
抗原の順序付けられた分子提示、提示の方法、および使用
本発明は、順序付けられかつ反復性の抗原または抗原決定基アレイの生成のための組成物およびプロセスを提供する。本発明の組成物は、感染性疾患の予防、アレルギーの処置および癌の処置のためのワクチンの生成に有用である。本発明の種々の実施形態は、特異的相互作用の結果として、非常に順序付けられかつ反復性の様式において、任意の所望の抗原で被覆した、ウイルス、ウイルス様粒子、ウイルスカプシド粒子、ファージ、またはそれらの組換え型形態を提供する。1つの特定の実施形態において、カセット型系(αVaccine Technology)に基づく多目的の新しい技術は、抗原で被覆したウイルス粒子の生成を可能にする。他の特定の実施形態は、抗原で被覆したB型肝炎ウイルス様粒子または抗原で被覆した麻疹ウイルス様粒子の生成を可能にする。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
(発明の背景)
(発明の分野)
本発明は、分子生物学、ウイルス学、免疫学および医学の分野に関する。本発明は、順序付けられそして反復性の抗原または抗原決定基のアレイを含む組成物を提供する。本発明はまた、順序付けられそして反復性のアレイにおいて抗原または抗原決定基を生成するためのプロセスを提供する。この順序付けられそして反復性の抗原または抗原決定基は、感染性疾患の処置、アレルギーの処置のためのワクチンの産生において、そして癌を予防または治療するための、および規定された自己特異的抗体を作製するためのファルマシン(pharmaccine)として有用である。
【0002】
(関連技術)
感染性疾患の予防のためのワクチンの開発は、任意の医学的発明のヒトの健康に対して最大の影響を有した。1年あたり300万人の死亡が、ワクチン接種によって世界中で予防されると見積もられている(Hillemann,Nature Medicine 4:507(1998))。最も通常のワクチン接種ストラテジーである、弱毒化(すなわち、毒性が低い)病原体または近縁の生物の使用は、危険性がより低い牛痘ウイルスの投与によって天然痘に対してワクチン接種したEdward Jennerによって1796年に最初に実証された。多数の生弱毒化ウイルス(例えば、麻疹、おたふくかぜ、風疹、水痘、アデノウイルス、ポリオ、インフルエンザ)および細菌(例えば、結核に対しての桿菌Calmette−Guerin(BCG))がワクチン接種について好首尾に投与されているが、「ワクチン」生物、特に免疫無防備状態の個体により毒性および感染へと後転することに関連した重篤な合併症を発症する危険性が存在する。
【0003】
弱毒化ウイルスの具体的設計は、欠失改変体または変異改変体の作製を介する組換えDNA技術(すなわち、遺伝子操作)によって現在可能にされている。例えば、nef遺伝子内に欠失を有する、操作されたサル免疫不全ウイルス(SIV)の投与は、マカクがその後の病原性SIV株での感染から保護されることを示した(Danielら,Science 258:1938−1941(1992))。しかし、弱毒化SIVを投与した動物における後天性免疫不全症候群(AIDS)様症状の進行は、安全性の懸念を生じる(Babaら,Science 267:1820−1825((1995))。
【0004】
代替的なアプローチとして、弱毒化ウイルスまたは細菌が、弱毒化形態で投与するにはあまりにも危険すぎると考えられる病原体(例えば、ヒト免疫不全ウイルス(HIV))の抗原コード遺伝子についてのキャリアとして用いられ得る。宿主への抗原コード遺伝子の送達の際に、この抗原は、インサイチュで合成される。ワクシニアおよび関連のアビポックスウイルス(avipox viruse)は、種々の疾患についての前臨床研究および臨床研究において種々の遺伝子についてのこのようなキャリアとして用いられている(例えば、Shenら,Science 252:440(1991))。このワクチン接種ストラテジーの1つの欠点は、これがビリオンの表面を模倣しないことである。なぜなら、この組換え型タンパク質は、宿主細胞の表面で発現されるからである。さらに、合併症は、生命を脅かす播種性ワクシニア感染によって証明されたように、免疫無防備状態の個体において発症し得る(Redfield,N.Eng.J.Med 316:673((1998))。
【0005】
第4のワクチン接種アプローチは、インビトロで増殖させた病原体(例えば、インフルエンザ血球凝集素またはノイラミニダーゼ)から精製されたかまたは単一のウイルスタンパク質(例えば、B型肝炎表面抗原)の異種発現後に発現されたかのいずれかの、単離された病原体成分の使用を含む。例えば、組換え型の、変異した毒素(無毒化された)は、ジフテリア、破傷風、コレラおよび百日咳の毒素に対するワクチン接種のために用いられ(Levineら,New generation Vaccines,第2版,Marcel Dekker,Inc.,New York 1997)、そしてHIVの組換え型タンパク質(gp120および全長gp160)を、HIVに対する中和抗体を誘導する手段として評価し、失望した結果となった(Connorら,J.Virol.72:1552(1998))。近年、HIV−1単離体に対するCTL応答を誘導し得、そしてHIV−1単離体に対する中和活性を有する惹起し得る、可溶性オリゴマーgp160を用いて有望な結果が得られた(Van Corttら,J.Virol.71:4319(1997))。さらに、抗原の公知のB細胞エピトープまたはT細胞エピトープが、T細胞の助けを刺激することによりエピトープの免疫原性を増加させるように設計されたキャリア分子に結合されている、ペプチドワクチンが用いられ得る。しかし、このアプローチに伴う1つの重大な問題は、このアプローチが、全体としてこのタンパク質に対して制限された免疫応答しか提供しないことである。さらに、ワクチンは、異なるMHCハプロタイプについて個々に設計されなければならない。この型のワクチンについての最も深刻な懸念は、防御性抗ウイルス抗体が、ペプチドによって模倣され得ない、複雑な三次元構造を認識することである。
【0006】
より新規のワクチン接種ストラテジーは、DNAワクチンの使用であり(Donnellyら,Ann.Rev.Immunol.15:617(1997))、これは、(生ベクターを使用することなく)MHCクラスI拘束CTL応答を生じ得る。これは、同じウイルスの多くの株に不変の保存された内部タンパク質由来のエピトープを標的化することにより、異なる株のウイルスに対する、より広範な防御を提供し得る。この抗原は、哺乳動物の翻訳後修飾、コンホメーションおよびオリゴマー形成を有して生成されるので、組換え型または化学的に改変されたタンパク質よりも、ウイルス感染によって生成される野生型タンパク質に類似するかまたは同一である可能性がより高い。しかし、この区別は、細菌抗原の適用については欠点となり得る。なぜなら、非ネイティブ翻訳後修飾が、減少した免疫原性を生じ得るからである。さらに、ウイルス表面タンパク質は、マトリックスタンパク質の非存在下において高度に組織化されない。
【0007】
感染性疾患の予防のための適用に加えて、ワクチン技術は、アレルギーと関連した免疫の問題と取り組むために現在利用される。アレルギー個体では、IgEアイソタイプの抗体は、特定の抗原(アレルゲン)に向けての不適切な体液性免疫応答において生成される。アレルギー免疫治療によるアレルギーの処置は、3〜5年までの期間にわたる、連続して漸増する用量の特定のアレルゲンの毎週の投与を必要とする。おそらく、脂肪細胞上のIgE抗体と反応する前に、鼻分泌物または呼吸分泌物中または膜中のアレルゲンを遮断する、「ブロッキング」IgG抗体が生成される。しかし、IgG力価と症状の軽減との間に一定の関係は存在しない。おそらく、これは、極めて時間がかかりかつコストのかかるプロセスであり、長期にわたって重篤な症状を有する患者について毎年でしか考えられない。
【0008】
精製されたタンパク質の単独投与は、強力な免疫応答を惹起するに通常充分でないことが充分に確立されている;単離された抗原は一般に、アジュバントと呼ばれるヘルパー物質と一緒に与えられねばならない。これらのアジュバント内では、投与された抗原は、迅速な分解から保護され、そしてアジュバントは、低レベルの抗原の長期の放出を提供する。
【0009】
単離されたタンパク質とは異なり、ウイルスは、T細胞の助けを伴って、または伴わずにの両方で、任意のアジュバントの非存在下で迅速かつ効率的な免疫応答を誘導する(BachmannおよびZinkernagel,Ann.Rev.Immunol.15:235−270(1997))。ウイルスはしばしばほんの少数のタンパク質からなっているが、これらは、単離された成分よりもずっと強力な免疫応答を誘発し得る。B細胞応答に関しては、ウイルスの免疫原性についての1つの重大な因子が、表面エピトープの反復性および順番であることが公知である。多くのウイルスは、B細胞上のエピトープ特異的免疫グロブリンを効率的に架橋するエピトープの規則的アレイを提示する準結晶表面を示す(BachmannおよびZinkernagel,Immunol.Today 17:553−558(1996))。B細胞上の表面免疫グロブリンのこの架橋は、細胞周期進行およびIgM抗体の生成を直接誘導する、強力な活性化シグナルである。さらに、このような誘発されたB細胞は、Tヘルパー細胞を活性化し得、これは次にB細胞におけるIgM抗体生成からIgG抗体生成へのスイッチおよび長期に生存するB細胞記憶の生成を誘導する(任意のワクチン接種の目的)(BachmannおよびZinkernagel,Ann.Rev.Immunol.15:235−270(1997))。ウイルスの構造は、自己免疫疾患における抗抗体の生成に均一に結びつけられ、そして病原体に対する自然な応答の一部である(Fehr,T.ら,J.Exp.Med 185:1785−1792(1997)を参照のこと)。従って、順序付けられかつ反復性のアレイにおいて組織化されたウイルス粒子上の抗原は、非常に免疫原性である。なぜなら、これらは、B細胞を直接活性化し得るからである。
【0010】
強力なB細胞応答に加えて、ウイルス粒子はまた、免疫系の別の重要なアームである、細胞傷害性T細胞応答の生成を誘導し得る。これらの細胞傷害性T細胞は、HIVまたはB型肝炎ウイルスのような非細胞変性性ウイルスの除去のために、および腫瘍の根絶のために特に重要である。細胞傷害性T細胞は、ネイティブな抗原を認識しないが、むしろ、MHCクラスI分子と会合したそれらの分解産物を認識する(TownsendおよびBodmer,Ann.Rev.Immunol.7:601−624(1989))。マクロファージおよび樹状細胞は、内因性ウイルス粒子を取り込み得、そしてプロセシングし得(しかし、それらの可溶性の単離された成分はそうでない)、そして生成された分解産物を細胞傷害性T細胞に対して提示し得、それらの活性化および増殖をもたらす(Kovacsovics−Bankowskiら,Proc.Natl.Acad.Sci.USA 90:4942−4946(1993);Bachmannら,Eur.J.Immunol.26:2595−2600(1996))。
【0011】
抗原としてのウイルス粒子は、それらの単離された成分に対して2つの利点を示す:(1)それらの高度の反復表面構造に起因して、これらは、B細胞を直接活性化し得、高い抗体力価および長期に永続するB細胞記憶をもたらす;および(2)ウイルスが非感染性であり、そしてアジュバントが存在しなくとも、ウイルス粒子は細胞傷害性T細胞応答を誘導し得るが、可溶性タンパク質は誘導し得ない。
【0012】
いくつかの新たなワクチンストラテジーは、ウイルスの固有の免疫原性を利用する。これらのアプローチのうちのいくつかは、ウイルス粒子の粒子状の性質に焦点を当てる;例えば、ラテックスビーズおよび抗原からなるワクチンを開示する、Harding,C.V.およびSong,R.(J.Immunology 153:4925(1994));酸化鉄ビーズおよび抗原からなるワクチンを開示する、Kovacsovics−Bankowski,M.ら(Proc.Natl.Acad.Sci.USA 90:4942−4946(1993));抗原で被覆したコア粒子を開示する、Kossovsky,N.らに対する米国特許第5,334,394号;表面に共有結合した1以上のタンパク質を表面に保有する合成ポリマー粒子を開示する、米国特許第5,871,747号;ならびにコア粒子の表面を少なくとも部分的に被覆する非共有結合した被覆および被覆したコア粒子と接触している少なくとも1つの生物学的活性剤を有するコア粒子(例えば、WO/94/15585を参照のこと)を参照のこと。
【0013】
しかし、これらのウイルス模倣システムの欠点は、ウイルス表面に見出される、順序付けられた抗原提示を回復できないことである。ランダムな方向で表面に結合された抗原は、CTL応答を誘導し、B細胞応答を全く誘導しないかまたは弱くしか誘導しないことが見出されている。効果的なワクチンについては、上記およびBachmannおよびZinkernagel,Ann.Rev.Immunol.15:235(1997)に記載されるように、免疫系の両方のアームが強力に活性化されなければならない。
【0014】
別の例では、組換えウイルスは、抗原送達のために利用される。カプシドタンパク質に融合された抗原を含む糸状ファージウイルスは、非常に免疫原性であることが見出された(Perham R.N.ら,FEMS Microbiol.Rev.17:25−31(1995);Willisら,Gene 128:85−88(1993);Minenkovaら,Gene 128:85−88(1993)を参照のこと)。しかし、この系は、融合タンパク質が高レベルで発現される場合(Iannoloら,J.Mol.Biol.248:835−844(1995))に非常に小さなペプチド(5または6アミノ酸残基)に制限されるか、またはより大きなタンパク質の低レベルの発現に制限される(de la Cruzら,J.Biol.Chem.263:4318−4322(1988))。低分子ペプチドについては、これまでCTL応答しか観察されておらず、そしてB細胞応答は観察されていないかまたは弱くしか観察されていない。
【0015】
なお別の系では、組換えアルファウイルスは、抗原送達の手段として提唱される(米国特許第5,766,602号;同第5,792,462号;同第5,739,026号;同第5,789,245号および同第5,814,482号を参照のこと)。これまでに記載された組換えウイルス系に伴う問題としては、ウイルス表面上での異種タンパク質の低密度発現および/または異なる適用のために新規でかつ異なる組換えウイルスを好首尾にかつ反復して作製することの困難性が挙げられる。
【0016】
さらなる開発では、ワクチン生成の領域においてウイルス様粒子(VLP)が利用される。なぜなら、それらの構造特性および非感染性性質の両方のためである。VLPは、対称様式で、1以上の型の多くのタンパク質分子から構築されたスーパー分子(supermolecular)構造である。これらはウイルスゲノムを欠き、それゆえ、非感染性である。VLPはしばしば、異種発現によって大量に生成され得、そして容易に精製され得る。
【0017】
VLPの例としては、以下のウイルスのカプシドタンパク質が挙げられる:B型肝炎ウイルス(Ulrichら,Virus Res.50:141−182(1998))、麻疹ウイルス(Warnesら,Gene 160:173−178(1995))、シンドビスウイルス、ロタウイルス(米国特許第5,071,651号および同第5,374,426号)、口蹄疫ウイルス(Twomeyら,Vaccine 13:1603−1610(1995))、ノーウォークウイルス(Jiang,X.ら,Science 250:1580−1583(1990);Matsui,S.M.,ら,J.Clin.Invest.87:1456−1461(1991))、レトロウイルスGAGタンパク質(PCT特許出願第WO 96/30523号)、レトロトランスポゾンTyタンパク質pl、B型肝炎ウイルスの表面タンパク質(WO 92/11291)およびヒトパピローマウイルス(WO 98/15631)。いくつかの例では、組換え型DNA技術は、VLPタンパク質に対して異種タンパク質を融合するために利用され得る(Kratz,P.A.ら,Proc.Natl.Acad.Sci.USA 96:19151920(1999))。
【0018】
従って、当該分野において、天然の病原体と同等に効率的な、強力なCTLおよびB細胞免疫応答を促進する、新規でかつ改善されたワクチンの開発についての必要性が存在する。
【0019】
(発明の要旨)
本発明は、任意の所望の抗原で被覆された粒子の産生が可能な、新たな融通が利く技術を提供する。この技術は、感染性疾患に対して高度に効果的なワクチンの作製ならびにアレルギーおよび癌の処置のためのワクチンの作製を可能にする。
【0020】
第1の実施形態において、本発明は、以下を含む新規な化合物を提供する:(A)非天然の分子足場、および(B)抗原または抗原決定基。
【0021】
非天然の分子足場は、以下を含む:(i)(1)非天然起源のコア粒子および(2)天然起源のコア粒子からなる群より選択される、コア粒子;ならびに(ii)少なくとも1つの第1付着部位を含む形成体であって、少なくとも1つの共有結合によって上記のコア粒子に連結されている、形成体。
【0022】
抗原または抗原決定基は、少なくとも1つの第2付着部位を有し、この部位は、以下からなる群より選択される:(i)その抗原または抗原決定基とともには天然に存在しない、付着部位;および(ii)その抗原または抗原決定とともに天然に存在する、付着部位。
【0023】
本発明は、少なくとも1つの非ペプチド結合による第1付着部位への第2付着部位の結合を介する順序づけられかつ反復性の抗原アレイを提供する。従って、抗原または抗原決定基と非天然の分子足場は、第1付着部位および第2付着部位のこの結合を通じて一緒にされて、順序づけられかつ反復性の抗原アレイを形成する。
【0024】
別の実施形態において、前述の組成物のコア粒子は、ウイルス、ウイルス様粒子、バクテリオファージ、ウイルスキャプシド粒子、またはそれらからの組換え体を含む。あるいは、このコア粒子は、合成ポリマーまたは金属であり得る。
【0025】
特定の実施形態において、形成体は、少なくとも1つの第1付着部位を含み得る。第1付着部位および第2付着部位は、本発明の組成物の特に重要なエレメントである。本発明の種々の実施形態において、第1付着部位および/または第2付着部位は、以下であり得る:抗原およびそれに対する抗体または抗体フラグメント;ビオチンおよびアビジン;ストレプトアビジンおよびビオチン;レセプターおよびそのリガンド;リガンド結合タンパク質およびそのリガンド;相互作用するロイシンジッパーポリペプチド;アミノ基およびそれに反応する化学基;カルボキシル基およびそれに反応する化学基;スルフヒドリル基およびそれに反応する化学基;またはこれらの組み合わせ。
【0026】
より好ましい実施形態において、本発明は、選択されたほとんどの任意の抗原の、ウイルス、バクテリオファージ、ウイルス様粒子またはウイルスキャプシド粒子の表面への結合を提供する、抗原を準結晶(quasi−crystallne)「ウイルス様」構造にすることによって、本発明は、提示された抗原に対する高度に有効な免疫応答の産生(すなわち、ワクチン接種)のための宿主の強力な抗ウイルス免疫反応を開発する。
【0027】
1つの好ましい実施形態において、コア粒子は、以下からなる群より選択され得る:ロタウイルスの組換えタンパク質、ノーウォークウイルスの組換えタンパク質、アルファウイルスの組換えタンパク質、口蹄疫ウイルスの組換えタンパク質、レトロウイルスの組換えタンパク質、B型肝炎ウイルスの組換えタンパク質、タバコモザイクウイルスの組換えタンパク質、フロックハウス(Flock House)ウイルスの組換えタンパク質、およびヒトパピローマウイルスの組換えタンパク質。
【0028】
別の好ましい実施形態において、抗原は、以下からなる群より選択され得る:(1)癌細胞に対する免疫応答を誘導するに適したタンパク質;(2)感染性疾患に対する免疫応答を誘導するに適したタンパク質;(3)アレルゲンに対する免疫応答を誘導するに適したタンパク質;および(4)家畜における免疫応答を誘導するに適したタンパク質。
【0029】
本発明の特に好ましい実施形態において、第1付着部位および/または第2付着部位は、相互作用するロイシンジッパーポリペプチドを含む。最も好ましい実施形態において、第1付着部位および/または第2付着部位は、以下からなる群より選択される:(1)JUNロイシンジッパータンパク質ドメイン;および(2)FOSロイシンジッパータンパク質ドメイン。
【0030】
別の好ましい実施形態において、第1付着部位および/または第2付着部位は、以下からなる群より選択される:(1)遺伝子操作されたリジン残基、および(2)遺伝子操作されたシステイン残基。これらの2つの残基は、ともに化学的に連結され得る。
【0031】
本発明の他の実施形態は、本発明の組成物の産生のためのプロセス、およびこの組成物を使用する医療処置の方法を含む。
【0032】
前述の一般的な記載および以下の詳細な説明の両方は、模範的および説明的であるのみであること、そして特許請求される本発明のさらなる説明を提供することが意図されることが、理解されるべきである。
【0033】
(好ましい実施形態の詳細な説明)
(1.定義)
以下の定義は、本発明者らが本発明であること考慮する主題を明確にするために提供される。
【0034】
(アルファウイルス):本明細書中で使用される場合、用語「アルファウイルス」とは、Alphavirus属内に含まれる任意のRNAウイルスをいう。この属のメンバーの記載は、StraussおよびStrauss,Microbiol.Rev.,58:491−562(1994)に含まれる。アルファウイルスの例としては、以下があげられる:アウラウイルス、ベバルウイルス、キャバッソウウイルス(Cabassou virus)、チクングンヤウイルス、東部ウマ脳脊髄炎ウイルス、フォートモーガンウイルス(Fort morgan virus)、ゲタウイルスクイジラガッハウイルス(Kyzylagach virus)、マヤロウイルス、ミッデルブルグウイルス、ムカンボウイルス、ヌヅムウイルス、ピクスナウイルス、トネートウイルス(Tonate virus)、トリニティウイルス、ユナウイルス、西部ウマ脳脊髄炎ウイルス、ワタロワウイルス、シンドビスウイルス(SIN)、セムリキ森林ウイルス(SFV)、ベネズエラウマ脳脊髄炎ウイルス(VEE),およびロスリバーウイルス。
【0035】
(抗原):本明細書中で使用される場合、用語「抗原」とは、抗体によって結合され得る分子である。抗原は、さらに、体液性免疫応答および/または細胞性免疫応答を誘導して、Bリンパ球および/またはTリンパ球の産生をもたらし得る。抗原は、1以上のエピトープ(BエピトープおよびTエピトープ)を有し得る。上記に言及される特定の反応は、抗原が、高度に選択的な様式で、その対応する抗体とは反応するが、他の抗原によって惹起され得る多数の他の抗体とは反応しないことを示すことが意味される。
【0036】
(抗原決定基):本明細書中で使用される場合、用語「抗原決定基」とは、Bリンパ球またはTリンパ球のいずれかによって特異的に認識される抗原の部分をいうことが意味される。Bリンパ球は抗体産生を介して外来抗原決定基に応答するのに対し、Tリンパ球は、細胞性免疫のメディエータである。従って、抗原決定基またはエピトープは、抗体によって、またはMHCの文脈においてはT細胞レセプターによって認識される抗原の部分である。
【0037】
(結合):本明細書中で使用される場合、第1付着部位および第2付着部位に適用されるような用語「結合」とは、少なくとも1つの非ペプチド結合をいうために使用される。結合の性質は、共有、イオン性、疎水性、極性、またはそれらの任意の組み合わせであり得る。
【0038】
(付着部位、第1):本明細書中で使用される場合、句「第1付着部位」とは、非ランダム様式でコア粒子にそれ自体が結合された「形成体」のエレメントをいい、この部位に、抗原または抗原決定基に位置する第2付着部位が結合し得る。第1付着部位は、タンパク質、ポリペプチド、ペプチド、糖、ポリヌクレオチド、天然ポリマーもしくは合成ポリマー、二次代謝産物もしくは化合物(ビオチン、フルオレセイン、レチノール、ジゴキシゲニン、金属イオン、フェニルメチルスルホニルフルオリド)、またはそれらの組み合わせ、あるいはそれらの化学的に反応な基であり得る。複数の第1付着部位が、反復性の立体配置にて、非天然の分子足場の表面に存在する。
【0039】
(付着部位、第2):本明細書中で使用される場合、句「第2付着部位」とは、非天然分子足場の表面上に位置する「形成体」の第1付着部位が結合し得る抗原または抗原決定基と結合したエレメントをいう。抗原または抗原決定基の第2着部位は、タンパク質、ポリペプチド、ペプチド、糖、ポリヌクレオチド、天然ポリマーもしくは合成ポリマー、二次代謝産物もしくは化合物(ビオチン、フルオレセイン、レチノール、ジゴキシゲニン、金属イオン、フェニルメチルスルホニルフルオリド)、またはそれらの組み合わせ、あるいはそれらの化学的に反応な基であり得る。少なくとも1つの第2付着部位が、抗原または抗原決定基に存在する。
【0040】
(コア粒子):本明細書中で使用される場合、用語「コア粒子」とは、「形成体」の付着のための基礎を提供する固有の反復性較正を有する剛性構造をいう。本明細書中で使用されるようなコア粒子は、合成プロセスの産物であってもよいし、生物学的プロセスの産物であってもよい。
【0041】
(シス作用性):本明細書中で使用される場合、句「シス作用性」配列とは、レプリカーゼが結合してRNA分子のRNA依存性複製を触媒する、核酸配列をいう。これらの複製事象は、全長RNA分子および部分RNA分子の複製を生じ、従って、アルファウイルスサブゲノムプロモーターはまた、「シス作用性」配列である。シス作用性配列は、核酸分子の5’末端、3’末端またはその両末端に位置しての、またはその付近に位置しても、そして内部に位置してもよい。
【0042】
(融合):本明細書中で使用される場合、用語「融合」とは、それらのコードヌクレオチド配列のインフレームの組み合わせによる1つのポリペプチド鎖における異なる起源のアミノ酸配列の組み合わせをいう。用語「融合」は、その末端の1つへの融合に加えて、内部融合(すなわち、ポリペプチド鎖内での異なる起源の配列の挿入)を明示的に包含する。
【0043】
(異種配列):本明細書中で使用される場合、用語「異種配列」とは、本発明のベクターに存在する第2のヌクレオチド配列をいう。用語「異種配列」とはまた、本発明のベクターに含まれる異種DNA配列によってコードされる任意のアミノ酸配列またはRNA配列をいう。異種ヌクレオチド配列は、それらが存在する細胞型において通常発現されるタンパク質またはRNA分子あるいは、その細胞壁では通常発現されない分子(例えば、シンドビス構造タンパク質)をコードし得る。
【0044】
(単離された):本明細書中で使用される場合、用語「単離された(されている)」が分子を参照して使用される場合、この用語は、その分子がそのネイティブ環境から取り除かれたことを意味する。例えば、生きている動物に天然に存在するポリヌクレオチドまたはポリペプチドは「単離されて」いないが、その天然状態の共存物質から分離された同じポリヌクレオチドまたはポリペプチドは、「単離されている」。さらに、ベクターに含まれる組換えDNA分子は、本発明の目的のために単離されるとみなされる。単離されたRNA分子は、DNAおよびRNA分子のインビボまたはインビトロRNA複製産物を含む。単離された核酸分子はさらに、合成的に産生された分子を含む。さらに、組換え宿主細胞に含まれるベクター分子もまた、単離されている。従って、全ての「単離された」分子が「精製されている」必要があるとは限らない。
【0045】
(免疫治療剤):本明細書中で使用される場合、用語「免疫治療剤」とは、疾患または障害の処置のための組成物である。より詳細には、この用語は、アレルギーの処置の方法または癌の処置の方法をいうために使用される。
【0046】
(個体):本明細書中で使用される場合、用語「個体」とは、多細胞生物をいい、そして植物および動物の両方を含む。好ましい多細胞生物は動物であり、より好ましくは脊椎動物であり、さらにより好ましくは哺乳動物であり、そして最も好ましくはヒトである。
【0047】
(低いまたは検出不可能):本明細書中で使用される場合、句「低いまたは検出不可能」とは、遺伝子の発現レベルに関して使用される場合、遺伝子が最大に誘導されるときに見られるより有意に低い(例えば、少なくとも5分の1)か、または以下の実施例の節で使用される方法によって容易には検出可能ではないかのいずれかである、発現のレベルをいう。
【0048】
レクチン:本明細書中で使用される場合、マメ科植物の種から特に得られるが、多くの他の植物供給源および動物供給源からもまた得られる、タンパク質であり、特定の単糖またはオリゴ糖への結合部位を有する。例としては、コンカナバリンAおよびコムギ胚芽凝集素が挙げられ、これらは、糖タンパク質の研究において、分析薬剤および調製薬剤として広く使用される。
【0049】
天然起源:本明細書中で使用される場合、用語「天然起源」とは、その全体または一部が、合成的ではなく、そして天然に存在するかまたは天然にて生成されることを、意味する。
【0050】
非天然:本明細書中で使用される場合、この用語は、一般的には、天然由来でないことを意味し、より詳細には、この用語は、ヒトの手によることを意味する。
【0051】
非天然起源:本明細書中で使用される場合、用語「非天然起源」は、一般的には、合成的であることまたは天然由来でないことを意味し、より詳細には、この用語は、ヒトの手によることを意味する。
【0052】
非天然の分子足場:本明細書中で使用される場合、句「非天然の分子足場」とは、第1の付着部位の固定した反復性のアレイを提供するように役立ち得る、ヒトの手によって作製された任意の産物をいう。必ずではないが理想的には、これらの第1の付着部位は、幾何学的順序になっている。この非天然の分子足場は、有機物であっても非有機物であってもよく、そして部分的にかまたは全体として、化学的に合成されても生物学的プロセスを介して合成されてもよい。この非天然の分子足場は、以下から構成される:(a)コア粒子(天然起源または非天然起源のいずれかである);および(b)形成体(それ自体が、少なくとも1つの第1の付着部位を含み、そして少なくとも1つの共有結合によってコア粒子に結合している)。特定の実施形態において、この非天然の分子足場は、ウイルスでも、ウイルス様粒子でも、ウイルスキャプシド粒子でも、ファージでも、その組換え形態でも、または合成ペプチドであってもよい。
【0053】
順序付けられそして反復性の抗原または抗原決定基のアレイ:本明細書中で使用される場合、用語「順序付けられそして反復性の抗原または抗原決定基のアレイ」とは、一般的には、抗原または抗原決定基の反復性パターンをいい、足場に関して、その抗原または抗原決定基の均一な空間配置によって特徴付けられる。本発明の1つの実施形態において、この反復性パターンは、幾何学的パターンであり得る。理想の順序付けられそして反復性の抗原または抗原決定基のアレイは、5〜15ナノメーターの間隔で、抗原または抗原決定基の厳密に反復性の準結晶性の序列を保有する。
【0054】
形成体:本明細書中で使用される場合、用語「形成体」とは、順序付けられそして反復性の抗原のアレイを作製するための核生成部位を提供する、ランダムでない様式でコア粒子に結合したエレメントをいうために使用される。形成体は、少なくとも1つの共有結合によりコア粒子に結合した、少なくとも1つの第1の付着部位を含む、任意のエレメントである。形成体は、タンパク質、ポリペプチド、ペプチド、アミノ酸(すなわち、タンパク質の残基、ポリペプチドの残基、またはペプチドの残基)、糖、ポリヌクレオチド、天然ポリマーまたは合成ポリマー、二次代謝物または化合物(ビオチン、フルオレセイン、レチノール、ジゴキシゲニン、金属イオン、フェニルメチルスルホニルフルオリド)、あるいはそれらの組み合わせ、あるいはそれらの化学反応基であり得る。
【0055】
許容温度:本明細書中で使用される場合、句「許容温度」とは、酵素が、比較的高レベルの触媒活性を有する温度をいう。
【0056】
精製された:本明細書中で使用される場合、用語「精製(された)」が分子に関して使用される場合、この用語は、精製される分子の濃度が、その分子の天然の環境中にてその分子と結合している分子と比較して、増加したことを意味する。天然に結合した分子とは、タンパク質、核酸、脂質および糖を含むが、一般的には、精製される分子の完全性を維持するためまたは精製を容易にするために添加された、水、緩衝液、および試薬を含まない。例えば、mRNAが、オリゴdTカラムクロマトグラフィーの間に水性溶媒で希釈されるとしても、天然に結合した核酸および他の生物学的分子がカラムに結合せず、そして対象RNA分子から分離している場合は、mRNA分子は、このクロマトグラフィーにより精製される。
【0057】
レセプター:本明細書中で使用される場合、用語「レセプター」とは、別の分子(リガンドと呼ばれる)と相互作用し得る、タンパク質または糖タンパク質、あるいはそれらのフラグメントをいう。このリガンドは、どの種類の生化学的化合物または化学的化合物にも属し得る。このレセプターは、必ずしも、膜結合タンパク質であることを必要とはしない。可溶性タンパク質(例えば、マルトース結合タンパク質またはレチノール結合タンパク質のようなもの)は、同様にレセプターである。
【0058】
残基:本明細書中で使用される場合、用語「残基」とは、ポリペプチド骨格または側鎖における、特定のアミノ酸を意味することが、意味される。
【0059】
温度感受性:本明細書中で使用される場合、句「温度感受性」とは、1つの温度で反応を容易に触媒するが、別の温度では、同じ反応を遅く触媒するか、または全く触媒しない、酵素をいう。温度感受性酵素の例は、pCYTtsベクターによりコードされるレプリカ−ゼタンパク質であり、これは、34℃より下の温度にて、容易に検出可能なレプリカ−ゼ活性を有し、そして37℃にて、低い活性または検出不能な活性を有する。
【0060】
転写:本明細書中で使用される場合、用語「転写」とは、RNAポリメラーゼにより触媒される、DNAテンプレートからのRNA分子の生成をいう。
【0061】
組換え宿主細胞:本明細書中で使用される場合、用語「組換え宿主細胞」とは、本発明の1つ以上の核酸分子が導入された、宿主細胞をいう。
【0062】
組換えウイルス:本明細書中で使用される場合、句「組換えウイルス」とは、ヒトの手によって遺伝子改変されたウイルスをいう。この句は、当該分野で公知の任意のウイルスを包含する。より詳細には、この句は、ヒトの手によって遺伝子改変されたアルファウイルスをいい、そして最も詳細には、この句は、ヒトの手によって遺伝子改変されたシンドビス(Sinbis)ウイルスをいう。
【0063】
制限温度:本明細書中で使用される場合、句「制限温度」とは、酵素が、低いレベルまたは検出不能なレベルの触媒活性を有する温度をいう。「熱さ」感受性変異体および「冷たさ」感受性変異体の両方が公知であり、従って、制限温度は、許容温度より高くても低くてもよい。
【0064】
RNA依存性RNA複製事象:本明細書中で使用される場合、句「RNA依存性RNA複製事象」とは、RNA分子をテンプレートとして使用して、RNA分子の形成を生じる、プロセスをいう。
【0065】
RNA依存性RNAポリメラーゼ:本明細書中で使用される場合、句「RNA依存性RNAポリメラーゼ」とは、別のRNA分子からのRNA分子の生成を触媒する、ポリメラーゼをいう。この用語は、本明細書中において、用語「レプリカ−ゼ」と同義に使用される。
【0066】
非翻訳RNA:本明細書中で使用される場合、句「非翻訳RNA」とは、オープンリーディングフレームをコードしないか、またはオープンリーディングフレームまたはその一部をコードするが、アミノ酸配列が生成しない様式(例えば、開始コドンが存在しない)でコードする、RNA配列またはRNA分子をいう。このような分子の例は、tRNA分子、rRNA分子、およびリボソームである。
【0067】
ベクター:本明細書中で使用される場合、用語「ベクター」とは、宿主細胞に遺伝子物質を伝達するために使用される因子(例えば、プラスミドまたはウイルス)をいう。ベクターは、DNAまたはRNAのいずれかから構成され得る。
【0068】
1つの、a、またはan:用語「1つの」、「a」、または「an」が、本開示にて使用される場合、これらは、他のように示さない限り、「少なくとも1つ」または「1つ以上」を意味する。
【0069】
(2.順序付けられそして反復性の抗原または抗原性決定基のアレイの組成物、およびこれを作製するための方法)
開示された発明は、順序付けられそして反復性の抗原または抗原決定基を含む、組成物を提供する。さらに、本発明は、実施者が、種々の処置目的のために、順序付けられそして反復性の抗原または抗原決定基のアレイを構築することを簡便に可能にする。この目的としては、感染性疾患の予防、アレルギーの処置および癌の処置が挙げられる。
【0070】
本発明の組成物は、本質的に以下2つの要素を含む:(1)非天然の分子足場;および(2)少なくとも1つの第2の付着部位を有する抗原または抗原決定基であって、この第2の付着部位は、少なくとも1つの非ペプチド結合を介して、第1の付着部位に結合し得る。
【0071】
この非天然の分子足場は、(a)コア粒子((1)非天然起源のコア粒子および(2)天然起源のコア粒子からなる群より選択される);ならびに(b)少なくとも1つの第1の付着部位を含む、形成体(この形成体は、少なくとも1つの共有結合によってこのコア粒子に結合している)を含む。
【0072】
この抗原または抗原決定基は、(a)この抗原または抗原決定基に天然に存在しない付着部位;および(b)この抗原または抗原決定基に天然に存在する付着部位、からなる群より選択される、少なくとも1つの第2の付着部位を有する。
【0073】
本発明は、少なくとも1つの非ペプチド結合によって第1の付着部位に第2の付着部位が結合することを介して、順序付けられそして反復性の抗原のアレイを提供する。従って、この抗原または抗原決定基と非天然の分子足場とは、第1の付着部位と第2の付着部位とのこの結合を介して結合されて、順序付けられそして反復性の抗原のアレイを形成する。
【0074】
実施者は、この抗原または抗原決定基および第2の付着部位を、非天然の分子足場に結合した抗原または抗原決定基すべての配列が均一であるように、特に設計し得る。例えば、カルボキシル末端またはアミノ末端に抗原または抗原決定基上の単一の第2の付着部位を配置し得、それにより、設計を介してその非天然の分子足場に結合した抗原または抗原決定基の分子すべてが均一な様式で位置することを確実にし得る。従って、本発明は、任意の抗原または抗原決定基を、規定された順序および反復性にて、非天然の分子足場上に配置する、簡便な手段を提供する。
【0075】
当業者に明らかであるように、本発明の特定の実施形態は、組換え核酸技術(例えば、クローニング、ポリメラーゼ連鎖反応、DNAおよびRNAの精製、原核生物細胞および真核生物細胞中での組換えタンパク質の発現など)の使用を含む。このような方法論は、当業者に周知であり、そして刊行された実験室方法マニュアル(例えば、Sambrook,J.ら編、MOLECULAR CLONING、A LABORATORY MANUAL、第2版、Cold Spring Harbor Laboratory Press、Cold Spring Harbor、N.Y.(1989):Ausubel,F.ら編、CURRENT PROTOCOLS IN MOLECULAR BIOLOGY、John,H.Wiley & Sons,Inc.(1997))において、簡単に見出され得る。組織培養細胞株(Celis,J.編、CELL BIOLOGY、Academic Press、第2版(1998))および抗体に基づく技術(Harlow,E.およびLane,D.「Antibodies:A Laboratory Manual」Cold Spring Harbor Laboratory、Cold Spring Harbor、N.Y.(1988);Deutscher,M.P.「Guide to Protein Purification」Meth.Enzymol.128、Academic Press San Diego(1990);Scopes,R.K.「Protein Purification Principles and Practice」第3版、Springer−Verlag、New York(1994))を用いて作業するための基本的実験室技術もまた、この文献に十分に記載されており、これらすべてが、本明細書中で参考として援用される。
【0076】
(A.非天然の分子足場の構築)
本発明の組成物中の1つの要素は、コア粒子および形成体を含む、非天然の分子足場である。本明細書中で使用される場合、句「非天然の分子足場」とは、第1の付着部位の堅固かつ反復性のアレイを提供するように役立ち得る、ヒトの手によって作製された任意の産物をいう。より詳細には、この非天然の分子足場は、(a)コア粒子((1)非天然起源のコア粒子および(2)天然起源のコア粒子からなる群より選択される);ならびに(b)少なくとも1つの第1の付着部位を含む、形成体(この形成体は、少なくとも1つの共有結合によって、このコア粒子と結合している)を含む。
【0077】
当業者に容易に明らかであるように、本発明の非天然の分子足場のコア粒子は、いかなる特定の形態にも限定されない。このコア粒子は、有機物であっても非有機物であってもよく、そして化学合成されてもよいし、生物プロセスを介して合成されてもよい。
【0078】
1つの実施形態において、非天然のコア粒子は、合成ポリマーであっても、脂質ミセルであっても、金属であってもよい。このようなコア粒子は、当該分野で公知であり、本発明の新規な、非天然の分子足場を構築するための基礎を提供する。例として、合成ポリマーまたは金属コア粒子は、米国特許第5,770,380号に記載される。この特許は、「抗体模倣物」の作製において複数のペプチドループが結合される、カリクスアーレン有機足場の使用を開示する。そして米国特許第5,334,394号は、ウイルスデコイとして使用される、ナノ結晶粒子を記載し、この粒子は、金属またはセラミックを含む、広範な種類の無機材料から構成される。この実施形態において好ましい金属としては、クロム、ルビジウム、鉄、亜鉛、セレン、ニッケル、金、銀、プラチナが挙げられる。この実施形態において好ましいセラミック材料としては、二酸化ケイ素、二酸化チタン、酸化アルミニウム、酸化ルテニウムおよび酸化スズが挙げられる。この実施形態のコア粒子は、炭素(ダイヤモンド)を含む有機材料から作製され得る。好ましいポリマーとしては、ポリスチレン、ナイロンおよびニトロセルロースが、挙げられる。この型のナノ結晶粒子について、酸化スズ、酸化チタンまたは炭素(ダイヤモンド)から作製された粒子が、特に好ましい。脂質ミセルは、当該分野で公知の任意の手段によって、調製され得る。例えば、ミセルは、以下の手順に従って調製され得る:BaiselleおよびMillar(Baiselle,C.J.およびMillar,D.B.、Biophys.Chem.4:355〜361(1975))またはCortiら(Corti,M.、Degriorgio,V.、Sonnino,S.、Ghidoni R.、Masserini,M.およびTettamanti,G.、Chem.Phys.Lipids 38:197〜214(1981))またはLopezら(Lopez,O.、de la Maza,A.、Coderch,L.、Lopez−Iglesias,C.、Wehrli,E.およびParra,J.L.、FEBS Lett.426:314〜318(1998))またはTopchievaおよびKarezin(Topchieva,I.およびKaraezin,K.、J.Colloid Interface Sci.213:29〜35(1999))またはMoreinら(Morein,B.、Sundquist,B.、Hoglund,S.、Dalsgaard K.およびOsterbaus,A.、Nature 308:457〜60(1984));これらはすべて、本明細書中で参考として援用される)。
【0079】
コア粒子はまた、生物学的プロセス(これは、天然であってもよいし、または非天然であってもよい)を通じて生成され得る。例の目的で、この型の実施形態としてはコア粒子(ウイルス、ウイルス様粒子、ファージ、ウイルスカプシド粒子、またはそれらの組換え形態を含む)が挙げられ得る。より詳細な実施形態において、このコア粒子としては、ロタウイルスの組換えタンパク質、ノーウォークウイルスの組換えタンパク質、アルファウイルスの組換えタンパク質、口蹄疫ウイルスの組換えタンパク質、レトロウイルスの組換えタンパク質、B型肝炎ウイルスの組換えタンパク質、タバコモザイクウイルスの組換えタンパク質、フロックハウスウイルス(Flock House Virus)の組換えタンパク質およびヒトパピローマウイルスの組換えタンパク質が挙げられ得る。
【0080】
天然であろうと非天然であろうと、本発明のコア粒子は、少なくとも1つの共有結合により天然または非天然のコア粒子に付着される形成体を含むことによって、特徴付けられる。この形成体は、順序付けられそして反復性の抗原アレイを作製するための核形成部位を提供する、ランダムでない様式でコア粒子に結合したエレメントである。理想的には(必要ではないが)、この形成体は、幾何学的順序でコア粒子と会合される。最小には、この形成体は、第1の付着部位を含む。
【0081】
先に述べたように、この形成体は、少なくとも1つの共有結合によってコア粒子に結合した、少なくとも1つの第1の付着部位を含む、任意のエレメントであり得る。形成体は、タンパク質、ポリペプチド、ペプチド、アミノ酸(すなわち、タンパク質の残基、ポリペプチドの残基またはペプチドの残基)、糖、ポリヌクレオチド、天然ポリマーまたは合成ポリマー、二次代謝物または化合物(ビオチン、フルオレセイン、レチノール、ジゴキシゲニン、金属イオン、フェニルメチルスルホニルフルオリド)、あるいはそれらの組合せ、あるいはそれらの化学反応基であり得る。より詳細な実施形態において、この形成体としては、抗原、抗体または抗体フラグメント、ビオチン、アビジン、ストレプトアビジン、レセプター、レセプターリガンド、リガンド、リガンド結合タンパク質、相互作用するロイシンジッパーポリペプチド、アミノ基、アミノ基に反応する化学基:カルボキシル基、カルボキシル基に反応する化学基、スルフヒドリル基、スルフヒドリル基に反応する化学基、あるいはそれらの組合せが挙げられ得る。
【0082】
好ましい実施形態において、非天然の分子足場のコア粒子としては、ウイルス、バクテリオファージ、ウイルス様粒子、ウイルスカプシド粒子、またはそれらの組換え形態が挙げられる。順序付けられそして反復性の被覆および/またはコアタンパク質構造を有する、当該分野で公知の任意のウイルスは、本発明の非天然の分子足場として選択され得る;適切なウイルスの例としては、以下が挙げられる:シンドビスウイルスおよび他のアルファウイルス;水疱性口内炎(vesicular somatitis)ウイルス;ラブドウイルス(例えば、水疱性口内炎ウイルス)、ピコルナウイルス、トガウイルス、オルトミクソウイルス、ポリオーマウイルス、パルボウイルス、ロタウイルス、ノーウォークウイルス、口蹄疫ウイルス、レトロウイルス、B型肝炎ウイルス、タバコモザイクウイルス、フロックハウスウイルス、ヒトパピローマウイルス(例えば、Bachman,M.F.およびZinkernagel,R.M.,Immunol.Today 17:553−558(1996)の表1を参照のこと)。
【0083】
1つの実施形態において、本発明は、ウイルスの遺伝的操作を利用して、順序付けられそして反復性のウイルスエンベロープタンパク質と形成体(異種タンパク質、ペプチド、抗原決定基または選り抜きの反応性アミノ酸残基を含む)との間の融合物を作製する。当業者に公知の他の遺伝的操作は、非天然の分子足場の構築に含まれ得る;例えば、遺伝的変異によって組換えウイルスの複製能力を制限することが望ましくあり得る。形成体(すなわち、第1の付着部位)タンパク質への融合のために選択されるウイルスタンパク質は、形成されそして反復性の構造、より好ましくは、このウイルスの表面上に5〜15nmの間隔を最適に有する、準結晶組織化を有するべきである。この型の融合タンパク質の作製は、このウイルスの表面上に、複数の、順序付けられそして反復性の形成体を生じる。従って、それにより生じる第1の付着部位の、順序付けられそして反復性の組織化は、ネイティブウイルスタンパク質の正常な組織化を反映する。
【0084】
本明細書中でより詳細に議論されるように、本発明の好ましい実施形態において、この足場は、組換えアルファウイルス、そしてより詳細には、組換えシンドビスウイルスである。アルファウイルスは、感染した細胞の細胞質において、およびDNA中間体を有さずに、それらのゲノムRNA全体を複製する、+鎖RNAである(Strauss,J.およびStrauss,E.,Microbiol.Rev.58:491〜562(1994))。アルファウイルスファミリーのいくつかのメンバー(シンドビス(Xiong,C.ら、Science 243:1188−1191(1989);Schlesinger,S.,Trends Biotechnol.11:18−22(1993))、セムリキ森林ウイルス(SFV)(Liljestrom,P.およびGaroff.H.,Bio/Technology 9:1356−1361(1991))およびその他(Davis,N.L.ら、Virology 171:189−204(1989)))は、種々の異なるタンパク質についてのウイルスに基づく発現ベクターとして(Lundstrom,K.,Curr.Opin.Biotechnol.8:578−582(1997);Liljestrom,P.,Curr.Opin.Biotechnol.5:495−500(1994))、およびワクチン開発の候補物としての使用に、かなりの注目を受けた。最近、異種タンパク質の発現およびワクチンの開発のためのアルファウイルスの使用に関して、多くの特許が発行されている(米国特許第5,766,602号;同第5,792,462号;同第5,739,026号;同第5;789,245号および同第5,814,482号を参照のこと)。本発明のアルファウイルス足場の構築は、前述の文献(これらは、本明細書中で参考として援用される)に記載されるような、組換えDNA技術の当該分野で一般に公知の手段によって行われ得る。
【0085】
種々の異なる組換え宿主細胞を利用して、抗原または抗原決定付着についてのウイルスに基づくコア粒子を生成し得る。例えば、アルファウイルスは、広範な宿主範囲を有することが公知である;シンドビスウイルスは、培養された哺乳動物細胞、爬虫類細胞および両生類細胞、ならびにいくつかの昆虫細胞に感染する(Clark,H.,J.Natl.Cancer Inst.51:645(1973);Leake,C.,J.Gen.Virol.35:335(1977);Stollar,V.in THE TOGAVIRUSES,R.W.Schlesinger編,Academic Press,(1980),583−621頁)。従って、多くの組換え宿主細胞は、本発明の実施において使用され得る。BHK細胞、COS細胞、Vero細胞、HeLa細胞およびCHO細胞は、異種タンパク質の生成のために特に適切である。なぜなら、これらは、ヒト細胞に類似の様式でグリコシル化異種タンパク質における可能性を有し(Watson,E.ら、Glycobiology 4:227,(1994))、そして選択され(Zang,M.ら、Bio/Technology 13:389(1995))、または遺伝的に操作されて(Renner W.ら、Biotech.Bioeng.4.476(1995);Lee K.ら、Biotech.Bioeng.50:336(1996))、無血清培地中ならびに懸濁液中で増殖され得るからである。
【0086】
ポリヌクレオチドベクターの宿主細胞中への導入は、標準的な研究室マニュアルに記載される方法によって果たされ得る(例えば、Sambrook,J.ら編、MOLECULAR CLONING,A LABORATORY MANUAL、第2版、Cold Spring Harbor Laboratory Press,Cold Spring Harbor,N.Y.(1989),第9章;Ausubel,F.ら編、CURRENT PROTOCOLS IN MOLECULAR BIOLOGY,John H.Wiley & Sons,Inc.(1997),第16章を参照のこと)。この方法としては、エレクトロポレーション、DEAEデキストラン媒介トランスフェクション、トランスフェクション、マイクロインジェクション、カチオン性脂質媒介トランスフェクション、形質導入、スクレープローディング(scrape loading)、弾道導入(ballistic introduction)、および感染のような方法が挙げられる。外因性DNA配列の宿主細胞への導入のための方法は、Felgner,P.ら、米国特許第5,580,859号において議論される。
【0087】
パッケージされたRNA配列はまた、宿主細胞を感染するために使用され得る。これらのパッケージされたRNA配列は、それらを培養培地に添加することによって宿主細胞に導入され得る。例えば、非感染アルファウイルス粒子の調製は、多くの出典(「Sindbis Expression System」、Version C,(Invitrogen Catalog No.K750−1)を含む)において記載される。
【0088】
哺乳動物細胞が、ウイルスに基づくコア粒子の生成のための組換え宿主細胞として使用される場合、これらの細胞は、一般に、組織培養中で増殖される。培養中で細胞を増殖するための方法は、当該分野で周知である(例えば、Celis,J.編、CELL BIOLOGY,Academic Press、第2版(1998);Sambrook,J.ら編、MOLECULAR CLONING,A LABORATORY MANUAL、第2版、Cold Spring Harbor Laboratory Press,Cold Spring Harbor,N.Y.(1989);Ausubel,F.ら編、CURRENT PROTOCOLS IN MOLECULAR BIOLOGY,John H.Wiley & Sons,Inc.(1997);Freshney,R.,CULTURE OF ANIMAL CELLS,Alan R.Liss,Inc.(1983)を参照のこと)。
【0089】
当業者によって理解されるように、第1の付着部位は、任意の適切なタンパク質、ポリペプチド、糖、ポリヌクレオチド、ペプチド(アミノ酸)、天然ポリマーもしくは合成ポリマー、二次代謝物、またはそれらの組合せであり得るか、あるいはそれらの一部であり得る。これらは、足場に対する選り抜きの抗原または抗原決定基を特異的に付着するように作用し得る。1つの実施形態において、この付着部位は、当該分野で公知のタンパク質またはペプチドから選択され得るタンパク質またはペプチドである。例えば、第1の付着部位は、以下の群から選択され得る:リガンド、レセプター、レクチン、アビジン、ストレプトアビジン、ビオチン、エピトープ(例えば、HAまたはT7タグ)、Myc、Max、免疫グロブリンドメイン、および第1の付着部位として有用な、当該分野で公知の任意の他のアミノ酸配列。
【0090】
本発明の別の実施形態において、この第1の付着部位が、カプシドタンパク質に対してインフレーム融合を構築することにおいて利用される形成体(すなわち、タンパク質またはポリペプチド)に対して二次的に作製され得ることは、当業者によってさらに理解されるべきである。例えば、タンパク質は、エンベロープタンパク質と、特定の様式においてグリコシル化されていることが公知のアミノ酸配列との融合のために利用され得、そして次いで、結果として付加された糖部分は、ウイルス足場の第1の付着部位で作用し得、これは、抗原の二次付着部位として作用するレクチンへの結合による。あるいは、この形成体配列は、インビボでビオチン化され得、そしてこのビオチン部分は、本発明の第1の付着部位として作用し得るか、またはこの形成体配列は、インビトロにおいて、別個のアミノ酸残基の化学的改変に供され得、この改変は、この第1の付着部位として作用する。
【0091】
本発明の1つの特定の実施形態は、シンドビスウイルスを利用する。シンドビスウイルスRNAゲノムは、E1、E2およびE3と呼ばれる3つのタンパク質を含む脂質二重層によって囲まれる、カプシドタンパク質中にパッケージされる。これらのいわゆるエンベロープタンパク質は、糖タンパク質であり、そしてこのグリコシル化タンパク質は、この脂質二重層の外側上に位置される。ここで、これらのタンパク質の複合体は、このウイルスの表面から外側へ突出することが電子顕微鏡写真において観察され得る「スパイク(spike)」を形成する。本発明の好ましい実施形態において、この第1の付着部位は、E2エンベロープタンパク質に対してインフレームで融合されるJUNロイシンジッパータンパク質ドメインまたはFOSロイシンジッパータンパク質ドメインであるように選択される。しかし、他のエンベロープタンパク質が、本発明の足場において第1の付着部位を位置付けるためにこの融合タンパク質構築物において利用され得ることは、当業者に明らかである。
【0092】
本発明の最も好ましい実施形態において、この第1の付着部位はB型肝炎カプシド(コア)タンパク質に対してインフレームで融合されるJUN−FOSロイシンジッパードメインであるように選択される。しかし、他のウイルスカプシドタンパク質が、本発明の足場において第1の付着部位を位置付けるためにこの融合タンパク質構築物において利用され得ることは、当業者に明らかである。
【0093】
本発明の別の好ましい実施形態において、この第1の付着部位は、肝炎コア(カプシド)タンパク質に対してインフレームで融合されるリジン残基またはシステイン残基であるように選択される。しかし、他のウイルスカプシド粒子またはウイルス様粒子が、本発明の足場において第1の付着部位を位置付けるためにこの融合タンパク質構築物において利用され得ることは、当該者に明らかである。
【0094】
実施例1は、Hahnら(Proc.Natl.Acad.Sci.USA 89:2679−2683(1992))のpTE5’2Jベクターを使用して、シンドビスウイルスE2エンベロープタンパク質とJUNロイシンジッパータンパク質ドメインとの間のインフレーム融合タンパク質の構築を示すために提供される。この第1の付着部位のために利用されるJUNアミノ酸配列は、以下の通りである:CGGRIARLEEKVKTLKAQNSELASTANMLREQVAQLKQKVMNHVGC(配列番号59)。この例において、抗原上の予測される第2の付着部位は、FOSロイシンジッパータンパク質ドメインであり、そしてこのアミノ酸配列は、以下の通りである:CGGLTDTLQAETDQVEDEKSALQTEIANLLKEKEKLEFILAAHGGC(配列番号60)。
【0095】
これらの配列は、各々、両側にシステイン残基を含む短い配列に隣接する、転写因子JUNおよびFOSに由来する。これらの配列は、互いに相互作用することが公知である。JUN−FOS二量体について提唱された元々のハイポセティカル構造は、1つの単量体の疎水性側鎖が、ジッパー様の様式で他の単量体のそれぞれの側鎖と嵌合することを仮定した(Landschulzら、Science 240:1759−1764 (1988))。しかし、この仮定は誤っていることが判明した。そしてこれらのタンパク質は、αヘリカルコイルドコイルを形成することが公知である(O’Sheaら、Science 243:538−542(1989);O’ Sheaら、Cell 68:699−708(1992);Cohen & Parry,Trends Biochem.Sci.11:245−248(1986))。従って、用語「ロイシンジッパー」は、頻繁に、構造的理由よりもより歴史的にこれらのタンパク質ドメインをいうために使用される。この特許にわたって、用語「ロイシンジッパー」は、上記の配列、または上記の配列に本質的に類似する配列をいうために使用される。用語JUNおよびFOSは、JUNタンパク質全体およびFOSタンパク質全体というよりもむしろ、それぞれのロイシンジッパードメインのために使用される。
【0096】
1つの実施形態において、本発明は、pCYTts発現系を使用する、シンドビスウイルスE2−JUN足場の生成を提供する(米国特許出願番号60/079,562;1998年3月27日出願)。このpCYTts発現系は、新規な発現ベクターを提供し、このベクターは、真核生物細胞において遺伝子発現の厳重な調節を可能にする。この系のDNAベクターは、転写されてRNA分子を形成し、次いで、温度感受性レプリカーゼによって複製されて、さらなるRNA分子を形成する。複製によって生成されるRNA分子は、目的のタンパク質を生成するために翻訳され得るヌクレオチド配列、または1つ以上の非翻訳RNA分子をコードするヌクレオチド配列を含む。従って、この発現系は、組換えシンドビスウイルス粒子の生成を可能にする。
【0097】
実施例2は、本発明の非天然のE2−JUNシンドビス分子足場の生成に対する詳細を提供する。さらに、実施例3は、実施例1において生成されたpTE5’2JE2:JUNベクターを使用して、組換えE2−JUNシンドビスウイルス足場の生成のための別の方法を提供する。従って、本発明は、2つの手段(pCYTts発現系(実施例2)およびpTE5’2Jベクター系(実施例3)を提供し、これによって、組換えシンドビスウイルスE2−JUNの非天然分子足場が生成され得る。各々の系において生成されたウイルス粒子の分析は、図1および図2において検証される。
【0098】
先に述べたように、本発明は、ウイルスに基づくコア粒子(これは、ウイルス、ウイルス様粒子、ファージ、ウイルスカプシド粒子またはそれらの組換え形態を含む)を含む。当業者は、このようなコア粒子を生成し、そしてそのコア粒子に形成体を付着させるための知識を有する。他の例を提供する目的で、本発明は、本明細書中で、コア粒子として、B型肝炎ウイルス様粒子および麻疹ウイルスカプシド粒子の生成を提供する(実施例17〜22)。このような実施形態において、JUNロイシンジッパータンパク質ドメインまたはFOSロイシンジッパータンパク質ドメインは、本発明の非天然の分子足場のための形成体として、そしてそれゆえに、第1の付着部位として、使用され得る。
【0099】
実施例23〜29は、それぞれ、第1の付着部位および第2の付着部位として、反応性リジン残基を有するインフレームで融合されたペプチドを保有するB型肝炎コア粒子、および遺伝的に融合されたシステイン残基を保有する抗原、の生成の詳細を提供する。
【0100】
(B.第2の付着部位を有する抗原または抗原決定基の構築)
本発明の組成物内の第2のエレメントは、非天然分子骨格の第1の付着部位と、少なくとも一つの非ペプチド結合を介して会合し得る少なくとも一つの第2の付着部位を保有する抗原または抗原決定基である。本発明は、所望の治療効果の考慮において選択された抗原または抗原決定基に従って変わる組成物を提供する。他の組成物が、第2の付着部位について選択される分子を変えることによって提供される。
【0101】
本発明の抗原は、以下からなる群から選択され得る:(a)癌細胞に対する免疫応答を誘導するのに適したタンパク質;(b)感染疾患に対する免疫応答を誘導するのに適したタンパク質;(c)アレルゲンに対する免疫応答を誘導するのに適したタンパク質;(d)家畜における免疫応答を誘導するのに適したタンパク質。
【0102】
本発明の一つの特定の実施形態において、抗原または抗原決定基は、感染疾患の予防に対して有用な実施形態である。そのような処置は、広い範囲の宿主(例えば、ヒト、ウシ、ヒツジ、ブタ、イヌ、ネコ、他の哺乳動物種および非哺乳動物種も同様)に影響を及ぼす広範に種々の感染疾患を処置するために有用である。処置可能な感染疾患は、当業者に周知であり、例としては、例えば、HIV、インフルエンザ、ヘルペス、ウイルス性肝炎、エプスタイン−バー、ポリオ、ウイルス脳炎、麻疹、水痘などのようなウイルス病因の感染;または例えば肺炎、結核、梅毒などの細菌病因感染;または例えばマラリア、トリパノソーマ症、リーシュマニア症、トリコモナス症、アメーバ症(amoebiasis)などの寄生的病因の感染が挙げられる。従って、本発明の組成物のために選択される抗原または抗原決定基は、医療分野における当業者に対して周知である;抗原または抗原決定基の例としては、以下が挙げられる:HIV抗原gp140およびgp160;インフルエンザ抗原赤血球凝集素およびノイラミニダーゼ、肺炎B表面抗原、マラリアのサーカムスポロゾイト(circumsporozoite)タンパク質。
【0103】
別の特定の実施形態において、本発明の組成物は、アレルギーまたは癌の処置のために使用され得る免疫治療法である。
【0104】
組成物のための抗原または抗原決定基の選択およびアレルギーに対する処置の方法は、そのような疾患を処置する医療分野の当業者に対して公知である;この型の抗原または抗原決定基の代表的な例としては、以下が挙げられる;ハチ毒ホスホリパーゼA2、Bet v I(カバノキ花粉アレルゲン)、5 Dol m V(ホワイトフェーススズメバチ(white−faced hornet)毒アレルゲン)、Der p I(イエダニアレルゲン)。
【0105】
組成物に対する抗原または抗原決定基の選択および癌に対する処置の方法は、そのような疾患を処置する医療分野における当業者に公知である;この型の抗原または抗原決定基の代表的な例としては、以下が挙げられる:Her2(乳癌)、GD2(神経芽細胞腫)、EGF−R(悪性グリア芽細胞腫)、CEA(髄質甲状腺(medullary thyroid)癌)、CD52(白血病)。
【0106】
本発明の特定の実施形態において、抗原または抗原決定基は、以下からなる群より選択される:(a)HIVの組換えタンパク質、(b)インフルエンザウイルスの組換えタンパク質(c)C型肝炎ウイルスの組換えタンパク質、(d)トキソプラスマの組換えタンパク質、(e)Plasmodium falciparumの組換えタンパク質、(f)Plasmodium vivaxの組換えタンパク質、(g)Plasmodium ovaleの組換えタンパク質、(h)Plasmodium malariaeの組換えタンパク質、(i)乳癌細胞の組換えタンパク質、(j)腎臓癌細胞の組換えタンパク質、(k)前立腺癌細胞の組換えタンパク質、(l)皮膚癌細胞の組換えタンパク質、(m)脳癌細胞の組換えタンパク質、(n)白血病細胞の組換えタンパク質、(o)組換えプローブファイリング(profiling)、(p)ハチ刺しアレルギーの組換えタンパク質、(q)堅果アレルギーの組換えタンパク質、(r)食物アレルギーの組換えタンパク質、喘息の組換えタンパク質およびクラミジアの組換えタンパク質。
【0107】
一旦、組成物の抗原または抗原決定基が選択されると、少なくとも一つの第2の付着部位が、本発明の非天然分子足場と会合した、組織化され、かつ反復性のアレイを構築するように調製する際に、その分子に添加され得る。適切な第2の付着部位を構成するものの知識は、当業者に公知である。第2の付着部位の代表的例としては、以下が挙げられるが、これらに限定されない:抗原、抗体または抗体フラグメント、ビオチン、アビジン、ストレプトアビジン(strepavidin)、レセプター、レセプターリガンド、リガンド、リガンド結合タンパク質、相互作用ロイシンジッパーポリペプチド、アミノ基、アミノ基に対して反応性の化学基;カルボキシル基、カルボキシル基に対して反応性の化学基、スルフヒドリル基、スルフヒドリル基に対して反応性の化学基、またはそれらの組み合わせ。
【0108】
第1の付着部位と第2の付着部位との間の結合は、選択されたそれぞれの分子の特徴によって決定されるが、少なくとも一つの非ペプチド結合を含む。第1の付着部位および第2の付着部位の組み合わせに依存して、天然の会合は、共有結合性、イオン性、疎水性、極性、またはその組み合わせであり得る。
【0109】
本発明の一つの実施形態において、第2の付着部位は、FOSロイシンジッパータンパク質ドメインまたはJUNロイシンジッパータンパク質ドメインであり得る。
【0110】
本発明の最も具体的な実施形態では、選択された第2の付着部位FOSロイシンジッパータンパク質ドメインであり、これは、本発明の非天然分子足場のJUNロイシンジッパータンパク質と特異的に会合する。JUNおよびFOSロイシンジッパータンパク質ドメインの会合は、足場の表面上に、組織化され、かつ反復性の抗原または抗原決定基アレイを形成するための基礎を提供する。このFOSロイシンジッパータンパク質ドメインは、アミノ末端、カルボキシル末端のいずれかにおける抗原または抗原決定基の選択に対してインフレームで融合され得るか、または所望される場合、そのタンパク質の内部に位置する。
【0111】
いくつかのFOS融合構築物が、例示的目的のために提供される。ヒト成長ホルモン(実施例4)、ハチ毒ホスホリパーゼA2(PLA)(実施例9)、オボアルブミン(実施例10)およびHIVgp140(実施例12)。
【0112】
FOS融合構築物の生成を簡略化するために、抗原または抗原決定基の設計および構築について選択を提供する、いくつかのベクターが開示される(実施例6を参照のこと)。ベクターpAV1−4を、E.coliにおけるFOS融合発現のために設計した;ベクターpAV5およびpAV6を、真核生物細胞におけるFOS融合タンパク質の発現のために設計した。これらのベクターの特性が、以下に手短に記載される;
1.pAV1:このベクターを、C末端にFOSを有する融合タンパク質のE.coliの細胞膜周辺腔への分泌のために設計した。目的の遺伝子(g.o.i)は、このベクターのStuI/NotI部位に連結され得る。
【0113】
2.pAV2:このベクターを、N末端にFOSを有する融合タンパク質のE.coliの細胞膜周辺腔への分泌のために設計した。目的の遺伝子(g.o.i)は、このベクターのNotI/EcoRV(またはNotI/HindIII)部位に連結され得る。
【0114】
3.pAV3:このベクターを、C末端にFOSを有する融合タンパク質のE.coliにおける細胞質産生のために設計した。目的の遺伝子(g.o.i)は、このベクターのEcoRV/NotI部位に連結され得る。
【0115】
4.pAV3:このベクターを、N末端にFOSを有する融合タンパク質をE.coliにおける細胞質産生のために設計した。目的の遺伝子(g.o.i)は、このベクターのNotI/EcoRV(またはNotI/HindIII)部位に連結され得る。そのN末端メチオニン残基は、タンパク質合成の際に酵素分解的に除去される(Hirelら、Proc.Natl.Acad.Sci.USA 86:8247−8251(1989))。
【0116】
5.pAV5:このベクターを、C末端にFOSを有する融合タンパク質の真核生物産生のために設計した。目的の遺伝子(g.o.i)は、このベクターのEco47III/NotI部位への連結によって、hGHシグナル配列をコードする配列とFOSドメインとの間に挿入され得る。あるいは、その自らのシグナル配列を含む遺伝子は、StuI/NotI部位への連結によって、FOSコード領域に融合され得る。
【0117】
6.pAV6:このベクターを、N末端にFOSを有する融合タンパク質の真核生物産生のために設計した。目的の遺伝子(g.o.i)は、このベクターのNotI/StuI(または、NotI/HindIII)部位へ連結され得る。
【0118】
当業者によって、理解されるように、FOS抗原またはFOS抗原決定基融合タンパク質の構築は、組換えタンパク質の産生を促進するような特定の遺伝子エレメントの付加を含み得る。実施例4は、翻訳のための特定のE.coli調節エレメントの添加についてのガイダンスを提供し、そして実施例7は、真核生物シグナル配列の添加についてのガイダンスを提供する。開業医の特定の必要性に依存して、他の遺伝子エレメントが、選択され得る。
【0119】
本発明はまた、細菌細胞(実施例5)または真核生物細胞(実施例8)のいずれかにおけるFOS抗原またはFOS抗原決定基融合タンパク質の産生を含むことが理解される。融合タンパク質を発現させるために、いずれの細胞型を選択するかは、当業者の知識内にあり、例えば、この組成物の設計において、翻訳後の改変が、重要な考察であるか否かというような因子に依存する。
【0120】
以前に注記されたように、本発明は、pAVベクターの使用を介するFOS抗原またはFOS抗原決定基融合タンパク質の構築のための種々の方法を開示する。原核生物発現および真核生物発現を可能にすることに加えて、これらのベクターは、開業医が、FOSロイシンジッパードメインの抗原への−とCの間の末端付加を選択すること可能にする。特定の例が、提供され、ここで−およびC末端FOS融合は、PLA(実施例9)およびオボアルブミン(実施例10)に対してなされる。実施例11は、PLAおよびオボアルブミンFOS融合タンパク質の精製を実証する。
【0121】
ほとんどの特定の実施形態において、本発明は、HIVゲノムによってコードされる抗原または抗原決定基に引き寄せられる。より具体的には、HIV抗原は、gp140である。実施例11〜15に提供されるように、HIVgp140は、FOSロイシンジッパータンパク質ドメインおよび合成された融合タンパク質とともに作製され得、そして本発明の非天然分子足場に対する付着のために精製され得る。当業者が公知のように、他のHIV抗原または抗原決定基が、本発明の組成物の作製の際に使用され得る。
【0122】
本発明の最も具体的な実施形態において、選択される第2の付着部位は、システイン残基であり、これは本発明の非天然分子足場のリジン残基と特異的に結合する。リジン残基(Lys)およびシステイン残基(Cys)のその化学結合は、足場の表面上に、組織化され、かつ反復性の抗原または抗原決定基アレイの形成のための基礎を提供する。システイン残基は、アミノ末端、カルボキシル末端のいずれかにおける抗原または抗原決定基の選択に対してインフレームで操作され得るか、または所望される場合、そのタンパク質の内部に位置され得る。例として、PLAおよびHIVgp140が、リジン残基の第1の付着部位に対する結合のためのシステイン残基とともに提供される。
【0123】
(C.αワクチン粒子の調製)
本発明は、順序付けされ、かつ反復性の抗原アレイの構築のための新規の組成物および方法を提供する。当業者が公知のように、順序付けされ、かつ反復性の抗原アレイのアセンブリのための条件は、大きな程度まで、非天然の足場の第1の付着部位の特異的な選択および抗原または抗原決定基の第2の付着部位の特異的な選択に依存する。従って、この組成物の設計における開業医の選択は(すなわち、第1の付着部位および第2の付着部位、抗原および非天然の足場の選択)、αワクチン粒子(順序付けされ、かつ反復性の抗原アレイおよび結合される非天然分子足場)の集合についての特定の条件を決定する。このαワクチン粒子の集合に関する情報は、十分に開業医の作業知識内であり、そして多くの参考文献が存在し、開業医を援助する(例えば、Sambork,Jら、編、MOLECULAR CLOONING、A LABORATORY MANUAL、第2版、Cold Spring Harbor Laboratory Press、Cold Spring Harbor、N.Y.(1989);Ausubel,Fら、編、CURRENT PROTOCOLS IN MOLECULAR BIOLOGY,John H.Wiley&Sons、Inc.(1997);Celis,J、編、CELL BIOLGY、Academic Press.第2版、(1988);Harlow,E.およびLane,D、「Antibodies:A Laboratory Manual、」Cold Spring Harbor Laboratory、Cold Spring Harbor,N.Y.(1988)。これらの全ては、本明細書中で参考として援用される。
【0124】
本発明の特定の実施形態において、JUNおよびFOSロイシンジッパータンパク質ドメインはそれぞれ、本発明の第1および第2の付着部位のために利用される。αワクチン粒子の調製において、抗原は、非天然足場上に順序付けされ、かつ反復性の抗原アレイのアセンブリを促進するような条件下で産生され、そして精製されなければならない。特定のJUN/FOSロイシンジッパータンパク質ドメインの実施形態において、FOS抗原またはFOS抗原決定基は、ジスルフィド結合形成の発生を減少または排除するために、還元剤(例えば、ジチオスレイトール(DTT))を用いて処理されるべきである(実施例15)。
【0125】
JUN/FOSロイシンジッパータンパク質ドメイン実施形態の非天然足場(すなわち、組換えSinbisウイルス)の調製のために、組換えE2−JUNウイルス粒子は、濃縮され、中和、そして還元剤を用いて処理されるべきである(実施例16を参照のこと)。
【0126】
JUN/FOS実施形態における、順序付けされ、かつ反復性の抗原アレイの集合は、酸化還元シャッフルの存在下でなされる。E2−JUNウイルス粒子は、240倍モル濃度超過のFOS抗原またはFOS抗原決定基と4℃で10時間結合される。次いで、そのαワクチン粒子は、クロマトグラフィーによって濃縮されて、精製される(実施例16)。
【0127】
本発明の別の実施形態において、非天然分子足場と抗原または抗原決定基の結合は、化学的架橋によって、達成され得る。最も好ましい実施形態において、その化学剤は、ヘテロ2官能基架橋剤(例えば、ε−マレイミドカプロン酸N−ヒドロキシスクシンイミドエステル(Tanimoriら、J.Pharm.Dyn.4:812(1981);Fujiwaraら、J.Immunol.Meth.45:195(1981))であり、これは、(1)アミノ基と反応するスクシンイミド基および(2)SH基と反応するマレイミド基を含む。第1の付着部位の異種タンパク質またはポリペプチドは、ヘテロ2官能基架橋剤のスクシンイミド部分に対する反応部分として役に立つ、一つ以上のリジン残基を含むように操作され得る。一旦、非天然分子足場の第1の付着部位に化学的に結合されると、ヘテロ2官能基架橋剤のマレイミド基は、抗原または抗原決定基上のシステイン残基のSH基と反応するために、利用可能となる。この場合における抗原または抗原決定基調製は、第2の付着部位として選択されたタンパク質またはポリペプチド内へシステイン残基を操作することを必要とし得、その結果システイン残基を、非天然分子足場の第1の付着部位に結合した架橋剤上の遊離マレイミド官能基と反応させ得る。
【0128】
(3.組成物、ワクチン、およびそれらの投与、ならびに処置方法)
1つの実施形態では、本発明は、広範な種々の種(特に、ヒト、サル、ウシ、イヌ、ネコ、ウマ、ブタなどのような哺乳動物種)における感染症の予防のためのワクチンを提供する。ワクチンは、ウイルス性病因の感染(例えば、HIV、インフルエンザ、ヘルペス、ウイルス性肝炎、エプスタイン−バー、ポリオ、ウイルス性脳炎、麻疹、水痘など);または、細菌性病因の感染(例えば、肺炎、結核、梅毒など);または寄生生物性病因の感染(例えば、マラリア、トリパノソーマ症、リーシュマニア症、トリコモナス症、アメーバ症など)を処置するように設計され得る。
【0129】
別の実施形態では、本発明は、広範な種々の種(特に、ヒト、サル、ウシ、イヌ、ネコ、ウマ、ブタなどのような哺乳動物種)における癌の予防のためのワクチンを提供する。ワクチンは、すべての型の癌(リンパ腫、癌腫、肉腫、黒色腫など)を処置するように設計され得る。
【0130】
本発明の別の実施形態では、本発明の組成物は、アレルギーの処置のためのワクチンの設計において使用され得る。IgEアイソタイプの抗体は、アレルギー反応において重要な成分である。肥満細胞は、それらの表面上でIgE抗体を結合し、そして肥満細胞表面上に結合したIgE分子に対する特異的抗原の結合に際して、ヒスタミンおよびアレルギー反応の他のメディエイタを放出する。従って、IgE抗体の産生を阻害することは、アレルギーに対して保護するための有望な標的である。これは、所望されるTヘルパー細胞応答を達成することによって可能であるはずである。Tヘルパー細胞応答は、1型(TH1)Tヘルパー細胞応答および2型(TH2)Tヘルパー細胞応答に分けられ得る(Romagnani、Immunol.Today 18:263−266(1997))。TH1細胞は、インターフェロン−γ、およびB細胞を誘発してIgG1−3抗体を産生する他のサイトカインを分泌する。対照的に、TH2細胞により産生される重要なサイトカインはIL−4であり、IL−4はB細胞を駆動してIgG4およびIgEを産生する。多くの実験系において、TH1応答およびTH2応答の発生は相互排他的である。なぜなら、TH1細胞はTH2細胞の誘導を抑制し、そして逆もまた同じであるからである。従って、強力なTH1応答を誘発する抗原は、同時に、TH2応答の発生を抑制し、従ってIgE抗体の産生を抑制する。興味深いことに、実質的にすべてのウイルスが、宿主においてTH1応答を誘導し、そしてIgE抗体の産生を誘発することに失敗する(Coutelierら、J.Exp.Med.165:64−69(1987))。このアイソタイプパターンは、生存しているウイルスに制限されず、不活化されたウイルス粒子または組換えウイルス粒子についても観察された(Lo−Manら、Eur.J.Immunol.28:1401−1407(1998))。従って、本発明のプロセス(例えば、αワクチン技術)を使用することにより、ウイルス粒子は種々のアレルゲンで修飾(decorate)され得、そして免疫のために使用され得る。生じるアレルゲンの「ウイルス構造」に起因して、TH1応答が誘発され、「保護的」IgG1−3抗体が産生され、そしてアレルギー反応を引き起こすIgE抗体の産生を妨げる。アレルゲンは、アレルゲン自体よりもヘルパーT細胞の異なるセットによって認識されるウイルス粒子によって提示されるので、アレルゲン特異的IgG1−3抗体は、このアレルゲンに対して特異的な既存のTH2細胞を保有するアレルギー性個体においてでさえ、誘導される可能性がある。高濃度のIgG抗体の存在は、肥満細胞結合IgEへのアレルゲンの結合を妨げ得、それによって、ヒスタミンの放出を阻害する。従って、IgG抗体の存在は、IgE媒介性アレルギー反応から保護し得る。アレルギーを引き起こす代表的な物質としては、以下が挙げられる:イネ科草本、ブタクサ、カバノキもしくはマウンテンシーダー(mountain cedar)の花粉、ハウスダスト、ダニ類、動物のフケ、カビ、昆虫毒液、または薬物(例えば、ペニシリン)。従って、アレルゲン修飾されたウイルス粒子での個体の免疫は、アレルギー発症の前のみならず後でも、有益である。
【0131】
当業者によって理解されるように、本発明の組成物が個体に投与される場合、それらは、組成物の効力を改善するために所望され得る塩、緩衝剤、アジュバント、または他の物質を含む組成物中であり得る。薬学的組成物を調製する際の使用に適切な物質の例は、REMINGTON’S PHARMACEUTICAL SCIENCES(Osol,A.編、Mack Publishing Co.(1980))を含む多数の供給源に提供される。
【0132】
本発明の組成物は、それらの投与がレシピエント個体によって耐えられ得る場合に、「薬理学的に受容可能」であるといわれる。さらに、本発明の組成物は、「治療的有効量」(すなわち、所望される生理学的効果を産生する量)で、投与される。
【0133】
本発明の組成物は、当該分野において公知の種々の方法によって投与され得るが、通常は、注射、注入、吸入、経口投与、または他の適切な物理的方法によって投与される。あるいは、組成物は、筋内、静脈内、または皮下に投与され得る。投与のための組成物の成分は、滅菌水溶液(例えば、生理的食塩水)または非水系溶液および懸濁液を含む。非水系溶媒の例は、プロピレングリコール、ポリエチレングリコール、植物油(例えば、オリーブオイル)、および注射可能な有機エステル(例えば、オレイン酸エチル)である。キャリアまたは閉鎖包帯が、皮膚の透過性を増大させるため、および抗原吸着を増強するために使用され得る。
【0134】
ワクチン技術に加えて、本発明の他の実施形態は、癌およびアレルギーの医学的処置の方法に向けられる。
【0135】
本明細書中で言及される、すべての特許および刊行物は、特に参考として援用される。
【0136】
(実施例)
続く実験において使用された酵素および試薬は、以下を含んだ:New England Biolabsから入手した、T4 DNAリガーゼ;QIAGENから入手した、Taq DNAポリメラーゼ、QIAprep Spin Plasmid Kit、QIAGEN Plasmid Midi Kit、QiaExII Gel Extraction Kit、QIAquick PCR Purification Kit;Pharmaciaから入手した、QuickPrep Micro mRNA Purification Kit;Gibco BRLから入手した、SuperScript One−step RT PCR Kit、ウシ胎仔血清(FCS)、バクト−トリプトン(bacto−tryptone)および酵母抽出物;Microsynth(Switzerland)から入手した、オリゴヌクレオチド;Boehringer Mannheim、New England BiolabsまたはMBI Fermentasから入手した、制限エンドヌクレアーゼ;Boehringer Mannheimから入手した、PwoポリメラーゼおよびdNTPs。HP−1培地を、Cell culture technologies(GlattBrugg、Switzerland)から入手した。すべての標準的な化学薬品を、Fluka−Sigma−Aldrichから入手した。そして、すべての細胞培養材料を、TPPから入手した。
【0137】
DNA操作を、標準的技術を使用して実施した。DNAを、製造業者の指示書に従って、QIAprep Spin Plasmid Kitを使用して2mlの細菌培養物から、またはQIAGEN Plasmid Midi Kitを使用して50mlの培養物からのいずれかから調製した。制限酵素消化のために、製造業者の推奨される条件(緩衝液および温度)下で、1mgのDNAあたり5〜10ユニット(U)の酵素濃度で、DNAを適切な制限酵素と共に少なくとも2時間インキュベートした。1つより多い酵素での消化を、反応条件がすべての酵素について適切である場合には同時に実施したが、そうでなければ連続して実施した。さらなる操作のために単離されたDNAフラグメントを、0.7〜1.5%アガロースゲルにおける電気泳動により分離し、このゲルから切除し、そして製造業者によって提供される指示書に従って、QiaExII Gel Extraction Kitを用いて精製した。DNAフラグメントの連結のために、100〜200pgの精製されたベクターDNAを、製造業者によって提供される緩衝液(総容量:10〜20μl)中で、1UのT4 DNAリガーゼの存在下において、3倍モル濃度過剰の挿入物フラグメントと共に、16℃にて一晩インキュベートした。連結反応物のアリコート(0.1〜0.5μl)を、E.coli EL1−Blue(Stratagene)の形質転換のために使用した。形質転換を、Gene Pulser(BioRAD)および0.1cmのGene Pulser Cuvettes(BioRAD)を、200Ω、25μF、1.7kVにて使用するエレクトロポレーションによって実施した。エレクトロポレーション後、選択的S.O.B.アガーにおけるプレーティングの前に、細胞を1mlのS.O.B.培地(Miller、1972)中で1時間振盪しながらインキュベートした。
【0138】
(実施例1)
(E2におけるJUN両親媒性ヘリックスドメインの挿入)
ベクターpTE5’2J(Hahnら、Proc.Natl.Acad.Sci.USA 89:2679−2683(1992))において、MlnIおよびBstEIIの制限酵素部位を、構造タンパク質E2コード配列のコドン71(Gln)と74(Thr)との間に導入し、ベクターpTE5’2JBMを得た。これらの制限酵素部位の導入を、以下のオリゴヌクレオチドを用いるPCRによって実施した。
【0139】
【化1】

PCR反応のために、100pmolの各オリゴを、100μlの反応混合物(4ユニットのTaqポリメラーゼまたはPwoポリメラーゼ、0.1mM dNTPs、および1.5mM MgSO4を含む)中で、鋳型DNA5ngと共に使用した。すべてのDNA濃度を、GeneQuant装置(Pharmacia)を使用して測光により決定した。ポリメラーゼを、PCR反応を開始する直前(開始点は、95℃)に添加した。温度サイクリングを、以下の様式および順序で実施した:95℃(2分間);95℃(45秒間)、53℃(60秒間)72℃(80秒間)の5サイクル;そして、95℃(45秒間)、57℃(60秒間)、72℃(80秒間)の25サイクル。
【0140】
2つのPCRフラグメントを、アガロースゲル電気泳動によって分析および精製した。増幅のためにオリゴ3および4を使用する、2つのPCRフラグメントのアセンブリPCR(assembly PCR)を、最終構築物を得るために実施した。
【0141】
アセンブリPCR反応のために、100pmolの各オリゴを、100μlの反応混合物(4ユニットのTaqポリメラーゼまたはPwoポリメラーゼ、0.1mM dNTPs、および1.5mM MgSO4を含む)中で、2ngの精製されたPCRフラグメントと共に使用した。すべてのDNA濃度を、GeneQuant装置(Pharmacia)を使用して測光により決定した。ポリメラーゼを、PCR反応を開始する直前(開始点は、95℃)に添加した。温度サイクリングを、以下の様式および順序で実施した:95℃(2分間);95℃(45秒間)、57℃(60秒間)72℃(90秒間)の5サイクル;そして、95℃(45秒間)、59℃(60秒間)、72℃(90秒間)の25サイクル。
【0142】
最終PCR産物を、Qia spin PCRカラム(Qiagen)を用いて精製し、そして適切な緩衝液中で、各10ユニットのBssHIIおよびStuI制限エンドヌクレアーゼを用いて、37℃にて12時間消化した。DNAフラグメントをゲル精製し、そしてBssHII/StuI消化されかつゲル精製されたpTE5’2Jベクター(Hahnら、Proc.Natl.Acad.Sci.USA 89:2679−2683)中に連結した。このPCR産物の正確な挿入を、まずBstEIIおよびMluI制限分析により分析し、次いで、PCRフラグメントのDNA配列決定により分析した。
【0143】
JUN両親媒性ヘリックスドメインをコードするDNA配列を、以下のオリゴヌクレオチドを用いて、ベクターpJuFo(CrameriおよびSuter、Gene 137:69(1993))からPCR増幅した。
【0144】
【化2】
PCR反応のために、100pmolの各オリゴを、100μlの反応混合物(4ユニットのTaqポリメラーゼまたはPwoポリメラーゼ、0.1mM dNTPs、および1.5mM MgSO4を含む)中で、5ngの鋳型DNAと共に使用した。すべてのDNA濃度を、GeneQuant装置(Pharmacia)を使用して測光により決定した。ポリメラーゼを、PCR反応を開始する直前(開始点は、95℃)に添加した。温度サイクリングを、以下の順序および様式で実施した:95℃(2分間);95℃(45秒間)、60℃(30秒間)72℃(25秒間)の5サイクル;そして、95℃(45秒間)、68℃(30秒間)、72℃(20秒間)の25サイクル。
【0145】
最終PCR産物をゲル精製し、そしてEcoRV消化されかつゲル精製されたpBluescript II(KS−)中に連結した。得られたベクターから、QiaExIIで精製されたMluI/BstEIIでの切断によって、JUN配列を単離し、そしてベクターpTE5’2JBM(予め、同一の制限酵素で切断されている)中に連結して、ベクターpTE5’2J:E2JUNを得た。
【0146】
(実施例2)
(pCYTts系を使用する、E2−JUN含有ウイルス粒子の産生)
構造タンパク質を、鋳型としてのpTE5’2J:E2JUN、ならびにオリゴヌクレオチドXbalStruct(ctatcaTCTAGAATGAATAGAGGATTCTTTAAC)(配列番号12)およびStructBsp1201(tcgaatGGGCCCTCATCTTCGTGTGCTAGTCAG)(配列番号87)を使用して、PCR増幅した。PCRのために、100pmolの各オリゴ(Ioligo)を使用し、そして5ngの鋳型DNAを、100μlの反応混合物(4ユニットのTaq(Tac)ポリメラーゼまたはPwoポリメラーゼ、0.1mM dNTPs、および1.5mM MgSO4を含む)中で使用した。すべてのDNA濃度を、GeneQuant装置(Pharmacia)を使用して測光により決定した。ポリメラーゼを、PCR反応を開始する直前(開始点は、95℃)に添加した。温度サイクルは、以下の通りであった:95℃(3分間)、次いで、92℃(30秒間)、54℃(35秒間)72℃(270秒間)の5サイクル、次いで、92℃(30秒間)、63℃(35秒間)、72℃(270秒間)の25サイクル。このPCR産物をゲル精製し、そして制限酵素XbaI/Bsp1201で消化し、そして予め同一の酵素で切断されたベクターpCYTts(米国特許出願番号60/079,562;1998年3月27日出願)中に連結した。
【0147】
20μgのpCYTtsE2:JUNを、37℃にて少なくとも4時間、適切な緩衝液中で30UのScaIと共にインキュベートした。この反応を、フェノール/クロロホルム抽出により停止し、次いで、直鎖化DNAのイソプロパノール沈殿を行った。制限反応を、アガロースゲル電気泳動によって確認した。トランスフェクションのために、30μlのH2O中において5.4μgの直鎖化pCYTtsE2:JUNを0.6μgの直鎖化pSV2Neoと混合し、そして30μlのCaCl2溶液を添加した。60μlのリン酸緩衝液(50mM HEPES、280mM NaCl、1.5mM Na2HPO4、pH7.05)の添加後、この溶液を5秒間ボルテックスし、次いで、室温で25秒間インキュベーションした。この溶液を、2%のFCSを含有する2mlのHP−1培地(2% FCS培地)に、すぐ添加した。次いで、6ウェルプレート中の80%コンフルエントなBHK21細胞培養の培地を、DNA含有培地と置換した。CO2インキュベーター内における37℃での5時間のインキュベーションの後、DNA含有培地を取り除き、そして2% FCS培地中の15%グリセロール2mlと置換した。このグリセロール含有培地を、30秒間のインキュベーション期の後に取り除き、そして10%のFCSを含有するHP−1培地5mlでリンスすることによって細胞を洗浄した。最後に、10%のFCSを含有する新鮮なHP−1培地2mlを添加した。
【0148】
安定にトランスフェクトされた細胞を、CO2インキュベーター内で37℃にて、選択培地(G418を補充した、HP−1培地)中で選択し、そして増殖させた。混合した集団をコンフルーエンシーまで増殖した時点で、培養物を2つのディッシュに分け、次いで、37℃での12時間の増殖期を行った。この細胞の一方のディッシュを、ウイルス粒子の発現を誘導するために30℃に移し;他方のディッシュを、37℃で維持した。
【0149】
ウイルス粒子の発現を、ウェスタンブロッティングにより決定した(図1)。培養培地(0.5ml)を、メタノール/クロロホルム沈殿し、そしてペレットを、SDS−PAGEサンプル緩衝液中に再懸濁した。サンプルを、15%アクリルアミドゲルに適用する前に、5分間、95℃で加熱した。SDS−PAGE後、BassおよびYang、Creighton,T.E.編、Protein Function:A Practical Approach、第2版、IRL Press、Oxford(1997)、29〜55頁によって記載されるように、タンパク質を、Protanニトロセルロースメンブレン(Schleicher&Schuell、Germany)に転写した。TBS(1リットルあたり10×TBS:87.7gのNaCl、66.1gのTrizma hydrochloride(Sigma)および9.7gのTrizma base(Sigma)、pH7.4)中の1%ウシアルブミン(Sigma)を用いて、室温にて1時間メンブレンをブロックし、次いで、抗E1/E2抗体(ポリクローナル血清)と共に1時間インキュベーションした。ブロットを、TBS−T(0.05% Tween20を有するTBS)を用いて10分間、3回洗浄し、そしてアルカリホスファターゼ−抗ウサギIgG結合体(0.1μg/ml、Amersham Life Science、England)を用いて、1時間インキュベートした。TBS−Tを用いた10分間の2回の洗浄およびTBSを用いた10分間の2回の洗浄の後、50μlのNBT溶液(70%ジメチルホルムアミド中の7.7% Nitro Blue Tetrazolium(Sigma))および37μlのX−Phosphate溶液(ジメチルホルムアミド中の5% 5−ブロモ−4−クロロ−3−インドリルホスフェート)と共に、アルカリホスファターゼ検出試薬(10ml AP緩衝液(100mM Tris/HCl、100mM NaCl、pH9.5))を用いて、発色反応を実施した。
【0150】
ウイルス粒子の産生を、図1に示す。ウェスタンブロットのパターンは、E2−JUN(レーン1)が、SDS−PAGEにおいて、野生型E2(レーン2)と比較してより大きな分子量に移動することを示した。そして、BHK21宿主細胞株は、バックグラウンドを全く示さなかった。
【0151】
(実施例3)
(pTE5’2JE2:JUNベクターを用いた、E2−JUNを含有するウイルス粒子の産生)
RNaseを含まないベクター(1.0μg)を、PvuI消化によって直鎖化した。続いて、SP6インビトロ転写キット(InvitroGen、Invitrogen BV、NV Leek、Netherlandsによる、InvitroscripCAP)を用いて、インビトロでの転写を実施した。生じた5’キャップ化されたmRNAを、還元アガロースゲルにおいて分析した。
【0152】
インビトロで転写されたmRNA(5μg)を、Invitrogenの説明書(Sindbis Expression system、Invitrogen BV、Netherlands)に従って、BHK21細胞(ATCC:CCL10)中にエレクトロポレーションした。37℃での10時間のインキュベーションの後、FCS含有培地を、FCSを有さないHP−1培地と交換し、次いで、37℃で10時間さらにインキュベーションした。上清を収集し、そして正確に実施例2に記載のように、ウイルス粒子の産生についてウェスタンブロット分析によって分析した。
【0153】
得られた結果は、図2に示されるようなpCYTtsE2:JUNで得られた結果と同一であった。
【0154】
(実施例4)
(FOSロイシンジッパードメイン(OmpAシグナル配列)へのヒト成長ホルモン(hGH)の融合)
ヒトリーダー配列を有さないhGH遺伝子を、もともとのプラスミド(ATCC 31389)から、PCRによって増幅した。内部XbaI部位を有するオリゴ7を、hGH遺伝子の5’末端でのアニーリングのために設計し、そして内部EcoRI部位を有するオリゴ9を、hGH遺伝子の3’末端でプライムした。PCR反応のために、100pmolの各オリゴおよび5ngのテンプレートDNAを、75μlの反応混合物(4ユニットのTaqポリメラーゼまたはPwoポリメラーゼ、0.1mM dNTPおよび1.5mM MgSO4)中で使用した。
【0155】
PCRサイクリングを、以下の様式で実施した:60℃のアニーリング温度および72℃での1分間の伸長時間の30サイクル。
【0156】
ゲル精製し、そして単離したPCR産物を、第2のPCR反応のためのテンプレートとして使用して、ompAシグナル配列およびシャイン−ダルガーノ配列を導入した。PCR反応のために、100pmolのオリゴ8および9、ならびに1ngのテンプレートPCRフラグメントを、75μlの反応混合液(4ユニットのTaqポリメラーゼまたはPwoポリメラーゼ、0.1mM dNTP、および1.5mM MgSO4)中で使用した。最初の5サイクルのアニーリング温度は55℃で、72℃での60秒間の伸長時間を伴い;別の25サイクルを、65℃のアニーリング温度および72℃で60秒間の伸長時間で実施した。
【0157】
【化3】
生じた組換えhGH遺伝子を、XbaI/EcoRIを介してpBluescriptにサブクローン化した。両鎖の正確な配列を、DNA配列決定によって確認した。
【0158】
FOS両親媒性ヘリックスドメインをコードするDNA配列を、ベクターpJuFo(Crameri&Suter Gene 137:69(1993))から、以下のオリゴヌクレオチドを用いてPCR増幅した。
【0159】
【化4】
PCR反応のために、100pmolの各オリゴおよび5ngのテンプレートDNAを、75μlの反応混合物(4ユニットのTaqポリメラーゼまたはPwoポリメラーゼ、0.1mM dNTP、および1.5mM MgSO4)中で使用した。温度サイクルは、以下の通りであった:
95℃で2分間、次いで、95℃(45秒間)、60℃(30秒間)、72℃(25秒間)の5サイクル、次いで、95℃(45秒間)、68℃(30秒間)、72℃(20秒間)の25サイクル。
【0160】
PCR産物を精製し、単離し、そしてStuI消化pBluescript−ompA−hGH中にクローン化した。次いで、ハイブリッド遺伝子を、pKK223−3プラスミド(Pharmacia)中にクローン化した。
【0161】
(実施例5)
(FOS−hGHの細菌性発現)
pkk223−3中のompA−FOS−hGHを、誘導性のIPTG依存性プロモーターの制御下で、E.coli宿主株としてJM101を使用して発現させた。発現を、振盪機フラスコ内で実施した。細胞を、0.5のOD600において、1mM IPTG(最終濃度)で誘導した。発現を、37℃にて10時間継続した。細胞を、10℃で15分間、3600での遠心分離によって収集した。細胞のペレットを、凍結し(−20℃または液体N2)、そして16時間保存した。次いで、ペレットを4℃にて融解し、そして600mMスクロースを含有する10mlの10mM Tris/HCl(pH7.4)中に再懸濁した。4℃での15分間の攪拌後、ペリプラズムのタンパク質を、浸透圧性ショック手順によって放出した。冷却(4℃)脱イオン水を添加し、そして懸濁液を4℃にて30分間攪拌した。スラッジを希釈し、再懸濁し、そしてリゾチームを添加して、細菌の細胞壁を破壊した。細胞およびペリプラズムの画分のスフェロプラストを、4℃で20分間、11000gでの遠心分離によって分離した。FOS−hGH含有上清を、還元性および非還元性SDS−Pageおよびドットブロットによって分析した。ドットブロットを、実施例8に記載のように、一次抗体として抗hGH抗体(Sigma)および二次抗体としてアルカリホスファターゼ(AP)−抗マウス抗体結合体を使用して、実施した。
【0162】
全長の正確にプロセスされたFOS−hGHが、還元条件および非還元条件下で検出され得た。FOS−hGHの一部は、FOS両親媒性ヘリックス中に存在する遊離システインに起因して、他の未同定のタンパク質に結合した。しかし、発現されたFOS−hGHの50%より多くが、そのネイティブなモノマー性コンフォメーションで存在した(図3)。
【0163】
精製されたFOS−hGHを使用して、JUN含有ウイルス粒子を用いる最初のドーピング実験を実施した。
【0164】
(実施例6)
(FOS融合タンパク質の発現のための、pAVベクター系列の構築)
−もしくはE.coliにおけるC末端FOS融合タンパク質の細胞質内産生もしくは分泌、または−もしくは真核生物細胞におけるC末端融合タンパク質の産生のいずれかを可能にする、汎用性のベクター系を構築した。E.coliにおけるFOS融合タンパク質の産生のために設計されたベクターpAV1〜pAV4は、以下に列挙されたDNAカセットを含む。このDNAカセットは、異なる順序で配列された以下の遺伝的エレメントを含む:(a)E.coli ompA遺伝子(aggaggtaaaaaacg)(配列番号13)由来の強力なリボソーム結合部位および5’非翻訳領域;(b)E.coli外膜タンパク質OmpA(MKKTAIAIAVALAGFATVAQA)(配列番号14)のシグナルペプチドをコードする配列;(c)2つのグリシン残基および1つのシステイン残基によって両側で隣接されたFOS二量体化ドメイン(CGGLTDTLQAETDQVEDEKSALQTEIANLLKEKEKLEFILAAHGGC)(配列番号15)をコードする配列;および(d)目的のタンパク質をFOS二量体化ドメインに連結する、短いペプチドリンカー(AAASGG)(配列番号16)またはGGSAAA(配列番号17))をコードする領域。関連するコード領域を、大文字で提供する。制限切断部位の配置は、シグナル配列を伴うかまたは伴わない、FOS融合遺伝子の容易な構築を可能にする。このカセットを、強力なtacプロモーターの制御下での融合遺伝子の発現のために、発現ベクターpKK223−3(Pharmacia)のEcoRI/HindIII制限部位にクローン化した。
【0165】
(pAV1)
このベクターは、C末端にFOSを有する融合タンパク質のE.coli細胞周辺腔への分泌のために設計された。目的の遺伝子(g.o.i.)は、このベクターのStuI/NotI部位に連結され得る。
【0166】
【化5】
。
【0167】
(pAV2)
このベクターは、N末端にFOSを有する融合タンパク質のE.coli細胞周辺腔への分泌のために設計された。目的の遺伝子(g.o.i.)は、このベクターのNotI/EcoRV(または、NotI/HindIII)部位に連結される
【0168】
【化6】
。
【0169】
(pAV3)
このベクターは、C末端にFOSを有する融合タンパク質のE.coliにおける細胞質内産生のために設計された。目的の遺伝子(g.o.i.)は、このベクターのEcoRV/NotI部位に連結され得る。
【0170】
【化7】
。
【0171】
(pAV4)
このベクターを、N末端にFOSを有する融合タンパク質のE.coli中での細胞質産生のために設計する。目的の遺伝子(g.o.i.)を、ベクターのNotI/EcoRV(またはNotI/HindIII)部位に連結し得る。N末端メチオニン残基を、タンパク質合成に際してタンパク質分解的に除去する(Hirelら、Proc.Natl.Acad.Sci.USA 86:8247−8251(1989))。
【0172】
【化8】
。
【0173】
ベクターpAV5およびpAV6(これらは、FOS融合タンパク質の真核生物産生のために設計される)は、異なる順番で配置された以下の遺伝的エレメントを含む:(a)ヒト成長ホルモンのリーダーペプチドをコードする領域(MATGSRTSLLLAFGLLCLPWLQEGSA)(配列番号26);(b)2つのグリシン残基および1つのシステイン残基が両側で隣接する、FOS二量体化ドメインをコードする配列(CGGLTDTLQAETDQVEDEKSALQTEIANLLKEKEKLEFILAAHGGC)(配列番号15);および(c)FOS二量体化ドメインに目的のタンパク質を接続する、短いペプチドリンカーをコードする領域(AAASGG(配列番号16)またはGGSAAA(配列番号17))。関連するコード領域を大文字で示す。制限切断部位の配列は、FOS融合遺伝子の容易な構築を可能にする。このカセットを、発現ベクターpMPSVEH(Arteltら、Gene 68:213−219(1988))のEcoRI/HindIII制限部位にクローニングする。
【0174】
(pAV5)
このベクターを、C末端にFOSを有する融合タンパク質の真核生物産生のために設計する。目的の遺伝子(g.o.i.)を、ベクターのEco47III/NotI部位への連結によって、hGHシグナル配列をコードする配列とFOSドメインをコードする配列との間に挿入し得る。あるいは、それ自体のシグナル配列を含む遺伝子を、StuI/NotI部位への連結によってFOSコード領域に融合し得る。
【0175】
【化9】
。
【0176】
(pAV6)
このベクターを、N末端にFOSを有する融合タンパク質の真核生物産生のために設計する。目的の遺伝子(g.o.i.)を、ベクターのNotI/StuI部位(またはNotI/HindIII部位)に連結し得る。
【0177】
【化10】
(発現ベクターpAV1−pAV6の構築)
以下のオリゴヌクレオチドを、発現ベクターpAV1−pAV6の構築のために合成した:
【0178】
【化11】
ベクターpAV2の構築のために、OmpAシグナル配列およびFOSドメインをコードする領域を、プライマー対OmpA−FOR1/FOS−REV2を使用して、ベクターpKK223−3中のompA−FOS−hGH融合遺伝子(実施例5を参照のこと)から増幅した。このPCR産物を、EcoRI/HindIIIで消化し、そしてベクターpKK223−3(Pharmacia)の同じ部位に連結した。
【0179】
ベクターpAV1の構築のために、FOSコード領域を、プライマー対FOS−FOR1/FOS−REV1を使用して、ベクターpKK223−3中のompA−FOS−hGH融合遺伝子(実施例5を参照のこと)から増幅した。このPCR産物を、HindIIIで消化し、そしてStuI/HindIII消化したベクターpAV2に連結した。
【0180】
ベクターpAV3の構築のために、FOSドメインをコードする領域を、プライマー対FOS−FOR2/FOS−REV1を使用して、ベクターpAV1から増幅した。このPCR産物を、EcoRI/HindIIIで消化し、そしてベクターpKK223−3(Pharmacia)の同じ部位に連結した。
【0181】
ベクターpAV4の構築のために、FOSドメインをコードする領域を、プライマー対FOS−FOR3/FOS−REV2を使用して、ベクターpKK223−3中のompA−FOS−hGH融合遺伝子(実施例5を参照のこと)から増幅した。このPCR産物を、EcoRI/HindIIIで消化し、そしてベクターpKK223−3(Pharmacia)の同じ部位に連結した。
【0182】
ベクターpAV5の構築のために、hGHシグナル配列をコードする領域を、プライマー対hGH−FOR1/hGHREV1を使用して、ベクターpSINrep5中のhGH−FOS−hGH融合遺伝子(実施例7を参照のこと)から増幅する。このPCR産物を、EcoRI/NotIで消化し、そしてベクターpAV1の同じ部位に連結する。次いで、hGHシグナル配列およびFOSドメインをコードする得られたカセットを、EcoRI/HindIII消化によって単離し、そして同じ酵素で消化したベクターpMPSVEH(Arteltら、Gene 68:213−219(1988))にクローニングする。
【0183】
ベクターpAV6の構築のために、FOSコード領域を、プライマー対FOS−FOR4/FOSREV3を使用して、ベクターpAV2から増幅する。このPCR産物をHindIIIで消化し、そしてEco47III/HindIII切断したベクターpAV5にクローニングする。次いで、hGHシグナル配列およびFOSドメインをコードする全体のカセットを、プライマー対hGH−FOR2/FOSREV3を使用して、得られるベクターから再増幅し、EcoRI/HindIIIで切断し、そして同じ酵素で切断したベクターpMPSVEH(Arteltら、Gene 68:213−219(1988))に連結する。
【0184】
(実施例7:ヒト(hGH)シグナル配列を有するFOS−hGHの構築)
FOS−hGH融合タンパク質の真核生物発現のために、OmpA−FOS−hGH融合遺伝子を、XbaI/Bsp120Iでの消化によって、pBluescript::OmpA−FOS−hGH(実施例4を参照のこと)から単離し、そして同じ酵素で切断したpSINrep5(Invitrogen)にクローニングした。hGHシグナル配列を、重複するオリゴヌクレオチド
【0185】
【化12】
を使用するPCR(反応混合物:50pモルの各プライマー、dATP、dGTP、dTTP、dCTP(各200μM)、2.5UのTaq DNAポリメラーゼ(Qiagen)、製造業者より供給された緩衝液中、50μlの総量;増幅;92℃で30秒間、55℃で30秒間、72℃で30秒間、30サイクル)によって合成した。このPCR産物を、QiaExllキットを使用して精製し、StuI/XbaIで消化し、そして同じ酵素で切断したベクターpSINrep5::OmpA−FOS−hGHに連結した。
【0186】
(実施例8:FOS−hGHの真核生物発現)
RNaseを含まないベクター(1.0μg)(pSINrep5::OmpA−FOS−hGH)および1.0μgのDHEB(Bredenbeekら、J.Virol.67:6439−6466(1993))を、ScaI制限消化によって線状化した。続いて、インビトロ転写を、SP6インビトロ転写キット(InvitroGen,Invitrogen BV,NV Leek,NetherlandsによるInvitroscripCAP)を使用して実行した。得られた5’キャップmRNAを、還元アガロースゲル上で分析した。
【0187】
インビトロで、Invitrogenのマニュアルに従って、転写された5μgのmRNAを、BHK 21細胞(ATCC:CCL10)にエレクトロポレーションした(Sindbis Expressionシステム、Invitrogen BV,Netherlands)。37℃で10時間のインキュベーション後、FCS含有培地を、FCSを含まないHP−1培地で交換し、続いて37℃で10時間さらなるインキュベーションを行った。その上清を収集し、そしてFOS−hghの産生についてドットブロット分析によって分析した。
【0188】
培養培地(2.5μl)を、ニトロセルロース膜上にスポットし、そして室温で10分間乾燥させた。その膜を、TBS(1リットルあたりの10×TBS:87.7g NaCl、66.1g Trizma塩酸塩(Sigma)、および9.7g Trizmaベース(Sigma)、pH 7.4)中の1%ウシアルブミン(Sigma)を用いて、室温で1時間ブロックし、続いて10mlのTBS−T(0.05% Tween20を有するTBS)中の、2μgのウサギ抗ヒトhGH抗体(Sigma)とともに1時間インキュベートした。そのブロットを、TBS−Tで10分間、3回洗浄し、そしてTBS−T中で1:5000希釈した、アルカリホスファターゼと結合体化した抗ウサギIgG(Jackson ImmunoResearch Laboratories,Inc.)とともに1時間インキュベートした。TBS−Tでの10分間、2回の洗浄およびTBSでの10分間、2回の洗浄の後、そのブロットを、実施例2に記載のようにAP染色によって染色した。結果を図3に示す。
【0189】
(実施例9:FOS−PLA(N末端およびC末端)の構築)
ハチ毒ホスホリパーゼA2(PLA)の触媒的に不活性な改変体(Foesterら、J.Allergy Clin.Immunol.95:1229−1235(1995))をコードする以下の遺伝子を、化学的な遺伝子合成によって構築する。
【0190】
【化13】
FOS二量体化ドメインのN末端へのPLAの融合のために、その領域を、オリゴヌクレオチドPLA−FOR1(CCATCATCTACCCAGGTAC)(配列番号45)およびPLA−REV1(CCCACACCCAGCGGCCGCGTATTTGCGCAGGTCG)(配列番号46)を使用して増幅する。そのPCR産物を、NotIで切断し、そして制限酵素StuI/NotIで事前に切断したベクターpAVIに連結する。FOS二量体化ドメインのC末端へのPLAの融合のために、その領域をオリゴヌクレオチドPLA−FOR2(CGGTGGTTCTGCGGCCGCTATCATCTACCCAGGTAC)(配列番号47)およびPLA−REV2(TTAGTATTTGCGCAGGTCG)(配列番号48)を使用して増幅する。そのPCR産物を、NotIで切断し、そして制限酵素NotI/EcoRVで事前に切断したベクターpAV2に連結する。
【0191】
(実施例10)
(FOS−オボアルブミン融合遺伝子(N末端およびC末端)の構築)
オボアルブミンコード配列のクローニングのために、ニワトリ卵管組織由来のmRNAを、QuickPrepTM Micro mRNA Purification Kit(Pharmacia)を用いて、製造業者の説明書に従って使用して、調製する。SuperScriptTM One−step RT PCR Kit(Gibco BRL)を使用して、オボアルブミンの成熟部分をコードするcDNA(そのmRNAのヌクレオチド68〜1222に対応する(McReynoldsら、Nature 273:723−728(1978))を、プライマーOva−FOR1(CCGGCTCCATCGGTGCAG)(配列番号49)およびOva−REV1(ACCACCAGAAGCGGCCGCAGGGGAAACACATCTGCC)(配列番号50)を使用して合成する。このPCR産物をNotIで消化し、そしてC末端でFOS二量体化ドメインを有する融合タンパク質の発現のために、StuI/NotI消化したベクターpAV1にクローニングする。N末端でFOS二量体化ドメインを有する融合タンパク質の産生のために、オボアルブミンコード領域を、プライマーOva−FOR2(CGGTGGTTCTGCGGCCGCTGGCTCCATCGGTGCAG)(配列番号51)およびOva−REV2(TTAAGGGGAAACACATCTGCC)(配列番号52)を使用して、その構築したベクター(pAV::Ova)から増幅させる。このPCR産物をNotIで消化し、NotI/EcoRV消化したベクターpAVにクローニングする。このクローニングしたフラグメントを、DNA配列分析によって確認する。
【0192】
(実施例11)
(FOS−PLAおよびFOSアルブミン融合タンパク質の産生および精製)
FOS融合タンパク質の細胞質内産生のために、適切なE.coli株を、ベクターpAV3::PLA、pAV4::PLA、pAV3::OvaまたはpAV4::Ovaで形質転換した。この培養物を、アンピシリン存在下の富化培地中で、37℃で振盪しながらインキュベートした。吸光度(550nm)1の時点で、1mM IPTGを添加し、そしてインキュベーションをさらに5時間続けた。この細胞を遠心分離によって収集し、DNase、RNaseおよびリゾチームを含む適切な緩衝液(例えば、トリス−HCl、pH7.2、150mM NaCl)中に再懸濁し、そしてフレンチプレッシャーセルを通過させて破壊した。遠心分離(Sorvall RC−5C、SS34ローター、15000rpm、10分、4℃)後、ペレットを、4℃で25mlの封入体洗浄緩衝液(20mM トリス−HCl、23%スクロース、0.5% Triton X−100、1mM EDTA、pH8)中に再懸濁し、上記のように再度遠心分離した。この手順を、遠心分離後の上清が実質的に清澄になるまで繰り返した。封入体を、室温で20mlの可溶化緩衝液(5.5M 塩酸グアニジウム、25mM トリス−HCl、pH7.5)中に再懸濁し、そして不溶性物質を、遠心分離、およびその後、上清を滅菌フィルター(0.45μm)に通過させることによって、除去した。タンパク質溶液を、10mM EDTAおよび100mM DTTの存在下で少なくとも10時間、4℃で維持し、次いで10容量の5.5M 塩酸グアニジウム、25mM トリス−HCl、10mM EDTA、pH6に対して3回透析した。この溶液を、適切な酸化還元混合物(redox shuffle)(酸化型グルタチオン/還元型グルタチオン;シスチン/システイン)の存在下の5リットルの2M 尿素、4mM EDTA、0.1M NH4Cl、20mM ホウ酸ナトリウム(pH8.3)に対して2回透析した。次いで、この再折り畳みされたタンパク質を、イオン交換クロマトグラフィーにアプライした。このタンパク質を、2〜10mM DTTの存在下の7より大きいpHを有する適切な適切な緩衝液中で保存し、FOSドメインに隣接するシステイン残基を還元形態に維持した。タンパク質のアルファウイルス粒子との結合の前に、このタンパク質溶液をSephadex G−25ゲル濾過カラムに通すことによって、DTTを除去した。
【0193】
(実施例12)
(gp140−FOSの構築)
内部プロテアーゼ切断部位を有さないgp140遺伝子(Swiss−Prot:P03375)を、以下のオリゴヌクレオチドを使用して、全長gp160遺伝子を含む元のプラスミドpAbT4674(ATCC40829)から、PCRによって増幅させた:
【0194】
【化14】
PCR Iについて、100pmolのオリゴHIV−1およびHIV−Cleav2ならびに5ngのテンプレートDNAを、75μlの反応混合物(4ユニットのTaqまたはPwoポリメラーゼ、0.1mM dNTPおよび1.5mM MgSO4)中で使用した。PCRサイクリングを以下の様式で行った:60℃のアニーリング温度および72℃で2分間の伸長時間での30サイクル。
【0195】
PCR IIについて、100pmolのオリゴHIV−endおよびHIV−Cleavならびに5ngのテンプレートDNAを、75μlの反応混合物(4ユニットのTaqまたはPwoポリメラーゼ、0.1mM dNTPおよび1.5mM MgSO4)中で使用した。PCRサイクリングを以下の様式で行った:60℃のアニーリング温度および72℃で50秒間の伸長時間での30サイクル。
【0196】
両方のPCRフラグメントを精製し、単離し、そしてアセンブリPCR反応系において使用した。アセンブリPCR反応について、100pmolのオリゴHIV−1およびHIV−endならびに2ngの各PCRフラグメント(PCR IおよびPCR II)を、75μlの反応(4ユニットのTaqまたはPwoポリメラーゼ、0.1mM dNTPおよび1.5mM MgSO4)中で使用した。PCRサイクリングを以下の様式で行った:60℃のアニーリング温度および72℃で2.5分間の伸長時間での30サイクル。このアセンブリPCR産物を、XbaIおよびNheIで消化した。FOS両親媒性ヘリックスを、gp−140のC末端にインフレームで融合した。
【0197】
FOS両親媒性ヘリックスドメインをコードするDNA配列を、以下のオリゴヌクレオチドを使用して、ベクターpJuFo(Crameri&Suter Gene 137:69(1993))からPCR増幅させた:
【0198】
【化15】
このPCR反応について、100pmolの各オリゴおよび5ngのテンプレートDNAを、75μlの反応混合物(4ユニットのTaqまたはPwoポリメラーゼ、0.1mM dNTPおよび1.5mM MgSO4)中で使用した。温度サイクリングを以下のように行った:95℃で2分間、その後の95℃(45秒)、60℃(30秒)、72℃(25秒)の5サイクル、そしてその後の95℃(45秒)、68℃(30秒)、72℃(20秒)の25サイクル。この得られたPCRフラグメントを、NheIおよびBsp120Lで消化した。
【0199】
GP140−FOSについての最終発現ベクターを、両PCRフラグメントのpSinRep5への3フラグメント連結で得た。この得られたベクターpSinRep5−GP140−FOSを、制限分析およびDNA配列決定によって評価した。
【0200】
GP140−FOSをまた、安定な誘導性GP140−FOS発現細胞株を得るために、XbaIおよびBsp120L消化したpCYTtsにクローニングした。
【0201】
(実施例13)
(pSinRep5−GP140FOSを使用するGP140FOSの発現)
RNaseを含まないベクター(1.0μg)(pSinRep5−GP140−FOS)および1.0μgのDHEB(Bredenbeekら、J.Virol.67:6439−6446(1993))を、制限消化によって直線化した。続いて、SP6インビトロ転写キット(InvitroGen,InvitrogenBV,NV Leek,NetherlandsによるInvitroscripCAP)を使用して、インビトロ転写を行った。この得られた5’キャップ化mRNAを、還元性アガロースゲル上で分析した。
【0202】
インビトロ転写したmRNA(5μg)を、Invitrogenのマニュアル(Sindbis Expression System,Invitrogen BV,Netherlands)に従って、BHK21細胞(ATCC:CCL10)内にエレクトロポレートした。37℃で10時間のインキュベーション後、FCS含有培地を、FCSを含まないHP−1培地で交換し、その後さらに10時間37℃でインキュベートした。この上清を収集し、そして実施例2の記載のように正確に、可溶性GP140−FOSの産生についてウェスタンブロット分析によって分析した。
【0203】
(実施例14)
(pCYTts−GP140FOSを使用するGP140FOSの発現)
pCYTts−GP140−FOS 20μgを、制限消化によって直線化した。この反応を、フェノール/クロロホルム抽出によって停止し、その後、この直線化DNAをイソプロパノール沈殿させた。この制限消化を、アガロースゲル電気泳動によって評価した。トランスフェクションのために、5.4μgの直線化pCYTtsGP140−FOSを、30μl H2O中の0.6μgの直線化pSV2Neoと混合し、そして30μlの1M CaCl2溶液を添加した。60μlのリン酸緩衝液(50mM HEPES、280mM NaCl、1.5mM Na2HPO4、pH7.05)の添加後、この溶液を5秒間ボルテックスし、その後、室温で25秒間インキュベートした。この溶液を、2% FCSを含む2mlのHP−1培地(2% FCS培地)に直ちに添加した。次いで、80%コンフルエントなBHK21細胞培養物の培地(6ウェルプレート)を、このDNA含有培地で置換した。CO2インキュベーター中、37℃で5時間のインキュベーション後、このDNA含有培地を除去し、そして2% FCS培地中の15%グリセロール(2mL)で置換した。このグリセロール含有培地を、30秒のインキュベーション段階の後で除去し、そしてこの細胞を、10% FCSを含む、5mlのHP−1培地でリンスすることによって洗浄した。最後に、10% FCSを含む、2mlの新鮮HP−1培地を添加した。
【0204】
安定にトランスフェクトされた細胞を選択し、CO2インキュベーター中、37℃で、選択培地(G418を補充したHP−1培地)中で増殖させた。この混合集団がコンフルエントに増殖された時に、この培養物を2つの皿に分割し、その後、37℃で12時間増殖させた。一方の細胞の皿を30℃に移動させて、可溶性GP140−FOSの発現を誘導した。もう一方の皿は、37℃で維持した。
【0205】
可溶性GP140−FOSの発現を、ウェスタンブロット分析によって決定した。培養培地(0.5ml)をメタノール/クロロホルム沈殿させ、そしてこのペレットを、SDS−PAGEサンプル緩衝液中に再懸濁した。サンプルを95℃で5分間加熱し、その後15%アクリルアミドゲルにアプライした。SDS−PAGE後、タンパク質を、BassおよびYang,Creighton,T.E.編、Protein Function:A Practical Approach、第2版、IRL Press,Oxford(1997)、29−55頁に記載のように、Protanニトロセルロース膜(Schleicher& Schuell,Germany)に転写した。この膜を、TBS(10×TBS 1リットルあたり:87.7g NaCl 66.1g 塩酸Trizma(Sigma)および9.7gのTrizma塩基(Sigma)、pH7.4)中の1%ウシアルブミン(Sigma)を用いて、室温で1時間ブロックし、その後、抗GP140抗体または抗Gp−160抗体と共に1時間インキュベートした。このブロットを、TBS−T(0.05% Tween 20を含むTBS)で10分間、3回洗浄し、アルカリホスファターゼ抗マウス/ウサギ/サル/ヒトIgG結合体と共に1時間インキュベートした。TBS−Tでの10分間の2回の洗浄およびTBSでの10分間の2回の洗浄後、アルカリホスファターゼ検出試薬(50μlのNBT溶液(70%ジメチルホルムアミド中の7.7% Nitro Blue Tetrazolium(Sigma))および37μlのX−Phosphate溶液(ジメチルホルムアミド中の5%の5−ブロモ−4−クロロ−3−インドリルホスフェート)を含む10mlのAP緩衝液(100mM Tris/HCl、100mM NaCl、pH9.5))を使用して、発色反応を行った。
【0206】
(実施例15)
(GP140FOSの産生および精製)
抗gp120抗体を、NHS/EDC活性化デキストランに共有結合させ、そしてクロマトグラフィーカラムに充填した。GP140FOSを含む上清を、このカラム上にロードし、そして十分な洗浄後、GP140FOSを、0.1M HClを使用して溶出した。この溶出物を、収集チューブ中の1M Tris pH7.2を用いて、収集中に直接中和した。
【0207】
ジスルフィド結合形成は、精製の間に生じ得、従って、この収集したサンプルを、10mM Tris、pH7.3中の10mM DTTで、25℃で2時間処理した。
【0208】
DTTを、10mM Mes;80mM NaCl、pH6.0に対する、その後の透析によって除去した。最後に、Gp140FOSを、実施例16の記載のように、E2においてJUNロイシンジッパーを含むアルファウイルス粒子と混合した。
【0209】
(実施例16)
(アルファワクチン粒子の調製)
100kDの分子量カットオフを有するMillipore Ultrafree Centrifugal Filter Devicesを、製造業者によって供給されるプロトコルに従って使用して、ウイルス粒子(実施例2および3を参照のこと)を濃縮した。あるいは、ウイルス粒子を、Sindbis Expression System(Invitrogen,San Diego,California)の説明マニュアルに記載のように、スクロース密度勾配遠心によって濃縮した。ウイルス懸濁液のpHを7.5に調整し、そしてウイルス粒子を、2〜10mM DTTの存在下で数時間インキュベートした。ウイルス粒子を、適切な緩衝液のSephacryl S−300カラム(Pharmacia)(ウイルス粒子を空隙容量で溶出する)上で、混在タンパク質から精製した。
【0210】
精製したウイルス粒子を、適切な酸化還元混合物(redox shuffle)(酸化型グルタチオン/還元型グルタチオン;シスチン/システイン)の存在下の適切な緩衝液(pH7.5〜8.5)中で、少なくとも240倍モル過剰のFOS−抗原融合タンパク質と共に、4℃で少なくとも10時間インキュベートした。100kDの分子量カットオフを有するMillipore Ultrafree Centrifugal Filter Deviceを使用して、この粒子を濃縮した後、この混合物を、Sephacryl S−300ゲル濾過カラム(Pharmacia)に通した。ウイルス粒子を、空隙容量で溶出させた。
【0211】
(実施例17:HBcAg(1〜144)のアミノ末端へのJUN両親媒性ヘリックスの融合)
JUNヘリックスを、HBcAgアミノ酸配列1〜144のアミノ末端に融合した(JUN−HBcAg構築物)。JUN−HBcAg DNA配列を構築するために、JUNヘリックスおよびHBcAg(1〜144)をコードする配列を、PCRにより別々に増幅した。JUN配列を、プライマーEcoRI−JUN(s)およびJUN−SacII(as)を用いて、pJuFoプラスミドから増幅した。EcoRI−JUN(s)プライマーには、EcoRI部位、次いで開始ATGコドンを導入した。JUN−SacII(as)プライマーには、アミノ酸配列GAAGSをコードするリンカーを導入した。HBcAg(1〜144)配列を、プライマーJUN−HBcAg(s)およびHBcAg(1〜144)Hind(as)を用いて、pEco63プラスミド(ATCC番号31518から得た)から増幅した。JUN−HBcAg(s)は、JUNヘリックスをコードする配列の3’末端に対応する配列、続いてGAAGSリンカーをコードする配列およびHBcAg配列の5’末端を含む。HBcAg(1〜144)Hind(as)に、HBcAg遺伝子の後ろに終止コドンおよびHindIII部位を導入する。PCR反応については、100pmolの各オリゴおよび50ngのテンプレートDNAを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含有する50μlの反応混合物において使用する。両方の反応については、温度サイクリングを以下のとおりに行った:94℃で2分間、および94℃(1分間)、50℃(1分間)、72℃(2分間)を30サイクル。
【0212】
【化16】
2つのPCRフラグメントの融合を、プライマーEcoRI−JUN(s)およびHBcAg(1〜144)Hind(as)を用いて、PCRにより行った。100pmolの各オリゴを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含有する50μlの反応混合物中、100ngの精製PCRフラグメントとともに用いた。PCRサイクリング条件は、以下であった:94℃で2分間、および94℃(1分間)、50℃(1分間)、72℃(2分間)を35サイクル。最終的なPCR産物を、アガロースゲル電気泳動により分析し、精製し、そしてEcoRIおよびHindIII制限酵素を含む適切な緩衝液中で16時間にわたり消化した。消化したDNAフラグメントをEcoRI/HindIII消化pKKベクターに連結して、pKK−JUN−HBcAg発現ベクターを生成した。PCR産物の挿入を、EcoRI/HindIII制限分析によりおよびインサートのDNA配列決定により分析した。
【0213】
(実施例18:HBcAg(1〜144)のカルボキシ末端へのJUN両親媒性ヘリックスの融合)
JUNヘリックスを、HBcAgアミノ酸配列1〜144に融合した(HBcAg−JUN構築物)。HBcAg−JUN DNA配列を構築するために、JUNヘリックスおよびHBcAg(1〜144)をコードする配列を、PCRにより別々に増幅した。JUN配列を、プライマーSacII−JUN(s)およびJUN−HindIII(as)を用いてpJuFoプラスミドから増幅した。SacII−JUN(s)には、アミノ酸LAAGをコードするリンカーを導入した。この配列はまた、SacII部位を含む。JUN−HindIII(as)には、終止コドン(TAA)、次にHindIII部位を導入した。HBcAg(1〜144)DNA配列を、プライマーEcoRI−HBcAg(s)およびHBcAg(1〜144)−JUN(as)を用いて、pEco63プラスミドから増幅した。EcoRI−HBcAg(s)には、HBcAgコード配列の開始ATGの前に、EcoRI部位を導入した。HBcAg(1〜144)−JUN(as)には、ペプチドリンカー(LAAG)をコードする配列を導入する。これはまた、SacII部位を含む。PCR反応については、100pmolの各オリゴおよび50ngのテンプレートDNAを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含有する50μlの反応混合物において使用した。温度サイクリングを以下のとおりに行った:94℃で2分間、および94℃(1分間)、50℃(1分間)、72℃(2分間)を30サイクル。
【0214】
【化17】
2つのPCRフラグメントの融合を、プライマーEcoRI−HBcAg(s)およびJUN−HindIII(as)を用いて、PCRにより行った。PCR融合については、100pmolの各オリゴを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含有する50μlの反応混合物中の、100ngの精製PCRフラグメントとともに用いた。PCRサイクリング条件は、以下であった:94℃で2分間;および94℃(1分間)、50℃(1分間)、72℃(2分間)を35サイクル。最終的なPCR産物を、アガロースゲル電気泳動により分析し、そしてEcoRIおよびHindIII制限酵素を含む適切な緩衝液中で16時間にわたり消化した。DNAフラグメントをゲル精製し、そしてEcoRI/HindIII消化pKKベクターに連結して、pKK−HBcAg−JUN発現ベクターを生成した。PCR産物の挿入を、EcoRI/HindIII制限分析によりおよびインサートのDNA配列決定により分析した。
【0215】
(実施例19:HBcAg(1〜144)のc/e1エピトープへのJUN両親媒性ヘリックスの挿入)
HBcAgのc/e1エピトープ(残基72〜88)は、B型肝炎ウイルスキャプシドの表面上の先端領域に位置することが公知である。タンパク質のこの領域の一部(残基76〜82)は、JUNヘリックスによって遺伝的に置換されて、抗原(HBcAg−JUNIns構築物)についての付着部位を提供した。このHBcAg−JUNIns DNA配列を、PCRにより生成した:JUNヘリックス配列および2つのHBcAgフラグメント(アミノ酸残基1〜75および83〜144)をコードする配列を、PCRにより別々に増幅した。JUN配列を、プライマーBamHI−JUN(s)およびJUN−SacII(as)を用いてpJuFoプラスミドから増幅した。BamHI−JUN(s)には、ペプチド配列GSGGGをコードするリンカー配列(BamHI部位もまた含む)を導入した。JUN−SacII(as)には、ペプチドリンカー(GAAGS)をコードする配列、次いで、JUNコード配列の3’末端に相補的な配列を導入した。HBcAg(1〜75)DNA配列を、プライマーEcoRIHBcAg(s)およびHBcAg75−JUN(as)を用いて、pEco63プラスミドから増幅した。EcoRIHBcAg(s)には、EcoRI部位、次いで、HBcAg配列の5’末端に対応する配列を導入した。HBcAg75−JUN(as)には、HBcAGのアミノ酸75の後にペプチドGSGGGをコードするリンカー、次いで、JUNヘリックスをコードする配列の5’末端に相補的な配列を導入した。HBcAg(83〜144)フラグメントを、プライマーJUN−HBcAg83(s)およびHBcAg(1〜144)Hind(as)を用いて増幅した。JUN−HBcAg83(s)は、JUNコード配列の3’末端に対応する配列、次いで、ペプチドGAAGSをコードするリンカー、およびHBcAg(83〜144)をコードする配列の5’末端に対応する配列を含んでいた。HBcAg(1〜144)Hind(as)には、HBcAG遺伝子のコドン144の後ろに、終止コドンおよびHindIII部位を導入した。PCR反応については、100pmolの各オリゴおよび50ngのテンプレートDNAを、50μlの反応混合物(2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4)において使用した。温度サイクリングを以下のとおりに行った:94℃で2分間、および94℃(1分間)、50℃(1分間)、72℃(2分間)を35サイクル。
【0216】
【化18】
3つのPCRフラグメントの融合を、以下のとおりに行った。最初に、HBcAg1〜75をコードするフラグメントを、プライマーEcoRIHBcAg(s)およびJUN−SacII(as)を用いて、PCRにより、JUNをコードする配列と融合した。第2に、得られた産物を、プライマーEcoRIHBcAg(s)およびHBcAgHindIII(as)を用いて、PCRにより、HBcAg(83〜144)フラグメントと融合した。PCR融合に関しては、100pmolの各オリゴを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含有する50μlの反応混合物中、100ngの精製PCRフラグメントとともに用いた。同じPCRサイクルを、個々のフラグメントの生成と同じように用いた。最終的なPCR産物を、EcoRIおよびHindIII制限酵素を含有する適切な緩衝液中で16時間消化した。DNAフラグメントをEcoRI/HindIIIで消化したpKKベクターに連結して、pKK−HBcAg−JUNInsベクターを得た。PCR産物の挿入を、EcoRI/HindIII制限分析およびインサートのDNA配列決定により分析した。
【0217】
(実施例20:麻疹ウイルスヌクレオキャプシド(N)タンパク質のカルボキシ末端へのJUN両親媒性ヘリックスの融合)
JUNヘリックスを、アミノ酸残基1〜473を含む短縮化麻疹ウイルスNタンパク質フラグメントのカルボキシ末端に融合した(N473−JUN構築物)。N473−JUNをコードするDNA配列を構築するために、JUNヘリックスをコードする配列およびN473−JUNをコードする配列を、PCRにより別々に増幅した。JUN配列を、プライマーSacII−JUN(s)およびJUN−HindIII(as)を用いて、pJuFoプラスミドから増幅した。SacII−JUN(s)には、ペプチドリンカーLAAGをコードする配列を導入した。この配列はまた、SacII部位を含んでいた。JUN−HindIII(as)アンチセンスプライマーには、終止コドン、次いで、HindIII部位を導入した。N(1〜473)配列を、プライマーEcoRI−Nmea(s)およびNmea−JUN(as)を用いて、完全な麻疹ウイルスNタンパク質コード配列を含むpSC−Nプラスミド(M.Billeter,Zurichから得た)から増幅した。EcoRI−N(mea)(s)には、Nコード配列の開始ATGの前にEcoRI部位を導入した。N(mea)−JUN(as)は、N(1〜473)コード配列の3’末端に相補的であり、次は、ペプチドリンカー(LAAG)についてのコード配列に相補的な配列であった。PCR反応に関しては、100pmolの各オリゴおよび50ngのテンプレートDNAを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含有する50μlの反応混合物中で用いた。温度サイクリングを以下のとおりに行った:94℃で2分間;および94℃(1分間)、55℃(1分間)、72℃(2分間)を35サイクル。
【0218】
【化19】
2つのPCRフラグメントの融合を、プライマーEcoRI−Nmea(s)およびNmea−JUN(as)を用いて、さらにPCRにおいて行った。PCR融合に関しては、100pmolの各オリゴを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含有する50μlの反応混合物中、100ngの精製PCRフラグメントとともに用いた。温度サイクリングを、以下のとおりに行った:94℃で2分間;および94℃(1分間)、50℃(1分間)、72℃(2分間)を35サイクル。PCR産物を、EcoRIおよびHindIII制限酵素を含有する適切な緩衝液中で16時間消化した。DNAフラグメントをゲル精製し、そしてEcoRI/HindIIIで消化したpKKベクターに連結して、pKK−N473−JUNプラスミドを得た。PCR産物の挿入を、EcoRI/HindIII制限分析およびインサートのDNA配列決定により分析した。
【0219】
(実施例21)
(HBcAg−JUNの発現および部分的精製)
E.coli株XL−1 blueを、pKK−HBcAg−JUNで形質転換した。細菌の一晩の培養物1mlを使用して、100μg/mlのアンピシリンを含む100mlのLB培地に接種(innoculate)した。この培養物を、600nmでおよそ0.8のODに達するまで、37℃で4時間増殖させた。HBcAg−JUNの合成の誘導を、1mMの最終濃度になるようにIPTGを添加することにより行った。誘導後、細菌を、37℃で16時間さらに振蕩した。細菌を、5000×gでの15分間の遠心分離によって回収した。このペレットを、−20℃で凍結した。このペレットを融解し、そして200μg/mlリゾチームおよび10μlのBenzonase(Merck)を補充した細菌溶解緩衝液(10mM Na2HPO4、pH7.0、30mM NaCl、0.25% Tween−20、10mM EDTA、10mM DTT)中に再懸濁した。細胞を、室温で30分間インキュベートし、そしてFrench pressure cellを使用して破壊した。Triton X−100を、溶解物に添加して、0.2%の最終濃度にして、そしてこの溶解物を、氷上で30分間インキュベートし、そして時々振蕩した。図4は、IPTGでの誘導の際のE.coliにおけるHBcAg−JUNタンパク質発現を示す。pKK−HBcAg−JUN発現プラスミドまたはコントロールプラスミドを保有するE.coli細胞を、IPTGによるHBcAg−JUN発現の誘導のために使用した。IPTGの添加前に、サンプルを、pKK−HBcAg−JUNプラスミドを保有する細菌培養物(レーン3)から、およびコントロールプラスミドを保有する培養物(レーン1)から取り除いた。IPTG添加の16時間後に、サンプルを再度、pKK−HBcAg−JUNを含む培養物(レーン4)から、およびコントロール培養物(レーン2)から除去した。タンパク質発現を、SDS−PAGE、続いてクーマシー染色によってモニターした。
【0220】
次いで、この溶解物を、不溶性細胞片を除去するために、12,000×gにて30分間遠心分離した。この上清およびペレットを、HBcAgに対するモノクローナル抗体(YVS1841、Accurate Chemical and Scientific Corp.,Westbury,NY,USAから購入)を使用して、ウエスタンブロッティングにより分析した。これは、有意な量のHBcAg−JUNタンパク質が可溶性であることを示した(図5)。簡潔には、HBcAg−JUNを発現しているE.coli細胞からの溶解物、およびコントロール細胞からの溶解物を、14,000×gで30分間遠心分離した。上清(=可溶性画分)およびペレット(=不溶性画分)を分離し、そしてSDSサンプル緩衝液で希釈して等しい容量にした。サンプルを、SDS−PAGE、続いて抗HBcAgモノクローナル抗体YVS1841でのウエスタンブロッティングによって分析した。レーン1:可溶性画分、コントロール細胞;レーン2:不溶性画分、コントロール細胞;レーン3:可溶性画分、HBcAg−JUNを発現している細胞;レーン4:不溶性画分、HbcAg−JUNを発現している細胞。
【0221】
清澄化した細胞溶解物を、3mlの15%スクロース溶液、続いて4mlの細菌溶解物を重層した4mlの65%スクロース溶液からなる、スクロース段階的勾配を用いる、段階的勾配遠心分離に使用した。このサンプルを、100,000×gにて4℃で3時間遠心分離した。遠心分離後、この勾配の頂端から1mlの画分を収集し、そしてSDS−PAGE、続いてクーマシー染色によって分析した(図6)。レーン1:遠心分離前の総E.coli溶解物、レーン1および2:勾配の頂端からの画分1および2、レーン4〜7:画分5〜8(15%スクロース)。HBcAg−JUNタンパク質を、クーマシー染色によって検出した。
【0222】
HBcAg−JUNタンパク質を、15%スクロースと65%スクロースとの間の界面に濃縮させた。このことは、このタンパク質がカプシド粒子を形成したことを示す。ほとんどの細菌タンパク質は、この勾配のスクロースを含まない上層に残存し、従って、HBcAG−JUN粒子の段階的勾配遠心分離は、この粒子を濃縮すること、および部分精製することの両方を導く。
【0223】
(実施例22)
(HBcAg−JUNへのhGH−FOSの共有結合)
HBcAg−JUN粒子へのタンパク質を結合の実証するために、本発明者らは、そのカルボキシ末端がFOSヘリックスに融合したヒト成長ホルモン(hGH)を、モデルタンパク質(hGH−FOS)として選択した。HBcAg−JUN粒子を、部分的に精製されたhGH−FOSと混合し、そして4℃で4時間インキュベートし、このタンパク質の結合を可能にした。次いで、この混合物を、HBcAg−JUN溶液およびhGH−FOS溶液の両方に存在するDTTを除去するために、3000倍容量の透析緩衝液(150mM NaCl、10mM Tris−HCl溶液(pH8.0))に対して一晩透析し、それによって、ジスルフィド結合の確立を通じて、タンパク質の共有結合を可能にする。コントロールとして、HBcAg−JUN溶液およびhGH−FOS溶液もまた、透析緩衝液に対して透析した。3つ全ての透析したタンパク質溶液からのサンプルを、非還元条件下でSDS−PAGEによって分析した。HBcAg−JUNへのhGH−FOSの結合を、抗hGHイムノブロットにおいて検出した(図7)。HBcAg−JUNに結合したhGH−FOSは、およそ53kDaの見かけの分子量で移動するはずであり、一方、非結合hGH−FOSは、31kDaの見かけの分子量で移動する。この透析液を、還元剤の非存在下(レーン3)、および還元剤の存在下(レーン2)でのSDS−PAGEにより分析し、そしてクーマシー染色によって検出した。コントロールとして、カプシド粒子と混合されていないhGH−FOSもまた、還元剤の存在下でゲルにロードした(レーン1)。
【0224】
およそ53kDaの分子量へのhGH−FOSの変動を、HBcAg−JUNカプシドタンパク質の存在下で観察した。このことは、HBcAg−JUNへのhGH−FOSの効率的な結合が生じたことを示唆する。
【0225】
(実施例23)
(HBcAg(1〜149)のc/e1エピトープへのリジン残基を含むペプチドの挿入)
HBcAgのc/e1エピトープ(残基72〜88)を、B型肝炎ウイルスカプシド(HBcAg)の表面上の先端領域(tip region)に配置した。この領域の一部(プロリン79およびアラニン80)を、ペプチドGly−Gly−Lys−Gly−Glyによって遺伝的に置換した(HBcAg−Lys構築物)。この導入されたリジン残基は、遊離システイン基を含む任意の抗原を有するHBcAg粒子の分子間化学的架橋のために使用され得る反応性アミノ基を、その側鎖に含む。
【0226】
HBcAg−Lys DNA配列を、PCRによって作製した:HBcAgフラグメントをコードする2つのフラグメント(アミノ酸残基1〜78および81〜149)を、PCRによって別々に増幅した。これらのPCRに使用されたプライマーもまた、Gly−Gly−Lys−Gly−GlyペプチドをコードするDNA配列を導入した。HBcAg(1〜78)フラグメントを、プライマーであるEcoRI HBcAg(s)およびLys−HBcAg(as)を使用して、pEco63から増幅した。HBcAg(81〜149)フラグメントを、プライマーであるLys−HBcAg(s)およびHBcAg(1〜149)Hind(as)を使用して、pEco63から増幅した。プライマーであるLys−HBcAg(as)およびLys−HBcAg(s)は、相補性DNA配列を、2つのPCR産物の末端に導入し、このことは、その後のアセンブリ(assembly)PCRにおいて2つのPCR産物の融合を可能にした。アセンブリしたフラグメントを、プライマーEcoRI HBcAg(s)およびHbcAg(1〜149)Hind(as)を使用して、PCRによって増幅した。
【0227】
PCRについては、100pmolの各オリゴおよび50ngのテンプレートDNAを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含む50μlの反応混合物において使用した。両方の反応について、温度のサイクリングを、以下の通りに行った:94℃で2分間;94℃(1分)、50℃(1分)、72℃(2分)を30サイクル。
【0228】
(プライマー配列)
【0229】
【化20】
PCRによる2つのPCRフラグメントの融合のために、100pmolのプライマーであるEcoRI HBcAg(s)およびHBcAg(1〜149)Hind(as)を、100ngの2つの精製されたPCRフラグメントとともに、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含む、50μlの反応混合物中で使用した。PCRサイクル条件は、以下であった:94℃で2分間;94℃(1分)、50℃(1分)、72℃(2分)を30サイクル。アセンブリされたPCR産物を、アガロースゲル電気泳動によって分析し、精製し、そしてEcoRI制限酵素およびHindIII制限酵素によって、適切な緩衝液中で19時間消化した。この消化されたDNAフラグメントを、EcoRI/HindIIIで消化したpKKベクター中に連結して、pKK−HBcAg−Lys発現ベクターを作製した。このベクターへのPCR産物の挿入を、EcoRI/HindIII制限分析およびこのインサートのDNA配列決定によって分析した。
【0230】
(実施例24)
(HBcAg−Lysの発現および部分精製)
E.coli株XL−1 blueを、pKK−HBcAg−Lysで形質転換した。細菌の一晩の培養物1mlを使用して、100μg/mlのアンピシリンを含む100mlのLB培地に接種した。この培養物を、600nmでおよそ0.8のODに達するまで、37℃で4時間増殖させた。HBcAg−Lysの合成の誘導を、1mMの最終濃度になるようにIPTGを添加することにより行った。誘導後、細菌を、37℃で16時間さらに振蕩した。細菌を、5000×gでの15分間の遠心分離によって回収した。このペレットを、−20℃で凍結した。このペレットを融解し、そして200μg/mlリゾチームおよび10μlのBenzonase(Merck)を補充した細菌溶解緩衝液(10mM Na2HPO4、pH7.0、30mM NaCl、0.25% Tween−20、10mM EDTA、10mM DTT)中に再懸濁した。細胞を、室温で30分間インキュベートし、そしてFrench pressure cellを使用して破壊した。Triton X−100を、溶解物に添加し、0.2%の最終濃度にして、そしてこの溶解物を、氷上で30分間インキュベートし、そして時々振蕩した。pKK−HBcAg−Lys発現プラスミドまたはコントロールプラスミドを保有するE.coli細胞を、IPTGによるHBcAg−Lys発現の誘導のために使用した。IPTGの添加前に、サンプルを、pKK−HBcAg−Lysプラスミドを保有する細菌培養物から、およびコントロールプラスミドを保有する培養物から取り除いた。IPTG添加の16時間後に、サンプルを再度、pKK−HBcAg−Lysを含む培養物から、およびコントロール培養物から除去した。タンパク質発現を、SDS−PAGE、続くクーマシー染色によってモニターした。
【0231】
次いで、この溶解物を、不溶性細胞片を除去するために、12,000×gにて30分間遠心分離した。この上清およびペレットを、HBcAgに対するモノクローナル抗体(YVS1841、Accurate Chemical and Scientific Corp.,Westbury,NY,USAから購入)を使用して、ウエスタンブロッティングにより分析した。これは、有意な量のHBcAg−Lysタンパク質が可溶性であることを示す(図5)。簡潔には、HBcAg−Lysを発現しているE.coli細胞からの溶解物、およびコントロール細胞からの溶解物を、14,000×gで30分間遠心分離した。上清(=可溶性画分)およびぺレット(=不溶性画分)を分離し、そしてSDSサンプル緩衝液で希釈して等しい容量にした。サンプルを、SDS−PAGE、続いて抗HBcAgモノクローナル抗体YVS1841でのウエスタンブロッティングによって分析した。
【0232】
清澄化した細胞溶解物を、3mlの15%スクロース溶液、続いて4mlの細菌溶解物を重層した4mlの65%スクロース溶液からなる、スクロース段階的勾配を用いる、段階的勾配遠心分離に使用した。このサンプルを、100,000×gにて4℃で3時間遠心分離した。遠心分離後、勾配の頂端から1mlの画分を収集し、そしてSDS−PAGE、続いてクーマシー染色によって分析した(図6)。HBcAg−Lysタンパク質を、クーマシー染色によって検出した。
【0233】
HBcAg−Lysタンパク質を、15%スクロースと65%スクロースとの間の界面に濃縮させた。このことは、このタンパク質がカプシド粒子を形成したことを示す。ほとんどの細菌タンパク質は、この勾配のスクロースを含まない上層に残存し、従って、HBcAg−Lys粒子の段階的勾配遠心分離は、この粒子を濃縮すること、および部分精製することの両方を導く。
【0234】
(実施例25)
(異種二機能性架橋剤SPDPを用いるFLAGペプチドのHBcAg−Lysへの化学的結合)
アミノ末端にシステイン残基を有する合成FLAGペプチド(アミノ酸配列CGGDYKDDDDK)を、FLAGペプチドに対する免疫応答を誘発するために、精製されたHBcAg−Lys粒子に化学的に結合した。HBcAg−Lys粒子の純度95%の溶液(2mg/ml) 600μlを、異種二機能性架橋剤N−Succinimidyl 3−(2−pyridyldithio)propionate(SPDP)(0.5mM)と共に30分間室温でインキュベートした。反応の終了後に、遊離のSPDPを除くために、混合物を、1リットルの50mM リン酸緩衝液(pH7.2)(150mM NaClを有する)に対して一晩透析した。次いで、500μlの誘導体化HBcAg−Lysカプシド(2mg/ml)を、金属触媒スフィドリル(sufhydryl)酸化を防ぐために10mM EDTAの存在下で0.1mM FLAGペプチド(アミノ末端システインを含む)と混合した。この反応を、ペプチドの遊離システインとの反応における、SPDPからのピリミジン−2−チオン(thione)の遊離に起因する343nMでの溶液の吸光度の増加により、モニターした。誘導体化Lys残基のペプチドとの反応は、約30分後に終了した。
【0235】
FLAG装飾された粒子を、マウスに注射した。
【0236】
(実施例26)
(pMPSV−gp140cysの構築)
gp140遺伝子を、オリゴgp140CysEcoRIおよびSalIgp140を用いて、pCytTSgp140FOSからPCRによって増幅した。PCRに関して、100pmolのそれぞれのオリゴおよび50ngの鋳型DNAを、50μlの反応混合液(2ユニットのPwoポリメラーゼ、0.1mMのdNTPおよび2mMのMgSO4を含む)中で使用した。両方の反応に対して、温度サイクルを以下のように実施した:94℃を2分間;94℃(0.5分間)、55度(0.5分間)、72℃(2分間)を30サイクル。
【0237】
PCR産物を、QiaEXIIキットを用いて精製し、SalI/EcoRIで消化し、そして同一の酵素で切断したベクターpMPSVHE中に連結した。(オリゴ配列)
【0238】
【化21】
(実施例27)
(pMPSVgp140Cysの発現)
pMPSVgp140Cys(20μg)を、制限消化によって直線化した。反応をフェノール/クロロホルム抽出によって停止し、その後直線化DNAをイソプロパノール沈殿する。制限消化を、アガロースゲル電気泳動により評価した。トランスフェクションのために、5.4μgの直線化pMPSVgp140−Cysを、30μlの水中で0.6μgの直線化pSV2Neoと混合し、そして30μlの1M CaCl2溶液を添加した。60μlのリン酸緩衝液(50mM HEPES、280mM NaCl、1.5mM Na2HPO4(pH7.05))の添加後に、この溶液を5秒間ボルテックスし、続いて室温で25秒間インキュベーションした。この溶液に、2%FCSを含むHP−1培地(2%FCS培地)を2ml、直ちに添加した。次いで、80%コンフルエントなBHK21細胞培養(6ウェルプレート)の培地を、DNA含有培地に置き換えた。37℃で5時間、CO2インキュベーター内でのインキュベートの後、DNA含有培地を取り出し、2mlの15%グリセロール含有2%FCS培地で置き換えた。グリセロール含有培地を、30秒間のインキュベート相の後に取り出し、そしてこの細胞を、5mlの10%FCS含有HP−1培地でのリンスにより洗浄した。最後に、2mlの新鮮なHP−1培地(10%FCSを含む)を加えた。
【0239】
安定にトランスフェクトされた細胞を選択し、そしてCO2インキュベーター内で、選択培地(G418を補充したHP−1培地)中で37℃で増殖した。混合集団がコンフルエントまで増殖したとき、培養物を2つのディッシュに分配し、その後、37℃で12時間の増殖期間を続ける。細胞の一方のディッシュを30℃に移動し、可溶性GP140−FOSの発現を誘導させた。他方のディッシュを37℃に維持した。
【0240】
可溶性GP140−Cysの発現を、ウエスタンブロット分析によって解析した。培養培地(0.5ml)を、メタノール/クロロホルム沈殿し、そしてこのペレットをSDS−PAGEサンプル緩衝液に再懸濁した。サンプルを、95℃で5分間加熱し、その後、15%アクリルアミドゲルにアプライした。SDS−PAGE後、BassおよびYang(Creighton,T.E.編、Protein Function:A Practical Approach、第2版、IRL Press、Oxford(1997)、29−55頁)に記載されたように、タンパク質をProtan nitrocellilose membrane(Shleicher&Schuell、Germany)に転写した。この膜を、TBS(10×TBS 1リットルあたり、87.7g NaCl、66.1g Trizma hydrochloride(Sigma)および9.7g Trizma base(Sigma)、pH7.4)中に1%のウシアルブミン(Sigma)を用いて、室温で一時間ブロックした後、抗GP140抗体または抗GP160抗体を用いて一時間インキュベートした。このブロットを、TBS−T(0.05%のTween20を含むTBS)で10分間3回洗浄し、アルカリホスファターゼ抗マウス/ウサギ/サル/ヒトIgG結合体を用いて一時間インキュベートした。TBS−Tで10分間2回、およびTBSで10分間2回洗浄した後、アルカリホスファターゼ検出試薬(50μlのNBT溶液(70%のジメチルホルムアミド中に7.7% Nitro Blue Tetrazolium(Sigma))および37μlのX−Phosphate溶液(ジメチルホルムアミド中に5%の5−ブロモ−4−クロロ−3−インドリルリン酸)を含む10mlのAP緩衝液(100mM Tris/HCl、100mM NaCl、pH9.5)を用いて現像反応を行った。
【0241】
(実施例28)
(gp140Cysの精製)
抗gp120抗体を、NHS/EDC活性化デキストランと共有結合し、そしてクロマトグラフィーカラムに充填した。GP140Cysを含む上清をカラムにロードし、そして十分な洗浄後に、GP140Cysを、0.1M HClで溶出した。溶出物を、回収チューブ中の1M Tris(pH7.2)を用いて回収中に直接中和した。
【0242】
精製中に、ジスフィルド結合形成が形成し得、したがって、回収サンプルを、10mMのDTT(10mM Tris(pH7.5)中)を用いて、25℃で2時間処理する。
【0243】
DTTを、10mM Mes;80mM NaCl(pH6.0)に対する引き続く分析によって除去する。最終的に、実施例16に記載されるように、GP140Cysを、E2中のJUN残基を含むアルファウイルス粒子と混合する。
【0244】
(実施例29)
(PLA2−Cysの構築)
PLA2遺伝子を、オリゴEcoRIPLAおよびオリゴPLA−Cys−hindを用いて、pAV3PLAfosからPCRによって増幅した。PCRについて、100pmolの各オリゴおよび50ngの鋳型DNAを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含む50μlの反応混合液中で用いた。両反応について、温度サイクルを、以下のように行った:94℃で2分間;94℃(0.5分間)、55℃(0.5分間)、72℃(2分間)を30サイクル。
【0245】
PCR産物を、QiaEXIIキットを用いて精製し、EcoRI/HindIIIで消化し、そして同一の酵素で消化されたベクターpAV3に連結した。(オリゴ)
【0246】
【化22】
(実施例30)
(PLA−cysの発現および精製)
Cysタグ化タンパク質の細胞質内産生のために、E.coli XL−1−Blue株をベクターpAV3::PLAおよびpPLA−Cysを用いて形質転換した。培養物を、37℃で振盪しながら、アンピシリン存在下の豊富な培地中でインキュベートした。培養物の吸光度(550nm)で、1mMのIPTGを添加し、インキュベーションをさらに5時間続けた。細胞を遠心分離によって回収し、DNase、RNaseおよびリゾチームを含む適切な緩衝液(例えば、Tris−HCl(pH7.2)、150mM NaCl)中に再懸濁し、そしてフレンチプレス(french pressure cell)を通す通過によって破壊した。遠心分離(Sorvall RC−5C、SS34ローター、15,000rpmで4℃10分間)の後、ペレットを、4℃で25mlの封入体洗浄緩衝液(20mM tris−HCl、23% スクロース、0.5% Triton X−100、1mM EDTA(pH8))中に再懸濁し、そして上記のように再遠心分離した。この手順を、遠心分離後の上清が本質的に清澄になるまで反復した。封入体を、20mlの可溶化緩衝液(5.5M 塩酸グアニジン、25mM tris−HCl(pH7.5))中に、室温で再懸濁し、そして不溶性の物質を、遠心分離および引き続く滅菌フィルター(0.45μm)を通す上清の通過で除去した。タンパク質溶液を、10mM EDTAおよび100mM DTTの存在下で、4℃で少なくとも10時間維持し、次いで、10容量の5.5M 塩酸グアニジン、25mM tris−HCl、10mM EDTA(pH6)に対して3回透析した。この溶液を、適切な酸化還元シャッフル(shuffle)(酸化型グルタチオン/還元型グルタチオン;シスチン/システイン)の存在下で、512Mの尿素、4mM EDTA、0.1M NH4Cl、20mM ホウ酸ナトリウム(pH8.3)に対して2回透析した。次いで、再フォールディングタンパク質をイオン交換クロマトグラフィーにアプライした。このタンパク質を、2〜10mM DTT存在下で適切な緩衝液(pH7以上)中に保存し、システイン残基を還元型で保持した。このタンパク質とαウイルス粒子との結合の前に、DTTを、Sephadex G−25ゲル濾過カラムを通すタンパク質溶液の通過によって除去した。
【図面の簡単な説明】
【図1】 pCYTts::E2JUN発現ベクターを使用するE2−JUN融合タンパク質を含むウイルス粒子の産生を示すウエスタンブロット。
【図2】 pTE5’2J::E2JUN発現ベクターから発現されるE2−JUN融合タンパク質を含むウイルス粒子の産生を示すウエスタンブロット。
【図3】 FOS−hgh抗原の細菌発現および真核生物発現を示すウエスタンブロット。
【図4】 E.coli細胞におけるHBcAg−JUNの発現。
【図5】 HBcAg−JUNがE.coli溶解物に可溶性であることを示すウエスタンブロット。
【図6】 スクロース密度勾配におけるHBcAg−JUNキャプシド粒子の濃縮のSDS−PAGE分析。
【図7】 hGH−FOS粒子およびHBcAg−JUN粒子の結合の非還元SDS−PAGE分析。
【発明の詳細な説明】
【0001】
(発明の背景)
(発明の分野)
本発明は、分子生物学、ウイルス学、免疫学および医学の分野に関する。本発明は、順序付けられそして反復性の抗原または抗原決定基のアレイを含む組成物を提供する。本発明はまた、順序付けられそして反復性のアレイにおいて抗原または抗原決定基を生成するためのプロセスを提供する。この順序付けられそして反復性の抗原または抗原決定基は、感染性疾患の処置、アレルギーの処置のためのワクチンの産生において、そして癌を予防または治療するための、および規定された自己特異的抗体を作製するためのファルマシン(pharmaccine)として有用である。
【0002】
(関連技術)
感染性疾患の予防のためのワクチンの開発は、任意の医学的発明のヒトの健康に対して最大の影響を有した。1年あたり300万人の死亡が、ワクチン接種によって世界中で予防されると見積もられている(Hillemann,Nature Medicine 4:507(1998))。最も通常のワクチン接種ストラテジーである、弱毒化(すなわち、毒性が低い)病原体または近縁の生物の使用は、危険性がより低い牛痘ウイルスの投与によって天然痘に対してワクチン接種したEdward Jennerによって1796年に最初に実証された。多数の生弱毒化ウイルス(例えば、麻疹、おたふくかぜ、風疹、水痘、アデノウイルス、ポリオ、インフルエンザ)および細菌(例えば、結核に対しての桿菌Calmette−Guerin(BCG))がワクチン接種について好首尾に投与されているが、「ワクチン」生物、特に免疫無防備状態の個体により毒性および感染へと後転することに関連した重篤な合併症を発症する危険性が存在する。
【0003】
弱毒化ウイルスの具体的設計は、欠失改変体または変異改変体の作製を介する組換えDNA技術(すなわち、遺伝子操作)によって現在可能にされている。例えば、nef遺伝子内に欠失を有する、操作されたサル免疫不全ウイルス(SIV)の投与は、マカクがその後の病原性SIV株での感染から保護されることを示した(Danielら,Science 258:1938−1941(1992))。しかし、弱毒化SIVを投与した動物における後天性免疫不全症候群(AIDS)様症状の進行は、安全性の懸念を生じる(Babaら,Science 267:1820−1825((1995))。
【0004】
代替的なアプローチとして、弱毒化ウイルスまたは細菌が、弱毒化形態で投与するにはあまりにも危険すぎると考えられる病原体(例えば、ヒト免疫不全ウイルス(HIV))の抗原コード遺伝子についてのキャリアとして用いられ得る。宿主への抗原コード遺伝子の送達の際に、この抗原は、インサイチュで合成される。ワクシニアおよび関連のアビポックスウイルス(avipox viruse)は、種々の疾患についての前臨床研究および臨床研究において種々の遺伝子についてのこのようなキャリアとして用いられている(例えば、Shenら,Science 252:440(1991))。このワクチン接種ストラテジーの1つの欠点は、これがビリオンの表面を模倣しないことである。なぜなら、この組換え型タンパク質は、宿主細胞の表面で発現されるからである。さらに、合併症は、生命を脅かす播種性ワクシニア感染によって証明されたように、免疫無防備状態の個体において発症し得る(Redfield,N.Eng.J.Med 316:673((1998))。
【0005】
第4のワクチン接種アプローチは、インビトロで増殖させた病原体(例えば、インフルエンザ血球凝集素またはノイラミニダーゼ)から精製されたかまたは単一のウイルスタンパク質(例えば、B型肝炎表面抗原)の異種発現後に発現されたかのいずれかの、単離された病原体成分の使用を含む。例えば、組換え型の、変異した毒素(無毒化された)は、ジフテリア、破傷風、コレラおよび百日咳の毒素に対するワクチン接種のために用いられ(Levineら,New generation Vaccines,第2版,Marcel Dekker,Inc.,New York 1997)、そしてHIVの組換え型タンパク質(gp120および全長gp160)を、HIVに対する中和抗体を誘導する手段として評価し、失望した結果となった(Connorら,J.Virol.72:1552(1998))。近年、HIV−1単離体に対するCTL応答を誘導し得、そしてHIV−1単離体に対する中和活性を有する惹起し得る、可溶性オリゴマーgp160を用いて有望な結果が得られた(Van Corttら,J.Virol.71:4319(1997))。さらに、抗原の公知のB細胞エピトープまたはT細胞エピトープが、T細胞の助けを刺激することによりエピトープの免疫原性を増加させるように設計されたキャリア分子に結合されている、ペプチドワクチンが用いられ得る。しかし、このアプローチに伴う1つの重大な問題は、このアプローチが、全体としてこのタンパク質に対して制限された免疫応答しか提供しないことである。さらに、ワクチンは、異なるMHCハプロタイプについて個々に設計されなければならない。この型のワクチンについての最も深刻な懸念は、防御性抗ウイルス抗体が、ペプチドによって模倣され得ない、複雑な三次元構造を認識することである。
【0006】
より新規のワクチン接種ストラテジーは、DNAワクチンの使用であり(Donnellyら,Ann.Rev.Immunol.15:617(1997))、これは、(生ベクターを使用することなく)MHCクラスI拘束CTL応答を生じ得る。これは、同じウイルスの多くの株に不変の保存された内部タンパク質由来のエピトープを標的化することにより、異なる株のウイルスに対する、より広範な防御を提供し得る。この抗原は、哺乳動物の翻訳後修飾、コンホメーションおよびオリゴマー形成を有して生成されるので、組換え型または化学的に改変されたタンパク質よりも、ウイルス感染によって生成される野生型タンパク質に類似するかまたは同一である可能性がより高い。しかし、この区別は、細菌抗原の適用については欠点となり得る。なぜなら、非ネイティブ翻訳後修飾が、減少した免疫原性を生じ得るからである。さらに、ウイルス表面タンパク質は、マトリックスタンパク質の非存在下において高度に組織化されない。
【0007】
感染性疾患の予防のための適用に加えて、ワクチン技術は、アレルギーと関連した免疫の問題と取り組むために現在利用される。アレルギー個体では、IgEアイソタイプの抗体は、特定の抗原(アレルゲン)に向けての不適切な体液性免疫応答において生成される。アレルギー免疫治療によるアレルギーの処置は、3〜5年までの期間にわたる、連続して漸増する用量の特定のアレルゲンの毎週の投与を必要とする。おそらく、脂肪細胞上のIgE抗体と反応する前に、鼻分泌物または呼吸分泌物中または膜中のアレルゲンを遮断する、「ブロッキング」IgG抗体が生成される。しかし、IgG力価と症状の軽減との間に一定の関係は存在しない。おそらく、これは、極めて時間がかかりかつコストのかかるプロセスであり、長期にわたって重篤な症状を有する患者について毎年でしか考えられない。
【0008】
精製されたタンパク質の単独投与は、強力な免疫応答を惹起するに通常充分でないことが充分に確立されている;単離された抗原は一般に、アジュバントと呼ばれるヘルパー物質と一緒に与えられねばならない。これらのアジュバント内では、投与された抗原は、迅速な分解から保護され、そしてアジュバントは、低レベルの抗原の長期の放出を提供する。
【0009】
単離されたタンパク質とは異なり、ウイルスは、T細胞の助けを伴って、または伴わずにの両方で、任意のアジュバントの非存在下で迅速かつ効率的な免疫応答を誘導する(BachmannおよびZinkernagel,Ann.Rev.Immunol.15:235−270(1997))。ウイルスはしばしばほんの少数のタンパク質からなっているが、これらは、単離された成分よりもずっと強力な免疫応答を誘発し得る。B細胞応答に関しては、ウイルスの免疫原性についての1つの重大な因子が、表面エピトープの反復性および順番であることが公知である。多くのウイルスは、B細胞上のエピトープ特異的免疫グロブリンを効率的に架橋するエピトープの規則的アレイを提示する準結晶表面を示す(BachmannおよびZinkernagel,Immunol.Today 17:553−558(1996))。B細胞上の表面免疫グロブリンのこの架橋は、細胞周期進行およびIgM抗体の生成を直接誘導する、強力な活性化シグナルである。さらに、このような誘発されたB細胞は、Tヘルパー細胞を活性化し得、これは次にB細胞におけるIgM抗体生成からIgG抗体生成へのスイッチおよび長期に生存するB細胞記憶の生成を誘導する(任意のワクチン接種の目的)(BachmannおよびZinkernagel,Ann.Rev.Immunol.15:235−270(1997))。ウイルスの構造は、自己免疫疾患における抗抗体の生成に均一に結びつけられ、そして病原体に対する自然な応答の一部である(Fehr,T.ら,J.Exp.Med 185:1785−1792(1997)を参照のこと)。従って、順序付けられかつ反復性のアレイにおいて組織化されたウイルス粒子上の抗原は、非常に免疫原性である。なぜなら、これらは、B細胞を直接活性化し得るからである。
【0010】
強力なB細胞応答に加えて、ウイルス粒子はまた、免疫系の別の重要なアームである、細胞傷害性T細胞応答の生成を誘導し得る。これらの細胞傷害性T細胞は、HIVまたはB型肝炎ウイルスのような非細胞変性性ウイルスの除去のために、および腫瘍の根絶のために特に重要である。細胞傷害性T細胞は、ネイティブな抗原を認識しないが、むしろ、MHCクラスI分子と会合したそれらの分解産物を認識する(TownsendおよびBodmer,Ann.Rev.Immunol.7:601−624(1989))。マクロファージおよび樹状細胞は、内因性ウイルス粒子を取り込み得、そしてプロセシングし得(しかし、それらの可溶性の単離された成分はそうでない)、そして生成された分解産物を細胞傷害性T細胞に対して提示し得、それらの活性化および増殖をもたらす(Kovacsovics−Bankowskiら,Proc.Natl.Acad.Sci.USA 90:4942−4946(1993);Bachmannら,Eur.J.Immunol.26:2595−2600(1996))。
【0011】
抗原としてのウイルス粒子は、それらの単離された成分に対して2つの利点を示す:(1)それらの高度の反復表面構造に起因して、これらは、B細胞を直接活性化し得、高い抗体力価および長期に永続するB細胞記憶をもたらす;および(2)ウイルスが非感染性であり、そしてアジュバントが存在しなくとも、ウイルス粒子は細胞傷害性T細胞応答を誘導し得るが、可溶性タンパク質は誘導し得ない。
【0012】
いくつかの新たなワクチンストラテジーは、ウイルスの固有の免疫原性を利用する。これらのアプローチのうちのいくつかは、ウイルス粒子の粒子状の性質に焦点を当てる;例えば、ラテックスビーズおよび抗原からなるワクチンを開示する、Harding,C.V.およびSong,R.(J.Immunology 153:4925(1994));酸化鉄ビーズおよび抗原からなるワクチンを開示する、Kovacsovics−Bankowski,M.ら(Proc.Natl.Acad.Sci.USA 90:4942−4946(1993));抗原で被覆したコア粒子を開示する、Kossovsky,N.らに対する米国特許第5,334,394号;表面に共有結合した1以上のタンパク質を表面に保有する合成ポリマー粒子を開示する、米国特許第5,871,747号;ならびにコア粒子の表面を少なくとも部分的に被覆する非共有結合した被覆および被覆したコア粒子と接触している少なくとも1つの生物学的活性剤を有するコア粒子(例えば、WO/94/15585を参照のこと)を参照のこと。
【0013】
しかし、これらのウイルス模倣システムの欠点は、ウイルス表面に見出される、順序付けられた抗原提示を回復できないことである。ランダムな方向で表面に結合された抗原は、CTL応答を誘導し、B細胞応答を全く誘導しないかまたは弱くしか誘導しないことが見出されている。効果的なワクチンについては、上記およびBachmannおよびZinkernagel,Ann.Rev.Immunol.15:235(1997)に記載されるように、免疫系の両方のアームが強力に活性化されなければならない。
【0014】
別の例では、組換えウイルスは、抗原送達のために利用される。カプシドタンパク質に融合された抗原を含む糸状ファージウイルスは、非常に免疫原性であることが見出された(Perham R.N.ら,FEMS Microbiol.Rev.17:25−31(1995);Willisら,Gene 128:85−88(1993);Minenkovaら,Gene 128:85−88(1993)を参照のこと)。しかし、この系は、融合タンパク質が高レベルで発現される場合(Iannoloら,J.Mol.Biol.248:835−844(1995))に非常に小さなペプチド(5または6アミノ酸残基)に制限されるか、またはより大きなタンパク質の低レベルの発現に制限される(de la Cruzら,J.Biol.Chem.263:4318−4322(1988))。低分子ペプチドについては、これまでCTL応答しか観察されておらず、そしてB細胞応答は観察されていないかまたは弱くしか観察されていない。
【0015】
なお別の系では、組換えアルファウイルスは、抗原送達の手段として提唱される(米国特許第5,766,602号;同第5,792,462号;同第5,739,026号;同第5,789,245号および同第5,814,482号を参照のこと)。これまでに記載された組換えウイルス系に伴う問題としては、ウイルス表面上での異種タンパク質の低密度発現および/または異なる適用のために新規でかつ異なる組換えウイルスを好首尾にかつ反復して作製することの困難性が挙げられる。
【0016】
さらなる開発では、ワクチン生成の領域においてウイルス様粒子(VLP)が利用される。なぜなら、それらの構造特性および非感染性性質の両方のためである。VLPは、対称様式で、1以上の型の多くのタンパク質分子から構築されたスーパー分子(supermolecular)構造である。これらはウイルスゲノムを欠き、それゆえ、非感染性である。VLPはしばしば、異種発現によって大量に生成され得、そして容易に精製され得る。
【0017】
VLPの例としては、以下のウイルスのカプシドタンパク質が挙げられる:B型肝炎ウイルス(Ulrichら,Virus Res.50:141−182(1998))、麻疹ウイルス(Warnesら,Gene 160:173−178(1995))、シンドビスウイルス、ロタウイルス(米国特許第5,071,651号および同第5,374,426号)、口蹄疫ウイルス(Twomeyら,Vaccine 13:1603−1610(1995))、ノーウォークウイルス(Jiang,X.ら,Science 250:1580−1583(1990);Matsui,S.M.,ら,J.Clin.Invest.87:1456−1461(1991))、レトロウイルスGAGタンパク質(PCT特許出願第WO 96/30523号)、レトロトランスポゾンTyタンパク質pl、B型肝炎ウイルスの表面タンパク質(WO 92/11291)およびヒトパピローマウイルス(WO 98/15631)。いくつかの例では、組換え型DNA技術は、VLPタンパク質に対して異種タンパク質を融合するために利用され得る(Kratz,P.A.ら,Proc.Natl.Acad.Sci.USA 96:19151920(1999))。
【0018】
従って、当該分野において、天然の病原体と同等に効率的な、強力なCTLおよびB細胞免疫応答を促進する、新規でかつ改善されたワクチンの開発についての必要性が存在する。
【0019】
(発明の要旨)
本発明は、任意の所望の抗原で被覆された粒子の産生が可能な、新たな融通が利く技術を提供する。この技術は、感染性疾患に対して高度に効果的なワクチンの作製ならびにアレルギーおよび癌の処置のためのワクチンの作製を可能にする。
【0020】
第1の実施形態において、本発明は、以下を含む新規な化合物を提供する:(A)非天然の分子足場、および(B)抗原または抗原決定基。
【0021】
非天然の分子足場は、以下を含む:(i)(1)非天然起源のコア粒子および(2)天然起源のコア粒子からなる群より選択される、コア粒子;ならびに(ii)少なくとも1つの第1付着部位を含む形成体であって、少なくとも1つの共有結合によって上記のコア粒子に連結されている、形成体。
【0022】
抗原または抗原決定基は、少なくとも1つの第2付着部位を有し、この部位は、以下からなる群より選択される:(i)その抗原または抗原決定基とともには天然に存在しない、付着部位;および(ii)その抗原または抗原決定とともに天然に存在する、付着部位。
【0023】
本発明は、少なくとも1つの非ペプチド結合による第1付着部位への第2付着部位の結合を介する順序づけられかつ反復性の抗原アレイを提供する。従って、抗原または抗原決定基と非天然の分子足場は、第1付着部位および第2付着部位のこの結合を通じて一緒にされて、順序づけられかつ反復性の抗原アレイを形成する。
【0024】
別の実施形態において、前述の組成物のコア粒子は、ウイルス、ウイルス様粒子、バクテリオファージ、ウイルスキャプシド粒子、またはそれらからの組換え体を含む。あるいは、このコア粒子は、合成ポリマーまたは金属であり得る。
【0025】
特定の実施形態において、形成体は、少なくとも1つの第1付着部位を含み得る。第1付着部位および第2付着部位は、本発明の組成物の特に重要なエレメントである。本発明の種々の実施形態において、第1付着部位および/または第2付着部位は、以下であり得る:抗原およびそれに対する抗体または抗体フラグメント;ビオチンおよびアビジン;ストレプトアビジンおよびビオチン;レセプターおよびそのリガンド;リガンド結合タンパク質およびそのリガンド;相互作用するロイシンジッパーポリペプチド;アミノ基およびそれに反応する化学基;カルボキシル基およびそれに反応する化学基;スルフヒドリル基およびそれに反応する化学基;またはこれらの組み合わせ。
【0026】
より好ましい実施形態において、本発明は、選択されたほとんどの任意の抗原の、ウイルス、バクテリオファージ、ウイルス様粒子またはウイルスキャプシド粒子の表面への結合を提供する、抗原を準結晶(quasi−crystallne)「ウイルス様」構造にすることによって、本発明は、提示された抗原に対する高度に有効な免疫応答の産生(すなわち、ワクチン接種)のための宿主の強力な抗ウイルス免疫反応を開発する。
【0027】
1つの好ましい実施形態において、コア粒子は、以下からなる群より選択され得る:ロタウイルスの組換えタンパク質、ノーウォークウイルスの組換えタンパク質、アルファウイルスの組換えタンパク質、口蹄疫ウイルスの組換えタンパク質、レトロウイルスの組換えタンパク質、B型肝炎ウイルスの組換えタンパク質、タバコモザイクウイルスの組換えタンパク質、フロックハウス(Flock House)ウイルスの組換えタンパク質、およびヒトパピローマウイルスの組換えタンパク質。
【0028】
別の好ましい実施形態において、抗原は、以下からなる群より選択され得る:(1)癌細胞に対する免疫応答を誘導するに適したタンパク質;(2)感染性疾患に対する免疫応答を誘導するに適したタンパク質;(3)アレルゲンに対する免疫応答を誘導するに適したタンパク質;および(4)家畜における免疫応答を誘導するに適したタンパク質。
【0029】
本発明の特に好ましい実施形態において、第1付着部位および/または第2付着部位は、相互作用するロイシンジッパーポリペプチドを含む。最も好ましい実施形態において、第1付着部位および/または第2付着部位は、以下からなる群より選択される:(1)JUNロイシンジッパータンパク質ドメイン;および(2)FOSロイシンジッパータンパク質ドメイン。
【0030】
別の好ましい実施形態において、第1付着部位および/または第2付着部位は、以下からなる群より選択される:(1)遺伝子操作されたリジン残基、および(2)遺伝子操作されたシステイン残基。これらの2つの残基は、ともに化学的に連結され得る。
【0031】
本発明の他の実施形態は、本発明の組成物の産生のためのプロセス、およびこの組成物を使用する医療処置の方法を含む。
【0032】
前述の一般的な記載および以下の詳細な説明の両方は、模範的および説明的であるのみであること、そして特許請求される本発明のさらなる説明を提供することが意図されることが、理解されるべきである。
【0033】
(好ましい実施形態の詳細な説明)
(1.定義)
以下の定義は、本発明者らが本発明であること考慮する主題を明確にするために提供される。
【0034】
(アルファウイルス):本明細書中で使用される場合、用語「アルファウイルス」とは、Alphavirus属内に含まれる任意のRNAウイルスをいう。この属のメンバーの記載は、StraussおよびStrauss,Microbiol.Rev.,58:491−562(1994)に含まれる。アルファウイルスの例としては、以下があげられる:アウラウイルス、ベバルウイルス、キャバッソウウイルス(Cabassou virus)、チクングンヤウイルス、東部ウマ脳脊髄炎ウイルス、フォートモーガンウイルス(Fort morgan virus)、ゲタウイルスクイジラガッハウイルス(Kyzylagach virus)、マヤロウイルス、ミッデルブルグウイルス、ムカンボウイルス、ヌヅムウイルス、ピクスナウイルス、トネートウイルス(Tonate virus)、トリニティウイルス、ユナウイルス、西部ウマ脳脊髄炎ウイルス、ワタロワウイルス、シンドビスウイルス(SIN)、セムリキ森林ウイルス(SFV)、ベネズエラウマ脳脊髄炎ウイルス(VEE),およびロスリバーウイルス。
【0035】
(抗原):本明細書中で使用される場合、用語「抗原」とは、抗体によって結合され得る分子である。抗原は、さらに、体液性免疫応答および/または細胞性免疫応答を誘導して、Bリンパ球および/またはTリンパ球の産生をもたらし得る。抗原は、1以上のエピトープ(BエピトープおよびTエピトープ)を有し得る。上記に言及される特定の反応は、抗原が、高度に選択的な様式で、その対応する抗体とは反応するが、他の抗原によって惹起され得る多数の他の抗体とは反応しないことを示すことが意味される。
【0036】
(抗原決定基):本明細書中で使用される場合、用語「抗原決定基」とは、Bリンパ球またはTリンパ球のいずれかによって特異的に認識される抗原の部分をいうことが意味される。Bリンパ球は抗体産生を介して外来抗原決定基に応答するのに対し、Tリンパ球は、細胞性免疫のメディエータである。従って、抗原決定基またはエピトープは、抗体によって、またはMHCの文脈においてはT細胞レセプターによって認識される抗原の部分である。
【0037】
(結合):本明細書中で使用される場合、第1付着部位および第2付着部位に適用されるような用語「結合」とは、少なくとも1つの非ペプチド結合をいうために使用される。結合の性質は、共有、イオン性、疎水性、極性、またはそれらの任意の組み合わせであり得る。
【0038】
(付着部位、第1):本明細書中で使用される場合、句「第1付着部位」とは、非ランダム様式でコア粒子にそれ自体が結合された「形成体」のエレメントをいい、この部位に、抗原または抗原決定基に位置する第2付着部位が結合し得る。第1付着部位は、タンパク質、ポリペプチド、ペプチド、糖、ポリヌクレオチド、天然ポリマーもしくは合成ポリマー、二次代謝産物もしくは化合物(ビオチン、フルオレセイン、レチノール、ジゴキシゲニン、金属イオン、フェニルメチルスルホニルフルオリド)、またはそれらの組み合わせ、あるいはそれらの化学的に反応な基であり得る。複数の第1付着部位が、反復性の立体配置にて、非天然の分子足場の表面に存在する。
【0039】
(付着部位、第2):本明細書中で使用される場合、句「第2付着部位」とは、非天然分子足場の表面上に位置する「形成体」の第1付着部位が結合し得る抗原または抗原決定基と結合したエレメントをいう。抗原または抗原決定基の第2着部位は、タンパク質、ポリペプチド、ペプチド、糖、ポリヌクレオチド、天然ポリマーもしくは合成ポリマー、二次代謝産物もしくは化合物(ビオチン、フルオレセイン、レチノール、ジゴキシゲニン、金属イオン、フェニルメチルスルホニルフルオリド)、またはそれらの組み合わせ、あるいはそれらの化学的に反応な基であり得る。少なくとも1つの第2付着部位が、抗原または抗原決定基に存在する。
【0040】
(コア粒子):本明細書中で使用される場合、用語「コア粒子」とは、「形成体」の付着のための基礎を提供する固有の反復性較正を有する剛性構造をいう。本明細書中で使用されるようなコア粒子は、合成プロセスの産物であってもよいし、生物学的プロセスの産物であってもよい。
【0041】
(シス作用性):本明細書中で使用される場合、句「シス作用性」配列とは、レプリカーゼが結合してRNA分子のRNA依存性複製を触媒する、核酸配列をいう。これらの複製事象は、全長RNA分子および部分RNA分子の複製を生じ、従って、アルファウイルスサブゲノムプロモーターはまた、「シス作用性」配列である。シス作用性配列は、核酸分子の5’末端、3’末端またはその両末端に位置しての、またはその付近に位置しても、そして内部に位置してもよい。
【0042】
(融合):本明細書中で使用される場合、用語「融合」とは、それらのコードヌクレオチド配列のインフレームの組み合わせによる1つのポリペプチド鎖における異なる起源のアミノ酸配列の組み合わせをいう。用語「融合」は、その末端の1つへの融合に加えて、内部融合(すなわち、ポリペプチド鎖内での異なる起源の配列の挿入)を明示的に包含する。
【0043】
(異種配列):本明細書中で使用される場合、用語「異種配列」とは、本発明のベクターに存在する第2のヌクレオチド配列をいう。用語「異種配列」とはまた、本発明のベクターに含まれる異種DNA配列によってコードされる任意のアミノ酸配列またはRNA配列をいう。異種ヌクレオチド配列は、それらが存在する細胞型において通常発現されるタンパク質またはRNA分子あるいは、その細胞壁では通常発現されない分子(例えば、シンドビス構造タンパク質)をコードし得る。
【0044】
(単離された):本明細書中で使用される場合、用語「単離された(されている)」が分子を参照して使用される場合、この用語は、その分子がそのネイティブ環境から取り除かれたことを意味する。例えば、生きている動物に天然に存在するポリヌクレオチドまたはポリペプチドは「単離されて」いないが、その天然状態の共存物質から分離された同じポリヌクレオチドまたはポリペプチドは、「単離されている」。さらに、ベクターに含まれる組換えDNA分子は、本発明の目的のために単離されるとみなされる。単離されたRNA分子は、DNAおよびRNA分子のインビボまたはインビトロRNA複製産物を含む。単離された核酸分子はさらに、合成的に産生された分子を含む。さらに、組換え宿主細胞に含まれるベクター分子もまた、単離されている。従って、全ての「単離された」分子が「精製されている」必要があるとは限らない。
【0045】
(免疫治療剤):本明細書中で使用される場合、用語「免疫治療剤」とは、疾患または障害の処置のための組成物である。より詳細には、この用語は、アレルギーの処置の方法または癌の処置の方法をいうために使用される。
【0046】
(個体):本明細書中で使用される場合、用語「個体」とは、多細胞生物をいい、そして植物および動物の両方を含む。好ましい多細胞生物は動物であり、より好ましくは脊椎動物であり、さらにより好ましくは哺乳動物であり、そして最も好ましくはヒトである。
【0047】
(低いまたは検出不可能):本明細書中で使用される場合、句「低いまたは検出不可能」とは、遺伝子の発現レベルに関して使用される場合、遺伝子が最大に誘導されるときに見られるより有意に低い(例えば、少なくとも5分の1)か、または以下の実施例の節で使用される方法によって容易には検出可能ではないかのいずれかである、発現のレベルをいう。
【0048】
レクチン:本明細書中で使用される場合、マメ科植物の種から特に得られるが、多くの他の植物供給源および動物供給源からもまた得られる、タンパク質であり、特定の単糖またはオリゴ糖への結合部位を有する。例としては、コンカナバリンAおよびコムギ胚芽凝集素が挙げられ、これらは、糖タンパク質の研究において、分析薬剤および調製薬剤として広く使用される。
【0049】
天然起源:本明細書中で使用される場合、用語「天然起源」とは、その全体または一部が、合成的ではなく、そして天然に存在するかまたは天然にて生成されることを、意味する。
【0050】
非天然:本明細書中で使用される場合、この用語は、一般的には、天然由来でないことを意味し、より詳細には、この用語は、ヒトの手によることを意味する。
【0051】
非天然起源:本明細書中で使用される場合、用語「非天然起源」は、一般的には、合成的であることまたは天然由来でないことを意味し、より詳細には、この用語は、ヒトの手によることを意味する。
【0052】
非天然の分子足場:本明細書中で使用される場合、句「非天然の分子足場」とは、第1の付着部位の固定した反復性のアレイを提供するように役立ち得る、ヒトの手によって作製された任意の産物をいう。必ずではないが理想的には、これらの第1の付着部位は、幾何学的順序になっている。この非天然の分子足場は、有機物であっても非有機物であってもよく、そして部分的にかまたは全体として、化学的に合成されても生物学的プロセスを介して合成されてもよい。この非天然の分子足場は、以下から構成される:(a)コア粒子(天然起源または非天然起源のいずれかである);および(b)形成体(それ自体が、少なくとも1つの第1の付着部位を含み、そして少なくとも1つの共有結合によってコア粒子に結合している)。特定の実施形態において、この非天然の分子足場は、ウイルスでも、ウイルス様粒子でも、ウイルスキャプシド粒子でも、ファージでも、その組換え形態でも、または合成ペプチドであってもよい。
【0053】
順序付けられそして反復性の抗原または抗原決定基のアレイ:本明細書中で使用される場合、用語「順序付けられそして反復性の抗原または抗原決定基のアレイ」とは、一般的には、抗原または抗原決定基の反復性パターンをいい、足場に関して、その抗原または抗原決定基の均一な空間配置によって特徴付けられる。本発明の1つの実施形態において、この反復性パターンは、幾何学的パターンであり得る。理想の順序付けられそして反復性の抗原または抗原決定基のアレイは、5〜15ナノメーターの間隔で、抗原または抗原決定基の厳密に反復性の準結晶性の序列を保有する。
【0054】
形成体:本明細書中で使用される場合、用語「形成体」とは、順序付けられそして反復性の抗原のアレイを作製するための核生成部位を提供する、ランダムでない様式でコア粒子に結合したエレメントをいうために使用される。形成体は、少なくとも1つの共有結合によりコア粒子に結合した、少なくとも1つの第1の付着部位を含む、任意のエレメントである。形成体は、タンパク質、ポリペプチド、ペプチド、アミノ酸(すなわち、タンパク質の残基、ポリペプチドの残基、またはペプチドの残基)、糖、ポリヌクレオチド、天然ポリマーまたは合成ポリマー、二次代謝物または化合物(ビオチン、フルオレセイン、レチノール、ジゴキシゲニン、金属イオン、フェニルメチルスルホニルフルオリド)、あるいはそれらの組み合わせ、あるいはそれらの化学反応基であり得る。
【0055】
許容温度:本明細書中で使用される場合、句「許容温度」とは、酵素が、比較的高レベルの触媒活性を有する温度をいう。
【0056】
精製された:本明細書中で使用される場合、用語「精製(された)」が分子に関して使用される場合、この用語は、精製される分子の濃度が、その分子の天然の環境中にてその分子と結合している分子と比較して、増加したことを意味する。天然に結合した分子とは、タンパク質、核酸、脂質および糖を含むが、一般的には、精製される分子の完全性を維持するためまたは精製を容易にするために添加された、水、緩衝液、および試薬を含まない。例えば、mRNAが、オリゴdTカラムクロマトグラフィーの間に水性溶媒で希釈されるとしても、天然に結合した核酸および他の生物学的分子がカラムに結合せず、そして対象RNA分子から分離している場合は、mRNA分子は、このクロマトグラフィーにより精製される。
【0057】
レセプター:本明細書中で使用される場合、用語「レセプター」とは、別の分子(リガンドと呼ばれる)と相互作用し得る、タンパク質または糖タンパク質、あるいはそれらのフラグメントをいう。このリガンドは、どの種類の生化学的化合物または化学的化合物にも属し得る。このレセプターは、必ずしも、膜結合タンパク質であることを必要とはしない。可溶性タンパク質(例えば、マルトース結合タンパク質またはレチノール結合タンパク質のようなもの)は、同様にレセプターである。
【0058】
残基:本明細書中で使用される場合、用語「残基」とは、ポリペプチド骨格または側鎖における、特定のアミノ酸を意味することが、意味される。
【0059】
温度感受性:本明細書中で使用される場合、句「温度感受性」とは、1つの温度で反応を容易に触媒するが、別の温度では、同じ反応を遅く触媒するか、または全く触媒しない、酵素をいう。温度感受性酵素の例は、pCYTtsベクターによりコードされるレプリカ−ゼタンパク質であり、これは、34℃より下の温度にて、容易に検出可能なレプリカ−ゼ活性を有し、そして37℃にて、低い活性または検出不能な活性を有する。
【0060】
転写:本明細書中で使用される場合、用語「転写」とは、RNAポリメラーゼにより触媒される、DNAテンプレートからのRNA分子の生成をいう。
【0061】
組換え宿主細胞:本明細書中で使用される場合、用語「組換え宿主細胞」とは、本発明の1つ以上の核酸分子が導入された、宿主細胞をいう。
【0062】
組換えウイルス:本明細書中で使用される場合、句「組換えウイルス」とは、ヒトの手によって遺伝子改変されたウイルスをいう。この句は、当該分野で公知の任意のウイルスを包含する。より詳細には、この句は、ヒトの手によって遺伝子改変されたアルファウイルスをいい、そして最も詳細には、この句は、ヒトの手によって遺伝子改変されたシンドビス(Sinbis)ウイルスをいう。
【0063】
制限温度:本明細書中で使用される場合、句「制限温度」とは、酵素が、低いレベルまたは検出不能なレベルの触媒活性を有する温度をいう。「熱さ」感受性変異体および「冷たさ」感受性変異体の両方が公知であり、従って、制限温度は、許容温度より高くても低くてもよい。
【0064】
RNA依存性RNA複製事象:本明細書中で使用される場合、句「RNA依存性RNA複製事象」とは、RNA分子をテンプレートとして使用して、RNA分子の形成を生じる、プロセスをいう。
【0065】
RNA依存性RNAポリメラーゼ:本明細書中で使用される場合、句「RNA依存性RNAポリメラーゼ」とは、別のRNA分子からのRNA分子の生成を触媒する、ポリメラーゼをいう。この用語は、本明細書中において、用語「レプリカ−ゼ」と同義に使用される。
【0066】
非翻訳RNA:本明細書中で使用される場合、句「非翻訳RNA」とは、オープンリーディングフレームをコードしないか、またはオープンリーディングフレームまたはその一部をコードするが、アミノ酸配列が生成しない様式(例えば、開始コドンが存在しない)でコードする、RNA配列またはRNA分子をいう。このような分子の例は、tRNA分子、rRNA分子、およびリボソームである。
【0067】
ベクター:本明細書中で使用される場合、用語「ベクター」とは、宿主細胞に遺伝子物質を伝達するために使用される因子(例えば、プラスミドまたはウイルス)をいう。ベクターは、DNAまたはRNAのいずれかから構成され得る。
【0068】
1つの、a、またはan:用語「1つの」、「a」、または「an」が、本開示にて使用される場合、これらは、他のように示さない限り、「少なくとも1つ」または「1つ以上」を意味する。
【0069】
(2.順序付けられそして反復性の抗原または抗原性決定基のアレイの組成物、およびこれを作製するための方法)
開示された発明は、順序付けられそして反復性の抗原または抗原決定基を含む、組成物を提供する。さらに、本発明は、実施者が、種々の処置目的のために、順序付けられそして反復性の抗原または抗原決定基のアレイを構築することを簡便に可能にする。この目的としては、感染性疾患の予防、アレルギーの処置および癌の処置が挙げられる。
【0070】
本発明の組成物は、本質的に以下2つの要素を含む:(1)非天然の分子足場;および(2)少なくとも1つの第2の付着部位を有する抗原または抗原決定基であって、この第2の付着部位は、少なくとも1つの非ペプチド結合を介して、第1の付着部位に結合し得る。
【0071】
この非天然の分子足場は、(a)コア粒子((1)非天然起源のコア粒子および(2)天然起源のコア粒子からなる群より選択される);ならびに(b)少なくとも1つの第1の付着部位を含む、形成体(この形成体は、少なくとも1つの共有結合によってこのコア粒子に結合している)を含む。
【0072】
この抗原または抗原決定基は、(a)この抗原または抗原決定基に天然に存在しない付着部位;および(b)この抗原または抗原決定基に天然に存在する付着部位、からなる群より選択される、少なくとも1つの第2の付着部位を有する。
【0073】
本発明は、少なくとも1つの非ペプチド結合によって第1の付着部位に第2の付着部位が結合することを介して、順序付けられそして反復性の抗原のアレイを提供する。従って、この抗原または抗原決定基と非天然の分子足場とは、第1の付着部位と第2の付着部位とのこの結合を介して結合されて、順序付けられそして反復性の抗原のアレイを形成する。
【0074】
実施者は、この抗原または抗原決定基および第2の付着部位を、非天然の分子足場に結合した抗原または抗原決定基すべての配列が均一であるように、特に設計し得る。例えば、カルボキシル末端またはアミノ末端に抗原または抗原決定基上の単一の第2の付着部位を配置し得、それにより、設計を介してその非天然の分子足場に結合した抗原または抗原決定基の分子すべてが均一な様式で位置することを確実にし得る。従って、本発明は、任意の抗原または抗原決定基を、規定された順序および反復性にて、非天然の分子足場上に配置する、簡便な手段を提供する。
【0075】
当業者に明らかであるように、本発明の特定の実施形態は、組換え核酸技術(例えば、クローニング、ポリメラーゼ連鎖反応、DNAおよびRNAの精製、原核生物細胞および真核生物細胞中での組換えタンパク質の発現など)の使用を含む。このような方法論は、当業者に周知であり、そして刊行された実験室方法マニュアル(例えば、Sambrook,J.ら編、MOLECULAR CLONING、A LABORATORY MANUAL、第2版、Cold Spring Harbor Laboratory Press、Cold Spring Harbor、N.Y.(1989):Ausubel,F.ら編、CURRENT PROTOCOLS IN MOLECULAR BIOLOGY、John,H.Wiley & Sons,Inc.(1997))において、簡単に見出され得る。組織培養細胞株(Celis,J.編、CELL BIOLOGY、Academic Press、第2版(1998))および抗体に基づく技術(Harlow,E.およびLane,D.「Antibodies:A Laboratory Manual」Cold Spring Harbor Laboratory、Cold Spring Harbor、N.Y.(1988);Deutscher,M.P.「Guide to Protein Purification」Meth.Enzymol.128、Academic Press San Diego(1990);Scopes,R.K.「Protein Purification Principles and Practice」第3版、Springer−Verlag、New York(1994))を用いて作業するための基本的実験室技術もまた、この文献に十分に記載されており、これらすべてが、本明細書中で参考として援用される。
【0076】
(A.非天然の分子足場の構築)
本発明の組成物中の1つの要素は、コア粒子および形成体を含む、非天然の分子足場である。本明細書中で使用される場合、句「非天然の分子足場」とは、第1の付着部位の堅固かつ反復性のアレイを提供するように役立ち得る、ヒトの手によって作製された任意の産物をいう。より詳細には、この非天然の分子足場は、(a)コア粒子((1)非天然起源のコア粒子および(2)天然起源のコア粒子からなる群より選択される);ならびに(b)少なくとも1つの第1の付着部位を含む、形成体(この形成体は、少なくとも1つの共有結合によって、このコア粒子と結合している)を含む。
【0077】
当業者に容易に明らかであるように、本発明の非天然の分子足場のコア粒子は、いかなる特定の形態にも限定されない。このコア粒子は、有機物であっても非有機物であってもよく、そして化学合成されてもよいし、生物プロセスを介して合成されてもよい。
【0078】
1つの実施形態において、非天然のコア粒子は、合成ポリマーであっても、脂質ミセルであっても、金属であってもよい。このようなコア粒子は、当該分野で公知であり、本発明の新規な、非天然の分子足場を構築するための基礎を提供する。例として、合成ポリマーまたは金属コア粒子は、米国特許第5,770,380号に記載される。この特許は、「抗体模倣物」の作製において複数のペプチドループが結合される、カリクスアーレン有機足場の使用を開示する。そして米国特許第5,334,394号は、ウイルスデコイとして使用される、ナノ結晶粒子を記載し、この粒子は、金属またはセラミックを含む、広範な種類の無機材料から構成される。この実施形態において好ましい金属としては、クロム、ルビジウム、鉄、亜鉛、セレン、ニッケル、金、銀、プラチナが挙げられる。この実施形態において好ましいセラミック材料としては、二酸化ケイ素、二酸化チタン、酸化アルミニウム、酸化ルテニウムおよび酸化スズが挙げられる。この実施形態のコア粒子は、炭素(ダイヤモンド)を含む有機材料から作製され得る。好ましいポリマーとしては、ポリスチレン、ナイロンおよびニトロセルロースが、挙げられる。この型のナノ結晶粒子について、酸化スズ、酸化チタンまたは炭素(ダイヤモンド)から作製された粒子が、特に好ましい。脂質ミセルは、当該分野で公知の任意の手段によって、調製され得る。例えば、ミセルは、以下の手順に従って調製され得る:BaiselleおよびMillar(Baiselle,C.J.およびMillar,D.B.、Biophys.Chem.4:355〜361(1975))またはCortiら(Corti,M.、Degriorgio,V.、Sonnino,S.、Ghidoni R.、Masserini,M.およびTettamanti,G.、Chem.Phys.Lipids 38:197〜214(1981))またはLopezら(Lopez,O.、de la Maza,A.、Coderch,L.、Lopez−Iglesias,C.、Wehrli,E.およびParra,J.L.、FEBS Lett.426:314〜318(1998))またはTopchievaおよびKarezin(Topchieva,I.およびKaraezin,K.、J.Colloid Interface Sci.213:29〜35(1999))またはMoreinら(Morein,B.、Sundquist,B.、Hoglund,S.、Dalsgaard K.およびOsterbaus,A.、Nature 308:457〜60(1984));これらはすべて、本明細書中で参考として援用される)。
【0079】
コア粒子はまた、生物学的プロセス(これは、天然であってもよいし、または非天然であってもよい)を通じて生成され得る。例の目的で、この型の実施形態としてはコア粒子(ウイルス、ウイルス様粒子、ファージ、ウイルスカプシド粒子、またはそれらの組換え形態を含む)が挙げられ得る。より詳細な実施形態において、このコア粒子としては、ロタウイルスの組換えタンパク質、ノーウォークウイルスの組換えタンパク質、アルファウイルスの組換えタンパク質、口蹄疫ウイルスの組換えタンパク質、レトロウイルスの組換えタンパク質、B型肝炎ウイルスの組換えタンパク質、タバコモザイクウイルスの組換えタンパク質、フロックハウスウイルス(Flock House Virus)の組換えタンパク質およびヒトパピローマウイルスの組換えタンパク質が挙げられ得る。
【0080】
天然であろうと非天然であろうと、本発明のコア粒子は、少なくとも1つの共有結合により天然または非天然のコア粒子に付着される形成体を含むことによって、特徴付けられる。この形成体は、順序付けられそして反復性の抗原アレイを作製するための核形成部位を提供する、ランダムでない様式でコア粒子に結合したエレメントである。理想的には(必要ではないが)、この形成体は、幾何学的順序でコア粒子と会合される。最小には、この形成体は、第1の付着部位を含む。
【0081】
先に述べたように、この形成体は、少なくとも1つの共有結合によってコア粒子に結合した、少なくとも1つの第1の付着部位を含む、任意のエレメントであり得る。形成体は、タンパク質、ポリペプチド、ペプチド、アミノ酸(すなわち、タンパク質の残基、ポリペプチドの残基またはペプチドの残基)、糖、ポリヌクレオチド、天然ポリマーまたは合成ポリマー、二次代謝物または化合物(ビオチン、フルオレセイン、レチノール、ジゴキシゲニン、金属イオン、フェニルメチルスルホニルフルオリド)、あるいはそれらの組合せ、あるいはそれらの化学反応基であり得る。より詳細な実施形態において、この形成体としては、抗原、抗体または抗体フラグメント、ビオチン、アビジン、ストレプトアビジン、レセプター、レセプターリガンド、リガンド、リガンド結合タンパク質、相互作用するロイシンジッパーポリペプチド、アミノ基、アミノ基に反応する化学基:カルボキシル基、カルボキシル基に反応する化学基、スルフヒドリル基、スルフヒドリル基に反応する化学基、あるいはそれらの組合せが挙げられ得る。
【0082】
好ましい実施形態において、非天然の分子足場のコア粒子としては、ウイルス、バクテリオファージ、ウイルス様粒子、ウイルスカプシド粒子、またはそれらの組換え形態が挙げられる。順序付けられそして反復性の被覆および/またはコアタンパク質構造を有する、当該分野で公知の任意のウイルスは、本発明の非天然の分子足場として選択され得る;適切なウイルスの例としては、以下が挙げられる:シンドビスウイルスおよび他のアルファウイルス;水疱性口内炎(vesicular somatitis)ウイルス;ラブドウイルス(例えば、水疱性口内炎ウイルス)、ピコルナウイルス、トガウイルス、オルトミクソウイルス、ポリオーマウイルス、パルボウイルス、ロタウイルス、ノーウォークウイルス、口蹄疫ウイルス、レトロウイルス、B型肝炎ウイルス、タバコモザイクウイルス、フロックハウスウイルス、ヒトパピローマウイルス(例えば、Bachman,M.F.およびZinkernagel,R.M.,Immunol.Today 17:553−558(1996)の表1を参照のこと)。
【0083】
1つの実施形態において、本発明は、ウイルスの遺伝的操作を利用して、順序付けられそして反復性のウイルスエンベロープタンパク質と形成体(異種タンパク質、ペプチド、抗原決定基または選り抜きの反応性アミノ酸残基を含む)との間の融合物を作製する。当業者に公知の他の遺伝的操作は、非天然の分子足場の構築に含まれ得る;例えば、遺伝的変異によって組換えウイルスの複製能力を制限することが望ましくあり得る。形成体(すなわち、第1の付着部位)タンパク質への融合のために選択されるウイルスタンパク質は、形成されそして反復性の構造、より好ましくは、このウイルスの表面上に5〜15nmの間隔を最適に有する、準結晶組織化を有するべきである。この型の融合タンパク質の作製は、このウイルスの表面上に、複数の、順序付けられそして反復性の形成体を生じる。従って、それにより生じる第1の付着部位の、順序付けられそして反復性の組織化は、ネイティブウイルスタンパク質の正常な組織化を反映する。
【0084】
本明細書中でより詳細に議論されるように、本発明の好ましい実施形態において、この足場は、組換えアルファウイルス、そしてより詳細には、組換えシンドビスウイルスである。アルファウイルスは、感染した細胞の細胞質において、およびDNA中間体を有さずに、それらのゲノムRNA全体を複製する、+鎖RNAである(Strauss,J.およびStrauss,E.,Microbiol.Rev.58:491〜562(1994))。アルファウイルスファミリーのいくつかのメンバー(シンドビス(Xiong,C.ら、Science 243:1188−1191(1989);Schlesinger,S.,Trends Biotechnol.11:18−22(1993))、セムリキ森林ウイルス(SFV)(Liljestrom,P.およびGaroff.H.,Bio/Technology 9:1356−1361(1991))およびその他(Davis,N.L.ら、Virology 171:189−204(1989)))は、種々の異なるタンパク質についてのウイルスに基づく発現ベクターとして(Lundstrom,K.,Curr.Opin.Biotechnol.8:578−582(1997);Liljestrom,P.,Curr.Opin.Biotechnol.5:495−500(1994))、およびワクチン開発の候補物としての使用に、かなりの注目を受けた。最近、異種タンパク質の発現およびワクチンの開発のためのアルファウイルスの使用に関して、多くの特許が発行されている(米国特許第5,766,602号;同第5,792,462号;同第5,739,026号;同第5;789,245号および同第5,814,482号を参照のこと)。本発明のアルファウイルス足場の構築は、前述の文献(これらは、本明細書中で参考として援用される)に記載されるような、組換えDNA技術の当該分野で一般に公知の手段によって行われ得る。
【0085】
種々の異なる組換え宿主細胞を利用して、抗原または抗原決定付着についてのウイルスに基づくコア粒子を生成し得る。例えば、アルファウイルスは、広範な宿主範囲を有することが公知である;シンドビスウイルスは、培養された哺乳動物細胞、爬虫類細胞および両生類細胞、ならびにいくつかの昆虫細胞に感染する(Clark,H.,J.Natl.Cancer Inst.51:645(1973);Leake,C.,J.Gen.Virol.35:335(1977);Stollar,V.in THE TOGAVIRUSES,R.W.Schlesinger編,Academic Press,(1980),583−621頁)。従って、多くの組換え宿主細胞は、本発明の実施において使用され得る。BHK細胞、COS細胞、Vero細胞、HeLa細胞およびCHO細胞は、異種タンパク質の生成のために特に適切である。なぜなら、これらは、ヒト細胞に類似の様式でグリコシル化異種タンパク質における可能性を有し(Watson,E.ら、Glycobiology 4:227,(1994))、そして選択され(Zang,M.ら、Bio/Technology 13:389(1995))、または遺伝的に操作されて(Renner W.ら、Biotech.Bioeng.4.476(1995);Lee K.ら、Biotech.Bioeng.50:336(1996))、無血清培地中ならびに懸濁液中で増殖され得るからである。
【0086】
ポリヌクレオチドベクターの宿主細胞中への導入は、標準的な研究室マニュアルに記載される方法によって果たされ得る(例えば、Sambrook,J.ら編、MOLECULAR CLONING,A LABORATORY MANUAL、第2版、Cold Spring Harbor Laboratory Press,Cold Spring Harbor,N.Y.(1989),第9章;Ausubel,F.ら編、CURRENT PROTOCOLS IN MOLECULAR BIOLOGY,John H.Wiley & Sons,Inc.(1997),第16章を参照のこと)。この方法としては、エレクトロポレーション、DEAEデキストラン媒介トランスフェクション、トランスフェクション、マイクロインジェクション、カチオン性脂質媒介トランスフェクション、形質導入、スクレープローディング(scrape loading)、弾道導入(ballistic introduction)、および感染のような方法が挙げられる。外因性DNA配列の宿主細胞への導入のための方法は、Felgner,P.ら、米国特許第5,580,859号において議論される。
【0087】
パッケージされたRNA配列はまた、宿主細胞を感染するために使用され得る。これらのパッケージされたRNA配列は、それらを培養培地に添加することによって宿主細胞に導入され得る。例えば、非感染アルファウイルス粒子の調製は、多くの出典(「Sindbis Expression System」、Version C,(Invitrogen Catalog No.K750−1)を含む)において記載される。
【0088】
哺乳動物細胞が、ウイルスに基づくコア粒子の生成のための組換え宿主細胞として使用される場合、これらの細胞は、一般に、組織培養中で増殖される。培養中で細胞を増殖するための方法は、当該分野で周知である(例えば、Celis,J.編、CELL BIOLOGY,Academic Press、第2版(1998);Sambrook,J.ら編、MOLECULAR CLONING,A LABORATORY MANUAL、第2版、Cold Spring Harbor Laboratory Press,Cold Spring Harbor,N.Y.(1989);Ausubel,F.ら編、CURRENT PROTOCOLS IN MOLECULAR BIOLOGY,John H.Wiley & Sons,Inc.(1997);Freshney,R.,CULTURE OF ANIMAL CELLS,Alan R.Liss,Inc.(1983)を参照のこと)。
【0089】
当業者によって理解されるように、第1の付着部位は、任意の適切なタンパク質、ポリペプチド、糖、ポリヌクレオチド、ペプチド(アミノ酸)、天然ポリマーもしくは合成ポリマー、二次代謝物、またはそれらの組合せであり得るか、あるいはそれらの一部であり得る。これらは、足場に対する選り抜きの抗原または抗原決定基を特異的に付着するように作用し得る。1つの実施形態において、この付着部位は、当該分野で公知のタンパク質またはペプチドから選択され得るタンパク質またはペプチドである。例えば、第1の付着部位は、以下の群から選択され得る:リガンド、レセプター、レクチン、アビジン、ストレプトアビジン、ビオチン、エピトープ(例えば、HAまたはT7タグ)、Myc、Max、免疫グロブリンドメイン、および第1の付着部位として有用な、当該分野で公知の任意の他のアミノ酸配列。
【0090】
本発明の別の実施形態において、この第1の付着部位が、カプシドタンパク質に対してインフレーム融合を構築することにおいて利用される形成体(すなわち、タンパク質またはポリペプチド)に対して二次的に作製され得ることは、当業者によってさらに理解されるべきである。例えば、タンパク質は、エンベロープタンパク質と、特定の様式においてグリコシル化されていることが公知のアミノ酸配列との融合のために利用され得、そして次いで、結果として付加された糖部分は、ウイルス足場の第1の付着部位で作用し得、これは、抗原の二次付着部位として作用するレクチンへの結合による。あるいは、この形成体配列は、インビボでビオチン化され得、そしてこのビオチン部分は、本発明の第1の付着部位として作用し得るか、またはこの形成体配列は、インビトロにおいて、別個のアミノ酸残基の化学的改変に供され得、この改変は、この第1の付着部位として作用する。
【0091】
本発明の1つの特定の実施形態は、シンドビスウイルスを利用する。シンドビスウイルスRNAゲノムは、E1、E2およびE3と呼ばれる3つのタンパク質を含む脂質二重層によって囲まれる、カプシドタンパク質中にパッケージされる。これらのいわゆるエンベロープタンパク質は、糖タンパク質であり、そしてこのグリコシル化タンパク質は、この脂質二重層の外側上に位置される。ここで、これらのタンパク質の複合体は、このウイルスの表面から外側へ突出することが電子顕微鏡写真において観察され得る「スパイク(spike)」を形成する。本発明の好ましい実施形態において、この第1の付着部位は、E2エンベロープタンパク質に対してインフレームで融合されるJUNロイシンジッパータンパク質ドメインまたはFOSロイシンジッパータンパク質ドメインであるように選択される。しかし、他のエンベロープタンパク質が、本発明の足場において第1の付着部位を位置付けるためにこの融合タンパク質構築物において利用され得ることは、当業者に明らかである。
【0092】
本発明の最も好ましい実施形態において、この第1の付着部位はB型肝炎カプシド(コア)タンパク質に対してインフレームで融合されるJUN−FOSロイシンジッパードメインであるように選択される。しかし、他のウイルスカプシドタンパク質が、本発明の足場において第1の付着部位を位置付けるためにこの融合タンパク質構築物において利用され得ることは、当業者に明らかである。
【0093】
本発明の別の好ましい実施形態において、この第1の付着部位は、肝炎コア(カプシド)タンパク質に対してインフレームで融合されるリジン残基またはシステイン残基であるように選択される。しかし、他のウイルスカプシド粒子またはウイルス様粒子が、本発明の足場において第1の付着部位を位置付けるためにこの融合タンパク質構築物において利用され得ることは、当該者に明らかである。
【0094】
実施例1は、Hahnら(Proc.Natl.Acad.Sci.USA 89:2679−2683(1992))のpTE5’2Jベクターを使用して、シンドビスウイルスE2エンベロープタンパク質とJUNロイシンジッパータンパク質ドメインとの間のインフレーム融合タンパク質の構築を示すために提供される。この第1の付着部位のために利用されるJUNアミノ酸配列は、以下の通りである:CGGRIARLEEKVKTLKAQNSELASTANMLREQVAQLKQKVMNHVGC(配列番号59)。この例において、抗原上の予測される第2の付着部位は、FOSロイシンジッパータンパク質ドメインであり、そしてこのアミノ酸配列は、以下の通りである:CGGLTDTLQAETDQVEDEKSALQTEIANLLKEKEKLEFILAAHGGC(配列番号60)。
【0095】
これらの配列は、各々、両側にシステイン残基を含む短い配列に隣接する、転写因子JUNおよびFOSに由来する。これらの配列は、互いに相互作用することが公知である。JUN−FOS二量体について提唱された元々のハイポセティカル構造は、1つの単量体の疎水性側鎖が、ジッパー様の様式で他の単量体のそれぞれの側鎖と嵌合することを仮定した(Landschulzら、Science 240:1759−1764 (1988))。しかし、この仮定は誤っていることが判明した。そしてこれらのタンパク質は、αヘリカルコイルドコイルを形成することが公知である(O’Sheaら、Science 243:538−542(1989);O’ Sheaら、Cell 68:699−708(1992);Cohen & Parry,Trends Biochem.Sci.11:245−248(1986))。従って、用語「ロイシンジッパー」は、頻繁に、構造的理由よりもより歴史的にこれらのタンパク質ドメインをいうために使用される。この特許にわたって、用語「ロイシンジッパー」は、上記の配列、または上記の配列に本質的に類似する配列をいうために使用される。用語JUNおよびFOSは、JUNタンパク質全体およびFOSタンパク質全体というよりもむしろ、それぞれのロイシンジッパードメインのために使用される。
【0096】
1つの実施形態において、本発明は、pCYTts発現系を使用する、シンドビスウイルスE2−JUN足場の生成を提供する(米国特許出願番号60/079,562;1998年3月27日出願)。このpCYTts発現系は、新規な発現ベクターを提供し、このベクターは、真核生物細胞において遺伝子発現の厳重な調節を可能にする。この系のDNAベクターは、転写されてRNA分子を形成し、次いで、温度感受性レプリカーゼによって複製されて、さらなるRNA分子を形成する。複製によって生成されるRNA分子は、目的のタンパク質を生成するために翻訳され得るヌクレオチド配列、または1つ以上の非翻訳RNA分子をコードするヌクレオチド配列を含む。従って、この発現系は、組換えシンドビスウイルス粒子の生成を可能にする。
【0097】
実施例2は、本発明の非天然のE2−JUNシンドビス分子足場の生成に対する詳細を提供する。さらに、実施例3は、実施例1において生成されたpTE5’2JE2:JUNベクターを使用して、組換えE2−JUNシンドビスウイルス足場の生成のための別の方法を提供する。従って、本発明は、2つの手段(pCYTts発現系(実施例2)およびpTE5’2Jベクター系(実施例3)を提供し、これによって、組換えシンドビスウイルスE2−JUNの非天然分子足場が生成され得る。各々の系において生成されたウイルス粒子の分析は、図1および図2において検証される。
【0098】
先に述べたように、本発明は、ウイルスに基づくコア粒子(これは、ウイルス、ウイルス様粒子、ファージ、ウイルスカプシド粒子またはそれらの組換え形態を含む)を含む。当業者は、このようなコア粒子を生成し、そしてそのコア粒子に形成体を付着させるための知識を有する。他の例を提供する目的で、本発明は、本明細書中で、コア粒子として、B型肝炎ウイルス様粒子および麻疹ウイルスカプシド粒子の生成を提供する(実施例17〜22)。このような実施形態において、JUNロイシンジッパータンパク質ドメインまたはFOSロイシンジッパータンパク質ドメインは、本発明の非天然の分子足場のための形成体として、そしてそれゆえに、第1の付着部位として、使用され得る。
【0099】
実施例23〜29は、それぞれ、第1の付着部位および第2の付着部位として、反応性リジン残基を有するインフレームで融合されたペプチドを保有するB型肝炎コア粒子、および遺伝的に融合されたシステイン残基を保有する抗原、の生成の詳細を提供する。
【0100】
(B.第2の付着部位を有する抗原または抗原決定基の構築)
本発明の組成物内の第2のエレメントは、非天然分子骨格の第1の付着部位と、少なくとも一つの非ペプチド結合を介して会合し得る少なくとも一つの第2の付着部位を保有する抗原または抗原決定基である。本発明は、所望の治療効果の考慮において選択された抗原または抗原決定基に従って変わる組成物を提供する。他の組成物が、第2の付着部位について選択される分子を変えることによって提供される。
【0101】
本発明の抗原は、以下からなる群から選択され得る:(a)癌細胞に対する免疫応答を誘導するのに適したタンパク質;(b)感染疾患に対する免疫応答を誘導するのに適したタンパク質;(c)アレルゲンに対する免疫応答を誘導するのに適したタンパク質;(d)家畜における免疫応答を誘導するのに適したタンパク質。
【0102】
本発明の一つの特定の実施形態において、抗原または抗原決定基は、感染疾患の予防に対して有用な実施形態である。そのような処置は、広い範囲の宿主(例えば、ヒト、ウシ、ヒツジ、ブタ、イヌ、ネコ、他の哺乳動物種および非哺乳動物種も同様)に影響を及ぼす広範に種々の感染疾患を処置するために有用である。処置可能な感染疾患は、当業者に周知であり、例としては、例えば、HIV、インフルエンザ、ヘルペス、ウイルス性肝炎、エプスタイン−バー、ポリオ、ウイルス脳炎、麻疹、水痘などのようなウイルス病因の感染;または例えば肺炎、結核、梅毒などの細菌病因感染;または例えばマラリア、トリパノソーマ症、リーシュマニア症、トリコモナス症、アメーバ症(amoebiasis)などの寄生的病因の感染が挙げられる。従って、本発明の組成物のために選択される抗原または抗原決定基は、医療分野における当業者に対して周知である;抗原または抗原決定基の例としては、以下が挙げられる:HIV抗原gp140およびgp160;インフルエンザ抗原赤血球凝集素およびノイラミニダーゼ、肺炎B表面抗原、マラリアのサーカムスポロゾイト(circumsporozoite)タンパク質。
【0103】
別の特定の実施形態において、本発明の組成物は、アレルギーまたは癌の処置のために使用され得る免疫治療法である。
【0104】
組成物のための抗原または抗原決定基の選択およびアレルギーに対する処置の方法は、そのような疾患を処置する医療分野の当業者に対して公知である;この型の抗原または抗原決定基の代表的な例としては、以下が挙げられる;ハチ毒ホスホリパーゼA2、Bet v I(カバノキ花粉アレルゲン)、5 Dol m V(ホワイトフェーススズメバチ(white−faced hornet)毒アレルゲン)、Der p I(イエダニアレルゲン)。
【0105】
組成物に対する抗原または抗原決定基の選択および癌に対する処置の方法は、そのような疾患を処置する医療分野における当業者に公知である;この型の抗原または抗原決定基の代表的な例としては、以下が挙げられる:Her2(乳癌)、GD2(神経芽細胞腫)、EGF−R(悪性グリア芽細胞腫)、CEA(髄質甲状腺(medullary thyroid)癌)、CD52(白血病)。
【0106】
本発明の特定の実施形態において、抗原または抗原決定基は、以下からなる群より選択される:(a)HIVの組換えタンパク質、(b)インフルエンザウイルスの組換えタンパク質(c)C型肝炎ウイルスの組換えタンパク質、(d)トキソプラスマの組換えタンパク質、(e)Plasmodium falciparumの組換えタンパク質、(f)Plasmodium vivaxの組換えタンパク質、(g)Plasmodium ovaleの組換えタンパク質、(h)Plasmodium malariaeの組換えタンパク質、(i)乳癌細胞の組換えタンパク質、(j)腎臓癌細胞の組換えタンパク質、(k)前立腺癌細胞の組換えタンパク質、(l)皮膚癌細胞の組換えタンパク質、(m)脳癌細胞の組換えタンパク質、(n)白血病細胞の組換えタンパク質、(o)組換えプローブファイリング(profiling)、(p)ハチ刺しアレルギーの組換えタンパク質、(q)堅果アレルギーの組換えタンパク質、(r)食物アレルギーの組換えタンパク質、喘息の組換えタンパク質およびクラミジアの組換えタンパク質。
【0107】
一旦、組成物の抗原または抗原決定基が選択されると、少なくとも一つの第2の付着部位が、本発明の非天然分子足場と会合した、組織化され、かつ反復性のアレイを構築するように調製する際に、その分子に添加され得る。適切な第2の付着部位を構成するものの知識は、当業者に公知である。第2の付着部位の代表的例としては、以下が挙げられるが、これらに限定されない:抗原、抗体または抗体フラグメント、ビオチン、アビジン、ストレプトアビジン(strepavidin)、レセプター、レセプターリガンド、リガンド、リガンド結合タンパク質、相互作用ロイシンジッパーポリペプチド、アミノ基、アミノ基に対して反応性の化学基;カルボキシル基、カルボキシル基に対して反応性の化学基、スルフヒドリル基、スルフヒドリル基に対して反応性の化学基、またはそれらの組み合わせ。
【0108】
第1の付着部位と第2の付着部位との間の結合は、選択されたそれぞれの分子の特徴によって決定されるが、少なくとも一つの非ペプチド結合を含む。第1の付着部位および第2の付着部位の組み合わせに依存して、天然の会合は、共有結合性、イオン性、疎水性、極性、またはその組み合わせであり得る。
【0109】
本発明の一つの実施形態において、第2の付着部位は、FOSロイシンジッパータンパク質ドメインまたはJUNロイシンジッパータンパク質ドメインであり得る。
【0110】
本発明の最も具体的な実施形態では、選択された第2の付着部位FOSロイシンジッパータンパク質ドメインであり、これは、本発明の非天然分子足場のJUNロイシンジッパータンパク質と特異的に会合する。JUNおよびFOSロイシンジッパータンパク質ドメインの会合は、足場の表面上に、組織化され、かつ反復性の抗原または抗原決定基アレイを形成するための基礎を提供する。このFOSロイシンジッパータンパク質ドメインは、アミノ末端、カルボキシル末端のいずれかにおける抗原または抗原決定基の選択に対してインフレームで融合され得るか、または所望される場合、そのタンパク質の内部に位置する。
【0111】
いくつかのFOS融合構築物が、例示的目的のために提供される。ヒト成長ホルモン(実施例4)、ハチ毒ホスホリパーゼA2(PLA)(実施例9)、オボアルブミン(実施例10)およびHIVgp140(実施例12)。
【0112】
FOS融合構築物の生成を簡略化するために、抗原または抗原決定基の設計および構築について選択を提供する、いくつかのベクターが開示される(実施例6を参照のこと)。ベクターpAV1−4を、E.coliにおけるFOS融合発現のために設計した;ベクターpAV5およびpAV6を、真核生物細胞におけるFOS融合タンパク質の発現のために設計した。これらのベクターの特性が、以下に手短に記載される;
1.pAV1:このベクターを、C末端にFOSを有する融合タンパク質のE.coliの細胞膜周辺腔への分泌のために設計した。目的の遺伝子(g.o.i)は、このベクターのStuI/NotI部位に連結され得る。
【0113】
2.pAV2:このベクターを、N末端にFOSを有する融合タンパク質のE.coliの細胞膜周辺腔への分泌のために設計した。目的の遺伝子(g.o.i)は、このベクターのNotI/EcoRV(またはNotI/HindIII)部位に連結され得る。
【0114】
3.pAV3:このベクターを、C末端にFOSを有する融合タンパク質のE.coliにおける細胞質産生のために設計した。目的の遺伝子(g.o.i)は、このベクターのEcoRV/NotI部位に連結され得る。
【0115】
4.pAV3:このベクターを、N末端にFOSを有する融合タンパク質をE.coliにおける細胞質産生のために設計した。目的の遺伝子(g.o.i)は、このベクターのNotI/EcoRV(またはNotI/HindIII)部位に連結され得る。そのN末端メチオニン残基は、タンパク質合成の際に酵素分解的に除去される(Hirelら、Proc.Natl.Acad.Sci.USA 86:8247−8251(1989))。
【0116】
5.pAV5:このベクターを、C末端にFOSを有する融合タンパク質の真核生物産生のために設計した。目的の遺伝子(g.o.i)は、このベクターのEco47III/NotI部位への連結によって、hGHシグナル配列をコードする配列とFOSドメインとの間に挿入され得る。あるいは、その自らのシグナル配列を含む遺伝子は、StuI/NotI部位への連結によって、FOSコード領域に融合され得る。
【0117】
6.pAV6:このベクターを、N末端にFOSを有する融合タンパク質の真核生物産生のために設計した。目的の遺伝子(g.o.i)は、このベクターのNotI/StuI(または、NotI/HindIII)部位へ連結され得る。
【0118】
当業者によって、理解されるように、FOS抗原またはFOS抗原決定基融合タンパク質の構築は、組換えタンパク質の産生を促進するような特定の遺伝子エレメントの付加を含み得る。実施例4は、翻訳のための特定のE.coli調節エレメントの添加についてのガイダンスを提供し、そして実施例7は、真核生物シグナル配列の添加についてのガイダンスを提供する。開業医の特定の必要性に依存して、他の遺伝子エレメントが、選択され得る。
【0119】
本発明はまた、細菌細胞(実施例5)または真核生物細胞(実施例8)のいずれかにおけるFOS抗原またはFOS抗原決定基融合タンパク質の産生を含むことが理解される。融合タンパク質を発現させるために、いずれの細胞型を選択するかは、当業者の知識内にあり、例えば、この組成物の設計において、翻訳後の改変が、重要な考察であるか否かというような因子に依存する。
【0120】
以前に注記されたように、本発明は、pAVベクターの使用を介するFOS抗原またはFOS抗原決定基融合タンパク質の構築のための種々の方法を開示する。原核生物発現および真核生物発現を可能にすることに加えて、これらのベクターは、開業医が、FOSロイシンジッパードメインの抗原への−とCの間の末端付加を選択すること可能にする。特定の例が、提供され、ここで−およびC末端FOS融合は、PLA(実施例9)およびオボアルブミン(実施例10)に対してなされる。実施例11は、PLAおよびオボアルブミンFOS融合タンパク質の精製を実証する。
【0121】
ほとんどの特定の実施形態において、本発明は、HIVゲノムによってコードされる抗原または抗原決定基に引き寄せられる。より具体的には、HIV抗原は、gp140である。実施例11〜15に提供されるように、HIVgp140は、FOSロイシンジッパータンパク質ドメインおよび合成された融合タンパク質とともに作製され得、そして本発明の非天然分子足場に対する付着のために精製され得る。当業者が公知のように、他のHIV抗原または抗原決定基が、本発明の組成物の作製の際に使用され得る。
【0122】
本発明の最も具体的な実施形態において、選択される第2の付着部位は、システイン残基であり、これは本発明の非天然分子足場のリジン残基と特異的に結合する。リジン残基(Lys)およびシステイン残基(Cys)のその化学結合は、足場の表面上に、組織化され、かつ反復性の抗原または抗原決定基アレイの形成のための基礎を提供する。システイン残基は、アミノ末端、カルボキシル末端のいずれかにおける抗原または抗原決定基の選択に対してインフレームで操作され得るか、または所望される場合、そのタンパク質の内部に位置され得る。例として、PLAおよびHIVgp140が、リジン残基の第1の付着部位に対する結合のためのシステイン残基とともに提供される。
【0123】
(C.αワクチン粒子の調製)
本発明は、順序付けされ、かつ反復性の抗原アレイの構築のための新規の組成物および方法を提供する。当業者が公知のように、順序付けされ、かつ反復性の抗原アレイのアセンブリのための条件は、大きな程度まで、非天然の足場の第1の付着部位の特異的な選択および抗原または抗原決定基の第2の付着部位の特異的な選択に依存する。従って、この組成物の設計における開業医の選択は(すなわち、第1の付着部位および第2の付着部位、抗原および非天然の足場の選択)、αワクチン粒子(順序付けされ、かつ反復性の抗原アレイおよび結合される非天然分子足場)の集合についての特定の条件を決定する。このαワクチン粒子の集合に関する情報は、十分に開業医の作業知識内であり、そして多くの参考文献が存在し、開業医を援助する(例えば、Sambork,Jら、編、MOLECULAR CLOONING、A LABORATORY MANUAL、第2版、Cold Spring Harbor Laboratory Press、Cold Spring Harbor、N.Y.(1989);Ausubel,Fら、編、CURRENT PROTOCOLS IN MOLECULAR BIOLOGY,John H.Wiley&Sons、Inc.(1997);Celis,J、編、CELL BIOLGY、Academic Press.第2版、(1988);Harlow,E.およびLane,D、「Antibodies:A Laboratory Manual、」Cold Spring Harbor Laboratory、Cold Spring Harbor,N.Y.(1988)。これらの全ては、本明細書中で参考として援用される。
【0124】
本発明の特定の実施形態において、JUNおよびFOSロイシンジッパータンパク質ドメインはそれぞれ、本発明の第1および第2の付着部位のために利用される。αワクチン粒子の調製において、抗原は、非天然足場上に順序付けされ、かつ反復性の抗原アレイのアセンブリを促進するような条件下で産生され、そして精製されなければならない。特定のJUN/FOSロイシンジッパータンパク質ドメインの実施形態において、FOS抗原またはFOS抗原決定基は、ジスルフィド結合形成の発生を減少または排除するために、還元剤(例えば、ジチオスレイトール(DTT))を用いて処理されるべきである(実施例15)。
【0125】
JUN/FOSロイシンジッパータンパク質ドメイン実施形態の非天然足場(すなわち、組換えSinbisウイルス)の調製のために、組換えE2−JUNウイルス粒子は、濃縮され、中和、そして還元剤を用いて処理されるべきである(実施例16を参照のこと)。
【0126】
JUN/FOS実施形態における、順序付けされ、かつ反復性の抗原アレイの集合は、酸化還元シャッフルの存在下でなされる。E2−JUNウイルス粒子は、240倍モル濃度超過のFOS抗原またはFOS抗原決定基と4℃で10時間結合される。次いで、そのαワクチン粒子は、クロマトグラフィーによって濃縮されて、精製される(実施例16)。
【0127】
本発明の別の実施形態において、非天然分子足場と抗原または抗原決定基の結合は、化学的架橋によって、達成され得る。最も好ましい実施形態において、その化学剤は、ヘテロ2官能基架橋剤(例えば、ε−マレイミドカプロン酸N−ヒドロキシスクシンイミドエステル(Tanimoriら、J.Pharm.Dyn.4:812(1981);Fujiwaraら、J.Immunol.Meth.45:195(1981))であり、これは、(1)アミノ基と反応するスクシンイミド基および(2)SH基と反応するマレイミド基を含む。第1の付着部位の異種タンパク質またはポリペプチドは、ヘテロ2官能基架橋剤のスクシンイミド部分に対する反応部分として役に立つ、一つ以上のリジン残基を含むように操作され得る。一旦、非天然分子足場の第1の付着部位に化学的に結合されると、ヘテロ2官能基架橋剤のマレイミド基は、抗原または抗原決定基上のシステイン残基のSH基と反応するために、利用可能となる。この場合における抗原または抗原決定基調製は、第2の付着部位として選択されたタンパク質またはポリペプチド内へシステイン残基を操作することを必要とし得、その結果システイン残基を、非天然分子足場の第1の付着部位に結合した架橋剤上の遊離マレイミド官能基と反応させ得る。
【0128】
(3.組成物、ワクチン、およびそれらの投与、ならびに処置方法)
1つの実施形態では、本発明は、広範な種々の種(特に、ヒト、サル、ウシ、イヌ、ネコ、ウマ、ブタなどのような哺乳動物種)における感染症の予防のためのワクチンを提供する。ワクチンは、ウイルス性病因の感染(例えば、HIV、インフルエンザ、ヘルペス、ウイルス性肝炎、エプスタイン−バー、ポリオ、ウイルス性脳炎、麻疹、水痘など);または、細菌性病因の感染(例えば、肺炎、結核、梅毒など);または寄生生物性病因の感染(例えば、マラリア、トリパノソーマ症、リーシュマニア症、トリコモナス症、アメーバ症など)を処置するように設計され得る。
【0129】
別の実施形態では、本発明は、広範な種々の種(特に、ヒト、サル、ウシ、イヌ、ネコ、ウマ、ブタなどのような哺乳動物種)における癌の予防のためのワクチンを提供する。ワクチンは、すべての型の癌(リンパ腫、癌腫、肉腫、黒色腫など)を処置するように設計され得る。
【0130】
本発明の別の実施形態では、本発明の組成物は、アレルギーの処置のためのワクチンの設計において使用され得る。IgEアイソタイプの抗体は、アレルギー反応において重要な成分である。肥満細胞は、それらの表面上でIgE抗体を結合し、そして肥満細胞表面上に結合したIgE分子に対する特異的抗原の結合に際して、ヒスタミンおよびアレルギー反応の他のメディエイタを放出する。従って、IgE抗体の産生を阻害することは、アレルギーに対して保護するための有望な標的である。これは、所望されるTヘルパー細胞応答を達成することによって可能であるはずである。Tヘルパー細胞応答は、1型(TH1)Tヘルパー細胞応答および2型(TH2)Tヘルパー細胞応答に分けられ得る(Romagnani、Immunol.Today 18:263−266(1997))。TH1細胞は、インターフェロン−γ、およびB細胞を誘発してIgG1−3抗体を産生する他のサイトカインを分泌する。対照的に、TH2細胞により産生される重要なサイトカインはIL−4であり、IL−4はB細胞を駆動してIgG4およびIgEを産生する。多くの実験系において、TH1応答およびTH2応答の発生は相互排他的である。なぜなら、TH1細胞はTH2細胞の誘導を抑制し、そして逆もまた同じであるからである。従って、強力なTH1応答を誘発する抗原は、同時に、TH2応答の発生を抑制し、従ってIgE抗体の産生を抑制する。興味深いことに、実質的にすべてのウイルスが、宿主においてTH1応答を誘導し、そしてIgE抗体の産生を誘発することに失敗する(Coutelierら、J.Exp.Med.165:64−69(1987))。このアイソタイプパターンは、生存しているウイルスに制限されず、不活化されたウイルス粒子または組換えウイルス粒子についても観察された(Lo−Manら、Eur.J.Immunol.28:1401−1407(1998))。従って、本発明のプロセス(例えば、αワクチン技術)を使用することにより、ウイルス粒子は種々のアレルゲンで修飾(decorate)され得、そして免疫のために使用され得る。生じるアレルゲンの「ウイルス構造」に起因して、TH1応答が誘発され、「保護的」IgG1−3抗体が産生され、そしてアレルギー反応を引き起こすIgE抗体の産生を妨げる。アレルゲンは、アレルゲン自体よりもヘルパーT細胞の異なるセットによって認識されるウイルス粒子によって提示されるので、アレルゲン特異的IgG1−3抗体は、このアレルゲンに対して特異的な既存のTH2細胞を保有するアレルギー性個体においてでさえ、誘導される可能性がある。高濃度のIgG抗体の存在は、肥満細胞結合IgEへのアレルゲンの結合を妨げ得、それによって、ヒスタミンの放出を阻害する。従って、IgG抗体の存在は、IgE媒介性アレルギー反応から保護し得る。アレルギーを引き起こす代表的な物質としては、以下が挙げられる:イネ科草本、ブタクサ、カバノキもしくはマウンテンシーダー(mountain cedar)の花粉、ハウスダスト、ダニ類、動物のフケ、カビ、昆虫毒液、または薬物(例えば、ペニシリン)。従って、アレルゲン修飾されたウイルス粒子での個体の免疫は、アレルギー発症の前のみならず後でも、有益である。
【0131】
当業者によって理解されるように、本発明の組成物が個体に投与される場合、それらは、組成物の効力を改善するために所望され得る塩、緩衝剤、アジュバント、または他の物質を含む組成物中であり得る。薬学的組成物を調製する際の使用に適切な物質の例は、REMINGTON’S PHARMACEUTICAL SCIENCES(Osol,A.編、Mack Publishing Co.(1980))を含む多数の供給源に提供される。
【0132】
本発明の組成物は、それらの投与がレシピエント個体によって耐えられ得る場合に、「薬理学的に受容可能」であるといわれる。さらに、本発明の組成物は、「治療的有効量」(すなわち、所望される生理学的効果を産生する量)で、投与される。
【0133】
本発明の組成物は、当該分野において公知の種々の方法によって投与され得るが、通常は、注射、注入、吸入、経口投与、または他の適切な物理的方法によって投与される。あるいは、組成物は、筋内、静脈内、または皮下に投与され得る。投与のための組成物の成分は、滅菌水溶液(例えば、生理的食塩水)または非水系溶液および懸濁液を含む。非水系溶媒の例は、プロピレングリコール、ポリエチレングリコール、植物油(例えば、オリーブオイル)、および注射可能な有機エステル(例えば、オレイン酸エチル)である。キャリアまたは閉鎖包帯が、皮膚の透過性を増大させるため、および抗原吸着を増強するために使用され得る。
【0134】
ワクチン技術に加えて、本発明の他の実施形態は、癌およびアレルギーの医学的処置の方法に向けられる。
【0135】
本明細書中で言及される、すべての特許および刊行物は、特に参考として援用される。
【0136】
(実施例)
続く実験において使用された酵素および試薬は、以下を含んだ:New England Biolabsから入手した、T4 DNAリガーゼ;QIAGENから入手した、Taq DNAポリメラーゼ、QIAprep Spin Plasmid Kit、QIAGEN Plasmid Midi Kit、QiaExII Gel Extraction Kit、QIAquick PCR Purification Kit;Pharmaciaから入手した、QuickPrep Micro mRNA Purification Kit;Gibco BRLから入手した、SuperScript One−step RT PCR Kit、ウシ胎仔血清(FCS)、バクト−トリプトン(bacto−tryptone)および酵母抽出物;Microsynth(Switzerland)から入手した、オリゴヌクレオチド;Boehringer Mannheim、New England BiolabsまたはMBI Fermentasから入手した、制限エンドヌクレアーゼ;Boehringer Mannheimから入手した、PwoポリメラーゼおよびdNTPs。HP−1培地を、Cell culture technologies(GlattBrugg、Switzerland)から入手した。すべての標準的な化学薬品を、Fluka−Sigma−Aldrichから入手した。そして、すべての細胞培養材料を、TPPから入手した。
【0137】
DNA操作を、標準的技術を使用して実施した。DNAを、製造業者の指示書に従って、QIAprep Spin Plasmid Kitを使用して2mlの細菌培養物から、またはQIAGEN Plasmid Midi Kitを使用して50mlの培養物からのいずれかから調製した。制限酵素消化のために、製造業者の推奨される条件(緩衝液および温度)下で、1mgのDNAあたり5〜10ユニット(U)の酵素濃度で、DNAを適切な制限酵素と共に少なくとも2時間インキュベートした。1つより多い酵素での消化を、反応条件がすべての酵素について適切である場合には同時に実施したが、そうでなければ連続して実施した。さらなる操作のために単離されたDNAフラグメントを、0.7〜1.5%アガロースゲルにおける電気泳動により分離し、このゲルから切除し、そして製造業者によって提供される指示書に従って、QiaExII Gel Extraction Kitを用いて精製した。DNAフラグメントの連結のために、100〜200pgの精製されたベクターDNAを、製造業者によって提供される緩衝液(総容量:10〜20μl)中で、1UのT4 DNAリガーゼの存在下において、3倍モル濃度過剰の挿入物フラグメントと共に、16℃にて一晩インキュベートした。連結反応物のアリコート(0.1〜0.5μl)を、E.coli EL1−Blue(Stratagene)の形質転換のために使用した。形質転換を、Gene Pulser(BioRAD)および0.1cmのGene Pulser Cuvettes(BioRAD)を、200Ω、25μF、1.7kVにて使用するエレクトロポレーションによって実施した。エレクトロポレーション後、選択的S.O.B.アガーにおけるプレーティングの前に、細胞を1mlのS.O.B.培地(Miller、1972)中で1時間振盪しながらインキュベートした。
【0138】
(実施例1)
(E2におけるJUN両親媒性ヘリックスドメインの挿入)
ベクターpTE5’2J(Hahnら、Proc.Natl.Acad.Sci.USA 89:2679−2683(1992))において、MlnIおよびBstEIIの制限酵素部位を、構造タンパク質E2コード配列のコドン71(Gln)と74(Thr)との間に導入し、ベクターpTE5’2JBMを得た。これらの制限酵素部位の導入を、以下のオリゴヌクレオチドを用いるPCRによって実施した。
【0139】
【化1】
PCR反応のために、100pmolの各オリゴを、100μlの反応混合物(4ユニットのTaqポリメラーゼまたはPwoポリメラーゼ、0.1mM dNTPs、および1.5mM MgSO4を含む)中で、鋳型DNA5ngと共に使用した。すべてのDNA濃度を、GeneQuant装置(Pharmacia)を使用して測光により決定した。ポリメラーゼを、PCR反応を開始する直前(開始点は、95℃)に添加した。温度サイクリングを、以下の様式および順序で実施した:95℃(2分間);95℃(45秒間)、53℃(60秒間)72℃(80秒間)の5サイクル;そして、95℃(45秒間)、57℃(60秒間)、72℃(80秒間)の25サイクル。
【0140】
2つのPCRフラグメントを、アガロースゲル電気泳動によって分析および精製した。増幅のためにオリゴ3および4を使用する、2つのPCRフラグメントのアセンブリPCR(assembly PCR)を、最終構築物を得るために実施した。
【0141】
アセンブリPCR反応のために、100pmolの各オリゴを、100μlの反応混合物(4ユニットのTaqポリメラーゼまたはPwoポリメラーゼ、0.1mM dNTPs、および1.5mM MgSO4を含む)中で、2ngの精製されたPCRフラグメントと共に使用した。すべてのDNA濃度を、GeneQuant装置(Pharmacia)を使用して測光により決定した。ポリメラーゼを、PCR反応を開始する直前(開始点は、95℃)に添加した。温度サイクリングを、以下の様式および順序で実施した:95℃(2分間);95℃(45秒間)、57℃(60秒間)72℃(90秒間)の5サイクル;そして、95℃(45秒間)、59℃(60秒間)、72℃(90秒間)の25サイクル。
【0142】
最終PCR産物を、Qia spin PCRカラム(Qiagen)を用いて精製し、そして適切な緩衝液中で、各10ユニットのBssHIIおよびStuI制限エンドヌクレアーゼを用いて、37℃にて12時間消化した。DNAフラグメントをゲル精製し、そしてBssHII/StuI消化されかつゲル精製されたpTE5’2Jベクター(Hahnら、Proc.Natl.Acad.Sci.USA 89:2679−2683)中に連結した。このPCR産物の正確な挿入を、まずBstEIIおよびMluI制限分析により分析し、次いで、PCRフラグメントのDNA配列決定により分析した。
【0143】
JUN両親媒性ヘリックスドメインをコードするDNA配列を、以下のオリゴヌクレオチドを用いて、ベクターpJuFo(CrameriおよびSuter、Gene 137:69(1993))からPCR増幅した。
【0144】
【化2】
PCR反応のために、100pmolの各オリゴを、100μlの反応混合物(4ユニットのTaqポリメラーゼまたはPwoポリメラーゼ、0.1mM dNTPs、および1.5mM MgSO4を含む)中で、5ngの鋳型DNAと共に使用した。すべてのDNA濃度を、GeneQuant装置(Pharmacia)を使用して測光により決定した。ポリメラーゼを、PCR反応を開始する直前(開始点は、95℃)に添加した。温度サイクリングを、以下の順序および様式で実施した:95℃(2分間);95℃(45秒間)、60℃(30秒間)72℃(25秒間)の5サイクル;そして、95℃(45秒間)、68℃(30秒間)、72℃(20秒間)の25サイクル。
【0145】
最終PCR産物をゲル精製し、そしてEcoRV消化されかつゲル精製されたpBluescript II(KS−)中に連結した。得られたベクターから、QiaExIIで精製されたMluI/BstEIIでの切断によって、JUN配列を単離し、そしてベクターpTE5’2JBM(予め、同一の制限酵素で切断されている)中に連結して、ベクターpTE5’2J:E2JUNを得た。
【0146】
(実施例2)
(pCYTts系を使用する、E2−JUN含有ウイルス粒子の産生)
構造タンパク質を、鋳型としてのpTE5’2J:E2JUN、ならびにオリゴヌクレオチドXbalStruct(ctatcaTCTAGAATGAATAGAGGATTCTTTAAC)(配列番号12)およびStructBsp1201(tcgaatGGGCCCTCATCTTCGTGTGCTAGTCAG)(配列番号87)を使用して、PCR増幅した。PCRのために、100pmolの各オリゴ(Ioligo)を使用し、そして5ngの鋳型DNAを、100μlの反応混合物(4ユニットのTaq(Tac)ポリメラーゼまたはPwoポリメラーゼ、0.1mM dNTPs、および1.5mM MgSO4を含む)中で使用した。すべてのDNA濃度を、GeneQuant装置(Pharmacia)を使用して測光により決定した。ポリメラーゼを、PCR反応を開始する直前(開始点は、95℃)に添加した。温度サイクルは、以下の通りであった:95℃(3分間)、次いで、92℃(30秒間)、54℃(35秒間)72℃(270秒間)の5サイクル、次いで、92℃(30秒間)、63℃(35秒間)、72℃(270秒間)の25サイクル。このPCR産物をゲル精製し、そして制限酵素XbaI/Bsp1201で消化し、そして予め同一の酵素で切断されたベクターpCYTts(米国特許出願番号60/079,562;1998年3月27日出願)中に連結した。
【0147】
20μgのpCYTtsE2:JUNを、37℃にて少なくとも4時間、適切な緩衝液中で30UのScaIと共にインキュベートした。この反応を、フェノール/クロロホルム抽出により停止し、次いで、直鎖化DNAのイソプロパノール沈殿を行った。制限反応を、アガロースゲル電気泳動によって確認した。トランスフェクションのために、30μlのH2O中において5.4μgの直鎖化pCYTtsE2:JUNを0.6μgの直鎖化pSV2Neoと混合し、そして30μlのCaCl2溶液を添加した。60μlのリン酸緩衝液(50mM HEPES、280mM NaCl、1.5mM Na2HPO4、pH7.05)の添加後、この溶液を5秒間ボルテックスし、次いで、室温で25秒間インキュベーションした。この溶液を、2%のFCSを含有する2mlのHP−1培地(2% FCS培地)に、すぐ添加した。次いで、6ウェルプレート中の80%コンフルエントなBHK21細胞培養の培地を、DNA含有培地と置換した。CO2インキュベーター内における37℃での5時間のインキュベーションの後、DNA含有培地を取り除き、そして2% FCS培地中の15%グリセロール2mlと置換した。このグリセロール含有培地を、30秒間のインキュベーション期の後に取り除き、そして10%のFCSを含有するHP−1培地5mlでリンスすることによって細胞を洗浄した。最後に、10%のFCSを含有する新鮮なHP−1培地2mlを添加した。
【0148】
安定にトランスフェクトされた細胞を、CO2インキュベーター内で37℃にて、選択培地(G418を補充した、HP−1培地)中で選択し、そして増殖させた。混合した集団をコンフルーエンシーまで増殖した時点で、培養物を2つのディッシュに分け、次いで、37℃での12時間の増殖期を行った。この細胞の一方のディッシュを、ウイルス粒子の発現を誘導するために30℃に移し;他方のディッシュを、37℃で維持した。
【0149】
ウイルス粒子の発現を、ウェスタンブロッティングにより決定した(図1)。培養培地(0.5ml)を、メタノール/クロロホルム沈殿し、そしてペレットを、SDS−PAGEサンプル緩衝液中に再懸濁した。サンプルを、15%アクリルアミドゲルに適用する前に、5分間、95℃で加熱した。SDS−PAGE後、BassおよびYang、Creighton,T.E.編、Protein Function:A Practical Approach、第2版、IRL Press、Oxford(1997)、29〜55頁によって記載されるように、タンパク質を、Protanニトロセルロースメンブレン(Schleicher&Schuell、Germany)に転写した。TBS(1リットルあたり10×TBS:87.7gのNaCl、66.1gのTrizma hydrochloride(Sigma)および9.7gのTrizma base(Sigma)、pH7.4)中の1%ウシアルブミン(Sigma)を用いて、室温にて1時間メンブレンをブロックし、次いで、抗E1/E2抗体(ポリクローナル血清)と共に1時間インキュベーションした。ブロットを、TBS−T(0.05% Tween20を有するTBS)を用いて10分間、3回洗浄し、そしてアルカリホスファターゼ−抗ウサギIgG結合体(0.1μg/ml、Amersham Life Science、England)を用いて、1時間インキュベートした。TBS−Tを用いた10分間の2回の洗浄およびTBSを用いた10分間の2回の洗浄の後、50μlのNBT溶液(70%ジメチルホルムアミド中の7.7% Nitro Blue Tetrazolium(Sigma))および37μlのX−Phosphate溶液(ジメチルホルムアミド中の5% 5−ブロモ−4−クロロ−3−インドリルホスフェート)と共に、アルカリホスファターゼ検出試薬(10ml AP緩衝液(100mM Tris/HCl、100mM NaCl、pH9.5))を用いて、発色反応を実施した。
【0150】
ウイルス粒子の産生を、図1に示す。ウェスタンブロットのパターンは、E2−JUN(レーン1)が、SDS−PAGEにおいて、野生型E2(レーン2)と比較してより大きな分子量に移動することを示した。そして、BHK21宿主細胞株は、バックグラウンドを全く示さなかった。
【0151】
(実施例3)
(pTE5’2JE2:JUNベクターを用いた、E2−JUNを含有するウイルス粒子の産生)
RNaseを含まないベクター(1.0μg)を、PvuI消化によって直鎖化した。続いて、SP6インビトロ転写キット(InvitroGen、Invitrogen BV、NV Leek、Netherlandsによる、InvitroscripCAP)を用いて、インビトロでの転写を実施した。生じた5’キャップ化されたmRNAを、還元アガロースゲルにおいて分析した。
【0152】
インビトロで転写されたmRNA(5μg)を、Invitrogenの説明書(Sindbis Expression system、Invitrogen BV、Netherlands)に従って、BHK21細胞(ATCC:CCL10)中にエレクトロポレーションした。37℃での10時間のインキュベーションの後、FCS含有培地を、FCSを有さないHP−1培地と交換し、次いで、37℃で10時間さらにインキュベーションした。上清を収集し、そして正確に実施例2に記載のように、ウイルス粒子の産生についてウェスタンブロット分析によって分析した。
【0153】
得られた結果は、図2に示されるようなpCYTtsE2:JUNで得られた結果と同一であった。
【0154】
(実施例4)
(FOSロイシンジッパードメイン(OmpAシグナル配列)へのヒト成長ホルモン(hGH)の融合)
ヒトリーダー配列を有さないhGH遺伝子を、もともとのプラスミド(ATCC 31389)から、PCRによって増幅した。内部XbaI部位を有するオリゴ7を、hGH遺伝子の5’末端でのアニーリングのために設計し、そして内部EcoRI部位を有するオリゴ9を、hGH遺伝子の3’末端でプライムした。PCR反応のために、100pmolの各オリゴおよび5ngのテンプレートDNAを、75μlの反応混合物(4ユニットのTaqポリメラーゼまたはPwoポリメラーゼ、0.1mM dNTPおよび1.5mM MgSO4)中で使用した。
【0155】
PCRサイクリングを、以下の様式で実施した:60℃のアニーリング温度および72℃での1分間の伸長時間の30サイクル。
【0156】
ゲル精製し、そして単離したPCR産物を、第2のPCR反応のためのテンプレートとして使用して、ompAシグナル配列およびシャイン−ダルガーノ配列を導入した。PCR反応のために、100pmolのオリゴ8および9、ならびに1ngのテンプレートPCRフラグメントを、75μlの反応混合液(4ユニットのTaqポリメラーゼまたはPwoポリメラーゼ、0.1mM dNTP、および1.5mM MgSO4)中で使用した。最初の5サイクルのアニーリング温度は55℃で、72℃での60秒間の伸長時間を伴い;別の25サイクルを、65℃のアニーリング温度および72℃で60秒間の伸長時間で実施した。
【0157】
【化3】
生じた組換えhGH遺伝子を、XbaI/EcoRIを介してpBluescriptにサブクローン化した。両鎖の正確な配列を、DNA配列決定によって確認した。
【0158】
FOS両親媒性ヘリックスドメインをコードするDNA配列を、ベクターpJuFo(Crameri&Suter Gene 137:69(1993))から、以下のオリゴヌクレオチドを用いてPCR増幅した。
【0159】
【化4】
PCR反応のために、100pmolの各オリゴおよび5ngのテンプレートDNAを、75μlの反応混合物(4ユニットのTaqポリメラーゼまたはPwoポリメラーゼ、0.1mM dNTP、および1.5mM MgSO4)中で使用した。温度サイクルは、以下の通りであった:
95℃で2分間、次いで、95℃(45秒間)、60℃(30秒間)、72℃(25秒間)の5サイクル、次いで、95℃(45秒間)、68℃(30秒間)、72℃(20秒間)の25サイクル。
【0160】
PCR産物を精製し、単離し、そしてStuI消化pBluescript−ompA−hGH中にクローン化した。次いで、ハイブリッド遺伝子を、pKK223−3プラスミド(Pharmacia)中にクローン化した。
【0161】
(実施例5)
(FOS−hGHの細菌性発現)
pkk223−3中のompA−FOS−hGHを、誘導性のIPTG依存性プロモーターの制御下で、E.coli宿主株としてJM101を使用して発現させた。発現を、振盪機フラスコ内で実施した。細胞を、0.5のOD600において、1mM IPTG(最終濃度)で誘導した。発現を、37℃にて10時間継続した。細胞を、10℃で15分間、3600での遠心分離によって収集した。細胞のペレットを、凍結し(−20℃または液体N2)、そして16時間保存した。次いで、ペレットを4℃にて融解し、そして600mMスクロースを含有する10mlの10mM Tris/HCl(pH7.4)中に再懸濁した。4℃での15分間の攪拌後、ペリプラズムのタンパク質を、浸透圧性ショック手順によって放出した。冷却(4℃)脱イオン水を添加し、そして懸濁液を4℃にて30分間攪拌した。スラッジを希釈し、再懸濁し、そしてリゾチームを添加して、細菌の細胞壁を破壊した。細胞およびペリプラズムの画分のスフェロプラストを、4℃で20分間、11000gでの遠心分離によって分離した。FOS−hGH含有上清を、還元性および非還元性SDS−Pageおよびドットブロットによって分析した。ドットブロットを、実施例8に記載のように、一次抗体として抗hGH抗体(Sigma)および二次抗体としてアルカリホスファターゼ(AP)−抗マウス抗体結合体を使用して、実施した。
【0162】
全長の正確にプロセスされたFOS−hGHが、還元条件および非還元条件下で検出され得た。FOS−hGHの一部は、FOS両親媒性ヘリックス中に存在する遊離システインに起因して、他の未同定のタンパク質に結合した。しかし、発現されたFOS−hGHの50%より多くが、そのネイティブなモノマー性コンフォメーションで存在した(図3)。
【0163】
精製されたFOS−hGHを使用して、JUN含有ウイルス粒子を用いる最初のドーピング実験を実施した。
【0164】
(実施例6)
(FOS融合タンパク質の発現のための、pAVベクター系列の構築)
−もしくはE.coliにおけるC末端FOS融合タンパク質の細胞質内産生もしくは分泌、または−もしくは真核生物細胞におけるC末端融合タンパク質の産生のいずれかを可能にする、汎用性のベクター系を構築した。E.coliにおけるFOS融合タンパク質の産生のために設計されたベクターpAV1〜pAV4は、以下に列挙されたDNAカセットを含む。このDNAカセットは、異なる順序で配列された以下の遺伝的エレメントを含む:(a)E.coli ompA遺伝子(aggaggtaaaaaacg)(配列番号13)由来の強力なリボソーム結合部位および5’非翻訳領域;(b)E.coli外膜タンパク質OmpA(MKKTAIAIAVALAGFATVAQA)(配列番号14)のシグナルペプチドをコードする配列;(c)2つのグリシン残基および1つのシステイン残基によって両側で隣接されたFOS二量体化ドメイン(CGGLTDTLQAETDQVEDEKSALQTEIANLLKEKEKLEFILAAHGGC)(配列番号15)をコードする配列;および(d)目的のタンパク質をFOS二量体化ドメインに連結する、短いペプチドリンカー(AAASGG)(配列番号16)またはGGSAAA(配列番号17))をコードする領域。関連するコード領域を、大文字で提供する。制限切断部位の配置は、シグナル配列を伴うかまたは伴わない、FOS融合遺伝子の容易な構築を可能にする。このカセットを、強力なtacプロモーターの制御下での融合遺伝子の発現のために、発現ベクターpKK223−3(Pharmacia)のEcoRI/HindIII制限部位にクローン化した。
【0165】
(pAV1)
このベクターは、C末端にFOSを有する融合タンパク質のE.coli細胞周辺腔への分泌のために設計された。目的の遺伝子(g.o.i.)は、このベクターのStuI/NotI部位に連結され得る。
【0166】
【化5】
。
【0167】
(pAV2)
このベクターは、N末端にFOSを有する融合タンパク質のE.coli細胞周辺腔への分泌のために設計された。目的の遺伝子(g.o.i.)は、このベクターのNotI/EcoRV(または、NotI/HindIII)部位に連結される
【0168】
【化6】
。
【0169】
(pAV3)
このベクターは、C末端にFOSを有する融合タンパク質のE.coliにおける細胞質内産生のために設計された。目的の遺伝子(g.o.i.)は、このベクターのEcoRV/NotI部位に連結され得る。
【0170】
【化7】
。
【0171】
(pAV4)
このベクターを、N末端にFOSを有する融合タンパク質のE.coli中での細胞質産生のために設計する。目的の遺伝子(g.o.i.)を、ベクターのNotI/EcoRV(またはNotI/HindIII)部位に連結し得る。N末端メチオニン残基を、タンパク質合成に際してタンパク質分解的に除去する(Hirelら、Proc.Natl.Acad.Sci.USA 86:8247−8251(1989))。
【0172】
【化8】
。
【0173】
ベクターpAV5およびpAV6(これらは、FOS融合タンパク質の真核生物産生のために設計される)は、異なる順番で配置された以下の遺伝的エレメントを含む:(a)ヒト成長ホルモンのリーダーペプチドをコードする領域(MATGSRTSLLLAFGLLCLPWLQEGSA)(配列番号26);(b)2つのグリシン残基および1つのシステイン残基が両側で隣接する、FOS二量体化ドメインをコードする配列(CGGLTDTLQAETDQVEDEKSALQTEIANLLKEKEKLEFILAAHGGC)(配列番号15);および(c)FOS二量体化ドメインに目的のタンパク質を接続する、短いペプチドリンカーをコードする領域(AAASGG(配列番号16)またはGGSAAA(配列番号17))。関連するコード領域を大文字で示す。制限切断部位の配列は、FOS融合遺伝子の容易な構築を可能にする。このカセットを、発現ベクターpMPSVEH(Arteltら、Gene 68:213−219(1988))のEcoRI/HindIII制限部位にクローニングする。
【0174】
(pAV5)
このベクターを、C末端にFOSを有する融合タンパク質の真核生物産生のために設計する。目的の遺伝子(g.o.i.)を、ベクターのEco47III/NotI部位への連結によって、hGHシグナル配列をコードする配列とFOSドメインをコードする配列との間に挿入し得る。あるいは、それ自体のシグナル配列を含む遺伝子を、StuI/NotI部位への連結によってFOSコード領域に融合し得る。
【0175】
【化9】
。
【0176】
(pAV6)
このベクターを、N末端にFOSを有する融合タンパク質の真核生物産生のために設計する。目的の遺伝子(g.o.i.)を、ベクターのNotI/StuI部位(またはNotI/HindIII部位)に連結し得る。
【0177】
【化10】
(発現ベクターpAV1−pAV6の構築)
以下のオリゴヌクレオチドを、発現ベクターpAV1−pAV6の構築のために合成した:
【0178】
【化11】
ベクターpAV2の構築のために、OmpAシグナル配列およびFOSドメインをコードする領域を、プライマー対OmpA−FOR1/FOS−REV2を使用して、ベクターpKK223−3中のompA−FOS−hGH融合遺伝子(実施例5を参照のこと)から増幅した。このPCR産物を、EcoRI/HindIIIで消化し、そしてベクターpKK223−3(Pharmacia)の同じ部位に連結した。
【0179】
ベクターpAV1の構築のために、FOSコード領域を、プライマー対FOS−FOR1/FOS−REV1を使用して、ベクターpKK223−3中のompA−FOS−hGH融合遺伝子(実施例5を参照のこと)から増幅した。このPCR産物を、HindIIIで消化し、そしてStuI/HindIII消化したベクターpAV2に連結した。
【0180】
ベクターpAV3の構築のために、FOSドメインをコードする領域を、プライマー対FOS−FOR2/FOS−REV1を使用して、ベクターpAV1から増幅した。このPCR産物を、EcoRI/HindIIIで消化し、そしてベクターpKK223−3(Pharmacia)の同じ部位に連結した。
【0181】
ベクターpAV4の構築のために、FOSドメインをコードする領域を、プライマー対FOS−FOR3/FOS−REV2を使用して、ベクターpKK223−3中のompA−FOS−hGH融合遺伝子(実施例5を参照のこと)から増幅した。このPCR産物を、EcoRI/HindIIIで消化し、そしてベクターpKK223−3(Pharmacia)の同じ部位に連結した。
【0182】
ベクターpAV5の構築のために、hGHシグナル配列をコードする領域を、プライマー対hGH−FOR1/hGHREV1を使用して、ベクターpSINrep5中のhGH−FOS−hGH融合遺伝子(実施例7を参照のこと)から増幅する。このPCR産物を、EcoRI/NotIで消化し、そしてベクターpAV1の同じ部位に連結する。次いで、hGHシグナル配列およびFOSドメインをコードする得られたカセットを、EcoRI/HindIII消化によって単離し、そして同じ酵素で消化したベクターpMPSVEH(Arteltら、Gene 68:213−219(1988))にクローニングする。
【0183】
ベクターpAV6の構築のために、FOSコード領域を、プライマー対FOS−FOR4/FOSREV3を使用して、ベクターpAV2から増幅する。このPCR産物をHindIIIで消化し、そしてEco47III/HindIII切断したベクターpAV5にクローニングする。次いで、hGHシグナル配列およびFOSドメインをコードする全体のカセットを、プライマー対hGH−FOR2/FOSREV3を使用して、得られるベクターから再増幅し、EcoRI/HindIIIで切断し、そして同じ酵素で切断したベクターpMPSVEH(Arteltら、Gene 68:213−219(1988))に連結する。
【0184】
(実施例7:ヒト(hGH)シグナル配列を有するFOS−hGHの構築)
FOS−hGH融合タンパク質の真核生物発現のために、OmpA−FOS−hGH融合遺伝子を、XbaI/Bsp120Iでの消化によって、pBluescript::OmpA−FOS−hGH(実施例4を参照のこと)から単離し、そして同じ酵素で切断したpSINrep5(Invitrogen)にクローニングした。hGHシグナル配列を、重複するオリゴヌクレオチド
【0185】
【化12】
を使用するPCR(反応混合物:50pモルの各プライマー、dATP、dGTP、dTTP、dCTP(各200μM)、2.5UのTaq DNAポリメラーゼ(Qiagen)、製造業者より供給された緩衝液中、50μlの総量;増幅;92℃で30秒間、55℃で30秒間、72℃で30秒間、30サイクル)によって合成した。このPCR産物を、QiaExllキットを使用して精製し、StuI/XbaIで消化し、そして同じ酵素で切断したベクターpSINrep5::OmpA−FOS−hGHに連結した。
【0186】
(実施例8:FOS−hGHの真核生物発現)
RNaseを含まないベクター(1.0μg)(pSINrep5::OmpA−FOS−hGH)および1.0μgのDHEB(Bredenbeekら、J.Virol.67:6439−6466(1993))を、ScaI制限消化によって線状化した。続いて、インビトロ転写を、SP6インビトロ転写キット(InvitroGen,Invitrogen BV,NV Leek,NetherlandsによるInvitroscripCAP)を使用して実行した。得られた5’キャップmRNAを、還元アガロースゲル上で分析した。
【0187】
インビトロで、Invitrogenのマニュアルに従って、転写された5μgのmRNAを、BHK 21細胞(ATCC:CCL10)にエレクトロポレーションした(Sindbis Expressionシステム、Invitrogen BV,Netherlands)。37℃で10時間のインキュベーション後、FCS含有培地を、FCSを含まないHP−1培地で交換し、続いて37℃で10時間さらなるインキュベーションを行った。その上清を収集し、そしてFOS−hghの産生についてドットブロット分析によって分析した。
【0188】
培養培地(2.5μl)を、ニトロセルロース膜上にスポットし、そして室温で10分間乾燥させた。その膜を、TBS(1リットルあたりの10×TBS:87.7g NaCl、66.1g Trizma塩酸塩(Sigma)、および9.7g Trizmaベース(Sigma)、pH 7.4)中の1%ウシアルブミン(Sigma)を用いて、室温で1時間ブロックし、続いて10mlのTBS−T(0.05% Tween20を有するTBS)中の、2μgのウサギ抗ヒトhGH抗体(Sigma)とともに1時間インキュベートした。そのブロットを、TBS−Tで10分間、3回洗浄し、そしてTBS−T中で1:5000希釈した、アルカリホスファターゼと結合体化した抗ウサギIgG(Jackson ImmunoResearch Laboratories,Inc.)とともに1時間インキュベートした。TBS−Tでの10分間、2回の洗浄およびTBSでの10分間、2回の洗浄の後、そのブロットを、実施例2に記載のようにAP染色によって染色した。結果を図3に示す。
【0189】
(実施例9:FOS−PLA(N末端およびC末端)の構築)
ハチ毒ホスホリパーゼA2(PLA)の触媒的に不活性な改変体(Foesterら、J.Allergy Clin.Immunol.95:1229−1235(1995))をコードする以下の遺伝子を、化学的な遺伝子合成によって構築する。
【0190】
【化13】
FOS二量体化ドメインのN末端へのPLAの融合のために、その領域を、オリゴヌクレオチドPLA−FOR1(CCATCATCTACCCAGGTAC)(配列番号45)およびPLA−REV1(CCCACACCCAGCGGCCGCGTATTTGCGCAGGTCG)(配列番号46)を使用して増幅する。そのPCR産物を、NotIで切断し、そして制限酵素StuI/NotIで事前に切断したベクターpAVIに連結する。FOS二量体化ドメインのC末端へのPLAの融合のために、その領域をオリゴヌクレオチドPLA−FOR2(CGGTGGTTCTGCGGCCGCTATCATCTACCCAGGTAC)(配列番号47)およびPLA−REV2(TTAGTATTTGCGCAGGTCG)(配列番号48)を使用して増幅する。そのPCR産物を、NotIで切断し、そして制限酵素NotI/EcoRVで事前に切断したベクターpAV2に連結する。
【0191】
(実施例10)
(FOS−オボアルブミン融合遺伝子(N末端およびC末端)の構築)
オボアルブミンコード配列のクローニングのために、ニワトリ卵管組織由来のmRNAを、QuickPrepTM Micro mRNA Purification Kit(Pharmacia)を用いて、製造業者の説明書に従って使用して、調製する。SuperScriptTM One−step RT PCR Kit(Gibco BRL)を使用して、オボアルブミンの成熟部分をコードするcDNA(そのmRNAのヌクレオチド68〜1222に対応する(McReynoldsら、Nature 273:723−728(1978))を、プライマーOva−FOR1(CCGGCTCCATCGGTGCAG)(配列番号49)およびOva−REV1(ACCACCAGAAGCGGCCGCAGGGGAAACACATCTGCC)(配列番号50)を使用して合成する。このPCR産物をNotIで消化し、そしてC末端でFOS二量体化ドメインを有する融合タンパク質の発現のために、StuI/NotI消化したベクターpAV1にクローニングする。N末端でFOS二量体化ドメインを有する融合タンパク質の産生のために、オボアルブミンコード領域を、プライマーOva−FOR2(CGGTGGTTCTGCGGCCGCTGGCTCCATCGGTGCAG)(配列番号51)およびOva−REV2(TTAAGGGGAAACACATCTGCC)(配列番号52)を使用して、その構築したベクター(pAV::Ova)から増幅させる。このPCR産物をNotIで消化し、NotI/EcoRV消化したベクターpAVにクローニングする。このクローニングしたフラグメントを、DNA配列分析によって確認する。
【0192】
(実施例11)
(FOS−PLAおよびFOSアルブミン融合タンパク質の産生および精製)
FOS融合タンパク質の細胞質内産生のために、適切なE.coli株を、ベクターpAV3::PLA、pAV4::PLA、pAV3::OvaまたはpAV4::Ovaで形質転換した。この培養物を、アンピシリン存在下の富化培地中で、37℃で振盪しながらインキュベートした。吸光度(550nm)1の時点で、1mM IPTGを添加し、そしてインキュベーションをさらに5時間続けた。この細胞を遠心分離によって収集し、DNase、RNaseおよびリゾチームを含む適切な緩衝液(例えば、トリス−HCl、pH7.2、150mM NaCl)中に再懸濁し、そしてフレンチプレッシャーセルを通過させて破壊した。遠心分離(Sorvall RC−5C、SS34ローター、15000rpm、10分、4℃)後、ペレットを、4℃で25mlの封入体洗浄緩衝液(20mM トリス−HCl、23%スクロース、0.5% Triton X−100、1mM EDTA、pH8)中に再懸濁し、上記のように再度遠心分離した。この手順を、遠心分離後の上清が実質的に清澄になるまで繰り返した。封入体を、室温で20mlの可溶化緩衝液(5.5M 塩酸グアニジウム、25mM トリス−HCl、pH7.5)中に再懸濁し、そして不溶性物質を、遠心分離、およびその後、上清を滅菌フィルター(0.45μm)に通過させることによって、除去した。タンパク質溶液を、10mM EDTAおよび100mM DTTの存在下で少なくとも10時間、4℃で維持し、次いで10容量の5.5M 塩酸グアニジウム、25mM トリス−HCl、10mM EDTA、pH6に対して3回透析した。この溶液を、適切な酸化還元混合物(redox shuffle)(酸化型グルタチオン/還元型グルタチオン;シスチン/システイン)の存在下の5リットルの2M 尿素、4mM EDTA、0.1M NH4Cl、20mM ホウ酸ナトリウム(pH8.3)に対して2回透析した。次いで、この再折り畳みされたタンパク質を、イオン交換クロマトグラフィーにアプライした。このタンパク質を、2〜10mM DTTの存在下の7より大きいpHを有する適切な適切な緩衝液中で保存し、FOSドメインに隣接するシステイン残基を還元形態に維持した。タンパク質のアルファウイルス粒子との結合の前に、このタンパク質溶液をSephadex G−25ゲル濾過カラムに通すことによって、DTTを除去した。
【0193】
(実施例12)
(gp140−FOSの構築)
内部プロテアーゼ切断部位を有さないgp140遺伝子(Swiss−Prot:P03375)を、以下のオリゴヌクレオチドを使用して、全長gp160遺伝子を含む元のプラスミドpAbT4674(ATCC40829)から、PCRによって増幅させた:
【0194】
【化14】
PCR Iについて、100pmolのオリゴHIV−1およびHIV−Cleav2ならびに5ngのテンプレートDNAを、75μlの反応混合物(4ユニットのTaqまたはPwoポリメラーゼ、0.1mM dNTPおよび1.5mM MgSO4)中で使用した。PCRサイクリングを以下の様式で行った:60℃のアニーリング温度および72℃で2分間の伸長時間での30サイクル。
【0195】
PCR IIについて、100pmolのオリゴHIV−endおよびHIV−Cleavならびに5ngのテンプレートDNAを、75μlの反応混合物(4ユニットのTaqまたはPwoポリメラーゼ、0.1mM dNTPおよび1.5mM MgSO4)中で使用した。PCRサイクリングを以下の様式で行った:60℃のアニーリング温度および72℃で50秒間の伸長時間での30サイクル。
【0196】
両方のPCRフラグメントを精製し、単離し、そしてアセンブリPCR反応系において使用した。アセンブリPCR反応について、100pmolのオリゴHIV−1およびHIV−endならびに2ngの各PCRフラグメント(PCR IおよびPCR II)を、75μlの反応(4ユニットのTaqまたはPwoポリメラーゼ、0.1mM dNTPおよび1.5mM MgSO4)中で使用した。PCRサイクリングを以下の様式で行った:60℃のアニーリング温度および72℃で2.5分間の伸長時間での30サイクル。このアセンブリPCR産物を、XbaIおよびNheIで消化した。FOS両親媒性ヘリックスを、gp−140のC末端にインフレームで融合した。
【0197】
FOS両親媒性ヘリックスドメインをコードするDNA配列を、以下のオリゴヌクレオチドを使用して、ベクターpJuFo(Crameri&Suter Gene 137:69(1993))からPCR増幅させた:
【0198】
【化15】
このPCR反応について、100pmolの各オリゴおよび5ngのテンプレートDNAを、75μlの反応混合物(4ユニットのTaqまたはPwoポリメラーゼ、0.1mM dNTPおよび1.5mM MgSO4)中で使用した。温度サイクリングを以下のように行った:95℃で2分間、その後の95℃(45秒)、60℃(30秒)、72℃(25秒)の5サイクル、そしてその後の95℃(45秒)、68℃(30秒)、72℃(20秒)の25サイクル。この得られたPCRフラグメントを、NheIおよびBsp120Lで消化した。
【0199】
GP140−FOSについての最終発現ベクターを、両PCRフラグメントのpSinRep5への3フラグメント連結で得た。この得られたベクターpSinRep5−GP140−FOSを、制限分析およびDNA配列決定によって評価した。
【0200】
GP140−FOSをまた、安定な誘導性GP140−FOS発現細胞株を得るために、XbaIおよびBsp120L消化したpCYTtsにクローニングした。
【0201】
(実施例13)
(pSinRep5−GP140FOSを使用するGP140FOSの発現)
RNaseを含まないベクター(1.0μg)(pSinRep5−GP140−FOS)および1.0μgのDHEB(Bredenbeekら、J.Virol.67:6439−6446(1993))を、制限消化によって直線化した。続いて、SP6インビトロ転写キット(InvitroGen,InvitrogenBV,NV Leek,NetherlandsによるInvitroscripCAP)を使用して、インビトロ転写を行った。この得られた5’キャップ化mRNAを、還元性アガロースゲル上で分析した。
【0202】
インビトロ転写したmRNA(5μg)を、Invitrogenのマニュアル(Sindbis Expression System,Invitrogen BV,Netherlands)に従って、BHK21細胞(ATCC:CCL10)内にエレクトロポレートした。37℃で10時間のインキュベーション後、FCS含有培地を、FCSを含まないHP−1培地で交換し、その後さらに10時間37℃でインキュベートした。この上清を収集し、そして実施例2の記載のように正確に、可溶性GP140−FOSの産生についてウェスタンブロット分析によって分析した。
【0203】
(実施例14)
(pCYTts−GP140FOSを使用するGP140FOSの発現)
pCYTts−GP140−FOS 20μgを、制限消化によって直線化した。この反応を、フェノール/クロロホルム抽出によって停止し、その後、この直線化DNAをイソプロパノール沈殿させた。この制限消化を、アガロースゲル電気泳動によって評価した。トランスフェクションのために、5.4μgの直線化pCYTtsGP140−FOSを、30μl H2O中の0.6μgの直線化pSV2Neoと混合し、そして30μlの1M CaCl2溶液を添加した。60μlのリン酸緩衝液(50mM HEPES、280mM NaCl、1.5mM Na2HPO4、pH7.05)の添加後、この溶液を5秒間ボルテックスし、その後、室温で25秒間インキュベートした。この溶液を、2% FCSを含む2mlのHP−1培地(2% FCS培地)に直ちに添加した。次いで、80%コンフルエントなBHK21細胞培養物の培地(6ウェルプレート)を、このDNA含有培地で置換した。CO2インキュベーター中、37℃で5時間のインキュベーション後、このDNA含有培地を除去し、そして2% FCS培地中の15%グリセロール(2mL)で置換した。このグリセロール含有培地を、30秒のインキュベーション段階の後で除去し、そしてこの細胞を、10% FCSを含む、5mlのHP−1培地でリンスすることによって洗浄した。最後に、10% FCSを含む、2mlの新鮮HP−1培地を添加した。
【0204】
安定にトランスフェクトされた細胞を選択し、CO2インキュベーター中、37℃で、選択培地(G418を補充したHP−1培地)中で増殖させた。この混合集団がコンフルエントに増殖された時に、この培養物を2つの皿に分割し、その後、37℃で12時間増殖させた。一方の細胞の皿を30℃に移動させて、可溶性GP140−FOSの発現を誘導した。もう一方の皿は、37℃で維持した。
【0205】
可溶性GP140−FOSの発現を、ウェスタンブロット分析によって決定した。培養培地(0.5ml)をメタノール/クロロホルム沈殿させ、そしてこのペレットを、SDS−PAGEサンプル緩衝液中に再懸濁した。サンプルを95℃で5分間加熱し、その後15%アクリルアミドゲルにアプライした。SDS−PAGE後、タンパク質を、BassおよびYang,Creighton,T.E.編、Protein Function:A Practical Approach、第2版、IRL Press,Oxford(1997)、29−55頁に記載のように、Protanニトロセルロース膜(Schleicher& Schuell,Germany)に転写した。この膜を、TBS(10×TBS 1リットルあたり:87.7g NaCl 66.1g 塩酸Trizma(Sigma)および9.7gのTrizma塩基(Sigma)、pH7.4)中の1%ウシアルブミン(Sigma)を用いて、室温で1時間ブロックし、その後、抗GP140抗体または抗Gp−160抗体と共に1時間インキュベートした。このブロットを、TBS−T(0.05% Tween 20を含むTBS)で10分間、3回洗浄し、アルカリホスファターゼ抗マウス/ウサギ/サル/ヒトIgG結合体と共に1時間インキュベートした。TBS−Tでの10分間の2回の洗浄およびTBSでの10分間の2回の洗浄後、アルカリホスファターゼ検出試薬(50μlのNBT溶液(70%ジメチルホルムアミド中の7.7% Nitro Blue Tetrazolium(Sigma))および37μlのX−Phosphate溶液(ジメチルホルムアミド中の5%の5−ブロモ−4−クロロ−3−インドリルホスフェート)を含む10mlのAP緩衝液(100mM Tris/HCl、100mM NaCl、pH9.5))を使用して、発色反応を行った。
【0206】
(実施例15)
(GP140FOSの産生および精製)
抗gp120抗体を、NHS/EDC活性化デキストランに共有結合させ、そしてクロマトグラフィーカラムに充填した。GP140FOSを含む上清を、このカラム上にロードし、そして十分な洗浄後、GP140FOSを、0.1M HClを使用して溶出した。この溶出物を、収集チューブ中の1M Tris pH7.2を用いて、収集中に直接中和した。
【0207】
ジスルフィド結合形成は、精製の間に生じ得、従って、この収集したサンプルを、10mM Tris、pH7.3中の10mM DTTで、25℃で2時間処理した。
【0208】
DTTを、10mM Mes;80mM NaCl、pH6.0に対する、その後の透析によって除去した。最後に、Gp140FOSを、実施例16の記載のように、E2においてJUNロイシンジッパーを含むアルファウイルス粒子と混合した。
【0209】
(実施例16)
(アルファワクチン粒子の調製)
100kDの分子量カットオフを有するMillipore Ultrafree Centrifugal Filter Devicesを、製造業者によって供給されるプロトコルに従って使用して、ウイルス粒子(実施例2および3を参照のこと)を濃縮した。あるいは、ウイルス粒子を、Sindbis Expression System(Invitrogen,San Diego,California)の説明マニュアルに記載のように、スクロース密度勾配遠心によって濃縮した。ウイルス懸濁液のpHを7.5に調整し、そしてウイルス粒子を、2〜10mM DTTの存在下で数時間インキュベートした。ウイルス粒子を、適切な緩衝液のSephacryl S−300カラム(Pharmacia)(ウイルス粒子を空隙容量で溶出する)上で、混在タンパク質から精製した。
【0210】
精製したウイルス粒子を、適切な酸化還元混合物(redox shuffle)(酸化型グルタチオン/還元型グルタチオン;シスチン/システイン)の存在下の適切な緩衝液(pH7.5〜8.5)中で、少なくとも240倍モル過剰のFOS−抗原融合タンパク質と共に、4℃で少なくとも10時間インキュベートした。100kDの分子量カットオフを有するMillipore Ultrafree Centrifugal Filter Deviceを使用して、この粒子を濃縮した後、この混合物を、Sephacryl S−300ゲル濾過カラム(Pharmacia)に通した。ウイルス粒子を、空隙容量で溶出させた。
【0211】
(実施例17:HBcAg(1〜144)のアミノ末端へのJUN両親媒性ヘリックスの融合)
JUNヘリックスを、HBcAgアミノ酸配列1〜144のアミノ末端に融合した(JUN−HBcAg構築物)。JUN−HBcAg DNA配列を構築するために、JUNヘリックスおよびHBcAg(1〜144)をコードする配列を、PCRにより別々に増幅した。JUN配列を、プライマーEcoRI−JUN(s)およびJUN−SacII(as)を用いて、pJuFoプラスミドから増幅した。EcoRI−JUN(s)プライマーには、EcoRI部位、次いで開始ATGコドンを導入した。JUN−SacII(as)プライマーには、アミノ酸配列GAAGSをコードするリンカーを導入した。HBcAg(1〜144)配列を、プライマーJUN−HBcAg(s)およびHBcAg(1〜144)Hind(as)を用いて、pEco63プラスミド(ATCC番号31518から得た)から増幅した。JUN−HBcAg(s)は、JUNヘリックスをコードする配列の3’末端に対応する配列、続いてGAAGSリンカーをコードする配列およびHBcAg配列の5’末端を含む。HBcAg(1〜144)Hind(as)に、HBcAg遺伝子の後ろに終止コドンおよびHindIII部位を導入する。PCR反応については、100pmolの各オリゴおよび50ngのテンプレートDNAを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含有する50μlの反応混合物において使用する。両方の反応については、温度サイクリングを以下のとおりに行った:94℃で2分間、および94℃(1分間)、50℃(1分間)、72℃(2分間)を30サイクル。
【0212】
【化16】
2つのPCRフラグメントの融合を、プライマーEcoRI−JUN(s)およびHBcAg(1〜144)Hind(as)を用いて、PCRにより行った。100pmolの各オリゴを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含有する50μlの反応混合物中、100ngの精製PCRフラグメントとともに用いた。PCRサイクリング条件は、以下であった:94℃で2分間、および94℃(1分間)、50℃(1分間)、72℃(2分間)を35サイクル。最終的なPCR産物を、アガロースゲル電気泳動により分析し、精製し、そしてEcoRIおよびHindIII制限酵素を含む適切な緩衝液中で16時間にわたり消化した。消化したDNAフラグメントをEcoRI/HindIII消化pKKベクターに連結して、pKK−JUN−HBcAg発現ベクターを生成した。PCR産物の挿入を、EcoRI/HindIII制限分析によりおよびインサートのDNA配列決定により分析した。
【0213】
(実施例18:HBcAg(1〜144)のカルボキシ末端へのJUN両親媒性ヘリックスの融合)
JUNヘリックスを、HBcAgアミノ酸配列1〜144に融合した(HBcAg−JUN構築物)。HBcAg−JUN DNA配列を構築するために、JUNヘリックスおよびHBcAg(1〜144)をコードする配列を、PCRにより別々に増幅した。JUN配列を、プライマーSacII−JUN(s)およびJUN−HindIII(as)を用いてpJuFoプラスミドから増幅した。SacII−JUN(s)には、アミノ酸LAAGをコードするリンカーを導入した。この配列はまた、SacII部位を含む。JUN−HindIII(as)には、終止コドン(TAA)、次にHindIII部位を導入した。HBcAg(1〜144)DNA配列を、プライマーEcoRI−HBcAg(s)およびHBcAg(1〜144)−JUN(as)を用いて、pEco63プラスミドから増幅した。EcoRI−HBcAg(s)には、HBcAgコード配列の開始ATGの前に、EcoRI部位を導入した。HBcAg(1〜144)−JUN(as)には、ペプチドリンカー(LAAG)をコードする配列を導入する。これはまた、SacII部位を含む。PCR反応については、100pmolの各オリゴおよび50ngのテンプレートDNAを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含有する50μlの反応混合物において使用した。温度サイクリングを以下のとおりに行った:94℃で2分間、および94℃(1分間)、50℃(1分間)、72℃(2分間)を30サイクル。
【0214】
【化17】
2つのPCRフラグメントの融合を、プライマーEcoRI−HBcAg(s)およびJUN−HindIII(as)を用いて、PCRにより行った。PCR融合については、100pmolの各オリゴを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含有する50μlの反応混合物中の、100ngの精製PCRフラグメントとともに用いた。PCRサイクリング条件は、以下であった:94℃で2分間;および94℃(1分間)、50℃(1分間)、72℃(2分間)を35サイクル。最終的なPCR産物を、アガロースゲル電気泳動により分析し、そしてEcoRIおよびHindIII制限酵素を含む適切な緩衝液中で16時間にわたり消化した。DNAフラグメントをゲル精製し、そしてEcoRI/HindIII消化pKKベクターに連結して、pKK−HBcAg−JUN発現ベクターを生成した。PCR産物の挿入を、EcoRI/HindIII制限分析によりおよびインサートのDNA配列決定により分析した。
【0215】
(実施例19:HBcAg(1〜144)のc/e1エピトープへのJUN両親媒性ヘリックスの挿入)
HBcAgのc/e1エピトープ(残基72〜88)は、B型肝炎ウイルスキャプシドの表面上の先端領域に位置することが公知である。タンパク質のこの領域の一部(残基76〜82)は、JUNヘリックスによって遺伝的に置換されて、抗原(HBcAg−JUNIns構築物)についての付着部位を提供した。このHBcAg−JUNIns DNA配列を、PCRにより生成した:JUNヘリックス配列および2つのHBcAgフラグメント(アミノ酸残基1〜75および83〜144)をコードする配列を、PCRにより別々に増幅した。JUN配列を、プライマーBamHI−JUN(s)およびJUN−SacII(as)を用いてpJuFoプラスミドから増幅した。BamHI−JUN(s)には、ペプチド配列GSGGGをコードするリンカー配列(BamHI部位もまた含む)を導入した。JUN−SacII(as)には、ペプチドリンカー(GAAGS)をコードする配列、次いで、JUNコード配列の3’末端に相補的な配列を導入した。HBcAg(1〜75)DNA配列を、プライマーEcoRIHBcAg(s)およびHBcAg75−JUN(as)を用いて、pEco63プラスミドから増幅した。EcoRIHBcAg(s)には、EcoRI部位、次いで、HBcAg配列の5’末端に対応する配列を導入した。HBcAg75−JUN(as)には、HBcAGのアミノ酸75の後にペプチドGSGGGをコードするリンカー、次いで、JUNヘリックスをコードする配列の5’末端に相補的な配列を導入した。HBcAg(83〜144)フラグメントを、プライマーJUN−HBcAg83(s)およびHBcAg(1〜144)Hind(as)を用いて増幅した。JUN−HBcAg83(s)は、JUNコード配列の3’末端に対応する配列、次いで、ペプチドGAAGSをコードするリンカー、およびHBcAg(83〜144)をコードする配列の5’末端に対応する配列を含んでいた。HBcAg(1〜144)Hind(as)には、HBcAG遺伝子のコドン144の後ろに、終止コドンおよびHindIII部位を導入した。PCR反応については、100pmolの各オリゴおよび50ngのテンプレートDNAを、50μlの反応混合物(2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4)において使用した。温度サイクリングを以下のとおりに行った:94℃で2分間、および94℃(1分間)、50℃(1分間)、72℃(2分間)を35サイクル。
【0216】
【化18】
3つのPCRフラグメントの融合を、以下のとおりに行った。最初に、HBcAg1〜75をコードするフラグメントを、プライマーEcoRIHBcAg(s)およびJUN−SacII(as)を用いて、PCRにより、JUNをコードする配列と融合した。第2に、得られた産物を、プライマーEcoRIHBcAg(s)およびHBcAgHindIII(as)を用いて、PCRにより、HBcAg(83〜144)フラグメントと融合した。PCR融合に関しては、100pmolの各オリゴを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含有する50μlの反応混合物中、100ngの精製PCRフラグメントとともに用いた。同じPCRサイクルを、個々のフラグメントの生成と同じように用いた。最終的なPCR産物を、EcoRIおよびHindIII制限酵素を含有する適切な緩衝液中で16時間消化した。DNAフラグメントをEcoRI/HindIIIで消化したpKKベクターに連結して、pKK−HBcAg−JUNInsベクターを得た。PCR産物の挿入を、EcoRI/HindIII制限分析およびインサートのDNA配列決定により分析した。
【0217】
(実施例20:麻疹ウイルスヌクレオキャプシド(N)タンパク質のカルボキシ末端へのJUN両親媒性ヘリックスの融合)
JUNヘリックスを、アミノ酸残基1〜473を含む短縮化麻疹ウイルスNタンパク質フラグメントのカルボキシ末端に融合した(N473−JUN構築物)。N473−JUNをコードするDNA配列を構築するために、JUNヘリックスをコードする配列およびN473−JUNをコードする配列を、PCRにより別々に増幅した。JUN配列を、プライマーSacII−JUN(s)およびJUN−HindIII(as)を用いて、pJuFoプラスミドから増幅した。SacII−JUN(s)には、ペプチドリンカーLAAGをコードする配列を導入した。この配列はまた、SacII部位を含んでいた。JUN−HindIII(as)アンチセンスプライマーには、終止コドン、次いで、HindIII部位を導入した。N(1〜473)配列を、プライマーEcoRI−Nmea(s)およびNmea−JUN(as)を用いて、完全な麻疹ウイルスNタンパク質コード配列を含むpSC−Nプラスミド(M.Billeter,Zurichから得た)から増幅した。EcoRI−N(mea)(s)には、Nコード配列の開始ATGの前にEcoRI部位を導入した。N(mea)−JUN(as)は、N(1〜473)コード配列の3’末端に相補的であり、次は、ペプチドリンカー(LAAG)についてのコード配列に相補的な配列であった。PCR反応に関しては、100pmolの各オリゴおよび50ngのテンプレートDNAを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含有する50μlの反応混合物中で用いた。温度サイクリングを以下のとおりに行った:94℃で2分間;および94℃(1分間)、55℃(1分間)、72℃(2分間)を35サイクル。
【0218】
【化19】
2つのPCRフラグメントの融合を、プライマーEcoRI−Nmea(s)およびNmea−JUN(as)を用いて、さらにPCRにおいて行った。PCR融合に関しては、100pmolの各オリゴを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含有する50μlの反応混合物中、100ngの精製PCRフラグメントとともに用いた。温度サイクリングを、以下のとおりに行った:94℃で2分間;および94℃(1分間)、50℃(1分間)、72℃(2分間)を35サイクル。PCR産物を、EcoRIおよびHindIII制限酵素を含有する適切な緩衝液中で16時間消化した。DNAフラグメントをゲル精製し、そしてEcoRI/HindIIIで消化したpKKベクターに連結して、pKK−N473−JUNプラスミドを得た。PCR産物の挿入を、EcoRI/HindIII制限分析およびインサートのDNA配列決定により分析した。
【0219】
(実施例21)
(HBcAg−JUNの発現および部分的精製)
E.coli株XL−1 blueを、pKK−HBcAg−JUNで形質転換した。細菌の一晩の培養物1mlを使用して、100μg/mlのアンピシリンを含む100mlのLB培地に接種(innoculate)した。この培養物を、600nmでおよそ0.8のODに達するまで、37℃で4時間増殖させた。HBcAg−JUNの合成の誘導を、1mMの最終濃度になるようにIPTGを添加することにより行った。誘導後、細菌を、37℃で16時間さらに振蕩した。細菌を、5000×gでの15分間の遠心分離によって回収した。このペレットを、−20℃で凍結した。このペレットを融解し、そして200μg/mlリゾチームおよび10μlのBenzonase(Merck)を補充した細菌溶解緩衝液(10mM Na2HPO4、pH7.0、30mM NaCl、0.25% Tween−20、10mM EDTA、10mM DTT)中に再懸濁した。細胞を、室温で30分間インキュベートし、そしてFrench pressure cellを使用して破壊した。Triton X−100を、溶解物に添加して、0.2%の最終濃度にして、そしてこの溶解物を、氷上で30分間インキュベートし、そして時々振蕩した。図4は、IPTGでの誘導の際のE.coliにおけるHBcAg−JUNタンパク質発現を示す。pKK−HBcAg−JUN発現プラスミドまたはコントロールプラスミドを保有するE.coli細胞を、IPTGによるHBcAg−JUN発現の誘導のために使用した。IPTGの添加前に、サンプルを、pKK−HBcAg−JUNプラスミドを保有する細菌培養物(レーン3)から、およびコントロールプラスミドを保有する培養物(レーン1)から取り除いた。IPTG添加の16時間後に、サンプルを再度、pKK−HBcAg−JUNを含む培養物(レーン4)から、およびコントロール培養物(レーン2)から除去した。タンパク質発現を、SDS−PAGE、続いてクーマシー染色によってモニターした。
【0220】
次いで、この溶解物を、不溶性細胞片を除去するために、12,000×gにて30分間遠心分離した。この上清およびペレットを、HBcAgに対するモノクローナル抗体(YVS1841、Accurate Chemical and Scientific Corp.,Westbury,NY,USAから購入)を使用して、ウエスタンブロッティングにより分析した。これは、有意な量のHBcAg−JUNタンパク質が可溶性であることを示した(図5)。簡潔には、HBcAg−JUNを発現しているE.coli細胞からの溶解物、およびコントロール細胞からの溶解物を、14,000×gで30分間遠心分離した。上清(=可溶性画分)およびペレット(=不溶性画分)を分離し、そしてSDSサンプル緩衝液で希釈して等しい容量にした。サンプルを、SDS−PAGE、続いて抗HBcAgモノクローナル抗体YVS1841でのウエスタンブロッティングによって分析した。レーン1:可溶性画分、コントロール細胞;レーン2:不溶性画分、コントロール細胞;レーン3:可溶性画分、HBcAg−JUNを発現している細胞;レーン4:不溶性画分、HbcAg−JUNを発現している細胞。
【0221】
清澄化した細胞溶解物を、3mlの15%スクロース溶液、続いて4mlの細菌溶解物を重層した4mlの65%スクロース溶液からなる、スクロース段階的勾配を用いる、段階的勾配遠心分離に使用した。このサンプルを、100,000×gにて4℃で3時間遠心分離した。遠心分離後、この勾配の頂端から1mlの画分を収集し、そしてSDS−PAGE、続いてクーマシー染色によって分析した(図6)。レーン1:遠心分離前の総E.coli溶解物、レーン1および2:勾配の頂端からの画分1および2、レーン4〜7:画分5〜8(15%スクロース)。HBcAg−JUNタンパク質を、クーマシー染色によって検出した。
【0222】
HBcAg−JUNタンパク質を、15%スクロースと65%スクロースとの間の界面に濃縮させた。このことは、このタンパク質がカプシド粒子を形成したことを示す。ほとんどの細菌タンパク質は、この勾配のスクロースを含まない上層に残存し、従って、HBcAG−JUN粒子の段階的勾配遠心分離は、この粒子を濃縮すること、および部分精製することの両方を導く。
【0223】
(実施例22)
(HBcAg−JUNへのhGH−FOSの共有結合)
HBcAg−JUN粒子へのタンパク質を結合の実証するために、本発明者らは、そのカルボキシ末端がFOSヘリックスに融合したヒト成長ホルモン(hGH)を、モデルタンパク質(hGH−FOS)として選択した。HBcAg−JUN粒子を、部分的に精製されたhGH−FOSと混合し、そして4℃で4時間インキュベートし、このタンパク質の結合を可能にした。次いで、この混合物を、HBcAg−JUN溶液およびhGH−FOS溶液の両方に存在するDTTを除去するために、3000倍容量の透析緩衝液(150mM NaCl、10mM Tris−HCl溶液(pH8.0))に対して一晩透析し、それによって、ジスルフィド結合の確立を通じて、タンパク質の共有結合を可能にする。コントロールとして、HBcAg−JUN溶液およびhGH−FOS溶液もまた、透析緩衝液に対して透析した。3つ全ての透析したタンパク質溶液からのサンプルを、非還元条件下でSDS−PAGEによって分析した。HBcAg−JUNへのhGH−FOSの結合を、抗hGHイムノブロットにおいて検出した(図7)。HBcAg−JUNに結合したhGH−FOSは、およそ53kDaの見かけの分子量で移動するはずであり、一方、非結合hGH−FOSは、31kDaの見かけの分子量で移動する。この透析液を、還元剤の非存在下(レーン3)、および還元剤の存在下(レーン2)でのSDS−PAGEにより分析し、そしてクーマシー染色によって検出した。コントロールとして、カプシド粒子と混合されていないhGH−FOSもまた、還元剤の存在下でゲルにロードした(レーン1)。
【0224】
およそ53kDaの分子量へのhGH−FOSの変動を、HBcAg−JUNカプシドタンパク質の存在下で観察した。このことは、HBcAg−JUNへのhGH−FOSの効率的な結合が生じたことを示唆する。
【0225】
(実施例23)
(HBcAg(1〜149)のc/e1エピトープへのリジン残基を含むペプチドの挿入)
HBcAgのc/e1エピトープ(残基72〜88)を、B型肝炎ウイルスカプシド(HBcAg)の表面上の先端領域(tip region)に配置した。この領域の一部(プロリン79およびアラニン80)を、ペプチドGly−Gly−Lys−Gly−Glyによって遺伝的に置換した(HBcAg−Lys構築物)。この導入されたリジン残基は、遊離システイン基を含む任意の抗原を有するHBcAg粒子の分子間化学的架橋のために使用され得る反応性アミノ基を、その側鎖に含む。
【0226】
HBcAg−Lys DNA配列を、PCRによって作製した:HBcAgフラグメントをコードする2つのフラグメント(アミノ酸残基1〜78および81〜149)を、PCRによって別々に増幅した。これらのPCRに使用されたプライマーもまた、Gly−Gly−Lys−Gly−GlyペプチドをコードするDNA配列を導入した。HBcAg(1〜78)フラグメントを、プライマーであるEcoRI HBcAg(s)およびLys−HBcAg(as)を使用して、pEco63から増幅した。HBcAg(81〜149)フラグメントを、プライマーであるLys−HBcAg(s)およびHBcAg(1〜149)Hind(as)を使用して、pEco63から増幅した。プライマーであるLys−HBcAg(as)およびLys−HBcAg(s)は、相補性DNA配列を、2つのPCR産物の末端に導入し、このことは、その後のアセンブリ(assembly)PCRにおいて2つのPCR産物の融合を可能にした。アセンブリしたフラグメントを、プライマーEcoRI HBcAg(s)およびHbcAg(1〜149)Hind(as)を使用して、PCRによって増幅した。
【0227】
PCRについては、100pmolの各オリゴおよび50ngのテンプレートDNAを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含む50μlの反応混合物において使用した。両方の反応について、温度のサイクリングを、以下の通りに行った:94℃で2分間;94℃(1分)、50℃(1分)、72℃(2分)を30サイクル。
【0228】
(プライマー配列)
【0229】
【化20】
PCRによる2つのPCRフラグメントの融合のために、100pmolのプライマーであるEcoRI HBcAg(s)およびHBcAg(1〜149)Hind(as)を、100ngの2つの精製されたPCRフラグメントとともに、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含む、50μlの反応混合物中で使用した。PCRサイクル条件は、以下であった:94℃で2分間;94℃(1分)、50℃(1分)、72℃(2分)を30サイクル。アセンブリされたPCR産物を、アガロースゲル電気泳動によって分析し、精製し、そしてEcoRI制限酵素およびHindIII制限酵素によって、適切な緩衝液中で19時間消化した。この消化されたDNAフラグメントを、EcoRI/HindIIIで消化したpKKベクター中に連結して、pKK−HBcAg−Lys発現ベクターを作製した。このベクターへのPCR産物の挿入を、EcoRI/HindIII制限分析およびこのインサートのDNA配列決定によって分析した。
【0230】
(実施例24)
(HBcAg−Lysの発現および部分精製)
E.coli株XL−1 blueを、pKK−HBcAg−Lysで形質転換した。細菌の一晩の培養物1mlを使用して、100μg/mlのアンピシリンを含む100mlのLB培地に接種した。この培養物を、600nmでおよそ0.8のODに達するまで、37℃で4時間増殖させた。HBcAg−Lysの合成の誘導を、1mMの最終濃度になるようにIPTGを添加することにより行った。誘導後、細菌を、37℃で16時間さらに振蕩した。細菌を、5000×gでの15分間の遠心分離によって回収した。このペレットを、−20℃で凍結した。このペレットを融解し、そして200μg/mlリゾチームおよび10μlのBenzonase(Merck)を補充した細菌溶解緩衝液(10mM Na2HPO4、pH7.0、30mM NaCl、0.25% Tween−20、10mM EDTA、10mM DTT)中に再懸濁した。細胞を、室温で30分間インキュベートし、そしてFrench pressure cellを使用して破壊した。Triton X−100を、溶解物に添加し、0.2%の最終濃度にして、そしてこの溶解物を、氷上で30分間インキュベートし、そして時々振蕩した。pKK−HBcAg−Lys発現プラスミドまたはコントロールプラスミドを保有するE.coli細胞を、IPTGによるHBcAg−Lys発現の誘導のために使用した。IPTGの添加前に、サンプルを、pKK−HBcAg−Lysプラスミドを保有する細菌培養物から、およびコントロールプラスミドを保有する培養物から取り除いた。IPTG添加の16時間後に、サンプルを再度、pKK−HBcAg−Lysを含む培養物から、およびコントロール培養物から除去した。タンパク質発現を、SDS−PAGE、続くクーマシー染色によってモニターした。
【0231】
次いで、この溶解物を、不溶性細胞片を除去するために、12,000×gにて30分間遠心分離した。この上清およびペレットを、HBcAgに対するモノクローナル抗体(YVS1841、Accurate Chemical and Scientific Corp.,Westbury,NY,USAから購入)を使用して、ウエスタンブロッティングにより分析した。これは、有意な量のHBcAg−Lysタンパク質が可溶性であることを示す(図5)。簡潔には、HBcAg−Lysを発現しているE.coli細胞からの溶解物、およびコントロール細胞からの溶解物を、14,000×gで30分間遠心分離した。上清(=可溶性画分)およびぺレット(=不溶性画分)を分離し、そしてSDSサンプル緩衝液で希釈して等しい容量にした。サンプルを、SDS−PAGE、続いて抗HBcAgモノクローナル抗体YVS1841でのウエスタンブロッティングによって分析した。
【0232】
清澄化した細胞溶解物を、3mlの15%スクロース溶液、続いて4mlの細菌溶解物を重層した4mlの65%スクロース溶液からなる、スクロース段階的勾配を用いる、段階的勾配遠心分離に使用した。このサンプルを、100,000×gにて4℃で3時間遠心分離した。遠心分離後、勾配の頂端から1mlの画分を収集し、そしてSDS−PAGE、続いてクーマシー染色によって分析した(図6)。HBcAg−Lysタンパク質を、クーマシー染色によって検出した。
【0233】
HBcAg−Lysタンパク質を、15%スクロースと65%スクロースとの間の界面に濃縮させた。このことは、このタンパク質がカプシド粒子を形成したことを示す。ほとんどの細菌タンパク質は、この勾配のスクロースを含まない上層に残存し、従って、HBcAg−Lys粒子の段階的勾配遠心分離は、この粒子を濃縮すること、および部分精製することの両方を導く。
【0234】
(実施例25)
(異種二機能性架橋剤SPDPを用いるFLAGペプチドのHBcAg−Lysへの化学的結合)
アミノ末端にシステイン残基を有する合成FLAGペプチド(アミノ酸配列CGGDYKDDDDK)を、FLAGペプチドに対する免疫応答を誘発するために、精製されたHBcAg−Lys粒子に化学的に結合した。HBcAg−Lys粒子の純度95%の溶液(2mg/ml) 600μlを、異種二機能性架橋剤N−Succinimidyl 3−(2−pyridyldithio)propionate(SPDP)(0.5mM)と共に30分間室温でインキュベートした。反応の終了後に、遊離のSPDPを除くために、混合物を、1リットルの50mM リン酸緩衝液(pH7.2)(150mM NaClを有する)に対して一晩透析した。次いで、500μlの誘導体化HBcAg−Lysカプシド(2mg/ml)を、金属触媒スフィドリル(sufhydryl)酸化を防ぐために10mM EDTAの存在下で0.1mM FLAGペプチド(アミノ末端システインを含む)と混合した。この反応を、ペプチドの遊離システインとの反応における、SPDPからのピリミジン−2−チオン(thione)の遊離に起因する343nMでの溶液の吸光度の増加により、モニターした。誘導体化Lys残基のペプチドとの反応は、約30分後に終了した。
【0235】
FLAG装飾された粒子を、マウスに注射した。
【0236】
(実施例26)
(pMPSV−gp140cysの構築)
gp140遺伝子を、オリゴgp140CysEcoRIおよびSalIgp140を用いて、pCytTSgp140FOSからPCRによって増幅した。PCRに関して、100pmolのそれぞれのオリゴおよび50ngの鋳型DNAを、50μlの反応混合液(2ユニットのPwoポリメラーゼ、0.1mMのdNTPおよび2mMのMgSO4を含む)中で使用した。両方の反応に対して、温度サイクルを以下のように実施した:94℃を2分間;94℃(0.5分間)、55度(0.5分間)、72℃(2分間)を30サイクル。
【0237】
PCR産物を、QiaEXIIキットを用いて精製し、SalI/EcoRIで消化し、そして同一の酵素で切断したベクターpMPSVHE中に連結した。(オリゴ配列)
【0238】
【化21】
(実施例27)
(pMPSVgp140Cysの発現)
pMPSVgp140Cys(20μg)を、制限消化によって直線化した。反応をフェノール/クロロホルム抽出によって停止し、その後直線化DNAをイソプロパノール沈殿する。制限消化を、アガロースゲル電気泳動により評価した。トランスフェクションのために、5.4μgの直線化pMPSVgp140−Cysを、30μlの水中で0.6μgの直線化pSV2Neoと混合し、そして30μlの1M CaCl2溶液を添加した。60μlのリン酸緩衝液(50mM HEPES、280mM NaCl、1.5mM Na2HPO4(pH7.05))の添加後に、この溶液を5秒間ボルテックスし、続いて室温で25秒間インキュベーションした。この溶液に、2%FCSを含むHP−1培地(2%FCS培地)を2ml、直ちに添加した。次いで、80%コンフルエントなBHK21細胞培養(6ウェルプレート)の培地を、DNA含有培地に置き換えた。37℃で5時間、CO2インキュベーター内でのインキュベートの後、DNA含有培地を取り出し、2mlの15%グリセロール含有2%FCS培地で置き換えた。グリセロール含有培地を、30秒間のインキュベート相の後に取り出し、そしてこの細胞を、5mlの10%FCS含有HP−1培地でのリンスにより洗浄した。最後に、2mlの新鮮なHP−1培地(10%FCSを含む)を加えた。
【0239】
安定にトランスフェクトされた細胞を選択し、そしてCO2インキュベーター内で、選択培地(G418を補充したHP−1培地)中で37℃で増殖した。混合集団がコンフルエントまで増殖したとき、培養物を2つのディッシュに分配し、その後、37℃で12時間の増殖期間を続ける。細胞の一方のディッシュを30℃に移動し、可溶性GP140−FOSの発現を誘導させた。他方のディッシュを37℃に維持した。
【0240】
可溶性GP140−Cysの発現を、ウエスタンブロット分析によって解析した。培養培地(0.5ml)を、メタノール/クロロホルム沈殿し、そしてこのペレットをSDS−PAGEサンプル緩衝液に再懸濁した。サンプルを、95℃で5分間加熱し、その後、15%アクリルアミドゲルにアプライした。SDS−PAGE後、BassおよびYang(Creighton,T.E.編、Protein Function:A Practical Approach、第2版、IRL Press、Oxford(1997)、29−55頁)に記載されたように、タンパク質をProtan nitrocellilose membrane(Shleicher&Schuell、Germany)に転写した。この膜を、TBS(10×TBS 1リットルあたり、87.7g NaCl、66.1g Trizma hydrochloride(Sigma)および9.7g Trizma base(Sigma)、pH7.4)中に1%のウシアルブミン(Sigma)を用いて、室温で一時間ブロックした後、抗GP140抗体または抗GP160抗体を用いて一時間インキュベートした。このブロットを、TBS−T(0.05%のTween20を含むTBS)で10分間3回洗浄し、アルカリホスファターゼ抗マウス/ウサギ/サル/ヒトIgG結合体を用いて一時間インキュベートした。TBS−Tで10分間2回、およびTBSで10分間2回洗浄した後、アルカリホスファターゼ検出試薬(50μlのNBT溶液(70%のジメチルホルムアミド中に7.7% Nitro Blue Tetrazolium(Sigma))および37μlのX−Phosphate溶液(ジメチルホルムアミド中に5%の5−ブロモ−4−クロロ−3−インドリルリン酸)を含む10mlのAP緩衝液(100mM Tris/HCl、100mM NaCl、pH9.5)を用いて現像反応を行った。
【0241】
(実施例28)
(gp140Cysの精製)
抗gp120抗体を、NHS/EDC活性化デキストランと共有結合し、そしてクロマトグラフィーカラムに充填した。GP140Cysを含む上清をカラムにロードし、そして十分な洗浄後に、GP140Cysを、0.1M HClで溶出した。溶出物を、回収チューブ中の1M Tris(pH7.2)を用いて回収中に直接中和した。
【0242】
精製中に、ジスフィルド結合形成が形成し得、したがって、回収サンプルを、10mMのDTT(10mM Tris(pH7.5)中)を用いて、25℃で2時間処理する。
【0243】
DTTを、10mM Mes;80mM NaCl(pH6.0)に対する引き続く分析によって除去する。最終的に、実施例16に記載されるように、GP140Cysを、E2中のJUN残基を含むアルファウイルス粒子と混合する。
【0244】
(実施例29)
(PLA2−Cysの構築)
PLA2遺伝子を、オリゴEcoRIPLAおよびオリゴPLA−Cys−hindを用いて、pAV3PLAfosからPCRによって増幅した。PCRについて、100pmolの各オリゴおよび50ngの鋳型DNAを、2ユニットのPwoポリメラーゼ、0.1mM dNTPおよび2mM MgSO4を含む50μlの反応混合液中で用いた。両反応について、温度サイクルを、以下のように行った:94℃で2分間;94℃(0.5分間)、55℃(0.5分間)、72℃(2分間)を30サイクル。
【0245】
PCR産物を、QiaEXIIキットを用いて精製し、EcoRI/HindIIIで消化し、そして同一の酵素で消化されたベクターpAV3に連結した。(オリゴ)
【0246】
【化22】
(実施例30)
(PLA−cysの発現および精製)
Cysタグ化タンパク質の細胞質内産生のために、E.coli XL−1−Blue株をベクターpAV3::PLAおよびpPLA−Cysを用いて形質転換した。培養物を、37℃で振盪しながら、アンピシリン存在下の豊富な培地中でインキュベートした。培養物の吸光度(550nm)で、1mMのIPTGを添加し、インキュベーションをさらに5時間続けた。細胞を遠心分離によって回収し、DNase、RNaseおよびリゾチームを含む適切な緩衝液(例えば、Tris−HCl(pH7.2)、150mM NaCl)中に再懸濁し、そしてフレンチプレス(french pressure cell)を通す通過によって破壊した。遠心分離(Sorvall RC−5C、SS34ローター、15,000rpmで4℃10分間)の後、ペレットを、4℃で25mlの封入体洗浄緩衝液(20mM tris−HCl、23% スクロース、0.5% Triton X−100、1mM EDTA(pH8))中に再懸濁し、そして上記のように再遠心分離した。この手順を、遠心分離後の上清が本質的に清澄になるまで反復した。封入体を、20mlの可溶化緩衝液(5.5M 塩酸グアニジン、25mM tris−HCl(pH7.5))中に、室温で再懸濁し、そして不溶性の物質を、遠心分離および引き続く滅菌フィルター(0.45μm)を通す上清の通過で除去した。タンパク質溶液を、10mM EDTAおよび100mM DTTの存在下で、4℃で少なくとも10時間維持し、次いで、10容量の5.5M 塩酸グアニジン、25mM tris−HCl、10mM EDTA(pH6)に対して3回透析した。この溶液を、適切な酸化還元シャッフル(shuffle)(酸化型グルタチオン/還元型グルタチオン;シスチン/システイン)の存在下で、512Mの尿素、4mM EDTA、0.1M NH4Cl、20mM ホウ酸ナトリウム(pH8.3)に対して2回透析した。次いで、再フォールディングタンパク質をイオン交換クロマトグラフィーにアプライした。このタンパク質を、2〜10mM DTT存在下で適切な緩衝液(pH7以上)中に保存し、システイン残基を還元型で保持した。このタンパク質とαウイルス粒子との結合の前に、DTTを、Sephadex G−25ゲル濾過カラムを通すタンパク質溶液の通過によって除去した。
【図面の簡単な説明】
【図1】 pCYTts::E2JUN発現ベクターを使用するE2−JUN融合タンパク質を含むウイルス粒子の産生を示すウエスタンブロット。
【図2】 pTE5’2J::E2JUN発現ベクターから発現されるE2−JUN融合タンパク質を含むウイルス粒子の産生を示すウエスタンブロット。
【図3】 FOS−hgh抗原の細菌発現および真核生物発現を示すウエスタンブロット。
【図4】 E.coli細胞におけるHBcAg−JUNの発現。
【図5】 HBcAg−JUNがE.coli溶解物に可溶性であることを示すウエスタンブロット。
【図6】 スクロース密度勾配におけるHBcAg−JUNキャプシド粒子の濃縮のSDS−PAGE分析。
【図7】 hGH−FOS粒子およびHBcAg−JUN粒子の結合の非還元SDS−PAGE分析。
【特許請求の範囲】
【請求項1】
以下を含む、組成物:
a)以下を含む、天然には存在しない分子足場:
(i)以下からなる群から選択されるコア粒子:
(1)非天然起源のコア粒子;および
(2)天然起源のコア粒子;ならびに
(ii)少なくとも1つの第1の付着部位を含む形成体であって、ここで、該形成体は、少なくとも1つの共有結合によって該コア粒子に連結している、形成体;ならびに、
b)少なくとも1つの第2の付着部位を有する抗原または抗原決定基であって、該第2の付着部位が、以下:
(i)該抗原または抗原決定基と一緒には天然に存在しない付着部位;および、
(ii)該抗原または抗原決定基と一緒に天然に存在する付着部位、
からなる群より選択され、ここで、該第2の付着部位が、該第1の付着部位に対して少なくとも1つの非ペプチド結合により会合し得、そして
ここで、該結合により該抗原または抗原決定基ならびに該足場が相互作用して、順序付けられかつ反復性の抗原アレイを形成する、抗原または抗原決定基。
【請求項2】
請求項1に記載の組成物であって、ここで;
a)前記コア粒子は、以下:
(i)ウイルス;
(ii)ウイルス様粒子;
(iii)バクテリオファージ;
(iv)ウイルスのカプシド粒子;および
(v)(i)、(ii)、(iii)または(iv)の組換え型、
からなる群より選択され;そして、
b)前記形成体は、ポリペプチドまたはその残基であり;そして
c)前記第2の付着部位が、ポリペプチドまたはその残基である、
組成物。
【請求項3】
請求項2の組成物であって、ここで、前記第1の付着部位および/または前記第2の付着部位が、以下:
a)抗原およびそれに対する抗体または抗体フラグメント;
b)ビオチンおよびアビジン;
c)ストレプトアビジンおよびビオチン;
d)レセプターおよびそのリガンド;
e)リガンド結合タンパク質およびそのリガンド;
f)相互作用するロイシンジッパーポリペプチド;
g)アミノ基およびそれに対して反応性の化学基;
h)カルボキシル基およびそれに対して反応性の化学基;
i)スルフヒドリル基およびそれに対して反応性の化学基;または
j)それらの組み合わせ、
を含む、組成物。
【請求項4】
前記第2の付着部位が、前記抗原または抗原決定基と一緒には天然に存在しない、請求項3に記載の組成物。
【請求項5】
前記コア粒子が、組換え型アルファウイルスである、請求項2に記載の組成物。
【請求項6】
請求項5に記載の組成物であって、ここで、前記組換え型アルファウイルスが、シンドビスウイルスであり、そして、前記第1の付着部位および前記第2の付着部位が、それぞれ、相互作用するロイシンジッパーポリペプチドを含む、組成物。
【請求項7】
前記第1の付着部位および前記第2の付着部位が、JUNロイシンジッパーポリペプチドおよび/またはFOSロイシンジッパーポリペプチドである、請求項6に記載の組成物。
【請求項8】
前記コア粒子が、ウイルス様粒子である、請求項2に記載の組成物。
【請求項9】
前記第1の付着部位がアミノ基であり、そして、前記第2の付着部位がスルフヒドリル基である、請求項8に記載の組成物。
【請求項10】
前記ウイルス様粒子が、B型肝炎ウイルスカプシドタンパク質である、請求項8に記載の組成物。
【請求項11】
前記第1の付着部位および前記第2の付着部位が、それぞれ、相互作用するロイシンジッパーポリペプチドを含む、請求項10に記載の組成物。
【請求項12】
前記第1の付着部位が、JUNポリペプチドであり、そして、前記第2の付着部位がFOポリペプチドである、請求項11に記載の組成物。
【請求項13】
前記第1の付着部位がリジン残基であり、そして、前記第2の付着部位がシステイン残基である、請求項10に記載の組成物。
【請求項14】
前記ウイルス様粒子が、麻疹ウイルスカプシドタンパク質である、請求項8に記載の組成物。
【請求項15】
前記第1の付着部位および前記第2の付着部位が、それぞれ、相互作用するロイシンジッパーポリペプチドを含む、請求項14の組成物。
【請求項16】
前記第1の付着部位および前記第2の付着部位が、JUNロイシンジッパーポリペプチドおよび/またはFOSロイシンジッパーポリペプチドである、請求項15に記載の組成物。
【請求項17】
請求項2の組成物であって、ここで、前記コア粒子が以下:
a)ロタウイルスの組換え型タンパク質、
b)ノーウォークウイルスの組換え型タンパク質、
c)アルファウイルスの組換え型タンパク質、
d)口蹄疫ウイルスの組換え型タンパク質、
e)レトロウイルスの組換え型タンパク質、
f)B型肝炎ウイルスの組換え型タンパク質、
g)タバコモザイクウイルスの組換え型タンパク質、
h)フロックハウスウイルスの組換え型タンパク質、および
i)ヒトパピローマウイルスの組換え型タンパク質、
からなる群より選択される、組成物。
【請求項18】
前記第1の付着部位および前記第2の付着部位が、それぞれ、相互作用するロイシンジッパーポリペプチドを含む、請求項17に記載の組成物。
【請求項19】
前記第1の付着部位がアミノ基であり、そして、前記第2の付着部位がスルフヒドリル基である、請求項17に記載の組成物。
【請求項20】
前記コア粒子が、非天然の起源である、請求項1に記載の組成物。
【請求項21】
請求項20に記載の組成物であって、ここで、前記コア粒子が、以下:
a)合成ポリマー、
b)脂質ミセル、および
c)金属、
からなる群より選択される、組成物。
【請求項22】
前記第1の付着部位および前記第2の付着部位が、それぞれ、相互作用するロイシンジッパーポリペプチドを含む、請求項21に記載の組成物。
【請求項23】
前記第1の付着部位および前記第2の付着部位が、JUNロイシンジッパーポリペプチドおよび/またはFOSロイシンジッパーポリペプチドである、請求項22に記載の組成物。
【請求項24】
請求項1に記載の組成物であって、ここで、前記抗原が、以下:
a)癌細胞に対して免疫応答を誘導するのに適しているタンパク質、
b)感染症に対して免疫応答を誘導するのに適しているタンパク質、
c)アレルゲンに対して免疫応答を誘導するのに適しているタンパク質、および
d)家畜の免疫応答を誘導することに適しているタンパク質、
からなる群より選択される、組成物。
【請求項25】
請求項24に記載の組成物であって、前記抗原が、以下:
a)HIVの組換え型タンパク質、
b)インフルエンザウイルスの組換え型タンパク質、
c)C型肝炎ウイルスの組換え型タンパク質、
d)トキソプラズマの組換え型タンパク質、
e)熱帯熱マラリア原虫の組換え型タンパク質、
f)三日熱マラリア原虫の組換え型タンパク質、
g)卵形マラリア原虫の組換え型タンパク質、
h)四日熱マラリア原虫の組換え型タンパク質、
i)乳がん細胞の組換え型タンパク質、
j)腎臓癌細胞の組換え型タンパク質、
k)前立腺癌細胞の組換え型タンパク質、
l)皮ふ癌細胞の組換え型タンパク質、
m)脳癌細胞の組換え型タンパク質、
n)白血病細胞の組換え型タンパク質、
o)組換え型プロファイリング、
p)ミツバチ針アレルギーの組換え型タンパク質、
q)堅果アレルギーの組換え型タンパク質、
r)食物アレルギーの組換え型タンパク質、
s)喘息の組換え型タンパク質、または
t)クラミジアの組換え型タンパク質、
である、組成物。
【請求項26】
前記第1の付着部位および前記第2の付着部位が、それぞれ、相互作用するロイシンジッパーポリペプチドを含む、請求項24に記載の組成物。
【請求項27】
天然に存在しない、順序付けられかつ反復性の抗原アレイを生産するためのプロセスであって、以下の工程:
a)天然には存在しない分子足場を提供する工程であって、該天然には存在しない分子足場は、以下:
(i)以下からなる群から選択されるコア粒子:
(1)非天然起源のコア粒子;および、
(2)天然起源のコア粒子;ならびに、
(ii)少なくとも1つの第1の付着部位を含む形成体であって、
ここで、該形成体は、少なくとも1つの共有結合によって該コア粒子に結合している、形成体、
を含む工程;ならびに
b)少なくとも1つの第2の付着部位を有する抗原または抗原決定基を提供する工程であって、該第2の付着部位が、以下:
(i)該抗原または抗原決定基と一緒には天然に存在しない付着部位;および、
(ii)該抗原または抗原決定基と一緒に天然に存在する付着部位、
からなる群より選択され、ここで、該第2の付着部位が、該第1の付着部位に少なくとも1つの非ペプチド結合により会合し得る、工程;ならびに
c)天然に存在しない該分子足場および該抗原または抗原決定基を組み合わせる工程であって、ここで、該会合により該抗原または抗原決定基ならびに該足場が相互作用して、順序付けられかつ反復性の抗原アレイを形成する、工程、
を包含するプロセス。
【請求項28】
請求項27に記載のプロセスであって、ここで、
a)前記コア粒子は、以下:
(i)ウイルス
(ii)ウイルス様粒子;
(iii)バクテリオファージ;
(iv)ウイルスのカプシド粒子;および
(v)(i)、(ii)、(iii)または(iv)の組換え型、
からなる群より選択され;そして、
b)前記形成体は、ポリペプチドまたはその残基であり;そして
c)前記第2の付着部位が、ポリペプチドまたはその残基である、
プロセス。
【請求項29】
請求項28のプロセスであって、ここで、前記第1の付着部位および/または前記第2の付着部位が、以下:
a)抗原およびそれに対する抗体または抗体フラグメント;
b)ビオチンおよびアビジン;
c)ストレプトアビジンおよびビオチン;
d)レセプターおよびそのリガンド;
e)リガンド結合タンパク質およびそのリガンド;
f)相互作用するロイシンジッパーポリペプチド;
g)アミノ基およびそれに対して反応性の化学基;
h)カルボキシル基およびそれに対して反応性の化学基;
i)スルフヒドリル基およびそれに対して反応性の化学基;または
j)それらの組み合わせ、
を含む、プロセス。
【請求項30】
前記第2の付着部位が、前記抗原または抗原決定基によって天然に存在しない、請求項29に記載のプロセス、
【請求項31】
単離された組換え型アルファウイルスであって、該アルファウイルスが以下:
a)RNAパッケージングシグナル配列の欠失;および
b)前記アルファウイルスのE2エンベロープタンパク質核酸配列とインフレームなJUNロイシンジッパータンパク質ドメイン核酸配列の天然に存在しない挿入、
をそのゲノム中に含む、アルファウイルス。
【請求項32】
請求項31に記載の組換え型アルファウイルスを含む、宿主細胞。
【請求項33】
請求項1に記載の組成物を、被験体に投与する工程を含む、医療処置の方法。
【請求項34】
薬学的組成物であって、以下:
a)請求項1に記載の組成物;および、
b)受容可能な薬学的キャリア、
を含む、薬学的組成物。
【請求項35】
以下を含む組成物を被験体に投与する工程を包含する免疫化の方法:
a)以下を含む、天然には存在しない分子足場:
(i)以下からなる群より選択されるコア粒子:
(1)非天然起源のコア粒子;および
(2)天然起源のコア粒子;ならびに
(ii)少なくとも1つの第1の付着部位を含む形成体であって、ここで、少なくとも1つの該形成体は、少なくとも1つの共有結合によって該コア粒子に結合している、形成体;ならびに、
b)少なくとも1つの第2の付着部位を有する抗原または抗原決定基であって、該第2の付着部位が、以下:
(i)該抗原または抗原決定基と一緒には天然に存在しない付着部位;および
(ii)該抗原または抗原決定基との天然に存在する付着部位、
からなる群より選択され、
ここで、該第2の付着部位が、該第1の付着部位に対する少なくとも1つの非ペプチド結合により会合し得;そして、
ここで、該会合を通じて該抗原または抗原決定基および該足場が相互作用して、順序付けられかつ反復性の抗原アレイを形成する、抗原または抗原決定基。
【請求項36】
前記免疫化が、免疫応答を生成する、請求項35に記載の方法。
【請求項37】
前記免疫化が、体液性免疫応答を生成する、請求項35に記載の方法。
【請求項38】
前記免疫化が、細胞免疫反応を生成する、請求項35に記載の方法。
【請求項39】
前記免疫化が、体液性免疫応答および細胞免疫反応を生成する、請求項35に記載の方法。
【請求項40】
前記免疫化が、防御応答を生成する、請求項35に記載の方法。
【請求項41】
以下を含むワクチン組成物:
a)以下を含む、天然には存在しない分子足場:
(i)以下からなる群より選択されるコア粒子:
(1)非天然起源のコア粒子;および
(2)天然起源のコア粒子;ならびに
(ii)少なくとも1つの第1の付着部位を含む形成体であって、
ここで、少なくとも1つの該形成体は、少なくとも1つの共有結合によって該コア粒子に連結している、形成体;ならびに、
b)少なくとも1つの第2の付着部位を有する抗原または抗原決定基であって、該第2の付着部位が、以下:
(i)該抗原または抗原決定基と一緒には天然に存在しない付着部位;および
(ii)該抗原または抗原決定基と一緒に天然に存在する付着部位、
からなる群より選択され:ここで、該第2の付着部位が、該第1の付着部位に対する少なくとも1つの非ペプチド結合により会合し得;そして、
ここで、該会合を通じて該抗原または抗原決定基および該足場が相互作用して、順序付けられかつ反復性の抗原アレイを形成する、抗原または抗原決定基。
【請求項42】
アジュバントをさらに含む、請求項41に記載のワクチン組成物。
【請求項43】
請求項41に記載のワクチン組成物であって、ここで:
a)前記コア粒子が、以下:
(i)ウイルス
(ii)ウイルス様粒子;
(iii)バクテリオファージ;
(iv)ウイルスのカプシド粒子;および
(v)(i)、(ii)、(iii)または(iv)の組換え型;からなる群より選択され;そして
b)前記形成体は、ポリペプチドまたはその残基であり;そして、
c)前記第2の付着部位が、ポリペプチドまたはその残基である、
ワクチン組成物。
【請求項44】
請求項43に記載のワクチン組成物であって、ここで、前記第1の付着部位および/または前記第2の付着部位が、以下:
a)抗原およびそれに対する抗体または抗体フラグメント;
b)ビオチンおよびアビジン;
c)ストレプトアビジンおよびビオチン;
d)レセプターおよびそのリガンド;
e)リガンド結合タンパク質およびそのリガンド;
f)相互作用するロイシンジッパーポリペプチド;
g)アミノ基およびそれに対して反応性の化学基;
h)カルボキシル基およびそれに対して反応性の化学基;
i)スルフヒドリル基およびそれに対して反応性の化学基;または
j)それらの組み合わせ、
を含む、ワクチン組成物。
【請求項45】
前記コア粒子が、ウイルス様粒子を含む、請求項43に記載のワクチン組成物。
【請求項46】
前記コア粒子が、B型肝炎ウイルス様粒子を含む、請求項45に記載のワクチン組成物。
【請求項47】
前記コア粒子が、麻疹ウイルス様粒子を含む、請求項45に記載のワクチン組成物。
【請求項48】
前記コア粒子が、ウイルスを含む、請求項43に記載のワクチン組成物。
【請求項49】
前記コア粒子が、シンドビスウイルスを含む、請求項48に記載のワクチン組成物。
【請求項1】
以下を含む、組成物:
a)以下を含む、天然には存在しない分子足場:
(i)以下からなる群から選択されるコア粒子:
(1)非天然起源のコア粒子;および
(2)天然起源のコア粒子;ならびに
(ii)少なくとも1つの第1の付着部位を含む形成体であって、ここで、該形成体は、少なくとも1つの共有結合によって該コア粒子に連結している、形成体;ならびに、
b)少なくとも1つの第2の付着部位を有する抗原または抗原決定基であって、該第2の付着部位が、以下:
(i)該抗原または抗原決定基と一緒には天然に存在しない付着部位;および、
(ii)該抗原または抗原決定基と一緒に天然に存在する付着部位、
からなる群より選択され、ここで、該第2の付着部位が、該第1の付着部位に対して少なくとも1つの非ペプチド結合により会合し得、そして
ここで、該結合により該抗原または抗原決定基ならびに該足場が相互作用して、順序付けられかつ反復性の抗原アレイを形成する、抗原または抗原決定基。
【請求項2】
請求項1に記載の組成物であって、ここで;
a)前記コア粒子は、以下:
(i)ウイルス;
(ii)ウイルス様粒子;
(iii)バクテリオファージ;
(iv)ウイルスのカプシド粒子;および
(v)(i)、(ii)、(iii)または(iv)の組換え型、
からなる群より選択され;そして、
b)前記形成体は、ポリペプチドまたはその残基であり;そして
c)前記第2の付着部位が、ポリペプチドまたはその残基である、
組成物。
【請求項3】
請求項2の組成物であって、ここで、前記第1の付着部位および/または前記第2の付着部位が、以下:
a)抗原およびそれに対する抗体または抗体フラグメント;
b)ビオチンおよびアビジン;
c)ストレプトアビジンおよびビオチン;
d)レセプターおよびそのリガンド;
e)リガンド結合タンパク質およびそのリガンド;
f)相互作用するロイシンジッパーポリペプチド;
g)アミノ基およびそれに対して反応性の化学基;
h)カルボキシル基およびそれに対して反応性の化学基;
i)スルフヒドリル基およびそれに対して反応性の化学基;または
j)それらの組み合わせ、
を含む、組成物。
【請求項4】
前記第2の付着部位が、前記抗原または抗原決定基と一緒には天然に存在しない、請求項3に記載の組成物。
【請求項5】
前記コア粒子が、組換え型アルファウイルスである、請求項2に記載の組成物。
【請求項6】
請求項5に記載の組成物であって、ここで、前記組換え型アルファウイルスが、シンドビスウイルスであり、そして、前記第1の付着部位および前記第2の付着部位が、それぞれ、相互作用するロイシンジッパーポリペプチドを含む、組成物。
【請求項7】
前記第1の付着部位および前記第2の付着部位が、JUNロイシンジッパーポリペプチドおよび/またはFOSロイシンジッパーポリペプチドである、請求項6に記載の組成物。
【請求項8】
前記コア粒子が、ウイルス様粒子である、請求項2に記載の組成物。
【請求項9】
前記第1の付着部位がアミノ基であり、そして、前記第2の付着部位がスルフヒドリル基である、請求項8に記載の組成物。
【請求項10】
前記ウイルス様粒子が、B型肝炎ウイルスカプシドタンパク質である、請求項8に記載の組成物。
【請求項11】
前記第1の付着部位および前記第2の付着部位が、それぞれ、相互作用するロイシンジッパーポリペプチドを含む、請求項10に記載の組成物。
【請求項12】
前記第1の付着部位が、JUNポリペプチドであり、そして、前記第2の付着部位がFOポリペプチドである、請求項11に記載の組成物。
【請求項13】
前記第1の付着部位がリジン残基であり、そして、前記第2の付着部位がシステイン残基である、請求項10に記載の組成物。
【請求項14】
前記ウイルス様粒子が、麻疹ウイルスカプシドタンパク質である、請求項8に記載の組成物。
【請求項15】
前記第1の付着部位および前記第2の付着部位が、それぞれ、相互作用するロイシンジッパーポリペプチドを含む、請求項14の組成物。
【請求項16】
前記第1の付着部位および前記第2の付着部位が、JUNロイシンジッパーポリペプチドおよび/またはFOSロイシンジッパーポリペプチドである、請求項15に記載の組成物。
【請求項17】
請求項2の組成物であって、ここで、前記コア粒子が以下:
a)ロタウイルスの組換え型タンパク質、
b)ノーウォークウイルスの組換え型タンパク質、
c)アルファウイルスの組換え型タンパク質、
d)口蹄疫ウイルスの組換え型タンパク質、
e)レトロウイルスの組換え型タンパク質、
f)B型肝炎ウイルスの組換え型タンパク質、
g)タバコモザイクウイルスの組換え型タンパク質、
h)フロックハウスウイルスの組換え型タンパク質、および
i)ヒトパピローマウイルスの組換え型タンパク質、
からなる群より選択される、組成物。
【請求項18】
前記第1の付着部位および前記第2の付着部位が、それぞれ、相互作用するロイシンジッパーポリペプチドを含む、請求項17に記載の組成物。
【請求項19】
前記第1の付着部位がアミノ基であり、そして、前記第2の付着部位がスルフヒドリル基である、請求項17に記載の組成物。
【請求項20】
前記コア粒子が、非天然の起源である、請求項1に記載の組成物。
【請求項21】
請求項20に記載の組成物であって、ここで、前記コア粒子が、以下:
a)合成ポリマー、
b)脂質ミセル、および
c)金属、
からなる群より選択される、組成物。
【請求項22】
前記第1の付着部位および前記第2の付着部位が、それぞれ、相互作用するロイシンジッパーポリペプチドを含む、請求項21に記載の組成物。
【請求項23】
前記第1の付着部位および前記第2の付着部位が、JUNロイシンジッパーポリペプチドおよび/またはFOSロイシンジッパーポリペプチドである、請求項22に記載の組成物。
【請求項24】
請求項1に記載の組成物であって、ここで、前記抗原が、以下:
a)癌細胞に対して免疫応答を誘導するのに適しているタンパク質、
b)感染症に対して免疫応答を誘導するのに適しているタンパク質、
c)アレルゲンに対して免疫応答を誘導するのに適しているタンパク質、および
d)家畜の免疫応答を誘導することに適しているタンパク質、
からなる群より選択される、組成物。
【請求項25】
請求項24に記載の組成物であって、前記抗原が、以下:
a)HIVの組換え型タンパク質、
b)インフルエンザウイルスの組換え型タンパク質、
c)C型肝炎ウイルスの組換え型タンパク質、
d)トキソプラズマの組換え型タンパク質、
e)熱帯熱マラリア原虫の組換え型タンパク質、
f)三日熱マラリア原虫の組換え型タンパク質、
g)卵形マラリア原虫の組換え型タンパク質、
h)四日熱マラリア原虫の組換え型タンパク質、
i)乳がん細胞の組換え型タンパク質、
j)腎臓癌細胞の組換え型タンパク質、
k)前立腺癌細胞の組換え型タンパク質、
l)皮ふ癌細胞の組換え型タンパク質、
m)脳癌細胞の組換え型タンパク質、
n)白血病細胞の組換え型タンパク質、
o)組換え型プロファイリング、
p)ミツバチ針アレルギーの組換え型タンパク質、
q)堅果アレルギーの組換え型タンパク質、
r)食物アレルギーの組換え型タンパク質、
s)喘息の組換え型タンパク質、または
t)クラミジアの組換え型タンパク質、
である、組成物。
【請求項26】
前記第1の付着部位および前記第2の付着部位が、それぞれ、相互作用するロイシンジッパーポリペプチドを含む、請求項24に記載の組成物。
【請求項27】
天然に存在しない、順序付けられかつ反復性の抗原アレイを生産するためのプロセスであって、以下の工程:
a)天然には存在しない分子足場を提供する工程であって、該天然には存在しない分子足場は、以下:
(i)以下からなる群から選択されるコア粒子:
(1)非天然起源のコア粒子;および、
(2)天然起源のコア粒子;ならびに、
(ii)少なくとも1つの第1の付着部位を含む形成体であって、
ここで、該形成体は、少なくとも1つの共有結合によって該コア粒子に結合している、形成体、
を含む工程;ならびに
b)少なくとも1つの第2の付着部位を有する抗原または抗原決定基を提供する工程であって、該第2の付着部位が、以下:
(i)該抗原または抗原決定基と一緒には天然に存在しない付着部位;および、
(ii)該抗原または抗原決定基と一緒に天然に存在する付着部位、
からなる群より選択され、ここで、該第2の付着部位が、該第1の付着部位に少なくとも1つの非ペプチド結合により会合し得る、工程;ならびに
c)天然に存在しない該分子足場および該抗原または抗原決定基を組み合わせる工程であって、ここで、該会合により該抗原または抗原決定基ならびに該足場が相互作用して、順序付けられかつ反復性の抗原アレイを形成する、工程、
を包含するプロセス。
【請求項28】
請求項27に記載のプロセスであって、ここで、
a)前記コア粒子は、以下:
(i)ウイルス
(ii)ウイルス様粒子;
(iii)バクテリオファージ;
(iv)ウイルスのカプシド粒子;および
(v)(i)、(ii)、(iii)または(iv)の組換え型、
からなる群より選択され;そして、
b)前記形成体は、ポリペプチドまたはその残基であり;そして
c)前記第2の付着部位が、ポリペプチドまたはその残基である、
プロセス。
【請求項29】
請求項28のプロセスであって、ここで、前記第1の付着部位および/または前記第2の付着部位が、以下:
a)抗原およびそれに対する抗体または抗体フラグメント;
b)ビオチンおよびアビジン;
c)ストレプトアビジンおよびビオチン;
d)レセプターおよびそのリガンド;
e)リガンド結合タンパク質およびそのリガンド;
f)相互作用するロイシンジッパーポリペプチド;
g)アミノ基およびそれに対して反応性の化学基;
h)カルボキシル基およびそれに対して反応性の化学基;
i)スルフヒドリル基およびそれに対して反応性の化学基;または
j)それらの組み合わせ、
を含む、プロセス。
【請求項30】
前記第2の付着部位が、前記抗原または抗原決定基によって天然に存在しない、請求項29に記載のプロセス、
【請求項31】
単離された組換え型アルファウイルスであって、該アルファウイルスが以下:
a)RNAパッケージングシグナル配列の欠失;および
b)前記アルファウイルスのE2エンベロープタンパク質核酸配列とインフレームなJUNロイシンジッパータンパク質ドメイン核酸配列の天然に存在しない挿入、
をそのゲノム中に含む、アルファウイルス。
【請求項32】
請求項31に記載の組換え型アルファウイルスを含む、宿主細胞。
【請求項33】
請求項1に記載の組成物を、被験体に投与する工程を含む、医療処置の方法。
【請求項34】
薬学的組成物であって、以下:
a)請求項1に記載の組成物;および、
b)受容可能な薬学的キャリア、
を含む、薬学的組成物。
【請求項35】
以下を含む組成物を被験体に投与する工程を包含する免疫化の方法:
a)以下を含む、天然には存在しない分子足場:
(i)以下からなる群より選択されるコア粒子:
(1)非天然起源のコア粒子;および
(2)天然起源のコア粒子;ならびに
(ii)少なくとも1つの第1の付着部位を含む形成体であって、ここで、少なくとも1つの該形成体は、少なくとも1つの共有結合によって該コア粒子に結合している、形成体;ならびに、
b)少なくとも1つの第2の付着部位を有する抗原または抗原決定基であって、該第2の付着部位が、以下:
(i)該抗原または抗原決定基と一緒には天然に存在しない付着部位;および
(ii)該抗原または抗原決定基との天然に存在する付着部位、
からなる群より選択され、
ここで、該第2の付着部位が、該第1の付着部位に対する少なくとも1つの非ペプチド結合により会合し得;そして、
ここで、該会合を通じて該抗原または抗原決定基および該足場が相互作用して、順序付けられかつ反復性の抗原アレイを形成する、抗原または抗原決定基。
【請求項36】
前記免疫化が、免疫応答を生成する、請求項35に記載の方法。
【請求項37】
前記免疫化が、体液性免疫応答を生成する、請求項35に記載の方法。
【請求項38】
前記免疫化が、細胞免疫反応を生成する、請求項35に記載の方法。
【請求項39】
前記免疫化が、体液性免疫応答および細胞免疫反応を生成する、請求項35に記載の方法。
【請求項40】
前記免疫化が、防御応答を生成する、請求項35に記載の方法。
【請求項41】
以下を含むワクチン組成物:
a)以下を含む、天然には存在しない分子足場:
(i)以下からなる群より選択されるコア粒子:
(1)非天然起源のコア粒子;および
(2)天然起源のコア粒子;ならびに
(ii)少なくとも1つの第1の付着部位を含む形成体であって、
ここで、少なくとも1つの該形成体は、少なくとも1つの共有結合によって該コア粒子に連結している、形成体;ならびに、
b)少なくとも1つの第2の付着部位を有する抗原または抗原決定基であって、該第2の付着部位が、以下:
(i)該抗原または抗原決定基と一緒には天然に存在しない付着部位;および
(ii)該抗原または抗原決定基と一緒に天然に存在する付着部位、
からなる群より選択され:ここで、該第2の付着部位が、該第1の付着部位に対する少なくとも1つの非ペプチド結合により会合し得;そして、
ここで、該会合を通じて該抗原または抗原決定基および該足場が相互作用して、順序付けられかつ反復性の抗原アレイを形成する、抗原または抗原決定基。
【請求項42】
アジュバントをさらに含む、請求項41に記載のワクチン組成物。
【請求項43】
請求項41に記載のワクチン組成物であって、ここで:
a)前記コア粒子が、以下:
(i)ウイルス
(ii)ウイルス様粒子;
(iii)バクテリオファージ;
(iv)ウイルスのカプシド粒子;および
(v)(i)、(ii)、(iii)または(iv)の組換え型;からなる群より選択され;そして
b)前記形成体は、ポリペプチドまたはその残基であり;そして、
c)前記第2の付着部位が、ポリペプチドまたはその残基である、
ワクチン組成物。
【請求項44】
請求項43に記載のワクチン組成物であって、ここで、前記第1の付着部位および/または前記第2の付着部位が、以下:
a)抗原およびそれに対する抗体または抗体フラグメント;
b)ビオチンおよびアビジン;
c)ストレプトアビジンおよびビオチン;
d)レセプターおよびそのリガンド;
e)リガンド結合タンパク質およびそのリガンド;
f)相互作用するロイシンジッパーポリペプチド;
g)アミノ基およびそれに対して反応性の化学基;
h)カルボキシル基およびそれに対して反応性の化学基;
i)スルフヒドリル基およびそれに対して反応性の化学基;または
j)それらの組み合わせ、
を含む、ワクチン組成物。
【請求項45】
前記コア粒子が、ウイルス様粒子を含む、請求項43に記載のワクチン組成物。
【請求項46】
前記コア粒子が、B型肝炎ウイルス様粒子を含む、請求項45に記載のワクチン組成物。
【請求項47】
前記コア粒子が、麻疹ウイルス様粒子を含む、請求項45に記載のワクチン組成物。
【請求項48】
前記コア粒子が、ウイルスを含む、請求項43に記載のワクチン組成物。
【請求項49】
前記コア粒子が、シンドビスウイルスを含む、請求項48に記載のワクチン組成物。
【図1】

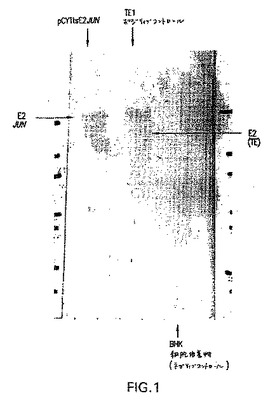
【図2】

【図3】
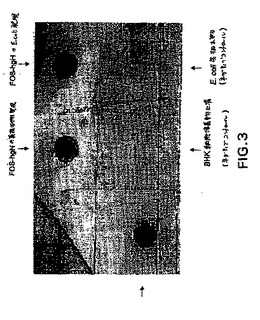
【図4】
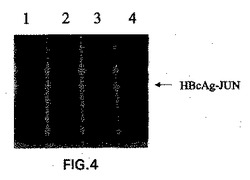
【図5】
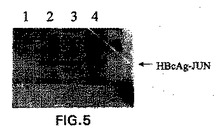
【図6】

【図7】
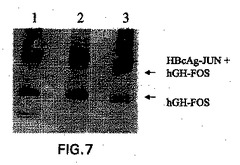
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公表番号】特表2010−510960(P2010−510960A)
【公表日】平成22年4月8日(2010.4.8)
【国際特許分類】
【出願番号】特願2000−584918(P2000−584918)
【出願日】平成11年11月30日(1999.11.30)
【国際出願番号】PCT/IB1999/001925
【国際公開番号】WO2000/032227
【国際公開日】平成12年6月8日(2000.6.8)
【出願人】(599118469)サイトス バイオテクノロジー アーゲー (1)
【Fターム(参考)】
【公表日】平成22年4月8日(2010.4.8)
【国際特許分類】
【出願日】平成11年11月30日(1999.11.30)
【国際出願番号】PCT/IB1999/001925
【国際公開番号】WO2000/032227
【国際公開日】平成12年6月8日(2000.6.8)
【出願人】(599118469)サイトス バイオテクノロジー アーゲー (1)
【Fターム(参考)】
[ Back to top ]