
抗原特異的T細胞、核酸、ベクター、PBMC、及び製薬組成物
【課題】抗原特異的T細胞を提供する。
【解決手段】抗原特異的T細胞は、患者由来MHC分子をエンコードする核酸、及び、抗原もしくは前記抗原をエンコードする核酸を準備する工程、これらの両方の化合物を、健康なドナーに由来する抗原提示細胞(APC)中にコトランスフェクションするか、または導入する工程、健康なドナーに由来する末梢血リンパ球(PBL)を、前記APCで初回抗原刺激する工程、及びMHC−抗原リガンドに特異的なT細胞を選択する工程を含む方法によって提供される。
【解決手段】抗原特異的T細胞は、患者由来MHC分子をエンコードする核酸、及び、抗原もしくは前記抗原をエンコードする核酸を準備する工程、これらの両方の化合物を、健康なドナーに由来する抗原提示細胞(APC)中にコトランスフェクションするか、または導入する工程、健康なドナーに由来する末梢血リンパ球(PBL)を、前記APCで初回抗原刺激する工程、及びMHC−抗原リガンドに特異的なT細胞を選択する工程を含む方法によって提供される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、抗原特異的T細胞の生成方法を目的とする。本発明はさらに、抗原特異的T細胞、単離トランスジェニックT細胞受容体(TCR)、これを含有する製薬組成物、および養子細胞療法におけるこれらの使用も目的とする。本発明は特に、非選択同種異系T細胞レパートリーからペプチド−特異的T細胞を誘発するための、同種異系MHC分子および抗原を共発現する細胞の使用に関する。
【背景技術】
【0002】
同種異系幹細胞移植(SCT)の設定におけるリンパ球の養子転移は、血液悪性腫瘍を根絶するための免疫系の力を実証している(Kolbら、1995年)。SCTはまた、いくつかの症例において、充実性腫瘍、例えば腎細胞癌(RCC)を取り除く作用も果たし得るように見える(Kolbら、2004年およびDudleyおよびRosenberg、2003年において概説されている)。SCT受容者において、悪性細胞の除去は、特異的T細胞が生体内で活性化されなければならず、次いで移植受容者における新しい造血系の発達後に適切な数まで拡張されなければならないという事実によって、数ヶ月から1年後にのみ発生し得る。あるいはまた、SCT受容者において耐性が確立される一定期間(約60日)後、非初回抗原刺激、非分離リンパ球の転移が、腫瘍細胞に対して向けられた免疫応答の生成を加速させることができる。ここでもまた、腫瘍細胞を攻撃し得る特異的リンパ球は活性化されなければならず、転移されるリンパ球の非選択集団中に存在する低周波数先駆物質リンパ球から拡張されなければならない。SCT後の非選択リンパ球集団のドナーリンパ球注入(DLI)は、ゆっくりと成長する慢性骨髄性白血病(CML)の除去のために良好に作用するが、部分的には、悪性細胞の成長が免疫細胞の拡張能力を凌ぐという事実によって、急性白血病の根絶において効果がより低い。この同じ拡張差はまた、急速に進行する充実性腫瘍の芳しくない免疫除去に対しても影響を与える。DLIにおける非選択リンパ球集団の使用における第二の不利な条件は、受容者の正常細胞および組織を攻撃する能力を有するT細胞もまた転移されることがあり、移植片対宿主疾患(GVHD)、すなわち高い罹患率および致死率を有する疾患に導くということである。
【0003】
最近の研究は、規定されたペプチド特異性を有する選択されたT細胞の養子転移が、特に患者が非骨髄機能廃絶方式で予め治療されているならば、自家設定における全身腫瘍組織量の大きな低減に導き得ることを実証している(Dudleyら、2002年、2003年)。このことは、腫瘍患者においてSCTを実施する必要性を取り除き、これによってGVHDの問題も回避する。効果的な免疫応答は、腫瘍浸潤リンパ球(TIL)の自家混合物を受けた、予め治療された黒色腫患者に見られた。CD4およびCD8の両方が陽性のT細胞を含有する細胞のこれらの混合物は、特定のMHC−腫瘍−関連抗原(TAA)リガンドについて特異的な多数の単一CD8T細胞クローンの養子転移よりも、臨床的にはより効果的に見える。この差に寄与する一つの因子は、CD4T細胞に長命CD8T細胞を維持させるための必要条件である。さらには、単一リガンドに対して向けられた免疫応答は、対応リガンドの発現を喪失した腫瘍細胞変異体の選択に導くことがあり、これによって免疫検出を免れることがある。一方で、T細胞の複合混合物の転移は、これらが腫瘍浸潤リンパ球の中に存在するので、CD4およびCD8細胞に多重特異性を与えることによってこれらの問題を克服し得るが、これらはまた、TILの混合物が、正常組織によって発現されたリガンドを認識するT細胞を含有するならば自己免疫に導き得る。このことは、例えばメラニン細胞中にも発現される黒色腫分化抗原を認識する細胞を用いた養子細胞療法(ACT)後に、黒色腫患者における白斑に導く正常メラニン細胞の攻撃によって実証される(Dudleyら、2002年)。
【0004】
より急速に成長する腫瘍を有する患者の治療のためにACTを用いる能力を拡大するために、正常組織の重大な攻撃を避けつつ、腫瘍細胞を効果的に攻撃するためのこれらのリガンド特異性のために選択された強化ペプチド−特異的エフェクターT細胞(CD4Tヘルパー細胞および細胞傷害性Tリンパ球の両方)を転移することが一つの目標である。これらの細胞は、生体外で急速に多数になるまで拡張させられ、ついでACTのために用いられることになる。あるいはまた、このようなリガンド−特異的T細胞のT細胞受容体(TCR)は、受容者末梢血リンパ球か、または十分に成長し、正常な宿主組織を攻撃する能力を有していない、規定された特異性を有する活性化されたT細胞クローンのどちらかを用いてクローンすることができ、活性化されたリンパ球中にTCR−トランスジーンとして発現させることができる。
【0005】
例として、受容者によって発現されないMHC分子について特異的な拡張アロ特異的T細胞クローン、またはウイルス、例えばサイトメガロウイルスまたはエプスタイン−バーウイルスについて特異的な拡張T細胞クローンは、トランスジェニックTCRへの受容体細胞として用いられることができるであろう。異なるMHC−ペプチドリガンドを認識するトランスジェニックTCRベクターのパネルの利用可能性は、CD4およびCD8亜類型の両方の多数の予め活性化されたT細胞を開発するために利用することができ、これによって多数のエフェクターリンパ球が迅速に調製され、対応TCRリガンドを発現する腫瘍を有する患者へ転移させることを可能にするであろう。このことは、腫瘍成長を制御するために必要とされる特異的T細胞の数の達成において時間を節約し、おそらくは、急速に進行する腫瘍のより効果的な腫瘍根絶に導くであろう。
【0006】
特異的T細胞が白血病およびリンパ腫、ならびに充実性腫瘍細胞に関して認識する決定因子は、自己−MHC分子によって提示される過剰発現タンパク質に由来する自己−ペプチドを代表することが多いことから、これらのT細胞受容体(TCR)の親和性は低い。それは、高い親和性受容体を担っているT細胞が、胸腺中のこれらの発達の間にリンパ球へ適用される負の選択プロセスを通して取り除かれているからである。これらのT細胞が、胸腺中のこれらの発達の間に自己−MHC分子に対して負の選択をされなかったリンパ球集団から生成されるならば、より効果的な腫瘍細胞認識が発生する。
【0007】
したがって、腫瘍細胞上のこれらのリガンドを認識する能力を有する高い機能的結合活性を有するTCRを担っているT細胞を迅速に生成する手段を発見する重要なニーズが存在する。このようなT細胞は、潜在的なACT受容者へのMHC−ミスマッチを有する同種異系個人のレパートリー中に存在する。
【0008】
Staussおよび共同研究者の研究(Gaoら、1999年、2000年)は、転写因子WT−1に由来し、かつ同種異系MHCクラスI分子によって提示されるペプチドを認識するために選択された細胞傷害性エフェクター細胞が高親和性TCRを有し、正常な幹細胞に損傷を与えずにWT−1陽性白血病を効果的に取り除きうることを証明している。部分的ミスマッチのSCTの設定において、このようなT細胞への耐性を確立することができ、これらの特異的養子転移を可能にする。Staussは、刺激細胞源として、WT−1ペプチドでパルスされたT2細胞株を用いて、このようなアロ制限CD8T細胞を得るという問題を解決した。T2細胞は、内因性ペプチドをこれらのMHCクラスI分子中に負荷するこれらの能力を損なう遺伝子欠失を有し、染色体6における欠失によって、これらの細胞は、HLA−A2分子を発現する能力が限定されている。ペプチドがT2細胞へ外因的に供給される時、これらは中空HLA−A2分子へ結合し、細胞表面において安定な複合体を形成し得る。これらのペプチド−パルスされた細胞は、次いでHLA−A2陰性個人からの末梢血リンパ球を刺激するために用いられることができる。このような初回抗原刺激条件下、いくつかの異なる活性化リンパ球集団が試験管内に現われる。HLA−A2分子を認識するT細胞が活性化され、これの一フラクションは、WT−1/HLA−A2リガンドについて特異的である。これらの探索されたT細胞は、ペプチドに関わりなくHLA−A2分子を認識するT細胞から分離されなければならない。残念ながら、T2細胞の、MHCクラスI分子の正常補体の発現の不成功、ならびに非MHC−制限細胞を活性化するその固有の能力は、NKおよびNK様T細胞の平行活性化に導く(Falkら、2002年)。これらの集団は多くの場合、これらの培養物を支配し、これらの細胞を取り除くため、および所望のアロ反応性細胞のためのT細胞集団を強化するために、冗漫な研究を必要とする。このことは、特異的T細胞の生成プロセスを遅延させ、したがって重大な臨床的制限を引起こす。さらには、これらの抗原からのペプチドは、前もって知られていなければならないが、それは、これらが外部からT2細胞上にパルスされなければならないからであり、その理由は、抗原提示細胞が、この細胞中に発現されたタンパク質からペプチドを生成することを可能にし、かつMHCクラスIおよびクラスII分子中でこれらの表面上にこれらを提示する抗原プロセシング関連輸送体(TAP)遺伝子をT2細胞が欠いているからである。さらには、T2のみが、内因性HLA−A2クラスI分子を発現する。
【0009】
米国特許出願第20020090362号明細書は、患者の治療方法であって、細胞の表面上のHLAクラスI(または同等)分子によって提示された時に、抗原分子の少なくとも一部を認識する細胞傷害性Tリンパ球(CTL)の治療的有効量を患者に投与する工程を含み、これらの細胞傷害性Tリンパ球が患者に由来しない方法を開示している。この出願は、CTLのドナーから見て同種異系であるか、または異種でさえある刺激細胞の使用について記載している。米国特許出願第20020090362号明細書はさらに、好ましくは選択された分子(ペプチド)の負荷が不可能である刺激細胞、および特にTAP欠乏刺激細胞の使用も開示している。
【0010】
米国特許第6,805,861号明細書は、選択された分子に対して反応性がある細胞傷害性Tリンパ球(CTL)のクローン集団の作製方法であって、(a)健康な個人に由来する(すなわち患者に由来しない)CTLを含有するサンプルを、HLAクラスI(または同等)分子をその表面上に発現し、かつ選択された(抗原)分子の少なくとも一部を促進剤細胞の表面上に提示する促進剤細胞とともに共培養する工程、および(b)前記分子の少なくとも一部が、細胞の表面上のHLAクラスI(または同等)分子によって提示される時に前記選択された分子に対して反応性のCTLクローンを選択する工程を含む方法について記載している。米国特許出願第20020090362号明細書について言及された考察が、ここでもまた適用される。
【発明の概要】
【発明が解決しようとする課題】
【0011】
したがって、病原性作用物質、例えば腫瘍細胞上にそのMHC−ペプチドリガンドを認識する能力を有する高い機能結合活性を有するTCRを担っているT細胞を生成させることが、本発明の基礎となる一つの問題である。養子細胞転移において用いることができる抗原特異的T細胞の迅速かつ効果的な生成方法を提供することが、本発明の基礎となるさらなる問題である。さらには、移植片対宿主疾患(GVHD)のリスクをともなわずに、病気を患っている患者の治療のために用いることができるT細胞ベースの製薬組成物を提供することが、本発明の基礎となる一つの問題である。
【課題を解決するための手段】
【0012】
本発明者らは、アロ制限ペプチド−特異的T細胞を得るための代替戦略を開発した。試験管内転写RNAの転移を通して(Nairら、1998年)、樹状細胞(DC)中に、または抗原提示細胞(APC)として機能し得る他の細胞中にタンパク質を発現させるための周知技術が用いられた。しかしながら、先行技術において通常であるように、抗原源をエンコードするRNAのみを発現する代わりに、本発明者らは、特異的標的分子、例えばモデルTAAとしてチロシナーゼをエンコードするRNA、および同種異系MHC分子、例えばHLA−A2をエンコードするRNAを、HLA−A2陰性ドナーに由来するDCまたは他の細胞中にコトランスフェクションした。
【0013】
これらのトランスフェクションされたDCは、次いでPBL、例えば自家PBLを初回抗原刺激するために用いられることができる。HLA−A2抗原リガンドは、HLA−A2陰性DCドナーのPBLについてのアロ決定因子を表し、従って高親和性TCRを担っているT細胞を得ることができる。DCが自己−MHC分子の完全補体を発現するので、リンパ球の非MHC−制限集団の出現は、陰性MHC調節を通して抑制される。いくつかの様々な戦略を、抗原に由来する特異的ペプチドとは独立してHLA−A2を認識するT細胞からペプチド−特異的/HLA−A2アロ制限T細胞を強化するために用いることができる。これは、サイトカイン捕獲、テトラマー選択、またはその後に拡張される個々のT細胞のクローニングによる戦略を包含する。
【0014】
本研究方法は、(Staussおよび共同研究者によって記載されているように)アロ制限ペプチド−特異的T細胞を得るために、T2系またはショウジョウバエ細胞に勝るいくつかの利点を提供する。
【0015】
第一に、あらゆるクローンされたMHCクラスIまたはクラスII対立遺伝子のために試験管内転写されたRNAを利用することができるので、多様なMHC分子についてのアロ制限を有するT細胞を開発することができる。従って、このようなAPCは、それぞれ転移された同種異系MHCクラスIIまたは同種異系MHCクラスI対立遺伝子を通して、所望の抗原の提示を介してアロ制限ペプチド−特異的CD4およびCD8T細胞の両方を生成するために用いられることができる。米国特許出願第20020090362号明細書に記載された研究方法は、CD8T細胞の生成に適しているだけであることが注目される。
【0016】
第二に、選択されたTAAの公知ペプチドのみに対する初回抗原刺激に制限されているわけではないが、それは、全抗原が、DC中のプロセシングおよび提示のために利用可能だからである。抗原は、タンパク質の形態で、またはAPC中の対応タンパク質を発現するためのテンプレートとしてその後用いられる核酸の形態のどちらかで提供されてもよい。ここでもまた、先行技術の研究方法との差が著しい:米国特許出願第20020090362号明細書において、ペプチドが刺激細胞上に負荷される(これらは、プロフェッショナルAPCではない)が、それは、刺激細胞が抗原を処理および提示することができないからである。このようにして、公知ペプチドのみを、この研究方法において用いることができ、本発明における場合のように、未知の構造の抗原は用いられることができない。
【0017】
第三に、高い機能的結合活性を有するこれらの選択されたT細胞のTCR配列は、それぞれのTCRリガンドを担っている腫瘍に対して高い機能的結合活性を有するTCRトランスジェニックT細胞を生成させるために自家PBMC中の発現用に用いられることができる。ACTは、試験管内でかなりの時間にわたって単離および拡張されなければならない特異的リンパ球の養子転移の代替方法として、TCRトランスジェニックT細胞を用いて実施することができる。このことは、SCTを受けない患者の治療、ならびにトランスジェニックTCRによって見られる対応MHC−ペプチドリガンドを発現する他の非血液悪性腫瘍を有する患者における適用を可能にするであろう。
【0018】
特に本発明は、抗原特異的T細胞の生成方法であって、健康なドナーに由来し、かつ同じ健康なドナーに由来するPBLに自家性のAPCが用いられる方法を提供する。しかしながら、前記APC中にトランスフェクションされたMHC分子は、患者由来である。この文脈における患者由来とは、これをエンコードする配列が、患者から直接得られるか、あるいはまた別の源、例えば、治療されることになる患者のMHC対立遺伝子と同一であるcDNAもしくはゲノムクローンに由来することを意味する。さらには、本発明は初めて、所望の抗原特異的T細胞を得るために、一般にこれらのAPC中にMHC分子および抗原をコトランスフェクションさせるための研究方法を利用する。
【0019】
本発明のトランスフェクションされたAPCは、APCから見て自家性であるPBLを初回抗原刺激するために用いられることができる。転移されたMHC−抗原リガンドは、APC中に転移された前記MHC分子に対応するMHC遺伝子を保有しないAPCドナーのPBLについてのアロ決定因子を表わす。従って、高親和性TCRを担っているT細胞を得ることができる。APCは、自己−MHC分子の完全補体を発現するので、リンパ球の非MHC制限集団の出現は、陰性MHC調節を通して抑制される。この発明におけるAPCの使用、特にDCの使用は特に重要であるが、その理由は、これらの細胞が、これらのMHCクラスIおよびクラスII分子においてペプチドを効率的に処理し、提示し得るからである。これに加えて、APCは、追加の共刺激性分子の発現を特徴とし、これらの分子によって、これらはTリンパ球上に追加受容体をシグナルすることが可能になり、これは、T細胞の最適な活性化、拡張、および生存に導く。さらには、APC、特にDCは、初回抗原刺激されたリンパ球の機能に影響を与える多様なサイトカインおよびケモカインを分泌する能力を有する。APCによって作製された因子に応じて、応答リンパ球は、これらの亜類型、ホーミング能力、および機能的能力に関して修飾され得る。例として、DCによって示されたMHC−ペプチドリガンドとこれらのTCRとの相互作用によって、所望の抗原特異性を有するが、APCとして用いられた特定のDCによって分泌されたサイトカイン/ケモカインに基づく所望の機能も有するT細胞を活性化するために、成熟DCを用いることができる。様々な型のサイトカイン/ケモカインを分泌するDCを試験管内で生成することができ、T細胞の所望の型を刺激するために用いることができる。このようにして、これらに多量のサイトカインIL−12を分泌するようにさせる成熟カクテルでの培養において生成された成熟DCは、抗腫瘍免疫において特に重要なTヘルパーI型のCD4T細胞を活性化させる良好な能力を有する(Napolitaniら、2005年)。これに対して、IL−10を分泌する未成熟DCは、他のリンパ球の活性を抑制し得る調節T細胞の活性化において特に強力であるように見える(LevingsおよびRoncarola、2005年)。このような調節T細胞は、これによって、自己免疫疾患、例えば1型糖尿病を患っている患者における自己攻撃性T細胞の制御において、またはあまりに圧倒的になり、例えばハンセン病のいくつかの形態におけるような免疫病を引起こす病原体への免疫応答の制御において臨床的メリットを有することができる。
【0020】
DCは、この細胞中の様々な区画において発現された抗原を処理し、提示する能力を有するので、所望の抗原特異性を有するT細胞を生成させるために、正確な抗原ペプチドを知る必要はない。例えばDCに、完全なタンパク質抗原、またはこのようなタンパク質をエンコードするRNAが備えられる時、これらは、全く同一なタンパク質とは異なるいくつかのペプチドを処理し、これらをMHCクラスIおよびII分子中に提示することができる。これらの様々なMHC−ペプチドリガンドは、今度はCD4およびCD8T細胞の様々な集団を初回抗原刺激することができる。同種異系MHCクラスIおよび同種異系MHCクラスII分子を同時に発現させるためにDCを修飾することができない理由はないが、それは、これらが抗原を発現させるために修飾され、これによって同じ培養物中において、CD4およびCD8アロ制限T細胞の両方の初回抗原刺激を可能にするからである。活性化されたCD4T細胞とDCとの相互作用は、今度は、これらがCD8T細胞を最適に活性化することを可能にするシグナルをこれらに与えることができる(Toesら、1998年)。DCはさらに、いくつかの様々な抗原を同時に発現させるために修飾されることができる。実際、DCは、何百ものRNA種を包含する、腫瘍細胞のRNA内容物全体を発現するためにトランスフェクションされることができる。DCは、多重MHCクラスIおよびクラスIIリガンドをこれらの表面上で同時に処理され、提示されることができ、多くの様々なT細胞型の活性化に導く(GilboaおよびViewig、2004年;Geigerら、2005年;Schaftら、2005年)。抗原についてのRNAまたはタンパク質の混合物と同時に、同種異系MHCクラスIおよび/またはクラスII分子を共発現することによって、アロ制限MHC−ペプチドリガンドを作成するためにこの同じ特性を獲得することができない理由はない。
【0021】
本発明は、以下の実験結果によって裏付けられる。
1)第一に、本発明者らは、同種異系MHC分子、例えばHLA−A2をエンコードするRNAが、HLA−A2陰性ドナーの細胞中に転移された時に、HLA−A2分子が、HLA−A2特異的モノクローナル抗体での染色後、フローサイトメトリーを用いて検出されるように、細胞表面に発現されることを実証した。さらには、これらの細胞は、HLA−A2アロ特異的T細胞クローン(JB4細胞)を活性化して、サイトカイン(IFN−ガンマ)を分泌することができ、これらの機能的能力を実証している。転移および発現は、DCならびに他の細胞、例えばK562細胞において達成されることができる。
【0022】
2)同様に、腫瘍関連抗原(TAA)、例えば黒色腫についてのTAAの一例としてチロシナーゼをエンコードするRNAが転移された時、細胞の内部でタンパク質発現を検出することができるが、それは、これが非膜タンパク質であるからである。このタンパク質発現は、フローサイトメトリー、およびチロシナーゼ−特異的抗体を用いた細胞内染色、および検出のための二次蛍光標識抗体を用いて実証されることができる。転移および発現は、DCおよび他の細胞、例えばK562において達成されることができる。
【0023】
3)RNAの両方の種(HLA−A2+チロシナーゼ)を同じ細胞中に転移する時、受容者細胞におけるタンパク質の両方の型の同時発現を検出することができる。これは、DCおよび他の細胞、例えばK562細胞において達成されることができる。
【0024】
4)MHC分子、例えばHLA−A2、およびTAA、例えばチロシナーゼを共発現するAPCを、MHC−ペプチド複合体を生成し、対応リガンドについてのTCRを担っているT細胞と相互作用することができるような方法でこれらをその表面上に示す。このことは、このような共発現APCが、サイトカイン放出によって測定されるような特異性を有するT細胞クローンを活性化することができるという事実によって実証される。このことは、DCまたは他の細胞、例えばHLA−A2−チロシナーゼ−ペプチド特異的CD8T細胞クローンのためのAPCとしてK562を用いて実証された(Tyr−F8細胞)。
【0025】
特に本発明は、次の態様および実施形態を目的とする。
第一の態様によれば、本発明は、抗原特異的T細胞の生成方法であって、
a)患者由来MHC分子をエンコードする核酸および抗原、または前記抗原をエンコードする核酸を準備する工程、
b)工程a)において規定されているような両方の化合物を、健康なドナーに由来する抗原提示細胞(APC)、好ましくは樹状細胞中にコトランスフェクションするか、または導入する工程、
c)健康なドナーに由来する末梢血リンパ球(PBL)を、前記APCで初回抗原刺激する工程、
d)MHC−抗原リガンドに特異的なT細胞を選択する工程、
を含む方法を提供する。
【0026】
これらのAPCは、好ましくは、樹状細胞、活性化B細胞、単球、マクロファージ、活性化T細胞、抗原提示能力を有する血液悪性腫瘍、および/またはEBV−形質転換リンパ芽球様細胞株から選択される。
【0027】
樹状細胞(DC)が特に好ましい。成熟樹状細胞(DC)は、非常に多様な共刺激性分子とともに、MHCクラスIおよびクラスII分子の両方を高いレベルで発現する。これらは、以前に抗原に遭遇しなかったナイーブT細胞に初回抗原刺激する完全な能力をこれらに与える。これらはまた、これらのMHCクラスIおよびクラスII分子中の細胞内タンパク質からの抗原をこれらが処理および提示すること可能にする、すべての必要な遺伝子/タンパク質も有する。このようにしてこれらは、CD4およびCD8T細胞応答の両方の誘発のための刺激細胞として用いるのに最適な抗原提示細胞(APC)である。自家DC中のTAAをエンコードするRNAの発現は、高い親和性を有する腫瘍抗原特異的T細胞が、同じ健康なドナーの末梢血リンパ球を用いて試験管内で初回抗原刺激されることを可能にした(Liaoら、2004年)。DCは、自己−エンコードされたMHCクラスI分子の正常補体を発現するので、これらは、T2細胞または他の種の細胞、例えばショウジョウバエの細胞に対して、NKおよびNK様T細胞の活性の負の調節を行なうことができる。
【0028】
一つの実施形態によれば、MHC分子および抗原は、抗原と、MHC分子をエンコードする核酸との混合物として用いられる。代替実施形態として、同種異系MHC分子をエンコードする核酸および抗原が、ビシストロンRNAとして準備される。
【0029】
特異的T細胞がこれに対して生成させられるべきである抗原は、好ましくは、ウイルス、細菌、原生動物、および寄生虫に由来する病原性作用物質、ならびに腫瘍細胞もしくは腫瘍細胞関連抗原、自己抗原、またはこれらの機能部分から選択される。
【0030】
これらのウイルスは、好ましくはインフルエンザウイルス、はしかおよび呼吸器合胞体ウイルス、デングウイルス、ヒト免疫不全ウイルス、ヒト肝炎ウイルス、ヘルペスウイルス、または乳頭腫ウイルスからなる群から選択される。原生動物は、熱帯マラリア原虫であってもよく、細菌は、結核を引起こすマイコバクテリアであってもよい。
【0031】
腫瘍関連抗原は、好ましくは、血液悪性腫瘍もしくは充実性腫瘍、より好ましくは結腸癌、乳癌、前立腺癌、腎細胞癌(RCC)、肺癌、肉腫、または黒色腫細胞から選択される。
【0032】
選択工程d)は、好ましくは、T細胞のサイトカイン放出の測定、またはT細胞活性化の他の測定値によって実施される。例えば活性化T細胞は、個々の細胞としてクローンされることができ、拡張後、これらのT細胞クローンは、これらのMHC−ペプチド特異性について分析されることができ、所望の特異性を有するものが、さらなる使用のために選択され得る(Schendelら、1979年、1997年)。あるいはまた、様々な形態の可溶性MHC−ペプチドリガンド、例えばテトラマーは、蛍光ラベルでマークされることができ、活性化T細胞でインキュベーションされることができる。テトラマーと相互作用するTCRを担っているこれらのT細胞は、次いでフローサイトメトリーによって検出されることができ、これらの蛍光に基づいてソートされることができる(Yeeら、1999年)。T細胞はさらに、これらが反応すべき腫瘍細胞で短時間刺激することができ、これらのインターフェロンガンマ分泌を、捕獲試薬によって、例えば公開されているように(Beckerら、2001年)検出することができる。
【0033】
好ましい実施形態によれば、本発明の方法はさらに、d)生体外で選択されたT細胞の拡張工程も含む。このことは、選択されたT細胞を、これらの当初初回抗原刺激のために用いられたのと同じ方法で生成されたAPCとともに共培養し、新しいAPCをT細胞培養物へ7〜10日毎に添加し、拡張しているT細胞の型に応じて、補足サイトカインを含有する新鮮な培地を規則的にこれらの細胞に供給することによって実施されることができ、これは、(Schendelら、1997年;Regnら、2001年;Suら、2001年、2002年)に記載されているように、IL−2、IL−4、IL−7、および/または中でも特にIL−15を包含する。
【0034】
さらに、本発明の方法はさらに、単離T細胞のT細胞受容体(TCR)のクローン工程、および/またはPBMC中にTCRトランスジーンを発現する工程も含む。これは、確立された方法、例えばEngelsら、2005年に記載された方法に従って行われることができる。
【0035】
これが特異的MHCクラスへ制限されないことが、本研究方法の一つの大きい利点である。このようにしてMHC分子は、これらが患者のMHC型に対応する限り、MHCクラスI、好ましくはHLA−A、HLA−B、HLA−C、もしくはHLA−E、またはMHCクラスII、好ましくはHLA−DP、HLA−DQ、HLA−DRから選択されてもよい。
【0036】
第二の態様によれば、本発明は、上記の方法によって得ることができる、抗原特異的T細胞を提供する。
前記T細胞は好ましくは、エフェクター細胞特徴を有するT細胞、より好ましくはサイトカイン生産T細胞、細胞傷害性T細胞、または調節T細胞、好ましくはCD4+もしくはCD8+T細胞である。
【0037】
第三の態様において、本発明は、上に説明されたような方法によって得ることができる、トランスジェニックTCRについてコードする核酸を目的とする。
追加の態様は、トランスジェニックTCRについてコードする核酸を含んでいるベクターを目的とする。このベクターは好ましくは、本発明による核酸、および1またはそれ以上の調節核酸配列を含有する発現ベクターである。好ましくはこのベクターは、プラスミドまたはレトロウイルスベクターである。
【0038】
本発明はさらに、上に規定されているようなベクターで形質転換されたPBMCも含んでいる。
さらなる態様において、本発明は、上に説明されているようなT細胞もしくはPBMC、および製薬的に許容可能な担体を含んでいる製薬組成物を提供する。
【0039】
本発明のこれらの活性成分は好ましくは、疾病が治療されるか、または少なくとも軽減されうるような製薬組成物において、許容可能な担体もしくは担体材料と混合された用量で用いられる。このような組成物は、(活性成分および担体に加えて)充填材料、塩、緩衝剤、安定剤、可溶化剤、および他の材料を包含する。これらは、公知の最新技術である。
【0040】
「製薬的に許容可能な」という用語は、活性成分の生物活性の有効性を妨げない、非毒性材料を規定する。担体の選択は用途に依存している。
製薬組成物は、活性成分の活性を強化するか、または治療を補足する追加成分を含有することができる。このような追加成分および/または因子は、相乗効果を得るため、または副作用もしくは望まれない作用を最小限にするためにこの製薬組成物の一部であってもよい。
【0041】
本発明の活性成分の配合または調製および適用/薬物適用の技術は、「レミントンの製薬科学(Remington’s Pharmaceutical Sciences)」、ペンシルベニア州イーストンのマック・パブリッシング社(Mack Publishing Co.,Easton, PA)の最新版に公開されている。適切な適用は、非経口適用、例えば筋肉内、皮下、骨髄内注射、ならびに鞘内、直接心室内、静脈内、結節内、腹膜内、または腫瘍内注射である。静脈内注射が、患者の好ましい治療である。
【0042】
好ましい実施形態によれば、この製薬組成物は、注入または注射またはワクチンである。
さらなる態様によれば、本発明は、養子細胞療法のため、および特に血液悪性腫瘍もしくは充実性腫瘍および急性もしくは慢性感染の治療のための薬剤の製造について上で説明されているような抗原特異的T細胞もしくはPBMCの使用を目的とする(同様に上記参照)。
【0043】
本発明は、以下において実施例および図面によって例証される。
【図面の簡単な説明】
【0044】
【図1】樹状細胞の成熟、および悪性細胞に対する従来の免疫応答における特異的細胞傷害性T細胞の活性化を示している。
【図2】RNA−パルスされた樹状細胞によるナイーブ腫瘍特異的T細胞のデノボ初回抗原刺激を例証している。
【図3】試験管内転写RNAの導入を示している。
【図4】試験管内転写RNAの導入を示している。
【図5】試験管内転写RNAの導入を示している。
【図6】試験管内転写RNAの導入を示している。
【図7】試験管内転写RNAの導入を示している。
【図8】試験管内転写RNAの導入を示している。
【図9】試験管内転写RNAの導入を示している。
【図10】RNA−発現DCによるTyrF8細胞の再刺激に対するRNA濃度の影響を示している。
【図11】RNA−パルスされた樹状細胞でのナイーブT細胞のデノボ初回抗原刺激プロトコルを例証する。
【図12】RNA−トランスフェクションされた樹状細胞上のMHC−ペプチドリガンドの発現を示している。
【図13】HLA−A2−陽性およびHLA−A2−陰性ドナーからの初回抗原刺激されたT細胞のテトラマー染色を描いている。
【図14】特異性およびT細胞受容体結合活性についてのテトラマー−ソートされたバルクT細胞培養物の評価を示している。
【図15】特異性、T細胞受容体結合活性、および機能についてのT細胞クローンの評価を描いている。
【発明を実施するための形態】
【0045】
実験および図面の説明
図1.樹状細胞の未成熟および成熟段階
樹状細胞(DC)は、抗原に遭遇したことがないT細胞(ナイーブT細胞)を活性化するこれらの能力のために、抗原提示細胞(APC)のうち最も「プロフェッショナル」であると考えられる。これは、DCについて既に発見されているいくつかの因子に応じており、同様に依然として同定されるべき特徴に依存することがある。DCの様々な形態が存在し、これらの独特の特徴は、継続的に調査中である。臨床研究のために最も普通に用いられているDCの型は、CD14−陽性血液単球から区別することができる骨髄DCである。サイトカイン、顆粒球−単球コロニー刺激因子(GM−CSF)、およびインターロイキン−4(IL−4)の存在下におけるこのような単球の試験管内培養を通して、その周囲から(異種抗原を含む)材料を取り上げるその高い能力を特徴とする未成熟骨髄DCが生成される。未成熟DCが、この段階の間に適切なシグナルを受け取る時、これは活性化されて、リンパ管を通って二次リンパ節へ移動する。この移動の間、およびリンパ節に達した時、これはさらなる変化を受け、これが完全成熟段階に導く。成熟段階において、DCは、高いレベルのMHCクラスIおよびクラスII分子を発現し、これらによってこれは、細胞内タンパク質に由来するペプチドの形態における抗原を、それぞれCD8−陽性およびCD−4陽性T細胞へ提示することが可能にされる。MHC分子の両方の型によって提示され、DCの表面上に示された抗原は、抗原プロセシングおよび提示の様々な工程を含む、複雑な細胞プロセスにおいてDC中に生成される。MHCクラスI分子中のDC細胞表面において提示されたペプチド(すなわち抗原−断片)の場合、APCが抗原プロセシング関連輸送体(TAP)遺伝子、TAP1およびTAP2によってエンコードされた分子を発現する必要がある。
【0046】
DC表面に示されたMHC−ペプチドリガンドは、Tリンパ球(すなわちT細胞受容体)上に存在する抗原特異的受容体と相互作用する。この相互作用は、T細胞へ第一シグナル(すなわちシグナル1)をもたらす。このシグナルは、各T細胞について独自のTCRを通してもたらされるので、これは、適合TCRを有するT細胞を選択することによって、結果として生じる応答の抗原特異性を決定する。成熟DCはまた、いくつか挙げればCD40、CD80、CD86を包含する非常に多様な共刺激性分子も発現し、これらは、Tリンパ球によって発現された追加受容体と相互作用し、これらのさらなる活性化(すなわちシグナル2)を引起こす。DCはさらに、サイトカインおよびケモカインを包含する多様な可溶性分子を分泌する能力を有する。これらは様々な亜型のリンパ球をこれらの近隣へ引き付けることができ、シグナル1およびシグナル2を受け取るT細胞の分化に対して影響を与えうる。リンパ球によって発現されたさらなる組の受容体への結合後に、これらの因子は、これらの分化および最終的機能に影響を与えうるリンパ球へさらに別のシグナルをもたらすことができる。これは時にはシグナル3と呼ばれる。
【0047】
試験管内で未成熟および成熟DCを生成することが可能である。例えば単球を、様々な手順によって(細胞選択用の商業キットを用いたCD14−陽性細胞の正の選択、プラスチック商品へのこれらの接着特性によって、または細胞溶出法を用いて、これらのサイズおよび密度によって)得ることができる。これらの単球は、次いでGM−CSFおよびIL−4とともに培養されて、未成熟DCの集団を生じる。試薬の様々な組み合わせが、DCの成熟を誘発するため、および平行して可溶性メディエーターのこれらの分泌を改変するために用いられることができる。未成熟/成熟DCのこれら様々な形態が、今度はDCによって示されたMHC−ペプチドリガンドとこれらのTCRとの相互作用によって所望の抗原特異性を有するのみならず、DCによって分泌されたサイトカインに基づいた所望の機能も有するT細胞を活性化するために用いられることができる。このようにして、生成されて多量のサイトカインIL−12を分泌する成熟DCは、抗腫瘍免疫において特に重要なTヘルパー1型のCD4T細胞を活性化する良好な能力を有する。
【0048】
適切に活性化され、選択されるならば、T細胞は、腫瘍細胞、または様々な病原体に感染した細胞を認識して破壊する能力を有する。腫瘍細胞の場合、APCによって処理され、提示された腫瘍関連抗原(TAA)は多くの場合、腫瘍細胞において過剰発現された自己−タンパク質である分子を表わす。自己分子に由来するMHC−ペプチドリガンドと相互作用するTCRを担っている大部分のT細胞は、「負の選択」として公知の方法で胸腺において取り除かれるので、このようなTAA−リガンドを発現する腫瘍細胞を認識するのに十分な結合活性をともなうTCRを有するT細胞は、レパートリー中にほとんど存在しない。
【0049】
図2.RNA−パルスされた樹状細胞での、ナイーブ腫瘍特異的T細胞のデノボ初回抗原刺激
腫瘍細胞または他の細胞、例えば病原体で感染された細胞上でMHC−ペプチドリガンドを認識するより良好な能力を有するT細胞を得るために、同種異系MHC分子を介して、選択された1または複数の抗原に由来するペプチドを提示することによって、健康な個人からのT細胞の非選択レパートリーを選ぶ(tap)ことができる。例えば、HLA−A2でない健康なドナーAからのT細胞は、胸腺においてHLA−A2−ペプチド複合体へ暴露されたことはなく、したがって、高い結合活性を有するHLA−A2−ペプチド複合体と相互作用することができるTCRを発現するこのような個人の末梢血単核細胞(PBMC)のうちで利用可能なT細胞が存在する。このような所望のTCRを担っているナイーブT細胞を刺激するために、前記ドナーAのDCは単球から生成され、次いで修飾されて、同種異系MHCクラスIもしくはクラスII分子を発現する。ここで、HLA−A2についての一例が与えられる。この分子は、HLA−A2でないドナーAのT細胞についての同種異系MHC分子である。HLA−A2エンコードRNAをドナーAのDC中に導入することによって、HLA−A2分子をその細胞表面において発現するDCの集団を作製することが可能である。これによって、DCは依然として、ドナーAの染色体MHC遺伝子によってエンコードされた自己−MHC分子のその正常な組を発現するが、これに加えてそれは、導入されたRNAから翻訳された同種異系MHC分子を発現する。同時にDCは、ペプチドがDCによって処理され、細胞表面において同種異系HLA−A2分子によって提示され得るタンパク質抗原をエンコードするRNAが供給されることができる。このような同種異系MHC−ペプチドリガンドをその細胞表面において発現するDCが、ナイーブT細胞を刺激するためにAPCとして用いられる時、これらは、所望の同種異系MHC−ペプチドリガンドと相互作用するTCRを有するT細胞を活性化することができる。同種異系MHC分子を認識するすべてのT細胞が、所望のペプチド特異性を有するわけではないであろう。したがって所望のTCRを有するこれらの細胞は、活性化されたT細胞の全部の集団から選択されなければならない。この選択を得るための、免疫学者に周知のいくつかの手順がある。例えば活性化されたT細胞は、個々の細胞としてクローンすることができ、拡張後にこれらのT細胞クローンは、これらのMHC−ペプチド特異性について分析することができ、所望の特異性を有するものが、さらなる使用のために選択されることができる。あるいはまた、様々な形態の可溶性MHC−ペプチドリガンド、例えばテトラマーは、蛍光ラベルでマークすることができ、活性化されたT細胞でインキュベーションされることができる。これらのテトラマーと相互作用するTCRを担っているこれらのT細胞は、次いでフローサイトメトリーによって検出されることができ、これらの蛍光に基づいてソートされることができる。
【0050】
所望のTCR特異性で選択されたT細胞は、次いで対応HLA−A2−ペプチドリガンドを発現する腫瘍または感染細胞を有する患者の養子細胞療法(ACT)における使用のために試験管内で拡張されてもよい。あるいはまた、TCR配列を単離し、このようなT細胞について決定することができ、リンパ球または他の細胞中に導入して、ACTのために用いることができるトランスジェニックTCR細胞を生成することができるTCR構成体を生成するために用いることができる。
【0051】
図3.K562細胞中への試験管内転写RNAの導入
この戦略の実行可能性を実証するための第一実験方法として、本発明者らは、RNA転移後の同種異系HLA−A2MHC分子の発現のために、受容者細胞株としてK562細胞株を用いた。この細胞株はエリスロミエロイド白血病(erythromyeloid
leukaemia)に由来し、細胞免疫学者には周知である。これは腫瘍系であるので、多数になるまで成長させることができ、実験的な取扱いが容易である。これはさらに、その細胞表面上にMHCクラスI分子をまったく発現させず、従って、同種異系MHC分子の発現は、MHCクラスI分子をエンコードする核酸の導入後、K562の細胞表面において容易に検出することができる。
【0052】
この図面に要約された実験において、HLA−A2−エンコードcDNAからの試験管内転写によって生成されたHLA−A2 mRNAが、エレクトロポレーションを介してK562細胞中に導入された。一時的にRNAトランスフェクションされた細胞は、K562−A2細胞と呼ばれる、HLA−A2をエンコードする遺伝子で安定的にトランスフェクションされたK562細胞の一系列と比較された。
【0053】
HLA−A2分子の表面発現は、HLA−A2分子について特異的なモノクローナル抗体でのこれらの細胞のインキュベーション後にフローサイトメトリーによって決定され、データは、陽性細胞のパーセンテージとして示されている(左側の図面)。模擬対照として、K562細胞は、HLA−A2 mRNAの代わりに水を収容しているキュベットにおいてエレクトロポレーションされた。エレクトロポレーションに続くすべての時点で、模擬対照において陽性細胞はまったく検出されなかった。HLA−A2 RNAの3つの異なる量が、同数のK562細胞のエレクトロポレーションのために用いられた。HLA−A2陽性細胞の最高パーセンテージが、24マイクログラムの量のRNAを用いて検出され、細胞の最高パーセンテージは、エレクトロポレーションの11時間後に見られた。RNAのエレクトロポレーション後のHLA−A2分子を発現する陽性細胞のパーセンテージは、11時間および24時間の時点で、HLA−A2遺伝子を安定的に発現する正の対照K562−A2細胞のパーセンテージに匹敵した。HLA−A2 mRNAの導入後のHLA−A2の一時的発現は、48時間の時点での陽性細胞のパーセンテージの減少、および120時間の時点での無検出によって実証される。
【0054】
アロ抗原としてHLA−A2分子を認識するTCRを担っているT細胞が、HLA−A2 RNA−発現細胞によって活性化されうるかどうかを決定するために、JB4と呼ばれるT細胞クローンが研究された。このT細胞クローンは、そのTCRがHLA−A2−ペプチドリガンドと相互作用する時に、サイトカインインターフェロン−ガンマを分泌する。この認識のために必要とされる正確なペプチドは公知でないが、K562細胞、およびDC、および本発明者らがテストした大部分の他の細胞中に発現され、このようにして、これは、遍在性タンパク質に由来するように見える。JB4細胞がK562模擬トランスフェクション細胞とともに共培養された時、IFN−ガンマ放出は検出されない(右側の図面)。K562−A2細胞の正の制御では強い放出が見られる。様々な量のHLA−A2 RNAでトランスフェクションされたすべてのK562細胞は、正の対照よりも低いレベルにおいてではあるが、JB4細胞によってサイトカインの分泌を誘発することができた。それにもかかわらず、これらはサイトカイン放出の実質的なレベルである。JB4細胞の添加を有しなかったK562またはDCの培養において、サイトカイン放出は測定されず、刺激細胞がインターフェロン−ガンマをそれ自体分泌しないことを実証した。刺激細胞をともなわないJB4細胞の最終対照は、サイトカイン放出を全く示さず、T細胞が、インターフェロン−ガンマを分泌するのに適切なMHC−ペプチドリガンドによって活性化されなければならないことを実証した。
【0055】
これらの研究は、一時的なRNA発現を介したK562細胞中への同種異系MHC分子の導入が、HLA−A2分子の細胞表面発現へ導き、これらの細胞が、HLA−A2アロ抗原特異的T細胞クローンを活性化することができることを証明した。
【0056】
図4.DC中への試験管内転写RNAの導入
HLA−A2 mRNAのエレクトロポレーション後にDC上のHLA−A2分子の表面発現を評価するため、およびJB4細胞からのサイトカイン分泌を刺激するこれらの能力を評価するために、同様な実験方法が、図3に記載されているように用いられた。ここで、24マイクログラムの量のHLA−A2 mRNAが全体を通して用いられたが、mRNAの3つの異なる源が比較された。これらは、RNA発現をDC内部で安定させる意図で、ヒトアルファ−グロブリン遺伝子の3’UTRがHLA−A2遺伝子構成体へ添加されている試験管内転写RNAの商業源を含んでいた。このRNAは、アンビオン(Ambion)からの商業的手順およびキットを用いて生成されたポリアデニル化HLA−A2 RNA対キュアヴァック(CureVac)からの商業的手順およびキットを用いて生成された同様なRNAと比較された。アンビオン手順を用いて生成され、HLA−A2陰性ドナーから培養されたDC中にエレクトロポレーションによって導入されたポリアデニル化mRNAが、最良の結果を示した。このmRNAの場合にHLA−A2陽性細胞の最高パーセンテージが見られ、DCの表面におけるHLA−A2分子の出現は、より早く(6時間の時点で)検出され、DC中の発現は、このmRNA源を用いて、なおも120時間の時点で見られた。従って、すべてのさらなる実験は、このmRNA源を利用した。
【0057】
DCは、図3に記載されているように、JB4細胞からのサイトカイン分泌を刺激するこれらの能力について研究された。模擬対照DC(最も左の棒)は、サイトカイン放出のバックグラウンドレベルのみを誘発した。より高いレベルは、mRNAの3つの異なる源を発現するDCを用いて見られたが、アンビオンmRNAでエレクトロポレーションされたこれらのDCが優れていた。JB4細胞を含有しないDC培養物は、インターフェロン−ガンマを有しておらず、これがHLA−A2−RNA発現DCでの刺激後にJB4細胞によって放出されるだけであることを実証した。
【0058】
図5.腫瘍関連抗原チロシナーゼをエンコードする試験管内転写RNAのK562−A2細胞中への導入
RNAは、アンビオン手順を用い、抗原チロシナーゼに対して特異的に生成された。これはK562−A2細胞中にエレクトロポレーションされ、HLA−A2の表面発現が、HLA−A2分子に特異的なモノクローナル抗体を用いて測定された。緑の曲線はイソタイプ対照抗体での染色を表わし、青色の曲線はHLA−A2−特異的抗体での染色を表わす。ここで、HLA−A2表面発現がすべての時点で見られたが、それは、K562−A2細胞が、HLA−A2遺伝子を構成的に発現するからである。HLA−A2の発現の減少は、12時間の時点で注目され、これは、エレクトロポレーションの結果としてのストレス誘発された衝撃と関連付けることができる。これらの細胞は、その後HLA−A2分子の高い発現を回復した。チロシナーゼのタンパク質発現は、チロシナーゼに特異的なモノクローナル抗体を用いて、タンパク質の細胞内検出後に測定された。オレンジ色の曲線は模擬対照の染色を表わし、青色の曲線は、これらの細胞内部のチロシナーゼタンパク質を表わす。染色におけるわずかなシフトを、いくつかの時点で見ることができ、タンパク質が、チロシナーゼRNAの導入後に作製されつつあることを示している。
【0059】
図6.K562−A2細胞におけるチロシナーゼの発現および機能
チロシナーゼRNAが、K562−A2細胞中にエレクトロポレーションされ、陽性細胞のパーセンテージ、ならびに平均蛍光強度(MFI)と呼ばれる染色強度が様々な時点で決定された(左上部分)。赤い棒はK562−A2細胞上の構成的HLA−A2発現を示しており、ここでもまた、11〜24時間のレベルにおける低下およびその後の回復をともなう。青い棒は、6時間の時点でピークがあるチロシナーゼタンパク質の細胞内染色を示している。6時間の時点での染色のシフトは、右下部分に図解され、ここでは模擬対照染色がオレンジ色の曲線によって示され、チロシナーゼ染色は青色の曲線によって示されている。模擬対照と比較した、経時的な発現の倍数増加(fold increase)は左下部分に示され、11時間の時点で最大を生じる。HLA−A2分子によって提示されたチロシナーゼ由来ペプチドが分かるT細胞クローンTyr−F8のインターフェロン−ガンマ分泌が右上部分に示されている。K562−A2細胞は、Tyr−F8T細胞によるインターフェロン−ガンマ選択を誘発しないが、これらは、チロシナーゼRNAでのエレクトロポレーション後に可能である。これらのT細胞は単独でサイトカインを分泌しないが、これらはまた、チロシナーゼからの合成ペプチドが負荷されたK562−A2細胞によって、またはチロシナーゼおよびHLA−A2を共発現する黒色腫細胞(MEL−IL2細胞)によって刺激されることができる。
【0060】
このことは、K562−A2細胞中へのチロシナーゼについてのRNAの導入によって、これらがTyr−F8細胞のTCRによって見られる適切なMHC−ペプチドリガンドを処理および提示することを可能にし、これらのT細胞を活性化して、インターフェロン−ガンマを分泌させることを実証する。エレクトロポレーションの12時間後および24時間後に用いられたRNAトランスフェクションされた細胞は、匹敵し得る刺激性能力を有していた。
【0061】
図7.試験管内転写チロシナーゼRNAの、HLA−A2陽性ドナーから生成されたDC中への導入
同様な一組の実験が、図6に記載されている実験のように行なわれたが、HLA−A2陽性ドナーからのDCがK562−A2細胞の代わりに用いられた。細胞内チロシナーゼタンパク質の発現が、RNAのエレクトロポレーション後の様々な時点でDC中に検出された(上の図)。模擬対照全体における最高の平均倍数発現が、DC中で3時間の時点で見られた。DC単独ではTyrF8細胞によるサイトカイン分泌を刺激することはできなかったが、これらは、チロシナーゼRNAでのエレクトロポレーション後では可能であった。エレクトロポレーションの12時間後または24時間後に用いられたDCは、Tyr−F8T細胞を活性化して、インターフェロン−ガンマを分泌させる能力を有していた。
【0062】
図8.K562細胞におけるHLA−A2およびチロシナーゼの共発現
HLA分子をまったく発現しないK562細胞が、HLA−A2およびチロシナーゼについてのRNAでエレクトロポレーションされた。RNAの各種が個々に導入されるか、または両方が組み合わされて導入された。右上図は、異なる時点におけるHLA−A2単独(最も左側の一組の棒)、チロシナーゼ単独のタンパク質発現、次いでHLA−A2およびチロシナーゼRNAの両方でエレクトロポレーションされた細胞におけるHLA−A2染色、およびRNAの両方の種を受け取っている細胞におけるチロシナーゼ染色を要約している。RNAの両方の種でエレクトロポレーションされた細胞中の両方のタンパク質の最適発現が、24時間の時点で見られた。両方のRNAを共発現するK562細胞は、エレクトロポレーションの12時間後および24時間後にTyrF8細胞からのインターフェロン−ガンマ分泌を刺激することができた。
【0063】
図9.HLA−A2陰性ドナーから作製されたDCにおけるHLA−A2およびチロシナーゼの共発現
HLA−A2陰性ドナーから調製されたDCにおけるHLA−A2タンパク質およびチロシナーゼタンパク質の共発現が、図8に記載されているように分析された。両方のタンパク質の共発現が、様々な時点においてDC中に見られた。HLA−A2発現のレベルは、チロシナーゼRNAの存在下に低下されるように思われた。それにもかかわらず、両方のRNAを共発現するDCは、TyrF8細胞によるインターフェロン−ガンマの有意レベルを、T細胞単独のもの、またはRNAの単独種でエレクトロポレーションされたDCでのインキュベーション後のもの以上に刺激することができた。
【0064】
図10.RNA発現DCによるTyrF8細胞の再刺激へのRNA濃度の影響
DCがTyrF8細胞からのサイトカイン分泌を刺激する能力が、様々な量のHLA−A2およびチロシナーゼRNAでトランスフェクションされたHLA−A2陰性ドナーから調製されたDCを用いて分析された。模擬対照DC(最も左側の棒)が、サイトカイン放出のバックグラウンドレベルのみを誘発した。T細胞単独ではサイトカインを分泌しないが、これらはまた、チロシナーゼおよびHLA−A2を共発現する黒色腫細胞(MEL−IL2細胞)によっても刺激されることができる。
【0065】
様々な量の両方のRNAでトランスフェクションされたすべてのDCは、TyrF8細胞によるインターフェロン−ガンマの分泌を誘発することができたが、一方で、24μgHLA−A2および24μgチロシナーゼRNA、または48μgRNA HLA−A2および24μgチロシナーゼRNAでエレクトロポレーションされたこれらのDCの方が優れており、T細胞の再刺激のためにはより少ないRNAがより良好であろうことを示している。
【0066】
図11.RNAパルスされた樹状細胞でのナイーブT細胞のデノボ初回抗原刺激プロトコル
RNA負荷されたDCがナイーブT細胞を初回抗原刺激して腫瘍特異的リンパ球になる能力を分析するために、3相初回抗原刺激プロトコルが開発された。初回抗原刺激の開始に先立つ8日前(−8日目)、単球が、記載されているようにHLA−A2陽性ドナーおよびHLA−A2陰性ドナーからプラスチック接着によって調製された。これらの精製された単球集団が未成熟DCを調製するために用いられ、これらは、次いで記載されているように成熟させられた。T細胞初回抗原刺激プロトコルの最初の日(0日目)に、DCは、記載されているようにエレクトロポレーションを介してRNAが負荷された。HLA−A2陽性ドナーに由来するDCに、モデル腫瘍抗原として24μgのチロシナーゼRNAが負荷された。これらのDCにおけるRNAから翻訳されたチロシナーゼタンパク質は、DC表面において自己−エンコードされたHLA−A2分子と関連してペプチドとして処理され、提示されるべきである。HLA−A2陰性ドナーに由来するDCは、HLA−A2(48μg)およびチロシナーゼ(24μg)をエンコードするRNAが負荷された。ここで、転移RNAによってエンコードされたHLA−A2分子は、同種異系MHC分子を表わす。チロシナーゼタンパク質に由来するペプチドは、DCへ転移されたHLA−A2エンコードRNAから翻訳されたHLA−A2分子によって処理および提示されるべきである。これらのペプチド−MHC複合体は、アロ制限−ペプチドリガンドを表す。初回抗原刺激プロトコルの0日目に、各ドナーからの自家CD8+Tリンパ球が、商業的キットおよび製造業者の指示(CD8+T細胞単離キットII(ヒト)、ドイツ国ベルギッシュ・グラードバッハのミルテニ(Miltenyi,Bergisch Gladbach))を用いて負の選択を介して>80%純度まで単離された。これらの手のつけられていない自家CD8+T細胞が、エレクトロポレーションの9時間後に、IL−7(10ng/mL)を含有するエイム(Aim)V培地(ドイツ国カールスルーエのギブコ社(Gibco BRL,Karlsruhe))中に10:1の比で、成熟RNA−パルスされたDCへ添加された。IL−2(20IU/mL)が2日後に、ついでその後3日目毎に添加された。第一回刺激の7日後(7日目)、初回抗原刺激培養物の第二回刺激が、第一回刺激についてと同じ方法で調製されたRNA−パルスされたDCを用い、初回抗原刺激の0日目に用いられたのと同じ培養条件を用いて実施された。さらに7日後(14日目)、HLA−A2制限チロシナーゼ特異的T細胞が、フィコエリトリン(PE)−標識されたHLA−A*0201/htyr369−377ペプチド/ヒトβ2mテトラマー(Wolflら、2004年;ドイツ国ミュンヘン、技術大学の医学微生物学、免疫学、および衛生研究所、(Institute of Medical Microbiology,Immunology and Hygiene,Technical University,Munich)のProf.D.Buschによって提供された)の助けを借りてソートされた。正の選択をされたT細胞のいくつかは、96ウエルV底プレート(スイス国トラサディンゲン(Trasadingen)のTPP)において、1細胞/ウエルで培養された。これらのウエルに、3日毎に50IU/mL IL−2(ドイツ国マールブルクのヒロン・ベーリンク(Chiron Behring,Marburg))を含有するエイムV培地、および7日毎に5ng/mL IL−7(ドイツ国ハイデルベルクのプロモキネ(Promokine))および10ng/mL IL−15(米国ニュージャージー州のペプロ・テック社(Pepro Tech Inc.))が供給された。選択されたクローン、および残りのテトラマー選択された非クローンT細胞が、IL−2、IL−7、IL−15で上記のようにエイムV培地中の培養に維持され、支持細胞が供給された。これらの細胞は、50Gyが照射された5ドナーのプールに由来する末梢血単核細胞からなっていた。これらのT細胞は、2週間毎に抗−CD3抗体(0.1μg/mL;ドイツ国ミュンヘンの分子免疫学研究所(Institute of Molecular Immunology,GSF)のDr.Elisabeth Kremmerから提供された)で非特異的に刺激された。
【0067】
図12.RNA−トランスフェクションされた樹状細胞上のMHC−ペプチドリガンドの発現
T細胞初回抗原刺激のために用いられたDCが、予想されたMHC−ペプチドリガンドを発現することを実証するために、細胞表面におけるHLA−A2分子および細胞内チロシナーゼタンパク質の発現が、既に記載されているフローサイトメトリーを用いて決定された。HLA−A2分子およびチロシナーゼタンパク質の共発現は、両方のドナーに由来するDCにおいて高レベルで検出された。細胞内チロシナーゼタンパク質のレベルは、HLA−A2陽性およびHLA−A2陰性の両方のドナーから調製されたDCにおいて匹敵しうるものであった。大部分の細胞上のHLA−A2表面発現のレベルは、HLA−A2陽性ドナーから調製されたDCにおいて、より高かった。この場合、これらは内因性遺伝子によってエンコードされている。一方、トランスジェニックHLA−A2分子のレベルは、HLA−A2陰性ドナーから調製されたDC上で、より変わりやすい。
【0068】
独立実験における同じプロトコルに従って調製された細胞を用いた、このようなDCがT細胞を刺激する能力は、これらが保有するペプチド(すなわちHLA−A2+xyzペプチド)には関わりなくHLA−A2分子を認識するHLA−A2−アロ応答性T細胞クローンを用いて決定された。DCの両方の集団は、以前に記載されているような標準的ELISAにおいて測定されたように、このT細胞クローン(JB4)を刺激してインターフェロン−ガンマを分泌させることができた。DCをともなわずにインキュベーションされたT細胞からのサイトカイン放出は、まったく見られなかった。チロシナーゼ由来ペプチドを提示するHLA−A2分子からなるリガンドの存在が、HLA−A2−チロシナーゼペプチドについて特異的なT細胞クローン(Tyr−F8)によるインターフェロン−ガンマ放出を測定することによって評価された。DCをともなわずにインキュベーションされたT細胞は、サイトカインのバックグラウンドレベルのみを放出した。この実験において、HLA−A2−陽性DCは、48μgのチロシナーゼが供給され、一方、HLA−A2−陰性DCは、24μgのチロシナーゼが供給された。
【0069】
これらの結果は、HLA−A2−陰性ドナーに由来するDC中へのRNA転移によって供給されたトランスジェニックHLA−A2分子が、特異的HLA−A2抗体で細胞表面において染色することができ、HLA−アロ特異的T細胞クローン(JB4)によって認識することができることを実証した。チロシナーゼRNAの転移は、両方のDC集団における細胞内タンパク質発現に導いた。HLA−A2およびチロシナーゼRNAの共転移は、内因的にエンコードされたHLA−A2分子を発現するDC中へのチロシナーゼRNAの転移のように、HLA−A2−チロシナーゼ特異的T細胞クローン(TyrF8)が活性化されることを可能にした。
【0070】
図13.HLA−A2陽性およびHLA−A2陰性ドナーからの初回抗原刺激されたT細胞のテトラマー染色
初回抗原刺激されたT細胞培養物の中に存在するHLA−A2−チロシナーゼペプチドリガンドについて特異的なT細胞受容体を担っているT細胞を選択するために、これらのT細胞は、HLA−A*0201/htyr369−377/Hβ2mテトラマーで染色
され、フローサイトメトリーによって分析された。テトラマー結合を示した2つの異なる初回抗原刺激培養物の各々からの細胞がゲートされ(黒い四角、□)、MoFlo(商標)高性能細胞ショーター(米国コロラド州フォートコリンズのダコ(Dako,Fort Collins,CO))において単離された。HLA−A2陽性ドナーのCD8+テトラマー+T細胞集団は、低い強度においてのみテトラマー結合を示したが、一方で、HLA−A2陰性ドナーからのCD8+テトラマー+細胞は、より高い強度において染色された。これらのソートされたテトラマー+T細胞は、単一細胞として培養され、残りの細胞は、非クローンバルク培養物として培養中に保持された。
【0071】
これらの結果は、初回抗原刺激プロトコルが、HLA−A2−チロシナーゼ特異的テトラマーを結合することができるTCRを保有するT細胞を生じたことを実証した。より多くのT細胞が、HLA−A2−陰性ドナーに由来するDCを用いて初回抗原刺激された培養物においてより高い強度でテトラマーを結合させ、HLA−A2同種異系ペプチドリガンドによるより良い刺激を示している。
【0072】
図14.特異性およびT細胞受容体結合活性についての、テトラマーソートされたバルクT細胞培養物の評価
バルク培養物として保持された、ソートされた細胞は、8日後テトラマー染色について再分析された。予想されたように、ソートされた細胞の大部分は、初回抗原刺激培養物の両方に由来する、ソートされたバルク培養物中のCD8+であった。テトラマー+T細胞は、HLA−A2陽性DCを用いて初回抗原刺激された培養物中に約8%の頻度で存在した。これらの細胞は、テトラマー結合の中間的強度(MFI=127.49)を有したが、一方で、HLA−A2陰性ドナーからのバルク培養T細胞の80%が、はるかに高い強度(MFI=3248.77)を有するテトラマーを結合させた。テトラマー結合の強度は、特定のMHC−ペプチドリガンドについてのT細胞受容体(TCR)結合活性を評価するために用いられる1つのパラメーターである(Yeeら、1999年)。テトラマー結合のより高い強度は、このリガンドとTCRとのより良い相互作用を示している。TCR結合活性の第二パラメーターは、テトラマーオフレート(off−rate)である(Palermoら、2005年)。T細胞は、テトラマーでインキュベーションされ、次いで洗浄され、テトラマーの不存在下、およびHLA−A2分子について特異的な抗体の存在下にインキュベーションされる。この抗体の存在は、これらのT細胞の表面から落ちたテトラマーが、細胞表面へ再結合するのを防ぐ。これらのT細胞上のテトラマー染色の強度は、洗浄後の様々な時点:0時間、1時間、2時間の時点で決定される。テトラマー染色が急速に失われるならば、このことは、TCRが、テトラマーのMHC−ペプチドリガンドに対して低い結合活性しか有していないことを示している。テトラマー結合が経時的により安定であるならば、このことは、MHC−ペプチドリガンドに対してのより高いTCR結合活性を示す。これら2つのバルク培養物の比較において、もとのテトラマー−結合細胞(1.7%対8.3%)の20%のみが、HLA−A2陽性ドナーに由来する細胞において2時間後に依然としてテトラマー+であることが発見された。これに対して、これらの細胞の75%が、HLA−A2陰性ドナーに由来するT細胞において2時間後に依然としてテトラマー+であった(60.5%対80.6%)。
【0073】
テトラマー結合の特異性が、サイトメガロウイルスに由来するペプチドを提示するHLA−B7分子からなるテトラマー(Prof.D.Buschによって提供されたB7−CMVテトラマー)を用いて決定された。バルクT細胞培養物はどちらも、低レベルの染色(0.3〜0.4%)しか示さなかった。
【0074】
これに加えて、HLA−A2−CMVペプチドテトラマーも用いられた。HLA−A2陽性ドナーからのT細胞の0.4%のみが、このテトラマーを結合させたが、一方で、第二ドナーからのバルクT細胞の12.7%が陽性であった。これらのT細胞は、これらが提示するペプチドとは関わりなく、アロ抗原としてHLA−A2分子を認識するTCRを有するT細胞を表わす可能性がある。テトラマー結合の強度は、HLA−A2−チロシナーゼ−テトラマー結合の強度(MFI=3248.77)と比較して低かった(MFI=166.98)。
【0075】
これらの結果は、テトラマー染色の強度として規定されるようなより高い結合活性およびより遅いテトラマーオフレートを有するTCRを担っているT細胞が、HLA−A2陰性DCを用いて、これらの培養物に由来することを実証しており、同種異系−ペプチドリガンドを用いて、非負選択されたT細胞レパートリーにおいて応答を誘発する優位性を実証している。
【0076】
図15.特異性、T細胞受容体結合活性、および機能についてのT細胞クローンの評価
単一細胞培養実験から、2つのクローンがさらなる特徴決定のために選択された。1つのクローン(PS P4D11)がHLA−A2陽性ドナーに由来し、1つのクローン(SW P12B10)がHLA−A2陰性ドナーに由来した。
【0077】
HLA−A2陽性ドナーからのPS P4D11クローンは、1時間および2時間の時点で測定された時の急速なテトラマー放出をともなって、テトラマー染色の中間的強度(MFI=166.98)を示した。このT細胞クローンは、特異的ペプチドが負荷されたT2細胞(コネチカット州ニューヘブン、イエール大学のPeter Cresswellによって提供されたT2細胞)でのインキュベーション後も、HLA−A2およびチロシナーゼを発現する腫瘍細胞(Mel 624.38およびMel IL−2)での刺激後も、インターフェロン−ガンマを分泌するよう刺激することはできなかった。しかしながらこれは、HLA−A2+チロシナーゼ+黒色腫腫瘍細胞を殺すことはできるが、HLA−A2+チロシナーゼ−腫瘍細胞を殺すことはできず、HLA−A2−チロシナーゼリガンドについてのその特異性を実証した。
【0078】
HLA−A2陰性ドナーに由来するSW P12B10クローンは、テトラマー結合のより高い強度(MFI=1009.04)を示し、テトラマー結合の安定性は、2時間後にテトラマー対して依然として陽性である細胞のすべての場合により高かった。このクローンは、チロシナーゼ由来ペプチドが負荷されたT2細胞での刺激後に特異的インターフェロン−ガンマ放出を示したが、ペプチドをともなわないT2細胞に対しては可能ではなく、ペプチド特異性を実証した。これはまた、HLA−A2+チロシナーゼ+黒色腫腫瘍細胞での刺激後インターフェロン−ガンマを放出するように刺激することもできたが、HLA−A2陰性、チロシナーゼ陽性黒色腫細胞(SK Mel 28)に対して、またはHLA−A2陽性、チロシナーゼ陰性黒色腫腫瘍細胞(Mel A375)に対しては可能ではなかった。このパターンは、このクローンがHLA−A2−チロシナーゼペプチドに特異的であることを実証した。このクローンはまた、HLA−A2陽性、チロシナーゼ陽性黒色腫腫瘍細胞を殺すこともできたが、HLA−A2陽性、チロシナーゼ陰性腫瘍細胞を殺すことができなかった。
【0079】
これらの結果は、これら2つの異なる初回抗原刺激培養物に由来するT細胞クローンが、テトラマー結合の強度およびテトラマーオフレートによって検出された場合、TCR結合活性に関して差を示し、アロ制限ペプチド−特異的T細胞クローンが、この定義によってより高い結合活性のTCRを有することを示している。さらにはこのT細胞クローンが機能的優位性を示したが、それは、これが黒色腫細胞を特異的に殺すことができるだけでなく、ペプチド刺激後、および腫瘍細胞での刺激後、HLA−A2、チロシナーゼ特異的にインターフェロン−ガンマを分泌するように活性化することもできたからである。
【0080】
材料および方法
接着性腫瘍細胞株、Mel A375、Mel IL−2、Mel 624.38、およびSK Mel 28
これらのヒト黒色腫細胞株が、12%ウシ胎児血清、1×非必須アミノ酸、2mM L−グルタミン、および1mMナトリウムピルベートが補足されたRPMI 1640培地中で培養された。中程度のサイズの培養フラスコ中の培地の容積は、10mLであった。ほぼ3〜4日毎に、細胞は密集成長した。個々の細胞株の成長率に応じて、これらは1:2〜1:10に分けられた。培地が除去され、細胞はPBSで一回洗浄され、ついで2mLトリプシン/EDTA2×で3分間、室温でインキュベーションされた。脱離細胞が、新鮮なRPMI 1640培地+補足物中に再縣濁された。
【0081】
T細胞クローン、JB4、およびTyr−F8
T細胞クローンがまず、標準的再刺激培養物中で拡張された。大きい保存材料(stock)がアリコートされて冷凍された。実験に必要とされる度に、T細胞は保存材料から取られて解凍され、24−ウエルプレートのウエル中に入れられ、標準化されたプロトコルにしたがって再刺激された。
【0082】
縣濁腫瘍細胞株、T2、K562、およびK562−A2細胞
T2、K562、およびK562−A2が、12%FBS、1×MEM、2mM L−グルタミン、および1mMナトリウムピルベートが補足されたRPMI 1640培地中で培養された。中程度のサイズの培養フラスコにおいてこの培地の容積は10mLであった。ほぼ4日毎に、この細胞縣濁液の3/4が除去され、同じ容積の新鮮な培地が添加された。K562−A2細胞について、この培地は、1mg/mLの選択抗生物質G418が補足された。
【0083】
DC生成および培養
ドナーからのPBMCが、フィコール密度勾配遠心分離によって単離された。PBMCは、T75培養フラスコ1あたり7.5×106細胞で1%自家血漿が補足されたRPMI 1640培地中に再縣濁された。これらのフラスコは37℃および5%CO2で1時間インキュベーションされた。非接着性細胞が注意深く洗い流された。接着性単球が、1%自家血漿、800U/mL GM−CSF、および800U/mL IL−4が補足された樹状細胞培地の1フラスコあたり10mL中に培養された。3日間の培養後、800U/mL GM−CSFおよび800U/mL IL−4が再び添加された。培養の6日目に、800U/mL GM−CSF、500U/mL IL−4、5ng/mL IL−1β、9ng/mL IL−6、9ng/mL TNF−α、および1mLあたり2μM PGE2が、成熟を誘発するために未成熟DCへ添加された。成熟DCが、培養の8日目に収穫された。
【0084】
フローサイトメトリー
選択された分子の細胞発現を測定するために、フローサイトメトリーが用いられた。これらの分子は、蛍光染料へ付着されたモノクローナル抗体で特異的に染色される。フローサイトメトリー分析は、蛍光−活性化細胞ソーター(FACS(商標))において実施された。FACSCalibur(商標)において、染色された細胞の縣濁液がノズルを強制的に通され、1つずつ間隔をあけて配置された細胞を含有する液体の細い流れを生じた。各細胞がレーザービームを通過する時、これはレーザー光を散乱させ、この細胞と関連した蛍光分子が励起された。感受性光電倍増管が、細胞のサイズおよび粒度についての情報を与える両方の散乱光、および標識されたモノクローナル抗体の結合、および従って各細胞による標的分子の発現についての情報を与える蛍光発光を検出した。該細胞のソートのために、MoFlo(商標)高性能細胞ソーター(米国コロラド州フォートコリンズのダコ)が、25,000事象(events)/sおよび最大1psiで用いられた。
【0085】
細胞表面分子の直接染色
表面上のHLA−A2分子の発現を測定するために、ハイブリドーマ上澄み液HB82(メリーランド州ベセスダ(Bethesda)のATCC)、およびPE(フィコエリトリン)と共役された二次ヤギ抗マウス抗体が、細胞表面上の直接染色のために用いられた。約1×105DC、K562、またはK562−A2細胞が、1回洗浄された。各洗浄工程は、600μLのFACS(商標)緩衝液中の細胞の再縣濁、300×gで4分間の遠心分離、および上澄み液の廃棄を包含していた。洗浄後、50μLのHB82がこれらのペレットへ別々に添加された。氷上の30分のインキュベーション時間後、細胞は1回洗浄された。ついで0.5μLの二次抗体(1/100希釈;50μLのFACS(商標)緩衝液中の0.5μL抗体)が、これらのペレットへ別々に添加された。氷上および暗室での30分のインキュベーション時間後、細胞は1回洗浄された。最後に、細胞は200μLのFACS(商標)緩衝液中に再縣濁され、フローサイトメトリー分析が実施された。
【0086】
細胞内分子の間接染色
RNA−トランスフェクションされたDCの内部のチロシナーゼの発現を測定するために、FACS(商標)緩衝液中の1%パラホルムアルデヒドを用いて、細胞がまず固定された。固定後、これらの細胞は、FACS(商標)緩衝液中の0.1%および0.25%サポニン(Saponin)を用いて透過性化(permeabilise)された。簡単に言えば、約3×105細胞が1回洗浄された。各洗浄工程は、500μLのFACS(商標)緩衝液中、またはFACS(商標)緩衝液中の0.1%サポニン中の細胞の再縣濁、300×gで4分間の遠心分離、および上澄み液の廃棄を包含していた。洗浄後、FACS(商標)緩衝液(固定培地)中の500μLの1%パラホルムアルデヒドがこれらのペレットへ添加され、静かに混合された。氷上の30分のインキュベーション時間後、細胞は、FACS(商標)緩衝液で2回洗浄された。これらの細胞を500μLのFACS(商標)緩衝液中に4℃で7日まで保存することが可能であった。これらのペレットは、FACS(商標)緩衝液中500μLの0.1%サポニンで1回洗浄された。その後、FACS(商標)緩衝液(透過性化培地)中の50μLの0.25%サポニン、および5μLのチロシナーゼ特異的一次抗体が、ペレットへ添加され、静かに混合された。室温で1時間のインキュベーション後、細胞は、FACS(商標)緩衝液中の0.1%サポニンで1回洗浄され、ついでFACS(商標)緩衝液中の50μLの0.25%サポニン中一
次抗体(1/100希釈)について特異的なCy5−共役二次抗体0.5μLが、ペレットへ添加され、静かに混合された。室温および暗室での30分のインキュベーション後、これらの細胞は、FACS(商標)緩衝液中の500μLの0.1%サポニンで洗浄された。最後に、ペレット化された細胞が、FACS(商標)緩衝液中の200μLの0.1%サポニン中に再縣濁され、フローサイトメトリー分析が実施された。
【0087】
テトラマー染色およびテトラマーオフレート
HLA−A2−制限チロシナーゼ特異的CD8+T細胞の結合活性を評価するため、およびMoFlo(商標)高性能細胞ソーター(米国コロラド州フォートコリンズのダコ)でのソートを実施するために、これらのT細胞が、PE−標識A*0201/htyr3
69−377/Hβ2mテトラマー(Prof.D.Buschによって供給された)で染色された。これらの細胞は2回洗浄された。各洗浄工程は、冷PBS+0.5%ヒト血清中の細胞の再縣濁、300×gで5分間の遠心分離、および上澄み液の廃棄を包含していた。インキュベーション容積は細胞数に依存している。106までの細胞が、50μL中25分、PE−標識テトラマーで、氷上で暗室においてインキュベーションされた(1/25希釈、50μLPBS+0.5%ヒト血清中の2μLテトラマー)。HLA−A2−制限チロシナーゼ特異的CD8+T細胞のソートのために、5×106細胞までが、100μLPBS+0.5%ヒト血清中の6μL A2/ペプチドテトラマーでインキュベーションされた。次いで、さらに25分間、1/50の比においてCD8−APC抗体(米国フランクリンレイクスのBDファーミンゲン(Pharmingen,Franklin Lakes))が添加された。染色後、これらの細胞は2回洗浄され、最後にFACS(商標)緩衝液中の1%パラホルムアルデヒドで固定され、フローサイトメトリーFACSCalibur(カリフォルニア州マウンテンビューのベクトン・ディッキンソン(Becton Dickinson))によって分析されるか、またはMoFlo(商標)高性能細胞ソーター(米国コロラド州フォートコリンズのダコ)においてテトラマー+CD8+T細胞のソートのためにPBS+0.5%ヒト血清中に希釈された。
【0088】
テトラマーオフレートアッセイのために、洗浄後、脱離テトラマーの再結合は、飽和量のハイブリドーマ上澄み液HB82(メリーランド州ベセスダのATCC)でのインキュベーションによって防がれた。これは、複合ペプチドとは独立してHLA−A2を結合する。時間間隔を増加した後、サンプルが採取されて固定され、テトラマー染色の強度についてフローサイトメトリーによって分析された。
【0089】
単一種cRNAの生産
ここではcRNAと呼ばれる、単一種試験管内転写RNAの生産は、4つの工程を含んでいた。すなわち、cDNA挿入断片を含有するプラスミドの線形化、プロモーター配列および線形化されたプラスミド中のcDNAテンプレートに基づく試験管内転写、合成cRNAのポリアデニル化、およびcRNA精製である。実質量のcDNAテンプレート、すなわちプラスミドDNA(pDNA)が試験管内転写反応に必要とされるので、このプラスミドは、コンピテント細菌中で増幅されなければならなかった。
【0090】
プラスミドDNAでのコンピテント細菌の形質転換
他の基から親切な贈り物(kind gift)として受け取られる、より多量のpDNAを得るために、コンピテント細菌は該pDNAで形質転換されて拡張されなければならなかった。一回大量トップ(One Shot TOP)10F’コンピテント細胞の50μLガラス瓶が、融解しつつある氷上でゆっくりと霜取りされ、小さい容積のpDNAが添加され、静かに栓を抜く(tap)ことによって細菌と混合された。これらの細胞は、氷上で30分間インキュベーションされ、次いで42℃で正確に30秒間加熱され、最後に氷上に載せられて2分間冷却された。コンピテント細胞とともに供給されたリッチなSOC培地が添加され、形質転換された細菌が、適切な選択抗生物質とともにLB培地寒天プレート上に培養された。プレートが細菌インキュベーター中に37℃で一晩入れられた。抗生物質ゼオシンが光感受性であり、その活性は高い塩濃度によって阻害されるので、pZeoSV2+/huTyrで形質転換された細菌用のプレートは低塩LB寒天培地を含有し、暗室に保存された。
【0091】
形質転換細菌の選択および拡張
抗生物質への耐性をエンコードする、pDNAで形質転換された細菌のみが、寒天培地中の対応抗生物質の存在にもかかわらず、選択プレート上で成長してコロニーを形成することができた。一晩の成長後、コロニーは、滅菌つま楊枝を用いてこれらのプレートから引き出された。各々の個々のコロニーは、15mLファルコン(Falcon)管において、適切な抗生物質を含有する5mLのLB培地中に接種された。予め選択され、凍結された形質転換細菌が拡張されることになるならば、凍結ガラス瓶が、−80℃フリーザーから、融解しつつある氷上へ移された。約5μLの一部解凍された細菌縣濁液が、次いで、適切な抗生物質とともに5mLのLB培地を収容している15mLファルコン管に移された。これらの管は、一晩(約12時間)37℃で激しい振とうをともなって、150rpmで、形質転換された細菌の効率的な成長のためにインキュベーションされた。
【0092】
形質転換細菌の凍結
細菌は、凍結培地が15%のグリセリンを含有するならば、凍結プロセスによって害されることが最も少ない。したがって400μLの一晩細菌縣濁液が、84μLのオートクレーブされた87%グリセリンへ添加され、渦巻き撹拌され、液体窒素中に急速凍結され、−80℃へ移された。
【0093】
形質転換細菌からのプラスミドDNA抽出
一晩の培養後、細菌縣濁液が、5300×g、4℃で10分間遠心分離された。細菌細胞からのプラスミドDNA抽出が、QIAwell(登録商標)8ウルトラプラスミドキットを用いて、製造業者の指示に従って実施された。簡単に言えば、ペレット化された細胞が溶解された。溶解物中の塩およびpH条件が、DNAのこの膜への独占的結合を保証したが、一方、分解されたRNAおよび細胞タンパク質は保持されなかった。この膜はついで、あらゆる核酸結合タンパク質およびpDNAからの他の残留汚染物質の除去を可能にする、あらゆるDNA−タンパク質相互作用を乱す緩衝液で洗浄された。追加の洗浄工程は、塩および他の非−DNA成分を除去した。精製pDNAは最終的にトリス緩衝液中に溶出された。抽出pDNAの収率は、プラスミドに応じて、5mLの一晩細菌培養あたり4〜8μgであった。
【0094】
cRNA中への単一種cDNAの試験管内転写
各プラスミドが、試験管内転写に適したテンプレートを生産するために、適切な制限酵素で線形化された。すべてのプラスミドが、所望のタンパク質をエンコードするcDNAの5’末端においてT7プロモーターを含有するので、転写は、mMESSAGE mMACHINE(商標)T7キットからのT7RNAポリメラーゼを用いて製造業者の指示に従って実施された。RNA分子の安定性、およびタンパク質中への翻訳用テンプレートとしてのその効率的な使用は、その5’末端におけるキャップおよび3’末端におけるポリ(A)テイルの存在に依存している。ポリ(A)テイルは常に、細胞によって合成されたmRNA分子の一部である。従って、上述された全細胞mRNA増幅手順は、試験管内転写反応においてキャップ類似体の添加のみを含んでいた。しかしながら、プラスミド中のcDNAを用いて生産された単一種cRNAは、テイルが構成体においてエンコードされるか否かに応じて、ポリ(A)テイルを含有してもよいし含有しなくてもよい。試験管内転写反応における5’末端でのキャップ類似体の統合に加えて、チロシナーゼ、HLA−A2 cRNAは、ポリ(A)テイリングキットを用いて3’末端においてポリアデニル化された。両方の反応は、製造業者によって提案された条件下で実施された。試験管内転写およびcRNA精製後、cRNAの測定された収率は、100μLのポリアデニル化反応混合物あたり60〜70μgであった。
【0095】
cRNAの精製
増幅された全細胞cDNAまたはプラスミド中の単一種cDNAのどちらかをテンプレートとして用いて、試験管内転写反応において得られたcRNAの精製は、RNeasy(登録商標)ミニキットを用いて、製造業者の指示にしたがって実施された。精製のために、この手順は、試験管内転写反応混合物のRNeasy(登録商標)カラム上への転移から開始された。RNAはDEPC水中に溶出され、アリコートが−80℃で保存された。
【0096】
DC中へのRNAトランスフェクション
細胞血漿膜は、細胞の分子内容物をその外部環境から分離するという生命維持機能を果たしている。これらの膜は、高度に絶縁性の二層中に自己集合する両親媒性脂質から大部分なっている。可能な限り多くの抗原を捕獲しようとして、未成熟DCは、RNAを包含する、これらの周囲からの様々な構造(サイズが小さい分子から、アポトーシス体までの範囲のもの)を取り上げることが周知である。これらは、マクロ飲作用およびエンドサイトーシスを通して可能である。RNAとの単純DCコインキュベーションは、T細胞初回抗原刺激を達成することが証明されてはいるが、より攻撃的な方法、例えばリポフェクションおよびエレクトロポレーションは、より良好なトランスフェクション方法であることが証明された。
【0097】
エレクトロポレーション
エレクトロポレーションは、細胞が、強い外部電場の短いパルスへ暴露された時、血漿膜の可逆性透過性を誘発する。親水性細孔の形成は、二層膜中の脂質の再配向の結果である。この現象の分子メカニズムは依然としてよく理解されていない。細孔数およびこれらの直径は、パルス振幅およびパルス持続時間の積とともに増加する。小さい分子が、これらの細孔を通って細胞質中に単純に拡散するが、より大きい分子、例えば核酸は、電気泳動力によって細胞中に駆動される。核酸の存在が細孔形成を促進することも証明されている。細孔は、電場への暴露の1マイクロ秒以内に現われるが、これらが再閉するには数分かかる。
【0098】
最初に、2〜3×106DCまたはK562細胞が、少なくとも170μL OptiMEM I培地中に再縣濁され、0.4cmエレクトロポレーションキュベット中に入れられ、氷上で約3分間インキュベーションされた。エレクトロポレーション前の縣濁液の冷却は有利であるが、その理由は、これが細胞代謝を遅くしたからである。これによって電気による過熱が避けられた。さらには電気ショックによって、および死んだ細胞から放出された分子によって引起こされた損傷が最小限にされた。その後、100μL以下のDEPC水中に再縣濁されたRNAが添加され、270μLの総エレクトロポレーション縣濁液容積を生じた。この縣濁液は、ピペットによって短時間混合され、ついで急速にエレクトロポレーションされた。エレクトロポレーションは、250Vおよび150μFで実施された。エレクトロポレーションの直後に、細胞が、2%自家血漿、10mM HEPES、800U/mL GM−CSF、および800U/mL IL−4が補足された樹状細胞培地中に移された。これらの細胞が計数され、縣濁液が、FACS(商標)分析、RNA単離、または機能的アッセイのために迅速にアリコートされ、37℃および5%CO2でインキュベーター中に入れられた。
【0099】
機能的アッセイ
機能的アッセイにおいて、RNA−トランスフェクションされたDCまたはK562細胞、ペプチド−パルスされたDCまたは腫瘍細胞が、促進剤として役立った。抗原特異的CTLクローンが、エフェクターとして役立った。促進剤の刺激能力を測定するために、DCまたは他の細胞がまず、T細胞とともにコインキュベーションされ、これらは収穫され、解凍および再刺激の8〜14日後にコインキュベーション培養において用いられた。活性化されたT細胞によって分泌されたIFN−γの量が、酵素結合免疫吸着アッセイ(ELISA)において測定された。
【0100】
T細胞とのペプチド−パルスされたT2細胞コインキュベーション
T2細胞のCTLとのコインキュベーションのために、より低い細胞数が利用された。外因性ペプチドパルシングのために、1×106T2細胞が、10μg/mL YMDチロシナーゼペプチド(YMDGTMSQV、ミュンヘンLMUのジーンセンター)および10μg/mLヒトβ2−マイクログロブリン(米国サンディエゴのカルビオケム(Calbiochem))でインキュベーションされた。20分毎に細胞縣濁液が振とうされた。37℃、5%CO2での2時間のインキュベーション後、細胞縣濁液が、非結合ペプチドを除去するために1回洗浄された。100μLのペプチド−パルスされたT2縣濁液(12%FCSが補足されたRPMI 1640中の1×104細胞)が、100μLのCTL縣濁液(15%ヒト血清が補足されたCTL培地中の2×103細胞)とコインキュベーションされ、5:1の促進剤対エフェクター比を生じた。
【0101】
エフェクターおよび促進剤の24時間のコインキュベーション後、150μLの各上澄み液が収穫され、−20℃で保存された。上澄み液が、酵素結合免疫吸着アッセイ(ELISA)において分析された。
【0102】
T細胞とのDCもしくはK562コインキュベーション
エレクトロポレーションの12時間もしくは24時間後、100μLのT細胞縣濁液(コインキュベーション培地中2×104細胞)が、100μLのトランスフェクションされた細胞縣濁液(2%自家血漿および800U/mL GM−CSFおよび800U/mL IL−4が補足された樹状細胞培地中の2×104または4×104細胞)へ添加され、96ウエルプレートの1ウエルあたり全部で200μL、および1:4、1:2、1:1、2:1、または4:1の促進剤対エフェクター比を生じた。
【0103】
非トランスフェクションDCが、10μLのペプチド溶液(100μL/mL)を100μLのDC縣濁液(2%自家血漿および800U/mL GM−CSFおよび800U/mL IL−4が補足された樹状細胞培地中の4×104細胞)へ添加することによって、ペプチドで外因的にパルスされた。37℃および5%CO2での2時間のインキュベーション後、100μLのCTL縣濁液(CTLコインキュベーション培地中の2×104細胞)が添加され、2:1の促進剤対エフェクター比を生じた。エフェクターおよび促進剤の24時間のコインキュベーション後、150μLの各上澄み液が収穫され、−20℃で保存された。
【0104】
インターフェロン−ガンマ(IFN−γ)ELISA
共培養上澄み液中のIFN−γの測定が、ELISAにおいてOptEIA(商標)ヒトIFN−γセットを用いて、製造業者の指示に従って実施された。簡単に言えば、ELISAプレートは、マウス抗−IFN−γ捕獲抗体でコーティングされ、次いでFBS−含有溶液で遮断された。いくつかの事例では、上澄み液は、コインキュベーション培地中において1:2.5で希釈された。同じ培地が、IFN−γ標準の連続希釈液を作るために用いられた。細胞培養上澄み液または標準溶液からのIFN−γが、これらのプレート中の捕獲抗体へ結合された後、ビオチチニル化マウス抗−IFN−γ検出抗体が、ホースラディッシュ−ペルオキシダーゼへ共役されたアビジンとともに添加された。捕獲抗体からなる複合体を視覚化するために、IFN−γ、検出抗体、ビオチン、アビジン、およびホースラディッシュ−ペルオキシダーゼ、H2O2(ペルオキシダーゼのための基質)が、テトラメチルベンジジン(基質試薬AおよびBミックス)とともに添加され、溶液の色彩を青色に変えた。酵素反応が、1Mオルト−リン酸で停止された。これによって、溶液の色彩が黄色に変わり、その強度は、処理された基質の量に正比例した。すなわち捕獲されたIFN−γの量に間接的に比例した。反応溶液中の光吸収が、450nmで測定された。不明なIFN−γ濃度が、IFN−γ標準の公知濃度および対応測定吸光度に基づいて描かれた標準曲線の助けを借りて計算された。
【0105】
細胞傷害性アッセイ
それぞれテトラマー+CD8+ソートされたバルク細胞株およびT細胞クローンの細胞溶解活性が、標準4時間クロム放出アッセイにおいて分析された。これら2つのHLA−マッチされた黒色腫細胞株Mel A375(HLA−A2−陽性、チロシナーゼ−陰性)およびMel IL−2(HLA−A2−陽性、チロシナーゼ−陽性)が用いられた。簡単に言えば、106標的黒色腫細胞が、200μCi51Crで1〜1.5時間標識された。51Cr−標識された標的細胞が、V底96ウエル組織培養プレート(ドイツ国ゾリンゲンのグライナー(Greiner,Solingen))において100μL/ウエルの12%のFCSを有するRPMI 1640中でT細胞とともに培養された。CTL−媒介溶解の有効性を評価するために、T細胞は連続的に希釈され、ついで段階的エフェクター細胞対標的細胞(E:T)比を生じるために103標的細胞/ウエルと共培養された。37℃で4時間の共培養後、50μLの上澄み液が収集され、放射能がガンマカウンターで測定された。特異的溶解のパーセンテージは、100×(実験放出−自然放出)/(最大放出−自然放出)として計算された。自然放出は、エフェクター細胞の不存在下に標的細胞をインキュベーションすることによって評価され、一般に14%未満であった。
【0106】
参照
Becker, C, Pohla, H., Frankenberger, B., Assenmacher, M., Schendel, D.J. and Th. Blankenstein. 2001. Adoptive tumor therapy with T lymphocytes enriched through an IFN capture assay. Nature Med. 7(10): 1159-1162.
Dudley, M. E., Wunderlich, J. R., Robbins, P. F., Yang, J. C., Hwu, P., Schwartzentruber, D. J., Topalian, S. L., Sherry, R., Restifo, N. P., Hubicki, A. M., Robinson, M. R., Raffeld, M., Duray, P., Seipp, C. A., Rogers-Freezer, L., Morton, K. E., Mavroukakis, S. A., White, D. E., Rosenberg, S. A. (2002). Cancer regression and autoimmunity in patients after clonalrepopulation with antitumor lymphocytes. Science 298:850-854.
Dudley, M.E. and Rosenberg, S.A.(2003). Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nature Reviews Cancer 3: 666-675.
Engels, B., Nossner, E., Frankenberger, B., Blankenstein, Th., Schendel, D. J., and W. Uckert. 2005. Redirecting human T lymphocytes towards renal cell carcinoma-specificity by retroviral transfer of T cell receptor genes. Human Gene Then, 16(7). -79.9-810
FaIk, C. S. and Schendel, D. J. (2002). Allogeneic MHC class I ligandsand their role in positive and negative regulation of human cytotoxiceffector cells. Human Immunol. 63:8-19.
Gao, L., Bellantuono, I., Elsasser, A., Marley, S. B., Gordon, M. Y., Goldmann, J. M., Stauss, H. J. (2000). Selective elimination of leukemic CD34+ progenitor cells by cytotoxicT lymphocytes specific for WTl . Blood 95: 2198-2203.
Gao, L., Yang, T. H., Tourdot, S., Sadovnikova, E., Hasserjian, R. and Stauss H. J. (1999). AlIo- major-histocompatibility complex restricted cytotoxic T lymphocytes engraft in bone marrow transplant recipients without causing graft- versus-host disease. Blood 94: 2999-3006.
Geiger, C, Regn, S., Noessner, E., Wilde, S., Frankenberger, B., Papier, B., Pohla, H., and D.J. Schendel. 2005. Development of a generic RNA-pulsed dendriticcell vaccine for renal cell carcinoma, J. Translational Medicine in press.
Gilboa-E and J. Viewig. 2004. Cancer immunotherapy with mRNA-transfected dendriticcells. Immunol. Rev. 199:251-63.
KoIb, H. J., Schmid, C, Barrett, A. J. and Schendel, D. J. (2004). Graft-versus-leukemia reactions in allogeneic chimeras. Blood 103:767-776.
KoIb, H.J., Schattenberg, A., Goldman, J.M., Hertenstein, B., Jacobsen, H., ArceseW., Ljungman, P., Ferrant, A., Verdonck, L. Niederwieser, B. et al. 1995. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood 86:2041.
Levings, M.K. and M.G. Roncarolo. 2005. Phenotypic and functional differences between human CD4+CD25+ and type 1 regulatory T cells. Curr. Top. Microbiol. Immunol. 293:303-326.
Liao, X., Yongging, L., Bonini, C, Nair, S., Gilboa, E., Greenberg, P. D., Yee, C. (2004). Transfection of RNA encoding tumor antigens following maturation of dendritic cells leads to prolonged presentation of antigen and the generation of high-affinity tumor-reactive cytotoxic T lymphocytes. MoI. Ther. 9: 757-764.
Nair, S. K., Boczkowski, D., Morse, M., Cumming, R.I., Lyerly, H. K. and Gilboa, E. (1998). Induction of primary carcinoembryonic antigen (CEA)-specific cytotoxic T lymphocytes in vitro using human dendritic cells transfectedwith RNA. Nat. Biotechnol. 16(4): 364-369.
Napolitani, G., Rinaldi, A., Bertoni, F., Salllusto, F. and A. Lanzavecchia. 2005. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type-1 polarizing programme in dendritic cells. Nat. Immunol. 6(8): 769-76.
Regn, S., Chen, X., Schendel, D., KoIb, H.-J. and M.A. Roskrow. 2001. The generation of monospecific and bispecific anti-viral cytotoxic T lymphocytes (CTL) for the prophylaxis of patients receiving an allogeneic bone marrow transplant. Bone Marrow Transplant. 27: 53-64.
Schaft, N, Dorrie, J., Thuman, P., Beck, V. E., Muller, I., Schultz, E. S., Kampgen, E., Dieckmann, D. and Schuler, G. 2005. Generation of an optimized polyvalent monocyte-derived dendritic cell vaccine by transfectingdefined RNA after rather than before maturation. J. Immunol. 174(5): 3087-97.
Schendel, D. J., Wank, R. and B. Dupont. 1979. Standardization of the human in vitro cell-mediated lympholysis technique. Tissue Antigens 13: 112-120.
Schendel, D. J., Maget, B., FaIk, C. S. and R. Wank. Human CD8+ T lymphocytes. 1997. In: The Immunology Methods Manual. (I. Lefkovits, Ed.) pp 670-690, Academic Press Ltd., London.
Su, Z., Peluso, M. V., Raffegerst, S. H., Schendel, D. J., and M. A. Roskrow. 2001. LMP2a-specific cytotoxic T lymphocytes for the treatment of patients with relapsed EBV-positive Hodgkin disease. Eur. J. Immunol. 31: 947-958.
Su, Z., Peluso, M. V., Raffergerst, S. H., Schendel, D. J. and Roskrow, M. 2002. Antigen presenting cells transfected with LMP2a RNA induce CD4+ LMP2a-specif[iota]c cytotoxic T lymphocytes which kill via a Fas-independent mechanism. Leuk. Lymphoma 43(8): 1651-62.
Toes, R. E., Schoenberger, S. P., van der Voort, E. I., Offringa, R. and Melief, C. J. 1998. CD40- CD40Ligand interactions and their role in cytotoxic T lymphocyte priming and anti-tumor immunity. Semin. Immunol. 10(6): 443-8.
Yee, C, Savage, P. A., Lee, P. P., Davis, M. M. and P. D. Greenberg. 1999. Isolation of high avidity melanoma-reactive CTL from heterogenouspopulations using peptide-MHC-tetramers. J. Immunol. 162(4): 2227-34.
更なる参照
Belinda Palermo, Silvia Garbelli, StefaniaMantovani, Elisabetta Scoccia, Gian Antonio Da Prada, Paola Bernabei, M. Antonietta Avanzini, Valeria Brazzelli, Giovanni Borroni and Claudia Giachino(2005). Qualitative difference between the cytotoxicT lymphocyte responses to melanocyte antigens in melanoma and vitiligo. Eur. J. Immunol. 35: 3153-3162.
Cassian Yee, Peter A. Savage, Peter P. Lee, Mark M. Davis, and Philip D. Greenberg (1999). Isolation of High Avidity Melanoma-Reactive CTL from Heterogeneous Populations Using Peptide-MHC Tetramers. The Journal of Immunology: 2227-2234.
Matthias Wolfl, Stefan Schalk, Martin Hellmich, Katharina M. Huster, Dirk H. Busch, and Frank Berthold(2004). Quantitation of MHC Tetramer-Positive Cells From Whole Blood: Evaluation of a Single-Platform, Six-Parameter Flow Cytometric Method. Wiley-Liss, Inc. Cytometry Part A 57A: 120-130.
【技術分野】
【0001】
本発明は、抗原特異的T細胞の生成方法を目的とする。本発明はさらに、抗原特異的T細胞、単離トランスジェニックT細胞受容体(TCR)、これを含有する製薬組成物、および養子細胞療法におけるこれらの使用も目的とする。本発明は特に、非選択同種異系T細胞レパートリーからペプチド−特異的T細胞を誘発するための、同種異系MHC分子および抗原を共発現する細胞の使用に関する。
【背景技術】
【0002】
同種異系幹細胞移植(SCT)の設定におけるリンパ球の養子転移は、血液悪性腫瘍を根絶するための免疫系の力を実証している(Kolbら、1995年)。SCTはまた、いくつかの症例において、充実性腫瘍、例えば腎細胞癌(RCC)を取り除く作用も果たし得るように見える(Kolbら、2004年およびDudleyおよびRosenberg、2003年において概説されている)。SCT受容者において、悪性細胞の除去は、特異的T細胞が生体内で活性化されなければならず、次いで移植受容者における新しい造血系の発達後に適切な数まで拡張されなければならないという事実によって、数ヶ月から1年後にのみ発生し得る。あるいはまた、SCT受容者において耐性が確立される一定期間(約60日)後、非初回抗原刺激、非分離リンパ球の転移が、腫瘍細胞に対して向けられた免疫応答の生成を加速させることができる。ここでもまた、腫瘍細胞を攻撃し得る特異的リンパ球は活性化されなければならず、転移されるリンパ球の非選択集団中に存在する低周波数先駆物質リンパ球から拡張されなければならない。SCT後の非選択リンパ球集団のドナーリンパ球注入(DLI)は、ゆっくりと成長する慢性骨髄性白血病(CML)の除去のために良好に作用するが、部分的には、悪性細胞の成長が免疫細胞の拡張能力を凌ぐという事実によって、急性白血病の根絶において効果がより低い。この同じ拡張差はまた、急速に進行する充実性腫瘍の芳しくない免疫除去に対しても影響を与える。DLIにおける非選択リンパ球集団の使用における第二の不利な条件は、受容者の正常細胞および組織を攻撃する能力を有するT細胞もまた転移されることがあり、移植片対宿主疾患(GVHD)、すなわち高い罹患率および致死率を有する疾患に導くということである。
【0003】
最近の研究は、規定されたペプチド特異性を有する選択されたT細胞の養子転移が、特に患者が非骨髄機能廃絶方式で予め治療されているならば、自家設定における全身腫瘍組織量の大きな低減に導き得ることを実証している(Dudleyら、2002年、2003年)。このことは、腫瘍患者においてSCTを実施する必要性を取り除き、これによってGVHDの問題も回避する。効果的な免疫応答は、腫瘍浸潤リンパ球(TIL)の自家混合物を受けた、予め治療された黒色腫患者に見られた。CD4およびCD8の両方が陽性のT細胞を含有する細胞のこれらの混合物は、特定のMHC−腫瘍−関連抗原(TAA)リガンドについて特異的な多数の単一CD8T細胞クローンの養子転移よりも、臨床的にはより効果的に見える。この差に寄与する一つの因子は、CD4T細胞に長命CD8T細胞を維持させるための必要条件である。さらには、単一リガンドに対して向けられた免疫応答は、対応リガンドの発現を喪失した腫瘍細胞変異体の選択に導くことがあり、これによって免疫検出を免れることがある。一方で、T細胞の複合混合物の転移は、これらが腫瘍浸潤リンパ球の中に存在するので、CD4およびCD8細胞に多重特異性を与えることによってこれらの問題を克服し得るが、これらはまた、TILの混合物が、正常組織によって発現されたリガンドを認識するT細胞を含有するならば自己免疫に導き得る。このことは、例えばメラニン細胞中にも発現される黒色腫分化抗原を認識する細胞を用いた養子細胞療法(ACT)後に、黒色腫患者における白斑に導く正常メラニン細胞の攻撃によって実証される(Dudleyら、2002年)。
【0004】
より急速に成長する腫瘍を有する患者の治療のためにACTを用いる能力を拡大するために、正常組織の重大な攻撃を避けつつ、腫瘍細胞を効果的に攻撃するためのこれらのリガンド特異性のために選択された強化ペプチド−特異的エフェクターT細胞(CD4Tヘルパー細胞および細胞傷害性Tリンパ球の両方)を転移することが一つの目標である。これらの細胞は、生体外で急速に多数になるまで拡張させられ、ついでACTのために用いられることになる。あるいはまた、このようなリガンド−特異的T細胞のT細胞受容体(TCR)は、受容者末梢血リンパ球か、または十分に成長し、正常な宿主組織を攻撃する能力を有していない、規定された特異性を有する活性化されたT細胞クローンのどちらかを用いてクローンすることができ、活性化されたリンパ球中にTCR−トランスジーンとして発現させることができる。
【0005】
例として、受容者によって発現されないMHC分子について特異的な拡張アロ特異的T細胞クローン、またはウイルス、例えばサイトメガロウイルスまたはエプスタイン−バーウイルスについて特異的な拡張T細胞クローンは、トランスジェニックTCRへの受容体細胞として用いられることができるであろう。異なるMHC−ペプチドリガンドを認識するトランスジェニックTCRベクターのパネルの利用可能性は、CD4およびCD8亜類型の両方の多数の予め活性化されたT細胞を開発するために利用することができ、これによって多数のエフェクターリンパ球が迅速に調製され、対応TCRリガンドを発現する腫瘍を有する患者へ転移させることを可能にするであろう。このことは、腫瘍成長を制御するために必要とされる特異的T細胞の数の達成において時間を節約し、おそらくは、急速に進行する腫瘍のより効果的な腫瘍根絶に導くであろう。
【0006】
特異的T細胞が白血病およびリンパ腫、ならびに充実性腫瘍細胞に関して認識する決定因子は、自己−MHC分子によって提示される過剰発現タンパク質に由来する自己−ペプチドを代表することが多いことから、これらのT細胞受容体(TCR)の親和性は低い。それは、高い親和性受容体を担っているT細胞が、胸腺中のこれらの発達の間にリンパ球へ適用される負の選択プロセスを通して取り除かれているからである。これらのT細胞が、胸腺中のこれらの発達の間に自己−MHC分子に対して負の選択をされなかったリンパ球集団から生成されるならば、より効果的な腫瘍細胞認識が発生する。
【0007】
したがって、腫瘍細胞上のこれらのリガンドを認識する能力を有する高い機能的結合活性を有するTCRを担っているT細胞を迅速に生成する手段を発見する重要なニーズが存在する。このようなT細胞は、潜在的なACT受容者へのMHC−ミスマッチを有する同種異系個人のレパートリー中に存在する。
【0008】
Staussおよび共同研究者の研究(Gaoら、1999年、2000年)は、転写因子WT−1に由来し、かつ同種異系MHCクラスI分子によって提示されるペプチドを認識するために選択された細胞傷害性エフェクター細胞が高親和性TCRを有し、正常な幹細胞に損傷を与えずにWT−1陽性白血病を効果的に取り除きうることを証明している。部分的ミスマッチのSCTの設定において、このようなT細胞への耐性を確立することができ、これらの特異的養子転移を可能にする。Staussは、刺激細胞源として、WT−1ペプチドでパルスされたT2細胞株を用いて、このようなアロ制限CD8T細胞を得るという問題を解決した。T2細胞は、内因性ペプチドをこれらのMHCクラスI分子中に負荷するこれらの能力を損なう遺伝子欠失を有し、染色体6における欠失によって、これらの細胞は、HLA−A2分子を発現する能力が限定されている。ペプチドがT2細胞へ外因的に供給される時、これらは中空HLA−A2分子へ結合し、細胞表面において安定な複合体を形成し得る。これらのペプチド−パルスされた細胞は、次いでHLA−A2陰性個人からの末梢血リンパ球を刺激するために用いられることができる。このような初回抗原刺激条件下、いくつかの異なる活性化リンパ球集団が試験管内に現われる。HLA−A2分子を認識するT細胞が活性化され、これの一フラクションは、WT−1/HLA−A2リガンドについて特異的である。これらの探索されたT細胞は、ペプチドに関わりなくHLA−A2分子を認識するT細胞から分離されなければならない。残念ながら、T2細胞の、MHCクラスI分子の正常補体の発現の不成功、ならびに非MHC−制限細胞を活性化するその固有の能力は、NKおよびNK様T細胞の平行活性化に導く(Falkら、2002年)。これらの集団は多くの場合、これらの培養物を支配し、これらの細胞を取り除くため、および所望のアロ反応性細胞のためのT細胞集団を強化するために、冗漫な研究を必要とする。このことは、特異的T細胞の生成プロセスを遅延させ、したがって重大な臨床的制限を引起こす。さらには、これらの抗原からのペプチドは、前もって知られていなければならないが、それは、これらが外部からT2細胞上にパルスされなければならないからであり、その理由は、抗原提示細胞が、この細胞中に発現されたタンパク質からペプチドを生成することを可能にし、かつMHCクラスIおよびクラスII分子中でこれらの表面上にこれらを提示する抗原プロセシング関連輸送体(TAP)遺伝子をT2細胞が欠いているからである。さらには、T2のみが、内因性HLA−A2クラスI分子を発現する。
【0009】
米国特許出願第20020090362号明細書は、患者の治療方法であって、細胞の表面上のHLAクラスI(または同等)分子によって提示された時に、抗原分子の少なくとも一部を認識する細胞傷害性Tリンパ球(CTL)の治療的有効量を患者に投与する工程を含み、これらの細胞傷害性Tリンパ球が患者に由来しない方法を開示している。この出願は、CTLのドナーから見て同種異系であるか、または異種でさえある刺激細胞の使用について記載している。米国特許出願第20020090362号明細書はさらに、好ましくは選択された分子(ペプチド)の負荷が不可能である刺激細胞、および特にTAP欠乏刺激細胞の使用も開示している。
【0010】
米国特許第6,805,861号明細書は、選択された分子に対して反応性がある細胞傷害性Tリンパ球(CTL)のクローン集団の作製方法であって、(a)健康な個人に由来する(すなわち患者に由来しない)CTLを含有するサンプルを、HLAクラスI(または同等)分子をその表面上に発現し、かつ選択された(抗原)分子の少なくとも一部を促進剤細胞の表面上に提示する促進剤細胞とともに共培養する工程、および(b)前記分子の少なくとも一部が、細胞の表面上のHLAクラスI(または同等)分子によって提示される時に前記選択された分子に対して反応性のCTLクローンを選択する工程を含む方法について記載している。米国特許出願第20020090362号明細書について言及された考察が、ここでもまた適用される。
【発明の概要】
【発明が解決しようとする課題】
【0011】
したがって、病原性作用物質、例えば腫瘍細胞上にそのMHC−ペプチドリガンドを認識する能力を有する高い機能結合活性を有するTCRを担っているT細胞を生成させることが、本発明の基礎となる一つの問題である。養子細胞転移において用いることができる抗原特異的T細胞の迅速かつ効果的な生成方法を提供することが、本発明の基礎となるさらなる問題である。さらには、移植片対宿主疾患(GVHD)のリスクをともなわずに、病気を患っている患者の治療のために用いることができるT細胞ベースの製薬組成物を提供することが、本発明の基礎となる一つの問題である。
【課題を解決するための手段】
【0012】
本発明者らは、アロ制限ペプチド−特異的T細胞を得るための代替戦略を開発した。試験管内転写RNAの転移を通して(Nairら、1998年)、樹状細胞(DC)中に、または抗原提示細胞(APC)として機能し得る他の細胞中にタンパク質を発現させるための周知技術が用いられた。しかしながら、先行技術において通常であるように、抗原源をエンコードするRNAのみを発現する代わりに、本発明者らは、特異的標的分子、例えばモデルTAAとしてチロシナーゼをエンコードするRNA、および同種異系MHC分子、例えばHLA−A2をエンコードするRNAを、HLA−A2陰性ドナーに由来するDCまたは他の細胞中にコトランスフェクションした。
【0013】
これらのトランスフェクションされたDCは、次いでPBL、例えば自家PBLを初回抗原刺激するために用いられることができる。HLA−A2抗原リガンドは、HLA−A2陰性DCドナーのPBLについてのアロ決定因子を表し、従って高親和性TCRを担っているT細胞を得ることができる。DCが自己−MHC分子の完全補体を発現するので、リンパ球の非MHC−制限集団の出現は、陰性MHC調節を通して抑制される。いくつかの様々な戦略を、抗原に由来する特異的ペプチドとは独立してHLA−A2を認識するT細胞からペプチド−特異的/HLA−A2アロ制限T細胞を強化するために用いることができる。これは、サイトカイン捕獲、テトラマー選択、またはその後に拡張される個々のT細胞のクローニングによる戦略を包含する。
【0014】
本研究方法は、(Staussおよび共同研究者によって記載されているように)アロ制限ペプチド−特異的T細胞を得るために、T2系またはショウジョウバエ細胞に勝るいくつかの利点を提供する。
【0015】
第一に、あらゆるクローンされたMHCクラスIまたはクラスII対立遺伝子のために試験管内転写されたRNAを利用することができるので、多様なMHC分子についてのアロ制限を有するT細胞を開発することができる。従って、このようなAPCは、それぞれ転移された同種異系MHCクラスIIまたは同種異系MHCクラスI対立遺伝子を通して、所望の抗原の提示を介してアロ制限ペプチド−特異的CD4およびCD8T細胞の両方を生成するために用いられることができる。米国特許出願第20020090362号明細書に記載された研究方法は、CD8T細胞の生成に適しているだけであることが注目される。
【0016】
第二に、選択されたTAAの公知ペプチドのみに対する初回抗原刺激に制限されているわけではないが、それは、全抗原が、DC中のプロセシングおよび提示のために利用可能だからである。抗原は、タンパク質の形態で、またはAPC中の対応タンパク質を発現するためのテンプレートとしてその後用いられる核酸の形態のどちらかで提供されてもよい。ここでもまた、先行技術の研究方法との差が著しい:米国特許出願第20020090362号明細書において、ペプチドが刺激細胞上に負荷される(これらは、プロフェッショナルAPCではない)が、それは、刺激細胞が抗原を処理および提示することができないからである。このようにして、公知ペプチドのみを、この研究方法において用いることができ、本発明における場合のように、未知の構造の抗原は用いられることができない。
【0017】
第三に、高い機能的結合活性を有するこれらの選択されたT細胞のTCR配列は、それぞれのTCRリガンドを担っている腫瘍に対して高い機能的結合活性を有するTCRトランスジェニックT細胞を生成させるために自家PBMC中の発現用に用いられることができる。ACTは、試験管内でかなりの時間にわたって単離および拡張されなければならない特異的リンパ球の養子転移の代替方法として、TCRトランスジェニックT細胞を用いて実施することができる。このことは、SCTを受けない患者の治療、ならびにトランスジェニックTCRによって見られる対応MHC−ペプチドリガンドを発現する他の非血液悪性腫瘍を有する患者における適用を可能にするであろう。
【0018】
特に本発明は、抗原特異的T細胞の生成方法であって、健康なドナーに由来し、かつ同じ健康なドナーに由来するPBLに自家性のAPCが用いられる方法を提供する。しかしながら、前記APC中にトランスフェクションされたMHC分子は、患者由来である。この文脈における患者由来とは、これをエンコードする配列が、患者から直接得られるか、あるいはまた別の源、例えば、治療されることになる患者のMHC対立遺伝子と同一であるcDNAもしくはゲノムクローンに由来することを意味する。さらには、本発明は初めて、所望の抗原特異的T細胞を得るために、一般にこれらのAPC中にMHC分子および抗原をコトランスフェクションさせるための研究方法を利用する。
【0019】
本発明のトランスフェクションされたAPCは、APCから見て自家性であるPBLを初回抗原刺激するために用いられることができる。転移されたMHC−抗原リガンドは、APC中に転移された前記MHC分子に対応するMHC遺伝子を保有しないAPCドナーのPBLについてのアロ決定因子を表わす。従って、高親和性TCRを担っているT細胞を得ることができる。APCは、自己−MHC分子の完全補体を発現するので、リンパ球の非MHC制限集団の出現は、陰性MHC調節を通して抑制される。この発明におけるAPCの使用、特にDCの使用は特に重要であるが、その理由は、これらの細胞が、これらのMHCクラスIおよびクラスII分子においてペプチドを効率的に処理し、提示し得るからである。これに加えて、APCは、追加の共刺激性分子の発現を特徴とし、これらの分子によって、これらはTリンパ球上に追加受容体をシグナルすることが可能になり、これは、T細胞の最適な活性化、拡張、および生存に導く。さらには、APC、特にDCは、初回抗原刺激されたリンパ球の機能に影響を与える多様なサイトカインおよびケモカインを分泌する能力を有する。APCによって作製された因子に応じて、応答リンパ球は、これらの亜類型、ホーミング能力、および機能的能力に関して修飾され得る。例として、DCによって示されたMHC−ペプチドリガンドとこれらのTCRとの相互作用によって、所望の抗原特異性を有するが、APCとして用いられた特定のDCによって分泌されたサイトカイン/ケモカインに基づく所望の機能も有するT細胞を活性化するために、成熟DCを用いることができる。様々な型のサイトカイン/ケモカインを分泌するDCを試験管内で生成することができ、T細胞の所望の型を刺激するために用いることができる。このようにして、これらに多量のサイトカインIL−12を分泌するようにさせる成熟カクテルでの培養において生成された成熟DCは、抗腫瘍免疫において特に重要なTヘルパーI型のCD4T細胞を活性化させる良好な能力を有する(Napolitaniら、2005年)。これに対して、IL−10を分泌する未成熟DCは、他のリンパ球の活性を抑制し得る調節T細胞の活性化において特に強力であるように見える(LevingsおよびRoncarola、2005年)。このような調節T細胞は、これによって、自己免疫疾患、例えば1型糖尿病を患っている患者における自己攻撃性T細胞の制御において、またはあまりに圧倒的になり、例えばハンセン病のいくつかの形態におけるような免疫病を引起こす病原体への免疫応答の制御において臨床的メリットを有することができる。
【0020】
DCは、この細胞中の様々な区画において発現された抗原を処理し、提示する能力を有するので、所望の抗原特異性を有するT細胞を生成させるために、正確な抗原ペプチドを知る必要はない。例えばDCに、完全なタンパク質抗原、またはこのようなタンパク質をエンコードするRNAが備えられる時、これらは、全く同一なタンパク質とは異なるいくつかのペプチドを処理し、これらをMHCクラスIおよびII分子中に提示することができる。これらの様々なMHC−ペプチドリガンドは、今度はCD4およびCD8T細胞の様々な集団を初回抗原刺激することができる。同種異系MHCクラスIおよび同種異系MHCクラスII分子を同時に発現させるためにDCを修飾することができない理由はないが、それは、これらが抗原を発現させるために修飾され、これによって同じ培養物中において、CD4およびCD8アロ制限T細胞の両方の初回抗原刺激を可能にするからである。活性化されたCD4T細胞とDCとの相互作用は、今度は、これらがCD8T細胞を最適に活性化することを可能にするシグナルをこれらに与えることができる(Toesら、1998年)。DCはさらに、いくつかの様々な抗原を同時に発現させるために修飾されることができる。実際、DCは、何百ものRNA種を包含する、腫瘍細胞のRNA内容物全体を発現するためにトランスフェクションされることができる。DCは、多重MHCクラスIおよびクラスIIリガンドをこれらの表面上で同時に処理され、提示されることができ、多くの様々なT細胞型の活性化に導く(GilboaおよびViewig、2004年;Geigerら、2005年;Schaftら、2005年)。抗原についてのRNAまたはタンパク質の混合物と同時に、同種異系MHCクラスIおよび/またはクラスII分子を共発現することによって、アロ制限MHC−ペプチドリガンドを作成するためにこの同じ特性を獲得することができない理由はない。
【0021】
本発明は、以下の実験結果によって裏付けられる。
1)第一に、本発明者らは、同種異系MHC分子、例えばHLA−A2をエンコードするRNAが、HLA−A2陰性ドナーの細胞中に転移された時に、HLA−A2分子が、HLA−A2特異的モノクローナル抗体での染色後、フローサイトメトリーを用いて検出されるように、細胞表面に発現されることを実証した。さらには、これらの細胞は、HLA−A2アロ特異的T細胞クローン(JB4細胞)を活性化して、サイトカイン(IFN−ガンマ)を分泌することができ、これらの機能的能力を実証している。転移および発現は、DCならびに他の細胞、例えばK562細胞において達成されることができる。
【0022】
2)同様に、腫瘍関連抗原(TAA)、例えば黒色腫についてのTAAの一例としてチロシナーゼをエンコードするRNAが転移された時、細胞の内部でタンパク質発現を検出することができるが、それは、これが非膜タンパク質であるからである。このタンパク質発現は、フローサイトメトリー、およびチロシナーゼ−特異的抗体を用いた細胞内染色、および検出のための二次蛍光標識抗体を用いて実証されることができる。転移および発現は、DCおよび他の細胞、例えばK562において達成されることができる。
【0023】
3)RNAの両方の種(HLA−A2+チロシナーゼ)を同じ細胞中に転移する時、受容者細胞におけるタンパク質の両方の型の同時発現を検出することができる。これは、DCおよび他の細胞、例えばK562細胞において達成されることができる。
【0024】
4)MHC分子、例えばHLA−A2、およびTAA、例えばチロシナーゼを共発現するAPCを、MHC−ペプチド複合体を生成し、対応リガンドについてのTCRを担っているT細胞と相互作用することができるような方法でこれらをその表面上に示す。このことは、このような共発現APCが、サイトカイン放出によって測定されるような特異性を有するT細胞クローンを活性化することができるという事実によって実証される。このことは、DCまたは他の細胞、例えばHLA−A2−チロシナーゼ−ペプチド特異的CD8T細胞クローンのためのAPCとしてK562を用いて実証された(Tyr−F8細胞)。
【0025】
特に本発明は、次の態様および実施形態を目的とする。
第一の態様によれば、本発明は、抗原特異的T細胞の生成方法であって、
a)患者由来MHC分子をエンコードする核酸および抗原、または前記抗原をエンコードする核酸を準備する工程、
b)工程a)において規定されているような両方の化合物を、健康なドナーに由来する抗原提示細胞(APC)、好ましくは樹状細胞中にコトランスフェクションするか、または導入する工程、
c)健康なドナーに由来する末梢血リンパ球(PBL)を、前記APCで初回抗原刺激する工程、
d)MHC−抗原リガンドに特異的なT細胞を選択する工程、
を含む方法を提供する。
【0026】
これらのAPCは、好ましくは、樹状細胞、活性化B細胞、単球、マクロファージ、活性化T細胞、抗原提示能力を有する血液悪性腫瘍、および/またはEBV−形質転換リンパ芽球様細胞株から選択される。
【0027】
樹状細胞(DC)が特に好ましい。成熟樹状細胞(DC)は、非常に多様な共刺激性分子とともに、MHCクラスIおよびクラスII分子の両方を高いレベルで発現する。これらは、以前に抗原に遭遇しなかったナイーブT細胞に初回抗原刺激する完全な能力をこれらに与える。これらはまた、これらのMHCクラスIおよびクラスII分子中の細胞内タンパク質からの抗原をこれらが処理および提示すること可能にする、すべての必要な遺伝子/タンパク質も有する。このようにしてこれらは、CD4およびCD8T細胞応答の両方の誘発のための刺激細胞として用いるのに最適な抗原提示細胞(APC)である。自家DC中のTAAをエンコードするRNAの発現は、高い親和性を有する腫瘍抗原特異的T細胞が、同じ健康なドナーの末梢血リンパ球を用いて試験管内で初回抗原刺激されることを可能にした(Liaoら、2004年)。DCは、自己−エンコードされたMHCクラスI分子の正常補体を発現するので、これらは、T2細胞または他の種の細胞、例えばショウジョウバエの細胞に対して、NKおよびNK様T細胞の活性の負の調節を行なうことができる。
【0028】
一つの実施形態によれば、MHC分子および抗原は、抗原と、MHC分子をエンコードする核酸との混合物として用いられる。代替実施形態として、同種異系MHC分子をエンコードする核酸および抗原が、ビシストロンRNAとして準備される。
【0029】
特異的T細胞がこれに対して生成させられるべきである抗原は、好ましくは、ウイルス、細菌、原生動物、および寄生虫に由来する病原性作用物質、ならびに腫瘍細胞もしくは腫瘍細胞関連抗原、自己抗原、またはこれらの機能部分から選択される。
【0030】
これらのウイルスは、好ましくはインフルエンザウイルス、はしかおよび呼吸器合胞体ウイルス、デングウイルス、ヒト免疫不全ウイルス、ヒト肝炎ウイルス、ヘルペスウイルス、または乳頭腫ウイルスからなる群から選択される。原生動物は、熱帯マラリア原虫であってもよく、細菌は、結核を引起こすマイコバクテリアであってもよい。
【0031】
腫瘍関連抗原は、好ましくは、血液悪性腫瘍もしくは充実性腫瘍、より好ましくは結腸癌、乳癌、前立腺癌、腎細胞癌(RCC)、肺癌、肉腫、または黒色腫細胞から選択される。
【0032】
選択工程d)は、好ましくは、T細胞のサイトカイン放出の測定、またはT細胞活性化の他の測定値によって実施される。例えば活性化T細胞は、個々の細胞としてクローンされることができ、拡張後、これらのT細胞クローンは、これらのMHC−ペプチド特異性について分析されることができ、所望の特異性を有するものが、さらなる使用のために選択され得る(Schendelら、1979年、1997年)。あるいはまた、様々な形態の可溶性MHC−ペプチドリガンド、例えばテトラマーは、蛍光ラベルでマークされることができ、活性化T細胞でインキュベーションされることができる。テトラマーと相互作用するTCRを担っているこれらのT細胞は、次いでフローサイトメトリーによって検出されることができ、これらの蛍光に基づいてソートされることができる(Yeeら、1999年)。T細胞はさらに、これらが反応すべき腫瘍細胞で短時間刺激することができ、これらのインターフェロンガンマ分泌を、捕獲試薬によって、例えば公開されているように(Beckerら、2001年)検出することができる。
【0033】
好ましい実施形態によれば、本発明の方法はさらに、d)生体外で選択されたT細胞の拡張工程も含む。このことは、選択されたT細胞を、これらの当初初回抗原刺激のために用いられたのと同じ方法で生成されたAPCとともに共培養し、新しいAPCをT細胞培養物へ7〜10日毎に添加し、拡張しているT細胞の型に応じて、補足サイトカインを含有する新鮮な培地を規則的にこれらの細胞に供給することによって実施されることができ、これは、(Schendelら、1997年;Regnら、2001年;Suら、2001年、2002年)に記載されているように、IL−2、IL−4、IL−7、および/または中でも特にIL−15を包含する。
【0034】
さらに、本発明の方法はさらに、単離T細胞のT細胞受容体(TCR)のクローン工程、および/またはPBMC中にTCRトランスジーンを発現する工程も含む。これは、確立された方法、例えばEngelsら、2005年に記載された方法に従って行われることができる。
【0035】
これが特異的MHCクラスへ制限されないことが、本研究方法の一つの大きい利点である。このようにしてMHC分子は、これらが患者のMHC型に対応する限り、MHCクラスI、好ましくはHLA−A、HLA−B、HLA−C、もしくはHLA−E、またはMHCクラスII、好ましくはHLA−DP、HLA−DQ、HLA−DRから選択されてもよい。
【0036】
第二の態様によれば、本発明は、上記の方法によって得ることができる、抗原特異的T細胞を提供する。
前記T細胞は好ましくは、エフェクター細胞特徴を有するT細胞、より好ましくはサイトカイン生産T細胞、細胞傷害性T細胞、または調節T細胞、好ましくはCD4+もしくはCD8+T細胞である。
【0037】
第三の態様において、本発明は、上に説明されたような方法によって得ることができる、トランスジェニックTCRについてコードする核酸を目的とする。
追加の態様は、トランスジェニックTCRについてコードする核酸を含んでいるベクターを目的とする。このベクターは好ましくは、本発明による核酸、および1またはそれ以上の調節核酸配列を含有する発現ベクターである。好ましくはこのベクターは、プラスミドまたはレトロウイルスベクターである。
【0038】
本発明はさらに、上に規定されているようなベクターで形質転換されたPBMCも含んでいる。
さらなる態様において、本発明は、上に説明されているようなT細胞もしくはPBMC、および製薬的に許容可能な担体を含んでいる製薬組成物を提供する。
【0039】
本発明のこれらの活性成分は好ましくは、疾病が治療されるか、または少なくとも軽減されうるような製薬組成物において、許容可能な担体もしくは担体材料と混合された用量で用いられる。このような組成物は、(活性成分および担体に加えて)充填材料、塩、緩衝剤、安定剤、可溶化剤、および他の材料を包含する。これらは、公知の最新技術である。
【0040】
「製薬的に許容可能な」という用語は、活性成分の生物活性の有効性を妨げない、非毒性材料を規定する。担体の選択は用途に依存している。
製薬組成物は、活性成分の活性を強化するか、または治療を補足する追加成分を含有することができる。このような追加成分および/または因子は、相乗効果を得るため、または副作用もしくは望まれない作用を最小限にするためにこの製薬組成物の一部であってもよい。
【0041】
本発明の活性成分の配合または調製および適用/薬物適用の技術は、「レミントンの製薬科学(Remington’s Pharmaceutical Sciences)」、ペンシルベニア州イーストンのマック・パブリッシング社(Mack Publishing Co.,Easton, PA)の最新版に公開されている。適切な適用は、非経口適用、例えば筋肉内、皮下、骨髄内注射、ならびに鞘内、直接心室内、静脈内、結節内、腹膜内、または腫瘍内注射である。静脈内注射が、患者の好ましい治療である。
【0042】
好ましい実施形態によれば、この製薬組成物は、注入または注射またはワクチンである。
さらなる態様によれば、本発明は、養子細胞療法のため、および特に血液悪性腫瘍もしくは充実性腫瘍および急性もしくは慢性感染の治療のための薬剤の製造について上で説明されているような抗原特異的T細胞もしくはPBMCの使用を目的とする(同様に上記参照)。
【0043】
本発明は、以下において実施例および図面によって例証される。
【図面の簡単な説明】
【0044】
【図1】樹状細胞の成熟、および悪性細胞に対する従来の免疫応答における特異的細胞傷害性T細胞の活性化を示している。
【図2】RNA−パルスされた樹状細胞によるナイーブ腫瘍特異的T細胞のデノボ初回抗原刺激を例証している。
【図3】試験管内転写RNAの導入を示している。
【図4】試験管内転写RNAの導入を示している。
【図5】試験管内転写RNAの導入を示している。
【図6】試験管内転写RNAの導入を示している。
【図7】試験管内転写RNAの導入を示している。
【図8】試験管内転写RNAの導入を示している。
【図9】試験管内転写RNAの導入を示している。
【図10】RNA−発現DCによるTyrF8細胞の再刺激に対するRNA濃度の影響を示している。
【図11】RNA−パルスされた樹状細胞でのナイーブT細胞のデノボ初回抗原刺激プロトコルを例証する。
【図12】RNA−トランスフェクションされた樹状細胞上のMHC−ペプチドリガンドの発現を示している。
【図13】HLA−A2−陽性およびHLA−A2−陰性ドナーからの初回抗原刺激されたT細胞のテトラマー染色を描いている。
【図14】特異性およびT細胞受容体結合活性についてのテトラマー−ソートされたバルクT細胞培養物の評価を示している。
【図15】特異性、T細胞受容体結合活性、および機能についてのT細胞クローンの評価を描いている。
【発明を実施するための形態】
【0045】
実験および図面の説明
図1.樹状細胞の未成熟および成熟段階
樹状細胞(DC)は、抗原に遭遇したことがないT細胞(ナイーブT細胞)を活性化するこれらの能力のために、抗原提示細胞(APC)のうち最も「プロフェッショナル」であると考えられる。これは、DCについて既に発見されているいくつかの因子に応じており、同様に依然として同定されるべき特徴に依存することがある。DCの様々な形態が存在し、これらの独特の特徴は、継続的に調査中である。臨床研究のために最も普通に用いられているDCの型は、CD14−陽性血液単球から区別することができる骨髄DCである。サイトカイン、顆粒球−単球コロニー刺激因子(GM−CSF)、およびインターロイキン−4(IL−4)の存在下におけるこのような単球の試験管内培養を通して、その周囲から(異種抗原を含む)材料を取り上げるその高い能力を特徴とする未成熟骨髄DCが生成される。未成熟DCが、この段階の間に適切なシグナルを受け取る時、これは活性化されて、リンパ管を通って二次リンパ節へ移動する。この移動の間、およびリンパ節に達した時、これはさらなる変化を受け、これが完全成熟段階に導く。成熟段階において、DCは、高いレベルのMHCクラスIおよびクラスII分子を発現し、これらによってこれは、細胞内タンパク質に由来するペプチドの形態における抗原を、それぞれCD8−陽性およびCD−4陽性T細胞へ提示することが可能にされる。MHC分子の両方の型によって提示され、DCの表面上に示された抗原は、抗原プロセシングおよび提示の様々な工程を含む、複雑な細胞プロセスにおいてDC中に生成される。MHCクラスI分子中のDC細胞表面において提示されたペプチド(すなわち抗原−断片)の場合、APCが抗原プロセシング関連輸送体(TAP)遺伝子、TAP1およびTAP2によってエンコードされた分子を発現する必要がある。
【0046】
DC表面に示されたMHC−ペプチドリガンドは、Tリンパ球(すなわちT細胞受容体)上に存在する抗原特異的受容体と相互作用する。この相互作用は、T細胞へ第一シグナル(すなわちシグナル1)をもたらす。このシグナルは、各T細胞について独自のTCRを通してもたらされるので、これは、適合TCRを有するT細胞を選択することによって、結果として生じる応答の抗原特異性を決定する。成熟DCはまた、いくつか挙げればCD40、CD80、CD86を包含する非常に多様な共刺激性分子も発現し、これらは、Tリンパ球によって発現された追加受容体と相互作用し、これらのさらなる活性化(すなわちシグナル2)を引起こす。DCはさらに、サイトカインおよびケモカインを包含する多様な可溶性分子を分泌する能力を有する。これらは様々な亜型のリンパ球をこれらの近隣へ引き付けることができ、シグナル1およびシグナル2を受け取るT細胞の分化に対して影響を与えうる。リンパ球によって発現されたさらなる組の受容体への結合後に、これらの因子は、これらの分化および最終的機能に影響を与えうるリンパ球へさらに別のシグナルをもたらすことができる。これは時にはシグナル3と呼ばれる。
【0047】
試験管内で未成熟および成熟DCを生成することが可能である。例えば単球を、様々な手順によって(細胞選択用の商業キットを用いたCD14−陽性細胞の正の選択、プラスチック商品へのこれらの接着特性によって、または細胞溶出法を用いて、これらのサイズおよび密度によって)得ることができる。これらの単球は、次いでGM−CSFおよびIL−4とともに培養されて、未成熟DCの集団を生じる。試薬の様々な組み合わせが、DCの成熟を誘発するため、および平行して可溶性メディエーターのこれらの分泌を改変するために用いられることができる。未成熟/成熟DCのこれら様々な形態が、今度はDCによって示されたMHC−ペプチドリガンドとこれらのTCRとの相互作用によって所望の抗原特異性を有するのみならず、DCによって分泌されたサイトカインに基づいた所望の機能も有するT細胞を活性化するために用いられることができる。このようにして、生成されて多量のサイトカインIL−12を分泌する成熟DCは、抗腫瘍免疫において特に重要なTヘルパー1型のCD4T細胞を活性化する良好な能力を有する。
【0048】
適切に活性化され、選択されるならば、T細胞は、腫瘍細胞、または様々な病原体に感染した細胞を認識して破壊する能力を有する。腫瘍細胞の場合、APCによって処理され、提示された腫瘍関連抗原(TAA)は多くの場合、腫瘍細胞において過剰発現された自己−タンパク質である分子を表わす。自己分子に由来するMHC−ペプチドリガンドと相互作用するTCRを担っている大部分のT細胞は、「負の選択」として公知の方法で胸腺において取り除かれるので、このようなTAA−リガンドを発現する腫瘍細胞を認識するのに十分な結合活性をともなうTCRを有するT細胞は、レパートリー中にほとんど存在しない。
【0049】
図2.RNA−パルスされた樹状細胞での、ナイーブ腫瘍特異的T細胞のデノボ初回抗原刺激
腫瘍細胞または他の細胞、例えば病原体で感染された細胞上でMHC−ペプチドリガンドを認識するより良好な能力を有するT細胞を得るために、同種異系MHC分子を介して、選択された1または複数の抗原に由来するペプチドを提示することによって、健康な個人からのT細胞の非選択レパートリーを選ぶ(tap)ことができる。例えば、HLA−A2でない健康なドナーAからのT細胞は、胸腺においてHLA−A2−ペプチド複合体へ暴露されたことはなく、したがって、高い結合活性を有するHLA−A2−ペプチド複合体と相互作用することができるTCRを発現するこのような個人の末梢血単核細胞(PBMC)のうちで利用可能なT細胞が存在する。このような所望のTCRを担っているナイーブT細胞を刺激するために、前記ドナーAのDCは単球から生成され、次いで修飾されて、同種異系MHCクラスIもしくはクラスII分子を発現する。ここで、HLA−A2についての一例が与えられる。この分子は、HLA−A2でないドナーAのT細胞についての同種異系MHC分子である。HLA−A2エンコードRNAをドナーAのDC中に導入することによって、HLA−A2分子をその細胞表面において発現するDCの集団を作製することが可能である。これによって、DCは依然として、ドナーAの染色体MHC遺伝子によってエンコードされた自己−MHC分子のその正常な組を発現するが、これに加えてそれは、導入されたRNAから翻訳された同種異系MHC分子を発現する。同時にDCは、ペプチドがDCによって処理され、細胞表面において同種異系HLA−A2分子によって提示され得るタンパク質抗原をエンコードするRNAが供給されることができる。このような同種異系MHC−ペプチドリガンドをその細胞表面において発現するDCが、ナイーブT細胞を刺激するためにAPCとして用いられる時、これらは、所望の同種異系MHC−ペプチドリガンドと相互作用するTCRを有するT細胞を活性化することができる。同種異系MHC分子を認識するすべてのT細胞が、所望のペプチド特異性を有するわけではないであろう。したがって所望のTCRを有するこれらの細胞は、活性化されたT細胞の全部の集団から選択されなければならない。この選択を得るための、免疫学者に周知のいくつかの手順がある。例えば活性化されたT細胞は、個々の細胞としてクローンすることができ、拡張後にこれらのT細胞クローンは、これらのMHC−ペプチド特異性について分析することができ、所望の特異性を有するものが、さらなる使用のために選択されることができる。あるいはまた、様々な形態の可溶性MHC−ペプチドリガンド、例えばテトラマーは、蛍光ラベルでマークすることができ、活性化されたT細胞でインキュベーションされることができる。これらのテトラマーと相互作用するTCRを担っているこれらのT細胞は、次いでフローサイトメトリーによって検出されることができ、これらの蛍光に基づいてソートされることができる。
【0050】
所望のTCR特異性で選択されたT細胞は、次いで対応HLA−A2−ペプチドリガンドを発現する腫瘍または感染細胞を有する患者の養子細胞療法(ACT)における使用のために試験管内で拡張されてもよい。あるいはまた、TCR配列を単離し、このようなT細胞について決定することができ、リンパ球または他の細胞中に導入して、ACTのために用いることができるトランスジェニックTCR細胞を生成することができるTCR構成体を生成するために用いることができる。
【0051】
図3.K562細胞中への試験管内転写RNAの導入
この戦略の実行可能性を実証するための第一実験方法として、本発明者らは、RNA転移後の同種異系HLA−A2MHC分子の発現のために、受容者細胞株としてK562細胞株を用いた。この細胞株はエリスロミエロイド白血病(erythromyeloid
leukaemia)に由来し、細胞免疫学者には周知である。これは腫瘍系であるので、多数になるまで成長させることができ、実験的な取扱いが容易である。これはさらに、その細胞表面上にMHCクラスI分子をまったく発現させず、従って、同種異系MHC分子の発現は、MHCクラスI分子をエンコードする核酸の導入後、K562の細胞表面において容易に検出することができる。
【0052】
この図面に要約された実験において、HLA−A2−エンコードcDNAからの試験管内転写によって生成されたHLA−A2 mRNAが、エレクトロポレーションを介してK562細胞中に導入された。一時的にRNAトランスフェクションされた細胞は、K562−A2細胞と呼ばれる、HLA−A2をエンコードする遺伝子で安定的にトランスフェクションされたK562細胞の一系列と比較された。
【0053】
HLA−A2分子の表面発現は、HLA−A2分子について特異的なモノクローナル抗体でのこれらの細胞のインキュベーション後にフローサイトメトリーによって決定され、データは、陽性細胞のパーセンテージとして示されている(左側の図面)。模擬対照として、K562細胞は、HLA−A2 mRNAの代わりに水を収容しているキュベットにおいてエレクトロポレーションされた。エレクトロポレーションに続くすべての時点で、模擬対照において陽性細胞はまったく検出されなかった。HLA−A2 RNAの3つの異なる量が、同数のK562細胞のエレクトロポレーションのために用いられた。HLA−A2陽性細胞の最高パーセンテージが、24マイクログラムの量のRNAを用いて検出され、細胞の最高パーセンテージは、エレクトロポレーションの11時間後に見られた。RNAのエレクトロポレーション後のHLA−A2分子を発現する陽性細胞のパーセンテージは、11時間および24時間の時点で、HLA−A2遺伝子を安定的に発現する正の対照K562−A2細胞のパーセンテージに匹敵した。HLA−A2 mRNAの導入後のHLA−A2の一時的発現は、48時間の時点での陽性細胞のパーセンテージの減少、および120時間の時点での無検出によって実証される。
【0054】
アロ抗原としてHLA−A2分子を認識するTCRを担っているT細胞が、HLA−A2 RNA−発現細胞によって活性化されうるかどうかを決定するために、JB4と呼ばれるT細胞クローンが研究された。このT細胞クローンは、そのTCRがHLA−A2−ペプチドリガンドと相互作用する時に、サイトカインインターフェロン−ガンマを分泌する。この認識のために必要とされる正確なペプチドは公知でないが、K562細胞、およびDC、および本発明者らがテストした大部分の他の細胞中に発現され、このようにして、これは、遍在性タンパク質に由来するように見える。JB4細胞がK562模擬トランスフェクション細胞とともに共培養された時、IFN−ガンマ放出は検出されない(右側の図面)。K562−A2細胞の正の制御では強い放出が見られる。様々な量のHLA−A2 RNAでトランスフェクションされたすべてのK562細胞は、正の対照よりも低いレベルにおいてではあるが、JB4細胞によってサイトカインの分泌を誘発することができた。それにもかかわらず、これらはサイトカイン放出の実質的なレベルである。JB4細胞の添加を有しなかったK562またはDCの培養において、サイトカイン放出は測定されず、刺激細胞がインターフェロン−ガンマをそれ自体分泌しないことを実証した。刺激細胞をともなわないJB4細胞の最終対照は、サイトカイン放出を全く示さず、T細胞が、インターフェロン−ガンマを分泌するのに適切なMHC−ペプチドリガンドによって活性化されなければならないことを実証した。
【0055】
これらの研究は、一時的なRNA発現を介したK562細胞中への同種異系MHC分子の導入が、HLA−A2分子の細胞表面発現へ導き、これらの細胞が、HLA−A2アロ抗原特異的T細胞クローンを活性化することができることを証明した。
【0056】
図4.DC中への試験管内転写RNAの導入
HLA−A2 mRNAのエレクトロポレーション後にDC上のHLA−A2分子の表面発現を評価するため、およびJB4細胞からのサイトカイン分泌を刺激するこれらの能力を評価するために、同様な実験方法が、図3に記載されているように用いられた。ここで、24マイクログラムの量のHLA−A2 mRNAが全体を通して用いられたが、mRNAの3つの異なる源が比較された。これらは、RNA発現をDC内部で安定させる意図で、ヒトアルファ−グロブリン遺伝子の3’UTRがHLA−A2遺伝子構成体へ添加されている試験管内転写RNAの商業源を含んでいた。このRNAは、アンビオン(Ambion)からの商業的手順およびキットを用いて生成されたポリアデニル化HLA−A2 RNA対キュアヴァック(CureVac)からの商業的手順およびキットを用いて生成された同様なRNAと比較された。アンビオン手順を用いて生成され、HLA−A2陰性ドナーから培養されたDC中にエレクトロポレーションによって導入されたポリアデニル化mRNAが、最良の結果を示した。このmRNAの場合にHLA−A2陽性細胞の最高パーセンテージが見られ、DCの表面におけるHLA−A2分子の出現は、より早く(6時間の時点で)検出され、DC中の発現は、このmRNA源を用いて、なおも120時間の時点で見られた。従って、すべてのさらなる実験は、このmRNA源を利用した。
【0057】
DCは、図3に記載されているように、JB4細胞からのサイトカイン分泌を刺激するこれらの能力について研究された。模擬対照DC(最も左の棒)は、サイトカイン放出のバックグラウンドレベルのみを誘発した。より高いレベルは、mRNAの3つの異なる源を発現するDCを用いて見られたが、アンビオンmRNAでエレクトロポレーションされたこれらのDCが優れていた。JB4細胞を含有しないDC培養物は、インターフェロン−ガンマを有しておらず、これがHLA−A2−RNA発現DCでの刺激後にJB4細胞によって放出されるだけであることを実証した。
【0058】
図5.腫瘍関連抗原チロシナーゼをエンコードする試験管内転写RNAのK562−A2細胞中への導入
RNAは、アンビオン手順を用い、抗原チロシナーゼに対して特異的に生成された。これはK562−A2細胞中にエレクトロポレーションされ、HLA−A2の表面発現が、HLA−A2分子に特異的なモノクローナル抗体を用いて測定された。緑の曲線はイソタイプ対照抗体での染色を表わし、青色の曲線はHLA−A2−特異的抗体での染色を表わす。ここで、HLA−A2表面発現がすべての時点で見られたが、それは、K562−A2細胞が、HLA−A2遺伝子を構成的に発現するからである。HLA−A2の発現の減少は、12時間の時点で注目され、これは、エレクトロポレーションの結果としてのストレス誘発された衝撃と関連付けることができる。これらの細胞は、その後HLA−A2分子の高い発現を回復した。チロシナーゼのタンパク質発現は、チロシナーゼに特異的なモノクローナル抗体を用いて、タンパク質の細胞内検出後に測定された。オレンジ色の曲線は模擬対照の染色を表わし、青色の曲線は、これらの細胞内部のチロシナーゼタンパク質を表わす。染色におけるわずかなシフトを、いくつかの時点で見ることができ、タンパク質が、チロシナーゼRNAの導入後に作製されつつあることを示している。
【0059】
図6.K562−A2細胞におけるチロシナーゼの発現および機能
チロシナーゼRNAが、K562−A2細胞中にエレクトロポレーションされ、陽性細胞のパーセンテージ、ならびに平均蛍光強度(MFI)と呼ばれる染色強度が様々な時点で決定された(左上部分)。赤い棒はK562−A2細胞上の構成的HLA−A2発現を示しており、ここでもまた、11〜24時間のレベルにおける低下およびその後の回復をともなう。青い棒は、6時間の時点でピークがあるチロシナーゼタンパク質の細胞内染色を示している。6時間の時点での染色のシフトは、右下部分に図解され、ここでは模擬対照染色がオレンジ色の曲線によって示され、チロシナーゼ染色は青色の曲線によって示されている。模擬対照と比較した、経時的な発現の倍数増加(fold increase)は左下部分に示され、11時間の時点で最大を生じる。HLA−A2分子によって提示されたチロシナーゼ由来ペプチドが分かるT細胞クローンTyr−F8のインターフェロン−ガンマ分泌が右上部分に示されている。K562−A2細胞は、Tyr−F8T細胞によるインターフェロン−ガンマ選択を誘発しないが、これらは、チロシナーゼRNAでのエレクトロポレーション後に可能である。これらのT細胞は単独でサイトカインを分泌しないが、これらはまた、チロシナーゼからの合成ペプチドが負荷されたK562−A2細胞によって、またはチロシナーゼおよびHLA−A2を共発現する黒色腫細胞(MEL−IL2細胞)によって刺激されることができる。
【0060】
このことは、K562−A2細胞中へのチロシナーゼについてのRNAの導入によって、これらがTyr−F8細胞のTCRによって見られる適切なMHC−ペプチドリガンドを処理および提示することを可能にし、これらのT細胞を活性化して、インターフェロン−ガンマを分泌させることを実証する。エレクトロポレーションの12時間後および24時間後に用いられたRNAトランスフェクションされた細胞は、匹敵し得る刺激性能力を有していた。
【0061】
図7.試験管内転写チロシナーゼRNAの、HLA−A2陽性ドナーから生成されたDC中への導入
同様な一組の実験が、図6に記載されている実験のように行なわれたが、HLA−A2陽性ドナーからのDCがK562−A2細胞の代わりに用いられた。細胞内チロシナーゼタンパク質の発現が、RNAのエレクトロポレーション後の様々な時点でDC中に検出された(上の図)。模擬対照全体における最高の平均倍数発現が、DC中で3時間の時点で見られた。DC単独ではTyrF8細胞によるサイトカイン分泌を刺激することはできなかったが、これらは、チロシナーゼRNAでのエレクトロポレーション後では可能であった。エレクトロポレーションの12時間後または24時間後に用いられたDCは、Tyr−F8T細胞を活性化して、インターフェロン−ガンマを分泌させる能力を有していた。
【0062】
図8.K562細胞におけるHLA−A2およびチロシナーゼの共発現
HLA分子をまったく発現しないK562細胞が、HLA−A2およびチロシナーゼについてのRNAでエレクトロポレーションされた。RNAの各種が個々に導入されるか、または両方が組み合わされて導入された。右上図は、異なる時点におけるHLA−A2単独(最も左側の一組の棒)、チロシナーゼ単独のタンパク質発現、次いでHLA−A2およびチロシナーゼRNAの両方でエレクトロポレーションされた細胞におけるHLA−A2染色、およびRNAの両方の種を受け取っている細胞におけるチロシナーゼ染色を要約している。RNAの両方の種でエレクトロポレーションされた細胞中の両方のタンパク質の最適発現が、24時間の時点で見られた。両方のRNAを共発現するK562細胞は、エレクトロポレーションの12時間後および24時間後にTyrF8細胞からのインターフェロン−ガンマ分泌を刺激することができた。
【0063】
図9.HLA−A2陰性ドナーから作製されたDCにおけるHLA−A2およびチロシナーゼの共発現
HLA−A2陰性ドナーから調製されたDCにおけるHLA−A2タンパク質およびチロシナーゼタンパク質の共発現が、図8に記載されているように分析された。両方のタンパク質の共発現が、様々な時点においてDC中に見られた。HLA−A2発現のレベルは、チロシナーゼRNAの存在下に低下されるように思われた。それにもかかわらず、両方のRNAを共発現するDCは、TyrF8細胞によるインターフェロン−ガンマの有意レベルを、T細胞単独のもの、またはRNAの単独種でエレクトロポレーションされたDCでのインキュベーション後のもの以上に刺激することができた。
【0064】
図10.RNA発現DCによるTyrF8細胞の再刺激へのRNA濃度の影響
DCがTyrF8細胞からのサイトカイン分泌を刺激する能力が、様々な量のHLA−A2およびチロシナーゼRNAでトランスフェクションされたHLA−A2陰性ドナーから調製されたDCを用いて分析された。模擬対照DC(最も左側の棒)が、サイトカイン放出のバックグラウンドレベルのみを誘発した。T細胞単独ではサイトカインを分泌しないが、これらはまた、チロシナーゼおよびHLA−A2を共発現する黒色腫細胞(MEL−IL2細胞)によっても刺激されることができる。
【0065】
様々な量の両方のRNAでトランスフェクションされたすべてのDCは、TyrF8細胞によるインターフェロン−ガンマの分泌を誘発することができたが、一方で、24μgHLA−A2および24μgチロシナーゼRNA、または48μgRNA HLA−A2および24μgチロシナーゼRNAでエレクトロポレーションされたこれらのDCの方が優れており、T細胞の再刺激のためにはより少ないRNAがより良好であろうことを示している。
【0066】
図11.RNAパルスされた樹状細胞でのナイーブT細胞のデノボ初回抗原刺激プロトコル
RNA負荷されたDCがナイーブT細胞を初回抗原刺激して腫瘍特異的リンパ球になる能力を分析するために、3相初回抗原刺激プロトコルが開発された。初回抗原刺激の開始に先立つ8日前(−8日目)、単球が、記載されているようにHLA−A2陽性ドナーおよびHLA−A2陰性ドナーからプラスチック接着によって調製された。これらの精製された単球集団が未成熟DCを調製するために用いられ、これらは、次いで記載されているように成熟させられた。T細胞初回抗原刺激プロトコルの最初の日(0日目)に、DCは、記載されているようにエレクトロポレーションを介してRNAが負荷された。HLA−A2陽性ドナーに由来するDCに、モデル腫瘍抗原として24μgのチロシナーゼRNAが負荷された。これらのDCにおけるRNAから翻訳されたチロシナーゼタンパク質は、DC表面において自己−エンコードされたHLA−A2分子と関連してペプチドとして処理され、提示されるべきである。HLA−A2陰性ドナーに由来するDCは、HLA−A2(48μg)およびチロシナーゼ(24μg)をエンコードするRNAが負荷された。ここで、転移RNAによってエンコードされたHLA−A2分子は、同種異系MHC分子を表わす。チロシナーゼタンパク質に由来するペプチドは、DCへ転移されたHLA−A2エンコードRNAから翻訳されたHLA−A2分子によって処理および提示されるべきである。これらのペプチド−MHC複合体は、アロ制限−ペプチドリガンドを表す。初回抗原刺激プロトコルの0日目に、各ドナーからの自家CD8+Tリンパ球が、商業的キットおよび製造業者の指示(CD8+T細胞単離キットII(ヒト)、ドイツ国ベルギッシュ・グラードバッハのミルテニ(Miltenyi,Bergisch Gladbach))を用いて負の選択を介して>80%純度まで単離された。これらの手のつけられていない自家CD8+T細胞が、エレクトロポレーションの9時間後に、IL−7(10ng/mL)を含有するエイム(Aim)V培地(ドイツ国カールスルーエのギブコ社(Gibco BRL,Karlsruhe))中に10:1の比で、成熟RNA−パルスされたDCへ添加された。IL−2(20IU/mL)が2日後に、ついでその後3日目毎に添加された。第一回刺激の7日後(7日目)、初回抗原刺激培養物の第二回刺激が、第一回刺激についてと同じ方法で調製されたRNA−パルスされたDCを用い、初回抗原刺激の0日目に用いられたのと同じ培養条件を用いて実施された。さらに7日後(14日目)、HLA−A2制限チロシナーゼ特異的T細胞が、フィコエリトリン(PE)−標識されたHLA−A*0201/htyr369−377ペプチド/ヒトβ2mテトラマー(Wolflら、2004年;ドイツ国ミュンヘン、技術大学の医学微生物学、免疫学、および衛生研究所、(Institute of Medical Microbiology,Immunology and Hygiene,Technical University,Munich)のProf.D.Buschによって提供された)の助けを借りてソートされた。正の選択をされたT細胞のいくつかは、96ウエルV底プレート(スイス国トラサディンゲン(Trasadingen)のTPP)において、1細胞/ウエルで培養された。これらのウエルに、3日毎に50IU/mL IL−2(ドイツ国マールブルクのヒロン・ベーリンク(Chiron Behring,Marburg))を含有するエイムV培地、および7日毎に5ng/mL IL−7(ドイツ国ハイデルベルクのプロモキネ(Promokine))および10ng/mL IL−15(米国ニュージャージー州のペプロ・テック社(Pepro Tech Inc.))が供給された。選択されたクローン、および残りのテトラマー選択された非クローンT細胞が、IL−2、IL−7、IL−15で上記のようにエイムV培地中の培養に維持され、支持細胞が供給された。これらの細胞は、50Gyが照射された5ドナーのプールに由来する末梢血単核細胞からなっていた。これらのT細胞は、2週間毎に抗−CD3抗体(0.1μg/mL;ドイツ国ミュンヘンの分子免疫学研究所(Institute of Molecular Immunology,GSF)のDr.Elisabeth Kremmerから提供された)で非特異的に刺激された。
【0067】
図12.RNA−トランスフェクションされた樹状細胞上のMHC−ペプチドリガンドの発現
T細胞初回抗原刺激のために用いられたDCが、予想されたMHC−ペプチドリガンドを発現することを実証するために、細胞表面におけるHLA−A2分子および細胞内チロシナーゼタンパク質の発現が、既に記載されているフローサイトメトリーを用いて決定された。HLA−A2分子およびチロシナーゼタンパク質の共発現は、両方のドナーに由来するDCにおいて高レベルで検出された。細胞内チロシナーゼタンパク質のレベルは、HLA−A2陽性およびHLA−A2陰性の両方のドナーから調製されたDCにおいて匹敵しうるものであった。大部分の細胞上のHLA−A2表面発現のレベルは、HLA−A2陽性ドナーから調製されたDCにおいて、より高かった。この場合、これらは内因性遺伝子によってエンコードされている。一方、トランスジェニックHLA−A2分子のレベルは、HLA−A2陰性ドナーから調製されたDC上で、より変わりやすい。
【0068】
独立実験における同じプロトコルに従って調製された細胞を用いた、このようなDCがT細胞を刺激する能力は、これらが保有するペプチド(すなわちHLA−A2+xyzペプチド)には関わりなくHLA−A2分子を認識するHLA−A2−アロ応答性T細胞クローンを用いて決定された。DCの両方の集団は、以前に記載されているような標準的ELISAにおいて測定されたように、このT細胞クローン(JB4)を刺激してインターフェロン−ガンマを分泌させることができた。DCをともなわずにインキュベーションされたT細胞からのサイトカイン放出は、まったく見られなかった。チロシナーゼ由来ペプチドを提示するHLA−A2分子からなるリガンドの存在が、HLA−A2−チロシナーゼペプチドについて特異的なT細胞クローン(Tyr−F8)によるインターフェロン−ガンマ放出を測定することによって評価された。DCをともなわずにインキュベーションされたT細胞は、サイトカインのバックグラウンドレベルのみを放出した。この実験において、HLA−A2−陽性DCは、48μgのチロシナーゼが供給され、一方、HLA−A2−陰性DCは、24μgのチロシナーゼが供給された。
【0069】
これらの結果は、HLA−A2−陰性ドナーに由来するDC中へのRNA転移によって供給されたトランスジェニックHLA−A2分子が、特異的HLA−A2抗体で細胞表面において染色することができ、HLA−アロ特異的T細胞クローン(JB4)によって認識することができることを実証した。チロシナーゼRNAの転移は、両方のDC集団における細胞内タンパク質発現に導いた。HLA−A2およびチロシナーゼRNAの共転移は、内因的にエンコードされたHLA−A2分子を発現するDC中へのチロシナーゼRNAの転移のように、HLA−A2−チロシナーゼ特異的T細胞クローン(TyrF8)が活性化されることを可能にした。
【0070】
図13.HLA−A2陽性およびHLA−A2陰性ドナーからの初回抗原刺激されたT細胞のテトラマー染色
初回抗原刺激されたT細胞培養物の中に存在するHLA−A2−チロシナーゼペプチドリガンドについて特異的なT細胞受容体を担っているT細胞を選択するために、これらのT細胞は、HLA−A*0201/htyr369−377/Hβ2mテトラマーで染色
され、フローサイトメトリーによって分析された。テトラマー結合を示した2つの異なる初回抗原刺激培養物の各々からの細胞がゲートされ(黒い四角、□)、MoFlo(商標)高性能細胞ショーター(米国コロラド州フォートコリンズのダコ(Dako,Fort Collins,CO))において単離された。HLA−A2陽性ドナーのCD8+テトラマー+T細胞集団は、低い強度においてのみテトラマー結合を示したが、一方で、HLA−A2陰性ドナーからのCD8+テトラマー+細胞は、より高い強度において染色された。これらのソートされたテトラマー+T細胞は、単一細胞として培養され、残りの細胞は、非クローンバルク培養物として培養中に保持された。
【0071】
これらの結果は、初回抗原刺激プロトコルが、HLA−A2−チロシナーゼ特異的テトラマーを結合することができるTCRを保有するT細胞を生じたことを実証した。より多くのT細胞が、HLA−A2−陰性ドナーに由来するDCを用いて初回抗原刺激された培養物においてより高い強度でテトラマーを結合させ、HLA−A2同種異系ペプチドリガンドによるより良い刺激を示している。
【0072】
図14.特異性およびT細胞受容体結合活性についての、テトラマーソートされたバルクT細胞培養物の評価
バルク培養物として保持された、ソートされた細胞は、8日後テトラマー染色について再分析された。予想されたように、ソートされた細胞の大部分は、初回抗原刺激培養物の両方に由来する、ソートされたバルク培養物中のCD8+であった。テトラマー+T細胞は、HLA−A2陽性DCを用いて初回抗原刺激された培養物中に約8%の頻度で存在した。これらの細胞は、テトラマー結合の中間的強度(MFI=127.49)を有したが、一方で、HLA−A2陰性ドナーからのバルク培養T細胞の80%が、はるかに高い強度(MFI=3248.77)を有するテトラマーを結合させた。テトラマー結合の強度は、特定のMHC−ペプチドリガンドについてのT細胞受容体(TCR)結合活性を評価するために用いられる1つのパラメーターである(Yeeら、1999年)。テトラマー結合のより高い強度は、このリガンドとTCRとのより良い相互作用を示している。TCR結合活性の第二パラメーターは、テトラマーオフレート(off−rate)である(Palermoら、2005年)。T細胞は、テトラマーでインキュベーションされ、次いで洗浄され、テトラマーの不存在下、およびHLA−A2分子について特異的な抗体の存在下にインキュベーションされる。この抗体の存在は、これらのT細胞の表面から落ちたテトラマーが、細胞表面へ再結合するのを防ぐ。これらのT細胞上のテトラマー染色の強度は、洗浄後の様々な時点:0時間、1時間、2時間の時点で決定される。テトラマー染色が急速に失われるならば、このことは、TCRが、テトラマーのMHC−ペプチドリガンドに対して低い結合活性しか有していないことを示している。テトラマー結合が経時的により安定であるならば、このことは、MHC−ペプチドリガンドに対してのより高いTCR結合活性を示す。これら2つのバルク培養物の比較において、もとのテトラマー−結合細胞(1.7%対8.3%)の20%のみが、HLA−A2陽性ドナーに由来する細胞において2時間後に依然としてテトラマー+であることが発見された。これに対して、これらの細胞の75%が、HLA−A2陰性ドナーに由来するT細胞において2時間後に依然としてテトラマー+であった(60.5%対80.6%)。
【0073】
テトラマー結合の特異性が、サイトメガロウイルスに由来するペプチドを提示するHLA−B7分子からなるテトラマー(Prof.D.Buschによって提供されたB7−CMVテトラマー)を用いて決定された。バルクT細胞培養物はどちらも、低レベルの染色(0.3〜0.4%)しか示さなかった。
【0074】
これに加えて、HLA−A2−CMVペプチドテトラマーも用いられた。HLA−A2陽性ドナーからのT細胞の0.4%のみが、このテトラマーを結合させたが、一方で、第二ドナーからのバルクT細胞の12.7%が陽性であった。これらのT細胞は、これらが提示するペプチドとは関わりなく、アロ抗原としてHLA−A2分子を認識するTCRを有するT細胞を表わす可能性がある。テトラマー結合の強度は、HLA−A2−チロシナーゼ−テトラマー結合の強度(MFI=3248.77)と比較して低かった(MFI=166.98)。
【0075】
これらの結果は、テトラマー染色の強度として規定されるようなより高い結合活性およびより遅いテトラマーオフレートを有するTCRを担っているT細胞が、HLA−A2陰性DCを用いて、これらの培養物に由来することを実証しており、同種異系−ペプチドリガンドを用いて、非負選択されたT細胞レパートリーにおいて応答を誘発する優位性を実証している。
【0076】
図15.特異性、T細胞受容体結合活性、および機能についてのT細胞クローンの評価
単一細胞培養実験から、2つのクローンがさらなる特徴決定のために選択された。1つのクローン(PS P4D11)がHLA−A2陽性ドナーに由来し、1つのクローン(SW P12B10)がHLA−A2陰性ドナーに由来した。
【0077】
HLA−A2陽性ドナーからのPS P4D11クローンは、1時間および2時間の時点で測定された時の急速なテトラマー放出をともなって、テトラマー染色の中間的強度(MFI=166.98)を示した。このT細胞クローンは、特異的ペプチドが負荷されたT2細胞(コネチカット州ニューヘブン、イエール大学のPeter Cresswellによって提供されたT2細胞)でのインキュベーション後も、HLA−A2およびチロシナーゼを発現する腫瘍細胞(Mel 624.38およびMel IL−2)での刺激後も、インターフェロン−ガンマを分泌するよう刺激することはできなかった。しかしながらこれは、HLA−A2+チロシナーゼ+黒色腫腫瘍細胞を殺すことはできるが、HLA−A2+チロシナーゼ−腫瘍細胞を殺すことはできず、HLA−A2−チロシナーゼリガンドについてのその特異性を実証した。
【0078】
HLA−A2陰性ドナーに由来するSW P12B10クローンは、テトラマー結合のより高い強度(MFI=1009.04)を示し、テトラマー結合の安定性は、2時間後にテトラマー対して依然として陽性である細胞のすべての場合により高かった。このクローンは、チロシナーゼ由来ペプチドが負荷されたT2細胞での刺激後に特異的インターフェロン−ガンマ放出を示したが、ペプチドをともなわないT2細胞に対しては可能ではなく、ペプチド特異性を実証した。これはまた、HLA−A2+チロシナーゼ+黒色腫腫瘍細胞での刺激後インターフェロン−ガンマを放出するように刺激することもできたが、HLA−A2陰性、チロシナーゼ陽性黒色腫細胞(SK Mel 28)に対して、またはHLA−A2陽性、チロシナーゼ陰性黒色腫腫瘍細胞(Mel A375)に対しては可能ではなかった。このパターンは、このクローンがHLA−A2−チロシナーゼペプチドに特異的であることを実証した。このクローンはまた、HLA−A2陽性、チロシナーゼ陽性黒色腫腫瘍細胞を殺すこともできたが、HLA−A2陽性、チロシナーゼ陰性腫瘍細胞を殺すことができなかった。
【0079】
これらの結果は、これら2つの異なる初回抗原刺激培養物に由来するT細胞クローンが、テトラマー結合の強度およびテトラマーオフレートによって検出された場合、TCR結合活性に関して差を示し、アロ制限ペプチド−特異的T細胞クローンが、この定義によってより高い結合活性のTCRを有することを示している。さらにはこのT細胞クローンが機能的優位性を示したが、それは、これが黒色腫細胞を特異的に殺すことができるだけでなく、ペプチド刺激後、および腫瘍細胞での刺激後、HLA−A2、チロシナーゼ特異的にインターフェロン−ガンマを分泌するように活性化することもできたからである。
【0080】
材料および方法
接着性腫瘍細胞株、Mel A375、Mel IL−2、Mel 624.38、およびSK Mel 28
これらのヒト黒色腫細胞株が、12%ウシ胎児血清、1×非必須アミノ酸、2mM L−グルタミン、および1mMナトリウムピルベートが補足されたRPMI 1640培地中で培養された。中程度のサイズの培養フラスコ中の培地の容積は、10mLであった。ほぼ3〜4日毎に、細胞は密集成長した。個々の細胞株の成長率に応じて、これらは1:2〜1:10に分けられた。培地が除去され、細胞はPBSで一回洗浄され、ついで2mLトリプシン/EDTA2×で3分間、室温でインキュベーションされた。脱離細胞が、新鮮なRPMI 1640培地+補足物中に再縣濁された。
【0081】
T細胞クローン、JB4、およびTyr−F8
T細胞クローンがまず、標準的再刺激培養物中で拡張された。大きい保存材料(stock)がアリコートされて冷凍された。実験に必要とされる度に、T細胞は保存材料から取られて解凍され、24−ウエルプレートのウエル中に入れられ、標準化されたプロトコルにしたがって再刺激された。
【0082】
縣濁腫瘍細胞株、T2、K562、およびK562−A2細胞
T2、K562、およびK562−A2が、12%FBS、1×MEM、2mM L−グルタミン、および1mMナトリウムピルベートが補足されたRPMI 1640培地中で培養された。中程度のサイズの培養フラスコにおいてこの培地の容積は10mLであった。ほぼ4日毎に、この細胞縣濁液の3/4が除去され、同じ容積の新鮮な培地が添加された。K562−A2細胞について、この培地は、1mg/mLの選択抗生物質G418が補足された。
【0083】
DC生成および培養
ドナーからのPBMCが、フィコール密度勾配遠心分離によって単離された。PBMCは、T75培養フラスコ1あたり7.5×106細胞で1%自家血漿が補足されたRPMI 1640培地中に再縣濁された。これらのフラスコは37℃および5%CO2で1時間インキュベーションされた。非接着性細胞が注意深く洗い流された。接着性単球が、1%自家血漿、800U/mL GM−CSF、および800U/mL IL−4が補足された樹状細胞培地の1フラスコあたり10mL中に培養された。3日間の培養後、800U/mL GM−CSFおよび800U/mL IL−4が再び添加された。培養の6日目に、800U/mL GM−CSF、500U/mL IL−4、5ng/mL IL−1β、9ng/mL IL−6、9ng/mL TNF−α、および1mLあたり2μM PGE2が、成熟を誘発するために未成熟DCへ添加された。成熟DCが、培養の8日目に収穫された。
【0084】
フローサイトメトリー
選択された分子の細胞発現を測定するために、フローサイトメトリーが用いられた。これらの分子は、蛍光染料へ付着されたモノクローナル抗体で特異的に染色される。フローサイトメトリー分析は、蛍光−活性化細胞ソーター(FACS(商標))において実施された。FACSCalibur(商標)において、染色された細胞の縣濁液がノズルを強制的に通され、1つずつ間隔をあけて配置された細胞を含有する液体の細い流れを生じた。各細胞がレーザービームを通過する時、これはレーザー光を散乱させ、この細胞と関連した蛍光分子が励起された。感受性光電倍増管が、細胞のサイズおよび粒度についての情報を与える両方の散乱光、および標識されたモノクローナル抗体の結合、および従って各細胞による標的分子の発現についての情報を与える蛍光発光を検出した。該細胞のソートのために、MoFlo(商標)高性能細胞ソーター(米国コロラド州フォートコリンズのダコ)が、25,000事象(events)/sおよび最大1psiで用いられた。
【0085】
細胞表面分子の直接染色
表面上のHLA−A2分子の発現を測定するために、ハイブリドーマ上澄み液HB82(メリーランド州ベセスダ(Bethesda)のATCC)、およびPE(フィコエリトリン)と共役された二次ヤギ抗マウス抗体が、細胞表面上の直接染色のために用いられた。約1×105DC、K562、またはK562−A2細胞が、1回洗浄された。各洗浄工程は、600μLのFACS(商標)緩衝液中の細胞の再縣濁、300×gで4分間の遠心分離、および上澄み液の廃棄を包含していた。洗浄後、50μLのHB82がこれらのペレットへ別々に添加された。氷上の30分のインキュベーション時間後、細胞は1回洗浄された。ついで0.5μLの二次抗体(1/100希釈;50μLのFACS(商標)緩衝液中の0.5μL抗体)が、これらのペレットへ別々に添加された。氷上および暗室での30分のインキュベーション時間後、細胞は1回洗浄された。最後に、細胞は200μLのFACS(商標)緩衝液中に再縣濁され、フローサイトメトリー分析が実施された。
【0086】
細胞内分子の間接染色
RNA−トランスフェクションされたDCの内部のチロシナーゼの発現を測定するために、FACS(商標)緩衝液中の1%パラホルムアルデヒドを用いて、細胞がまず固定された。固定後、これらの細胞は、FACS(商標)緩衝液中の0.1%および0.25%サポニン(Saponin)を用いて透過性化(permeabilise)された。簡単に言えば、約3×105細胞が1回洗浄された。各洗浄工程は、500μLのFACS(商標)緩衝液中、またはFACS(商標)緩衝液中の0.1%サポニン中の細胞の再縣濁、300×gで4分間の遠心分離、および上澄み液の廃棄を包含していた。洗浄後、FACS(商標)緩衝液(固定培地)中の500μLの1%パラホルムアルデヒドがこれらのペレットへ添加され、静かに混合された。氷上の30分のインキュベーション時間後、細胞は、FACS(商標)緩衝液で2回洗浄された。これらの細胞を500μLのFACS(商標)緩衝液中に4℃で7日まで保存することが可能であった。これらのペレットは、FACS(商標)緩衝液中500μLの0.1%サポニンで1回洗浄された。その後、FACS(商標)緩衝液(透過性化培地)中の50μLの0.25%サポニン、および5μLのチロシナーゼ特異的一次抗体が、ペレットへ添加され、静かに混合された。室温で1時間のインキュベーション後、細胞は、FACS(商標)緩衝液中の0.1%サポニンで1回洗浄され、ついでFACS(商標)緩衝液中の50μLの0.25%サポニン中一
次抗体(1/100希釈)について特異的なCy5−共役二次抗体0.5μLが、ペレットへ添加され、静かに混合された。室温および暗室での30分のインキュベーション後、これらの細胞は、FACS(商標)緩衝液中の500μLの0.1%サポニンで洗浄された。最後に、ペレット化された細胞が、FACS(商標)緩衝液中の200μLの0.1%サポニン中に再縣濁され、フローサイトメトリー分析が実施された。
【0087】
テトラマー染色およびテトラマーオフレート
HLA−A2−制限チロシナーゼ特異的CD8+T細胞の結合活性を評価するため、およびMoFlo(商標)高性能細胞ソーター(米国コロラド州フォートコリンズのダコ)でのソートを実施するために、これらのT細胞が、PE−標識A*0201/htyr3
69−377/Hβ2mテトラマー(Prof.D.Buschによって供給された)で染色された。これらの細胞は2回洗浄された。各洗浄工程は、冷PBS+0.5%ヒト血清中の細胞の再縣濁、300×gで5分間の遠心分離、および上澄み液の廃棄を包含していた。インキュベーション容積は細胞数に依存している。106までの細胞が、50μL中25分、PE−標識テトラマーで、氷上で暗室においてインキュベーションされた(1/25希釈、50μLPBS+0.5%ヒト血清中の2μLテトラマー)。HLA−A2−制限チロシナーゼ特異的CD8+T細胞のソートのために、5×106細胞までが、100μLPBS+0.5%ヒト血清中の6μL A2/ペプチドテトラマーでインキュベーションされた。次いで、さらに25分間、1/50の比においてCD8−APC抗体(米国フランクリンレイクスのBDファーミンゲン(Pharmingen,Franklin Lakes))が添加された。染色後、これらの細胞は2回洗浄され、最後にFACS(商標)緩衝液中の1%パラホルムアルデヒドで固定され、フローサイトメトリーFACSCalibur(カリフォルニア州マウンテンビューのベクトン・ディッキンソン(Becton Dickinson))によって分析されるか、またはMoFlo(商標)高性能細胞ソーター(米国コロラド州フォートコリンズのダコ)においてテトラマー+CD8+T細胞のソートのためにPBS+0.5%ヒト血清中に希釈された。
【0088】
テトラマーオフレートアッセイのために、洗浄後、脱離テトラマーの再結合は、飽和量のハイブリドーマ上澄み液HB82(メリーランド州ベセスダのATCC)でのインキュベーションによって防がれた。これは、複合ペプチドとは独立してHLA−A2を結合する。時間間隔を増加した後、サンプルが採取されて固定され、テトラマー染色の強度についてフローサイトメトリーによって分析された。
【0089】
単一種cRNAの生産
ここではcRNAと呼ばれる、単一種試験管内転写RNAの生産は、4つの工程を含んでいた。すなわち、cDNA挿入断片を含有するプラスミドの線形化、プロモーター配列および線形化されたプラスミド中のcDNAテンプレートに基づく試験管内転写、合成cRNAのポリアデニル化、およびcRNA精製である。実質量のcDNAテンプレート、すなわちプラスミドDNA(pDNA)が試験管内転写反応に必要とされるので、このプラスミドは、コンピテント細菌中で増幅されなければならなかった。
【0090】
プラスミドDNAでのコンピテント細菌の形質転換
他の基から親切な贈り物(kind gift)として受け取られる、より多量のpDNAを得るために、コンピテント細菌は該pDNAで形質転換されて拡張されなければならなかった。一回大量トップ(One Shot TOP)10F’コンピテント細胞の50μLガラス瓶が、融解しつつある氷上でゆっくりと霜取りされ、小さい容積のpDNAが添加され、静かに栓を抜く(tap)ことによって細菌と混合された。これらの細胞は、氷上で30分間インキュベーションされ、次いで42℃で正確に30秒間加熱され、最後に氷上に載せられて2分間冷却された。コンピテント細胞とともに供給されたリッチなSOC培地が添加され、形質転換された細菌が、適切な選択抗生物質とともにLB培地寒天プレート上に培養された。プレートが細菌インキュベーター中に37℃で一晩入れられた。抗生物質ゼオシンが光感受性であり、その活性は高い塩濃度によって阻害されるので、pZeoSV2+/huTyrで形質転換された細菌用のプレートは低塩LB寒天培地を含有し、暗室に保存された。
【0091】
形質転換細菌の選択および拡張
抗生物質への耐性をエンコードする、pDNAで形質転換された細菌のみが、寒天培地中の対応抗生物質の存在にもかかわらず、選択プレート上で成長してコロニーを形成することができた。一晩の成長後、コロニーは、滅菌つま楊枝を用いてこれらのプレートから引き出された。各々の個々のコロニーは、15mLファルコン(Falcon)管において、適切な抗生物質を含有する5mLのLB培地中に接種された。予め選択され、凍結された形質転換細菌が拡張されることになるならば、凍結ガラス瓶が、−80℃フリーザーから、融解しつつある氷上へ移された。約5μLの一部解凍された細菌縣濁液が、次いで、適切な抗生物質とともに5mLのLB培地を収容している15mLファルコン管に移された。これらの管は、一晩(約12時間)37℃で激しい振とうをともなって、150rpmで、形質転換された細菌の効率的な成長のためにインキュベーションされた。
【0092】
形質転換細菌の凍結
細菌は、凍結培地が15%のグリセリンを含有するならば、凍結プロセスによって害されることが最も少ない。したがって400μLの一晩細菌縣濁液が、84μLのオートクレーブされた87%グリセリンへ添加され、渦巻き撹拌され、液体窒素中に急速凍結され、−80℃へ移された。
【0093】
形質転換細菌からのプラスミドDNA抽出
一晩の培養後、細菌縣濁液が、5300×g、4℃で10分間遠心分離された。細菌細胞からのプラスミドDNA抽出が、QIAwell(登録商標)8ウルトラプラスミドキットを用いて、製造業者の指示に従って実施された。簡単に言えば、ペレット化された細胞が溶解された。溶解物中の塩およびpH条件が、DNAのこの膜への独占的結合を保証したが、一方、分解されたRNAおよび細胞タンパク質は保持されなかった。この膜はついで、あらゆる核酸結合タンパク質およびpDNAからの他の残留汚染物質の除去を可能にする、あらゆるDNA−タンパク質相互作用を乱す緩衝液で洗浄された。追加の洗浄工程は、塩および他の非−DNA成分を除去した。精製pDNAは最終的にトリス緩衝液中に溶出された。抽出pDNAの収率は、プラスミドに応じて、5mLの一晩細菌培養あたり4〜8μgであった。
【0094】
cRNA中への単一種cDNAの試験管内転写
各プラスミドが、試験管内転写に適したテンプレートを生産するために、適切な制限酵素で線形化された。すべてのプラスミドが、所望のタンパク質をエンコードするcDNAの5’末端においてT7プロモーターを含有するので、転写は、mMESSAGE mMACHINE(商標)T7キットからのT7RNAポリメラーゼを用いて製造業者の指示に従って実施された。RNA分子の安定性、およびタンパク質中への翻訳用テンプレートとしてのその効率的な使用は、その5’末端におけるキャップおよび3’末端におけるポリ(A)テイルの存在に依存している。ポリ(A)テイルは常に、細胞によって合成されたmRNA分子の一部である。従って、上述された全細胞mRNA増幅手順は、試験管内転写反応においてキャップ類似体の添加のみを含んでいた。しかしながら、プラスミド中のcDNAを用いて生産された単一種cRNAは、テイルが構成体においてエンコードされるか否かに応じて、ポリ(A)テイルを含有してもよいし含有しなくてもよい。試験管内転写反応における5’末端でのキャップ類似体の統合に加えて、チロシナーゼ、HLA−A2 cRNAは、ポリ(A)テイリングキットを用いて3’末端においてポリアデニル化された。両方の反応は、製造業者によって提案された条件下で実施された。試験管内転写およびcRNA精製後、cRNAの測定された収率は、100μLのポリアデニル化反応混合物あたり60〜70μgであった。
【0095】
cRNAの精製
増幅された全細胞cDNAまたはプラスミド中の単一種cDNAのどちらかをテンプレートとして用いて、試験管内転写反応において得られたcRNAの精製は、RNeasy(登録商標)ミニキットを用いて、製造業者の指示にしたがって実施された。精製のために、この手順は、試験管内転写反応混合物のRNeasy(登録商標)カラム上への転移から開始された。RNAはDEPC水中に溶出され、アリコートが−80℃で保存された。
【0096】
DC中へのRNAトランスフェクション
細胞血漿膜は、細胞の分子内容物をその外部環境から分離するという生命維持機能を果たしている。これらの膜は、高度に絶縁性の二層中に自己集合する両親媒性脂質から大部分なっている。可能な限り多くの抗原を捕獲しようとして、未成熟DCは、RNAを包含する、これらの周囲からの様々な構造(サイズが小さい分子から、アポトーシス体までの範囲のもの)を取り上げることが周知である。これらは、マクロ飲作用およびエンドサイトーシスを通して可能である。RNAとの単純DCコインキュベーションは、T細胞初回抗原刺激を達成することが証明されてはいるが、より攻撃的な方法、例えばリポフェクションおよびエレクトロポレーションは、より良好なトランスフェクション方法であることが証明された。
【0097】
エレクトロポレーション
エレクトロポレーションは、細胞が、強い外部電場の短いパルスへ暴露された時、血漿膜の可逆性透過性を誘発する。親水性細孔の形成は、二層膜中の脂質の再配向の結果である。この現象の分子メカニズムは依然としてよく理解されていない。細孔数およびこれらの直径は、パルス振幅およびパルス持続時間の積とともに増加する。小さい分子が、これらの細孔を通って細胞質中に単純に拡散するが、より大きい分子、例えば核酸は、電気泳動力によって細胞中に駆動される。核酸の存在が細孔形成を促進することも証明されている。細孔は、電場への暴露の1マイクロ秒以内に現われるが、これらが再閉するには数分かかる。
【0098】
最初に、2〜3×106DCまたはK562細胞が、少なくとも170μL OptiMEM I培地中に再縣濁され、0.4cmエレクトロポレーションキュベット中に入れられ、氷上で約3分間インキュベーションされた。エレクトロポレーション前の縣濁液の冷却は有利であるが、その理由は、これが細胞代謝を遅くしたからである。これによって電気による過熱が避けられた。さらには電気ショックによって、および死んだ細胞から放出された分子によって引起こされた損傷が最小限にされた。その後、100μL以下のDEPC水中に再縣濁されたRNAが添加され、270μLの総エレクトロポレーション縣濁液容積を生じた。この縣濁液は、ピペットによって短時間混合され、ついで急速にエレクトロポレーションされた。エレクトロポレーションは、250Vおよび150μFで実施された。エレクトロポレーションの直後に、細胞が、2%自家血漿、10mM HEPES、800U/mL GM−CSF、および800U/mL IL−4が補足された樹状細胞培地中に移された。これらの細胞が計数され、縣濁液が、FACS(商標)分析、RNA単離、または機能的アッセイのために迅速にアリコートされ、37℃および5%CO2でインキュベーター中に入れられた。
【0099】
機能的アッセイ
機能的アッセイにおいて、RNA−トランスフェクションされたDCまたはK562細胞、ペプチド−パルスされたDCまたは腫瘍細胞が、促進剤として役立った。抗原特異的CTLクローンが、エフェクターとして役立った。促進剤の刺激能力を測定するために、DCまたは他の細胞がまず、T細胞とともにコインキュベーションされ、これらは収穫され、解凍および再刺激の8〜14日後にコインキュベーション培養において用いられた。活性化されたT細胞によって分泌されたIFN−γの量が、酵素結合免疫吸着アッセイ(ELISA)において測定された。
【0100】
T細胞とのペプチド−パルスされたT2細胞コインキュベーション
T2細胞のCTLとのコインキュベーションのために、より低い細胞数が利用された。外因性ペプチドパルシングのために、1×106T2細胞が、10μg/mL YMDチロシナーゼペプチド(YMDGTMSQV、ミュンヘンLMUのジーンセンター)および10μg/mLヒトβ2−マイクログロブリン(米国サンディエゴのカルビオケム(Calbiochem))でインキュベーションされた。20分毎に細胞縣濁液が振とうされた。37℃、5%CO2での2時間のインキュベーション後、細胞縣濁液が、非結合ペプチドを除去するために1回洗浄された。100μLのペプチド−パルスされたT2縣濁液(12%FCSが補足されたRPMI 1640中の1×104細胞)が、100μLのCTL縣濁液(15%ヒト血清が補足されたCTL培地中の2×103細胞)とコインキュベーションされ、5:1の促進剤対エフェクター比を生じた。
【0101】
エフェクターおよび促進剤の24時間のコインキュベーション後、150μLの各上澄み液が収穫され、−20℃で保存された。上澄み液が、酵素結合免疫吸着アッセイ(ELISA)において分析された。
【0102】
T細胞とのDCもしくはK562コインキュベーション
エレクトロポレーションの12時間もしくは24時間後、100μLのT細胞縣濁液(コインキュベーション培地中2×104細胞)が、100μLのトランスフェクションされた細胞縣濁液(2%自家血漿および800U/mL GM−CSFおよび800U/mL IL−4が補足された樹状細胞培地中の2×104または4×104細胞)へ添加され、96ウエルプレートの1ウエルあたり全部で200μL、および1:4、1:2、1:1、2:1、または4:1の促進剤対エフェクター比を生じた。
【0103】
非トランスフェクションDCが、10μLのペプチド溶液(100μL/mL)を100μLのDC縣濁液(2%自家血漿および800U/mL GM−CSFおよび800U/mL IL−4が補足された樹状細胞培地中の4×104細胞)へ添加することによって、ペプチドで外因的にパルスされた。37℃および5%CO2での2時間のインキュベーション後、100μLのCTL縣濁液(CTLコインキュベーション培地中の2×104細胞)が添加され、2:1の促進剤対エフェクター比を生じた。エフェクターおよび促進剤の24時間のコインキュベーション後、150μLの各上澄み液が収穫され、−20℃で保存された。
【0104】
インターフェロン−ガンマ(IFN−γ)ELISA
共培養上澄み液中のIFN−γの測定が、ELISAにおいてOptEIA(商標)ヒトIFN−γセットを用いて、製造業者の指示に従って実施された。簡単に言えば、ELISAプレートは、マウス抗−IFN−γ捕獲抗体でコーティングされ、次いでFBS−含有溶液で遮断された。いくつかの事例では、上澄み液は、コインキュベーション培地中において1:2.5で希釈された。同じ培地が、IFN−γ標準の連続希釈液を作るために用いられた。細胞培養上澄み液または標準溶液からのIFN−γが、これらのプレート中の捕獲抗体へ結合された後、ビオチチニル化マウス抗−IFN−γ検出抗体が、ホースラディッシュ−ペルオキシダーゼへ共役されたアビジンとともに添加された。捕獲抗体からなる複合体を視覚化するために、IFN−γ、検出抗体、ビオチン、アビジン、およびホースラディッシュ−ペルオキシダーゼ、H2O2(ペルオキシダーゼのための基質)が、テトラメチルベンジジン(基質試薬AおよびBミックス)とともに添加され、溶液の色彩を青色に変えた。酵素反応が、1Mオルト−リン酸で停止された。これによって、溶液の色彩が黄色に変わり、その強度は、処理された基質の量に正比例した。すなわち捕獲されたIFN−γの量に間接的に比例した。反応溶液中の光吸収が、450nmで測定された。不明なIFN−γ濃度が、IFN−γ標準の公知濃度および対応測定吸光度に基づいて描かれた標準曲線の助けを借りて計算された。
【0105】
細胞傷害性アッセイ
それぞれテトラマー+CD8+ソートされたバルク細胞株およびT細胞クローンの細胞溶解活性が、標準4時間クロム放出アッセイにおいて分析された。これら2つのHLA−マッチされた黒色腫細胞株Mel A375(HLA−A2−陽性、チロシナーゼ−陰性)およびMel IL−2(HLA−A2−陽性、チロシナーゼ−陽性)が用いられた。簡単に言えば、106標的黒色腫細胞が、200μCi51Crで1〜1.5時間標識された。51Cr−標識された標的細胞が、V底96ウエル組織培養プレート(ドイツ国ゾリンゲンのグライナー(Greiner,Solingen))において100μL/ウエルの12%のFCSを有するRPMI 1640中でT細胞とともに培養された。CTL−媒介溶解の有効性を評価するために、T細胞は連続的に希釈され、ついで段階的エフェクター細胞対標的細胞(E:T)比を生じるために103標的細胞/ウエルと共培養された。37℃で4時間の共培養後、50μLの上澄み液が収集され、放射能がガンマカウンターで測定された。特異的溶解のパーセンテージは、100×(実験放出−自然放出)/(最大放出−自然放出)として計算された。自然放出は、エフェクター細胞の不存在下に標的細胞をインキュベーションすることによって評価され、一般に14%未満であった。
【0106】
参照
Becker, C, Pohla, H., Frankenberger, B., Assenmacher, M., Schendel, D.J. and Th. Blankenstein. 2001. Adoptive tumor therapy with T lymphocytes enriched through an IFN capture assay. Nature Med. 7(10): 1159-1162.
Dudley, M. E., Wunderlich, J. R., Robbins, P. F., Yang, J. C., Hwu, P., Schwartzentruber, D. J., Topalian, S. L., Sherry, R., Restifo, N. P., Hubicki, A. M., Robinson, M. R., Raffeld, M., Duray, P., Seipp, C. A., Rogers-Freezer, L., Morton, K. E., Mavroukakis, S. A., White, D. E., Rosenberg, S. A. (2002). Cancer regression and autoimmunity in patients after clonalrepopulation with antitumor lymphocytes. Science 298:850-854.
Dudley, M.E. and Rosenberg, S.A.(2003). Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nature Reviews Cancer 3: 666-675.
Engels, B., Nossner, E., Frankenberger, B., Blankenstein, Th., Schendel, D. J., and W. Uckert. 2005. Redirecting human T lymphocytes towards renal cell carcinoma-specificity by retroviral transfer of T cell receptor genes. Human Gene Then, 16(7). -79.9-810
FaIk, C. S. and Schendel, D. J. (2002). Allogeneic MHC class I ligandsand their role in positive and negative regulation of human cytotoxiceffector cells. Human Immunol. 63:8-19.
Gao, L., Bellantuono, I., Elsasser, A., Marley, S. B., Gordon, M. Y., Goldmann, J. M., Stauss, H. J. (2000). Selective elimination of leukemic CD34+ progenitor cells by cytotoxicT lymphocytes specific for WTl . Blood 95: 2198-2203.
Gao, L., Yang, T. H., Tourdot, S., Sadovnikova, E., Hasserjian, R. and Stauss H. J. (1999). AlIo- major-histocompatibility complex restricted cytotoxic T lymphocytes engraft in bone marrow transplant recipients without causing graft- versus-host disease. Blood 94: 2999-3006.
Geiger, C, Regn, S., Noessner, E., Wilde, S., Frankenberger, B., Papier, B., Pohla, H., and D.J. Schendel. 2005. Development of a generic RNA-pulsed dendriticcell vaccine for renal cell carcinoma, J. Translational Medicine in press.
Gilboa-E and J. Viewig. 2004. Cancer immunotherapy with mRNA-transfected dendriticcells. Immunol. Rev. 199:251-63.
KoIb, H. J., Schmid, C, Barrett, A. J. and Schendel, D. J. (2004). Graft-versus-leukemia reactions in allogeneic chimeras. Blood 103:767-776.
KoIb, H.J., Schattenberg, A., Goldman, J.M., Hertenstein, B., Jacobsen, H., ArceseW., Ljungman, P., Ferrant, A., Verdonck, L. Niederwieser, B. et al. 1995. Graft-versus-leukemia effect of donor lymphocyte transfusions in marrow grafted patients. Blood 86:2041.
Levings, M.K. and M.G. Roncarolo. 2005. Phenotypic and functional differences between human CD4+CD25+ and type 1 regulatory T cells. Curr. Top. Microbiol. Immunol. 293:303-326.
Liao, X., Yongging, L., Bonini, C, Nair, S., Gilboa, E., Greenberg, P. D., Yee, C. (2004). Transfection of RNA encoding tumor antigens following maturation of dendritic cells leads to prolonged presentation of antigen and the generation of high-affinity tumor-reactive cytotoxic T lymphocytes. MoI. Ther. 9: 757-764.
Nair, S. K., Boczkowski, D., Morse, M., Cumming, R.I., Lyerly, H. K. and Gilboa, E. (1998). Induction of primary carcinoembryonic antigen (CEA)-specific cytotoxic T lymphocytes in vitro using human dendritic cells transfectedwith RNA. Nat. Biotechnol. 16(4): 364-369.
Napolitani, G., Rinaldi, A., Bertoni, F., Salllusto, F. and A. Lanzavecchia. 2005. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type-1 polarizing programme in dendritic cells. Nat. Immunol. 6(8): 769-76.
Regn, S., Chen, X., Schendel, D., KoIb, H.-J. and M.A. Roskrow. 2001. The generation of monospecific and bispecific anti-viral cytotoxic T lymphocytes (CTL) for the prophylaxis of patients receiving an allogeneic bone marrow transplant. Bone Marrow Transplant. 27: 53-64.
Schaft, N, Dorrie, J., Thuman, P., Beck, V. E., Muller, I., Schultz, E. S., Kampgen, E., Dieckmann, D. and Schuler, G. 2005. Generation of an optimized polyvalent monocyte-derived dendritic cell vaccine by transfectingdefined RNA after rather than before maturation. J. Immunol. 174(5): 3087-97.
Schendel, D. J., Wank, R. and B. Dupont. 1979. Standardization of the human in vitro cell-mediated lympholysis technique. Tissue Antigens 13: 112-120.
Schendel, D. J., Maget, B., FaIk, C. S. and R. Wank. Human CD8+ T lymphocytes. 1997. In: The Immunology Methods Manual. (I. Lefkovits, Ed.) pp 670-690, Academic Press Ltd., London.
Su, Z., Peluso, M. V., Raffegerst, S. H., Schendel, D. J., and M. A. Roskrow. 2001. LMP2a-specific cytotoxic T lymphocytes for the treatment of patients with relapsed EBV-positive Hodgkin disease. Eur. J. Immunol. 31: 947-958.
Su, Z., Peluso, M. V., Raffergerst, S. H., Schendel, D. J. and Roskrow, M. 2002. Antigen presenting cells transfected with LMP2a RNA induce CD4+ LMP2a-specif[iota]c cytotoxic T lymphocytes which kill via a Fas-independent mechanism. Leuk. Lymphoma 43(8): 1651-62.
Toes, R. E., Schoenberger, S. P., van der Voort, E. I., Offringa, R. and Melief, C. J. 1998. CD40- CD40Ligand interactions and their role in cytotoxic T lymphocyte priming and anti-tumor immunity. Semin. Immunol. 10(6): 443-8.
Yee, C, Savage, P. A., Lee, P. P., Davis, M. M. and P. D. Greenberg. 1999. Isolation of high avidity melanoma-reactive CTL from heterogenouspopulations using peptide-MHC-tetramers. J. Immunol. 162(4): 2227-34.
更なる参照
Belinda Palermo, Silvia Garbelli, StefaniaMantovani, Elisabetta Scoccia, Gian Antonio Da Prada, Paola Bernabei, M. Antonietta Avanzini, Valeria Brazzelli, Giovanni Borroni and Claudia Giachino(2005). Qualitative difference between the cytotoxicT lymphocyte responses to melanocyte antigens in melanoma and vitiligo. Eur. J. Immunol. 35: 3153-3162.
Cassian Yee, Peter A. Savage, Peter P. Lee, Mark M. Davis, and Philip D. Greenberg (1999). Isolation of High Avidity Melanoma-Reactive CTL from Heterogeneous Populations Using Peptide-MHC Tetramers. The Journal of Immunology: 2227-2234.
Matthias Wolfl, Stefan Schalk, Martin Hellmich, Katharina M. Huster, Dirk H. Busch, and Frank Berthold(2004). Quantitation of MHC Tetramer-Positive Cells From Whole Blood: Evaluation of a Single-Platform, Six-Parameter Flow Cytometric Method. Wiley-Liss, Inc. Cytometry Part A 57A: 120-130.
【特許請求の範囲】
【請求項1】
抗原特異的T細胞であって、
a)患者由来MHC分子をエンコードする核酸、および、抗原もしくは前記抗原をエンコードする核酸を準備する工程、
b)工程a)において規定される両方の化合物を、健康なドナーに由来する抗原提示細胞(APC)中にコトランスフェクションするか、または導入する工程、
c)健康なドナーに由来する末梢血リンパ球(PBL)を、前記APCで初回抗原刺激する工程、
d)MHC−抗原リガンドに特異的なT細胞を選択する工程
を含む方法によって得られる抗原特異的T細胞。
【請求項2】
エフェクター細胞特徴を有するT細胞、サイトカイン生産T細胞、細胞傷害性T細胞、および調節T細胞からなる群から選択される、請求項1に記載のT細胞。
【請求項3】
前記調節T細胞は、CD4+もしくはCD8+T細胞である、請求項2に記載のT細胞。
【請求項4】
トランスジェニックTCRをコードする核酸であって、
a)患者由来MHC分子をエンコードする核酸、および、抗原もしくは前記抗原をエンコードする核酸を準備する工程、
b)工程a)において規定される両方の化合物を、健康なドナーに由来する抗原提示細胞(APC)中にコトランスフェクションするか、または導入する工程、
c)健康なドナーに由来する末梢血リンパ球(PBL)を、前記APCで初回抗原刺激する工程、
d)MHC−抗原リガンドに特異的なT細胞を選択する工程、
e)単離T細胞のT細胞受容体(TCR)のクローン工程
を含む方法によって得られる核酸。
【請求項5】
請求項4に記載の核酸を含むベクター。
【請求項6】
プラスミドまたはレトロウイルスベクターである、請求項5に記載のベクター。
【請求項7】
請求項5または請求項6に記載のベクターで形質転換されているPBMC。
【請求項8】
請求項1もしくは請求項2に記載の該T細胞、または請求項7に記載のPBMC、および製薬的に許容可能な担体を含む製薬組成物。
【請求項9】
注入、注射、またはワクチンである、請求項8に記載の製薬組成物。
【請求項1】
抗原特異的T細胞であって、
a)患者由来MHC分子をエンコードする核酸、および、抗原もしくは前記抗原をエンコードする核酸を準備する工程、
b)工程a)において規定される両方の化合物を、健康なドナーに由来する抗原提示細胞(APC)中にコトランスフェクションするか、または導入する工程、
c)健康なドナーに由来する末梢血リンパ球(PBL)を、前記APCで初回抗原刺激する工程、
d)MHC−抗原リガンドに特異的なT細胞を選択する工程
を含む方法によって得られる抗原特異的T細胞。
【請求項2】
エフェクター細胞特徴を有するT細胞、サイトカイン生産T細胞、細胞傷害性T細胞、および調節T細胞からなる群から選択される、請求項1に記載のT細胞。
【請求項3】
前記調節T細胞は、CD4+もしくはCD8+T細胞である、請求項2に記載のT細胞。
【請求項4】
トランスジェニックTCRをコードする核酸であって、
a)患者由来MHC分子をエンコードする核酸、および、抗原もしくは前記抗原をエンコードする核酸を準備する工程、
b)工程a)において規定される両方の化合物を、健康なドナーに由来する抗原提示細胞(APC)中にコトランスフェクションするか、または導入する工程、
c)健康なドナーに由来する末梢血リンパ球(PBL)を、前記APCで初回抗原刺激する工程、
d)MHC−抗原リガンドに特異的なT細胞を選択する工程、
e)単離T細胞のT細胞受容体(TCR)のクローン工程
を含む方法によって得られる核酸。
【請求項5】
請求項4に記載の核酸を含むベクター。
【請求項6】
プラスミドまたはレトロウイルスベクターである、請求項5に記載のベクター。
【請求項7】
請求項5または請求項6に記載のベクターで形質転換されているPBMC。
【請求項8】
請求項1もしくは請求項2に記載の該T細胞、または請求項7に記載のPBMC、および製薬的に許容可能な担体を含む製薬組成物。
【請求項9】
注入、注射、またはワクチンである、請求項8に記載の製薬組成物。
【図1】


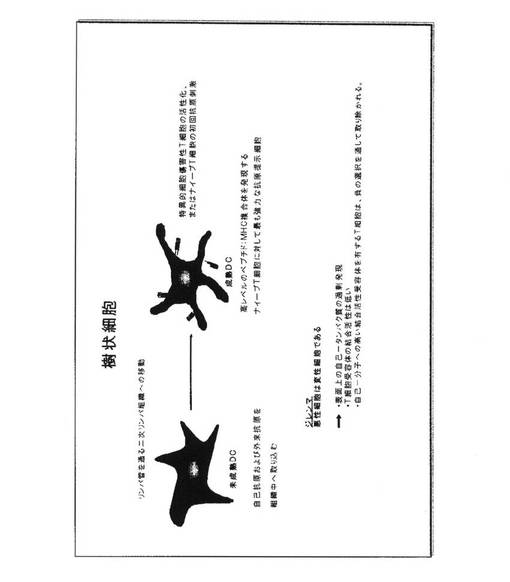
【図2】
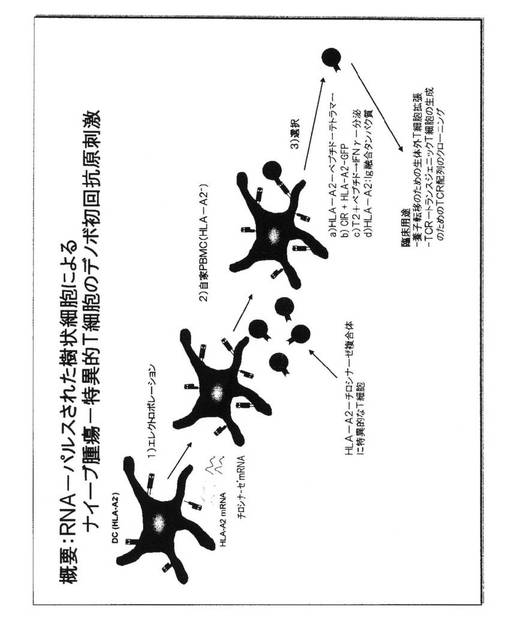
【図3】
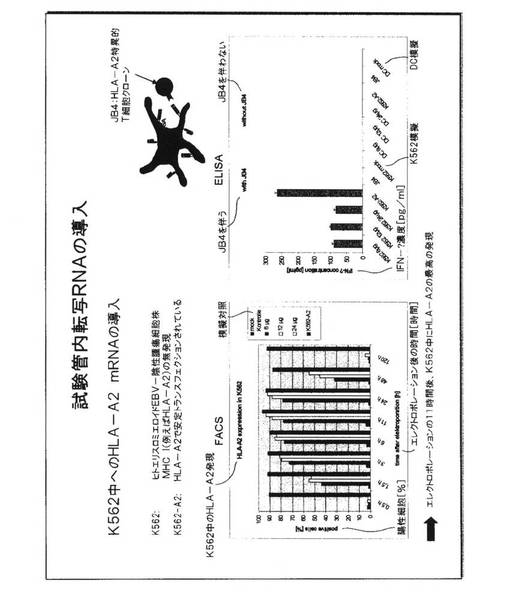
【図4】
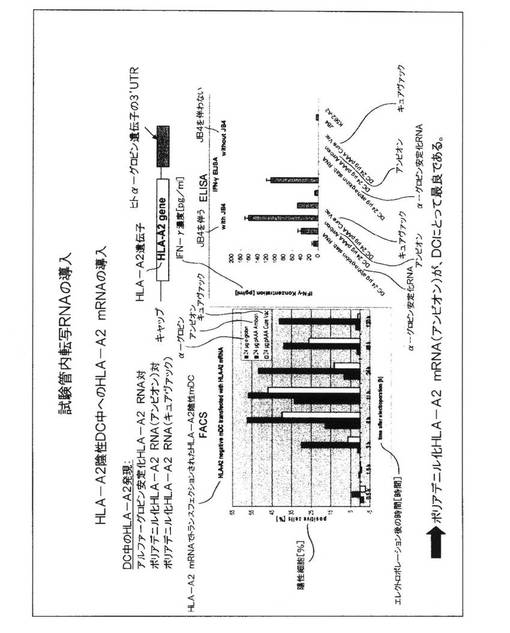
【図5】

【図6】
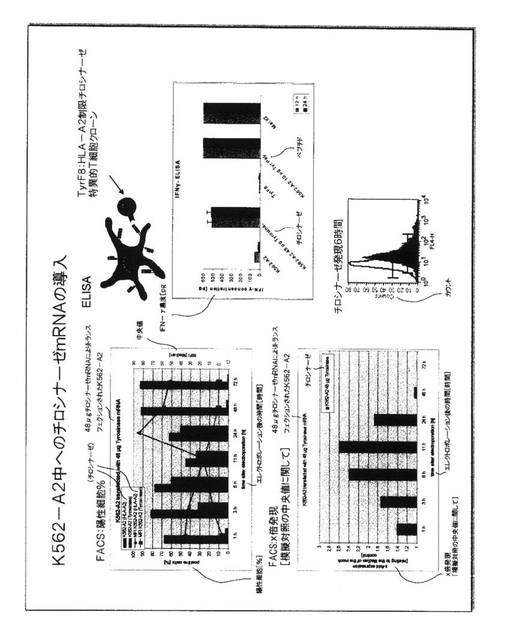
【図7】

【図8】
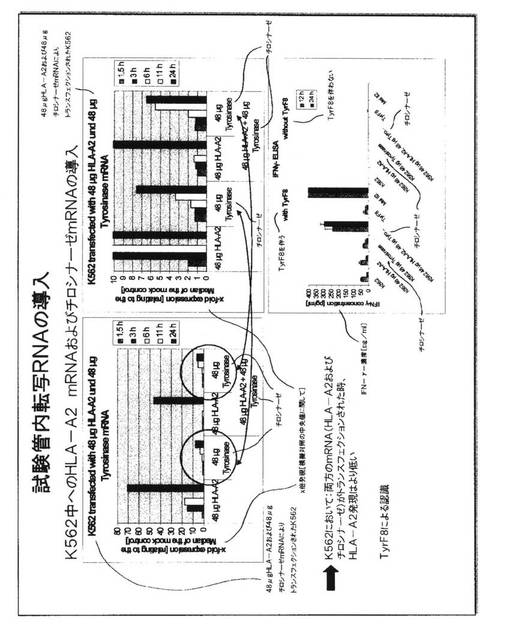
【図9】
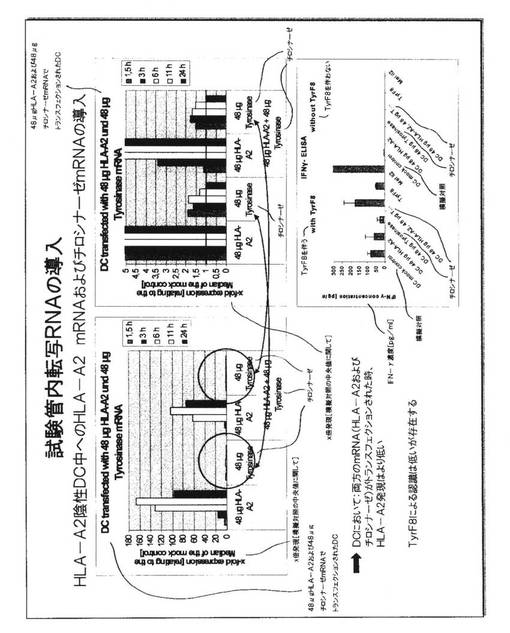
【図10】
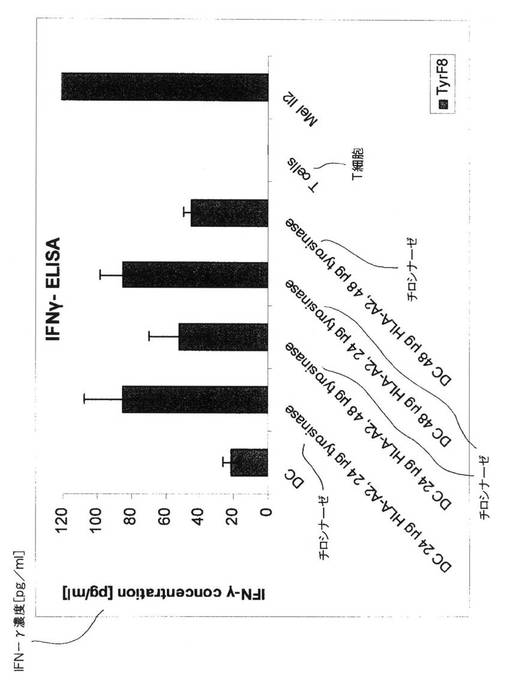
【図11】
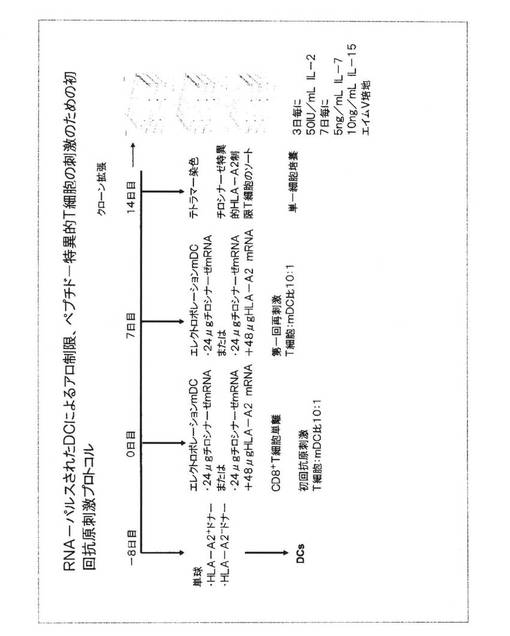
【図12】
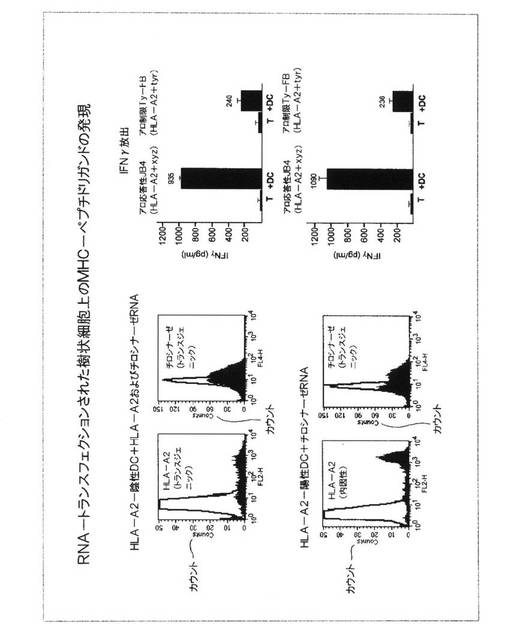
【図13】

【図14】
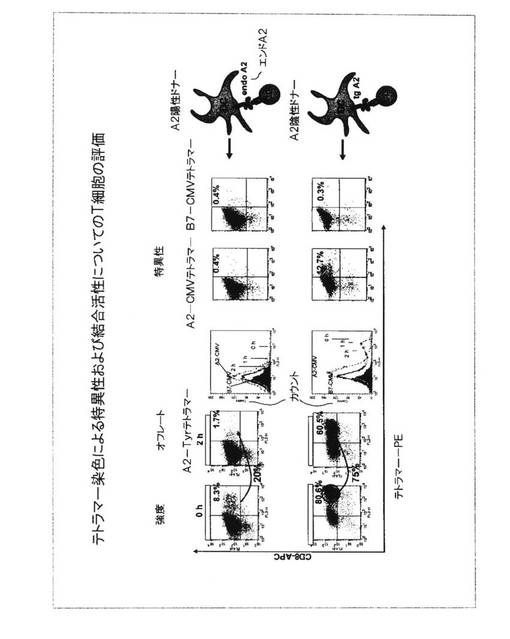
【図15】
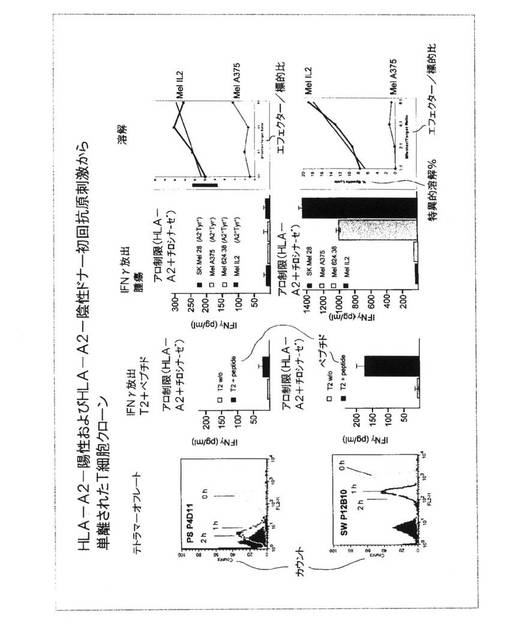
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【公開番号】特開2012−213402(P2012−213402A)
【公開日】平成24年11月8日(2012.11.8)
【国際特許分類】
【出願番号】特願2012−154420(P2012−154420)
【出願日】平成24年7月10日(2012.7.10)
【分割の表示】特願2008−524450(P2008−524450)の分割
【原出願日】平成18年8月4日(2006.8.4)
【出願人】(597121382)ヘルムホルツ ツェントラム ミュンヘン ドイチェス フォーシュングスツェントラム フュール ゲズントハイト ウント ウンヴェルト ゲーエムベーハー (4)
【氏名又は名称原語表記】HELMHOLTZ ZENTRUM MUENCHEN DEUTSCHES FORSCHUNGSZENTRUM FUER GESUNDHEIT UND UMWELT GMBH
【出願人】(507393665)
【Fターム(参考)】
【公開日】平成24年11月8日(2012.11.8)
【国際特許分類】
【出願日】平成24年7月10日(2012.7.10)
【分割の表示】特願2008−524450(P2008−524450)の分割
【原出願日】平成18年8月4日(2006.8.4)
【出願人】(597121382)ヘルムホルツ ツェントラム ミュンヘン ドイチェス フォーシュングスツェントラム フュール ゲズントハイト ウント ウンヴェルト ゲーエムベーハー (4)
【氏名又は名称原語表記】HELMHOLTZ ZENTRUM MUENCHEN DEUTSCHES FORSCHUNGSZENTRUM FUER GESUNDHEIT UND UMWELT GMBH
【出願人】(507393665)
【Fターム(参考)】
[ Back to top ]