
抗体依存性細胞傷害性を改善するための抗体のグリコシル化操作
【課題】改善された治療価値を生じる改変されたグリコシル化パターンを有するタンパク質を生成するグリコシル化操作を提供する。
【解決手段】調節されたレベルにおいて糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸を発現するために操作された、宿主細胞であり、バイセクティングGlcNAcを有するN結合型複合型オリゴ糖を増加する、糖タンパク質改変グリコシルトランスフェラーゼ活性の好ましい範囲を発現し得るように操作された宿主細胞。また、糖タンパク質、例えば、抗体(抗体分子全体、抗体フラグメント、または免疫グロブリンのFc領域に等価な領域を含む融合タンパク質を含み、増強されたFc媒介細胞傷害性を有する)の改変された糖形態を生成するための方法、およびそのように生成された糖タンパク質。
【解決手段】調節されたレベルにおいて糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸を発現するために操作された、宿主細胞であり、バイセクティングGlcNAcを有するN結合型複合型オリゴ糖を増加する、糖タンパク質改変グリコシルトランスフェラーゼ活性の好ましい範囲を発現し得るように操作された宿主細胞。また、糖タンパク質、例えば、抗体(抗体分子全体、抗体フラグメント、または免疫グロブリンのFc領域に等価な領域を含む融合タンパク質を含み、増強されたFc媒介細胞傷害性を有する)の改変された糖形態を生成するための方法、およびそのように生成された糖タンパク質。
【発明の詳細な説明】
【技術分野】
【0001】
(I.他の出願との関連)
本出願は、1998年4月20日に出願された米国仮特許出願第60/082,581号(これは、本明細書中にその全体が参考として援用される)の優先権を主張する。
【0002】
(II.発明の分野)
本発明は、タンパク質のグリコシル化操作の分野に関する。さらに詳細には、本発明は、改善された治療特性を有するタンパク質(増強された抗体依存性細胞傷害性を有する抗体を含む)を生成するグリコシル化操作に関する。
【背景技術】
【0003】
(III.発明の背景)
糖タンパク質は、ヒト、他の真核生物、およびいくつかの原核生物における多くの必須の機能(触媒、シグナル伝達、細胞間コミュニケーション、および分子認識および分子会合を含む)を媒介する。それらは、真核生物において非細胞質ゾルタンパク質の大部分を構成している。非特許文献1。治療目的のために多くの糖タンパク質が探索されており、そして最近20年の間には、天然に存在する分泌糖タンパク質の組換えバージョンが生物工学産業の主要な産物になった。例としては、エリスロポエチン(EPO)、治療的モノクローナル抗体(治療的mAb)、組織プラスミノーゲンアクチベーター(tPA)、インターフェロン−β(IFN−β)、顆粒球−マクロファージコロニー刺激因子(GM−CSF)、およびヒト絨毛性ゴナドトロピン(hCH)が挙げられる。非特許文献2。
【0004】
オリゴ糖成分は、治療的糖タンパク質の有効性(物理学的安定性、プロテアーゼの攻撃に対する耐性、免疫系との相互作用、薬物動態、および特異的生物学的活性を含む)に関する特性に、有意に影響を及ぼし得る。このような特性は、オリゴ糖の存在または非存在に依存し得るだけでなく、その特異的構造にも依存し得る。オリゴ糖構造と糖タンパク質機能との間のいくつかの一般化がなされ得る。例えば、特定のオリゴ糖構造は、特定の糖質結合タンパク質との相互作用を介した血流からのこの糖タンパク質の迅速なクリアランスを媒介する一方、他のオリゴ糖構造は、抗体に結合し、所望されない免疫応答を誘発し得る。非特許文献3。
【0005】
哺乳動物細胞は、ヒト適用のために最も適合した形態でタンパク質をグリコシル化する能力のために、治療的糖タンパク質の生成のために好ましい宿主である。非特許文献2;非特許文献3。細菌はタンパク質をほとんどグリコシル化しない。そして同様に他の型の一般的な宿主(例えば、酵母、糸状菌、昆虫細胞および植物細胞)は、血流からの迅速なクリアランス、所望されない免疫応答、およびいくつかの特定の場合では、減少した生物学的活性と関連したグリコシル化パターンを生じる。哺乳動物細胞の中でも、チャイニーズハムスター卵巣(CHO)細胞が、最近20年の間に最も一般的に使用された。適切なグリコシル化パターンを与えることに加えて、これらの細胞は、遺伝的に安定な、高度に生産的なクローン性細胞株の一貫した生成を可能にする。それらは、無血清培地を使用して単純なバイオリアクター中で高密度に培養され得、そして安全かつ再現可能な生物プロセスの開発を可能にする。他の一般的に使用された動物細胞としては、新生仔ハムスター腎臓(BHK)細胞、NS0マウスミエローマ細胞およびSP2/0マウスミエローマ細胞が挙げられる。さらに最近、トランスジェニック動物からの生成もまた、試験されている。非特許文献3。
【0006】
動物細胞で生成した組換え治療的タンパク質のグリコシル化は、宿主細胞中のグリコシルトランスフェラーゼ遺伝子の過剰発現によって操作され得る。非特許文献4。しかし、この分野の以前の研究では、糖タンパク質改変グリコシルトランスフェラーゼ遺伝子の構成性発現が使用されてきたに過ぎず、発現レベルについては、ほとんど注意が払われてこなかった。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】LisおよびSharon、Eur.J.Biochem.(1993)218:1−27
【非特許文献2】Cummingら、Glycobiology(1991)1:115−130
【非特許文献3】Jenkinsら、Nature Biotechn.(1996)14:975−981
【非特許文献4】Bailey、Science(1991)252:1668−1675
【発明の概要】
【課題を解決するための手段】
【0008】
(IV.発明の要旨)
本発明は、一般に、改善された治療価値を生じる改変されたグリコシル化パターンを有するタンパク質の生成のための宿主細胞および方法に関する。1つの特定の実施態様において、本発明は、バイセクティングGlcNAcを有するN結合型複合型オリゴ糖を増加する、糖タンパク質改変グリコシルトランスフェラーゼ活性の好ましい範囲を発現し得るように操作された宿主細胞に関する。他の実施態様において、本発明は、糖タンパク質、例えば、抗体(抗体分子全体、抗体フラグメント、または免疫グロブリンのFc領域に等価な領域を含む融合タンパク質を含み、増強されたFc媒介細胞傷害性を有する)の改変された糖形態を生成するための方法、およびそのように生成された糖タンパク質に関する。本発明は、バイセクティングGlcNAcを有するN結合型複合型オリゴ糖の最大化のための糖タンパク質改変グリコシルトランスフェラーゼ発現の最適な範囲が存在するという本発明者らの発見に一部基づく。
【0009】
より詳細には、本発明は、改善した治療的価値を有する改変された糖形態のタンパク質、例えば、増強された抗体依存性細胞傷害性(ADCC)を有する抗体を宿主細胞において生成する方法に関する。本発明は、目的のタンパク質(例えば、抗体)をコードする核酸、および糖タンパク質改変グリコシル化トランスフェラーゼをコードする少なくとも1つの核酸を有する宿主細胞を提供する。さらに、本発明は、上記の目的のタンパク質(例えば、抗体依存性細胞傷害性を増強した抗体)の発現を可能にする条件下でこのような宿主細胞を培養する方法およびプロトコルを提供する。さらに、このように生成された、改変されたグリコシル化パターンを有するタンパク質(例えば、増強された抗体依存性細胞傷害性を有する抗体)を単離する方法が記載される。
【0010】
さらに、本発明は、改善した治療特性を有するタンパク質の別の糖形態を提供する。本発明のタンパク質は、増強された抗体依存性細胞傷害性(ADCC)を有する抗体を含み、これは、開示された方法および宿主細胞を使用して生成される。
【0011】
(VI.定義)
用語は、以下に定義されなければ、当該分野で一般に使用されるように本明細書中で使用される。
【0012】
本明細書中で使用される用語「抗体」は、抗体分子全体、抗体フラグメント、または免疫グロブリンのFc領域と等価な領域を含む融合タンパク質を含むことが意図される。
【0013】
本明細書中で使用される用語「糖タンパク質改変グリコシルトランスフェラーゼ」とは、糖タンパク質のグリコシル化パターンの改変をもたらす酵素をいう。糖タンパク質改変グリコシルトランスフェラーゼの例としては、例えば、GnT III、GnT V、GalT、およびManIIのようなグリコシルトランスフェラーゼが挙げられるが、これらに限定されない。
【0014】
本明細書中で使用される用語「グリコシル化操作」は、天然に存在するポリペプチドまたはそのフラグメントのグリコシル化パターンに対して任意の種類の変化を含むとみなされる。グリコシル化操作としては、細胞のグリコシル化機構の代謝的操作(細胞中で発現された糖タンパク質の改変されたグリコシル化を達成する、オリゴ糖合成経路の遺伝子操作を含む)が挙げられる。さらに、グリコシル化操作としては、グリコシル化に対する変異および細胞環境の効果が挙げられる。
【0015】
本明細書中で使用される用語「宿主細胞」は、タンパク質、タンパク質フラグメント、または目的のペプチド(抗体または抗体フラグメントを含む)の改変された糖形態を生成するように操作され得る任意の種類の細胞系を含む。代表的には、宿主細胞は、少なくとも1つの、糖タンパク質改変グリコシルトランスフェラーゼの最適化されたレベルを発現するように操作されており、これらのグリコシルトランスフェラーゼとしては、GnT III、GnT V、GalT、およびManII、ならびに/または少なくとも1つのグリコシダーゼが挙げられるが、これらに限定されない。宿主細胞としては、培養細胞、例えば、哺乳動物培養細胞(例えば、CHO細胞、BHK細胞、NS0細胞、SP2/0細胞またはハイブリドーマ細胞)、酵母細胞および昆虫細胞が挙げられ、トランスジェニック動物または培養組織内に含まれる細胞もまた挙げられる。
【0016】
本明細書中で使用される用語「Fc媒介細胞傷害性」は、抗体依存性細胞傷害性(ADCC)、および免疫グロブリンGのFc領域またはそれに等価な領域をその細胞表面で発現するように操作された細胞に対する細胞傷害性、ならびに免疫グロブリンGのFc領域またはそれに等価な領域のN末端に融合された標的タンパク質ドメインからなる可溶性融合タンパク質により媒介される細胞傷害性を含むことが意図される。
したがって本発明は、以下の項目を提供する
(項目1) 調節されたレベルにおいて糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸を発現するために操作された、宿主細胞。
(項目2) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子を含む核酸分子が、上記宿主細胞に導入されている、項目1に記載の宿主細胞。
(項目3) 内因性の糖タンパク質改変グリコシルトランスフェラーゼが活性化されるように、上記宿主細胞が操作されている、項目1に記載の宿主細胞。
(項目4) 上記宿主細胞が、CHO細胞、BHK細胞、NS0細胞、SP2/0細胞、またはハイブリドーマ細胞である、項目2または3に記載の宿主細胞。
(項目5) 上記内因性の糖タンパク質改変グリコシルトランスフェラーゼが、調節性プロモーターエレメントを上記宿主細胞染色体へ挿入することによって活性化されている、項目3に記載の宿主細胞。
(項目6) 上記糖タンパク質改変グリコシルトランスフェラーゼが、GnT III、GnT V、Man II、またはGal Tである、項目2または3に記載の宿主細胞。
(項目7) 上記宿主細胞が、GnT III、GnT V、Man II、およびGal Tからなる群から選択される少なくとも2つの異なる、糖タンパク質改変グリコシルトランスフェラーゼを発現するために操作された、項目2または3に記載の宿主細胞。
(項目8) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子が、構成性プロモーターエレメントに作動可能に連結されている、項目7に記載の宿主細胞。
(項目9) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子が、調節性プロモーターエレメントに作動可能に連結されている、項目2に記載の宿主細胞。
(項目10) 上記調節性プロモーターエレメントが、テトラサイクリンに調節性プロモーター系、エクジソン誘導性プロモーター系、lacスイッチプロモーター系、グルココルチコイド誘導性プロモーター系、温度誘導性プロモーター系、またはメタロチオネイン金属誘導性プロモーター系である、項目5または9に記載の宿主細胞。
(項目11) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸分子を発現するために操作された宿主細胞であって、ここでその宿主細胞が、増強したFc媒介細胞傷害性を有するタンパク質を産生し得る、宿主細胞。
(項目12) 上記タンパク質が、抗体分子全体、抗体フラグメント、または免疫グロブリンのFc領域に等価な領域を含む融合タンパク質である、項目11に記載の宿主細胞。
(項目13) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子を含む核酸分子が、上記宿主細胞染色体へ導入されている、項目12に記載の宿主細胞。
(項目14) 上記宿主細胞が、内因性の糖タンパク質改変グリコシルトランスフェラーゼの変異誘発発現を保有するために選択されている、項目12に記載の宿主細胞。
(項目15) 上記宿主細胞が、変異lec10である、項目14に記載の宿主細胞。
(項目16) 内因性の糖タンパク質改変グリコシルトランスフェラーゼが活性化されるように、上記宿主細胞が操作されている、項目12に記載の宿主細胞。
(項目17) 上記内因性の糖タンパク質改変グリコシルトランスフェラーゼが、調節性プロモーターエレメントを上記宿主細胞染色体へ挿入することによって活性化されている、項目16に記載の宿主細胞。
(項目18) 上記内因性の糖タンパク質改変グリコシルトランスフェラーゼが、構成性プロモーターエレメント、トランスポゾン、またはレトロウイルスエレメントを上記宿主細胞染色体へ挿入することによって活性化されている、項目16に記載の宿主細胞。
(項目19) 抗体分子、抗体フラグメント、または免疫グロブリンのFc領域に等価な領域を含む融合タンパク質をコードする少なくとも1つのトランスフェクトされた核酸をさらに含む、項目11または13に記載の宿主細胞。
(項目20) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子が、構成性プロモーターエレメントに作動可能に連結される、項目13に記載の宿主細胞。
(項目21) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子が、調節性プロモーターエレメントに作動可能に連結される、項目13に記載の宿主細胞。
(項目22) 上記調節性プロモーターエレメントが、テトラサイクリンに調節性プロモーター系、エクジソン誘導性プロモーター系、lacスイッチプロモーター系、グルココルチコイド誘導性プロモーター系、温度誘導性プロモーター系、またはメタロチオネイン金属誘導性プロモーター系である、項目21に記載の宿主細胞。
(項目23) 上記宿主細胞がハイブリドーマ細胞である、項目11に記載の宿主細胞。
(項目24) 上記操作された宿主細胞が、操作されたCHO細胞、操作されたBHK細胞、操作されたNS0細胞、または操作されたSP2/0細胞である、項目11に記載の宿主細胞。
(項目25) 上記宿主細胞が、キメラ抗CD20モノクローナル抗体(C2B8)をコードする少なくとも1つのトランスフェクトされた核酸を含む、項目11に記載の宿主細胞。
(項目26) 上記宿主細胞が、キメラ抗ヒト神経芽腫モノクローナル抗体(chCE7)をコードする少なくとも1つのトランスフェクトされた核酸を含む、項目11に記載の宿主細胞。
(項目27) 上記宿主細胞が、キメラ抗ヒト腎細胞癌モノクローナル抗体(ch−G250)、ヒト化抗HER2モノクローナル抗体、キメラ抗ヒト結腸癌、肺癌、および乳癌モノクローナル抗体(ING−1)、ヒト化抗ヒト17−1A抗原モノクローナル抗体(3622W94)、ヒト化抗ヒト結腸直腸腫瘍抗体(A33)、GD3ガングリオシドに対する抗ヒト黒色腫抗体(R24)、またはキメラ抗ヒト扁平上皮癌モノクローナル抗体(SF−25)をコードする少なくとも1つのトランスフェクトされた核酸を含む、項目11に記載の宿主細胞。
(項目28) 少なくとも1つの核酸分子が、β(1,4)−N−アセチルグルコサミニルトランスフェラーゼIII(GnT III)をコードする、項目11に記載の宿主細胞。
(項目29) β(1,4)−ガラクトシルトランスフェラーゼ(GalT)をコードする少なくとも1つの核酸をさらに含む、項目28に記載の宿主細胞。
(項目30) マンノシダーゼII(Man II)をコードする少なくとも1つの核酸をさらに含む、項目28に記載の宿主細胞。
(項目31) β(1,4)−ガラクトシルトランスフェラーゼ(GalT)をコードする少なくとも1つの核酸およびマンノシダーゼII(Man II)をコードする少なくとも1つの核酸をさらに含む、項目28に記載の宿主細胞。
(項目32) 宿主細胞において、増強したFc媒介細胞傷害性を有するタンパク質化合物を産生するための方法であって、以下の工程:
(a)調節されたレベルにおいて、目的のタンパク質化合物のグリコシル化を改善するように選択された、糖タンパク質改変グリコシルトランスフェラーゼを発現するために操作された宿主細胞を提供する工程であって、ここで、その宿主細胞が、抗体、抗体フラグメント、または免疫グロブリンのFc領域に等価な領域を含む融合タンパク質をコードする少なくとも1つの核酸を発現する、工程;
(b)増強したFc媒介性依存性細胞傷害性を有するそのタンパク質化合物の産生を可能にする条件下で、その宿主細胞を培養する工程;および
(c)増強したFc媒介細胞傷害性を有するそのタンパク質化合物を単離する工程、
を包含する、方法。
(項目33) 工程(a)において、上記宿主細胞が、抗体全体をコードする少なくとも1つの核酸を含む、項目32に記載の方法。
(項目34) 工程(a)において、上記宿主細胞が、抗体フラグメントをコードする少なくとも1つの核酸を含む、項目32に記載の方法。
(項目35) 工程(a)において、上記宿主細胞が、免疫グロブリンのグルコシル化Fc領域に類似の領域を含む融合タンパク質をコードする少なくとも1つの核酸を含む、項目32に記載の方法。
(項目36) 上記宿主細胞が、キメラ抗CD20モノクローナル抗体(C2B8)をコードする少なくとも1つのトランスフェクトされた核酸を含む、項目32に記載の方法。
(項目37) 上記宿主細胞が、キメラ抗ヒト神経芽腫モノクローナル抗体(chCE7)をコードする少なくとも1つのトランスフェクトされた核酸を含む、項目32に記載の方法。
(項目38) 上記宿主細胞が、キメラ抗ヒト腎細胞癌モノクローナル抗体(ch−G250)、ヒト化抗HER2モノクローナル抗体、キメラ抗ヒト結腸癌、肺癌、および乳癌モノクローナル抗体(ING−1)、ヒト化抗ヒト17−1A抗原モノクローナル抗体(3622W94)、ヒト化抗ヒト結腸直腸腫瘍抗体(A33)、GD3ガングリオシドに対する抗ヒト黒色腫抗体(R24)、またはキメラ抗ヒト扁平上皮癌モノクローナル抗体(SF−25)をコードする少なくとも1つのトランスフェクトされた核酸を含む、
項目32に記載の方法。
(項目39) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子が、上記宿主細胞に導入されている、項目32に記載の方法。
(項目40) 上記宿主細胞が、内因性の糖タンパク質改変グリコシルトランスフェラーゼの変異誘発発現を保有するために選択されている、項目32に記載の宿主細胞。
(項目41) 上記宿主細胞が、変異lec10である、項目40に記載の宿主細胞。
(項目42) 内因性の糖タンパク質改変グリコシルトランスフェラーゼが活性化されるように、上記宿主細胞が操作されている、項目32に記載の宿主細胞。
(項目43) 上記グリコシルトランスフェラーゼが、β(1,4)−N−アセチルグルコサミニルトランスフェラーゼIII(GnT III)である、項目32に記載の方法。
(項目44) 上記GnT IIIが、構成性プロモーター系を使用して発現される、項目43に記載の方法。
(項目45) 上記GnT IIIが、調節性プロモーター系を使用して発現される、項目43に記載の方法。
(項目46) 上記調節性プロモーター系が、テトラサイクリンに調節性プロモーター系、エクジソン誘導性プロモーター系、lacスイッチプロモーター系、グルココルチコイド誘導性プロモーター系、温度誘導性プロモーター系、またはメタロチオネイン金属誘導性プロモーター系である、項目45に記載の方法。
(項目47) 上記グリコシルトランスフェラーゼが、β(1,4)−ガラクトシルトランスフェラーゼ(GalT)である、項目32に記載の方法。
(項目48) 上記GalTが、構成性プロモーター系を使用して発現される、項目47に記載の方法。
(項目49) 上記GalTが、調節性プロモーター系を使用して発現される、項目47に記載の方法。
(項目50) 上記調節性プロモーター系が、テトラサイクリンに調節性プロモーター系、エクジソン誘導性プロモーター系、lacスイッチプロモーター系、グルココルチコイド誘導性プロモーター系、温度誘導性プロモーター系、またはメタロチオネイン金属誘導性プロモーター系である、項目49に記載の方法。
(項目51) 上記宿主細胞が、調節されたレベルにおいて、目的のタンパク質化合物のグリコシル化を改善するように選択された、グリコシルトランスフェラーゼをコードする複数の核酸を発現するために操作され、ここで、少なくとも1つの核酸がGnT IIIをコードし、そして少なくとも1つの核酸がβ(1,4)−ガラクトシルトランスフェラーゼ(GalT)をコードする、項目32に記載の方法。
(項目52) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子を含む核酸分子が、上記宿主細胞に導入されている、項目51に記載の宿主細胞。
(項目53) 上記宿主細胞が、少なくとも1つの、内因性の糖タンパク質改変グリコシルトランスフェラーゼの変異誘発発現を保有するために選択されている、項目51に記載の宿主細胞。
(項目54) 内因性の糖タンパク質改変グリコシルトランスフェラーゼが活性化されるように、上記宿主細胞が操作されている、項目51、52、または53に記載の宿主細胞。
(項目55) 上記宿主細胞が、調節されたレベルにおいて、目的のタンパク質化合物のグリコシル化を改善するように選択された、糖タンパク質改変グリコシルトランスフェラーゼをコードする複数の核酸を含み、ここで、少なくとも1つの核酸がGnT IIIをコードし、そして少なくとも1つの核酸がマンノシダーゼII(Man II)をコードする、項目32に記載の方法。
(項目56) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子を含む核酸分子が、上記宿主細胞に導入されている、項目55に記載の宿主細胞。
(項目57) 上記宿主細胞が、少なくとも1つの、内因性の糖タンパク質改変グリコシルトランスフェラーゼの変異誘発発現を保有するために選択されている、項目55に記載の宿主細胞。
(項目58) 内因性の糖タンパク質改変グリコシルトランスフェラーゼが活性化されるように、上記宿主細胞が操作されている、項目55、56、または57に記載の宿主細胞。
(項目59) 上記宿主細胞が、調節されたレベルにおいて、目的のタンパク質のグリコシル化を改善するように選択された、糖タンパク質改変グリコシルトランスフェラーゼをコードする複数の核酸を含み、ここで、少なくとも1つの核酸がGnT IIIをコードし、少なくとも1つの核酸がβ(1,4)−ガラクトシルトランスフェラーゼ(GalT)をコードし、そして少なくとも1つの核酸がマンノシダーゼII(Man II)をコードする、項目32に記載の方法。
(項目60) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子を含む核酸分子が、上記宿主細胞に導入されている、項目59に記載の宿主細胞。
(項目61) 上記宿主細胞が、少なくとも1つの、内因性の糖タンパク質改変グリコシルトランスフェラーゼの変異誘発発現を保有するために選択されている、項目59に記載の宿主細胞。
(項目62) 内因性の糖タンパク質改変グリコシルトランスフェラーゼが活性化されるように、上記宿主細胞が操作されている、項目59、60、または61に記載の宿主細胞。
(項目63) 少なくとも1つの糖タンパク質改変グリコシルトランスフェラーゼの発現レベルが、抗体分子、抗体フラグメント、または免疫グロブリンのFc領域に等価な領域を含む融合タンパク質を産生するように選択され、その糖タンパク質改変グリコシルトランスフェラーゼが、異なる発現レベルの同一のグリコシルトランスフェラーゼ遺伝子から得られるFc媒介細胞傷害性よりも、より高いレベルにおいて増強したFc媒介細胞傷害性を有する、項目32に記載の方法。
(項目64) 上記発現レベルが、グリコシルトランスフェラーゼ特異的抗体を使用するウエスタンブロット分析によって決定される、項目63に記載の方法。
(項目65) 上記発現レベルが、グリコシルトランスフェラーゼ特異的プローブを使用するノーザンブロット分析によって決定される、項目63に記載の方法。
(項目66) 上記発現レベルが、グリコシルトランスフェラーゼの酵素活性を測定することによって決定される、項目63に記載の方法。
(項目67) 上記発現レベルが、糖タンパク質改変グリコシルトランスフェラーゼの生合成産物に結合するレクチンを使用して決定される、項目63に記載の方法。
(項目68) 上記レクチンがE4−PHAレクチンである、項目67に記載の方法
。
(項目69) 上記糖タンパク質改変グリコシルトランスフェラーゼをコードする上記核酸が、レポーター遺伝子に作動可能に連結され、そしてここで、そのグリコシルトランスフェラーゼの上記発現レベルが、そのレポーター遺伝子の発現レベルと相関するシグナルを測定することによって決定される、項目63に記載の方法。
(項目70) 上記レポーター遺伝子が、単一のRNA分子として、上記糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸とともに転写され、そしてそれらの各コード配列が、内部リボソーム侵入部位(IRES)またはキャップ独立性翻訳エンハンサー(CITE)のいずれかによって連結される、項目69に記載の方法。
(項目71) 単一のポリペプチド鎖が形成されるように、上記レポーター遺伝子が、上記糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸とともに翻訳される、項目69に記載の方法。
(項目72) 上記糖タンパク質改変グリコシルトランスフェラーゼをコードする上記核酸が、単一のプロモーターの制御下でレポーター遺伝子に作動可能に連結され、ここでその糖タンパク質改変グリコシルトランスフェラーゼをコードするその核酸およびそのレポーター遺伝子が、2つの別個のメッセンジャーRNA(mRNA)分子に選択的スプライスされるRNA分子へ転写され、ここで得られたmRNAの一方がそのレポータータンパク質に翻訳され、そして他方がその糖タンパク質改変グリコシルトランスフェラーゼに翻訳される、項目63に記載の方法。
(項目73) 上記宿主細胞が、グリコシダーゼをコードする核酸をさらに含む、項目32に記載の方法。
(項目74) 項目11に記載の宿主細胞によって産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、抗体。
(項目75) 項目25に記載の宿主細胞によって産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、キメラ抗CD20モノクローナル抗体(C2D8)。
(項目76) 項目26に記載の宿主細胞を使用して産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、キメラ抗ヒト神経芽腫モノクローナル抗体(chCE7)。
(項目77) 項目27に記載の宿主細胞を使用して産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、キメラ抗ヒト腎細胞癌モノクローナル抗体(ch−G250)。
(項目78) 項目27に記載の宿主細胞を使用して産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、ヒト化抗HER2モノクローナル抗体。
(項目79) 項目27に記載の宿主細胞を使用して産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、キメラ抗ヒト結腸癌、肺癌、および乳癌モノクローナル抗体(ING−1)。
(項目80) 項目27に記載の宿主細胞を使用して産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、ヒト化抗ヒト17−1A抗原モノクローナル抗体(3622W94)。
(項目81) 項目27に記載の宿主細胞を使用して産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、キメラ抗ヒト扁平上皮癌モノクローナル抗体(SF−25)。
(項目82) 項目27に記載の宿主細胞を使用して産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、ヒト化抗ヒト結腸直腸腫瘍抗体(A33)。
(項目83) 項目27に記載の宿主細胞を使用して産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、GD3ガングリオシドに対する抗ヒト黒色腫抗体(R24)。
(項目84) 項目11に記載の宿主細胞を使用して産生された、増強したFc媒介細胞傷害性を有する、免疫グロブリンのFc領域に等価な領域を含む、抗体フラグメント。
(項目85) 項目11に記載の宿主細胞を使用して産生された、増強したFc媒介細胞傷害性を有する、免疫グロブリンのFc領域に等価な領域を含む、融合タンパク質。
【図面の簡単な説明】
【0017】
【図1】図1は、典型的なFc関連オリゴ糖構造の代表を記載する。
【図2】図2は、2つの異なるtTA生成CHOクローンにおけるGnT IIIのテトラサイクリン調節性発現のウェスタンブロット分析を示す。CHOt2細胞(レーンAおよびB)およびCHOt17細胞(レーンCおよびD)は、pUDH10−3GnTIIIm発現ベクターでトランスフェクトされ、そしてテトラサイクリンの非存在下(レーンAおよびC)または400ng/mlの濃度の存在下(レーンBおよびD)で36時間培養した。次いで、細胞溶解物を、抗体(9E10)でプローブするウェスタンブロット分析のために調製した。この抗体は、そのカルボキシ末端でGnT IIIに付加されたc−mycタグを特異的に認識する。
【図3】図3は、mycタグ化GnT III発現が制御され得るテトラサイクリン濃度の範囲の決定を示す。CHOt17細胞を、pUDH10−3−GnTIIIm発現ベクターでトランスフェクトし、次いで、示された濃度のテトラサイクリンの存在下で48時間培養した。これらの培養物の細胞溶解物のGnT IIIレベルは、ウェスタンブロット分析を使用して比較した。GnT IIIを、9E10抗体を使用するc−mycタグを介して検出した。
【図4】図4A〜4Bは、GnT V(図4A)またはmycタグ化GnT III(図4B)グリコシルトランスフェラーゼの安定な、テトラサイクリン調節性発現についてのCHOクローンのウェスタンブロット分析によるスクリーニングを示す。CHOt17細胞は、プロマイシン耐性(pPUR)の発現のためのベクターと、pUHD10−3GnTV(図4A)またはpUDH10−3GnTIIIm(図4B)のいずれかとで同時トランスフェクトされ、そして安定なCHOクローンを、テトラサイクリンの存在下(2μg/ml)で、プロマイシン(7.5μg/ml)に対する耐性について選択した。各グリコシルトランスフェラーゼについての8つのクローン(1〜8)を、テトラサイクリンの非存在下または存在下(+)(2μg/ml)で48時間培養し、そして抗GnT V抗体(図4A)または抗myc抗体(9E10)抗体(図4B)のいずれかを用いてウェスタンブロットによって分析した。
【図5】図5Aおよび5Bは、レクチンブロット分析によるインビボでの異種GnT V(図5A)およびGnT III(図5B)のグリコシルトランスフェラーゼの活性の検証を示す。テトラサイクリン(2μg/ml)の非存在下または存在下(+)で培養した種々の安定なクローンからの細胞糖タンパク質(図4と同様に番号付けした)をSDS−PAGEで分離し、膜にブロットし、そしてL−PHAレクチン(図5A)およびE−PHAレクチン(図5B)のいずれかでプローブした。これらのレクチンは、それぞれ、GnT VおよびGnT IIIによって触媒された反応のオリゴ糖生成物に対して、これらの反応物のオリゴ糖基質に対するより、より高い親和性で結合する。分子量マーカー(MWM)を、並行して泳動した。図5Aおよび5Bのレクチンブロットの比較によって、内因性CHO細胞糖タンパク質の中で、GnT V(図5A)についてより、GnT III(図5B)についてより広範囲の基質が示される。
【図6】図6A〜6Dは、グリコシルトランスフェラーゼの過剰発現の際の細胞増殖の阻害を示す、CHO−tet−GnTIIIm細胞を、5〜10%コンフルエントで播種し、そしてテトラサイクリンの非存在下(図6Aおよび6B)または存在下(図6Cおよび6D)で培養した。培養物を播種して45時間後(図6Aおよび6C)ならびに85時間後(図6Bおよび6D)に写真を撮った。
【図7】図7は、chCE7重鎖遺伝子の構築のためにPCRで使用したオリゴヌクレオチドプライマーの配列を示す。正方向および逆方向プライマーは、それぞれ、「.fwd」および「.rev」を接尾辞としてつけることによって同定する。テンプレートとして最初のPCR工程の産物を使用して、第2のPCR工程を行うために必要な、異なるプライマー間の重複を示す。導入された制限部位、CE7キメラゲノムDNAにアニーリングする配列、および導入された合成リーダー配列もまた示す。
【図8】図8は、chCE7軽鎖遺伝子の構築のためにPCRで使用したオリゴヌクレオチドプライマーの配列を示す。正方向プライマーおよび逆方向プライマーは、それぞれ、「.fwd」および「.rev」を接尾辞としてつけることによって同定する。テンプレートとして最初のPCR工程の産物を使用して、第2のPCR工程を行うために必要な、異なるプライマー間の重複を示す。導入された制限部位、CE7キメラゲノムDNAにアニーリングする配列、および導入されたリーダー配列もまた示す。
【図9A】図9は、SP2/0マウスミエローマ細胞(図9A、50μgのCE7−SP2/0からのオリゴ糖)、または培地に添加したテトラサイクリンの濃度が異なるため、GnT III遺伝子を異なるレベルで発現するCHO−tetGnTIII−chCE7細胞培養物のいずれかにより生成されたchCE7サンプルからの中性オリゴ糖混合物のMALDI/TOF−MSスペクトルを示す。テトラサイクリン濃度の大きさを低減する、すなわち、GnT III遺伝子発現のレベルを増加させることにおいて、後者のサンプルは、CE7−2000t(図9B、37.5μgの抗体からのオリゴ糖)、CE7−60t(図9C、37.5μgの抗体からのオリゴ糖)、CE7−30t(図9D、25μgの抗体からのオリゴ糖)およびCE7−15t(図9E、10μgの抗体からのオリゴ糖)である。
【図9B】図9は、SP2/0マウスミエローマ細胞(図9A、50μgのCE7−SP2/0からのオリゴ糖)、または培地に添加したテトラサイクリンの濃度が異なるため、GnTIII遺伝子を異なるレベルで発現するCHO−tetGnTIII−chCE7細胞培養物のいずれかにより生成されたchCE7サンプルからの中性オリゴ糖混合物のMALDI/TOF−MSスペクトルを示す。テトラサイクリン濃度の大きさを低減する、すなわち、GnT III遺伝子発現のレベルを増加させることにおいて、後者のサンプルは、CE7−2000t(図9B、37.5μgの抗体からのオリゴ糖)、CE7−60t(図9C、37.5μgの抗体からのオリゴ糖)、CE7−30t(図9D、25μgの抗体からのオリゴ糖)およびCE7−15t(図9E、10μgの抗体からのオリゴ糖)である。
【図9C】図9は、SP2/0マウスミエローマ細胞(図9A、50μgのCE7−SP2/0からのオリゴ糖)、または培地に添加したテトラサイクリンの濃度が異なるため、GnTIII遺伝子を異なるレベルで発現するCHO−tetGnTIII−chCE7細胞培養物のいずれかにより生成されたchCE7サンプルからの中性オリゴ糖混合物のMALDI/TOF−MSスペクトルを示す。テトラサイクリン濃度の大きさを低減する、すなわち、GnT III遺伝子発現のレベルを増加させることにおいて、後者のサンプルは、CE7−2000t(図9B、37.5μgの抗体からのオリゴ糖)、CE7−60t(図9C、37.5μgの抗体からのオリゴ糖)、CE7−30t(図9D、25μgの抗体からのオリゴ糖)およびCE7−15t(図9E、10μgの抗体からのオリゴ糖)である。
【図9D】図9は、SP2/0マウスミエローマ細胞(図9A、50μgのCE7−SP2/0からのオリゴ糖)、または培地に添加したテトラサイクリンの濃度が異なるため、GnTIII遺伝子を異なるレベルで発現するCHO−tetGnTIII−chCE7細胞培養物のいずれかにより生成されたchCE7サンプルからの中性オリゴ糖混合物のMALDI/TOF−MSスペクトルを示す。テトラサイクリン濃度の大きさを低減する、すなわち、GnT III遺伝子発現のレベルを増加させることにおいて、後者のサンプルは、CE7−2000t(図9B、37.5μgの抗体からのオリゴ糖)、CE7−60t(図9C、37.5μgの抗体からのオリゴ糖)、CE7−30t(図9D、25μgの抗体からのオリゴ糖)およびCE7−15t(図9E、10μgの抗体からのオリゴ糖)である。
【図9E】図9は、SP2/0マウスミエローマ細胞(図9A、50μgのCE7−SP2/0からのオリゴ糖)、または培地に添加したテトラサイクリンの濃度が異なるため、GnTIII遺伝子を異なるレベルで発現するCHO−tetGnTIII−chCE7細胞培養物のいずれかにより生成されたchCE7サンプルからの中性オリゴ糖混合物のMALDI/TOF−MSスペクトルを示す。テトラサイクリン濃度の大きさを低減する、すなわち、GnT III遺伝子発現のレベルを増加させることにおいて、後者のサンプルは、CE7−2000t(図9B、37.5μgの抗体からのオリゴ糖)、CE7−60t(図9C、37.5μgの抗体からのオリゴ糖)、CE7−30t(図9D、25μgの抗体からのオリゴ糖)およびCE7−15t(図9E、10μgの抗体からのオリゴ糖)である。
【図10】図10は、GnT III触媒された反応を介する、複合型バイセクト型オリゴ糖を導く、N結合型オリゴ糖生合成経路を記載する。Mはマンノース;GnはN−アセチルグルコサミン(GlcNAc);Gはガラクトース;GnbはバイセクティングGlcNAc;fはフコースを示す。オリゴ糖命名法は、コアオリゴ糖に結合されたM、GnおよびG残基を数えること、およびGnbを含めることによってバイセクティングGlcNAcの存在を示すことからなる。オリゴ糖コアは、それ自体が2つのGn残基から構成され、そしてフコースを含んでも、含まなくてもよい。オリゴ糖の主要なクラスは、破線で囲んだ枠の中に示す。Man Iはゴルジマンノシダーゼ;GnTはGlcNAcトランスフェラーゼ;およびGalTはガラクトシルトランスフェラーゼを表す。MALDI/TOF−MS分析で観察される、主要なナトリウムと会合したオリゴ糖イオンに関連する質量は、各オリゴ糖の隣に示す。潜在的にコアがフコシル化され得るオリゴ糖については、フコシル化形態(+f)および非フコシル化形態(−f)の両方に関連する質量を示す。
【図11】図11は、GnT III触媒反応を介して、複合型バイセクト型オリゴ糖および混成型バイセクト型オリゴ糖を導くN結合型オリゴ糖生合成経路を示す。Mはマンノース;GnはN−アセチルグルコサミン(GlcNAc);Gはガラクトース;GnbはバイセクティングGlcNAc;fはフコースを示す。オリゴ糖命名法は、共通のオリゴ糖に結合されたM、GnおよびG残基を数えること、およびGnbを含めることによってバイセクティングGlcNAcの存在を示すことからなる。オリゴ糖コアは、それ自体が2つのGn残基から構成され、そしてフコースを含んでも、含まなくてもよい。オリゴ糖の主要なクラスは、破線で囲んだ枠の中に示す。Man Iはゴルジマンノシダーゼ;GnTはGlcNAcトランスフェラーゼ;およびGalTはガラクトシルトランスフェラーゼを表す。MALDI/TOF−MS分析で観察される、主要なナトリウムと会合したオリゴ糖イオンと関連する質量は、各オリゴ糖の隣に示す。潜在的にコアがフコシル化され得るオリゴ糖については、フコシル化形態(+f)および非フコシル化形態(−f)の両方に関連する質量を示す。
【図12】図12は、異なるchCE7サンプルのADCC活性を示す。ヒトリンパ節によるIMR−32神経芽腫細胞の溶解(標的:エフェクター比は1:19、37℃で16時間のインキュベーション)を、chCE7サンプルの異なる濃度で媒介し、蛍光色素の保持によって測定した。細胞傷害性の割合は、抗体の非存在下でのシグナルを差し引いた後に、総溶解コントロール(界面活性剤による)に対して算出した。
【図13】図13は、異なるテトラサイクリン濃度で増殖させた、CHO−tet−GnT IIIの異なる培養物のGnT III発現を使用して、異なるC2B8抗体サンプルを生成したことを記載する。2000ng/ml(レーンC)および25ng/ml(レーンD)のテトラサイクリン濃度で増殖させた各培養物からの細胞溶解物をSDS−PAGEで分離し、メンブラン上にブロットし、そして1次抗体および2次抗体として、それぞれ、9E10(前出を参照のこと)および抗マウス西洋ワサビペルオキシダーゼを用いてプローブした。レーンAは、陰性コントロールを示す。
【図14】図14は、懸濁物中での細胞を用いた間接免疫蛍光アッセイを使用するC2B8抗CD20モノクローナル抗体の抗原結合の特異性を示す。CD20陽性細胞(SB細胞;ATCC受託番号ATCC CCL120)およびCD20陰性細胞(HSB細胞;ATCC受託番号ATCC CCL120.1)(それぞれ、図14Aおよび14B)を使用した。各型の細胞を、1次抗体として25ng/mlテトラサイクリンで生成したC2B8抗体とともにインキュベートした。陰性コントロールは、1次抗体の代わりに、HBSSBを含めた。抗ヒトIgG Fc特異的ポリクローナルFITC結合体化抗体を2次抗体として全てのサンプルで使用した。
【図15】図15は、異なる抗体濃度(0.04〜5μg/ml)での異なるC2B8抗体サンプルのADCC活性を示す。サンプルC2B8−ntは、GnT III発現なしの細胞株で生成されたC2B8抗体のADCC活性を表す。サンプルC2B8−2000t、C2B8−50tおよびC2B8−25tは、漸減テトラサイクリン濃度(すなわち、漸増GnT III発現)で生成した3つの抗体サンプルのADCC活性を示す。
【発明を実施するための形態】
【0018】
(VII.発明の詳細な説明)
(A.一般的な概要)
本発明の目的は、タンパク質(特に抗体(抗体分子全体、抗体フラグメント、または免疫グロブリンのFc領域に等価な領域を含む融合タンパク質を含む))の糖形態を提供して、治療的タンパク質の新たな改変体を生成することである。本発明は、細胞のグリコシル化反応ネットワークが、その集団内での特定の糖形態の割合を最大化するように操作され得、そして特定の糖形態が改善された治療特性を有するという本発明者らの発見に一部基づく。本発明はさらに、改善された治療価値を有するタンパク質の糖形態を同定する方法、およびそれらを再現可能に生成する方法の発見に一部基づく。本発明はさらに、バイセクティングGlcNAcを保有するN結合型複合型オリゴ糖を増加させるために、抗体生成細胞での糖タンパク質改変グリコシルトランスフェラーゼ発現の好ましい範囲が存在するという発見に一部基づく。
【0019】
そのように、本発明は、一般に、治療特性を改変し、そして改善するようにタンパク質をグリコシル化操作するための方法に関する。より詳細には、本発明は、改変されたグリコシル化パターンを有し、その結果、増強された抗体依存性細胞傷害性(ADCC)を生じる抗体を宿主細胞において生成するための方法を記載する。方法の実施のために、本発明は、抗体をコードする核酸および糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸を有する宿主細胞を提供する。さらに、本発明は、改変されたグリコシル化パターンを有し、その結果、増強された抗体依存性細胞傷害性を生じる所望の抗体の発現を可能にする条件下でこのような宿主細胞を培養する方法およびプロトコルを提供する。さらに、増強された抗体依存性細胞傷害性を有する、そのように生成された抗体を単離する方法を記載する。
【0020】
本発明のさらに特定の実施態様において、2つのモノクローナル抗体(すなわち、抗神経芽腫抗体chCE7および抗CD20抗体C2B8)はモデル治療糖タンパク質として使用され、そして標的糖形態は、特定のクラスの糖質(すなわち、バイセクティングN−アセチルグルコサミン(GlcNAc)で改変されたN結合型複合型バイアンテナリーオリゴ糖)を有する糖形態である。本発明によって提供されるモデル系においては、CHO細胞が宿主細胞として使用されるが、多くの他の細胞系が宿主細胞系として企図され得る。種々の型のN結合型オリゴ糖にバイセクティングGlcNAcを付加するグリコシルトランスフェラーゼであるGlcNAcトランスフェラーゼIII(GnT III)は、通常は、CHO細胞によって生成されない。StanleyおよびCampell、1984、J.Biol.Chem.261:13370−13378。
【0021】
GnT III過剰発現の効果を実験的に調査するために、ラットGnT III cDNAのテトラサイクリン調節性過剰発現を有するCHO細胞株が樹立された。この実験系を使用して、本発明者らは、高レベルへのGnT IIIの過剰発現が増殖阻害を導き、そして細胞に対して毒性であることを発見した。GnT V(これは、異なるグリコシルトランスフェラーゼである)のテトラサイクリン調節性過剰発現を有する別のCHO細胞株は、同じ阻害効果を示した。このことは、糖タンパク質改変グリコシルトランスフェラーゼの過剰発現の一般的特徴であり得ることを示す。細胞増殖に対する酵素発現の効果は、糖タンパク質改変グリコシルトランスフェラーゼ過剰発現のレベルに上限を設定し、従って、接近しにくいグリコシル化部位が、非調節性発現ベクターを使用するグリコシル化経路およびグリコシル化パターンの操作によって改変され得る程度もまた制限し得る。
【0022】
基本レベルと毒性レベルとの間の範囲でGnT III発現を制御することによって糖形態分布が異なるchCE7 mAbおよびC2B8サンプルのセットの生成が開示される。chCE7 mAbサンプルのADCC活性の測定によって、インビトロでの最大chCE7生物学的活性についてのGnT III発現の最適範囲が示された。この活性は、Fcと関連する複合型バイセクト型オリゴ糖のレベルと相関した。実際の範囲内(すなわち、有意な増殖阻害も毒性も見られない)でのGnT IIIの発現は、chCE7サンプルのこのセットについて標的の複合型バイセクト型構造の増加を導いた。高レベルのGnT IIIで産生されたchCE7サンプルのMALDI/TOF質量分析におけるオリゴ糖ピークのパターンは、潜在的なGnT III基質の有意な割合が混成型バイセクト型オリゴ糖副生成物に転換されることを示す。従って、経路のさらなる操作による、これらの副生成物の最小化は、有益であり得る。
【0023】
(B.グリコシル化パターンの改変を指向するタンパク質をコードする核酸の同定および生成)
本発明は、グリコシル化パターンの変化が指向される、目的の任意のタンパク質、タンパク質フラグメントまたはペプチドの変化した糖形態の生成に適切な宿主細胞系を、提供する。そのような目的のタンパク質、タンパク質フラグメントまたはペプチドをコードする核酸は、一般に当該分野において公知の方法によって得られ得る。例えば、核酸は、cDNAライブラリーまたはゲノムライブラリーから単離され得る。使用され得るクローニング戦略のための総説について、例えば、Maniatis、1989、Molecular Cloning、A Laboratory Manual、Cold Springs Harbor Press、N.Y.;およびAusubelら、1989、Current Protocols in Molecular Biology、(Green Publishing Associates and Wiley Interscience、N.Y.)を参照のこと。
【0024】
本発明の別の実施態様において、目的のタンパク質、タンパク質フラグメントまたはペプチドのコード配列は、当該分野において周知の化学的方法を使用して、全体的にか、または部分的に、合成され得る。例えば、Caruthersら、1980、Nuc.Acids Res.Symp.Ser.7:215〜233;CreaおよびHorn、1980、Nuc.Acids Res.USA 9:2331;MatteucciおよびCaruthers、1980、Tetrahedron Letters 21:719;ChowおよびKempe、1981、Nuc.Acids Res.9:2807〜2817を参照のこと。あるいは、そのタンパク質自体が、そのアミノ酸配列の全体または部分を合成する化学的方法を使用して生成され得る。例えば、ペプチドは、固相技術によって合成され、樹脂から切断され、そして調製用高速液体クロマトグラフィーによって精製され得る。例えば、Creighton、1983、Protein Structures And Molecular Principles、W.H.Freeman and Co.、N.Y.,50〜60頁を参照のこと。合成ペプチドの組成は、アミノ酸分析またはアミノ酸配列決定(例えば、エドマン分解手順;Creighton、1983、Proteins、Structures and Molecular
Principles、W.H.Freeman and Co.、N.Y.34〜49頁を参照のこと)によって確認され得る。
【0025】
好ましい実施態様において、本発明は、抗体または抗体フラグメントあるいは増強した抗体依存性細胞傷害性を有する抗体フラグメントを含有する融合タンパク質の糖形態の産生のための宿主細胞系の生成および使用のための方法を提供する。標的エピトープの同定、およびグリコシル化パターンの改変が所望される、可能性のある治療的価値を有する抗体の生成、ならびにその対応するコード核酸配列の単離は、本発明の範囲内である。
【0026】
当該分野において公知の種々の手順が、目的の標的エピトープに対する抗体の産生のために使用され得る。そのような抗体としては、ポリクローナル抗体、モノクローナル抗体、キメラ抗体、単鎖抗体、Fabフラグメント、およびFab発現ライブラリーによって産生されたフラグメントが挙げられるが、これらに限定されない。そのような抗体は、例えば、診断剤および治療剤として有用であり得る。治療剤として、中和抗体(すなわち、リガンド、基質またはアダプター分子との結合に競合する抗体)が、特に好ましく興味深い。
【0027】
抗体の産生のために、種々の宿主動物(ウサギ、マウス、ラットなどを含むがこれらに限定されない)が、目的の標的タンパク質を用いて、注射によって免疫される。種々のアジュバント(宿主の種に依存して、フロイント(完全および不完全)、水酸化アルミニウムのようなミネラルゲル、リゾレシチンのような界面活性剤、プルロニックポリオール(pluronic polyols)、ポリアニオン、ペプチド、懸濁オイル、キーホールリンペットヘモシアニン、ジニトロフェノール、およびBCG(bacille Calmette−Guerin)およびCorynebacterium parvumのような可能性のある有用なヒトアジュバントが挙げられるが、これらに限定されない)を使用して、免疫応答を増加し得る。
【0028】
目的の標的に対するモノクローナル抗体が、培養物中の連続的細胞株による抗体分子産生のために提供される任意の技術を使用して、調製され得る。この技術には、KohlerおよびMilstein(1975、Nature 256:495〜497)によって最初に記載されたハイブリドーマ技術、ヒトB細胞ハイブリドーマ技術(Kosborら、1983、Immunology Today、4:72;Coteら、1983、Proc.Natl.Acad.Sci.USA 80:2026〜2030)およびEBVハイブリドーマ技術(Coleら、1985、Monoclonal Antibodies and Cancer Therapy、Alan R.Liss、Inc.77〜96頁)が挙げられるが、これらに限定されない。さらに、適切な抗原特異性のマウス抗体分子由来の遺伝子を、適切な生物学的活性のヒト抗体分子由来の遺伝子とともに、スプライシングすることによる、「キメラ抗体」の産生のために開発された技術(Morrisonら、1984、Proc.Natl.Acad.Sci.USA 81:6851〜6855;Neubergerら、1984、Nature 312:604〜608;Takedaら、1985、Nature 314:452〜454)が、使用され得る。あるいは、単鎖抗体の産生について記載される技術(米国特許第4,946,778号)が、所望の特異性を有する単鎖抗体の産生に適用され得る。
【0029】
目的の標的タンパク質の特異的結合部位を含有する抗体フラグメントが、公知の技術によって生成され得る。例えば、そのようなフラグメントとしては、抗体分子のペプシン消化によって生成され得るF(ab’)2フラグメント、およびF(ab’)2フラグメントのジスルフィド架橋の還元によって生成され得るFabフラグメントが挙げられるが、これらに限定されない。あるいは、Fab発現ライブラリーが、構築され得(Huseら、1989、Science 246:1275〜1281)、目的の標的タンパク質に対する所望の特異性を有するモノクローナルFabフラグメントの迅速かつ容易な同定を可能にする。
【0030】
一旦、グリコシル化パターンにおける改変が所望される抗体または抗体フラグメントが同定されると、コード核酸配列が、当該分野において周知の技術を用いて、同定および単離される。上記を参照のこと。
【0031】
(C.変化したグリコシル化パターンを有するタンパク質の産生のための細胞株の生成)
本発明は、改変されたグリコシル化パターンを有するタンパク質の生成のための宿主細胞発現系を提供する。具体的には、本発明は、改善された治療的価値を有するタンパク質の糖形態を生成するための宿主細胞系を提供する。従って、本発明は、糖タンパク質改変グリコシルトランスフェラーゼの発現を増加するように選択されるか、または操作された宿主細胞発現系を提供する。具体的には、そのような宿主細胞発現系を操作して、構成性プロモーター系または調節性プロモーター系と作動可能に連結された、糖タンパク質改変グリコシルトランスフェラーゼをコードする組換え核酸分子を含有し得る。あるいは、糖タンパク質改変グリコシルトランスフェラーゼを、天然に産生する、産生するように誘導される、および/または産生するように選択される、宿主発現系が、使用され得る。
【0032】
1つの特定の実施態様において、本発明は、糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸を発現するように操作された宿主細胞を提供する。1つの局面において、宿主細胞は、糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子を含有する核酸分子で、形質転換されるか、またはトランスフェクションされる。代替的な局面において、この宿主細胞は、内因性の糖タンパク質改変グリコシルトランスフェラーゼが活性化されるように、操作および/または選択される。例えば、宿主細胞は、内因性の糖タンパク質改変グリコシルトランスフェラーゼの発現を誘発するような変異を含有するように選択され得る。この局面は、宿主細胞がCHO lec10変異株である1つの特定の実施態様において例示される。あるいは、宿主細胞は、内因性の糖タンパク質改変グリコシルトランスフェラーゼが活性化されるように操作され得る。再度別の代替物において、宿主細胞は、内因性の糖タンパク質改変グリコシルトランスフェラーゼが宿主細胞染色体中に調節性プロモーターエレメントを挿入することによって活性化されるように操作される。さらなる代替物において、宿主細胞は、内因性の糖タンパク質改変グリコシルトランスフェラーゼが宿主細胞染色体中に構成性プロモーターエレメント、トランスポゾン、またはレトロウイルスエレメントを挿入することによって活性化されるように操作される。
【0033】
一般に、任意のタイプの培養細胞株を、本発明の宿主細胞株を操作するバックグラウンドとして使用し得る。好ましい実施態様において、CHO細胞、BHK細胞、NS0細胞、SP2/0細胞またはハイブリドーマ細胞株を、本発明の操作された宿主細胞を生成するための、バックグラウンド細胞株として使用する。
【0034】
本発明は、本明細書において規定される糖タンパク質改変グリコシルトランスフェラーゼの任意のタイプを発現する操作された宿主細胞を包含することが意図される。しかし、好ましい実施態様において、本発明の宿主細胞によって発現される少なくとも1つの糖タンパク質改変グリコシルトランスフェラーゼは、GnT IIIであるか、あるいは、β(1,4)−N−アセチルグルコサミニルトランスフェラーゼV(β(1,4)−N−acetylglucosaminyltrasferase V)(GnT V)である。しかし、代表的にGnT IIIまたはGnT Vに加えて、β(1,4)−ガラクトシルトランスフェラーゼ(GalT)、およびマンノシダーゼII(ManII)を含む、糖タンパク質改変グリコシルトランスフェラーゼの別のタイプもまた、宿主系において発現され得る。本発明の1つの実施態様において、GnT IIIは、GalTと同時に発現される。本発明の別の実施態様において、GnT IIIは、ManIIと同時に発現される。本発明のさらなる実施態様において、GnT IIIは、GalTおよびManIIと同時に発現される。しかし、糖タンパク質改変グリコシルトランスフェラーゼの任意の他の変更は、本発明の範囲内である。さらに、宿主細胞系内でのグリコシダーゼの発現が所望され得る。
【0035】
糖タンパク質改変グリコシルトランスフェラーゼをコードする核酸の1つまたはいくつかは、構成性プロモーターの制御下、あるいは調節性発現系の制御下において発現され得る。適切な調節性発現系としては、テトラサイクリン−調節性発現系、エクジソン誘導性発現系、lacスイッチ発現系、グルココルチコイド誘導性発現系、温度誘導性プロモーター系、およびメタロチオネイン金属誘導性発現系が挙げられるが、これらに限定されない。糖タンパク質改変グリコシルトランスフェラーゼをコードするいくつかの異なる核酸が、宿主細胞系内に含有される場合、それらのいくつかは、構成性プロモーターの制御下において発現され得るが、一方、その他は、調節性プロモーターの制御下で発現される。最適な発現レベルは、目的の各タンパク質について異なり、そして慣用的な実験を使用して決定される。発現レベルは、当該分野において一般に公知の方法(グリコシルトランスフェラーゼ特異的抗体を使用するウエスタンブロット分析、グリコシルトランスフェラーゼ特異的核酸プローブを使用するノザンブロット分析、または酵素活性の測定を含む)によって決定される。あるいは、グリコシルトランスフェラーゼの生合成産物と結合するレクチン(例えば、E4−PHAレクチン)を使用し得る。さらなる代替物において、核酸
は、レポーター遺伝子と作動可能に連結され得る;糖タンパク質改変グリコシルトランスフェラーゼの発現レベルは、レポーター遺伝子の発現レベルと相関するシグナルを測定することによって決定される。このレポーター遺伝子は、この糖タンパク質改変グリコシルトランスフェラーゼをコードする核酸とともに、単一のmRNA分子として転写され得る;その対応するコード配列は、内部リボゾーム進入部位(IRES)またはキャップ非依存性翻訳エンハンサー(CITE)のいずれかと連結され得る。このレポーター遺伝子は、この糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸とともに翻訳され得、その結果単一のポリペプチド鎖が形成される。糖タンパク質改変グリコシルトランスフェラーゼをコードする核酸は、単一のプロモーターの制御下のレポーター遺伝子に作動可能に連結され得、その結果糖タンパク質改変グリコシルトランスフェラーゼおよびレポーター遺伝子をコードする核酸は、2つの別のメッセンジャーRNA(mRNA)分子に選択的スプライシングされるRNA分子に転写される;生じるRNAの一方は、そのレポータータンパク質に翻訳され、そして他方は糖タンパク質改変グリコシルトランスフェラーゼに翻訳される。
【0036】
糖タンパク質改変グリコシルトランスフェラーゼをコードするいくつかの異なる核酸が発現する場合、これらの核酸は、1つかまたはいくつかのmRNA分子として転写されるように配列され得る。これらの核酸が、単一のmRNA分子として転写される場合、その対応するコード配列は、内部リボゾーム進入部位(IRES)またはキャップ非依存性翻訳エンハンサー(CITE)のいずれかと連結され得る。これらの核酸は、単一のプロモーターから1つのRNA分子に転写され、いくつかの別々のメッセンジャーRNA(mRNA)分子に選択的スプライシングされ得る。次いで、これらmRNAの各々は、対応するコードされた糖タンパク質改変グリコシルトランスフェラーゼに翻訳される。
【0037】
他の実施態様において、本発明は、治療的タンパク質(例えば、増強された抗体依存性細胞傷害性を有する抗体、およびFc媒介細胞傷害性を促進する表面にIgG Fc領域を提示する細胞)の生成のための宿主細胞発現系を提供する。一般に、宿主細胞発現系は、操作および/または選択され、糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸ともに、改変された糖形態の生成が所望されるタンパク質をコードする核酸を発現する。1つの実施態様において、宿主細胞系は、糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子でトランスフェクトされる。代表的には、トランスフェクトされた細胞を選択して、糖タンパク質改変グリコシルトランスフェラーゼを安定に発現するクローンを同定および単離する。別の実施態様において、この宿主細胞は、内因性グリコシルトランスフェラーゼの発現について選択された。例えば、そうでなければサイレントな糖タンパク質改変グリコシルトランスフェラーゼの発現を誘発する変異を有する細胞が、選択され得る。例えば、CHO細胞は、特定の変異体において(例えば、変異体Lec10において)活性な、サイレントなGnT III遺伝子を保有することが公知である。さらに、サイレントな糖タンパク質改変グリコシルトランスフェラーゼ遺伝子を活性化するために、当該分野において公知の方法(調節性プロモーターまたは構成性プロモーターの挿入、あるいはトランスポゾン、レトロウイルスエレメントの使用などを含む)が使用され得る。遺伝子ノックアウト技術の使用、またはリボザイム方法の使用を用いて、宿主細胞のグリコシルトランスフェラーゼおよび/またはグリコシダーゼ発現レベルを適応させ得る。この使用も本発明の範囲内である。
【0038】
培養細胞株の任意のタイプが、本発明の宿主細胞株を操作するためのバックグラウンドとして使用され得る。好ましい実施態様において、CHO細胞、BHK細胞、NS0細胞、SP2/0細胞である。代表的には、そのような細胞株は操作され、抗体分子全体、抗体フラグメント、または免疫グロブリンのFc領域と等価の領域を含む融合タンパク質をコードする少なくとも1つのトランスフェクトされた核酸をさらに含む。別の実施態様において、目的の特定の抗体を発現するハイブリドーマ細胞株を、本発明の操作された宿主細胞を生成するためのバックグラウンド細胞株として使用する。
【0039】
代表的には、宿主細胞系内の少なくとも1つの核酸は、GnT III、あるいはGnT Vをコードする。しかし、他のタイプの糖タンパク質改変グリコシルトランスフェラーゼ(代表的にはGnT IIIまたはGnT Vに加え、GalTおよびManIIを含む)が、宿主系内で発現され得る。本発明の1つの実施態様において、GnT IIIは、GalTと同時に発現される。本発明の別の実施態様において、GnT IIIは、ManIIと同時に発現される。本発明のさらなる実施態様において、GnT IIIは、GalTおよびManIIと同時に発現される。しかし、糖タンパク質改変グリコシルトランスフェラーゼの任意の他の変更は、本発明の範囲内である。さらに、宿主細胞系内でのグリコシダーゼの発現が所望され得る。
【0040】
糖タンパク質改変グリコシルトランスフェラーゼをコードする核酸の1つまたはいくつかは、構成性プロモーターの制御下、あるいは調節性発現系の制御下において発現され得る。適切な調節性発現系としては、テトラサイクリン−調節性発現系、エクジソン誘導性発現系、lacスイッチ発現系、グルココルチコイド誘導性発現系、温度誘導性プロモーター系、およびメタロチオネイン金属誘導性発現系が挙げられるがこれらに限定されない。糖タンパク質改変グリコシルトランスフェラーゼをコードするいくつかの異なる核酸が、宿主細胞系内に含有される場合、それらのいくつかは、構成性プロモーターの制御下において発現され得るが、一方、その他は、調節性プロモーターの制御下で発現される。最適な発現レベルは、目的の各タンパク質について異なり、そして慣用的な実験を使用して決定される。発現レベルは、当該分野において一般に公知の方法(グリコシルトランスフェラーゼ特異的抗体を使用するウエスタンブロット分析、グリコシルトランスフェラーゼ特異的核酸プローブを使用するノザンブロット分析、または酵素活性の測定を含む)によって決定される。あるいは、グリコシルトランスフェラーゼの生合成産物と結合するレクチン(例えば、E4−PHAレクチン)を使用し得る。さらなる代替物において、核酸は
、レポーター遺伝子と作動可能に連結され得る;糖タンパク質改変グリコシルトランスフェラーゼの発現レベルは、レポーター遺伝子の発現レベルと相関するシグナルを測定することによって決定される。このレポーター遺伝子は、この糖タンパク質改変グリコシルトランスフェラーゼをコードする核酸とともに、単一のmRNA分子として転写され得る;その対応するコード配列は、内部リボゾーム進入部位(IRES)またはキャップ非依存性翻訳エンハンサー(CITE)のいずれかと連結され得る。このレポーター遺伝子は、この糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸とともに翻訳され得、その結果単一のポリペプチド鎖が形成される。糖タンパク質改変グリコシルトランスフェラーゼをコードする核酸は、単一のプロモーターの制御下のレポーター遺伝子に作動可能に連結され得、その結果糖タンパク質改変グリコシルトランスフェラーゼおよびレポーター遺伝子をコードする核酸は、2つの別々のメッセンジャーRNA(mRNA)分子に選択的スプライシングされるRNA分子に転写される;生じるRNAの1つは、そのレポータータンパク質に翻訳され、そして他方は糖タンパク質改変グリコシルトランスフェラーゼに翻訳される。
【0041】
糖タンパク質改変グリコシルトランスフェラーゼをコードするいくつかの異なる核酸が発現する場合、これらの核酸は、1つかまたはいくつかのmRNA分子として転写されるように配列され得る。これらの核酸が、単一のmRNA分子として転写される場合、その対応するコード配列は、内部リボゾーム進入部位(IRES)またはキャップ非依存性翻訳エンハンサー(CITE)のいずれかと連結され得る。これらの核酸は、単一のプロモーターから1つのRNA分子に転写され、いくつかの別々のメッセンジャーRNA(mRNA)分子に選択的スプライシングされ得る。次いで、これらmRNAの各々は、対応するコードされた糖タンパク質改変グリコシルトランスフェラーゼに翻訳される。
【0042】
(1.発現系)
当業者に周知の方法を使用して、目的のタンパク質のコード配列および糖タンパク質改変グリコシルトランスフェラーゼのコード配列、および適切な転写/翻訳制御シグナルを含有する発現ベクターを構築し得る。これらの方法としては、インビトロ組換えDNA技術、合成技術およびインビボ組換え/遺伝子組換えが挙げられる。例えば、Maniatisら、1989、Molecular Cloning A Laboratory Manual、Cold Spring Harbor Laboratory、N.Y.およびAusubelら、1989、Current Protocols in Molecular Biology、Greene Publishing Associates and Wiley Interscience、N.Y.に記載される技術を参照のこと。
【0043】
種々の宿主−発現ベクター系を使用して、目的のタンパク質のコード配列、および糖タンパク質改変グリコシルトランスフェラーゼのコード配列を発現し得る。好ましくは、哺乳動物細胞を、目的のタンパク質のコード配列、および糖タンパク質改変グリコシルトランスフェラーゼのコード配列を含有する、組換えプラスミドDNAまたはコスミドDNA発現ベクターでトランスフェクトされた宿主細胞系として使用する。最も好ましくは、CHO細胞、BHK細胞、NS0細胞、またはSP2/0細胞あるいはハイブリドーマ細胞株を、宿主細胞系として使用する。代替的な実施態様において、他の真核生物宿主細胞系(目的のタンパク質のコード配列、および糖タンパク質改変グリコシルトランスフェラーゼのコード配列を含む組換え酵母発現ベクターで形質転換された酵母細胞;目的のタンパク質のコード配列、および糖タンパク質改変グリコシルトランスフェラーゼのコード配列を含む組換えウイルス発現ベクター(例えば、バキュロウイルス)で感染された昆虫細胞系;目的のタンパク質のコード配列、および糖タンパク質改変グリコシルトランスフェラーゼのコード配列を含む、組換えウイルス発現ベクター(例えば、カリフラワーモザイクウイルス、CaMV;タバコモザイクウイルス、TMV)で感染されたか、または組換えプラスミド発現ベクター(例えば、Tiプラスミド)で形質転換された植物細胞系;あるいは目的のタンパク質をコードするDNA、および糖タンパク質改変グリコシルトランスフェラーゼのコード配列の複数のコピー(安定に増幅された(CHO/dhfr)か、もしくは二重微小染色体中で不安定に増幅された(例えば、マウス細胞株))を含有するように操作された細胞株を含む、組換えウイルス発現ベクター(例えば、アデノウイルス、ワクシニアウイルス)で感染された動物細胞系を含む)が意図され得る。
【0044】
本発明の方法のために、安定な発現が、一般には一過性の発現よりも好ましい。なぜなら、安定な発現は、代表的により再現性のある結果を達成し、そしてまた、大スケール産生により適合しやすいからである。ウイルスの複製起点を含有する発現ベクターを使用するよりむしろ、宿主細胞を、適切な発現制御エレメント(例えば、プロモーター、エンハンサー配列、転写ターミネーター、ポリアデニル化部位など)によって制御されるそれぞれのコード核酸、および選択マーカーで形質転換し得る。外来DNAの導入後、操作された細胞を、富化された培地中で1〜2日間増殖させ得、次に、選択培地に変更する。組換えプラスミド中の選択マーカーは、選択に対する耐性を付与し、そしてプラスミドをその染色体中に安定に組み込み、そして増殖してコロニーを形成し、その結果クローン化され、細胞株に拡大される細胞の選択を可能にする。
【0045】
それぞれ、tk-、hgprt-、またはaprt-細胞において使用され得る、単純ヘ
ルペスウイルスチミジンキナーゼ(Wiglerら、1977、Cell 11:223)、ヒポキサンチン−グアニンホスホリボシルトランスフェラーゼ(SzybalskaおよびSzybalski、1962、Proc.Natl.Acad.Sci.USA
48:2026)、およびアデニンホスホリボシルトランスフェラーゼ(Lowyら、1980、Cell 22:817)遺伝子を含むが、これらに限定されない多数の選択系が使用され得る。抗代謝産物耐性が、メトトレキサートに対する耐性を付与するdhfr(Wiglerら、1980、Proc.Natl.Acad.Sci.USA 77:3567;O’Hareら、1981、Proc.Natl.Acad.Sci.USA 78:1527)、ミコフェノール酸に対する耐性を付与するgpt(MulliganおよびBerg、1981、Proc.Natl.Acad.Sci.USA 78:2072);アミノグリコシドG−418に対する耐性を付与するneo(Colberre−Garapinら、1981、J.Mol.Biol.150:1);およびハイグロマイシンに対する耐性を付与するhygro(Santerreら、1984、Gene 30:147)遺伝子についての選択の基礎として使用され得る。近年、細胞にトリプトファンの代わりにインドールを利用させ得るtrpB;細胞にヒスチジンの代わりにヒスチノールを利用させ得るhisD(HartmenおよびMulligan、1988、Proc.Natl.Acad.Sci.USA 85:8047);グルタミンシンターゼ系;およびオルニチンデカルボキシラーゼインヒビターである2−(ジフルオロメチル)−DL−オルニチン、DFMOに対する耐性を付与するODC(オルニチンデカルボキシラーゼ)(McConlogue、1987、Current Communications in Molecular Biology、Cold Spring Harbor Laboratory編)と称するさらなる選択遺伝子が記載されている。
【0046】
(2.改変グリコシル化パターンを有するタンパク質を発現するトランスフェクト体および形質転換体の同定)
コード配列を含む宿主細胞および生物学的に活性な遺伝子産物を発現する宿主細胞は、少なくとも4つの一般的なアプローチにより同定され得る;(a)DNA−DNAハイブリダイゼーションまたはDNA−RNAハイブリダイゼーション;(b)「マーカー」遺伝子機能の存在または非存在;(c)宿主細胞におけるそれぞれのmRNA転写物の発現により測定されるような、転写レベルの評価;および(d)イムノアッセイまたはその生物学的活性により測定されるような、遺伝子産物の検出。
【0047】
第1のアプローチにおいて、発現ベクター中に挿入された目的のタンパク質のコード配列および糖タンパク質改変グリコシルトランスフェラーゼのコード配列の存在は、それぞれのコード配列、またはそれらのそれぞれの部分もしくは誘導体に相同であるヌクレオチド配列を含むプローブを用いて、DNA−DNAハイブリダイゼーションまたはDNA−RNAハイブリダイゼーションにより検出され得る。
【0048】
第2のアプローチにおいて、組換え発現ベクター/宿主系は同定され、そして特定の「マーカー」遺伝子機能(例えば、チミジンキナーゼ活性、抗生物質に対する耐性、メトトレキサートに対する耐性、形質転換表現型、バキュロウイルス中の封入体形成など)の存在または非存在に基づき、選択され得る。例えば、目的のタンパク質のコード配列および糖タンパク質改変グリコシルトランスフェラーゼのコード配列が、ベクターのマーカー遺伝子配列内に挿入されている場合、それぞれのコード配列を含む組換え体は、マーカー遺伝子機能の非存在により同定され得る。あるいは、マーカー遺伝子は、コード配列の発現を制御するために使用される同じプロモーターまたは異なるプロモーターの制御下で、コード配列と縦列して配置され得る。誘導または選択に応じたマーカーの発現は、目的のタンパク質のコード配列および糖タンパク質改変グリコシルトランスフェラーゼのコード配列の発現を示す。
【0049】
第3のアプローチにおいて、目的のタンパク質のコード領域および糖タンパク質改変グリコシルトランスフェラーゼのコード配列の転写活性は、ハイブリダイゼーションアッセイにより評価され得る。例えば、RNAは、単離されそして目的のタンパク質のコード配列および糖タンパク質改変グリコシルトランスフェラーゼのコード配列またはそれらの特定部分に相同なプローブを用いたノーザンブロットにより分析され得る。あるいは、宿主細胞の全核酸が抽出され、そしてこのようなプローブへのハイブリダイゼーションについて評価され得る。
【0050】
第4のアプローチにおいて、目的のタンパク質のタンパク質産物および糖タンパク質改変グリコシルトランスフェラーゼのコード配列の発現は、免疫学的に、例えば、ウェスタンブロット、イムノアッセイ(例えば、ラジオイムノ沈降、酵素結合イムノアッセイなど)により評価され得る。しかし、発現系の最終的な試験の成功は、生物学的に活性化な遺伝子産物の検出を含む。
【0051】
(D.変化したグリコシル化パターンを有するタンパク質およびタンパク質フラグメントの生成および使用)
(1.増強した抗体依存性細胞傷害性を有する抗体の生成および使用)
好ましい実施態様において、本発明は、増強した抗体依存性細胞傷害性を有する抗体および抗体フラグメントの糖形態を提供する。
【0052】
いくつかの型の癌の処置のための非結合体化モノクローナル抗体(mAb)の臨床的試行は、近年進んだ結果を生じた。Dillman、1997、Cancer Biother.&Radiopharm.12:223−225;Deoら、1997、Immunology Today 18:127。キメラの非結合体化IgG1が、低級なまたは小胞性のB細胞非ホジキンリンパ腫について認可されている(Dillman、1997、前出)が、一方別の非結合体化mAbである、ヒト化IgG1標的固形乳腫瘍もまた、第III相臨床試験において有望な結果を示しつつあった。Deoら、1997、前出。これらの2つのmAbの抗原は、それらの各々の腫瘍細胞において高度に発現され、そしてそれらの抗体は、インビトロおよびインビボでエフェクター細胞により強力な腫瘍破壊を媒介する。対照的に、良好な腫瘍特異性を有する多くの他の非結合体化mAbは、臨床的に有用である程十分な効力のエフェクター機能を引き起こし得ない。Frostら、1997、Cancer 80:317−333;Surfusら、1996、J.Immunother.19:184−191。これらのより弱いmAbのいくつかについて、補助的なサイトカイン治療が、現在も試験されている。サイトカインの添加は、その活性および循環リンパ球の数を増加することによって抗体依存性細胞傷害性(ADCC)を刺激し得る。Frostら、1997、前出;Surfusら、1996、前出。溶解物が抗体標的化細胞に対して攻撃するADCCは、リンパ球レセプターの、抗体の定常境域(Fc)への結合の際、引き起こされる。Deoら、1997、前出。
【0053】
非結合体化IgG1のADCC活性を増大させる、異なるが相補的であるアプローチは、抗体のFc領域を、リンパ球レセプターに対する親和性(FcγR)を増大させるように操作することである。タンパク質工学研究は、FcγRが、IgG CH2ドメインのより低いヒンジ領域と相互作用することを示した。Lundら、1996、J.Immunol.157:4963−4969。しかし、FcγR結合はまた、CH2領域における保存されたAsn297で共有結合的に付着したオリゴ糖の存在を必要とする。Lundら、1996、前出;WrightおよびMorrison、1997、Tibtech 15:26−31は、オリゴ糖およびポリペプチドの両方が直接相互作用部位に寄与するか、またはオリゴ糖が活性なCH2ポリペプチドコンフォメーションを維持するために必要とされるかのいずれかであることを示唆している。従って、オリゴ糖の構造の改変は、相互作用の親和性を増大させるための手段として、探索され得る。
【0054】
IgG分子は、そのFc領域中に2つのN結合オリゴ糖を保有し、一つは、各重鎖にある。任意の糖タンパク質と同様に、抗体は、糖形態の集団として産生される。この糖形態は、同じポリペプチド骨格を共有するが、グリコシル化部位に付着した異なるオリゴ糖を有する。血清IgGのFc領域中に通常見出されるオリゴ糖は、低いレベルの末端シアル酸およびバイセクティングN−アセチルグルコサミン(GlcNAc)、ならびに程度が可変し得る末端ガラクトシル化およびコアフルコシル化を有する、バイアンテナリー複合型オリゴ糖である(Wormaldら、1997、Biochemistry 36:130−1380)(図1)。いくつかの研究は、FcγR結合を必要とする最小の糖質構
造は、オリゴ糖コア内にあることを示唆する。Lundら、1996、前出。末端ガラクトースの除去は、ADCC活性において約2倍の減少をもたらす。このことは、FcγRレセプター結合におけるこれらの残基についての役割を示す。Lundら、1996、前出。
【0055】
非結合体化治療用mAbの生成のために産業および教育において使用される マウスまたはハムスター誘導細胞株は、通常、必要とされるオリゴ糖決定因子をFc部位に付着させる。しかし、これらの細胞株において発現されるIgGは、血清IgGにおいて低量で見出されるバイセクティングGlcNAcを欠く。Lifelyら、1995、Glycobiology 318:813−822。対照的には、ラット骨髄腫産生ヒト化IgG1(CAMPATH−1H)が、その糖形態のいくつかにおいてバイセクティングGlcNAcを保有することが、最近観察された。Lifelyら、1995、前出。ラット細胞誘導化抗体は、標準的な細胞株において産生されるCAMPATH−1H抗体と類似したインビトロのADCC活性に、しかし有意により低い抗体濃度で、達した。
【0056】
CAMPATH抗原は、通常、リンパ腫細胞に高いレベルで存在し、そしてこのキメラmAbは、バイセクティングGlcNAcの非存在下で高いADCC活性を有する。Lifelyら、1995、前出。Lifelyら、1995、前出の研究において、たとえ最大のインビトロADCC活性が、グリコシル化パターンの変化により増大しなかったとしても、この活性レベルが、バイセクト型オリゴ糖を保有する抗体の比較的低い抗体濃度で得られたという事実は、バイセクト型オリゴ糖の重要な役割を示唆する。Fc領域においてバイセクト型オリゴ糖を保有するこれらの抗体の糖形態を生成することによって、低い基底活性レベルを有するIgG1のADCC活性を増大するようなアプローチが発達した。
【0057】
N結合グリコシル化経路において、バイセクティングGlcNAcは、酵素β(1,4)−N−アセチルグルコサミニルトランスフェラーゼIII(GnT III)により添加される。Schachter、1986、Biochem.Cell Biol.64:163−181。Lifelyら、1995、前出は、内因的な一定のレベルでGnT
IIIを発現するラット骨髄腫細胞株を含む、異なるが非操作のグリコシル化機構を有する異なる細胞株において抗体を産生することによって、同じ抗体の異なるグリコシル化パターンを得た。対照的に、本発明者らは、単一の抗体産生CHO細胞株を使用した。これは、外部調節した様式で、異なるレベルのクローン化GnT III遺伝子を発現するようにあらかじめ操作されていた。このアプローチにより、本発明者らは、GnT IIIの発現と改変した抗体のADCC活性との間の厳密な相関を初めて確立し得た。
【0058】
本明細書中に実証されるように(実施例4(後述)を参照のこと)、開示される方法に従って改変されたC2B8抗体は、同一の細胞培養および精製条件下で産生された、標準的な非改変C2B8抗体よりも約16倍高いADCC活性を有した。簡単に言うと、GnT III発現を有さないCHO−tTA−C2B8細胞において発現されたC2B8抗体サンプルは、ヒトリンパ球によりSB細胞(CD20+)のインビトロ溶解として測定された、約31%の細胞傷害性活性を示した(1μg/ml抗体濃度で)。対照的に、基底の主な抑制レベルでGnT IIIを発現するCHO細胞培養物由来のC2B8抗体は、1μg/ml抗体濃度で、同じ抗体濃度でのコントロールに対してADCC活性の33%増大を示した。さらに、GnT IIIの発現の増大は、同じ抗体濃度でのコントロールと比較して、最大のADCC活性においてほぼ80%の大きな増加を生じた(1μg/ml抗体濃度で)。実施例4(後述)を参照のこと。
【0059】
増強した抗体依存性細胞傷害性を有する本発明のさらなる抗体は、以下を含むが、これらに限定されない:本発明の方法により産生される抗ヒト神経芽腫モノクローナル抗体(chCE7)、本発明の方法により産生されるキメラ抗ヒト腎細胞癌腫モノクローナル抗体(ch−G250)、本発明の方法により産生されるヒト化抗HER2モノクローナル抗体、本発明の方法により産生されるキメラ抗ヒト結腸、肺および乳癌腫モノクローナル抗体(ING−1)、本発明の方法により産生されるヒト化抗ヒト17−1A抗原モノクローナル抗体(3622W94)、本発明の方法により産生されるヒト化抗ヒト結腸直腸腫瘍抗体(A33)、本発明の方法により産生されるGD3ガングリオシドに対する抗ヒト骨髄腫抗体(R24)、ならびに本発明の方法により産生されるキメラ抗ヒト扁平上皮細胞癌モノクローナル抗体(SF−25)。さらに、本発明は、抗体フラグメント、および免疫グロブリンのFc領域と等価である領域を含む融合タンパク質に対して指向される。後述を参照のこと。
【0060】
(2.Fc媒介細胞傷害性を促進する免疫グロブリンのFc領域と等価な領域を含む融合タンパク質の生成および使用)
上記で議論されるように、本発明は、治療用抗体のADCC活性を増強するための方法に関する。これは、このような抗体のFc領域のグリコシル化パターンを操作することによって、特に、それらのFc領域で変換されたグリコシル化部位にN結合した複合型バイセクト型オリゴ糖を保有する抗体分子の割合を最大にすることによって達成される。この戦略は、治療用抗体によるのみならず、免疫グロブリンのFc領域に等価な領域を保有する任意の分子により媒介される、望ましくない細胞に対するFc媒介性細胞細胞傷害を、増大させるために適用され得る。なぜならば、グリコシル化の操作により導入された変化は、Fc領域のみに影響を与え、従って、エフェクター細胞の表面上のFcレセプターとのその相互作用は、ADCC機構に関与したからである。現在開示される方法で適用され得るFc含有分子としては以下が挙げられるが、これらに限定されない:(a)Fc領域のN末端に融合された標的タンパク質ドメインで作製された可溶性融合タンパク質(ChamovおよびAshkenazi、1996、TIBTECH 14:52)、および(b)Fc領域のN末端に融合された原形質膜に局在するII型膜貫通ドメインで作製された原形質膜固定化融合タンパク質(Stabila,P.E.、1998、Nature Biotech.16:1357)。
【0061】
可溶性融合タンパク質(a)の場合、標的ドメインは、融合タンパク質の望ましくない細胞(例えば、癌細胞)への結合、すなわち治療用抗体と類似した様式で、指向させる。従って、これらの分子によって媒介されるFc媒介細胞傷害性活性を増強するための、近年開示された方法の適用は、治療用抗体に適用される方法と同一である。米国特許仮出願番号第60/082,581号(本明細書中で参考として援用される)の実施例2を参照のこと。
【0062】
膜固定化融合タンパク質(b)の場合、身体中の望ましくない細胞が、融合タンパク質をコードする遺伝子を発現する必要がある。これは、遺伝子治療アプローチ、すなわち、融合タンパク質コード遺伝子の発現を望ましくない細胞に指向させるプラスミドベクターもしくはウイルスベクターを用いて、インビボで細胞をトランスフェクトすることによるか、または細胞表面上に融合タンパク質を発現するように遺伝子的に操作された細胞の身体中への移植によるかのいずれかで達成され得る。後者の細胞は、通常、それらの細胞が、Fc媒介細胞傷害性機構によって破壊され得ないポリマーカプセルに内包されて身体内へ移植される(カプセル化細胞治療)。しかし、カプセル送達が失敗し、そして逸脱細胞が望ましくないようになれば、それらはFc媒介細胞傷害性により除去され得る。Stabilaら、1998、Nature、Biotech.16:1357。この場合、現在開示される方法は、遺伝子治療用ベクターに、十分なもしくは最適なGnT IIIの発現レベルを指向するさらなる遺伝子発現カセットを組込むことによってか、または十分なもしくは最適なレベルのGnT IIIを発現するように、移植されるべき細胞を操作することによってかのいずれかで適用され得る。両方の場合において、開示される方法の目的は、複合型バイセクト型オリゴ糖を保有する表面提示Fc領域の割合を増大または最大にすることである。
【0063】
以下の実施例は、本発明をより詳細に説明する。以下の調製物および実施例は、当業者が本発明をより明瞭に理解し、そして実施することを可能にするために、与えられる。しかし、本発明は、例示される実施態様および本発明の範囲内にある機能的に等価である方法によって範囲を限定されることなく、本発明の単なる単一の局面の例証として意図される。実際、本明細書中に記載されるものに加え、本発明の種々の改変は、前述の説明および添付している図面より当業者に明らかである。このような改変は、添付された特許請求の範囲の範囲内に含まれることが意図される。
【実施例】
【0064】
(VIII.実施例)
(A.実施例1:チャイニーズハムスター卵巣細胞におけるグリコシルトランスフェラーゼのテトラサイクリン調節過剰発現)
GnT IIIの発現が外部制御され得る細胞株を確立するために、テトラサイクリン調節発現系を使用した。Gossen,M.およびBujard,H.、1992、Proc.Nat.Acad.Sci.USA、89:5547−5551。これらの細胞中のGnT IIIの量は、培養培地中のテトラサイクリン濃度を単に操作することによって制御され得る。この系を用いて、高いレベルまでのGnT IIIの過剰発現が増殖阻害を導き、そして細胞に対して毒性であることを見出した。GnT V(異なる糖タンパク質改変グリコシルトランスフェラーゼ)のテトラサイクリン調節過剰発現を有する別のCHO細胞株は、同様の阻害効果を示した。このことは、これが、糖タンパク質改変グリコシルトランスフェラーゼ過剰発現の一般的特徴であり得ることを示す。この現象は、以前に報告されておらず、おそらく発明者は、一般的には、関連実験については構成的なプロモーターを使用したという事実に起因する。増殖効果は、糖タンパク質改変グリコシルトランスフェラーゼ過剰発現のレベルの上方制限を設定し、それによってまた貧弱な到達可能な糖タンパク質部位の改変の最大範囲を制限し得る。
【0065】
(1.材料および方法)
グリコシルトランスフェラーゼのテトラサイクリン調節発現を有するCHO細胞の樹立。第1の工程において、テトラサイクリン制御トランスアクチベーター(tTA)を、調節系に十分なレベルで、構成的に発現する中間のCHO細胞株(CHO−tTA)をまず生成した。リポフェクタミン試薬(Gibco,Eggenfelden,Germany)を使用して、CHO(DUKX)細胞を、tTA遺伝子の構成的発現のためのベクターであるpUHD15−1(GossenおよびBujard、1992、Proc.Nat.Acad.Sci.USA、89:5547−5551)およびネオマイシン耐性遺伝子の構成的発現のためのベクターであるpSV2Neo(Clontech、Palo Alto、CA)とともに同時トランスフェクトした。安定した薬物耐性クローンを選択し、そしてテトラサイクリン調節β−ガラクトシダーゼ発現ベクターであるpUHG16−3を用いた一過性トランスフェクションによる十分なレベルのtTA発現についてスクリーニングした。c−mycエピトープコードDNAを、PCR増幅によってラットGnT III cDNA(Nishikawaら、1992、J.Biol.Chem.267:18199−18204)の3’末端に付加した。Nilssonら、1993、J.Cell Biol.120:5−13。この産物を配列決定し、そしてテトラサイクリン調節発現のためのベクターであるpUHD10−3(GossenおよびBujard、前出)にサブクローニングし、ベクターpUHD10−3−GnT IIImを生成した。ヒトGnT V cDNA(Saitoら、1995、Eur.J.Biochem.233:18−26)を、pUHD10−3に直接サブクローニングし、プラスミドベクターpUHD10−3−GnT Vを生成した。CHO−tTA細胞を、リン酸カルシウムトランスフェクション法(JordanおよびWurm.1996、Nucleic Acids Res.24:596−601)を用いて、ピューロマイシン耐性の構成的発現のためのベクターであるpPur(Clontech、Palo Alto、CA)、およびベクターpUHD10−3−GnT IIImまたはベクターpUHD10−3−GnT Vのいずれかで、同時トランスフェクトした。ピューロマイシン耐性クローンをテトラサイクリンの存在下で選択し、単離し、次いでウェスタンブロット分析によりGnT IIIまたはGnT Vのテトラサイクリン調節発現について分析した。後述を参照のこと。
【0066】
ウェスタンブロッティングおよびレクチンブロッティング。GnT IIIまたはGnT Vのウェスタンブロット分析のために、細胞溶解物を、SDS−PAGEにより分離し、そしてPVDF膜(Millipore、Bedford、MA)に電気ブロットした。抗c−mycモノクローナル抗体9E10(Nilssonら、1993、J.Cell Biol.120:5−13)を用いてGnT IIIを検出し、そして抗GnT
Vウサギポリクローナル抗体(Chenら、1995、Glycoconjugate
J.12:813−823)を用いてGnT Vを検出した。二次抗体として、抗マウスまたは抗ウサギIgG西洋ワサビペルオキシダーゼ(Amersham、Arlington、IL)を使用した。結合した二次抗体を、増強化学発光キット(ECLキット、Amersham、Arlington、IL)を使用して検出した。
【0067】
GnT III触媒反応またはGnT V触媒反応のいずれかにより改変される糖タンパク質のレクチンブロット分析について、ビオチン化E−PHA(Oxford Glycosciences、Oxford、United Kingdom)またはL−PHA−ジゴキシゲニン(Boehringer Mannheim、Mannheim、Germany)をそれぞれ使用した。MerkleおよびCummings、1987、Methods Enzymol.138:232−259。
【0068】
(2.結果および考察)
グリコシルトランスフェラーゼのテトラサイクリン調節過剰発現を有するCHO細胞株の樹立。グリコシルトランスフェラーゼ過剰発現細胞株の樹立のために使用される戦略は、最初に、この系が働くのに十分なレベルで、テトラサイクリン制御トランスアクチベーター(tTA)を構成的に発現する中間体CHO細胞株をまず生成することからなった。Yinら、1996、Anal.Biochem.235:195−201。このレベルは、テトラサイクリン非存在下で、グリコシルトランスフェラーゼ遺伝子の上流の最小プロモータからの転写の高いレベルを活性化するのに十分高くなくてはいけなかった。CHO細胞を、tTAの構成的発現のためのベクター(これは、ヒトサイトメガロウイルス(hCMV)プロモーター/エンハンサーによって駆動される)、およびネオマイシン耐性(NeoR)遺伝子の発現のためのベクターとともに同時トランスフェクトした。過剰な
tTA発現ベクターを使用し、そしてネオマイシン耐性クローンを単離した。
【0069】
哺乳動物細胞において、同時トランスフェクトされたDNAは、染色体内のランダムな位置で隣接して組込まれ、そして発現は、組込み部位に大部分依存し、そしてインタクトな発現カセットのコピー数にもまた依存する。トランスフェクトされた遺伝子の異なる発現レベルを有するクローンの混合集団が生成される。Yinら、1996、前出。ネオマイシン耐性についての選択は、単に、インタクトなNeoR発現カセットの組込みについ
て選択し、一方過剰なtTA発現ベクターの使用は、tTAの良好な発現を有するクローンを見出す確率を増大させる。クローンの混合集団は、tTA発現についての機能的アッセイを用いてスクリーニングされなければならない。GossenおよびBujard、1992、前出;Yinら、1996、前出。これは、tetプロモーターの制御下でレポーター遺伝子lacZを保有する第2のベクターによる各クローンのトランスフェクション、およびβ−ガラクトシダーゼ活性の、テトラサイクリン調節(tet調節)一過的発現(すなわち、トランスフェクション後1〜3日後)についてのスクリーニングによりなされた。CHOt17(これは、20のスクリーニングされたクローンの間で最も高いレベルのtet調節β−ガラクトシダーゼ活性を示した)を、さらなる研究のために選択した。
【0070】
CHOt17細胞を、ベクターpUHDIO−3−GnT IIImで細胞をトランスフェクトし、そしてテトラサイクリンの存在下または非存在下での36時間、細胞をインキュベートした後のGnT IIIの相対レベルを比較することにより、Gnt IIIのtet調節発現について試験した。GnT IIIレベルを、GnT IIIのカルボキシ末端でc−mycペプチドエピトープタグを認識するモノクローナル抗体(9E10)を用いて、ウェスタンブロット分析により比較した。このタグを、PCR増幅を用いたグリコシルトランスフェラーゼ遺伝子の改変により導入した。種々の報告は、局在または活性を崩壊することなく、同じトポロジーを共有する酵素群であるグリコシルトランスフェラーゼのカルボキシ末端へペプチドエピトープの付加することを実証した。Nilssonら、1993、前出;Rabouilleら、1995、J.Cell Science 108:1617−1627。図2は、クローンCHOt17において、GnT III蓄積が、テトラサイクリンの存在下よりも非存在においての方が、有意に高いことを示す。β−ガラクトシダーゼ活性のアッセイにおいてより弱い転写活性化を与えるさらなるクローン、CHOt2を、並行して試験した(図2)。GnT IIIおよびβ−ガラクトシダーゼの発現レベルは、これらの両方のクローンについて同じパターンのテトラサイクリン調節を伴う。GnT III発現が量的に制御され得るテトラサイクリン濃度の範囲は、0〜100ng/mlと見出された(図3)。この結果は、異なる細胞株と遺伝子を用いた前述の研究に一致する(Yinら、1996、前出)。
【0071】
GnT IIIのtet調節発現を有する安定な細胞株を生成するために、CHOt17細胞を、ベクターpUHDIO−3−GnT IIImおよびプロマイシン耐性遺伝子の発現のためのベクターpPURで同時トランスフェクトした。並行して、CHOt17細胞を、pUHDIO−3−GnT VベクターおよびpPURベクターで同時トランスフェクトし、この他のグリコシルトランスフェラーゼについて類似の細胞株を生成した。高度に効率的なリン酸カルシウムトランスフェクション方法を使用して、そして真核生物発現カセットの外側の唯一の制限部位でDNAを線状化し、組込みの際にこれらを崩壊する確率を減少させた。発現したtTAのレベルが最初に十分であると証明されている宿主を用いることによって、テトラサイクリン非存在下でグリコシルトランスフェラーゼの高度な発現を有するクローンを見出す確率が増大する。
【0072】
安定な組込み体を、ピューロマイシン耐性により選択し、クローン選択を行った培地中にテトラサイクリンを保ち、基底レベルでのグリコシルトランスフェラーゼの発現を維持した。各グリコシルトランスフェラーゼについて、16のピューロマイシン耐性クローンをテトラサイクリンの存在および非存在下で増殖し、それぞれの8つをウェスタンブロット分析により分析した(図4)。大多数のクローンは、グリコシルトランスフェラーゼ発現の良好な調節を示した。GnT III発現クローンのうちの1つは、テトラサイクリンの存在下で比較的高い基底レベルを示した(図4B、クローン3)。このことは、発現カセットの組込みが内因性CHO細胞エンハンサーに近接することを示唆する;一方、2つのピューロマイシン耐性クローンは、テトラサイクリンの非存在下でGnT IIIの発現を示さなかった(図4B、クローン6および8)。発現の良好な調節を示すクローンの間で、異なる最大レベルのグリコシルトランスフェラーゼが観察された。これは、組込み部位または組込まれたコピー数におけるバリエーションに起因し得る。グリコシルトランスフェラーゼの活性を、テトラサイクリンの存在および非存在において増殖させた種々のクローン由来の内因性細胞糖タンパク質へのE−PHAおよびL−PHAレクチン結合により確認した(図5)。レクチンは、特異的なオリゴ糖構造に結合するタンパク質である。E−PHAレクチンは、GnT III触媒反応の産物であるバイセクト型オリゴ糖に結合し、そしてL−PHAは、GnT V触媒化反応により産生されるトリアンテナリー型オリゴ糖およびテトラアンテナリー型オリゴ糖に結合する(MerkleおよびCummings、1987、Methods Enzymol.138:232−259)。各グリコシルトランスフェラーゼについて、テトラサイクリンの非存在において高度に発現し、しかしテトラサイクリン存在下では検出不可能であるクローン(クローン6、図4A、CHO−tet−GnT Vおよびクローン4、図4B、CHO−tet−GnT
IIIm)を、さらなる研究のために選択した。
【0073】
(B.実施例2:グリコシルトランスフェラーゼ過剰発現によってもたらされる細胞増殖の阻害)
テトラサイクリンの非存在下でのGnT IIIを発現するクローンおよびGnT Vを発現するクローンのスクリーニングの間(上記の実施例1を参照のこと)、クローンの各セットのおよそ半分が増殖の強力な阻害を示した。増殖阻害の程度は、クローンにより変動し、そしてウェスタンブロット分析から見積もった発現レベルとの比較(図4)は、増殖阻害の程度と、グリコシルトランスフェラーゼ過剰発現との間の相関を示唆した。この相関は、最後のクローンCHO−tet−GnT IIImおよびCHO−tet−GnT Vを、種々の濃度のテトラサイクリンにおいて増殖させることによって強固に確立された。増殖の強力な阻害は、低レベルのテトラサイクリンでの培養の2日後に明確になった(図6)。増殖阻害した細胞は、接着性CHO細胞の代表的な細長い形状の代わりに小さく丸い形態を提示した。数日後、有意な細胞死が、増殖阻害された細胞の形態から明らかであった。
【0074】
グリコシルトランスフェラーゼ過剰発現に起因する増殖阻害は、これまで、文献には報告されていない。これはおそらく、構成性のプロモーターの広汎な使用に起因する。増殖阻害レベルでのグリコシルトランスフェラーゼの構成性発現を与えるこれらのクローンは、選択手順の間に消失する。これは、選択の間に、培地中のテトラサイクリン(すなわち、基底発現レベル)を維持することによってここで回避された。選択前に、構成性hCMVプロモーター/エンハンサーに基づく伝統的な哺乳動物ベクターを用いて増殖阻害レベルにまでグリコシルトランスフェラーゼを発現し得るクローンの頻度は、より低いと予測される。これは、任意の所定の遺伝子について、高度の構成性レベルのtTAについて選択されたCHO細胞株におけるpUHDIO−3ベクターが、他者らによって観察されるような、構成性のhCMVプロモーター/エンハンサーに基づくベクターよりも有意に高い発現レベルを与えるという事実に起因する。Yinら、1996、前出。
【0075】
細胞増殖の阻害は、それらのインビボでの触媒活性には非依存性の、膜結合型の、ゴルジ体内在性グリコシルトランスフェラーゼの過剰発現の直接の効果(例えば、小胞体(ER)における折り畳みの誤りを介し、ERにおけるタンパク質折り畳みを補助するエレメントの飽和を生じる)に起因し得る。これは、おそらく、他の必須の細胞タンパク質の折り畳みおよび分泌に影響を与え得る。あるいは、増殖阻害は、グリコシルトランスフェラーゼのインビボ活性の増加に関連し得る。この活性増加は、機能を破壊する様式で、標準的なインビトロ培養条件下での増殖にとって必要な1セットの内因性糖タンパク質のグリコシル化パターンの変化をもたらす。
【0076】
根底をなす機構とは独立して、この増殖阻害効果は、動物細胞のグリコシル化を操作することについて2つの結果を有する。第一に、この効果は、標的となる糖タンパク質産物のためのベクターとともに構成性グリコシルトランスフェラーゼ発現ベクターを同時トランスフェクトすることは、貧弱な戦略であることを示唆する。これらの2つのクラスのタンパク質の連結した発現の他の方法(例えば、類似の強度の複数の構成性プロモーターの使用または複数シストロン性の構成性発現ベクターの使用による)もまた、回避されるはずである。これらの場合、経済的なバイオプロセスにとって必要である、標的糖タンパク質の非常に高度で構成性の発現を有するクローンもまた、グリコシルトランスフェラーゼの高度な発現を有し、そしてこの選択プロセスにおいて排除され得る。連結された誘導性の発現もまた、産業的バイオプロセスについて問題であり得る。なぜなら、増殖停止した細胞の生存性は、グリコシルトランスフェラーゼの過剰発現によって減弱化されるからである。
【0077】
第二の結果は、これが、グリコシル化操作のアプローチについてグリコシルトランスフェラーゼ過剰発現に上限を課すことである。明らかに、細胞において、グリコシルトランスフェラーゼの内因性レベルでの、多くのグリコシルトランスフェラーゼ触媒性の反応の変換は、いくつかのグリコシル化部位について非常に高い。しかし、そのオリゴ糖がいくらかアクセス不能であるか、または特定のグリコシルトランスフェラーゼについて有利でないコンホメーションにおいて安定化されている、グリコシル化部位もまた存在する。例えば、バイセクティングGlcNAcの添加は、ヒトIgG抗体の可変領域に配置されるオリゴ糖よりもそのFc領域に付着するオリゴ糖に、より制限されることが観察されている。Savvidouら、1984、Biochemistry 23:3736−3740。これらの制限された部位のグリコシル化操作は、グリコシルトランスフェラーゼ発現におけるそのような限定によって影響を受け得る。これは、糖形態の「非天然の」分布について目的化することを示唆するが、これらは、糖タンパク質の特定の治療適用について利益があり得る。
【0078】
(C.実施例3:チャイニーズハムスター卵巣細胞の抗ヒト神経芽腫抗体のグリコシル化の操作)
そのグリコシル化パターンを改変することによって治療抗体を操作する概念を確認するために、キメラ抗ヒト神経芽腫IgG1(chCE7)を選択した。このIgG1は、SP2/0組換えマウスミエローマ細胞によって産生される場合有意ではないADCC活性を有する。chCE7は、腫瘍に会合する190kDaの膜糖タンパク質を認識し、そしてこれまでに試験されている神経芽腫腫瘍のすべてと強力に反応する。chCE7は、その抗原について高度の親和性(1010M-1のKd)を有し、そしてその高度の腫瘍特異性
のために、chCE7は、臨床病理において診断ツールとして慣用される。Amstutzら、1993.Int.J Cancer 53:147−152。近年の研究において、放射標識されたchCE7は、ヒト患者において良好な腫瘍局在を示している。Durr、1993、Eur.J.Nucl.Med.20:858。抗神経芽腫治療モノクローナル抗体(mAb)であるchCE7のグリコシル化パターンをCHO細胞において、GnT IIIのテトラサイクリン調節発現を用いて操作した。その糖形態分布において異なる1セットのmAbサンプルを、基底レベルと毒性レベルとの間の範囲においてGnT III発現を制御することによって産生した。そして、そのグリコシル化プロフィールを中性オリゴ糖のMALDI/TOF−MSによって分析した。これらのサンプルのADCC活性の測定によって、インビトロ生物学的活性における最大のchCE7についてのGnT III発現の最適範囲が示された。そして、この活性は、Fcに関連し、複合型バイセクト型オリゴ糖のレベルと相関していた。
【0079】
(1.材料および方法)
chCE7発現ベクターの構築。抗ヒト神経芽腫キメラ抗体chCE7の、重鎖(IgG1)および軽鎖(κ)の発現のための、それぞれ、プラスミドベクター10CE7VHおよび98CE7VL(これらは、マウス免疫グロブリンプロモーター/エンハンサー、マウス抗体可変領域、およびヒト抗体定常領域を含むキメラゲノムDNAを含む)(Amstutzら、1993、Int.J.Cancer 53:147−152)を、最終の発現ベクターpchCE7HおよびpchCE7Lの構築のための出発材料として使用した。キメラ重鎖および軽鎖chCE7の遺伝子を、再アセンブリし、そしてpcDNA3.1(+)ベクター中にサブクローニングした。再アセンブリの間、全てのイントロンを除去し、リーダー配列を、合成したものに置き換えた(Reffら、1994、Blood 83:435−445)。そして、可変領域および定常領域の配列を接合する独特な制限部位を導入した。重鎖の定常領域からイントロンを、重複伸長PCRを用いてスプライシングすることによって除去した(Clacksonら、1991、General
Applications of PCR to Gene Cloning and
Manipulation、p.187−214、in:McPhersonら(編)、PCR a Practical
Approach、Oxford University Press、Oxford)。
【0080】
異なるレベルのGnT IIIを発現するchCE7のCHO細胞における産生。CHO−tet−GnT IIIm(前出を参照のこと)細胞を、リン酸カルシウムトランスフェクション方法を使用して、ベクターpchCE7H、pchCE7L、およびpZeoSV2(Zeocin耐性のため、Invitrogen、Groningen、The Netherlands)を用いて同時トランスフェクトした。Zeocin耐性クローンを、96ウェル細胞培養プレートに移した。そして、ヒトIgG定常領域について特異的なELISAアッセイを用いてキメラ抗体発現についてアッセイした。Lifelyら、1995、前出。4つのchCE7抗体サンプルは、10% FCSを補充したFMX‐8細胞培養培地中で増殖させた選択したクローン(CHO−tet−GnT IIIm−chCE7)の並行培養物に由来した;各培養物は、異なるレベルのテトラサイクリンを含み、従って、異なるレベルのGnT IIIを発現した。CHO−tet−GnT IIIm−chCE7細胞を増大させ、そして7日間、異なる濃度のテトラサイクリンに予備適合させた。テトラサイクリンのレベルは、2000ng/ml、60ng/ml、30ng/ml、および15ng/mlであった。
【0081】
chCE7抗体サンプルの精製。抗体を、20mMリン酸ナトリウム、20mMクエン酸ナトリウム、500mM塩化ナトリウム、0.01% Tween20、1M尿素、pH 7.5(緩衝液A)から緩衝液B(リン酸ナトリウムのない緩衝液A、pH 2.5)へのpH線形勾配溶出を用いて、1mlのHiTrapプロテインAカラム(Pharmacia Biotech、Uppsala、Sweden)におけるプロテインAアフィニティークロマトグラフィーによって培養培地から精製した。アフィニティー精製したchCE7サンプルを、1mlのResourceSカチオン交換カラム(Pharmacia Biotech、Uppsala、Sweden)においてPBSへと緩衝液交換した。最終純度は、SDS−PAGEおよびクーマシーブルー染色から95%より高いと判断した。各サンプルの濃度を、280nmでの吸光度から見積もった。
【0082】
抗体の神経芽腫細胞への結合。ヒト神経芽腫細胞への結合親和性を、CHO産生したサンプルによる125I標識したchCE7の変位量から見積もった。Amstutzら、1
993、前出。
【0083】
MALDI/TOF−MSによるオリゴ糖分析。CE7−2000t,CE7−60t,CE7−30t、およびCE7−15tのサンプルを、A.urefaciensシアリダーゼ(Oxford Glycosciences、Oxford,United Kingdom)を、製造業者の指示に従って用いて処理して、任意のシアル酸モノサッカリド残基を除いた。次いで、シアリダーゼ消化物を、ペプチドN−グリコシダーゼF(PNGaseF、Oxford Glycosciences、Oxford、United Kingdom)を製造業者の指示に従って用いて処理して、N結合型オリゴ糖を放出させた。タンパク質、界面活性剤および塩を、その消化物を上部から底部まで、20mlのSapPak C18逆相マトリクス(Waters,Milford、MA)を含むマイクロカラム、20mlのDowex AG 50W X8カチオン交換マトリクス(BioRad,Hercules、CA)を含むマイクロカラム、および20mlのAG 4X4アニオン交換マトリクス(BioRad,Hercules、CA)を含むマイクロカラムに通すことによって除去した。これらのマイクロカラムを、これらのマトリクスを、エタノールを満たしたGel Loaderチップ(Eppendorf,Basel、Switzerland)中に充填すること、続いて水を用いて平衡化することによって作製した。Kusterら、1997、Anal.Biochem.250:82−101。フロースルーの液体および300mlの水での洗浄物をプールし、濾過し、エバポレートして、室温で乾燥させ、そして2mlの脱イオン水中に再懸濁した。1μlをMALDI−MSサンプルプレート(Perseptive Biosystems、Farmingham、MA)に適用し、そしてアセトニトリル中の1mlの10mg/mlデヒドロ安息香酸(DHB、Aldrich、Milwakee、Wisconsin)溶液と混合した。これらのサンプルを風乾し、そして得られた結晶を0.2mlのエタノール中に溶解し、そして風乾により再結晶させた。Harvey、1993、Rapid Mass、Spectrom.7:614−619。次いで、このオリゴ糖サンプルを、陽イオンおよび反射様式で、20kVの加速電圧を用いて、遅延イオン抽出MALDIイオン供給源を装着したElite Voyager400分光器(Perseptive Biosystems、Farmingham,MA)を用いるマトリクス支持レーザー脱離イオン化/飛行時間重量スペクトル測定(MALDI/TOF−MS)によって分析した。128の走査を平均化した。バイアンテナリー複合型バイセクト型オリゴ糖構造を、5つのHexNAcに関連するピークに割り当てた。代替の5つのHexNAcを含むアイソマーであるN結合型トリアンテナリー非バイセクト型オリゴ糖は、IgGのFc領域においては決して見出されず、そしてそれらの合成は、GnT IIIとは別のグリコシルトランスフェラーゼによって触媒された。
【0084】
ADCC活性アッセイ。異なる濃度のchCE7サンプルの存在下で、1:19での標的:エフェクター比にて、37℃16時間のインキュベーションの間のIMR−32ヒト神経芽腫細胞(標的)のヒトリンパ球(エフェクター)による溶解を、蛍光色素の保持を介して測定した。Kolberら、1988、J.Immunol.Methods 108:255−264。IMR−32細胞を、蛍光色素Calcein AMを用いて20分間標識した(最終濃度3.3μM)。標識した細胞(80,000細胞/ウェル)を、1時間、異なる濃度のCE7抗体とインキュベートした。次いで、単球を涸渇した単核細胞を添加した(1,500,000細胞/ウェル)。そして、細胞混合物を、5%CO2雰囲気中で37℃で16時間インキュベートした。この上清を捨て、そしてその細胞を
HBSSで一回洗浄し、そしてTriton X−100 (0.1%)中で溶解した。IMR−32細胞における蛍光色素の保持を、蛍光測定器(Perkin Elmer,Luminscence Spectrometer LS 50B、(Foster City、CA))を用いて測定し、そして特定の溶解物を、その標的を抗体への曝露の代わりに界面活性剤に対する曝露から得られた総溶解物コントロールに対して相対的に算出した。抗体の非存在下でのシグナルを、0%の細胞傷害性に設定した。各抗体濃度を、三連にて分析し、そしてそのアッセイを、3回の別個の回数反復した。
【0085】
(2.結果および考察)
異なるレベルのGnT IIIを発現するCHO細胞におけるchCE7の産生。ChCE7の重鎖および軽鎖の発現ベクターを、ヒトサイトメガロウイルス(hCMV)プロモーター、ウシ成長ホルモン終結配列およびポリアデニル化配列を組み込み、そして全ての重鎖および軽鎖のイントロンを除去して構築した。このベクター設計は、CHO細胞における組換えIgG遺伝子の再現性の良い高レベル発現の報告に基づいた。Reffら、1994、前出;Trillら、1995、Current Opinion Biotechnol.6:553−560。さらに、独特な制限部位を、可変領域と定常領域との間の接合部にて各鎖に導入した。これらの部位は、リーディングフレームを保存し、そしてアミノ酸配列を変化させない。これらは、他のマウス−ヒトキメラ抗体の産生のための、マウス可変領域の単純な交換を可能にするはずである。Reffら,1994、前出。DNA配列決定によって、所望の遺伝子が適切にアセンブルされているかを確認した。そして、トランスフェクトされたCHO細胞におけるキメラ抗体の産生を、ヒトFc−ELISAアッセイを用いて確認した。
【0086】
GnT IIIの安定なテトラサイクリン制御された発現およびchCE7の安定な構成的発現を伴うCHO−tet−GnT IIIm−chCE7細胞を確立し、そして1セットのchCE7サンプルの産生のために規模拡大した。規模拡大の間、同じCHOクローンに由来する4つの並行培養物を、各々、異なるレベルのテトラサイクリンおよびすなわちGnT III遺伝子の発現レベルのみにおいて異なるように増殖させた。この手順は、N結合型糖形態の生合成に影響を与える他の可変物からの任意のクローン性効果を除去し、GnT III遺伝子発現とグリコシル化抗体の生物学的活性との間の厳格な相関付けの確立を可能にする。テトラサイクリンの濃度は、2000ng/ml(すなわち、GnT III発現の基底レベル)から15ng/ml(この濃度において、グリコシルトランスフェラーゼの過剰発現に起因する有意な増殖阻害および毒性が観察された(前出を参照のこと))までに及んだ。実際、ほんのわずかの量の抗体が後者の培養物から回収され得た。テトラサイクリンを30ng/mlの濃度で用いた第二の最高レベルのGnT III発現は、緩和な増殖阻害のみをもたらした。この培養物から精製した抗体の収率は、GnT III遺伝子過剰発現の残りの2つのより低いレベル由来のものの約70%であった。
【0087】
4つの抗体サンプルCE7−2000t、CE7−60t、CE7−30t、CE7−15t(数字は、テトラサイクリンの会合濃度を示す)を、プロテインA上でアフィニティークロマトグラフィーにより精製し、そしてカチオン交換カラムを用いてPBSへと緩衝液交換した。純度は、クーマシーブルー染色を用いたSDS−PAGEから判断した場合95%を超えた。ヒト神経芽腫細胞に対する結合アッセイは、細胞に対する高度の親和性および異なるサンプル間で抗原結合が有意に相違しないことを示した(2.0Mと2.7×10-10Mとの間で変動させた推定平衡解離定数)。このことは予測どおりであった
。なぜならCE7可変領域には、推定N結合型グリコシル化部位が存在しないからである。
【0088】
異なるchCE7サンプルの複合型バイセクト型オリゴ糖の分布およびレベル。オリゴ糖プロフィールを、飛行時間におけるマトリクス支持レーザ脱離/イオン化質量測定(MALDI/TOF−MS)によって得た。4つのCHO産生した抗体サンプルの各々およびSP2/0マウスミエローマ由来のchCE7(CE7−SP2/0)サンプルに由来する中性のN結合型オリゴ糖の混合物を、2,5−デヒドロ安息香酸(2,5−DHB)を用いてマトリクスとして分析した(図9)。これらの条件下で、中性のオリゴ糖は、本質的に単一の[M+ Na+]イオンであるようであり、これは、ときに、そのマトリク
スのカリウム含有量に依存してより小さな[M+ K+]イオンを伴う。Bergwef
fら、1995、Glycoconjugate J.12:318−330。
【0089】
この型の分析は、異なる質量の中性オリゴ糖の相対比(これは、相対ピーク高さに反映される)、および各ピークのモノサッカリドの荷電組成の両方を得た。Kusterら、1997、前出;NavenおよびHarvey、1996、Rapid Commun.Mass Spectrom.10:1361−1366。一時的な構造を、そのモノサッカリドの組成、生合成経路の知識および同じ宿主によって産生された同じ糖タンパク質由来のオリゴ糖についてのこれまでの構造データに基づくピークにより割り当てる。なぜなら、タンパク質バックボーンおよび細胞型は、オリゴ糖分布に対して強力な影響を有し得るからである。Fieldら、1996、Anal.Biochem.239:92−98。Fcが会合するオリゴ糖の場合、バイアンテナリー複合型オリゴ糖のみがヒト血清に存在するIgGにおいて検出されるか、または正常条件下での哺乳動物細胞培養物によって産生された。Wormaldら、1997、Biochemistry 36:1370−1380;WrightおよびMorrison、1997,Tibtech 15:26−31。これらの化合物へ導く経路を、各オリゴ糖に対応する[M+ Na+
]イオンの質量を含めて図10に示す。高マンノースオリゴ糖もまた、バッチ細胞培養物の安定期および死期において産生された抗体において検出された。Yu Ipら、1994、Arch.Biochem.Biophys 308:387−399。
【0090】
CE7−SP2/0サンプルにおける2つの主要なピーク(図9A)は、4つのN−アセチルヘキソサミン(HexNAc)を伴うフコシル化オリゴ糖の質量に対応する(これらは、3つ(m/z1486)または4つ(m/z1648)のいずれかのヘキソースを含む)。図10を参照のこと。しかし、この図におけるオリゴ糖についてまとめた注記は、コアの2つのGlcNAcを計数していないことに注意されたい。この組成は、それぞれ、0個または1個のガラクトース残基を有する、コアがフコシル化バイアンテナリー複合型オリゴ糖構造(これは、Fcが会合したオリゴ糖に代表的であり、そしてSP2/0細胞において発現されたキメラIgG1に由来するFcオリゴ糖のNMR分析においてこれまでに観察されている通りである)と一致する。Bergweffら、1995、前出。
【0091】
GnT IIIに触媒される、バイセクティングGlcNAcのこれらのバイアンテナリー化合物(この化合物は、好ましいGnT IIIアクセプターである)への移入は、5つのHexNAcを伴うオリゴ糖をもたらす(m/z1689および1851、それぞれ、非ガラクトシル化およびモノガラクトシル化、図10)。これらのオリゴ糖は、CE7−SP2/0サンプルにおいては明らかに存在しなかった。後者のピークは、chCE7がCHO−tet−GnTIIIm細胞において発現される場合に現れる。CHOが発現する抗体において、4つのHexNAcを含有するピークもまた、主にフコシル化されている。しかし、少量のフコシル化されていない構造も、m/z1339でのピークから明らかである(図10を参照のこと)。ガラクトシル化のレベルはまた、CHO由来材料とSP2/0由来材料との間であまり異ならない。GnT III発現の基底レベルでは(CE7−2000tサンプル.図9B)、5つのHexNAcを伴う分子は、4つのHexNAcを伴うものよりも低い割合で存在する。より高いレベルのGnT III発現(CE7−60tサンプル、図9C)は、5つのHexNAcを伴うオリゴ糖に有利に比の逆転をもたらした。この傾向に基づいて、バイアンテナリー複合型バイセクト型オリゴ糖構造は、これらのサンプルにおいて5つのHexNAcを伴う化合物であると割り当てられた。三アンテナ性N結合型オリゴ糖である別の5HexNAc含有アイソマーは、IgGのFc領域においては決して見出されず、そしてその合成は、GnT IIIとは異なるGlcNAcトランスフェラーゼによって触媒される。
【0092】
GnT III発現におけるさらなる増加(CE7−30tサンプル、図9D)は、複合型バイセクト型オリゴ糖のレベルにおいてなんらの有意な変化をももたらさなかった。5つのHexNAcを含む別のピーク(m/z1543)は、CHO−GnTIIIサンプルにおいて低いが比較的安定なレベルで現れ、そして質量において非フコシル化複合型バイセクト型オリゴ糖質量に対応する(図10)。m/z1705およびm/z1867での小さなピークもまた、5つのHexNAc含有バイアンテナリー複合型オリゴ糖に対応する。それらは、m/z1689およびm/z1851でのピークのカリウム付加物(ナトリウム付加物に関する16Daの質量の相違)(Kusterら、1997、前出)か、またはフコースを伴わないモノガラクトシル化および二ガラクトシル化複合型バイセクト型オリゴ糖のいずれかに割り当てられ得る(図10)。まとめると、複合型バイセクト型オリゴ糖は、サンプルCE7−2000tにおける合計の約25%の量であり、そしてサンプルCE7−60tおよびCE7−30tにおいて約45%〜50%に達する。
【0093】
chCE7サンプルのオリゴ糖プロフィールからのさらなる情報。複合型バイセクト型オリゴ糖のレベルは、サンプルCE730tにおいてより高くなかったが、GnT IIIの過剰発現の増加は、少量ではあるが、バイアンテナリー複合型オリゴ糖基質の基質比を継続して減少させた。このことは、2つの異なる4つのHexNAc含有ピーク(m/z1664およびm/z1810)において中程度の増加を伴った。後者の2つのピークは、ガラクトシル化バイアンテナリー複合型オリゴ糖、または混成型バイセクト型化合物のいずれかに対応し得る(図11)。両方のクラスの構造の組合せもまた、可能である。これらのピークにおける相対的な増加は、GnT III過剰発現の混成型バイセクト型の副産物の蓄積と一致する。実際、GnT III過剰発現の最高レベルで産生されたサンプルであるCE7−15tは、m/z1664におけるピークの大きな増加、m/z1810におけるピークの減少、およびバイセクト型複合オリゴ糖の約25%のレベルへの同時減少を示した。図9Eにおけるm/z1689およびm/z1851のピークならびに図11における対応する構造を参照のこと。フコシル化されたもの(m/z1810)に代わる、非フコシル化(m/z1664)の混成型バイセクト型副産物のより高度な蓄積は、GnT IIIによってまず改変されているオリゴ糖がもはや、コアのα1,6−フコシルトランスフェラーゼについての生合成基質ではあり得ないという事実に一致する。Schachter、1986、Biochem.Cell Biol.64:163−181。
【0094】
m/z1257でのピークは、CHOに由来するサンプルにおける合計の10〜15%のレベルで、およびCE7−SP2/0よりも低いレベルで存在する(図9)。これは、5つのヘキソースおよび2つのHexNAcに対応する。この組成を有する唯一公知であるN結合型オリゴ糖構造は、高マンノース型の5つのマンノースを含有する化合物である。別の高マンノースオリゴ糖である6マンノースのもの(m/z1420)もまた、はるかに低いレベルで存在する。上記のように、このようなオリゴ糖は、バッチ細胞培養物の後期において発現されるIgGのFcにおいて検出されている。Yu Ipら、1994、前出。
【0095】
chCE7サンプルの抗体依存性細胞傷害性。chCE7は、最小レベルのGnTIII過剰発現をとともにCHO−tet−GnTIIIm細胞中で発現されるとき、ヒトリンパ球による神経芽腫細胞のインビトロ溶解として測定される、ある程度のADCC活性を示す(図12、サンプルCE7−2000t)。GnTIIIのレベルが上昇すると、ADCC活性における大きな増加が生じた(図12、サンプルCE7−60t)。GnTIIIのさらなる過剰発現は、活性のさらなる増加をともなわず(図12,サンプルCE7−30t)、そして最高レベルの発現は、実際に、減少したADCCに至った(図12、サンプルCE7−15t)。最高のADCC活性を示すことに加えて、CE7−60tおよびCE7−30tの両方のサンプルの両方は、非常に低い抗体濃度で、有意なレベルの細胞傷害性を示す。これらの結果は、ADCC活性についてCHO細胞における最適範囲のGnTIII過剰発現があることを示し、そしてオリゴ糖プロフィールとの比較は、活性が、Fcに結合した複合型バイセクト型オリゴ糖のレベルと相関することを示す。
【0096】
ADCC活性についての複合型バイセクト型オリゴ糖の重要性を考慮すると、これら化合物の比率をさらに増加させるために、この経路を操作することが有用である。GnTIIIの、サンプルCE7−30tについて用いられたレベルに接近するレベルまでの過剰発現は、有意な傷害性および増殖阻害が観察されない生物工学的に実施可能な範囲内にある。このレベルの発現では、非ガラクトシル化された、バイアンテナリー複合型非バイセクト型オリゴ糖、すなわち、好適な潜在的GnTIII基質は、全体の10%より少なくなるまで減少する。m/z 1486のピーク、図9Dを参照のこと。しかし、わずか50%が、所望のバイアンテナリー複合型バイセクト型構造に転換されるだけである。残りは、混成型バイセクト型オリゴ糖副産物に転用されるか、または競合酵素β1,4−ガラクトシルトランスフェラーゼ、GalTにより消費されるかのいずれかである(図11)。
【0097】
相補的構造分析による、混成型バイセクト型およびガラクトシル化複合型非バイセクト型オリゴ糖ピークの分離は、各々の潜在的な所望されない経路がいくら消費されるかを決定する。高いGnTIII過剰発現レベルにおけるm/z 1664および1810ピークの増大は、これらピークの少なくとも一部分は、混成型バイセクト型オリゴ糖に対応することを示唆する(図11)。理論的には、混成型バイセクト型化合物に行くフラックスは、GnTIIIと一緒のマンノシダーゼIIのような経路中のより先の酵素の同時過剰発現により低減され得る。その一方、GnTIIIとGalTとの間の複合型バイセクト型オリゴ糖基質に対する競合は、GnTIIIを過剰発現する間に、UDP−GlcNAcのゴルジ体内濃度を増加させることにより、GnTIII触媒反応に向かって潜在的に片寄り得る。GnTIIIは、GlcNAcを、同時基質(co−subtrate)UDP−GlcNAcから異なるオリゴ糖に転移させる。UDP−GlcNAc同時基質のゴルジ体内濃度が、GnTIIIに対して準飽和である場合、それを、培養培地組成の操作によるか、またはゴルジ体中への糖−ヌクレオチド輸送の遺伝的操作により増加させて、オリゴ糖についてGalTとの競合でGnTIIIに有利にし得る。
【0098】
ADCC活性における増加が、ガラクトシル化および非ガラクトシル化の両方の複合型バイセクト型オリゴ糖における増加から生じるか、またはこれらの形態の1つのみにおける増加から生じるか否かは決定されるべきままである。図9におけるm/z 1689および1851のピークを参照のこと。ガラクトシル化バイアンテナリー複合型バイセクト型オリゴ糖が、増加したADCC活性について最適構造であることが見出される場合、Fc領域におけるこれら化合物の画分を最大にすることは、GnTIIIおよびGalTの両方の過剰発現を必要とする。先に論議された競合シナリオを考慮すると、両方の遺伝子の発現レベルは、注意深く調節されなければならない。さらに、可能な限り、トランス−ゴルジ体嚢の代わりにTGNに向かって、過剰発現GalTを再分配する試みは、価値がある。後者の戦略は、GalTの膜貫通領域コード配列を、α2,6−シアリルトランスフェラーゼの膜貫通領域(ChegeおよびPfeffer、1990、J.Cell.Biol.111:893〜899)と交換することにより実現され得る。
【0099】
(D.実施例4:抗CD20モノクローナル抗体C2B8のグリコシル化の操作)
C2B8は、抗ヒトCD20キメラ抗体である。Reff、M.E.ら、1994、前述。C2B8は、1997年にFDA認可を受け、そして現在、合衆国において、非ホジキンリンパ腫の処置のために、RituxanTMの商品名の下で用いられている。C2B8は、CHO細胞培養物に由来し、そしてそれ故、バイセクト型オリゴ糖を保持しないはずである。前述を参照のこと。この抗体の改良型を生成するために、chCE7抗神経芽腫抗体について先に示された方法が適用された。前述を参照のこと。開示された方法に従って改変されたC2B8抗体は、同じ細胞培養物および精製条件の下で生成された、標準的な非改変C2B8抗体よりも高いADCC活性を有していた。
【0100】
(1.材料および方法)
キメラ抗CD20モノクローナル抗体(C2B8)の可変軽鎖領域および可変重鎖領域の合成。C2B8抗体のVH遺伝子およびVL遺伝子を、PCRを用いる1工程プロセス(Kobayashiら、1997、Biotechniques 23:500〜503)で、1セットの重複する一本鎖オリゴヌクレオチド(プライマー)を用いて合成的に
アセンブルした。抗CD20抗体のマウス免疫グロブリンの軽鎖および重鎖の可変領域(それぞれVLおよびVH)をコードする配列データを、公開された国際特許出願(国際公開番号:WO 94/11026)から得た。アセンブルされたDNAフラグメントを、pBluescriptIIKS(+)中にサブクローン化し、そしてDNAサイクルシークエンシングにより配列決定し、変異が導入されなかったことを確認した。
【0101】
キメラ抗CD20モノクローナル抗体(C2B8)の発現のためのベクターの構築。C2B8モノクローナル抗体のVHおよびVLのコード領域を、pchCE7HおよびpchCE7L中にそれぞれサブクローン化した。このサブクローニングでは、抗神経芽腫CE7の可変重鎖および可変軽鎖をコードする配列(前述)を、C2B8の合成的にアセンブルされた可変重鎖領域および可変軽鎖領域と交換した。
【0102】
C2B8抗体を発現するCHO−tet−GnTIIIm細胞の生成。C2B8抗体を発現するCHO−tet−GnTIIIm細胞株の生成のための方法は、CHO−tet−GnTIIIm−CE7に対するのと正確に同じであった。前述を参照のこと。さらなる作業のために選択されたクローンを、CHO−tet−GnTIIIm−C2B8と名づけた。
【0103】
C2B8抗体を発現するCHO−tTAの生成。CHO−tTAは、CHO−tet−GnTIIImの親細胞株である。前述を参照のこと。GnTIIIを発現せず、C2B8抗体を発現するCHO−tTA細胞株の生成のための方法は、CHO−tet−GnTIIIm−C2B8およびCHO−tet−GnTIIIm−chCE7に対するのと正確に同じであった。前述を参照のこと。さらなる作業のために選択されたクローンを、CHO−tTA−C2B8と名づけた。
【0104】
C2B8抗体サンプルの生成。2つのC2B8抗体サンプルは、平行なCHO−tet−GnTIIIm−C2B8培養物に由来した;各培養物は、種々のレベルのテトラサイクリンを含み、そしてそれ故GnTIIIを種々のレベルで発現すると予想される。テトラサイクリンのレベルは、2000ng/ml、50ng/ml、および25ng/mlであった。これらの培養に由来するC2B8抗体サンプルを、C2B8−2000t、C2B8−50t、およびC2B8−25tとそれぞれ称した。平行して、1つの抗体サンプル(C2B8−nt)を、CHO−tTA−C2B8培養から作成した。この細胞株は、GnTIIIを発現しない。CHO−tTA−C2B8細胞を、テトラサイクリンなしで培養した。
【0105】
GnTIII発現の分析。GnTIIIのウェスタンブロット分析については、生産培養物の各々の細胞溶解産物を、SDS−PAGEにより分離し、そしてポリビニリデンジフルオライド膜に電気ブロットした。抗c−mycモノクローナル抗体9E10および抗マウスIgG−西洋ワサビペルオキシダーゼ(Amersham、Arlington、IL)を、一次抗体および二次抗体としてそれぞれ用いた。結合抗体を、増感化学発光キット(Amersham、Arlington、IL)を用いて検出した。
【0106】
C2B8抗体サンプルの精製。抗体サンプルを、chCE7抗体サンプルについてと同じ手順を用いて精製した。前述を参照のこと。濃度を、Molecular Probes(Leiden、The Netherlands)からの蛍光に基づくキットを用いて測定した。
【0107】
特異的C2B8抗原結合の確認。C2B8抗CD20モノクローナル抗体の抗原結合の特異性を、懸濁液中の細胞について間接免疫蛍光アッセイを用いて確認した。この研究には、CD20陽性細胞(SB細胞;ATCC寄託番号ATCC CCL120)およびCD陰性細胞(HSB細胞;ATCC寄託番号ATCC CCL120.1)を利用した。各タイプの細胞を、一次抗体としての、25ng/mlテトラサイクリンで生成したC2B8抗体とともにインキュベートした。陰性のコントロールは、一次抗体の代わりにHBSSBを含んだ。抗ヒトIgG Fc特異的なポリクローナルFITC結合体化抗体を、すべてのサンプルについて二次抗体として用いた(SIGMA、St.Louis、MO)。細胞を、Leica(Bensheim、Germany)蛍光顕微鏡を用いて調べた。
【0108】
ADCC活性アッセイ。異なる濃度のC2B8サンプルの存在下の、ヒト単球枯渇末梢血単核細胞(エフェクター細胞)によるSB細胞(CD20+標的細胞;ATCC寄託番号ATCC CCL120)の溶解を、Brunnerら、1968、Immunology 14:181−189に記載されるのと同じ手順に基本的に従って実施した。エフェクター細胞の標的細胞に対する比率は、100:1であった。
【0109】
(2.結果および考察)
GnTIIIは、種々の細胞株および培養物において種々のレベルで発現される。平行したCHO−tet−GnTIIIm−C2B8培養物(各培養物は種々のレベルのテトラサイクリン(2000、50、および25ng/ml)を含み、そしてそれ故、種々のレベルでGnTIIIを発現すると期待される)の細胞を溶解し、そして細胞溶解産物をSDS−PAGEにより分離し、そしてウェスタンブロッティングにより検出した。25ng/mlテトラサイクリンで増殖させた培養物の溶解産物は、GnTIIIの対応する分子量に強いバンドを示し、その一方、50および2000ng/mlで増殖させた培養物は、図13に示されるように、GnTIIIのかなりより少ない発現を有していた。
【0110】
特異的C2B8抗原結合の確認。種々のレベルのGnTIIIを発現する細胞の平行培養物から産生されたC2B8サンプルを、アフィニティークロマトグラフィーにより培養上清から精製し、そしてカチオン交換カラム上でPBSに緩衝液交換した。純度は、還元条件下のSDS−PAGEのクマシーブルー染色から95%より高いと推定された。これらの抗体サンプルは、可変領域がPCRアセンブリ法により合成された抗体遺伝子の発現に由来した。この合成cDNAフラグメントの配列決定は、先に公開された国際特許出願(国際公開番号WO94/11026)中の元のC2B8可変領域配列とは差異を示さなかった。このサンプルのヒトCD20(C2B8の標的抗原である)への特異的結合は、その表面上にCD20を発現するヒトリンパ芽球様細胞株SB、およびこの抗原を欠くHSBリンパ芽球様細胞株を用いる、間接免疫蛍光により証明された。抗体サンプルC2B8−25tは、SB細胞の陽性染色を与えたが(図14A)、同一の実験条件下でHSB細胞の陽性染色は与えなかった(図14Bを参照のこと)。SB細胞からなるさらなる陰性コントロールを、C2B8−25t抗体の代わりにPBS緩衝液とともにインキュベートした。それは、染色が全くないことを示した。
【0111】
C2B8サンプルのインビトロADCC活性。GntIII発現を有さないCHO−tTA−C2B8細胞(前述を参照のこと)中で発現した抗体サンプルC2B8−ntは、ヒトリンパ球によるSB細胞(CD20+)のインビトロ溶解で測定したとき、(1μg/mlの抗体濃度で)31%の細胞傷害性活性を示した(図15、サンプルC2B8−nt)。2000ng/mlのテトラサイクリンで増殖したCHO−tet−GnTIII培養物(すなわち、クローン化したGnTIII発現の基底レベル)由来のC2B8−2000t抗体は、同じ抗体濃度におけるC2B8−ntサンプルに関してADCC活性において、1μg/mlの抗体濃度で33%の増加を示した。テトラサイクリンの濃度を25ng/mlまで低減させると(サンプルC2B8−25t)、これはGnTIII発現を有意に増大させ、同じ抗体濃度におけるC2B8−nt抗体サンプルに関して最大ADCC活性において(1μg/mlの抗体濃度で)ほぼ80%の大きな増加を生じた(図15、サンプルC2B8−25t)。
【0112】
最も高いADCC活性を示すことに加えて、C2B8−25tは、非常に低抗体濃度で、有意なレベルの細胞傷害性を示した。0.06μg/mlにおけるC2B8−25tサンプルは、1μg/mlにおけるC2B8−ntの最大ADCC活性に類似のADCC活性を示した。この結果は、サンプルC2B8−25tが、C2B8−ntより16倍低い抗体濃度で、C2B8−ntと同じADCC活性に到達したことを示した。この結果は、GnTIIIを活性に発現する細胞株で生産されたキメラ抗CD20抗体C2B8が、GnTIIIを発現しなかった細胞株で生産された同じ抗体より有意に活性であったことを示す。
【0113】
本発明の方法を用いるこの抗体の1つの利点は、(1)同じ治療効果に到達するために注入されなけばならない抗体のより低い用量が、抗体産生の経済学で有利なインパクトを有すること、または(2)同じ用量の抗体を用いてより良い治療効果が得られることである。
【0114】
(E.実施例5:最大ADCC活性に至る最適レベルでグリコシルトランスフェラーゼ遺伝子の構成的発現を有するCHO細胞株の確立)
ADCCを増大するための方法のいくつかの適用では、それ自体で、または他のクローン化されたグリコシルトランスフェラーゼおよび/またはグリコシダーゼとともに、GnTIIIの調節された発現よりも構成的発現を用いることが所望され得る。しかし、本発明者らは、改変された抗体のADCC活性が、GnTIIIの発現レベルに依存することを証明した。前述を参照のこと。従って、GnTIII単独の、またはその他のグリコシルトランスフェラーゼおよび/またはグリコシダーゼ遺伝子とともに、最適レベルまたは最適レベル近傍でその構成的発現を有するクローンを選択することが重要である。単独またはβ(1,4)−ガラクトシルトランスフェラーゼ(GalT)のような他のグリコシルトランスフェラーゼとともにのいずれかで、GnTIII発現の最適レベルは、グリコシルトランスフェラーゼの調節性発現を有する細胞株を用いて最初に決定される。次いで、GnTIIIおよび任意のその他のクローン化されたグリコシルトランスフェラーゼの構成的発現を有する安定クローンを、最適近傍の発現レベルについてスクリーニングする。
【0115】
(1.調節されたGnTIIIのためのベクターの最適近傍発現レベル構成の決定)
GFP発現に連鎖する発現。各グリコシルトランスフェラーゼ遺伝子は、IRES配列を経由して、細胞中に保持されるタンパク質、例えば、グリーン蛍光タンパク質(GFP)または利用可能な抗体により認識され得るペプチドでタグ化される原形質膜タンパク質をコードするレポーター遺伝子に連結される。1つより多くのグリコシルトランスフェラーゼが試験されている場合、種々のマーカーが各々のグリコシルトランスフェラーゼと連結される。例えば、GnTIIIがGFPに連結され得、そしてGalTがブルー蛍光タンパク質(BFP)に連結され得る。GFP cDNAの上流のIRESエレメントの上流のGnTIII cDNAからなる真核生物発現カセットは、まず、標準的なサブクローニングおよび/またはPCR工程によりアセンブルされる。次いでこのカセットを、テトラサイクリン調節発現ベクターpUHD10−3(前述を参照のこと)中、tet−プロモーターの下流でかつ終止配列およびポリアデニル化配列の上流にサブクローン化し、ベクターpUHD10−3−GnTIII−GFPを得る。
【0116】
GFP発現に連鎖した調節されたGnTIII発現を持つCHO細胞の確立、および構成的chCE7抗体発現。テトラサイクリン応答性トランスアクチベーターを発現するCHO−tTA細胞(前述を参照のこと)を、ベクターpUHD10−3−GnTIII−GFPおよびピューロマイシン耐性遺伝子の発現のためのベクターpPurで同時トランスフェクトする。前述を参照のこと。ピューロマイシン耐性クローンを、テトラサイクリンの存在下で選択する。個々のクローンを、テトラサイクリンの存在下(2μg/ml)または非存在下で2連で培養する。グリコシルトランスフェラーゼ過剰発現に起因して(前述を参照のこと)、テトラサイクリンの非存在下で増殖の阻害を示す6つのクローンを選択し、そしてGFP関連シグナルの検出のために、蛍光活性化細胞ソーティング(FACS)により分析する。最高の誘導比(テトラサイクリンの存在下の蛍光に対するテトラサイクリンの非存在下の蛍光の比として規定される)を与えるクローンを、さらなる作業のために選択し、そしてCHO−tet−GnTIII−GFPと称する。CHO−tet−GnTIII−GFPを、抗体chCE7についての発現ベクターでトランスフェクトし、そしてこの抗体の高い構成的発現を有するクローンは、選択されたCHO−tet−GnTIII−GFP−chCE7である。前述を参照のこと。
【0117】
chCE7サンプルの生産、ADCC活性の測定および最適GnTIII発現レベルの決定。CHO−tet−GnTIII−GFP−chCE7の平行培養物を、種々のレベルのテトラサイクリン、そしてそれ故、GFPとともにGnTIIIを種々のレベルで発現して増殖させる。chCE7抗体サンプルを、アフィニティークロマトグラフィーにより培養上清から精製する。平行して、各培養物からの細胞をFACSにより分析して、各培養物の、GnTIIIの発現レベルに相関する、GFP関連蛍光の平均レベルを決定する。各chCE7抗体サンプルのインビトロADCC活性を決定し(前述を参照のこと)、そして各サンプルの最大インビトロADCC活性を、それを生成するために用いた細胞の平均蛍光に対してプロットする。
【0118】
(2.最適近傍レベルで構成的GnTIII発現を持つCHO細胞株の確立)
GnTIII−IRES−GFPカセット(前述を参照のこと)を、構成的発現ベクター中にサブクローン化する。CHO細胞を、このベクターおよびピューロマイシン耐性のためのベクターで安定に同時トランスフェクトする。ピューロマイシン耐性細胞を選択する。次いで、この安定にトランスフェクトされた細胞の集団を、FACSによりソートし、そして最適または最適近傍のADCC活性が達成される範囲内近傍のレポーターGFP遺伝子レベルを発現するクローンを選択する。前述を参照のこと。この最終のトランスフェクション工程は、すでに治療抗体を安定に発現するCHO細胞、または空のCHO細胞(例えば、DUKXまたはDG44 dhfr−CHO細胞)のいずれかに対してなされ得る。後者の場合には、上記の手順から得られたクローンは、最終の抗体産生細胞株を作製するために、治療抗体発現ベクターでトランスフェクトされる。
【0119】
(F.実施例6:最適化されたグリコシル化を有するヒトIgG Fcキメラの細胞表面発現)
現在、カプセル化細胞治療が多くの疾患に対して試験されている。カプセル化細胞インプラントを、身体内に外科的に配置されるように設計し、所望の治療物質をそれが必要である場所に直接送達する。しかし、移植されたカプセル化デバイスが、一旦機械的破壊を有する場合、細胞は、逃れそして所望でなくなり得る。身体内で、逃れた、所望でない細胞を破壊する1つの方法は、Fc媒介細胞傷害性メカニズムを経由することである。この目的のために、カプセル化されるべき細胞を先に操作して、Fc領域のN末端に融合した原形質膜に局在化するII型膜貫通ドメインからなる原形質膜係留融合タンパク質を発現し得る。Stabila、P.F.、1998、前述。カプセル内側の細胞は、Fc媒介細胞傷害性に対してカプセルによって保護され、その一方、逃れた細胞は、表面に提示されたFc領域を認識するリンパ球による破壊、すなわち、Fc媒介細胞傷害性メカニズムによる破壊に接近可能である。この実施例は、このFc媒介細胞傷害性活性が、提示されたFc領域のグリコシル化操作によってどのように増大するかを説明する。
【0120】
(1.Fcキメラをその表面上に発現し、およびGnTIIIを発現する細胞の確立)
特定の治療のために移植されるべき細胞、例えば、表面に提示されたFcキメラおよび分泌性治療タンパク質を既に生産する、ベビーハムスター腎臓(BHK)細胞を、まず、IRESエレメントを経由してGFPの発現に連鎖したGnTIIIの構成的発現のためのベクターで安定にトランスフェクトする。前述を参照のこと。安定なトランスフェクタントを、ベクター中に取り込んだマーカーにより、例えば、薬物耐性マーカーにより選択し、そしてその薬物の存在下の生存について選択する。
【0121】
(2.種々のレベルのGnTIIIを発現する細胞のスクリーニングおよび測定)
安定なトランスフェクタントを、蛍光活性化細胞ソーティング(FACS)により分析し、そして種々の平均蛍光レベルを有する一連のクローンを、さらなる研究のために選択する。各選択されたクローンを増殖させ、そしてFACSにより再分析し、GFPの安定性、そしてそれ故、関連したGnTIII発現を確実にする。
【0122】
(3.提示されたFc領域上の複合型バイセクト型オリゴ糖の種々のレベルの確認)
種々のレベルのGFP関連蛍光を有する3つのクローンから、およびGnTIII−IRES−GFPベクターでトランスフェクトしていない元のBHK細胞からのFc領域を、界面活性剤により膜から可溶化し、次いでアフィニティクロマトグラフィーにより精製する。次いで、オリゴ糖を取り出し、精製し、そしてMALDI−TOF/MSにより分析する。前述を参照のこと。得られるMALDI−TOF/MSプロフィールは、改変された、蛍光クローンのFc領域が、種々の比率の複合型バイセクト型オリゴ糖を保持することを示す。改変されていない細胞からのMALDIプロフィールは、バイセクト型オリゴ糖にともなう任意のピークを示さない。提示されたFc領域上で最高レベルの複合型バイセクト型オリゴ糖を保持するクローンを、さらなる作業のために選択する。
【0123】
(4.インビトロのFcに媒介される細胞の細胞傷害性活性アッセイ)
次いで、Fcに媒介される細胞の細胞傷害性活性アッセイを2つ、平行して実施する。1つのアッセイでは、標的細胞は、上記で選択されたクローンに由来する。平行するアッセイでは、標的細胞は、カプセル化され、そしてGnTIIIを発現するように改変されていない元の細胞である。このアッセイを、先に記載された手順を用いるが(前述を参照のこと)、任意のさらなる抗体の非存在下で実施する。なぜなら、標的細胞は、既にFc領域を提示しているからである。この実験は、GnTIIIを発現する細胞に対するFc媒介細胞傷害性活性が、このグリコシルトランスフェラーゼを発現しない細胞に対するFc媒介細胞傷害性活性よりも高いことを示す。
【0124】
本明細書の本体内で引用されるすべての参考文献は、その全体が参考として本明細書に援用される。
【技術分野】
【0001】
(I.他の出願との関連)
本出願は、1998年4月20日に出願された米国仮特許出願第60/082,581号(これは、本明細書中にその全体が参考として援用される)の優先権を主張する。
【0002】
(II.発明の分野)
本発明は、タンパク質のグリコシル化操作の分野に関する。さらに詳細には、本発明は、改善された治療特性を有するタンパク質(増強された抗体依存性細胞傷害性を有する抗体を含む)を生成するグリコシル化操作に関する。
【背景技術】
【0003】
(III.発明の背景)
糖タンパク質は、ヒト、他の真核生物、およびいくつかの原核生物における多くの必須の機能(触媒、シグナル伝達、細胞間コミュニケーション、および分子認識および分子会合を含む)を媒介する。それらは、真核生物において非細胞質ゾルタンパク質の大部分を構成している。非特許文献1。治療目的のために多くの糖タンパク質が探索されており、そして最近20年の間には、天然に存在する分泌糖タンパク質の組換えバージョンが生物工学産業の主要な産物になった。例としては、エリスロポエチン(EPO)、治療的モノクローナル抗体(治療的mAb)、組織プラスミノーゲンアクチベーター(tPA)、インターフェロン−β(IFN−β)、顆粒球−マクロファージコロニー刺激因子(GM−CSF)、およびヒト絨毛性ゴナドトロピン(hCH)が挙げられる。非特許文献2。
【0004】
オリゴ糖成分は、治療的糖タンパク質の有効性(物理学的安定性、プロテアーゼの攻撃に対する耐性、免疫系との相互作用、薬物動態、および特異的生物学的活性を含む)に関する特性に、有意に影響を及ぼし得る。このような特性は、オリゴ糖の存在または非存在に依存し得るだけでなく、その特異的構造にも依存し得る。オリゴ糖構造と糖タンパク質機能との間のいくつかの一般化がなされ得る。例えば、特定のオリゴ糖構造は、特定の糖質結合タンパク質との相互作用を介した血流からのこの糖タンパク質の迅速なクリアランスを媒介する一方、他のオリゴ糖構造は、抗体に結合し、所望されない免疫応答を誘発し得る。非特許文献3。
【0005】
哺乳動物細胞は、ヒト適用のために最も適合した形態でタンパク質をグリコシル化する能力のために、治療的糖タンパク質の生成のために好ましい宿主である。非特許文献2;非特許文献3。細菌はタンパク質をほとんどグリコシル化しない。そして同様に他の型の一般的な宿主(例えば、酵母、糸状菌、昆虫細胞および植物細胞)は、血流からの迅速なクリアランス、所望されない免疫応答、およびいくつかの特定の場合では、減少した生物学的活性と関連したグリコシル化パターンを生じる。哺乳動物細胞の中でも、チャイニーズハムスター卵巣(CHO)細胞が、最近20年の間に最も一般的に使用された。適切なグリコシル化パターンを与えることに加えて、これらの細胞は、遺伝的に安定な、高度に生産的なクローン性細胞株の一貫した生成を可能にする。それらは、無血清培地を使用して単純なバイオリアクター中で高密度に培養され得、そして安全かつ再現可能な生物プロセスの開発を可能にする。他の一般的に使用された動物細胞としては、新生仔ハムスター腎臓(BHK)細胞、NS0マウスミエローマ細胞およびSP2/0マウスミエローマ細胞が挙げられる。さらに最近、トランスジェニック動物からの生成もまた、試験されている。非特許文献3。
【0006】
動物細胞で生成した組換え治療的タンパク質のグリコシル化は、宿主細胞中のグリコシルトランスフェラーゼ遺伝子の過剰発現によって操作され得る。非特許文献4。しかし、この分野の以前の研究では、糖タンパク質改変グリコシルトランスフェラーゼ遺伝子の構成性発現が使用されてきたに過ぎず、発現レベルについては、ほとんど注意が払われてこなかった。
【先行技術文献】
【非特許文献】
【0007】
【非特許文献1】LisおよびSharon、Eur.J.Biochem.(1993)218:1−27
【非特許文献2】Cummingら、Glycobiology(1991)1:115−130
【非特許文献3】Jenkinsら、Nature Biotechn.(1996)14:975−981
【非特許文献4】Bailey、Science(1991)252:1668−1675
【発明の概要】
【課題を解決するための手段】
【0008】
(IV.発明の要旨)
本発明は、一般に、改善された治療価値を生じる改変されたグリコシル化パターンを有するタンパク質の生成のための宿主細胞および方法に関する。1つの特定の実施態様において、本発明は、バイセクティングGlcNAcを有するN結合型複合型オリゴ糖を増加する、糖タンパク質改変グリコシルトランスフェラーゼ活性の好ましい範囲を発現し得るように操作された宿主細胞に関する。他の実施態様において、本発明は、糖タンパク質、例えば、抗体(抗体分子全体、抗体フラグメント、または免疫グロブリンのFc領域に等価な領域を含む融合タンパク質を含み、増強されたFc媒介細胞傷害性を有する)の改変された糖形態を生成するための方法、およびそのように生成された糖タンパク質に関する。本発明は、バイセクティングGlcNAcを有するN結合型複合型オリゴ糖の最大化のための糖タンパク質改変グリコシルトランスフェラーゼ発現の最適な範囲が存在するという本発明者らの発見に一部基づく。
【0009】
より詳細には、本発明は、改善した治療的価値を有する改変された糖形態のタンパク質、例えば、増強された抗体依存性細胞傷害性(ADCC)を有する抗体を宿主細胞において生成する方法に関する。本発明は、目的のタンパク質(例えば、抗体)をコードする核酸、および糖タンパク質改変グリコシル化トランスフェラーゼをコードする少なくとも1つの核酸を有する宿主細胞を提供する。さらに、本発明は、上記の目的のタンパク質(例えば、抗体依存性細胞傷害性を増強した抗体)の発現を可能にする条件下でこのような宿主細胞を培養する方法およびプロトコルを提供する。さらに、このように生成された、改変されたグリコシル化パターンを有するタンパク質(例えば、増強された抗体依存性細胞傷害性を有する抗体)を単離する方法が記載される。
【0010】
さらに、本発明は、改善した治療特性を有するタンパク質の別の糖形態を提供する。本発明のタンパク質は、増強された抗体依存性細胞傷害性(ADCC)を有する抗体を含み、これは、開示された方法および宿主細胞を使用して生成される。
【0011】
(VI.定義)
用語は、以下に定義されなければ、当該分野で一般に使用されるように本明細書中で使用される。
【0012】
本明細書中で使用される用語「抗体」は、抗体分子全体、抗体フラグメント、または免疫グロブリンのFc領域と等価な領域を含む融合タンパク質を含むことが意図される。
【0013】
本明細書中で使用される用語「糖タンパク質改変グリコシルトランスフェラーゼ」とは、糖タンパク質のグリコシル化パターンの改変をもたらす酵素をいう。糖タンパク質改変グリコシルトランスフェラーゼの例としては、例えば、GnT III、GnT V、GalT、およびManIIのようなグリコシルトランスフェラーゼが挙げられるが、これらに限定されない。
【0014】
本明細書中で使用される用語「グリコシル化操作」は、天然に存在するポリペプチドまたはそのフラグメントのグリコシル化パターンに対して任意の種類の変化を含むとみなされる。グリコシル化操作としては、細胞のグリコシル化機構の代謝的操作(細胞中で発現された糖タンパク質の改変されたグリコシル化を達成する、オリゴ糖合成経路の遺伝子操作を含む)が挙げられる。さらに、グリコシル化操作としては、グリコシル化に対する変異および細胞環境の効果が挙げられる。
【0015】
本明細書中で使用される用語「宿主細胞」は、タンパク質、タンパク質フラグメント、または目的のペプチド(抗体または抗体フラグメントを含む)の改変された糖形態を生成するように操作され得る任意の種類の細胞系を含む。代表的には、宿主細胞は、少なくとも1つの、糖タンパク質改変グリコシルトランスフェラーゼの最適化されたレベルを発現するように操作されており、これらのグリコシルトランスフェラーゼとしては、GnT III、GnT V、GalT、およびManII、ならびに/または少なくとも1つのグリコシダーゼが挙げられるが、これらに限定されない。宿主細胞としては、培養細胞、例えば、哺乳動物培養細胞(例えば、CHO細胞、BHK細胞、NS0細胞、SP2/0細胞またはハイブリドーマ細胞)、酵母細胞および昆虫細胞が挙げられ、トランスジェニック動物または培養組織内に含まれる細胞もまた挙げられる。
【0016】
本明細書中で使用される用語「Fc媒介細胞傷害性」は、抗体依存性細胞傷害性(ADCC)、および免疫グロブリンGのFc領域またはそれに等価な領域をその細胞表面で発現するように操作された細胞に対する細胞傷害性、ならびに免疫グロブリンGのFc領域またはそれに等価な領域のN末端に融合された標的タンパク質ドメインからなる可溶性融合タンパク質により媒介される細胞傷害性を含むことが意図される。
したがって本発明は、以下の項目を提供する
(項目1) 調節されたレベルにおいて糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸を発現するために操作された、宿主細胞。
(項目2) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子を含む核酸分子が、上記宿主細胞に導入されている、項目1に記載の宿主細胞。
(項目3) 内因性の糖タンパク質改変グリコシルトランスフェラーゼが活性化されるように、上記宿主細胞が操作されている、項目1に記載の宿主細胞。
(項目4) 上記宿主細胞が、CHO細胞、BHK細胞、NS0細胞、SP2/0細胞、またはハイブリドーマ細胞である、項目2または3に記載の宿主細胞。
(項目5) 上記内因性の糖タンパク質改変グリコシルトランスフェラーゼが、調節性プロモーターエレメントを上記宿主細胞染色体へ挿入することによって活性化されている、項目3に記載の宿主細胞。
(項目6) 上記糖タンパク質改変グリコシルトランスフェラーゼが、GnT III、GnT V、Man II、またはGal Tである、項目2または3に記載の宿主細胞。
(項目7) 上記宿主細胞が、GnT III、GnT V、Man II、およびGal Tからなる群から選択される少なくとも2つの異なる、糖タンパク質改変グリコシルトランスフェラーゼを発現するために操作された、項目2または3に記載の宿主細胞。
(項目8) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子が、構成性プロモーターエレメントに作動可能に連結されている、項目7に記載の宿主細胞。
(項目9) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子が、調節性プロモーターエレメントに作動可能に連結されている、項目2に記載の宿主細胞。
(項目10) 上記調節性プロモーターエレメントが、テトラサイクリンに調節性プロモーター系、エクジソン誘導性プロモーター系、lacスイッチプロモーター系、グルココルチコイド誘導性プロモーター系、温度誘導性プロモーター系、またはメタロチオネイン金属誘導性プロモーター系である、項目5または9に記載の宿主細胞。
(項目11) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸分子を発現するために操作された宿主細胞であって、ここでその宿主細胞が、増強したFc媒介細胞傷害性を有するタンパク質を産生し得る、宿主細胞。
(項目12) 上記タンパク質が、抗体分子全体、抗体フラグメント、または免疫グロブリンのFc領域に等価な領域を含む融合タンパク質である、項目11に記載の宿主細胞。
(項目13) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子を含む核酸分子が、上記宿主細胞染色体へ導入されている、項目12に記載の宿主細胞。
(項目14) 上記宿主細胞が、内因性の糖タンパク質改変グリコシルトランスフェラーゼの変異誘発発現を保有するために選択されている、項目12に記載の宿主細胞。
(項目15) 上記宿主細胞が、変異lec10である、項目14に記載の宿主細胞。
(項目16) 内因性の糖タンパク質改変グリコシルトランスフェラーゼが活性化されるように、上記宿主細胞が操作されている、項目12に記載の宿主細胞。
(項目17) 上記内因性の糖タンパク質改変グリコシルトランスフェラーゼが、調節性プロモーターエレメントを上記宿主細胞染色体へ挿入することによって活性化されている、項目16に記載の宿主細胞。
(項目18) 上記内因性の糖タンパク質改変グリコシルトランスフェラーゼが、構成性プロモーターエレメント、トランスポゾン、またはレトロウイルスエレメントを上記宿主細胞染色体へ挿入することによって活性化されている、項目16に記載の宿主細胞。
(項目19) 抗体分子、抗体フラグメント、または免疫グロブリンのFc領域に等価な領域を含む融合タンパク質をコードする少なくとも1つのトランスフェクトされた核酸をさらに含む、項目11または13に記載の宿主細胞。
(項目20) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子が、構成性プロモーターエレメントに作動可能に連結される、項目13に記載の宿主細胞。
(項目21) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子が、調節性プロモーターエレメントに作動可能に連結される、項目13に記載の宿主細胞。
(項目22) 上記調節性プロモーターエレメントが、テトラサイクリンに調節性プロモーター系、エクジソン誘導性プロモーター系、lacスイッチプロモーター系、グルココルチコイド誘導性プロモーター系、温度誘導性プロモーター系、またはメタロチオネイン金属誘導性プロモーター系である、項目21に記載の宿主細胞。
(項目23) 上記宿主細胞がハイブリドーマ細胞である、項目11に記載の宿主細胞。
(項目24) 上記操作された宿主細胞が、操作されたCHO細胞、操作されたBHK細胞、操作されたNS0細胞、または操作されたSP2/0細胞である、項目11に記載の宿主細胞。
(項目25) 上記宿主細胞が、キメラ抗CD20モノクローナル抗体(C2B8)をコードする少なくとも1つのトランスフェクトされた核酸を含む、項目11に記載の宿主細胞。
(項目26) 上記宿主細胞が、キメラ抗ヒト神経芽腫モノクローナル抗体(chCE7)をコードする少なくとも1つのトランスフェクトされた核酸を含む、項目11に記載の宿主細胞。
(項目27) 上記宿主細胞が、キメラ抗ヒト腎細胞癌モノクローナル抗体(ch−G250)、ヒト化抗HER2モノクローナル抗体、キメラ抗ヒト結腸癌、肺癌、および乳癌モノクローナル抗体(ING−1)、ヒト化抗ヒト17−1A抗原モノクローナル抗体(3622W94)、ヒト化抗ヒト結腸直腸腫瘍抗体(A33)、GD3ガングリオシドに対する抗ヒト黒色腫抗体(R24)、またはキメラ抗ヒト扁平上皮癌モノクローナル抗体(SF−25)をコードする少なくとも1つのトランスフェクトされた核酸を含む、項目11に記載の宿主細胞。
(項目28) 少なくとも1つの核酸分子が、β(1,4)−N−アセチルグルコサミニルトランスフェラーゼIII(GnT III)をコードする、項目11に記載の宿主細胞。
(項目29) β(1,4)−ガラクトシルトランスフェラーゼ(GalT)をコードする少なくとも1つの核酸をさらに含む、項目28に記載の宿主細胞。
(項目30) マンノシダーゼII(Man II)をコードする少なくとも1つの核酸をさらに含む、項目28に記載の宿主細胞。
(項目31) β(1,4)−ガラクトシルトランスフェラーゼ(GalT)をコードする少なくとも1つの核酸およびマンノシダーゼII(Man II)をコードする少なくとも1つの核酸をさらに含む、項目28に記載の宿主細胞。
(項目32) 宿主細胞において、増強したFc媒介細胞傷害性を有するタンパク質化合物を産生するための方法であって、以下の工程:
(a)調節されたレベルにおいて、目的のタンパク質化合物のグリコシル化を改善するように選択された、糖タンパク質改変グリコシルトランスフェラーゼを発現するために操作された宿主細胞を提供する工程であって、ここで、その宿主細胞が、抗体、抗体フラグメント、または免疫グロブリンのFc領域に等価な領域を含む融合タンパク質をコードする少なくとも1つの核酸を発現する、工程;
(b)増強したFc媒介性依存性細胞傷害性を有するそのタンパク質化合物の産生を可能にする条件下で、その宿主細胞を培養する工程;および
(c)増強したFc媒介細胞傷害性を有するそのタンパク質化合物を単離する工程、
を包含する、方法。
(項目33) 工程(a)において、上記宿主細胞が、抗体全体をコードする少なくとも1つの核酸を含む、項目32に記載の方法。
(項目34) 工程(a)において、上記宿主細胞が、抗体フラグメントをコードする少なくとも1つの核酸を含む、項目32に記載の方法。
(項目35) 工程(a)において、上記宿主細胞が、免疫グロブリンのグルコシル化Fc領域に類似の領域を含む融合タンパク質をコードする少なくとも1つの核酸を含む、項目32に記載の方法。
(項目36) 上記宿主細胞が、キメラ抗CD20モノクローナル抗体(C2B8)をコードする少なくとも1つのトランスフェクトされた核酸を含む、項目32に記載の方法。
(項目37) 上記宿主細胞が、キメラ抗ヒト神経芽腫モノクローナル抗体(chCE7)をコードする少なくとも1つのトランスフェクトされた核酸を含む、項目32に記載の方法。
(項目38) 上記宿主細胞が、キメラ抗ヒト腎細胞癌モノクローナル抗体(ch−G250)、ヒト化抗HER2モノクローナル抗体、キメラ抗ヒト結腸癌、肺癌、および乳癌モノクローナル抗体(ING−1)、ヒト化抗ヒト17−1A抗原モノクローナル抗体(3622W94)、ヒト化抗ヒト結腸直腸腫瘍抗体(A33)、GD3ガングリオシドに対する抗ヒト黒色腫抗体(R24)、またはキメラ抗ヒト扁平上皮癌モノクローナル抗体(SF−25)をコードする少なくとも1つのトランスフェクトされた核酸を含む、
項目32に記載の方法。
(項目39) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子が、上記宿主細胞に導入されている、項目32に記載の方法。
(項目40) 上記宿主細胞が、内因性の糖タンパク質改変グリコシルトランスフェラーゼの変異誘発発現を保有するために選択されている、項目32に記載の宿主細胞。
(項目41) 上記宿主細胞が、変異lec10である、項目40に記載の宿主細胞。
(項目42) 内因性の糖タンパク質改変グリコシルトランスフェラーゼが活性化されるように、上記宿主細胞が操作されている、項目32に記載の宿主細胞。
(項目43) 上記グリコシルトランスフェラーゼが、β(1,4)−N−アセチルグルコサミニルトランスフェラーゼIII(GnT III)である、項目32に記載の方法。
(項目44) 上記GnT IIIが、構成性プロモーター系を使用して発現される、項目43に記載の方法。
(項目45) 上記GnT IIIが、調節性プロモーター系を使用して発現される、項目43に記載の方法。
(項目46) 上記調節性プロモーター系が、テトラサイクリンに調節性プロモーター系、エクジソン誘導性プロモーター系、lacスイッチプロモーター系、グルココルチコイド誘導性プロモーター系、温度誘導性プロモーター系、またはメタロチオネイン金属誘導性プロモーター系である、項目45に記載の方法。
(項目47) 上記グリコシルトランスフェラーゼが、β(1,4)−ガラクトシルトランスフェラーゼ(GalT)である、項目32に記載の方法。
(項目48) 上記GalTが、構成性プロモーター系を使用して発現される、項目47に記載の方法。
(項目49) 上記GalTが、調節性プロモーター系を使用して発現される、項目47に記載の方法。
(項目50) 上記調節性プロモーター系が、テトラサイクリンに調節性プロモーター系、エクジソン誘導性プロモーター系、lacスイッチプロモーター系、グルココルチコイド誘導性プロモーター系、温度誘導性プロモーター系、またはメタロチオネイン金属誘導性プロモーター系である、項目49に記載の方法。
(項目51) 上記宿主細胞が、調節されたレベルにおいて、目的のタンパク質化合物のグリコシル化を改善するように選択された、グリコシルトランスフェラーゼをコードする複数の核酸を発現するために操作され、ここで、少なくとも1つの核酸がGnT IIIをコードし、そして少なくとも1つの核酸がβ(1,4)−ガラクトシルトランスフェラーゼ(GalT)をコードする、項目32に記載の方法。
(項目52) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子を含む核酸分子が、上記宿主細胞に導入されている、項目51に記載の宿主細胞。
(項目53) 上記宿主細胞が、少なくとも1つの、内因性の糖タンパク質改変グリコシルトランスフェラーゼの変異誘発発現を保有するために選択されている、項目51に記載の宿主細胞。
(項目54) 内因性の糖タンパク質改変グリコシルトランスフェラーゼが活性化されるように、上記宿主細胞が操作されている、項目51、52、または53に記載の宿主細胞。
(項目55) 上記宿主細胞が、調節されたレベルにおいて、目的のタンパク質化合物のグリコシル化を改善するように選択された、糖タンパク質改変グリコシルトランスフェラーゼをコードする複数の核酸を含み、ここで、少なくとも1つの核酸がGnT IIIをコードし、そして少なくとも1つの核酸がマンノシダーゼII(Man II)をコードする、項目32に記載の方法。
(項目56) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子を含む核酸分子が、上記宿主細胞に導入されている、項目55に記載の宿主細胞。
(項目57) 上記宿主細胞が、少なくとも1つの、内因性の糖タンパク質改変グリコシルトランスフェラーゼの変異誘発発現を保有するために選択されている、項目55に記載の宿主細胞。
(項目58) 内因性の糖タンパク質改変グリコシルトランスフェラーゼが活性化されるように、上記宿主細胞が操作されている、項目55、56、または57に記載の宿主細胞。
(項目59) 上記宿主細胞が、調節されたレベルにおいて、目的のタンパク質のグリコシル化を改善するように選択された、糖タンパク質改変グリコシルトランスフェラーゼをコードする複数の核酸を含み、ここで、少なくとも1つの核酸がGnT IIIをコードし、少なくとも1つの核酸がβ(1,4)−ガラクトシルトランスフェラーゼ(GalT)をコードし、そして少なくとも1つの核酸がマンノシダーゼII(Man II)をコードする、項目32に記載の方法。
(項目60) 糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子を含む核酸分子が、上記宿主細胞に導入されている、項目59に記載の宿主細胞。
(項目61) 上記宿主細胞が、少なくとも1つの、内因性の糖タンパク質改変グリコシルトランスフェラーゼの変異誘発発現を保有するために選択されている、項目59に記載の宿主細胞。
(項目62) 内因性の糖タンパク質改変グリコシルトランスフェラーゼが活性化されるように、上記宿主細胞が操作されている、項目59、60、または61に記載の宿主細胞。
(項目63) 少なくとも1つの糖タンパク質改変グリコシルトランスフェラーゼの発現レベルが、抗体分子、抗体フラグメント、または免疫グロブリンのFc領域に等価な領域を含む融合タンパク質を産生するように選択され、その糖タンパク質改変グリコシルトランスフェラーゼが、異なる発現レベルの同一のグリコシルトランスフェラーゼ遺伝子から得られるFc媒介細胞傷害性よりも、より高いレベルにおいて増強したFc媒介細胞傷害性を有する、項目32に記載の方法。
(項目64) 上記発現レベルが、グリコシルトランスフェラーゼ特異的抗体を使用するウエスタンブロット分析によって決定される、項目63に記載の方法。
(項目65) 上記発現レベルが、グリコシルトランスフェラーゼ特異的プローブを使用するノーザンブロット分析によって決定される、項目63に記載の方法。
(項目66) 上記発現レベルが、グリコシルトランスフェラーゼの酵素活性を測定することによって決定される、項目63に記載の方法。
(項目67) 上記発現レベルが、糖タンパク質改変グリコシルトランスフェラーゼの生合成産物に結合するレクチンを使用して決定される、項目63に記載の方法。
(項目68) 上記レクチンがE4−PHAレクチンである、項目67に記載の方法
。
(項目69) 上記糖タンパク質改変グリコシルトランスフェラーゼをコードする上記核酸が、レポーター遺伝子に作動可能に連結され、そしてここで、そのグリコシルトランスフェラーゼの上記発現レベルが、そのレポーター遺伝子の発現レベルと相関するシグナルを測定することによって決定される、項目63に記載の方法。
(項目70) 上記レポーター遺伝子が、単一のRNA分子として、上記糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸とともに転写され、そしてそれらの各コード配列が、内部リボソーム侵入部位(IRES)またはキャップ独立性翻訳エンハンサー(CITE)のいずれかによって連結される、項目69に記載の方法。
(項目71) 単一のポリペプチド鎖が形成されるように、上記レポーター遺伝子が、上記糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸とともに翻訳される、項目69に記載の方法。
(項目72) 上記糖タンパク質改変グリコシルトランスフェラーゼをコードする上記核酸が、単一のプロモーターの制御下でレポーター遺伝子に作動可能に連結され、ここでその糖タンパク質改変グリコシルトランスフェラーゼをコードするその核酸およびそのレポーター遺伝子が、2つの別個のメッセンジャーRNA(mRNA)分子に選択的スプライスされるRNA分子へ転写され、ここで得られたmRNAの一方がそのレポータータンパク質に翻訳され、そして他方がその糖タンパク質改変グリコシルトランスフェラーゼに翻訳される、項目63に記載の方法。
(項目73) 上記宿主細胞が、グリコシダーゼをコードする核酸をさらに含む、項目32に記載の方法。
(項目74) 項目11に記載の宿主細胞によって産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、抗体。
(項目75) 項目25に記載の宿主細胞によって産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、キメラ抗CD20モノクローナル抗体(C2D8)。
(項目76) 項目26に記載の宿主細胞を使用して産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、キメラ抗ヒト神経芽腫モノクローナル抗体(chCE7)。
(項目77) 項目27に記載の宿主細胞を使用して産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、キメラ抗ヒト腎細胞癌モノクローナル抗体(ch−G250)。
(項目78) 項目27に記載の宿主細胞を使用して産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、ヒト化抗HER2モノクローナル抗体。
(項目79) 項目27に記載の宿主細胞を使用して産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、キメラ抗ヒト結腸癌、肺癌、および乳癌モノクローナル抗体(ING−1)。
(項目80) 項目27に記載の宿主細胞を使用して産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、ヒト化抗ヒト17−1A抗原モノクローナル抗体(3622W94)。
(項目81) 項目27に記載の宿主細胞を使用して産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、キメラ抗ヒト扁平上皮癌モノクローナル抗体(SF−25)。
(項目82) 項目27に記載の宿主細胞を使用して産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、ヒト化抗ヒト結腸直腸腫瘍抗体(A33)。
(項目83) 項目27に記載の宿主細胞を使用して産生された、増強した抗体依存性細胞傷害性(ADCC)を有する、GD3ガングリオシドに対する抗ヒト黒色腫抗体(R24)。
(項目84) 項目11に記載の宿主細胞を使用して産生された、増強したFc媒介細胞傷害性を有する、免疫グロブリンのFc領域に等価な領域を含む、抗体フラグメント。
(項目85) 項目11に記載の宿主細胞を使用して産生された、増強したFc媒介細胞傷害性を有する、免疫グロブリンのFc領域に等価な領域を含む、融合タンパク質。
【図面の簡単な説明】
【0017】
【図1】図1は、典型的なFc関連オリゴ糖構造の代表を記載する。
【図2】図2は、2つの異なるtTA生成CHOクローンにおけるGnT IIIのテトラサイクリン調節性発現のウェスタンブロット分析を示す。CHOt2細胞(レーンAおよびB)およびCHOt17細胞(レーンCおよびD)は、pUDH10−3GnTIIIm発現ベクターでトランスフェクトされ、そしてテトラサイクリンの非存在下(レーンAおよびC)または400ng/mlの濃度の存在下(レーンBおよびD)で36時間培養した。次いで、細胞溶解物を、抗体(9E10)でプローブするウェスタンブロット分析のために調製した。この抗体は、そのカルボキシ末端でGnT IIIに付加されたc−mycタグを特異的に認識する。
【図3】図3は、mycタグ化GnT III発現が制御され得るテトラサイクリン濃度の範囲の決定を示す。CHOt17細胞を、pUDH10−3−GnTIIIm発現ベクターでトランスフェクトし、次いで、示された濃度のテトラサイクリンの存在下で48時間培養した。これらの培養物の細胞溶解物のGnT IIIレベルは、ウェスタンブロット分析を使用して比較した。GnT IIIを、9E10抗体を使用するc−mycタグを介して検出した。
【図4】図4A〜4Bは、GnT V(図4A)またはmycタグ化GnT III(図4B)グリコシルトランスフェラーゼの安定な、テトラサイクリン調節性発現についてのCHOクローンのウェスタンブロット分析によるスクリーニングを示す。CHOt17細胞は、プロマイシン耐性(pPUR)の発現のためのベクターと、pUHD10−3GnTV(図4A)またはpUDH10−3GnTIIIm(図4B)のいずれかとで同時トランスフェクトされ、そして安定なCHOクローンを、テトラサイクリンの存在下(2μg/ml)で、プロマイシン(7.5μg/ml)に対する耐性について選択した。各グリコシルトランスフェラーゼについての8つのクローン(1〜8)を、テトラサイクリンの非存在下または存在下(+)(2μg/ml)で48時間培養し、そして抗GnT V抗体(図4A)または抗myc抗体(9E10)抗体(図4B)のいずれかを用いてウェスタンブロットによって分析した。
【図5】図5Aおよび5Bは、レクチンブロット分析によるインビボでの異種GnT V(図5A)およびGnT III(図5B)のグリコシルトランスフェラーゼの活性の検証を示す。テトラサイクリン(2μg/ml)の非存在下または存在下(+)で培養した種々の安定なクローンからの細胞糖タンパク質(図4と同様に番号付けした)をSDS−PAGEで分離し、膜にブロットし、そしてL−PHAレクチン(図5A)およびE−PHAレクチン(図5B)のいずれかでプローブした。これらのレクチンは、それぞれ、GnT VおよびGnT IIIによって触媒された反応のオリゴ糖生成物に対して、これらの反応物のオリゴ糖基質に対するより、より高い親和性で結合する。分子量マーカー(MWM)を、並行して泳動した。図5Aおよび5Bのレクチンブロットの比較によって、内因性CHO細胞糖タンパク質の中で、GnT V(図5A)についてより、GnT III(図5B)についてより広範囲の基質が示される。
【図6】図6A〜6Dは、グリコシルトランスフェラーゼの過剰発現の際の細胞増殖の阻害を示す、CHO−tet−GnTIIIm細胞を、5〜10%コンフルエントで播種し、そしてテトラサイクリンの非存在下(図6Aおよび6B)または存在下(図6Cおよび6D)で培養した。培養物を播種して45時間後(図6Aおよび6C)ならびに85時間後(図6Bおよび6D)に写真を撮った。
【図7】図7は、chCE7重鎖遺伝子の構築のためにPCRで使用したオリゴヌクレオチドプライマーの配列を示す。正方向および逆方向プライマーは、それぞれ、「.fwd」および「.rev」を接尾辞としてつけることによって同定する。テンプレートとして最初のPCR工程の産物を使用して、第2のPCR工程を行うために必要な、異なるプライマー間の重複を示す。導入された制限部位、CE7キメラゲノムDNAにアニーリングする配列、および導入された合成リーダー配列もまた示す。
【図8】図8は、chCE7軽鎖遺伝子の構築のためにPCRで使用したオリゴヌクレオチドプライマーの配列を示す。正方向プライマーおよび逆方向プライマーは、それぞれ、「.fwd」および「.rev」を接尾辞としてつけることによって同定する。テンプレートとして最初のPCR工程の産物を使用して、第2のPCR工程を行うために必要な、異なるプライマー間の重複を示す。導入された制限部位、CE7キメラゲノムDNAにアニーリングする配列、および導入されたリーダー配列もまた示す。
【図9A】図9は、SP2/0マウスミエローマ細胞(図9A、50μgのCE7−SP2/0からのオリゴ糖)、または培地に添加したテトラサイクリンの濃度が異なるため、GnT III遺伝子を異なるレベルで発現するCHO−tetGnTIII−chCE7細胞培養物のいずれかにより生成されたchCE7サンプルからの中性オリゴ糖混合物のMALDI/TOF−MSスペクトルを示す。テトラサイクリン濃度の大きさを低減する、すなわち、GnT III遺伝子発現のレベルを増加させることにおいて、後者のサンプルは、CE7−2000t(図9B、37.5μgの抗体からのオリゴ糖)、CE7−60t(図9C、37.5μgの抗体からのオリゴ糖)、CE7−30t(図9D、25μgの抗体からのオリゴ糖)およびCE7−15t(図9E、10μgの抗体からのオリゴ糖)である。
【図9B】図9は、SP2/0マウスミエローマ細胞(図9A、50μgのCE7−SP2/0からのオリゴ糖)、または培地に添加したテトラサイクリンの濃度が異なるため、GnTIII遺伝子を異なるレベルで発現するCHO−tetGnTIII−chCE7細胞培養物のいずれかにより生成されたchCE7サンプルからの中性オリゴ糖混合物のMALDI/TOF−MSスペクトルを示す。テトラサイクリン濃度の大きさを低減する、すなわち、GnT III遺伝子発現のレベルを増加させることにおいて、後者のサンプルは、CE7−2000t(図9B、37.5μgの抗体からのオリゴ糖)、CE7−60t(図9C、37.5μgの抗体からのオリゴ糖)、CE7−30t(図9D、25μgの抗体からのオリゴ糖)およびCE7−15t(図9E、10μgの抗体からのオリゴ糖)である。
【図9C】図9は、SP2/0マウスミエローマ細胞(図9A、50μgのCE7−SP2/0からのオリゴ糖)、または培地に添加したテトラサイクリンの濃度が異なるため、GnTIII遺伝子を異なるレベルで発現するCHO−tetGnTIII−chCE7細胞培養物のいずれかにより生成されたchCE7サンプルからの中性オリゴ糖混合物のMALDI/TOF−MSスペクトルを示す。テトラサイクリン濃度の大きさを低減する、すなわち、GnT III遺伝子発現のレベルを増加させることにおいて、後者のサンプルは、CE7−2000t(図9B、37.5μgの抗体からのオリゴ糖)、CE7−60t(図9C、37.5μgの抗体からのオリゴ糖)、CE7−30t(図9D、25μgの抗体からのオリゴ糖)およびCE7−15t(図9E、10μgの抗体からのオリゴ糖)である。
【図9D】図9は、SP2/0マウスミエローマ細胞(図9A、50μgのCE7−SP2/0からのオリゴ糖)、または培地に添加したテトラサイクリンの濃度が異なるため、GnTIII遺伝子を異なるレベルで発現するCHO−tetGnTIII−chCE7細胞培養物のいずれかにより生成されたchCE7サンプルからの中性オリゴ糖混合物のMALDI/TOF−MSスペクトルを示す。テトラサイクリン濃度の大きさを低減する、すなわち、GnT III遺伝子発現のレベルを増加させることにおいて、後者のサンプルは、CE7−2000t(図9B、37.5μgの抗体からのオリゴ糖)、CE7−60t(図9C、37.5μgの抗体からのオリゴ糖)、CE7−30t(図9D、25μgの抗体からのオリゴ糖)およびCE7−15t(図9E、10μgの抗体からのオリゴ糖)である。
【図9E】図9は、SP2/0マウスミエローマ細胞(図9A、50μgのCE7−SP2/0からのオリゴ糖)、または培地に添加したテトラサイクリンの濃度が異なるため、GnTIII遺伝子を異なるレベルで発現するCHO−tetGnTIII−chCE7細胞培養物のいずれかにより生成されたchCE7サンプルからの中性オリゴ糖混合物のMALDI/TOF−MSスペクトルを示す。テトラサイクリン濃度の大きさを低減する、すなわち、GnT III遺伝子発現のレベルを増加させることにおいて、後者のサンプルは、CE7−2000t(図9B、37.5μgの抗体からのオリゴ糖)、CE7−60t(図9C、37.5μgの抗体からのオリゴ糖)、CE7−30t(図9D、25μgの抗体からのオリゴ糖)およびCE7−15t(図9E、10μgの抗体からのオリゴ糖)である。
【図10】図10は、GnT III触媒された反応を介する、複合型バイセクト型オリゴ糖を導く、N結合型オリゴ糖生合成経路を記載する。Mはマンノース;GnはN−アセチルグルコサミン(GlcNAc);Gはガラクトース;GnbはバイセクティングGlcNAc;fはフコースを示す。オリゴ糖命名法は、コアオリゴ糖に結合されたM、GnおよびG残基を数えること、およびGnbを含めることによってバイセクティングGlcNAcの存在を示すことからなる。オリゴ糖コアは、それ自体が2つのGn残基から構成され、そしてフコースを含んでも、含まなくてもよい。オリゴ糖の主要なクラスは、破線で囲んだ枠の中に示す。Man Iはゴルジマンノシダーゼ;GnTはGlcNAcトランスフェラーゼ;およびGalTはガラクトシルトランスフェラーゼを表す。MALDI/TOF−MS分析で観察される、主要なナトリウムと会合したオリゴ糖イオンに関連する質量は、各オリゴ糖の隣に示す。潜在的にコアがフコシル化され得るオリゴ糖については、フコシル化形態(+f)および非フコシル化形態(−f)の両方に関連する質量を示す。
【図11】図11は、GnT III触媒反応を介して、複合型バイセクト型オリゴ糖および混成型バイセクト型オリゴ糖を導くN結合型オリゴ糖生合成経路を示す。Mはマンノース;GnはN−アセチルグルコサミン(GlcNAc);Gはガラクトース;GnbはバイセクティングGlcNAc;fはフコースを示す。オリゴ糖命名法は、共通のオリゴ糖に結合されたM、GnおよびG残基を数えること、およびGnbを含めることによってバイセクティングGlcNAcの存在を示すことからなる。オリゴ糖コアは、それ自体が2つのGn残基から構成され、そしてフコースを含んでも、含まなくてもよい。オリゴ糖の主要なクラスは、破線で囲んだ枠の中に示す。Man Iはゴルジマンノシダーゼ;GnTはGlcNAcトランスフェラーゼ;およびGalTはガラクトシルトランスフェラーゼを表す。MALDI/TOF−MS分析で観察される、主要なナトリウムと会合したオリゴ糖イオンと関連する質量は、各オリゴ糖の隣に示す。潜在的にコアがフコシル化され得るオリゴ糖については、フコシル化形態(+f)および非フコシル化形態(−f)の両方に関連する質量を示す。
【図12】図12は、異なるchCE7サンプルのADCC活性を示す。ヒトリンパ節によるIMR−32神経芽腫細胞の溶解(標的:エフェクター比は1:19、37℃で16時間のインキュベーション)を、chCE7サンプルの異なる濃度で媒介し、蛍光色素の保持によって測定した。細胞傷害性の割合は、抗体の非存在下でのシグナルを差し引いた後に、総溶解コントロール(界面活性剤による)に対して算出した。
【図13】図13は、異なるテトラサイクリン濃度で増殖させた、CHO−tet−GnT IIIの異なる培養物のGnT III発現を使用して、異なるC2B8抗体サンプルを生成したことを記載する。2000ng/ml(レーンC)および25ng/ml(レーンD)のテトラサイクリン濃度で増殖させた各培養物からの細胞溶解物をSDS−PAGEで分離し、メンブラン上にブロットし、そして1次抗体および2次抗体として、それぞれ、9E10(前出を参照のこと)および抗マウス西洋ワサビペルオキシダーゼを用いてプローブした。レーンAは、陰性コントロールを示す。
【図14】図14は、懸濁物中での細胞を用いた間接免疫蛍光アッセイを使用するC2B8抗CD20モノクローナル抗体の抗原結合の特異性を示す。CD20陽性細胞(SB細胞;ATCC受託番号ATCC CCL120)およびCD20陰性細胞(HSB細胞;ATCC受託番号ATCC CCL120.1)(それぞれ、図14Aおよび14B)を使用した。各型の細胞を、1次抗体として25ng/mlテトラサイクリンで生成したC2B8抗体とともにインキュベートした。陰性コントロールは、1次抗体の代わりに、HBSSBを含めた。抗ヒトIgG Fc特異的ポリクローナルFITC結合体化抗体を2次抗体として全てのサンプルで使用した。
【図15】図15は、異なる抗体濃度(0.04〜5μg/ml)での異なるC2B8抗体サンプルのADCC活性を示す。サンプルC2B8−ntは、GnT III発現なしの細胞株で生成されたC2B8抗体のADCC活性を表す。サンプルC2B8−2000t、C2B8−50tおよびC2B8−25tは、漸減テトラサイクリン濃度(すなわち、漸増GnT III発現)で生成した3つの抗体サンプルのADCC活性を示す。
【発明を実施するための形態】
【0018】
(VII.発明の詳細な説明)
(A.一般的な概要)
本発明の目的は、タンパク質(特に抗体(抗体分子全体、抗体フラグメント、または免疫グロブリンのFc領域に等価な領域を含む融合タンパク質を含む))の糖形態を提供して、治療的タンパク質の新たな改変体を生成することである。本発明は、細胞のグリコシル化反応ネットワークが、その集団内での特定の糖形態の割合を最大化するように操作され得、そして特定の糖形態が改善された治療特性を有するという本発明者らの発見に一部基づく。本発明はさらに、改善された治療価値を有するタンパク質の糖形態を同定する方法、およびそれらを再現可能に生成する方法の発見に一部基づく。本発明はさらに、バイセクティングGlcNAcを保有するN結合型複合型オリゴ糖を増加させるために、抗体生成細胞での糖タンパク質改変グリコシルトランスフェラーゼ発現の好ましい範囲が存在するという発見に一部基づく。
【0019】
そのように、本発明は、一般に、治療特性を改変し、そして改善するようにタンパク質をグリコシル化操作するための方法に関する。より詳細には、本発明は、改変されたグリコシル化パターンを有し、その結果、増強された抗体依存性細胞傷害性(ADCC)を生じる抗体を宿主細胞において生成するための方法を記載する。方法の実施のために、本発明は、抗体をコードする核酸および糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸を有する宿主細胞を提供する。さらに、本発明は、改変されたグリコシル化パターンを有し、その結果、増強された抗体依存性細胞傷害性を生じる所望の抗体の発現を可能にする条件下でこのような宿主細胞を培養する方法およびプロトコルを提供する。さらに、増強された抗体依存性細胞傷害性を有する、そのように生成された抗体を単離する方法を記載する。
【0020】
本発明のさらに特定の実施態様において、2つのモノクローナル抗体(すなわち、抗神経芽腫抗体chCE7および抗CD20抗体C2B8)はモデル治療糖タンパク質として使用され、そして標的糖形態は、特定のクラスの糖質(すなわち、バイセクティングN−アセチルグルコサミン(GlcNAc)で改変されたN結合型複合型バイアンテナリーオリゴ糖)を有する糖形態である。本発明によって提供されるモデル系においては、CHO細胞が宿主細胞として使用されるが、多くの他の細胞系が宿主細胞系として企図され得る。種々の型のN結合型オリゴ糖にバイセクティングGlcNAcを付加するグリコシルトランスフェラーゼであるGlcNAcトランスフェラーゼIII(GnT III)は、通常は、CHO細胞によって生成されない。StanleyおよびCampell、1984、J.Biol.Chem.261:13370−13378。
【0021】
GnT III過剰発現の効果を実験的に調査するために、ラットGnT III cDNAのテトラサイクリン調節性過剰発現を有するCHO細胞株が樹立された。この実験系を使用して、本発明者らは、高レベルへのGnT IIIの過剰発現が増殖阻害を導き、そして細胞に対して毒性であることを発見した。GnT V(これは、異なるグリコシルトランスフェラーゼである)のテトラサイクリン調節性過剰発現を有する別のCHO細胞株は、同じ阻害効果を示した。このことは、糖タンパク質改変グリコシルトランスフェラーゼの過剰発現の一般的特徴であり得ることを示す。細胞増殖に対する酵素発現の効果は、糖タンパク質改変グリコシルトランスフェラーゼ過剰発現のレベルに上限を設定し、従って、接近しにくいグリコシル化部位が、非調節性発現ベクターを使用するグリコシル化経路およびグリコシル化パターンの操作によって改変され得る程度もまた制限し得る。
【0022】
基本レベルと毒性レベルとの間の範囲でGnT III発現を制御することによって糖形態分布が異なるchCE7 mAbおよびC2B8サンプルのセットの生成が開示される。chCE7 mAbサンプルのADCC活性の測定によって、インビトロでの最大chCE7生物学的活性についてのGnT III発現の最適範囲が示された。この活性は、Fcと関連する複合型バイセクト型オリゴ糖のレベルと相関した。実際の範囲内(すなわち、有意な増殖阻害も毒性も見られない)でのGnT IIIの発現は、chCE7サンプルのこのセットについて標的の複合型バイセクト型構造の増加を導いた。高レベルのGnT IIIで産生されたchCE7サンプルのMALDI/TOF質量分析におけるオリゴ糖ピークのパターンは、潜在的なGnT III基質の有意な割合が混成型バイセクト型オリゴ糖副生成物に転換されることを示す。従って、経路のさらなる操作による、これらの副生成物の最小化は、有益であり得る。
【0023】
(B.グリコシル化パターンの改変を指向するタンパク質をコードする核酸の同定および生成)
本発明は、グリコシル化パターンの変化が指向される、目的の任意のタンパク質、タンパク質フラグメントまたはペプチドの変化した糖形態の生成に適切な宿主細胞系を、提供する。そのような目的のタンパク質、タンパク質フラグメントまたはペプチドをコードする核酸は、一般に当該分野において公知の方法によって得られ得る。例えば、核酸は、cDNAライブラリーまたはゲノムライブラリーから単離され得る。使用され得るクローニング戦略のための総説について、例えば、Maniatis、1989、Molecular Cloning、A Laboratory Manual、Cold Springs Harbor Press、N.Y.;およびAusubelら、1989、Current Protocols in Molecular Biology、(Green Publishing Associates and Wiley Interscience、N.Y.)を参照のこと。
【0024】
本発明の別の実施態様において、目的のタンパク質、タンパク質フラグメントまたはペプチドのコード配列は、当該分野において周知の化学的方法を使用して、全体的にか、または部分的に、合成され得る。例えば、Caruthersら、1980、Nuc.Acids Res.Symp.Ser.7:215〜233;CreaおよびHorn、1980、Nuc.Acids Res.USA 9:2331;MatteucciおよびCaruthers、1980、Tetrahedron Letters 21:719;ChowおよびKempe、1981、Nuc.Acids Res.9:2807〜2817を参照のこと。あるいは、そのタンパク質自体が、そのアミノ酸配列の全体または部分を合成する化学的方法を使用して生成され得る。例えば、ペプチドは、固相技術によって合成され、樹脂から切断され、そして調製用高速液体クロマトグラフィーによって精製され得る。例えば、Creighton、1983、Protein Structures And Molecular Principles、W.H.Freeman and Co.、N.Y.,50〜60頁を参照のこと。合成ペプチドの組成は、アミノ酸分析またはアミノ酸配列決定(例えば、エドマン分解手順;Creighton、1983、Proteins、Structures and Molecular
Principles、W.H.Freeman and Co.、N.Y.34〜49頁を参照のこと)によって確認され得る。
【0025】
好ましい実施態様において、本発明は、抗体または抗体フラグメントあるいは増強した抗体依存性細胞傷害性を有する抗体フラグメントを含有する融合タンパク質の糖形態の産生のための宿主細胞系の生成および使用のための方法を提供する。標的エピトープの同定、およびグリコシル化パターンの改変が所望される、可能性のある治療的価値を有する抗体の生成、ならびにその対応するコード核酸配列の単離は、本発明の範囲内である。
【0026】
当該分野において公知の種々の手順が、目的の標的エピトープに対する抗体の産生のために使用され得る。そのような抗体としては、ポリクローナル抗体、モノクローナル抗体、キメラ抗体、単鎖抗体、Fabフラグメント、およびFab発現ライブラリーによって産生されたフラグメントが挙げられるが、これらに限定されない。そのような抗体は、例えば、診断剤および治療剤として有用であり得る。治療剤として、中和抗体(すなわち、リガンド、基質またはアダプター分子との結合に競合する抗体)が、特に好ましく興味深い。
【0027】
抗体の産生のために、種々の宿主動物(ウサギ、マウス、ラットなどを含むがこれらに限定されない)が、目的の標的タンパク質を用いて、注射によって免疫される。種々のアジュバント(宿主の種に依存して、フロイント(完全および不完全)、水酸化アルミニウムのようなミネラルゲル、リゾレシチンのような界面活性剤、プルロニックポリオール(pluronic polyols)、ポリアニオン、ペプチド、懸濁オイル、キーホールリンペットヘモシアニン、ジニトロフェノール、およびBCG(bacille Calmette−Guerin)およびCorynebacterium parvumのような可能性のある有用なヒトアジュバントが挙げられるが、これらに限定されない)を使用して、免疫応答を増加し得る。
【0028】
目的の標的に対するモノクローナル抗体が、培養物中の連続的細胞株による抗体分子産生のために提供される任意の技術を使用して、調製され得る。この技術には、KohlerおよびMilstein(1975、Nature 256:495〜497)によって最初に記載されたハイブリドーマ技術、ヒトB細胞ハイブリドーマ技術(Kosborら、1983、Immunology Today、4:72;Coteら、1983、Proc.Natl.Acad.Sci.USA 80:2026〜2030)およびEBVハイブリドーマ技術(Coleら、1985、Monoclonal Antibodies and Cancer Therapy、Alan R.Liss、Inc.77〜96頁)が挙げられるが、これらに限定されない。さらに、適切な抗原特異性のマウス抗体分子由来の遺伝子を、適切な生物学的活性のヒト抗体分子由来の遺伝子とともに、スプライシングすることによる、「キメラ抗体」の産生のために開発された技術(Morrisonら、1984、Proc.Natl.Acad.Sci.USA 81:6851〜6855;Neubergerら、1984、Nature 312:604〜608;Takedaら、1985、Nature 314:452〜454)が、使用され得る。あるいは、単鎖抗体の産生について記載される技術(米国特許第4,946,778号)が、所望の特異性を有する単鎖抗体の産生に適用され得る。
【0029】
目的の標的タンパク質の特異的結合部位を含有する抗体フラグメントが、公知の技術によって生成され得る。例えば、そのようなフラグメントとしては、抗体分子のペプシン消化によって生成され得るF(ab’)2フラグメント、およびF(ab’)2フラグメントのジスルフィド架橋の還元によって生成され得るFabフラグメントが挙げられるが、これらに限定されない。あるいは、Fab発現ライブラリーが、構築され得(Huseら、1989、Science 246:1275〜1281)、目的の標的タンパク質に対する所望の特異性を有するモノクローナルFabフラグメントの迅速かつ容易な同定を可能にする。
【0030】
一旦、グリコシル化パターンにおける改変が所望される抗体または抗体フラグメントが同定されると、コード核酸配列が、当該分野において周知の技術を用いて、同定および単離される。上記を参照のこと。
【0031】
(C.変化したグリコシル化パターンを有するタンパク質の産生のための細胞株の生成)
本発明は、改変されたグリコシル化パターンを有するタンパク質の生成のための宿主細胞発現系を提供する。具体的には、本発明は、改善された治療的価値を有するタンパク質の糖形態を生成するための宿主細胞系を提供する。従って、本発明は、糖タンパク質改変グリコシルトランスフェラーゼの発現を増加するように選択されるか、または操作された宿主細胞発現系を提供する。具体的には、そのような宿主細胞発現系を操作して、構成性プロモーター系または調節性プロモーター系と作動可能に連結された、糖タンパク質改変グリコシルトランスフェラーゼをコードする組換え核酸分子を含有し得る。あるいは、糖タンパク質改変グリコシルトランスフェラーゼを、天然に産生する、産生するように誘導される、および/または産生するように選択される、宿主発現系が、使用され得る。
【0032】
1つの特定の実施態様において、本発明は、糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸を発現するように操作された宿主細胞を提供する。1つの局面において、宿主細胞は、糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子を含有する核酸分子で、形質転換されるか、またはトランスフェクションされる。代替的な局面において、この宿主細胞は、内因性の糖タンパク質改変グリコシルトランスフェラーゼが活性化されるように、操作および/または選択される。例えば、宿主細胞は、内因性の糖タンパク質改変グリコシルトランスフェラーゼの発現を誘発するような変異を含有するように選択され得る。この局面は、宿主細胞がCHO lec10変異株である1つの特定の実施態様において例示される。あるいは、宿主細胞は、内因性の糖タンパク質改変グリコシルトランスフェラーゼが活性化されるように操作され得る。再度別の代替物において、宿主細胞は、内因性の糖タンパク質改変グリコシルトランスフェラーゼが宿主細胞染色体中に調節性プロモーターエレメントを挿入することによって活性化されるように操作される。さらなる代替物において、宿主細胞は、内因性の糖タンパク質改変グリコシルトランスフェラーゼが宿主細胞染色体中に構成性プロモーターエレメント、トランスポゾン、またはレトロウイルスエレメントを挿入することによって活性化されるように操作される。
【0033】
一般に、任意のタイプの培養細胞株を、本発明の宿主細胞株を操作するバックグラウンドとして使用し得る。好ましい実施態様において、CHO細胞、BHK細胞、NS0細胞、SP2/0細胞またはハイブリドーマ細胞株を、本発明の操作された宿主細胞を生成するための、バックグラウンド細胞株として使用する。
【0034】
本発明は、本明細書において規定される糖タンパク質改変グリコシルトランスフェラーゼの任意のタイプを発現する操作された宿主細胞を包含することが意図される。しかし、好ましい実施態様において、本発明の宿主細胞によって発現される少なくとも1つの糖タンパク質改変グリコシルトランスフェラーゼは、GnT IIIであるか、あるいは、β(1,4)−N−アセチルグルコサミニルトランスフェラーゼV(β(1,4)−N−acetylglucosaminyltrasferase V)(GnT V)である。しかし、代表的にGnT IIIまたはGnT Vに加えて、β(1,4)−ガラクトシルトランスフェラーゼ(GalT)、およびマンノシダーゼII(ManII)を含む、糖タンパク質改変グリコシルトランスフェラーゼの別のタイプもまた、宿主系において発現され得る。本発明の1つの実施態様において、GnT IIIは、GalTと同時に発現される。本発明の別の実施態様において、GnT IIIは、ManIIと同時に発現される。本発明のさらなる実施態様において、GnT IIIは、GalTおよびManIIと同時に発現される。しかし、糖タンパク質改変グリコシルトランスフェラーゼの任意の他の変更は、本発明の範囲内である。さらに、宿主細胞系内でのグリコシダーゼの発現が所望され得る。
【0035】
糖タンパク質改変グリコシルトランスフェラーゼをコードする核酸の1つまたはいくつかは、構成性プロモーターの制御下、あるいは調節性発現系の制御下において発現され得る。適切な調節性発現系としては、テトラサイクリン−調節性発現系、エクジソン誘導性発現系、lacスイッチ発現系、グルココルチコイド誘導性発現系、温度誘導性プロモーター系、およびメタロチオネイン金属誘導性発現系が挙げられるが、これらに限定されない。糖タンパク質改変グリコシルトランスフェラーゼをコードするいくつかの異なる核酸が、宿主細胞系内に含有される場合、それらのいくつかは、構成性プロモーターの制御下において発現され得るが、一方、その他は、調節性プロモーターの制御下で発現される。最適な発現レベルは、目的の各タンパク質について異なり、そして慣用的な実験を使用して決定される。発現レベルは、当該分野において一般に公知の方法(グリコシルトランスフェラーゼ特異的抗体を使用するウエスタンブロット分析、グリコシルトランスフェラーゼ特異的核酸プローブを使用するノザンブロット分析、または酵素活性の測定を含む)によって決定される。あるいは、グリコシルトランスフェラーゼの生合成産物と結合するレクチン(例えば、E4−PHAレクチン)を使用し得る。さらなる代替物において、核酸
は、レポーター遺伝子と作動可能に連結され得る;糖タンパク質改変グリコシルトランスフェラーゼの発現レベルは、レポーター遺伝子の発現レベルと相関するシグナルを測定することによって決定される。このレポーター遺伝子は、この糖タンパク質改変グリコシルトランスフェラーゼをコードする核酸とともに、単一のmRNA分子として転写され得る;その対応するコード配列は、内部リボゾーム進入部位(IRES)またはキャップ非依存性翻訳エンハンサー(CITE)のいずれかと連結され得る。このレポーター遺伝子は、この糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸とともに翻訳され得、その結果単一のポリペプチド鎖が形成される。糖タンパク質改変グリコシルトランスフェラーゼをコードする核酸は、単一のプロモーターの制御下のレポーター遺伝子に作動可能に連結され得、その結果糖タンパク質改変グリコシルトランスフェラーゼおよびレポーター遺伝子をコードする核酸は、2つの別のメッセンジャーRNA(mRNA)分子に選択的スプライシングされるRNA分子に転写される;生じるRNAの一方は、そのレポータータンパク質に翻訳され、そして他方は糖タンパク質改変グリコシルトランスフェラーゼに翻訳される。
【0036】
糖タンパク質改変グリコシルトランスフェラーゼをコードするいくつかの異なる核酸が発現する場合、これらの核酸は、1つかまたはいくつかのmRNA分子として転写されるように配列され得る。これらの核酸が、単一のmRNA分子として転写される場合、その対応するコード配列は、内部リボゾーム進入部位(IRES)またはキャップ非依存性翻訳エンハンサー(CITE)のいずれかと連結され得る。これらの核酸は、単一のプロモーターから1つのRNA分子に転写され、いくつかの別々のメッセンジャーRNA(mRNA)分子に選択的スプライシングされ得る。次いで、これらmRNAの各々は、対応するコードされた糖タンパク質改変グリコシルトランスフェラーゼに翻訳される。
【0037】
他の実施態様において、本発明は、治療的タンパク質(例えば、増強された抗体依存性細胞傷害性を有する抗体、およびFc媒介細胞傷害性を促進する表面にIgG Fc領域を提示する細胞)の生成のための宿主細胞発現系を提供する。一般に、宿主細胞発現系は、操作および/または選択され、糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸ともに、改変された糖形態の生成が所望されるタンパク質をコードする核酸を発現する。1つの実施態様において、宿主細胞系は、糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの遺伝子でトランスフェクトされる。代表的には、トランスフェクトされた細胞を選択して、糖タンパク質改変グリコシルトランスフェラーゼを安定に発現するクローンを同定および単離する。別の実施態様において、この宿主細胞は、内因性グリコシルトランスフェラーゼの発現について選択された。例えば、そうでなければサイレントな糖タンパク質改変グリコシルトランスフェラーゼの発現を誘発する変異を有する細胞が、選択され得る。例えば、CHO細胞は、特定の変異体において(例えば、変異体Lec10において)活性な、サイレントなGnT III遺伝子を保有することが公知である。さらに、サイレントな糖タンパク質改変グリコシルトランスフェラーゼ遺伝子を活性化するために、当該分野において公知の方法(調節性プロモーターまたは構成性プロモーターの挿入、あるいはトランスポゾン、レトロウイルスエレメントの使用などを含む)が使用され得る。遺伝子ノックアウト技術の使用、またはリボザイム方法の使用を用いて、宿主細胞のグリコシルトランスフェラーゼおよび/またはグリコシダーゼ発現レベルを適応させ得る。この使用も本発明の範囲内である。
【0038】
培養細胞株の任意のタイプが、本発明の宿主細胞株を操作するためのバックグラウンドとして使用され得る。好ましい実施態様において、CHO細胞、BHK細胞、NS0細胞、SP2/0細胞である。代表的には、そのような細胞株は操作され、抗体分子全体、抗体フラグメント、または免疫グロブリンのFc領域と等価の領域を含む融合タンパク質をコードする少なくとも1つのトランスフェクトされた核酸をさらに含む。別の実施態様において、目的の特定の抗体を発現するハイブリドーマ細胞株を、本発明の操作された宿主細胞を生成するためのバックグラウンド細胞株として使用する。
【0039】
代表的には、宿主細胞系内の少なくとも1つの核酸は、GnT III、あるいはGnT Vをコードする。しかし、他のタイプの糖タンパク質改変グリコシルトランスフェラーゼ(代表的にはGnT IIIまたはGnT Vに加え、GalTおよびManIIを含む)が、宿主系内で発現され得る。本発明の1つの実施態様において、GnT IIIは、GalTと同時に発現される。本発明の別の実施態様において、GnT IIIは、ManIIと同時に発現される。本発明のさらなる実施態様において、GnT IIIは、GalTおよびManIIと同時に発現される。しかし、糖タンパク質改変グリコシルトランスフェラーゼの任意の他の変更は、本発明の範囲内である。さらに、宿主細胞系内でのグリコシダーゼの発現が所望され得る。
【0040】
糖タンパク質改変グリコシルトランスフェラーゼをコードする核酸の1つまたはいくつかは、構成性プロモーターの制御下、あるいは調節性発現系の制御下において発現され得る。適切な調節性発現系としては、テトラサイクリン−調節性発現系、エクジソン誘導性発現系、lacスイッチ発現系、グルココルチコイド誘導性発現系、温度誘導性プロモーター系、およびメタロチオネイン金属誘導性発現系が挙げられるがこれらに限定されない。糖タンパク質改変グリコシルトランスフェラーゼをコードするいくつかの異なる核酸が、宿主細胞系内に含有される場合、それらのいくつかは、構成性プロモーターの制御下において発現され得るが、一方、その他は、調節性プロモーターの制御下で発現される。最適な発現レベルは、目的の各タンパク質について異なり、そして慣用的な実験を使用して決定される。発現レベルは、当該分野において一般に公知の方法(グリコシルトランスフェラーゼ特異的抗体を使用するウエスタンブロット分析、グリコシルトランスフェラーゼ特異的核酸プローブを使用するノザンブロット分析、または酵素活性の測定を含む)によって決定される。あるいは、グリコシルトランスフェラーゼの生合成産物と結合するレクチン(例えば、E4−PHAレクチン)を使用し得る。さらなる代替物において、核酸は
、レポーター遺伝子と作動可能に連結され得る;糖タンパク質改変グリコシルトランスフェラーゼの発現レベルは、レポーター遺伝子の発現レベルと相関するシグナルを測定することによって決定される。このレポーター遺伝子は、この糖タンパク質改変グリコシルトランスフェラーゼをコードする核酸とともに、単一のmRNA分子として転写され得る;その対応するコード配列は、内部リボゾーム進入部位(IRES)またはキャップ非依存性翻訳エンハンサー(CITE)のいずれかと連結され得る。このレポーター遺伝子は、この糖タンパク質改変グリコシルトランスフェラーゼをコードする少なくとも1つの核酸とともに翻訳され得、その結果単一のポリペプチド鎖が形成される。糖タンパク質改変グリコシルトランスフェラーゼをコードする核酸は、単一のプロモーターの制御下のレポーター遺伝子に作動可能に連結され得、その結果糖タンパク質改変グリコシルトランスフェラーゼおよびレポーター遺伝子をコードする核酸は、2つの別々のメッセンジャーRNA(mRNA)分子に選択的スプライシングされるRNA分子に転写される;生じるRNAの1つは、そのレポータータンパク質に翻訳され、そして他方は糖タンパク質改変グリコシルトランスフェラーゼに翻訳される。
【0041】
糖タンパク質改変グリコシルトランスフェラーゼをコードするいくつかの異なる核酸が発現する場合、これらの核酸は、1つかまたはいくつかのmRNA分子として転写されるように配列され得る。これらの核酸が、単一のmRNA分子として転写される場合、その対応するコード配列は、内部リボゾーム進入部位(IRES)またはキャップ非依存性翻訳エンハンサー(CITE)のいずれかと連結され得る。これらの核酸は、単一のプロモーターから1つのRNA分子に転写され、いくつかの別々のメッセンジャーRNA(mRNA)分子に選択的スプライシングされ得る。次いで、これらmRNAの各々は、対応するコードされた糖タンパク質改変グリコシルトランスフェラーゼに翻訳される。
【0042】
(1.発現系)
当業者に周知の方法を使用して、目的のタンパク質のコード配列および糖タンパク質改変グリコシルトランスフェラーゼのコード配列、および適切な転写/翻訳制御シグナルを含有する発現ベクターを構築し得る。これらの方法としては、インビトロ組換えDNA技術、合成技術およびインビボ組換え/遺伝子組換えが挙げられる。例えば、Maniatisら、1989、Molecular Cloning A Laboratory Manual、Cold Spring Harbor Laboratory、N.Y.およびAusubelら、1989、Current Protocols in Molecular Biology、Greene Publishing Associates and Wiley Interscience、N.Y.に記載される技術を参照のこと。
【0043】
種々の宿主−発現ベクター系を使用して、目的のタンパク質のコード配列、および糖タンパク質改変グリコシルトランスフェラーゼのコード配列を発現し得る。好ましくは、哺乳動物細胞を、目的のタンパク質のコード配列、および糖タンパク質改変グリコシルトランスフェラーゼのコード配列を含有する、組換えプラスミドDNAまたはコスミドDNA発現ベクターでトランスフェクトされた宿主細胞系として使用する。最も好ましくは、CHO細胞、BHK細胞、NS0細胞、またはSP2/0細胞あるいはハイブリドーマ細胞株を、宿主細胞系として使用する。代替的な実施態様において、他の真核生物宿主細胞系(目的のタンパク質のコード配列、および糖タンパク質改変グリコシルトランスフェラーゼのコード配列を含む組換え酵母発現ベクターで形質転換された酵母細胞;目的のタンパク質のコード配列、および糖タンパク質改変グリコシルトランスフェラーゼのコード配列を含む組換えウイルス発現ベクター(例えば、バキュロウイルス)で感染された昆虫細胞系;目的のタンパク質のコード配列、および糖タンパク質改変グリコシルトランスフェラーゼのコード配列を含む、組換えウイルス発現ベクター(例えば、カリフラワーモザイクウイルス、CaMV;タバコモザイクウイルス、TMV)で感染されたか、または組換えプラスミド発現ベクター(例えば、Tiプラスミド)で形質転換された植物細胞系;あるいは目的のタンパク質をコードするDNA、および糖タンパク質改変グリコシルトランスフェラーゼのコード配列の複数のコピー(安定に増幅された(CHO/dhfr)か、もしくは二重微小染色体中で不安定に増幅された(例えば、マウス細胞株))を含有するように操作された細胞株を含む、組換えウイルス発現ベクター(例えば、アデノウイルス、ワクシニアウイルス)で感染された動物細胞系を含む)が意図され得る。
【0044】
本発明の方法のために、安定な発現が、一般には一過性の発現よりも好ましい。なぜなら、安定な発現は、代表的により再現性のある結果を達成し、そしてまた、大スケール産生により適合しやすいからである。ウイルスの複製起点を含有する発現ベクターを使用するよりむしろ、宿主細胞を、適切な発現制御エレメント(例えば、プロモーター、エンハンサー配列、転写ターミネーター、ポリアデニル化部位など)によって制御されるそれぞれのコード核酸、および選択マーカーで形質転換し得る。外来DNAの導入後、操作された細胞を、富化された培地中で1〜2日間増殖させ得、次に、選択培地に変更する。組換えプラスミド中の選択マーカーは、選択に対する耐性を付与し、そしてプラスミドをその染色体中に安定に組み込み、そして増殖してコロニーを形成し、その結果クローン化され、細胞株に拡大される細胞の選択を可能にする。
【0045】
それぞれ、tk-、hgprt-、またはaprt-細胞において使用され得る、単純ヘ
ルペスウイルスチミジンキナーゼ(Wiglerら、1977、Cell 11:223)、ヒポキサンチン−グアニンホスホリボシルトランスフェラーゼ(SzybalskaおよびSzybalski、1962、Proc.Natl.Acad.Sci.USA
48:2026)、およびアデニンホスホリボシルトランスフェラーゼ(Lowyら、1980、Cell 22:817)遺伝子を含むが、これらに限定されない多数の選択系が使用され得る。抗代謝産物耐性が、メトトレキサートに対する耐性を付与するdhfr(Wiglerら、1980、Proc.Natl.Acad.Sci.USA 77:3567;O’Hareら、1981、Proc.Natl.Acad.Sci.USA 78:1527)、ミコフェノール酸に対する耐性を付与するgpt(MulliganおよびBerg、1981、Proc.Natl.Acad.Sci.USA 78:2072);アミノグリコシドG−418に対する耐性を付与するneo(Colberre−Garapinら、1981、J.Mol.Biol.150:1);およびハイグロマイシンに対する耐性を付与するhygro(Santerreら、1984、Gene 30:147)遺伝子についての選択の基礎として使用され得る。近年、細胞にトリプトファンの代わりにインドールを利用させ得るtrpB;細胞にヒスチジンの代わりにヒスチノールを利用させ得るhisD(HartmenおよびMulligan、1988、Proc.Natl.Acad.Sci.USA 85:8047);グルタミンシンターゼ系;およびオルニチンデカルボキシラーゼインヒビターである2−(ジフルオロメチル)−DL−オルニチン、DFMOに対する耐性を付与するODC(オルニチンデカルボキシラーゼ)(McConlogue、1987、Current Communications in Molecular Biology、Cold Spring Harbor Laboratory編)と称するさらなる選択遺伝子が記載されている。
【0046】
(2.改変グリコシル化パターンを有するタンパク質を発現するトランスフェクト体および形質転換体の同定)
コード配列を含む宿主細胞および生物学的に活性な遺伝子産物を発現する宿主細胞は、少なくとも4つの一般的なアプローチにより同定され得る;(a)DNA−DNAハイブリダイゼーションまたはDNA−RNAハイブリダイゼーション;(b)「マーカー」遺伝子機能の存在または非存在;(c)宿主細胞におけるそれぞれのmRNA転写物の発現により測定されるような、転写レベルの評価;および(d)イムノアッセイまたはその生物学的活性により測定されるような、遺伝子産物の検出。
【0047】
第1のアプローチにおいて、発現ベクター中に挿入された目的のタンパク質のコード配列および糖タンパク質改変グリコシルトランスフェラーゼのコード配列の存在は、それぞれのコード配列、またはそれらのそれぞれの部分もしくは誘導体に相同であるヌクレオチド配列を含むプローブを用いて、DNA−DNAハイブリダイゼーションまたはDNA−RNAハイブリダイゼーションにより検出され得る。
【0048】
第2のアプローチにおいて、組換え発現ベクター/宿主系は同定され、そして特定の「マーカー」遺伝子機能(例えば、チミジンキナーゼ活性、抗生物質に対する耐性、メトトレキサートに対する耐性、形質転換表現型、バキュロウイルス中の封入体形成など)の存在または非存在に基づき、選択され得る。例えば、目的のタンパク質のコード配列および糖タンパク質改変グリコシルトランスフェラーゼのコード配列が、ベクターのマーカー遺伝子配列内に挿入されている場合、それぞれのコード配列を含む組換え体は、マーカー遺伝子機能の非存在により同定され得る。あるいは、マーカー遺伝子は、コード配列の発現を制御するために使用される同じプロモーターまたは異なるプロモーターの制御下で、コード配列と縦列して配置され得る。誘導または選択に応じたマーカーの発現は、目的のタンパク質のコード配列および糖タンパク質改変グリコシルトランスフェラーゼのコード配列の発現を示す。
【0049】
第3のアプローチにおいて、目的のタンパク質のコード領域および糖タンパク質改変グリコシルトランスフェラーゼのコード配列の転写活性は、ハイブリダイゼーションアッセイにより評価され得る。例えば、RNAは、単離されそして目的のタンパク質のコード配列および糖タンパク質改変グリコシルトランスフェラーゼのコード配列またはそれらの特定部分に相同なプローブを用いたノーザンブロットにより分析され得る。あるいは、宿主細胞の全核酸が抽出され、そしてこのようなプローブへのハイブリダイゼーションについて評価され得る。
【0050】
第4のアプローチにおいて、目的のタンパク質のタンパク質産物および糖タンパク質改変グリコシルトランスフェラーゼのコード配列の発現は、免疫学的に、例えば、ウェスタンブロット、イムノアッセイ(例えば、ラジオイムノ沈降、酵素結合イムノアッセイなど)により評価され得る。しかし、発現系の最終的な試験の成功は、生物学的に活性化な遺伝子産物の検出を含む。
【0051】
(D.変化したグリコシル化パターンを有するタンパク質およびタンパク質フラグメントの生成および使用)
(1.増強した抗体依存性細胞傷害性を有する抗体の生成および使用)
好ましい実施態様において、本発明は、増強した抗体依存性細胞傷害性を有する抗体および抗体フラグメントの糖形態を提供する。
【0052】
いくつかの型の癌の処置のための非結合体化モノクローナル抗体(mAb)の臨床的試行は、近年進んだ結果を生じた。Dillman、1997、Cancer Biother.&Radiopharm.12:223−225;Deoら、1997、Immunology Today 18:127。キメラの非結合体化IgG1が、低級なまたは小胞性のB細胞非ホジキンリンパ腫について認可されている(Dillman、1997、前出)が、一方別の非結合体化mAbである、ヒト化IgG1標的固形乳腫瘍もまた、第III相臨床試験において有望な結果を示しつつあった。Deoら、1997、前出。これらの2つのmAbの抗原は、それらの各々の腫瘍細胞において高度に発現され、そしてそれらの抗体は、インビトロおよびインビボでエフェクター細胞により強力な腫瘍破壊を媒介する。対照的に、良好な腫瘍特異性を有する多くの他の非結合体化mAbは、臨床的に有用である程十分な効力のエフェクター機能を引き起こし得ない。Frostら、1997、Cancer 80:317−333;Surfusら、1996、J.Immunother.19:184−191。これらのより弱いmAbのいくつかについて、補助的なサイトカイン治療が、現在も試験されている。サイトカインの添加は、その活性および循環リンパ球の数を増加することによって抗体依存性細胞傷害性(ADCC)を刺激し得る。Frostら、1997、前出;Surfusら、1996、前出。溶解物が抗体標的化細胞に対して攻撃するADCCは、リンパ球レセプターの、抗体の定常境域(Fc)への結合の際、引き起こされる。Deoら、1997、前出。
【0053】
非結合体化IgG1のADCC活性を増大させる、異なるが相補的であるアプローチは、抗体のFc領域を、リンパ球レセプターに対する親和性(FcγR)を増大させるように操作することである。タンパク質工学研究は、FcγRが、IgG CH2ドメインのより低いヒンジ領域と相互作用することを示した。Lundら、1996、J.Immunol.157:4963−4969。しかし、FcγR結合はまた、CH2領域における保存されたAsn297で共有結合的に付着したオリゴ糖の存在を必要とする。Lundら、1996、前出;WrightおよびMorrison、1997、Tibtech 15:26−31は、オリゴ糖およびポリペプチドの両方が直接相互作用部位に寄与するか、またはオリゴ糖が活性なCH2ポリペプチドコンフォメーションを維持するために必要とされるかのいずれかであることを示唆している。従って、オリゴ糖の構造の改変は、相互作用の親和性を増大させるための手段として、探索され得る。
【0054】
IgG分子は、そのFc領域中に2つのN結合オリゴ糖を保有し、一つは、各重鎖にある。任意の糖タンパク質と同様に、抗体は、糖形態の集団として産生される。この糖形態は、同じポリペプチド骨格を共有するが、グリコシル化部位に付着した異なるオリゴ糖を有する。血清IgGのFc領域中に通常見出されるオリゴ糖は、低いレベルの末端シアル酸およびバイセクティングN−アセチルグルコサミン(GlcNAc)、ならびに程度が可変し得る末端ガラクトシル化およびコアフルコシル化を有する、バイアンテナリー複合型オリゴ糖である(Wormaldら、1997、Biochemistry 36:130−1380)(図1)。いくつかの研究は、FcγR結合を必要とする最小の糖質構
造は、オリゴ糖コア内にあることを示唆する。Lundら、1996、前出。末端ガラクトースの除去は、ADCC活性において約2倍の減少をもたらす。このことは、FcγRレセプター結合におけるこれらの残基についての役割を示す。Lundら、1996、前出。
【0055】
非結合体化治療用mAbの生成のために産業および教育において使用される マウスまたはハムスター誘導細胞株は、通常、必要とされるオリゴ糖決定因子をFc部位に付着させる。しかし、これらの細胞株において発現されるIgGは、血清IgGにおいて低量で見出されるバイセクティングGlcNAcを欠く。Lifelyら、1995、Glycobiology 318:813−822。対照的には、ラット骨髄腫産生ヒト化IgG1(CAMPATH−1H)が、その糖形態のいくつかにおいてバイセクティングGlcNAcを保有することが、最近観察された。Lifelyら、1995、前出。ラット細胞誘導化抗体は、標準的な細胞株において産生されるCAMPATH−1H抗体と類似したインビトロのADCC活性に、しかし有意により低い抗体濃度で、達した。
【0056】
CAMPATH抗原は、通常、リンパ腫細胞に高いレベルで存在し、そしてこのキメラmAbは、バイセクティングGlcNAcの非存在下で高いADCC活性を有する。Lifelyら、1995、前出。Lifelyら、1995、前出の研究において、たとえ最大のインビトロADCC活性が、グリコシル化パターンの変化により増大しなかったとしても、この活性レベルが、バイセクト型オリゴ糖を保有する抗体の比較的低い抗体濃度で得られたという事実は、バイセクト型オリゴ糖の重要な役割を示唆する。Fc領域においてバイセクト型オリゴ糖を保有するこれらの抗体の糖形態を生成することによって、低い基底活性レベルを有するIgG1のADCC活性を増大するようなアプローチが発達した。
【0057】
N結合グリコシル化経路において、バイセクティングGlcNAcは、酵素β(1,4)−N−アセチルグルコサミニルトランスフェラーゼIII(GnT III)により添加される。Schachter、1986、Biochem.Cell Biol.64:163−181。Lifelyら、1995、前出は、内因的な一定のレベルでGnT
IIIを発現するラット骨髄腫細胞株を含む、異なるが非操作のグリコシル化機構を有する異なる細胞株において抗体を産生することによって、同じ抗体の異なるグリコシル化パターンを得た。対照的に、本発明者らは、単一の抗体産生CHO細胞株を使用した。これは、外部調節した様式で、異なるレベルのクローン化GnT III遺伝子を発現するようにあらかじめ操作されていた。このアプローチにより、本発明者らは、GnT IIIの発現と改変した抗体のADCC活性との間の厳密な相関を初めて確立し得た。
【0058】
本明細書中に実証されるように(実施例4(後述)を参照のこと)、開示される方法に従って改変されたC2B8抗体は、同一の細胞培養および精製条件下で産生された、標準的な非改変C2B8抗体よりも約16倍高いADCC活性を有した。簡単に言うと、GnT III発現を有さないCHO−tTA−C2B8細胞において発現されたC2B8抗体サンプルは、ヒトリンパ球によりSB細胞(CD20+)のインビトロ溶解として測定された、約31%の細胞傷害性活性を示した(1μg/ml抗体濃度で)。対照的に、基底の主な抑制レベルでGnT IIIを発現するCHO細胞培養物由来のC2B8抗体は、1μg/ml抗体濃度で、同じ抗体濃度でのコントロールに対してADCC活性の33%増大を示した。さらに、GnT IIIの発現の増大は、同じ抗体濃度でのコントロールと比較して、最大のADCC活性においてほぼ80%の大きな増加を生じた(1μg/ml抗体濃度で)。実施例4(後述)を参照のこと。
【0059】
増強した抗体依存性細胞傷害性を有する本発明のさらなる抗体は、以下を含むが、これらに限定されない:本発明の方法により産生される抗ヒト神経芽腫モノクローナル抗体(chCE7)、本発明の方法により産生されるキメラ抗ヒト腎細胞癌腫モノクローナル抗体(ch−G250)、本発明の方法により産生されるヒト化抗HER2モノクローナル抗体、本発明の方法により産生されるキメラ抗ヒト結腸、肺および乳癌腫モノクローナル抗体(ING−1)、本発明の方法により産生されるヒト化抗ヒト17−1A抗原モノクローナル抗体(3622W94)、本発明の方法により産生されるヒト化抗ヒト結腸直腸腫瘍抗体(A33)、本発明の方法により産生されるGD3ガングリオシドに対する抗ヒト骨髄腫抗体(R24)、ならびに本発明の方法により産生されるキメラ抗ヒト扁平上皮細胞癌モノクローナル抗体(SF−25)。さらに、本発明は、抗体フラグメント、および免疫グロブリンのFc領域と等価である領域を含む融合タンパク質に対して指向される。後述を参照のこと。
【0060】
(2.Fc媒介細胞傷害性を促進する免疫グロブリンのFc領域と等価な領域を含む融合タンパク質の生成および使用)
上記で議論されるように、本発明は、治療用抗体のADCC活性を増強するための方法に関する。これは、このような抗体のFc領域のグリコシル化パターンを操作することによって、特に、それらのFc領域で変換されたグリコシル化部位にN結合した複合型バイセクト型オリゴ糖を保有する抗体分子の割合を最大にすることによって達成される。この戦略は、治療用抗体によるのみならず、免疫グロブリンのFc領域に等価な領域を保有する任意の分子により媒介される、望ましくない細胞に対するFc媒介性細胞細胞傷害を、増大させるために適用され得る。なぜならば、グリコシル化の操作により導入された変化は、Fc領域のみに影響を与え、従って、エフェクター細胞の表面上のFcレセプターとのその相互作用は、ADCC機構に関与したからである。現在開示される方法で適用され得るFc含有分子としては以下が挙げられるが、これらに限定されない:(a)Fc領域のN末端に融合された標的タンパク質ドメインで作製された可溶性融合タンパク質(ChamovおよびAshkenazi、1996、TIBTECH 14:52)、および(b)Fc領域のN末端に融合された原形質膜に局在するII型膜貫通ドメインで作製された原形質膜固定化融合タンパク質(Stabila,P.E.、1998、Nature Biotech.16:1357)。
【0061】
可溶性融合タンパク質(a)の場合、標的ドメインは、融合タンパク質の望ましくない細胞(例えば、癌細胞)への結合、すなわち治療用抗体と類似した様式で、指向させる。従って、これらの分子によって媒介されるFc媒介細胞傷害性活性を増強するための、近年開示された方法の適用は、治療用抗体に適用される方法と同一である。米国特許仮出願番号第60/082,581号(本明細書中で参考として援用される)の実施例2を参照のこと。
【0062】
膜固定化融合タンパク質(b)の場合、身体中の望ましくない細胞が、融合タンパク質をコードする遺伝子を発現する必要がある。これは、遺伝子治療アプローチ、すなわち、融合タンパク質コード遺伝子の発現を望ましくない細胞に指向させるプラスミドベクターもしくはウイルスベクターを用いて、インビボで細胞をトランスフェクトすることによるか、または細胞表面上に融合タンパク質を発現するように遺伝子的に操作された細胞の身体中への移植によるかのいずれかで達成され得る。後者の細胞は、通常、それらの細胞が、Fc媒介細胞傷害性機構によって破壊され得ないポリマーカプセルに内包されて身体内へ移植される(カプセル化細胞治療)。しかし、カプセル送達が失敗し、そして逸脱細胞が望ましくないようになれば、それらはFc媒介細胞傷害性により除去され得る。Stabilaら、1998、Nature、Biotech.16:1357。この場合、現在開示される方法は、遺伝子治療用ベクターに、十分なもしくは最適なGnT IIIの発現レベルを指向するさらなる遺伝子発現カセットを組込むことによってか、または十分なもしくは最適なレベルのGnT IIIを発現するように、移植されるべき細胞を操作することによってかのいずれかで適用され得る。両方の場合において、開示される方法の目的は、複合型バイセクト型オリゴ糖を保有する表面提示Fc領域の割合を増大または最大にすることである。
【0063】
以下の実施例は、本発明をより詳細に説明する。以下の調製物および実施例は、当業者が本発明をより明瞭に理解し、そして実施することを可能にするために、与えられる。しかし、本発明は、例示される実施態様および本発明の範囲内にある機能的に等価である方法によって範囲を限定されることなく、本発明の単なる単一の局面の例証として意図される。実際、本明細書中に記載されるものに加え、本発明の種々の改変は、前述の説明および添付している図面より当業者に明らかである。このような改変は、添付された特許請求の範囲の範囲内に含まれることが意図される。
【実施例】
【0064】
(VIII.実施例)
(A.実施例1:チャイニーズハムスター卵巣細胞におけるグリコシルトランスフェラーゼのテトラサイクリン調節過剰発現)
GnT IIIの発現が外部制御され得る細胞株を確立するために、テトラサイクリン調節発現系を使用した。Gossen,M.およびBujard,H.、1992、Proc.Nat.Acad.Sci.USA、89:5547−5551。これらの細胞中のGnT IIIの量は、培養培地中のテトラサイクリン濃度を単に操作することによって制御され得る。この系を用いて、高いレベルまでのGnT IIIの過剰発現が増殖阻害を導き、そして細胞に対して毒性であることを見出した。GnT V(異なる糖タンパク質改変グリコシルトランスフェラーゼ)のテトラサイクリン調節過剰発現を有する別のCHO細胞株は、同様の阻害効果を示した。このことは、これが、糖タンパク質改変グリコシルトランスフェラーゼ過剰発現の一般的特徴であり得ることを示す。この現象は、以前に報告されておらず、おそらく発明者は、一般的には、関連実験については構成的なプロモーターを使用したという事実に起因する。増殖効果は、糖タンパク質改変グリコシルトランスフェラーゼ過剰発現のレベルの上方制限を設定し、それによってまた貧弱な到達可能な糖タンパク質部位の改変の最大範囲を制限し得る。
【0065】
(1.材料および方法)
グリコシルトランスフェラーゼのテトラサイクリン調節発現を有するCHO細胞の樹立。第1の工程において、テトラサイクリン制御トランスアクチベーター(tTA)を、調節系に十分なレベルで、構成的に発現する中間のCHO細胞株(CHO−tTA)をまず生成した。リポフェクタミン試薬(Gibco,Eggenfelden,Germany)を使用して、CHO(DUKX)細胞を、tTA遺伝子の構成的発現のためのベクターであるpUHD15−1(GossenおよびBujard、1992、Proc.Nat.Acad.Sci.USA、89:5547−5551)およびネオマイシン耐性遺伝子の構成的発現のためのベクターであるpSV2Neo(Clontech、Palo Alto、CA)とともに同時トランスフェクトした。安定した薬物耐性クローンを選択し、そしてテトラサイクリン調節β−ガラクトシダーゼ発現ベクターであるpUHG16−3を用いた一過性トランスフェクションによる十分なレベルのtTA発現についてスクリーニングした。c−mycエピトープコードDNAを、PCR増幅によってラットGnT III cDNA(Nishikawaら、1992、J.Biol.Chem.267:18199−18204)の3’末端に付加した。Nilssonら、1993、J.Cell Biol.120:5−13。この産物を配列決定し、そしてテトラサイクリン調節発現のためのベクターであるpUHD10−3(GossenおよびBujard、前出)にサブクローニングし、ベクターpUHD10−3−GnT IIImを生成した。ヒトGnT V cDNA(Saitoら、1995、Eur.J.Biochem.233:18−26)を、pUHD10−3に直接サブクローニングし、プラスミドベクターpUHD10−3−GnT Vを生成した。CHO−tTA細胞を、リン酸カルシウムトランスフェクション法(JordanおよびWurm.1996、Nucleic Acids Res.24:596−601)を用いて、ピューロマイシン耐性の構成的発現のためのベクターであるpPur(Clontech、Palo Alto、CA)、およびベクターpUHD10−3−GnT IIImまたはベクターpUHD10−3−GnT Vのいずれかで、同時トランスフェクトした。ピューロマイシン耐性クローンをテトラサイクリンの存在下で選択し、単離し、次いでウェスタンブロット分析によりGnT IIIまたはGnT Vのテトラサイクリン調節発現について分析した。後述を参照のこと。
【0066】
ウェスタンブロッティングおよびレクチンブロッティング。GnT IIIまたはGnT Vのウェスタンブロット分析のために、細胞溶解物を、SDS−PAGEにより分離し、そしてPVDF膜(Millipore、Bedford、MA)に電気ブロットした。抗c−mycモノクローナル抗体9E10(Nilssonら、1993、J.Cell Biol.120:5−13)を用いてGnT IIIを検出し、そして抗GnT
Vウサギポリクローナル抗体(Chenら、1995、Glycoconjugate
J.12:813−823)を用いてGnT Vを検出した。二次抗体として、抗マウスまたは抗ウサギIgG西洋ワサビペルオキシダーゼ(Amersham、Arlington、IL)を使用した。結合した二次抗体を、増強化学発光キット(ECLキット、Amersham、Arlington、IL)を使用して検出した。
【0067】
GnT III触媒反応またはGnT V触媒反応のいずれかにより改変される糖タンパク質のレクチンブロット分析について、ビオチン化E−PHA(Oxford Glycosciences、Oxford、United Kingdom)またはL−PHA−ジゴキシゲニン(Boehringer Mannheim、Mannheim、Germany)をそれぞれ使用した。MerkleおよびCummings、1987、Methods Enzymol.138:232−259。
【0068】
(2.結果および考察)
グリコシルトランスフェラーゼのテトラサイクリン調節過剰発現を有するCHO細胞株の樹立。グリコシルトランスフェラーゼ過剰発現細胞株の樹立のために使用される戦略は、最初に、この系が働くのに十分なレベルで、テトラサイクリン制御トランスアクチベーター(tTA)を構成的に発現する中間体CHO細胞株をまず生成することからなった。Yinら、1996、Anal.Biochem.235:195−201。このレベルは、テトラサイクリン非存在下で、グリコシルトランスフェラーゼ遺伝子の上流の最小プロモータからの転写の高いレベルを活性化するのに十分高くなくてはいけなかった。CHO細胞を、tTAの構成的発現のためのベクター(これは、ヒトサイトメガロウイルス(hCMV)プロモーター/エンハンサーによって駆動される)、およびネオマイシン耐性(NeoR)遺伝子の発現のためのベクターとともに同時トランスフェクトした。過剰な
tTA発現ベクターを使用し、そしてネオマイシン耐性クローンを単離した。
【0069】
哺乳動物細胞において、同時トランスフェクトされたDNAは、染色体内のランダムな位置で隣接して組込まれ、そして発現は、組込み部位に大部分依存し、そしてインタクトな発現カセットのコピー数にもまた依存する。トランスフェクトされた遺伝子の異なる発現レベルを有するクローンの混合集団が生成される。Yinら、1996、前出。ネオマイシン耐性についての選択は、単に、インタクトなNeoR発現カセットの組込みについ
て選択し、一方過剰なtTA発現ベクターの使用は、tTAの良好な発現を有するクローンを見出す確率を増大させる。クローンの混合集団は、tTA発現についての機能的アッセイを用いてスクリーニングされなければならない。GossenおよびBujard、1992、前出;Yinら、1996、前出。これは、tetプロモーターの制御下でレポーター遺伝子lacZを保有する第2のベクターによる各クローンのトランスフェクション、およびβ−ガラクトシダーゼ活性の、テトラサイクリン調節(tet調節)一過的発現(すなわち、トランスフェクション後1〜3日後)についてのスクリーニングによりなされた。CHOt17(これは、20のスクリーニングされたクローンの間で最も高いレベルのtet調節β−ガラクトシダーゼ活性を示した)を、さらなる研究のために選択した。
【0070】
CHOt17細胞を、ベクターpUHDIO−3−GnT IIImで細胞をトランスフェクトし、そしてテトラサイクリンの存在下または非存在下での36時間、細胞をインキュベートした後のGnT IIIの相対レベルを比較することにより、Gnt IIIのtet調節発現について試験した。GnT IIIレベルを、GnT IIIのカルボキシ末端でc−mycペプチドエピトープタグを認識するモノクローナル抗体(9E10)を用いて、ウェスタンブロット分析により比較した。このタグを、PCR増幅を用いたグリコシルトランスフェラーゼ遺伝子の改変により導入した。種々の報告は、局在または活性を崩壊することなく、同じトポロジーを共有する酵素群であるグリコシルトランスフェラーゼのカルボキシ末端へペプチドエピトープの付加することを実証した。Nilssonら、1993、前出;Rabouilleら、1995、J.Cell Science 108:1617−1627。図2は、クローンCHOt17において、GnT III蓄積が、テトラサイクリンの存在下よりも非存在においての方が、有意に高いことを示す。β−ガラクトシダーゼ活性のアッセイにおいてより弱い転写活性化を与えるさらなるクローン、CHOt2を、並行して試験した(図2)。GnT IIIおよびβ−ガラクトシダーゼの発現レベルは、これらの両方のクローンについて同じパターンのテトラサイクリン調節を伴う。GnT III発現が量的に制御され得るテトラサイクリン濃度の範囲は、0〜100ng/mlと見出された(図3)。この結果は、異なる細胞株と遺伝子を用いた前述の研究に一致する(Yinら、1996、前出)。
【0071】
GnT IIIのtet調節発現を有する安定な細胞株を生成するために、CHOt17細胞を、ベクターpUHDIO−3−GnT IIImおよびプロマイシン耐性遺伝子の発現のためのベクターpPURで同時トランスフェクトした。並行して、CHOt17細胞を、pUHDIO−3−GnT VベクターおよびpPURベクターで同時トランスフェクトし、この他のグリコシルトランスフェラーゼについて類似の細胞株を生成した。高度に効率的なリン酸カルシウムトランスフェクション方法を使用して、そして真核生物発現カセットの外側の唯一の制限部位でDNAを線状化し、組込みの際にこれらを崩壊する確率を減少させた。発現したtTAのレベルが最初に十分であると証明されている宿主を用いることによって、テトラサイクリン非存在下でグリコシルトランスフェラーゼの高度な発現を有するクローンを見出す確率が増大する。
【0072】
安定な組込み体を、ピューロマイシン耐性により選択し、クローン選択を行った培地中にテトラサイクリンを保ち、基底レベルでのグリコシルトランスフェラーゼの発現を維持した。各グリコシルトランスフェラーゼについて、16のピューロマイシン耐性クローンをテトラサイクリンの存在および非存在下で増殖し、それぞれの8つをウェスタンブロット分析により分析した(図4)。大多数のクローンは、グリコシルトランスフェラーゼ発現の良好な調節を示した。GnT III発現クローンのうちの1つは、テトラサイクリンの存在下で比較的高い基底レベルを示した(図4B、クローン3)。このことは、発現カセットの組込みが内因性CHO細胞エンハンサーに近接することを示唆する;一方、2つのピューロマイシン耐性クローンは、テトラサイクリンの非存在下でGnT IIIの発現を示さなかった(図4B、クローン6および8)。発現の良好な調節を示すクローンの間で、異なる最大レベルのグリコシルトランスフェラーゼが観察された。これは、組込み部位または組込まれたコピー数におけるバリエーションに起因し得る。グリコシルトランスフェラーゼの活性を、テトラサイクリンの存在および非存在において増殖させた種々のクローン由来の内因性細胞糖タンパク質へのE−PHAおよびL−PHAレクチン結合により確認した(図5)。レクチンは、特異的なオリゴ糖構造に結合するタンパク質である。E−PHAレクチンは、GnT III触媒反応の産物であるバイセクト型オリゴ糖に結合し、そしてL−PHAは、GnT V触媒化反応により産生されるトリアンテナリー型オリゴ糖およびテトラアンテナリー型オリゴ糖に結合する(MerkleおよびCummings、1987、Methods Enzymol.138:232−259)。各グリコシルトランスフェラーゼについて、テトラサイクリンの非存在において高度に発現し、しかしテトラサイクリン存在下では検出不可能であるクローン(クローン6、図4A、CHO−tet−GnT Vおよびクローン4、図4B、CHO−tet−GnT
IIIm)を、さらなる研究のために選択した。
【0073】
(B.実施例2:グリコシルトランスフェラーゼ過剰発現によってもたらされる細胞増殖の阻害)
テトラサイクリンの非存在下でのGnT IIIを発現するクローンおよびGnT Vを発現するクローンのスクリーニングの間(上記の実施例1を参照のこと)、クローンの各セットのおよそ半分が増殖の強力な阻害を示した。増殖阻害の程度は、クローンにより変動し、そしてウェスタンブロット分析から見積もった発現レベルとの比較(図4)は、増殖阻害の程度と、グリコシルトランスフェラーゼ過剰発現との間の相関を示唆した。この相関は、最後のクローンCHO−tet−GnT IIImおよびCHO−tet−GnT Vを、種々の濃度のテトラサイクリンにおいて増殖させることによって強固に確立された。増殖の強力な阻害は、低レベルのテトラサイクリンでの培養の2日後に明確になった(図6)。増殖阻害した細胞は、接着性CHO細胞の代表的な細長い形状の代わりに小さく丸い形態を提示した。数日後、有意な細胞死が、増殖阻害された細胞の形態から明らかであった。
【0074】
グリコシルトランスフェラーゼ過剰発現に起因する増殖阻害は、これまで、文献には報告されていない。これはおそらく、構成性のプロモーターの広汎な使用に起因する。増殖阻害レベルでのグリコシルトランスフェラーゼの構成性発現を与えるこれらのクローンは、選択手順の間に消失する。これは、選択の間に、培地中のテトラサイクリン(すなわち、基底発現レベル)を維持することによってここで回避された。選択前に、構成性hCMVプロモーター/エンハンサーに基づく伝統的な哺乳動物ベクターを用いて増殖阻害レベルにまでグリコシルトランスフェラーゼを発現し得るクローンの頻度は、より低いと予測される。これは、任意の所定の遺伝子について、高度の構成性レベルのtTAについて選択されたCHO細胞株におけるpUHDIO−3ベクターが、他者らによって観察されるような、構成性のhCMVプロモーター/エンハンサーに基づくベクターよりも有意に高い発現レベルを与えるという事実に起因する。Yinら、1996、前出。
【0075】
細胞増殖の阻害は、それらのインビボでの触媒活性には非依存性の、膜結合型の、ゴルジ体内在性グリコシルトランスフェラーゼの過剰発現の直接の効果(例えば、小胞体(ER)における折り畳みの誤りを介し、ERにおけるタンパク質折り畳みを補助するエレメントの飽和を生じる)に起因し得る。これは、おそらく、他の必須の細胞タンパク質の折り畳みおよび分泌に影響を与え得る。あるいは、増殖阻害は、グリコシルトランスフェラーゼのインビボ活性の増加に関連し得る。この活性増加は、機能を破壊する様式で、標準的なインビトロ培養条件下での増殖にとって必要な1セットの内因性糖タンパク質のグリコシル化パターンの変化をもたらす。
【0076】
根底をなす機構とは独立して、この増殖阻害効果は、動物細胞のグリコシル化を操作することについて2つの結果を有する。第一に、この効果は、標的となる糖タンパク質産物のためのベクターとともに構成性グリコシルトランスフェラーゼ発現ベクターを同時トランスフェクトすることは、貧弱な戦略であることを示唆する。これらの2つのクラスのタンパク質の連結した発現の他の方法(例えば、類似の強度の複数の構成性プロモーターの使用または複数シストロン性の構成性発現ベクターの使用による)もまた、回避されるはずである。これらの場合、経済的なバイオプロセスにとって必要である、標的糖タンパク質の非常に高度で構成性の発現を有するクローンもまた、グリコシルトランスフェラーゼの高度な発現を有し、そしてこの選択プロセスにおいて排除され得る。連結された誘導性の発現もまた、産業的バイオプロセスについて問題であり得る。なぜなら、増殖停止した細胞の生存性は、グリコシルトランスフェラーゼの過剰発現によって減弱化されるからである。
【0077】
第二の結果は、これが、グリコシル化操作のアプローチについてグリコシルトランスフェラーゼ過剰発現に上限を課すことである。明らかに、細胞において、グリコシルトランスフェラーゼの内因性レベルでの、多くのグリコシルトランスフェラーゼ触媒性の反応の変換は、いくつかのグリコシル化部位について非常に高い。しかし、そのオリゴ糖がいくらかアクセス不能であるか、または特定のグリコシルトランスフェラーゼについて有利でないコンホメーションにおいて安定化されている、グリコシル化部位もまた存在する。例えば、バイセクティングGlcNAcの添加は、ヒトIgG抗体の可変領域に配置されるオリゴ糖よりもそのFc領域に付着するオリゴ糖に、より制限されることが観察されている。Savvidouら、1984、Biochemistry 23:3736−3740。これらの制限された部位のグリコシル化操作は、グリコシルトランスフェラーゼ発現におけるそのような限定によって影響を受け得る。これは、糖形態の「非天然の」分布について目的化することを示唆するが、これらは、糖タンパク質の特定の治療適用について利益があり得る。
【0078】
(C.実施例3:チャイニーズハムスター卵巣細胞の抗ヒト神経芽腫抗体のグリコシル化の操作)
そのグリコシル化パターンを改変することによって治療抗体を操作する概念を確認するために、キメラ抗ヒト神経芽腫IgG1(chCE7)を選択した。このIgG1は、SP2/0組換えマウスミエローマ細胞によって産生される場合有意ではないADCC活性を有する。chCE7は、腫瘍に会合する190kDaの膜糖タンパク質を認識し、そしてこれまでに試験されている神経芽腫腫瘍のすべてと強力に反応する。chCE7は、その抗原について高度の親和性(1010M-1のKd)を有し、そしてその高度の腫瘍特異性
のために、chCE7は、臨床病理において診断ツールとして慣用される。Amstutzら、1993.Int.J Cancer 53:147−152。近年の研究において、放射標識されたchCE7は、ヒト患者において良好な腫瘍局在を示している。Durr、1993、Eur.J.Nucl.Med.20:858。抗神経芽腫治療モノクローナル抗体(mAb)であるchCE7のグリコシル化パターンをCHO細胞において、GnT IIIのテトラサイクリン調節発現を用いて操作した。その糖形態分布において異なる1セットのmAbサンプルを、基底レベルと毒性レベルとの間の範囲においてGnT III発現を制御することによって産生した。そして、そのグリコシル化プロフィールを中性オリゴ糖のMALDI/TOF−MSによって分析した。これらのサンプルのADCC活性の測定によって、インビトロ生物学的活性における最大のchCE7についてのGnT III発現の最適範囲が示された。そして、この活性は、Fcに関連し、複合型バイセクト型オリゴ糖のレベルと相関していた。
【0079】
(1.材料および方法)
chCE7発現ベクターの構築。抗ヒト神経芽腫キメラ抗体chCE7の、重鎖(IgG1)および軽鎖(κ)の発現のための、それぞれ、プラスミドベクター10CE7VHおよび98CE7VL(これらは、マウス免疫グロブリンプロモーター/エンハンサー、マウス抗体可変領域、およびヒト抗体定常領域を含むキメラゲノムDNAを含む)(Amstutzら、1993、Int.J.Cancer 53:147−152)を、最終の発現ベクターpchCE7HおよびpchCE7Lの構築のための出発材料として使用した。キメラ重鎖および軽鎖chCE7の遺伝子を、再アセンブリし、そしてpcDNA3.1(+)ベクター中にサブクローニングした。再アセンブリの間、全てのイントロンを除去し、リーダー配列を、合成したものに置き換えた(Reffら、1994、Blood 83:435−445)。そして、可変領域および定常領域の配列を接合する独特な制限部位を導入した。重鎖の定常領域からイントロンを、重複伸長PCRを用いてスプライシングすることによって除去した(Clacksonら、1991、General
Applications of PCR to Gene Cloning and
Manipulation、p.187−214、in:McPhersonら(編)、PCR a Practical
Approach、Oxford University Press、Oxford)。
【0080】
異なるレベルのGnT IIIを発現するchCE7のCHO細胞における産生。CHO−tet−GnT IIIm(前出を参照のこと)細胞を、リン酸カルシウムトランスフェクション方法を使用して、ベクターpchCE7H、pchCE7L、およびpZeoSV2(Zeocin耐性のため、Invitrogen、Groningen、The Netherlands)を用いて同時トランスフェクトした。Zeocin耐性クローンを、96ウェル細胞培養プレートに移した。そして、ヒトIgG定常領域について特異的なELISAアッセイを用いてキメラ抗体発現についてアッセイした。Lifelyら、1995、前出。4つのchCE7抗体サンプルは、10% FCSを補充したFMX‐8細胞培養培地中で増殖させた選択したクローン(CHO−tet−GnT IIIm−chCE7)の並行培養物に由来した;各培養物は、異なるレベルのテトラサイクリンを含み、従って、異なるレベルのGnT IIIを発現した。CHO−tet−GnT IIIm−chCE7細胞を増大させ、そして7日間、異なる濃度のテトラサイクリンに予備適合させた。テトラサイクリンのレベルは、2000ng/ml、60ng/ml、30ng/ml、および15ng/mlであった。
【0081】
chCE7抗体サンプルの精製。抗体を、20mMリン酸ナトリウム、20mMクエン酸ナトリウム、500mM塩化ナトリウム、0.01% Tween20、1M尿素、pH 7.5(緩衝液A)から緩衝液B(リン酸ナトリウムのない緩衝液A、pH 2.5)へのpH線形勾配溶出を用いて、1mlのHiTrapプロテインAカラム(Pharmacia Biotech、Uppsala、Sweden)におけるプロテインAアフィニティークロマトグラフィーによって培養培地から精製した。アフィニティー精製したchCE7サンプルを、1mlのResourceSカチオン交換カラム(Pharmacia Biotech、Uppsala、Sweden)においてPBSへと緩衝液交換した。最終純度は、SDS−PAGEおよびクーマシーブルー染色から95%より高いと判断した。各サンプルの濃度を、280nmでの吸光度から見積もった。
【0082】
抗体の神経芽腫細胞への結合。ヒト神経芽腫細胞への結合親和性を、CHO産生したサンプルによる125I標識したchCE7の変位量から見積もった。Amstutzら、1
993、前出。
【0083】
MALDI/TOF−MSによるオリゴ糖分析。CE7−2000t,CE7−60t,CE7−30t、およびCE7−15tのサンプルを、A.urefaciensシアリダーゼ(Oxford Glycosciences、Oxford,United Kingdom)を、製造業者の指示に従って用いて処理して、任意のシアル酸モノサッカリド残基を除いた。次いで、シアリダーゼ消化物を、ペプチドN−グリコシダーゼF(PNGaseF、Oxford Glycosciences、Oxford、United Kingdom)を製造業者の指示に従って用いて処理して、N結合型オリゴ糖を放出させた。タンパク質、界面活性剤および塩を、その消化物を上部から底部まで、20mlのSapPak C18逆相マトリクス(Waters,Milford、MA)を含むマイクロカラム、20mlのDowex AG 50W X8カチオン交換マトリクス(BioRad,Hercules、CA)を含むマイクロカラム、および20mlのAG 4X4アニオン交換マトリクス(BioRad,Hercules、CA)を含むマイクロカラムに通すことによって除去した。これらのマイクロカラムを、これらのマトリクスを、エタノールを満たしたGel Loaderチップ(Eppendorf,Basel、Switzerland)中に充填すること、続いて水を用いて平衡化することによって作製した。Kusterら、1997、Anal.Biochem.250:82−101。フロースルーの液体および300mlの水での洗浄物をプールし、濾過し、エバポレートして、室温で乾燥させ、そして2mlの脱イオン水中に再懸濁した。1μlをMALDI−MSサンプルプレート(Perseptive Biosystems、Farmingham、MA)に適用し、そしてアセトニトリル中の1mlの10mg/mlデヒドロ安息香酸(DHB、Aldrich、Milwakee、Wisconsin)溶液と混合した。これらのサンプルを風乾し、そして得られた結晶を0.2mlのエタノール中に溶解し、そして風乾により再結晶させた。Harvey、1993、Rapid Mass、Spectrom.7:614−619。次いで、このオリゴ糖サンプルを、陽イオンおよび反射様式で、20kVの加速電圧を用いて、遅延イオン抽出MALDIイオン供給源を装着したElite Voyager400分光器(Perseptive Biosystems、Farmingham,MA)を用いるマトリクス支持レーザー脱離イオン化/飛行時間重量スペクトル測定(MALDI/TOF−MS)によって分析した。128の走査を平均化した。バイアンテナリー複合型バイセクト型オリゴ糖構造を、5つのHexNAcに関連するピークに割り当てた。代替の5つのHexNAcを含むアイソマーであるN結合型トリアンテナリー非バイセクト型オリゴ糖は、IgGのFc領域においては決して見出されず、そしてそれらの合成は、GnT IIIとは別のグリコシルトランスフェラーゼによって触媒された。
【0084】
ADCC活性アッセイ。異なる濃度のchCE7サンプルの存在下で、1:19での標的:エフェクター比にて、37℃16時間のインキュベーションの間のIMR−32ヒト神経芽腫細胞(標的)のヒトリンパ球(エフェクター)による溶解を、蛍光色素の保持を介して測定した。Kolberら、1988、J.Immunol.Methods 108:255−264。IMR−32細胞を、蛍光色素Calcein AMを用いて20分間標識した(最終濃度3.3μM)。標識した細胞(80,000細胞/ウェル)を、1時間、異なる濃度のCE7抗体とインキュベートした。次いで、単球を涸渇した単核細胞を添加した(1,500,000細胞/ウェル)。そして、細胞混合物を、5%CO2雰囲気中で37℃で16時間インキュベートした。この上清を捨て、そしてその細胞を
HBSSで一回洗浄し、そしてTriton X−100 (0.1%)中で溶解した。IMR−32細胞における蛍光色素の保持を、蛍光測定器(Perkin Elmer,Luminscence Spectrometer LS 50B、(Foster City、CA))を用いて測定し、そして特定の溶解物を、その標的を抗体への曝露の代わりに界面活性剤に対する曝露から得られた総溶解物コントロールに対して相対的に算出した。抗体の非存在下でのシグナルを、0%の細胞傷害性に設定した。各抗体濃度を、三連にて分析し、そしてそのアッセイを、3回の別個の回数反復した。
【0085】
(2.結果および考察)
異なるレベルのGnT IIIを発現するCHO細胞におけるchCE7の産生。ChCE7の重鎖および軽鎖の発現ベクターを、ヒトサイトメガロウイルス(hCMV)プロモーター、ウシ成長ホルモン終結配列およびポリアデニル化配列を組み込み、そして全ての重鎖および軽鎖のイントロンを除去して構築した。このベクター設計は、CHO細胞における組換えIgG遺伝子の再現性の良い高レベル発現の報告に基づいた。Reffら、1994、前出;Trillら、1995、Current Opinion Biotechnol.6:553−560。さらに、独特な制限部位を、可変領域と定常領域との間の接合部にて各鎖に導入した。これらの部位は、リーディングフレームを保存し、そしてアミノ酸配列を変化させない。これらは、他のマウス−ヒトキメラ抗体の産生のための、マウス可変領域の単純な交換を可能にするはずである。Reffら,1994、前出。DNA配列決定によって、所望の遺伝子が適切にアセンブルされているかを確認した。そして、トランスフェクトされたCHO細胞におけるキメラ抗体の産生を、ヒトFc−ELISAアッセイを用いて確認した。
【0086】
GnT IIIの安定なテトラサイクリン制御された発現およびchCE7の安定な構成的発現を伴うCHO−tet−GnT IIIm−chCE7細胞を確立し、そして1セットのchCE7サンプルの産生のために規模拡大した。規模拡大の間、同じCHOクローンに由来する4つの並行培養物を、各々、異なるレベルのテトラサイクリンおよびすなわちGnT III遺伝子の発現レベルのみにおいて異なるように増殖させた。この手順は、N結合型糖形態の生合成に影響を与える他の可変物からの任意のクローン性効果を除去し、GnT III遺伝子発現とグリコシル化抗体の生物学的活性との間の厳格な相関付けの確立を可能にする。テトラサイクリンの濃度は、2000ng/ml(すなわち、GnT III発現の基底レベル)から15ng/ml(この濃度において、グリコシルトランスフェラーゼの過剰発現に起因する有意な増殖阻害および毒性が観察された(前出を参照のこと))までに及んだ。実際、ほんのわずかの量の抗体が後者の培養物から回収され得た。テトラサイクリンを30ng/mlの濃度で用いた第二の最高レベルのGnT III発現は、緩和な増殖阻害のみをもたらした。この培養物から精製した抗体の収率は、GnT III遺伝子過剰発現の残りの2つのより低いレベル由来のものの約70%であった。
【0087】
4つの抗体サンプルCE7−2000t、CE7−60t、CE7−30t、CE7−15t(数字は、テトラサイクリンの会合濃度を示す)を、プロテインA上でアフィニティークロマトグラフィーにより精製し、そしてカチオン交換カラムを用いてPBSへと緩衝液交換した。純度は、クーマシーブルー染色を用いたSDS−PAGEから判断した場合95%を超えた。ヒト神経芽腫細胞に対する結合アッセイは、細胞に対する高度の親和性および異なるサンプル間で抗原結合が有意に相違しないことを示した(2.0Mと2.7×10-10Mとの間で変動させた推定平衡解離定数)。このことは予測どおりであった
。なぜならCE7可変領域には、推定N結合型グリコシル化部位が存在しないからである。
【0088】
異なるchCE7サンプルの複合型バイセクト型オリゴ糖の分布およびレベル。オリゴ糖プロフィールを、飛行時間におけるマトリクス支持レーザ脱離/イオン化質量測定(MALDI/TOF−MS)によって得た。4つのCHO産生した抗体サンプルの各々およびSP2/0マウスミエローマ由来のchCE7(CE7−SP2/0)サンプルに由来する中性のN結合型オリゴ糖の混合物を、2,5−デヒドロ安息香酸(2,5−DHB)を用いてマトリクスとして分析した(図9)。これらの条件下で、中性のオリゴ糖は、本質的に単一の[M+ Na+]イオンであるようであり、これは、ときに、そのマトリク
スのカリウム含有量に依存してより小さな[M+ K+]イオンを伴う。Bergwef
fら、1995、Glycoconjugate J.12:318−330。
【0089】
この型の分析は、異なる質量の中性オリゴ糖の相対比(これは、相対ピーク高さに反映される)、および各ピークのモノサッカリドの荷電組成の両方を得た。Kusterら、1997、前出;NavenおよびHarvey、1996、Rapid Commun.Mass Spectrom.10:1361−1366。一時的な構造を、そのモノサッカリドの組成、生合成経路の知識および同じ宿主によって産生された同じ糖タンパク質由来のオリゴ糖についてのこれまでの構造データに基づくピークにより割り当てる。なぜなら、タンパク質バックボーンおよび細胞型は、オリゴ糖分布に対して強力な影響を有し得るからである。Fieldら、1996、Anal.Biochem.239:92−98。Fcが会合するオリゴ糖の場合、バイアンテナリー複合型オリゴ糖のみがヒト血清に存在するIgGにおいて検出されるか、または正常条件下での哺乳動物細胞培養物によって産生された。Wormaldら、1997、Biochemistry 36:1370−1380;WrightおよびMorrison、1997,Tibtech 15:26−31。これらの化合物へ導く経路を、各オリゴ糖に対応する[M+ Na+
]イオンの質量を含めて図10に示す。高マンノースオリゴ糖もまた、バッチ細胞培養物の安定期および死期において産生された抗体において検出された。Yu Ipら、1994、Arch.Biochem.Biophys 308:387−399。
【0090】
CE7−SP2/0サンプルにおける2つの主要なピーク(図9A)は、4つのN−アセチルヘキソサミン(HexNAc)を伴うフコシル化オリゴ糖の質量に対応する(これらは、3つ(m/z1486)または4つ(m/z1648)のいずれかのヘキソースを含む)。図10を参照のこと。しかし、この図におけるオリゴ糖についてまとめた注記は、コアの2つのGlcNAcを計数していないことに注意されたい。この組成は、それぞれ、0個または1個のガラクトース残基を有する、コアがフコシル化バイアンテナリー複合型オリゴ糖構造(これは、Fcが会合したオリゴ糖に代表的であり、そしてSP2/0細胞において発現されたキメラIgG1に由来するFcオリゴ糖のNMR分析においてこれまでに観察されている通りである)と一致する。Bergweffら、1995、前出。
【0091】
GnT IIIに触媒される、バイセクティングGlcNAcのこれらのバイアンテナリー化合物(この化合物は、好ましいGnT IIIアクセプターである)への移入は、5つのHexNAcを伴うオリゴ糖をもたらす(m/z1689および1851、それぞれ、非ガラクトシル化およびモノガラクトシル化、図10)。これらのオリゴ糖は、CE7−SP2/0サンプルにおいては明らかに存在しなかった。後者のピークは、chCE7がCHO−tet−GnTIIIm細胞において発現される場合に現れる。CHOが発現する抗体において、4つのHexNAcを含有するピークもまた、主にフコシル化されている。しかし、少量のフコシル化されていない構造も、m/z1339でのピークから明らかである(図10を参照のこと)。ガラクトシル化のレベルはまた、CHO由来材料とSP2/0由来材料との間であまり異ならない。GnT III発現の基底レベルでは(CE7−2000tサンプル.図9B)、5つのHexNAcを伴う分子は、4つのHexNAcを伴うものよりも低い割合で存在する。より高いレベルのGnT III発現(CE7−60tサンプル、図9C)は、5つのHexNAcを伴うオリゴ糖に有利に比の逆転をもたらした。この傾向に基づいて、バイアンテナリー複合型バイセクト型オリゴ糖構造は、これらのサンプルにおいて5つのHexNAcを伴う化合物であると割り当てられた。三アンテナ性N結合型オリゴ糖である別の5HexNAc含有アイソマーは、IgGのFc領域においては決して見出されず、そしてその合成は、GnT IIIとは異なるGlcNAcトランスフェラーゼによって触媒される。
【0092】
GnT III発現におけるさらなる増加(CE7−30tサンプル、図9D)は、複合型バイセクト型オリゴ糖のレベルにおいてなんらの有意な変化をももたらさなかった。5つのHexNAcを含む別のピーク(m/z1543)は、CHO−GnTIIIサンプルにおいて低いが比較的安定なレベルで現れ、そして質量において非フコシル化複合型バイセクト型オリゴ糖質量に対応する(図10)。m/z1705およびm/z1867での小さなピークもまた、5つのHexNAc含有バイアンテナリー複合型オリゴ糖に対応する。それらは、m/z1689およびm/z1851でのピークのカリウム付加物(ナトリウム付加物に関する16Daの質量の相違)(Kusterら、1997、前出)か、またはフコースを伴わないモノガラクトシル化および二ガラクトシル化複合型バイセクト型オリゴ糖のいずれかに割り当てられ得る(図10)。まとめると、複合型バイセクト型オリゴ糖は、サンプルCE7−2000tにおける合計の約25%の量であり、そしてサンプルCE7−60tおよびCE7−30tにおいて約45%〜50%に達する。
【0093】
chCE7サンプルのオリゴ糖プロフィールからのさらなる情報。複合型バイセクト型オリゴ糖のレベルは、サンプルCE730tにおいてより高くなかったが、GnT IIIの過剰発現の増加は、少量ではあるが、バイアンテナリー複合型オリゴ糖基質の基質比を継続して減少させた。このことは、2つの異なる4つのHexNAc含有ピーク(m/z1664およびm/z1810)において中程度の増加を伴った。後者の2つのピークは、ガラクトシル化バイアンテナリー複合型オリゴ糖、または混成型バイセクト型化合物のいずれかに対応し得る(図11)。両方のクラスの構造の組合せもまた、可能である。これらのピークにおける相対的な増加は、GnT III過剰発現の混成型バイセクト型の副産物の蓄積と一致する。実際、GnT III過剰発現の最高レベルで産生されたサンプルであるCE7−15tは、m/z1664におけるピークの大きな増加、m/z1810におけるピークの減少、およびバイセクト型複合オリゴ糖の約25%のレベルへの同時減少を示した。図9Eにおけるm/z1689およびm/z1851のピークならびに図11における対応する構造を参照のこと。フコシル化されたもの(m/z1810)に代わる、非フコシル化(m/z1664)の混成型バイセクト型副産物のより高度な蓄積は、GnT IIIによってまず改変されているオリゴ糖がもはや、コアのα1,6−フコシルトランスフェラーゼについての生合成基質ではあり得ないという事実に一致する。Schachter、1986、Biochem.Cell Biol.64:163−181。
【0094】
m/z1257でのピークは、CHOに由来するサンプルにおける合計の10〜15%のレベルで、およびCE7−SP2/0よりも低いレベルで存在する(図9)。これは、5つのヘキソースおよび2つのHexNAcに対応する。この組成を有する唯一公知であるN結合型オリゴ糖構造は、高マンノース型の5つのマンノースを含有する化合物である。別の高マンノースオリゴ糖である6マンノースのもの(m/z1420)もまた、はるかに低いレベルで存在する。上記のように、このようなオリゴ糖は、バッチ細胞培養物の後期において発現されるIgGのFcにおいて検出されている。Yu Ipら、1994、前出。
【0095】
chCE7サンプルの抗体依存性細胞傷害性。chCE7は、最小レベルのGnTIII過剰発現をとともにCHO−tet−GnTIIIm細胞中で発現されるとき、ヒトリンパ球による神経芽腫細胞のインビトロ溶解として測定される、ある程度のADCC活性を示す(図12、サンプルCE7−2000t)。GnTIIIのレベルが上昇すると、ADCC活性における大きな増加が生じた(図12、サンプルCE7−60t)。GnTIIIのさらなる過剰発現は、活性のさらなる増加をともなわず(図12,サンプルCE7−30t)、そして最高レベルの発現は、実際に、減少したADCCに至った(図12、サンプルCE7−15t)。最高のADCC活性を示すことに加えて、CE7−60tおよびCE7−30tの両方のサンプルの両方は、非常に低い抗体濃度で、有意なレベルの細胞傷害性を示す。これらの結果は、ADCC活性についてCHO細胞における最適範囲のGnTIII過剰発現があることを示し、そしてオリゴ糖プロフィールとの比較は、活性が、Fcに結合した複合型バイセクト型オリゴ糖のレベルと相関することを示す。
【0096】
ADCC活性についての複合型バイセクト型オリゴ糖の重要性を考慮すると、これら化合物の比率をさらに増加させるために、この経路を操作することが有用である。GnTIIIの、サンプルCE7−30tについて用いられたレベルに接近するレベルまでの過剰発現は、有意な傷害性および増殖阻害が観察されない生物工学的に実施可能な範囲内にある。このレベルの発現では、非ガラクトシル化された、バイアンテナリー複合型非バイセクト型オリゴ糖、すなわち、好適な潜在的GnTIII基質は、全体の10%より少なくなるまで減少する。m/z 1486のピーク、図9Dを参照のこと。しかし、わずか50%が、所望のバイアンテナリー複合型バイセクト型構造に転換されるだけである。残りは、混成型バイセクト型オリゴ糖副産物に転用されるか、または競合酵素β1,4−ガラクトシルトランスフェラーゼ、GalTにより消費されるかのいずれかである(図11)。
【0097】
相補的構造分析による、混成型バイセクト型およびガラクトシル化複合型非バイセクト型オリゴ糖ピークの分離は、各々の潜在的な所望されない経路がいくら消費されるかを決定する。高いGnTIII過剰発現レベルにおけるm/z 1664および1810ピークの増大は、これらピークの少なくとも一部分は、混成型バイセクト型オリゴ糖に対応することを示唆する(図11)。理論的には、混成型バイセクト型化合物に行くフラックスは、GnTIIIと一緒のマンノシダーゼIIのような経路中のより先の酵素の同時過剰発現により低減され得る。その一方、GnTIIIとGalTとの間の複合型バイセクト型オリゴ糖基質に対する競合は、GnTIIIを過剰発現する間に、UDP−GlcNAcのゴルジ体内濃度を増加させることにより、GnTIII触媒反応に向かって潜在的に片寄り得る。GnTIIIは、GlcNAcを、同時基質(co−subtrate)UDP−GlcNAcから異なるオリゴ糖に転移させる。UDP−GlcNAc同時基質のゴルジ体内濃度が、GnTIIIに対して準飽和である場合、それを、培養培地組成の操作によるか、またはゴルジ体中への糖−ヌクレオチド輸送の遺伝的操作により増加させて、オリゴ糖についてGalTとの競合でGnTIIIに有利にし得る。
【0098】
ADCC活性における増加が、ガラクトシル化および非ガラクトシル化の両方の複合型バイセクト型オリゴ糖における増加から生じるか、またはこれらの形態の1つのみにおける増加から生じるか否かは決定されるべきままである。図9におけるm/z 1689および1851のピークを参照のこと。ガラクトシル化バイアンテナリー複合型バイセクト型オリゴ糖が、増加したADCC活性について最適構造であることが見出される場合、Fc領域におけるこれら化合物の画分を最大にすることは、GnTIIIおよびGalTの両方の過剰発現を必要とする。先に論議された競合シナリオを考慮すると、両方の遺伝子の発現レベルは、注意深く調節されなければならない。さらに、可能な限り、トランス−ゴルジ体嚢の代わりにTGNに向かって、過剰発現GalTを再分配する試みは、価値がある。後者の戦略は、GalTの膜貫通領域コード配列を、α2,6−シアリルトランスフェラーゼの膜貫通領域(ChegeおよびPfeffer、1990、J.Cell.Biol.111:893〜899)と交換することにより実現され得る。
【0099】
(D.実施例4:抗CD20モノクローナル抗体C2B8のグリコシル化の操作)
C2B8は、抗ヒトCD20キメラ抗体である。Reff、M.E.ら、1994、前述。C2B8は、1997年にFDA認可を受け、そして現在、合衆国において、非ホジキンリンパ腫の処置のために、RituxanTMの商品名の下で用いられている。C2B8は、CHO細胞培養物に由来し、そしてそれ故、バイセクト型オリゴ糖を保持しないはずである。前述を参照のこと。この抗体の改良型を生成するために、chCE7抗神経芽腫抗体について先に示された方法が適用された。前述を参照のこと。開示された方法に従って改変されたC2B8抗体は、同じ細胞培養物および精製条件の下で生成された、標準的な非改変C2B8抗体よりも高いADCC活性を有していた。
【0100】
(1.材料および方法)
キメラ抗CD20モノクローナル抗体(C2B8)の可変軽鎖領域および可変重鎖領域の合成。C2B8抗体のVH遺伝子およびVL遺伝子を、PCRを用いる1工程プロセス(Kobayashiら、1997、Biotechniques 23:500〜503)で、1セットの重複する一本鎖オリゴヌクレオチド(プライマー)を用いて合成的に
アセンブルした。抗CD20抗体のマウス免疫グロブリンの軽鎖および重鎖の可変領域(それぞれVLおよびVH)をコードする配列データを、公開された国際特許出願(国際公開番号:WO 94/11026)から得た。アセンブルされたDNAフラグメントを、pBluescriptIIKS(+)中にサブクローン化し、そしてDNAサイクルシークエンシングにより配列決定し、変異が導入されなかったことを確認した。
【0101】
キメラ抗CD20モノクローナル抗体(C2B8)の発現のためのベクターの構築。C2B8モノクローナル抗体のVHおよびVLのコード領域を、pchCE7HおよびpchCE7L中にそれぞれサブクローン化した。このサブクローニングでは、抗神経芽腫CE7の可変重鎖および可変軽鎖をコードする配列(前述)を、C2B8の合成的にアセンブルされた可変重鎖領域および可変軽鎖領域と交換した。
【0102】
C2B8抗体を発現するCHO−tet−GnTIIIm細胞の生成。C2B8抗体を発現するCHO−tet−GnTIIIm細胞株の生成のための方法は、CHO−tet−GnTIIIm−CE7に対するのと正確に同じであった。前述を参照のこと。さらなる作業のために選択されたクローンを、CHO−tet−GnTIIIm−C2B8と名づけた。
【0103】
C2B8抗体を発現するCHO−tTAの生成。CHO−tTAは、CHO−tet−GnTIIImの親細胞株である。前述を参照のこと。GnTIIIを発現せず、C2B8抗体を発現するCHO−tTA細胞株の生成のための方法は、CHO−tet−GnTIIIm−C2B8およびCHO−tet−GnTIIIm−chCE7に対するのと正確に同じであった。前述を参照のこと。さらなる作業のために選択されたクローンを、CHO−tTA−C2B8と名づけた。
【0104】
C2B8抗体サンプルの生成。2つのC2B8抗体サンプルは、平行なCHO−tet−GnTIIIm−C2B8培養物に由来した;各培養物は、種々のレベルのテトラサイクリンを含み、そしてそれ故GnTIIIを種々のレベルで発現すると予想される。テトラサイクリンのレベルは、2000ng/ml、50ng/ml、および25ng/mlであった。これらの培養に由来するC2B8抗体サンプルを、C2B8−2000t、C2B8−50t、およびC2B8−25tとそれぞれ称した。平行して、1つの抗体サンプル(C2B8−nt)を、CHO−tTA−C2B8培養から作成した。この細胞株は、GnTIIIを発現しない。CHO−tTA−C2B8細胞を、テトラサイクリンなしで培養した。
【0105】
GnTIII発現の分析。GnTIIIのウェスタンブロット分析については、生産培養物の各々の細胞溶解産物を、SDS−PAGEにより分離し、そしてポリビニリデンジフルオライド膜に電気ブロットした。抗c−mycモノクローナル抗体9E10および抗マウスIgG−西洋ワサビペルオキシダーゼ(Amersham、Arlington、IL)を、一次抗体および二次抗体としてそれぞれ用いた。結合抗体を、増感化学発光キット(Amersham、Arlington、IL)を用いて検出した。
【0106】
C2B8抗体サンプルの精製。抗体サンプルを、chCE7抗体サンプルについてと同じ手順を用いて精製した。前述を参照のこと。濃度を、Molecular Probes(Leiden、The Netherlands)からの蛍光に基づくキットを用いて測定した。
【0107】
特異的C2B8抗原結合の確認。C2B8抗CD20モノクローナル抗体の抗原結合の特異性を、懸濁液中の細胞について間接免疫蛍光アッセイを用いて確認した。この研究には、CD20陽性細胞(SB細胞;ATCC寄託番号ATCC CCL120)およびCD陰性細胞(HSB細胞;ATCC寄託番号ATCC CCL120.1)を利用した。各タイプの細胞を、一次抗体としての、25ng/mlテトラサイクリンで生成したC2B8抗体とともにインキュベートした。陰性のコントロールは、一次抗体の代わりにHBSSBを含んだ。抗ヒトIgG Fc特異的なポリクローナルFITC結合体化抗体を、すべてのサンプルについて二次抗体として用いた(SIGMA、St.Louis、MO)。細胞を、Leica(Bensheim、Germany)蛍光顕微鏡を用いて調べた。
【0108】
ADCC活性アッセイ。異なる濃度のC2B8サンプルの存在下の、ヒト単球枯渇末梢血単核細胞(エフェクター細胞)によるSB細胞(CD20+標的細胞;ATCC寄託番号ATCC CCL120)の溶解を、Brunnerら、1968、Immunology 14:181−189に記載されるのと同じ手順に基本的に従って実施した。エフェクター細胞の標的細胞に対する比率は、100:1であった。
【0109】
(2.結果および考察)
GnTIIIは、種々の細胞株および培養物において種々のレベルで発現される。平行したCHO−tet−GnTIIIm−C2B8培養物(各培養物は種々のレベルのテトラサイクリン(2000、50、および25ng/ml)を含み、そしてそれ故、種々のレベルでGnTIIIを発現すると期待される)の細胞を溶解し、そして細胞溶解産物をSDS−PAGEにより分離し、そしてウェスタンブロッティングにより検出した。25ng/mlテトラサイクリンで増殖させた培養物の溶解産物は、GnTIIIの対応する分子量に強いバンドを示し、その一方、50および2000ng/mlで増殖させた培養物は、図13に示されるように、GnTIIIのかなりより少ない発現を有していた。
【0110】
特異的C2B8抗原結合の確認。種々のレベルのGnTIIIを発現する細胞の平行培養物から産生されたC2B8サンプルを、アフィニティークロマトグラフィーにより培養上清から精製し、そしてカチオン交換カラム上でPBSに緩衝液交換した。純度は、還元条件下のSDS−PAGEのクマシーブルー染色から95%より高いと推定された。これらの抗体サンプルは、可変領域がPCRアセンブリ法により合成された抗体遺伝子の発現に由来した。この合成cDNAフラグメントの配列決定は、先に公開された国際特許出願(国際公開番号WO94/11026)中の元のC2B8可変領域配列とは差異を示さなかった。このサンプルのヒトCD20(C2B8の標的抗原である)への特異的結合は、その表面上にCD20を発現するヒトリンパ芽球様細胞株SB、およびこの抗原を欠くHSBリンパ芽球様細胞株を用いる、間接免疫蛍光により証明された。抗体サンプルC2B8−25tは、SB細胞の陽性染色を与えたが(図14A)、同一の実験条件下でHSB細胞の陽性染色は与えなかった(図14Bを参照のこと)。SB細胞からなるさらなる陰性コントロールを、C2B8−25t抗体の代わりにPBS緩衝液とともにインキュベートした。それは、染色が全くないことを示した。
【0111】
C2B8サンプルのインビトロADCC活性。GntIII発現を有さないCHO−tTA−C2B8細胞(前述を参照のこと)中で発現した抗体サンプルC2B8−ntは、ヒトリンパ球によるSB細胞(CD20+)のインビトロ溶解で測定したとき、(1μg/mlの抗体濃度で)31%の細胞傷害性活性を示した(図15、サンプルC2B8−nt)。2000ng/mlのテトラサイクリンで増殖したCHO−tet−GnTIII培養物(すなわち、クローン化したGnTIII発現の基底レベル)由来のC2B8−2000t抗体は、同じ抗体濃度におけるC2B8−ntサンプルに関してADCC活性において、1μg/mlの抗体濃度で33%の増加を示した。テトラサイクリンの濃度を25ng/mlまで低減させると(サンプルC2B8−25t)、これはGnTIII発現を有意に増大させ、同じ抗体濃度におけるC2B8−nt抗体サンプルに関して最大ADCC活性において(1μg/mlの抗体濃度で)ほぼ80%の大きな増加を生じた(図15、サンプルC2B8−25t)。
【0112】
最も高いADCC活性を示すことに加えて、C2B8−25tは、非常に低抗体濃度で、有意なレベルの細胞傷害性を示した。0.06μg/mlにおけるC2B8−25tサンプルは、1μg/mlにおけるC2B8−ntの最大ADCC活性に類似のADCC活性を示した。この結果は、サンプルC2B8−25tが、C2B8−ntより16倍低い抗体濃度で、C2B8−ntと同じADCC活性に到達したことを示した。この結果は、GnTIIIを活性に発現する細胞株で生産されたキメラ抗CD20抗体C2B8が、GnTIIIを発現しなかった細胞株で生産された同じ抗体より有意に活性であったことを示す。
【0113】
本発明の方法を用いるこの抗体の1つの利点は、(1)同じ治療効果に到達するために注入されなけばならない抗体のより低い用量が、抗体産生の経済学で有利なインパクトを有すること、または(2)同じ用量の抗体を用いてより良い治療効果が得られることである。
【0114】
(E.実施例5:最大ADCC活性に至る最適レベルでグリコシルトランスフェラーゼ遺伝子の構成的発現を有するCHO細胞株の確立)
ADCCを増大するための方法のいくつかの適用では、それ自体で、または他のクローン化されたグリコシルトランスフェラーゼおよび/またはグリコシダーゼとともに、GnTIIIの調節された発現よりも構成的発現を用いることが所望され得る。しかし、本発明者らは、改変された抗体のADCC活性が、GnTIIIの発現レベルに依存することを証明した。前述を参照のこと。従って、GnTIII単独の、またはその他のグリコシルトランスフェラーゼおよび/またはグリコシダーゼ遺伝子とともに、最適レベルまたは最適レベル近傍でその構成的発現を有するクローンを選択することが重要である。単独またはβ(1,4)−ガラクトシルトランスフェラーゼ(GalT)のような他のグリコシルトランスフェラーゼとともにのいずれかで、GnTIII発現の最適レベルは、グリコシルトランスフェラーゼの調節性発現を有する細胞株を用いて最初に決定される。次いで、GnTIIIおよび任意のその他のクローン化されたグリコシルトランスフェラーゼの構成的発現を有する安定クローンを、最適近傍の発現レベルについてスクリーニングする。
【0115】
(1.調節されたGnTIIIのためのベクターの最適近傍発現レベル構成の決定)
GFP発現に連鎖する発現。各グリコシルトランスフェラーゼ遺伝子は、IRES配列を経由して、細胞中に保持されるタンパク質、例えば、グリーン蛍光タンパク質(GFP)または利用可能な抗体により認識され得るペプチドでタグ化される原形質膜タンパク質をコードするレポーター遺伝子に連結される。1つより多くのグリコシルトランスフェラーゼが試験されている場合、種々のマーカーが各々のグリコシルトランスフェラーゼと連結される。例えば、GnTIIIがGFPに連結され得、そしてGalTがブルー蛍光タンパク質(BFP)に連結され得る。GFP cDNAの上流のIRESエレメントの上流のGnTIII cDNAからなる真核生物発現カセットは、まず、標準的なサブクローニングおよび/またはPCR工程によりアセンブルされる。次いでこのカセットを、テトラサイクリン調節発現ベクターpUHD10−3(前述を参照のこと)中、tet−プロモーターの下流でかつ終止配列およびポリアデニル化配列の上流にサブクローン化し、ベクターpUHD10−3−GnTIII−GFPを得る。
【0116】
GFP発現に連鎖した調節されたGnTIII発現を持つCHO細胞の確立、および構成的chCE7抗体発現。テトラサイクリン応答性トランスアクチベーターを発現するCHO−tTA細胞(前述を参照のこと)を、ベクターpUHD10−3−GnTIII−GFPおよびピューロマイシン耐性遺伝子の発現のためのベクターpPurで同時トランスフェクトする。前述を参照のこと。ピューロマイシン耐性クローンを、テトラサイクリンの存在下で選択する。個々のクローンを、テトラサイクリンの存在下(2μg/ml)または非存在下で2連で培養する。グリコシルトランスフェラーゼ過剰発現に起因して(前述を参照のこと)、テトラサイクリンの非存在下で増殖の阻害を示す6つのクローンを選択し、そしてGFP関連シグナルの検出のために、蛍光活性化細胞ソーティング(FACS)により分析する。最高の誘導比(テトラサイクリンの存在下の蛍光に対するテトラサイクリンの非存在下の蛍光の比として規定される)を与えるクローンを、さらなる作業のために選択し、そしてCHO−tet−GnTIII−GFPと称する。CHO−tet−GnTIII−GFPを、抗体chCE7についての発現ベクターでトランスフェクトし、そしてこの抗体の高い構成的発現を有するクローンは、選択されたCHO−tet−GnTIII−GFP−chCE7である。前述を参照のこと。
【0117】
chCE7サンプルの生産、ADCC活性の測定および最適GnTIII発現レベルの決定。CHO−tet−GnTIII−GFP−chCE7の平行培養物を、種々のレベルのテトラサイクリン、そしてそれ故、GFPとともにGnTIIIを種々のレベルで発現して増殖させる。chCE7抗体サンプルを、アフィニティークロマトグラフィーにより培養上清から精製する。平行して、各培養物からの細胞をFACSにより分析して、各培養物の、GnTIIIの発現レベルに相関する、GFP関連蛍光の平均レベルを決定する。各chCE7抗体サンプルのインビトロADCC活性を決定し(前述を参照のこと)、そして各サンプルの最大インビトロADCC活性を、それを生成するために用いた細胞の平均蛍光に対してプロットする。
【0118】
(2.最適近傍レベルで構成的GnTIII発現を持つCHO細胞株の確立)
GnTIII−IRES−GFPカセット(前述を参照のこと)を、構成的発現ベクター中にサブクローン化する。CHO細胞を、このベクターおよびピューロマイシン耐性のためのベクターで安定に同時トランスフェクトする。ピューロマイシン耐性細胞を選択する。次いで、この安定にトランスフェクトされた細胞の集団を、FACSによりソートし、そして最適または最適近傍のADCC活性が達成される範囲内近傍のレポーターGFP遺伝子レベルを発現するクローンを選択する。前述を参照のこと。この最終のトランスフェクション工程は、すでに治療抗体を安定に発現するCHO細胞、または空のCHO細胞(例えば、DUKXまたはDG44 dhfr−CHO細胞)のいずれかに対してなされ得る。後者の場合には、上記の手順から得られたクローンは、最終の抗体産生細胞株を作製するために、治療抗体発現ベクターでトランスフェクトされる。
【0119】
(F.実施例6:最適化されたグリコシル化を有するヒトIgG Fcキメラの細胞表面発現)
現在、カプセル化細胞治療が多くの疾患に対して試験されている。カプセル化細胞インプラントを、身体内に外科的に配置されるように設計し、所望の治療物質をそれが必要である場所に直接送達する。しかし、移植されたカプセル化デバイスが、一旦機械的破壊を有する場合、細胞は、逃れそして所望でなくなり得る。身体内で、逃れた、所望でない細胞を破壊する1つの方法は、Fc媒介細胞傷害性メカニズムを経由することである。この目的のために、カプセル化されるべき細胞を先に操作して、Fc領域のN末端に融合した原形質膜に局在化するII型膜貫通ドメインからなる原形質膜係留融合タンパク質を発現し得る。Stabila、P.F.、1998、前述。カプセル内側の細胞は、Fc媒介細胞傷害性に対してカプセルによって保護され、その一方、逃れた細胞は、表面に提示されたFc領域を認識するリンパ球による破壊、すなわち、Fc媒介細胞傷害性メカニズムによる破壊に接近可能である。この実施例は、このFc媒介細胞傷害性活性が、提示されたFc領域のグリコシル化操作によってどのように増大するかを説明する。
【0120】
(1.Fcキメラをその表面上に発現し、およびGnTIIIを発現する細胞の確立)
特定の治療のために移植されるべき細胞、例えば、表面に提示されたFcキメラおよび分泌性治療タンパク質を既に生産する、ベビーハムスター腎臓(BHK)細胞を、まず、IRESエレメントを経由してGFPの発現に連鎖したGnTIIIの構成的発現のためのベクターで安定にトランスフェクトする。前述を参照のこと。安定なトランスフェクタントを、ベクター中に取り込んだマーカーにより、例えば、薬物耐性マーカーにより選択し、そしてその薬物の存在下の生存について選択する。
【0121】
(2.種々のレベルのGnTIIIを発現する細胞のスクリーニングおよび測定)
安定なトランスフェクタントを、蛍光活性化細胞ソーティング(FACS)により分析し、そして種々の平均蛍光レベルを有する一連のクローンを、さらなる研究のために選択する。各選択されたクローンを増殖させ、そしてFACSにより再分析し、GFPの安定性、そしてそれ故、関連したGnTIII発現を確実にする。
【0122】
(3.提示されたFc領域上の複合型バイセクト型オリゴ糖の種々のレベルの確認)
種々のレベルのGFP関連蛍光を有する3つのクローンから、およびGnTIII−IRES−GFPベクターでトランスフェクトしていない元のBHK細胞からのFc領域を、界面活性剤により膜から可溶化し、次いでアフィニティクロマトグラフィーにより精製する。次いで、オリゴ糖を取り出し、精製し、そしてMALDI−TOF/MSにより分析する。前述を参照のこと。得られるMALDI−TOF/MSプロフィールは、改変された、蛍光クローンのFc領域が、種々の比率の複合型バイセクト型オリゴ糖を保持することを示す。改変されていない細胞からのMALDIプロフィールは、バイセクト型オリゴ糖にともなう任意のピークを示さない。提示されたFc領域上で最高レベルの複合型バイセクト型オリゴ糖を保持するクローンを、さらなる作業のために選択する。
【0123】
(4.インビトロのFcに媒介される細胞の細胞傷害性活性アッセイ)
次いで、Fcに媒介される細胞の細胞傷害性活性アッセイを2つ、平行して実施する。1つのアッセイでは、標的細胞は、上記で選択されたクローンに由来する。平行するアッセイでは、標的細胞は、カプセル化され、そしてGnTIIIを発現するように改変されていない元の細胞である。このアッセイを、先に記載された手順を用いるが(前述を参照のこと)、任意のさらなる抗体の非存在下で実施する。なぜなら、標的細胞は、既にFc領域を提示しているからである。この実験は、GnTIIIを発現する細胞に対するFc媒介細胞傷害性活性が、このグリコシルトランスフェラーゼを発現しない細胞に対するFc媒介細胞傷害性活性よりも高いことを示す。
【0124】
本明細書の本体内で引用されるすべての参考文献は、その全体が参考として本明細書に援用される。
【特許請求の範囲】
【請求項1】
本願明細書に記載された発明。
【請求項1】
本願明細書に記載された発明。
【図1】


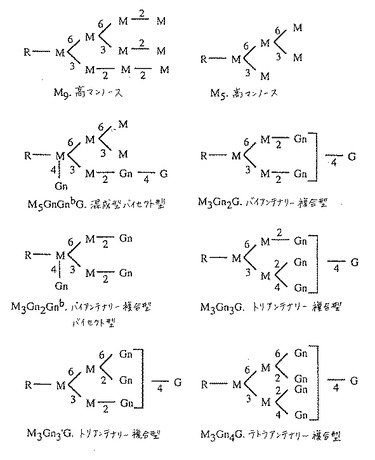
【図2】
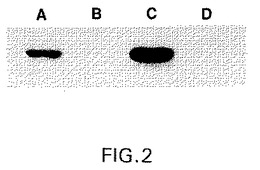
【図3】
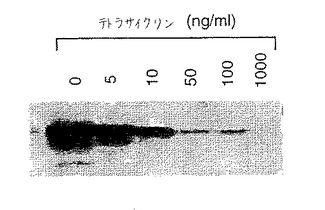
【図4】
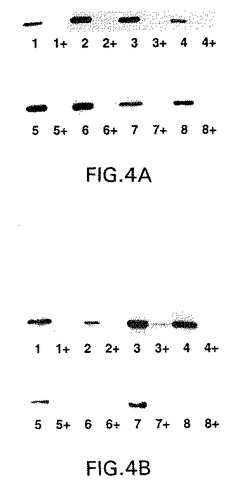
【図5】
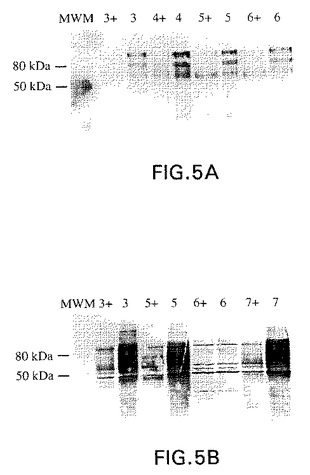
【図6】

【図7】

【図8】
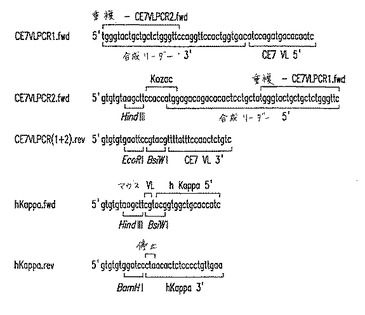
【図9A】
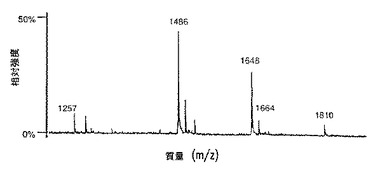
【図9B】
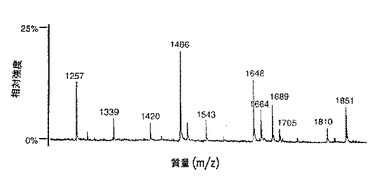
【図9C】
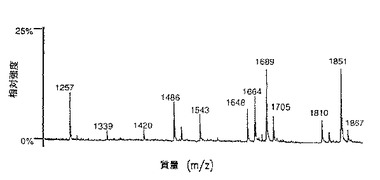
【図9D】
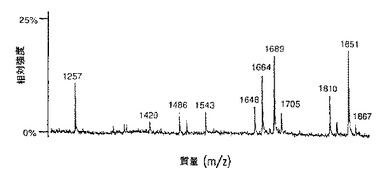
【図9E】
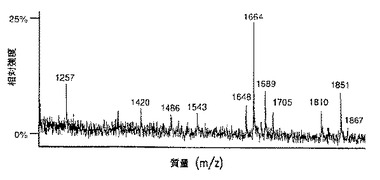
【図10】
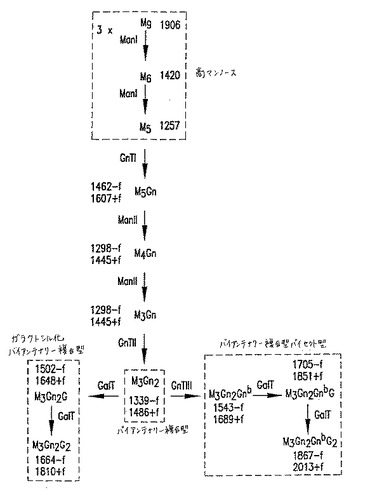
【図11】
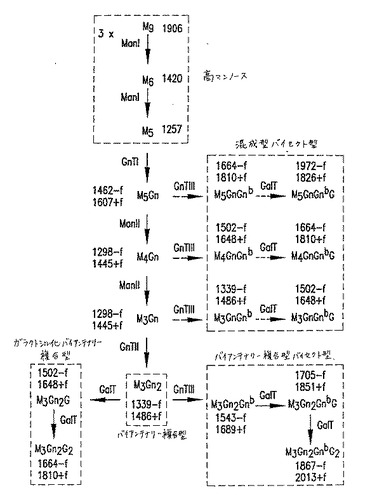
【図12】
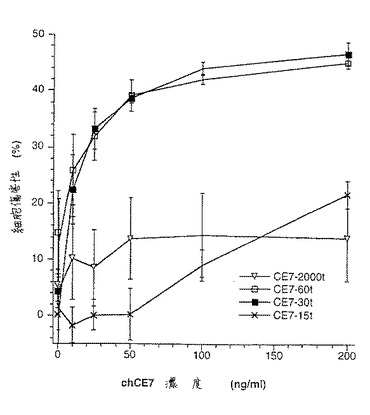
【図13】
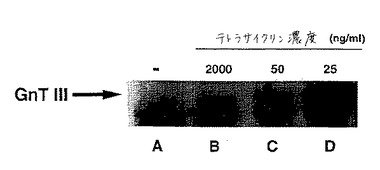
【図14】

【図15】
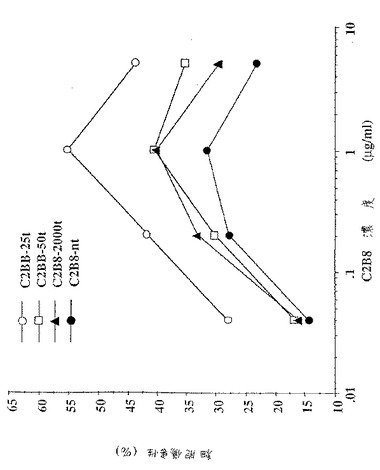
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9A】
【図9B】
【図9C】
【図9D】
【図9E】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【公開番号】特開2012−210209(P2012−210209A)
【公開日】平成24年11月1日(2012.11.1)
【国際特許分類】
【出願番号】特願2012−74560(P2012−74560)
【出願日】平成24年3月28日(2012.3.28)
【分割の表示】特願2008−300179(P2008−300179)の分割
【原出願日】平成11年4月20日(1999.4.20)
【出願人】(505275354)グリカート バイオテクノロジー アクチェンゲゼルシャフト (4)
【Fターム(参考)】
【公開日】平成24年11月1日(2012.11.1)
【国際特許分類】
【出願日】平成24年3月28日(2012.3.28)
【分割の表示】特願2008−300179(P2008−300179)の分割
【原出願日】平成11年4月20日(1999.4.20)
【出願人】(505275354)グリカート バイオテクノロジー アクチェンゲゼルシャフト (4)
【Fターム(参考)】
[ Back to top ]