
抗凝固薬の解毒剤
本発明は、抗凝固薬、特にダビガトランに対する抗体分子、およびそのような抗凝固薬の解毒剤としてのそれらの使用に関する。

【発明の詳細な説明】
【技術分野】
【0001】
発明の背景
技術分野
本発明は、医学の分野、特に抗凝固治療の分野に関するものである。
【0002】
背景の情報
抗凝固薬は、血液凝固を防止する物質であって;すなわち抗凝固薬は血液が凝固するのを妨げる。抗凝固薬は、血栓性障害のための薬物療法としてのヒトの治療、例えば素因をもつ者における深部静脈血栓、肺塞栓、心筋梗塞および脳卒中の一次および二次予防に広く使用される。
【0003】
重要なクラスの経口抗凝固薬、例えばワルファリンを含めたクマリンは、ビタミンKの効果と拮抗することによって作用する。第二のクラスの化合物は、アンチトロンビンIIIまたはヘパリンコファクターIIなどのコファクターを介して間接的に血液凝固を阻害する。これには、アンチトロンビンIIIを介して主に第Xa因子(および低い程度にはトロンビン)の阻害を触媒するいくつかの低分子量ヘパリン製品(ベミパリン、セルトパリン、ダルテパリン、エノキサパリン、ナドロパリン、パルナパリン、レビパリン、チンザパリン)が含まれる。より小さなオリゴ糖(フォンダパリヌクス、イドラパリヌクス)は、アンチトロンビンIIIを介して第Xa因子だけを阻害する。ヘパリノイド(ダナパロイド、スロデキシド、デルマタン硫酸)は、両方のコファクターを介して作用し、第Xa因子とトロンビンの両方を阻害する。第三のクラスは、血液凝固の直接阻害剤に相当する。直接第Xa因子阻害剤には、アピキサバン、エドキサバン、オタミキサバン、リバロキサバンが含まれ、直接トロンビン阻害剤には、二価ヒルジン(ビバリルジン、レピルジン、デシルジン)、ならびに一価化合物であるアルガトロバンおよびダビガトランが含まれる。
【0004】
血液凝固は出血を止める生物学的メカニズムであるので、抗凝固治療の副作用は、望まれない出血事象のことがある。したがって、そのような抗凝固薬関連出血事象が起こったときにそれを止めることができる解毒剤を提供することが望ましい(Zikria and Ansell, Current Opinion in Hematology 2009, 16(5): 347-356)。これを達成するための一方法は、投与後に患者に存在する抗凝固化合物の活性を中和することによる。
【0005】
抗凝固薬の現在入手可能な解毒剤は、プロタミン(ヘパリンの中和用)、およびワルファリンのようなビタミンKアンタゴニストを中和するためのビタミンKである。新鮮凍結血漿および組換え第VIIa因子もまた、大外傷または重度出血を被った、低分子量ヘパリン処置下の患者における非特異的解毒剤として使用されている(Lauritzen, B. et al, Blood, 2005, 607A-608A.)。ヘパリンまたは低分子量ヘパリン解毒剤としてのプロタミンフラグメント(米国特許第6,624,141号)および小型合成ペプチド(米国特許第6,200,955号);ならびにトロンビン阻害剤用の解毒剤としてのトロンビンムテイン(米国特許第6,060,300号)も報告されている。プロトロンビンの中間体および誘導体は、ヒルジンおよび合成トロンビン阻害剤に対する解毒剤として報告されている(米国特許第5,817,309号および第6,086,871号)。直接第Xa因子阻害剤については、不活性第Xa因子アナログが解毒剤として提案されている(国際公開公報第2009042962号)。さらに、組換え第VIIa因子が、フォンダパリヌクスおよびイドラパリヌクスなどの間接アンチトロンビンIII依存性第Xa因子阻害剤の効果をリバースするために使用されている(Bijsterveld, NR et al, Circulation, 2002, 106: 2550-2554; Bijsterveld, NR et al, British J. of Haematology, 2004 (124): 653-658)。抗凝固薬の効果をリバースする方法の総説は、Schulman and Bijsterveld, Transfusion Medicine Reviews 2007、21(1): 37-48に提供されている。
【0006】
抗凝固治療についての改良された解毒剤、特に特異的解毒剤がこれまでのところ開示されていないダビガトランのような直接トロンビン阻害剤のための解毒剤を提供する必要性がある。
【0007】
発明の簡単な概要
一局面では、本発明は、抗凝固薬の活性を中和可能な抗体分子に関する。
【0008】
さらなる局面では、抗体分子は、抗凝固薬に対する結合特異性を有する。
【0009】
さらなる局面では、抗凝固薬は、直接トロンビン阻害剤、第Xa因子阻害剤、またはビタミンKアンタゴニストである。
【0010】
さらなる局面では、抗凝固薬は、ダビガトラン、アルガトロバン、メラガトラン、キシメラガトラン、ヒルジン、ビバリルジン、レピルジン、デシルジン、アピキサバン、オタミキサバン、エドキサバン、リバロキサバン、デフィブロチド、ラマトロバン、アンチトロンビンIII、またはドロトレコギンアルファである。
【0011】
さらなる態様では、抗凝固薬は、一般式
【0012】
【化1】
[式中、
Aは、基Hetのベンゾ、ピリドまたはチエノ部分に結合されたカルボニルまたはスルホニル基を表し、
Bは、基Arに結合されたメチレン基が酸素もしくは硫黄原子によって、または−NR1−基によって置き換えられていてもよいエチレン基を表し、
ここで、R1は、水素原子またはC1−4−アルキル基を表し、
Eは、RbNH−C(=NH)−基を表し、
ここで、Rbは、水素原子、ヒドロキシ、C1−9−アルコキシカルボニル、シクロヘキシルオキシカルボニル、フェニル−C1−3−アルコキシカルボニル、ベンゾイル、p−C1−3−アルキル−ベンゾイルまたはピリジノイル基を表し、一方で上記C1−9−アルコキシカルボニル基の2位のエトキシ部分は、C1−3−アルキルスルホニルまたは2−(C1−3−アルコキシ)−エチル基によって更に置換されていてもよく、
Arは、場合により塩素原子によって、またはメチル、エチルもしくはメトキシ基によって置換されている1,4−フェニレン基を表すか、あるいはArは2,5−チエニレン基を表し、
Hetは、1−(C1−3−アルキル)−2,5−ベンゾイミダゾリレン、1−シクロプロピル−2,5−ベンゾイミダゾリレン、2,5−ベンゾチアゾリレン、1−(C1−3−アルキル)−2,5−インドリレン、1−(C1−3−アルキル)−2,5−イミダゾ[4,5−b]ピリジニレン、3−(C1−3−アルキル)−2,7−イミダゾ[1,2−a]ピリジニレンまたは1−(C1−3−アルキル)−2,5−チエノ[2,3−d]イミダゾリレン基を表し、そして
Raは、R2NR3−基を表し、
ここで、R2は、カルボキシ、C1−6−アルキルオキシカルボニル、ベンジルオキシカルボニル、C1−3−アルキルスルホニルアミノカルボニルもしくは1H−テトラゾール−5−イル基によって置換されていてもよいC1−4−アルキル基、
ヒドロキシ、ベンジルオキシ、カルボキシ−C1−3−アルキルアミノ、C1−3−アルコキシカルボニル−C1−3−アルキルアミノ、N−(C1−3−アルキル)−カルボキシ−C1−3−アルキルアミノもしくはN−(C1−3−アルキル)−C1−3−アルコキシカルボニル−C1−3−アルキルアミノ基によって置換されているC2−4−アルキル基であり、一方で上記基において隣接窒素原子に対してα−位にある炭素原子は、置換されていなくてもよく、
R3は、C3−7−シクロアルキル基、不飽和部分がR2NR3基の窒素原子に直接結合されていなくてもよいプロパルギル基、フッ素もしくは塩素原子によって、またはメチルもしくはメトキシ基によって場合により置換されているフェニル基、あるいはメチル基によって場合により置換されているピラゾリル、ピリダゾリルまたはピリジニル基を表すか、あるいは
R2およびR3は、それらの間の窒素原子と一緒になって、カルボキシまたはC1−4−アルコキシカルボニル基によって場合により置換されている5〜7員シクロアルキレンイミノ基を表し、フェニル環が更に縮合されていてもよい]
で示される二置換二環式複素環、その互変異性体、立体異性体および塩である。
【0013】
さらなる態様では、抗凝固薬は、
(a) 2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾチアゾール−5−カルボン酸−N−フェニル−N−(2−カルボキシエチル)−アミド、
(b) 2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾチアゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(c) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(d) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(3−ヒドロキシカルボニルプロピル)−アミド、
(e) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(ヒドロキシカルボニルメチル)−アミド、
(f) 1−メチル−2−[2−(2−アミジノチオフェン−5−イル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(g) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(h) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(i) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(j) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[2−(1H−テトラゾール−5−イル)エチル]−アミド、
(k) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[2−(1H−テトラゾール−5−イル)エチル]−アミド、
(l) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(m) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(3−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(n) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(o) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[(N−ヒドロキシカルボニルエチル−N−メチル)−2−アミノエチル]−アミド、
(p) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(3−フルオロフェニル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(q) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(4−フルオロフェニル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(r) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(s) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(t) 1−メチル−2−[N−(4−アミジノフェニル)アミノメチル]−インドール−5−イル−カルボン酸−N−フェニル−N−(2−メトキシカルボニルエチル)−アミド、
(u) 1−メチル−2−[N−(4−アミジノフェニル)アミノメチル]−チエノ[2.3−d]イミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(v) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(w) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(x) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(y) 1−メチル−2−[N−[4−(N−n−ヘキシルオキシカルボニルアミジノ)フェニル]−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−エトキシカルボニルエチル)−アミド、
より選択される化合物、ならびにその互変異性体、立体異性体および塩である。
【0014】
別の局面では、本発明は、ダビガトラン、ダビガトランエテキシラート、および/またはダビガトランのO−アシルグルクロニドに対する抗体分子に関する。
【0015】
さらなる局面では、抗体分子は、ポリクローナル抗体、モノクローナル抗体、ヒト抗体、ヒト化抗体、キメラ抗体、抗体のフラグメント、特にFab、Fab’、もしくはF(ab’)2フラグメント、単鎖抗体、特に単鎖可変フラグメント(scFv)、ドメイン抗体、ナノボディ(nanobody)、ダイアボディ(diabody)、またはDARPinである。
【0016】
さらなる局面では、本発明は、医学に使用するための上記抗体分子に関する。
【0017】
さらなる局面では、本発明は、抗凝固治療の副作用の治療または予防に使用するための上記抗体分子に関する。
【0018】
さらなる局面では、副作用は出血事象である。
【0019】
さらなる局面では、本発明は、抗凝固治療の副作用の治療または予防方法であって、それを必要とする患者に上記抗体分子の有効量を投与することを含む方法に関する。
【0020】
別の局面では、本発明は、容器およびラベルと一緒に前記抗体分子を含むキットに関する。
【図面の簡単な説明】
【0021】
【図1】トロンビン凝固時間アッセイを用いて、ダビガトランの濃度の増加とともに凝固時間の増加が見られたことを示す図である。濃度200nMは、凝固時間にベースラインからの5倍の延長を招き、この濃度を第1および第2セットの実験に使用した。最終セットの実験には濃度500nM(治療濃度を超える)を使用した。
【図2】ダビガトランに対する4種の異なる抗体(A〜D)全てが、ヒト血漿でのダビガトランの凝固時間延長を中和したことを示す図である。ヒト血漿でのベースライン凝固は10.9秒であり、200nMダビガトランを血漿と一緒に予備インキュベーションした場合、凝固は51秒に延長した。200nMダビガトランと一緒に予備インキュベーションされた血漿に各抗体を添加し、さらに5分間インキュベーションした。次に、トロンビンの添加によってトロンビン凝固時間を開始させた。各抗体は、ダビガトランの凝固時間を異なる程度にリバースすることができた。最も濃い溶液が抗凝固活性の最大のリバースを招いた。
【図3】200nMダビガトランと予備インキュベーションされたヒト血漿に添加される、漸増するポリクローナル抗体(抗体D)の濃度の作用を測定したことを示す図である。ベースライン凝固時間は11秒であり、ダビガトランの添加は、凝固を63.7秒に延長した。次に、抗体の漸増する希釈が、ダビガトランで延長されたトロンビン凝固時間のリバースに及ぼす作用を試験した。最低濃度はトロンビン凝固時間を43.9秒に短縮した。より高い濃度は、トロンビン凝固時間をベースラインレベルに完全に減らし、ダビガトランの抗凝固作用の完全な中和をもたらした。非特異的ウサギポリクローナル抗体(四角)の添加は、ダビガトランの抗凝固作用のリバースに効果を有さなかった。
【図4】500nMダビガトランと予備インキュベーションされたヒト血漿に添加された、漸増するポリクローナル抗体(抗体D)の濃度の作用を測定したことを示す図である。ベースライン凝固時間は10.9秒であり、このより高い濃度のダビガトランの添加は、凝固を111.7秒に延長した(約10倍の増加)。抗体または原液の1:2希釈溶液の作用は、ダビガトランを用いたトロンビン凝固時間延長を濃度依存的にリバースした。最も高い濃度もまた、トロンビン凝固時間をベースラインレベルに完全にリバースさせ、たとえ治療濃度を超えるダビガトランの抗凝固作用であっても完全な中和を招いた。
【図5】抗ダビガトラン抗体分子重鎖の可変領域の配列を示す図である。
【図6】抗ダビガトラン抗体分子軽鎖の可変領域の配列を示す図である。
【図7】マウスモノクローナル抗体(クローン22)が、ヒト血漿およびヒト全血でのダビガトランの抗凝固作用をリバースすることを示す図である。漸増する濃度のマウス抗体を、30nMダビガトランと予備インキュベーションされたヒト血漿または全血に添加した。1.5〜2U/mLトロンビンの添加によってアッセイを開始し、凝固時間を測定した。100%のダビガトラン活性を、化合物の存在下と不在下の凝固時間の差として定義した。抗体は、凝固時間のダビガトラン介在性延長を用量依存的に阻害した。
【図8】クローン22抗体から生成されたマウスFabが、ヒト血漿でのダビガトランの抗凝固作用をリバースすることを示す図である。漸増する濃度のマウスFabを、7nMダビガトランと予備インキュベーションされたヒト血漿に添加した。インタクトな抗体も陽性対照として試験した。0.4U/mLトロンビンの添加によってアッセイを開始し、凝固時間を測定した。100%阻害を、凝固時間のダビガトラン介在性増加の完全遮断として定義した。Fabは、ヒト血漿での凝固時間におけるダビガトラン誘導延長を用量依存的に阻害した。
【図9】マウスモノクローナル抗体(クローン22)が、ヒト血漿でのダビガトランアシルグルクロニドの抗凝固作用をリバースすることを示す図である。漸増する濃度のマウス抗体を、7nMダビガトランアシルグルクロニドまたはダビガトランと予備インキュベーションされたヒト血漿に添加した。0.4U/mLトロンビンの添加によってアッセイを開始し、凝固時間を測定した。100%阻害を凝固時間における化合物介在性増加の完全遮断として定義した。抗体は、ヒト血漿での凝固時間におけるダビガトランアシルグルクロニド誘導延長を用量依存的に阻害した。
【図10】ヒト化Fab(Fab18/15)が、ヒト血漿でのダビガトランの抗凝固作用をリバースすることを示す図である。漸増する濃度のFab18/15を、7nMダビガトランと予備インキュベーションされたヒト血漿に添加した。0.4U/mLトロンビンの添加によってアッセイを開始し、凝固時間を測定した。100%阻害を、凝固時間におけるダビガトラン介在性増加の完全遮断として定義した。Fabは、ヒト血漿での凝固時間におけるダビガトラン誘導延長を用量依存的に阻害した。
【図11】t=0での等モル濃度のFabのボーラス投与とダビガトランを連続注入として受けている、ラットにおけるex vivo全血トロンビン凝固時間(3.0U/mLトロンビン)を示す図である。無地丸との線は、薬物なしのビヒクル処置を表す。無地四角の線は、Fabなしのダビガトランの抗凝固活性を表す。無地三角との線は、Fab投与後の抗凝固活性を表す。データを平均±SE(一処置群あたり動物数n=4)として表す。
【図12】t=0で等モル濃度のFabのボーラス投与とダビガトランを連続注入として受けている、投与されているラットにおけるex vivo全血aPTTを示す図である。無地丸は、薬物なしのビヒクル処置を表す。無地四角との線は、Fabなしのダビガトランの抗凝固活性を表す。無地三角との線は、Fab投与後の抗凝固活性を表す。データを平均±SE(一処置群あたり動物数n=4)として表す。
【図13】t=0での漸増する用量のFabのボーラス投与とダビガトランを連続注入として受けている、ラットにおけるex vivo全血トロンビン凝固時間(3.0U/mLトロンビン)を示す図である。無地丸との線は、薬物なしのビヒクル処置を表す。無地四角とのの線は、Fabなしのダビガトランの抗凝固活性を表す。無地三角との線は、等モルのFabを投与後の、破線は、等モル用量の50%を投与後の抗凝固活性を表す。データを平均±SE(一処置群あたり動物数n=4)として表す。
【図14】t=0での漸増する用量のFabのボーラス投与とダビガトランを連続注入として受けている、ラットにおけるex vivo全血aPTTを示す図である。無地丸は、薬物なしのビヒクル処置を表す。無地四角との線は、Fabなしのダビガトランの抗凝固活性を表す。無地三角との線は、等モルのFabを投与後の、破線は、等モル用量の50%を投与後の抗凝固活性を表す。データを平均±SE(一処置群あたり動物数n=4)として表す。
【0022】
発明の詳細な説明
一局面では、本発明は、抗凝固薬の活性を中和可能な抗体分子に関する。
【0023】
抗体(免疫グロブリン、略してIgとしても知られている)は、脊椎動物の血液または他の体液中に見出されるガンマグロブリンタンパク質であって、細菌およびウイルスなどの外来物を同定および中和するために免疫系によって使用される。抗体は、典型的には基本構造ユニットでできており、それぞれが2本の大きな重鎖および2本の小さな軽鎖を有し、例えば1個のユニットを有するモノマー、2個のユニットを有するダイマーまたは5個のユニットを有するペンタマーを形成する。抗体は、抗原として知られている他の分子または構造に非共有相互作用によって結合することができる。この結合は、抗体が高親和性で特異構造だけに結合するという意味で特異的である。抗体によって認識される抗原の独特な部分は、エピトープまたは抗原決定基と呼ばれる。抗体のエピトープ結合性部分は、時にパラトープと呼ばれ、抗体のいわゆる可変ドメイン、または可変領域(Fv)に位置する。可変ドメインは、フレームワーク領域(FR)によって間隔が空けられた3個のいわゆる相補性決定領域(CDR)を含む。
【0024】
本発明の状況の範囲内で、CDRへの参照は、Chothia(Chothia and Lesk, J. Mol. Biol. 1987, 196: 901-917)ならびにKabat(E.A. Kabat, T.T. Wu, H. Bilofsky, M. Reid-Miller and H. Perry, Sequence of Proteins of Immunological Interest, National Institutes of Health, Bethesda (1983))の定義に基づく。
【0025】
当該技術は、さらに抗体を開発し、それらを医学および工学における多用途の道具にしている。したがって、本発明の状況で「抗体分子」または「抗体」という用語(本明細書において同義語として使用される)には、例えば2本の軽鎖および2本の重鎖、またはラクダ科の種のように2本の重鎖だけを含む、自然界に見出されうる抗体が含まれるだけではなく、さらには抗原への結合特異性および免疫グロブリンの可変ドメインとの構造類似性を有する少なくとも1個のパラトープを含む全ての分子も包含する。
【0026】
したがって、本発明による抗体分子は、ポリクローナル抗体、モノクローナル抗体、ヒト抗体、ヒト化抗体、キメラ抗体、抗体のフラグメント、特にFv、Fab、Fab’、またはF(ab’)2フラグメント、単鎖抗体、特に単鎖可変フラグメント(scFv)、小モジュラー免疫医薬(Small Modular Immunopharmaceutical)(SMIP)、ドメイン抗体、ナノボディ、ダイアボディでありうる。
【0027】
ポリクローナル抗体は、異なるアミノ酸配列を有する抗体分子の集合を表し、当技術分野で周知のプロセスにより抗原で免疫処置した後に、脊椎動物の血液から得ることができる。
【0028】
モノクローナル抗体(mAbまたはmoAb)は、アミノ酸配列が同一の単一特異性抗体である。それらは、ハイブリドーマ技法によって、特異抗体産生性B細胞と骨髄腫(B細胞ガン)細胞の融合体のクローンを意味するハイブリッド細胞系(ハイブリドーマと呼ばれる)から製造することができる(Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975;256:495-7.)。または、モノクローナル抗体は、宿主細胞での組換え発現によって製造することができる(Norderhaug L, Olafsen T, Michaelsen TE, Sandlie I. (May 1997). "Versatile vectors for transient and stable expression of recombinant antibody molecules in mammalian cells.". J Immunol Methods 204 (1): 77-87;下記も参照されたい)。
【0029】
人間に応用するために、マウスのような本来他の種由来の抗体の免疫原性を減少させることが望ましいことが多い。これは、キメラ抗体の構築によって、または「ヒト化」と呼ばれるプロセスによって行うことができる。この状況で「キメラ抗体」は、ある種(例えばマウス)由来の配列部分(例えば可変ドメイン)が別の種(例えばヒト)由来の配列部分(例えば定常ドメイン)と融合したものを含む抗体であると理解される。「ヒト化抗体」は、本来非ヒト種由来の可変ドメインを含む抗体であって、その可変ドメインの全体的な配列がヒト可変ドメインの配列により密接に類似するように、あるアミノ酸が突然変異された抗体である。抗体のキメラ化方法およびヒト化方法は、当技術分野において周知である(Billetta R, Lobuglio AF. "Chimeric antibodies". Int Rev Immunol. 1993;10(2-3):165-76; Riechmann L, ClarkM, Waldmann H, Winter G (1988). "Reshaping human antibodies for therapy". Nature: 332:323.)。
【0030】
さらに、例えばファージディスプレイによって、またはトランスジェニック動物を使用してヒトゲノム由来の配列に基づき抗体を作出するための技法が開発されている(国際公開公報第90/05144号; D. Marks, H.R. Hoogenboom, T.P. Bonnert, J. McCafferty, A.D. Griffiths and G. Winter (1991) "By-passing immunisation. Human antibodies from V-gene libraries displayed on phage." J.Mol.Biol., 222, 581-597; Knappik et al., J. Mol. Biol. 296: 57-86, 2000; S. Carmen and L. Jermutus, "Concepts in antibody phage display". Briefings in Functional Genomics and Proteomics 2002 1(2):189-203; Lonberg N, Huszar D. "Human antibodies from transgenic mice". Int Rev Immunol. 1995;13(1):65-93.; Brueggemann M, Taussig MJ. "Production of human antibody repertoires in transgenic mice". Curr Opin Biotechnol. 1997 Aug;8(4):455-8.)。そのような抗体は、本発明の状況で「ヒト抗体」である。
【0031】
本発明による抗体分子には、Fab、Fab’、またはF(ab’)2フラグメントのような、抗原結合特性を保持する免疫グロブリンのフラグメントも含まれる。そのようなフラグメントは、免疫グロブリンのフラグメント化によって、例えばタンパク質分解消化によって、またはそのようなフラグメントの組換え発現によって得ることができる。例えば、免疫グロブリンの消化は、日常的な技法によって、例えばパパインもしくはペプシン(国際公開公報第94/29348号)、またはエンドプロテイナーゼLys−C(Kleemann, et al, Anal. Chem. 80, 2001-2009, 2008)を使用して達成することができる。抗体のパパインまたはLys−C消化によって、典型的にはそれぞれ単一の抗原結合部位を有する二つの同一の抗原結合フラグメント、いわゆるFabフラグメントおよび残余のFcフラグメントが製造される。ペプシン処理によってF(ab’)2が回収される。宿主細胞での組換え発現によってFab分子を製造する方法を下記により詳細に概要する。
【0032】
免疫グロブリンの可変ドメインまたはそのような可変ドメイン由来の分子を異なる分子状況に配置するためにいくつかの技法が開発されている。それらもまた、本発明による「抗体分子」と見なすべきである。一般にこれらの抗体分子は、免疫グロブリンよりもサイズが小さく、単一のアミノ酸鎖を含むか、または数本のアミノ酸鎖から構成されることがある。例えば、単鎖可変フラグメント(scFv)は、短いリンカー、通常はセリン(S)またはグリシン(G)で一緒に結合されている、免疫グロブリンの重鎖及び軽鎖可変領域との融合体である(国際公開公報第88/01649号;国際公開公報第91/17271号; Huston et al; International Reviews of Immunology, Volume 10, 1993, 195 - 217)。「単一ドメイン抗体」または「ナノボディ」は、単一のIg様ドメインに抗原結合部位を有する(国際公開公報第94/04678号;国際公開公報第03/050531号、Ward et al., Nature. 1989 Oct 12;341(6242):544-6; Revets et al., Expert Opin Biol Ther. 5(1):111-24, 2005)。同一または異なる抗原に対して結合特異性を有する一つ以上の単一ドメイン抗体が一緒に結合されることがある。ダイアボティは、2個の可変ドメインを含む2本のアミノ酸鎖から成る二価抗体分子である(国際公開公報第94/13804号、Holliger et al., Proc Natl Acad Sci U S A. 1993 Jul 15;90(14):6444-8)。抗体様分子の他の例は、免疫グロブリンスーパーファミリー抗体である(IgSF; Srinivasan and Roeske, Current Protein Pept. Sci. 2005, 6(2): 185-96)。異なる考え方が、定常ドメインCH1を欠如する単鎖ヒンジおよびエフェクタードメインに結合したFvドメインを含む、いわゆる小モジュラー免疫医薬(SMIP)につながっている(国際公開公報第02/056910号)。
【0033】
さらなる局面では、本発明の抗体分子は、免疫グロブリン可変ドメインに匹敵するある一定の結合特異性および親和性を有する限り、免疫グロブリン可変ドメインとかけ離れた構造関連性だけさえ有していればよいし、またそのような関係を全く有していなくてもよい。そのような非免疫グロブリン「抗体模倣物」は、時に「足場タンパク質」と呼ばれ、プロテインA、リポカリン、フィブロネクチンドメイン、アンキリンコンセンサスリピートドメイン、およびチオレドキシンの遺伝子に基づくことがある(Skerra, Current Opinion in Biotechnology 2007, 18(4): 295-304)。本発明の状況における好ましい態様は、設計されたアンキリンリピートタンパク質である(DARPin; Steiner et al., J Mol Biol. 2008 Oct 24;382(5): 1211-27; Stumpp MT, Amstutz P. Curr Opin Drug Discov Devel. 2007 Mar;10(2):153-9)。
【0034】
抗体分子は、その抗体分子の性質に所望の影響を有する他の分子実体に融合されるか(融合タンパク質として)、または別の方法で結合される(共有または非共有結合により)ことがある。例えば、特に単鎖抗体またはドメイン抗体の場合、抗体分子の薬物動態特性、例えば血液のような体液中での安定性を改善することが望ましいことがある。これに関して、特に循環中でのそのような抗体分子の半減期を延長するために、PEG化(国際公開公報第98/25971号;国際公開公報第98/48837号;国際公開公報第2004081026号)、その抗体分子を、アルブミンのような血清タンパク質に対して親和性を有する別の抗体分子と融合もしくは別の方法で共有結合させること(国際公開公報第2004041865号;国際公開公報第2004003019号)、またはアルブミンもしくはトランスフェリンのような血清タンパク質の全てまたは一部との融合タンパク質としてその抗体分子を発現させること(国際公開公報第01/79258号)などのいくつかの技法が開発されている。
【0035】
さらなる局面では、抗体分子は、抗凝固薬に対する結合特異性を有する。「結合特異性」は、抗体分子が、構造的に無関係の分子よりも抗凝固薬に対して有意に高い結合親和性を有することを意味する。
【0036】
親和性は、抗体分子上の単一抗原結合部位と単一エピトープの間の相互作用である。親和性は、結合定数KA=kass/kdiss、または解離定数KD=kdiss/kassによって表される。
【0037】
本発明の一局面では、抗体は、例えば表面プラズモン共鳴分析によって決定されるとき、0.1pM〜100μM、好ましくは1pM〜100μM、好ましくは1pM〜1μMの範囲のKD値を有する親和性で抗凝固薬に結合する(Malmqvist M., "Surface plasmon resonance for detection and measurement of antibody-antigen affinity and kinetics.", Curr Opin Immunol. 1993 Apr;5(2):282-6.)。抗体親和性は、また、動力学排除アッセイ(kinetic exclusion assay)(KinExA)技法を用いて測定することもできる(Darling, R.J., and Brault P-A., "Kinetic exclusion assay technology: Characterization of Molecular Interactions." ASSAY and Drug Development Technologies. 2004, Dec 2(6): 647-657)。
【0038】
抗体分子の結合親和性は、親和性成熟として知られているプロセスによって高めることができる(Marks et al., 1992, Biotechnology 10:779-783; Barbas, et al., 1994, Proc. Nat. Acad. Sci, USA91:3809-3813; Shier et al., 1995, Gene 169:147-155)。したがって、親和性成熟された抗体もまた、本発明に包含される。
【0039】
本発明のさらなる局面では、抗体分子は、抗凝固薬の活性を中和可能である。すなわち抗凝固薬は、抗体分子に結合すると、もはやその抗凝固活性を発揮することができないか、または有意に減少した大きさでこの活性を発揮する。好ましくは抗凝固活性は、当該抗凝固薬にとって適切な活性アッセイで、特にエカリン凝固時間またはトロンビン凝固時間などの、トロンビンに感受性の凝固アッセイで決定されるとき、抗体の結合について、少なくとも2倍、5倍、10倍、100倍で減少している(H. Bounameaux, Marbet GA, Lammle B, et al. "Monitoring of heparin treatment. Comparison of thrombin time, activated partial thromboplastin time, and plasma heparin concentration, and analysis of the behaviour of antithormbin III". American Journal of Clinical Pathology 1980 74(1): 68-72)。
【0040】
本発明の抗体分子を製造するために、当業者は、当技術分野で周知の多様な方法より選択することができる(Norderhaug et al., J Immunol Methods 1997, 204 (1): 77-87; Kipriyanow and Le Gall, Molecular Biotechnology 26: 39- 60, 2004; Shukla et al., 2007, J. Chromatography B, 848(1): 28-39)。
【0041】
抗凝固薬は、上記に概要するように当技術分野において周知である。本発明のさらなる局面では、抗凝固薬は、直接トロンビン阻害剤、第Xa因子阻害剤、またはビタミンKアンタゴニストである。ビタミンKアンタゴニストの例は、ワルファリンが含まれるクマリンである。主に第Xa因子の間接阻害剤の例は、アンチトロンビンIIIの活性化を介して作用するヘパリン群の物質であって、それらには、いくつかの低分子量ヘパリン製品(ベミパリン、セルトパリン、ダルテパリン、エノキサパリン、ナドロパリン、パルナパリン、レビパリン、チンザパリン)、ある種のオリゴ糖(フォンダパリヌクス、イドラパリヌクス)、ヘパリノイド(ダナパロイド、スロデキシド、デルマタン硫酸)、および直接第Xa因子阻害剤(アピキサバン、オタミキサバン、リバロキサバン)が含まれる。トロンビン阻害剤の例には、二価ヒルジン(ビバリルジン、レピルジン、デシルジン)、ならびに一価化合物アルガトロバンおよびダビガトランが含まれる。
【0042】
したがってさらなる局面では、抗凝固薬は、ダビガトラン、アルガトロバン、メラガトラン、キシメラガトラン、ヒルジン、ビバリルジン、レピルジン、デシルジン、アピキサバン、エドキサバン、オタミキサバン、リバロキサバン、デフィブロチド、ラマトロバン、アンチトロンビンIII、またはドロトレコギンアルファである。
【0043】
さらなる態様では、抗凝固薬は、一般式
【0044】
【化2】
[式中、
Aは、基Hetのベンゾ、ピリドまたはチエノ部分に結合したカルボニルまたはスルホニル基を表し、
Bは、基Arに結合されたメチレン基が酸素もしくは硫黄原子によって、または−NR1−基によって置き換えられていてもよいエチレン基を表し、
ここで、R1は、水素原子またはC1−4−アルキル基を表し、
Eは、RbNH−C(=NH)−基を表し、
ここで、Rbは、水素原子、ヒドロキシ、C1−9−アルコキシカルボニル、シクロヘキシルオキシカルボニル、フェニル−C1−3−アルコキシカルボニル、ベンゾイル、p−C1−3−アルキル−ベンゾイルまたはピリジノイル基を表し、一方で上記C1−9−アルコキシカルボニル基の2位のエトキシ部分は、C1−3−アルキルスルホニルまたは2−(C1−3−アルコキシ)−エチル基によって更に置換されていてもよく、
Arは、塩素原子によって、またはメチル、エチルもしくはメトキシ基によって場合により置換されている1,4−フェニレン基を表すか、もしくはそれは2,5−チエニレン基を表し、
Hetは、1−(C1−3−アルキル)−2,5−ベンゾイミダゾリレン、1−シクロプロピル−2,5−ベンゾイミダゾリレン、2,5−ベンゾチアゾリレン、1−(C1−3−アルキル)−2,5−インドリレン、1−(C1−3−アルキル)−2,5−イミダゾ[4,5−b]ピリジニレン、3−(C1−3−アルキル)−2,7−イミダゾ[1,2−a]ピリジニレンまたは1−(C1−3−アルキル)−2,5−チエノ[2,3−d]イミダゾリレン基を表し、そして
Raは、R2NR3−基を表し、
ここで、R2は、カルボキシ、C1−6−アルキルオキシカルボニル、ベンジルオキシカルボニル、C1−3−アルキルスルホニルアミノカルボニルもしくは1H−テトラゾール−5−イル基によって置換されていてもよいC1−4−アルキル基、
ヒドロキシ、ベンジルオキシ、カルボキシ−C1−3−アルキルアミノ、C1−3−アルコキシカルボニル−C1−3−アルキルアミノ、N−(C1−3−アルキル)−カルボキシ−C1−3−アルキルアミノもしくはN−(C1−3−アルキル)−C1−3−アルコキシカルボニル−C1−3−アルキルアミノ基によって置換されているC2−4−アルキル基であり、一方で上記基において隣接窒素原子に対してα−位にある炭素原子は、置換されていなくてもよく、
R3は、C3−7−シクロアルキル基、不飽和部分がR2NR3基の窒素原子に直接結合されていなくてもよいプロパルギル基、フッ素もしくは塩素原子によって、またはメチルもしくはメトキシ基によって場合により置換されているフェニル基、あるいはメチル基によって場合により置換されているピラゾリル、ピリダゾリルまたはピリジニル基を表すか、あるいは
R2およびR3は、それらの間の窒素原子と一緒になって、カルボキシまたはC1−4−アルコキシカルボニル基によって場合により置換されている5〜7員シクロアルキレンイミノ基を表し、フェニル環が更に縮合されていてもよい]
で示される二置換二環式複素環、その互変異性体、立体異性体および塩である。式(I)で示される化合物、これらの化合物の調製および抗凝固薬としてのそれらの使用は、国際公開公報第98/37075号に記載されている。
【0045】
さらなる態様では、抗凝固薬は、
(a) 2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾチアゾール−5−カルボン酸−N−フェニル−N−(2−カルボキシエチル)−アミド、
(b) 2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾチアゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(c) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(d) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(3−ヒドロキシカルボニルプロピル)−アミド、
(e) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(ヒドロキシカルボニルメチル)−アミド、
(f) 1−メチル−2−[2−(2−アミジノチオフェン−5−イル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(g) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(h) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(i) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(j) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[2−(1H−テトラゾール−5−イル)エチル]−アミド、
(k) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[2−(1H−テトラゾール−5−イル)エチル]−アミド、
(l) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(m) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(3−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(n) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(o) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[(N−ヒドロキシカルボニルエチル−N−メチル)−2−アミノエチル]−アミド、
(p) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(3−フルオロフェニル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(q) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(4−フルオロフェニル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(r) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(s) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(t) 1−メチル−2−[N−(4−アミジノフェニル)アミノメチル]−インドール−5−イル−カルボン酸−N−フェニル−N−(2−メトキシカルボニルエチル)−アミド、
(u) 1−メチル−2−[N−(4−アミジノフェニル)アミノメチル]−チエノ[2.3−d]イミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(v) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(w) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(x) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(y) 1−メチル−2−[N−[4−(N−n−ヘキシルオキシカルボニルアミジノ)フェニル]−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−エトキシカルボニルエチル)−アミド、
より選択される化合物、その互変異性体、立体異性体および塩であり、それらの全ては、国際公開公報第98/37075号に記載されている。
【0046】
本発明の状況で好ましい抗凝固薬は、化学式(II):
【化3】
を有するダビガトラン(CAS211914−51−1、N−[2−(4−アミジノフェニルアミノメチル)−1−メチル−1H−ベンゾイミダゾール−5−イルカルボニル]−N−(2−ピリジル)−β−アラニン)である。
【0047】
ダビガトランは、トロンビン阻害作用およびトロンビン時間延長作用を有する化合物を1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミドの名称で開示している国際公開公報第98/37075号から公知である。Hauel et al. J Med Chem 2002, 45 (9): 1757-66も参照されたい。
【0048】
ダビガトランは、式(III):
【化4】
で示されるプロドラッグとして適用される。
【0049】
式IIIで示される化合物(名称ダビガトランエテキシラート、CAS211915−06−9;エチル3−[(2−{[4−(ヘキシルオキシカルボニルアミノ−イミノ−メチル)−フェニルアミノ]−メチル}−1−メチル−1H−ベンゾイミダゾール−5−カルボニル)−ピリジン−2−イル−アミノ]−プロピオナート)は、体内に入った後で活性化合物(II)に変換される。ダビガトランエテキシラートの好ましい多形は、ダビガトランエテキシラートメシラート塩である。
【0050】
ダビガトランの主要効能は、深部静脈血栓の術後予防、確立した深部静脈血栓の処置および心房細動を有する患者における発作の予防である(Eriksson et al., Lancet 2007, 370 (9591): 949-56; Schulman S et al, N Engl J Med 2009, 361 (24): 2342-52; Connolly S et al., N Engl J Med 2009, 361 (12): 1139-51; Wallentin et al., Lancet 2010, 376 (9745): 975-983)。
【0051】
ヒトの身体では、カルボキシラート部分のグルクロン酸抱合がダビガトランの主要なヒト代謝経路である(Ebner et al., Drug Metab. Dispos. 2010, 38(9):1567-75)。それは、1−O−アシルグルクロニド(βアノマー)の形成をもたらす。1−O−アシルグルクロニドは、アグリコンへのマイナー加水分解(minor hydrolysis)に加えて、水溶液中で非酵素的アシル転移を受けて、2−O−、3−O−、および4−O−アシルグルクロニドの形成をもたらすことがある。精製1−O−アシルグルクロニドおよびその異性体性転位産物を用いた実験は、ダビガトランに比べて等しい効力の活性化部分トロンボプラスチン時間の延長を明らかにした。
【0052】
本発明の別の局面では、抗体分子は、ダビガトランとダビガトランエテキシラートの両方に結合する。
【0053】
本発明の別の局面では、抗体分子は、ダビガトランとダビガトランのO−アシルグルクロニドの両方に、特にダビガトランの1−O−アシルグルクロニドに結合する。
【0054】
本発明の別の局面では、抗体分子は、さらにダビガトランの2−O−、3−O−、および4−O−アシルグルクロニドに結合する。
【0055】
本発明の別の局面では、抗体分子は、ダビガトランおよびダビガトランのO−アシルグルクロニド、特にダビガトランの1−O−アシルグルクロニドの活性を中和可能である。
【0056】
本発明の別の局面では、抗体分子は、ダビガトランに対する結合特異性を有し、図5および6に示されるCDR配列を含む。
【0057】
本発明の別の局面では、抗体分子は、図5に示される重鎖CDR配列、および図6に示される軽鎖CDR配列を含む。
【0058】
本発明の別の局面では、抗体分子は、図5に示される重鎖可変ドメイン配列、および図6に示される軽鎖可変ドメイン配列を含む。
【0059】
本発明の別の局面では、抗体分子は、図5において名称DBG 13 VHで示される重鎖可変ドメイン配列、および図6において名称DBG 13 VKで示される軽鎖可変ドメイン配列を含む。
【0060】
本発明の別の局面では、抗体分子は、図5において名称DBG 14 VHで示される重鎖可変ドメイン配列、および図6において名称DBG 14 VKで示される軽鎖可変ドメイン配列を含む。
【0061】
本発明の別の局面では、抗体分子は、図5において名称DBG 22 VHで示される重鎖可変ドメイン配列、および図6において名称DBG 22 VKで示される軽鎖可変ドメイン配列を含む。
【0062】
本発明の別の局面では、抗体分子は、図5において名称Eng VH# 14で示される重鎖可変ドメイン配列、および図6において名称Eng VK# 11で示される軽鎖可変ドメイン配列を含む。
【0063】
本発明の別の局面では、抗体分子は、図5において名称Eng VH# 15で示される重鎖可変ドメイン配列、および図6において名称Eng VK# 17で示される軽鎖可変ドメイン配列を含む。
【0064】
本発明の別の局面では、抗体分子は、図5において名称Eng VH# 15で示される重鎖可変ドメイン配列、および図6において名称Eng VK# 18で示される軽鎖可変ドメイン配列を含む。
【0065】
本発明の別の局面では、抗体分子は、図5において名称Eng VH# 31で示される重鎖可変ドメイン配列、および図6において名称Eng VK# 18で示される軽鎖可変ドメイン配列を含む。
【0066】
本発明の別の局面では、抗体分子は、ダビガトランに対する結合特異性を有し、配列番号:1および配列番号:2から成る群より選択されるCDR1、配列番号:3、配列番号:4、配列番号:5、配列番号:6、配列番号:7、および配列番号:8から成る群より選択されるCDR2、ならびに配列番号:9および配列番号:10から成る群より選択されるCDR3を有する重鎖可変ドメインと、配列番号:11、配列番号:12、および配列番号:13から成る群より選択されるCDR1、配列番号:14で示されるCDR2、ならびに配列番号:15で示されるCDR3を有する軽鎖可変ドメインとを含む。
【0067】
本発明の別の局面では、抗体分子は、配列番号:1で示されるCDR1、配列番号:6、配列番号:7、および配列番号:8から成る群より選択されるCDR2、ならびに配列番号:10で示されるCDR3を有する重鎖可変ドメインと、配列番号:13で示されるCDR1、配列番号:14で示されるCDR2、および配列番号:15で示されるCDR3を有する軽鎖可変ドメインとを含む。
【0068】
本発明の別の局面では、抗体分子は、配列番号:1で示されるCDR1、配列番号:7で示されるCDR2、および配列番号:10で示されるCDR3を有する重鎖可変ドメイン、ならびに配列番号:13で示されるCDR1、配列番号:14で示されるCDR2、および配列番号:15で示されるCDR3を有する軽鎖可変ドメインを含む。
【0069】
本発明の別の局面では、抗体分子は、配列番号:1で示されるCDR1、配列番号:5で示されるCDR2、および配列番号:10で示されるCDR3を有する重鎖可変ドメイン、ならびに配列番号:11で示されるCDR1、配列番号:14で示されるCDR2、および配列番号:15で示されるCDR3を有する軽鎖可変ドメインを含む。
【0070】
本発明の別の局面では、抗体分子は、配列番号:16、18、20、22、24、および26から成る群より選択される重鎖可変ドメイン、ならびに配列番号:17、19、21、23、25、および27から成る群より選択される軽鎖可変ドメインを含む。
【0071】
本発明の別の局面では、抗体分子は、配列番号:16で示される重鎖可変ドメイン、および配列番号:17で示される軽鎖可変ドメインを含む。
【0072】
本発明の別の局面では、抗体分子は、配列番号:18で示される重鎖可変ドメイン、および配列番号:19で示される軽鎖可変ドメインを含む。
【0073】
本発明の別の局面では、抗体分子は、配列番号:20で示される重鎖可変ドメイン、および配列番号:21で示される軽鎖可変ドメインを含む。
【0074】
本発明の別の局面では、抗体分子は、配列番号:22で示される重鎖可変ドメイン、および配列番号:23で示される軽鎖可変ドメインを含む。
【0075】
本発明の別の局面では、抗体分子は、配列番号:24で示される重鎖可変ドメイン、および配列番号:25で示される軽鎖可変ドメインを含む。
【0076】
本発明の別の局面では、抗体分子は、配列番号:24で示される重鎖可変ドメイン、および配列番号:27で示される軽鎖可変ドメインを含む。
【0077】
本発明の別の局面では、抗体分子は、配列番号:26で示される重鎖可変ドメイン、および配列番号:27で示される軽鎖可変ドメインを含む。
【0078】
本発明の別の局面では、抗体分子は、scFv分子である。この形式では、本明細書に開示された可変ドメインは、例えば配列番号:28、29、30、または31から成る群より選択される、適切なリンカーペプチドで相互に融合されていることがある。その構築物は、N末端からC末端にかけて(重鎖可変ドメイン)−(リンカーペプチド)−(軽鎖可変ドメイン)、または(軽鎖可変ドメイン)−(リンカーペプチド)−(重鎖可変ドメイン)の順序でこれらの要素を含むことがある。
【0079】
本発明の別の局面では、抗体分子は、配列番号:32または配列番号:33を含むscFv分子である。本発明の別の局面では、抗体分子は、配列番号:32または配列番号:33から成るscFv分子である。
【0080】
sFv構築物をコードする核酸を、宿主細胞(E. coli、Pichia pastoris、または哺乳動物細胞系、例えばCHOもしくはNS0など)に、組換え発現させて、機能的scFv分子を得るプロセスは、当技術分野において公知である(例えばRippmann et al., Applied and Environmental Microbiology 1998, 64(12): 4862-4869; Yamawaki et al., J. Biosci. Bioeng. 2007, 104(5): 403-407; Sonoda et al., Protein Expr. Purif. 2010, 70(2): 248-253を参照されたい)。
【0081】
特に、本発明のscFv抗体分子は、以下のように製造することができる。構築物は、W3110、TG1、BL21、BL21(DE3)、HMS174、HMS174(DE3)、MM294のような異なるE. coliに誘導性プロモーターのコントロール下で発現させることができる。このプロモーターは、lacUV5、tac、T7、trp、trc、T5、araBより選択することができる。培養培地は、好ましくはWilmsら、2001(Wilms et al., Biotechnology and Bioengineering 2001, 73(2): 95-103)、DeLisaら、1999(DeLisa et al., Biotechnology and Bioengineering 1999, 65(1): 54-64)にしたがって十分に定義されているか、または同等である。しかしながら、バッチ培地および/または供給培地へのイソロイシン、ロイシン、リシン、メチオニン、フェニルアラニン、トレオニン、トリプトファンおよびバリンなどのアミノ酸、またはダイズペプトンまたは酵母エキスなどの複合培地成分の補給が有益なことがある。発酵のためのプロセスは、流加モードで行う。条件:温度20〜40℃、pH5.5〜7.5、DOは20%よりも高く保つ。最初の炭素源の消費後に、培養物に上記供給培地(または同等物)を供給する。発酵槽中で40〜100g/Lの乾燥細胞重量に達したときに、使用されるプロモーター系に対応する適切な誘導物質(例えばIPTG、ラクトース、アラビノース)で培養物を誘導する。誘導は、パルス完全誘導として、またはそれぞれの誘導物質を発酵槽中に長時間供給することによる部分誘導もしくはその組み合わせとしてのいずれかで行うことができる。生産期は、少なくとも4時間持続すべきである。細胞は、ボウル遠心分離機、管状ボウル遠心分離機またはディスクスタック遠心分離機での遠心分離によって回収され、培養上清は捨てられる。
【0082】
E. coli細胞塊を、4〜8倍量の溶解緩衝液(リン酸緩衝液またはトリス緩衝液、pH7〜8.5)中に再懸濁する。細胞溶解を、好ましくは、高圧ホモジナイズに続いて、ボウル遠心分離機、管状ボウル遠心分離機またはディスクスタック遠心分離機での遠心分離によるペレットの回収によって行う。scFv封入体を含有するペレットを、20mMトリス、150mM NaCl、5mM EDTA、2M尿素、0.5%トリトンX−100(pH8.0)で2〜3回洗浄し、20mMトリス、150mM NaCl、5mM EDTA(pH8.0)を使用する2回の洗浄段階が続く。scFv封入体は、ボウル遠心分離機、管状ボウル遠心分離機またはディスクスタック遠心分離機での遠心分離によって最終的に回収する。scFv封入体の可溶化は、6Mグアニジン−HClまたは8〜10mM尿素などのカオトロピック剤を含有する100mMグリシン/NaOH、5mM EDTA、20mMジチオトレイトール(pH9.5〜10.5)中で行うことができる。30〜60分間インキュベーション後に、溶液を遠心分離し、その後の再フォールディングのためにターゲットタンパク質を含有する上清を回収する。再フォールディングは、好ましくは流加モードでタンパク質溶液を再フォールディング緩衝液に1:10〜1:50希釈して0.1〜0.5mg/mlの最終タンパク質濃度にすることによって行われる。再フォールディング緩衝液は、50〜100mMトリスおよび/または50〜100mMグリシン、50〜150mM NaCl、1〜3M尿素、0.5〜1Mアルギニン、例えばシテイン(cytein)/シスチンまたは酸化/還元型グルタチオンなどの2〜6mMの酸化還元系(pH9.5〜10.5)を含有することがある。4℃で24〜72hインキュベーション後に、再フォールディング溶液を、場合により0.22μmフィルターを使用して濾過し、希釈し、pHをpH7.0〜8.0に調整する。タンパク質を、結合モードの陽イオン交換クロマトグラフィー(例えばToyopearl GigaCap S-650M、SPセファロースFFまたはS HyperCel(商標))を介してpH7.0〜8.5で分離する。溶離を、漸増するNaClの直線勾配によって行う。ターゲットタンパク質を含有する画分をプールし、次に非結合モードの陰イオン交換カラム(例えばToyopearl GigaCap Q-650M、Q−セファロースFF、Q HyperCel(商標))に続く陽イオン交換仕上げステップ(例えばSPセファロースHP)で分離する。最低限で90%の純度レベルのターゲットタンパク質を含有する画分をプールし、ダイアフィルトレーションまたはPBSを用いたサイズ排除クロマトグラフィーによって製剤化する。製造されたscFv分子の同一性および製品品質を、還元SDS−PAGEによって分析し、その場合scFvは、約26kDaの1本の主バンドの形で検出することができる。scFvの特徴づけのためのさらなるアッセイには、質量分析、RP−HPLCおよびSE−HPLCが挙げられる。
【0083】
本発明の別の局面では、抗体分子は、免疫グロブリン、好ましくはIgG1またはそのエフェクター機能ノックアウトまたはIgG4型の免疫グロブリンである。本発明の別の局面では、抗体分子は、配列番号:34、配列番号:40、または配列番号:42を含む重鎖、および配列番号:35または配列番号:43を含む軽鎖を有する免疫グロブリンである。
【0084】
本発明の別の局面では、抗体分子は、配列番号:34を含む重鎖および配列番号:35を含む軽鎖、または配列番号:40を含む重鎖および配列番号:35を含む軽鎖を有する免疫グロブリンである。本発明の別の局面では、抗体分子は、配列番号:42を含む重鎖および配列番号:43を含む軽鎖を有する免疫グロブリンである。
【0085】
本発明の別の局面では、抗体分子は、配列番号:34から成る重鎖および配列番号:35から成る軽鎖、または配列番号:40から成る重鎖および配列番号:35から成る軽鎖を有する免疫グロブリンである。本発明の別の局面では、抗体分子は、配列番号:42から成る重鎖および配列番号:43から成る軽鎖を有する免疫グロブリンである。
【0086】
本発明の別の局面では、抗体分子はFab分子である。その形式では、上に開示された可変ドメインは、それぞれ免疫グロブリン定常ドメイン、好ましくはヒト起源に融合されていてもよい。したがって、重鎖可変ドメインは、CH1ドメインに融合されていることがあり(いわゆるFdフラグメント)、軽鎖可変ドメインはCLドメインに融合されていることがある。
【0087】
本発明の別の局面では、抗体分子は、配列番号:36または配列番号:38を含むFdフラグメントおよび配列番号:37または配列番号:39を含む軽鎖を有するFab分子である。本発明の別の局面では、抗体分子は、配列番号:36を含むFdフラグメントおよび配列番号:37を含む軽鎖を有するFab分子である。本発明の別の局面では、抗体分子は、配列番号:38を含むFdフラグメントおよび配列番号:39を含む軽鎖を有するFab分子である。本発明の別の局面では、抗体分子は、配列番号:41を含むFdフラグメントおよび配列番号:37を含む軽鎖を有するFab分子である。本発明の別の局面では、抗体分子は、配列番号:36から成るFdフラグメントおよび配列番号:37から成る軽鎖を有するFab分子である。本発明の別の局面では、抗体分子は、配列番号:38から成るFdフラグメントおよび配列番号:39から成る軽鎖を有するFab分子である。本発明の別の局面では、抗体分子は、配列番号:41から成るFdフラグメントおよび配列番号:37から成る軽鎖を有するFab分子である。
【0088】
E. coli、Pichia pastoris、または哺乳動物細胞系(例えばCHOまたはNS0)のような宿主細胞にそのような重鎖および軽鎖を発現させるために、Fab構築物をコードする核酸を使用することができる。Fdフラグメントおよび軽鎖を含む機能的Fab分子に、これらの鎖を適切にフォールディング、会合、およびジスルフィド結合させる方法は、当技術分野において公知である(Burtet et al., J. Biochem. 2007, 142(6), 665-669; Ning et al., Biochem. Mol. Biol. 2005, 38: 204-299; Quintero-Hernandez et al., Mol. Immunol. 2007, 44: 1307-1315; Willems et al. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2003;786:161-176.)。
【0089】
特に、本発明のFab分子は、以下のようにCHO細胞から製造することができる。無血清培地中に懸濁状態で成長しているCHO−DG44細胞(Urlaub,G., Kas,E., Carothers,A.M., and Chasin,L.A. (1983). Deletion of the diploid dihydrofolate reductase locus from cultured mammalian cells. Cell 33, 405-412.)に、Lipofectamine(商標) and Plus(商標)試薬(Invitrogen)を製造業者の説明書にしたがって使用して、Fab分子の重鎖および軽鎖をコードする発現構築物をトランスフェクションする。48時間後に、200μg/mLの抗生物質G418を含有し、ヒポキサンチンおよびチミジンを有さない培地中で細胞を選択に供し、安定的にトランスフェクションされた細胞集団を発生させる。続いて、漸増する濃度(最大100または400nM)のメトトレキサート(MTX)を培養培地に添加することによって、これらの安定トランスフェクタントを遺伝子増幅に供する。いったん細胞が適応したならば、それらを10〜11日間流加発酵に供し、Fabタンパク質材料を製造する。
【0090】
化学的に限定された無血清培養培地中でCHO−DG44細胞およびその安定トランスフェクタントの懸濁培養物をインキュベーションする。播種用保存培養物を2〜3日毎にそれぞれ細胞3×105〜2×105個/mLの播種密度で継代培養する。細胞を振盪フラスコ中、Multitron HTインキュベーター(Infors)で5% CO2、37℃および120rpmで成長させる。流加実験のために、細胞を振盪フラスコの中で抗生物質もMTXも有さないBI独自の生産培地中に細胞3×105個/mLとなるように播種する。培養物を120rpmで37℃および5% CO2にて撹拌し、細胞数が増加するにつれてCO2をその後2%に減少させる。細胞数、生存率、pH、グルコースおよび乳酸濃度を含めた培養パラメーターを毎日決定し、必要に応じて炭酸塩を使用してpHをpH7.0に調整する。BI独自の栄養溶液を24時間毎に添加する。異なる時点で上清から試料を採取し、ELISAによってFab産物濃度を決定する。10〜11日後に、細胞培養液を遠心分離によって回収し、精製研究室に移送する。
【0091】
Fab分子を、流加培養の上清からクロマトグラフィーおよび濾過によって精製する。一次捕捉ステップとして、アフィニティークロマトグラフィー、例えばプロテインGまたはプロテインLを適用する。または、低結合親和性および低結合能の場合、Fabを、分子のpIを利用する陽イオン交換クロマトグラフィー(CEX)によって捕捉する。追加的な直交性の精製ステップによって、宿主細胞タンパク質および混入物、例えばDNAまたはウイルスを除去する。
【0092】
製造されたFab分子の同一性および製品品質を、電気泳動法、例えばSDS−PAGEによって分析し、それによって約50kDaの1本の主バンドとしてFabを検出することができる。Fab産物の特徴づけのためのさらなるアッセイには、質量分析、等電点電気泳動およびサイズ排除クロマトグラフィーが挙げられる。結合活性をBIAcore分析によって追跡する。
【0093】
細胞培養上清中のFabまたは完全長IgG分子の定量を、サンドイッチ酵素結合免疫吸着アッセイ(ELISA)により行う。完全長IgGは、ヒト−Fcフラグメント(Jackson Immuno Research Laboratories)およびヒトκ軽鎖に対して産生された抗体(ペルオキシダーゼ−コンジュゲーション型、Sigma)を使用して検出することができる。Fabフラグメントを、ヤギポリクローナル抗ヒトIgG(HおよびL、Novus)によって固定化し、ヒトIgGに対して産生されたヒツジポリクローナル抗体(ペルオキシダーゼ−コンジュゲーション型、結合部位)によって検出する。
【0094】
Fab分子は、また、酵素的切断によって完全長抗体分子から生成させることができる。このアプローチの利点は、所望の製品品質でのスケールアップおよび高収量が容易な、堅調で効率的な発酵および精製のための足場となるプロセスが適用可能であることである。精製のために、組換えプロテインA樹脂を使用するアフィニティークロマトグラフィーを一次捕捉ステップとして使用することができ、それは通常、高い純度をもたらす。
【0095】
この目的のために、Fab配列をコードする重鎖を、ヒトIgG抗体分子のFc領域に融合する。次に、得られた発現構築物を、無血清培地中に懸濁状態で成長しているCHO−DG44細胞にリポフェクションを用いてトランスフェクションする。48時間後に200μg/mLの抗生物質G418を含有しヒポキサンチンおよびチミジンを有さない培地中で細胞を選択に供し、安定トランスフェクションされた細胞集団を発生させた。続いて、メトトレキサート(MTX)を漸増する濃度(最大100または400nM)で培養培地に添加することによって、これらの安定トランスフェクタントを遺伝子増幅に供する。いったん細胞が適応したならば、それらを10〜11日間流加発酵に供し、IgGタンパク質材料を製造する。IgGタンパク質を、組換えプロテインA−アフィニティークロマトグラフィーを使用することによって培養上清から精製する。次に、所望の中和Fabフラグメントを得るために、ヒンジ領域内でIgGを切断するパパインの存在下で完全長IgGをインキュベーションし、それによって2本のFabフラグメントおよびFc部分を放出させる。配列番号:41のFd鎖を含むFab分子は、完全長IgGタンパク質のパパイン消化によって得られるFab分子の一例である。
【0096】
Fab分子を、アフィニティークロマトグラフィー、例えばプロテインGまたはプロテインLによって単離する。または、低結合親和性および低結合能の場合、Fabを、分子のpIを利用する陽イオン交換クロマトグラフィー(CEX)によって捕捉する。追加的な直交性の精製ステップによって宿主細胞タンパク質および混入物、例えばパパイン、DNAまたはウイルスを除去する。
【0097】
本発明の別の局面では、抗体分子は、本明細書記載の抗体分子のアミノ酸配列変異体である。
【0098】
抗体のアミノ酸配列変異体は、抗体DNAに適切なヌクレオチド変化を導入することによって、またはペプチド合成によって調製することができる。そのような変異体には、例えば本明細書における例に示される抗体のアミノ酸配列内の残基からの欠失および/またはその配列への挿入および/またはその配列の置換が挙げられる。最終構築物が所望の性質を有するという条件で、その最終構築物に到達するために欠失、挿入、および置換の任意の組み合わせが作られる。アミノ酸変化は、また、ヒト化または変異抗体の翻訳後プロセスを変え、グリコシル化部位の数または位置などを変化させることがある。
【0099】
突然変異誘発のために好ましい位置である、抗体のある種の残基または領域を同定するための有用な方法は、CunninghamおよびWells(Science, 244:1081-1085 (1989))によって記載されたような「アラニンスキャニング突然変異誘発」と呼ばれる。ここで、ターゲット残基のうちの一残基または群(例えばarg、asp、his、lys、およびgluなどの荷電残基)が同定され、アミノ酸と抗原の相互作用に影響するように中性または負電荷アミノ酸(典型的にはアラニン)によって置き換えられる。次に、置換に機能的感受性を示しているアミノ酸位置は、置換部位で、または置換部位の代わりにさらなる変異体または他の変異体を導入することによって洗練される。したがって、アミノ酸配列変異を導入するための部位が予め決定されている一方で、突然変異自体の性質が予め決定されている必要はない。例えば、所与の部位での突然変異の性能を分析するために、アラニンスキャニングまたはランダム突然変異誘発がターゲットのコドンまたは領域で行われ、発現された抗体変異体は、所望の活性についてスクリーニングされる。
【0100】
アミノ酸配列の挿入には、1個の残基から100個以上の残基を有するポリペプチドまでの長さの範囲のアミノ末端融合および/またはカルボキシル末端融合、ならびに一つ以上のアミノ酸残基の配列内挿入が挙げられる。末端挿入の例には、エピトープタグに融合された抗体が挙げられる。抗体分子の他の挿入変異体には、抗体のN末端もしくはC末端と、抗体の血清半減期を増加させる酵素またはポリペプチドとの融合体が挙げられる。
【0101】
別の型の変異体は、アミノ酸置換変異体である。これらの変異体は、抗体分子から少なくとも1個のアミノ酸残基が除去され、その位置に異なる残基が挿入されている。置換突然変異誘発のための最大に関心がもたれる部位には、超可変領域が挙げられるが、FRの変化も考えられる。保存的置換を、下表に「好ましい置換」の見出しのもとに示す。そのような置換が生物学的活性の変化を招くならば、「例示的な置換」と呼ばれるより実質的な変化またはアミノ酸クラスに関して下にさらに説明するような変化を導入して、その産物をスクリーニングすることができる。
【表1】
【0102】
タンパク質化学において、抗体の生物学的性質は、(a)置換領域におけるポリペプチド主鎖の、例えばシートまたはヘリカルコンフォメーションとしての構造、(b)ターゲット部位での分子の荷電もしくは疎水性、または(c)側鎖のかさを維持することに及ぼす作用が顕著に異なる置換を選択することによって達成できることが、一般に受け入れられている。天然残基は、共通の側鎖性質に基づき群に分類される:
(1)疎水性:ノルロイシン、met、ala、val、leu、ile;
(2)中性親水性:cys、ser、thr;
(3)酸性:asp、glu;
(4)塩基性:asn、gin、his、lys、arg;
(5)鎖の配向に影響する残基:gly、pro;および
(6)芳香族:trp、tyr、phe。
【0103】
非保存的置換は、これらのクラスのうち一つの一メンバーを別のクラスに交換することを伴うものである。
【0104】
ヒト化抗体または変異型抗体の適切なコンフォメーションを維持することに関与しない任意のシステイン残基も一般にセリンで置換して、分子の酸化的安定性を改善するか、異常な架橋を防止するか、または細胞毒性化合物もしくは細胞増殖抑制性化合物への所定の結合点を与えることができる。逆に、抗体にシステイン結合を加えて、その安定性を改善することができる(特にその抗体がFvフラグメントなどの抗体フラグメントである場合)。
【0105】
置換変異体の1種は、親抗体(例えばヒト化またはヒト抗体)の一つ以上の超可変領域残基を置換することを伴う。一般に、さらなる開発のために選択された、得られる変異体は、それらが生成された親抗体よりも改善された生物学的性質を有するものである。そのような置換変異体を生成させるために好都合な方法は、ファージディスプレイを用いたアフィニティー成熟である。簡潔には、いくつかの超可変領域部位(例えば6〜7個の部位)を突然変異させて、各部位で全ての可能なアミノ置換を生成させる。そのように生成された抗体変異体は、線維状ファージ粒子から、各粒子内にパッケージングされたM13の遺伝子III産物との融合物として一価的にディスプレイされる。次に、ファージディスプレイされた変異体をそれらの生物学的活性(例えば結合親和性)についてスクリーニングする。改変のための候補となる超可変領域部位を同定するために、アラニンスキャニング突然変異誘発を行って、抗原結合に顕著に寄与する超可変領域残基を同定することができる。または、あるいは加えて、抗原−抗体複合体の結晶構造を分析して、抗体とヒトダビガトランの間の接触点を同定することが有益なことがある。そのような接触残基および隣接残基は、本明細書に詳述される技法による置換のための候補である。そのような変異体がいったん生成されたならば、そのパネルの変異体を本明細書記載のスクリーニングに供し、一つ以上の関連するアッセイで優れた性質を有する抗体をさらなる開発のために選択することができる。
【0106】
抗体の別の種類のアミノ酸変異体は、抗体の本来のグリコシル化パターンを変える。「変えること」によって、抗体に見られる一つ以上の糖質部分を除去すること、および/またはその抗体に存在しない一つ以上のグリコシル化部位を付加することが意味される。
【0107】
いくつかの態様では、本発明の抗体を改変してグリコシル化部位を付加することが望ましいことがある。抗体のグリコシル化は、典型的にはN−結合型またはO−結合型のいずれかである。N−結合型は、アスパラギン残基側鎖に糖質部分が結合していることを表す。トリペプチド配列アスパラギン−X−セリンおよびアスパラギン−X−トレオニン(式中、Xはプロリン以外の任意のアミノ酸である)は、アスパラギン側鎖に糖質部分を酵素的に結合させるための認識配列である。したがって、ポリペプチド中にこれらのトリペプチド配列のいずれかが存在することで、潜在的グリコシル化部位が生じる。O−結合型グリコシル化は、ヒドロキシアミノ酸、最も一般的にはセリンまたはトレオニン(もっとも5−ヒドロキシプロリンまたは5−ヒドロキシリシンも使用することができる)に糖であるN−アセチルガラクトサミン、ガラクトース、またはキシロースの一つを結合させることを表す。したがって、所与のタンパク質、例えば抗体をグリコシル化するために、タンパク質のアミノ酸配列を操作して(N−結合型グリコシル化部位のために)一つ以上の上記トリペプチド配列を有するようにされる。その変化は、また、(O−結合型グリコシル化部位のための)本来の抗体配列への一つ以上のセリンまたはトレオニン残基の付加またはその残基による置換によって行うことができる。
【0108】
抗体のアミノ酸配列変異体をコードする核酸分子は、当技術分野で公知の多様な方法によって調製される。これらの方法には、非限定的に、天然起源からの単離(天然アミノ酸配列変異体の場合)または初期に調製された変異体もしくは非変異体版の本明細書記載の抗体分子のオリゴヌクレオチド介在性(もしくは部位特異的)突然変異誘発、PCR突然変異誘発、およびカセット突然変異誘発による調製が挙げられる。上記に概要するように、本発明の抗体分子の抗原は、抗凝固薬である。抗原は、動物の免疫処置によって、またはファージディスプレイ法を用いるような、配列ライブラリーから抗体配列を選択することによってのいずれかで抗体分子を生成させるために使用される。
【0109】
動物のための免疫処置プロトコールは、当技術分野において周知である。適切な免疫応答を達成するために、抗原をリン酸アルミニウム、水酸化アルミニウム、スクアレン、またはフロイントの完全/不完全アジュバントなどのアジュバントと結合することが必要なことがある。ダビガトランのような本発明に関連する抗原は、主に比較的小型の有機分子であって、動物に投与されたときに抗体の形成を刺激しないことがある有機分子である。したがって、抗原をハプテンとしての巨大分子に結合させることが必要なことがある。
【0110】
さらなる局面では、本発明は、医学に使用するための上記抗体分子に関する。
【0111】
さらなる局面では、本発明は、前記抗体分子および医薬担体を含む医薬組成物に関する。
【0112】
治療に使用するために、抗体分子は、動物またはヒトへの投与を容易にするために適切な医薬組成物に含まれる。抗体分子の典型的な製剤は、抗体分子を生理学的に許容されうる担体、賦形剤または安定化剤を混合することによって、凍結乾燥もしくはその他の方法で乾燥された製剤、あるいは水性溶液または水性もしくは非水性懸濁液の剤形に調製することができる。担体、賦形剤、改質剤(modifier)または安定化剤は、採用される薬用量および濃度で無毒である。それらには、リン酸、クエン酸、酢酸ならびに他の無機酸または有機酸およびそれらの塩などの緩衝系;アスコルビン酸およびメチオニンを含めた抗酸化剤;オクタデシルジメチルベンジルアンモニウムクロリド;塩化ヘキサメトニウム;塩化ベンザルコニウム、塩化ベンゼトニウム;フェノール、ブチルまたはベンジルアルコール;メチルまたはプロピルパラベンなどのアルキルパラベン;カテコール;レゾルシノール;シクロヘキサノール;3−ペンタノール;およびm−クレゾールなどの保存料);血清アルブミン、ゼラチン、または免疫グロブリンなどのタンパク質;ポリビニルピロリドンまたはポリエチレングリコール(PEG)などの親水性ポリマー;グリシン、グルタミン、アスパラギン、ヒスチジン、アルギニン、またはリシンなどのアミノ酸;グルコース、マンノース、スクロース、トレハロース、デキストリンまたはデキストランを含めた単糖、二糖、オリゴ糖または多糖および他の糖質;EDTAなどのキレート剤;マンニトールまたはソルビトールなどの糖アルコール;ナトリウムなどの塩形成性対イオン;金属錯体(例えば、Zn−タンパク質錯体);および/またはTWEEN(商標)(ポリソルベート)、PLURONICS(商標)もしくは脂肪酸エステル、脂肪酸エーテルもしくは糖エステルなどのイオン性もしくは非イオン性界面活性剤が挙げられる。有機溶媒もエタノールまたはイソプロパノールなどの抗体製剤に含有されることがある。賦形剤は、また、放出改変または吸収改変機能を有することがある。
【0113】
一局面では、医薬組成物は、水性緩衝液中に10〜20mg/mlの濃度で抗体分子を含むか、またはそのような溶液から作られた凍結乾燥物を含む。
【0114】
好ましい適用様式は、注入または注射(静脈内、筋肉内、皮下、腹腔内、皮内)による非経口的であるが、吸入、経皮、鼻腔内、口腔、経口などの他の適用様式もまた適用可能なことがある。
【0115】
さらなる局面では、本発明は、抗凝固治療の副作用、特に出血事象の治療または予防に使用するための上記抗体分子に関する。さらなる局面では、本発明は、抗凝固薬、特にダビガトランまたはダビガトランエテキシラートの過剰投与のリバースに使用するための上記抗体分子に関する。
【0116】
さらなる局面では、本発明は、抗凝固薬、特にダビガトランまたはダビガトランエテキシラートの解毒剤として使用するための上記抗体分子に関する。
【0117】
さらなる局面では、本発明は、抗凝固治療の副作用の治療または予防方法であって、それを必要とする患者に上記抗体分子の有効量を投与することを含む方法に関する。
【0118】
さらなる局面では、本発明は、抗凝固治療における過剰投与事象の処置方法であって、それを必要とする患者に上記抗体分子の有効量を投与することを含む方法に関する。
【0119】
投与されるべき抗体の「治療有効量」は、抗凝固治療の副作用を予防、改善、または治療するために必要な最小量、特に出血を止めるために有効な最小量である。これは、抗体分子の化学量論量で達成することができる。
【0120】
ダビガトランは、例えば、推奨用量で与えられた場合に200nMの大きさの血漿中濃度に達することがある。分子量約50kDの一価抗体分子が使用される場合、ボーラスとして静脈内に与えたとき例えば約1mg/kgの用量で中和に達することができる。別の態様では、ヒト患者に適用されたFab分子の用量は、適用1回あたり50〜1000mg、例えば100、200、500、750、または1000mgのことがある。状況に応じて、例えばダビガトランが患者に過剰投与されている場合、より高い用量、例えば適用1回あたり1250、1500、1750または2000mgを適用することさえ妥当なことがある。適切な用量は、投与される抗凝固薬の種類および用量;そのような投与から経過した時間、抗原分子の性質、患者の状態、ならびに他の要因に応じて異なることがある。熟練の専門家は、治療的に有効かつ安全な用量を確立するための方法を知っている。
【0121】
さらなる局面では、本発明は、ダビガトランおよび/またはダビガトランエテキシラートに対する結合親和性を有する抗体分子に関する。好ましくは、抗体分子は、例えば表面プラズモン共鳴分析(Malmqvist M., "Surface plasmon resonance for detection and measurement of antibody-antigen affinity and kinetics. "Curr Opin Immunol. 1993 Apr;5(2):282-6.)または動力学排除アッセイ(KinExA)技法(Darling, R.J., and Brault P-A., "Kinetic exclusion assay technology: Characterization of Molecular Interactions." ASSAY and Drug Development Technologies. 2004, Dec 2(6): 647-657)により決定されるとき、0.1pM〜100μM、好ましくは1pM〜100μM、より好ましくは1pM〜1μMの範囲のKD値を有する親和性でダビガトランおよび/またはダビガトランエテキシラートに結合する。
【0122】
本発明の抗体分子を、また、分析および診断手順のために使用して、例えば血漿、血清、または他の体液などの試料中の抗原濃度を決定することができる。例えば、抗原分子は、実施例に記載されたような酵素結合免疫吸着アッセイ(ELISA)に使用することができる。したがって、さらなる局面では、本発明は、本明細書記載の抗体分子を含む分析キットおよび診断キットならびにそれぞれの分析方法および診断方法に関する。
【0123】
さらなる局面では、本発明は、前記請求項のいずれか一項記載の抗体分子を製造する方法であって、
(a)発現制御配列と機能的に関連して該抗体分子をコードする一つ以上の核酸を含む宿主細胞を提供すること、
(b)該宿主細胞を培養すること、および
(c)細胞培養物から抗体分子を回収すること
を含む方法に関する。
【0124】
本発明は、さらに、経口抗凝固薬、特に直接トロンビン阻害剤の中和に有用な材料が入っている製品およびキットを提供する。製品は、ラベル付きの容器を含む。適切な容器には、例えばボトル、バイアル、および試験管が挙げられる。容器は、ガラス、金属、プラスチックまたはその組み合わせなどの多様な材料から形成されていることがある。容器は、本明細書記載の抗体またはダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩を含む医薬組成物を収容する。医薬組成物中の活性薬剤は、特定の抗体またはダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩である。抗体の容器上のラベルは、その医薬組成物がダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩を中和または部分的に中和するためにin vivoで使用されることを表示している。
【0125】
本発明のキットは、一つ以上の上記容器を含む。そのキットは、さらに、他の緩衝液、希釈剤、フィルター、針、シリンジ、および使用法を掲載した添付文書を含めた、商業的および利用者の観点から望ましい他の材料を包含することができる。
【0126】
本発明の一態様では、キットは、本明細書記載の任意の一つの抗体である抗体またはその医薬組成物を含む。例えば、キットは、(1)本明細書記載の任意の一つの抗体またはその医薬組成物、(2)容器および(3)ラベルを含むことができる。
【0127】
別の態様では、キットは、本明細書記載の任意の一つの抗体である抗体またはその医薬組成物、およびダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩を含む。ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の形態は、固体、液体またはゲルの形態のことがある。好ましい態様では、ダビガトランエテキシラートの薬学的に許容されうる塩は、メシラート塩である。なお別の好ましい態様では、投薬ユニットあたりのダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の量は、約50mg〜約400mg、約75mg〜約300mg、約75mg〜150mg、または約110mg〜約150mgの間であって、1日1回(QD)または1日2回(BID)与えられる。例えば、キットは、(1)本明細書記載の任意の一つの抗体またはその医薬組成物、(2)ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の医薬組成物、(3)容器および(4)ラベルを含むことがある。
【0128】
代替の態様では、キットは、(1)ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩を含む第1の医薬組成物、(2)本明細書記載の抗体の任意の一つまたはその組み合わせを含む第2の医薬組成物、(3)患者に該第1および第2の医薬組成物を別々に投与するための説明書を含み、ここで、該第1および第2の医薬組成物は、別々の容器に入っており、該第2の医薬組成物は、ダビガトランまたはダビガトランの1−O−アシルグルクロニドの中和または部分中和を必要とする患者に投与される。本発明は、また、ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩で処置されている患者においてダビガトランまたはダビガトランの1−O−アシルグルクロニドを中和または部分中和するための診断方法であって、本明細書記載の抗体の任意の一つ、その組み合わせまたはその医薬組成物を投与することを含む方法を提供する。具体的には、本発明は、患者においてダビガトランまたはダビガトランの1−O−アシルグルクロニドを中和または部分中和するための方法であって、(a)患者がダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩で処置されていたこと、およびその患者によって取り込まれた量を確認するステップ;(b)ダビガトランまたはダビガトランの1−O−アシルグルクロニドが試験またはアッセイの結果の正確な読み出しを妨害するであろう試験またはアッセイを行う前に、本明細書記載の抗体の任意の一つまたはその組み合わせでダビガトランまたは1−O−アシルグルクロニドを中和するステップ;(c)ダビガトランもダビガトランの1−O−アシルグルクロニドも存在しないときの血餅形成レベルを決定するために、患者から採取された試料に凝固または血液凝固の試験またはアッセイを行うステップ;ならびに(d)患者における血餅形成と分解の間に適切なバランスを達成するために、患者に投与されるダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の量を調整するステップを含む方法を提供する。ダビガトランまたはダビガトランの1−O−アシルグルクロニドに対する抗体のモル比は、0.1〜100の間、好ましくは0.1〜10の間のモル比である。試験またはアッセイの結果の正確な読み出しは、フィブリノーゲンレベル、活性化プロテインC抵抗性または関連試験の正確な読み出しのことがある。
【0129】
実施例
I. ポリクローナル抗ダビガトラン抗体の製造
ポリクローナル抗ダビガトラン抗体の製造のために、2種の異なるハプテンおよびハプテンと担体タンパク質(BSA)の異なるモル投入量比で、3種の異なる免疫原を製造した。
【0130】
スクリーニングのために、酵素ホースラディッシュペルオキシダーゼ(HRP)−コンジュゲートを製造し、酵素免疫吸着アッセイ(ELISA)を開発した。
【0131】
ポリクローナル抗体のさらなる精製は、プロテインAセファロース FFを用いたアフィニティークロマトグラフィーによって行った。
【0132】
1. 材料および方法
【表2】
【0133】
1.1 免疫原およびトレーサーを合成するために使用されたハプテン
【表3】
【0134】
【表4】
【0135】
1.2 ハプテンの合成
ハプテンである、ハプテン1およびハプテン2を以下のように合成した:
ハプテン1 2−[(4−カルバムイミドイル−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボン酸[2−(4−アミノ−ブチルカルバモイル)−エチル]−フェニル−アミド
【化5】
【0136】
1a 3−[(4−メチルアミノ−3−ニトロ−ベンゾイル)−フェニル−アミノ]−プロピオン酸メチルエステル
【化6】
4−メチルアミノ−3−ニトロ−安息香酸クロリド(23.3mmol)および3−フェニル−アミノ−プロピオン酸メチルエステル(23.3mmol)の無水テトラヒドロフラン(THF)80mL中の溶液に、トリエチルアミン(50.2mmol)を撹拌しながら室温で滴下により加えた。3時間後に反応混合物を蒸発乾固し、残りの固体を水で粉砕し、固体生成物を濾過により単離した。
収率:99%
C18H19N3O5(357.36)
TLC(シリカゲル;ジクロロメタン/エタノール 19:1):Rf=0.48
【0137】
1b 3−[(3−アミノ−4−メチルアミノ−ベンゾイル)−フェニル−アミノ]−プロピオン酸メチルエステル
【化7】
触媒としてPd(炭上に10%)を用いてエタノール中で室温で水素化することによって生成物1aのニトロ基を還元した。
収率:99%
C18H21N3O3(327.38)
TLC(シリカゲル;ジクロロメタン/エタノール 9:1):Rf=0.23
質量スペクトル(ESI):[M+H]+=328
【0138】
1c 3−({3−[2−(4−シアノ−フェニルアミノ)−アセチルアミノ]−4−メチルアミノ−ベンゾイル}−フェニル−アミノ)−プロピオン酸メチルエステル
【化8】
生成物1b(23.2mmol)およびN−(4−シアノ−フェニル)−グリシン(23.2mmol)を無水THF中で室温にてCDI(23.2mmol)でカップリングさせた。反応の完了後に、その混合物を蒸発乾固させ、粗生成物をさらに精製せずに使用した。
収率:97%
C27H27N5O4(485.54)
質量スペクトル(ESI):[M+H]+=486
【0139】
1d 3−({2−[(4−シアノ−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボニル}−フェニル−アミノ)−プロピオン酸メチルエステル
【化9】
生成物1c(22.6mmol)の濃酢酸100mL中の溶液を1時間加熱還流した。次に、その溶液を蒸発乾固し、残った固体を水で粉砕し、撹拌しながらpHを約8〜9に調整した。酢酸エチルで抽出することによって粗生成物を単離し、シリカゲルを用いたクロマトグラフィー(溶離液:ジクロロメタン/エタノール 1:1)によって精製した。
収率:58%
C27H25N5O3(467.52)
TLC(シリカゲル;ジクロロメタン/エタノール 9:1):Rf=0.71
質量スペクトル(ESI):[M+H]+=468
【0140】
1e 3−({2−[(4−シアノ−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボニル}−フェニル−アミノ)−プロピオン酸
【化10】
生成物1d(13.0mmol)の100mLメタノール中の溶液に、水酸化ナトリウム(20.0mmol)を添加した。その混合物を40℃で2.5時間撹拌し、次に蒸発乾固した。残った固体を水100mLと一緒に撹拌し、そして濃酢酸でpHを約6に調整した。沈殿した生成物を濾過によって単離し、水で洗浄し、60℃で乾燥させた。
収率:88%
C26H23N5O3(453.49)
TLC(シリカゲル;ジクロロメタン/エタノール 9:1):Rf=0.33
質量スペクトル(ESI):[M+H]+=454
【0141】
1f {4−[3−({2−[(4−シアノ−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボニル}−フェニル−アミノ)−プロピオニルアミノ]−ブチル}−カルバミン酸tert−ブチルエステル
【化11】
生成物1e(5.23mmol)、2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウム テトラフルオロボレート(TBTU、5.23mmol)およびN−メチル−モルホリン(5.23mmol)のDMF 20mL中の溶液を室温で30分間撹拌した。次に、(4−アミノ−ブチル)−カルバミン酸tert−ブチルエステル(5.23mmol)を添加し、その混合物を室温で新たに24時間撹拌した。次に、その混合物を水(100mL)で希釈し、酢酸エチルで抽出することによって生成物を単離した。
収率:92%
C35H41N7O4(623.75)
TLC(シリカゲル;ジクロロメタン/エタノール 9:1):Rf=0.51
【0142】
1g 2−[(4−カルバムイミドイル−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボン酸[2−(4−アミノ−ブチルカルバモイル)−エチル]−フェニル−アミド
【化12】
生成物1f(4.81mmol)をHClの飽和エタノール溶液(250mL)に溶解させ、その混合物を室温で一晩撹拌し、次に30℃で蒸発乾固した。残った原材料を無水エタノール200mLに溶解させ、次に炭酸アンモニウム(48.1mmol)を添加し、その混合物を室温で一晩撹拌した。溶媒の蒸発後に、残った原材料をエタノール約5mLと一緒に摩砕し、不溶性物質を濾過によって分離し、溶媒を30℃で蒸発させた。次に、生成物を水30mLに溶解させ、溶液を約2gの炭と一緒に撹拌し、濾過し、蒸発乾固した。
収率:90%
C30H36N8O2(540.67)
TLC(逆相RP−8;メタノール/5%NaCl水溶液 9:1):Rf=0.79
質量スペクトル(ESI):[M+H]+=541
[M+Cl]−=575/7
【0143】
ハプテン2 2−[(4−カルバムイミドイル−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボン酸[2−(2−アミノ−エチルカルバモイル)−エチル]−ピリジン−2−イル−アミド
【化13】
【0144】
2a 3−({2−[(4−シアノ−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボニル}−ピリジン−2−イル−アミノ)−プロピオン酸
【化14】
水酸化ナトリウム(50.0mmol)のエタノール500mLおよび水50mL中の溶液に、3−({2−[(4−シアノ−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボニル}−ピリジン−2−イル−アミノ)−プロピオン酸エチルエステル(41.4mmol)を添加した。その混合物を室温で3時間撹拌し、次にエタノール約350mLを留出させ、水約100mLを添加し、pHを6に調整した。次にジエチルエーテル(50mL)を添加し、その混合物を一晩撹拌した。生成物を濾過により単離し、さらに精製せずに使用した。
収率:78%
C25H22N6O3(454.48)
【0145】
2b {2−[3−({2−[(4−シアノ−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボニル}−ピリジン−2−イル−アミノ)−プロピオニルアミノ]−エチル}−カルバミン酸tert−ブチルエステル
【化15】
生成物2a(2.20mmol)、2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムテトラフルオロボレート(TBTU、2.20mmol)およびN−メチル−モルホリン(2.20mmol)の無水テトラヒドロフラン(100mL)中の溶液を室温で15分間撹拌した。次に(2−アミノ−エチル)−カルバミン酸tert−ブチルエステル(2.20mmol)を添加し、そしてその混合物を室温で新たに24時間撹拌した。次にその混合物を水40mLで希釈し、酢酸エチルで抽出することによって生成物を単離し、クロマトグラフィー(シリカゲル;ジクロロメタン/メタノール 15:1)によって精製した。
収率:61%
C32H36N8O4(596.68)
質量スペクトル(ESI):[M+H]+=597
[M+H]−=595
【0146】
2c 2−[(4−カルバムイミドイル−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボン酸[2−(2−アミノ−エチルカルバモイル)−エチル]−ピリジン−2−イル−アミド
【化16】
生成物2b(1.34mmol)をHClの飽和無水エタノール溶液(30mL)に添加した。その溶液を室温で5時間撹拌し、次に30℃で蒸発乾固した。エタノール(30mL)および炭酸アンモニウム(13.0mmol)を添加し、そしてその混合物を室温で一晩撹拌した。次に溶媒を蒸発させ、無機塩からの生成物を分離するために残った物質をジクロロメタン/メタノール(30:1)混合物約4mLで5回粉砕し、濾過し、蒸発させた。
収率:27%
C27H31N9O2(513.61)
質量スペクトル(ESI):[M+Cl]−=548/50
[M+HCl+Cl]−=584/6
[M+H]+=514
【0147】
2. 化学物質
2.1 試薬合成用の化学物質
【表5】
【0148】
2.2 ELISA用化学物質
【表6】
【0149】
2.3 ELISA用緩衝液
【表7】
【0150】
3. 免疫原の合成
ウサギの免疫系を刺激してダビガトランに対するポリクローナル抗体を生成するために、カップリング試薬として1,4−ベンゾキノンまたは1,1’−カルボニル−ジ−(1,2,4−トリアゾール)を使用して、ハプテンであるハプテン1およびハプテン2を担体タンパク質であるウシ血清アルブミン(BSA)にカップリングさせることによって、3種の免疫原(ロット番号GL256、GL258、およびGL262)を合成した。
【0151】
GL256の合成のために、2個の反応部位を有するホモ二官能性化合物として1,4−ベンゾキノンを使用した。まず、それは酸性pHで2個の部位のうち一方のアミノ基だけと反応し、そして最小限のポリマー化で、アルカリ性pHでもう一方の部位で反応する。GL258およびGL262は、ハプテンに対する担体タンパク質の異なる投入量比で、1,1’−カルボニル−ジ−(1,2,4−トリアゾール)をカップリング試薬として使用して合成した。
【0152】
3.1 GL256の合成
0.1M KH2PO4緩衝液8.5mL中の0.75μMol BSAの溶液(pH=4.5)に、0.416mMol 1,4−ベンゾキノン(エタノール1.5mL中)を添加し、そして室温、暗中で1.5時間インキュベーションした。その後0.15M NaClで平衡化したセファデックスG25カラムにその溶液を通過させ、過剰の1,4−ベンゾキノン(最終体積12.5mL)を除去した。
【0153】
0.1M NaHCO3/Na2CO3緩衝液(pH=8.5)2mLに溶解された525μMolハプテンであるハプテン1の溶液に、撹拌しながら精製BSA溶液2.5mL(0.15μMol)をゆっくりと添加した。BSA溶液を添加する間に、pHを約8.0に調整した。ハプテンと担体タンパク質のモル投入量比は、3500:1であった。
【0154】
室温で一晩インキュベーション後に、免疫原を1リットルの蒸留水で6回透析した。薄層クロマトグラフィーは、ハプテン−担体コンジュゲート中に残った非結合ハプテンのスポットを示さなかった。
【0155】
免疫原を小分けして−20℃で凍結保存した。免疫原の上清中のハプテンによるBSAの置換度は、302nmでUV吸収分光法によって決定したとおり、約1:18であった。最終溶液中の免疫原の含量は、GL256/mL 0.75mgであった。
【0156】
3.2 GL258の合成
N,N−ジメチルホルムアミド(DMF)6.3mL中の158μMolハプテン2の溶液を室温で調製した。158μMol 1,1’−カルボニル−ジ−(1,2,4−トリアゾール)を添加し、そしてまず10℃で4時間、そしてその後室温で30分間インキュベーションした。薄層クロマトグラフィーで化学反応をチェックし、そしてそれは約20〜25%であった。次に、0.75μMol BSAを0.13M NaHCO3 2mLに溶解させ、撹拌しながらN,N−ジメチルホルムアミド(DMF)1mLを滴下により加えた。pHを約8.3に調整した。その後、撹拌しながらハプテン溶液(6.3mL)および0.13M NaHCO3 4mLをBSA溶液に滴下により加え、pHを8.4に調整した。免疫原GL258についてハプテンと担体タンパク質のモル投入量比は210:1であった。
【0157】
室温で一晩、撹拌条件下でインキュベーション後に、免疫原を1リットルの蒸留水で6回透析した。薄層クロマトグラフィーは、ハプテン−担体コンジュゲート中に残った非結合ハプテンのスポットを示さなかった。
【0158】
免疫原を小分けにして−20℃で凍結保存した。免疫原の上清中のハプテンによるBSAの置換度は、302nmでUV吸収分光法によって決定したとき約1:5であった。最終溶液中の免疫原の含量は、GL258/mL 0.28mgであった。
【0159】
3.3 GL262の合成
N,N−ジメチルホルムアミド(DMF)8.75mL中の225μMolハプテン2の溶液を室温で調製した。225μMol 1,1’−カルボニル−ジ−(1,2,4−トリアゾール)を添加し、そして10℃で4時間インキュベーションした。薄層クロマトグラフィーで化学反応をチェックし、それは約20〜25%であった。
【0160】
次に0.49μMol BSAを0.13M NaHCO3 2mLに溶解させ、撹拌しながらN,N−ジメチルホルムアミド(DMF)1mLを滴下により加えた。pHを約8.2に調整した。その後、撹拌しながらハプテン溶液(8.75mL)および0.13M NaHCO3 6mLをBSA溶液に滴下により加え、pHを8.3に調整した。免疫原GL262についてハプテンと担体タンパク質のモル投入量比は、460:1であった。
【0161】
室温で一晩、撹拌条件下でインキュベーション後に、免疫原を1リットルの蒸留水で6回透析した。薄層クロマトグラフィーは、ハプテン−担体コンジュゲート中に残った非結合ハプテンのスポットを示さなかった。
【0162】
免疫原を小分けにして−20℃で凍結保存した。免疫原の上清中のハプテンによるBSAの置換度は、302nmでUV吸収分光法によって決定したとき約1:32であった。最終溶液中の免疫原の含量は、GL262/mL 0.71mgであった。
【0163】
4. コンジュゲートの合成
4.1 GL261の合成
N,N−ジメチルホルムアミド(DMF)1.5mL中の37.4μMolハプテン2の溶液を室温で調製した。37.5μMol 1,1’−カルボニル−ジ−(1,2,4−トリアゾール)を添加し、そしてまず10℃で4時間、そしてその後室温で30分間インキュベーションした。薄層クロマトグラフィーで化学反応をチェックし、それは約20〜25%であった。
【0164】
次に1.125μMol酵素ホースラディッシュペルオキシダーゼ(HRP)を0.13M NaHCO3 0.4mLに溶解させ、撹拌しながらN,N−ジメチルホルムアミド(DMF)0.267mLを滴下により加えた。pHを約8.2に調整した。その後、撹拌しながらハプテン溶液(22.5μMol)0.9mLおよび0.13M NaHCO3 0.57mLをHRP溶液に滴下により加え、そしてpHを8.4に調整した。HRPコンジュゲートGL261についてハプテンとHRPのモル投入量比は、20:1であった。
【0165】
室温で一晩、撹拌条件下でインキュベーション後に、HRPコンジュゲートを有機溶媒および過剰のハプテンからゲルクロマトグラフィーによって分離した。0.1Mリン酸緩衝液(pH7.0)で平衡化したセファデックスG25カラムにその溶液を通過させた。最終濃度のハプテン−HRPコンジュゲート(トレーサー、5.64mg/mL)は、BSAによりスパイクされ、濃度約10mg/mLで得られ、凍結防止のために等体積のグリセリン、および細菌の成長を防止するためにチモールの結晶を加えた。トレーサー溶液をロット番号GL261とラベルして、小分けして−20℃で保存した。
【0166】
ハプテンによるHRPの置換度は、302nmでUV吸収分光法によって決定したとき1:0.2であった。
【0167】
BSAでブロッキングされたマイクロタイタープレート中、基質としてo−フェニレン−ジアミン(OPD)および対照物質としてネイティブなHRPを使用してトレーサーの比活性を測定した。希釈されたHRP標準、またはハプテン−HRPコンジュゲート及び基質溶液との混合物を暗中で、30分間インキュベーションし、硫酸で停止させ、吸光度を490nmで測定した。残りの活性はネイティブなHRPの94%であって、グリセリン中でのコンジュゲート形成の比活性は611U/mLであった。
【0168】
トレーサーの特定の概要:
【0169】
【表8】
【0170】
5 免疫処置および抗体の製造
5.1 ウサギの免疫処置
12匹の雌チンチラウサギ(3月齢)を、100μg免疫原GL256、GL258およびGL262の、0.9% NaCl溶液0.5mLおよびフロイント完全アジュバント(CFA)0.5mLのエマルションで免疫化した。何回かの追加免疫処置を翌月に続けた。3回目の免疫処置については、フロイント不完全アジュバント(IFA)0.5mLを使用した。4箇所の皮下部位および4箇所の筋肉内部位に各免疫処置を行った。
【0171】
A群 − 免疫原GL256
ウサギ1 #50
ウサギ2 #51
ウサギ3 #52
ウサギ4 #53
B群 − 免疫原GL258
ウサギ5 #54
ウサギ6 #55
ウサギ7 #56
ウサギ8 #57
C群 − 免疫原GL262
ウサギ9 #46
ウサギ10 #47
ウサギ11 #48
ウサギ12 #49
【0172】
【表9】
*ウサギ番号1〜12を5回目の免疫処置の10日後に完全に瀉血した。
キシラジン(Rompun(登録商標), Bayer, Leverkusen, Germany)および塩酸ケタミン(Ketavet(登録商標), Parke-Davis, Freiburg, Germany)での麻酔下で、頸動脈を介して瀉血を行った。
【0173】
5.2 ウサギ血清の分析
凝固したウサギ血液の遠心分離によって血清を調製した。硫酸アンモニウム沈殿およびセファデックスG25カラム内に通過させる脱塩によってタンパク質画分を得た。
【0174】
ウサギ血清からの個別のタンパク質画分を標準ELISA法によって抗ダビガトラン力価についてスクリーニングした。
【0175】
スクリーニング−ELISA
【表10】
【0176】
5.3 ウサギ血清中の抗ダビガトラン抗体の検出
最後の3個の欄:値はダビガトランに関するものである。
【0177】
【表11】
【0178】
採血2からの全ウサギのタンパク質画分のスクリーニング後に、ウサギ番号5(#54)が好ましいハプテンであるハプテン2で、最も高い力価の抗ダビガトラン抗体を有することが明らかとなった。さらに、低濃度の分析物(ダビガトラン)だけで抗体結合部位からトレーサーを置換させることが可能であった。
【0179】
最終の採血3のスクリーニングについて、使用された免疫原に関する情報が欠落しているので、低濃度の分析物(ダビガトラン)を用いた抗体結合部位からのトレーサーの置換を主な決定基準として使用した。したがって、ウサギ番号2、3および5をさらなる精製のために使用した。
【0180】
5.4 ポリクローナル抗体の精製
ウサギ番号5(#54)、採血番号2およびウサギ番号2、3および5採血番号3(最後の採血)の抗血清を硫酸アンモニウムで沈殿させた。沈殿を4500U/min、10℃で30分間遠心分離し、溶液から分離し、トリス緩衝液中に再溶解させた。この手順を繰り返した。プロテインAセファロースFFを用いたアフィニティークロマトグラフィーによってさらなる精製を行った。カラム緩衝液は0.01Mトリス(pH=7.5)であり、そして0.1Mグリシン(pH=3.0)を溶離のために使用した。ウサギIgGを含有する画分を合わせた。タンパク質濃度は、280nmでのUV分光法によって決定した。
【0181】
抗体の特定の概要:
【表12】
【0182】
II. ダビガトランの中和
2系列の実験を行って、ダビガトラン抗凝固活性に対する抗体のin vitro作用を示した。4種のポリクローナル抗体を実験室に受け入れ、ヒト血漿でさらに試験した。これを機能的アッセイであるトロンビン凝固時間で試験した。
【0183】
アッセイの説明:
簡潔には、3.13%クエン酸ナトリウムへ全血を入れて、ヒト血漿を得る。次に、これを遠心分離して無血小板血漿を得て、そして別個のチューブに移し、アッセイの日に必要とされるまで凍結する。アッセイの日に血漿を37℃で解凍する。
【0184】
トロンビン凝固時間は以下のように行う。最初に、提供された緩衝液(Dade Behring Test kit)中に、製造業者の説明書通りにトロンビンを希釈し(3IU/mLトロンビン)、37℃に予熱する。それは、調製してから2時間以内に使用する。全てのアッセイを市販のCL4凝固測定機(Behnk Electronics, Norderstadt, Germany)で行った。磁気スターラーの入った、提供されたキュベットに血漿50μLをピペットで入れ、CL4機で37℃に予熱されたウェルの中で2分間撹拌させる。この時点でトロンビン溶液100μLを添加し、血漿試料が凝固するために必要な時間をCL4によって自動的に記録する。提供されたキュベット中の血漿に入れてダビガトランを5分間予備インキュベーションしてからトロンビンを添加し、測定を開始する。抗体も試験する場合(原液50μLまで)、37℃でさらに5分間インキュベーションしてから凝固を開始する(すなわちダビガトランと一緒に合計10分間インキュベーション、抗体と一緒に合計5分間インキュベーション、次にトロンビンで凝固を開始)。
【0185】
最初に、ヒト血漿に漸増する濃度のダビガトランを添加し、トロンビンを添加後に凝固までの時間を測定することによって、ダビガトランの標準曲線を作成した(図1)。漸増する濃度のダビガトランと一緒にトロンビン凝固時間の濃度依存的増加があった。
【0186】
最初のセットの中和実験について、臨床的に関連する濃度である200nMのダビガトランを中和のために全血漿試料に添加した。4個の抗体調製物は、全て、ダビガトランを含有する血漿での凝固時間を短縮することができた(図2)。中和度は、各抗体調製物中のタンパク質濃度に関連した。次に、最も高い濃度を有する抗体溶液(D)を系列希釈し、別々の実験セットで200nMダビガトランの抗凝固活性を中和する能力について試験した。図3から、抗体濃度が増加するとともに、ダビガトラン誘導抗凝固活性の濃度依存的阻害があった。加えて、非特異的ウサギポリクローナル抗体(青色四角)をダビガトラン含有血漿に添加した場合、それは、抗凝固活性を中和する能力を有さなかった。濃度依存性および非特異的抗体の中和欠如は、抗体による抗血液凝固のリバースがダビガトランに特異的であることを示している。
【0187】
しかし、これらの濃度のダビガトランは臨床的に意義があるものであって、出血または過剰投与は、おそらくより高い濃度で起こるものである。したがって、抗体が、図1の標準曲線における最も高い濃度のダビガトラン(500nM)の抗凝固活性を阻害する能力も試験した。図4に、抗体Dが高濃度のダビガトランも阻害することができたことを図示する。
【0188】
III. モノクローナル抗ダビガトラン抗体の製造および特徴づけ
1. モノクローナル抗ダビガトラン抗体およびFabの製造
ヘモシアニンおよび免疫グロブリンなどの担体タンパク質にコンジュゲーションしたハプテン1(実施例1.1参照)をマウスに免疫処置し、標準的な手順によりハイブリドーマを発生させた。培養上清から精製されたモノクローナル抗体は、ダビガトラン−タンパク質コンジュゲートに結合し、そしてこの結合を溶液中のダビガトランと1〜10nMの範囲の濃度の最大半値阻害で競合することができた。モノクローナル抗体のパパイン切断に続く、プロテインAによるFcドメインの除去によってFabを生成させた。
【0189】
マウス抗体の重鎖および軽鎖の可変領域をクローニングし、標準法を用いて配列決定した。抗体の質量分析およびN−末端配列決定によるタンパク質分析によって配列を確認した。特異的マウス可変領域およびヒトIgG定常領域を含むキメラ抗体をコードするDNA構築物を生成させ、HEK293T細胞からタンパク質を発現させ、それを精製した。
【0190】
2. モノクローナル抗ダビガトラン抗体およびFabの特徴づけ
3種のモノクローナル抗体クローンの可変ドメイン配列を図5および6に示す。クローン13の可変ドメインのアミノ酸配列を図5(DBG 13 VH、重鎖、配列番号:16)および図6(DBG 13 VK、軽鎖、配列番号:17)に示す。クローン14の可変ドメインのアミノ酸配列を図5(DBG 14 VH、重鎖、配列番号:18)および図6(DBG 14 VK、軽鎖、配列番号:19)に示す。クローン22の可変ドメインのアミノ酸配列を図5(DBG 22 VH、重鎖、配列番号:20)および図6(DBG 22 VK、軽鎖、配列番号:21)に示す。
【0191】
マウスモノクローナル抗体クローン22が、実施例IIに概要されたトロンビン凝固時間アッセイにおいてヒト血漿中のダビガトランの抗凝固活性を中和する能力について、試験した。その抗体は、ヒト血漿中のトロンビン依存性凝固のダビガトラン介在性延長を用量依存的に完全にリバースした(図7)。その抗体は、また、ヒト全血中のダビガトランの機能を効果的に阻害した。この抗体から生成されたFabは、ヒト血漿におけるダビガトラン活性を遮断したが、このことは、一価の抗原結合ドメインが化合物の抗凝固活性を中和することができることを示している(図8)。
【0192】
ヒトでのダビガトランの主要代謝経路は、カルボン酸部分のグルクロン酸抱合による。ダビガトランアシルグルクロニドは、薬理学的に活性であることが示されている(Ebner et al., Drug Metab. Dispos. 2010, 38(9):1567-75)。マウスモノクローナル抗体クローン22がこれらの代謝物を中和できるかどうかを試験するために、ダビガトランで処置されたアカゲザルの尿からダビガトランアシルグルクロニドを精製し、トロンビン凝固時間アッセイで評価した。抗体は、ヒト血漿におけるトロンビン依存性凝固のダビガトランアシルグルクロニド介在性延長を、ダビガトランで見られたものと類似の効力で用量依存的にリバースした(図9)。したがって、抗体は、ヒトで見られるダビガトラン代謝物の抗凝固活性の遮断に有効である。
【0193】
Fabと、クローン22、13および14の可変ドメインならびにヒト免疫グロブリン定常領域(軽鎖定常領域:配列番号:44;重鎖定常領域:配列番号:45)を含むマウス−ヒトキメラ抗体との親和性は、Kinexa技法を用いて決定した。平衡に到達するまで様々な濃度のダビガトランで、一定濃度のFabまたはキメラ抗体をインキュベーションした。このインキュベーションの後に、ビオチン−コンジュゲーション型ダビガトランアナログとカップリングされたニュートラアビジンビーズ上に抗体を捕捉することによって、遊離抗体の濃度を決定した。捕捉されたFabを、FITCでラベルされた抗マウスIgG(Fab特異的)F(ab’)2フラグメントを用いて検出した。捕捉されたキメラ抗体を、Cy5とコンジュゲーションされた抗ヒトIgGを用いて検出した。解離定数は1:1結合モデルを用いて計算した。これらの実験からの結果を下記表に概要する。
【0194】
抗ダビガトラン抗体の親和性
【表13】
【0195】
Fabとキメラ抗体の両方が高親和性でダビガトランと結合する。
【0196】
3. ヒト化モノクローナル抗ダビガトラン抗体およびFabの生成
人間に投与後の潜在的免疫原性を減少させるために、マウスモノクローナル抗体を「ヒト化」した。マウス先導物のために、フレームワークの相同性、CDRの構造、保存されたカノニカル残基、保存された界面充填残基および他のパラメーターに基づきヒトフレームワーク配列を選択した。これらのフレームワーク位置におけるアミノ酸残基の特異的置換は、結合親和性および/または安定性を含めた抗体の性能の様々な局面を、ヒト生殖細胞系フレームワーク領域へのCDRまたはHVLの「直接スワップ」によって形成されたヒト化抗体において実証されたものよりも改善することができる。キメラ親Fabに比べて、より良いまたは同等の結合と改善された発現とを示したFabを、さらなる特徴づけのために選択した。ヒト化Fabの可変ドメインのアミノ酸配列を、図5(Eng VH 14、配列番号:22;ENG VH 15、配列番号:24;およびENG VH 31、配列番号:26)および図6(Eng VK 11、配列番号:23;ENG VK 17、配列番号:25;およびENG VK 18、配列番号:27)に示す。Eng VH 15およびEng VK 18を含むFab(軽鎖:配列番号:37;重鎖:配列番号:36)をCHO細胞に直接発現させ、カッパー選択およびプロテインG樹脂を使用して精製した。
【0197】
Eng VH 15およびEng VK 18を含むFabもまた、IgG1KO形式での完全長IgG(軽鎖:配列番号:35;重鎖:配列番号:40)に変換した。IgG1KO(エフェクター機能のノックアウト)は、FcgRおよび補体の結合などのエフェクター機能を減少させる、Fc領域中の2個の突然変異、Leu234AlaおよびLeu235Alaを有する。IgG形式は、文献に記載されている(例えばHezareh et al. (2001) Journal of Virology 75: 12161-12168参照)。ヒト化抗ダビガトラン抗体は、場合により、コンセンサス領域または生殖系フレームワーク領域に特異的アミノ酸置換を含む。18/15抗体をHEK293T細胞またはCHO細胞に発現させ、精製した。インタクトな抗体のLys−Cまたはパパイン切断のいずれかによってFabフラグメントを生成させ、プロテインAによりFcドメインを除去して精製した。
【0198】
4. 抗ダビガトランFabの特徴づけ
インタクトな抗体のLys−C切断によって生成された18/15 Fabフラグメントを、実施例IIに概要されたトロンビン凝固時間アッセイにおいてそれがヒト血漿でのダビガトラン抗凝固活性を中和する能力について試験した。Fabは、ヒト血漿におけるトロンビン依存性凝固のダビガトラン介在性延長を2.6nMのIC50で用量依存的に完全にリバースした(図10)。直接発現されたFabフラグメントおよびインタクトな抗体のパパイン切断によって生成されたFabフラグメントもまた、それぞれ2.6および2.7nMのIC50でダビガトランの抗凝固活性を中和した。
【0199】
インタクトな抗体のLys−C切断によって生成された18/15 Fabおよび直接発現されたFabの親和性を、SPR技法を利用してBIAcore装置で決定した。Fabを漸増する濃度のダビガトランと一緒に室温で30分間予備インキュベーションした。固定化ビオチン−コンジュゲーション型ダビガトランアナログをコーティングされたセンサーチップの上にその混合物を流し、遊離Fabの結合をモニターした。この溶液競合アッセイ設計を使用して、ダビガトランに対するFabのKD値をLys−Cで生成されたFabについて0.16pM、直接発現されたFabについて0.45pMと決定した。
【0200】
パパイン切断によって生成されたFabを用いたin vivo実験
ラット(雄性Wistar、約300g)を、ペントバルビタールでボーラス(60mg/kg i.p.)および維持麻酔のための連続注入(20mg/kg/hr i.p.)として麻酔し、内部体温を維持するために37℃の温熱パッド上に置いた。採血のために頸動脈を、物質投与のために右頸静脈を分離し、カニューレを挿入した。ダビガトランをボーラスとして開始し(0.3μM/kg)、続いて20分間かけて注入し(0.1μM/kg/hr)、定常状態の血漿レベルを達成した。20分後に、左頸静脈を介してFabを等モル濃度または等モル濃度の半分のいずれかで単回ボーラスとしてi.v.注射した。血液試料(3.13%クエン酸ナトリウム中に1/10希釈)をベースライン(−20、−2min)で、そしてFab注射後に様々な間隔で30分間採取する。
【0201】
ダビガトランの抗凝固作用を、トロンビン時間(TT)および活性化部分トロンボプラスチン時間(aPTT)を含めた全血凝固時間として測定した。簡潔には、トロンビン時間は、37℃に予熱されたウェルに全血50μLを添加することによって凝固計で行う。濃度3.0U/mLのトロンビン(Siemens Healthcare, Marburg, Germany)を添加し(体積100μL)、試料を凝固させるために必要な時間を測定する。凝固計で全血50μLを37℃に予熱し、aPTT試薬(Roche Diagnostics, Mannheim, Germany)50μLを3分間添加することによって全血aPTTを行う。0.025M予熱(37℃)塩化カルシウム50μLの添加によって凝固時間を開始する。次に、全血試料を凝固させるために必要な時間を記録する。
【0202】
ダビガトランと等しいモル用量での単回i.v.注射として与えられた18/15Fab(軽鎖:配列番号:37、重鎖Fdフラグメント:配列番号:41;CHO細胞で発現された完全免疫グロブリンのパパイン切断によって製造)の結果を図11および12に示す。このモデルにおいて、TT(図11)とaPTT(図12)の両方として測定された、ダビガトラン抗凝固活性の迅速でほぼ即時の阻害があった。注射から1分以内で、ダビガトランの抗凝固活性は、完全に中和されてベースラインレベルに戻った。これは、ダビガトランの継続中のi.v.連続注入にかかわらず、20分よりも長く維持された。
【0203】
より低い用量である半分のモル用量のダビガトランが与えられた場合も、TT(図13)とaPTT(図14)の両方に初回減少があった。しかしこれは、ダビガトランの継続中の連続注入の条件下で、より高い用量ほど長くは維持されなかった。
【0204】
したがって、これらの結果から、この動物モデルにおいて抗ダビガトランFabの単回i.v.投与後のダビガトラン抗凝固活性の、予測可能で、用量依存的で、非常に迅速な中和が実証される。
【技術分野】
【0001】
発明の背景
技術分野
本発明は、医学の分野、特に抗凝固治療の分野に関するものである。
【0002】
背景の情報
抗凝固薬は、血液凝固を防止する物質であって;すなわち抗凝固薬は血液が凝固するのを妨げる。抗凝固薬は、血栓性障害のための薬物療法としてのヒトの治療、例えば素因をもつ者における深部静脈血栓、肺塞栓、心筋梗塞および脳卒中の一次および二次予防に広く使用される。
【0003】
重要なクラスの経口抗凝固薬、例えばワルファリンを含めたクマリンは、ビタミンKの効果と拮抗することによって作用する。第二のクラスの化合物は、アンチトロンビンIIIまたはヘパリンコファクターIIなどのコファクターを介して間接的に血液凝固を阻害する。これには、アンチトロンビンIIIを介して主に第Xa因子(および低い程度にはトロンビン)の阻害を触媒するいくつかの低分子量ヘパリン製品(ベミパリン、セルトパリン、ダルテパリン、エノキサパリン、ナドロパリン、パルナパリン、レビパリン、チンザパリン)が含まれる。より小さなオリゴ糖(フォンダパリヌクス、イドラパリヌクス)は、アンチトロンビンIIIを介して第Xa因子だけを阻害する。ヘパリノイド(ダナパロイド、スロデキシド、デルマタン硫酸)は、両方のコファクターを介して作用し、第Xa因子とトロンビンの両方を阻害する。第三のクラスは、血液凝固の直接阻害剤に相当する。直接第Xa因子阻害剤には、アピキサバン、エドキサバン、オタミキサバン、リバロキサバンが含まれ、直接トロンビン阻害剤には、二価ヒルジン(ビバリルジン、レピルジン、デシルジン)、ならびに一価化合物であるアルガトロバンおよびダビガトランが含まれる。
【0004】
血液凝固は出血を止める生物学的メカニズムであるので、抗凝固治療の副作用は、望まれない出血事象のことがある。したがって、そのような抗凝固薬関連出血事象が起こったときにそれを止めることができる解毒剤を提供することが望ましい(Zikria and Ansell, Current Opinion in Hematology 2009, 16(5): 347-356)。これを達成するための一方法は、投与後に患者に存在する抗凝固化合物の活性を中和することによる。
【0005】
抗凝固薬の現在入手可能な解毒剤は、プロタミン(ヘパリンの中和用)、およびワルファリンのようなビタミンKアンタゴニストを中和するためのビタミンKである。新鮮凍結血漿および組換え第VIIa因子もまた、大外傷または重度出血を被った、低分子量ヘパリン処置下の患者における非特異的解毒剤として使用されている(Lauritzen, B. et al, Blood, 2005, 607A-608A.)。ヘパリンまたは低分子量ヘパリン解毒剤としてのプロタミンフラグメント(米国特許第6,624,141号)および小型合成ペプチド(米国特許第6,200,955号);ならびにトロンビン阻害剤用の解毒剤としてのトロンビンムテイン(米国特許第6,060,300号)も報告されている。プロトロンビンの中間体および誘導体は、ヒルジンおよび合成トロンビン阻害剤に対する解毒剤として報告されている(米国特許第5,817,309号および第6,086,871号)。直接第Xa因子阻害剤については、不活性第Xa因子アナログが解毒剤として提案されている(国際公開公報第2009042962号)。さらに、組換え第VIIa因子が、フォンダパリヌクスおよびイドラパリヌクスなどの間接アンチトロンビンIII依存性第Xa因子阻害剤の効果をリバースするために使用されている(Bijsterveld, NR et al, Circulation, 2002, 106: 2550-2554; Bijsterveld, NR et al, British J. of Haematology, 2004 (124): 653-658)。抗凝固薬の効果をリバースする方法の総説は、Schulman and Bijsterveld, Transfusion Medicine Reviews 2007、21(1): 37-48に提供されている。
【0006】
抗凝固治療についての改良された解毒剤、特に特異的解毒剤がこれまでのところ開示されていないダビガトランのような直接トロンビン阻害剤のための解毒剤を提供する必要性がある。
【0007】
発明の簡単な概要
一局面では、本発明は、抗凝固薬の活性を中和可能な抗体分子に関する。
【0008】
さらなる局面では、抗体分子は、抗凝固薬に対する結合特異性を有する。
【0009】
さらなる局面では、抗凝固薬は、直接トロンビン阻害剤、第Xa因子阻害剤、またはビタミンKアンタゴニストである。
【0010】
さらなる局面では、抗凝固薬は、ダビガトラン、アルガトロバン、メラガトラン、キシメラガトラン、ヒルジン、ビバリルジン、レピルジン、デシルジン、アピキサバン、オタミキサバン、エドキサバン、リバロキサバン、デフィブロチド、ラマトロバン、アンチトロンビンIII、またはドロトレコギンアルファである。
【0011】
さらなる態様では、抗凝固薬は、一般式
【0012】
【化1】
[式中、
Aは、基Hetのベンゾ、ピリドまたはチエノ部分に結合されたカルボニルまたはスルホニル基を表し、
Bは、基Arに結合されたメチレン基が酸素もしくは硫黄原子によって、または−NR1−基によって置き換えられていてもよいエチレン基を表し、
ここで、R1は、水素原子またはC1−4−アルキル基を表し、
Eは、RbNH−C(=NH)−基を表し、
ここで、Rbは、水素原子、ヒドロキシ、C1−9−アルコキシカルボニル、シクロヘキシルオキシカルボニル、フェニル−C1−3−アルコキシカルボニル、ベンゾイル、p−C1−3−アルキル−ベンゾイルまたはピリジノイル基を表し、一方で上記C1−9−アルコキシカルボニル基の2位のエトキシ部分は、C1−3−アルキルスルホニルまたは2−(C1−3−アルコキシ)−エチル基によって更に置換されていてもよく、
Arは、場合により塩素原子によって、またはメチル、エチルもしくはメトキシ基によって置換されている1,4−フェニレン基を表すか、あるいはArは2,5−チエニレン基を表し、
Hetは、1−(C1−3−アルキル)−2,5−ベンゾイミダゾリレン、1−シクロプロピル−2,5−ベンゾイミダゾリレン、2,5−ベンゾチアゾリレン、1−(C1−3−アルキル)−2,5−インドリレン、1−(C1−3−アルキル)−2,5−イミダゾ[4,5−b]ピリジニレン、3−(C1−3−アルキル)−2,7−イミダゾ[1,2−a]ピリジニレンまたは1−(C1−3−アルキル)−2,5−チエノ[2,3−d]イミダゾリレン基を表し、そして
Raは、R2NR3−基を表し、
ここで、R2は、カルボキシ、C1−6−アルキルオキシカルボニル、ベンジルオキシカルボニル、C1−3−アルキルスルホニルアミノカルボニルもしくは1H−テトラゾール−5−イル基によって置換されていてもよいC1−4−アルキル基、
ヒドロキシ、ベンジルオキシ、カルボキシ−C1−3−アルキルアミノ、C1−3−アルコキシカルボニル−C1−3−アルキルアミノ、N−(C1−3−アルキル)−カルボキシ−C1−3−アルキルアミノもしくはN−(C1−3−アルキル)−C1−3−アルコキシカルボニル−C1−3−アルキルアミノ基によって置換されているC2−4−アルキル基であり、一方で上記基において隣接窒素原子に対してα−位にある炭素原子は、置換されていなくてもよく、
R3は、C3−7−シクロアルキル基、不飽和部分がR2NR3基の窒素原子に直接結合されていなくてもよいプロパルギル基、フッ素もしくは塩素原子によって、またはメチルもしくはメトキシ基によって場合により置換されているフェニル基、あるいはメチル基によって場合により置換されているピラゾリル、ピリダゾリルまたはピリジニル基を表すか、あるいは
R2およびR3は、それらの間の窒素原子と一緒になって、カルボキシまたはC1−4−アルコキシカルボニル基によって場合により置換されている5〜7員シクロアルキレンイミノ基を表し、フェニル環が更に縮合されていてもよい]
で示される二置換二環式複素環、その互変異性体、立体異性体および塩である。
【0013】
さらなる態様では、抗凝固薬は、
(a) 2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾチアゾール−5−カルボン酸−N−フェニル−N−(2−カルボキシエチル)−アミド、
(b) 2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾチアゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(c) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(d) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(3−ヒドロキシカルボニルプロピル)−アミド、
(e) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(ヒドロキシカルボニルメチル)−アミド、
(f) 1−メチル−2−[2−(2−アミジノチオフェン−5−イル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(g) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(h) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(i) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(j) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[2−(1H−テトラゾール−5−イル)エチル]−アミド、
(k) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[2−(1H−テトラゾール−5−イル)エチル]−アミド、
(l) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(m) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(3−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(n) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(o) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[(N−ヒドロキシカルボニルエチル−N−メチル)−2−アミノエチル]−アミド、
(p) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(3−フルオロフェニル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(q) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(4−フルオロフェニル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(r) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(s) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(t) 1−メチル−2−[N−(4−アミジノフェニル)アミノメチル]−インドール−5−イル−カルボン酸−N−フェニル−N−(2−メトキシカルボニルエチル)−アミド、
(u) 1−メチル−2−[N−(4−アミジノフェニル)アミノメチル]−チエノ[2.3−d]イミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(v) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(w) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(x) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(y) 1−メチル−2−[N−[4−(N−n−ヘキシルオキシカルボニルアミジノ)フェニル]−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−エトキシカルボニルエチル)−アミド、
より選択される化合物、ならびにその互変異性体、立体異性体および塩である。
【0014】
別の局面では、本発明は、ダビガトラン、ダビガトランエテキシラート、および/またはダビガトランのO−アシルグルクロニドに対する抗体分子に関する。
【0015】
さらなる局面では、抗体分子は、ポリクローナル抗体、モノクローナル抗体、ヒト抗体、ヒト化抗体、キメラ抗体、抗体のフラグメント、特にFab、Fab’、もしくはF(ab’)2フラグメント、単鎖抗体、特に単鎖可変フラグメント(scFv)、ドメイン抗体、ナノボディ(nanobody)、ダイアボディ(diabody)、またはDARPinである。
【0016】
さらなる局面では、本発明は、医学に使用するための上記抗体分子に関する。
【0017】
さらなる局面では、本発明は、抗凝固治療の副作用の治療または予防に使用するための上記抗体分子に関する。
【0018】
さらなる局面では、副作用は出血事象である。
【0019】
さらなる局面では、本発明は、抗凝固治療の副作用の治療または予防方法であって、それを必要とする患者に上記抗体分子の有効量を投与することを含む方法に関する。
【0020】
別の局面では、本発明は、容器およびラベルと一緒に前記抗体分子を含むキットに関する。
【図面の簡単な説明】
【0021】
【図1】トロンビン凝固時間アッセイを用いて、ダビガトランの濃度の増加とともに凝固時間の増加が見られたことを示す図である。濃度200nMは、凝固時間にベースラインからの5倍の延長を招き、この濃度を第1および第2セットの実験に使用した。最終セットの実験には濃度500nM(治療濃度を超える)を使用した。
【図2】ダビガトランに対する4種の異なる抗体(A〜D)全てが、ヒト血漿でのダビガトランの凝固時間延長を中和したことを示す図である。ヒト血漿でのベースライン凝固は10.9秒であり、200nMダビガトランを血漿と一緒に予備インキュベーションした場合、凝固は51秒に延長した。200nMダビガトランと一緒に予備インキュベーションされた血漿に各抗体を添加し、さらに5分間インキュベーションした。次に、トロンビンの添加によってトロンビン凝固時間を開始させた。各抗体は、ダビガトランの凝固時間を異なる程度にリバースすることができた。最も濃い溶液が抗凝固活性の最大のリバースを招いた。
【図3】200nMダビガトランと予備インキュベーションされたヒト血漿に添加される、漸増するポリクローナル抗体(抗体D)の濃度の作用を測定したことを示す図である。ベースライン凝固時間は11秒であり、ダビガトランの添加は、凝固を63.7秒に延長した。次に、抗体の漸増する希釈が、ダビガトランで延長されたトロンビン凝固時間のリバースに及ぼす作用を試験した。最低濃度はトロンビン凝固時間を43.9秒に短縮した。より高い濃度は、トロンビン凝固時間をベースラインレベルに完全に減らし、ダビガトランの抗凝固作用の完全な中和をもたらした。非特異的ウサギポリクローナル抗体(四角)の添加は、ダビガトランの抗凝固作用のリバースに効果を有さなかった。
【図4】500nMダビガトランと予備インキュベーションされたヒト血漿に添加された、漸増するポリクローナル抗体(抗体D)の濃度の作用を測定したことを示す図である。ベースライン凝固時間は10.9秒であり、このより高い濃度のダビガトランの添加は、凝固を111.7秒に延長した(約10倍の増加)。抗体または原液の1:2希釈溶液の作用は、ダビガトランを用いたトロンビン凝固時間延長を濃度依存的にリバースした。最も高い濃度もまた、トロンビン凝固時間をベースラインレベルに完全にリバースさせ、たとえ治療濃度を超えるダビガトランの抗凝固作用であっても完全な中和を招いた。
【図5】抗ダビガトラン抗体分子重鎖の可変領域の配列を示す図である。
【図6】抗ダビガトラン抗体分子軽鎖の可変領域の配列を示す図である。
【図7】マウスモノクローナル抗体(クローン22)が、ヒト血漿およびヒト全血でのダビガトランの抗凝固作用をリバースすることを示す図である。漸増する濃度のマウス抗体を、30nMダビガトランと予備インキュベーションされたヒト血漿または全血に添加した。1.5〜2U/mLトロンビンの添加によってアッセイを開始し、凝固時間を測定した。100%のダビガトラン活性を、化合物の存在下と不在下の凝固時間の差として定義した。抗体は、凝固時間のダビガトラン介在性延長を用量依存的に阻害した。
【図8】クローン22抗体から生成されたマウスFabが、ヒト血漿でのダビガトランの抗凝固作用をリバースすることを示す図である。漸増する濃度のマウスFabを、7nMダビガトランと予備インキュベーションされたヒト血漿に添加した。インタクトな抗体も陽性対照として試験した。0.4U/mLトロンビンの添加によってアッセイを開始し、凝固時間を測定した。100%阻害を、凝固時間のダビガトラン介在性増加の完全遮断として定義した。Fabは、ヒト血漿での凝固時間におけるダビガトラン誘導延長を用量依存的に阻害した。
【図9】マウスモノクローナル抗体(クローン22)が、ヒト血漿でのダビガトランアシルグルクロニドの抗凝固作用をリバースすることを示す図である。漸増する濃度のマウス抗体を、7nMダビガトランアシルグルクロニドまたはダビガトランと予備インキュベーションされたヒト血漿に添加した。0.4U/mLトロンビンの添加によってアッセイを開始し、凝固時間を測定した。100%阻害を凝固時間における化合物介在性増加の完全遮断として定義した。抗体は、ヒト血漿での凝固時間におけるダビガトランアシルグルクロニド誘導延長を用量依存的に阻害した。
【図10】ヒト化Fab(Fab18/15)が、ヒト血漿でのダビガトランの抗凝固作用をリバースすることを示す図である。漸増する濃度のFab18/15を、7nMダビガトランと予備インキュベーションされたヒト血漿に添加した。0.4U/mLトロンビンの添加によってアッセイを開始し、凝固時間を測定した。100%阻害を、凝固時間におけるダビガトラン介在性増加の完全遮断として定義した。Fabは、ヒト血漿での凝固時間におけるダビガトラン誘導延長を用量依存的に阻害した。
【図11】t=0での等モル濃度のFabのボーラス投与とダビガトランを連続注入として受けている、ラットにおけるex vivo全血トロンビン凝固時間(3.0U/mLトロンビン)を示す図である。無地丸との線は、薬物なしのビヒクル処置を表す。無地四角の線は、Fabなしのダビガトランの抗凝固活性を表す。無地三角との線は、Fab投与後の抗凝固活性を表す。データを平均±SE(一処置群あたり動物数n=4)として表す。
【図12】t=0で等モル濃度のFabのボーラス投与とダビガトランを連続注入として受けている、投与されているラットにおけるex vivo全血aPTTを示す図である。無地丸は、薬物なしのビヒクル処置を表す。無地四角との線は、Fabなしのダビガトランの抗凝固活性を表す。無地三角との線は、Fab投与後の抗凝固活性を表す。データを平均±SE(一処置群あたり動物数n=4)として表す。
【図13】t=0での漸増する用量のFabのボーラス投与とダビガトランを連続注入として受けている、ラットにおけるex vivo全血トロンビン凝固時間(3.0U/mLトロンビン)を示す図である。無地丸との線は、薬物なしのビヒクル処置を表す。無地四角とのの線は、Fabなしのダビガトランの抗凝固活性を表す。無地三角との線は、等モルのFabを投与後の、破線は、等モル用量の50%を投与後の抗凝固活性を表す。データを平均±SE(一処置群あたり動物数n=4)として表す。
【図14】t=0での漸増する用量のFabのボーラス投与とダビガトランを連続注入として受けている、ラットにおけるex vivo全血aPTTを示す図である。無地丸は、薬物なしのビヒクル処置を表す。無地四角との線は、Fabなしのダビガトランの抗凝固活性を表す。無地三角との線は、等モルのFabを投与後の、破線は、等モル用量の50%を投与後の抗凝固活性を表す。データを平均±SE(一処置群あたり動物数n=4)として表す。
【0022】
発明の詳細な説明
一局面では、本発明は、抗凝固薬の活性を中和可能な抗体分子に関する。
【0023】
抗体(免疫グロブリン、略してIgとしても知られている)は、脊椎動物の血液または他の体液中に見出されるガンマグロブリンタンパク質であって、細菌およびウイルスなどの外来物を同定および中和するために免疫系によって使用される。抗体は、典型的には基本構造ユニットでできており、それぞれが2本の大きな重鎖および2本の小さな軽鎖を有し、例えば1個のユニットを有するモノマー、2個のユニットを有するダイマーまたは5個のユニットを有するペンタマーを形成する。抗体は、抗原として知られている他の分子または構造に非共有相互作用によって結合することができる。この結合は、抗体が高親和性で特異構造だけに結合するという意味で特異的である。抗体によって認識される抗原の独特な部分は、エピトープまたは抗原決定基と呼ばれる。抗体のエピトープ結合性部分は、時にパラトープと呼ばれ、抗体のいわゆる可変ドメイン、または可変領域(Fv)に位置する。可変ドメインは、フレームワーク領域(FR)によって間隔が空けられた3個のいわゆる相補性決定領域(CDR)を含む。
【0024】
本発明の状況の範囲内で、CDRへの参照は、Chothia(Chothia and Lesk, J. Mol. Biol. 1987, 196: 901-917)ならびにKabat(E.A. Kabat, T.T. Wu, H. Bilofsky, M. Reid-Miller and H. Perry, Sequence of Proteins of Immunological Interest, National Institutes of Health, Bethesda (1983))の定義に基づく。
【0025】
当該技術は、さらに抗体を開発し、それらを医学および工学における多用途の道具にしている。したがって、本発明の状況で「抗体分子」または「抗体」という用語(本明細書において同義語として使用される)には、例えば2本の軽鎖および2本の重鎖、またはラクダ科の種のように2本の重鎖だけを含む、自然界に見出されうる抗体が含まれるだけではなく、さらには抗原への結合特異性および免疫グロブリンの可変ドメインとの構造類似性を有する少なくとも1個のパラトープを含む全ての分子も包含する。
【0026】
したがって、本発明による抗体分子は、ポリクローナル抗体、モノクローナル抗体、ヒト抗体、ヒト化抗体、キメラ抗体、抗体のフラグメント、特にFv、Fab、Fab’、またはF(ab’)2フラグメント、単鎖抗体、特に単鎖可変フラグメント(scFv)、小モジュラー免疫医薬(Small Modular Immunopharmaceutical)(SMIP)、ドメイン抗体、ナノボディ、ダイアボディでありうる。
【0027】
ポリクローナル抗体は、異なるアミノ酸配列を有する抗体分子の集合を表し、当技術分野で周知のプロセスにより抗原で免疫処置した後に、脊椎動物の血液から得ることができる。
【0028】
モノクローナル抗体(mAbまたはmoAb)は、アミノ酸配列が同一の単一特異性抗体である。それらは、ハイブリドーマ技法によって、特異抗体産生性B細胞と骨髄腫(B細胞ガン)細胞の融合体のクローンを意味するハイブリッド細胞系(ハイブリドーマと呼ばれる)から製造することができる(Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975;256:495-7.)。または、モノクローナル抗体は、宿主細胞での組換え発現によって製造することができる(Norderhaug L, Olafsen T, Michaelsen TE, Sandlie I. (May 1997). "Versatile vectors for transient and stable expression of recombinant antibody molecules in mammalian cells.". J Immunol Methods 204 (1): 77-87;下記も参照されたい)。
【0029】
人間に応用するために、マウスのような本来他の種由来の抗体の免疫原性を減少させることが望ましいことが多い。これは、キメラ抗体の構築によって、または「ヒト化」と呼ばれるプロセスによって行うことができる。この状況で「キメラ抗体」は、ある種(例えばマウス)由来の配列部分(例えば可変ドメイン)が別の種(例えばヒト)由来の配列部分(例えば定常ドメイン)と融合したものを含む抗体であると理解される。「ヒト化抗体」は、本来非ヒト種由来の可変ドメインを含む抗体であって、その可変ドメインの全体的な配列がヒト可変ドメインの配列により密接に類似するように、あるアミノ酸が突然変異された抗体である。抗体のキメラ化方法およびヒト化方法は、当技術分野において周知である(Billetta R, Lobuglio AF. "Chimeric antibodies". Int Rev Immunol. 1993;10(2-3):165-76; Riechmann L, ClarkM, Waldmann H, Winter G (1988). "Reshaping human antibodies for therapy". Nature: 332:323.)。
【0030】
さらに、例えばファージディスプレイによって、またはトランスジェニック動物を使用してヒトゲノム由来の配列に基づき抗体を作出するための技法が開発されている(国際公開公報第90/05144号; D. Marks, H.R. Hoogenboom, T.P. Bonnert, J. McCafferty, A.D. Griffiths and G. Winter (1991) "By-passing immunisation. Human antibodies from V-gene libraries displayed on phage." J.Mol.Biol., 222, 581-597; Knappik et al., J. Mol. Biol. 296: 57-86, 2000; S. Carmen and L. Jermutus, "Concepts in antibody phage display". Briefings in Functional Genomics and Proteomics 2002 1(2):189-203; Lonberg N, Huszar D. "Human antibodies from transgenic mice". Int Rev Immunol. 1995;13(1):65-93.; Brueggemann M, Taussig MJ. "Production of human antibody repertoires in transgenic mice". Curr Opin Biotechnol. 1997 Aug;8(4):455-8.)。そのような抗体は、本発明の状況で「ヒト抗体」である。
【0031】
本発明による抗体分子には、Fab、Fab’、またはF(ab’)2フラグメントのような、抗原結合特性を保持する免疫グロブリンのフラグメントも含まれる。そのようなフラグメントは、免疫グロブリンのフラグメント化によって、例えばタンパク質分解消化によって、またはそのようなフラグメントの組換え発現によって得ることができる。例えば、免疫グロブリンの消化は、日常的な技法によって、例えばパパインもしくはペプシン(国際公開公報第94/29348号)、またはエンドプロテイナーゼLys−C(Kleemann, et al, Anal. Chem. 80, 2001-2009, 2008)を使用して達成することができる。抗体のパパインまたはLys−C消化によって、典型的にはそれぞれ単一の抗原結合部位を有する二つの同一の抗原結合フラグメント、いわゆるFabフラグメントおよび残余のFcフラグメントが製造される。ペプシン処理によってF(ab’)2が回収される。宿主細胞での組換え発現によってFab分子を製造する方法を下記により詳細に概要する。
【0032】
免疫グロブリンの可変ドメインまたはそのような可変ドメイン由来の分子を異なる分子状況に配置するためにいくつかの技法が開発されている。それらもまた、本発明による「抗体分子」と見なすべきである。一般にこれらの抗体分子は、免疫グロブリンよりもサイズが小さく、単一のアミノ酸鎖を含むか、または数本のアミノ酸鎖から構成されることがある。例えば、単鎖可変フラグメント(scFv)は、短いリンカー、通常はセリン(S)またはグリシン(G)で一緒に結合されている、免疫グロブリンの重鎖及び軽鎖可変領域との融合体である(国際公開公報第88/01649号;国際公開公報第91/17271号; Huston et al; International Reviews of Immunology, Volume 10, 1993, 195 - 217)。「単一ドメイン抗体」または「ナノボディ」は、単一のIg様ドメインに抗原結合部位を有する(国際公開公報第94/04678号;国際公開公報第03/050531号、Ward et al., Nature. 1989 Oct 12;341(6242):544-6; Revets et al., Expert Opin Biol Ther. 5(1):111-24, 2005)。同一または異なる抗原に対して結合特異性を有する一つ以上の単一ドメイン抗体が一緒に結合されることがある。ダイアボティは、2個の可変ドメインを含む2本のアミノ酸鎖から成る二価抗体分子である(国際公開公報第94/13804号、Holliger et al., Proc Natl Acad Sci U S A. 1993 Jul 15;90(14):6444-8)。抗体様分子の他の例は、免疫グロブリンスーパーファミリー抗体である(IgSF; Srinivasan and Roeske, Current Protein Pept. Sci. 2005, 6(2): 185-96)。異なる考え方が、定常ドメインCH1を欠如する単鎖ヒンジおよびエフェクタードメインに結合したFvドメインを含む、いわゆる小モジュラー免疫医薬(SMIP)につながっている(国際公開公報第02/056910号)。
【0033】
さらなる局面では、本発明の抗体分子は、免疫グロブリン可変ドメインに匹敵するある一定の結合特異性および親和性を有する限り、免疫グロブリン可変ドメインとかけ離れた構造関連性だけさえ有していればよいし、またそのような関係を全く有していなくてもよい。そのような非免疫グロブリン「抗体模倣物」は、時に「足場タンパク質」と呼ばれ、プロテインA、リポカリン、フィブロネクチンドメイン、アンキリンコンセンサスリピートドメイン、およびチオレドキシンの遺伝子に基づくことがある(Skerra, Current Opinion in Biotechnology 2007, 18(4): 295-304)。本発明の状況における好ましい態様は、設計されたアンキリンリピートタンパク質である(DARPin; Steiner et al., J Mol Biol. 2008 Oct 24;382(5): 1211-27; Stumpp MT, Amstutz P. Curr Opin Drug Discov Devel. 2007 Mar;10(2):153-9)。
【0034】
抗体分子は、その抗体分子の性質に所望の影響を有する他の分子実体に融合されるか(融合タンパク質として)、または別の方法で結合される(共有または非共有結合により)ことがある。例えば、特に単鎖抗体またはドメイン抗体の場合、抗体分子の薬物動態特性、例えば血液のような体液中での安定性を改善することが望ましいことがある。これに関して、特に循環中でのそのような抗体分子の半減期を延長するために、PEG化(国際公開公報第98/25971号;国際公開公報第98/48837号;国際公開公報第2004081026号)、その抗体分子を、アルブミンのような血清タンパク質に対して親和性を有する別の抗体分子と融合もしくは別の方法で共有結合させること(国際公開公報第2004041865号;国際公開公報第2004003019号)、またはアルブミンもしくはトランスフェリンのような血清タンパク質の全てまたは一部との融合タンパク質としてその抗体分子を発現させること(国際公開公報第01/79258号)などのいくつかの技法が開発されている。
【0035】
さらなる局面では、抗体分子は、抗凝固薬に対する結合特異性を有する。「結合特異性」は、抗体分子が、構造的に無関係の分子よりも抗凝固薬に対して有意に高い結合親和性を有することを意味する。
【0036】
親和性は、抗体分子上の単一抗原結合部位と単一エピトープの間の相互作用である。親和性は、結合定数KA=kass/kdiss、または解離定数KD=kdiss/kassによって表される。
【0037】
本発明の一局面では、抗体は、例えば表面プラズモン共鳴分析によって決定されるとき、0.1pM〜100μM、好ましくは1pM〜100μM、好ましくは1pM〜1μMの範囲のKD値を有する親和性で抗凝固薬に結合する(Malmqvist M., "Surface plasmon resonance for detection and measurement of antibody-antigen affinity and kinetics.", Curr Opin Immunol. 1993 Apr;5(2):282-6.)。抗体親和性は、また、動力学排除アッセイ(kinetic exclusion assay)(KinExA)技法を用いて測定することもできる(Darling, R.J., and Brault P-A., "Kinetic exclusion assay technology: Characterization of Molecular Interactions." ASSAY and Drug Development Technologies. 2004, Dec 2(6): 647-657)。
【0038】
抗体分子の結合親和性は、親和性成熟として知られているプロセスによって高めることができる(Marks et al., 1992, Biotechnology 10:779-783; Barbas, et al., 1994, Proc. Nat. Acad. Sci, USA91:3809-3813; Shier et al., 1995, Gene 169:147-155)。したがって、親和性成熟された抗体もまた、本発明に包含される。
【0039】
本発明のさらなる局面では、抗体分子は、抗凝固薬の活性を中和可能である。すなわち抗凝固薬は、抗体分子に結合すると、もはやその抗凝固活性を発揮することができないか、または有意に減少した大きさでこの活性を発揮する。好ましくは抗凝固活性は、当該抗凝固薬にとって適切な活性アッセイで、特にエカリン凝固時間またはトロンビン凝固時間などの、トロンビンに感受性の凝固アッセイで決定されるとき、抗体の結合について、少なくとも2倍、5倍、10倍、100倍で減少している(H. Bounameaux, Marbet GA, Lammle B, et al. "Monitoring of heparin treatment. Comparison of thrombin time, activated partial thromboplastin time, and plasma heparin concentration, and analysis of the behaviour of antithormbin III". American Journal of Clinical Pathology 1980 74(1): 68-72)。
【0040】
本発明の抗体分子を製造するために、当業者は、当技術分野で周知の多様な方法より選択することができる(Norderhaug et al., J Immunol Methods 1997, 204 (1): 77-87; Kipriyanow and Le Gall, Molecular Biotechnology 26: 39- 60, 2004; Shukla et al., 2007, J. Chromatography B, 848(1): 28-39)。
【0041】
抗凝固薬は、上記に概要するように当技術分野において周知である。本発明のさらなる局面では、抗凝固薬は、直接トロンビン阻害剤、第Xa因子阻害剤、またはビタミンKアンタゴニストである。ビタミンKアンタゴニストの例は、ワルファリンが含まれるクマリンである。主に第Xa因子の間接阻害剤の例は、アンチトロンビンIIIの活性化を介して作用するヘパリン群の物質であって、それらには、いくつかの低分子量ヘパリン製品(ベミパリン、セルトパリン、ダルテパリン、エノキサパリン、ナドロパリン、パルナパリン、レビパリン、チンザパリン)、ある種のオリゴ糖(フォンダパリヌクス、イドラパリヌクス)、ヘパリノイド(ダナパロイド、スロデキシド、デルマタン硫酸)、および直接第Xa因子阻害剤(アピキサバン、オタミキサバン、リバロキサバン)が含まれる。トロンビン阻害剤の例には、二価ヒルジン(ビバリルジン、レピルジン、デシルジン)、ならびに一価化合物アルガトロバンおよびダビガトランが含まれる。
【0042】
したがってさらなる局面では、抗凝固薬は、ダビガトラン、アルガトロバン、メラガトラン、キシメラガトラン、ヒルジン、ビバリルジン、レピルジン、デシルジン、アピキサバン、エドキサバン、オタミキサバン、リバロキサバン、デフィブロチド、ラマトロバン、アンチトロンビンIII、またはドロトレコギンアルファである。
【0043】
さらなる態様では、抗凝固薬は、一般式
【0044】
【化2】
[式中、
Aは、基Hetのベンゾ、ピリドまたはチエノ部分に結合したカルボニルまたはスルホニル基を表し、
Bは、基Arに結合されたメチレン基が酸素もしくは硫黄原子によって、または−NR1−基によって置き換えられていてもよいエチレン基を表し、
ここで、R1は、水素原子またはC1−4−アルキル基を表し、
Eは、RbNH−C(=NH)−基を表し、
ここで、Rbは、水素原子、ヒドロキシ、C1−9−アルコキシカルボニル、シクロヘキシルオキシカルボニル、フェニル−C1−3−アルコキシカルボニル、ベンゾイル、p−C1−3−アルキル−ベンゾイルまたはピリジノイル基を表し、一方で上記C1−9−アルコキシカルボニル基の2位のエトキシ部分は、C1−3−アルキルスルホニルまたは2−(C1−3−アルコキシ)−エチル基によって更に置換されていてもよく、
Arは、塩素原子によって、またはメチル、エチルもしくはメトキシ基によって場合により置換されている1,4−フェニレン基を表すか、もしくはそれは2,5−チエニレン基を表し、
Hetは、1−(C1−3−アルキル)−2,5−ベンゾイミダゾリレン、1−シクロプロピル−2,5−ベンゾイミダゾリレン、2,5−ベンゾチアゾリレン、1−(C1−3−アルキル)−2,5−インドリレン、1−(C1−3−アルキル)−2,5−イミダゾ[4,5−b]ピリジニレン、3−(C1−3−アルキル)−2,7−イミダゾ[1,2−a]ピリジニレンまたは1−(C1−3−アルキル)−2,5−チエノ[2,3−d]イミダゾリレン基を表し、そして
Raは、R2NR3−基を表し、
ここで、R2は、カルボキシ、C1−6−アルキルオキシカルボニル、ベンジルオキシカルボニル、C1−3−アルキルスルホニルアミノカルボニルもしくは1H−テトラゾール−5−イル基によって置換されていてもよいC1−4−アルキル基、
ヒドロキシ、ベンジルオキシ、カルボキシ−C1−3−アルキルアミノ、C1−3−アルコキシカルボニル−C1−3−アルキルアミノ、N−(C1−3−アルキル)−カルボキシ−C1−3−アルキルアミノもしくはN−(C1−3−アルキル)−C1−3−アルコキシカルボニル−C1−3−アルキルアミノ基によって置換されているC2−4−アルキル基であり、一方で上記基において隣接窒素原子に対してα−位にある炭素原子は、置換されていなくてもよく、
R3は、C3−7−シクロアルキル基、不飽和部分がR2NR3基の窒素原子に直接結合されていなくてもよいプロパルギル基、フッ素もしくは塩素原子によって、またはメチルもしくはメトキシ基によって場合により置換されているフェニル基、あるいはメチル基によって場合により置換されているピラゾリル、ピリダゾリルまたはピリジニル基を表すか、あるいは
R2およびR3は、それらの間の窒素原子と一緒になって、カルボキシまたはC1−4−アルコキシカルボニル基によって場合により置換されている5〜7員シクロアルキレンイミノ基を表し、フェニル環が更に縮合されていてもよい]
で示される二置換二環式複素環、その互変異性体、立体異性体および塩である。式(I)で示される化合物、これらの化合物の調製および抗凝固薬としてのそれらの使用は、国際公開公報第98/37075号に記載されている。
【0045】
さらなる態様では、抗凝固薬は、
(a) 2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾチアゾール−5−カルボン酸−N−フェニル−N−(2−カルボキシエチル)−アミド、
(b) 2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾチアゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(c) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(d) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(3−ヒドロキシカルボニルプロピル)−アミド、
(e) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(ヒドロキシカルボニルメチル)−アミド、
(f) 1−メチル−2−[2−(2−アミジノチオフェン−5−イル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(g) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(h) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(i) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(j) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[2−(1H−テトラゾール−5−イル)エチル]−アミド、
(k) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[2−(1H−テトラゾール−5−イル)エチル]−アミド、
(l) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(m) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(3−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(n) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(o) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[(N−ヒドロキシカルボニルエチル−N−メチル)−2−アミノエチル]−アミド、
(p) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(3−フルオロフェニル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(q) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(4−フルオロフェニル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(r) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(s) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(t) 1−メチル−2−[N−(4−アミジノフェニル)アミノメチル]−インドール−5−イル−カルボン酸−N−フェニル−N−(2−メトキシカルボニルエチル)−アミド、
(u) 1−メチル−2−[N−(4−アミジノフェニル)アミノメチル]−チエノ[2.3−d]イミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(v) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(w) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(x) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(y) 1−メチル−2−[N−[4−(N−n−ヘキシルオキシカルボニルアミジノ)フェニル]−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−エトキシカルボニルエチル)−アミド、
より選択される化合物、その互変異性体、立体異性体および塩であり、それらの全ては、国際公開公報第98/37075号に記載されている。
【0046】
本発明の状況で好ましい抗凝固薬は、化学式(II):
【化3】
を有するダビガトラン(CAS211914−51−1、N−[2−(4−アミジノフェニルアミノメチル)−1−メチル−1H−ベンゾイミダゾール−5−イルカルボニル]−N−(2−ピリジル)−β−アラニン)である。
【0047】
ダビガトランは、トロンビン阻害作用およびトロンビン時間延長作用を有する化合物を1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミドの名称で開示している国際公開公報第98/37075号から公知である。Hauel et al. J Med Chem 2002, 45 (9): 1757-66も参照されたい。
【0048】
ダビガトランは、式(III):
【化4】
で示されるプロドラッグとして適用される。
【0049】
式IIIで示される化合物(名称ダビガトランエテキシラート、CAS211915−06−9;エチル3−[(2−{[4−(ヘキシルオキシカルボニルアミノ−イミノ−メチル)−フェニルアミノ]−メチル}−1−メチル−1H−ベンゾイミダゾール−5−カルボニル)−ピリジン−2−イル−アミノ]−プロピオナート)は、体内に入った後で活性化合物(II)に変換される。ダビガトランエテキシラートの好ましい多形は、ダビガトランエテキシラートメシラート塩である。
【0050】
ダビガトランの主要効能は、深部静脈血栓の術後予防、確立した深部静脈血栓の処置および心房細動を有する患者における発作の予防である(Eriksson et al., Lancet 2007, 370 (9591): 949-56; Schulman S et al, N Engl J Med 2009, 361 (24): 2342-52; Connolly S et al., N Engl J Med 2009, 361 (12): 1139-51; Wallentin et al., Lancet 2010, 376 (9745): 975-983)。
【0051】
ヒトの身体では、カルボキシラート部分のグルクロン酸抱合がダビガトランの主要なヒト代謝経路である(Ebner et al., Drug Metab. Dispos. 2010, 38(9):1567-75)。それは、1−O−アシルグルクロニド(βアノマー)の形成をもたらす。1−O−アシルグルクロニドは、アグリコンへのマイナー加水分解(minor hydrolysis)に加えて、水溶液中で非酵素的アシル転移を受けて、2−O−、3−O−、および4−O−アシルグルクロニドの形成をもたらすことがある。精製1−O−アシルグルクロニドおよびその異性体性転位産物を用いた実験は、ダビガトランに比べて等しい効力の活性化部分トロンボプラスチン時間の延長を明らかにした。
【0052】
本発明の別の局面では、抗体分子は、ダビガトランとダビガトランエテキシラートの両方に結合する。
【0053】
本発明の別の局面では、抗体分子は、ダビガトランとダビガトランのO−アシルグルクロニドの両方に、特にダビガトランの1−O−アシルグルクロニドに結合する。
【0054】
本発明の別の局面では、抗体分子は、さらにダビガトランの2−O−、3−O−、および4−O−アシルグルクロニドに結合する。
【0055】
本発明の別の局面では、抗体分子は、ダビガトランおよびダビガトランのO−アシルグルクロニド、特にダビガトランの1−O−アシルグルクロニドの活性を中和可能である。
【0056】
本発明の別の局面では、抗体分子は、ダビガトランに対する結合特異性を有し、図5および6に示されるCDR配列を含む。
【0057】
本発明の別の局面では、抗体分子は、図5に示される重鎖CDR配列、および図6に示される軽鎖CDR配列を含む。
【0058】
本発明の別の局面では、抗体分子は、図5に示される重鎖可変ドメイン配列、および図6に示される軽鎖可変ドメイン配列を含む。
【0059】
本発明の別の局面では、抗体分子は、図5において名称DBG 13 VHで示される重鎖可変ドメイン配列、および図6において名称DBG 13 VKで示される軽鎖可変ドメイン配列を含む。
【0060】
本発明の別の局面では、抗体分子は、図5において名称DBG 14 VHで示される重鎖可変ドメイン配列、および図6において名称DBG 14 VKで示される軽鎖可変ドメイン配列を含む。
【0061】
本発明の別の局面では、抗体分子は、図5において名称DBG 22 VHで示される重鎖可変ドメイン配列、および図6において名称DBG 22 VKで示される軽鎖可変ドメイン配列を含む。
【0062】
本発明の別の局面では、抗体分子は、図5において名称Eng VH# 14で示される重鎖可変ドメイン配列、および図6において名称Eng VK# 11で示される軽鎖可変ドメイン配列を含む。
【0063】
本発明の別の局面では、抗体分子は、図5において名称Eng VH# 15で示される重鎖可変ドメイン配列、および図6において名称Eng VK# 17で示される軽鎖可変ドメイン配列を含む。
【0064】
本発明の別の局面では、抗体分子は、図5において名称Eng VH# 15で示される重鎖可変ドメイン配列、および図6において名称Eng VK# 18で示される軽鎖可変ドメイン配列を含む。
【0065】
本発明の別の局面では、抗体分子は、図5において名称Eng VH# 31で示される重鎖可変ドメイン配列、および図6において名称Eng VK# 18で示される軽鎖可変ドメイン配列を含む。
【0066】
本発明の別の局面では、抗体分子は、ダビガトランに対する結合特異性を有し、配列番号:1および配列番号:2から成る群より選択されるCDR1、配列番号:3、配列番号:4、配列番号:5、配列番号:6、配列番号:7、および配列番号:8から成る群より選択されるCDR2、ならびに配列番号:9および配列番号:10から成る群より選択されるCDR3を有する重鎖可変ドメインと、配列番号:11、配列番号:12、および配列番号:13から成る群より選択されるCDR1、配列番号:14で示されるCDR2、ならびに配列番号:15で示されるCDR3を有する軽鎖可変ドメインとを含む。
【0067】
本発明の別の局面では、抗体分子は、配列番号:1で示されるCDR1、配列番号:6、配列番号:7、および配列番号:8から成る群より選択されるCDR2、ならびに配列番号:10で示されるCDR3を有する重鎖可変ドメインと、配列番号:13で示されるCDR1、配列番号:14で示されるCDR2、および配列番号:15で示されるCDR3を有する軽鎖可変ドメインとを含む。
【0068】
本発明の別の局面では、抗体分子は、配列番号:1で示されるCDR1、配列番号:7で示されるCDR2、および配列番号:10で示されるCDR3を有する重鎖可変ドメイン、ならびに配列番号:13で示されるCDR1、配列番号:14で示されるCDR2、および配列番号:15で示されるCDR3を有する軽鎖可変ドメインを含む。
【0069】
本発明の別の局面では、抗体分子は、配列番号:1で示されるCDR1、配列番号:5で示されるCDR2、および配列番号:10で示されるCDR3を有する重鎖可変ドメイン、ならびに配列番号:11で示されるCDR1、配列番号:14で示されるCDR2、および配列番号:15で示されるCDR3を有する軽鎖可変ドメインを含む。
【0070】
本発明の別の局面では、抗体分子は、配列番号:16、18、20、22、24、および26から成る群より選択される重鎖可変ドメイン、ならびに配列番号:17、19、21、23、25、および27から成る群より選択される軽鎖可変ドメインを含む。
【0071】
本発明の別の局面では、抗体分子は、配列番号:16で示される重鎖可変ドメイン、および配列番号:17で示される軽鎖可変ドメインを含む。
【0072】
本発明の別の局面では、抗体分子は、配列番号:18で示される重鎖可変ドメイン、および配列番号:19で示される軽鎖可変ドメインを含む。
【0073】
本発明の別の局面では、抗体分子は、配列番号:20で示される重鎖可変ドメイン、および配列番号:21で示される軽鎖可変ドメインを含む。
【0074】
本発明の別の局面では、抗体分子は、配列番号:22で示される重鎖可変ドメイン、および配列番号:23で示される軽鎖可変ドメインを含む。
【0075】
本発明の別の局面では、抗体分子は、配列番号:24で示される重鎖可変ドメイン、および配列番号:25で示される軽鎖可変ドメインを含む。
【0076】
本発明の別の局面では、抗体分子は、配列番号:24で示される重鎖可変ドメイン、および配列番号:27で示される軽鎖可変ドメインを含む。
【0077】
本発明の別の局面では、抗体分子は、配列番号:26で示される重鎖可変ドメイン、および配列番号:27で示される軽鎖可変ドメインを含む。
【0078】
本発明の別の局面では、抗体分子は、scFv分子である。この形式では、本明細書に開示された可変ドメインは、例えば配列番号:28、29、30、または31から成る群より選択される、適切なリンカーペプチドで相互に融合されていることがある。その構築物は、N末端からC末端にかけて(重鎖可変ドメイン)−(リンカーペプチド)−(軽鎖可変ドメイン)、または(軽鎖可変ドメイン)−(リンカーペプチド)−(重鎖可変ドメイン)の順序でこれらの要素を含むことがある。
【0079】
本発明の別の局面では、抗体分子は、配列番号:32または配列番号:33を含むscFv分子である。本発明の別の局面では、抗体分子は、配列番号:32または配列番号:33から成るscFv分子である。
【0080】
sFv構築物をコードする核酸を、宿主細胞(E. coli、Pichia pastoris、または哺乳動物細胞系、例えばCHOもしくはNS0など)に、組換え発現させて、機能的scFv分子を得るプロセスは、当技術分野において公知である(例えばRippmann et al., Applied and Environmental Microbiology 1998, 64(12): 4862-4869; Yamawaki et al., J. Biosci. Bioeng. 2007, 104(5): 403-407; Sonoda et al., Protein Expr. Purif. 2010, 70(2): 248-253を参照されたい)。
【0081】
特に、本発明のscFv抗体分子は、以下のように製造することができる。構築物は、W3110、TG1、BL21、BL21(DE3)、HMS174、HMS174(DE3)、MM294のような異なるE. coliに誘導性プロモーターのコントロール下で発現させることができる。このプロモーターは、lacUV5、tac、T7、trp、trc、T5、araBより選択することができる。培養培地は、好ましくはWilmsら、2001(Wilms et al., Biotechnology and Bioengineering 2001, 73(2): 95-103)、DeLisaら、1999(DeLisa et al., Biotechnology and Bioengineering 1999, 65(1): 54-64)にしたがって十分に定義されているか、または同等である。しかしながら、バッチ培地および/または供給培地へのイソロイシン、ロイシン、リシン、メチオニン、フェニルアラニン、トレオニン、トリプトファンおよびバリンなどのアミノ酸、またはダイズペプトンまたは酵母エキスなどの複合培地成分の補給が有益なことがある。発酵のためのプロセスは、流加モードで行う。条件:温度20〜40℃、pH5.5〜7.5、DOは20%よりも高く保つ。最初の炭素源の消費後に、培養物に上記供給培地(または同等物)を供給する。発酵槽中で40〜100g/Lの乾燥細胞重量に達したときに、使用されるプロモーター系に対応する適切な誘導物質(例えばIPTG、ラクトース、アラビノース)で培養物を誘導する。誘導は、パルス完全誘導として、またはそれぞれの誘導物質を発酵槽中に長時間供給することによる部分誘導もしくはその組み合わせとしてのいずれかで行うことができる。生産期は、少なくとも4時間持続すべきである。細胞は、ボウル遠心分離機、管状ボウル遠心分離機またはディスクスタック遠心分離機での遠心分離によって回収され、培養上清は捨てられる。
【0082】
E. coli細胞塊を、4〜8倍量の溶解緩衝液(リン酸緩衝液またはトリス緩衝液、pH7〜8.5)中に再懸濁する。細胞溶解を、好ましくは、高圧ホモジナイズに続いて、ボウル遠心分離機、管状ボウル遠心分離機またはディスクスタック遠心分離機での遠心分離によるペレットの回収によって行う。scFv封入体を含有するペレットを、20mMトリス、150mM NaCl、5mM EDTA、2M尿素、0.5%トリトンX−100(pH8.0)で2〜3回洗浄し、20mMトリス、150mM NaCl、5mM EDTA(pH8.0)を使用する2回の洗浄段階が続く。scFv封入体は、ボウル遠心分離機、管状ボウル遠心分離機またはディスクスタック遠心分離機での遠心分離によって最終的に回収する。scFv封入体の可溶化は、6Mグアニジン−HClまたは8〜10mM尿素などのカオトロピック剤を含有する100mMグリシン/NaOH、5mM EDTA、20mMジチオトレイトール(pH9.5〜10.5)中で行うことができる。30〜60分間インキュベーション後に、溶液を遠心分離し、その後の再フォールディングのためにターゲットタンパク質を含有する上清を回収する。再フォールディングは、好ましくは流加モードでタンパク質溶液を再フォールディング緩衝液に1:10〜1:50希釈して0.1〜0.5mg/mlの最終タンパク質濃度にすることによって行われる。再フォールディング緩衝液は、50〜100mMトリスおよび/または50〜100mMグリシン、50〜150mM NaCl、1〜3M尿素、0.5〜1Mアルギニン、例えばシテイン(cytein)/シスチンまたは酸化/還元型グルタチオンなどの2〜6mMの酸化還元系(pH9.5〜10.5)を含有することがある。4℃で24〜72hインキュベーション後に、再フォールディング溶液を、場合により0.22μmフィルターを使用して濾過し、希釈し、pHをpH7.0〜8.0に調整する。タンパク質を、結合モードの陽イオン交換クロマトグラフィー(例えばToyopearl GigaCap S-650M、SPセファロースFFまたはS HyperCel(商標))を介してpH7.0〜8.5で分離する。溶離を、漸増するNaClの直線勾配によって行う。ターゲットタンパク質を含有する画分をプールし、次に非結合モードの陰イオン交換カラム(例えばToyopearl GigaCap Q-650M、Q−セファロースFF、Q HyperCel(商標))に続く陽イオン交換仕上げステップ(例えばSPセファロースHP)で分離する。最低限で90%の純度レベルのターゲットタンパク質を含有する画分をプールし、ダイアフィルトレーションまたはPBSを用いたサイズ排除クロマトグラフィーによって製剤化する。製造されたscFv分子の同一性および製品品質を、還元SDS−PAGEによって分析し、その場合scFvは、約26kDaの1本の主バンドの形で検出することができる。scFvの特徴づけのためのさらなるアッセイには、質量分析、RP−HPLCおよびSE−HPLCが挙げられる。
【0083】
本発明の別の局面では、抗体分子は、免疫グロブリン、好ましくはIgG1またはそのエフェクター機能ノックアウトまたはIgG4型の免疫グロブリンである。本発明の別の局面では、抗体分子は、配列番号:34、配列番号:40、または配列番号:42を含む重鎖、および配列番号:35または配列番号:43を含む軽鎖を有する免疫グロブリンである。
【0084】
本発明の別の局面では、抗体分子は、配列番号:34を含む重鎖および配列番号:35を含む軽鎖、または配列番号:40を含む重鎖および配列番号:35を含む軽鎖を有する免疫グロブリンである。本発明の別の局面では、抗体分子は、配列番号:42を含む重鎖および配列番号:43を含む軽鎖を有する免疫グロブリンである。
【0085】
本発明の別の局面では、抗体分子は、配列番号:34から成る重鎖および配列番号:35から成る軽鎖、または配列番号:40から成る重鎖および配列番号:35から成る軽鎖を有する免疫グロブリンである。本発明の別の局面では、抗体分子は、配列番号:42から成る重鎖および配列番号:43から成る軽鎖を有する免疫グロブリンである。
【0086】
本発明の別の局面では、抗体分子はFab分子である。その形式では、上に開示された可変ドメインは、それぞれ免疫グロブリン定常ドメイン、好ましくはヒト起源に融合されていてもよい。したがって、重鎖可変ドメインは、CH1ドメインに融合されていることがあり(いわゆるFdフラグメント)、軽鎖可変ドメインはCLドメインに融合されていることがある。
【0087】
本発明の別の局面では、抗体分子は、配列番号:36または配列番号:38を含むFdフラグメントおよび配列番号:37または配列番号:39を含む軽鎖を有するFab分子である。本発明の別の局面では、抗体分子は、配列番号:36を含むFdフラグメントおよび配列番号:37を含む軽鎖を有するFab分子である。本発明の別の局面では、抗体分子は、配列番号:38を含むFdフラグメントおよび配列番号:39を含む軽鎖を有するFab分子である。本発明の別の局面では、抗体分子は、配列番号:41を含むFdフラグメントおよび配列番号:37を含む軽鎖を有するFab分子である。本発明の別の局面では、抗体分子は、配列番号:36から成るFdフラグメントおよび配列番号:37から成る軽鎖を有するFab分子である。本発明の別の局面では、抗体分子は、配列番号:38から成るFdフラグメントおよび配列番号:39から成る軽鎖を有するFab分子である。本発明の別の局面では、抗体分子は、配列番号:41から成るFdフラグメントおよび配列番号:37から成る軽鎖を有するFab分子である。
【0088】
E. coli、Pichia pastoris、または哺乳動物細胞系(例えばCHOまたはNS0)のような宿主細胞にそのような重鎖および軽鎖を発現させるために、Fab構築物をコードする核酸を使用することができる。Fdフラグメントおよび軽鎖を含む機能的Fab分子に、これらの鎖を適切にフォールディング、会合、およびジスルフィド結合させる方法は、当技術分野において公知である(Burtet et al., J. Biochem. 2007, 142(6), 665-669; Ning et al., Biochem. Mol. Biol. 2005, 38: 204-299; Quintero-Hernandez et al., Mol. Immunol. 2007, 44: 1307-1315; Willems et al. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 2003;786:161-176.)。
【0089】
特に、本発明のFab分子は、以下のようにCHO細胞から製造することができる。無血清培地中に懸濁状態で成長しているCHO−DG44細胞(Urlaub,G., Kas,E., Carothers,A.M., and Chasin,L.A. (1983). Deletion of the diploid dihydrofolate reductase locus from cultured mammalian cells. Cell 33, 405-412.)に、Lipofectamine(商標) and Plus(商標)試薬(Invitrogen)を製造業者の説明書にしたがって使用して、Fab分子の重鎖および軽鎖をコードする発現構築物をトランスフェクションする。48時間後に、200μg/mLの抗生物質G418を含有し、ヒポキサンチンおよびチミジンを有さない培地中で細胞を選択に供し、安定的にトランスフェクションされた細胞集団を発生させる。続いて、漸増する濃度(最大100または400nM)のメトトレキサート(MTX)を培養培地に添加することによって、これらの安定トランスフェクタントを遺伝子増幅に供する。いったん細胞が適応したならば、それらを10〜11日間流加発酵に供し、Fabタンパク質材料を製造する。
【0090】
化学的に限定された無血清培養培地中でCHO−DG44細胞およびその安定トランスフェクタントの懸濁培養物をインキュベーションする。播種用保存培養物を2〜3日毎にそれぞれ細胞3×105〜2×105個/mLの播種密度で継代培養する。細胞を振盪フラスコ中、Multitron HTインキュベーター(Infors)で5% CO2、37℃および120rpmで成長させる。流加実験のために、細胞を振盪フラスコの中で抗生物質もMTXも有さないBI独自の生産培地中に細胞3×105個/mLとなるように播種する。培養物を120rpmで37℃および5% CO2にて撹拌し、細胞数が増加するにつれてCO2をその後2%に減少させる。細胞数、生存率、pH、グルコースおよび乳酸濃度を含めた培養パラメーターを毎日決定し、必要に応じて炭酸塩を使用してpHをpH7.0に調整する。BI独自の栄養溶液を24時間毎に添加する。異なる時点で上清から試料を採取し、ELISAによってFab産物濃度を決定する。10〜11日後に、細胞培養液を遠心分離によって回収し、精製研究室に移送する。
【0091】
Fab分子を、流加培養の上清からクロマトグラフィーおよび濾過によって精製する。一次捕捉ステップとして、アフィニティークロマトグラフィー、例えばプロテインGまたはプロテインLを適用する。または、低結合親和性および低結合能の場合、Fabを、分子のpIを利用する陽イオン交換クロマトグラフィー(CEX)によって捕捉する。追加的な直交性の精製ステップによって、宿主細胞タンパク質および混入物、例えばDNAまたはウイルスを除去する。
【0092】
製造されたFab分子の同一性および製品品質を、電気泳動法、例えばSDS−PAGEによって分析し、それによって約50kDaの1本の主バンドとしてFabを検出することができる。Fab産物の特徴づけのためのさらなるアッセイには、質量分析、等電点電気泳動およびサイズ排除クロマトグラフィーが挙げられる。結合活性をBIAcore分析によって追跡する。
【0093】
細胞培養上清中のFabまたは完全長IgG分子の定量を、サンドイッチ酵素結合免疫吸着アッセイ(ELISA)により行う。完全長IgGは、ヒト−Fcフラグメント(Jackson Immuno Research Laboratories)およびヒトκ軽鎖に対して産生された抗体(ペルオキシダーゼ−コンジュゲーション型、Sigma)を使用して検出することができる。Fabフラグメントを、ヤギポリクローナル抗ヒトIgG(HおよびL、Novus)によって固定化し、ヒトIgGに対して産生されたヒツジポリクローナル抗体(ペルオキシダーゼ−コンジュゲーション型、結合部位)によって検出する。
【0094】
Fab分子は、また、酵素的切断によって完全長抗体分子から生成させることができる。このアプローチの利点は、所望の製品品質でのスケールアップおよび高収量が容易な、堅調で効率的な発酵および精製のための足場となるプロセスが適用可能であることである。精製のために、組換えプロテインA樹脂を使用するアフィニティークロマトグラフィーを一次捕捉ステップとして使用することができ、それは通常、高い純度をもたらす。
【0095】
この目的のために、Fab配列をコードする重鎖を、ヒトIgG抗体分子のFc領域に融合する。次に、得られた発現構築物を、無血清培地中に懸濁状態で成長しているCHO−DG44細胞にリポフェクションを用いてトランスフェクションする。48時間後に200μg/mLの抗生物質G418を含有しヒポキサンチンおよびチミジンを有さない培地中で細胞を選択に供し、安定トランスフェクションされた細胞集団を発生させた。続いて、メトトレキサート(MTX)を漸増する濃度(最大100または400nM)で培養培地に添加することによって、これらの安定トランスフェクタントを遺伝子増幅に供する。いったん細胞が適応したならば、それらを10〜11日間流加発酵に供し、IgGタンパク質材料を製造する。IgGタンパク質を、組換えプロテインA−アフィニティークロマトグラフィーを使用することによって培養上清から精製する。次に、所望の中和Fabフラグメントを得るために、ヒンジ領域内でIgGを切断するパパインの存在下で完全長IgGをインキュベーションし、それによって2本のFabフラグメントおよびFc部分を放出させる。配列番号:41のFd鎖を含むFab分子は、完全長IgGタンパク質のパパイン消化によって得られるFab分子の一例である。
【0096】
Fab分子を、アフィニティークロマトグラフィー、例えばプロテインGまたはプロテインLによって単離する。または、低結合親和性および低結合能の場合、Fabを、分子のpIを利用する陽イオン交換クロマトグラフィー(CEX)によって捕捉する。追加的な直交性の精製ステップによって宿主細胞タンパク質および混入物、例えばパパイン、DNAまたはウイルスを除去する。
【0097】
本発明の別の局面では、抗体分子は、本明細書記載の抗体分子のアミノ酸配列変異体である。
【0098】
抗体のアミノ酸配列変異体は、抗体DNAに適切なヌクレオチド変化を導入することによって、またはペプチド合成によって調製することができる。そのような変異体には、例えば本明細書における例に示される抗体のアミノ酸配列内の残基からの欠失および/またはその配列への挿入および/またはその配列の置換が挙げられる。最終構築物が所望の性質を有するという条件で、その最終構築物に到達するために欠失、挿入、および置換の任意の組み合わせが作られる。アミノ酸変化は、また、ヒト化または変異抗体の翻訳後プロセスを変え、グリコシル化部位の数または位置などを変化させることがある。
【0099】
突然変異誘発のために好ましい位置である、抗体のある種の残基または領域を同定するための有用な方法は、CunninghamおよびWells(Science, 244:1081-1085 (1989))によって記載されたような「アラニンスキャニング突然変異誘発」と呼ばれる。ここで、ターゲット残基のうちの一残基または群(例えばarg、asp、his、lys、およびgluなどの荷電残基)が同定され、アミノ酸と抗原の相互作用に影響するように中性または負電荷アミノ酸(典型的にはアラニン)によって置き換えられる。次に、置換に機能的感受性を示しているアミノ酸位置は、置換部位で、または置換部位の代わりにさらなる変異体または他の変異体を導入することによって洗練される。したがって、アミノ酸配列変異を導入するための部位が予め決定されている一方で、突然変異自体の性質が予め決定されている必要はない。例えば、所与の部位での突然変異の性能を分析するために、アラニンスキャニングまたはランダム突然変異誘発がターゲットのコドンまたは領域で行われ、発現された抗体変異体は、所望の活性についてスクリーニングされる。
【0100】
アミノ酸配列の挿入には、1個の残基から100個以上の残基を有するポリペプチドまでの長さの範囲のアミノ末端融合および/またはカルボキシル末端融合、ならびに一つ以上のアミノ酸残基の配列内挿入が挙げられる。末端挿入の例には、エピトープタグに融合された抗体が挙げられる。抗体分子の他の挿入変異体には、抗体のN末端もしくはC末端と、抗体の血清半減期を増加させる酵素またはポリペプチドとの融合体が挙げられる。
【0101】
別の型の変異体は、アミノ酸置換変異体である。これらの変異体は、抗体分子から少なくとも1個のアミノ酸残基が除去され、その位置に異なる残基が挿入されている。置換突然変異誘発のための最大に関心がもたれる部位には、超可変領域が挙げられるが、FRの変化も考えられる。保存的置換を、下表に「好ましい置換」の見出しのもとに示す。そのような置換が生物学的活性の変化を招くならば、「例示的な置換」と呼ばれるより実質的な変化またはアミノ酸クラスに関して下にさらに説明するような変化を導入して、その産物をスクリーニングすることができる。
【表1】
【0102】
タンパク質化学において、抗体の生物学的性質は、(a)置換領域におけるポリペプチド主鎖の、例えばシートまたはヘリカルコンフォメーションとしての構造、(b)ターゲット部位での分子の荷電もしくは疎水性、または(c)側鎖のかさを維持することに及ぼす作用が顕著に異なる置換を選択することによって達成できることが、一般に受け入れられている。天然残基は、共通の側鎖性質に基づき群に分類される:
(1)疎水性:ノルロイシン、met、ala、val、leu、ile;
(2)中性親水性:cys、ser、thr;
(3)酸性:asp、glu;
(4)塩基性:asn、gin、his、lys、arg;
(5)鎖の配向に影響する残基:gly、pro;および
(6)芳香族:trp、tyr、phe。
【0103】
非保存的置換は、これらのクラスのうち一つの一メンバーを別のクラスに交換することを伴うものである。
【0104】
ヒト化抗体または変異型抗体の適切なコンフォメーションを維持することに関与しない任意のシステイン残基も一般にセリンで置換して、分子の酸化的安定性を改善するか、異常な架橋を防止するか、または細胞毒性化合物もしくは細胞増殖抑制性化合物への所定の結合点を与えることができる。逆に、抗体にシステイン結合を加えて、その安定性を改善することができる(特にその抗体がFvフラグメントなどの抗体フラグメントである場合)。
【0105】
置換変異体の1種は、親抗体(例えばヒト化またはヒト抗体)の一つ以上の超可変領域残基を置換することを伴う。一般に、さらなる開発のために選択された、得られる変異体は、それらが生成された親抗体よりも改善された生物学的性質を有するものである。そのような置換変異体を生成させるために好都合な方法は、ファージディスプレイを用いたアフィニティー成熟である。簡潔には、いくつかの超可変領域部位(例えば6〜7個の部位)を突然変異させて、各部位で全ての可能なアミノ置換を生成させる。そのように生成された抗体変異体は、線維状ファージ粒子から、各粒子内にパッケージングされたM13の遺伝子III産物との融合物として一価的にディスプレイされる。次に、ファージディスプレイされた変異体をそれらの生物学的活性(例えば結合親和性)についてスクリーニングする。改変のための候補となる超可変領域部位を同定するために、アラニンスキャニング突然変異誘発を行って、抗原結合に顕著に寄与する超可変領域残基を同定することができる。または、あるいは加えて、抗原−抗体複合体の結晶構造を分析して、抗体とヒトダビガトランの間の接触点を同定することが有益なことがある。そのような接触残基および隣接残基は、本明細書に詳述される技法による置換のための候補である。そのような変異体がいったん生成されたならば、そのパネルの変異体を本明細書記載のスクリーニングに供し、一つ以上の関連するアッセイで優れた性質を有する抗体をさらなる開発のために選択することができる。
【0106】
抗体の別の種類のアミノ酸変異体は、抗体の本来のグリコシル化パターンを変える。「変えること」によって、抗体に見られる一つ以上の糖質部分を除去すること、および/またはその抗体に存在しない一つ以上のグリコシル化部位を付加することが意味される。
【0107】
いくつかの態様では、本発明の抗体を改変してグリコシル化部位を付加することが望ましいことがある。抗体のグリコシル化は、典型的にはN−結合型またはO−結合型のいずれかである。N−結合型は、アスパラギン残基側鎖に糖質部分が結合していることを表す。トリペプチド配列アスパラギン−X−セリンおよびアスパラギン−X−トレオニン(式中、Xはプロリン以外の任意のアミノ酸である)は、アスパラギン側鎖に糖質部分を酵素的に結合させるための認識配列である。したがって、ポリペプチド中にこれらのトリペプチド配列のいずれかが存在することで、潜在的グリコシル化部位が生じる。O−結合型グリコシル化は、ヒドロキシアミノ酸、最も一般的にはセリンまたはトレオニン(もっとも5−ヒドロキシプロリンまたは5−ヒドロキシリシンも使用することができる)に糖であるN−アセチルガラクトサミン、ガラクトース、またはキシロースの一つを結合させることを表す。したがって、所与のタンパク質、例えば抗体をグリコシル化するために、タンパク質のアミノ酸配列を操作して(N−結合型グリコシル化部位のために)一つ以上の上記トリペプチド配列を有するようにされる。その変化は、また、(O−結合型グリコシル化部位のための)本来の抗体配列への一つ以上のセリンまたはトレオニン残基の付加またはその残基による置換によって行うことができる。
【0108】
抗体のアミノ酸配列変異体をコードする核酸分子は、当技術分野で公知の多様な方法によって調製される。これらの方法には、非限定的に、天然起源からの単離(天然アミノ酸配列変異体の場合)または初期に調製された変異体もしくは非変異体版の本明細書記載の抗体分子のオリゴヌクレオチド介在性(もしくは部位特異的)突然変異誘発、PCR突然変異誘発、およびカセット突然変異誘発による調製が挙げられる。上記に概要するように、本発明の抗体分子の抗原は、抗凝固薬である。抗原は、動物の免疫処置によって、またはファージディスプレイ法を用いるような、配列ライブラリーから抗体配列を選択することによってのいずれかで抗体分子を生成させるために使用される。
【0109】
動物のための免疫処置プロトコールは、当技術分野において周知である。適切な免疫応答を達成するために、抗原をリン酸アルミニウム、水酸化アルミニウム、スクアレン、またはフロイントの完全/不完全アジュバントなどのアジュバントと結合することが必要なことがある。ダビガトランのような本発明に関連する抗原は、主に比較的小型の有機分子であって、動物に投与されたときに抗体の形成を刺激しないことがある有機分子である。したがって、抗原をハプテンとしての巨大分子に結合させることが必要なことがある。
【0110】
さらなる局面では、本発明は、医学に使用するための上記抗体分子に関する。
【0111】
さらなる局面では、本発明は、前記抗体分子および医薬担体を含む医薬組成物に関する。
【0112】
治療に使用するために、抗体分子は、動物またはヒトへの投与を容易にするために適切な医薬組成物に含まれる。抗体分子の典型的な製剤は、抗体分子を生理学的に許容されうる担体、賦形剤または安定化剤を混合することによって、凍結乾燥もしくはその他の方法で乾燥された製剤、あるいは水性溶液または水性もしくは非水性懸濁液の剤形に調製することができる。担体、賦形剤、改質剤(modifier)または安定化剤は、採用される薬用量および濃度で無毒である。それらには、リン酸、クエン酸、酢酸ならびに他の無機酸または有機酸およびそれらの塩などの緩衝系;アスコルビン酸およびメチオニンを含めた抗酸化剤;オクタデシルジメチルベンジルアンモニウムクロリド;塩化ヘキサメトニウム;塩化ベンザルコニウム、塩化ベンゼトニウム;フェノール、ブチルまたはベンジルアルコール;メチルまたはプロピルパラベンなどのアルキルパラベン;カテコール;レゾルシノール;シクロヘキサノール;3−ペンタノール;およびm−クレゾールなどの保存料);血清アルブミン、ゼラチン、または免疫グロブリンなどのタンパク質;ポリビニルピロリドンまたはポリエチレングリコール(PEG)などの親水性ポリマー;グリシン、グルタミン、アスパラギン、ヒスチジン、アルギニン、またはリシンなどのアミノ酸;グルコース、マンノース、スクロース、トレハロース、デキストリンまたはデキストランを含めた単糖、二糖、オリゴ糖または多糖および他の糖質;EDTAなどのキレート剤;マンニトールまたはソルビトールなどの糖アルコール;ナトリウムなどの塩形成性対イオン;金属錯体(例えば、Zn−タンパク質錯体);および/またはTWEEN(商標)(ポリソルベート)、PLURONICS(商標)もしくは脂肪酸エステル、脂肪酸エーテルもしくは糖エステルなどのイオン性もしくは非イオン性界面活性剤が挙げられる。有機溶媒もエタノールまたはイソプロパノールなどの抗体製剤に含有されることがある。賦形剤は、また、放出改変または吸収改変機能を有することがある。
【0113】
一局面では、医薬組成物は、水性緩衝液中に10〜20mg/mlの濃度で抗体分子を含むか、またはそのような溶液から作られた凍結乾燥物を含む。
【0114】
好ましい適用様式は、注入または注射(静脈内、筋肉内、皮下、腹腔内、皮内)による非経口的であるが、吸入、経皮、鼻腔内、口腔、経口などの他の適用様式もまた適用可能なことがある。
【0115】
さらなる局面では、本発明は、抗凝固治療の副作用、特に出血事象の治療または予防に使用するための上記抗体分子に関する。さらなる局面では、本発明は、抗凝固薬、特にダビガトランまたはダビガトランエテキシラートの過剰投与のリバースに使用するための上記抗体分子に関する。
【0116】
さらなる局面では、本発明は、抗凝固薬、特にダビガトランまたはダビガトランエテキシラートの解毒剤として使用するための上記抗体分子に関する。
【0117】
さらなる局面では、本発明は、抗凝固治療の副作用の治療または予防方法であって、それを必要とする患者に上記抗体分子の有効量を投与することを含む方法に関する。
【0118】
さらなる局面では、本発明は、抗凝固治療における過剰投与事象の処置方法であって、それを必要とする患者に上記抗体分子の有効量を投与することを含む方法に関する。
【0119】
投与されるべき抗体の「治療有効量」は、抗凝固治療の副作用を予防、改善、または治療するために必要な最小量、特に出血を止めるために有効な最小量である。これは、抗体分子の化学量論量で達成することができる。
【0120】
ダビガトランは、例えば、推奨用量で与えられた場合に200nMの大きさの血漿中濃度に達することがある。分子量約50kDの一価抗体分子が使用される場合、ボーラスとして静脈内に与えたとき例えば約1mg/kgの用量で中和に達することができる。別の態様では、ヒト患者に適用されたFab分子の用量は、適用1回あたり50〜1000mg、例えば100、200、500、750、または1000mgのことがある。状況に応じて、例えばダビガトランが患者に過剰投与されている場合、より高い用量、例えば適用1回あたり1250、1500、1750または2000mgを適用することさえ妥当なことがある。適切な用量は、投与される抗凝固薬の種類および用量;そのような投与から経過した時間、抗原分子の性質、患者の状態、ならびに他の要因に応じて異なることがある。熟練の専門家は、治療的に有効かつ安全な用量を確立するための方法を知っている。
【0121】
さらなる局面では、本発明は、ダビガトランおよび/またはダビガトランエテキシラートに対する結合親和性を有する抗体分子に関する。好ましくは、抗体分子は、例えば表面プラズモン共鳴分析(Malmqvist M., "Surface plasmon resonance for detection and measurement of antibody-antigen affinity and kinetics. "Curr Opin Immunol. 1993 Apr;5(2):282-6.)または動力学排除アッセイ(KinExA)技法(Darling, R.J., and Brault P-A., "Kinetic exclusion assay technology: Characterization of Molecular Interactions." ASSAY and Drug Development Technologies. 2004, Dec 2(6): 647-657)により決定されるとき、0.1pM〜100μM、好ましくは1pM〜100μM、より好ましくは1pM〜1μMの範囲のKD値を有する親和性でダビガトランおよび/またはダビガトランエテキシラートに結合する。
【0122】
本発明の抗体分子を、また、分析および診断手順のために使用して、例えば血漿、血清、または他の体液などの試料中の抗原濃度を決定することができる。例えば、抗原分子は、実施例に記載されたような酵素結合免疫吸着アッセイ(ELISA)に使用することができる。したがって、さらなる局面では、本発明は、本明細書記載の抗体分子を含む分析キットおよび診断キットならびにそれぞれの分析方法および診断方法に関する。
【0123】
さらなる局面では、本発明は、前記請求項のいずれか一項記載の抗体分子を製造する方法であって、
(a)発現制御配列と機能的に関連して該抗体分子をコードする一つ以上の核酸を含む宿主細胞を提供すること、
(b)該宿主細胞を培養すること、および
(c)細胞培養物から抗体分子を回収すること
を含む方法に関する。
【0124】
本発明は、さらに、経口抗凝固薬、特に直接トロンビン阻害剤の中和に有用な材料が入っている製品およびキットを提供する。製品は、ラベル付きの容器を含む。適切な容器には、例えばボトル、バイアル、および試験管が挙げられる。容器は、ガラス、金属、プラスチックまたはその組み合わせなどの多様な材料から形成されていることがある。容器は、本明細書記載の抗体またはダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩を含む医薬組成物を収容する。医薬組成物中の活性薬剤は、特定の抗体またはダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩である。抗体の容器上のラベルは、その医薬組成物がダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩を中和または部分的に中和するためにin vivoで使用されることを表示している。
【0125】
本発明のキットは、一つ以上の上記容器を含む。そのキットは、さらに、他の緩衝液、希釈剤、フィルター、針、シリンジ、および使用法を掲載した添付文書を含めた、商業的および利用者の観点から望ましい他の材料を包含することができる。
【0126】
本発明の一態様では、キットは、本明細書記載の任意の一つの抗体である抗体またはその医薬組成物を含む。例えば、キットは、(1)本明細書記載の任意の一つの抗体またはその医薬組成物、(2)容器および(3)ラベルを含むことができる。
【0127】
別の態様では、キットは、本明細書記載の任意の一つの抗体である抗体またはその医薬組成物、およびダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩を含む。ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の形態は、固体、液体またはゲルの形態のことがある。好ましい態様では、ダビガトランエテキシラートの薬学的に許容されうる塩は、メシラート塩である。なお別の好ましい態様では、投薬ユニットあたりのダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の量は、約50mg〜約400mg、約75mg〜約300mg、約75mg〜150mg、または約110mg〜約150mgの間であって、1日1回(QD)または1日2回(BID)与えられる。例えば、キットは、(1)本明細書記載の任意の一つの抗体またはその医薬組成物、(2)ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の医薬組成物、(3)容器および(4)ラベルを含むことがある。
【0128】
代替の態様では、キットは、(1)ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩を含む第1の医薬組成物、(2)本明細書記載の抗体の任意の一つまたはその組み合わせを含む第2の医薬組成物、(3)患者に該第1および第2の医薬組成物を別々に投与するための説明書を含み、ここで、該第1および第2の医薬組成物は、別々の容器に入っており、該第2の医薬組成物は、ダビガトランまたはダビガトランの1−O−アシルグルクロニドの中和または部分中和を必要とする患者に投与される。本発明は、また、ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩で処置されている患者においてダビガトランまたはダビガトランの1−O−アシルグルクロニドを中和または部分中和するための診断方法であって、本明細書記載の抗体の任意の一つ、その組み合わせまたはその医薬組成物を投与することを含む方法を提供する。具体的には、本発明は、患者においてダビガトランまたはダビガトランの1−O−アシルグルクロニドを中和または部分中和するための方法であって、(a)患者がダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩で処置されていたこと、およびその患者によって取り込まれた量を確認するステップ;(b)ダビガトランまたはダビガトランの1−O−アシルグルクロニドが試験またはアッセイの結果の正確な読み出しを妨害するであろう試験またはアッセイを行う前に、本明細書記載の抗体の任意の一つまたはその組み合わせでダビガトランまたは1−O−アシルグルクロニドを中和するステップ;(c)ダビガトランもダビガトランの1−O−アシルグルクロニドも存在しないときの血餅形成レベルを決定するために、患者から採取された試料に凝固または血液凝固の試験またはアッセイを行うステップ;ならびに(d)患者における血餅形成と分解の間に適切なバランスを達成するために、患者に投与されるダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の量を調整するステップを含む方法を提供する。ダビガトランまたはダビガトランの1−O−アシルグルクロニドに対する抗体のモル比は、0.1〜100の間、好ましくは0.1〜10の間のモル比である。試験またはアッセイの結果の正確な読み出しは、フィブリノーゲンレベル、活性化プロテインC抵抗性または関連試験の正確な読み出しのことがある。
【0129】
実施例
I. ポリクローナル抗ダビガトラン抗体の製造
ポリクローナル抗ダビガトラン抗体の製造のために、2種の異なるハプテンおよびハプテンと担体タンパク質(BSA)の異なるモル投入量比で、3種の異なる免疫原を製造した。
【0130】
スクリーニングのために、酵素ホースラディッシュペルオキシダーゼ(HRP)−コンジュゲートを製造し、酵素免疫吸着アッセイ(ELISA)を開発した。
【0131】
ポリクローナル抗体のさらなる精製は、プロテインAセファロース FFを用いたアフィニティークロマトグラフィーによって行った。
【0132】
1. 材料および方法
【表2】
【0133】
1.1 免疫原およびトレーサーを合成するために使用されたハプテン
【表3】
【0134】
【表4】
【0135】
1.2 ハプテンの合成
ハプテンである、ハプテン1およびハプテン2を以下のように合成した:
ハプテン1 2−[(4−カルバムイミドイル−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボン酸[2−(4−アミノ−ブチルカルバモイル)−エチル]−フェニル−アミド
【化5】
【0136】
1a 3−[(4−メチルアミノ−3−ニトロ−ベンゾイル)−フェニル−アミノ]−プロピオン酸メチルエステル
【化6】
4−メチルアミノ−3−ニトロ−安息香酸クロリド(23.3mmol)および3−フェニル−アミノ−プロピオン酸メチルエステル(23.3mmol)の無水テトラヒドロフラン(THF)80mL中の溶液に、トリエチルアミン(50.2mmol)を撹拌しながら室温で滴下により加えた。3時間後に反応混合物を蒸発乾固し、残りの固体を水で粉砕し、固体生成物を濾過により単離した。
収率:99%
C18H19N3O5(357.36)
TLC(シリカゲル;ジクロロメタン/エタノール 19:1):Rf=0.48
【0137】
1b 3−[(3−アミノ−4−メチルアミノ−ベンゾイル)−フェニル−アミノ]−プロピオン酸メチルエステル
【化7】
触媒としてPd(炭上に10%)を用いてエタノール中で室温で水素化することによって生成物1aのニトロ基を還元した。
収率:99%
C18H21N3O3(327.38)
TLC(シリカゲル;ジクロロメタン/エタノール 9:1):Rf=0.23
質量スペクトル(ESI):[M+H]+=328
【0138】
1c 3−({3−[2−(4−シアノ−フェニルアミノ)−アセチルアミノ]−4−メチルアミノ−ベンゾイル}−フェニル−アミノ)−プロピオン酸メチルエステル
【化8】
生成物1b(23.2mmol)およびN−(4−シアノ−フェニル)−グリシン(23.2mmol)を無水THF中で室温にてCDI(23.2mmol)でカップリングさせた。反応の完了後に、その混合物を蒸発乾固させ、粗生成物をさらに精製せずに使用した。
収率:97%
C27H27N5O4(485.54)
質量スペクトル(ESI):[M+H]+=486
【0139】
1d 3−({2−[(4−シアノ−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボニル}−フェニル−アミノ)−プロピオン酸メチルエステル
【化9】
生成物1c(22.6mmol)の濃酢酸100mL中の溶液を1時間加熱還流した。次に、その溶液を蒸発乾固し、残った固体を水で粉砕し、撹拌しながらpHを約8〜9に調整した。酢酸エチルで抽出することによって粗生成物を単離し、シリカゲルを用いたクロマトグラフィー(溶離液:ジクロロメタン/エタノール 1:1)によって精製した。
収率:58%
C27H25N5O3(467.52)
TLC(シリカゲル;ジクロロメタン/エタノール 9:1):Rf=0.71
質量スペクトル(ESI):[M+H]+=468
【0140】
1e 3−({2−[(4−シアノ−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボニル}−フェニル−アミノ)−プロピオン酸
【化10】
生成物1d(13.0mmol)の100mLメタノール中の溶液に、水酸化ナトリウム(20.0mmol)を添加した。その混合物を40℃で2.5時間撹拌し、次に蒸発乾固した。残った固体を水100mLと一緒に撹拌し、そして濃酢酸でpHを約6に調整した。沈殿した生成物を濾過によって単離し、水で洗浄し、60℃で乾燥させた。
収率:88%
C26H23N5O3(453.49)
TLC(シリカゲル;ジクロロメタン/エタノール 9:1):Rf=0.33
質量スペクトル(ESI):[M+H]+=454
【0141】
1f {4−[3−({2−[(4−シアノ−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボニル}−フェニル−アミノ)−プロピオニルアミノ]−ブチル}−カルバミン酸tert−ブチルエステル
【化11】
生成物1e(5.23mmol)、2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウム テトラフルオロボレート(TBTU、5.23mmol)およびN−メチル−モルホリン(5.23mmol)のDMF 20mL中の溶液を室温で30分間撹拌した。次に、(4−アミノ−ブチル)−カルバミン酸tert−ブチルエステル(5.23mmol)を添加し、その混合物を室温で新たに24時間撹拌した。次に、その混合物を水(100mL)で希釈し、酢酸エチルで抽出することによって生成物を単離した。
収率:92%
C35H41N7O4(623.75)
TLC(シリカゲル;ジクロロメタン/エタノール 9:1):Rf=0.51
【0142】
1g 2−[(4−カルバムイミドイル−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボン酸[2−(4−アミノ−ブチルカルバモイル)−エチル]−フェニル−アミド
【化12】
生成物1f(4.81mmol)をHClの飽和エタノール溶液(250mL)に溶解させ、その混合物を室温で一晩撹拌し、次に30℃で蒸発乾固した。残った原材料を無水エタノール200mLに溶解させ、次に炭酸アンモニウム(48.1mmol)を添加し、その混合物を室温で一晩撹拌した。溶媒の蒸発後に、残った原材料をエタノール約5mLと一緒に摩砕し、不溶性物質を濾過によって分離し、溶媒を30℃で蒸発させた。次に、生成物を水30mLに溶解させ、溶液を約2gの炭と一緒に撹拌し、濾過し、蒸発乾固した。
収率:90%
C30H36N8O2(540.67)
TLC(逆相RP−8;メタノール/5%NaCl水溶液 9:1):Rf=0.79
質量スペクトル(ESI):[M+H]+=541
[M+Cl]−=575/7
【0143】
ハプテン2 2−[(4−カルバムイミドイル−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボン酸[2−(2−アミノ−エチルカルバモイル)−エチル]−ピリジン−2−イル−アミド
【化13】
【0144】
2a 3−({2−[(4−シアノ−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボニル}−ピリジン−2−イル−アミノ)−プロピオン酸
【化14】
水酸化ナトリウム(50.0mmol)のエタノール500mLおよび水50mL中の溶液に、3−({2−[(4−シアノ−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボニル}−ピリジン−2−イル−アミノ)−プロピオン酸エチルエステル(41.4mmol)を添加した。その混合物を室温で3時間撹拌し、次にエタノール約350mLを留出させ、水約100mLを添加し、pHを6に調整した。次にジエチルエーテル(50mL)を添加し、その混合物を一晩撹拌した。生成物を濾過により単離し、さらに精製せずに使用した。
収率:78%
C25H22N6O3(454.48)
【0145】
2b {2−[3−({2−[(4−シアノ−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボニル}−ピリジン−2−イル−アミノ)−プロピオニルアミノ]−エチル}−カルバミン酸tert−ブチルエステル
【化15】
生成物2a(2.20mmol)、2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムテトラフルオロボレート(TBTU、2.20mmol)およびN−メチル−モルホリン(2.20mmol)の無水テトラヒドロフラン(100mL)中の溶液を室温で15分間撹拌した。次に(2−アミノ−エチル)−カルバミン酸tert−ブチルエステル(2.20mmol)を添加し、そしてその混合物を室温で新たに24時間撹拌した。次にその混合物を水40mLで希釈し、酢酸エチルで抽出することによって生成物を単離し、クロマトグラフィー(シリカゲル;ジクロロメタン/メタノール 15:1)によって精製した。
収率:61%
C32H36N8O4(596.68)
質量スペクトル(ESI):[M+H]+=597
[M+H]−=595
【0146】
2c 2−[(4−カルバムイミドイル−フェニルアミノ)−メチル]−1−メチル−1H−ベンゾイミダゾール−5−カルボン酸[2−(2−アミノ−エチルカルバモイル)−エチル]−ピリジン−2−イル−アミド
【化16】
生成物2b(1.34mmol)をHClの飽和無水エタノール溶液(30mL)に添加した。その溶液を室温で5時間撹拌し、次に30℃で蒸発乾固した。エタノール(30mL)および炭酸アンモニウム(13.0mmol)を添加し、そしてその混合物を室温で一晩撹拌した。次に溶媒を蒸発させ、無機塩からの生成物を分離するために残った物質をジクロロメタン/メタノール(30:1)混合物約4mLで5回粉砕し、濾過し、蒸発させた。
収率:27%
C27H31N9O2(513.61)
質量スペクトル(ESI):[M+Cl]−=548/50
[M+HCl+Cl]−=584/6
[M+H]+=514
【0147】
2. 化学物質
2.1 試薬合成用の化学物質
【表5】
【0148】
2.2 ELISA用化学物質
【表6】
【0149】
2.3 ELISA用緩衝液
【表7】
【0150】
3. 免疫原の合成
ウサギの免疫系を刺激してダビガトランに対するポリクローナル抗体を生成するために、カップリング試薬として1,4−ベンゾキノンまたは1,1’−カルボニル−ジ−(1,2,4−トリアゾール)を使用して、ハプテンであるハプテン1およびハプテン2を担体タンパク質であるウシ血清アルブミン(BSA)にカップリングさせることによって、3種の免疫原(ロット番号GL256、GL258、およびGL262)を合成した。
【0151】
GL256の合成のために、2個の反応部位を有するホモ二官能性化合物として1,4−ベンゾキノンを使用した。まず、それは酸性pHで2個の部位のうち一方のアミノ基だけと反応し、そして最小限のポリマー化で、アルカリ性pHでもう一方の部位で反応する。GL258およびGL262は、ハプテンに対する担体タンパク質の異なる投入量比で、1,1’−カルボニル−ジ−(1,2,4−トリアゾール)をカップリング試薬として使用して合成した。
【0152】
3.1 GL256の合成
0.1M KH2PO4緩衝液8.5mL中の0.75μMol BSAの溶液(pH=4.5)に、0.416mMol 1,4−ベンゾキノン(エタノール1.5mL中)を添加し、そして室温、暗中で1.5時間インキュベーションした。その後0.15M NaClで平衡化したセファデックスG25カラムにその溶液を通過させ、過剰の1,4−ベンゾキノン(最終体積12.5mL)を除去した。
【0153】
0.1M NaHCO3/Na2CO3緩衝液(pH=8.5)2mLに溶解された525μMolハプテンであるハプテン1の溶液に、撹拌しながら精製BSA溶液2.5mL(0.15μMol)をゆっくりと添加した。BSA溶液を添加する間に、pHを約8.0に調整した。ハプテンと担体タンパク質のモル投入量比は、3500:1であった。
【0154】
室温で一晩インキュベーション後に、免疫原を1リットルの蒸留水で6回透析した。薄層クロマトグラフィーは、ハプテン−担体コンジュゲート中に残った非結合ハプテンのスポットを示さなかった。
【0155】
免疫原を小分けして−20℃で凍結保存した。免疫原の上清中のハプテンによるBSAの置換度は、302nmでUV吸収分光法によって決定したとおり、約1:18であった。最終溶液中の免疫原の含量は、GL256/mL 0.75mgであった。
【0156】
3.2 GL258の合成
N,N−ジメチルホルムアミド(DMF)6.3mL中の158μMolハプテン2の溶液を室温で調製した。158μMol 1,1’−カルボニル−ジ−(1,2,4−トリアゾール)を添加し、そしてまず10℃で4時間、そしてその後室温で30分間インキュベーションした。薄層クロマトグラフィーで化学反応をチェックし、そしてそれは約20〜25%であった。次に、0.75μMol BSAを0.13M NaHCO3 2mLに溶解させ、撹拌しながらN,N−ジメチルホルムアミド(DMF)1mLを滴下により加えた。pHを約8.3に調整した。その後、撹拌しながらハプテン溶液(6.3mL)および0.13M NaHCO3 4mLをBSA溶液に滴下により加え、pHを8.4に調整した。免疫原GL258についてハプテンと担体タンパク質のモル投入量比は210:1であった。
【0157】
室温で一晩、撹拌条件下でインキュベーション後に、免疫原を1リットルの蒸留水で6回透析した。薄層クロマトグラフィーは、ハプテン−担体コンジュゲート中に残った非結合ハプテンのスポットを示さなかった。
【0158】
免疫原を小分けにして−20℃で凍結保存した。免疫原の上清中のハプテンによるBSAの置換度は、302nmでUV吸収分光法によって決定したとき約1:5であった。最終溶液中の免疫原の含量は、GL258/mL 0.28mgであった。
【0159】
3.3 GL262の合成
N,N−ジメチルホルムアミド(DMF)8.75mL中の225μMolハプテン2の溶液を室温で調製した。225μMol 1,1’−カルボニル−ジ−(1,2,4−トリアゾール)を添加し、そして10℃で4時間インキュベーションした。薄層クロマトグラフィーで化学反応をチェックし、それは約20〜25%であった。
【0160】
次に0.49μMol BSAを0.13M NaHCO3 2mLに溶解させ、撹拌しながらN,N−ジメチルホルムアミド(DMF)1mLを滴下により加えた。pHを約8.2に調整した。その後、撹拌しながらハプテン溶液(8.75mL)および0.13M NaHCO3 6mLをBSA溶液に滴下により加え、pHを8.3に調整した。免疫原GL262についてハプテンと担体タンパク質のモル投入量比は、460:1であった。
【0161】
室温で一晩、撹拌条件下でインキュベーション後に、免疫原を1リットルの蒸留水で6回透析した。薄層クロマトグラフィーは、ハプテン−担体コンジュゲート中に残った非結合ハプテンのスポットを示さなかった。
【0162】
免疫原を小分けにして−20℃で凍結保存した。免疫原の上清中のハプテンによるBSAの置換度は、302nmでUV吸収分光法によって決定したとき約1:32であった。最終溶液中の免疫原の含量は、GL262/mL 0.71mgであった。
【0163】
4. コンジュゲートの合成
4.1 GL261の合成
N,N−ジメチルホルムアミド(DMF)1.5mL中の37.4μMolハプテン2の溶液を室温で調製した。37.5μMol 1,1’−カルボニル−ジ−(1,2,4−トリアゾール)を添加し、そしてまず10℃で4時間、そしてその後室温で30分間インキュベーションした。薄層クロマトグラフィーで化学反応をチェックし、それは約20〜25%であった。
【0164】
次に1.125μMol酵素ホースラディッシュペルオキシダーゼ(HRP)を0.13M NaHCO3 0.4mLに溶解させ、撹拌しながらN,N−ジメチルホルムアミド(DMF)0.267mLを滴下により加えた。pHを約8.2に調整した。その後、撹拌しながらハプテン溶液(22.5μMol)0.9mLおよび0.13M NaHCO3 0.57mLをHRP溶液に滴下により加え、そしてpHを8.4に調整した。HRPコンジュゲートGL261についてハプテンとHRPのモル投入量比は、20:1であった。
【0165】
室温で一晩、撹拌条件下でインキュベーション後に、HRPコンジュゲートを有機溶媒および過剰のハプテンからゲルクロマトグラフィーによって分離した。0.1Mリン酸緩衝液(pH7.0)で平衡化したセファデックスG25カラムにその溶液を通過させた。最終濃度のハプテン−HRPコンジュゲート(トレーサー、5.64mg/mL)は、BSAによりスパイクされ、濃度約10mg/mLで得られ、凍結防止のために等体積のグリセリン、および細菌の成長を防止するためにチモールの結晶を加えた。トレーサー溶液をロット番号GL261とラベルして、小分けして−20℃で保存した。
【0166】
ハプテンによるHRPの置換度は、302nmでUV吸収分光法によって決定したとき1:0.2であった。
【0167】
BSAでブロッキングされたマイクロタイタープレート中、基質としてo−フェニレン−ジアミン(OPD)および対照物質としてネイティブなHRPを使用してトレーサーの比活性を測定した。希釈されたHRP標準、またはハプテン−HRPコンジュゲート及び基質溶液との混合物を暗中で、30分間インキュベーションし、硫酸で停止させ、吸光度を490nmで測定した。残りの活性はネイティブなHRPの94%であって、グリセリン中でのコンジュゲート形成の比活性は611U/mLであった。
【0168】
トレーサーの特定の概要:
【0169】
【表8】
【0170】
5 免疫処置および抗体の製造
5.1 ウサギの免疫処置
12匹の雌チンチラウサギ(3月齢)を、100μg免疫原GL256、GL258およびGL262の、0.9% NaCl溶液0.5mLおよびフロイント完全アジュバント(CFA)0.5mLのエマルションで免疫化した。何回かの追加免疫処置を翌月に続けた。3回目の免疫処置については、フロイント不完全アジュバント(IFA)0.5mLを使用した。4箇所の皮下部位および4箇所の筋肉内部位に各免疫処置を行った。
【0171】
A群 − 免疫原GL256
ウサギ1 #50
ウサギ2 #51
ウサギ3 #52
ウサギ4 #53
B群 − 免疫原GL258
ウサギ5 #54
ウサギ6 #55
ウサギ7 #56
ウサギ8 #57
C群 − 免疫原GL262
ウサギ9 #46
ウサギ10 #47
ウサギ11 #48
ウサギ12 #49
【0172】
【表9】
*ウサギ番号1〜12を5回目の免疫処置の10日後に完全に瀉血した。
キシラジン(Rompun(登録商標), Bayer, Leverkusen, Germany)および塩酸ケタミン(Ketavet(登録商標), Parke-Davis, Freiburg, Germany)での麻酔下で、頸動脈を介して瀉血を行った。
【0173】
5.2 ウサギ血清の分析
凝固したウサギ血液の遠心分離によって血清を調製した。硫酸アンモニウム沈殿およびセファデックスG25カラム内に通過させる脱塩によってタンパク質画分を得た。
【0174】
ウサギ血清からの個別のタンパク質画分を標準ELISA法によって抗ダビガトラン力価についてスクリーニングした。
【0175】
スクリーニング−ELISA
【表10】
【0176】
5.3 ウサギ血清中の抗ダビガトラン抗体の検出
最後の3個の欄:値はダビガトランに関するものである。
【0177】
【表11】
【0178】
採血2からの全ウサギのタンパク質画分のスクリーニング後に、ウサギ番号5(#54)が好ましいハプテンであるハプテン2で、最も高い力価の抗ダビガトラン抗体を有することが明らかとなった。さらに、低濃度の分析物(ダビガトラン)だけで抗体結合部位からトレーサーを置換させることが可能であった。
【0179】
最終の採血3のスクリーニングについて、使用された免疫原に関する情報が欠落しているので、低濃度の分析物(ダビガトラン)を用いた抗体結合部位からのトレーサーの置換を主な決定基準として使用した。したがって、ウサギ番号2、3および5をさらなる精製のために使用した。
【0180】
5.4 ポリクローナル抗体の精製
ウサギ番号5(#54)、採血番号2およびウサギ番号2、3および5採血番号3(最後の採血)の抗血清を硫酸アンモニウムで沈殿させた。沈殿を4500U/min、10℃で30分間遠心分離し、溶液から分離し、トリス緩衝液中に再溶解させた。この手順を繰り返した。プロテインAセファロースFFを用いたアフィニティークロマトグラフィーによってさらなる精製を行った。カラム緩衝液は0.01Mトリス(pH=7.5)であり、そして0.1Mグリシン(pH=3.0)を溶離のために使用した。ウサギIgGを含有する画分を合わせた。タンパク質濃度は、280nmでのUV分光法によって決定した。
【0181】
抗体の特定の概要:
【表12】
【0182】
II. ダビガトランの中和
2系列の実験を行って、ダビガトラン抗凝固活性に対する抗体のin vitro作用を示した。4種のポリクローナル抗体を実験室に受け入れ、ヒト血漿でさらに試験した。これを機能的アッセイであるトロンビン凝固時間で試験した。
【0183】
アッセイの説明:
簡潔には、3.13%クエン酸ナトリウムへ全血を入れて、ヒト血漿を得る。次に、これを遠心分離して無血小板血漿を得て、そして別個のチューブに移し、アッセイの日に必要とされるまで凍結する。アッセイの日に血漿を37℃で解凍する。
【0184】
トロンビン凝固時間は以下のように行う。最初に、提供された緩衝液(Dade Behring Test kit)中に、製造業者の説明書通りにトロンビンを希釈し(3IU/mLトロンビン)、37℃に予熱する。それは、調製してから2時間以内に使用する。全てのアッセイを市販のCL4凝固測定機(Behnk Electronics, Norderstadt, Germany)で行った。磁気スターラーの入った、提供されたキュベットに血漿50μLをピペットで入れ、CL4機で37℃に予熱されたウェルの中で2分間撹拌させる。この時点でトロンビン溶液100μLを添加し、血漿試料が凝固するために必要な時間をCL4によって自動的に記録する。提供されたキュベット中の血漿に入れてダビガトランを5分間予備インキュベーションしてからトロンビンを添加し、測定を開始する。抗体も試験する場合(原液50μLまで)、37℃でさらに5分間インキュベーションしてから凝固を開始する(すなわちダビガトランと一緒に合計10分間インキュベーション、抗体と一緒に合計5分間インキュベーション、次にトロンビンで凝固を開始)。
【0185】
最初に、ヒト血漿に漸増する濃度のダビガトランを添加し、トロンビンを添加後に凝固までの時間を測定することによって、ダビガトランの標準曲線を作成した(図1)。漸増する濃度のダビガトランと一緒にトロンビン凝固時間の濃度依存的増加があった。
【0186】
最初のセットの中和実験について、臨床的に関連する濃度である200nMのダビガトランを中和のために全血漿試料に添加した。4個の抗体調製物は、全て、ダビガトランを含有する血漿での凝固時間を短縮することができた(図2)。中和度は、各抗体調製物中のタンパク質濃度に関連した。次に、最も高い濃度を有する抗体溶液(D)を系列希釈し、別々の実験セットで200nMダビガトランの抗凝固活性を中和する能力について試験した。図3から、抗体濃度が増加するとともに、ダビガトラン誘導抗凝固活性の濃度依存的阻害があった。加えて、非特異的ウサギポリクローナル抗体(青色四角)をダビガトラン含有血漿に添加した場合、それは、抗凝固活性を中和する能力を有さなかった。濃度依存性および非特異的抗体の中和欠如は、抗体による抗血液凝固のリバースがダビガトランに特異的であることを示している。
【0187】
しかし、これらの濃度のダビガトランは臨床的に意義があるものであって、出血または過剰投与は、おそらくより高い濃度で起こるものである。したがって、抗体が、図1の標準曲線における最も高い濃度のダビガトラン(500nM)の抗凝固活性を阻害する能力も試験した。図4に、抗体Dが高濃度のダビガトランも阻害することができたことを図示する。
【0188】
III. モノクローナル抗ダビガトラン抗体の製造および特徴づけ
1. モノクローナル抗ダビガトラン抗体およびFabの製造
ヘモシアニンおよび免疫グロブリンなどの担体タンパク質にコンジュゲーションしたハプテン1(実施例1.1参照)をマウスに免疫処置し、標準的な手順によりハイブリドーマを発生させた。培養上清から精製されたモノクローナル抗体は、ダビガトラン−タンパク質コンジュゲートに結合し、そしてこの結合を溶液中のダビガトランと1〜10nMの範囲の濃度の最大半値阻害で競合することができた。モノクローナル抗体のパパイン切断に続く、プロテインAによるFcドメインの除去によってFabを生成させた。
【0189】
マウス抗体の重鎖および軽鎖の可変領域をクローニングし、標準法を用いて配列決定した。抗体の質量分析およびN−末端配列決定によるタンパク質分析によって配列を確認した。特異的マウス可変領域およびヒトIgG定常領域を含むキメラ抗体をコードするDNA構築物を生成させ、HEK293T細胞からタンパク質を発現させ、それを精製した。
【0190】
2. モノクローナル抗ダビガトラン抗体およびFabの特徴づけ
3種のモノクローナル抗体クローンの可変ドメイン配列を図5および6に示す。クローン13の可変ドメインのアミノ酸配列を図5(DBG 13 VH、重鎖、配列番号:16)および図6(DBG 13 VK、軽鎖、配列番号:17)に示す。クローン14の可変ドメインのアミノ酸配列を図5(DBG 14 VH、重鎖、配列番号:18)および図6(DBG 14 VK、軽鎖、配列番号:19)に示す。クローン22の可変ドメインのアミノ酸配列を図5(DBG 22 VH、重鎖、配列番号:20)および図6(DBG 22 VK、軽鎖、配列番号:21)に示す。
【0191】
マウスモノクローナル抗体クローン22が、実施例IIに概要されたトロンビン凝固時間アッセイにおいてヒト血漿中のダビガトランの抗凝固活性を中和する能力について、試験した。その抗体は、ヒト血漿中のトロンビン依存性凝固のダビガトラン介在性延長を用量依存的に完全にリバースした(図7)。その抗体は、また、ヒト全血中のダビガトランの機能を効果的に阻害した。この抗体から生成されたFabは、ヒト血漿におけるダビガトラン活性を遮断したが、このことは、一価の抗原結合ドメインが化合物の抗凝固活性を中和することができることを示している(図8)。
【0192】
ヒトでのダビガトランの主要代謝経路は、カルボン酸部分のグルクロン酸抱合による。ダビガトランアシルグルクロニドは、薬理学的に活性であることが示されている(Ebner et al., Drug Metab. Dispos. 2010, 38(9):1567-75)。マウスモノクローナル抗体クローン22がこれらの代謝物を中和できるかどうかを試験するために、ダビガトランで処置されたアカゲザルの尿からダビガトランアシルグルクロニドを精製し、トロンビン凝固時間アッセイで評価した。抗体は、ヒト血漿におけるトロンビン依存性凝固のダビガトランアシルグルクロニド介在性延長を、ダビガトランで見られたものと類似の効力で用量依存的にリバースした(図9)。したがって、抗体は、ヒトで見られるダビガトラン代謝物の抗凝固活性の遮断に有効である。
【0193】
Fabと、クローン22、13および14の可変ドメインならびにヒト免疫グロブリン定常領域(軽鎖定常領域:配列番号:44;重鎖定常領域:配列番号:45)を含むマウス−ヒトキメラ抗体との親和性は、Kinexa技法を用いて決定した。平衡に到達するまで様々な濃度のダビガトランで、一定濃度のFabまたはキメラ抗体をインキュベーションした。このインキュベーションの後に、ビオチン−コンジュゲーション型ダビガトランアナログとカップリングされたニュートラアビジンビーズ上に抗体を捕捉することによって、遊離抗体の濃度を決定した。捕捉されたFabを、FITCでラベルされた抗マウスIgG(Fab特異的)F(ab’)2フラグメントを用いて検出した。捕捉されたキメラ抗体を、Cy5とコンジュゲーションされた抗ヒトIgGを用いて検出した。解離定数は1:1結合モデルを用いて計算した。これらの実験からの結果を下記表に概要する。
【0194】
抗ダビガトラン抗体の親和性
【表13】
【0195】
Fabとキメラ抗体の両方が高親和性でダビガトランと結合する。
【0196】
3. ヒト化モノクローナル抗ダビガトラン抗体およびFabの生成
人間に投与後の潜在的免疫原性を減少させるために、マウスモノクローナル抗体を「ヒト化」した。マウス先導物のために、フレームワークの相同性、CDRの構造、保存されたカノニカル残基、保存された界面充填残基および他のパラメーターに基づきヒトフレームワーク配列を選択した。これらのフレームワーク位置におけるアミノ酸残基の特異的置換は、結合親和性および/または安定性を含めた抗体の性能の様々な局面を、ヒト生殖細胞系フレームワーク領域へのCDRまたはHVLの「直接スワップ」によって形成されたヒト化抗体において実証されたものよりも改善することができる。キメラ親Fabに比べて、より良いまたは同等の結合と改善された発現とを示したFabを、さらなる特徴づけのために選択した。ヒト化Fabの可変ドメインのアミノ酸配列を、図5(Eng VH 14、配列番号:22;ENG VH 15、配列番号:24;およびENG VH 31、配列番号:26)および図6(Eng VK 11、配列番号:23;ENG VK 17、配列番号:25;およびENG VK 18、配列番号:27)に示す。Eng VH 15およびEng VK 18を含むFab(軽鎖:配列番号:37;重鎖:配列番号:36)をCHO細胞に直接発現させ、カッパー選択およびプロテインG樹脂を使用して精製した。
【0197】
Eng VH 15およびEng VK 18を含むFabもまた、IgG1KO形式での完全長IgG(軽鎖:配列番号:35;重鎖:配列番号:40)に変換した。IgG1KO(エフェクター機能のノックアウト)は、FcgRおよび補体の結合などのエフェクター機能を減少させる、Fc領域中の2個の突然変異、Leu234AlaおよびLeu235Alaを有する。IgG形式は、文献に記載されている(例えばHezareh et al. (2001) Journal of Virology 75: 12161-12168参照)。ヒト化抗ダビガトラン抗体は、場合により、コンセンサス領域または生殖系フレームワーク領域に特異的アミノ酸置換を含む。18/15抗体をHEK293T細胞またはCHO細胞に発現させ、精製した。インタクトな抗体のLys−Cまたはパパイン切断のいずれかによってFabフラグメントを生成させ、プロテインAによりFcドメインを除去して精製した。
【0198】
4. 抗ダビガトランFabの特徴づけ
インタクトな抗体のLys−C切断によって生成された18/15 Fabフラグメントを、実施例IIに概要されたトロンビン凝固時間アッセイにおいてそれがヒト血漿でのダビガトラン抗凝固活性を中和する能力について試験した。Fabは、ヒト血漿におけるトロンビン依存性凝固のダビガトラン介在性延長を2.6nMのIC50で用量依存的に完全にリバースした(図10)。直接発現されたFabフラグメントおよびインタクトな抗体のパパイン切断によって生成されたFabフラグメントもまた、それぞれ2.6および2.7nMのIC50でダビガトランの抗凝固活性を中和した。
【0199】
インタクトな抗体のLys−C切断によって生成された18/15 Fabおよび直接発現されたFabの親和性を、SPR技法を利用してBIAcore装置で決定した。Fabを漸増する濃度のダビガトランと一緒に室温で30分間予備インキュベーションした。固定化ビオチン−コンジュゲーション型ダビガトランアナログをコーティングされたセンサーチップの上にその混合物を流し、遊離Fabの結合をモニターした。この溶液競合アッセイ設計を使用して、ダビガトランに対するFabのKD値をLys−Cで生成されたFabについて0.16pM、直接発現されたFabについて0.45pMと決定した。
【0200】
パパイン切断によって生成されたFabを用いたin vivo実験
ラット(雄性Wistar、約300g)を、ペントバルビタールでボーラス(60mg/kg i.p.)および維持麻酔のための連続注入(20mg/kg/hr i.p.)として麻酔し、内部体温を維持するために37℃の温熱パッド上に置いた。採血のために頸動脈を、物質投与のために右頸静脈を分離し、カニューレを挿入した。ダビガトランをボーラスとして開始し(0.3μM/kg)、続いて20分間かけて注入し(0.1μM/kg/hr)、定常状態の血漿レベルを達成した。20分後に、左頸静脈を介してFabを等モル濃度または等モル濃度の半分のいずれかで単回ボーラスとしてi.v.注射した。血液試料(3.13%クエン酸ナトリウム中に1/10希釈)をベースライン(−20、−2min)で、そしてFab注射後に様々な間隔で30分間採取する。
【0201】
ダビガトランの抗凝固作用を、トロンビン時間(TT)および活性化部分トロンボプラスチン時間(aPTT)を含めた全血凝固時間として測定した。簡潔には、トロンビン時間は、37℃に予熱されたウェルに全血50μLを添加することによって凝固計で行う。濃度3.0U/mLのトロンビン(Siemens Healthcare, Marburg, Germany)を添加し(体積100μL)、試料を凝固させるために必要な時間を測定する。凝固計で全血50μLを37℃に予熱し、aPTT試薬(Roche Diagnostics, Mannheim, Germany)50μLを3分間添加することによって全血aPTTを行う。0.025M予熱(37℃)塩化カルシウム50μLの添加によって凝固時間を開始する。次に、全血試料を凝固させるために必要な時間を記録する。
【0202】
ダビガトランと等しいモル用量での単回i.v.注射として与えられた18/15Fab(軽鎖:配列番号:37、重鎖Fdフラグメント:配列番号:41;CHO細胞で発現された完全免疫グロブリンのパパイン切断によって製造)の結果を図11および12に示す。このモデルにおいて、TT(図11)とaPTT(図12)の両方として測定された、ダビガトラン抗凝固活性の迅速でほぼ即時の阻害があった。注射から1分以内で、ダビガトランの抗凝固活性は、完全に中和されてベースラインレベルに戻った。これは、ダビガトランの継続中のi.v.連続注入にかかわらず、20分よりも長く維持された。
【0203】
より低い用量である半分のモル用量のダビガトランが与えられた場合も、TT(図13)とaPTT(図14)の両方に初回減少があった。しかしこれは、ダビガトランの継続中の連続注入の条件下で、より高い用量ほど長くは維持されなかった。
【0204】
したがって、これらの結果から、この動物モデルにおいて抗ダビガトランFabの単回i.v.投与後のダビガトラン抗凝固活性の、予測可能で、用量依存的で、非常に迅速な中和が実証される。
【特許請求の範囲】
【請求項1】
抗凝固薬の活性を中和可能な抗体分子。
【請求項2】
抗凝固薬に対して結合特異性を有する、請求項1記載の抗体分子。
【請求項3】
抗凝固薬が、直接トロンビン阻害剤、第Xa因子阻害剤、またはビタミンKアンタゴニストである、請求項1または2記載の抗体分子。
【請求項4】
抗凝固薬が、ダビガトラン、アルガトロバン、メラガトラン、キシメラガトラン、ヒルジン、ビバリルジン、レピルジン、デシルジン、アピキサバン、オタミキサバン、リバロキサバン、デフィブロチド、ラマトロバン、アンチトロンビンIII、またはドロトレコギンアルファである、前記請求項のいずれか一項記載の抗体分子。
【請求項5】
抗凝固薬が、一般式
【化17】
[式中、
Aは、基Hetのベンゾ、ピリドまたはチエノ部分に結合されたカルボニルまたはスルホニル基を表し、
Bは、基Arに結合されたメチレン基が酸素もしくは硫黄原子によって、または−NR1−基によって置き換えられていてもよいエチレン基を表し、
ここで、R1は、水素原子またはC1−4−アルキル基を表し、
Eは、RbNH−C(=NH)−基を表し、
ここで、Rbは、水素原子、ヒドロキシ、C1−9−アルコキシカルボニル、シクロヘキシルオキシカルボニル、フェニル−C1−3−アルコキシカルボニル、ベンゾイル、p−C1−3−アルキル−ベンゾイルまたはピリジノイル基を表し、一方で該C1−9−アルコキシカルボニル基の2位のエトキシ部分は、C1−3−アルキルスルホニルまたは2−(C1−3−アルコキシ)−エチル基によって更に置換されていてもよく、
Arは、塩素原子によって、またはメチル、エチルもしくはメトキシ基によって場合により置換されている1,4−フェニレン基を表すか、あるいはArは2,5−チエニレン基を表し、
Hetは、1−(C1−3−アルキル)−2,5−ベンゾイミダゾリレン、1−シクロプロピル−2,5−ベンゾイミダゾリレン、2,5−ベンゾチアゾリレン、1−(C1−3−アルキル)−2,5−インドリレン、1−(C1−3−アルキル)−2,5−イミダゾ[4,5−b]ピリジニレン、3−(C1−3−アルキル)−2,7−イミダゾ[1,2−a]ピリジニレンまたは1−(C1−3−アルキル)−2,5−チエノ[2,3−d]イミダゾリレン基を表し、そして
Raは、R2NR3−基を表し、
ここで、R2は、カルボキシ、C1−6−アルキルオキシカルボニル、ベンジルオキシカルボニル、C1−3−アルキルスルホニルアミノカルボニルもしくは1H−テトラゾール−5−イル基によって置換されていてもよいC1−4−アルキル基、
ヒドロキシ、ベンジルオキシ、カルボキシ−C1−3−アルキルアミノ、C1−3−アルコキシカルボニル−C1−3−アルキルアミノ、N−(C1−3−アルキル)−カルボキシ−C1−3−アルキルアミノもしくはN−(C1−3−アルキル)−C1−3−アルコキシカルボニル−C1−3−アルキルアミノ基によって置換されているC2−4−アルキル基であり、一方で上記基において隣接窒素原子に対してα−位にある炭素原子は、置換されていなくてもよく、
R3は、C3−7−シクロアルキル基、不飽和部分がR2NR3基の窒素原子に直接結合されていなくてもよいプロパルギル基、フッ素もしくは塩素原子によって、またはメチルもしくはメトキシ基によって場合により置換されているフェニル基、メチル基によって場合により置換されているピラゾリル、ピリダゾリルまたはピリジニル基を表すか、あるいは
R2およびR3は、それらの間の窒素原子と一緒になって、カルボキシまたはC1−4−アルコキシカルボニル基によって場合により置換されている5〜7員シクロアルキレンイミノ基を表し、フェニル環が更に縮合されていてもよい]
で示される二置換二環式複素環、その互変異性体、立体異性体および塩である、前記請求項のいずれか一項記載の抗体分子。
【請求項6】
抗凝固薬が、
(a) 2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾチアゾール−5−カルボン酸−N−フェニル−N−(2−カルボキシエチル)−アミド、
(b) 2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾチアゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(c) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(d) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(3−ヒドロキシカルボニルプロピル)−アミド、
(e) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(ヒドロキシカルボニルメチル)−アミド、
(f) 1−メチル−2−[2−(2−アミジノチオフェン−5−イル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(g) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(h) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(i) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(j) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[2−(1H−テトラゾール−5−イル)エチル]−アミド、
(k) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[2−(1H−テトラゾール−5−イル)エチル]−アミド、
(l) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(m) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(3−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(n) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(o) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[(N−ヒドロキシカルボニルエチル−N−メチル)−2−アミノエチル]−アミド、
(p) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(3−フルオロフェニル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(q) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(4−フルオロフェニル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(r) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(s) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(t) 1−メチル−2−[N−(4−アミジノフェニル)アミノメチル]−インドール−5−イル−カルボン酸−N−フェニル−N−(2−メトキシカルボニルエチル)−アミド及び
(u) 1−メチル−2−[N−(4−アミジノフェニル)アミノメチル]−チエノ[2.3−d]イミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(v) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(w) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(x) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(y) 1−メチル−2−[N−[4−(N−n−ヘキシルオキシカルボニルアミジノ)フェニル]−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−エトキシカルボニルエチル)−アミド、
より選択される化合物、その互変異性体、立体異性体および塩である、請求項5記載の抗体分子。
【請求項7】
抗凝固薬がダビガトランである、請求項2記載の抗体分子。
【請求項8】
ダビガトランおよびダビガトランの1−O−アシルグルクロニドの活性を中和可能である、請求項7記載の抗体分子。
【請求項9】
配列番号:1および配列番号:2から成る群より選択されるCDR1、配列番号:3、配列番号:4、配列番号:5、配列番号:6、配列番号:7、および配列番号:8から成る群より選択されるCDR2、ならびに配列番号:9および配列番号:10から成る群より選択されるCDR3を有する重鎖可変ドメインと、配列番号:11、配列番号:12、および配列番号:13から成る群より選択されるCDR1、配列番号:14で示されるCDR2、ならびに配列番号:15で示されるCDR3を有する軽鎖可変ドメインとを含む、請求項5〜8のいずれか一項記載の抗体分子。
【請求項10】
配列番号:1で示されるCDR1、配列番号:6、配列番号:7、および配列番号:8から成る群より選択されるCDR2、ならびに配列番号:10で示されるCDR3を有する重鎖可変ドメインと、配列番号:13で示されるCDR1、配列番号:14で示されるCDR2、ならびに配列番号:15で示されるCDR3を有する軽鎖可変ドメインとを含む、請求項9記載の抗体分子。
【請求項11】
配列番号:16、18、20、22、24、および26から成る群より選択される重鎖可変ドメイン、ならびに配列番号:17、19、21、23、25、および27から成る群より選択される軽鎖可変ドメインを含む、請求項9または10記載の抗体分子。
【請求項12】
配列番号:16で示される重鎖可変ドメインおよび配列番号:17で示される軽鎖可変ドメイン、または配列番号:18で示される重鎖可変ドメインおよび配列番号:19で示される軽鎖可変ドメイン、または配列番号:20で示される重鎖可変ドメインおよび配列番号:21で示される軽鎖可変ドメイン、または配列番号:22で示される重鎖可変ドメインおよび配列番号:23で示される軽鎖可変ドメイン、または配列番号:24で示される重鎖可変ドメインおよび配列番号:25で示される軽鎖可変ドメイン、または配列番号:24で示される重鎖可変ドメインおよび配列番号:27で示される軽鎖可変ドメイン、または配列番号:26で示される重鎖可変ドメインおよび配列番号:27で示される軽鎖可変ドメインを含む、請求項11記載の抗体分子。
【請求項13】
ポリクローナル抗体、モノクローナル抗体、ヒト抗体、ヒト化抗体、キメラ抗体、抗体のフラグメント、特にFab、Fab’、もしくはF(ab’)2フラグメント、単鎖抗体、特に単鎖可変フラグメント(scFv)、小モジュラー免疫医薬(SMIP)、ドメイン抗体、ナノボディ、ダイアボディ、または設計されたアンキリン反復タンパク(Designed Ankyrin Repeat Protein)(DARPin)である、前記請求項のいずれか一項記載の抗体分子。
【請求項14】
重鎖可変ドメインおよび軽鎖可変ドメインが、配列番号:28、配列番号:29、配列番号:30、および配列番号:31から成る群より選択されるリンカーペプチドを介して相互に結合されている、scFvである、請求項13記載の抗体分子。
【請求項15】
配列番号:32、または配列番号:33を含む、請求項14記載の抗体分子。
【請求項16】
配列番号:34または配列番号:40を含む重鎖および配列番号:35を含む軽鎖、または配列番号:42を含む重鎖および配列番号:43を含む軽鎖を有する、請求項13記載の抗体分子。
【請求項17】
配列番号:36、配列番号:38、または配列番号:41を含むFdフラグメントおよび配列番号:37または配列番号:39を含む軽鎖を有するFab分子である、請求項13記載の抗体分子。
【請求項18】
医学に使用するための、前記請求項のいずれか一項記載の抗体分子。
【請求項19】
抗凝固治療の副作用の治療もしくは予防に使用するための、および/または抗凝固薬の過剰投与をリバースするための、前記請求項のいずれか一項記載の抗体分子。
【請求項20】
副作用が出血事象である、請求項19記載の抗体分子。
【請求項21】
抗凝固治療の副作用、または抗凝固治療での過剰投与事象の治療または予防方法であって、それを必要とする患者に前記請求項のいずれか一項記載の抗体分子の有効量を投与することを含む方法。
【請求項22】
前記請求項のいずれか一項記載の抗体分子を製造する方法であって、
(a)発現制御配列と機能的に関連して、該抗体分子をコードする一つ以上の核酸を含む宿主細胞を提供すること、
(b)該宿主細胞を培養すること、および
(c)細胞培養物から該抗体分子を回収すること
を含む方法。
【請求項23】
請求項1〜20のいずれか一項記載の抗体またはその医薬組成物を含むキット。
【請求項24】
(a)請求項1〜20のいずれか一項記載の抗体またはその医薬組成物;
(b)容器;および
(c)ラベル
を含むキット。
【請求項25】
請求項7〜20のいずれか一項記載の抗体、およびダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩を含むキット。
【請求項26】
ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の形態が、固体、液体またはゲルの形態である、請求項25記載のキット。
【請求項27】
ダビガトランエテキシラートの薬学的に許容されうる塩がメシラート塩である、請求項25記載のキット。
【請求項28】
用量単位あたりのダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の強度が、QDまたはBIDのいずれかで75mg〜300mgの間である、請求項25または27記載のキット。
【請求項29】
(a)請求項7〜20のいずれか一項記載の抗体、またはその医薬組成物;
(b)ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の医薬組成物;
(c)容器;および
(d)ラベル
を含むキット。
【請求項30】
ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の形態が、固体、液体またはゲルの形態である、請求項29記載のキット。
【請求項31】
ダビガトランエテキシラートの薬学的に許容されうる塩がメシラート塩である、請求項29記載のキット。
【請求項32】
用量単位あたりのダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の強度が、QDまたはBIDのいずれかで75mg〜300mgの間である、請求項29または31記載のキット。
【請求項33】
(a)ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩を含む第1の医薬組成物;
(b)請求項7〜20のいずれか一項記載の抗体を含む第2の医薬組成物;
(c)患者に該第1および第2の医薬組成物を別々に投与するための説明書
を含み、該第1および第2の医薬組成物が、別々の容器に入っており、そして該第2の医薬組成物が、ダビガトランまたはダビガトランの1−O−アシルグルクロニドの中和または部分中和を必要とする患者に投与されるキット。
【請求項34】
請求項7〜20のいずれか一項記載の抗体またはその医薬組成物を投与することを含む、ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩で処置されている患者におけるダビガトランまたはダビガトランの1−O−アシルグルクロニドを中和または部分中和する方法。
【請求項35】
患者におけるダビガトランまたはダビガトランの1−O−アシルグルクロニドを中和または部分中和するため:
(a)患者がダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩で処置されていたこと、および該患者によって取り込まれた量を確認すること;
(b)ダビガトランまたはダビガトランの1−O−アシルグルクロニドが、凝固または血液凝固の試験またはアッセイの結果の正確な読み出しを妨害するであろう該試験またはアッセイを行う前に、請求項7〜20のいずれか一項記載の抗体でダビガトランまたは1−O−アシルグルクロニドを中和すること;
(c)ダビガトランまたはダビガトランの1−O−アシルグルクロニドも存在しないときの血餅形成レベルを決定するために、該患者から採取された試料の凝固または血液凝固の試験またはアッセイを行うこと;ならびに
(d)患者における血餅形成と分解の間に適切なバランスを達成するために、該患者に投与されるダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の量を調整すること
を含む方法。
【請求項36】
ダビガトランまたはダビガトランの1−O−アシルグルクロニドに対する抗体の量が、0.1〜100の間のモル比である、請求項34または35記載の方法。
【請求項37】
ダビガトランまたはダビガトランの1−O−アシルグルクロニドに対する抗体の量が、0.1〜10の間のモル比である、請求項36記載の方法。
【請求項38】
試験またはアッセイの結果の正確な読み出しが、フィブリノーゲンレベル、活性化プロテインC抵抗性または関連試験の正確な読み出しである、請求項35記載の方法。
【請求項1】
抗凝固薬の活性を中和可能な抗体分子。
【請求項2】
抗凝固薬に対して結合特異性を有する、請求項1記載の抗体分子。
【請求項3】
抗凝固薬が、直接トロンビン阻害剤、第Xa因子阻害剤、またはビタミンKアンタゴニストである、請求項1または2記載の抗体分子。
【請求項4】
抗凝固薬が、ダビガトラン、アルガトロバン、メラガトラン、キシメラガトラン、ヒルジン、ビバリルジン、レピルジン、デシルジン、アピキサバン、オタミキサバン、リバロキサバン、デフィブロチド、ラマトロバン、アンチトロンビンIII、またはドロトレコギンアルファである、前記請求項のいずれか一項記載の抗体分子。
【請求項5】
抗凝固薬が、一般式
【化17】
[式中、
Aは、基Hetのベンゾ、ピリドまたはチエノ部分に結合されたカルボニルまたはスルホニル基を表し、
Bは、基Arに結合されたメチレン基が酸素もしくは硫黄原子によって、または−NR1−基によって置き換えられていてもよいエチレン基を表し、
ここで、R1は、水素原子またはC1−4−アルキル基を表し、
Eは、RbNH−C(=NH)−基を表し、
ここで、Rbは、水素原子、ヒドロキシ、C1−9−アルコキシカルボニル、シクロヘキシルオキシカルボニル、フェニル−C1−3−アルコキシカルボニル、ベンゾイル、p−C1−3−アルキル−ベンゾイルまたはピリジノイル基を表し、一方で該C1−9−アルコキシカルボニル基の2位のエトキシ部分は、C1−3−アルキルスルホニルまたは2−(C1−3−アルコキシ)−エチル基によって更に置換されていてもよく、
Arは、塩素原子によって、またはメチル、エチルもしくはメトキシ基によって場合により置換されている1,4−フェニレン基を表すか、あるいはArは2,5−チエニレン基を表し、
Hetは、1−(C1−3−アルキル)−2,5−ベンゾイミダゾリレン、1−シクロプロピル−2,5−ベンゾイミダゾリレン、2,5−ベンゾチアゾリレン、1−(C1−3−アルキル)−2,5−インドリレン、1−(C1−3−アルキル)−2,5−イミダゾ[4,5−b]ピリジニレン、3−(C1−3−アルキル)−2,7−イミダゾ[1,2−a]ピリジニレンまたは1−(C1−3−アルキル)−2,5−チエノ[2,3−d]イミダゾリレン基を表し、そして
Raは、R2NR3−基を表し、
ここで、R2は、カルボキシ、C1−6−アルキルオキシカルボニル、ベンジルオキシカルボニル、C1−3−アルキルスルホニルアミノカルボニルもしくは1H−テトラゾール−5−イル基によって置換されていてもよいC1−4−アルキル基、
ヒドロキシ、ベンジルオキシ、カルボキシ−C1−3−アルキルアミノ、C1−3−アルコキシカルボニル−C1−3−アルキルアミノ、N−(C1−3−アルキル)−カルボキシ−C1−3−アルキルアミノもしくはN−(C1−3−アルキル)−C1−3−アルコキシカルボニル−C1−3−アルキルアミノ基によって置換されているC2−4−アルキル基であり、一方で上記基において隣接窒素原子に対してα−位にある炭素原子は、置換されていなくてもよく、
R3は、C3−7−シクロアルキル基、不飽和部分がR2NR3基の窒素原子に直接結合されていなくてもよいプロパルギル基、フッ素もしくは塩素原子によって、またはメチルもしくはメトキシ基によって場合により置換されているフェニル基、メチル基によって場合により置換されているピラゾリル、ピリダゾリルまたはピリジニル基を表すか、あるいは
R2およびR3は、それらの間の窒素原子と一緒になって、カルボキシまたはC1−4−アルコキシカルボニル基によって場合により置換されている5〜7員シクロアルキレンイミノ基を表し、フェニル環が更に縮合されていてもよい]
で示される二置換二環式複素環、その互変異性体、立体異性体および塩である、前記請求項のいずれか一項記載の抗体分子。
【請求項6】
抗凝固薬が、
(a) 2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾチアゾール−5−カルボン酸−N−フェニル−N−(2−カルボキシエチル)−アミド、
(b) 2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾチアゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(c) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(d) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(3−ヒドロキシカルボニルプロピル)−アミド、
(e) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(ヒドロキシカルボニルメチル)−アミド、
(f) 1−メチル−2−[2−(2−アミジノチオフェン−5−イル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(g) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(h) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(i) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(j) 1−メチル−2−[2−(4−アミジノフェニル)エチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[2−(1H−テトラゾール−5−イル)エチル]−アミド、
(k) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[2−(1H−テトラゾール−5−イル)エチル]−アミド、
(l) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(m) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(3−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(n) 1−メチル−2−[N−(4−アミジノフェニル)−N−メチル−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(o) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−[(N−ヒドロキシカルボニルエチル−N−メチル)−2−アミノエチル]−アミド、
(p) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(3−フルオロフェニル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(q) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(4−フルオロフェニル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(r) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(s) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(t) 1−メチル−2−[N−(4−アミジノフェニル)アミノメチル]−インドール−5−イル−カルボン酸−N−フェニル−N−(2−メトキシカルボニルエチル)−アミド及び
(u) 1−メチル−2−[N−(4−アミジノフェニル)アミノメチル]−チエノ[2.3−d]イミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(v) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−フェニル−N−(2−ヒドロキシカルボニルエチル)−アミド、
(w) 1−メチル−2−[N−(4−アミジノフェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(x) 1−メチル−2−[N−(4−アミジノ−2−メトキシ−フェニル)−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−ヒドロキシカルボニルエチル)−アミド、
(y) 1−メチル−2−[N−[4−(N−n−ヘキシルオキシカルボニルアミジノ)フェニル]−アミノメチル]−ベンゾイミダゾール−5−イル−カルボン酸−N−(2−ピリジル)−N−(2−エトキシカルボニルエチル)−アミド、
より選択される化合物、その互変異性体、立体異性体および塩である、請求項5記載の抗体分子。
【請求項7】
抗凝固薬がダビガトランである、請求項2記載の抗体分子。
【請求項8】
ダビガトランおよびダビガトランの1−O−アシルグルクロニドの活性を中和可能である、請求項7記載の抗体分子。
【請求項9】
配列番号:1および配列番号:2から成る群より選択されるCDR1、配列番号:3、配列番号:4、配列番号:5、配列番号:6、配列番号:7、および配列番号:8から成る群より選択されるCDR2、ならびに配列番号:9および配列番号:10から成る群より選択されるCDR3を有する重鎖可変ドメインと、配列番号:11、配列番号:12、および配列番号:13から成る群より選択されるCDR1、配列番号:14で示されるCDR2、ならびに配列番号:15で示されるCDR3を有する軽鎖可変ドメインとを含む、請求項5〜8のいずれか一項記載の抗体分子。
【請求項10】
配列番号:1で示されるCDR1、配列番号:6、配列番号:7、および配列番号:8から成る群より選択されるCDR2、ならびに配列番号:10で示されるCDR3を有する重鎖可変ドメインと、配列番号:13で示されるCDR1、配列番号:14で示されるCDR2、ならびに配列番号:15で示されるCDR3を有する軽鎖可変ドメインとを含む、請求項9記載の抗体分子。
【請求項11】
配列番号:16、18、20、22、24、および26から成る群より選択される重鎖可変ドメイン、ならびに配列番号:17、19、21、23、25、および27から成る群より選択される軽鎖可変ドメインを含む、請求項9または10記載の抗体分子。
【請求項12】
配列番号:16で示される重鎖可変ドメインおよび配列番号:17で示される軽鎖可変ドメイン、または配列番号:18で示される重鎖可変ドメインおよび配列番号:19で示される軽鎖可変ドメイン、または配列番号:20で示される重鎖可変ドメインおよび配列番号:21で示される軽鎖可変ドメイン、または配列番号:22で示される重鎖可変ドメインおよび配列番号:23で示される軽鎖可変ドメイン、または配列番号:24で示される重鎖可変ドメインおよび配列番号:25で示される軽鎖可変ドメイン、または配列番号:24で示される重鎖可変ドメインおよび配列番号:27で示される軽鎖可変ドメイン、または配列番号:26で示される重鎖可変ドメインおよび配列番号:27で示される軽鎖可変ドメインを含む、請求項11記載の抗体分子。
【請求項13】
ポリクローナル抗体、モノクローナル抗体、ヒト抗体、ヒト化抗体、キメラ抗体、抗体のフラグメント、特にFab、Fab’、もしくはF(ab’)2フラグメント、単鎖抗体、特に単鎖可変フラグメント(scFv)、小モジュラー免疫医薬(SMIP)、ドメイン抗体、ナノボディ、ダイアボディ、または設計されたアンキリン反復タンパク(Designed Ankyrin Repeat Protein)(DARPin)である、前記請求項のいずれか一項記載の抗体分子。
【請求項14】
重鎖可変ドメインおよび軽鎖可変ドメインが、配列番号:28、配列番号:29、配列番号:30、および配列番号:31から成る群より選択されるリンカーペプチドを介して相互に結合されている、scFvである、請求項13記載の抗体分子。
【請求項15】
配列番号:32、または配列番号:33を含む、請求項14記載の抗体分子。
【請求項16】
配列番号:34または配列番号:40を含む重鎖および配列番号:35を含む軽鎖、または配列番号:42を含む重鎖および配列番号:43を含む軽鎖を有する、請求項13記載の抗体分子。
【請求項17】
配列番号:36、配列番号:38、または配列番号:41を含むFdフラグメントおよび配列番号:37または配列番号:39を含む軽鎖を有するFab分子である、請求項13記載の抗体分子。
【請求項18】
医学に使用するための、前記請求項のいずれか一項記載の抗体分子。
【請求項19】
抗凝固治療の副作用の治療もしくは予防に使用するための、および/または抗凝固薬の過剰投与をリバースするための、前記請求項のいずれか一項記載の抗体分子。
【請求項20】
副作用が出血事象である、請求項19記載の抗体分子。
【請求項21】
抗凝固治療の副作用、または抗凝固治療での過剰投与事象の治療または予防方法であって、それを必要とする患者に前記請求項のいずれか一項記載の抗体分子の有効量を投与することを含む方法。
【請求項22】
前記請求項のいずれか一項記載の抗体分子を製造する方法であって、
(a)発現制御配列と機能的に関連して、該抗体分子をコードする一つ以上の核酸を含む宿主細胞を提供すること、
(b)該宿主細胞を培養すること、および
(c)細胞培養物から該抗体分子を回収すること
を含む方法。
【請求項23】
請求項1〜20のいずれか一項記載の抗体またはその医薬組成物を含むキット。
【請求項24】
(a)請求項1〜20のいずれか一項記載の抗体またはその医薬組成物;
(b)容器;および
(c)ラベル
を含むキット。
【請求項25】
請求項7〜20のいずれか一項記載の抗体、およびダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩を含むキット。
【請求項26】
ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の形態が、固体、液体またはゲルの形態である、請求項25記載のキット。
【請求項27】
ダビガトランエテキシラートの薬学的に許容されうる塩がメシラート塩である、請求項25記載のキット。
【請求項28】
用量単位あたりのダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の強度が、QDまたはBIDのいずれかで75mg〜300mgの間である、請求項25または27記載のキット。
【請求項29】
(a)請求項7〜20のいずれか一項記載の抗体、またはその医薬組成物;
(b)ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の医薬組成物;
(c)容器;および
(d)ラベル
を含むキット。
【請求項30】
ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の形態が、固体、液体またはゲルの形態である、請求項29記載のキット。
【請求項31】
ダビガトランエテキシラートの薬学的に許容されうる塩がメシラート塩である、請求項29記載のキット。
【請求項32】
用量単位あたりのダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の強度が、QDまたはBIDのいずれかで75mg〜300mgの間である、請求項29または31記載のキット。
【請求項33】
(a)ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩を含む第1の医薬組成物;
(b)請求項7〜20のいずれか一項記載の抗体を含む第2の医薬組成物;
(c)患者に該第1および第2の医薬組成物を別々に投与するための説明書
を含み、該第1および第2の医薬組成物が、別々の容器に入っており、そして該第2の医薬組成物が、ダビガトランまたはダビガトランの1−O−アシルグルクロニドの中和または部分中和を必要とする患者に投与されるキット。
【請求項34】
請求項7〜20のいずれか一項記載の抗体またはその医薬組成物を投与することを含む、ダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩で処置されている患者におけるダビガトランまたはダビガトランの1−O−アシルグルクロニドを中和または部分中和する方法。
【請求項35】
患者におけるダビガトランまたはダビガトランの1−O−アシルグルクロニドを中和または部分中和するため:
(a)患者がダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩で処置されていたこと、および該患者によって取り込まれた量を確認すること;
(b)ダビガトランまたはダビガトランの1−O−アシルグルクロニドが、凝固または血液凝固の試験またはアッセイの結果の正確な読み出しを妨害するであろう該試験またはアッセイを行う前に、請求項7〜20のいずれか一項記載の抗体でダビガトランまたは1−O−アシルグルクロニドを中和すること;
(c)ダビガトランまたはダビガトランの1−O−アシルグルクロニドも存在しないときの血餅形成レベルを決定するために、該患者から採取された試料の凝固または血液凝固の試験またはアッセイを行うこと;ならびに
(d)患者における血餅形成と分解の間に適切なバランスを達成するために、該患者に投与されるダビガトラン、ダビガトランエテキシラート、ダビガトランのプロドラッグまたはその薬学的に許容されうる塩の量を調整すること
を含む方法。
【請求項36】
ダビガトランまたはダビガトランの1−O−アシルグルクロニドに対する抗体の量が、0.1〜100の間のモル比である、請求項34または35記載の方法。
【請求項37】
ダビガトランまたはダビガトランの1−O−アシルグルクロニドに対する抗体の量が、0.1〜10の間のモル比である、請求項36記載の方法。
【請求項38】
試験またはアッセイの結果の正確な読み出しが、フィブリノーゲンレベル、活性化プロテインC抵抗性または関連試験の正確な読み出しである、請求項35記載の方法。
【図5】


【図6】

【図1】
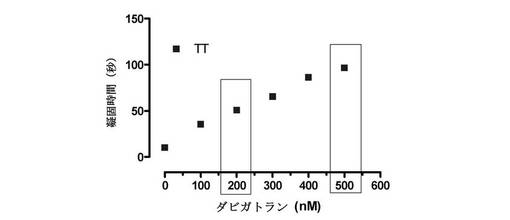
【図2】
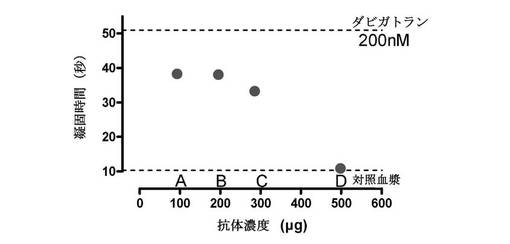
【図3】
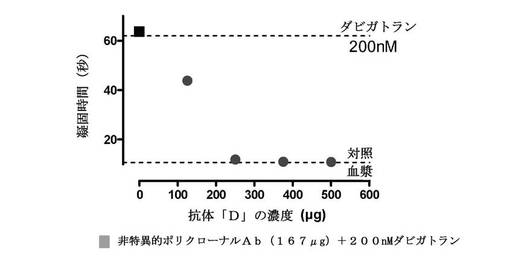
【図4】
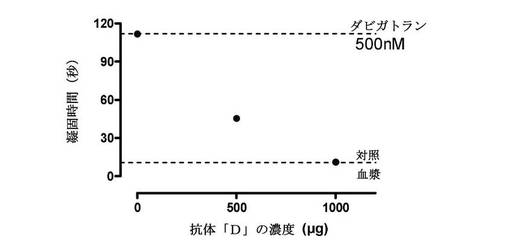
【図7】
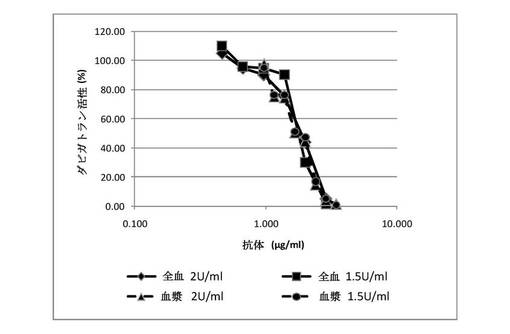
【図8】
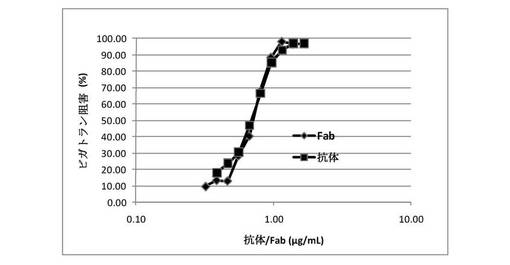
【図9】
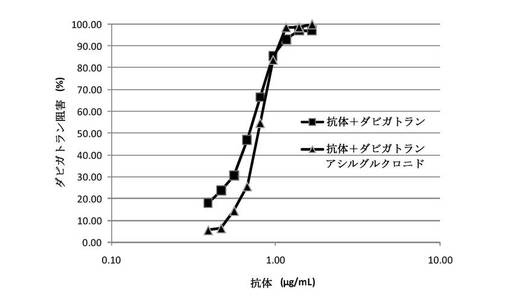
【図10】
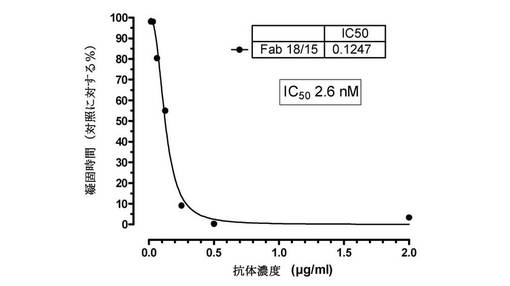
【図11】
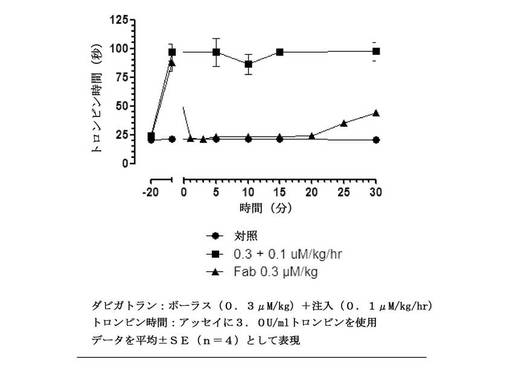
【図12】
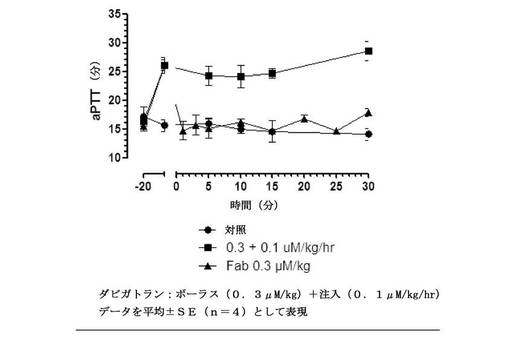
【図13】
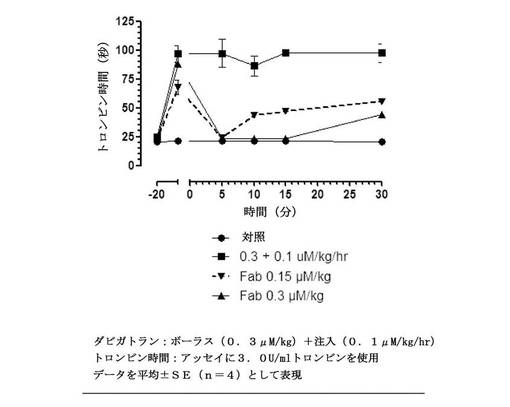
【図14】
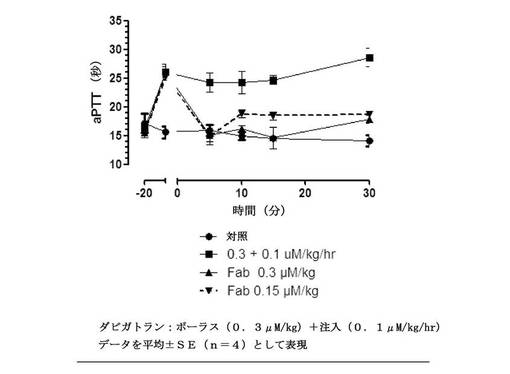
【図6】
【図1】
【図2】
【図3】
【図4】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公表番号】特表2013−517317(P2013−517317A)
【公表日】平成25年5月16日(2013.5.16)
【国際特許分類】
【出願番号】特願2012−549355(P2012−549355)
【出願日】平成23年1月20日(2011.1.20)
【国際出願番号】PCT/EP2011/050749
【国際公開番号】WO2011/089183
【国際公開日】平成23年7月28日(2011.7.28)
【出願人】(503385923)ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング (976)
【Fターム(参考)】
【公表日】平成25年5月16日(2013.5.16)
【国際特許分類】
【出願日】平成23年1月20日(2011.1.20)
【国際出願番号】PCT/EP2011/050749
【国際公開番号】WO2011/089183
【国際公開日】平成23年7月28日(2011.7.28)
【出願人】(503385923)ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング (976)
【Fターム(参考)】
[ Back to top ]