
抗原に対する免疫を生じさせる方法
抗原に対する免疫応答を生じさせる方法を提供する。方法は抗原をコードする発現ベクターを投与して個体をプライミングすることを含む。ベクターは分泌可能な融合タンパク質をコードする転写ユニットを含み、融合タンパク質は抗原およびCD40リガンドを含む。抗原およびCD40リガンドを含む融合タンパク質の投与を用いて、ベクター投与のみで得られるより高く免疫応答を増強させる。本発明の方法を使用して癌が発現する腫瘍抗原(例えばムチンまたはヒト乳頭腫ウイルス腫瘍抗原)に対する免疫応答を生成し、感染性物質に対する免疫応答を生成してもよい。発現ベクターおよび融合タンパク質を同時に生成する方法も提供する。

【発明の詳細な説明】
【技術分野】
【0001】
発明の属する分野
本発明は、CD40リガンドに融合した抗原を含む分泌可能な融合タンパク質を発現する発現ベクターを用いて抗原に対する免疫を確立させる方法に関する。本発明はまた、発現ベクターでのプライミングおよびタンパク質抗原でのブースティングの免疫化スキームにも関する。本発明はまた、生成細胞系においてベクターとタンパク質抗原を同時に生成する方法にも関する。
【背景技術】
【0002】
発明の背景
以下の本発明の背景に関する記載は読者の本発明の理解を容易にするために提供するものであり、本発明に対する先行技術を記述または構成すると認めるものではない。本出願は米国特許出願第60/529,016号(2003年12月11日出願)(図面を含めてその全体が本明細書に組み込まれる)に対する優先権を主張するものである。本願に関連する出願はPCT/US03/36237(2003年11月12日出願)“アデノウイルスベクターワクチン”、および米国特許仮出願60/524,925号(2003年11月24日出願)、 60/525,552号(2003年11月25日出願)、および60/529,015号(2003年12月11日出願)(これらは全て、図面も含めてその全体が参照により本明細書に組み込まれる)である。
【0003】
樹状細胞(DC)を含む抗原提示細胞(APC)の活性化およびそれに続く抗原提示細胞による相当抗原の担持は、癌細胞に対するT細胞依存型免疫応答の生成に必須の段階である。腫瘍抗原での活性化および担持が起こると、DCは所属リンパ節(LN)に移動し、T細胞に対して抗原を提示する。非常に多くの場合、これらのAPCは、腫瘍抗原を認識する能力のあるT細胞クローンが最適に活性化および増殖するのに必要な表面活性化分子を不十分な量しか発現しない。Shortman, ら, Stem Cells 15:409-419, 1997参照。
【0004】
APCの表面上で共刺激分子が発現していない状態でナイーブT細胞へ抗原が提示されると、T細胞は不顕性(anergy)となる。Steinbrinkら, Blood 99: 2468-2476, 2002参照。更に、CD4+T細胞の助力なしでのDCによるクロスプレゼンテーションでは、末梢での所属LNにおけるAg特異的T細胞の欠失が起こる。Kusuharaら, Eur J Immunol 32:1035-1043, 2002参照。これに対して、CD4+T細胞の助力があれば、DCはT細胞をクロスプライム(cross-prime)する機能的能力を習得し、それによってエフェクターT細胞のクローン増殖が起こる。Gunzerら, Semin Immunol 13:291-302, 2001参照。このCD4+T細胞の助力はCD40-CD40リガンド(CD40L)相互作用で置き換えることができる。Luftら, Int Immunol 14:367-380, 2002参照。CD40Lは33kDaのII型膜タンパク質で、TNF遺伝子ファミリーのメンバーであり、TCRの活性化(engagement)後にCD4+T細胞上で一時的に発現される。Skovら, J Immunol. 164: 3500-3505, 2000参照。
【0005】
インビボにおいてDCが抗腫瘍免疫応答を生じる能力は多くの動物腫瘍モデルで報告されている。Pagliaら, J Exp Med 183: 317-322, 1996;Zitvogelら, J Exp Med. 183: 87-97, 1996参照。しかしながら、DCが介在する免疫誘導には多くの治療上の困難がある。抗原提示細胞が好適な接着分子およびケモカイン受容体を発現して第2のリンパ器官にDCを誘引し、T細胞をプライミングするようにさせることを確実にすることは困難であると考えられている。Fongら, J Immunol. 166: 4254-4259, 2001;Markowiczら, J Clin Invest. 85: 955-961, 1990;Hsuら, Nat Med. 2: 52-58, 1996;Nestleら, Nat Med. 4: 328-332, 1998; Murphyら, Prostate 38: 73-78, 1999;Dhodapkarら, J Clin Invest. 104: 173-180, 1999参照。
【発明の開示】
【課題を解決するための手段】
【0006】
発明の概要
第一の観点では、本発明は抗原に対する免疫応答の確立における融合タンパク質の使用に関する。好ましい態様では、分泌可能な融合タンパク質をコードする発現ベクターを投与することによって抗原に対する免疫応答を得る。ベクターは抗原およびCD40リガンドを含む分泌可能な融合タンパク質をコードする転写ユニットを含む。融合タンパク質の投与はベクター投与の前、同時、または後に行う。好ましくは融合タンパク質はベクター投与後に投与する。
【0007】
ある方法では、融合タンパク質転写ユニット中の抗原をコードする配列はCD40リガンドをコードする配列の5'側にある。別の方法では、融合タンパク質転写ユニット中のCD40リガンドをコードする配列は抗原をコードする配列の5'側にある。好ましい態様では、CD40リガンドはその膜貫通ドメインの全部または一部を欠失している。
【0008】
抗原は、個体において免疫応答が生じうる抗原のいずれでもよい。好ましい態様では、抗原は腫瘍抗原である;腫瘍抗原はヒト乳頭腫ウイルスのE6またはE7タンパク質である;腫瘍抗原はムチン抗原であり、MUC1、MUC2、MUC3A、MUC3B、MUC4、MUC5AC、MUC5B、MUC6、MUC7、MUC8、MUC9、MUC12、MUC13、MUC15、およびMUC16からなる群から選択することができる;ムチン抗原はMUC1由来である;ヒト上皮増殖因子(EGF)様受容体(例えばHER1、HER2、HER3、およびHER4)、抗原は感染性物質抗原である;感染性物質抗原はウイルス抗原である;感染性物質ウイルス抗原はヒト乳頭腫ウイルス由来である;ウイルス抗原はヒト乳頭腫ウイルスのE6またはE7タンパク質である。
【0009】
別の観点では、本発明は腫瘍抗原を発現する癌に罹患した個体を治療する方法を提供する。方法は腫瘍抗原およびCD40リガンドを含む分泌可能な融合タンパク質をコードする転写ユニットを含む発現ベクターを投与することを含む。融合タンパク質はベクター投与の前、同時、または後に投与する。好ましくは融合タンパク質はベクター投与後に投与する。
【0010】
更なる観点では、本発明は被験体において感染性物質に対する免疫を生じさせる方法を提供する。方法は感染性物質抗原およびCD40リガンドを含む分泌可能な融合タンパク質をコードする転写ユニットを含む発現ベクターを投与することを含む。融合タンパク質はベクター投与の前、同時、または後に投与する。好ましくは融合タンパク質はベクター投与後に投与する。
【0011】
更に別の観点では、本発明は同じホスト細胞の生成系においてベクターおよび融合タンパク質を共に生成する方法に関する。好ましい態様では、融合タンパク質はワクチン接種によって免疫を生じさせるのに使用されるのと同じベクターから発現される。この方法において、ベクターおよび融合タンパク質の両方を、一つの生成系で同時に生成できる。
【0012】
好ましい態様では、発現ベクターはウイルス発現ベクターまたは非ウイルス発現ベクターであってもよい;発現ベクターはアデノウイルスベクターであってもよい;ベクターを便宜に皮下投与してもよい;ベクターを連続的に投与して免疫応答を増加させてもよい;シグナル配列は融合タンパク質の分泌のために融合タンパク質の上流に位置してもよい;抗原に対する免疫は長期にわたって持続し、抗原発現細胞に対する細胞傷害性CD8+ T細胞の生成および抗原に対する抗体の産生を伴ってもよい;転写ユニットは抗原とCD40リガンドとの間にリンカーをコードする配列を含んでもよい;好適なリンカーの長さおよび組成は種々であってもよい;発現ベクターは転写ユニットの転写を制御するためのヒト・サイトメガロウイルス・プロモーター/エンハンサーを含んでもよい;そしてCD40リガンドはヒトCD40リガンドであってもよい。
【0013】
本明細書で使用する略号には以下がある:“Ad”(アデノウイルス);“sig”(シグナル配列);および“ecd”(細胞外ドメイン)。
【0014】
これらおよび他の態様を以下に詳述する。
【発明を実施するための最良の形態】
【0015】
図面の簡単な説明
図1は、ヒトMUC1をコードするヌクレオチド配列である(SEQ ID NO : 1)。
【0016】
図2は、ヒトMUC1のアミノ酸配列である(SEQ ID NO : 2)。
【0017】
図3は、ELISAスポットアッセイで得られたインターフェロンガンマのレベルを示し、これはアデノウイルス発現ベクターAd-K/ecdhMUC1-ΔCtΔTmCD40Lでプライミングし、発現ベクターまたは成熟融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lのいずれかを皮下にブースト投与したMUC-1トランスジェニック動物(hMUC-1.Tg)由来の脾臓細胞を使用して行った。種々の処理群には、タンパク質のブースト投与を週1回のベクター注射を2回行った7日後(T1)、週1回のベクター注射2回の2週間後(T2)、1回のベクター注射の1週間後(T3)、および1回のベクター注射の2週間後(T4)に行ったものがある。T5では1回のベクター注射の7日後に開始して2回のタンパク質の皮下注射(2週間間隔で投与)を行った。T1では2回のベクター注射を行い、タンパク質は注射しなかった。
【0018】
図4は、アデノウイルス発現ベクターAd-K/ecdhMUC1-ΔCtΔTmCD40Lでプライミングし、発現ベクターまたは成熟融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lのいずれかを皮下にブースト投与したMUC-1トランスジェニック動物(hMUC-1.Tg)由来のT細胞傷害性のレベルを示す。種々の処理群を図3に示す。
【0019】
図5は、アデノウイルス発現ベクターAd-K/ecdhMUC1-ΔCtΔTmCD40Lでプライミングし、発現ベクターまたは成熟融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lのいずれかを皮下にブースト投与したMUC-1トランスジェニック動物(hMUC-1.Tg)の血清中の融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lに対する抗体のレベルを示す。種々の処理群は図3に記載するものである。ELISAで抗体を検出した。融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lをコーティングしたマイクロウェルプレートを血清と反応させ、洗浄し、結合したマウス抗体をホースラディッシュペルオキシダーゼとコンジュゲートさせたラット抗マウス抗体を用いて検出した。
【0020】
図6は、アデノウイルス発現ベクターAd-K/ecdhMUC1-ΔCtΔTmCD40Lを投与したMUC-1トランスジェニック動物(hMUC-1.Tg)におけるMUC1発現腫瘍細胞(LL2/LL2hMUC-1)の増殖レベルを、融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lの1回または2回の連続投与と比較して示す。
【0021】
図7は、Ad-sig-ecdhMUC-1/ΔCtΔTmCD40LベクターおよびecdhMUC-1/ΔCtΔTm CD40Lタンパク質を接種した動物における腫瘍の予防を示す。VVV= Ad-sig-ecdhMUC-1/ΔCtΔTm CD40Lベクターの皮下注射3回(1日目、7日目、および21日目に投与);PPP=ecdhMUC-1/ΔCtΔTm CD40Lタンパク質の皮下注射3回(1日目、7日目、および21日目に投与);またはVPP= Ad-sig-ecdhMUC-1/ΔCtΔTm CD40Lベクターを1回皮下注射後、7日目および21日目にecdhMUC-1/ΔCtΔTm CD40Lタンパク質を皮下注射。1週間後(28日目)、マウスに500,000のLL2/LL1hMUC-1肺癌細胞を皮下注射した。2週間後(42日目)、500,000のLL2/LL1hMUC-1腫瘍細胞を静脈内投与し、尾静脈を介してマウスを試験した。複数回のベクター投与(単独で、またはその後タンパク質をブースター投与)はマウスにおけるヒト腫瘍の樹立の予防に有効であった。
【0022】
図8は、ワクチン接種開始の63日後のワクチン接種マウスにおけるhMUC-1特異的抗体のレベルを示す。VVV= Ad-sig-ecdhMUC-1/ΔCtΔTm CD40Lベクターの皮下注射3回(1日目、7日目、および21日目に投与);PPP= ecdhMUC-1/ΔCtΔTm CD40Lタンパク質の皮下注射3回(1日目、7日目、および21日目に投与);またはVPP= Ad-sig-ecdhMUC-1/ΔCtΔTm CD40Lベクターを1回皮下注射後、7日目および21日目にecdhMUC-1/ΔCtΔTm CD40Lタンパク質を皮下注射。Ad-sig-ecdhMUC-1/ΔCtΔTmCD40Lベクターを1回皮下注射した後、ecdhMUC-1/ΔCtΔTmCD40Lタンパク質を連続2回、7および21日目に皮下注射するというスケジュールで、最も高いレベルのhMUC-1特異的抗体が誘導された。
【0023】
図9は、Ad-sig-ecdhMUC-1/ΔCtΔTmCD40LベクターおよびecdhMUC-1/ΔCtΔTm CD40Lタンパク質で免疫化した動物における(定着後の)皮下腫瘍療法を示す。VVV= Ad-sig-ecdhMUC-1/ΔCtΔTm CD40Lベクターの皮下注射3回(5日目、12日目、および26日目に投与);PPP= ecdhMUC-1/ΔCtΔTm CD40Lタンパク質の皮下注射3回(5日目、12日目、および26日目に投与);またはVPP= Ad-sig-ecdhMUC-1/ΔCtΔTm CD40Lベクターを1回皮下注射後、12日目および26日目にecdhMUC-1/ΔCtΔTm CD40Lタンパク質を皮下注射。皮下腫瘍(500,000のLL2/LL1hMUC-1)を1日目に投与し、ワクチン接種を5日目に実施した。40日目に腫瘍を静脈内投与し、54日目に腫瘍の発生(皮下および肺)を評価した。
【0024】
図10は、図9に記載するように処理した動物における(定着後の)肺転移腫瘍結節療法を示す。左のパネル:結果は皮下腫瘍の予防と同様であり、スケジュールVVVおよびVPPが最も有効であった。右のパネル:1回のベクター注射およびその後の2回のタンパク質注射の組み合わせ(VPP)では、定着したhMUC-1ポジティブ癌細胞の肺結節の増殖が完全に抑制された。
【0025】
図11は、MUC1発現腫瘍の発生に抵抗する動物の能力に関する、Ad-sig-ecdhMUC-1/ecdCD40Lベクターの1回皮下投与後の種々のブースト計画の比較である。細菌抽出物中のecdhMUC-1/ecdCD40Lタンパク質;(フロイドの不完全アジュバントと共に、または無しで)キーホールリンペットヘモシアニン(KLH)に結合したecdhMUC-1;PBS(リン酸バッファー);およびコントロール細菌抽出物(Ad-sig-ecdhMUC-1/ecdCD40Lベクターに感染していない細菌ホスト株)。
【0026】
発明の詳細な説明
本発明のある観点によれば、発現ベクターを用いて抗原に対する免疫応答を生じさせる方法を提供する。ベクターは抗原およびCD40リガンドを含む分泌可能な融合タンパク質をコードする転写ユニットを含む。好ましい態様では、転写ユニットはアミノ末端側から、分泌シグナル配列、抗原、リンカー、および分泌型CD40リガンドを含む。好ましい態様では、分泌型CD40リガンドはその膜貫通ドメインの全部または一部を欠失している。
【0027】
好ましい方法では、まず個体にベクターを1回以上投与し、初期免疫応答を生成させる。ベクター投与後、有効量の融合タンパク質を投与し、抗原に対する免疫応答をブーストし、ベクターの単独投与で得られるより高いものとする。
【0028】
融合タンパク質の投与に関する“有効量”という用語は発現ベクター単独で得られるものより高い免疫応答となる量をいう。最適な結果を得るためには、一般に投与間の時間間隔が必要である。免疫応答の増加はT細胞活性または抗体産生の増加として測定してもよい(例えば図3-5参照)。一般に、ベクター投与とタンパク質ブースト投与間は少なくとも1週間が有効であるが、より短い間隔でもよい。投与間の有効な間隔は1週間から12週間、またはそれより長くてもよい。複数回のブースト投与を、1-12週間、またはそれ以上の期間をおいて行ってもよい。
【0029】
免疫応答をブーストするために融合タンパク質を使用することにより、複数回の注射に対する過敏性を引き起こしうる発現ベクターの連続的な投与の必要が回避される。融合タンパク質の抗原部分は好ましくは最初の投与に使用される発現ベクターの転写ユニットにコードされる融合タンパク質である。しかしながら、融合タンパク質の抗原部分は、発現ベクターの抗原およびブーストに使用される融合タンパク質の抗原に共通する抗原決定基またはエピトープを少なくとも一つ共有すれば、コードされる抗原と異なっていてもよい。
【0030】
融合タンパク質は哺乳動物細胞系で調製してもよく、これはベクターに相補的である。例えばアデノウイルスの場合、細胞系は初期領域1(Early Region 1;E1)遺伝子を含む293細胞であってもよく、またE1置換型組換えアデノウイルスの増殖をサポートしうる。アデノウイルスベクターが生成細胞に感染すると、ウイルスベクターはウイルスの複製サイクル後、自己増殖する。しかしながら、発現カセット中のウイルスベクターによって行われる目的の遺伝子はウイルスの増殖過程で発現する。これを利用して、ベクターを生成するのと同じ系においてベクターにコードされる融合タンパク質の調製を行うことができる。ベクターおよび融合タンパク質の生成を生成系で同時に行う。このように生成されたベクターおよびタンパク質を異なる過程で更に単離および精製できる。
【0031】
融合タンパク質の投与は非経口、例えば血管内、静脈内、動脈内、筋肉内、皮下などで行ってもよい。投与は経口、経鼻、直腸内、経皮、またはエアロゾルによる吸入で行ってもよい。タンパク質ブーストはボーラス、または点滴(slowly infused)で投与してもよい。タンパク質ブーストは皮下投与が好ましい。
【0032】
融合タンパク質ブーストは、生じる免疫応答を増強させるためにアジュバントと共に調製してもよい。本明細書で使用する“アジュバント”という用語は、ワクチンと共に投与するとワクチンに対する免疫応答が増強するような化学物質を意味する。アジュバントは、アジュバントが免疫原または抗原と化学的に結合しない点においてキャリアータンパク質とは区別される。アジュバントは当該分野で知られており、例えば以下がある:鉱油エマルジョン(米国特許第4,608,251号。上記)、例えばフロイドの完全または不完全アジュバント(Freund, Adv. Tuberc. Res. 7:130 (1956); Calbiochem, San Diego Calif.)、アルミニウム塩、特に水酸化アルミニウムまたはALLOHYDROGEL(米国食品医薬品庁によってヒトへの使用が承認されている)、ムラミルジペプチド(MDP)およびその類似体、例えば[Thr1 ]-MDP(ByersおよびAllison, Vaccine 5:223 (1987))、モノホスホリルリピッドA(Johnsonら, Rev. Infect. Dis. 9:S512 (1987))など。
【0033】
当該分野で知られる方法を用いて、融合タンパク質をマイクロカプセル化またはマクロカプセル化した形態で投与することができる。融合タンパク質をカプセル化して例えばリポソーム(例えばGarconおよびSix, J. Immunol. 146:3697 (1991)参照)、ウシロタウイルスの内部キャプシドタンパク質(Redmondら, Mol. Immunol. 28:269 (1991))、Quil Aのようなサポニンから構成される免疫刺激分子(ISCOMS)(Moreinら, Nature 308:457 (1984);Moreinら, Immunological Adjuvants and Vaccines (G. Gregoriadisら編) pp.153-162, Plenum Press, NY (1987))、または(例えばラクチド/グリコリドコポリマーから構成される)徐放生分解性マイクロスフェア(O'Haganら, Immunology 73:239 (1991);O'Haganら, Vaccine 11:149 (1993))としてもよい。
【0034】
L-121のようなPLURONICブロックコポリマーと共に調製し、TWEEN80のような界面活性剤で安定化させたスクワレンまたはスクワランエマルジョンを含むリピッドマイクロスフェアの表面に融合タンパク質を吸収させてもよい(AllisonおよびByers, Vaccines: New Approaches to Immunological Problems (R. Ellis編) pp. 431-449, Butterworth-Hinemann, Stoneman N.Y. (1992)参照)。マイクロカプセル化またはマクロカプセル化した融合タンパク質はアジュバントを含有してもよい。
【0035】
融合タンパク質をキャリアーまたは外来分子、例えばタンパク質ブーストを投与すべき個体にとって外来性であるキャリアータンパク質にコンジュゲートさせてもよい。免疫応答を活性化し、本明細書に記載する融合タンパク質にコンジュゲートすることができる外来タンパク質には分子量が少なくとも約20,000ダルトン、好ましくは少なくとも約40,000ダルトン、より好ましくは少なくとも約60,000ダルトンであるタンパク質または他の分子がある。本発明に有用なキャリアータンパク質には、例えばGST、(例えばキーホールリンペット由来の)ヘモシアニン、血清アルブミンまたはカチオン化した血清アルブミン、チログロブリン、卵白アルブミン、種々のトキソイドタンパク質(例えば破傷風トキソイドまたはジフテリアトキソイド)、免疫グロブリン、熱ショックタンパク質などがある。
【0036】
あるタンパク質を別の(キャリアー)タンパク質に化学的に結合させる方法は当該分野で知られており、それらには例えば以下がある:水溶性カルボジイミド(例えば1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩)によるコンジュゲート、(例えばNHSエステル基またはスルホ-NHSエステル類似体を有する)ホモ2価性架橋リンカーによるコンジュゲート、(例えばNHSエステルおよびマレイミド基、例えばスルホスクシンイミジル-4-(N-マレイミドメチル)シクロヘキサン-1-カルボキシレートを有する)ヘテロ2価性架橋リンカーによるコンジュゲート、およびグルタールアルデヒドによるコンジュゲート(例えばHermanson, Bioconjugate Techniques, Academic Press, San Diego, Calif. (1996);米国特許第4,608,251号および4,161,519号参照)。
【0037】
本明細書で使用する転写ユニットを含む“ベクター”(“発現ベクター”としても知られる)という用語は、インビボで投与された際に標的細胞に侵入してコードされたタンパク質を発現できるウイルス性および非ウイルス性の発現ベクターをいう。インビボでの運搬および外因性タンパク質の発現に好適なウイルスベクターは知られており、それらにはアデノウイルスベクター、アデノ関連ウイルスベクター、レトロウイルスベクター、単純疱疹ウイルスベクターなどがある。ウイルスベクターは好ましくは正常細胞内で複製欠陥を起こす。米国特許第6,669,942号;6,566,128号;6,794,188号;6,110,744号;6,133,029号参照。
【0038】
本明細書で使用する“細胞”という用語は広義に使用し、哺乳動物細胞、植物細胞、真核細胞、原核細胞など、いずれの生細胞も含む。
【0039】
本明細書で使用する“アデノウイルス発現ベクター”という用語は、ポリペプチドをコードする、そのゲノム中に挿入された外因性DNAを含むアデノウイルス由来の任意のベクターをいう。ベクターは、欠陥のある必須遺伝子がトランスで提供された際に複製しおよび封入されうる能力を有さなければならない。好ましくは、アデノウイルスベクターはウイルスDNAの複製を支持するのに必要な各末端反復の少なくとも一部、好ましくは完全長ITR配列の少なくとも約90%、およびウイルスキャップシドにゲノムを封入するのに必要なDNAを含む。多くの好適なアデノウイルスベクターが当該分野で報告されている。米国特許第6,440,944号および6,040,174号参照(複製欠陥E1欠損ベクターおよび特殊化パッケージング細胞系)。好ましいアデノウイルス発現ベクターは、正常細胞において複製欠陥のあるものである。
【0040】
アデノ関連ウイルスとは、その遺伝物質を19番染色体上の特定の部位に挿入できる小型1本鎖DNAウイルスのクラスを表す。遺伝子運搬のためのアデノ関連ウイルスベクターの調製および使用は米国特許第5,658,785号に記載されている。
【0041】
遺伝子運搬のための非ウイルス性ベクターには、運搬の際にベクターのDNAを保護するために脂質、タンパク質、および他の分子(またはそれらを組み合わせたもの)と合わせた種々の型の発現ベクター(例えばプラスミド)が含まれる。融合性の(fusigenic)非ウイルス性ベクター粒子は、ウイルス性融合タンパク質を記載の発現ベクターと組み合わせることによって構築できる。Kaneda, Curr Drug Targets (2003) 4(8):599-602。再構築したHVJ(hemagglutinating virus of Japan;センダイウイルス)-リポソームを使用して発現ベクターを運搬するか、またはリポソームを使用せずに不活性化HVJ粒子にベクターを直接導入することもできる。Kaneda, Curr Drug Targets (2003) 4(8):599-602参照。DMRIE/DOPE脂質混合物は非ウイルス性発現ベクターの媒体として有用である。米国特許第6,147,055号参照。ポリカチオン-DNA複合体を非ウイルス性遺伝子運搬媒体として使用してもよい。Thomasら, Appl Microbiol Biotechnol (2003) 62(1):27-34参照。
【0042】
本明細書において発現ベクターに関連して使用する“転写ユニット”という用語は、RNAポリメラーゼによって1本の連続するmRNA鎖として転写される一続きのDNAであって、転写の開始および停止のためのシグナルを含むものをいう。例えばある態様では、本発明の転写ユニットは5’から3’方向に、分泌シグナル配列、抗原、およびCD40リガンドをコードする核酸を含む。転写ユニットは転写および/または翻訳発現調節要素、例えばプロモーター、そして必要により任意の上流または下流エンハンサー要素と、機能的に結合している。有用なプロモーター/エンハンサーはサイトメガロウイルス(CMV)IE(immediate-early)プロモーター/エンハンサーである。米国特許第5,849,522号および 6,218,140号参照。
【0043】
本明細書で使用する“分泌シグナル配列”(“シグナル配列”、“シグナルペプチド”、“リーダー配列”、または“リーダーペプチド”としても知られる)という用語は(in charter)一般に疎水性である約20から30アミノ酸を含む短鎖ペプチド配列をいい、これはポリペプチドのN末端で合成され、ポリペプチドを小胞体に誘導する。分泌シグナル配列は一般に、ポリペプチドの小胞体へのトランスロケーションの際に開裂される。真核生物の分泌シグナル配列は、発現ベクターの外因性遺伝子産物の分泌を誘導するのに好ましい。種々のそのような好適な配列が当該分野で知られており、それらにはヒト成長ホルモン、免疫グロブリンのκ鎖の分泌シグナル配列などがある。ある態様では、外来性腫瘍抗原シグナル配列を用いて分泌を起こさせてもよい。
【0044】
本明細書で使用する“抗原”という用語は広範に、個体が免疫応答を起こすことができるいずれの抗原をもさす。本明細書で使用する“抗原”は広範に、免疫応答が標的としうる抗原決定基を少なくとも1つ含む分子をいう。免疫応答は細胞性、体液性、またはその両方であってもよい。
【0045】
当該分野で知られているように、抗原は天然の(in nature)タンパク質、天然の炭化水素、天然の脂質、もしくは天然の核酸、またはこれらの生体分子を組み合わせたものであってもよい。抗原は非天然分子、例えばポリマーなどを含んでもよい。抗原には自己抗原および外来抗原、例えば別の動物が生成した抗原または感染性物質由来の抗原が含まれる。感染性物質抗原は細菌性、ウイルス性、真菌性、原核動物性などであってもよい。
【0046】
本明細書で使用する“腫瘍関連抗原”(TAA)という用語は腫瘍細胞上、および胎児期の正常細胞上(癌胎児性抗原)に存在するタンパク質をいい、これは出産後は選択された臓器中、または多くの正常細胞上に存在するがその濃度は腫瘍細胞上よりかなり低い濃度である。種々のTAAが報告されている。代表的なTAAはムチン(例えばMUC1。以下に更に詳細に記載する)またはHER2(neu)(これも以下に記載する)である。これに対して腫瘍特異抗原(TSA)(腫瘍特異移植抗原またはTSTAともいう)は正常細胞には存在しないタンパク質をいう。TSAは通常、感染するウイルスによって細胞が不死化し、ウイルス抗原が発現される際に出現する。
【0047】
代表的なウイルスTSAはHPV16型のE6またはE7タンパク質である。ウイルスによって誘導されないTSAにはB細胞リンパ腫に関係する免疫グロブリンまたはT細胞リンパ腫のT細胞受容体(TCR)のイディオタイプがある(TSAs not induced by viruses include idiotypes the immunoglobulin idiotypes associated with B cell lymphomas or the T cell receptor (TCR) on T cell lymphomas)。
【0048】
代表的なウイルスTSAはHPV16型のE6またはE7タンパク質である。HPVは種々の皮膚および生殖管の上皮病変を発生させうる。HPVが関係する生殖管疾患は、世界中の女性の2番目に高い癌死亡の原因となっている。それらには生殖器疣贅、頚部上皮内新生物(CIN)、および子宮頚癌がある。最も一般的に高度(high grade)CINおよび子宮頚癌と関係するHPVの型はHPV16型である。子宮頚癌の大半は非構造HPV16誘導型遺伝子産物E6およびE7腫瘍性タンパク質を発現する。HPV誘導型子宮頚癌モデルでは、E6/E7腫瘍性タンパク質が悪性フェノタイプの保持に必要とされ、それらの発現はHPV16の形質転換の可能性と相関関係を有する。E6またはE7を腫瘍抗原として使用するのに加え、これらのタンパク質の抗原性フラグメントを代わりに使用してもよい。抗原性フラグメントは、周知のコンピューター演算(例えばHoppおよびWoods疎水性分析)を用いてエピトープを保有すると予想されるような分子の部分との免疫応答を試験することによって決定してもよい。
【0049】
ウイルスによって誘導されないTSAはB細胞リンパ腫の免疫グロブリンまたはT細胞リンパ腫のT細胞受容体(TCR)のイディオタイプであってもよい。腫瘍関連抗原(TAA)はTSAより一般的である。
【0050】
TAAおよびTSAは共に、発現ベクターワクチンの免疫学的標的となりうる。特に記載しない限り、本明細書において“腫瘍抗原”という用語は集合的にTAAおよびTSAを示す。
【0051】
本明細書で使用する“ムチン”という用語は、スレオニン、セリン、およびプロリン含量の高いタンデムリピート構造を持つペプチド配列にO-グリコシド結合した高含量のクラスター化したオリゴ糖を有する、任意のクラスの高分子量糖タンパク質をいう。ムチンは細胞防御において役割を果たし、伸張構造上に露出された多くの糖で、種々の型の細胞(白血球および感染性物質を含む)との複数の相互作用に影響を与える。ムチン抗原には以下と同定されているものもある:CD227、腫瘍関連上皮膜抗原(EMA)、多型上皮性ムチン(PEM)、ピーナツ反応性尿ムチン(Peanut-reactive Urinary Mucin;PUM)、エピシアリン(episialin)、乳癌関連抗原DF3、H23抗原、ムチン1、エピシアリン(Episialin)、腫瘍関連ムチン、癌関連ムチン。また、CA15-3抗原、M344抗原、Sialosyl Lewis抗原(SLA)、CA19-9、CA195、およびこれまでにモノクローナル抗体によって同定された他のムチン抗原(例えば米国特許第5,849,876号参照)もある。ムチンという用語はプロテオグリカン(タンパク質バックボーンに共有結合したグリコサミノグリカン鎖を特徴とする糖タンパク質である)を含まない。
【0052】
少なくとも15の異なるムチンが報告されており、それらにはMUC1、MUC2、MUC3A、MUC3B、MUC4、MUC5AC、MUC5B、MUC6、MUC7、MUC8、MUC9、MUC12、MUC13、MUC15、およびMUC16が含まれる(これらは“MUC”と数字の間にハイフンを入れて示してもよい)。これらのムチンのヌクレオチド配列およびアミノ酸配列は知られている。これらムチンのNCBIおよびSwiss Protアクセッション番号は以下の通りである:MUC1(NCBI NM002456, Swiss Prot P15941)、MUC2(NCBI NM002457, Swiss Prot Q02817) 、MUC3A(NCBI AF113616, Swiss Prot Q02505)、MUC3B(NCBI AJ291390, Swiss Prot Q9H195)、MUC4(NCBI NM138299, Swiss Prot Q99102)、MUC5AC(NCBI AF043909, Swiss Prot Q8WWQ5)、MUC5B(Swiss Prot Q9HC84)、MUC6(NCBI U97698, Swiss Prot Q8N8I1)、MUC7(NCBI L42983, Swiss Prot Q8TAX7)、MUC8(NCBI U14383, Swiss Prot Q12964)、MUC9(NCBI U09550, Swiss Prot Q12889)、 MUC12(Swiss Prot Q9UKN1)、MUC13(NCBI NM017648, Swiss Prot Q9H3R2)、MUC15(NCBI NM145650, Swiss Prot Q8WW41)、およびMUC16(NCBI AF361486, Swiss Prot Q8WXI7; CA125ともいう)。
【0053】
ムチンには構造的および機能的に異なる2つのクラス:分泌型ゲル形成ムチン(MUC2、MUC5AC、MUC5B、およびMUC6)および膜貫通ムチン(MUC1、MUC3A、MUC3B、MUC4、MUC12、MUC17)がある。いくつかのMUC遺伝子の産物はいずれのクラスにも該当しない(MUC7、MUC8、MUC9、MUC13、MUC15、MUC16)。
【0054】
特定の癌におけるTAAとしての特定のムチンの特徴は、前腫瘍性病変および腫瘍性病変に伴う発現および構造の変化によって確認される(Filipe MI: Invest Cell Pathol 1979, 2:195-216;Filipe MI, Acta Med Port 1979, 1:351-365)。例えば、正常な胃粘膜はMUC1、MUC5A/C、MUC6 mRNA、およびコードされる免疫反応性タンパク質の発現を特徴とする。また、高レベルのMUC2、MUC3ムチンmRNA、およびコードされる免疫反応性タンパク質は腸上皮化生に関係する。胃癌は隣接する正常な粘膜と比較して顕著に変化した分泌型ムチンmRNAレベルを示し、MUC5およびMUC6 mRNAレベルは低下し、MUC3およびMUC4 mRNAレベルは上昇する。高レベルのMUC2およびMUC3 mRNAおよびタンパク質は小腸で検出可能であり、MUC2は最も存在量の多い結腸ムチンである。
【0055】
ムチンは膵臓癌および他の細胞型の早期検出のための診断マーカーである。研究で明らかになったところによれば、腺癌(ductal adenocarcinomas;DAC)および腫瘍細胞系は通常、MUC1ムチンを過剰発現する。Andrianifahananaら, Clin Cancer Res 2001, 7:4033-4040参照。このムチンは、ほとんどの慢性膵炎および正常な膵臓組織では低レベルでしか検出されないが、膵臓癌の全段階で過剰発現される。膵臓癌および細胞系におけるMUC4のデノボ発現が報告されている(Hollingsworthら, Int J Cancer 1994, 57:198-203)。MUC4 mRNA発現は大多数の膵臓癌および確立された膵臓癌細胞系で観察されているが、正常な膵臓または慢性膵炎組織では観察されていない。MUC4発現は肺癌との関係も見いだされている(Nguyenら, 1996 Tumor Biol. 17:176-192参照)。MUC5は非小細胞肺癌において転移と関係する(Yuら, 1996 Int. J. Cancer 69:457-465参照)。胃型および浸潤性DACではMUC6が過剰発現され、MUC5ACがデノボ発現される(Kimら, Gastroenterology 2002, 123:1052-1060)。MUC7は浸潤性膀胱癌のマーカーとして報告されている(Retzら, 1998 Cancer Res. 58:5662-5666)。
【0056】
MUC2分泌型ゲル形成ムチンの発現は一般に大腸腺癌で減少し、粘液癌(マイクロサテライト不安定性を伴う異なるサブタイプの大腸癌)では保存されている。MUC2は咽頭癌で上昇する(Jeannonら, 2001 Otolaryngol Head Neck Surg. 124:199-202)。別の分泌型ゲル形成ムチン、MUC5AC(正常な胃粘膜の産物)は正常な結腸には存在しないが、大腸腺癌および大腸癌では高頻度で存在する。
【0057】
MUC1(エピシアリンとしても知られる)、多型上皮性ムチン(PEM)、ムチン様癌関連抗原(MCA)、CA27.29、ピーナツ反応性尿ムチン(PUM)、腫瘍関連上皮ムチン、上皮膜抗原(EMA)、ヒト乳脂肪球(HMFG)抗原、MUC1/REP、MUC1/SEC、MUC1/Y、CD227は最もよく知られたムチンである。MUC1をコードする遺伝子はlq21-q24にマッピングされる。MUC1遺伝子は7つのエクソンを含み、7つの異なるスプライス変異体を生成する。タンデムリピートドメインは高度にO-グリコシル化されており、グリコシル化の変化が上皮癌細胞において明らかになっている。
【0058】
MUC1 mRNAはサイズ多型である。現在のところ、スプライシングの変化に基づいて9つのMUC1のアイソフォームがある(アイソフォーム番号:NCBIアクセッション番号;1:ID P15941-1、2:ID P15941-2、3:ID P15941-3、4:ID P15941-4、5:P15941-5、6:ID P15941-6、7:ID P15941-7、8:ID P15941-8、9:ID P15941-9)。
【0059】
MUC1アイソフォーム1(MUC1/REPともいう)は多型のI型膜貫通タンパク質であり、以下を含む:1)主に20-アミノ酸(aa)反復モチーフ(可変数(30-100)タンデムリピート(Variable Number of tandem repeats-VNTR)として知られる領域)から成る大型の細胞外ドメイン;2)膜貫通ドメイン;および3)72-aa細胞質尾部。生合成の際、MUC1/REPタンパク質は大幅に修飾され、相当な数のO-結合糖部分は成熟タンパク質上にムチン様の特徴を与える。翻訳後まもなくMUC1/REPは開裂されて2つの産物となり、これらは強く結合したヘテロダイマー複合体を形成するが、これは大型の細胞外ドメインが細胞質ドメインおよび膜貫通ドメインを含むそれよりかなり小型のタンパク質に非共有結合したものから構成される。細胞外ドメインは細胞から離脱できる。参照としてSwiss Prot P15941を用い(図1参照)、MUC1アイソフォーム1の細胞外ドメイン(ecm)はアミノ酸24から1158であり、膜貫通ドメインは1159-1181であり、そして細胞質ドメインは1182-1255である。SEAドメインは1034-1151であり、細胞外ドメインと呼ばれるC末端部分である。ムチンのSEAドメインは一般にタンパク分解による開裂の標的であり、2つのサブユニットを生成し、小さい方は細胞膜と結合する。
【0060】
MUC1アイソフォーム5(MUC1/SECともいう)は細胞によって分泌されるMUC1の型である。アイソフォーム1(MUC1/REP)と同じ細胞外ドメインを有するが、細胞膜にタンパク質を係留するための膜貫通ドメインを有さない。MUC1アイソフォーム7(MUC1/Yともいう)はアイソフォーム1(MUC1/REP)および5(MUC1/SEC)で観察される細胞質および膜貫通ドメインを含むが、MUC1より小さい細胞外ドメインを有し、反復モチーフおよびその隣接領域を有さない(Baruch A.ら, 1999 Cancer Res. 59, 1552-1561参照)。アイソフォーム7は受容体として作用し、分泌型アイソフォーム5に結合する。結合によりアイソフォーム7のリン酸エステル化が誘導され、細胞の形態学に変化が起き、GRB2のような第2のメッセンジャータンパク質を通して細胞シグナリングが開始される(Zrihan-Licht S.ら, 1995 FEBS Lett. 356, 130-136参照)。β-カテニンはMUC1の細胞質ドメインと反応することが明らかになっている(Yamamoto M.ら, 1997 J. Biol. Chem. 272, 12492-12494)。
【0061】
MUC1は正常な上皮細胞表面上で局所的に低レベルで発現される。15参照。Greenleeら, Cancer Statistics CA Cancer J. 50, 7-33 (2000);Renら, J. Biol. Chem. 277, 17616-17622 (2002);Kontaniら, Br. J. Cancer 84, 1258-1264 (2001);Rowseら, Cancer Res. 58, 315 (1998)。MUC1は乳癌、卵巣癌、膵臓癌、並びに他の癌で過剰発現される(Gendler S.J.ら, 1990 J. Biol. Chem. 265, 15286-15293も参照されたい)。MUC1遺伝子の更なるコピーの獲得と高mRNAレベルとの間には相関関係が見られ(p<0.0001)、これはMUC1遺伝子の過剰発現の原因である遺伝子機構を明らかにしており、乳癌の病因におけるMUC1遺伝子投与(dosage)の役割を支持している((Bieche I.ら, 1997 Cancer Genet. Cytogenet. 98, 75-80参照)。MUC1ムチンは免疫学的検出によれば、結腸癌(これは予後の悪さと関係する)および卵巣癌で発現が上昇する。
【0062】
MUC1抗原の高レベル発現は、足場依存性増殖の調節を混乱させる(E-カドヘリン機能を混乱させる)ことによって腫瘍性上皮粘膜細胞の増殖において役割を果たし、これによって転移が起こる。Greenleeら, Cancer Statistics CA Cancer J. 50, 7-33 (2000);Renら, J. Biol. Chem. 277, 17616-17622 (2002)参照。MUC1に対する非MHC制限細胞傷害性T細胞の反応が乳癌患者において報告されている。Kontaniら, Br. J. Cancer 84, 1258-1264 (2001)参照。ヒトMUC1トランスジェニックマウス(“MUC1.Tg”)はヒトMUC1抗原での刺激に反応しないことが報告されている。Rowseら, Cancer Res. 58, 315 (1998)参照。ヒトMUC1トランスジェニックマウスは自己抗原としてのMUC1に対する免疫の確立を評価するのに有用である。
【0063】
MUC1タンパク質およびmRNAはER陽性MCF-7およびBT-474細胞、並びにER陰性MDA-MB-231およびSK-BR-3 BCC細胞において観察されている。mRNA転写レベルはER-よりER+細胞系の方が高い。MUC1は細胞内接着分子-1(ICAM-1)と反応する。MUC1の少なくとも6つのタンデムリピートが必要である(Regimbaldら, 1996 Cancer Res. 56,4244-4249)。T-47D BCC由来のMUC1のタンデムリピートペプチドは高度にO-グリコシル化されており、ミルク由来のムチンのリピート当たり2.6グリコシル化部位に比較してリピート当たり4.8部位である。
【0064】
本明細書で使用する“ムチン抗原”という用語は完全長のムチン、または本明細書に記載するようにインビボで投与されたMUC-CD40L発現ベクターを用いて細胞免疫を誘導できることを特徴とするエピトープを含むムチンの一部をいう。“ムチン抗原”はムチンの細胞外ドメインからの1つ以上のエピトープ、例えばVNTRに関連する1つ以上のタンデムリピートモチーフまたはSEA領域を含む。ムチン抗原は細胞外ドメイン全体を含んでもよい。また、“ムチン抗原”の意味には、保存的アミノ酸変化など、天然のムチン配列と交差反応する免疫応答を誘導する抗原の能力を変更しない、配列の変種も含まれる。
【0065】
VNTRは遺伝子多型およびムチンの性質に従って長さ(および組成)の異なる可変数のタンデムリピートペプチド配列から成る。VNTRは変性タンデムリピートを含有する5’および3’領域も含んでもよい。例えばMUC1では、北欧人集団においてリピートの数は21から125まで種々である。米国では、最も頻度の低い対立遺伝子は41および85リピートを含み、より一般的な対立遺伝子は60-84リピートを有する。MUC1リピートは一般的なリピートペプチド配列PDTRPAPGSTAPPAHGVTSA(SEQ ID NO : 3)を有する。基礎となるMUC1タンデムリピートは太線および下線で示す3つの位置(位置2、3、および13)における遺伝子配列多型である。協奏的置換(concerted replacement)DT→ES(配列変種1)および単一置換P→Q(配列変種2)、P→A(配列変種3)、およびP→T(配列変種4)が同定されており、ドメイン中の位置は種々である(Engelmannら, 2001 J. Biol. Chem. 276:27764-27769参照)。最も頻度の高い置換DT→ESは50%までのリピートで起こる。表1に代表的なタンデムリピート配列を示す。
【0066】
【表1】
【0067】
本明細書で使用するムチン抗原は1つのタンデムリピート配列モチーフだけを含んでもよいが、ベクターが複数のそのようなリピートを含めば、一般に生成される免疫応答はより強く、より有効であることは理解されるべきである。本発明のベクターは好ましくは2-4、より好ましくは5-9、より好ましくは10-19、より好ましくは20-29、更に好ましくは30-39、そして更により好ましくは40-50のムチンタンデムリピートをコードする。50より大きいタンデムリピートが可能であり、天然ムチンで見られるリピート数を含んでもよい。
【0068】
本明細書で使用するムチン抗原という用語は種々のタイプのムチン由来のタンデムリピートを含んでもよい。例えば発現ベクターは2つの異なるムチン(例えばMUC1およびMUC2)由来のタンデムリピートをコードしてもよい。それらのベクターは複数の型のSEAドメインを同様にコードしてもよく、またはタンデムリピートと1つ以上のSEAドメインを組み合わせて含んでもよい。
【0069】
分泌型の抗原は、その膜貫通ドメイン(成熟タンパク質中に存在する場合)の全部または実質的に全部が欠失したものである。例えばムチンの場合、膜貫通ドメインは、もし存在するなら、一般に約24アミノ酸長で、ムチンまたはムチンのフラグメントを細胞膜に係留する機能を果たす。全部の膜貫通ドメインが欠失した分泌型のMUC1は残基1159-1181が欠失したMUC1である。実質的に全部の膜貫通ドメインが欠失したムチン(または抗原)は、ドメインが膜貫通ドメインの一端の配列の6以下の残基、より好ましくは膜貫通ドメインの一端の配列の約4未満の残基、更に好ましくは膜貫通ドメインの一端の配列の約2未満の残基、そして最も好ましくは膜貫通ドメインの一端の1以下の残基を含むものである。好ましい態様では、ワクチンベクターの転写ユニットは膜貫通ドメイン全体が欠失した分泌型のムチン(または抗原)をコードする。膜貫通ドメインの実質的に全部が欠失して分泌可能となったムチンは、ドメインの一端における配列の6を超えない残基を含むものである。本明細書ではMUC1のようなヒトムチンの細胞外ドメインを “ecdhMUC1”と記載する。
【0070】
機能性膜貫通ドメインが欠失したムチンは、(もし存在する場合は)細胞質ドメインの全部または一部およびSEAの全部または一部をなおも含みうることは理解されるべきである。
【0071】
種々のムチンおよびムチン抗原をコードするDNA源は、市販のcDNA合成キットおよび増幅(確立されたムチンDNA配列から設計できる好適なPCRプライマー対を使用)を用いてムチン発現細胞系から得てもよい。例えばMUC1またはMUC2をコードする核酸はAmerican Type Culture Collectionから入手できるCRL-1500細胞から得てもよい。ムチンをコードするDNAは、ヒトまたは他の動物組織から採取または調製したRNAまたはcDNAから増幅して得てもよい。さほど大きくないDNAセグメントでは、DNAは自動オリゴヌクレオチド合成装置を用いて合成してもよい。
【0072】
本明細書で発現ベクターの転写ユニットに関して使用する“リンカー”という用語は、抗原のカルボキシ末端とCD40リガンドのアミノ末端との間にある1つ以上のアミノ酸残基をいう。リンカーの組成および長さは当該分野で知られる方法に従って決定してもよく、有効性に関して試験してもよい。例えばAraiら, "Design of the linkers which effectively separate domains of a bifunctional fusion protein" Protein Engineering, Vol. 14, No. 8, 529-532, August 2001参照。リンカーは一般に約3から約15アミノ酸長、より好ましくは約5から約10アミノ酸長であるが、より長い、もしくはより短いリンカーを使用してもよく、またはリンカーが全く無くてもよい。より長いリンカーは約50アミノ酸まで、または約100アミノ酸まで及んでもよい。ムチン抗原がCD40リガンドのN-末端側にある場合は、10残基未満の短いリンカーが好ましい。
【0073】
本明細書で使用する“CD40リガンド”(CD40L)という用語はCD154またはTNF5としても知られる分子の全長または一部をいう。CD40LはN-末端に細胞質ドメイン、膜貫通領域、そして細胞外ドメインをC-末端に有するII型膜ポリペプチドである。特に記載しない限り、本明細書では完全長のCD40Lを“CD40L”、“wtCD40L”、または“wtTmCD40L”と記載する。細胞質ドメインが欠失した型のCD40Lを、本明細書では“ΔCtCD40L”と記載する。膜貫通ドメインが欠失した型のCD40Lを、本明細書では“ΔTmCD40L”と記載する。細胞質ドメインおよび膜貫通ドメインの両方が欠失した型のCD40Lを、本明細書では“ΔCtΔTmCD40L”と記載する。マウスおよびヒト由来のCD40Lのヌクレオチドおよびアミノ酸配列は当該分野で知られており、例えば米国特許第5,962,406号(Armitageら)に報告されている。本発明の融合タンパク質に関連して、ムチンに対する免疫応答を誘導するリガンドの能力に変化を与えないような、保存的アミノ酸変化などを含む配列変異体もCD40リガンドの意味に含まれる。
【0074】
マウスCD40L(mCD40L)は260アミノ酸長である。mCD40Lの細胞質(Ct)ドメインはおおよそ1-22位、膜貫通ドメインはおおよそ23-46位、細胞外ドメインはおおよそ47-260位にわたる。
【0075】
ヒトCD40L(hCD40L)は261アミノ酸長である。hCD40Lの細胞質ドメインはおおよそ1-22位、膜貫通ドメインはおおよそ23-46位、細胞外ドメインはおおよそ47-261位にわたる。
【0076】
本明細書で使用する“CD40リガンドが膜貫通ドメインの全部または実質的に全部を欠失して分泌可能となっている”というフレーズは、細胞から分泌されることが可能な組換え型のCD40リガンドをいう。約24アミノ酸長を含むCD40Lの膜貫通ドメインは細胞膜においてCD40リガンドを係留する機能を果たす。膜貫通ドメインの全部が欠失したCD40Lは、残基23-46が欠失したCD40リガンドである。実質的に全部の膜貫通ドメインが欠失したCD40リガンドは、膜貫通ドメインの一端の配列の6残基以下、より好ましくは膜貫通ドメインの一端の配列の約4残基未満、更に好ましくは膜貫通ドメインの一端の配列の約2残基未満、そして最も好ましくは膜貫通ドメインの一端の1残基以下を保有するものである。従って、膜貫通ドメインの実質的に全部が欠失して分泌可能となったCD40Lは、ドメインの一端の配列の6を超えない残基を保有するものである。そのように、CD40Lは、細胞外ドメインおよび必要により細胞質ドメインに加え、CD40Lの膜貫通ドメインに位置するアミノ酸41-46または23-28を超えないアミノ酸を含む(Such as CD40L would contain, in addition to the extracellular domain and optionally the cytoplasmic domain, and no more than amino acids 41-46 or 23-28 located in the transmembrane domain of CD40L.)。 好ましい態様では、ワクチンベクター転写ユニットは、膜貫通ドメインの10%未満を含む分泌型CD40をコードする。更に好ましくは、CD40Lは膜貫通ドメインを含まない。
【0077】
機能性膜貫通ドメインが欠失したCD40Lは細胞質ドメインの全部または一部をなおも含みうることは理解すべきである。同様に、機能性膜貫通ドメインが欠失したCD40Lは細胞外ドメインの全部または実質的に一部を含みうる。
【0078】
本明細書における使用では、発現ベクターおよび融合タンパク質ブーストをワクチンとして投与し、腫瘍抗原に対する免疫を誘導する。発現ベクターおよび融合タンパク質ブーストは必要により、好適な医薬的に許容しうるキャリアーと共に調製してもよい。従って、ベクターまたはタンパク質ブーストを薬剤または医薬組成物の製造に使用してもよい。発現ベクターおよび融合タンパク質を非経口投与のための溶液または凍結乾燥粉末として調製してもよい。使用の前に好適な希釈剤または他の医薬的に許容されるキャリアーを添加して粉末を再調製してもよい。液体製剤は緩衝された等張の水溶液であってもよい。粉末を乾燥状態でスプレーしてもよい。好適な希釈剤の例は通常の等張食塩水、標準的な5%デキストロース水溶液、または酢酸ナトリウムもしくはアンモニウム緩衝液である。それらの製剤は非経口投与に特に好適であるが、経口投与に使用するか、または吸入のための定量噴霧式吸入器もしくはネブライザーに含有させてもよい。ポリビニルピロリドン、ゼラチン、ヒドロキシセルロース、アカシアゴム、ポリエチレングリコール、マンニトール、塩化ナトリウム、クエン酸ナトリウムなどのような賦形剤の添加が所望されうる。
【0079】
あるいはまた、発現ベクターおよび融合タンパク質を経口投与用に調製してもよい。医薬的に許容される固体または液体キャリアーを添加して組成物を増強もしくは安定化させるか、またはベクターの調製を容易にしてもよい。固体キャリアーにはデンプン、ラクトース、硫酸カルシウム2水和物、白土(terra alba)、ステアリン酸マグネシウムもしくはステアリン酸、タルク、ペクチン、アカシアガム、寒天、またはゼラチンがある。液体キャリアーにはシロップ、落花生油、オリーブ油、食塩水、および水がある。キャリアーはモノステアリン酸グリセリンまたはジステアリン酸グリセリンのような持続放出物質を単独で、またはワックスと共に含有してもよい。固体キャリアーの量は種々であるが、好ましくは投与ユニット当たり約20mgから約1gの間である。液体キャリアーを使用する場合、製剤はシロップ、エリキシル、エマルジョン、または水性もしくは非水性懸濁液の形態であってもよい。
【0080】
発現ベクターおよび融合タンパク質は、他の医薬的に有用な薬剤または生物学的物質を含有させて調製してもよい。ベクターを、本発明の化合物が目的とする疾病または症状に有用な他の薬剤または生物学的物質の投与と併せて投与してもよい。
【0081】
本明細書で使用する“有効量”というフレーズは、そのレシピエントにおいて免疫応答を生じさせる(または生成に関与する)のに十分高い濃度となるような投与量をいう。特定の被験体についての固有の有効量は種々の因子に依存し、それらには治療すべき障害、障害の重篤度、特定の化合物の活性、投与経路、ウイルスベクターのクリアランス速度、治療期間、ウイルスベクターと併用する、または同時に使用する薬剤、年齢、体重、性別、食事、および被験体の全身の健康状態、そして医学および科学の分野で周知の因子がある。“治療的有効量”を決定するための種々の一般的に考慮される点は当該分野で周知であり、例えば以下に記載されている:Gilmanら編, Goodman And Gilman's: The Pharmacological Bases of Therapeutics, 第8版, Pergamon Press, 1990;およびRemington's Pharmaceutical Sciences,第17版, Mack Publishing Co., Easton, Pa., 1990。ベクターの投与では、投与当たりの粒子の範囲は一般に約1x107から1x1011、より好ましくは1x108から5x1010、そして更に好ましくは5x108から2x1010である。ベクターは非経口で、例えば血管内、静脈内、動脈内、筋肉内、皮下などで投与してもよい。投与は経口、経鼻、直腸内、経皮、またはエアロゾルを介した吸入であってもよい。ベクターはボーラスまたは点滴(slowly infused)で投与してもよい。好ましくはベクターを皮下投与する。
【0082】
本明細書に示すように、腫瘍関連抗原をコードするベクターは、それらの抗原(すでに寛容が確立されたものを含む)に対する防御細胞および体液免疫を誘導する。いずれの理論に拘束されることも望まないが、本発明のワクチンを投与すると、分泌型のCD40リガンドに結合した分泌型の抗原から構成される融合タンパク質の継続的な局所放出が起こると考えられる。本明細書に示すように、これによってDCの成熟が促進され、有効な抗原特異的免疫の生成が促進される。また、本明細書に示すように、アデノウイルスベクターから生成された膜貫通ドメインおよび細胞質ドメインが欠失したヒトMUC1およびマウスCD40Lの細胞外ドメイン(すなわちsighMUC1-ΔCtΔTmCD40L)をコードする分泌可能な融合タンパク質は、MUC1発現腫瘍細胞に対する細胞免疫応答の有効性を劇的に増強した。いずれの理論に拘束されることも望まないが、Ad-K-ecdhMUC1-ΔCtΔTmCD40Lベクターの皮下注射によって強いMUC1特異的CD8+ T細胞介在型免疫が誘導され、これによってMUC1腫瘍関連抗原を発現する癌細胞の生着が予防されると考えられる。
【0083】
本発明の方法を使用して抗原に対して生じさせた免疫は長期にわたって持続される。本明細書で使用する長期持続という用語は、ベクターにコードされる抗原によって誘導された免疫が最終投与から6ヶ月まで、好ましくは8ヶ月まで、より好ましくは1年まで、より好ましくは1.5年まで、そして更に好ましくは少なくとも2年間示されることを意味する。
【0084】
ある態様では、ムチンTAAに対する免疫を生じさせるために、膜貫通ドメインおよび細胞質ドメインを欠失させたCD40リガンドのアミノ末端に融合させたMUC1の細胞外ドメインを含む融合タンパク質を生成してもよい。それらのベクターの構築を実施例に開示する。本明細書で観察されたように、このアデノウイルスベクタームチンワクチンの皮下投与によって、MUC1抗原を保持する癌細胞に対する非常に強力な長期持続性のCD8+ 細胞傷害性T細胞依存型全身免疫が誘導された。ムチンワクチンは長期持続性免疫の基礎となる記憶細胞の生成を誘導した。
【0085】
マウスにアデノウイルスベクター Ad-sig-ecdhMUC1/ecdmCD40Lをワクチン接種すると、100%のhMUC1トランスジェニックマウス(すなわちこれらのマウスはベクター注射以前はhMUC1抗原に対して不顕性である)でヒトMUC1(hMUC1)抗原陽性腫瘍細胞の増殖を抑制する免疫応答が誘導された。Rowseら, Cancer Res. 58, 315 (1998)参照。Ad-sig-ecdhMUC1/ecdmCD40Lベクターに対する免疫応答は1年まで持続し、抗原特異性を示した。これらの結果は、MUC1を発現する上皮性悪性病変の治療にAd-sig-ecdhMUC1-ecd/ecdCD40Lベクターを使用できることを示している。
【0086】
アデノウイルスMUC1発現ベクターの皮下注射によって、注射したhMUC1トランスジェニックマウスの脾臓におけるhMUC1特異的T細胞のレベルが250倍上昇した。トランスジェニックマウスはベクター注射以前はhMUC1抗原に対して不顕性であった。従って、ベクター注入によって不顕性が克服され、抗原特異的かつHLA制限的(HLA restricted)であるCD8+ T細胞依存的全身Th1免疫応答が誘導される。アデノウイルスMUC1発現ベクターの接種で観察されたような不顕性を克服する能力は、トランスジェニックマウスに精製したecdhMUC1/ecdCD40L-HISタンパク質を接種した場合には観察されなかった。
【0087】
いずれの理論にも拘束されることを望まないが、ベクターの皮下注射部位の近傍の感染した細胞が腫瘍抗原/CD40リガンド分泌物を放出し、これが感染細胞の近傍の抗原表示細胞(例えばDC)に取り込まれると考えられる。内在化された腫瘍抗原はプロテオソームで消化され、生じるムチン抗原ペプチドは小胞体に移動し、そこでMHCクラスI分子と結合する。最終的にDCはMHCクラスI分子において表面上に腫瘍抗原を提示する。活性化された腫瘍抗原を担持する抗原提示細胞はリンパ球を保有する第2の器官、例えば所属リンパ節または脾臓に移動する。2週間にわたる腫瘍抗原/CD40融合タンパク質の連続放出の間、腫瘍関連抗原を運搬する細胞を認識および死滅させる能力のあるCD8細胞傷害性T細胞は、活性化された、抗原を担持する樹状細胞の存在によってリンパ節および脾臓で増殖する。ベクターに感染させた細胞からの連続的放出による腫瘍抗原特異的細胞傷害性T細胞の刺激および増殖の連続性により、非分泌性である腫瘍抗原/CD40リガンドを運搬するベクターを用いて得られるより強い免疫反応が生ずると考えられる。
【0088】
従って、本発明の方法を使用して個体において自己抗原である抗原に対する免疫を生じさせることができる。例えば、MUC1由来のムチン抗原をコードするベクターを使用して、MUC1ムチン抗原が自己抗原であるヒトにおいてCD8+ 免疫を生じさせることができる。本発明の方法を使用して、自己抗原である抗原に対する免疫学的不顕性状態を克服することもできる。
【実施例】
【0089】
以下の実施例は本発明を例証するためのものである。これらの実施例は本発明の範囲を制限することを意図しない。
1.アデノウイルス発現ベクターの構築
アデノウイルスベクターの転写ユニットsig-ecdhMUC1-ΔCtΔTmCD40Lはシグナル配列(Igカッパ鎖由来)、次いでヒトMUC1の細胞外ドメインをコードし、これは膜貫通ドメインまたは細胞質ドメインを有さず細胞外ドメインを含有するCD40リガンド(ヒトまたはマウス)のフラグメントにリンカーを介して結合している。カッパ鎖シグナル配列を融合タンパク質のアミノ末端に添加することにより、ベクター感染細胞から融合タンパク質が分泌されるように遺伝子操作した。
【0090】
ヒトMUC1のアミノ酸配列およびコードするヌクレオチド配列を、それぞれ図2および1に示す。コードされたMUC1タンパク質はSEQ ID NO : 1のヌクレオチド74から3,841にコードされる1255アミノ酸に相当する。最初の23アミノ酸(SEQ ID NO : 1の74から142にコードされる)はMUC1シグナル配列であり、成熟ムチンからは除去される。細胞外ドメインは24位から1158位の約1135アミノ酸(ヌクレオチド143から3547にコードされる)である。タンデムリピート領域は約900アミノ酸に相当する。アミノ酸74から126(SEQ ID NO : 1の229から451にコードされる)は5’変性タンデムリピート領域、アミノ酸127から945はタンデムリピート領域(SEQ ID NO : 1の452から2,908にコードされる)であり、アミノ酸946から962は3’変性タンデムリピート領域(SEQ ID NO : 1の2809から2959にコードされる)に相当する。SEAドメインはアミノ酸1034から1151、膜貫通ドメインは1159から1181、そして細胞質ドメインは1181から1255である(SEQ ID NO : 2参照)。
【0091】
転写ユニットをアデノウイルスベクターのバックボーンのE1遺伝子領域に導入した。アデノウイルスベクター粒子をHEK293細胞で生成させた後、塩化セシウム密度勾配遠心分離法によってベクターDNAを精製した。アデノウイルスベクター中のシグナルペプチドの存在を、制限酵素分析およびDNAシーケンシングによって確認した。
【0092】
ヒトMUC-1をコードするDNAの上流にマウスIgGカッパ鎖遺伝子のシグナル配列をコードするDNAを含む転写ユニット(“sig-ecdhMUC-1”)をPCRで生成したが、これにはプラスミドpcDNA3-hMUC-1(Finn O.J., University of Pittsburgh School of Medicineより供与)および以下のプライマーを用いた:マウスIgGカッパ鎖METDTLLLWVLLLWVPGSTGD(1文字アミノ酸コード)(SEQ ID NO : 11)をコードするDNAをPCR増幅で調製し(SEQ ID NO : 12、13、および14)、完全長の21アミノ酸マウスIgGカッパ鎖シグナル配列を生成した(SEQ ID NO : 12において、開始コドン“ATG”を太字(下線)で示す)。
5'-CCACC ATG GAG ACAGAC ACA CTC CTG CTA TGG GTA CTG CTG-3'
(SEQ ID NO : 12)
5'- TC CTG CTA TGG GTA CTG CTG CTC TGG GTT CCAGGT TC-3'
(SEQ ID NO : 13)
5'- TG CTC TGG GTT CCAGGT TCC ACT GGT GAC GAT G -3'
(SEQ ID NO : 14)
5'- GGT TCC ACT GGT GAC GAT GTC ACC TCG GTC CCAGTC-3'
(SEQ ID NO : 15)(MUC-1リピート領域の順向きプライマー)
5'- GAGCTCGAG ATT GTG GAC TGG AGG GGC GGT G-3'
(SEQ ID NO : 16)(MUC-1リピート領域の逆向きプライマー)
【0093】
上流にカッパシグナル配列を有するsig-ecdhMUC-1を4ラウンドのPCR増幅で生成した(第1ラウンド:プライマーSEQ ID NO : 15および16;第2ラウンド:プライマーSEQ ID NO : 14および16;第3ラウンド:プライマーSEQ ID NO : 13および16;第4ラウンド:プライマーSEQ ID NO : 12および16)。sig-ecdhMUC-1をコードするDNAをpcDNATM 3.1 TOPOベクター(Invitrogen, San Diego, CA)にクローニングしてpcDNA-sig-ecdhMUC-1を構築した。
【0094】
pShuttle-ΔCtΔTmCD40L(シグナル配列が無く、マウスCD40Lを含む)を以下のように調製した:プラスミドpDC406-mCD40LはAmerican Type Culture Collectionから購入した。1組のPCRプライマー(SEQ ID NO : 17および18)を、マウスCD40リガンドの52位から260位(すなわち細胞質ドメインおよび膜貫通ドメインを含まない)を増幅し、増幅子の5’末端にリンカーをコードする配列(“+スペーサー”と表示)が含まれるように設計した。
マウスΔCtΔTmCD40L+スペーサー順向きプライマー(MCD40LSPF)(CD40L配列はイタリック体(小文字);クローニング部位は下線および太線):
5'- CCGCTCGAGAACGACGCACAAGCACCAAAATCAAAGGTCGAAGAGGAAGTA -3'
(SEQ ID NO : 17)
マウスCD40L逆向きプライマー(MCD40LR;クローニング部位は下線):
5'-GCGGGCC CGCGGCCGCCGCTAG TCTAGAGAG TTT GAG TAAGCC AAAAGA TGAG-3'
(SEQ ID NO : 18)
【0095】
順向きプライマーMCD40LSPFは転写ユニットのムチンとCD40Lリガンド(mCD40L)との間に位置する10残基のスペーサー(LENDAQAPKS;1文字コード;SEQ ID NO : 19)をコードする。順向きおよび逆向きプライマー(SEQ ID NO : 17および18)およびテンプレートとしてプラスミドpDC406-mCD40Lを使用してPCRを行い、PCRフラグメント“スペース+ΔCtΔTMCD40L”を得、これをXbaI(TCTAGA)およびXho I(CTCGAG)で制限エンドヌクレアーゼ消化した後、プラスミドpcDNA-sig-ecdhMUC1に挿入した。このベクターをpcDNA-sig- ecdhMUC1/ΔCtΔTmCD40Lで消化する。切断型ではなく完全長のCD40Lをコードすること以外は同じであるベクターを構築した。このベクターの生成は、CD40順向きプライマーを用い、マウスCD40Lの開始コドンにアニールさせて行った。このベクターをpShuttlsig40Lと記載する(シグナル配列なし)。
【0096】
HindIII-XbaI 制限を用いてpCDNA3TOPO ベクターからsig-ecdhMUC1/ΔCtΔTmCD40LをコードするDNAを切り出し、pShuttle-CMV(Murphyら, Prostate 38: 73-78, 1999参照)のCMVプロモーターの下流に挿入した。このプラスミドをpShuttle-sig-ecdhMUC1-ΔCtΔTmCD40Lと記載する。従って転写ユニットsig-ecdhMUC1-ΔCtΔTmCD40LはマウスIgGカッパ鎖分泌シグナル、次いでヒトMUC1の細胞外ドメイン、次いで10アミノ酸リンカー(NDAQAPK;SEQ ID NO : 19)、次いでマウスCD40リガンド残基52-260をコードする。
【0097】
あるベクターでは、マウスHSF1 3量体ドメインをecdhMUC1をコードするDNAとΔCtΔTm CD40Lとの間に加えたが、これはプラスミドpcDNA-sig-ecdhMUC1/ΔCtΔTmCD40Lおよび以下のプライマーを用いたPCRで行った:
5'-AAC AAG CTC ATT CAG TTC CTG ATC TCA CTG GTG GGATCC AAC GAC GCA CAAGCA CCAAAA TC-3'
(SEQ ID NO : 20)
5'- AGC CTT CGG CAG AAG CAT GCC CAG CAA CAG AAAGTC GTC AAC AAG CTC ATT CAG TTC CTG-3'
(SEQ ID NO : 22)
5'GAT ATC CTC AGG CTC GAG aac gac gca caagca ccaaaagAG AAT GAG GCT CTG TGG CGG G-3'
(SEQ ID NO : 23)
5'-GCGGGCC CGCGGCCGCCGCTAG TCTAGA GAG TTT GAG TAAGCC AAAAGA TGAG-3'
(SEQ ID NO : 18)
【0098】
3量体ドメイン配列を持つHSF1/ΔCtΔTm CD40Lを4ラウンドのPCR増幅によって生成した(第1ラウンド:プライマーSEQ ID NO : 23および18;第2ラウンド:プライマーSEQ ID NO : 22および18;第3ラウンド:プライマー21および18;第4ラウンド:プライマーSEQ ID NO : 20および18)。HSF1/ΔCtΔTm CD40LをコードするDNAをpcDNA-sig- hMUC-1制限部位XbaI(TCTAGA)およびXho I(CTCGAG)にクローニングした。MUC1およびmCD40L間の配列は以下である:
L E N D A Q A P K E N E A L W R E V A S F R Q K H A Q Q Q K V V N K L I Q F L I S L V G S N D A Q A P K S(SEQ ID NO : 24)
ここで、下線のセグメントは3量体配列であり、これがリンカーLENDAQAPK (SEQ ID NO : 25)およびNDAQAPKS(SEQ ID NO : 26)に結合する。
【0099】
あるベクターでは、Hisタグをコードする配列をΔCtΔTm CD40Lの末端に添加し、プラスミドpDC406-mCD40L(American Type Culture Collectionより購入)および以下のプライマーを用いたPCRで生成した:
5'-CCG CTCGAG AACGACGCACAAGCACCAAAATCAAaggtcgaagaggaagta -3'(SEQ ID NO : 27)(順向きプライマー)
5'-ATG GTG ATG ATG ACC GGT ACG GAG TTT GAG TAA GCC AAA AGA TGA GAA GCC-3' (SEQ ID NO: 28)(逆向きプライマー)
5'-GTGC tctaga TCAgaattc <ATG GTG ATG GTG ATG ATG> ACC GGT ACG GAG -3'(SEQ ID NO : 29)(ポリHis領域は囲み線内(括弧内)のヌクレオチドにコードされる)。
【0100】
Hisタグ配列を持つベクター/ΔCtΔTm CD40L/Hisを2ラウンドのPCR増幅で生成した(第1ラウンド:プライマー1+2;第2ラウンド:プライマー1+3)。/ΔCtΔTmCD40L/HisをコードするDNAをpcDNA-sig-ecdhMUC-1 制限部位XbaI(TCTAGA)およびXho I(CTCGAG)にクローニングした。
【0101】
組み換えアデノウイルスベクターをAdEasyベクター系(Stratagene, San Diego, CA)を用いて生成した。簡潔に記載すると、得られたプラスミドpShuttle-sig- ecdhMUC1-ΔCtΔTmCD40Lおよび他のコントロールアデノウイルスベクターをPme Iで直線化し、ウイルスDNAプラスミドpadEasy-1と共にE. coli株BJ5183に共トランスフォームした。組換え体をカナマイシンで選択し、制限酵素分析によってスクリーニングした。その後、組換えアデノウイルスコンストラクトをPac Iで開裂してその逆向き末端反復配列(ITR)を暴露し、293A細胞にトランスフェクトしてウイルス粒子を生成した。組換えアデノウイルスの力価を組織培養感染価(TCID50)法で測定した。
【0102】
ヒトCD40リガンドcDNAテンプレートを用いるヒトΔCtΔTmCD40L+スペーサーの増幅のためのプライマーを以下に記載する。
ヒトΔCtΔTmCD40L+スペーサー順向きプライマー(HCD40LSPF)(イタリック体(小文字)はCD40L配列):
5'- CCG CTCGAGAACGACGCACAAGCACCAAAATCAGTGTATCTTcatagaaggttggacaag-3'
(SEQ ID NO : 30)
ヒトCD40L逆向きプライマー(HCD40LR)
5'-CCCTCTAGA TCAGAGTTTGAGTAAGCCAAAGGAC-3'(SEQ ID NO : 31)
【0103】
これらのプライマーはヒトCD40Lの47-261をコードするΔCtΔTmCD40L+スペーサーを増幅する。順向きプライマーHCD40LSPFは転写ユニットの腫瘍抗原とCD40リガンド(hCD40L)との間に位置する10残基スペーサー(LENDAQAPKS;1文字コード;SEQ ID NO : 19)をコードする。順向きおよび逆向きプライマー(SEQ ID NO : 30および31)、およびテンプレートとしてプラスミドpDC406-hCD40Lを使用してPCRを実施してPCRフラグメント“space+ΔCtΔTmCD40L(ヒト)”を得、これをXbaI(TCTAGA)およびXho I(CTCGAG)での制限エンドヌクレアーゼ消化後、プラスミドpcDNA-sig-ecdhMUC1に挿入した。HindIII-XbaI 制限を用いてpCDNA3TOPO からsig-ecdhMUC1/ΔCtΔTmCD40L(ヒト)をコードするDNAを切り出し、これをpShuttle-CMV(Murphyら, Prostate 38: 73-78, 1999)のCMVプロモーターの下流に挿入した。このベクターをpShuttle sig-ecdhMUC1/ΔCtΔTmCD40L(ヒト)と記載する。ヒトCD40リガンド配列の上流にecdhMUC1を含有させるためのpShuttle sig-ecdhMUC1/ΔCtΔTmCD40L(ヒト)の修飾を、本質的にマウスCD40リガンドをコードするベクターに関して前記したようにして行った。従って転写ユニットsig-ecdhMUC1-ΔCtΔTmCD40L(ヒト)はカッパ分泌シグナル、次いでヒトMUC1の細胞外ドメイン、次いで10アミノ酸リンカー(NDAQAPK;SEQ ID NO : 19)、次いでヒトCD40リガンド残基47-261をコードする。
【0104】
別の方法では、ヒト成長ホルモンシグナル配列MATGSRTSLLLAFGLLCLPWLQEGSA(1文字アミノ酸コード)(SEQ ID NO : 32)をコードするDNAをカッパ鎖シグナル配列の代わりに使用してもよい。
【0105】
2.MUC1トランスジェニックマウスにおけるMUC1に対する不顕性の克服
a)アデノウイルス感染DCのサイトカイン生成
骨髄由来DCをhMUC-.Tgトランスジェニックマウスから、アデノウイルスベクターへの暴露の48時間後に回収した。MOI 100で細胞をベクターに暴露し、24ウェルプレートに2×105細胞/mlで播種した。37℃で24時間インキュベートした後、上清(1ml)を回収し、遠心分離してデブリを除去した。培養液に放出されたマウスIL-12またはIFN-ガンマのレベルの評価を、マウスIL-12 p70またはIFN-ガンマR & Dシステムキットを用いる酵素結合免疫吸着アッセイ(ELISA)によって行った。
【0106】
Ad-sig-ecdmMUC1-ΔCtΔTCD40L(マウス)ベクターと接触させた骨髄由来DCにより、ベクターへの暴露の48時間後にhMUC-.Tgトランスジェニックマウスから回収したDCからのインターフェロン・ガンマおよびIL-12サイトカインのレベルが有意に上昇した。これと対照的に、cMUC1の細胞外ドメインをコードするが分泌型のCD40Lに融合させていないアデノウイルスベクターで免疫化した動物由来の再刺激DCからは実質的にサイトカインは検出されなかった。これらの結果はecdhMUC1/ecdmCD40L(マウス)融合タンパク質が機能性3量体を形成し、DC上のCD40受容体に結合することを示している。
【0107】
b)Ad-sig-ecdhMUC1-HSF1-ΔCtΔTmCD40L-HISから発現されるecdhMUC1-HSF1-ΔCtΔTmCD40L融合タンパク質による3量体形成の評価
ecdhMUC1-HSF1-ΔCtΔTmCD40L-HIS融合タンパク質の3量体化を、Ad-sig-ecdhMUC1-HSF1-ΔCtΔTmCD40L-HISベクターで形質転換した細胞からの放出に従って評価した。発現された融合タンパク質を、Hisタグ精製キットを用いてベクターに暴露した293細胞の上清から精製した。非変性ゲル電気泳動は、分子量が3量体の形成と矛盾しないことを示した。
【0108】
c)MUC1発現癌細胞の確立に対するAd-sig-ecdhMUC1-ΔCtΔTmCD40Lベクター注射の影響
Ad-sig-ecdhMUC1-ΔCtΔTmCD40L (マウス)ベクターを皮下注射したhMUC1.TgマウスはhMUC1ポジティブLL2/LL1hMUC1マウス癌細胞による移植に対して耐性を示した。ベクターを注射しないコントロール動物は同じ細胞の増殖に対して耐性を示さなかった。また、Ad-sig-ecdhMUC1/ecdCD40L (マウス)ベクターを注射したhMUC1.Tgマウスは、MUC1を発現しない親細胞系(LL2/LL1)による移植に対して耐性を示さなかった。
【0109】
ecdhMUC1-ΔCtΔTmCD40L(マウス)タンパク質を静脈内注射したhMUC1.TgマウスはhMUC1ポジティブLL2/LL1hMUC1マウス癌細胞による移植に対して耐性を示さなかった。更に、Ad-sig-ecdhMUC1-ΔCtΔTmCD40L(マウス)ベクターを注射したhMUC1.Tgマウスはコントロールベクター注射し、その後LL2/LL1hMUC1細胞系を投与したマウスより長く生存した。
【0110】
3.不顕性の崩壊の根底にある細胞機構
a)ワクチン接種マウスvs非ワクチン接種マウスからのサイトカイン放出
脾臓CD8+ Tリンパ球の集団を、Ad-sig-ecdhMUC1-ΔCtΔTmCD40L(マウス)ベクター投与の7日後、CD4+抗体をコーティングしたビーズを用いてCD4+ Tリンパ球を涸渇させることによって得た。単離したCD8+ Tリンパ球は、コントロールベクター(MUC1未含有)を投与したMUC1.Tgマウス由来のCD8+ T細胞の2,000倍高いレベルのインターフェロンガンマを放出した。
【0111】
b)細胞傷害性アッセイ
Ad-sig-ecdhMUC1-ΔCtΔTmCD40L(マウス)ベクターの投与7日後のhMUC1.Tgマウスから回収した脾臓T細胞をhMUC1抗原ポジティブLL2/LL1hMUC1癌細胞と共にインビトロで7日間培養した。刺激した膵臓T細胞を種々の比率のhMUC1ポジティブLL2/LL1hMUC1癌細胞またはhMUC1ネガティブLL2/LL1癌細胞と混合した。結果は、Ad-sig-ecdhMUC1-ΔCtΔTmCD40L(マウス)ベクター接種マウス由来のT細胞がhMUC1を発現する癌細胞にだけ細胞傷害性を有することを示した。
【0112】
c)Ad-sig-ecdhMUC1-ΔCtΔTmCD40Lベクター注射によってhMUC1特異的T細胞の増殖に対する耐性が克服される
インビトロで骨髄細胞から得たDCをAd-sig-ecdhMUC1-ΔCtΔTmCD40L(マウス)ベクターに48時間暴露した。脾臓CD8+ T細胞(ベクターを注射しない、またはAd- sig-ecdhMUC1-ΔCtΔTmCD40L(マウス)ベクターを皮下注射した7日後のhMUC1.Tgトランスジェニックマウスから採取)を1/1の割合でAd- sig-ecdhMUC1/ecdCD40L(マウス)ベクター感染DCと混合した。ERK1/EK2タンパク質(Ras/MAPKシグナリング経路の終点)は、Ad-sig-ecdhMUC1-ΔCtΔTmCD40L(マウス)ベクター感染DCへのインビトロでの45分間の暴露後にAd-sig-ecdhMUC1-ΔCtΔTmCD40Lベクター注射hMUC1.Tgトランスジェニックマウスから単離したCD8+ T細胞においてリン酸化された。これに対して未接種hMUC1.Tgマウス由来のCD8ポジティブT細胞ではERK1およびERK2タンパク質のリン酸化の上昇は観察されなかった。これらの結果は、Ad-sig-ecdhMUC1-ΔCtΔTmCD40L(マウス)ベクターを接種したMUC1.Tgトランスジェニックマウス由来のCD8ポジティブT細胞がもはやMUC1に対して不顕性ではなくなったことを示している。
【0113】
4.融合タンパク質およびベクターの生成
腫瘍抗原融合タンパク質を、融合タンパク質をコードする融合遺伝子の発現カセットを保持するアデノウイルスベクターから直接生成した。増殖培地中の80%の細胞密度の生成細胞(例えば293細胞系)を、細胞当たり10-100ウイルス粒子の比率でウイルスベクターに感染させた。感染した細胞を更に48-72時間培養したが、この時点でウイルスベクターは細胞中で増殖し、腫瘍抗原融合タンパク質が細胞中で発現され、培養液中に分泌された。感染した細胞の70-90%が細胞変性効果(CPE)を示した時点でそれらを回収した。細胞培養液を分離回収した。細胞溶解産物を3回の凍結/解凍サイクル処理した。標準的な方法で(19)ウイルス粒子を単離した。アフィニティークロマトグラフィーによって、回収した細胞培養液から腫瘍抗原融合タンパク質を精製した。
【0114】
5.タンパク質ブースト投与による免疫応答の増幅
腫瘍抗原融合タンパク質のブースト投与とアデノウイルス発現ベクターのブースト投与の相対値を評価した。
【0115】
記載のようにhMUC-1.Tg動物にAd-K/ecdhMUC1-ΔCtΔTmCD40Lベクターを皮下投与してプライミングした。10マイクログラムのecdhMUC-1/ecdCD40L融合タンパク質から構成されるタンパク質ブーストを皮下注射した。タンパク質ブースト投与の時期およびベクターとの比較を表2に示す種々の処理群で評価した。
【0116】
【表2】
【0117】
異なる群からの脾臓細胞を単離し、インターフェロンガンマ陽性率(positivity)についてELISPOTアッセイによって評価した。図3に見られるように、最初のベクター注射の1週間後から開始して14日間隔で2回タンパク質を皮下注射した場合、未処理、またはタンパク質ブースト投与を行わずに1もしくは2回ベクターを注射した場合に比較してポジティブT細胞の頻度が大きく上昇した。インターフェロンガンマポジティブT細胞の頻度が次に高かったのはT3群(最初のベクター注射の7日後に1回タンパク質を注射)であった。
【0118】
種々の免疫群における細胞傷害性T細胞の樹立を評価した(図4)。種々の処理群からの脾臓細胞をインビトロで、hMUC-1ポジティブ細胞系(LL1/LL2hMUC-1)を用いて5日間刺激した。CD8 T細胞を単離し、標的細胞(LL1/LL2hMUC-1)と50/1の割合で混合した。細胞傷害性は概ねELISPOTアッセイの結果に従い、T5群が最も高い上昇レベルのLL1/LL2hMUC-1特異的細胞傷害性T細胞活性を示した。T5群からのT細胞で観察された細胞傷害性のレベルはネガティブコントロール群で観察されたものの9倍であった。
【0119】
ELISAで種々の処理群の動物由来の血清を抗ecdhMUC1-ΔCtΔTmCD40L特異的抗体について評価した。簡潔に記載すると、ecdhMUC1-ΔCtΔTmCD40Lタンパク質をコーティングしたマイクロウェルを試験マウス血清と共にインキュベートし、洗浄し、ホースラディッシュペルオキシダーゼにコンジュゲートさせたラット抗マウス2次抗体を用いて、結合したマウス抗体を同定した。
【0120】
図5は、1回のベクター注射および2回のタンパク質注射(14日間隔)での処理によって生じるecdhMUC1-ΔCtΔTmCD40L融合タンパク質に対する抗体のレベルが劇的に上昇することを示している。T5処理後の抗ecdhMUC1-ΔCtΔTmCD40L抗体の増加は他のいずれの処理群のものより2倍高かった。
【0121】
これらのアッセイからの結果は、タンパク質のブースト投与が腫瘍抗原発現細胞に対する細胞傷害性T細胞活性の生成および腫瘍抗原に対する抗体の反応において、ベクターのブースト投与より優れていることを示している。タンパク質のブースト投与に関して総体的に最良の結果が得られるのは、アデノウイルス発現ベクター注射を1回行い、その1週間後にタンパク質を皮下注射してブーストし、2週間後にタンパク質ブーストをもう1度反復した場合であった。
【0122】
ワクチン接種したhMUC-1.Tgマウス由来の血清中の抗体を、癌バイオプシー組織標本への結合に関して評価した。正常な乳組織切片および乳癌組織切片を含む組織マイクロアレイを購入した。Ad-K/ecdhMUC-1//ΔCtΔTm CD40Lベクターで免疫化し、その後ecdhMUC-1//ΔCtΔTm CD40Lタンパク質をブースト投与したトランスジェニックマウス由来の血清と組織を接触させた。アレイを洗浄した後、マウスIgG抗体を認識するホースラディッシュペルオキシダーゼ(HRP)2次抗体に暴露した。コントロールとして血清をまずhMUC-1ドメインの抗原性リピートからのhMUC-1ペプチドに暴露した(タンパク質ブーストに使用したものと同じもの)。
【0123】
ワクチン接種マウス由来の血清は癌性上皮細胞のバイオプシー標本からの乳上皮細胞に結合した。間にある線維芽細胞または間質細胞への結合は観察されなかった。正常マウス由来の血清は反応を示さなかった。
【0124】
Ad-sig-hMUC-1/ecdCD40L で免疫化し、その後タンパク質sc-hMUC-1/ecdCD40L を2回連続して皮下投与したhMUC-1.Tgマウス由来の血清は組織マイクロアレイスライド上でヒト前立腺癌由来のバイオプシー標本と反応した。
【0125】
hMUC-1リピートに対して血清から産生された抗体の特異性を確認するために、hMUC-1リピートからのアミノ酸配列を含有するペプチドを、量を漸増させて、上記のワクチン接種動物由来の血清と混合した。次いで混合物をマイクロアレイスライドに適用し、反応性を評価した。コントロールとして、hMUC-1リピートと同じアミノ酸を有するが配列順序が異なるペプチド(“スクランブルペプチド”)をワクチン接種した動物由来の血清に添加した。hMUC-1ペプチドによって、ワクチン接種動物血清中の抗体の乳癌上皮細胞への結合が阻害された。スクランブルペプチドでは阻害は観察されなかった。これらのことは、ベクタープライム/タンパク質ブーストワクチン接種によって、ヒト乳癌上皮細胞のバイオプシー標本によって発現されるMUC-1と反応性のあるhMUC-1特異的体液性反応が誘導されたことを示している。
【0126】
タンパク質のブースト投与を行ったマウスにおける腫瘍免疫を評価した。記載のようにhMUC-1.Tg動物にAd-K/ecdhMUC1-ΔCtΔTmCD40Lベクターを皮下投与してプライミングするか、またはecdhMUC1-ΔCtΔTmCD40L融合タンパク質を1もしくは2回投与して免疫化した。その後動物をLL2/LL1hMUC-1腫瘍細胞で攻撃感染させた。
【0127】
図6に示すように、Ad-K/ecdhMUC1-ΔCtΔTmCD40Lベクターを接種したマウスは120日より長く生存したが(実線)、Ad-sig-ecdhMUC-1/ecdCD40Lベクターを接種しなかったマウスは全て50日までに死亡した(波線)。これらの結果は、ベクター注射によりhMUC-1.TgマウスにおけるLL2/LL1hMUC-1細胞系増殖の抑制が誘導されたことを示している。
【0128】
hMUC-1抗原に関する腫瘍増殖抑制の特異性を評価するために、LL2/LL1hMUC-1細胞系(hMUC-1抗原陽性)の拒絶をLL2/LL1細胞系(hMUC-1抗原が存在しないこと以外は同じ)と比較した。結果は、アデノウイルスベクターの皮下注射によってLL2/LL1hMUC-1細胞系の増殖は完全に抑制されるが、同じ細胞でMUC-1を発現しないものは抑制されなかったことを示した。
【0129】
ベクターおよびタンパク質投与の組み合わせによって腫瘍増殖抑制を評価した。Ad-sig-ecdhMUC-1/ecdCD40LベクターおよびecdhMUC-1/ecdCD40Lタンパク質を3種の組み合わせでhMUC-1.Tgマウスに投与した後、LL2/LL1hMUC-1腫瘍細胞で攻撃感染した。VVV= Ad-sig-ecdhMUC-1/ΔCtΔTm CD40Lベクターを3回、1、7、および21日目に皮下注射;PPP= ecdhMUC-1/ΔCtΔTm CD40Lタンパク質を3回、1、7、および21日目に皮下注射;またはVPP=Ad-sig-ecdhMUC-1/ΔCtΔTm CD40Lベクターを1回皮下注射した後、7および21日目にecdhMUC-1/ΔCtΔTm CD40Lタンパク質を皮下注射。更に詳細には図7参照。1週間後、マウスに500,000の LL2/LL1hMUC-1肺癌細胞を皮下注射して攻撃感染し、2週間後に500,000のLL2/LL1hMUC-1腫瘍細胞を静脈内注射した。カリパーを用いて複数の時点でその日の皮下腫瘍結節のサイズを測定し、LL2/LL1hMUC-1細胞の増殖に対する種々のワクチンスケジュールの効果を皮下結節として測定した。殺処理後、肺の総重量によって転移を測定した。
【0130】
図7に示すように、あらかじめAd-sig-ecdhMUC-1/ecdCD40Lベクター注射をせずに融合タンパク質を3回注射したところ(PPP)、皮下LL2/LL1hMUC-1腫瘍の発生に対する完全な耐性は誘導されなかった。それに対し、3回連続でベクターを注射するか(VVV)、または1回のベクター注射後にタンパク質を2回注射する(VPP)スケジュールでは、皮下LL2/LL1hMUC-1腫瘍の発生が完全に抑制された。
【0131】
これらのマウスにおけるワクチン接種開始の63日後のhMUC-1特異的抗体のレベルを測定した(図8)。1回のベクター注射後2回連続で融合タンパク質をブースト注射するというスケジュール(VPP)では最も高いレベルのhMUC-1特異的抗体が誘導され、スケジュールVVVでは中程度、そしてVPPでは実質的に効果がなかった。従って、これらの動物における癌治療は抗体反応と幾分逆の関係にある。
【0132】
(定着後の)腫瘍治療プロトコールも評価した。このスケジュールでは、皮下腫瘍(500,000のLL2/LL1hMUC-1)を1日目に投与した。3種のスケジュール(PPP、VPP、およびVVV)を5、12、および26日目実施した。35日目に腫瘍を静脈内投与し、腫瘍の発生(皮下および肺)を49日目に評価した。更なる詳細を図9の説明書きに記載する。
【0133】
図9に示すように、1回のベクター注射とその後2回のタンパク質注射の組み合わせ(VPP)では、定着後の皮下hMUC-1ポジティブ癌細胞腫瘍の増殖が完全に抑制された。3回連続のベクター投与(VVV)ではわずかな治療効果が見られたが、3回連続のタンパク質注射(PPP)ではほとんど、または全く効果が無かった。
【0134】
処置前および処置後(定着前)癌モデルにおける肺転移結節の増殖を図10に示す。図10の前処理の結果(左側のパネル)は、3回連続の融合タンパク質注射(PPP)が肺結節の増殖を抑制しなかったと考えられることを示している。これに対し、スケジュールVVVおよびスケジュールVPPはワクチン接種動物の肺における肺癌の定着を完全に抑制すると考えられた。
【0135】
図10の後処理の結果(右側のパネル)は、1回のベクター注射とその後2回のタンパク質注射の組み合わせ(VPP)では定着したhMUC-1ポジティブ癌細胞の肺結節の増殖が完全に抑制されたことを示している。これに対し、3回連続のベクター投与(VVV)および3回連続のタンパク質注射(PPP)はいくらかの治療効果は示したものの、VPPプロトコールより小さなものだった。
【0136】
これらの結果は、総括的に最良の治療スケジュールはVPPスケジュールであることを示しており、このスケジュールは1回のAd-sig-ecdhMUC-1/ecdCD40Lベクター注射を行い、その1週間後に2回連続のecdhMUC-1/ecdCD40L タンパク質皮下注射を2週間の間隔をおいて行うものである。このプロトコールはムチン抗原に対する抗体(体液性免疫)およびT細胞免疫(細胞性免疫)の誘導を特徴とする。
【0137】
同じタンパク質をコードするアデノウイルス発現ベクターを初期投与した後のブースティングをecdMUC-1/ecdCD40L可溶性タンパク質で行う場合と他の可溶性タンパク質で行う場合を比較するために、MUC-1発現腫瘍(LL2/LL1hMUC-1細胞系)に攻撃感染させたhMUC-1.Tg動物で評価を行った。以下を含有する細菌抽出物で動物をブースティングした: ecdMUC-1/ecdCD40(Ad-sig-ecdMUC-1/ecdCD40Lベクターに感染させた細菌ホスト株由来);キーホールリンペットヘモシアニン(KLH)に結合したecdMUC-1(フロイドの不完全アジュバントと共に、または無しで);PBS;およびコントロール細菌抽出物(Ad-sig-ecdMUC-1/ecdCD40Lベクターに感染していない細菌ホスト株由来)。2回目のタンパク質ブースト投与完了の7日後に腫瘍細胞を投与した。図12に示す結果は、ecdMUC-1/ecdCD40L可溶性タンパク質でのブースティングが他の全ての方法より優れていることを示している。
【0138】
6.HPV E7-CD40リガンド融合タンパク質をコードするアデノウイルスベクターの構築
分泌型CD40リガンドに結合したE7から構成されうる転写ユニット融合タンパク質を発現するアデノウイルスベクターの投与によって免疫を生じさせる方法が近年報告された(Methods of generating immunity by administering and adenoviral vector expressing a transcription unit fusion protein constituting E7 linked to a secretable form of CD40 ligand was recently reported)。Ziangら, "An adenoviral vector cancer vaccine that delivers a tumor-associated antigen/CD40-ligand fusion protein to dendritic cells" Proc. Natl. Acad. Sci (USA)(2003年11月25日発行), 10.1073/pnas.2135379100 (vol. 100(25):15101)。
【0139】
転写ユニットは、ΔCtΔTmCD40Lの上流に完全長のHPV 16型 E7タンパク質をコードするDNA、その上流にHGH遺伝子由来のシグナルペプチドをコードするDNAを含む。ヒト成長ホルモンシグナル配列MATGSRTSLLLAFGLLCLPWLQEGSA(1文字アミノ酸コード)(SEQ ID NO:32)をコードするDNAを調製するために、ホスホリル化したオリゴヌクレオチド(SEQ ID NO:33および34)をアニールさせ、Bgl IIおよびNot1オーバーハングを有する完全長の26アミノ酸HGH配列を生成した。
成長ホルモンシグナル上流鎖(イタリック体(小文字)のコード配列):
5'-GATCT CCACC atg gct aca ggc tcc cgg acg tcc ctg ctc ctg gct ttt ggc ctg ctc tgc ctg ccc tgg ctt caa gag ggc agt gcc GGC -3'(SEQ ID NO: 33)
成長ホルモンシグナル下流鎖:
3'-A GGTGG TAC CGA TGT CCG AGG GCC TGC AGG GAC GAG GAC CGA AAA CCG GAC GAG ACG GAC GGG ACC GAA GTT CTC CCG TCA CGG CCGCCGG -5'(SEQ ID NO:34)
【0140】
上記の上流および下流オリゴ鎖をアニールさせて合成HGHシグナル配列を調製した。オリゴ鎖を50μlのH2O(約3mg/ml)に溶解した。各オリゴ鎖(上流および下流鎖)からの1μlを48μlのアニーリングバッファー(100mM 酢酸カリウム、30mM HEPES-KOH pH 7.4、および2mM Mg-酢酸)に添加し、95℃で4分間、70℃で10分間インキュベートし、約4℃まで徐々に冷却した。標準的な条件下でT4 PNK(ポリヌクレオチドキナーゼ)を用いてアニールさせたDNAをホスホリル化した。
【0141】
Bgl IIおよびNot Iオーバーハングを有するHGHシグナル配列をBgl IIおよびNot Iを介してpShuttle-E7-ΔCtΔTmCD40L(シグナル配列未含有)に挿入し、pshuttle-HGH/E7-ΔCtΔTmCD40Lを得た。以下のように、CD40リガンド配列の上流にHPV-16 E7を挿入し、pShuttle-E7-ΔCtΔTmCD40L(シグナル配列未含有)を調製した:完全長のHPV-16 E7タンパク質をコードする配列を、以下のプライマーを用いてHPVウイルスゲノムからのPCR増幅によって得た:
HPV 16 E7順向きプライマー(SEQ ID NO: 35)
5'-ATTT GCGGCCGC TGTAATCATGCATGGAGA-3'
HPV E7逆向きプライマー(SEQ ID NO: 36)
5-CC CTCGAG TTATGGTTTCTGAGAACAGAT-3'
【0142】
得られた増幅子は5'末端にNot I、3'末端にXho I制限部位を有する、HPV 16 E7をコードするDNAであった。Not I(GCGGCCGC)およびXho I(CTCGAG)を用いて、pShuttleΔCtΔTmCD40L のCMVプロモーターとΔCtΔTMCD40L配列のスペーサーのすぐ5'側との間にE7 DNAを挿入した。プラスミドをpShuttle-E7-ΔCtΔTmCD40L(シグナル配列未含有)と記載し、これを用いてE7の上流にHGHシグナル配列を挿入し、既に記載したようにHGH/E7-ΔCtΔTmCD40Lを生成した。従って転写ユニットHGH/E7-ΔCtΔTmCD40LはHGH分泌シグナル、次いで完全長のHPV16型E7、次いで10アミノ酸リンカー(FENDAQAPKS; SEQ ID NO: 37)、次いでマウスCD40リガンド残基52-260をコードする。
【0143】
完全長HPV16型 E7タンパク質をコードするDNAの上流にマウスIgGカッパ鎖遺伝子のシグナル配列をコードするDNAを含む転写ユニット(“K/E7”)を、HPV16プラスミドおよび以下のプライマーを使用してPCRで生成した:
(プライマー1)5'-ACG ATG GAG ACA GAC ACA Ctc ctg cta tgg gta ctg ctg-3'(SEQ ID NO: 38);
(プライマー2)5'- TC CTG CTA TGG GTA CTG CTG CTC tgg gtt cca ggt tc-3'(SEQ ID NO: 39);
(プライマー3)5'- TG CTC TGG GTT CCA ggt tcc act ggt gac atg cat g-3'
(SEQ ID NO: 40);
(プライマー4)5'- TGG GTT CCA GGT TCC ACT GGT GAC ATG CAT GGA G AT ACA CCT AC-3'(SEQ ID NO: 41);および
(プライマー5)5'- CCG CTC GAG TGG TTT CTG AGA ACA GAT GGG GCA C -3.'(SEQ ID NO: 42)
【0144】
上流カッパシグナル配列を有するK/E7を4ラウンドのPCR増幅によって生成した(第1ラウンド:プライマー4+5;第2ラウンド:プライマー3を添加;第3ラウンド:プライマー2を添加;第4ラウンド:プライマー1を添加)。K/E7をコードするDNAをpcDNATM 3.1 TOPOベクター(Invitrogen, San Diego, CA)にクローニングしてpcDNA-K/E7を生成した。
【0145】
膜貫通ドメインおよび細胞質ドメインを欠失させたマウスCD40リガンドを含むDNAフラグメント(ΔCtΔTmCD40L)を、以下のPCRプライマーを用いてマウスCD40リガンドcDNAプラスミド(pDC406-mCD40L;ATCC)から生成した:
5'-CCG CTCGAG aac gac gca caa gca cca aaa agc AAG GTC GAA GAG GAA GTA AAC CTT C-3'(SEQ ID NO: 43);および
5'-CGCGCCGCGCGCTAGtctagaGAGTTTGAGTAAGCCAAAAGATGAG-3'(SEQ ID NO: 44)(ハイファイデリティー(high fidelity)PCRキット, Roche)
フラグメントΔCtΔTmCD40LをXba IおよびXho I制限エンドヌクレアーゼで消化し、pcDNA-E7にライゲートした。K/E7-ΔCtΔTmCD40LフラグメントをpcDNAベクターから切り出し、Hind IIIおよびXba I部位を用いてpShuttleプラスミドに挿入した(pShuttle K/E7-ΔCtΔTmCD40L)。従って、K/E7-ΔCtΔTmCD40Lフラグメントはカッパ鎖分泌シグナル、次いで完全長HPV16型E7、次いで10アミノ酸リンカー(LQNDAQAPKS; SEQ ID NO: 31)、次いでマウスCD40リガンド残基52-260を含む。
【0146】
膜貫通ドメインを欠失したヒトCD40リガンドに融合したE7をコードするベクターを調製するために、“スペース+ΔCtΔTmCD40L(ヒト)”(上記のように調製)をプラスミドpShuttle-CMV(13)に、Hind III(AAGCTT)およびXho I (CTCGAG)での制限エンドヌクレアーゼ消化後に挿入した。このベクターをpShuttleΔCtΔTmCD40L(ヒト)と記載する。pShuttleΔCtΔTmCD40L(ヒト)を修飾してヒトCD40リガンド配列の上流にHPV-16 E7を含有させたが、これは本質的に上でマウスCD40リガンドをコードするベクターに関して記載したようにして行った。得られたプラスミドをpShuttle-E7-ΔCtΔTmCD40L(ヒト)(シグナル配列未含有)と記載し、これを用いてE7の上流にHGHシグナル配列を挿入し、HGH/E7-ΔCtΔTmCD40L(ヒト)を生成する。従って、転写ユニットHGH/E7-ΔCtΔTmCD40L(ヒト)はHGH分泌シグナル、次いで完全長HPV16型E7、次いで10アミノ酸リンカー(FENDAQAPKS; SEQ ID NO:19)、次いでヒトCD40リガンド残基47-261をコードする。
【0147】
7.ラットHER2(Neu)/CD40Lをコードするアデノウイルスベクターの構築
乳癌の30%で、Her-2-Neu(H2N)増殖因子受容体の過剰発現が術後の再発頻度の増加および生存期間の短縮と関連している。HER2(“H2N”または“rH2N”)遺伝子のラット相当物のトランスジェニックであり、そのためこの遺伝子に寛容であるマウス(Mullerら, Cell 54: 105-115, (1998);Gutら Proc. Natl. Acad. Sci. USA 89: 10578-10582, (1992))を実験用ホストとして使用し、Ad-sig-rH2N/ecdCD40Lベクターでの免疫を評価した。このモデルにおいて、乳特異的転写プロモーター(例えばMMTVプロモーター)の制御下でマウスを正常な非活性化ラットHer-2-Neu遺伝子のトランスジェニックとした。MMTVプロモーターは非変異ラットHer-2-Neu受容体の過剰発現を起こすが、これはヒト乳癌で起こるものと類似している。このモデルでは24週間で原発組織(乳)に触知可能な腫瘍結節、32週間で肺転移が発生する。乳癌は自然発生する。癌はクローン性事象(clonal event)として乳上皮組織で段階的な過程を通して局所的に開始される(同上)。異形成は生後12週で検出できる。乳腺における触知可能な腫瘍は25週で検出でき、乳癌の肺転移は32週までに70%のマウスで確認される(同上)。
【0148】
Ad-sig-rH2N/ecdCD40Lベクターを1または2回(7日間隔)、トランスジェニック動物に皮下投与し、ラットHer-2-Neu抗原に対して免疫応答が誘導されるかどうか試験した。2回のAd-sig-rH2N/ecdCD40Lベクターの皮下注射によりN202(rH2Nポジティブ)マウス乳癌細胞系の増殖に対する完全な耐性が誘導されたが、同じベクターの1回の皮下注射ではrH2NポジティブN202細胞系の増殖を完全に抑えるのに十分な免疫応答は誘導されなかった。ELISPOTアッセイでは、7日間隔で2回Ad-sig-rH2N/ecdCD40Lベクターを皮下注射することにより、マウスにおいてAd-sig-rH2N/ecdCD40Lベクターを1回注射した後に誘導されるrH2N特異的T細胞のレベルより10倍高いレベルのrH2N特異的T細胞が、ワクチン接種したマウスの脾臓において誘導されたことが明らかになった。最終的に、Ad-sig-rH2N/ecdCD40Lベクターのプライム免疫によってNT2細胞に対して誘導される免疫抵抗は、放射線照射したサイトカインポジティブ腫瘍細胞(GMCSF転写ユニットをトランスフェクトしたNTW細胞をミトマイシン処理したもの)を接種したトランスジェニック動物で得られる応答より良好であった。
【0149】
Ad-sig-rH2N/ecdCD40Lベクターの皮下注射を1または2回行ったマウスにおけるrH2N特異的抗体レベルも測定した。以下の図11に示すように、rH2N特異的抗体のレベルはAd-sig-rH2N/ecdCD40Lベクターを2回皮下注射した後の方が1回皮下注射した後より高かった。
【0150】
8.huHER2/CD40Lをコードするアデノウイルスベクターの構築
sig ecdhuHER2/CD40Lをコードするアデノウイルスベクターを以下のように調製した。マウスIgGカッパ鎖METDTLLLWVLLLWVPGSTGD(1文字アミノ酸コード)(SEQ ID NO: 11)をPCR増幅(SEQ ID NO: 12、13、および45)によって調製し、完全長の21アミノ酸マウスIgGカッパ鎖シグナル配列を生成した(SEQ ID NO:12で、開始コドン“ATG”を太字(下線)で示す)。
5'-CCACC ATG GAG ACA GAC ACA CTC CTG CTA TGG GTA CTG CTG-3'
(SEQ ID NO: 12)
5'- TC CTG CTA TGG GTA CTG CTG CTC TGG GTT CCA GGT TC-3'
(SEQ ID NO:13)
順向きプライマー
5'-5'- TG CTC TGG GTT CCA GGT TCC ACT GGT GAC GAA CTC -3'
(SEQ ID NO:45)
ヒトHER2細胞外ドメインの順向きプライマー
5'- TCC ACT GGT GAC GAACTCACCTACCTGCCCACCAATGC-3'
(SEQ ID NO:46)
ヒトHER2細胞外ドメインの逆向きプライマー
5'- GGAGCTCGAG GGCTGGGTCCCCATCAAAGCTCTC-3'
(SEQ ID NO:47)
【0151】
上流カッパシグナル配列を有するsig-ecdhHER2を4ラウンドのPCR増幅によって生成する(第1ラウンド:プライマーSEQ ID NO:46および47;第2ラウンド:プライマーSEQ ID NO:45および47;第3ラウンドSEQ ID NO:13および47;第4ラウンド:プライマーSEQ ID NO:12および47)。sig-ecdhHER2をコードするDNAをpcDNATM 3.1 TOPOベクター(Invitrogen, San Diego, CA)にクローニングしてpcDNA-sig-ecdhHER2を生成できる。MUC-1/CD40リガンド発現ベクターに関して記載した更なるクローニング段階をHER2/CD40リガンド発現ベクターにも適用する。
【0152】
この領域、CD40リガンドに融合させるべきHER2細胞外ドメインは2つのCTLエピトープを含有する;1つはHLA-A2ペプチド、K I F G S L A F L(SEQ ID NO:48)であり、アミノ酸369-377に相当する。このペプチドはHER2発現癌患者において一時的なペプチド特異的免疫を引き起こした。Knutsonら, Immunization of cancer patients with a HER-2/neu, HLA-A2 peptide, Clin Cancer Res. 2002 May;8(5):1014-8p369-377参照。第2のエピトープはE L T Y L P T N A S(SEQ ID NO :49)(HER2残基63-71)であり、HER2発現腫瘍細胞に対する免疫の生成にも有用であった。Wang ら, Essential roles of tumor-derived helper T cell epitopes for an effective peptide-based tumor vaccine, Cancer Immun. 2003 Nov 21;3:16参照。HER2 ecdの領域はB細胞エピトープ、P L H N Q E V T A E D G T Q R C E K C S K P C(SEQ ID NO: 50)(HER2の316-339位)も含有する。Dakappagari ら, Chimeric multi-human epidermal growth factor receptor-2 B cell epitope peptide vaccine mediates superior antitumor responses, J Immunol. 2003 Apr 15;170(8):4242-53参照。
【0153】
本明細書に記載する全ての特許および文献は本発明が属する分野の技術者のレベルを示すものである。全ての特許および文献は、個々の文献が特定かつ個別に参照によって組み込まれることが示されるのと同じ程度まで、参照により本明細書に組み込まれる。
【0154】
本明細書に例証的に記載する本発明は、任意の要素(単数または複数)、制限(単数または複数)無しで好適に実施してもよいが、それらについては本明細書で特に開示しない。従って、例えば本明細書においてそれぞれの場合で、“〜を含む”、“本質的に〜からなる”、および“〜からなる”という用語の任意のものを他の2つのいずれかで置き換えてもよい。使用した用語および表現は制限ではなく説明のための用語として使用されるものであり、それらの用語および表現の使用に際して明示および記載する特徴と同等のものまたはそれらの一部のいずれを排除することも意図しないが、認識されるように、種々の修飾が請求する本発明の範囲内で可能である。従って、好ましい態様および必要に応じた(optional)特徴によって本発明についての特定の開示を行ったが、当業者は本明細書に開示するコンセプトの修飾および変更を行ってもよく、それらの修飾および変更は添付する請求の範囲に定義する本発明の範囲内にあると見なされることは理解すべきである。
【0155】
他の態様を添付の請求の範囲内に記載する。
【図面の簡単な説明】
【0156】
図面の簡単な説明
【図1A】図1は、ヒトMUC1をコードするヌクレオチド配列である(SEQ ID NO : 1)。
【図1B】図1は、ヒトMUC1をコードするヌクレオチド配列である(SEQ ID NO : 1)。
【図2】図2は、ヒトMUC1のアミノ酸配列である(SEQ ID NO : 2)。
【図3】図3は、ELISAスポットアッセイで得られたインターフェロンガンマのレベルを示す。
【図4】図4は、アデノウイルス発現ベクターAd-K/ecdhMUC1-ΔCtΔTmCD40Lでプライミングし、発現ベクターまたは成熟融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lのいずれかを皮下にブースト投与したMUC-1トランスジェニック動物(hMUC-1.Tg)由来のT細胞傷害性のレベルを示す。
【図5】図5は、アデノウイルス発現ベクターAd-K/ecdhMUC1-ΔCtΔTmCD40Lでプライミングし、発現ベクターまたは成熟融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lのいずれかを皮下にブースト投与したMUC-1トランスジェニック動物(hMUC-1.Tg)の血清中の融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lに対する抗体のレベルを示す。
【図6】図6は、アデノウイルス発現ベクターAd-K/ecdhMUC1-ΔCtΔTmCD40Lを投与したMUC-1トランスジェニック動物(hMUC-1.Tg)におけるMUC1発現腫瘍細胞(LL2/LL2hMUC-1)の増殖レベルを示す。
【図7】図7は、Ad-sig-ecdhMUC-1/ΔCtΔTmCD40LベクターおよびecdhMUC-1/ΔCtΔTm CD40Lタンパク質を接種した動物における腫瘍の予防を示す。
【図8】図8は、ワクチン接種開始の63日後のワクチン接種マウスにおけるhMUC-1特異的抗体のレベルを示す。
【図9】図9は、Ad-sig-ecdhMUC-1/ΔCtΔTmCD40LベクターおよびecdhMUC-1/ΔCtΔTm CD40Lタンパク質で免疫化した動物における(定着後の)皮下腫瘍療法を示す。
【図10】図10は、図9に記載するように処理した動物における(定着後の)肺転移腫瘍結節療法を示す。
【図11】図11は、MUC1発現腫瘍の発生に抵抗する動物の能力に関する、Ad-sig-ecdhMUC-1/ecdCD40Lベクターの1回皮下投与後の種々のブースト計画の比較である。
【技術分野】
【0001】
発明の属する分野
本発明は、CD40リガンドに融合した抗原を含む分泌可能な融合タンパク質を発現する発現ベクターを用いて抗原に対する免疫を確立させる方法に関する。本発明はまた、発現ベクターでのプライミングおよびタンパク質抗原でのブースティングの免疫化スキームにも関する。本発明はまた、生成細胞系においてベクターとタンパク質抗原を同時に生成する方法にも関する。
【背景技術】
【0002】
発明の背景
以下の本発明の背景に関する記載は読者の本発明の理解を容易にするために提供するものであり、本発明に対する先行技術を記述または構成すると認めるものではない。本出願は米国特許出願第60/529,016号(2003年12月11日出願)(図面を含めてその全体が本明細書に組み込まれる)に対する優先権を主張するものである。本願に関連する出願はPCT/US03/36237(2003年11月12日出願)“アデノウイルスベクターワクチン”、および米国特許仮出願60/524,925号(2003年11月24日出願)、 60/525,552号(2003年11月25日出願)、および60/529,015号(2003年12月11日出願)(これらは全て、図面も含めてその全体が参照により本明細書に組み込まれる)である。
【0003】
樹状細胞(DC)を含む抗原提示細胞(APC)の活性化およびそれに続く抗原提示細胞による相当抗原の担持は、癌細胞に対するT細胞依存型免疫応答の生成に必須の段階である。腫瘍抗原での活性化および担持が起こると、DCは所属リンパ節(LN)に移動し、T細胞に対して抗原を提示する。非常に多くの場合、これらのAPCは、腫瘍抗原を認識する能力のあるT細胞クローンが最適に活性化および増殖するのに必要な表面活性化分子を不十分な量しか発現しない。Shortman, ら, Stem Cells 15:409-419, 1997参照。
【0004】
APCの表面上で共刺激分子が発現していない状態でナイーブT細胞へ抗原が提示されると、T細胞は不顕性(anergy)となる。Steinbrinkら, Blood 99: 2468-2476, 2002参照。更に、CD4+T細胞の助力なしでのDCによるクロスプレゼンテーションでは、末梢での所属LNにおけるAg特異的T細胞の欠失が起こる。Kusuharaら, Eur J Immunol 32:1035-1043, 2002参照。これに対して、CD4+T細胞の助力があれば、DCはT細胞をクロスプライム(cross-prime)する機能的能力を習得し、それによってエフェクターT細胞のクローン増殖が起こる。Gunzerら, Semin Immunol 13:291-302, 2001参照。このCD4+T細胞の助力はCD40-CD40リガンド(CD40L)相互作用で置き換えることができる。Luftら, Int Immunol 14:367-380, 2002参照。CD40Lは33kDaのII型膜タンパク質で、TNF遺伝子ファミリーのメンバーであり、TCRの活性化(engagement)後にCD4+T細胞上で一時的に発現される。Skovら, J Immunol. 164: 3500-3505, 2000参照。
【0005】
インビボにおいてDCが抗腫瘍免疫応答を生じる能力は多くの動物腫瘍モデルで報告されている。Pagliaら, J Exp Med 183: 317-322, 1996;Zitvogelら, J Exp Med. 183: 87-97, 1996参照。しかしながら、DCが介在する免疫誘導には多くの治療上の困難がある。抗原提示細胞が好適な接着分子およびケモカイン受容体を発現して第2のリンパ器官にDCを誘引し、T細胞をプライミングするようにさせることを確実にすることは困難であると考えられている。Fongら, J Immunol. 166: 4254-4259, 2001;Markowiczら, J Clin Invest. 85: 955-961, 1990;Hsuら, Nat Med. 2: 52-58, 1996;Nestleら, Nat Med. 4: 328-332, 1998; Murphyら, Prostate 38: 73-78, 1999;Dhodapkarら, J Clin Invest. 104: 173-180, 1999参照。
【発明の開示】
【課題を解決するための手段】
【0006】
発明の概要
第一の観点では、本発明は抗原に対する免疫応答の確立における融合タンパク質の使用に関する。好ましい態様では、分泌可能な融合タンパク質をコードする発現ベクターを投与することによって抗原に対する免疫応答を得る。ベクターは抗原およびCD40リガンドを含む分泌可能な融合タンパク質をコードする転写ユニットを含む。融合タンパク質の投与はベクター投与の前、同時、または後に行う。好ましくは融合タンパク質はベクター投与後に投与する。
【0007】
ある方法では、融合タンパク質転写ユニット中の抗原をコードする配列はCD40リガンドをコードする配列の5'側にある。別の方法では、融合タンパク質転写ユニット中のCD40リガンドをコードする配列は抗原をコードする配列の5'側にある。好ましい態様では、CD40リガンドはその膜貫通ドメインの全部または一部を欠失している。
【0008】
抗原は、個体において免疫応答が生じうる抗原のいずれでもよい。好ましい態様では、抗原は腫瘍抗原である;腫瘍抗原はヒト乳頭腫ウイルスのE6またはE7タンパク質である;腫瘍抗原はムチン抗原であり、MUC1、MUC2、MUC3A、MUC3B、MUC4、MUC5AC、MUC5B、MUC6、MUC7、MUC8、MUC9、MUC12、MUC13、MUC15、およびMUC16からなる群から選択することができる;ムチン抗原はMUC1由来である;ヒト上皮増殖因子(EGF)様受容体(例えばHER1、HER2、HER3、およびHER4)、抗原は感染性物質抗原である;感染性物質抗原はウイルス抗原である;感染性物質ウイルス抗原はヒト乳頭腫ウイルス由来である;ウイルス抗原はヒト乳頭腫ウイルスのE6またはE7タンパク質である。
【0009】
別の観点では、本発明は腫瘍抗原を発現する癌に罹患した個体を治療する方法を提供する。方法は腫瘍抗原およびCD40リガンドを含む分泌可能な融合タンパク質をコードする転写ユニットを含む発現ベクターを投与することを含む。融合タンパク質はベクター投与の前、同時、または後に投与する。好ましくは融合タンパク質はベクター投与後に投与する。
【0010】
更なる観点では、本発明は被験体において感染性物質に対する免疫を生じさせる方法を提供する。方法は感染性物質抗原およびCD40リガンドを含む分泌可能な融合タンパク質をコードする転写ユニットを含む発現ベクターを投与することを含む。融合タンパク質はベクター投与の前、同時、または後に投与する。好ましくは融合タンパク質はベクター投与後に投与する。
【0011】
更に別の観点では、本発明は同じホスト細胞の生成系においてベクターおよび融合タンパク質を共に生成する方法に関する。好ましい態様では、融合タンパク質はワクチン接種によって免疫を生じさせるのに使用されるのと同じベクターから発現される。この方法において、ベクターおよび融合タンパク質の両方を、一つの生成系で同時に生成できる。
【0012】
好ましい態様では、発現ベクターはウイルス発現ベクターまたは非ウイルス発現ベクターであってもよい;発現ベクターはアデノウイルスベクターであってもよい;ベクターを便宜に皮下投与してもよい;ベクターを連続的に投与して免疫応答を増加させてもよい;シグナル配列は融合タンパク質の分泌のために融合タンパク質の上流に位置してもよい;抗原に対する免疫は長期にわたって持続し、抗原発現細胞に対する細胞傷害性CD8+ T細胞の生成および抗原に対する抗体の産生を伴ってもよい;転写ユニットは抗原とCD40リガンドとの間にリンカーをコードする配列を含んでもよい;好適なリンカーの長さおよび組成は種々であってもよい;発現ベクターは転写ユニットの転写を制御するためのヒト・サイトメガロウイルス・プロモーター/エンハンサーを含んでもよい;そしてCD40リガンドはヒトCD40リガンドであってもよい。
【0013】
本明細書で使用する略号には以下がある:“Ad”(アデノウイルス);“sig”(シグナル配列);および“ecd”(細胞外ドメイン)。
【0014】
これらおよび他の態様を以下に詳述する。
【発明を実施するための最良の形態】
【0015】
図面の簡単な説明
図1は、ヒトMUC1をコードするヌクレオチド配列である(SEQ ID NO : 1)。
【0016】
図2は、ヒトMUC1のアミノ酸配列である(SEQ ID NO : 2)。
【0017】
図3は、ELISAスポットアッセイで得られたインターフェロンガンマのレベルを示し、これはアデノウイルス発現ベクターAd-K/ecdhMUC1-ΔCtΔTmCD40Lでプライミングし、発現ベクターまたは成熟融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lのいずれかを皮下にブースト投与したMUC-1トランスジェニック動物(hMUC-1.Tg)由来の脾臓細胞を使用して行った。種々の処理群には、タンパク質のブースト投与を週1回のベクター注射を2回行った7日後(T1)、週1回のベクター注射2回の2週間後(T2)、1回のベクター注射の1週間後(T3)、および1回のベクター注射の2週間後(T4)に行ったものがある。T5では1回のベクター注射の7日後に開始して2回のタンパク質の皮下注射(2週間間隔で投与)を行った。T1では2回のベクター注射を行い、タンパク質は注射しなかった。
【0018】
図4は、アデノウイルス発現ベクターAd-K/ecdhMUC1-ΔCtΔTmCD40Lでプライミングし、発現ベクターまたは成熟融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lのいずれかを皮下にブースト投与したMUC-1トランスジェニック動物(hMUC-1.Tg)由来のT細胞傷害性のレベルを示す。種々の処理群を図3に示す。
【0019】
図5は、アデノウイルス発現ベクターAd-K/ecdhMUC1-ΔCtΔTmCD40Lでプライミングし、発現ベクターまたは成熟融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lのいずれかを皮下にブースト投与したMUC-1トランスジェニック動物(hMUC-1.Tg)の血清中の融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lに対する抗体のレベルを示す。種々の処理群は図3に記載するものである。ELISAで抗体を検出した。融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lをコーティングしたマイクロウェルプレートを血清と反応させ、洗浄し、結合したマウス抗体をホースラディッシュペルオキシダーゼとコンジュゲートさせたラット抗マウス抗体を用いて検出した。
【0020】
図6は、アデノウイルス発現ベクターAd-K/ecdhMUC1-ΔCtΔTmCD40Lを投与したMUC-1トランスジェニック動物(hMUC-1.Tg)におけるMUC1発現腫瘍細胞(LL2/LL2hMUC-1)の増殖レベルを、融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lの1回または2回の連続投与と比較して示す。
【0021】
図7は、Ad-sig-ecdhMUC-1/ΔCtΔTmCD40LベクターおよびecdhMUC-1/ΔCtΔTm CD40Lタンパク質を接種した動物における腫瘍の予防を示す。VVV= Ad-sig-ecdhMUC-1/ΔCtΔTm CD40Lベクターの皮下注射3回(1日目、7日目、および21日目に投与);PPP=ecdhMUC-1/ΔCtΔTm CD40Lタンパク質の皮下注射3回(1日目、7日目、および21日目に投与);またはVPP= Ad-sig-ecdhMUC-1/ΔCtΔTm CD40Lベクターを1回皮下注射後、7日目および21日目にecdhMUC-1/ΔCtΔTm CD40Lタンパク質を皮下注射。1週間後(28日目)、マウスに500,000のLL2/LL1hMUC-1肺癌細胞を皮下注射した。2週間後(42日目)、500,000のLL2/LL1hMUC-1腫瘍細胞を静脈内投与し、尾静脈を介してマウスを試験した。複数回のベクター投与(単独で、またはその後タンパク質をブースター投与)はマウスにおけるヒト腫瘍の樹立の予防に有効であった。
【0022】
図8は、ワクチン接種開始の63日後のワクチン接種マウスにおけるhMUC-1特異的抗体のレベルを示す。VVV= Ad-sig-ecdhMUC-1/ΔCtΔTm CD40Lベクターの皮下注射3回(1日目、7日目、および21日目に投与);PPP= ecdhMUC-1/ΔCtΔTm CD40Lタンパク質の皮下注射3回(1日目、7日目、および21日目に投与);またはVPP= Ad-sig-ecdhMUC-1/ΔCtΔTm CD40Lベクターを1回皮下注射後、7日目および21日目にecdhMUC-1/ΔCtΔTm CD40Lタンパク質を皮下注射。Ad-sig-ecdhMUC-1/ΔCtΔTmCD40Lベクターを1回皮下注射した後、ecdhMUC-1/ΔCtΔTmCD40Lタンパク質を連続2回、7および21日目に皮下注射するというスケジュールで、最も高いレベルのhMUC-1特異的抗体が誘導された。
【0023】
図9は、Ad-sig-ecdhMUC-1/ΔCtΔTmCD40LベクターおよびecdhMUC-1/ΔCtΔTm CD40Lタンパク質で免疫化した動物における(定着後の)皮下腫瘍療法を示す。VVV= Ad-sig-ecdhMUC-1/ΔCtΔTm CD40Lベクターの皮下注射3回(5日目、12日目、および26日目に投与);PPP= ecdhMUC-1/ΔCtΔTm CD40Lタンパク質の皮下注射3回(5日目、12日目、および26日目に投与);またはVPP= Ad-sig-ecdhMUC-1/ΔCtΔTm CD40Lベクターを1回皮下注射後、12日目および26日目にecdhMUC-1/ΔCtΔTm CD40Lタンパク質を皮下注射。皮下腫瘍(500,000のLL2/LL1hMUC-1)を1日目に投与し、ワクチン接種を5日目に実施した。40日目に腫瘍を静脈内投与し、54日目に腫瘍の発生(皮下および肺)を評価した。
【0024】
図10は、図9に記載するように処理した動物における(定着後の)肺転移腫瘍結節療法を示す。左のパネル:結果は皮下腫瘍の予防と同様であり、スケジュールVVVおよびVPPが最も有効であった。右のパネル:1回のベクター注射およびその後の2回のタンパク質注射の組み合わせ(VPP)では、定着したhMUC-1ポジティブ癌細胞の肺結節の増殖が完全に抑制された。
【0025】
図11は、MUC1発現腫瘍の発生に抵抗する動物の能力に関する、Ad-sig-ecdhMUC-1/ecdCD40Lベクターの1回皮下投与後の種々のブースト計画の比較である。細菌抽出物中のecdhMUC-1/ecdCD40Lタンパク質;(フロイドの不完全アジュバントと共に、または無しで)キーホールリンペットヘモシアニン(KLH)に結合したecdhMUC-1;PBS(リン酸バッファー);およびコントロール細菌抽出物(Ad-sig-ecdhMUC-1/ecdCD40Lベクターに感染していない細菌ホスト株)。
【0026】
発明の詳細な説明
本発明のある観点によれば、発現ベクターを用いて抗原に対する免疫応答を生じさせる方法を提供する。ベクターは抗原およびCD40リガンドを含む分泌可能な融合タンパク質をコードする転写ユニットを含む。好ましい態様では、転写ユニットはアミノ末端側から、分泌シグナル配列、抗原、リンカー、および分泌型CD40リガンドを含む。好ましい態様では、分泌型CD40リガンドはその膜貫通ドメインの全部または一部を欠失している。
【0027】
好ましい方法では、まず個体にベクターを1回以上投与し、初期免疫応答を生成させる。ベクター投与後、有効量の融合タンパク質を投与し、抗原に対する免疫応答をブーストし、ベクターの単独投与で得られるより高いものとする。
【0028】
融合タンパク質の投与に関する“有効量”という用語は発現ベクター単独で得られるものより高い免疫応答となる量をいう。最適な結果を得るためには、一般に投与間の時間間隔が必要である。免疫応答の増加はT細胞活性または抗体産生の増加として測定してもよい(例えば図3-5参照)。一般に、ベクター投与とタンパク質ブースト投与間は少なくとも1週間が有効であるが、より短い間隔でもよい。投与間の有効な間隔は1週間から12週間、またはそれより長くてもよい。複数回のブースト投与を、1-12週間、またはそれ以上の期間をおいて行ってもよい。
【0029】
免疫応答をブーストするために融合タンパク質を使用することにより、複数回の注射に対する過敏性を引き起こしうる発現ベクターの連続的な投与の必要が回避される。融合タンパク質の抗原部分は好ましくは最初の投与に使用される発現ベクターの転写ユニットにコードされる融合タンパク質である。しかしながら、融合タンパク質の抗原部分は、発現ベクターの抗原およびブーストに使用される融合タンパク質の抗原に共通する抗原決定基またはエピトープを少なくとも一つ共有すれば、コードされる抗原と異なっていてもよい。
【0030】
融合タンパク質は哺乳動物細胞系で調製してもよく、これはベクターに相補的である。例えばアデノウイルスの場合、細胞系は初期領域1(Early Region 1;E1)遺伝子を含む293細胞であってもよく、またE1置換型組換えアデノウイルスの増殖をサポートしうる。アデノウイルスベクターが生成細胞に感染すると、ウイルスベクターはウイルスの複製サイクル後、自己増殖する。しかしながら、発現カセット中のウイルスベクターによって行われる目的の遺伝子はウイルスの増殖過程で発現する。これを利用して、ベクターを生成するのと同じ系においてベクターにコードされる融合タンパク質の調製を行うことができる。ベクターおよび融合タンパク質の生成を生成系で同時に行う。このように生成されたベクターおよびタンパク質を異なる過程で更に単離および精製できる。
【0031】
融合タンパク質の投与は非経口、例えば血管内、静脈内、動脈内、筋肉内、皮下などで行ってもよい。投与は経口、経鼻、直腸内、経皮、またはエアロゾルによる吸入で行ってもよい。タンパク質ブーストはボーラス、または点滴(slowly infused)で投与してもよい。タンパク質ブーストは皮下投与が好ましい。
【0032】
融合タンパク質ブーストは、生じる免疫応答を増強させるためにアジュバントと共に調製してもよい。本明細書で使用する“アジュバント”という用語は、ワクチンと共に投与するとワクチンに対する免疫応答が増強するような化学物質を意味する。アジュバントは、アジュバントが免疫原または抗原と化学的に結合しない点においてキャリアータンパク質とは区別される。アジュバントは当該分野で知られており、例えば以下がある:鉱油エマルジョン(米国特許第4,608,251号。上記)、例えばフロイドの完全または不完全アジュバント(Freund, Adv. Tuberc. Res. 7:130 (1956); Calbiochem, San Diego Calif.)、アルミニウム塩、特に水酸化アルミニウムまたはALLOHYDROGEL(米国食品医薬品庁によってヒトへの使用が承認されている)、ムラミルジペプチド(MDP)およびその類似体、例えば[Thr1 ]-MDP(ByersおよびAllison, Vaccine 5:223 (1987))、モノホスホリルリピッドA(Johnsonら, Rev. Infect. Dis. 9:S512 (1987))など。
【0033】
当該分野で知られる方法を用いて、融合タンパク質をマイクロカプセル化またはマクロカプセル化した形態で投与することができる。融合タンパク質をカプセル化して例えばリポソーム(例えばGarconおよびSix, J. Immunol. 146:3697 (1991)参照)、ウシロタウイルスの内部キャプシドタンパク質(Redmondら, Mol. Immunol. 28:269 (1991))、Quil Aのようなサポニンから構成される免疫刺激分子(ISCOMS)(Moreinら, Nature 308:457 (1984);Moreinら, Immunological Adjuvants and Vaccines (G. Gregoriadisら編) pp.153-162, Plenum Press, NY (1987))、または(例えばラクチド/グリコリドコポリマーから構成される)徐放生分解性マイクロスフェア(O'Haganら, Immunology 73:239 (1991);O'Haganら, Vaccine 11:149 (1993))としてもよい。
【0034】
L-121のようなPLURONICブロックコポリマーと共に調製し、TWEEN80のような界面活性剤で安定化させたスクワレンまたはスクワランエマルジョンを含むリピッドマイクロスフェアの表面に融合タンパク質を吸収させてもよい(AllisonおよびByers, Vaccines: New Approaches to Immunological Problems (R. Ellis編) pp. 431-449, Butterworth-Hinemann, Stoneman N.Y. (1992)参照)。マイクロカプセル化またはマクロカプセル化した融合タンパク質はアジュバントを含有してもよい。
【0035】
融合タンパク質をキャリアーまたは外来分子、例えばタンパク質ブーストを投与すべき個体にとって外来性であるキャリアータンパク質にコンジュゲートさせてもよい。免疫応答を活性化し、本明細書に記載する融合タンパク質にコンジュゲートすることができる外来タンパク質には分子量が少なくとも約20,000ダルトン、好ましくは少なくとも約40,000ダルトン、より好ましくは少なくとも約60,000ダルトンであるタンパク質または他の分子がある。本発明に有用なキャリアータンパク質には、例えばGST、(例えばキーホールリンペット由来の)ヘモシアニン、血清アルブミンまたはカチオン化した血清アルブミン、チログロブリン、卵白アルブミン、種々のトキソイドタンパク質(例えば破傷風トキソイドまたはジフテリアトキソイド)、免疫グロブリン、熱ショックタンパク質などがある。
【0036】
あるタンパク質を別の(キャリアー)タンパク質に化学的に結合させる方法は当該分野で知られており、それらには例えば以下がある:水溶性カルボジイミド(例えば1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩)によるコンジュゲート、(例えばNHSエステル基またはスルホ-NHSエステル類似体を有する)ホモ2価性架橋リンカーによるコンジュゲート、(例えばNHSエステルおよびマレイミド基、例えばスルホスクシンイミジル-4-(N-マレイミドメチル)シクロヘキサン-1-カルボキシレートを有する)ヘテロ2価性架橋リンカーによるコンジュゲート、およびグルタールアルデヒドによるコンジュゲート(例えばHermanson, Bioconjugate Techniques, Academic Press, San Diego, Calif. (1996);米国特許第4,608,251号および4,161,519号参照)。
【0037】
本明細書で使用する転写ユニットを含む“ベクター”(“発現ベクター”としても知られる)という用語は、インビボで投与された際に標的細胞に侵入してコードされたタンパク質を発現できるウイルス性および非ウイルス性の発現ベクターをいう。インビボでの運搬および外因性タンパク質の発現に好適なウイルスベクターは知られており、それらにはアデノウイルスベクター、アデノ関連ウイルスベクター、レトロウイルスベクター、単純疱疹ウイルスベクターなどがある。ウイルスベクターは好ましくは正常細胞内で複製欠陥を起こす。米国特許第6,669,942号;6,566,128号;6,794,188号;6,110,744号;6,133,029号参照。
【0038】
本明細書で使用する“細胞”という用語は広義に使用し、哺乳動物細胞、植物細胞、真核細胞、原核細胞など、いずれの生細胞も含む。
【0039】
本明細書で使用する“アデノウイルス発現ベクター”という用語は、ポリペプチドをコードする、そのゲノム中に挿入された外因性DNAを含むアデノウイルス由来の任意のベクターをいう。ベクターは、欠陥のある必須遺伝子がトランスで提供された際に複製しおよび封入されうる能力を有さなければならない。好ましくは、アデノウイルスベクターはウイルスDNAの複製を支持するのに必要な各末端反復の少なくとも一部、好ましくは完全長ITR配列の少なくとも約90%、およびウイルスキャップシドにゲノムを封入するのに必要なDNAを含む。多くの好適なアデノウイルスベクターが当該分野で報告されている。米国特許第6,440,944号および6,040,174号参照(複製欠陥E1欠損ベクターおよび特殊化パッケージング細胞系)。好ましいアデノウイルス発現ベクターは、正常細胞において複製欠陥のあるものである。
【0040】
アデノ関連ウイルスとは、その遺伝物質を19番染色体上の特定の部位に挿入できる小型1本鎖DNAウイルスのクラスを表す。遺伝子運搬のためのアデノ関連ウイルスベクターの調製および使用は米国特許第5,658,785号に記載されている。
【0041】
遺伝子運搬のための非ウイルス性ベクターには、運搬の際にベクターのDNAを保護するために脂質、タンパク質、および他の分子(またはそれらを組み合わせたもの)と合わせた種々の型の発現ベクター(例えばプラスミド)が含まれる。融合性の(fusigenic)非ウイルス性ベクター粒子は、ウイルス性融合タンパク質を記載の発現ベクターと組み合わせることによって構築できる。Kaneda, Curr Drug Targets (2003) 4(8):599-602。再構築したHVJ(hemagglutinating virus of Japan;センダイウイルス)-リポソームを使用して発現ベクターを運搬するか、またはリポソームを使用せずに不活性化HVJ粒子にベクターを直接導入することもできる。Kaneda, Curr Drug Targets (2003) 4(8):599-602参照。DMRIE/DOPE脂質混合物は非ウイルス性発現ベクターの媒体として有用である。米国特許第6,147,055号参照。ポリカチオン-DNA複合体を非ウイルス性遺伝子運搬媒体として使用してもよい。Thomasら, Appl Microbiol Biotechnol (2003) 62(1):27-34参照。
【0042】
本明細書において発現ベクターに関連して使用する“転写ユニット”という用語は、RNAポリメラーゼによって1本の連続するmRNA鎖として転写される一続きのDNAであって、転写の開始および停止のためのシグナルを含むものをいう。例えばある態様では、本発明の転写ユニットは5’から3’方向に、分泌シグナル配列、抗原、およびCD40リガンドをコードする核酸を含む。転写ユニットは転写および/または翻訳発現調節要素、例えばプロモーター、そして必要により任意の上流または下流エンハンサー要素と、機能的に結合している。有用なプロモーター/エンハンサーはサイトメガロウイルス(CMV)IE(immediate-early)プロモーター/エンハンサーである。米国特許第5,849,522号および 6,218,140号参照。
【0043】
本明細書で使用する“分泌シグナル配列”(“シグナル配列”、“シグナルペプチド”、“リーダー配列”、または“リーダーペプチド”としても知られる)という用語は(in charter)一般に疎水性である約20から30アミノ酸を含む短鎖ペプチド配列をいい、これはポリペプチドのN末端で合成され、ポリペプチドを小胞体に誘導する。分泌シグナル配列は一般に、ポリペプチドの小胞体へのトランスロケーションの際に開裂される。真核生物の分泌シグナル配列は、発現ベクターの外因性遺伝子産物の分泌を誘導するのに好ましい。種々のそのような好適な配列が当該分野で知られており、それらにはヒト成長ホルモン、免疫グロブリンのκ鎖の分泌シグナル配列などがある。ある態様では、外来性腫瘍抗原シグナル配列を用いて分泌を起こさせてもよい。
【0044】
本明細書で使用する“抗原”という用語は広範に、個体が免疫応答を起こすことができるいずれの抗原をもさす。本明細書で使用する“抗原”は広範に、免疫応答が標的としうる抗原決定基を少なくとも1つ含む分子をいう。免疫応答は細胞性、体液性、またはその両方であってもよい。
【0045】
当該分野で知られているように、抗原は天然の(in nature)タンパク質、天然の炭化水素、天然の脂質、もしくは天然の核酸、またはこれらの生体分子を組み合わせたものであってもよい。抗原は非天然分子、例えばポリマーなどを含んでもよい。抗原には自己抗原および外来抗原、例えば別の動物が生成した抗原または感染性物質由来の抗原が含まれる。感染性物質抗原は細菌性、ウイルス性、真菌性、原核動物性などであってもよい。
【0046】
本明細書で使用する“腫瘍関連抗原”(TAA)という用語は腫瘍細胞上、および胎児期の正常細胞上(癌胎児性抗原)に存在するタンパク質をいい、これは出産後は選択された臓器中、または多くの正常細胞上に存在するがその濃度は腫瘍細胞上よりかなり低い濃度である。種々のTAAが報告されている。代表的なTAAはムチン(例えばMUC1。以下に更に詳細に記載する)またはHER2(neu)(これも以下に記載する)である。これに対して腫瘍特異抗原(TSA)(腫瘍特異移植抗原またはTSTAともいう)は正常細胞には存在しないタンパク質をいう。TSAは通常、感染するウイルスによって細胞が不死化し、ウイルス抗原が発現される際に出現する。
【0047】
代表的なウイルスTSAはHPV16型のE6またはE7タンパク質である。ウイルスによって誘導されないTSAにはB細胞リンパ腫に関係する免疫グロブリンまたはT細胞リンパ腫のT細胞受容体(TCR)のイディオタイプがある(TSAs not induced by viruses include idiotypes the immunoglobulin idiotypes associated with B cell lymphomas or the T cell receptor (TCR) on T cell lymphomas)。
【0048】
代表的なウイルスTSAはHPV16型のE6またはE7タンパク質である。HPVは種々の皮膚および生殖管の上皮病変を発生させうる。HPVが関係する生殖管疾患は、世界中の女性の2番目に高い癌死亡の原因となっている。それらには生殖器疣贅、頚部上皮内新生物(CIN)、および子宮頚癌がある。最も一般的に高度(high grade)CINおよび子宮頚癌と関係するHPVの型はHPV16型である。子宮頚癌の大半は非構造HPV16誘導型遺伝子産物E6およびE7腫瘍性タンパク質を発現する。HPV誘導型子宮頚癌モデルでは、E6/E7腫瘍性タンパク質が悪性フェノタイプの保持に必要とされ、それらの発現はHPV16の形質転換の可能性と相関関係を有する。E6またはE7を腫瘍抗原として使用するのに加え、これらのタンパク質の抗原性フラグメントを代わりに使用してもよい。抗原性フラグメントは、周知のコンピューター演算(例えばHoppおよびWoods疎水性分析)を用いてエピトープを保有すると予想されるような分子の部分との免疫応答を試験することによって決定してもよい。
【0049】
ウイルスによって誘導されないTSAはB細胞リンパ腫の免疫グロブリンまたはT細胞リンパ腫のT細胞受容体(TCR)のイディオタイプであってもよい。腫瘍関連抗原(TAA)はTSAより一般的である。
【0050】
TAAおよびTSAは共に、発現ベクターワクチンの免疫学的標的となりうる。特に記載しない限り、本明細書において“腫瘍抗原”という用語は集合的にTAAおよびTSAを示す。
【0051】
本明細書で使用する“ムチン”という用語は、スレオニン、セリン、およびプロリン含量の高いタンデムリピート構造を持つペプチド配列にO-グリコシド結合した高含量のクラスター化したオリゴ糖を有する、任意のクラスの高分子量糖タンパク質をいう。ムチンは細胞防御において役割を果たし、伸張構造上に露出された多くの糖で、種々の型の細胞(白血球および感染性物質を含む)との複数の相互作用に影響を与える。ムチン抗原には以下と同定されているものもある:CD227、腫瘍関連上皮膜抗原(EMA)、多型上皮性ムチン(PEM)、ピーナツ反応性尿ムチン(Peanut-reactive Urinary Mucin;PUM)、エピシアリン(episialin)、乳癌関連抗原DF3、H23抗原、ムチン1、エピシアリン(Episialin)、腫瘍関連ムチン、癌関連ムチン。また、CA15-3抗原、M344抗原、Sialosyl Lewis抗原(SLA)、CA19-9、CA195、およびこれまでにモノクローナル抗体によって同定された他のムチン抗原(例えば米国特許第5,849,876号参照)もある。ムチンという用語はプロテオグリカン(タンパク質バックボーンに共有結合したグリコサミノグリカン鎖を特徴とする糖タンパク質である)を含まない。
【0052】
少なくとも15の異なるムチンが報告されており、それらにはMUC1、MUC2、MUC3A、MUC3B、MUC4、MUC5AC、MUC5B、MUC6、MUC7、MUC8、MUC9、MUC12、MUC13、MUC15、およびMUC16が含まれる(これらは“MUC”と数字の間にハイフンを入れて示してもよい)。これらのムチンのヌクレオチド配列およびアミノ酸配列は知られている。これらムチンのNCBIおよびSwiss Protアクセッション番号は以下の通りである:MUC1(NCBI NM002456, Swiss Prot P15941)、MUC2(NCBI NM002457, Swiss Prot Q02817) 、MUC3A(NCBI AF113616, Swiss Prot Q02505)、MUC3B(NCBI AJ291390, Swiss Prot Q9H195)、MUC4(NCBI NM138299, Swiss Prot Q99102)、MUC5AC(NCBI AF043909, Swiss Prot Q8WWQ5)、MUC5B(Swiss Prot Q9HC84)、MUC6(NCBI U97698, Swiss Prot Q8N8I1)、MUC7(NCBI L42983, Swiss Prot Q8TAX7)、MUC8(NCBI U14383, Swiss Prot Q12964)、MUC9(NCBI U09550, Swiss Prot Q12889)、 MUC12(Swiss Prot Q9UKN1)、MUC13(NCBI NM017648, Swiss Prot Q9H3R2)、MUC15(NCBI NM145650, Swiss Prot Q8WW41)、およびMUC16(NCBI AF361486, Swiss Prot Q8WXI7; CA125ともいう)。
【0053】
ムチンには構造的および機能的に異なる2つのクラス:分泌型ゲル形成ムチン(MUC2、MUC5AC、MUC5B、およびMUC6)および膜貫通ムチン(MUC1、MUC3A、MUC3B、MUC4、MUC12、MUC17)がある。いくつかのMUC遺伝子の産物はいずれのクラスにも該当しない(MUC7、MUC8、MUC9、MUC13、MUC15、MUC16)。
【0054】
特定の癌におけるTAAとしての特定のムチンの特徴は、前腫瘍性病変および腫瘍性病変に伴う発現および構造の変化によって確認される(Filipe MI: Invest Cell Pathol 1979, 2:195-216;Filipe MI, Acta Med Port 1979, 1:351-365)。例えば、正常な胃粘膜はMUC1、MUC5A/C、MUC6 mRNA、およびコードされる免疫反応性タンパク質の発現を特徴とする。また、高レベルのMUC2、MUC3ムチンmRNA、およびコードされる免疫反応性タンパク質は腸上皮化生に関係する。胃癌は隣接する正常な粘膜と比較して顕著に変化した分泌型ムチンmRNAレベルを示し、MUC5およびMUC6 mRNAレベルは低下し、MUC3およびMUC4 mRNAレベルは上昇する。高レベルのMUC2およびMUC3 mRNAおよびタンパク質は小腸で検出可能であり、MUC2は最も存在量の多い結腸ムチンである。
【0055】
ムチンは膵臓癌および他の細胞型の早期検出のための診断マーカーである。研究で明らかになったところによれば、腺癌(ductal adenocarcinomas;DAC)および腫瘍細胞系は通常、MUC1ムチンを過剰発現する。Andrianifahananaら, Clin Cancer Res 2001, 7:4033-4040参照。このムチンは、ほとんどの慢性膵炎および正常な膵臓組織では低レベルでしか検出されないが、膵臓癌の全段階で過剰発現される。膵臓癌および細胞系におけるMUC4のデノボ発現が報告されている(Hollingsworthら, Int J Cancer 1994, 57:198-203)。MUC4 mRNA発現は大多数の膵臓癌および確立された膵臓癌細胞系で観察されているが、正常な膵臓または慢性膵炎組織では観察されていない。MUC4発現は肺癌との関係も見いだされている(Nguyenら, 1996 Tumor Biol. 17:176-192参照)。MUC5は非小細胞肺癌において転移と関係する(Yuら, 1996 Int. J. Cancer 69:457-465参照)。胃型および浸潤性DACではMUC6が過剰発現され、MUC5ACがデノボ発現される(Kimら, Gastroenterology 2002, 123:1052-1060)。MUC7は浸潤性膀胱癌のマーカーとして報告されている(Retzら, 1998 Cancer Res. 58:5662-5666)。
【0056】
MUC2分泌型ゲル形成ムチンの発現は一般に大腸腺癌で減少し、粘液癌(マイクロサテライト不安定性を伴う異なるサブタイプの大腸癌)では保存されている。MUC2は咽頭癌で上昇する(Jeannonら, 2001 Otolaryngol Head Neck Surg. 124:199-202)。別の分泌型ゲル形成ムチン、MUC5AC(正常な胃粘膜の産物)は正常な結腸には存在しないが、大腸腺癌および大腸癌では高頻度で存在する。
【0057】
MUC1(エピシアリンとしても知られる)、多型上皮性ムチン(PEM)、ムチン様癌関連抗原(MCA)、CA27.29、ピーナツ反応性尿ムチン(PUM)、腫瘍関連上皮ムチン、上皮膜抗原(EMA)、ヒト乳脂肪球(HMFG)抗原、MUC1/REP、MUC1/SEC、MUC1/Y、CD227は最もよく知られたムチンである。MUC1をコードする遺伝子はlq21-q24にマッピングされる。MUC1遺伝子は7つのエクソンを含み、7つの異なるスプライス変異体を生成する。タンデムリピートドメインは高度にO-グリコシル化されており、グリコシル化の変化が上皮癌細胞において明らかになっている。
【0058】
MUC1 mRNAはサイズ多型である。現在のところ、スプライシングの変化に基づいて9つのMUC1のアイソフォームがある(アイソフォーム番号:NCBIアクセッション番号;1:ID P15941-1、2:ID P15941-2、3:ID P15941-3、4:ID P15941-4、5:P15941-5、6:ID P15941-6、7:ID P15941-7、8:ID P15941-8、9:ID P15941-9)。
【0059】
MUC1アイソフォーム1(MUC1/REPともいう)は多型のI型膜貫通タンパク質であり、以下を含む:1)主に20-アミノ酸(aa)反復モチーフ(可変数(30-100)タンデムリピート(Variable Number of tandem repeats-VNTR)として知られる領域)から成る大型の細胞外ドメイン;2)膜貫通ドメイン;および3)72-aa細胞質尾部。生合成の際、MUC1/REPタンパク質は大幅に修飾され、相当な数のO-結合糖部分は成熟タンパク質上にムチン様の特徴を与える。翻訳後まもなくMUC1/REPは開裂されて2つの産物となり、これらは強く結合したヘテロダイマー複合体を形成するが、これは大型の細胞外ドメインが細胞質ドメインおよび膜貫通ドメインを含むそれよりかなり小型のタンパク質に非共有結合したものから構成される。細胞外ドメインは細胞から離脱できる。参照としてSwiss Prot P15941を用い(図1参照)、MUC1アイソフォーム1の細胞外ドメイン(ecm)はアミノ酸24から1158であり、膜貫通ドメインは1159-1181であり、そして細胞質ドメインは1182-1255である。SEAドメインは1034-1151であり、細胞外ドメインと呼ばれるC末端部分である。ムチンのSEAドメインは一般にタンパク分解による開裂の標的であり、2つのサブユニットを生成し、小さい方は細胞膜と結合する。
【0060】
MUC1アイソフォーム5(MUC1/SECともいう)は細胞によって分泌されるMUC1の型である。アイソフォーム1(MUC1/REP)と同じ細胞外ドメインを有するが、細胞膜にタンパク質を係留するための膜貫通ドメインを有さない。MUC1アイソフォーム7(MUC1/Yともいう)はアイソフォーム1(MUC1/REP)および5(MUC1/SEC)で観察される細胞質および膜貫通ドメインを含むが、MUC1より小さい細胞外ドメインを有し、反復モチーフおよびその隣接領域を有さない(Baruch A.ら, 1999 Cancer Res. 59, 1552-1561参照)。アイソフォーム7は受容体として作用し、分泌型アイソフォーム5に結合する。結合によりアイソフォーム7のリン酸エステル化が誘導され、細胞の形態学に変化が起き、GRB2のような第2のメッセンジャータンパク質を通して細胞シグナリングが開始される(Zrihan-Licht S.ら, 1995 FEBS Lett. 356, 130-136参照)。β-カテニンはMUC1の細胞質ドメインと反応することが明らかになっている(Yamamoto M.ら, 1997 J. Biol. Chem. 272, 12492-12494)。
【0061】
MUC1は正常な上皮細胞表面上で局所的に低レベルで発現される。15参照。Greenleeら, Cancer Statistics CA Cancer J. 50, 7-33 (2000);Renら, J. Biol. Chem. 277, 17616-17622 (2002);Kontaniら, Br. J. Cancer 84, 1258-1264 (2001);Rowseら, Cancer Res. 58, 315 (1998)。MUC1は乳癌、卵巣癌、膵臓癌、並びに他の癌で過剰発現される(Gendler S.J.ら, 1990 J. Biol. Chem. 265, 15286-15293も参照されたい)。MUC1遺伝子の更なるコピーの獲得と高mRNAレベルとの間には相関関係が見られ(p<0.0001)、これはMUC1遺伝子の過剰発現の原因である遺伝子機構を明らかにしており、乳癌の病因におけるMUC1遺伝子投与(dosage)の役割を支持している((Bieche I.ら, 1997 Cancer Genet. Cytogenet. 98, 75-80参照)。MUC1ムチンは免疫学的検出によれば、結腸癌(これは予後の悪さと関係する)および卵巣癌で発現が上昇する。
【0062】
MUC1抗原の高レベル発現は、足場依存性増殖の調節を混乱させる(E-カドヘリン機能を混乱させる)ことによって腫瘍性上皮粘膜細胞の増殖において役割を果たし、これによって転移が起こる。Greenleeら, Cancer Statistics CA Cancer J. 50, 7-33 (2000);Renら, J. Biol. Chem. 277, 17616-17622 (2002)参照。MUC1に対する非MHC制限細胞傷害性T細胞の反応が乳癌患者において報告されている。Kontaniら, Br. J. Cancer 84, 1258-1264 (2001)参照。ヒトMUC1トランスジェニックマウス(“MUC1.Tg”)はヒトMUC1抗原での刺激に反応しないことが報告されている。Rowseら, Cancer Res. 58, 315 (1998)参照。ヒトMUC1トランスジェニックマウスは自己抗原としてのMUC1に対する免疫の確立を評価するのに有用である。
【0063】
MUC1タンパク質およびmRNAはER陽性MCF-7およびBT-474細胞、並びにER陰性MDA-MB-231およびSK-BR-3 BCC細胞において観察されている。mRNA転写レベルはER-よりER+細胞系の方が高い。MUC1は細胞内接着分子-1(ICAM-1)と反応する。MUC1の少なくとも6つのタンデムリピートが必要である(Regimbaldら, 1996 Cancer Res. 56,4244-4249)。T-47D BCC由来のMUC1のタンデムリピートペプチドは高度にO-グリコシル化されており、ミルク由来のムチンのリピート当たり2.6グリコシル化部位に比較してリピート当たり4.8部位である。
【0064】
本明細書で使用する“ムチン抗原”という用語は完全長のムチン、または本明細書に記載するようにインビボで投与されたMUC-CD40L発現ベクターを用いて細胞免疫を誘導できることを特徴とするエピトープを含むムチンの一部をいう。“ムチン抗原”はムチンの細胞外ドメインからの1つ以上のエピトープ、例えばVNTRに関連する1つ以上のタンデムリピートモチーフまたはSEA領域を含む。ムチン抗原は細胞外ドメイン全体を含んでもよい。また、“ムチン抗原”の意味には、保存的アミノ酸変化など、天然のムチン配列と交差反応する免疫応答を誘導する抗原の能力を変更しない、配列の変種も含まれる。
【0065】
VNTRは遺伝子多型およびムチンの性質に従って長さ(および組成)の異なる可変数のタンデムリピートペプチド配列から成る。VNTRは変性タンデムリピートを含有する5’および3’領域も含んでもよい。例えばMUC1では、北欧人集団においてリピートの数は21から125まで種々である。米国では、最も頻度の低い対立遺伝子は41および85リピートを含み、より一般的な対立遺伝子は60-84リピートを有する。MUC1リピートは一般的なリピートペプチド配列PDTRPAPGSTAPPAHGVTSA(SEQ ID NO : 3)を有する。基礎となるMUC1タンデムリピートは太線および下線で示す3つの位置(位置2、3、および13)における遺伝子配列多型である。協奏的置換(concerted replacement)DT→ES(配列変種1)および単一置換P→Q(配列変種2)、P→A(配列変種3)、およびP→T(配列変種4)が同定されており、ドメイン中の位置は種々である(Engelmannら, 2001 J. Biol. Chem. 276:27764-27769参照)。最も頻度の高い置換DT→ESは50%までのリピートで起こる。表1に代表的なタンデムリピート配列を示す。
【0066】
【表1】
【0067】
本明細書で使用するムチン抗原は1つのタンデムリピート配列モチーフだけを含んでもよいが、ベクターが複数のそのようなリピートを含めば、一般に生成される免疫応答はより強く、より有効であることは理解されるべきである。本発明のベクターは好ましくは2-4、より好ましくは5-9、より好ましくは10-19、より好ましくは20-29、更に好ましくは30-39、そして更により好ましくは40-50のムチンタンデムリピートをコードする。50より大きいタンデムリピートが可能であり、天然ムチンで見られるリピート数を含んでもよい。
【0068】
本明細書で使用するムチン抗原という用語は種々のタイプのムチン由来のタンデムリピートを含んでもよい。例えば発現ベクターは2つの異なるムチン(例えばMUC1およびMUC2)由来のタンデムリピートをコードしてもよい。それらのベクターは複数の型のSEAドメインを同様にコードしてもよく、またはタンデムリピートと1つ以上のSEAドメインを組み合わせて含んでもよい。
【0069】
分泌型の抗原は、その膜貫通ドメイン(成熟タンパク質中に存在する場合)の全部または実質的に全部が欠失したものである。例えばムチンの場合、膜貫通ドメインは、もし存在するなら、一般に約24アミノ酸長で、ムチンまたはムチンのフラグメントを細胞膜に係留する機能を果たす。全部の膜貫通ドメインが欠失した分泌型のMUC1は残基1159-1181が欠失したMUC1である。実質的に全部の膜貫通ドメインが欠失したムチン(または抗原)は、ドメインが膜貫通ドメインの一端の配列の6以下の残基、より好ましくは膜貫通ドメインの一端の配列の約4未満の残基、更に好ましくは膜貫通ドメインの一端の配列の約2未満の残基、そして最も好ましくは膜貫通ドメインの一端の1以下の残基を含むものである。好ましい態様では、ワクチンベクターの転写ユニットは膜貫通ドメイン全体が欠失した分泌型のムチン(または抗原)をコードする。膜貫通ドメインの実質的に全部が欠失して分泌可能となったムチンは、ドメインの一端における配列の6を超えない残基を含むものである。本明細書ではMUC1のようなヒトムチンの細胞外ドメインを “ecdhMUC1”と記載する。
【0070】
機能性膜貫通ドメインが欠失したムチンは、(もし存在する場合は)細胞質ドメインの全部または一部およびSEAの全部または一部をなおも含みうることは理解されるべきである。
【0071】
種々のムチンおよびムチン抗原をコードするDNA源は、市販のcDNA合成キットおよび増幅(確立されたムチンDNA配列から設計できる好適なPCRプライマー対を使用)を用いてムチン発現細胞系から得てもよい。例えばMUC1またはMUC2をコードする核酸はAmerican Type Culture Collectionから入手できるCRL-1500細胞から得てもよい。ムチンをコードするDNAは、ヒトまたは他の動物組織から採取または調製したRNAまたはcDNAから増幅して得てもよい。さほど大きくないDNAセグメントでは、DNAは自動オリゴヌクレオチド合成装置を用いて合成してもよい。
【0072】
本明細書で発現ベクターの転写ユニットに関して使用する“リンカー”という用語は、抗原のカルボキシ末端とCD40リガンドのアミノ末端との間にある1つ以上のアミノ酸残基をいう。リンカーの組成および長さは当該分野で知られる方法に従って決定してもよく、有効性に関して試験してもよい。例えばAraiら, "Design of the linkers which effectively separate domains of a bifunctional fusion protein" Protein Engineering, Vol. 14, No. 8, 529-532, August 2001参照。リンカーは一般に約3から約15アミノ酸長、より好ましくは約5から約10アミノ酸長であるが、より長い、もしくはより短いリンカーを使用してもよく、またはリンカーが全く無くてもよい。より長いリンカーは約50アミノ酸まで、または約100アミノ酸まで及んでもよい。ムチン抗原がCD40リガンドのN-末端側にある場合は、10残基未満の短いリンカーが好ましい。
【0073】
本明細書で使用する“CD40リガンド”(CD40L)という用語はCD154またはTNF5としても知られる分子の全長または一部をいう。CD40LはN-末端に細胞質ドメイン、膜貫通領域、そして細胞外ドメインをC-末端に有するII型膜ポリペプチドである。特に記載しない限り、本明細書では完全長のCD40Lを“CD40L”、“wtCD40L”、または“wtTmCD40L”と記載する。細胞質ドメインが欠失した型のCD40Lを、本明細書では“ΔCtCD40L”と記載する。膜貫通ドメインが欠失した型のCD40Lを、本明細書では“ΔTmCD40L”と記載する。細胞質ドメインおよび膜貫通ドメインの両方が欠失した型のCD40Lを、本明細書では“ΔCtΔTmCD40L”と記載する。マウスおよびヒト由来のCD40Lのヌクレオチドおよびアミノ酸配列は当該分野で知られており、例えば米国特許第5,962,406号(Armitageら)に報告されている。本発明の融合タンパク質に関連して、ムチンに対する免疫応答を誘導するリガンドの能力に変化を与えないような、保存的アミノ酸変化などを含む配列変異体もCD40リガンドの意味に含まれる。
【0074】
マウスCD40L(mCD40L)は260アミノ酸長である。mCD40Lの細胞質(Ct)ドメインはおおよそ1-22位、膜貫通ドメインはおおよそ23-46位、細胞外ドメインはおおよそ47-260位にわたる。
【0075】
ヒトCD40L(hCD40L)は261アミノ酸長である。hCD40Lの細胞質ドメインはおおよそ1-22位、膜貫通ドメインはおおよそ23-46位、細胞外ドメインはおおよそ47-261位にわたる。
【0076】
本明細書で使用する“CD40リガンドが膜貫通ドメインの全部または実質的に全部を欠失して分泌可能となっている”というフレーズは、細胞から分泌されることが可能な組換え型のCD40リガンドをいう。約24アミノ酸長を含むCD40Lの膜貫通ドメインは細胞膜においてCD40リガンドを係留する機能を果たす。膜貫通ドメインの全部が欠失したCD40Lは、残基23-46が欠失したCD40リガンドである。実質的に全部の膜貫通ドメインが欠失したCD40リガンドは、膜貫通ドメインの一端の配列の6残基以下、より好ましくは膜貫通ドメインの一端の配列の約4残基未満、更に好ましくは膜貫通ドメインの一端の配列の約2残基未満、そして最も好ましくは膜貫通ドメインの一端の1残基以下を保有するものである。従って、膜貫通ドメインの実質的に全部が欠失して分泌可能となったCD40Lは、ドメインの一端の配列の6を超えない残基を保有するものである。そのように、CD40Lは、細胞外ドメインおよび必要により細胞質ドメインに加え、CD40Lの膜貫通ドメインに位置するアミノ酸41-46または23-28を超えないアミノ酸を含む(Such as CD40L would contain, in addition to the extracellular domain and optionally the cytoplasmic domain, and no more than amino acids 41-46 or 23-28 located in the transmembrane domain of CD40L.)。 好ましい態様では、ワクチンベクター転写ユニットは、膜貫通ドメインの10%未満を含む分泌型CD40をコードする。更に好ましくは、CD40Lは膜貫通ドメインを含まない。
【0077】
機能性膜貫通ドメインが欠失したCD40Lは細胞質ドメインの全部または一部をなおも含みうることは理解すべきである。同様に、機能性膜貫通ドメインが欠失したCD40Lは細胞外ドメインの全部または実質的に一部を含みうる。
【0078】
本明細書における使用では、発現ベクターおよび融合タンパク質ブーストをワクチンとして投与し、腫瘍抗原に対する免疫を誘導する。発現ベクターおよび融合タンパク質ブーストは必要により、好適な医薬的に許容しうるキャリアーと共に調製してもよい。従って、ベクターまたはタンパク質ブーストを薬剤または医薬組成物の製造に使用してもよい。発現ベクターおよび融合タンパク質を非経口投与のための溶液または凍結乾燥粉末として調製してもよい。使用の前に好適な希釈剤または他の医薬的に許容されるキャリアーを添加して粉末を再調製してもよい。液体製剤は緩衝された等張の水溶液であってもよい。粉末を乾燥状態でスプレーしてもよい。好適な希釈剤の例は通常の等張食塩水、標準的な5%デキストロース水溶液、または酢酸ナトリウムもしくはアンモニウム緩衝液である。それらの製剤は非経口投与に特に好適であるが、経口投与に使用するか、または吸入のための定量噴霧式吸入器もしくはネブライザーに含有させてもよい。ポリビニルピロリドン、ゼラチン、ヒドロキシセルロース、アカシアゴム、ポリエチレングリコール、マンニトール、塩化ナトリウム、クエン酸ナトリウムなどのような賦形剤の添加が所望されうる。
【0079】
あるいはまた、発現ベクターおよび融合タンパク質を経口投与用に調製してもよい。医薬的に許容される固体または液体キャリアーを添加して組成物を増強もしくは安定化させるか、またはベクターの調製を容易にしてもよい。固体キャリアーにはデンプン、ラクトース、硫酸カルシウム2水和物、白土(terra alba)、ステアリン酸マグネシウムもしくはステアリン酸、タルク、ペクチン、アカシアガム、寒天、またはゼラチンがある。液体キャリアーにはシロップ、落花生油、オリーブ油、食塩水、および水がある。キャリアーはモノステアリン酸グリセリンまたはジステアリン酸グリセリンのような持続放出物質を単独で、またはワックスと共に含有してもよい。固体キャリアーの量は種々であるが、好ましくは投与ユニット当たり約20mgから約1gの間である。液体キャリアーを使用する場合、製剤はシロップ、エリキシル、エマルジョン、または水性もしくは非水性懸濁液の形態であってもよい。
【0080】
発現ベクターおよび融合タンパク質は、他の医薬的に有用な薬剤または生物学的物質を含有させて調製してもよい。ベクターを、本発明の化合物が目的とする疾病または症状に有用な他の薬剤または生物学的物質の投与と併せて投与してもよい。
【0081】
本明細書で使用する“有効量”というフレーズは、そのレシピエントにおいて免疫応答を生じさせる(または生成に関与する)のに十分高い濃度となるような投与量をいう。特定の被験体についての固有の有効量は種々の因子に依存し、それらには治療すべき障害、障害の重篤度、特定の化合物の活性、投与経路、ウイルスベクターのクリアランス速度、治療期間、ウイルスベクターと併用する、または同時に使用する薬剤、年齢、体重、性別、食事、および被験体の全身の健康状態、そして医学および科学の分野で周知の因子がある。“治療的有効量”を決定するための種々の一般的に考慮される点は当該分野で周知であり、例えば以下に記載されている:Gilmanら編, Goodman And Gilman's: The Pharmacological Bases of Therapeutics, 第8版, Pergamon Press, 1990;およびRemington's Pharmaceutical Sciences,第17版, Mack Publishing Co., Easton, Pa., 1990。ベクターの投与では、投与当たりの粒子の範囲は一般に約1x107から1x1011、より好ましくは1x108から5x1010、そして更に好ましくは5x108から2x1010である。ベクターは非経口で、例えば血管内、静脈内、動脈内、筋肉内、皮下などで投与してもよい。投与は経口、経鼻、直腸内、経皮、またはエアロゾルを介した吸入であってもよい。ベクターはボーラスまたは点滴(slowly infused)で投与してもよい。好ましくはベクターを皮下投与する。
【0082】
本明細書に示すように、腫瘍関連抗原をコードするベクターは、それらの抗原(すでに寛容が確立されたものを含む)に対する防御細胞および体液免疫を誘導する。いずれの理論に拘束されることも望まないが、本発明のワクチンを投与すると、分泌型のCD40リガンドに結合した分泌型の抗原から構成される融合タンパク質の継続的な局所放出が起こると考えられる。本明細書に示すように、これによってDCの成熟が促進され、有効な抗原特異的免疫の生成が促進される。また、本明細書に示すように、アデノウイルスベクターから生成された膜貫通ドメインおよび細胞質ドメインが欠失したヒトMUC1およびマウスCD40Lの細胞外ドメイン(すなわちsighMUC1-ΔCtΔTmCD40L)をコードする分泌可能な融合タンパク質は、MUC1発現腫瘍細胞に対する細胞免疫応答の有効性を劇的に増強した。いずれの理論に拘束されることも望まないが、Ad-K-ecdhMUC1-ΔCtΔTmCD40Lベクターの皮下注射によって強いMUC1特異的CD8+ T細胞介在型免疫が誘導され、これによってMUC1腫瘍関連抗原を発現する癌細胞の生着が予防されると考えられる。
【0083】
本発明の方法を使用して抗原に対して生じさせた免疫は長期にわたって持続される。本明細書で使用する長期持続という用語は、ベクターにコードされる抗原によって誘導された免疫が最終投与から6ヶ月まで、好ましくは8ヶ月まで、より好ましくは1年まで、より好ましくは1.5年まで、そして更に好ましくは少なくとも2年間示されることを意味する。
【0084】
ある態様では、ムチンTAAに対する免疫を生じさせるために、膜貫通ドメインおよび細胞質ドメインを欠失させたCD40リガンドのアミノ末端に融合させたMUC1の細胞外ドメインを含む融合タンパク質を生成してもよい。それらのベクターの構築を実施例に開示する。本明細書で観察されたように、このアデノウイルスベクタームチンワクチンの皮下投与によって、MUC1抗原を保持する癌細胞に対する非常に強力な長期持続性のCD8+ 細胞傷害性T細胞依存型全身免疫が誘導された。ムチンワクチンは長期持続性免疫の基礎となる記憶細胞の生成を誘導した。
【0085】
マウスにアデノウイルスベクター Ad-sig-ecdhMUC1/ecdmCD40Lをワクチン接種すると、100%のhMUC1トランスジェニックマウス(すなわちこれらのマウスはベクター注射以前はhMUC1抗原に対して不顕性である)でヒトMUC1(hMUC1)抗原陽性腫瘍細胞の増殖を抑制する免疫応答が誘導された。Rowseら, Cancer Res. 58, 315 (1998)参照。Ad-sig-ecdhMUC1/ecdmCD40Lベクターに対する免疫応答は1年まで持続し、抗原特異性を示した。これらの結果は、MUC1を発現する上皮性悪性病変の治療にAd-sig-ecdhMUC1-ecd/ecdCD40Lベクターを使用できることを示している。
【0086】
アデノウイルスMUC1発現ベクターの皮下注射によって、注射したhMUC1トランスジェニックマウスの脾臓におけるhMUC1特異的T細胞のレベルが250倍上昇した。トランスジェニックマウスはベクター注射以前はhMUC1抗原に対して不顕性であった。従って、ベクター注入によって不顕性が克服され、抗原特異的かつHLA制限的(HLA restricted)であるCD8+ T細胞依存的全身Th1免疫応答が誘導される。アデノウイルスMUC1発現ベクターの接種で観察されたような不顕性を克服する能力は、トランスジェニックマウスに精製したecdhMUC1/ecdCD40L-HISタンパク質を接種した場合には観察されなかった。
【0087】
いずれの理論にも拘束されることを望まないが、ベクターの皮下注射部位の近傍の感染した細胞が腫瘍抗原/CD40リガンド分泌物を放出し、これが感染細胞の近傍の抗原表示細胞(例えばDC)に取り込まれると考えられる。内在化された腫瘍抗原はプロテオソームで消化され、生じるムチン抗原ペプチドは小胞体に移動し、そこでMHCクラスI分子と結合する。最終的にDCはMHCクラスI分子において表面上に腫瘍抗原を提示する。活性化された腫瘍抗原を担持する抗原提示細胞はリンパ球を保有する第2の器官、例えば所属リンパ節または脾臓に移動する。2週間にわたる腫瘍抗原/CD40融合タンパク質の連続放出の間、腫瘍関連抗原を運搬する細胞を認識および死滅させる能力のあるCD8細胞傷害性T細胞は、活性化された、抗原を担持する樹状細胞の存在によってリンパ節および脾臓で増殖する。ベクターに感染させた細胞からの連続的放出による腫瘍抗原特異的細胞傷害性T細胞の刺激および増殖の連続性により、非分泌性である腫瘍抗原/CD40リガンドを運搬するベクターを用いて得られるより強い免疫反応が生ずると考えられる。
【0088】
従って、本発明の方法を使用して個体において自己抗原である抗原に対する免疫を生じさせることができる。例えば、MUC1由来のムチン抗原をコードするベクターを使用して、MUC1ムチン抗原が自己抗原であるヒトにおいてCD8+ 免疫を生じさせることができる。本発明の方法を使用して、自己抗原である抗原に対する免疫学的不顕性状態を克服することもできる。
【実施例】
【0089】
以下の実施例は本発明を例証するためのものである。これらの実施例は本発明の範囲を制限することを意図しない。
1.アデノウイルス発現ベクターの構築
アデノウイルスベクターの転写ユニットsig-ecdhMUC1-ΔCtΔTmCD40Lはシグナル配列(Igカッパ鎖由来)、次いでヒトMUC1の細胞外ドメインをコードし、これは膜貫通ドメインまたは細胞質ドメインを有さず細胞外ドメインを含有するCD40リガンド(ヒトまたはマウス)のフラグメントにリンカーを介して結合している。カッパ鎖シグナル配列を融合タンパク質のアミノ末端に添加することにより、ベクター感染細胞から融合タンパク質が分泌されるように遺伝子操作した。
【0090】
ヒトMUC1のアミノ酸配列およびコードするヌクレオチド配列を、それぞれ図2および1に示す。コードされたMUC1タンパク質はSEQ ID NO : 1のヌクレオチド74から3,841にコードされる1255アミノ酸に相当する。最初の23アミノ酸(SEQ ID NO : 1の74から142にコードされる)はMUC1シグナル配列であり、成熟ムチンからは除去される。細胞外ドメインは24位から1158位の約1135アミノ酸(ヌクレオチド143から3547にコードされる)である。タンデムリピート領域は約900アミノ酸に相当する。アミノ酸74から126(SEQ ID NO : 1の229から451にコードされる)は5’変性タンデムリピート領域、アミノ酸127から945はタンデムリピート領域(SEQ ID NO : 1の452から2,908にコードされる)であり、アミノ酸946から962は3’変性タンデムリピート領域(SEQ ID NO : 1の2809から2959にコードされる)に相当する。SEAドメインはアミノ酸1034から1151、膜貫通ドメインは1159から1181、そして細胞質ドメインは1181から1255である(SEQ ID NO : 2参照)。
【0091】
転写ユニットをアデノウイルスベクターのバックボーンのE1遺伝子領域に導入した。アデノウイルスベクター粒子をHEK293細胞で生成させた後、塩化セシウム密度勾配遠心分離法によってベクターDNAを精製した。アデノウイルスベクター中のシグナルペプチドの存在を、制限酵素分析およびDNAシーケンシングによって確認した。
【0092】
ヒトMUC-1をコードするDNAの上流にマウスIgGカッパ鎖遺伝子のシグナル配列をコードするDNAを含む転写ユニット(“sig-ecdhMUC-1”)をPCRで生成したが、これにはプラスミドpcDNA3-hMUC-1(Finn O.J., University of Pittsburgh School of Medicineより供与)および以下のプライマーを用いた:マウスIgGカッパ鎖METDTLLLWVLLLWVPGSTGD(1文字アミノ酸コード)(SEQ ID NO : 11)をコードするDNAをPCR増幅で調製し(SEQ ID NO : 12、13、および14)、完全長の21アミノ酸マウスIgGカッパ鎖シグナル配列を生成した(SEQ ID NO : 12において、開始コドン“ATG”を太字(下線)で示す)。
5'-CCACC ATG GAG ACAGAC ACA CTC CTG CTA TGG GTA CTG CTG-3'
(SEQ ID NO : 12)
5'- TC CTG CTA TGG GTA CTG CTG CTC TGG GTT CCAGGT TC-3'
(SEQ ID NO : 13)
5'- TG CTC TGG GTT CCAGGT TCC ACT GGT GAC GAT G -3'
(SEQ ID NO : 14)
5'- GGT TCC ACT GGT GAC GAT GTC ACC TCG GTC CCAGTC-3'
(SEQ ID NO : 15)(MUC-1リピート領域の順向きプライマー)
5'- GAGCTCGAG ATT GTG GAC TGG AGG GGC GGT G-3'
(SEQ ID NO : 16)(MUC-1リピート領域の逆向きプライマー)
【0093】
上流にカッパシグナル配列を有するsig-ecdhMUC-1を4ラウンドのPCR増幅で生成した(第1ラウンド:プライマーSEQ ID NO : 15および16;第2ラウンド:プライマーSEQ ID NO : 14および16;第3ラウンド:プライマーSEQ ID NO : 13および16;第4ラウンド:プライマーSEQ ID NO : 12および16)。sig-ecdhMUC-1をコードするDNAをpcDNATM 3.1 TOPOベクター(Invitrogen, San Diego, CA)にクローニングしてpcDNA-sig-ecdhMUC-1を構築した。
【0094】
pShuttle-ΔCtΔTmCD40L(シグナル配列が無く、マウスCD40Lを含む)を以下のように調製した:プラスミドpDC406-mCD40LはAmerican Type Culture Collectionから購入した。1組のPCRプライマー(SEQ ID NO : 17および18)を、マウスCD40リガンドの52位から260位(すなわち細胞質ドメインおよび膜貫通ドメインを含まない)を増幅し、増幅子の5’末端にリンカーをコードする配列(“+スペーサー”と表示)が含まれるように設計した。
マウスΔCtΔTmCD40L+スペーサー順向きプライマー(MCD40LSPF)(CD40L配列はイタリック体(小文字);クローニング部位は下線および太線):
5'- CCGCTCGAGAACGACGCACAAGCACCAAAATCAAAGGTCGAAGAGGAAGTA -3'
(SEQ ID NO : 17)
マウスCD40L逆向きプライマー(MCD40LR;クローニング部位は下線):
5'-GCGGGCC CGCGGCCGCCGCTAG TCTAGAGAG TTT GAG TAAGCC AAAAGA TGAG-3'
(SEQ ID NO : 18)
【0095】
順向きプライマーMCD40LSPFは転写ユニットのムチンとCD40Lリガンド(mCD40L)との間に位置する10残基のスペーサー(LENDAQAPKS;1文字コード;SEQ ID NO : 19)をコードする。順向きおよび逆向きプライマー(SEQ ID NO : 17および18)およびテンプレートとしてプラスミドpDC406-mCD40Lを使用してPCRを行い、PCRフラグメント“スペース+ΔCtΔTMCD40L”を得、これをXbaI(TCTAGA)およびXho I(CTCGAG)で制限エンドヌクレアーゼ消化した後、プラスミドpcDNA-sig-ecdhMUC1に挿入した。このベクターをpcDNA-sig- ecdhMUC1/ΔCtΔTmCD40Lで消化する。切断型ではなく完全長のCD40Lをコードすること以外は同じであるベクターを構築した。このベクターの生成は、CD40順向きプライマーを用い、マウスCD40Lの開始コドンにアニールさせて行った。このベクターをpShuttlsig40Lと記載する(シグナル配列なし)。
【0096】
HindIII-XbaI 制限を用いてpCDNA3TOPO ベクターからsig-ecdhMUC1/ΔCtΔTmCD40LをコードするDNAを切り出し、pShuttle-CMV(Murphyら, Prostate 38: 73-78, 1999参照)のCMVプロモーターの下流に挿入した。このプラスミドをpShuttle-sig-ecdhMUC1-ΔCtΔTmCD40Lと記載する。従って転写ユニットsig-ecdhMUC1-ΔCtΔTmCD40LはマウスIgGカッパ鎖分泌シグナル、次いでヒトMUC1の細胞外ドメイン、次いで10アミノ酸リンカー(NDAQAPK;SEQ ID NO : 19)、次いでマウスCD40リガンド残基52-260をコードする。
【0097】
あるベクターでは、マウスHSF1 3量体ドメインをecdhMUC1をコードするDNAとΔCtΔTm CD40Lとの間に加えたが、これはプラスミドpcDNA-sig-ecdhMUC1/ΔCtΔTmCD40Lおよび以下のプライマーを用いたPCRで行った:
5'-AAC AAG CTC ATT CAG TTC CTG ATC TCA CTG GTG GGATCC AAC GAC GCA CAAGCA CCAAAA TC-3'
(SEQ ID NO : 20)
5'- AGC CTT CGG CAG AAG CAT GCC CAG CAA CAG AAAGTC GTC AAC AAG CTC ATT CAG TTC CTG-3'
(SEQ ID NO : 22)
5'GAT ATC CTC AGG CTC GAG aac gac gca caagca ccaaaagAG AAT GAG GCT CTG TGG CGG G-3'
(SEQ ID NO : 23)
5'-GCGGGCC CGCGGCCGCCGCTAG TCTAGA GAG TTT GAG TAAGCC AAAAGA TGAG-3'
(SEQ ID NO : 18)
【0098】
3量体ドメイン配列を持つHSF1/ΔCtΔTm CD40Lを4ラウンドのPCR増幅によって生成した(第1ラウンド:プライマーSEQ ID NO : 23および18;第2ラウンド:プライマーSEQ ID NO : 22および18;第3ラウンド:プライマー21および18;第4ラウンド:プライマーSEQ ID NO : 20および18)。HSF1/ΔCtΔTm CD40LをコードするDNAをpcDNA-sig- hMUC-1制限部位XbaI(TCTAGA)およびXho I(CTCGAG)にクローニングした。MUC1およびmCD40L間の配列は以下である:
L E N D A Q A P K E N E A L W R E V A S F R Q K H A Q Q Q K V V N K L I Q F L I S L V G S N D A Q A P K S(SEQ ID NO : 24)
ここで、下線のセグメントは3量体配列であり、これがリンカーLENDAQAPK (SEQ ID NO : 25)およびNDAQAPKS(SEQ ID NO : 26)に結合する。
【0099】
あるベクターでは、Hisタグをコードする配列をΔCtΔTm CD40Lの末端に添加し、プラスミドpDC406-mCD40L(American Type Culture Collectionより購入)および以下のプライマーを用いたPCRで生成した:
5'-CCG CTCGAG AACGACGCACAAGCACCAAAATCAAaggtcgaagaggaagta -3'(SEQ ID NO : 27)(順向きプライマー)
5'-ATG GTG ATG ATG ACC GGT ACG GAG TTT GAG TAA GCC AAA AGA TGA GAA GCC-3' (SEQ ID NO: 28)(逆向きプライマー)
5'-GTGC tctaga TCAgaattc <ATG GTG ATG GTG ATG ATG> ACC GGT ACG GAG -3'(SEQ ID NO : 29)(ポリHis領域は囲み線内(括弧内)のヌクレオチドにコードされる)。
【0100】
Hisタグ配列を持つベクター/ΔCtΔTm CD40L/Hisを2ラウンドのPCR増幅で生成した(第1ラウンド:プライマー1+2;第2ラウンド:プライマー1+3)。/ΔCtΔTmCD40L/HisをコードするDNAをpcDNA-sig-ecdhMUC-1 制限部位XbaI(TCTAGA)およびXho I(CTCGAG)にクローニングした。
【0101】
組み換えアデノウイルスベクターをAdEasyベクター系(Stratagene, San Diego, CA)を用いて生成した。簡潔に記載すると、得られたプラスミドpShuttle-sig- ecdhMUC1-ΔCtΔTmCD40Lおよび他のコントロールアデノウイルスベクターをPme Iで直線化し、ウイルスDNAプラスミドpadEasy-1と共にE. coli株BJ5183に共トランスフォームした。組換え体をカナマイシンで選択し、制限酵素分析によってスクリーニングした。その後、組換えアデノウイルスコンストラクトをPac Iで開裂してその逆向き末端反復配列(ITR)を暴露し、293A細胞にトランスフェクトしてウイルス粒子を生成した。組換えアデノウイルスの力価を組織培養感染価(TCID50)法で測定した。
【0102】
ヒトCD40リガンドcDNAテンプレートを用いるヒトΔCtΔTmCD40L+スペーサーの増幅のためのプライマーを以下に記載する。
ヒトΔCtΔTmCD40L+スペーサー順向きプライマー(HCD40LSPF)(イタリック体(小文字)はCD40L配列):
5'- CCG CTCGAGAACGACGCACAAGCACCAAAATCAGTGTATCTTcatagaaggttggacaag-3'
(SEQ ID NO : 30)
ヒトCD40L逆向きプライマー(HCD40LR)
5'-CCCTCTAGA TCAGAGTTTGAGTAAGCCAAAGGAC-3'(SEQ ID NO : 31)
【0103】
これらのプライマーはヒトCD40Lの47-261をコードするΔCtΔTmCD40L+スペーサーを増幅する。順向きプライマーHCD40LSPFは転写ユニットの腫瘍抗原とCD40リガンド(hCD40L)との間に位置する10残基スペーサー(LENDAQAPKS;1文字コード;SEQ ID NO : 19)をコードする。順向きおよび逆向きプライマー(SEQ ID NO : 30および31)、およびテンプレートとしてプラスミドpDC406-hCD40Lを使用してPCRを実施してPCRフラグメント“space+ΔCtΔTmCD40L(ヒト)”を得、これをXbaI(TCTAGA)およびXho I(CTCGAG)での制限エンドヌクレアーゼ消化後、プラスミドpcDNA-sig-ecdhMUC1に挿入した。HindIII-XbaI 制限を用いてpCDNA3TOPO からsig-ecdhMUC1/ΔCtΔTmCD40L(ヒト)をコードするDNAを切り出し、これをpShuttle-CMV(Murphyら, Prostate 38: 73-78, 1999)のCMVプロモーターの下流に挿入した。このベクターをpShuttle sig-ecdhMUC1/ΔCtΔTmCD40L(ヒト)と記載する。ヒトCD40リガンド配列の上流にecdhMUC1を含有させるためのpShuttle sig-ecdhMUC1/ΔCtΔTmCD40L(ヒト)の修飾を、本質的にマウスCD40リガンドをコードするベクターに関して前記したようにして行った。従って転写ユニットsig-ecdhMUC1-ΔCtΔTmCD40L(ヒト)はカッパ分泌シグナル、次いでヒトMUC1の細胞外ドメイン、次いで10アミノ酸リンカー(NDAQAPK;SEQ ID NO : 19)、次いでヒトCD40リガンド残基47-261をコードする。
【0104】
別の方法では、ヒト成長ホルモンシグナル配列MATGSRTSLLLAFGLLCLPWLQEGSA(1文字アミノ酸コード)(SEQ ID NO : 32)をコードするDNAをカッパ鎖シグナル配列の代わりに使用してもよい。
【0105】
2.MUC1トランスジェニックマウスにおけるMUC1に対する不顕性の克服
a)アデノウイルス感染DCのサイトカイン生成
骨髄由来DCをhMUC-.Tgトランスジェニックマウスから、アデノウイルスベクターへの暴露の48時間後に回収した。MOI 100で細胞をベクターに暴露し、24ウェルプレートに2×105細胞/mlで播種した。37℃で24時間インキュベートした後、上清(1ml)を回収し、遠心分離してデブリを除去した。培養液に放出されたマウスIL-12またはIFN-ガンマのレベルの評価を、マウスIL-12 p70またはIFN-ガンマR & Dシステムキットを用いる酵素結合免疫吸着アッセイ(ELISA)によって行った。
【0106】
Ad-sig-ecdmMUC1-ΔCtΔTCD40L(マウス)ベクターと接触させた骨髄由来DCにより、ベクターへの暴露の48時間後にhMUC-.Tgトランスジェニックマウスから回収したDCからのインターフェロン・ガンマおよびIL-12サイトカインのレベルが有意に上昇した。これと対照的に、cMUC1の細胞外ドメインをコードするが分泌型のCD40Lに融合させていないアデノウイルスベクターで免疫化した動物由来の再刺激DCからは実質的にサイトカインは検出されなかった。これらの結果はecdhMUC1/ecdmCD40L(マウス)融合タンパク質が機能性3量体を形成し、DC上のCD40受容体に結合することを示している。
【0107】
b)Ad-sig-ecdhMUC1-HSF1-ΔCtΔTmCD40L-HISから発現されるecdhMUC1-HSF1-ΔCtΔTmCD40L融合タンパク質による3量体形成の評価
ecdhMUC1-HSF1-ΔCtΔTmCD40L-HIS融合タンパク質の3量体化を、Ad-sig-ecdhMUC1-HSF1-ΔCtΔTmCD40L-HISベクターで形質転換した細胞からの放出に従って評価した。発現された融合タンパク質を、Hisタグ精製キットを用いてベクターに暴露した293細胞の上清から精製した。非変性ゲル電気泳動は、分子量が3量体の形成と矛盾しないことを示した。
【0108】
c)MUC1発現癌細胞の確立に対するAd-sig-ecdhMUC1-ΔCtΔTmCD40Lベクター注射の影響
Ad-sig-ecdhMUC1-ΔCtΔTmCD40L (マウス)ベクターを皮下注射したhMUC1.TgマウスはhMUC1ポジティブLL2/LL1hMUC1マウス癌細胞による移植に対して耐性を示した。ベクターを注射しないコントロール動物は同じ細胞の増殖に対して耐性を示さなかった。また、Ad-sig-ecdhMUC1/ecdCD40L (マウス)ベクターを注射したhMUC1.Tgマウスは、MUC1を発現しない親細胞系(LL2/LL1)による移植に対して耐性を示さなかった。
【0109】
ecdhMUC1-ΔCtΔTmCD40L(マウス)タンパク質を静脈内注射したhMUC1.TgマウスはhMUC1ポジティブLL2/LL1hMUC1マウス癌細胞による移植に対して耐性を示さなかった。更に、Ad-sig-ecdhMUC1-ΔCtΔTmCD40L(マウス)ベクターを注射したhMUC1.Tgマウスはコントロールベクター注射し、その後LL2/LL1hMUC1細胞系を投与したマウスより長く生存した。
【0110】
3.不顕性の崩壊の根底にある細胞機構
a)ワクチン接種マウスvs非ワクチン接種マウスからのサイトカイン放出
脾臓CD8+ Tリンパ球の集団を、Ad-sig-ecdhMUC1-ΔCtΔTmCD40L(マウス)ベクター投与の7日後、CD4+抗体をコーティングしたビーズを用いてCD4+ Tリンパ球を涸渇させることによって得た。単離したCD8+ Tリンパ球は、コントロールベクター(MUC1未含有)を投与したMUC1.Tgマウス由来のCD8+ T細胞の2,000倍高いレベルのインターフェロンガンマを放出した。
【0111】
b)細胞傷害性アッセイ
Ad-sig-ecdhMUC1-ΔCtΔTmCD40L(マウス)ベクターの投与7日後のhMUC1.Tgマウスから回収した脾臓T細胞をhMUC1抗原ポジティブLL2/LL1hMUC1癌細胞と共にインビトロで7日間培養した。刺激した膵臓T細胞を種々の比率のhMUC1ポジティブLL2/LL1hMUC1癌細胞またはhMUC1ネガティブLL2/LL1癌細胞と混合した。結果は、Ad-sig-ecdhMUC1-ΔCtΔTmCD40L(マウス)ベクター接種マウス由来のT細胞がhMUC1を発現する癌細胞にだけ細胞傷害性を有することを示した。
【0112】
c)Ad-sig-ecdhMUC1-ΔCtΔTmCD40Lベクター注射によってhMUC1特異的T細胞の増殖に対する耐性が克服される
インビトロで骨髄細胞から得たDCをAd-sig-ecdhMUC1-ΔCtΔTmCD40L(マウス)ベクターに48時間暴露した。脾臓CD8+ T細胞(ベクターを注射しない、またはAd- sig-ecdhMUC1-ΔCtΔTmCD40L(マウス)ベクターを皮下注射した7日後のhMUC1.Tgトランスジェニックマウスから採取)を1/1の割合でAd- sig-ecdhMUC1/ecdCD40L(マウス)ベクター感染DCと混合した。ERK1/EK2タンパク質(Ras/MAPKシグナリング経路の終点)は、Ad-sig-ecdhMUC1-ΔCtΔTmCD40L(マウス)ベクター感染DCへのインビトロでの45分間の暴露後にAd-sig-ecdhMUC1-ΔCtΔTmCD40Lベクター注射hMUC1.Tgトランスジェニックマウスから単離したCD8+ T細胞においてリン酸化された。これに対して未接種hMUC1.Tgマウス由来のCD8ポジティブT細胞ではERK1およびERK2タンパク質のリン酸化の上昇は観察されなかった。これらの結果は、Ad-sig-ecdhMUC1-ΔCtΔTmCD40L(マウス)ベクターを接種したMUC1.Tgトランスジェニックマウス由来のCD8ポジティブT細胞がもはやMUC1に対して不顕性ではなくなったことを示している。
【0113】
4.融合タンパク質およびベクターの生成
腫瘍抗原融合タンパク質を、融合タンパク質をコードする融合遺伝子の発現カセットを保持するアデノウイルスベクターから直接生成した。増殖培地中の80%の細胞密度の生成細胞(例えば293細胞系)を、細胞当たり10-100ウイルス粒子の比率でウイルスベクターに感染させた。感染した細胞を更に48-72時間培養したが、この時点でウイルスベクターは細胞中で増殖し、腫瘍抗原融合タンパク質が細胞中で発現され、培養液中に分泌された。感染した細胞の70-90%が細胞変性効果(CPE)を示した時点でそれらを回収した。細胞培養液を分離回収した。細胞溶解産物を3回の凍結/解凍サイクル処理した。標準的な方法で(19)ウイルス粒子を単離した。アフィニティークロマトグラフィーによって、回収した細胞培養液から腫瘍抗原融合タンパク質を精製した。
【0114】
5.タンパク質ブースト投与による免疫応答の増幅
腫瘍抗原融合タンパク質のブースト投与とアデノウイルス発現ベクターのブースト投与の相対値を評価した。
【0115】
記載のようにhMUC-1.Tg動物にAd-K/ecdhMUC1-ΔCtΔTmCD40Lベクターを皮下投与してプライミングした。10マイクログラムのecdhMUC-1/ecdCD40L融合タンパク質から構成されるタンパク質ブーストを皮下注射した。タンパク質ブースト投与の時期およびベクターとの比較を表2に示す種々の処理群で評価した。
【0116】
【表2】
【0117】
異なる群からの脾臓細胞を単離し、インターフェロンガンマ陽性率(positivity)についてELISPOTアッセイによって評価した。図3に見られるように、最初のベクター注射の1週間後から開始して14日間隔で2回タンパク質を皮下注射した場合、未処理、またはタンパク質ブースト投与を行わずに1もしくは2回ベクターを注射した場合に比較してポジティブT細胞の頻度が大きく上昇した。インターフェロンガンマポジティブT細胞の頻度が次に高かったのはT3群(最初のベクター注射の7日後に1回タンパク質を注射)であった。
【0118】
種々の免疫群における細胞傷害性T細胞の樹立を評価した(図4)。種々の処理群からの脾臓細胞をインビトロで、hMUC-1ポジティブ細胞系(LL1/LL2hMUC-1)を用いて5日間刺激した。CD8 T細胞を単離し、標的細胞(LL1/LL2hMUC-1)と50/1の割合で混合した。細胞傷害性は概ねELISPOTアッセイの結果に従い、T5群が最も高い上昇レベルのLL1/LL2hMUC-1特異的細胞傷害性T細胞活性を示した。T5群からのT細胞で観察された細胞傷害性のレベルはネガティブコントロール群で観察されたものの9倍であった。
【0119】
ELISAで種々の処理群の動物由来の血清を抗ecdhMUC1-ΔCtΔTmCD40L特異的抗体について評価した。簡潔に記載すると、ecdhMUC1-ΔCtΔTmCD40Lタンパク質をコーティングしたマイクロウェルを試験マウス血清と共にインキュベートし、洗浄し、ホースラディッシュペルオキシダーゼにコンジュゲートさせたラット抗マウス2次抗体を用いて、結合したマウス抗体を同定した。
【0120】
図5は、1回のベクター注射および2回のタンパク質注射(14日間隔)での処理によって生じるecdhMUC1-ΔCtΔTmCD40L融合タンパク質に対する抗体のレベルが劇的に上昇することを示している。T5処理後の抗ecdhMUC1-ΔCtΔTmCD40L抗体の増加は他のいずれの処理群のものより2倍高かった。
【0121】
これらのアッセイからの結果は、タンパク質のブースト投与が腫瘍抗原発現細胞に対する細胞傷害性T細胞活性の生成および腫瘍抗原に対する抗体の反応において、ベクターのブースト投与より優れていることを示している。タンパク質のブースト投与に関して総体的に最良の結果が得られるのは、アデノウイルス発現ベクター注射を1回行い、その1週間後にタンパク質を皮下注射してブーストし、2週間後にタンパク質ブーストをもう1度反復した場合であった。
【0122】
ワクチン接種したhMUC-1.Tgマウス由来の血清中の抗体を、癌バイオプシー組織標本への結合に関して評価した。正常な乳組織切片および乳癌組織切片を含む組織マイクロアレイを購入した。Ad-K/ecdhMUC-1//ΔCtΔTm CD40Lベクターで免疫化し、その後ecdhMUC-1//ΔCtΔTm CD40Lタンパク質をブースト投与したトランスジェニックマウス由来の血清と組織を接触させた。アレイを洗浄した後、マウスIgG抗体を認識するホースラディッシュペルオキシダーゼ(HRP)2次抗体に暴露した。コントロールとして血清をまずhMUC-1ドメインの抗原性リピートからのhMUC-1ペプチドに暴露した(タンパク質ブーストに使用したものと同じもの)。
【0123】
ワクチン接種マウス由来の血清は癌性上皮細胞のバイオプシー標本からの乳上皮細胞に結合した。間にある線維芽細胞または間質細胞への結合は観察されなかった。正常マウス由来の血清は反応を示さなかった。
【0124】
Ad-sig-hMUC-1/ecdCD40L で免疫化し、その後タンパク質sc-hMUC-1/ecdCD40L を2回連続して皮下投与したhMUC-1.Tgマウス由来の血清は組織マイクロアレイスライド上でヒト前立腺癌由来のバイオプシー標本と反応した。
【0125】
hMUC-1リピートに対して血清から産生された抗体の特異性を確認するために、hMUC-1リピートからのアミノ酸配列を含有するペプチドを、量を漸増させて、上記のワクチン接種動物由来の血清と混合した。次いで混合物をマイクロアレイスライドに適用し、反応性を評価した。コントロールとして、hMUC-1リピートと同じアミノ酸を有するが配列順序が異なるペプチド(“スクランブルペプチド”)をワクチン接種した動物由来の血清に添加した。hMUC-1ペプチドによって、ワクチン接種動物血清中の抗体の乳癌上皮細胞への結合が阻害された。スクランブルペプチドでは阻害は観察されなかった。これらのことは、ベクタープライム/タンパク質ブーストワクチン接種によって、ヒト乳癌上皮細胞のバイオプシー標本によって発現されるMUC-1と反応性のあるhMUC-1特異的体液性反応が誘導されたことを示している。
【0126】
タンパク質のブースト投与を行ったマウスにおける腫瘍免疫を評価した。記載のようにhMUC-1.Tg動物にAd-K/ecdhMUC1-ΔCtΔTmCD40Lベクターを皮下投与してプライミングするか、またはecdhMUC1-ΔCtΔTmCD40L融合タンパク質を1もしくは2回投与して免疫化した。その後動物をLL2/LL1hMUC-1腫瘍細胞で攻撃感染させた。
【0127】
図6に示すように、Ad-K/ecdhMUC1-ΔCtΔTmCD40Lベクターを接種したマウスは120日より長く生存したが(実線)、Ad-sig-ecdhMUC-1/ecdCD40Lベクターを接種しなかったマウスは全て50日までに死亡した(波線)。これらの結果は、ベクター注射によりhMUC-1.TgマウスにおけるLL2/LL1hMUC-1細胞系増殖の抑制が誘導されたことを示している。
【0128】
hMUC-1抗原に関する腫瘍増殖抑制の特異性を評価するために、LL2/LL1hMUC-1細胞系(hMUC-1抗原陽性)の拒絶をLL2/LL1細胞系(hMUC-1抗原が存在しないこと以外は同じ)と比較した。結果は、アデノウイルスベクターの皮下注射によってLL2/LL1hMUC-1細胞系の増殖は完全に抑制されるが、同じ細胞でMUC-1を発現しないものは抑制されなかったことを示した。
【0129】
ベクターおよびタンパク質投与の組み合わせによって腫瘍増殖抑制を評価した。Ad-sig-ecdhMUC-1/ecdCD40LベクターおよびecdhMUC-1/ecdCD40Lタンパク質を3種の組み合わせでhMUC-1.Tgマウスに投与した後、LL2/LL1hMUC-1腫瘍細胞で攻撃感染した。VVV= Ad-sig-ecdhMUC-1/ΔCtΔTm CD40Lベクターを3回、1、7、および21日目に皮下注射;PPP= ecdhMUC-1/ΔCtΔTm CD40Lタンパク質を3回、1、7、および21日目に皮下注射;またはVPP=Ad-sig-ecdhMUC-1/ΔCtΔTm CD40Lベクターを1回皮下注射した後、7および21日目にecdhMUC-1/ΔCtΔTm CD40Lタンパク質を皮下注射。更に詳細には図7参照。1週間後、マウスに500,000の LL2/LL1hMUC-1肺癌細胞を皮下注射して攻撃感染し、2週間後に500,000のLL2/LL1hMUC-1腫瘍細胞を静脈内注射した。カリパーを用いて複数の時点でその日の皮下腫瘍結節のサイズを測定し、LL2/LL1hMUC-1細胞の増殖に対する種々のワクチンスケジュールの効果を皮下結節として測定した。殺処理後、肺の総重量によって転移を測定した。
【0130】
図7に示すように、あらかじめAd-sig-ecdhMUC-1/ecdCD40Lベクター注射をせずに融合タンパク質を3回注射したところ(PPP)、皮下LL2/LL1hMUC-1腫瘍の発生に対する完全な耐性は誘導されなかった。それに対し、3回連続でベクターを注射するか(VVV)、または1回のベクター注射後にタンパク質を2回注射する(VPP)スケジュールでは、皮下LL2/LL1hMUC-1腫瘍の発生が完全に抑制された。
【0131】
これらのマウスにおけるワクチン接種開始の63日後のhMUC-1特異的抗体のレベルを測定した(図8)。1回のベクター注射後2回連続で融合タンパク質をブースト注射するというスケジュール(VPP)では最も高いレベルのhMUC-1特異的抗体が誘導され、スケジュールVVVでは中程度、そしてVPPでは実質的に効果がなかった。従って、これらの動物における癌治療は抗体反応と幾分逆の関係にある。
【0132】
(定着後の)腫瘍治療プロトコールも評価した。このスケジュールでは、皮下腫瘍(500,000のLL2/LL1hMUC-1)を1日目に投与した。3種のスケジュール(PPP、VPP、およびVVV)を5、12、および26日目実施した。35日目に腫瘍を静脈内投与し、腫瘍の発生(皮下および肺)を49日目に評価した。更なる詳細を図9の説明書きに記載する。
【0133】
図9に示すように、1回のベクター注射とその後2回のタンパク質注射の組み合わせ(VPP)では、定着後の皮下hMUC-1ポジティブ癌細胞腫瘍の増殖が完全に抑制された。3回連続のベクター投与(VVV)ではわずかな治療効果が見られたが、3回連続のタンパク質注射(PPP)ではほとんど、または全く効果が無かった。
【0134】
処置前および処置後(定着前)癌モデルにおける肺転移結節の増殖を図10に示す。図10の前処理の結果(左側のパネル)は、3回連続の融合タンパク質注射(PPP)が肺結節の増殖を抑制しなかったと考えられることを示している。これに対し、スケジュールVVVおよびスケジュールVPPはワクチン接種動物の肺における肺癌の定着を完全に抑制すると考えられた。
【0135】
図10の後処理の結果(右側のパネル)は、1回のベクター注射とその後2回のタンパク質注射の組み合わせ(VPP)では定着したhMUC-1ポジティブ癌細胞の肺結節の増殖が完全に抑制されたことを示している。これに対し、3回連続のベクター投与(VVV)および3回連続のタンパク質注射(PPP)はいくらかの治療効果は示したものの、VPPプロトコールより小さなものだった。
【0136】
これらの結果は、総括的に最良の治療スケジュールはVPPスケジュールであることを示しており、このスケジュールは1回のAd-sig-ecdhMUC-1/ecdCD40Lベクター注射を行い、その1週間後に2回連続のecdhMUC-1/ecdCD40L タンパク質皮下注射を2週間の間隔をおいて行うものである。このプロトコールはムチン抗原に対する抗体(体液性免疫)およびT細胞免疫(細胞性免疫)の誘導を特徴とする。
【0137】
同じタンパク質をコードするアデノウイルス発現ベクターを初期投与した後のブースティングをecdMUC-1/ecdCD40L可溶性タンパク質で行う場合と他の可溶性タンパク質で行う場合を比較するために、MUC-1発現腫瘍(LL2/LL1hMUC-1細胞系)に攻撃感染させたhMUC-1.Tg動物で評価を行った。以下を含有する細菌抽出物で動物をブースティングした: ecdMUC-1/ecdCD40(Ad-sig-ecdMUC-1/ecdCD40Lベクターに感染させた細菌ホスト株由来);キーホールリンペットヘモシアニン(KLH)に結合したecdMUC-1(フロイドの不完全アジュバントと共に、または無しで);PBS;およびコントロール細菌抽出物(Ad-sig-ecdMUC-1/ecdCD40Lベクターに感染していない細菌ホスト株由来)。2回目のタンパク質ブースト投与完了の7日後に腫瘍細胞を投与した。図12に示す結果は、ecdMUC-1/ecdCD40L可溶性タンパク質でのブースティングが他の全ての方法より優れていることを示している。
【0138】
6.HPV E7-CD40リガンド融合タンパク質をコードするアデノウイルスベクターの構築
分泌型CD40リガンドに結合したE7から構成されうる転写ユニット融合タンパク質を発現するアデノウイルスベクターの投与によって免疫を生じさせる方法が近年報告された(Methods of generating immunity by administering and adenoviral vector expressing a transcription unit fusion protein constituting E7 linked to a secretable form of CD40 ligand was recently reported)。Ziangら, "An adenoviral vector cancer vaccine that delivers a tumor-associated antigen/CD40-ligand fusion protein to dendritic cells" Proc. Natl. Acad. Sci (USA)(2003年11月25日発行), 10.1073/pnas.2135379100 (vol. 100(25):15101)。
【0139】
転写ユニットは、ΔCtΔTmCD40Lの上流に完全長のHPV 16型 E7タンパク質をコードするDNA、その上流にHGH遺伝子由来のシグナルペプチドをコードするDNAを含む。ヒト成長ホルモンシグナル配列MATGSRTSLLLAFGLLCLPWLQEGSA(1文字アミノ酸コード)(SEQ ID NO:32)をコードするDNAを調製するために、ホスホリル化したオリゴヌクレオチド(SEQ ID NO:33および34)をアニールさせ、Bgl IIおよびNot1オーバーハングを有する完全長の26アミノ酸HGH配列を生成した。
成長ホルモンシグナル上流鎖(イタリック体(小文字)のコード配列):
5'-GATCT CCACC atg gct aca ggc tcc cgg acg tcc ctg ctc ctg gct ttt ggc ctg ctc tgc ctg ccc tgg ctt caa gag ggc agt gcc GGC -3'(SEQ ID NO: 33)
成長ホルモンシグナル下流鎖:
3'-A GGTGG TAC CGA TGT CCG AGG GCC TGC AGG GAC GAG GAC CGA AAA CCG GAC GAG ACG GAC GGG ACC GAA GTT CTC CCG TCA CGG CCGCCGG -5'(SEQ ID NO:34)
【0140】
上記の上流および下流オリゴ鎖をアニールさせて合成HGHシグナル配列を調製した。オリゴ鎖を50μlのH2O(約3mg/ml)に溶解した。各オリゴ鎖(上流および下流鎖)からの1μlを48μlのアニーリングバッファー(100mM 酢酸カリウム、30mM HEPES-KOH pH 7.4、および2mM Mg-酢酸)に添加し、95℃で4分間、70℃で10分間インキュベートし、約4℃まで徐々に冷却した。標準的な条件下でT4 PNK(ポリヌクレオチドキナーゼ)を用いてアニールさせたDNAをホスホリル化した。
【0141】
Bgl IIおよびNot Iオーバーハングを有するHGHシグナル配列をBgl IIおよびNot Iを介してpShuttle-E7-ΔCtΔTmCD40L(シグナル配列未含有)に挿入し、pshuttle-HGH/E7-ΔCtΔTmCD40Lを得た。以下のように、CD40リガンド配列の上流にHPV-16 E7を挿入し、pShuttle-E7-ΔCtΔTmCD40L(シグナル配列未含有)を調製した:完全長のHPV-16 E7タンパク質をコードする配列を、以下のプライマーを用いてHPVウイルスゲノムからのPCR増幅によって得た:
HPV 16 E7順向きプライマー(SEQ ID NO: 35)
5'-ATTT GCGGCCGC TGTAATCATGCATGGAGA-3'
HPV E7逆向きプライマー(SEQ ID NO: 36)
5-CC CTCGAG TTATGGTTTCTGAGAACAGAT-3'
【0142】
得られた増幅子は5'末端にNot I、3'末端にXho I制限部位を有する、HPV 16 E7をコードするDNAであった。Not I(GCGGCCGC)およびXho I(CTCGAG)を用いて、pShuttleΔCtΔTmCD40L のCMVプロモーターとΔCtΔTMCD40L配列のスペーサーのすぐ5'側との間にE7 DNAを挿入した。プラスミドをpShuttle-E7-ΔCtΔTmCD40L(シグナル配列未含有)と記載し、これを用いてE7の上流にHGHシグナル配列を挿入し、既に記載したようにHGH/E7-ΔCtΔTmCD40Lを生成した。従って転写ユニットHGH/E7-ΔCtΔTmCD40LはHGH分泌シグナル、次いで完全長のHPV16型E7、次いで10アミノ酸リンカー(FENDAQAPKS; SEQ ID NO: 37)、次いでマウスCD40リガンド残基52-260をコードする。
【0143】
完全長HPV16型 E7タンパク質をコードするDNAの上流にマウスIgGカッパ鎖遺伝子のシグナル配列をコードするDNAを含む転写ユニット(“K/E7”)を、HPV16プラスミドおよび以下のプライマーを使用してPCRで生成した:
(プライマー1)5'-ACG ATG GAG ACA GAC ACA Ctc ctg cta tgg gta ctg ctg-3'(SEQ ID NO: 38);
(プライマー2)5'- TC CTG CTA TGG GTA CTG CTG CTC tgg gtt cca ggt tc-3'(SEQ ID NO: 39);
(プライマー3)5'- TG CTC TGG GTT CCA ggt tcc act ggt gac atg cat g-3'
(SEQ ID NO: 40);
(プライマー4)5'- TGG GTT CCA GGT TCC ACT GGT GAC ATG CAT GGA G AT ACA CCT AC-3'(SEQ ID NO: 41);および
(プライマー5)5'- CCG CTC GAG TGG TTT CTG AGA ACA GAT GGG GCA C -3.'(SEQ ID NO: 42)
【0144】
上流カッパシグナル配列を有するK/E7を4ラウンドのPCR増幅によって生成した(第1ラウンド:プライマー4+5;第2ラウンド:プライマー3を添加;第3ラウンド:プライマー2を添加;第4ラウンド:プライマー1を添加)。K/E7をコードするDNAをpcDNATM 3.1 TOPOベクター(Invitrogen, San Diego, CA)にクローニングしてpcDNA-K/E7を生成した。
【0145】
膜貫通ドメインおよび細胞質ドメインを欠失させたマウスCD40リガンドを含むDNAフラグメント(ΔCtΔTmCD40L)を、以下のPCRプライマーを用いてマウスCD40リガンドcDNAプラスミド(pDC406-mCD40L;ATCC)から生成した:
5'-CCG CTCGAG aac gac gca caa gca cca aaa agc AAG GTC GAA GAG GAA GTA AAC CTT C-3'(SEQ ID NO: 43);および
5'-CGCGCCGCGCGCTAGtctagaGAGTTTGAGTAAGCCAAAAGATGAG-3'(SEQ ID NO: 44)(ハイファイデリティー(high fidelity)PCRキット, Roche)
フラグメントΔCtΔTmCD40LをXba IおよびXho I制限エンドヌクレアーゼで消化し、pcDNA-E7にライゲートした。K/E7-ΔCtΔTmCD40LフラグメントをpcDNAベクターから切り出し、Hind IIIおよびXba I部位を用いてpShuttleプラスミドに挿入した(pShuttle K/E7-ΔCtΔTmCD40L)。従って、K/E7-ΔCtΔTmCD40Lフラグメントはカッパ鎖分泌シグナル、次いで完全長HPV16型E7、次いで10アミノ酸リンカー(LQNDAQAPKS; SEQ ID NO: 31)、次いでマウスCD40リガンド残基52-260を含む。
【0146】
膜貫通ドメインを欠失したヒトCD40リガンドに融合したE7をコードするベクターを調製するために、“スペース+ΔCtΔTmCD40L(ヒト)”(上記のように調製)をプラスミドpShuttle-CMV(13)に、Hind III(AAGCTT)およびXho I (CTCGAG)での制限エンドヌクレアーゼ消化後に挿入した。このベクターをpShuttleΔCtΔTmCD40L(ヒト)と記載する。pShuttleΔCtΔTmCD40L(ヒト)を修飾してヒトCD40リガンド配列の上流にHPV-16 E7を含有させたが、これは本質的に上でマウスCD40リガンドをコードするベクターに関して記載したようにして行った。得られたプラスミドをpShuttle-E7-ΔCtΔTmCD40L(ヒト)(シグナル配列未含有)と記載し、これを用いてE7の上流にHGHシグナル配列を挿入し、HGH/E7-ΔCtΔTmCD40L(ヒト)を生成する。従って、転写ユニットHGH/E7-ΔCtΔTmCD40L(ヒト)はHGH分泌シグナル、次いで完全長HPV16型E7、次いで10アミノ酸リンカー(FENDAQAPKS; SEQ ID NO:19)、次いでヒトCD40リガンド残基47-261をコードする。
【0147】
7.ラットHER2(Neu)/CD40Lをコードするアデノウイルスベクターの構築
乳癌の30%で、Her-2-Neu(H2N)増殖因子受容体の過剰発現が術後の再発頻度の増加および生存期間の短縮と関連している。HER2(“H2N”または“rH2N”)遺伝子のラット相当物のトランスジェニックであり、そのためこの遺伝子に寛容であるマウス(Mullerら, Cell 54: 105-115, (1998);Gutら Proc. Natl. Acad. Sci. USA 89: 10578-10582, (1992))を実験用ホストとして使用し、Ad-sig-rH2N/ecdCD40Lベクターでの免疫を評価した。このモデルにおいて、乳特異的転写プロモーター(例えばMMTVプロモーター)の制御下でマウスを正常な非活性化ラットHer-2-Neu遺伝子のトランスジェニックとした。MMTVプロモーターは非変異ラットHer-2-Neu受容体の過剰発現を起こすが、これはヒト乳癌で起こるものと類似している。このモデルでは24週間で原発組織(乳)に触知可能な腫瘍結節、32週間で肺転移が発生する。乳癌は自然発生する。癌はクローン性事象(clonal event)として乳上皮組織で段階的な過程を通して局所的に開始される(同上)。異形成は生後12週で検出できる。乳腺における触知可能な腫瘍は25週で検出でき、乳癌の肺転移は32週までに70%のマウスで確認される(同上)。
【0148】
Ad-sig-rH2N/ecdCD40Lベクターを1または2回(7日間隔)、トランスジェニック動物に皮下投与し、ラットHer-2-Neu抗原に対して免疫応答が誘導されるかどうか試験した。2回のAd-sig-rH2N/ecdCD40Lベクターの皮下注射によりN202(rH2Nポジティブ)マウス乳癌細胞系の増殖に対する完全な耐性が誘導されたが、同じベクターの1回の皮下注射ではrH2NポジティブN202細胞系の増殖を完全に抑えるのに十分な免疫応答は誘導されなかった。ELISPOTアッセイでは、7日間隔で2回Ad-sig-rH2N/ecdCD40Lベクターを皮下注射することにより、マウスにおいてAd-sig-rH2N/ecdCD40Lベクターを1回注射した後に誘導されるrH2N特異的T細胞のレベルより10倍高いレベルのrH2N特異的T細胞が、ワクチン接種したマウスの脾臓において誘導されたことが明らかになった。最終的に、Ad-sig-rH2N/ecdCD40Lベクターのプライム免疫によってNT2細胞に対して誘導される免疫抵抗は、放射線照射したサイトカインポジティブ腫瘍細胞(GMCSF転写ユニットをトランスフェクトしたNTW細胞をミトマイシン処理したもの)を接種したトランスジェニック動物で得られる応答より良好であった。
【0149】
Ad-sig-rH2N/ecdCD40Lベクターの皮下注射を1または2回行ったマウスにおけるrH2N特異的抗体レベルも測定した。以下の図11に示すように、rH2N特異的抗体のレベルはAd-sig-rH2N/ecdCD40Lベクターを2回皮下注射した後の方が1回皮下注射した後より高かった。
【0150】
8.huHER2/CD40Lをコードするアデノウイルスベクターの構築
sig ecdhuHER2/CD40Lをコードするアデノウイルスベクターを以下のように調製した。マウスIgGカッパ鎖METDTLLLWVLLLWVPGSTGD(1文字アミノ酸コード)(SEQ ID NO: 11)をPCR増幅(SEQ ID NO: 12、13、および45)によって調製し、完全長の21アミノ酸マウスIgGカッパ鎖シグナル配列を生成した(SEQ ID NO:12で、開始コドン“ATG”を太字(下線)で示す)。
5'-CCACC ATG GAG ACA GAC ACA CTC CTG CTA TGG GTA CTG CTG-3'
(SEQ ID NO: 12)
5'- TC CTG CTA TGG GTA CTG CTG CTC TGG GTT CCA GGT TC-3'
(SEQ ID NO:13)
順向きプライマー
5'-5'- TG CTC TGG GTT CCA GGT TCC ACT GGT GAC GAA CTC -3'
(SEQ ID NO:45)
ヒトHER2細胞外ドメインの順向きプライマー
5'- TCC ACT GGT GAC GAACTCACCTACCTGCCCACCAATGC-3'
(SEQ ID NO:46)
ヒトHER2細胞外ドメインの逆向きプライマー
5'- GGAGCTCGAG GGCTGGGTCCCCATCAAAGCTCTC-3'
(SEQ ID NO:47)
【0151】
上流カッパシグナル配列を有するsig-ecdhHER2を4ラウンドのPCR増幅によって生成する(第1ラウンド:プライマーSEQ ID NO:46および47;第2ラウンド:プライマーSEQ ID NO:45および47;第3ラウンドSEQ ID NO:13および47;第4ラウンド:プライマーSEQ ID NO:12および47)。sig-ecdhHER2をコードするDNAをpcDNATM 3.1 TOPOベクター(Invitrogen, San Diego, CA)にクローニングしてpcDNA-sig-ecdhHER2を生成できる。MUC-1/CD40リガンド発現ベクターに関して記載した更なるクローニング段階をHER2/CD40リガンド発現ベクターにも適用する。
【0152】
この領域、CD40リガンドに融合させるべきHER2細胞外ドメインは2つのCTLエピトープを含有する;1つはHLA-A2ペプチド、K I F G S L A F L(SEQ ID NO:48)であり、アミノ酸369-377に相当する。このペプチドはHER2発現癌患者において一時的なペプチド特異的免疫を引き起こした。Knutsonら, Immunization of cancer patients with a HER-2/neu, HLA-A2 peptide, Clin Cancer Res. 2002 May;8(5):1014-8p369-377参照。第2のエピトープはE L T Y L P T N A S(SEQ ID NO :49)(HER2残基63-71)であり、HER2発現腫瘍細胞に対する免疫の生成にも有用であった。Wang ら, Essential roles of tumor-derived helper T cell epitopes for an effective peptide-based tumor vaccine, Cancer Immun. 2003 Nov 21;3:16参照。HER2 ecdの領域はB細胞エピトープ、P L H N Q E V T A E D G T Q R C E K C S K P C(SEQ ID NO: 50)(HER2の316-339位)も含有する。Dakappagari ら, Chimeric multi-human epidermal growth factor receptor-2 B cell epitope peptide vaccine mediates superior antitumor responses, J Immunol. 2003 Apr 15;170(8):4242-53参照。
【0153】
本明細書に記載する全ての特許および文献は本発明が属する分野の技術者のレベルを示すものである。全ての特許および文献は、個々の文献が特定かつ個別に参照によって組み込まれることが示されるのと同じ程度まで、参照により本明細書に組み込まれる。
【0154】
本明細書に例証的に記載する本発明は、任意の要素(単数または複数)、制限(単数または複数)無しで好適に実施してもよいが、それらについては本明細書で特に開示しない。従って、例えば本明細書においてそれぞれの場合で、“〜を含む”、“本質的に〜からなる”、および“〜からなる”という用語の任意のものを他の2つのいずれかで置き換えてもよい。使用した用語および表現は制限ではなく説明のための用語として使用されるものであり、それらの用語および表現の使用に際して明示および記載する特徴と同等のものまたはそれらの一部のいずれを排除することも意図しないが、認識されるように、種々の修飾が請求する本発明の範囲内で可能である。従って、好ましい態様および必要に応じた(optional)特徴によって本発明についての特定の開示を行ったが、当業者は本明細書に開示するコンセプトの修飾および変更を行ってもよく、それらの修飾および変更は添付する請求の範囲に定義する本発明の範囲内にあると見なされることは理解すべきである。
【0155】
他の態様を添付の請求の範囲内に記載する。
【図面の簡単な説明】
【0156】
図面の簡単な説明
【図1A】図1は、ヒトMUC1をコードするヌクレオチド配列である(SEQ ID NO : 1)。
【図1B】図1は、ヒトMUC1をコードするヌクレオチド配列である(SEQ ID NO : 1)。
【図2】図2は、ヒトMUC1のアミノ酸配列である(SEQ ID NO : 2)。
【図3】図3は、ELISAスポットアッセイで得られたインターフェロンガンマのレベルを示す。
【図4】図4は、アデノウイルス発現ベクターAd-K/ecdhMUC1-ΔCtΔTmCD40Lでプライミングし、発現ベクターまたは成熟融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lのいずれかを皮下にブースト投与したMUC-1トランスジェニック動物(hMUC-1.Tg)由来のT細胞傷害性のレベルを示す。
【図5】図5は、アデノウイルス発現ベクターAd-K/ecdhMUC1-ΔCtΔTmCD40Lでプライミングし、発現ベクターまたは成熟融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lのいずれかを皮下にブースト投与したMUC-1トランスジェニック動物(hMUC-1.Tg)の血清中の融合タンパク質ecdhMUC1-ΔCtΔTmCD40Lに対する抗体のレベルを示す。
【図6】図6は、アデノウイルス発現ベクターAd-K/ecdhMUC1-ΔCtΔTmCD40Lを投与したMUC-1トランスジェニック動物(hMUC-1.Tg)におけるMUC1発現腫瘍細胞(LL2/LL2hMUC-1)の増殖レベルを示す。
【図7】図7は、Ad-sig-ecdhMUC-1/ΔCtΔTmCD40LベクターおよびecdhMUC-1/ΔCtΔTm CD40Lタンパク質を接種した動物における腫瘍の予防を示す。
【図8】図8は、ワクチン接種開始の63日後のワクチン接種マウスにおけるhMUC-1特異的抗体のレベルを示す。
【図9】図9は、Ad-sig-ecdhMUC-1/ΔCtΔTmCD40LベクターおよびecdhMUC-1/ΔCtΔTm CD40Lタンパク質で免疫化した動物における(定着後の)皮下腫瘍療法を示す。
【図10】図10は、図9に記載するように処理した動物における(定着後の)肺転移腫瘍結節療法を示す。
【図11】図11は、MUC1発現腫瘍の発生に抵抗する動物の能力に関する、Ad-sig-ecdhMUC-1/ecdCD40Lベクターの1回皮下投与後の種々のブースト計画の比較である。
【特許請求の範囲】
【請求項1】
個体において抗原に対する免疫応答を生成させる方法であって、
a)個体に有効量の発現ベクターを投与し、ここで、該ベクターは分泌可能な融合タンパク質をコードする転写ユニットを含み、該融合タンパク質は抗原およびCD40リガンドを含み;そして
b)抗原およびCD40リガンドを含む有効量の融合タンパク質を投与する、
ことを含む上記方法。
【請求項2】
該タンパク質がベクターの投与後に投与される、請求項1記載の方法。
【請求項3】
該抗原がポリペプチド抗原である、請求項1記載の方法。
【請求項4】
該抗原が感染物質抗原である、請求項1記載の方法。
【請求項5】
該免疫応答が抗原を発現する細胞に対するものである、請求項1記載の方法。
【請求項6】
該免疫応答が抗原を発現する微生物に対するものである、請求項1記載の方法。
【請求項7】
該抗原が腫瘍抗原である、請求項1記載の方法。
【請求項8】
該腫瘍抗原がHER-2由来である、請求項1記載の方法。
【請求項9】
該腫瘍抗原がムチンである、請求項1記載の方法。
【請求項10】
該腫瘍抗原がMUC1、MUC2、MUC3A、MUC3B、MUC4、MUC5AC、MUC5B、MUC6、MUC7、MUC8、MUC9、MUC12、MUC13、MUC15、およびMUC16からなる群から選択されるムチン由来である、請求項5記載の方法。
【請求項11】
該ムチン抗原がMUC1由来である、請求項9記載の方法。
【請求項12】
該ムチン抗原がムチンの細胞外ドメインを含む、請求項9記載の方法。
【請求項13】
該ムチン抗原がムチンの少なくとも1つのタンデムリピートを含む、請求項9記載の方法。
【請求項14】
該ムチン抗原がMUC1の細胞外ドメインを含む、請求項9記載の方法。
【請求項15】
該抗原が個体中の自己抗原である、請求項1記載の方法。
【請求項16】
該抗原がヒト乳頭腫ウイルスのE7タンパク質である、請求項1記載の方法。
【請求項17】
該抗原が上皮癌細胞由来である、請求項1記載の方法。
【請求項18】
該転写ユニットが該抗原と該CD40リガンドとの間のリンカーをコードする、請求項1記載の方法。
【請求項19】
該ベクターが転写ユニットの転写を制御するためのヒトサイトメガロウイルスプロモーター/エンハンサーを含む、請求項1記載の方法。
【請求項20】
該ベクターがウイルスベクターである、請求項1記載の方法。
【請求項21】
該ウイルスベクターがアデノウイルスベクターである、請求項20記載の方法。
【請求項22】
該CD40リガンドがヒトCD40リガンドである、請求項1記載の方法。
【請求項23】
該CD40リガンドが細胞質ドメインを欠失している、請求項1記載の方法。
【請求項24】
該ベクターが膜貫通ドメインのいずれかの末端からの6以下の残基を含むCD40Lをコードする、請求項1記載の方法。
【請求項25】
該ベクターがCD40リガンドの膜貫通ドメインをコードしない、請求項1記載の方法。
【請求項26】
該CD40リガンドがその膜貫通ドメインの全部または実質的に全部を欠失している、請求項1記載の方法。
【請求項27】
該CD40リガンドが残基47-261を含む、請求項1記載の方法。
【請求項28】
該CD40リガンドが残基1-23および47-261を含む、請求項1記載の方法。
【請求項29】
該ベクターが正常なヒト細胞において非複製性とされている、請求項1記載の方法。
【請求項30】
後の時点で、段階b)の反復である段階c)を含む、請求項1記載の方法。
【請求項31】
該免疫応答が該抗原に対する細胞傷害性CD8+ T細胞の生成を含む、請求項1記載の方法。
【請求項32】
該免疫応答が該抗原に対する抗体の産生を含む、請求項1記載の方法。
【請求項33】
該融合タンパク質がアジュバントと共に投与される、請求項1記載の方法。
【請求項34】
該融合タンパク質が皮下投与される、請求項1記載の方法。
【請求項35】
該ベクターにコードされるCD40リガンドの配列と融合タンパク質として投与されるCD40リガンドの配列とが異なる、請求項1記載の方法。
【請求項36】
該ベクターにコードされる抗原の配列と融合タンパク質として投与される抗原の配列とが異なる、請求項1記載の方法。
【請求項37】
転写ユニットが分泌シグナル配列をコードする、請求項1記載の方法。
【請求項38】
抗原がCD40リガンド由来ではない、請求項1記載の方法。
【請求項39】
腫瘍抗原を発現する癌に罹患した個体を治療する方法であって、
a)個体に有効量の発現ベクターを投与し、ここで、該ベクターは分泌可能な融合タンパク質をコードする転写ユニットを含み、該融合タンパク質は腫瘍抗原およびCD40リガンドを含み;そして
b)腫瘍抗原およびCD40リガンドを含む有効量の融合タンパク質を投与する、
ことを含む上記方法。
【請求項40】
該タンパク質がベクターの投与後に投与される、請求項39記載の方法。
【請求項41】
該腫瘍抗原がムチン抗原である、請求項39記載の方法。
【請求項42】
該腫瘍抗原がMUC1、MUC2、MUC3A、MUC3B、MUC4、MUC5AC、MUC5B、MUC6、MUC7、MUC8、MUC9、MUC12、MUC13、MUC15、およびMUC16からなる群から選択されるムチン由来である、請求項41記載の方法。
【請求項43】
該ムチン抗原がMUC1由来である、請求項41記載の方法。
【請求項44】
該ムチン抗原がムチンの細胞外ドメインを含む、請求項41記載の方法。
【請求項45】
該ムチン抗原がムチンのタンデムリピートを少なくとも1つ含む、請求項41記載の方法。
【請求項46】
該ムチン抗原がMUC1の細胞外ドメインを含む、請求項41記載の方法。
【請求項47】
該腫瘍抗原がヒト乳頭腫ウイルスのE7タンパク質である、請求項39記載の方法。
【請求項48】
該癌細胞が上皮癌細胞である、請求項39記載の方法。
【請求項49】
該癌細胞が子宮頸癌細胞である、請求項41記載の方法。
【請求項50】
該ベクターが転写ユニットの転写を制御するためのヒトサイトメガロウイルスプロモーター/エンハンサーを含む、請求項41記載の方法。
【請求項51】
該ベクターがウイルスベクターである、請求項41記載の方法。
【請求項52】
該ウイルスベクターがアデノウイルスベクターである、請求項51記載の方法。
【請求項53】
該CD40リガンドがヒトCD40リガンドである、請求項39記載の方法。
【請求項54】
該CD40リガンドが細胞質ドメインを欠失している、請求項39記載の方法。
【請求項55】
該ベクターが膜貫通ドメインのいずれかの末端からの6以下の残基を含むCD40Lをコードする、請求項39記載の方法。
【請求項56】
該ベクターがCD40リガンドの膜貫通ドメインをコードしない、請求項39記載の方法。
【請求項57】
該CD40リガンドがその膜貫通ドメインの全部または実質的に全部を欠失している、請求項39記載の方法。
【請求項58】
該CD40リガンドが残基47-261を含む、請求項39記載の方法。
【請求項59】
該CD40リガンドが残基1-23および47-261を含む、請求項39記載の方法。
【請求項60】
該ベクターが正常なヒト細胞において非複製性とされている、請求項39記載の方法。
【請求項61】
後の時点で、段階b)の反復である段階c)を含む、請求項39記載の方法。
【請求項62】
該免疫応答が該腫瘍抗原に対する細胞傷害性CD8+ T細胞の生成を含む、請求項39記載の方法。
【請求項63】
該免疫応答が該腫瘍抗原に対する抗体の産生を含む、請求項39記載の方法。
【請求項64】
該融合タンパク質がアジュバントと共に投与される、請求項39記載の方法。
【請求項65】
該融合タンパク質が皮下投与される、請求項39記載の方法。
【請求項66】
該ベクターにコードされるCD40リガンドの配列と融合タンパク質として投与されるCD40リガンドの配列とが異なる、請求項39記載の方法。
【請求項67】
該ベクターにコードされる腫瘍抗原の配列と融合タンパク質として投与される腫瘍抗原の配列とが異なる、請求項39記載の方法。
【請求項68】
転写ユニットが分泌シグナル配列をコードする、請求項41記載の方法。
【請求項69】
抗原がCD40リガンド由来ではない、請求項41記載の方法。
【請求項70】
被験体において感染性物質に対する免疫を生じさせる方法であって、
a)個体に有効量の発現ベクターを投与し、ここで、該ベクターは分泌可能な融合タンパク質をコードする転写ユニットを含み、該融合タンパク質は感染性物質抗原およびCD40リガンドを含み;そして
b)感染性物質抗原およびCD40リガンドを含む有効量の融合タンパク質を投与する、
ことを含む上記方法。
【請求項71】
該タンパク質がベクターの投与後に投与される、請求項70記載の方法。
【請求項72】
該感染性物質抗原がウイルス抗原、細菌抗原、真菌抗原、および原生動物抗原からなる群から選択される、請求項70記載の方法。
【請求項73】
該感染性物質抗原がウイルス抗原である、請求項70記載の方法。
【請求項74】
該ウイルス抗原がヒト乳頭腫ウイルスのE6またはE7タンパク質である、請求項73記載の方法。
【請求項75】
該ベクターが転写ユニットの転写を制御するためのヒトサイトメガロウイルスプロモーター/エンハンサーを含む、請求項70記載の方法。
【請求項76】
該ベクターがウイルスベクターである、請求項70記載の方法。
【請求項77】
該ウイルスベクターがアデノウイルスベクターである、請求項70記載の方法。
【請求項78】
該CD40リガンドがヒトCD40リガンドである、請求項70記載の方法。
【請求項79】
該CD40リガンドが細胞質ドメインを欠失している、請求項70記載の方法。
【請求項80】
該ベクターが膜貫通ドメインのいずれかの末端からの6以下の残基を含む、請求項70記載の方法。
【請求項81】
該ベクターがCD40リガンドの膜貫通ドメインをコードしない、請求項70記載の方法。
【請求項82】
該CD40リガンドがその膜貫通ドメインの全部または実質的に全部を欠失している、請求項70記載の方法。
【請求項83】
該CD40リガンドが残基47-261を含む、請求項70記載の方法。
【請求項84】
該CD40リガンドが残基1-23および47-261を含む、請求項70記載の方法。
【請求項85】
該ベクターが正常なヒト細胞において非複製性とされている、請求項70記載の方法。
【請求項86】
後の時点で、段階b)の反復である段階c)を含む、請求項70記載の方法。
【請求項87】
該免疫応答が該腫瘍抗原に対する細胞傷害性CD8+ T細胞の生成を含む、請求項70記載の方法。
【請求項88】
該免疫応答が該腫瘍抗原に対する抗体の産生を含む、請求項70記載の方法。
【請求項89】
該融合タンパク質がアジュバントと共に投与される、請求項70記載の方法。
【請求項90】
該融合タンパク質が皮下投与される、請求項70記載の方法。
【請求項91】
該ベクターにコードされるCD40リガンドの配列と融合タンパク質として投与されるCD40リガンドの配列とが異なる、請求項70記載の方法。
【請求項92】
該ベクターにコードされる感染性物質抗原の配列と融合タンパク質として投与される感染性物質抗原の配列とが異なる、請求項70記載の方法。
【請求項93】
転写ユニットが分泌シグナル配列をコードする、請求項70記載の方法。
【請求項94】
抗原がCD40リガンド由来ではない、請求項70記載の方法。
【請求項95】
融合タンパク質をコードする発現ベクターと融合タンパク質とを同時に生成する方法であって、培養液中で発現ベクターを含む細胞を培養することを含み、ここで、該細胞はベクターを複製し、増殖の間にベクターから発現されるタンパク質を培養液中に生成する上記方法。
【請求項96】
該ベクターがアデノウイルスベクターである、請求項95記載の方法。
【請求項97】
該発現された融合タンパク質を培養液から精製する、請求項95記載の方法。
【請求項98】
分泌可能な融合タンパク質をコードする転写ユニットを含むタンパク質であって、該融合タンパク質が抗原およびCD40リガンドを含む上記タンパク質。
【請求項99】
該抗原が腫瘍抗原である、請求項98記載のタンパク質。
【請求項100】
該抗原が感染性物質抗原である、請求項98記載のタンパク質。
【請求項101】
該感染性疾患抗原がヒト乳頭腫ウイルスのE7タンパク質である、請求項98記載のタンパク質。
【請求項102】
該抗原がHER2由来である、請求項98記載のタンパク質。
【請求項103】
該腫瘍抗原がムチン抗原である、請求項98記載のタンパク質。
【請求項104】
該腫瘍抗原がMUC1、MUC2、MUC3A、MUC3B、MUC4、MUC5AC、MUC5B、MUC6、MUC7、MUC8、MUC9、MUC12、MUC13、MUC15、およびMUC16からなる群から選択されるムチン由来である、請求項98記載のタンパク質。
【請求項105】
該ムチン抗原がMUC1由来である、請求項98記載のタンパク質。
【請求項106】
該ムチン抗原がムチンの細胞外ドメインを含む、請求項98記載のタンパク質。
【請求項107】
該抗原が個体中の自己抗原である、請求項98記載のタンパク質。
【請求項108】
該CD40リガンドがヒトCD40リガンドである、請求項98記載のタンパク質。
【請求項109】
該CD40リガンドが細胞質ドメインを欠失している、請求項98記載のタンパク質。
【請求項110】
該ベクターが膜貫通ドメインのいずれかの末端からの6以下の残基を含むCD40Lをコードする、請求項98記載のタンパク質。
【請求項111】
該ベクターがCD40リガンドの膜貫通ドメインをコードしない、請求項98記載のタンパク質。
【請求項112】
該CD40リガンドが膜貫通ドメインの全部または実質的に全部を欠失している、請求項98記載のタンパク質。
【請求項113】
該CD40リガンドが残基47-261を含む、請求項98記載のタンパク質。
【請求項114】
該CD40リガンドが残基1-23および47-261を含む、請求項98記載のタンパク質。
【請求項1】
個体において抗原に対する免疫応答を生成させる方法であって、
a)個体に有効量の発現ベクターを投与し、ここで、該ベクターは分泌可能な融合タンパク質をコードする転写ユニットを含み、該融合タンパク質は抗原およびCD40リガンドを含み;そして
b)抗原およびCD40リガンドを含む有効量の融合タンパク質を投与する、
ことを含む上記方法。
【請求項2】
該タンパク質がベクターの投与後に投与される、請求項1記載の方法。
【請求項3】
該抗原がポリペプチド抗原である、請求項1記載の方法。
【請求項4】
該抗原が感染物質抗原である、請求項1記載の方法。
【請求項5】
該免疫応答が抗原を発現する細胞に対するものである、請求項1記載の方法。
【請求項6】
該免疫応答が抗原を発現する微生物に対するものである、請求項1記載の方法。
【請求項7】
該抗原が腫瘍抗原である、請求項1記載の方法。
【請求項8】
該腫瘍抗原がHER-2由来である、請求項1記載の方法。
【請求項9】
該腫瘍抗原がムチンである、請求項1記載の方法。
【請求項10】
該腫瘍抗原がMUC1、MUC2、MUC3A、MUC3B、MUC4、MUC5AC、MUC5B、MUC6、MUC7、MUC8、MUC9、MUC12、MUC13、MUC15、およびMUC16からなる群から選択されるムチン由来である、請求項5記載の方法。
【請求項11】
該ムチン抗原がMUC1由来である、請求項9記載の方法。
【請求項12】
該ムチン抗原がムチンの細胞外ドメインを含む、請求項9記載の方法。
【請求項13】
該ムチン抗原がムチンの少なくとも1つのタンデムリピートを含む、請求項9記載の方法。
【請求項14】
該ムチン抗原がMUC1の細胞外ドメインを含む、請求項9記載の方法。
【請求項15】
該抗原が個体中の自己抗原である、請求項1記載の方法。
【請求項16】
該抗原がヒト乳頭腫ウイルスのE7タンパク質である、請求項1記載の方法。
【請求項17】
該抗原が上皮癌細胞由来である、請求項1記載の方法。
【請求項18】
該転写ユニットが該抗原と該CD40リガンドとの間のリンカーをコードする、請求項1記載の方法。
【請求項19】
該ベクターが転写ユニットの転写を制御するためのヒトサイトメガロウイルスプロモーター/エンハンサーを含む、請求項1記載の方法。
【請求項20】
該ベクターがウイルスベクターである、請求項1記載の方法。
【請求項21】
該ウイルスベクターがアデノウイルスベクターである、請求項20記載の方法。
【請求項22】
該CD40リガンドがヒトCD40リガンドである、請求項1記載の方法。
【請求項23】
該CD40リガンドが細胞質ドメインを欠失している、請求項1記載の方法。
【請求項24】
該ベクターが膜貫通ドメインのいずれかの末端からの6以下の残基を含むCD40Lをコードする、請求項1記載の方法。
【請求項25】
該ベクターがCD40リガンドの膜貫通ドメインをコードしない、請求項1記載の方法。
【請求項26】
該CD40リガンドがその膜貫通ドメインの全部または実質的に全部を欠失している、請求項1記載の方法。
【請求項27】
該CD40リガンドが残基47-261を含む、請求項1記載の方法。
【請求項28】
該CD40リガンドが残基1-23および47-261を含む、請求項1記載の方法。
【請求項29】
該ベクターが正常なヒト細胞において非複製性とされている、請求項1記載の方法。
【請求項30】
後の時点で、段階b)の反復である段階c)を含む、請求項1記載の方法。
【請求項31】
該免疫応答が該抗原に対する細胞傷害性CD8+ T細胞の生成を含む、請求項1記載の方法。
【請求項32】
該免疫応答が該抗原に対する抗体の産生を含む、請求項1記載の方法。
【請求項33】
該融合タンパク質がアジュバントと共に投与される、請求項1記載の方法。
【請求項34】
該融合タンパク質が皮下投与される、請求項1記載の方法。
【請求項35】
該ベクターにコードされるCD40リガンドの配列と融合タンパク質として投与されるCD40リガンドの配列とが異なる、請求項1記載の方法。
【請求項36】
該ベクターにコードされる抗原の配列と融合タンパク質として投与される抗原の配列とが異なる、請求項1記載の方法。
【請求項37】
転写ユニットが分泌シグナル配列をコードする、請求項1記載の方法。
【請求項38】
抗原がCD40リガンド由来ではない、請求項1記載の方法。
【請求項39】
腫瘍抗原を発現する癌に罹患した個体を治療する方法であって、
a)個体に有効量の発現ベクターを投与し、ここで、該ベクターは分泌可能な融合タンパク質をコードする転写ユニットを含み、該融合タンパク質は腫瘍抗原およびCD40リガンドを含み;そして
b)腫瘍抗原およびCD40リガンドを含む有効量の融合タンパク質を投与する、
ことを含む上記方法。
【請求項40】
該タンパク質がベクターの投与後に投与される、請求項39記載の方法。
【請求項41】
該腫瘍抗原がムチン抗原である、請求項39記載の方法。
【請求項42】
該腫瘍抗原がMUC1、MUC2、MUC3A、MUC3B、MUC4、MUC5AC、MUC5B、MUC6、MUC7、MUC8、MUC9、MUC12、MUC13、MUC15、およびMUC16からなる群から選択されるムチン由来である、請求項41記載の方法。
【請求項43】
該ムチン抗原がMUC1由来である、請求項41記載の方法。
【請求項44】
該ムチン抗原がムチンの細胞外ドメインを含む、請求項41記載の方法。
【請求項45】
該ムチン抗原がムチンのタンデムリピートを少なくとも1つ含む、請求項41記載の方法。
【請求項46】
該ムチン抗原がMUC1の細胞外ドメインを含む、請求項41記載の方法。
【請求項47】
該腫瘍抗原がヒト乳頭腫ウイルスのE7タンパク質である、請求項39記載の方法。
【請求項48】
該癌細胞が上皮癌細胞である、請求項39記載の方法。
【請求項49】
該癌細胞が子宮頸癌細胞である、請求項41記載の方法。
【請求項50】
該ベクターが転写ユニットの転写を制御するためのヒトサイトメガロウイルスプロモーター/エンハンサーを含む、請求項41記載の方法。
【請求項51】
該ベクターがウイルスベクターである、請求項41記載の方法。
【請求項52】
該ウイルスベクターがアデノウイルスベクターである、請求項51記載の方法。
【請求項53】
該CD40リガンドがヒトCD40リガンドである、請求項39記載の方法。
【請求項54】
該CD40リガンドが細胞質ドメインを欠失している、請求項39記載の方法。
【請求項55】
該ベクターが膜貫通ドメインのいずれかの末端からの6以下の残基を含むCD40Lをコードする、請求項39記載の方法。
【請求項56】
該ベクターがCD40リガンドの膜貫通ドメインをコードしない、請求項39記載の方法。
【請求項57】
該CD40リガンドがその膜貫通ドメインの全部または実質的に全部を欠失している、請求項39記載の方法。
【請求項58】
該CD40リガンドが残基47-261を含む、請求項39記載の方法。
【請求項59】
該CD40リガンドが残基1-23および47-261を含む、請求項39記載の方法。
【請求項60】
該ベクターが正常なヒト細胞において非複製性とされている、請求項39記載の方法。
【請求項61】
後の時点で、段階b)の反復である段階c)を含む、請求項39記載の方法。
【請求項62】
該免疫応答が該腫瘍抗原に対する細胞傷害性CD8+ T細胞の生成を含む、請求項39記載の方法。
【請求項63】
該免疫応答が該腫瘍抗原に対する抗体の産生を含む、請求項39記載の方法。
【請求項64】
該融合タンパク質がアジュバントと共に投与される、請求項39記載の方法。
【請求項65】
該融合タンパク質が皮下投与される、請求項39記載の方法。
【請求項66】
該ベクターにコードされるCD40リガンドの配列と融合タンパク質として投与されるCD40リガンドの配列とが異なる、請求項39記載の方法。
【請求項67】
該ベクターにコードされる腫瘍抗原の配列と融合タンパク質として投与される腫瘍抗原の配列とが異なる、請求項39記載の方法。
【請求項68】
転写ユニットが分泌シグナル配列をコードする、請求項41記載の方法。
【請求項69】
抗原がCD40リガンド由来ではない、請求項41記載の方法。
【請求項70】
被験体において感染性物質に対する免疫を生じさせる方法であって、
a)個体に有効量の発現ベクターを投与し、ここで、該ベクターは分泌可能な融合タンパク質をコードする転写ユニットを含み、該融合タンパク質は感染性物質抗原およびCD40リガンドを含み;そして
b)感染性物質抗原およびCD40リガンドを含む有効量の融合タンパク質を投与する、
ことを含む上記方法。
【請求項71】
該タンパク質がベクターの投与後に投与される、請求項70記載の方法。
【請求項72】
該感染性物質抗原がウイルス抗原、細菌抗原、真菌抗原、および原生動物抗原からなる群から選択される、請求項70記載の方法。
【請求項73】
該感染性物質抗原がウイルス抗原である、請求項70記載の方法。
【請求項74】
該ウイルス抗原がヒト乳頭腫ウイルスのE6またはE7タンパク質である、請求項73記載の方法。
【請求項75】
該ベクターが転写ユニットの転写を制御するためのヒトサイトメガロウイルスプロモーター/エンハンサーを含む、請求項70記載の方法。
【請求項76】
該ベクターがウイルスベクターである、請求項70記載の方法。
【請求項77】
該ウイルスベクターがアデノウイルスベクターである、請求項70記載の方法。
【請求項78】
該CD40リガンドがヒトCD40リガンドである、請求項70記載の方法。
【請求項79】
該CD40リガンドが細胞質ドメインを欠失している、請求項70記載の方法。
【請求項80】
該ベクターが膜貫通ドメインのいずれかの末端からの6以下の残基を含む、請求項70記載の方法。
【請求項81】
該ベクターがCD40リガンドの膜貫通ドメインをコードしない、請求項70記載の方法。
【請求項82】
該CD40リガンドがその膜貫通ドメインの全部または実質的に全部を欠失している、請求項70記載の方法。
【請求項83】
該CD40リガンドが残基47-261を含む、請求項70記載の方法。
【請求項84】
該CD40リガンドが残基1-23および47-261を含む、請求項70記載の方法。
【請求項85】
該ベクターが正常なヒト細胞において非複製性とされている、請求項70記載の方法。
【請求項86】
後の時点で、段階b)の反復である段階c)を含む、請求項70記載の方法。
【請求項87】
該免疫応答が該腫瘍抗原に対する細胞傷害性CD8+ T細胞の生成を含む、請求項70記載の方法。
【請求項88】
該免疫応答が該腫瘍抗原に対する抗体の産生を含む、請求項70記載の方法。
【請求項89】
該融合タンパク質がアジュバントと共に投与される、請求項70記載の方法。
【請求項90】
該融合タンパク質が皮下投与される、請求項70記載の方法。
【請求項91】
該ベクターにコードされるCD40リガンドの配列と融合タンパク質として投与されるCD40リガンドの配列とが異なる、請求項70記載の方法。
【請求項92】
該ベクターにコードされる感染性物質抗原の配列と融合タンパク質として投与される感染性物質抗原の配列とが異なる、請求項70記載の方法。
【請求項93】
転写ユニットが分泌シグナル配列をコードする、請求項70記載の方法。
【請求項94】
抗原がCD40リガンド由来ではない、請求項70記載の方法。
【請求項95】
融合タンパク質をコードする発現ベクターと融合タンパク質とを同時に生成する方法であって、培養液中で発現ベクターを含む細胞を培養することを含み、ここで、該細胞はベクターを複製し、増殖の間にベクターから発現されるタンパク質を培養液中に生成する上記方法。
【請求項96】
該ベクターがアデノウイルスベクターである、請求項95記載の方法。
【請求項97】
該発現された融合タンパク質を培養液から精製する、請求項95記載の方法。
【請求項98】
分泌可能な融合タンパク質をコードする転写ユニットを含むタンパク質であって、該融合タンパク質が抗原およびCD40リガンドを含む上記タンパク質。
【請求項99】
該抗原が腫瘍抗原である、請求項98記載のタンパク質。
【請求項100】
該抗原が感染性物質抗原である、請求項98記載のタンパク質。
【請求項101】
該感染性疾患抗原がヒト乳頭腫ウイルスのE7タンパク質である、請求項98記載のタンパク質。
【請求項102】
該抗原がHER2由来である、請求項98記載のタンパク質。
【請求項103】
該腫瘍抗原がムチン抗原である、請求項98記載のタンパク質。
【請求項104】
該腫瘍抗原がMUC1、MUC2、MUC3A、MUC3B、MUC4、MUC5AC、MUC5B、MUC6、MUC7、MUC8、MUC9、MUC12、MUC13、MUC15、およびMUC16からなる群から選択されるムチン由来である、請求項98記載のタンパク質。
【請求項105】
該ムチン抗原がMUC1由来である、請求項98記載のタンパク質。
【請求項106】
該ムチン抗原がムチンの細胞外ドメインを含む、請求項98記載のタンパク質。
【請求項107】
該抗原が個体中の自己抗原である、請求項98記載のタンパク質。
【請求項108】
該CD40リガンドがヒトCD40リガンドである、請求項98記載のタンパク質。
【請求項109】
該CD40リガンドが細胞質ドメインを欠失している、請求項98記載のタンパク質。
【請求項110】
該ベクターが膜貫通ドメインのいずれかの末端からの6以下の残基を含むCD40Lをコードする、請求項98記載のタンパク質。
【請求項111】
該ベクターがCD40リガンドの膜貫通ドメインをコードしない、請求項98記載のタンパク質。
【請求項112】
該CD40リガンドが膜貫通ドメインの全部または実質的に全部を欠失している、請求項98記載のタンパク質。
【請求項113】
該CD40リガンドが残基47-261を含む、請求項98記載のタンパク質。
【請求項114】
該CD40リガンドが残基1-23および47-261を含む、請求項98記載のタンパク質。
【図1A】


【図1B】

【図2】

【図3】
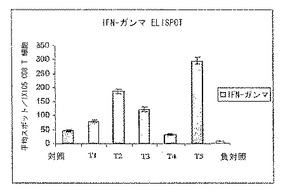
【図4】
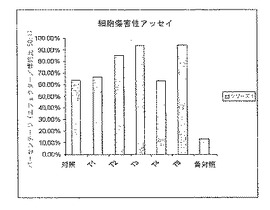
【図5】
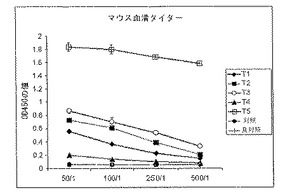
【図6】
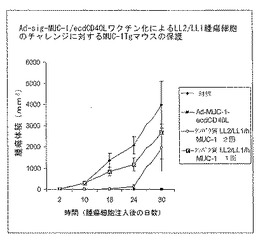
【図7】
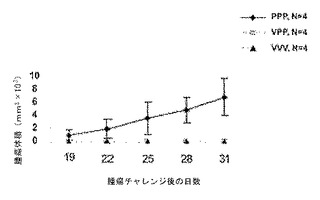
【図8】
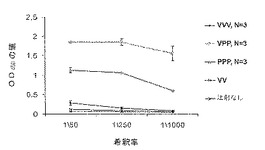
【図9】
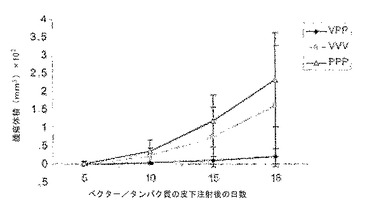
【図10】
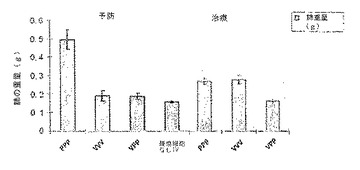
【図11】
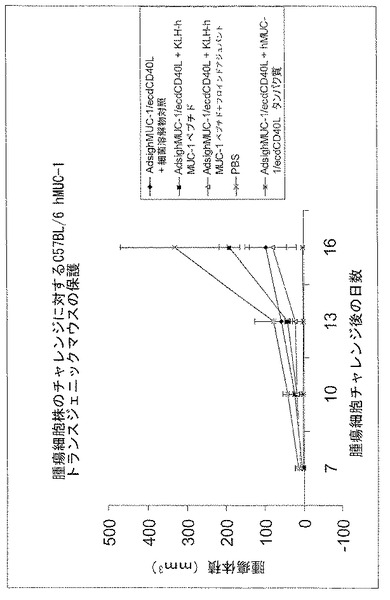
【図1B】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公表番号】特表2008−504219(P2008−504219A)
【公表日】平成20年2月14日(2008.2.14)
【国際特許分類】
【出願番号】特願2006−544078(P2006−544078)
【出願日】平成16年12月10日(2004.12.10)
【国際出願番号】PCT/US2004/041690
【国際公開番号】WO2005/058950
【国際公開日】平成17年6月30日(2005.6.30)
【出願人】(506197923)
【Fターム(参考)】
【公表日】平成20年2月14日(2008.2.14)
【国際特許分類】
【出願日】平成16年12月10日(2004.12.10)
【国際出願番号】PCT/US2004/041690
【国際公開番号】WO2005/058950
【国際公開日】平成17年6月30日(2005.6.30)
【出願人】(506197923)
【Fターム(参考)】
[ Back to top ]