
抗癌剤としての複素環式化合物および使用
チアゾール環で置換された縮合二環式ヘテロ芳香環系を有する新規な化合物を開示する。該化合物は、いろいろなタイプの癌細胞の成長を阻害するので、癌を処置するのに有用である。これら化合物の効力は、それらが癌細胞の成長および/または移動の強力な阻害剤であることを示す、細胞成長/移動を監視するシステムで示されている。更に、本発明の化合物は、腫瘍の成長を in vivo で止めるおよび腫瘍のサイズを in vivo で減少させることが分かった。これら化合物を含む組成物およびこれら化合物および組成物を癌の処置に用いる方法を開示する。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願のクロス・リファレンス
本出願は、2010年2月19日出願の米国仮特許出願第61/306,416号および2010年3月16日出願の同第61/314,510号に優先権を主張するが、それらの開示は、本明細書中にそのまま援用される。
【0002】
本発明の分野は、複素環式化合物、医薬組成物および方法であって、特に、癌および関連疾患の処置および予防のための組成物および方法に関するものである。
【背景技術】
【0003】
癌は、先進諸国において二番目に大きい死因である。したがって、癌は、依然として、人類への最も重要な未だかなえられていない医学的挑戦の一つである。腫瘍を処置するには、外科手術、放射線、化学療法またはこれらアプローチのいずれかの組み合わせを含めた多数の選択肢が利用可能である。これらの中で、化学療法は、全てのタイプの癌に、具体的には、手術不能のものまたは転移特性を有するものに広く用いられる。異なったヒト癌の生存率の改善のために臨床で用いられているいろいろな化学療法用化合物にもかかわらず、化学療法は、概して、治癒的ではなく、疾患進行を遅らせるだけである。
【0004】
一般的に、腫瘍およびそれらの転移は、腫瘍細胞が多剤耐性の能力を発現するにつれて、化学療法に不応性になる。いくつかの場合、腫瘍は、本質的に、いくつかのクラスの化学療法薬に耐性である。他の場合、化学療法薬に対して獲得される耐性は、化学療法的介入中に発現する。したがって、異なったクラスの腫瘍を処置する場合、利用可能な化学療法用化合物の効力への有意の限界が、依然として存在する。更に、腫瘍の化学療法的処置に用いられる多くの細胞障害性剤または細胞分裂抑制性剤は、重症の副作用を有して、幾人かの患者に化学療法の停止をもたらす。
【発明の概要】
【発明が解決しようとする課題】
【0005】
したがって、癌を処置するための新しい化学療法薬への要求が、依然として存在する。
【課題を解決するための手段】
【0006】
本発明は、アニリン置換チアゾール環に連結した二環式ヘテロアリール環系を有する新規な化合物、これら化合物を含有する医薬組成物、およびこれら化合物および組成物を使用する方法に関する。本明細書中に記載の化合物は、抗腫瘍、抗癌、抗炎症、抗感染および抗増殖の活性を示す。それらは、多くの異なったタイプの癌を含めた癌細胞への選択的毒性によって示されるように、癌の処置に特に有用である。本発明は、更に、腫瘍、癌、および感染性および/または増殖性疾患を処置するのに用いることができるこのような化合物を含有する医薬組成物に関する。
【0007】
本発明の内容の一つの側面において、新規な複素環式化合物は、式Iまたは式II:
【0008】
【化1】

【0009】
[式中、Zは、次の置換されたフェニル環または複素環式環(これら構造中の破線が交差している結合は、式Iまたは式II中のNHへのZ基の結合点である):
【0010】
【化2】
【0011】
より選択されるが、これに制限されるわけではない]
に従う構造を有するか、またはその薬学的に許容しうる塩若しくはアシル化プロドラッグである。
【0012】
本明細書中に記載のそれらに似ている化合物は、癌を処置するためのこのような化合物の使用を含めて、報告された(WO2009/023402号)。しかしながら、本明細書中に記載の新規な化合物は、当該技術分野において知られている化合物よりも予想外に優れている。
【0013】
式I〜IIの化合物は、中性化合物として、または無機および有機対イオンを含むそれらの薬学的に適する塩として用いることができる。それら塩には、ハロゲン化物(Cl−、Br−、I−)、ニトレート、メシレート、p−トルエンスルホネート/トシレート、オキサレート、シトレート、マレート、マレエート、タータレート、フマレート、ホルメート、アセテートおよびこれらクラスの類似の陰イオンなどがあるがこれに制限されるわけではない薬学的に許容しうる対イオンを含む酸付加塩が含まれる。
【0014】
上記の複素環式化合物には、それら化合物自体、更には、適用可能ならば、それらの塩およびそれらのプロドラッグが含まれる。このような塩は、例えば、化合物上の陽電荷置換基(例えば、プロトン化されている複素環式環または芳香環上のアミノ基)と薬学的に適する陰イオンとの間で、または式Iまたは式IIの化合物の塩基性複素環式基への酸の付加によって形成することができる。適する陰イオンには、塩化物、臭化物、ヨウ化物、スルフェート、ニトレート、ホスフェート、シトレート、ベンゼンスルホネート、メタンスルホネート、トリフルオロアセテート、マレイエートおよびアセテートが含まれるが、これに制限されるわけではない。同様に、化合物上の陰電荷置換基(例えば、複素環式環または芳香環上のカルボン酸基)は、薬学的に許容しうる陽イオンと塩を形成することができる。適する陽イオンの非制限例は、ナトリウムイオン、カリウムイオン、マグネシウムイオン、カルシウムイオン、およびテトラメチルアンモニウムイオン、テトラブチルアンモニウムイオンなどの有機アンモニウムイオンおよび他の有機陽イオンである。
【0015】
適するプロドラッグは、式Iまたは式IIのNH基のアシル化によって形成することができる。代表的なプロドラッグには、NHがNC(O)−R*へアシル化された式Iまたは式IIの化合物が含まれるが、この場合、C(O)R*は、ホルミル、アセチル、クロロアセチル、トリクロロアセチル、トリフルオロアセチル等などの置換されていてよいアシル基である。他のプロドラッグには、NHがスルホニル化されて、例えば、N−SO2−R’を形成した式Iの化合物が含まれるが、この場合、R’は、例えば、メチル、フルオロメタンスルホニルまたはトリフルオロメタンスルホニル(trifluormethanesulfonyl)でありうる。
【0016】
本発明の化合物は、光学異性体、幾何異性体、互変異性体、およびアトロプ異性体を含めた回転異性体を含めた異性体として存在してよい。本発明は、式I〜IIの化合物のこのような異性体各々およびそれらの混合物を包含する。化合物が、例えば、キラル中心を有する場合、本発明は、各々の個々の異性体、更には、等量の双方の異性体を有するラセミ混合物を含めた、いろいろな量の双方の異性体の混合物を包含する。本発明の化合物は、ビアリールであるので、それらは、ビアリール結合についての回転異性体として存在することもでき、そして各々の異性体並びにこのような異性体の混合物は、本発明の範囲内に包含される。
【0017】
本発明の化合物およびそれら化合物を含む組成物は、望ましくない細胞増殖を特徴とする状態を処置するのに有用である。具体的には、それら化合物は、肉腫、類表皮癌、線維肉腫、子宮頸癌、胃癌、皮膚癌、白血病、リンパ腫、肺癌、非小細胞性肺癌、結腸癌、CNS癌、黒色腫、卵巣癌、腎癌、前立腺癌、乳癌、肝癌、頭頸部癌、膵癌および他のタイプの増殖性疾患を処置するのに有用である。
【図面の簡単な説明】
【0018】
【図1】図1は、免疫不全ヌードマウスの皮下植込みによって異種移植片移植されたMKN45ヒト胃腸癌へのCOMPOUND Oの in vivo 抗腫瘍効力を示す。
【図2】図2は、免疫不全ヌードマウスの皮下植込みによって異種移植片移植されたH460ヒト非小細胞性肺癌へのCOMPOUND Oの in vivo 抗腫瘍効力を示す。
【図3】図3は、免疫不全ヌードマウスの皮下植込みによって異種移植片移植されたA549ヒト非小細胞性肺癌へのCOMPOUND Oの in vivo 抗腫瘍効力を示す。
【図4A】図4Aは、Real-Time Cell Electronic Sensing(Roche 製のxCELLigenceシステムと同じである、ACEA Biosciences 製のRT−CESシステム)で決定される、異なった濃度のCOMPOUND OへのA549ヒト非小細胞性肺癌細胞系の動的応答プロフィールを示す。
【図4B】図4Bは、Real-Time Cell Electronic Sensing(Roche 製のxCELLigenceシステムと同じである、ACEA Biosciences 製のRT−CESシステム)で決定される、異なった濃度のパクリタキセルへのA549ヒト非小細胞性肺癌細胞系の動的応答プロフィールを示す。
【図4C】図4Cは、Real-Time Cell Electronic Sensing(Roche 製のxCELLigenceシステムと同じである、ACEA Biosciences 製のRT−CESシステム)で決定される、異なった濃度のビンクリスチンへのA549ヒト非小細胞性肺癌細胞系の動的応答プロフィールを示す。
【図5】図5は、Real-Time Cell Electronic Sensing(RT−CES)システム(ACEA Biosciences)と同じであるxCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのH596ヒト腺扁平上皮肺癌細胞系の動的応答プロフィールを示す。
【図6】図6は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのH292ヒト肺性肺癌細胞系の動的応答プロフィールを示す。
【図7】図7は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのH460ヒト大細胞肺癌細胞系の動的応答プロフィールを示す。
【図8】図8は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのH1993ヒト非小細胞性肺癌細胞系の動的応答プロフィールを示す。
【図9】図9は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのH1838ヒト非小細胞性肺癌細胞系の動的応答プロフィールを示す。
【図10】図10は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのH2347ヒト非小細胞性肺癌細胞系の動的応答プロフィールを示す。
【図11】図11は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのSW620ヒト結腸癌細胞系の動的応答プロフィールを示す。
【図12】図12は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのGTL16ヒト胃癌細胞系(MKN45ヒト胃癌細胞系に由来した)の動的応答プロフィールを示す。
【図13】図13は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのHT29ヒト結腸癌細胞系の動的応答プロフィールを示す。
【図14】図14は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのA172ヒト脳癌細胞系の動的応答プロフィールを示す。
【図15】図15は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのU138MGヒト脳癌細胞系の動的応答プロフィールを示す。
【図16】図16は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのU118MGヒト脳癌細胞系の動的応答プロフィールを示す。
【図17】図17は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのSW1088ヒト脳癌細胞系の動的応答プロフィールを示す。
【図18】図18は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのHT1080ヒト結合組織癌細胞系の動的応答プロフィールを示す。
【図19】図19は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのBxPC3ヒト膵癌細胞系の動的応答プロフィールを示す。
【図20】図20は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのHepG2ヒト肝癌細胞系の動的応答プロフィールを示す。
【図21】図21は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのSKOV3ヒト卵巣癌細胞系の動的応答プロフィールを示す。
【図22】図22は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのMCF7ヒト乳癌細胞系の動的応答プロフィールを示す。
【図23】図23は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのMDA−MB−231ヒト乳癌細胞系の動的応答プロフィールを示す。
【図24A】図24Aは、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのKBヒト子宮頸癌細胞系の動的応答プロフィールを示す。
【図24B】図24Bは、xCELLigenceシステム(Roche)で決定される、異なった濃度のパクリタキセルへのKBヒト子宮頸癌細胞系の動的応答プロフィールを示す。
【図24C】図24Cは、xCELLigenceシステム(Roche)で決定される、異なった濃度のビンブラスチンへのKBヒト子宮頸癌細胞系の動的応答プロフィールを示す。
【図24D】図24Dは、xCELLigenceシステム(Roche)で決定される、異なった濃度のコルヒチンへのKBヒト子宮頸癌細胞系の動的応答プロフィールを示す。
【図24E】図24Eは、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのKB200ヒト子宮頸癌細胞系(多剤耐性MDR遺伝子を発現する)の動的応答プロフィールを示す。
【図24F】図24Fは、xCELLigenceシステム(Roche)で決定される、異なった濃度のパクリタキセルへのKB200ヒト子宮頸癌細胞系(多剤耐性MDR遺伝子を発現する)の動的応答プロフィールを示す。
【図24G】図24Gは、xCELLigenceシステム(Roche)で決定される、異なった濃度のビンブラスチンへのKB200ヒト子宮頸癌細胞系(多剤耐性MDR遺伝子を発現する)の動的応答プロフィールを示す。
【図24H】図24Hは、xCELLigenceシステム(Roche)で決定される、異なった濃度のコルヒチンへのKB200ヒト子宮頸癌細胞系(多剤耐性MDR遺伝子を発現する)の動的応答プロフィールを示す。
【図25】図25は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOPMOUND OへのNIH3T3正常組織細胞系の動的応答プロフィールを示す。
【図26A】図26Aは、MAPの多いチューブリンを用いた in vitro の微小管集合(assembly)へのCOPMOUND Oの作用を示す。
【図26B】図26Bは、COMPOUND O、パクリタキセルおよびビンクリスチンの20時間処置によるA549細胞中の微小管体制の阻害作用を示す。
【図26C】図26Cは、スピンカラム検定を用いたコルヒチン結合部位を経るチューブリンとCOMPOUND Oの相互作用を示す。
【図27A】図27Aは、37nM COMPOUND Oおよび37nMパクリタキセルの24時間、48時間および72時間処置によって誘発される、A549ヒト肺癌細胞のアポトーシスを示す。
【図27B】図27Bは、37nM COMPOUND Oおよび37nMパクリタキセルの72時間処置によって誘発される、A549、H596およびH292ヒト肺癌細胞のアポトーシスを示す。
【図28A】図28Aは、有糸分裂指数で定量される、パクリタキセルおよびCOMPOUND OによるA549ヒト肺癌細胞の有糸分裂阻止の程度を示す。
【図28B】図28Bは、COMPOUND Oで24時間処置後のA549ヒト肺癌細胞の細胞周期分布を示す。
【発明を実施するための形態】
【0019】
開示の明快さのために、制限としてではなく、本発明の選択された態様について次の説明を与える。便宜上、次の小区分に分けるが、それら区分は、本発明の範囲を制限すると解釈されるべきではない。
【0020】
A.定義
それ以外に定義されない限り、本明細書中で用いられる専門用語および科学用語は全て、本発明が属する当該技術分野の業者によって一般的に理解されるのと同じ意味を有する。本明細書中に挙げられる特許、出願、公開出願および他の公報は全て、そのまま援用される。この部分に示されている定義が、本明細書中に援用される特許、出願、公開出願および他の公報に示されている定義に反するまたはそれ以外にそれと一致しない場合、この部分に示されている定義は、本明細書中に援用される定義に優先する。
【0021】
本明細書中で用いられる「ある(a または an)」は、「少なくとも一つ」または「一つまたはそれを超える」を意味する。
本明細書中で用いられる「アルキル」という用語は、直鎖状、分枝状または環状の立体配置の飽和炭化水素基を意味し、具体的に考えられるアルキル基には、低級アルキル基(すなわち、10個またはそれ未満の炭素原子を有するもの)が含まれる。代表的なアルキル基は、メチル、エチル、プロピル、イソプロピル、ブチル、sec−ブチル、第三級ブチル、ペンチル、イソペンチル、ヘキシル等である。本明細書中で用いられる「アルケニル」という用語は、上に定義のような且つ少なくとも一つの二重結合を有するアルキルを意味する。したがって、具体的に考えられるアルケニル基には、2〜10個の炭素原子を有する直鎖状、分枝状または環状のアルケニル基(例えば、エテニル、プロペニル、ブテニル、ペンテニル等)が含まれる。同様に、本明細書中で用いられる「アルキニル」という用語は、上に定義のような且つ少なくとも一つの三重結合を有するアルキルまたはアルケニルを意味する。特に考えられるアルキニルには、2〜10個の全炭素原子を有する直鎖状、分枝状または環状のアルキン(例えば、エチニル、プロピニル、ブチニル等)が含まれる。
【0022】
本明細書中で用いられる「シクロアルキル」という用語は、好ましくは、3〜8個の炭素原子を包含する環状アルカン(すなわち、炭化水素の炭素原子鎖が環を形成しているもの)を意味する。したがって、代表的なシクロアルカンには、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチルおよびシクロオクチルが含まれる。シクロアルキルは、更に、一つまたは二つの二重結合を包含し、それは、「シクロアルケニル」基を形成する。シクロアルキル基は、更に、アルキル、アルケニル、アルキニル、ハロおよび他の一般的な基で置換されてもいる。
【0023】
本明細書中で用いられる「アリール」または「芳香族部分」という用語は、芳香族環系を意味し、それは、1個またはそれを超える非炭素原子を更に包含してよい。したがって、考えられるアリール基には、(例えば、フェニル、ナフチル等)およびピリジルが含まれる。更に考えられるアリール基は、一つまたは二つの5員または6員アリール基または複素環式基と縮合(すなわち、第一芳香環上の2原子と共有結合)していてよく、したがって、「縮合アリール」または「縮合芳香族」と称される。
【0024】
本明細書中で更に用いられる「複素環」、「シクロヘテロアルキル」および「複素環式部分」という用語は、本明細書中において同じ意味に用いられ、複数の原子が複数の共有結合によって環を形成しているいずれかの化合物を意味し、この場合、その環は、炭素原子以外の少なくとも一つの原子を包含する。具体的に考えられる複素環式塩基には、非炭素原子として窒素、硫黄または酸素を含む5員および6員環(例えば、イミダゾール、ピロール、トリアゾール、ジヒドロピリミジン、インドール、ピリジン、チアゾール、テトラゾール等)が含まれる。更に考えられる複素環は、一つまたは二つの環または複素環に縮合(すなわち、第一複素環式環上の2個の原子と共有結合)していてよく、したがって、本明細書中で用いられる「縮合複素環」または「縮合複素環式塩基」または「縮合複素環式部分」と称される。
【0025】
本明細書中で用いられる「ハロゲン」という用語は、フッ素、塩素、臭素およびヨウ素を意味する。
上に定義の基はいずれも、一つまたはそれを超える置換基で更に置換されていてよいし、それが順次、更に置換されていてよいということは更に理解されるはずである。例えば、アルキルまたはアリール中の水素原子は、アミノ、ハロまたは他の基で置換されている。
【0026】
本明細書中で用いられる「置換された」という用語は、H原子の別の原子または基での代用を意味する。アルキル基、アルケニル基およびアルキニル基は、しばしば、このような置換が化学的に意味を成す程度に置換されている。典型的な置換基には、ハロ、=O、=N−CN、=N−OR、=NR、OR、NR2、SR、SO2R、SO2NR2、NRSO2R、NRCONR2、NRCOOR、NRCOR、CN、COOR、CONR2、OOCR、CORおよびNO2が含まれるが、これに制限されるわけではなく、ここにおいて、Rは、各々独立して、H、C1−C8アルキル、C2−C8ヘテロアルキル、C1−C8アシル、C2−C8ヘテロアシル、C2−C8アルケニル、C2−C8ヘテロアルケニル、C2−C8アルキニル、C2−C8ヘテロアルキニル、C6−C10アリールまたはC5−C10ヘテロアリールであり、そしてRは各々、ハロ、=O、=N−CN、=N−OR’、=NR’、OR’、NR’2、SR’、SO2R’、SO2NR’2、NR’SO2R’、NR’CONR’2、NR’COOR’、NR’COR’、CN、COOR’、CONR’2、OOCR’、COR’およびNO2で置換されていてよく、ここにおいて、R’は、各々独立して、H、C1−C8アルキル、C2−C8ヘテロアルキル、C1−C8アシル、C2−C8ヘテロアシル、C6−C10アリールまたはC5−C10ヘテロアリールである。アルキル基、アルケニル基およびアルキニル基は、C1−C8アシル、C2−C8ヘテロアシル、C6−C10アリールまたはC5−C10ヘテロアリールで置換されていることもありうるし、それらは各々、具体的な基について適当である置換基で置換されうる。
【0027】
「ヘテロアルキル」、「ヘテロアルケニル」および「ヘテロアルキニル」等は、該当するヒドロカルビル(アルキル、アルケニルおよびアルキニル)基と同様に定義されるが、「ヘテロ」という用語は、1〜3個のO、SまたはNヘテロ原子またはその組み合わせを主鎖残基中に含有する基を意味する;したがって、該当するアルキル基、アルケニル基またはアルキニル基の少なくとも一つの炭素原子は、指定のヘテロ原子の一つで代用されて、ヘテロアルキル基、ヘテロアルケニル基またはヘテロアルキニル基を形成する。アルキル基、アルケニル基およびアルキニル基のヘテロ形に典型的な且つ好ましいサイズは、概して、該当するヒドロカルビル基の場合と同じであり、そしてヘテロ形上に存在してよい置換基は、ヒドロカルビル基について上に記載されたものと同じである。化学的安定性の理由で、特に断らない限り、このような基は、ニトロ基またはスルホニル基の場合のようにNまたはS上にオキソ基が存在する場合を除いて、2個以下の隣接するヘテロ原子を包含するということも理解される。
【0028】
本明細書中で用いられる「アルキル」が、シクロアルキル基およびシクロアルキルアルキル基を包含する場合、「シクロアルキル」という用語は、本明細書中において、環炭素原子によって連結している炭素環式非芳香族基を記載するのに用いることができ、そして「シクロアルキルアルキル」は、アルキルリンカーによって分子に連結している炭素環式非芳香族基を記載するのに用いることができる。同様に、「ヘテロシクリル」は、少なくとも一つのヘテロ原子を環員として含有する且つCまたはNであってよい環原子によって分子に連結している非芳香族環状基を記載するのに用いることができる;そして「ヘテロシクリルアルキル」は、リンカーによって別の分子に連結しているこのような基を記載するのに用いることができる。シクロアルキル基、シクロアルキルアルキル基、ヘテロシクリル基およびヘテロシクリルアルキル基に適するサイズおよび置換基は、アルキル基について上に記載されたものと同じである。本明細書中で用いられるように、これら用語は、その環が芳香族でない限り、一つまたは二つの二重結合を含有する環も包含する。
【0029】
本明細書中で用いられる「アシル」は、カルボニル炭素原子の二つの利用可能な原子価位置の一つに付いたアルキル、アルケニル、アルキニル、アリールまたはアリールアルキルのラジカルを含む基を包含し、そしてヘテロアシルは、カルボニル炭素以外の少なくとも一つの炭素が、N、OおよびSより選択されるヘテロ原子で代用された該当する基を意味する。したがって、ヘテロアシルには、例えば、−C(=O)ORおよび−C(=O)NR2、更には、−C(=O)−ヘテロアリールが含まれる。
【0030】
概して、いずれのアルキル基、アルケニル基、アルキニル基、アシル基またはアリール基またはアリールアルキル基も、または置換基中にあるこれら基の一つのいずれのヘテロ形も、それ自体、追加の置換基で置換されていてもよい。これら置換基の性状は、それら置換基がそれ以外に記載されていない場合、本来の置換基自体に関して挙げられたものと似ている。したがって、例えば、R7の態様がアルキルである場合、このアルキルは、R7の態様として挙げられた残りの置換基で置換されていてもよく、この場合、これは、化学的意味を成し、そしてこれは、アルキル自体に与えられたサイズ限界を害することはない;例えば、アルキルまたはアルケニルで置換されたアルキルは、これら態様についての炭素原子の上限を簡単に広げると考えられ、包含されない。しかしながら、アリール、アミノ、アルコキシ、=O等で置換されたアルキルは、本発明の範囲内に包含されると考えられ、これら置換基の原子は、アルキル、アルケニル等の基を記載するのに用いられる記載されている数に入れられない。置換基の数が指定されない場合、このようなアルキル基、アルケニル基、アルキニル基、アシル基またはアリール基は各々、その利用可能な原子価にしたがった数の置換基で置換されていよい;具体的には、これら基はいずれも、例えば、その利用可能な原子価のいずれかまたは全てにおいてフッ素原子で置換されていよい。
【0031】
具体的に考えられる官能基には、求核基(例えば、−NH2、−OH、−SH、−NC等)、求電子基(例えば、C(O)OR、C(X)OH等)、極性基(例えば、−OH)、無極性基(例えば、複素環、アリール、アルキル、アルケニル、アルキニル等)、イオン性基(例えば、−NH3+)およびハロゲン(例えば、−F、−Cl)、NHCOR、NHCONH2、OCH2COOH、OCH2CONH2、OCH2CONHR、NHCH2COOH、NHCH2CONH2、NHSO2R、OCH2−複素環、PO3H、SO3H、アミノ酸、およびそれらの化学的に妥当な組み合わせ全てが含まれる。更に、「置換された」という用語は、置換の多重度も包含し、そして多数の置換基が開示されているまたは請求の範囲に記載されている場合、置換された化合物は、開示されたまたは請求の範囲に記載された一つまたはそれを超える置換基部分で独立して置換されうる。更に、本明細書中で用いられる「一置換/二置換/三置換/四置換された」という用語は、芳香族または複素環式部分上に置換されたまたは芳香族または複素環式部分に縮合した上記の1個または2個または3個または4個の官能基を意味し、ここにおいて、このような多官能基は、芳香族または複素環式部分のオルト位またはパラ位またはメタ位のいずれかの組み合わせに置換されている。
【0032】
更に、本明細書中に与えられている式はいずれも、このような化合物の水和物、溶媒和化合物および多形およびそれらの組み合わせを示すものである。
本明細書中で用いられる「薬学的に許容しうる」または「薬理学的に許容しうる」とは、生物学的にまたはそれ以外に望ましくないことがない物質を意味し、例えば、その物質は、有意の望ましくない生物学的作用を全く引き起こすことなくまたはそれが入っている組成物の他の成分のいずれとも有害に相互作用することなく、患者に投与される医薬組成物中に包含されていてよい。薬学的に許容しうる担体または賦形剤は、好ましくは、毒性試験および製造試験の必要な基準を満たしているおよび/または米国食品医薬品局(U.S. Food and Drug administration)が作成したInactive Ingredient Guide に包含される。「薬学的に許容しうる塩」は、遊離(非塩)化合物の生物活性を少なくとも若干保持する且つ薬物または医薬品として個体に投与することができるそれら塩である。薬学的に許容しうる塩は、イオン相互作用を表し、共有結合ではない。それとして、N−オキシドは、塩とは考えられない。このような塩には、例えば、(1)塩酸、臭化水素酸、硫酸、硝酸、リン酸等などの無機酸で形成される;または酢酸、シュウ酸、プロピオン酸、コハク酸、マレイン酸、酒石酸等などの有機酸で形成される酸付加塩;(2)親化合物中に存在する酸性プロトンが、金属イオン、例えば、アルカリ金属イオン、アルカリ土類金属イオンまたはアルミニウムイオンで代用されているか;または有機塩基で配位している場合に形成される塩が含まれる。許容しうる有機塩基には、エタノールアミン、ジエタノールアミン、トリエタノールアミン等が含まれる。許容しうる無機塩基には、水酸化アルミニウム、水酸化カルシウム、水酸化カリウム、炭酸ナトリウム、水酸化ナトリウム等が含まれる。薬学的に許容しうる塩の追加の例には、Berge et al, Pharmaceutical Salts, J. Pharm. Sci. 66(1):1-19, 1977 に挙げられたものが含まれる。薬学的に許容しうる塩は、製造プロセス中に現場で、または遊離酸または塩基の形の本発明の精製済み化合物を適する有機または無機の塩基または酸とそれぞれ別々に反応させ、そしてこのようにして形成された塩を引き続きの精製中に単離することによって製造することができる。薬学的に許容しうる塩の意味が、その溶媒付加形または結晶形、具体的には、溶媒和化合物または多形を包含するということは理解されるはずである。溶媒和化合物は、化学量論的量かまたは非化学量論的量の溶媒を含有し、そしてしばしば、結晶化プロセス中に形成される。水和物は、溶媒が水である場合に形成され、またはアルコラートは、溶媒がアルコールである場合に形成される。多形には、ある化合物の同じ元素組成を有する異なった結晶充填配置が含まれる。多形は、通常、異なったX線回折図形、赤外スペクトル、融点、密度、硬度、結晶形状、光学的および電気的性質、安定性および溶解性を有する。再結晶溶媒、結晶化速度および貯蔵温度などのいろいろな因子は、単結晶形を優勢にさせることがありうる。
【0033】
本明細書中で用いられる「賦形剤」という用語は、本発明の化合物を活性成分として含有する錠剤などの薬物または医薬品の製造に用いることができる反応不活性または不活性(inert or inactive)物質を意味する。賦形剤という用語には、結合剤、崩壊剤、コーティング、圧縮/封入助剤、クリームまたはローション剤、滑沢剤、非経口投与用溶液、チュアブル錠用材料、甘味剤または着香剤、懸濁化/ゲル化剤、または湿式造粒剤として用いられるいずれの物質も制限されることなく含めたいろいろな物質を包含することができる。結合剤には、例えば、カルボマー、ポビドン、キサンタンガム等が含まれ;コーティングには、例えば、酢酸フタル酸セルロース、エチルセルロース、ゲランガム(gellan gum)、マルトデキストリン、腸溶コーティング等が含まれ;圧縮/封入助剤には、例えば、炭酸カルシウム、デキストロース、フルクトースdc(dc=「直接圧縮性(directly compressible)」、ハチミツdc、ラクトース(無水物または一水和物;場合により、アスパルテーム、セルロースまたは微結晶性セルロースとの組み合わせで)、デンプンdc、スクロース等が含まれ;崩壊剤には、例えば、クロスカルメロースナトリウム、ゲランガム、ナトリウムデンプングリコラート等が含まれ;クリーム剤またはローション剤には、例えば、マルトデキストリン、カラゲナン等が含まれ;滑沢剤には、例えば、ステアリン酸マグネシウム、ステアリン酸、ステアリルフマル酸ナトリウム等が含まれ;チュアブル錠用材料には、例えば、デキストロース、フルクトースdc、ラクトース(一水和物、場合により、アスパルテームまたはセルロースとの組み合わせで)等が含まれ;懸濁化/ゲル化剤には、例えば、カラゲナン、ナトリウムデンプングリコラート、キサンタンガム等が含まれ;甘味剤には、例えば、アスパルテーム、デキストロース、フルクトースdc、ソルビトール、スクロースdc等が含まれ;そして湿式造粒剤には、例えば、炭酸カルシウム、マルトデキストリン、微結晶性セルロース等が含まれる。
【0034】
本明細書中で用いられるように、そして特に断らない限り、「対象」という用語は、本明細書中において、霊長類(例えば、ヒト)、雌牛、ヒツジ、ヤギ、ウマ、イヌ、ネコ、ウサギ、ラット、マウス等が含まれるがこれに制限されるわけではない哺乳動物などの動物を包含すると定義される。特定の態様において、対象はヒトである。
【0035】
B.複素環式化合物およびそれらの医薬組成物
B.1.代表する化合物:
本発明のいくつかの代表する化合物を、表1に挙げる。
【0036】
【表1A】
【0037】
【表1B】
【0038】
B.2.代表的な合成方法
代表的な化合物を、スキームIおよびスキームIIに示される経路によって合成した。当該技術分野により知られている方法は、当業者が用い且つ修飾して、利用可能な出発物質から本発明の化合物を製造することができる。いくつか有用な方法は、公開PCT出願WO2009/023402号に開示されている。本発明の範囲内の化合物を製造するのに有用でありうる追加の合成方法は、例えば、Hayakawa et al., Biorg. Med. Chem. Vol. 15, 403-12 (2007); Ermolat’ev, et al., J. Comb. Chem. Vol. 8, 659-63 (2006); Carballares, et al., Tetrahedron Lett. vol. 48, 2041-45 (2007); および Rupert, et al., Biorg. Med. Chem. Lett., vol. 13, 347-50 (2003) に開示されている。
【0039】
下のスキームは、本明細書中に与えられている化合物の製造のための代表的な合成方法を与える。当業者は、類似の方法を用いて、本明細書中に与えられている化合物を製造することができるということを理解するであろう。言い換えると、当業者は、試薬、保護基、反応条件および反応配列順序への適する調整を用いて、所望の態様を製造することができるということを理解するであろう。それら反応は、製造される物質の量に適合するように規模を上向きまたは下向きにすることができる。
【0040】
本明細書中に与えられている化合物を製造するための具体的なスキームを、下に示す。詳細な反応条件を、いろいろな具体的実施例について本明細書中の下に与える。当業者は、次のスキームを、適当な試薬、保護基、条件、出発物質または反応配列順序で修飾して、本明細書中に与えられている他の態様の製造に適合させることができるということを理解するであろう。
【0041】
本明細書中に開示の参考文献は全て、そのまま援用される。
化合物名は、ChemDraw Ultra 10.0で得た;そして実施例中で用いられる中間体および試薬の名称も、ChemDraw Ultra 10.0で得た。
【0042】
2a.式Iの化合物の一般的な合成スキーム/方法:
スキームI
【0043】
【化3】
【0044】
チアゾール−2−アミン1を、EtOH中の3−クロロペンタン−2,4−ジオン2で、還流温度において処理して、環化生成物3を生じ、それを、HOAc中の臭素で更に臭素化して、α−ブロモケトン4を生じた。置換されたアニリン5を、アセトン中のイソチオシアン酸ベンゾイル6と反応させて、N−(フェニルカルバモチオイル)ベンズアミド7を生じ、それを、80℃において5%水性NaOHで加水分解して、置換フェニルチオ尿素8を生じた。次に、二つの不可欠な中間体7および8を、EtOH中で加熱して、チアゾール9(式I)をHBr塩として高収率で形成させたが、それは、in vitro および in vivo 研究用に他の薬学的に許容しうる塩(例えば、HCl塩)または遊離塩基形へ変換しうると考えられる。
【0045】
本発明の化合物の製造に必要とされる式5の置換アニリンは、商業的に入手可能であるまたは既知の方法によって合成することができる。
2b.式IIの化合物の一般的な合成スキーム/方法:
スキームII
【0046】
【化4】
【0047】
ピリミジン−2−アミン10を、EtOH中の3−クロロペンタン−2,4−ジオン2で、還流温度において処理して、環化生成物11を生じ、それを、HOAc中の臭素で更に臭素化して、α−ブロモケトン12を生じた。置換アニリン5を、アセトン中のイソチオシアン酸ベンゾイル6と反応させて、N−(フェニルカルバモチオイル)ベンズアミド7を生じ、それを、80℃において5%水性NaOHで加水分解して、置換フェニルチオ尿素8を生じた。次に、二つの不可欠な中間体12および8を、EtOH中で加熱して、チアゾール13(式II)をHBr塩として高収率で形成させたが、それは、in vitro および in vivo 研究用に他の薬学的に許容しうる塩(例えば、HCl塩)または遊離塩基形へ変換しうると考えられる。
【0048】
本発明の化合物の製造に必要とされる式5の置換アニリンは、商業的に入手可能であるまたは既知の方法によって合成することができる。
【実施例】
【0049】
B.3.実施例:
以下の実施例は、本発明を詳しく説明するために与えるが、制限するためではない。
スキームIおよびスキームIIに記載の合成方法/スキームにしたがって、表1の化合物を合成した。いくつか代表的な合成を、本明細書中に与える。
【0050】
実施例B(1):2−(4−エトキシフェニルアミノ)−4−(2−メチルイミダゾ[1,2−a]ピリミジン−3−イル)チアゾール一臭化水素酸塩(a)の合成:
化合物aの合成を、スキームIIIに示す:
スキームIII
【0051】
【化5】
【0052】
1−(2−メチルイミダゾール[1,2−a]ピリミジン−3−イル)エタノン(4)の合成。
3−クロロ−2,5−ペンタンジオン(2)(106mL,119g,887mmol,1.2eq)を、650mLの無水エタノール中に溶解させた。2−アミノピリミジン(1)(71.5gx97%=69.36g,729mmol)を、上の撹拌溶液に加えた。得られた混合物を、100〜105℃の油浴温度で40時間還流した。黒色反応混合物を冷却し、減圧下で濃縮した。残留物を、飽和重炭酸ナトリウム溶液(約500mL)で少量ずつ処理し、フラスコを波立たさせて十分に混合した。その混合物を、ジクロロメタン(x6)で抽出した。抽出物を、重炭酸ナトリウム溶液およびブラインで洗浄した。有機相を乾燥させ、濃縮した。残留物を、シリカゲルカラム(7x30cm)上のフラッシュクロマトグラフィーにより、n−ヘキサン−酢酸エチル(3:1、2:1、1:1、1:2および0:1)、そして次にジクロロメタン−メタノール(30:1、20:1、10:1および5:1)を用いた勾配溶離で精製した。生成物画分を集め(TLC、Rf0.36、100%酢酸エチル)、濃縮して、淡黒色固体を与えた。生成物が入っている他の画分を集め、再度同様に再精製した。27.84g(21.8%)の最終生成物を得た。生成物の一部分を、少量のアセトニトリルから再結晶させて、赤〜淡褐色結晶4、m.p.255.6〜256.6℃を生じた。
【0053】
1H NMR (CDCl3) δ 2.65 (s, 3H, 3-COCH3), 2.86 (s, 3H, 2-CH3), 7.04-7.12 (m, 1H, 6-H), 7.70-7.74 (m, 1H), 9.96-10.00 (m, 1H)。
2−ブロモ−1−(2−メチルイミダゾ[1,2−a]ピリミジン−3−イル)エタノン一臭化水素酸塩(5)の合成(臭素化)。
【0054】
1−(2−メチルイミダゾール[1,2−a]ピリミジン−3−イル)エタノン(4)(1.75g,10mmol)を、20mLの氷酢酸中に、フラスコを静かに暖めることによって溶解させた後、それを室温に冷却した。4mLの酢酸中の臭素(0.6mL,1.85g,11.5mmol,1.15eq)の溶液を、上の撹拌反応混合物に室温で30分間を超えて徐々に加えた。[注意:臭素は、極めて腐食性である。臭素の取り扱いは、十分に換気されたヒュームフード中で極めて注意深く行う必要がある。長手袋または二重手袋を操作に必要とする。皮膚上へ跳ねかけるまたは蒸気を吸うのを絶対に避けられたい]。若干の固体が、添加を終える前に析出した。反応混合物を、100〜110℃の油浴温度で3時間撹拌後、室温で一晩撹拌した。固体を濾過し、そしてアセトンで数回洗浄し、アセトン洗浄の途中に時々、無水エタノールで洗浄した。淡褐色固体を、少量のエタノールを含有するアセトンで吸収させ、その混合物を、室温で5時間を超えて撹拌した。固体を濾過し、その固体を上述のように洗浄した(もう一回吸収させ、より大きい規模用に洗浄サイクルを提示する)。真空下で乾燥後、2.43g(72.5%)の淡褐色粉末固体生成物5を、一臭化水素酸塩として得た。250℃より上で分解。
【0055】
TLC、Rf0.42(100%酢酸エチル)。
1HNMR (DMSO-d6) δ 2.82 (s, 3H, 2-CH3), 4.83 (s, 2H, CH2Br), 7.40-7.50 (m, 1H), 8.80-8.90 (m, 1H), 9.82-9.90 (m, 1H)。
【0056】
1−ベンゾイル−3−[4−(エトキシフェニル)]チオ尿素(9)の合成。
[参考文献:ARKIVOC 2003, 434-442; Bioorg. Med. Chem. 2000, 2663]。塩化ベンゾイル(6)(14.0mL,16.95g,120mmol)を、100mLのアセトン中のチオシアン酸アンモニウム(10.26 g,135mmol,1.125eq)の撹拌溶液に室温で滴加した。若干の白色固体を析出した。反応混合物を、加熱して5分間還流した(約65〜70℃の油温度)。このようにして得られたイソチオシアン酸ベンゾイル(7)を、精製することなく、次の工程に直接的に用いた。25mLのアセトン中の4−エトキシアニリン(8)(17.0mL,18.1g,132mmol,1.1eq)の溶液を、油浴(65〜70℃)中に静置している上の撹拌反応混合物に徐々に加えた。添加は、発熱反応を考慮して、極めて徐々に約1時間行う必要がある。多量の白色固体が析出した。反応混合物を、手動で波立たせ、そして更に、5分間還流した。冷却した反応混合物を、氷水中に注いだ。固体を濾過し、水で3回洗浄した。その固体を、エタノール(約1.6L)から再結晶させて、所望の生成物9を淡黄色の長針状結晶、36g収量(99%)、m.p.151.0〜153.5℃として与えた。
【0057】
(p−エトキシフェニル)チオ尿素(10)の合成。
水酸化ナトリウム水溶液(1M,60mL,60mmol,1.2eq)を、350mLのエタノール中の1−ベンゾイル−3−[4−(エトキシフェニル)]チオ尿素(9)(16.5g,55mmol)の撹拌混合物に加えた。反応混合物を、1時間還流し、冷却し、濃縮した。その白色固体を、水(約200mL)で処理した。固体を濾過し、水で洗浄した。粗製結晶性生成物を、エタノールから再結晶させ、濾過し、真空下で乾燥させて、7.66g(71.0%)の所望の生成物10、m.p.176.5〜178.5℃を与えた。
【0058】
TLC、Rf0.45(n−ヘキサン−酢酸:1:1)。
1HNMR (DMSO-d6) δ 1.31 (t, 3H, J = 6.8Hz), 4.00 (q, 2H, J = 6.8Hz), 6.80-6.90 (m, 2H), 7.15-7.25 (m, 2H), 9.50 (s, 1H, NH)。
【0059】
2−(4−エトキシフェニルアミノ)−4−(2−メチルイミダゾ[1,2−a]ピリミジン−3−イル)チアゾール一臭化水素酸塩(a)の合成(チアゾール環への環化)。
140mLの無水エタノール中の2−ブロモ−1−(2−メチルイミダゾ[1,2−a]ピリミジン−3−イル)エタノン一臭化水素酸塩(5)(2.43g,7.25mmol)および(p−エトキシフェニル)チオ尿素(10)(1.40g,7.1mmol)の混合物を、撹拌しながら15時間還流した(約105℃の油浴温度)。次に、それを室温で6時間または一晩撹拌した。固体を濾過し、アセトンで洗浄した。粗製軟質結晶を、アセトン−エタノール(3:1)で吸収させ、室温で6時間を超えてまたは一晩撹拌した。固体を濾過し、上のように洗浄した。粗生成物を、アセトン−エタノール(3:1)で吸収させ、室温で6時間を超えて撹拌した。粗生成物を、濾過し、洗浄し、メタノールから再結晶させた。そのメタノール溶液を、熱い内に濾過して、黒色微粉を除去後、加熱して溶液とした。黄色結晶を濾過し、洗浄した。それを、メタノールから更に2回再結晶させ、真空下で乾燥させて、所望の生成物11を長い軟質黄色針状結晶、1.55g収量(50.5%)として与え、240℃より上で分解した。
【0060】
TLC Rf0.32(ジクロロメタン−メタノール:20:1);Rf0.46(ジクロロメタン−メタノール含有:20:1、1%水酸化アンモニウム水溶液を含有);Rf0.30(100%酢酸エチルX2)。
【0061】
HPLC純度:99%。
1HNMR (DMSO-d6) δ 1.32 (t, 3H, J = 6.8Hz), 2.67 (s, 3H), 4.00 (q, 2H, J = 6.8Hz), 6.90-6.95 (m, 2H), 7.35 (s, 1H), 7.49-7.52 (m, 2H), 7.61-7.67 (m, 1H), 8.94-8.97 (m, 1H), 9.57 (d, 1H, J = 6.8Hz), 10.28 (s, 1H, NH)。
【0062】
ESI−MS,m/z352(M+1)+。
実施例B(2):2−(4−ブロモフェニル)アミノ−4−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)−チアゾール一臭化水素酸塩(u)の合成。
【0063】
化合物uの合成を、スキームIVに示す。
スキームIV
【0064】
【化6】
【0065】
(p−ブロモフェニル)チオ尿素(14)の合成:p−ブロモアニリンをp−エトキシルアニリンの代わりに用いる、化合物10の合成と類似の方法。
1−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)エタノン(17)の合成。2−アミノチアゾールを、無水エタノールから再結晶させ、濾過し、そして乾燥後に使用した。180mLの無水エタノール中の2−アミノチアゾール(16)(20.9g,202.4mmoL)および3−クロロ−2,5−ペンタンジオン(2)(33.7g,97%,242.9mmol,1.2eq)の溶液を、油浴中で72時間還流した。黒色反応混合物を、冷却し、減圧下で濃縮した。残留物を、飽和重炭酸ナトリウム溶液で少量ずつ処理後、ジクロロメタンで抽出した。有機相を乾燥させ、濃縮した。残留物を、シリカゲルカラム上のフラッシュクロマトグラフィーにより、ジクロロメタン−メタノール(80:1)を用いて精製した。生成物画分を集め(TLC、Rf0.60中性形;Rf=0.5塩形、40:1のジクロロメタン−メタノール)、濃縮して、白色固体生成物17を、11.7%収率、1.6gの中性形および3.2gの塩形で与えた。
【0066】
1H NMR (CDCl3) δ 2.55 (s, 3H, 5-COCH3), 2.70 (s, 3H, 6-CH3), 6.78 (d, 1H, J = 4.8 Hz), 8.39 (d, 1H, J = 4.8 Hz)。
2−ブロモ−1−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)エタノン臭化水素酸塩(18)の合成。1−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)エタノン(17)(0.54g,3.0mmol)を、7mLの氷酢酸中に溶解させた。3mLの氷酢酸中の臭素(0.18mL,0.56g,3.5mmol)の溶液を、上の撹拌溶液に30分間で徐々に加えた。若干の黄色固体が現れた。その混合物を、加熱して撹拌しながら3時間還流後、室温で一晩撹拌した。固体を濾過し、アセトンで3回洗浄し、そして各々の洗浄に3〜5時間の撹拌を必要とした。固体を濾過し、真空下で乾燥させて、所望の生成物18である0.73g(71.6%)の白色固体を与えた。
【0067】
2−(4−ブロモフェニル)アミノ−4−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)−チアゾール一臭化水素酸塩(u)の合成。10mLの無水エタノール中の2−ブロモ−1−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)エタノン臭化水素酸塩(18)(0.73g,2.0mmol)および(p−ブロモフェニル)チオ尿素(14)(0.50g,2.0mmol)の混合物を、撹拌しながら20時間還流後、室温に冷却した。固体を濾過した。その粗製白色固体生成物を、メタノールから再結晶させた。メタノール溶液を、まだ熱い内に濾過して、可能性のある微粉を除去後、加熱して溶液とした。それを、メタノールから更に2回再結晶させ、真空下で乾燥させて、所望の生成物uを、白色固体、0.34g収量(36%)、HPLC純度98.49%、mp>250℃として与えた。
【0068】
1HNMR (CD3OD) δ 2.68 (s, 3H), 7.45 (s, 1H), 7.18 (s, 1H), 7.18-7.47 (m, 2H), 7.54-7.66 (m, 2H), 7.66 (d, 1H, J = 4.4Hz), 8.94 (d, 1H, J = 4.4Hz)。
実施例B(3):2−(4−エチルフェニル)アミノ−4−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)−チアゾール一臭化水素酸塩(o)の合成。
【0069】
化合物(o)の合成は、スキームIVに示している。
(p−ブロモフェニル)チオ尿素(21)の合成:p−エチルアニリンをp−エトキシルアニリンの代わりに用いる、化合物10の合成と類似の方法。
【0070】
2−(4−エチルフェニル)アミノ−4−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)−チアゾール一臭化水素酸塩(o)の合成。40mLの無水エタノール中の2−ブロモ−1−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)エタノン臭化水素酸塩(18)(20g,91%,53.5mmol)および(p−エチルフェニル)チオ尿素(10)(10.1g,56.2mmol)の混合物を、撹拌しながら3時間還流後、室温に冷却した。固体を濾過し、エタノールで洗浄した。粗製白色固体生成物を、メタノールから再結晶させた。メタノール溶液を、まだ熱い内に濾過して、可能性のある微粉を除去後、加熱して溶液とした。それを、メタノールからもう1回再結晶させ、真空下で乾燥させて、所望の生成物(o)を白色固体として与えた。このHBr塩を、遊離塩基へ変換後、HCl塩へ変換し、それを、50%エタノール/水から再結晶させて、オフホワイト結晶生成物(14.8g,73.4%)を生じた。
【0071】
HPLC純度99%、mp=181〜183℃。
1HNMR (CD3OD) δ 1.11 (t, 3H, J = 7.6 Hz), 2.52 (s, 3H), 2.58 (m, 2H), 7.11 (d, 2H, J = 8Hz), 7.22 (s, 1H), 7.52 (d, 2H, J = 8.4Hz), 7.75 (d, 1H, J = 4.4Hz), 8.42 (d, 1H, J = 4.4Hz)。
【0072】
ESI−MSm/z341.5(M+1)+。
式Iまたは式IIの化合物(HBr塩)からの遊離塩基化合物の製造。
HBr塩を、メタノール中に懸濁させ、過剰量の重炭酸ナトリウムを、懸濁した化合物塩が完全に溶解するまで激しく撹拌しながら加えた。過剰量の無機塩を濾去した。溶液を濃縮し、そして残留物を、メタノールまたはエタノールまたはエタノール/水またはいずれか他の一つまたは複数の有機溶媒または溶媒混合物から再結晶させて、結晶質を遊離塩基化合物として与えた。
【0073】
異なった塩の製造。得られた遊離塩基化合物および1.1当量の選択された酸。その溶液を濃縮し、そして残留物を、メタノールまたはエタノールまたはエタノール/水またはいずれか他の一つまたは複数の有機溶媒または溶媒混合物から再結晶させて、上記のような選択された陰イオンを含む所望の塩の結晶質/多形物質を与えた。
【0074】
記載の化合物の遊離塩基または塩の可能性のある結晶形/多形は全て、本発明に包含される。
B.3.追加の活性剤
本明細書中の化合物は、本発明の方法および組成物において、他の薬理学的に活性な化合物(「追加の活性剤」)と組み合わせることができる。特定の組み合わせは、特定のタイプの癌、および望ましくない血管新生に関連したまたはそれを特徴とする特定の疾患および状態の処置において共力的に働くと考えられる。免疫調節性化合物も、特定の二次活性剤に関連した副作用を軽減するように働くことができるし、そしていくつかの二次活性剤は、免疫調節性化合物に関連した副作用を軽減するのに用いることができる。
【0075】
一つまたはそれを超える活性成分または活性剤は、本発明の方法および組成物において一緒に用いることができる。追加の活性剤は、高分子(例えば、タンパク質)または低分子(例えば、合成の無機分子または有機金属分子または有機分子)でありうる。
【0076】
高分子活性剤の例には、造血増殖因子、サイトカイン、および単クローン性および多クローン性抗体が含まれるが、これに制限されるわけではない。典型的な高分子活性剤は、天然に存在するまたは人工的に作られたタンパク質などの生体分子である。本発明において特に有用であるタンパク質には、造血前駆細胞および免疫学的に活性な生産性細胞(poietic cell)の in vitro または in vivo の生存および/または増殖を刺激するタンパク質が含まれる。その他は、in vitro または in vivo の細胞における拘束された(committed)赤血球前駆体の分裂および分化を刺激する。具体的なタンパク質には、IL−2(リコンビナントIL−II(「rIL2」)およびカナリア痘IL−2を含めた)、IL−10、IL−12およびIL−18などのインターロイキン;インターフェロンα−2a、インターフェロンα−2b、インターフェロンα−n1、インターフェロンα−n3、インターフェロンβ−Iaおよびインターフェロンγ−Ibなどのインターフェロン;GM−CFおよびGM−CSF;およびEPOが含まれるが、これに制限されるわけではない。
【0077】
本発明の方法および組成物において用いることができる具体的なタンパク質には、米国において Neupogen.RTM.(Amgen, Thousand Oaks, Calif.)という商品名で販売されているフィルグラスティム;米国において Leukine.RTM.(Immunex, Seattle, Wash.)という商品名で販売されているサルグラモスチン;および米国において Epogen.RTM.(Amgen, Thousand Oaks, Calif.)という商品名で販売されているリコンビナントEPOが含まれるが、これに制限されるわけではない。
【0078】
GM−CSFのリコンビナントおよび突然変異形は、米国特許第5,391,485号;同第5,393,870号;および同第5,229,496号に記載のように製造することができる;それらは全て、本明細書中に援用される。G−CSFのリコンビナントおよび突然変異形は、米国特許第4,810,643号;同第4,999,291号;同第5,528,823号;および同第5,580,755号に記載のように製造することができる;それらは全て、本明細書中に援用される。
【0079】
本発明は、自然のままの、天然に存在するおよびリコンビナントのタンパク質の使用を包含する。本発明は、更に、天然に存在するタンパク質の突然変異体および誘導体(例えば、修飾された形)であって、それらが基づいているタンパク質の少なくともいくつかの薬理活性を in vivo で示すものを包含する。突然変異体の例には、それらタンパク質の天然に存在する形の該当する残基とは異なる一つまたはそれを超えるアミノ残基を有するタンパク質が含まれるが、これに制限されるわけではない。「突然変異体」という用語によって更に包含されるのは、それら天然に存在する形(例えば、非グリコシル化形)に普通に存在する炭水化物部分を欠いているタンパク質が含まれる。誘導体の例には、ペグ化誘導体;およびIgG1またはIgG3をタンパク質または目的のタンパク質の活性部分に融合することによって形成されるタンパク質などの融合タンパク質が含まれるが、これに制限されるわけではない。例えば、Penichet, M. L. and Morrison, S. L., J. Immunol. Methods 248:91-101 (2001) を参照されたい。
【0080】
本発明の化合物との組み合わせで用いることができる抗体には、単クローン性抗体および多クローン性抗体が含まれる。抗体の例には、トラスツズマブ(Herceptin.RTM.)、リツキシマブ(rituximab)(Rituxan.RTM.)、ベバシズマブ(bevacizumab)(Avastin.TM.)、ペルツズマブ(pertuzumab)(Omnitarg.TM.)、トシツモマブ(tositumomab)(Bexxar.RTM.)、エドレコロマブ(edrecolomab)(Panorex.RTM.)およびG250が含まれるが、これに制限されるわけではない。本発明の化合物は、更に、抗TNF−α抗体と一緒にすることができるまたはそれと組み合わせて用いることができる。
【0081】
高分子活性剤は、抗癌ワクチンの形で投与することができる。例えば、IL−2、G−CSFおよびGM−CSFなどのサイトカインを分泌するまたは分泌を引き起こすワクチンは、本発明の方法、医薬組成物およびキットに用いることができる。例えば、Emens, L. A., et al., Curr. Opinion Mol. Ther. 3(1):77-84 (2001) を参照されたい。
【0082】
本発明の一つの態様において、高分子活性剤は、免疫調節性化合物の投与に関連した副作用を減少させる、排除するまたは予防する。具体的な免疫調節性化合物および処置されている(begin treated)疾患または障害に依存して、副作用には、嗜眠状態および傾眠、めまい感および起立性低血圧、好中球減少症、好中球減少症に起因する感染、増加したHIVウイルス量、徐脈、スティーブンズ−ジョンソン症候群および中毒性表皮壊死症、および発作(例えば、大発作痙攣)が含まれうるが、これに制限されるわけではない。具体的な副作用は、好中球減少症である。
【0083】
低分子である追加の活性剤も、免疫調節性化合物の投与に関連した副作用を軽減するのに用いることができる。低分子活性剤の例には、抗癌剤、抗生物質、免疫抑制剤およびステロイドが含まれるが、これに制限されるわけではない。
【0084】
抗癌剤の例には、アシビシン(acivicin);アクラルビシン;アコダゾール(acodazole)塩酸塩;アクロニン(acronine);アドゼレシン(adozelesin);アルデスロイキン(aldesleukin);アルトレタミン(altretamine);アンボマイシン(ambomycin);酢酸アメタントロン(ametantrone);アムサクリン;アナストロゾール(anastrozole);アントラマイシン;アスパラギナーゼ;アスペルリン(asperlin);アザシチジン;アゼテパ(azetepa);アゾトマイシン(azotomycin);バチマスタト(batimastat);ベンゾデパ(benzodepa);ビカルタミド(bicalutamide);ビスアントレン(bisantrene)塩酸塩;ビスナフィド(bisnafide)ジメシレート;ビゼレシン(bizelesin);硫酸ブレオマイシン;ブレキナル(brequinar)ナトリウム;ブロピリミン(bropirimine);ブスルファン;カクチノマイシン;カルステロン(calusterone);カラセミド(caracemide);カルベチマー(carbetimer);カルボプラチン;カルムスチン;カルビシン(carubicin)塩酸塩;カルゼレシン(carzelesin);セデフィンゴール(cedefingol);セレコキシブ(celecoxib)(COX−2阻害剤);クロラムブシル(chlorambucil);シロレマイシン(cirolemycin);シスプラチン(cisplatin);クラドリビン(cladribine);クリスナトール(crisnatol)メシレート;シクロホスファミド;シタラビン(cytarabine);ダカルバジン;ダクチノマイシン;ダウノルビシン塩酸塩;デシタビン(decitabine);デキソルマプラチン(dexormaplatin);デザグアニン(dezaguanine);デザグアニンメシレート;ジアジクオン(diaziquone);ドセタキセル(docetaxel);ドキソルビシン;ドキソルビシン塩酸塩;ドロロキシフェン(droloxifene);クエン酸ドロロキシフェン;プロピオン酸ドロモスタノロン;ドゥアゾマイシン(duazomycin);エダトレキサート(edatrexate);エフロルニチン(eflornithine)塩酸塩;エルサミトルシン(elsamitrucin);エンロプラチン(enloplatin);エンプロメート(enpromate);エピプロピジン(epipropidine);エピルビシン(epirubicin)塩酸塩;エルブロゾール(erbulozole);エソルビシン(esorubicin)塩酸塩;エストラムスチン;エストラムスチンリン酸ナトリウム;エタニダゾール(etanidazole);エトポシド;リン酸エトポシド;エトプリン(etoprine);ファドロゾール(fadrozole)塩酸塩;ファザラビン(fazarabine);フェンレチニド(fenretinide);フロクスウリジン(floxuridine);リン酸フルダラビン(fludarabine);フルオロウラシル;フルオロシタビン(fluorocitabine);ホスキドン(fosquidone);ホストリエシン(fostriecin)ナトリウム;ジェムシタビン(gemcitabine);ジェムシタビン塩酸塩;ヒドロキシ尿素;イダルビシン(idarubicin)塩酸塩;イホスファミド;イルモホシン(ilmofosine);イプロプラチン(iproplatin);イリノテカン(irinotecan);イリノテカン塩酸塩;酢酸ランレオチド(lanreotide);レトロゾール(letrozole);酢酸ロイプロリド;リアロゾール(liarozole)塩酸塩;ロメトレキソール(lometrexol)ナトリウム;ロムスチン;ロソキサントロン(losoxantrone)塩酸塩;マソプロコール(masoprocol);メイタンシン;メクロレタミン(mechlorethamine)塩酸塩;酢酸メゲストロール(megestrol);酢酸メレンゲストロール;メルファラン;メノガリル(menogaril);メルカプトプリン;メトトレキサート;メトトレキサートナトリウム;メトプリン(metoprine);メツレデパ(meturedepa);ミチンドミド(mitindomide);ミトカルシン(mitocarcin);ミトクロミン(mitocromin);ミトジリン(mitogillin);ミトマルシン(mitomalcin);マイトマイシン;ミトスペル(mitosper);ミトタン(mitotane);ミトザントロン塩酸塩;ミコフェノール酸;ノコダゾール(nocodazole);ノガラマイシン(nogalamycin);オルマプラチン(ormaplatin);オキシスラン(oxisuran);パクリタキセル;ペガスパルガーゼ(pegaspargase);ペリオマイシン(peliomycin);ペンタムスチン(pentamustine);硫酸ペプロマイシン(peplomycin);ペルホスファミド(perfosfamide);ピポブロマン;ピポスルファン(piposulfan);ピロキサントロン(piroxantrone)塩酸塩;プリカマイシン(plicamycin);プロメスタン(plomestane);ポルフィマー(porfimer)ナトリウム;ポルフィロマイシン(porfiromycin);プレドニムスチン;プロカルバジン塩酸塩;ピュロマイシン;ピュロマイシン塩酸塩;ピラゾフリン;リボプリン(riboprine);サフィンゴール(safingol);サフィンゴール塩酸塩;セムスチン;シムトラゼン(simtrazene);スパルホセート(sparfosate)ナトリウム;スパルソマイシン(sparsomycin);スピロゲルマニウム塩酸塩;スピロムスチン(spiromustine);スピロプラチン(spiroplatin);ストレプトニグリン(streptonigrin);ストレプトゾシン;スロフェヌル(sulofenur);タリソマイシン(talisomycin);テコガラン(tecogalan)ナトリウム;タキソテール(taxotere);テガフル;テロキサントロン(teloxantrone)塩酸塩;テモポルフィン(temoporfin);テニポシド(teniposide);テロキシロン(teroxirone);テストラクトン(testolactone);チアミプリン(thiamiprine);チオグアニン;チオテパ;チアゾフリン(tiazofurin);チラパザミン(tirapazamine);クエン酸トレミフェン(toremifene);酢酸トレストロン(trestolone);リン酸トリシリビン(triciribine);トリメトレキサート(trimetrexate);グルクロン酸トリメトレキサート;トリプトレリン(triptorelin);ツブロゾール(tubulozole)塩酸塩;ウラシルマスタード;ウレデパ(uredepa);バプレオチド(vapreotide);ベルテポルフィン;硫酸ビンブラスチン;硫酸ビンクリスチン;ビンデシン;硫酸ビンデシン;硫酸ビネピジン(vinepidine);硫酸ビングリシネート(vinglycinate);硫酸ビンロイロシン(vinleurosine);酒石酸ビノレルビン(vinorelbine);硫酸ビンロシジン(vinrosidine);硫酸ビンゾリジン(vinzolidine);ボロゾール(vorozole);ゼニプラチン(zeniplatin);ジノスタチン(zinostatin);およびゾルビシン塩酸塩が含まれるが、これに制限されるわけではない。
【0085】
他の抗癌薬には、20−エピ−1,25ジヒドロキシビタミンD3;5−エチニルウラシル;アビラテロン(abiraterone);アクラルビシン;アシルフルベン(acylfulvene);アデシペノール(adecypenol);アドゼレシン;アルデスロイキン;ALL−TKアンタゴニスト;アルトレタミン;アンバムスチン(ambamustine);アミドクス(amidox);アミホスチン(amifostine);アミノレブリン酸;アムルビシン(amrubicin);アムサクリン;アナグレリド(anagrelide);アナストロゾール;アンドログラホリド;血管新生阻害剤;アンタゴニストD;アンタゴニストG;アンタレリクス(antarelix);抗背方化形態形成プロテイン−1(anti-dorsalizing morphogenetic protein-1);抗アンドロゲン、前立腺癌;抗エストロゲン;アンチネオプラストン;アンチセンスオリゴヌクレオチド;アフィジコリングリシネート;アポトーシス遺伝子モジュレーター;アポトーシスレギュレーター;アプリン酸;ara−CDP−DL−PTBA;アルギニンデアミナーゼ;アスラクリン(asulacrine);アタメスタン(atamestane);アトリムスチン(atrimustine);アクシナスタチン1(axinastatin 1);アクシナスタチン2;アクシナスタチン3;アザステロン(azasetron);アザトキシン(azatoxin);アザチロシン(azatyrosine);バカチンIII(baccatin III)誘導体;バラノール(balanol);バチマスタト;BCR/ABLアンタゴニスト;ベンゾクロリン(benzochlorins);ベンゾイルスタウロスポリン(benzoylstaurosporine);βラクタム誘導体;β−アレチン(alethine);βクラマイシンB(clamycin B);ベツリン酸(betulinic acid);bFGF阻害剤;ビカルタミド;ビスアントレン;ビスアジリジニルスペルミン(bisaziridinylspermine);ビスナフィド;ビストラテン(bistratene)A;ビゼレシン;ブレフレート(breflate);ブロピリミン;ブドチタン(budotitane);ブチオニンスルホキシイミン(buthionine sulfoximine);カルシポトリオール(calcipotriol);カルホスチンC(calphostin C);カンプトテシン誘導体;カペシタビン;カルボキサミドアミノトリアゾール;カルボキシアミドトリアゾール;CaRest M3;CARN700;軟骨由来阻害剤;カルゼレシン;カゼインキナーゼ阻害剤(ICOS);カスタノスペルミン(castanospermine);セクロピンB;セトロレリクス(cetrorelix);クロリン;クロロキノキサリンスルホンアミド;シカプロスト(cicaprost);cis−ポルフィリン;クラドリビン(cladribine);クロミフェン類似体;クロトリマゾール;コリスマイシンA(collismycin A);コリスマイシンB;コンブレタスタチンA4(combretastatin A4);コンブレタスタチン類似体;コナゲニン(conagenin);クラムベシジン816(crambescidin 816);クリスナトール;クリプトフィシン8(cryptophycin 8);クリプトフィシンA誘導体;クラシンA(curacin A);シクロペンタアントラキノン(cyclopentanthraquinones);シクロプラタム(cycloplatam);シペマイシン(cypemycin);シタラビンオクホスフェート(cytarabine ocfosfate);細胞溶解性因子;シトスタチン(cytostatin);ダクリキシマブ(dacliximab);デシタビン;デヒドロジデムニンB(dehydrodidemnin B);デスロレリン(deslorelin);デキサメタゾン;デキシホスファミド(dexifosfamide);デクスラゾキサン(dexrazoxane);デクスベラパミル(dexverapamil);ジアジクオン;ジデムニンB(didemnin B);ジドクス(didox);ジエチルノルスペルミン(diethylnorspermine);ジヒドロ−5−アザシチジン;ジヒドロタキソール、9−;ジオキサマイシン(dioxamycin);ジフェニルスピロムスチン;ドセタキセル;ドコサノール(docosanol);ドラセトロン(dolasetron);ドキシフルリジン(doxifluridine);ドキソルビシン;ドロロキシフェン;ドロナビノール;ズオカルマイシンSA(duocarmycin SA);エブセレン(ebselen);エコムスチン(ecomustine);エデルホシン(edelfosine);エドレコロマブ;エフロルニチン;エレメン(elemene);エミテフル(emitefur);エピルビシン;エプリステリド(epristeride);エストラムスチン類似体;エストロゲンアゴニスト;エストロゲンアンタゴニスト;エタニダゾール;リン酸エトポシド;エクセメスタン(exemestane);ファドロゾール;ファザラビン;フェンレチニド;フィルグラスティム;フィナステリド(finasteride);フラボピリドール(flavopiridol);フレゼラスチン(flezelastine);フルアステロン(fluasterone);フルダラビン;フルオロダウノルニシン(fluorodaunorunicin)塩酸塩;ホルフェニメクス(forfenimex);ホルメスタン(formestane);ホストリエシン;ホテムスチン(fotemustine);ガドリニウムテキサフィリン(gadolinium texaphyrin);硝酸ガリウム;ガロシタビン(galocitabine);ガニレリクス(ganirelix);ゼラチナーゼ阻害剤;ジェムシタビン;グルタチオン阻害剤;ヘプスルファム(hepsulfam);ヘレグリン(heregulin);ヘキサメチレンビスアセトアミド;ヒペリシン;イバンドロン酸(ibandronic acid);イダルビシン;イドキシフェン(idoxifene);イドラマントン(idramantone);イルモホシン;イロマスタト(ilomastat);イマチニブ(例えば、Gleevec.RTM.)、イミキモッド;免疫刺激ペプチド;インスリン様増殖因子−1受容体阻害剤;インターフェロンアゴニスト;インターフェロン;インターロイキン;イオベングアン(iobenguane);ヨードドキソルビシン;イポメアノール(ipomeanol)、4−;イロプラクト(iroplact);イルソグラジン(irsogladine);イソベンガゾール(isobengazole);イソホモハリコンドリンB(isohomohalicondrin B);イタセトロン(itasetron);ジャスプラキノリド(jasplakinolide);カハラリドF(kahalalide F);ラメラリン(lamellarin)−Nトリアセテート;ランレオチド;リーナマイシン(leinamycin);レノグラスチム(lenograstim);硫酸レンチナン(lentinan);レプトルスタチン(leptolstatin);レトロゾール;白血病阻害因子;白血球αインターフェロン;ロイプロリド+エストロゲン+プロゲステロン;ロイプロレリン(leuprorelin);レバミゾール;リアロゾール(liarozole);直鎖状ポリアミン類似体;親油性二糖ペプチド;親油性白金化合物;リソクリナミド7(lissoclinamide 7);ロバプラチン(lobaplatin);ロムブリシン(lombricine);ロメトレキソール;ロニダミン(lonidamine);ロソキサントロン;ロクソリビン(loxoribine);ルルトテカン(lurtotecan);ルテチウムテキサフィリン(lutetium texaphyrin);リソフィリン(lysofylline);溶菌ペプチド;メイタンシン;マンノスタチンA(mannostatin A);マリマスタト(marimastat);マソプロコール(masoprocol);マスピン(maspin);マトリライシン阻害剤;マトリックスメタロプロテイナーゼ阻害剤;メノガリル;メルバロン(merbarone);メテレリン(meterelin);メチオニナーゼ;メトクロプラミド;MIF阻害剤;ミフェプリストン;ミルテホシン(miltefosine);ミリモスチム(mirimostim);ミトグアゾン(mitoguazone);ミトラクトール(mitolactol);マイトマイシン類似体;ミトナフィド(mitonafide);マイトトキシン(mitotoxin)線維芽細胞増殖因子−サポリン(saporin);ミトザントロン;モファロテン(mofarotene);モルグラモスチム(molgramostim);Erbitux、ヒト絨毛性ゴナドトロピン(gonadotrophin);モノホスホリルリピドA+ミオバクテリウム(myobacterium)細胞壁sk;モピダモール(mopidamol);マスタード抗癌剤;ミカペルオキシドB(mycaperoxide B);ミコバクテリア細胞壁抽出物;ミリアポロン(myriaporone);N−アセチルジナリン(N-acetyldinaline);N−置換ベンズアミド;ナファレリン(nafarelin);ナグレスチプ(nagrestip);ナロキソン+ペンタゾシン;ナパビン(napavin);ナフテルピン(naphterpin);ナルトグラスチム(nartograstim);ネダプラチン(nedaplatin);ネモルビシン(nemorubicin);ネリドロン酸(neridronic acid);ニルタミド(nilutamide);ニサマイシン(nisamycin);酸化窒素モジュレーター;ニトロオキシド抗酸化剤;ニトルリン(nitrullyn);オブリメルセン(oblimersen)(Genasense.RTM.);O.sup.6−ベンジルグアニン;オクトレオチド(octreotide);オキセノン(okicenone);オリゴヌクレオチド;オナプリストン(onapristone);オンダンセトロン(ondansetron);オンダンセトロン;オラシン(oracin);経口サイトカインインデューサー;オルマプラチン;オサテロン(osaterone);オキサリプラチン(oxaliplatin);オキサウノマイシン(oxaunomycin);パクリタキセル;パクリタキセル類似体;パクリタキセル誘導体;パラウアミン(palauamine);パルミトイルリゾキシン(palmitoylrhizoxin);パミドロン酸(pamidronic acid);パナキシトリオール(panaxytriol);パノミフェン(panomifene);パラバクチン(parabactin);パゼリプチン(pazelliptine);ペガスパルガーゼ;ペルデシン(peldesine);ペントサンポリスルフェートナトリウム;ペントスタチン(pentostatin);ペントロゾール(pentrozole);ペルフルブロン(perflubron);ペルホスファミド;ペリリルアルコール;フェナジノマイシン(phenazinomycin);フェニルアセテート;ホスファターゼ阻害剤;ピシバニル(picibanil);ピロカルピン塩酸塩;ピラルビシン(pirarubicin);ピリトレキシム(piritrexim);プラセチンA(placetin A);プラセチンB;プラスミノーゲンアクチベーター阻害剤;白金錯体;白金化合物;白金−トリアミン錯体;ポルフィマーナトリウム;ポルフィロマイシン;プレドニソン;プロピルビスアクリドン(propyl bis-acridone);プロスタグランジンJ2;プロテアソーム阻害剤;プロテインA基剤免疫モジュレーター;プロテインキナーゼC阻害剤;プロテインキナーゼC阻害剤、ミクロアルガル(microalgal);プロテインチロシンホスファターゼ阻害剤;プリンヌクレオシドホスホリラーゼ阻害剤;プルプリン;ピラゾロアクリジン;ピリドキシル化ヘモグロビンポリオキシエチレンコンジュゲート;rafアンタゴニスト;ラルチトレキセド(raltitrexed);ラモセトロン(ramosetron);rasファルネシルプロテイントランスフェラーゼ阻害剤;ras阻害剤;ras−GAP阻害剤;脱メチルレテリプチン(retelliptine demethylated);レニウムRe186エチドロネート(etidronate);リゾキシン(rhizoxin);リボザイム;RIIレチナミド(RII retinamide);ロヒツキン(rohitukine);ロムルチド(romurtide);ロキニメクス(roquinimex);ルビジノンB1(rubiginone B1);ルボキシル(ruboxyl);サフィンゴール;セイントピン(saintopin);SarCNU;サルコフィトールA(sarcophytol A);サルグラモスチン;Sdi1模擬体;セムスチン;老化由来(senescence derived)阻害剤1;センスオリゴヌクレオチド;シグナル伝達阻害剤;シゾフィラン(sizofiran);ソブゾキサン(sobuzoxane);ナトリウムボロカプテート(borocaptate);ナトリウムフェニルアセテート;ソルベロール(solverol);ソマトメジン結合タンパク質;ソネルミン(sonermin);スパルホス酸(sparfosic acid);スピカマイシンD(spicamycin D);スピロムスチン;スプレノペンチン(splenopentin);スポンジスタチン1(spongistatin 1);スクアラミン(squalamine);スチピアミド(stipiamide);ストロメライシン阻害剤;スルフィノシン(sulfinosine);スーパーアクティブバソアクティブインテスティナルペプチド(superactive vasoactive intestinal peptide)アンタゴニスト;スラジスタ(suradista);スラミン(suramin);スワインソニン(swainsonine);タリムスチン(tallimustine);タモキシフェンメチオジド(tamoxifen methiodide);タウロムスチン(tauromustine);タザロテン(tazarotene)
;テコガラン(tecogalan)ナトリウム;テガフル(tegafur);テルラピリリウム(tellurapyrylium);テロメラーゼ阻害剤;テモポルフィン;テニポシド;テトラクロロデカオキシド;テトラゾミン(tetrazomine);タリブラスチン(thaliblastine);チオコラリン(thiocoraline);トロンボポエチン;トロンボポエチン模擬体;チマルファシン(thymalfasin);チモポエチン受容体アゴニスト;チモトリナン(thymotrinan);甲状腺刺激ホルモン;スズエチルエチオプルプリン(tin ethyl etiopurpurin);チラパザミン(tirapazamine);二塩化チタノセン(titanocene bichloride);トプセンチン(topsentin);トレミフェン;翻訳阻害剤;トレチノイン(tretinoin);トリアセチルウリジン;トリシリビン(triciribine);トリメトレキサート;トリプトレリン;トロピセトロン(tropisetron);ツロステリド(turosteride);チロシンキナーゼ阻害剤;チルホスチン(tyrphostins);UBC阻害剤;ユベニメクス(ubenimex);尿生殖洞由来増殖阻害因子;ウロキナーゼ受容体アンタゴニスト;バプレオチド;バリオリンB(variolin B);ベラレソール(velaresol);ベラミン(veramine);ベルジン;ベルテポルフィン;ビノレルビン(vinorelbine);ビンキサルチン(vinxaltine);ビタキシン(vitaxin);ボロゾール;ザノテロン(zanoterone);ゼニプラチン;ジラスコルブ(zilascorb);およびジノスタチンスティマラマー(zinostatin stimalamer)が含まれるが、これに制限されるわけではない。
【0086】
具体的な追加の活性剤には、オブリメルセン(Genasense.RTM.)、レミケード(remicade)、ドセタキセル、セレコキシブ、メルファラン、デキサメタゾン(Decadron.RTM.)、ステロイド、ジェムシタビン、シスプラチン、テモゾロミド(temozolomide)、エトポシド、シクロホスファミド、テモダール(temodar)、カルボプラチン、プロカルバジン、グリアデル(gliadel)、タモキシフェン、トポテカン(topotecan)、メトトレキサート、Arisa.RTM.、タキソール、タキソテール、フルオロウラシル、ロイコボリン、イリノテカン、キセロダ(xeloda)、CPT−11、インターフェロンα、ペグ化インターフェロンα(例えば、PEG INTRON−A)、カペシタビン、シスプラチン、チオテパ、フルダラビン、カルボプラチン、リポソームダウノルビシン、シタラビン、ドキセタキソール(doxetaxol)、パシリタキセル(pacilitaxel)、ビンブラスチン、IL−2、GM−CSF、ダカルバジン、ビノレルビン、ゾレドロン酸(zoledronic acid)、パルミトロネート(palmitronate)、ビアキシン(biaxin)、ブスルファン、プレドニソン、ビスホスホネート、三酸化ヒ素、ビンクリスチン、ドキソルビシン(Doxil.RTM.)、パクリタキセル、ガンシクロビル(ganciclovir)、アドリアマイシン、エストラムスチンリン酸ナトリウム(Emcyt.RTM.)、スリンダクおよびエトポシドが含まれるが、これに制限されるわけではない。
【0087】
B.4.製剤
本明細書中に記載の化合物の適する製剤はいずれも、製造することができる。化合物が、安定な無毒性の酸または塩基塩を形成するのに十分に塩基性または酸性である場合、それら化合物の塩としての投与は、適当でありうる。薬学的に許容しうる塩の例は、生理学的に許容しうる陰イオンを与える酸で形成される有機酸付加塩、例えば、トシレート、メタンスルホン酸塩、酢酸塩、クエン酸塩、マロン酸塩、酒石酸塩、コハク酸塩、安息香酸塩、アスコルビン酸塩、α−ケトグルタル酸塩およびα−グリセロリン酸塩である。適する無機塩も形成することができ、塩酸塩、硫酸塩、硝酸塩、重炭酸塩および炭酸塩が含まれる。薬学的に許容しうる塩は、当該技術分野において周知の標準的な手順を用いて、例えば、アミンなどの十分に塩基性の化合物を、適する酸と接触させ、生理学的に許容しうる塩を与えることによって得られる。カルボン酸のアルカリ金属(例えば、ナトリウム、カリウムまたはリチウム)塩またはアルカリ土類金属(例えば、カルシウム)塩も包含され、慣用的な方法によって製造される。
【0088】
本発明は、更に、医薬組成物であって、少なくとも一つの本発明の化合物を、少なくとも一つの薬学的に許容しうる賦形剤と混合した状態で含む医薬組成物を包含する。好ましくは、少なくとも一つのこのような賦形剤は、水以外の賦形剤またはC1−C3アルコールまたはジメチルスルホキシドである。
【0089】
本発明の化合物は、経口、局所、経皮でまたは吸入または注射によるものを含めた慣用的な経路によって投与することができる。本発明の化合物は、当業者が、既知の方法を参照して製剤化することができ、そして製剤は、予定の投与経路にしたがって作ることができる。有機化合物を製剤化するのに適する方法は、例えば、REMINGTON’S PHARMACEUTICAL SCIENCES, 18th ed. (1990) に記載され、それは、本明細書中に援用される。
【0090】
これら化合物を薬理学的組成物中で投与する場合、その化合物は、薬学的に許容しうる担体との混合物で製剤することができると考えられる。例えば、考えられる化合物は、薬理学的に許容しうる塩として経口でまたは生理食塩水溶液中で静脈内に投与することができる。リン酸塩、重炭酸塩またはクエン酸塩などの慣用的な緩衝剤は、この目的に用いることができる。当然ながら、当業者は、それら製剤を、本明細書の内容の範囲内で修飾して、特定の投与経路について多数の製剤を与えることができる。具体的には、考えられる化合物は、水または他のビヒクル中にそれらを一層可溶性にするように修飾することができるが、それは、例えば、当該技術の十分に範囲内である僅かな修飾(塩製剤化、エステル化等)で容易に達成することができる。患者の最大限有益な作用について本発明の化合物の薬物動態を管理するために、特定の化合物の投与経路およびおよび投薬計画を修飾することも、当該技術の十分に範囲内である。
【0091】
本明細書中に記載の式Iまたは式IIを有する化合物は、概して、クロロホルム、ジクロロメタン、酢酸エチル、エタノール、メタノール、イソプロパノール、アセトニトリル、グリセロール、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、ジメチルスルホキシド等などの有機溶媒中に可溶性である。一つの態様において、本発明は、式I〜IIを有する化合物を、薬学的に許容しうる担体と混合することによって製造された製剤を提供する。一つの側面において、その製剤は、(a)記載の化合物を、水溶性有機溶媒、非イオン性溶媒、水溶性脂質、シクロデキストリン、トコフェロールなどのビタミン、脂肪酸、脂肪酸エステル、リン脂質またはそれらの組み合わせ中に溶解させて、溶液を与え;そして(b)1〜10%の炭水化物溶液を含有する緩衝剤または生理食塩水を加えることを含む方法を用いて製造することができる。一例において、炭水化物は、デキストロースを含む。本方法を用いて得られた医薬組成物は、動物用途および臨床用途に安定且つ有用である。
【0092】
本方法で用いるための水溶性有機溶媒の代表例には、ポリエチレングリコール(PEG)、アルコール、アセトニトリル、N−メチル−2−ピロリドン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、ジメチルスルホキシドまたはそれらの組み合わせが含まれるが、これに制限されるわけではない。適するアルコールの例には、メタノール、エタノール、イソプロパノール、グリセロールまたはプロピレングリコールが含まれるが、これに制限されるわけではない。
【0093】
本方法で用いるための水溶性非イオン界面活性剤の代表例には、CREMOPHOR(登録商標)EL、ポリエチレングリコール修飾CREMOPHOR(登録商標)(ポリオキシエチレングリセロールトリシノレート35(polyoxyethyleneglyceroltriricinoleat 35))、水素化CREMOPHOR(登録商標)RH40、水素化CREMOPHOR(登録商標)RH60、PEG−スクシネート、ポリソルベート20、ポリソルベート80、SOLUTOL(登録商標)HS(ポリエチレングリコール660 12−ヒドロキシステアレート)、ソルビタンモノオレアート、ポロキサマー、LABRAFIL(登録商標)(エトキシル化杏仁油)、LABRASOL(登録商標)(カプリルカプロイルマクロゴール−8−グリセリド(capryl-caproyl macrogol-8-glyceride))、GELUCIRE(登録商標)(グリセロールエステル)、SOFTIGEN(登録商標)(PEG6カプリル酸グリセリド)、グリセリン、グリコールポリソルベートまたはそれらの組み合わせが含まれるが、これに制限されるわけではない。
【0094】
本方法で用いるための水溶性脂質の代表例には、植物油、トリグリセリド、プラント油またはそれらの組み合わせが含まれるが、これに制限されるわけではない。脂質油の例には、ヒマシ油、ポリオキシルヒマシ油、トウモロコシ油、オリーブ油、綿実油、ラッカセイ油、ハッカ油、ベニバナ油、ゴマ油、ダイズ油、水素化植物油、水素化ダイズ油、ヤシ油のトリグリセリド、パーム種子油およびそれらの水素化形またはそれらの組み合わせが含まれるが、これに制限されるわけではない。
【0095】
本方法で用いるための脂肪酸および脂肪酸エステルの代表例には、オレイン酸、モノグリセリド、ジグリセリド、PEGの一または二脂肪酸エステル、またはそれらの組み合わせが含まれるが、これに制限されるわけではない。
【0096】
本方法で用いるためのシクロデキストリンの代表例には、α−シクロデキストリン、β−シクロデキストリン、ヒドロキシプロピル−β−シクロデキストリンまたはスルホブチルエーテル−β−シクロデキストリンが含まれるが、これに制限されるわけではない。
【0097】
本方法で用いるためのリン脂質の代表例には、ダイズホスファチジルコリンまたはジステアロイルホスファチジルグリセロールおよびそれらの水素化形、またはそれらの組み合わせが含まれるが、これに制限されるわけではない。
【0098】
当業者は、それら製剤を、本明細書の内容の範囲内で修飾して、特定の投与経路について多数の製剤を与えることができる。具体的には、それら化合物は、水または他のビヒクル中にそれらを一層可溶性にするように修飾することができる。患者の最大限有益な作用について本発明の化合物の薬物動態を管理するために、特定の化合物の投与経路およびおよび投薬計画を修飾することも、当該技術の十分に範囲内である。
【0099】
B.5.医薬組成物
本発明は、更に、本発明の新規な処置方法で用いるのに適する局所、経口、全身および非経口用の医薬製剤を提供する目的を有する。本発明の化合物を活性成分として含有するそれら組成物は、経口、全身または標的部位投与用のビヒクル中の広範囲の治療的剤形で投与することができる。
【0100】
本発明は、更に、一つまたはそれを超える本発明の化合物を、薬学的に許容しうる担体と一緒に含む医薬組成物を提供する。好ましくは、これら組成物は、経口、非経口、鼻腔内、舌下または直腸投与用の、または吸入または吹入による投与用の、錠剤、丸剤、カプセル剤、散剤、顆粒剤、懸濁剤、ゲル剤、軟ゲル剤、滅菌非経口液剤、乳剤、エアゾル剤、液状噴霧剤、滴剤、アンプル剤、自己注射装置または坐剤などの単位剤形である。
【0101】
錠剤またはカプセル剤などの固形組成物を製造するには、主要活性成分を、医薬担体、例えば、トウモロコシデンプン、ラクトース、スクロース、ソルビトール、タルク、ステアリン酸、ステアリン酸マグネシウム、リン酸二カルシウムまたはガムなどの慣用的な錠剤成形成分、および他の医薬希釈剤、例えば、水と混合して、本発明の化合物またはその薬学的に許容しうる塩の均一混合物を含有する固形組成物を形成する。更に、主要活性成分は、一つまたはそれを超える医薬担体と混合して、改善されたバイオアベイラビリティーまたは他の薬物動態学的性質を有する剤形を与えることができる。このような系の例には、ヒドロキシプロピルメチルセルロース(HPMC)、ヒプロメロースアセテートスクシネート(hypromellose acetate succinate)(HPMCAS)などの成分を含む噴霧乾燥分散(Spray Dried Dispersion)固形剤形;低粘度ヒドロキシプロピルセルロース(Low viscosity hydroxypropylcellulos)(HPC−SL)、ドクセートナトリウム(docusate sodium)、プルロニク(pluronics)、ホスファチジルコリン、レシチンおよびコレステロールなどの成分を含むナノパーティクル(Nano-particles)製剤;ホスファチジルコリン、ポリビニルピロリドン、レシチンおよびコレステロールなどの成分を含む脂質基剤製剤(Lipid-base formulation);スルホブチルエーテル−β−シクロデキストリン(SBECD)および2−ヒドロキシプロピル−β−シクロデキストリン(HPCD)などの成分を含むシクロデキストリン製剤が含まれるが、これに制限されるわけではない。これら組成物を均一と言及する場合、それは、活性成分が、組成物中に一様に分散していることを意味するので、組成物は、錠剤、丸剤およびカプセル剤などの等しく有効な単位剤形および脂質基剤製剤中に容易に小分けすることができる。
【0102】
新規な組成物の錠剤または丸剤は、コーティングしてまたはそれ以外に配合して、持続性作用の利点を与える剤形を提供することができる。例えば、錠剤または丸剤は、内部投与成分および外部投与成分を含むことができるが、後者は、前者の上のエンベロープの形である。腸溶層は、それら二つの成分を隔てることができる。その腸溶層は、胃内の崩壊に耐えるのに役立ち且つ内部成分を十二指腸中に無傷で通過させるまたは放出を遅らせる。いろいろな材料を、このような腸溶層またはコーティングに用いることができ、このような材料には、シェラック、アセチルアルコールおよび酢酸セルロースのような、多数のポリマー性酸およびポリマー性酸とこのような材料との混合物が含まれる。
【0103】
本発明の新規な組成物を、経口または注射での投与用に包含することができる液状形には、水性液剤;好適に着香したシロップ剤;水性または油状懸濁剤;綿実油、ゴマ油、ヤシ油またはラッカセイ油などの食用油との乳剤;ポリソルベート80、トコフェリルポリエチレングリコールスクシネート(TPGS)、Cremophor、capmul MCM、ポリエチレングリコールなどの界面活性剤または補助溶媒を含むマイクロエマルジョンまたは自己乳化性系;ホスファチジルコリン、コレステロール、レシチン、HPC−SL、Docusate Soldium などの成分を含むリポソームまたはナノパーティクル製剤;溶解性を増強するSBECD、HPCDなどの成分を含むシクロデキストリン複合体製剤が含まれる。水性懸濁剤に適する分散助剤または懸濁化剤には、デキストラン、ナトリウムカルボキシメチルセルロース、メチルセルロース、ポリビニルピロリドンまたはゼラチンなどの合成および天然のガムが含まれる。
【0104】
本発明の化合物は、前述の組成物のいずれでも、そして効力研究において有効である投薬計画にしたがって投与することができる。
本発明の化合物は、最適抗癌作用を得るために、常套試験によって規定される適当な投薬量で単独に用いることができる。更に、他の腫瘍学物質の共投与または連続投与が望まれる。
【0105】
C.新規な化合物および医薬組成物を使用する方法
C.1.新規な化合物を使用する方法
本発明の化合物は、癌または他のタイプの増殖性疾患を処置する場合に、細胞障害性剤および/または細胞分裂抑制性剤として用いることができる。これら化合物は、いずれかのタイプの作用機構によって機能することができる。例えば、それら化合物は、分子および/またはシグナル伝達経路を阻害して、G2/M期で細胞周期の阻止をもたらすことができるが、それは、ついには、腫瘍細胞のアポトーシスを引き起こすかもしれない(例えば、Weung et al. (1997) Biochim. Biophys. Res. Comm., vol: 263, pp 398-404 を参照されたい)。別の例において、それら化合物は、チューブリン集合/脱集合を妨げることができ、それは、細胞有糸分裂を阻害し且つ細胞アポトーシスを引き起こすことができる(例えば、Panda et al., (1997) Proc. Natl. Acad. Sci. USA, vol: 94, 10560-10564 を参照されたい)。それら化合物は、更に、内皮細胞増殖および血管新生作用を阻害することができる(例えば、Witte et al., 1998, Cancer Metastasis Rev. vol. 17: 155-161 を参照されたい)。本発明の化合物は、いろいろな癌細胞系に活性であることが分かっている。
【0106】
別の側面において、本発明は、哺乳動物の肉腫、類表皮癌、線維肉腫、子宮頸癌、白血病、リンパ腫、肺癌、非小細胞性肺癌、結腸癌、CNS癌、黒色腫、卵巣癌、腎癌、前立腺癌、乳癌、頭頸部癌、膵癌および他のタイプの増殖性疾患が含まれるがこれに制限されるわけではない全ての組織または器官由来の癌の処置方法であって、このような処置を必要としている対象に、治療的有効量の式I〜IIを有する化合物を細胞障害性剤および/または細胞分裂抑制性剤として少なくとも一つの処置で投与することを含む方法に関する。
【0107】
また別の側面において、本発明は、白血病、リンパ腫、肺癌、結腸癌、CNS癌、黒色腫、卵巣癌、腎癌、前立腺癌または乳癌および他のタイプの増殖性疾患が含まれるがこれに制限されるわけではない全ての組織または器官由来の癌の処置用の医薬製剤を製造する方法であって、治療的有効量の式I〜IIを有する化合物を、薬学的に許容しうる担体と混合することを含む方法に関する。
【0108】
本発明の方法を実施するために、式I〜IIを有する化合物およびそれらの薬学的に許容しうる塩は、経口、非経口、吸入スプレーで、局所、直腸、鼻腔内、口腔内、膣内、植込みレザバーによって、または他の薬物投与方法で投与することができる。本明細書中で用いられる「非経口」という用語は、皮下、皮内、静脈内、筋肉内、関節内、動脈内、滑液嚢内、胸骨内、髄腔内、病巣内および頭蓋内の注射または注入技法を包含する。いくつかの態様において、本発明の化合物は、注射によって、すなわち、非経口で送達される。いくつかの態様において、好ましい投与経路は、静脈内または腹腔内注射による。
【0109】
滅菌注射可能水性または油脂性懸濁剤などの滅菌注射可能組成物は、当該技術分野において知られている技法にしたがって、適する分散助剤または湿潤剤および懸濁化剤を用いて製剤化することができる。滅菌注射可能製剤は、無毒性の非経口に許容しうる希釈剤または溶媒中の滅菌注射可能溶液または懸濁液であってもよい。用いることができる許容しうるビヒクルおよび溶媒の中には、マンニトール、水、リンガー液および等張塩化ナトリウム溶液が含まれる。更に、滅菌固定油は、溶媒または懸濁媒として好都合に用いられる(例えば、合成モノグリセリドまたはジグリセリド)。オレイン酸などの脂肪酸およびそのグリセリド誘導体は、オリーブ油またはヒマシ油などの薬学的に許容しうる油剤であるような、特に、それらのポリオキシエチル化型の注射可能なものの製造において有用である。これら油状液剤または懸濁剤は、更に、長鎖アルコール希釈剤または分散剤、またはカルボキシメチルセルロースまたは類似の分散助剤を含有することができる。薬学的に許容しうる固体、液体または他の剤形の製造において一般的に用いられるいろいろな乳化剤またはバイオアベイラビリティー増強剤も、製剤目的に用いることができる。
【0110】
経口投与用組成物は、錠剤、カプセル剤、乳剤および水性懸濁剤、分散剤および液剤が含まれるがこれに制限されるわけではないいずれかの経口に許容しうる剤形であってよい。経口使用のための錠剤の場合、一般的に用いられる担体には、ラクトースおよびトウモロコシデンプンが含まれる。ステアリン酸マグネシウムなどの滑沢剤も、加えることができる。カプセル形での経口投与用に有用な希釈剤には、ラクトースおよび乾燥トウモロコシデンプンが含まれる。水性懸濁剤または乳剤を経口投与する場合、活性成分は、乳化剤または懸濁化剤と混合した油状相中に懸濁させるまたは溶解させることができる。必要ならば、特定の甘味剤、着香剤または着色剤を加えることができる。鼻内エアゾールまたは吸入組成物は、当医薬製剤技術分野において周知の技法にしたがって製造することができるし、しかも液剤として、例えば、生理食塩水中で、適する保存剤(例えば、ベンジルアルコール)、バイオアベイラビリティーを増強する吸収促進剤、および/または当該技術分野において知られている他の可溶化剤または分散助剤を用いて製造することができる。
【0111】
本発明の化合物の有効量は、当該技術分野において知られている常套実験によって決定することができる。典型的に、これは、十分に許容されることが分かっている量の投与;および症状の減少、腫瘍サイズの減少または腫瘍成長の停止などの所望の作用が得られるまで投薬量を漸増することを必要とする。いくつかの態様において、約5〜10mg/kgの開始投薬量を用い、そしてその投薬量を、所望の作用が認められるまたは許容性問題が認められるまで、週に1回、約50%ずつ漸増増加させる。いくつかの態様において、適する投薬量は、約5〜250mg/kg;または約10〜150mg/kgである。10〜100mg/kgの投薬量は、時々好適である。投与は、1回、週に1回、1日1回または1日1回より多く行うことができる。いくつかの態様において、処置を必要としている対象に、1日に1〜4用量を送達する。
【0112】
更に、式I〜IIを有する化合物は、いろいろな癌または状態の処置のために、単独でまたは他の抗癌剤との組み合わせで投与することができる。本発明による組み合わせ療法は、少なくとも一つの本発明の化合物またはその機能性誘導体および少なくとも一つの他の薬学的に活性な成分の投与を含む。一つまたは複数の活性成分および薬学的活性剤は、別々にまたは一緒に投与することができる。一つまたは複数の活性成分および一つまたは複数の薬学的活性剤の量および相対的な投与のタイミングは、所望の組み合わせの治療的作用を達成するために選択されるであろう。
【0113】
また別の側面において、本発明は、冠状動脈疾患の患者のための冠状ステント処置後の再狭窄の、式I〜IIを有する化合物での処置方法に関する。
冠状動脈疾患の患者のための冠状ステント処置後の再狭窄の主因は、平滑筋細胞の増殖および移動および細胞外マトリックス生産によって生じる新生内膜増殖である(例えば、“Pathology of acute and chronic coronary stenting in humans”, by Farb, A., Sangiorgi, G., Certer, A.J., et al, in Circulation, vol. 99, pp 44-52, 1999 を参照されたい)。抗増殖能力を有する化合物は、このような化合物が適する手段で送達された時の臨床的および血管造影的再狭窄のリスクを減少させる場合に作用を有することができる(例えば、“A polymer-based, paclitaxel-eluting stent in patients with coronary artery disease”, by Stone, G.W., Ellis, S.G., Cox, D.A, et al, in New England Journal of Medicine, vol. 350: pp 221-231, 2004 を参照されたい)。したがって、腫瘍を処置する場合の式I〜IXを有する化合物について、それらは、新生内膜増殖に関与する細胞の増殖を阻害する場合に、したがって、新生内膜増殖および再狭窄の発生を減少させる場合に、有用でもありうる。いろいろな方法を、それら化合物をこれら細胞へ有効に送達する場合に用いることができる。例えば、式I〜Xを有する上記の化合物を含む組成物は、経口、非経口または植込みレザバーによって投与することができる。他の例において、次の論文に記載のアプローチを用いることもできる。“A polymer-based, paclitaxel-eluting stent in patients with coronary artery disease”, by Stone, G.W., Ellis, S.G., Cox, D.A. et al, in New England Journal of Medicine, vol. 350: pp 221-231, 2004; “A randomized comparison of a sirolimus-eluting stent with a standard stent for coronary revascularization”, by Morice, M.-C., Serruys, P.W., Sousa, J.E., et al, in New England Journal of Medicine, vol. 346: pp 1773-1780, 2002; “Sirolimus-eluting stents versus standard stents in patients with stenosis in a native coronary artery”, by Moses, J.W., Leon, M.B., Popma, J.J., et al, in New England Journal of Medicine, vol. 349: pp 1315-1323, 2003。
【0114】
処置および予防の方法
本発明の方法は、いろいろなタイプの癌を処置する、予防するおよび/または管理する方法を包含する。本明細書中で用いられるように、特に断らない限り、「処置する」という用語は、本発明の化合物または他の追加の活性剤の投与を意味する。本明細書中で用いられるように、特に断らない限り、「予防する」という用語は、症状の開始前の、特に、癌のリスクがある患者への投与を意味する。「予防」という用語は、特定の疾患または障害の症状の阻害を包含する。癌の家族歴を有する患者は、予防計画に好ましい候補である。本明細書中で用いられるように且つ特に断らない限り、「管理する」という用語は、特定の癌に罹患した患者の癌の再発を予防すること、および/または癌に罹患した患者が寛解状態のままである期間を延長することを包含する。
【0115】
本明細書中で用いられる「癌」という用語には、充実性腫瘍および血液由来腫瘍が含まれるが、これに制限されるわけではない。「癌」という用語は、膀胱、骨または血液、脳、乳房、子宮頸部、胸部、結腸、子宮内膜(endrometrium)、食道、眼、頭部、腎、肝、リンパ節、肺、口、頸部、卵巣、膵臓、前立腺、直腸、胃、精巣、咽頭および子宮の癌が含まれるがこれに制限されるわけではない、皮膚組織、器官、血液および血管の疾患を意味する。具体的な癌には、進行悪性疾患、アミロイドーシス、神経芽細胞腫、髄膜腫、血管周囲細胞腫、多発性脳転移、多形性グリア芽細胞腫、グリア芽細胞腫、脳幹グリオーム、予後のよくない悪性脳腫瘍、悪性グリオーム、再発性悪性グリオーム(giolma)、退形成性星状細胞腫、退形成性希突起グリオーム、神経内分泌腫瘍、直腸腺癌、Dukes C&D結腸直腸癌、切除不可能な結腸直腸癌、転移性肝細胞癌、カポジ肉腫、カロータイプ(karotype)急性骨髄芽球性白血病、ホジキンリンパ腫、非ホジキンリンパ腫、皮膚T細胞リンパ腫、皮膚B細胞リンパ腫、びまん性大B細胞リンパ腫、低級濾胞性リンパ腫、悪性黒色腫、悪性中皮腫、悪性胸膜浸出中皮腫症候群、腹膜癌、乳頭状漿液性癌、婦人科肉腫、軟組織肉腫、強皮症、皮膚血管炎、ランゲルハンス細胞組織球症、平滑筋肉腫、進行性骨化性線維異形成、ホルモン不応性前立腺癌、切除済みハイリスク軟組織肉腫、切除不可能な肝細胞癌、ヴァルデンストレームマクログロブリン血症、くすぶり型骨髄腫、無症候性骨髄腫、ファロピウス管癌、アンドロゲン非依存性前立腺癌、アンドロゲン依存性IV期非転移性前立腺癌、ホルモン不感性前立腺癌、化学療法不感性前立腺癌、乳頭状甲状腺癌、濾胞性甲状腺癌、髄様甲状腺癌および平滑筋腫が含まれるが、これに制限されるわけではない。特定の態様において、癌は転移性である。別の態様において、癌は、化学療法または放射線に不応性または耐性である;具体的には、サリドマイドに不応性である。
【0116】
C.2.処置および予防の方法
本発明の方法は、いろいろなタイプの癌を処置する、予防するおよび/または管理する方法を包含する。本明細書中で用いられるように、特に断らない限り、「処置する」という用語は、本発明の化合物または他の追加の活性剤の投与を意味する。本明細書中で用いられるように、特に断らない限り、「予防する」という用語は、症状の開始前の、特に、癌のリスクがある患者への投与を意味する。「予防」という用語は、特定の疾患または障害の症状の阻害を包含する。癌の家族歴を有する患者は、予防計画に好ましい候補である。本明細書中で用いられるように且つ特に断らない限り、「管理する」という用語は、特定の癌に罹患した患者の癌の再発を予防すること、および/または癌に罹患した患者が寛解状態のままである期間を延長することを包含する。
【0117】
本明細書中で用いられる「癌」という用語には、充実性腫瘍および血液由来腫瘍が含まれるが、これに制限されるわけではない。「癌」という用語は、膀胱、骨または血液、脳、乳房、子宮頸部、胸部、結腸、子宮内膜(endrometrium)、食道、眼、頭部、腎、肝、リンパ節、肺、口、頸部、卵巣、膵臓、前立腺、直腸、胃、精巣、咽頭および子宮の癌が含まれるがこれに制限されるわけではない、皮膚組織、器官、血液および血管の疾患を意味する。具体的な癌には、進行悪性疾患、アミロイドーシス、神経芽細胞腫、髄膜腫、血管周囲細胞腫、多発性脳転移、多形性グリア芽細胞腫、グリア芽細胞腫、脳幹グリオーム、予後のよくない悪性脳腫瘍、悪性グリオーム、再発性悪性グリオーム(giolma)、退形成性星状細胞腫、退形成性希突起グリオーム、神経内分泌腫瘍、直腸腺癌、Dukes C&D結腸直腸癌、切除不可能な結腸直腸癌、転移性肝細胞癌、カポジ肉腫、カロータイプ(karotype)急性骨髄芽球性白血病、ホジキンリンパ腫、非ホジキンリンパ腫、皮膚T細胞リンパ腫、皮膚B細胞リンパ腫、びまん性大B細胞リンパ腫、低級濾胞性リンパ腫、悪性黒色腫、悪性中皮腫、悪性胸膜浸出中皮腫症候群、腹膜癌、乳頭状漿液性癌、婦人科肉腫、軟組織肉腫、強皮症、皮膚血管炎、ランゲルハンス細胞組織球症、平滑筋肉腫、進行性骨化性線維異形成、ホルモン不応性前立腺癌、切除済みハイリスク軟組織肉腫、切除不可能な肝細胞癌、ヴァルデンストレームマクログロブリン血症、くすぶり型骨髄腫、無症候性骨髄腫、ファロピウス管癌、アンドロゲン非依存性前立腺癌、アンドロゲン依存性IV期非転移性前立腺癌、ホルモン不感性前立腺癌、化学療法不感性前立腺癌、乳頭状甲状腺癌、濾胞性甲状腺癌、髄様甲状腺癌および平滑筋腫が含まれるが、これに制限されるわけではない。特定の態様において、癌は転移性である。別の態様において、癌は、化学療法または放射線に不応性または耐性である;具体的には、サリドマイドに不応性である。
【0118】
C.3.生物学的スクリーニングおよび抗癌活性:
リアルタイム細胞電子検知(Real-Time Cell Electronic Sensing)(RT−CES)を用いた in vitro の細胞に基づくスクリーニング
本明細書中に開示の化合物の生物学的活性を、ACEA Biosciences, Inc. 製の Real-Time Cell Electronic Sensing(RT−CES(登録商標))システムを用いて監視し且つプロフィールを集めた。RT−CESシステムは、細胞−基質インピーダンス技術を利用して、微量滴定プレート形式の組織培養ウェル内部の細胞挙動を監視する。その技術は、ミクロ電子工学での分子・細胞生物学の組込みを特徴とし、生物学的検定法の電子検出に基づいている。この細胞電子検知技術および関連装置、システムおよび使用方法の詳細は、米国特許第7,167,585号;米国特許第7,468,255号;PCT公開WO2004/010102号;米国特許第7,470,533号;および米国特許第7,459,303号に記載されていて、それらは各々、本明細書中に援用される。RT−CES技術の更なる詳細は、米国特許第7,468,255号に更に開示されている。
【0119】
RT−CES技術を用いた細胞−基質または細胞−電極インピーダンスの測定には、適当な幾何学的形を有する微小電極を、微量滴定プレートまたは類似の装置の底面上に製作して、ウェル中に向ける。細胞を、それら装置のウェル中に入れ、電極表面に接触させ且つ付着させる。細胞の存在、不存在または性状の変化は、電極センサー表面上の電子およびイオンの通過に影響する。電極の間または中でのインピーダンスを測定することは、センサー上に存在する細胞の生物学的状態について重要な情報を与える。それら細胞の生物学的状態に変化がある場合、アナログ電子読み取りシグナルを、自動的に且つリアルタイムで測定し、そして処理用および分析用のディジタルシグナルへ変換する。RT−CESシステムの場合、細胞指数(インピーダンス変化の任意表示)は、測定された電極インピーダンス値に基づいて自動的に導かれ且つ与えられる。一定のウェルについて得られた細胞指数は、(1)このウェル中の電極表面に、いかに多くの細胞が付着しているか;(2)このウェル中の電極表面に、いかに十分に細胞が付着しているかを反映している。したがって、同様の生理学的状態にある同じタイプの細胞が、より多く電極表面に付着してほど、細胞指数は大きい。そして、細胞が、より十分に電極表面に付着しているほど(例えば、細胞が、より大きい接触面積を有するように一層広がっている、または細胞が、よりしっかりと電極表面に付着しているほど)、細胞指数は大きい。
【0120】
RT−CESシステムは、三つの成分、すなわち、電子センサー分析器、装置ステーションおよび16Xまたは96X微量滴定装置を含む。微小電極センサーアレイは、リソグラフィーによる微小製作方法でガラススライド上に製作したが、電極含有スライドは、プラスチックトレーへ組み立てられて、電極含有ウェルを形成している。装置ステーションには、16Xまたは96X微量滴定プレート装置を与え、それは、ウェルのいずれか一つを、インピーダンス測定用のセンサー分析器へ電子的に切り替えることができる。操作中に、ウェル中で培養された細胞を含む装置を、インキュベーター内部に位置する装置ステーション中に入れる。電気ケーブルは、装置ステーションをセンサー分析器へ連通する。RT−CESソフトウェア制御下において、センサー分析器は、測定されるウェルを自動的に選択し且つ連続的にインピーダンス測定を行うことができる。分析器からのインピーダンスデータを、コンピューターへ転送し、組み込まれたソフトウェアで分析し且つ処理する。
【0121】
個々のウェル中の電極間で測定されたインピーダンスは、電極幾何学的形、ウェル中のイオン濃度、および電極に付着した細胞が存在するかどうかに依存する。細胞が不存在の場合、電極インピーダンスは、主に、電極/溶液界面およびバルク溶液中の双方におけるイオン環境によって決定される。細胞の存在下において、電極センサー表面に付着した細胞は、電極/溶液界面における局部イオン環境を変更して、インピーダンスの増加をもたらすであろう。電極上に存在する細胞が多いほど、細胞−電極インピーダンスの増加は大きい。更に、インピーダンス変化も、細胞形態、および細胞が電極に付着している程度に依存する。
【0122】
測定された細胞−電極インピーダンスに基づいて細胞状態を定量するために、細胞指数(Cell Index)と称するパラメーターを導く。細胞指数は、電極含有ウェル中の細胞の状態の定量的尺度である。同じ生理学的条件下において、電極上に付着したより多くの細胞は、より大きい細胞−電極抵抗値をもたらして、細胞指数についてより大きい値をもたらす。更に、ウェル中に存在する同数の細胞については、形態などの細胞状態の変化が、細胞指数の変化をもたらすであろう。例えば、細胞接着または細胞広がり(spreading)の増加は、より大きい細胞−電極接触面積をもたらし、それが、細胞−電極抵抗の増加を、したがって、細胞指数についてより大きい値をもたらすであろう。
【0123】
生物学的に活性な化合物と、Eプレートのウェル内部の細胞成長との相互作用は、化合物自体の生物学的機構、濃度、インキュベーション長さおよび細胞タイプに依存する独特の活性パターン(すなわち、化合物処置に応答した独特の細胞インピーダンス曲線または細胞指数曲線)を生じる。各々の化合物への「記名(signature)」細胞応答パターンは、細胞周期阻止、形態変化および細胞死などの特定の生物学的現象と相関する。RT−CESシステムで細胞応答プロフィールを集めることは、有効であると証明されており、本発明者は、同様の作用機構を有する化合物が、同様のパターンを示すということを示した。したがって、化合物処置への細胞応答パターンの類似性は、作用機構、耐性の様式、そしておそらくは、分子標的の類似性を示すことができる。本発明者は、抗有糸分裂剤での処置に応答して有糸分裂阻止を行っている細胞について独特のRT−CES記名パターンを識別した。一例として、図4Bおよび図4Cは、異なった濃度の周知の抗有糸分裂剤パクリタキセルおよびビンクリスチンで処置されたA549肺癌細胞の特異的プロフィールを示している。
【0124】
本発明者は、RT−CESシステム(ACEA Biosciences)またはxCelligenceシステム(Roche)を用いて、いくつかの新規な化合物への多数の癌細胞系の応答を評価した。RT−CESシステムおよびxCelligenceシステムは、本質的に同じであり、同じACEAリアルタイム細胞インピーダンス測定技術を用いていることに注目されたい。いくつかの本発明の化合物の(特定の濃度での)時間依存性細胞応答パターンは、パクリタキセルおよびビンクリスチンの(特定の濃度での)それらと幾分同様であった。したがって、これら化合物は、パクリタキセルおよびビンブラスチンの場合と同様の抗癌作用機構を有することができる。もう一方において、これら化合物は、これら本発明の化合物の時間依存性細胞応答パターンが、パクリタキセルおよびビンクリスチンの場合と同様であるとしても、パクリタキセルおよびビンブラスチンの場合とは異なった他の作用機構によって癌細胞に作用することができる。これら化合物が、パクリタキセルおよびビンブラスチンの場合と同様の作用機構を含めた多数の作用機構によって癌細胞に作用するということも考えられる。
【0125】
本発明者は、本明細書中に記載のRT−CES方法で測定される本発明の化合物の活性を調べた。そのデータは、それら化合物が、広範囲の癌細胞系に活性であり、そして正常細胞へは活性がはるかに少ないということを示している。ここで、本発明者は、一例として、活性化合物の一つであるCOMPOUND Oを用いる。図4〜24は、いろいろな濃度でのCOMPOUND Oの添加前および後の多数のヒト癌細胞系の時間依存性細胞指数を示す。これら細胞系には、A549(非小細胞性肺癌細胞系)、NCI−H460(大細胞肺癌細胞系)、H1993細胞(非小細胞性肺癌細胞系)、H1838細胞(非小細胞性肺癌細胞系)、H2347細胞(非小細胞性肺癌細胞系)、SW620細胞(結腸癌細胞系)、GTL16細胞(胃癌細胞系)、HT29細胞(結腸癌細胞系)、A172細胞(脳癌細胞系)、U138細胞(脳癌細胞系)、U118細胞(脳癌細胞系)、SW1088細胞(脳癌細胞系)、HT1080細胞(結合組織癌細胞系)、BxPC3細胞(膵癌細胞系)、HepG2細胞(肝癌細胞系)、SKOV3細胞(卵巣癌細胞系)、MCF7細胞(乳癌細胞系)、MDA−MB−231(乳癌細胞系)およびKB(子宮頸癌細胞系)が含まれる。それら結果は、COMPOUND Oが、これら癌細胞系の増殖に阻害剤作用を示したということを示している。これら細胞系に対するCOMPOUND OのIC50は、慣用的な化学療法薬であるパクリタキセルおよびビンクリスチンのそれと同様である。
【0126】
本発明者は、更に、多剤耐性(MDR)遺伝子を過発現するKB200へのCOMPOUND Oの作用を調べた。この遺伝子は、臨床の化学療法薬、例えば、パクリタキセルおよびビンクリスチン/ビンブラスチンの耐性に応答性である。図24A〜Dは、親KB細胞系の成長が、COMPOUND O(IC50=33.1nM)、パクリタキセル(IC50=7.19nM)、ビンブラスチン(IC50=4.74nM)およびコルヒチン(IC50=8.20nM)によって阻害されたということを示している。これら4種類の化合物のIC50は、同様の範囲内である。図24Eは、KB200細胞の成長が、COMPOUND O(IC50=9.84nM)による阻害に感受性であったことを示している。対照的に、KB200細胞の成長は、パクリタキセル(IC50>1uM)、ビンブラスチン(IC50=135nM)およびコルヒチン(IC50=116nM)による阻害への感受性がはるかに少なかった(図24F〜Hに示される)。これは、COMPOUND Oが、パクリタキセルおよびビンブラスチン処置に失敗した患者に対する大きなセカンドライン療法でありうるということを示唆している。
【0127】
図25は、非癌性細胞系NIH−3T3が、COMPOUND O(IC50=6uM)による阻害への感受性がはるかに少なかったことを示している。したがって、COMPOUND Oは、癌細胞に対して、正常細胞に対するよりも高い細胞障害作用を示す。
【0128】
抗癌活性についての in vivo スクリーニング
COMPOUND Oの in vivo 抗癌効力を評価するために、3種類のヒト腫瘍異種移植片モデルを用いた。それには、免疫不全ヌードマウスにおけるMKN45ヒト胃腸癌、H460ヒト非小細胞性肺癌およびA549ヒト肺癌の異種移植片モデルが含まれた。COMPOUND Oの in vivo 抗癌効力の詳細を、実施例1〜3に与える。
【0129】
実施例1
ヌードマウスにおけるMKN45ヒト胃腸癌へのCOMPOUND Oの in vivo 抗癌活性
COMPOUND Oの in vivo 抗癌効力を評価するために、免疫不全ヌードマウスにおけるMKN45ヒト胃腸癌異種移植片モデルを調べた。マウスモデルは全て、Pharmacology Lab of ACEA Bio (Hangzhou) CO., Ltd. で維持する。BALB/c免疫不全ヌードマウスは、Shanghai SLAC Laboratory Animal、証明番号:SCXKA(Shanghai)2007−0005より購入した。マウス体重は、19±1gであった。雌マウスのみを、この研究に用いた。被験動物数は、次の通りであった。各用量群に7匹、正対照群に7匹、そして負対照群に7匹。
【0130】
試験対照。負対照には、各々のマウスに、COMPOUND O試験に用いられたのと同容量および同濃度を有する溶媒のみを、120mg/kgで1日おきに1回(qod)18日間経口投与した。正対照には、経口抗癌化合物であるエトポシド(Etoposide)を、50mg/kgで4日毎に1回、18日間経口投与した。
【0131】
試験化合物の調製・投与。COMPOUND Oを、25%リン脂質(S75)および75%ポリビニルピロリドン(PL−PVP)中に溶解させた後、更に希釈して、0.9%NaCl水溶液中に8mg/mlとした。120mg/kg qod(1日おきに1回)〜160mg/kg q3d(3日毎に1回)の異なった投薬量のCOMPOUND Oを、研究に用いた。
【0132】
移植および化合物効力決定のための腫瘍細胞の調製。MKN45ヒト胃腸癌異種移植片モデルのための腫瘍細胞を調製するために、急速成長した腫瘍を、被移植腫瘍マウスから最初に取り出し、それら腫瘍組織を、1〜2mm3寸法に切開した。次に、これら微小腫瘍を、各々のマウスの補助領域(右側)中に皮下注射した。接種された腫瘍が、ヌードマウスにおいて一定サイズ(60〜80mm3)に成長後、それらマウスを無作為化して、異なった投薬群とし、そして化合物処置を施した。最初の投薬後2〜3週間に、マウスを屠殺し、そして移植された腫瘍を、実験用マウスから取り出した。各々取り出された充実性腫瘍を秤量した;各々の投薬群の腫瘍阻害率を、次の式にしたがって計算した。
【0133】
腫瘍阻害率%=(負対照群の平均腫瘍重量−化合物処置群の平均腫瘍重量)/負対照群の平均腫瘍重量x100 (1)
動物用飼料、動物用ケージ、支持材料および被験動物が接触する装置を含めた用いられる材料は全て、高圧滅菌した。ヌードマウスは、SPF条件下の層流棚で維持した。腫瘍移植後、各々の化合物投薬群のマウス体重および腫瘍サイズを、動的に監視し且つプロットした。腫瘍サイズは、腫瘍の長軸(a)および短軸(b)を測定することによって決定し、そして腫瘍体積は、次の式にしたがって計算した。
【0134】
腫瘍体積=axb2/2 (2)
結果。ヌードマウスのMKN45ヒト胃腸癌異種移植片モデルにおいて、COMPOUND Oは、120mg/kg qodおよび160mg/kg q3d投薬群において、それぞれ、58.0%および55.7%の平均 in vivo 腫瘍阻害率を示した。同じ実験において、Etoposide は、50mg/kg q3dの経路投与投薬量について48.4%の平均 in vivo 腫瘍阻害率を示した。それら詳細を、表2に与える。腫瘍サイズの動的変化は、図1に要約する。キャリアマウスの体重の動的変化は、表3に要約する。MKN45異種移植片モデルにおけるCOMPOUND O(120mg/kg qod)の抗癌作用は、同じ薬物投与経路の下での Etoposide(30mg/kg q3d)の場合と同様である。
【0135】
【表2】
【0136】
【表3】
【0137】
実施例2
ヌードマウスにおけるH460ヒト非小細胞性肺癌へのCOMPOUND Oの in vivo 抗癌活性
COMPOUND Oの in vivo 抗癌効力を評価するために、免疫不全ヌードマウスにおけるH460ヒト非小細胞性肺癌異種移植片モデルを用いた。細胞系およびマウスは、Pharmacology Lab of ACEA Bio (Hangzhou) CO., Ltd. で維持した。BALB/c免疫不全ヌードマウスは、Shanghai SLAC Laboratory Animal、証明番号:SCXKA(Shanghai)2007−0005より購入した。マウス体重は、19±2gの間であった。雌マウスのみを、この研究に用いた。被験動物数は、次の通りであった。各用量群に7匹、正対照群に7匹、そして負対照群に7匹。
【0138】
試験対照。負対照には、各々のマウスに、COMPOUND O試験に用いられたのと同容量および同濃度を有する溶媒のみを、120mg/kgで1日おきに1回(qod)25日間経口投与した。正対照には、経口抗癌化合物である Etoposide を、30mg/kg qodで25日間経口投与した。
【0139】
試験化合物の調製・投与。COMPOUND Oを、25%リン脂質(S75)および75%ポリビニルピロリドン(PL−PVP)中に溶解させた後、更に希釈して、0.9%NaCl水溶液中に8mg/mlとした。各々のマウスに、化合物溶液を経口投与した。異なった投薬量のCOMPOUND O(120mg/kg qodおよび160mg/kg q3d)を、研究に用いた。
【0140】
移植および化合物効力決定のための腫瘍細胞の調製。H460ヒト非小細胞性肺癌異種移植片モデルのための癌細胞を調製するために、フラスコ中の対数期増殖性細胞を、トリプシン処理し、そして細胞を、PBS(pH7.2)中に1.5x107個細胞/mlで再懸濁させた。細胞懸濁液(3x106個細胞)を、各々のマウスの補助領域(右側)中に皮下注射した。接種された癌細胞が、ヌードマウスにおいて一定サイズ(150mm3)の腫瘍に成長後、それらマウスを無作為化して、異なった投薬群とし、そして化合物処置を施した。最初の投薬後3〜4週間に、マウスを屠殺し、そして移植された腫瘍を、実験用マウスから取り出した。各々取り出された充実性腫瘍を秤量した;各々の投薬群の腫瘍阻害率を、実施例1の方程式(1)にしたがって計算した。
【0141】
動物用飼料、動物用ケージ、支持材料および被験動物が接触する装置を含めた用いられる材料は全て、高圧滅菌した。ヌードマウスは、SPF条件下の層流棚で維持した。腫瘍移植後、各々の化合物投薬群のマウス体重および腫瘍サイズを、動的に監視し且つプロットした。腫瘍サイズは、腫瘍の長軸(a)および短軸(b)を測定することによって決定し、そして腫瘍体積は、実施例1の式(2)にしたがって計算した。
【0142】
結果。ヌードマウスのH460ヒト非小細胞性肺癌異種移植片モデルにおいて、COMPOUND Oは、120mg/kg qodおよび160mg/kg q3d投薬群において、それぞれ、47.1%および31.3%の平均 in vivo 腫瘍阻害率を示した。同じ実験において、Etoposide は、30mg/kg qodの経路投与投薬量について48.4%の平均 in vivo 腫瘍阻害率を示した。それら詳細を、表4に与える。腫瘍サイズの動的変化は、図2に要約する。キャリアマウスの体重の動的変化は、表5に要約する。H460ヒト非小細胞性肺癌異種移植片モデルにおけるCOMPOUND O(120mg/kg)の抗癌作用は、同じ薬物投与手順の下での Etoposide(30mg/kg)の場合と同様である。
【0143】
【表4】
【0144】
【表5】
【0145】
実施例3
ヌードマウスにおけるA549ヒト非小細胞性肺癌へのCOMPOUND Oの in vivo 抗癌活性
COMPOUND Oの in vivo 抗癌効力を評価するために、免疫不全ヌードマウスにおけるA549ヒト非小細胞性肺癌異種移植片モデルを用いた。細胞系およびマウスは、Pharmacology Lab of ACEA Bio (Hangzhou) CO., Ltd. で維持した。BALB/c免疫不全ヌードマウスは、Shanghai SLAC Laboratory Animal、証明番号:SCXKA(Shanghai)2007−0005より購入した。マウス体重は、21±1gの間であった。雌マウスのみを、この研究に用いた。被験動物数は、次の通りであった。各用量群に7〜8匹、正対照群に6匹、そして負対照群に7匹。
【0146】
試験対照。負対照には、各々のマウスに、COMPOUND O試験に用いられたのと同容量および同濃度を有する溶媒のみを、120mg/kgで1日おきに1回(qod)25日間経口投与した。正対照には、経口抗癌化合物である Etoposide を、30mg/kg qodで25日間経口投与した。
【0147】
試験化合物の調製・投与。COMPOUND Oを、25%リン脂質(S75)および75%ポリビニルピロリドン(PL−PVP)中に溶解させた後、更に希釈して、0.9%NaCl水溶液中に8mg/mlとした。各々のマウスに、化合物溶液を経口投与した。異なった投薬量のCOMPOUND O(120mg/kg qodおよび160mg/kg q2*2(2日間続けて投薬後、2日間続けて休む)を、研究に用いた。
【0148】
移植および化合物効力決定のための腫瘍細胞の調製。A549ヒト非小細胞性肺癌異種移植片モデルのための癌細胞を調製するために、フラスコ中の対数期増殖性細胞を、トリプシン処理し、そして細胞を、PBS(pH7.2)中に2.5x107個細胞/mlで再懸濁させた。細胞懸濁液(5x106個細胞)を、各々のマウスの補助領域(右側)中に皮下注射した。接種された癌細胞が、ヌードマウスにおいて一定サイズ(50〜60mm3)の腫瘍に成長後(接種後約15日)、それらマウスを無作為化して、異なった投薬群とし、そして化合物処置を施した。最初の投薬後3〜4週間に、マウスを屠殺し、そして移植された腫瘍を、実験用マウスから取り出した。各々取り出された充実性腫瘍を秤量した;各々の投薬群の腫瘍阻害率を、実施例1の方程式(1)にしたがって計算した。
【0149】
動物用飼料、動物用ケージ、支持材料および被験動物が接触する装置を含めた用いられる材料は全て、高圧滅菌した。ヌードマウスは、SPF条件下の層流棚で維持した。腫瘍移植後、各々の化合物投薬群のマウス体重および腫瘍サイズを、動的に監視し且つプロットした。腫瘍サイズは、腫瘍の長軸(a)および短軸(b)を測定することによって決定し、そして腫瘍体積は、実施例1の式(2)にしたがって計算した。
【0150】
結果。ヌードマウスのA549ヒト非小細胞性肺癌異種移植片モデルにおいて、COMPOUND Oは、120mg/kg qodおよび160mg/kg q2*2投薬群において、それぞれ、67.8%および62.5%の平均 in vivo 腫瘍阻害率を示した。同じ実験において、Etoposide は、30mg/kg qodの経路投与投薬量について59.9%の平均 in vivo 腫瘍阻害率を示した。それら詳細を、表6に与える。腫瘍サイズの動的変化は、図3に要約する。キャリアマウスの体重の動的変化は、表7に要約する。A549ヒト非小細胞性肺癌異種移植片モデルにおけるCOMPOUND O(120mg/kg)の抗癌作用は、同じ薬物投与手順の下での Etoposide(30mg/kg)の場合と同様である。
【0151】
【表6】
【0152】
【表7】
【0153】
実施例4
A549細胞におけるCOMPOUND O、パクリタキセルおよびビンクリスチンによる細胞増殖の阻害
A549細胞(ヒト肺癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、RT−CESシステム(ACEA Biosciences)を用いて監視した。RT−CESシステムは、xCelligenceシステム(Roche より現在入手可能)と同じである。図4A〜Cは、化合物添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND O、パクリタキセルおよびビンクリスチンについてそれぞれ、10.9nM、5.3nMおよび61.8nMである。
【0154】
実施例5
H596細胞におけるCOMPOUND Oによる細胞増殖の阻害
H596細胞(ヒト腺扁平上皮肺癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。xCelligenceシステムは、ACEABiosciences 製のRT−CESシステムと同じである。図5は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて37.9nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルおよびビンクリスチンについてそれぞれ、12.5nMおよび36.9nMである。
【0155】
実施例6
H292細胞におけるCOMPOUND Oによる細胞増殖の阻害
H292細胞(ヒト肺性肺癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図6は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて10.8nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルおよびビンクリスチンについてそれぞれ、2.59nMおよび2.63nMである。
【0156】
実施例7
H460細胞におけるCOMPOUND Oによる細胞増殖の阻害
NCI−H460細胞(ヒト大細胞肺癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図7は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて10.9nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルおよびビンクリスチンについてそれぞれ、2.68nMおよび10.4nMである。
【0157】
実施例8
H1993細胞におけるCOMPOUND Oによる細胞増殖の阻害
H1993細胞(ヒト非小細胞性肺癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図8は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて60.2nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルおよびビンクリスチンについてそれぞれ、5.12nMおよび2.43nMである。
【0158】
実施例9
H1838細胞におけるCOMPOUND Oによる細胞増殖の阻害
H1838細胞(ヒト非小細胞性肺癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図9は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて11.8nMである。比較において、計算されたIC50(化合物処置後72時間)は、ビンクリスチンについて3.16nMである。
【0159】
実施例10
H2347細胞におけるCOMPOUND Oによる細胞増殖の阻害
H2347細胞(ヒト非小細胞性肺癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図10は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて1.76nMである。比較において、計算されたIC50(化合物処置後72時間)は、ビンクリスチンについて5.05nMである。
【0160】
実施例11
SW620細胞におけるCOMPOUND Oによる細胞増殖の阻害
SW620細胞(ヒト結腸癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図11は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて2.65nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルについて0.998nMである。
【0161】
実施例12
GTL16細胞におけるCOMPOUND Oによる細胞増殖の阻害
GTL16細胞(MKN45ヒト胃癌細胞系に由来したヒト胃癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図12は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて67.0nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルおよびビンクリスチンについてそれぞれ、1.02nMおよび3.75nMである。
【0162】
実施例13
HT29細胞におけるCOMPOUND Oによる細胞増殖の阻害
HT29細胞(ヒト結腸癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図13は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて25.4nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルおよびビンクリスチンについてそれぞれ、2.52nMおよび12.7nMである。
【0163】
実施例14
A172細胞におけるCOMPOUND Oによる細胞増殖の阻害
A172細胞(ヒト脳癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図14は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND O、パクリタキセルおよびビンクリスチンについてそれぞれ、11.6nM、4.46nMおよび1.30nMである。
【0164】
実施例15
U138MG細胞におけるCOMPOUND Oによる細胞増殖の阻害
U138MG細胞(ヒト脳癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図15は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND O、パクリタキセルおよびビンクリスチンについてそれぞれ、3.41nM、18.1nMおよび0.641nMである。
【0165】
実施例16
U118MG細胞におけるCOMPOUND Oによる細胞増殖の阻害
U118MG細胞(ヒト脳癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図16は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND O、パクリタキセルおよびビンクリスチンについてそれぞれ、13.1nM、12.3nMおよび4.31nMである。
【0166】
実施例17
SW1088細胞におけるCOMPOUND Oによる細胞増殖の阻害
SW1088細胞(ヒト脳癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図17は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND O、パクリタキセルおよびビンクリスチンについてそれぞれ、21.2nM、13.1nMおよび4.96nMである。
【0167】
実施例18
HT1080細胞におけるCOMPOUND Oによる細胞増殖の阻害
HT1080細胞(ヒト結合組織癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図18は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oおよびビンクリスチンについてそれぞれ、7.63nMおよび2.01nMである。
【0168】
実施例19
BxPC3細胞におけるCOMPOUND Oによる細胞増殖の阻害
BxPC3細胞(ヒト膵癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図19は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて6.60nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルおよびビンクリスチンについてそれぞれ、8.86nMおよび4.06nMである。
【0169】
実施例20
HepG2細胞におけるCOMPOUND Oによる細胞増殖の阻害
HepG2細胞(ヒト肝癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、10,000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図20は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて25.9nMである。
【0170】
実施例21
SKOV3細胞におけるCOMPOUND Oによる細胞増殖の阻害
SKOV3細胞(ヒト卵巣癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、3000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図21は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて5.47nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルについて5.34nMである。
【0171】
実施例22
MCF7細胞におけるCOMPOUND Oによる細胞増殖の阻害
MCF7細胞(ヒト乳癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図22は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて74.4nMである。比較において、計算されたIC50(化合物処置後72時間)は、11.3nMパクリタキセルである。
【0172】
実施例23
MDA−MB−231細胞におけるCOMPOUND Oによる細胞増殖の阻害
MDA−MB−231細胞(ヒト乳癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図23は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて32.4nMである。
【0173】
実施例24
多剤耐性(MDR)遺伝子を過発現するKB200細胞におけるCOMPOUND Oによる細胞増殖の阻害。
【0174】
親としての子宮頸癌細胞系であるKB細胞を、最初に調べた。KB細胞を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセル、ビンブラスチンおよびコルヒチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図24A〜Dは、化合物添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND O、パクリタキセル、ビンブラスチンおよびコルヒチンについてそれぞれ、33.1nM、7.19nM、4.74nMおよび8.20nMである。
【0175】
これら化合物の効力を、多剤耐性(MDR)遺伝子を過発現するKB200細胞に対して更に調べた。同様に、KB200細胞を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセル、ビンブラスチンおよびコルヒチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図24E〜Hは、化合物添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND O、ビンブラスチンおよびコルヒチンについてそれぞれ、9.84nM、0.135μMおよび0.116μMである。パクリタキセルについて計算されたIC50(化合物処置後72時間)は、>1μMである。COMPOUND Oは、慣用的な化学療法(例えば、パクリタキセルおよびビンブラスチン)に耐性である細胞系に対して十分な効力を示す。これは、COMPOUND Oが、パクリタキセルおよびビンブラスチン処置に失敗した患者に対する大きなセカンドライン療法でありうるということを示唆している。
【0176】
実施例25
NIH3T3正常組織細胞系におけるCOMPOUND Oによる細胞増殖の阻害
NIH3T3細胞(正常組織細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND Oを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図25は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて6.0μMである。対照的に、試験された癌細胞系全てに対するCOMPOUND OのIC50は、低nM範囲内である。COMPOUND Oは、癌細胞に対して、正常細胞に対するよりも高い細胞障害性作用を示す。
【0177】
実施例26
COMPOUND Oによる in vitro および in vivo チューブリン重合の阻害
微小管は、重複した染色体が、二つの娘細胞への細胞の開裂前に二つの同一のセットに分離する時の有糸分裂を含めた数多くの細胞過程において重要である。微小管の不可欠な役割、および有糸分裂および細胞分裂におけるそれらの動力は、微小管を、抗癌薬の重要な標的にする。間期中の細胞において、微小管は、それらのチューブリンを、約3分〜数時間のハーフタイムで、細胞質プール中の可溶性チューブリンと交換する。有糸分裂の開始で、間期微小管ネットワークは脱集合し、そして有糸分裂紡錘体を形成し且つ染色体を動かす極めて動的な微小管の集団によって置き換えられる。有糸分裂紡錘体微小管は、間期細胞中の微小管よりも20〜50倍動的であり、そしてより多くの紡錘体微小管が、それらのチューブリンを、15秒もの速いハーフタイムで、可溶性プール中のチューブリンと交換する。
【0178】
有糸分裂紡錘体微小管の動力は、レギュレーターによるモジュレーションに、および微小管活性薬による破裂に、鋭く感受性である。微小管標的薬は、微小管重合および動力を広範囲の方法で変更することができる。ここで、本発明者は、COMPOUND Oの作用機構、すなわち、(1)それは、in vitro の微小管集合を阻害する、(2)それは、培養細胞中の微小管ネットワークに影響を与える、そして(3)それは、Colchicine と同様の機構によってチューブリン集合を阻害するということを証明する。
【0179】
方法
in vitro 微小管集合検定。微小管重合は、96ウェル微量滴定プレート中において、MAPの多いチューブリンと、HTS−チューブリン重合検定キットプロトコル(Cytoskeleton)に基づく、80mM PIPES pH6.9、0.5mM EGTA、2mM MgCl2、1mM GTP、10%グリセロールおよび4%(v/v)ジメチルスルホキシド(DMSO)を含有する緩衝液中のいろいろな濃度のCOMPOUND Oで行った。吸光度の増加は、37℃において405nm、Beckman Multimode DTX880プレートリーダーで測定し、60秒毎に30分間記録した。
【0180】
スピンカラム検定。チューブリンを、[[3H]ビンブラスチンまたは[3H]コルヒチンと一緒に、0.05M PIPES、pH6.9、1mM MgCl2および1mM GTPを含有する緩衝液中の異なった濃度の未標識ビンブラスチンか、コルヒチンかまたはCOMPOUND Oの存在下でインキュベートした。反応混合物を、37℃で1時間インキュベートした。それら試料を、緩衝溶液で予め平衡した illustraTM MicroSpinTMG−50カラム(GE Healthcare)上に装填した。それらカラムを、1.5ml管中に入れ、室温において750gで2分間回転させ、そしてフロースルー(flow-through)中の放射能を、シンチレーションカウンターによって分析した。
【0181】
結果
図26Aは、MAPの多いチューブリンを用いた in vitro の微小管集合へのCOMPOUND Oの作用を示す。負対照試料において、405nMでの吸光度(A405)は、時間で増加した。A405の増加は、10分でプラトーに達する。5μMビンクリスチンの存在下において、チューブリン重合は、対照試料の場合と比較して50%を超えて阻害された。COMPOUND Oの存在下において、チューブリン重合は、濃度依存方式で阻害された。0.04μMでのCOMPOUND Oは、負対照の場合と同様である阻害を示さなかった。5μMでのCOMPOUND Oは、正対照(5μMでのビンクリスチン)の場合と同様の阻害パターンを示した。5μMパクリタキセル(チューブリン重合増強剤)の存在下において、チューブリン重合は、負対照の場合と比較して更に増強された。
【0182】
図26Bは、A549細胞中において、COMPOUND Oの20時間処置による微小管体制の阻害を示す。対照細胞における微小管ネットワークは、正常な体制および配置を示した(図24B DMSO)。対照的に、パクリタキセル処置は、細胞微小管の密度の増加を伴う微小管重合を引き起こした(図24B Paclitaxel)。更に、COMPOUND O処置は、ビンクリスチンで誘発される微小管変化の場合と同様の知見をもたらした(図24B COMPOUND O&ビンクリスチン)。
【0183】
図26Cは、スピンカラム検定を用いて、COMPOUND Oが、コルヒチン結合部位によってチューブリンと相互作用するということを示している。図26C(上部パネル)に示されるように、未標識コルヒチンの存在下における[3H]コルヒチンとのチューブリンインキュベーションは、フロースルー中で見出される[3H]コルヒチンの量を濃度依存方式で減少させた。同様に、COMPOUND Oは、フロースルー中の[3H]コルヒチンの量を濃度依存方式で減少させた。負対照として、ビンブラスチンは、フロースルー中の[3H]コルヒチンの量に影響を与えなかった。本発明者は、更に、スピンカラム検定を用いて、COMPOUND Oが、チューブリン上のビンブラスチン結合部位へ結合することによってチューブリンと相互作用するかどうかを調べた。図26C(下部パネル)に示されるように、未標識ビンブラスチンの存在下における[3H]ビンブラスチンとのチューブリンインキュベーションは、フロースルー中で見出される[3H]ビンブラスチンの量を濃度依存方式で減少させた。対照すると、コルヒチンもCOMPOUND Oも、フロースルー中の[3H]ビンブラスチンの量に影響することはなかった。
【0184】
実施例27
COMPOUND Oは、癌細胞においてアポトーシスを誘発する
COMPOUND O化合物が、癌細胞においてアポトーシスを誘発するかどうかを調べるために、A549ヒト肺癌細胞を、37nM COMPOUND Oおよび37nMパクリタキセルで処置した。細胞アポトーシスおよび細胞死の検出は、Cell Death Detection ELISAキット(Roche Applied Sciences)により、キットによる検定プロトコルにしたがって測定した。簡単にいうと、5000個細胞を、96ウェルプレートの各々のウェル中に播種した。24時間インキュベーション後、それら細胞を、細胞増殖のIC50値に近い濃度である37nMのCOMPOUND Oおよびパクリタキセルで処置した。細胞を採取し、そして薬物処置後24時間、48時間または72時間に溶解させた。20マイクロリットルの溶解産物を取り出し、ストレプトアビジン被覆マイクロプレートに移した後、抗ヒストン−ビオチン抗体および抗DNA−POD抗体と一緒に2時間インキュベート後、発色用に2,2’−アジノビス−3−エチルベンズチアゾリンスルホン酸基質を加えた。そのプレートを、Beckman Multimode DTX880プレートリーダーにおいて405nmおよび490nmの吸光度で測定した。
【0185】
図27Aに示されるように、37nM COMPOUND Oおよび37nMパクリタキセルで処置された細胞は、(薬物処置後48時間から開始する)強いアポトーシスシグナルを示すが、DMSOで処置されただけの対照細胞は、最小のシグナルを示した。これは、COMPOUND Oが、A549肺癌細胞において時間依存性アポトーシスを誘発したことを示している。更に、本発明者は、二つの他の癌細胞系H596およびH292も調べた。図27Bは、37nM COMPOUND Oが、これら二つの細胞系において37nMパクリタキセルと同様のレベルのアポトーシスを引き起こしたことを示している。総合すると、COMPOUND Oは、癌細胞においてアポトーシスを誘発した。
【0186】
実施例28
COMPOUND Oは、癌細胞においてG2/M細胞周期停止を誘発する
微小管は、細胞の重複した染色体が、二つの娘細胞への細胞の開裂前に二つの同一のセットに分離する際の有糸分裂の過程において極めて重要である。パクリタキセルおよびビンブラスチンなどの、微小管を標的とする化合物は、微小管動力を抑制し且つ有糸分裂過程をブロックする。結果として、細胞は、G2/M期で阻止されるであろう。COMPOUND Oが、癌細胞分裂の場合の有糸分裂過程に影響を与えるかどうかを調べるために、A549ヒト肺癌細胞を、33nMのCOMPOUND Oおよびパクリタキセルで、または負対照として役立つ0.2%DMSOで処置した。被処置細胞を、有糸分裂および有糸分裂阻止を行う細胞についてのマーカーである、ホスホヒストンH3に対する抗体で染色した。図28Aは、パクリタキセルおよびCOMPOUND Oによる有糸分裂阻止の程度を有糸分裂指数で定量したものを示す。それは、パクリタキセル、COMPOUND OおよびDMSOで処置されたA549についてそれぞれ、44±8%、45±3%および3±1%であった。
【0187】
更に、本発明者は、フローサイトメトリーを用いて、細胞周期へのCOMPOUND Oの作用も調べた。簡単にいうと、A549細胞を、6ウェル組織培養プレート中に400,000個/ウェルの細胞密度で播種した。約24時間後、それら細胞を、37nM COMPOUND Oで処置し、そして更に24時間インキュベートさせた。細胞を、PBS中で洗浄し、トリプシン処理し、計数し、そして氷冷70%メタノール中で固定し且つ4℃で貯蔵した。細胞を、PBSで洗浄し、ヨウ化プロピジウムで染色し、そしてフローサイトメトリー分析まで氷上で保持した。図28Bに示されるように、G2/M期での細胞集団は、COMPOUND Oで処置された細胞において、DMSOのみで処置された細胞と比較して有意に増加した。
【0188】
公報、特許、特許出願および公表された特許出願などの参考文献は、全てを通して、本明細書中にそのまま援用される。
前述の発明は、理解の明瞭さの目的で、図および実施例によってある程度詳細に記載してきたが、特定の僅かな変化および修飾が実施されるであろうということは当業者に明らかである。したがって、説明および実施例は、本発明の範囲を制限すると解釈されるべきではない。
【技術分野】
【0001】
関連出願のクロス・リファレンス
本出願は、2010年2月19日出願の米国仮特許出願第61/306,416号および2010年3月16日出願の同第61/314,510号に優先権を主張するが、それらの開示は、本明細書中にそのまま援用される。
【0002】
本発明の分野は、複素環式化合物、医薬組成物および方法であって、特に、癌および関連疾患の処置および予防のための組成物および方法に関するものである。
【背景技術】
【0003】
癌は、先進諸国において二番目に大きい死因である。したがって、癌は、依然として、人類への最も重要な未だかなえられていない医学的挑戦の一つである。腫瘍を処置するには、外科手術、放射線、化学療法またはこれらアプローチのいずれかの組み合わせを含めた多数の選択肢が利用可能である。これらの中で、化学療法は、全てのタイプの癌に、具体的には、手術不能のものまたは転移特性を有するものに広く用いられる。異なったヒト癌の生存率の改善のために臨床で用いられているいろいろな化学療法用化合物にもかかわらず、化学療法は、概して、治癒的ではなく、疾患進行を遅らせるだけである。
【0004】
一般的に、腫瘍およびそれらの転移は、腫瘍細胞が多剤耐性の能力を発現するにつれて、化学療法に不応性になる。いくつかの場合、腫瘍は、本質的に、いくつかのクラスの化学療法薬に耐性である。他の場合、化学療法薬に対して獲得される耐性は、化学療法的介入中に発現する。したがって、異なったクラスの腫瘍を処置する場合、利用可能な化学療法用化合物の効力への有意の限界が、依然として存在する。更に、腫瘍の化学療法的処置に用いられる多くの細胞障害性剤または細胞分裂抑制性剤は、重症の副作用を有して、幾人かの患者に化学療法の停止をもたらす。
【発明の概要】
【発明が解決しようとする課題】
【0005】
したがって、癌を処置するための新しい化学療法薬への要求が、依然として存在する。
【課題を解決するための手段】
【0006】
本発明は、アニリン置換チアゾール環に連結した二環式ヘテロアリール環系を有する新規な化合物、これら化合物を含有する医薬組成物、およびこれら化合物および組成物を使用する方法に関する。本明細書中に記載の化合物は、抗腫瘍、抗癌、抗炎症、抗感染および抗増殖の活性を示す。それらは、多くの異なったタイプの癌を含めた癌細胞への選択的毒性によって示されるように、癌の処置に特に有用である。本発明は、更に、腫瘍、癌、および感染性および/または増殖性疾患を処置するのに用いることができるこのような化合物を含有する医薬組成物に関する。
【0007】
本発明の内容の一つの側面において、新規な複素環式化合物は、式Iまたは式II:
【0008】
【化1】
【0009】
[式中、Zは、次の置換されたフェニル環または複素環式環(これら構造中の破線が交差している結合は、式Iまたは式II中のNHへのZ基の結合点である):
【0010】
【化2】
【0011】
より選択されるが、これに制限されるわけではない]
に従う構造を有するか、またはその薬学的に許容しうる塩若しくはアシル化プロドラッグである。
【0012】
本明細書中に記載のそれらに似ている化合物は、癌を処置するためのこのような化合物の使用を含めて、報告された(WO2009/023402号)。しかしながら、本明細書中に記載の新規な化合物は、当該技術分野において知られている化合物よりも予想外に優れている。
【0013】
式I〜IIの化合物は、中性化合物として、または無機および有機対イオンを含むそれらの薬学的に適する塩として用いることができる。それら塩には、ハロゲン化物(Cl−、Br−、I−)、ニトレート、メシレート、p−トルエンスルホネート/トシレート、オキサレート、シトレート、マレート、マレエート、タータレート、フマレート、ホルメート、アセテートおよびこれらクラスの類似の陰イオンなどがあるがこれに制限されるわけではない薬学的に許容しうる対イオンを含む酸付加塩が含まれる。
【0014】
上記の複素環式化合物には、それら化合物自体、更には、適用可能ならば、それらの塩およびそれらのプロドラッグが含まれる。このような塩は、例えば、化合物上の陽電荷置換基(例えば、プロトン化されている複素環式環または芳香環上のアミノ基)と薬学的に適する陰イオンとの間で、または式Iまたは式IIの化合物の塩基性複素環式基への酸の付加によって形成することができる。適する陰イオンには、塩化物、臭化物、ヨウ化物、スルフェート、ニトレート、ホスフェート、シトレート、ベンゼンスルホネート、メタンスルホネート、トリフルオロアセテート、マレイエートおよびアセテートが含まれるが、これに制限されるわけではない。同様に、化合物上の陰電荷置換基(例えば、複素環式環または芳香環上のカルボン酸基)は、薬学的に許容しうる陽イオンと塩を形成することができる。適する陽イオンの非制限例は、ナトリウムイオン、カリウムイオン、マグネシウムイオン、カルシウムイオン、およびテトラメチルアンモニウムイオン、テトラブチルアンモニウムイオンなどの有機アンモニウムイオンおよび他の有機陽イオンである。
【0015】
適するプロドラッグは、式Iまたは式IIのNH基のアシル化によって形成することができる。代表的なプロドラッグには、NHがNC(O)−R*へアシル化された式Iまたは式IIの化合物が含まれるが、この場合、C(O)R*は、ホルミル、アセチル、クロロアセチル、トリクロロアセチル、トリフルオロアセチル等などの置換されていてよいアシル基である。他のプロドラッグには、NHがスルホニル化されて、例えば、N−SO2−R’を形成した式Iの化合物が含まれるが、この場合、R’は、例えば、メチル、フルオロメタンスルホニルまたはトリフルオロメタンスルホニル(trifluormethanesulfonyl)でありうる。
【0016】
本発明の化合物は、光学異性体、幾何異性体、互変異性体、およびアトロプ異性体を含めた回転異性体を含めた異性体として存在してよい。本発明は、式I〜IIの化合物のこのような異性体各々およびそれらの混合物を包含する。化合物が、例えば、キラル中心を有する場合、本発明は、各々の個々の異性体、更には、等量の双方の異性体を有するラセミ混合物を含めた、いろいろな量の双方の異性体の混合物を包含する。本発明の化合物は、ビアリールであるので、それらは、ビアリール結合についての回転異性体として存在することもでき、そして各々の異性体並びにこのような異性体の混合物は、本発明の範囲内に包含される。
【0017】
本発明の化合物およびそれら化合物を含む組成物は、望ましくない細胞増殖を特徴とする状態を処置するのに有用である。具体的には、それら化合物は、肉腫、類表皮癌、線維肉腫、子宮頸癌、胃癌、皮膚癌、白血病、リンパ腫、肺癌、非小細胞性肺癌、結腸癌、CNS癌、黒色腫、卵巣癌、腎癌、前立腺癌、乳癌、肝癌、頭頸部癌、膵癌および他のタイプの増殖性疾患を処置するのに有用である。
【図面の簡単な説明】
【0018】
【図1】図1は、免疫不全ヌードマウスの皮下植込みによって異種移植片移植されたMKN45ヒト胃腸癌へのCOMPOUND Oの in vivo 抗腫瘍効力を示す。
【図2】図2は、免疫不全ヌードマウスの皮下植込みによって異種移植片移植されたH460ヒト非小細胞性肺癌へのCOMPOUND Oの in vivo 抗腫瘍効力を示す。
【図3】図3は、免疫不全ヌードマウスの皮下植込みによって異種移植片移植されたA549ヒト非小細胞性肺癌へのCOMPOUND Oの in vivo 抗腫瘍効力を示す。
【図4A】図4Aは、Real-Time Cell Electronic Sensing(Roche 製のxCELLigenceシステムと同じである、ACEA Biosciences 製のRT−CESシステム)で決定される、異なった濃度のCOMPOUND OへのA549ヒト非小細胞性肺癌細胞系の動的応答プロフィールを示す。
【図4B】図4Bは、Real-Time Cell Electronic Sensing(Roche 製のxCELLigenceシステムと同じである、ACEA Biosciences 製のRT−CESシステム)で決定される、異なった濃度のパクリタキセルへのA549ヒト非小細胞性肺癌細胞系の動的応答プロフィールを示す。
【図4C】図4Cは、Real-Time Cell Electronic Sensing(Roche 製のxCELLigenceシステムと同じである、ACEA Biosciences 製のRT−CESシステム)で決定される、異なった濃度のビンクリスチンへのA549ヒト非小細胞性肺癌細胞系の動的応答プロフィールを示す。
【図5】図5は、Real-Time Cell Electronic Sensing(RT−CES)システム(ACEA Biosciences)と同じであるxCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのH596ヒト腺扁平上皮肺癌細胞系の動的応答プロフィールを示す。
【図6】図6は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのH292ヒト肺性肺癌細胞系の動的応答プロフィールを示す。
【図7】図7は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのH460ヒト大細胞肺癌細胞系の動的応答プロフィールを示す。
【図8】図8は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのH1993ヒト非小細胞性肺癌細胞系の動的応答プロフィールを示す。
【図9】図9は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのH1838ヒト非小細胞性肺癌細胞系の動的応答プロフィールを示す。
【図10】図10は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのH2347ヒト非小細胞性肺癌細胞系の動的応答プロフィールを示す。
【図11】図11は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのSW620ヒト結腸癌細胞系の動的応答プロフィールを示す。
【図12】図12は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのGTL16ヒト胃癌細胞系(MKN45ヒト胃癌細胞系に由来した)の動的応答プロフィールを示す。
【図13】図13は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのHT29ヒト結腸癌細胞系の動的応答プロフィールを示す。
【図14】図14は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのA172ヒト脳癌細胞系の動的応答プロフィールを示す。
【図15】図15は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのU138MGヒト脳癌細胞系の動的応答プロフィールを示す。
【図16】図16は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのU118MGヒト脳癌細胞系の動的応答プロフィールを示す。
【図17】図17は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのSW1088ヒト脳癌細胞系の動的応答プロフィールを示す。
【図18】図18は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのHT1080ヒト結合組織癌細胞系の動的応答プロフィールを示す。
【図19】図19は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのBxPC3ヒト膵癌細胞系の動的応答プロフィールを示す。
【図20】図20は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのHepG2ヒト肝癌細胞系の動的応答プロフィールを示す。
【図21】図21は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのSKOV3ヒト卵巣癌細胞系の動的応答プロフィールを示す。
【図22】図22は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのMCF7ヒト乳癌細胞系の動的応答プロフィールを示す。
【図23】図23は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのMDA−MB−231ヒト乳癌細胞系の動的応答プロフィールを示す。
【図24A】図24Aは、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのKBヒト子宮頸癌細胞系の動的応答プロフィールを示す。
【図24B】図24Bは、xCELLigenceシステム(Roche)で決定される、異なった濃度のパクリタキセルへのKBヒト子宮頸癌細胞系の動的応答プロフィールを示す。
【図24C】図24Cは、xCELLigenceシステム(Roche)で決定される、異なった濃度のビンブラスチンへのKBヒト子宮頸癌細胞系の動的応答プロフィールを示す。
【図24D】図24Dは、xCELLigenceシステム(Roche)で決定される、異なった濃度のコルヒチンへのKBヒト子宮頸癌細胞系の動的応答プロフィールを示す。
【図24E】図24Eは、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOMPOUND OへのKB200ヒト子宮頸癌細胞系(多剤耐性MDR遺伝子を発現する)の動的応答プロフィールを示す。
【図24F】図24Fは、xCELLigenceシステム(Roche)で決定される、異なった濃度のパクリタキセルへのKB200ヒト子宮頸癌細胞系(多剤耐性MDR遺伝子を発現する)の動的応答プロフィールを示す。
【図24G】図24Gは、xCELLigenceシステム(Roche)で決定される、異なった濃度のビンブラスチンへのKB200ヒト子宮頸癌細胞系(多剤耐性MDR遺伝子を発現する)の動的応答プロフィールを示す。
【図24H】図24Hは、xCELLigenceシステム(Roche)で決定される、異なった濃度のコルヒチンへのKB200ヒト子宮頸癌細胞系(多剤耐性MDR遺伝子を発現する)の動的応答プロフィールを示す。
【図25】図25は、xCELLigenceシステム(Roche)で決定される、異なった濃度のCOPMOUND OへのNIH3T3正常組織細胞系の動的応答プロフィールを示す。
【図26A】図26Aは、MAPの多いチューブリンを用いた in vitro の微小管集合(assembly)へのCOPMOUND Oの作用を示す。
【図26B】図26Bは、COMPOUND O、パクリタキセルおよびビンクリスチンの20時間処置によるA549細胞中の微小管体制の阻害作用を示す。
【図26C】図26Cは、スピンカラム検定を用いたコルヒチン結合部位を経るチューブリンとCOMPOUND Oの相互作用を示す。
【図27A】図27Aは、37nM COMPOUND Oおよび37nMパクリタキセルの24時間、48時間および72時間処置によって誘発される、A549ヒト肺癌細胞のアポトーシスを示す。
【図27B】図27Bは、37nM COMPOUND Oおよび37nMパクリタキセルの72時間処置によって誘発される、A549、H596およびH292ヒト肺癌細胞のアポトーシスを示す。
【図28A】図28Aは、有糸分裂指数で定量される、パクリタキセルおよびCOMPOUND OによるA549ヒト肺癌細胞の有糸分裂阻止の程度を示す。
【図28B】図28Bは、COMPOUND Oで24時間処置後のA549ヒト肺癌細胞の細胞周期分布を示す。
【発明を実施するための形態】
【0019】
開示の明快さのために、制限としてではなく、本発明の選択された態様について次の説明を与える。便宜上、次の小区分に分けるが、それら区分は、本発明の範囲を制限すると解釈されるべきではない。
【0020】
A.定義
それ以外に定義されない限り、本明細書中で用いられる専門用語および科学用語は全て、本発明が属する当該技術分野の業者によって一般的に理解されるのと同じ意味を有する。本明細書中に挙げられる特許、出願、公開出願および他の公報は全て、そのまま援用される。この部分に示されている定義が、本明細書中に援用される特許、出願、公開出願および他の公報に示されている定義に反するまたはそれ以外にそれと一致しない場合、この部分に示されている定義は、本明細書中に援用される定義に優先する。
【0021】
本明細書中で用いられる「ある(a または an)」は、「少なくとも一つ」または「一つまたはそれを超える」を意味する。
本明細書中で用いられる「アルキル」という用語は、直鎖状、分枝状または環状の立体配置の飽和炭化水素基を意味し、具体的に考えられるアルキル基には、低級アルキル基(すなわち、10個またはそれ未満の炭素原子を有するもの)が含まれる。代表的なアルキル基は、メチル、エチル、プロピル、イソプロピル、ブチル、sec−ブチル、第三級ブチル、ペンチル、イソペンチル、ヘキシル等である。本明細書中で用いられる「アルケニル」という用語は、上に定義のような且つ少なくとも一つの二重結合を有するアルキルを意味する。したがって、具体的に考えられるアルケニル基には、2〜10個の炭素原子を有する直鎖状、分枝状または環状のアルケニル基(例えば、エテニル、プロペニル、ブテニル、ペンテニル等)が含まれる。同様に、本明細書中で用いられる「アルキニル」という用語は、上に定義のような且つ少なくとも一つの三重結合を有するアルキルまたはアルケニルを意味する。特に考えられるアルキニルには、2〜10個の全炭素原子を有する直鎖状、分枝状または環状のアルキン(例えば、エチニル、プロピニル、ブチニル等)が含まれる。
【0022】
本明細書中で用いられる「シクロアルキル」という用語は、好ましくは、3〜8個の炭素原子を包含する環状アルカン(すなわち、炭化水素の炭素原子鎖が環を形成しているもの)を意味する。したがって、代表的なシクロアルカンには、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチルおよびシクロオクチルが含まれる。シクロアルキルは、更に、一つまたは二つの二重結合を包含し、それは、「シクロアルケニル」基を形成する。シクロアルキル基は、更に、アルキル、アルケニル、アルキニル、ハロおよび他の一般的な基で置換されてもいる。
【0023】
本明細書中で用いられる「アリール」または「芳香族部分」という用語は、芳香族環系を意味し、それは、1個またはそれを超える非炭素原子を更に包含してよい。したがって、考えられるアリール基には、(例えば、フェニル、ナフチル等)およびピリジルが含まれる。更に考えられるアリール基は、一つまたは二つの5員または6員アリール基または複素環式基と縮合(すなわち、第一芳香環上の2原子と共有結合)していてよく、したがって、「縮合アリール」または「縮合芳香族」と称される。
【0024】
本明細書中で更に用いられる「複素環」、「シクロヘテロアルキル」および「複素環式部分」という用語は、本明細書中において同じ意味に用いられ、複数の原子が複数の共有結合によって環を形成しているいずれかの化合物を意味し、この場合、その環は、炭素原子以外の少なくとも一つの原子を包含する。具体的に考えられる複素環式塩基には、非炭素原子として窒素、硫黄または酸素を含む5員および6員環(例えば、イミダゾール、ピロール、トリアゾール、ジヒドロピリミジン、インドール、ピリジン、チアゾール、テトラゾール等)が含まれる。更に考えられる複素環は、一つまたは二つの環または複素環に縮合(すなわち、第一複素環式環上の2個の原子と共有結合)していてよく、したがって、本明細書中で用いられる「縮合複素環」または「縮合複素環式塩基」または「縮合複素環式部分」と称される。
【0025】
本明細書中で用いられる「ハロゲン」という用語は、フッ素、塩素、臭素およびヨウ素を意味する。
上に定義の基はいずれも、一つまたはそれを超える置換基で更に置換されていてよいし、それが順次、更に置換されていてよいということは更に理解されるはずである。例えば、アルキルまたはアリール中の水素原子は、アミノ、ハロまたは他の基で置換されている。
【0026】
本明細書中で用いられる「置換された」という用語は、H原子の別の原子または基での代用を意味する。アルキル基、アルケニル基およびアルキニル基は、しばしば、このような置換が化学的に意味を成す程度に置換されている。典型的な置換基には、ハロ、=O、=N−CN、=N−OR、=NR、OR、NR2、SR、SO2R、SO2NR2、NRSO2R、NRCONR2、NRCOOR、NRCOR、CN、COOR、CONR2、OOCR、CORおよびNO2が含まれるが、これに制限されるわけではなく、ここにおいて、Rは、各々独立して、H、C1−C8アルキル、C2−C8ヘテロアルキル、C1−C8アシル、C2−C8ヘテロアシル、C2−C8アルケニル、C2−C8ヘテロアルケニル、C2−C8アルキニル、C2−C8ヘテロアルキニル、C6−C10アリールまたはC5−C10ヘテロアリールであり、そしてRは各々、ハロ、=O、=N−CN、=N−OR’、=NR’、OR’、NR’2、SR’、SO2R’、SO2NR’2、NR’SO2R’、NR’CONR’2、NR’COOR’、NR’COR’、CN、COOR’、CONR’2、OOCR’、COR’およびNO2で置換されていてよく、ここにおいて、R’は、各々独立して、H、C1−C8アルキル、C2−C8ヘテロアルキル、C1−C8アシル、C2−C8ヘテロアシル、C6−C10アリールまたはC5−C10ヘテロアリールである。アルキル基、アルケニル基およびアルキニル基は、C1−C8アシル、C2−C8ヘテロアシル、C6−C10アリールまたはC5−C10ヘテロアリールで置換されていることもありうるし、それらは各々、具体的な基について適当である置換基で置換されうる。
【0027】
「ヘテロアルキル」、「ヘテロアルケニル」および「ヘテロアルキニル」等は、該当するヒドロカルビル(アルキル、アルケニルおよびアルキニル)基と同様に定義されるが、「ヘテロ」という用語は、1〜3個のO、SまたはNヘテロ原子またはその組み合わせを主鎖残基中に含有する基を意味する;したがって、該当するアルキル基、アルケニル基またはアルキニル基の少なくとも一つの炭素原子は、指定のヘテロ原子の一つで代用されて、ヘテロアルキル基、ヘテロアルケニル基またはヘテロアルキニル基を形成する。アルキル基、アルケニル基およびアルキニル基のヘテロ形に典型的な且つ好ましいサイズは、概して、該当するヒドロカルビル基の場合と同じであり、そしてヘテロ形上に存在してよい置換基は、ヒドロカルビル基について上に記載されたものと同じである。化学的安定性の理由で、特に断らない限り、このような基は、ニトロ基またはスルホニル基の場合のようにNまたはS上にオキソ基が存在する場合を除いて、2個以下の隣接するヘテロ原子を包含するということも理解される。
【0028】
本明細書中で用いられる「アルキル」が、シクロアルキル基およびシクロアルキルアルキル基を包含する場合、「シクロアルキル」という用語は、本明細書中において、環炭素原子によって連結している炭素環式非芳香族基を記載するのに用いることができ、そして「シクロアルキルアルキル」は、アルキルリンカーによって分子に連結している炭素環式非芳香族基を記載するのに用いることができる。同様に、「ヘテロシクリル」は、少なくとも一つのヘテロ原子を環員として含有する且つCまたはNであってよい環原子によって分子に連結している非芳香族環状基を記載するのに用いることができる;そして「ヘテロシクリルアルキル」は、リンカーによって別の分子に連結しているこのような基を記載するのに用いることができる。シクロアルキル基、シクロアルキルアルキル基、ヘテロシクリル基およびヘテロシクリルアルキル基に適するサイズおよび置換基は、アルキル基について上に記載されたものと同じである。本明細書中で用いられるように、これら用語は、その環が芳香族でない限り、一つまたは二つの二重結合を含有する環も包含する。
【0029】
本明細書中で用いられる「アシル」は、カルボニル炭素原子の二つの利用可能な原子価位置の一つに付いたアルキル、アルケニル、アルキニル、アリールまたはアリールアルキルのラジカルを含む基を包含し、そしてヘテロアシルは、カルボニル炭素以外の少なくとも一つの炭素が、N、OおよびSより選択されるヘテロ原子で代用された該当する基を意味する。したがって、ヘテロアシルには、例えば、−C(=O)ORおよび−C(=O)NR2、更には、−C(=O)−ヘテロアリールが含まれる。
【0030】
概して、いずれのアルキル基、アルケニル基、アルキニル基、アシル基またはアリール基またはアリールアルキル基も、または置換基中にあるこれら基の一つのいずれのヘテロ形も、それ自体、追加の置換基で置換されていてもよい。これら置換基の性状は、それら置換基がそれ以外に記載されていない場合、本来の置換基自体に関して挙げられたものと似ている。したがって、例えば、R7の態様がアルキルである場合、このアルキルは、R7の態様として挙げられた残りの置換基で置換されていてもよく、この場合、これは、化学的意味を成し、そしてこれは、アルキル自体に与えられたサイズ限界を害することはない;例えば、アルキルまたはアルケニルで置換されたアルキルは、これら態様についての炭素原子の上限を簡単に広げると考えられ、包含されない。しかしながら、アリール、アミノ、アルコキシ、=O等で置換されたアルキルは、本発明の範囲内に包含されると考えられ、これら置換基の原子は、アルキル、アルケニル等の基を記載するのに用いられる記載されている数に入れられない。置換基の数が指定されない場合、このようなアルキル基、アルケニル基、アルキニル基、アシル基またはアリール基は各々、その利用可能な原子価にしたがった数の置換基で置換されていよい;具体的には、これら基はいずれも、例えば、その利用可能な原子価のいずれかまたは全てにおいてフッ素原子で置換されていよい。
【0031】
具体的に考えられる官能基には、求核基(例えば、−NH2、−OH、−SH、−NC等)、求電子基(例えば、C(O)OR、C(X)OH等)、極性基(例えば、−OH)、無極性基(例えば、複素環、アリール、アルキル、アルケニル、アルキニル等)、イオン性基(例えば、−NH3+)およびハロゲン(例えば、−F、−Cl)、NHCOR、NHCONH2、OCH2COOH、OCH2CONH2、OCH2CONHR、NHCH2COOH、NHCH2CONH2、NHSO2R、OCH2−複素環、PO3H、SO3H、アミノ酸、およびそれらの化学的に妥当な組み合わせ全てが含まれる。更に、「置換された」という用語は、置換の多重度も包含し、そして多数の置換基が開示されているまたは請求の範囲に記載されている場合、置換された化合物は、開示されたまたは請求の範囲に記載された一つまたはそれを超える置換基部分で独立して置換されうる。更に、本明細書中で用いられる「一置換/二置換/三置換/四置換された」という用語は、芳香族または複素環式部分上に置換されたまたは芳香族または複素環式部分に縮合した上記の1個または2個または3個または4個の官能基を意味し、ここにおいて、このような多官能基は、芳香族または複素環式部分のオルト位またはパラ位またはメタ位のいずれかの組み合わせに置換されている。
【0032】
更に、本明細書中に与えられている式はいずれも、このような化合物の水和物、溶媒和化合物および多形およびそれらの組み合わせを示すものである。
本明細書中で用いられる「薬学的に許容しうる」または「薬理学的に許容しうる」とは、生物学的にまたはそれ以外に望ましくないことがない物質を意味し、例えば、その物質は、有意の望ましくない生物学的作用を全く引き起こすことなくまたはそれが入っている組成物の他の成分のいずれとも有害に相互作用することなく、患者に投与される医薬組成物中に包含されていてよい。薬学的に許容しうる担体または賦形剤は、好ましくは、毒性試験および製造試験の必要な基準を満たしているおよび/または米国食品医薬品局(U.S. Food and Drug administration)が作成したInactive Ingredient Guide に包含される。「薬学的に許容しうる塩」は、遊離(非塩)化合物の生物活性を少なくとも若干保持する且つ薬物または医薬品として個体に投与することができるそれら塩である。薬学的に許容しうる塩は、イオン相互作用を表し、共有結合ではない。それとして、N−オキシドは、塩とは考えられない。このような塩には、例えば、(1)塩酸、臭化水素酸、硫酸、硝酸、リン酸等などの無機酸で形成される;または酢酸、シュウ酸、プロピオン酸、コハク酸、マレイン酸、酒石酸等などの有機酸で形成される酸付加塩;(2)親化合物中に存在する酸性プロトンが、金属イオン、例えば、アルカリ金属イオン、アルカリ土類金属イオンまたはアルミニウムイオンで代用されているか;または有機塩基で配位している場合に形成される塩が含まれる。許容しうる有機塩基には、エタノールアミン、ジエタノールアミン、トリエタノールアミン等が含まれる。許容しうる無機塩基には、水酸化アルミニウム、水酸化カルシウム、水酸化カリウム、炭酸ナトリウム、水酸化ナトリウム等が含まれる。薬学的に許容しうる塩の追加の例には、Berge et al, Pharmaceutical Salts, J. Pharm. Sci. 66(1):1-19, 1977 に挙げられたものが含まれる。薬学的に許容しうる塩は、製造プロセス中に現場で、または遊離酸または塩基の形の本発明の精製済み化合物を適する有機または無機の塩基または酸とそれぞれ別々に反応させ、そしてこのようにして形成された塩を引き続きの精製中に単離することによって製造することができる。薬学的に許容しうる塩の意味が、その溶媒付加形または結晶形、具体的には、溶媒和化合物または多形を包含するということは理解されるはずである。溶媒和化合物は、化学量論的量かまたは非化学量論的量の溶媒を含有し、そしてしばしば、結晶化プロセス中に形成される。水和物は、溶媒が水である場合に形成され、またはアルコラートは、溶媒がアルコールである場合に形成される。多形には、ある化合物の同じ元素組成を有する異なった結晶充填配置が含まれる。多形は、通常、異なったX線回折図形、赤外スペクトル、融点、密度、硬度、結晶形状、光学的および電気的性質、安定性および溶解性を有する。再結晶溶媒、結晶化速度および貯蔵温度などのいろいろな因子は、単結晶形を優勢にさせることがありうる。
【0033】
本明細書中で用いられる「賦形剤」という用語は、本発明の化合物を活性成分として含有する錠剤などの薬物または医薬品の製造に用いることができる反応不活性または不活性(inert or inactive)物質を意味する。賦形剤という用語には、結合剤、崩壊剤、コーティング、圧縮/封入助剤、クリームまたはローション剤、滑沢剤、非経口投与用溶液、チュアブル錠用材料、甘味剤または着香剤、懸濁化/ゲル化剤、または湿式造粒剤として用いられるいずれの物質も制限されることなく含めたいろいろな物質を包含することができる。結合剤には、例えば、カルボマー、ポビドン、キサンタンガム等が含まれ;コーティングには、例えば、酢酸フタル酸セルロース、エチルセルロース、ゲランガム(gellan gum)、マルトデキストリン、腸溶コーティング等が含まれ;圧縮/封入助剤には、例えば、炭酸カルシウム、デキストロース、フルクトースdc(dc=「直接圧縮性(directly compressible)」、ハチミツdc、ラクトース(無水物または一水和物;場合により、アスパルテーム、セルロースまたは微結晶性セルロースとの組み合わせで)、デンプンdc、スクロース等が含まれ;崩壊剤には、例えば、クロスカルメロースナトリウム、ゲランガム、ナトリウムデンプングリコラート等が含まれ;クリーム剤またはローション剤には、例えば、マルトデキストリン、カラゲナン等が含まれ;滑沢剤には、例えば、ステアリン酸マグネシウム、ステアリン酸、ステアリルフマル酸ナトリウム等が含まれ;チュアブル錠用材料には、例えば、デキストロース、フルクトースdc、ラクトース(一水和物、場合により、アスパルテームまたはセルロースとの組み合わせで)等が含まれ;懸濁化/ゲル化剤には、例えば、カラゲナン、ナトリウムデンプングリコラート、キサンタンガム等が含まれ;甘味剤には、例えば、アスパルテーム、デキストロース、フルクトースdc、ソルビトール、スクロースdc等が含まれ;そして湿式造粒剤には、例えば、炭酸カルシウム、マルトデキストリン、微結晶性セルロース等が含まれる。
【0034】
本明細書中で用いられるように、そして特に断らない限り、「対象」という用語は、本明細書中において、霊長類(例えば、ヒト)、雌牛、ヒツジ、ヤギ、ウマ、イヌ、ネコ、ウサギ、ラット、マウス等が含まれるがこれに制限されるわけではない哺乳動物などの動物を包含すると定義される。特定の態様において、対象はヒトである。
【0035】
B.複素環式化合物およびそれらの医薬組成物
B.1.代表する化合物:
本発明のいくつかの代表する化合物を、表1に挙げる。
【0036】
【表1A】
【0037】
【表1B】
【0038】
B.2.代表的な合成方法
代表的な化合物を、スキームIおよびスキームIIに示される経路によって合成した。当該技術分野により知られている方法は、当業者が用い且つ修飾して、利用可能な出発物質から本発明の化合物を製造することができる。いくつか有用な方法は、公開PCT出願WO2009/023402号に開示されている。本発明の範囲内の化合物を製造するのに有用でありうる追加の合成方法は、例えば、Hayakawa et al., Biorg. Med. Chem. Vol. 15, 403-12 (2007); Ermolat’ev, et al., J. Comb. Chem. Vol. 8, 659-63 (2006); Carballares, et al., Tetrahedron Lett. vol. 48, 2041-45 (2007); および Rupert, et al., Biorg. Med. Chem. Lett., vol. 13, 347-50 (2003) に開示されている。
【0039】
下のスキームは、本明細書中に与えられている化合物の製造のための代表的な合成方法を与える。当業者は、類似の方法を用いて、本明細書中に与えられている化合物を製造することができるということを理解するであろう。言い換えると、当業者は、試薬、保護基、反応条件および反応配列順序への適する調整を用いて、所望の態様を製造することができるということを理解するであろう。それら反応は、製造される物質の量に適合するように規模を上向きまたは下向きにすることができる。
【0040】
本明細書中に与えられている化合物を製造するための具体的なスキームを、下に示す。詳細な反応条件を、いろいろな具体的実施例について本明細書中の下に与える。当業者は、次のスキームを、適当な試薬、保護基、条件、出発物質または反応配列順序で修飾して、本明細書中に与えられている他の態様の製造に適合させることができるということを理解するであろう。
【0041】
本明細書中に開示の参考文献は全て、そのまま援用される。
化合物名は、ChemDraw Ultra 10.0で得た;そして実施例中で用いられる中間体および試薬の名称も、ChemDraw Ultra 10.0で得た。
【0042】
2a.式Iの化合物の一般的な合成スキーム/方法:
スキームI
【0043】
【化3】
【0044】
チアゾール−2−アミン1を、EtOH中の3−クロロペンタン−2,4−ジオン2で、還流温度において処理して、環化生成物3を生じ、それを、HOAc中の臭素で更に臭素化して、α−ブロモケトン4を生じた。置換されたアニリン5を、アセトン中のイソチオシアン酸ベンゾイル6と反応させて、N−(フェニルカルバモチオイル)ベンズアミド7を生じ、それを、80℃において5%水性NaOHで加水分解して、置換フェニルチオ尿素8を生じた。次に、二つの不可欠な中間体7および8を、EtOH中で加熱して、チアゾール9(式I)をHBr塩として高収率で形成させたが、それは、in vitro および in vivo 研究用に他の薬学的に許容しうる塩(例えば、HCl塩)または遊離塩基形へ変換しうると考えられる。
【0045】
本発明の化合物の製造に必要とされる式5の置換アニリンは、商業的に入手可能であるまたは既知の方法によって合成することができる。
2b.式IIの化合物の一般的な合成スキーム/方法:
スキームII
【0046】
【化4】
【0047】
ピリミジン−2−アミン10を、EtOH中の3−クロロペンタン−2,4−ジオン2で、還流温度において処理して、環化生成物11を生じ、それを、HOAc中の臭素で更に臭素化して、α−ブロモケトン12を生じた。置換アニリン5を、アセトン中のイソチオシアン酸ベンゾイル6と反応させて、N−(フェニルカルバモチオイル)ベンズアミド7を生じ、それを、80℃において5%水性NaOHで加水分解して、置換フェニルチオ尿素8を生じた。次に、二つの不可欠な中間体12および8を、EtOH中で加熱して、チアゾール13(式II)をHBr塩として高収率で形成させたが、それは、in vitro および in vivo 研究用に他の薬学的に許容しうる塩(例えば、HCl塩)または遊離塩基形へ変換しうると考えられる。
【0048】
本発明の化合物の製造に必要とされる式5の置換アニリンは、商業的に入手可能であるまたは既知の方法によって合成することができる。
【実施例】
【0049】
B.3.実施例:
以下の実施例は、本発明を詳しく説明するために与えるが、制限するためではない。
スキームIおよびスキームIIに記載の合成方法/スキームにしたがって、表1の化合物を合成した。いくつか代表的な合成を、本明細書中に与える。
【0050】
実施例B(1):2−(4−エトキシフェニルアミノ)−4−(2−メチルイミダゾ[1,2−a]ピリミジン−3−イル)チアゾール一臭化水素酸塩(a)の合成:
化合物aの合成を、スキームIIIに示す:
スキームIII
【0051】
【化5】
【0052】
1−(2−メチルイミダゾール[1,2−a]ピリミジン−3−イル)エタノン(4)の合成。
3−クロロ−2,5−ペンタンジオン(2)(106mL,119g,887mmol,1.2eq)を、650mLの無水エタノール中に溶解させた。2−アミノピリミジン(1)(71.5gx97%=69.36g,729mmol)を、上の撹拌溶液に加えた。得られた混合物を、100〜105℃の油浴温度で40時間還流した。黒色反応混合物を冷却し、減圧下で濃縮した。残留物を、飽和重炭酸ナトリウム溶液(約500mL)で少量ずつ処理し、フラスコを波立たさせて十分に混合した。その混合物を、ジクロロメタン(x6)で抽出した。抽出物を、重炭酸ナトリウム溶液およびブラインで洗浄した。有機相を乾燥させ、濃縮した。残留物を、シリカゲルカラム(7x30cm)上のフラッシュクロマトグラフィーにより、n−ヘキサン−酢酸エチル(3:1、2:1、1:1、1:2および0:1)、そして次にジクロロメタン−メタノール(30:1、20:1、10:1および5:1)を用いた勾配溶離で精製した。生成物画分を集め(TLC、Rf0.36、100%酢酸エチル)、濃縮して、淡黒色固体を与えた。生成物が入っている他の画分を集め、再度同様に再精製した。27.84g(21.8%)の最終生成物を得た。生成物の一部分を、少量のアセトニトリルから再結晶させて、赤〜淡褐色結晶4、m.p.255.6〜256.6℃を生じた。
【0053】
1H NMR (CDCl3) δ 2.65 (s, 3H, 3-COCH3), 2.86 (s, 3H, 2-CH3), 7.04-7.12 (m, 1H, 6-H), 7.70-7.74 (m, 1H), 9.96-10.00 (m, 1H)。
2−ブロモ−1−(2−メチルイミダゾ[1,2−a]ピリミジン−3−イル)エタノン一臭化水素酸塩(5)の合成(臭素化)。
【0054】
1−(2−メチルイミダゾール[1,2−a]ピリミジン−3−イル)エタノン(4)(1.75g,10mmol)を、20mLの氷酢酸中に、フラスコを静かに暖めることによって溶解させた後、それを室温に冷却した。4mLの酢酸中の臭素(0.6mL,1.85g,11.5mmol,1.15eq)の溶液を、上の撹拌反応混合物に室温で30分間を超えて徐々に加えた。[注意:臭素は、極めて腐食性である。臭素の取り扱いは、十分に換気されたヒュームフード中で極めて注意深く行う必要がある。長手袋または二重手袋を操作に必要とする。皮膚上へ跳ねかけるまたは蒸気を吸うのを絶対に避けられたい]。若干の固体が、添加を終える前に析出した。反応混合物を、100〜110℃の油浴温度で3時間撹拌後、室温で一晩撹拌した。固体を濾過し、そしてアセトンで数回洗浄し、アセトン洗浄の途中に時々、無水エタノールで洗浄した。淡褐色固体を、少量のエタノールを含有するアセトンで吸収させ、その混合物を、室温で5時間を超えて撹拌した。固体を濾過し、その固体を上述のように洗浄した(もう一回吸収させ、より大きい規模用に洗浄サイクルを提示する)。真空下で乾燥後、2.43g(72.5%)の淡褐色粉末固体生成物5を、一臭化水素酸塩として得た。250℃より上で分解。
【0055】
TLC、Rf0.42(100%酢酸エチル)。
1HNMR (DMSO-d6) δ 2.82 (s, 3H, 2-CH3), 4.83 (s, 2H, CH2Br), 7.40-7.50 (m, 1H), 8.80-8.90 (m, 1H), 9.82-9.90 (m, 1H)。
【0056】
1−ベンゾイル−3−[4−(エトキシフェニル)]チオ尿素(9)の合成。
[参考文献:ARKIVOC 2003, 434-442; Bioorg. Med. Chem. 2000, 2663]。塩化ベンゾイル(6)(14.0mL,16.95g,120mmol)を、100mLのアセトン中のチオシアン酸アンモニウム(10.26 g,135mmol,1.125eq)の撹拌溶液に室温で滴加した。若干の白色固体を析出した。反応混合物を、加熱して5分間還流した(約65〜70℃の油温度)。このようにして得られたイソチオシアン酸ベンゾイル(7)を、精製することなく、次の工程に直接的に用いた。25mLのアセトン中の4−エトキシアニリン(8)(17.0mL,18.1g,132mmol,1.1eq)の溶液を、油浴(65〜70℃)中に静置している上の撹拌反応混合物に徐々に加えた。添加は、発熱反応を考慮して、極めて徐々に約1時間行う必要がある。多量の白色固体が析出した。反応混合物を、手動で波立たせ、そして更に、5分間還流した。冷却した反応混合物を、氷水中に注いだ。固体を濾過し、水で3回洗浄した。その固体を、エタノール(約1.6L)から再結晶させて、所望の生成物9を淡黄色の長針状結晶、36g収量(99%)、m.p.151.0〜153.5℃として与えた。
【0057】
(p−エトキシフェニル)チオ尿素(10)の合成。
水酸化ナトリウム水溶液(1M,60mL,60mmol,1.2eq)を、350mLのエタノール中の1−ベンゾイル−3−[4−(エトキシフェニル)]チオ尿素(9)(16.5g,55mmol)の撹拌混合物に加えた。反応混合物を、1時間還流し、冷却し、濃縮した。その白色固体を、水(約200mL)で処理した。固体を濾過し、水で洗浄した。粗製結晶性生成物を、エタノールから再結晶させ、濾過し、真空下で乾燥させて、7.66g(71.0%)の所望の生成物10、m.p.176.5〜178.5℃を与えた。
【0058】
TLC、Rf0.45(n−ヘキサン−酢酸:1:1)。
1HNMR (DMSO-d6) δ 1.31 (t, 3H, J = 6.8Hz), 4.00 (q, 2H, J = 6.8Hz), 6.80-6.90 (m, 2H), 7.15-7.25 (m, 2H), 9.50 (s, 1H, NH)。
【0059】
2−(4−エトキシフェニルアミノ)−4−(2−メチルイミダゾ[1,2−a]ピリミジン−3−イル)チアゾール一臭化水素酸塩(a)の合成(チアゾール環への環化)。
140mLの無水エタノール中の2−ブロモ−1−(2−メチルイミダゾ[1,2−a]ピリミジン−3−イル)エタノン一臭化水素酸塩(5)(2.43g,7.25mmol)および(p−エトキシフェニル)チオ尿素(10)(1.40g,7.1mmol)の混合物を、撹拌しながら15時間還流した(約105℃の油浴温度)。次に、それを室温で6時間または一晩撹拌した。固体を濾過し、アセトンで洗浄した。粗製軟質結晶を、アセトン−エタノール(3:1)で吸収させ、室温で6時間を超えてまたは一晩撹拌した。固体を濾過し、上のように洗浄した。粗生成物を、アセトン−エタノール(3:1)で吸収させ、室温で6時間を超えて撹拌した。粗生成物を、濾過し、洗浄し、メタノールから再結晶させた。そのメタノール溶液を、熱い内に濾過して、黒色微粉を除去後、加熱して溶液とした。黄色結晶を濾過し、洗浄した。それを、メタノールから更に2回再結晶させ、真空下で乾燥させて、所望の生成物11を長い軟質黄色針状結晶、1.55g収量(50.5%)として与え、240℃より上で分解した。
【0060】
TLC Rf0.32(ジクロロメタン−メタノール:20:1);Rf0.46(ジクロロメタン−メタノール含有:20:1、1%水酸化アンモニウム水溶液を含有);Rf0.30(100%酢酸エチルX2)。
【0061】
HPLC純度:99%。
1HNMR (DMSO-d6) δ 1.32 (t, 3H, J = 6.8Hz), 2.67 (s, 3H), 4.00 (q, 2H, J = 6.8Hz), 6.90-6.95 (m, 2H), 7.35 (s, 1H), 7.49-7.52 (m, 2H), 7.61-7.67 (m, 1H), 8.94-8.97 (m, 1H), 9.57 (d, 1H, J = 6.8Hz), 10.28 (s, 1H, NH)。
【0062】
ESI−MS,m/z352(M+1)+。
実施例B(2):2−(4−ブロモフェニル)アミノ−4−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)−チアゾール一臭化水素酸塩(u)の合成。
【0063】
化合物uの合成を、スキームIVに示す。
スキームIV
【0064】
【化6】
【0065】
(p−ブロモフェニル)チオ尿素(14)の合成:p−ブロモアニリンをp−エトキシルアニリンの代わりに用いる、化合物10の合成と類似の方法。
1−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)エタノン(17)の合成。2−アミノチアゾールを、無水エタノールから再結晶させ、濾過し、そして乾燥後に使用した。180mLの無水エタノール中の2−アミノチアゾール(16)(20.9g,202.4mmoL)および3−クロロ−2,5−ペンタンジオン(2)(33.7g,97%,242.9mmol,1.2eq)の溶液を、油浴中で72時間還流した。黒色反応混合物を、冷却し、減圧下で濃縮した。残留物を、飽和重炭酸ナトリウム溶液で少量ずつ処理後、ジクロロメタンで抽出した。有機相を乾燥させ、濃縮した。残留物を、シリカゲルカラム上のフラッシュクロマトグラフィーにより、ジクロロメタン−メタノール(80:1)を用いて精製した。生成物画分を集め(TLC、Rf0.60中性形;Rf=0.5塩形、40:1のジクロロメタン−メタノール)、濃縮して、白色固体生成物17を、11.7%収率、1.6gの中性形および3.2gの塩形で与えた。
【0066】
1H NMR (CDCl3) δ 2.55 (s, 3H, 5-COCH3), 2.70 (s, 3H, 6-CH3), 6.78 (d, 1H, J = 4.8 Hz), 8.39 (d, 1H, J = 4.8 Hz)。
2−ブロモ−1−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)エタノン臭化水素酸塩(18)の合成。1−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)エタノン(17)(0.54g,3.0mmol)を、7mLの氷酢酸中に溶解させた。3mLの氷酢酸中の臭素(0.18mL,0.56g,3.5mmol)の溶液を、上の撹拌溶液に30分間で徐々に加えた。若干の黄色固体が現れた。その混合物を、加熱して撹拌しながら3時間還流後、室温で一晩撹拌した。固体を濾過し、アセトンで3回洗浄し、そして各々の洗浄に3〜5時間の撹拌を必要とした。固体を濾過し、真空下で乾燥させて、所望の生成物18である0.73g(71.6%)の白色固体を与えた。
【0067】
2−(4−ブロモフェニル)アミノ−4−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)−チアゾール一臭化水素酸塩(u)の合成。10mLの無水エタノール中の2−ブロモ−1−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)エタノン臭化水素酸塩(18)(0.73g,2.0mmol)および(p−ブロモフェニル)チオ尿素(14)(0.50g,2.0mmol)の混合物を、撹拌しながら20時間還流後、室温に冷却した。固体を濾過した。その粗製白色固体生成物を、メタノールから再結晶させた。メタノール溶液を、まだ熱い内に濾過して、可能性のある微粉を除去後、加熱して溶液とした。それを、メタノールから更に2回再結晶させ、真空下で乾燥させて、所望の生成物uを、白色固体、0.34g収量(36%)、HPLC純度98.49%、mp>250℃として与えた。
【0068】
1HNMR (CD3OD) δ 2.68 (s, 3H), 7.45 (s, 1H), 7.18 (s, 1H), 7.18-7.47 (m, 2H), 7.54-7.66 (m, 2H), 7.66 (d, 1H, J = 4.4Hz), 8.94 (d, 1H, J = 4.4Hz)。
実施例B(3):2−(4−エチルフェニル)アミノ−4−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)−チアゾール一臭化水素酸塩(o)の合成。
【0069】
化合物(o)の合成は、スキームIVに示している。
(p−ブロモフェニル)チオ尿素(21)の合成:p−エチルアニリンをp−エトキシルアニリンの代わりに用いる、化合物10の合成と類似の方法。
【0070】
2−(4−エチルフェニル)アミノ−4−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)−チアゾール一臭化水素酸塩(o)の合成。40mLの無水エタノール中の2−ブロモ−1−(6−メチルイミダゾ[2,1−b]チアゾール−5−イル)エタノン臭化水素酸塩(18)(20g,91%,53.5mmol)および(p−エチルフェニル)チオ尿素(10)(10.1g,56.2mmol)の混合物を、撹拌しながら3時間還流後、室温に冷却した。固体を濾過し、エタノールで洗浄した。粗製白色固体生成物を、メタノールから再結晶させた。メタノール溶液を、まだ熱い内に濾過して、可能性のある微粉を除去後、加熱して溶液とした。それを、メタノールからもう1回再結晶させ、真空下で乾燥させて、所望の生成物(o)を白色固体として与えた。このHBr塩を、遊離塩基へ変換後、HCl塩へ変換し、それを、50%エタノール/水から再結晶させて、オフホワイト結晶生成物(14.8g,73.4%)を生じた。
【0071】
HPLC純度99%、mp=181〜183℃。
1HNMR (CD3OD) δ 1.11 (t, 3H, J = 7.6 Hz), 2.52 (s, 3H), 2.58 (m, 2H), 7.11 (d, 2H, J = 8Hz), 7.22 (s, 1H), 7.52 (d, 2H, J = 8.4Hz), 7.75 (d, 1H, J = 4.4Hz), 8.42 (d, 1H, J = 4.4Hz)。
【0072】
ESI−MSm/z341.5(M+1)+。
式Iまたは式IIの化合物(HBr塩)からの遊離塩基化合物の製造。
HBr塩を、メタノール中に懸濁させ、過剰量の重炭酸ナトリウムを、懸濁した化合物塩が完全に溶解するまで激しく撹拌しながら加えた。過剰量の無機塩を濾去した。溶液を濃縮し、そして残留物を、メタノールまたはエタノールまたはエタノール/水またはいずれか他の一つまたは複数の有機溶媒または溶媒混合物から再結晶させて、結晶質を遊離塩基化合物として与えた。
【0073】
異なった塩の製造。得られた遊離塩基化合物および1.1当量の選択された酸。その溶液を濃縮し、そして残留物を、メタノールまたはエタノールまたはエタノール/水またはいずれか他の一つまたは複数の有機溶媒または溶媒混合物から再結晶させて、上記のような選択された陰イオンを含む所望の塩の結晶質/多形物質を与えた。
【0074】
記載の化合物の遊離塩基または塩の可能性のある結晶形/多形は全て、本発明に包含される。
B.3.追加の活性剤
本明細書中の化合物は、本発明の方法および組成物において、他の薬理学的に活性な化合物(「追加の活性剤」)と組み合わせることができる。特定の組み合わせは、特定のタイプの癌、および望ましくない血管新生に関連したまたはそれを特徴とする特定の疾患および状態の処置において共力的に働くと考えられる。免疫調節性化合物も、特定の二次活性剤に関連した副作用を軽減するように働くことができるし、そしていくつかの二次活性剤は、免疫調節性化合物に関連した副作用を軽減するのに用いることができる。
【0075】
一つまたはそれを超える活性成分または活性剤は、本発明の方法および組成物において一緒に用いることができる。追加の活性剤は、高分子(例えば、タンパク質)または低分子(例えば、合成の無機分子または有機金属分子または有機分子)でありうる。
【0076】
高分子活性剤の例には、造血増殖因子、サイトカイン、および単クローン性および多クローン性抗体が含まれるが、これに制限されるわけではない。典型的な高分子活性剤は、天然に存在するまたは人工的に作られたタンパク質などの生体分子である。本発明において特に有用であるタンパク質には、造血前駆細胞および免疫学的に活性な生産性細胞(poietic cell)の in vitro または in vivo の生存および/または増殖を刺激するタンパク質が含まれる。その他は、in vitro または in vivo の細胞における拘束された(committed)赤血球前駆体の分裂および分化を刺激する。具体的なタンパク質には、IL−2(リコンビナントIL−II(「rIL2」)およびカナリア痘IL−2を含めた)、IL−10、IL−12およびIL−18などのインターロイキン;インターフェロンα−2a、インターフェロンα−2b、インターフェロンα−n1、インターフェロンα−n3、インターフェロンβ−Iaおよびインターフェロンγ−Ibなどのインターフェロン;GM−CFおよびGM−CSF;およびEPOが含まれるが、これに制限されるわけではない。
【0077】
本発明の方法および組成物において用いることができる具体的なタンパク質には、米国において Neupogen.RTM.(Amgen, Thousand Oaks, Calif.)という商品名で販売されているフィルグラスティム;米国において Leukine.RTM.(Immunex, Seattle, Wash.)という商品名で販売されているサルグラモスチン;および米国において Epogen.RTM.(Amgen, Thousand Oaks, Calif.)という商品名で販売されているリコンビナントEPOが含まれるが、これに制限されるわけではない。
【0078】
GM−CSFのリコンビナントおよび突然変異形は、米国特許第5,391,485号;同第5,393,870号;および同第5,229,496号に記載のように製造することができる;それらは全て、本明細書中に援用される。G−CSFのリコンビナントおよび突然変異形は、米国特許第4,810,643号;同第4,999,291号;同第5,528,823号;および同第5,580,755号に記載のように製造することができる;それらは全て、本明細書中に援用される。
【0079】
本発明は、自然のままの、天然に存在するおよびリコンビナントのタンパク質の使用を包含する。本発明は、更に、天然に存在するタンパク質の突然変異体および誘導体(例えば、修飾された形)であって、それらが基づいているタンパク質の少なくともいくつかの薬理活性を in vivo で示すものを包含する。突然変異体の例には、それらタンパク質の天然に存在する形の該当する残基とは異なる一つまたはそれを超えるアミノ残基を有するタンパク質が含まれるが、これに制限されるわけではない。「突然変異体」という用語によって更に包含されるのは、それら天然に存在する形(例えば、非グリコシル化形)に普通に存在する炭水化物部分を欠いているタンパク質が含まれる。誘導体の例には、ペグ化誘導体;およびIgG1またはIgG3をタンパク質または目的のタンパク質の活性部分に融合することによって形成されるタンパク質などの融合タンパク質が含まれるが、これに制限されるわけではない。例えば、Penichet, M. L. and Morrison, S. L., J. Immunol. Methods 248:91-101 (2001) を参照されたい。
【0080】
本発明の化合物との組み合わせで用いることができる抗体には、単クローン性抗体および多クローン性抗体が含まれる。抗体の例には、トラスツズマブ(Herceptin.RTM.)、リツキシマブ(rituximab)(Rituxan.RTM.)、ベバシズマブ(bevacizumab)(Avastin.TM.)、ペルツズマブ(pertuzumab)(Omnitarg.TM.)、トシツモマブ(tositumomab)(Bexxar.RTM.)、エドレコロマブ(edrecolomab)(Panorex.RTM.)およびG250が含まれるが、これに制限されるわけではない。本発明の化合物は、更に、抗TNF−α抗体と一緒にすることができるまたはそれと組み合わせて用いることができる。
【0081】
高分子活性剤は、抗癌ワクチンの形で投与することができる。例えば、IL−2、G−CSFおよびGM−CSFなどのサイトカインを分泌するまたは分泌を引き起こすワクチンは、本発明の方法、医薬組成物およびキットに用いることができる。例えば、Emens, L. A., et al., Curr. Opinion Mol. Ther. 3(1):77-84 (2001) を参照されたい。
【0082】
本発明の一つの態様において、高分子活性剤は、免疫調節性化合物の投与に関連した副作用を減少させる、排除するまたは予防する。具体的な免疫調節性化合物および処置されている(begin treated)疾患または障害に依存して、副作用には、嗜眠状態および傾眠、めまい感および起立性低血圧、好中球減少症、好中球減少症に起因する感染、増加したHIVウイルス量、徐脈、スティーブンズ−ジョンソン症候群および中毒性表皮壊死症、および発作(例えば、大発作痙攣)が含まれうるが、これに制限されるわけではない。具体的な副作用は、好中球減少症である。
【0083】
低分子である追加の活性剤も、免疫調節性化合物の投与に関連した副作用を軽減するのに用いることができる。低分子活性剤の例には、抗癌剤、抗生物質、免疫抑制剤およびステロイドが含まれるが、これに制限されるわけではない。
【0084】
抗癌剤の例には、アシビシン(acivicin);アクラルビシン;アコダゾール(acodazole)塩酸塩;アクロニン(acronine);アドゼレシン(adozelesin);アルデスロイキン(aldesleukin);アルトレタミン(altretamine);アンボマイシン(ambomycin);酢酸アメタントロン(ametantrone);アムサクリン;アナストロゾール(anastrozole);アントラマイシン;アスパラギナーゼ;アスペルリン(asperlin);アザシチジン;アゼテパ(azetepa);アゾトマイシン(azotomycin);バチマスタト(batimastat);ベンゾデパ(benzodepa);ビカルタミド(bicalutamide);ビスアントレン(bisantrene)塩酸塩;ビスナフィド(bisnafide)ジメシレート;ビゼレシン(bizelesin);硫酸ブレオマイシン;ブレキナル(brequinar)ナトリウム;ブロピリミン(bropirimine);ブスルファン;カクチノマイシン;カルステロン(calusterone);カラセミド(caracemide);カルベチマー(carbetimer);カルボプラチン;カルムスチン;カルビシン(carubicin)塩酸塩;カルゼレシン(carzelesin);セデフィンゴール(cedefingol);セレコキシブ(celecoxib)(COX−2阻害剤);クロラムブシル(chlorambucil);シロレマイシン(cirolemycin);シスプラチン(cisplatin);クラドリビン(cladribine);クリスナトール(crisnatol)メシレート;シクロホスファミド;シタラビン(cytarabine);ダカルバジン;ダクチノマイシン;ダウノルビシン塩酸塩;デシタビン(decitabine);デキソルマプラチン(dexormaplatin);デザグアニン(dezaguanine);デザグアニンメシレート;ジアジクオン(diaziquone);ドセタキセル(docetaxel);ドキソルビシン;ドキソルビシン塩酸塩;ドロロキシフェン(droloxifene);クエン酸ドロロキシフェン;プロピオン酸ドロモスタノロン;ドゥアゾマイシン(duazomycin);エダトレキサート(edatrexate);エフロルニチン(eflornithine)塩酸塩;エルサミトルシン(elsamitrucin);エンロプラチン(enloplatin);エンプロメート(enpromate);エピプロピジン(epipropidine);エピルビシン(epirubicin)塩酸塩;エルブロゾール(erbulozole);エソルビシン(esorubicin)塩酸塩;エストラムスチン;エストラムスチンリン酸ナトリウム;エタニダゾール(etanidazole);エトポシド;リン酸エトポシド;エトプリン(etoprine);ファドロゾール(fadrozole)塩酸塩;ファザラビン(fazarabine);フェンレチニド(fenretinide);フロクスウリジン(floxuridine);リン酸フルダラビン(fludarabine);フルオロウラシル;フルオロシタビン(fluorocitabine);ホスキドン(fosquidone);ホストリエシン(fostriecin)ナトリウム;ジェムシタビン(gemcitabine);ジェムシタビン塩酸塩;ヒドロキシ尿素;イダルビシン(idarubicin)塩酸塩;イホスファミド;イルモホシン(ilmofosine);イプロプラチン(iproplatin);イリノテカン(irinotecan);イリノテカン塩酸塩;酢酸ランレオチド(lanreotide);レトロゾール(letrozole);酢酸ロイプロリド;リアロゾール(liarozole)塩酸塩;ロメトレキソール(lometrexol)ナトリウム;ロムスチン;ロソキサントロン(losoxantrone)塩酸塩;マソプロコール(masoprocol);メイタンシン;メクロレタミン(mechlorethamine)塩酸塩;酢酸メゲストロール(megestrol);酢酸メレンゲストロール;メルファラン;メノガリル(menogaril);メルカプトプリン;メトトレキサート;メトトレキサートナトリウム;メトプリン(metoprine);メツレデパ(meturedepa);ミチンドミド(mitindomide);ミトカルシン(mitocarcin);ミトクロミン(mitocromin);ミトジリン(mitogillin);ミトマルシン(mitomalcin);マイトマイシン;ミトスペル(mitosper);ミトタン(mitotane);ミトザントロン塩酸塩;ミコフェノール酸;ノコダゾール(nocodazole);ノガラマイシン(nogalamycin);オルマプラチン(ormaplatin);オキシスラン(oxisuran);パクリタキセル;ペガスパルガーゼ(pegaspargase);ペリオマイシン(peliomycin);ペンタムスチン(pentamustine);硫酸ペプロマイシン(peplomycin);ペルホスファミド(perfosfamide);ピポブロマン;ピポスルファン(piposulfan);ピロキサントロン(piroxantrone)塩酸塩;プリカマイシン(plicamycin);プロメスタン(plomestane);ポルフィマー(porfimer)ナトリウム;ポルフィロマイシン(porfiromycin);プレドニムスチン;プロカルバジン塩酸塩;ピュロマイシン;ピュロマイシン塩酸塩;ピラゾフリン;リボプリン(riboprine);サフィンゴール(safingol);サフィンゴール塩酸塩;セムスチン;シムトラゼン(simtrazene);スパルホセート(sparfosate)ナトリウム;スパルソマイシン(sparsomycin);スピロゲルマニウム塩酸塩;スピロムスチン(spiromustine);スピロプラチン(spiroplatin);ストレプトニグリン(streptonigrin);ストレプトゾシン;スロフェヌル(sulofenur);タリソマイシン(talisomycin);テコガラン(tecogalan)ナトリウム;タキソテール(taxotere);テガフル;テロキサントロン(teloxantrone)塩酸塩;テモポルフィン(temoporfin);テニポシド(teniposide);テロキシロン(teroxirone);テストラクトン(testolactone);チアミプリン(thiamiprine);チオグアニン;チオテパ;チアゾフリン(tiazofurin);チラパザミン(tirapazamine);クエン酸トレミフェン(toremifene);酢酸トレストロン(trestolone);リン酸トリシリビン(triciribine);トリメトレキサート(trimetrexate);グルクロン酸トリメトレキサート;トリプトレリン(triptorelin);ツブロゾール(tubulozole)塩酸塩;ウラシルマスタード;ウレデパ(uredepa);バプレオチド(vapreotide);ベルテポルフィン;硫酸ビンブラスチン;硫酸ビンクリスチン;ビンデシン;硫酸ビンデシン;硫酸ビネピジン(vinepidine);硫酸ビングリシネート(vinglycinate);硫酸ビンロイロシン(vinleurosine);酒石酸ビノレルビン(vinorelbine);硫酸ビンロシジン(vinrosidine);硫酸ビンゾリジン(vinzolidine);ボロゾール(vorozole);ゼニプラチン(zeniplatin);ジノスタチン(zinostatin);およびゾルビシン塩酸塩が含まれるが、これに制限されるわけではない。
【0085】
他の抗癌薬には、20−エピ−1,25ジヒドロキシビタミンD3;5−エチニルウラシル;アビラテロン(abiraterone);アクラルビシン;アシルフルベン(acylfulvene);アデシペノール(adecypenol);アドゼレシン;アルデスロイキン;ALL−TKアンタゴニスト;アルトレタミン;アンバムスチン(ambamustine);アミドクス(amidox);アミホスチン(amifostine);アミノレブリン酸;アムルビシン(amrubicin);アムサクリン;アナグレリド(anagrelide);アナストロゾール;アンドログラホリド;血管新生阻害剤;アンタゴニストD;アンタゴニストG;アンタレリクス(antarelix);抗背方化形態形成プロテイン−1(anti-dorsalizing morphogenetic protein-1);抗アンドロゲン、前立腺癌;抗エストロゲン;アンチネオプラストン;アンチセンスオリゴヌクレオチド;アフィジコリングリシネート;アポトーシス遺伝子モジュレーター;アポトーシスレギュレーター;アプリン酸;ara−CDP−DL−PTBA;アルギニンデアミナーゼ;アスラクリン(asulacrine);アタメスタン(atamestane);アトリムスチン(atrimustine);アクシナスタチン1(axinastatin 1);アクシナスタチン2;アクシナスタチン3;アザステロン(azasetron);アザトキシン(azatoxin);アザチロシン(azatyrosine);バカチンIII(baccatin III)誘導体;バラノール(balanol);バチマスタト;BCR/ABLアンタゴニスト;ベンゾクロリン(benzochlorins);ベンゾイルスタウロスポリン(benzoylstaurosporine);βラクタム誘導体;β−アレチン(alethine);βクラマイシンB(clamycin B);ベツリン酸(betulinic acid);bFGF阻害剤;ビカルタミド;ビスアントレン;ビスアジリジニルスペルミン(bisaziridinylspermine);ビスナフィド;ビストラテン(bistratene)A;ビゼレシン;ブレフレート(breflate);ブロピリミン;ブドチタン(budotitane);ブチオニンスルホキシイミン(buthionine sulfoximine);カルシポトリオール(calcipotriol);カルホスチンC(calphostin C);カンプトテシン誘導体;カペシタビン;カルボキサミドアミノトリアゾール;カルボキシアミドトリアゾール;CaRest M3;CARN700;軟骨由来阻害剤;カルゼレシン;カゼインキナーゼ阻害剤(ICOS);カスタノスペルミン(castanospermine);セクロピンB;セトロレリクス(cetrorelix);クロリン;クロロキノキサリンスルホンアミド;シカプロスト(cicaprost);cis−ポルフィリン;クラドリビン(cladribine);クロミフェン類似体;クロトリマゾール;コリスマイシンA(collismycin A);コリスマイシンB;コンブレタスタチンA4(combretastatin A4);コンブレタスタチン類似体;コナゲニン(conagenin);クラムベシジン816(crambescidin 816);クリスナトール;クリプトフィシン8(cryptophycin 8);クリプトフィシンA誘導体;クラシンA(curacin A);シクロペンタアントラキノン(cyclopentanthraquinones);シクロプラタム(cycloplatam);シペマイシン(cypemycin);シタラビンオクホスフェート(cytarabine ocfosfate);細胞溶解性因子;シトスタチン(cytostatin);ダクリキシマブ(dacliximab);デシタビン;デヒドロジデムニンB(dehydrodidemnin B);デスロレリン(deslorelin);デキサメタゾン;デキシホスファミド(dexifosfamide);デクスラゾキサン(dexrazoxane);デクスベラパミル(dexverapamil);ジアジクオン;ジデムニンB(didemnin B);ジドクス(didox);ジエチルノルスペルミン(diethylnorspermine);ジヒドロ−5−アザシチジン;ジヒドロタキソール、9−;ジオキサマイシン(dioxamycin);ジフェニルスピロムスチン;ドセタキセル;ドコサノール(docosanol);ドラセトロン(dolasetron);ドキシフルリジン(doxifluridine);ドキソルビシン;ドロロキシフェン;ドロナビノール;ズオカルマイシンSA(duocarmycin SA);エブセレン(ebselen);エコムスチン(ecomustine);エデルホシン(edelfosine);エドレコロマブ;エフロルニチン;エレメン(elemene);エミテフル(emitefur);エピルビシン;エプリステリド(epristeride);エストラムスチン類似体;エストロゲンアゴニスト;エストロゲンアンタゴニスト;エタニダゾール;リン酸エトポシド;エクセメスタン(exemestane);ファドロゾール;ファザラビン;フェンレチニド;フィルグラスティム;フィナステリド(finasteride);フラボピリドール(flavopiridol);フレゼラスチン(flezelastine);フルアステロン(fluasterone);フルダラビン;フルオロダウノルニシン(fluorodaunorunicin)塩酸塩;ホルフェニメクス(forfenimex);ホルメスタン(formestane);ホストリエシン;ホテムスチン(fotemustine);ガドリニウムテキサフィリン(gadolinium texaphyrin);硝酸ガリウム;ガロシタビン(galocitabine);ガニレリクス(ganirelix);ゼラチナーゼ阻害剤;ジェムシタビン;グルタチオン阻害剤;ヘプスルファム(hepsulfam);ヘレグリン(heregulin);ヘキサメチレンビスアセトアミド;ヒペリシン;イバンドロン酸(ibandronic acid);イダルビシン;イドキシフェン(idoxifene);イドラマントン(idramantone);イルモホシン;イロマスタト(ilomastat);イマチニブ(例えば、Gleevec.RTM.)、イミキモッド;免疫刺激ペプチド;インスリン様増殖因子−1受容体阻害剤;インターフェロンアゴニスト;インターフェロン;インターロイキン;イオベングアン(iobenguane);ヨードドキソルビシン;イポメアノール(ipomeanol)、4−;イロプラクト(iroplact);イルソグラジン(irsogladine);イソベンガゾール(isobengazole);イソホモハリコンドリンB(isohomohalicondrin B);イタセトロン(itasetron);ジャスプラキノリド(jasplakinolide);カハラリドF(kahalalide F);ラメラリン(lamellarin)−Nトリアセテート;ランレオチド;リーナマイシン(leinamycin);レノグラスチム(lenograstim);硫酸レンチナン(lentinan);レプトルスタチン(leptolstatin);レトロゾール;白血病阻害因子;白血球αインターフェロン;ロイプロリド+エストロゲン+プロゲステロン;ロイプロレリン(leuprorelin);レバミゾール;リアロゾール(liarozole);直鎖状ポリアミン類似体;親油性二糖ペプチド;親油性白金化合物;リソクリナミド7(lissoclinamide 7);ロバプラチン(lobaplatin);ロムブリシン(lombricine);ロメトレキソール;ロニダミン(lonidamine);ロソキサントロン;ロクソリビン(loxoribine);ルルトテカン(lurtotecan);ルテチウムテキサフィリン(lutetium texaphyrin);リソフィリン(lysofylline);溶菌ペプチド;メイタンシン;マンノスタチンA(mannostatin A);マリマスタト(marimastat);マソプロコール(masoprocol);マスピン(maspin);マトリライシン阻害剤;マトリックスメタロプロテイナーゼ阻害剤;メノガリル;メルバロン(merbarone);メテレリン(meterelin);メチオニナーゼ;メトクロプラミド;MIF阻害剤;ミフェプリストン;ミルテホシン(miltefosine);ミリモスチム(mirimostim);ミトグアゾン(mitoguazone);ミトラクトール(mitolactol);マイトマイシン類似体;ミトナフィド(mitonafide);マイトトキシン(mitotoxin)線維芽細胞増殖因子−サポリン(saporin);ミトザントロン;モファロテン(mofarotene);モルグラモスチム(molgramostim);Erbitux、ヒト絨毛性ゴナドトロピン(gonadotrophin);モノホスホリルリピドA+ミオバクテリウム(myobacterium)細胞壁sk;モピダモール(mopidamol);マスタード抗癌剤;ミカペルオキシドB(mycaperoxide B);ミコバクテリア細胞壁抽出物;ミリアポロン(myriaporone);N−アセチルジナリン(N-acetyldinaline);N−置換ベンズアミド;ナファレリン(nafarelin);ナグレスチプ(nagrestip);ナロキソン+ペンタゾシン;ナパビン(napavin);ナフテルピン(naphterpin);ナルトグラスチム(nartograstim);ネダプラチン(nedaplatin);ネモルビシン(nemorubicin);ネリドロン酸(neridronic acid);ニルタミド(nilutamide);ニサマイシン(nisamycin);酸化窒素モジュレーター;ニトロオキシド抗酸化剤;ニトルリン(nitrullyn);オブリメルセン(oblimersen)(Genasense.RTM.);O.sup.6−ベンジルグアニン;オクトレオチド(octreotide);オキセノン(okicenone);オリゴヌクレオチド;オナプリストン(onapristone);オンダンセトロン(ondansetron);オンダンセトロン;オラシン(oracin);経口サイトカインインデューサー;オルマプラチン;オサテロン(osaterone);オキサリプラチン(oxaliplatin);オキサウノマイシン(oxaunomycin);パクリタキセル;パクリタキセル類似体;パクリタキセル誘導体;パラウアミン(palauamine);パルミトイルリゾキシン(palmitoylrhizoxin);パミドロン酸(pamidronic acid);パナキシトリオール(panaxytriol);パノミフェン(panomifene);パラバクチン(parabactin);パゼリプチン(pazelliptine);ペガスパルガーゼ;ペルデシン(peldesine);ペントサンポリスルフェートナトリウム;ペントスタチン(pentostatin);ペントロゾール(pentrozole);ペルフルブロン(perflubron);ペルホスファミド;ペリリルアルコール;フェナジノマイシン(phenazinomycin);フェニルアセテート;ホスファターゼ阻害剤;ピシバニル(picibanil);ピロカルピン塩酸塩;ピラルビシン(pirarubicin);ピリトレキシム(piritrexim);プラセチンA(placetin A);プラセチンB;プラスミノーゲンアクチベーター阻害剤;白金錯体;白金化合物;白金−トリアミン錯体;ポルフィマーナトリウム;ポルフィロマイシン;プレドニソン;プロピルビスアクリドン(propyl bis-acridone);プロスタグランジンJ2;プロテアソーム阻害剤;プロテインA基剤免疫モジュレーター;プロテインキナーゼC阻害剤;プロテインキナーゼC阻害剤、ミクロアルガル(microalgal);プロテインチロシンホスファターゼ阻害剤;プリンヌクレオシドホスホリラーゼ阻害剤;プルプリン;ピラゾロアクリジン;ピリドキシル化ヘモグロビンポリオキシエチレンコンジュゲート;rafアンタゴニスト;ラルチトレキセド(raltitrexed);ラモセトロン(ramosetron);rasファルネシルプロテイントランスフェラーゼ阻害剤;ras阻害剤;ras−GAP阻害剤;脱メチルレテリプチン(retelliptine demethylated);レニウムRe186エチドロネート(etidronate);リゾキシン(rhizoxin);リボザイム;RIIレチナミド(RII retinamide);ロヒツキン(rohitukine);ロムルチド(romurtide);ロキニメクス(roquinimex);ルビジノンB1(rubiginone B1);ルボキシル(ruboxyl);サフィンゴール;セイントピン(saintopin);SarCNU;サルコフィトールA(sarcophytol A);サルグラモスチン;Sdi1模擬体;セムスチン;老化由来(senescence derived)阻害剤1;センスオリゴヌクレオチド;シグナル伝達阻害剤;シゾフィラン(sizofiran);ソブゾキサン(sobuzoxane);ナトリウムボロカプテート(borocaptate);ナトリウムフェニルアセテート;ソルベロール(solverol);ソマトメジン結合タンパク質;ソネルミン(sonermin);スパルホス酸(sparfosic acid);スピカマイシンD(spicamycin D);スピロムスチン;スプレノペンチン(splenopentin);スポンジスタチン1(spongistatin 1);スクアラミン(squalamine);スチピアミド(stipiamide);ストロメライシン阻害剤;スルフィノシン(sulfinosine);スーパーアクティブバソアクティブインテスティナルペプチド(superactive vasoactive intestinal peptide)アンタゴニスト;スラジスタ(suradista);スラミン(suramin);スワインソニン(swainsonine);タリムスチン(tallimustine);タモキシフェンメチオジド(tamoxifen methiodide);タウロムスチン(tauromustine);タザロテン(tazarotene)
;テコガラン(tecogalan)ナトリウム;テガフル(tegafur);テルラピリリウム(tellurapyrylium);テロメラーゼ阻害剤;テモポルフィン;テニポシド;テトラクロロデカオキシド;テトラゾミン(tetrazomine);タリブラスチン(thaliblastine);チオコラリン(thiocoraline);トロンボポエチン;トロンボポエチン模擬体;チマルファシン(thymalfasin);チモポエチン受容体アゴニスト;チモトリナン(thymotrinan);甲状腺刺激ホルモン;スズエチルエチオプルプリン(tin ethyl etiopurpurin);チラパザミン(tirapazamine);二塩化チタノセン(titanocene bichloride);トプセンチン(topsentin);トレミフェン;翻訳阻害剤;トレチノイン(tretinoin);トリアセチルウリジン;トリシリビン(triciribine);トリメトレキサート;トリプトレリン;トロピセトロン(tropisetron);ツロステリド(turosteride);チロシンキナーゼ阻害剤;チルホスチン(tyrphostins);UBC阻害剤;ユベニメクス(ubenimex);尿生殖洞由来増殖阻害因子;ウロキナーゼ受容体アンタゴニスト;バプレオチド;バリオリンB(variolin B);ベラレソール(velaresol);ベラミン(veramine);ベルジン;ベルテポルフィン;ビノレルビン(vinorelbine);ビンキサルチン(vinxaltine);ビタキシン(vitaxin);ボロゾール;ザノテロン(zanoterone);ゼニプラチン;ジラスコルブ(zilascorb);およびジノスタチンスティマラマー(zinostatin stimalamer)が含まれるが、これに制限されるわけではない。
【0086】
具体的な追加の活性剤には、オブリメルセン(Genasense.RTM.)、レミケード(remicade)、ドセタキセル、セレコキシブ、メルファラン、デキサメタゾン(Decadron.RTM.)、ステロイド、ジェムシタビン、シスプラチン、テモゾロミド(temozolomide)、エトポシド、シクロホスファミド、テモダール(temodar)、カルボプラチン、プロカルバジン、グリアデル(gliadel)、タモキシフェン、トポテカン(topotecan)、メトトレキサート、Arisa.RTM.、タキソール、タキソテール、フルオロウラシル、ロイコボリン、イリノテカン、キセロダ(xeloda)、CPT−11、インターフェロンα、ペグ化インターフェロンα(例えば、PEG INTRON−A)、カペシタビン、シスプラチン、チオテパ、フルダラビン、カルボプラチン、リポソームダウノルビシン、シタラビン、ドキセタキソール(doxetaxol)、パシリタキセル(pacilitaxel)、ビンブラスチン、IL−2、GM−CSF、ダカルバジン、ビノレルビン、ゾレドロン酸(zoledronic acid)、パルミトロネート(palmitronate)、ビアキシン(biaxin)、ブスルファン、プレドニソン、ビスホスホネート、三酸化ヒ素、ビンクリスチン、ドキソルビシン(Doxil.RTM.)、パクリタキセル、ガンシクロビル(ganciclovir)、アドリアマイシン、エストラムスチンリン酸ナトリウム(Emcyt.RTM.)、スリンダクおよびエトポシドが含まれるが、これに制限されるわけではない。
【0087】
B.4.製剤
本明細書中に記載の化合物の適する製剤はいずれも、製造することができる。化合物が、安定な無毒性の酸または塩基塩を形成するのに十分に塩基性または酸性である場合、それら化合物の塩としての投与は、適当でありうる。薬学的に許容しうる塩の例は、生理学的に許容しうる陰イオンを与える酸で形成される有機酸付加塩、例えば、トシレート、メタンスルホン酸塩、酢酸塩、クエン酸塩、マロン酸塩、酒石酸塩、コハク酸塩、安息香酸塩、アスコルビン酸塩、α−ケトグルタル酸塩およびα−グリセロリン酸塩である。適する無機塩も形成することができ、塩酸塩、硫酸塩、硝酸塩、重炭酸塩および炭酸塩が含まれる。薬学的に許容しうる塩は、当該技術分野において周知の標準的な手順を用いて、例えば、アミンなどの十分に塩基性の化合物を、適する酸と接触させ、生理学的に許容しうる塩を与えることによって得られる。カルボン酸のアルカリ金属(例えば、ナトリウム、カリウムまたはリチウム)塩またはアルカリ土類金属(例えば、カルシウム)塩も包含され、慣用的な方法によって製造される。
【0088】
本発明は、更に、医薬組成物であって、少なくとも一つの本発明の化合物を、少なくとも一つの薬学的に許容しうる賦形剤と混合した状態で含む医薬組成物を包含する。好ましくは、少なくとも一つのこのような賦形剤は、水以外の賦形剤またはC1−C3アルコールまたはジメチルスルホキシドである。
【0089】
本発明の化合物は、経口、局所、経皮でまたは吸入または注射によるものを含めた慣用的な経路によって投与することができる。本発明の化合物は、当業者が、既知の方法を参照して製剤化することができ、そして製剤は、予定の投与経路にしたがって作ることができる。有機化合物を製剤化するのに適する方法は、例えば、REMINGTON’S PHARMACEUTICAL SCIENCES, 18th ed. (1990) に記載され、それは、本明細書中に援用される。
【0090】
これら化合物を薬理学的組成物中で投与する場合、その化合物は、薬学的に許容しうる担体との混合物で製剤することができると考えられる。例えば、考えられる化合物は、薬理学的に許容しうる塩として経口でまたは生理食塩水溶液中で静脈内に投与することができる。リン酸塩、重炭酸塩またはクエン酸塩などの慣用的な緩衝剤は、この目的に用いることができる。当然ながら、当業者は、それら製剤を、本明細書の内容の範囲内で修飾して、特定の投与経路について多数の製剤を与えることができる。具体的には、考えられる化合物は、水または他のビヒクル中にそれらを一層可溶性にするように修飾することができるが、それは、例えば、当該技術の十分に範囲内である僅かな修飾(塩製剤化、エステル化等)で容易に達成することができる。患者の最大限有益な作用について本発明の化合物の薬物動態を管理するために、特定の化合物の投与経路およびおよび投薬計画を修飾することも、当該技術の十分に範囲内である。
【0091】
本明細書中に記載の式Iまたは式IIを有する化合物は、概して、クロロホルム、ジクロロメタン、酢酸エチル、エタノール、メタノール、イソプロパノール、アセトニトリル、グリセロール、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、ジメチルスルホキシド等などの有機溶媒中に可溶性である。一つの態様において、本発明は、式I〜IIを有する化合物を、薬学的に許容しうる担体と混合することによって製造された製剤を提供する。一つの側面において、その製剤は、(a)記載の化合物を、水溶性有機溶媒、非イオン性溶媒、水溶性脂質、シクロデキストリン、トコフェロールなどのビタミン、脂肪酸、脂肪酸エステル、リン脂質またはそれらの組み合わせ中に溶解させて、溶液を与え;そして(b)1〜10%の炭水化物溶液を含有する緩衝剤または生理食塩水を加えることを含む方法を用いて製造することができる。一例において、炭水化物は、デキストロースを含む。本方法を用いて得られた医薬組成物は、動物用途および臨床用途に安定且つ有用である。
【0092】
本方法で用いるための水溶性有機溶媒の代表例には、ポリエチレングリコール(PEG)、アルコール、アセトニトリル、N−メチル−2−ピロリドン、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、ジメチルスルホキシドまたはそれらの組み合わせが含まれるが、これに制限されるわけではない。適するアルコールの例には、メタノール、エタノール、イソプロパノール、グリセロールまたはプロピレングリコールが含まれるが、これに制限されるわけではない。
【0093】
本方法で用いるための水溶性非イオン界面活性剤の代表例には、CREMOPHOR(登録商標)EL、ポリエチレングリコール修飾CREMOPHOR(登録商標)(ポリオキシエチレングリセロールトリシノレート35(polyoxyethyleneglyceroltriricinoleat 35))、水素化CREMOPHOR(登録商標)RH40、水素化CREMOPHOR(登録商標)RH60、PEG−スクシネート、ポリソルベート20、ポリソルベート80、SOLUTOL(登録商標)HS(ポリエチレングリコール660 12−ヒドロキシステアレート)、ソルビタンモノオレアート、ポロキサマー、LABRAFIL(登録商標)(エトキシル化杏仁油)、LABRASOL(登録商標)(カプリルカプロイルマクロゴール−8−グリセリド(capryl-caproyl macrogol-8-glyceride))、GELUCIRE(登録商標)(グリセロールエステル)、SOFTIGEN(登録商標)(PEG6カプリル酸グリセリド)、グリセリン、グリコールポリソルベートまたはそれらの組み合わせが含まれるが、これに制限されるわけではない。
【0094】
本方法で用いるための水溶性脂質の代表例には、植物油、トリグリセリド、プラント油またはそれらの組み合わせが含まれるが、これに制限されるわけではない。脂質油の例には、ヒマシ油、ポリオキシルヒマシ油、トウモロコシ油、オリーブ油、綿実油、ラッカセイ油、ハッカ油、ベニバナ油、ゴマ油、ダイズ油、水素化植物油、水素化ダイズ油、ヤシ油のトリグリセリド、パーム種子油およびそれらの水素化形またはそれらの組み合わせが含まれるが、これに制限されるわけではない。
【0095】
本方法で用いるための脂肪酸および脂肪酸エステルの代表例には、オレイン酸、モノグリセリド、ジグリセリド、PEGの一または二脂肪酸エステル、またはそれらの組み合わせが含まれるが、これに制限されるわけではない。
【0096】
本方法で用いるためのシクロデキストリンの代表例には、α−シクロデキストリン、β−シクロデキストリン、ヒドロキシプロピル−β−シクロデキストリンまたはスルホブチルエーテル−β−シクロデキストリンが含まれるが、これに制限されるわけではない。
【0097】
本方法で用いるためのリン脂質の代表例には、ダイズホスファチジルコリンまたはジステアロイルホスファチジルグリセロールおよびそれらの水素化形、またはそれらの組み合わせが含まれるが、これに制限されるわけではない。
【0098】
当業者は、それら製剤を、本明細書の内容の範囲内で修飾して、特定の投与経路について多数の製剤を与えることができる。具体的には、それら化合物は、水または他のビヒクル中にそれらを一層可溶性にするように修飾することができる。患者の最大限有益な作用について本発明の化合物の薬物動態を管理するために、特定の化合物の投与経路およびおよび投薬計画を修飾することも、当該技術の十分に範囲内である。
【0099】
B.5.医薬組成物
本発明は、更に、本発明の新規な処置方法で用いるのに適する局所、経口、全身および非経口用の医薬製剤を提供する目的を有する。本発明の化合物を活性成分として含有するそれら組成物は、経口、全身または標的部位投与用のビヒクル中の広範囲の治療的剤形で投与することができる。
【0100】
本発明は、更に、一つまたはそれを超える本発明の化合物を、薬学的に許容しうる担体と一緒に含む医薬組成物を提供する。好ましくは、これら組成物は、経口、非経口、鼻腔内、舌下または直腸投与用の、または吸入または吹入による投与用の、錠剤、丸剤、カプセル剤、散剤、顆粒剤、懸濁剤、ゲル剤、軟ゲル剤、滅菌非経口液剤、乳剤、エアゾル剤、液状噴霧剤、滴剤、アンプル剤、自己注射装置または坐剤などの単位剤形である。
【0101】
錠剤またはカプセル剤などの固形組成物を製造するには、主要活性成分を、医薬担体、例えば、トウモロコシデンプン、ラクトース、スクロース、ソルビトール、タルク、ステアリン酸、ステアリン酸マグネシウム、リン酸二カルシウムまたはガムなどの慣用的な錠剤成形成分、および他の医薬希釈剤、例えば、水と混合して、本発明の化合物またはその薬学的に許容しうる塩の均一混合物を含有する固形組成物を形成する。更に、主要活性成分は、一つまたはそれを超える医薬担体と混合して、改善されたバイオアベイラビリティーまたは他の薬物動態学的性質を有する剤形を与えることができる。このような系の例には、ヒドロキシプロピルメチルセルロース(HPMC)、ヒプロメロースアセテートスクシネート(hypromellose acetate succinate)(HPMCAS)などの成分を含む噴霧乾燥分散(Spray Dried Dispersion)固形剤形;低粘度ヒドロキシプロピルセルロース(Low viscosity hydroxypropylcellulos)(HPC−SL)、ドクセートナトリウム(docusate sodium)、プルロニク(pluronics)、ホスファチジルコリン、レシチンおよびコレステロールなどの成分を含むナノパーティクル(Nano-particles)製剤;ホスファチジルコリン、ポリビニルピロリドン、レシチンおよびコレステロールなどの成分を含む脂質基剤製剤(Lipid-base formulation);スルホブチルエーテル−β−シクロデキストリン(SBECD)および2−ヒドロキシプロピル−β−シクロデキストリン(HPCD)などの成分を含むシクロデキストリン製剤が含まれるが、これに制限されるわけではない。これら組成物を均一と言及する場合、それは、活性成分が、組成物中に一様に分散していることを意味するので、組成物は、錠剤、丸剤およびカプセル剤などの等しく有効な単位剤形および脂質基剤製剤中に容易に小分けすることができる。
【0102】
新規な組成物の錠剤または丸剤は、コーティングしてまたはそれ以外に配合して、持続性作用の利点を与える剤形を提供することができる。例えば、錠剤または丸剤は、内部投与成分および外部投与成分を含むことができるが、後者は、前者の上のエンベロープの形である。腸溶層は、それら二つの成分を隔てることができる。その腸溶層は、胃内の崩壊に耐えるのに役立ち且つ内部成分を十二指腸中に無傷で通過させるまたは放出を遅らせる。いろいろな材料を、このような腸溶層またはコーティングに用いることができ、このような材料には、シェラック、アセチルアルコールおよび酢酸セルロースのような、多数のポリマー性酸およびポリマー性酸とこのような材料との混合物が含まれる。
【0103】
本発明の新規な組成物を、経口または注射での投与用に包含することができる液状形には、水性液剤;好適に着香したシロップ剤;水性または油状懸濁剤;綿実油、ゴマ油、ヤシ油またはラッカセイ油などの食用油との乳剤;ポリソルベート80、トコフェリルポリエチレングリコールスクシネート(TPGS)、Cremophor、capmul MCM、ポリエチレングリコールなどの界面活性剤または補助溶媒を含むマイクロエマルジョンまたは自己乳化性系;ホスファチジルコリン、コレステロール、レシチン、HPC−SL、Docusate Soldium などの成分を含むリポソームまたはナノパーティクル製剤;溶解性を増強するSBECD、HPCDなどの成分を含むシクロデキストリン複合体製剤が含まれる。水性懸濁剤に適する分散助剤または懸濁化剤には、デキストラン、ナトリウムカルボキシメチルセルロース、メチルセルロース、ポリビニルピロリドンまたはゼラチンなどの合成および天然のガムが含まれる。
【0104】
本発明の化合物は、前述の組成物のいずれでも、そして効力研究において有効である投薬計画にしたがって投与することができる。
本発明の化合物は、最適抗癌作用を得るために、常套試験によって規定される適当な投薬量で単独に用いることができる。更に、他の腫瘍学物質の共投与または連続投与が望まれる。
【0105】
C.新規な化合物および医薬組成物を使用する方法
C.1.新規な化合物を使用する方法
本発明の化合物は、癌または他のタイプの増殖性疾患を処置する場合に、細胞障害性剤および/または細胞分裂抑制性剤として用いることができる。これら化合物は、いずれかのタイプの作用機構によって機能することができる。例えば、それら化合物は、分子および/またはシグナル伝達経路を阻害して、G2/M期で細胞周期の阻止をもたらすことができるが、それは、ついには、腫瘍細胞のアポトーシスを引き起こすかもしれない(例えば、Weung et al. (1997) Biochim. Biophys. Res. Comm., vol: 263, pp 398-404 を参照されたい)。別の例において、それら化合物は、チューブリン集合/脱集合を妨げることができ、それは、細胞有糸分裂を阻害し且つ細胞アポトーシスを引き起こすことができる(例えば、Panda et al., (1997) Proc. Natl. Acad. Sci. USA, vol: 94, 10560-10564 を参照されたい)。それら化合物は、更に、内皮細胞増殖および血管新生作用を阻害することができる(例えば、Witte et al., 1998, Cancer Metastasis Rev. vol. 17: 155-161 を参照されたい)。本発明の化合物は、いろいろな癌細胞系に活性であることが分かっている。
【0106】
別の側面において、本発明は、哺乳動物の肉腫、類表皮癌、線維肉腫、子宮頸癌、白血病、リンパ腫、肺癌、非小細胞性肺癌、結腸癌、CNS癌、黒色腫、卵巣癌、腎癌、前立腺癌、乳癌、頭頸部癌、膵癌および他のタイプの増殖性疾患が含まれるがこれに制限されるわけではない全ての組織または器官由来の癌の処置方法であって、このような処置を必要としている対象に、治療的有効量の式I〜IIを有する化合物を細胞障害性剤および/または細胞分裂抑制性剤として少なくとも一つの処置で投与することを含む方法に関する。
【0107】
また別の側面において、本発明は、白血病、リンパ腫、肺癌、結腸癌、CNS癌、黒色腫、卵巣癌、腎癌、前立腺癌または乳癌および他のタイプの増殖性疾患が含まれるがこれに制限されるわけではない全ての組織または器官由来の癌の処置用の医薬製剤を製造する方法であって、治療的有効量の式I〜IIを有する化合物を、薬学的に許容しうる担体と混合することを含む方法に関する。
【0108】
本発明の方法を実施するために、式I〜IIを有する化合物およびそれらの薬学的に許容しうる塩は、経口、非経口、吸入スプレーで、局所、直腸、鼻腔内、口腔内、膣内、植込みレザバーによって、または他の薬物投与方法で投与することができる。本明細書中で用いられる「非経口」という用語は、皮下、皮内、静脈内、筋肉内、関節内、動脈内、滑液嚢内、胸骨内、髄腔内、病巣内および頭蓋内の注射または注入技法を包含する。いくつかの態様において、本発明の化合物は、注射によって、すなわち、非経口で送達される。いくつかの態様において、好ましい投与経路は、静脈内または腹腔内注射による。
【0109】
滅菌注射可能水性または油脂性懸濁剤などの滅菌注射可能組成物は、当該技術分野において知られている技法にしたがって、適する分散助剤または湿潤剤および懸濁化剤を用いて製剤化することができる。滅菌注射可能製剤は、無毒性の非経口に許容しうる希釈剤または溶媒中の滅菌注射可能溶液または懸濁液であってもよい。用いることができる許容しうるビヒクルおよび溶媒の中には、マンニトール、水、リンガー液および等張塩化ナトリウム溶液が含まれる。更に、滅菌固定油は、溶媒または懸濁媒として好都合に用いられる(例えば、合成モノグリセリドまたはジグリセリド)。オレイン酸などの脂肪酸およびそのグリセリド誘導体は、オリーブ油またはヒマシ油などの薬学的に許容しうる油剤であるような、特に、それらのポリオキシエチル化型の注射可能なものの製造において有用である。これら油状液剤または懸濁剤は、更に、長鎖アルコール希釈剤または分散剤、またはカルボキシメチルセルロースまたは類似の分散助剤を含有することができる。薬学的に許容しうる固体、液体または他の剤形の製造において一般的に用いられるいろいろな乳化剤またはバイオアベイラビリティー増強剤も、製剤目的に用いることができる。
【0110】
経口投与用組成物は、錠剤、カプセル剤、乳剤および水性懸濁剤、分散剤および液剤が含まれるがこれに制限されるわけではないいずれかの経口に許容しうる剤形であってよい。経口使用のための錠剤の場合、一般的に用いられる担体には、ラクトースおよびトウモロコシデンプンが含まれる。ステアリン酸マグネシウムなどの滑沢剤も、加えることができる。カプセル形での経口投与用に有用な希釈剤には、ラクトースおよび乾燥トウモロコシデンプンが含まれる。水性懸濁剤または乳剤を経口投与する場合、活性成分は、乳化剤または懸濁化剤と混合した油状相中に懸濁させるまたは溶解させることができる。必要ならば、特定の甘味剤、着香剤または着色剤を加えることができる。鼻内エアゾールまたは吸入組成物は、当医薬製剤技術分野において周知の技法にしたがって製造することができるし、しかも液剤として、例えば、生理食塩水中で、適する保存剤(例えば、ベンジルアルコール)、バイオアベイラビリティーを増強する吸収促進剤、および/または当該技術分野において知られている他の可溶化剤または分散助剤を用いて製造することができる。
【0111】
本発明の化合物の有効量は、当該技術分野において知られている常套実験によって決定することができる。典型的に、これは、十分に許容されることが分かっている量の投与;および症状の減少、腫瘍サイズの減少または腫瘍成長の停止などの所望の作用が得られるまで投薬量を漸増することを必要とする。いくつかの態様において、約5〜10mg/kgの開始投薬量を用い、そしてその投薬量を、所望の作用が認められるまたは許容性問題が認められるまで、週に1回、約50%ずつ漸増増加させる。いくつかの態様において、適する投薬量は、約5〜250mg/kg;または約10〜150mg/kgである。10〜100mg/kgの投薬量は、時々好適である。投与は、1回、週に1回、1日1回または1日1回より多く行うことができる。いくつかの態様において、処置を必要としている対象に、1日に1〜4用量を送達する。
【0112】
更に、式I〜IIを有する化合物は、いろいろな癌または状態の処置のために、単独でまたは他の抗癌剤との組み合わせで投与することができる。本発明による組み合わせ療法は、少なくとも一つの本発明の化合物またはその機能性誘導体および少なくとも一つの他の薬学的に活性な成分の投与を含む。一つまたは複数の活性成分および薬学的活性剤は、別々にまたは一緒に投与することができる。一つまたは複数の活性成分および一つまたは複数の薬学的活性剤の量および相対的な投与のタイミングは、所望の組み合わせの治療的作用を達成するために選択されるであろう。
【0113】
また別の側面において、本発明は、冠状動脈疾患の患者のための冠状ステント処置後の再狭窄の、式I〜IIを有する化合物での処置方法に関する。
冠状動脈疾患の患者のための冠状ステント処置後の再狭窄の主因は、平滑筋細胞の増殖および移動および細胞外マトリックス生産によって生じる新生内膜増殖である(例えば、“Pathology of acute and chronic coronary stenting in humans”, by Farb, A., Sangiorgi, G., Certer, A.J., et al, in Circulation, vol. 99, pp 44-52, 1999 を参照されたい)。抗増殖能力を有する化合物は、このような化合物が適する手段で送達された時の臨床的および血管造影的再狭窄のリスクを減少させる場合に作用を有することができる(例えば、“A polymer-based, paclitaxel-eluting stent in patients with coronary artery disease”, by Stone, G.W., Ellis, S.G., Cox, D.A, et al, in New England Journal of Medicine, vol. 350: pp 221-231, 2004 を参照されたい)。したがって、腫瘍を処置する場合の式I〜IXを有する化合物について、それらは、新生内膜増殖に関与する細胞の増殖を阻害する場合に、したがって、新生内膜増殖および再狭窄の発生を減少させる場合に、有用でもありうる。いろいろな方法を、それら化合物をこれら細胞へ有効に送達する場合に用いることができる。例えば、式I〜Xを有する上記の化合物を含む組成物は、経口、非経口または植込みレザバーによって投与することができる。他の例において、次の論文に記載のアプローチを用いることもできる。“A polymer-based, paclitaxel-eluting stent in patients with coronary artery disease”, by Stone, G.W., Ellis, S.G., Cox, D.A. et al, in New England Journal of Medicine, vol. 350: pp 221-231, 2004; “A randomized comparison of a sirolimus-eluting stent with a standard stent for coronary revascularization”, by Morice, M.-C., Serruys, P.W., Sousa, J.E., et al, in New England Journal of Medicine, vol. 346: pp 1773-1780, 2002; “Sirolimus-eluting stents versus standard stents in patients with stenosis in a native coronary artery”, by Moses, J.W., Leon, M.B., Popma, J.J., et al, in New England Journal of Medicine, vol. 349: pp 1315-1323, 2003。
【0114】
処置および予防の方法
本発明の方法は、いろいろなタイプの癌を処置する、予防するおよび/または管理する方法を包含する。本明細書中で用いられるように、特に断らない限り、「処置する」という用語は、本発明の化合物または他の追加の活性剤の投与を意味する。本明細書中で用いられるように、特に断らない限り、「予防する」という用語は、症状の開始前の、特に、癌のリスクがある患者への投与を意味する。「予防」という用語は、特定の疾患または障害の症状の阻害を包含する。癌の家族歴を有する患者は、予防計画に好ましい候補である。本明細書中で用いられるように且つ特に断らない限り、「管理する」という用語は、特定の癌に罹患した患者の癌の再発を予防すること、および/または癌に罹患した患者が寛解状態のままである期間を延長することを包含する。
【0115】
本明細書中で用いられる「癌」という用語には、充実性腫瘍および血液由来腫瘍が含まれるが、これに制限されるわけではない。「癌」という用語は、膀胱、骨または血液、脳、乳房、子宮頸部、胸部、結腸、子宮内膜(endrometrium)、食道、眼、頭部、腎、肝、リンパ節、肺、口、頸部、卵巣、膵臓、前立腺、直腸、胃、精巣、咽頭および子宮の癌が含まれるがこれに制限されるわけではない、皮膚組織、器官、血液および血管の疾患を意味する。具体的な癌には、進行悪性疾患、アミロイドーシス、神経芽細胞腫、髄膜腫、血管周囲細胞腫、多発性脳転移、多形性グリア芽細胞腫、グリア芽細胞腫、脳幹グリオーム、予後のよくない悪性脳腫瘍、悪性グリオーム、再発性悪性グリオーム(giolma)、退形成性星状細胞腫、退形成性希突起グリオーム、神経内分泌腫瘍、直腸腺癌、Dukes C&D結腸直腸癌、切除不可能な結腸直腸癌、転移性肝細胞癌、カポジ肉腫、カロータイプ(karotype)急性骨髄芽球性白血病、ホジキンリンパ腫、非ホジキンリンパ腫、皮膚T細胞リンパ腫、皮膚B細胞リンパ腫、びまん性大B細胞リンパ腫、低級濾胞性リンパ腫、悪性黒色腫、悪性中皮腫、悪性胸膜浸出中皮腫症候群、腹膜癌、乳頭状漿液性癌、婦人科肉腫、軟組織肉腫、強皮症、皮膚血管炎、ランゲルハンス細胞組織球症、平滑筋肉腫、進行性骨化性線維異形成、ホルモン不応性前立腺癌、切除済みハイリスク軟組織肉腫、切除不可能な肝細胞癌、ヴァルデンストレームマクログロブリン血症、くすぶり型骨髄腫、無症候性骨髄腫、ファロピウス管癌、アンドロゲン非依存性前立腺癌、アンドロゲン依存性IV期非転移性前立腺癌、ホルモン不感性前立腺癌、化学療法不感性前立腺癌、乳頭状甲状腺癌、濾胞性甲状腺癌、髄様甲状腺癌および平滑筋腫が含まれるが、これに制限されるわけではない。特定の態様において、癌は転移性である。別の態様において、癌は、化学療法または放射線に不応性または耐性である;具体的には、サリドマイドに不応性である。
【0116】
C.2.処置および予防の方法
本発明の方法は、いろいろなタイプの癌を処置する、予防するおよび/または管理する方法を包含する。本明細書中で用いられるように、特に断らない限り、「処置する」という用語は、本発明の化合物または他の追加の活性剤の投与を意味する。本明細書中で用いられるように、特に断らない限り、「予防する」という用語は、症状の開始前の、特に、癌のリスクがある患者への投与を意味する。「予防」という用語は、特定の疾患または障害の症状の阻害を包含する。癌の家族歴を有する患者は、予防計画に好ましい候補である。本明細書中で用いられるように且つ特に断らない限り、「管理する」という用語は、特定の癌に罹患した患者の癌の再発を予防すること、および/または癌に罹患した患者が寛解状態のままである期間を延長することを包含する。
【0117】
本明細書中で用いられる「癌」という用語には、充実性腫瘍および血液由来腫瘍が含まれるが、これに制限されるわけではない。「癌」という用語は、膀胱、骨または血液、脳、乳房、子宮頸部、胸部、結腸、子宮内膜(endrometrium)、食道、眼、頭部、腎、肝、リンパ節、肺、口、頸部、卵巣、膵臓、前立腺、直腸、胃、精巣、咽頭および子宮の癌が含まれるがこれに制限されるわけではない、皮膚組織、器官、血液および血管の疾患を意味する。具体的な癌には、進行悪性疾患、アミロイドーシス、神経芽細胞腫、髄膜腫、血管周囲細胞腫、多発性脳転移、多形性グリア芽細胞腫、グリア芽細胞腫、脳幹グリオーム、予後のよくない悪性脳腫瘍、悪性グリオーム、再発性悪性グリオーム(giolma)、退形成性星状細胞腫、退形成性希突起グリオーム、神経内分泌腫瘍、直腸腺癌、Dukes C&D結腸直腸癌、切除不可能な結腸直腸癌、転移性肝細胞癌、カポジ肉腫、カロータイプ(karotype)急性骨髄芽球性白血病、ホジキンリンパ腫、非ホジキンリンパ腫、皮膚T細胞リンパ腫、皮膚B細胞リンパ腫、びまん性大B細胞リンパ腫、低級濾胞性リンパ腫、悪性黒色腫、悪性中皮腫、悪性胸膜浸出中皮腫症候群、腹膜癌、乳頭状漿液性癌、婦人科肉腫、軟組織肉腫、強皮症、皮膚血管炎、ランゲルハンス細胞組織球症、平滑筋肉腫、進行性骨化性線維異形成、ホルモン不応性前立腺癌、切除済みハイリスク軟組織肉腫、切除不可能な肝細胞癌、ヴァルデンストレームマクログロブリン血症、くすぶり型骨髄腫、無症候性骨髄腫、ファロピウス管癌、アンドロゲン非依存性前立腺癌、アンドロゲン依存性IV期非転移性前立腺癌、ホルモン不感性前立腺癌、化学療法不感性前立腺癌、乳頭状甲状腺癌、濾胞性甲状腺癌、髄様甲状腺癌および平滑筋腫が含まれるが、これに制限されるわけではない。特定の態様において、癌は転移性である。別の態様において、癌は、化学療法または放射線に不応性または耐性である;具体的には、サリドマイドに不応性である。
【0118】
C.3.生物学的スクリーニングおよび抗癌活性:
リアルタイム細胞電子検知(Real-Time Cell Electronic Sensing)(RT−CES)を用いた in vitro の細胞に基づくスクリーニング
本明細書中に開示の化合物の生物学的活性を、ACEA Biosciences, Inc. 製の Real-Time Cell Electronic Sensing(RT−CES(登録商標))システムを用いて監視し且つプロフィールを集めた。RT−CESシステムは、細胞−基質インピーダンス技術を利用して、微量滴定プレート形式の組織培養ウェル内部の細胞挙動を監視する。その技術は、ミクロ電子工学での分子・細胞生物学の組込みを特徴とし、生物学的検定法の電子検出に基づいている。この細胞電子検知技術および関連装置、システムおよび使用方法の詳細は、米国特許第7,167,585号;米国特許第7,468,255号;PCT公開WO2004/010102号;米国特許第7,470,533号;および米国特許第7,459,303号に記載されていて、それらは各々、本明細書中に援用される。RT−CES技術の更なる詳細は、米国特許第7,468,255号に更に開示されている。
【0119】
RT−CES技術を用いた細胞−基質または細胞−電極インピーダンスの測定には、適当な幾何学的形を有する微小電極を、微量滴定プレートまたは類似の装置の底面上に製作して、ウェル中に向ける。細胞を、それら装置のウェル中に入れ、電極表面に接触させ且つ付着させる。細胞の存在、不存在または性状の変化は、電極センサー表面上の電子およびイオンの通過に影響する。電極の間または中でのインピーダンスを測定することは、センサー上に存在する細胞の生物学的状態について重要な情報を与える。それら細胞の生物学的状態に変化がある場合、アナログ電子読み取りシグナルを、自動的に且つリアルタイムで測定し、そして処理用および分析用のディジタルシグナルへ変換する。RT−CESシステムの場合、細胞指数(インピーダンス変化の任意表示)は、測定された電極インピーダンス値に基づいて自動的に導かれ且つ与えられる。一定のウェルについて得られた細胞指数は、(1)このウェル中の電極表面に、いかに多くの細胞が付着しているか;(2)このウェル中の電極表面に、いかに十分に細胞が付着しているかを反映している。したがって、同様の生理学的状態にある同じタイプの細胞が、より多く電極表面に付着してほど、細胞指数は大きい。そして、細胞が、より十分に電極表面に付着しているほど(例えば、細胞が、より大きい接触面積を有するように一層広がっている、または細胞が、よりしっかりと電極表面に付着しているほど)、細胞指数は大きい。
【0120】
RT−CESシステムは、三つの成分、すなわち、電子センサー分析器、装置ステーションおよび16Xまたは96X微量滴定装置を含む。微小電極センサーアレイは、リソグラフィーによる微小製作方法でガラススライド上に製作したが、電極含有スライドは、プラスチックトレーへ組み立てられて、電極含有ウェルを形成している。装置ステーションには、16Xまたは96X微量滴定プレート装置を与え、それは、ウェルのいずれか一つを、インピーダンス測定用のセンサー分析器へ電子的に切り替えることができる。操作中に、ウェル中で培養された細胞を含む装置を、インキュベーター内部に位置する装置ステーション中に入れる。電気ケーブルは、装置ステーションをセンサー分析器へ連通する。RT−CESソフトウェア制御下において、センサー分析器は、測定されるウェルを自動的に選択し且つ連続的にインピーダンス測定を行うことができる。分析器からのインピーダンスデータを、コンピューターへ転送し、組み込まれたソフトウェアで分析し且つ処理する。
【0121】
個々のウェル中の電極間で測定されたインピーダンスは、電極幾何学的形、ウェル中のイオン濃度、および電極に付着した細胞が存在するかどうかに依存する。細胞が不存在の場合、電極インピーダンスは、主に、電極/溶液界面およびバルク溶液中の双方におけるイオン環境によって決定される。細胞の存在下において、電極センサー表面に付着した細胞は、電極/溶液界面における局部イオン環境を変更して、インピーダンスの増加をもたらすであろう。電極上に存在する細胞が多いほど、細胞−電極インピーダンスの増加は大きい。更に、インピーダンス変化も、細胞形態、および細胞が電極に付着している程度に依存する。
【0122】
測定された細胞−電極インピーダンスに基づいて細胞状態を定量するために、細胞指数(Cell Index)と称するパラメーターを導く。細胞指数は、電極含有ウェル中の細胞の状態の定量的尺度である。同じ生理学的条件下において、電極上に付着したより多くの細胞は、より大きい細胞−電極抵抗値をもたらして、細胞指数についてより大きい値をもたらす。更に、ウェル中に存在する同数の細胞については、形態などの細胞状態の変化が、細胞指数の変化をもたらすであろう。例えば、細胞接着または細胞広がり(spreading)の増加は、より大きい細胞−電極接触面積をもたらし、それが、細胞−電極抵抗の増加を、したがって、細胞指数についてより大きい値をもたらすであろう。
【0123】
生物学的に活性な化合物と、Eプレートのウェル内部の細胞成長との相互作用は、化合物自体の生物学的機構、濃度、インキュベーション長さおよび細胞タイプに依存する独特の活性パターン(すなわち、化合物処置に応答した独特の細胞インピーダンス曲線または細胞指数曲線)を生じる。各々の化合物への「記名(signature)」細胞応答パターンは、細胞周期阻止、形態変化および細胞死などの特定の生物学的現象と相関する。RT−CESシステムで細胞応答プロフィールを集めることは、有効であると証明されており、本発明者は、同様の作用機構を有する化合物が、同様のパターンを示すということを示した。したがって、化合物処置への細胞応答パターンの類似性は、作用機構、耐性の様式、そしておそらくは、分子標的の類似性を示すことができる。本発明者は、抗有糸分裂剤での処置に応答して有糸分裂阻止を行っている細胞について独特のRT−CES記名パターンを識別した。一例として、図4Bおよび図4Cは、異なった濃度の周知の抗有糸分裂剤パクリタキセルおよびビンクリスチンで処置されたA549肺癌細胞の特異的プロフィールを示している。
【0124】
本発明者は、RT−CESシステム(ACEA Biosciences)またはxCelligenceシステム(Roche)を用いて、いくつかの新規な化合物への多数の癌細胞系の応答を評価した。RT−CESシステムおよびxCelligenceシステムは、本質的に同じであり、同じACEAリアルタイム細胞インピーダンス測定技術を用いていることに注目されたい。いくつかの本発明の化合物の(特定の濃度での)時間依存性細胞応答パターンは、パクリタキセルおよびビンクリスチンの(特定の濃度での)それらと幾分同様であった。したがって、これら化合物は、パクリタキセルおよびビンブラスチンの場合と同様の抗癌作用機構を有することができる。もう一方において、これら化合物は、これら本発明の化合物の時間依存性細胞応答パターンが、パクリタキセルおよびビンクリスチンの場合と同様であるとしても、パクリタキセルおよびビンブラスチンの場合とは異なった他の作用機構によって癌細胞に作用することができる。これら化合物が、パクリタキセルおよびビンブラスチンの場合と同様の作用機構を含めた多数の作用機構によって癌細胞に作用するということも考えられる。
【0125】
本発明者は、本明細書中に記載のRT−CES方法で測定される本発明の化合物の活性を調べた。そのデータは、それら化合物が、広範囲の癌細胞系に活性であり、そして正常細胞へは活性がはるかに少ないということを示している。ここで、本発明者は、一例として、活性化合物の一つであるCOMPOUND Oを用いる。図4〜24は、いろいろな濃度でのCOMPOUND Oの添加前および後の多数のヒト癌細胞系の時間依存性細胞指数を示す。これら細胞系には、A549(非小細胞性肺癌細胞系)、NCI−H460(大細胞肺癌細胞系)、H1993細胞(非小細胞性肺癌細胞系)、H1838細胞(非小細胞性肺癌細胞系)、H2347細胞(非小細胞性肺癌細胞系)、SW620細胞(結腸癌細胞系)、GTL16細胞(胃癌細胞系)、HT29細胞(結腸癌細胞系)、A172細胞(脳癌細胞系)、U138細胞(脳癌細胞系)、U118細胞(脳癌細胞系)、SW1088細胞(脳癌細胞系)、HT1080細胞(結合組織癌細胞系)、BxPC3細胞(膵癌細胞系)、HepG2細胞(肝癌細胞系)、SKOV3細胞(卵巣癌細胞系)、MCF7細胞(乳癌細胞系)、MDA−MB−231(乳癌細胞系)およびKB(子宮頸癌細胞系)が含まれる。それら結果は、COMPOUND Oが、これら癌細胞系の増殖に阻害剤作用を示したということを示している。これら細胞系に対するCOMPOUND OのIC50は、慣用的な化学療法薬であるパクリタキセルおよびビンクリスチンのそれと同様である。
【0126】
本発明者は、更に、多剤耐性(MDR)遺伝子を過発現するKB200へのCOMPOUND Oの作用を調べた。この遺伝子は、臨床の化学療法薬、例えば、パクリタキセルおよびビンクリスチン/ビンブラスチンの耐性に応答性である。図24A〜Dは、親KB細胞系の成長が、COMPOUND O(IC50=33.1nM)、パクリタキセル(IC50=7.19nM)、ビンブラスチン(IC50=4.74nM)およびコルヒチン(IC50=8.20nM)によって阻害されたということを示している。これら4種類の化合物のIC50は、同様の範囲内である。図24Eは、KB200細胞の成長が、COMPOUND O(IC50=9.84nM)による阻害に感受性であったことを示している。対照的に、KB200細胞の成長は、パクリタキセル(IC50>1uM)、ビンブラスチン(IC50=135nM)およびコルヒチン(IC50=116nM)による阻害への感受性がはるかに少なかった(図24F〜Hに示される)。これは、COMPOUND Oが、パクリタキセルおよびビンブラスチン処置に失敗した患者に対する大きなセカンドライン療法でありうるということを示唆している。
【0127】
図25は、非癌性細胞系NIH−3T3が、COMPOUND O(IC50=6uM)による阻害への感受性がはるかに少なかったことを示している。したがって、COMPOUND Oは、癌細胞に対して、正常細胞に対するよりも高い細胞障害作用を示す。
【0128】
抗癌活性についての in vivo スクリーニング
COMPOUND Oの in vivo 抗癌効力を評価するために、3種類のヒト腫瘍異種移植片モデルを用いた。それには、免疫不全ヌードマウスにおけるMKN45ヒト胃腸癌、H460ヒト非小細胞性肺癌およびA549ヒト肺癌の異種移植片モデルが含まれた。COMPOUND Oの in vivo 抗癌効力の詳細を、実施例1〜3に与える。
【0129】
実施例1
ヌードマウスにおけるMKN45ヒト胃腸癌へのCOMPOUND Oの in vivo 抗癌活性
COMPOUND Oの in vivo 抗癌効力を評価するために、免疫不全ヌードマウスにおけるMKN45ヒト胃腸癌異種移植片モデルを調べた。マウスモデルは全て、Pharmacology Lab of ACEA Bio (Hangzhou) CO., Ltd. で維持する。BALB/c免疫不全ヌードマウスは、Shanghai SLAC Laboratory Animal、証明番号:SCXKA(Shanghai)2007−0005より購入した。マウス体重は、19±1gであった。雌マウスのみを、この研究に用いた。被験動物数は、次の通りであった。各用量群に7匹、正対照群に7匹、そして負対照群に7匹。
【0130】
試験対照。負対照には、各々のマウスに、COMPOUND O試験に用いられたのと同容量および同濃度を有する溶媒のみを、120mg/kgで1日おきに1回(qod)18日間経口投与した。正対照には、経口抗癌化合物であるエトポシド(Etoposide)を、50mg/kgで4日毎に1回、18日間経口投与した。
【0131】
試験化合物の調製・投与。COMPOUND Oを、25%リン脂質(S75)および75%ポリビニルピロリドン(PL−PVP)中に溶解させた後、更に希釈して、0.9%NaCl水溶液中に8mg/mlとした。120mg/kg qod(1日おきに1回)〜160mg/kg q3d(3日毎に1回)の異なった投薬量のCOMPOUND Oを、研究に用いた。
【0132】
移植および化合物効力決定のための腫瘍細胞の調製。MKN45ヒト胃腸癌異種移植片モデルのための腫瘍細胞を調製するために、急速成長した腫瘍を、被移植腫瘍マウスから最初に取り出し、それら腫瘍組織を、1〜2mm3寸法に切開した。次に、これら微小腫瘍を、各々のマウスの補助領域(右側)中に皮下注射した。接種された腫瘍が、ヌードマウスにおいて一定サイズ(60〜80mm3)に成長後、それらマウスを無作為化して、異なった投薬群とし、そして化合物処置を施した。最初の投薬後2〜3週間に、マウスを屠殺し、そして移植された腫瘍を、実験用マウスから取り出した。各々取り出された充実性腫瘍を秤量した;各々の投薬群の腫瘍阻害率を、次の式にしたがって計算した。
【0133】
腫瘍阻害率%=(負対照群の平均腫瘍重量−化合物処置群の平均腫瘍重量)/負対照群の平均腫瘍重量x100 (1)
動物用飼料、動物用ケージ、支持材料および被験動物が接触する装置を含めた用いられる材料は全て、高圧滅菌した。ヌードマウスは、SPF条件下の層流棚で維持した。腫瘍移植後、各々の化合物投薬群のマウス体重および腫瘍サイズを、動的に監視し且つプロットした。腫瘍サイズは、腫瘍の長軸(a)および短軸(b)を測定することによって決定し、そして腫瘍体積は、次の式にしたがって計算した。
【0134】
腫瘍体積=axb2/2 (2)
結果。ヌードマウスのMKN45ヒト胃腸癌異種移植片モデルにおいて、COMPOUND Oは、120mg/kg qodおよび160mg/kg q3d投薬群において、それぞれ、58.0%および55.7%の平均 in vivo 腫瘍阻害率を示した。同じ実験において、Etoposide は、50mg/kg q3dの経路投与投薬量について48.4%の平均 in vivo 腫瘍阻害率を示した。それら詳細を、表2に与える。腫瘍サイズの動的変化は、図1に要約する。キャリアマウスの体重の動的変化は、表3に要約する。MKN45異種移植片モデルにおけるCOMPOUND O(120mg/kg qod)の抗癌作用は、同じ薬物投与経路の下での Etoposide(30mg/kg q3d)の場合と同様である。
【0135】
【表2】
【0136】
【表3】
【0137】
実施例2
ヌードマウスにおけるH460ヒト非小細胞性肺癌へのCOMPOUND Oの in vivo 抗癌活性
COMPOUND Oの in vivo 抗癌効力を評価するために、免疫不全ヌードマウスにおけるH460ヒト非小細胞性肺癌異種移植片モデルを用いた。細胞系およびマウスは、Pharmacology Lab of ACEA Bio (Hangzhou) CO., Ltd. で維持した。BALB/c免疫不全ヌードマウスは、Shanghai SLAC Laboratory Animal、証明番号:SCXKA(Shanghai)2007−0005より購入した。マウス体重は、19±2gの間であった。雌マウスのみを、この研究に用いた。被験動物数は、次の通りであった。各用量群に7匹、正対照群に7匹、そして負対照群に7匹。
【0138】
試験対照。負対照には、各々のマウスに、COMPOUND O試験に用いられたのと同容量および同濃度を有する溶媒のみを、120mg/kgで1日おきに1回(qod)25日間経口投与した。正対照には、経口抗癌化合物である Etoposide を、30mg/kg qodで25日間経口投与した。
【0139】
試験化合物の調製・投与。COMPOUND Oを、25%リン脂質(S75)および75%ポリビニルピロリドン(PL−PVP)中に溶解させた後、更に希釈して、0.9%NaCl水溶液中に8mg/mlとした。各々のマウスに、化合物溶液を経口投与した。異なった投薬量のCOMPOUND O(120mg/kg qodおよび160mg/kg q3d)を、研究に用いた。
【0140】
移植および化合物効力決定のための腫瘍細胞の調製。H460ヒト非小細胞性肺癌異種移植片モデルのための癌細胞を調製するために、フラスコ中の対数期増殖性細胞を、トリプシン処理し、そして細胞を、PBS(pH7.2)中に1.5x107個細胞/mlで再懸濁させた。細胞懸濁液(3x106個細胞)を、各々のマウスの補助領域(右側)中に皮下注射した。接種された癌細胞が、ヌードマウスにおいて一定サイズ(150mm3)の腫瘍に成長後、それらマウスを無作為化して、異なった投薬群とし、そして化合物処置を施した。最初の投薬後3〜4週間に、マウスを屠殺し、そして移植された腫瘍を、実験用マウスから取り出した。各々取り出された充実性腫瘍を秤量した;各々の投薬群の腫瘍阻害率を、実施例1の方程式(1)にしたがって計算した。
【0141】
動物用飼料、動物用ケージ、支持材料および被験動物が接触する装置を含めた用いられる材料は全て、高圧滅菌した。ヌードマウスは、SPF条件下の層流棚で維持した。腫瘍移植後、各々の化合物投薬群のマウス体重および腫瘍サイズを、動的に監視し且つプロットした。腫瘍サイズは、腫瘍の長軸(a)および短軸(b)を測定することによって決定し、そして腫瘍体積は、実施例1の式(2)にしたがって計算した。
【0142】
結果。ヌードマウスのH460ヒト非小細胞性肺癌異種移植片モデルにおいて、COMPOUND Oは、120mg/kg qodおよび160mg/kg q3d投薬群において、それぞれ、47.1%および31.3%の平均 in vivo 腫瘍阻害率を示した。同じ実験において、Etoposide は、30mg/kg qodの経路投与投薬量について48.4%の平均 in vivo 腫瘍阻害率を示した。それら詳細を、表4に与える。腫瘍サイズの動的変化は、図2に要約する。キャリアマウスの体重の動的変化は、表5に要約する。H460ヒト非小細胞性肺癌異種移植片モデルにおけるCOMPOUND O(120mg/kg)の抗癌作用は、同じ薬物投与手順の下での Etoposide(30mg/kg)の場合と同様である。
【0143】
【表4】
【0144】
【表5】
【0145】
実施例3
ヌードマウスにおけるA549ヒト非小細胞性肺癌へのCOMPOUND Oの in vivo 抗癌活性
COMPOUND Oの in vivo 抗癌効力を評価するために、免疫不全ヌードマウスにおけるA549ヒト非小細胞性肺癌異種移植片モデルを用いた。細胞系およびマウスは、Pharmacology Lab of ACEA Bio (Hangzhou) CO., Ltd. で維持した。BALB/c免疫不全ヌードマウスは、Shanghai SLAC Laboratory Animal、証明番号:SCXKA(Shanghai)2007−0005より購入した。マウス体重は、21±1gの間であった。雌マウスのみを、この研究に用いた。被験動物数は、次の通りであった。各用量群に7〜8匹、正対照群に6匹、そして負対照群に7匹。
【0146】
試験対照。負対照には、各々のマウスに、COMPOUND O試験に用いられたのと同容量および同濃度を有する溶媒のみを、120mg/kgで1日おきに1回(qod)25日間経口投与した。正対照には、経口抗癌化合物である Etoposide を、30mg/kg qodで25日間経口投与した。
【0147】
試験化合物の調製・投与。COMPOUND Oを、25%リン脂質(S75)および75%ポリビニルピロリドン(PL−PVP)中に溶解させた後、更に希釈して、0.9%NaCl水溶液中に8mg/mlとした。各々のマウスに、化合物溶液を経口投与した。異なった投薬量のCOMPOUND O(120mg/kg qodおよび160mg/kg q2*2(2日間続けて投薬後、2日間続けて休む)を、研究に用いた。
【0148】
移植および化合物効力決定のための腫瘍細胞の調製。A549ヒト非小細胞性肺癌異種移植片モデルのための癌細胞を調製するために、フラスコ中の対数期増殖性細胞を、トリプシン処理し、そして細胞を、PBS(pH7.2)中に2.5x107個細胞/mlで再懸濁させた。細胞懸濁液(5x106個細胞)を、各々のマウスの補助領域(右側)中に皮下注射した。接種された癌細胞が、ヌードマウスにおいて一定サイズ(50〜60mm3)の腫瘍に成長後(接種後約15日)、それらマウスを無作為化して、異なった投薬群とし、そして化合物処置を施した。最初の投薬後3〜4週間に、マウスを屠殺し、そして移植された腫瘍を、実験用マウスから取り出した。各々取り出された充実性腫瘍を秤量した;各々の投薬群の腫瘍阻害率を、実施例1の方程式(1)にしたがって計算した。
【0149】
動物用飼料、動物用ケージ、支持材料および被験動物が接触する装置を含めた用いられる材料は全て、高圧滅菌した。ヌードマウスは、SPF条件下の層流棚で維持した。腫瘍移植後、各々の化合物投薬群のマウス体重および腫瘍サイズを、動的に監視し且つプロットした。腫瘍サイズは、腫瘍の長軸(a)および短軸(b)を測定することによって決定し、そして腫瘍体積は、実施例1の式(2)にしたがって計算した。
【0150】
結果。ヌードマウスのA549ヒト非小細胞性肺癌異種移植片モデルにおいて、COMPOUND Oは、120mg/kg qodおよび160mg/kg q2*2投薬群において、それぞれ、67.8%および62.5%の平均 in vivo 腫瘍阻害率を示した。同じ実験において、Etoposide は、30mg/kg qodの経路投与投薬量について59.9%の平均 in vivo 腫瘍阻害率を示した。それら詳細を、表6に与える。腫瘍サイズの動的変化は、図3に要約する。キャリアマウスの体重の動的変化は、表7に要約する。A549ヒト非小細胞性肺癌異種移植片モデルにおけるCOMPOUND O(120mg/kg)の抗癌作用は、同じ薬物投与手順の下での Etoposide(30mg/kg)の場合と同様である。
【0151】
【表6】
【0152】
【表7】
【0153】
実施例4
A549細胞におけるCOMPOUND O、パクリタキセルおよびビンクリスチンによる細胞増殖の阻害
A549細胞(ヒト肺癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、RT−CESシステム(ACEA Biosciences)を用いて監視した。RT−CESシステムは、xCelligenceシステム(Roche より現在入手可能)と同じである。図4A〜Cは、化合物添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND O、パクリタキセルおよびビンクリスチンについてそれぞれ、10.9nM、5.3nMおよび61.8nMである。
【0154】
実施例5
H596細胞におけるCOMPOUND Oによる細胞増殖の阻害
H596細胞(ヒト腺扁平上皮肺癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。xCelligenceシステムは、ACEABiosciences 製のRT−CESシステムと同じである。図5は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて37.9nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルおよびビンクリスチンについてそれぞれ、12.5nMおよび36.9nMである。
【0155】
実施例6
H292細胞におけるCOMPOUND Oによる細胞増殖の阻害
H292細胞(ヒト肺性肺癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図6は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて10.8nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルおよびビンクリスチンについてそれぞれ、2.59nMおよび2.63nMである。
【0156】
実施例7
H460細胞におけるCOMPOUND Oによる細胞増殖の阻害
NCI−H460細胞(ヒト大細胞肺癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図7は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて10.9nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルおよびビンクリスチンについてそれぞれ、2.68nMおよび10.4nMである。
【0157】
実施例8
H1993細胞におけるCOMPOUND Oによる細胞増殖の阻害
H1993細胞(ヒト非小細胞性肺癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図8は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて60.2nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルおよびビンクリスチンについてそれぞれ、5.12nMおよび2.43nMである。
【0158】
実施例9
H1838細胞におけるCOMPOUND Oによる細胞増殖の阻害
H1838細胞(ヒト非小細胞性肺癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図9は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて11.8nMである。比較において、計算されたIC50(化合物処置後72時間)は、ビンクリスチンについて3.16nMである。
【0159】
実施例10
H2347細胞におけるCOMPOUND Oによる細胞増殖の阻害
H2347細胞(ヒト非小細胞性肺癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図10は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて1.76nMである。比較において、計算されたIC50(化合物処置後72時間)は、ビンクリスチンについて5.05nMである。
【0160】
実施例11
SW620細胞におけるCOMPOUND Oによる細胞増殖の阻害
SW620細胞(ヒト結腸癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図11は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて2.65nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルについて0.998nMである。
【0161】
実施例12
GTL16細胞におけるCOMPOUND Oによる細胞増殖の阻害
GTL16細胞(MKN45ヒト胃癌細胞系に由来したヒト胃癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図12は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて67.0nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルおよびビンクリスチンについてそれぞれ、1.02nMおよび3.75nMである。
【0162】
実施例13
HT29細胞におけるCOMPOUND Oによる細胞増殖の阻害
HT29細胞(ヒト結腸癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図13は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて25.4nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルおよびビンクリスチンについてそれぞれ、2.52nMおよび12.7nMである。
【0163】
実施例14
A172細胞におけるCOMPOUND Oによる細胞増殖の阻害
A172細胞(ヒト脳癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図14は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND O、パクリタキセルおよびビンクリスチンについてそれぞれ、11.6nM、4.46nMおよび1.30nMである。
【0164】
実施例15
U138MG細胞におけるCOMPOUND Oによる細胞増殖の阻害
U138MG細胞(ヒト脳癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図15は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND O、パクリタキセルおよびビンクリスチンについてそれぞれ、3.41nM、18.1nMおよび0.641nMである。
【0165】
実施例16
U118MG細胞におけるCOMPOUND Oによる細胞増殖の阻害
U118MG細胞(ヒト脳癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図16は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND O、パクリタキセルおよびビンクリスチンについてそれぞれ、13.1nM、12.3nMおよび4.31nMである。
【0166】
実施例17
SW1088細胞におけるCOMPOUND Oによる細胞増殖の阻害
SW1088細胞(ヒト脳癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図17は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND O、パクリタキセルおよびビンクリスチンについてそれぞれ、21.2nM、13.1nMおよび4.96nMである。
【0167】
実施例18
HT1080細胞におけるCOMPOUND Oによる細胞増殖の阻害
HT1080細胞(ヒト結合組織癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図18は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oおよびビンクリスチンについてそれぞれ、7.63nMおよび2.01nMである。
【0168】
実施例19
BxPC3細胞におけるCOMPOUND Oによる細胞増殖の阻害
BxPC3細胞(ヒト膵癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図19は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて6.60nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルおよびビンクリスチンについてそれぞれ、8.86nMおよび4.06nMである。
【0169】
実施例20
HepG2細胞におけるCOMPOUND Oによる細胞増殖の阻害
HepG2細胞(ヒト肝癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、10,000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図20は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて25.9nMである。
【0170】
実施例21
SKOV3細胞におけるCOMPOUND Oによる細胞増殖の阻害
SKOV3細胞(ヒト卵巣癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、3000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図21は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて5.47nMである。比較において、計算されたIC50(化合物処置後72時間)は、パクリタキセルについて5.34nMである。
【0171】
実施例22
MCF7細胞におけるCOMPOUND Oによる細胞増殖の阻害
MCF7細胞(ヒト乳癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図22は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて74.4nMである。比較において、計算されたIC50(化合物処置後72時間)は、11.3nMパクリタキセルである。
【0172】
実施例23
MDA−MB−231細胞におけるCOMPOUND Oによる細胞増殖の阻害
MDA−MB−231細胞(ヒト乳癌細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセルおよびビンクリスチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図23は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて32.4nMである。
【0173】
実施例24
多剤耐性(MDR)遺伝子を過発現するKB200細胞におけるCOMPOUND Oによる細胞増殖の阻害。
【0174】
親としての子宮頸癌細胞系であるKB細胞を、最初に調べた。KB細胞を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセル、ビンブラスチンおよびコルヒチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図24A〜Dは、化合物添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND O、パクリタキセル、ビンブラスチンおよびコルヒチンについてそれぞれ、33.1nM、7.19nM、4.74nMおよび8.20nMである。
【0175】
これら化合物の効力を、多剤耐性(MDR)遺伝子を過発現するKB200細胞に対して更に調べた。同様に、KB200細胞を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND O、パクリタキセル、ビンブラスチンおよびコルヒチンを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図24E〜Hは、化合物添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND O、ビンブラスチンおよびコルヒチンについてそれぞれ、9.84nM、0.135μMおよび0.116μMである。パクリタキセルについて計算されたIC50(化合物処置後72時間)は、>1μMである。COMPOUND Oは、慣用的な化学療法(例えば、パクリタキセルおよびビンブラスチン)に耐性である細胞系に対して十分な効力を示す。これは、COMPOUND Oが、パクリタキセルおよびビンブラスチン処置に失敗した患者に対する大きなセカンドライン療法でありうるということを示唆している。
【0176】
実施例25
NIH3T3正常組織細胞系におけるCOMPOUND Oによる細胞増殖の阻害
NIH3T3細胞(正常組織細胞系)を、96ウェルEプレート装置(Roche)のウェル中に、5000個/ウェルの初期播種細胞密度で播種し、そしてインキュベーター中において標準的な細胞培養条件下で約24時間プレインキュベートした。COMPOUND Oを、DMSO中の異なった濃度で、インキュベーション時間後にウェル中に加えた。細胞状態を、化合物添加の前・後に、xCelligenceシステム(Roche)を用いて監視した。図25は、COMPOUND O添加の前・後の時間の関数として規格化された細胞指数を示す。その細胞指数は、化合物添加直前の時点での細胞指数値に対して規格化した。計算されたIC50(化合物処置後72時間)は、COMPOUND Oについて6.0μMである。対照的に、試験された癌細胞系全てに対するCOMPOUND OのIC50は、低nM範囲内である。COMPOUND Oは、癌細胞に対して、正常細胞に対するよりも高い細胞障害性作用を示す。
【0177】
実施例26
COMPOUND Oによる in vitro および in vivo チューブリン重合の阻害
微小管は、重複した染色体が、二つの娘細胞への細胞の開裂前に二つの同一のセットに分離する時の有糸分裂を含めた数多くの細胞過程において重要である。微小管の不可欠な役割、および有糸分裂および細胞分裂におけるそれらの動力は、微小管を、抗癌薬の重要な標的にする。間期中の細胞において、微小管は、それらのチューブリンを、約3分〜数時間のハーフタイムで、細胞質プール中の可溶性チューブリンと交換する。有糸分裂の開始で、間期微小管ネットワークは脱集合し、そして有糸分裂紡錘体を形成し且つ染色体を動かす極めて動的な微小管の集団によって置き換えられる。有糸分裂紡錘体微小管は、間期細胞中の微小管よりも20〜50倍動的であり、そしてより多くの紡錘体微小管が、それらのチューブリンを、15秒もの速いハーフタイムで、可溶性プール中のチューブリンと交換する。
【0178】
有糸分裂紡錘体微小管の動力は、レギュレーターによるモジュレーションに、および微小管活性薬による破裂に、鋭く感受性である。微小管標的薬は、微小管重合および動力を広範囲の方法で変更することができる。ここで、本発明者は、COMPOUND Oの作用機構、すなわち、(1)それは、in vitro の微小管集合を阻害する、(2)それは、培養細胞中の微小管ネットワークに影響を与える、そして(3)それは、Colchicine と同様の機構によってチューブリン集合を阻害するということを証明する。
【0179】
方法
in vitro 微小管集合検定。微小管重合は、96ウェル微量滴定プレート中において、MAPの多いチューブリンと、HTS−チューブリン重合検定キットプロトコル(Cytoskeleton)に基づく、80mM PIPES pH6.9、0.5mM EGTA、2mM MgCl2、1mM GTP、10%グリセロールおよび4%(v/v)ジメチルスルホキシド(DMSO)を含有する緩衝液中のいろいろな濃度のCOMPOUND Oで行った。吸光度の増加は、37℃において405nm、Beckman Multimode DTX880プレートリーダーで測定し、60秒毎に30分間記録した。
【0180】
スピンカラム検定。チューブリンを、[[3H]ビンブラスチンまたは[3H]コルヒチンと一緒に、0.05M PIPES、pH6.9、1mM MgCl2および1mM GTPを含有する緩衝液中の異なった濃度の未標識ビンブラスチンか、コルヒチンかまたはCOMPOUND Oの存在下でインキュベートした。反応混合物を、37℃で1時間インキュベートした。それら試料を、緩衝溶液で予め平衡した illustraTM MicroSpinTMG−50カラム(GE Healthcare)上に装填した。それらカラムを、1.5ml管中に入れ、室温において750gで2分間回転させ、そしてフロースルー(flow-through)中の放射能を、シンチレーションカウンターによって分析した。
【0181】
結果
図26Aは、MAPの多いチューブリンを用いた in vitro の微小管集合へのCOMPOUND Oの作用を示す。負対照試料において、405nMでの吸光度(A405)は、時間で増加した。A405の増加は、10分でプラトーに達する。5μMビンクリスチンの存在下において、チューブリン重合は、対照試料の場合と比較して50%を超えて阻害された。COMPOUND Oの存在下において、チューブリン重合は、濃度依存方式で阻害された。0.04μMでのCOMPOUND Oは、負対照の場合と同様である阻害を示さなかった。5μMでのCOMPOUND Oは、正対照(5μMでのビンクリスチン)の場合と同様の阻害パターンを示した。5μMパクリタキセル(チューブリン重合増強剤)の存在下において、チューブリン重合は、負対照の場合と比較して更に増強された。
【0182】
図26Bは、A549細胞中において、COMPOUND Oの20時間処置による微小管体制の阻害を示す。対照細胞における微小管ネットワークは、正常な体制および配置を示した(図24B DMSO)。対照的に、パクリタキセル処置は、細胞微小管の密度の増加を伴う微小管重合を引き起こした(図24B Paclitaxel)。更に、COMPOUND O処置は、ビンクリスチンで誘発される微小管変化の場合と同様の知見をもたらした(図24B COMPOUND O&ビンクリスチン)。
【0183】
図26Cは、スピンカラム検定を用いて、COMPOUND Oが、コルヒチン結合部位によってチューブリンと相互作用するということを示している。図26C(上部パネル)に示されるように、未標識コルヒチンの存在下における[3H]コルヒチンとのチューブリンインキュベーションは、フロースルー中で見出される[3H]コルヒチンの量を濃度依存方式で減少させた。同様に、COMPOUND Oは、フロースルー中の[3H]コルヒチンの量を濃度依存方式で減少させた。負対照として、ビンブラスチンは、フロースルー中の[3H]コルヒチンの量に影響を与えなかった。本発明者は、更に、スピンカラム検定を用いて、COMPOUND Oが、チューブリン上のビンブラスチン結合部位へ結合することによってチューブリンと相互作用するかどうかを調べた。図26C(下部パネル)に示されるように、未標識ビンブラスチンの存在下における[3H]ビンブラスチンとのチューブリンインキュベーションは、フロースルー中で見出される[3H]ビンブラスチンの量を濃度依存方式で減少させた。対照すると、コルヒチンもCOMPOUND Oも、フロースルー中の[3H]ビンブラスチンの量に影響することはなかった。
【0184】
実施例27
COMPOUND Oは、癌細胞においてアポトーシスを誘発する
COMPOUND O化合物が、癌細胞においてアポトーシスを誘発するかどうかを調べるために、A549ヒト肺癌細胞を、37nM COMPOUND Oおよび37nMパクリタキセルで処置した。細胞アポトーシスおよび細胞死の検出は、Cell Death Detection ELISAキット(Roche Applied Sciences)により、キットによる検定プロトコルにしたがって測定した。簡単にいうと、5000個細胞を、96ウェルプレートの各々のウェル中に播種した。24時間インキュベーション後、それら細胞を、細胞増殖のIC50値に近い濃度である37nMのCOMPOUND Oおよびパクリタキセルで処置した。細胞を採取し、そして薬物処置後24時間、48時間または72時間に溶解させた。20マイクロリットルの溶解産物を取り出し、ストレプトアビジン被覆マイクロプレートに移した後、抗ヒストン−ビオチン抗体および抗DNA−POD抗体と一緒に2時間インキュベート後、発色用に2,2’−アジノビス−3−エチルベンズチアゾリンスルホン酸基質を加えた。そのプレートを、Beckman Multimode DTX880プレートリーダーにおいて405nmおよび490nmの吸光度で測定した。
【0185】
図27Aに示されるように、37nM COMPOUND Oおよび37nMパクリタキセルで処置された細胞は、(薬物処置後48時間から開始する)強いアポトーシスシグナルを示すが、DMSOで処置されただけの対照細胞は、最小のシグナルを示した。これは、COMPOUND Oが、A549肺癌細胞において時間依存性アポトーシスを誘発したことを示している。更に、本発明者は、二つの他の癌細胞系H596およびH292も調べた。図27Bは、37nM COMPOUND Oが、これら二つの細胞系において37nMパクリタキセルと同様のレベルのアポトーシスを引き起こしたことを示している。総合すると、COMPOUND Oは、癌細胞においてアポトーシスを誘発した。
【0186】
実施例28
COMPOUND Oは、癌細胞においてG2/M細胞周期停止を誘発する
微小管は、細胞の重複した染色体が、二つの娘細胞への細胞の開裂前に二つの同一のセットに分離する際の有糸分裂の過程において極めて重要である。パクリタキセルおよびビンブラスチンなどの、微小管を標的とする化合物は、微小管動力を抑制し且つ有糸分裂過程をブロックする。結果として、細胞は、G2/M期で阻止されるであろう。COMPOUND Oが、癌細胞分裂の場合の有糸分裂過程に影響を与えるかどうかを調べるために、A549ヒト肺癌細胞を、33nMのCOMPOUND Oおよびパクリタキセルで、または負対照として役立つ0.2%DMSOで処置した。被処置細胞を、有糸分裂および有糸分裂阻止を行う細胞についてのマーカーである、ホスホヒストンH3に対する抗体で染色した。図28Aは、パクリタキセルおよびCOMPOUND Oによる有糸分裂阻止の程度を有糸分裂指数で定量したものを示す。それは、パクリタキセル、COMPOUND OおよびDMSOで処置されたA549についてそれぞれ、44±8%、45±3%および3±1%であった。
【0187】
更に、本発明者は、フローサイトメトリーを用いて、細胞周期へのCOMPOUND Oの作用も調べた。簡単にいうと、A549細胞を、6ウェル組織培養プレート中に400,000個/ウェルの細胞密度で播種した。約24時間後、それら細胞を、37nM COMPOUND Oで処置し、そして更に24時間インキュベートさせた。細胞を、PBS中で洗浄し、トリプシン処理し、計数し、そして氷冷70%メタノール中で固定し且つ4℃で貯蔵した。細胞を、PBSで洗浄し、ヨウ化プロピジウムで染色し、そしてフローサイトメトリー分析まで氷上で保持した。図28Bに示されるように、G2/M期での細胞集団は、COMPOUND Oで処置された細胞において、DMSOのみで処置された細胞と比較して有意に増加した。
【0188】
公報、特許、特許出願および公表された特許出願などの参考文献は、全てを通して、本明細書中にそのまま援用される。
前述の発明は、理解の明瞭さの目的で、図および実施例によってある程度詳細に記載してきたが、特定の僅かな変化および修飾が実施されるであろうということは当業者に明らかである。したがって、説明および実施例は、本発明の範囲を制限すると解釈されるべきではない。
【特許請求の範囲】
【請求項1】
式(I)または式(II):
【化1】
(式中、Zは、次の置換されたフェニル環または複素環式環:
【化2】
より選択されるが、これに制限されるわけではない)
を有する化合物またはその薬学的に許容しうる塩。
【請求項2】
式(I)を有する化合物である、請求項1に記載の化合物。
【請求項3】
式(II)を有する化合物である、請求項1に記載の化合物。
【請求項4】
化合物であって、
【化3】
より選択される、式(I)を有する化合物。
【請求項5】
化合物であって、
【化4】
より選択される、式(II)を有する化合物。
【請求項6】
請求項1〜5のいずれか1項に記載の化合物および薬学的に許容しうる賦形剤または担体を含む医薬組成物。
【請求項7】
追加の活性剤を更に含む、請求項6に記載の医薬組成物。
【請求項8】
癌を処置する、予防するまたは管理する方法であって、このような処置を必要としている対象に、有効量の請求項1〜5に記載の化合物または請求項6または請求項7に記載の医薬組成物を投与することを含む方法。
【請求項9】
癌が、肉腫、類表皮癌、線維肉腫、子宮頸癌、胃癌、皮膚癌、白血病、リンパ腫、肺癌、非小細胞性肺癌、結腸癌、CNS癌、黒色腫、卵巣癌、腎癌、前立腺癌、乳癌、肝癌、頭部癌、頸部癌または膵癌である、請求項8に記載の方法。
【請求項10】
対象がヒトである、請求項8に記載の方法。
【請求項11】
癌を処置するのに用いるための、請求項1〜7のいずれか1項に記載の化合物。
【請求項12】
癌の処置用の薬剤の製造における、請求項1〜7のいずれか1項に記載の化合物またはその薬学的に許容しうる塩の使用。
【請求項13】
癌が、肉腫、類表皮癌、線維肉腫、子宮頸癌、胃癌、皮膚癌、白血病、リンパ腫、肺癌、非小細胞性肺癌、結腸癌、CNS癌、黒色腫、卵巣癌、腎癌、前立腺癌、乳癌、肝癌、頭部癌、頸部癌または膵癌である、請求項11または請求項12に記載の化合物。
【請求項1】
式(I)または式(II):
【化1】
(式中、Zは、次の置換されたフェニル環または複素環式環:
【化2】
より選択されるが、これに制限されるわけではない)
を有する化合物またはその薬学的に許容しうる塩。
【請求項2】
式(I)を有する化合物である、請求項1に記載の化合物。
【請求項3】
式(II)を有する化合物である、請求項1に記載の化合物。
【請求項4】
化合物であって、
【化3】
より選択される、式(I)を有する化合物。
【請求項5】
化合物であって、
【化4】
より選択される、式(II)を有する化合物。
【請求項6】
請求項1〜5のいずれか1項に記載の化合物および薬学的に許容しうる賦形剤または担体を含む医薬組成物。
【請求項7】
追加の活性剤を更に含む、請求項6に記載の医薬組成物。
【請求項8】
癌を処置する、予防するまたは管理する方法であって、このような処置を必要としている対象に、有効量の請求項1〜5に記載の化合物または請求項6または請求項7に記載の医薬組成物を投与することを含む方法。
【請求項9】
癌が、肉腫、類表皮癌、線維肉腫、子宮頸癌、胃癌、皮膚癌、白血病、リンパ腫、肺癌、非小細胞性肺癌、結腸癌、CNS癌、黒色腫、卵巣癌、腎癌、前立腺癌、乳癌、肝癌、頭部癌、頸部癌または膵癌である、請求項8に記載の方法。
【請求項10】
対象がヒトである、請求項8に記載の方法。
【請求項11】
癌を処置するのに用いるための、請求項1〜7のいずれか1項に記載の化合物。
【請求項12】
癌の処置用の薬剤の製造における、請求項1〜7のいずれか1項に記載の化合物またはその薬学的に許容しうる塩の使用。
【請求項13】
癌が、肉腫、類表皮癌、線維肉腫、子宮頸癌、胃癌、皮膚癌、白血病、リンパ腫、肺癌、非小細胞性肺癌、結腸癌、CNS癌、黒色腫、卵巣癌、腎癌、前立腺癌、乳癌、肝癌、頭部癌、頸部癌または膵癌である、請求項11または請求項12に記載の化合物。
【図1】

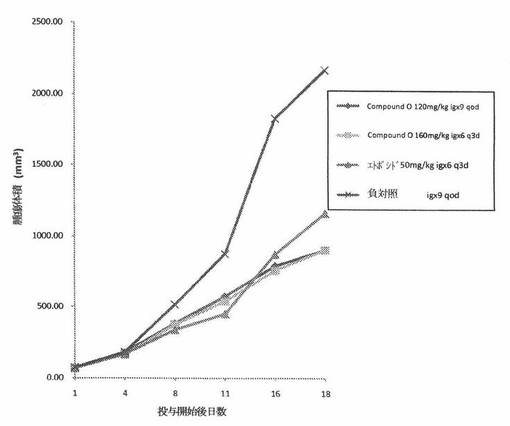
【図2】
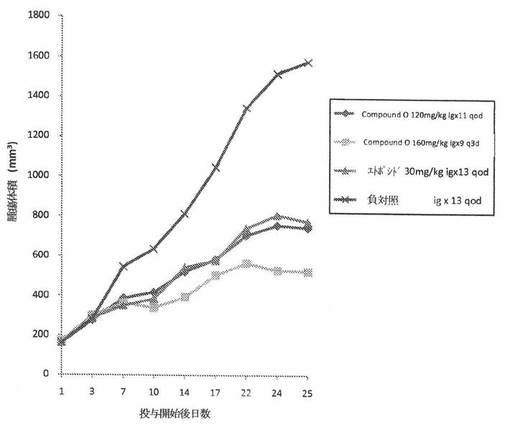
【図3】
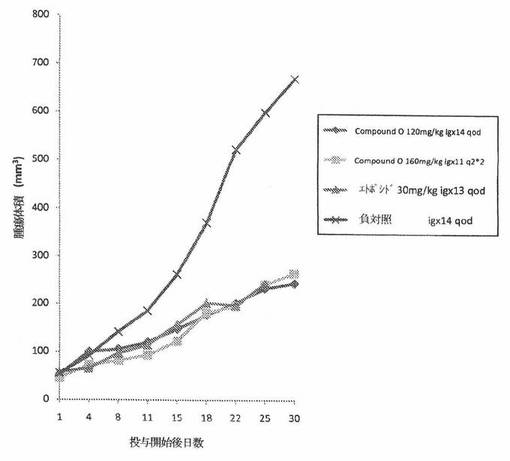
【図4A】
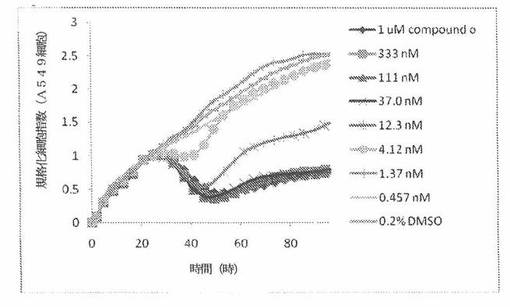
【図4B】
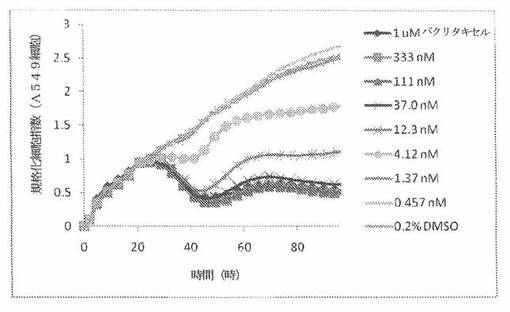
【図4C】
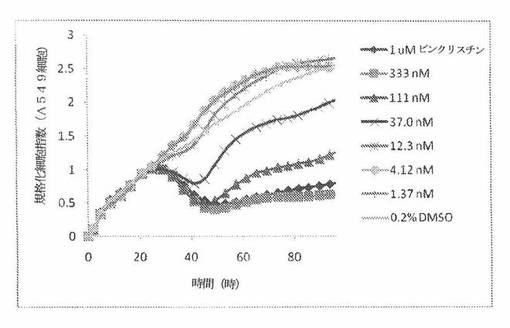
【図5】
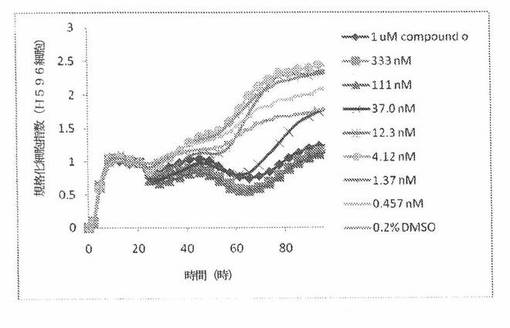
【図6】
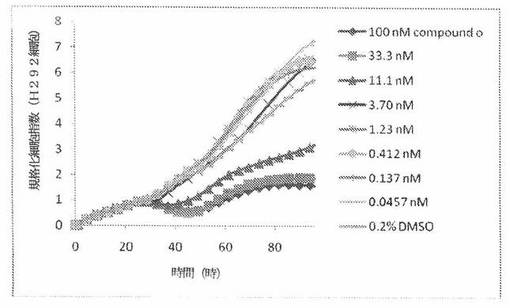
【図7】
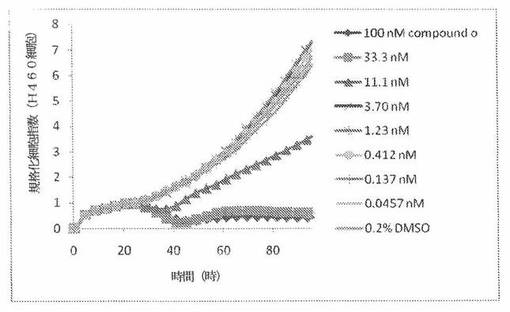
【図8】
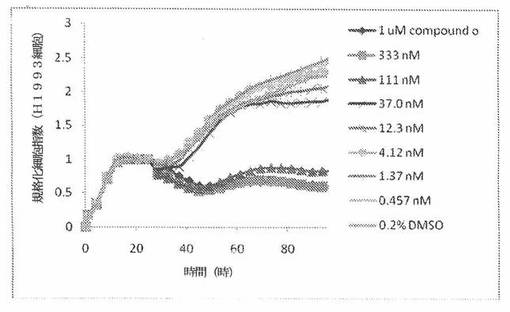
【図9】
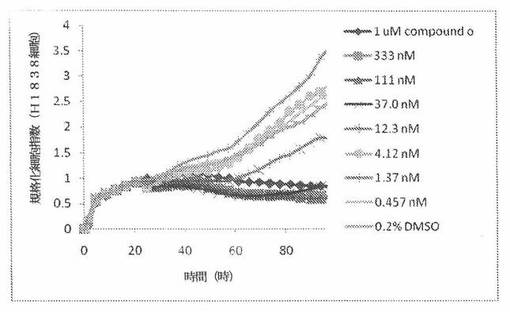
【図10】
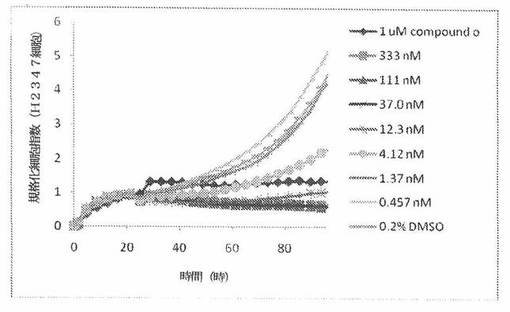
【図11】
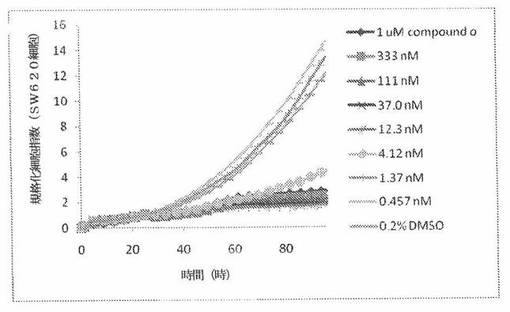
【図12】
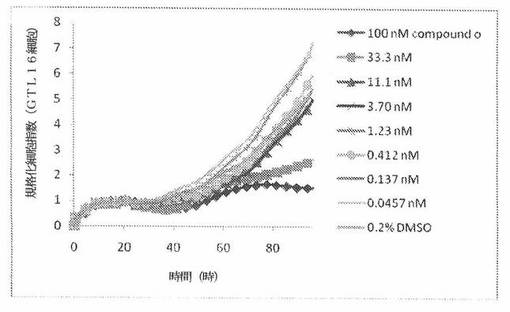
【図13】
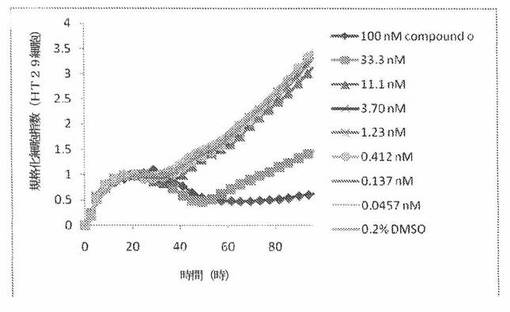
【図14】
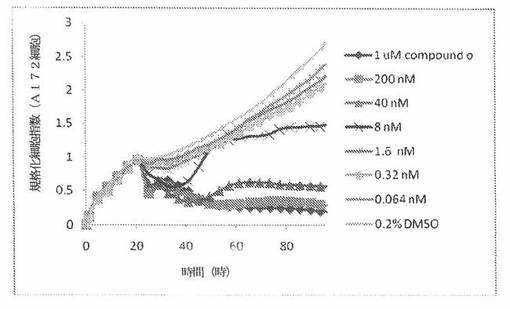
【図15】
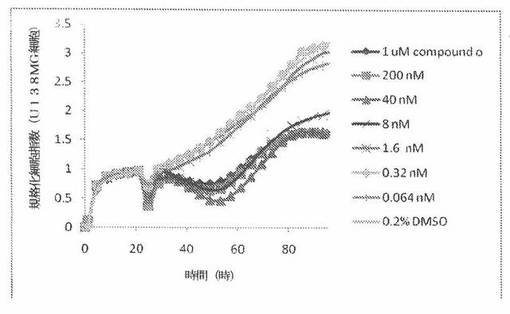
【図16】
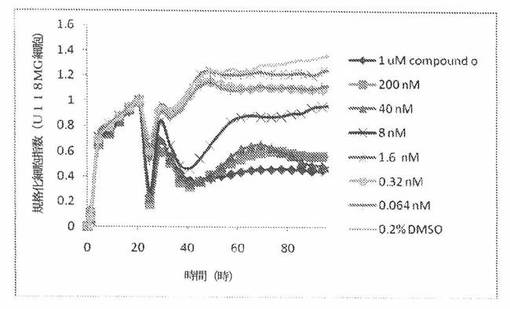
【図17】
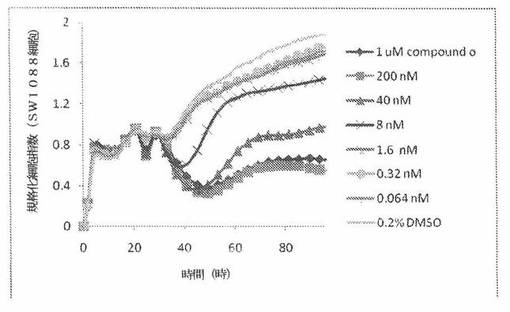
【図18】
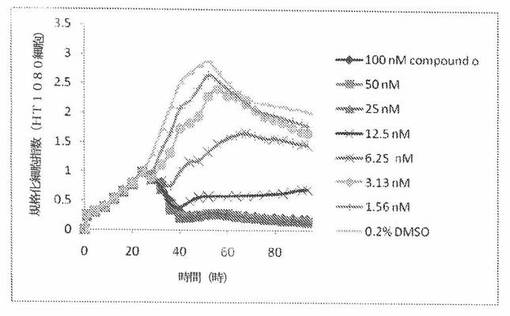
【図19】
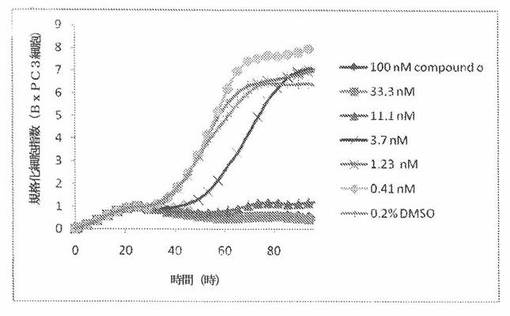
【図20】
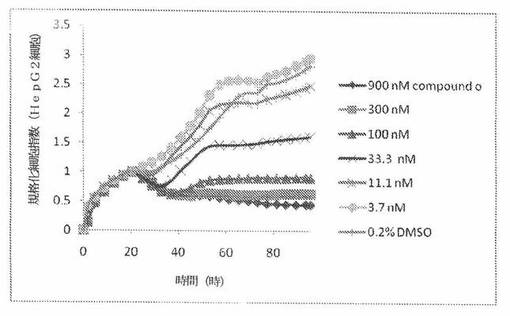
【図21】
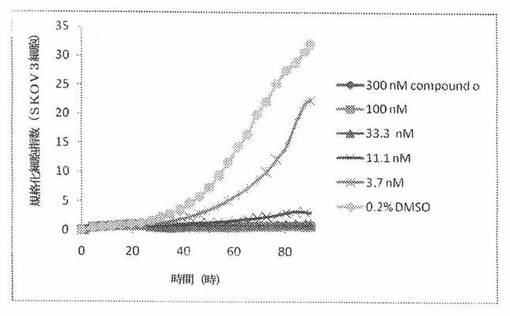
【図22】
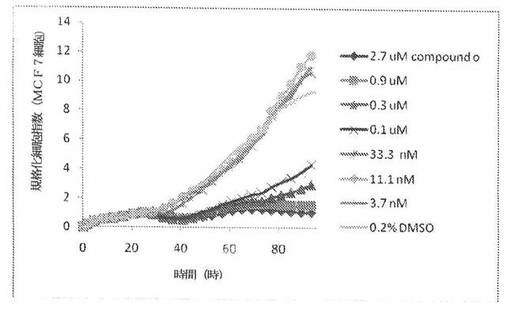
【図23】
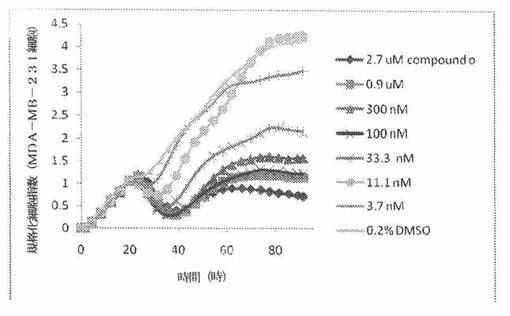
【図24A】
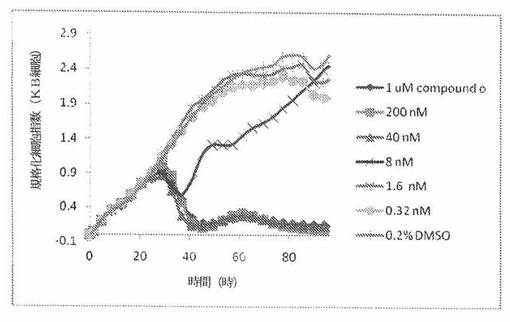
【図24B】
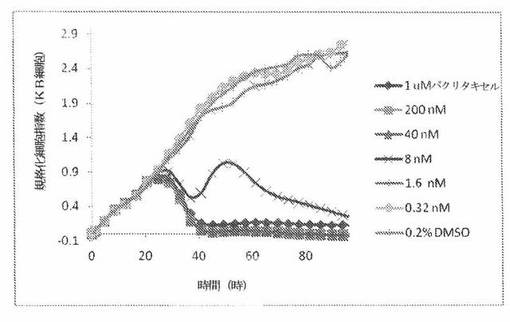
【図24C】
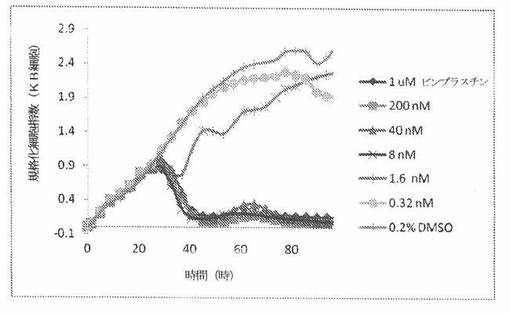
【図24D】
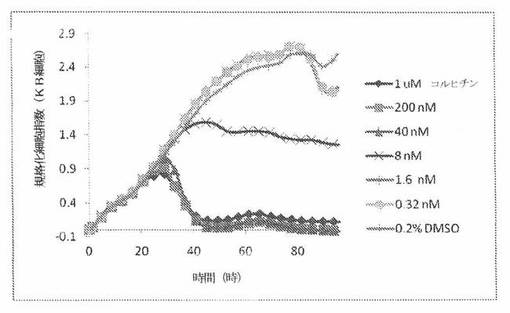
【図24E】
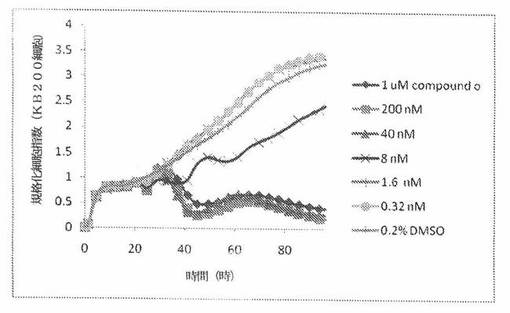
【図24F】
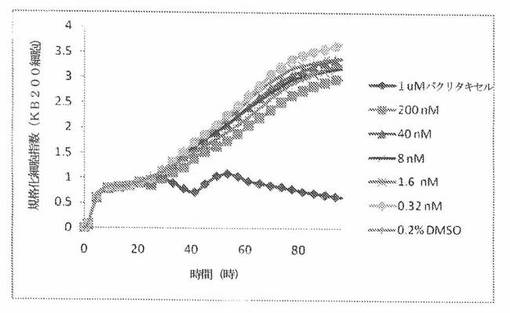
【図24G】
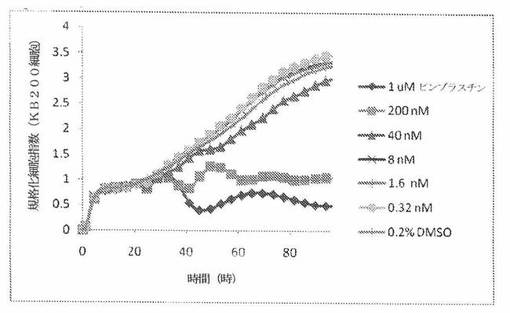
【図24H】
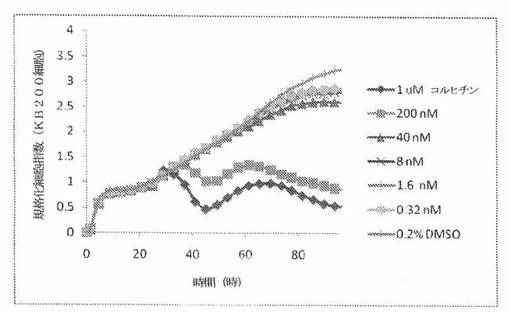
【図25】
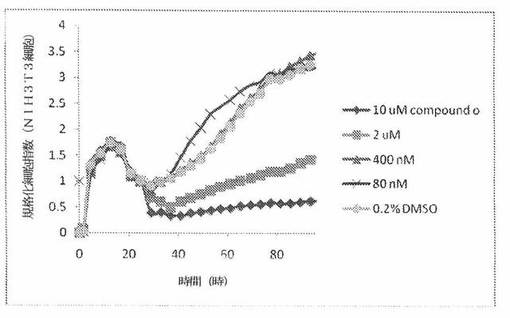
【図26A】
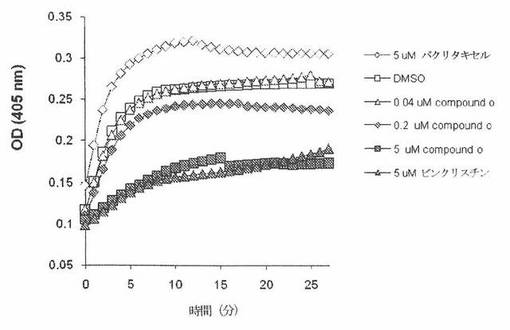
【図26B】
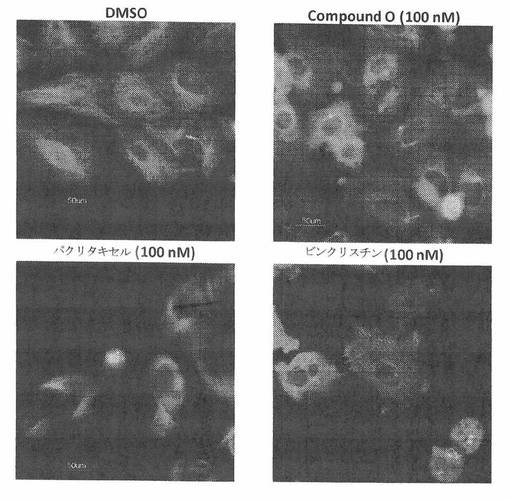
【図26C】
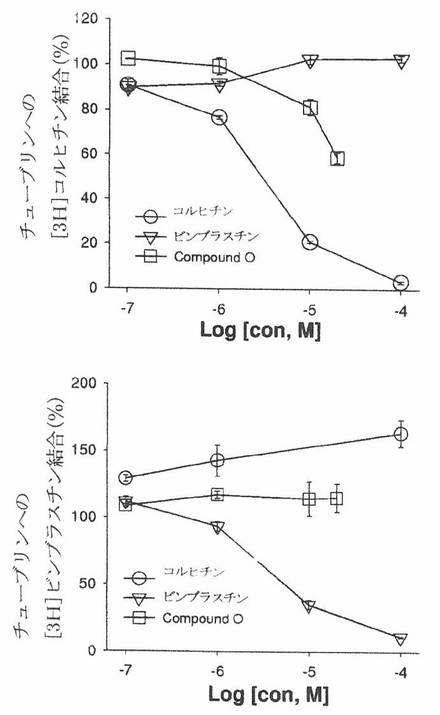
【図27A】
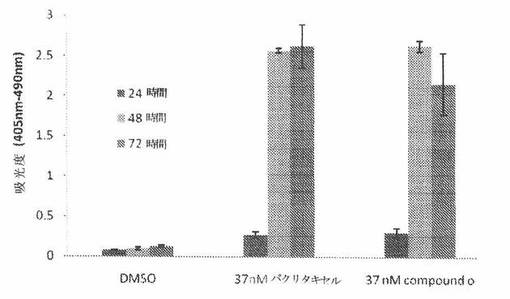
【図27B】
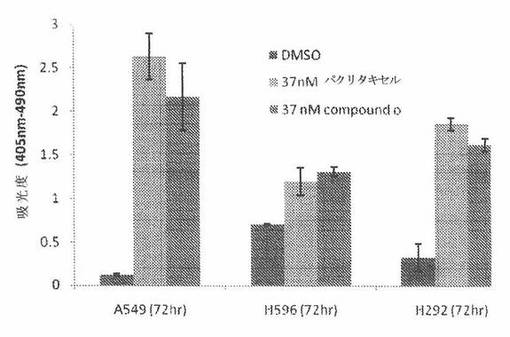
【図28A】
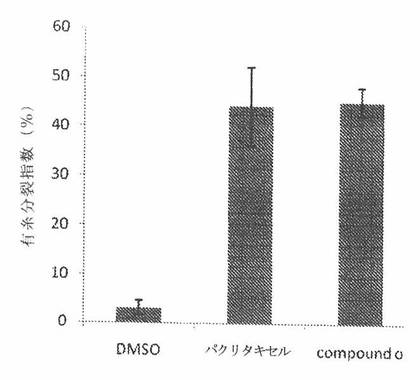
【図28B】
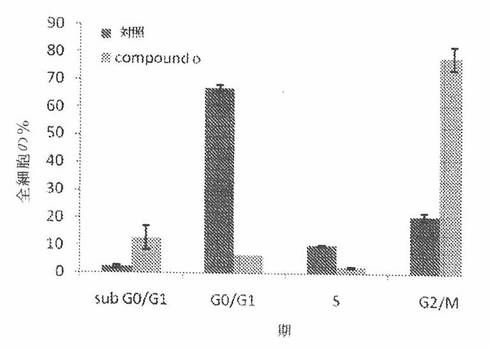
【図2】
【図3】
【図4A】
【図4B】
【図4C】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24A】
【図24B】
【図24C】
【図24D】
【図24E】
【図24F】
【図24G】
【図24H】
【図25】
【図26A】
【図26B】
【図26C】
【図27A】
【図27B】
【図28A】
【図28B】
【公表番号】特表2013−520439(P2013−520439A)
【公表日】平成25年6月6日(2013.6.6)
【国際特許分類】
【出願番号】特願2012−554073(P2012−554073)
【出願日】平成23年2月18日(2011.2.18)
【国際出願番号】PCT/US2011/025550
【国際公開番号】WO2011/103509
【国際公開日】平成23年8月25日(2011.8.25)
【出願人】(510014674)アセア バイオサイエンシズ インコーポレイテッド (2)
【氏名又は名称原語表記】ACEA BIOSCIENCES INC.
【Fターム(参考)】
【公表日】平成25年6月6日(2013.6.6)
【国際特許分類】
【出願日】平成23年2月18日(2011.2.18)
【国際出願番号】PCT/US2011/025550
【国際公開番号】WO2011/103509
【国際公開日】平成23年8月25日(2011.8.25)
【出願人】(510014674)アセア バイオサイエンシズ インコーポレイテッド (2)
【氏名又は名称原語表記】ACEA BIOSCIENCES INC.
【Fターム(参考)】
[ Back to top ]