
抗体
【課題】苦味受容体T2Rに対してアンタゴニスト活性を有する新規な抗体を提供する。
【解決手段】苦味受容体(T2R)タンパクを抗原として、鳥類又は哺乳類に免疫して得られる抗体であって、該苦味受容体に対するアンタゴニスト活性を有することを特徴とする抗体。前記抗原は、ヒト苦味受容体であるhT2R16タンパクであることが好ましく、前記抗原を鶏に免疫して得られる卵から調製された抗体であることがより好ましい。当該抗体は、苦味受容体の研究や機能性食品の原料などとして利用することができる。
【解決手段】苦味受容体(T2R)タンパクを抗原として、鳥類又は哺乳類に免疫して得られる抗体であって、該苦味受容体に対するアンタゴニスト活性を有することを特徴とする抗体。前記抗原は、ヒト苦味受容体であるhT2R16タンパクであることが好ましく、前記抗原を鶏に免疫して得られる卵から調製された抗体であることがより好ましい。当該抗体は、苦味受容体の研究や機能性食品の原料などとして利用することができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、苦味受容体T2Rに対してアンタゴニスト活性を有する抗体に関するものである。
【背景技術】
【0002】
哺乳動物は、少なくとも5つの基本的味覚、すなわち、甘味、苦味、酸味、塩味、及び旨味を持つと考えられている。苦味、甘味、及び旨味の感知は、舌の表面にある味覚受容体細胞で発現するGタンパク共役受容体(GPCR)を介して行なわれ、酸味、塩味の感知は、イオンチャネルを介して行なわれると考えられている。
【0003】
味覚は、生物にとって食物の種類や質に関する情報を得るための重要な役割を持っている。例えば、甘味(糖のシグナル)、旨味(タンパク質や核酸のシグナル)、塩味(ミネラルのシグナル)は、生きていくために必要な栄養素を含む食物であることを知らせ、酸味(腐敗物のシグナル)や苦味(毒物のシグナル)は、有毒な物質や有害な物質を含むことを知らせる。
【0004】
しかし、ヒトにとって苦味や酸味はその強さの程度によってはおいしさの重要な要素となり得るものである。特に嗜好性の強い食品においては苦味や酸味は重要な要素となっている。
【0005】
苦味物質は味蕾の味細胞膜上に特異的に発現する苦味受容体T2Rによって受容される。T2Rは細胞膜を貫通する領域を7つ持った「7回膜貫通型タンパク質」と呼ばれる膜タンパク質であり、細胞内に位置する部分でGタンパク質と共役して苦味刺激を伝達する。ヒトでは約30種類存在していることが知られているが、各T2Rはそれぞれ異なるリガンド特異性を持っており、多くのT2Rのリガンドは未だ不明のままである。
【0006】
これまで、味覚受容体の機能を研究するためにアゴニストやアンタゴニストの検索が行なわれているが、その多くはペプチドや化合物であり、アゴニスト活性やアンタゴニスト活性を有する抗体は報告されていない(特許文献1〜5)。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特表2003−530098号公報
【特許文献2】特開2005−501519号公報
【特許文献3】特表2006−515157号公報
【特許文献4】特開2009−145349号公報
【特許文献5】特開2010−115201号公報
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明は、苦味受容体T2Rに対してアンタゴニスト活性を有する新規な抗体を提供することを目的とする。
【課題を解決するための手段】
【0009】
本発明は、上記課題を解決するために、以下の各発明を包含する。
[1]苦味受容体(T2R)タンパクを抗原として、鳥類又は哺乳類に免疫して得られる抗体であって、該苦味受容体に対するアンタゴニスト活性を有することを特徴とする抗体。
[2]前記抗原は、ヒト苦味受容体であるhT2R16タンパクである[1]記載の抗体。
[3]前記抗原は、hT2R16タンパクのC末端側に緑色蛍光タンパク質(GFP)及び/又はタンパク質タグを付加したものである前記[3]に記載の抗体。
[4]前記抗原を鶏に免疫して得られる卵から調製されたものである前記[1]〜[3]のいずれか一つに記載の抗体。
【発明の効果】
【0010】
本発明によれば、苦味受容体T2Rに対してアンタゴニスト活性を有する抗体を提供することができる。この抗体は、苦味受容体の研究や機能性食品の原料などとして利用することができる。
【図面の簡単な説明】
【0011】
【図1】hT2r16遺伝子の塩基配列及びアミノ酸配列を示す図である。
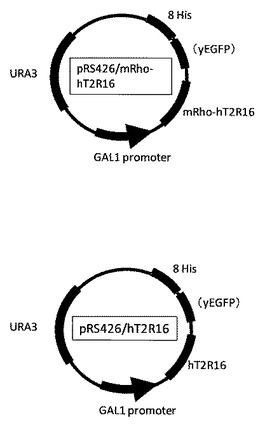
【図2】hT2r16遺伝子を組み込んだ発現ベクター(S.cerevisiae用)の模式図である。
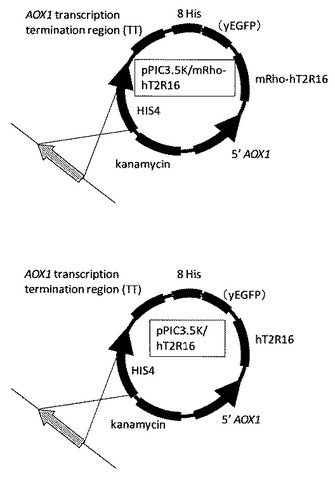
【図3】hT2r16遺伝子を組み込んだ発現ベクター(P.pastris用)の模式図である。
【図4】形質転換体におけるhT2R16融合タンパク質の発現量を調べた結果を示す図である。
【図5】精製したhT2R16融合タンパク質のSDS−PAGEの結果を示す図である。
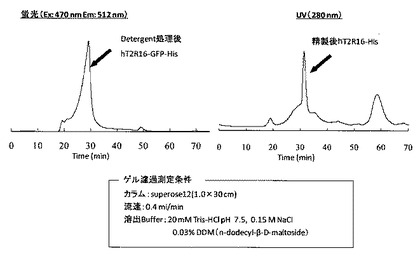
【図6】精製したhT2R16融合タンパク質のゲル濾過クロマトグラフィーの結果を示す図である。
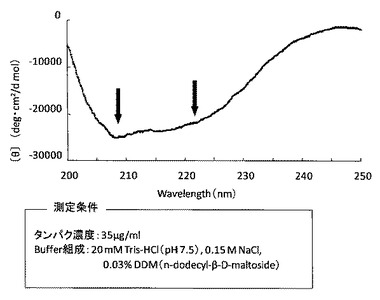
【図7】精製したhT2R16融合タンパク質のCDスペクトル(円偏光2色性)測定の結果を示した図である。
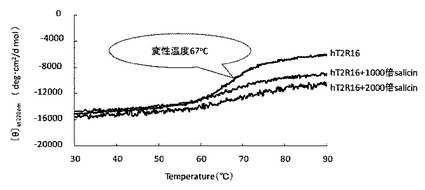
【図8】単離したhT2R16−His融合タンパク質のリガンド結合能をCDスペクトルによって測定した結果を示す図である。
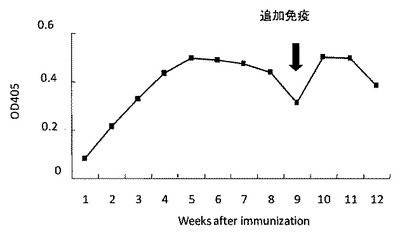
【図9】hT2R16を基質としたELISAによる血中の抗体量の推移を調べた結果を示す図である。
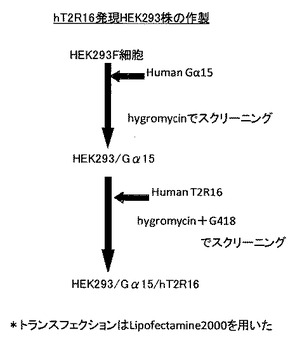
【図10】hT2R16発現HEK293株の作製方法を示した図である。
【図11】hT2R16発現HEK293細胞を用いて、hT2R16抗体添加後の細胞内カルシウム濃度の推移を調べた結果を示す図である。
【図12】サリシン及びhT2R16抗体を用いた官能評価の結果を示す図である。
【発明を実施するための形態】
【0012】
[抗原の調製]
抗原として用いられるT2Rタンパクは、7回膜貫通型タンパク質と呼ばれる膜タンパク質であり、Gタンパク質と共役することから、「Gタンパク質共役受容体」という受容体のグループに属している。T2Rファミリーには多くの種類があり、ヒトでは25種類(hT2R1、hT2R3、hT2R4、hT2R5、hT2R7、hT2R8、hT2R9、hT2R10、hT2R13、hT2R14、hT2R16、hT2R38、hT2R39、hT2R40、hT2R41、hT2R42、hT2R43、hT2R44、hT2R45、hT2R46、hT2R47、hT2R48、hT2R49、hT2R50、hT2R60)が存在し、マウスでは35種類(T2R3、T2R4、T2R9、T2R10、T2R11、T2R12、T2R13、T2R14、T2R15、T2R16、T2R17、T2R18、T2R20、T2R21、T2R22、T2R23、T2R24、T2R26、T2R27、T2R29、T2R30、T2R31、T2R34、T2R35、T2R36、T2R37、T2R38、T2R40、T2R43,T2R104、T2R106、T2R108、T2R109、T2R119、T2R122、T2R139)が存在する(Chandrashekar J.,Hoon M.A. Ryba N.J.et al:The receptors and cells for mammalian taste.Nature,2006;444;288−294)。
【0013】
本発明において抗原として用いるT2Rタンパクは、得られる抗体のアゴニスト・アンタゴニスト活性を調べる必要があるため、リガンドが判明しているものが好ましい。このようなT2Rタンパクとしては、T2R4,44(リガンド:デナトニウム)、マウスT2R5(リガンド:シクロヘキシミド)、T2R10(リガンド:ストリキニーネ)、T2R16(リガンド:サリシン)、T2R38(リガンド:フェニルチオカルバミド)、T2R43,44(リガンド:サッカリン)が例示できる。本発明においては、hT2R16タンパク(図1)が好ましく例示できる。
【0014】
上記T2Rタンパクは、公知の方法でT2R遺伝子をクローニングして、種々の発現系において発現させることで得ることができる。その際、マーカーペプチド(例えば、緑色蛍光タンパク質GFP(Green Fluorescent Protein)など)、タグ配列(例えば、ポリヒスチジンタグ、Mycタグ、FLAGタグなど)、マウスロドプシンなどのアミノ酸配列を付加し、融合タンパク質として発現させてもよい。例えば、マーカーペプチドを付加した場合、形質転換体のスクリーニングが容易になり、タグ配列を付加した場合、目的タンパクの精製が容易になり、マウスロドプシンを付加した場合、目的タンパクの局在性の改善効果が期待できる。
【0015】
上記各T2R遺伝子の塩基配列は公知であり、例えば、hT2Rの場合、hT2R1(GenBank Accession No.AY724812)、hT2R3(GenBank Accession No.AY724961)、hT2R4(GenBank Accession No.AY724958)、hT2R16 (GenBank Accession No.BC095524)である。その塩基配列に基づいてPCRプライマーを作製し、常法に従ってクローニングすることができる。
【0016】
クローニングしたT2R遺伝子を導入する発現ベクターは、宿主により適宜選択すればよく、例えば、宿主が哺乳動物細胞等の場合はpME18SFL3(Genbank Accession AB009864)等、大腸菌の場合はpET3、pET11(ストラタジーン社製)、pGEX(アマシャムファルマシアバイオテク社製)等、酵母の場合はpESP−Iエクスプレッションベクター(ストラタジーン社製)、pRS426(Addgene社製)、pPIC3.5K(Invitrogen社製)等、昆虫細胞の場合はBacPAK6(クロンテック社製)等が用いられる。
【0017】
本発明においては、宿主は特に限定されるものではなく、従来公知の各種細胞(例えば、大腸菌等の細菌、酵母(Saccharomyces cerevisiae、Schizosaccharomyces pombe、Pichia pastoris、Pichia methanolica)、線虫(Caenorhabditis elegans)、アフリカツメガエル(Xenopus laevis)の卵母細胞、動物細胞(例えば、CHO細胞、COS細胞、及びBowes黒色腫細胞)など)を用いることができるが、中でも酵母が好ましく用いられる。
【0018】
上記発現ベクターを宿主細胞に導入する方法、すなわち形質転換法も特に限定されるものではなく、電気穿孔法、リン酸カルシウム法、リポソーム法、DEAEデキストラン法等の公知の方法を用いることができる。
【0019】
得られた形質転換体は、培養・発現誘導した後、培養物などから、濾過、遠心分離、破砕、ゲル濾過クロマトグラフィー、イオン交換クロマトグラフィーなどの慣用的な手法を組み合わせて、T2Rタンパクを回収、精製することができる。
【0020】
[抗体の調製]
上記で得られたT2Rタンパクを抗原として、鳥類又は哺乳類に免疫することによって本発明の抗体を得ることができる。鳥類としては、家禽(鶏、鶉、アヒル等)の他、ダチョウが挙げられるが、鶏(特に産卵種)を用いるのが好ましい。一方、哺乳類としては、マウス、ラット、ウサギ、ウシ、ヤギ、ヒツジ等が挙げられるが、好ましくは畜産に適する産乳動物(特に乳牛)を用いるのが好ましい。本発明においては、抗体の量産性などの点から、鳥類に免疫して、その卵から抗体を調製することが好ましい。
【0021】
鳥類又は哺乳類への免疫は、抗原を免疫増強剤(アジュバント)と共に接種(皮下注射、筋肉注射など)することにより行うことが好ましい。アジュバントとしては、水酸化ナトリウム、水酸化アルミニウム、リン酸カルシウム、リン酸アルミニウム、ミョウバン、ペペス、カルボキシビニルポリマーなどの沈降性アジュバンドや、流動パラフィン、ラノリン、CFA(完全フロイントアジュバント)、IFA(不完全フロイントアジュバント)などの油性アジュバントが例示できる。
【0022】
抗原の接種量は、目的とする抗体が体内に適当量形成され、かつ動物に対して過度の毒性が発揮されないように適宜決定すればよい。また、抗原の接種は数回に分けて行い、高力価が持続するように追加接種することが好ましい。
【0023】
例えば、鳥類への免疫は、Makoto S.et al,Biosci.Biotech.Biochem.,56(2):270−274,1992に記載された方法、Production and Characterization of Anti−human Insulin Antibodies in the Hen’s Egg(Agric.Biol.Chem.,55(8).2141−2143.1991)に記載された方法等が挙げられる。
【0024】
抗原を免疫された鶏が生産した卵から抗体を調製する方法は、公知の方法で行なうことができる。例えば、免疫した鶏の卵中に抗体が適当量生成したことを確認した後、卵を採取し、卵から卵黄液を分離し、得られた卵黄液を粉末化した後、その粉末をエタノール等を用いて脱脂した粉末中から緩衝液を用いて抽出方法、カラギーナンを使用した方法(H.Hatta.et al.,Agric.Biol.chem.,54(10),2531−2535,1990)などによって抗体を調製することができる。
【0025】
上記で得られた抗体は、適宜、塩析やカラムを用いた方法などにより更に精製することもできる。
【0026】
一方、乳牛への免疫は、特開昭57−188523号公報等に記載された方法が挙げられる。抗原を免疫された乳牛が産生する乳から抗体を調製する方法は、公知の方法で行なうことができる。例えば、免疫した哺乳類の乳中に抗体が適当量生成したことを確認した後、乳を採取し、ヒドロキシプロピルメチルセルロースフタレート、ポリエチレングリコールなどを用いる方法により脂質成分を除去した後、適宜、塩析などの方法により抗体を精製することができる。
【実施例】
【0027】
以下、実施例により本発明を詳細に説明するが、本発明はこれらに限定されるものではない。
【0028】
〔実施例1:抗原の調製〕
(1)ヒトT2R16(hT2R16)遺伝子のクローニング
hT2R16遺伝子は、ヒトゲノムDNA(クロンテック社製)を鋳型に用いて表1に示すF1及びR1プライマーを用いてPCR法により増幅しクローニングした。図1にhT2R16遺伝子の塩基配列、hT2R16タンパク質のアミノ酸配列を示す。
【0029】
マウス・ロドプシンのN末端38残基配列遺伝子(mRho)は、Dr.LiquanHuang(モネル研究所、USA)から供与された遺伝子を鋳型にして表1のF2及びR2プライマーを用いてPCR法により増幅した。表1のF3及びR3プライマーを用いて再度PCR法により増幅したhT2R16とmRhoを混合し、これを鋳型として表1のF2及びR3プライマーを用いてPCR法にてmRho−hT2R16融合遺伝子を増幅した後、pcDNA3.1ベクター(Invitrogen社製)にクローニングした。
【0030】
【表1】

【0031】
(2)hT2R16とGFP及び/又はHisタグ融合遺伝子の増幅
pcDNA3.5/mRho−hTAS2R16と表1に示す各プライマーを用いてPCR法により、hT2R16とGFP及び/又はHisタグ融合遺伝子の増幅を行なった。
【0032】
なお、各プライマーは30bpの組み換え配列(下線部)と30bpの苦味受容体遺伝子と同じ配列を持つように設計した(表2)。
【0033】
【表2】
【0034】
得られた各PCR産物は、1%アガロースゲル電気泳動を行い、目的のバンドを確認した。
【0035】
(3)形質転換(S.cerevisiae)の作製
YPD培地で前日から培養しておいた酵母(S.cerevisiae)を、YPD培地30mLでO.D.600nm=0.4になるように調製し、30℃で4時間培養した。培養後、培養液を遠心して菌体を回収し、1Mソルビトールで3回洗浄後、1Mソルビトール:500μLに再懸濁した。
【0036】
pRS426 GAL1 GFP(Proc Natl Acad Sci USA.,104(35),13936−13941(2007)記載の方法で調製)をSmaIで処理(30℃で数時間反応させた後、60℃で30分反応)し、直線化した。
【0037】
酵母培養液:100μL、PCR産物:1μL、SmaI処理したpRS426 GAL1 GFP:1μLをキュベット電極(エレクトロポレーションキュベット:ネッパジーン社)に分注し、エレクトロポレーション(エレクトロポレーション条件:1.5kボルト、200〜700Ω、25μファラデー)を行った。
【0038】
その後、SC−Uプレートに撒布し、約3日後まで30℃でインキュベートし、コロニーが複数できていることを確認した。
【0039】
なお、酵母(S.cerevisiae)は、YPH499株、BJ5464株、BJ2168株、FGY217株(酵母遺伝資源センターより分譲)を用い、それぞれの株で形質転換体を作製した。
【0040】
(4)酵母からのプラスミド抽出
コロニーをピックアップして、一晩液体培養した酵母培養液2〜10mlを1.5mLエッペンに移し、8000rpmで1分間遠心(2回)し、上清を綺麗に除いた。次いで、3.50mM EDTA:293μLと「zymolyase 100T(3mg/mL)」(生化学バイオビジネス社製)7.5μLを加え37℃で1時間反応させた後、8000rpmで1分間遠心(2回)し、上清を綺麗に除いた。そして、「QIAprepSpin Miniprep kit」(QIAGEN社製)を用いてプラスミド抽出を行った。
【0041】
(5)コンピテントセルを使ってのプラスミド増幅
E.coli(DH5α株)をSOB培地10mlでO.D.610nm=0.5になるまで培養した後、10分間冷却し、4℃で冷却しながら集菌した。集菌した菌は氷冷したTB3.2mLに再懸濁し、10分間冷却し、4℃で冷却しながら集菌した。その後、氷冷したTB0.8mLに再懸濁し、DMSO:0.06mLを加え、10分間冷却してから、50μLずつ分注し、液体窒素で急冷し、コンピテントセルを調製した。コンピテントセルは−80℃で保存した。
【0042】
コンピテントセルを氷上で溶解し、上記(4)で得られたプラスミド0.01μg加えて1時間氷冷した後、42℃でヒートショックを30秒与え、すぐに氷冷を2分間行った。
【0043】
その後、SOC培地2mLに懸濁し、37℃で培養し、集菌後、LB培地200μlに懸濁し、プレーティングした。その際、滅菌したコンラージ棒でプレートに均等に伸ばした。
【0044】
37℃で1日培養し、出てきたコロニーを1つ取り、LB培地にて37℃で12〜18時間培養した後、「Sigma GenElute Plasmid Miniprep kit」(シグマ社製)を用いて、プラスミドを精製した。得られた各プラスミドは、以下の通りである。
【0045】
【表3】
【0046】
pRS426 mRho−hT2R16−GFP−Hisタグ、pRS426 hT2R16−GFP−Hisタグの模式図を図2に示す。
【0047】
(6)形質転換体(Pichia pastris)の作製
P.pastris(KM71株、GS115株、SMD1163株(酵母遺伝資源センターより分譲)を用いた。
【0048】
上記(5)で得られたpRS426 mRho−hT2R16又はpRS426 hT2R16と、pPIC3.5K(Invitorogen社製)を、BamHI及びEcoRIを用いて処理(37℃で3時間)し、直線化した。
【0049】
直線化したpRS426 mRho−hT2R16又はpRS426 hT2R16とpPIC3.5Kの各47μLにTE buffer:153μLを加えてそれぞれ合計200μLにした。これにフェノール・クロロホルム溶液:200μLを加えて、ボルテックスをかけて撹拌した後、14000rpmで5分遠心し、上清だけ195μL取り除き、4M NaCl:10μLとエタノール400μLを混ぜ合わせ、−20℃で1時間冷やした。そして、14000rpmで20分遠心し、上清を捨て乾かした後、70%エタノールを加え、14000rpmで10分遠心した。上清を捨て、デシケーターで低圧にし、乾燥させた。なお、酵素処理したpPIC3.5Kのみ常法に従ってCIAP処理を行い、フェノール抽出・エタノール沈殿した後、TE buffer:12μLに溶解させた。
【0050】
目的配列のDNAと直線化したpPIC3.5Kを、「Ligation high Ver.2」(TOYOBO社製)を用いてライゲーションした後、上記(5)と同様の方法にてライゲーションしたプラスミドを増幅した。
【0051】
得られたプラスミド(pPIC3.5K mRho−hT2R16、pPIC3.5KhT2R16)の模式図を図3に示す。
【0052】
SalI処理したpPIC3.5K mRho−hT2R16及びpPIC3.5K hT2R16を用いて、上記(3)と同様にエレクトロポレーション法を用により形質転換を行ない、形質転換体を作製した。
【0053】
(7)得られた形質転換体の培養条件、誘導条件の検討
酵母を培養、誘導する際の温度条件(20℃と30℃)、日数(2日もしくは3日)、培地(SC−U、SD−CAA、YPD)を組み合わせた。誘導後、蛍光分光光度計「FP−6500」(日本分光社)を用いて、whole−cellスペクトル波長測定(励起光:470nm、測定スペクトル:480nm〜600nm)を行った。
【0054】
その結果、S.cerevisiaeの形質転換体においては、2%ガラクトース添加SD−CAA培地で20℃48時間誘導することにした。
【0055】
また、P.pastrisの形質転換体においては、2%ガラクトース添加SD−CAA培地で30℃、24時間後、20℃にして24時間誘導した。
【0056】
(8)hT2R16融合タンパク質の発現の確認
8−1. 走査型共焦点レーザー顕微鏡による局在性確認
上記(7)の条件にて誘導をかけた各酵母の培養液3μLをスライドガラスにのせてカバーガラスを被せた後、走査型共焦点レーザー顕微鏡「Fluovire FV300」(Olympus社製)にて、488nmの励起光を当て、GFPを発光させて、細胞内の局在性を確認したところ、mRho(マウスロドプシン)の有無による局在性の改善は見られなかった。そのため、酵母の発現系ではマウスロドプシンを付加する必要がないことが分かった。
【0057】
8−2. 蛍光分光光度計によるGFPの蛍光測定
GFP buffer(50mM Tris−HCl(pH7.6)、50mM EDTA、10%glycerol、0.12Mソルビトール、Protease inhibitor)を調製した。酵母の培地をGFPbufferに交換して実験に供した。
【0058】
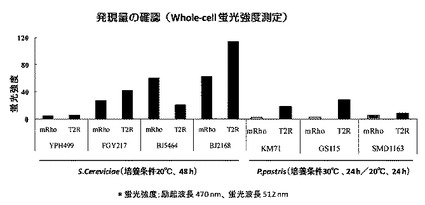
蛍光分光光度計「FP−6500」(日本分光社製)を用いて、スペクトル波長測定(励起光470nm、測定スペクトル:480nm〜600nm)を行った(図4)。図4から、BJ2168株を用いた形質転換体の発現量が最も多いことが分かった。
【0059】
以上の結果から、BJ2168株(pRS426 hT2R16−GFP−Hisタグ)、BJ2168株(pRS426 hT2R16−Hisタグ)を抗原調製に用いることに決定した。
【0060】
(9)hT2R16融合タンパクの調製
9−1.形質転換体の培養
BJ2168株(pRS426 hT2R16−GFP−Hisタグ)を2%グルコース添加SD−CAA培地400mLで30度48時間培養したのち、2%ガラクトース添加SD−CAA培地に交換し、20℃48時間誘導をかけた。
【0061】
9−2.膜画分の調製・hT2R16融合タンパクの精製
得られた培養液を遠心(600rpm、10分で2回)し、上清を除いて酵母を回収し、40mLのcell suspension buffer(50mM Tris−HCl(p7.5)、0.6M Sorbitol、1mM EDTA、Protease inhibitor)に懸濁した。
【0062】
cell suspension bufferと等量のglass beadsをコーニングに入れ、「FastPrep FP100A」(MP−Biomedicals社)もしくは「FastPrep 24 Instrument」(MP−Biomedicals社製)で破砕した後、遠心(6400rpm、10分)し、上清を超遠心管に入れた。また、glass beadsをcell suspension buffer 20mLで洗浄、遠心(6400rpm、10分)して得られた上清を同じく超遠心管に入れた。これらの上清を、遠心(30000rpm、4℃、1時時間)した。遠心後、上清を捨て、沈殿物(膜画分)を4mLのmembrane suspension buffer(20mM Tris−HCl(pH7.5)、0.15M NaCl)に再懸濁し、BCA法によりタンパク濃度を測定した。
【0063】
膜画分タンパク質濃度が3.5mg/mLになるように、dilution membrane buffer(50mM Tris−HCl(pH7.5)、0.15M NaCl、20mM Imidazole、Protease inhibitor)で希釈してから、10倍濃度のdetergent(1%Anzergent、0.2%Cholesteryl hemisuccinate)を加え、4℃で1時間反応させた。
【0064】
30000rpmで4℃1時時間遠心し、上清を回収し、これにNi−NTA agarose樹脂(QIAGEN社)を加え、2時間吸着させた後、hT2R16が吸着したNi−NTA agarose樹脂を混和し、液ごとミニカラム容器にアプライし、カラムの下から出てくるflow throughフラクションを分取した。
【0065】
次にWash Buffer(50mM Tris−HCl(pH7.5)、0.15MNaCl、20mM Imidazole、0.03%DDM)をミニカラムにアプライし、カラムから出てくる溶出フラクションを分取した。
【0066】
その後、Elution Buffer(50mM Tris−HCl(pH7.5)、0.15M NaCl、250mM Imidazole、0.03% DDM)をミニカラムにアプライし、溶出フラクションを5mLずつ同じく6本の試験管に分取した。
【0067】
得られた各フラクションは、ゲル濾過クロマトグラフィー(FSEC)を行い、hT2R16−GFP−Hisタグタンパクの精製を行なった。
【0068】
あらかじめゲル濾過buffer(20mM Tris−HCl(pH7.5)、0.15M NaCl、0.03%DDM)で平衡化しておいた「superrose12(1.0×30cm)」カラム(ファルマシア社製)にサンプルをアプライしてフラクションを分取し、UV波長測定器と蛍光波長測定器(島津社製)により測定してhT2R16−GFP−Hisタグタンパク質を回収した。
【0069】
上記と同様の方法で、BJ2168株(pRS426 hT2R16−Hisタグ)を培養し、膜画分からhT2R16−Hisタグタンパク質を精製した。
【0070】
なお、GPCRは発現量が少ないことが問題となっているが、上記形質転換体を用いることで、T2R16−GFP−Hisタグ融合タンパク質は2〜3mg/1L培養液、T2R16−Hisタグ融合タンパク質は0.5mg/1L培養液のタンパク質を精製することができることが分かった。
【0071】
9−3.精製した各hT2R16融合タンパク質の確認
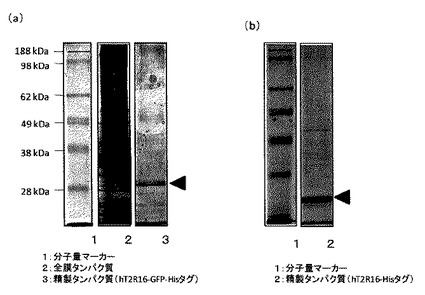
回収した各hT2R16融合タンパク質は、SDS−PAGEにより、単一バンドであることを確認した(図5)。
【0072】
また、目的タンパク質がDetergent処理や精製の過程で重合をおこしていないか調べるために、ゲルろ過クロマトグラフィーを行った(図6)。図6から、Detergent処理後又は精製後のhT2R16融合タンパク質は単一のピークであることが確認でき、Deterdentによって可溶化しても、精製の過程を経ても単量体のまま存在しているものが多いことが分かった。
【0073】
さらに、CDスペクトル(円偏光2色性)測定(円二色性分散計「J−820」、日本分光社製)を行なった(図7)。図7から、精製したタンパク質は208nm及び222nmに負のピークを示しており、α−ヘリックス含量が約64%と推定することができた。この値は、hT2R16アミノ酸配列からの二次構造予測の値に近く、精製したタンパク質が目的のタンパク質であることが示唆された。
【0074】
次に、単離したhT2R16−Hisタグ融合タンパク質のリガンド結合能を調べるために、CDスペクトルによる温度変化測定を行った。温度を30〜90℃まで変化させ、α−ヘリックスのピークである220nmのモル平均楕円率を測定した(図8)。その結果、hT2R16の変性温度が約67℃であることが分かった。
【0075】
また、hT2R16のリガンドであるサリシンを加え、再度CDスペクトル測定を行ったところ、α−ヘリックスの減少がサリシン濃度に依存して抑制された(図8中、1000倍というのは、モル濃度でhT2R16の1000倍ということを表す。)。
【0076】
この結果から、単離したhT2R16はリガンドに対して結合能を持つことが示唆され、結合定数と解離定数を計算すると、解離定数がKd=1.2×10−3〜1.45×10−3M、結合定数がKb=0.69×103〜0.83×103Mとなった。
【0077】
〔実施例2:抗体の調製〕
(1)免疫
実施例1で調製したhT2R16−GFP−Hisタグ融合タンパク質を抗原として用い、以下の条件で産卵鶏(2羽)のももの筋肉に注射して免疫を行なった。
・1回目免疫
hT2R16−GFP−Hisタグ融合タンパク質:0.2mg/羽
アジュバント:CFA(完全フロイントアジュバント)
・2回目免疫(9週間後)
hT2R16−GFP−Hisタグ融合タンパク質:0.5mg/羽
アジュバント:IFA(不完全フロイントアジュバント)
【0078】
なお、免疫期間中は、鶏が抗原抗体反応を示しているかを確認するため、T2R16を基質としたELISAを行い、血中の抗体量の推移を調べ、血中の抗体濃度が上昇していることを確認した(図9)。
【0079】
(2)鶏卵からの抗体精製
2回目免疫後、十分に抗体濃度が上昇した10日目の卵から卵黄を分離した。得られた卵黄を均質化した後、この卵黄液100gを用い、等量の水(100g)を加え、均質になるよう撹拌した。更に、0.15%のλ−カラギーナン水溶液400mLを加え、撹拌後、30分間静置した。静置後、遠心機を用い、8,000rpm、30分間遠心分離し、その上清を回収した。回収した上清液に、硫酸アンモニウムを加え、塩析を行った。塩析を3回実施し、純度が90%以上の抗体を得た。
【0080】
(3)抗体の評価
作製したhT2R16抗体が、hT2R16に対して活性を持っているか否かをG蛋白質の1種であるGα15とhT2R16を発現したHEK293細胞(ヒト胎児腎細胞由来の培養細胞)を用いたカルシウムイメージングとヒトによる官能検査によって評価した。
【0081】
3−1.カルシウムイメージング法による評価
カルシウムイメージング法とは細胞にカルシウム感受性の試薬を負荷し、細胞内のカルシウムイオン濃度を測定する方法であり、味溶液によって苦味受容体が活性化すると細胞内のカルシウムイオン濃度が上昇する。
【0082】
まず、Gα15とhT2R16を発現したHEK293細胞を、図10に示す方法で作製した。作製したhT2R安定発現細胞株は、RT−PCRと免疫染色により細胞表面にhT2R16が発現しているか確認した。
【0083】
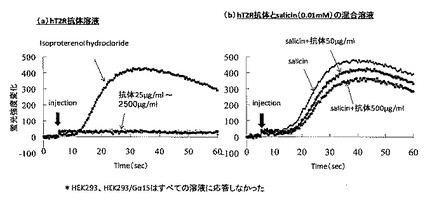
hT2R16を発現したHEK293細胞を用いて、hT2R16抗体添加後の細胞内カルシウム濃度の推移を調べた結果を図11に示す。図11(a)はhT2R16抗体溶液に対する応答を示した結果で、イソプロテノールハイドロクロライドはコントロールである。コントロールに対しては反応するものの、hT2R16抗体溶液に対しては細胞内のカルシウム濃度は全く変化しないことが分かる。この結果から、hT2R16抗体はhT2R16を活性化させないことが分かった。
【0084】
一方、図11(b)はhT2R16抗体とT2R16に対するリガンドであるサリシンの混合溶液に対する応答を示した結果である。hT2R16抗体の濃度依存的にサリシンに対する細胞内のカルシウムイオン濃度の上昇が抑制される傾向があることが分かる。
【0085】
以上の結果から、精製したhT2R16抗体は、hT2R16を活性化しなかったが、サリシンの苦味を抑制する傾向があることが分かった。なお、HEK293と、Gα15のみを発現したHEK293は全てのサンプルに応答しなかった
【0086】
3−2.官能評価
味刺激であるサリシン(0.5、1、3mM)溶液、hT2R16抗体溶液(250μg/mL)、hT2R16抗体(250μg/mL)とサリシン(0.5、1、3mM)の混合溶液を、被験者である13名の成人男女(28.4±2.3才)に呈示した。なお、各溶液の濃度は予備実験を行い、最も効果が大きいと考えられる濃度を選んだ。
【0087】
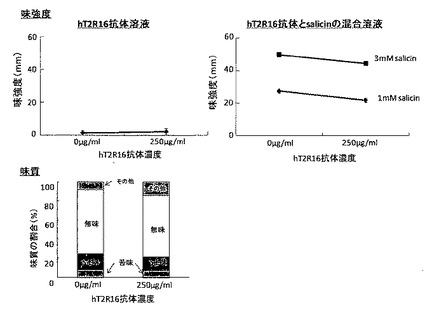
被験者に、感じた味強度をLMS(ラベルドマグニチュードスケール)にチェックしてもらい、スケール下端からのチェックまでの距離を測り、被験者間の相乗平均を求め、平均呈味強度とした。また、各溶液の味質も回答してもらった。その結果を図12に示す。
【0088】
図12から、精製したhT2R16抗体は無味であったが、サリシンの苦味を抑制する傾向があることが分かった。
【0089】
以上の結果から、hT2R16抗体はhT2R16に対するアゴニスト活性はなかったものの、アンタゴニスト活性を有していることが分かった。
【技術分野】
【0001】
本発明は、苦味受容体T2Rに対してアンタゴニスト活性を有する抗体に関するものである。
【背景技術】
【0002】
哺乳動物は、少なくとも5つの基本的味覚、すなわち、甘味、苦味、酸味、塩味、及び旨味を持つと考えられている。苦味、甘味、及び旨味の感知は、舌の表面にある味覚受容体細胞で発現するGタンパク共役受容体(GPCR)を介して行なわれ、酸味、塩味の感知は、イオンチャネルを介して行なわれると考えられている。
【0003】
味覚は、生物にとって食物の種類や質に関する情報を得るための重要な役割を持っている。例えば、甘味(糖のシグナル)、旨味(タンパク質や核酸のシグナル)、塩味(ミネラルのシグナル)は、生きていくために必要な栄養素を含む食物であることを知らせ、酸味(腐敗物のシグナル)や苦味(毒物のシグナル)は、有毒な物質や有害な物質を含むことを知らせる。
【0004】
しかし、ヒトにとって苦味や酸味はその強さの程度によってはおいしさの重要な要素となり得るものである。特に嗜好性の強い食品においては苦味や酸味は重要な要素となっている。
【0005】
苦味物質は味蕾の味細胞膜上に特異的に発現する苦味受容体T2Rによって受容される。T2Rは細胞膜を貫通する領域を7つ持った「7回膜貫通型タンパク質」と呼ばれる膜タンパク質であり、細胞内に位置する部分でGタンパク質と共役して苦味刺激を伝達する。ヒトでは約30種類存在していることが知られているが、各T2Rはそれぞれ異なるリガンド特異性を持っており、多くのT2Rのリガンドは未だ不明のままである。
【0006】
これまで、味覚受容体の機能を研究するためにアゴニストやアンタゴニストの検索が行なわれているが、その多くはペプチドや化合物であり、アゴニスト活性やアンタゴニスト活性を有する抗体は報告されていない(特許文献1〜5)。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】特表2003−530098号公報
【特許文献2】特開2005−501519号公報
【特許文献3】特表2006−515157号公報
【特許文献4】特開2009−145349号公報
【特許文献5】特開2010−115201号公報
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明は、苦味受容体T2Rに対してアンタゴニスト活性を有する新規な抗体を提供することを目的とする。
【課題を解決するための手段】
【0009】
本発明は、上記課題を解決するために、以下の各発明を包含する。
[1]苦味受容体(T2R)タンパクを抗原として、鳥類又は哺乳類に免疫して得られる抗体であって、該苦味受容体に対するアンタゴニスト活性を有することを特徴とする抗体。
[2]前記抗原は、ヒト苦味受容体であるhT2R16タンパクである[1]記載の抗体。
[3]前記抗原は、hT2R16タンパクのC末端側に緑色蛍光タンパク質(GFP)及び/又はタンパク質タグを付加したものである前記[3]に記載の抗体。
[4]前記抗原を鶏に免疫して得られる卵から調製されたものである前記[1]〜[3]のいずれか一つに記載の抗体。
【発明の効果】
【0010】
本発明によれば、苦味受容体T2Rに対してアンタゴニスト活性を有する抗体を提供することができる。この抗体は、苦味受容体の研究や機能性食品の原料などとして利用することができる。
【図面の簡単な説明】
【0011】
【図1】hT2r16遺伝子の塩基配列及びアミノ酸配列を示す図である。
【図2】hT2r16遺伝子を組み込んだ発現ベクター(S.cerevisiae用)の模式図である。
【図3】hT2r16遺伝子を組み込んだ発現ベクター(P.pastris用)の模式図である。
【図4】形質転換体におけるhT2R16融合タンパク質の発現量を調べた結果を示す図である。
【図5】精製したhT2R16融合タンパク質のSDS−PAGEの結果を示す図である。
【図6】精製したhT2R16融合タンパク質のゲル濾過クロマトグラフィーの結果を示す図である。
【図7】精製したhT2R16融合タンパク質のCDスペクトル(円偏光2色性)測定の結果を示した図である。
【図8】単離したhT2R16−His融合タンパク質のリガンド結合能をCDスペクトルによって測定した結果を示す図である。
【図9】hT2R16を基質としたELISAによる血中の抗体量の推移を調べた結果を示す図である。
【図10】hT2R16発現HEK293株の作製方法を示した図である。
【図11】hT2R16発現HEK293細胞を用いて、hT2R16抗体添加後の細胞内カルシウム濃度の推移を調べた結果を示す図である。
【図12】サリシン及びhT2R16抗体を用いた官能評価の結果を示す図である。
【発明を実施するための形態】
【0012】
[抗原の調製]
抗原として用いられるT2Rタンパクは、7回膜貫通型タンパク質と呼ばれる膜タンパク質であり、Gタンパク質と共役することから、「Gタンパク質共役受容体」という受容体のグループに属している。T2Rファミリーには多くの種類があり、ヒトでは25種類(hT2R1、hT2R3、hT2R4、hT2R5、hT2R7、hT2R8、hT2R9、hT2R10、hT2R13、hT2R14、hT2R16、hT2R38、hT2R39、hT2R40、hT2R41、hT2R42、hT2R43、hT2R44、hT2R45、hT2R46、hT2R47、hT2R48、hT2R49、hT2R50、hT2R60)が存在し、マウスでは35種類(T2R3、T2R4、T2R9、T2R10、T2R11、T2R12、T2R13、T2R14、T2R15、T2R16、T2R17、T2R18、T2R20、T2R21、T2R22、T2R23、T2R24、T2R26、T2R27、T2R29、T2R30、T2R31、T2R34、T2R35、T2R36、T2R37、T2R38、T2R40、T2R43,T2R104、T2R106、T2R108、T2R109、T2R119、T2R122、T2R139)が存在する(Chandrashekar J.,Hoon M.A. Ryba N.J.et al:The receptors and cells for mammalian taste.Nature,2006;444;288−294)。
【0013】
本発明において抗原として用いるT2Rタンパクは、得られる抗体のアゴニスト・アンタゴニスト活性を調べる必要があるため、リガンドが判明しているものが好ましい。このようなT2Rタンパクとしては、T2R4,44(リガンド:デナトニウム)、マウスT2R5(リガンド:シクロヘキシミド)、T2R10(リガンド:ストリキニーネ)、T2R16(リガンド:サリシン)、T2R38(リガンド:フェニルチオカルバミド)、T2R43,44(リガンド:サッカリン)が例示できる。本発明においては、hT2R16タンパク(図1)が好ましく例示できる。
【0014】
上記T2Rタンパクは、公知の方法でT2R遺伝子をクローニングして、種々の発現系において発現させることで得ることができる。その際、マーカーペプチド(例えば、緑色蛍光タンパク質GFP(Green Fluorescent Protein)など)、タグ配列(例えば、ポリヒスチジンタグ、Mycタグ、FLAGタグなど)、マウスロドプシンなどのアミノ酸配列を付加し、融合タンパク質として発現させてもよい。例えば、マーカーペプチドを付加した場合、形質転換体のスクリーニングが容易になり、タグ配列を付加した場合、目的タンパクの精製が容易になり、マウスロドプシンを付加した場合、目的タンパクの局在性の改善効果が期待できる。
【0015】
上記各T2R遺伝子の塩基配列は公知であり、例えば、hT2Rの場合、hT2R1(GenBank Accession No.AY724812)、hT2R3(GenBank Accession No.AY724961)、hT2R4(GenBank Accession No.AY724958)、hT2R16 (GenBank Accession No.BC095524)である。その塩基配列に基づいてPCRプライマーを作製し、常法に従ってクローニングすることができる。
【0016】
クローニングしたT2R遺伝子を導入する発現ベクターは、宿主により適宜選択すればよく、例えば、宿主が哺乳動物細胞等の場合はpME18SFL3(Genbank Accession AB009864)等、大腸菌の場合はpET3、pET11(ストラタジーン社製)、pGEX(アマシャムファルマシアバイオテク社製)等、酵母の場合はpESP−Iエクスプレッションベクター(ストラタジーン社製)、pRS426(Addgene社製)、pPIC3.5K(Invitrogen社製)等、昆虫細胞の場合はBacPAK6(クロンテック社製)等が用いられる。
【0017】
本発明においては、宿主は特に限定されるものではなく、従来公知の各種細胞(例えば、大腸菌等の細菌、酵母(Saccharomyces cerevisiae、Schizosaccharomyces pombe、Pichia pastoris、Pichia methanolica)、線虫(Caenorhabditis elegans)、アフリカツメガエル(Xenopus laevis)の卵母細胞、動物細胞(例えば、CHO細胞、COS細胞、及びBowes黒色腫細胞)など)を用いることができるが、中でも酵母が好ましく用いられる。
【0018】
上記発現ベクターを宿主細胞に導入する方法、すなわち形質転換法も特に限定されるものではなく、電気穿孔法、リン酸カルシウム法、リポソーム法、DEAEデキストラン法等の公知の方法を用いることができる。
【0019】
得られた形質転換体は、培養・発現誘導した後、培養物などから、濾過、遠心分離、破砕、ゲル濾過クロマトグラフィー、イオン交換クロマトグラフィーなどの慣用的な手法を組み合わせて、T2Rタンパクを回収、精製することができる。
【0020】
[抗体の調製]
上記で得られたT2Rタンパクを抗原として、鳥類又は哺乳類に免疫することによって本発明の抗体を得ることができる。鳥類としては、家禽(鶏、鶉、アヒル等)の他、ダチョウが挙げられるが、鶏(特に産卵種)を用いるのが好ましい。一方、哺乳類としては、マウス、ラット、ウサギ、ウシ、ヤギ、ヒツジ等が挙げられるが、好ましくは畜産に適する産乳動物(特に乳牛)を用いるのが好ましい。本発明においては、抗体の量産性などの点から、鳥類に免疫して、その卵から抗体を調製することが好ましい。
【0021】
鳥類又は哺乳類への免疫は、抗原を免疫増強剤(アジュバント)と共に接種(皮下注射、筋肉注射など)することにより行うことが好ましい。アジュバントとしては、水酸化ナトリウム、水酸化アルミニウム、リン酸カルシウム、リン酸アルミニウム、ミョウバン、ペペス、カルボキシビニルポリマーなどの沈降性アジュバンドや、流動パラフィン、ラノリン、CFA(完全フロイントアジュバント)、IFA(不完全フロイントアジュバント)などの油性アジュバントが例示できる。
【0022】
抗原の接種量は、目的とする抗体が体内に適当量形成され、かつ動物に対して過度の毒性が発揮されないように適宜決定すればよい。また、抗原の接種は数回に分けて行い、高力価が持続するように追加接種することが好ましい。
【0023】
例えば、鳥類への免疫は、Makoto S.et al,Biosci.Biotech.Biochem.,56(2):270−274,1992に記載された方法、Production and Characterization of Anti−human Insulin Antibodies in the Hen’s Egg(Agric.Biol.Chem.,55(8).2141−2143.1991)に記載された方法等が挙げられる。
【0024】
抗原を免疫された鶏が生産した卵から抗体を調製する方法は、公知の方法で行なうことができる。例えば、免疫した鶏の卵中に抗体が適当量生成したことを確認した後、卵を採取し、卵から卵黄液を分離し、得られた卵黄液を粉末化した後、その粉末をエタノール等を用いて脱脂した粉末中から緩衝液を用いて抽出方法、カラギーナンを使用した方法(H.Hatta.et al.,Agric.Biol.chem.,54(10),2531−2535,1990)などによって抗体を調製することができる。
【0025】
上記で得られた抗体は、適宜、塩析やカラムを用いた方法などにより更に精製することもできる。
【0026】
一方、乳牛への免疫は、特開昭57−188523号公報等に記載された方法が挙げられる。抗原を免疫された乳牛が産生する乳から抗体を調製する方法は、公知の方法で行なうことができる。例えば、免疫した哺乳類の乳中に抗体が適当量生成したことを確認した後、乳を採取し、ヒドロキシプロピルメチルセルロースフタレート、ポリエチレングリコールなどを用いる方法により脂質成分を除去した後、適宜、塩析などの方法により抗体を精製することができる。
【実施例】
【0027】
以下、実施例により本発明を詳細に説明するが、本発明はこれらに限定されるものではない。
【0028】
〔実施例1:抗原の調製〕
(1)ヒトT2R16(hT2R16)遺伝子のクローニング
hT2R16遺伝子は、ヒトゲノムDNA(クロンテック社製)を鋳型に用いて表1に示すF1及びR1プライマーを用いてPCR法により増幅しクローニングした。図1にhT2R16遺伝子の塩基配列、hT2R16タンパク質のアミノ酸配列を示す。
【0029】
マウス・ロドプシンのN末端38残基配列遺伝子(mRho)は、Dr.LiquanHuang(モネル研究所、USA)から供与された遺伝子を鋳型にして表1のF2及びR2プライマーを用いてPCR法により増幅した。表1のF3及びR3プライマーを用いて再度PCR法により増幅したhT2R16とmRhoを混合し、これを鋳型として表1のF2及びR3プライマーを用いてPCR法にてmRho−hT2R16融合遺伝子を増幅した後、pcDNA3.1ベクター(Invitrogen社製)にクローニングした。
【0030】
【表1】
【0031】
(2)hT2R16とGFP及び/又はHisタグ融合遺伝子の増幅
pcDNA3.5/mRho−hTAS2R16と表1に示す各プライマーを用いてPCR法により、hT2R16とGFP及び/又はHisタグ融合遺伝子の増幅を行なった。
【0032】
なお、各プライマーは30bpの組み換え配列(下線部)と30bpの苦味受容体遺伝子と同じ配列を持つように設計した(表2)。
【0033】
【表2】
【0034】
得られた各PCR産物は、1%アガロースゲル電気泳動を行い、目的のバンドを確認した。
【0035】
(3)形質転換(S.cerevisiae)の作製
YPD培地で前日から培養しておいた酵母(S.cerevisiae)を、YPD培地30mLでO.D.600nm=0.4になるように調製し、30℃で4時間培養した。培養後、培養液を遠心して菌体を回収し、1Mソルビトールで3回洗浄後、1Mソルビトール:500μLに再懸濁した。
【0036】
pRS426 GAL1 GFP(Proc Natl Acad Sci USA.,104(35),13936−13941(2007)記載の方法で調製)をSmaIで処理(30℃で数時間反応させた後、60℃で30分反応)し、直線化した。
【0037】
酵母培養液:100μL、PCR産物:1μL、SmaI処理したpRS426 GAL1 GFP:1μLをキュベット電極(エレクトロポレーションキュベット:ネッパジーン社)に分注し、エレクトロポレーション(エレクトロポレーション条件:1.5kボルト、200〜700Ω、25μファラデー)を行った。
【0038】
その後、SC−Uプレートに撒布し、約3日後まで30℃でインキュベートし、コロニーが複数できていることを確認した。
【0039】
なお、酵母(S.cerevisiae)は、YPH499株、BJ5464株、BJ2168株、FGY217株(酵母遺伝資源センターより分譲)を用い、それぞれの株で形質転換体を作製した。
【0040】
(4)酵母からのプラスミド抽出
コロニーをピックアップして、一晩液体培養した酵母培養液2〜10mlを1.5mLエッペンに移し、8000rpmで1分間遠心(2回)し、上清を綺麗に除いた。次いで、3.50mM EDTA:293μLと「zymolyase 100T(3mg/mL)」(生化学バイオビジネス社製)7.5μLを加え37℃で1時間反応させた後、8000rpmで1分間遠心(2回)し、上清を綺麗に除いた。そして、「QIAprepSpin Miniprep kit」(QIAGEN社製)を用いてプラスミド抽出を行った。
【0041】
(5)コンピテントセルを使ってのプラスミド増幅
E.coli(DH5α株)をSOB培地10mlでO.D.610nm=0.5になるまで培養した後、10分間冷却し、4℃で冷却しながら集菌した。集菌した菌は氷冷したTB3.2mLに再懸濁し、10分間冷却し、4℃で冷却しながら集菌した。その後、氷冷したTB0.8mLに再懸濁し、DMSO:0.06mLを加え、10分間冷却してから、50μLずつ分注し、液体窒素で急冷し、コンピテントセルを調製した。コンピテントセルは−80℃で保存した。
【0042】
コンピテントセルを氷上で溶解し、上記(4)で得られたプラスミド0.01μg加えて1時間氷冷した後、42℃でヒートショックを30秒与え、すぐに氷冷を2分間行った。
【0043】
その後、SOC培地2mLに懸濁し、37℃で培養し、集菌後、LB培地200μlに懸濁し、プレーティングした。その際、滅菌したコンラージ棒でプレートに均等に伸ばした。
【0044】
37℃で1日培養し、出てきたコロニーを1つ取り、LB培地にて37℃で12〜18時間培養した後、「Sigma GenElute Plasmid Miniprep kit」(シグマ社製)を用いて、プラスミドを精製した。得られた各プラスミドは、以下の通りである。
【0045】
【表3】
【0046】
pRS426 mRho−hT2R16−GFP−Hisタグ、pRS426 hT2R16−GFP−Hisタグの模式図を図2に示す。
【0047】
(6)形質転換体(Pichia pastris)の作製
P.pastris(KM71株、GS115株、SMD1163株(酵母遺伝資源センターより分譲)を用いた。
【0048】
上記(5)で得られたpRS426 mRho−hT2R16又はpRS426 hT2R16と、pPIC3.5K(Invitorogen社製)を、BamHI及びEcoRIを用いて処理(37℃で3時間)し、直線化した。
【0049】
直線化したpRS426 mRho−hT2R16又はpRS426 hT2R16とpPIC3.5Kの各47μLにTE buffer:153μLを加えてそれぞれ合計200μLにした。これにフェノール・クロロホルム溶液:200μLを加えて、ボルテックスをかけて撹拌した後、14000rpmで5分遠心し、上清だけ195μL取り除き、4M NaCl:10μLとエタノール400μLを混ぜ合わせ、−20℃で1時間冷やした。そして、14000rpmで20分遠心し、上清を捨て乾かした後、70%エタノールを加え、14000rpmで10分遠心した。上清を捨て、デシケーターで低圧にし、乾燥させた。なお、酵素処理したpPIC3.5Kのみ常法に従ってCIAP処理を行い、フェノール抽出・エタノール沈殿した後、TE buffer:12μLに溶解させた。
【0050】
目的配列のDNAと直線化したpPIC3.5Kを、「Ligation high Ver.2」(TOYOBO社製)を用いてライゲーションした後、上記(5)と同様の方法にてライゲーションしたプラスミドを増幅した。
【0051】
得られたプラスミド(pPIC3.5K mRho−hT2R16、pPIC3.5KhT2R16)の模式図を図3に示す。
【0052】
SalI処理したpPIC3.5K mRho−hT2R16及びpPIC3.5K hT2R16を用いて、上記(3)と同様にエレクトロポレーション法を用により形質転換を行ない、形質転換体を作製した。
【0053】
(7)得られた形質転換体の培養条件、誘導条件の検討
酵母を培養、誘導する際の温度条件(20℃と30℃)、日数(2日もしくは3日)、培地(SC−U、SD−CAA、YPD)を組み合わせた。誘導後、蛍光分光光度計「FP−6500」(日本分光社)を用いて、whole−cellスペクトル波長測定(励起光:470nm、測定スペクトル:480nm〜600nm)を行った。
【0054】
その結果、S.cerevisiaeの形質転換体においては、2%ガラクトース添加SD−CAA培地で20℃48時間誘導することにした。
【0055】
また、P.pastrisの形質転換体においては、2%ガラクトース添加SD−CAA培地で30℃、24時間後、20℃にして24時間誘導した。
【0056】
(8)hT2R16融合タンパク質の発現の確認
8−1. 走査型共焦点レーザー顕微鏡による局在性確認
上記(7)の条件にて誘導をかけた各酵母の培養液3μLをスライドガラスにのせてカバーガラスを被せた後、走査型共焦点レーザー顕微鏡「Fluovire FV300」(Olympus社製)にて、488nmの励起光を当て、GFPを発光させて、細胞内の局在性を確認したところ、mRho(マウスロドプシン)の有無による局在性の改善は見られなかった。そのため、酵母の発現系ではマウスロドプシンを付加する必要がないことが分かった。
【0057】
8−2. 蛍光分光光度計によるGFPの蛍光測定
GFP buffer(50mM Tris−HCl(pH7.6)、50mM EDTA、10%glycerol、0.12Mソルビトール、Protease inhibitor)を調製した。酵母の培地をGFPbufferに交換して実験に供した。
【0058】
蛍光分光光度計「FP−6500」(日本分光社製)を用いて、スペクトル波長測定(励起光470nm、測定スペクトル:480nm〜600nm)を行った(図4)。図4から、BJ2168株を用いた形質転換体の発現量が最も多いことが分かった。
【0059】
以上の結果から、BJ2168株(pRS426 hT2R16−GFP−Hisタグ)、BJ2168株(pRS426 hT2R16−Hisタグ)を抗原調製に用いることに決定した。
【0060】
(9)hT2R16融合タンパクの調製
9−1.形質転換体の培養
BJ2168株(pRS426 hT2R16−GFP−Hisタグ)を2%グルコース添加SD−CAA培地400mLで30度48時間培養したのち、2%ガラクトース添加SD−CAA培地に交換し、20℃48時間誘導をかけた。
【0061】
9−2.膜画分の調製・hT2R16融合タンパクの精製
得られた培養液を遠心(600rpm、10分で2回)し、上清を除いて酵母を回収し、40mLのcell suspension buffer(50mM Tris−HCl(p7.5)、0.6M Sorbitol、1mM EDTA、Protease inhibitor)に懸濁した。
【0062】
cell suspension bufferと等量のglass beadsをコーニングに入れ、「FastPrep FP100A」(MP−Biomedicals社)もしくは「FastPrep 24 Instrument」(MP−Biomedicals社製)で破砕した後、遠心(6400rpm、10分)し、上清を超遠心管に入れた。また、glass beadsをcell suspension buffer 20mLで洗浄、遠心(6400rpm、10分)して得られた上清を同じく超遠心管に入れた。これらの上清を、遠心(30000rpm、4℃、1時時間)した。遠心後、上清を捨て、沈殿物(膜画分)を4mLのmembrane suspension buffer(20mM Tris−HCl(pH7.5)、0.15M NaCl)に再懸濁し、BCA法によりタンパク濃度を測定した。
【0063】
膜画分タンパク質濃度が3.5mg/mLになるように、dilution membrane buffer(50mM Tris−HCl(pH7.5)、0.15M NaCl、20mM Imidazole、Protease inhibitor)で希釈してから、10倍濃度のdetergent(1%Anzergent、0.2%Cholesteryl hemisuccinate)を加え、4℃で1時間反応させた。
【0064】
30000rpmで4℃1時時間遠心し、上清を回収し、これにNi−NTA agarose樹脂(QIAGEN社)を加え、2時間吸着させた後、hT2R16が吸着したNi−NTA agarose樹脂を混和し、液ごとミニカラム容器にアプライし、カラムの下から出てくるflow throughフラクションを分取した。
【0065】
次にWash Buffer(50mM Tris−HCl(pH7.5)、0.15MNaCl、20mM Imidazole、0.03%DDM)をミニカラムにアプライし、カラムから出てくる溶出フラクションを分取した。
【0066】
その後、Elution Buffer(50mM Tris−HCl(pH7.5)、0.15M NaCl、250mM Imidazole、0.03% DDM)をミニカラムにアプライし、溶出フラクションを5mLずつ同じく6本の試験管に分取した。
【0067】
得られた各フラクションは、ゲル濾過クロマトグラフィー(FSEC)を行い、hT2R16−GFP−Hisタグタンパクの精製を行なった。
【0068】
あらかじめゲル濾過buffer(20mM Tris−HCl(pH7.5)、0.15M NaCl、0.03%DDM)で平衡化しておいた「superrose12(1.0×30cm)」カラム(ファルマシア社製)にサンプルをアプライしてフラクションを分取し、UV波長測定器と蛍光波長測定器(島津社製)により測定してhT2R16−GFP−Hisタグタンパク質を回収した。
【0069】
上記と同様の方法で、BJ2168株(pRS426 hT2R16−Hisタグ)を培養し、膜画分からhT2R16−Hisタグタンパク質を精製した。
【0070】
なお、GPCRは発現量が少ないことが問題となっているが、上記形質転換体を用いることで、T2R16−GFP−Hisタグ融合タンパク質は2〜3mg/1L培養液、T2R16−Hisタグ融合タンパク質は0.5mg/1L培養液のタンパク質を精製することができることが分かった。
【0071】
9−3.精製した各hT2R16融合タンパク質の確認
回収した各hT2R16融合タンパク質は、SDS−PAGEにより、単一バンドであることを確認した(図5)。
【0072】
また、目的タンパク質がDetergent処理や精製の過程で重合をおこしていないか調べるために、ゲルろ過クロマトグラフィーを行った(図6)。図6から、Detergent処理後又は精製後のhT2R16融合タンパク質は単一のピークであることが確認でき、Deterdentによって可溶化しても、精製の過程を経ても単量体のまま存在しているものが多いことが分かった。
【0073】
さらに、CDスペクトル(円偏光2色性)測定(円二色性分散計「J−820」、日本分光社製)を行なった(図7)。図7から、精製したタンパク質は208nm及び222nmに負のピークを示しており、α−ヘリックス含量が約64%と推定することができた。この値は、hT2R16アミノ酸配列からの二次構造予測の値に近く、精製したタンパク質が目的のタンパク質であることが示唆された。
【0074】
次に、単離したhT2R16−Hisタグ融合タンパク質のリガンド結合能を調べるために、CDスペクトルによる温度変化測定を行った。温度を30〜90℃まで変化させ、α−ヘリックスのピークである220nmのモル平均楕円率を測定した(図8)。その結果、hT2R16の変性温度が約67℃であることが分かった。
【0075】
また、hT2R16のリガンドであるサリシンを加え、再度CDスペクトル測定を行ったところ、α−ヘリックスの減少がサリシン濃度に依存して抑制された(図8中、1000倍というのは、モル濃度でhT2R16の1000倍ということを表す。)。
【0076】
この結果から、単離したhT2R16はリガンドに対して結合能を持つことが示唆され、結合定数と解離定数を計算すると、解離定数がKd=1.2×10−3〜1.45×10−3M、結合定数がKb=0.69×103〜0.83×103Mとなった。
【0077】
〔実施例2:抗体の調製〕
(1)免疫
実施例1で調製したhT2R16−GFP−Hisタグ融合タンパク質を抗原として用い、以下の条件で産卵鶏(2羽)のももの筋肉に注射して免疫を行なった。
・1回目免疫
hT2R16−GFP−Hisタグ融合タンパク質:0.2mg/羽
アジュバント:CFA(完全フロイントアジュバント)
・2回目免疫(9週間後)
hT2R16−GFP−Hisタグ融合タンパク質:0.5mg/羽
アジュバント:IFA(不完全フロイントアジュバント)
【0078】
なお、免疫期間中は、鶏が抗原抗体反応を示しているかを確認するため、T2R16を基質としたELISAを行い、血中の抗体量の推移を調べ、血中の抗体濃度が上昇していることを確認した(図9)。
【0079】
(2)鶏卵からの抗体精製
2回目免疫後、十分に抗体濃度が上昇した10日目の卵から卵黄を分離した。得られた卵黄を均質化した後、この卵黄液100gを用い、等量の水(100g)を加え、均質になるよう撹拌した。更に、0.15%のλ−カラギーナン水溶液400mLを加え、撹拌後、30分間静置した。静置後、遠心機を用い、8,000rpm、30分間遠心分離し、その上清を回収した。回収した上清液に、硫酸アンモニウムを加え、塩析を行った。塩析を3回実施し、純度が90%以上の抗体を得た。
【0080】
(3)抗体の評価
作製したhT2R16抗体が、hT2R16に対して活性を持っているか否かをG蛋白質の1種であるGα15とhT2R16を発現したHEK293細胞(ヒト胎児腎細胞由来の培養細胞)を用いたカルシウムイメージングとヒトによる官能検査によって評価した。
【0081】
3−1.カルシウムイメージング法による評価
カルシウムイメージング法とは細胞にカルシウム感受性の試薬を負荷し、細胞内のカルシウムイオン濃度を測定する方法であり、味溶液によって苦味受容体が活性化すると細胞内のカルシウムイオン濃度が上昇する。
【0082】
まず、Gα15とhT2R16を発現したHEK293細胞を、図10に示す方法で作製した。作製したhT2R安定発現細胞株は、RT−PCRと免疫染色により細胞表面にhT2R16が発現しているか確認した。
【0083】
hT2R16を発現したHEK293細胞を用いて、hT2R16抗体添加後の細胞内カルシウム濃度の推移を調べた結果を図11に示す。図11(a)はhT2R16抗体溶液に対する応答を示した結果で、イソプロテノールハイドロクロライドはコントロールである。コントロールに対しては反応するものの、hT2R16抗体溶液に対しては細胞内のカルシウム濃度は全く変化しないことが分かる。この結果から、hT2R16抗体はhT2R16を活性化させないことが分かった。
【0084】
一方、図11(b)はhT2R16抗体とT2R16に対するリガンドであるサリシンの混合溶液に対する応答を示した結果である。hT2R16抗体の濃度依存的にサリシンに対する細胞内のカルシウムイオン濃度の上昇が抑制される傾向があることが分かる。
【0085】
以上の結果から、精製したhT2R16抗体は、hT2R16を活性化しなかったが、サリシンの苦味を抑制する傾向があることが分かった。なお、HEK293と、Gα15のみを発現したHEK293は全てのサンプルに応答しなかった
【0086】
3−2.官能評価
味刺激であるサリシン(0.5、1、3mM)溶液、hT2R16抗体溶液(250μg/mL)、hT2R16抗体(250μg/mL)とサリシン(0.5、1、3mM)の混合溶液を、被験者である13名の成人男女(28.4±2.3才)に呈示した。なお、各溶液の濃度は予備実験を行い、最も効果が大きいと考えられる濃度を選んだ。
【0087】
被験者に、感じた味強度をLMS(ラベルドマグニチュードスケール)にチェックしてもらい、スケール下端からのチェックまでの距離を測り、被験者間の相乗平均を求め、平均呈味強度とした。また、各溶液の味質も回答してもらった。その結果を図12に示す。
【0088】
図12から、精製したhT2R16抗体は無味であったが、サリシンの苦味を抑制する傾向があることが分かった。
【0089】
以上の結果から、hT2R16抗体はhT2R16に対するアゴニスト活性はなかったものの、アンタゴニスト活性を有していることが分かった。
【特許請求の範囲】
【請求項1】
苦味受容体(T2R)タンパクを抗原として、鳥類又は哺乳類に免疫して得られる抗体であって、該苦味受容体に対するアンタゴニスト活性を有することを特徴とする抗体。
【請求項2】
前記抗原は、ヒト苦味受容体であるhT2R16タンパクである請求項1記載の抗体。
【請求項3】
前記抗原は、hT2R16タンパクのC末端側に緑色蛍光タンパク質(GFP)及び/又はタンパク質タグを付加したものである請求項2記載の抗体。
【請求項4】
前記抗原を鶏に免疫して得られる卵から調製されたものである請求項1から3のいずれか一つに記載の抗体。
【請求項1】
苦味受容体(T2R)タンパクを抗原として、鳥類又は哺乳類に免疫して得られる抗体であって、該苦味受容体に対するアンタゴニスト活性を有することを特徴とする抗体。
【請求項2】
前記抗原は、ヒト苦味受容体であるhT2R16タンパクである請求項1記載の抗体。
【請求項3】
前記抗原は、hT2R16タンパクのC末端側に緑色蛍光タンパク質(GFP)及び/又はタンパク質タグを付加したものである請求項2記載の抗体。
【請求項4】
前記抗原を鶏に免疫して得られる卵から調製されたものである請求項1から3のいずれか一つに記載の抗体。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公開番号】特開2012−51867(P2012−51867A)
【公開日】平成24年3月15日(2012.3.15)
【国際特許分類】
【出願番号】特願2010−208503(P2010−208503)
【出願日】平成22年8月31日(2010.8.31)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成22年3月5日 社団法人日本農芸化学会発行の「大会講演要旨集 2010年度(平成22年度)大会[東京]」に発表
【出願人】(500101243)株式会社ファーマフーズ (30)
【出願人】(510249667)
【出願人】(598096991)学校法人東京農業大学 (85)
【Fターム(参考)】
【公開日】平成24年3月15日(2012.3.15)
【国際特許分類】
【出願日】平成22年8月31日(2010.8.31)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成22年3月5日 社団法人日本農芸化学会発行の「大会講演要旨集 2010年度(平成22年度)大会[東京]」に発表
【出願人】(500101243)株式会社ファーマフーズ (30)
【出願人】(510249667)
【出願人】(598096991)学校法人東京農業大学 (85)
【Fターム(参考)】
[ Back to top ]