/mLとなるよう添加した。
【0062】
(5)抗原とアジュバントの混合
(A)〜(D)のそれぞれのアジュバントと抗原溶液を1mL容シリンジ(テルモ)および18Gの試薬混合針(三商)により体積比1:1で混合し、油中水型エマルジョンを作製して免疫に使用した。菌体を混合したアジュバントは初回免疫時のみ使用し、2回目以降はオイルベースのみのアジュバントを使用した。
【0063】
2.抗体の産生
EGFP−Hisタンパク質を抗原とする試験区A〜E、精製GA−Hisを抗原とする試験区F〜J、培養上清GA−Hisを抗原とする試験区K,Lおよびコントロールとして試験区Mを作成し、各試験区における水泡眼は6匹とした。各試験区の抗原、アジュバントの組合せを表1に示した。
EGFP−Hisタンパク質を抗原とする試験区A〜E、およびコントロールの試験区Mの合計36匹は、6L水槽に一匹ずつ、同水系において飼育した。また、精製GA−Hisを抗原とする試験区F〜J、および培養上清GA−Hisを抗原とする試験区K,Lの合計36匹は個体識別のためのタグ(図4(A))を水泡眼の尾柄部分に装着し(図4(B))、60L水槽に1試験区(6匹)ごと飼育した。
【0064】
水泡眼への免疫は、試験区ごとに抗原のみ、またはアジュバントと混合してエマルジョン化した抗原を水泡眼の右側水泡に1匹当たり100μg注射することによって行った。免疫は14日に1回行い、70日間継続した。
免疫前および免疫と同日に水泡眼の左側水泡から1匹当たり50〜100μLの水泡液を採取した。免疫と同日に水泡液を採取する場合は、先に免疫した後、水泡液を採取した。
採取した水泡液は、4℃で1500xg 10分間遠心分離して上清を回収し、抗体価測定のためのサンプルとした。なお、調製したサンプルは抗原特異的な抗体の検出に使用するまで、−20℃で保管した。
【0065】
【表1】
【0066】
3.ドットブロット法による抗体検出
1)サンプル
上記2.において採取した水泡液を解凍した後、4℃で10000xg 10分間遠心分離して沈殿を除去してサンプルとした。
また、抗myc tag抗体(SANTA CRUZ)をネガティブコントロールとし、抗His抗体(Amersham Biosciences)をPBSで1/100〜1/5000の範囲に希釈したものをポジティブコントロールとした。
2)抗原
上記I.で調製したEGFP−Hisタンパク質および精製GA−HisをそれぞれPBSに置換したものを抗原とした。
【0067】
3)ドットブロット
抗原抗体反応の検出は、次の(1)〜(6)の手順により、サンドイッチ法によるドットブロット法によって行った。
(1)PVDF膜(ATTO)をメタノールに浸漬して5分間振盪しながら置換し、その後超純水に浸漬して10分間x2回振盪しながら置換した。最後に、PBSに浸漬して10分間以上振盪しながら置換することによって平衡化した。サンプル滴下直前にPVDF膜をプロワイプ(エリエール)に挟み込むことによって余分なPBSを除去し、パラフィルム上のPVDF膜へサンプルを2μLずつ滴下して風乾した。
(2)乾燥させたPVDF膜を5% スキムミルク/0.05% Tween 20 in TBS{20mM Tris−HCl、150mM NaCl(pH7.5)(0.05% TBST)}で平衡化およびブロッキングした。
続いて、PVDF膜を0.05% TBSTにより2回リンスし、10分間振盪しながら洗浄した。さらに、5分間x2回振盪しながら洗浄することによって、余分なスキムミルク溶液を除去した。
【0068】
(3)次に、Can Get Signals Solution 1(TOYOBO)によって抗原となるEGFP−Hisタンパク質あるいは精製GA−Hisをそれぞれ5ng/μLあるいは10ng/μLに希釈し、PVDF膜を浸漬して室温で2時間振盪しながら反応させた。反応後のPVDF膜は0.05% TBSTにより10分間x3回洗浄し、余分な抗原溶液を除去した。
(4)次に、Can Get Signals Solution 1によって抗GFP抗体(MBL)あるいは抗GA抗体(月桂冠より供与)をともに1/3,000に希釈し、PVDF膜を浸漬して室温で1時間振盪しながら反応させた。反応後のPVDF膜は0.05% TBSTにより10分間x3回洗浄し、余分な抗体溶液を除去した。
【0069】
(5)続いて、Can Get Signals Solution 2によって抗ラビットIgG HRP標識二次抗体を1/100,000に希釈し、PVDF膜を浸漬して室温で1時間振盪しながら反応させた。反応後のPVDF膜は0.05% TBSTにより10分間x3回洗浄し、余分な抗体溶液を除去した。
(6)反応後のPVDF膜をPBSTで10分間x3回洗浄し、発光基質液(Amersham ECL plus Western Blotting Detection Reagents、GE ヘルスケア)に浸漬して室温で5分間反応させた後、CCDカメラによって発光を検出した。
EGFP−Hisタンパク質を投与した試験区の結果を図5および表2に示し、精製GA−His、培養上清GA−Hisを投与した試験区の結果を図6および表3に示した。
【0070】
【表2】
【0071】
その結果、図5および表2に示されるように、抗原とともにアジュバントとしてオイルベースを投与した試験区Bの2匹と、オイルベースに不活化大腸菌を加えて免疫した試験区Cの2匹において、EGFP−Hisタンパク質(図5、EGFP−His、以下同様である)に対する抗体が検出され、さらに抗体価の増加が見られた。
しかしながら、抗原とともにオイルベースと結核菌を加えて免疫した試験区D、麹菌を加えて免疫した試験区EではEGFP−Hisタンパク質に対する抗体価の増加が認められなかった。また同様に、抗原のみを投与した試験区Aにおいても抗体価の増加は見られなかった。
【0072】
試験区BにおいてEGFP−Hisタンパク質に対する抗体価の増加が認められた2匹のうち、識別個体3では初回免疫から42日以降の水泡液において抗体価が増加し、特に56日後から70日後にかけて抗体価が徐々に増加していた。また、識別個体6では42日後に抗体価がわずかに増加し、56日後にさらに増加していた。
一方、試験区CにおいてEGFP−Hisタンパク質に対する抗体価の増加がみられた2匹のうち、識別個体3では42日後から抗体価が大きく増加し、70日後まで継続しており、また識別個体6でも42日後から抗体価が増加し、70日後まで継続していた。
【0073】
なお、図5では、左上に抗His抗体を反応させたポジティブコントロール(+)および抗myc tag抗体を反応させたネガティブコントロール(−)の結果を示している(希釈倍率:1/100〜1/1800)。
図5、各PVDF膜上部のアルファベットは各試験区、1〜6までの数字は水泡眼の個体識別番号を示し、アルファベットの右に各試験群の免疫条件(OB:オイルベース)を示している。また、右側には各列にはサンプル(水泡液)の採取日(pre:免疫前、その他は初回免疫から採取日までの日数)を示している。
【0074】
【表3】
【0075】
また、図6および表3に示されるように抗原とともにアジュバントとしてオイルベースを免疫した試験区Gの3匹において精製GA−Hisに対する抗体が検出され、抗体価も増加していた。
しかしながら、オイルベースに大腸菌を加えて免疫した試験区H、結核菌を加えて免疫した試験区I、麹菌を加えて免疫した試験区Jでは、精製GA−Hisに対する抗体価の増加は見られなかった。また同様に、抗原のみを投与した試験区Fにおいても抗体価の増加は検出されなかった。
【0076】
培養上清GA−His(図6、GA麹菌培養上清、以下同様である)を免疫した試験区KおよびLでは、抗原のみを免疫した試験区Kの3匹、抗原とともにオイルベースを免疫した試験区Lの1匹において培養上清GA−Hisに対する抗体価の増加が見られた。
試験区Kにおいて培養上清GA−Hisに対する抗体価の増加が検出された3匹のうち、識別個体1では初回免疫から42日後の水泡液から抗体価が増加し、70日後まで継続した。また、識別個体4では70日後の抗体価がわずかに増加し、識別個体6では28日後の抗体価が増加した。試験区Lについては、培養上清GA−Hisに対する抗体価の増加が検出された識別個体6では、初回免疫から42日後の水泡液における抗体価がわずかに増加し、56日後にさらに増加して70日後まで継続した。
コントロールとして金魚用リンガー液とともにオイルベースを免疫した試験区Mでは、EGFP−Hisタンパク質、精製GA−Hisおよび培養上清GA−Hisのいずれの抗原に対しても抗体価の増加は認められなかった。
【0077】
なお、図6では、左上に抗His抗体を反応させたポジティブコントロール(+)および抗myc tag抗体を反応させたネガティブコントロール(−)の結果を示している(希釈倍率:1/100〜1/5000)。
図6、各PVDF膜上部のアルファベットは各試験区、1〜6までの数字は水泡眼の個体識別番号を示し、アルファベットの右に各試験群の免疫条件(OB:オイルベース)を示している。また、右側には各列にはサンプル(水泡液)の採取日(pre:免疫前、その他は初回免疫から採取日までの日数)を示している。
【実施例2】
【0078】
I.抗原の調製
<抗原> LGR3
1−1.ベクターの作製(プラスミドの構築〜目的遺伝子の組み込み)
pCold TF DNA(タカラバイオ)を次のように改変することにより、発現ベクターpCold−TEE−His−hLGR3−LRRを作製した。
pCold TF DNA(タカラバイオ)上のトリガーファクター(TF)配列を除去するために、pCold TF DNAを鋳型とし、制限酵素SmaIおよびSfiI認識配列を持つプライマーによるPCRを行い、ベクターを増幅した。
同様に、pCR4−TOPO−hLGR3(インビトロジェン)を鋳型とし、制限酵素SmaIおよびSfiI認識配列を持つプライマーによるPCRを行い、hLGR3(human leucine−rich repeat containing G protein−coupled receptor 3;hLGR3)遺伝子のleucine−rich region(LRR)部分(配列表配列番23)(図7、hLGRマーカー部分(塩基:1695〜2173))を増幅した。
増幅産物を精製後、制限酵素処理に供し、DNA Ligation Kit(タカラバイオ)により、発現ベクターpCold−TEE−His−hLGR3−LRRを構築した。Escherichia coli HST08株によりコンストラクトを作製し、上記にて構築したpCold−TEE−His−hLGR3−LRRの塩基配列を確認したところ、構築前に予想した塩基配列と完全に一致した。従って、この結果より、コンストラクトが正しく構築されていることが確認できた。
【0079】
II.抗原抗体反応の確認
上記I.で作製したコンストラクト(発現ベクターpCold−TEE−His−hLGR3−LRR)を大腸菌origami株に形質転換した。
ダイレクトPCR法によって形質転換が確認された株を、5ml LB培地(アンピシリンを終濃度100μg/mlとなるように添加)に植菌し、37℃、180rpmで6時間前培養した。
前培養液1mlを250ml 2×YT培地に接種し、OD600=0.45−0.5になるまで37℃、130rpmで培養した後、速やかに15℃に急冷し30分間保冷した。30分後、終濃度が0.1mMになるようにIPTGを添加し、15℃、130rpm、24時間培養した。
菌体を集菌後、20mM リン酸バッファー(pH7.4、20mM イミダゾール、1.5M NaCl)に懸濁し、ソニケーション後、4℃、20,000×g、30分間遠心分離した。
上清を可溶性画分とし、また沈殿を6M 尿素を用いて可溶化不溶性画分とした。それぞれの画分をNi Sepharoseカラムを用いて精製し、Amicon Ultra(ミリポア)によって濃縮した後、SDS−PAGEで精製度を確認した。
【0080】
精製・濃縮した可溶性画分および可溶化不溶性画分をSDS−PAGEで分離後、抗His抗体(Anti−His Antibody(Amersham Biosciences))を用いて検出した結果を図8に示した。
その結果、可溶性画分および可溶化不溶性画分のいずれにおいても、目的分子量の位置にバンドが検出されたことから(図8、矢印部分)、目的タンパク質であるhLGR3タンパク質のLRR部分が発現していることが確認できた。なお、図8において、Mはマーカー、Sは可溶性画分、Pか可溶化不溶性画分のことを示している。
【0081】
III.水泡を有する魚類による抗体産生
1.試料
実施例1と同様の方法によって入手し2か月以上飼育した水泡眼(体長約8cmから10cmの健康な個体5匹)、実施例1と同様の試薬を試料とした。
また、上記II.で調製した可溶性画分(hLGR3タンパク質のLRR部分)を0.6μg/μlに調製したもの、または可溶化不溶性画分(hLGR3タンパク質のLRR部分)を0.1μg/μlに調製したものをそれぞれ抗原とし、表4の成分が均一になるまで混合したオイルベースをアジュバントとした。
【0082】
【表4】
【0083】
2.抗体の産生
次の(1)〜(5)の手順により、抗体の産生を行った。
(1)比較として免疫前の水泡眼の水泡液を100μl 注射器によりサンプリングした。
(2)上記(1)の後、免疫1回目として、上記II.で調製した抗原(0.6μg/μlの可溶性画分)90μlを等量のオイルベースと混合してエマルジョンとしたもの(合計180μl)を水泡眼の各個体の水泡内に注射した。
(3)上記(2)の免疫1回目から1週間後に、各個体から水泡液を100μl採取(サンプリング 1回目)した直後、免疫2回目として、上記II.で調製した抗原(0.1μg/μlの可溶化不溶性画分)を100μlを等量のオイルベースと混合してエマルジョンとしたもの(合計200μl)を水泡眼の各個体の水泡内に注射した。
(4)上記(3)の免疫2回目から1週間後に、各個体から水泡液を100μl採取(サンプリング 2回目)した。
(5)上記(4)のサンプリング 2回目から1週間後に、さらに、各個体から水泡液を100μl採取(サンプリング 3回目)した。
採取した水泡液はいずれも4℃で1,500×g 10分間遠心分離して上清を回収し、抗体価測定のためのサンプルとした。調製したサンプルは抗原特異的な抗体の検出に使用するまで、4℃で保存した。なお、この免疫期間中、死亡した個体はいなかった。
【0084】
3.ドットブロット法による抗体検出
1)サンプル
各個体において、上記1.において採取した水泡液を、1/2、1/10、1/20、1/100、1/200、1/1000、または1/2000にそれぞれ希釈したものをサンプルとした。
また、抗His抗体を検出用一次抗体とし、抗TF抗体(GenScript)をポジティブコントロールとし、抗AIF抗体(ラビット−ポリクローナル抗体)(PromoKine)をネガティブコントロールとした。
【0085】
2)抗原
上記I.II.と同様の手法によって、調製したHis−TF−hLGR3(図7、His部分(塩基:278〜295、Trigger Factor部分(塩基:296〜1657)、およびhLGRマーカー部分(塩基:1695〜2173)を含む)を終濃度5ng/μlとなるようにPBSに置換したものを抗原とした。
【0086】
3)ドットブロット
抗原抗体反応の検出は、実施例1と同様の手順により、サンドイッチ法によるドットブロット法によって行った。
サンプルは、上記1)において調製したものを各2μl、図9に示したようにPVDF膜に滴下した。これに上記2)の抗原を反応させた後、上記1)の検出用一次抗体、ネガティブコントロール、もしくはポジティブコントロールを反応させた。その後、続いて、PBSによって抗マウスIgG HRP標識二次抗体(Cell Signaling)を1/25,000に希釈して反応させた。
反応後、洗浄等したPVDF膜を発光基質液(Amersham ECL plus Western Blotting Detection Reagents、GE ヘルスケア)に浸漬して室温で5分間反応させた後、CCDカメラによって発光を検出した。
【0087】
その結果、図9に示したように、抗His抗体を検出用一次抗体とした場合、水泡液を1/2希釈したサンプルにおいて、免疫前よりもサンプリング 2回目、さらにサンプリング 3回目のサンプルのシグナルが強くなったことが確認できた。従って、この結果より、水泡眼に抗原を投与したことにより、水泡液中に抗原に対する抗体が産生されていることが示唆された。
なお、図9において、下のB1〜B5は各サンプルの由来となる水泡眼の個体を示している。上の0は免疫前の水泡液、2はサンプリング 2回目の水泡液、3はサンプリング 3回目の水泡液を示しており、これらの水泡液の希釈倍率(1/2〜1/2000)を縦に示している。さらに、上のPはポジティブコントロール、Nはネガティブコントロールを示しており、これらのコントロールについては、上から1/100、1/200、1/500、1/1000、1/2000、1/5000に希釈したものを固相化している。
【0088】
以上の結果より、水泡を有する魚類によって抗体を製造することが可能であることが示された。水泡を有する魚類における抗体の製造では、水泡を有する魚類に対し、抗原を水泡内へ直接投与することができ、また、産生された抗体を水泡から直接回収できることも確認できた。
水泡から1回で直接回収できるリンパ液は1〜5mlであるため、1度の抗原の投与で産生された抗体を大量に回収することができる。また、抗体を含むリンパ液を採取後も、水泡眼が生きたままの状態であり、繰り返し水泡眼から抗体を含むリンパ液を継続的に採取することも可能であった。
【産業上の利用可能性】
【0089】
本発明により、大量かつ、魚類を生かしたままの状態で繰り返し、魚類に産生させた抗体を得ることが可能となった。本発明によって得られる抗体は、糖タンパク質等を抗原とするものも含まれ、哺乳動物等を対象とする抗体と同様に幅広く活用することができる。
【特許請求の範囲】
【請求項1】
水泡を有する魚類に抗原を投与し、抗体を産生させる工程を含む、抗体の製造方法。
【請求項2】
抗原を投与する部位が水泡である請求項1に記載の製造方法。
【請求項3】
抗原をオイルベースとともに、またはオイルベースと不活化大腸菌とともに投与する請求項1または2に記載の製造方法。
【請求項4】
魚類に産生させた抗体を、魚類の水泡から採取する工程を含む、請求項1〜3のいずれかに記載の製造方法。
【請求項5】
水泡を有する魚類が水泡眼またはらんちゅうである請求項1〜4のいずれかに記載の製造方法。
【請求項6】
抗原がタンパク質または糖タンパク質である請求項1〜5のいずれかに記載の製造方法。
【請求項7】
抗原がEGFP、グルコアミラーゼまたはLGR3である請求項6に記載の製造方法。
【請求項8】
請求項1〜7のいずれかに記載の製造方法によって製造される抗体。
【請求項1】
水泡を有する魚類に抗原を投与し、抗体を産生させる工程を含む、抗体の製造方法。
【請求項2】
抗原を投与する部位が水泡である請求項1に記載の製造方法。
【請求項3】
抗原をオイルベースとともに、またはオイルベースと不活化大腸菌とともに投与する請求項1または2に記載の製造方法。
【請求項4】
魚類に産生させた抗体を、魚類の水泡から採取する工程を含む、請求項1〜3のいずれかに記載の製造方法。
【請求項5】
水泡を有する魚類が水泡眼またはらんちゅうである請求項1〜4のいずれかに記載の製造方法。
【請求項6】
抗原がタンパク質または糖タンパク質である請求項1〜5のいずれかに記載の製造方法。
【請求項7】
抗原がEGFP、グルコアミラーゼまたはLGR3である請求項6に記載の製造方法。
【請求項8】
請求項1〜7のいずれかに記載の製造方法によって製造される抗体。
【図1】

【図2】
【図3】

【図4】
【図5】
【図6】
【図7】

【図8】

【図9】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2013−82664(P2013−82664A)
【公開日】平成25年5月9日(2013.5.9)
【国際特許分類】
【出願番号】特願2012−58566(P2012−58566)
【出願日】平成24年3月15日(2012.3.15)
【出願人】(304026696)国立大学法人三重大学 (270)
【出願人】(000165251)月桂冠株式会社 (88)
【Fターム(参考)】
【公開日】平成25年5月9日(2013.5.9)
【国際特許分類】
【出願日】平成24年3月15日(2012.3.15)
【出願人】(304026696)国立大学法人三重大学 (270)
【出願人】(000165251)月桂冠株式会社 (88)
【Fターム(参考)】
[ Back to top ]