
抗体を生成し抗体レパートリーをスクリーニングするための方法とコンパウンド
本発明は、一般に1)少なくとも1つの標的抗原を保持する高度に免疫原性の小胞を提供すること、および2)前記抗原を保持する小胞で動物を免疫化して抗原特異的抗体反応を誘起すること、を含む組成物と方法に関する。本発明はまた、抗体レパートリーをスクリーニングする方法であって、1)少なくとも1つの標的抗原と1つのマーカーを保持する小胞を提供すること、および2)前記抗原とマーカーを保持する小胞を用いて、所定の抗原特異性を有する抗体産生細胞または粒子を単離することを含む方法に関する。次に、所定の抗原特異性を持つ抗体を、単離した抗体産生細胞から周知の方法を用いて調製することができる。本発明は、実験、研究、治療、予防、または診断の分野で使用することができる。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、抗体を生成するための組成物および方法であって、一般に1)少なくとも1つの標的抗原を有する高度に免疫原性の小胞を提供すること、および2)前記抗原を有する小胞を用いて動物を免疫化して、抗原特異的な抗体応答を引き起こすこと、を含む組成物および方法に関する。本発明はまた、抗体レパートリーをスクリーニングする方法であって、1)少なくとも1つの標的抗原および1つのマーカーを有する小胞を提供すること、および2)前記抗原およびマーカーを有する小胞を用いて、所定の抗原特異性を有する抗体産生細胞または粒子を単離すること、を含む方法に関する。次に、所定の抗原特異性を有する抗体を、単離した抗体産生細胞から周知の方法を用いて調製することができる。本発明は、実験、研究、治療、予防、または診断の分野で使用することができる。
【背景技術】
【0002】
抗体は、微生物を含む環境からの侵入者に対する防衛の第1線において決定的な役割を果たす、かなめのタンパク質である。それは研究と診断の道具であり、最も重要なことに、治療用複合物として用いることができる。多数の企業および研究センターが、今日、抗体ベースの治療候補開発の様々な段階にあり、いくつかの抗体薬物が既に市場に出荷されている。
【0003】
抗体は、ポリクローナルであっても、モノクローナルであってもよい。マウス、齧歯類、霊長類、ウマ、ブタ、ウサギ、家禽などを含む様々な種からポリクローナル抗体を産生する方法が、例えば、(1)において見出される。手短に言えば、抗原をアジュバント(例えば、完全または不完全アジュバント、例えばフロイントアジュバント)存在下で注入する。動物に一般には、皮下、腹膜内、静脈内、筋肉内注入によって投与する。反復注入を行っても良い。血液試料を収集し、免疫グロブリンまたは血清を分離する。
【0004】
モノクローナル抗体を産生する最初の方法は、KohlerとMilstein(2)により確立された。この方法はまた、例えば(3)に詳細に記載されている。手短に言えば、この方法は、アジュバント存在下で抗原により動物を免疫化し、続いて脾臓細胞またはリンパ節細胞を採取し、これを次にミエローマ細胞などの不死の細胞と融合させることを含む。その結果得られるハイブリドーマがモノクローナル抗体を産生し、これを、限界希釈によって選択して個々のクローンを単離する。
【0005】
抗体の調製が、新しいタンパク質の機能の評価に必須となっており、多くの場合、抗体は治療薬に成長している。治療化合物としての抗体に対する興味は、ヒト抗体およびヒト化抗体生成用の技術の発展により、最近復活した。進行中の多くのゲノム配列決定計画が情報を豊富に提供しつつあり、これに伴って新しい推定タンパク質薬物標的に対する抗体の系統的調製が必要となっている。これが、新しい薬物標的の同定が進展する中で、経費のかさむボトルネックを作り出し、モノクローナル抗体調製方法を合理化する新しい手法の必要性が強調されている。
【0006】
実際、モノクローナル抗体調製の基本的方法にはいくつかの限界がある。例えば、ハイブリドーマの調製は、時間がかかり冗長で非効率的な、ハイブリッド細胞生成、薬物選択、スクリーニング、およびクローン増殖の過程を必要とし、その間に希に存在する所望の抗原特異性を有する抗体産生細胞が失われる可能性がある。もう1つの困難は、ハイブリド−マの大きなプール中のユニークな抗体産生ハイブリドーマを同定するために、急速スクリーニング法を発展させる必要があることである。この方法は、標的抗原の性質とその入手可能性によって異なる。さらにもう1つの困難は、ハイブリドーマが主に、マウスまたは数少ない他の非霊長類動物の細胞を用いて生成されることである。それ故、ヒト抗体を発現する遺伝子組換えマウスを用いない限り、ハイブリドーマはヒト以外の抗体を産生するので、これは組換えDNA法を用いて、ヒト化した抗体に転換する必要がある。後者の過程が医療適用に必要であり、これは一旦所望の特異性を有する抗体を産生するクローンが得られた後にのみ開始することができる。
【0007】
これらの限界に対処するために、2つのタイプの戦略が出現した:それは1)上述の古くからある手法の特定の工程を改良すること、および2)組換えDNA技術を用いて抗体レパートリーを開発すること、である。第1の戦略が、例えば、SLAM技術(4)を生み出し、それによると、ハイブリドーマの調製とスクリーニングの必要性が除かれる。第2の戦略は、例えば、ファージディスプレイ技術を生み出し、それによると、標的タンパク質が、抗体またはその断片を発現しているバクテリオファージのライブラリーと、バクテリオファージパニング法を用いて反応する(5)。より最近、HuMY技術が GeneTastix(US patent 6,406,863)により開発され、それによると、標的タンパク質の断片が抗体レパートリーのライブラリーと、酵母のツーハイブリッド法を用いて反応する。
【0008】
改良が成されたけれども、これまでに開発された各方法は依然として限界を有している。特に、大部分の方法の主要な限界が、それらが動物を免疫化するためおよび/またはハイブリドーマもしくはバクテリオファージライブラリーのスクリーニングに大量の標的タンパク質を必要とすることである。ハイブリドーマの調製とスクリーニングを必要としないSLAM技術の場合においてさえも、著者たちが記述しているように、抗体産生細胞を単離するためのプラーク分析を行うために、やはり大量の精製した標的タンパク質が必要である。組換えタンパク質を大量に精製する必要性が、多くの予想薬物標的タンパク質の迅速な確認を妨げている。多くの例で、精製過程はタンパク質毎に異なり、不十分量の材料、機能しない又は変性した産物を産み出す。精製過程は、膜タンパク質または複数ポリペプチド複合体から成る対象を取り扱う時には、より問題となる。
【0009】
あるファージディスプレイ法は、ツーハイブリッド法と共に抗原ペプチドのライブラリーを用いており、それによって大規模な抗原精製の必要性を軽減している。しかし、これらの方法の1つの欠点は、立体構造のエピトープに対する抗体を得ることが困難であって、例えば、抗−MHC/ペプチド複合体抗体などの、複数ポリペプチドからなる単位から生ずるエピトープに限定された抗体を得ることができないことである。もう1つの限界は、使用するペプチドライブラリーと抗体ライブラリーの大きさと質によっては、必ず高い抗原特異的親和性を有する抗体を単離できるとは限らないことである。これは、抗体の親和性の成熟は、第1世代の抗体が得られる場合にのみ実行され得るからである。対照的に、哺乳動物中に見出される無制限の抗体配列のレパートリーは、これらの動物を繰り返し免疫化することにより起こる自然の親和性成熟過程に結びついて、いかなるエピトープに対しても抗体を生成する潜在能力を持つ最も効率的な方法を提供する。
【0010】
本発明は、新しい効果的な手法を提供することにより、抗体調製法の限界に対処する。本発明はまた、現存する抗体調製法を改良するための新しい価値ある手段を提供する。
【発明の開示】
【0011】
本発明はここで、エキソソーム上の抗原、アジュバントおよび/またはマーカーの表示、ならびにエキソソームの強力な液性免疫応答の誘導ためのおよび/または抗体レパートリーをスクリーニングするための媒体としての使用、に関連した2つの技術を組み合わせた、抗体を生成するための新しい方法を開示する。
【0012】
エキソソームはエンドソーム起源の小胞であって、後期エンドソーム多胞体の原形質膜との融合に続いて細胞外環境に分泌される(6,7)。種々のタイプの組織の細胞、例えば、樹状細胞、Bリンパ球、腫瘍細胞および肥満細胞などが、エキソソームを分泌することが示されてきた。異なる起源のエキソソームは、別々のセットのタンパク質と脂質部分を表示する(8,9)。エキソソームは、抗原提示と免疫モジュレーションに含まれるタンパク質を特に含み、エキソソームが免疫応答のモジュレーションを引き起こす細胞間信号伝達において役割を果たしていることを示唆している。実際、腫瘍抗原由来のペプチドをパルス投与した樹状細胞(DC)から得られたエキソソームが、適合する腫瘍を用いた動物モデルにおいて抗腫瘍反応を誘発する(10,11)。治療目的または研究手段としてエキソソームを産生、精製あるいは使用する方法が、例えば、WO99/03499、WO00/44389、およびWO97/05900中に記載されており、これらを参照してここに取り入れる。組換えエキソソームについては、従来技術において既に記述されており、それは、組換えタンパク質をコードするプラスミドによりトランスフェクションされた細胞に由来するものである。そのような組換えエキソソームは、プラスミドがコードした組換えタンパク質を含む(WO00/28001)。
【0013】
治療目的でまたは研究手段として、エキソソームのタンパク質含量を操作し、抗原、アジュバントおよびマーカーを表示する方法が、WO03/016522に記載されている。
【0014】
本発明は、選ばれた抗原に対して特異性を有する単一の抗体産生粒子を単離する方法であって:
1)選択した抗原とマーカーを表示している小胞、好ましくはエキソソーム、を調製すること;
2)工程1の前記小胞を抗体レパートリーと接触させる、または懸濁させること;および
3)前記小胞と反応している単一の抗体産生粒子を同定および単離すること;
を含む方法に関する。
【0015】
ある実施態様においては、前記抗体産生粒子は抗体産生細胞であり、前記抗体レパートリーは抗体産生細胞のレパートリーである。前記抗体産生細胞は、プラズマ細胞、ハイブリドーマ、またはリンパ球でよい。好ましくは、前記抗体産生細胞は産生した抗体を表示する。前記抗体レパートリーは、標準的な組換えDNAの手法を用いて調製され、例えば、ファージまたは酵母のディスプレーライブラリー中に見出される。したがって、別の実施態様においては、前記抗体産生粒子は特定の抗体を提示しているファージまたは酵母の集合である。前記抗体産生細胞はまた抗体分泌細胞でもよい。
【0016】
本発明は、ある抗原に対して特異性を有する単一の抗体産生細胞を同定する方法であって:
1)抗体産生細胞を提供すること;
2)前記抗原およびマーカーを表示している小胞、好ましくはエキソソームを調製すること;
3)工程1の抗体産生細胞を、工程2の小胞と懸濁させること;および
3)前記小胞と反応している単一の抗体産生細胞を同定および単離すること;
を含む方法に関する。
好ましくは、前記抗体産生細胞はリンパ球である。ある実施態様においては、前記リンパ球を前記抗原によって免疫化されたヒト以外の動物から収集する。
【0017】
もし抗体産生細胞が抗体分泌細胞であるなら、方法はさらに、抗体産生細胞を小胞と懸濁させる工程の前に、抗体産生細胞をCD81、CD45などの遍在する細胞表面マーカーに対する第1のビオチン化抗体、ストレプトアビジンおよび前記抗体分泌細胞により産生される免疫グロブリンに向けられた第2のビオチン化抗体とインキュベートする工程を含む。場合によっては、前記工程は:1)抗体分泌細胞を、遍在する細胞表面マーカーに対する第1のビオチン化抗体およびストレプトアビジンとインキュベートすること;および、2)その結果得られた抗体分泌細胞を、前記抗体分泌細胞の免疫グロブリンに向けられた第2のビオチン化抗体とインキュベートすること;を含む。またはそれに代えて、前記工程は:1)ストレプトアビジンを、遍在する細胞表面マーカーに対する第1のビオチン化抗体および前記抗体分泌細胞の免疫グロブリンに向けられた第2のビオチン化抗体とインキュベートすること;および2)その結果得られた第1と第2の抗体を保持しているストレプトアビジンを、抗体分泌細胞とインキュベートすること;を含む。前記抗体分泌細胞は、リンパ球、ハイブリドーマ、プラズマ細胞から成る群から選択するのが好ましい。
【0018】
本発明は、単一の抗体産生細胞を単離する方法であって:
1)少なくとも1つの抗原またはそのエピト−プを表示している免疫原性の小胞、好ましくはエキソソームを提供すること;
2)ヒト以外の動物を前記免疫原性小胞で免疫化することによって抗体反応を生起させること;
3)免疫化した動物からリンパ球を収集すること;
4)工程1の前記抗原またはそのエピトープとマーカーを表示している小胞、好ましくはエキソソームを調製すること;
5)工程3のリンパ球を工程4の小胞と共に懸濁すること;および、
6)工程4の小胞と反応している単一の抗体産生細胞を同定および単離すること;
を含む方法を開示する。
【0019】
場合によっては、前記動物は、抗原を表示している組換えエキソソーム以外の免疫原により免疫化されている。これらの免疫原には、アジュバント中の精製した組換え抗原、ヌクレオチドベースの免疫原(裸のDNA、ウイルスDNA)などの常用の抗原調合物、および抗原を含む細胞または細胞断片が含まれる。
【0020】
本発明は、変異抗原に特異的な単一抗体を産生している粒子を、抗体レパートリーから単離する方法であって:
1)前記変異抗原とマーカーを表示している小胞、好ましくはエキソソーム、の第1の集団を調製すること;
2)自然抗原を表示しているが前記マーカーは表示していない小胞、好ましくはエキソソーム、の第2の集団を調製すること;
3)前記抗体レパートリーを小胞の第1および第2の集団と共に、第2集団は過剰に、懸濁すること;
4)工程1の小胞と反応している単一の抗体産生粒子を同定および単離すること;
を含む方法に関する。
【0021】
本発明は、変異抗原に特異的な単一抗体を産生している細胞を同定する方法であって:
1)抗体産生細胞を提供すること;
2)動物の免疫化に用いた前記変異抗原とマーカーを表示している小胞、好ましくはエキソソーム、を調製すること;
3)自然抗原を表示し、前記マーカーを表示していない小胞、好ましくはエキソソーム、を調製すること;
4)工程1の抗体産生細胞を、工程2の前記変異抗原とマーカーを表示している小胞および工程3の前記自然抗原を表示している過剰量の小胞、と共に懸濁すること;および、
5)工程2の小胞と反応している単一の抗体産生細胞を同定および単離すること;
を含む方法に関する。
好ましくは、前記抗体産生細胞はリンパ球である。ある実施態様においては、前記リンパ球は前記抗原で免疫化したヒト以外の動物から収集される。
【0022】
本発明はさらに、変異抗原に特異的な単一抗体を産生している細胞を単離する方法であって:
1)変異抗原を表示している免疫原性小胞、好ましくはエキソソーム、を提供すること;
2)ヒト以外の動物を前記免疫原性小胞によって免疫化することにより、抗体反応を生起すること;
3)免疫化した動物からリンパ球を収集すること;
4)工程1の前記変異抗原およびマーカーを表示している小胞、好ましくはエキソソームを調製すること;
5)自然抗原を表示し、前記マーカーを表示していない小胞、好ましくはエキソソーム、を調製すること;
6)工程3のリンパ球を、工程4の前記変異抗原とマーカーを表示している小胞および工程5の前記自然抗原を表示している過剰量の小胞と懸濁すること;および、
7)工程4の小胞と反応している抗原変異体に特異的な単一の抗体を産生している細胞を同定および単離すること;
を含む方法を開示する。
【0023】
場合によっては、前記方法はさらに次の工程を含む:a)前記の選択された抗体産生粒子からDNAまたはRNAを回収すること、b)免疫グロブリン配列またはその一部をコードする核酸配列を増幅すること、c)増幅された核酸配列を発現ベクターへクローニングして、所望の抗原特異性を有するタンパク質を生成すること。
【0024】
本発明に従って抗体産生細胞を単離する上記の方法のある実施態様においては、前記小胞によって表示される前記抗原を、エキソソームを標的として指向する(エキソソーム指向)ポリペプチドに融合する。別の実施態様においては、前記小胞によって表示される前記抗原を、エキソソームターゲティングポリペプチドに架橋する。
【0025】
本発明に従って抗体産生細胞を単離する上で開示した方法の他の実施態様において、前記抗原は少なくとも1つの膜貫通ドメインを有するポリペプチドであり、また前記抗原はエキソソーム産生細胞中に過剰に発現され、それによって、前記抗原を表示する組換えエキソソームの生成が可能になる。好ましくは、前記少なくとも1つの膜貫通ドメインを有するポリペプチドはレセプターである。より好ましくは、前記レセプターは、SSTR2、CCR7、CXCR4、およびCCR5などのGPCR(Gタンパク質共役レセプター)である。
【0026】
前記抗原は、例えばレセプターまたは酵素のような任意のタンパク質でよく、あるいは、糖脂質、多糖類、薬剤、および有機化学物質などの、ポリペプチド以外の化合物でもよい。場合によっては、前記抗原はオーファンレセプターである。場合によっては、前記抗原は腫瘍、ウイルス、または微生物抗原である。あるいは、前記抗原はMHC複合体、より具体的にはMHC I/ペプチド複合体でもよい。
【0027】
本発明により変異抗原に特異的な単一抗体を産生する細胞を単離する上に開示した方法の好ましい実施態様において、前記変異抗原は突然変異抗原であり、前記自然抗原は野生型抗原である。場合によっては、前記変異抗原は、ポリペプチド、脂質、DNAまたは小分子から成る群から選ばれる分子と接触している抗原であり、前記自然抗原はフリーの抗原である。好ましくは、前記分子と接触している抗原はMHC/ペプチド複合体である。したがって、前記変異抗原はMHC/ペプチド複合体であり、前記自然抗原は負荷されていないMHC、または異なるペプチドを負荷されたMHCである。ある実施態様において、前記変異抗原はHLA-C/HIVペプチド複合体であり、自然抗原は負荷されていないHLA-Cまたは異なるペプチドを負荷されたHLA-Cである。異なるペプチドとは第1のHIVペプチドとは異なるペプチド(好ましくは50、30、20または10%より少ない同一性を有する)、例えばHIV由来ではないペプチドを意味する。あるいは、前記変異抗原はリガンド-レセプター複合体であり、自然抗原はフリーのリガンドまたはフリーのレセプターである。ある実施態様において、前記変異抗原は、gp120、CXCR4およびCD4、またはgp120、CCR5およびCD4を含み、前記自然抗原はgp120である。また、前記変異抗原および自然抗原は、酵素を含む任意のタンパク質の異なる立体構造状態であってもよい。
【0028】
本発明に従い抗体産生細胞を単離する上に開示した方法の別の実施態様において、前記免疫原性小胞はさらに免疫アクセサリー分子を表示する。好ましくは、前記免疫アクセサリー分子はアジュバントポリペプチドである。より好ましくは、前記アジュバントポリペプチドは、GM-CSF、IL-2およびCD40Lなどのサイトカインである。前記CD40Lは、好ましくは突然変異CD40Lであり、前記突然変異は、可溶性CD40Lの切断と放出を阻止する。場合によっては、前記免疫アクセサリー分子も、エキソソームターゲティングポリペプチドに融合または架橋させる。好ましくは、GM-CSFまたはIL-2などの前記可溶性免疫アクセサリー分子を、エキソソームターゲティングポリペプチドに融合させる。あるいは、少なくとも1つの膜貫通ドメインを有する前記免疫アクセサリー分子が、エキソソーム産生細胞中への過剰発現により免疫原性小胞に取込まれる。場合によっては、前記免疫アクセサリー分子は、抗原提示細胞へ特異的抗原を送達するためのリガンドである。
【0029】
場合によっては、前記マーカーは、タグ、酵素、ビオチン、蛍光分子などの検出可能な分子である。場合によっては、前記マーカーをエキソソームターゲティングポリペプチドに融合または架橋させる。あるいは、前記マーカーは、エキソソーム産生細胞中に過剰発現させることによって免疫原性小胞に取込まれる膜貫通ドメインを有する。さらに、前記マーカーは小胞、好ましくはエキソソームに優先して取込まれるラベルされた脂質である。好ましくは、前記ラベルされた脂質は、ローダミン-DOPEまたはフルオレセイン-DOPEなどの蛍光団結合脂質である。
【0030】
場合によっては、小胞、好ましくはエキソソームを調製する工程は次の工程を含む:
a)前記抗原をコードする遺伝子コンストラクトを提供すること;
b)場合によっては、前記マーカーをコードするコンストラクトを提供すること;
c)前記コンストラクトをエキソソーム産生細胞に導入して、組換えエキソソームを生成すること;および、
d)前記組換えエキソソームを収集すること。ここで、前記エキソソームは、その表面に前記遺伝子コンストラクトによりコードされる抗原および場合によっては前記マーカーを所持する。
【0031】
場合によっては、互いに異なる抗原をコードするいくつかの互いに異なる遺伝子コンストラクトを、前記エキソソーム産生細胞に導入する。好ましくは、前記エキソソーム産生細胞は哺乳動物細胞である。より好ましくは、前記哺乳動物エキソソーム産生細胞はマウスの細胞である。
【0032】
あるいは小胞、好ましくはエキソソームの調製工程は次の工程を含む:
a)エキソソームターゲティングポリペプチドに融合された前記抗原を含む分子を提供すること;
b)場合によっては、エキソソームターゲティングポリペプチドに融合された前記マーカーを含む分子を提供すること;および
c)前記抗原を含む前記分子、および場合によっては前記マーカーを含む前記分子を、フォスファチジルセリンまたはエキソソームに自然に含まれる他の脂質を含む脂質小胞と接触させて、表面に前記抗原および場合によっては前記マーカーを提示している機能化された脂質小胞を生成すること。
【0033】
本発明はさらに、単離された抗体産生粒子、好ましくは細胞、およびそれらの抗体産生への使用、および前記抗体産生粒子、好ましくは細胞によって産生された抗体に関する。本発明はまた、前記単離された抗体産生細胞または前記抗体産生細胞によって産生された前記抗体および薬学的に許容できる賦形剤または坦体を含む組成物に関する。
【0034】
本発明はさらに、エキソソームの表面に少なくとも1つの膜貫通ドメインを有するポリペプチドを発現させる方法であって、:
1)前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを含むその一部をコードする遺伝子コンストラクトを提供すること;
2)エキソソーム産生細胞に前記コンストラクトを導入して、前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを含むその一部をエキソソーム産生細胞中に過剰発現させ、組換えエキソソームを生成すること;および、
3)前記組換えエキソソームを収集すること(ここで前記エキソソームは、その表面に前記遺伝子コンストラクトによりコードされるポリペプチドを所持している);
方法に関する。
【0035】
この方法の好ましい実施態様において、少なくとも1つの膜貫通ドメインを有する前記ポリペプチドは、レセプターである。より好ましくは、前記レセプターはSSTR2、CCR7、CXCR4、およびCCR5などのGPCR(Gタンパク質共役レセプター)である。あるいは、前記ポリペプチドはCD40L、好ましくは 突然変異CD40Lであり、前記突然変異は可溶性CD40Lの切断と放出を阻害する。
【0036】
本発明は、上記の方法によって調製された機能化されたエキソソーム、前記機能化されたエキソソームを含む組成物およびそれの使用に関する。より詳細には、本発明は、前記機能化されたエキソソームおよび薬学的に許容できる賦形剤または坦体を含む組成物に関する。本発明はまた、抗体を産生し、または対象に抗原を送達するための前記機能化されたエキソソームの使用に関する。
【0037】
したがって、本発明は、少なくとも1つの膜貫通ドメインを有するポリペプチドまたはそのエピトープを結合する抗体を産生する方法であって:
a)前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを含むその一部をコードする遺伝子コンストラクトを提供すること;
b)エキソソーム産生細胞中に、前記コンストラクトを導入し、前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを含むその一部をエキソソーム産生細胞中に過剰発現させ、前記ポリペプチドまたはそのエピトープを表面に提示している組換えエキソソームを生成すること:
c)前記組換えエキソソームを収集し、前記エキソソームまたはその一部を、ヒト以外の哺乳動物に注入して、前記ポリペプチドまたはそのエピトープを結合する抗体を生成すること;および、
d)抗体または抗体産生細胞を前記哺乳動物から収集すること;
を含む方法に関する。
【0038】
この方法の好ましい実施態様においては、少なくとも1つの膜貫通ドメインを有する前記ポリペプチドはレセプターである。より好ましくは、前記レセプターはSSTR2、CCR7、CXCR4、およびCCR5などのGPCR(Gタンパク質共役レセプター)である。好ましくは、前記エキソソームはさらに、突然変異CD40Lを表示し、前記突然変異は可溶性CD40Lの切断と放出を阻害する。次に抗体を、ポリクローナル抗体調製のための動物の血清から直接単離することができる。モノクローナル抗体もまた、ハイブリドーマの生成またはSLAMなどの方法による単一の抗体産生細胞の単離を含む、古くからある手法を用いて、これらの動物から調製することができる。
【0039】
本発明はまた、少なくとも1つの膜貫通ドメインを有する抗原または少なくとも1つの膜貫通ドメインを含むその一部を、対象に送達する方法であって:
a)前記抗原または少なくとも1つの膜貫通ドメインを有するその一部をコードする遺伝子コンストラクトを提供すること;
b)前記コンストラクトをエキソソーム産生細胞中に導入し、前記抗原または少なくとも1つの膜貫通ドメインを有するその一部をエキソソーム産生細胞中に過剰発現させて、前記抗原または前記その一部を表面に提示している組換えエキソソームを生成すること;
c)前記組換えエキソソームを収集し、前記エキソソームまたはその一部を、前記対象に注入すること;
を含む方法に関する。この方法の好ましい実施態様において、前記抗原はレセプターである。より好ましくは、前記レセプターはSSTR2、CCR7、CXCR4、およびCCR5などのGPCR(Gタンパク質共役レセプター)である。好ましくは、前記エキソソームはさらに、突然変異したCD40Lを表示し、前記突然変異は可溶性CD40Lの切断と放出を阻害する。
【0040】
本発明は、少なくとも1つの膜貫通ドメインを有する特異的な抗原または少なくとも1つの膜貫通ドメインを含むその一部に対して、対象中に免疫応答を作り出す方法であって:
a)前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを含むその一部をコードする遺伝子コンストラクトを提供すること;
b)前記コンストラクトをエキソソーム産生細胞中に導入し、前記抗原または少なくとも1つの膜貫通ドメインを含むその一部をエキソソーム産生細胞中に過剰発現させて、前記抗原または前記その一部を表面に提示している組換えエキソソームを生成すること;
c)前記組換えエキソソームを収集し、前記エキソソームまたはその一部を、前記対象に注入すること;
を含む方法に関する。この方法の好ましい実施態様において、前記抗原はレセプターである。より好ましくは、前記レセプターはSSTR2、CCR7、CXCR4、およびCCR5などのGPCR(Gタンパク質共役レセプター)である。好ましくは、前記エキソソームはさらに、突然変異CD40Lを提示し、前記突然変異は可溶性CD40Lの切断と放出を阻害する。
【0041】
本発明はさらに、生合成的にエキソソームを蛍光団結合脂質でラベルする方法、およびクローニング部位に突然変異を導入することなく挿入をプラスミドにクローニングするための方法を考察する。
【0042】
発明の詳細な説明
本発明は、抗体を生成し抗体レパートリーをスクリーニングする新しい方法を開示する。本発明は、より詳細には標的抗原、共刺激分子および/またはマーカーを表示している自然または合成した膜小胞、好ましくはエキソソームを用いる。本発明を、実験、研究、治療、予防または診断の分野において用いることができる。
【0043】
本発明は、エキソソームターゲティングドメインを含む組換えキメラタンパク質および組換え膜レセプターの、新しい予想外の性質の発見に由来する。より詳細には、エキソソーム指向抗原が、高度に免疫原性であり、強力な液性免疫応答を生み出すことを本発明が示す。さらに、抗原とマーカーを表示するエキソソームを用いることにより、応答細胞の出現度数が低い場合でさえも、抗原特異的な抗体産生細胞を都合良く単離することができることも示す。
【0044】
本発明は、従来の抗体調製方法に比べて、多くの利点を有する。本発明は、免疫原性が弱い抗原、膜タンパク質、または多成分ポリペプチド複合体を取り扱う場合に最も有利である。従来の方法を用いた抗体調製では失敗したか、または有用な抗体を産生しない難しい標的を取り扱う場合に、本発明が特に適切である。また、抗体調製を、大量の抗原を精製する必要なく達成できる。実際、エキソソームを単離するための単一の小規模精製方法(US patent 09/780,748号)を、エキソソームの表面に発現されている外来性抗原の性質に関わらず、用いることができる。このように、抗原調製段階を非常に急速に、即ち通常12時間以内に完了できる。この方法は、速く、多数の試料について平行して実施できる。したがって免疫化のための複数の抗原の同時調製が可能となる。抗原を自然に存在する小胞中に発現させることが、穏和な精製方法と組み合わせて、抗原の自然な立体構造を保つ助けになり、これが有力な医療適用ができる重要な抗体の生成を可能にする。さらに、本発明は、表面に高密度の抗原を含む脂質小胞を生成する。この高密度は、抗原結合活性を増加させることにより、抗体産生を非常に有利にするポリマー状態と比較できる。本発明のさらなる利点は、ポリペプチドがエキソソーム産生細胞によって発現されるため、プロセッシングおよび翻訳後修飾(グリコシル化など)の、自然の経路にゆだねられ得ることである。もう1つの利点は、抗体産生細胞を、応答細胞の生成頻度が低い場合でさえも、単離できることである。このような低い生成頻度は、一般に免疫原性に乏しい抗原または少量の抗原による免疫化の結果起こる。抗原を保持するエキソソームを用いてB細胞を選別することにより、極めて希な抗原特異的抗体産生細胞の単離が可能になる。抗原を保持するエキソソームを用いるB細胞選別の追加の利点は、固有のエピトープに対する抗体や立体構造的エピトープに対する抗体を産生しているB細胞を選択的に単離するために、選別の厳密性と特異性を調整できることである。例えば、あるレセプターを保持する過剰のエキソソームおよび生理的なリガンドに結合している同じレセプターを保持する少量で追跡可能なエキソソームの存在下で実施される減算的選別によって、レセプター/リガンド複合体のみと反応する抗体を産生するB細胞の単離が可能になる。したがって本発明は、他の化合物との相互作用または突然変異/欠失による標的抗原の微妙な立体構造変化を識別できる、高性能の機能的抗体の生成を可能にする。後者の特徴は、他の抗体調製方法に比較して相当な利点である。
【0045】
高度に免疫原性の複合物による免疫化が、独特で希な抗体産生細胞の高度に選択的な選別と組み合わされて、所定のエピトープに対する抗体を生成する効率的で新しい方法を提供する。
【0046】
抗体生成法
この方法では、少なくとも1つの標的抗原を発現している免疫原性小胞、好ましくはエキソソーム、を動物に導入したときに、抗体が生成される。抗体産生は、免疫化された動物の血清を標準的な方法によってテストすることにより評価できる。抗体の調製は、ポリクローナル抗体に対しては血清から抗体をアフィニティー精製することにより、またモノクローナル抗体に対しては抗原特異的な抗体産生細胞を単離することにより、達成できる。後者は好ましくは、下記の抗体レパートリーをスクリーニングするための方法を用いて実施できる。あるいは、モノクローナル抗体を、ハイブリドーマ・スクリーニングおよびSLAMなどの、様々な既知の免疫された動物からのモノクローナル抗体の調製方法を用いて、調製することができる。
【0047】
抗原を表示している免疫原性小胞の調製
抗原を提示している免疫原性小胞は、一般に抗原配列を、エキソソームターゲティングドメイン、好ましくはラクトアドヘリンのC1C2ドメイン、と融合させることにより調製する。免疫原性小胞は、エキソソームやリポソームなどの、自然のまたは合成された小胞である。エキソソームターゲティングドメインを含むキメラタンパク質を保持する小胞の調製方法およびその使用が、WO03/016522に記載されている。
【0048】
ラクトアドヘリンは、殆ど必ずエキソソームに結合して見出される。ラクトアドヘリンのC1/C2ドメインは、エキソソーム表面に対する高度に特異的なターゲティングモチーフを含む。したがって、ラクトアドヘリンのC1および/またはC2ドメインの一部または全体またはその機能的同等物をタンパク質へ導入することにより、その結果得られるキメラタンパク質が、エキソソームや他の脂質構造物を標的とすることが可能になる。
【0049】
したがって、前記エキソソームターゲティングポリペプチドは、ラクトアドヘリンまたは機能的C1および/またはC2ドメインを含むその一部を含む。本発明の実施において、様々な供給源または起源から得られるラクトアドヘリンを用いることが可能である。より好ましくは、前記ラクトアドヘリンまたはその一部は、ヒト以外の哺乳動物ラクトアドヘリンまたはその一部である。哺乳動物ラクトアドヘリンは例えば、マウス、ラット、ウシ、ブタ、ウマのラクトアドヘリンである。より詳細には、前記ヒト以外の哺乳動物ラクトアドヘリン又はその一部は:(i)マウスラクトアドヘリン、(ii)機能的C1および/またはC2ドメインを含むマウスラクトアドヘリンの断片、および(iii)(i)または(ii)のポリペプチドと少なくとも50%の一次構造同一性を有するポリペプチド、から選ばれる。より好ましくは、前記タンパク質が、機能的C1/C2ドメインと融合されている。
【0050】
上記のように、ターゲティング部分は、上の(i)または(ii)のポリペプチドと少なくとも50%の一次構造同一性を有するポリペプチドでよい。同一性は、コンピュータプログラム、好ましくはCLUSTAL法などの様々な既知の技法により決定される。より好ましくは、標的ターゲティングポリペプチドは(i)または(ii)のポリペプチドと少なくとも60%の同一性を、有利には少なくとも70%の同一性を有する。そのようなラクトアドヘリン変異体(または機能的同等物)は、ポリペプチドをエキソソームを標的とする能力を保持しているいるに違いない。この性質は、実施例に記載されているように、例えば、マーカーポリペプチドに融合された前記変異体を含むキメラ遺伝子を作り、この遺伝子をエキソソーム産生細胞中に発現させ、エキソソーム表面のマーカーポリペプチドの存在を確認することにより立証できる。好ましいラクトアドヘリン変異体は、上の(i)または(ii)のポリペプチドと少なくとも85%の同一性を有する。アミノ酸欠失、置換、突然変異および/または付加を含む変異体も可能である。
【0051】
マウスラクトアドヘリンのアミノ酸配列が記載されている(配列番号1;(12)とGenbank Accession n°M38337も参照のこと)。場合によっては、前記ラクトアドヘリンが、配列番号1のアミノ酸配列、または機能的C1および/またはC2ドメインを含むその断片を有する。好ましくは、前記ラクトアドヘリンは、配列番号1の機能的C1/C2ドメインを含むアミノ酸配列を有する。より好ましくは、前記ラクトアドヘリンは、配列番号1のアミノ酸残基111〜266、109〜266、271〜426、111〜426または109〜426を含むアミノ酸配列を有する。
【0052】
場合によっては、前記エキソソームターゲティングポリペプチドは、Del-1、ニューロピリン-1、凝固因子5、または凝集素8の機能的C1および/またはC2ドメインを含む。場合によっては、前記抗原は、標的ターゲティングポリペプチドの上流、下流、または任意の内部ドメイン結合部に融合される。
【0053】
別のある実施態様において、エキソソーム産生細胞および/またはエキソソームターゲティングポリペプチド(好ましくはラクトアドヘリン、または機能的C1/C2ドメインを含むその一部もしくは変異体)が、免疫化に用いた哺乳動物と同じ種に由来する。実際、そのような系においては、エキソソームとターゲティングポリペプチドは免疫原性でなく、抗体はほとんど選択した抗原に対してのみ産生される。
【0054】
ある実施態様においては、エキソソーム産生細胞がマウスの細胞であり、ラクトアドヘリンがマウスのラクトアドヘリンあるいは機能的C1および/もしくはC2ドメインを含むその一部または変異体であり、ヒト以外の哺乳動物がマウスであり、抗原またはエピトープは、例えばヒト起源などの異なる種由来である。マウスがヒト化マウスであって、ヒト化抗体を産生できることがさらにより好ましい。
【0055】
その趣旨で、タンパク質(抗原またはエピトープ)のヌクレオチド配列をマウスのラクトアドヘリンのC1および/またはC2ドメインと融合させることができ、その結果物のキメラ配列を、標準分子生物学技術を用いて真核生物発現ベクター中にクローニングする。キメラタンパク質をコードするプラスミドを、エキソソーム産生マウス細胞系統へトランスフェクションし、トランスフェクションされた細胞を数日培養した後、組換えエキソソームを採取する。組換えエキソソームを、次に蔗糖密度勾配により精製する(US pattent 09/780,748)。組換えエキソソーム上のキメラタンパク質の存在を、ウエスタンブロット分析またはELISAにより確認する。キメラタンパク質を保持する組換えエキソソームを、次に同系のマウスに注入して抗体を生成する。この状況において、キメラタンパク質生成に用いたタンパク質配列中に含まれる抗原決定基のみが、免疫化されるマウス中の外来抗原である。
【0056】
代りのエキソソームターゲティングドメインを、次の工程を含む方法によりスクリーニング、同定、または選択することができる:
− 候補ポリペプチド、好ましくは候補の膜貫通ポリペプチドをコードする第1の遺伝子コンストラクトを提供すること;
− 第1の遺伝子コンストラクトをエキソソーム産生細胞中に導入し、候補ポリペプチドのエキソソームにおける発現をテストすること;
− エキソソームで発現された候補ポリペプチドを選択し、標的ポリペプチドに融合した前記選択されたポリペプチドをコードする第2の遺伝子コンストラクトを調製すること;
− 第2の遺伝子コンストラクトをエキソソーム産生細胞中に導入し、融合ポリペプチドのエキソソームにおける発現をテストすること;および、
− 標的ポリペプチドをエキソソーム中に効率的に発現させるポリペプチドを選択すること。
【0057】
われわれの結果は、エキソソーム上での発現を導く特異的なターゲティング信号を含む種々のタンパク質またはポリペプチドを、上記方法を用いて、同定、選択および/または改良できることを示している。これらのポリペプチドには、エキソソーム中に発現される能力および他の分子をそのような小胞を標的とする能力の双方が必要である。これらのポリペプチドは膜貫通タンパク質に由来し、そのようなタンパク質のすべてまたは一部、一般には少なくとも膜貫通ドメインを含む部分を含めばよい。これらのコンストラクトが、エキソソームへの抗原、特にレセプターおよび膜貫通タンパク質の送達に特に適している。候補の標的ターゲティングポリペプチドは、実際上、レセプター、チャネルなどのような膜貫通ドメインを有するいかなるタンパク質に由来するものでもよい。そのような標的ターゲティングポリペプチドの具体的な例には、MelanA/MARTl、CD40L、CD81などまたはそれらの一部が挙げられる。標的ターゲティングポリペプチドは、膜貫通タンパク質の全体を含んでも、または少なくとも1つの膜貫通ドメインを含むその一部のみを含んでもよい。
【0058】
前記抗原は、例えば、レセプターもしくは酵素のような任意のタンパク質であっても、または、糖脂質、多糖類、薬物、および有機化学物質などのポリペプチド以外の化合物であってもよい。第1の実施態様において、前記抗原は、特性が決定されていないタンパク質である。前記抗原に対する抗体が、前記タンパク質の機能を同定し特性を決定するために必要である。そのような研究法が、ポストゲノムの分野で有用である。実際、全ゲノム配列が知られているが、コードされたタンパク質の機能を見出すことが、今や必須である。さらに、前記抗原はオーファンレセプターであってもよい。第2の実施態様において、前記抗原は稀なものであってもよい。したがって、前記抗原に対する抗体の調製が極めて効率的でなければならない。本発明の方法は、そのような応用に特に適合している。
【0059】
抗原の別の例には、例えば、腫瘍抗原、ウイルス抗原、および微生物抗原がある。腫瘍抗原の実例には、MAGE、BAGE、前立腺腫瘍抗原、および癌遺伝子などがある。これらの抗原のアミノ酸配列それ自体が知られており、組換え技術または合成により生成できる。本発明で標的にされ、または提示される特定の抗原には、可溶性抗原およびレセプターの細胞外ドメインが挙げられる。さらに抗原の例としてリンフォカイン(IL-2、IL-4、IL-13)、栄養因子(TNF、IFN、GM-CSF、G-CSF、など)、酵素、凝固因子、ホルモン、リポタンパク質などがある。
【0060】
特に興味深いタイプの抗原は、少なくとも1つの膜貫通ドメインを有するレセプター、より好ましくはGPCRまたはその一部である。実際、本発明はここで、エキソソームターゲティングポリペプチドを有するかまたは有しない膜貫通ポリペプチドを表示しているエキソソームの調製を可能にしている。小胞内におけるGPCRの発現が、GPCRの精製、特性評価、リガンド(合成の、または自然の)のスクリーニング、抗体の生成などを可能にする。GPCRの具体的な例としては、例えば、SSTR2、CCR7、CXCR4およびCCR5がある。しかし、本発明は他のレセプターにも同様に用いることができる。
【0061】
別の実施態様において、少なくとも1つの膜貫通ドメインを含む抗原を表示している自然の免疫原性小胞を、エキソソーム産生細胞に前記膜貫通タンパク質をトランスフェクションし過剰発現させることにより、調製することができる。実際、われわれは、組換え膜貫通タンパク質がエキソソーム産生細胞により過剰発現される場合には、組換え膜貫通タンパク質がエキソソーム区画に移入されて発現されることを発見した。この予想外の現象が、自然にはエキソソーム上には存在せず、特殊な細胞系統に限られているように見える膜貫通タンパク質について証明された(実施例2に記載の通り)。これは、可溶性タンパク質またはレセプターの細胞外ドメインのエキソソーム上への発現には、タンパク質がエキソソームターゲティングドメインへ融合することが必要であることを証明した以前の結果と対照的である。また、エキソソームを標的とする信号が無い場合には、組換え膜貫通タンパク質は、主に細胞表面に見出され、発現されたタンパク質の一部のみがエキソソーム上に見出される。エキソソーム上に見出される量は、細胞表面に見出されるタンパク質量に正比例しているように見える。
【0062】
さらに別の実施態様においては、組換えエキソソーム上に発現されるタンパク質のエピトーププロフィールが、エキソソームを可溶性化合物と反応させることにより修飾される可能性がある。これは例えば、特異的なペプチドを負荷されたMHC分子またはレセプター−リガンド複合体などの多量体を生み出すことができる。ここでの目標は、MHC/ペプチド複合体には反応するが負荷されていないMHC分子には反応しない制限抗体(restricted antibody)のような抗体を生成することである。MHC分子にペプチドを負荷する方法(直接負荷)が、最近WO01/82958に記載された。制限抗体は、癌細胞および感染細胞のマーカーと反応するので、探し求められている診断用および治療用薬剤である。古典的なMHC分子に加えて、制限抗体を、CD1および古典的でないMHC分子などの他の多形体を含む複合体に対して生成することができる。しかし制限抗体を古くから知られた抗体調製方法によって獲得することは非常に困難である。制限抗体の応用の一例が、HIV感染細胞を殺すことのできる複合物の生成である。より具体的には、これは、HLA-C/HIVペプチド複合体と反応する制限抗体を用いることにより達成できる。実際、HIVが、ウイルスによる養子免疫回避のための多くの手段の1つとして、HLA-AおよびBをダウンレギュレーションすることが示されている。しかし、もしHLA-Cを、HLA-AおよびBサブタイプと共にダウンレギュレーションすると、NK細胞本来の応答をトリガーするので、HLA-Cを保持することもまた、ウイルスの生存には決定的に重要である。したがって、大部分のHIV感染細胞が、HLA-C/HIVペプチド複合体を発現している見込みがあるので、感染細胞上のこれらの標識に対する制限抗体が、HIV患者のHIVの蓄積を除去する強力な手段となる可能性がある。したがって本発明はまた、HIV感染の治療および/または防止のための医薬品組成物を調製するための、HLA-C/HIVに対する制限抗体の使用に関する。本発明はさらに、前記抗体を含む医薬品組成物、および対象にHLA-C/HIVに対する効果的な量の制限抗体を投与することを含む、前記対象のHIV感染の治療および/または予防方法に関する。
【0063】
別の実施例は、レセプターまたはリガンドの活性型には反応し(レセプター/リガンド相互作用による立体構造変化によってトリガーされる)、不活性型のあるいは空のレセプターまたはリガンドには反応しない、また突然変異型抗原に反応して野生型抗原には反応しない抗体を生成するものである。ここでの典型的な実施例は、HIVを無効化する抗体を調製することである。実際、HIV gp120上には、この抗原が標的細胞上のレセプターに結合しているときにのみ、無効化エピトープが見出されている。したがって、gpl20、CXCR4およびCD4、またはgpl20、CCR5およびCD4を含むレセプター/リガンド複合体の生成が、レセプターが仲介するHIVの細胞への結合を阻害する無効化抗体の生成のために必要である。これに関連して本発明は、自然な機能的立体構造を保持している複合体から成る適切な免疫原を調製する効果的な手段、および複合体中に存在する特異的立体構造エピトープに対しては抗体を生成するが、そこに含まれる個々の構成要素に対しては抗体を生成しない効果的な手段を提供する。
【0064】
稀なエピトープに反応する抗体の生成と単離が、さらに下記のような対側性免疫化(contralateral immunization)とB細胞の減算的選別(subtractive sorting)の実行により可能となる。
【0065】
さらに別の実施態様において、抗原を表示している免疫原性小胞はまた、抗原提示細胞へ特異的に抗原を送達するために、アジュバントおよび/またはリガンドを表示することもできる。これはさらに小胞の免疫原性を高める手段となる可能性があり、これが液性応答の強度を増加し、それにより、抗原特異的な抗体産生細胞の度数を増加させる結果となる可能性がある。これに加えて、これが検出可能な液性応答を誘導するのに必要な抗原の量をより低下させもする可能性がある。免疫原性が増加した小胞を、GM-CSF/C1C2などの、エキソソームターゲティングドメインに融合されたアジュバントをコードするプラスミドでエキソソーム産生細胞をトランスフェクションすることにより、調製することができる。エキソソームに連結された最適のアジュバント活性を有するように選択された安定した細胞系統を、薬物選択を用いた標準的な方法により確立することができ、また標的抗原の受容細胞として用いることができる。増加した免疫原性を有し、エキソソームターゲティングドメインを含むキメラのアジュバントを保持する小胞の調製方法とその使用が、WO03/016522に記載されている。
【0066】
あるいは、膜貫通ドメインを有するアジュバントが、上記のように、エキソソーム産生細胞内の過剰発現に続いて、エキソソーム上に表示されてもよい。膜貫通ドメインを含むアジュバントのクラスには、例えば、CD40リガンド(CD40L)発現細胞の表面に膜貫通型に、または循環する可溶型に生成される、強力な免疫刺激剤であるCD40Lが含まれる。われわれは、細胞系統をCD40Lをコードするプラスミドによりトランスフェクションすると、このアジュバントがトランスフェクションされた細胞により生成されるエキソソーム上に発現することを見出した。そのような組換えエキソソームが、実施例4に記載されているように、増大した免疫能力を示した。われわれはまた、このアジュバントの可溶性型を生み出すCD40Lの切断部位を突然変異させることにより、エキソソーム表面から可溶性CD40Lが切断されて放出されることを阻害することができることを見出した。これが、CD40Lの膜貫通型のみを発現しているエキソソームをもたらし、これは野生型のCD40Lを発現しているエキソソームよりもさらに強力な免疫活性を示す。この予想外の現象に対しては、野生型は、CD40Lの完全長型と可溶性型ならびにCD40Lの残りの膜貫通ドメインの混合物を含んでいて、これがCD40Lの機能的な3量体型の効果的な形成を妨げるためである、という説明が可能である。本発明は、切断モチーフが突然変異したCD40Lを含むエキソソームに関する。
【0067】
したがって、本発明は、少なくとも1つの膜貫通ドメインを有するポリペプチドをエキソソームの表面に発現させる新しい方法であって、
1)前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを含むその一部をコードする遺伝子コンストラクトを提供すること;
2)エキソソーム産生細胞中に前記コンストラクトを導入し、前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを含むその一部を過剰発現させて、組換えエキソソームを生成すること;および、
3)その表面に前記遺伝子コンストラクトによりコードされるポリペプチドを保持する前記組換えエキソソームを収集すること;
を含む方法に関する。
【0068】
この方法の好ましい実施態様において、少なくとも1つの膜貫通ドメインを有する前記ポリペプチドは、レセプターである。より好ましくは前記レセプターはSSTR2、CCR7、CXCR4およびCCR5などのGPCR(Gタンパク質共役レセプター)である。あるいは前記ポリペプチドはCD40L、好ましくは突然変異CD40Lであって、前記突然変異が可溶性CD40Lの切断と放出を阻害する。
【0069】
特異的な抗原送達のためのリガンドの例が免疫グロブリンまたはその断片、例えば免疫グロブリンのFc断片である。そのようなFc断片は、エキソソームの表面に発現すると、そのようなFc断片のレセプターを発現している細胞、例えば抗原提示細胞などを、エキソソームが標的とするように作用することができる。そのようなFc断片の発現は、単独であっても、または抗原の発現との組合せであっても、抗原提示細胞、特に樹状細胞によるエキソソームの認識を促進し増強して、そのような抗原のクロスプライミングを増大させる。
【0070】
融合
キメラのポリペプチドまたは複合物を、遺伝的または化学的融合により調製することができる。
【0071】
遺伝子融合のためには、キメラ遺伝子の関心のあるポリペプチドをコードするドメインを、ラクトアドヘリンまたはターゲティングペプチドの上流、下流または任意の内部ドメイン接合部に融合さればよい。さらに、ドメイン同士が互いに直接融合しても、またはキメラポリペプチドの性質を変えないスペーサー領域で隔てられてもよい。そのようなスペーサー領域には、クローニング部位、切断部位、フレキシブルドメインなどが含まれる。その上、キメラの遺伝子コンストラクトがさらにリーダー シグナル配列を含み、コードされたキメラポリペプチドをエキソソーム産生細胞の小胞体へと分泌しやすくしてもよい。その上、キメラ遺伝子が、mycタグ、ポリヒスチジンタグなどの、精製またはモニターを容易にするタグをさらに含んでもよい。
【0072】
化学的融合のために、部分的または全長のラクトアドヘリン配列を選択または修飾して、末端にチオール、アミノ、カルボキシル基などのフリーの反応基を付与し、可溶性のポリペプチド、糖脂質、または任意の小分子を架橋することができる。好ましい実施態様においてラクトアドヘリンのコンストラクトが、少なくとも配列番号1のアミノ酸1〜271をコードする。この配列においてC1ドメイン(アミノ酸111〜266)が、エキソソームターゲティングモチーフを提供し、システイン271が、他の分子と化学的に架橋するための、フリーのチオール残基を提供する。ペプチド、化学物質のSH基との架橋は、十分に確立された方法(13)により実施できる。この方法の利点は、それが、本発明の有効範囲を、糖脂質、薬物および有機化学物質などのポリペプチド以外の化合物に対する抗体の調製へ拡張することである。それは、新しい抗原決定基となる可能性のある基を導入することなく、ポリペプチドおよび化合物をエキソソームにターゲティングさせる手段もまた提供する。選択した架橋試薬は、免疫学的に非活性であることが示されている(13)。新しい抗原決定基がときにはキメラ遺伝子の結合部位で生じ、キメラ遺伝子産物を特異的な予防的および治療的なヒトへの応用に用いることを制限する可能性がある。配列番号1などの適切なラクトアドヘリンコンストラクトを提示しているエキソソーム(またはリポソーム)を産生し、次にそれを連結したい産物と反応させることにより、修飾されたエキソソームまたは脂質小胞(例えばリポソーム)を調製することができる。あるいは、産物に架橋したラクトアドヘリン断片を調製し、続いて精製したエキソソームまたはリポソームに加えてもよい。
【0073】
ベクター
本発明は上記のようなキメラの遺伝子コンストラクトまたはベクターを含む組換え細胞はもちろん、さらに、上記のようなキメラの遺伝子コンストラクトを含むベクターも含む。ベクターは、プラスミド、ファージ、ウイルス、人工染色体などである。代表的な例には、商業的に購入できるプラスミド、特にpUC、pcDNA、pBRなどに由来するプラスミドが挙げられる。他の好ましいベクターが、複製欠損レトロウイルス、アデノウイルス、AAV、バキュロウイルスまたはワクシニアウイルスなどのウイルスに由来する。ベクターの選択は、前記ベクターを使用するべき組換え宿主細胞に従って当業者が決定できる。これに関しては、哺乳動物細胞にトランスフェクションまたは感染させることができるベクターを用いるのが好ましい。実際、好ましい組換え宿主細胞は、哺乳動物細胞である。これは初代細胞、または確立した細胞系統でよい。代表例には、線維芽細胞、筋細胞、肝細胞、免疫細胞など、およびそれらの祖先または前駆細胞が含まれる。最も好ましい哺乳動物細胞が、エキソソーム産生哺乳動物細胞である。これらには、例えば腫瘍細胞、樹状細胞、BおよびTリンパ球または肥満細胞が含まれる。
【0074】
エキソソーム産生細胞
エキソソーム産生細胞には任意の、好ましくは哺乳動物起源の、後期エンドソーム多胞体が原形質膜と融合して生ずるエンドソーム起源の膜小胞を生成し分泌する細胞が含まれる(6)。様々なタイプの組織から得られた細胞、例えば樹状細胞、Bリンパ球、腫瘍細胞、Tリンパ球および肥満細胞などが、エキソソームを分泌することが示されている。治療目的で、または研究用具としてエキソソームを生成し、精製しまたは使用する方法が、例えばWO99/03499、WO00/44389、WO97/05900中に記載されており、参照によりここに取り入れる。本発明の好ましいエキソソーム産生細胞は、哺乳動物腫瘍細胞、哺乳動物BおよびTリンパ球、および哺乳動物樹状細胞であり、一般にはマウスまたはヒト起源のものである。これに関して、細胞は、好ましくは不死化した樹状細胞(WO94/28113)、未成熟樹状細、または腫瘍細胞(WO99/03499)である。さらに、抗体産生のためには、Bリンパ球をエキソソーム産生細胞として用いるのが有利である。何故ならば、その結果得られるエキソソームが、抗体産生を促進するMHCクラスII分子などの、補助的な機能および分子を含むからである。さらに、B細胞由来のエキソソームが、濾胞樹状細胞と結合できることが示された。これは抗体誘導のための、もう1つの重要な特徴である(14)。
【0075】
細胞は、RPMI、DMEM、AIM Vなどの任意の適当な培地中で、好ましくは、培地由来のタンパク質によるエキソソームの汚染を避けるために、タンパク質を含まない培地中で、培養し維持できる。培養は、プレート、シャーレ、チューブ、フラスコなどの任意の適当な装置で実施できる。
【0076】
遺伝子コンストラクト(またはベクター)を、エキソソーム産生細胞に、裸のDNA技術、陽イオン性脂質媒介トランスフェクション、ポリマー媒介トランスフェクション、ペプチド媒介トランスフェクション、ウイルス媒介トランスフェクション、物理的もしくは化学的な試薬または処理、エレクトロポレーションなどの、通常の方法のいずれかを用いて導入することができる。これに関しては、適切なキメラ遺伝子を発現させるには一時的なトランスフェクションで十分であり、したがって安定した細胞系統を作製することが不必要であることに注意するべきである。そのような細胞によって産生されるエキソソームを、遠心、クロマトグラフィーなどの従来知られた方法に従って、収集および/または精製することができる。好ましい技術が、WO00/44389、およびUS patent 09/780,748中に記載されており、参照によりここに取り入れる。
【0077】
接種材料および注入経路
抗原を、好ましくは上記のように調製した組換えエキソソームの形で、動物へ接種すると、抗体が生成される。あるいは、抗原をDNAの形で、または組換えタンパク質の形で投与してもよい。さらに別の可能性は、組換えエキソソーム産生細胞全体を投与することである。
【0078】
抗体は、ポリクローナルでも、モノクローナルでもよい。動物は、マウス、齧歯類、霊長類、ウマ、ブタ、ウサギ、家禽、などを含む様々な種でよい。好ましい動物はマウスである。
【0079】
遺伝子免疫化を、ワクシニア、ポックスウイルス、アデノウイルス、アデノ関連ウイルスなどの様々なウイルスベクター、または種々の脂質性またはタンパク質性組成物を組み合わせたDNAなどの非ウイルスベクターを用いて、または純粋な(例えば裸の)DNAを用いて、実施できる。遺伝子銃またはエレクトロポレーションを含む様々なベクター送達装置または技術が、遺伝子接種のために使用できる。上に指摘したように、遺伝子コンストラクトは、任意のDNAまたはRNA分子、一般にはプラスミド、ウイルスベクター、ウイルス粒子、裸のDNAまたは前記細胞を含む任意の細胞でよい。様々な遺伝子コンストラクトが、単一のベクター中もしくは別々のベクター中に、または任意の組み合わせ中に、含まれることが可能である。
【0080】
タンパク質免疫化もまた同様に用いることができる。この点で、組換えキメラ抗原を精製した形で動物への投与に使用してもよい。
【0081】
そのような投与に続いて、エキソソームターゲティングドメイン、好ましくはラクトアドヘリンのC1および/またはC2ドメインを有するキメラ抗原が生体内で動物自身の循環しているエキソソームに負荷され、それによって免疫応答を誘導することになる。
【0082】
接種組成物は一般に希釈液、緩衝液、等張液などの薬学的に許容できる賦形剤または媒体をさらに含む。組成物はまた上記のような遺伝子免疫化のために、トランスフェクションを容易にする試薬を含んでもよい。組成物はさらにアジュバントを含んでもよい。
【0083】
接種材の投与は例えば静脈内、筋肉内、腹膜内、腫瘍内、皮下、脾臓内、リンパ節内などの全身性注入などの様々な経路により実施することができる。
【0084】
所定のエピトープに反応する抗体を産生する細胞の局所的な度数をさらに増加させるために、対側性免疫化を実施してもよい。この手法は、例えば、MHC/ペプチド複合体には見出されるが、空のMHC分子には見出されない、またはレセプター/リガンド複合体には見出されるがレセプターのみには見出されない特異的なエピトープに対する抗体の調製および単離には、極めて有利である。この手法の別の例は、活性型の化合物には見出されるが不活性型のその化合物には見出されない、または突然変異型の抗原には見出されるが正常型には見出されないエピトープから成る。対側性免疫化は最初、細胞抽出物などのタンパク質混合物中に見出される抗原に向けられた抗体産生細胞を単離する手段として記載された(15)。それは、例えば動物の一方の体側に細胞抽出物のみを注入し、次に後日細胞抽出物のみを同側に、標的抗原を含む同じ細胞抽出物を反対側に同時に注入して動物を増強することにより実施する。この手法で一般に用いられる注入の経路は、動物の左右の足蹠の皮下である。この場合、抗体産生細胞は、動物の標的抗原を受けた体側に位置する膝窩部リンパ節から単離される。本発明においては、動物の一方の体側に抗原を、他方の体側にその抗原の立体構造変異体を注入することにより、対側性免疫化をエピトープ特異的な抗体の調製と単離に適用する。立体構造変異体は、タンパク質タンパク質相互作用、突然変異/欠失、または、新しい抗原決定基を形成する結果となる任意の抗原の修飾の結果として生ずる。本発明においては、対側性免疫化を、以下に記載するB細胞の減算的選別と組み合わせて用いるのが最も良い。
【0085】
抗体レパートリーをスクリーニングする方法
本発明は、抗体レパートリーをスクリーニングする方法であって:1)標的抗原とマーカーを保持する追跡可能な組換えエキソソームを抗体レパートリーと接触させること、および2)抗原特異的な抗体を、抗原を保持するエキソソームを介した前記マーカーとの結合に基づき単離すること、を含む方法をここで開示する。抗体レパートリーの細胞ベースのライブラリー由来の所望の特異性を有する抗体を、組換えDNAとハイブリドーマを含む標準の抗体調製方法により調製することができる。この方法は、上記の抗体生成法に従って免疫化した動物からの抗体の調製に最も適している。
【0086】
「抗体レパートリー」とは、様々な抗体を表示している抗体産生粒子の集団を意味する。「抗体産生粒子」とは、粒子が表示している抗体をコードする核酸を含む粒子を意味する。好ましくは、前記粒子は細胞、酵母またはファージである。より詳細には、前記抗体産生細胞は、プラズマ細胞、ハイブリドーマまたはリンパ球でよい。前記抗体産生細胞はまた抗体分泌細胞であってもよい。
【0087】
抗原とマーカーを表示している追跡可能な小胞の調製
追跡可能な小胞は、上記のように合成のまたは自然の小胞であって、好ましくはエキソソームである。それらは標的抗原を表示し、また例えばタグ、ビオチン、酵素マーカーまたは蛍光団などを介して標準の免疫学的、生化学的、または物理化学的方法によって検出することができる。
【0088】
抗原表示小胞の調製方法は、既に上に記載した。キメラタンパク質を免疫化に用いた場合には、同じタンパク質抗原配列を、伸長したエキソソームターゲティングドメインに融合した第2のキメラタンパク質を調製する。あるいは、タンパク質抗原を異なる種由来のエキソソームターゲティングドメインのホモログに融合してもよい。ここでの目標は、新しい連結部配列を有するキメラタンパク質を作り出し、これにより免疫化に用いるキメラタンパク質の連結部に見出される可能性のある新しい抗原決定基と反応する抗体の検出/選択を回避することである。
【0089】
追跡可能なエキソソームは、抗原表示エキソソームと同様に、エキソソームターゲティングドメインに融合させたマーカー(例えばタグ、酵素、および蛍光タンパク質)をコードするベクターを細胞にトランスフェクションすることにより、またはマーカーが膜貫通部分を含む場合にはより単純に、マーカーを過剰発現させることにより調製できる。抗原とマーカーの両方を表示しているエキソソームを、追跡可能なエキソソームのみを産生している予め確立した安定な細胞系統を用いて調製することができる。ここで用いる安定な細胞系統は、293細胞またはCHOなどの組換えタンパク質を大量に産生する能力のために一般に用いられている任意の実験室細胞系統でよい。この細胞系統は、好ましくは大量のエキソソームを産生し、また免疫化に用いた抗原表示エキソソームを産生する細胞系統とは異なる種に由来するべきである。換言すれば、免疫化された動物例えばマウスに由来する抗体産生細胞を用いて抗体レパートリーのスクリーニングを実施し、そしてこの動物の抗体応答が、マウスの細胞系統由来の組換えエキソソームによる免疫化により誘導された場合には、抗体レパートリーのスクリーニングに用いる標的抗原とマーカーを表示している追跡可能なエキソソームは、マウス以外の種に由来する細胞系統により産生されるべきである。これは、マウス由来のエキソソームタンパク質に対して誘導された可能性がある抗体の選択/単離を回避するためであり、また追跡可能なエキソソームと抗体発現細胞間の抗原抗体相互作用でない相互作用、例えばリガンド−レセプターを介した相互作用を回避するためである。
【0090】
もう1つの実施形態において、追跡可能なエキソソームを、ビオチンなどの活性な試薬をエキソソームに結合させることにより調製できる。
【0091】
別の実施形態において、追跡可能なエキソソームを生合成経路により、例えばエキソソームを含むエンドソーム小胞に優先的に取り込まれるラベルされた脂質を用いて調製できる。最近、網状赤血球を蛍光団結合脂質であるリサミンローダミンBジオレイル-ホスファチジルエタノールアミン(Rh-DOPE)とインキュベーションすると、その結果エキソソームよりも類似した密度を持つローダミンを含む小胞が放出されることが示された(16)。実施例6に示すように、293細胞をRh-DOPEまたはフルオレセイン-DOPEを補充した培地で培養すると、蛍光を有する293細胞由来のエキソソームが生成されることを、我々は見出した。対照的に、樹状細胞の類似の培養は、蛍光性エキソソームを生成しなかった。この不一致は、樹状細胞由来と腫瘍細胞系統由来のエキソソームの異なる脂質組成により説明できる。
【0092】
したがって、本発明は追跡可能なエキソソームを調製する方法であって、次の工程:
1)エキソソーム産生細胞を、蛍光団結合脂質とインキュベーションすること;および、
2)前記蛍光団結合脂質を取り込んだ前記細胞からエキソソームを生成させ単離すること;
を含む方法に関する。
【0093】
好ましくは、前記蛍光団結合脂質は蛍光団結合DOPE(ジオレイル-ホスファチジル エタノールアミン)である。より好ましくは、前記蛍光団結合DOPEはRh-DOPEまたはフルオレセイン-DOPEである。場合によっては、前記エキソソーム産生細胞は293細胞である。
【0094】
抗原特異的な抗体の単離
抗原特異的なモノクローナル抗体の単離を、抗体レパートリーのスクリーニングにより実行する。実行に際しては、抗体産生細胞により発現される細胞表面抗体を、上記のように調製した前記抗原およびマーカーを保持する追跡可能なエキソソームに接触させる。抗体産生細胞は、好ましくは一次のまたは不死化したBリンパ球である。不死化は、Bリンパ球をウイルスおよび癌遺伝子を含む形質転換物質により形質転換するか、または不死化された細胞と融合させてハイブリドーマを作ることにより達成される。抗体産生細胞はまた抗体、好ましくはヒト抗体を発現している原核または真核の組換え細胞のライブラリーでもよい。Bリンパ球は、免疫化した動物、好ましくはヒト免疫グロブリンを発現しているトランスジェニック動物に由来するものでよい。免疫化した動物は、上記の抗体生成のための本発明を用いて、または抗原を表示している組換えエキソソームとは異なる免疫原により調製できる。これらには、アジュバント中の精製された組換え抗原、ヌクレオチド・ベースの免疫原(裸のDNA、ウイルスDNA)および抗原を含む細胞または細胞分画などの、普通に用いられる抗原製剤が含まれる。
【0095】
追跡可能なエキソソームを保持する抗体産生細胞の個体または集団(2以上のプール)の検出および単離が、公知の技術を用い、抗体発現細胞と使用するエキソソーム上のマーカーの性質に従って実行できる。例えば、蛍光性エキソソームと接触している単一のBリンパ球またはBリンパ球の集団を迅速に効率的に単離する方法は、蛍光活性化セルソーター(FACS)を用いて細胞を選別することである。そうするためには、Bリンパ球を含むことが知られている組織または器官、例えば血液、脾臓、リンパ節またはそれらの分画などの細胞を免疫化した動物から収集し、免疫化に用いた抗原を保持する蛍光性組換えエキソソームとインキュベートする。過剰量の無蛍光の親のエキソソームも加えて、Bリンパ球上の抗原特異的部位以外の結合部位をブロックする。次に蛍光性細胞をFACSで分析し、装置の設定を適当に調整することにより単一またはプール(2以上の)の蛍光細胞を単離することができる。
【0096】
別の実施形態において、プラズマ細胞またはハイブリドーマのような、抗体を細胞表面に発現しておらず、細胞外環境に放出される可溶性の抗体のみを産生している細胞もまた、追跡可能なエキソソームを用いて単離することができる。この場合、抗体発現細胞を追跡可能なエキソソームとインキュベートする前に、細胞表面に抗体トラップを形成して、分泌された抗体が放出されたときに捕獲する。抗体トラップは、例えばCD81またはCD45などの遍在する細胞表面マーカーに対する第1のビオチン化抗体、ストレプトアビジンおよびリンパ球の種類に特異的な免疫グロブリンに向けられた第2のビオチン化抗体を含めばよい。例えば、抗体産生細胞がマウス由来であれば、第2のビオチン化抗体はマウスの免疫グロブリンに対するものである。トラップは、細胞を第1のビオチン化抗−細胞表面マーカー抗体およびストレプトアビジンと次々にインキュベーションして調製する。つぎにストレプトアビジン上の残りのフリーのビオチン結合部位を用いて、抗体産生細胞の表面と抗体捕獲部分、即ち第2のビオチン化抗−免疫グロブリン抗体とを架橋することができる。あるいは抗体トラップを、両方のビオチン化抗体を保持するストレプトアビジンを含む予め作られた架橋を用いて調製することができる。抗体トラップは、細胞の培養が細胞内で産生された抗体の放出及び産生細胞の細胞表面でのその捕獲をすることができる間に起こる。抗体分泌細胞の個体または集団(2以上のプール)を検出し単離することが、上記のようにして追跡可能なエキソソームを用いて実行できる。もちろんこの方法を、ビオチン・ストレプトアビジンの組み合わせと同等な手段を用いても実行できる。
【0097】
本発明はまた、免疫化された動物の血液またはその分画に由来する可溶性抗体の単離に関する。可溶性抗体は、個々に、または様々な抗原特異性を有する組換え抗体のプールもしくはライブラリーとして、任意の組換え抗体を産生している発現系から導出できる。ここでは、抗原を発現している組換えエキソソームを、古くから知られたアフィニティー精製法によって抗体を単離する手段として用いる。
【0098】
別の実施形態において、本発明はさらに、エピトープ特異的または立体構造特異的な抗体の単離に関する。そのような抗体は、特定の抗原の変異体型には特異的に反応するが、その抗原の自然型には反応しない。変異抗原および自然抗原は、突然変異した抗原および野生型の抗原、または別のポリペプチド、脂質、DNA、または小分子と接触している抗原およびフリーの抗原によって代表される。後者の例が、MHC/ペプチド複合体および負荷されていないMHCまたは別のペプチドを負荷されたMHCである。本発明の好ましいMHC/ペプチド複合体は、HLA-C/HIVペプチドである。そのような変異抗原および自然抗原の別の類別が、リガンド−レセプター複合体およびフリーのリガンドまたはフリーのレセプターである。本発明の好ましいリガンド−レセプター複合体は、gpl20、CCR5、CD4複合体およびgpl20、CXCR4、CD4複合体である。抗原の変異体はまた、特定の抗原の非恒常型を生み出す生理的過程の結果である場合がある。これらにはキナーゼ、プロテアーゼ、グリコシラーゼ、およびホスファターゼなどの修飾酵素を含んでいる活性化/不活性化経路により誘導される、抗原の一時的な立体構造状態が含まれる。エピトープ特異的または立体構造特異的抗体の単離を減算的スクリーニングにより実行できる。この場合、標的となるエピトープを保持する変異型抗原が追跡可能なエキソソーム上に発現されており、標的となるエピトープを欠く前記抗原の自然型が追跡不可能なエキソソーム上に発現されている。後者は抗体産生細胞に過剰に加えられると、前記抗原の変異型と自然型に共通のエピトープに反応する抗体を産生している細胞上の結合部位をブロックすることができる。したがって、変異型抗原を発現している追跡可能なエキソソームおよび過剰量の自然型抗原を発現している追跡不可能なエキソソームを同時に加えると、変異型抗原に見出される特異的なエピトープに向けた抗体を産生している細胞が追跡可能なエキソソームと反応する可能性がさらに増す。こうして、エピトープ特異的または立体構造特異的、を選択的に単離することができる。エピトープ特異的または立体構造特異的抗体の減算的単離を、抗体産生細胞が上記の対側性注入を受けて免疫化された動物由来である場合に用いると、さらに良好である。何故なら、これらの動物から収集された細胞集団はまた、エピトープ特異的または立体構造特異的な抗体産生細胞に富むからである。
【0099】
抗原特異的抗体の生成
次に、上記のようにして単離された抗体産生細胞により産生された免疫グロブリンの重鎖および軽鎖の少なくとも可変ドメインをコードするヌクレオチド配列を、組換え抗体の全長または断片として産生させるために、発現ベクター中にクローニングすることができる。これを行うために、選択した1個またはプール(2個以上)の抗体産生細胞からRNAを抽出し、抗体をコードしているヌクレオチド配列を、標準のRT-PCR法により適当なプライマーを用いて増幅する。PCR産物を次に発現ベクター中にクローニングする。免疫グロブリンの可変ドメインのみを増幅するときは、クローニングは、好ましくはヒト由来の免疫グロブリンの適合するリーダー配列ならびに重鎖および軽鎖の定常ドメインの全長または一部を既に含む受入れベクターに行われる。次に組換え抗体またはその断片の生成が、適合する免疫グロブリンの重鎖および軽鎖をコードするDNAプラスミドの混合物を細胞にトランスフェクションすることにより実行される。あるいは、両鎖を、2つの遺伝子転写物を同時に発現できる独特のプラスミド中にクローニングしてもよい。重鎖および軽鎖のPCR産物が2個以上の抗体産生細胞から得られるときには、重鎖および軽鎖DNAプラスミドの種々の組合せのマトリックスができるため、適切な抗原特異性を有する免疫グロブリンを再構成することができる。組換え抗体を発現させるための受容細胞は、CHO、293または骨髄細胞系統などの、組換えタンパク質を生成できる任意の細胞系統でよい。組換え免疫グロブリンの発現を、ELISAなどの標準的方法によって測定することができる。組換え免疫グロブリンの抗原特異性もまた、標的抗原の性質と利用可能性に従う標準の免疫学的方法により確認される。例えば、ELIZAを上記の抗原を発現している組換えエキソソームを用いて実施することができる。抗体配列をさらに組換えDNA技術により操作して、標的抗原に対する抗体の親和性を増すことや(抗体熟成として知られる過程)、医療適用のために抗体をヒト化することもできる。
【0100】
別の実施形態において、本発明は免疫グロブリンの可変配列をクローニングするためのレシピエントベクターの使用に関する。このベクターは、BsmB IなどのII型の制限酵素によって認識される2つの制限部位を備えるカセットを含む。これらのII型の酵素は、その認識部位の外側で2重鎖DNAを切断し、これが挿入配列のオーバーラップした塩基を用いたDNA断片の挿入を可能にする。他方、従来のクローニングは通常、挿入の両端に付加される制限部位のオーバーラップした塩基を用いる。したがって、本発明により、免疫グロブリンのリーダー配列と定常ドメインの間に可変配列をクローニングすることが、クローニング部位に突然変異を導入することなく可能になる。対照的に、大部分の従来の可変配列のクローニング方法は、組換え免疫グロブリンの親和性を修飾する可能性のある突然変異を導入する。実施例10がさらに、受入れベクターおよび本発明の利点を詳述する。
【0101】
したがって、本発明は、クローニング部位に突然変異を導入することなく、免疫グロブリンのリーダー配列と定常ドメインの間に可変配列をクローニングする方法であって、次の工程:
1)前記リーダー配列、II型制限酵素により認識される2つの制限部位を備え、制限酵素切断後に制限部位が前記ベクターから除去されるように構成されたクローニングカセット、および前記定常ドメイン、を順番に含むベクターを提供すること;
2)前記可変配列を提供すること;
3)前記ベクターを前記制限酵素とインキュベーションすること;
4)前記可変配列と工程3で得られた前記ベクターをライゲーションすること;
5)前記リーダー配列と前記定常配列の間に前記可変配列を含む前記ベクターを単離すること;
を含む方法に関する。
【0102】
別の実施形態において、単離された抗体産生細胞を、適当な培地中で培養することにより、生体外で増殖させることができる。あるいは、これらの細胞を上記の形質転換または融合により不死化することもできる。これにより生成される抗体を、次にProtein-Gビーズを用いたクロマトグラフィーなどの公知の方法により、培養上清から精製する。
【0103】
さらに別の実施形態において、ポリクローナル抗体を、例えば固体支持体に結合した組換えエキソソームを用いるアフィニティー精製により、血清から精製できる。抗原特異的な相互作用により固体支持体に結合した抗体は、標準の抗体溶出法、すなわち、低pHを用いて溶出できる。
【0104】
より一般的な観点から、本発明はクローニング部位に突然変異を導入することなくベクター中に配列をクローニングする方法であって、次の工程:
1)II型制限酵素により認識される2つの制限部位を備え、制限酵素切断後に制限部位が前記ベクターから除去されるように構成されたクローニングカセットを提供すること;
2)適合する末端を有する前記配列を提供すること;
3)前記ベクターを前記制限酵素とインキュベートすること;
4)前記配列と工程3で得られた前記ベクターをライゲーションすること;
5)前記配列を含む前記ベクターを単離すること;
を含む方法に関する。
【0105】
次の実施例は、説明のために提供されるのであって、制限のためではない。
【実施例1】
【0106】
エキソソームは抗体生成のための有力な新しい媒体である。
【0107】
9匹のBalb/Cマウスを、3匹のマウスからなる3つの免疫化グループに分けた。各マウスを、PBS中の〜20 ng組換えヒトラクトアドヘリン(グループ1)、1:1 PBS/完全フロイントアジュバント混合物中の〜20 ng組換えヒトラクトアドヘリン(グループ2)、またはPBS中の〜20 ngヒトラクトアドヘリンを含む組換えWEHIエキソソーム(グループ3)の腹腔内投与により免疫化した。
【0108】
ヒト組換えラクトアドヘリンを、(His)6タグ(配列番号2)と融合した全長の組換えヒトラクトアドヘリンをコードするpcDNA6hLactlf/HisプラスミドによりトランスフェクションされたCHO細胞が生成した組換えエキソソームから調製した。pcDNA6hLactlf/Hisを次のようにして調製した。ヒトラクトアドヘリンcDNAの2つのオーバーラップした断片を、造血細胞由来のcDNAから、それぞれプライマー対LTDNfl5(配列番号3)/LTDNr8(配列番号4)およびLTDNf2(配列番号5)/LTDNrl3(配列番号6)を用いて増幅した。LTDN/fl5およびLTDNrl3の5’端を、Hind IIIおよびAge I制限部位を含めるために延長した。一方LTDNr8およびLTDNf2はEcoR I部位を含んでいた。LTDNf2/LTDNrl3を用いたラクトアドヘリンcDNAの3’端断片の増幅によって複数の生成物が生み出され、最長のものはラクトアドヘリンcDNAと一致した。5’端断片をHind IIIとEcoR Iで消化し、一方3’端断片をAge IとEcoR Iで消化した。5’端と3’端断片を互いにライゲーションし、そしてpcDNA6hLactlf/Hisを生じるようにHind IIIとAge Iによって予め切断しておいたpcDNA6A-His(Invitrogen)にライゲーションした。このプラスミドを、リポフェクタミン(Invitrogen)を用いて、ハムスター卵巣細胞系統(ATCC)であるCHO細胞にトランスフェクションした。完全培地(2 mM L-グルタミン、100 U/mlペニシリン、0.1 mg/mlストレプトマイシン、および2%ウシ胎児血清(FBS)を補充したCHO-SFM)中、37℃、5% CO2雰囲気下での培養1日目に、安定してトランスフェクションされた細胞を、2μg/mlブラストサイジンを補充した培地中に選別した。4日間培養後、安定なクローンを限界希釈技術により単離した。大量のラクトアドヘリンを生成しているクローンを、エキソソームに発現されている組換えラクトアドヘリンのウェスタンブロット分析により、次のようにして選択した。培養上清を集め、続いて200 gで遠心し、0.2μmフィルターで濾過して細胞片を除いた。次に澄んだ上清を4℃で、100,000 g 90分間遠心し、エキソソームを沈殿させた。沈殿を100 μl氷冷PBSに再懸濁した。8μlのSDS-PAGE試料緩衝液5X(SB)を、PBS中のエキソソーム32μlに加え、100℃で5分インキュベートし、次にSDS-PAGEで分析した。ゲル上のタンパク質を、半湿式エレクトロトランスファーにより、PVDFメンブレンに移動させた。試料中のヒトラクトアドヘリンの存在を、ヒトラクトアドヘリンのRGDモチーフに対するポリクローナル抗体(Dr.Sebastian Amigorenaから寄贈)の1/2500希釈液を用いた免疫検出により確かめた。ラクトアドヘリンに結合した抗体を、西洋わさびペルオキシダーゼと結合した二次抗−ウサギIgG抗体(Jackson ImmunoResearch)の1/5000希釈液と比色用基質(CN/DAB, Pierce)を用いて検出した。CHO-3.2クローンをラクトアドヘリン生成用に選択し、1Lスピナーフラスコ中のFBSを含まない完全培地に移して、ラクトアドヘリンの大量産生のために増殖させた。7日間細胞培養した上清を、250 ml遠心ボトルに移し、2000 rpmで5分間遠心して細胞を沈殿させた。次に、上清を0.2μmフィルターで濾過し、500 KDカットオフサイズのファイバーカートリッジを用いて100 mlに濃縮した。濃縮した上清を次に、100,000 g、1時間15分間4℃で遠心した。エキソソームを含む沈殿を、1mlのMLB II(50mM NaP04 pH 8/300mM NaCl/lOmMイミダゾール/0.5% Tween)に再懸濁し、2 mlのNi-NTAスラリー(予めEtOHを除くため遠心した)を含むチューブに移した。2〜3時間4℃でシェ−カーによりインキュベーションした後、試料をBioRadカラムに注ぎ、4℃で保持した。カラムを10 ml MWBI(50mM NaP04 pH 8/300mM NaCl/20mMイミダゾール/0.05% Tween)で、次に20 ml MWBII(50mM NaP04 pH 8/500mM NaCl/20mMイミダゾール)で洗浄した。カラムに結合したタンパク質を、8 ml MEBII(50mM NaP04 pH 8/300mM NaCl/250mMイミダゾール)で溶出した。溶出したタンパク質を、Millipore Ultrafree-4 10,000 MWCO装置を使用して濃縮し、緩衝液をPBS pH 7.4に交換した。タンパク質試料を小分し、-20℃で保存した。この過程によって、高度に精製された組換えラクトアドヘリンが得られる。
【0109】
マウスの細胞系統WEHIに由来し、組換えヒトラクトアドヘリンを発現しているエキソソームの調製を、CHO細胞系統を用いて、一時的にトランスフェクションした細胞を4日培養した後にエキソソームを収集した点を除いては正確に上記の通りに行った。免疫化グループ1〜3に注入する試料を、マウス当たり注入される組換えラクトアドヘリン量に対して正規化した。正規化を、組換えラクトアドヘリンと組換えエキソソームの順次希釈を利用した、上記のウエスタンブロット分析により確かめた。
【0110】
動物は、最初の注入の2週間後にグループ2を除いて同じ試料を用いて1回のブーストを受けた。グループ2では抗原を1:1 PBS/不完全フロイントアジュバント混合液に再懸濁した試料を用いた。2回目の免疫化後に、動物から採血し、ELISAによって抗ヒトラクトアドヘリン抗体をテストした。ELISAのために、PBSに溶かした50 ngヒトラクトアドヘリンでマイクロ滴定プレートのウエルを37℃で1時間コートした。PBS中に0.05%Tween-20と6%脱脂粉乳を含むブロッキング緩衝液を1時間室温(RT)でウエルに加え、残っているフリーの結合部位を飽和させた。次にウエルを1時間RTで、ブロッキング緩衝液で1/1000希釈した免疫化マウスの血清とインキュベーションした。ウエルを3回ブロッキング緩衝液で洗浄した後、結合した抗体を、1/10000希釈の西洋わさびペルオキシダーゼ結合二次抗−マウスIgG抗体(Jackson ImmunoResearch)とECL基質(Amersham)を用いて検出した。このELISAの結果を図1に示す。
【0111】
結果:
抗ラクトアドヘリン抗体が、ヒトラクトアドヘリンで覆われたマウスエキソソームを用いて免疫化したマウスの血清中で検出され、一方ヒトラクトアドヘリンを単独で、またはフロイントアジュバント中の乳濁液として与えた場合は、抗体応答が生成されなかった。フロイントアジュバントを用いた場合は、4回の接種材料注入後でさえも抗体が検出されなかったが、一方でラクトアドヘリンを保持するエキソソームを与えられたマウスの血清中の抗体力価は、以降の注入により増加した(データを示さず)。
【0112】
結論:
抗原を保持するエキソソームは、アジュバント無しで高度に免疫原性であり、極めて少量であってフロイントアジュバントなどの古くから知られた強力なアジュバントでは効果がない程度の量の抗原を用いて、抗体応答を誘導することができる。したがって、抗原を表示している組換えエキソソームは、前記抗原に対する抗体を生成するための強力な手段である。
【実施例2】
【0113】
組換え膜タンパク質を発現しているエキソソームを生成するための方法
【0114】
Gタンパク質共役レセプター(GPCR)の1つであるソマトスタチンレセプター(SSTR2)をコードし、ヒト脳RNA(Clontech、CA)に由来するcDNAを、プライマーSSTR2f1(配列番号7)およびSSTR2r2(配列番号8)を用いたPCRにより増幅した。プライマーは、SSTR2f1についてはHind III制限部位を有し、SSTR2r2についてはNot I制限部位を有する5’端の延長部分を含んでいた。Hind IIIおよびNot Iによる消化に続いて、PCR産物を、修飾されたpcDNA3.1(Invitrogen, CA)であるHApC3.1にクローニングし、発現ベクターHApC3.1/SSTR2(配列番号9)を調製した。HApC3.1は、合成により得られ(Genset, CA)、pcDNA3.1ベクターのNhe I部位とHind III部位の間にクローニングされたHAタグ挿入(配列番号11)を含む。HAタグは、開始コドンおよびそれに続くヘマグルチニン抗原配列由来の一連のコドンをコードし、そしてHApC3.1/SSTR2は、N-末端におけるHAタグとの組換えSSTR2であるHA-SSTR2(配列番号10)をコードする。HApC3.1/SSTR2を、マウスの細胞系統EL-4およびB3-Zに、エレクトロポレーション(BioRad GenePulserエレクトロポレーターで、220V、950μF条件下)によりトランスフェクションした。トランスフェクションされた細胞を、1 mg/mlの抗生物質G418(Invitrogen)を補足した選択培地(RPMI 1640−2 mM L-グルタミン、100 U/mlペニシリン、0.1 mg/mlストレプトマイシン、1 mMピルビン酸ナトリウム、および10%ウシ胎児血清(FBS))中で、37℃、5%CO2雰囲気下、7日に亘り培養した。細胞をFACS分析のために集め、一方培養上清をエキソソーム調製用に収集した。FACS分析のために、106のトランスフェクションされた細胞と親細胞を1時間4℃下1 mlのPBS/5% FBS中で、FITC(Roche, CA)に結合された抗−HA抗体とインキュベーションした。PBS/5% FBS中で数回洗浄した後、細胞をFACSにより分析し、その表面のHA-SSTR2の発現を評価した。EL-4およびB3-Z細胞上のHA-SSTR2発現のプロフィールが、それぞれ図2Aおよび2Bに示されている。
【0115】
エキソソームを、培地上清から実施例1に記載の通りに調製した。エキソソーム産生とHA-SSTR2の存在もまた、実施例1に記載した通りに、抗体を検出する際に抗アクチン抗体(Sigma、MO)および抗HA抗体(HRP-結合体、Roche、CA)を用いた、ウエスタンブロット分析により測定した。組換えEL-4およびB3-Zから得られたエキソソームを、図3のレーン1と2で分析した。検出用抗体は、図3のパネルAでは抗アクチン抗体で、パネルBでは抗HA抗体であった。
【0116】
CXCR4、CCR5、およびCCR7を含む他のGPCRをコードする一団のcDNAのもまた、SSTR2に対する上記の同じクローニング戦略を用いて、HApcDNA3.1中にクローニングした。プライマー対CXCR4fl(配列番号10)/CXCR4r2(配列番号11)、CCR5fl(配列番号12)/CCR5r2(配列番号13)およびCCR7f7(配列番号14)/CCR7r8(配列番号15)を用いて、それぞれCXCR4、CCR5およびCCR7をコードするPCR産物を、ヒト造血細胞培養物由来のcDNAを使用して生成した。その結果得られるプラスミドHApcDNA3.1/CXCR4(配列番号16)、HApcDNA3.1/CCR5(配列番号20)およびHApcDNA3.1/CCR7(配列番号22)は、それぞれ組換えタンパク質:HA-CXCR4(配列番号19)、HA-CCR5(配列番号21)、およびHA-CCR7(配列番号22)をコードする。HApcDNA3.1/SSTR2はもちろん、これらのプラスミドを、ヒトの胚細胞系統293細胞へ、エレクトロポレーション(220V, 950μF、BioRad GenePulserを使用)により、トランスフェクションした。安定したトランスフェクションされた細胞が、4 mM L-グルタミン、100 U/ml ペニシリン、0.1 mg/mlストレプトマイシン、2% FBSおよび、250μg/ml G418を補充した、293 -SFM培地の培養物中に確立された。HAタグを有する組換えタンパク質の、トランスフェクションされた細胞の細胞表面およびエキソソーム上における発現を、それぞれ、上記のFACS分析および捕獲ELISAによりモニターした。トランスフェクションされた293細胞上のHA-GPCR発現のプロフィールを、図4に示す。捕獲ELISAのために、マイクロ滴定プレートのウエルを、100 ngの抗−CD81捕獲抗体(PharMingen)で、37℃下1時間コートした。プレートを200μl/ウエルのPBS/0.05% Tween20で3回洗浄し、次に室温(RT)で1時間200μL/ウエルのDPBS/1% BSAとインキュベートした。洗浄工程に続いて、実施例1記載のようにして調製した精製エキソソームをウエルに加えた。終夜RTで振盪インキュベーションの後、ウエルを3回洗浄緩衝液で洗浄した。次に検出用抗体をウエルに加え、引き続いて、2次抗体−HRP結合体、またはビオチンに結合した検出用抗体を用いる場合はストレプトアビジン-HRPを加え、その後化学発光基質(Amersham)を加えて、測定した。エキソソームを定量化するための正規化定量を、エキソソーム標品の逐次希釈およびHRPに結合した検出用抗−CD81抗体を用いたCD81の測定に基づいて行った。次に第2の定量を実行し、等量で非飽和量のエキソソームと抗−HA抗体とを用いて組換え抗原の発現を評価した。第2の定量の結果を、図5に示す。
【0117】
結果
図2が、抗−HA抗体が、HApcDNA3.1/SSTR2をトランスフェクションされたEL4およびB3Zの両方の表面において、HAタグを含む組換えタンパク質を特異的に検出したが、一方でこの抗体は親のEL4およびB3Z細胞には結合しなかったことを明らかにしている。HA-タグを含むタンパク質はまたウエスタンブロットによって、安定にトランスフェクションされたEL-4細胞由来のエキソソーム中に検出することができた(図3A、レーン1)。これは、エキソソー指向ドメインとの融合を必要とせずに、組換えSSTR2がエキソソーム上に発現され得ることを裏付けている。対照的に、同じ分析によって、安定にトランスフェクションされたB3Z細胞由来のエキソソーム中にHA-SSTR2を検出することができなかった(図3A,レーン2)。両細胞系統によってエキソソームが生成されることが、エキソソームの恒常的成分であるアクチンに対する抗体を用いたウエスタンブロット分析によって証明された(図3B、レーン1および2)。類似の結果が、細胞上とエキソソーム上のHA-SSTR2発現を検出するために、抗−SSTR2抗体を用いた時にも得られた(データを示さず)。
【0118】
293細胞のHA-pcDNA3.1/GPCRによるトランスフェクションもまた、トランスフェクションされた細胞の細胞表面にHAタグを含むタンパク質を発現させる結果となった。図4が、それぞれ、HA-SSTR2、HA-CXCR4、HA-CCR5、およびHA-CCR7を発現している、4つの安定なトランスフェクションされた細胞の染色プロフィールを示す。4つのプロフィールの平均の蛍光が、発現の相対レベルは:SSTR2 > CXCR4 > CCR7 > CCR5 であることを示している。捕獲ELISAが、EL-4細胞と同様に、トランスフェクションされた293も、HA-GPCRを保持するエキソソームを生成することを明らかにした(図5)。抗−CD81抗体についての結果が示すように、様々なトランスフェクションされた細胞によって生成される正規化した量のエキソソームを用いた。したがって、検出されたGPCRの量が、エキソソーム当たりに発現されたGPCRの量に正比例した。注目すべきことに、細胞表面と、エキソソーム上に発現されるGPCRの相対レベルに関して、SSTR2 CXCR4>CCR7>CCR5という、同じ順序が見出された。
【0119】
結論
自然にはエキソソームに存在しないGPCRを含む膜貫通タンパク質を、膜タンパク質をコードするDNAで細胞がトランスフェクションされた後にエキソソーム上に検出できる。この現象は、膜貫通タンパク質配列のエキソソームを標的とする配列との融合を必要とせず、受容細胞に依存するように見える。これは組換え膜貫通タンパク質の過剰発現による可能性があり、過剰のレセプターの分泌の別の経路として働く可能性がある。トランスフェクションされた細胞によって産生される組換えタンパク質のわずかな一部が、エキソソーム上に発現され、大部分のタンパク質が細胞表面に向けられていることに注意すべきである。それにも関わらず、この方法が、検出できる量の全長のレセプターをエキソソーム上に発現させ、したがって、そうでなければエキソソーム上に見出されない膜貫通タンパク質をエキソソームに表示させるために、または既知のエキソソーム膜貫通タンパク質をさらに豊富にするために、この方法を利用することができる。
【実施例3】
【0120】
組換えMHC I/ペプチド複合体を発現するエキソソームを生成するための方法
【0121】
A2-陽性ヒト白血球から逆転写によって導出されるヒト白血球抗原(HLA)A201のα鎖のcDNAを、プライマーHLA2f1(配列番号17)およびHLA2r2(配列番号18)を用いるPCRにより増幅した。プライマーの5’端を延長し、HLA2f1に対してはHind III制限部位を、HLA2r2に対してはBstB I制限部位を含むようにした。PCR産物をHind IIIおよびBstB I酵素で消化し、Hind IIIおよびBstB Iで予め切断したpcDNA6-Myc/Hisにライゲーションして、pcDNA6-A2-Myc/His(配列番号19)を生成した。
【0122】
同様に、HLAのβ鎖(β2-ミクログロブリン即ちβ2M)配列を、RT-PCRによりプライマーβ2-MICfl(配列番号20)およびβ2-MICr3(配列番号21)を用いて増幅し、そしてpcDNA6-Myc/His中にクローニングしてpcDNA6-β2M(配列番号22)を生成した。次に、pcDNA6-β2M中のβ2M挿入を、pcDNA6とは異なる抗生物質耐性遺伝子を有するpcDNA3中にサブクローニングした。この目的のために、挿入を、プライマーβ2-MICflおよびNot I5’端延長をもつプライマーであるpcDNA6r4(配列番号23)を用いて増幅した。Hind III/Not I消化に続いて、PCR産物を、同じ酵素で予め切断したpcDNA3にライゲーションして、pcDNA3-β2Mを生成した。
【0123】
ヒト293細胞を、pcDNA6-A2-Myc/HisおよびpcDNA3-β2Mの両方により、上述のようにエレクトロポレーションを用いてトランスフェクションした。トランスフェクション2日後、A2およびβ2Mの両方を発現している安定な二重トランスフェクション細胞を確立するために、細胞を二重の抗生物質、即ち2μg/mlブラストサイジンおよび250μg/mlネオマイシン(G418)の選択下に置いた。選択下に2週間置いた後、細胞の全体集団を、上記のようにエキソソームを調製するために増大させた。
【0124】
規格化したエキソソーム試料上の、ヒトA2およびβ2Mの発現を、実施例2に記載したように捕獲ELISAにより評価した。293由来のエキソソーム上のHLA-A2へ、ペプチドが負荷されることが、直接ロード法を用いて証明された。この方法により、機能的MHCクラス1/ペプチド複合体が生成されることが示されている(WO01/82958)。手短に述べると、試料を、pH5で、100μg/mlビオチン化基準肝炎BペプチドFLPSDCFPSVおよび20μg/mlのβ2M(Sigma)と1時間インキュベーショした。次にエキソソームを1%NP4Oに溶解し、抗−MHCクラス1抗体を捕獲抗体として用い、またストレプトアビジンでラベルされたユーロピウムを、結合したビオチン化標準ペプチドの検出に用いて、捕獲ELISAにより分析した。捕獲ELISAによる組換えタンパク質発現およびペプチド負荷の測定結果を、それぞれ図6と7に示す。
【0125】
結果
293細胞を、HLA-A2をコードしているプラスミドでトランスフェクションした結果、トランスフェクションされた細胞が生成したエキソソームに組換えA2が発現した(図6)。期待通りに、親の293由来のエキソソームもまた、293がヒトHLA-A2+細胞系統であるので、内在性のHLA-A2を含んでいた。しかし、正規化した試料を用いた分析が、組換えエキソソームが、親のエキソソームに比べて〜5倍多くHLA-A2を含んでいたことを示す。対照的に、組換え293細胞によるβ2Mの全発現は有意に増加したけれども(データを示さず)、β2Mの量は、親のエキソソームと組換えエキソソームで同程度であった。MHCクラスI複合体のβ鎖は可溶性タンパク質であるため、そのエキソソームへの結合は、そのα鎖との相互作用のみに仲介される。したがって、われわれの結果は、エキソソーム上のαβ鎖複合体の総量は一定のままであるが、α鎖のサブタイプは組換えHLA-A2サブタイプの方へシフトするということを示唆している。図7に示すように、A2特異的標準ペプチドを組換えエキソソーム上には検出できたが、親のエキソソームを用いた場合は、測定値はバックグラウンドよりわずかに上に過ぎなかった。この定量におけるペプチド検出のバックグラウンドレベルを、A2-のドナー由来のDCが生成するエキソソームを用いて測定した(図7の、A2陰性Dex)。このデータは、直接ロード法を用いて組換えHLA-A2にA2特異的ペプチドを負荷できることを示している。
【0126】
結論
エキソソーム産生細胞にMHCクラスIをコードするプラスミドをトランスフェクションすると、組換えMHCクラスI分子がエキソソームに発現される。この方法が、独特のMHCクラスIサブタイプを保持するエキソソームの調製を可能にする。直接ロード法と組み合わせることにより、それが、サブタイプ特異的なMHCクラスI/ペプチド複合体が豊富なエキソソームを生成する強力な手段を提供する。これらのエキソソームは、特異的なMHCクラスI/ペプチド複合体に限定された抗体を調製するための有力な媒体である。
【実施例4】
【0127】
CD40L活性を表示する強化されたエキソソームを生成するための方法
【0128】
ヒトCD40L cDNAを、活性化されたT細胞RNAのRT-PCRにより、プライマーCD40Lf8(配列番号24)およびCD40LrlO(配列番号25)を用いて増幅した。プライマーCD40Lf8を、5’端で延長してBamH I制限部位を含めた。一方CD40Lr1Oは、BstB Iの5’端延長を含んでいた。PCR産物をBamH IおよびBstB Iで消化し、同じ制限酵素で予め切断したpcDNA6-Myc/Hisにライゲーションして、pcDNA6-CD40L(配列番号26)を作製した。次に、pcDNA6-CD40LのCD40L挿入を鋳型として用いて、5’端および3’端のオーバーラップした断片を生成した。5’端断片をプライマーCD40Lf8およびCD40Lr21(配列番号35)を用いて生成し、一方3’端断片をプライマーCD40Lf20(配列番号36)およびCD40LrlOを用いて生成した。CD40Lf20とCD40Lr21は、互いに相補的であって、CD40Lの112と113番目の残基をコードする配列を含むCD40Lのセンスおよびアンチセンスドメインに由来する。この部位のタンパク質分解切断が、可溶性CD40Lの放出をもたらす。プライマーを、野生型CD40Lではグルタミン酸とメチオニンである112と113番目の残基を、2個のグリシンに変化するように設計して、この部位で切断できずに膜貫通型のまま残るCD40Lの突然変異型を作製した。112と113位置の突然変異型CD40L(mutCD40L)をコードする全長の配列を、5’端と3’端断片の混合物を鋳型とし、プライマー対CD40Lf8/CD40LrlOを用いたPCRにより生成した。その結果得られた生成物を、上のようにpcDNA6の中にクローニングして、pcDNA6-mutCD40L(配列番号37)を生成した。pcDNA6-CD40LおよびpcDNA6-mutCD40Lは、それぞれ全長のCD40L(配列番号34)およびmutCD40L(配列番号27)をコードする。トランスフェクションされた293細胞由来の組換えエキソソームを、実施例2および実施例3に記載したようにして調製した。
【0129】
組換えCD40Lおよび突然変異CD40Lの発現を、ウサギ抗−CD40L抗体(Santa Cruz Biotechnology)を検出用抗体として用いて実施例1と2に記載の通り、ウエスタンブロット分析によりモニターした。また定量を実行して、組換え293がCD40L様活性を獲得したかどうかを、樹状細胞(DC)の成熟を刺激することにより評価した。エキソソームを、それが発現した、1)CD81の量、2)CD40Lの量に基づき、実施例2に記載した交差捕獲ELISAを用いて正規化した。正規化した量のエキソソームを、IL4/GM-CSFを含むAIM V培地で成育した7日目のDC培養液に加えた。終夜培養後、細胞表面のCD83発現を測定してDCの成熟を評価した。これは実施例2に記載したように、FITCを結合したモノクローナル抗−CD83抗体を用い、標準FACS分析により実施した。抗−CD83および抗−CD86抗体による二重染色を、DC細胞(CD86陽性細胞)の中でCD83も発現している数を特定するために、実行した。CD40L発現のためのウエスタンブロット分析およびDC成熟実験のためのFACS分析の結果を、それぞれ図8および9に示す。
【0130】
結果
組換えエキソソームのウエスタンブロット分析が、293細胞をpcDNA6-CD40LおよびpcDNA6-mutCD40Lでトランスフェクションすると、CD40Lがエキソソームに発現されたことを明らかにした(図8)。CD40Lを発現している293細胞由来のエキソソームは、全長のCD40L(レーン2のFL)を、CD40Lの可溶性型(レーン2のSF)およびCD40Lの膜貫通ドメインを含む残存N端末端部(レーン2のTM)から成るそのタンパク切断質分解産物と共に、含んでいた。対照的に、mutCD50Lを発現している293細胞由来のエキソソームは、全長型(レーン3のFL)のみを含んでいた。親の293細胞由来のエキソソームでは、タンパク質が検出されなかった(レーン1)。
【0131】
細胞染色およびFACS分析により、DC成熟のマーカーであるCD83陽性DCの数が、DCをCD40LおよびmutCD40Lを保持するエキソソームと培養すると増加したが、親のエキソソームは無効であったことが明らかになった(図9)。さらに、mutCD40Lを保持するエキソソームの方が、分析に同じ量のエキソソームを用いるか、エキソソームに結合した同じ量のCD40Lを用いるかによらず、より優れた活性を示した。実際、エキソソームに結合した200 ng/mlのmutCD40Lを測定に用いたとき、DC成熟が検出された。対照的に、同量の、エキソソームに結合したCD40L、または親のエキソソームに混合した可溶性CD40Lは、検出できる効果を持たなかった。可溶性CD40Lの活性を、1μg/ml以上のCD40L濃度での測定においてのみ検出することができた。
【0132】
結論
CD40Lが、他の膜貫通タンパク質(実施例2および3)と同様に、トランスフェクションされた細胞の過剰発現に続いて、エキソソームを標的とするドメインへの融合の必要なく、エキソソーム上に発現される。CD40Lを発現している組換えエキソソームはまた、CD40L活性を示す。したがって、本発明を、エキソソームの免疫能力を増強するために使用できる。CD40Lのタンパク質分解切断部位の突然変異が、エキソソームの免疫能力をさらに増大させる結果になった。この性質の可能な説明は、エキソソーム上の野生型CD40Lは、CD40Lの残りの膜貫通ドメインはもちろん、全長のCD40Lと可溶性型のCD40Lの混合物から成り、これが機能的なCD40Lの3量体の効果的な形成を妨げている可能性がある、ということである。このようにして、CD40Lが断片に切断されることを妨げると、その結果、より高度な特異的活性を示す強化エキソソームが生成されることになる。これが、組換えエキソソームを用いるワクチンおよび他の研究への応用に必要である。何故なら、エキソソーム注入のすぐ後で循環しているタンパク質分解酵素がCD40Lを切断して、エキソソームを不活性化する可能性があるからである。
【実施例5】
【0133】
GFP/C1C2キメラタンパク質を用いて追跡可能なエキソソームを生成するための方法
【0134】
蛍光タンパク質GFPをコードするcDNAから開始コドンを除いたものを、ラクトアドヘリンのリーダー配列とC1/C2ドメインの間にクローニングして、LS-GFP-C1/C2(配列番号40)を生成した。GFPのcDNAを、pEGFP(Clontech,CA)を鋳型とし、プライマーGFPfl(配列番号41)およびGFPr2(配列番号42)を用いて、PCRにより増幅した。プライマーGFPf1を5’端で延長してEcoR I制限部位を含むようにしたが、一方GFPr2はNot Iの5’端延長を含んでいた。PCR産物をEcoR VとNot Iで消化し、予めNot IとBstB 1で切断したC1/C2 PCR産物にライゲーションした。C1/C2 cDNA断片は、pcDNA6-hLact1f/Hisを鋳型として用い、プライマーLTDNf40(配列番号43)およびLTDNr35(配列番号44)を用い、PCRにより生成したものである。ライゲーション混合物を次に、予めEcoR VとBstB Iで切断しておいたpcDNA6-hLact28-Myc/His(配列番号45)に加えた。pcDNA6-hLact28-Myc/Hisは、リーダー配列とEGFドメインを含むヒトラクトアドヘリンのN末端をコードする。それは、鋳型としてpcDNA6-hLact1f/Hisを用いプライマーLTDNfl5およびLTDNr28(配列番号46)を用いたPCR産物をライゲーションして調製したものである。pcDNA6-hLact28-Myc/HisをEcoR VおよびBstB Iで消化すると、ラクトアドヘリンのリーダー配列とMyc/Hisタグの間のDNA断片が放出され、それがGFP-C1/C2 DNAで置き換えられてpcDNA6-LS-GFP-Cl/C2-Myc/His(配列番号39)が生成された。LS-GFP-Myc/His(配列番号29)をコードする対照のプラスミド、pcDNA6-LS-GFP-Myc/His(配列番号28)もまた類似の手法で調製した。すなわち、GFPr2はBstB I延長を含むように修飾し、またGFP配列はC1/C2 PCR産物と予めライゲーションする工程を除外して、予め切断しておいたpcDNA6-hLact28-Myc/Hisに挿入した。GFPプラスミドでトランスフェクションされた安定な細胞系統由来のクローンを調製し、エキソソームを、他のコンストラクション(実施例1〜4)について上述したようにして精製した。エキソソームについて、その蛍光発光に基づいてGFPの存在を評価した。測定は、96ウエルプレートのウエルに入れたエキソソームについて、Wallac 1420 Victor2 プレートリーダーを用いた蛍光測定によって行った。この測定結果を図10に示す。
【0135】
結果
蛍光が、pcDNA6-LS-GFP-Cl/C2-Myc/Hisをトランスフェクションした細胞由来のエキソソームで検出された一方で、バックグラウンドレベルがpcDNA6-LS-GFP-Myc/Hisをトランスフェクションした細胞が産生したエキソソームで測定された(図10)。ウエスタンブロット分析が、予想通り後者が、可溶性タンパク質として培養上清液に放出される組換えGFPを産生することを明らかにした(データは示さず)。
【0136】
結論
これらのデータがさらに、可溶性タンパク質をコードする配列をエキソソームターゲティングドメインに融合させると、タンパク質がエキソソーム上に発現することを確認する。エキソソームに結びついたGFPの発現を、抗原特異的抗体産生細胞の選別を含む様々な応用ができる、追跡可能な手段を調製するために使用できる(下の実施例7参照)。
【実施例6】
【0137】
蛍光脂質を用いて代謝ラベルすることにより追跡可能なエキソソームを生成するための方法
【0138】
この実施例では、追跡可能なエキソソームを、エキソソームを代謝的にラベルする蛍光脂質を用いて調製した。脂質1,2-ジオレイル-sn-グリセロ-3-ホスホエタノールアミン-N-(リサミンローダミンBスルフォニル)(Rh-DOPE, Avanti Polar Lipids)を、脂質のエタノール溶液を滅菌PBSに希釈し、最終濃度を40μMとして調製した。次に、40μMの原液を293細胞培養液に徐々に加えて、3.0μM蛍光脂質とした。培養7日目の培養液からエキソソームを収集し、実施例1〜4記載のように精製した。精製したエキソソームに結合した蛍光を、抗−CD81抗体で予めコートした96ウエルプレートのウエルに試料を加えて行う捕獲ELISAにより評価した。よく洗浄した後に、ウエルを560 nmで励起したときの600nmの蛍光放射を、Wallac 1420 Victor2プレートリーダーを用いて測定した。この実験に対する対照試料は、蛍光脂質を含まない培地由来のエキソソーム(Exo)、蛍光脂質を含まない培地由来であって、24時間蛍光脂質を含む培地でインキュベートしたエキソソーム(Exo+Rh)、蛍光脂質を含む培地のみ(Rh)、およびPBSである。結果を図11に示す。
【0139】
結果
Rh-DOPE脂質にさらした細胞によるエキソソーム産生(図11のRh/Exo)が、強い蛍光性のエキソソームをもたらした。対照的に、エキソソームを直接蛍光脂質とインキュベートした場合(図11のExo+Rh)、またはエキソソームが蛍光脂質無しで生成された場合(図11のExo)にはバックグラウンドレベルに近く低い蛍光が検出された。測定した蛍光は、直接エキソソームに結合していた。なぜなら、Rh-DOPEを含む培地のみをウエルに加えた定量(図11のRh)においては、バックグラウンド蛍光のみが検出されたからである。1,2-ジオレイル-sn-グリセロ-3-ホスホエタノールアミン-N-(カルボキシフルオレセイン)(Fl-DOPE)などの、他の蛍光脂質にさらした細胞によるエキソソーム産生もまた、強い蛍光性のエキソソームをもたらした(データを示さず)。
【0140】
結論
ここに記載した方法が、追跡可能なエキソソームの産生をもたらす。これらのエキソソームに結合した蛍光の強度は、蛍光脂質により直接にラベルされたエキソソームの強度より、はるかに上である。それらを利用することにより、多くの研究とスクリーニングへの応用において、増大した感度で読み取りができることになるはずである。蛍光脂質を含む追跡可能なエキソソームは、例えば、抗原特異的な抗体産生細胞を選別単離するための極めて強力な手段である(下の実施例7参照)。
【実施例7】
【0141】
抗原特異的B細胞を同定および単離するための方法
【0142】
組換えGPCR発現エキソソームによって免疫化されたマウスの膝窩リンパ節細胞(PLNC)を、接種物を足蹠に1週間毎に4回連続注入した後に収集した。接種物は、GPCRをコードするプラスミドをトランスフェクションされたマウス細胞に由来し、実施例2に記載のように調製した精製エキソソームから成っていた。PLNCを4℃で2回PBS/5% FBSで洗浄し、106のPLNCを分取したもの2つを、500μl PBS/5% FBSに再懸濁した。正規化した量の、GFPを発現しているエキソソーム、GFPとGPCRを発現しているエキソソームおよび親のエキソソームを、実施例2と5に記載のように調製した。同量の追跡可能な対照エキソソーム(GFP-293)および追跡可能なテストエキソソーム(GFP/GPCR-293)を、それぞれ、第1と第2のPLNC試料に加えた。エキソソームのPLNCとの非特異的な相互作用を遮断するために、両試料に10×過剰の親のエキソソームを加えた。フィコエリトリンに結合された抗−CD19抗体も、PLNC中のB細胞サブ集団を特異的に選択するために、試料に加えた。試料をPBS/5% FBSで調整して最終1mlにし、1時間4℃でインキュベートした。次に細胞を、GFPとCD19に二重陽性であることを識別するために、FACSで分析した。二重陽性細胞が、抗体産生細胞即ちB細胞であり、GFPエキソソーム上の組換えGPCRと特異的に反応する抗体をその表面に発現していると予想される。CCR7をモデルとして用いて行った実験の結果を図12に示す。
【0143】
結果
免疫化された細胞由来のPLNC集団中のGFP陽性細胞の百分率が、細胞をGFP/CCR7-293エキソソームとインキュベートしたときは、対照のGFP-293エキソソームとインキュベートしたときよりも、有意に高かった(図12、パネルA:0.13%陽性細胞 対 パネルB:0.36%陽性細胞)。この百分率は低いが、類似の傾向が、CXCR4、CCR5、およびSSTR2などの他のGPCRを発現しているエキソソームにより免疫化したマウスから得られたPLNCと適合する追跡可能な組換えエキソソームを用いた場合に観察された(データを示さず)。
【0144】
結論
追跡可能なエキソソームは、抗原特異的抗体産生細胞を同定する便利な手段である。FACS分析と選別を組み合わせると、応答する細胞の頻度が極めて低い場合にさえも、所定の抗原特異性を有する個々の細胞を単離することができるに違いない。
【実施例8】
【0145】
エピトープ特異的抗体を産生しているB細胞を同定および単離するための減算的方法
【0146】
この実施例において、抗体産生細胞の選別を、エピトープ特異的抗体産生細胞へ拡張する。方法は、次の点を除いて実施例7記載の方法と同様である。即ち、免疫化マウスのPLNCを過剰の親のエキソソームとインキュベートする代わりに、ここでは過剰の抗原発現エキソソームとインキュベートして、抗原上の望ましくないエピトープに反応するB細胞を遮断する。そのような減算的選別において、突然変異した抗原または抗原の立体構造変異体に見出される新しいエピトープに反応するB細胞は、自由に、抗原変異体を発現している追跡可能なGFP-293と反応できる。この手法の最も良い例が、MHC/ペプチド複合体と反応する制限抗体の調製である。HLA-A2/ペプチド複合体を発現するエキソソームの調製を、実施例3においてすでに記述した。PLNCを、エピトープ特異的抗体産生細胞に富むようにするために、対側注入を用いてマウスを組換えエキソソームで免疫化した。ここでは、マウスの右の足蹠に、組換えA2を発現しているマウス由来のエキソソームの6回の接種を、−3、0、3、10、17、および24日目に行った。左の足蹠に、MART1由来のペプチド(配列番号30)を負荷した同じエキソソームの5回の接種を、0、3、10、17、および24日目に行った。右と左のPNLCを、別々に実施例7に記載のように染色用に調製した。等量の追跡可能な対照エキソソーム(GFP/A2-293)または追跡可能なテストエキソソーム(GFP/A2/MART1-293)を、それぞれ両方のPLNC集団に加えた。A2のみに反応する抗体を産生するB細胞をマスクするために、10×過剰のA2-293を全ての試料に加えた。これらの試料についてのFACS分析の結果を図13に示す。
【0147】
結果
マウスのA2/MART1エキソソームで免疫化した側由来の、左のPLNC集団中のGFT-陽性細胞の百分率が、細胞をGFP/A2/MART1-293エキソソームとインキュベートした時には、GFP/A2-293エキソソームとインキュベートした時よりも有意に高かった(図13、パネルA:0.83%陽性細胞 対 パネルB:0.33%陽性細胞)。対照的に、右のPLNCについて、GFP/A2/MART1-293およびGFP/A2-293とのインキュベートを比較したときには、有意な差は検出されなかった(図13、パネルC:0.49%陽性細胞 対 パネルD:0.39%陽性細胞)。
【0148】
結論
ここで述べた追跡可能なエキソソームを用いる減算的方法は、エピトープ特異的な抗体産生細胞を同定するための強力な手法である。FACS分析および選別と組み合わせると、所定のエピトープ特異性を有する個々の細胞の単離同定を可能にする。減算的選別は、例えば、MHC/ペプチド複合体と反応する制限抗体を産生するB細胞の単離に適切である。
【実施例9】
【0149】
RT-PCRによって免疫グロブリン配列を取り出すための方法
【0150】
所定の抗原またはエピトープ特異性を有する抗体を産生し、実施例7および8に記載されたようにFACSによって同定された細胞を、個々に、96ウエルプレートの25μlの氷冷溶解緩衝液(Cells to cDNA IIキット、Ambion)を含むウエルに選別した。96ウエルプレートを75℃15分間インキュベートしてRNaseを熱失活させた。次にゲノムDNAを除去するために、試料を1単位のDNase Iで処理した。得られたRNAを含む試料を新しいプレートに移して、逆転写酵素とオリゴdT(SuperScript II, Invitrogen)を用いたcDNA合成を行った。抗原との接触に含まれる相補性決定ドメインを含む免疫グロブリンの軽鎖と重鎖の可変ドメインをコードするcDNAを、ネストPCRにより増幅した。IGLKfl(配列番号31)、IGLKf2(配列番号32)、IGLKf3(配列番号33)、IGLKf4(配列番号34)、IGLKf5(配列番号35)を含む順方向プライマーおよび逆方向プライマーIGKrl5(配列番号36)の混合物を、軽鎖配列の1回目のPCRに用いた。2回目のPCR(ネストPCR)を、小分けにした第1回目のPCRを鋳型とし、縮重プライマーIGKf6(配列番号37)およびIGKrl5を用いて実行した。同様に、IGLHfl(配列番号38)、IGLHf2(配列番号39)、およびIGLHf3(配列番号40)を含む順方向プライマー混合物および逆方向プライマーIGHr5(配列番号41)を、重鎖配列の1回目のPCRに用いた。2回目のPCR(ネストPCR)を、小分けにした第1回目のPCRを鋳型として、また縮重プライマーIGHfll(配列番号42)およびIGHrl3(配列番号43)を用いて実行した。蛍光性エキソソームに抗原特異的態様で結合できる能力に基づいて個別に選別した細胞由来のcDNAを用いた軽鎖配列増幅の結果を、図14に示す。
【0151】
結果
2回のPCRにより、いくつかの単一細胞cDNAに由来する検出可能な量のPCR産物が生成された(図14, レーン 2、5、6、8、および13)。これらのPCR産物の配列決定により、PCR産物が免疫グロブリンの可変ドメインをコードすることが確認された。いくつかの可能性により、図14の空白のレーンにPCR産物を欠くことを説明できる。1つの可能性が、PCRのサイクリング条件およびここで用いたプライマー対が、元のウエルの単一細胞により発現される免疫グロブリンの可変配列を効率的に増幅するために最適ではないことである。実際、各可変ドメインが独特で、増幅のために異なるPCR条件を必要とする可能性がある。他のプライマーの組み合わせおよびサイクリング条件を、最適の結果を得るために、各cDNAに試すのがよい。元のウエルに選別した細胞が、非B細胞または休止B細胞であったという別の可能性がある。追跡可能なエキソソームとPLNCとの非特異的な相互作用(実施例7と8のバックグラウンド染色を参照のこと)のために選別されたこれらの細胞は、RT-PCRによって検出するための免疫グロブリンをコードするRNAを含んでいないか、低すぎるレベルしか含んでいない。このことは、ここで提案する免疫グロブリンの可変ドメインを取り出すための方法の感度によって、非抗体産生細胞からのcDNAのさらなる持ち越しが回避できる可能性があることを際立たせる。
【0152】
結論
追跡可能なエキソソームと抗原特異的態様で相互作用できる能力に基づいて単離された単一細胞から得られた免疫グロブリンの可変配列を、古典的PT-PCRにより首尾よく取り出すことができる。次にPCR産物を、下の実施例10に記載する通り、発現ベクターにクローニングできる。
【実施例10】
【0153】
免疫グロブリンの可変配列を発現ベクターにクローニングするための方法
【0154】
実施例9で生成された可変ドメインPCR産物を受け入れるために、抗体配列の重鎖および軽鎖部分を埋め込んだ発現ベクターを構成した。可変ドメインPCR産物のこれらのベクターへのクローニングにより、全長の重鎖および軽鎖配列が生成される。挿入クローニングの古くからある方法と異なり、ここで用いるクローニング戦略は、クローニング部位に突然変異を導入しない。免疫グロブリンの可変ドメインのいずれの端で突然変異が生じても、抗原の親和性と特異性を修飾する可能性があるので、この方法が有利である。この方法は、配列認識部位の外側で切断して切断部の一方側に認識配列を残す、BfuAI、BsaI、およびBsmBIなどの「II」型制限酵素を用いる。2つのそのような制限部位を含むカセットにより、2つの部位に縁取られた挿入部を放出し、埋め込まれた抗体配列のみを残すことが可能になる。クローニング用の挿入部を、埋め込まれた抗体配列と適合性のある配列端を有する同様の部位をその両端に含むように生成する。それにより、挿入部のライゲーションが、遺伝子特異的配列のアニーリングを介して起こり、制限酵素の認識部位に適合するための塩基の挿入または修飾を必要としない。
【0155】
抗体の重鎖および軽鎖のために、別々のベクターを構成した。これらは、リンカーの無いクローニングカセットの両側に接して、リーダー配列と原型の重鎖と軽鎖の定常ドメインを、それぞれ含んでいた。軽鎖配列をコードするマウスの脾臓cDNAを、上流リーダー配列断片を生成するためにプライマーIGKfl(配列番号44)およびIGKr2(配列番号45)を用い、また下流定常ドメイン断片を生成するためにプライマーIGKfl2(配列番号46)およびIGKr4(配列番号47)を用いて増幅した。IGKr2とIGKfl2を、リンカーの無いクローニングカセットを包含するオーバーラップした配列を含むように設計した。リーダー配列、リンカーの無いクローニングカセット、および定常ドメインを含む完全なDNA断片を、上流と下流の断片を鋳型とし、プライマーIGKflとIGKr4を用い第2のPCRにより生成した。これらのプライマーは、IGKflはHind III制限部位を有し、IGKr4はBstB Iを有する5’端延長を含んでいた。PCR産物を、Hind IIIおよびBstB Iによる消化に続いて予め同じ酵素で切断したpcDNA6-Myc/Hisへクローニングして、pcDNA6-LCJ-Myc/His(配列番号48)を生成した。pcDNA6-LCJ-Myc/Hisの等価物で重鎖をコードするpcDNA6-HCJ-Myc/His(配列番号49)を、同じ手法を用いて調製した。ここで、1回目のPCRを、プライマー対IGHfl(配列番号50)/IGHr2(配列番号51)、およびIGHfl5(配列番号52)/IGHr4(配列番号53)を用いて実行した。2回目のPCRを、IGHflおよびIGHr4を用いて行った。
【0156】
別のコンストラクションを、抗体の重鎖と軽鎖を単一のベクター中に発現させるために調製した。これは、いずれのトランスフェクションされた細胞も重鎖と軽鎖の両方を産生することを保証するためである。この目的のために、内部リボソーム進入部位(IRES)およびpIRES(Clontech)由来の第2の多重クローニング部位(mcs2)を、pcDNA6-LCJ-Myc/His中のLCJの下流にクローニングしたIRES/mcs2挿入を、pIRESを鋳型に用い、プライマーpIRfl(配列番号54)およびpIRr2(配列番号55)を用いて、PCRにより生成した。PCR産物をBstBIとAge Iで消化後、予め同じ酵素で切断したpcDNA6-LCJ-Myc/Hisにライゲーションして、pcDNA6-LCJ-IRESを生成した。次に、重鎖および軽鎖の可変部位のpcDNA6-LCJ-IRESへのクローニングを、2工程で行った。第1に、軽鎖と重鎖を、それぞれpcDNA6-LCJ-IRESとpcDNA6-HCJ-Myc/Hisへクローニングした。第2に、第1工程で調製したpcDNA6-HCJ-Myc/His中の全長の重鎖を、これも第1工程で調製して、すでに全長の軽鎖をコードしているpcDNA6-L-IRESにサブクローニングした。
【0157】
実施例9で生成され、ここで記述した方法を用いて発現ベクターにクローニングされたPCR産物が、免疫グロブリンを産生したことを証明するめの実験を行った。実施例8記載のようにして単離した単一細胞由来の、重鎖および軽鎖配列の可変ドメインをそれぞれコードしているPCR産物HV-M90/12(配列番号56)およびLV-M90/12(配列番号57)をモデルとして用いた。BsmB I制限部位を、LV-M90/12に対してはプライマーIGKf6および 1: 1混合のIGKrlO(配列番号58):IGKrll(配列番号59)を、またHV-M90/12に対してはプライマーIGHfllおよび 1: 1: 1 混合のIGHrl6(配列番号60)/IGHrl7(配列番号61)/IGHrl8(配列番号62)を用いた10サイクルのPCRによって、各DNA断片の両端に導入した。これらのプライマーを、受け入れ側発現ベクター中の取り込まれた抗体配列の接続部に相補的な突出末端を残すように設計した。LV-M90/12を、BsmB Iによる消化に続いて予め同じ酵素で切断したpcDNA6-LCJ-IRESおよびpcDNA6-LCJ-Myc/Hisにクローニングして、それぞれpcDNA6-L-M90/12-IRES(配列番号63)およびpcDNA6-L-M90/12-Myc/His(配列番号64)、を生成した。HV-M90/12をはじめに、予めBsmB Iで切断したpcDNA6-HCJ-Myc/Hisにクローニングして、pcDNA6-H-M90/12-Myc/His(配列番号65)を生成した。全長のH-M90/12重鎖配列を次に、pcDNA6-L-M90/12-IRESのmcs2にサブクローニングした。そうするために、pcDNA6-H-M90/12-Myc/Hisを鋳型に用い、プライマーpIRHfl(配列番号66)およびpIRHr2(配列番号67)を用いて、PCRにより、H-M90/12の5’端と3’端に、それぞれ、Xba IとNot Iの制限部位を導入した。H-M90/12を、Xba IとNot Iによる消化に続いて、予め同じ酵素で切断したpcDNA6-L-M90/12-IRESにクローニングして、pcDNA6-L+H-M90/12を生成した。CHO細胞を、空のプラスミド、pcDNA6-L+H-M90/12、または1:1 pcDNA6-L-M90/12-Myc/His:pcDNA6-H-M90/12-Myc/His混合物によりトランスフェクションした。培養3日目の上清を収集し、免疫グロブリン産生を、ロバの抗−マウスIgG抗体(Jackson ImmunoResearch)を捕獲抗体として用い、またウサギ抗−マウスIgGを検出抗体として用いて、実施例2に述べた捕獲ELISAによりテストした。標準曲線を、精製したマウスIgGの量を次第に増加させて作製した。このELISAの結果を図15に示す。
【0158】
結果
組換え免疫グロブリンが、トランスフェクションされたCHO細胞の上清に首尾よく検出された(図15)。さらに、抗体の軽鎖と重鎖の両方をコードする発現ベクターをトランスフェクションした場合の方が、各鎖を別々にコードするプラスミドを共にトランスフェクションした場合より優れていた。
【0159】
結論
ここで述べた、全長の免疫グロブリンをコードするプラスミドを構成するためのクローニング戦略が、産生される抗体の可変ドメイン中に挿入または突然変異を導入することなく、大量の組換え免疫グロブリンを産生する発現ベクターをもたらす。全体として、実施例7〜10によって、所定の特異性を有する抗体を産生する個々の細胞に由来する抗体配列を、本発明に記載された方法を用いて取り出し、クローニングし、そして組換えタンパク質として発現させることができることが、裏付けられる。次に、産生された組換え抗体の抗原特異性を結合実験により、好ましくは抗原源として組換えエキソソームを用いて評価できる。
【実施例11】
【0160】
ビオチン化エキソソームを用いた抗体分泌ハイブリドーマの単離
【0161】
実施例8記載のように免疫化したマウスの脾臓細胞を、ハイブリドーマ調製の標準的方法を用いて、マウス骨髄腫細胞系統Sp2/0に融合させた。融合に続いて、ハイブリドーマを選別するために、アザセリンを補充した培地中で細胞を大量培養して成長させた。培養8日目に細胞を収集し、ビオチン化抗−CD45抗体およびストレプトアビジンと、逐次インキュベートした。次に、細胞を同等の2分画、即ち抗体トラップ陽性(AbTrap+)分画と陰性(AbTrap-)分画に分けた。AbTrap+分画の抗体トラップを、細胞をビオチン化抗−マウスIgG抗体とインキュベートすることにより完成させた。次に、両細胞分画を培養液中で終夜37℃10% CO2雰囲気下でインキュベートして、ハイブリドーマに抗体を分泌させ、これを抗体トラップによって捕獲させた。細胞表面で捕獲されて望みの抗原特異性を示した抗体を分泌しているハイブリドーマを、ビオチン化エキソソームとのインキュベーションに続くFACSにより単離した。この追跡可能なエキソソームは、酸化ポリエチレン(PEO)リンカーを介してビオチンに結合したテトラフルオロフェノール(TFP)を有する化学的反応性ビオチンとエキソソームをインキュベートして調製した。TFPにより活性化されたビオチンが、エキソソーム上の第一級アミンと反応して、共有結合を形成する。表面にビオチン化エキソソームを有するハイブリドーマを、蛍光団Alexa488と結合したストレプトアビジンを用いて、検出した。抗−エキソソーム抗体を産生するAbTrap+およびAbTrap-ハイブリドーマのFACS分析を図16に示す。
【0162】
結果
ビオチン化エキソソームをAbTrap-細胞とインキュベートすると、これらの細胞に結合した蛍光はバックグラウンドのみであった(図16、パネル1)。対照的に、完全なAbTrapを有する場合は、〜25%の細胞が陽性であった(図16、パネル2)。
【0163】
結論
ここで設計した抗体トラップが、首尾よくハイブリドーマが分泌する抗体を捕獲した。これによって、FACSによるハイブリドーマの単離が可能となる。さらに、この結果は、ビオチン化エキソソームが、抗体産生細胞を抗原特異的な方法で単離するための、適切な追跡可能エキソソームであることを示している。
【0164】
【表1】
【図面の簡単な説明】
【0165】
【図1】ラクトアドヘリンで免疫化したマウス血清中の抗ラクトアドヘリン抗体を検出するELISA。
【図2】HApC3.1/SSTR2によってトランスフェクションされ、抗HAタグ抗体でラベルされた組換え細胞のFACS分析。
【図3】HApC3.1/SSTR2をトランスフェクションされた細胞により生成されたエキソソームに発現されているHA-SSTR2を検出するウエスタンブロット分析。
【図4】様々なGCPRをコ−ドするHApC3.1でトランスフェクションされ、抗HA-タグ抗体でラベルされた一団の293細胞のFACS分析。
【図5】様々なGPCRをコードするHApC3.1をトランスフェクションされた一団の293細胞由来のエキソソーム上のHA-GPCR発現を検出する捕獲ELISA。
【図6】293エキソソーム上の、HLA-A2およびβ2-ミクログロブリン発現を検出するの捕獲ELISA。
【図7】エキソソーム上のHLA-A2に結合した標準ペプチドを検出する捕獲ELISA。
【図8】組換え293エキソソーム上での、CD40LおよびmutCD40L発現を検出するウエスタンブロット分析。
【図9】CD40Lを表示している組換えエキソソームの生物学的活性を測定するDC-成熟分析。
【図10】GFP/C1C2キメラタンパク質を発現している組換えエキソソームに結合した蛍光の測定。
【図11】Rh-DOPEで代謝的にラベルされた細胞由来のエキソソームに結合した蛍光を検出する捕獲ELISA。
【図12】GFP/CCR7-293エキソソームを有するPLNCを検出するFACS分析。
【図13】GFP/A2/MART1-293エキソソームを有するPLNCを検出するFACS分析。
【図14】単一細胞由来のRT-PCR産物のゲル分析。
【図15】CHO培養上清中の組換えIgGを検出する捕獲ELISA。
【図16】ビオチン化エキソソームとインキュベートしたAbTrap-およびAbTrap+細胞のFACS分析。
【技術分野】
【0001】
本発明は、抗体を生成するための組成物および方法であって、一般に1)少なくとも1つの標的抗原を有する高度に免疫原性の小胞を提供すること、および2)前記抗原を有する小胞を用いて動物を免疫化して、抗原特異的な抗体応答を引き起こすこと、を含む組成物および方法に関する。本発明はまた、抗体レパートリーをスクリーニングする方法であって、1)少なくとも1つの標的抗原および1つのマーカーを有する小胞を提供すること、および2)前記抗原およびマーカーを有する小胞を用いて、所定の抗原特異性を有する抗体産生細胞または粒子を単離すること、を含む方法に関する。次に、所定の抗原特異性を有する抗体を、単離した抗体産生細胞から周知の方法を用いて調製することができる。本発明は、実験、研究、治療、予防、または診断の分野で使用することができる。
【背景技術】
【0002】
抗体は、微生物を含む環境からの侵入者に対する防衛の第1線において決定的な役割を果たす、かなめのタンパク質である。それは研究と診断の道具であり、最も重要なことに、治療用複合物として用いることができる。多数の企業および研究センターが、今日、抗体ベースの治療候補開発の様々な段階にあり、いくつかの抗体薬物が既に市場に出荷されている。
【0003】
抗体は、ポリクローナルであっても、モノクローナルであってもよい。マウス、齧歯類、霊長類、ウマ、ブタ、ウサギ、家禽などを含む様々な種からポリクローナル抗体を産生する方法が、例えば、(1)において見出される。手短に言えば、抗原をアジュバント(例えば、完全または不完全アジュバント、例えばフロイントアジュバント)存在下で注入する。動物に一般には、皮下、腹膜内、静脈内、筋肉内注入によって投与する。反復注入を行っても良い。血液試料を収集し、免疫グロブリンまたは血清を分離する。
【0004】
モノクローナル抗体を産生する最初の方法は、KohlerとMilstein(2)により確立された。この方法はまた、例えば(3)に詳細に記載されている。手短に言えば、この方法は、アジュバント存在下で抗原により動物を免疫化し、続いて脾臓細胞またはリンパ節細胞を採取し、これを次にミエローマ細胞などの不死の細胞と融合させることを含む。その結果得られるハイブリドーマがモノクローナル抗体を産生し、これを、限界希釈によって選択して個々のクローンを単離する。
【0005】
抗体の調製が、新しいタンパク質の機能の評価に必須となっており、多くの場合、抗体は治療薬に成長している。治療化合物としての抗体に対する興味は、ヒト抗体およびヒト化抗体生成用の技術の発展により、最近復活した。進行中の多くのゲノム配列決定計画が情報を豊富に提供しつつあり、これに伴って新しい推定タンパク質薬物標的に対する抗体の系統的調製が必要となっている。これが、新しい薬物標的の同定が進展する中で、経費のかさむボトルネックを作り出し、モノクローナル抗体調製方法を合理化する新しい手法の必要性が強調されている。
【0006】
実際、モノクローナル抗体調製の基本的方法にはいくつかの限界がある。例えば、ハイブリドーマの調製は、時間がかかり冗長で非効率的な、ハイブリッド細胞生成、薬物選択、スクリーニング、およびクローン増殖の過程を必要とし、その間に希に存在する所望の抗原特異性を有する抗体産生細胞が失われる可能性がある。もう1つの困難は、ハイブリド−マの大きなプール中のユニークな抗体産生ハイブリドーマを同定するために、急速スクリーニング法を発展させる必要があることである。この方法は、標的抗原の性質とその入手可能性によって異なる。さらにもう1つの困難は、ハイブリドーマが主に、マウスまたは数少ない他の非霊長類動物の細胞を用いて生成されることである。それ故、ヒト抗体を発現する遺伝子組換えマウスを用いない限り、ハイブリドーマはヒト以外の抗体を産生するので、これは組換えDNA法を用いて、ヒト化した抗体に転換する必要がある。後者の過程が医療適用に必要であり、これは一旦所望の特異性を有する抗体を産生するクローンが得られた後にのみ開始することができる。
【0007】
これらの限界に対処するために、2つのタイプの戦略が出現した:それは1)上述の古くからある手法の特定の工程を改良すること、および2)組換えDNA技術を用いて抗体レパートリーを開発すること、である。第1の戦略が、例えば、SLAM技術(4)を生み出し、それによると、ハイブリドーマの調製とスクリーニングの必要性が除かれる。第2の戦略は、例えば、ファージディスプレイ技術を生み出し、それによると、標的タンパク質が、抗体またはその断片を発現しているバクテリオファージのライブラリーと、バクテリオファージパニング法を用いて反応する(5)。より最近、HuMY技術が GeneTastix(US patent 6,406,863)により開発され、それによると、標的タンパク質の断片が抗体レパートリーのライブラリーと、酵母のツーハイブリッド法を用いて反応する。
【0008】
改良が成されたけれども、これまでに開発された各方法は依然として限界を有している。特に、大部分の方法の主要な限界が、それらが動物を免疫化するためおよび/またはハイブリドーマもしくはバクテリオファージライブラリーのスクリーニングに大量の標的タンパク質を必要とすることである。ハイブリドーマの調製とスクリーニングを必要としないSLAM技術の場合においてさえも、著者たちが記述しているように、抗体産生細胞を単離するためのプラーク分析を行うために、やはり大量の精製した標的タンパク質が必要である。組換えタンパク質を大量に精製する必要性が、多くの予想薬物標的タンパク質の迅速な確認を妨げている。多くの例で、精製過程はタンパク質毎に異なり、不十分量の材料、機能しない又は変性した産物を産み出す。精製過程は、膜タンパク質または複数ポリペプチド複合体から成る対象を取り扱う時には、より問題となる。
【0009】
あるファージディスプレイ法は、ツーハイブリッド法と共に抗原ペプチドのライブラリーを用いており、それによって大規模な抗原精製の必要性を軽減している。しかし、これらの方法の1つの欠点は、立体構造のエピトープに対する抗体を得ることが困難であって、例えば、抗−MHC/ペプチド複合体抗体などの、複数ポリペプチドからなる単位から生ずるエピトープに限定された抗体を得ることができないことである。もう1つの限界は、使用するペプチドライブラリーと抗体ライブラリーの大きさと質によっては、必ず高い抗原特異的親和性を有する抗体を単離できるとは限らないことである。これは、抗体の親和性の成熟は、第1世代の抗体が得られる場合にのみ実行され得るからである。対照的に、哺乳動物中に見出される無制限の抗体配列のレパートリーは、これらの動物を繰り返し免疫化することにより起こる自然の親和性成熟過程に結びついて、いかなるエピトープに対しても抗体を生成する潜在能力を持つ最も効率的な方法を提供する。
【0010】
本発明は、新しい効果的な手法を提供することにより、抗体調製法の限界に対処する。本発明はまた、現存する抗体調製法を改良するための新しい価値ある手段を提供する。
【発明の開示】
【0011】
本発明はここで、エキソソーム上の抗原、アジュバントおよび/またはマーカーの表示、ならびにエキソソームの強力な液性免疫応答の誘導ためのおよび/または抗体レパートリーをスクリーニングするための媒体としての使用、に関連した2つの技術を組み合わせた、抗体を生成するための新しい方法を開示する。
【0012】
エキソソームはエンドソーム起源の小胞であって、後期エンドソーム多胞体の原形質膜との融合に続いて細胞外環境に分泌される(6,7)。種々のタイプの組織の細胞、例えば、樹状細胞、Bリンパ球、腫瘍細胞および肥満細胞などが、エキソソームを分泌することが示されてきた。異なる起源のエキソソームは、別々のセットのタンパク質と脂質部分を表示する(8,9)。エキソソームは、抗原提示と免疫モジュレーションに含まれるタンパク質を特に含み、エキソソームが免疫応答のモジュレーションを引き起こす細胞間信号伝達において役割を果たしていることを示唆している。実際、腫瘍抗原由来のペプチドをパルス投与した樹状細胞(DC)から得られたエキソソームが、適合する腫瘍を用いた動物モデルにおいて抗腫瘍反応を誘発する(10,11)。治療目的または研究手段としてエキソソームを産生、精製あるいは使用する方法が、例えば、WO99/03499、WO00/44389、およびWO97/05900中に記載されており、これらを参照してここに取り入れる。組換えエキソソームについては、従来技術において既に記述されており、それは、組換えタンパク質をコードするプラスミドによりトランスフェクションされた細胞に由来するものである。そのような組換えエキソソームは、プラスミドがコードした組換えタンパク質を含む(WO00/28001)。
【0013】
治療目的でまたは研究手段として、エキソソームのタンパク質含量を操作し、抗原、アジュバントおよびマーカーを表示する方法が、WO03/016522に記載されている。
【0014】
本発明は、選ばれた抗原に対して特異性を有する単一の抗体産生粒子を単離する方法であって:
1)選択した抗原とマーカーを表示している小胞、好ましくはエキソソーム、を調製すること;
2)工程1の前記小胞を抗体レパートリーと接触させる、または懸濁させること;および
3)前記小胞と反応している単一の抗体産生粒子を同定および単離すること;
を含む方法に関する。
【0015】
ある実施態様においては、前記抗体産生粒子は抗体産生細胞であり、前記抗体レパートリーは抗体産生細胞のレパートリーである。前記抗体産生細胞は、プラズマ細胞、ハイブリドーマ、またはリンパ球でよい。好ましくは、前記抗体産生細胞は産生した抗体を表示する。前記抗体レパートリーは、標準的な組換えDNAの手法を用いて調製され、例えば、ファージまたは酵母のディスプレーライブラリー中に見出される。したがって、別の実施態様においては、前記抗体産生粒子は特定の抗体を提示しているファージまたは酵母の集合である。前記抗体産生細胞はまた抗体分泌細胞でもよい。
【0016】
本発明は、ある抗原に対して特異性を有する単一の抗体産生細胞を同定する方法であって:
1)抗体産生細胞を提供すること;
2)前記抗原およびマーカーを表示している小胞、好ましくはエキソソームを調製すること;
3)工程1の抗体産生細胞を、工程2の小胞と懸濁させること;および
3)前記小胞と反応している単一の抗体産生細胞を同定および単離すること;
を含む方法に関する。
好ましくは、前記抗体産生細胞はリンパ球である。ある実施態様においては、前記リンパ球を前記抗原によって免疫化されたヒト以外の動物から収集する。
【0017】
もし抗体産生細胞が抗体分泌細胞であるなら、方法はさらに、抗体産生細胞を小胞と懸濁させる工程の前に、抗体産生細胞をCD81、CD45などの遍在する細胞表面マーカーに対する第1のビオチン化抗体、ストレプトアビジンおよび前記抗体分泌細胞により産生される免疫グロブリンに向けられた第2のビオチン化抗体とインキュベートする工程を含む。場合によっては、前記工程は:1)抗体分泌細胞を、遍在する細胞表面マーカーに対する第1のビオチン化抗体およびストレプトアビジンとインキュベートすること;および、2)その結果得られた抗体分泌細胞を、前記抗体分泌細胞の免疫グロブリンに向けられた第2のビオチン化抗体とインキュベートすること;を含む。またはそれに代えて、前記工程は:1)ストレプトアビジンを、遍在する細胞表面マーカーに対する第1のビオチン化抗体および前記抗体分泌細胞の免疫グロブリンに向けられた第2のビオチン化抗体とインキュベートすること;および2)その結果得られた第1と第2の抗体を保持しているストレプトアビジンを、抗体分泌細胞とインキュベートすること;を含む。前記抗体分泌細胞は、リンパ球、ハイブリドーマ、プラズマ細胞から成る群から選択するのが好ましい。
【0018】
本発明は、単一の抗体産生細胞を単離する方法であって:
1)少なくとも1つの抗原またはそのエピト−プを表示している免疫原性の小胞、好ましくはエキソソームを提供すること;
2)ヒト以外の動物を前記免疫原性小胞で免疫化することによって抗体反応を生起させること;
3)免疫化した動物からリンパ球を収集すること;
4)工程1の前記抗原またはそのエピトープとマーカーを表示している小胞、好ましくはエキソソームを調製すること;
5)工程3のリンパ球を工程4の小胞と共に懸濁すること;および、
6)工程4の小胞と反応している単一の抗体産生細胞を同定および単離すること;
を含む方法を開示する。
【0019】
場合によっては、前記動物は、抗原を表示している組換えエキソソーム以外の免疫原により免疫化されている。これらの免疫原には、アジュバント中の精製した組換え抗原、ヌクレオチドベースの免疫原(裸のDNA、ウイルスDNA)などの常用の抗原調合物、および抗原を含む細胞または細胞断片が含まれる。
【0020】
本発明は、変異抗原に特異的な単一抗体を産生している粒子を、抗体レパートリーから単離する方法であって:
1)前記変異抗原とマーカーを表示している小胞、好ましくはエキソソーム、の第1の集団を調製すること;
2)自然抗原を表示しているが前記マーカーは表示していない小胞、好ましくはエキソソーム、の第2の集団を調製すること;
3)前記抗体レパートリーを小胞の第1および第2の集団と共に、第2集団は過剰に、懸濁すること;
4)工程1の小胞と反応している単一の抗体産生粒子を同定および単離すること;
を含む方法に関する。
【0021】
本発明は、変異抗原に特異的な単一抗体を産生している細胞を同定する方法であって:
1)抗体産生細胞を提供すること;
2)動物の免疫化に用いた前記変異抗原とマーカーを表示している小胞、好ましくはエキソソーム、を調製すること;
3)自然抗原を表示し、前記マーカーを表示していない小胞、好ましくはエキソソーム、を調製すること;
4)工程1の抗体産生細胞を、工程2の前記変異抗原とマーカーを表示している小胞および工程3の前記自然抗原を表示している過剰量の小胞、と共に懸濁すること;および、
5)工程2の小胞と反応している単一の抗体産生細胞を同定および単離すること;
を含む方法に関する。
好ましくは、前記抗体産生細胞はリンパ球である。ある実施態様においては、前記リンパ球は前記抗原で免疫化したヒト以外の動物から収集される。
【0022】
本発明はさらに、変異抗原に特異的な単一抗体を産生している細胞を単離する方法であって:
1)変異抗原を表示している免疫原性小胞、好ましくはエキソソーム、を提供すること;
2)ヒト以外の動物を前記免疫原性小胞によって免疫化することにより、抗体反応を生起すること;
3)免疫化した動物からリンパ球を収集すること;
4)工程1の前記変異抗原およびマーカーを表示している小胞、好ましくはエキソソームを調製すること;
5)自然抗原を表示し、前記マーカーを表示していない小胞、好ましくはエキソソーム、を調製すること;
6)工程3のリンパ球を、工程4の前記変異抗原とマーカーを表示している小胞および工程5の前記自然抗原を表示している過剰量の小胞と懸濁すること;および、
7)工程4の小胞と反応している抗原変異体に特異的な単一の抗体を産生している細胞を同定および単離すること;
を含む方法を開示する。
【0023】
場合によっては、前記方法はさらに次の工程を含む:a)前記の選択された抗体産生粒子からDNAまたはRNAを回収すること、b)免疫グロブリン配列またはその一部をコードする核酸配列を増幅すること、c)増幅された核酸配列を発現ベクターへクローニングして、所望の抗原特異性を有するタンパク質を生成すること。
【0024】
本発明に従って抗体産生細胞を単離する上記の方法のある実施態様においては、前記小胞によって表示される前記抗原を、エキソソームを標的として指向する(エキソソーム指向)ポリペプチドに融合する。別の実施態様においては、前記小胞によって表示される前記抗原を、エキソソームターゲティングポリペプチドに架橋する。
【0025】
本発明に従って抗体産生細胞を単離する上で開示した方法の他の実施態様において、前記抗原は少なくとも1つの膜貫通ドメインを有するポリペプチドであり、また前記抗原はエキソソーム産生細胞中に過剰に発現され、それによって、前記抗原を表示する組換えエキソソームの生成が可能になる。好ましくは、前記少なくとも1つの膜貫通ドメインを有するポリペプチドはレセプターである。より好ましくは、前記レセプターは、SSTR2、CCR7、CXCR4、およびCCR5などのGPCR(Gタンパク質共役レセプター)である。
【0026】
前記抗原は、例えばレセプターまたは酵素のような任意のタンパク質でよく、あるいは、糖脂質、多糖類、薬剤、および有機化学物質などの、ポリペプチド以外の化合物でもよい。場合によっては、前記抗原はオーファンレセプターである。場合によっては、前記抗原は腫瘍、ウイルス、または微生物抗原である。あるいは、前記抗原はMHC複合体、より具体的にはMHC I/ペプチド複合体でもよい。
【0027】
本発明により変異抗原に特異的な単一抗体を産生する細胞を単離する上に開示した方法の好ましい実施態様において、前記変異抗原は突然変異抗原であり、前記自然抗原は野生型抗原である。場合によっては、前記変異抗原は、ポリペプチド、脂質、DNAまたは小分子から成る群から選ばれる分子と接触している抗原であり、前記自然抗原はフリーの抗原である。好ましくは、前記分子と接触している抗原はMHC/ペプチド複合体である。したがって、前記変異抗原はMHC/ペプチド複合体であり、前記自然抗原は負荷されていないMHC、または異なるペプチドを負荷されたMHCである。ある実施態様において、前記変異抗原はHLA-C/HIVペプチド複合体であり、自然抗原は負荷されていないHLA-Cまたは異なるペプチドを負荷されたHLA-Cである。異なるペプチドとは第1のHIVペプチドとは異なるペプチド(好ましくは50、30、20または10%より少ない同一性を有する)、例えばHIV由来ではないペプチドを意味する。あるいは、前記変異抗原はリガンド-レセプター複合体であり、自然抗原はフリーのリガンドまたはフリーのレセプターである。ある実施態様において、前記変異抗原は、gp120、CXCR4およびCD4、またはgp120、CCR5およびCD4を含み、前記自然抗原はgp120である。また、前記変異抗原および自然抗原は、酵素を含む任意のタンパク質の異なる立体構造状態であってもよい。
【0028】
本発明に従い抗体産生細胞を単離する上に開示した方法の別の実施態様において、前記免疫原性小胞はさらに免疫アクセサリー分子を表示する。好ましくは、前記免疫アクセサリー分子はアジュバントポリペプチドである。より好ましくは、前記アジュバントポリペプチドは、GM-CSF、IL-2およびCD40Lなどのサイトカインである。前記CD40Lは、好ましくは突然変異CD40Lであり、前記突然変異は、可溶性CD40Lの切断と放出を阻止する。場合によっては、前記免疫アクセサリー分子も、エキソソームターゲティングポリペプチドに融合または架橋させる。好ましくは、GM-CSFまたはIL-2などの前記可溶性免疫アクセサリー分子を、エキソソームターゲティングポリペプチドに融合させる。あるいは、少なくとも1つの膜貫通ドメインを有する前記免疫アクセサリー分子が、エキソソーム産生細胞中への過剰発現により免疫原性小胞に取込まれる。場合によっては、前記免疫アクセサリー分子は、抗原提示細胞へ特異的抗原を送達するためのリガンドである。
【0029】
場合によっては、前記マーカーは、タグ、酵素、ビオチン、蛍光分子などの検出可能な分子である。場合によっては、前記マーカーをエキソソームターゲティングポリペプチドに融合または架橋させる。あるいは、前記マーカーは、エキソソーム産生細胞中に過剰発現させることによって免疫原性小胞に取込まれる膜貫通ドメインを有する。さらに、前記マーカーは小胞、好ましくはエキソソームに優先して取込まれるラベルされた脂質である。好ましくは、前記ラベルされた脂質は、ローダミン-DOPEまたはフルオレセイン-DOPEなどの蛍光団結合脂質である。
【0030】
場合によっては、小胞、好ましくはエキソソームを調製する工程は次の工程を含む:
a)前記抗原をコードする遺伝子コンストラクトを提供すること;
b)場合によっては、前記マーカーをコードするコンストラクトを提供すること;
c)前記コンストラクトをエキソソーム産生細胞に導入して、組換えエキソソームを生成すること;および、
d)前記組換えエキソソームを収集すること。ここで、前記エキソソームは、その表面に前記遺伝子コンストラクトによりコードされる抗原および場合によっては前記マーカーを所持する。
【0031】
場合によっては、互いに異なる抗原をコードするいくつかの互いに異なる遺伝子コンストラクトを、前記エキソソーム産生細胞に導入する。好ましくは、前記エキソソーム産生細胞は哺乳動物細胞である。より好ましくは、前記哺乳動物エキソソーム産生細胞はマウスの細胞である。
【0032】
あるいは小胞、好ましくはエキソソームの調製工程は次の工程を含む:
a)エキソソームターゲティングポリペプチドに融合された前記抗原を含む分子を提供すること;
b)場合によっては、エキソソームターゲティングポリペプチドに融合された前記マーカーを含む分子を提供すること;および
c)前記抗原を含む前記分子、および場合によっては前記マーカーを含む前記分子を、フォスファチジルセリンまたはエキソソームに自然に含まれる他の脂質を含む脂質小胞と接触させて、表面に前記抗原および場合によっては前記マーカーを提示している機能化された脂質小胞を生成すること。
【0033】
本発明はさらに、単離された抗体産生粒子、好ましくは細胞、およびそれらの抗体産生への使用、および前記抗体産生粒子、好ましくは細胞によって産生された抗体に関する。本発明はまた、前記単離された抗体産生細胞または前記抗体産生細胞によって産生された前記抗体および薬学的に許容できる賦形剤または坦体を含む組成物に関する。
【0034】
本発明はさらに、エキソソームの表面に少なくとも1つの膜貫通ドメインを有するポリペプチドを発現させる方法であって、:
1)前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを含むその一部をコードする遺伝子コンストラクトを提供すること;
2)エキソソーム産生細胞に前記コンストラクトを導入して、前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを含むその一部をエキソソーム産生細胞中に過剰発現させ、組換えエキソソームを生成すること;および、
3)前記組換えエキソソームを収集すること(ここで前記エキソソームは、その表面に前記遺伝子コンストラクトによりコードされるポリペプチドを所持している);
方法に関する。
【0035】
この方法の好ましい実施態様において、少なくとも1つの膜貫通ドメインを有する前記ポリペプチドは、レセプターである。より好ましくは、前記レセプターはSSTR2、CCR7、CXCR4、およびCCR5などのGPCR(Gタンパク質共役レセプター)である。あるいは、前記ポリペプチドはCD40L、好ましくは 突然変異CD40Lであり、前記突然変異は可溶性CD40Lの切断と放出を阻害する。
【0036】
本発明は、上記の方法によって調製された機能化されたエキソソーム、前記機能化されたエキソソームを含む組成物およびそれの使用に関する。より詳細には、本発明は、前記機能化されたエキソソームおよび薬学的に許容できる賦形剤または坦体を含む組成物に関する。本発明はまた、抗体を産生し、または対象に抗原を送達するための前記機能化されたエキソソームの使用に関する。
【0037】
したがって、本発明は、少なくとも1つの膜貫通ドメインを有するポリペプチドまたはそのエピトープを結合する抗体を産生する方法であって:
a)前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを含むその一部をコードする遺伝子コンストラクトを提供すること;
b)エキソソーム産生細胞中に、前記コンストラクトを導入し、前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを含むその一部をエキソソーム産生細胞中に過剰発現させ、前記ポリペプチドまたはそのエピトープを表面に提示している組換えエキソソームを生成すること:
c)前記組換えエキソソームを収集し、前記エキソソームまたはその一部を、ヒト以外の哺乳動物に注入して、前記ポリペプチドまたはそのエピトープを結合する抗体を生成すること;および、
d)抗体または抗体産生細胞を前記哺乳動物から収集すること;
を含む方法に関する。
【0038】
この方法の好ましい実施態様においては、少なくとも1つの膜貫通ドメインを有する前記ポリペプチドはレセプターである。より好ましくは、前記レセプターはSSTR2、CCR7、CXCR4、およびCCR5などのGPCR(Gタンパク質共役レセプター)である。好ましくは、前記エキソソームはさらに、突然変異CD40Lを表示し、前記突然変異は可溶性CD40Lの切断と放出を阻害する。次に抗体を、ポリクローナル抗体調製のための動物の血清から直接単離することができる。モノクローナル抗体もまた、ハイブリドーマの生成またはSLAMなどの方法による単一の抗体産生細胞の単離を含む、古くからある手法を用いて、これらの動物から調製することができる。
【0039】
本発明はまた、少なくとも1つの膜貫通ドメインを有する抗原または少なくとも1つの膜貫通ドメインを含むその一部を、対象に送達する方法であって:
a)前記抗原または少なくとも1つの膜貫通ドメインを有するその一部をコードする遺伝子コンストラクトを提供すること;
b)前記コンストラクトをエキソソーム産生細胞中に導入し、前記抗原または少なくとも1つの膜貫通ドメインを有するその一部をエキソソーム産生細胞中に過剰発現させて、前記抗原または前記その一部を表面に提示している組換えエキソソームを生成すること;
c)前記組換えエキソソームを収集し、前記エキソソームまたはその一部を、前記対象に注入すること;
を含む方法に関する。この方法の好ましい実施態様において、前記抗原はレセプターである。より好ましくは、前記レセプターはSSTR2、CCR7、CXCR4、およびCCR5などのGPCR(Gタンパク質共役レセプター)である。好ましくは、前記エキソソームはさらに、突然変異したCD40Lを表示し、前記突然変異は可溶性CD40Lの切断と放出を阻害する。
【0040】
本発明は、少なくとも1つの膜貫通ドメインを有する特異的な抗原または少なくとも1つの膜貫通ドメインを含むその一部に対して、対象中に免疫応答を作り出す方法であって:
a)前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを含むその一部をコードする遺伝子コンストラクトを提供すること;
b)前記コンストラクトをエキソソーム産生細胞中に導入し、前記抗原または少なくとも1つの膜貫通ドメインを含むその一部をエキソソーム産生細胞中に過剰発現させて、前記抗原または前記その一部を表面に提示している組換えエキソソームを生成すること;
c)前記組換えエキソソームを収集し、前記エキソソームまたはその一部を、前記対象に注入すること;
を含む方法に関する。この方法の好ましい実施態様において、前記抗原はレセプターである。より好ましくは、前記レセプターはSSTR2、CCR7、CXCR4、およびCCR5などのGPCR(Gタンパク質共役レセプター)である。好ましくは、前記エキソソームはさらに、突然変異CD40Lを提示し、前記突然変異は可溶性CD40Lの切断と放出を阻害する。
【0041】
本発明はさらに、生合成的にエキソソームを蛍光団結合脂質でラベルする方法、およびクローニング部位に突然変異を導入することなく挿入をプラスミドにクローニングするための方法を考察する。
【0042】
発明の詳細な説明
本発明は、抗体を生成し抗体レパートリーをスクリーニングする新しい方法を開示する。本発明は、より詳細には標的抗原、共刺激分子および/またはマーカーを表示している自然または合成した膜小胞、好ましくはエキソソームを用いる。本発明を、実験、研究、治療、予防または診断の分野において用いることができる。
【0043】
本発明は、エキソソームターゲティングドメインを含む組換えキメラタンパク質および組換え膜レセプターの、新しい予想外の性質の発見に由来する。より詳細には、エキソソーム指向抗原が、高度に免疫原性であり、強力な液性免疫応答を生み出すことを本発明が示す。さらに、抗原とマーカーを表示するエキソソームを用いることにより、応答細胞の出現度数が低い場合でさえも、抗原特異的な抗体産生細胞を都合良く単離することができることも示す。
【0044】
本発明は、従来の抗体調製方法に比べて、多くの利点を有する。本発明は、免疫原性が弱い抗原、膜タンパク質、または多成分ポリペプチド複合体を取り扱う場合に最も有利である。従来の方法を用いた抗体調製では失敗したか、または有用な抗体を産生しない難しい標的を取り扱う場合に、本発明が特に適切である。また、抗体調製を、大量の抗原を精製する必要なく達成できる。実際、エキソソームを単離するための単一の小規模精製方法(US patent 09/780,748号)を、エキソソームの表面に発現されている外来性抗原の性質に関わらず、用いることができる。このように、抗原調製段階を非常に急速に、即ち通常12時間以内に完了できる。この方法は、速く、多数の試料について平行して実施できる。したがって免疫化のための複数の抗原の同時調製が可能となる。抗原を自然に存在する小胞中に発現させることが、穏和な精製方法と組み合わせて、抗原の自然な立体構造を保つ助けになり、これが有力な医療適用ができる重要な抗体の生成を可能にする。さらに、本発明は、表面に高密度の抗原を含む脂質小胞を生成する。この高密度は、抗原結合活性を増加させることにより、抗体産生を非常に有利にするポリマー状態と比較できる。本発明のさらなる利点は、ポリペプチドがエキソソーム産生細胞によって発現されるため、プロセッシングおよび翻訳後修飾(グリコシル化など)の、自然の経路にゆだねられ得ることである。もう1つの利点は、抗体産生細胞を、応答細胞の生成頻度が低い場合でさえも、単離できることである。このような低い生成頻度は、一般に免疫原性に乏しい抗原または少量の抗原による免疫化の結果起こる。抗原を保持するエキソソームを用いてB細胞を選別することにより、極めて希な抗原特異的抗体産生細胞の単離が可能になる。抗原を保持するエキソソームを用いるB細胞選別の追加の利点は、固有のエピトープに対する抗体や立体構造的エピトープに対する抗体を産生しているB細胞を選択的に単離するために、選別の厳密性と特異性を調整できることである。例えば、あるレセプターを保持する過剰のエキソソームおよび生理的なリガンドに結合している同じレセプターを保持する少量で追跡可能なエキソソームの存在下で実施される減算的選別によって、レセプター/リガンド複合体のみと反応する抗体を産生するB細胞の単離が可能になる。したがって本発明は、他の化合物との相互作用または突然変異/欠失による標的抗原の微妙な立体構造変化を識別できる、高性能の機能的抗体の生成を可能にする。後者の特徴は、他の抗体調製方法に比較して相当な利点である。
【0045】
高度に免疫原性の複合物による免疫化が、独特で希な抗体産生細胞の高度に選択的な選別と組み合わされて、所定のエピトープに対する抗体を生成する効率的で新しい方法を提供する。
【0046】
抗体生成法
この方法では、少なくとも1つの標的抗原を発現している免疫原性小胞、好ましくはエキソソーム、を動物に導入したときに、抗体が生成される。抗体産生は、免疫化された動物の血清を標準的な方法によってテストすることにより評価できる。抗体の調製は、ポリクローナル抗体に対しては血清から抗体をアフィニティー精製することにより、またモノクローナル抗体に対しては抗原特異的な抗体産生細胞を単離することにより、達成できる。後者は好ましくは、下記の抗体レパートリーをスクリーニングするための方法を用いて実施できる。あるいは、モノクローナル抗体を、ハイブリドーマ・スクリーニングおよびSLAMなどの、様々な既知の免疫された動物からのモノクローナル抗体の調製方法を用いて、調製することができる。
【0047】
抗原を表示している免疫原性小胞の調製
抗原を提示している免疫原性小胞は、一般に抗原配列を、エキソソームターゲティングドメイン、好ましくはラクトアドヘリンのC1C2ドメイン、と融合させることにより調製する。免疫原性小胞は、エキソソームやリポソームなどの、自然のまたは合成された小胞である。エキソソームターゲティングドメインを含むキメラタンパク質を保持する小胞の調製方法およびその使用が、WO03/016522に記載されている。
【0048】
ラクトアドヘリンは、殆ど必ずエキソソームに結合して見出される。ラクトアドヘリンのC1/C2ドメインは、エキソソーム表面に対する高度に特異的なターゲティングモチーフを含む。したがって、ラクトアドヘリンのC1および/またはC2ドメインの一部または全体またはその機能的同等物をタンパク質へ導入することにより、その結果得られるキメラタンパク質が、エキソソームや他の脂質構造物を標的とすることが可能になる。
【0049】
したがって、前記エキソソームターゲティングポリペプチドは、ラクトアドヘリンまたは機能的C1および/またはC2ドメインを含むその一部を含む。本発明の実施において、様々な供給源または起源から得られるラクトアドヘリンを用いることが可能である。より好ましくは、前記ラクトアドヘリンまたはその一部は、ヒト以外の哺乳動物ラクトアドヘリンまたはその一部である。哺乳動物ラクトアドヘリンは例えば、マウス、ラット、ウシ、ブタ、ウマのラクトアドヘリンである。より詳細には、前記ヒト以外の哺乳動物ラクトアドヘリン又はその一部は:(i)マウスラクトアドヘリン、(ii)機能的C1および/またはC2ドメインを含むマウスラクトアドヘリンの断片、および(iii)(i)または(ii)のポリペプチドと少なくとも50%の一次構造同一性を有するポリペプチド、から選ばれる。より好ましくは、前記タンパク質が、機能的C1/C2ドメインと融合されている。
【0050】
上記のように、ターゲティング部分は、上の(i)または(ii)のポリペプチドと少なくとも50%の一次構造同一性を有するポリペプチドでよい。同一性は、コンピュータプログラム、好ましくはCLUSTAL法などの様々な既知の技法により決定される。より好ましくは、標的ターゲティングポリペプチドは(i)または(ii)のポリペプチドと少なくとも60%の同一性を、有利には少なくとも70%の同一性を有する。そのようなラクトアドヘリン変異体(または機能的同等物)は、ポリペプチドをエキソソームを標的とする能力を保持しているいるに違いない。この性質は、実施例に記載されているように、例えば、マーカーポリペプチドに融合された前記変異体を含むキメラ遺伝子を作り、この遺伝子をエキソソーム産生細胞中に発現させ、エキソソーム表面のマーカーポリペプチドの存在を確認することにより立証できる。好ましいラクトアドヘリン変異体は、上の(i)または(ii)のポリペプチドと少なくとも85%の同一性を有する。アミノ酸欠失、置換、突然変異および/または付加を含む変異体も可能である。
【0051】
マウスラクトアドヘリンのアミノ酸配列が記載されている(配列番号1;(12)とGenbank Accession n°M38337も参照のこと)。場合によっては、前記ラクトアドヘリンが、配列番号1のアミノ酸配列、または機能的C1および/またはC2ドメインを含むその断片を有する。好ましくは、前記ラクトアドヘリンは、配列番号1の機能的C1/C2ドメインを含むアミノ酸配列を有する。より好ましくは、前記ラクトアドヘリンは、配列番号1のアミノ酸残基111〜266、109〜266、271〜426、111〜426または109〜426を含むアミノ酸配列を有する。
【0052】
場合によっては、前記エキソソームターゲティングポリペプチドは、Del-1、ニューロピリン-1、凝固因子5、または凝集素8の機能的C1および/またはC2ドメインを含む。場合によっては、前記抗原は、標的ターゲティングポリペプチドの上流、下流、または任意の内部ドメイン結合部に融合される。
【0053】
別のある実施態様において、エキソソーム産生細胞および/またはエキソソームターゲティングポリペプチド(好ましくはラクトアドヘリン、または機能的C1/C2ドメインを含むその一部もしくは変異体)が、免疫化に用いた哺乳動物と同じ種に由来する。実際、そのような系においては、エキソソームとターゲティングポリペプチドは免疫原性でなく、抗体はほとんど選択した抗原に対してのみ産生される。
【0054】
ある実施態様においては、エキソソーム産生細胞がマウスの細胞であり、ラクトアドヘリンがマウスのラクトアドヘリンあるいは機能的C1および/もしくはC2ドメインを含むその一部または変異体であり、ヒト以外の哺乳動物がマウスであり、抗原またはエピトープは、例えばヒト起源などの異なる種由来である。マウスがヒト化マウスであって、ヒト化抗体を産生できることがさらにより好ましい。
【0055】
その趣旨で、タンパク質(抗原またはエピトープ)のヌクレオチド配列をマウスのラクトアドヘリンのC1および/またはC2ドメインと融合させることができ、その結果物のキメラ配列を、標準分子生物学技術を用いて真核生物発現ベクター中にクローニングする。キメラタンパク質をコードするプラスミドを、エキソソーム産生マウス細胞系統へトランスフェクションし、トランスフェクションされた細胞を数日培養した後、組換えエキソソームを採取する。組換えエキソソームを、次に蔗糖密度勾配により精製する(US pattent 09/780,748)。組換えエキソソーム上のキメラタンパク質の存在を、ウエスタンブロット分析またはELISAにより確認する。キメラタンパク質を保持する組換えエキソソームを、次に同系のマウスに注入して抗体を生成する。この状況において、キメラタンパク質生成に用いたタンパク質配列中に含まれる抗原決定基のみが、免疫化されるマウス中の外来抗原である。
【0056】
代りのエキソソームターゲティングドメインを、次の工程を含む方法によりスクリーニング、同定、または選択することができる:
− 候補ポリペプチド、好ましくは候補の膜貫通ポリペプチドをコードする第1の遺伝子コンストラクトを提供すること;
− 第1の遺伝子コンストラクトをエキソソーム産生細胞中に導入し、候補ポリペプチドのエキソソームにおける発現をテストすること;
− エキソソームで発現された候補ポリペプチドを選択し、標的ポリペプチドに融合した前記選択されたポリペプチドをコードする第2の遺伝子コンストラクトを調製すること;
− 第2の遺伝子コンストラクトをエキソソーム産生細胞中に導入し、融合ポリペプチドのエキソソームにおける発現をテストすること;および、
− 標的ポリペプチドをエキソソーム中に効率的に発現させるポリペプチドを選択すること。
【0057】
われわれの結果は、エキソソーム上での発現を導く特異的なターゲティング信号を含む種々のタンパク質またはポリペプチドを、上記方法を用いて、同定、選択および/または改良できることを示している。これらのポリペプチドには、エキソソーム中に発現される能力および他の分子をそのような小胞を標的とする能力の双方が必要である。これらのポリペプチドは膜貫通タンパク質に由来し、そのようなタンパク質のすべてまたは一部、一般には少なくとも膜貫通ドメインを含む部分を含めばよい。これらのコンストラクトが、エキソソームへの抗原、特にレセプターおよび膜貫通タンパク質の送達に特に適している。候補の標的ターゲティングポリペプチドは、実際上、レセプター、チャネルなどのような膜貫通ドメインを有するいかなるタンパク質に由来するものでもよい。そのような標的ターゲティングポリペプチドの具体的な例には、MelanA/MARTl、CD40L、CD81などまたはそれらの一部が挙げられる。標的ターゲティングポリペプチドは、膜貫通タンパク質の全体を含んでも、または少なくとも1つの膜貫通ドメインを含むその一部のみを含んでもよい。
【0058】
前記抗原は、例えば、レセプターもしくは酵素のような任意のタンパク質であっても、または、糖脂質、多糖類、薬物、および有機化学物質などのポリペプチド以外の化合物であってもよい。第1の実施態様において、前記抗原は、特性が決定されていないタンパク質である。前記抗原に対する抗体が、前記タンパク質の機能を同定し特性を決定するために必要である。そのような研究法が、ポストゲノムの分野で有用である。実際、全ゲノム配列が知られているが、コードされたタンパク質の機能を見出すことが、今や必須である。さらに、前記抗原はオーファンレセプターであってもよい。第2の実施態様において、前記抗原は稀なものであってもよい。したがって、前記抗原に対する抗体の調製が極めて効率的でなければならない。本発明の方法は、そのような応用に特に適合している。
【0059】
抗原の別の例には、例えば、腫瘍抗原、ウイルス抗原、および微生物抗原がある。腫瘍抗原の実例には、MAGE、BAGE、前立腺腫瘍抗原、および癌遺伝子などがある。これらの抗原のアミノ酸配列それ自体が知られており、組換え技術または合成により生成できる。本発明で標的にされ、または提示される特定の抗原には、可溶性抗原およびレセプターの細胞外ドメインが挙げられる。さらに抗原の例としてリンフォカイン(IL-2、IL-4、IL-13)、栄養因子(TNF、IFN、GM-CSF、G-CSF、など)、酵素、凝固因子、ホルモン、リポタンパク質などがある。
【0060】
特に興味深いタイプの抗原は、少なくとも1つの膜貫通ドメインを有するレセプター、より好ましくはGPCRまたはその一部である。実際、本発明はここで、エキソソームターゲティングポリペプチドを有するかまたは有しない膜貫通ポリペプチドを表示しているエキソソームの調製を可能にしている。小胞内におけるGPCRの発現が、GPCRの精製、特性評価、リガンド(合成の、または自然の)のスクリーニング、抗体の生成などを可能にする。GPCRの具体的な例としては、例えば、SSTR2、CCR7、CXCR4およびCCR5がある。しかし、本発明は他のレセプターにも同様に用いることができる。
【0061】
別の実施態様において、少なくとも1つの膜貫通ドメインを含む抗原を表示している自然の免疫原性小胞を、エキソソーム産生細胞に前記膜貫通タンパク質をトランスフェクションし過剰発現させることにより、調製することができる。実際、われわれは、組換え膜貫通タンパク質がエキソソーム産生細胞により過剰発現される場合には、組換え膜貫通タンパク質がエキソソーム区画に移入されて発現されることを発見した。この予想外の現象が、自然にはエキソソーム上には存在せず、特殊な細胞系統に限られているように見える膜貫通タンパク質について証明された(実施例2に記載の通り)。これは、可溶性タンパク質またはレセプターの細胞外ドメインのエキソソーム上への発現には、タンパク質がエキソソームターゲティングドメインへ融合することが必要であることを証明した以前の結果と対照的である。また、エキソソームを標的とする信号が無い場合には、組換え膜貫通タンパク質は、主に細胞表面に見出され、発現されたタンパク質の一部のみがエキソソーム上に見出される。エキソソーム上に見出される量は、細胞表面に見出されるタンパク質量に正比例しているように見える。
【0062】
さらに別の実施態様においては、組換えエキソソーム上に発現されるタンパク質のエピトーププロフィールが、エキソソームを可溶性化合物と反応させることにより修飾される可能性がある。これは例えば、特異的なペプチドを負荷されたMHC分子またはレセプター−リガンド複合体などの多量体を生み出すことができる。ここでの目標は、MHC/ペプチド複合体には反応するが負荷されていないMHC分子には反応しない制限抗体(restricted antibody)のような抗体を生成することである。MHC分子にペプチドを負荷する方法(直接負荷)が、最近WO01/82958に記載された。制限抗体は、癌細胞および感染細胞のマーカーと反応するので、探し求められている診断用および治療用薬剤である。古典的なMHC分子に加えて、制限抗体を、CD1および古典的でないMHC分子などの他の多形体を含む複合体に対して生成することができる。しかし制限抗体を古くから知られた抗体調製方法によって獲得することは非常に困難である。制限抗体の応用の一例が、HIV感染細胞を殺すことのできる複合物の生成である。より具体的には、これは、HLA-C/HIVペプチド複合体と反応する制限抗体を用いることにより達成できる。実際、HIVが、ウイルスによる養子免疫回避のための多くの手段の1つとして、HLA-AおよびBをダウンレギュレーションすることが示されている。しかし、もしHLA-Cを、HLA-AおよびBサブタイプと共にダウンレギュレーションすると、NK細胞本来の応答をトリガーするので、HLA-Cを保持することもまた、ウイルスの生存には決定的に重要である。したがって、大部分のHIV感染細胞が、HLA-C/HIVペプチド複合体を発現している見込みがあるので、感染細胞上のこれらの標識に対する制限抗体が、HIV患者のHIVの蓄積を除去する強力な手段となる可能性がある。したがって本発明はまた、HIV感染の治療および/または防止のための医薬品組成物を調製するための、HLA-C/HIVに対する制限抗体の使用に関する。本発明はさらに、前記抗体を含む医薬品組成物、および対象にHLA-C/HIVに対する効果的な量の制限抗体を投与することを含む、前記対象のHIV感染の治療および/または予防方法に関する。
【0063】
別の実施例は、レセプターまたはリガンドの活性型には反応し(レセプター/リガンド相互作用による立体構造変化によってトリガーされる)、不活性型のあるいは空のレセプターまたはリガンドには反応しない、また突然変異型抗原に反応して野生型抗原には反応しない抗体を生成するものである。ここでの典型的な実施例は、HIVを無効化する抗体を調製することである。実際、HIV gp120上には、この抗原が標的細胞上のレセプターに結合しているときにのみ、無効化エピトープが見出されている。したがって、gpl20、CXCR4およびCD4、またはgpl20、CCR5およびCD4を含むレセプター/リガンド複合体の生成が、レセプターが仲介するHIVの細胞への結合を阻害する無効化抗体の生成のために必要である。これに関連して本発明は、自然な機能的立体構造を保持している複合体から成る適切な免疫原を調製する効果的な手段、および複合体中に存在する特異的立体構造エピトープに対しては抗体を生成するが、そこに含まれる個々の構成要素に対しては抗体を生成しない効果的な手段を提供する。
【0064】
稀なエピトープに反応する抗体の生成と単離が、さらに下記のような対側性免疫化(contralateral immunization)とB細胞の減算的選別(subtractive sorting)の実行により可能となる。
【0065】
さらに別の実施態様において、抗原を表示している免疫原性小胞はまた、抗原提示細胞へ特異的に抗原を送達するために、アジュバントおよび/またはリガンドを表示することもできる。これはさらに小胞の免疫原性を高める手段となる可能性があり、これが液性応答の強度を増加し、それにより、抗原特異的な抗体産生細胞の度数を増加させる結果となる可能性がある。これに加えて、これが検出可能な液性応答を誘導するのに必要な抗原の量をより低下させもする可能性がある。免疫原性が増加した小胞を、GM-CSF/C1C2などの、エキソソームターゲティングドメインに融合されたアジュバントをコードするプラスミドでエキソソーム産生細胞をトランスフェクションすることにより、調製することができる。エキソソームに連結された最適のアジュバント活性を有するように選択された安定した細胞系統を、薬物選択を用いた標準的な方法により確立することができ、また標的抗原の受容細胞として用いることができる。増加した免疫原性を有し、エキソソームターゲティングドメインを含むキメラのアジュバントを保持する小胞の調製方法とその使用が、WO03/016522に記載されている。
【0066】
あるいは、膜貫通ドメインを有するアジュバントが、上記のように、エキソソーム産生細胞内の過剰発現に続いて、エキソソーム上に表示されてもよい。膜貫通ドメインを含むアジュバントのクラスには、例えば、CD40リガンド(CD40L)発現細胞の表面に膜貫通型に、または循環する可溶型に生成される、強力な免疫刺激剤であるCD40Lが含まれる。われわれは、細胞系統をCD40Lをコードするプラスミドによりトランスフェクションすると、このアジュバントがトランスフェクションされた細胞により生成されるエキソソーム上に発現することを見出した。そのような組換えエキソソームが、実施例4に記載されているように、増大した免疫能力を示した。われわれはまた、このアジュバントの可溶性型を生み出すCD40Lの切断部位を突然変異させることにより、エキソソーム表面から可溶性CD40Lが切断されて放出されることを阻害することができることを見出した。これが、CD40Lの膜貫通型のみを発現しているエキソソームをもたらし、これは野生型のCD40Lを発現しているエキソソームよりもさらに強力な免疫活性を示す。この予想外の現象に対しては、野生型は、CD40Lの完全長型と可溶性型ならびにCD40Lの残りの膜貫通ドメインの混合物を含んでいて、これがCD40Lの機能的な3量体型の効果的な形成を妨げるためである、という説明が可能である。本発明は、切断モチーフが突然変異したCD40Lを含むエキソソームに関する。
【0067】
したがって、本発明は、少なくとも1つの膜貫通ドメインを有するポリペプチドをエキソソームの表面に発現させる新しい方法であって、
1)前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを含むその一部をコードする遺伝子コンストラクトを提供すること;
2)エキソソーム産生細胞中に前記コンストラクトを導入し、前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを含むその一部を過剰発現させて、組換えエキソソームを生成すること;および、
3)その表面に前記遺伝子コンストラクトによりコードされるポリペプチドを保持する前記組換えエキソソームを収集すること;
を含む方法に関する。
【0068】
この方法の好ましい実施態様において、少なくとも1つの膜貫通ドメインを有する前記ポリペプチドは、レセプターである。より好ましくは前記レセプターはSSTR2、CCR7、CXCR4およびCCR5などのGPCR(Gタンパク質共役レセプター)である。あるいは前記ポリペプチドはCD40L、好ましくは突然変異CD40Lであって、前記突然変異が可溶性CD40Lの切断と放出を阻害する。
【0069】
特異的な抗原送達のためのリガンドの例が免疫グロブリンまたはその断片、例えば免疫グロブリンのFc断片である。そのようなFc断片は、エキソソームの表面に発現すると、そのようなFc断片のレセプターを発現している細胞、例えば抗原提示細胞などを、エキソソームが標的とするように作用することができる。そのようなFc断片の発現は、単独であっても、または抗原の発現との組合せであっても、抗原提示細胞、特に樹状細胞によるエキソソームの認識を促進し増強して、そのような抗原のクロスプライミングを増大させる。
【0070】
融合
キメラのポリペプチドまたは複合物を、遺伝的または化学的融合により調製することができる。
【0071】
遺伝子融合のためには、キメラ遺伝子の関心のあるポリペプチドをコードするドメインを、ラクトアドヘリンまたはターゲティングペプチドの上流、下流または任意の内部ドメイン接合部に融合さればよい。さらに、ドメイン同士が互いに直接融合しても、またはキメラポリペプチドの性質を変えないスペーサー領域で隔てられてもよい。そのようなスペーサー領域には、クローニング部位、切断部位、フレキシブルドメインなどが含まれる。その上、キメラの遺伝子コンストラクトがさらにリーダー シグナル配列を含み、コードされたキメラポリペプチドをエキソソーム産生細胞の小胞体へと分泌しやすくしてもよい。その上、キメラ遺伝子が、mycタグ、ポリヒスチジンタグなどの、精製またはモニターを容易にするタグをさらに含んでもよい。
【0072】
化学的融合のために、部分的または全長のラクトアドヘリン配列を選択または修飾して、末端にチオール、アミノ、カルボキシル基などのフリーの反応基を付与し、可溶性のポリペプチド、糖脂質、または任意の小分子を架橋することができる。好ましい実施態様においてラクトアドヘリンのコンストラクトが、少なくとも配列番号1のアミノ酸1〜271をコードする。この配列においてC1ドメイン(アミノ酸111〜266)が、エキソソームターゲティングモチーフを提供し、システイン271が、他の分子と化学的に架橋するための、フリーのチオール残基を提供する。ペプチド、化学物質のSH基との架橋は、十分に確立された方法(13)により実施できる。この方法の利点は、それが、本発明の有効範囲を、糖脂質、薬物および有機化学物質などのポリペプチド以外の化合物に対する抗体の調製へ拡張することである。それは、新しい抗原決定基となる可能性のある基を導入することなく、ポリペプチドおよび化合物をエキソソームにターゲティングさせる手段もまた提供する。選択した架橋試薬は、免疫学的に非活性であることが示されている(13)。新しい抗原決定基がときにはキメラ遺伝子の結合部位で生じ、キメラ遺伝子産物を特異的な予防的および治療的なヒトへの応用に用いることを制限する可能性がある。配列番号1などの適切なラクトアドヘリンコンストラクトを提示しているエキソソーム(またはリポソーム)を産生し、次にそれを連結したい産物と反応させることにより、修飾されたエキソソームまたは脂質小胞(例えばリポソーム)を調製することができる。あるいは、産物に架橋したラクトアドヘリン断片を調製し、続いて精製したエキソソームまたはリポソームに加えてもよい。
【0073】
ベクター
本発明は上記のようなキメラの遺伝子コンストラクトまたはベクターを含む組換え細胞はもちろん、さらに、上記のようなキメラの遺伝子コンストラクトを含むベクターも含む。ベクターは、プラスミド、ファージ、ウイルス、人工染色体などである。代表的な例には、商業的に購入できるプラスミド、特にpUC、pcDNA、pBRなどに由来するプラスミドが挙げられる。他の好ましいベクターが、複製欠損レトロウイルス、アデノウイルス、AAV、バキュロウイルスまたはワクシニアウイルスなどのウイルスに由来する。ベクターの選択は、前記ベクターを使用するべき組換え宿主細胞に従って当業者が決定できる。これに関しては、哺乳動物細胞にトランスフェクションまたは感染させることができるベクターを用いるのが好ましい。実際、好ましい組換え宿主細胞は、哺乳動物細胞である。これは初代細胞、または確立した細胞系統でよい。代表例には、線維芽細胞、筋細胞、肝細胞、免疫細胞など、およびそれらの祖先または前駆細胞が含まれる。最も好ましい哺乳動物細胞が、エキソソーム産生哺乳動物細胞である。これらには、例えば腫瘍細胞、樹状細胞、BおよびTリンパ球または肥満細胞が含まれる。
【0074】
エキソソーム産生細胞
エキソソーム産生細胞には任意の、好ましくは哺乳動物起源の、後期エンドソーム多胞体が原形質膜と融合して生ずるエンドソーム起源の膜小胞を生成し分泌する細胞が含まれる(6)。様々なタイプの組織から得られた細胞、例えば樹状細胞、Bリンパ球、腫瘍細胞、Tリンパ球および肥満細胞などが、エキソソームを分泌することが示されている。治療目的で、または研究用具としてエキソソームを生成し、精製しまたは使用する方法が、例えばWO99/03499、WO00/44389、WO97/05900中に記載されており、参照によりここに取り入れる。本発明の好ましいエキソソーム産生細胞は、哺乳動物腫瘍細胞、哺乳動物BおよびTリンパ球、および哺乳動物樹状細胞であり、一般にはマウスまたはヒト起源のものである。これに関して、細胞は、好ましくは不死化した樹状細胞(WO94/28113)、未成熟樹状細、または腫瘍細胞(WO99/03499)である。さらに、抗体産生のためには、Bリンパ球をエキソソーム産生細胞として用いるのが有利である。何故ならば、その結果得られるエキソソームが、抗体産生を促進するMHCクラスII分子などの、補助的な機能および分子を含むからである。さらに、B細胞由来のエキソソームが、濾胞樹状細胞と結合できることが示された。これは抗体誘導のための、もう1つの重要な特徴である(14)。
【0075】
細胞は、RPMI、DMEM、AIM Vなどの任意の適当な培地中で、好ましくは、培地由来のタンパク質によるエキソソームの汚染を避けるために、タンパク質を含まない培地中で、培養し維持できる。培養は、プレート、シャーレ、チューブ、フラスコなどの任意の適当な装置で実施できる。
【0076】
遺伝子コンストラクト(またはベクター)を、エキソソーム産生細胞に、裸のDNA技術、陽イオン性脂質媒介トランスフェクション、ポリマー媒介トランスフェクション、ペプチド媒介トランスフェクション、ウイルス媒介トランスフェクション、物理的もしくは化学的な試薬または処理、エレクトロポレーションなどの、通常の方法のいずれかを用いて導入することができる。これに関しては、適切なキメラ遺伝子を発現させるには一時的なトランスフェクションで十分であり、したがって安定した細胞系統を作製することが不必要であることに注意するべきである。そのような細胞によって産生されるエキソソームを、遠心、クロマトグラフィーなどの従来知られた方法に従って、収集および/または精製することができる。好ましい技術が、WO00/44389、およびUS patent 09/780,748中に記載されており、参照によりここに取り入れる。
【0077】
接種材料および注入経路
抗原を、好ましくは上記のように調製した組換えエキソソームの形で、動物へ接種すると、抗体が生成される。あるいは、抗原をDNAの形で、または組換えタンパク質の形で投与してもよい。さらに別の可能性は、組換えエキソソーム産生細胞全体を投与することである。
【0078】
抗体は、ポリクローナルでも、モノクローナルでもよい。動物は、マウス、齧歯類、霊長類、ウマ、ブタ、ウサギ、家禽、などを含む様々な種でよい。好ましい動物はマウスである。
【0079】
遺伝子免疫化を、ワクシニア、ポックスウイルス、アデノウイルス、アデノ関連ウイルスなどの様々なウイルスベクター、または種々の脂質性またはタンパク質性組成物を組み合わせたDNAなどの非ウイルスベクターを用いて、または純粋な(例えば裸の)DNAを用いて、実施できる。遺伝子銃またはエレクトロポレーションを含む様々なベクター送達装置または技術が、遺伝子接種のために使用できる。上に指摘したように、遺伝子コンストラクトは、任意のDNAまたはRNA分子、一般にはプラスミド、ウイルスベクター、ウイルス粒子、裸のDNAまたは前記細胞を含む任意の細胞でよい。様々な遺伝子コンストラクトが、単一のベクター中もしくは別々のベクター中に、または任意の組み合わせ中に、含まれることが可能である。
【0080】
タンパク質免疫化もまた同様に用いることができる。この点で、組換えキメラ抗原を精製した形で動物への投与に使用してもよい。
【0081】
そのような投与に続いて、エキソソームターゲティングドメイン、好ましくはラクトアドヘリンのC1および/またはC2ドメインを有するキメラ抗原が生体内で動物自身の循環しているエキソソームに負荷され、それによって免疫応答を誘導することになる。
【0082】
接種組成物は一般に希釈液、緩衝液、等張液などの薬学的に許容できる賦形剤または媒体をさらに含む。組成物はまた上記のような遺伝子免疫化のために、トランスフェクションを容易にする試薬を含んでもよい。組成物はさらにアジュバントを含んでもよい。
【0083】
接種材の投与は例えば静脈内、筋肉内、腹膜内、腫瘍内、皮下、脾臓内、リンパ節内などの全身性注入などの様々な経路により実施することができる。
【0084】
所定のエピトープに反応する抗体を産生する細胞の局所的な度数をさらに増加させるために、対側性免疫化を実施してもよい。この手法は、例えば、MHC/ペプチド複合体には見出されるが、空のMHC分子には見出されない、またはレセプター/リガンド複合体には見出されるがレセプターのみには見出されない特異的なエピトープに対する抗体の調製および単離には、極めて有利である。この手法の別の例は、活性型の化合物には見出されるが不活性型のその化合物には見出されない、または突然変異型の抗原には見出されるが正常型には見出されないエピトープから成る。対側性免疫化は最初、細胞抽出物などのタンパク質混合物中に見出される抗原に向けられた抗体産生細胞を単離する手段として記載された(15)。それは、例えば動物の一方の体側に細胞抽出物のみを注入し、次に後日細胞抽出物のみを同側に、標的抗原を含む同じ細胞抽出物を反対側に同時に注入して動物を増強することにより実施する。この手法で一般に用いられる注入の経路は、動物の左右の足蹠の皮下である。この場合、抗体産生細胞は、動物の標的抗原を受けた体側に位置する膝窩部リンパ節から単離される。本発明においては、動物の一方の体側に抗原を、他方の体側にその抗原の立体構造変異体を注入することにより、対側性免疫化をエピトープ特異的な抗体の調製と単離に適用する。立体構造変異体は、タンパク質タンパク質相互作用、突然変異/欠失、または、新しい抗原決定基を形成する結果となる任意の抗原の修飾の結果として生ずる。本発明においては、対側性免疫化を、以下に記載するB細胞の減算的選別と組み合わせて用いるのが最も良い。
【0085】
抗体レパートリーをスクリーニングする方法
本発明は、抗体レパートリーをスクリーニングする方法であって:1)標的抗原とマーカーを保持する追跡可能な組換えエキソソームを抗体レパートリーと接触させること、および2)抗原特異的な抗体を、抗原を保持するエキソソームを介した前記マーカーとの結合に基づき単離すること、を含む方法をここで開示する。抗体レパートリーの細胞ベースのライブラリー由来の所望の特異性を有する抗体を、組換えDNAとハイブリドーマを含む標準の抗体調製方法により調製することができる。この方法は、上記の抗体生成法に従って免疫化した動物からの抗体の調製に最も適している。
【0086】
「抗体レパートリー」とは、様々な抗体を表示している抗体産生粒子の集団を意味する。「抗体産生粒子」とは、粒子が表示している抗体をコードする核酸を含む粒子を意味する。好ましくは、前記粒子は細胞、酵母またはファージである。より詳細には、前記抗体産生細胞は、プラズマ細胞、ハイブリドーマまたはリンパ球でよい。前記抗体産生細胞はまた抗体分泌細胞であってもよい。
【0087】
抗原とマーカーを表示している追跡可能な小胞の調製
追跡可能な小胞は、上記のように合成のまたは自然の小胞であって、好ましくはエキソソームである。それらは標的抗原を表示し、また例えばタグ、ビオチン、酵素マーカーまたは蛍光団などを介して標準の免疫学的、生化学的、または物理化学的方法によって検出することができる。
【0088】
抗原表示小胞の調製方法は、既に上に記載した。キメラタンパク質を免疫化に用いた場合には、同じタンパク質抗原配列を、伸長したエキソソームターゲティングドメインに融合した第2のキメラタンパク質を調製する。あるいは、タンパク質抗原を異なる種由来のエキソソームターゲティングドメインのホモログに融合してもよい。ここでの目標は、新しい連結部配列を有するキメラタンパク質を作り出し、これにより免疫化に用いるキメラタンパク質の連結部に見出される可能性のある新しい抗原決定基と反応する抗体の検出/選択を回避することである。
【0089】
追跡可能なエキソソームは、抗原表示エキソソームと同様に、エキソソームターゲティングドメインに融合させたマーカー(例えばタグ、酵素、および蛍光タンパク質)をコードするベクターを細胞にトランスフェクションすることにより、またはマーカーが膜貫通部分を含む場合にはより単純に、マーカーを過剰発現させることにより調製できる。抗原とマーカーの両方を表示しているエキソソームを、追跡可能なエキソソームのみを産生している予め確立した安定な細胞系統を用いて調製することができる。ここで用いる安定な細胞系統は、293細胞またはCHOなどの組換えタンパク質を大量に産生する能力のために一般に用いられている任意の実験室細胞系統でよい。この細胞系統は、好ましくは大量のエキソソームを産生し、また免疫化に用いた抗原表示エキソソームを産生する細胞系統とは異なる種に由来するべきである。換言すれば、免疫化された動物例えばマウスに由来する抗体産生細胞を用いて抗体レパートリーのスクリーニングを実施し、そしてこの動物の抗体応答が、マウスの細胞系統由来の組換えエキソソームによる免疫化により誘導された場合には、抗体レパートリーのスクリーニングに用いる標的抗原とマーカーを表示している追跡可能なエキソソームは、マウス以外の種に由来する細胞系統により産生されるべきである。これは、マウス由来のエキソソームタンパク質に対して誘導された可能性がある抗体の選択/単離を回避するためであり、また追跡可能なエキソソームと抗体発現細胞間の抗原抗体相互作用でない相互作用、例えばリガンド−レセプターを介した相互作用を回避するためである。
【0090】
もう1つの実施形態において、追跡可能なエキソソームを、ビオチンなどの活性な試薬をエキソソームに結合させることにより調製できる。
【0091】
別の実施形態において、追跡可能なエキソソームを生合成経路により、例えばエキソソームを含むエンドソーム小胞に優先的に取り込まれるラベルされた脂質を用いて調製できる。最近、網状赤血球を蛍光団結合脂質であるリサミンローダミンBジオレイル-ホスファチジルエタノールアミン(Rh-DOPE)とインキュベーションすると、その結果エキソソームよりも類似した密度を持つローダミンを含む小胞が放出されることが示された(16)。実施例6に示すように、293細胞をRh-DOPEまたはフルオレセイン-DOPEを補充した培地で培養すると、蛍光を有する293細胞由来のエキソソームが生成されることを、我々は見出した。対照的に、樹状細胞の類似の培養は、蛍光性エキソソームを生成しなかった。この不一致は、樹状細胞由来と腫瘍細胞系統由来のエキソソームの異なる脂質組成により説明できる。
【0092】
したがって、本発明は追跡可能なエキソソームを調製する方法であって、次の工程:
1)エキソソーム産生細胞を、蛍光団結合脂質とインキュベーションすること;および、
2)前記蛍光団結合脂質を取り込んだ前記細胞からエキソソームを生成させ単離すること;
を含む方法に関する。
【0093】
好ましくは、前記蛍光団結合脂質は蛍光団結合DOPE(ジオレイル-ホスファチジル エタノールアミン)である。より好ましくは、前記蛍光団結合DOPEはRh-DOPEまたはフルオレセイン-DOPEである。場合によっては、前記エキソソーム産生細胞は293細胞である。
【0094】
抗原特異的な抗体の単離
抗原特異的なモノクローナル抗体の単離を、抗体レパートリーのスクリーニングにより実行する。実行に際しては、抗体産生細胞により発現される細胞表面抗体を、上記のように調製した前記抗原およびマーカーを保持する追跡可能なエキソソームに接触させる。抗体産生細胞は、好ましくは一次のまたは不死化したBリンパ球である。不死化は、Bリンパ球をウイルスおよび癌遺伝子を含む形質転換物質により形質転換するか、または不死化された細胞と融合させてハイブリドーマを作ることにより達成される。抗体産生細胞はまた抗体、好ましくはヒト抗体を発現している原核または真核の組換え細胞のライブラリーでもよい。Bリンパ球は、免疫化した動物、好ましくはヒト免疫グロブリンを発現しているトランスジェニック動物に由来するものでよい。免疫化した動物は、上記の抗体生成のための本発明を用いて、または抗原を表示している組換えエキソソームとは異なる免疫原により調製できる。これらには、アジュバント中の精製された組換え抗原、ヌクレオチド・ベースの免疫原(裸のDNA、ウイルスDNA)および抗原を含む細胞または細胞分画などの、普通に用いられる抗原製剤が含まれる。
【0095】
追跡可能なエキソソームを保持する抗体産生細胞の個体または集団(2以上のプール)の検出および単離が、公知の技術を用い、抗体発現細胞と使用するエキソソーム上のマーカーの性質に従って実行できる。例えば、蛍光性エキソソームと接触している単一のBリンパ球またはBリンパ球の集団を迅速に効率的に単離する方法は、蛍光活性化セルソーター(FACS)を用いて細胞を選別することである。そうするためには、Bリンパ球を含むことが知られている組織または器官、例えば血液、脾臓、リンパ節またはそれらの分画などの細胞を免疫化した動物から収集し、免疫化に用いた抗原を保持する蛍光性組換えエキソソームとインキュベートする。過剰量の無蛍光の親のエキソソームも加えて、Bリンパ球上の抗原特異的部位以外の結合部位をブロックする。次に蛍光性細胞をFACSで分析し、装置の設定を適当に調整することにより単一またはプール(2以上の)の蛍光細胞を単離することができる。
【0096】
別の実施形態において、プラズマ細胞またはハイブリドーマのような、抗体を細胞表面に発現しておらず、細胞外環境に放出される可溶性の抗体のみを産生している細胞もまた、追跡可能なエキソソームを用いて単離することができる。この場合、抗体発現細胞を追跡可能なエキソソームとインキュベートする前に、細胞表面に抗体トラップを形成して、分泌された抗体が放出されたときに捕獲する。抗体トラップは、例えばCD81またはCD45などの遍在する細胞表面マーカーに対する第1のビオチン化抗体、ストレプトアビジンおよびリンパ球の種類に特異的な免疫グロブリンに向けられた第2のビオチン化抗体を含めばよい。例えば、抗体産生細胞がマウス由来であれば、第2のビオチン化抗体はマウスの免疫グロブリンに対するものである。トラップは、細胞を第1のビオチン化抗−細胞表面マーカー抗体およびストレプトアビジンと次々にインキュベーションして調製する。つぎにストレプトアビジン上の残りのフリーのビオチン結合部位を用いて、抗体産生細胞の表面と抗体捕獲部分、即ち第2のビオチン化抗−免疫グロブリン抗体とを架橋することができる。あるいは抗体トラップを、両方のビオチン化抗体を保持するストレプトアビジンを含む予め作られた架橋を用いて調製することができる。抗体トラップは、細胞の培養が細胞内で産生された抗体の放出及び産生細胞の細胞表面でのその捕獲をすることができる間に起こる。抗体分泌細胞の個体または集団(2以上のプール)を検出し単離することが、上記のようにして追跡可能なエキソソームを用いて実行できる。もちろんこの方法を、ビオチン・ストレプトアビジンの組み合わせと同等な手段を用いても実行できる。
【0097】
本発明はまた、免疫化された動物の血液またはその分画に由来する可溶性抗体の単離に関する。可溶性抗体は、個々に、または様々な抗原特異性を有する組換え抗体のプールもしくはライブラリーとして、任意の組換え抗体を産生している発現系から導出できる。ここでは、抗原を発現している組換えエキソソームを、古くから知られたアフィニティー精製法によって抗体を単離する手段として用いる。
【0098】
別の実施形態において、本発明はさらに、エピトープ特異的または立体構造特異的な抗体の単離に関する。そのような抗体は、特定の抗原の変異体型には特異的に反応するが、その抗原の自然型には反応しない。変異抗原および自然抗原は、突然変異した抗原および野生型の抗原、または別のポリペプチド、脂質、DNA、または小分子と接触している抗原およびフリーの抗原によって代表される。後者の例が、MHC/ペプチド複合体および負荷されていないMHCまたは別のペプチドを負荷されたMHCである。本発明の好ましいMHC/ペプチド複合体は、HLA-C/HIVペプチドである。そのような変異抗原および自然抗原の別の類別が、リガンド−レセプター複合体およびフリーのリガンドまたはフリーのレセプターである。本発明の好ましいリガンド−レセプター複合体は、gpl20、CCR5、CD4複合体およびgpl20、CXCR4、CD4複合体である。抗原の変異体はまた、特定の抗原の非恒常型を生み出す生理的過程の結果である場合がある。これらにはキナーゼ、プロテアーゼ、グリコシラーゼ、およびホスファターゼなどの修飾酵素を含んでいる活性化/不活性化経路により誘導される、抗原の一時的な立体構造状態が含まれる。エピトープ特異的または立体構造特異的抗体の単離を減算的スクリーニングにより実行できる。この場合、標的となるエピトープを保持する変異型抗原が追跡可能なエキソソーム上に発現されており、標的となるエピトープを欠く前記抗原の自然型が追跡不可能なエキソソーム上に発現されている。後者は抗体産生細胞に過剰に加えられると、前記抗原の変異型と自然型に共通のエピトープに反応する抗体を産生している細胞上の結合部位をブロックすることができる。したがって、変異型抗原を発現している追跡可能なエキソソームおよび過剰量の自然型抗原を発現している追跡不可能なエキソソームを同時に加えると、変異型抗原に見出される特異的なエピトープに向けた抗体を産生している細胞が追跡可能なエキソソームと反応する可能性がさらに増す。こうして、エピトープ特異的または立体構造特異的、を選択的に単離することができる。エピトープ特異的または立体構造特異的抗体の減算的単離を、抗体産生細胞が上記の対側性注入を受けて免疫化された動物由来である場合に用いると、さらに良好である。何故なら、これらの動物から収集された細胞集団はまた、エピトープ特異的または立体構造特異的な抗体産生細胞に富むからである。
【0099】
抗原特異的抗体の生成
次に、上記のようにして単離された抗体産生細胞により産生された免疫グロブリンの重鎖および軽鎖の少なくとも可変ドメインをコードするヌクレオチド配列を、組換え抗体の全長または断片として産生させるために、発現ベクター中にクローニングすることができる。これを行うために、選択した1個またはプール(2個以上)の抗体産生細胞からRNAを抽出し、抗体をコードしているヌクレオチド配列を、標準のRT-PCR法により適当なプライマーを用いて増幅する。PCR産物を次に発現ベクター中にクローニングする。免疫グロブリンの可変ドメインのみを増幅するときは、クローニングは、好ましくはヒト由来の免疫グロブリンの適合するリーダー配列ならびに重鎖および軽鎖の定常ドメインの全長または一部を既に含む受入れベクターに行われる。次に組換え抗体またはその断片の生成が、適合する免疫グロブリンの重鎖および軽鎖をコードするDNAプラスミドの混合物を細胞にトランスフェクションすることにより実行される。あるいは、両鎖を、2つの遺伝子転写物を同時に発現できる独特のプラスミド中にクローニングしてもよい。重鎖および軽鎖のPCR産物が2個以上の抗体産生細胞から得られるときには、重鎖および軽鎖DNAプラスミドの種々の組合せのマトリックスができるため、適切な抗原特異性を有する免疫グロブリンを再構成することができる。組換え抗体を発現させるための受容細胞は、CHO、293または骨髄細胞系統などの、組換えタンパク質を生成できる任意の細胞系統でよい。組換え免疫グロブリンの発現を、ELISAなどの標準的方法によって測定することができる。組換え免疫グロブリンの抗原特異性もまた、標的抗原の性質と利用可能性に従う標準の免疫学的方法により確認される。例えば、ELIZAを上記の抗原を発現している組換えエキソソームを用いて実施することができる。抗体配列をさらに組換えDNA技術により操作して、標的抗原に対する抗体の親和性を増すことや(抗体熟成として知られる過程)、医療適用のために抗体をヒト化することもできる。
【0100】
別の実施形態において、本発明は免疫グロブリンの可変配列をクローニングするためのレシピエントベクターの使用に関する。このベクターは、BsmB IなどのII型の制限酵素によって認識される2つの制限部位を備えるカセットを含む。これらのII型の酵素は、その認識部位の外側で2重鎖DNAを切断し、これが挿入配列のオーバーラップした塩基を用いたDNA断片の挿入を可能にする。他方、従来のクローニングは通常、挿入の両端に付加される制限部位のオーバーラップした塩基を用いる。したがって、本発明により、免疫グロブリンのリーダー配列と定常ドメインの間に可変配列をクローニングすることが、クローニング部位に突然変異を導入することなく可能になる。対照的に、大部分の従来の可変配列のクローニング方法は、組換え免疫グロブリンの親和性を修飾する可能性のある突然変異を導入する。実施例10がさらに、受入れベクターおよび本発明の利点を詳述する。
【0101】
したがって、本発明は、クローニング部位に突然変異を導入することなく、免疫グロブリンのリーダー配列と定常ドメインの間に可変配列をクローニングする方法であって、次の工程:
1)前記リーダー配列、II型制限酵素により認識される2つの制限部位を備え、制限酵素切断後に制限部位が前記ベクターから除去されるように構成されたクローニングカセット、および前記定常ドメイン、を順番に含むベクターを提供すること;
2)前記可変配列を提供すること;
3)前記ベクターを前記制限酵素とインキュベーションすること;
4)前記可変配列と工程3で得られた前記ベクターをライゲーションすること;
5)前記リーダー配列と前記定常配列の間に前記可変配列を含む前記ベクターを単離すること;
を含む方法に関する。
【0102】
別の実施形態において、単離された抗体産生細胞を、適当な培地中で培養することにより、生体外で増殖させることができる。あるいは、これらの細胞を上記の形質転換または融合により不死化することもできる。これにより生成される抗体を、次にProtein-Gビーズを用いたクロマトグラフィーなどの公知の方法により、培養上清から精製する。
【0103】
さらに別の実施形態において、ポリクローナル抗体を、例えば固体支持体に結合した組換えエキソソームを用いるアフィニティー精製により、血清から精製できる。抗原特異的な相互作用により固体支持体に結合した抗体は、標準の抗体溶出法、すなわち、低pHを用いて溶出できる。
【0104】
より一般的な観点から、本発明はクローニング部位に突然変異を導入することなくベクター中に配列をクローニングする方法であって、次の工程:
1)II型制限酵素により認識される2つの制限部位を備え、制限酵素切断後に制限部位が前記ベクターから除去されるように構成されたクローニングカセットを提供すること;
2)適合する末端を有する前記配列を提供すること;
3)前記ベクターを前記制限酵素とインキュベートすること;
4)前記配列と工程3で得られた前記ベクターをライゲーションすること;
5)前記配列を含む前記ベクターを単離すること;
を含む方法に関する。
【0105】
次の実施例は、説明のために提供されるのであって、制限のためではない。
【実施例1】
【0106】
エキソソームは抗体生成のための有力な新しい媒体である。
【0107】
9匹のBalb/Cマウスを、3匹のマウスからなる3つの免疫化グループに分けた。各マウスを、PBS中の〜20 ng組換えヒトラクトアドヘリン(グループ1)、1:1 PBS/完全フロイントアジュバント混合物中の〜20 ng組換えヒトラクトアドヘリン(グループ2)、またはPBS中の〜20 ngヒトラクトアドヘリンを含む組換えWEHIエキソソーム(グループ3)の腹腔内投与により免疫化した。
【0108】
ヒト組換えラクトアドヘリンを、(His)6タグ(配列番号2)と融合した全長の組換えヒトラクトアドヘリンをコードするpcDNA6hLactlf/HisプラスミドによりトランスフェクションされたCHO細胞が生成した組換えエキソソームから調製した。pcDNA6hLactlf/Hisを次のようにして調製した。ヒトラクトアドヘリンcDNAの2つのオーバーラップした断片を、造血細胞由来のcDNAから、それぞれプライマー対LTDNfl5(配列番号3)/LTDNr8(配列番号4)およびLTDNf2(配列番号5)/LTDNrl3(配列番号6)を用いて増幅した。LTDN/fl5およびLTDNrl3の5’端を、Hind IIIおよびAge I制限部位を含めるために延長した。一方LTDNr8およびLTDNf2はEcoR I部位を含んでいた。LTDNf2/LTDNrl3を用いたラクトアドヘリンcDNAの3’端断片の増幅によって複数の生成物が生み出され、最長のものはラクトアドヘリンcDNAと一致した。5’端断片をHind IIIとEcoR Iで消化し、一方3’端断片をAge IとEcoR Iで消化した。5’端と3’端断片を互いにライゲーションし、そしてpcDNA6hLactlf/Hisを生じるようにHind IIIとAge Iによって予め切断しておいたpcDNA6A-His(Invitrogen)にライゲーションした。このプラスミドを、リポフェクタミン(Invitrogen)を用いて、ハムスター卵巣細胞系統(ATCC)であるCHO細胞にトランスフェクションした。完全培地(2 mM L-グルタミン、100 U/mlペニシリン、0.1 mg/mlストレプトマイシン、および2%ウシ胎児血清(FBS)を補充したCHO-SFM)中、37℃、5% CO2雰囲気下での培養1日目に、安定してトランスフェクションされた細胞を、2μg/mlブラストサイジンを補充した培地中に選別した。4日間培養後、安定なクローンを限界希釈技術により単離した。大量のラクトアドヘリンを生成しているクローンを、エキソソームに発現されている組換えラクトアドヘリンのウェスタンブロット分析により、次のようにして選択した。培養上清を集め、続いて200 gで遠心し、0.2μmフィルターで濾過して細胞片を除いた。次に澄んだ上清を4℃で、100,000 g 90分間遠心し、エキソソームを沈殿させた。沈殿を100 μl氷冷PBSに再懸濁した。8μlのSDS-PAGE試料緩衝液5X(SB)を、PBS中のエキソソーム32μlに加え、100℃で5分インキュベートし、次にSDS-PAGEで分析した。ゲル上のタンパク質を、半湿式エレクトロトランスファーにより、PVDFメンブレンに移動させた。試料中のヒトラクトアドヘリンの存在を、ヒトラクトアドヘリンのRGDモチーフに対するポリクローナル抗体(Dr.Sebastian Amigorenaから寄贈)の1/2500希釈液を用いた免疫検出により確かめた。ラクトアドヘリンに結合した抗体を、西洋わさびペルオキシダーゼと結合した二次抗−ウサギIgG抗体(Jackson ImmunoResearch)の1/5000希釈液と比色用基質(CN/DAB, Pierce)を用いて検出した。CHO-3.2クローンをラクトアドヘリン生成用に選択し、1Lスピナーフラスコ中のFBSを含まない完全培地に移して、ラクトアドヘリンの大量産生のために増殖させた。7日間細胞培養した上清を、250 ml遠心ボトルに移し、2000 rpmで5分間遠心して細胞を沈殿させた。次に、上清を0.2μmフィルターで濾過し、500 KDカットオフサイズのファイバーカートリッジを用いて100 mlに濃縮した。濃縮した上清を次に、100,000 g、1時間15分間4℃で遠心した。エキソソームを含む沈殿を、1mlのMLB II(50mM NaP04 pH 8/300mM NaCl/lOmMイミダゾール/0.5% Tween)に再懸濁し、2 mlのNi-NTAスラリー(予めEtOHを除くため遠心した)を含むチューブに移した。2〜3時間4℃でシェ−カーによりインキュベーションした後、試料をBioRadカラムに注ぎ、4℃で保持した。カラムを10 ml MWBI(50mM NaP04 pH 8/300mM NaCl/20mMイミダゾール/0.05% Tween)で、次に20 ml MWBII(50mM NaP04 pH 8/500mM NaCl/20mMイミダゾール)で洗浄した。カラムに結合したタンパク質を、8 ml MEBII(50mM NaP04 pH 8/300mM NaCl/250mMイミダゾール)で溶出した。溶出したタンパク質を、Millipore Ultrafree-4 10,000 MWCO装置を使用して濃縮し、緩衝液をPBS pH 7.4に交換した。タンパク質試料を小分し、-20℃で保存した。この過程によって、高度に精製された組換えラクトアドヘリンが得られる。
【0109】
マウスの細胞系統WEHIに由来し、組換えヒトラクトアドヘリンを発現しているエキソソームの調製を、CHO細胞系統を用いて、一時的にトランスフェクションした細胞を4日培養した後にエキソソームを収集した点を除いては正確に上記の通りに行った。免疫化グループ1〜3に注入する試料を、マウス当たり注入される組換えラクトアドヘリン量に対して正規化した。正規化を、組換えラクトアドヘリンと組換えエキソソームの順次希釈を利用した、上記のウエスタンブロット分析により確かめた。
【0110】
動物は、最初の注入の2週間後にグループ2を除いて同じ試料を用いて1回のブーストを受けた。グループ2では抗原を1:1 PBS/不完全フロイントアジュバント混合液に再懸濁した試料を用いた。2回目の免疫化後に、動物から採血し、ELISAによって抗ヒトラクトアドヘリン抗体をテストした。ELISAのために、PBSに溶かした50 ngヒトラクトアドヘリンでマイクロ滴定プレートのウエルを37℃で1時間コートした。PBS中に0.05%Tween-20と6%脱脂粉乳を含むブロッキング緩衝液を1時間室温(RT)でウエルに加え、残っているフリーの結合部位を飽和させた。次にウエルを1時間RTで、ブロッキング緩衝液で1/1000希釈した免疫化マウスの血清とインキュベーションした。ウエルを3回ブロッキング緩衝液で洗浄した後、結合した抗体を、1/10000希釈の西洋わさびペルオキシダーゼ結合二次抗−マウスIgG抗体(Jackson ImmunoResearch)とECL基質(Amersham)を用いて検出した。このELISAの結果を図1に示す。
【0111】
結果:
抗ラクトアドヘリン抗体が、ヒトラクトアドヘリンで覆われたマウスエキソソームを用いて免疫化したマウスの血清中で検出され、一方ヒトラクトアドヘリンを単独で、またはフロイントアジュバント中の乳濁液として与えた場合は、抗体応答が生成されなかった。フロイントアジュバントを用いた場合は、4回の接種材料注入後でさえも抗体が検出されなかったが、一方でラクトアドヘリンを保持するエキソソームを与えられたマウスの血清中の抗体力価は、以降の注入により増加した(データを示さず)。
【0112】
結論:
抗原を保持するエキソソームは、アジュバント無しで高度に免疫原性であり、極めて少量であってフロイントアジュバントなどの古くから知られた強力なアジュバントでは効果がない程度の量の抗原を用いて、抗体応答を誘導することができる。したがって、抗原を表示している組換えエキソソームは、前記抗原に対する抗体を生成するための強力な手段である。
【実施例2】
【0113】
組換え膜タンパク質を発現しているエキソソームを生成するための方法
【0114】
Gタンパク質共役レセプター(GPCR)の1つであるソマトスタチンレセプター(SSTR2)をコードし、ヒト脳RNA(Clontech、CA)に由来するcDNAを、プライマーSSTR2f1(配列番号7)およびSSTR2r2(配列番号8)を用いたPCRにより増幅した。プライマーは、SSTR2f1についてはHind III制限部位を有し、SSTR2r2についてはNot I制限部位を有する5’端の延長部分を含んでいた。Hind IIIおよびNot Iによる消化に続いて、PCR産物を、修飾されたpcDNA3.1(Invitrogen, CA)であるHApC3.1にクローニングし、発現ベクターHApC3.1/SSTR2(配列番号9)を調製した。HApC3.1は、合成により得られ(Genset, CA)、pcDNA3.1ベクターのNhe I部位とHind III部位の間にクローニングされたHAタグ挿入(配列番号11)を含む。HAタグは、開始コドンおよびそれに続くヘマグルチニン抗原配列由来の一連のコドンをコードし、そしてHApC3.1/SSTR2は、N-末端におけるHAタグとの組換えSSTR2であるHA-SSTR2(配列番号10)をコードする。HApC3.1/SSTR2を、マウスの細胞系統EL-4およびB3-Zに、エレクトロポレーション(BioRad GenePulserエレクトロポレーターで、220V、950μF条件下)によりトランスフェクションした。トランスフェクションされた細胞を、1 mg/mlの抗生物質G418(Invitrogen)を補足した選択培地(RPMI 1640−2 mM L-グルタミン、100 U/mlペニシリン、0.1 mg/mlストレプトマイシン、1 mMピルビン酸ナトリウム、および10%ウシ胎児血清(FBS))中で、37℃、5%CO2雰囲気下、7日に亘り培養した。細胞をFACS分析のために集め、一方培養上清をエキソソーム調製用に収集した。FACS分析のために、106のトランスフェクションされた細胞と親細胞を1時間4℃下1 mlのPBS/5% FBS中で、FITC(Roche, CA)に結合された抗−HA抗体とインキュベーションした。PBS/5% FBS中で数回洗浄した後、細胞をFACSにより分析し、その表面のHA-SSTR2の発現を評価した。EL-4およびB3-Z細胞上のHA-SSTR2発現のプロフィールが、それぞれ図2Aおよび2Bに示されている。
【0115】
エキソソームを、培地上清から実施例1に記載の通りに調製した。エキソソーム産生とHA-SSTR2の存在もまた、実施例1に記載した通りに、抗体を検出する際に抗アクチン抗体(Sigma、MO)および抗HA抗体(HRP-結合体、Roche、CA)を用いた、ウエスタンブロット分析により測定した。組換えEL-4およびB3-Zから得られたエキソソームを、図3のレーン1と2で分析した。検出用抗体は、図3のパネルAでは抗アクチン抗体で、パネルBでは抗HA抗体であった。
【0116】
CXCR4、CCR5、およびCCR7を含む他のGPCRをコードする一団のcDNAのもまた、SSTR2に対する上記の同じクローニング戦略を用いて、HApcDNA3.1中にクローニングした。プライマー対CXCR4fl(配列番号10)/CXCR4r2(配列番号11)、CCR5fl(配列番号12)/CCR5r2(配列番号13)およびCCR7f7(配列番号14)/CCR7r8(配列番号15)を用いて、それぞれCXCR4、CCR5およびCCR7をコードするPCR産物を、ヒト造血細胞培養物由来のcDNAを使用して生成した。その結果得られるプラスミドHApcDNA3.1/CXCR4(配列番号16)、HApcDNA3.1/CCR5(配列番号20)およびHApcDNA3.1/CCR7(配列番号22)は、それぞれ組換えタンパク質:HA-CXCR4(配列番号19)、HA-CCR5(配列番号21)、およびHA-CCR7(配列番号22)をコードする。HApcDNA3.1/SSTR2はもちろん、これらのプラスミドを、ヒトの胚細胞系統293細胞へ、エレクトロポレーション(220V, 950μF、BioRad GenePulserを使用)により、トランスフェクションした。安定したトランスフェクションされた細胞が、4 mM L-グルタミン、100 U/ml ペニシリン、0.1 mg/mlストレプトマイシン、2% FBSおよび、250μg/ml G418を補充した、293 -SFM培地の培養物中に確立された。HAタグを有する組換えタンパク質の、トランスフェクションされた細胞の細胞表面およびエキソソーム上における発現を、それぞれ、上記のFACS分析および捕獲ELISAによりモニターした。トランスフェクションされた293細胞上のHA-GPCR発現のプロフィールを、図4に示す。捕獲ELISAのために、マイクロ滴定プレートのウエルを、100 ngの抗−CD81捕獲抗体(PharMingen)で、37℃下1時間コートした。プレートを200μl/ウエルのPBS/0.05% Tween20で3回洗浄し、次に室温(RT)で1時間200μL/ウエルのDPBS/1% BSAとインキュベートした。洗浄工程に続いて、実施例1記載のようにして調製した精製エキソソームをウエルに加えた。終夜RTで振盪インキュベーションの後、ウエルを3回洗浄緩衝液で洗浄した。次に検出用抗体をウエルに加え、引き続いて、2次抗体−HRP結合体、またはビオチンに結合した検出用抗体を用いる場合はストレプトアビジン-HRPを加え、その後化学発光基質(Amersham)を加えて、測定した。エキソソームを定量化するための正規化定量を、エキソソーム標品の逐次希釈およびHRPに結合した検出用抗−CD81抗体を用いたCD81の測定に基づいて行った。次に第2の定量を実行し、等量で非飽和量のエキソソームと抗−HA抗体とを用いて組換え抗原の発現を評価した。第2の定量の結果を、図5に示す。
【0117】
結果
図2が、抗−HA抗体が、HApcDNA3.1/SSTR2をトランスフェクションされたEL4およびB3Zの両方の表面において、HAタグを含む組換えタンパク質を特異的に検出したが、一方でこの抗体は親のEL4およびB3Z細胞には結合しなかったことを明らかにしている。HA-タグを含むタンパク質はまたウエスタンブロットによって、安定にトランスフェクションされたEL-4細胞由来のエキソソーム中に検出することができた(図3A、レーン1)。これは、エキソソー指向ドメインとの融合を必要とせずに、組換えSSTR2がエキソソーム上に発現され得ることを裏付けている。対照的に、同じ分析によって、安定にトランスフェクションされたB3Z細胞由来のエキソソーム中にHA-SSTR2を検出することができなかった(図3A,レーン2)。両細胞系統によってエキソソームが生成されることが、エキソソームの恒常的成分であるアクチンに対する抗体を用いたウエスタンブロット分析によって証明された(図3B、レーン1および2)。類似の結果が、細胞上とエキソソーム上のHA-SSTR2発現を検出するために、抗−SSTR2抗体を用いた時にも得られた(データを示さず)。
【0118】
293細胞のHA-pcDNA3.1/GPCRによるトランスフェクションもまた、トランスフェクションされた細胞の細胞表面にHAタグを含むタンパク質を発現させる結果となった。図4が、それぞれ、HA-SSTR2、HA-CXCR4、HA-CCR5、およびHA-CCR7を発現している、4つの安定なトランスフェクションされた細胞の染色プロフィールを示す。4つのプロフィールの平均の蛍光が、発現の相対レベルは:SSTR2 > CXCR4 > CCR7 > CCR5 であることを示している。捕獲ELISAが、EL-4細胞と同様に、トランスフェクションされた293も、HA-GPCRを保持するエキソソームを生成することを明らかにした(図5)。抗−CD81抗体についての結果が示すように、様々なトランスフェクションされた細胞によって生成される正規化した量のエキソソームを用いた。したがって、検出されたGPCRの量が、エキソソーム当たりに発現されたGPCRの量に正比例した。注目すべきことに、細胞表面と、エキソソーム上に発現されるGPCRの相対レベルに関して、SSTR2 CXCR4>CCR7>CCR5という、同じ順序が見出された。
【0119】
結論
自然にはエキソソームに存在しないGPCRを含む膜貫通タンパク質を、膜タンパク質をコードするDNAで細胞がトランスフェクションされた後にエキソソーム上に検出できる。この現象は、膜貫通タンパク質配列のエキソソームを標的とする配列との融合を必要とせず、受容細胞に依存するように見える。これは組換え膜貫通タンパク質の過剰発現による可能性があり、過剰のレセプターの分泌の別の経路として働く可能性がある。トランスフェクションされた細胞によって産生される組換えタンパク質のわずかな一部が、エキソソーム上に発現され、大部分のタンパク質が細胞表面に向けられていることに注意すべきである。それにも関わらず、この方法が、検出できる量の全長のレセプターをエキソソーム上に発現させ、したがって、そうでなければエキソソーム上に見出されない膜貫通タンパク質をエキソソームに表示させるために、または既知のエキソソーム膜貫通タンパク質をさらに豊富にするために、この方法を利用することができる。
【実施例3】
【0120】
組換えMHC I/ペプチド複合体を発現するエキソソームを生成するための方法
【0121】
A2-陽性ヒト白血球から逆転写によって導出されるヒト白血球抗原(HLA)A201のα鎖のcDNAを、プライマーHLA2f1(配列番号17)およびHLA2r2(配列番号18)を用いるPCRにより増幅した。プライマーの5’端を延長し、HLA2f1に対してはHind III制限部位を、HLA2r2に対してはBstB I制限部位を含むようにした。PCR産物をHind IIIおよびBstB I酵素で消化し、Hind IIIおよびBstB Iで予め切断したpcDNA6-Myc/Hisにライゲーションして、pcDNA6-A2-Myc/His(配列番号19)を生成した。
【0122】
同様に、HLAのβ鎖(β2-ミクログロブリン即ちβ2M)配列を、RT-PCRによりプライマーβ2-MICfl(配列番号20)およびβ2-MICr3(配列番号21)を用いて増幅し、そしてpcDNA6-Myc/His中にクローニングしてpcDNA6-β2M(配列番号22)を生成した。次に、pcDNA6-β2M中のβ2M挿入を、pcDNA6とは異なる抗生物質耐性遺伝子を有するpcDNA3中にサブクローニングした。この目的のために、挿入を、プライマーβ2-MICflおよびNot I5’端延長をもつプライマーであるpcDNA6r4(配列番号23)を用いて増幅した。Hind III/Not I消化に続いて、PCR産物を、同じ酵素で予め切断したpcDNA3にライゲーションして、pcDNA3-β2Mを生成した。
【0123】
ヒト293細胞を、pcDNA6-A2-Myc/HisおよびpcDNA3-β2Mの両方により、上述のようにエレクトロポレーションを用いてトランスフェクションした。トランスフェクション2日後、A2およびβ2Mの両方を発現している安定な二重トランスフェクション細胞を確立するために、細胞を二重の抗生物質、即ち2μg/mlブラストサイジンおよび250μg/mlネオマイシン(G418)の選択下に置いた。選択下に2週間置いた後、細胞の全体集団を、上記のようにエキソソームを調製するために増大させた。
【0124】
規格化したエキソソーム試料上の、ヒトA2およびβ2Mの発現を、実施例2に記載したように捕獲ELISAにより評価した。293由来のエキソソーム上のHLA-A2へ、ペプチドが負荷されることが、直接ロード法を用いて証明された。この方法により、機能的MHCクラス1/ペプチド複合体が生成されることが示されている(WO01/82958)。手短に述べると、試料を、pH5で、100μg/mlビオチン化基準肝炎BペプチドFLPSDCFPSVおよび20μg/mlのβ2M(Sigma)と1時間インキュベーショした。次にエキソソームを1%NP4Oに溶解し、抗−MHCクラス1抗体を捕獲抗体として用い、またストレプトアビジンでラベルされたユーロピウムを、結合したビオチン化標準ペプチドの検出に用いて、捕獲ELISAにより分析した。捕獲ELISAによる組換えタンパク質発現およびペプチド負荷の測定結果を、それぞれ図6と7に示す。
【0125】
結果
293細胞を、HLA-A2をコードしているプラスミドでトランスフェクションした結果、トランスフェクションされた細胞が生成したエキソソームに組換えA2が発現した(図6)。期待通りに、親の293由来のエキソソームもまた、293がヒトHLA-A2+細胞系統であるので、内在性のHLA-A2を含んでいた。しかし、正規化した試料を用いた分析が、組換えエキソソームが、親のエキソソームに比べて〜5倍多くHLA-A2を含んでいたことを示す。対照的に、組換え293細胞によるβ2Mの全発現は有意に増加したけれども(データを示さず)、β2Mの量は、親のエキソソームと組換えエキソソームで同程度であった。MHCクラスI複合体のβ鎖は可溶性タンパク質であるため、そのエキソソームへの結合は、そのα鎖との相互作用のみに仲介される。したがって、われわれの結果は、エキソソーム上のαβ鎖複合体の総量は一定のままであるが、α鎖のサブタイプは組換えHLA-A2サブタイプの方へシフトするということを示唆している。図7に示すように、A2特異的標準ペプチドを組換えエキソソーム上には検出できたが、親のエキソソームを用いた場合は、測定値はバックグラウンドよりわずかに上に過ぎなかった。この定量におけるペプチド検出のバックグラウンドレベルを、A2-のドナー由来のDCが生成するエキソソームを用いて測定した(図7の、A2陰性Dex)。このデータは、直接ロード法を用いて組換えHLA-A2にA2特異的ペプチドを負荷できることを示している。
【0126】
結論
エキソソーム産生細胞にMHCクラスIをコードするプラスミドをトランスフェクションすると、組換えMHCクラスI分子がエキソソームに発現される。この方法が、独特のMHCクラスIサブタイプを保持するエキソソームの調製を可能にする。直接ロード法と組み合わせることにより、それが、サブタイプ特異的なMHCクラスI/ペプチド複合体が豊富なエキソソームを生成する強力な手段を提供する。これらのエキソソームは、特異的なMHCクラスI/ペプチド複合体に限定された抗体を調製するための有力な媒体である。
【実施例4】
【0127】
CD40L活性を表示する強化されたエキソソームを生成するための方法
【0128】
ヒトCD40L cDNAを、活性化されたT細胞RNAのRT-PCRにより、プライマーCD40Lf8(配列番号24)およびCD40LrlO(配列番号25)を用いて増幅した。プライマーCD40Lf8を、5’端で延長してBamH I制限部位を含めた。一方CD40Lr1Oは、BstB Iの5’端延長を含んでいた。PCR産物をBamH IおよびBstB Iで消化し、同じ制限酵素で予め切断したpcDNA6-Myc/Hisにライゲーションして、pcDNA6-CD40L(配列番号26)を作製した。次に、pcDNA6-CD40LのCD40L挿入を鋳型として用いて、5’端および3’端のオーバーラップした断片を生成した。5’端断片をプライマーCD40Lf8およびCD40Lr21(配列番号35)を用いて生成し、一方3’端断片をプライマーCD40Lf20(配列番号36)およびCD40LrlOを用いて生成した。CD40Lf20とCD40Lr21は、互いに相補的であって、CD40Lの112と113番目の残基をコードする配列を含むCD40Lのセンスおよびアンチセンスドメインに由来する。この部位のタンパク質分解切断が、可溶性CD40Lの放出をもたらす。プライマーを、野生型CD40Lではグルタミン酸とメチオニンである112と113番目の残基を、2個のグリシンに変化するように設計して、この部位で切断できずに膜貫通型のまま残るCD40Lの突然変異型を作製した。112と113位置の突然変異型CD40L(mutCD40L)をコードする全長の配列を、5’端と3’端断片の混合物を鋳型とし、プライマー対CD40Lf8/CD40LrlOを用いたPCRにより生成した。その結果得られた生成物を、上のようにpcDNA6の中にクローニングして、pcDNA6-mutCD40L(配列番号37)を生成した。pcDNA6-CD40LおよびpcDNA6-mutCD40Lは、それぞれ全長のCD40L(配列番号34)およびmutCD40L(配列番号27)をコードする。トランスフェクションされた293細胞由来の組換えエキソソームを、実施例2および実施例3に記載したようにして調製した。
【0129】
組換えCD40Lおよび突然変異CD40Lの発現を、ウサギ抗−CD40L抗体(Santa Cruz Biotechnology)を検出用抗体として用いて実施例1と2に記載の通り、ウエスタンブロット分析によりモニターした。また定量を実行して、組換え293がCD40L様活性を獲得したかどうかを、樹状細胞(DC)の成熟を刺激することにより評価した。エキソソームを、それが発現した、1)CD81の量、2)CD40Lの量に基づき、実施例2に記載した交差捕獲ELISAを用いて正規化した。正規化した量のエキソソームを、IL4/GM-CSFを含むAIM V培地で成育した7日目のDC培養液に加えた。終夜培養後、細胞表面のCD83発現を測定してDCの成熟を評価した。これは実施例2に記載したように、FITCを結合したモノクローナル抗−CD83抗体を用い、標準FACS分析により実施した。抗−CD83および抗−CD86抗体による二重染色を、DC細胞(CD86陽性細胞)の中でCD83も発現している数を特定するために、実行した。CD40L発現のためのウエスタンブロット分析およびDC成熟実験のためのFACS分析の結果を、それぞれ図8および9に示す。
【0130】
結果
組換えエキソソームのウエスタンブロット分析が、293細胞をpcDNA6-CD40LおよびpcDNA6-mutCD40Lでトランスフェクションすると、CD40Lがエキソソームに発現されたことを明らかにした(図8)。CD40Lを発現している293細胞由来のエキソソームは、全長のCD40L(レーン2のFL)を、CD40Lの可溶性型(レーン2のSF)およびCD40Lの膜貫通ドメインを含む残存N端末端部(レーン2のTM)から成るそのタンパク切断質分解産物と共に、含んでいた。対照的に、mutCD50Lを発現している293細胞由来のエキソソームは、全長型(レーン3のFL)のみを含んでいた。親の293細胞由来のエキソソームでは、タンパク質が検出されなかった(レーン1)。
【0131】
細胞染色およびFACS分析により、DC成熟のマーカーであるCD83陽性DCの数が、DCをCD40LおよびmutCD40Lを保持するエキソソームと培養すると増加したが、親のエキソソームは無効であったことが明らかになった(図9)。さらに、mutCD40Lを保持するエキソソームの方が、分析に同じ量のエキソソームを用いるか、エキソソームに結合した同じ量のCD40Lを用いるかによらず、より優れた活性を示した。実際、エキソソームに結合した200 ng/mlのmutCD40Lを測定に用いたとき、DC成熟が検出された。対照的に、同量の、エキソソームに結合したCD40L、または親のエキソソームに混合した可溶性CD40Lは、検出できる効果を持たなかった。可溶性CD40Lの活性を、1μg/ml以上のCD40L濃度での測定においてのみ検出することができた。
【0132】
結論
CD40Lが、他の膜貫通タンパク質(実施例2および3)と同様に、トランスフェクションされた細胞の過剰発現に続いて、エキソソームを標的とするドメインへの融合の必要なく、エキソソーム上に発現される。CD40Lを発現している組換えエキソソームはまた、CD40L活性を示す。したがって、本発明を、エキソソームの免疫能力を増強するために使用できる。CD40Lのタンパク質分解切断部位の突然変異が、エキソソームの免疫能力をさらに増大させる結果になった。この性質の可能な説明は、エキソソーム上の野生型CD40Lは、CD40Lの残りの膜貫通ドメインはもちろん、全長のCD40Lと可溶性型のCD40Lの混合物から成り、これが機能的なCD40Lの3量体の効果的な形成を妨げている可能性がある、ということである。このようにして、CD40Lが断片に切断されることを妨げると、その結果、より高度な特異的活性を示す強化エキソソームが生成されることになる。これが、組換えエキソソームを用いるワクチンおよび他の研究への応用に必要である。何故なら、エキソソーム注入のすぐ後で循環しているタンパク質分解酵素がCD40Lを切断して、エキソソームを不活性化する可能性があるからである。
【実施例5】
【0133】
GFP/C1C2キメラタンパク質を用いて追跡可能なエキソソームを生成するための方法
【0134】
蛍光タンパク質GFPをコードするcDNAから開始コドンを除いたものを、ラクトアドヘリンのリーダー配列とC1/C2ドメインの間にクローニングして、LS-GFP-C1/C2(配列番号40)を生成した。GFPのcDNAを、pEGFP(Clontech,CA)を鋳型とし、プライマーGFPfl(配列番号41)およびGFPr2(配列番号42)を用いて、PCRにより増幅した。プライマーGFPf1を5’端で延長してEcoR I制限部位を含むようにしたが、一方GFPr2はNot Iの5’端延長を含んでいた。PCR産物をEcoR VとNot Iで消化し、予めNot IとBstB 1で切断したC1/C2 PCR産物にライゲーションした。C1/C2 cDNA断片は、pcDNA6-hLact1f/Hisを鋳型として用い、プライマーLTDNf40(配列番号43)およびLTDNr35(配列番号44)を用い、PCRにより生成したものである。ライゲーション混合物を次に、予めEcoR VとBstB Iで切断しておいたpcDNA6-hLact28-Myc/His(配列番号45)に加えた。pcDNA6-hLact28-Myc/Hisは、リーダー配列とEGFドメインを含むヒトラクトアドヘリンのN末端をコードする。それは、鋳型としてpcDNA6-hLact1f/Hisを用いプライマーLTDNfl5およびLTDNr28(配列番号46)を用いたPCR産物をライゲーションして調製したものである。pcDNA6-hLact28-Myc/HisをEcoR VおよびBstB Iで消化すると、ラクトアドヘリンのリーダー配列とMyc/Hisタグの間のDNA断片が放出され、それがGFP-C1/C2 DNAで置き換えられてpcDNA6-LS-GFP-Cl/C2-Myc/His(配列番号39)が生成された。LS-GFP-Myc/His(配列番号29)をコードする対照のプラスミド、pcDNA6-LS-GFP-Myc/His(配列番号28)もまた類似の手法で調製した。すなわち、GFPr2はBstB I延長を含むように修飾し、またGFP配列はC1/C2 PCR産物と予めライゲーションする工程を除外して、予め切断しておいたpcDNA6-hLact28-Myc/Hisに挿入した。GFPプラスミドでトランスフェクションされた安定な細胞系統由来のクローンを調製し、エキソソームを、他のコンストラクション(実施例1〜4)について上述したようにして精製した。エキソソームについて、その蛍光発光に基づいてGFPの存在を評価した。測定は、96ウエルプレートのウエルに入れたエキソソームについて、Wallac 1420 Victor2 プレートリーダーを用いた蛍光測定によって行った。この測定結果を図10に示す。
【0135】
結果
蛍光が、pcDNA6-LS-GFP-Cl/C2-Myc/Hisをトランスフェクションした細胞由来のエキソソームで検出された一方で、バックグラウンドレベルがpcDNA6-LS-GFP-Myc/Hisをトランスフェクションした細胞が産生したエキソソームで測定された(図10)。ウエスタンブロット分析が、予想通り後者が、可溶性タンパク質として培養上清液に放出される組換えGFPを産生することを明らかにした(データは示さず)。
【0136】
結論
これらのデータがさらに、可溶性タンパク質をコードする配列をエキソソームターゲティングドメインに融合させると、タンパク質がエキソソーム上に発現することを確認する。エキソソームに結びついたGFPの発現を、抗原特異的抗体産生細胞の選別を含む様々な応用ができる、追跡可能な手段を調製するために使用できる(下の実施例7参照)。
【実施例6】
【0137】
蛍光脂質を用いて代謝ラベルすることにより追跡可能なエキソソームを生成するための方法
【0138】
この実施例では、追跡可能なエキソソームを、エキソソームを代謝的にラベルする蛍光脂質を用いて調製した。脂質1,2-ジオレイル-sn-グリセロ-3-ホスホエタノールアミン-N-(リサミンローダミンBスルフォニル)(Rh-DOPE, Avanti Polar Lipids)を、脂質のエタノール溶液を滅菌PBSに希釈し、最終濃度を40μMとして調製した。次に、40μMの原液を293細胞培養液に徐々に加えて、3.0μM蛍光脂質とした。培養7日目の培養液からエキソソームを収集し、実施例1〜4記載のように精製した。精製したエキソソームに結合した蛍光を、抗−CD81抗体で予めコートした96ウエルプレートのウエルに試料を加えて行う捕獲ELISAにより評価した。よく洗浄した後に、ウエルを560 nmで励起したときの600nmの蛍光放射を、Wallac 1420 Victor2プレートリーダーを用いて測定した。この実験に対する対照試料は、蛍光脂質を含まない培地由来のエキソソーム(Exo)、蛍光脂質を含まない培地由来であって、24時間蛍光脂質を含む培地でインキュベートしたエキソソーム(Exo+Rh)、蛍光脂質を含む培地のみ(Rh)、およびPBSである。結果を図11に示す。
【0139】
結果
Rh-DOPE脂質にさらした細胞によるエキソソーム産生(図11のRh/Exo)が、強い蛍光性のエキソソームをもたらした。対照的に、エキソソームを直接蛍光脂質とインキュベートした場合(図11のExo+Rh)、またはエキソソームが蛍光脂質無しで生成された場合(図11のExo)にはバックグラウンドレベルに近く低い蛍光が検出された。測定した蛍光は、直接エキソソームに結合していた。なぜなら、Rh-DOPEを含む培地のみをウエルに加えた定量(図11のRh)においては、バックグラウンド蛍光のみが検出されたからである。1,2-ジオレイル-sn-グリセロ-3-ホスホエタノールアミン-N-(カルボキシフルオレセイン)(Fl-DOPE)などの、他の蛍光脂質にさらした細胞によるエキソソーム産生もまた、強い蛍光性のエキソソームをもたらした(データを示さず)。
【0140】
結論
ここに記載した方法が、追跡可能なエキソソームの産生をもたらす。これらのエキソソームに結合した蛍光の強度は、蛍光脂質により直接にラベルされたエキソソームの強度より、はるかに上である。それらを利用することにより、多くの研究とスクリーニングへの応用において、増大した感度で読み取りができることになるはずである。蛍光脂質を含む追跡可能なエキソソームは、例えば、抗原特異的な抗体産生細胞を選別単離するための極めて強力な手段である(下の実施例7参照)。
【実施例7】
【0141】
抗原特異的B細胞を同定および単離するための方法
【0142】
組換えGPCR発現エキソソームによって免疫化されたマウスの膝窩リンパ節細胞(PLNC)を、接種物を足蹠に1週間毎に4回連続注入した後に収集した。接種物は、GPCRをコードするプラスミドをトランスフェクションされたマウス細胞に由来し、実施例2に記載のように調製した精製エキソソームから成っていた。PLNCを4℃で2回PBS/5% FBSで洗浄し、106のPLNCを分取したもの2つを、500μl PBS/5% FBSに再懸濁した。正規化した量の、GFPを発現しているエキソソーム、GFPとGPCRを発現しているエキソソームおよび親のエキソソームを、実施例2と5に記載のように調製した。同量の追跡可能な対照エキソソーム(GFP-293)および追跡可能なテストエキソソーム(GFP/GPCR-293)を、それぞれ、第1と第2のPLNC試料に加えた。エキソソームのPLNCとの非特異的な相互作用を遮断するために、両試料に10×過剰の親のエキソソームを加えた。フィコエリトリンに結合された抗−CD19抗体も、PLNC中のB細胞サブ集団を特異的に選択するために、試料に加えた。試料をPBS/5% FBSで調整して最終1mlにし、1時間4℃でインキュベートした。次に細胞を、GFPとCD19に二重陽性であることを識別するために、FACSで分析した。二重陽性細胞が、抗体産生細胞即ちB細胞であり、GFPエキソソーム上の組換えGPCRと特異的に反応する抗体をその表面に発現していると予想される。CCR7をモデルとして用いて行った実験の結果を図12に示す。
【0143】
結果
免疫化された細胞由来のPLNC集団中のGFP陽性細胞の百分率が、細胞をGFP/CCR7-293エキソソームとインキュベートしたときは、対照のGFP-293エキソソームとインキュベートしたときよりも、有意に高かった(図12、パネルA:0.13%陽性細胞 対 パネルB:0.36%陽性細胞)。この百分率は低いが、類似の傾向が、CXCR4、CCR5、およびSSTR2などの他のGPCRを発現しているエキソソームにより免疫化したマウスから得られたPLNCと適合する追跡可能な組換えエキソソームを用いた場合に観察された(データを示さず)。
【0144】
結論
追跡可能なエキソソームは、抗原特異的抗体産生細胞を同定する便利な手段である。FACS分析と選別を組み合わせると、応答する細胞の頻度が極めて低い場合にさえも、所定の抗原特異性を有する個々の細胞を単離することができるに違いない。
【実施例8】
【0145】
エピトープ特異的抗体を産生しているB細胞を同定および単離するための減算的方法
【0146】
この実施例において、抗体産生細胞の選別を、エピトープ特異的抗体産生細胞へ拡張する。方法は、次の点を除いて実施例7記載の方法と同様である。即ち、免疫化マウスのPLNCを過剰の親のエキソソームとインキュベートする代わりに、ここでは過剰の抗原発現エキソソームとインキュベートして、抗原上の望ましくないエピトープに反応するB細胞を遮断する。そのような減算的選別において、突然変異した抗原または抗原の立体構造変異体に見出される新しいエピトープに反応するB細胞は、自由に、抗原変異体を発現している追跡可能なGFP-293と反応できる。この手法の最も良い例が、MHC/ペプチド複合体と反応する制限抗体の調製である。HLA-A2/ペプチド複合体を発現するエキソソームの調製を、実施例3においてすでに記述した。PLNCを、エピトープ特異的抗体産生細胞に富むようにするために、対側注入を用いてマウスを組換えエキソソームで免疫化した。ここでは、マウスの右の足蹠に、組換えA2を発現しているマウス由来のエキソソームの6回の接種を、−3、0、3、10、17、および24日目に行った。左の足蹠に、MART1由来のペプチド(配列番号30)を負荷した同じエキソソームの5回の接種を、0、3、10、17、および24日目に行った。右と左のPNLCを、別々に実施例7に記載のように染色用に調製した。等量の追跡可能な対照エキソソーム(GFP/A2-293)または追跡可能なテストエキソソーム(GFP/A2/MART1-293)を、それぞれ両方のPLNC集団に加えた。A2のみに反応する抗体を産生するB細胞をマスクするために、10×過剰のA2-293を全ての試料に加えた。これらの試料についてのFACS分析の結果を図13に示す。
【0147】
結果
マウスのA2/MART1エキソソームで免疫化した側由来の、左のPLNC集団中のGFT-陽性細胞の百分率が、細胞をGFP/A2/MART1-293エキソソームとインキュベートした時には、GFP/A2-293エキソソームとインキュベートした時よりも有意に高かった(図13、パネルA:0.83%陽性細胞 対 パネルB:0.33%陽性細胞)。対照的に、右のPLNCについて、GFP/A2/MART1-293およびGFP/A2-293とのインキュベートを比較したときには、有意な差は検出されなかった(図13、パネルC:0.49%陽性細胞 対 パネルD:0.39%陽性細胞)。
【0148】
結論
ここで述べた追跡可能なエキソソームを用いる減算的方法は、エピトープ特異的な抗体産生細胞を同定するための強力な手法である。FACS分析および選別と組み合わせると、所定のエピトープ特異性を有する個々の細胞の単離同定を可能にする。減算的選別は、例えば、MHC/ペプチド複合体と反応する制限抗体を産生するB細胞の単離に適切である。
【実施例9】
【0149】
RT-PCRによって免疫グロブリン配列を取り出すための方法
【0150】
所定の抗原またはエピトープ特異性を有する抗体を産生し、実施例7および8に記載されたようにFACSによって同定された細胞を、個々に、96ウエルプレートの25μlの氷冷溶解緩衝液(Cells to cDNA IIキット、Ambion)を含むウエルに選別した。96ウエルプレートを75℃15分間インキュベートしてRNaseを熱失活させた。次にゲノムDNAを除去するために、試料を1単位のDNase Iで処理した。得られたRNAを含む試料を新しいプレートに移して、逆転写酵素とオリゴdT(SuperScript II, Invitrogen)を用いたcDNA合成を行った。抗原との接触に含まれる相補性決定ドメインを含む免疫グロブリンの軽鎖と重鎖の可変ドメインをコードするcDNAを、ネストPCRにより増幅した。IGLKfl(配列番号31)、IGLKf2(配列番号32)、IGLKf3(配列番号33)、IGLKf4(配列番号34)、IGLKf5(配列番号35)を含む順方向プライマーおよび逆方向プライマーIGKrl5(配列番号36)の混合物を、軽鎖配列の1回目のPCRに用いた。2回目のPCR(ネストPCR)を、小分けにした第1回目のPCRを鋳型とし、縮重プライマーIGKf6(配列番号37)およびIGKrl5を用いて実行した。同様に、IGLHfl(配列番号38)、IGLHf2(配列番号39)、およびIGLHf3(配列番号40)を含む順方向プライマー混合物および逆方向プライマーIGHr5(配列番号41)を、重鎖配列の1回目のPCRに用いた。2回目のPCR(ネストPCR)を、小分けにした第1回目のPCRを鋳型として、また縮重プライマーIGHfll(配列番号42)およびIGHrl3(配列番号43)を用いて実行した。蛍光性エキソソームに抗原特異的態様で結合できる能力に基づいて個別に選別した細胞由来のcDNAを用いた軽鎖配列増幅の結果を、図14に示す。
【0151】
結果
2回のPCRにより、いくつかの単一細胞cDNAに由来する検出可能な量のPCR産物が生成された(図14, レーン 2、5、6、8、および13)。これらのPCR産物の配列決定により、PCR産物が免疫グロブリンの可変ドメインをコードすることが確認された。いくつかの可能性により、図14の空白のレーンにPCR産物を欠くことを説明できる。1つの可能性が、PCRのサイクリング条件およびここで用いたプライマー対が、元のウエルの単一細胞により発現される免疫グロブリンの可変配列を効率的に増幅するために最適ではないことである。実際、各可変ドメインが独特で、増幅のために異なるPCR条件を必要とする可能性がある。他のプライマーの組み合わせおよびサイクリング条件を、最適の結果を得るために、各cDNAに試すのがよい。元のウエルに選別した細胞が、非B細胞または休止B細胞であったという別の可能性がある。追跡可能なエキソソームとPLNCとの非特異的な相互作用(実施例7と8のバックグラウンド染色を参照のこと)のために選別されたこれらの細胞は、RT-PCRによって検出するための免疫グロブリンをコードするRNAを含んでいないか、低すぎるレベルしか含んでいない。このことは、ここで提案する免疫グロブリンの可変ドメインを取り出すための方法の感度によって、非抗体産生細胞からのcDNAのさらなる持ち越しが回避できる可能性があることを際立たせる。
【0152】
結論
追跡可能なエキソソームと抗原特異的態様で相互作用できる能力に基づいて単離された単一細胞から得られた免疫グロブリンの可変配列を、古典的PT-PCRにより首尾よく取り出すことができる。次にPCR産物を、下の実施例10に記載する通り、発現ベクターにクローニングできる。
【実施例10】
【0153】
免疫グロブリンの可変配列を発現ベクターにクローニングするための方法
【0154】
実施例9で生成された可変ドメインPCR産物を受け入れるために、抗体配列の重鎖および軽鎖部分を埋め込んだ発現ベクターを構成した。可変ドメインPCR産物のこれらのベクターへのクローニングにより、全長の重鎖および軽鎖配列が生成される。挿入クローニングの古くからある方法と異なり、ここで用いるクローニング戦略は、クローニング部位に突然変異を導入しない。免疫グロブリンの可変ドメインのいずれの端で突然変異が生じても、抗原の親和性と特異性を修飾する可能性があるので、この方法が有利である。この方法は、配列認識部位の外側で切断して切断部の一方側に認識配列を残す、BfuAI、BsaI、およびBsmBIなどの「II」型制限酵素を用いる。2つのそのような制限部位を含むカセットにより、2つの部位に縁取られた挿入部を放出し、埋め込まれた抗体配列のみを残すことが可能になる。クローニング用の挿入部を、埋め込まれた抗体配列と適合性のある配列端を有する同様の部位をその両端に含むように生成する。それにより、挿入部のライゲーションが、遺伝子特異的配列のアニーリングを介して起こり、制限酵素の認識部位に適合するための塩基の挿入または修飾を必要としない。
【0155】
抗体の重鎖および軽鎖のために、別々のベクターを構成した。これらは、リンカーの無いクローニングカセットの両側に接して、リーダー配列と原型の重鎖と軽鎖の定常ドメインを、それぞれ含んでいた。軽鎖配列をコードするマウスの脾臓cDNAを、上流リーダー配列断片を生成するためにプライマーIGKfl(配列番号44)およびIGKr2(配列番号45)を用い、また下流定常ドメイン断片を生成するためにプライマーIGKfl2(配列番号46)およびIGKr4(配列番号47)を用いて増幅した。IGKr2とIGKfl2を、リンカーの無いクローニングカセットを包含するオーバーラップした配列を含むように設計した。リーダー配列、リンカーの無いクローニングカセット、および定常ドメインを含む完全なDNA断片を、上流と下流の断片を鋳型とし、プライマーIGKflとIGKr4を用い第2のPCRにより生成した。これらのプライマーは、IGKflはHind III制限部位を有し、IGKr4はBstB Iを有する5’端延長を含んでいた。PCR産物を、Hind IIIおよびBstB Iによる消化に続いて予め同じ酵素で切断したpcDNA6-Myc/Hisへクローニングして、pcDNA6-LCJ-Myc/His(配列番号48)を生成した。pcDNA6-LCJ-Myc/Hisの等価物で重鎖をコードするpcDNA6-HCJ-Myc/His(配列番号49)を、同じ手法を用いて調製した。ここで、1回目のPCRを、プライマー対IGHfl(配列番号50)/IGHr2(配列番号51)、およびIGHfl5(配列番号52)/IGHr4(配列番号53)を用いて実行した。2回目のPCRを、IGHflおよびIGHr4を用いて行った。
【0156】
別のコンストラクションを、抗体の重鎖と軽鎖を単一のベクター中に発現させるために調製した。これは、いずれのトランスフェクションされた細胞も重鎖と軽鎖の両方を産生することを保証するためである。この目的のために、内部リボソーム進入部位(IRES)およびpIRES(Clontech)由来の第2の多重クローニング部位(mcs2)を、pcDNA6-LCJ-Myc/His中のLCJの下流にクローニングしたIRES/mcs2挿入を、pIRESを鋳型に用い、プライマーpIRfl(配列番号54)およびpIRr2(配列番号55)を用いて、PCRにより生成した。PCR産物をBstBIとAge Iで消化後、予め同じ酵素で切断したpcDNA6-LCJ-Myc/Hisにライゲーションして、pcDNA6-LCJ-IRESを生成した。次に、重鎖および軽鎖の可変部位のpcDNA6-LCJ-IRESへのクローニングを、2工程で行った。第1に、軽鎖と重鎖を、それぞれpcDNA6-LCJ-IRESとpcDNA6-HCJ-Myc/Hisへクローニングした。第2に、第1工程で調製したpcDNA6-HCJ-Myc/His中の全長の重鎖を、これも第1工程で調製して、すでに全長の軽鎖をコードしているpcDNA6-L-IRESにサブクローニングした。
【0157】
実施例9で生成され、ここで記述した方法を用いて発現ベクターにクローニングされたPCR産物が、免疫グロブリンを産生したことを証明するめの実験を行った。実施例8記載のようにして単離した単一細胞由来の、重鎖および軽鎖配列の可変ドメインをそれぞれコードしているPCR産物HV-M90/12(配列番号56)およびLV-M90/12(配列番号57)をモデルとして用いた。BsmB I制限部位を、LV-M90/12に対してはプライマーIGKf6および 1: 1混合のIGKrlO(配列番号58):IGKrll(配列番号59)を、またHV-M90/12に対してはプライマーIGHfllおよび 1: 1: 1 混合のIGHrl6(配列番号60)/IGHrl7(配列番号61)/IGHrl8(配列番号62)を用いた10サイクルのPCRによって、各DNA断片の両端に導入した。これらのプライマーを、受け入れ側発現ベクター中の取り込まれた抗体配列の接続部に相補的な突出末端を残すように設計した。LV-M90/12を、BsmB Iによる消化に続いて予め同じ酵素で切断したpcDNA6-LCJ-IRESおよびpcDNA6-LCJ-Myc/Hisにクローニングして、それぞれpcDNA6-L-M90/12-IRES(配列番号63)およびpcDNA6-L-M90/12-Myc/His(配列番号64)、を生成した。HV-M90/12をはじめに、予めBsmB Iで切断したpcDNA6-HCJ-Myc/Hisにクローニングして、pcDNA6-H-M90/12-Myc/His(配列番号65)を生成した。全長のH-M90/12重鎖配列を次に、pcDNA6-L-M90/12-IRESのmcs2にサブクローニングした。そうするために、pcDNA6-H-M90/12-Myc/Hisを鋳型に用い、プライマーpIRHfl(配列番号66)およびpIRHr2(配列番号67)を用いて、PCRにより、H-M90/12の5’端と3’端に、それぞれ、Xba IとNot Iの制限部位を導入した。H-M90/12を、Xba IとNot Iによる消化に続いて、予め同じ酵素で切断したpcDNA6-L-M90/12-IRESにクローニングして、pcDNA6-L+H-M90/12を生成した。CHO細胞を、空のプラスミド、pcDNA6-L+H-M90/12、または1:1 pcDNA6-L-M90/12-Myc/His:pcDNA6-H-M90/12-Myc/His混合物によりトランスフェクションした。培養3日目の上清を収集し、免疫グロブリン産生を、ロバの抗−マウスIgG抗体(Jackson ImmunoResearch)を捕獲抗体として用い、またウサギ抗−マウスIgGを検出抗体として用いて、実施例2に述べた捕獲ELISAによりテストした。標準曲線を、精製したマウスIgGの量を次第に増加させて作製した。このELISAの結果を図15に示す。
【0158】
結果
組換え免疫グロブリンが、トランスフェクションされたCHO細胞の上清に首尾よく検出された(図15)。さらに、抗体の軽鎖と重鎖の両方をコードする発現ベクターをトランスフェクションした場合の方が、各鎖を別々にコードするプラスミドを共にトランスフェクションした場合より優れていた。
【0159】
結論
ここで述べた、全長の免疫グロブリンをコードするプラスミドを構成するためのクローニング戦略が、産生される抗体の可変ドメイン中に挿入または突然変異を導入することなく、大量の組換え免疫グロブリンを産生する発現ベクターをもたらす。全体として、実施例7〜10によって、所定の特異性を有する抗体を産生する個々の細胞に由来する抗体配列を、本発明に記載された方法を用いて取り出し、クローニングし、そして組換えタンパク質として発現させることができることが、裏付けられる。次に、産生された組換え抗体の抗原特異性を結合実験により、好ましくは抗原源として組換えエキソソームを用いて評価できる。
【実施例11】
【0160】
ビオチン化エキソソームを用いた抗体分泌ハイブリドーマの単離
【0161】
実施例8記載のように免疫化したマウスの脾臓細胞を、ハイブリドーマ調製の標準的方法を用いて、マウス骨髄腫細胞系統Sp2/0に融合させた。融合に続いて、ハイブリドーマを選別するために、アザセリンを補充した培地中で細胞を大量培養して成長させた。培養8日目に細胞を収集し、ビオチン化抗−CD45抗体およびストレプトアビジンと、逐次インキュベートした。次に、細胞を同等の2分画、即ち抗体トラップ陽性(AbTrap+)分画と陰性(AbTrap-)分画に分けた。AbTrap+分画の抗体トラップを、細胞をビオチン化抗−マウスIgG抗体とインキュベートすることにより完成させた。次に、両細胞分画を培養液中で終夜37℃10% CO2雰囲気下でインキュベートして、ハイブリドーマに抗体を分泌させ、これを抗体トラップによって捕獲させた。細胞表面で捕獲されて望みの抗原特異性を示した抗体を分泌しているハイブリドーマを、ビオチン化エキソソームとのインキュベーションに続くFACSにより単離した。この追跡可能なエキソソームは、酸化ポリエチレン(PEO)リンカーを介してビオチンに結合したテトラフルオロフェノール(TFP)を有する化学的反応性ビオチンとエキソソームをインキュベートして調製した。TFPにより活性化されたビオチンが、エキソソーム上の第一級アミンと反応して、共有結合を形成する。表面にビオチン化エキソソームを有するハイブリドーマを、蛍光団Alexa488と結合したストレプトアビジンを用いて、検出した。抗−エキソソーム抗体を産生するAbTrap+およびAbTrap-ハイブリドーマのFACS分析を図16に示す。
【0162】
結果
ビオチン化エキソソームをAbTrap-細胞とインキュベートすると、これらの細胞に結合した蛍光はバックグラウンドのみであった(図16、パネル1)。対照的に、完全なAbTrapを有する場合は、〜25%の細胞が陽性であった(図16、パネル2)。
【0163】
結論
ここで設計した抗体トラップが、首尾よくハイブリドーマが分泌する抗体を捕獲した。これによって、FACSによるハイブリドーマの単離が可能となる。さらに、この結果は、ビオチン化エキソソームが、抗体産生細胞を抗原特異的な方法で単離するための、適切な追跡可能エキソソームであることを示している。
【0164】
【表1】
【図面の簡単な説明】
【0165】
【図1】ラクトアドヘリンで免疫化したマウス血清中の抗ラクトアドヘリン抗体を検出するELISA。
【図2】HApC3.1/SSTR2によってトランスフェクションされ、抗HAタグ抗体でラベルされた組換え細胞のFACS分析。
【図3】HApC3.1/SSTR2をトランスフェクションされた細胞により生成されたエキソソームに発現されているHA-SSTR2を検出するウエスタンブロット分析。
【図4】様々なGCPRをコ−ドするHApC3.1でトランスフェクションされ、抗HA-タグ抗体でラベルされた一団の293細胞のFACS分析。
【図5】様々なGPCRをコードするHApC3.1をトランスフェクションされた一団の293細胞由来のエキソソーム上のHA-GPCR発現を検出する捕獲ELISA。
【図6】293エキソソーム上の、HLA-A2およびβ2-ミクログロブリン発現を検出するの捕獲ELISA。
【図7】エキソソーム上のHLA-A2に結合した標準ペプチドを検出する捕獲ELISA。
【図8】組換え293エキソソーム上での、CD40LおよびmutCD40L発現を検出するウエスタンブロット分析。
【図9】CD40Lを表示している組換えエキソソームの生物学的活性を測定するDC-成熟分析。
【図10】GFP/C1C2キメラタンパク質を発現している組換えエキソソームに結合した蛍光の測定。
【図11】Rh-DOPEで代謝的にラベルされた細胞由来のエキソソームに結合した蛍光を検出する捕獲ELISA。
【図12】GFP/CCR7-293エキソソームを有するPLNCを検出するFACS分析。
【図13】GFP/A2/MART1-293エキソソームを有するPLNCを検出するFACS分析。
【図14】単一細胞由来のRT-PCR産物のゲル分析。
【図15】CHO培養上清中の組換えIgGを検出する捕獲ELISA。
【図16】ビオチン化エキソソームとインキュベートしたAbTrap-およびAbTrap+細胞のFACS分析。
【特許請求の範囲】
【請求項1】
選択した抗原に対して特異性を有する単一の抗体産生粒子を単離する方法であって:
1)選択した抗原およびマーカーを表示している小胞、好ましくはエキソソームを調製すること;
2)工程1の前記小胞を抗体レパートリーと接触させること;および
3)前記小胞と反応している単一の抗体産生粒子を同定および単離すること;
を含む方法。
【請求項2】
前記抗体産生粒子が抗体産生細胞であり、前記抗体レパートリーが抗体産生細胞のレパートリーである、請求項1記載の方法。
【請求項3】
前記抗体産生細胞がプラズマ細胞、ハイブリドーマ、またはリンパ球から成る群より選択される、請求項2記載の方法。
【請求項4】
請求項2または3記載の方法であって:
1)抗体産生細胞を提供すること;
2)前記抗原およびマーカーを表示している小胞、好ましくはエキソソームを調製すること;
3)工程1のリンパ球を、工程2の小胞と懸濁すること;および
4)前記小胞と反応している単一の抗体産生細胞を同定および単離すること;
を含む方法。
【請求項5】
前記抗体産生細胞が、前記抗原により免疫化されたヒト以外の動物から収集したリンパ球である、請求項4記載の方法。
【請求項6】
請求項4記載の方法であって、前記抗体産生細胞が抗体分泌細胞であり、さらに、抗体分泌細胞を小胞と懸濁する工程の前に、抗体分泌細胞を、遍在する細胞表面マーカーに対する第1のビオチン化抗体、ストレプトアビジンおよび前記抗体分泌細胞の免疫グロブリンに向けられた第2のビオチン化抗体とインキュベーションする工程を含む方法。
【請求項7】
請求項5記載の方法であって:
1)少なくとも1つの抗原またはそのエピト−プを表示している免疫原性の小胞、好ましくはエキソソームを調製すること;
2)ヒト以外の動物を前記免疫原性小胞で免疫化することにより、抗体反応を生起させること;
3)免疫化した動物からリンパ球を収集すること;
4)工程1の前記抗原またはそのエピトープおよびマーカーを表示している小胞、好ましくはエキソソームを調製すること;
5)工程3のリンパ球を工程4の小胞と懸濁すること;および、
6)工程4の小胞と反応している単一の抗体産生細胞を同定および単離すること;
を含む方法。
【請求項8】
変異抗原に特異的な単一抗体を産生している粒子を抗体レパートリーから単離する方法であって:
1)前記変異抗原およびマーカーを表示している第1集団の小胞、好ましくはエキソソームを調製すること;
2)自然抗原を表示し、前記マーカーを表示していない第2集団の小胞、好ましくはエキソソームを調製すること;
3)前記抗体レパートリーを第1および第2集団の小胞と、第2集団を過剰にして、懸濁すること;および、
4)工程1の小胞と反応している単一の抗体産生粒子を同定および単離すること;
を含む方法。
【請求項9】
請求項8記載の方法であって:
1)前記変異抗原によって免疫化されたヒト以外の動物からリンパ球を収集すること;
2)動物の免疫化に用いた前記変異抗原およびマーカーを表示している小胞、好ましくはエキソソームを調製すること;
3)自然抗原を表示し、前記マーカーを表示していない小胞、好ましくはエキソソームを調製すること;
4)工程1のリンパ球を、工程2の前記変異抗原およびマーカーを表示している小胞ならびに工程3の前記自然抗原を表示している過剰量の小胞と共に懸濁すること;および、
5)工程2の小胞と反応している単一の抗体産生細胞を同定および単離すること;
を含む方法。
【請求項10】
請求項9の方法であって:
1)変異抗原を表示している免疫原性の小胞、好ましくはエキソソームを調製すること;
2)ヒト以外の動物を前記免疫原性小胞で免疫化すことにより抗体反応を生起させること;
3)免疫化した動物からリンパ球を収集すること;
4)工程1の前記変異抗原およびマーカーを表示している小胞、好ましくはエキソソームを調製すること;
5)自然抗原を表示し前記マーカーを表示していない小胞、好ましくはエキソソームを調製すること;
6)工程3のリンパ球を、工程4の前記変異抗原およびマーカーを表示している小胞、ならびに工程5の前記自然抗原を表示している過剰量の小胞と懸濁すること;および
7)工程4の小胞と反応している、抗原変異体に特異的な単一抗体を産生している細胞を同定および単離すること;
を含む方法。
【請求項11】
前記抗体産生粒子が抗体を産生しているファージまたは酵母であり、前記抗体レパートリーが抗体を産生するファージまたは酵母のレパートリーである、請求項1または8記載の方法。
【請求項12】
請求項1〜11のいずれか1項記載の方法であって、前記方法がさらに次の工程:
a)前記の選択された抗体産生粒子からDNAまたはRNAを回収すること、b)免疫グロブリン配列またはその一部をコードする核酸配列を増幅すること、c)増幅した核酸配列を発現ベクターへクローニングして、所望の抗原特異性を有するタンパク質を産生すること、を含む方法。
【請求項13】
前記免疫原性小胞がさらにアジュバントポリペプチドなどの免疫アクセサリー分子を表示する、請求項7または10のいずれかに記載の方法。
【請求項14】
前記アジュバントポリペプチドが、GM-CSF、IL-2およびCD40Lなどのサイトカインから、特に突然変異CD40Lから選択され、前記突然変異が可溶性CD40Lの切断と放出を阻害する、請求項13記載の方法。
【請求項15】
前記免疫アクセサリー分子がエキソソームターゲティングポリペプチドに融合または架橋している、請求項13記載の方法。
【請求項16】
前記免疫アクセサリー分子が少なくとも1つの膜貫通ドメインを有し、エキソソーム産生細胞中への過剰発現により免疫原性小胞に取り込まれる、請求項13記載の方法。
【請求項17】
前記変異抗原が突然変異抗原であり、前記自然抗原が野生型抗原である、請求項8〜10のいずれか1項記載の方法。
【請求項18】
前記変異抗原が、ポリペプチド、脂質、DNA、または小分子から成る群より選択される分子と接触している抗原であり、前記自然抗原がフリーの抗原である、請求項8〜10のいずれか1項記載の方法。
【請求項19】
前記変異抗原がMHC/ペプチド複合体であり、前記自然抗原が負荷されていないMHCであるかまたは別のペプチドを負荷されているMHCである、請求項18記載の方法。
【請求項20】
前記変異抗原がリガンド-レセプター複合体であり、前記自然抗原がフリーのリガンドまたはフリーのレセプターである、請求項19記載の方法。
【請求項21】
前記変異抗原および前記自然抗原が、酵素を含む任意の異なる立体構造状態のタンパク質である、請求項8〜10のいずれか1項記載の方法。
【請求項22】
前記小胞により表示されている前記抗原がエキソソームターゲティングポリペプチドに融合している、請求項1〜12のいずれか1項記載の方法。
【請求項23】
前記小胞によって表示されている前記抗原がエキソソームターゲティングポリペプチドに架橋されている、請求項1〜12のいずれか1項記載の方法。
【請求項24】
前記マーカーが、タグ、ビオチン、酵素、蛍光分子などの検出可能な分子である、請求項1〜23のいずれか1項記載の方法。
【請求項25】
前記マーカーがエキソソームターゲティングポリペプチドに融合または架橋されている、請求項24記載の方法。
【請求項26】
前記マーカーが、エキソソーム産生細胞中への過剰発現により免疫原性小胞に取り込まれる膜貫通ドメインを有している、請求項24記載の方法。
【請求項27】
前記マーカーが優先的に小胞へ、好ましくはエキソソームへ取り込まれるラベルされた脂質である、請求項24記載の方法。
【請求項28】
前記ラベルされた脂質がRh-DOPEまたはフルオレセイン-DOPEなどの蛍光団結合脂質である、請求項27記載の方法。
【請求項29】
前記抗原が少なくとも1つの膜貫通ドメインを有するポリペプチドであり、前記抗原がエキソソーム産生細胞中に過剰発現され、それにより前記抗原を表示している前記小胞を生成することができる、請求項1〜21のいずれか1項記載の方法。
【請求項30】
少なくとも1つの膜貫通ドメインを有する前記ポリペプチドがレセプターである、請求項29記載の方法。
【請求項31】
前記レセプターがGPCR(Gタンパク質共役レセプター)である、請求項30記載の方法。
【請求項32】
前記抗原が、例えばレセプターまたは酵素などの任意のタンパク質、あるいは糖脂質、多糖類、薬物および有機化学物質などのポリペプチド以外の化合物である、請求項1〜31のいずれか1項記載の方法。
【請求項33】
前記エキソソームターゲティングポリペプチドが、ラクトアドヘリンあるいは機能的なC1および/またはC2ドメインを含有するその一部を含む、請求項15、22、23または25のいずれか1項記載の方法。
【請求項34】
前記ラクトアドヘリンまたはその一部が、ヒト以外の哺乳動物のラクトアドヘリンまたはその一部である、請求項33記載の方法。
【請求項35】
前記ヒト以外の哺乳動物のラクトアドヘリンまたはその一部が、(i)マウスラクトアドヘリン、(ii)機能的なC1および/またはC2ドメインを含有するマウスラクトアドヘリンの断片、および(iii)(i)または(ii)のポリペプチドと少なくとも50%の一次構造同一性を有するポリペプチド、から選択される、請求項34記載の方法。
【請求項36】
前記ラクトアドヘリンが、配列番号1を含むアミノ酸配列あるいは機能的なC1および/またはC2ドメインを含有するその断片を有する、請求項35記載の方法。
【請求項37】
前記ラクトアドヘリンが、配列番号1の機能的C1/C2ドメインを含むアミノ酸配列を有する、請求項36記載の方法。
【請求項38】
前記ラクトアドヘリンが、配列番号1のアミノ酸残基111〜266、109〜266、271〜426、111〜426、または109〜426を含むアミノ酸配列を有する、請求項36記載の方法。
【請求項39】
前記エキソソームターゲティングポリペプチドが、Del-1、ニューロピリン-1、凝固因子5または凝固因子8の機能的なC1および/またはC2ドメインを含む、請求項15、22、23,または25のいずれか1項記載の方法。
【請求項40】
前記変異抗原がHLA-C/HIVペプチド複合体であり、前記自然抗原が負荷されていないHLA-Cまたは異なるペプチドを負荷されたHLA-Cである、請求項19記載の方法。
【請求項41】
請求項1〜39のいずれか1項記載の方法により得られる抗体産生細胞。
【請求項42】
請求項41記載の抗体産生細胞により産生される抗体。
【請求項43】
請求項41記載の抗体産生細胞および/または請求項42記載の抗体および薬学的に許容できる賦形剤または担体を含む組成物。
【請求項44】
少なくとも1つの膜貫通ドメインを有するポリペプチドをエキソソームの表面に発現させる方法であって:
1)前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを有するその一部をコードする遺伝子コンストラクトを提供すること;
2)エキソソーム産生細胞中に前記コンストラクトを導入し、前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを有するその一部を過剰発現させて、組換えエキソソームを生成すること;および、
3)前記遺伝子コンストラクトによってコードされるポリペプチドを表面に担持する前記組換えエキソソームを収集すること;
を含む方法。
【請求項45】
少なくとも1つの膜貫通ドメインを有する前記ポリペプチドがCD40Lである、請求項44記載の方法。
【請求項46】
前記CD40Lが突然変異CD40Lであって、前記突然変異が可溶性CD40Lの切断と放出を阻害する、請求項45記載の方法。
【請求項47】
少なくとも1つの膜貫通ドメインを有するポリペプチドまたはそのエピトープを結合する抗体を産生する方法であって:
1)前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを有するその一部をコードする遺伝子コンストラクトを提供すること;
2)エキソソーム産生細胞中に前記コンストラクトを導入し、前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを有するその一部を過剰発現させて、表面に前記ポリペプチドまたはエピトープを提示している組換えエキソソームを生成すること;
3)前記組換えエキソソームを収集し、前記エキソソームまたはその一部をヒト以外の哺乳動物に注入して、前記ポリペプチドまたはエピトープを結合する抗体を生成すること;および、
4)前記哺乳動物から抗体または抗体産生細胞を収集すること;
を含む方法。
【請求項48】
前記エキソソームがさらに突然変異CD40Lを表示し、前記突然変異が可溶性CD40Lの切断と放出を阻害する、請求項47記載の方法。
【請求項49】
前記少なくとも1つの膜貫通ドメインを有するポリペプチドがレセプターである、請求項44または47記載の方法。
【請求項50】
前記レセプターがGPCR(Gタンパク質共役レセプター)である、請求項49記載の方法。
【請求項51】
少なくとも1つの膜貫通ドメインを有する抗原、または少なくとも1つの膜貫通ドメインを有するその一部を、対象に送達する方法であって:
a)前記抗原または少なくとも1つの膜貫通ドメインを有するその一部をコードする遺伝子コンストラクトを提供すること;
b)エキソソーム産生細胞中に前記コンストラクトを導入し、前記抗原または少なくとも1つの膜貫通ドメインを有するその一部を過剰発現させて、前記抗原または前記その一部を表面に提示している組換えエキソソームを生成すること:
c)前記組換えエキソソームを収集し、前記エキソソームまたはその一部を前記対象に注入すること;
を含む方法。
【請求項52】
少なくとも1つの膜貫通ドメインを有する特異的な抗原、または少なくとも1つの膜貫通ドメインを含むその一部に対して、対象中に免疫応答を引き起こす方法であって、:
a)前記抗原または少なくとも1つの膜貫通ドメインを含むその一部をコードする遺伝子コンストラクトを提供すること;
b)エキソソーム産生細胞中に前記コンストラクトを導入し、前記抗原または少なくとも1つの膜貫通ドメインを含むその一部を過剰発現させて、前記抗原または前記その一部を表面に提示している組換えエキソソームを生成すること;
c)前記組換えエキソソームを収集し、前記エキソソームまたはその一部を、前記対象に注入すること;
を含む方法。
【請求項53】
前記抗原がレセプターである、請求項51または52に記載の方法。
【請求項54】
前記レセプターがGPCR(Gタンパク質共役レセプター)である、請求項53記載の方法。
【請求項55】
追跡可能なエキソソームを調製する方法であって、次の工程:
1)エキソソーム産生細胞を蛍光団結合脂質とインキュベーションすること;および、
2)前記蛍光団結合脂質を取り込んだ前記細胞からエキソソームを産成し単離すること;
を含む方法。
【請求項56】
前記蛍光団結合脂質が蛍光団結合DOPE(ジオレオイル-ホスファチジルエタノールアミン)である、請求項55記載の方法。
【請求項57】
前記蛍光団結合DOPEがRh-DOPEまたはフルオレセイン-DOPEである、請求項56記載の方法。
【請求項58】
クローニング部位に突然変異を導入することなく、配列をベクター中にクローニングする方法であって、次の工程:
1)II型制限酵素により認識される2つの制限部位を備え、制限酵素切断後に制限部位が前記ベクターから除去されるように構成されたクローニングカセットを含むベクターを提供すること;
2)前記配列に、互換性のある末端を与えること;
3)前記ベクターを前記制限酵素とインキュベーションすること;
4)前記配列と工程3で得られた前記ベクターをライゲーションすること;
5)前記配列を含む前記ベクターを単離すること;
を含む方法。
【請求項59】
前記配列が免疫グロブリンの可変配列であり、前記ベクターの前記クローニングカセットが、その一端を免疫グロブリンのリーダー配列に、他端を定常ドメインに接している請求項58記載の方法。
【請求項1】
選択した抗原に対して特異性を有する単一の抗体産生粒子を単離する方法であって:
1)選択した抗原およびマーカーを表示している小胞、好ましくはエキソソームを調製すること;
2)工程1の前記小胞を抗体レパートリーと接触させること;および
3)前記小胞と反応している単一の抗体産生粒子を同定および単離すること;
を含む方法。
【請求項2】
前記抗体産生粒子が抗体産生細胞であり、前記抗体レパートリーが抗体産生細胞のレパートリーである、請求項1記載の方法。
【請求項3】
前記抗体産生細胞がプラズマ細胞、ハイブリドーマ、またはリンパ球から成る群より選択される、請求項2記載の方法。
【請求項4】
請求項2または3記載の方法であって:
1)抗体産生細胞を提供すること;
2)前記抗原およびマーカーを表示している小胞、好ましくはエキソソームを調製すること;
3)工程1のリンパ球を、工程2の小胞と懸濁すること;および
4)前記小胞と反応している単一の抗体産生細胞を同定および単離すること;
を含む方法。
【請求項5】
前記抗体産生細胞が、前記抗原により免疫化されたヒト以外の動物から収集したリンパ球である、請求項4記載の方法。
【請求項6】
請求項4記載の方法であって、前記抗体産生細胞が抗体分泌細胞であり、さらに、抗体分泌細胞を小胞と懸濁する工程の前に、抗体分泌細胞を、遍在する細胞表面マーカーに対する第1のビオチン化抗体、ストレプトアビジンおよび前記抗体分泌細胞の免疫グロブリンに向けられた第2のビオチン化抗体とインキュベーションする工程を含む方法。
【請求項7】
請求項5記載の方法であって:
1)少なくとも1つの抗原またはそのエピト−プを表示している免疫原性の小胞、好ましくはエキソソームを調製すること;
2)ヒト以外の動物を前記免疫原性小胞で免疫化することにより、抗体反応を生起させること;
3)免疫化した動物からリンパ球を収集すること;
4)工程1の前記抗原またはそのエピトープおよびマーカーを表示している小胞、好ましくはエキソソームを調製すること;
5)工程3のリンパ球を工程4の小胞と懸濁すること;および、
6)工程4の小胞と反応している単一の抗体産生細胞を同定および単離すること;
を含む方法。
【請求項8】
変異抗原に特異的な単一抗体を産生している粒子を抗体レパートリーから単離する方法であって:
1)前記変異抗原およびマーカーを表示している第1集団の小胞、好ましくはエキソソームを調製すること;
2)自然抗原を表示し、前記マーカーを表示していない第2集団の小胞、好ましくはエキソソームを調製すること;
3)前記抗体レパートリーを第1および第2集団の小胞と、第2集団を過剰にして、懸濁すること;および、
4)工程1の小胞と反応している単一の抗体産生粒子を同定および単離すること;
を含む方法。
【請求項9】
請求項8記載の方法であって:
1)前記変異抗原によって免疫化されたヒト以外の動物からリンパ球を収集すること;
2)動物の免疫化に用いた前記変異抗原およびマーカーを表示している小胞、好ましくはエキソソームを調製すること;
3)自然抗原を表示し、前記マーカーを表示していない小胞、好ましくはエキソソームを調製すること;
4)工程1のリンパ球を、工程2の前記変異抗原およびマーカーを表示している小胞ならびに工程3の前記自然抗原を表示している過剰量の小胞と共に懸濁すること;および、
5)工程2の小胞と反応している単一の抗体産生細胞を同定および単離すること;
を含む方法。
【請求項10】
請求項9の方法であって:
1)変異抗原を表示している免疫原性の小胞、好ましくはエキソソームを調製すること;
2)ヒト以外の動物を前記免疫原性小胞で免疫化すことにより抗体反応を生起させること;
3)免疫化した動物からリンパ球を収集すること;
4)工程1の前記変異抗原およびマーカーを表示している小胞、好ましくはエキソソームを調製すること;
5)自然抗原を表示し前記マーカーを表示していない小胞、好ましくはエキソソームを調製すること;
6)工程3のリンパ球を、工程4の前記変異抗原およびマーカーを表示している小胞、ならびに工程5の前記自然抗原を表示している過剰量の小胞と懸濁すること;および
7)工程4の小胞と反応している、抗原変異体に特異的な単一抗体を産生している細胞を同定および単離すること;
を含む方法。
【請求項11】
前記抗体産生粒子が抗体を産生しているファージまたは酵母であり、前記抗体レパートリーが抗体を産生するファージまたは酵母のレパートリーである、請求項1または8記載の方法。
【請求項12】
請求項1〜11のいずれか1項記載の方法であって、前記方法がさらに次の工程:
a)前記の選択された抗体産生粒子からDNAまたはRNAを回収すること、b)免疫グロブリン配列またはその一部をコードする核酸配列を増幅すること、c)増幅した核酸配列を発現ベクターへクローニングして、所望の抗原特異性を有するタンパク質を産生すること、を含む方法。
【請求項13】
前記免疫原性小胞がさらにアジュバントポリペプチドなどの免疫アクセサリー分子を表示する、請求項7または10のいずれかに記載の方法。
【請求項14】
前記アジュバントポリペプチドが、GM-CSF、IL-2およびCD40Lなどのサイトカインから、特に突然変異CD40Lから選択され、前記突然変異が可溶性CD40Lの切断と放出を阻害する、請求項13記載の方法。
【請求項15】
前記免疫アクセサリー分子がエキソソームターゲティングポリペプチドに融合または架橋している、請求項13記載の方法。
【請求項16】
前記免疫アクセサリー分子が少なくとも1つの膜貫通ドメインを有し、エキソソーム産生細胞中への過剰発現により免疫原性小胞に取り込まれる、請求項13記載の方法。
【請求項17】
前記変異抗原が突然変異抗原であり、前記自然抗原が野生型抗原である、請求項8〜10のいずれか1項記載の方法。
【請求項18】
前記変異抗原が、ポリペプチド、脂質、DNA、または小分子から成る群より選択される分子と接触している抗原であり、前記自然抗原がフリーの抗原である、請求項8〜10のいずれか1項記載の方法。
【請求項19】
前記変異抗原がMHC/ペプチド複合体であり、前記自然抗原が負荷されていないMHCであるかまたは別のペプチドを負荷されているMHCである、請求項18記載の方法。
【請求項20】
前記変異抗原がリガンド-レセプター複合体であり、前記自然抗原がフリーのリガンドまたはフリーのレセプターである、請求項19記載の方法。
【請求項21】
前記変異抗原および前記自然抗原が、酵素を含む任意の異なる立体構造状態のタンパク質である、請求項8〜10のいずれか1項記載の方法。
【請求項22】
前記小胞により表示されている前記抗原がエキソソームターゲティングポリペプチドに融合している、請求項1〜12のいずれか1項記載の方法。
【請求項23】
前記小胞によって表示されている前記抗原がエキソソームターゲティングポリペプチドに架橋されている、請求項1〜12のいずれか1項記載の方法。
【請求項24】
前記マーカーが、タグ、ビオチン、酵素、蛍光分子などの検出可能な分子である、請求項1〜23のいずれか1項記載の方法。
【請求項25】
前記マーカーがエキソソームターゲティングポリペプチドに融合または架橋されている、請求項24記載の方法。
【請求項26】
前記マーカーが、エキソソーム産生細胞中への過剰発現により免疫原性小胞に取り込まれる膜貫通ドメインを有している、請求項24記載の方法。
【請求項27】
前記マーカーが優先的に小胞へ、好ましくはエキソソームへ取り込まれるラベルされた脂質である、請求項24記載の方法。
【請求項28】
前記ラベルされた脂質がRh-DOPEまたはフルオレセイン-DOPEなどの蛍光団結合脂質である、請求項27記載の方法。
【請求項29】
前記抗原が少なくとも1つの膜貫通ドメインを有するポリペプチドであり、前記抗原がエキソソーム産生細胞中に過剰発現され、それにより前記抗原を表示している前記小胞を生成することができる、請求項1〜21のいずれか1項記載の方法。
【請求項30】
少なくとも1つの膜貫通ドメインを有する前記ポリペプチドがレセプターである、請求項29記載の方法。
【請求項31】
前記レセプターがGPCR(Gタンパク質共役レセプター)である、請求項30記載の方法。
【請求項32】
前記抗原が、例えばレセプターまたは酵素などの任意のタンパク質、あるいは糖脂質、多糖類、薬物および有機化学物質などのポリペプチド以外の化合物である、請求項1〜31のいずれか1項記載の方法。
【請求項33】
前記エキソソームターゲティングポリペプチドが、ラクトアドヘリンあるいは機能的なC1および/またはC2ドメインを含有するその一部を含む、請求項15、22、23または25のいずれか1項記載の方法。
【請求項34】
前記ラクトアドヘリンまたはその一部が、ヒト以外の哺乳動物のラクトアドヘリンまたはその一部である、請求項33記載の方法。
【請求項35】
前記ヒト以外の哺乳動物のラクトアドヘリンまたはその一部が、(i)マウスラクトアドヘリン、(ii)機能的なC1および/またはC2ドメインを含有するマウスラクトアドヘリンの断片、および(iii)(i)または(ii)のポリペプチドと少なくとも50%の一次構造同一性を有するポリペプチド、から選択される、請求項34記載の方法。
【請求項36】
前記ラクトアドヘリンが、配列番号1を含むアミノ酸配列あるいは機能的なC1および/またはC2ドメインを含有するその断片を有する、請求項35記載の方法。
【請求項37】
前記ラクトアドヘリンが、配列番号1の機能的C1/C2ドメインを含むアミノ酸配列を有する、請求項36記載の方法。
【請求項38】
前記ラクトアドヘリンが、配列番号1のアミノ酸残基111〜266、109〜266、271〜426、111〜426、または109〜426を含むアミノ酸配列を有する、請求項36記載の方法。
【請求項39】
前記エキソソームターゲティングポリペプチドが、Del-1、ニューロピリン-1、凝固因子5または凝固因子8の機能的なC1および/またはC2ドメインを含む、請求項15、22、23,または25のいずれか1項記載の方法。
【請求項40】
前記変異抗原がHLA-C/HIVペプチド複合体であり、前記自然抗原が負荷されていないHLA-Cまたは異なるペプチドを負荷されたHLA-Cである、請求項19記載の方法。
【請求項41】
請求項1〜39のいずれか1項記載の方法により得られる抗体産生細胞。
【請求項42】
請求項41記載の抗体産生細胞により産生される抗体。
【請求項43】
請求項41記載の抗体産生細胞および/または請求項42記載の抗体および薬学的に許容できる賦形剤または担体を含む組成物。
【請求項44】
少なくとも1つの膜貫通ドメインを有するポリペプチドをエキソソームの表面に発現させる方法であって:
1)前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを有するその一部をコードする遺伝子コンストラクトを提供すること;
2)エキソソーム産生細胞中に前記コンストラクトを導入し、前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを有するその一部を過剰発現させて、組換えエキソソームを生成すること;および、
3)前記遺伝子コンストラクトによってコードされるポリペプチドを表面に担持する前記組換えエキソソームを収集すること;
を含む方法。
【請求項45】
少なくとも1つの膜貫通ドメインを有する前記ポリペプチドがCD40Lである、請求項44記載の方法。
【請求項46】
前記CD40Lが突然変異CD40Lであって、前記突然変異が可溶性CD40Lの切断と放出を阻害する、請求項45記載の方法。
【請求項47】
少なくとも1つの膜貫通ドメインを有するポリペプチドまたはそのエピトープを結合する抗体を産生する方法であって:
1)前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを有するその一部をコードする遺伝子コンストラクトを提供すること;
2)エキソソーム産生細胞中に前記コンストラクトを導入し、前記ポリペプチドまたは少なくとも1つの膜貫通ドメインを有するその一部を過剰発現させて、表面に前記ポリペプチドまたはエピトープを提示している組換えエキソソームを生成すること;
3)前記組換えエキソソームを収集し、前記エキソソームまたはその一部をヒト以外の哺乳動物に注入して、前記ポリペプチドまたはエピトープを結合する抗体を生成すること;および、
4)前記哺乳動物から抗体または抗体産生細胞を収集すること;
を含む方法。
【請求項48】
前記エキソソームがさらに突然変異CD40Lを表示し、前記突然変異が可溶性CD40Lの切断と放出を阻害する、請求項47記載の方法。
【請求項49】
前記少なくとも1つの膜貫通ドメインを有するポリペプチドがレセプターである、請求項44または47記載の方法。
【請求項50】
前記レセプターがGPCR(Gタンパク質共役レセプター)である、請求項49記載の方法。
【請求項51】
少なくとも1つの膜貫通ドメインを有する抗原、または少なくとも1つの膜貫通ドメインを有するその一部を、対象に送達する方法であって:
a)前記抗原または少なくとも1つの膜貫通ドメインを有するその一部をコードする遺伝子コンストラクトを提供すること;
b)エキソソーム産生細胞中に前記コンストラクトを導入し、前記抗原または少なくとも1つの膜貫通ドメインを有するその一部を過剰発現させて、前記抗原または前記その一部を表面に提示している組換えエキソソームを生成すること:
c)前記組換えエキソソームを収集し、前記エキソソームまたはその一部を前記対象に注入すること;
を含む方法。
【請求項52】
少なくとも1つの膜貫通ドメインを有する特異的な抗原、または少なくとも1つの膜貫通ドメインを含むその一部に対して、対象中に免疫応答を引き起こす方法であって、:
a)前記抗原または少なくとも1つの膜貫通ドメインを含むその一部をコードする遺伝子コンストラクトを提供すること;
b)エキソソーム産生細胞中に前記コンストラクトを導入し、前記抗原または少なくとも1つの膜貫通ドメインを含むその一部を過剰発現させて、前記抗原または前記その一部を表面に提示している組換えエキソソームを生成すること;
c)前記組換えエキソソームを収集し、前記エキソソームまたはその一部を、前記対象に注入すること;
を含む方法。
【請求項53】
前記抗原がレセプターである、請求項51または52に記載の方法。
【請求項54】
前記レセプターがGPCR(Gタンパク質共役レセプター)である、請求項53記載の方法。
【請求項55】
追跡可能なエキソソームを調製する方法であって、次の工程:
1)エキソソーム産生細胞を蛍光団結合脂質とインキュベーションすること;および、
2)前記蛍光団結合脂質を取り込んだ前記細胞からエキソソームを産成し単離すること;
を含む方法。
【請求項56】
前記蛍光団結合脂質が蛍光団結合DOPE(ジオレオイル-ホスファチジルエタノールアミン)である、請求項55記載の方法。
【請求項57】
前記蛍光団結合DOPEがRh-DOPEまたはフルオレセイン-DOPEである、請求項56記載の方法。
【請求項58】
クローニング部位に突然変異を導入することなく、配列をベクター中にクローニングする方法であって、次の工程:
1)II型制限酵素により認識される2つの制限部位を備え、制限酵素切断後に制限部位が前記ベクターから除去されるように構成されたクローニングカセットを含むベクターを提供すること;
2)前記配列に、互換性のある末端を与えること;
3)前記ベクターを前記制限酵素とインキュベーションすること;
4)前記配列と工程3で得られた前記ベクターをライゲーションすること;
5)前記配列を含む前記ベクターを単離すること;
を含む方法。
【請求項59】
前記配列が免疫グロブリンの可変配列であり、前記ベクターの前記クローニングカセットが、その一端を免疫グロブリンのリーダー配列に、他端を定常ドメインに接している請求項58記載の方法。
【図1】

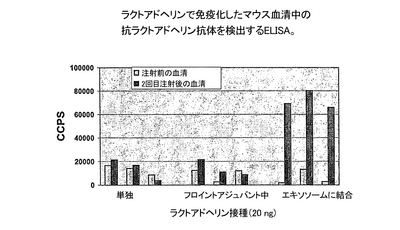
【図2】
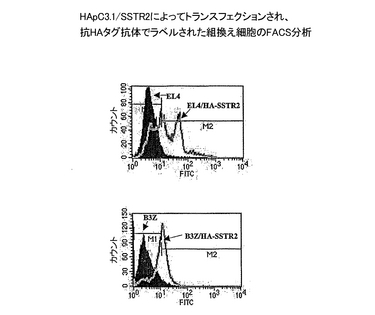
【図3】
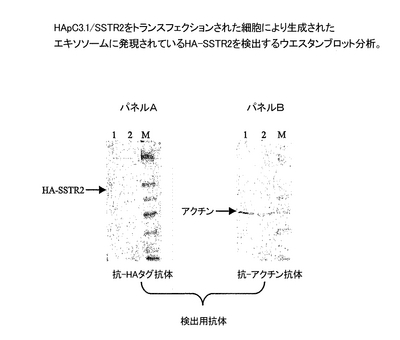
【図4】
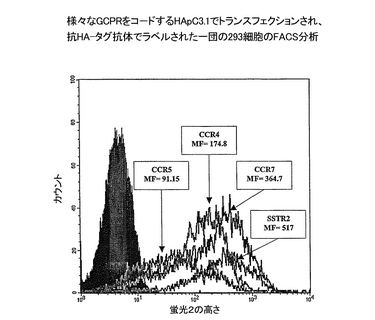
【図5】
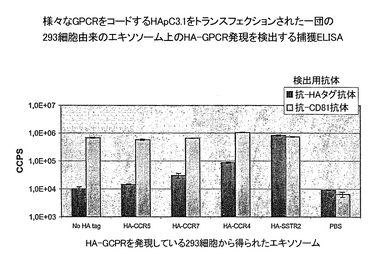
【図6】
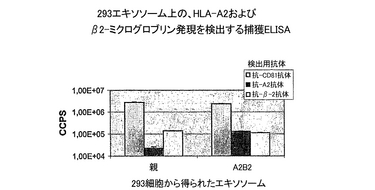
【図7】
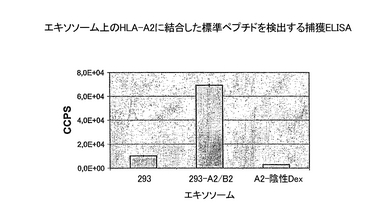
【図8】
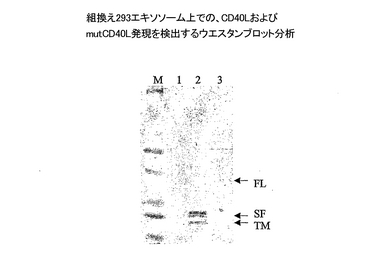
【図9】
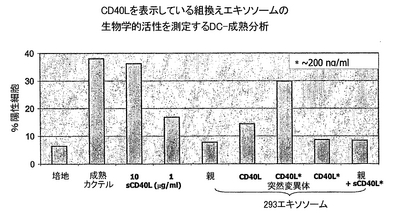
【図10】
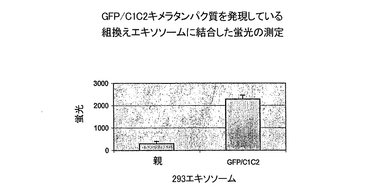
【図11】

【図12】
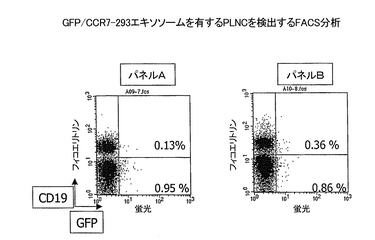
【図13】
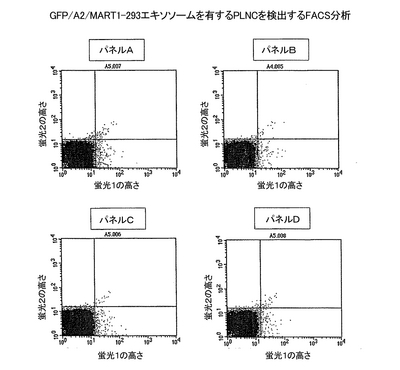
【図14】
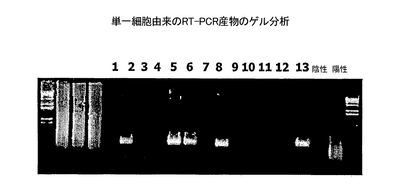
【図15】
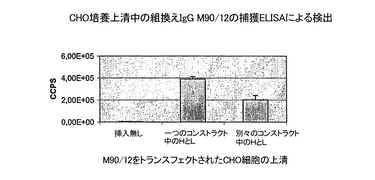
【図16】
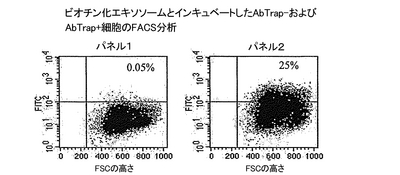
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【公表番号】特表2006−518212(P2006−518212A)
【公表日】平成18年8月10日(2006.8.10)
【国際特許分類】
【出願番号】特願2006−502494(P2006−502494)
【出願日】平成16年2月13日(2004.2.13)
【国際出願番号】PCT/IB2004/000888
【国際公開番号】WO2004/073319
【国際公開日】平成16年8月26日(2004.8.26)
【出願人】(501278766)アノシス・インコーポレーテッド (1)
【氏名又は名称原語表記】ANOSYS,INC.
【Fターム(参考)】
【公表日】平成18年8月10日(2006.8.10)
【国際特許分類】
【出願日】平成16年2月13日(2004.2.13)
【国際出願番号】PCT/IB2004/000888
【国際公開番号】WO2004/073319
【国際公開日】平成16年8月26日(2004.8.26)
【出願人】(501278766)アノシス・インコーポレーテッド (1)
【氏名又は名称原語表記】ANOSYS,INC.
【Fターム(参考)】
[ Back to top ]