
抗寄生虫活性を有する化合物及びそれを含有する医薬
本発明は、抗寄生虫活性を有する化合物、より詳細には、抗マラリア活性及び抗バベシア病活性を有する新規な化合物である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明の主題は、抗寄生虫活性を有する化合物、より詳細には、抗マラリア活性及び抗バベシア病活性を有する化合物である。
【背景技術】
【0002】
いろいろな疾病を引き起こす寄生虫、特にPlasmodium属、なかでも、熱帯熱マラリア原虫(Plasmodium falciparum)に対して活性をもつ医薬の研究が行われている。
【発明の開示】
【発明が解決しようとする課題】
【0003】
本発明者は、あるカテゴリーの化学化合物が、認容することができる毒性と高いバイオアベイラビリティ特性を示し、高い活性をもつことを観察した。それらの化合物は、好ましくは、経口経路により投与される。
【0004】
したがって、本発明の目的は、抗寄生虫活性、特に抗マラリア活性を有する新規な化合物を提供することである。
【0005】
また、本発明は、該化合物の製造方法に関する。
【0006】
さらに、本発明は、活性成分として、それらの化合物を含有する医薬、及び該化合物の使用と上記活性をもつ医薬を製造するための誘導体の製造方法に関する。
【課題を解決するための手段】
【0007】
本発明に係る化合物は、一般式(I)で表される点に特徴があり、

但し、Xは式(II)の基であり、
Zは−(CH2)mの基であり、m=8−21,n=0又は1,及びY=R3であり、
R1及びR'1は、互いに同一又は異なり、H,アルキル,OH,O−アルキル,O−アリール,O−CO−アルキル,O−CO−アリール,OSO2−アルキル,OSO2−アリール,OSO2−ヘテロ環,O−CO−O(又はS又はNH)−アルキル,O−CO−O(又はS又はNH)−アリール,PO(O−アルキル又はO−アリール)2,CO−O−CH2−アリール,シクロアルキル,から選択され、
R2及びR'2は、互いに同一又は異なり、H,アルキル,CO−O−CH2−アリール,CO−O−アルキル,シクロアルキル,から選択され、
R3及びR'3は、互いに同一又は異なり、H,アルキル,CO−O−アルキル,CO−O−アリール,COO− CH(R)−O−CO−アルキル,PO(O−アルキル又はO−アリール又はONa)2,CO−O−CH(R)
−アリール,から選択され、RはH又はアルキルである、
あるいは、
R1及びR2,及び/又はR'1及びR'2,又はR2及びR3,及び/又はR'2及びR'3,は共に、それぞれが結合する1又は複数の窒素原子をもつ非芳香族モノ(non
aromatic mono)ヘテロ環を形成し、
あるいは、さらに、
R2及びR3及び/又はR'2及びR'3は、同一の置換基であり、窒素に二重結合しており、それぞれ、R1又はR'1と環化してヘテロ環を形成し、Raで置換される場合は、それは、H,アルキル,1,2又は3つのハロゲン元素で置換したアルキル,アリール,CO−O−アルキル(又はアリール),−CO−OH,−CO−NH2,−CN,−CO−NH−アルキル(又はアリール),−CO−N−(アルキル)2,窒素及び/又は酸素を有する−CO−ヘテロ環,NH(H又はアルキル),N(アルキル)2,窒素及び/又は酸素を有するヘテロ環,−O−アルキル(又はアリール),−O−CH2−アリール,CH2N[H,(H,アルキル),(ジアルキル),アリール],窒素及び/又は酸素を有する−CH2−ヘテロ環,CH2−CO−OH,から選択され、
あるいは、X=R4、Yは式(III)の基であり、
n及びZは上記したのと同様であり、
R1及びR'1は互いに同一又は異なり、H,アルキル,OH,O−アルキル,O−アリール,O−CO−アルキル,O−CO−アリール,OSO2−アルキル,OSO2−アリール,OSO2−ヘテロ環,O−CO−O(又はS又はNH)−アルキル,O−CO−O(又はS又はNH)−アリール,PO(O−アルキル又はO−アリール)2,CO−O−CH2−アリール,シクロアルキル,から選択され、
R4及びR'4は、H,アルキル,又はアリールであり、これはOH,O−アルキル,O−アリール,NH(H又はアルキル),窒素又は酸素へテロ環で代替することが出来、
R2及びR'2は、互いに同一又は異なり、H,アルキル,CO−O−CH2−アリール,CO−O−アルキル,シクロアルキル,から選択され、
あるいは、水素の1又は複数の原子は低級アルキルに置き換えることができ、R1及びR4及び/又はR'1及びR'4は共に−(CH2)pの基を形成し、pは3−5の整数、
R2及びR'2はHを表し、又はR4及びR2及び/又はR'4及びR'2は、共に−(CH2)pの基を形成し、1又は複数の水素原子は低級アルキルに置き換えることができ、R1及びR'1はHを表す、及びこれらの化合物の薬理学的に許容できる塩である。
【0008】
特に定義されていない場合は、“アリール”は、フェニル又は芳香族性を有するあらゆる環又はヘテロ環を意味し、例えば、ピリジン,オキサゾール,チアゾール環,置換している場合は、特に塩素,−NO2,−NH2,N(H,アルキル)又は(ジアルキル)による置換体を意味する。
【0009】
“窒素及び/又は酸素を有するヘテロ環”は、ピロリジン,ピペリジン,モルホリン,ピペラジン,メチルピペラジン環のような5又は6の頂点をもつ環を意味する。
【0010】
“アルキル”は、直鎖又は分枝したC1−C5のアルキル、置換している場合は、1又はそれ以上のハロゲン原子、−NH2,N(H,アルキル)又は(ジアルキル)アミノ基による置換体を意味する。
【0011】
本発明の好適な誘導体のファミリー、すなわち、ファミリーAは、式(IV)で表され、
n,Z,R1,R'1,R2,R'2,R3及びR'3は、式(II)で前記規定したのと同様である。
【0012】
このファミリーの好適な化合物は、n=0の場合であり、式(V)で表される。
【0013】
好ましいグループ、すなわち、グループa1においては、R1,R2及びR3及び/又はR'1,R'2及びR'3は、互いに独立している。
【0014】
サブグループにおいては、R1及び/又はR'1,及びR2及び/又はR'2は、ハロゲン原子を表し、R3及び/又はR'3は、前記したのと同様であるが、水素原子とは異なる。
【0015】
他のサブグループにおいては、R1及び/又はR'1は、前記したのと同様であるが、水素原子とは異なり、一方、R3及び/又はR'3,R2及び/又はR'2は、水素原子を表す。
【0016】
他の好ましい式(V)の化合物のグループは、n=0であり、
R1及びR2,及び/又はR'1及びR'2は、式(VI)、すなわち、グループa2に該当する、
あるいは、
R2及びR3及び/又はR'2及びR'3は、式(VII)、すなわち、グループa3に該当する。
【0017】
式(VI)に該当するサブグループにおいては、R1及びR2及び/又はR'1及びR2は、共に−O−CO−,O−SO−,O−CS,S−CO又は−S−CS基を形成し、R3及び/又はR'3は、水素原子を表す。
【0018】
式(VI)に該当するもう1つのサブグループにおいては、R1及びR2,及び/又はR'1及びR'2は、選択的に、分枝したアルキレン基を表し、R3及び/又はR'3は、−CO−O−アルキル(又はアリール),−CO−O−CH2−アリール,CO−O−CH(アルキル)−O−CO−アルキル,PO(O−アルキル又はアリール)2,アルキル又はHを表す。
【0019】
式(VII)に該当するサブグループにおいては、R1及び/又はR'1は、水素原子を表し、R2及びR3,及び/又はR'2及び/又はR'3は、−(CH2)p−の基を表す。
【0020】
他の好ましいファミリーAのグループ、すなわち、グループa4は、R2及びR3及び/又はR'2及びR'3は、同一の置換基を形成し、それぞれR1又はR'1と共に、式(VIII)のビス−オキサジアゾールを形成する。
但し、Raは、前記規定したのと同様である。
【0021】
このグループの好ましい化合物においては、ハロゲンはF又はClが好ましく、アルキルはメチル又はエチル、アリールはフェニルである。
【0022】
本発明の他の好ましいファミリー、すなわち、ファミリーBは、式(IX)で表される。
【0023】
このファミリーの好ましい化合物においては、Z=−(CH2)m及びn=0であり、式(X)に該当するものである。
【0024】
ファミリーBの好ましいグループ、すなわち、グループb1においては、置換基は互いに独立している。
【0025】
サブグループにおいては、R1及びR4及び/又はR'1及び/又はR'4は、前記したとおりであり、R2は水素原子を表す。
【0026】
他のサブグループにおいては、R1及びR2及び/又はR'1及び/又はR'2は、共にオキシカルボニル鎖−OCO−を表し、R4及びR'4は、上記の様に定義される。
【0027】
他のサブグループにおいては、R1及びR4及び/又はR'1及び/又はR'4は、共に−(CH2)p−の基を表し、nは3−5の整数であり、R2及びR'2は、Hを表す。
【0028】
さらなる他のサブグループにおいては、R1及びR'1は、Hを表し、R4及びR2及び/又はR'4及びR'2は、共に−(CH2)p−の基を表し、pは3−5の整数であり、一またはそれ以上の水素原子が低級アルキルに置き換えられることができる。
【0029】
他のサブグループは、式(XI)に該当する。
【0030】
ファミリーAとBの好ましい化合物は、以下の表1−3に示している。
【0031】
【表1】
【0032】
【表2】
【0033】
【表3】
【0034】
本発明に係る化合物は、適当な場合には、塩の形をとる。例として、塩酸塩、クエン酸塩、酒石酸塩、マレイン酸塩、酪酸塩、酢酸塩及びトリフルオロ酢酸塩が挙げられる。
【0035】
本発明においては、前記した一般式(V)及び(VI)のカルバメート及びN−ホスホリル誘導体は、一般式(V)及び(VI)のビスアミジン化合物の2相溶媒中での反応を含むことを特徴とする方法によって製造することができ、該(V)及び(VI)では、R3及びR'3=HとCl−R3(又はR'3)誘導体であり、R3及びR'3は前記したのと同様であり、実施例に示したようにHとは異なる。
【0036】
前記規定した一般式(V)及び(X)のアミドキシム誘導体は、R1及びR'1=OHである一般式(V)及び(X)のビスアミドキシムと実施例に示したような適当な試薬との塩基性溶媒中での反応を含むことを特徴とする方法によって製造することができる。
【0037】
好ましくは、前記した一般式(VI)のグループa2及び(VIII)のグループa4は、実施例で示した適当な試薬の存在下、前記した一般式(V)のグループa1で表されるアミドキシム又はアミドキシム誘導体の分子内環化で調製される。
【0038】
寄生虫、特にPlasmodium属に関する本発明の生成物の活性についての研究により、それらの生成物が高いin vitro活性を有することが示された。
【0039】
その結果、熱帯熱マラリア原虫に関して、IC50値(該寄生虫の50%阻止濃度)は、nMからμMの単位である。
【0040】
したがって、本発明は、医薬組成物を製造するために、該化合物の特性の利用に関するものである。
【0041】
本発明の医薬組成物は、前記した少なくとも1つの化合物の有効量を、不活性な薬剤学的担体と結合して含有することを特徴とする。
【0042】
また、本発明は、感染症、特にマラリアの治療薬を製造するための、前記化合物の少なくとも1つの利用に関するものである。
【0043】
これらの組成物は、他の医薬の活性成分を含むことができる。特に薬理学的共同作用又は耐性回避のために、他の抗マラリア薬(例えば、向リソゾーム薬、アトバコン、抗葉酸薬又は抗フォリニン酸薬、アルテミシニン又はその誘導体の1つ)との組み合わせが挙げられる。
【0044】
また、それらの組成物は、同化を促進する化合物と組み合わせて用いることが好ましい。
【0045】
本発明の医薬組成物は、種々の形態で、特に経口経路又は注射経路あるいは又、直腸経路で投与することができる。
【0046】
経口経路には、特に錠剤、丸剤、ゼラチンカプセル、ドロップが利用できる。
【0047】
他の投与形態には、静脈内経路、皮下経路、又は筋肉内経路で注射される溶液を含む。これらは、無菌又は滅菌可能な溶液から調製される。また、該溶液には、懸濁剤あるいは乳剤も含む。
【0048】
坐剤も他の投与形態として利用することができる。
【0049】
本発明の組成物は、特にヒト及び動物の感染症に適しており、特にマラリア、バベシア病の治療に適している。
【0050】
例を挙げると、ヒトに使用し得る用量は、次の用量である。例えば、1回又はそれ以上の用量につき、患者に対し1−90mg/kgを投与する。
【0051】
また、本発明は、上記した化合物を活性成分として含有する生物学的試薬に関する。
【0052】
この試薬は、あらゆる抗寄生虫活性の研究において、対照あるいは標準として使用することができる。
【0053】
本発明の他の特徴及び利点は、上記化合物の合成及び抗寄生虫活性の研究に関する以下に述べる実施例により明らかになるであろう。これらの実施例において、図1を参照として設けた。これは、薬物濃度の関数として、Desjardins’test(Desjardins
R.E. et al., Antimicrob. Agents Chemother.1979,16,710−718)により、化合物6.0の抗マラリア活性を表す。
【実施例】
【0054】
実施例1:
中間体の合成
1,12−ジシアノドデカン:
3.3g(67.10mmol)のシアン化ナトリウムを含むジメチルスルホキシド30mlの懸濁液を、完全に溶解するまで、激しく攪拌しながら90−95℃の間で加熱する。1,12−ジブロモドデカン10g(30.49mmol)を、ごく少量ずつ、ゆっくりと得られた該溶液に添加し、50℃以下の温度に冷却する。周囲温度にて2時間攪拌した後、得られた該懸濁液にジクロロメタン90mlを添加して溶解し、塩化ナトリウム飽和水溶液で数回洗浄する。その後、有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。低温油状の残渣をエーテルに溶解し、次いで、蒸発乾固して晶出し、白色結晶の生成物6.44g(96%)を得る。
融点:<40℃
NMR1H(CDCl3,100MHz),δ(ppm):1.24(s,16H);1.60(m,4H);2.31(t,4H)
FT−IR,ν(cm-1):2246(CN)
【0055】
テトラデカンジイミド酸ジエチルニ塩酸塩:
無水エタノール20ml、無水エーテル60ml及び1,12−ジシアノドデカン5g(22.73mmol)の0℃に冷却した溶液に塩酸ガスを泡状にして2時間通じ、次いで、反応混合物を一晩攪拌させる。その後、減圧下で溶媒を蒸発させる。得られた固形物の残渣を晶出し、エーテルで数回洗浄した後、デシケーターで乾燥させる。白色粉末7.7g(82%)を得る。
融点:118−120℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.26(s,16H);1.35(t,6H);1.60(m,4H);2.63(t,4H);4.55(q,4H);11.16及び12.08(2s,4H)
FT−IR,ν(cm-1):1097(C−O);1651.50(C=N);3032及び3118(NH2,Cl)
【0056】
N,N’−ジ−ベンジルオキシカルボニル−S−メチルイソチオ尿素:
2ml(14.4mol)のクロロギ酸ベンジルを、1g(3.60mmol)のS−メチルイソチオ尿素硫酸塩を含むジクロロメタン/炭酸水素ナトリウム飽和水溶液(1:1)の2相混合物40mlの溶液に激しく攪拌しながら滴下する。周囲温度にて25時間攪拌する。その後、ジクロロメタンで水相を3回抽出する。次いで、結合した有機相を水で洗浄し、その後、硫酸ナトリウムで乾燥し、減圧下、蒸発させる。得られた残渣を、シリカカラム(DCM)によるクロマトグラフィーで精製し、黄色油状の生成物1.06g(82%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):2.46(s,3H);5.24(s,4H);7.42(s,10H);11.91(s,1H)
FT−IR,ν(cm-1):1022及び1173(C−O);1647(C=N);1751(NCO);3169(NHCO)
【0057】
N,N’−ジ−tert−ブチルオキシカルボニル−S−メチルイソチオ尿素:
5.81g(26.60 mmol)の炭酸ジ−tert−ブチル及び2.53g(18.20mmol)のS−メチルイソチオ尿素ヘミ硫酸塩を含むDCM/NaHCO3の飽和2相水溶液(100ml)を48時間激しく攪拌する。その後、分離した水相を2×100mlのDCMで抽出する。次に、結合した有機相を2×200mlの水で洗浄し、その後、減圧下、蒸発させる。次いで、残渣を0.55g(3.85
mmol)のS−メチルイソチオ尿素ヘミ硫酸塩を加えた100mlの飽和水性DCM/NaHCO3の2相混合物に溶解させる。反応混合物を再び72時間激しく攪拌する。結合した有機相は、前記と同様の処理を行った後、硫酸ナトリウムで乾燥させ、減圧下、蒸発させる。最後に、残渣をシリカ(ヘキサン/CHCl35%次いでCHCl3)で精製して、白色粉末の生成物3.46g(90%)を得る。
融点:123−124℃
NMR1H(CDCl3,100MHz),δ(ppm):1.49(s,18H);2.38(s,3H);11.60(s,1H)
FT−IR,ν(cm-1):1043及び1252(C−O);1667(C=N);1765(NCO);3337(NHCO)
【0058】
チオ炭酸O−クロロメチル−S−エチル
37ml(500mmol)のエタンチオールと69.3 ml(500mmol)のトリエチルアミンを含むエーテル溶液200mlを、44ml(500mmol)のクロロギ酸クロロメチル含むエーテル溶液900mlに攪拌下、2時間滴下し、0−5℃に冷却した。その後、処理状態を30分間保つ。その後、反応混合物を周囲温度で16時間攪拌し、次いで、形成した沈殿物をろ過し、エーテルで洗浄する。結合したエーテル相を蒸発させ、残渣を蒸留して精製して、液状の生成物57g(73%)を得る。
沸点:99−100℃/.18mbar
NMR1H(CDCl3,100MHz),δ(ppm):);1.30(t,3H);2.89(q,2H);5.96(s,2H)
FT−IR,ν(cm-1):1719(S−C−O)
【0059】
チオ炭酸S−エチル−O−ヨードエチル:
55g(356mmol)のチオ炭酸O−クロロメチル−S−エチルを、106.8g(712mmol)のヨウ化ナトリウム及び3g(35.6mmol)の重炭酸ナトリウムを含むアセトン450mlの攪拌溶液に直接加える。その後、反応混合物を、攪拌したまま、40℃で4時間放置する。形成される沈殿物をろ過し、アセトンとエーテルで洗浄する。有機相を蒸発させ、残渣を、1100mlの0℃に冷却されたヘキサンと50mlの冷水に分ける。その後、分離された有機相を、5%の水性重炭酸ナトリウム200ml、1%のチオ硫酸ナトリウム200ml(溶液の脱色まで)及び水2×200mlで連続的に洗浄する。ヘキサン相を硫酸ナトリウム上で乾燥させ、減圧下で蒸発させた後、81g(92%)の未精製の黄色がかった液体の製品を得る。
NMR 1H(CDCl3, 100MHz),δ(ppm): ); 1.30 (t, 3H); 2.89 (q,
2H,); 5.96 (s, 2H).
FT−IR,ν(cm-1): 1715 (S-C-O)
【0060】
チオ炭酸O−アセチルオキシメチル−S−エチル:
80g(325.2mmol)のチオ炭酸S−エチル−O−ヨードメチルを、26.7g(325.2mmol)の酢酸ナトリウムを含む無水ホルムアミドジメチル420mlの溶液に攪拌下、2時間以上滴下し、−20℃に冷却した。その後、反応混合物を周囲温度で16時間攪拌し、次いで、形成した沈殿物をろ過し、20mlのホルムアミドジエチルと40mlのエーテルで洗浄する。
【0061】
850mlのエーテルと350mlの冷水を、分離漏斗で有機相に添加する。水相を単離し、350mlの水で抽出する。次いで、結合したエーテル相を、220mlの5%水性炭酸水素ナトリウム、220mlの水、2×220mlの0.01N塩酸及び220mlの水で続けて洗浄する。有機相を硫酸ナトリウムで乾燥させた後、残渣を蒸留して精製し、黄色液状の生成物35g(60%)を得る。
沸点:82−83℃/.0.25mbar
NMR1H(CDCl3,100MHz),δ(ppm):);1.30(t,3H);2.09(s,3H);2.87(q,2H);5.76(s,2H)
FT−IR,ν(cm-1):1717(S−CO−O);1767(CO−O)
【0062】
クロロギ酸アセチルオキシメチル
14.90ml(185.4mmol)の塩化スルフリルを、33g(185.4mmol)のチオ炭酸O−アセチルオキシメチル−S−エチルの溶液に攪拌下添加し、0−5℃に冷却した。処理状態を15分間保った後、反応混合物を周囲温度で45分間攪拌する。次いで、形成されたS−エチルクロリド溶液を周囲温度、20mbarで一晩蒸発させる。次いで、得られた残渣を抽出して精製し、オレンジ色液状の生成物15.50g(56%)を得る。
沸点:75−76℃/.17mbar
NMR1H(CDCl3,100MHz),δ(ppm):2.12(s,3H);5.76(s,2H)
FT−IR,ν(cm-1):1724(CO−O);1773(Cl−CO)
【0063】
3,4−ジヒドロ−5−メトキシ−2H−ピロール:
23ml(1当量)の硫酸ジメチルを、23.03g(0.235mol)のピロリジン−2−オンのベンゼン溶液85mlに滴下し、70℃に加熱する。この反応混合物を3時間還流下、加熱する。周囲温度に冷却した後、15Nソーダ溶液19mlを反応混合物に添加する。反応混合物を分離漏斗に注ぎ、ベンゼンで3回水相を抽出する。結合した有機相を硫酸ナトリウムで乾燥させ、減圧下蒸発させる。残渣を減圧して抽出し、無色液体の生成物を収率58%で得る。
融点:10-2mbar下、37℃
NMR1H(CDCl3,100MHz),δ(ppm):1.97(m,2H);2.41(t,2H);3.61(t,2H);3.75(s,3H)
【0064】
2−メチルスルファニル−3,4,5,6−テトラヒドロピリミジニウムヨージド:
メタノール(チオ尿素10ml/g)に1当量の3,4,5,6−テトラヒドロ(1H)ピリミジン−2−チオンと1.2当量のヨウ化メタンを含む反応混合物を還流下5時間加熱する。冷却した後、メタノールを減圧下蒸発させて、定量的に生成物を得る。沈殿物は、その後、アセトン又は石油エーテルで洗浄することができる。
NMR1H(CDCl3,100MHz):0.95(s,6H);2.62(s,3H);3.11(s,4H);8.76(s,2H)ppm
【0065】
5,5−ジメチル−3,4,5,6−テトラヒドロ(1H)ピリミジン−2−チオン:
10ml(167.2mol;2当量)の二硫化炭素を、250mlフラスコ中の10ml(83.6mol)の2,2−ジメチル−1,3−ジアミノプロパンを含む無水エタノール50mlの溶液に添加する。次いで、16.02g(83.6mol)のEDCを該反応混合物に添加し、周囲温度で3時間攪拌する。エタノールを蒸発させ、残渣を水に溶解し、ジクロロメタンで抽出する。硫酸ナトリウムで乾燥し、有機相を蒸発させた後、生成物11.2g(93%)を得る。これは付加的に精製することなく使用される。
融点:225−228℃
NMR1H(CDCl3,100MHz):0.91(s,6H);2.88(d,4H);7.46(s,2H)ppm
【0066】
5,5−ジメチル−2−メチルスルファニル−3,4,5,6−テトラヒドロピリミジニウム塩酸塩:
メタノール(チオ尿素10ml/g)に1当量の5,5−ジメチル−3,4,5,6−テトラヒドロ(1H)ピリミジン−2−チオンと1.2当量のヨウ化メタンを含む反応混合物を還流下5時間加熱する。冷却した後、メタノールを減圧下蒸発させて、定量的に生成物を得る。沈殿物は、その後、アセトン又は石油エーテルで洗浄することができる。
融点:225−228℃
NMR1H(CDCl3,100MHz):0.95(s,6H);2.62(s,3H);3.11(s,4H),8.76(s,2H)ppm
【0067】
1−(N−tert−ブチルオキシカルボニルアミノ)ドデカン−12−アンモニウム塩酸塩:
4.02g(18.4mol)のジ炭酸ジ−tert−ブチルのジオキサン溶液60mlを、15.01g(75mmol,4.1当量)の1,12−ドデカンジアミンを含むジオキサン/水(150/70ml)混合物溶液にゆっくり滴下(約2時間以上)する。反応混合物を周囲温度で24時間攪拌し、過剰のジアミンはろ過し、溶媒は減圧下蒸発させる。残渣は1NのHCl溶液とジクロロメタンに溶解させる。水相に形成した沈殿物はろ過して、モノ−Boc誘導体と少量のジアミンを共に塩酸塩の形で生成する。
この固形物をエタノールとエーテルで再結晶して、モノ−保護ジアミン塩の生成物4.56g(収率74%)を白色粉末の形で得る。
融点:153−155℃
NMR1H(CD3OD,100MHz),δ(ppm):1.28(m,20H);1.38(s,9H);2.91(m,4H)
MS−FAB+:[M+H]+:301
【0068】
[1−[N−(5,5−ジメチル−3,4,5,6−テトラヒドロピリミジン−2−イル)アミノ]−12−(N’−tert−ブチルオキシカルボニルアミノ)]ドデカン塩酸塩:
0.85g(3mmol)の5,5−ジメチル−2−メチルスルファニル−3,4,5,6−テトラヒドロピリミジニウムヨージドと0.85ml(2当量)のトリエチルアミンを、1.02g(3mmol)の12−(N−tert−ブチルオキシカルボニルアミノ)ドデカン−1−アンモニウムクロリドを含むアセトニトリル15mlの懸濁液に添加する。この反応混合物を24時間還流下、加熱する。周囲温度に冷却した後、反応しなかったアミンをろ過する。ろ過物は減圧下蒸発させ、シリカカラム(溶出溶媒:CH2Cl2/CH3OH/NH4OH
80:16:4)でクロマトグラフィーを行い、生成物1.36g(85%)を得る。
NMR1H(CD3OD,100MHz),δ(ppm):1.07(s,6H);1.37(m,29H);3.05(m,8H)
【0069】
[1−[N−(5,5−ジメチル−3,4,5,6−テトラヒドロピリミジン−2−イル)アミノ]ドデカン−12−アンモニウムジトリフルオロアセテート:
1.06g(2mmol)の[1−[N−(5,5−ジメチル−3,4,5,6−テトラヒドロピリミジン−2−イル)アミノ]−12−(N’−tert−ブチルオキシカルボニルアミノ)]ドデカンヨウ化水素塩を15mlのTFA/CH2Cl2(3/1)溶液に溶解する。この溶液を周囲温度で3時間攪拌し、過剰のトリフルオロ酢酸を減圧下蒸発させ、定量的に生成物0.98gを得る。
NMR1H(DMSO−d6,100MHz),δ(ppm):1.0(s,6H);1.3(m,20H);3.0(m,8H);8.0(bs,2H);9.0(s,3H)
MS−ES+:[M+H]+:311;[M+2H]++/2:156
【0070】
合成
1,12−ビス(アミジル)ドデカンニ塩酸塩:1.0, 2HCl
45mlの無水エタノールと5g(13mmol)のジエチルテトラデカンジイミド酸塩酸塩の溶液に、アンモニアガスを泡にして2時間通じ、氷浴で10℃以下の温度に冷却する。その後、溶媒を減圧下蒸発させる。次いで、得られた残渣は、エタノールで数回洗浄し、デシケーターで乾燥させる。白色粉末状の生成物3.53g(83%)を得る。
融点:170−172℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.25(s,16H);1.58(m,4H);2.37(t,4H);8.82(s,4H);9.10(s,4H)
FT−IR,ν(cm-1):1688(C=N);3081(NH2,Cl);3245(NH2)
【0071】
1,12−ビス[N,N’−(メチルオキシカルボニル)アミジニル]ドデカン:1.1
60ml(7.65mmol)のクロロギ酸メチルを、1g(3.06mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液60mlに激しく攪拌しながら、4N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、100mlの水を加える。次いで、形成された沈殿物を分離し、水その後エーテルで数回洗浄し、デシケーターで乾燥させ、白色粉末状の生成物1.30g(77%)を得る。
融点:116−117℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.69(s,16H);1.96(m,4H);2.61(t,4H);3.97(s,6H);9.06(s,4H)
FT−IR,ν(cm-1):1256(C−O−C);1633(C=N);1670(NHCO);3343(NH)
MS−ES+:[M+H]+:371
25
【0072】
1,12−ビス[N,N’−(エチルオキシカルボニル)アミジニル]ドデカン:1.2
0.73ml(7.65mmol)のクロロギ酸エチルを、0.1g(3.06mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液60mlに激しく攪拌しながら、4N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、100mlの水を加える。次いで、形成された沈殿物を分離し、水その後エーテルで数回洗浄し、デシケーターで乾燥させ、白色粉末状の生成物0.98g(80%)を得る。
融点:95−96℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.63(t,6H);1.71(m,16H);1.93(m,4H);2.63(t,4H);4.49(q,4H);8.95及び9.12(2s,4H)
FT−IR,ν(cm-1):1251(C−O−C);1667(C=N);1757(CO);3186(NHCO);3330(NH)
MS−ES+:[M+H]+:399;[M+H]2+/2:200(100%)
【0073】
1,12−ビス[N,N’−(ブチルオキシカルボニル)アミジニル]ドデカン:1.3
0.99ml(7.65mmol)のクロロギ酸ブチルを、1g(3.06mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液60mlに激しく攪拌しながら、4N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、100mlの水を加える。次いで、形成された沈殿物を分離し、水その後エーテルで数回洗浄し、デシケーターで乾燥させ、白色粉末状の生成物1.12g(80%)を得る。
融点:87−88℃
NMR1H(CDCl3,100MHz),δ(ppm):0.88(t,6H);1.21(s,16H);1.48−1.70(m,8H);1.88(m,4H);2.24(t,4H);4.03(t,4H);6.21(s,2H);9.24(s,2H)
FT−IR,ν(cm-1):1084及び1234(C−O−C);1594(C=N);1660(CO);3313(NHCO);3441(NH)
MS−ES+:[M+H]+:455
【0074】
1,12−ビス[N,N’−(イソブチルオキシカルボニル)アミジニル]ドデカン:1.4
1ml(7.65mmol)のクロロギ酸イソブチルを、1g(3.06mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液60mlに激しく攪拌しながら、4N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、100mlの水を加える。次いで、形成された沈殿物を分離し、水その後エーテルで数回洗浄し、デシケーターで乾燥させ、白色粉末状の生成物1.27g(91%)を得る。
融点:102−103℃
NMR1H(CDCl3,100MHz),δ(ppm):0.92(d,12H);1.22(s,16H)1.62(m,4H);1.96(m,2H);2.26(t,4H);3.82(d,4H);6.19(s,2H);9.27(s,2H)
FT−IR,ν(cm-1):1069及び1237(C−O−C);1599(C=N);1661(CO);3314(NHCO);3440(NH)
MS−ES+:[M+H]+:455
【0075】
1,12−ビス[N,N’−(ベンジルオキシカルボニル)アミジニル]ドデカン:1.5
1.1ml(7.65mmol)のクロロギ酸ベンジルを、1g(3.06mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液60mlに激しく攪拌しながら、4N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、100mlの水を加える。次いで、形成された沈殿物を分離し、水その後エーテルで数回洗浄し、デシケーターで乾燥させ、白色粉末状の生成物1.2g(75%)を得る。
融点:118−119℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.19(s,16);1.47(m,4H);2.13(t,H);4.96(s,4H);7.30(s,10H);8.29(s,2H);8.50(s,2H)
FT−IR,ν(cm-1):693及び745(芳香族性C−H);1236(C−O−C);1648(C=N);1666(CO);3311(NHCO);3436(NH)
MS−ES+:[M+H]+:523;[M+2H]2+/2:262(100%)
【0076】
1,12−ビス[N,N’−(4−ニトロベンジルオキシカルボニル)アミジニル]ドデカン:1.6
1.65g(7.65mmol)のクロロギ酸4−ニトロベンジルのジオキサン溶液5mlを、1g(3.06mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液60mlに激しく攪拌しながら滴下し、氷浴で冷却する。該溶液のpHは、4N炭酸ナトリウム水溶液で10−12に保つ。次いで、混合物を周囲温度で一晩攪拌する。その後、100mlの水を加える。次いで、形成された沈殿物を分離し、水その後エーテルで数回洗浄し、デシケーターで乾燥させ、白色粉末状の生成物1.37g(74%)を得る。
融点:87−88℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.22(s,16H);1.51(m,4H);2.18(t,4H);5.15(s,4H);7.59(dd,4H);8.23(dd,4H);8.69(s,4H)
FT−IR,ν(cm-1):1248(C−O−C);1344及び1512(NO2);1621(C=N);1658(CO);3316(NHCO);3406(NH)
MS−ES+:[M+H]+:623
【0077】
1,12−ビス[N,N’−(フェニルオキシカルボニル)アミジニル]ドデカン:1.7
1.44ml(11.47mmol)のクロロギ酸フェニルを、1.5g(4.59mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液80mlに激しく攪拌しながら、2N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、150mlの水を加え、該溶液を3×60mlのDCMで抽出する。次いで、有機相を水で数回洗浄し、減圧下蒸発させる。液状の生成物1.77g(78%)を得る。
融点:87−88℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.73(s,16H);2.05(m,4H);2.72(t,4H);7.17−7.90(m,10H);9.20(s,4H)
FT−IR,ν(cm-1): 1623(C=N);1682(COO);3378(NH及びNHCO)
MS−ES+:[M+H]+:495
【0078】
1,12−ビス[N,N’−(4−フルオロフェニルオキシカルボニル)アミジニル]ドデカン:1.8
1.50ml(11.47mmol)のクロロギ酸4−フルオロフェニルを、1.5g(4.59mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液80mlに激しく攪拌しながら、2N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、150mlの水を加える。次いで、形成された沈殿物を分離し、水その後エーテルで数回洗浄し、デシケーターで乾燥させ、白色粉末の生成物1.55g(64%)を得る。
融点:℃
NMR1H(CDCl3,100MHz),δ(ppm):1.26(s,16H);1.70(m,4H);2.35(t,4H);6.62−7.14(m,8H);4.20(s,2H);9.23(s,2H)
FT−IR,ν(cm-1): 1177(C−O−C);1253(C−F);1622(C=N);1679(COO);3327(NH及びNHCO)
MS−ES+:[M+H]+:531
【0079】
1,12−ビス[N,N’−(4−フルオロフェニルオキシカルボニル)アミジニル]ドデカンニ塩酸塩:1.8, 2HCl
1gの1.8を、塩酸ガスを飽和したエタノール溶液20mlに添加する。次いで、反応混合物を50℃で2時間激しく攪拌しながら加熱する。エーテル150mlを該低温溶液に添加した後、該混合物を冷蔵庫に一晩放置する。デカンテーションした後、形成した油層を100mlの蒸留水に溶解し、その後、ろ過する。ろ過物は最後、凍結乾燥して、白色粉末の塩を得る。
NMR1H(DMSO−d6,100MHz),δ(ppm):1.75(s,16H);2.15(m,4H);3.06(t,4H);7.20(s,2H);7.70及び7.78(d,8H);7.85及び8.21(2s,4H)
FT−IR,ν(cm-1):1684(C=N);1753(NCO);3324(NH,HCl)
【0080】
1,12−ビス[N,N’−(4−メトキシフェニルオキシカルボニル)アミジニル]ドデカン:1.9
1.70ml(11.47mmol)のクロロギ酸4−メトキシフェニルを、1.5g(4.59mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液80mlに激しく攪拌しながら、2N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、150mlの水を加え、該溶液を3×60mlのDCMで抽出する。その後、有機相を水で数回洗浄し、次いで、減圧下蒸発させ、液状の生成物1.97g(78%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.62(m,4H);2.28(t,4H);3.73(s,6H);6.71−7.06(m,8H);6.18(s,2H);9.21(s,2H)
FT−IR,ν(cm-1): 1176及び1189(C−O−C);1504(芳香族性C−H);1623(C=N);1678(COO);3374(NH及びNHCO)
MS−ES+:[M+H]+:555
【0081】
[1,12−ビス(アミジニル)ドデカン]−1,12−ビス−N,N’−ホスホン酸ジフェニル:1.12
2.85ml(13.76mmol)のクロロホスホン酸ジフェニルを、1.5g(4.59mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液80mlに激しく攪拌しながら、4N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、50mlの水を加える。次いで、形成した沈殿物を分離し、水その後エーテルで数回洗浄し、デシケーターで乾燥して、白色粉末の生成物2.70g(82%)を得る。
融点:100−101℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.65(s,16H);1.93(m,4H);2.79(t,4H);7.68−7.81(m,20H);8.31(s,2H);8.71(s,2H)
NMR31P(DMSO−d6,81MHz),δ(ppm):−1.64
FT−IR,ν(cm-1):935(P=O);1230(C−O);1675 (C=N);3169(NHPO);3324(NH)
MS−FAB+:[M+H]+:719;[M+2H]++/2:360(100%)
【0082】
[1,12−ビス(アミジニル)ドデカン]−1,12−ビス−N,N’−ホスホン酸ジエチル:1.13
1.11ml(7.65mmol)のクロロホスホン酸ジエチルを、1g(3.06mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液40mlに激しく攪拌しながら、4N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で一晩攪拌する。その後、溶液をジクロロメタン(3×30ml)で抽出する。次いで、有機相を水で数回洗浄し、硫酸ナトリウムで乾燥する。減圧下溶液を蒸発させて油状残渣を生成する。最小量のエーテルに溶解させた該残渣を−4℃、一晩で晶出して、白色粉末の生成物1.08g(67%)を得る。
融点:68−69℃
NMR1H(CDCl3,100MHz),δ(ppm):1.20(s,16H);1.26(t,12H);1.53(m,4H);2.21(t,4H);3.99(quintuplet,8H);6.05(s,2H);7.78(s,2H)
NMR31P(DMSO−d6,81MHz),δ(ppm):8.76
FT−IR,ν(cm-1):793及び1031(P−O− CH2CH3);958(P=O);1647
(C=N);3187(NHPO);1581及び3386(NH)
MS−ES+:[M+H]+:527
【0083】
[1,12−ビス(アミジニル)ドデカン]−1,12−ビス−N,N’−ホスホン酸ナトリウム:1.14
a)[1,12−ビス(アミジニル)ドデカン]−1,12−ビス−N,N’−ホスホン酸:
1.36ml(9.51mmol)のトリメチルシランヨージドを、窒素雰囲気下で、0℃に冷却した10mlの無水ジクロロメタンに、1g(1.9 mmol)の1.13の攪拌下、滴下する。このように操作状態を1時間保つ。その後、溶液を減圧下蒸発させる。次いで、3mlの水を含むアセトン10mlに低温残渣を溶解し、24時間攪拌する。その後、形成した沈殿物を分離し、アセトンで数回洗浄し、エタノールで再結晶して、白色粉末の生成物0.52g(66%)を得る。
融点:164−165℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.24(s,16H);1.56(m,4H);2.32(t,4H);9.26及び9.63(2s,8H)
NMR31P(CDCl3,81MHz),δ(ppm):−5.82
FT−IR,ν(cm-1):1044及び1211(P−OH);939(P=O);1667 (C=N);2324(POH);3019(NHPO);1557及び3322(N−H)
MS−ES+:[M+H]+:415
【0084】
b )[1,12−ビス(アミジニル)ドデカン]−1,12−ビス−N,N’−ホスホン酸ナトリウム:1.14
0.1N炭酸ナトリウム溶液(約21ml)を、水20mlに[1,12−ビス(アミジニル)ドデカン]−1,12−ビス−N,N’−ホスホン酸1gを含む懸濁液に攪拌下、pHが7.4になるまで滴下する。その後、溶液を凍結乾燥して、白色粉末を得る。
NMR31P(81MHz),δ(ppm):−3.16
【0085】
1,12−ビス(N,N’−ヒドロキシアミジニル)ドデカン:1.15
13.70g(196.91mmol)のヒドロキシルアミン塩酸塩を、水−アルコール性炭酸ナトリウム溶液[炭酸ナトリウム8.22g、水36ml、及び95%エチルアルコール138mlから調製]に添加する。15分間攪拌した後、20g(90.91mmol)の1,12−ジシアノドデカンを添加する。その後、反応混合物を、還流下、72時間加熱し、次いで、減圧下、蒸発させる。次に、得られた残渣を、水に溶解させ、攪拌し、ろ過する。沈殿物を分離し、水と石油エーテルで数回洗浄する。エタノールで再結晶し、デシケーターで一晩乾燥させた後、白色粉末の生成物25g(96%)を得る。
融点:170−171℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.70(s,16H);1.90(m,4H);2.39(t,4H);5.75(s,4H);9.13(s,2H)
FT−IR,ν(cm-1):1661(C=N);3244(N−OH);3315及び3400(NH2)
+TOF MS:287(M+H);254(M−32);144(M+H/2)
【0086】
1,12−ビス(N,N’−メトキシアミジニル)ドデカン:1.16
1.70ml(17.48mmol)の硫酸ジメチルを、氷浴で冷却したジオキサン/NaOH 0.7N2相混合物50mlに1.15の2g(7mmol)を含む懸濁駅に攪拌下、滴下する。周囲温度で一晩攪拌する。その後、反応混合物をDCMで抽出し、ろ過し、ろ過物を3×100mlの水で洗浄する。有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。石油エーテルでの添加で低温残渣は晶出し、デシケーターで乾燥させた後、白色粉末の生成物1.05g(48%)を得る。
融点:85−86℃
NMR1H(CDCl3,100MHz),δ(ppm):1.23(s,16H);1.50(m,4H);2.10(t,4H);3.74(s,6H);4.44(s,4H)
FT−IR,ν(cm-1):1048(N−O−C);1643(C=N);3288及び3433(NH2)
+TOF MS:315(M+H)
【0087】
1,12−ビス(N,N’−エトキシアミジニル)ドデカン:1.17
1.40ml(14.68mmol)のクロロギ酸エチルのクロロホルム5mlを、クロロホルム45mlに1.15の2g(7mmol)とトリエチルアミン2.65ml(18.9mmol)を含む懸濁駅に攪拌下、滴下する。周囲温度で3時間攪拌する。その後、反応混合物をろ過し、ろ過物を3×100mlの水で洗浄する。有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。低温残渣を晶出した後、結晶を石油エーテルで洗浄し、デシケーターで乾燥して、白色粉末の生成物2.53g(84.33%)を得る。
融点:100−101℃
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.29(t,6H);1.53(m,4H);2.19(t,4H);4.23(quartet,4H).4.79(s,4H)
FT−IR,ν(cm-1):1240(C−O−C);1620(C=N);1741(OCOO);3374及び3508(NH2)
+TOF MS:431(M+H)
【0088】
1,12−ビス(N,N’−エトキシカルボニルオキシアミジニル)ドデカン:1.18
1.14ml(14.68mmol)のクロロギ酸メチルのクロロホルム5mlを、クロロホルム45mlに1.15の2g(7mmol)とトリエチルアミン2.65ml(18.9mmol)を含む懸濁駅に攪拌下、滴下する。周囲温度で3時間攪拌する。その後、反応混合物をろ過し、ろ過物を3×100mlの水で洗浄する。有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。低温残渣を石油エーテルで洗浄し、その後、分離し、デシケーターで乾燥して、白色粉末の生成物2.05g(73%)を得る。
融点:79−80℃
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.53(m,4H);2.19(t,4H);3.81(s,6H).4.81(s,4H)
FT−IR,ν(cm-1):1247(C−O−C);1622(C=N);1750(OCOO);3380及び3497(NH2)
+TOF MS:403(M+H)
【0089】
1,12−ビス(N,N’−フェノキシカルボニルオキシアミジニル)ドデカン:1.19
1.85ml(14.68mmol)のクロロギ酸フェニルを、DMF30mlに1.15の2g(7mmol)とトリエチルアミン2.65ml(18.9mmol)を含む懸濁液に攪拌下、滴下し、冷水浴で冷却する。周囲温度で4時間攪拌し、その後、反応混合物をろ過し、ろ過物を150mlの酢酸エチルで希釈する。次いで、溶液を水(100ml)と飽和塩化ナトリウム溶液(2×100ml)で洗浄する。有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。低温残渣を石油エーテルで洗浄し、その後、分離し、デシケーターで乾燥して、白色粉末の生成物3.02g(82%)を得る。
融点:108−109℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.72(s,16H);1.98(m,4H);2.52(t,4H);6.98(s,4H);7.65−7.98(m,10H)
FT−IR,ν(cm-1):1197及び1241(C−O−C);1633(C=N);1770(OCOO);3309及び3473(NH2)
+TOF MS:(M+H)
【0090】
1,12−ビス(N,N’−チオメチルカルボニルオキシアミジニル)ドデカン:1.20
1.55ml(18.06mmol)のクロロギ酸チオメチルのクロロホルム5mlを、クロロホルム45mlに1.15の2.46g(8.60mmol)とトリエチルアミン3.26ml(23.22mmol)を含む懸濁液に攪拌下、滴下する。周囲温度で2時間攪拌する。その後、反応混合物をろ過し、ろ過物を3×100mlの水で洗浄する。有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。低温残渣を石油エーテルで洗浄し、その後、分離し、デシケーターで乾燥して、白色粉末の生成物3.45g(92%)を得る。
融点:89−90℃
NMR1H(CDCl3,100MHz),δ(ppm):1.24(s,16H);1.56(m,4H);2.20(t,4H);2.31(s,6H);4.79(s,4H)
FT−IR,n(cm-1):1131(C−O−C);1605(C=N);1719(OCOS);3384及び3496(NH2)
ES+MS:435(M+H)
【0091】
1,12−ビス(N,N’−チオエチルカルボニルオキシアミジニル)ドデカン:1.21
1.53ml(14.68mmol)のクロロギ酸チオエチルのクロロホルム5mlを、クロロホルム45mlに1.15の2g(7mmol)とトリエチルアミン2.65ml(18.9mmol)を含む懸濁液に攪拌下、滴下する。周囲温度で3時間攪拌する。その後、反応混合物をろ過し、ろ過物を3×100mlの水で洗浄する。有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。低温残渣を石油エーテルで洗浄し、その後、分離し、デシケーターで乾燥して、白色粉末の生成物2.70g(83%)を得る。
融点:59−60℃
NMR1H(CDCl3,100MHz),δ(ppm):1.23(s,16H);1.30(t,6H);1.56(m,4H);2.20(t,4H);2.86(quartet,4H);4.80(s,4H)
FT−IR, n(cm-1):1127(C−O−C);1608(C=N);1718(OCOS);3386及び3496(NH2)
ES+MS:462(M+H)
【0092】
1,12−ビス(N,N’−アセトキシアミジニル)ドデカン:1.22
2g(7mmol)の1.15を、一部ずつ26ml(280mmol)の酢酸無水物に攪拌下添加し、氷水浴で冷却する。周囲温度で2時間攪拌する。100mlのクロロホルムを反応混合物に添加する。その後、溶液を2×100mlの飽和塩化ナトリウム水溶液、3×100mlの3N炭酸ナトリウム溶液、100mlの水で続けて洗浄する。有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。低温残渣を晶出した後、結晶を石油エーテルで洗浄し、デシケーターで乾燥して、白色粉末の生成物2.33g(90%)を得る。
融点:128−129℃
NMR1H(CDCl3,100MHz),δ(ppm):1.23(s,16H);1.55(m,4H);2.12(s,6H);2.23(t,4H);8.74(s,4H)
FT−IR, ν(cm-1):1232(C−O−C);1633(C=N);1736(OCO);3319及び3425(NH2)
+TOF MS:371(M+H)
【0093】
1,12−ビス(N,N’−ベンゾイルオキシアミジニル)ドデカン:1.23
1.28ml(14.68mmol)のベンゾイルクロリドを、DMF30mlに1.15の1.5g(5.24mmol)とトリエチルアミン2ml(14.16mmol)を含む懸濁液に攪拌下、滴下する。周囲温度で3時間攪拌し、その後、反応混合物をろ過し、ろ過物を200mlの冷水に沈殿させる。次いで、水と石油エーテルで洗浄し、デシケーターで乾燥して、白色粉末の生成物2.25g(87%)を得る。
融点:158−159℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.73(s,16H);2.01(m,4H);2.56(t,4H);6.94(s,4H);7.96−8.11(m,6H);8.54−8.61(m,4H)
FT−IR, ν(cm-1):684及び699(芳香族性C−H);1266(C−O−C);1625(C=N);1721(OCO);3315及び3452(NH2)
【0094】
1,12−ビス(N,N’−エチルカルバモイルオキシアミジニル)ドデカン:1.24
1.16ml(14.68mmol)のエチルイソシアン酸を、クロロホルム80mlに1.15の2g(7mmol)と炭酸カリウム1.01g(7.34mmol)を含む懸濁液に攪拌下、滴下する。周囲温度で一晩攪拌する。その後、反応混合物をろ過し、ろ過物を2×100mlの水で洗浄する。次いで、有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させて、有色油状の生成物2.22g(74%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.14(t,6H);1.22(s,16H);1.51(m,4H);2.12(t,4H);3.26(quintuplet,4H);5.00(s,4H);6.44(t,2H)
FT−IR, ν(cm-1): 1650(C=N);1701(OCONH);3335(NH);3374及び3490(NH2)
+TOF MS:429(M+H)
【0095】
1,12−ビス(N,N’−フェニルカルバモイルオキシアミジニル)ドデカン:1.25
1.6ml(14.68mmol)のフェニルイソシアン酸を、DMF40mlに1.15の2g(7mmol)と炭酸カリウム1.01g(7.34mmol)を含む懸濁液に攪拌下、滴下し、冷水浴で0℃に冷却する。周囲温度で2時間30分攪拌する。その後、反応混合物をろ過し、ろ過物を150mlの水に沈殿させる。次いで、沈殿物を水、アセトン、エーテルで続けて洗浄する。デシケーターで乾燥した後、白色粉末の生成物2.95g(80%)を得る。
融点:134−135℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.72(s,16H);2.0(m,4H);2.55(t,4H);6.83(s,4H);7.54(t,2H);7.76(t,4H);7.93(t,49.65(s,2H)
FT−IR, ν(cm-1): 1018及び1220(C−O−C);1628(C=N);1708(CON);3249(OCONH);3345及び3455(NH2)
ES+MS:525(M+H);263(M+H/2)
【0096】
[1,12−ビス(アミジニル)ドデカン]−1,12−ビス−N,N’−リン酸ジエチル:1.26
2.08ml(14.33mmol)のクロロホスホン酸ジエチルを、DMF30mlに1.15の2g(7mmol)とトリエチルアミン2.06ml(14.68mmol)を含む懸濁液に攪拌下、滴下し、冷水浴で0℃に冷却する。周囲温度で16時間攪拌する。その後、反応混合物をろ過し、ろ過物を150mlの酢酸エチルに溶解させる。次いで、有機相を3×200mlの水で洗浄し、その後、硫酸ナトリウムで乾燥し、減圧下、蒸発させて、有色油状の生成物2.88g(74%)を得る。
融点:134−135℃
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.31(t,12H);1.51(m,4H);2.15(t,4H);4.15(quintuplet,8H);4.30(s,4H)
FT−IR, ν(cm-1): 837及び1027(OP−O−CH2CH3);969(OP=O);1648(C=N);3323及び3487(NH2)
ES+MS:559(M+H)
【0097】
1,12−ビス[(1,2,4−オキサジアゾール−5(4H)−オン)−3−イル]ドデカン:1.27
キシレン70mlに1.17の4g(9.30mmol)を含む懸濁液を攪拌下、150℃、2時間加熱する(有色の油層が形成されるまで)。その後、反応混合物を減圧下、蒸発させる。次に、得られた固体残渣を50mlのDMSOに溶解し、そして、200mlの冷水に沈殿させる。次いで、形成された沈殿物を分離し、アセトンに再溶解し、ろ過する。ろ過物を硫酸ナトリウムで乾燥し、減圧下、蒸発させ、デシケーターで乾燥して、有色結晶の生成物2.97g(94%)を得る。
融点:150−151℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.71(s,16H);2.03(m,4H);2.93(t,4H)
FT−IR, n(cm-1): 1718(C=N);1783(OCONH);3163(NHCO)
ES+MS:339(M+H)
【0098】
1,12−ビス[(1,2,3,5−オキサチアジアゾール−2(3H)−オキシド)−4−イル]ドデカン:1.28
1.1ml(15.03mmol)の塩化チオニルを、0℃に冷却されたDMF30mlに1.15の2g(7mmol)とピリジン2.82ml(34.96mmol)を含む懸濁液に攪拌下、滴下する。低温状態で、45分間攪拌し、その後、反応溶液を150mlの冷水に沈殿させる。沈殿物をろ過し、デシケーターで乾燥して、有色粉末の生成物1.65g(62%)を得る。
融点:94−95℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.71(s,16H);2.05(m,4H);2.99(t,4H)
FT−IR, ν(cm-1): 1146(NSCO);1615(C−N);1655(C=N);3219及び3323(NHSO)
ES+MS:379(M+H)
【0099】
1,12−ビス[(1,2,4−オキサジアゾール−5(4H)−チオン)−3−イル]ドデカン:1.29
1.32g(32.97mmol)の60%水素化ナトリウムを、0℃に冷却されたDMF50mlに1.22の2g(5.4mmol)と硫化炭素2.5ml(34.96mmol)を含む懸濁液にゆっくり攪拌下、滴下する。低温状態で、45分間攪拌し、その後、周囲温度で2時間攪拌する。次いで、反応溶液を150mlの冷水に沈殿させ、2NのHClでpH5に酸性化し、その後、3×50mlの酢酸エチルで抽出する。有機相を水で洗浄し、その後、硫酸ナトリウムで乾燥し、減圧下、蒸発させて、固体残渣を得る。その後、それをエーテルで洗浄し、デシケーターで乾燥して、有色粉末の生成物1.45g(72%)を得る。
NMR1H(DMSO−d6,100MHz),δ(ppm):1.71(s,16H);2.06(m,4H);3.02(t,4H);9.25(s,2H)
FT−IR, ν(cm-1): 1644(C=N);1737(SCONH);3090(NHCO)
ES+MS:371(M+H);313[M+2H−60(SCO)]
【0100】
1,12−ビス[(1,2,4−チアジアゾール−5(4H)−オン)−3−イル]ドデカン:1.30
THF70mlに1.15の2g(7mmol)と1,1’−チオカルボニルジイミダゾール4g(21mmol)を含む懸濁液を周囲温度で16時間攪拌する。反応混合物を150mlの水で希釈し、3×70mlの酢酸エチルで抽出する。その後、有機相を水で洗浄し、硫酸ナトリウムで乾燥し、減圧下、蒸発させる。油状残渣を50mlのTHFに溶解し、BF3,
Et2Oを5.32ml(41.96mmol)添加する。得られた反応混合物は、次いで、周囲温度で一晩攪拌する。溶液を水で希釈し、酢酸エチルで抽出し、水で洗浄し、乾燥し、その後、減圧下で蒸発させる。得られた低温残渣は、次いで、50mlのエタノールに溶解させ、200mlの冷水に沈殿させる。単離した沈殿物を水で洗浄し、乾燥して、オレンジ色粉末の生成物1.62g(62.55%)を得る。
NMR1H(DMSO−d6,100MHz),δ(ppm):1.70(s,16H);2.04(m,4H);2.93(t,4H);9.30(s,2H)
FT−IR, ν(cm-1): 1666(C=N);1707(SCONH);3192(NHCO)
+TOF MS:313[M+2H−60(SCO)]
【0101】
1,14−ジ(ピロリジン−1−イル)テトラデカン−1,14−ジアミン:2.0, 2HCl
テトラデカンイミド酸ジエチルニ塩酸塩1.16g(3mmol)、ピロリジン0.51ml、及びエタノール15mlからなる混合物を還流下、24時間加熱する。蒸発させた後、得られた残渣をメタノール−エーテル混合物で再結晶して、ベージュ色粉末の生成物0.97g(75%)を得る。
融点:171℃
NMR1H(CD3OD,250MHz),δ(ppm):1.35(s,16H);1.68(m,4H);2.08(m,8H);2.55(t,4H);3.42(t,4H);3.70(t,4H);9.12(s,4H)
【0102】
1,14−ジ(ピペリジン−1−イル)テトラデカン−1,14−ジイミン:3.0
1.68ml(20mmol)の無水ピペリジンを、1,12−ジシアノドデカン2.2g(10mmol)とCuCl1.98g(20mmol)に添加する。最初、緑色の混合物は、青色に変化する。その後、80℃で20時間加熱し、生じた赤色溶液をエーテル125mlに注ぎ、12mlのNaOH(30%水性)を加え、2分間攪拌する。オレンジ色の相を単離し、硫酸ナトリウムで乾燥し、ろ過して蒸発させる。得られた残渣をエーテルから再結晶する(収率50%)。
融点:149−150℃
NMR1H(CDCl3,100MHz),δ(ppm):1.3(m,20H);1.4−1.6(m,12H);2.2(t,4H);3.3(m,8H);6.5(m,2H)
【0103】
N1,N1, N14, N14−テトラメチルテトラデカンジイミドアミド:4.0,
2HCl
1.16g(3mmol)のテトラデカンジイミド酸ジエチルニ塩酸塩とジエチルアミンのエタノール溶液(5.6M)1.1mlをエタノール15mlと混合し、還流下26時間加熱する。その後、溶媒を留去し、得られた残渣をエタノール−エーテル混合物から再結晶して、緑色がかった粉末の生成物0.98g(85%)を得る。
融点:209℃
NMR1H(CDCl3,250MHz),δ(ppm):1.35(m,16H);1.68(m,4H);2.59(t,4H);3.12(s,6H);9.10(s,4H)
【0104】
N1,N1, N14, N14−テトラメチルオクタデカンジイミドアミド:5.0
アンモニアのメタノール溶液(2M)5.3mlと0.46g(1.2mmol)のオクタデカンジイミド酸ジエチルニ塩酸塩の混合物を−10℃で1時間冷却し、その後、周囲温度で15時間放置する。溶媒を蒸発させた後、得られた残渣をメタノール−エーテル混合物で再結晶し、ろ過し、デシケーターで乾燥して、生成物0.25g(54%)を得る。
融点:196−198℃
NMR1H(DMSO−d6,250MHz),δ(ppm):1.24(m,24H);1.60(m,4H);2.37(t,4H);8.82(s,4H);9.10(s,4H)
【0105】
1,12−ビス(イミダゾリン−2−イル)ドデカン:6.0
a)1,12−ビス(イミダゾリン−2−イル)ドデカンニ塩酸塩:6.0, 2HCl
1.64ml(24.55mmol)のエチレンジアミンを、4.5g(11.69mmol)の1,14−ジエトキシテトラデカン−1,14−ジアミン塩酸塩の無水エタノール溶液100mlにゆっくりと低温状態で添加する。次いで、反応混合物を還流下、4時間加熱する。減圧下、蒸発させて最小量の溶媒とした後、得られた低温残渣を、攪拌下、エーテルを添加して晶出させる。その後、ろ過して単離した沈殿物をエーテルで洗浄し、デシケーターで乾燥して、白色粉末の生成物3.9g(88%)を得る。
融点:191−192℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.22(s,16H);1.57(m,4H);2.45(t,4H);3.75(s,8H);9.28(m,4H)
FT−IR, ν(cm-1): 1600(C=N);3031(C=NH,HCl);3200(N−H)
【0106】
b)1,12−ビス(イミダゾリン−2−イル)ドデカン:6.0
2g(5.28mmol)の6.0,2HClの水溶液(20ml)に、pHが14になるまで、トリエチルアミンを滴下する。形成した沈殿物を分離し、水、アセトン、その後エーテルで洗浄し、デシケーターで乾燥して、遊離塩基の白色粉末の生成物1.4g(86%)を得る。その後、この粉末をメタノールから再結晶させて、白色結晶の生成物1.26g(90%)を得る。
融点:176−178℃
NMR1H(CD3OD,100MHz),δ(ppm):1.29(s,16H);1.55(m,4H);2.20(t,4H);3.52(s,8H)
FT−IR, ν(cm-1): 1611(C=N);3177(N−H)
【0107】
1,12−ビス[N,N’−(メチルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.1
0.80ml(10.29mmol)のクロロギ酸メチルのクロロホルム溶液5mlを、1.5g(4.90mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、1.5ml(10.78mmol)のトリエチルアミンを含むクロロホルム25mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。次いで、反応混合物をろ過し、ろ過物を120mlの水、120mlの飽和塩化ナトリウム水溶液、及び2×120mlの水で続けて洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発して油状の残渣を生成する。これを石油エーテルに溶解し、−4℃で放置することにより、白色液状の生成物1.51g(73%)を得る。
融点:134−135℃
NMR1H(CDCl3,100MHz),δ(ppm):1.20(s,16H);1.58(m,4H);2.64(t,4H);3.70(s,8H);3.76(s,6H)
FT−IR, ν(cm-1):1142及び1194(C−O−C);1642(C=N);1723(NCO)
【0108】
1,12−ビス[N,N’−(エチルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.2
0.98ml(10.29mmol)のクロロギ酸エチルのクロロホルム溶液5mlを、1.5g(4.90mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、1.5ml(10.78mmol)のトリエチルアミンを含むクロロホルム25mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。次いで、反応混合物をろ過し、ろ過物を120mlの水、120mlの飽和塩化ナトリウム水溶液、及び2×120mlの水で続けて洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発して、油状の生成物1.78g(81%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.22(s,16H);1.30(t,6H);1.59(m,4H);2.65(t,4H);3.72(s,8H);4.16(q,4H)
FT−IR, ν(cm-1):1073及び1099(C−O−C);1644(C=N);1720(NCO)
【0109】
1,12−ビス[N,N’−(ブチルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.3
1.33ml(10.29mmol)のクロロギ酸ブチルのクロロホルム溶液5mlを、1.5g(4.90mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、1.5ml(10.78mmol)のトリエチルアミンを含むクロロホルム25mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。次いで、反応混合物をろ過し、ろ過物を120mlの水、120mlの飽和塩化ナトリウム水溶液、及び2×120mlの水で続けて洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発して、油状の生成物1.78g(81%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):0.88(t,6H);1.20(s,16H);1.36−1.64(m,12H);2.63(t,4H);3.71(s,8H);4.08(t,4H)
FT−IR, ν(cm-1):1073及び1152(C−O−C);1644(C=N);1722(NCO)
【0110】
1,12−ビス[N,N’−(イソブチルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.4
1.35ml(10.29mmol)のクロロギ酸イソブチルのクロロホルム溶液5mlを、1.5g(4.90mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、1.5ml(10.78mmol)のトリエチルアミンを含むクロロホルム25mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。次いで、反応混合物をろ過し、ろ過物を120mlの水、120mlの飽和塩化ナトリウム水溶液、及び2×120mlの水で続けて洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発して、油状の残渣を生成する。これを石油エーテルに溶解し、−4℃で置き、白色固形の生成物2.26g(91%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):0.87(d,12H);1.17(s,16H);1.55(m,4H);1.89(quint,2H);2.62(t,4H);3.70(s,8H);3.84(d,4H)
FT−IR, ν(cm-1):1072及び1142(C−O−C);1642(C=N);1722(NCO)
【0111】
1,12−ビス[N,N’−(ベンジルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.5
0.94ml(6.54mmol)のクロロギ酸ベンジルのクロロホルム溶液5mlを、1g(3.27mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、0.85ml(6.54mmol)のトリエチルアミンを含むクロロホルム30mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。得られた溶液を減圧下、蒸発させる。次いで、単離した沈殿物をエーテルで数回抽出する。エーテル相を減圧下、蒸発させて、油状の生成物を得、−4℃でエーテルから晶出して白色粉末の生成物1.02g(54%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.23(s,16H);1.60(m,4H);2.68(t,4H);3.77(s,8H);5.16(s,4H);7.35(s,10H)
FT−IR, ν(cm-1):1142及び1299(C−O−C);1646(C=N);1714(NCO)
MS−ES+:[M+H]+:575
【0112】
1,12−ビス[N,N’−(4−ニトロベンジルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.6
1.44g(6.70mmol)のクロロギ酸4−ニトロベンジルのクロロホルム溶液10mlを、1g(3.27mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、0.87ml(6.70mmol)のトリエチルアミンを含むクロロホルム20mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。次いで、反応混合物を30mlのクロロホルムで希釈した後、120mlの水、120mlの飽和塩化ナトリウム水溶液、及び2×120mlの水で続けて洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発させる。残渣を冷却し、最小量のクロロホルムに溶解して、最終的に−4℃でヘキサンから晶出して白色粉末の生成物2.08g(96%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.60(m,4H);2.67(t,4H);3.80(s,8H);5.24(s,4H);7.46及び7.55(dd,4H);8.17及び8.25(dd,4H)
FT−IR, ν(cm-1):1005及び1154(C−O−C);1343及び1517(NO2);1645(C=N);1724(NCO)
MS−ES+:[M+H]+:665
【0113】
1,12−ビス[N,N’−(フェニルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.7
1.26g(10.05mmol)のクロロギ酸フェニルのクロロホルム溶液5mlを、1.5g(4.90mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、1.3ml(10.05mmol)のトリエチルアミンを含むクロロホルム25mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。次いで、反応混合物を30mlのクロロホルムで希釈した後、120mlの水、120mlの飽和塩化ナトリウム水溶液、及び2×120mlの水で続けて洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発させて油状の生成物2.42g(90%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.64(m,4H);2.72(t,4H);3.89(s,8H);7.06−7.43(m,10H)
FT−IR, ν(cm-1):1162及び1188(C−O−C);1646(C=N);1737(NCO)
MS−ES+:[M+H]+:547;[M+H]2+/2:274(100%)
【0114】
1,12−ビス[N,N’−(4−フルオロフェニルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.8
0.89ml(6.70mmol)のクロロギ酸4−フルオロフェニルのクロロホルム溶液5mlを、1g(3.27mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、0.89ml(6.86mmol)のトリエチルアミンを含むクロロホルム20mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。次いで、反応混合物を20mlのクロロホルムで希釈した後、60mlの水、60mlの飽和塩化ナトリウム水溶液、及び2×60mlの水で続けて洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発させる。残渣を冷却し、最終的に−4℃でヘキサンから晶出して、白色粉末の生成物1.70g(85%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.64(m,4H);2.71(t,4H);3.90(s,8H);7.03及び7.36(d,8H)
FT−IR, ν(cm-1):1100(C−F);1179(C−O−C);1655(C=N);1733(NCO)
MS−ES+:[M+H]+:583;[M+H]2+/2:292(100%)
【0115】
1,12−ビス[N,N’−(4−フルオロフェニルオキシカルボニル)イミダゾリン−2−イル]ドデカンニ塩酸塩:6.8, 2HCl
1gの6.8を、塩酸ガスで飽和したエタノール溶液20mlに添加する。次いで、反応混合物を50℃に加熱し、4時間激しく攪拌する。エーテル150mlを低温溶液に添加し、その後、混合物を一晩冷蔵庫に静置する。デカンテーションした後、形成された油層を蒸留水100mlに溶解し、そして、ろ過する。ろ過物を最終的に凍結乾燥して、白色粉末の塩生成物を得る。
NMR1H(DMSO−d6,100MHz),δ(ppm):1.70(s,16H);2.13(m,4H);3.46(t,4H);4.55(t,4H);4.80(t,4H);7.78及び7.85(d,8H);8.71(s,2H)
FT−IR, ν(cm-1):1692(C=N);1760(NCO);3350(N,HCl)
【0116】
1,12−ビス[N,N’−(4−メトキシフェニルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.9
1.02ml(6.86mmol)のクロロギ酸4−メトキシフェニルのクロロホルム溶液5mlを、1g(3.27mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、0.89ml(6.86mmol)のトリエチルアミンを含むクロロホルム20mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。次いで、単離した沈殿物(トリエチルアミン塩酸塩)を数回エーテルで抽出する。エーテル相を減圧下、蒸発させて、油状の生成物1.31g(66%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.63(s,4H);2.71(t,4H);3.75(s,8H);3.88(s,2H);6.61−7.10(m,8H)
FT−IR, ν(cm-1):1177及び1248(C−O−C);1646(C=N);1735(NCO)
MS−ES+:[M+H]+:607;[M+2H]2+/2:304(100%)
【0117】
1,12−ビス[N,N’−(4−ニトロフェニルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.10
1.38g(6.86mmol)のクロロギ酸4−ニトロフェニルのクロロホルム溶液5mlを、1g(3.27mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、0.89ml(6.86mmol)のトリエチルアミンを含むクロロホルム25mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で5時間攪拌する。溶液を減圧下、蒸発させた後、形成された沈殿物を水、次いでエーテルで洗浄する。得られた黄色固体物は、次いで、アセトンをゆっくり添加し、最小量のジクロロホルムに溶解させて晶出する。白色結晶の生成物74%(1.54g)を単離する。
融点:121−122℃
NMR1H(CDCl3,100MHz),δ(ppm):1.23(s,16H);1.67(m,4H);2.73(t,4H);3.95(s,8H);7.25及び7.36(dd,4H);8.24及び8.32(dd,4H)
FT−IR, ν(cm-1):1185及び1196(C−O−C);1349及び1524(NO2);1656(C=N);1748(NCO)
MS−ES+:[M+H]+:637;[M+2H]2+/2:319(100%)
【0118】
1,12−ビス[N,N’−(アセトキシメトキシカルボニル)イミダゾリン−2−イル]ドデカン:6.11
1g(6.54mmol)のクロロギ酸アセトキシメチルのクロロホルム溶液5mlを、1g(3.27mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、0.92ml(6.54mmol)のトリエチルアミンを含むクロロホルム20mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。その後、反応混合物を、20mlの水、20mlの5%炭酸水素ナトリウム水溶液、次いで20mlの飽和塩化ナトリウム水溶液で洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発させて、油状の生成物2.95g(84%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.58(m,4H);2.08(s,6H);2.65(t,4H);3.74(s,8H);5.74(s,4H)
FT−IR, ν(cm-1):1643(C=N);1682(NCO);1736(OCO)
MS−ES+:[M+H]+:539
【0119】
1,12−ビス[N,N’−((1−アセトキシエトキシ)カルボニル)イミダゾリン−2−イル]ドデカン:6.12
a)1,12−ビス[N,N’−((1−クロロエトキシ)カルボニル)イミダゾリン−2−イル]ドデカン
1.8ml(16.75mmol)のクロロギ酸1−クロロエチルのクロロホルム溶液10mlを、2.50g(8.17mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、2.16ml(16.75mmol)のトリエチルアミンを含むクロロホルム40mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。その後、反応混合物を、100mlの水、100mlの飽和塩化ナトリウム水溶液、及び3×100
mlの水で続けて洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発させる。残渣を冷却し、その後、ヘキサンから−4℃で晶出して、白色粉末の生成物3.91g(92%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.19(s,16H);1.58(m,4H);1.77(d,6H);2.64(t,4H);3.74(s,8H);6.49(q,2H)
FT−IR, ν(cm-1):1087(C−O−C);1377(CH3;C−H);1648(C=N);1737(NCO)
【0120】
b)1,12−ビス[N,N’−((1−アセトキシエトキシ)カルボニル)イミダゾリン−2−イル]ドデカン:6.12
1,12−ビス[N,N’−((1−クロロエトキシ)カルボニル)イミダゾリン−2−イル]ドデカンニ塩酸塩3.20g(6.17mmol)と酢酸水銀5.9g(18.50mmol)を含む酢酸溶液を攪拌下、周囲温度で72時間放置する。減圧下、溶媒を蒸発させた後、反応残渣をクロロホルム150mlに希釈し、次いで、ろ過する。その後、ろ過物を3×300mlの飽和塩化ナトリウム水溶液で洗浄する。有機相を硫酸ナトリウムで乾燥させ、減圧下、蒸発させて、有色油状の生成物2.40g(68%)を得る。
NMR1H(DMSO−d6,100MHz),δ(ppm):1.24(s,16H);1.44(d,6H);1.52(m,4H);2.03(s,6H);2.61(t,4H);3.79(s,8H);6.69(q,2H)
FT−IR, ν(cm-1):1072及び1224(C−O−C);1645(C=N);1684(NCO);1733(CO)
【0121】
[1,12−ビス(イミダゾリン−2−イル)ドデカン]−1,12−ビス−N,N’−ホスホン酸ジフェニル:6.13
1.40g(6.70mmol)のクロロホスホン酸ジフェニルのクロロホルム溶液5mlを、1g(3.26mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、0.96ml(6.86mmol)のトリエチルアミンを含むクロロホルム25mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩攪拌する。次いで、反応混合物をろ過し、ろ過物を100mlの水、100mlの飽和塩化ナトリウム水溶液、及び2×100mlの水で続けて洗浄する。硫酸ナトリウムで乾燥した後、有機相を減圧下、蒸発させて、油状の生成物2.37g(94%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.20(s,16H);1.62(m,4H);2.58(t,4H);3.79(s,8H);7.17−7.34(m,20H)
FT−IR, ν(cm-1):925(P=O);1160及び1184(C−O);1646(C=N)
【0122】
[1,12−ビス(イミダゾリン−2−イル)ドデカン]−1,12−ビス−N,N’−ホスホン酸ジエチル:6.14
1.49ml(10.29mmol)のクロロホスホン酸ジエチルのクロロホルム溶液5mlを、1.5g(4.90mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、1.5ml(10.78mmol)のトリエチルアミンを含むクロロホルム25mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩攪拌する。次いで、反応混合物をろ過し、ろ過物を100mlの水、100mlの飽和塩化ナトリウム水溶液、及び100
mlの水で続けて洗浄する。硫酸ナトリウムで乾燥した後、有機相を減圧下、蒸発させて、油状の生成物2.38g(84%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.15(s,16H);1.23(t,12H);1.54(m,4H);2.37(t,4H);3.60(t,8H);4.00(quintuplet,8H)
FT−IR, ν(cm-1):963(P=O);1016及び1268(C−O);1640(C=N)
MS−ES+:[M+H]+:579;[M+H]2+/2:290(100%)
【0123】
1,12−ビス(1−メチルイミダゾリン−2−イル)ドデカンニ塩酸塩:7.0, 2HCl
3.9g(10mmol)のテトラデカンジイミド酸ジエチルニ塩酸塩を過剰のN−メチルエチレンジアミンと共に24時間還流する。溶液を冷却した後、溶液にエチルエーテルを添加する。得られた結晶をろ過して単離し、エーテルで洗浄し、デシケーターで乾燥して、白色結晶の生成物2.9g(71%)を得る。
融点:166−170℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.38(m,16H);1.67(m,4H);2.47(t,4H);2.90(s,6H);3.55(s,8H);9.28(m,2H)
FT−IR, ν(cm-1):1600(C=N);3031(C=NH,HCl);3200(N−H)
【0124】
1,12−ビス(1,4,5,6−テトラヒドロピリミジン−2−イル)ドデカンニ塩酸塩:8.0, 2HCl
0.51ml(6.12mmol)の1,3−ジアミノプロパンを、テトラデカンジイミド酸ジエチルニ塩酸塩1.16g(3mmol)の無水エタノール15mlの溶液に添加する。次いで、還流下、反応混合物を加熱する。4.5mlの水を加えた後、反応混合物を95℃で4時間放置する。蒸発させて乾燥した後、得られた残渣にエーテルを加えて晶出し、その後、48時間撹拌する。次いで、ろ過して単離した沈殿物を激しく攪拌して数回エーテルで洗浄し、デシケーターで乾燥して、白色結晶の生成物0.83g(68%)を得る。
融点:117−119℃
NMR1H(DMSO−d6,250MHz),δ(ppm):1.22(s,16H);1.55(m,4H);1.80(m,4H);2.45(t,4H);3.25(s,8H);9.28(m,2H)
【0125】
1,12−ビス(4−メチルイミダゾリン−2−イル)ドデカンニ塩酸塩:9.0, 2HCl
0.44ml(5.1mmol)の1,2−ジアミノプロパンを、テトラデカンジイミド酸ジエチルニ塩酸塩0.96g(2.5mmol)の無水エタノール12mlの溶液に攪拌下、滴下する。反応混合物を、その後、48時間、90℃の温度で加熱する。溶媒を蒸発させた後、得られた残渣をエタノールに溶解し、その後、ろ過する。ろ過物を蒸発させ、次いで、エーテルを加え、減圧下、脱離反応を行い、残渣溶媒を除去する。そして、白色粉末の生成物0.71g(70%)を得る。
融点:117−119℃
NMR1H(DMSO−d6,250MHz),δ(ppm):1.30(m,22H);1.64(m,4H);2.52(t,4H);3.40(t,2H);3.95(t,2H);3.95(m,2H);9.32(m,2H)
【0126】
1,12−ビス(4,4−ジメチルイミダゾリン−2−イル)ドデカンニ塩酸塩:10.0,2HCl
0.96g(2.5mmol)のテトラデカンジイミド酸ジエチルニ塩酸塩の無水エタノール12mlと0.54ml(5.1mmol)の1,2−ジアミノ−2−メチルプロパンの反応混合物を90℃で48時間加熱する。溶媒を蒸発させ、残渣を高温の状態でエタノールに溶解する。周囲温度に冷却した後、形成された固形物をろ過し、その後、化合物9.0,2HClのように、エーテルと処理して、ベージュ色粉末の生成物0.76g(70%)を得る。
NMR1H(DMSO−d6,250MHz),δ(ppm):1.24(s,12H);1.32(s,16H);1.62(m,4H);2.50(t,4H);3.54(s,4H);9.30(m,2H)
【0127】
1,12−ジ(3a,4,5,6,7,7a−ヘキサヒドロ−1H−ベンゾイミダゾ−2−イル)ドデカンニ塩酸塩:11.0, 2HCl
0.96g(2.5mmol)のテトラデカンジイミド酸ジエチルニ塩酸塩の無水エタノール12mlと1,2−ジアミノシクロヘキサン0.63mlを48時間還流(90℃)する。混合物を蒸発させた後、エタノールを溶液に加え、蒸発したろ過物を化合物9.0,2HClで述べたのと同様に処理して、結晶0.82g(67%)を得る。
NMR1H(DMSO−d6,250MHz),δ(ppm):1.25(s,16H);1.30−1.65(m,16H);1.70(m,4H);2.50(t,4H);3.34(m,2H);4.10(m,2H);
9.32(m,2H)
【0128】
1,12−ビス[(1,2,4−オキサジアゾール)−3−イル]ドデカン:18.0
0.6ml(4.9mmol)のトリフルオロ炭酸ジエチルを、2g(7mmol)のビス−アミドキシム1.15を含むオルトギ酸エチル23.26ml(139.86mmol)の懸濁液に攪拌下、添加する。反応混合物を周囲温度で15分間攪拌し、次いで、1時間還流下、加熱する。得られた溶液に150mlの酢酸エチルを添加し、水(100ml)、飽和炭酸水素ナトリウム溶液(100ml)、飽和塩化ナトリウム溶液(100
ml)で続けて洗浄する。次いで、有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。得られた残渣を冷エーテルで洗浄し、その後、乾燥させて、白色粉末の生成物1.50g(70%)を得る。
融点:92−93℃
NMR1H(CDCl3,100MHz),δ(ppm):1.24(s,16H);1.73(m,4H);2.77(t,4H);8.61(s,2H)
FT−IR, ν(cm-1): 1111(C−O);1558(C=N);3123(=CH)
【0129】
1,12−ビス[(5−メチル−1,2,4−オキサジアゾール)−3−イル]ドデカン:18.1
1.22の1.5g(4.05mmol)を含むキシレン30mlの懸濁液を攪拌下、150℃で2時間加熱する。次いで、反応混合物を減圧下、蒸発させる。得られた固体残渣を、次いで、石油エーテルから再結晶し、デシケーターで乾燥した後、白色粉末の生成物1.10g(81%)を得る。
融点:63−64℃
NMR1H(CDCl3,100MHz),δ(ppm):1.23(s,16H);1.69(m,4H);2.53(s,6H);2.66(t,4H)
FT−IR, ν(cm-1): 1581(C−N);1640(C=N)
【0130】
1,12−ビス[(5−トリフルオロメチル−1,2,4−オキサジアゾール)−3−イル]ドデカン:18.4
1g(3.50mmol)のビス−アミドキシム1.15を一部ずつ、トリフルオロ無水酢酸20ml(139.86mmol)に攪拌下、添加し、氷水浴で冷却する。1.15が完全に溶解するまで、冷却状態で30分間攪拌する。反応混合物にエーテル50ml及び非常にゆっくりと冷水100mlを添加する(発熱反応)。その後、エーテル相を、2×100mlの水、2×100mlの1N炭酸ナトリウム溶液、及び100mlの水で続けて洗浄する。次いで、有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。冷却した後、残渣を晶出して、白色結晶の生成物1.39g(90%)を得る。
融点:<40℃
NMR1H(CDCl3,100MHz),δ(ppm):1.26(s,16H);1.75(m,4H);2.81(t,4H)
FT−IR, ν(cm-1): 766及び1150(CF3);1519(C−N);1608(C=N)
【0131】
1,12−ビス[(5−トリクロロメチル−1,2,4−オキサジアゾール)−3−イル]ドデカン:18.5
3g(10.49mmol)のビス−アミドキシム1.15と、13.71g(83.92mmol)のトリクロロ無水酢酸を含むクロロホルム10mlの懸濁液を攪拌下、溶液となるまで、85℃で加熱する。3等分にして、溶液に4.70ml(41.96mmol)のトリクロロ塩化アセチルを添加し、次いで、混合物を、飽和炭酸ナトリウム溶液(2×100ml)、飽和塩化ナトリウム溶液(2×100
ml)、及び水(100ml)で続けて洗浄する。次いで、有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。得られた残渣を乾燥させて、黄色粉末の生成物5.19g(91%)を得る。
融点:55−56℃
NMR1H(CDCl3,100MHz),δ(ppm):1.26(s,16H);1.76(t,4H);2.78(t,4H)
FT−IR, ν(cm-1): 798及び830(CCl3);1573(C=N)
【0132】
1,12−ビス[(5−フェニル−1,2,4−オキサジアゾール)−3−イル]ドデカン:18.6
1.23の1.5g(3.04mmol)を含むキシレン30mlの懸濁液を攪拌下、150℃で2時間加熱する。次いで、反応溶液をデカンテーションし、その後、減圧下、蒸発させる。得られた残渣を続いて石油エーテルから再結晶して、デシケーターで乾燥した後、有色粉末の生成物1.12g(80%)を得る。
融点:97−98℃
NMR1H(CDCl3,100MHz),δ(ppm):1.25(s,16H);1.78(m,4H);2.77(t,4H);7.48−7.54(m,6H);8.06−8.14(m,4H)
FT−IR, ν(cm-1):1581(C−N);1640(C=N)
【0133】
1,12−ビス[(5−エチルオキシカルボニル−1,2,4−オキサジアゾール)−3−イル]ドデカン:18.7
2.35ml(21mmol)の塩化オキサリルエチルを、2g(7mmol)のビス−アミドキシム1.15とピリジン4ml(49.50 mmol)と4Åモレキュラーシーブ4gを含むクロロホルム50mlの懸濁液に攪拌下、添加する。その後、反応混合物を80℃で16時間加熱し、次いで、ろ過し、減圧下、蒸発させる。残渣に酢酸エチル100mlを加え、その後、溶液を、飽和炭酸ナトリウム溶液(2×50ml)、飽和塩化ナトリウム溶液(2×50ml)、及び水50mlで続けて洗浄する。次いで、有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。得られた残渣をエタノールで洗浄し、その後、乾燥させて、粉末の生成物1.96g(62%)を得る。
融点:70−71℃
NMR1H(CDCl3,100MHz),δ(ppm):1.24(s,16H);1.44(t,6H);1.77(m,4H);2.80(t,4H);4.51(quartet,4H)
FT−IR, ν(cm-1):1029及び1197(C−O−C);1584(C=N);1760(COO)
【0134】
1,12−ビス[(5−カルバモイル−1,2,4−オキサジアゾール)−3−イル]ドデカン:18.9
2g(4.44mmol)の18.7を、10%アンモニウムのエタノール溶液(90ml)に添加する。反応混合物を密封して、周囲温度で24時間攪拌下、放置する。次いで、懸濁液をろ過し、冷エタノールで洗浄し、乾燥する。こうして黄色粉末の生成物1.58g(91%)を得る。
融点:196−197℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.71(s,16H);2.14(m,4H);3.23(t,4H);8.98(s,2H)
FT−IR, ν(cm-1):1573(C−N);1604(C=N);1673(CON);3233及び3432(NH2)
【0135】
1,12−ビス[(5−シアノ−1,2,4−オキサジアゾール)−3−イル]ドデカン:18.10
1.3ml(9.18mmol)のトリフルオロ無水酢酸を、1.5g(3.83mmol)の18.9とピリジン1.55ml(19.13mmol)を含むジオキサン60mlの懸濁液に添加し、0℃に冷却する。反応混合物を周囲温度で16時間攪拌下、放置する。次いで、得られた溶液を酢酸エチル150mlで希釈し、その後、水(100ml)及び飽和塩化ナトリウム溶液(2×10ml)で続けて洗浄する。有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させて、有色油状の生成物0.83g(61%)を得る。
融点:70−71℃
NMR1H(CDCl3,100MHz),δ(ppm):1.24(s,16H);1.73(m,4H)
FT−IR, ν(cm-1):1561(C=N);2260(CN)
【0136】
1,12−ビス(N,N’−ジベンゾイルオキシカルボニルグアニジノ)ドデカン:12.1
1g(5mmol)の1,12−ジアミノドデカンと3.76g(10.5mmol)のN,N’−ジベンゾイルオキシカルボニル−S−メチルイソチオ尿素を含むテトラヒドロフラン70mlの溶液を60℃から70℃の間で24時間加熱する。その後、減圧下、溶媒を蒸発させる。次いで、得られた残渣をジクロロメタンに溶解し、5%炭酸水素ナトリウム溶液、飽和塩化ナトリウム水溶液及び水で続けて洗浄する。次いで、有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させ、シリカカラム(DCM)のクロマトグラフィーで精製する。異なる分画を合わせ、減圧下、蒸発させた黄色の油をエーテルから晶出して、白色粉末の生成物2.4g(58%)を得る。
融点:97−98℃
NMR1H(CDCl3,100MHz),δ(ppm):1.24(s,16H);1.54(s,4H);3.40(q,4H);5.11(s,4H);5.16(s,4H);7.15−7.54(m,20H);8.28(t,2H);11.73(s,2H)
FT−IR, ν(cm-1):1054及び1130(C−O);1651(C=N);1736(NCO);3130(NHCO);3333(NH)
MS−FAB+:[M+H]+:821;[M+2H]++/2:412
【0137】
1,12−ビス(N,N’−ジ−tert−ブチルオキシカルボニルグアニジノ)ドデカン:12.2
1g(5mmol)の1,12−ジアミノドデカン1gと3.19g(11mmol)のN,N’−ジ−tert−ブチルオキシカルボニル−S−メチルイソチオ尿素の混合物を含むエタノール100mlを50℃から60℃の間で48時間加熱する。減圧下、溶液を蒸発させた後、得られた残渣をDCM100mlに溶解し、5%炭酸水素ナトリウム水溶液(2×100ml)、水(2×100ml)、及び飽和塩化ナトリウム溶液100mlで続けて洗浄する。次いで、有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させ、シリカ(DCM/MeOH
98%)で精製し、低温状態で石油エーテルから晶出させて、粉末状の生成物2.50g(73%)を得る。
融点:114−116℃
NMR1H(CDCl3,100MHz),δ(ppm):1.24(s,16H);1.47(s,36H);1.61(m,4H);3.35(q,4H);8.27(t,2H);11.48(s,2H)
FT−IR, ν(cm-1):1028及び1138(C−O);1670(C=N);1740(NCO);3132(NHCO);3314(NH)
MS−ES+:[M+H]+:685
【0138】
1,12−ビス[N,N’−(2−アミノ−3,4,5,6−テトラヒドロピリミジル)]ドデカンニ塩酸塩:16.0, 2HCl
0.72g(3.6mmol)の1,12−ジアミノドデカン、1.86g(7.2mmol)の2−メチルスルファニル−3,4,5,6−テトラヒドロピリミジニウムヨージド、及び0.5ml(3.6mmol)のトリエチルアミンを含む反応混合物のアセトニトリル20mlを還流下22時間加熱する。周囲温度に冷却した後、反応溶媒を蒸発させ、残渣をシリカカラム(CH2Cl2/CH3OH/NH4OH
89:10:1)でクロマトグラフィーを行う。グアニジン塩0.91g、すなわち収率41%を得る。
NMR1H(CD3OD,100MHz),δ(ppm):1.49(m,20H);2.13(m,4H);3.33(m,4H);3.56(m,8H)
FT−IR, ν(cm-1):1028及び1138(C−O);1670(C=N);1740(NCO);3132(NHCO);3314(NH)
MS−FAB+:[M+H]+:365;[M+H+HI]+:493
【0139】
1,12−ビス[N,N’−(5,5−ジメチル−3,4,5,6−テトラヒドロピリミジン−2−イル)アミノ]ドデカンニ塩酸塩:17.0, 2HCl
0.72g(3.6mmol)の1,12−ジアミノドデカン、2.06g(7.2mmol)の5,5−ジメチル−2−メチルスルファニル−3,4,5,6−テトラヒドロピリミジニウムヨージド、及び0.5ml(3.6mmol)のトリエチルアミンを含む反応混合物のアセトニトリル20mlを還流下22時間加熱する。周囲温度に冷却した後、反応溶媒を蒸発させ、残渣をシリカカラム(CH2Cl2/CH3OH/NH4OH
89:10:1)でクロマトグラフィーを行う。塩0.91g、すなわち収率36%を得る。
NMR1H(CD3OD,100MHz),δ(ppm):1.17(s,12H);1.49(m,20H);3.07(s,8H);3.23(m,4H)
【0140】
[1−[N−(5,5−ジメチル−3,4,5,6−テトラヒドロピリミジン−2−イル)アミノ]−12−[N’−(3,4−ジヒドロ−2H−ピロール−5−イル)アミノ]]ドデカンジトリフルオロ酢酸塩:25.0,
2TFA
1mmolの1−[N−(5,5−ジメチル−3,4,5,6−テトラヒドロピリミジニウム−2−イル)アミノ]ドデカン−12−アンモニウムジトリフルオロ酢酸塩、1mmol(1当量)の3,4−ジヒドロ−5−メトキシ−2H−ピロール及び0.5mlのトリエチルアミンを含む反応混合物の無水エタノール10mlを還流下20時間加熱する。周囲温度に冷却した後、反応混合物を蒸発させて乾燥し、シリカカラム(CH2Cl2/CH3OH/NH4OH
85:13:2)でクロマトグラフィーを行い、油状の生成物を収率65%で得る。
NMR1H(CD3OD,360MHz),δ(ppm):1.12(s,12H);1.36(m,16H);1.60(m,2H);1.69(m,2H);2.28(m,2H);2.93(m,2H);3.09(s,4H);3.22(m,2H);3.31(m,2H);3.75(m,2H)
MS−ES+:[M+H]+:378;[M+H+TFA]+:492;[M+2H]++/2:189.5
【0141】
1,12−ビス[N, N’−(3,4−ジヒドロ−2H−ピロール−5−イル)アミノ]ドデカン:21.0
2.5g(25.25mmol)の2−メトキシピロリンを、2.76g(10.1mmol)の1,12−ジアミノドデカン塩酸塩を含む無水エタノール70mlに加える。反応混合物を周囲温度で24時間攪拌下、放置する。次に、溶液を減圧下蒸発させ、冷却した後の残渣を水100mlに溶解し、その後、0.1N炭酸ナトリウム溶液でアルカリ性にする。その後、形成した沈殿物を分離し、水その後エーテルで洗浄し、デシケーターで乾燥して、白色粉末の予定生成物3.27g(97%)を得る。
融点:155−156℃
NMR1H(CDCl3,100MHz),δ(ppm):1.23(s,16H);1.49(m,4H);1.89(quint.,4H);2.38(t,4H);3.21(t,4H);3.43(s,2H);3.62(t,4H)
FT−IR, ν(cm-1):1630(C=N);3049及び3221(NH)
【0142】
1,12−ビス(N, N’−アセトアミジニル)ドデカン:20.0
2g(10mmol)の1,12−ジアミノドデカンと2.47g(20mmol)のエチルアセトアミド塩酸塩の混合物を含む無水ジオキサン25mlを還流下24時間加熱する。周囲温度に冷却した後、1N水酸化カリウム水溶液を加えて、沈殿物を形成させる。この沈殿物を高温状態でメタノールで洗浄し、その後、ろ過する。デシケーターに数時間置いた後、生成物2.06g(7.3
mmol)、すなわち収率73%を得る。
融点:92−97℃
NMR1H(DMSO−d6,250MHz),δ(ppm):1.3(m,16H);1.5(m,4H);1.8(s,6H);2.9(t,4H);5.5(m,2H)
【0143】
1,12−ビス(N, N’−ヒドロキシアセトアミジニル)ドデカン:20.1及びその二塩酸塩 20.1,2HCL
10g(50mmol)の1,12−ジアミノドデカンを、12.9g(125mmol)のエチル N−ヒドロキシアセトイミダートを含むエタノール125mlに加える。反応混合物を80℃まで4日間加熱する。冷却後、結果として得られる沈殿物を分離し、エタノールで、次いでエーテルで数回洗浄し、乾燥器で乾燥させ、エタノール中で再結晶化して、白色粉末の7.26g(46%)のヒドロキシアセトアミジニル20.1(融点150−151℃)を得る。
【0144】
1gのヒドロキシアセトアミジニル20.1を、塩酸(塩化水素酸)ガスで飽和した20mlの無水エタノール中に溶解させる。混合物を、激しく攪拌して50℃まで2時間加熱する。100mlの無水エーテルを低温溶液に加え、この混合物を放置して沈殿させる。結果として得られる固形物を分離し、乾燥させ、100mlの蒸留水に溶解させて、その後ろ過する。水性ろ液は、凍結乾燥して白色粉末の製品を得る。
1H NMR(DMSO−d6 100MHz): 1.72(s. 16H, H3−H10);
1.96 (s, 4H, H2 and H11); 2.62 (s, 6H, 2H2:
3.86 (q, 4H, H1 and H12); 9.30 (t, 2H, 2NH); 11.40 (s,
2H, 2NOH); 12.92 (s, 2H, 2C=NOH,HCL).
FT−IR: 1683 (C=N); 3002; 3142 and 3202 (NH; C=NOH, HCL and NOH
【0145】
1,12−ビス[N,N’−メトキシアセトアミジニル]ドデカン,20.2
1g (3.18mmol) のヒドロキシアセトアミジニル20.1を、0.45g(11.15mmol)のソーダ及び25mlのエタノール/水(4:1)から調整されたヒドロキシル−アルコールソーダ溶液に加える。30分攪拌後、0.42ml
(6.68mmol)よう化メチルを、反応懸濁液に滴下する。混合物を室温で24時間攪拌する。その後、不透明な溶液を過して、ろ液を減圧下で蒸発させる。得られた残渣を50mlのクロロホルム中で再溶解させ、塩化ナトリムの飽和水性溶液(2×100ml)で洗浄する。有機相を硫酸ナトリウム上で乾燥させ、減圧下で蒸発させて、1.05g(96%)の有色オイルのメトキシアセトアミジニルを得る。
1H NMR(CDCl3, 100MHz): 1.24(s, 16H, H3-H10);
1.45 9m, 4H, H2 and H11; 1.83 (s, 6H, 2H2'):
3.05 (q, 4H, H1 and H12); 3.70 (s, 6H, 2H3'):
5.03 (t, 2H, 2NH).
ES+ SM: 343 [M+H+]; 172 (100%) [(M+2H+)/2].
FT−IR: 1637 (C=N); 3264 (NH).
【0146】
1−12−ビス[N,N’−アセトキシアセトアミジニル]ドデカン:20.8
1g(3.18mmol)のヒドロキシアセトアミジニル20.1を、12ml(127.4mmol)の0℃に冷却された無水酢酸の攪拌溶液に少量加える。混合物を、室温で2時間攪拌する。100mlのクロロホルムを反応混合物に加える。この溶液を、2×100mlの水性飽和塩化ナトリウム溶液、3×100mlのソーダ溶液2N及び2×100mlの水で連続的に洗浄する。その後、有機相を硫酸ナトリウム上で乾燥させて、減圧下で蒸発させる。30mlのエーテルを含油低温残渣(oily
cold residue)に加え、その溶液を16時間冷蔵庫で放置する。その後、結果として得られる固形物をエーテル中で粉砕して分離する。得られた粉末をエーテルで最終的に洗浄し、乾燥させて、1.12g(89%)の白色粉末のアセトキシアセトアミジニルを得る。
融点68-69℃。
1NMR(CDCl3, 100MHz): 1.24(s, 16H, H3−H10);
1.44 (m, 4H, H2 and H11); 1.92 (s, 6H, 2H2');
2.11 (s, 6H, 2H4'); 3.10 9q. 4H, H1 and H12);
5.04 9t, 2H, 2NH).
ES+ SM:399 (100%)[M+H+};200[(M+2H+)/2
FT−IR: 1623 (C=N); 1742 (OCO): 3333 (NH)
【0147】
1,12−ビス[N,N’−ベンゾイルオキシアセトアミジニル]ドデカン:20.9
0.78ml (6.68mmol)の塩化ベンゾイルを含む5 mlのクロロホルムを、0℃に冷却して、1g(3.18mmol)のヒドロキシアセトアミジニル20.1及び0.94ml(6.68mmol)のトリエチルアミンを含むクロロホルム30mlの攪拌された懸濁液に滴下する。混合物を3時間室温で撹拌する。その後、反応混合物を2×100mlの水で、次いで100
mlの塩化ナトリウムの飽和溶液で洗浄する。その後、有機相を硫酸ナトリウム上で乾燥させて、減圧下で蒸発させる。含油残渣を冷蔵庫中で一晩放置する。結果として得られる固形物を低温エーテルで粉砕し、エーテルで洗浄し、その後、分離し乾燥させて1.27g(77%)の白色粉末のベンゾイルオキシアセトアミジニルを得る。
融点95−96℃。
1H NMR(CDCl3, 200MHz): 1.23(s. 16H, H3−H10);
1.51 (m, 4H, H2 and H11); 2.02 (sm, 6H, 2H2');
3.15 (q, 4H, H1 and H12); 5.19 (t, 2H, 2NH); 7.34−7.54
[m, 6H, 2(6H,2(H6−H8)]; 7.93−8.03 [dd, 4H, 2(H5
and H9)]
FT−IR: 1279 (C−O−C); 1622 (C=N); 1714 (OCO); 3385 (NH)
ES+ SM: 523 (100%) [M+H+]; 262[M+2H+)/2
【0148】
1,12−ビス(N,N’−エチルカルバモイルオキシアミジニル)ドデカン:20.10
0.53 ml (6.69mmol)のエチルイソシアン酸塩を、1g(3.18mmol)のヒドロキシアセトアミジニル20.1及び0.45g(3.18mmol)の炭酸カリウムを含むクロロホルム40
mlの攪拌された懸濁液に滴下する。混合物を一晩室温で攪拌する。反応混合物をろ過し、ろ過液を3×100mlの水で洗浄する。その後、有機相を硫酸ナトリウム上で乾燥させて、減圧下で蒸発させて、有色オイルのエチルカルバモイルオキシアミジニルを得る。
1H NMR(CDCl3, 100MHz): 1.14(t, 6H, 2H5'); 1.22 (s, 16H, H3−H10);
1.44 (m, 4H, H2 and H11); 1.84 (s, 6H, 2H2');
3.07 (q, 4H, H1 and H12); 3.26 (quintuplet, 4H, 2H4);
5.33 (t, 2H, 2NH); 6.49 (t, 2H, 2NHCO).
FT−IR: 1215 (C−O); 1641 (C=N); 1705 (OCON); 3345 (NHCO); 3398 (NH).
【0149】
1,12−ビス(N,N’−フェニルカルバモイルオキシアミジニル)ドデカン:20.11
0.73ml (6.69mmol)のフェニルイソシアン酸塩を、1g(3.18mmol)のヒドロキシアセトアミジニル20.1及び0.45g(3.18mmol)の炭酸カリウムを含むクロロホルム40mlの攪拌された懸濁液に滴下する。混合物を一晩室温で攪拌する。その後、反応混合物をろ過し、ろ過液を3×100mlの水で洗浄する。その後、有機相を硫酸ナトリウム上で乾燥させて、減圧下で蒸発させる。得られる固形物残渣をエーテルで数回洗浄し、乾燥器中で乾燥させて、1.53g(87%)の白色粉末のフェニルカルバモイルオキシアミジニルを得る。
1H NMR(CDCl3, 100 MHz): 1.26(s, 16H, H3−H10);
1.51 (m, 4H, H2 and H11); 1.94 (s, 6H, 2H2');
3.13 (q, 4H, H1 and H12); 5.46 (t, 2H, 2NH); 7.0−7.52 (m,
10H, H aromatique); 8.61 (s, 2H, 2NHCO).
FT−IR: 1663 (C=N); 1717 (OCON); 3291 (NHCO); 3443 (NH).
ES+ SM: 457 (100%) [M+H+]; 299 [(M+2H+)/2.
【0150】
1,12−ビス(N,N’−メチルスルホニルオキシアセトアミジニル)ドデカン:20.12
0.52ml (6.68mmol)の塩化メチルスルフォニル(methylsulfonyl chloride)を含む5mlのクロロホルムを、氷槽で0〜5℃間に冷却した1g(3.18mmol)のヒドロキシアセトアミジニル20.1及び0.55ml(6.68mmol)のピリジンを含むクロロホルム30mlの攪拌された懸濁液に滴下する。混合物を、10〜15℃の間で4時間攪拌する。その後、反応混合物を3×100
mlの水で洗浄する。その後、有機相を硫酸ナトリウム上で乾燥させて、減圧下で蒸発させる。含油残渣を冷蔵庫中で一晩放置する。結果として得られる固形物を低温エーテルで粉砕し、エーテルで洗浄し、その後、分離し、乾燥させて、1.33g(89%)の白色粉末のメチルスルホニルオキシアセトアミジニルを得る。
1H NMR(CDCl3, 100 MHz): 1.25(s. 16H, H3−H10);
1.51 (m, 4H, H2 and H11); 1.91 (s, 6H, 2H2');
3.11 (q, 6H, 2H3'); 3.15 (q, 4H, H1−H12); 5.23
(t, 2H, 2NH).
FT−IR: 1637 (C=N); 3302 (NH).
ES+ SM: 471 (100%) [M+H+].
【0151】
(1,12−ビス(アセトアミジニル)ドデカン)−1,12−ビス−N,N’−リン酸ジエチル:20.13
0.97ml (6.68mmol)のクロロホスホン酸ジエチルを含む5mlのクロロホルムを、0~5℃までに冷却した、1g(3.18mmol)のヒドロキシアセトアミジニル20.1及び0.94ml(6.68mmol)のトリエチルアミン含むDMF
30mlの攪拌された懸濁液に滴下する。混合物を一晩攪拌する。反応混合物をろ過し、ろ過液を酢酸エチル100ml中で再溶解させる。有機相を、3×100mlの水で洗浄し、その後、硫酸ナトリウム上で乾燥させて、減圧下で蒸発させる。残渣を、100mlのクロロホルムで再溶解させ、3×100mlの水で再洗浄し、硫酸ナトリウム上で乾燥させて、減圧下で蒸発させる。残渣を、100mlのクロロホルムで再溶解させ、3×100mlの水で再洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で蒸発させて、有色オイルの20.13を1.13g(61%)得る。
1H NMR(CDCl3, 100 MHz): 1.25(s. 16H, H3−H10);
1.33 (t, 12H, 4H4); 1.43 (m, 4H, H2 and H11);
1.87 (s, 6H, 2H2'); 3.08 (q, 4H, H1 and H12);
4.18 (quint, 8H, 4H3') 5.28 (t, 2H, 2NH).
FT−IR: 1634 (C=N); 3316 (NH).
【0152】
1,12−ビス(N,N’(3−メチル−1,2,4−oxadiazol−5(4H)−one)−3−yl)
ドデカン:20.14
0.50 ml (6.68mmol)のクロロギ酸メチルを含む 5mlのクロロホルムを、1g(3.18mmol)のヒドロキシアセトアミジニル20.1及び0.45g(3.18mmol)の炭酸カリウムを含むクロロホルム30mlの攪拌された懸濁液に滴下する。混合物を、室温で1時間攪拌し、その後、懸濁液をおよそ50℃まで45分間加熱する。反応混合物を冷却し、その後、ろ過し、3×100mlの水で洗浄する。その後、有機相を硫酸ナトリウム上で乾燥させて、減圧下で蒸発させる。残渣を、冷蔵庫の中で一晩放置する。結果として得られる固形物を最終的に低温のエーテル中で破砕し、エーテルで洗浄し、その後、分離し、乾燥して、白色粉末のオキサジアゾロン
0.92g (79%)を得る。
溶融点:53-54℃。
1H NMR(CDCl3, 100 MHz): 1.24(s. 16H, H3−H10);
1.64 (s, 4H, H2 and H11); 2.24 (s, 6H, 2H2');
3.52 (t, 4H, H1 and H12).
FT−IR: 1559 (C=N); 1752 (OCO).
【0153】
1,12−ビス(N, N’−2−ヒドロキシアセトアミジニル)ドデカン:26.0
a)ヒドロキシアセトニトリル又はグリコール酸ニトリル
22.73g (303mmol)のホルムアルデヒドのナトリウム(40%水溶液)を、0℃に冷却した、15g(306mmol)のシアン化ナトリウム水溶液(60ml)に滴下する。混合物を、この温度で30分間攪拌する。溶液のpHは、H2SO4
7.5N(45ml)で2に、かつNa2CO3で5に、連続的に調整する。硫酸ナトリウムは結局生成されるが、加わえられる水で再溶解され、その後、エーテル(4×200ml)で抽出する。有機相を無水硫酸ナトリウム上で乾燥させ、減圧下で蒸発させて、有色オイルのヒドロキシアセトニトリル
13.13g(76%)を得る。この精製されていないオイルをそのまま使用する。このオイルは、24時間後、室温で分解(変質)する。
1H NMR(CDCl3, 100 MHz): 2.30(s, 1H, OH); 4.34 (s, 2H, CH2).
FT−IR: 2259 (C=N); 3425 (OH).
【0154】
b)ヒドロキシアセトイミダート塩酸塩
HCLガスを、1時間半冷却し、20mlの無水エタノール、40mlの無水エーテル及び9g(158mmol)のヒドロキシアセトニトリルの溶液を通して発泡させる。この反応沈殿物を分離し、エーテルで数回洗浄し、その後、乾燥器中で乾燥させ、白色粉末のヒドロキシアセトイミダート塩酸塩19.52g
(89%)を得る。
1H NMR(DMSO−d6, 100 MHz): 1.80(t, 3H, CH3);
4.80 (s, 2H, CH2); 4.85 (q, 2H, CH2); 11.73 (sl, 2H, NH,
HCl).
FT−IR: 1645 (C=N); 3119 (OH); 3229 (NH, HCl).
【0155】
1,12−ビス(N, N’2−ヒドロキシアセトアミジニル)ドデカン:26.0
2g (10mmol)の1,12−ジアミノドデカンを、3.07g(22mmol)のエチルヒドロキシアセトイミダート塩酸塩を含むメタノール(50ml)の溶液に加える。反応混合物を室温で24時間攪拌する。その後、この溶液を減圧下で蒸発させる。その後、低温残渣を100mlの水に再溶解させ、その後、NaOH
0.5N でアルカリ性(塩基性)化冷却(bastified cold)させる。結果として得られる沈殿物を分離し、水で、アセトンで数回、それからエーテルで洗浄し、その後、分離し、乾燥後、白色粉末の
2-ヒドロキシアセトアミジニル26,0 2.55gを得る。
溶融点:99−100℃。
1H NMR(CD3OD, 250MHz): 1.00 (s, 16H, H3−H10);
1.27 (m, 4H, H2 and H11); 2.84 (t, 4H, H1 and
H12): 2.98 (s, 2H, 2 OH); 3.70 (s, 4H, 2H2')
FT−IR: 1605 (C=N); 3072 (OH); 3290 and 3388 (2NH)
ES+ SM: 315 [M+H+]
【0156】
1,12−ビス[N, N’−(2−イミノピロリジニル)]ドデカン:24.0
1.64g(5mmol)の1,12−ジブロモドデカンを、窒素下、90℃に加熱した2.21g(26mmol)のピロリジン−2−オンと0.23g(10mmol)のナトリウムの混合物に少量ずつ加える。その後、この反応混合物を攪拌下、120℃で4時間加熱する。周囲温度に冷却した後、水40mlを加え、40mlのDCMで抽出する。次いで、有機相を塩化ナトリウムの飽和水溶液で洗浄し、硫酸マグネシウムで乾燥し、その後、減圧下、蒸発させる。シリカカラム(溶離液DCM−エタノール,[18−1])で精製した後、エーテル−ヘキサン混合物で周囲温度にて共蒸発させて、1,12−ビス[N,
N’−(ピロリジン−2−オン−1−イル)]ドデカン0.655g、収率39%の生成物を得る。0.18ml(2mmol)のスルホニルクロリドイソシアン酸塩を含むクロロホルム3mlを、この生成物0.336g(1mmol)を含むクロロホルム3mlに窒素下、滴下する。反応混合物を77℃で6時間加熱する。4.5mlの水を加えた後、反応混合物の温度を95℃で4時間置く。その後、5mlの水を加え、溶液を2×5mlのDCMで洗浄する。次いで、水性相を1Nの水酸化カリウム水溶液で中性化し、固形物をろ過し、エーテルで洗浄し、その後、デシケーターで1時間30分乾燥して、生成物0.135g(40%)を得る。
融点:92−97℃
NMR1H(DMSO−d6,250MHz),δ(ppm):1.3(m,16H);1.5(m,4H);1.8(q,4H);2.3(t,4H);3.2(t,4H);3.3(t,4H);8.0(s,2H)
【0157】
本発明に係る化合物の薬理学的活性の研究
A.P.falciparumに対するin vivoの抗マラリア活性及びP.vinckeに感染させたマウスに対するin vitroの抗マラリア活性
表4及び5に、以下、本発明の化合物について得られたIC50値(μM)及びED50値(mg/kg)の結果を示す。
72
【0158】
【表4】
【0159】
【表5】
【0160】
IC50はin vitroでP.falciparumの成長を50%阻害する濃度であり(IC50の測定は、核酸に組み込んだ[3H]ヒポキサンチンがcell
viability indexとしての役割を果たすDesjardinsの方法に従って行った。)(Desjardins RE,Canfield CJ,Haynes
JD,Chulay JD.Quantitative assessment of antimalarial activity in vitro by a
semiautomated microdilution technique. Antimicrob. Agents Chemother. 1979; 16:
710−718)(図1)、ED50は4日間の抑制試験によりin vitroでP.vinckeの成長を50%阻害する有効濃度である (Peters
W, Portus JH, Robinson BL. The chemotherapy of rodent malaria. XXII. The value
of drug-resistant strains of P.berghei in screening for blood schizonticidal
activity. Ann. Trop. Med. Parasitol. 1975; 69: 155-17)。
TIは治療係数、TI=LD50(半慢性) / ED50;ip:腹腔内投与;po:経口投与を表す。
これらの結果は、本発明の化合物が良好な耐性及び良好な吸収性だけでなく、invitro及びin vivoで強力な抗マラリア活性及び抗バベシア病活性を有することを示している。
【0161】
B.マウスにおける薬力学的パラメーター
化合物6.0をマウスに腹腔内又は経口経路で投与した後の薬力学的パラメーターの結果を以下に示す。血清レベルで決定するため、バイオテストをex vivoで用いた、簡潔に言えば、医薬を動物に投与し、その後、繰り返し採取する。血清は56℃、30分間にて脱補体化する。次いで、活性な代謝物の含量は、[3H]ヒポキサンチンを用いるDesjardinsの方法に従い、P.falciparumに感染させた赤血球の懸濁液の存在下、異なる濃度(二分希釈)の各血清をインキュベートして決定する。結果はIS50で表し、これはP.falciparumの成長を50%阻害することができる血清濃度(活性化した代謝物質を含む)に相当する。
この値は、次にP.falciparumに感染した同じ懸濁液で活性化合物を直接試験する(動物を通すことなく)ことにより、また、そのIC50値(ng/ml)で決定することにより、血清濃度(通例ng/mlで表す)に変換する[血清総計=IC50](ng/ml)×100/IS50(%)]。
結果は時間の関数としてlog(医薬の血清総計)で表し、これにより血清コンパートメントへの分布に対する半減時間t1/2(d);血清コンパートメントの消失に対する半減時間t1/2(e);消失相での最初の血清濃度の推定値であるC0,AUC(血流中で循環する薬物量を示す);経口経路による吸収の程度を表す、経口経路による投与方法対腹腔内経路による投与方法[AUC(po)/AUC(ip)]における比較バイオアベイラビリティの評価が可能となる。
【0162】
6.0の薬力学
LD50/3に相当する17mg/kg及び300 mg/kgの用量の6.0を、腹腔内経路及び経口経路で投与した。化合物は10%DMSOに溶解させる。結果は表4に示す。半対数表記により、2つの投与経路に対する活性代謝物の主要な薬力学パラメーターを決定することができる。薬力学パラメーターは、C0=50ng/ml,t1/2=16時間,AUC=170ng.h/ml(300mg/kg経口投与後)である。
【0163】
21.0の薬力学
15mg/kg(LD50/3腹腔内)及び100 mg/kg(LD50/4経口)の用量の化合物21.0を、それぞれマウスに腹腔内経路及び経口経路で投与した。15mg/kgの腹腔内投与後、C0は24ng/ml、t1/2は約35時間となる。100
mg/kgの経口経路では、C0は16ng/ml、見かけのt1/2は36時間となる。
【0164】
C.化合物の抗バベシア病活性
化合物6.0,22.0,及び2.0については、また、Babesia divergens及びB.canisに対するそれらの活性をin vitroで評価した。いずれに対しても、化合物6.0,22.0,及び2.0は、顕著に活性がある(IC50<50nM)ことがわかった。これらの結果は、このタイプの化合物の強力な抗バベシア病活性を示している。
【図面の簡単な説明】
【0165】
【図1】図1は、本発明に係る化合物の薬理学的活性の研究を示す説明図である。
【技術分野】
【0001】
本発明の主題は、抗寄生虫活性を有する化合物、より詳細には、抗マラリア活性及び抗バベシア病活性を有する化合物である。
【背景技術】
【0002】
いろいろな疾病を引き起こす寄生虫、特にPlasmodium属、なかでも、熱帯熱マラリア原虫(Plasmodium falciparum)に対して活性をもつ医薬の研究が行われている。
【発明の開示】
【発明が解決しようとする課題】
【0003】
本発明者は、あるカテゴリーの化学化合物が、認容することができる毒性と高いバイオアベイラビリティ特性を示し、高い活性をもつことを観察した。それらの化合物は、好ましくは、経口経路により投与される。
【0004】
したがって、本発明の目的は、抗寄生虫活性、特に抗マラリア活性を有する新規な化合物を提供することである。
【0005】
また、本発明は、該化合物の製造方法に関する。
【0006】
さらに、本発明は、活性成分として、それらの化合物を含有する医薬、及び該化合物の使用と上記活性をもつ医薬を製造するための誘導体の製造方法に関する。
【課題を解決するための手段】
【0007】
本発明に係る化合物は、一般式(I)で表される点に特徴があり、
但し、Xは式(II)の基であり、
Zは−(CH2)mの基であり、m=8−21,n=0又は1,及びY=R3であり、
R1及びR'1は、互いに同一又は異なり、H,アルキル,OH,O−アルキル,O−アリール,O−CO−アルキル,O−CO−アリール,OSO2−アルキル,OSO2−アリール,OSO2−ヘテロ環,O−CO−O(又はS又はNH)−アルキル,O−CO−O(又はS又はNH)−アリール,PO(O−アルキル又はO−アリール)2,CO−O−CH2−アリール,シクロアルキル,から選択され、
R2及びR'2は、互いに同一又は異なり、H,アルキル,CO−O−CH2−アリール,CO−O−アルキル,シクロアルキル,から選択され、
R3及びR'3は、互いに同一又は異なり、H,アルキル,CO−O−アルキル,CO−O−アリール,COO− CH(R)−O−CO−アルキル,PO(O−アルキル又はO−アリール又はONa)2,CO−O−CH(R)
−アリール,から選択され、RはH又はアルキルである、
あるいは、
R1及びR2,及び/又はR'1及びR'2,又はR2及びR3,及び/又はR'2及びR'3,は共に、それぞれが結合する1又は複数の窒素原子をもつ非芳香族モノ(non
aromatic mono)ヘテロ環を形成し、
あるいは、さらに、
R2及びR3及び/又はR'2及びR'3は、同一の置換基であり、窒素に二重結合しており、それぞれ、R1又はR'1と環化してヘテロ環を形成し、Raで置換される場合は、それは、H,アルキル,1,2又は3つのハロゲン元素で置換したアルキル,アリール,CO−O−アルキル(又はアリール),−CO−OH,−CO−NH2,−CN,−CO−NH−アルキル(又はアリール),−CO−N−(アルキル)2,窒素及び/又は酸素を有する−CO−ヘテロ環,NH(H又はアルキル),N(アルキル)2,窒素及び/又は酸素を有するヘテロ環,−O−アルキル(又はアリール),−O−CH2−アリール,CH2N[H,(H,アルキル),(ジアルキル),アリール],窒素及び/又は酸素を有する−CH2−ヘテロ環,CH2−CO−OH,から選択され、
あるいは、X=R4、Yは式(III)の基であり、
n及びZは上記したのと同様であり、
R1及びR'1は互いに同一又は異なり、H,アルキル,OH,O−アルキル,O−アリール,O−CO−アルキル,O−CO−アリール,OSO2−アルキル,OSO2−アリール,OSO2−ヘテロ環,O−CO−O(又はS又はNH)−アルキル,O−CO−O(又はS又はNH)−アリール,PO(O−アルキル又はO−アリール)2,CO−O−CH2−アリール,シクロアルキル,から選択され、
R4及びR'4は、H,アルキル,又はアリールであり、これはOH,O−アルキル,O−アリール,NH(H又はアルキル),窒素又は酸素へテロ環で代替することが出来、
R2及びR'2は、互いに同一又は異なり、H,アルキル,CO−O−CH2−アリール,CO−O−アルキル,シクロアルキル,から選択され、
あるいは、水素の1又は複数の原子は低級アルキルに置き換えることができ、R1及びR4及び/又はR'1及びR'4は共に−(CH2)pの基を形成し、pは3−5の整数、
R2及びR'2はHを表し、又はR4及びR2及び/又はR'4及びR'2は、共に−(CH2)pの基を形成し、1又は複数の水素原子は低級アルキルに置き換えることができ、R1及びR'1はHを表す、及びこれらの化合物の薬理学的に許容できる塩である。
【0008】
特に定義されていない場合は、“アリール”は、フェニル又は芳香族性を有するあらゆる環又はヘテロ環を意味し、例えば、ピリジン,オキサゾール,チアゾール環,置換している場合は、特に塩素,−NO2,−NH2,N(H,アルキル)又は(ジアルキル)による置換体を意味する。
【0009】
“窒素及び/又は酸素を有するヘテロ環”は、ピロリジン,ピペリジン,モルホリン,ピペラジン,メチルピペラジン環のような5又は6の頂点をもつ環を意味する。
【0010】
“アルキル”は、直鎖又は分枝したC1−C5のアルキル、置換している場合は、1又はそれ以上のハロゲン原子、−NH2,N(H,アルキル)又は(ジアルキル)アミノ基による置換体を意味する。
【0011】
本発明の好適な誘導体のファミリー、すなわち、ファミリーAは、式(IV)で表され、
n,Z,R1,R'1,R2,R'2,R3及びR'3は、式(II)で前記規定したのと同様である。
【0012】
このファミリーの好適な化合物は、n=0の場合であり、式(V)で表される。
【0013】
好ましいグループ、すなわち、グループa1においては、R1,R2及びR3及び/又はR'1,R'2及びR'3は、互いに独立している。
【0014】
サブグループにおいては、R1及び/又はR'1,及びR2及び/又はR'2は、ハロゲン原子を表し、R3及び/又はR'3は、前記したのと同様であるが、水素原子とは異なる。
【0015】
他のサブグループにおいては、R1及び/又はR'1は、前記したのと同様であるが、水素原子とは異なり、一方、R3及び/又はR'3,R2及び/又はR'2は、水素原子を表す。
【0016】
他の好ましい式(V)の化合物のグループは、n=0であり、
R1及びR2,及び/又はR'1及びR'2は、式(VI)、すなわち、グループa2に該当する、
あるいは、
R2及びR3及び/又はR'2及びR'3は、式(VII)、すなわち、グループa3に該当する。
【0017】
式(VI)に該当するサブグループにおいては、R1及びR2及び/又はR'1及びR2は、共に−O−CO−,O−SO−,O−CS,S−CO又は−S−CS基を形成し、R3及び/又はR'3は、水素原子を表す。
【0018】
式(VI)に該当するもう1つのサブグループにおいては、R1及びR2,及び/又はR'1及びR'2は、選択的に、分枝したアルキレン基を表し、R3及び/又はR'3は、−CO−O−アルキル(又はアリール),−CO−O−CH2−アリール,CO−O−CH(アルキル)−O−CO−アルキル,PO(O−アルキル又はアリール)2,アルキル又はHを表す。
【0019】
式(VII)に該当するサブグループにおいては、R1及び/又はR'1は、水素原子を表し、R2及びR3,及び/又はR'2及び/又はR'3は、−(CH2)p−の基を表す。
【0020】
他の好ましいファミリーAのグループ、すなわち、グループa4は、R2及びR3及び/又はR'2及びR'3は、同一の置換基を形成し、それぞれR1又はR'1と共に、式(VIII)のビス−オキサジアゾールを形成する。
但し、Raは、前記規定したのと同様である。
【0021】
このグループの好ましい化合物においては、ハロゲンはF又はClが好ましく、アルキルはメチル又はエチル、アリールはフェニルである。
【0022】
本発明の他の好ましいファミリー、すなわち、ファミリーBは、式(IX)で表される。
【0023】
このファミリーの好ましい化合物においては、Z=−(CH2)m及びn=0であり、式(X)に該当するものである。
【0024】
ファミリーBの好ましいグループ、すなわち、グループb1においては、置換基は互いに独立している。
【0025】
サブグループにおいては、R1及びR4及び/又はR'1及び/又はR'4は、前記したとおりであり、R2は水素原子を表す。
【0026】
他のサブグループにおいては、R1及びR2及び/又はR'1及び/又はR'2は、共にオキシカルボニル鎖−OCO−を表し、R4及びR'4は、上記の様に定義される。
【0027】
他のサブグループにおいては、R1及びR4及び/又はR'1及び/又はR'4は、共に−(CH2)p−の基を表し、nは3−5の整数であり、R2及びR'2は、Hを表す。
【0028】
さらなる他のサブグループにおいては、R1及びR'1は、Hを表し、R4及びR2及び/又はR'4及びR'2は、共に−(CH2)p−の基を表し、pは3−5の整数であり、一またはそれ以上の水素原子が低級アルキルに置き換えられることができる。
【0029】
他のサブグループは、式(XI)に該当する。
【0030】
ファミリーAとBの好ましい化合物は、以下の表1−3に示している。
【0031】
【表1】
【0032】
【表2】
【0033】
【表3】
【0034】
本発明に係る化合物は、適当な場合には、塩の形をとる。例として、塩酸塩、クエン酸塩、酒石酸塩、マレイン酸塩、酪酸塩、酢酸塩及びトリフルオロ酢酸塩が挙げられる。
【0035】
本発明においては、前記した一般式(V)及び(VI)のカルバメート及びN−ホスホリル誘導体は、一般式(V)及び(VI)のビスアミジン化合物の2相溶媒中での反応を含むことを特徴とする方法によって製造することができ、該(V)及び(VI)では、R3及びR'3=HとCl−R3(又はR'3)誘導体であり、R3及びR'3は前記したのと同様であり、実施例に示したようにHとは異なる。
【0036】
前記規定した一般式(V)及び(X)のアミドキシム誘導体は、R1及びR'1=OHである一般式(V)及び(X)のビスアミドキシムと実施例に示したような適当な試薬との塩基性溶媒中での反応を含むことを特徴とする方法によって製造することができる。
【0037】
好ましくは、前記した一般式(VI)のグループa2及び(VIII)のグループa4は、実施例で示した適当な試薬の存在下、前記した一般式(V)のグループa1で表されるアミドキシム又はアミドキシム誘導体の分子内環化で調製される。
【0038】
寄生虫、特にPlasmodium属に関する本発明の生成物の活性についての研究により、それらの生成物が高いin vitro活性を有することが示された。
【0039】
その結果、熱帯熱マラリア原虫に関して、IC50値(該寄生虫の50%阻止濃度)は、nMからμMの単位である。
【0040】
したがって、本発明は、医薬組成物を製造するために、該化合物の特性の利用に関するものである。
【0041】
本発明の医薬組成物は、前記した少なくとも1つの化合物の有効量を、不活性な薬剤学的担体と結合して含有することを特徴とする。
【0042】
また、本発明は、感染症、特にマラリアの治療薬を製造するための、前記化合物の少なくとも1つの利用に関するものである。
【0043】
これらの組成物は、他の医薬の活性成分を含むことができる。特に薬理学的共同作用又は耐性回避のために、他の抗マラリア薬(例えば、向リソゾーム薬、アトバコン、抗葉酸薬又は抗フォリニン酸薬、アルテミシニン又はその誘導体の1つ)との組み合わせが挙げられる。
【0044】
また、それらの組成物は、同化を促進する化合物と組み合わせて用いることが好ましい。
【0045】
本発明の医薬組成物は、種々の形態で、特に経口経路又は注射経路あるいは又、直腸経路で投与することができる。
【0046】
経口経路には、特に錠剤、丸剤、ゼラチンカプセル、ドロップが利用できる。
【0047】
他の投与形態には、静脈内経路、皮下経路、又は筋肉内経路で注射される溶液を含む。これらは、無菌又は滅菌可能な溶液から調製される。また、該溶液には、懸濁剤あるいは乳剤も含む。
【0048】
坐剤も他の投与形態として利用することができる。
【0049】
本発明の組成物は、特にヒト及び動物の感染症に適しており、特にマラリア、バベシア病の治療に適している。
【0050】
例を挙げると、ヒトに使用し得る用量は、次の用量である。例えば、1回又はそれ以上の用量につき、患者に対し1−90mg/kgを投与する。
【0051】
また、本発明は、上記した化合物を活性成分として含有する生物学的試薬に関する。
【0052】
この試薬は、あらゆる抗寄生虫活性の研究において、対照あるいは標準として使用することができる。
【0053】
本発明の他の特徴及び利点は、上記化合物の合成及び抗寄生虫活性の研究に関する以下に述べる実施例により明らかになるであろう。これらの実施例において、図1を参照として設けた。これは、薬物濃度の関数として、Desjardins’test(Desjardins
R.E. et al., Antimicrob. Agents Chemother.1979,16,710−718)により、化合物6.0の抗マラリア活性を表す。
【実施例】
【0054】
実施例1:
中間体の合成
1,12−ジシアノドデカン:
3.3g(67.10mmol)のシアン化ナトリウムを含むジメチルスルホキシド30mlの懸濁液を、完全に溶解するまで、激しく攪拌しながら90−95℃の間で加熱する。1,12−ジブロモドデカン10g(30.49mmol)を、ごく少量ずつ、ゆっくりと得られた該溶液に添加し、50℃以下の温度に冷却する。周囲温度にて2時間攪拌した後、得られた該懸濁液にジクロロメタン90mlを添加して溶解し、塩化ナトリウム飽和水溶液で数回洗浄する。その後、有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。低温油状の残渣をエーテルに溶解し、次いで、蒸発乾固して晶出し、白色結晶の生成物6.44g(96%)を得る。
融点:<40℃
NMR1H(CDCl3,100MHz),δ(ppm):1.24(s,16H);1.60(m,4H);2.31(t,4H)
FT−IR,ν(cm-1):2246(CN)
【0055】
テトラデカンジイミド酸ジエチルニ塩酸塩:
無水エタノール20ml、無水エーテル60ml及び1,12−ジシアノドデカン5g(22.73mmol)の0℃に冷却した溶液に塩酸ガスを泡状にして2時間通じ、次いで、反応混合物を一晩攪拌させる。その後、減圧下で溶媒を蒸発させる。得られた固形物の残渣を晶出し、エーテルで数回洗浄した後、デシケーターで乾燥させる。白色粉末7.7g(82%)を得る。
融点:118−120℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.26(s,16H);1.35(t,6H);1.60(m,4H);2.63(t,4H);4.55(q,4H);11.16及び12.08(2s,4H)
FT−IR,ν(cm-1):1097(C−O);1651.50(C=N);3032及び3118(NH2,Cl)
【0056】
N,N’−ジ−ベンジルオキシカルボニル−S−メチルイソチオ尿素:
2ml(14.4mol)のクロロギ酸ベンジルを、1g(3.60mmol)のS−メチルイソチオ尿素硫酸塩を含むジクロロメタン/炭酸水素ナトリウム飽和水溶液(1:1)の2相混合物40mlの溶液に激しく攪拌しながら滴下する。周囲温度にて25時間攪拌する。その後、ジクロロメタンで水相を3回抽出する。次いで、結合した有機相を水で洗浄し、その後、硫酸ナトリウムで乾燥し、減圧下、蒸発させる。得られた残渣を、シリカカラム(DCM)によるクロマトグラフィーで精製し、黄色油状の生成物1.06g(82%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):2.46(s,3H);5.24(s,4H);7.42(s,10H);11.91(s,1H)
FT−IR,ν(cm-1):1022及び1173(C−O);1647(C=N);1751(NCO);3169(NHCO)
【0057】
N,N’−ジ−tert−ブチルオキシカルボニル−S−メチルイソチオ尿素:
5.81g(26.60 mmol)の炭酸ジ−tert−ブチル及び2.53g(18.20mmol)のS−メチルイソチオ尿素ヘミ硫酸塩を含むDCM/NaHCO3の飽和2相水溶液(100ml)を48時間激しく攪拌する。その後、分離した水相を2×100mlのDCMで抽出する。次に、結合した有機相を2×200mlの水で洗浄し、その後、減圧下、蒸発させる。次いで、残渣を0.55g(3.85
mmol)のS−メチルイソチオ尿素ヘミ硫酸塩を加えた100mlの飽和水性DCM/NaHCO3の2相混合物に溶解させる。反応混合物を再び72時間激しく攪拌する。結合した有機相は、前記と同様の処理を行った後、硫酸ナトリウムで乾燥させ、減圧下、蒸発させる。最後に、残渣をシリカ(ヘキサン/CHCl35%次いでCHCl3)で精製して、白色粉末の生成物3.46g(90%)を得る。
融点:123−124℃
NMR1H(CDCl3,100MHz),δ(ppm):1.49(s,18H);2.38(s,3H);11.60(s,1H)
FT−IR,ν(cm-1):1043及び1252(C−O);1667(C=N);1765(NCO);3337(NHCO)
【0058】
チオ炭酸O−クロロメチル−S−エチル
37ml(500mmol)のエタンチオールと69.3 ml(500mmol)のトリエチルアミンを含むエーテル溶液200mlを、44ml(500mmol)のクロロギ酸クロロメチル含むエーテル溶液900mlに攪拌下、2時間滴下し、0−5℃に冷却した。その後、処理状態を30分間保つ。その後、反応混合物を周囲温度で16時間攪拌し、次いで、形成した沈殿物をろ過し、エーテルで洗浄する。結合したエーテル相を蒸発させ、残渣を蒸留して精製して、液状の生成物57g(73%)を得る。
沸点:99−100℃/.18mbar
NMR1H(CDCl3,100MHz),δ(ppm):);1.30(t,3H);2.89(q,2H);5.96(s,2H)
FT−IR,ν(cm-1):1719(S−C−O)
【0059】
チオ炭酸S−エチル−O−ヨードエチル:
55g(356mmol)のチオ炭酸O−クロロメチル−S−エチルを、106.8g(712mmol)のヨウ化ナトリウム及び3g(35.6mmol)の重炭酸ナトリウムを含むアセトン450mlの攪拌溶液に直接加える。その後、反応混合物を、攪拌したまま、40℃で4時間放置する。形成される沈殿物をろ過し、アセトンとエーテルで洗浄する。有機相を蒸発させ、残渣を、1100mlの0℃に冷却されたヘキサンと50mlの冷水に分ける。その後、分離された有機相を、5%の水性重炭酸ナトリウム200ml、1%のチオ硫酸ナトリウム200ml(溶液の脱色まで)及び水2×200mlで連続的に洗浄する。ヘキサン相を硫酸ナトリウム上で乾燥させ、減圧下で蒸発させた後、81g(92%)の未精製の黄色がかった液体の製品を得る。
NMR 1H(CDCl3, 100MHz),δ(ppm): ); 1.30 (t, 3H); 2.89 (q,
2H,); 5.96 (s, 2H).
FT−IR,ν(cm-1): 1715 (S-C-O)
【0060】
チオ炭酸O−アセチルオキシメチル−S−エチル:
80g(325.2mmol)のチオ炭酸S−エチル−O−ヨードメチルを、26.7g(325.2mmol)の酢酸ナトリウムを含む無水ホルムアミドジメチル420mlの溶液に攪拌下、2時間以上滴下し、−20℃に冷却した。その後、反応混合物を周囲温度で16時間攪拌し、次いで、形成した沈殿物をろ過し、20mlのホルムアミドジエチルと40mlのエーテルで洗浄する。
【0061】
850mlのエーテルと350mlの冷水を、分離漏斗で有機相に添加する。水相を単離し、350mlの水で抽出する。次いで、結合したエーテル相を、220mlの5%水性炭酸水素ナトリウム、220mlの水、2×220mlの0.01N塩酸及び220mlの水で続けて洗浄する。有機相を硫酸ナトリウムで乾燥させた後、残渣を蒸留して精製し、黄色液状の生成物35g(60%)を得る。
沸点:82−83℃/.0.25mbar
NMR1H(CDCl3,100MHz),δ(ppm):);1.30(t,3H);2.09(s,3H);2.87(q,2H);5.76(s,2H)
FT−IR,ν(cm-1):1717(S−CO−O);1767(CO−O)
【0062】
クロロギ酸アセチルオキシメチル
14.90ml(185.4mmol)の塩化スルフリルを、33g(185.4mmol)のチオ炭酸O−アセチルオキシメチル−S−エチルの溶液に攪拌下添加し、0−5℃に冷却した。処理状態を15分間保った後、反応混合物を周囲温度で45分間攪拌する。次いで、形成されたS−エチルクロリド溶液を周囲温度、20mbarで一晩蒸発させる。次いで、得られた残渣を抽出して精製し、オレンジ色液状の生成物15.50g(56%)を得る。
沸点:75−76℃/.17mbar
NMR1H(CDCl3,100MHz),δ(ppm):2.12(s,3H);5.76(s,2H)
FT−IR,ν(cm-1):1724(CO−O);1773(Cl−CO)
【0063】
3,4−ジヒドロ−5−メトキシ−2H−ピロール:
23ml(1当量)の硫酸ジメチルを、23.03g(0.235mol)のピロリジン−2−オンのベンゼン溶液85mlに滴下し、70℃に加熱する。この反応混合物を3時間還流下、加熱する。周囲温度に冷却した後、15Nソーダ溶液19mlを反応混合物に添加する。反応混合物を分離漏斗に注ぎ、ベンゼンで3回水相を抽出する。結合した有機相を硫酸ナトリウムで乾燥させ、減圧下蒸発させる。残渣を減圧して抽出し、無色液体の生成物を収率58%で得る。
融点:10-2mbar下、37℃
NMR1H(CDCl3,100MHz),δ(ppm):1.97(m,2H);2.41(t,2H);3.61(t,2H);3.75(s,3H)
【0064】
2−メチルスルファニル−3,4,5,6−テトラヒドロピリミジニウムヨージド:
メタノール(チオ尿素10ml/g)に1当量の3,4,5,6−テトラヒドロ(1H)ピリミジン−2−チオンと1.2当量のヨウ化メタンを含む反応混合物を還流下5時間加熱する。冷却した後、メタノールを減圧下蒸発させて、定量的に生成物を得る。沈殿物は、その後、アセトン又は石油エーテルで洗浄することができる。
NMR1H(CDCl3,100MHz):0.95(s,6H);2.62(s,3H);3.11(s,4H);8.76(s,2H)ppm
【0065】
5,5−ジメチル−3,4,5,6−テトラヒドロ(1H)ピリミジン−2−チオン:
10ml(167.2mol;2当量)の二硫化炭素を、250mlフラスコ中の10ml(83.6mol)の2,2−ジメチル−1,3−ジアミノプロパンを含む無水エタノール50mlの溶液に添加する。次いで、16.02g(83.6mol)のEDCを該反応混合物に添加し、周囲温度で3時間攪拌する。エタノールを蒸発させ、残渣を水に溶解し、ジクロロメタンで抽出する。硫酸ナトリウムで乾燥し、有機相を蒸発させた後、生成物11.2g(93%)を得る。これは付加的に精製することなく使用される。
融点:225−228℃
NMR1H(CDCl3,100MHz):0.91(s,6H);2.88(d,4H);7.46(s,2H)ppm
【0066】
5,5−ジメチル−2−メチルスルファニル−3,4,5,6−テトラヒドロピリミジニウム塩酸塩:
メタノール(チオ尿素10ml/g)に1当量の5,5−ジメチル−3,4,5,6−テトラヒドロ(1H)ピリミジン−2−チオンと1.2当量のヨウ化メタンを含む反応混合物を還流下5時間加熱する。冷却した後、メタノールを減圧下蒸発させて、定量的に生成物を得る。沈殿物は、その後、アセトン又は石油エーテルで洗浄することができる。
融点:225−228℃
NMR1H(CDCl3,100MHz):0.95(s,6H);2.62(s,3H);3.11(s,4H),8.76(s,2H)ppm
【0067】
1−(N−tert−ブチルオキシカルボニルアミノ)ドデカン−12−アンモニウム塩酸塩:
4.02g(18.4mol)のジ炭酸ジ−tert−ブチルのジオキサン溶液60mlを、15.01g(75mmol,4.1当量)の1,12−ドデカンジアミンを含むジオキサン/水(150/70ml)混合物溶液にゆっくり滴下(約2時間以上)する。反応混合物を周囲温度で24時間攪拌し、過剰のジアミンはろ過し、溶媒は減圧下蒸発させる。残渣は1NのHCl溶液とジクロロメタンに溶解させる。水相に形成した沈殿物はろ過して、モノ−Boc誘導体と少量のジアミンを共に塩酸塩の形で生成する。
この固形物をエタノールとエーテルで再結晶して、モノ−保護ジアミン塩の生成物4.56g(収率74%)を白色粉末の形で得る。
融点:153−155℃
NMR1H(CD3OD,100MHz),δ(ppm):1.28(m,20H);1.38(s,9H);2.91(m,4H)
MS−FAB+:[M+H]+:301
【0068】
[1−[N−(5,5−ジメチル−3,4,5,6−テトラヒドロピリミジン−2−イル)アミノ]−12−(N’−tert−ブチルオキシカルボニルアミノ)]ドデカン塩酸塩:
0.85g(3mmol)の5,5−ジメチル−2−メチルスルファニル−3,4,5,6−テトラヒドロピリミジニウムヨージドと0.85ml(2当量)のトリエチルアミンを、1.02g(3mmol)の12−(N−tert−ブチルオキシカルボニルアミノ)ドデカン−1−アンモニウムクロリドを含むアセトニトリル15mlの懸濁液に添加する。この反応混合物を24時間還流下、加熱する。周囲温度に冷却した後、反応しなかったアミンをろ過する。ろ過物は減圧下蒸発させ、シリカカラム(溶出溶媒:CH2Cl2/CH3OH/NH4OH
80:16:4)でクロマトグラフィーを行い、生成物1.36g(85%)を得る。
NMR1H(CD3OD,100MHz),δ(ppm):1.07(s,6H);1.37(m,29H);3.05(m,8H)
【0069】
[1−[N−(5,5−ジメチル−3,4,5,6−テトラヒドロピリミジン−2−イル)アミノ]ドデカン−12−アンモニウムジトリフルオロアセテート:
1.06g(2mmol)の[1−[N−(5,5−ジメチル−3,4,5,6−テトラヒドロピリミジン−2−イル)アミノ]−12−(N’−tert−ブチルオキシカルボニルアミノ)]ドデカンヨウ化水素塩を15mlのTFA/CH2Cl2(3/1)溶液に溶解する。この溶液を周囲温度で3時間攪拌し、過剰のトリフルオロ酢酸を減圧下蒸発させ、定量的に生成物0.98gを得る。
NMR1H(DMSO−d6,100MHz),δ(ppm):1.0(s,6H);1.3(m,20H);3.0(m,8H);8.0(bs,2H);9.0(s,3H)
MS−ES+:[M+H]+:311;[M+2H]++/2:156
【0070】
合成
1,12−ビス(アミジル)ドデカンニ塩酸塩:1.0, 2HCl
45mlの無水エタノールと5g(13mmol)のジエチルテトラデカンジイミド酸塩酸塩の溶液に、アンモニアガスを泡にして2時間通じ、氷浴で10℃以下の温度に冷却する。その後、溶媒を減圧下蒸発させる。次いで、得られた残渣は、エタノールで数回洗浄し、デシケーターで乾燥させる。白色粉末状の生成物3.53g(83%)を得る。
融点:170−172℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.25(s,16H);1.58(m,4H);2.37(t,4H);8.82(s,4H);9.10(s,4H)
FT−IR,ν(cm-1):1688(C=N);3081(NH2,Cl);3245(NH2)
【0071】
1,12−ビス[N,N’−(メチルオキシカルボニル)アミジニル]ドデカン:1.1
60ml(7.65mmol)のクロロギ酸メチルを、1g(3.06mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液60mlに激しく攪拌しながら、4N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、100mlの水を加える。次いで、形成された沈殿物を分離し、水その後エーテルで数回洗浄し、デシケーターで乾燥させ、白色粉末状の生成物1.30g(77%)を得る。
融点:116−117℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.69(s,16H);1.96(m,4H);2.61(t,4H);3.97(s,6H);9.06(s,4H)
FT−IR,ν(cm-1):1256(C−O−C);1633(C=N);1670(NHCO);3343(NH)
MS−ES+:[M+H]+:371
25
【0072】
1,12−ビス[N,N’−(エチルオキシカルボニル)アミジニル]ドデカン:1.2
0.73ml(7.65mmol)のクロロギ酸エチルを、0.1g(3.06mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液60mlに激しく攪拌しながら、4N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、100mlの水を加える。次いで、形成された沈殿物を分離し、水その後エーテルで数回洗浄し、デシケーターで乾燥させ、白色粉末状の生成物0.98g(80%)を得る。
融点:95−96℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.63(t,6H);1.71(m,16H);1.93(m,4H);2.63(t,4H);4.49(q,4H);8.95及び9.12(2s,4H)
FT−IR,ν(cm-1):1251(C−O−C);1667(C=N);1757(CO);3186(NHCO);3330(NH)
MS−ES+:[M+H]+:399;[M+H]2+/2:200(100%)
【0073】
1,12−ビス[N,N’−(ブチルオキシカルボニル)アミジニル]ドデカン:1.3
0.99ml(7.65mmol)のクロロギ酸ブチルを、1g(3.06mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液60mlに激しく攪拌しながら、4N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、100mlの水を加える。次いで、形成された沈殿物を分離し、水その後エーテルで数回洗浄し、デシケーターで乾燥させ、白色粉末状の生成物1.12g(80%)を得る。
融点:87−88℃
NMR1H(CDCl3,100MHz),δ(ppm):0.88(t,6H);1.21(s,16H);1.48−1.70(m,8H);1.88(m,4H);2.24(t,4H);4.03(t,4H);6.21(s,2H);9.24(s,2H)
FT−IR,ν(cm-1):1084及び1234(C−O−C);1594(C=N);1660(CO);3313(NHCO);3441(NH)
MS−ES+:[M+H]+:455
【0074】
1,12−ビス[N,N’−(イソブチルオキシカルボニル)アミジニル]ドデカン:1.4
1ml(7.65mmol)のクロロギ酸イソブチルを、1g(3.06mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液60mlに激しく攪拌しながら、4N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、100mlの水を加える。次いで、形成された沈殿物を分離し、水その後エーテルで数回洗浄し、デシケーターで乾燥させ、白色粉末状の生成物1.27g(91%)を得る。
融点:102−103℃
NMR1H(CDCl3,100MHz),δ(ppm):0.92(d,12H);1.22(s,16H)1.62(m,4H);1.96(m,2H);2.26(t,4H);3.82(d,4H);6.19(s,2H);9.27(s,2H)
FT−IR,ν(cm-1):1069及び1237(C−O−C);1599(C=N);1661(CO);3314(NHCO);3440(NH)
MS−ES+:[M+H]+:455
【0075】
1,12−ビス[N,N’−(ベンジルオキシカルボニル)アミジニル]ドデカン:1.5
1.1ml(7.65mmol)のクロロギ酸ベンジルを、1g(3.06mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液60mlに激しく攪拌しながら、4N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、100mlの水を加える。次いで、形成された沈殿物を分離し、水その後エーテルで数回洗浄し、デシケーターで乾燥させ、白色粉末状の生成物1.2g(75%)を得る。
融点:118−119℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.19(s,16);1.47(m,4H);2.13(t,H);4.96(s,4H);7.30(s,10H);8.29(s,2H);8.50(s,2H)
FT−IR,ν(cm-1):693及び745(芳香族性C−H);1236(C−O−C);1648(C=N);1666(CO);3311(NHCO);3436(NH)
MS−ES+:[M+H]+:523;[M+2H]2+/2:262(100%)
【0076】
1,12−ビス[N,N’−(4−ニトロベンジルオキシカルボニル)アミジニル]ドデカン:1.6
1.65g(7.65mmol)のクロロギ酸4−ニトロベンジルのジオキサン溶液5mlを、1g(3.06mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液60mlに激しく攪拌しながら滴下し、氷浴で冷却する。該溶液のpHは、4N炭酸ナトリウム水溶液で10−12に保つ。次いで、混合物を周囲温度で一晩攪拌する。その後、100mlの水を加える。次いで、形成された沈殿物を分離し、水その後エーテルで数回洗浄し、デシケーターで乾燥させ、白色粉末状の生成物1.37g(74%)を得る。
融点:87−88℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.22(s,16H);1.51(m,4H);2.18(t,4H);5.15(s,4H);7.59(dd,4H);8.23(dd,4H);8.69(s,4H)
FT−IR,ν(cm-1):1248(C−O−C);1344及び1512(NO2);1621(C=N);1658(CO);3316(NHCO);3406(NH)
MS−ES+:[M+H]+:623
【0077】
1,12−ビス[N,N’−(フェニルオキシカルボニル)アミジニル]ドデカン:1.7
1.44ml(11.47mmol)のクロロギ酸フェニルを、1.5g(4.59mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液80mlに激しく攪拌しながら、2N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、150mlの水を加え、該溶液を3×60mlのDCMで抽出する。次いで、有機相を水で数回洗浄し、減圧下蒸発させる。液状の生成物1.77g(78%)を得る。
融点:87−88℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.73(s,16H);2.05(m,4H);2.72(t,4H);7.17−7.90(m,10H);9.20(s,4H)
FT−IR,ν(cm-1): 1623(C=N);1682(COO);3378(NH及びNHCO)
MS−ES+:[M+H]+:495
【0078】
1,12−ビス[N,N’−(4−フルオロフェニルオキシカルボニル)アミジニル]ドデカン:1.8
1.50ml(11.47mmol)のクロロギ酸4−フルオロフェニルを、1.5g(4.59mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液80mlに激しく攪拌しながら、2N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、150mlの水を加える。次いで、形成された沈殿物を分離し、水その後エーテルで数回洗浄し、デシケーターで乾燥させ、白色粉末の生成物1.55g(64%)を得る。
融点:℃
NMR1H(CDCl3,100MHz),δ(ppm):1.26(s,16H);1.70(m,4H);2.35(t,4H);6.62−7.14(m,8H);4.20(s,2H);9.23(s,2H)
FT−IR,ν(cm-1): 1177(C−O−C);1253(C−F);1622(C=N);1679(COO);3327(NH及びNHCO)
MS−ES+:[M+H]+:531
【0079】
1,12−ビス[N,N’−(4−フルオロフェニルオキシカルボニル)アミジニル]ドデカンニ塩酸塩:1.8, 2HCl
1gの1.8を、塩酸ガスを飽和したエタノール溶液20mlに添加する。次いで、反応混合物を50℃で2時間激しく攪拌しながら加熱する。エーテル150mlを該低温溶液に添加した後、該混合物を冷蔵庫に一晩放置する。デカンテーションした後、形成した油層を100mlの蒸留水に溶解し、その後、ろ過する。ろ過物は最後、凍結乾燥して、白色粉末の塩を得る。
NMR1H(DMSO−d6,100MHz),δ(ppm):1.75(s,16H);2.15(m,4H);3.06(t,4H);7.20(s,2H);7.70及び7.78(d,8H);7.85及び8.21(2s,4H)
FT−IR,ν(cm-1):1684(C=N);1753(NCO);3324(NH,HCl)
【0080】
1,12−ビス[N,N’−(4−メトキシフェニルオキシカルボニル)アミジニル]ドデカン:1.9
1.70ml(11.47mmol)のクロロギ酸4−メトキシフェニルを、1.5g(4.59mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液80mlに激しく攪拌しながら、2N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、150mlの水を加え、該溶液を3×60mlのDCMで抽出する。その後、有機相を水で数回洗浄し、次いで、減圧下蒸発させ、液状の生成物1.97g(78%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.62(m,4H);2.28(t,4H);3.73(s,6H);6.71−7.06(m,8H);6.18(s,2H);9.21(s,2H)
FT−IR,ν(cm-1): 1176及び1189(C−O−C);1504(芳香族性C−H);1623(C=N);1678(COO);3374(NH及びNHCO)
MS−ES+:[M+H]+:555
【0081】
[1,12−ビス(アミジニル)ドデカン]−1,12−ビス−N,N’−ホスホン酸ジフェニル:1.12
2.85ml(13.76mmol)のクロロホスホン酸ジフェニルを、1.5g(4.59mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液80mlに激しく攪拌しながら、4N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で3時間攪拌する。その後、50mlの水を加える。次いで、形成した沈殿物を分離し、水その後エーテルで数回洗浄し、デシケーターで乾燥して、白色粉末の生成物2.70g(82%)を得る。
融点:100−101℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.65(s,16H);1.93(m,4H);2.79(t,4H);7.68−7.81(m,20H);8.31(s,2H);8.71(s,2H)
NMR31P(DMSO−d6,81MHz),δ(ppm):−1.64
FT−IR,ν(cm-1):935(P=O);1230(C−O);1675 (C=N);3169(NHPO);3324(NH)
MS−FAB+:[M+H]+:719;[M+2H]++/2:360(100%)
【0082】
[1,12−ビス(アミジニル)ドデカン]−1,12−ビス−N,N’−ホスホン酸ジエチル:1.13
1.11ml(7.65mmol)のクロロホスホン酸ジエチルを、1g(3.06mmol)の1,12−ビス(アミジニル)ドデカンニ塩酸塩のジオキサン/水(3:1)2相混合物の溶液40mlに激しく攪拌しながら、4N炭酸ナトリウム水溶液でpH10−12に保持しつつ滴下し、氷浴で冷却する。次いで、混合物を周囲温度で一晩攪拌する。その後、溶液をジクロロメタン(3×30ml)で抽出する。次いで、有機相を水で数回洗浄し、硫酸ナトリウムで乾燥する。減圧下溶液を蒸発させて油状残渣を生成する。最小量のエーテルに溶解させた該残渣を−4℃、一晩で晶出して、白色粉末の生成物1.08g(67%)を得る。
融点:68−69℃
NMR1H(CDCl3,100MHz),δ(ppm):1.20(s,16H);1.26(t,12H);1.53(m,4H);2.21(t,4H);3.99(quintuplet,8H);6.05(s,2H);7.78(s,2H)
NMR31P(DMSO−d6,81MHz),δ(ppm):8.76
FT−IR,ν(cm-1):793及び1031(P−O− CH2CH3);958(P=O);1647
(C=N);3187(NHPO);1581及び3386(NH)
MS−ES+:[M+H]+:527
【0083】
[1,12−ビス(アミジニル)ドデカン]−1,12−ビス−N,N’−ホスホン酸ナトリウム:1.14
a)[1,12−ビス(アミジニル)ドデカン]−1,12−ビス−N,N’−ホスホン酸:
1.36ml(9.51mmol)のトリメチルシランヨージドを、窒素雰囲気下で、0℃に冷却した10mlの無水ジクロロメタンに、1g(1.9 mmol)の1.13の攪拌下、滴下する。このように操作状態を1時間保つ。その後、溶液を減圧下蒸発させる。次いで、3mlの水を含むアセトン10mlに低温残渣を溶解し、24時間攪拌する。その後、形成した沈殿物を分離し、アセトンで数回洗浄し、エタノールで再結晶して、白色粉末の生成物0.52g(66%)を得る。
融点:164−165℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.24(s,16H);1.56(m,4H);2.32(t,4H);9.26及び9.63(2s,8H)
NMR31P(CDCl3,81MHz),δ(ppm):−5.82
FT−IR,ν(cm-1):1044及び1211(P−OH);939(P=O);1667 (C=N);2324(POH);3019(NHPO);1557及び3322(N−H)
MS−ES+:[M+H]+:415
【0084】
b )[1,12−ビス(アミジニル)ドデカン]−1,12−ビス−N,N’−ホスホン酸ナトリウム:1.14
0.1N炭酸ナトリウム溶液(約21ml)を、水20mlに[1,12−ビス(アミジニル)ドデカン]−1,12−ビス−N,N’−ホスホン酸1gを含む懸濁液に攪拌下、pHが7.4になるまで滴下する。その後、溶液を凍結乾燥して、白色粉末を得る。
NMR31P(81MHz),δ(ppm):−3.16
【0085】
1,12−ビス(N,N’−ヒドロキシアミジニル)ドデカン:1.15
13.70g(196.91mmol)のヒドロキシルアミン塩酸塩を、水−アルコール性炭酸ナトリウム溶液[炭酸ナトリウム8.22g、水36ml、及び95%エチルアルコール138mlから調製]に添加する。15分間攪拌した後、20g(90.91mmol)の1,12−ジシアノドデカンを添加する。その後、反応混合物を、還流下、72時間加熱し、次いで、減圧下、蒸発させる。次に、得られた残渣を、水に溶解させ、攪拌し、ろ過する。沈殿物を分離し、水と石油エーテルで数回洗浄する。エタノールで再結晶し、デシケーターで一晩乾燥させた後、白色粉末の生成物25g(96%)を得る。
融点:170−171℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.70(s,16H);1.90(m,4H);2.39(t,4H);5.75(s,4H);9.13(s,2H)
FT−IR,ν(cm-1):1661(C=N);3244(N−OH);3315及び3400(NH2)
+TOF MS:287(M+H);254(M−32);144(M+H/2)
【0086】
1,12−ビス(N,N’−メトキシアミジニル)ドデカン:1.16
1.70ml(17.48mmol)の硫酸ジメチルを、氷浴で冷却したジオキサン/NaOH 0.7N2相混合物50mlに1.15の2g(7mmol)を含む懸濁駅に攪拌下、滴下する。周囲温度で一晩攪拌する。その後、反応混合物をDCMで抽出し、ろ過し、ろ過物を3×100mlの水で洗浄する。有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。石油エーテルでの添加で低温残渣は晶出し、デシケーターで乾燥させた後、白色粉末の生成物1.05g(48%)を得る。
融点:85−86℃
NMR1H(CDCl3,100MHz),δ(ppm):1.23(s,16H);1.50(m,4H);2.10(t,4H);3.74(s,6H);4.44(s,4H)
FT−IR,ν(cm-1):1048(N−O−C);1643(C=N);3288及び3433(NH2)
+TOF MS:315(M+H)
【0087】
1,12−ビス(N,N’−エトキシアミジニル)ドデカン:1.17
1.40ml(14.68mmol)のクロロギ酸エチルのクロロホルム5mlを、クロロホルム45mlに1.15の2g(7mmol)とトリエチルアミン2.65ml(18.9mmol)を含む懸濁駅に攪拌下、滴下する。周囲温度で3時間攪拌する。その後、反応混合物をろ過し、ろ過物を3×100mlの水で洗浄する。有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。低温残渣を晶出した後、結晶を石油エーテルで洗浄し、デシケーターで乾燥して、白色粉末の生成物2.53g(84.33%)を得る。
融点:100−101℃
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.29(t,6H);1.53(m,4H);2.19(t,4H);4.23(quartet,4H).4.79(s,4H)
FT−IR,ν(cm-1):1240(C−O−C);1620(C=N);1741(OCOO);3374及び3508(NH2)
+TOF MS:431(M+H)
【0088】
1,12−ビス(N,N’−エトキシカルボニルオキシアミジニル)ドデカン:1.18
1.14ml(14.68mmol)のクロロギ酸メチルのクロロホルム5mlを、クロロホルム45mlに1.15の2g(7mmol)とトリエチルアミン2.65ml(18.9mmol)を含む懸濁駅に攪拌下、滴下する。周囲温度で3時間攪拌する。その後、反応混合物をろ過し、ろ過物を3×100mlの水で洗浄する。有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。低温残渣を石油エーテルで洗浄し、その後、分離し、デシケーターで乾燥して、白色粉末の生成物2.05g(73%)を得る。
融点:79−80℃
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.53(m,4H);2.19(t,4H);3.81(s,6H).4.81(s,4H)
FT−IR,ν(cm-1):1247(C−O−C);1622(C=N);1750(OCOO);3380及び3497(NH2)
+TOF MS:403(M+H)
【0089】
1,12−ビス(N,N’−フェノキシカルボニルオキシアミジニル)ドデカン:1.19
1.85ml(14.68mmol)のクロロギ酸フェニルを、DMF30mlに1.15の2g(7mmol)とトリエチルアミン2.65ml(18.9mmol)を含む懸濁液に攪拌下、滴下し、冷水浴で冷却する。周囲温度で4時間攪拌し、その後、反応混合物をろ過し、ろ過物を150mlの酢酸エチルで希釈する。次いで、溶液を水(100ml)と飽和塩化ナトリウム溶液(2×100ml)で洗浄する。有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。低温残渣を石油エーテルで洗浄し、その後、分離し、デシケーターで乾燥して、白色粉末の生成物3.02g(82%)を得る。
融点:108−109℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.72(s,16H);1.98(m,4H);2.52(t,4H);6.98(s,4H);7.65−7.98(m,10H)
FT−IR,ν(cm-1):1197及び1241(C−O−C);1633(C=N);1770(OCOO);3309及び3473(NH2)
+TOF MS:(M+H)
【0090】
1,12−ビス(N,N’−チオメチルカルボニルオキシアミジニル)ドデカン:1.20
1.55ml(18.06mmol)のクロロギ酸チオメチルのクロロホルム5mlを、クロロホルム45mlに1.15の2.46g(8.60mmol)とトリエチルアミン3.26ml(23.22mmol)を含む懸濁液に攪拌下、滴下する。周囲温度で2時間攪拌する。その後、反応混合物をろ過し、ろ過物を3×100mlの水で洗浄する。有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。低温残渣を石油エーテルで洗浄し、その後、分離し、デシケーターで乾燥して、白色粉末の生成物3.45g(92%)を得る。
融点:89−90℃
NMR1H(CDCl3,100MHz),δ(ppm):1.24(s,16H);1.56(m,4H);2.20(t,4H);2.31(s,6H);4.79(s,4H)
FT−IR,n(cm-1):1131(C−O−C);1605(C=N);1719(OCOS);3384及び3496(NH2)
ES+MS:435(M+H)
【0091】
1,12−ビス(N,N’−チオエチルカルボニルオキシアミジニル)ドデカン:1.21
1.53ml(14.68mmol)のクロロギ酸チオエチルのクロロホルム5mlを、クロロホルム45mlに1.15の2g(7mmol)とトリエチルアミン2.65ml(18.9mmol)を含む懸濁液に攪拌下、滴下する。周囲温度で3時間攪拌する。その後、反応混合物をろ過し、ろ過物を3×100mlの水で洗浄する。有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。低温残渣を石油エーテルで洗浄し、その後、分離し、デシケーターで乾燥して、白色粉末の生成物2.70g(83%)を得る。
融点:59−60℃
NMR1H(CDCl3,100MHz),δ(ppm):1.23(s,16H);1.30(t,6H);1.56(m,4H);2.20(t,4H);2.86(quartet,4H);4.80(s,4H)
FT−IR, n(cm-1):1127(C−O−C);1608(C=N);1718(OCOS);3386及び3496(NH2)
ES+MS:462(M+H)
【0092】
1,12−ビス(N,N’−アセトキシアミジニル)ドデカン:1.22
2g(7mmol)の1.15を、一部ずつ26ml(280mmol)の酢酸無水物に攪拌下添加し、氷水浴で冷却する。周囲温度で2時間攪拌する。100mlのクロロホルムを反応混合物に添加する。その後、溶液を2×100mlの飽和塩化ナトリウム水溶液、3×100mlの3N炭酸ナトリウム溶液、100mlの水で続けて洗浄する。有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。低温残渣を晶出した後、結晶を石油エーテルで洗浄し、デシケーターで乾燥して、白色粉末の生成物2.33g(90%)を得る。
融点:128−129℃
NMR1H(CDCl3,100MHz),δ(ppm):1.23(s,16H);1.55(m,4H);2.12(s,6H);2.23(t,4H);8.74(s,4H)
FT−IR, ν(cm-1):1232(C−O−C);1633(C=N);1736(OCO);3319及び3425(NH2)
+TOF MS:371(M+H)
【0093】
1,12−ビス(N,N’−ベンゾイルオキシアミジニル)ドデカン:1.23
1.28ml(14.68mmol)のベンゾイルクロリドを、DMF30mlに1.15の1.5g(5.24mmol)とトリエチルアミン2ml(14.16mmol)を含む懸濁液に攪拌下、滴下する。周囲温度で3時間攪拌し、その後、反応混合物をろ過し、ろ過物を200mlの冷水に沈殿させる。次いで、水と石油エーテルで洗浄し、デシケーターで乾燥して、白色粉末の生成物2.25g(87%)を得る。
融点:158−159℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.73(s,16H);2.01(m,4H);2.56(t,4H);6.94(s,4H);7.96−8.11(m,6H);8.54−8.61(m,4H)
FT−IR, ν(cm-1):684及び699(芳香族性C−H);1266(C−O−C);1625(C=N);1721(OCO);3315及び3452(NH2)
【0094】
1,12−ビス(N,N’−エチルカルバモイルオキシアミジニル)ドデカン:1.24
1.16ml(14.68mmol)のエチルイソシアン酸を、クロロホルム80mlに1.15の2g(7mmol)と炭酸カリウム1.01g(7.34mmol)を含む懸濁液に攪拌下、滴下する。周囲温度で一晩攪拌する。その後、反応混合物をろ過し、ろ過物を2×100mlの水で洗浄する。次いで、有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させて、有色油状の生成物2.22g(74%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.14(t,6H);1.22(s,16H);1.51(m,4H);2.12(t,4H);3.26(quintuplet,4H);5.00(s,4H);6.44(t,2H)
FT−IR, ν(cm-1): 1650(C=N);1701(OCONH);3335(NH);3374及び3490(NH2)
+TOF MS:429(M+H)
【0095】
1,12−ビス(N,N’−フェニルカルバモイルオキシアミジニル)ドデカン:1.25
1.6ml(14.68mmol)のフェニルイソシアン酸を、DMF40mlに1.15の2g(7mmol)と炭酸カリウム1.01g(7.34mmol)を含む懸濁液に攪拌下、滴下し、冷水浴で0℃に冷却する。周囲温度で2時間30分攪拌する。その後、反応混合物をろ過し、ろ過物を150mlの水に沈殿させる。次いで、沈殿物を水、アセトン、エーテルで続けて洗浄する。デシケーターで乾燥した後、白色粉末の生成物2.95g(80%)を得る。
融点:134−135℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.72(s,16H);2.0(m,4H);2.55(t,4H);6.83(s,4H);7.54(t,2H);7.76(t,4H);7.93(t,49.65(s,2H)
FT−IR, ν(cm-1): 1018及び1220(C−O−C);1628(C=N);1708(CON);3249(OCONH);3345及び3455(NH2)
ES+MS:525(M+H);263(M+H/2)
【0096】
[1,12−ビス(アミジニル)ドデカン]−1,12−ビス−N,N’−リン酸ジエチル:1.26
2.08ml(14.33mmol)のクロロホスホン酸ジエチルを、DMF30mlに1.15の2g(7mmol)とトリエチルアミン2.06ml(14.68mmol)を含む懸濁液に攪拌下、滴下し、冷水浴で0℃に冷却する。周囲温度で16時間攪拌する。その後、反応混合物をろ過し、ろ過物を150mlの酢酸エチルに溶解させる。次いで、有機相を3×200mlの水で洗浄し、その後、硫酸ナトリウムで乾燥し、減圧下、蒸発させて、有色油状の生成物2.88g(74%)を得る。
融点:134−135℃
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.31(t,12H);1.51(m,4H);2.15(t,4H);4.15(quintuplet,8H);4.30(s,4H)
FT−IR, ν(cm-1): 837及び1027(OP−O−CH2CH3);969(OP=O);1648(C=N);3323及び3487(NH2)
ES+MS:559(M+H)
【0097】
1,12−ビス[(1,2,4−オキサジアゾール−5(4H)−オン)−3−イル]ドデカン:1.27
キシレン70mlに1.17の4g(9.30mmol)を含む懸濁液を攪拌下、150℃、2時間加熱する(有色の油層が形成されるまで)。その後、反応混合物を減圧下、蒸発させる。次に、得られた固体残渣を50mlのDMSOに溶解し、そして、200mlの冷水に沈殿させる。次いで、形成された沈殿物を分離し、アセトンに再溶解し、ろ過する。ろ過物を硫酸ナトリウムで乾燥し、減圧下、蒸発させ、デシケーターで乾燥して、有色結晶の生成物2.97g(94%)を得る。
融点:150−151℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.71(s,16H);2.03(m,4H);2.93(t,4H)
FT−IR, n(cm-1): 1718(C=N);1783(OCONH);3163(NHCO)
ES+MS:339(M+H)
【0098】
1,12−ビス[(1,2,3,5−オキサチアジアゾール−2(3H)−オキシド)−4−イル]ドデカン:1.28
1.1ml(15.03mmol)の塩化チオニルを、0℃に冷却されたDMF30mlに1.15の2g(7mmol)とピリジン2.82ml(34.96mmol)を含む懸濁液に攪拌下、滴下する。低温状態で、45分間攪拌し、その後、反応溶液を150mlの冷水に沈殿させる。沈殿物をろ過し、デシケーターで乾燥して、有色粉末の生成物1.65g(62%)を得る。
融点:94−95℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.71(s,16H);2.05(m,4H);2.99(t,4H)
FT−IR, ν(cm-1): 1146(NSCO);1615(C−N);1655(C=N);3219及び3323(NHSO)
ES+MS:379(M+H)
【0099】
1,12−ビス[(1,2,4−オキサジアゾール−5(4H)−チオン)−3−イル]ドデカン:1.29
1.32g(32.97mmol)の60%水素化ナトリウムを、0℃に冷却されたDMF50mlに1.22の2g(5.4mmol)と硫化炭素2.5ml(34.96mmol)を含む懸濁液にゆっくり攪拌下、滴下する。低温状態で、45分間攪拌し、その後、周囲温度で2時間攪拌する。次いで、反応溶液を150mlの冷水に沈殿させ、2NのHClでpH5に酸性化し、その後、3×50mlの酢酸エチルで抽出する。有機相を水で洗浄し、その後、硫酸ナトリウムで乾燥し、減圧下、蒸発させて、固体残渣を得る。その後、それをエーテルで洗浄し、デシケーターで乾燥して、有色粉末の生成物1.45g(72%)を得る。
NMR1H(DMSO−d6,100MHz),δ(ppm):1.71(s,16H);2.06(m,4H);3.02(t,4H);9.25(s,2H)
FT−IR, ν(cm-1): 1644(C=N);1737(SCONH);3090(NHCO)
ES+MS:371(M+H);313[M+2H−60(SCO)]
【0100】
1,12−ビス[(1,2,4−チアジアゾール−5(4H)−オン)−3−イル]ドデカン:1.30
THF70mlに1.15の2g(7mmol)と1,1’−チオカルボニルジイミダゾール4g(21mmol)を含む懸濁液を周囲温度で16時間攪拌する。反応混合物を150mlの水で希釈し、3×70mlの酢酸エチルで抽出する。その後、有機相を水で洗浄し、硫酸ナトリウムで乾燥し、減圧下、蒸発させる。油状残渣を50mlのTHFに溶解し、BF3,
Et2Oを5.32ml(41.96mmol)添加する。得られた反応混合物は、次いで、周囲温度で一晩攪拌する。溶液を水で希釈し、酢酸エチルで抽出し、水で洗浄し、乾燥し、その後、減圧下で蒸発させる。得られた低温残渣は、次いで、50mlのエタノールに溶解させ、200mlの冷水に沈殿させる。単離した沈殿物を水で洗浄し、乾燥して、オレンジ色粉末の生成物1.62g(62.55%)を得る。
NMR1H(DMSO−d6,100MHz),δ(ppm):1.70(s,16H);2.04(m,4H);2.93(t,4H);9.30(s,2H)
FT−IR, ν(cm-1): 1666(C=N);1707(SCONH);3192(NHCO)
+TOF MS:313[M+2H−60(SCO)]
【0101】
1,14−ジ(ピロリジン−1−イル)テトラデカン−1,14−ジアミン:2.0, 2HCl
テトラデカンイミド酸ジエチルニ塩酸塩1.16g(3mmol)、ピロリジン0.51ml、及びエタノール15mlからなる混合物を還流下、24時間加熱する。蒸発させた後、得られた残渣をメタノール−エーテル混合物で再結晶して、ベージュ色粉末の生成物0.97g(75%)を得る。
融点:171℃
NMR1H(CD3OD,250MHz),δ(ppm):1.35(s,16H);1.68(m,4H);2.08(m,8H);2.55(t,4H);3.42(t,4H);3.70(t,4H);9.12(s,4H)
【0102】
1,14−ジ(ピペリジン−1−イル)テトラデカン−1,14−ジイミン:3.0
1.68ml(20mmol)の無水ピペリジンを、1,12−ジシアノドデカン2.2g(10mmol)とCuCl1.98g(20mmol)に添加する。最初、緑色の混合物は、青色に変化する。その後、80℃で20時間加熱し、生じた赤色溶液をエーテル125mlに注ぎ、12mlのNaOH(30%水性)を加え、2分間攪拌する。オレンジ色の相を単離し、硫酸ナトリウムで乾燥し、ろ過して蒸発させる。得られた残渣をエーテルから再結晶する(収率50%)。
融点:149−150℃
NMR1H(CDCl3,100MHz),δ(ppm):1.3(m,20H);1.4−1.6(m,12H);2.2(t,4H);3.3(m,8H);6.5(m,2H)
【0103】
N1,N1, N14, N14−テトラメチルテトラデカンジイミドアミド:4.0,
2HCl
1.16g(3mmol)のテトラデカンジイミド酸ジエチルニ塩酸塩とジエチルアミンのエタノール溶液(5.6M)1.1mlをエタノール15mlと混合し、還流下26時間加熱する。その後、溶媒を留去し、得られた残渣をエタノール−エーテル混合物から再結晶して、緑色がかった粉末の生成物0.98g(85%)を得る。
融点:209℃
NMR1H(CDCl3,250MHz),δ(ppm):1.35(m,16H);1.68(m,4H);2.59(t,4H);3.12(s,6H);9.10(s,4H)
【0104】
N1,N1, N14, N14−テトラメチルオクタデカンジイミドアミド:5.0
アンモニアのメタノール溶液(2M)5.3mlと0.46g(1.2mmol)のオクタデカンジイミド酸ジエチルニ塩酸塩の混合物を−10℃で1時間冷却し、その後、周囲温度で15時間放置する。溶媒を蒸発させた後、得られた残渣をメタノール−エーテル混合物で再結晶し、ろ過し、デシケーターで乾燥して、生成物0.25g(54%)を得る。
融点:196−198℃
NMR1H(DMSO−d6,250MHz),δ(ppm):1.24(m,24H);1.60(m,4H);2.37(t,4H);8.82(s,4H);9.10(s,4H)
【0105】
1,12−ビス(イミダゾリン−2−イル)ドデカン:6.0
a)1,12−ビス(イミダゾリン−2−イル)ドデカンニ塩酸塩:6.0, 2HCl
1.64ml(24.55mmol)のエチレンジアミンを、4.5g(11.69mmol)の1,14−ジエトキシテトラデカン−1,14−ジアミン塩酸塩の無水エタノール溶液100mlにゆっくりと低温状態で添加する。次いで、反応混合物を還流下、4時間加熱する。減圧下、蒸発させて最小量の溶媒とした後、得られた低温残渣を、攪拌下、エーテルを添加して晶出させる。その後、ろ過して単離した沈殿物をエーテルで洗浄し、デシケーターで乾燥して、白色粉末の生成物3.9g(88%)を得る。
融点:191−192℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.22(s,16H);1.57(m,4H);2.45(t,4H);3.75(s,8H);9.28(m,4H)
FT−IR, ν(cm-1): 1600(C=N);3031(C=NH,HCl);3200(N−H)
【0106】
b)1,12−ビス(イミダゾリン−2−イル)ドデカン:6.0
2g(5.28mmol)の6.0,2HClの水溶液(20ml)に、pHが14になるまで、トリエチルアミンを滴下する。形成した沈殿物を分離し、水、アセトン、その後エーテルで洗浄し、デシケーターで乾燥して、遊離塩基の白色粉末の生成物1.4g(86%)を得る。その後、この粉末をメタノールから再結晶させて、白色結晶の生成物1.26g(90%)を得る。
融点:176−178℃
NMR1H(CD3OD,100MHz),δ(ppm):1.29(s,16H);1.55(m,4H);2.20(t,4H);3.52(s,8H)
FT−IR, ν(cm-1): 1611(C=N);3177(N−H)
【0107】
1,12−ビス[N,N’−(メチルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.1
0.80ml(10.29mmol)のクロロギ酸メチルのクロロホルム溶液5mlを、1.5g(4.90mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、1.5ml(10.78mmol)のトリエチルアミンを含むクロロホルム25mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。次いで、反応混合物をろ過し、ろ過物を120mlの水、120mlの飽和塩化ナトリウム水溶液、及び2×120mlの水で続けて洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発して油状の残渣を生成する。これを石油エーテルに溶解し、−4℃で放置することにより、白色液状の生成物1.51g(73%)を得る。
融点:134−135℃
NMR1H(CDCl3,100MHz),δ(ppm):1.20(s,16H);1.58(m,4H);2.64(t,4H);3.70(s,8H);3.76(s,6H)
FT−IR, ν(cm-1):1142及び1194(C−O−C);1642(C=N);1723(NCO)
【0108】
1,12−ビス[N,N’−(エチルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.2
0.98ml(10.29mmol)のクロロギ酸エチルのクロロホルム溶液5mlを、1.5g(4.90mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、1.5ml(10.78mmol)のトリエチルアミンを含むクロロホルム25mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。次いで、反応混合物をろ過し、ろ過物を120mlの水、120mlの飽和塩化ナトリウム水溶液、及び2×120mlの水で続けて洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発して、油状の生成物1.78g(81%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.22(s,16H);1.30(t,6H);1.59(m,4H);2.65(t,4H);3.72(s,8H);4.16(q,4H)
FT−IR, ν(cm-1):1073及び1099(C−O−C);1644(C=N);1720(NCO)
【0109】
1,12−ビス[N,N’−(ブチルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.3
1.33ml(10.29mmol)のクロロギ酸ブチルのクロロホルム溶液5mlを、1.5g(4.90mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、1.5ml(10.78mmol)のトリエチルアミンを含むクロロホルム25mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。次いで、反応混合物をろ過し、ろ過物を120mlの水、120mlの飽和塩化ナトリウム水溶液、及び2×120mlの水で続けて洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発して、油状の生成物1.78g(81%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):0.88(t,6H);1.20(s,16H);1.36−1.64(m,12H);2.63(t,4H);3.71(s,8H);4.08(t,4H)
FT−IR, ν(cm-1):1073及び1152(C−O−C);1644(C=N);1722(NCO)
【0110】
1,12−ビス[N,N’−(イソブチルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.4
1.35ml(10.29mmol)のクロロギ酸イソブチルのクロロホルム溶液5mlを、1.5g(4.90mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、1.5ml(10.78mmol)のトリエチルアミンを含むクロロホルム25mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。次いで、反応混合物をろ過し、ろ過物を120mlの水、120mlの飽和塩化ナトリウム水溶液、及び2×120mlの水で続けて洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発して、油状の残渣を生成する。これを石油エーテルに溶解し、−4℃で置き、白色固形の生成物2.26g(91%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):0.87(d,12H);1.17(s,16H);1.55(m,4H);1.89(quint,2H);2.62(t,4H);3.70(s,8H);3.84(d,4H)
FT−IR, ν(cm-1):1072及び1142(C−O−C);1642(C=N);1722(NCO)
【0111】
1,12−ビス[N,N’−(ベンジルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.5
0.94ml(6.54mmol)のクロロギ酸ベンジルのクロロホルム溶液5mlを、1g(3.27mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、0.85ml(6.54mmol)のトリエチルアミンを含むクロロホルム30mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。得られた溶液を減圧下、蒸発させる。次いで、単離した沈殿物をエーテルで数回抽出する。エーテル相を減圧下、蒸発させて、油状の生成物を得、−4℃でエーテルから晶出して白色粉末の生成物1.02g(54%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.23(s,16H);1.60(m,4H);2.68(t,4H);3.77(s,8H);5.16(s,4H);7.35(s,10H)
FT−IR, ν(cm-1):1142及び1299(C−O−C);1646(C=N);1714(NCO)
MS−ES+:[M+H]+:575
【0112】
1,12−ビス[N,N’−(4−ニトロベンジルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.6
1.44g(6.70mmol)のクロロギ酸4−ニトロベンジルのクロロホルム溶液10mlを、1g(3.27mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、0.87ml(6.70mmol)のトリエチルアミンを含むクロロホルム20mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。次いで、反応混合物を30mlのクロロホルムで希釈した後、120mlの水、120mlの飽和塩化ナトリウム水溶液、及び2×120mlの水で続けて洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発させる。残渣を冷却し、最小量のクロロホルムに溶解して、最終的に−4℃でヘキサンから晶出して白色粉末の生成物2.08g(96%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.60(m,4H);2.67(t,4H);3.80(s,8H);5.24(s,4H);7.46及び7.55(dd,4H);8.17及び8.25(dd,4H)
FT−IR, ν(cm-1):1005及び1154(C−O−C);1343及び1517(NO2);1645(C=N);1724(NCO)
MS−ES+:[M+H]+:665
【0113】
1,12−ビス[N,N’−(フェニルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.7
1.26g(10.05mmol)のクロロギ酸フェニルのクロロホルム溶液5mlを、1.5g(4.90mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、1.3ml(10.05mmol)のトリエチルアミンを含むクロロホルム25mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。次いで、反応混合物を30mlのクロロホルムで希釈した後、120mlの水、120mlの飽和塩化ナトリウム水溶液、及び2×120mlの水で続けて洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発させて油状の生成物2.42g(90%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.64(m,4H);2.72(t,4H);3.89(s,8H);7.06−7.43(m,10H)
FT−IR, ν(cm-1):1162及び1188(C−O−C);1646(C=N);1737(NCO)
MS−ES+:[M+H]+:547;[M+H]2+/2:274(100%)
【0114】
1,12−ビス[N,N’−(4−フルオロフェニルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.8
0.89ml(6.70mmol)のクロロギ酸4−フルオロフェニルのクロロホルム溶液5mlを、1g(3.27mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、0.89ml(6.86mmol)のトリエチルアミンを含むクロロホルム20mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。次いで、反応混合物を20mlのクロロホルムで希釈した後、60mlの水、60mlの飽和塩化ナトリウム水溶液、及び2×60mlの水で続けて洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発させる。残渣を冷却し、最終的に−4℃でヘキサンから晶出して、白色粉末の生成物1.70g(85%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.64(m,4H);2.71(t,4H);3.90(s,8H);7.03及び7.36(d,8H)
FT−IR, ν(cm-1):1100(C−F);1179(C−O−C);1655(C=N);1733(NCO)
MS−ES+:[M+H]+:583;[M+H]2+/2:292(100%)
【0115】
1,12−ビス[N,N’−(4−フルオロフェニルオキシカルボニル)イミダゾリン−2−イル]ドデカンニ塩酸塩:6.8, 2HCl
1gの6.8を、塩酸ガスで飽和したエタノール溶液20mlに添加する。次いで、反応混合物を50℃に加熱し、4時間激しく攪拌する。エーテル150mlを低温溶液に添加し、その後、混合物を一晩冷蔵庫に静置する。デカンテーションした後、形成された油層を蒸留水100mlに溶解し、そして、ろ過する。ろ過物を最終的に凍結乾燥して、白色粉末の塩生成物を得る。
NMR1H(DMSO−d6,100MHz),δ(ppm):1.70(s,16H);2.13(m,4H);3.46(t,4H);4.55(t,4H);4.80(t,4H);7.78及び7.85(d,8H);8.71(s,2H)
FT−IR, ν(cm-1):1692(C=N);1760(NCO);3350(N,HCl)
【0116】
1,12−ビス[N,N’−(4−メトキシフェニルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.9
1.02ml(6.86mmol)のクロロギ酸4−メトキシフェニルのクロロホルム溶液5mlを、1g(3.27mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、0.89ml(6.86mmol)のトリエチルアミンを含むクロロホルム20mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。次いで、単離した沈殿物(トリエチルアミン塩酸塩)を数回エーテルで抽出する。エーテル相を減圧下、蒸発させて、油状の生成物1.31g(66%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.63(s,4H);2.71(t,4H);3.75(s,8H);3.88(s,2H);6.61−7.10(m,8H)
FT−IR, ν(cm-1):1177及び1248(C−O−C);1646(C=N);1735(NCO)
MS−ES+:[M+H]+:607;[M+2H]2+/2:304(100%)
【0117】
1,12−ビス[N,N’−(4−ニトロフェニルオキシカルボニル)イミダゾリン−2−イル]ドデカン:6.10
1.38g(6.86mmol)のクロロギ酸4−ニトロフェニルのクロロホルム溶液5mlを、1g(3.27mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、0.89ml(6.86mmol)のトリエチルアミンを含むクロロホルム25mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で5時間攪拌する。溶液を減圧下、蒸発させた後、形成された沈殿物を水、次いでエーテルで洗浄する。得られた黄色固体物は、次いで、アセトンをゆっくり添加し、最小量のジクロロホルムに溶解させて晶出する。白色結晶の生成物74%(1.54g)を単離する。
融点:121−122℃
NMR1H(CDCl3,100MHz),δ(ppm):1.23(s,16H);1.67(m,4H);2.73(t,4H);3.95(s,8H);7.25及び7.36(dd,4H);8.24及び8.32(dd,4H)
FT−IR, ν(cm-1):1185及び1196(C−O−C);1349及び1524(NO2);1656(C=N);1748(NCO)
MS−ES+:[M+H]+:637;[M+2H]2+/2:319(100%)
【0118】
1,12−ビス[N,N’−(アセトキシメトキシカルボニル)イミダゾリン−2−イル]ドデカン:6.11
1g(6.54mmol)のクロロギ酸アセトキシメチルのクロロホルム溶液5mlを、1g(3.27mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、0.92ml(6.54mmol)のトリエチルアミンを含むクロロホルム20mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。その後、反応混合物を、20mlの水、20mlの5%炭酸水素ナトリウム水溶液、次いで20mlの飽和塩化ナトリウム水溶液で洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発させて、油状の生成物2.95g(84%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.21(s,16H);1.58(m,4H);2.08(s,6H);2.65(t,4H);3.74(s,8H);5.74(s,4H)
FT−IR, ν(cm-1):1643(C=N);1682(NCO);1736(OCO)
MS−ES+:[M+H]+:539
【0119】
1,12−ビス[N,N’−((1−アセトキシエトキシ)カルボニル)イミダゾリン−2−イル]ドデカン:6.12
a)1,12−ビス[N,N’−((1−クロロエトキシ)カルボニル)イミダゾリン−2−イル]ドデカン
1.8ml(16.75mmol)のクロロギ酸1−クロロエチルのクロロホルム溶液10mlを、2.50g(8.17mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、2.16ml(16.75mmol)のトリエチルアミンを含むクロロホルム40mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩放置する。その後、反応混合物を、100mlの水、100mlの飽和塩化ナトリウム水溶液、及び3×100
mlの水で続けて洗浄する。硫酸ナトリウムで乾燥させた後、有機相を減圧下、蒸発させる。残渣を冷却し、その後、ヘキサンから−4℃で晶出して、白色粉末の生成物3.91g(92%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.19(s,16H);1.58(m,4H);1.77(d,6H);2.64(t,4H);3.74(s,8H);6.49(q,2H)
FT−IR, ν(cm-1):1087(C−O−C);1377(CH3;C−H);1648(C=N);1737(NCO)
【0120】
b)1,12−ビス[N,N’−((1−アセトキシエトキシ)カルボニル)イミダゾリン−2−イル]ドデカン:6.12
1,12−ビス[N,N’−((1−クロロエトキシ)カルボニル)イミダゾリン−2−イル]ドデカンニ塩酸塩3.20g(6.17mmol)と酢酸水銀5.9g(18.50mmol)を含む酢酸溶液を攪拌下、周囲温度で72時間放置する。減圧下、溶媒を蒸発させた後、反応残渣をクロロホルム150mlに希釈し、次いで、ろ過する。その後、ろ過物を3×300mlの飽和塩化ナトリウム水溶液で洗浄する。有機相を硫酸ナトリウムで乾燥させ、減圧下、蒸発させて、有色油状の生成物2.40g(68%)を得る。
NMR1H(DMSO−d6,100MHz),δ(ppm):1.24(s,16H);1.44(d,6H);1.52(m,4H);2.03(s,6H);2.61(t,4H);3.79(s,8H);6.69(q,2H)
FT−IR, ν(cm-1):1072及び1224(C−O−C);1645(C=N);1684(NCO);1733(CO)
【0121】
[1,12−ビス(イミダゾリン−2−イル)ドデカン]−1,12−ビス−N,N’−ホスホン酸ジフェニル:6.13
1.40g(6.70mmol)のクロロホスホン酸ジフェニルのクロロホルム溶液5mlを、1g(3.26mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、0.96ml(6.86mmol)のトリエチルアミンを含むクロロホルム25mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩攪拌する。次いで、反応混合物をろ過し、ろ過物を100mlの水、100mlの飽和塩化ナトリウム水溶液、及び2×100mlの水で続けて洗浄する。硫酸ナトリウムで乾燥した後、有機相を減圧下、蒸発させて、油状の生成物2.37g(94%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.20(s,16H);1.62(m,4H);2.58(t,4H);3.79(s,8H);7.17−7.34(m,20H)
FT−IR, ν(cm-1):925(P=O);1160及び1184(C−O);1646(C=N)
【0122】
[1,12−ビス(イミダゾリン−2−イル)ドデカン]−1,12−ビス−N,N’−ホスホン酸ジエチル:6.14
1.49ml(10.29mmol)のクロロホスホン酸ジエチルのクロロホルム溶液5mlを、1.5g(4.90mmol)の1,12−ビス(イミダゾリン−2−イル)ドデカンと、1.5ml(10.78mmol)のトリエチルアミンを含むクロロホルム25mlの氷浴で冷却した懸濁液に攪拌下、滴下する。その後、周囲温度で一晩攪拌する。次いで、反応混合物をろ過し、ろ過物を100mlの水、100mlの飽和塩化ナトリウム水溶液、及び100
mlの水で続けて洗浄する。硫酸ナトリウムで乾燥した後、有機相を減圧下、蒸発させて、油状の生成物2.38g(84%)を得る。
NMR1H(CDCl3,100MHz),δ(ppm):1.15(s,16H);1.23(t,12H);1.54(m,4H);2.37(t,4H);3.60(t,8H);4.00(quintuplet,8H)
FT−IR, ν(cm-1):963(P=O);1016及び1268(C−O);1640(C=N)
MS−ES+:[M+H]+:579;[M+H]2+/2:290(100%)
【0123】
1,12−ビス(1−メチルイミダゾリン−2−イル)ドデカンニ塩酸塩:7.0, 2HCl
3.9g(10mmol)のテトラデカンジイミド酸ジエチルニ塩酸塩を過剰のN−メチルエチレンジアミンと共に24時間還流する。溶液を冷却した後、溶液にエチルエーテルを添加する。得られた結晶をろ過して単離し、エーテルで洗浄し、デシケーターで乾燥して、白色結晶の生成物2.9g(71%)を得る。
融点:166−170℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.38(m,16H);1.67(m,4H);2.47(t,4H);2.90(s,6H);3.55(s,8H);9.28(m,2H)
FT−IR, ν(cm-1):1600(C=N);3031(C=NH,HCl);3200(N−H)
【0124】
1,12−ビス(1,4,5,6−テトラヒドロピリミジン−2−イル)ドデカンニ塩酸塩:8.0, 2HCl
0.51ml(6.12mmol)の1,3−ジアミノプロパンを、テトラデカンジイミド酸ジエチルニ塩酸塩1.16g(3mmol)の無水エタノール15mlの溶液に添加する。次いで、還流下、反応混合物を加熱する。4.5mlの水を加えた後、反応混合物を95℃で4時間放置する。蒸発させて乾燥した後、得られた残渣にエーテルを加えて晶出し、その後、48時間撹拌する。次いで、ろ過して単離した沈殿物を激しく攪拌して数回エーテルで洗浄し、デシケーターで乾燥して、白色結晶の生成物0.83g(68%)を得る。
融点:117−119℃
NMR1H(DMSO−d6,250MHz),δ(ppm):1.22(s,16H);1.55(m,4H);1.80(m,4H);2.45(t,4H);3.25(s,8H);9.28(m,2H)
【0125】
1,12−ビス(4−メチルイミダゾリン−2−イル)ドデカンニ塩酸塩:9.0, 2HCl
0.44ml(5.1mmol)の1,2−ジアミノプロパンを、テトラデカンジイミド酸ジエチルニ塩酸塩0.96g(2.5mmol)の無水エタノール12mlの溶液に攪拌下、滴下する。反応混合物を、その後、48時間、90℃の温度で加熱する。溶媒を蒸発させた後、得られた残渣をエタノールに溶解し、その後、ろ過する。ろ過物を蒸発させ、次いで、エーテルを加え、減圧下、脱離反応を行い、残渣溶媒を除去する。そして、白色粉末の生成物0.71g(70%)を得る。
融点:117−119℃
NMR1H(DMSO−d6,250MHz),δ(ppm):1.30(m,22H);1.64(m,4H);2.52(t,4H);3.40(t,2H);3.95(t,2H);3.95(m,2H);9.32(m,2H)
【0126】
1,12−ビス(4,4−ジメチルイミダゾリン−2−イル)ドデカンニ塩酸塩:10.0,2HCl
0.96g(2.5mmol)のテトラデカンジイミド酸ジエチルニ塩酸塩の無水エタノール12mlと0.54ml(5.1mmol)の1,2−ジアミノ−2−メチルプロパンの反応混合物を90℃で48時間加熱する。溶媒を蒸発させ、残渣を高温の状態でエタノールに溶解する。周囲温度に冷却した後、形成された固形物をろ過し、その後、化合物9.0,2HClのように、エーテルと処理して、ベージュ色粉末の生成物0.76g(70%)を得る。
NMR1H(DMSO−d6,250MHz),δ(ppm):1.24(s,12H);1.32(s,16H);1.62(m,4H);2.50(t,4H);3.54(s,4H);9.30(m,2H)
【0127】
1,12−ジ(3a,4,5,6,7,7a−ヘキサヒドロ−1H−ベンゾイミダゾ−2−イル)ドデカンニ塩酸塩:11.0, 2HCl
0.96g(2.5mmol)のテトラデカンジイミド酸ジエチルニ塩酸塩の無水エタノール12mlと1,2−ジアミノシクロヘキサン0.63mlを48時間還流(90℃)する。混合物を蒸発させた後、エタノールを溶液に加え、蒸発したろ過物を化合物9.0,2HClで述べたのと同様に処理して、結晶0.82g(67%)を得る。
NMR1H(DMSO−d6,250MHz),δ(ppm):1.25(s,16H);1.30−1.65(m,16H);1.70(m,4H);2.50(t,4H);3.34(m,2H);4.10(m,2H);
9.32(m,2H)
【0128】
1,12−ビス[(1,2,4−オキサジアゾール)−3−イル]ドデカン:18.0
0.6ml(4.9mmol)のトリフルオロ炭酸ジエチルを、2g(7mmol)のビス−アミドキシム1.15を含むオルトギ酸エチル23.26ml(139.86mmol)の懸濁液に攪拌下、添加する。反応混合物を周囲温度で15分間攪拌し、次いで、1時間還流下、加熱する。得られた溶液に150mlの酢酸エチルを添加し、水(100ml)、飽和炭酸水素ナトリウム溶液(100ml)、飽和塩化ナトリウム溶液(100
ml)で続けて洗浄する。次いで、有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。得られた残渣を冷エーテルで洗浄し、その後、乾燥させて、白色粉末の生成物1.50g(70%)を得る。
融点:92−93℃
NMR1H(CDCl3,100MHz),δ(ppm):1.24(s,16H);1.73(m,4H);2.77(t,4H);8.61(s,2H)
FT−IR, ν(cm-1): 1111(C−O);1558(C=N);3123(=CH)
【0129】
1,12−ビス[(5−メチル−1,2,4−オキサジアゾール)−3−イル]ドデカン:18.1
1.22の1.5g(4.05mmol)を含むキシレン30mlの懸濁液を攪拌下、150℃で2時間加熱する。次いで、反応混合物を減圧下、蒸発させる。得られた固体残渣を、次いで、石油エーテルから再結晶し、デシケーターで乾燥した後、白色粉末の生成物1.10g(81%)を得る。
融点:63−64℃
NMR1H(CDCl3,100MHz),δ(ppm):1.23(s,16H);1.69(m,4H);2.53(s,6H);2.66(t,4H)
FT−IR, ν(cm-1): 1581(C−N);1640(C=N)
【0130】
1,12−ビス[(5−トリフルオロメチル−1,2,4−オキサジアゾール)−3−イル]ドデカン:18.4
1g(3.50mmol)のビス−アミドキシム1.15を一部ずつ、トリフルオロ無水酢酸20ml(139.86mmol)に攪拌下、添加し、氷水浴で冷却する。1.15が完全に溶解するまで、冷却状態で30分間攪拌する。反応混合物にエーテル50ml及び非常にゆっくりと冷水100mlを添加する(発熱反応)。その後、エーテル相を、2×100mlの水、2×100mlの1N炭酸ナトリウム溶液、及び100mlの水で続けて洗浄する。次いで、有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。冷却した後、残渣を晶出して、白色結晶の生成物1.39g(90%)を得る。
融点:<40℃
NMR1H(CDCl3,100MHz),δ(ppm):1.26(s,16H);1.75(m,4H);2.81(t,4H)
FT−IR, ν(cm-1): 766及び1150(CF3);1519(C−N);1608(C=N)
【0131】
1,12−ビス[(5−トリクロロメチル−1,2,4−オキサジアゾール)−3−イル]ドデカン:18.5
3g(10.49mmol)のビス−アミドキシム1.15と、13.71g(83.92mmol)のトリクロロ無水酢酸を含むクロロホルム10mlの懸濁液を攪拌下、溶液となるまで、85℃で加熱する。3等分にして、溶液に4.70ml(41.96mmol)のトリクロロ塩化アセチルを添加し、次いで、混合物を、飽和炭酸ナトリウム溶液(2×100ml)、飽和塩化ナトリウム溶液(2×100
ml)、及び水(100ml)で続けて洗浄する。次いで、有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。得られた残渣を乾燥させて、黄色粉末の生成物5.19g(91%)を得る。
融点:55−56℃
NMR1H(CDCl3,100MHz),δ(ppm):1.26(s,16H);1.76(t,4H);2.78(t,4H)
FT−IR, ν(cm-1): 798及び830(CCl3);1573(C=N)
【0132】
1,12−ビス[(5−フェニル−1,2,4−オキサジアゾール)−3−イル]ドデカン:18.6
1.23の1.5g(3.04mmol)を含むキシレン30mlの懸濁液を攪拌下、150℃で2時間加熱する。次いで、反応溶液をデカンテーションし、その後、減圧下、蒸発させる。得られた残渣を続いて石油エーテルから再結晶して、デシケーターで乾燥した後、有色粉末の生成物1.12g(80%)を得る。
融点:97−98℃
NMR1H(CDCl3,100MHz),δ(ppm):1.25(s,16H);1.78(m,4H);2.77(t,4H);7.48−7.54(m,6H);8.06−8.14(m,4H)
FT−IR, ν(cm-1):1581(C−N);1640(C=N)
【0133】
1,12−ビス[(5−エチルオキシカルボニル−1,2,4−オキサジアゾール)−3−イル]ドデカン:18.7
2.35ml(21mmol)の塩化オキサリルエチルを、2g(7mmol)のビス−アミドキシム1.15とピリジン4ml(49.50 mmol)と4Åモレキュラーシーブ4gを含むクロロホルム50mlの懸濁液に攪拌下、添加する。その後、反応混合物を80℃で16時間加熱し、次いで、ろ過し、減圧下、蒸発させる。残渣に酢酸エチル100mlを加え、その後、溶液を、飽和炭酸ナトリウム溶液(2×50ml)、飽和塩化ナトリウム溶液(2×50ml)、及び水50mlで続けて洗浄する。次いで、有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させる。得られた残渣をエタノールで洗浄し、その後、乾燥させて、粉末の生成物1.96g(62%)を得る。
融点:70−71℃
NMR1H(CDCl3,100MHz),δ(ppm):1.24(s,16H);1.44(t,6H);1.77(m,4H);2.80(t,4H);4.51(quartet,4H)
FT−IR, ν(cm-1):1029及び1197(C−O−C);1584(C=N);1760(COO)
【0134】
1,12−ビス[(5−カルバモイル−1,2,4−オキサジアゾール)−3−イル]ドデカン:18.9
2g(4.44mmol)の18.7を、10%アンモニウムのエタノール溶液(90ml)に添加する。反応混合物を密封して、周囲温度で24時間攪拌下、放置する。次いで、懸濁液をろ過し、冷エタノールで洗浄し、乾燥する。こうして黄色粉末の生成物1.58g(91%)を得る。
融点:196−197℃
NMR1H(DMSO−d6,100MHz),δ(ppm):1.71(s,16H);2.14(m,4H);3.23(t,4H);8.98(s,2H)
FT−IR, ν(cm-1):1573(C−N);1604(C=N);1673(CON);3233及び3432(NH2)
【0135】
1,12−ビス[(5−シアノ−1,2,4−オキサジアゾール)−3−イル]ドデカン:18.10
1.3ml(9.18mmol)のトリフルオロ無水酢酸を、1.5g(3.83mmol)の18.9とピリジン1.55ml(19.13mmol)を含むジオキサン60mlの懸濁液に添加し、0℃に冷却する。反応混合物を周囲温度で16時間攪拌下、放置する。次いで、得られた溶液を酢酸エチル150mlで希釈し、その後、水(100ml)及び飽和塩化ナトリウム溶液(2×10ml)で続けて洗浄する。有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させて、有色油状の生成物0.83g(61%)を得る。
融点:70−71℃
NMR1H(CDCl3,100MHz),δ(ppm):1.24(s,16H);1.73(m,4H)
FT−IR, ν(cm-1):1561(C=N);2260(CN)
【0136】
1,12−ビス(N,N’−ジベンゾイルオキシカルボニルグアニジノ)ドデカン:12.1
1g(5mmol)の1,12−ジアミノドデカンと3.76g(10.5mmol)のN,N’−ジベンゾイルオキシカルボニル−S−メチルイソチオ尿素を含むテトラヒドロフラン70mlの溶液を60℃から70℃の間で24時間加熱する。その後、減圧下、溶媒を蒸発させる。次いで、得られた残渣をジクロロメタンに溶解し、5%炭酸水素ナトリウム溶液、飽和塩化ナトリウム水溶液及び水で続けて洗浄する。次いで、有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させ、シリカカラム(DCM)のクロマトグラフィーで精製する。異なる分画を合わせ、減圧下、蒸発させた黄色の油をエーテルから晶出して、白色粉末の生成物2.4g(58%)を得る。
融点:97−98℃
NMR1H(CDCl3,100MHz),δ(ppm):1.24(s,16H);1.54(s,4H);3.40(q,4H);5.11(s,4H);5.16(s,4H);7.15−7.54(m,20H);8.28(t,2H);11.73(s,2H)
FT−IR, ν(cm-1):1054及び1130(C−O);1651(C=N);1736(NCO);3130(NHCO);3333(NH)
MS−FAB+:[M+H]+:821;[M+2H]++/2:412
【0137】
1,12−ビス(N,N’−ジ−tert−ブチルオキシカルボニルグアニジノ)ドデカン:12.2
1g(5mmol)の1,12−ジアミノドデカン1gと3.19g(11mmol)のN,N’−ジ−tert−ブチルオキシカルボニル−S−メチルイソチオ尿素の混合物を含むエタノール100mlを50℃から60℃の間で48時間加熱する。減圧下、溶液を蒸発させた後、得られた残渣をDCM100mlに溶解し、5%炭酸水素ナトリウム水溶液(2×100ml)、水(2×100ml)、及び飽和塩化ナトリウム溶液100mlで続けて洗浄する。次いで、有機相を硫酸ナトリウムで乾燥し、減圧下、蒸発させ、シリカ(DCM/MeOH
98%)で精製し、低温状態で石油エーテルから晶出させて、粉末状の生成物2.50g(73%)を得る。
融点:114−116℃
NMR1H(CDCl3,100MHz),δ(ppm):1.24(s,16H);1.47(s,36H);1.61(m,4H);3.35(q,4H);8.27(t,2H);11.48(s,2H)
FT−IR, ν(cm-1):1028及び1138(C−O);1670(C=N);1740(NCO);3132(NHCO);3314(NH)
MS−ES+:[M+H]+:685
【0138】
1,12−ビス[N,N’−(2−アミノ−3,4,5,6−テトラヒドロピリミジル)]ドデカンニ塩酸塩:16.0, 2HCl
0.72g(3.6mmol)の1,12−ジアミノドデカン、1.86g(7.2mmol)の2−メチルスルファニル−3,4,5,6−テトラヒドロピリミジニウムヨージド、及び0.5ml(3.6mmol)のトリエチルアミンを含む反応混合物のアセトニトリル20mlを還流下22時間加熱する。周囲温度に冷却した後、反応溶媒を蒸発させ、残渣をシリカカラム(CH2Cl2/CH3OH/NH4OH
89:10:1)でクロマトグラフィーを行う。グアニジン塩0.91g、すなわち収率41%を得る。
NMR1H(CD3OD,100MHz),δ(ppm):1.49(m,20H);2.13(m,4H);3.33(m,4H);3.56(m,8H)
FT−IR, ν(cm-1):1028及び1138(C−O);1670(C=N);1740(NCO);3132(NHCO);3314(NH)
MS−FAB+:[M+H]+:365;[M+H+HI]+:493
【0139】
1,12−ビス[N,N’−(5,5−ジメチル−3,4,5,6−テトラヒドロピリミジン−2−イル)アミノ]ドデカンニ塩酸塩:17.0, 2HCl
0.72g(3.6mmol)の1,12−ジアミノドデカン、2.06g(7.2mmol)の5,5−ジメチル−2−メチルスルファニル−3,4,5,6−テトラヒドロピリミジニウムヨージド、及び0.5ml(3.6mmol)のトリエチルアミンを含む反応混合物のアセトニトリル20mlを還流下22時間加熱する。周囲温度に冷却した後、反応溶媒を蒸発させ、残渣をシリカカラム(CH2Cl2/CH3OH/NH4OH
89:10:1)でクロマトグラフィーを行う。塩0.91g、すなわち収率36%を得る。
NMR1H(CD3OD,100MHz),δ(ppm):1.17(s,12H);1.49(m,20H);3.07(s,8H);3.23(m,4H)
【0140】
[1−[N−(5,5−ジメチル−3,4,5,6−テトラヒドロピリミジン−2−イル)アミノ]−12−[N’−(3,4−ジヒドロ−2H−ピロール−5−イル)アミノ]]ドデカンジトリフルオロ酢酸塩:25.0,
2TFA
1mmolの1−[N−(5,5−ジメチル−3,4,5,6−テトラヒドロピリミジニウム−2−イル)アミノ]ドデカン−12−アンモニウムジトリフルオロ酢酸塩、1mmol(1当量)の3,4−ジヒドロ−5−メトキシ−2H−ピロール及び0.5mlのトリエチルアミンを含む反応混合物の無水エタノール10mlを還流下20時間加熱する。周囲温度に冷却した後、反応混合物を蒸発させて乾燥し、シリカカラム(CH2Cl2/CH3OH/NH4OH
85:13:2)でクロマトグラフィーを行い、油状の生成物を収率65%で得る。
NMR1H(CD3OD,360MHz),δ(ppm):1.12(s,12H);1.36(m,16H);1.60(m,2H);1.69(m,2H);2.28(m,2H);2.93(m,2H);3.09(s,4H);3.22(m,2H);3.31(m,2H);3.75(m,2H)
MS−ES+:[M+H]+:378;[M+H+TFA]+:492;[M+2H]++/2:189.5
【0141】
1,12−ビス[N, N’−(3,4−ジヒドロ−2H−ピロール−5−イル)アミノ]ドデカン:21.0
2.5g(25.25mmol)の2−メトキシピロリンを、2.76g(10.1mmol)の1,12−ジアミノドデカン塩酸塩を含む無水エタノール70mlに加える。反応混合物を周囲温度で24時間攪拌下、放置する。次に、溶液を減圧下蒸発させ、冷却した後の残渣を水100mlに溶解し、その後、0.1N炭酸ナトリウム溶液でアルカリ性にする。その後、形成した沈殿物を分離し、水その後エーテルで洗浄し、デシケーターで乾燥して、白色粉末の予定生成物3.27g(97%)を得る。
融点:155−156℃
NMR1H(CDCl3,100MHz),δ(ppm):1.23(s,16H);1.49(m,4H);1.89(quint.,4H);2.38(t,4H);3.21(t,4H);3.43(s,2H);3.62(t,4H)
FT−IR, ν(cm-1):1630(C=N);3049及び3221(NH)
【0142】
1,12−ビス(N, N’−アセトアミジニル)ドデカン:20.0
2g(10mmol)の1,12−ジアミノドデカンと2.47g(20mmol)のエチルアセトアミド塩酸塩の混合物を含む無水ジオキサン25mlを還流下24時間加熱する。周囲温度に冷却した後、1N水酸化カリウム水溶液を加えて、沈殿物を形成させる。この沈殿物を高温状態でメタノールで洗浄し、その後、ろ過する。デシケーターに数時間置いた後、生成物2.06g(7.3
mmol)、すなわち収率73%を得る。
融点:92−97℃
NMR1H(DMSO−d6,250MHz),δ(ppm):1.3(m,16H);1.5(m,4H);1.8(s,6H);2.9(t,4H);5.5(m,2H)
【0143】
1,12−ビス(N, N’−ヒドロキシアセトアミジニル)ドデカン:20.1及びその二塩酸塩 20.1,2HCL
10g(50mmol)の1,12−ジアミノドデカンを、12.9g(125mmol)のエチル N−ヒドロキシアセトイミダートを含むエタノール125mlに加える。反応混合物を80℃まで4日間加熱する。冷却後、結果として得られる沈殿物を分離し、エタノールで、次いでエーテルで数回洗浄し、乾燥器で乾燥させ、エタノール中で再結晶化して、白色粉末の7.26g(46%)のヒドロキシアセトアミジニル20.1(融点150−151℃)を得る。
【0144】
1gのヒドロキシアセトアミジニル20.1を、塩酸(塩化水素酸)ガスで飽和した20mlの無水エタノール中に溶解させる。混合物を、激しく攪拌して50℃まで2時間加熱する。100mlの無水エーテルを低温溶液に加え、この混合物を放置して沈殿させる。結果として得られる固形物を分離し、乾燥させ、100mlの蒸留水に溶解させて、その後ろ過する。水性ろ液は、凍結乾燥して白色粉末の製品を得る。
1H NMR(DMSO−d6 100MHz): 1.72(s. 16H, H3−H10);
1.96 (s, 4H, H2 and H11); 2.62 (s, 6H, 2H2:
3.86 (q, 4H, H1 and H12); 9.30 (t, 2H, 2NH); 11.40 (s,
2H, 2NOH); 12.92 (s, 2H, 2C=NOH,HCL).
FT−IR: 1683 (C=N); 3002; 3142 and 3202 (NH; C=NOH, HCL and NOH
【0145】
1,12−ビス[N,N’−メトキシアセトアミジニル]ドデカン,20.2
1g (3.18mmol) のヒドロキシアセトアミジニル20.1を、0.45g(11.15mmol)のソーダ及び25mlのエタノール/水(4:1)から調整されたヒドロキシル−アルコールソーダ溶液に加える。30分攪拌後、0.42ml
(6.68mmol)よう化メチルを、反応懸濁液に滴下する。混合物を室温で24時間攪拌する。その後、不透明な溶液を過して、ろ液を減圧下で蒸発させる。得られた残渣を50mlのクロロホルム中で再溶解させ、塩化ナトリムの飽和水性溶液(2×100ml)で洗浄する。有機相を硫酸ナトリウム上で乾燥させ、減圧下で蒸発させて、1.05g(96%)の有色オイルのメトキシアセトアミジニルを得る。
1H NMR(CDCl3, 100MHz): 1.24(s, 16H, H3-H10);
1.45 9m, 4H, H2 and H11; 1.83 (s, 6H, 2H2'):
3.05 (q, 4H, H1 and H12); 3.70 (s, 6H, 2H3'):
5.03 (t, 2H, 2NH).
ES+ SM: 343 [M+H+]; 172 (100%) [(M+2H+)/2].
FT−IR: 1637 (C=N); 3264 (NH).
【0146】
1−12−ビス[N,N’−アセトキシアセトアミジニル]ドデカン:20.8
1g(3.18mmol)のヒドロキシアセトアミジニル20.1を、12ml(127.4mmol)の0℃に冷却された無水酢酸の攪拌溶液に少量加える。混合物を、室温で2時間攪拌する。100mlのクロロホルムを反応混合物に加える。この溶液を、2×100mlの水性飽和塩化ナトリウム溶液、3×100mlのソーダ溶液2N及び2×100mlの水で連続的に洗浄する。その後、有機相を硫酸ナトリウム上で乾燥させて、減圧下で蒸発させる。30mlのエーテルを含油低温残渣(oily
cold residue)に加え、その溶液を16時間冷蔵庫で放置する。その後、結果として得られる固形物をエーテル中で粉砕して分離する。得られた粉末をエーテルで最終的に洗浄し、乾燥させて、1.12g(89%)の白色粉末のアセトキシアセトアミジニルを得る。
融点68-69℃。
1NMR(CDCl3, 100MHz): 1.24(s, 16H, H3−H10);
1.44 (m, 4H, H2 and H11); 1.92 (s, 6H, 2H2');
2.11 (s, 6H, 2H4'); 3.10 9q. 4H, H1 and H12);
5.04 9t, 2H, 2NH).
ES+ SM:399 (100%)[M+H+};200[(M+2H+)/2
FT−IR: 1623 (C=N); 1742 (OCO): 3333 (NH)
【0147】
1,12−ビス[N,N’−ベンゾイルオキシアセトアミジニル]ドデカン:20.9
0.78ml (6.68mmol)の塩化ベンゾイルを含む5 mlのクロロホルムを、0℃に冷却して、1g(3.18mmol)のヒドロキシアセトアミジニル20.1及び0.94ml(6.68mmol)のトリエチルアミンを含むクロロホルム30mlの攪拌された懸濁液に滴下する。混合物を3時間室温で撹拌する。その後、反応混合物を2×100mlの水で、次いで100
mlの塩化ナトリウムの飽和溶液で洗浄する。その後、有機相を硫酸ナトリウム上で乾燥させて、減圧下で蒸発させる。含油残渣を冷蔵庫中で一晩放置する。結果として得られる固形物を低温エーテルで粉砕し、エーテルで洗浄し、その後、分離し乾燥させて1.27g(77%)の白色粉末のベンゾイルオキシアセトアミジニルを得る。
融点95−96℃。
1H NMR(CDCl3, 200MHz): 1.23(s. 16H, H3−H10);
1.51 (m, 4H, H2 and H11); 2.02 (sm, 6H, 2H2');
3.15 (q, 4H, H1 and H12); 5.19 (t, 2H, 2NH); 7.34−7.54
[m, 6H, 2(6H,2(H6−H8)]; 7.93−8.03 [dd, 4H, 2(H5
and H9)]
FT−IR: 1279 (C−O−C); 1622 (C=N); 1714 (OCO); 3385 (NH)
ES+ SM: 523 (100%) [M+H+]; 262[M+2H+)/2
【0148】
1,12−ビス(N,N’−エチルカルバモイルオキシアミジニル)ドデカン:20.10
0.53 ml (6.69mmol)のエチルイソシアン酸塩を、1g(3.18mmol)のヒドロキシアセトアミジニル20.1及び0.45g(3.18mmol)の炭酸カリウムを含むクロロホルム40
mlの攪拌された懸濁液に滴下する。混合物を一晩室温で攪拌する。反応混合物をろ過し、ろ過液を3×100mlの水で洗浄する。その後、有機相を硫酸ナトリウム上で乾燥させて、減圧下で蒸発させて、有色オイルのエチルカルバモイルオキシアミジニルを得る。
1H NMR(CDCl3, 100MHz): 1.14(t, 6H, 2H5'); 1.22 (s, 16H, H3−H10);
1.44 (m, 4H, H2 and H11); 1.84 (s, 6H, 2H2');
3.07 (q, 4H, H1 and H12); 3.26 (quintuplet, 4H, 2H4);
5.33 (t, 2H, 2NH); 6.49 (t, 2H, 2NHCO).
FT−IR: 1215 (C−O); 1641 (C=N); 1705 (OCON); 3345 (NHCO); 3398 (NH).
【0149】
1,12−ビス(N,N’−フェニルカルバモイルオキシアミジニル)ドデカン:20.11
0.73ml (6.69mmol)のフェニルイソシアン酸塩を、1g(3.18mmol)のヒドロキシアセトアミジニル20.1及び0.45g(3.18mmol)の炭酸カリウムを含むクロロホルム40mlの攪拌された懸濁液に滴下する。混合物を一晩室温で攪拌する。その後、反応混合物をろ過し、ろ過液を3×100mlの水で洗浄する。その後、有機相を硫酸ナトリウム上で乾燥させて、減圧下で蒸発させる。得られる固形物残渣をエーテルで数回洗浄し、乾燥器中で乾燥させて、1.53g(87%)の白色粉末のフェニルカルバモイルオキシアミジニルを得る。
1H NMR(CDCl3, 100 MHz): 1.26(s, 16H, H3−H10);
1.51 (m, 4H, H2 and H11); 1.94 (s, 6H, 2H2');
3.13 (q, 4H, H1 and H12); 5.46 (t, 2H, 2NH); 7.0−7.52 (m,
10H, H aromatique); 8.61 (s, 2H, 2NHCO).
FT−IR: 1663 (C=N); 1717 (OCON); 3291 (NHCO); 3443 (NH).
ES+ SM: 457 (100%) [M+H+]; 299 [(M+2H+)/2.
【0150】
1,12−ビス(N,N’−メチルスルホニルオキシアセトアミジニル)ドデカン:20.12
0.52ml (6.68mmol)の塩化メチルスルフォニル(methylsulfonyl chloride)を含む5mlのクロロホルムを、氷槽で0〜5℃間に冷却した1g(3.18mmol)のヒドロキシアセトアミジニル20.1及び0.55ml(6.68mmol)のピリジンを含むクロロホルム30mlの攪拌された懸濁液に滴下する。混合物を、10〜15℃の間で4時間攪拌する。その後、反応混合物を3×100
mlの水で洗浄する。その後、有機相を硫酸ナトリウム上で乾燥させて、減圧下で蒸発させる。含油残渣を冷蔵庫中で一晩放置する。結果として得られる固形物を低温エーテルで粉砕し、エーテルで洗浄し、その後、分離し、乾燥させて、1.33g(89%)の白色粉末のメチルスルホニルオキシアセトアミジニルを得る。
1H NMR(CDCl3, 100 MHz): 1.25(s. 16H, H3−H10);
1.51 (m, 4H, H2 and H11); 1.91 (s, 6H, 2H2');
3.11 (q, 6H, 2H3'); 3.15 (q, 4H, H1−H12); 5.23
(t, 2H, 2NH).
FT−IR: 1637 (C=N); 3302 (NH).
ES+ SM: 471 (100%) [M+H+].
【0151】
(1,12−ビス(アセトアミジニル)ドデカン)−1,12−ビス−N,N’−リン酸ジエチル:20.13
0.97ml (6.68mmol)のクロロホスホン酸ジエチルを含む5mlのクロロホルムを、0~5℃までに冷却した、1g(3.18mmol)のヒドロキシアセトアミジニル20.1及び0.94ml(6.68mmol)のトリエチルアミン含むDMF
30mlの攪拌された懸濁液に滴下する。混合物を一晩攪拌する。反応混合物をろ過し、ろ過液を酢酸エチル100ml中で再溶解させる。有機相を、3×100mlの水で洗浄し、その後、硫酸ナトリウム上で乾燥させて、減圧下で蒸発させる。残渣を、100mlのクロロホルムで再溶解させ、3×100mlの水で再洗浄し、硫酸ナトリウム上で乾燥させて、減圧下で蒸発させる。残渣を、100mlのクロロホルムで再溶解させ、3×100mlの水で再洗浄し、硫酸ナトリウム上で乾燥させ、減圧下で蒸発させて、有色オイルの20.13を1.13g(61%)得る。
1H NMR(CDCl3, 100 MHz): 1.25(s. 16H, H3−H10);
1.33 (t, 12H, 4H4); 1.43 (m, 4H, H2 and H11);
1.87 (s, 6H, 2H2'); 3.08 (q, 4H, H1 and H12);
4.18 (quint, 8H, 4H3') 5.28 (t, 2H, 2NH).
FT−IR: 1634 (C=N); 3316 (NH).
【0152】
1,12−ビス(N,N’(3−メチル−1,2,4−oxadiazol−5(4H)−one)−3−yl)
ドデカン:20.14
0.50 ml (6.68mmol)のクロロギ酸メチルを含む 5mlのクロロホルムを、1g(3.18mmol)のヒドロキシアセトアミジニル20.1及び0.45g(3.18mmol)の炭酸カリウムを含むクロロホルム30mlの攪拌された懸濁液に滴下する。混合物を、室温で1時間攪拌し、その後、懸濁液をおよそ50℃まで45分間加熱する。反応混合物を冷却し、その後、ろ過し、3×100mlの水で洗浄する。その後、有機相を硫酸ナトリウム上で乾燥させて、減圧下で蒸発させる。残渣を、冷蔵庫の中で一晩放置する。結果として得られる固形物を最終的に低温のエーテル中で破砕し、エーテルで洗浄し、その後、分離し、乾燥して、白色粉末のオキサジアゾロン
0.92g (79%)を得る。
溶融点:53-54℃。
1H NMR(CDCl3, 100 MHz): 1.24(s. 16H, H3−H10);
1.64 (s, 4H, H2 and H11); 2.24 (s, 6H, 2H2');
3.52 (t, 4H, H1 and H12).
FT−IR: 1559 (C=N); 1752 (OCO).
【0153】
1,12−ビス(N, N’−2−ヒドロキシアセトアミジニル)ドデカン:26.0
a)ヒドロキシアセトニトリル又はグリコール酸ニトリル
22.73g (303mmol)のホルムアルデヒドのナトリウム(40%水溶液)を、0℃に冷却した、15g(306mmol)のシアン化ナトリウム水溶液(60ml)に滴下する。混合物を、この温度で30分間攪拌する。溶液のpHは、H2SO4
7.5N(45ml)で2に、かつNa2CO3で5に、連続的に調整する。硫酸ナトリウムは結局生成されるが、加わえられる水で再溶解され、その後、エーテル(4×200ml)で抽出する。有機相を無水硫酸ナトリウム上で乾燥させ、減圧下で蒸発させて、有色オイルのヒドロキシアセトニトリル
13.13g(76%)を得る。この精製されていないオイルをそのまま使用する。このオイルは、24時間後、室温で分解(変質)する。
1H NMR(CDCl3, 100 MHz): 2.30(s, 1H, OH); 4.34 (s, 2H, CH2).
FT−IR: 2259 (C=N); 3425 (OH).
【0154】
b)ヒドロキシアセトイミダート塩酸塩
HCLガスを、1時間半冷却し、20mlの無水エタノール、40mlの無水エーテル及び9g(158mmol)のヒドロキシアセトニトリルの溶液を通して発泡させる。この反応沈殿物を分離し、エーテルで数回洗浄し、その後、乾燥器中で乾燥させ、白色粉末のヒドロキシアセトイミダート塩酸塩19.52g
(89%)を得る。
1H NMR(DMSO−d6, 100 MHz): 1.80(t, 3H, CH3);
4.80 (s, 2H, CH2); 4.85 (q, 2H, CH2); 11.73 (sl, 2H, NH,
HCl).
FT−IR: 1645 (C=N); 3119 (OH); 3229 (NH, HCl).
【0155】
1,12−ビス(N, N’2−ヒドロキシアセトアミジニル)ドデカン:26.0
2g (10mmol)の1,12−ジアミノドデカンを、3.07g(22mmol)のエチルヒドロキシアセトイミダート塩酸塩を含むメタノール(50ml)の溶液に加える。反応混合物を室温で24時間攪拌する。その後、この溶液を減圧下で蒸発させる。その後、低温残渣を100mlの水に再溶解させ、その後、NaOH
0.5N でアルカリ性(塩基性)化冷却(bastified cold)させる。結果として得られる沈殿物を分離し、水で、アセトンで数回、それからエーテルで洗浄し、その後、分離し、乾燥後、白色粉末の
2-ヒドロキシアセトアミジニル26,0 2.55gを得る。
溶融点:99−100℃。
1H NMR(CD3OD, 250MHz): 1.00 (s, 16H, H3−H10);
1.27 (m, 4H, H2 and H11); 2.84 (t, 4H, H1 and
H12): 2.98 (s, 2H, 2 OH); 3.70 (s, 4H, 2H2')
FT−IR: 1605 (C=N); 3072 (OH); 3290 and 3388 (2NH)
ES+ SM: 315 [M+H+]
【0156】
1,12−ビス[N, N’−(2−イミノピロリジニル)]ドデカン:24.0
1.64g(5mmol)の1,12−ジブロモドデカンを、窒素下、90℃に加熱した2.21g(26mmol)のピロリジン−2−オンと0.23g(10mmol)のナトリウムの混合物に少量ずつ加える。その後、この反応混合物を攪拌下、120℃で4時間加熱する。周囲温度に冷却した後、水40mlを加え、40mlのDCMで抽出する。次いで、有機相を塩化ナトリウムの飽和水溶液で洗浄し、硫酸マグネシウムで乾燥し、その後、減圧下、蒸発させる。シリカカラム(溶離液DCM−エタノール,[18−1])で精製した後、エーテル−ヘキサン混合物で周囲温度にて共蒸発させて、1,12−ビス[N,
N’−(ピロリジン−2−オン−1−イル)]ドデカン0.655g、収率39%の生成物を得る。0.18ml(2mmol)のスルホニルクロリドイソシアン酸塩を含むクロロホルム3mlを、この生成物0.336g(1mmol)を含むクロロホルム3mlに窒素下、滴下する。反応混合物を77℃で6時間加熱する。4.5mlの水を加えた後、反応混合物の温度を95℃で4時間置く。その後、5mlの水を加え、溶液を2×5mlのDCMで洗浄する。次いで、水性相を1Nの水酸化カリウム水溶液で中性化し、固形物をろ過し、エーテルで洗浄し、その後、デシケーターで1時間30分乾燥して、生成物0.135g(40%)を得る。
融点:92−97℃
NMR1H(DMSO−d6,250MHz),δ(ppm):1.3(m,16H);1.5(m,4H);1.8(q,4H);2.3(t,4H);3.2(t,4H);3.3(t,4H);8.0(s,2H)
【0157】
本発明に係る化合物の薬理学的活性の研究
A.P.falciparumに対するin vivoの抗マラリア活性及びP.vinckeに感染させたマウスに対するin vitroの抗マラリア活性
表4及び5に、以下、本発明の化合物について得られたIC50値(μM)及びED50値(mg/kg)の結果を示す。
72
【0158】
【表4】
【0159】
【表5】
【0160】
IC50はin vitroでP.falciparumの成長を50%阻害する濃度であり(IC50の測定は、核酸に組み込んだ[3H]ヒポキサンチンがcell
viability indexとしての役割を果たすDesjardinsの方法に従って行った。)(Desjardins RE,Canfield CJ,Haynes
JD,Chulay JD.Quantitative assessment of antimalarial activity in vitro by a
semiautomated microdilution technique. Antimicrob. Agents Chemother. 1979; 16:
710−718)(図1)、ED50は4日間の抑制試験によりin vitroでP.vinckeの成長を50%阻害する有効濃度である (Peters
W, Portus JH, Robinson BL. The chemotherapy of rodent malaria. XXII. The value
of drug-resistant strains of P.berghei in screening for blood schizonticidal
activity. Ann. Trop. Med. Parasitol. 1975; 69: 155-17)。
TIは治療係数、TI=LD50(半慢性) / ED50;ip:腹腔内投与;po:経口投与を表す。
これらの結果は、本発明の化合物が良好な耐性及び良好な吸収性だけでなく、invitro及びin vivoで強力な抗マラリア活性及び抗バベシア病活性を有することを示している。
【0161】
B.マウスにおける薬力学的パラメーター
化合物6.0をマウスに腹腔内又は経口経路で投与した後の薬力学的パラメーターの結果を以下に示す。血清レベルで決定するため、バイオテストをex vivoで用いた、簡潔に言えば、医薬を動物に投与し、その後、繰り返し採取する。血清は56℃、30分間にて脱補体化する。次いで、活性な代謝物の含量は、[3H]ヒポキサンチンを用いるDesjardinsの方法に従い、P.falciparumに感染させた赤血球の懸濁液の存在下、異なる濃度(二分希釈)の各血清をインキュベートして決定する。結果はIS50で表し、これはP.falciparumの成長を50%阻害することができる血清濃度(活性化した代謝物質を含む)に相当する。
この値は、次にP.falciparumに感染した同じ懸濁液で活性化合物を直接試験する(動物を通すことなく)ことにより、また、そのIC50値(ng/ml)で決定することにより、血清濃度(通例ng/mlで表す)に変換する[血清総計=IC50](ng/ml)×100/IS50(%)]。
結果は時間の関数としてlog(医薬の血清総計)で表し、これにより血清コンパートメントへの分布に対する半減時間t1/2(d);血清コンパートメントの消失に対する半減時間t1/2(e);消失相での最初の血清濃度の推定値であるC0,AUC(血流中で循環する薬物量を示す);経口経路による吸収の程度を表す、経口経路による投与方法対腹腔内経路による投与方法[AUC(po)/AUC(ip)]における比較バイオアベイラビリティの評価が可能となる。
【0162】
6.0の薬力学
LD50/3に相当する17mg/kg及び300 mg/kgの用量の6.0を、腹腔内経路及び経口経路で投与した。化合物は10%DMSOに溶解させる。結果は表4に示す。半対数表記により、2つの投与経路に対する活性代謝物の主要な薬力学パラメーターを決定することができる。薬力学パラメーターは、C0=50ng/ml,t1/2=16時間,AUC=170ng.h/ml(300mg/kg経口投与後)である。
【0163】
21.0の薬力学
15mg/kg(LD50/3腹腔内)及び100 mg/kg(LD50/4経口)の用量の化合物21.0を、それぞれマウスに腹腔内経路及び経口経路で投与した。15mg/kgの腹腔内投与後、C0は24ng/ml、t1/2は約35時間となる。100
mg/kgの経口経路では、C0は16ng/ml、見かけのt1/2は36時間となる。
【0164】
C.化合物の抗バベシア病活性
化合物6.0,22.0,及び2.0については、また、Babesia divergens及びB.canisに対するそれらの活性をin vitroで評価した。いずれに対しても、化合物6.0,22.0,及び2.0は、顕著に活性がある(IC50<50nM)ことがわかった。これらの結果は、このタイプの化合物の強力な抗バベシア病活性を示している。
【図面の簡単な説明】
【0165】
【図1】図1は、本発明に係る化合物の薬理学的活性の研究を示す説明図である。
【特許請求の範囲】
【請求項1】
一般式(I)で表されることを特徴とする、抗寄生虫活性、特に抗マラリア活性を有する化合物。
但し、Xは式(II)の基であり、
Zは−(CH2)mの基であり、m=8−21,n=0又は1,及びY=R3であり、
R1及びR'1は、互いに同一又は異なり、H,アルキル,OH,O−アルキル,O−アリール,O−CO−アルキル,O− CO−アリール,OSO2−アルキル,OSO2−アリール,OSO2−ヘテロ環,O−CO−O(又はS又はNH)−アルキル,O−CO−O(又はS又はNH)−アリール,PO(O−アルキル又はO−アリール)2,CO−O−CH2−アリール,シクロアルキル,から選択され、
R2及びR'2は、互いに同一又は異なり、H,アルキル,CO−O−CH2−アリール,CO−O−アルキル,シクロアルキル,から選択され、
R3及びR'3は、互いに同一又は異なり、H,アルキル,アリール,CO−O−アルキル,CO−O−アリール,COO−CH(R)
−O−CO−アルキル,PO(O−アルキル又はO−アリール又はONa)2,CO−O− CH(R) −アリール,から選択され、RはH又はアルキルである、
あるいは、
R1及びR2,及び/又はR'1及びR'2,又はR2及びR3,及び/又はR'2及びR'3,は共に、それぞれが結合する1又は複数の窒素原子をもつモノヘテロ環を形成し、
あるいは、さらに、
R2及びR3及び/又はR'2及びR'3は、同一の置換基であり、窒素に二重結合しており、それぞれ、R1又はR'1と環化してヘテロ環を形成し、Raで置換される場合は、それは、H,アルキル,1,2又は3つのハロゲン元素で置換したアルキル,アリール,CO−O−アルキル(又はアリール),−CO−OH,−CO−NH2,−CN,−CO−NH−アルキル(又はアリール),−CO−N−(アルキル)2,窒素及び/又は酸素を有する−CO−ヘテロ環,NH(H又はアルキル),N(アルキル)2,窒素及び/又は酸素を有するヘテロ環,−O−アルキル(又はアリール),−O−CH2−アリール,CH2N[H,(H,アルキル),(ジアルキル),アリール],窒素及び/又は酸素を有する−CH2−ヘテロ環,CH2−CO−OH,から選択され、
あるいは、X=R4,Yは式(III)の基であり、
n及びZは上記したのと同様であり、
R1及びR'1は、互いに同一又は異なり、H,アルキル,OH,O−アルキル,O−アリール,O−CO−アルキル,O−CO−アリール,OSO2−アルキル,OSO2−アリール,OSO2−ヘテロ環,O−CO−O(又はS又はNH)−アルキル,O−CO−O(又はS又はNH)−アリール,PO(O−アルキル又はO−アリール)2,CO−O−CH2−アリール,シクロアルキル,から選択され、
R4及びR'4は、H,アルキル又はアリールであり、OH,O−アルキル,O−アリール,NH(H又はアルキル),窒素又は酸素ヘテロ環により代替可能で、また、
R2及びR'2は、互いに同一又は異なり、H,アルキル,CO−O−CH2−アリール,CO−O−アルキル,シクロアルキル,から選択され、
あるいは、
R1及びR4及び/又はR'1及びR'4は、共に−(CH2)pの基を形成し、pは1−5の整数、1又は複数の水素原子は低級アルキルに選択的に変更され、かつR2及びR'2はHを表し、又はR4及びR2及び/又はR'4及びR'2は、共に−(CH2)pの基を形成し、1又は複数の水素原子は低級アルキルに選択的に変更され、かつR1及びR'1はHを表す、及びこれらの化合物の薬理学的に許容できる塩である。
【請求項2】
式(IV)で表されることを特徴とする請求項1記載の化合物。
但し、n,Z,R1,R'1,R2,R'2,R3及びR'3は、請求項1で規定したのと同様である。
【請求項3】
式(V)で表されることを特徴とする請求項2記載の化合物。
但し、Z,R1,R'1,R2,R'2,R3及びR'3は、請求項1で規定したのと同様である。
【請求項4】
R1,R'1,R2,R'2,R3及びR'3は、互いに独立していることを特徴とする請求項3記載の化合物。
【請求項5】
R1及び(/又は)R'1は、前記したのと同様であるが、水素原子とは異なり、一方、R3及び/又はR'3,R2及び/又はR'2は、水素原子,R1,R2及びR3を表すことを特徴とする請求項4記載の化合物。
【請求項6】
R1及び/又はR'1,及びR2及び/又はR'2は、水素原子を表し、一方、R3及び/又はR'3は、前記したのと同様であるが、水素原子とは異なることを特徴とする請求項5記載の化合物。
【請求項7】
R1及びR2,及び/又はR'1及びR'2,又はR2及びR3,及び/又はR'2及びR'3,又はR1,R2及びR3,及び/又はR'1,
R'2及びR'3は、共にヘテロ環を形成することを特徴とする請求項3記載の化合物。
【請求項8】
R1及びR2並びにR'1及びR'2は、式(VI)で表されるヘテロ環を形成することを特徴とする請求項7記載の化合物。
【請求項9】
式(VII)で表されることを特徴とする請求項7記載の化合物。
【請求項10】
式(VI)において、R1及びR2及び/又はR'1及びR'2は、共に−O−CO−,O−SO−,−O−CS,S−CO又は−S−CS基を形成し、R3及びR'3は、水素原子を表すことを特徴とする請求項8記載の化合物。
【請求項11】
R1及びR2,及び/又はR'1及びR'2は、選択的に、分枝したアルキレン基を表し、R3及び/又はR'3は、−CO−O−アルキル(又はアリール),−CO−O−CH2−アリール,CO−O−CH(アルキル)−O−CO−アルキル,PO(O−アルキル又はアリール)2,アルキル又はHを表すことを特徴とする請求項8記載の化合物。
【請求項12】
R1及び/又はR'1は、水素原子を表し、R2及びR3,及び/又はR'2及び/又はR'3は、−(CH2)p−の基を表すことを特徴とする請求項9記載の化合物。
【請求項13】
R2及びR3及び/又はR'2及びR'3は、同一の置換基を形成し、それぞれR1又はR'1と共に、式(VIII)のビス−オキサジアゾールを形成することを特徴とする請求項2記載の化合物。
但し、Raは、前記規定したのと同様である。
【請求項14】
式(IX)で表されることを特徴とする請求項1記載の化合物。
【請求項15】
Z=−(CH2)m及びn=0であり、式(X)で表されることを特徴とする請求項14記載の化合物。
【請求項16】
置換基は互いに独立していることを特徴とする請求項15記載の化合物。
【請求項17】
R1及びR4及び/又はR'1及び/又はR'4は、前記したとおりであり、R2及びR'2は水素原子を表すことを特徴とする請求項16記載の化合物。
【請求項18】
R1及びR2及び/又はR'1及び/又はR'2は、共にオキシカルボニル(−OCO−)鎖を表し、R4及びR'4は前記したとおりであることを特徴とする請求項16記載の化合物。
【請求項19】
R1及びR4及び/又はR'1及び/又はR'4は、共に−(CH2)p−の基を表し、pは3−5の整数であり、R2及びR'2は、Hを表すことを特徴とする請求項16記載の化合物。
【請求項20】
R1及びR'1は、Hを表し、R4及びR2及び/又はR'4及びR'2は、共に−(CH2)p−の基を表し、pは3−5の整数であり、及び一またはそれ以上の水素原子が低級アルキルに置き換えられることができることを特徴とする請求項16記載の化合物。
【請求項21】
式(XI)で表されることを特徴とする請求項16記載の化合物。
【請求項22】
一般式(V)のビスアミジン化合物の2相溶媒中での反応を含むことを特徴とする一般式(V)のカルバメート及びN−ホスホリル誘導体の製造方法。
但し、一般式(V)において、R3及びR'3=HとCl− R3(又はR'3)誘導体であり、R3及びR'3は前記したのと同様であり、Hとは異なる。
【請求項23】
R1及びR'1=OHである一般式(X)のビスアミドキシムと適当な試薬の塩基性溶媒中での反応を含むことを特徴とする一般式(X)のアミドキシム誘導体の製造方法。
【請求項24】
前記した一般式(VI)のグループa2及び(VIII)のグループa4の化合物を得るために、適当な試薬の存在下、前記した一般式(V)のグループa1で表されるアミドキシム又はアミドキシム誘導体の分子内環化を行うことを特徴とする請求項23記載の方法。
【請求項25】
請求項1〜20のいずれか1項記載の少なくとも1つの化合物の有効量を、不活性な薬剤学的担体と結合して含有することを特徴とする医薬組成物。
【請求項26】
経口経路又は注射経路あるいは又、直腸経路で投与することができる請求項25記載の医薬組成物。
【請求項27】
請求項1〜21のいずれか1項記載の化合物の有効量を含むことを特徴とする、感染症、特にマラリア治療のための請求項25又は26記載の組成物。
【請求項28】
抗寄生虫病薬、特にマラリア治療薬を製造するための請求項1〜21のいずれか1項記載の化合物の使用。
【請求項29】
抗寄生虫病薬、特にマラリア治療薬及びバベシア病治療薬を製造するための請求項1〜21のいずれか1項記載の少なくとも1つの化合物の使用。
【請求項1】
一般式(I)で表されることを特徴とする、抗寄生虫活性、特に抗マラリア活性を有する化合物。
但し、Xは式(II)の基であり、
Zは−(CH2)mの基であり、m=8−21,n=0又は1,及びY=R3であり、
R1及びR'1は、互いに同一又は異なり、H,アルキル,OH,O−アルキル,O−アリール,O−CO−アルキル,O− CO−アリール,OSO2−アルキル,OSO2−アリール,OSO2−ヘテロ環,O−CO−O(又はS又はNH)−アルキル,O−CO−O(又はS又はNH)−アリール,PO(O−アルキル又はO−アリール)2,CO−O−CH2−アリール,シクロアルキル,から選択され、
R2及びR'2は、互いに同一又は異なり、H,アルキル,CO−O−CH2−アリール,CO−O−アルキル,シクロアルキル,から選択され、
R3及びR'3は、互いに同一又は異なり、H,アルキル,アリール,CO−O−アルキル,CO−O−アリール,COO−CH(R)
−O−CO−アルキル,PO(O−アルキル又はO−アリール又はONa)2,CO−O− CH(R) −アリール,から選択され、RはH又はアルキルである、
あるいは、
R1及びR2,及び/又はR'1及びR'2,又はR2及びR3,及び/又はR'2及びR'3,は共に、それぞれが結合する1又は複数の窒素原子をもつモノヘテロ環を形成し、
あるいは、さらに、
R2及びR3及び/又はR'2及びR'3は、同一の置換基であり、窒素に二重結合しており、それぞれ、R1又はR'1と環化してヘテロ環を形成し、Raで置換される場合は、それは、H,アルキル,1,2又は3つのハロゲン元素で置換したアルキル,アリール,CO−O−アルキル(又はアリール),−CO−OH,−CO−NH2,−CN,−CO−NH−アルキル(又はアリール),−CO−N−(アルキル)2,窒素及び/又は酸素を有する−CO−ヘテロ環,NH(H又はアルキル),N(アルキル)2,窒素及び/又は酸素を有するヘテロ環,−O−アルキル(又はアリール),−O−CH2−アリール,CH2N[H,(H,アルキル),(ジアルキル),アリール],窒素及び/又は酸素を有する−CH2−ヘテロ環,CH2−CO−OH,から選択され、
あるいは、X=R4,Yは式(III)の基であり、
n及びZは上記したのと同様であり、
R1及びR'1は、互いに同一又は異なり、H,アルキル,OH,O−アルキル,O−アリール,O−CO−アルキル,O−CO−アリール,OSO2−アルキル,OSO2−アリール,OSO2−ヘテロ環,O−CO−O(又はS又はNH)−アルキル,O−CO−O(又はS又はNH)−アリール,PO(O−アルキル又はO−アリール)2,CO−O−CH2−アリール,シクロアルキル,から選択され、
R4及びR'4は、H,アルキル又はアリールであり、OH,O−アルキル,O−アリール,NH(H又はアルキル),窒素又は酸素ヘテロ環により代替可能で、また、
R2及びR'2は、互いに同一又は異なり、H,アルキル,CO−O−CH2−アリール,CO−O−アルキル,シクロアルキル,から選択され、
あるいは、
R1及びR4及び/又はR'1及びR'4は、共に−(CH2)pの基を形成し、pは1−5の整数、1又は複数の水素原子は低級アルキルに選択的に変更され、かつR2及びR'2はHを表し、又はR4及びR2及び/又はR'4及びR'2は、共に−(CH2)pの基を形成し、1又は複数の水素原子は低級アルキルに選択的に変更され、かつR1及びR'1はHを表す、及びこれらの化合物の薬理学的に許容できる塩である。
【請求項2】
式(IV)で表されることを特徴とする請求項1記載の化合物。
但し、n,Z,R1,R'1,R2,R'2,R3及びR'3は、請求項1で規定したのと同様である。
【請求項3】
式(V)で表されることを特徴とする請求項2記載の化合物。
但し、Z,R1,R'1,R2,R'2,R3及びR'3は、請求項1で規定したのと同様である。
【請求項4】
R1,R'1,R2,R'2,R3及びR'3は、互いに独立していることを特徴とする請求項3記載の化合物。
【請求項5】
R1及び(/又は)R'1は、前記したのと同様であるが、水素原子とは異なり、一方、R3及び/又はR'3,R2及び/又はR'2は、水素原子,R1,R2及びR3を表すことを特徴とする請求項4記載の化合物。
【請求項6】
R1及び/又はR'1,及びR2及び/又はR'2は、水素原子を表し、一方、R3及び/又はR'3は、前記したのと同様であるが、水素原子とは異なることを特徴とする請求項5記載の化合物。
【請求項7】
R1及びR2,及び/又はR'1及びR'2,又はR2及びR3,及び/又はR'2及びR'3,又はR1,R2及びR3,及び/又はR'1,
R'2及びR'3は、共にヘテロ環を形成することを特徴とする請求項3記載の化合物。
【請求項8】
R1及びR2並びにR'1及びR'2は、式(VI)で表されるヘテロ環を形成することを特徴とする請求項7記載の化合物。
【請求項9】
式(VII)で表されることを特徴とする請求項7記載の化合物。
【請求項10】
式(VI)において、R1及びR2及び/又はR'1及びR'2は、共に−O−CO−,O−SO−,−O−CS,S−CO又は−S−CS基を形成し、R3及びR'3は、水素原子を表すことを特徴とする請求項8記載の化合物。
【請求項11】
R1及びR2,及び/又はR'1及びR'2は、選択的に、分枝したアルキレン基を表し、R3及び/又はR'3は、−CO−O−アルキル(又はアリール),−CO−O−CH2−アリール,CO−O−CH(アルキル)−O−CO−アルキル,PO(O−アルキル又はアリール)2,アルキル又はHを表すことを特徴とする請求項8記載の化合物。
【請求項12】
R1及び/又はR'1は、水素原子を表し、R2及びR3,及び/又はR'2及び/又はR'3は、−(CH2)p−の基を表すことを特徴とする請求項9記載の化合物。
【請求項13】
R2及びR3及び/又はR'2及びR'3は、同一の置換基を形成し、それぞれR1又はR'1と共に、式(VIII)のビス−オキサジアゾールを形成することを特徴とする請求項2記載の化合物。
但し、Raは、前記規定したのと同様である。
【請求項14】
式(IX)で表されることを特徴とする請求項1記載の化合物。
【請求項15】
Z=−(CH2)m及びn=0であり、式(X)で表されることを特徴とする請求項14記載の化合物。
【請求項16】
置換基は互いに独立していることを特徴とする請求項15記載の化合物。
【請求項17】
R1及びR4及び/又はR'1及び/又はR'4は、前記したとおりであり、R2及びR'2は水素原子を表すことを特徴とする請求項16記載の化合物。
【請求項18】
R1及びR2及び/又はR'1及び/又はR'2は、共にオキシカルボニル(−OCO−)鎖を表し、R4及びR'4は前記したとおりであることを特徴とする請求項16記載の化合物。
【請求項19】
R1及びR4及び/又はR'1及び/又はR'4は、共に−(CH2)p−の基を表し、pは3−5の整数であり、R2及びR'2は、Hを表すことを特徴とする請求項16記載の化合物。
【請求項20】
R1及びR'1は、Hを表し、R4及びR2及び/又はR'4及びR'2は、共に−(CH2)p−の基を表し、pは3−5の整数であり、及び一またはそれ以上の水素原子が低級アルキルに置き換えられることができることを特徴とする請求項16記載の化合物。
【請求項21】
式(XI)で表されることを特徴とする請求項16記載の化合物。
【請求項22】
一般式(V)のビスアミジン化合物の2相溶媒中での反応を含むことを特徴とする一般式(V)のカルバメート及びN−ホスホリル誘導体の製造方法。
但し、一般式(V)において、R3及びR'3=HとCl− R3(又はR'3)誘導体であり、R3及びR'3は前記したのと同様であり、Hとは異なる。
【請求項23】
R1及びR'1=OHである一般式(X)のビスアミドキシムと適当な試薬の塩基性溶媒中での反応を含むことを特徴とする一般式(X)のアミドキシム誘導体の製造方法。
【請求項24】
前記した一般式(VI)のグループa2及び(VIII)のグループa4の化合物を得るために、適当な試薬の存在下、前記した一般式(V)のグループa1で表されるアミドキシム又はアミドキシム誘導体の分子内環化を行うことを特徴とする請求項23記載の方法。
【請求項25】
請求項1〜20のいずれか1項記載の少なくとも1つの化合物の有効量を、不活性な薬剤学的担体と結合して含有することを特徴とする医薬組成物。
【請求項26】
経口経路又は注射経路あるいは又、直腸経路で投与することができる請求項25記載の医薬組成物。
【請求項27】
請求項1〜21のいずれか1項記載の化合物の有効量を含むことを特徴とする、感染症、特にマラリア治療のための請求項25又は26記載の組成物。
【請求項28】
抗寄生虫病薬、特にマラリア治療薬を製造するための請求項1〜21のいずれか1項記載の化合物の使用。
【請求項29】
抗寄生虫病薬、特にマラリア治療薬及びバベシア病治療薬を製造するための請求項1〜21のいずれか1項記載の少なくとも1つの化合物の使用。
【図1】

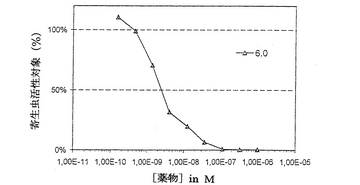
【公表番号】特表2006−504652(P2006−504652A)
【公表日】平成18年2月9日(2006.2.9)
【国際特許分類】
【出願番号】特願2004−522264(P2004−522264)
【出願日】平成15年7月18日(2003.7.18)
【国際出願番号】PCT/FR2003/002283
【国際公開番号】WO2004/009068
【国際公開日】平成16年1月29日(2004.1.29)
【出願人】(502071218)サーントル ナシオナル ドゥ ラ ルシェルシェ シャーンティフィク(セーエンヌエールエス) (8)
【Fターム(参考)】
【公表日】平成18年2月9日(2006.2.9)
【国際特許分類】
【出願日】平成15年7月18日(2003.7.18)
【国際出願番号】PCT/FR2003/002283
【国際公開番号】WO2004/009068
【国際公開日】平成16年1月29日(2004.1.29)
【出願人】(502071218)サーントル ナシオナル ドゥ ラ ルシェルシェ シャーンティフィク(セーエンヌエールエス) (8)
【Fターム(参考)】
[ Back to top ]