
抗体ライブラリー
【課題】イムノグロブリンの軽鎖可変領域遺伝子と重鎖可変領域遺伝子の組み合わせからなる遺伝子ライブラリーを提供する。
【解決手段】軽鎖可変領域遺伝子として、重鎖可変領域遺伝子の発現産物との機能的なコンフォーメーションの再構成が可能な軽鎖分子をコードするものを選択し、得られる軽鎖可変領域遺伝子の集合である遺伝子のライブラリーを調製する。本抗体ライブラリーは、重鎖可変領域の多様性を生体外において高度に維持することができる。そのため、様々な結合活性を持つ抗体の取得が期待できる。
【解決手段】軽鎖可変領域遺伝子として、重鎖可変領域遺伝子の発現産物との機能的なコンフォーメーションの再構成が可能な軽鎖分子をコードするものを選択し、得られる軽鎖可変領域遺伝子の集合である遺伝子のライブラリーを調製する。本抗体ライブラリーは、重鎖可変領域の多様性を生体外において高度に維持することができる。そのため、様々な結合活性を持つ抗体の取得が期待できる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、抗体可変領域をコードするDNAを含む抗体ライブラリーに関する。
【背景技術】
【0002】
動物個体は体液中に侵入する様々な異物に対して、その物質の表面に位置する種々の構造(エピトープ)を認識して特異的に結合する抗体を産生する能力がある。その抗体レパートリーの大きさ(異なる抗原に結合する違ったアミノ酸配列を持つ抗体の種類の総数)は、動物個体あたり100万種〜1億種と見積もられている。このような巨大な抗体レパートリーは、骨髄幹細胞が抗体産生細胞であるBリンパ細胞へ分化する途上に、抗体遺伝子座で重鎖ではVH-D-JH、軽鎖ではVL-JLというDNA再編成を起こすことによって生じる。このDNA再編成はB細胞毎に独立に起こるプロセスである。したがって一組のVH-D-JH、VL-JL遺伝子を持つ1個のB細胞は1種類の抗体しか産生しない。しかし個体を構成するB細胞全体では、多様な抗体の産生が可能となる。
【0003】
従来行われてきた抗血清調製技術、および細胞融合によるモノクローン抗体作製技術は、この動物の持つ抗体産生機構を利用している。すなわち、抗原物質をアジュバンドと共に動物(ウサギ、ヤギ、マウス等)に一定間隔をおいて数回注入する。動物の免疫系がその物質を異物と認識すると、その抗原物質に結合する抗体を発現したB細胞が増殖分化刺激を受け、大量の抗体の体液中への分泌を開始する。抗原物質の表面には様々な構造が存在し、精製された抗原といえどもそれに結合する抗体は、通常は多種類の抗体の混合物として分泌される。このような抗体を含む血清(抗血清)はポリクローン抗体と呼ばれる。現在もポリクローン抗体は、研究試薬として有効に利用されている。しかしポリクローン抗体は、目的とする抗原物質以外にも、部分的に(抗体にとって)類似した構造を持つ分子とも交叉反応性(cross-reactivity)を示すことが多い。交叉反応は、ポリクローン抗体を抗原検出試薬として用いるときの問題点となっていた。
【0004】
細胞融合技術の確立がこの状況を一変させた。抗原物質で免疫した動物の脾臓には抗原と結合する抗体を産生するBリンパ球は多数存在する。しかしその細胞を試験管内で長期間維持培養し続けることは困難である。そこで永代培養が可能なように株化した腫瘍細胞と抗体産生細胞を融合することにより、抗体を産生し永代培養可能な細胞を作製するというアイデアが生まれ、方法として確立した。このようにして確立した融合株(ハイブリドーマ)は1個の抗体産生細胞と1個の腫瘍細胞に由来するので産生される抗体は1種類であり、モノクローン抗体と呼ばれる。これはケラーとミルスタインにより1975年に開発された技術である。モノクローン抗体は、均一な抗体分子の集合体であることから、交叉反応を生じにくい特異性に優れた抗体として利用されている。しかしこの方法にも次のような問題点が指摘されている。
(1)抗原物質として必要充分量の精製標品を有していること
(2)その物質が免疫される動物に対して免疫原性を示す必要があること
(3)モノクローン抗体を得るまでに多大な労力と日数を要すること
【0005】
ポリクローン抗体やモノクローン抗体の作製技術の有効性は、多くの有用な抗体を提供してきたことによって証明されている。しかし、これらの方法によっては解決が困難な多くの課題が今なお存在することも事実である。たとえば、多種類の抗原に対する抗体を短期間に得ること、あるいは特殊な構造をしたエピトープに特異的に結合する抗体を選択的に得ること、といった要求には、これらの方法では応えることはできない。希望する抗体を短期間で得ることができるように、多様な抗体分子で構成された抗体ライブラリーの作製が望まれていた。このような抗体ライブラリーに含まれる抗体の種類は、理論的には動物個体の有する抗体レパートリーの大きさに匹敵する必要がある。しかし現実には、動物細胞を用いてそのような巨大なライブラリーを作製することは不可能である。モノクローン抗体の作製は、動物の持つ抗体産生細胞のライブラリーから、期待する反応性を持つ抗体をスクリーニングする作業に他ならない。しかし、そのライブラリーを構成するレパートリーは、細胞融合などの操作を通じて大幅に失われてしまう。
【0006】
そこで、大腸菌による抗体遺伝子の発現系が提案された。大腸菌中で抗原結合力のある抗体を発現させるのに初めて成功したのはBetter et al(1988)(Better M, Chang CP, Robinson RR, Horwitz AH Science 1988, 240:4855 1041-3:非特許文献1)とSkerraとPlukthun(1988) (Skerra A, Plukthun A Science 1988, 240:4855 1038-41:非特許文献2)である。彼らは大腸菌で分泌シグナルとして機能する配列を抗体のN末端に付加することによりFab型とFv型抗体を大腸菌中での産生、それに続く分泌に成功した。
更に、1988年のPCRの開発は、直ちに抗体可変領域をコードする遺伝子の増幅に応用される。動物(とりわけヒト)中で発現されている全てのVHDJH、VLJL遺伝子を増幅するためのプライマー配列が提案された(Orlandi R et al. Proc Natl Acad Sci USA 1989 86:10 3833-7, Sastry L et al. Proc Natl Acad Sci USA 1989 86:15 5728-32:非特許文献3)。そしてこれらのプライマーによって増幅した抗体遺伝子を利用して、大腸菌中で抗体を産生させるためのベクターが構築された(Huse WD et al.Science 1989 246:4935 1275-81, Ward GE et al.J Clin Microbiol 1989 27:12 2717-23:非特許文献4)。この段階で、抗体ライブラリーのレパートリーサイズは飛躍的に向上した。しかし、大腸菌中で産生される微量の抗体について抗原との結合活性を指標とするスクリーニングを行うことは、容易なことではなかった。スクリーニングを能率的に行うには、ファージディスプレー法が抗体ライブラリー作製に応用されるのを待つ必要があった。
【0007】
ファージディスプレー法はSmithにより1985年(Smith GP Science 1985 228:4075 1315-7:非特許文献5)に考案されたもので、M13ファージのような一本鎖環状DNAを持つ線状のバクテリオファージが用いられる。ファージ粒子はDNAの周囲を取り囲んでファージ粒子の大部分を構成するcp8というタンパクと、ファージが大腸菌に感染する時に機能する5個のcp3と呼ばれるタンパクからなっている。このcp3もしくはcp8と融合した形でポリペプチドをコードするように遺伝子を構築し、ファージ粒子表面にそのタンパクを発現させるシステムがファージディスプレーシステムである。結合性のタンパク質を表面に保持したファージ粒子は、そのリガンドとの結合活性を利用して濃縮することができる。こうして目的とするDNAを濃縮する方法は、パニング法と呼ばれている。濃縮されたファージ粒子には、必要な結合活性を持つタンパク質をコードするDNAがパッケージングされている。このように繊維状ファージの利用によって、結合活性に基づくスクリーニングと、DNAのクローニングとをきわめて効率的に行うことができるシステムが実現した(特表平5-508076:特許文献1)。繊維状ファージを使ったライブラリーには、Fab分子として発現が可能な方法も報告された(特表平6-506836:特許文献2)。この報告において、cp3等のN末端を欠損させて可変領域を融合させる方法が試みられた。
【0008】
ファージディスプレー系が抗体に応用され、VHドメインのみ、scFv、Fv、Fab型抗体がcp3又はcp8と融合された形で発現された。抗原と結合するファージ抗体は同時に抗体をコードする遺伝子を含む。しかしファージディスプレー系を用いて作製された初期に抗体ライブラリーから単離された抗体は、抗原結合力の低いものが多かった。結合力を高める試みの一つとして、人為的に遺伝子に変異を与える方法が提案された。Winterらは(1994)、単離した全てのVH、VL遺伝子と、JH、JL遺伝子の間にランダムな配列を挿入する半人工的配列を持つ抗体ライブラリーを作製することにより、親和性に優れた抗体の取得を可能とする抗体ライブラリーを得た(Nissim A, Winter G et al. EMBO J 1994 13:3 692-8:非特許文献6)。De Kruifら(1995)も基本的に同じ原理に基づいて抗体ライブラリーを作製している(de Kruif J, Boel E, Logtenberg T J Mol Biol 1995 248:1 97-105:非特許文献7)。Vaughanら(1996)は、ライブラリーの大きさを拡大することで充分な大きさの抗体レパートリーを確保しようとしている(Vaughan TJ et al. Nat Biotechnol 1996 14:3 309-14:非特許文献8)。このような試みは、確かに限られた抗原に対しては成功を収めた。しかし必要とする抗体を得ることができる可能性は未だに不充分である。たとえば、マウスにおいてモノクローン抗体を得ることができるのと同じような確率でヒト抗体を得ることができるライブラリーを現在の技術で構築することは不可能である。したがって、より多様な抗体から構成されるライブラリーの提供が望まれている。
【0009】
ヒトが産生することが可能な抗体を完全にin vitroでライブラリー化し、様々な抗原に特異的に結合する抗体が得られるようにするには、ヒトが抗体を産生する過程を試験管内で忠実に反映できれば理想的である。抗体が抗原と結合する部位はHL両鎖のN末端に位置する可変(V)ドメインの中の各々抗原相補性決定領域(complementarity determining region;以下CDRと省略する)I、II、IIIの計6ヶ所である。CDRを構成するアミノ酸配列の多様性(長さに関する多様性も含めて)の総数が、そのまま抗体レパートリーの大きさを反映していると考えてよい。
抗体レパートリーを考える時に、体内への抗原侵入前の「ナイーブレパートリー」と抗原侵入後に起こる「抗体の成熟」を考慮する必要がある。抗体をコードする活性型抗体遺伝子はDNA再編成によって作り出される。軽鎖にはλ鎖とκ鎖があるが、そのV領域をコードする遺伝子はVL遺伝子とJL遺伝子に分かれている。λ鎖においてVλ遺伝子は36個存在し、Jλ遺伝子は7個存在する。B細胞への分化途上、軽鎖に関してはκ鎖かλ鎖でVL遺伝子とJL遺伝子の近傍でDNAの切断と再結合が起こり、VL-JL遺伝子ができる。多くの例(2/3)で1〜96番目のアミノ酸がVL遺伝子由来、97〜110番目のアミノ酸がJL遺伝子由来となる。しかしV(D)J DNA組換え酵素(群)は、DNAを切断後、つなぐべき末端をエキソヌクレアーゼで少し除いたのち再結合させる。その結果、VL-JL遺伝子でコードされたVLドメインの大きさに±3アミノ酸程度の差異が生じる。軽鎖のCDR1は24-34番目のアミノ酸に、CDR2は50-56に、そしてCDR3は89-97番目に相当するので、その作り出す多様性の総数はλ鎖の場合(Vλ遺伝子の数)x(Jλ遺伝子の数)x(ズレの総数)となる。ただしJλ遺伝子の場合その配列は相互に似ており、結合点のズレに関しても67%が一定で27%以上が±1であるので、実際のVλ-Jλ遺伝子レパートリーは大きく見積もっても200を越えない。κ遺伝子もλ遺伝子に状況が似ている。Vκ遺伝子総数は37個、Jκ遺伝子数は4個である。そこでVκ-Jκ遺伝子レパートリーも200種を越えない。
【0010】
軽鎖可変領域の多様性の度合いが比較的小さいのに比べると、重鎖可変領域では大きく状況が異なる。CDR1(31-35b番目のアミノ酸)とCDR2(50-65番目のアミノ酸)については36個のVH遺伝子にコードされる部分なので多様性は限られている。問題はCDR3である。抗体の抗原結合面全体を眺めた時も、HL両鎖のCDR1、2が両側に位置し、CDR3が真ん中に位置する。重鎖のCDR1,2,3が全体の60%ぐらいを占め、軽鎖が40%ぐらいの面積を占める。重鎖CDR3を除く残りの部分は軽鎖の多様性(多くて数百種)x重鎖CDR1,2の多様性(36)=計約1万種程度のレパートリーである。重鎖CDR3は独立したD遺伝子にコードされる領域だが、D遺伝子は26個存在する。CDR3をコードする領域はD-JH、VH-Dという2回のDNA再編成後VH-D-JHとなって完成する。
【0011】
問題はこのDNA再編成過程で、次に示すような過程が存在することである。
(1)VH、D、JHの近傍に存在するシグナル配列真横のDNAの切断、
(2)エキソヌクレアーゼによるDNA末端部分切除、
(3)末端添加酵素によるランダム配列(Nと呼ばれる)の挿入、
(4)DNAの修復と連結
これらの過程において、重鎖では(2)の過程のバリエーションが軽鎖に比べて大きい。更により決定的なことは、軽鎖ではみられなかった(3)の過程が存在することである。重鎖CDR3(アミノ酸95-102番目に相当)は、92番目のシステインと103番目のトリプトファンに挟まれた領域として存在する。その長さは5アミノ酸から20アミノ酸を越えるものまで、まちまちの長さを持つことに加えて、その配列も多種多様である。このような特徴のために、CDR3は事実上独立して分化したB細胞毎に異なるほど多様である。
【0012】
B細胞分化途上に抗体遺伝子座で起こる重鎖のVH-D-JH、軽鎖のVL-JL DNA再編成は、抗原の有無に関係なく起こる。1個のB細胞が1組のVH-D-JH、VL-JL遺伝子を発現し、B細胞全体で形造る抗体集団のことを抗体のナイーブレパートリーと呼ぶ。抗原侵入後、抗原と結合できる性質を持つ抗体を発現した細胞は増殖、分化刺激を受ける。抗体を分泌すると同時にその抗体可変領域をコードする遺伝子(VH-D-JH、VL-JL)に高頻度に突然変異が導入される。その変異の導入によって抗原に対する結合力の高まった抗体を産生する細胞が生き残り、優れた性能の抗体を分泌し続けると同時に記憶細胞として残る。これが「抗体の成熟」である。ここで重要なことは突然変異の役割である。もともと抗体ナイーブレパートリーの中に存在しなかった抗原特異性が、突然変異の導入で生まれることはない。したがってin vitroで抗体をライブラリー化する場合に、生体内における抗体産生過程を再現するのであれば、ナイーブレパートリーに存在しない抗原特異性を持つクローンを排除する機構が必要である。
【0013】
以下に、in vitroで抗体ライブラリーを作製する上での問題点を示す。
(1)個々の抗体遺伝子は、たとえば大腸菌中で大量に発現され、フォールディングし、重鎖可変領域と軽鎖可変領域ドメインが会合して抗体分子となる。これらの段階のいずれかが正しく起こらなかったクローンは、イムノグロブリンとしての正常な構造を再現することができないので意味を持たない。
(2)in vivoでは個々の細胞中で重鎖可変領域と軽鎖可変領域がペアをつくって1つの抗原特異性を示すが、in vitroでライブラリー化する場合には重鎖可変領域集団と軽鎖可変領域集団を別個にライブラリー化した後に組み合わせざるを得ない。たとえば1万種からなるB細胞のレパートリーは、理論的には重鎖可変領域1万×軽鎖可変領域1万=合計1億種のライブラリーでやっとその全てをカバーすることが可能となる。しかし単純に組み合わせの数を増やすだけでは、ライブラリーの規模を大きくする一方で、ライブラリーに占める抗体としての活性を持たないクローンの割合も増加する。
(3)抗体遺伝子のソースとしてヒト血液を用いると、各人の免疫学的経歴によって発現されている抗体遺伝子に大きな偏りがあるはずである。
【0014】
以上の3つの問題点は、いずれもライブラリーのレパートリーに偏りを与える原因となる。つまり、ライブラリーを構成する理論的なクローン数に比べて、現実のライブラリーに含まれる機能的なファージ抗体の種類が著しく少ないというギャップを生じることになる。
より具体的には、特定の抗原に対して免疫応答が亢進しているために、使用頻度に於いて非常に偏りのあるクローンからなるライブラリーが考えられる。あるいは、十分な抗原結合力を持たない抗体をコードする多数のクローンを含む抗体ライブラリー等も生成されるであろう。たとえば軽鎖可変領域遺伝子と重鎖可変領域遺伝子の、それぞれ50%が抗体として活性な分子を発現できるとすると、両者の組み合わせによって活性な抗体が構成される割合はわずかに25%である。
【0015】
このようなライブラリーの問題点は、実際のレパートリーサイズが理論的なサイズよりも遙かに小さいことに留まらない。たとえば、スクリーニングにおいては抗原結合力の弱い抗体分子は、免疫反応を妨害する。すなわち、抗体―抗原複合体形成は平衡反応であるために、多数のクローンと少数のクローンが混在すると、その抗原結合力の差を越えて多数派が少数派を駆逐することになる。
また不活性な抗体をコードするクローンは、クローニングにおいても障害となる。つまり、不活性な抗体をコードするクローンが多ければ、その中には増殖速度が特に速いクローンが生じる可能性も高まる。増殖速度に優れるクローンは、スクリーニング過程で選択的に選ばれてしまうので、非常に高いバックグラウンドとなる。
今までに報告されたライブラリーの問題点は、ライブラリーを構成するクローンの中で、実際にどれだけのクローンが有効であるのかを解析できる形で作製されていないことにある。したがって、ライブラリーやスクリーニングの効率についての評価ができない。
【先行技術文献】
【特許文献】
【0016】
【特許文献1】特表平5-508076
【特許文献2】特表平6-506836
【非特許文献】
【0017】
【非特許文献1】Better et al(1988)(Better M, Chang CP, Robinson RR, Horwitz AH Science 1988, 240:4855 1041-3)
【非特許文献2】Skerra A, Plukthun A Science 1988, 240:4855 1038-41
【非特許文献3】Orlandi R et al. Proc Natl Acad Sci USA 1989 86:10 3833-7, Sastry L et al. Proc Natl Acad Sci USA 1989 86:15 5728-32
【非特許文献4】Huse WD et al.Science 1989 246:4935 1275-81, Ward GE et al.J Clin Microbiol 1989 27:12 2717-23
【非特許文献5】Smith GP Science 1985 228:4075 1315-7
【非特許文献6】Nissim A, Winter G et al. EMBO J 1994 13:3 692-8
【非特許文献7】de Kruif J, Boel E, Logtenberg T J Mol Biol 1995 248:1 97-105
【非特許文献8】Vaughan TJ et al. Nat Biotechnol 1996 14:3 309-14
【発明の概要】
【0018】
本発明の課題は、機能的なコンフォーメーションを保持した抗体分子を高い割合で含む抗体ライブラリーを提供することである。本発明は、このような抗体ライブラリーと、その製造方法、ならびにそのライブラリーを使った抗体のスクリーニング方法の提供を課題とする。また本発明は、機能的なコンフォーメーションを保持しうる抗体分子を与えるイムノグロブリンの軽鎖をコードする遺伝子を単離する方法の提供を課題としている。
【0019】
本発明者らは、公知の抗体レパートリーから必要な抗体をスクリーニングすることを妨げている要因について研究を重ねた。そして、現在までに知られている抗体ライブラリーにおいては、そのレパートリーに占める、機能的なコンフォーメーションを保持した抗体分子の割合が少ないことが、有用な抗体の単離を妨げているのではないかと考えた。公知の抗体ライブラリーの製造方法においては、生体内に保持されている抗体レパートリーを忠実にin vitroで構成することを目標として様々な工夫が提案された。
【0020】
ある場合には、多様性を高めるために人為的な変異をランダムに挿入する方法さえ試みられた。しかし、こういった試みの多くは、機能的なコンフォーメーションを維持した抗体分子を生成する一方で、明らかに抗体としての活性が不充分な抗体分子の生成も伴っていた。その結果、得られた抗体ライブラリーには、生体内では用いられることのないレパートリーや、多くの不活性な抗体分子が含まれ、機能的な抗体のスクリーニングを妨げる原因となってしまう。たとえば軽鎖可変領域遺伝子を105個の独立したクローンとし、これに109個の重鎖可変領域遺伝子を組み合わせてライブラリーを構成したとする。数の上では重鎖可変領域遺伝子は十分な多様性を与えたことになるが、軽鎖可変領域遺伝子に不活性ものが多く含まれているとすると、重鎖可変領域遺伝子の一部はイムノグロブリンとしての活性を得ることができずスクリーニングの段階で失われしまう。
生体内においては、多様な抗体分子をランダムに生成する機構とともに、抗体としての活性に優れる抗体を産生するクローンが選択的に増殖する機構が働いている。in vitroにおいても、不活性な抗体を除くステップを設けなければ、単に多様性を追求するのみでは、真に生体内の抗体レパートリーを再現することにはつながらない。
【0021】
このような背景の下で、本発明者らは、抗体ライブラリーに占める機能的な抗体分子の割合を高めることによって、より効率的な抗体の単離が可能になると考えた。そのためには、公知の抗体ライブラリーにおいて不活性な抗体分子をもたらす原因を明らかにしなければならない。本発明者らは、抗体活性の維持に果たす軽鎖の役割に着目した。そして、まず、抗体分子に機能的なコンフォーメーションを与えることができる軽鎖をスクリーニングする方法を確立した。
【0022】
更に本発明者らは、こうして選択された軽鎖可変領域遺伝子の構造を丹念に解析することによって、限られた構造の軽鎖可変領域遺伝子のみを利用することによって、機能的なコンフォーメーションを維持した抗体分子を高い割合で含む抗体ライブラリーの構築が可能となることを見出し本発明を完成した。軽鎖可変領域遺伝子の選択工程を経ることによって、抗体レパートリーを狭めてしまう可能性が心配される。しかしながら本発明者らは、本発明の選択方法によって選択された軽鎖可変領域遺伝子の構造を丹念に解析した結果、重鎖と機能的なコンフォーメーションを再構成しうる軽鎖の構造は、現実には限られた範囲に収束することを見出した。機能的な抗体分子を構成する軽鎖の構造が、一定のレパートリーに集約されることを明らかにし、これをライブラリーの調製に応用した点に本発明の最大の特徴がある。すなわち本発明は、以下の工程を含んでなるイムノグロブリンの遺伝子ライブラリーの調製方法と、このライブラリーに基づくrgdpライブラリー、更にはこれらのライブラリーから特定の抗原を認識する抗体をコードする遺伝子をスクリーニングする方法に関する。また本発明は、以下に示す工程からなる、重鎖と機能的なコンフォーメーションを再構成することができる軽鎖をコードする遺伝子の選択方法を提供する。
【0023】
〔1〕 以下の工程を含む、イムノグロブリンの軽鎖可変領域遺伝子と重鎖可変領域遺伝子の組み合わせからなる遺伝子ライブラリーの調製方法。
a)軽鎖可変領域遺伝子として、重鎖可変領域遺伝子の発現産物との機能的なコンフォーメーションの再構成が可能な軽鎖分子をコードするものを選択し、
b)工程a)によって得られる軽鎖可変領域遺伝子の集合である遺伝子のライブラリーを調製し、
c)工程b)のライブラリーに重鎖可変領域をコードする遺伝子のライブラリーを組み合わせる
〔2〕 工程c)において、VHファミリーごとにそれぞれ独立して調製した重鎖可変領域遺伝子のライブラリーを、生体内におけるVHファミリーの割合に応じて組み合わせる〔1〕に記載の方法。
〔3〕 工程c)における遺伝子ライブラリーの組み合わせを同一のベクター上で行う〔1〕に記載の方法。
〔4〕 更に次の工程d)を含む、〔1〕に記載の方法。
d)重鎖可変領域に標識ペプチドが融合しており、この標識ペプチドを指標として重鎖を発現するクローンを選択する工程
〔5〕 〔1〕に記載の方法によって得ることができる遺伝子ライブラリー。
〔6〕 少なくともイムノグロブリンの軽鎖可変領域をコードする遺伝子からなる遺伝子ライブラリーであって、イムノグロブリンの重鎖可変領域と機能的なコンフォーメーションを再構成できない軽鎖可変領域をコードする遺伝子が実質的に排除されている遺伝子ライブラリー。
〔7〕 ライブラリーが重鎖可変領域をコードする遺伝子のライブラリーを伴っている〔6〕に記載のライブラリー。
〔8〕 VHファミリーのそれぞれの重鎖可変領域遺伝子のライブラリーが、生体内の多様性を包含するのに十分なクローン数を有する〔7〕に記載のライブラリー。
〔9〕 ライブラリーを構成する遺伝子が、細菌、酵母、および植物細胞からなる群から選択されるいずれかの宿主細胞に導入されたものである〔6〕に記載のライブラリー。
〔10〕 ライブラリーを構成する遺伝子が、哺乳動物細胞に導入されたものである〔6〕に記載のライブラリー。
〔11〕 ライブラリーを構成する少なくとも一部の遺伝子が、繊維状ファージに組み込まれている〔6〕に記載のライブラリー。
〔12〕 ライブラリーを構成する遺伝子によってコードされる重鎖可変領域と軽鎖可変領域の断片を繊維状ファージ表面に発現し、かつそれらが機能的に再構成されている〔11〕に記載のライブラリー。
〔13〕 重鎖可変領域に標識ペプチドをコードする遺伝子が融合されている〔12〕に記載のライブラリー。
〔14〕 イムノグロブリン軽鎖可変領域遺伝子がヒトに由来するものである〔6〕に記載のライブラリー。
〔15〕 イムノグロブリン軽鎖超可変部位がシステイン残基を含まないアミノ酸配列からなる〔14〕に記載のライブラリー。
〔16〕 〔5〕に記載の遺伝子ライブラリーを構成する各クローンが、そのクローンによって保持される遺伝子によってコードされる抗体タンパク質を伴っているrgdpライブラリー。
〔17〕 〔5〕に記載の遺伝子ライブラリーを構成する各クローンについて、そのクローンによって保持される遺伝子によってコードされる抗体タンパク質から構成される抗体ライブラリー。
〔18〕 次の工程を含む、重鎖可変領域と機能的なコンフォーメーションを再構成することができる軽鎖可変領域をコードする遺伝子の選択方法。
a)軽鎖可変領域をコードする、1つまたは複数の遺伝子を取得する工程、
b)軽鎖可変領域と機能的なコンフォーメーションを構成することが確認されているイムノグロブリン重鎖可変領域をコードする遺伝子を取得する工程、
c)工程a)によって得られた軽鎖可変領域をコードする遺伝子の任意の1つを選択し、工程b)で取得した重鎖可変領域をコードする遺伝子とイムノグロブリンの機能的なコンフォーメーションを再構成が可能な条件下でタンパク質に翻訳する工程、
d)工程c)において翻訳されたタンパク質の抗原結合領域の形成を検出する工程、および
e)抗原結合領域の形成が検出されたタンパク質を構成する軽鎖可変領域をコードする遺伝子を選択する工程
〔19〕 更に、次の工程f)を含む〔18〕に記載の方法。
f)軽鎖可変領域遺伝子の塩基配列を決定し、その塩基配列によってコードされるアミノ酸配列を比較して同一のアミノ酸配列をコードする遺伝子、およびアミノ酸の欠損を生じている遺伝子を排除する工程
〔20〕 イムノグロブリンの軽鎖可変領域が、繊維状ファージの表面に発現している〔18〕に記載の方法。
〔21〕 イムノグロブリンの軽鎖可変領域をコードする遺伝子と重鎖可変領域をコードする遺伝子が挿入されたファージミドを感染させた宿主微生物の培養上清そのものを、工程c)のための軽鎖を含む試料として利用する〔18〕に記載の方法。
〔22〕 次の工程を含む、特定の抗原と結合するイムノグロブリン可変領域の検出方法。
a)〔5〕または〔7〕に記載のライブラリー、またはその発現産物と、該抗原とを抗原抗体反応に適した条件下に接触させる工程
b)該抗原とイムノグロブリン可変領域との結合を検出する工程
〔23〕 ライブラリーが〔12〕に記載のライブラリーである〔22〕に記載の方法。
〔24〕 〔22〕に記載の方法の後、更に次の工程c)を行うことを特徴とする、特定の抗原と結合するイムノグロブリン可変領域の取得方法。
c)前記抗原と結合するイムノグロブリン可変領域を発現するクローンを選択する工程
〔25〕 更に次の工程d)−e)を含む〔24〕に記載の方法。
d)工程c)で選択したファージクローンを増幅して2次的なライブラリーを得る工程、
e)工程c)において選択されるクローンの回収率が上昇するまで、2次的なライブラリーについて、工程a)−d)を繰り返す工程
〔26〕 〔24〕に記載の方法によって得られたクローン、またはイムノグロブリン断片、もしくはそれをコードする遺伝子。
〔27〕 配列番号:61〜配列番号:78のいずれかに記載の塩基配列を含む、ポリヌクレオチド。
〔28〕 配列番号:79〜配列番号:96のいずれかに記載のアミノ酸配列を含む蛋白質。
〔29〕 次の要素からなる抗体ライブラリーの調製用キット。
a)重鎖可変領域と機能的なコンフォーメーションを再構成することができない軽鎖可変領域をコードする遺伝子を実質的に排除した軽鎖可変領域遺伝子ライブラリー、および
b)重鎖可変領域をコードする遺伝子を増幅することができるプライマーセット
〔30〕 以下の工程からなる抗体ライブラリーの調製方法。
a)ファージミドに、イムノグロブリンの少なくとも可変領域を含む領域をコードする遺伝子を組み込む工程
b)工程a)で得られたファージミドを宿主微生物に感染させる工程
c)ヘルパーファージを感染させること無く、工程b)の宿主微生物の培養上清を回収して抗体ライブラリーとする工程
あるいは本発明は、以下の病原体を認識する抗体の取得方法に関する。
〔31〕 前記抗原が、病原体に由来する抗原である〔24〕に記載の方法。
〔32〕 〔24〕に記載の方法の後、更に次の工程f)を行うことを特徴とする、病原体の中和活性を有する抗体の取得方法。
f)工程c)で選択した抗体可変領域の病原体に対する中和活性を評価し、中和活性を有する抗体可変領域を選択する工程
〔33〕 病原体に由来する抗原が、インフルエンザウイルスHA抗原、ジフテリア菌毒素、破傷風毒素、および水痘ウイルス由来の糖蛋白質からなる群から選択されるいずれかの抗原である〔31〕に記載の方法。
〔34〕 工程a)の前に、次の工程a’)を行う、〔31〕に記載の方法。a’)ライブラリーを吸収用抗原と接触させ、吸収用抗原に結合する抗体をライブラリーから除去する工程;ここで吸収用抗原とは、前記抗原と同じ病原体に由来するが、吸収用抗原との反応性を有する抗体の取得を望まない抗原を意味する
〔35〕 〔12〕に記載のライブラリーの、病原体中和抗体の調製における使用。
【0024】
本発明において、イムノグロブリンとは、動物種やクラスを問わず、重鎖と軽鎖とで構成される全てのイムノグロブリン分子を意味する。また、抗原との結合が可能な領域のみからなる断片や、あるいはイムノグロブリンを構成する領域が異なる動物種からなるキメラ抗体であることもできる。哺乳動物の重鎖可変領域をコードする遺伝子は、一般に遺伝子の構造的な特徴に基づいていくつかのVHファミリーに分類されている。たとえば、ヒトではVH1〜VH7の7つのファミリーに分類されている。各ファミリーは、ファミリー内で保存性の高い塩基配列を含み、この保存性の高さを利用して各ファミリー用のPCRプライマーが提案されている。軽鎖可変領域も、重鎖と同様に構造的な特徴に基づいてファミリーに分類することができる。
重鎖可変領域をコードする遺伝子は、V(Variable)、D(Diversity)、およびJ(Junction)の3つの遺伝子群から構成される。V、D、そしてJの各遺伝子群が、それぞれ複数の遺伝子からなっており、それらがランダムに組み合わさり、更に変異が加わることによって抗体の多様性が生まれる。これに対して、軽鎖可変領域を構成しているのは、V、およびJの2つの遺伝子群である。軽鎖可変領域においても重鎖可変領域と同様に、複数の遺伝子群の組み合わせと変異によって多様性がもたらされている。
【0025】
本明細書では、用語「ライブラリー」を用いる。ライブラリーは、多様なレパートリーからなる構成要素を含む集合体を意味する。遺伝子は遺伝子ライブラリーを、抗体分子は抗体ライブラリーを、あるいはファージやファージミドはファージライブラリーをそれぞれ構成する。ファージが内部に保持した抗体遺伝子を表面に発現している場合には、遺伝子ライブラリーであると同時に抗体ライブラリーでもある。ただし本明細書における用語ファージライブラリーは、ファージが抗体分子の発現を伴わない場合も含む。すなわち、ファージミドを感染させた宿主微生物や、抗体可変領域をコードする遺伝子をゲノムに保持したファージが溶原化している場合もファージライブラリーである。
【0026】
更に本明細書においては、用語「rgdp ライブラリー」(replicable genetic display package library;複製可能な遺伝的表示パッケージのライブラリー)を用いる。rgdpライブラリーとは、すなわち、遺伝子を保持するとともに、その遺伝子の発現生成物を表面に提示したもので構成されるライブラリーを呼ぶ。前記ファージライブラリーが、抗体タンパク質を表面に発現している場合には、rgdpライブラリーに含まれる。rgdpライブラリーには、ファージライブラリーのほか、外来タンパク質をその表面に発現している形質転換細胞やリボゾームからなるライブラリーを示すことができる。
【0027】
また本発明においては、用語「コンフォーメーション(conformation)」を用いる。イムノグロブリンが、重鎖と軽鎖の会合(holding)によって構成されることは既に述べた。この会合の結果として生じる重鎖と軽鎖の結合物の構造が、コンフォーメーションである。コンフォーメーションは、一般的に定常領域における-SS-結合によって成立する。このとき、常に抗原との結合活性を獲得するとは限らない。本発明において、あるイムノグロブリンが抗原との結合活性を持つとき、そのイムノグロブリンのコンフォーメーションが機能的(functional)であると言う。そして、ある軽鎖との組み合わせにおいて機能的なコンフォーメーションを与える重鎖が、他の軽鎖との組み合わせによっても機能的なコンフォーメーションを与えるとき、両者の結合を特に再構成(re-holding)と呼ぶ。再構成を構成する他の軽鎖とは、いったん別々のクローンとして単離された同一の細胞に由来する軽鎖を含む。また、本発明におけるコンフォーメーションの再構成とは、あくまでもイムノグロブリンの抗原との結合に必要な領域における再構成を意味する。したがって、定常領域の有無に関わらず、可変領域における分子構造がイムノグロブリンとして再構成されていれば、機能的なコンフォーメーションを再構成したと見なす。更に、軽鎖や重鎖をコードする遺伝子への人為的な塩基配列やファージの構成タンパク質の融合などを伴う場合であっても、可変領域における再構成が達成されている限り、本発明においては機能的なコンフォーメーションを再構成したと見なす。より具体的には、本発明における分子構造の再構成とは、たとえば重鎖可変領域と軽鎖可変領域とが異なるタンパク質として翻訳された場合には、定常領域において形成される-SS-結合によって重鎖可変領域と軽鎖可変領域とがイムノグロブリンの可変領域を構成することと言い換えることができる。
ところでsingle chain Fv抗体(scFv)のように、人工的なリンカーによって重鎖と軽鎖がはじめから結合されている抗体分子も存在する。この種の特殊な抗体においては、コンフォーメーションは-SS-結合ではなく、ペプチド結合によって構成される場合がある。したがってscFv抗体においては、定常領域を介することなくコンフォーメーションが再構成される。
【0028】
まず本発明は、機能的なイムノグロブリンを再構成することができる軽鎖をコードする遺伝子の選択と、この軽鎖可変領域遺伝子に重鎖をコードする遺伝子のライブラリーを組み合わせることによる遺伝子ライブラリーの調製方法に関する。本発明における軽鎖可変領域遺伝子の選択は、以下の工程によって行うことができる。すなわち本発明は、次の工程を含む、重鎖と機能的なコンフォーメーションを再構成することができる軽鎖をコードする遺伝子の選択方法に関する。
a)軽鎖可変領域をコードする、1つまたは複数の遺伝子を取得する工程、
b)軽鎖可変領域と機能的なコンフォーメーションを再構成することが確認されているイムノグロブリン重鎖可変領域をコードする遺伝子を取得する工程、
c)工程a)によって得られた軽鎖可変領域をコードする遺伝子の任意の1つを選択し、工程b)で取得した重鎖可変領域をコードする遺伝子とイムノグロブリンの機能的なコンフォーメーションを再構成が可能な条件下でタンパク質に翻訳する工程、
d)工程c)において翻訳されたタンパク質の抗原結合部位の形成を検出する工程、および
e)抗原結合部位の形成が検出されたタンパク質を構成する軽鎖可変領域をコードする遺伝子を選択する工程
【0029】
本発明において、軽鎖可変領域あるいは重鎖可変領域とは、少なくとも抗原との結合に必要な領域を含む任意の領域とすることができる。言いかえれば、3つのCDRとそれを保持するフレーム(FR)で構成される領域を含む任意の領域を本発明の可変領域として用いることができる。したがって、たとえば定常領域をも含む断片であっても、抗原との結合に必要な領域を含んでおれば、本発明の可変領域として利用することができる。抗体の可変領域としてしばしば用いられる、FabやFab'は、もともとイムノグロブリンの酵素的な切断によって得られる断片に対して与えられた名称である。本発明においては、Fabを可変領域を特定するための用語として理解すべきではない。
【0030】
本発明の軽鎖可変領域遺伝子の選択方法において、対象となる軽鎖可変領域遺伝子は、任意の抗体産生細胞より得ることができる。抗体産生細胞としては、たとえば、末梢血リンパ球や脾細胞等を挙げることができる。軽鎖可変領域遺伝子の単離にはRT-PCRを利用するのが有利である。たとえばヒトの場合、VLJL遺伝子を増幅することができるプライマーが明らかにされている(特表平3-502801あるいは特表平4-500607。また、MRC社はホームページ[「V-base」:http://www.mrc-cpe.cam.ac.uk/imt-doc/restricted/ok.htmlでプライマーを公開している)。したがって、これらのプライマーを用いてRT-PCRを行えば、工程a)に必要な軽鎖可変領域をコードする遺伝子を得ることができる。得られた遺伝子を工程c)に用いる。
【0031】
次に工程b)において、軽鎖可変領域と機能的なコンフォーメーションを再構成することが確認されているイムノグロブリン重鎖可変領域をコードする遺伝子を取得する。このとき取得する重鎖可変領域は、工程a)で得た軽鎖可変領域と同じ動物種に由来し、かつ軽鎖可変領域と機能的なコンフォーメーションを再構成することができるものであれば、抗原結合特異性などは任意であって良い。このような重鎖可変領域をコードする遺伝子は、たとえば、抗体活性を持つことが明らかなイムノグロブリン分子をコードする遺伝子から得ることができる。工程b)の重鎖可変領域としては、κ鎖およびλ鎖との再構成が可能なものを用意するのが望ましい。このような重鎖可変領域としては、実際に軽鎖との会合効率を確認して、最も効率の高い重鎖可変領域を選ぶのが望ましい。たとえば後に述べる実施例においては、重鎖に対応する各種クローンについて、軽鎖との会合効率を検討したところ、次のような構造を持つVH3-4が最も会合の効率が高かったことから、VH3-4(配列番号:1)を選択した。VH3-4は、以下に示す構造を持っている。
FR1:EVQLVESGGGLVQPGRSLRLSCAASGFTFD
CDR1:DYAMH
FR2:WVRQAPGKGLEWVS
CDR2:GISWNSGSIGYADSVKG
FR3:RFTISRDNAKNSLYLQMNSLRAEDTALYYCAK
CDR3:GPSGSFDAFDI
FR4:WGQGTTVTVSS
【0032】
続いて工程c)において、工程a)によって得られた軽鎖可変領域をコードする遺伝子の任意の1つと工程b)で得た重鎖可変領域をコードする遺伝子とを、イムノグロブリンの機能的なコンフォーメーションを再構成が可能な条件下でタンパク質に翻訳する。工程b)では、機能的なコンフォーメーションを構成しうる重鎖可変領域をコードする遺伝子を選択しているので、工程c)においてはイムノグロブリン分子の可変領域としての構造を再構成しているものは、機能的なコンフォーメーションを再構成したと見なすことができる。なお、ここでいうイムノグロブリン分子とは、イムノグロブリンにおける抗原との結合に必須の部分を含む限り、あらゆる構成であることができる。したがって、定常領域の有無に関わらず、抗原結合部位を再構成しているものは、イムノグロブリン分子として再構成されたと見なすことができる。
工程c)におけるイムノグロブリンの再構成が可能な条件下とは、-SS-結合によって重鎖可変領域と軽鎖可変領域の会合(holding)が可能となる条件を意味する。より具体的には、たとえば前述のように大腸菌のペリプラズムなど生体内の還元環境でのFabタンパク質の発現は、イムノグロブリンの再構成が可能な条件と言える。抗体のコンフォメーション形成に必要な還元的微小環境としては、ヒトなどの哺乳動物細胞では小胞体などのオルガネラを挙げることもできる。更に、重鎖と軽鎖の可変領域が人工的なアミノ酸配列(リンカー)で結合されたscFvタイプの抗体であれば、イムノグロブリンとしての再構成に必ずしも還元的な環境を必要としない場合もある。
工程c)における軽鎖可変領域と重鎖可変領域の発現には、外来遺伝子を表面に発現するファージを用いるのが有利である。例えば繊維状ファージは、その表面にcp3やcp8などのファージの構成タンパク質との融合タンパク質として外来遺伝子にコードされるタンパク質を発現する。
【0033】
さて、通常ファージライブラリーのスクリーニングは、ファージを粒子として回収するステップを含む。したがって、たとえば外来遺伝子をファージミドとして感染させた場合には、ヘルパーファージを感染させることによりファージ粒子として回収される。ところが本発明者らは、cp3との融合タンパク質としてFab遺伝子を挿入したファージミドを感染させた大腸菌をヘルパーファージを加えないで培養すると、その培養上清中に、Fabとcp3の融合タンパク質が分泌されることを見出した。ファージミドを感染させた大腸菌によって分泌されるFabとcp3の融合タンパク質は、20時間の培養後でさえ微量ではあったが、本発明による軽鎖の選択方法を実施するには十分な量であった。したがって、本発明による軽鎖可変領域遺伝子の選択方法のための試料としては、ファージミドを感染させた宿主微生物の培養上清を利用することもできる。ヘルパーファージを感染させてファージ粒子として回収する工程を不要とするこの方法は、実験操作上、きわめて簡便である。本発明の方法に基づいてファージミドを感染させた宿主微生物の培養上清をスクリーニングのための試料とするためには、宿主微生物において動作可能なプロモーターと、シグナル配列とを備えたベクターを用いる。たとえば大腸菌を宿主とする場合には、シグナル配列としてpelB配列などを挿入した繊維状ファージ用のファージミドベクターを利用することができる。
【0034】
もしも軽鎖可変領域が重鎖可変領域との機能的なコンフォーメーションを再構成しうるものであれば、イムノグロブリンの可変領域が形成される。この可変領域の形成を検出することによって、選択すべき軽鎖可変領域を知ることができる。可変領域の形成は例えば、イムノアッセイの原理を利用して検出することができる。すなわち、κ鎖(あるいはλ鎖)に対する抗体を固相化抗体としてコートしたプレートに、重鎖可変領域と軽鎖可変領域の発現生成物を含む試料を加えて、軽鎖可変領域をプレート上に捕捉する。もしも重鎖可変領域が軽鎖可変領域と会合していれば、重鎖可変領域は軽鎖可変領域とともにプレート上に捕捉されるはずである。次いで重鎖やFabに対する標識抗体を加えれば、両者が会合できたときに限り、標識抗体がプレート上に捕捉されることになる。適当な時間インキュベーションしてプレートを洗浄し、標識抗体を検出すれば、機能的なコンフォーメーションを構成する軽鎖可変領域を検出することができる。標識抗体と固相化抗体は、逆の組合せとすることもできる。あるいは、重鎖可変領域を予めビオチン化しておき、標識アビジンによって検出することもできる。前述のとおり、ここで検出に用いる試料は、ファージミドを感染させた大腸菌の培養上清を用いることができることを我々は見出している。
こうして重鎖可変領域との会合が確認された軽鎖可変領域を、重鎖可変領域との機能的なコンフォーメーションが可能な軽鎖可変領域として選択することができる。軽鎖可変領域をコードする遺伝子がファージライブラリーとして保持されている場合には、ファージを回収することによって軽鎖可変領域の遺伝子を選択することができる。
【0035】
以上に述べた過程を経て得られた軽鎖可変領域遺伝子は、重鎖可変領域との再構成が可能であるのみならず、スクリーニングに利用した発現系における発現が証明されたものとなる。たとえばファージによる発現を利用した場合には、大腸菌において十分な発現が見られるものが選択される。したがって、哺乳動物細胞では発現するものの、大腸菌では発現量が少なくなる遺伝子をこの段階で除くことができる。このような特徴は、本発明における軽鎖可変領域遺伝子の選択方法において期待することができる新規な利点である。これに対して従来の抗体ライブラリーの作製技術においては、軽鎖の選択工程を含まないため、発現レベルが不充分な軽鎖遺伝子の混入を防ぐことはできなかった。
【0036】
選択された軽鎖可変領域をコードする遺伝子は、そのまま本発明による遺伝子ライブラリーの調製に用いることができる。しかしこの段階では、選択した軽鎖可変領域遺伝子の間に重複が存在している可能性がある。したがって、好ましくは軽鎖可変領域遺伝子の構造を解析し、重複を除いた上で、遺伝子ライブラリーの調製に用いる。遺伝子の重複は、たとえば次のような方法によって除くことができる。
まず前記工程d)の後に、あるいは先だって軽鎖可変領域遺伝子の塩基配列を決定し、その塩基配列によってコードされるアミノ酸配列を推定する。推定されたアミノ酸配列を比較し、同じアミノ酸配列をコードする遺伝子は除く。この段階で更に、欠損(deletion)のチェックも行うのが望ましい。このため、塩基配列の決定を行って読みとり枠のずれているものを除いた。
【0037】
なお、遺伝子の取捨選択は、実際には、類似の遺伝子をグルーピングし、各グループから代表的な配列を選択することになる。このとき、選択された遺伝子を偏りなくカバーできるように、そしてVL-JL結合点のズレに関しても天然に存在する抗体で見られる分布を損なわないように選択する。実際には、遺伝子のデータベースから実際の抗体で使用されている軽鎖可変領域遺伝子をリストアップし、結合点のズレに関して集計した結果を元に分布を決定した。表1に、V-Jの結合点(アミノ酸配列91-96)にいくつのアミノ酸を持つかについて調査した結果を示す。
【0038】
【表1】

【0039】
以上の結果を総合したところ、本発明者らの解析結果によれば、ヒトのイムノグロブリンの場合、このような選択方法に基づいて選択される代表的な軽鎖可変領域のレパートリーサイズは、κ鎖で101、並びにλ鎖でも99である。
つまり、ヒトの機能的なイムノグロブリンを代表しうる軽鎖可変領域のレパートリーサイズは、せいぜい200にすぎないことが明らかとなったのである。なお、本発明者らが明らかにしたおよそ100というレパートリーサイズを、限定的に解釈すべきではない。すなわち、本発明の選択方法に基づいて、たとえばヒトの軽鎖可変領域遺伝子を選択するときに選択すべき軽鎖可変領域遺伝子の数は、必ずしも100になるとは限らない。あくまでも、得られたアミノ酸配列に基づいて、ここに述べた選択方法を実施することによって選択された軽鎖の遺伝子を以降の工程に用いることが重要である。
ここで、スクリーニングによって得られたファージ抗体のVL遺伝子を分類した。結果は図1に示した。VL遺伝子の使用は特定の遺伝子に偏っていることがわかる。この結果から、イムノグロブリンのコンフォーメーションを再構成しうる軽鎖を多く含むセットを揃えることにより、機能的なイムノグロブリンの割合の高い良質なライブラリーを作製できることが示された。
【0040】
ここで、生体内における抗体の多様性を維持するには、アミノ酸配列の解析の対象をできるだけ大きく取ることが望ましい。
1人のヒトの軽鎖を構成するゲノム遺伝子は、36種類のVl、7種類のJl、37種類のVk、そして4種類のJkで構成される。V遺伝子とJ遺伝子の組み合わせによって軽鎖遺伝子が作られるので、単純計算では36×7=252と37×4=148の和、つまり400種類となる。加えて、遺伝子の結合の際にその結合点のアミノ酸の数には、変異が伴う。つまり、前述のように抗体遺伝子の再構成に特有の現象により、平均±1アミノ酸程度(最大で±5アミノ酸程度)の多様性が生まれる。この組み合わせから更に不要な遺伝子は削除されて、個人の抗体遺伝子のレパートリーとなる。さらに、個人によって遺伝子は少しずつ異なっている(これを多型;polymorphismという)。全人類が持っている抗体遺伝子の種類を調べ尽くすことは現実的ではないが、総計して1000種類程度であると推定される。これらのことから、1人のヒトがあらゆる抗原に対応する抗体のセットを作ることができるとすれば、理論的には好ましくは400種類以上1000種類程度までのアミノ酸配列について本発明による解析方法を実施することによって、軽鎖可変領域遺伝子のレパートリーを十分に再現することができることになる。
【0041】
本発明者らがヒトにおいて明らかにしたこのような軽鎖のレパートリーサイズ(200種類)は、約1000種類のアミノ酸配列の解析の結果得られた数字である。したがって、理論的には、あらゆる抗体セットにおいて機能的なコンフォーメーションを再構成することができる軽鎖可変領域が選択されていると言うことができる。実験的にも、本発明による200種類と言う軽鎖可変領域遺伝子のレパートリーサイズは、生体内における抗体の多様性をin vitroで再現するのに十分有効であることを示すことができた。しかし、より多くの塩基配列を決定し、そのアミノ酸配列についても解析を行えば、レパートリーサイズが大きくなる可能性は否定できない。
【0042】
そこで、本発明によって選択された軽鎖可変領域ライブラリーのレパートリーサイズを、軽鎖遺伝子ライブラリーとの混合によって補うことができる。すなわち、軽鎖可変領域についても重鎖と同じく選択をかけない全ての軽鎖遺伝子(VLライブラリーと呼ぶ)を用いてライブラリーを作製するのである。こうして作製したVLライブラリーを200種の軽鎖可変領域のみを用いたライブラリー(KL200ライブラリーと呼ぶ)と混合することによって、相補的に欠点を補うライブラリーとすることができる。各ライブラリーは以下に述べる特徴を持つ。
KL200ライブラリーは、重鎖とのコンフォメーション形成が確認されている。ただし数を限定したために、特定の特異性を持つクローンを形成するのに必要な軽鎖が除外されている可能性も否定できない。
VLライブラリーは、独立クローン数が109に達しているため必要なクローンはもらしていない。しかし発現と重鎖とのコンフォメーション形成率はKL200ライブラリーに比較して低い。
【0043】
なお、本発明において、アミノ酸配列の解析結果に基づいて分類した各グループから、いずれの遺伝子を選択するかは本来任意である。したがって、重鎖との機能的なコンフォーメーションの再構成が可能な軽鎖をコードする遺伝子の構造をここに明らかにすることには大きな意味はない。重要なのは、このような選択方法によって、重鎖とともに機能的なコンフォーメーションを再構成することができる軽鎖の選択工程を実施することである。いずれにせよ、このような操作を通じて、ヒトであれば、101種類のκ鎖遺伝子ライブラリーと、99種類のλ鎖遺伝子ライブラリーからなる軽鎖可変領域遺伝子ライブラリーが得られることになる。
【0044】
さて、本発明において選択された軽鎖可変領域の遺伝子ライブラリーは、後に述べるように重鎖可変領域をコードする遺伝子を組み合わせることによってイムノグロブリンの遺伝子ライブラリーを与える。したがって、本発明によって選択された軽鎖可変領域の遺伝子ライブラリーは、イムノグロブリンの遺伝子ライブラリー調製用のライブラリーとして有用である。すなわち本発明は、少なくともイムノグロブリンの軽鎖可変領域をコードする遺伝子からなる遺伝子ライブラリーであって、イムノグロブリンの重鎖と機能的なコンフォーメーションを再構成できない軽鎖可変領域をコードする遺伝子が実質的に排除されている遺伝子ライブラリーに関する。
【0045】
本発明において、イムノグロブリンの重鎖可変領域と機能的なコンフォーメーションを再構成できない軽鎖可変領域をコードする遺伝子とは、先に述べた軽鎖可変領域の選択方法によって排除することができる。重鎖可変領域と機能的なコンフォーメーションを再構成できない軽鎖可変領域をコードする遺伝子を、本発明においてはdefective遺伝子と呼ぶ。本発明においてdefective遺伝子が実質的に排除されたライブラリーとは、defective遺伝子の完全な排除までも要求するものではない。たとえばdefective遺伝子が混入したライブラリーであっても、免疫学的な反応に基づく抗体のスクリーニングを妨げない範囲であれば、defective遺伝子が実質的に排除されたライブラリーと言うことができる。
【0046】
抗体のスクリーニングを妨げない範囲とは、ライブラリーに占めるdefective遺伝子の割合が、たとえば0−50%、望ましくは0−25%であることを意味する。defective遺伝子の割合は低いほどスクリーニングの効率が高まり、有用なクローンをスクリーニングの過程で失う危険性が低くなることは言うまでも無い。しかし発明者らが試作したライブラリーにおいて、defectiveな遺伝子を50%含むと考えられるVLライブラリーをdefectiveな遺伝子を完璧に排除したKL200ライブラリーに様々な割合で混合したところ、50%までの混合では有効なスクリーニングができることを確認している。この事実により、defectiveな遺伝子の割合がより好ましくは25%以下であれば、defective遺伝子が実質的に排除されていると言うことができる。
【0047】
本発明による軽鎖のみからなるライブラリーは、重鎖の可変領域をコードする遺伝子ライブラリーを組み合わせるための材料として有用である。このようなライブラリーとしては、たとえばファージのcp3をコードする遺伝子にdefective遺伝子が実質的に排除された軽鎖ライブラリーを組み込むとともに、重鎖の可変領域を挿入するためのクローニングサイトを設けたファージライブラリーを挙げることができる。一般にファージライブラリーは、ファージの宿主微生物に対する感染性を損なうことがないように、ファージミド上に外来遺伝子を保持させ、これをヘルパーファージを用いてファージ化する方法が用いられる。本発明のファージライブラリーも、ファージミド上に構築することができる。すなわち、宿主において機能するプロモーターの制御下に、シグナル配列と連結した前記軽鎖可変領域をコードする遺伝子と、重鎖可変領域をコードする遺伝子のクローニングサイトを設ける。重鎖可変領域遺伝子のためのクローニングサイトは、目的とする遺伝子に見出される頻度の低い制限酵素とするのが有利である。本発明によるファージライブラリーは、ファージミドのみならずファージのゲノムを利用することもできる。クローニングサイトを付加したプライマーによって重鎖可変領域遺伝子をPCRによって合成し、ファージライブラリーに挿入すればイムノグロブリンの可変領域を発現するファージライブラリーを完成することができる。
【0048】
あるいは、defective遺伝子が実質的に排除された軽鎖ライブラリーを大腸菌用の発現ベクターに組み込んで、大腸菌に形質転換することによって軽鎖可変領域ライブラリー分泌発現株とすることもできる。この大腸菌に、PCRによって得た重鎖可変領域を組み込んだファージを感染させれば、Fabをその表面に再構成したファージ粒子を得ることができる。重鎖可変領域遺伝子を様々な免疫履歴を持つ個体から選択することによって、目的とする抗体に合った抗体ライブラリーを得ることができる。
【0049】
このような方法に基づいて、ファージライブラリー作成用キットを提供することができる。このキットは、defective遺伝子が実質的に排除された軽鎖ライブラリーと、重鎖可変領域遺伝子増幅用のプライマーとからなる。使用者は、重鎖可変領域遺伝子増幅用のプライマーを用いて、目的とする抗体に合った免疫履歴を持つ遺伝子ソースからPCR生成物を得ることができる。たとえば、がんの宿主からは、腫瘍関連抗原を認識する抗体ポピュレーションに富むライブラリーが期待できる。
【0050】
こうして選択された軽鎖可変領域遺伝子を利用し、本発明によるライブラリーを調製する。すなわち本発明は、以下の工程を含む、イムノグロブリンの軽鎖可変領域遺伝子と重鎖可変領域遺伝子の組み合わせからなる遺伝子ライブラリーの調製方法に関する。
a)軽鎖可変領域遺伝子として、重鎖可変領域遺伝子の発現産物との機能的なコンフォーメーションの再構成が可能な軽鎖分子をコードするものを選択し、
b)工程a)によって得られる軽鎖可変領域遺伝子の集合である遺伝子のライブラリーを調製し、
c)工程b)のライブラリーに重鎖をコードする遺伝子のライブラリーを組み合わせる
【0051】
工程a)の軽鎖の選択工程は、先に述べたとおりである。先に得られた軽鎖をコードする遺伝子を集めて、工程b)のライブラリーとすることができる。軽鎖可変領域遺伝子が繊維状ファージに保持されている場合には、このファージを増殖させ、回収してライブラリーとすることができる。次いで工程c)として、前記軽鎖の遺伝子ライブラリーに重鎖の遺伝子ライブラリーを組み合わせる。重鎖可変領域遺伝子を、末梢血リンパ球や脾細胞のような抗体産生細胞より取得する方法は公知である。たとえばヒトのイムノグロブリンは、VH1〜VH7の7つのVHファミリーからなる。
各ファミリーの遺伝子を増幅することができるプライマーは公知である(Campbell, M. J., Zelenetz, A. D., Levy, S. & Levy, R. (1992). Use of family-specific primers for PCR amplification of the human heavy chain variable gene repertoire. Mol. Immunol., 29, 193-203.また、MRC社はホームページ「V-base」:http://www.mrc-cpe.cam.ac.uk/imt-doc/restricted/ok.htmlでプライマーを公開している)。したがって、このようなプライマーに基づいてRT-PCRを行えば、各ファミリー毎に重鎖可変領域遺伝子を増幅することができる。
増幅生成物として得られた重鎖可変領域遺伝子は、適当なベクターに挿入することによって遺伝子ライブラリーとすることができる。このとき、VHファミリー毎に重鎖可変領域遺伝子ライブラリーを調製し、生体内における各ファミリーの構成割合に応じて混合することによって、本発明の遺伝子ライブラリーをより生体内の抗体レパートリーに近い状態にすることができる。具体的には、たとえばヒトの場合、各ポピュレーションはおよそ次のような割合で存在することが明らかにされている。生体における抗体レパートリーを模倣することによって、スクリーニングを通じて必要なクローンを失う機会を少なくすることができる。
VH1:25%
VH2: 6.6%
VH3:40%
VH4:19%
VH5: 5%
VH6: 3.8%
VH7: 1.2%
【0052】
以下に重鎖可変領域遺伝子の取得について、より具体的に述べる。7種のVHファミリー毎にプライマーを設定し、6個のJH遺伝子に共通に働くプライマーと組み合わせてRT-PCRを行う。ヒトの各VHファミリーをファミリーごとに幅広く増幅することができるプライマーは公知である(Marks JD et al. J. Mol. Biol. (1991) 222, 581-597あるいはCampbell, M. J., Zelenetz, A. D., Levy, S. & Levy, R. (1992). Use of family-specific primers for PCR amplification of the human heavy chain variable gene repertoire. Mol. Immunol., 29, 193-203.また、MRC社はホームページ「V-base」: http://www.mrc-cpe.cam.ac.uk/imt-doc/restricted/ok.htmlでプライマーを公開している)。用いたプライマーによって各ファミリーがきちんと増幅されているかどうかを確認する。すなわち、増幅されたVHDJH構造を有するバンドについて各ファミリー毎に数10種のクローンを得て塩基配列を決定し、どの重鎖可変領域遺伝子が増幅されているかを解析する。もしも増幅されていない遺伝子が存在する場合には更に新しくプライマーを設計し、追加する。たとえば、それまでに報告されているプライマーでは増幅することができなかった遺伝子の増幅を可能とする新たなプライマーが報告されている。
in frameでVHDJH構造を持つクローンについては、適当なベクターに組み込んで大腸菌中での発現と軽鎖可変領域遺伝子との会合-folding-を解析する。もしどこかの段階が正しく起こらないクローンが存在すればその理由を推定し、ライブラリー全体の中でそのクローンの占める率を推定する。
個体のイムノグロブリンのポピュレーションは、免疫履歴によって影響を受けている。したがって、なるべく多数の個体の免疫的経歴を反映できるように、多様なB細胞から重鎖可変領域遺伝子を調製するようにする。実際には、臍帯血、扁桃、末梢血、あるいは骨髄等、入手可能な重鎖可変領域遺伝子ソースの種類を増やす。
更に、免疫原(自己の抗原も含む)と一度も接触していないnaiveなB細胞集団からB細胞を調製することも重要である。なぜならば、自己の成分と反応するクローンは免疫系の成熟時に排除されるからである。自己抗原に対する抗体レパートリーをも含む遺伝子ライブラリーとするには、nativeなB細胞は重要である。そして混合に際してはリンパ球数とクローン数が比例するように心がける。最終的には109オーダー(109〜1010)の独立したVHDJHライブラリーを作製する。
【0053】
このような操作は、次のような意義を持つ。抗体の抗原結合面を構成するアミノ酸配列は、軽鎖CDR1、CDR2、CDR3、重鎖CDR1、CDR2についてはゲノム上に存在する遺伝子によって規定されており(進化的な選別も含めて)、その多様性の総数は1万種程度である。その上に重鎖CDR3の極端に高い多様性が上積みされる。そこで抗体ライブラリーとしてはこの1万種の多様性はできる限り一様に偏り無い形で含んだ上で、重鎖CDR3(これは個々人、個々のB細胞でランダムなプロセスで作られている)についてはなるべく広範囲にライブラリー化される必要がある。上記の方法はこの要求を満たしている。
以上のような解析を通じて、本発明者らは、CDRの中にシステイン残基を含む場合には、VH遺伝子の発現が正しく起こらなかった等の知見を得た。また、作製した重鎖可変領域ライブラリーに関して、その70%以上が大腸菌中での発現と重鎖可変領域ドメインとの会合-folding-が正しく起きていることが確認できた。
【0054】
あるいは免疫履歴を逆用し、重鎖可変領域遺伝子のソースを選択することによって、あるていどライブラリーを特徴付けることができる。たとえばある感染症の既往歴を持つ場合には、その病原体に対して親和性の高いイムノグロブリンを得られる可能性が高まる。あるいはがん患者の抗体産生細胞を重鎖可変領域遺伝子のソースとすることにより、腫瘍抗原を認識するイムノグロブリンを得ることもできる。
【0055】
更に、VH遺伝子に対して人為的な変異を加えることによって、ライブラリーの多様性を高めることができる。人為的な変異を与える方法としては、error-prone PCR(Winter,G. et.al.,Annu.Rev.Immunol.,12,433-455,1994)が公知である。本発明においては、軽鎖可変領域におけるdefective遺伝子が実質的に排除されているため、重鎖可変領域遺伝子の多様性は、そのままライブラリーの多様性として反映される。したがって、公知のerror-prone PCRを適用した場合であっても、はるかに高度な多様化を達成することができる。error-prone PCRは、以下のように行うことができる。
error-prone PCRは、無作為に点突然変異を導入する方法として用いられる手法である。具体的には、PCRに使用するDNAポリメラーゼの以下の生化学的性質を用いる。
(1) Taq DNA polymeraseは通常Mg2+イオン存在下で用いられるが、Mn2+イオンの存在下では塩基の取り込みに誤りを起こしやすくなる。
(2) PCR反応時に塩基の供給源としてdATP, dCTP, dTTP, dGTPを通常は等量で混合するが、この濃度を突然変異を起こしやすいように変更する。
(3) 上述の塩基の供給源としてdITPを混合する。dITPはTaq DNA polymeraseによってDNA鎖にとりこまれると塩基イノシンを形成する。イノシンはどの塩基とも塩基対を形成しないので、PCR反応が進行するとイノシンに相補的な塩基の部分には無作為の塩基が挿入される。
以上のような3つの条件の相乗的効果により、突然変異が無作為に導入される。具体的には、たとえば7.5mM MgCl2、0.5mM MnCl2、0.2mM dATP/dGTP、1.0mM dCTP/dTTP、0.1-1.0mM dITPのような条件で反応を行う。実際には、実験の目的に合わせて更に濃度条件などを調整する。温度などの反応条件は通常のPCRと同様に設定する。
【0056】
本発明の遺伝子ライブラリーの調製には、公知のベクターを用いることができる。本発明に用いることができるベクターとしては、たとえば先に調製した軽鎖可変領域遺伝子ライブラリーを保持したファージライブラリーを示すことができる。すなわち、ファージミドが保持している軽鎖可変領域遺伝子の上流に重鎖可変領域遺伝子を挿入するのである(Gene 194 (1997) 35-46 Iba Y et al)。重鎖可変領域と軽鎖可変領域を同時に表面に発現するファージライブラリーを用いれば、抗原との結合を指標としてパニング法(後述)を行うのに有利である。すなわち、本発明の遺伝子ライブラリーをファージライブラリーとして構成することによって、rgdpライブラリーとすることができる。本発明の遺伝子ライブラリーは、ファージのみならず、リボゾームや大腸菌鞭毛タンパク質の融合タンパク質として外来遺伝子を発現するシステムを用いることによってrgdpライブラリーとすることもできる。
【0057】
代表的なrgdpライブラリーとして、ファージライブラリーが挙げられる。本発明の抗体ライブラリーに基づくファージライブラリーは、次のようにして得ることができる。ファージ表面に外来タンパク質を発現させるには、一般にファージミドとヘルパーファージが用いられる。例えば、pTZ19R(ファルマシア製)などのファージミドベクターが市販されている。ファージミドには、cp3やcp8等のファージの構成タンパク質をコードする遺伝子に、発現させたい外来タンパク質をコードする遺伝子を連結する。
ファージミドは、大腸菌などの宿主に感染させることによって増幅できる。しかしこの状態ではファージ粒子として回収することはできない。いわば、一般の遺伝子ライブラリーと同じ状態にある。ファージミドが保持する外来タンパク質を表面に提示したファージ粒子とするには、ファージミドを感染させた宿主にヘルパーファージを重感染させる。たとえばファージミドベクターpTZ19Rは、ヘルパーファージM13K07の重感染によってファージ粒子として回収することができる。このとき利用されるファージミドのcp3タンパク質が外来タンパク質と融合されていれば、完成するファージの表面には外来タンパク質が提示されることになる。
市販のファージミドに抗体の遺伝子のクローニングに好適な制限酵素サイトを導入しておけば、本発明による軽鎖可変領域遺伝子ライブラリーを重鎖可変領域遺伝子ライブラリーとともに組み込むことができる。ファージミドベクターpTZ19Rに適当な制限酵素サイトを導入し、PCRで増幅した抗体遺伝子ライブラリーを組み込む方法が公知である(Gene 194,35-46,1997)。以下に述べる実施例においては、ファージミドベクターのシグナル配列PelBの下流にSfiIサイトとAscIサイトを導入した。一方、軽鎖可変領域遺伝子の増幅には、同じサイトを導入したプライマーを用いる。PCRの増幅生成物を当該制限酵素で消化してこのサイトに組み込むことにより、PelBの下流に抗体可変領域遺伝子が挿入され、更にその下流に位置するcp3との融合タンパク質をコードするファージミドベクター(図2のpFCAH9-E8d)とすることができる。
【0058】
あるいは、重鎖可変領域遺伝子を細菌宿主発現用のベクターに挿入することもできる。この場合には、ベクターを細菌に形質転換することによって重鎖可変領域遺伝子を発現させる。得られた形質転換細胞に、更に軽鎖可変領域遺伝子を保持したファージライブラリーを感染させることにより、結果的に本発明による遺伝子ライブラリーが完成する。大腸菌の形質転換用ベクターとしては、pFK等を示すことができる。なお細菌に形質転換するには、重鎖可変領域遺伝子を適当な分泌シグナルの下流に連結することによって、重鎖可変領域タンパク質をペリプラズムに分泌させることができる。pFKは、分泌シグナルとしてpelBを備えたベクターである。
この他、動物細胞発現用のベクターに、先に調製した軽鎖可変領域遺伝子ライブラリーと、重鎖可変領域遺伝子ライブラリーを挿入することによって、本発明のライブラリーを調製することもできる。このようなベクターとしてはpcDNAI等を示すことができる。
【0059】
本発明の遺伝子ライブラリーを構成する重鎖可変領域遺伝子は、その3'末端に予め標識ペプチドをコードする塩基配列を融合しておくことができる。標識ペプチドとしては、たとえばヒスチジンタグ(His x 6)、myc-tag、あるいはHA-tag等を示すことができる。ヒスチジンタグは金属イオンとの結合活性を持つタグである。本発明のライブラリーを構成する重鎖にヒスチジンタグを融合させておくことにより、たとえば重鎖が発現しているファージをニッケルカラムに捕捉することができる。ニッケルカラムに結合できなかったファージやヘルパーファージをニッケルカラムから洗い去ることにより、重鎖が発現しているクローンを濃縮、および精製することができる。
【0060】
本発明の遺伝子ライブラリーは、様々な形態の抗体ライブラリーとすることもできる。具体的には、ファージライブラリー、大腸菌ライブラリー、あるいはリボゾームライブラリー等を示すことができる。本発明による抗体ライブラリーは、本発明によるライブラリーを構成するクローンが保持しているイムノグロブリン遺伝子を発現させ、発現生成物を回収することによって得ることができる。本発明者らは、軽鎖可変領域遺伝子と重鎖可変領域遺伝子をcp3に融合させたファージミドを感染させた大腸菌を、ヘルパーファージを加えないで長期間培養するとFab-cp3融合タンパクが菌上清に分泌されることを発見した。この現象を利用することによって、ファージミドが有する抗体遺伝子の特性を容易に試験することができる。更に各クローンについて、軽鎖可変領域と重鎖可変領域とが再構成されているかどうかを確認し、再構成に失敗しているクローンを排除することができる。この操作を行うことによって、本発明の抗体ライブラリーをより生体内のポピュレーションに近づけることができる。
本発明の抗体ライブラリーは、抗体のスクリーニングを通じて必要な可変領域遺伝子のスクリーニングを行うために用いることができる。本発明の抗体ライブラリーを利用することによって、可変領域遺伝子のライブラリーを手元に置き、抗体ライブラリーのみを供給して必要な抗体のスクリーニングを第三者に実施させることもできる。本発明は、本発明のライブラリーから、目的とする反応性を有する抗体を検出する方法に関する。抗体の反応性は、抗原との結合活性によって評価することができる。すなわち、次の工程によって目的とする抗原との結合活性を指標として、必要な抗体を検出することができる。
【0061】
a)本発明の抗体ライブラリーと、該抗原とを抗原抗体反応に適した条件下に接触させる工程
b)該抗原とイムノグロブリン可変領域との結合を検出する工程
更に本発明は、本発明のライブラリーを用いる、特定の抗原を結合するイムノグロブリン可変領域の取得方法に関する。まず本発明の抗体可変領域の検出方法は、以下の工程を含む。
また本発明による、特定の抗原と結合するイムノグロブリン可変領域の取得方法は、前記工程a)およびb)に加えて次の工程c)を含む。
c)前記抗原と結合するイムノグロブリン可変領域を発現するクローンを選択する工程
工程a)で用いるライブラリーには、軽鎖可変領域と重鎖可変領域の両方を発現可能なライブラリー、あるいはこのライブラリーの発現生成物からなる抗体ライブラリーを用いる。特に有用なライブラリーは、繊維状ファージ表面にライブラリーを構成する遺伝子によってコードされる重鎖と軽鎖の断片を発現し、かつそれらがファージ粒子表面上で機能的に再構成しているライブラリーである。このようなファージライブラリーは、rgdpライブラリーとして機能し、パニング法によって容易に必要な反応性を持つクローンを濃縮することができる。本発明に基づくパニング法は、次のように実施される。
まず、目的とする抗原にrgdpライブラリーを接触させ、この抗原に結合するクローンを回収する。回収したクローンを増幅し、再び目的の抗原と接触させて、結合するクローンを回収する工程を繰り返す。クローンの増幅は、ファージを大腸菌に感染させ、回収することによって行われる。この工程を繰り返すことによって、目的とする反応性を持つ可変領域が濃縮される。抗原との結合性に基づくスクリーニングは、一般にクローンの回収率が増加するまで行われる。ここで回収率とは、抗原に対してチャージしたクローンに数に対する、抗原への結合性を有するものとして回収されたクローンの数の割合である。回収率がその前のスクリーニングに比較して明らかに増加するとき、目的とする反応性を持つ抗体を提示したファージが濃縮されつつあることを意味する。
【0062】
この他、ライブラリーを構成するクローンの発現生成物をクローン毎に回収した抗体ライブラリーに基づくスクリーニングを行うこともできる。各クローンの発現生成物を目的とする抗原に接触させることによって、必要な反応性を持つイムノグロブリンを直接選び出すこともできる。抗原との結合が観察されたイムノグロブリンをコードするクローンを選択すれば、目的とする可変領域を得ることができる。
【0063】
本発明においては、抗体可変領域の検出や取得に用いる抗原として、抗原決定基を有するあらゆる化合物を用いることができる。幅広い蛋白質、糖、核酸、有機化合物、あるいは無機化合物が、抗原決定基を有することが知られている。これらの化合物は、生体に由来するものであっても良いし、人工的に合成されたものであってもよい。具体的には、動植物、細菌や真菌等の微生物、あるいはウイルス等の細胞や粒子、これらを構成する蛋白質、糖鎖、あるいは脂質等が抗原として用いられる。この他、非蛋白質性の、各種の薬物、ホルモン、ビタミン、サイトカイン、ケモカイン、環境汚染の原因となる化学物質等も、抗原決定基を有するものであれば、本発明に用いることができる。
【0064】
また、目的とする抗体が結合すべき抗原分子の全体を用いることもできるし、あるいは抗原分子の一部を用いることもできる。特に断らない限り、本発明における抗原は、その抗原の部分的な構造を保持した化合物を含む。たとえば、抗原分子の一部として、その抗原分子に固有の構造からなる抗原決定基を含む領域を用いることにより、特異性に優れた抗体を取得することができる。
【0065】
更に、複数の分子が結合した複合体を抗原とすることもできる。特に断らない限り、本発明における抗原は、複数の分子からなる複合体を含む。複合体を抗原とすることにより、単独の分子と複合体とを識別することができる抗体を取得することができる。
【0066】
本発明のライブラリーから、たとえば病原体に対する中和抗体として有用な抗体分子を検出し、それをコードする遺伝子を取得することができる。本発明において病原体とは、病原性を有するあらゆる生命体、あるいは生命体に由来する物質を含む。より具体的には、ウイルス、細菌、真菌、マイコプラズマ、あるいは多細胞の寄生性生命体などを示すことができる。また微生物や動植物に由来する毒素も、本発明の病原体に含まれる。したがって、コレラ菌や腸管出血性大腸菌等が産生する毒素は、本発明の病原体に含まれる。あるいは、蛇毒や蜂毒などの生物毒素もまた本発明の病原体に含まれる。他方、本発明の中和抗体とは、病原体の、病原性や感染性を抑制する働きを有する抗体を言う。
【0067】
本発明による中和抗体の検出方法は、病原体に由来する抗原に本発明のライブラリーを接触させ、この抗原に結合するイムノグロブリン可変領域を発現するクローンを検出する。更に本発明は、抗原に結合する可変領域を発現するクローンを選択することにより、中和抗体をスクリーニングする方法を提供する。
【0068】
本発明による中和抗体のスクリーニング方法においては、目的としない反応性を有する抗体可変領域を抗体ライブラリーから吸収することができる。たとえば、インフルエンザウイルスのHA抗原に対する抗体は、その感染性の抑制に有効とされている。一方、同じインフルエンザウイルス粒子に対する抗体であっても、核蛋白質(NP)に対する抗体は、ウイルスを中和する活性はない。しかし、インフルエンザウイルス粒子を抗原として用いると、しばしばNPに対する抗体も検出されるため、結果として中和抗体のスクリーニング効率を低下させる原因となってしまう。このようなときには、あらかじめ抗NP抗体を吸収しておくことにより、抗HA抗体を効率的に検出することができる。抗NP抗体は、本発明のライブラリーをインフルエンザウイルス粒子のNP抗原と抗原抗体反応が可能な条件下で接触させ、NP抗原に結合した抗体可変領域を除去することにより、吸収することができる。HA抗原に対する抗体を効率的に取得するためには、たとえばNP抗原を含まない、精製HA抗原との結合活性を指標とすることも可能である。しかし、予め抗NP抗体を吸収する工程を利用する方法は、表面構造を維持したウイルス粒子に対する抗体の反応性を確認できる点で有利である。
【0069】
このようにして得られたイムノグロブリン可変領域をコードする遺伝子、並びにその発現産物であるイムノグロブリン可変領域は本発明に含まれる。得られたイムノグロブリン可変領域は、診断や治療に有用である。特に、ヒトのイムノグロブリンである場合には、ヒトへの投与が可能である。具体的には、各種感染症、腫瘍、あるいは動脈硬化等の診断や治療に利用することができる。イムノグロブリンを用いた、診断や治療については様々なバリエーションが公知である。
本発明によって得られたイムノグロブリン可変領域を完全なヒト・イムノグロブリン分子ととすることによって、単なる親和性リガンドとしてではなく、抗体分子として用いることができる。すなわち、本発明によって得られた重鎖可変領域、ならびに軽鎖可変領域遺伝子を、定常領域をコードする遺伝子CH、およびCLと接合する。定常領域をコードする遺伝子として、IgGに由来するものを用いることにより、オプソニン作用に優れるイムノグロブリンとすることができる。
なお本明細書において引用された全ての先行技術文献は、参照として本明細書に組み入れられる。
【図面の簡単な説明】
【0070】
【図1】図1は、本発明によって得られたファージ抗体のVL遺伝子を分類した結果を示すグラフ。グラフの横軸:軽鎖のGermline(染色体)可変領域遺伝子63種類。グラフの縦軸:ファージ抗体スクリーニング時の抗原奥から、1. 破傷風毒素(TET)2. インフルエンザウイルス抗原(IFL)3. 水痘帯状疱疹ウイルス抗原(VZGH)4. ジフテリア毒素(DTD)5. 6+7の合計(total)6. 感作抗原合計(1+2+3+4)(imunized)7. 非感作抗原合計(C.elegansなど人間が一生出会わないと考えられる抗原)(no imunized)グラフの高さ:ファージ抗体の数。
【図2】図2は、本発明による可変領域ライブラリーの作製に用いた各種のベクターの構造を模式的に示す図。1)pAALFab:D1.3 mutation用ベクター。2)pFCAH3-E8T:E8発現用ベクター。pAALFabをもとに、制限酵素サイトを改変した。新たにPstI、XbaI、およびKpnIサイトを付加し、EcoRI、およびXhoIサイトの位置を変更した。3)pFvCA-E8VHd:重鎖可変領域遺伝子クローニング用ベクター。pFCAH3-E8Tをもとに、制限酵素サイトを改変した。XbaI-EcoRI間を欠落させ、新たにKpnI、SfiI、NcoI、およびSpeIサイトを付加した。重鎖可変領域遺伝子をSfiI-XhoIサイトにクローニング可能。4)pFCAH9-E8d:重鎖可変領域遺伝子クローニング用ベクター。pFCAH3-E8T、およびpFvCA-E8VHdをもとにDNA配列を改変した。マウスγCH1をヒトγCH1で置きかえた。新たに、SfiI、NcoI、およびAscIサイトを付加した。軽鎖可変領域をSfiI-AscIサイトにクローニング可能。
【図3】図3は、pFCAH9-E8dのインサートの塩基配列を示す図。
【図4】図4は、pFCAH9-E8dのインサートの制限酵素サイトと塩基配列によってコードされるアミノ酸配列を示す図(1)。
【図5】図5は、pFCAH9-E8dのインサートの制限酵素サイトと塩基配列によってコードされるアミノ酸配列を示す図(2)。
【図6】図6は、pFCAH9-E8dのインサートの制限酵素サイトと塩基配列によってコードされるアミノ酸配列を示す図(3)。
【図7】図7は、pscFvCA-E8VHdのインサートの塩基配列を示す図。
【図8】図8は、pscFvCA-E8VHdのインサートの制限酵素サイトと塩基配列によってコードされるアミノ酸配列を示す図(1)。
【図9】図9は、pscFvCA-E8VHdのインサートの制限酵素サイトと塩基配列によってコードされるアミノ酸配列を示す図(2)。
【図10】図10は、本発明による抗体ライブラリーから選択された抗体のアフィニティーの測定結果を示すグラフ。縦軸は492nmにおける吸光度を、横軸はクローンの番号を示す。横軸のPCは陽性コントロールを示す。
【図11】図11は、表15の各クローンのELISAにおけるgHあるいはgEに対する反応性を示すグラフ。
【発明を実施するための形態】
【0071】
以下、実施例に基づいて本発明を更に具体的に説明する。
【実施例】
【0072】
1. ライブラリー作製用ファージミドベクターの作製
1-1 重鎖および軽鎖の組合せライブラリーを作製するためのベクターの作製。
図2に概念的に示すように、pTZ19Rファージミドベクター(ファルマシア)にM13ファージのpelB(シグナル配列)、His6タグ配列、M13ファージのcp3タンパク質(Δcp3(198aa-406aa)N端欠失キャプシドタンパク質3)配列、およびproteinAのアミノ酸配列をコードするDNAを適当な制限酵素部位で組み込みベクターpFCAH9-E8dを作成した(GENE 194(1997)35-46 Iba Y et al参照)。軽鎖λ5, λ6遺伝子に存在するBstPIでその遺伝子が切断されることを避けるためにpFCAH9-E8dはXhoI部位が設けられている。pFCAH9-E8dのインサートの塩基配列を図3に、制限酵素サイトと塩基配列によってコードされるアミノ酸配列を図4〜図6に示した。
このベクターの所定の位置に重鎖と軽鎖の遺伝子を挿入することにより、実際の抗体タンパク質発現ベクターが完成することとなる。完成したベクターによって発現される抗体の形状はFab型であり、重鎖軽鎖はN末の可変領域に続いて定常領域CH1, CLをそれぞれ有している。定常領域同士の-SS-結合によって、重鎖と軽鎖は結合されることになる。軽鎖定常領域CL遺伝子は前述のcp3遺伝子と結合されており、結果として発現タンパク質はFab-cp3の形状となる。
【0073】
具体的には、以下のような操作を行った。
用いたプライマー:
527 Reverse(配列番号:2):
5'-CAGGAAACAGCTATGAC-3'
599 E8VHf-PstR:(配列番号:3)
3'-CGGCTCCAAGTCGACGTCGTCA-5'
544 E8VHf-PstF:(配列番号:4)
5'-CAGCTGCAGCAGTCTGGGGCAGAGCTTGTGAAGCCAGGGGCCTCAGTCAAGTTGTCCTGCACAGCTTCTGGCTTCAACATTAA-3'
545 E8VHf-XbaR:(配列番号:5)
3'-AGACCGAAGTTGTAATTTCTGTGGATATACGTGACCCACTTCGTCTCCGGACTTTTCCCAGATCTCACCTAACCTTCCTAA-5'
546 E8VHf-XbaF:(配列番号:6)
5'-AAGGGTCTAGAGTGGATTGGAAGGATTGATCCTGCGAGTGGTAATACTAAATATGACCCGAAGGACAAGGCCACTATAACAGCA-3'
547 E8VHf-EcoR(配列番号:7)
3'-TTCCTGTTCCGGTGATATTGTCGTCTGTGTAGGAGGTTGTGTCGGATGGATGTCGACTTAAGGGAC-5'
548 E8VHf-EcoF(配列番号:8)
5'-CAGCTGAATTCCCTGACATCTGAGGACACTGCCGTCTATTACTGTGCTGGT-3'
549 E8VHf-BstR(配列番号:9):
3'-CAGATAATGACACGACCAATACTAATGCCGTTGAAACTGATGACCCCGGTTCCGTGGTGCCAGTGGCACAAGG-5'
590 His6-SmaR(配列番号:10):
3'-GGTTCTCTAACAGTAGTGGTAGTAGTGGTAATTATTCTCGATAGGGCCCTCGAA-5'
542 E8VLf-SacF(配列番号:11):
5'-GACATCGAGCTCACCCAGTCTCCAGCCTCCCTTTCTGCGTCTGTGGGAGAAACTGTCACCATCACATGT-3'
539 E8VLf-KpnR(配列番号:12):
3'-TGACAGTGGTAGTGTACAGCTCGTTCACCCTTATAAGTGTTAATAAATCGTACCATGGTCGTC-5'
542 E8VLf-KpnF(配列番号:13):
5'-GCATGGTACCAGCAGAAACCAGGGAAATCTCCTCAGCTCCTGGTCTAT-3'
543 E8VLf-BamR(配列番号:14):
3'-GGAGTCGAGGACCAGATATTACGTTTTTGGAATCGTCTACCACACGGTAGTTCCAAGTCACCGTCACCTAGGCCTTGTGTT-5'
562 E8VLf-XhoR(配列番号:15):
3'-TCATGAGGCACCTGCAAGCCACCTCCGTGGTTCGAGCTCTAGTTT-5'
563 E8VLf-XhoF(配列番号:16):
5'-AGTACTCCGTGGACGTTCGGTGGAGGCACCAAGCTCGAGATCAAA-3'
613 NheR(配列番号:17):
3'-ATCGACAGCT-5'
600 E8VLKpnXhoR(配列番号:18):
3'-AAGCCACCTCCATGGTTCGAGCTCTAGTTT-5'
LCP3ASC(配列番号:19):
3'-TCGAAGTTGTCCTTACTCACAAGCCGCGCGGTCAGCTGAGGTAA-5'
hCH1Bst(配列番号:20):
5'-ACCCTGGTCACCGTCTCCTCAGCCTCCACCAAGGGCCCATCGGTCTTCCCCCTGG-3'
hCH1midAS(配列番号:21):
3'-GGGAGTCGTCGCAGCACTGGCACGGGAGGTCGTCGAA-5'
hCH1midS(配列番号:22):
5'-GGACTCTACTCCCTCAGCAGCGTCGTGACCGTGCCC-3'
hCH1H6(配列番号:23):
3'-GGGTCGTTGTGGTTCCACCTGTTCTTTCAACTCGGGTTTAGAACAGTAGTGGTAGTAGTGGTA-5'
hCH1H6Sma(配列番号:24):
3'-GGGTTTAGAACAGTAGTGGTAGTAGTGGTAATTATTCTCGATAGGGCCCTCGAACG-5'
702 BstXhoF(配列番号:25):
5'-GGCACCACGGTCACCGTCTCGAGCGCCTCCACC-3'
pFCAH3-E8T 重鎖部分の作製
1)pAALFabを鋳型にして527-599を用いたPCR, 547-590を用いたPCRを行いDNA断片を作製した。
2)544-545, 546-547, 548-549にてPCRを行いDNA断片を作製した
3)1)2)を混合し527, 590によるPCRを行い、これをpAALFabのHindIII-SmaI siteにクローニングした
pFCAH3-E8T 軽鎖部分
4)542-562, 561-613を用いたPCRを行いDNA断片を作製した
5)538-539, 542-543にてPCRを行いDNA断片を作製した
6)4)5)を混合し538, 562によるPCRを行い、これをpAALFabのSacI-NheI siteにクローニングした
pFCAH9-E8d
6)VH stuffer部分の作製
pFCAH3-E8TをXbaI,EcoRIにて消化、klenow fragmentを作用させて平滑末端に変えた後self ligationさせてVH部分のstufferを作製した。
7)VH stuffer部分の作製
pFCAH3-E8Tを鋳型にして527-600にてPCR。6)のHindIII-XhoI siteにクローニングした。
8)これをKpnIにて消化、self ligationさせてVL部分のstufferを作製
9)SfiI,NcoI,SpeI siteの導入
pFCAH3-E8Tを鋳型にして527-663にてPCR。1)のHindIII-SacI siteにクローニングした。
10)AscI siteの導入
pFCAH3-E8Tを鋳型にして527-LCP3ASCにてPCRし、それをSacI完全消化、SalI部分消化した2)にクローニングした。
11)gammaCH1部分をヒト遺伝子に変換
ヒトgammaCH1部分にはBstPI siteが存在するためこれをなくす設計でクローニングを行った。扁桃cDNAを鋳型にしてhCH1Bst-hCH1midS, hCH1midAS-hCH1H6にてPCRしたのち、これを混合してhCH1Bst-hCH16SmaにてPCRし、そのDNA断片を3)のBstPI-Sma siteにクローニングした
12)Xho siteの導入
11)を鋳型に702-663にてPCRを行い、これを11)のBstPI-SacI siteにクローニングした。
【0074】
1-2 重鎖可変領域を一時的にクローニングするためのベクターの作製
公知の手法(GENE 194(1997)35-46 Iba Y et al参照)に従って、まずpAALFabベクター(図2)を作製した。pAALFabベクターのXbaIからEcoRIの間を欠落させ、新たに制限酵素切断部位Kpn I, Sfi I, Nco I, Spe Iを付加して、pFCAH3-E8Tを経て、VH(重鎖可変領域)をクローニング可能としたベクターpscFvCA-E8VHd(図2)を作製し、重鎖可変領域を一時的にクローニングするためのベクターとした。pscFvCA-E8VHdのインサートの塩基配列を図7に、制限酵素サイトと塩基配列によってコードされるアミノ酸配列を図8〜図9に示した。
具体的には
610 scBstSpeSacF(配列番号:26):
5'-CACCACGGTCACCGTCTCCTCAGGCGGTGGCGGATCAGGTGGCGGTGGAAGTGGCGGTGGTGGGTCTACTAGTGACATCGAGCTCACCCAG-3'
611 scBstSpeSacR(配列番号:27):
3'-GTGGTGCCAGTGGCAGAGGAGTCCGCCACCGCCTAGTCCACCGCCACCTTCACCGCCACCACCCAGATGATCACTGTAGCTCGAGTGGGTC-5'
527 Reverse(配列番号:28):
5'-CAGGAAACAGCTATGAC-3'
619 E8VHf-SfiNcoPstR(配列番号:29):
3'-GACGCCGGGTCGGCCGGTACCGGCTCCAAGTCGACGTCGTCA-5'
primer610とprimer611をアニールさせ、それをpFCAH3-E8TのBstPI-SacI siteにクローニングしてsingle chainの作製を行なった。さらに、primer527とprimer619にてPCRを行い、これをさらにHindIII-PstI siteにクローニングし、SfiI,NcoI siteへ導入した。
【0075】
2. イムノグロブリン軽鎖ライブラリーの作製
2-1 PCRを用いたイムノグロブリン軽鎖遺伝子の単離
骨髄細胞(検体No.59)4×107 cells、および臍帯血と末梢血のリンパ球から、市販のキット(Pharmacia Biotech社製 QuickPrep Micro mRNA Purification Kit)を用いて、2.6μgのmRNAを得た。このmRNAからcDNAを作製した。cDNAは、GibcoBRL社製 SuperScript Preamplification Systemによって作製した。プライマーには、オリゴdTを用いた。得られたcDNAを鋳型にして、軽鎖遺伝子の取得用5'プライマー(κ1 〜κ6、λ1〜λ6 )と3'プライマー(hCKASCプライマーまたはhCLASCプライマー)を用いて、PCRを行った。PCR産物は、フェノール処理後、エタノール沈殿して10μlのTEバッファーに懸濁した。用いたプライマーの塩基配列とPCRの条件は以下のとおりである。軽鎖遺伝子取得用プライマーの塩基配列中、下線部はSfiIサイト、AscIサイトを示す。
5'-プライマーκ1〜κ6
hVK1a(配列番号:30):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC GACATCCAGATGACCCAGTCTCC
hVK2a(配列番号:31):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC GATGTTGTGATGACTCAGTCTCC
hVK3a(配列番号:32):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC GAAATTGTGTTGACGCAGTCTCC
hVK4a(配列番号:33):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC GACATCGTGATGACCCAGTCTCC
hVK5a(配列番号:34):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC GAAACGACACTCACGCAGTCTCC
hVK6a(配列番号:35):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC GAAATTGTGCTGACTCAGTCTCC
5'-プライマーλ1〜λ6
hVL1(配列番号:36):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGTCTGTGTTGACGCAGCCGCC
hVL2(配列番号:37):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGTCTGCCCTGACTCAGCCTGC
hVK3a(配列番号:38):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC TCCTATGTGCTGACTCAGCCACC
hVL3b(配列番号:39):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC TCTTCTGAGCTGACTCAGGACCC
hVL4(配列番号:40):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CACGTTATACTGACTCAACCGCC
hVL5(配列番号:41):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGGCTGTGCTCACTCAGCCGCC
hVL6(配列番号:42):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC AATTTTATGCTGACTCAGCCCCA
3'-プライマーhCKASC(配列番号:43):
TCGACTGGCGCGCCGAACACTCTCCCCTGTTGAAGCTCTTTGTG
3'-プライマーHCLASC(配列番号:44):
TCGACTGGCGCGCCGAACATTCTGTAGGGGCCACTGTCTTCTC
【0076】
PCRの条件
cDNA 2μl
10× buffer ♯1(KODに添付) 10μl
dNTP mix(2.0mM) 10μl
25mM MgCl2 4μl
5'側プライマー(100pmol/μl) 1μl
3'側プライマー(100pmol/μl) 1μl
滅菌済MilliQ 71μl
KOD DNA polymerase(東洋紡2.5U/μl) 1μl
94℃ 1分、55℃ 2分、74℃ 1分を35サイクル
【0077】
2-2 ライブラリー作製に適した軽鎖を選択して軽鎖遺伝子ライブラリーを作製する方法
2-2-1 軽鎖遺伝子のファージミドへの組込み
1で得たPCR産物を以下の条件で制限酵素処理した。
PCR産物 10μl
10×NEB4(AscIに添付) 5μl
10×BSA(SfiIに添付) 5μl
滅菌済MilliQ 28μl
AscI (NEW ENGLAND Biolabs Inc. 10 U/μl) 1μl
SfiI (NEW ENGLAND Biolabs Inc. 20 U/μl) 1μl
37℃で1時間、50℃で1時間反応後、そのうち10μl分をアガロース電気泳動し、600bp付近のバンドを切り出して、ジーンクリーンIIキット(フナコシ株式会社)で精製した。PCR産物と同様に制限酵素処理したpFCAH9-E8d(図2)をジーンクリーンIIキットで精製し、制限酵素処理したPCR産物と以下の条件で16℃で4時間〜一晩反応させることによりライゲーションした。
制限酵素処理したpFCAH9-E8d 2μl
制限酵素処理したPCR産物 1μl
10×ligation buffer 1.5μl
(T4 DNA ligaseに添付)
10mM ATP 1.5μl
滅菌済MilliQ 8μl
T4 DNA ligase (宝酒造 10 U/μl) 1μl
【0078】
2-2-2 ファージミドの大腸菌への導入
得られたligated DNAを用いて以下のように大腸菌DH12Sを形質転換した。即ち、ligated DNA を一旦エタノール沈殿し、1/5TE(TEを滅菌済MilliQで5倍希釈したもの)3μlに溶解した。そのうち、1.5μlをコンピテントセルDH12S(GIBCO BRL製)20μlに懸濁し、以下の条件でエレクトロポレーションを行った。
エレクトロポレーター
BRL社Cell-Porator(Cat.series 1600)
設定条件;voltage booster 4kΩ
capacitance 330μF
DC volts LowΩ
charge rate Fast
【0079】
2-2-3 ファージミドで形質転換した大腸菌からのFab-cp3型抗体培地中への分泌
形質転換した上記の大腸菌を形質転換用培地(SOB)2mlに植え、37℃で1時間振盪培養したあと、一部を寒天培地(Ampプレート)にまき、残りは、0.1%グルコース、100μg/mlアンピシリン含有2×TY培地で培養し、グリセリンストックした。寒天培地は30℃でincubateし、生えてきたコロニーを楊枝でつついて分離し、それぞれプラスミドを調製し、軽鎖遺伝子の塩基配列を調べた。
SOB培地:950mLの精製水に次の成分を加えて振とうし、完全に溶解した後250mMのKCl溶液10mLを加え、5N NaOHでpH7.0に調製した。精製水を加えて1000mLに調整した後、オートクレーブで20分間滅菌し、使用直前に滅菌した2MのMgCl2を5mL加えた。
bacto-tryptone 20g
bacto-yeast extract 5g
NaCl 0.5g
2×YT培地:900mLの精製水に次の成分を加えて振とうし、完全に溶解した後5N NaOHでpHを7.0に調製し、精製水を加えて1000mLとした。オートクレーブで20分間滅菌して使用した。
bacto-tryptone 16g
bacto-yeast extract 10g
NaCl 5g
その他の試薬は以下から購入した。
メーカー 品名
シグマ アンピシリンナトリウム
和光純薬 フェノール
シグマ BSA
DIFCO 2×YT培地
和光純薬 カナマイシン硫酸塩
ナカライテスク ポリエチレングリコール6000
ナカライテスク Tween20
片山化学 NaCl
和光純薬 IPTG
和光純薬 スキムミルク
和光純薬 アジ化ナトリウム
和光純薬 トリエチルアミン
和光純薬 過酸化水素
和光純薬 OPD錠
和光純薬 エタノール
【0080】
κ1、κ2、κ3、κ4、κ5、およびκ6、並びにλ1、λ2、λ3a、λ3b、λ4、λ5、λ6、λ7、λ8、λ9、およびλ10の全てについて以上の操作を行い、目的のクローンが得られているかどうか確認した。続いてκ1、κ2などの各グループのクローンをin vivoでの使用頻度に近い比率になるように混合した。これら軽鎖の各グループは、それぞれ実際の生体内でどのような割合で発現しているのかが既に知られている。PCR法で増幅してベクターに組み込んだこれらの遺伝子クローンを、in vivoでの使用頻度に近い比率になるように混合しVLライブラリーとした。VLライブラリーにおける各familyの構成比率を以下に示す。
【0081】
【表2】
【0082】
【表3】
【0083】
次に、VLライブラリーから無作為に選んだ約1000個の軽鎖遺伝子の塩基配列を確認した。すなわち、蛍光プライマーhuCH1J(5'-ATTAATAAGAGCTATCCCGG-3'/配列番号:45)を用い、サーモシークエンスキット(アマシャム・ファルマシア製)とアロカ社製L1-COR4200L(S)-2を使用したジデオキシ法によって塩基配列を決定した。得られた塩基配列を比較して重複するクローンを除いた。更にデータベースと照合し、deletionが無いと確認されたクローンについて、予め発現することがわかっている重鎖遺伝子のクローンの一つVH3-4と組み合わせて、発現実験を行った。操作は以下のとおりである。VH3-4のアミノ酸配列を配列番号:1に示した。
まずVH3-4をHindIIIとXhoIで消化し、重鎖遺伝子を切り出して、ジーンクリーンIIキットで精製した。一方、deletionが無いと確認された軽鎖遺伝子クローンについてもHindIIIとXhoIで消化し、軽鎖遺伝子を切り出して、ジーンクリーンIIキットで精製し、VH3-4の重鎖遺伝子とライゲーションすることにより、組み合わせた。得られたligated DNAを用いて大腸菌DH12Sを形質転換した。生えてきたコロニーを試験管にいれた培地にうえ、IPTGで発現を誘導することにより、Fab-cp3型の抗体分子を培養上清中に発現させた。20時間程度の培養により、ヘルパーファージの感染無しでもFab-cp3型の抗体分子が培養上清に発現される。この培養上清を用いて以下のようなELISAを行った。
【0084】
2-2-4 ELISA法による重鎖と軽鎖の正しい発現と会合の検定
1)抗体結合96wellマイクロタイタープレートの作製
抗κ抗体(MBL code No.159)を0.01Mナトリウム−リン酸緩衝液 pH8.0, 0.1% NaN3で1.25μg/mlで希釈し、100μLずつマイクロタイタープレートに添加した。4℃で一晩静置することにより、ウエルに抗κ抗体を吸着させた。反応液を捨て、5% BSA in 0.01Mナトリウム−リン酸緩衝液 pH8.0, 0.1% NaN3を200μLずつマイクロタイタープレートに添加し、37℃で2時間静置することにより、非特異的な吸着を防ぐためにブロッキングした。
次に、非特異的活性吸収済の抗λ抗体(MBL code No.159)を0.01Mナトリウムリン酸緩衝液 pH8.0, 0.1% NaN3で2.5μg/mlに希釈し、100μLずつマイクロタイタープレートに添加し氷室で一晩静置した。反応液を捨て、5% BSA in 0.01Mナトリウム−リン酸緩衝液 pH8.0, 0.1% NaN3を200μLずつマイクロタイタープレートに添加し37℃で2時間静置し、非特異的な結合を防ぐためにブロッキングを行った。
【0085】
2)1次反応
positive contorolとして、ヒトFab(10μg/ml)を、negative controlとして、PBS/0.1%NaN3をそれぞれ100μlずつマイクロタイタープレートに添加した。IPTGでFab-cp3型の抗体分子の発現を誘導した培養上清の原液を100μlずつマイクロタイタープレートに添加し37℃で1時間反応させた。
【0086】
3)2次反応
1次反応を終了したマイクロタイタープレートを0.05%Tween20-PBSで5回洗浄した。次いでPBS/0.1%NaN3で希釈した抗Fd抗体(1μg/ml)を100μlずつマイクロタイタープレートに添加し37℃で1時間反応させた。
【0087】
4)3次反応
2次反応を終了したマイクロタイタープレートを0.05%Tween20-PBSで5回洗浄した。次いでPBS/0.1%NaN3で希釈したアルカリフォスファターゼ標識抗ヒツジIgG抗体(4000倍希釈)を100μlずつマイクロタイタープレートに添加し37℃で1時間反応させた。
【0088】
5)発色反応および吸光度測定
3次反応を終了したマイクロタイタープレートを0.05%Tween20-PBSで5回洗浄した。次いで発色基質溶液(SIGMA 1040 phosphatase substrate tablets 1粒あたり5mlの50mMジエタノールアミンPH9.8に溶解したもの)を100μlずつマイクロタイタープレートに添加した。室温で反応させ、405nmの吸光度が0.5以上になったと思われる時点で、停止液を添加し、プレートリーダー(タイターテック マルチスキャンMCC)で吸光度測定した。
このELISAで陽性(吸光度0.5以上)となったクローンは、Fab-cp3型の抗体分子の発現と会合がうまく行われているとし、κ鎖遺伝子、λ鎖遺伝子それぞれ反応性の高いものから100個ずつ選択した。両者を混合してFab-cp3型の抗体分子の発現と会合がうまく行われているクローンを集めたライブラリーKL200とした。
【0089】
3. 軽鎖遺伝子ライブラリーと重鎖遺伝子ライブラリーの組み合わせライブラリーの作製
3-1-1 PCRを用いたイムノグロブリン重鎖遺伝子の単離
2-1と同様の手順を用いて臍帯血、骨髄液、および末梢血のリンパ球、並びに扁桃からhuman μ primer(以下に示すプライマーの634)あるいはrandom hexamerを用いてcDNAを調製し、このcDNAを鋳型にして、以下に示すヒト抗体重鎖遺伝子の取得用5'プライマー(VH1〜VH7)と3'プライマー(human JHプライマー4種を等量混合したもの、以下に示すプライマーの697〜700)、または、humanμプライマー(以下に示すプライマーの634)を用いて、PCRを行った。表中、下線をつけた部分はSfiIサイトを示す。hVH2aはgerm line VH2 familyに対応していないため、新たにVH2a-2を設計した。またhVH4aではVH4ファミリー全体に対応していないため、新たにhVH4a-2を設計した。VH5aもgerm line VH5 subfamilyに対応していなかったため新たにVH5a-2を設計した。またVH7に対応するprimerとしてhVH7を設計した。これらについても遺伝子増幅を行い、pscFvCA-E8VHd(0−2)に組み込み、その塩基配列を決定することによって、どのような遺伝子が増幅されたのかを確認した。hVH5a-2についてはhVH1aと配列が酷似しているため、hVH1aで増幅させたものと同様の遺伝子産物が得られることが予想されるためこれについては使用しなかった。PCR産物は、フェノール処理後、エタノール沈殿して10μLのTEバッファーに懸濁した。
【0090】
634 humμCH1R(配列番号:46):
ATGGAGTCGGGAAGGAAGTC
各VH familyの増幅に使用したprimer
Human VH primer SfiI siteを下線で示す
628 hVH1a(配列番号:47):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGGTGCAGCTGGTGCAGTCTGG
629 hVH2a(配列番号:48):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGGTCAACTTAAGGGAGTCTGG
630 hVH3a(配列番号:49):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC GAGGTGCAGCTGGTGGAGTCTGG
631 hVH4a(配列番号:50):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGGTGCAGCTGCAGGAGTCGGG
632 hVH5a(配列番号:51):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGGTGCAGCTGTTGCAGTCTGC
633 hVH6a(配列番号:52):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGGTACAGCTGCAGCAGTCAGG
629-2 hVH2a-2(配列番号:53):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGRTCACCTTGAAGGAGTCTGGTCC
631-2 hVH4a-2(配列番号:54):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGGTGCAGCTACAGCAGTGGGG
632-2 hVH5a-2(配列番号:55):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC GAGGTGCAGCTGGTGCAGTCTGG
712 hVH7(配列番号:56):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGGTGCAGCTGGTGCAATCTGGGTCTGAGT
Human JH primer BstPI, XhoI siteを下線で示す
697 hJH1-2(配列番号:57):
GGTGGAGGCACTCGAGACGGTGACCAGGGTGC
698 hJH3(配列番号:58):
GGTGGAGGCACTCGAGACGGTGACCATTGTCC
699 hJH4-5(配列番号:59):
GGTGGAGGCACTCGAGACGGTGACCAGGGTTC
700 hJH6(配列番号:60):
GGTGGAGGCACTCGAGACGGTGACCGTGGTCC
【0091】
cDNA 2μL
10× buffer ♯1(KODに添付) 10μL
dNTP mix(2.0mM) 10μL
25mM MgCl2 4μL
5'側プライマー(100pmol/μL) 1μL
3'側プライマー(100pmol/μL) 1μL
滅菌済MilliQ 71μL
KOD DNA polymerase(東洋紡2.5U/μL) 1μL
PCR条件:94℃ 1分、55℃ 2分、74℃ 1分を35サイクル
【0092】
3-1-2 重鎖遺伝子ライブラリーの作製
3-1-1で得たPCR産物を以下の条件で制限酵素処理した。
PCR産物 10μL
10×K buffer(宝酒造) 5μL
滅菌済MilliQ 33μL
HindIII (宝酒造15 U/μL) 1μL
XhoI (宝酒造12 U/μL) 1μL
37℃で2時間反応後、そのうち10μL分をアガロース電気泳動し、400 bp付近のバンドを切り出して、ジーンクリーンIIキット(フナコシ株式会社)で精製した。PCR産物と同様に制限酵素処理したpscFvCA-E8VHd(図2)をジーンクリーンIIキットで精製し、制限酵素処理したPCR産物と以下の条件で16℃で4時間〜一晩反応させることによりライゲーションした。
制限酵素処理したpscFvCA-E8VHd 2μl
制限酵素処理したPCR産物 1μl
10×ligation buffer 1.5μl
(T4 DNA ligaseに添付)
10mM ATP 1.5μl
滅菌済MilliQ 8μl
T4 DNA ligase (宝酒造 10 U/μl) 1μl
【0093】
3-1-3 ファージミドの大腸菌への導入
得られたDNAを大腸菌DH12Sに形質転換した。具体的にはDNAを一旦エタノール沈殿し、1/5TE(TEを滅菌済MilliQで5倍希釈したもの)3μLに溶解する。そのうち、1.5μLをコンピテントセルDH12S(GIBCO BRL製)20μLに懸濁し、エレクトロポレーション法により形質転換した。
エレクトロポレーター
BRL社Cell-Porator(Cat.series 1600)
設定条件;voltage booster 4kΩ
capacitance 330μF
DC volts LowΩ
charge rate Fast
【0094】
形質転換用培地(SOB)2mlに上記操作の終了した形質転換大腸菌を植え、37℃で1時間振盪培養したあと、一部を寒天培地(Ampプレート)にまき、残りは、0.1%グルコース、100μg/mlアンピシリン含有2×YT培地で培養し、グリセリンストックした。寒天培地は30℃でインキュベートし、生えてきたコロニーを楊枝でつついて分離し、それぞれプラスミドを調製し、重鎖遺伝子の塩基配列を調べた。VH1〜VH7の全てについてこれらのことを行い、目的のクローンが得られているかどうか確認した。これらの各グループ(ファミリー)のクローンをin vivoでの使用頻度に近い比率になるように混合してVHライブラリーとした。VHライブラリーにおける各ファミリーの構成比率を以下に示す。
【0095】
【表4】
【0096】
3-2 組み合わせ遺伝子ライブラリーの作製
VHライブラリー200μgを下記条件でHindIIIとXhoIで消化し、重鎖遺伝子を切り出して、ジーンクリーンIIキットで精製した。
VHライブラリー200μg 100μL
10×K buffer(宝酒造) 40μL
滅菌済MilliQ 205μL
HindIII (宝酒造40 U/μL) 30μL
XhoI (宝酒造50 U/μL) 25μL
deletionが無いと確認された軽鎖遺伝子クローンKL200、およびVLライブラリーの挿入されたベクターpFCAH9-E8dについても下記条件でHindIIIとXhoIで消化し、軽鎖遺伝子を含む断片を、ジーンクリーンIIキットで精製した。
KL200またはVLライブラリー
を挿入したpFCAH9-E8d 100μg 100μL
10×K buffer(宝酒造) 40μL
滅菌済Milli-Q 230μL
HindIII (宝酒造40 U/μL) 15μL
XhoI (宝酒造50 U/μL) 15μL
【0097】
次に、VH遺伝子ライブラリー断片と軽鎖遺伝子の挿入されたpFCAH9-E8dベクターを、次の条件下、16℃で一晩反応させてライゲーションした。
制限酵素処理した
VHライブラリー断片 10μg 50μL
制限酵素処理したKL200または
VLライブラリーの断片
を含むpFCAH9-E8d 40μg 50μL
10×ligation buffer
(T4 DNA ligaseに添付) 100μL
10mM ATP 100μL
滅菌済MilliQ 670μL
T4 DNA ligase (宝酒造 10 U/μL) 30μL
【0098】
反応の終了したDNAを用いて大腸菌DH12Sを形質転換した。具体的にはDNAを一旦エタノール沈殿し、1/5TE(TEを滅菌済MilliQで5倍希釈したもの)30μLに溶解した。これをコンピテントセルDH12S(GIBCO BRL製)500μLに懸濁し、エレクトロポレーションを行った。
エレクトロポレーター
BRL社Cell-Porator(Cat.series 1600)
設定条件;voltage booster 4kΩ
capacitance 330μF
DC volts LowΩ
charge rate Fast
【0099】
形質転換用培地(SOB)12mlに上記操作の終了した大腸菌を植え、37℃で1時間振盪培養したあと、一部を寒天培地(Ampプレート)にまき、残りは、0.1%グルコース、100μg/mlアンピシリン含有2×YT培地500 mlで培養し、グリセリンストックした。寒天培地は30℃でインキュベートし、生えてきたコロニーの数から得られたクローンの数を推定した。それぞれ5×1010クローンが得られた。
扁桃mRNAよりrandam hexamerにて合成したcDNAをもとに得た各VH familyをpscFvCA-E8VHdベクターにクローニングし、KL200と組み合わせたライブラリーをAIMS1とした。(1.28×1010の独立したクローン)
臍帯血、骨髄液、末梢血、扁桃mRNAよりhuman m primerにて合成したcDNAをもとに得た各VH familyをpscFvCA-E8VHdベクターにクローニングし、KL200と組み合わせた遺伝子ライブラリーをAIMS2とした. (3.20×1010の独立したクローン)
臍帯血、骨髄液、末梢血、扁桃mRNAよりhuman m primerにて合成したcDNAをもとに得た各VH familyをVL libraryと組み合わせたライブラリーをAIMS3とした。(4.50×1010の独立したクローン)
更に(AIMS1+AIMS2) : AIMS3 = 1 : 1で混合し、1×1011の独立したクローンからなるファージ抗体ライブラリーとした(AIMS4と呼ぶ)。
【0100】
3-3 組み合わせ遺伝子ライブラリーによるファージライブラリーの作製
3-3-1 ファージライブラリーの調製
1%グルコース及び100μg/mLのアンピシリンを加えた2×YT培地300mLを入れた5リットルのフラスコにAIMS4懸濁液を2.5mLを加え、37℃で振とう培養し1時間おきに波長600nmにおける吸光度を測定しながら、吸光度が1.0になるまで増殖させた。培養液にヘルパーファージ液(M13KO7)をフラスコ当たり12mL加えてヘルパーファージを感染させ、37℃で2時間培養し、ヘルパーファージ感染済みDH12Sとした。
3リットルのフラスコ24本に2×YT培地600mLと100μg/mLのアンピシリン1.2mL、50μg/mLのカナマイシン0.8mL、ヘルパーファージ感染済みDH12S 200mLを加えて37℃で2時間毎に波長600nmの吸光度を測定しながら振とう培養した。アンピシリンは、測定毎に200μg/mLとなるよう追加した。培養は波長600nmにおける吸光度が1.3になるまで行った。
菌体は4℃で8000rpm、10分間遠心し、上清を集めた。上清に20%のポリエチレングリコール/2.5M NaCl 4Lを加えて約20分間静かに攪拌した後、4℃で8000rpm、20分間遠心、沈殿を1LのPBSで溶かし、20%のポリエチレングリコール/2.5M NaCl 200mLを加えて約20分間静かに攪拌した後、4℃で8000rpm、20分間遠心した。上清を捨ててさらに4℃で8000rpm、3分間遠心して沈殿を回収した。沈殿は0.05% NaN3を加えたPBSで溶解し、4℃で1000rpm、15分間遠心し、上清を回収した後、4℃で8000rpm、3分間さらに遠心して上清を回収した。
【0101】
回収したファージ溶液の力価は以下のようにチェックした。すなわち、ファージ溶液をPBSで106、107、108希釈し、その10μLをDH12S 990μLに感染させ、37℃で1時間培養した。これをLBGAプレートに100μL播いて30℃で18時間培養した。コロニーの数をカウントすることにより希釈前の原液の力価を算出した。ファージ溶液原液を2%スキムミルク及び0.05% NaN3を含むPBSに2×1014/mlになるよう懸濁した。
【0102】
3-3-2 Fab-cp3を表面に発現しているファージを濃縮する方法
以上のようにして調製したライブラリーには、Fab-cp3をその表面に発現しているファージを選択的に濃縮し、ヘルパーファージやFab-cp3の発現していないファージの混入割合を減少させるための工夫が施されている。すなわち上記ライブラリーを構成するファージが発現する重鎖のC末端には、His6ペプチド(ヒスチジンタグ)が付加されている。ヒスチジンタグを発現したファージは、ニッケルイオンなどに吸着することを利用して容易に回収することができる。具体的にはニッケルイオンが付加されたゲル(Ni-NTAアガロース等)を用いる。操作は以下のとおりである。
Ni-NTAアガロースを2%スキムミルク及び0.1%Tween20を含むPBS(以下ブロッキング緩衝液)で室温で30分ブロッキングした。次に、重鎖にHis-Tagが付加されていないFabを表面に発現しているファージ(pFCA-E9HLφ;ファージHis-)と重鎖にHis-Tagが付加されているFabを表面に発現しているファージ(pFCAH6-D1.3HLφ;ファージHis+)をファージHis-:ファージHis+ = 100:1となるようにブロッキング緩衝液中で混合した。これらの合計1×1010CFUのファージ溶液250μLをNi-NTAアガロースと混合し、室温1時間反応させた。Ni-NTAアガロースをブロッキング緩衝液で洗浄し、次いで0.5Mイミダゾール(pH 7.55)500μLを加えてNi-NTAアガロースに結合していたファージを溶出した。
溶出したファージを後(4-3)で述べる操作にしたがって回収し、回収したクローンを調べてみたところ、23クローン中15クローンがファージHis+であった(表3)。このことはNi-NTAアガロースにより、His6ペプチドが付加されたファージが53倍に濃縮されたことを示している。
この操作を加えることにより、ライブラリーの性能を向上させ、あるいはスクリーニングの能率を上昇させることが可能であることが以上(4)によって示された。
【0103】
【表5】
【0104】
4. ファージライブラリーからの特定の抗原に特異的に結合するファージの選択
ファージライブラリーから特定の抗原に特異的に結合するファージを選択する操作を、本明細書ではスクリーニングと呼ぶ。抗原として破傷風トキソイドを用い、本発明による抗体ライブラリーのスクリーニングを行った。
【0105】
4-1 スクリーニング用試験管の作製
抗原(破傷風トキソイド)をPBSで20μg/mLに調製し、試験管3本(Nunc社製Maxisorp)に3mLずつ添加して4℃で18時間インキュベートして、試験管内表面へ抗原を吸着させた。吸着後、抗原溶液を捨て、2%スキムミルク含有PBS溶液3mLずつを加えて25℃で1時間反応させ、ファージ抗体が非特異的に試験管に結合することを防ぐためにブロッキングを行った。
【0106】
4-2 スクリーニング操作
作製した抗原吸着済試験管にAIMS4ファージライブラリーを2%スキムミルク、0.1%Tween20含有PBSになるよう溶解して1×1014CFU/9mLに調製し、この液を試験管3本に3mLずつ添加して25℃で2時間反応させた後、0.1%Tween20を加えたPBSで4回、PBSで4回、および滅菌した超純水(Milli-Qにて作製)で1回洗浄した。
続いて抗原結合試験管に結合したファージを以下のように回収した。すなわち、0.1Mトリエチルアミン(pH12.3)を試験管1本当たり3ml添加し、ローテーターを用いて室温で20分間反応させ乖離させた後、1M Tris-HCl緩衝液(pH6.8)1.1mLを加えて中和し、この液を回収した。
【0107】
4-3 回収したファージの増幅
回収した液は(ファージの大腸菌への感染)(ヘルパーファージの感染)(ファージの回収)の処理を行い、含まれているファージを精製・増幅した。
1) ファージの大腸菌への感染
大腸菌(DH12S)を2×YT培地50mlで培養し、波長600nmの吸光度が0.5になるよう増殖させ、上記で回収したファージ液を加えて37℃で1時間振とう培養した。
2) ヘルパーファージの感染
1)の培養液62.3mLをとり、2×YT培地425mL、40%グルコース12.5ml、および100μg/mLアンピシリン0.5mLを加えて37℃で波長600nmにおける吸光度が0.5になるまで培養した後、4℃、5000rpmで10分間遠心して菌体を沈殿させ、回収して100mg/mLアンピシリン0.3mLを加えた2×YT培地300mLに懸濁した。これにヘルパーファージM13K07を1/100量加え、37℃で1時間振とう培養した。
培養液を予め37℃に暖めた培地(2×YT培地に100μg/mLアンピシリンと70μg/mLのカナマイシンを加えた液)900mLに加えて37℃で一晩培養した。
3) ファージの回収
2)の培養液を4℃で7000rpm、10分間遠心し、その上清に2.5Mの塩化ナトリウムを加えた20%のポリエチレングリコールを1/5量加えて室温で20分間静置した後、4℃で8000rpm、15分間遠心して沈殿を回収し、培養液の1/10量の滅菌PBSを加えて溶解し、再度2.5Mの塩化ナトリウムを加えた20%のポリエチレングリコールを1/5量加えて4℃で10000rpm、20分間遠心して上清を捨て、さらにスピンダウンして4℃で10000rpm、2分間遠心した。これに0.05%のNaN3を加えたPBSを培養液の1/100量加えて沈殿を溶解し、ファージを回収した。
【0108】
4-4 増幅したファージによる再スクリーニング
増幅したファージを用いて4-2と同様に抗原結合試験管を用いてスクリーニングを繰り返した。スクリーニングでの洗浄は、非特異に吸着したファージを乖離し、結合力の高いファージを選択する上で重要なステップであることから、2回目以降のスクリーニングにおける洗浄条件は以下のようにした。
2回目;PBS+0.1% Tween20で6回、PBSで6回、滅菌した超純水で1回
3回目;PBS+0.1% Tween20で13回、PBSで13回、滅菌した超純水で1回
【0109】
4-5 ファージのスクリーニング手技の評価法
4-4の方法でスクリーニングを繰り返すとき、(抗原吸着済試験管に投入したファージの総数)÷(抗原吸着済試験管から回収したファージの総数)が前回のスクリーニングに比べて明らかに小さくなれば、目的の抗体を提示しているファージが濃縮されつつあると推測できる。溶液に含まれるファージ数は、以下のように計算した。
1)ファージの希釈系列を以下のように作製した。
[1] 1×10-2希釈:ファージ液10μL+PBS990μL
[2] 1×10-4希釈:[1]の希釈液10μL+PBS990μL
[3] 1×10-6希釈:[2]の希釈液10μL+PBS990μL
[4] 1×10-8希釈:[3]の希釈液10μL+PBS990μL
[5] 1×10-9希釈:[4]の希釈液100μL+PBS900μL
[6] 1×10-10希釈:[5]の希釈液100μL+PBS900μL
[4]、[5]、および[6]の希釈系列10μLにDH12S 990μLを加えたものを作り、37℃で1時間感染させ、これをLBGAプレートに100μLまいて30℃で18〜24時間培養し、コロニーを計数した。上記希釈系列のうち、通常[4]のプレートが50個以上のコロニーを作る。mL当たりのファージ数は[4]のプレートのコロニー数に基づいて、以下のように算出される。
原液のファージ数=(コロニー数/プレート)×(1×108)×103 cfu/mL
【0110】
また、回収されたファージ数も同様に計算し、本抗原に対する抗体を提示するファージの数をスクリーニングごとに求めたところ、表6のようになった。
【0111】
【表6】
【0112】
4-6 スクリーニングによって得られた抗体の抗原結合活性(アフィニティー)の測定
以上のスクリーニングによって選択された抗体について、抗原結合活性(アフィニティー)を測定した。アフィニティーの測定には、ファージ型の抗体ではなく、Fab-cp3型抗体をサンプルとして用いた。測定方法は、96ウェルマイクロタイタープレートを用いたELISA法とした。Fab-cp3型抗体の発現誘導法については4-7で述べる。
まずELISA用のプレートを以下のように調製した。抗原20μg/mLを96ウェルマイクロタイタープレート(Nunc社製Maxisorp)の各ウエルに100μL添加して4℃で18時間結合させたのち、5%BSA(ブロッキング液)を各ウエルに200μL添加して37℃で1時間ブロッキングした。ブロッキング液を捨てた後、PBSで1回洗浄してアフィニティーの測定に用いた。4-7の手順で採取した培養上清を各ウエルに100μL加え、25℃1時間反応させた。反応後、PBSで4回洗浄し、5000倍希釈したペルオキシダーゼ標識抗ヒトIgG((株)医学生物学研究所製)を100μL加えて25℃で1時間反応させた。再度PBSで4回洗浄し、オルトフェニレンジアミンと過酸化水素の溶液100μLを加えて暫時反応させた後、1.5Nリン酸100μLを加えて反応を停止し、波長492nmにおける吸光度を測定した。その結果96クローン中77クローンに活性が確認された(図10)
更にこれら77クローンのうち、37クローンについて中和活性を調べた。表7の○が、中和活性を調べたクローンである。中和活性を調べた段階では、まだ塩基配列を確認していなかったので、これらのクローンの中には同じDNA配列の組み合わせであるクローンが存在する可能性があった。これらのクローンの塩基配列を決定して照合し、同じ配列の組み合わせを持つものは表7では同じ枠内にまとめた。あとで照合してみると同じDNA配列の組み合わせを持つクローンはELISAにおいても同程度のアフィニティーを示していたことが確認された。
【0113】
4-7 Fab-cp3型抗体の発現誘導
ファージの感染した大腸菌を1%グルコースと100μg/mLのアンピシリンを加えた2×YTで30℃18時間培養した後、0.1%グルコースと100μg/mLのアンピシリンを加えた2×YT 1.5mLに上記培養液を5μL加えて30℃で4時間培養した。このときの大腸菌の濃度は波長600nmの吸光度を測定するとき、約0.5であった。
これに1mMになるようIPTG(イソプロピル-1-チオ-β-D-ガラクトシド)を加えてさらに30℃で18時間培養した後、培養液1.5mLをエッペンドルフチューブにとり、4℃で10000r.p.m.5分間遠心してその培養上清をとり、0.1%となるようアジ化ナトリウムを添加して検体とした。
Fab-cp3型抗体が発現しているか否かは2-3で示したELISA法で確認した。発現が認められたクローンのうち、アガロース電気泳動によってdeletionを確認したところ、8クローンにdeletionが認められたが、これらはいずれも抗原結合活性が認められなかった。
【0114】
4-8 モノクローナル化とファージミドの精製
4-5「ファージのスクリーニングの評価」の項で得たシングルコロニーのプレートから大腸菌クローンを選択し、LBGA中で30℃18時間培養した後、倉敷紡績社製DNA分離装置PI-50 File No.50を用いてファージミドを精製した。
【0115】
4-9 モノクローナル抗体の同定
本発明の抗体ライブラリーからスクリーニングによって選択された抗破傷風毒素抗体について、その遺伝子の配列を確認した。配列は、重鎖、軽鎖について以下のプライマーを用い、サーモシークエンスキット(アマシャム・ファルマシア製)とアロカ社製L1-COR4200L(S)-2を使用したジデオキシ法によって決定した。結果を表7にまとめた。表7では、塩基配列決定の結果、同じクローンであることが判明したクローンをまとめて示した。表中のH-chainに示した番号とL-chainに示した番号の組み合わせが同一となっているクローンが、同じ塩基配列を含むクローンである。表7に示すように、36種類のクローンが得られていたことがわかった。
重鎖のシークエンシング用プライマー:蛍光T7プライマー(アロカ製)
軽鎖のシークエンシング用プライマー:蛍光プライマーhuCH1J(配列番号:45)
【0116】
【表7】
【0117】
4-10 中和活性検定用 Fab-cp3型抗体の採取
本発明の抗体ライブラリーからスクリーニングによって選択された抗体の、破傷風毒素に対する中和活性の有無を検討した。まず抗体活性のあった77クローンについて、発現前の培養液と、1% グルコース, 100mg/mLアンピシリンを加えた2×YT培地(YTA)で30℃で一晩培養した後、0.1% グルコース YTAで30℃3時間培養したのち、1M IPTGを加え、30℃で20時間培養した。10000rpmで5分間遠心して上清をとり、60%になるよう硫安を加えて1時間振とうした。さらに12000rpmで10分間遠心して上清を除き、沈殿にPBSを加えて溶解後、50倍量以上のPBSに対して2時間透析した。これを3回繰り返した後、エッペンドルフチューブに移して15000rpmで4℃、10分間遠心し、上清をろ過した後、破傷風毒素に対する中和活性を検討した。
【0118】
4-11 中和活性の検討
まず、抗体自体の毒性を確認するため、上記で得た抗体溶液0.2mLをマウスの腹腔と尾静脈内に注射した。その結果、抗体溶液には毒性は認められなかった。次に、抗体注射後約1時間で、マウス最小致死量(1MLDまたは約2LD50)の10倍量の破傷風毒素を後肢内股部皮下に注射した。その結果、各抗体を注射したマウスは約半数において部分的中和能(毒素による発症時期の遅延、4〜7日)が認められた。中和抗体(標準破傷風抗毒素)または中和能のない抗体を注射した マウスは、破傷風毒素による特異的麻痺が観察された後、約2日で死亡した。
部分的中和活性を示したクローンは、TETM1, TETM13, TETM26, TETM96であった。したがって、本発明の抗体ライブラリーをスクリーニングすることによって、中和活性を持つ有効な抗体を得られることが証明されたと言える。
【0119】
4-12 抗体可変領域の配列決定
上記のとおり、本発明者らは破傷風トキソイド(毒素)に結合し、これを中和する抗体可変領域を発現するファージを得ることができた。
さらに、上記工程を繰り返すことにより、中和活性を持つが互いに異なる塩基配列を持つ別のファージクローンを得ることもできた。具体的にはクローン番号TETM13とTETK36が強い中和活性を持つと判断された。これらファージクローンが有する重鎖と軽鎖の塩基配列、およびこの塩基配列によってコードされるアミノ酸配列を以下の配列番号として記載する。
クローン番号 重鎖 軽鎖
TETM13(塩基配列) 63 64
TETM13(アミノ酸配列) 81 82
TETK36(塩基配列) 65 66
TETK36(アミノ酸配列) 83 84
【0120】
5. 選択をかけない軽鎖遺伝子ライブラリーと、KL200ライブラリーの比較
選択をかけない軽鎖遺伝子ライブラリーを使用して作製した重鎖との組合せライブラリー(AIMS3)と、選択した軽鎖遺伝子ライブラリーKL200を使用して作製した重鎖との組合せライブラリー(AIMS1+AIMS2)とでライブラリーの性能を比較した。
まず選択されたクローンの数の比較においては、本発明による抗体ライブラリーであるAIMS1+AIMS2が、圧倒的に有利であることが明らかとなった。選択をかけた軽鎖遺伝子ライブラリーであるAIMS1+AIMS2から選択されたクローンは、前記表7における、KL200のカラムにKL200におけるクローンの名称が記載された29クローンである。一方、AIMS3からは7クローン(前記表7における、KL200のカラムがVL Libraryとなっているクローン)が選択された。つまり、等量で混合したライブラリーを用いたのにもかかわらず選択されたクローンの80%以上(29/36)が本発明によるライブラリーに由来するクローンであった。このように、スクリーニングで得られたファージ数の比較によりKL200を用いて作製したファージライブラリーが有利であることを示すことができた。
【0121】
5-2 スクリーニングによって得られたファージの抗体活性の比較
次に、選択された抗体の活性について比較した。4-11で述べた中和活性の試験において部分的中和能が確認されたクローンは、次の4クローンである。
TETM1 (AIMS3)
TETM13 (AIMS1+AIMS2)
TETM26 (AIMS1+AIMS2)
TETM96 (AIMS1+AIMS2)
つまり、AIMS1+AIMS2から3クローン、AIMS3から1クローンが選択されていた。この割合は、スクリーニングで得られたクローンの数の割合(29:7)とほぼ同様である。しかも、本発明によるライブラリーに由来するAIMS1+AIMS2から選択された3クローンには、最も中和活性の強いクローンであるTETM13が含まれていた。したがって、抗体の機能の面からみても、選択された軽鎖ライブラリーKL200を用いて作製したファージライブラリーのスクリーニングによって優れた抗体を効率的に選択できることが確認できた。
【0122】
6. ファージライブラリーからの特定の抗原に特異的に結合するファージの選択−2
抗原としてジフテリアトキソイドを用い、本発明による抗体ライブラリーから、抗体のスクリーニングを試みた。スクリーニングに用いたライブラリーは、4と同じAIMS4ファージライブラリーである。このライブラリーは、AIMS3(選択をかけない軽鎖遺伝子ライブラリーと重鎖遺伝子ライブラリーを組み合わせたライブラリー)と、AIMS1+AIMS2(選択をかけた軽鎖遺伝子ライブラリーと重鎖遺伝子ライブラリーを組み合わせたライブラリー)の混合液からなっている。
【0123】
6-1 スクリーニング用試験管の作製
抗原(ジフテリアトキソイド)をPBSで20μg/mLに調製し、試験管3本(Nunc社製Maxisorp)に3mLずつ添加して4℃で18時間感作した。抗原感作後、抗原溶液を捨て、2%スキムミルク含有PBS溶液3mLずつを加えて25℃で1時間反応させ、ブロッキングした。
【0124】
6-2 スクリーニング操作
作製した抗原結合試験管に、4と同じファージライブラリーを2%スキムミルク、0.1%Tween20含有PBSになるよう溶解して1×1014CFU/9mLに調製し、この液を試験管3本に3mLずつ添加して25℃で2時間反応させた後、0.1%Tween20を加えたPBSで4回、PBSで4回及び滅菌した超純水(MilliQにて作製)で1回洗浄した。
続いて抗原結合試験管に結合したファージを以下のように回収した。すなわち、0.1Mトリエチルアミン(pH12.3)を試験管1本当たり3mL添加し、ローテーターを用いて室温で20分間反応させ乖離させた後、1M Tris-HCl緩衝液(pH6.8)1.1mLを加えて中和し、この液を回収した。
【0125】
6-3 回収したファージの増幅
回収した液は実施例4-3および4-4と同じ(ファージの大腸菌への感染)(ヘルパーファージの感染)(ファージの回収)の操作を繰り返した。洗浄条件も実施例4-4の(スクリーニング)と同様に行った。
【0126】
6-4 ファージのスクリーニング手法の評価
ここで得られた発現の認められたクローンについて、軽鎖の塩基配列を決定してその由来を確認したところ、20クローンは(AIMS1+AIMS2)由来であったのに対し、(AIMS4)からは16クローンが選択された。
さらに、これらのクローンの中に実際に使用可能な活性の高いクローンが含まれているか否かについて検討した。ジフテリア毒素に対する抗体の中和活性は、培養細胞を用いたCell Culture Method(CCM)によって定量した。CCM法は以下のように行った。
標準抗毒素の溶液は、国内標準ジフテリア抗毒素Lot.9の1アンプルを0.2w/v%ゼラチン加PBSで溶解し、更に3%cs培地(精製水1,000mLにイーグルMEM9.4g、L-グルタミン0.3g、ペニシリン20万単位/2mL、ストレプトマイシン0.1g力価/0.5mL、グルコース3.0g、子牛血清30mL、1%フェノールレッド3mL、及び7w/v%重炭酸ソーダ20mLを含む)で10IU/mLに調製し、使用した。
【0127】
試験毒素の溶液はジフテリア試験毒素(Lot.M59)1バイアル(Lf/vial =2.5 、CD50/vial=1.6×105)を0.2w/v%ゼラチン加PBSで溶解し、3%cs培地で1.6×104CD50/mL)に調製した後、更に3%cs培地で希釈して用いた。細胞は7% cs培地(精製水1,000mLにイーグルMEM9.4g、L-グルタミン0.3g、ペニシリン20万単位/2mL、ストレプトマイシン0.1g力価/0.5mL、子牛血清70mL、1%フェノールレッド3mL、及び7w/v%重炭酸ソーダ20mLを含む)で継代培養したVERO細胞を用いた。
96穴マイクロプレート中に3%cs培地で標準抗毒素、被験抗体の希釈系列(各25μL)を作製し、これに調製した上記ジフテリア毒素(25μL)を加え37℃30分処理した後、3% cs培地で細胞数を3×105cells/mLに調製したVERO細胞50μLと、さらに3%cs培地100μLを加えてシールし、37℃のインキュベーターで4日間培養した。pH指示薬の色調の変化(オレンジを目安とする)から、本測定系における標準抗毒素のエンドポイントを求め、被験抗体のエンドポイントを示した希釈倍数をエンドポイントの抗毒素価に乗じて、被験抗体の抗毒素価とした。本測定系における標準抗毒素のエンドポイントは0.0046IU/mLであった。
その結果、CCM法では試験した43クローン中1クローンが0.0520IU/mLと、ジフテリアトキソイド(毒素)に対して中和活性を示した。しかし、AIMS3(選択をかけない軽鎖遺伝子用いたライブラリー)からは活性のあるクローンは得られなかった。これらの結果から、本発明に基づく抗体ライブラリーAIMS1+AIMS2は、生体内の抗体の多様性を高度に再現した優れたライブラリーを与えることが明らかである。
【0128】
本発明者らはジフテリア毒素に結合し、これを中和する能力をもつファージクローン(DTD10)も得ることができた。これらファージクローンが有する重鎖と軽鎖の塩基配列、およびこの塩基配列によってコードされるアミノ酸配列を以下の配列番号として記載する。
DTD10の重鎖 塩基配列/配列番号:61、アミノ酸配列/配列番号:79
DTD10の軽鎖塩基配列/配列番号:62、アミノ酸配列/配列番号:80
【0129】
7. ファージライブラリーからの特定の抗原に特異的に結合するファージの選択−3
抗原としてインフルエンザウイルス由来の抗原を用い、本発明による抗体ライブラリーから、抗体のスクリーニングを試みた。これ以降インフルエンザウイルス株名を以下のように省略して表記する。ただし、( )内はウイルスの亜型を示す。
ウイルス株名称 省略名称
A/New Caledonia/20/99株(H1N1) ニューカレドニア株(H1N1)
A/Sydney/5/97株(H3N2) シドニー株(H3N2)
A/Okuda/57株(H2N2) 奥田株(H2N2)
A/北京/262/95株(H1N1) 北京株(H1N1)
【0130】
7-1 インフルエンザウイルス由来抗原の精製
当業者には衆知の方法を用いて以下のようにインフルエンザウイルス抗原の精製を行なった。国立感染症研究所から購入したワクチン株を孵化鶏卵に接種し、33〜35℃で2日間培養した。培養後、4℃の冷蔵室で1晩放置し、常法に従い感染尿膜腔液を採取した。次いで、限外ろ過法・通常の生化学的方法などで濃縮し、ショ糖密度勾配遠心法で精製した。すなわち、0−60%のショ糖の密度勾配中で濃縮ウイルスを回転数35000rpmで超遠心し、ショ糖密度が40%前後の画分を採取した。このウイルス精製画分をエーテルで処理した(エーテル不活化ウイルス)。こののちにホルマリンを添加しショ糖密度勾配遠心法でさらに精製してインフルエンザ精製抗原を得た。
【0131】
7-2 北京株(H1N1)を用いたファージ抗体の単離
7-2-1 スクリーニング用試験管の作製
【0132】
北京株(H1N1)から7-1の手順で抽出したインフルエンザウイルス抗原をPBS中で12.5μg/mlの濃度で溶かし、イムノチューブ(Polysorp)に4.5ml入れて4℃18時間あるいは25℃2時間、穏やかに転倒混和してインフルエンザウイルス抗原をイムノチューブ内表面に結合させた。インフルエンザウイルス抗原溶液を除いたのち、2%の濃度でPBSに解かしたスキムミルクを4.5mlチューブに入れ、4℃で18時間あるいは25℃で1時間反応させ非特異的反応を防ぐブロッキング処理を行なった。
【0133】
7-2-2 スクリーニング操作
上記のようにして作製した抗原結合済みのイムノチューブに2%スキムミルクin PBSに懸濁した実施例4と同じAIMS4ファージライブラリー4.5mLを入れ、25℃で2時間転倒混和して反応させ、チューブ上に結合したインフルエンザウイルス抗原とAIMS4ファージライブラリーを接触させた。
上記の接触が終わったのち、ファージ懸濁液を除き、以下の表8に示す緩衝液で表に示す回数洗浄し、チューブに結合しているファージ以外のファージを取り除いた。
【0134】
【表8】
次に0.1MトリエチルアミンpH12.3を4.5mlチューブに添加し、20分室温で転倒混和して、ファージをチューブから乖離させて別の新たなチューブに移し、直ちに1M Tris−HCl pH6.8を1.1ml加えて中和した。
【0135】
7-2-3 回収したファージの増幅
大腸菌DH12S(2xYT培地)50ml(2ndスクリーニング以降は30mL)をOD600nm= 0.5になるように予め増やしておき、上記の過程で得たファージ液を加え、抗原結合ファージを37℃1時間振とう培養し感染させた。
前述の大腸菌液を全体で500mlになるよう以下の如く希釈した。(3Lフラスコ使用)
大腸菌液 61.25ml
2xYT 425ml
40%グルコース 12.5ml(終濃度1%)
100mg/ml Ampicillin 0.5ml(終濃度100μg/ml)
上記の希釈済み大腸菌液をさらに37℃1〜2時間培養したのち、4℃に移し終夜保存した。
終夜保存したフラスコを再び、37℃に戻し振盪培養した。OD600nm=0.5になったら、5000 rpm 10分、4℃にて滅菌遠心管を用いて遠心し、菌体を回収した。
回収した菌体に2xYT培地1〜2mlを加え溶かしたのち、一部をとって、グリセロール・ストックを作製した。(最終グリセロール濃度20%)
残りの菌体には2xYT培地300mL(ただし、3rd, 4thスクリーニングの場合はそれぞれ150mL, 50mL)に終濃度100μg/mlのアンピシリンを加え培養した。OD600nm=0.5に到達したとき、さらにヘルパーファージM13K07を培養液の1/100量加え、37℃で1時間振盪培養し、ヘルパーファージを感染させた。
上記の培養液の一部を取り、2×TY寒天培地上に播き、ヘルパーファージの感染した大腸菌数の計測に供した。残りの培養液には新たな培養液(100μg/mlのAmpicillinを含有し、70μg/mlのKanamycineを含有した900ml 2×YT培地、37℃にあらかじめ保温しておく)を加え、37℃で終夜振盪培養することで、ヘルパーファージが感染した大腸菌を選択した。
終夜培養後、培養液を7000r.p.m 10分4℃(滅菌ずみ遠心管1000ml用を使う)で遠心分離して菌体を沈殿させ、上清を回収した。上清に1/5体積の2.5M NaCl含有20%ポリエチレングリコール溶液を加え、20分室温で静置したのち、8000 r.p.m 15分4℃で遠心分離して沈殿させる事によってファージを回収した。
沈殿を崩さないように上清を除き、培養液の1/10体積の滅菌PBSで沈殿を溶かした。さらに、2.5M NaCl含有20%ポリエチレングリコールをPBSの1/5体積加え、10000r.p.m 20分4℃(滅菌ずみ遠心管200ml用を使う)で遠心してファージを沈殿させることにより不純物を取り除いた。沈殿したファージに0.05% NaN3 PBSを培養液の1/100加え、シェイカーを使用して、約2時間振盪することにより懸濁した。
以上の工程によって得られたファージは、さらに同様の2回のスクリーニング工程に供した。しかし、この方法ではインフルエンザウイルスのNPというタンパク質に反応する抗体しか得られず、インフルエンザウイルスを中和する能力は無かった。
【0136】
7-3 ニューカレドニア株(H1N1)を用いた亜型特異的に反応するファージ抗体の単離
インフルエンザを中和する抗体のエピトープはウイルス表面のHAタンパク質にあると言われており、また、HAタンパク質は各ウイルス株間で変異の大きい箇所でもある。そこで、ウイルス株に特異的に反応する抗体を単離することで中和抗体を得ることを試みた。
【0137】
7-3-1 抗NP抗体吸収済ライブラリーの準備
インフルエンザウイルス奥田株(H2N2)を用いて7-1の手順に従いエーテル処理不活化ウイルスを作製した。不活化ウイルス 20μg/mlをPBSに溶解し、この溶液を反応用チューブ3ml/本、3本に添加し、4℃18時間反応させた。反応後液を捨て、2%スキムミルクin PBSで1時間反応させた。AIMS4ライブラリー LOT000614/2%スキムミルクin PBSを1x1014CFU/9mlを上記3本のtubeに加えた。室温2時間25℃攪拌しながら反応させた。この反応終了後のライブラリーを抗NP抗体吸収済AIMS4ライブラリーとした。
【0138】
7-3-2 目的抗原ニューカレドニア株(H1N1)由来抗原を用いたスクリーニング操作
上記と同様に作製したニューカレドニア株(H1N1)ホルマリン処理不活化ウイルスを100μg/mlになるようにPBSに溶解した。この溶液を反応用チューブ3ml/本、3本に添加し、4℃18時間反応させた。反応後、液をすて、非特異的反応を防止するため2%スキムミルクin PBSを加え1時間インキュベートした。保温終了後、7-3-2で作製した抗NP抗体・吸収済AIMS4ライブラリーを上記3本のチューブに加えた。2時間25℃にて攪拌しながら反応させた。反応後PBSで洗浄した。洗浄回数は以下の表9を参照。
洗浄後、0.1MトリエチルアミンpH12.3を3ml/tube加え、室温20分、ローテーターで攪拌しながら反応させファージを抗原と乖離し、1M Tris−HCl pH6.8を1ml加えて中和した(抗原結合ファージの回収)。大腸菌DH12S(2xYT培地)50mlをOD600nm 0.5〜1.0になるように予め増やしておき、抗原結合ファージを37℃1時間振とう培養し、感染させた。
7-2-3と同様に回収したファージの増幅工程を行ない、増幅後抗NP抗体を7-3-1と同様に吸収し、ファージ液の力価を以下のように測定した。力価を算出した後、同様のスクリーニング過程に増幅ファージ液を用いた。スクリーニング操作は3回繰返した。
【0139】
7-3-3 ファージの力価測定
次回のスクリーニングに用いるファージ液の力価をあらかじめ以下の要領で調べた。
希釈系列を下記のように作製した。
1. 1x10−2希釈 :ファージ液10μl+ PBS 990μl
2. 1x10−4希釈 :1の希釈液10μl+ PBS 990μl
3. 1x10−6希釈 :2の希釈液10μl+ PBS 990μl
4. 1x10−8希釈 :3の希釈液10μl+ PBS 990μl
5. 1x10−10希釈 :4の希釈液10μl+ PBS 990μl
6. 1x10−12希釈 :5の希釈液10μl+ PBS 990μl
4,5,6の希釈系列10μlにDH12S990μlを加えたものをつくり、37℃1時間感染させ、これをLBGAプレートに100μlまき30℃18〜24時間培養後コロニーをカウントした。
4,5,6のうち、50個/プレート以上コロニーをつくったプレートをデータとして採用した。
次回のスクリーニングに用いるファージ力価の計算式(4.の場合)
=(コロニー数/プレート)x(1x108)x103 ファージ/ml
【0140】
【表9】
【0141】
7-4 H3N2亜型特異的中和抗体の単離方法
7-4-1 目的抗原シドニー株(H3N2)由来抗原を用いたスクリーニング
インフルエンザウイルスシドニー株(H3N2)・ホルマリン処理不活化ウイルス100μg/mlをPBSに溶解し、この溶液をNunc Polysorp 4.5ml/本、3本に添加し、4℃ 18時間反応させた。反応後溶液を捨て、2%スキムミルクin PBSで1時間ブロッキングした。その後、7-3-1で作製した抗NP抗体・吸収済AIMS4ライブラリーを上記3本のtubeに加え、室温2時間25℃ローテーターを用いて振盪反応させた。
反応後PBSで以下の表10に示す回数洗浄した。
洗浄後、0.1MトリエチルアミンpH12.3を3ml/tube加え、室温20分、ローテーターで攪拌しながら反応させファージを抗原と乖離した。1M Tris−HCl pH6.8を1ml加えて中和した(抗原結合ファージの回収)。
大腸菌DH12S(2xYT培地)50mlをOD600nm 0.5〜1.0になるように予め増やしておき、抗原結合ファージを37℃1時間振とう培養し、感染させた。
7-2-3と同様に回収したファージの増幅工程を行ない、増幅後抗NP抗体を7-3-1と同様に吸収し、ファージ液の力価を7-3-3と同様に測定した(表11)。力価を算出した後、同様のスクリーニング過程に増幅ファージ液を用いた。スクリーニング操作は3回繰返した。
【0142】
【表10】
【0143】
7-5 インフルエンザウイルスを中和するファージ抗体の評価
7-5-1 Fab型抗体の誘導
ファージの感染した大腸菌を発現誘導した後長期培養することにより、ファージではなく、Fab型の抗体が大腸菌から培養液中に分泌されることを本発明者らは見出した。
上記の工程7-2-3、7-3-3、および7-4-1から得られたファージを含む大腸菌のコロニーから大腸菌を2xYT+0.1%グルコース+100μg/ml Ampicillinに植菌して30℃で培養し、OD600nmが0.5付近になったところでIPTGを最終濃度1mMとなるよう添加し、30℃ 18時間培養することによりファージ抗体を誘導した。
また、別にIPTGを入れないで18時間培養したものを準備し、終濃度30%のグリセロールを加えて保存用大腸菌を作製した。
【0144】
7-5-2 ELISA法によるFab型抗体の特性の確認
Fab型抗体を発現させた7-5-1の培養液を1.5mlチューブにいれ、10000rpm. 4℃5分遠心した。培養上清をとり、終濃度0.1%のNaN3を加えELISAサンプルとした。
次に、スクリーニングに用いた際に用いた濃度と同じ濃度の抗原を96well ELISAプレートに100μl/well加え4℃18時間保温することにより抗原をプレートに結合した。その後、2.5%BSA in PBSを200μl/well加え、4℃18時間保温することによりブロッキングした。
ELISA試験実施前に、ブロッキング液をすて、PBSで一回洗った。7-5-1で誘導した培養上清を100μl/well加え、25℃1時間反応させた。終了後、PBSで4回ウェルを洗浄し、POD結合抗ヒトIgG(MBL社Cat#206 LOT150)を5000倍希釈し100μl/well加え、25℃1時間(または、37℃1時間、室温250rpm. 振盪しながら30分)反応させた。終了後、PBSで4回洗浄し、基質溶液を100μl/well加えた。基質溶液は以下のように作製した:0.1Mクエン酸−リン酸水素2ナトリウムpH5.1に終濃度0.01% H2O212mlを加え、さらにOPD錠(和光純薬社、生化学用Code# 158-01671)を1錠加えた。15〜30分後OD492nmをプレートリーダーによって吸光度を測定した。
その結果、ELISAにおいてニューカレドニア株(H1N1)から1クローン、シドニー株(H3N2)から3クローンの株特異的な反応性を示す中和抗体が得られたことが分かった。これらの4クローンの反応性を以下の表12に示した。
【0145】
【表11】
【0146】
【表12】
【0147】
7-5-3 プラスミドの精製とシークエンシング
2xYT培地(0.1%グルコース+200μg/ml Ampicillin含有)で一晩培養し、DNA分離装置(倉敷紡績社 PI-50)にて操作法に従って、プラスミドを精製した。精製したプラスミドには最終濃度20μg/mlのRNAseを添加してフェノール・クロロホルム処理し、-20℃以下にて保存した。
得られたファージ抗体が有している抗体遺伝子はシークエンシングを行なう事によって解読した。シークエンシングはDNAシークエンサ(アロカ社 L1−COR4200L(S)−2)を用い、試薬はサーモシークエンスキット(アマシャム・ファルマシアUS78500)を使用し、ダイデオキシ法にておこなった。
重鎖、軽鎖のシークエンスプライマーは以下のものを使用した。
重鎖:T7Promoter IDR700ダイラベルプライマー 50μl
5'TAATACgACTCACTATAggg3'(20mer)(配列番号:97)
軽鎖:カスタム・プライマー 3’huCH1J
IDR800ダイラベルプライマー 50μl
5'ATTAATAAgAgCTATCCCgg3'(20mer)(配列番号:98)
遺伝子の塩基配列解読の結果、配列の欠失が生じていないものの数は表11に示した。また、株特異的なクローンの遺伝子およびアミノ酸配列については、配列表に示した。
【0148】
7-5-4 インフルエンザウイルス中和試験
MDCK細胞を96ウェル平底プレート(コーニング社cat#3596)の各ウェルに(約104 cells/well)分注し、CO2インキュベータにてウェルの底にモノレイヤーシートを形成するように37℃で培養した。次の日0.2% BSA(fraction V, Sigma Chemical Co.)を含むMEM培地により96well丸底プレートの各ウェル内に試験する各抗体を4倍希釈して入れた。0.2% BSAを含むMEM培地により希釈したインフルエンザウイルス液(4×104 FFU/mL)25μlと、希釈した抗体あるいはコントロールの溶液25μlを混合し、37℃60min反応させた。
それらの混合液を先に述べた96穴プレート中のMDCK細胞に25μl添加し、37℃60min.保温することにより、ウイルスを細胞に吸着させた。
吸着後PBSで洗浄し100μL/wellの0.5% tragacanth gumと5μg/mlのトリプシンを含むMEM培地を添加し、さらに37℃24時間培養した。
培養後、PBSでウェルを洗浄した後、100%エタノールをウェルに添加し、室温10分処理し、ウェルをヘアドライヤーで乾燥させた。PAP染色後Focusを計数した。
抗体の中和能の強さは、何も入れない場合(陽性コントロール)からのフォーカスの減少率で表わされる。
以下の表13に示すようにFab型抗体クローン番号NC1、SY39およびSY47は有為なインフルエンザウイルスに対する中和活性を示した。
【0149】
【表13】
【0150】
7-5-5 評価の結論
予測した通りにウイルス株に特異的なファージクローンはインフルエンザウイルスを中和する高い活性を有していた。すなわちニューカレドニア株(H1N1)に対するクローンNC1またはシドニー株(H3N2)に対するクローンSY39、およびクローンSY47である。これらファージクローンが有する重鎖と軽鎖の塩基配列、およびこの塩基配列によってコードされるアミノ酸配列を以下の配列番号として記載する。
クローン番号 重鎖 軽鎖
NC1(塩基配列) 67 68
NC1(アミノ酸配列) 85 86
SY39(塩基配列) 69 70
SY39(アミノ酸配列) 87 88
SY47(塩基配列) 71 72
SY47(アミノ酸配列) 89 90
これらの可変領域を実際に医療用に使用するにあたっては、抗体遺伝子が得られている特性を生かし、完全イムノグロブリン型への変換や酵素の結合なども比較的簡単に実施することができると考えられる。また、突然変異を人工的に起こすことによってより活性を上昇させることができると期待される。
【0151】
8. ファージライブラリーからの特定の抗原に特異的に結合するファージの選択−4
抗原として水痘帯状疱疹ウイルス(VZV)由来の抗原を用い、本発明による抗体ライブラリーから、抗体のスクリーニングを試みた。
8-1 VZVに反応性を持つファージ抗体の単離
8-1-1 スクリーニング用試験管の作製
公的機関で入手可能であるVZVの感染したヒト胎児肺細胞(HEL)をPBS中で超音波処理によって破砕し、Shiraki et al (1982) (Shiraki K, Takahashi M J Gen Virol 1982, 61 : 271-5) に従いgEとgHタンパク質を調製し、そのうちgHタンパク質を25μg/mlの濃度に調整しVZV抗原溶液とした。VZV抗原溶液をイムノチューブ(Maxisorp)に4.5ml入れて4℃18時間あるいは25℃2時間、穏やかに転倒混和してVZV抗原をイムノチューブ内表面に結合させた。VZV抗原溶液を除いたのち、2%の濃度でPBSに解かしたスキムミルクを4.5mlチューブに入れ、4℃で18時間あるいは25℃で1時間反応させ非特異的反応を防ぐブロッキング処理を行なった。
【0152】
8-1-2 スクリーニング操作
上記のようにして作製した抗原結合済みのイムノチューブに2%スキムミルクin PBSに懸濁したAIMS4ファージライブラリー4.5mLを入れ、25℃で2時間転倒混和して反応させ、チューブ上に結合したVZV抗原とAIMS4ファージライブラリーを接触させた。
上記の接触が終わったのち、ファージ懸濁液を除き、以下の表に示す緩衝液で表14に示す回数洗浄し(洗浄とは緩衝液をチューブに入れ転倒混和した後、排出することを指す)、チューブに結合しているファージ以外のファージを取り除いた。
【0153】
【表14】
次に0.1MトリエチルアミンpH12.3を4.5mlチューブに添加し、20分室温で転倒混和して、ファージをチューブから乖離させて別の新たなチューブに移し、直ちに1M Tris−HCl pH6.8を1.1ml加えて中和した。
7-2-3と同様に回収したファージの増幅工程を行ない、増幅後回収したファージ液の力価を7-3-3と同様に測定した。
以上のスクリーニングを3回繰返した。
【0154】
8-2 結合活性を大腸菌の培養上清を用いてELISA法により確認する工程
7-5-2と同様にELISA法を用いて結合活性の確認を行なった。VZV抗原をPBS中で12.5μg/mlの濃度で溶かし100μl/well 96wellプレートの各ウェルに添加し、4℃18時間保温し、96ウェルプレートに抗原を結合させた。次に、2.5%BSAを200μl/well加え、4℃18時間保温することによりブロッキングした。
ブロッキングの終了した96wellプレートのブロッキング液をすて、PBSで一回洗ったのち、8-5-2と同様の手順で調製したファージが感染した大腸菌の培養上清を100μl/well加え、25℃1時間で反応させた。これ以降の過程は8-5-2と同様に実施した。
上記の表に示すように、3回目のスクリーニング操作にて回収率(output)の上昇が見られたため、12クローンについてELISAを行ったところ活性の強い1クローンを得た。そのまま、4回目のスクリーニングを行って48クローンにつきELISAを行ったところ、活性は弱いが陽性であった。
【0155】
8-3 gEとgHの反応性の比較とシークエンシング
糖鎖に対する抗体はウイルスの他の部分にも反応し、特異性が低く、中和活性も低いことが予測された。そこで、豊富な糖蛋白を持つgEに対するELISAも実施し、gEに高い反応性を持つ抗体は検討から除くこととした。
この基準により、gHの活性が高い29クローン(表15)を選択し、7-5-3と同様に塩基配列を解析(シークエンシング)した。シークエンシングによって16種類の塩基配列が確認された。それぞれ代表的なクローンにつき培養上清をとり、飽和硫安による濃縮、透析の後、中和活性の試験を実施した。
【0156】
【表15】
【0157】
8-4 得られた抗体のVZVウイルス中和試験
8-4-1 試験に用いるcell-free水痘帯状疱疹ウイルス(VZV岡株)の作製
ヒト胎児肺細胞(HEL)の培養細胞の約50%に感染細胞特有の変化(CPE:cytopathic effect)を認めたときに,EDTA(0.04−0.1%)を含むPBS(トリプシンは使用しない)によって細胞を回収し、低速遠心により細胞を集め,SPGC保存液(5% sucrose,0.1%,Na−グルタミン酸含有滅菌PBSに牛胎児血清を10%添加したもの)に懸濁した。
超音波または,凍結融解+超音波にて,細胞を破砕して細胞内のウイルスを遊離させ、3000 rpm,10分の遠心上清をウイルス液とし、-85℃で保存した。一部をHEL細胞にて感染力価を測定した。すなわち、ウイルス液をSPGC保存液で適度に希釈し、HEL細胞に0.2mlをあらかじめ用意された6cmシャーレのHEL細胞に振り掛けることにより接種した。HEL表面が乾かないように,ウイルス液が全体に均一になるように何度も細胞表面をウイルス液で覆うように液を往復させながら1時間感染させた。感染後培養液を入れて4−6日間培養し、その後培溶液を除き、定法に従って5%ホルマリンで細胞を固定し,メチレンブルー染色し,実体顕微鏡下でプラック数を測定した。単位溶液あたりのプラック数を(PFU/ml)として感染力のあるウイルスの指標(力価)とした。
【0158】
8-4-2 中和能試験
上記で作製したウイルス液をSPGC保存液で希釈して,100PFU/0.1mlとした。希釈したウイルス液とSPGCで10倍希釈した抗体溶液を0.3mlずつ混合し、37度で1時間反応させた。その中から,0.2mlをあらかじめ用意された6cmシャーレのHEL細胞に振り掛けることにより接種した。HEL表面が乾かないように,ウイルス液が全体に均一になるように何度も細胞表面をウイルス液で覆うように液を往復させながら1時間感染させた。
感染後培養液を5ml入れて4−6日間培養した。培養後、培養液を除き、定法に従って5%ホルマリンで細胞を固定し,メチレンブルー染色し,実体顕微鏡下でプラック数を測定した。
抗体なし(PBSのみ)で形成されるプラック数を対照として、抗体によってどれだけプラック数が減少できるかによって、抗体のウイルス中和活性を測定した。以下の表16のように、No.10, 24, 94に強い中和活性が見られた。
【0159】
【表16】
【0160】
8-4-3 中和試験の結論
本発明により、プラック形成を非常に高い効率で抑制したファージクローンが3つ得られた。すなわち、VZ10とVZ24とVZ94である。これらファージクローンが有する重鎖と軽鎖の塩基配列、およびこの塩基配列によってコードされるアミノ酸配列を以下の配列番号として記載する。
クローン番号 重鎖 軽鎖
VZ10(塩基配列) 73 74
VZ10(アミノ酸配列) 91 92
VZ24(塩基配列) 75 76
VZ24(アミノ酸配列) 93 94
VZ94(塩基配列) 77 78
VZ94(アミノ酸配列) 95 96
これらは、異なる塩基配列をもち、実際に医療用に使用する場合には、その中和活性が相乗的に作用する可能性がある。実際の使用にあたっては、抗体遺伝子が得られている特性を生かし、定常領域を備えた完全なイムノグロブリン分子への変換や酵素の結合なども比較的簡単に実施することができる。
【産業上の利用可能性】
【0161】
本発明によって、機能的なコンフォーメーションを維持した抗体分子を高い割合で含む抗体ライブラリー、あるいはそれをコードする遺伝子のライブラリーの提供が可能となった。本発明よる遺伝子ライブラリーは、まず重鎖可変領域と機能的なコンフォーメーションを再構成することができる軽鎖可変領域をコードする遺伝子の選択に始まる。こうして選択された軽鎖可変領域遺伝子に、重鎖可変領域遺伝子を組み合わせることによって構成された本発明よる遺伝子ライブラリーからは、機能的な抗体からなる抗体レパートリーの再現が可能である。機能的な抗体分子の選択工程を経て生成される本発明の抗体ライブラリーは、生体内における抗体レパートリーをin vitroで再現したものと言うこともできる。
【0162】
本発明による遺伝子ライブラリーは、機能的な抗体を高い割合で含む抗体ライブラリーを生成することができる。このような抗体ライブラリーを用いれば、機能的な抗体を効率的にスクリーニングすることができる。公知の抗体ライブラリーが、単に多様性の追求にとどまっていたのに対して、本発明はライブラリーを構成する抗体の質に着目し、その質を高める手段を確立した。この点において、本発明はin vitroにおける抗体ライブラリーの構築と、抗体のスクリーニング方法に、まったく新たな視点を与える。
【0163】
本発明によって得られる抗体ライブラリーにおいては、重鎖の多様性を十分に生かすことができる。この利点は、単に生体内の抗体の多様性をin vitroで維持できるということにとどまらない。本発明に基づけば、たとえばerror-prone PCRのような人為的な変異の導入方法のメリットを最大限に生かすことができる。すなわち本発明においては、変異を与えた重鎖の多様性は、理論的には完全に抗体の多様性として再構成される。なぜならば、重鎖との機能的な再構成が可能な軽鎖が選択されているからである。公知のライブラリーにおいては、軽鎖が機能的な抗体分子を再構成できないものである場合には、有意義な重鎖の変異が抗体活性の発現に至らず、スクリーニングすることができないのと対称的である。
【技術分野】
【0001】
本発明は、抗体可変領域をコードするDNAを含む抗体ライブラリーに関する。
【背景技術】
【0002】
動物個体は体液中に侵入する様々な異物に対して、その物質の表面に位置する種々の構造(エピトープ)を認識して特異的に結合する抗体を産生する能力がある。その抗体レパートリーの大きさ(異なる抗原に結合する違ったアミノ酸配列を持つ抗体の種類の総数)は、動物個体あたり100万種〜1億種と見積もられている。このような巨大な抗体レパートリーは、骨髄幹細胞が抗体産生細胞であるBリンパ細胞へ分化する途上に、抗体遺伝子座で重鎖ではVH-D-JH、軽鎖ではVL-JLというDNA再編成を起こすことによって生じる。このDNA再編成はB細胞毎に独立に起こるプロセスである。したがって一組のVH-D-JH、VL-JL遺伝子を持つ1個のB細胞は1種類の抗体しか産生しない。しかし個体を構成するB細胞全体では、多様な抗体の産生が可能となる。
【0003】
従来行われてきた抗血清調製技術、および細胞融合によるモノクローン抗体作製技術は、この動物の持つ抗体産生機構を利用している。すなわち、抗原物質をアジュバンドと共に動物(ウサギ、ヤギ、マウス等)に一定間隔をおいて数回注入する。動物の免疫系がその物質を異物と認識すると、その抗原物質に結合する抗体を発現したB細胞が増殖分化刺激を受け、大量の抗体の体液中への分泌を開始する。抗原物質の表面には様々な構造が存在し、精製された抗原といえどもそれに結合する抗体は、通常は多種類の抗体の混合物として分泌される。このような抗体を含む血清(抗血清)はポリクローン抗体と呼ばれる。現在もポリクローン抗体は、研究試薬として有効に利用されている。しかしポリクローン抗体は、目的とする抗原物質以外にも、部分的に(抗体にとって)類似した構造を持つ分子とも交叉反応性(cross-reactivity)を示すことが多い。交叉反応は、ポリクローン抗体を抗原検出試薬として用いるときの問題点となっていた。
【0004】
細胞融合技術の確立がこの状況を一変させた。抗原物質で免疫した動物の脾臓には抗原と結合する抗体を産生するBリンパ球は多数存在する。しかしその細胞を試験管内で長期間維持培養し続けることは困難である。そこで永代培養が可能なように株化した腫瘍細胞と抗体産生細胞を融合することにより、抗体を産生し永代培養可能な細胞を作製するというアイデアが生まれ、方法として確立した。このようにして確立した融合株(ハイブリドーマ)は1個の抗体産生細胞と1個の腫瘍細胞に由来するので産生される抗体は1種類であり、モノクローン抗体と呼ばれる。これはケラーとミルスタインにより1975年に開発された技術である。モノクローン抗体は、均一な抗体分子の集合体であることから、交叉反応を生じにくい特異性に優れた抗体として利用されている。しかしこの方法にも次のような問題点が指摘されている。
(1)抗原物質として必要充分量の精製標品を有していること
(2)その物質が免疫される動物に対して免疫原性を示す必要があること
(3)モノクローン抗体を得るまでに多大な労力と日数を要すること
【0005】
ポリクローン抗体やモノクローン抗体の作製技術の有効性は、多くの有用な抗体を提供してきたことによって証明されている。しかし、これらの方法によっては解決が困難な多くの課題が今なお存在することも事実である。たとえば、多種類の抗原に対する抗体を短期間に得ること、あるいは特殊な構造をしたエピトープに特異的に結合する抗体を選択的に得ること、といった要求には、これらの方法では応えることはできない。希望する抗体を短期間で得ることができるように、多様な抗体分子で構成された抗体ライブラリーの作製が望まれていた。このような抗体ライブラリーに含まれる抗体の種類は、理論的には動物個体の有する抗体レパートリーの大きさに匹敵する必要がある。しかし現実には、動物細胞を用いてそのような巨大なライブラリーを作製することは不可能である。モノクローン抗体の作製は、動物の持つ抗体産生細胞のライブラリーから、期待する反応性を持つ抗体をスクリーニングする作業に他ならない。しかし、そのライブラリーを構成するレパートリーは、細胞融合などの操作を通じて大幅に失われてしまう。
【0006】
そこで、大腸菌による抗体遺伝子の発現系が提案された。大腸菌中で抗原結合力のある抗体を発現させるのに初めて成功したのはBetter et al(1988)(Better M, Chang CP, Robinson RR, Horwitz AH Science 1988, 240:4855 1041-3:非特許文献1)とSkerraとPlukthun(1988) (Skerra A, Plukthun A Science 1988, 240:4855 1038-41:非特許文献2)である。彼らは大腸菌で分泌シグナルとして機能する配列を抗体のN末端に付加することによりFab型とFv型抗体を大腸菌中での産生、それに続く分泌に成功した。
更に、1988年のPCRの開発は、直ちに抗体可変領域をコードする遺伝子の増幅に応用される。動物(とりわけヒト)中で発現されている全てのVHDJH、VLJL遺伝子を増幅するためのプライマー配列が提案された(Orlandi R et al. Proc Natl Acad Sci USA 1989 86:10 3833-7, Sastry L et al. Proc Natl Acad Sci USA 1989 86:15 5728-32:非特許文献3)。そしてこれらのプライマーによって増幅した抗体遺伝子を利用して、大腸菌中で抗体を産生させるためのベクターが構築された(Huse WD et al.Science 1989 246:4935 1275-81, Ward GE et al.J Clin Microbiol 1989 27:12 2717-23:非特許文献4)。この段階で、抗体ライブラリーのレパートリーサイズは飛躍的に向上した。しかし、大腸菌中で産生される微量の抗体について抗原との結合活性を指標とするスクリーニングを行うことは、容易なことではなかった。スクリーニングを能率的に行うには、ファージディスプレー法が抗体ライブラリー作製に応用されるのを待つ必要があった。
【0007】
ファージディスプレー法はSmithにより1985年(Smith GP Science 1985 228:4075 1315-7:非特許文献5)に考案されたもので、M13ファージのような一本鎖環状DNAを持つ線状のバクテリオファージが用いられる。ファージ粒子はDNAの周囲を取り囲んでファージ粒子の大部分を構成するcp8というタンパクと、ファージが大腸菌に感染する時に機能する5個のcp3と呼ばれるタンパクからなっている。このcp3もしくはcp8と融合した形でポリペプチドをコードするように遺伝子を構築し、ファージ粒子表面にそのタンパクを発現させるシステムがファージディスプレーシステムである。結合性のタンパク質を表面に保持したファージ粒子は、そのリガンドとの結合活性を利用して濃縮することができる。こうして目的とするDNAを濃縮する方法は、パニング法と呼ばれている。濃縮されたファージ粒子には、必要な結合活性を持つタンパク質をコードするDNAがパッケージングされている。このように繊維状ファージの利用によって、結合活性に基づくスクリーニングと、DNAのクローニングとをきわめて効率的に行うことができるシステムが実現した(特表平5-508076:特許文献1)。繊維状ファージを使ったライブラリーには、Fab分子として発現が可能な方法も報告された(特表平6-506836:特許文献2)。この報告において、cp3等のN末端を欠損させて可変領域を融合させる方法が試みられた。
【0008】
ファージディスプレー系が抗体に応用され、VHドメインのみ、scFv、Fv、Fab型抗体がcp3又はcp8と融合された形で発現された。抗原と結合するファージ抗体は同時に抗体をコードする遺伝子を含む。しかしファージディスプレー系を用いて作製された初期に抗体ライブラリーから単離された抗体は、抗原結合力の低いものが多かった。結合力を高める試みの一つとして、人為的に遺伝子に変異を与える方法が提案された。Winterらは(1994)、単離した全てのVH、VL遺伝子と、JH、JL遺伝子の間にランダムな配列を挿入する半人工的配列を持つ抗体ライブラリーを作製することにより、親和性に優れた抗体の取得を可能とする抗体ライブラリーを得た(Nissim A, Winter G et al. EMBO J 1994 13:3 692-8:非特許文献6)。De Kruifら(1995)も基本的に同じ原理に基づいて抗体ライブラリーを作製している(de Kruif J, Boel E, Logtenberg T J Mol Biol 1995 248:1 97-105:非特許文献7)。Vaughanら(1996)は、ライブラリーの大きさを拡大することで充分な大きさの抗体レパートリーを確保しようとしている(Vaughan TJ et al. Nat Biotechnol 1996 14:3 309-14:非特許文献8)。このような試みは、確かに限られた抗原に対しては成功を収めた。しかし必要とする抗体を得ることができる可能性は未だに不充分である。たとえば、マウスにおいてモノクローン抗体を得ることができるのと同じような確率でヒト抗体を得ることができるライブラリーを現在の技術で構築することは不可能である。したがって、より多様な抗体から構成されるライブラリーの提供が望まれている。
【0009】
ヒトが産生することが可能な抗体を完全にin vitroでライブラリー化し、様々な抗原に特異的に結合する抗体が得られるようにするには、ヒトが抗体を産生する過程を試験管内で忠実に反映できれば理想的である。抗体が抗原と結合する部位はHL両鎖のN末端に位置する可変(V)ドメインの中の各々抗原相補性決定領域(complementarity determining region;以下CDRと省略する)I、II、IIIの計6ヶ所である。CDRを構成するアミノ酸配列の多様性(長さに関する多様性も含めて)の総数が、そのまま抗体レパートリーの大きさを反映していると考えてよい。
抗体レパートリーを考える時に、体内への抗原侵入前の「ナイーブレパートリー」と抗原侵入後に起こる「抗体の成熟」を考慮する必要がある。抗体をコードする活性型抗体遺伝子はDNA再編成によって作り出される。軽鎖にはλ鎖とκ鎖があるが、そのV領域をコードする遺伝子はVL遺伝子とJL遺伝子に分かれている。λ鎖においてVλ遺伝子は36個存在し、Jλ遺伝子は7個存在する。B細胞への分化途上、軽鎖に関してはκ鎖かλ鎖でVL遺伝子とJL遺伝子の近傍でDNAの切断と再結合が起こり、VL-JL遺伝子ができる。多くの例(2/3)で1〜96番目のアミノ酸がVL遺伝子由来、97〜110番目のアミノ酸がJL遺伝子由来となる。しかしV(D)J DNA組換え酵素(群)は、DNAを切断後、つなぐべき末端をエキソヌクレアーゼで少し除いたのち再結合させる。その結果、VL-JL遺伝子でコードされたVLドメインの大きさに±3アミノ酸程度の差異が生じる。軽鎖のCDR1は24-34番目のアミノ酸に、CDR2は50-56に、そしてCDR3は89-97番目に相当するので、その作り出す多様性の総数はλ鎖の場合(Vλ遺伝子の数)x(Jλ遺伝子の数)x(ズレの総数)となる。ただしJλ遺伝子の場合その配列は相互に似ており、結合点のズレに関しても67%が一定で27%以上が±1であるので、実際のVλ-Jλ遺伝子レパートリーは大きく見積もっても200を越えない。κ遺伝子もλ遺伝子に状況が似ている。Vκ遺伝子総数は37個、Jκ遺伝子数は4個である。そこでVκ-Jκ遺伝子レパートリーも200種を越えない。
【0010】
軽鎖可変領域の多様性の度合いが比較的小さいのに比べると、重鎖可変領域では大きく状況が異なる。CDR1(31-35b番目のアミノ酸)とCDR2(50-65番目のアミノ酸)については36個のVH遺伝子にコードされる部分なので多様性は限られている。問題はCDR3である。抗体の抗原結合面全体を眺めた時も、HL両鎖のCDR1、2が両側に位置し、CDR3が真ん中に位置する。重鎖のCDR1,2,3が全体の60%ぐらいを占め、軽鎖が40%ぐらいの面積を占める。重鎖CDR3を除く残りの部分は軽鎖の多様性(多くて数百種)x重鎖CDR1,2の多様性(36)=計約1万種程度のレパートリーである。重鎖CDR3は独立したD遺伝子にコードされる領域だが、D遺伝子は26個存在する。CDR3をコードする領域はD-JH、VH-Dという2回のDNA再編成後VH-D-JHとなって完成する。
【0011】
問題はこのDNA再編成過程で、次に示すような過程が存在することである。
(1)VH、D、JHの近傍に存在するシグナル配列真横のDNAの切断、
(2)エキソヌクレアーゼによるDNA末端部分切除、
(3)末端添加酵素によるランダム配列(Nと呼ばれる)の挿入、
(4)DNAの修復と連結
これらの過程において、重鎖では(2)の過程のバリエーションが軽鎖に比べて大きい。更により決定的なことは、軽鎖ではみられなかった(3)の過程が存在することである。重鎖CDR3(アミノ酸95-102番目に相当)は、92番目のシステインと103番目のトリプトファンに挟まれた領域として存在する。その長さは5アミノ酸から20アミノ酸を越えるものまで、まちまちの長さを持つことに加えて、その配列も多種多様である。このような特徴のために、CDR3は事実上独立して分化したB細胞毎に異なるほど多様である。
【0012】
B細胞分化途上に抗体遺伝子座で起こる重鎖のVH-D-JH、軽鎖のVL-JL DNA再編成は、抗原の有無に関係なく起こる。1個のB細胞が1組のVH-D-JH、VL-JL遺伝子を発現し、B細胞全体で形造る抗体集団のことを抗体のナイーブレパートリーと呼ぶ。抗原侵入後、抗原と結合できる性質を持つ抗体を発現した細胞は増殖、分化刺激を受ける。抗体を分泌すると同時にその抗体可変領域をコードする遺伝子(VH-D-JH、VL-JL)に高頻度に突然変異が導入される。その変異の導入によって抗原に対する結合力の高まった抗体を産生する細胞が生き残り、優れた性能の抗体を分泌し続けると同時に記憶細胞として残る。これが「抗体の成熟」である。ここで重要なことは突然変異の役割である。もともと抗体ナイーブレパートリーの中に存在しなかった抗原特異性が、突然変異の導入で生まれることはない。したがってin vitroで抗体をライブラリー化する場合に、生体内における抗体産生過程を再現するのであれば、ナイーブレパートリーに存在しない抗原特異性を持つクローンを排除する機構が必要である。
【0013】
以下に、in vitroで抗体ライブラリーを作製する上での問題点を示す。
(1)個々の抗体遺伝子は、たとえば大腸菌中で大量に発現され、フォールディングし、重鎖可変領域と軽鎖可変領域ドメインが会合して抗体分子となる。これらの段階のいずれかが正しく起こらなかったクローンは、イムノグロブリンとしての正常な構造を再現することができないので意味を持たない。
(2)in vivoでは個々の細胞中で重鎖可変領域と軽鎖可変領域がペアをつくって1つの抗原特異性を示すが、in vitroでライブラリー化する場合には重鎖可変領域集団と軽鎖可変領域集団を別個にライブラリー化した後に組み合わせざるを得ない。たとえば1万種からなるB細胞のレパートリーは、理論的には重鎖可変領域1万×軽鎖可変領域1万=合計1億種のライブラリーでやっとその全てをカバーすることが可能となる。しかし単純に組み合わせの数を増やすだけでは、ライブラリーの規模を大きくする一方で、ライブラリーに占める抗体としての活性を持たないクローンの割合も増加する。
(3)抗体遺伝子のソースとしてヒト血液を用いると、各人の免疫学的経歴によって発現されている抗体遺伝子に大きな偏りがあるはずである。
【0014】
以上の3つの問題点は、いずれもライブラリーのレパートリーに偏りを与える原因となる。つまり、ライブラリーを構成する理論的なクローン数に比べて、現実のライブラリーに含まれる機能的なファージ抗体の種類が著しく少ないというギャップを生じることになる。
より具体的には、特定の抗原に対して免疫応答が亢進しているために、使用頻度に於いて非常に偏りのあるクローンからなるライブラリーが考えられる。あるいは、十分な抗原結合力を持たない抗体をコードする多数のクローンを含む抗体ライブラリー等も生成されるであろう。たとえば軽鎖可変領域遺伝子と重鎖可変領域遺伝子の、それぞれ50%が抗体として活性な分子を発現できるとすると、両者の組み合わせによって活性な抗体が構成される割合はわずかに25%である。
【0015】
このようなライブラリーの問題点は、実際のレパートリーサイズが理論的なサイズよりも遙かに小さいことに留まらない。たとえば、スクリーニングにおいては抗原結合力の弱い抗体分子は、免疫反応を妨害する。すなわち、抗体―抗原複合体形成は平衡反応であるために、多数のクローンと少数のクローンが混在すると、その抗原結合力の差を越えて多数派が少数派を駆逐することになる。
また不活性な抗体をコードするクローンは、クローニングにおいても障害となる。つまり、不活性な抗体をコードするクローンが多ければ、その中には増殖速度が特に速いクローンが生じる可能性も高まる。増殖速度に優れるクローンは、スクリーニング過程で選択的に選ばれてしまうので、非常に高いバックグラウンドとなる。
今までに報告されたライブラリーの問題点は、ライブラリーを構成するクローンの中で、実際にどれだけのクローンが有効であるのかを解析できる形で作製されていないことにある。したがって、ライブラリーやスクリーニングの効率についての評価ができない。
【先行技術文献】
【特許文献】
【0016】
【特許文献1】特表平5-508076
【特許文献2】特表平6-506836
【非特許文献】
【0017】
【非特許文献1】Better et al(1988)(Better M, Chang CP, Robinson RR, Horwitz AH Science 1988, 240:4855 1041-3)
【非特許文献2】Skerra A, Plukthun A Science 1988, 240:4855 1038-41
【非特許文献3】Orlandi R et al. Proc Natl Acad Sci USA 1989 86:10 3833-7, Sastry L et al. Proc Natl Acad Sci USA 1989 86:15 5728-32
【非特許文献4】Huse WD et al.Science 1989 246:4935 1275-81, Ward GE et al.J Clin Microbiol 1989 27:12 2717-23
【非特許文献5】Smith GP Science 1985 228:4075 1315-7
【非特許文献6】Nissim A, Winter G et al. EMBO J 1994 13:3 692-8
【非特許文献7】de Kruif J, Boel E, Logtenberg T J Mol Biol 1995 248:1 97-105
【非特許文献8】Vaughan TJ et al. Nat Biotechnol 1996 14:3 309-14
【発明の概要】
【0018】
本発明の課題は、機能的なコンフォーメーションを保持した抗体分子を高い割合で含む抗体ライブラリーを提供することである。本発明は、このような抗体ライブラリーと、その製造方法、ならびにそのライブラリーを使った抗体のスクリーニング方法の提供を課題とする。また本発明は、機能的なコンフォーメーションを保持しうる抗体分子を与えるイムノグロブリンの軽鎖をコードする遺伝子を単離する方法の提供を課題としている。
【0019】
本発明者らは、公知の抗体レパートリーから必要な抗体をスクリーニングすることを妨げている要因について研究を重ねた。そして、現在までに知られている抗体ライブラリーにおいては、そのレパートリーに占める、機能的なコンフォーメーションを保持した抗体分子の割合が少ないことが、有用な抗体の単離を妨げているのではないかと考えた。公知の抗体ライブラリーの製造方法においては、生体内に保持されている抗体レパートリーを忠実にin vitroで構成することを目標として様々な工夫が提案された。
【0020】
ある場合には、多様性を高めるために人為的な変異をランダムに挿入する方法さえ試みられた。しかし、こういった試みの多くは、機能的なコンフォーメーションを維持した抗体分子を生成する一方で、明らかに抗体としての活性が不充分な抗体分子の生成も伴っていた。その結果、得られた抗体ライブラリーには、生体内では用いられることのないレパートリーや、多くの不活性な抗体分子が含まれ、機能的な抗体のスクリーニングを妨げる原因となってしまう。たとえば軽鎖可変領域遺伝子を105個の独立したクローンとし、これに109個の重鎖可変領域遺伝子を組み合わせてライブラリーを構成したとする。数の上では重鎖可変領域遺伝子は十分な多様性を与えたことになるが、軽鎖可変領域遺伝子に不活性ものが多く含まれているとすると、重鎖可変領域遺伝子の一部はイムノグロブリンとしての活性を得ることができずスクリーニングの段階で失われしまう。
生体内においては、多様な抗体分子をランダムに生成する機構とともに、抗体としての活性に優れる抗体を産生するクローンが選択的に増殖する機構が働いている。in vitroにおいても、不活性な抗体を除くステップを設けなければ、単に多様性を追求するのみでは、真に生体内の抗体レパートリーを再現することにはつながらない。
【0021】
このような背景の下で、本発明者らは、抗体ライブラリーに占める機能的な抗体分子の割合を高めることによって、より効率的な抗体の単離が可能になると考えた。そのためには、公知の抗体ライブラリーにおいて不活性な抗体分子をもたらす原因を明らかにしなければならない。本発明者らは、抗体活性の維持に果たす軽鎖の役割に着目した。そして、まず、抗体分子に機能的なコンフォーメーションを与えることができる軽鎖をスクリーニングする方法を確立した。
【0022】
更に本発明者らは、こうして選択された軽鎖可変領域遺伝子の構造を丹念に解析することによって、限られた構造の軽鎖可変領域遺伝子のみを利用することによって、機能的なコンフォーメーションを維持した抗体分子を高い割合で含む抗体ライブラリーの構築が可能となることを見出し本発明を完成した。軽鎖可変領域遺伝子の選択工程を経ることによって、抗体レパートリーを狭めてしまう可能性が心配される。しかしながら本発明者らは、本発明の選択方法によって選択された軽鎖可変領域遺伝子の構造を丹念に解析した結果、重鎖と機能的なコンフォーメーションを再構成しうる軽鎖の構造は、現実には限られた範囲に収束することを見出した。機能的な抗体分子を構成する軽鎖の構造が、一定のレパートリーに集約されることを明らかにし、これをライブラリーの調製に応用した点に本発明の最大の特徴がある。すなわち本発明は、以下の工程を含んでなるイムノグロブリンの遺伝子ライブラリーの調製方法と、このライブラリーに基づくrgdpライブラリー、更にはこれらのライブラリーから特定の抗原を認識する抗体をコードする遺伝子をスクリーニングする方法に関する。また本発明は、以下に示す工程からなる、重鎖と機能的なコンフォーメーションを再構成することができる軽鎖をコードする遺伝子の選択方法を提供する。
【0023】
〔1〕 以下の工程を含む、イムノグロブリンの軽鎖可変領域遺伝子と重鎖可変領域遺伝子の組み合わせからなる遺伝子ライブラリーの調製方法。
a)軽鎖可変領域遺伝子として、重鎖可変領域遺伝子の発現産物との機能的なコンフォーメーションの再構成が可能な軽鎖分子をコードするものを選択し、
b)工程a)によって得られる軽鎖可変領域遺伝子の集合である遺伝子のライブラリーを調製し、
c)工程b)のライブラリーに重鎖可変領域をコードする遺伝子のライブラリーを組み合わせる
〔2〕 工程c)において、VHファミリーごとにそれぞれ独立して調製した重鎖可変領域遺伝子のライブラリーを、生体内におけるVHファミリーの割合に応じて組み合わせる〔1〕に記載の方法。
〔3〕 工程c)における遺伝子ライブラリーの組み合わせを同一のベクター上で行う〔1〕に記載の方法。
〔4〕 更に次の工程d)を含む、〔1〕に記載の方法。
d)重鎖可変領域に標識ペプチドが融合しており、この標識ペプチドを指標として重鎖を発現するクローンを選択する工程
〔5〕 〔1〕に記載の方法によって得ることができる遺伝子ライブラリー。
〔6〕 少なくともイムノグロブリンの軽鎖可変領域をコードする遺伝子からなる遺伝子ライブラリーであって、イムノグロブリンの重鎖可変領域と機能的なコンフォーメーションを再構成できない軽鎖可変領域をコードする遺伝子が実質的に排除されている遺伝子ライブラリー。
〔7〕 ライブラリーが重鎖可変領域をコードする遺伝子のライブラリーを伴っている〔6〕に記載のライブラリー。
〔8〕 VHファミリーのそれぞれの重鎖可変領域遺伝子のライブラリーが、生体内の多様性を包含するのに十分なクローン数を有する〔7〕に記載のライブラリー。
〔9〕 ライブラリーを構成する遺伝子が、細菌、酵母、および植物細胞からなる群から選択されるいずれかの宿主細胞に導入されたものである〔6〕に記載のライブラリー。
〔10〕 ライブラリーを構成する遺伝子が、哺乳動物細胞に導入されたものである〔6〕に記載のライブラリー。
〔11〕 ライブラリーを構成する少なくとも一部の遺伝子が、繊維状ファージに組み込まれている〔6〕に記載のライブラリー。
〔12〕 ライブラリーを構成する遺伝子によってコードされる重鎖可変領域と軽鎖可変領域の断片を繊維状ファージ表面に発現し、かつそれらが機能的に再構成されている〔11〕に記載のライブラリー。
〔13〕 重鎖可変領域に標識ペプチドをコードする遺伝子が融合されている〔12〕に記載のライブラリー。
〔14〕 イムノグロブリン軽鎖可変領域遺伝子がヒトに由来するものである〔6〕に記載のライブラリー。
〔15〕 イムノグロブリン軽鎖超可変部位がシステイン残基を含まないアミノ酸配列からなる〔14〕に記載のライブラリー。
〔16〕 〔5〕に記載の遺伝子ライブラリーを構成する各クローンが、そのクローンによって保持される遺伝子によってコードされる抗体タンパク質を伴っているrgdpライブラリー。
〔17〕 〔5〕に記載の遺伝子ライブラリーを構成する各クローンについて、そのクローンによって保持される遺伝子によってコードされる抗体タンパク質から構成される抗体ライブラリー。
〔18〕 次の工程を含む、重鎖可変領域と機能的なコンフォーメーションを再構成することができる軽鎖可変領域をコードする遺伝子の選択方法。
a)軽鎖可変領域をコードする、1つまたは複数の遺伝子を取得する工程、
b)軽鎖可変領域と機能的なコンフォーメーションを構成することが確認されているイムノグロブリン重鎖可変領域をコードする遺伝子を取得する工程、
c)工程a)によって得られた軽鎖可変領域をコードする遺伝子の任意の1つを選択し、工程b)で取得した重鎖可変領域をコードする遺伝子とイムノグロブリンの機能的なコンフォーメーションを再構成が可能な条件下でタンパク質に翻訳する工程、
d)工程c)において翻訳されたタンパク質の抗原結合領域の形成を検出する工程、および
e)抗原結合領域の形成が検出されたタンパク質を構成する軽鎖可変領域をコードする遺伝子を選択する工程
〔19〕 更に、次の工程f)を含む〔18〕に記載の方法。
f)軽鎖可変領域遺伝子の塩基配列を決定し、その塩基配列によってコードされるアミノ酸配列を比較して同一のアミノ酸配列をコードする遺伝子、およびアミノ酸の欠損を生じている遺伝子を排除する工程
〔20〕 イムノグロブリンの軽鎖可変領域が、繊維状ファージの表面に発現している〔18〕に記載の方法。
〔21〕 イムノグロブリンの軽鎖可変領域をコードする遺伝子と重鎖可変領域をコードする遺伝子が挿入されたファージミドを感染させた宿主微生物の培養上清そのものを、工程c)のための軽鎖を含む試料として利用する〔18〕に記載の方法。
〔22〕 次の工程を含む、特定の抗原と結合するイムノグロブリン可変領域の検出方法。
a)〔5〕または〔7〕に記載のライブラリー、またはその発現産物と、該抗原とを抗原抗体反応に適した条件下に接触させる工程
b)該抗原とイムノグロブリン可変領域との結合を検出する工程
〔23〕 ライブラリーが〔12〕に記載のライブラリーである〔22〕に記載の方法。
〔24〕 〔22〕に記載の方法の後、更に次の工程c)を行うことを特徴とする、特定の抗原と結合するイムノグロブリン可変領域の取得方法。
c)前記抗原と結合するイムノグロブリン可変領域を発現するクローンを選択する工程
〔25〕 更に次の工程d)−e)を含む〔24〕に記載の方法。
d)工程c)で選択したファージクローンを増幅して2次的なライブラリーを得る工程、
e)工程c)において選択されるクローンの回収率が上昇するまで、2次的なライブラリーについて、工程a)−d)を繰り返す工程
〔26〕 〔24〕に記載の方法によって得られたクローン、またはイムノグロブリン断片、もしくはそれをコードする遺伝子。
〔27〕 配列番号:61〜配列番号:78のいずれかに記載の塩基配列を含む、ポリヌクレオチド。
〔28〕 配列番号:79〜配列番号:96のいずれかに記載のアミノ酸配列を含む蛋白質。
〔29〕 次の要素からなる抗体ライブラリーの調製用キット。
a)重鎖可変領域と機能的なコンフォーメーションを再構成することができない軽鎖可変領域をコードする遺伝子を実質的に排除した軽鎖可変領域遺伝子ライブラリー、および
b)重鎖可変領域をコードする遺伝子を増幅することができるプライマーセット
〔30〕 以下の工程からなる抗体ライブラリーの調製方法。
a)ファージミドに、イムノグロブリンの少なくとも可変領域を含む領域をコードする遺伝子を組み込む工程
b)工程a)で得られたファージミドを宿主微生物に感染させる工程
c)ヘルパーファージを感染させること無く、工程b)の宿主微生物の培養上清を回収して抗体ライブラリーとする工程
あるいは本発明は、以下の病原体を認識する抗体の取得方法に関する。
〔31〕 前記抗原が、病原体に由来する抗原である〔24〕に記載の方法。
〔32〕 〔24〕に記載の方法の後、更に次の工程f)を行うことを特徴とする、病原体の中和活性を有する抗体の取得方法。
f)工程c)で選択した抗体可変領域の病原体に対する中和活性を評価し、中和活性を有する抗体可変領域を選択する工程
〔33〕 病原体に由来する抗原が、インフルエンザウイルスHA抗原、ジフテリア菌毒素、破傷風毒素、および水痘ウイルス由来の糖蛋白質からなる群から選択されるいずれかの抗原である〔31〕に記載の方法。
〔34〕 工程a)の前に、次の工程a’)を行う、〔31〕に記載の方法。a’)ライブラリーを吸収用抗原と接触させ、吸収用抗原に結合する抗体をライブラリーから除去する工程;ここで吸収用抗原とは、前記抗原と同じ病原体に由来するが、吸収用抗原との反応性を有する抗体の取得を望まない抗原を意味する
〔35〕 〔12〕に記載のライブラリーの、病原体中和抗体の調製における使用。
【0024】
本発明において、イムノグロブリンとは、動物種やクラスを問わず、重鎖と軽鎖とで構成される全てのイムノグロブリン分子を意味する。また、抗原との結合が可能な領域のみからなる断片や、あるいはイムノグロブリンを構成する領域が異なる動物種からなるキメラ抗体であることもできる。哺乳動物の重鎖可変領域をコードする遺伝子は、一般に遺伝子の構造的な特徴に基づいていくつかのVHファミリーに分類されている。たとえば、ヒトではVH1〜VH7の7つのファミリーに分類されている。各ファミリーは、ファミリー内で保存性の高い塩基配列を含み、この保存性の高さを利用して各ファミリー用のPCRプライマーが提案されている。軽鎖可変領域も、重鎖と同様に構造的な特徴に基づいてファミリーに分類することができる。
重鎖可変領域をコードする遺伝子は、V(Variable)、D(Diversity)、およびJ(Junction)の3つの遺伝子群から構成される。V、D、そしてJの各遺伝子群が、それぞれ複数の遺伝子からなっており、それらがランダムに組み合わさり、更に変異が加わることによって抗体の多様性が生まれる。これに対して、軽鎖可変領域を構成しているのは、V、およびJの2つの遺伝子群である。軽鎖可変領域においても重鎖可変領域と同様に、複数の遺伝子群の組み合わせと変異によって多様性がもたらされている。
【0025】
本明細書では、用語「ライブラリー」を用いる。ライブラリーは、多様なレパートリーからなる構成要素を含む集合体を意味する。遺伝子は遺伝子ライブラリーを、抗体分子は抗体ライブラリーを、あるいはファージやファージミドはファージライブラリーをそれぞれ構成する。ファージが内部に保持した抗体遺伝子を表面に発現している場合には、遺伝子ライブラリーであると同時に抗体ライブラリーでもある。ただし本明細書における用語ファージライブラリーは、ファージが抗体分子の発現を伴わない場合も含む。すなわち、ファージミドを感染させた宿主微生物や、抗体可変領域をコードする遺伝子をゲノムに保持したファージが溶原化している場合もファージライブラリーである。
【0026】
更に本明細書においては、用語「rgdp ライブラリー」(replicable genetic display package library;複製可能な遺伝的表示パッケージのライブラリー)を用いる。rgdpライブラリーとは、すなわち、遺伝子を保持するとともに、その遺伝子の発現生成物を表面に提示したもので構成されるライブラリーを呼ぶ。前記ファージライブラリーが、抗体タンパク質を表面に発現している場合には、rgdpライブラリーに含まれる。rgdpライブラリーには、ファージライブラリーのほか、外来タンパク質をその表面に発現している形質転換細胞やリボゾームからなるライブラリーを示すことができる。
【0027】
また本発明においては、用語「コンフォーメーション(conformation)」を用いる。イムノグロブリンが、重鎖と軽鎖の会合(holding)によって構成されることは既に述べた。この会合の結果として生じる重鎖と軽鎖の結合物の構造が、コンフォーメーションである。コンフォーメーションは、一般的に定常領域における-SS-結合によって成立する。このとき、常に抗原との結合活性を獲得するとは限らない。本発明において、あるイムノグロブリンが抗原との結合活性を持つとき、そのイムノグロブリンのコンフォーメーションが機能的(functional)であると言う。そして、ある軽鎖との組み合わせにおいて機能的なコンフォーメーションを与える重鎖が、他の軽鎖との組み合わせによっても機能的なコンフォーメーションを与えるとき、両者の結合を特に再構成(re-holding)と呼ぶ。再構成を構成する他の軽鎖とは、いったん別々のクローンとして単離された同一の細胞に由来する軽鎖を含む。また、本発明におけるコンフォーメーションの再構成とは、あくまでもイムノグロブリンの抗原との結合に必要な領域における再構成を意味する。したがって、定常領域の有無に関わらず、可変領域における分子構造がイムノグロブリンとして再構成されていれば、機能的なコンフォーメーションを再構成したと見なす。更に、軽鎖や重鎖をコードする遺伝子への人為的な塩基配列やファージの構成タンパク質の融合などを伴う場合であっても、可変領域における再構成が達成されている限り、本発明においては機能的なコンフォーメーションを再構成したと見なす。より具体的には、本発明における分子構造の再構成とは、たとえば重鎖可変領域と軽鎖可変領域とが異なるタンパク質として翻訳された場合には、定常領域において形成される-SS-結合によって重鎖可変領域と軽鎖可変領域とがイムノグロブリンの可変領域を構成することと言い換えることができる。
ところでsingle chain Fv抗体(scFv)のように、人工的なリンカーによって重鎖と軽鎖がはじめから結合されている抗体分子も存在する。この種の特殊な抗体においては、コンフォーメーションは-SS-結合ではなく、ペプチド結合によって構成される場合がある。したがってscFv抗体においては、定常領域を介することなくコンフォーメーションが再構成される。
【0028】
まず本発明は、機能的なイムノグロブリンを再構成することができる軽鎖をコードする遺伝子の選択と、この軽鎖可変領域遺伝子に重鎖をコードする遺伝子のライブラリーを組み合わせることによる遺伝子ライブラリーの調製方法に関する。本発明における軽鎖可変領域遺伝子の選択は、以下の工程によって行うことができる。すなわち本発明は、次の工程を含む、重鎖と機能的なコンフォーメーションを再構成することができる軽鎖をコードする遺伝子の選択方法に関する。
a)軽鎖可変領域をコードする、1つまたは複数の遺伝子を取得する工程、
b)軽鎖可変領域と機能的なコンフォーメーションを再構成することが確認されているイムノグロブリン重鎖可変領域をコードする遺伝子を取得する工程、
c)工程a)によって得られた軽鎖可変領域をコードする遺伝子の任意の1つを選択し、工程b)で取得した重鎖可変領域をコードする遺伝子とイムノグロブリンの機能的なコンフォーメーションを再構成が可能な条件下でタンパク質に翻訳する工程、
d)工程c)において翻訳されたタンパク質の抗原結合部位の形成を検出する工程、および
e)抗原結合部位の形成が検出されたタンパク質を構成する軽鎖可変領域をコードする遺伝子を選択する工程
【0029】
本発明において、軽鎖可変領域あるいは重鎖可変領域とは、少なくとも抗原との結合に必要な領域を含む任意の領域とすることができる。言いかえれば、3つのCDRとそれを保持するフレーム(FR)で構成される領域を含む任意の領域を本発明の可変領域として用いることができる。したがって、たとえば定常領域をも含む断片であっても、抗原との結合に必要な領域を含んでおれば、本発明の可変領域として利用することができる。抗体の可変領域としてしばしば用いられる、FabやFab'は、もともとイムノグロブリンの酵素的な切断によって得られる断片に対して与えられた名称である。本発明においては、Fabを可変領域を特定するための用語として理解すべきではない。
【0030】
本発明の軽鎖可変領域遺伝子の選択方法において、対象となる軽鎖可変領域遺伝子は、任意の抗体産生細胞より得ることができる。抗体産生細胞としては、たとえば、末梢血リンパ球や脾細胞等を挙げることができる。軽鎖可変領域遺伝子の単離にはRT-PCRを利用するのが有利である。たとえばヒトの場合、VLJL遺伝子を増幅することができるプライマーが明らかにされている(特表平3-502801あるいは特表平4-500607。また、MRC社はホームページ[「V-base」:http://www.mrc-cpe.cam.ac.uk/imt-doc/restricted/ok.htmlでプライマーを公開している)。したがって、これらのプライマーを用いてRT-PCRを行えば、工程a)に必要な軽鎖可変領域をコードする遺伝子を得ることができる。得られた遺伝子を工程c)に用いる。
【0031】
次に工程b)において、軽鎖可変領域と機能的なコンフォーメーションを再構成することが確認されているイムノグロブリン重鎖可変領域をコードする遺伝子を取得する。このとき取得する重鎖可変領域は、工程a)で得た軽鎖可変領域と同じ動物種に由来し、かつ軽鎖可変領域と機能的なコンフォーメーションを再構成することができるものであれば、抗原結合特異性などは任意であって良い。このような重鎖可変領域をコードする遺伝子は、たとえば、抗体活性を持つことが明らかなイムノグロブリン分子をコードする遺伝子から得ることができる。工程b)の重鎖可変領域としては、κ鎖およびλ鎖との再構成が可能なものを用意するのが望ましい。このような重鎖可変領域としては、実際に軽鎖との会合効率を確認して、最も効率の高い重鎖可変領域を選ぶのが望ましい。たとえば後に述べる実施例においては、重鎖に対応する各種クローンについて、軽鎖との会合効率を検討したところ、次のような構造を持つVH3-4が最も会合の効率が高かったことから、VH3-4(配列番号:1)を選択した。VH3-4は、以下に示す構造を持っている。
FR1:EVQLVESGGGLVQPGRSLRLSCAASGFTFD
CDR1:DYAMH
FR2:WVRQAPGKGLEWVS
CDR2:GISWNSGSIGYADSVKG
FR3:RFTISRDNAKNSLYLQMNSLRAEDTALYYCAK
CDR3:GPSGSFDAFDI
FR4:WGQGTTVTVSS
【0032】
続いて工程c)において、工程a)によって得られた軽鎖可変領域をコードする遺伝子の任意の1つと工程b)で得た重鎖可変領域をコードする遺伝子とを、イムノグロブリンの機能的なコンフォーメーションを再構成が可能な条件下でタンパク質に翻訳する。工程b)では、機能的なコンフォーメーションを構成しうる重鎖可変領域をコードする遺伝子を選択しているので、工程c)においてはイムノグロブリン分子の可変領域としての構造を再構成しているものは、機能的なコンフォーメーションを再構成したと見なすことができる。なお、ここでいうイムノグロブリン分子とは、イムノグロブリンにおける抗原との結合に必須の部分を含む限り、あらゆる構成であることができる。したがって、定常領域の有無に関わらず、抗原結合部位を再構成しているものは、イムノグロブリン分子として再構成されたと見なすことができる。
工程c)におけるイムノグロブリンの再構成が可能な条件下とは、-SS-結合によって重鎖可変領域と軽鎖可変領域の会合(holding)が可能となる条件を意味する。より具体的には、たとえば前述のように大腸菌のペリプラズムなど生体内の還元環境でのFabタンパク質の発現は、イムノグロブリンの再構成が可能な条件と言える。抗体のコンフォメーション形成に必要な還元的微小環境としては、ヒトなどの哺乳動物細胞では小胞体などのオルガネラを挙げることもできる。更に、重鎖と軽鎖の可変領域が人工的なアミノ酸配列(リンカー)で結合されたscFvタイプの抗体であれば、イムノグロブリンとしての再構成に必ずしも還元的な環境を必要としない場合もある。
工程c)における軽鎖可変領域と重鎖可変領域の発現には、外来遺伝子を表面に発現するファージを用いるのが有利である。例えば繊維状ファージは、その表面にcp3やcp8などのファージの構成タンパク質との融合タンパク質として外来遺伝子にコードされるタンパク質を発現する。
【0033】
さて、通常ファージライブラリーのスクリーニングは、ファージを粒子として回収するステップを含む。したがって、たとえば外来遺伝子をファージミドとして感染させた場合には、ヘルパーファージを感染させることによりファージ粒子として回収される。ところが本発明者らは、cp3との融合タンパク質としてFab遺伝子を挿入したファージミドを感染させた大腸菌をヘルパーファージを加えないで培養すると、その培養上清中に、Fabとcp3の融合タンパク質が分泌されることを見出した。ファージミドを感染させた大腸菌によって分泌されるFabとcp3の融合タンパク質は、20時間の培養後でさえ微量ではあったが、本発明による軽鎖の選択方法を実施するには十分な量であった。したがって、本発明による軽鎖可変領域遺伝子の選択方法のための試料としては、ファージミドを感染させた宿主微生物の培養上清を利用することもできる。ヘルパーファージを感染させてファージ粒子として回収する工程を不要とするこの方法は、実験操作上、きわめて簡便である。本発明の方法に基づいてファージミドを感染させた宿主微生物の培養上清をスクリーニングのための試料とするためには、宿主微生物において動作可能なプロモーターと、シグナル配列とを備えたベクターを用いる。たとえば大腸菌を宿主とする場合には、シグナル配列としてpelB配列などを挿入した繊維状ファージ用のファージミドベクターを利用することができる。
【0034】
もしも軽鎖可変領域が重鎖可変領域との機能的なコンフォーメーションを再構成しうるものであれば、イムノグロブリンの可変領域が形成される。この可変領域の形成を検出することによって、選択すべき軽鎖可変領域を知ることができる。可変領域の形成は例えば、イムノアッセイの原理を利用して検出することができる。すなわち、κ鎖(あるいはλ鎖)に対する抗体を固相化抗体としてコートしたプレートに、重鎖可変領域と軽鎖可変領域の発現生成物を含む試料を加えて、軽鎖可変領域をプレート上に捕捉する。もしも重鎖可変領域が軽鎖可変領域と会合していれば、重鎖可変領域は軽鎖可変領域とともにプレート上に捕捉されるはずである。次いで重鎖やFabに対する標識抗体を加えれば、両者が会合できたときに限り、標識抗体がプレート上に捕捉されることになる。適当な時間インキュベーションしてプレートを洗浄し、標識抗体を検出すれば、機能的なコンフォーメーションを構成する軽鎖可変領域を検出することができる。標識抗体と固相化抗体は、逆の組合せとすることもできる。あるいは、重鎖可変領域を予めビオチン化しておき、標識アビジンによって検出することもできる。前述のとおり、ここで検出に用いる試料は、ファージミドを感染させた大腸菌の培養上清を用いることができることを我々は見出している。
こうして重鎖可変領域との会合が確認された軽鎖可変領域を、重鎖可変領域との機能的なコンフォーメーションが可能な軽鎖可変領域として選択することができる。軽鎖可変領域をコードする遺伝子がファージライブラリーとして保持されている場合には、ファージを回収することによって軽鎖可変領域の遺伝子を選択することができる。
【0035】
以上に述べた過程を経て得られた軽鎖可変領域遺伝子は、重鎖可変領域との再構成が可能であるのみならず、スクリーニングに利用した発現系における発現が証明されたものとなる。たとえばファージによる発現を利用した場合には、大腸菌において十分な発現が見られるものが選択される。したがって、哺乳動物細胞では発現するものの、大腸菌では発現量が少なくなる遺伝子をこの段階で除くことができる。このような特徴は、本発明における軽鎖可変領域遺伝子の選択方法において期待することができる新規な利点である。これに対して従来の抗体ライブラリーの作製技術においては、軽鎖の選択工程を含まないため、発現レベルが不充分な軽鎖遺伝子の混入を防ぐことはできなかった。
【0036】
選択された軽鎖可変領域をコードする遺伝子は、そのまま本発明による遺伝子ライブラリーの調製に用いることができる。しかしこの段階では、選択した軽鎖可変領域遺伝子の間に重複が存在している可能性がある。したがって、好ましくは軽鎖可変領域遺伝子の構造を解析し、重複を除いた上で、遺伝子ライブラリーの調製に用いる。遺伝子の重複は、たとえば次のような方法によって除くことができる。
まず前記工程d)の後に、あるいは先だって軽鎖可変領域遺伝子の塩基配列を決定し、その塩基配列によってコードされるアミノ酸配列を推定する。推定されたアミノ酸配列を比較し、同じアミノ酸配列をコードする遺伝子は除く。この段階で更に、欠損(deletion)のチェックも行うのが望ましい。このため、塩基配列の決定を行って読みとり枠のずれているものを除いた。
【0037】
なお、遺伝子の取捨選択は、実際には、類似の遺伝子をグルーピングし、各グループから代表的な配列を選択することになる。このとき、選択された遺伝子を偏りなくカバーできるように、そしてVL-JL結合点のズレに関しても天然に存在する抗体で見られる分布を損なわないように選択する。実際には、遺伝子のデータベースから実際の抗体で使用されている軽鎖可変領域遺伝子をリストアップし、結合点のズレに関して集計した結果を元に分布を決定した。表1に、V-Jの結合点(アミノ酸配列91-96)にいくつのアミノ酸を持つかについて調査した結果を示す。
【0038】
【表1】
【0039】
以上の結果を総合したところ、本発明者らの解析結果によれば、ヒトのイムノグロブリンの場合、このような選択方法に基づいて選択される代表的な軽鎖可変領域のレパートリーサイズは、κ鎖で101、並びにλ鎖でも99である。
つまり、ヒトの機能的なイムノグロブリンを代表しうる軽鎖可変領域のレパートリーサイズは、せいぜい200にすぎないことが明らかとなったのである。なお、本発明者らが明らかにしたおよそ100というレパートリーサイズを、限定的に解釈すべきではない。すなわち、本発明の選択方法に基づいて、たとえばヒトの軽鎖可変領域遺伝子を選択するときに選択すべき軽鎖可変領域遺伝子の数は、必ずしも100になるとは限らない。あくまでも、得られたアミノ酸配列に基づいて、ここに述べた選択方法を実施することによって選択された軽鎖の遺伝子を以降の工程に用いることが重要である。
ここで、スクリーニングによって得られたファージ抗体のVL遺伝子を分類した。結果は図1に示した。VL遺伝子の使用は特定の遺伝子に偏っていることがわかる。この結果から、イムノグロブリンのコンフォーメーションを再構成しうる軽鎖を多く含むセットを揃えることにより、機能的なイムノグロブリンの割合の高い良質なライブラリーを作製できることが示された。
【0040】
ここで、生体内における抗体の多様性を維持するには、アミノ酸配列の解析の対象をできるだけ大きく取ることが望ましい。
1人のヒトの軽鎖を構成するゲノム遺伝子は、36種類のVl、7種類のJl、37種類のVk、そして4種類のJkで構成される。V遺伝子とJ遺伝子の組み合わせによって軽鎖遺伝子が作られるので、単純計算では36×7=252と37×4=148の和、つまり400種類となる。加えて、遺伝子の結合の際にその結合点のアミノ酸の数には、変異が伴う。つまり、前述のように抗体遺伝子の再構成に特有の現象により、平均±1アミノ酸程度(最大で±5アミノ酸程度)の多様性が生まれる。この組み合わせから更に不要な遺伝子は削除されて、個人の抗体遺伝子のレパートリーとなる。さらに、個人によって遺伝子は少しずつ異なっている(これを多型;polymorphismという)。全人類が持っている抗体遺伝子の種類を調べ尽くすことは現実的ではないが、総計して1000種類程度であると推定される。これらのことから、1人のヒトがあらゆる抗原に対応する抗体のセットを作ることができるとすれば、理論的には好ましくは400種類以上1000種類程度までのアミノ酸配列について本発明による解析方法を実施することによって、軽鎖可変領域遺伝子のレパートリーを十分に再現することができることになる。
【0041】
本発明者らがヒトにおいて明らかにしたこのような軽鎖のレパートリーサイズ(200種類)は、約1000種類のアミノ酸配列の解析の結果得られた数字である。したがって、理論的には、あらゆる抗体セットにおいて機能的なコンフォーメーションを再構成することができる軽鎖可変領域が選択されていると言うことができる。実験的にも、本発明による200種類と言う軽鎖可変領域遺伝子のレパートリーサイズは、生体内における抗体の多様性をin vitroで再現するのに十分有効であることを示すことができた。しかし、より多くの塩基配列を決定し、そのアミノ酸配列についても解析を行えば、レパートリーサイズが大きくなる可能性は否定できない。
【0042】
そこで、本発明によって選択された軽鎖可変領域ライブラリーのレパートリーサイズを、軽鎖遺伝子ライブラリーとの混合によって補うことができる。すなわち、軽鎖可変領域についても重鎖と同じく選択をかけない全ての軽鎖遺伝子(VLライブラリーと呼ぶ)を用いてライブラリーを作製するのである。こうして作製したVLライブラリーを200種の軽鎖可変領域のみを用いたライブラリー(KL200ライブラリーと呼ぶ)と混合することによって、相補的に欠点を補うライブラリーとすることができる。各ライブラリーは以下に述べる特徴を持つ。
KL200ライブラリーは、重鎖とのコンフォメーション形成が確認されている。ただし数を限定したために、特定の特異性を持つクローンを形成するのに必要な軽鎖が除外されている可能性も否定できない。
VLライブラリーは、独立クローン数が109に達しているため必要なクローンはもらしていない。しかし発現と重鎖とのコンフォメーション形成率はKL200ライブラリーに比較して低い。
【0043】
なお、本発明において、アミノ酸配列の解析結果に基づいて分類した各グループから、いずれの遺伝子を選択するかは本来任意である。したがって、重鎖との機能的なコンフォーメーションの再構成が可能な軽鎖をコードする遺伝子の構造をここに明らかにすることには大きな意味はない。重要なのは、このような選択方法によって、重鎖とともに機能的なコンフォーメーションを再構成することができる軽鎖の選択工程を実施することである。いずれにせよ、このような操作を通じて、ヒトであれば、101種類のκ鎖遺伝子ライブラリーと、99種類のλ鎖遺伝子ライブラリーからなる軽鎖可変領域遺伝子ライブラリーが得られることになる。
【0044】
さて、本発明において選択された軽鎖可変領域の遺伝子ライブラリーは、後に述べるように重鎖可変領域をコードする遺伝子を組み合わせることによってイムノグロブリンの遺伝子ライブラリーを与える。したがって、本発明によって選択された軽鎖可変領域の遺伝子ライブラリーは、イムノグロブリンの遺伝子ライブラリー調製用のライブラリーとして有用である。すなわち本発明は、少なくともイムノグロブリンの軽鎖可変領域をコードする遺伝子からなる遺伝子ライブラリーであって、イムノグロブリンの重鎖と機能的なコンフォーメーションを再構成できない軽鎖可変領域をコードする遺伝子が実質的に排除されている遺伝子ライブラリーに関する。
【0045】
本発明において、イムノグロブリンの重鎖可変領域と機能的なコンフォーメーションを再構成できない軽鎖可変領域をコードする遺伝子とは、先に述べた軽鎖可変領域の選択方法によって排除することができる。重鎖可変領域と機能的なコンフォーメーションを再構成できない軽鎖可変領域をコードする遺伝子を、本発明においてはdefective遺伝子と呼ぶ。本発明においてdefective遺伝子が実質的に排除されたライブラリーとは、defective遺伝子の完全な排除までも要求するものではない。たとえばdefective遺伝子が混入したライブラリーであっても、免疫学的な反応に基づく抗体のスクリーニングを妨げない範囲であれば、defective遺伝子が実質的に排除されたライブラリーと言うことができる。
【0046】
抗体のスクリーニングを妨げない範囲とは、ライブラリーに占めるdefective遺伝子の割合が、たとえば0−50%、望ましくは0−25%であることを意味する。defective遺伝子の割合は低いほどスクリーニングの効率が高まり、有用なクローンをスクリーニングの過程で失う危険性が低くなることは言うまでも無い。しかし発明者らが試作したライブラリーにおいて、defectiveな遺伝子を50%含むと考えられるVLライブラリーをdefectiveな遺伝子を完璧に排除したKL200ライブラリーに様々な割合で混合したところ、50%までの混合では有効なスクリーニングができることを確認している。この事実により、defectiveな遺伝子の割合がより好ましくは25%以下であれば、defective遺伝子が実質的に排除されていると言うことができる。
【0047】
本発明による軽鎖のみからなるライブラリーは、重鎖の可変領域をコードする遺伝子ライブラリーを組み合わせるための材料として有用である。このようなライブラリーとしては、たとえばファージのcp3をコードする遺伝子にdefective遺伝子が実質的に排除された軽鎖ライブラリーを組み込むとともに、重鎖の可変領域を挿入するためのクローニングサイトを設けたファージライブラリーを挙げることができる。一般にファージライブラリーは、ファージの宿主微生物に対する感染性を損なうことがないように、ファージミド上に外来遺伝子を保持させ、これをヘルパーファージを用いてファージ化する方法が用いられる。本発明のファージライブラリーも、ファージミド上に構築することができる。すなわち、宿主において機能するプロモーターの制御下に、シグナル配列と連結した前記軽鎖可変領域をコードする遺伝子と、重鎖可変領域をコードする遺伝子のクローニングサイトを設ける。重鎖可変領域遺伝子のためのクローニングサイトは、目的とする遺伝子に見出される頻度の低い制限酵素とするのが有利である。本発明によるファージライブラリーは、ファージミドのみならずファージのゲノムを利用することもできる。クローニングサイトを付加したプライマーによって重鎖可変領域遺伝子をPCRによって合成し、ファージライブラリーに挿入すればイムノグロブリンの可変領域を発現するファージライブラリーを完成することができる。
【0048】
あるいは、defective遺伝子が実質的に排除された軽鎖ライブラリーを大腸菌用の発現ベクターに組み込んで、大腸菌に形質転換することによって軽鎖可変領域ライブラリー分泌発現株とすることもできる。この大腸菌に、PCRによって得た重鎖可変領域を組み込んだファージを感染させれば、Fabをその表面に再構成したファージ粒子を得ることができる。重鎖可変領域遺伝子を様々な免疫履歴を持つ個体から選択することによって、目的とする抗体に合った抗体ライブラリーを得ることができる。
【0049】
このような方法に基づいて、ファージライブラリー作成用キットを提供することができる。このキットは、defective遺伝子が実質的に排除された軽鎖ライブラリーと、重鎖可変領域遺伝子増幅用のプライマーとからなる。使用者は、重鎖可変領域遺伝子増幅用のプライマーを用いて、目的とする抗体に合った免疫履歴を持つ遺伝子ソースからPCR生成物を得ることができる。たとえば、がんの宿主からは、腫瘍関連抗原を認識する抗体ポピュレーションに富むライブラリーが期待できる。
【0050】
こうして選択された軽鎖可変領域遺伝子を利用し、本発明によるライブラリーを調製する。すなわち本発明は、以下の工程を含む、イムノグロブリンの軽鎖可変領域遺伝子と重鎖可変領域遺伝子の組み合わせからなる遺伝子ライブラリーの調製方法に関する。
a)軽鎖可変領域遺伝子として、重鎖可変領域遺伝子の発現産物との機能的なコンフォーメーションの再構成が可能な軽鎖分子をコードするものを選択し、
b)工程a)によって得られる軽鎖可変領域遺伝子の集合である遺伝子のライブラリーを調製し、
c)工程b)のライブラリーに重鎖をコードする遺伝子のライブラリーを組み合わせる
【0051】
工程a)の軽鎖の選択工程は、先に述べたとおりである。先に得られた軽鎖をコードする遺伝子を集めて、工程b)のライブラリーとすることができる。軽鎖可変領域遺伝子が繊維状ファージに保持されている場合には、このファージを増殖させ、回収してライブラリーとすることができる。次いで工程c)として、前記軽鎖の遺伝子ライブラリーに重鎖の遺伝子ライブラリーを組み合わせる。重鎖可変領域遺伝子を、末梢血リンパ球や脾細胞のような抗体産生細胞より取得する方法は公知である。たとえばヒトのイムノグロブリンは、VH1〜VH7の7つのVHファミリーからなる。
各ファミリーの遺伝子を増幅することができるプライマーは公知である(Campbell, M. J., Zelenetz, A. D., Levy, S. & Levy, R. (1992). Use of family-specific primers for PCR amplification of the human heavy chain variable gene repertoire. Mol. Immunol., 29, 193-203.また、MRC社はホームページ「V-base」:http://www.mrc-cpe.cam.ac.uk/imt-doc/restricted/ok.htmlでプライマーを公開している)。したがって、このようなプライマーに基づいてRT-PCRを行えば、各ファミリー毎に重鎖可変領域遺伝子を増幅することができる。
増幅生成物として得られた重鎖可変領域遺伝子は、適当なベクターに挿入することによって遺伝子ライブラリーとすることができる。このとき、VHファミリー毎に重鎖可変領域遺伝子ライブラリーを調製し、生体内における各ファミリーの構成割合に応じて混合することによって、本発明の遺伝子ライブラリーをより生体内の抗体レパートリーに近い状態にすることができる。具体的には、たとえばヒトの場合、各ポピュレーションはおよそ次のような割合で存在することが明らかにされている。生体における抗体レパートリーを模倣することによって、スクリーニングを通じて必要なクローンを失う機会を少なくすることができる。
VH1:25%
VH2: 6.6%
VH3:40%
VH4:19%
VH5: 5%
VH6: 3.8%
VH7: 1.2%
【0052】
以下に重鎖可変領域遺伝子の取得について、より具体的に述べる。7種のVHファミリー毎にプライマーを設定し、6個のJH遺伝子に共通に働くプライマーと組み合わせてRT-PCRを行う。ヒトの各VHファミリーをファミリーごとに幅広く増幅することができるプライマーは公知である(Marks JD et al. J. Mol. Biol. (1991) 222, 581-597あるいはCampbell, M. J., Zelenetz, A. D., Levy, S. & Levy, R. (1992). Use of family-specific primers for PCR amplification of the human heavy chain variable gene repertoire. Mol. Immunol., 29, 193-203.また、MRC社はホームページ「V-base」: http://www.mrc-cpe.cam.ac.uk/imt-doc/restricted/ok.htmlでプライマーを公開している)。用いたプライマーによって各ファミリーがきちんと増幅されているかどうかを確認する。すなわち、増幅されたVHDJH構造を有するバンドについて各ファミリー毎に数10種のクローンを得て塩基配列を決定し、どの重鎖可変領域遺伝子が増幅されているかを解析する。もしも増幅されていない遺伝子が存在する場合には更に新しくプライマーを設計し、追加する。たとえば、それまでに報告されているプライマーでは増幅することができなかった遺伝子の増幅を可能とする新たなプライマーが報告されている。
in frameでVHDJH構造を持つクローンについては、適当なベクターに組み込んで大腸菌中での発現と軽鎖可変領域遺伝子との会合-folding-を解析する。もしどこかの段階が正しく起こらないクローンが存在すればその理由を推定し、ライブラリー全体の中でそのクローンの占める率を推定する。
個体のイムノグロブリンのポピュレーションは、免疫履歴によって影響を受けている。したがって、なるべく多数の個体の免疫的経歴を反映できるように、多様なB細胞から重鎖可変領域遺伝子を調製するようにする。実際には、臍帯血、扁桃、末梢血、あるいは骨髄等、入手可能な重鎖可変領域遺伝子ソースの種類を増やす。
更に、免疫原(自己の抗原も含む)と一度も接触していないnaiveなB細胞集団からB細胞を調製することも重要である。なぜならば、自己の成分と反応するクローンは免疫系の成熟時に排除されるからである。自己抗原に対する抗体レパートリーをも含む遺伝子ライブラリーとするには、nativeなB細胞は重要である。そして混合に際してはリンパ球数とクローン数が比例するように心がける。最終的には109オーダー(109〜1010)の独立したVHDJHライブラリーを作製する。
【0053】
このような操作は、次のような意義を持つ。抗体の抗原結合面を構成するアミノ酸配列は、軽鎖CDR1、CDR2、CDR3、重鎖CDR1、CDR2についてはゲノム上に存在する遺伝子によって規定されており(進化的な選別も含めて)、その多様性の総数は1万種程度である。その上に重鎖CDR3の極端に高い多様性が上積みされる。そこで抗体ライブラリーとしてはこの1万種の多様性はできる限り一様に偏り無い形で含んだ上で、重鎖CDR3(これは個々人、個々のB細胞でランダムなプロセスで作られている)についてはなるべく広範囲にライブラリー化される必要がある。上記の方法はこの要求を満たしている。
以上のような解析を通じて、本発明者らは、CDRの中にシステイン残基を含む場合には、VH遺伝子の発現が正しく起こらなかった等の知見を得た。また、作製した重鎖可変領域ライブラリーに関して、その70%以上が大腸菌中での発現と重鎖可変領域ドメインとの会合-folding-が正しく起きていることが確認できた。
【0054】
あるいは免疫履歴を逆用し、重鎖可変領域遺伝子のソースを選択することによって、あるていどライブラリーを特徴付けることができる。たとえばある感染症の既往歴を持つ場合には、その病原体に対して親和性の高いイムノグロブリンを得られる可能性が高まる。あるいはがん患者の抗体産生細胞を重鎖可変領域遺伝子のソースとすることにより、腫瘍抗原を認識するイムノグロブリンを得ることもできる。
【0055】
更に、VH遺伝子に対して人為的な変異を加えることによって、ライブラリーの多様性を高めることができる。人為的な変異を与える方法としては、error-prone PCR(Winter,G. et.al.,Annu.Rev.Immunol.,12,433-455,1994)が公知である。本発明においては、軽鎖可変領域におけるdefective遺伝子が実質的に排除されているため、重鎖可変領域遺伝子の多様性は、そのままライブラリーの多様性として反映される。したがって、公知のerror-prone PCRを適用した場合であっても、はるかに高度な多様化を達成することができる。error-prone PCRは、以下のように行うことができる。
error-prone PCRは、無作為に点突然変異を導入する方法として用いられる手法である。具体的には、PCRに使用するDNAポリメラーゼの以下の生化学的性質を用いる。
(1) Taq DNA polymeraseは通常Mg2+イオン存在下で用いられるが、Mn2+イオンの存在下では塩基の取り込みに誤りを起こしやすくなる。
(2) PCR反応時に塩基の供給源としてdATP, dCTP, dTTP, dGTPを通常は等量で混合するが、この濃度を突然変異を起こしやすいように変更する。
(3) 上述の塩基の供給源としてdITPを混合する。dITPはTaq DNA polymeraseによってDNA鎖にとりこまれると塩基イノシンを形成する。イノシンはどの塩基とも塩基対を形成しないので、PCR反応が進行するとイノシンに相補的な塩基の部分には無作為の塩基が挿入される。
以上のような3つの条件の相乗的効果により、突然変異が無作為に導入される。具体的には、たとえば7.5mM MgCl2、0.5mM MnCl2、0.2mM dATP/dGTP、1.0mM dCTP/dTTP、0.1-1.0mM dITPのような条件で反応を行う。実際には、実験の目的に合わせて更に濃度条件などを調整する。温度などの反応条件は通常のPCRと同様に設定する。
【0056】
本発明の遺伝子ライブラリーの調製には、公知のベクターを用いることができる。本発明に用いることができるベクターとしては、たとえば先に調製した軽鎖可変領域遺伝子ライブラリーを保持したファージライブラリーを示すことができる。すなわち、ファージミドが保持している軽鎖可変領域遺伝子の上流に重鎖可変領域遺伝子を挿入するのである(Gene 194 (1997) 35-46 Iba Y et al)。重鎖可変領域と軽鎖可変領域を同時に表面に発現するファージライブラリーを用いれば、抗原との結合を指標としてパニング法(後述)を行うのに有利である。すなわち、本発明の遺伝子ライブラリーをファージライブラリーとして構成することによって、rgdpライブラリーとすることができる。本発明の遺伝子ライブラリーは、ファージのみならず、リボゾームや大腸菌鞭毛タンパク質の融合タンパク質として外来遺伝子を発現するシステムを用いることによってrgdpライブラリーとすることもできる。
【0057】
代表的なrgdpライブラリーとして、ファージライブラリーが挙げられる。本発明の抗体ライブラリーに基づくファージライブラリーは、次のようにして得ることができる。ファージ表面に外来タンパク質を発現させるには、一般にファージミドとヘルパーファージが用いられる。例えば、pTZ19R(ファルマシア製)などのファージミドベクターが市販されている。ファージミドには、cp3やcp8等のファージの構成タンパク質をコードする遺伝子に、発現させたい外来タンパク質をコードする遺伝子を連結する。
ファージミドは、大腸菌などの宿主に感染させることによって増幅できる。しかしこの状態ではファージ粒子として回収することはできない。いわば、一般の遺伝子ライブラリーと同じ状態にある。ファージミドが保持する外来タンパク質を表面に提示したファージ粒子とするには、ファージミドを感染させた宿主にヘルパーファージを重感染させる。たとえばファージミドベクターpTZ19Rは、ヘルパーファージM13K07の重感染によってファージ粒子として回収することができる。このとき利用されるファージミドのcp3タンパク質が外来タンパク質と融合されていれば、完成するファージの表面には外来タンパク質が提示されることになる。
市販のファージミドに抗体の遺伝子のクローニングに好適な制限酵素サイトを導入しておけば、本発明による軽鎖可変領域遺伝子ライブラリーを重鎖可変領域遺伝子ライブラリーとともに組み込むことができる。ファージミドベクターpTZ19Rに適当な制限酵素サイトを導入し、PCRで増幅した抗体遺伝子ライブラリーを組み込む方法が公知である(Gene 194,35-46,1997)。以下に述べる実施例においては、ファージミドベクターのシグナル配列PelBの下流にSfiIサイトとAscIサイトを導入した。一方、軽鎖可変領域遺伝子の増幅には、同じサイトを導入したプライマーを用いる。PCRの増幅生成物を当該制限酵素で消化してこのサイトに組み込むことにより、PelBの下流に抗体可変領域遺伝子が挿入され、更にその下流に位置するcp3との融合タンパク質をコードするファージミドベクター(図2のpFCAH9-E8d)とすることができる。
【0058】
あるいは、重鎖可変領域遺伝子を細菌宿主発現用のベクターに挿入することもできる。この場合には、ベクターを細菌に形質転換することによって重鎖可変領域遺伝子を発現させる。得られた形質転換細胞に、更に軽鎖可変領域遺伝子を保持したファージライブラリーを感染させることにより、結果的に本発明による遺伝子ライブラリーが完成する。大腸菌の形質転換用ベクターとしては、pFK等を示すことができる。なお細菌に形質転換するには、重鎖可変領域遺伝子を適当な分泌シグナルの下流に連結することによって、重鎖可変領域タンパク質をペリプラズムに分泌させることができる。pFKは、分泌シグナルとしてpelBを備えたベクターである。
この他、動物細胞発現用のベクターに、先に調製した軽鎖可変領域遺伝子ライブラリーと、重鎖可変領域遺伝子ライブラリーを挿入することによって、本発明のライブラリーを調製することもできる。このようなベクターとしてはpcDNAI等を示すことができる。
【0059】
本発明の遺伝子ライブラリーを構成する重鎖可変領域遺伝子は、その3'末端に予め標識ペプチドをコードする塩基配列を融合しておくことができる。標識ペプチドとしては、たとえばヒスチジンタグ(His x 6)、myc-tag、あるいはHA-tag等を示すことができる。ヒスチジンタグは金属イオンとの結合活性を持つタグである。本発明のライブラリーを構成する重鎖にヒスチジンタグを融合させておくことにより、たとえば重鎖が発現しているファージをニッケルカラムに捕捉することができる。ニッケルカラムに結合できなかったファージやヘルパーファージをニッケルカラムから洗い去ることにより、重鎖が発現しているクローンを濃縮、および精製することができる。
【0060】
本発明の遺伝子ライブラリーは、様々な形態の抗体ライブラリーとすることもできる。具体的には、ファージライブラリー、大腸菌ライブラリー、あるいはリボゾームライブラリー等を示すことができる。本発明による抗体ライブラリーは、本発明によるライブラリーを構成するクローンが保持しているイムノグロブリン遺伝子を発現させ、発現生成物を回収することによって得ることができる。本発明者らは、軽鎖可変領域遺伝子と重鎖可変領域遺伝子をcp3に融合させたファージミドを感染させた大腸菌を、ヘルパーファージを加えないで長期間培養するとFab-cp3融合タンパクが菌上清に分泌されることを発見した。この現象を利用することによって、ファージミドが有する抗体遺伝子の特性を容易に試験することができる。更に各クローンについて、軽鎖可変領域と重鎖可変領域とが再構成されているかどうかを確認し、再構成に失敗しているクローンを排除することができる。この操作を行うことによって、本発明の抗体ライブラリーをより生体内のポピュレーションに近づけることができる。
本発明の抗体ライブラリーは、抗体のスクリーニングを通じて必要な可変領域遺伝子のスクリーニングを行うために用いることができる。本発明の抗体ライブラリーを利用することによって、可変領域遺伝子のライブラリーを手元に置き、抗体ライブラリーのみを供給して必要な抗体のスクリーニングを第三者に実施させることもできる。本発明は、本発明のライブラリーから、目的とする反応性を有する抗体を検出する方法に関する。抗体の反応性は、抗原との結合活性によって評価することができる。すなわち、次の工程によって目的とする抗原との結合活性を指標として、必要な抗体を検出することができる。
【0061】
a)本発明の抗体ライブラリーと、該抗原とを抗原抗体反応に適した条件下に接触させる工程
b)該抗原とイムノグロブリン可変領域との結合を検出する工程
更に本発明は、本発明のライブラリーを用いる、特定の抗原を結合するイムノグロブリン可変領域の取得方法に関する。まず本発明の抗体可変領域の検出方法は、以下の工程を含む。
また本発明による、特定の抗原と結合するイムノグロブリン可変領域の取得方法は、前記工程a)およびb)に加えて次の工程c)を含む。
c)前記抗原と結合するイムノグロブリン可変領域を発現するクローンを選択する工程
工程a)で用いるライブラリーには、軽鎖可変領域と重鎖可変領域の両方を発現可能なライブラリー、あるいはこのライブラリーの発現生成物からなる抗体ライブラリーを用いる。特に有用なライブラリーは、繊維状ファージ表面にライブラリーを構成する遺伝子によってコードされる重鎖と軽鎖の断片を発現し、かつそれらがファージ粒子表面上で機能的に再構成しているライブラリーである。このようなファージライブラリーは、rgdpライブラリーとして機能し、パニング法によって容易に必要な反応性を持つクローンを濃縮することができる。本発明に基づくパニング法は、次のように実施される。
まず、目的とする抗原にrgdpライブラリーを接触させ、この抗原に結合するクローンを回収する。回収したクローンを増幅し、再び目的の抗原と接触させて、結合するクローンを回収する工程を繰り返す。クローンの増幅は、ファージを大腸菌に感染させ、回収することによって行われる。この工程を繰り返すことによって、目的とする反応性を持つ可変領域が濃縮される。抗原との結合性に基づくスクリーニングは、一般にクローンの回収率が増加するまで行われる。ここで回収率とは、抗原に対してチャージしたクローンに数に対する、抗原への結合性を有するものとして回収されたクローンの数の割合である。回収率がその前のスクリーニングに比較して明らかに増加するとき、目的とする反応性を持つ抗体を提示したファージが濃縮されつつあることを意味する。
【0062】
この他、ライブラリーを構成するクローンの発現生成物をクローン毎に回収した抗体ライブラリーに基づくスクリーニングを行うこともできる。各クローンの発現生成物を目的とする抗原に接触させることによって、必要な反応性を持つイムノグロブリンを直接選び出すこともできる。抗原との結合が観察されたイムノグロブリンをコードするクローンを選択すれば、目的とする可変領域を得ることができる。
【0063】
本発明においては、抗体可変領域の検出や取得に用いる抗原として、抗原決定基を有するあらゆる化合物を用いることができる。幅広い蛋白質、糖、核酸、有機化合物、あるいは無機化合物が、抗原決定基を有することが知られている。これらの化合物は、生体に由来するものであっても良いし、人工的に合成されたものであってもよい。具体的には、動植物、細菌や真菌等の微生物、あるいはウイルス等の細胞や粒子、これらを構成する蛋白質、糖鎖、あるいは脂質等が抗原として用いられる。この他、非蛋白質性の、各種の薬物、ホルモン、ビタミン、サイトカイン、ケモカイン、環境汚染の原因となる化学物質等も、抗原決定基を有するものであれば、本発明に用いることができる。
【0064】
また、目的とする抗体が結合すべき抗原分子の全体を用いることもできるし、あるいは抗原分子の一部を用いることもできる。特に断らない限り、本発明における抗原は、その抗原の部分的な構造を保持した化合物を含む。たとえば、抗原分子の一部として、その抗原分子に固有の構造からなる抗原決定基を含む領域を用いることにより、特異性に優れた抗体を取得することができる。
【0065】
更に、複数の分子が結合した複合体を抗原とすることもできる。特に断らない限り、本発明における抗原は、複数の分子からなる複合体を含む。複合体を抗原とすることにより、単独の分子と複合体とを識別することができる抗体を取得することができる。
【0066】
本発明のライブラリーから、たとえば病原体に対する中和抗体として有用な抗体分子を検出し、それをコードする遺伝子を取得することができる。本発明において病原体とは、病原性を有するあらゆる生命体、あるいは生命体に由来する物質を含む。より具体的には、ウイルス、細菌、真菌、マイコプラズマ、あるいは多細胞の寄生性生命体などを示すことができる。また微生物や動植物に由来する毒素も、本発明の病原体に含まれる。したがって、コレラ菌や腸管出血性大腸菌等が産生する毒素は、本発明の病原体に含まれる。あるいは、蛇毒や蜂毒などの生物毒素もまた本発明の病原体に含まれる。他方、本発明の中和抗体とは、病原体の、病原性や感染性を抑制する働きを有する抗体を言う。
【0067】
本発明による中和抗体の検出方法は、病原体に由来する抗原に本発明のライブラリーを接触させ、この抗原に結合するイムノグロブリン可変領域を発現するクローンを検出する。更に本発明は、抗原に結合する可変領域を発現するクローンを選択することにより、中和抗体をスクリーニングする方法を提供する。
【0068】
本発明による中和抗体のスクリーニング方法においては、目的としない反応性を有する抗体可変領域を抗体ライブラリーから吸収することができる。たとえば、インフルエンザウイルスのHA抗原に対する抗体は、その感染性の抑制に有効とされている。一方、同じインフルエンザウイルス粒子に対する抗体であっても、核蛋白質(NP)に対する抗体は、ウイルスを中和する活性はない。しかし、インフルエンザウイルス粒子を抗原として用いると、しばしばNPに対する抗体も検出されるため、結果として中和抗体のスクリーニング効率を低下させる原因となってしまう。このようなときには、あらかじめ抗NP抗体を吸収しておくことにより、抗HA抗体を効率的に検出することができる。抗NP抗体は、本発明のライブラリーをインフルエンザウイルス粒子のNP抗原と抗原抗体反応が可能な条件下で接触させ、NP抗原に結合した抗体可変領域を除去することにより、吸収することができる。HA抗原に対する抗体を効率的に取得するためには、たとえばNP抗原を含まない、精製HA抗原との結合活性を指標とすることも可能である。しかし、予め抗NP抗体を吸収する工程を利用する方法は、表面構造を維持したウイルス粒子に対する抗体の反応性を確認できる点で有利である。
【0069】
このようにして得られたイムノグロブリン可変領域をコードする遺伝子、並びにその発現産物であるイムノグロブリン可変領域は本発明に含まれる。得られたイムノグロブリン可変領域は、診断や治療に有用である。特に、ヒトのイムノグロブリンである場合には、ヒトへの投与が可能である。具体的には、各種感染症、腫瘍、あるいは動脈硬化等の診断や治療に利用することができる。イムノグロブリンを用いた、診断や治療については様々なバリエーションが公知である。
本発明によって得られたイムノグロブリン可変領域を完全なヒト・イムノグロブリン分子ととすることによって、単なる親和性リガンドとしてではなく、抗体分子として用いることができる。すなわち、本発明によって得られた重鎖可変領域、ならびに軽鎖可変領域遺伝子を、定常領域をコードする遺伝子CH、およびCLと接合する。定常領域をコードする遺伝子として、IgGに由来するものを用いることにより、オプソニン作用に優れるイムノグロブリンとすることができる。
なお本明細書において引用された全ての先行技術文献は、参照として本明細書に組み入れられる。
【図面の簡単な説明】
【0070】
【図1】図1は、本発明によって得られたファージ抗体のVL遺伝子を分類した結果を示すグラフ。グラフの横軸:軽鎖のGermline(染色体)可変領域遺伝子63種類。グラフの縦軸:ファージ抗体スクリーニング時の抗原奥から、1. 破傷風毒素(TET)2. インフルエンザウイルス抗原(IFL)3. 水痘帯状疱疹ウイルス抗原(VZGH)4. ジフテリア毒素(DTD)5. 6+7の合計(total)6. 感作抗原合計(1+2+3+4)(imunized)7. 非感作抗原合計(C.elegansなど人間が一生出会わないと考えられる抗原)(no imunized)グラフの高さ:ファージ抗体の数。
【図2】図2は、本発明による可変領域ライブラリーの作製に用いた各種のベクターの構造を模式的に示す図。1)pAALFab:D1.3 mutation用ベクター。2)pFCAH3-E8T:E8発現用ベクター。pAALFabをもとに、制限酵素サイトを改変した。新たにPstI、XbaI、およびKpnIサイトを付加し、EcoRI、およびXhoIサイトの位置を変更した。3)pFvCA-E8VHd:重鎖可変領域遺伝子クローニング用ベクター。pFCAH3-E8Tをもとに、制限酵素サイトを改変した。XbaI-EcoRI間を欠落させ、新たにKpnI、SfiI、NcoI、およびSpeIサイトを付加した。重鎖可変領域遺伝子をSfiI-XhoIサイトにクローニング可能。4)pFCAH9-E8d:重鎖可変領域遺伝子クローニング用ベクター。pFCAH3-E8T、およびpFvCA-E8VHdをもとにDNA配列を改変した。マウスγCH1をヒトγCH1で置きかえた。新たに、SfiI、NcoI、およびAscIサイトを付加した。軽鎖可変領域をSfiI-AscIサイトにクローニング可能。
【図3】図3は、pFCAH9-E8dのインサートの塩基配列を示す図。
【図4】図4は、pFCAH9-E8dのインサートの制限酵素サイトと塩基配列によってコードされるアミノ酸配列を示す図(1)。
【図5】図5は、pFCAH9-E8dのインサートの制限酵素サイトと塩基配列によってコードされるアミノ酸配列を示す図(2)。
【図6】図6は、pFCAH9-E8dのインサートの制限酵素サイトと塩基配列によってコードされるアミノ酸配列を示す図(3)。
【図7】図7は、pscFvCA-E8VHdのインサートの塩基配列を示す図。
【図8】図8は、pscFvCA-E8VHdのインサートの制限酵素サイトと塩基配列によってコードされるアミノ酸配列を示す図(1)。
【図9】図9は、pscFvCA-E8VHdのインサートの制限酵素サイトと塩基配列によってコードされるアミノ酸配列を示す図(2)。
【図10】図10は、本発明による抗体ライブラリーから選択された抗体のアフィニティーの測定結果を示すグラフ。縦軸は492nmにおける吸光度を、横軸はクローンの番号を示す。横軸のPCは陽性コントロールを示す。
【図11】図11は、表15の各クローンのELISAにおけるgHあるいはgEに対する反応性を示すグラフ。
【発明を実施するための形態】
【0071】
以下、実施例に基づいて本発明を更に具体的に説明する。
【実施例】
【0072】
1. ライブラリー作製用ファージミドベクターの作製
1-1 重鎖および軽鎖の組合せライブラリーを作製するためのベクターの作製。
図2に概念的に示すように、pTZ19Rファージミドベクター(ファルマシア)にM13ファージのpelB(シグナル配列)、His6タグ配列、M13ファージのcp3タンパク質(Δcp3(198aa-406aa)N端欠失キャプシドタンパク質3)配列、およびproteinAのアミノ酸配列をコードするDNAを適当な制限酵素部位で組み込みベクターpFCAH9-E8dを作成した(GENE 194(1997)35-46 Iba Y et al参照)。軽鎖λ5, λ6遺伝子に存在するBstPIでその遺伝子が切断されることを避けるためにpFCAH9-E8dはXhoI部位が設けられている。pFCAH9-E8dのインサートの塩基配列を図3に、制限酵素サイトと塩基配列によってコードされるアミノ酸配列を図4〜図6に示した。
このベクターの所定の位置に重鎖と軽鎖の遺伝子を挿入することにより、実際の抗体タンパク質発現ベクターが完成することとなる。完成したベクターによって発現される抗体の形状はFab型であり、重鎖軽鎖はN末の可変領域に続いて定常領域CH1, CLをそれぞれ有している。定常領域同士の-SS-結合によって、重鎖と軽鎖は結合されることになる。軽鎖定常領域CL遺伝子は前述のcp3遺伝子と結合されており、結果として発現タンパク質はFab-cp3の形状となる。
【0073】
具体的には、以下のような操作を行った。
用いたプライマー:
527 Reverse(配列番号:2):
5'-CAGGAAACAGCTATGAC-3'
599 E8VHf-PstR:(配列番号:3)
3'-CGGCTCCAAGTCGACGTCGTCA-5'
544 E8VHf-PstF:(配列番号:4)
5'-CAGCTGCAGCAGTCTGGGGCAGAGCTTGTGAAGCCAGGGGCCTCAGTCAAGTTGTCCTGCACAGCTTCTGGCTTCAACATTAA-3'
545 E8VHf-XbaR:(配列番号:5)
3'-AGACCGAAGTTGTAATTTCTGTGGATATACGTGACCCACTTCGTCTCCGGACTTTTCCCAGATCTCACCTAACCTTCCTAA-5'
546 E8VHf-XbaF:(配列番号:6)
5'-AAGGGTCTAGAGTGGATTGGAAGGATTGATCCTGCGAGTGGTAATACTAAATATGACCCGAAGGACAAGGCCACTATAACAGCA-3'
547 E8VHf-EcoR(配列番号:7)
3'-TTCCTGTTCCGGTGATATTGTCGTCTGTGTAGGAGGTTGTGTCGGATGGATGTCGACTTAAGGGAC-5'
548 E8VHf-EcoF(配列番号:8)
5'-CAGCTGAATTCCCTGACATCTGAGGACACTGCCGTCTATTACTGTGCTGGT-3'
549 E8VHf-BstR(配列番号:9):
3'-CAGATAATGACACGACCAATACTAATGCCGTTGAAACTGATGACCCCGGTTCCGTGGTGCCAGTGGCACAAGG-5'
590 His6-SmaR(配列番号:10):
3'-GGTTCTCTAACAGTAGTGGTAGTAGTGGTAATTATTCTCGATAGGGCCCTCGAA-5'
542 E8VLf-SacF(配列番号:11):
5'-GACATCGAGCTCACCCAGTCTCCAGCCTCCCTTTCTGCGTCTGTGGGAGAAACTGTCACCATCACATGT-3'
539 E8VLf-KpnR(配列番号:12):
3'-TGACAGTGGTAGTGTACAGCTCGTTCACCCTTATAAGTGTTAATAAATCGTACCATGGTCGTC-5'
542 E8VLf-KpnF(配列番号:13):
5'-GCATGGTACCAGCAGAAACCAGGGAAATCTCCTCAGCTCCTGGTCTAT-3'
543 E8VLf-BamR(配列番号:14):
3'-GGAGTCGAGGACCAGATATTACGTTTTTGGAATCGTCTACCACACGGTAGTTCCAAGTCACCGTCACCTAGGCCTTGTGTT-5'
562 E8VLf-XhoR(配列番号:15):
3'-TCATGAGGCACCTGCAAGCCACCTCCGTGGTTCGAGCTCTAGTTT-5'
563 E8VLf-XhoF(配列番号:16):
5'-AGTACTCCGTGGACGTTCGGTGGAGGCACCAAGCTCGAGATCAAA-3'
613 NheR(配列番号:17):
3'-ATCGACAGCT-5'
600 E8VLKpnXhoR(配列番号:18):
3'-AAGCCACCTCCATGGTTCGAGCTCTAGTTT-5'
LCP3ASC(配列番号:19):
3'-TCGAAGTTGTCCTTACTCACAAGCCGCGCGGTCAGCTGAGGTAA-5'
hCH1Bst(配列番号:20):
5'-ACCCTGGTCACCGTCTCCTCAGCCTCCACCAAGGGCCCATCGGTCTTCCCCCTGG-3'
hCH1midAS(配列番号:21):
3'-GGGAGTCGTCGCAGCACTGGCACGGGAGGTCGTCGAA-5'
hCH1midS(配列番号:22):
5'-GGACTCTACTCCCTCAGCAGCGTCGTGACCGTGCCC-3'
hCH1H6(配列番号:23):
3'-GGGTCGTTGTGGTTCCACCTGTTCTTTCAACTCGGGTTTAGAACAGTAGTGGTAGTAGTGGTA-5'
hCH1H6Sma(配列番号:24):
3'-GGGTTTAGAACAGTAGTGGTAGTAGTGGTAATTATTCTCGATAGGGCCCTCGAACG-5'
702 BstXhoF(配列番号:25):
5'-GGCACCACGGTCACCGTCTCGAGCGCCTCCACC-3'
pFCAH3-E8T 重鎖部分の作製
1)pAALFabを鋳型にして527-599を用いたPCR, 547-590を用いたPCRを行いDNA断片を作製した。
2)544-545, 546-547, 548-549にてPCRを行いDNA断片を作製した
3)1)2)を混合し527, 590によるPCRを行い、これをpAALFabのHindIII-SmaI siteにクローニングした
pFCAH3-E8T 軽鎖部分
4)542-562, 561-613を用いたPCRを行いDNA断片を作製した
5)538-539, 542-543にてPCRを行いDNA断片を作製した
6)4)5)を混合し538, 562によるPCRを行い、これをpAALFabのSacI-NheI siteにクローニングした
pFCAH9-E8d
6)VH stuffer部分の作製
pFCAH3-E8TをXbaI,EcoRIにて消化、klenow fragmentを作用させて平滑末端に変えた後self ligationさせてVH部分のstufferを作製した。
7)VH stuffer部分の作製
pFCAH3-E8Tを鋳型にして527-600にてPCR。6)のHindIII-XhoI siteにクローニングした。
8)これをKpnIにて消化、self ligationさせてVL部分のstufferを作製
9)SfiI,NcoI,SpeI siteの導入
pFCAH3-E8Tを鋳型にして527-663にてPCR。1)のHindIII-SacI siteにクローニングした。
10)AscI siteの導入
pFCAH3-E8Tを鋳型にして527-LCP3ASCにてPCRし、それをSacI完全消化、SalI部分消化した2)にクローニングした。
11)gammaCH1部分をヒト遺伝子に変換
ヒトgammaCH1部分にはBstPI siteが存在するためこれをなくす設計でクローニングを行った。扁桃cDNAを鋳型にしてhCH1Bst-hCH1midS, hCH1midAS-hCH1H6にてPCRしたのち、これを混合してhCH1Bst-hCH16SmaにてPCRし、そのDNA断片を3)のBstPI-Sma siteにクローニングした
12)Xho siteの導入
11)を鋳型に702-663にてPCRを行い、これを11)のBstPI-SacI siteにクローニングした。
【0074】
1-2 重鎖可変領域を一時的にクローニングするためのベクターの作製
公知の手法(GENE 194(1997)35-46 Iba Y et al参照)に従って、まずpAALFabベクター(図2)を作製した。pAALFabベクターのXbaIからEcoRIの間を欠落させ、新たに制限酵素切断部位Kpn I, Sfi I, Nco I, Spe Iを付加して、pFCAH3-E8Tを経て、VH(重鎖可変領域)をクローニング可能としたベクターpscFvCA-E8VHd(図2)を作製し、重鎖可変領域を一時的にクローニングするためのベクターとした。pscFvCA-E8VHdのインサートの塩基配列を図7に、制限酵素サイトと塩基配列によってコードされるアミノ酸配列を図8〜図9に示した。
具体的には
610 scBstSpeSacF(配列番号:26):
5'-CACCACGGTCACCGTCTCCTCAGGCGGTGGCGGATCAGGTGGCGGTGGAAGTGGCGGTGGTGGGTCTACTAGTGACATCGAGCTCACCCAG-3'
611 scBstSpeSacR(配列番号:27):
3'-GTGGTGCCAGTGGCAGAGGAGTCCGCCACCGCCTAGTCCACCGCCACCTTCACCGCCACCACCCAGATGATCACTGTAGCTCGAGTGGGTC-5'
527 Reverse(配列番号:28):
5'-CAGGAAACAGCTATGAC-3'
619 E8VHf-SfiNcoPstR(配列番号:29):
3'-GACGCCGGGTCGGCCGGTACCGGCTCCAAGTCGACGTCGTCA-5'
primer610とprimer611をアニールさせ、それをpFCAH3-E8TのBstPI-SacI siteにクローニングしてsingle chainの作製を行なった。さらに、primer527とprimer619にてPCRを行い、これをさらにHindIII-PstI siteにクローニングし、SfiI,NcoI siteへ導入した。
【0075】
2. イムノグロブリン軽鎖ライブラリーの作製
2-1 PCRを用いたイムノグロブリン軽鎖遺伝子の単離
骨髄細胞(検体No.59)4×107 cells、および臍帯血と末梢血のリンパ球から、市販のキット(Pharmacia Biotech社製 QuickPrep Micro mRNA Purification Kit)を用いて、2.6μgのmRNAを得た。このmRNAからcDNAを作製した。cDNAは、GibcoBRL社製 SuperScript Preamplification Systemによって作製した。プライマーには、オリゴdTを用いた。得られたcDNAを鋳型にして、軽鎖遺伝子の取得用5'プライマー(κ1 〜κ6、λ1〜λ6 )と3'プライマー(hCKASCプライマーまたはhCLASCプライマー)を用いて、PCRを行った。PCR産物は、フェノール処理後、エタノール沈殿して10μlのTEバッファーに懸濁した。用いたプライマーの塩基配列とPCRの条件は以下のとおりである。軽鎖遺伝子取得用プライマーの塩基配列中、下線部はSfiIサイト、AscIサイトを示す。
5'-プライマーκ1〜κ6
hVK1a(配列番号:30):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC GACATCCAGATGACCCAGTCTCC
hVK2a(配列番号:31):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC GATGTTGTGATGACTCAGTCTCC
hVK3a(配列番号:32):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC GAAATTGTGTTGACGCAGTCTCC
hVK4a(配列番号:33):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC GACATCGTGATGACCCAGTCTCC
hVK5a(配列番号:34):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC GAAACGACACTCACGCAGTCTCC
hVK6a(配列番号:35):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC GAAATTGTGCTGACTCAGTCTCC
5'-プライマーλ1〜λ6
hVL1(配列番号:36):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGTCTGTGTTGACGCAGCCGCC
hVL2(配列番号:37):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGTCTGCCCTGACTCAGCCTGC
hVK3a(配列番号:38):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC TCCTATGTGCTGACTCAGCCACC
hVL3b(配列番号:39):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC TCTTCTGAGCTGACTCAGGACCC
hVL4(配列番号:40):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CACGTTATACTGACTCAACCGCC
hVL5(配列番号:41):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGGCTGTGCTCACTCAGCCGCC
hVL6(配列番号:42):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC AATTTTATGCTGACTCAGCCCCA
3'-プライマーhCKASC(配列番号:43):
TCGACTGGCGCGCCGAACACTCTCCCCTGTTGAAGCTCTTTGTG
3'-プライマーHCLASC(配列番号:44):
TCGACTGGCGCGCCGAACATTCTGTAGGGGCCACTGTCTTCTC
【0076】
PCRの条件
cDNA 2μl
10× buffer ♯1(KODに添付) 10μl
dNTP mix(2.0mM) 10μl
25mM MgCl2 4μl
5'側プライマー(100pmol/μl) 1μl
3'側プライマー(100pmol/μl) 1μl
滅菌済MilliQ 71μl
KOD DNA polymerase(東洋紡2.5U/μl) 1μl
94℃ 1分、55℃ 2分、74℃ 1分を35サイクル
【0077】
2-2 ライブラリー作製に適した軽鎖を選択して軽鎖遺伝子ライブラリーを作製する方法
2-2-1 軽鎖遺伝子のファージミドへの組込み
1で得たPCR産物を以下の条件で制限酵素処理した。
PCR産物 10μl
10×NEB4(AscIに添付) 5μl
10×BSA(SfiIに添付) 5μl
滅菌済MilliQ 28μl
AscI (NEW ENGLAND Biolabs Inc. 10 U/μl) 1μl
SfiI (NEW ENGLAND Biolabs Inc. 20 U/μl) 1μl
37℃で1時間、50℃で1時間反応後、そのうち10μl分をアガロース電気泳動し、600bp付近のバンドを切り出して、ジーンクリーンIIキット(フナコシ株式会社)で精製した。PCR産物と同様に制限酵素処理したpFCAH9-E8d(図2)をジーンクリーンIIキットで精製し、制限酵素処理したPCR産物と以下の条件で16℃で4時間〜一晩反応させることによりライゲーションした。
制限酵素処理したpFCAH9-E8d 2μl
制限酵素処理したPCR産物 1μl
10×ligation buffer 1.5μl
(T4 DNA ligaseに添付)
10mM ATP 1.5μl
滅菌済MilliQ 8μl
T4 DNA ligase (宝酒造 10 U/μl) 1μl
【0078】
2-2-2 ファージミドの大腸菌への導入
得られたligated DNAを用いて以下のように大腸菌DH12Sを形質転換した。即ち、ligated DNA を一旦エタノール沈殿し、1/5TE(TEを滅菌済MilliQで5倍希釈したもの)3μlに溶解した。そのうち、1.5μlをコンピテントセルDH12S(GIBCO BRL製)20μlに懸濁し、以下の条件でエレクトロポレーションを行った。
エレクトロポレーター
BRL社Cell-Porator(Cat.series 1600)
設定条件;voltage booster 4kΩ
capacitance 330μF
DC volts LowΩ
charge rate Fast
【0079】
2-2-3 ファージミドで形質転換した大腸菌からのFab-cp3型抗体培地中への分泌
形質転換した上記の大腸菌を形質転換用培地(SOB)2mlに植え、37℃で1時間振盪培養したあと、一部を寒天培地(Ampプレート)にまき、残りは、0.1%グルコース、100μg/mlアンピシリン含有2×TY培地で培養し、グリセリンストックした。寒天培地は30℃でincubateし、生えてきたコロニーを楊枝でつついて分離し、それぞれプラスミドを調製し、軽鎖遺伝子の塩基配列を調べた。
SOB培地:950mLの精製水に次の成分を加えて振とうし、完全に溶解した後250mMのKCl溶液10mLを加え、5N NaOHでpH7.0に調製した。精製水を加えて1000mLに調整した後、オートクレーブで20分間滅菌し、使用直前に滅菌した2MのMgCl2を5mL加えた。
bacto-tryptone 20g
bacto-yeast extract 5g
NaCl 0.5g
2×YT培地:900mLの精製水に次の成分を加えて振とうし、完全に溶解した後5N NaOHでpHを7.0に調製し、精製水を加えて1000mLとした。オートクレーブで20分間滅菌して使用した。
bacto-tryptone 16g
bacto-yeast extract 10g
NaCl 5g
その他の試薬は以下から購入した。
メーカー 品名
シグマ アンピシリンナトリウム
和光純薬 フェノール
シグマ BSA
DIFCO 2×YT培地
和光純薬 カナマイシン硫酸塩
ナカライテスク ポリエチレングリコール6000
ナカライテスク Tween20
片山化学 NaCl
和光純薬 IPTG
和光純薬 スキムミルク
和光純薬 アジ化ナトリウム
和光純薬 トリエチルアミン
和光純薬 過酸化水素
和光純薬 OPD錠
和光純薬 エタノール
【0080】
κ1、κ2、κ3、κ4、κ5、およびκ6、並びにλ1、λ2、λ3a、λ3b、λ4、λ5、λ6、λ7、λ8、λ9、およびλ10の全てについて以上の操作を行い、目的のクローンが得られているかどうか確認した。続いてκ1、κ2などの各グループのクローンをin vivoでの使用頻度に近い比率になるように混合した。これら軽鎖の各グループは、それぞれ実際の生体内でどのような割合で発現しているのかが既に知られている。PCR法で増幅してベクターに組み込んだこれらの遺伝子クローンを、in vivoでの使用頻度に近い比率になるように混合しVLライブラリーとした。VLライブラリーにおける各familyの構成比率を以下に示す。
【0081】
【表2】
【0082】
【表3】
【0083】
次に、VLライブラリーから無作為に選んだ約1000個の軽鎖遺伝子の塩基配列を確認した。すなわち、蛍光プライマーhuCH1J(5'-ATTAATAAGAGCTATCCCGG-3'/配列番号:45)を用い、サーモシークエンスキット(アマシャム・ファルマシア製)とアロカ社製L1-COR4200L(S)-2を使用したジデオキシ法によって塩基配列を決定した。得られた塩基配列を比較して重複するクローンを除いた。更にデータベースと照合し、deletionが無いと確認されたクローンについて、予め発現することがわかっている重鎖遺伝子のクローンの一つVH3-4と組み合わせて、発現実験を行った。操作は以下のとおりである。VH3-4のアミノ酸配列を配列番号:1に示した。
まずVH3-4をHindIIIとXhoIで消化し、重鎖遺伝子を切り出して、ジーンクリーンIIキットで精製した。一方、deletionが無いと確認された軽鎖遺伝子クローンについてもHindIIIとXhoIで消化し、軽鎖遺伝子を切り出して、ジーンクリーンIIキットで精製し、VH3-4の重鎖遺伝子とライゲーションすることにより、組み合わせた。得られたligated DNAを用いて大腸菌DH12Sを形質転換した。生えてきたコロニーを試験管にいれた培地にうえ、IPTGで発現を誘導することにより、Fab-cp3型の抗体分子を培養上清中に発現させた。20時間程度の培養により、ヘルパーファージの感染無しでもFab-cp3型の抗体分子が培養上清に発現される。この培養上清を用いて以下のようなELISAを行った。
【0084】
2-2-4 ELISA法による重鎖と軽鎖の正しい発現と会合の検定
1)抗体結合96wellマイクロタイタープレートの作製
抗κ抗体(MBL code No.159)を0.01Mナトリウム−リン酸緩衝液 pH8.0, 0.1% NaN3で1.25μg/mlで希釈し、100μLずつマイクロタイタープレートに添加した。4℃で一晩静置することにより、ウエルに抗κ抗体を吸着させた。反応液を捨て、5% BSA in 0.01Mナトリウム−リン酸緩衝液 pH8.0, 0.1% NaN3を200μLずつマイクロタイタープレートに添加し、37℃で2時間静置することにより、非特異的な吸着を防ぐためにブロッキングした。
次に、非特異的活性吸収済の抗λ抗体(MBL code No.159)を0.01Mナトリウムリン酸緩衝液 pH8.0, 0.1% NaN3で2.5μg/mlに希釈し、100μLずつマイクロタイタープレートに添加し氷室で一晩静置した。反応液を捨て、5% BSA in 0.01Mナトリウム−リン酸緩衝液 pH8.0, 0.1% NaN3を200μLずつマイクロタイタープレートに添加し37℃で2時間静置し、非特異的な結合を防ぐためにブロッキングを行った。
【0085】
2)1次反応
positive contorolとして、ヒトFab(10μg/ml)を、negative controlとして、PBS/0.1%NaN3をそれぞれ100μlずつマイクロタイタープレートに添加した。IPTGでFab-cp3型の抗体分子の発現を誘導した培養上清の原液を100μlずつマイクロタイタープレートに添加し37℃で1時間反応させた。
【0086】
3)2次反応
1次反応を終了したマイクロタイタープレートを0.05%Tween20-PBSで5回洗浄した。次いでPBS/0.1%NaN3で希釈した抗Fd抗体(1μg/ml)を100μlずつマイクロタイタープレートに添加し37℃で1時間反応させた。
【0087】
4)3次反応
2次反応を終了したマイクロタイタープレートを0.05%Tween20-PBSで5回洗浄した。次いでPBS/0.1%NaN3で希釈したアルカリフォスファターゼ標識抗ヒツジIgG抗体(4000倍希釈)を100μlずつマイクロタイタープレートに添加し37℃で1時間反応させた。
【0088】
5)発色反応および吸光度測定
3次反応を終了したマイクロタイタープレートを0.05%Tween20-PBSで5回洗浄した。次いで発色基質溶液(SIGMA 1040 phosphatase substrate tablets 1粒あたり5mlの50mMジエタノールアミンPH9.8に溶解したもの)を100μlずつマイクロタイタープレートに添加した。室温で反応させ、405nmの吸光度が0.5以上になったと思われる時点で、停止液を添加し、プレートリーダー(タイターテック マルチスキャンMCC)で吸光度測定した。
このELISAで陽性(吸光度0.5以上)となったクローンは、Fab-cp3型の抗体分子の発現と会合がうまく行われているとし、κ鎖遺伝子、λ鎖遺伝子それぞれ反応性の高いものから100個ずつ選択した。両者を混合してFab-cp3型の抗体分子の発現と会合がうまく行われているクローンを集めたライブラリーKL200とした。
【0089】
3. 軽鎖遺伝子ライブラリーと重鎖遺伝子ライブラリーの組み合わせライブラリーの作製
3-1-1 PCRを用いたイムノグロブリン重鎖遺伝子の単離
2-1と同様の手順を用いて臍帯血、骨髄液、および末梢血のリンパ球、並びに扁桃からhuman μ primer(以下に示すプライマーの634)あるいはrandom hexamerを用いてcDNAを調製し、このcDNAを鋳型にして、以下に示すヒト抗体重鎖遺伝子の取得用5'プライマー(VH1〜VH7)と3'プライマー(human JHプライマー4種を等量混合したもの、以下に示すプライマーの697〜700)、または、humanμプライマー(以下に示すプライマーの634)を用いて、PCRを行った。表中、下線をつけた部分はSfiIサイトを示す。hVH2aはgerm line VH2 familyに対応していないため、新たにVH2a-2を設計した。またhVH4aではVH4ファミリー全体に対応していないため、新たにhVH4a-2を設計した。VH5aもgerm line VH5 subfamilyに対応していなかったため新たにVH5a-2を設計した。またVH7に対応するprimerとしてhVH7を設計した。これらについても遺伝子増幅を行い、pscFvCA-E8VHd(0−2)に組み込み、その塩基配列を決定することによって、どのような遺伝子が増幅されたのかを確認した。hVH5a-2についてはhVH1aと配列が酷似しているため、hVH1aで増幅させたものと同様の遺伝子産物が得られることが予想されるためこれについては使用しなかった。PCR産物は、フェノール処理後、エタノール沈殿して10μLのTEバッファーに懸濁した。
【0090】
634 humμCH1R(配列番号:46):
ATGGAGTCGGGAAGGAAGTC
各VH familyの増幅に使用したprimer
Human VH primer SfiI siteを下線で示す
628 hVH1a(配列番号:47):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGGTGCAGCTGGTGCAGTCTGG
629 hVH2a(配列番号:48):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGGTCAACTTAAGGGAGTCTGG
630 hVH3a(配列番号:49):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC GAGGTGCAGCTGGTGGAGTCTGG
631 hVH4a(配列番号:50):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGGTGCAGCTGCAGGAGTCGGG
632 hVH5a(配列番号:51):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGGTGCAGCTGTTGCAGTCTGC
633 hVH6a(配列番号:52):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGGTACAGCTGCAGCAGTCAGG
629-2 hVH2a-2(配列番号:53):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGRTCACCTTGAAGGAGTCTGGTCC
631-2 hVH4a-2(配列番号:54):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGGTGCAGCTACAGCAGTGGGG
632-2 hVH5a-2(配列番号:55):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC GAGGTGCAGCTGGTGCAGTCTGG
712 hVH7(配列番号:56):
GTCCTCGCAACTGCGGCCCAGCCGGCCATGGCC CAGGTGCAGCTGGTGCAATCTGGGTCTGAGT
Human JH primer BstPI, XhoI siteを下線で示す
697 hJH1-2(配列番号:57):
GGTGGAGGCACTCGAGACGGTGACCAGGGTGC
698 hJH3(配列番号:58):
GGTGGAGGCACTCGAGACGGTGACCATTGTCC
699 hJH4-5(配列番号:59):
GGTGGAGGCACTCGAGACGGTGACCAGGGTTC
700 hJH6(配列番号:60):
GGTGGAGGCACTCGAGACGGTGACCGTGGTCC
【0091】
cDNA 2μL
10× buffer ♯1(KODに添付) 10μL
dNTP mix(2.0mM) 10μL
25mM MgCl2 4μL
5'側プライマー(100pmol/μL) 1μL
3'側プライマー(100pmol/μL) 1μL
滅菌済MilliQ 71μL
KOD DNA polymerase(東洋紡2.5U/μL) 1μL
PCR条件:94℃ 1分、55℃ 2分、74℃ 1分を35サイクル
【0092】
3-1-2 重鎖遺伝子ライブラリーの作製
3-1-1で得たPCR産物を以下の条件で制限酵素処理した。
PCR産物 10μL
10×K buffer(宝酒造) 5μL
滅菌済MilliQ 33μL
HindIII (宝酒造15 U/μL) 1μL
XhoI (宝酒造12 U/μL) 1μL
37℃で2時間反応後、そのうち10μL分をアガロース電気泳動し、400 bp付近のバンドを切り出して、ジーンクリーンIIキット(フナコシ株式会社)で精製した。PCR産物と同様に制限酵素処理したpscFvCA-E8VHd(図2)をジーンクリーンIIキットで精製し、制限酵素処理したPCR産物と以下の条件で16℃で4時間〜一晩反応させることによりライゲーションした。
制限酵素処理したpscFvCA-E8VHd 2μl
制限酵素処理したPCR産物 1μl
10×ligation buffer 1.5μl
(T4 DNA ligaseに添付)
10mM ATP 1.5μl
滅菌済MilliQ 8μl
T4 DNA ligase (宝酒造 10 U/μl) 1μl
【0093】
3-1-3 ファージミドの大腸菌への導入
得られたDNAを大腸菌DH12Sに形質転換した。具体的にはDNAを一旦エタノール沈殿し、1/5TE(TEを滅菌済MilliQで5倍希釈したもの)3μLに溶解する。そのうち、1.5μLをコンピテントセルDH12S(GIBCO BRL製)20μLに懸濁し、エレクトロポレーション法により形質転換した。
エレクトロポレーター
BRL社Cell-Porator(Cat.series 1600)
設定条件;voltage booster 4kΩ
capacitance 330μF
DC volts LowΩ
charge rate Fast
【0094】
形質転換用培地(SOB)2mlに上記操作の終了した形質転換大腸菌を植え、37℃で1時間振盪培養したあと、一部を寒天培地(Ampプレート)にまき、残りは、0.1%グルコース、100μg/mlアンピシリン含有2×YT培地で培養し、グリセリンストックした。寒天培地は30℃でインキュベートし、生えてきたコロニーを楊枝でつついて分離し、それぞれプラスミドを調製し、重鎖遺伝子の塩基配列を調べた。VH1〜VH7の全てについてこれらのことを行い、目的のクローンが得られているかどうか確認した。これらの各グループ(ファミリー)のクローンをin vivoでの使用頻度に近い比率になるように混合してVHライブラリーとした。VHライブラリーにおける各ファミリーの構成比率を以下に示す。
【0095】
【表4】
【0096】
3-2 組み合わせ遺伝子ライブラリーの作製
VHライブラリー200μgを下記条件でHindIIIとXhoIで消化し、重鎖遺伝子を切り出して、ジーンクリーンIIキットで精製した。
VHライブラリー200μg 100μL
10×K buffer(宝酒造) 40μL
滅菌済MilliQ 205μL
HindIII (宝酒造40 U/μL) 30μL
XhoI (宝酒造50 U/μL) 25μL
deletionが無いと確認された軽鎖遺伝子クローンKL200、およびVLライブラリーの挿入されたベクターpFCAH9-E8dについても下記条件でHindIIIとXhoIで消化し、軽鎖遺伝子を含む断片を、ジーンクリーンIIキットで精製した。
KL200またはVLライブラリー
を挿入したpFCAH9-E8d 100μg 100μL
10×K buffer(宝酒造) 40μL
滅菌済Milli-Q 230μL
HindIII (宝酒造40 U/μL) 15μL
XhoI (宝酒造50 U/μL) 15μL
【0097】
次に、VH遺伝子ライブラリー断片と軽鎖遺伝子の挿入されたpFCAH9-E8dベクターを、次の条件下、16℃で一晩反応させてライゲーションした。
制限酵素処理した
VHライブラリー断片 10μg 50μL
制限酵素処理したKL200または
VLライブラリーの断片
を含むpFCAH9-E8d 40μg 50μL
10×ligation buffer
(T4 DNA ligaseに添付) 100μL
10mM ATP 100μL
滅菌済MilliQ 670μL
T4 DNA ligase (宝酒造 10 U/μL) 30μL
【0098】
反応の終了したDNAを用いて大腸菌DH12Sを形質転換した。具体的にはDNAを一旦エタノール沈殿し、1/5TE(TEを滅菌済MilliQで5倍希釈したもの)30μLに溶解した。これをコンピテントセルDH12S(GIBCO BRL製)500μLに懸濁し、エレクトロポレーションを行った。
エレクトロポレーター
BRL社Cell-Porator(Cat.series 1600)
設定条件;voltage booster 4kΩ
capacitance 330μF
DC volts LowΩ
charge rate Fast
【0099】
形質転換用培地(SOB)12mlに上記操作の終了した大腸菌を植え、37℃で1時間振盪培養したあと、一部を寒天培地(Ampプレート)にまき、残りは、0.1%グルコース、100μg/mlアンピシリン含有2×YT培地500 mlで培養し、グリセリンストックした。寒天培地は30℃でインキュベートし、生えてきたコロニーの数から得られたクローンの数を推定した。それぞれ5×1010クローンが得られた。
扁桃mRNAよりrandam hexamerにて合成したcDNAをもとに得た各VH familyをpscFvCA-E8VHdベクターにクローニングし、KL200と組み合わせたライブラリーをAIMS1とした。(1.28×1010の独立したクローン)
臍帯血、骨髄液、末梢血、扁桃mRNAよりhuman m primerにて合成したcDNAをもとに得た各VH familyをpscFvCA-E8VHdベクターにクローニングし、KL200と組み合わせた遺伝子ライブラリーをAIMS2とした. (3.20×1010の独立したクローン)
臍帯血、骨髄液、末梢血、扁桃mRNAよりhuman m primerにて合成したcDNAをもとに得た各VH familyをVL libraryと組み合わせたライブラリーをAIMS3とした。(4.50×1010の独立したクローン)
更に(AIMS1+AIMS2) : AIMS3 = 1 : 1で混合し、1×1011の独立したクローンからなるファージ抗体ライブラリーとした(AIMS4と呼ぶ)。
【0100】
3-3 組み合わせ遺伝子ライブラリーによるファージライブラリーの作製
3-3-1 ファージライブラリーの調製
1%グルコース及び100μg/mLのアンピシリンを加えた2×YT培地300mLを入れた5リットルのフラスコにAIMS4懸濁液を2.5mLを加え、37℃で振とう培養し1時間おきに波長600nmにおける吸光度を測定しながら、吸光度が1.0になるまで増殖させた。培養液にヘルパーファージ液(M13KO7)をフラスコ当たり12mL加えてヘルパーファージを感染させ、37℃で2時間培養し、ヘルパーファージ感染済みDH12Sとした。
3リットルのフラスコ24本に2×YT培地600mLと100μg/mLのアンピシリン1.2mL、50μg/mLのカナマイシン0.8mL、ヘルパーファージ感染済みDH12S 200mLを加えて37℃で2時間毎に波長600nmの吸光度を測定しながら振とう培養した。アンピシリンは、測定毎に200μg/mLとなるよう追加した。培養は波長600nmにおける吸光度が1.3になるまで行った。
菌体は4℃で8000rpm、10分間遠心し、上清を集めた。上清に20%のポリエチレングリコール/2.5M NaCl 4Lを加えて約20分間静かに攪拌した後、4℃で8000rpm、20分間遠心、沈殿を1LのPBSで溶かし、20%のポリエチレングリコール/2.5M NaCl 200mLを加えて約20分間静かに攪拌した後、4℃で8000rpm、20分間遠心した。上清を捨ててさらに4℃で8000rpm、3分間遠心して沈殿を回収した。沈殿は0.05% NaN3を加えたPBSで溶解し、4℃で1000rpm、15分間遠心し、上清を回収した後、4℃で8000rpm、3分間さらに遠心して上清を回収した。
【0101】
回収したファージ溶液の力価は以下のようにチェックした。すなわち、ファージ溶液をPBSで106、107、108希釈し、その10μLをDH12S 990μLに感染させ、37℃で1時間培養した。これをLBGAプレートに100μL播いて30℃で18時間培養した。コロニーの数をカウントすることにより希釈前の原液の力価を算出した。ファージ溶液原液を2%スキムミルク及び0.05% NaN3を含むPBSに2×1014/mlになるよう懸濁した。
【0102】
3-3-2 Fab-cp3を表面に発現しているファージを濃縮する方法
以上のようにして調製したライブラリーには、Fab-cp3をその表面に発現しているファージを選択的に濃縮し、ヘルパーファージやFab-cp3の発現していないファージの混入割合を減少させるための工夫が施されている。すなわち上記ライブラリーを構成するファージが発現する重鎖のC末端には、His6ペプチド(ヒスチジンタグ)が付加されている。ヒスチジンタグを発現したファージは、ニッケルイオンなどに吸着することを利用して容易に回収することができる。具体的にはニッケルイオンが付加されたゲル(Ni-NTAアガロース等)を用いる。操作は以下のとおりである。
Ni-NTAアガロースを2%スキムミルク及び0.1%Tween20を含むPBS(以下ブロッキング緩衝液)で室温で30分ブロッキングした。次に、重鎖にHis-Tagが付加されていないFabを表面に発現しているファージ(pFCA-E9HLφ;ファージHis-)と重鎖にHis-Tagが付加されているFabを表面に発現しているファージ(pFCAH6-D1.3HLφ;ファージHis+)をファージHis-:ファージHis+ = 100:1となるようにブロッキング緩衝液中で混合した。これらの合計1×1010CFUのファージ溶液250μLをNi-NTAアガロースと混合し、室温1時間反応させた。Ni-NTAアガロースをブロッキング緩衝液で洗浄し、次いで0.5Mイミダゾール(pH 7.55)500μLを加えてNi-NTAアガロースに結合していたファージを溶出した。
溶出したファージを後(4-3)で述べる操作にしたがって回収し、回収したクローンを調べてみたところ、23クローン中15クローンがファージHis+であった(表3)。このことはNi-NTAアガロースにより、His6ペプチドが付加されたファージが53倍に濃縮されたことを示している。
この操作を加えることにより、ライブラリーの性能を向上させ、あるいはスクリーニングの能率を上昇させることが可能であることが以上(4)によって示された。
【0103】
【表5】
【0104】
4. ファージライブラリーからの特定の抗原に特異的に結合するファージの選択
ファージライブラリーから特定の抗原に特異的に結合するファージを選択する操作を、本明細書ではスクリーニングと呼ぶ。抗原として破傷風トキソイドを用い、本発明による抗体ライブラリーのスクリーニングを行った。
【0105】
4-1 スクリーニング用試験管の作製
抗原(破傷風トキソイド)をPBSで20μg/mLに調製し、試験管3本(Nunc社製Maxisorp)に3mLずつ添加して4℃で18時間インキュベートして、試験管内表面へ抗原を吸着させた。吸着後、抗原溶液を捨て、2%スキムミルク含有PBS溶液3mLずつを加えて25℃で1時間反応させ、ファージ抗体が非特異的に試験管に結合することを防ぐためにブロッキングを行った。
【0106】
4-2 スクリーニング操作
作製した抗原吸着済試験管にAIMS4ファージライブラリーを2%スキムミルク、0.1%Tween20含有PBSになるよう溶解して1×1014CFU/9mLに調製し、この液を試験管3本に3mLずつ添加して25℃で2時間反応させた後、0.1%Tween20を加えたPBSで4回、PBSで4回、および滅菌した超純水(Milli-Qにて作製)で1回洗浄した。
続いて抗原結合試験管に結合したファージを以下のように回収した。すなわち、0.1Mトリエチルアミン(pH12.3)を試験管1本当たり3ml添加し、ローテーターを用いて室温で20分間反応させ乖離させた後、1M Tris-HCl緩衝液(pH6.8)1.1mLを加えて中和し、この液を回収した。
【0107】
4-3 回収したファージの増幅
回収した液は(ファージの大腸菌への感染)(ヘルパーファージの感染)(ファージの回収)の処理を行い、含まれているファージを精製・増幅した。
1) ファージの大腸菌への感染
大腸菌(DH12S)を2×YT培地50mlで培養し、波長600nmの吸光度が0.5になるよう増殖させ、上記で回収したファージ液を加えて37℃で1時間振とう培養した。
2) ヘルパーファージの感染
1)の培養液62.3mLをとり、2×YT培地425mL、40%グルコース12.5ml、および100μg/mLアンピシリン0.5mLを加えて37℃で波長600nmにおける吸光度が0.5になるまで培養した後、4℃、5000rpmで10分間遠心して菌体を沈殿させ、回収して100mg/mLアンピシリン0.3mLを加えた2×YT培地300mLに懸濁した。これにヘルパーファージM13K07を1/100量加え、37℃で1時間振とう培養した。
培養液を予め37℃に暖めた培地(2×YT培地に100μg/mLアンピシリンと70μg/mLのカナマイシンを加えた液)900mLに加えて37℃で一晩培養した。
3) ファージの回収
2)の培養液を4℃で7000rpm、10分間遠心し、その上清に2.5Mの塩化ナトリウムを加えた20%のポリエチレングリコールを1/5量加えて室温で20分間静置した後、4℃で8000rpm、15分間遠心して沈殿を回収し、培養液の1/10量の滅菌PBSを加えて溶解し、再度2.5Mの塩化ナトリウムを加えた20%のポリエチレングリコールを1/5量加えて4℃で10000rpm、20分間遠心して上清を捨て、さらにスピンダウンして4℃で10000rpm、2分間遠心した。これに0.05%のNaN3を加えたPBSを培養液の1/100量加えて沈殿を溶解し、ファージを回収した。
【0108】
4-4 増幅したファージによる再スクリーニング
増幅したファージを用いて4-2と同様に抗原結合試験管を用いてスクリーニングを繰り返した。スクリーニングでの洗浄は、非特異に吸着したファージを乖離し、結合力の高いファージを選択する上で重要なステップであることから、2回目以降のスクリーニングにおける洗浄条件は以下のようにした。
2回目;PBS+0.1% Tween20で6回、PBSで6回、滅菌した超純水で1回
3回目;PBS+0.1% Tween20で13回、PBSで13回、滅菌した超純水で1回
【0109】
4-5 ファージのスクリーニング手技の評価法
4-4の方法でスクリーニングを繰り返すとき、(抗原吸着済試験管に投入したファージの総数)÷(抗原吸着済試験管から回収したファージの総数)が前回のスクリーニングに比べて明らかに小さくなれば、目的の抗体を提示しているファージが濃縮されつつあると推測できる。溶液に含まれるファージ数は、以下のように計算した。
1)ファージの希釈系列を以下のように作製した。
[1] 1×10-2希釈:ファージ液10μL+PBS990μL
[2] 1×10-4希釈:[1]の希釈液10μL+PBS990μL
[3] 1×10-6希釈:[2]の希釈液10μL+PBS990μL
[4] 1×10-8希釈:[3]の希釈液10μL+PBS990μL
[5] 1×10-9希釈:[4]の希釈液100μL+PBS900μL
[6] 1×10-10希釈:[5]の希釈液100μL+PBS900μL
[4]、[5]、および[6]の希釈系列10μLにDH12S 990μLを加えたものを作り、37℃で1時間感染させ、これをLBGAプレートに100μLまいて30℃で18〜24時間培養し、コロニーを計数した。上記希釈系列のうち、通常[4]のプレートが50個以上のコロニーを作る。mL当たりのファージ数は[4]のプレートのコロニー数に基づいて、以下のように算出される。
原液のファージ数=(コロニー数/プレート)×(1×108)×103 cfu/mL
【0110】
また、回収されたファージ数も同様に計算し、本抗原に対する抗体を提示するファージの数をスクリーニングごとに求めたところ、表6のようになった。
【0111】
【表6】
【0112】
4-6 スクリーニングによって得られた抗体の抗原結合活性(アフィニティー)の測定
以上のスクリーニングによって選択された抗体について、抗原結合活性(アフィニティー)を測定した。アフィニティーの測定には、ファージ型の抗体ではなく、Fab-cp3型抗体をサンプルとして用いた。測定方法は、96ウェルマイクロタイタープレートを用いたELISA法とした。Fab-cp3型抗体の発現誘導法については4-7で述べる。
まずELISA用のプレートを以下のように調製した。抗原20μg/mLを96ウェルマイクロタイタープレート(Nunc社製Maxisorp)の各ウエルに100μL添加して4℃で18時間結合させたのち、5%BSA(ブロッキング液)を各ウエルに200μL添加して37℃で1時間ブロッキングした。ブロッキング液を捨てた後、PBSで1回洗浄してアフィニティーの測定に用いた。4-7の手順で採取した培養上清を各ウエルに100μL加え、25℃1時間反応させた。反応後、PBSで4回洗浄し、5000倍希釈したペルオキシダーゼ標識抗ヒトIgG((株)医学生物学研究所製)を100μL加えて25℃で1時間反応させた。再度PBSで4回洗浄し、オルトフェニレンジアミンと過酸化水素の溶液100μLを加えて暫時反応させた後、1.5Nリン酸100μLを加えて反応を停止し、波長492nmにおける吸光度を測定した。その結果96クローン中77クローンに活性が確認された(図10)
更にこれら77クローンのうち、37クローンについて中和活性を調べた。表7の○が、中和活性を調べたクローンである。中和活性を調べた段階では、まだ塩基配列を確認していなかったので、これらのクローンの中には同じDNA配列の組み合わせであるクローンが存在する可能性があった。これらのクローンの塩基配列を決定して照合し、同じ配列の組み合わせを持つものは表7では同じ枠内にまとめた。あとで照合してみると同じDNA配列の組み合わせを持つクローンはELISAにおいても同程度のアフィニティーを示していたことが確認された。
【0113】
4-7 Fab-cp3型抗体の発現誘導
ファージの感染した大腸菌を1%グルコースと100μg/mLのアンピシリンを加えた2×YTで30℃18時間培養した後、0.1%グルコースと100μg/mLのアンピシリンを加えた2×YT 1.5mLに上記培養液を5μL加えて30℃で4時間培養した。このときの大腸菌の濃度は波長600nmの吸光度を測定するとき、約0.5であった。
これに1mMになるようIPTG(イソプロピル-1-チオ-β-D-ガラクトシド)を加えてさらに30℃で18時間培養した後、培養液1.5mLをエッペンドルフチューブにとり、4℃で10000r.p.m.5分間遠心してその培養上清をとり、0.1%となるようアジ化ナトリウムを添加して検体とした。
Fab-cp3型抗体が発現しているか否かは2-3で示したELISA法で確認した。発現が認められたクローンのうち、アガロース電気泳動によってdeletionを確認したところ、8クローンにdeletionが認められたが、これらはいずれも抗原結合活性が認められなかった。
【0114】
4-8 モノクローナル化とファージミドの精製
4-5「ファージのスクリーニングの評価」の項で得たシングルコロニーのプレートから大腸菌クローンを選択し、LBGA中で30℃18時間培養した後、倉敷紡績社製DNA分離装置PI-50 File No.50を用いてファージミドを精製した。
【0115】
4-9 モノクローナル抗体の同定
本発明の抗体ライブラリーからスクリーニングによって選択された抗破傷風毒素抗体について、その遺伝子の配列を確認した。配列は、重鎖、軽鎖について以下のプライマーを用い、サーモシークエンスキット(アマシャム・ファルマシア製)とアロカ社製L1-COR4200L(S)-2を使用したジデオキシ法によって決定した。結果を表7にまとめた。表7では、塩基配列決定の結果、同じクローンであることが判明したクローンをまとめて示した。表中のH-chainに示した番号とL-chainに示した番号の組み合わせが同一となっているクローンが、同じ塩基配列を含むクローンである。表7に示すように、36種類のクローンが得られていたことがわかった。
重鎖のシークエンシング用プライマー:蛍光T7プライマー(アロカ製)
軽鎖のシークエンシング用プライマー:蛍光プライマーhuCH1J(配列番号:45)
【0116】
【表7】
【0117】
4-10 中和活性検定用 Fab-cp3型抗体の採取
本発明の抗体ライブラリーからスクリーニングによって選択された抗体の、破傷風毒素に対する中和活性の有無を検討した。まず抗体活性のあった77クローンについて、発現前の培養液と、1% グルコース, 100mg/mLアンピシリンを加えた2×YT培地(YTA)で30℃で一晩培養した後、0.1% グルコース YTAで30℃3時間培養したのち、1M IPTGを加え、30℃で20時間培養した。10000rpmで5分間遠心して上清をとり、60%になるよう硫安を加えて1時間振とうした。さらに12000rpmで10分間遠心して上清を除き、沈殿にPBSを加えて溶解後、50倍量以上のPBSに対して2時間透析した。これを3回繰り返した後、エッペンドルフチューブに移して15000rpmで4℃、10分間遠心し、上清をろ過した後、破傷風毒素に対する中和活性を検討した。
【0118】
4-11 中和活性の検討
まず、抗体自体の毒性を確認するため、上記で得た抗体溶液0.2mLをマウスの腹腔と尾静脈内に注射した。その結果、抗体溶液には毒性は認められなかった。次に、抗体注射後約1時間で、マウス最小致死量(1MLDまたは約2LD50)の10倍量の破傷風毒素を後肢内股部皮下に注射した。その結果、各抗体を注射したマウスは約半数において部分的中和能(毒素による発症時期の遅延、4〜7日)が認められた。中和抗体(標準破傷風抗毒素)または中和能のない抗体を注射した マウスは、破傷風毒素による特異的麻痺が観察された後、約2日で死亡した。
部分的中和活性を示したクローンは、TETM1, TETM13, TETM26, TETM96であった。したがって、本発明の抗体ライブラリーをスクリーニングすることによって、中和活性を持つ有効な抗体を得られることが証明されたと言える。
【0119】
4-12 抗体可変領域の配列決定
上記のとおり、本発明者らは破傷風トキソイド(毒素)に結合し、これを中和する抗体可変領域を発現するファージを得ることができた。
さらに、上記工程を繰り返すことにより、中和活性を持つが互いに異なる塩基配列を持つ別のファージクローンを得ることもできた。具体的にはクローン番号TETM13とTETK36が強い中和活性を持つと判断された。これらファージクローンが有する重鎖と軽鎖の塩基配列、およびこの塩基配列によってコードされるアミノ酸配列を以下の配列番号として記載する。
クローン番号 重鎖 軽鎖
TETM13(塩基配列) 63 64
TETM13(アミノ酸配列) 81 82
TETK36(塩基配列) 65 66
TETK36(アミノ酸配列) 83 84
【0120】
5. 選択をかけない軽鎖遺伝子ライブラリーと、KL200ライブラリーの比較
選択をかけない軽鎖遺伝子ライブラリーを使用して作製した重鎖との組合せライブラリー(AIMS3)と、選択した軽鎖遺伝子ライブラリーKL200を使用して作製した重鎖との組合せライブラリー(AIMS1+AIMS2)とでライブラリーの性能を比較した。
まず選択されたクローンの数の比較においては、本発明による抗体ライブラリーであるAIMS1+AIMS2が、圧倒的に有利であることが明らかとなった。選択をかけた軽鎖遺伝子ライブラリーであるAIMS1+AIMS2から選択されたクローンは、前記表7における、KL200のカラムにKL200におけるクローンの名称が記載された29クローンである。一方、AIMS3からは7クローン(前記表7における、KL200のカラムがVL Libraryとなっているクローン)が選択された。つまり、等量で混合したライブラリーを用いたのにもかかわらず選択されたクローンの80%以上(29/36)が本発明によるライブラリーに由来するクローンであった。このように、スクリーニングで得られたファージ数の比較によりKL200を用いて作製したファージライブラリーが有利であることを示すことができた。
【0121】
5-2 スクリーニングによって得られたファージの抗体活性の比較
次に、選択された抗体の活性について比較した。4-11で述べた中和活性の試験において部分的中和能が確認されたクローンは、次の4クローンである。
TETM1 (AIMS3)
TETM13 (AIMS1+AIMS2)
TETM26 (AIMS1+AIMS2)
TETM96 (AIMS1+AIMS2)
つまり、AIMS1+AIMS2から3クローン、AIMS3から1クローンが選択されていた。この割合は、スクリーニングで得られたクローンの数の割合(29:7)とほぼ同様である。しかも、本発明によるライブラリーに由来するAIMS1+AIMS2から選択された3クローンには、最も中和活性の強いクローンであるTETM13が含まれていた。したがって、抗体の機能の面からみても、選択された軽鎖ライブラリーKL200を用いて作製したファージライブラリーのスクリーニングによって優れた抗体を効率的に選択できることが確認できた。
【0122】
6. ファージライブラリーからの特定の抗原に特異的に結合するファージの選択−2
抗原としてジフテリアトキソイドを用い、本発明による抗体ライブラリーから、抗体のスクリーニングを試みた。スクリーニングに用いたライブラリーは、4と同じAIMS4ファージライブラリーである。このライブラリーは、AIMS3(選択をかけない軽鎖遺伝子ライブラリーと重鎖遺伝子ライブラリーを組み合わせたライブラリー)と、AIMS1+AIMS2(選択をかけた軽鎖遺伝子ライブラリーと重鎖遺伝子ライブラリーを組み合わせたライブラリー)の混合液からなっている。
【0123】
6-1 スクリーニング用試験管の作製
抗原(ジフテリアトキソイド)をPBSで20μg/mLに調製し、試験管3本(Nunc社製Maxisorp)に3mLずつ添加して4℃で18時間感作した。抗原感作後、抗原溶液を捨て、2%スキムミルク含有PBS溶液3mLずつを加えて25℃で1時間反応させ、ブロッキングした。
【0124】
6-2 スクリーニング操作
作製した抗原結合試験管に、4と同じファージライブラリーを2%スキムミルク、0.1%Tween20含有PBSになるよう溶解して1×1014CFU/9mLに調製し、この液を試験管3本に3mLずつ添加して25℃で2時間反応させた後、0.1%Tween20を加えたPBSで4回、PBSで4回及び滅菌した超純水(MilliQにて作製)で1回洗浄した。
続いて抗原結合試験管に結合したファージを以下のように回収した。すなわち、0.1Mトリエチルアミン(pH12.3)を試験管1本当たり3mL添加し、ローテーターを用いて室温で20分間反応させ乖離させた後、1M Tris-HCl緩衝液(pH6.8)1.1mLを加えて中和し、この液を回収した。
【0125】
6-3 回収したファージの増幅
回収した液は実施例4-3および4-4と同じ(ファージの大腸菌への感染)(ヘルパーファージの感染)(ファージの回収)の操作を繰り返した。洗浄条件も実施例4-4の(スクリーニング)と同様に行った。
【0126】
6-4 ファージのスクリーニング手法の評価
ここで得られた発現の認められたクローンについて、軽鎖の塩基配列を決定してその由来を確認したところ、20クローンは(AIMS1+AIMS2)由来であったのに対し、(AIMS4)からは16クローンが選択された。
さらに、これらのクローンの中に実際に使用可能な活性の高いクローンが含まれているか否かについて検討した。ジフテリア毒素に対する抗体の中和活性は、培養細胞を用いたCell Culture Method(CCM)によって定量した。CCM法は以下のように行った。
標準抗毒素の溶液は、国内標準ジフテリア抗毒素Lot.9の1アンプルを0.2w/v%ゼラチン加PBSで溶解し、更に3%cs培地(精製水1,000mLにイーグルMEM9.4g、L-グルタミン0.3g、ペニシリン20万単位/2mL、ストレプトマイシン0.1g力価/0.5mL、グルコース3.0g、子牛血清30mL、1%フェノールレッド3mL、及び7w/v%重炭酸ソーダ20mLを含む)で10IU/mLに調製し、使用した。
【0127】
試験毒素の溶液はジフテリア試験毒素(Lot.M59)1バイアル(Lf/vial =2.5 、CD50/vial=1.6×105)を0.2w/v%ゼラチン加PBSで溶解し、3%cs培地で1.6×104CD50/mL)に調製した後、更に3%cs培地で希釈して用いた。細胞は7% cs培地(精製水1,000mLにイーグルMEM9.4g、L-グルタミン0.3g、ペニシリン20万単位/2mL、ストレプトマイシン0.1g力価/0.5mL、子牛血清70mL、1%フェノールレッド3mL、及び7w/v%重炭酸ソーダ20mLを含む)で継代培養したVERO細胞を用いた。
96穴マイクロプレート中に3%cs培地で標準抗毒素、被験抗体の希釈系列(各25μL)を作製し、これに調製した上記ジフテリア毒素(25μL)を加え37℃30分処理した後、3% cs培地で細胞数を3×105cells/mLに調製したVERO細胞50μLと、さらに3%cs培地100μLを加えてシールし、37℃のインキュベーターで4日間培養した。pH指示薬の色調の変化(オレンジを目安とする)から、本測定系における標準抗毒素のエンドポイントを求め、被験抗体のエンドポイントを示した希釈倍数をエンドポイントの抗毒素価に乗じて、被験抗体の抗毒素価とした。本測定系における標準抗毒素のエンドポイントは0.0046IU/mLであった。
その結果、CCM法では試験した43クローン中1クローンが0.0520IU/mLと、ジフテリアトキソイド(毒素)に対して中和活性を示した。しかし、AIMS3(選択をかけない軽鎖遺伝子用いたライブラリー)からは活性のあるクローンは得られなかった。これらの結果から、本発明に基づく抗体ライブラリーAIMS1+AIMS2は、生体内の抗体の多様性を高度に再現した優れたライブラリーを与えることが明らかである。
【0128】
本発明者らはジフテリア毒素に結合し、これを中和する能力をもつファージクローン(DTD10)も得ることができた。これらファージクローンが有する重鎖と軽鎖の塩基配列、およびこの塩基配列によってコードされるアミノ酸配列を以下の配列番号として記載する。
DTD10の重鎖 塩基配列/配列番号:61、アミノ酸配列/配列番号:79
DTD10の軽鎖塩基配列/配列番号:62、アミノ酸配列/配列番号:80
【0129】
7. ファージライブラリーからの特定の抗原に特異的に結合するファージの選択−3
抗原としてインフルエンザウイルス由来の抗原を用い、本発明による抗体ライブラリーから、抗体のスクリーニングを試みた。これ以降インフルエンザウイルス株名を以下のように省略して表記する。ただし、( )内はウイルスの亜型を示す。
ウイルス株名称 省略名称
A/New Caledonia/20/99株(H1N1) ニューカレドニア株(H1N1)
A/Sydney/5/97株(H3N2) シドニー株(H3N2)
A/Okuda/57株(H2N2) 奥田株(H2N2)
A/北京/262/95株(H1N1) 北京株(H1N1)
【0130】
7-1 インフルエンザウイルス由来抗原の精製
当業者には衆知の方法を用いて以下のようにインフルエンザウイルス抗原の精製を行なった。国立感染症研究所から購入したワクチン株を孵化鶏卵に接種し、33〜35℃で2日間培養した。培養後、4℃の冷蔵室で1晩放置し、常法に従い感染尿膜腔液を採取した。次いで、限外ろ過法・通常の生化学的方法などで濃縮し、ショ糖密度勾配遠心法で精製した。すなわち、0−60%のショ糖の密度勾配中で濃縮ウイルスを回転数35000rpmで超遠心し、ショ糖密度が40%前後の画分を採取した。このウイルス精製画分をエーテルで処理した(エーテル不活化ウイルス)。こののちにホルマリンを添加しショ糖密度勾配遠心法でさらに精製してインフルエンザ精製抗原を得た。
【0131】
7-2 北京株(H1N1)を用いたファージ抗体の単離
7-2-1 スクリーニング用試験管の作製
【0132】
北京株(H1N1)から7-1の手順で抽出したインフルエンザウイルス抗原をPBS中で12.5μg/mlの濃度で溶かし、イムノチューブ(Polysorp)に4.5ml入れて4℃18時間あるいは25℃2時間、穏やかに転倒混和してインフルエンザウイルス抗原をイムノチューブ内表面に結合させた。インフルエンザウイルス抗原溶液を除いたのち、2%の濃度でPBSに解かしたスキムミルクを4.5mlチューブに入れ、4℃で18時間あるいは25℃で1時間反応させ非特異的反応を防ぐブロッキング処理を行なった。
【0133】
7-2-2 スクリーニング操作
上記のようにして作製した抗原結合済みのイムノチューブに2%スキムミルクin PBSに懸濁した実施例4と同じAIMS4ファージライブラリー4.5mLを入れ、25℃で2時間転倒混和して反応させ、チューブ上に結合したインフルエンザウイルス抗原とAIMS4ファージライブラリーを接触させた。
上記の接触が終わったのち、ファージ懸濁液を除き、以下の表8に示す緩衝液で表に示す回数洗浄し、チューブに結合しているファージ以外のファージを取り除いた。
【0134】
【表8】
次に0.1MトリエチルアミンpH12.3を4.5mlチューブに添加し、20分室温で転倒混和して、ファージをチューブから乖離させて別の新たなチューブに移し、直ちに1M Tris−HCl pH6.8を1.1ml加えて中和した。
【0135】
7-2-3 回収したファージの増幅
大腸菌DH12S(2xYT培地)50ml(2ndスクリーニング以降は30mL)をOD600nm= 0.5になるように予め増やしておき、上記の過程で得たファージ液を加え、抗原結合ファージを37℃1時間振とう培養し感染させた。
前述の大腸菌液を全体で500mlになるよう以下の如く希釈した。(3Lフラスコ使用)
大腸菌液 61.25ml
2xYT 425ml
40%グルコース 12.5ml(終濃度1%)
100mg/ml Ampicillin 0.5ml(終濃度100μg/ml)
上記の希釈済み大腸菌液をさらに37℃1〜2時間培養したのち、4℃に移し終夜保存した。
終夜保存したフラスコを再び、37℃に戻し振盪培養した。OD600nm=0.5になったら、5000 rpm 10分、4℃にて滅菌遠心管を用いて遠心し、菌体を回収した。
回収した菌体に2xYT培地1〜2mlを加え溶かしたのち、一部をとって、グリセロール・ストックを作製した。(最終グリセロール濃度20%)
残りの菌体には2xYT培地300mL(ただし、3rd, 4thスクリーニングの場合はそれぞれ150mL, 50mL)に終濃度100μg/mlのアンピシリンを加え培養した。OD600nm=0.5に到達したとき、さらにヘルパーファージM13K07を培養液の1/100量加え、37℃で1時間振盪培養し、ヘルパーファージを感染させた。
上記の培養液の一部を取り、2×TY寒天培地上に播き、ヘルパーファージの感染した大腸菌数の計測に供した。残りの培養液には新たな培養液(100μg/mlのAmpicillinを含有し、70μg/mlのKanamycineを含有した900ml 2×YT培地、37℃にあらかじめ保温しておく)を加え、37℃で終夜振盪培養することで、ヘルパーファージが感染した大腸菌を選択した。
終夜培養後、培養液を7000r.p.m 10分4℃(滅菌ずみ遠心管1000ml用を使う)で遠心分離して菌体を沈殿させ、上清を回収した。上清に1/5体積の2.5M NaCl含有20%ポリエチレングリコール溶液を加え、20分室温で静置したのち、8000 r.p.m 15分4℃で遠心分離して沈殿させる事によってファージを回収した。
沈殿を崩さないように上清を除き、培養液の1/10体積の滅菌PBSで沈殿を溶かした。さらに、2.5M NaCl含有20%ポリエチレングリコールをPBSの1/5体積加え、10000r.p.m 20分4℃(滅菌ずみ遠心管200ml用を使う)で遠心してファージを沈殿させることにより不純物を取り除いた。沈殿したファージに0.05% NaN3 PBSを培養液の1/100加え、シェイカーを使用して、約2時間振盪することにより懸濁した。
以上の工程によって得られたファージは、さらに同様の2回のスクリーニング工程に供した。しかし、この方法ではインフルエンザウイルスのNPというタンパク質に反応する抗体しか得られず、インフルエンザウイルスを中和する能力は無かった。
【0136】
7-3 ニューカレドニア株(H1N1)を用いた亜型特異的に反応するファージ抗体の単離
インフルエンザを中和する抗体のエピトープはウイルス表面のHAタンパク質にあると言われており、また、HAタンパク質は各ウイルス株間で変異の大きい箇所でもある。そこで、ウイルス株に特異的に反応する抗体を単離することで中和抗体を得ることを試みた。
【0137】
7-3-1 抗NP抗体吸収済ライブラリーの準備
インフルエンザウイルス奥田株(H2N2)を用いて7-1の手順に従いエーテル処理不活化ウイルスを作製した。不活化ウイルス 20μg/mlをPBSに溶解し、この溶液を反応用チューブ3ml/本、3本に添加し、4℃18時間反応させた。反応後液を捨て、2%スキムミルクin PBSで1時間反応させた。AIMS4ライブラリー LOT000614/2%スキムミルクin PBSを1x1014CFU/9mlを上記3本のtubeに加えた。室温2時間25℃攪拌しながら反応させた。この反応終了後のライブラリーを抗NP抗体吸収済AIMS4ライブラリーとした。
【0138】
7-3-2 目的抗原ニューカレドニア株(H1N1)由来抗原を用いたスクリーニング操作
上記と同様に作製したニューカレドニア株(H1N1)ホルマリン処理不活化ウイルスを100μg/mlになるようにPBSに溶解した。この溶液を反応用チューブ3ml/本、3本に添加し、4℃18時間反応させた。反応後、液をすて、非特異的反応を防止するため2%スキムミルクin PBSを加え1時間インキュベートした。保温終了後、7-3-2で作製した抗NP抗体・吸収済AIMS4ライブラリーを上記3本のチューブに加えた。2時間25℃にて攪拌しながら反応させた。反応後PBSで洗浄した。洗浄回数は以下の表9を参照。
洗浄後、0.1MトリエチルアミンpH12.3を3ml/tube加え、室温20分、ローテーターで攪拌しながら反応させファージを抗原と乖離し、1M Tris−HCl pH6.8を1ml加えて中和した(抗原結合ファージの回収)。大腸菌DH12S(2xYT培地)50mlをOD600nm 0.5〜1.0になるように予め増やしておき、抗原結合ファージを37℃1時間振とう培養し、感染させた。
7-2-3と同様に回収したファージの増幅工程を行ない、増幅後抗NP抗体を7-3-1と同様に吸収し、ファージ液の力価を以下のように測定した。力価を算出した後、同様のスクリーニング過程に増幅ファージ液を用いた。スクリーニング操作は3回繰返した。
【0139】
7-3-3 ファージの力価測定
次回のスクリーニングに用いるファージ液の力価をあらかじめ以下の要領で調べた。
希釈系列を下記のように作製した。
1. 1x10−2希釈 :ファージ液10μl+ PBS 990μl
2. 1x10−4希釈 :1の希釈液10μl+ PBS 990μl
3. 1x10−6希釈 :2の希釈液10μl+ PBS 990μl
4. 1x10−8希釈 :3の希釈液10μl+ PBS 990μl
5. 1x10−10希釈 :4の希釈液10μl+ PBS 990μl
6. 1x10−12希釈 :5の希釈液10μl+ PBS 990μl
4,5,6の希釈系列10μlにDH12S990μlを加えたものをつくり、37℃1時間感染させ、これをLBGAプレートに100μlまき30℃18〜24時間培養後コロニーをカウントした。
4,5,6のうち、50個/プレート以上コロニーをつくったプレートをデータとして採用した。
次回のスクリーニングに用いるファージ力価の計算式(4.の場合)
=(コロニー数/プレート)x(1x108)x103 ファージ/ml
【0140】
【表9】
【0141】
7-4 H3N2亜型特異的中和抗体の単離方法
7-4-1 目的抗原シドニー株(H3N2)由来抗原を用いたスクリーニング
インフルエンザウイルスシドニー株(H3N2)・ホルマリン処理不活化ウイルス100μg/mlをPBSに溶解し、この溶液をNunc Polysorp 4.5ml/本、3本に添加し、4℃ 18時間反応させた。反応後溶液を捨て、2%スキムミルクin PBSで1時間ブロッキングした。その後、7-3-1で作製した抗NP抗体・吸収済AIMS4ライブラリーを上記3本のtubeに加え、室温2時間25℃ローテーターを用いて振盪反応させた。
反応後PBSで以下の表10に示す回数洗浄した。
洗浄後、0.1MトリエチルアミンpH12.3を3ml/tube加え、室温20分、ローテーターで攪拌しながら反応させファージを抗原と乖離した。1M Tris−HCl pH6.8を1ml加えて中和した(抗原結合ファージの回収)。
大腸菌DH12S(2xYT培地)50mlをOD600nm 0.5〜1.0になるように予め増やしておき、抗原結合ファージを37℃1時間振とう培養し、感染させた。
7-2-3と同様に回収したファージの増幅工程を行ない、増幅後抗NP抗体を7-3-1と同様に吸収し、ファージ液の力価を7-3-3と同様に測定した(表11)。力価を算出した後、同様のスクリーニング過程に増幅ファージ液を用いた。スクリーニング操作は3回繰返した。
【0142】
【表10】
【0143】
7-5 インフルエンザウイルスを中和するファージ抗体の評価
7-5-1 Fab型抗体の誘導
ファージの感染した大腸菌を発現誘導した後長期培養することにより、ファージではなく、Fab型の抗体が大腸菌から培養液中に分泌されることを本発明者らは見出した。
上記の工程7-2-3、7-3-3、および7-4-1から得られたファージを含む大腸菌のコロニーから大腸菌を2xYT+0.1%グルコース+100μg/ml Ampicillinに植菌して30℃で培養し、OD600nmが0.5付近になったところでIPTGを最終濃度1mMとなるよう添加し、30℃ 18時間培養することによりファージ抗体を誘導した。
また、別にIPTGを入れないで18時間培養したものを準備し、終濃度30%のグリセロールを加えて保存用大腸菌を作製した。
【0144】
7-5-2 ELISA法によるFab型抗体の特性の確認
Fab型抗体を発現させた7-5-1の培養液を1.5mlチューブにいれ、10000rpm. 4℃5分遠心した。培養上清をとり、終濃度0.1%のNaN3を加えELISAサンプルとした。
次に、スクリーニングに用いた際に用いた濃度と同じ濃度の抗原を96well ELISAプレートに100μl/well加え4℃18時間保温することにより抗原をプレートに結合した。その後、2.5%BSA in PBSを200μl/well加え、4℃18時間保温することによりブロッキングした。
ELISA試験実施前に、ブロッキング液をすて、PBSで一回洗った。7-5-1で誘導した培養上清を100μl/well加え、25℃1時間反応させた。終了後、PBSで4回ウェルを洗浄し、POD結合抗ヒトIgG(MBL社Cat#206 LOT150)を5000倍希釈し100μl/well加え、25℃1時間(または、37℃1時間、室温250rpm. 振盪しながら30分)反応させた。終了後、PBSで4回洗浄し、基質溶液を100μl/well加えた。基質溶液は以下のように作製した:0.1Mクエン酸−リン酸水素2ナトリウムpH5.1に終濃度0.01% H2O212mlを加え、さらにOPD錠(和光純薬社、生化学用Code# 158-01671)を1錠加えた。15〜30分後OD492nmをプレートリーダーによって吸光度を測定した。
その結果、ELISAにおいてニューカレドニア株(H1N1)から1クローン、シドニー株(H3N2)から3クローンの株特異的な反応性を示す中和抗体が得られたことが分かった。これらの4クローンの反応性を以下の表12に示した。
【0145】
【表11】
【0146】
【表12】
【0147】
7-5-3 プラスミドの精製とシークエンシング
2xYT培地(0.1%グルコース+200μg/ml Ampicillin含有)で一晩培養し、DNA分離装置(倉敷紡績社 PI-50)にて操作法に従って、プラスミドを精製した。精製したプラスミドには最終濃度20μg/mlのRNAseを添加してフェノール・クロロホルム処理し、-20℃以下にて保存した。
得られたファージ抗体が有している抗体遺伝子はシークエンシングを行なう事によって解読した。シークエンシングはDNAシークエンサ(アロカ社 L1−COR4200L(S)−2)を用い、試薬はサーモシークエンスキット(アマシャム・ファルマシアUS78500)を使用し、ダイデオキシ法にておこなった。
重鎖、軽鎖のシークエンスプライマーは以下のものを使用した。
重鎖:T7Promoter IDR700ダイラベルプライマー 50μl
5'TAATACgACTCACTATAggg3'(20mer)(配列番号:97)
軽鎖:カスタム・プライマー 3’huCH1J
IDR800ダイラベルプライマー 50μl
5'ATTAATAAgAgCTATCCCgg3'(20mer)(配列番号:98)
遺伝子の塩基配列解読の結果、配列の欠失が生じていないものの数は表11に示した。また、株特異的なクローンの遺伝子およびアミノ酸配列については、配列表に示した。
【0148】
7-5-4 インフルエンザウイルス中和試験
MDCK細胞を96ウェル平底プレート(コーニング社cat#3596)の各ウェルに(約104 cells/well)分注し、CO2インキュベータにてウェルの底にモノレイヤーシートを形成するように37℃で培養した。次の日0.2% BSA(fraction V, Sigma Chemical Co.)を含むMEM培地により96well丸底プレートの各ウェル内に試験する各抗体を4倍希釈して入れた。0.2% BSAを含むMEM培地により希釈したインフルエンザウイルス液(4×104 FFU/mL)25μlと、希釈した抗体あるいはコントロールの溶液25μlを混合し、37℃60min反応させた。
それらの混合液を先に述べた96穴プレート中のMDCK細胞に25μl添加し、37℃60min.保温することにより、ウイルスを細胞に吸着させた。
吸着後PBSで洗浄し100μL/wellの0.5% tragacanth gumと5μg/mlのトリプシンを含むMEM培地を添加し、さらに37℃24時間培養した。
培養後、PBSでウェルを洗浄した後、100%エタノールをウェルに添加し、室温10分処理し、ウェルをヘアドライヤーで乾燥させた。PAP染色後Focusを計数した。
抗体の中和能の強さは、何も入れない場合(陽性コントロール)からのフォーカスの減少率で表わされる。
以下の表13に示すようにFab型抗体クローン番号NC1、SY39およびSY47は有為なインフルエンザウイルスに対する中和活性を示した。
【0149】
【表13】
【0150】
7-5-5 評価の結論
予測した通りにウイルス株に特異的なファージクローンはインフルエンザウイルスを中和する高い活性を有していた。すなわちニューカレドニア株(H1N1)に対するクローンNC1またはシドニー株(H3N2)に対するクローンSY39、およびクローンSY47である。これらファージクローンが有する重鎖と軽鎖の塩基配列、およびこの塩基配列によってコードされるアミノ酸配列を以下の配列番号として記載する。
クローン番号 重鎖 軽鎖
NC1(塩基配列) 67 68
NC1(アミノ酸配列) 85 86
SY39(塩基配列) 69 70
SY39(アミノ酸配列) 87 88
SY47(塩基配列) 71 72
SY47(アミノ酸配列) 89 90
これらの可変領域を実際に医療用に使用するにあたっては、抗体遺伝子が得られている特性を生かし、完全イムノグロブリン型への変換や酵素の結合なども比較的簡単に実施することができると考えられる。また、突然変異を人工的に起こすことによってより活性を上昇させることができると期待される。
【0151】
8. ファージライブラリーからの特定の抗原に特異的に結合するファージの選択−4
抗原として水痘帯状疱疹ウイルス(VZV)由来の抗原を用い、本発明による抗体ライブラリーから、抗体のスクリーニングを試みた。
8-1 VZVに反応性を持つファージ抗体の単離
8-1-1 スクリーニング用試験管の作製
公的機関で入手可能であるVZVの感染したヒト胎児肺細胞(HEL)をPBS中で超音波処理によって破砕し、Shiraki et al (1982) (Shiraki K, Takahashi M J Gen Virol 1982, 61 : 271-5) に従いgEとgHタンパク質を調製し、そのうちgHタンパク質を25μg/mlの濃度に調整しVZV抗原溶液とした。VZV抗原溶液をイムノチューブ(Maxisorp)に4.5ml入れて4℃18時間あるいは25℃2時間、穏やかに転倒混和してVZV抗原をイムノチューブ内表面に結合させた。VZV抗原溶液を除いたのち、2%の濃度でPBSに解かしたスキムミルクを4.5mlチューブに入れ、4℃で18時間あるいは25℃で1時間反応させ非特異的反応を防ぐブロッキング処理を行なった。
【0152】
8-1-2 スクリーニング操作
上記のようにして作製した抗原結合済みのイムノチューブに2%スキムミルクin PBSに懸濁したAIMS4ファージライブラリー4.5mLを入れ、25℃で2時間転倒混和して反応させ、チューブ上に結合したVZV抗原とAIMS4ファージライブラリーを接触させた。
上記の接触が終わったのち、ファージ懸濁液を除き、以下の表に示す緩衝液で表14に示す回数洗浄し(洗浄とは緩衝液をチューブに入れ転倒混和した後、排出することを指す)、チューブに結合しているファージ以外のファージを取り除いた。
【0153】
【表14】
次に0.1MトリエチルアミンpH12.3を4.5mlチューブに添加し、20分室温で転倒混和して、ファージをチューブから乖離させて別の新たなチューブに移し、直ちに1M Tris−HCl pH6.8を1.1ml加えて中和した。
7-2-3と同様に回収したファージの増幅工程を行ない、増幅後回収したファージ液の力価を7-3-3と同様に測定した。
以上のスクリーニングを3回繰返した。
【0154】
8-2 結合活性を大腸菌の培養上清を用いてELISA法により確認する工程
7-5-2と同様にELISA法を用いて結合活性の確認を行なった。VZV抗原をPBS中で12.5μg/mlの濃度で溶かし100μl/well 96wellプレートの各ウェルに添加し、4℃18時間保温し、96ウェルプレートに抗原を結合させた。次に、2.5%BSAを200μl/well加え、4℃18時間保温することによりブロッキングした。
ブロッキングの終了した96wellプレートのブロッキング液をすて、PBSで一回洗ったのち、8-5-2と同様の手順で調製したファージが感染した大腸菌の培養上清を100μl/well加え、25℃1時間で反応させた。これ以降の過程は8-5-2と同様に実施した。
上記の表に示すように、3回目のスクリーニング操作にて回収率(output)の上昇が見られたため、12クローンについてELISAを行ったところ活性の強い1クローンを得た。そのまま、4回目のスクリーニングを行って48クローンにつきELISAを行ったところ、活性は弱いが陽性であった。
【0155】
8-3 gEとgHの反応性の比較とシークエンシング
糖鎖に対する抗体はウイルスの他の部分にも反応し、特異性が低く、中和活性も低いことが予測された。そこで、豊富な糖蛋白を持つgEに対するELISAも実施し、gEに高い反応性を持つ抗体は検討から除くこととした。
この基準により、gHの活性が高い29クローン(表15)を選択し、7-5-3と同様に塩基配列を解析(シークエンシング)した。シークエンシングによって16種類の塩基配列が確認された。それぞれ代表的なクローンにつき培養上清をとり、飽和硫安による濃縮、透析の後、中和活性の試験を実施した。
【0156】
【表15】
【0157】
8-4 得られた抗体のVZVウイルス中和試験
8-4-1 試験に用いるcell-free水痘帯状疱疹ウイルス(VZV岡株)の作製
ヒト胎児肺細胞(HEL)の培養細胞の約50%に感染細胞特有の変化(CPE:cytopathic effect)を認めたときに,EDTA(0.04−0.1%)を含むPBS(トリプシンは使用しない)によって細胞を回収し、低速遠心により細胞を集め,SPGC保存液(5% sucrose,0.1%,Na−グルタミン酸含有滅菌PBSに牛胎児血清を10%添加したもの)に懸濁した。
超音波または,凍結融解+超音波にて,細胞を破砕して細胞内のウイルスを遊離させ、3000 rpm,10分の遠心上清をウイルス液とし、-85℃で保存した。一部をHEL細胞にて感染力価を測定した。すなわち、ウイルス液をSPGC保存液で適度に希釈し、HEL細胞に0.2mlをあらかじめ用意された6cmシャーレのHEL細胞に振り掛けることにより接種した。HEL表面が乾かないように,ウイルス液が全体に均一になるように何度も細胞表面をウイルス液で覆うように液を往復させながら1時間感染させた。感染後培養液を入れて4−6日間培養し、その後培溶液を除き、定法に従って5%ホルマリンで細胞を固定し,メチレンブルー染色し,実体顕微鏡下でプラック数を測定した。単位溶液あたりのプラック数を(PFU/ml)として感染力のあるウイルスの指標(力価)とした。
【0158】
8-4-2 中和能試験
上記で作製したウイルス液をSPGC保存液で希釈して,100PFU/0.1mlとした。希釈したウイルス液とSPGCで10倍希釈した抗体溶液を0.3mlずつ混合し、37度で1時間反応させた。その中から,0.2mlをあらかじめ用意された6cmシャーレのHEL細胞に振り掛けることにより接種した。HEL表面が乾かないように,ウイルス液が全体に均一になるように何度も細胞表面をウイルス液で覆うように液を往復させながら1時間感染させた。
感染後培養液を5ml入れて4−6日間培養した。培養後、培養液を除き、定法に従って5%ホルマリンで細胞を固定し,メチレンブルー染色し,実体顕微鏡下でプラック数を測定した。
抗体なし(PBSのみ)で形成されるプラック数を対照として、抗体によってどれだけプラック数が減少できるかによって、抗体のウイルス中和活性を測定した。以下の表16のように、No.10, 24, 94に強い中和活性が見られた。
【0159】
【表16】
【0160】
8-4-3 中和試験の結論
本発明により、プラック形成を非常に高い効率で抑制したファージクローンが3つ得られた。すなわち、VZ10とVZ24とVZ94である。これらファージクローンが有する重鎖と軽鎖の塩基配列、およびこの塩基配列によってコードされるアミノ酸配列を以下の配列番号として記載する。
クローン番号 重鎖 軽鎖
VZ10(塩基配列) 73 74
VZ10(アミノ酸配列) 91 92
VZ24(塩基配列) 75 76
VZ24(アミノ酸配列) 93 94
VZ94(塩基配列) 77 78
VZ94(アミノ酸配列) 95 96
これらは、異なる塩基配列をもち、実際に医療用に使用する場合には、その中和活性が相乗的に作用する可能性がある。実際の使用にあたっては、抗体遺伝子が得られている特性を生かし、定常領域を備えた完全なイムノグロブリン分子への変換や酵素の結合なども比較的簡単に実施することができる。
【産業上の利用可能性】
【0161】
本発明によって、機能的なコンフォーメーションを維持した抗体分子を高い割合で含む抗体ライブラリー、あるいはそれをコードする遺伝子のライブラリーの提供が可能となった。本発明よる遺伝子ライブラリーは、まず重鎖可変領域と機能的なコンフォーメーションを再構成することができる軽鎖可変領域をコードする遺伝子の選択に始まる。こうして選択された軽鎖可変領域遺伝子に、重鎖可変領域遺伝子を組み合わせることによって構成された本発明よる遺伝子ライブラリーからは、機能的な抗体からなる抗体レパートリーの再現が可能である。機能的な抗体分子の選択工程を経て生成される本発明の抗体ライブラリーは、生体内における抗体レパートリーをin vitroで再現したものと言うこともできる。
【0162】
本発明による遺伝子ライブラリーは、機能的な抗体を高い割合で含む抗体ライブラリーを生成することができる。このような抗体ライブラリーを用いれば、機能的な抗体を効率的にスクリーニングすることができる。公知の抗体ライブラリーが、単に多様性の追求にとどまっていたのに対して、本発明はライブラリーを構成する抗体の質に着目し、その質を高める手段を確立した。この点において、本発明はin vitroにおける抗体ライブラリーの構築と、抗体のスクリーニング方法に、まったく新たな視点を与える。
【0163】
本発明によって得られる抗体ライブラリーにおいては、重鎖の多様性を十分に生かすことができる。この利点は、単に生体内の抗体の多様性をin vitroで維持できるということにとどまらない。本発明に基づけば、たとえばerror-prone PCRのような人為的な変異の導入方法のメリットを最大限に生かすことができる。すなわち本発明においては、変異を与えた重鎖の多様性は、理論的には完全に抗体の多様性として再構成される。なぜならば、重鎖との機能的な再構成が可能な軽鎖が選択されているからである。公知のライブラリーにおいては、軽鎖が機能的な抗体分子を再構成できないものである場合には、有意義な重鎖の変異が抗体活性の発現に至らず、スクリーニングすることができないのと対称的である。
【特許請求の範囲】
【請求項1】
以下の工程を含む、イムノグロブリンの軽鎖可変領域遺伝子と重鎖可変領域遺伝子の組み合わせからなる遺伝子ライブラリーの調製方法。
a)軽鎖可変領域遺伝子として、重鎖可変領域遺伝子の発現産物との機能的なコンフォーメーションの再構成が可能な軽鎖分子をコードするものを選択し、
b)工程a)によって得られる軽鎖可変領域遺伝子の集合である遺伝子のライブラリーを調製し、
c)工程b)のライブラリーに重鎖可変領域をコードする遺伝子のライブラリーを組み合わせる
【請求項2】
工程c)において、VHファミリーごとにそれぞれ独立して調製した重鎖可変領域遺伝子のライブラリーを、生体内におけるVHファミリーの割合に応じて組み合わせる請求項1に記載の方法。
【請求項3】
工程c)における遺伝子ライブラリーの組み合わせを同一のベクター上で行う請求項1に記載の方法。
【請求項4】
更に次の工程d)を含む、請求項1に記載の方法。
d)重鎖可変領域に標識ペプチドが融合しており、この標識ペプチドを指標として重鎖を発現するクローンを選択する工程
【請求項5】
請求項1に記載の方法によって得ることができる遺伝子ライブラリー。
【請求項6】
少なくともイムノグロブリンの軽鎖可変領域をコードする遺伝子からなる遺伝子ライブラリーであって、イムノグロブリンの重鎖可変領域と機能的なコンフォーメーションを再構成できない軽鎖可変領域をコードする遺伝子が実質的に排除されている遺伝子ライブラリー。
【請求項7】
ライブラリーが重鎖可変領域をコードする遺伝子のライブラリーを伴っている請求項6に記載のライブラリー。
【請求項8】
VHファミリーのそれぞれの重鎖可変領域遺伝子のライブラリーが、生体内の多様性を包含するのに十分なクローン数を有する請求項7に記載のライブラリー。
【請求項9】
ライブラリーを構成する遺伝子が、細菌、酵母、および植物細胞からなる群から選択されるいずれかの宿主細胞に導入されたものである請求項6に記載のライブラリー。
【請求項10】
ライブラリーを構成する遺伝子が、哺乳動物細胞に導入されたものである請求項6に記載のライブラリー。
【請求項11】
ライブラリーを構成する少なくとも一部の遺伝子が、繊維状ファージに組み込まれている請求項6に記載のライブラリー。
【請求項12】
ライブラリーを構成する遺伝子によってコードされる重鎖可変領域と軽鎖可変領域の断片を繊維状ファージ表面に発現し、かつそれらが機能的に再構成されている請求項11に記載のライブラリー。
【請求項13】
重鎖可変領域に標識ペプチドをコードする遺伝子が融合されている請求項12に記載のライブラリー。
【請求項14】
イムノグロブリン軽鎖可変領域遺伝子がヒトに由来するものである請求項6に記載のライブラリー。
【請求項15】
イムノグロブリン軽鎖超可変部位がシステイン残基を含まないアミノ酸配列からなる請求項14に記載のライブラリー。
【請求項16】
請求項5に記載の遺伝子ライブラリーを構成する各クローンが、そのクローンによって保持される遺伝子によってコードされる抗体タンパク質を伴っているrgdpライブラリー。
【請求項17】
請求項5に記載の遺伝子ライブラリーを構成する各クローンについて、そのクローンによって保持される遺伝子によってコードされる抗体タンパク質から構成される抗体ライブラリー。
【請求項18】
次の工程を含む、重鎖可変領域と機能的なコンフォーメーションを再構成することができる軽鎖可変領域をコードする遺伝子の選択方法。
a)軽鎖可変領域をコードする、1つまたは複数の遺伝子を取得する工程、
b)軽鎖可変領域と機能的なコンフォーメーションを構成することが確認されているイムノグロブリン重鎖可変領域をコードする遺伝子を取得する工程、
c)工程a)によって得られた軽鎖可変領域をコードする遺伝子の任意の1つを選択し、工程b)で取得した重鎖可変領域をコードする遺伝子とイムノグロブリンの機能的なコンフォーメーションを再構成が可能な条件下でタンパク質に翻訳する工程、
d)工程c)において翻訳されたタンパク質の抗原結合領域の形成を検出する工程、および
e)抗原結合領域の形成が検出されたタンパク質を構成する軽鎖可変領域をコードする遺伝子を選択する工程
【請求項19】
更に、次の工程f)を含む請求項18に記載の方法。
f)軽鎖可変領域遺伝子の塩基配列を決定し、その塩基配列によってコードされるアミノ酸配列を比較して同一のアミノ酸配列をコードする遺伝子、およびアミノ酸の欠損を生じている遺伝子を排除する工程
【請求項20】
イムノグロブリンの軽鎖可変領域が、繊維状ファージの表面に発現している請求項18に記載の方法。
【請求項21】
イムノグロブリンの軽鎖可変領域をコードする遺伝子と重鎖可変領域をコードする遺伝子が挿入されたファージミドを感染させた宿主微生物の培養上清そのものを、工程c)のための軽鎖を含む試料として利用する請求項18に記載の方法。
【請求項22】
次の工程を含む、特定の抗原と結合するイムノグロブリン可変領域の検出方法。
a)請求項5または請求項7に記載のライブラリー、またはその発現産物と、該抗原とを抗原抗体反応に適した条件下に接触させる工程
b)該抗原とイムノグロブリン可変領域との結合を検出する工程
【請求項23】
ライブラリーが請求項12に記載のライブラリーである請求項22に記載の方法。
【請求項24】
請求項22に記載の方法の後、更に次の工程c)を行うことを特徴とする、特定の抗原と結合するイムノグロブリン可変領域の取得方法。
c)前記抗原と結合するイムノグロブリン可変領域を発現するクローンを選択する工程
【請求項25】
更に次の工程d)−e)を含む請求項24に記載の方法。
d)工程c)で選択したファージクローンを増幅して2次的なライブラリーを得る工程、
e)工程c)において選択されるクローンの回収率が上昇するまで、2次的なライブラリーについて、工程a)−d)を繰り返す工程
【請求項26】
請求項24に記載の方法によって得られたクローン、またはイムノグロブリン断片、もしくはそれをコードする遺伝子。
【請求項27】
配列番号:61〜配列番号:78のいずれかに記載の塩基配列を含む、ポリヌクレオチド。
【請求項28】
配列番号:79〜配列番号:96のいずれかに記載のアミノ酸配列を含む蛋白質。
【請求項29】
次の要素からなる抗体ライブラリーの調製用キット。
a)重鎖可変領域と機能的なコンフォーメーションを再構成することができない軽鎖可変領域をコードする遺伝子を実質的に排除した軽鎖可変領域遺伝子ライブラリー、および
b)重鎖可変領域をコードする遺伝子を増幅することができるプライマーセット
【請求項30】
以下の工程からなる抗体ライブラリーの調製方法。
a)ファージミドに、イムノグロブリンの少なくとも可変領域を含む領域をコードする遺伝子を組み込む工程
b)工程a)で得られたファージミドを宿主微生物に感染させる工程
c)ヘルパーファージを感染させること無く、工程b)の宿主微生物の培養上清を回収して抗体ライブラリーとする工程
【請求項1】
以下の工程を含む、イムノグロブリンの軽鎖可変領域遺伝子と重鎖可変領域遺伝子の組み合わせからなる遺伝子ライブラリーの調製方法。
a)軽鎖可変領域遺伝子として、重鎖可変領域遺伝子の発現産物との機能的なコンフォーメーションの再構成が可能な軽鎖分子をコードするものを選択し、
b)工程a)によって得られる軽鎖可変領域遺伝子の集合である遺伝子のライブラリーを調製し、
c)工程b)のライブラリーに重鎖可変領域をコードする遺伝子のライブラリーを組み合わせる
【請求項2】
工程c)において、VHファミリーごとにそれぞれ独立して調製した重鎖可変領域遺伝子のライブラリーを、生体内におけるVHファミリーの割合に応じて組み合わせる請求項1に記載の方法。
【請求項3】
工程c)における遺伝子ライブラリーの組み合わせを同一のベクター上で行う請求項1に記載の方法。
【請求項4】
更に次の工程d)を含む、請求項1に記載の方法。
d)重鎖可変領域に標識ペプチドが融合しており、この標識ペプチドを指標として重鎖を発現するクローンを選択する工程
【請求項5】
請求項1に記載の方法によって得ることができる遺伝子ライブラリー。
【請求項6】
少なくともイムノグロブリンの軽鎖可変領域をコードする遺伝子からなる遺伝子ライブラリーであって、イムノグロブリンの重鎖可変領域と機能的なコンフォーメーションを再構成できない軽鎖可変領域をコードする遺伝子が実質的に排除されている遺伝子ライブラリー。
【請求項7】
ライブラリーが重鎖可変領域をコードする遺伝子のライブラリーを伴っている請求項6に記載のライブラリー。
【請求項8】
VHファミリーのそれぞれの重鎖可変領域遺伝子のライブラリーが、生体内の多様性を包含するのに十分なクローン数を有する請求項7に記載のライブラリー。
【請求項9】
ライブラリーを構成する遺伝子が、細菌、酵母、および植物細胞からなる群から選択されるいずれかの宿主細胞に導入されたものである請求項6に記載のライブラリー。
【請求項10】
ライブラリーを構成する遺伝子が、哺乳動物細胞に導入されたものである請求項6に記載のライブラリー。
【請求項11】
ライブラリーを構成する少なくとも一部の遺伝子が、繊維状ファージに組み込まれている請求項6に記載のライブラリー。
【請求項12】
ライブラリーを構成する遺伝子によってコードされる重鎖可変領域と軽鎖可変領域の断片を繊維状ファージ表面に発現し、かつそれらが機能的に再構成されている請求項11に記載のライブラリー。
【請求項13】
重鎖可変領域に標識ペプチドをコードする遺伝子が融合されている請求項12に記載のライブラリー。
【請求項14】
イムノグロブリン軽鎖可変領域遺伝子がヒトに由来するものである請求項6に記載のライブラリー。
【請求項15】
イムノグロブリン軽鎖超可変部位がシステイン残基を含まないアミノ酸配列からなる請求項14に記載のライブラリー。
【請求項16】
請求項5に記載の遺伝子ライブラリーを構成する各クローンが、そのクローンによって保持される遺伝子によってコードされる抗体タンパク質を伴っているrgdpライブラリー。
【請求項17】
請求項5に記載の遺伝子ライブラリーを構成する各クローンについて、そのクローンによって保持される遺伝子によってコードされる抗体タンパク質から構成される抗体ライブラリー。
【請求項18】
次の工程を含む、重鎖可変領域と機能的なコンフォーメーションを再構成することができる軽鎖可変領域をコードする遺伝子の選択方法。
a)軽鎖可変領域をコードする、1つまたは複数の遺伝子を取得する工程、
b)軽鎖可変領域と機能的なコンフォーメーションを構成することが確認されているイムノグロブリン重鎖可変領域をコードする遺伝子を取得する工程、
c)工程a)によって得られた軽鎖可変領域をコードする遺伝子の任意の1つを選択し、工程b)で取得した重鎖可変領域をコードする遺伝子とイムノグロブリンの機能的なコンフォーメーションを再構成が可能な条件下でタンパク質に翻訳する工程、
d)工程c)において翻訳されたタンパク質の抗原結合領域の形成を検出する工程、および
e)抗原結合領域の形成が検出されたタンパク質を構成する軽鎖可変領域をコードする遺伝子を選択する工程
【請求項19】
更に、次の工程f)を含む請求項18に記載の方法。
f)軽鎖可変領域遺伝子の塩基配列を決定し、その塩基配列によってコードされるアミノ酸配列を比較して同一のアミノ酸配列をコードする遺伝子、およびアミノ酸の欠損を生じている遺伝子を排除する工程
【請求項20】
イムノグロブリンの軽鎖可変領域が、繊維状ファージの表面に発現している請求項18に記載の方法。
【請求項21】
イムノグロブリンの軽鎖可変領域をコードする遺伝子と重鎖可変領域をコードする遺伝子が挿入されたファージミドを感染させた宿主微生物の培養上清そのものを、工程c)のための軽鎖を含む試料として利用する請求項18に記載の方法。
【請求項22】
次の工程を含む、特定の抗原と結合するイムノグロブリン可変領域の検出方法。
a)請求項5または請求項7に記載のライブラリー、またはその発現産物と、該抗原とを抗原抗体反応に適した条件下に接触させる工程
b)該抗原とイムノグロブリン可変領域との結合を検出する工程
【請求項23】
ライブラリーが請求項12に記載のライブラリーである請求項22に記載の方法。
【請求項24】
請求項22に記載の方法の後、更に次の工程c)を行うことを特徴とする、特定の抗原と結合するイムノグロブリン可変領域の取得方法。
c)前記抗原と結合するイムノグロブリン可変領域を発現するクローンを選択する工程
【請求項25】
更に次の工程d)−e)を含む請求項24に記載の方法。
d)工程c)で選択したファージクローンを増幅して2次的なライブラリーを得る工程、
e)工程c)において選択されるクローンの回収率が上昇するまで、2次的なライブラリーについて、工程a)−d)を繰り返す工程
【請求項26】
請求項24に記載の方法によって得られたクローン、またはイムノグロブリン断片、もしくはそれをコードする遺伝子。
【請求項27】
配列番号:61〜配列番号:78のいずれかに記載の塩基配列を含む、ポリヌクレオチド。
【請求項28】
配列番号:79〜配列番号:96のいずれかに記載のアミノ酸配列を含む蛋白質。
【請求項29】
次の要素からなる抗体ライブラリーの調製用キット。
a)重鎖可変領域と機能的なコンフォーメーションを再構成することができない軽鎖可変領域をコードする遺伝子を実質的に排除した軽鎖可変領域遺伝子ライブラリー、および
b)重鎖可変領域をコードする遺伝子を増幅することができるプライマーセット
【請求項30】
以下の工程からなる抗体ライブラリーの調製方法。
a)ファージミドに、イムノグロブリンの少なくとも可変領域を含む領域をコードする遺伝子を組み込む工程
b)工程a)で得られたファージミドを宿主微生物に感染させる工程
c)ヘルパーファージを感染させること無く、工程b)の宿主微生物の培養上清を回収して抗体ライブラリーとする工程
【図1】

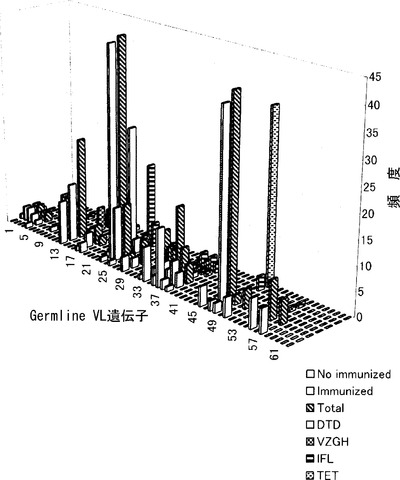
【図2】
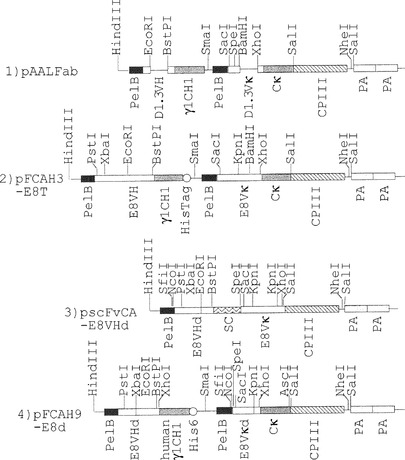
【図3】

【図4】

【図5】

【図6】

【図7】

【図8】

【図9】

【図10】
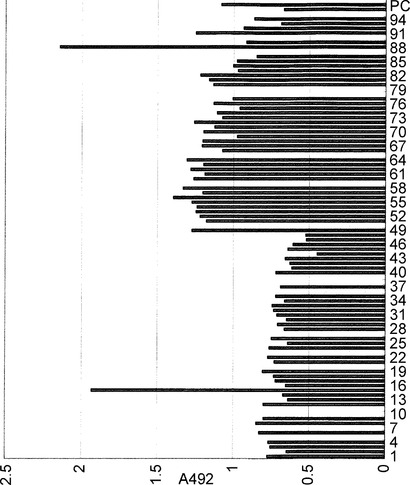
【図11】
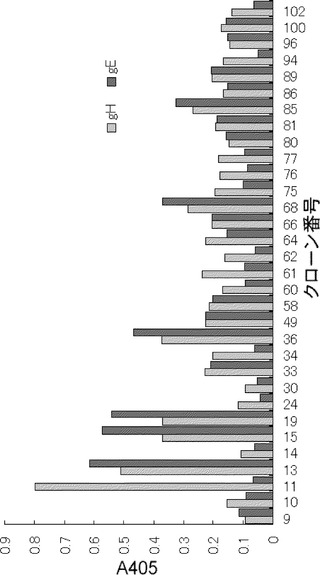
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公開番号】特開2011−87586(P2011−87586A)
【公開日】平成23年5月6日(2011.5.6)
【国際特許分類】
【出願番号】特願2010−261910(P2010−261910)
【出願日】平成22年11月25日(2010.11.25)
【分割の表示】特願2001−562681(P2001−562681)の分割
【原出願日】平成13年2月22日(2001.2.22)
【出願人】(390004097)株式会社医学生物学研究所 (41)
【Fターム(参考)】
【公開日】平成23年5月6日(2011.5.6)
【国際特許分類】
【出願日】平成22年11月25日(2010.11.25)
【分割の表示】特願2001−562681(P2001−562681)の分割
【原出願日】平成13年2月22日(2001.2.22)
【出願人】(390004097)株式会社医学生物学研究所 (41)
【Fターム(参考)】
[ Back to top ]