
抗癲癇誘発剤
【課題】癲癇を含む痙攣疾患の抑制に有用な方法および化合物の提示。
【解決手段】α,α-二置換β-アラニン、α,β-二置換β-アラニン、β,β-二置換β-アラニン、α,β,α-三置換β-アラニン、α,β,β-三置換β-アラニン、α,α,N-三置換β-アラニン、α,β,N-三置換β-アラニン、β,β,N-三置換β-アラニン、α,α,N,N-四置換β-アラニン、α,β,N,N-四置換β-アラニン、β,β,N,N-四置換β-アラニン、α,α,β,β-四置換β-アラニン、α,α,β,N-四置換β-アラニン、α,β,β,N-四置換β-アラニン、α,α,β,N,N-五置換β-アラニン、α,β,β,N,N-五置換β-アラニン、α,α,β,β,N-五置換β-アラニン、α,α,β,β,N,N-六置換β-アラニン、およびその薬学的に許容される塩もしくはエステルからなる群より選択される化合物の有効量を投与する段階を含む、癲癇誘発を抑制する方法。
【解決手段】α,α-二置換β-アラニン、α,β-二置換β-アラニン、β,β-二置換β-アラニン、α,β,α-三置換β-アラニン、α,β,β-三置換β-アラニン、α,α,N-三置換β-アラニン、α,β,N-三置換β-アラニン、β,β,N-三置換β-アラニン、α,α,N,N-四置換β-アラニン、α,β,N,N-四置換β-アラニン、β,β,N,N-四置換β-アラニン、α,α,β,β-四置換β-アラニン、α,α,β,N-四置換β-アラニン、α,β,β,N-四置換β-アラニン、α,α,β,N,N-五置換β-アラニン、α,β,β,N,N-五置換β-アラニン、α,α,β,β,N-五置換β-アラニン、α,α,β,β,N,N-六置換β-アラニン、およびその薬学的に許容される塩もしくはエステルからなる群より選択される化合物の有効量を投与する段階を含む、癲癇誘発を抑制する方法。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願
本出願は、2001年3月13日に提出された米国仮特許出願第60/275,618号の優先権を主張し;本出願は、さらに、1998年3月11日に提出された米国特許出願第09/041,371号(現在は、その全内容が参照として本明細書に組み入れられる米国特許第6,306,909号)に関し、その内容を開示する。
【背景技術】
【0002】
発明の背景
癲癇は、世界中の何十万人もの人が罹患している、発作を伴う重度の神経学的病態である。臨床的には、発作は、脳においてニューロンの集合体から突然電気が放電されることから生じる。結果として生じる神経細胞活性は、制御不可能な動きなどの症状となって顕在化する。
【0003】
発作は、「発作発生(ictogenesis)」と称される過程を通してニューロン集合体からの過剰な電気放電により引き起こされる単一の独立した臨床事象である。従って、発作は、単なる癲癇の症状である。癲癇は、正常な脳が変化し、「癲癇誘発」と称される過程を通じて反復性発作に感受性となる、根底にある一連の病的変換を特徴とする動的でしばしば進行的な過程である。発作発生および癲癇誘発は共通の生化学的経路を有すると考えられているが、二つの過程は同一ではない。発作発生(時間的および空間的な発作の開始および伝播)は、数秒間または数分間におよび起こる急速で明確な電気的/化学的事象である。癲癇誘発(正常な脳が、「癲癇誘発焦点」の開始および成熟により、自発的で偶発的で時間制限的な反復性発作に感受性の状態に転換される漸進的な過程)は、一般的に数ヶ月から数年間かけて生じる、緩徐な生化学的過程および/または組織学的過程である。癲癇誘発は二相過程である。第1相癲癇誘発は、最初の発作前の癲癇誘発過程の開始であり、発作、疾病(例えば髄膜炎)、または頭部への事故的打撃または脳に行われた外科手術などの外傷の結果であることが多い。第2相癲癇誘発は、発作にすでに感受性である脳が、依然として、頻度および/または重度の増加中の発作にさらに一層感受性となる過程を意味する。癲癇誘発に関与する過程は明確に確定されていないが、N-メチル-D-アスパルテート(NMDA)受容体により媒介されるニューロン間の興奮連関のアップレギュレーションが関与すると考える研究者もいる。また、γ-アミノ-酪酸(GABA)受容体により媒介されるニューロン間の抑制連関のダウンレギュレートを示唆する研究者もいる。
【0004】
癲癇発作が致死的であることは稀であるが、多数の患者が、発作の破壊的で危険性のある結果を避けるための薬物療法を必要としている。多くの場合、薬物療法は長期間必要とされ、生涯、処方薬を服用し続けなければならない場合もある。さらに、癲癇の管理に使用される薬物には、長期使用に伴う副作用があり、薬価がかなり高い場合もある。
【0005】
古い抗痙攣薬(例えばフェニトイン、バルプロ酸塩、およびカルバマゼピン)(イオンチャネル遮断剤)ならびに新しい薬剤(例えばフェルバメート、ガバペンチン、およびチアガビン)を含む、多種多様の薬物が癲癇発作の管理に使用できる。β-アラニンは、抗痙攣活性ならびにNMDA抑制活性およびGABA作動性刺激活性を示すと報告されているが、臨床的には使用されていない。現在入手可能な承認されている癲癇薬は抗痙攣剤であり、本明細書において「抗痙攣剤」という用語は、「抗発作」または「抗発作発生」と同義語であり;これらの薬物は、発作発生を遮断することにより発作を抑止できるが、癲癇誘発を遮断しないことから、癲癇には影響を及ぼさないと考えられている。従って、癲癇の治療に使用できる薬物は多数あるにも関わらず(すなわち、癲癇発作に伴う痙攣抑止により)、癲癇誘発を特徴とする病的変化の治療に一般的に承認されている薬物は全くない。癲癇誘発過程を抑制する一般的に承認されている方法は全くなく、抗癲癇誘発剤として認識されている一般的に承認されている薬物は全くない。
【発明の開示】
【0006】
発明の概要
本発明は、癲癇を含む痙攣疾患の治療および/または予防に有用である、方法および化合物、例えば抗発作発生化合物および/または抗癲癇誘発化合物に関する。
【0007】
一つの局面において、本発明は、被験者における癲癇誘発を抑制する方法を提供する。本方法は、それを必要とする被験者に、被験者における癲癇誘発が抑制されるように癲癇誘発に関連した経路中の一過程を変調する薬剤の有効量を投与する段階を含む。
【0008】
別の局面において、被験者における癲癇誘発抑制法を提供する。NMDA受容体に拮抗し、内因性GABA抑制を増強する薬剤の有効量を、被験者における癲癇誘発が抑制されるように、それを必要とする被験者に投与する。好ましい態様において、該薬剤は、NMDA受容体のグリシン結合部位に結合することにより、NMDA受容体に拮抗する。好ましい態様において、該薬剤は、グリア細胞のGABA取り込みを減少させることにより、GABA抑制を増強する。ある好ましい態様において、該薬剤は、NMDA受容体に拮抗し且つ内因性GABA抑制を増強するファルマコフォアを含む。該薬剤は経口投与でき、ある態様において、経口投与段階後に、該薬剤は、能動輸送シャトル機序により被験者の神経系に輸送され得る。好ましい態様において、抗癲癇誘発剤は、β-アミノアニオン化合物であり、アニオン部分は、カルボキシレート、サルフェート、スルホネート、スルフィネート、スルファメート、テトラゾリル、ホスフェート、ホスホネート、ホスフィネート、およびホスホロチオエートからなる群より選択される。ある態様において、該薬剤はβ-アミノ酸であるが、好ましくはβ-アラニンではない。
【0009】
別の局面において、本発明は、被験者における癲癇誘発を抑制する方法を提供する。該方法は、それを必要とする被験者に、癲癇誘発が抑制されるように、下記式
【化1】

(式中、Aは、生理的pHにおいて陰性基であり;R1は、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、またはアミノカルボニルであり;R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、もしくはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、環の原子数が3〜7である非置換もしくは置換の複素環を形成している)
で示される化合物;
またはその薬学的に許容される塩もしくはエステルの有効量を投与する段階を含む。
【0010】
別の局面において、本発明は、被験者における癲癇誘発を抑制する方法を提供する。該方法は、それを必要とする被験者に、癲癇誘発が抑制されるように、下記式
【化2】
(式中、点線は、選択的な単結合/二重結合(EまたはZ立体配置の)を示し;Aは、生理的pHにおいて陰性基であり;R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、もしくはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、環の原子数が3〜7である非置換または置換の複素環を形成しており;R4およびR5は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、アルコキシ、アリールオキシ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニルであるか;または、R4およびR5は、一緒に、環の原子数が5〜15である置換または非置換の炭素環または複素環を形成している)
で示される化合物;またはその薬学的に許容される塩もしくはエステルの有効量を投与する段階を含む。
【0011】
別の局面において、本発明は、被験者における痙攣疾患を抑制する方法を提供する。該方法は、痙攣疾患が抑制されるよう、それを必要とする被験者に、β-アミノアニオン化合物はβ-アラニンまたはタウリンではないという条件で、有効量のβ-アミノアニオン化合物を投与する段階を含む。
【0012】
別の局面において、本発明は、下記式:
【化3】
(式中、Aは、生理的pHにおいて陰性基であり;R1は、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、ニトロ、チオール、チオアルキル、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、またはアミノカルボニルであり;R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、もしくはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、環の原子数が3〜7である非置換または置換の複素環を形成している)
で示される抗癲癇誘発化合物;またはその薬学的に許容される塩もしくはエステルを提供し、抗癲癇誘発化合物は抗癲癇誘発活性を有する。好ましい態様において、Aはカルボキシレートを示す。
【0013】
ある好ましい態様において、該化合物は、α-シクロヘキシル-β-アラニン、α-(4-tert-ブチルシクロヘキシル)-β-アラニン、α-(4-フェニルシクロヘキシル)-β-アラニン、α-シクロドデシル-β-アラニン、β-(p-メトキシフェネチル)-β-アラニン、およびβ-(p-メチルフェネチル)-β-アラニン、およびその薬学的に許容される塩からなる群より選択されるか;または、該化合物は、β-(4-トリフルオロメチルフェニル)-β-アラニンおよびβ-[2-(4-ヒドロキシ-3-メトキシフェニル)エチル]-β-アラニン、およびその薬学的に許容される塩からなる群より選択されるか;または、該化合物は、β-(3-ペンチル)-β-アラニンおよびβ-(4-メチルシクロヘキシル)-β-アラニン、およびその薬学的に許容される塩からなる群より選択される。
【0014】
さらに別の局面において、本発明は、下記式
【化4】
(式中、Arは、非置換または置換のアリール基を示し;R6およびR6*は、各々独立的に、水素、アルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yは水素またはチアゾリル、トリアゾリル、およびイミダゾリルからなる群より選択される複素環式部分であり、ただし、Arが非置換フェニル基である場合、R7は水素、メチル、またはフェニルではない)
で示されるジオキサピペラジン化合物;またはその薬学的に許容される塩を提供する。
【0015】
被験者における痙攣疾患を抑制する方法も開示する。有効量の薬剤を、被験者における癲癇誘発および発作発生が抑制されるように、それを必要とする被験者に投与する。該薬剤は、ナトリウムまたはカルシウムイオンチャネルを遮断するか、あるいは、カリウムまたは塩化物イオンチャネルを開口し;少なくとも一つの活性、例えばNMDA受容体拮抗作用、内因性GABA抑制増強、カルシウム結合、鉄結合、亜鉛結合、NOシンターゼ阻害、および抗酸化活性を有する。望ましい態様において、該薬剤は、NMDA受容体に結合することにより、例えばNMDA受容体のグリシン結合部位に結合することによりNMDA受容体に拮抗する、および/または、グリア細胞のGABA取り込みを減少することによりGABA抑制を増強する。
【0016】
別の局面において、本発明は、痙攣疾患を抑制する方法を提供する。該方法は、それを必要とする被験者に、痙攣疾患が抑制されるように、下記式:
【化5】
(式中、Arは、非置換または置換のアリール基を示し;R6およびR6*は、各々独立的に、水素、アルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yは水素または複素環式部分、例えばチアゾリル、トリアゾリル、およびイミダゾリルであり、ただし、Arが非置換フェニル基である場合、R7は水素、メチル、またはフェニルではない)
で示される化合物;またはその薬学的に許容される塩もしくはエステルの有効量を投与する段階を含む。
【0017】
別の局面において、本発明は、下記式
【化6】
(式中、Arは、非置換または置換のアリール基を示し;R6は、水素またはアルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R6*は、抗酸化部分、NMDAアンタゴニスト、NOシンターゼ阻害剤、鉄キレート部分、Ca(II)キレート部分、またはZn(II)キレート部分であってよく;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yは複素環式部分、例えばチアゾリル、トリアゾリル、またはイミダゾリルである)
で示される化合物;またはその薬学的に許容される塩を提供する。好ましい態様において、R6*は、D-α-アミノアジピルである、および/またはR7はメルカプトメチルである。
【0018】
別の局面において、本発明は、それを必要とする被験者に、癲癇誘発が抑制されるように、下記式
【化7】
(式中、Arは、非置換または置換のアリール基を示し;R6は、水素またはアルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R6*は、抗酸化部分、NMDAアンタゴニスト、NOシンターゼ阻害剤、鉄キレート部分、Ca(II)キレート部分、またはZn(II)キレート部分であり得;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yは、チアゾリル、トリアゾリル、およびイミダゾリルからなる群より選択された複素環式部分である)
で示される化合物;またはその薬学的に許容される塩の有効量を投与する段階を含む、癲癇誘発および発作発生を同時に抑制する方法を提供する。
【0019】
別の局面において、本発明は、それを必要とする被験者に、NMDA受容体拮抗作用に関連した疾患が治療されるように、下記式
【化8】
(式中、Arは、非置換または置換のアリール基を示し;R6は、水素またはアルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R6*はNMDAアンタゴニスト部分であり;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yは、チアゾリル、トリアゾリル、およびイミダゾリルからなる群より選択された複素環式部分である)
で示される化合物;またはその薬学的に許容される塩の有効量を投与する段階を含む、NMDA受容体拮抗作用に関連した疾患を治療する方法を提供する。
【0020】
別の局面において、本発明は、下記式
【化9】
(式中、点線は、選択的な単結合/二重結合(EまたはZ立体配置の)を示し;R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、複素環の原子数が3〜7である非置換または置換の複素環を形成しており;R4およびR5は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、カルボキシル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R4およびR5は一緒に環の原子数が5〜15である置換または非置換の炭素環または複素環を形成し;R8は、水素、アルキル、アリール、または有機塩もしくは有機塩形成カチオンである)
で示されるβ-アミノカルボキシル化合物を調製する方法を提供する。該方法は、下記式
【化10】
(式中、点線は、各々、選択的な単結合を示し;Xは、ニトロ、アジド、またはNR2R3であり、ここでのR2およびR3は前記に定義した通りであり;Wは、-CNまたは-COORであり;R4およびR5は前記に定義した通りであり;R8は、水素、アルキル、アリール、または有機塩もしくは無機塩形成カチオンである)
で示される化合物を、還元的脱硫条件下で反応させて、β-アミノカルボキシル化合物を形成する段階を含む。
【0021】
別の局面において、本発明は、下記式
【化11】
(式中、R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、複素環の原子数が3〜7である非置換または置換の複素環を形成しており;R4およびR5は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、アルコキシ、アリールオキシ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニルであるか;または、R4およびR5は、一緒に、環の原子数が5〜15である置換または非置換の炭素環または複素環を形成しており;R8は、水素、アルキル、アリール、または有機塩もしくは無機塩形成カチオンである)
で示されるβ-アミノカルボキシル化合物を調製する方法を提供する。該方法は、下記式
【化12】
(式中、点線は、各々、選択的な単結合/二重結合を示し;Xは、ニトロ、アジド、またはNR2R3であり、R2およびR3は前記に定義した通りであり;Wは-CNまたは-COOR8であり;R8は、水素、アルキル、アリール、または有機塩もしくは無機塩形成カチオンであり;R4およびR5は前記に定義した通りである)
で示される化合物を、還元的脱硫条件下で反応させ、前記式のβ-アミノカルボキシル化合物を形成する段階を含み、ただし、Wが-CNである場合、該方法はさらに酸性化段階を含む。
【0022】
本発明はまた、必要とする被験者に、被験者における癲癇誘発が抑制されるように、式A-Bにより示される有効量の薬剤を投与することを含む、被験者における癲癇誘発および発作発生を抑制する方法を提供し、ここでのAは、ナトリウムまたはカルシウムイオンチャネル遮断活性を有するドメインであるか、あるいは、Aは、カリウムまたは塩化物チャネル開口活性を有し;Bは、少なくとも一つの活性、例えばNMDA受容体拮抗作用;内因性GABA抑制増強、カルシウム結合、鉄結合、亜鉛結合、NOシンターゼ阻害、および抗酸化活性を有するドメインである。好ましい態様において、薬剤のドメインAおよびBは共有結合している。好ましい態様において、Aはジオキサピペラジン部分である。
【0023】
さらに別の局面において、本発明は、それを必要とする被験者に、癲癇誘発が抑制されるように、下記式
【化13】
(式中、Aは生理的pHにおいて陰性基であり;R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、複素環の原子数が3〜7である非置換または置換の複素環を形成しており;R4およびR5は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、アルコキシ、アリールオキシ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニルであるか;または、R4およびR5は、一緒に、環の原子数が5〜15である置換または非置換の炭素環または複素環を形成している)
で示される化合物;またはその薬学的に許容される塩もしくはエステルの有効量を投与する段階を含む、癲癇誘発を抑制する方法を提供する。
【0024】
被験者における神経学的状態を抑制する方法には、それを必要とする被験者に、被験者における神経学的状態が抑制されるように、NMDA受容体に拮抗し、内因性GABA抑制を増強する薬剤の有効量を投与する段階が含まれる。神経学的状態は、例えば、卒中、アルツハイマー病、癌、および神経変性性疾患であり得る。
【0025】
β-アリール-β-アラニン化合物の調製法を提示し、これには、アリールアルデヒドをマロネート化合物およびアンモニウム化合物と、β-アリール-β-アラニン化合物が形成されるような条件下で反応させる段階が含まれる。
【0026】
癲癇誘発を抑制する他の方法には、それを必要とする被験者に、癲癇誘発が抑制されるように、下記式
【化14】
(式中、R9およびR10は、各々独立的に、水素、アルキル、アルケニル、アルキニル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、チオール、アルキルチオール、ニトロ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、およびアミノカルボニルであり得るか;または、R9およびR10は、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成し;R11は、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R10およびR11は、それらがそれぞれ結合している炭素原子および窒素原子と共に連結して、環の員数が4〜8である複素環を形成し;R12は、水素、アルキル、アリール、および炭水化物からなる群より選択される)
で示される化合物;またはその薬学的に許容される塩もしくはエステルの有効量を投与する段階を含む。
【0027】
別の局面において、癲癇誘発を抑制する方法には、それを必要とする被験者に、癲癇誘発が抑制されるように、下記式:
【化15】
(式中、R9a、R9b、R10a、R10bは、各々独立的に、水素、アルキル、アルケニル、アルキニル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、チオール、アルキルチオール、ニトロ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、およびアミノカルボニルであり得るか;または、R9aおよびR9bは、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成しているか;または、R10aおよびR10bは、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成しているか;または、R9aおよびR9bの一方は、R10aおよびR10bの一方と、それらが結合している2炭素単位と共に連結して、環の員数が4〜8である炭素環または複素環を形成し;R11は、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R10aおよびR10bの一方が、R11と、それらがそれぞれ結合している炭素原子および窒素原子と共に連結して、環の員数が4〜8である複素環を形成し;R12は、水素、アルキル、アリール、および炭水化物(例えば、糖、例えばリボースまたはデオキシリボース)からなる群より選択される)
で示される化合物;またはその薬学的に許容される塩もしくはエステルの有効量を投与する段階が含まれる。
【0028】
被験者における癲癇誘発を予防および/または抑制できる化合物を同定するためのファルマコフォアモデリング法は、本発明の一部であり、癲癇誘発に関与するタンパク質または分子に対して直接的または間接的薬理作用を引き起こすことが知られる二つ以上の化合物の構造の検証を特色とする。癲癇誘発に関与するこれらのタンパク質および分子には、細胞表面受容体分子(例えばNMDA受容体)または神経伝達物質輸送に関与する分子(例えばGABA輸送体)が含まれる。好ましくは、これらの化合物の構造には各々、化合物の薬理作用の少なくともいくつかを示すことのできる、一つまたは複数のファルマコフォアが含まれる。本発明の方法にはまた、二つ以上の化合物のファルマコフォア構造に基づいた平均的なファルマコフォア構造(例えば炭素骨格構造および/または3次元空間充填構造)の決定が含まれる。一つまたは複数の平均的なファルマコフォア構造を有する新規化合物を、実施例1に示したようなこれらの方法を使用して選択できる。
【0029】
関連した態様において、これらの方法は、癲癇誘発に関与する二つ以上のタンパク質または分子に対して直接的または間接的な薬理作用を引き起こすことが知られる、二つ以上の化合物の構造の検証を特徴とする。選択する新規化合物は、好ましくは、癲癇誘発に関与する異なるタンパク質または分子に対して活性である、一つ以上のファルマコフォアを有する。
【0030】
好ましい態様において、本発明の方法により選択される(例えば設計される)新規化合物は、被験者における癲癇誘発を抑制する。本発明のさらに他の目的は、卒中、アルツハイマー病、および神経変性性疾患の治療用の化合物および方法を提供することである。本発明のさらに他の目的は、新規の抗痙攣剤を提供することである。本発明のさらに他の目的は、卒中および疼痛を治療する化合物および方法を提供することである。本発明のこれらおよびその他の目的、特徴、および利点は、以下の記載および特許請求の範囲から明らかである。
【0031】
発明の詳細な説明
本発明は、癲癇および痙攣疾患の治療、癲癇誘発の抑制、および発作発生の抑制に有用な方法および薬剤;ならびに、本発明の抗痙攣剤および抗癲癇誘発剤の調製法に関する。本発明はさらに、痙攣疾患治療のための薬学的組成物、および本発明の抗痙攣化合物を含むキットにも関する。
【0032】
定義
便宜上、明細書、実施例および添付の特許請求の範囲に使用される特定の用語をここに集める。
【0033】
「癲癇誘発に関連した経路中の一過程」という用語には、第1相または第2相の癲癇誘発中に起こり、組織、すなわち中枢神経系(CNS)、例えば脳の組織における癲癇誘発変化をもたらす、生化学的過程または事象が含まれる。癲癇誘発に関連した経路中の過程の例は、下記に、より詳細に考察する。
【0034】
「NMDA受容体拮抗作用に関連した疾患」という用語には、NMDA受容体の異常(例えば過剰)活性を、NMDA受容体の拮抗作用により治療できる、被験者の疾患が含まれる。癲癇は、過剰なNMDA系の活性に関連した疾患である。過剰なNMDA系の活性に関連した疾患のその他の非制限的な例には、疼痛、卒中、不安、精神分裂病、その他の精神病、脳虚血、ハンチントン舞踏病、運動神経疾患、アルツハイマー病、AIDS痴呆、ならびに、過剰なNMDA受容体活性が疾患の少なくとも原因の一部であるその他の疾患(ヒトおよび動物における)が含まれる。例えば、Schoeppら、Eur J.Pharmacol.203:237-243(1991);Leesonら、J.Med.Chem 34:1243-1252(1991);Kulagowskiら、J.Med.Chem.37:1402-1405(1994);Mallamoら、J.Med.Chem.37:4438-4448(1994);およびそれに引用されている文献を参照されたい。
【0035】
「痙攣疾患」という用語には、被験者が、痙攣、例えば癲癇発作に起因する痙攣に患っている、疾患が含まれる。痙攣疾患には、癲癇および非癲癇性痙攣、例えば、被験者への痙攣剤の投与に起因する痙攣が含まれるがこれに限定されない。
【0036】
「癲癇誘発抑制」という用語には、癲癇誘発過程の予防、遅延、停止、または回復が含まれる。
【0037】
「抗癲癇誘発剤」という用語には、薬剤を被験者に投与した場合に、癲癇誘発を抑制できる薬剤が含まれる。
【0038】
「抗痙攣剤」という用語には、薬剤を被験者に投与した場合に、発作発生を抑制(例えば予防、遅延、停止、または回復)できる薬剤が含まれる。
【0039】
「ファルマコフォア」という用語は当技術分野において既知であり、これには、選択した生化学的作用、例えば酵素の阻害、受容体への結合、イオンのキレート形成などを果たすことのできる分子部分が含まれる。選択したファルマコフォアは、二つ以上の生化学的作用を有することができ、例えば、ある酵素の阻害剤であり、同時に第二酵素のアゴニストであることができる。治療剤は、同じまたは異なる生化学活性を有することのできる、一つ以上のファルマコフォアを含むことができる。当業者は、類似の構造および/または特性(例えば生物学的作用)を有する多くのファルマコフォアを合わせて、最適化または「平均ファルマコフォア」構造を予測または設計し得ることを認識していると考えられる。このような平均的なファルマコフォア構造は、個々のファルマコフォアを使用して平均的な構造を作製するよりも望ましいレベルの生物学的作用を与え得る。
【0040】
「陰性基」は、生理的pHにおいて負に荷電した基を意味する。好ましい陰性基には、カルボキシレート、サルフェート、スルホネート、スルフィネート、スルファメート、テトラゾリル、ホスフェート、ホスホネート、ホスフィネート、またはホスホロチオエートもしくはその機能的等価体が含まれる。陰性基の「機能的等価体」という用語には、生物学的等価体、例えばカルボキシレート基の生物学的等価体が含まれる。生物学的等価体には、古典的な生物学的に等価な同等体および非古典的な生物学的に等価な同等体の両方が含まれる。古典的および非古典的な生物学的等価体は当技術分野において既知である。例えば、Silverman,R.B. The Organic Chemistry of Drug Design and Drug Action、アカデミックプレス社:CA州サンディエゴ所在、1992、19〜23頁を参照されたい。特に好ましい陰性基はカルボキシレートである。
【0041】
「β-アミノアニオン化合物」という用語には、2炭素スペーサー単位により陰性基から離れている、例えば-NRaRb(RaおよびRbは、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、もしくはアリールオキシカルボニルであり得るか、または、RaおよびRbは、それらが結合している窒素原子と共に、環の原子数が3〜8である環式部分を形成している)などの、アミノ基を有する化合物が含まれる。従って、例えば、β-アミノアニオン化合物は、亜構造式A-C-C-NRaRb(Aは陰性基である)により示すことができる。好ましいβ-アミノアニオン化合物には、β-アミノ酸およびその類似体が含まれる。ある好ましい態様において、β-アミノアニオン化合物はβ-アラニンまたはタウリンではない。
【0042】
「還元的脱硫」という用語は当技術分野において既知であり、化合物から硫黄を還元的に除去する過程を意味する。還元的脱硫条件は当技術分野において既知であり、例えば、TiCl4/LiAlH4またはラネーニッケル/H2による処理が含まれる。一般に、Kharash,NおよびMeyers,C.Y.「The Chemistry of Organic Sulfur Compounds」Pergamon Press、ニューヨーク(1996)第2巻を参照されたい。
【0043】
「被験者」という用語は当技術分野において既知であり、恒温動物、より好ましくは、類人猿、サルおよびヒトの他に、ヒト以外の動物(例えば、ラット、マウス、ネコ、イヌ、ヒツジ、ウマ、ウシ)を含む哺乳動物を意味する。好ましい態様において、被験者はヒトである。
【0044】
特記しない限り、本発明の化学基は、置換されていても非置換でもよい。さらに、特記しない限り、この化学的置換基も置換されていても非置換でもよい。さらに、複数の置換基が、化学基または置換基上に存在してもよい。置換基の例には、アルケニル、アルキニル、ハロゲン、ヒドロキシル、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、カルボキシル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルアミノカルボニル、ジアルキルアミノカルボニル、アルキルチオカルボニル、アルコキシル、ホルミル、トリメチルシリル、ホスフェート、ホスホナート、ホスフィナート、シアノ、アミノ(アルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、およびアルキルアリールアミノを含む)、アシルアミノ(アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイルおよびウレイドを含む)、アミド、イミノ、スルフヒドリル、アルキルチオ、アリールチオ、チオカルボキシレート、サルフェート、アルキルスルフィニル、スルホナート、スルファモイル、スルホンアミド、ニトロ、トリフルオロメチル、シアノ、アジド、ヘテロサイクリル、アルキルアリール、および芳香族部分もしくはへテロ芳香族部分が含まれる。
【0045】
「アルキル」という用語は、直鎖アルキル基、分岐鎖アルキル基、シクロアルキル、ヘテロサイクリル、シクロアルキル(脂環式)基、アルキルで置換されたシクロアルキル基、およびシクロアルキルで置換されたアルキル基を含む、飽和脂肪族基を意味する。好ましい態様において、直鎖または分岐鎖アルキルは、その骨格に30以下の炭素原子を有し(例えば直鎖ではC1〜C30、分岐鎖ではC3〜C30)、より好ましくは骨格に20以下の炭素原子を有する。同様に、好ましいシクロアルキルは、その環構造に4〜10個の炭素原子、より好ましくは環構造に5、6または7個の炭素を有する。
【0046】
さらに、アルキル(例えばメチル、エチル、プロピル、ブチル、ペンチル、ヘキシルなど)は、「非置換アルキル」および「置換アルキル」の両方を含み、後者は、炭化水素骨格の一つ以上の炭素上の水素が置換基で置換されているアルキル部分を意味する。このような置換基には、例えば、ハロゲン、ヒドロキシル、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、カルボキシレート、アルキルカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルチオカルボニル、アルコキシル、ホスフェート、ホスホナート、ホスフィナート、シアノ、アミノ(アルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、およびアルキルアリールアミノを含む)、アシルアミノ(アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイル、およびウレイドを含む)、アミジノ、イミノ、スルフヒドリル、アルキルチオ、アリールチオ、チオカルボキシレート、サルフェート、スルホナート、スルファモイル、スルホンアミド、ニトロ、トリフルオロメチル、シアノ、アジド、ヘテロサイクリル、または芳香族部分もしくはヘテロ芳香族部分が含まれ得る。炭化水素鎖上の部分は、それ自体、適宜、置換されていてもよいことが当業者には理解される。シクロアルキルはさらに、例えば、前記した置換基で置換されていてもよい。「アラルキル」部分は、アリールで置換されたアルキルである(例えばフェニルメチル(すなわちベンジル))。
【0047】
「アリール」という用語は、0〜4個のヘテロ原子を含み得る5員環および6員環の単環芳香族基、例えば、ベンゼン、ピロール、フラン、チオフェン、イミダゾール、オキサゾール、チアゾール、トリアゾール、ピラゾール、ピリジン、ピラジン、ピリダジン、およびピリミジンなどが含まれる。アリール基にはまた、多環縮合芳香族基、例えばナフチル、キノリル、インドリルなどが含まれる。環構造にヘテロ原子を有するこのようなアリール基はまた、「アリール複素環」、「ヘテロアリール」、または「ヘテロ芳香族」とも称し得る。芳香族環(例えばフェニル、インドール、チオフェン)は、前記したような置換基(例えば、ハロゲン、ヒドロキシル、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、カルボキシレート、アルキルカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルチオカルボニル、アルコキシル、ホスフェート、ホスホナート、ホスフィナート、シアノ、アミノ(アルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、およびアルキルアリールアミノを含む)、アシルアミノ(アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイル、およびウレイドを含む)、アミジノ、イミノ、スルフヒドリル、アルキルチオ、アリールチオ、チオカルボキシレート、サルフェート、スルホナート、スルファモイル、スルホンアミド、ニトロ、トリフルオロメチル、シアノ、アジド、ヘテロサイクリル、または芳香族部分もしくはヘテロ芳香族部分)により、一つ以上の環位置において置換されていてもよい。アリール基はまた、脂環式または芳香族でない複素環式環と縮合または架橋して、テトラリンなどの多環を形成できる。
【0048】
「アルケニル」および「アルキニル」という用語には、前記したアルキルに対して、長さおよびあり得る置換基の点から、不飽和脂肪族基の類似体が含まれるが、それぞれ少なくとも一つの二重結合または三重結合および少なくとも二つの隣接炭素原子が含まれる。
【0049】
明細書および図面に使用したような「選択的な単結合/二重結合」は、点線を伴う実線で示され、適宜EまたはZ立体配置の単結合または二重結合であり得る二つの炭素原子の間の共有結合を意味する。例えば、下記構造はシクロヘキサンまたはシクロヘキセンを示すことができる:
【化16】
【0050】
炭素数を特記しない限り、「低級アルキル」とは、前記したようなアルキル基を意味するが、1〜10個の炭素原子、より好ましくは1〜6個の炭素原子をその骨格構造に有する。同様に、「低級アルケニル」および「低級アルキニル」は、類似した鎖長を有する。好ましいアルキル基は低級アルキルである。
【0051】
「ヘテロサイクリル」または「複素環式基」という用語は、3〜10員環、より好ましくは4〜7員環構造を意味し、環構造には、一つ以上のヘテロ原子、例えば2、3、または4個のヘテロ原子が含まれる。複素環式基は、ピロリジン、オキソラン、チオラン、ピペリジン、ピペラジン、モルホリン、ラクトン、ラクタム、例えばアゼチジノンおよびピロリジノン、スルタム、スルトンなどが含まれる。複素環式環は、一つ以上の位置において、前記したような置換基(ハロゲン、ヒドロキシル、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、カルボキシレート、アルキルカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルチオカルボニル、アルコキシル、ホスフェート、ホスホナート、ホスフィナート、シアノ、アミノ(アルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、およびアルキルアリールアミノを含む)、アシルアミノ(アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイル、およびウレイドを含む)、アミジノ、イミノ、スルフヒドリル、アルキルチオ、アリールチオ、チオカルボキシレート、サルフェート、スルホナート、スルファモイル、スルホンアミド、ニトロ、トリフルオロメチル、シアノ、アジド、ヘテロサイクリル、または芳香族もしくはヘテロ芳香族部分を含む)で置換されていてもよい。
【0052】
「ポリサイクリル」または「多環式基」という用語は、二つ以上の炭素が二つの隣接している環に共通している、例えば環が「縮合環」である、二つ以上の環式環(例えばシクロアルキル、シクロアルケニル、シクロアルキニル、アリールおよび/または複素環)を意味する。隣接していない原子により連結している環は、「架橋」環と称される。多環の各々の環は、前記したような置換基、例えば、ハロゲン、ヒドロキシル、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、カルボキシレート、アルキルカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルチオカルボニル、アルコキシル、ホスフェート、ホスホナート、ホスフィナート、シアノ、アミノ(アルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、およびアルキルアリールアミノを含む)、アシルアミノ(アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイル、およびウレイドを含む)、アミジノ、イミノ、スルフヒドリル、アルキルチオ、アリールチオ、チオカルボキシレート、サルフェート、スルホナート、スルファモイル、スルホンアミド、ニトロ、トリフルオロメチル、シアノ、アジド、ヘテロサイクリル、または芳香族部分もしくはヘテロ芳香族部分)により置換されていてもよい。
【0053】
本明細書に使用したような「ヘテロ原子」という用語は、炭素または水素以外のあらゆる元素の原子を意味する。好ましいヘテロ原子は、窒素、酸素、硫黄およびリンである。
【0054】
本明細書に使用したような「アリールアルデヒド」という用語は、式Ar-C(O)Hにより示される化合物を意味し、ここでのArは、アリール部分(前記したような)であり、-C(O)Hはホルミル基またはアルデヒド基である。好ましい態様において、アリールアルデヒドは、(置換または非置換)ベンズアルデヒドである。多種多様なアリールアルデヒドが商業的に入手可能であるか、または、市販の前駆体から慣用的な手順により調製することもできる。アリールアルデヒドの調製手順には、Vilsmeier-Haack反応(例えばJutz.Adv.Org.Chem.9、pt.1、225-342(1976)を参照されたい)、Gatterman反応(Truce、Org.React.9、37-72(1957))、Gatterman-Koch反応(Crounse、Org.React.5、290-300(1949))、およびReimer-Tieman反応(WynbergおよびMeijer、Org.React.28、1-36(1982))が含まれる。
【0055】
本発明のいくつかの化合物の構造は、不斉炭素原子を含むことに留意されたい。従って、このような不斉から生じる異性体(例えば全ての鏡像異性体およびジアステレオマー)が、特記しない限り本発明の範囲内に含まれると理解する。すなわち、特記しない限り、あらゆるキラル炭素中心は(R)または(S)立体化学であり得る。このような異性体は、実質的に純粋な形で、古典的な分離技術により、および立体化学的に制御された合成により得ることができる。さらに、アルケンは、適宜、E幾何またはZ幾何を含むことができる。
【0056】
I.痙攣疾患を治療する方法
一つの局面において、本発明は、痙攣疾患(癲癇を含む)を治療する方法を提供する。
【0057】
一つの局面において、本発明は、被験者における癲癇誘発を抑制する方法を提供する。該方法には、それを必要とする被験者に、被験者における癲癇誘発が抑制されるように、癲癇誘発に関連した経路中の一過程を変調する薬剤の有効量を投与する段階が含まれる。
【0058】
前記したように、N-メチル-D-アスパルテート(NMDA)受容体により媒介されるニューロン間の興奮連関のアップレギュレーション、および、γ-アミノ-酪酸(GABA)受容体により媒介されるニューロン間の抑制連関のダウンレギュレートは両方共、癲癇誘発に関与している。癲癇誘発に関連した経路中の他の過程には、癲癇誘発に関与する神経伝達物質である一酸化窒素(NO)遊離;過剰に遊離された場合にニューロンへの傷害を媒介し得るカルシウム(Ca2+)の遊離;過剰な亜鉛(Zn2+)に起因する神経毒性;過剰な鉄(Fe2+)に起因する神経毒性;および酸化的細胞傷害に起因する神経毒性が含まれる。従って、好ましい態様において、癲癇誘発を抑制するために被験者に投与できる薬剤は、好ましくは、癲癇誘発に関連した少なくとも一つの経路中の一つ以上の過程を抑制できる。例えば、癲癇誘発の抑制に有用な薬剤は、脳組織中のNOの遊離を減少できるか、NOの癲癇誘発作用を減弱でき;NMDA受容体に拮抗でき;内因性GABA抑制を増大でき;電位開口型イオンチャネルを遮断でき;Ca2+、Zn2+、またはFe2+を含むカチオンの遊離を減少できるか、該カチオンの遊離濃度を減少できるか(例えばキレート形成により)、またはその他の方法で該カチオンの癲癇誘発作用を低減できる。ある好ましい態様において、癲癇誘発を抑制するために被験者に投与できる薬剤は、癲癇誘発に関連した少なくとも一つの経路中の少なくとも二つの過程を抑制できる。
【0059】
癲癇誘発に関連した経路中の過程を変調できるファルマコフォアの非制限的な例には以下が含まれる:
・NOシンターゼ阻害剤、例えばL-アルギニンおよびそのアルキル化誘導体
・NMDA受容体のアンタゴニスト、例えば(R)-α-アミノ酸。例えば、NMDA受容体阻害剤の一般的総説に関する、Leeson,PおよびIverson,L.L.、J.Med.Chem.(1994)37:4053-4067を参照されたい;
・内因性GABA抑制増強剤、例えばGABAアミノトランスフェラーゼ失活剤、例えばγ-ビニル-GABA。例えば、GABA受容体アゴニストおよびアンタゴニストの総説に関する、Krogsgaard-Larsen,Pら、J.Med.Chem.(1994)37:2489-2505を参照されたい;
・Ca2+、Zn2+、またはFe2+のキレート化剤、例えばEDTA、EGTA、TNTA、2,2-ビピリジン-4,4-ジカルボキシレート、エンテロバクチン、ポルフィリン、クラウンエーテル、アザクラウンエーテルなど;
・抗酸化剤、例えばビタミンCおよびE、カロテノイド、例えばβ-カロテン、ブチル化フェノール、トロロクス(トコフェロール類似体)、セレニウムおよびグルタチオン。
【0060】
一つの好ましい態様において、該薬剤はNMDA受容体に拮抗し、内因性GABA抑制を増強する。ある好ましい態様において、該薬剤は経口投与する。好ましくは、経口投与後に、該薬剤は、能動輸送シャトル機序により被験者の神経系に輸送される。能動輸送シャトルの非制限的な例は、アミノ酸を血液脳関門(BBB)を通過して輸送できる、大きな中性アミノ酸輸送体である。
【0061】
別の態様において、本発明は癲癇誘発を抑制する方法を提供する。該方法には、それを必要とする被験者に、癲癇誘発が抑制されるように、式(式I):
【化17】
(式中、Aは、生理的pHにおいて陰性基であり;R1は、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、またはアミノカルボニルであり;R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、複素環の原子数が3〜7である非置換または置換の複素環を形成している)
で示される化合物;またはその薬学的に許容される塩もしくはエステルの有効量を投与する段階が含まれる。
【0062】
ある態様において、式Iの化合物は、癲癇誘発が抑制されるような、式(式II):
【化18】
(式中、点線は、選択的な単結合を示し;R4およびR5は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、アルコキシ、アリールオキシ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、複素環であるか;または、R4およびR5は、一緒に、環の原子数が5〜15(より好ましくは原子数5〜8)である置換または非置換の炭素環または複素環を形成しており;A、R2およびR3は前記に定義した通りである)
またはその薬学的に許容される塩もしくはエステルにより示すことができる。
【0063】
別の態様において、本発明は、癲癇誘発を抑制する方法を提供する。該方法には、それを必要とする被験者に、癲癇誘発が抑制されるように、式(式III)
【化19】
(式中、A、R2、R3、R4およびR5は前記に定義した通りである)
で示される化合物;またはその薬学的に許容される塩もしくはエステルの有効量を投与する段階が含まれる。好ましい態様において、Aはカルボキシレートである。特に好ましい態様において、Aはカルボキシレートであり、R4は水素であり、R5は(置換または非置換)アリール基である。別の好ましい態様において、R4およびR5は一緒に6員環、例えば2-、3-、または4-アミノ安息香酸、特にアントラニル酸を形成する。
【0064】
別の態様において、本発明は、癲癇誘発を抑制する方法を提供する。該方法には、それを必要とする被験者に、癲癇誘発が抑制されるように、α,α-二置換β-アラニン、α,β-二置換β-アラニン、β,β-二置換β-アラニン、α,β,α-三置換β-アラニン、α,β,β-三置換β-アラニン、α,α,N-三置換β-アラニン、α,β,N-三置換β-アラニン、β,β,N-三置換β-アラニン、α,α,N,N-四置換β-アラニン、α,β,N,N-四置換β-アラニン、β,β,N,N-四置換β-アラニン、α,α,β,β-四置換β-アラニン、α,α,β,N-四置換β-アラニン、α,β,β,N-四置換β-アラニン、α,α,β,N,N-五置換β-アラニン、α,β,β,N,N-五置換β-アラニン、α,α,β,β,N-五置換β-アラニン、α,α,β,β,N,N-六置換β-アラニン(全ての立体異性体を含む);またはその薬学的に許容される塩もしくはエステルを投与する段階が含まれる。
【0065】
被験者に投与する段階には、本発明の抗痙攣化合物および/または抗癲癇誘発化合物へと代謝される化合物の被験者への投与が含まれ得る。例えば、本発明の方法には、インビボで本発明の治療化合物へと変換されるプロドラッグの使用が含まれる。例えば、前記で引用したSilverman、第8章を参照されたい。このようなプロドラッグを使用すると、一般的には血液脳関門を通過しない化合物が血液脳関門を通過できるようになるように体内分布を変化させたり、または治療化合物の薬物動態を変化させることができる。例えば、陰性基(例えばカルボキシレート基)は、エチル基または脂肪基でエステル化して、カルボン酸エステルを生成できる。カルボン酸エステルを被験者に投与する場合、エステルは、酵素的または非酵素的に切断され、陰性基を顕現できる。
【0066】
別の例示的態様において、本発明の方法には、被験者に、ウラシルまたはその類似体(置換ピリミジン、UMPおよびウリジン、またはその類似体を含む)を投与する段階が含まれる。ウラシル化合物またはその代謝物(例えばジヒドロウラシルまたはβ-ウレイドプロピオネート)の投与により、本発明の活性化合物をインビボで形成することができる。従って、好ましい態様において、本発明の方法には、それを必要とする被験者に、例えば、ウラシル、ジヒドロウラシルまたはβ-ウレイドプロピオネート化合物を痙攣疾患を治療または予防するのに有効であるβ-アミノ酸化合物へとインビボで変換することにより、痙攣疾患を治療するおよび/または癲癇誘発を抑制するに有効な量の、有効量の置換または非置換のウラシル、ジヒドロウラシルまたはβ-ウレイドプロピオネート化合物、またはその誘導体もしくは類似体(またはその薬学的に許容される塩もしくはエステル)を投与する段階が含まれ得る。
【0067】
従って、ある態様において、被験者への投与に好ましい化合物には、ピリミジン、例えば置換ウラシル(インビボでβ-アミノアニオン化合物に変換され得る)が含まれる。好ましい態様において、該化合物は、式(式V):
【化20】
(式中、R9およびR10は、各々独立的に、水素、アルキル(シクロアルキル、ヘテロサイクリル、およびアラルキルを含む)、アルケニル、アルキニル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ(非置換および置換のアミノを含む)、ヒドロキシ、チオール、アルキルチオール、ニトロ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、またはアミノカルボニルであり得るか;または、R9およびR10は、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成し;R11は、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R10およびR11は、それらがそれぞれ結合している炭素原子および窒素原子と共に連結して、環の員数が4〜8である複素環を形成し;R12は、水素、アルキル、アリール、および炭水化物(例えば糖、例えばリボースまたはデオキシリボース)からなる群より選択される)
またはその薬学的に許容される塩もしくはエステルにより示すことができる。別の態様において、該化合物は、式(式Va):
【化21】
(式中、R9a、R9b、R10a、R10bは、各々独立的に、水素、アルキル(シクロアルキル、ヘテロサイクリル、およびアラルキルを含む)、アルケニル、アルキニル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ(非置換および置換のアミノを含む)、ヒドロキシ、チオール、アルキルチオール、ニトロ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、およびアミノカルボニルであり得るか;または、R9aおよびR9bは、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成しているか;または、R10aおよびR10bは、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成しているか;または、R9aおよびR9bの一方は、R10aおよびR10bの一方と、それらが結合している2炭素単位と共に連結して、環の員数が4〜8である炭素環または複素環を形成し;R11は、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R10aおよびR10bの一方が、R11と、それらがそれぞれ結合している炭素原子および窒素原子と共に連結して、環の員数が4〜8である複素環を形成し;R12は、水素、アルキル、アリール、および炭水化物(例えば、糖、例えばリボースまたはデオキシリボース)からなる群より選択される)
またはその薬学的に許容される塩もしくはエステルにより示すことができる。
【0068】
ピリミジン化合物、例えば5-フルオロウラシル(5FU)は、抗新生物剤として使用されてきた。5FUおよび類似化合物の抗癌活性は、5FUがチミジル酸シンターゼ(DNA合成に重要な酵素)を阻害する「自殺基質」機序に起因すると考えられている。好ましい態様において、痙攣疾患の治療(癲癇誘発の抑制)のために本発明に従って投与したピリミジン化合物およびジヒドロピリミジン化合物は、チミジル酸シンターゼを有意に阻害しない。理論により限定したくはないが、ピリミジン化合物によるチミジル酸シンターゼ阻害は、ピリミジン環の5位(すなわち式VaのR9)における電気陰性基の存在により増加すると考えられ、それ故、ピリミジン環の5位(すなわち式VaのR9)における非電気陰性基を該化合物に与えることにより減少させることができる。ピリミジン化合物のチミジル酸シンターゼへの結合能を減少させるために、十分な立体的なかさを有する置換基を与えることにより、チミジル酸シンターゼ阻害を減少させることができると考えられている。従って、好ましい態様において、本発明に従って投与する式Vの化合物において、R9は非電気陰性(すなわち中性または電気陽性)基(例えばアルキル、アリールまたはその他)である。好ましい態様において、式VのR9およびR10の少なくとも一方は立体的にかさ高い基(例えば長鎖または分岐アルキル、置換アリール、またはその他)であるか、または、R9およびR10は連結して、炭素環または複素環式環を形成している。
【0069】
本発明に従って使用するピリミジン化合物およびジヒドロピリミジン化合物の非制限的な例を、その活性代謝物の例と共に、図1に示す。
【0070】
置換または非置換のウラシル、またはその誘導体もしくは類似体の使用は、ラットの抗発作モデルで試験した場合に、あるウラシル化合物が抗発作発生特性(のみ)を有することが判明したので、特に有益であり得る。例えば、Medicinal Chemistry、Volume V;W.J.Close、L.Doub、M.A.Spielman;W.H.Hartung著;John Wiley and Sons 1961を参照されたい。従って、プロドラッグ型の化合物(ウラシル)は、抗発作活性を示すことができるが、代謝により生成されたβ-アミノアニオン化合物は抗癲癇誘発活性および/または抗痙攣活性を示すことができる。これらの活性は、独立的におよび組合せて、哺乳動物(ヒトを含む)の痙攣疾患の効果的療法を提供できる。
【0071】
ある態様において、本発明の活性剤は、NMDA受容体のグリシン結合部位に結合することによりNMDA受容体に拮抗する。ある好ましい態様において、該薬剤は、グリア細胞のGABA取り込みを減少させることによりGABA抑制を増強する。ある他の態様において、該薬剤は経口投与する。さらに他の態様において、該方法にはさらに、薬学的に許容されるビヒクル中の薬剤の投与が含まれる。
【0072】
さらに別の態様において、本発明は、痙攣疾患を抑制する方法を提供する。該方法には、それを必要とする被験者に、痙攣疾患が抑制されるように、有効量のβ-アミノアニオン化合物を投与する段階が含まれ;ただし、β-アミノアニオン化合物はβ-アラニンまたはタウリンではない。
【0073】
別の態様において、本発明は、被験者における痙攣疾患および癲癇誘発の両方を抑制する方法を提供する。該方法には、それを必要とする被験者に、被験者における癲癇誘発が抑制されるように、ナトリウムもしくはカルシウムイオンチャネルを遮断するか、または、カリウムもしくは塩化物イオンチャネルを開口し;NMDA受容体拮抗作用、内因性GABA抑制増強、カルシウム結合、鉄結合、NOシンターゼ阻害、および抗酸化活性からなる群より選択される少なくとも一つの活性を有する薬剤の有効量を投与する段階が含まれる。
【0074】
ナトリウムおよび/またはカルシウムイオンチャネル活性の遮断剤は当技術分野において公知であり、本発明の化合物および方法のA部分として使用できる。同様に、カリウムまたは塩化物チャネルを開口するあらゆる化合物を、本発明の化合物および方法におけるA部分として使用できる。NMDA受容体のアンタゴニストおよび内因性GABA抑制増強剤は当業者には公知であり、本発明の方法および化合物に使用できる。例えば、2,3-キノキサリジノンが、NMDA受容体アンタゴニスト活性を有すると報告されている(例えば、米国特許第5,721,234号を参照されたい)。カルシウムおよび亜鉛キレート剤の例には、前記したものの他に、エチレンジアミンテトラ酢酸(EDTA)、エチレングリコールビス(β-アミノエチルエーテル)-N,N,N',N'-テトラ酢酸などの二価カチオンのキレート形成に関して当技術分野において既知である部分が含まれる。鉄キレート剤の例には、エンテロバクチン、ピリドキサールイソニコチニルヒドラゾン、N,N'-ビス(2-ヒドロキシベンゾイル)-エチレンジアミン-N,N'-ジ酢酸(HBED)、および1-置換-2-アルキル-3-ヒドロキシ-4-ピリドン(1-(2’-カルボキシエチル)-2-メチル-3-ヒドロキシ-4-ピリドンを含む)、および鉄にキレートすることが当技術分野において既知である他の部分が含まれる。NOシンターゼ活性を阻害する化合物は当技術分野において既知であり、例えば、Nγ-置換アルギニン類似体、特にL立体配置のもの(L-Nγ-ニトロ-アルギニン(脳NOシンターゼの特異的阻害剤)、L-Nγ-アミノ-アルギニン、およびL-Nγ-アルキル-アルギニン);またはそのエステル、好ましくはメチルエステルが含まれる。抗酸化剤の例には、アスコルビン酸、トコフェロール(α-トコフェロールを含む)などが含まれる。
【0075】
別の態様において、本発明は、痙攣疾患を抑制する方法を提供する。該方法には、それを必要とする被験者に、痙攣疾患が抑制されるように、式(式IV):
【化22】
(式中、Arは、非置換または置換のアリール基を示し;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yはチアゾリル、トリアゾリル、およびイミダゾリルからなる群より選択される複素環式部分であり;R6およびR6*は、各々独立的に、水素、アルキル、アルキルカルボニル、またはアリールカルボニルである)
で示されるジオキサピペラジン(ジケトピペラジンとしても知られる)化合物;またはその薬学的に許容される塩の有効量を投与する段階が含まれる。好ましい態様において、R7は水素、メチルまたはフェニルではない。好ましい態様において、該化合物は、シクロ-D-フェニルグリシル-(S-Me)-L-システインである。ジオキサピペラジンの合成については、例えば、Kopple,K.D.ら、J.Org.Chem.33:862(1968);Slater,G.P.Chem.Ind.(ロンドン)32:1092(1969);Grahl-Nielsen,O. Tetrahedron Lett.1969:2827(1969)を参照されたい。選択したジオキサピペラジン化合物の合成は下記の実施例に記載する。
【0076】
別の態様において、本発明は、癲癇誘発および発作発生を同時に抑制する方法を提供し、該方法には、それを必要とする被験者に、癲癇誘発が抑制されるように、下記式:
【化23】
(式中、Arは、非置換または置換のアリール基を示し;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yはチアゾリル、トリアゾリル、およびイミダゾリルからなる群より選択される複素環式部分であり;R6は水素またはアルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R6*は、抗酸化部分、NMDAアンタゴニスト、NOシンターゼ阻害剤、鉄キレート部分、Ca(II)キレート部分、Zn(II)キレート部分、および抗酸化部分からなる群より選択される)
で示される化合物;またはその薬学的に許容される塩の有効量を投与する段階が含まれる。ある態様において、R7は、水素、メチル、またはフェニルではない。
【0077】
別の態様において、本発明は、NMDA受容体アンタゴニスト作用に関連した疾患を治療する方法を提供する。該方法には、それを必要とする被験者に、NMDA受容体アンタゴニスト作用に関連した疾患が治療されるように、下記式:
【化24】
(式中、Arは、非置換または置換のアリール基を示し;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yはチアゾリル、トリアゾリル、およびイミダゾリルからなる群より選択される複素環式部分であり;R6は水素またはアルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R6*は、NMDAアンタゴニスト部分である)
で示される化合物;またはその薬学的に許容される塩を投与する段階が含まれる。ある態様において、R7は、水素、メチル、またはフェニルである。
【0078】
さらに別の態様において、本発明は、被験者における発作発生および癲癇誘発を抑制する方法を提供する。該方法には、それを必要とする被験者に、被験者における癲癇誘発が抑制されるように、式A-Bにより示される薬剤の有効量を投与する段階が含まれ、ここでのAは、ナトリウムイオンチャネル遮断活性を有するドメインであり;Bは、NMDA受容体拮抗作用、GABA抑制増強、カルシウム結合、鉄結合、亜鉛結合、NOシンターゼ阻害、および抗酸化活性からなる群より選択される少なくとも一つの活性を有するドメインである。ある好ましい態様において、薬剤のドメインAおよびB(例えばファルマコフォア)は共有結合している。ある好ましい態様において、Aはジオキサピペラジン部分、フェニトイン部分、またはカルバマゼピン部分である。
【0079】
別の態様において、本発明は、被験者における発作発生および癲癇誘発を抑制する方法を提供する。該方法には、それを必要とする被験者に、被験者における癲癇誘発が抑制されるように、式A-Bにより示される薬剤の有効量を投与する段階が含まれ、ここでのAは、抗発作発生活性を有するドメインであり;Bは、NMDA受容体拮抗作用、GABA抑制増強;カルシウム結合;鉄結合;亜鉛結合;NOシンターゼ阻害;および抗酸化活性からなる群より選択された少なくとも一つの活性を有するドメインである。ある好ましい態様において、薬剤のドメインAおよびB(例えばファルマコフォア)は共有結合している。ある好ましい態様において、Aはジオキサピペラジン部分、フェニトイン部分、またはカルバマゼピン部分である。
【0080】
本発明に記載のハイブリッド薬物は、抗発作発生部分と、抗癲癇誘発部分を、好ましくはアミド結合またはエステル結合などの共有結合を介して接続することによる、二機能性分子であり得る。該連鎖は、選択的に、インビボで切断され得る。該連鎖は、A部分とB部分の間に屈曲性または十分な空間を与えるリンカー部分またはスペーサー部分を含んでいてもよく、これによりAおよびBが結合するまたはAおよびBが相互作用するそれぞれの部分との相互作用が可能となる。リンカーの例には、アミノ基含有AおよびB部分を連結するための二酸(例えばアジピン酸);または例えばカルボキシル基含有AおよびB部分を連結するためのジアミン(例えば1,6-ヘキサンジアミン);または、アミノ官能基化B部分をカルボキシ官能基化A部分に連結(またはその逆)するためのアミノ酸が含まれる。リンカーは、当業者に公知の考慮に従って所望の特性を与えるように選択できる。よって、二機能性分子は発作発生および癲癇誘発の両方を標的化する。当業者は、ハイブリッド薬物が、一つ以上の所望の平均的なファルマコフォアを含み得ることを認識していると考えられる。
【0081】
別の態様において、被験者における癲癇誘発および/または発作発生を抑制する方法には、癲癇誘発が抑制されるように、有効量の化合物を被験者に投与することが含まれ、ここでの化合物は、式A:
【化25】
(式中、R1は、水素、アルキル、アルケニル、アルキニル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R2はアルキル、アルケニル、アルキニル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;Aは生理的pHにおいて陰性基である)
およびその薬学的に許容される塩もしくはエステルである。
【0082】
式Aの好ましい態様において、Aはカルボン酸またはエステルである。式Aの別の好ましい態様において、R1は水素である。式Aのさらに別の好ましい態様において、R2はアルキル、例えばアリールアルキル、例えばフェニルアルキルである。
【0083】
式Aの化合物の例には、
【化26】
【化27】
【化28】
【化29】
【化30】
【化31】
およびその薬学的に許容される塩もしくはエステルが含まれる。
【0084】
別の態様において、被験者における癲癇誘発および/または発作発生を抑制する方法には、被験者に、癲癇誘発が抑制されるように、有効量の化合物を投与する段階が含まれ、ここでの該化合物は、式B:
【化32】
(式中、Aは生理的pHにおいて陰性基であり;Bはフェノキシで置換されたフェニル基である)
およびその薬学的に許容される塩もしくはエステルである。
【0085】
式Bの好ましい態様において、Aはカルボキシル基である。式Bの好ましい態様において、Bはアルキルフェノキシで置換されたフェニル基、例えばメチルフェノキシで置換されたフェニル基、またはハロフェノキシで置換されたフェニル基、例えばクロロフェノキシで置換されたフェニル基である。式Bの好ましい化合物は、以下に例示したような、単一の立体異性体である。
【0086】
式Bの化合物の例には
【化33】
【化34】
【化35】
およびその薬学的に許容される塩もしくはエステルが含まれる。
【0087】
式Bの化合物のさらに他の好ましい態様を、表5および以下に提示する:
【化36】
【化37】
【0088】
別の態様において、被験者における癲癇誘発および/または発作発生を抑制する方法には、被験者に、癲癇誘発が抑制されるように、有効量の化合物を投与することが含まれ、ここでの化合物は、式C:
【化38】
(式中、Aは生理的pHにおいて陰性基であり;Dは、二つ以上のアルコキシ部分またはアリールオキシ部分で置換されたアリール基である)
およびその薬学的に許容される塩もしくはエステルである。
【0089】
式Cの好ましい態様において、Aはカルボキシル基である。式Cの別の好ましい態様において、Dは、二つ以上のアルコキシ部分またはアリールオキシ部分で置換されたフェニル基である。式Cの別の好ましい態様において、Dは、二つ以上のアルコキシ(例えばメトキシ)基で置換されたフェニル基である。
【0090】
式Cの化合物の例には
【化39】
【化40】
【化41】
およびその薬学的に許容される塩が含まれる。
【0091】
別の態様において、被験者における癲癇誘発および/または発作発生を抑制する方法には、被験者に、癲癇誘発が抑制されるように、有効量の化合物を投与する段階が含まれ、ここでの化合物は、式D
【化42】
(式中、Aは生理的pHにおいて陰性基であり;mおよびnは1〜3であり;Eは、置換または非置換のフェニルである)
およびその薬学的に許容される塩もしくはエステルである。
【0092】
式Dの好ましい態様において、Aはカルボキシル基である。式Dの別の好ましい態様において、nは1であり、Eはジフェニルで置換されたメチルである。
【0093】
式Dの化合物の例には
【化43】
【化44】
【化45】
【化46】
およびその薬学的に許容される塩もしくはエステルが含まれる。
【0094】
さらに別の態様において、被験者における癲癇誘発および/または発作発生を抑制する方法には、癲癇誘発が抑制されるように、有効量の化合物を被験者に投与する段階が含まれ、ここでの化合物は、式E
【化47】
(式中、R13は、水素、アルキル、アリール、または有機塩もしくは無機塩形成カチオンであり;nは1〜5であり;tは1〜2(好ましい)であり;各Xは、独立的に、ハロゲン、ニトロ、シアノ、および置換もしくは非置換のアルキル基およびアルコキシ基からなる群より選択される)
およびその薬学的に許容される塩もしくはエステルである。
【0095】
式Eの好ましい態様において、R13は水素であり、tは2である。
【0096】
式Eの好ましい化合物の例には、以下が含まれる:
3-アミノ-3-(4-ニトロフェニル)プロピオン酸
【化48】
3-アミノ-3-(4-メチルフェニル)-2-カルボキシプロピオン酸
【化49】
3-アミノ-3-(4-メトキシフェニル)-2-カルボキシプロピオン酸
【化50】
3-アミノ-3-(4-ニトロフェニル)-2-カルボキシプロピオン酸
【化51】
【0097】
本発明の治療法に有用である化合物は、慣用的なスクリーニングアッセイ法により決定できる。例えば、下記の実施例2に記載した第1相癲癇誘発動物モデルを使用して、特定の化合物が、第1相癲癇誘発に対して抗癲癇誘発活性を示すかどうかを決定できる。慢性的な癲癇誘発は、ラットにおいて、Silverら(Ann.Neurol.(1991)29:356)により記載されたキンドリングアッセイ法(およびこれでスクリーニングした候補化合物)を用いてモデリングできる。同様に、抗痙攣剤として有用な化合物を、従来の動物モデル、例えばHorton,R.W.ら、Eur.J.Pharmacol.(1979)59:75-83に記載のマウスモデルでスクリーニングできる。例えば受容体または酵素の結合または阻害に有用な化合物またはファルマコフォアは、当業者に既知の従来の方法に従ってスクリーニングできる。例えば、GABA取り込み受容体への結合は、Schlewer(Schlewer,J.ら、J.Med.Chem.(1991)34:2547)により改変されたRamseyらの方法により定量できる。NMDA受容体上のグリシン部位への結合は、例えば、Kemp,Aら、Proc.Natl.Acad.Sci.USA(1988)85:6547に記載の方法に従って定量できる。電位開口型Na+チャネルに対する作用は、ラット海馬片における電位固定アッセイ法で評価できる。
【0098】
マウスまたはラットにおける抗痙攣活性および/または抗癲癇誘発活性について候補化合物をスクリーニングするのに適したアッセイ法を、下記の実施例4および5に記載する。
【0099】
II.化合物および化合物を同定するための方法
別の局面において、本発明は、癲癇および痙攣疾患を治療するのに有用な化合物を提供する。
【0100】
一つの態様において、本発明は、式(式I)
【化52】
(式中、Aは、生理的pHにおいて陰性基であり;R1は、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、またはアミノカルボニルであり;R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、複素環の原子数が3〜7である非置換または置換の複素環を形成している)
で示される抗癲癇誘発化合物(ここでの抗癲癇誘発化合物は、抗癲癇誘発活性を示す);またはその薬学的に許容される塩もしくはエステルを提供する。
【0101】
ある好ましい態様において、Aはカルボキシレートを示す。ある好ましい態様において、該化合物は、α-シクロヘキシル-β-アラニン、α-(4-tert-ブチルシクロヘキシル)-β-アラニン、α-(4-フェニルシクロヘキシル)-β-アラニン、α-シクロドデシル-β-アラニン、β-(p-メトキシフェネチル)-β-アラニン、β-(p-メチルフェネチル)-β-アラニン、およびその薬学的に許容される塩からなる群より選択される。他の好ましい態様において、該化合物は、β-(4-トリフルオロメチルフェニル)-β-アラニンおよびβ-[2-(4-ヒドロキシ-3-メトキシフェニル)エチル]-β-アラニンおよびその薬学的に許容される塩からなる群より選択される。さらに他の態様において、該化合物は、β-(3-ペンチル)-β-アラニンおよびβ-(4-メチルシクロヘキシル)-β-アラニンおよびその薬学的に許容される塩からなる群より選択される。
【0102】
別の態様において、本発明は、式(式IV)
【化53】
(式中、Arは、非置換または置換のアリール基を示し;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yは水素またはチアゾリル、トリアゾリル、およびイミダゾリルからなる群より選択される複素環式部分であり;R6およびR6*は、各々独立的に、水素、アルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルからなる群より選択される)
で示されるジオキサピペラジン化合物またはその薬学的に許容される塩を提供する。ある好ましい態様において、Ar基が結合している炭素原子は「D」または「R」立体化学配置を有する。ある態様において、Arは非置換または置換のフェニル基である。ある態様において、Yは水素である。ある好ましい態様において、R6およびR6*の少なくとも一方は、抗酸化部分、NMDAアンタゴニスト、NOシンターゼ阻害剤、鉄キレート部分、Ca(II)キレート部分、およびZn(II)キレート部分からなる群より選択される。ある好ましい態様において、R7はメチルまたはメルカプトメチルである。
【0103】
ある好ましい態様において、R6およびR6*は、両方共に水素である。ある特に好ましい態様において、該化合物は、シクロフェニルグリシル-2-(アミノ-3-メルカプトブタン酸)、より好ましくはシクロ-D-フェニルグリシル-L-[2-(アミノ-3-メルカプトブタン酸)]である。関連した態様において、該化合物は、シクロ-D-フェニルグリシル-(S-Me)-L-システインである。いくつかの好ましい態様において、Arは非置換フェニル基である。ある態様において、R7は、水素、メチル、またはフェニルではない。
【0104】
別の態様において、本発明は、式(式IV)
【化54】
(式中、Arは、非置換または置換のアリール基を示し;R7は、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yは水素またはチアゾリル、トリアゾリル、およびイミダゾリルからなる群より選択される複素環式部分であり;R6は、水素またはアルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R6*は、抗酸化部分、NMDAアンタゴニスト、NOシンターゼ阻害剤、鉄キレート部分、Ca(II)キレート部分、およびZn(II)キレート部分からなる群より選択されるか;または、R6およびR6*の両方が、抗酸化部分、NMDAアンタゴニスト、NOシンターゼ阻害剤、鉄キレート部分、Ca(II)キレート部分、およびZn(II)キレート部分からなる群より選択される)
で示される化合物;またはその薬学的に許容される塩を提供する。ある好ましい態様において、R6*はD-α-アミノアジピルである。ある好ましい態様において、R7はメルカプトメチルである。ある態様において、R7は、水素、メチルまたはフェニルではない。ある好ましい態様において、R6*はさらに、切断可能な連鎖を含む。一つの態様において、該化合物はシクロ-D-フェニルグリシル-L-アラニンを含む。
【0105】
当業者には理解されるように、本発明の化合物には、単一のファルマコフォア(例えばジオキサピペラジン、ここでのジオキサピペラジン部分は単独のファルマコフォアである);または、β-アミノアニオン部分(ここでのβ-アミノアニオン部分は化合物の生化学的活性に関与している)を有し得る化合物が含まれる。本発明のある化合物には、二つの別個のファルマコフォアが含まれ、A-Bにより示される構造を有し、ここでのAおよびBは、生化学活性を有する各ドメインまたはファルマコフォア(例えば、独立した抗酸化部分を有する抗痙攣ジオキサピペラジン部分、例えばR6*)(本明細書ではハイブリッド」薬物とも称する)である。二つのファルマコフォアを含む化合物は、二つ以上の別個の受容体と相互作用できる。本発明の化合物が二つ以上のファルマコフォアを含む場合、ファルマコフォアは、当業者に既知の多種多様な技術により互いに連結させることができる。例えば、R6*により示されるファルマコフォアは、ジオキサピペラジン環の窒素へのアミド連鎖を介して、ジオキサピペラジン部分に共有結合できる。二つのファルマコフォア間の連鎖は、二つのファルマコフォアが互いにインビボで切断されるように(すなわち、インビボで不安定である連鎖の選択により)選択できる。このような生物学的に不安定な連鎖の例は、当技術分野において公知である。例えば、前記に引用したSilvermanを参照されたい。有利には、このような「ハイブリッド」の二つのファルマコフォア薬物は、体内に輸送され、脳などの部位または臓器に到達するように設計でき、そこで一つ以上のファルマコフォア部分が生物学的作用を示し、この部位でハイブリッド薬物は切断されて二つの活性薬物部分を与え得る。ハイブリッド薬物のいくつかの例は前記している。
【0106】
本発明は、さらに、本発明の治療化合物にインビボで変換されるプロドラッグの使用に関する。このようなプロドラッグを使用して、治療化合物の体内分布を変化(例えば、典型的には血液脳関門を通らない化合物が血液脳関門を通過できるようにするために)または薬物動態を変化させることができる。例えば、陰性基、例えばカルボキシレートまたはスルホネートは、例えばメチル基またはフェニル基でエステル化すると、カルボン酸エステルまたはスルホン酸エステルを生成することができる。カルボン酸エステルまたはスルホン酸エステルを被験者に投与した場合、エステルが酵素的または非酵素的に切断され、陰性基が出現する。エステルは環式(例えばラクトンまたはスルトン)でもよく、または、二つ以上のアニオン部分を連結基を通してエステル化してもよい。陰性基を部分(例えばアシルオキシメチルエステル)を用いてエステル化し、これは切断されると中間化合物が出現し、これはその後分解されると、活性化合物を生じることができる。別法として、アニオン部分は、インビボで能動的に輸送される基か、または標的臓器により選択的に取り込まれる基へとエステル化できる。エステルは、治療部分を特定の臓器へと特異的に標的化できるように選択できる。別の態様において、プロドラッグは、インビボで酸化されて治療化合物となる、還元形の陰性基、例えばカルボキシレートまたはスルホネート、例えばアルコールまたはチオールである。
【0107】
従って、前記したように、好ましい化合物には、インビボでβ-アミノアニオン化合物へと変換されることのできる、ピリミジン、例えば置換ウラシルが含まれる。好ましい態様において、該化合物は、式(式V):
【化55】
(式中、R9およびR10は、各々独立的に、水素、アルキル(シクロアルキル、ヘテロサイクリル、およびアラルキルを含む)、アルケニル、アルキニル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ(非置換および置換のアミノを含む)、ヒドロキシ、チオール、アルキルチオール、ニトロ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、またはアミノカルボニルからなる群より選択されるか;または、R9およびR10は、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成し;R11は、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R10およびR11は、それらがそれぞれ結合している炭素原子および窒素原子と共に連結して、環の員数が4〜8である複素環を形成し;R12は、水素、アルキル、アリール、および炭水化物(例えば糖、例えばリボースまたはデオキシリボース)からなる群より選択される)
またはその薬学的に許容される塩もしくはエステルにより示すことができる。別の態様において、該化合物は、式(式Va):
【化56】
(式中、R9a、R9b、R10a、R10bは、各々独立的に、水素、アルキル(シクロアルキル、ヘテロサイクリル、およびアラルキルを含む)、アルケニル、アルキニル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ(置換および非置換のアミノを含む)、ヒドロキシ、チオール、アルキルチオール、ニトロ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、またはアミノカルボニルからなる群より選択されるか;または、R9aおよびR9bは、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成しているか;または、R10aおよびR10bは、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成しているか;または、R9aおよびR9bの一方は、R10aおよびR10bの一方と、それらが結合している2炭素単位と共に連結して、環の員数が4〜8である炭素環または複素環を形成し;R11は、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R10aおよびR10bの一方が、R11と、それらがそれぞれ結合している炭素原子および窒素原子と共に連結して、環の員数が4〜8である複素環を形成し;R12は、水素、アルキル、アリール、および炭水化物(例えば、糖、例えばリボースまたはデオキシリボース)からなる群より選択される)
またはその薬学的に許容される塩もしくはエステルにより示すことができる。
【0108】
式VおよびVaの化合物は、種々の合成技術(そのいくつかは当技術分野において既知である)に従って調製できる。シンターゼの例を図2に示す。例えば、図2に示したように、バルビツール酸化合物を改変(例えばメシルクロリドおよびアミン塩基を用いてのメシル化により)して化合物を得、これはさらに官能基化できるか(例えば適切な求核試薬のミカエル付加により);または還元的に脱スルホン化して、適切な求ジエン体を用いてのその後のディールス・アルダー環化付加用の求ジエン体を得ることができる。
【0109】
本発明に有用な化合物はまた、治療化合物を標的臓器または臓器群に選択的に送達することを可能とする、担体または標的化部分も含み得る。例えば、治療化合物の脳への送達が望まれる場合、該化合物には、該化合物を脳に、能動輸送または受動輸送により標的化できる部分(「標的化部分」)が含まれ得る。例として、担体分子には、米国特許第4,450,564号および第5,389,623号に記載のような、酸化還元部分が含まれ得る。これらの特許は、脳に進入でき、そこで荷電したピリジニウム種へと酸化され、脳内に補足される、ジヒドロピリジン部分に連結した薬物を開示する。従って、薬物は脳に蓄積する。他の担体部分には、インビボで能動的または受動的に輸送され得る、アミノ酸またはチロキシンなどの化合物が含まれる。このような担体部分は、インビボで代謝的に除去されるか、または、活性化合物の一部として無傷で留まることができる。多くの標的化部分は既知であり、例えば、アシアログリコタンパク質(例えば、米国特許第5,166,320号を参照されたい)、および、受容体により媒介されるエンドサイトーシスを介して細胞に輸送される他のリガンドが含まれる。
【0110】
前記した標的化戦略およびプロドラッグ戦略を合わせて、プロドラッグとして所望の作用部位に輸送でき、その後、アンマスクされると、活性化合物が出現する化合物を作成することができる。
【0111】
別の局面において、本発明は、被験者における癲癇誘発を抑制できる化合物を同定するためのファルマコフォアモデリング法を提供する。これらの方法は、癲癇誘発に関与するタンパク質または分子に対して直接的または間接的薬理作用を引き起こすことが知られる二つ以上の化合物の構造の検証を特徴とする。癲癇誘発に関与するこれらのタンパク質および分子は、細胞表面受容体分子(例えばNMDA受容体)または神経伝達物質の輸送に関与する分子(例えばGABA輸送体)を含むと考えられている。好ましくは、これらの化合物の構造は各々、化合物の薬理作用の少なくとも一部を示す一つ以上のファルマコフォアを含む。本発明の方法にはまた、二つ以上の化合物のファルマコフォア構造に基づいた、平均的なファルマコフォア構造(例えば、炭素骨格構造および/または3次元空間充填構造)の決定が含まれる。一つ以上の平均的なファルマコフォア構造を有する新規化合物を、これらの方法を使用して選択できる。
【0112】
関連した態様において、これらの方法は、癲癇誘発に関与する二つ以上のタンパク質または分子に対して直接的または間接的薬理作用を引き起こすことが知られる二つ以上の化合物の構造の検証を特色とする。このような態様において、当業者は、選択した新規化合物が、好ましくは、癲癇誘発に関与する異なるタンパク質または分子に対して活性である一つ以上のファルマコフォアを有することを理解していると考えられる。
【0113】
好ましい態様において、本発明のこれらの方法により選択した(例えば設計した)新規化合物は、被験者における癲癇誘発を抑制する。
【0114】
化合物の同定法はさらに、癲癇誘発に関与するタンパク質または分子の少なくとも一部(例えば「偽受容体」)を刺激する他の相補的モデルの作成に依拠し得る。このような刺激を使用してさらに、一つ以上の平均的なファルマコフォアを含む新規候補化合物を評価できる。相補モデルは、癲癇誘発に関与するタンパク質分子と相互作用するファルマコフォアまたは化合物全体の構造に依拠した、アルゴリズムおよび/または方法を使用して作成できる。このような刺激の作成におけるアルゴリズムは当業者に公知であり、MM2分子力学力場が含まれる(例えば、Allinger(1977)J.Am.Chem.Soc.99:8127-8134、Allingerら(1988)J.Comp Chem.9:591-595、Liiら(1989)J.Comp.Chem.10:503-513、Cornellら(1995)J.Am.Chem.Soc.117:5179-5197、Wienerら(1986)J.Comp.Chem.7:230-252を参照されたい)。
【0115】
本発明はさらに、本発明の化合物の容器、および、治療有効量の化合物をそれを必要とする被験者に、被験者における痙攣疾患(例えば癲癇誘発)が抑制されるように使用するための説明書を含む、キットを提供する。本発明のキットは、本発明の化合物を使用、例えば投与するための便利な手段を提供する。特に好ましい態様において、キットは、治療有効量の化合物を、好ましくは単位投与形で含む。
【0116】
本発明はまた、検出マーカーで標識した本発明の化合物(例えば後記した化合物1〜14およびA1〜A32)を被験者に投与し;該被験者の脳のニューロンのNMDA受容体への該化合物の結合の増加を測定し、これにより該被験者における癲癇誘発容態を診断する段階を含む、被験者における癲癇誘発容態を診断する方法を提供する。
【0117】
本発明はさらに、検出マーカーで標識した本発明の化合物(例えば後記した化合物1〜14およびA1〜A32)を被験者に投与し;該被験者の脳のニューロンのGABA受容体への該化合物の結合の減少を測定し、これにより該被験者における癲癇誘発容態を診断する段階を含む、被験者における癲癇誘発容態を診断する方法を提供する。
【0118】
本明細書に使用したような「検出マーカーで標識した化合物」には、検出手段により標識した化合物が含まれ、放射能、蛍光、化学発光、および/または生物発光で標識した抗体が含まれる。
【0119】
標識として使用できる酵素の例には、リンゴ酸デヒドロゲナーゼ、スタフィロコッカスヌクレアーゼ、デルタ-V-ステロイドイソメラーゼ、酵母アルコールデヒドロゲナーゼ、α-グリセロリン酸デヒドロゲナーゼ、トリオースリン酸イソメラーゼ、セイヨウワサビペルオキシダーゼ、アルカリホスファターゼ、アスパラギナーゼ、グルコースオキシダーゼ、β-ガラクトシダーゼ、リボヌクレアーゼ、ウレアーゼ、カタラーゼ、グルコース-VI-リン酸デヒドロゲナーゼ、グルコアミラーゼ、およびアセチルコリンエステラーゼが含まれる。
【0120】
放射性標識の例には、3H、125I、131I、35S、14C、および好ましくは125Iが含まれる。蛍光標識の例には、フルオレセインイソチオシアネート、ローダミン、フィコエリトリン、フィコシアニン、アロフィコシアニン、o-フタルアルデヒド、およびフルオレサミンが含まれる。化学発光標識の例には、ルミノール、ルシフェリン、イソルミノール、理論量のアクリジニウムエステル、イミダゾール、アクリジニウム塩、およびそのシュウ酸エステルが含まれる。生物発光標識の例には、ルシフェリン、ルシフェラーゼ、およびエクオリンが含まれる。
【0121】
III.β-アミノアニオン化合物の調製法
本発明はさらに、β-アミノアニオン化合物の調製法を提供する。
【0122】
一つの態様において、本発明は、式(式VI):
【化57】
(式中、点線は、選択的な単結合/二重結合(EまたはZ立体配置の)を示し;R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、複素環の原子数が3〜7である非置換または置換の複素環を形成しており;R4およびR5は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、アルコキシ、アリールオキシ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、ヘテロサイクリルであるか;または、R4およびR5は、共に、環の原子数が5〜15(より好ましくは5〜8)である置換または非置換の炭素環または複素環を形成している)
で示されるβ-アミノカルボキシル化合物の調製法を提供する。該方法には、式VI
【化58】
(式中、点線は、選択的な単結合/二重結合を示し;Xは、ニトロ、アジド、またはNR2R3であり、ここでのR2およびR3は前記に定義した通りであり;Wは、-CNまたは-COOR8であり;R8は、水素、アルキル、アリール、または有機塩もしくは無機塩形成カチオンであり;R4およびR5は前記に定義した通りである)
で示される化合物を、β-アミノカルボキシルまたはβ-アミノ二トリル化合物が形成されるような、還元的脱硫条件下で反応させる段階が含まれる。ある好ましい態様において、R7は、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり、R3は水素である。
【0123】
式VIIの化合物は、当技術分野で公知の方法に従って調製できる。例えば、アミノチオフェンカルボキシレート(すなわち、式VIの化合物、ここでのWは-COOR8であり、R8はカチオンであり、Xはアミノ基であり、各点線は単結合である)の合成が、いくつかの方法により報告されている。例えば、Beck、J.Org.Chem(1972)37:3224;Meth-Cohn、J.Chem.Res.(1977)(S)294、(M)3262を参照されたい。還元的脱硫条件下におけるアミノチオフェンカルボキシレート(またはアミノチオフェンニトリル)の還元は、現在、良好な収率でβ-アミノ酸を生成することが判明している(アミノチオフェンニトリルにはニトリル基の加水分解も必要であり、これは公知の方法に従って達成できる、例えばLarock、Comprehensive Organic Transformations、VCH Publishers(1989)およびそれに引用されている文献を参照されたい)。好ましい態様において、還元的脱硫条件には、アミノチオフェンカルボキシレートが脱硫するように、アミノチオフェンカルボキシレートをラネーニッケルと反応させる段階が含まれる。
【0124】
別の態様において、本発明は、式VIII:
【化59】
(式中、R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、複素環の原子数が3〜7である、非置換または置換の複素環を形成しており;R4およびR5は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、アルコキシ、アリールオキシ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、ヘテロサイクリルであるか;または、R4およびR5は、一緒に、環の原子数が5〜15(より好ましくは5〜8)である置換または非置換の炭素環または複素環を形成しており;R8は、水素、アルキル、アリール、または有機塩もしくは有機塩形成カチオンである)
により示されるβ-アミノカルボキシル化合物の調製法を提供する。該方法には、式IX
【化60】
(式中、点線は、各々、選択的な単結合を示し;Xは、ニトロ、アジド、またはNR2R3であり、ここでのR2およびR3は前記に定義した通りであり;Wは、-CNまたは-COOR8であり、R8は、水素、アルキル、アリール、または有機塩もしくは無機塩形成カチオンであり;R4およびR5は、前記に定義した通りである)
で示される化合物を、式VIII(ここでのW=-CNである、カルボキシレートは還元的脱硫化および酸性化の後に形成される)で示されるβ-アミノカルボキシル化合物が形成されるような、還元的脱硫条件下で反応させる段階が含まれる。ある好ましい態様において、R2はアルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり、R3は水素である。
【0125】
式IXの化合物(またはそのエステル、これを既知の方法に従って加水分解すると、式IXの化合物を得ることができる)は、当技術分野において既知の方法に従って調製できる。例えば、米国特許第4,029,647号;HenriksenおよびAutrup、Acta Chem.Scand.26:3342(1972);またはHartkeおよびPeshkar、Pharm Zentralhalle 107:348(1968)を参照されたい。
【0126】
本発明の合成法は、以前に報告されたβ-アミノ酸の合成よりも優れた利点を提供する。例えば、本発明の方法により、2炭素骨格のどちらかの炭素または両方の炭素の置換された多種多様なβ-アミノ酸が得られ;生成される具体的なβ-アミノ酸は、出発物質のアミノチオフェンカルボキシレート(多種多様な置換基を用いて調製できる)により決定される。実施例1に下記したように、本発明の方法は、温和な条件下で、市販の試薬からほんの少数の段階で良好な収率でβ-アミノ酸を提供する。この方法により調製された化合物の例を実施例1に提示する。従って、本発明の方法は、一般的で迅速で簡単で高収率のβ-アミノ酸への経路を提供する。
【0127】
別の態様において、本発明は、β-アリール-β-アラニン化合物の調製法を提供する。この態様において、本発明は、しばしば容易に入手できる前駆体を使用して、多種多様の置換および非置換のβ-アリール-β-アラニン化合物を製造できる簡単でワンポットの反応を提供する。本明細書に使用した方法は、β-アラニン類似体を製造するための改法である。該方法には、アリールアルデヒドをマロネート化合物およびアンモニウム化合物と、β-アリール-β-アラニン化合物が形成される条件下で反応させる段階が含まれる。好ましい態様において、アリールアルデヒドは置換または非置換のベンズアルデヒドである。好ましい態様において、マロネート化合物はマロン酸である。好ましい態様において、アンモニウム化合物は、アンモニア、一級アミン、および二級アミンからなる群より選択される化合物のアンモニウム塩である。特に好ましいアンモニウム化合物は、アンモニアの塩、最も好ましくは酢酸アンモニウムである。好ましい態様において、溶媒は、エタノールなどの極性溶媒である。本発明に記載の合成例は、実施例3に記載する。
【0128】
β-アミノ酸は、本明細書に記載した抗癲癇誘発特性の他に、例えば合成中間体として、および商品化学物質としてのその他の用途を有することが理解される。例えば、β-ラクタム構造は、例えば、ペニシリン、カルバペネム、ノルカルジン(norcardins)、モノバクタムなどを含む、多くの商業的に価値ある抗生物質に存在する。β-アミノ酸をβ-ラクタムに変換する多種多様な方法が報告されている。例えば、Wang,W.-BおよびRoskamp,E.J.、J.Am.Chem.Soc.(1993)115:9417-9420およびそれに引用されている文献を参照されたい。従って、本発明は、さらに、β-ラクタムの合成法を提供する。該方法には、式VII(または式IX)の化合物を還元的脱硫条件にかけ、式VI(またはIもしくはVIII)の化合物を生成し、その後、式VI(またはIもしくはVIII)の化合物を環化してβ-ラクタムを形成する段階を含む。さらに、β-アミノ酸は、特定の癌患者の容態を改善することが示されている(例えば、Rougereau,Aら、Ann.Gastroenterol.Hepatol.(パリ)29(2):99-102(1993)を参照されたい)。従って、本発明は、癌の治療に有用な化合物の調製法を提供する。
【0129】
IV.ライブラリー
別の局面において、本発明は、式IV、式VI、または式VIIIの化合物のライブラリー、およびこのようなライブラリーの調製法を提供する。
【0130】
コンビナトリアルライブラリーの合成は当技術分野において公知であり、総説されている(例えば、E.M.Gordonら、J.Med.Chem.37:1385-1401(1994)を参照されたい)。従って、本発明は、式IV、式VI、または式VIIIの化合物のコンビナトリアルライブラリーの合成法を含む。このようなライブラリーは、多種多様な方法に従って合成できる。例えば、「スプリット・プール(split-pool)」戦略を実行して、化合物ライブラリーを作成できる。その後、固定された化合物ライブラリーを洗浄して不純物を除去できる。特定の態様において、固定された化合物を固相支持体から切断して、式IV、VIまたはVIIIの化合物を得ることができる。
【0131】
別のコンビナトリアル合成法の例において、「ディバーソマー(diversomer)ライブラリー」を、Hobbs,De Wittら(Proc.Natl.Acad.Sci.U.S.A.90:6909(1993))の方法により創造する。化合物ライブラリーの創製後、精製および後処理により、式IV、VIまたはVIIIで示される可溶性の置換化合物のライブラリーが得られる。
【0132】
Houghtenら、Nature 354:84-86(1991)の「ティーバッグ」技術を含む他の合成法を使用しても、該当発明に従って化合物のライブラリーを合成できる。
【0133】
コンビナトリアルライブラリーをスクリーニングして、ライブラリーのどのメンバーが所望の活性を有するかを決定でき、活性を有する場合には、活性種を同定できる。コンビナトリアルライブラリーのスクリーニング法が記載されている(例えば、Gordonら、J.Med.Chem.手順引用を参照されたい)。可溶性の化合物ライブラリーは、適切な受容体を用いての親和性アフィニティークロマトグラフィーによりスクリーニングして、受容体に対するリガンドを単離でき、その後、従来の技術(例えば質量分析法、NMRなど)により単離リガンドを同定できる。固定された化合物は、該化合物を可溶性受容体と接触させることによりスクリーニングでき;好ましくは、可溶性受容体は、リガンド結合を検出し指示できる標識(例えばフルオロフォア、比色定量酵素、放射性同位体、発光化合物など)にコンジュゲートしている。別法として、固定された化合物を選択的に遊離させ、膜中を拡散させ、受容体と相互作用させることができる。本発明のライブラリーのスクリーニングに有用なアッセイ法の例は当技術分野で既知である(例えば、E.M.Gordonら、J.Med.Chem.37:1385-1401(1994)を参照されたい)。
【0134】
化合物のコンビナトリアルライブラリーも、ライブラリーの各メンバーの実体をコードする「タグ」を用いて合成できる。例えば、米国特許第5,565,324号およびPCT公開公報WO94/08051号を参照されたい。一般に、この方法は、固相支持体または化合物に結合した、不活性であるが、容易に検出可能なタグの使用を特色とする。活性化合物が、前記した技術の一つにより検出される場合に、化合物の実体は、独特な随伴するタグの同定により決定される。このタギング法により、大きな化合物ライブラリーの合成が可能となり、これは非常に低いレベルで同定できる。
【0135】
好ましい態様において、本発明の化合物ライブラリーは、少なくとも30個の化合物、より好ましくは少なくとも100個の化合物、さらにより好ましくは少なくとも500個の化合物を含む。好ましい態様において、本発明の化合物ライブラリーは、109個より少ない化合物、より好ましくは108個より少ない化合物、さらにより好ましくは107個より少ない化合物を含む。
【0136】
化合物ライブラリーは、好ましくは、実質的に純粋である、すなわち、目的の生成物、例えばライブラリーのメンバー以外の化合物は実質的に含まない。好ましい態様において、本発明の方法に従って作成したライブラリーの純度は、少なくとも約50%、より好ましくは少なくとも約70%、さらにより好ましくは少なくとも約90%、最も好ましくは少なくとも約95%である。
【0137】
本発明のライブラリーは、本明細書に記載したように調製できる。一般に、本発明のライブラリーの合成に使用した少なくとも一つの出発物質は、雑種個体群として提供される。本明細書に使用したような「雑種個体群」という用語は、少なくとも二つの異なる化学実体、例えば異なる化学構造の化学実体を含む、個体群を意味する。例えば、式VIIの化合物の「雑種個体群」は、式VIIの少なくとも二つの異なる化合物を含む。化合物を固相支持体に固定するためのリンカーの雑種個体群の使用により、リンカー切断時に多種多様な化合物を作成できる。
【0138】
本発明のライブラリーは、とりわけ、薬物発見に有用である。例えば、本発明のライブラリーをスクリーニングして、ライブラリーが、予め選択された活性、例えば抗癲癇誘発活性または抗痙攣活性を有する化合物を含むかどうかを決定できる。
【0139】
V.薬学的組成物
別の局面において、本発明は、一つ以上の薬学的に許容される担体(添加剤)および/または希釈剤と共に製剤化された、治療有効量の一つ以上の前記した化合物を含む、薬学的に許容される組成物を提供する。本発明の薬学的組成物は、以下に適合したものを含む、固体型または液体型で投与するために特に製剤化され得る:(1)経口投与、例えば、舌に施薬するための水薬(水性または非水性の溶液または懸濁液)、錠剤、ボーラス、粉末、顆粒、ペースト;(2)非経口投与、例えば、滅菌溶液または懸濁液として、皮下、筋肉内または静脈内注射により;(3)局所適用、例えば、皮膚に塗るクリーム、軟膏、またはスプレーとして;または(4)膣内または直腸内、例えば膣座薬、クリーム、気泡として。好ましい態様において、治療化合物は経口投与する。本発明の化合物は、ヒトを含む被験者に、例えば哺乳動物に投与するための薬学的組成物として製剤化できる。
【0140】
本発明の化合物は、インビボでの医薬投与に適した生物学的に適合性の形態で被験者に投与する。「インビボで投与するに適した生物学的に適合性の形態」は、抗体の治療作用が毒性作用を上回る、投与化合物を意味する。被験者という用語は、免疫応答が誘発され得る生物、例えば哺乳動物を含むものとする。被験者の例には、ヒト、イヌ、ネコ、齧歯類(例えばマウスまたはラット)、およびそのトランスジェニック種が含まれる。治療有効量の本発明の治療組成物の投与は、所望の結果を達成するに必要な用量および投与期間において効果的な量として定義する。例えば、治療有効量の本発明の化合物は、個体の疾病状態、年齢、性別、および体重、ならびに、個体における所望の応答を誘発する抗体の能力などの因子に応じて変化し得る。投与方式は、最適な治療応答が得られるように調整し得る。例えば、数回に分割した用量を毎日投与しても、または、治療状況の緊急性に応じて比例的に減少させてもよい。
【0141】
活性化合物は、注射(皮下、静脈内など)、経口投与、吸入、経皮塗布、または直腸投与などの従来の様式で投与し得る。投与経路に応じて、活性化合物を材料でコーティングして、酵素、酸、および化合物を失活させ得る他の天然条件の作用から化合物を防御し得る。
【0142】
本発明の化合物は、酵素阻害剤と同時投与する適切な担体または希釈剤に含めて、またはリポソームなどの適切な担体に含めて被験者に投与できる。本明細書に使用したような「薬学的に許容される担体」は、生理食塩水または緩衝水溶液などの希釈剤を含むものとする。本発明の化合物を非経口投与以外で投与するために、その失活を防ぐ材料で抗体をコーティングするか、またはその材料と該化合物を同時投与する必要があり得る。リポソームには、水/油/水型乳剤ならびに従来のリポソームが含まれる(Strejanら、(1984)J.Neuroimmunol 7:27)。活性化合物はまた非経口的にまたは腹腔内に投与し得る。分散液は、グリセロール、液体ポリエチレングリコール、およびその混合物中で、および油中で調製できる。通常の貯蔵および使用の条件下で、これらの調製物は、微生物の増殖を防ぐための保存剤を含み得る。
【0143】
注射に使用するのに適した薬学的組成物には、滅菌水溶液(水溶性である場合)または分散液、ならびに、無菌注射溶液または分散液の即時調製用の滅菌粉末が含まれる。全ての場合において、組成物は無菌でなければならず、シリンジが容易に扱えるほど十分に流動性でなければならない。製造および貯蔵の条件下で安定でなければならず、細菌および真菌などの微生物の汚染作用に対して防御されていなければならない。薬学的に許容される担体は、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、および液体ポリエチレングリコールなど)およびその適切な混合物を含む、溶媒または分散媒体であり得る。適切な流動性が、例えば、レシチンなどのコーティングの使用により、分散液の場合には必要な粒子サイズの維持により、および界面活性剤の使用により維持できる。微生物の作用の防御は、種々の抗細菌剤および抗真菌剤、例えば、パラベン、クロロブタノール、フェノール、アスコルビン酸、チメロサールなどにより行なうことができる。多くの場合、等張剤、例えば、糖、ポリアルコール、例えばマンニトール、ソルビトール、塩化ナトリウムが組成物に含まれることが好ましい。注射用組成物の吸収延長は、組成物中に、吸収を遅延する薬剤、例えばモノステアリン酸アルミニウムおよびゼラチンを含めることにより行なうことができる。
【0144】
無菌注射溶液は、必要量の活性化合物を適切な溶媒中に、必要である場合には、前記に列挙した成分の一つまたは組合せて共に取り込み、その後、滅菌濾過することにより調製できる。一般に、分散液は、活性化合物を、基本分散媒体および前記に列挙したものからの他の必要な成分を含む、無菌ビヒクルに取り込むことにより調製する。無菌注射溶液の調製用の無菌粉末の場合、好ましい調製法は、真空乾燥法および凍結乾燥法であり、これにより活性成分の粉末と、以前に滅菌濾過したその溶液からの任意のその他の所望の成分が得られる。
【0145】
活性化合物を、前記したように適切に保護した場合、組成物は、例えば、不活性希釈剤または同化可能な食用担体と共に経口投与し得る。本明細書に使用したような「薬学的に許容される担体」には、任意および全ての溶媒、分散媒体、コーティング、抗細菌剤および抗真菌剤、等張剤および吸収遅延剤などが含まれる。薬学的に活性な物質のためにこのような媒体および薬剤を使用することは、当技術分野において公知である。任意の従来の媒体または薬剤が活性化合物と不適合性でない限り、治療組成物におけるその使用が考えられる。補助的活性化合物も組成物に取り込むことができる。
【0146】
投与を容易にし、用量を均一にするために、投与単位形で非経口組成物を製剤化することが特に有利である。本明細書に使用したような投与単位形は、治療したい哺乳動物被験者の単位量として適した物理的に独立した単位を意味し;各単位は、必要な薬学的担体と合わせて所望の治療効果が得られるように計算された、予め決定された量の活性化合物を含む。本発明の投与単位形の詳細は、活性化合物の独特な特徴および達成したい具体的な治療効果、および個体の治療的処置における活性化合物などの配合の分野に固有の限界により影響されこれに直接依存する。
【0147】
実施例
実施例1:ファルマコフォアモデルに基づく化合物の同定
2つの異なるクラスの化合物:(1)GABA摂取受容体の阻害物質および(2)NMDA受容体の共作用物質(co-agonist)の構造パラメータおよび特徴を組み入れたファルマコフォアモデルを開発した。
【0148】
以前のモデル(Murali Dharら、(1994) J. Med. Hem.37:2234、FalchおよびKrogsgaard-Larson (1991) Eur.J Med.Chem.26:69、N'Goka(1991)J. Med. Chem 34:2457)では、GABA摂取阻害物質が以下のものを含むことが示唆されている:
i)アミン基(好ましくは二級アミン)
ii)カルボキシル基
iii)親油基、好ましくは芳香族基
iv)アミンと親油基との間に配置された電子に富む官能基(二重結合または酸素)
v)アミン基と二重結合または酸素原子との間の2炭素鎖長。
【0149】
NMDA受容体錯体のグリシン共作用物質部位の拮抗物質に焦点が合わされた他の以前のモデル(例えば、LeesonおよびIverson(1994)J.Med.Chem 37:4053)では、NMDA受容体の共作用物質は望ましくは以下のものを含むことが示唆されている:
i)アミン基(好ましくは二級アミン)
ii)カルボキシル基
iii)2つの小さな親油基
iv)大きな親油基。
【0150】
この情報に基づき、クラスとしてはβ-アミノ酸およびその類似体であると考えられる平均のファルマコフォアモデル化合物を調製した。これらの化合物の重要なパラメータは以下のものを含む:
i)アミン基
ii)カルボキシル基
iii)β-アラニン骨格
iv)フレキシブルな脂肪親和性部分。
【0151】
所望の化合物のプロファイルをさらに精密化するために、一連の分枝モデル化計算(MM2分子機構力場)を使用して「平均的な受容体部位」の3次元視覚化を構成した。最初に、NMDA受容体上のグリシンサブサイトに結合することが知られている様々なプローブ分子を使用して、相補モデリングアプローチにより「擬似受容体」モデルを作成した。これを達成するために、周知のNMDA受容体サイトペプチドの断片を幾つかのプローブ分子(例えば、受容体に結合することが知られている化合物)付近に機械的に配置し、受容体をシミュレートした。すなわち、プローブ分子を鋳型として使用しその周りに受容体モデルを構成した。例えば、グルタミン酸塩の側鎖を使用してプローブ分子内の塩基性アンモニウム基に「合体」させた。脂肪親和性ポケットはフェニルアラニンの側鎖を用いてシミュレートした。そうすることにより、NMDA受容体上のグリシンサブサイトの「受容体」を数学的にモデル化した。次に同じ手順をグリアGABA摂取受容体に対し実施した。2つのモデル受容体をオーバーラップさせ、モデルハイブリッド受容体(平均的な受容体サイト)を設計した。このモデルハイブリッド受容体サイトは3つの「ポケット」を含んだ。アニオンポケットはカチオンポケットから7.7オングストローム離れて位置し、それぞれアンモニウム基およびカルボキシル基と相互作用することができる。移動性の脂肪親和性ポケットをアニオンポケットから5.2から8.1オングストロームの範囲の可変位置に配置した。上記条件を含むβ-アミノ酸類似体をモデルハイブリッド受容体中に挿入した。短い(2〜3炭素)フレキシブルアーム上に芳香族環を有するβ-置換β-アミノ酸を用いると最適適合が得られた。フレキシブルアームにより移動性の脂肪親和性ポケットとの相互作用が可能になると考えた。
【0152】
これらの方法により同定された候補化合物のリストを以下に示す。
【化61】
【0153】
多くのβ-アリールβ-アミノ酸化合物を、容易な「ワンポット」合成法によりさらに製造した。簡単に説明すると、置換ベンズアルデヒドの無水エチルアルコール溶液にマロン酸および過剰の酢酸アンモニウムを添加し、反応混合物を加熱して還流させた。反応混合物を冷却しβ-アリールβ-アラニンおよび(一定の場合)桂皮酸誘導体の混合物を得た。桂皮酸は(存在すれば)混合物の酸/塩基抽出により除去し、β-アリールβ-アラニンを得た。しばしば中程度から良好な収率であった。この方法により得られた候補化合物のリストを以下に列挙する。
【化62】
【化63】
【0154】
実施例2 候補化合物の癲癇誘発阻害に対する薬理学的有用性のインビボ評価
2つの群の候補類似体のインビボ試験を抗発作活性および神経毒性の両方に対し実施した。1つの発作モデルを、カナダ動物実験評議会のガイドラインに従い、クィーンズ大学動物倫理委員会の監督下、雄の成体SDラットを用いて実施した。この試験手順は、Turskiら(1984)Brain Res.321:237による以前の研究から採用されている。腹腔内(i.p.)注射により100mg/kgで試験化合物を投与した。塩酸ピロカルピンの腹腔内投与(350mg/kg)により20分後に発作が誘発された。防護は、ピロカルピン投与後30分の観察期間にわたり、慢性痙攣が起こらないものとして規定した。化合物1、2、3、5、8、10、11、13、A1、A4、A5、A11、A13、A14、A15、A16、A21、A26、A28、A29およびA31はこのアッセイ法で有意な抗発作活性を示した。抗発作活性を示す化合物のクラスとしては、N置換β-アミノ酸酸類似体(化合物1、2、3および10);β-置換β-アミノ酸類似体(化合物5、11、A1、A4、A5、A11、A13、A14、A15、A16、A21、A26、A28、A29およびA31);α-置換β-アミノ酸類似体(すなわち、化合物8および13)が挙げられる。
【0155】
候補化合物の抗発作および神経毒性を試験するための更なるアッセイ法には、最大電気ショック発作(MES)モデル、皮下ペンチレンテトラゾール(PTZ)-誘発発作モデル、およびロトロッド(rotorod)神経毒性試験が含まれた。全てのアッセイ法はNIHの癲癇部門における抗痙攣薬開発(ADD)プログラムにより実施した(例えば、StablesおよびKupferberg(1997)、NIH抗痙攣薬開発(ADD)プログラム:前臨床抗痙攣スクリーニングプロジェクト(The NIH anticonvulsant Drug Development (ADD) Program:Preclinical Anticonvulsant Screening Project、Libby & Sonsを参照のこと)。全ての化合物は雄のCarworth Farms #1マウスまたは雄のSDラットのいずれかを用いて試験した。各試験化合物は300、100および30mg/kgの腹腔内注射により投与した。
【0156】
MES-誘発発作モデルでは、試験化合物の抗発作活性は30分の観察期間にわたる後脚緊張性伸展の完全停止として規定した(例えば、「抗癲癇薬のための分子および細胞標的」G.Avanziniら(1997) John Libbey&Company Ltd.,pp191-198;James P. StablesおよびHarvey J. Kupferbergによる第16章「NIH抗痙攣薬開発(ADD)プログラム:前臨床抗痙攣スクリーニングプロジェクト」を参照のこと)。化合物9、10およびA3はこのアッセイ法で有意な抗発作活性を示した。
【0157】
PTZ-誘発発作モデルでは、発作は典型的にはPTZの腹腔内注射(マウスでは85mg/kgおよびラットでは70mg/kg)による試験化合物投与後0.5および4時間で誘発された。防護は、30分の観察期間にわたり慢性痙攣が阻害されることとして規定した。化合物9、10、A3、A7、A17、A22、A23、A24およびA25はこのアッセイ法では有意な抗発作活性を示した。
【0158】
ロトロッド神経毒性試験では、試験化合物の投与後、マウスを6rpmの速度で回転する直径1インチのぎざぎざのついたプラスチックロッド上に置いた。神経毒性は、1分の観察期間にわたりマウスが平衡を維持することができないものとして規定した。化合物1、2、4〜9、11、12、14、A3、A4、A6、A8、A9、A10、A17、A21、A22、A23、A26、A27、A28、A29、A30、A31およびA32はこのアッセイ法では神経毒性を示さなかった。しかしながら、いくらかの神経毒性を示した残りの化合物の毒性レベルは、カルバマジンおよびバルプロ酸などの抗発作薬に比べ低かった。
【0159】
実施例3:β-アミノ酸の合成:方法A
一般手順
無水酢酸によるN-アセチル保護
アセトアミドチオフェンカルボン酸エステルを、無水AcOH中で1時間、対応するアミノ化合物を過剰のAc2O(4当量)と還流させることにより調製した。混合物を冷水中に注ぎ入れ、生成物を濾過により単離し、水で洗浄し、EtOHから再結晶化した。
【0160】
レーニーニッケル触媒の合成
水(1.2 L)に溶解したNaOH(320.0g、8mol)溶液を2.0Lフラスコ中で機械的に撹拌した。氷浴で10℃まで冷却した後、ニッケルアルミニウム合金(250g)を少量ずつ90分にわたり添加した。得られた懸濁液を室温で1時間、50℃でさらに8時間撹拌した。懸濁液を目盛付きシリンダに移し、水性の上清のデカンテーションを行った。得られたスラリーを2.5MのNaOH水溶液(200mL)と共に振り混ぜ、その後にデカンテーションを行った。ニッケル触媒を水(150ml)中に懸濁させることにより30回洗浄し、その後にデカンテーションを行った。無水EtOH(100mL)を用いて洗浄を3度繰り返し、得られたレーニーニッケルを無水EtOH下で保存した。
【0161】
レーニーニッケル還元脱硫
アセトアミドチオフェンカルボン酸アルキル(20mmol)および新たに調製したレーニーニッケル(8当量)をEtOH(75mL)中で激しく撹拌しながら16時間還流させた。高温混合物を珪藻土(セライト)を通して濾過し、ニッケル残渣を熱EtOH(50mL)で洗浄した。濾液を濃縮すると、純粋なN-アセチル-β-アラニンアルキルエステルが透明油、ゴムまたは白色結晶として得られた。
【0162】
酸分解を介するN-アセチルおよびアルキルエステル脱保護
二重に保護したα-またはβ-置換β-アラニンを6M HCl中で5時間還流させた。溶液を蒸発させ(H2O、HCl、MeOHおよびAcOHを除去し)、残渣を蒸留H2O中に2度溶解し、濃縮した(残留HClを除去)。生成物をEtOHから再結晶化させ、塩酸塩を白色結晶として得た。または、粗生成物を最小量の熱H2Oに溶解し、NH4OHを滴下し遊離β-アミノ酸を沈澱させた。二体積のEtOHまたはMeOHを添加し生成物の分離を支援させ、凝集を防止させた。混合物を24時間冷却し(4℃)、さらに沈澱させ、その後濾過した。生成物を氷冷H2OおよびEtOHで洗浄し、その後MeOHまたはEtOHから再結晶化させ、純粋な置換β-アラニンを白色結晶として得た。
【0163】
TLC解析
下記の実験手順では、薄層クロマトグラフィー分析のために使用する溶媒は以下のように略す。
溶媒B:塩化メチレン:アセトン:酢酸100:100:0.5
溶媒I:酢酸エチル:メタノール9:1
溶媒J:クロロホルム:アセトン:水88:12:15
溶媒K:メタノール:酢酸5:1
溶媒L:エタノール:酢酸50:1
【0164】
アセトアミドチオフェンカルボン酸アルキルの合成
3-アセトアミドベンゾ[b]チオフェン-2-カルボン酸メチル
上記手順を用いて、3-アミドベンゾ[b]チオフェン-2-カルボン酸メチル(1.8596g、8.97mmol)をアセチル化し、EtOH再結晶化により精製し、微細な白色結晶として純粋生成物を得た(1.4723g、5.91mmol、65.9%);融点:178〜180℃;TLC:Rf=0.63(溶媒I)、0.55(溶媒J)、0.80(溶媒L);IR(cm-1):3271(NH)、3021(CH)、1716(エステルC=O)、1670(アミドC=O)、746(=CH)。
【0165】
3-アセトアミド-6-(トリフルオロメチル)ベンゾ[b]チオフェン-2-カルボン酸メチル
3-アミノ-6-(トリフルオロメチル)ベンゾ[b]チオフェン-2-カルボン酸メチル(1.4944g、5.43mmol)をアセチル化し、EtOH再結晶化により精製し、綿毛状淡黄色結晶として純粋生成物を得た(1.5261g、4.81mmol、88.6%);融点:204〜205℃;TLC:Rf=0.72(溶媒I)、0.78(溶媒L);IR(cm-1):3274(NH)、3069(CH芳香族)、2962(CH脂肪族)、1720(エステルC=O)、1676(アミノC=O)。
【0166】
2-アセトアミド-4,5,6,7-テトラヒドロベンゾ[b]チオフェン-3-カルボン酸メチル
2-アミノ-4,5,6,7-テトラヒドロベンゾ[b]チオフェン-3-カルボン酸メチル(3.0004g、14.20mmol)を、上述したようにアセチル化し、EtOH再結晶化により精製し、淡茶色結晶として純粋生成物を得た(3.3823g、13.35mmol、94.0%);融点:103〜106℃;TLC:Rf=0.68(溶媒I)、0.66(溶媒J)、0.76(溶媒L);IR(cm-1):3248(NH)、2932(CH)、1698(エステルC=O)、1668(アミドC=O)。
【0167】
2-アセトアミド-6-tert-ブチル-4,5,6,7-テトラヒドロベンゾ[b]チオフェン-3-カルボン酸メチル
2-アミノ-6-tert-ブチル-4,5,6,7-テトラヒドロベンゾ[b]チオフェン-3-カルボン酸メチル(1.3693g、5.12mmol)を、上述したようにアセチル化し、EtOH再結晶化により精製し、白色微細結晶として純粋生成物を得た(0.9312g、3.01mmol、58.8%);融点:117〜118℃;TLC:Rf=0.74(溶媒I)、0.70(溶媒J);IR(cm-1):3271(NH)、2953(CH)、1674(C=O)。
【0168】
2-アセトアミドシクロドデカ[b]チオフェン-3-カルボン酸エチル
2-アミノシクロドデカ[b]チオフェン-3-カルボン酸エチル(4.9236g、15.91mmol)を、上述したようにアセチル化し、EtOH再結晶化により精製し、淡茶色結晶として純粋生成物を得た(4.6058g、13.10mmol、82.3%);融点:54〜74℃;TLC:Rf=0.73(溶媒I);IR(cm-1):3358(NH)、2929(CH)、1710(エステルC=O)、1678(アミドC=O)。
【0169】
2-アセトアミド-4,5,6,7-テトラヒドロ-6-フェニルベンゾ[b]チオフェン-3-カルボン酸メチル
2-アミノ-4,5,6,7-テトラヒドロ-6-フェニルベンゾ[b]チオフェン-3-カルボン酸メチル(2.5046g、8.71mmol)を、上述したようにアセチル化し、EtOH再結晶化により精製し、オフホワイト微細粉末として純粋生成物を得た(2.3763g、7.21mmol、82.8%);融点:116〜117℃;TLC:Rf=0.79(溶媒I)、0.78(溶媒J);IR(cm-1):3255(NH)、3029(CH)、2925(CH)、1686(エステルC=O)、1668(アミドC=O)、703(=CH)。
【0170】
3-アセトアミド-5-フェニルチオフェン-2-カルボン酸メチル
3-アミノ-5-フェニルチオフェン-2-カルボン酸メチル(2.5031g、10.73mmol)を、上述したようにアセチル化し、EtOH再結晶化により精製し、白色結晶として純粋生成物を得た(2.7726g、10.07mmol、93.8%);融点:115℃;TLC:Rf=0.70(溶媒I)、0.70(溶媒J);IR(cm-1):3319(NH)、3122(CH)、2950(CH)、1715(エステルC=O)、1680(アミドC=O)、765(=CH)。
【0171】
3-アセトアミド-5-(4-メトキシフェニル)チオフェン-2-カルボン酸メチル
3-アミノ-5-(4-メトキシフェニル)チオフェン-2-カルボン酸メチル(2.5004g、9.50mmol)を、アセチル化し、EtOH再結晶化により精製し、白色微細結晶として純粋生成物を得た(2.7173g、8.90mmol、93.7%);融点:148〜149℃;TLC:Rf=0.68(溶媒I)、0.65(溶媒J);IR(cm-1):3303(NH)、3143(CH)、2943(CH)、1705(エステルC=O)、1663(アミドC=O)、817(=CH)。
【0172】
3-アセトアミド-5-(4-メチルフェニル)チオフェン-2-カルボン酸メチル
3-アミノ-5-(4-メチルフェニル)チオフェン-2-カルボン酸メチル(1.5098g、6.10mmol)を、上述したようにアセチル化し、EtOH再結晶化により精製し、綿毛状白色結晶として純粋生成物を得た(1.6694g、5.77mmol、94.6%);融点:127〜129℃;TLC:Rf=0.70(溶媒I)、0.64(溶媒J)、0.75(溶媒K);IR(cm-1):3316(NH)、2953(CH)、1710(エステルC=O)、1675(アミドC=O)、812(=CH)。
【0173】
3-アセトアミド-5-[3-メトキシ-4-(4-ニトロベンジルオキシ)フェニル]チオフェン-2-カルボン酸メチル
3-アミノ-5-[3-メトキシ-4-(4-ニトロベンジルオキシ)フェニル]チオフェン-2-カルボン酸メチル(1.5174g、3.66mmol)を、上述したようにアセチル化し、EtOH再結晶化により精製し、黄色結晶として純粋生成物を得た(1.5487g、3.39mmol、92.6%);融点:193〜194℃;TLC:Rf=0.68(溶媒I)、0.65(溶媒J);IR(cm-1):3326(NH)、3072(CH)、2944(CH)、1705(エステルC=O)、1671(アミドC=O)、836(=CH)。
【0174】
N-アセチル-α-置換-β-アラニンアルキルエステルの合成
N-アセチル-α-シクロヘキシル-β-アラニンメチルおよびエチルエステル
2-アセトアミド-4,5,6,7-テトラヒドロベンゾ[b]チオフェン-3-カルボン酸メチル(0.8125g、3.37mmol)を、レーニーニッケルを用いて還元脱硫し、淡黄色油として標題化合物を得た(0.6051g、2.81mmol、83.4%);TLC: Rf=0.80(溶媒I)、0.81(溶媒L);IR(cm-1):2894(CH脂肪族)、1738(エステルC=O)、1674(アミドC=O)。
【0175】
N-アセチル-α-シクロドデシル-β-アラニンエチルエステル
2-アセトアミドシクロドデカ[b]チオフェン-3-カルボン酸エチル(2.3366g、6.65mmol)を、レーニーニッケルを用いて還元脱硫し、黄色油として標題化合物を得た(2.1314g、6.55mmol、98.5%);TLC: Rf=0.75(溶媒I)、0.46(溶媒J);IR(cm-1):3316(NH)、2903(CH脂肪族)、1725(エステルC=O)、1661(アミドC=O)。
【0176】
N-アセチル-α-(4-tert-ブチルシクロヘキシル)-β-アラニンメチルエステル
2-アセトアミド-6-tert-ブチル-4,5,6,7-テトラヒドロベンゾ[b]チオフェン-3-カルボン酸メチル(0.8286g、2.68mmol)を、レーニーニッケルを用いて還元脱硫し、粘着性白色固体として標題化合物を得た(0.7466g、2.63mmol、98.3%);融点:73〜75℃;TLC: Rf=0.70(溶媒I)、0.33(溶媒J);IR(cm-1):3261(NH)、2943(CH脂肪族)、1735(エステルC=O)、1648(アミドC=O)。
【0177】
N-アセチル-α-(4-フェニルシクロヘキシル)-β-アラニンメチルエステル
2-アセトアミド-4,5,6,7-テトラヒドロ-6-フェニルベンゾ[b]チオフェン-3-カルボン酸メチル(2.0292g、6.16mmol)を、レーニーニッケルを用いて還元脱硫し、白色固体として標題化合物を得た(1.7908g、5.90mmol、95.8%);融点:75〜80℃;TLC: Rf=0.58(溶媒J)、0.79(溶媒L);IR(cm-1):3259(NH)、3079(=CH)、2929(CH脂肪族)、1730(エステルC=O)、1647(アミドC=O)、698(=CH)。
【0178】
N-アセチル-β-置換-β-アラニンメチルエステルの合成
N-アセチル-β-フェニル-β-アラニンメチルエステル
3-アセトアミドベンゾ[b]チオフェン-2-カルボン酸メチル(1.3742g、5.51mmol)を、レーニーニッケルを用いて還元脱硫し、淡黄茶色固体として標題化合物を得た(1.1876g、5.37mmol、97.4%);融点:58〜61℃;TLC: Rf=0.42(溶媒I)、0.24(溶媒J);IR(cm-1):3322(NH)、3061(CH芳香族)、2955(CH脂肪族)、1741(エステルC=O)、1649(アミドC=O)。
【0179】
N-アセチル-β-(4-トリフルオロメチルフェニル)-β-アラニンメチルエステル
3-アセトアミド-6-(トリフルオロメチル)ベンゾ[b]チオフェン-2-カルボン酸メチル(0.7014g、2.21mmol)を、レーニーニッケルを用いて還元脱硫し、透明油として標題化合物を得た(0.5961g、2.05mmol、92.6%);TLC: Rf=0.52(溶媒I)、0.86(溶媒L);IR(cm-1):3340(NH)、1736(エステルC=O)、1654(アミドC=O)。
【0180】
N-アセチル-β-フェネチル-β-アラニンメチルエステル
3-アセトアミド-5-フェニルチオフェン-2-カルボン酸メチル(2.3660g、8.59mmol)を、レーニーニッケルを用いて還元脱硫し、オフホワイトゴムとして標題化合物を得た(2.1108g、8.47mmol、98.6%);TLC: Rf=0.68(溶媒I)、0.65(溶媒J);IR(cm-1):3475(NH)、2893(CH脂肪族)、1735(エステルC=O)、1654(アミドC=O)。
【0181】
N-アセチル-β-(p-メトキシフェネチル)-β-アラニンメチルエステル
3-アセトアミド-5-(4-メトキシフェニル)チオフェン-2-カルボン酸メチル(1.8100g、5.93mmol)を、レーニーニッケルを用いて還元脱硫し、黄色油として標題化合物を得た(1.5544g、5.56mmol、93.8%);TLC: Rf=0.54(溶媒I)、0.25(溶媒J);IR(cm-1):3285(NH)、2944(CH)、1735(エステルC=O)、1651(アミドC=O)、728(=CH)。
【0182】
N-アセチル-β-[2-(4-メチルフェニル)エチル]-β-アラニンメチルエステル
3-アセトアミド-5-(4-メチルフェニル)チオフェン-2-カルボン酸メチル(1.4905g、5.15mmol)を、レーニーニッケルを用いて還元脱硫し、白色ゴムとして標題化合物を得た(1.3434g、5.10mmol、99.1%);融点:50〜51℃;TLC: Rf=0.63(溶媒I)、0.85(溶媒L);IR(cm-1):3288(NH)、2906(CH脂肪族)、1731(エステルC=O)、1639(アミドC=O)、807(=CH)。
【0183】
N-アセチル-β-[2-(3-メトキシ-4-ヒドロキシフェニル)エチル]-β-アラニンメチルエステル
3-アセトアミド-5-[3-メトキシ-4-(4-ニトロベンジルオキシ)フェニル]チオフェン-2-カルボン酸メチル(1.4481g、3.17mmol)を、レーニーニッケルを用いて還元脱硫した。濾過した溶液を熱EtOAcに溶解し、その後0.5N HCl(2×30mL)およびH2Oで洗浄した。有機層を乾燥させ(MgSO4)、濾過、濃縮し、黄色油として標題化合物を得た(0.5620g、1.90mmol、60.0%);TLC: Rf=0.80(溶媒L);IR(cm-1):3498(OH)、2905(CH脂肪族)、1743(エステルC=O)、1663(アミドC=O)、726(=CH)。
【0184】
α-置換-β-アラニンの合成
α-シクロヘキシル-β-アラニン
N-アセチル-α-シクロヘキシル-β-アラニンエチルおよびメチルエステル(2.4499g、10.77mmol)を脱保護し、白色微細結晶として標題化合物を得た(0.9573g、5.59mmol、51.9%);融点:238〜240℃;TLC: Rf=0.75(溶媒I);IR(cm-1):3300〜2700(OH)、2207、1635(カルボン酸C=O)。
【0185】
α-シクロドデシル-β-アラニン塩酸塩
N-アセチル-α-シクロドデシル-β-アラニンエチルエステル(2.1268g、6.83mmol)を脱保護し、白色結晶として標題化合物を得た(0.7322g、2.51mmol、36.7%);融点:201〜204℃;TLC: Rf=0.79(溶媒I)、0.80(溶媒L);IR(cm-1):3400〜2700(OH)、1722(カルボン酸C=O)。
【0186】
α-(4-tert-ブチルシクロヘキシル)-β-アラニン塩酸塩
N-アセチル-α-(4-tert-ブチルシクロヘキシル)-β-アラニンメチルエステル(0.7463g、2.63mmol)を脱保護し、白色微細結晶として標題化合物を得た(0.4347g、1.65mmol、62.7%);融点:230℃(dec);TLC: Rf=0.91(溶媒K);IR(cm-1):3400〜2700(OH)、1732(カルボン酸C=O)。
【0187】
α-(4-フェニルシクロヘキシル)-β-アラニン塩酸塩
N-アセチル-α-(4-フェニルシクロヘキシル)-β-アラニンメチルエステル(1.6699g、5.50mmol)を脱保護し、白色微細結晶として標題化合物を得た(0.5235g、1.84mmol、33.5%);融点:268℃(dec);TLC: Rf=0.74(溶媒I)、0.64(溶媒K);IR(cm-1):3300〜2500(OH)、1701(カルボン酸C=O)。
【0188】
β-置換-β-アラニンの合成
β-フェニル-β-アラニン
N-アセチル-β-フェニル-β-アラニンメチルエステル(1.1561g、5.23mmol)を脱保護し、白色微細結晶として標題化合物を得た(0.5275g、3.19mmol、61.1%);融点:220〜221℃;TLC: Rf=0.75(溶媒I);IR(cm-1):3305(鋭い:OH、H結合せず)、2195、1627(カルボン酸C=O)。
【0189】
β-(4-トリフルオロメチルフェニル)-β-アラニン塩酸塩
N-アセチル-β-(4-トリフルオロメチルフェニル)-β-アラニンメチルエステル(0.5850g、2.01mmol)を脱保護し、白色粉末として標題化合物を得た(0.5076g、1.87mmol、93.0%);融点:203℃(dec);TLC: Rf=0.60(溶媒H);IR(cm-1):3500〜2900(OH)、1715(カルボン酸C=O)。
【0190】
β-フェネチル-β-アラニン
N-アセチル-β-2-フェネチル-β-アラニンメチルエステル(1.5322g、6.15mmol)を脱保護し、白色結晶として標題化合物を得た(0.4709g、2.44mmol、39.6%);融点:211〜214℃;TLC: Rf=0.37(溶媒I)、0.74(溶媒L);IR(cm-1):3496、3310(鋭い:OH、H結合せず)、3028(CH)、2932(CH)、2162、1663(カルボン酸C=O)、702(=CH)。
【0191】
β-(p-メトキシフェネチル)-β-アラニン
N-アセチル-β-(p-メトキシフェネチル)-β-アラニンメチルエステル(1.1244g、4.03mmol)を脱保護し、MeOHから再結晶化し、オフホワイト結晶として標題化合物を得た(0.2761g、1.25mmol、31.0%);融点:180〜184℃;TLC: Rf=0.34(溶媒I)、0.74(溶媒K);IR(cm-1):3400〜2500(OH)、2171、1632(カルボン酸C=O)。
【0192】
β-(p-メチルフェネチル)-β-アラニン
N-アセチル-β-[2-(4-メチルフェニル)エチル]-β-アラニンメチルエステル(1.2884g、4.89mmol)を脱保護し、綿毛状白色結晶として標題化合物を得た(0.6779g、3.27mmol、66.9%);融点:206〜207℃;TLC: Rf=0.89(溶媒K);IR(cm-1):3530、3280(鋭い:OH、H結合せず)、3017(CH)、2166、1706(カルボン酸C=O)、810(=CH)。
【0193】
β-[2-(4-ヒドロキシ-3-メトキシフェニル)エチル]-β-アラニン塩酸塩
N-アセチル-β-[2-(4-ヒドロキシ-3-メトキシフェニル)エチル]-β-アラニンメチルエステル(0.5281g、1.79mmol)を脱保護し黄色油として標題化合物を得た(0.4852g、1.76mmol、98.4%);TLC: Rf=0.32(溶媒I);IR(cm-1):3447(OH)、1718(カルボン酸C=O)。
【0194】
2-アゼチジノンの合成
N-置換β-アミノ酸からのN-置換2-アゼチジノンの調製
CCl4(1.0mL、10mmol)およびトリエチルアミン(TEA)(1.7mL、12mmol)を、N-置換β-アミノ酸(10mmol)および(C6H5)3P(1.56g、1.2mmol)のMeCN(100mL)撹拌溶液に添加した。反応混合物を1.5時間還流させ、その後真空下で濃縮した。残渣をCH2Cl2(100mL)に溶解し、水および塩類溶液で洗浄した。有機層を乾燥させ(MgSO4)、蒸発乾燥させた。生成物を溶離液としてEtOAC/ヘキサン(1:2)を用いてシリカゲルフラッシュクロマトグラフィにより単離した。
【0195】
N-非置換β-アミノ酸からのN-シリル2-アゼチジノンの調製
N-ブロモスクシンイミド(2.14g、12mmol)およびTEA(1.7mL、12mmol)を、N-非置換β-アミノ酸(10mmol)および(C6H5)3P(1.56g、1.2mmol)のMeCN(100mL)撹拌溶液に添加した。反応混合物を室温で10時間撹拌し、その後真空下で濃縮した。残渣をCH2Cl2(60mL)に溶解し、t-ブチルジメチルシリルクロリド(2.25g、15mmol)およびジイソプロピルアミン(2.8mL、15mmol)で処理し、室温で5時間撹拌した。その後溶液をCH2Cl2(100mL)で希釈し、水および塩類溶液で洗浄した。有機層を乾燥させ(MgSO4)、蒸発乾燥させた。生成物を溶離液としてEtOAC/ヘキサン(1:7)を用いてシリカゲルフラッシュクロマトグラフィにより単離した。
【0196】
実施例4:β-アリールβ-アラニンの合成
β-アリールβ-アラニンをワンポット反応で調製した。簡単に説明すると、置換ベンズアルデヒドの無水エタノール溶液に、マロン酸および過剰の酢酸アンモニウムを添加し、反応混合物を加熱し還流させた。反応混合物を冷却しβ-アリールβ-アラニンおよび(一定の場合には)桂皮酸誘導体の混合物を得た。桂皮酸は(存在すれば)混合物の酸/塩基抽出により除去し、β-アリールβ-アラニンを得た。収率はしばしば中程度か良好であった。過程を図3に示した。ある一定のβ-アリールβ-アラニン化合物を合成するための実験手順の更なる詳細は以下に示す。化合物を精製するための代表的な精製スキームを図4に示す。本明細書で説明したように調製したある一定の化合物を以下の表1に示す。収率データは2列で示してあり、第2番目は以下の表2のものと同一である。
【0197】
(表1) ベンズアルデヒドから調製したβ-アリールβ-アラニンの平均収率
(反応条件は最適化していない)
【0198】
本方法により合成し選択した化合物を表1に示す。本発明のある一定のこれらの化合物および他の化合物の代表的な合成を以下で示す。
【0199】
対応するベンズアルデヒド誘導体を過剰の酢酸アンモニウム(〜2当量)およびマロン酸(1当量)と共に無水エタノール中で還流させ、反応を完了させる(TLCおよびNMRにより決定)ことによりβ-置換-β-アミノ酸を調製した。桂皮酸誘導体が副産物として生成した。反応混合物をその後、例えば図4で説明するように標準手順により処理した。
【0200】
β-3(3,4-ジクロロフェノキシ)フェニル-β-アラニン塩酸塩
上記手順を用いて、3-(3,4-ジクロロフェノキシ)ベンズアルデヒド(10g、37.4mmol)、酢酸アンモニウム(3.8437g、49.8mmol)およびマロン酸(3.8923g、37.4mmol)を無水エタノール(30mL)中で5時間(ゆっくりと)還流させた。白色固体のβ-3(3,4-ジクロロフェノキシ)フェニル-β-アラニンを濾過し、10mLの無水エタノールで2度洗浄した。続いて、このβ-3(3,4-ジクロロフェノキシ)フェニル-β-アラニンに10mLの3N HClを添加し、β-3(3,4-ジクロロフェノキシ)フェニル-β-アラニン塩酸塩(4.44g、12.2mmol、32.6%)を得た;融点164〜165℃;IR(KBr):3193、1609cm-1、Rf=0.55(溶媒24)、0.72(溶媒25)。
【0201】
β-4-ブロモフェニル-β-アラニン
4-ブロモベンズアルデヒド(10g、54mmol)、酢酸アンモニウム(8.663g、112.4mmol)およびマロン酸(5.6762g、54.5mmol)を無水エタノール(45mL)中で150時間(ゆっくりと)還流させた。白色固体を濾過し、50mLのNa2CO3および50mLのH2Oの温かい溶液(70℃)に溶解した。その後、この溶液を100mLのジエチルエーテルで3度抽出した。水層をさらに酸性化してpH7とし、白色固体β-4-ブロモフェニル-β-アラニン(4.5140g、18.49mmol、34.2%)を得た;融点:234℃;IR(KBr):3061、1594cm-1;TLC:Rf=0.35(溶媒24)、0.32(溶媒25)。
【0202】
β-4-フルオロフェニル-β-アラニン
4-フルオロベンズアルデヒド(10g、80mmol)、酢酸アンモニウム(8.2487g、107mmol)およびマロン酸(8.3285g、80mmol)を無水エタノール(60mL)中で48時間(ゆっくりと)還流させた。白色固体を濾過し、エタノール再結晶化により精製し、β-4-フルオロフェニル-β-アラニン(10.04g、54.8mmol、68.5%)を得た;融点:216〜217℃;IR(KBr):3160、1606cm-1;TLC:Rf=0.41(溶媒24)、0.42(溶媒25)。
【0203】
β-2,5-ジメトキシフェニル-β-アラニン
2,5-ジメトキシベンズアルデヒド(4.1437g、25mmol)、酢酸アンモニウム(3.1200g、40.47mmol)およびマロン酸(3.1244g、30.02mmol)を無水エタノール(60mL)中で6時間(ゆっくりと)還流させた。白色固体を濾過し、メタノール再結晶化により精製し、β-2,5-ジメトキシフェニル-β-アラニン(1.239g、5.5mmol、22.0%)を得た;融点:206〜208℃;IR(KBr):2944、1630cm-1;TLC:Rf=0.29(溶媒21)、0.66(溶媒23)。
【0204】
β-3-ブロモ-4-メトキシフェニル-β-アラニン
3-ブロモ-4-メトキシベンズアルデヒド(9.9835g、46.42mmol)、酢酸アンモニウム(7.2984g、94.69mmol)およびマロン酸(4.9124g、47.21mmol)を無水エタノール(110mL)中で281時間(ゆっくりと)還流させた。白色固体を濾過し、50mLのNa2CO3および50mLのH2Oの温かい溶液(70℃)に溶解した。その後、この溶液を100mLのジエチルエーテルで3度抽出した。水層をさらに酸性化してpH1とし、100mLの酢酸エチルで2度抽出した。続いて、水層を蒸発乾燥させ、その後白色残渣に30mLの無水エタノールを添加し、15分間撹拌し、濾過した。その後、同じ手順を2度繰り返した。最終混合物を濾過し、濾液を蒸発乾燥した。プロピレンオキシド(9.75mL、139.3mmol)をエタノール部分に添加した。溶液を撹拌し、50℃まで加熱し、β-3-ブロモ-4-メトキシフェニル-β-アラニン(3.0284g、11.05mmol、23.8%)を得た;融点:213℃;IR(KBr):2945、1604cm-1;TLC:Rf=0.26(溶媒24)、0.28(溶媒25)。
【0205】
前の段落に従い一般に合成される他の化合物およびそれらの分析データを以下の表2に示す。
【0206】
(表2) ベンズアルデヒドから調製されたβ-アリール-β-アラニン
【0207】
TLC解析
上記実験手順では、薄層クロマトグラフィー分析用に使用する溶媒は以下のように略記される:
溶媒21:アセトニトリル:酢酸:水 8:1:1
溶媒23:メタノール:酢酸 7:1
溶媒24:n-ブタノール:酢酸:水 4:1:1
溶媒25:メタノール:クロロホルム:酢酸 7:7:1
【0208】
β-アリール-β-アラニン、β-フェネチル-β-アラニン、α-シクロヘキシル-β-アラニン、およびα-置換-β-アラニン(およびそれらのある一定のエステルおよびアミド)、ならびに4'-置換N-アセチル-α-ピペリジニル-β-アラニンに対する他の分析データおよび生物学的データを表3-1から3-3において示す。
【0209】
(表3−1) 解析および生物活性データ
A. β-アリール-β-アラニンおよび前駆体
a.再結晶化のためにEtOH、H2Oまたは混合物を使用した。
b.ピロカルピンを使用すると、化合物はラットでは100mg/kgで活性であるか、または不活性である。
B. アリール置換β-フェネチル-β-アラニンおよび前駆体
a.可能であれば、再結晶化のためにEtOH、H2O、または混合物を使用した。
b.ピロカルピンを使用すると、化合物はラットでは100mg/kgで活性であるか、または不活性である。
【0210】
(表3−2) 解析および生物活性データ
C. 4'-置換α-シクロヘキシル-β-アラニンおよび前駆体
a.再結晶化のためにEtOH、H2Oまたは混合物を使用した。
b.ピロカルピンを使用すると、化合物はラットでは100mg/kgで活性であるか、または不活性である。
D. 4'-置換N-アセチル-α-ピペリジニル-β-アラニンメチルエステル
【0211】
(表3−3) 解析および生物活性データ
E. N-アセチル-α-置換-β-アラニンメチルエステルおよびα-置換-β-アラニン
a.最終合成段階の収率
b.ピロカルピンを使用すると、化合物はラットでは100mg/kgで活性であるか、または不活性である。
【0212】
実施例5
癲癇の「自発反復性発作」(SRS)モデルを使用して、I相癲癇誘発に対するモデルにおいて候補化合物を評価した(例えば、Mello,E.ら、Epilepsia(1993)34:985;Cavalheiro,J.ら、Epilepsia(1991)32:778を参照のこと)。SRSモデルでは、成体の雄SDラット(c.260g)に注射によりピロカルピンを投与する(380mg/kg、腹腔内)。25分以内に、動物は癲癇重積持続状態になり、これが典型的には15〜20時間続く(この段階で動物の約10%が死亡する)。ラットは自発的に回復させ、食物と水を自由に与え、16時間/8時間の明/暗サイクルで維持する。ラットは通常4つの群で研究する。約13〜15日に始めると、ラットは自発反復性発作を発症する。これは1週間につき約4〜5の割合で起こる。ラットは1日につき16時間ビデオテープ録画し、重要な発作挙動(点頭、前肢クロヌスおよび立ち上がり)に対しビデオテープを再検討する。動物は3ヶ月観察し、十分な数の発作の評価を可能とする。評価用の実験化合物は2つの時のうちのいずれかで投与することができる:時間1、1日目、癲癇重積持続状態の停止後SRSの発症前;または時間2、30日目、ラットは約2週間の間SRSを経験した時。時間Iに候補化合物を投与すると、抗癲癇誘発特性(発作の発症の阻害能力)を評価することができ;時間2に化合物を投与すると、定着した発作を抑制する能力を有する抗発作発生としての薬物評価が可能となる。
【0213】
参照として、時間1または時間2のいずれかで、標準抗痙攣薬フェニトインを投与した(20mg/kg/日、静脈内、10日間)。予測通り、フェニトインは時間1で投与した場合発作の発症を阻害する効果が無かったが、時間2で投与すると発作頻度を50%以上減少させるのに75%の効果を示した。
【0214】
対照的に、β-アラニンおよび類似(α-(4-tert-ブチルシクロヘキシル)-アラニン(実施例3を参照のこと)を、上記で概要を示した同じプロトコルを用い時間1または時間2のいずれかで相当する用量(20mg/kg/日、静脈内、10日間)で投与した。時間1では、これらの化合物のそれぞれが、少なくとも50%発作を減少させるのに75%の効果を示し、時間2では、各化合物は少なくとも50%発作を減少させるのに50%の効果を示した。
【0215】
前の表2および3で列挙した本発明の化合物について、実施例7に従う生物活性試験を行った。以下の化合物は少なくとも弱い活性を有することがわかった:塩酸β-p-メチルフェニル-β-アラニン、β-2-ヒドロキシ-3-メトキシフェニル-β-アラニン、β-3-メチル-4-メトキシフェニル-β-アラニン(わずか)、塩酸β-3-(3,4-ジクロロフェノキシ)フェニル-β-アラニン(中程度)、β-2,5-ジメチル-4-メトキシフェニル-β-アラニン、β-p-(トリフルオロメトキシ)フェニル-β-アラニン、およびβ-2-フルオロ-3-(トリフルオロメチル)フェニル-β-アラニン(中程度)。
【0216】
このように、β-アミノ酸は抗癲癇誘発化合物および抗発作発生化合物の両方としての活性を示す。
【0217】
実施例6
標準方法に従いジオキサピペラジン化合物を合成し、NMR、FAB-MS、融点およびHPLCによりキャラクタリゼーションを行った。幾つかの化合物の結晶構造を決定した。
【0218】
例示的な手順は以下の通りである。
Boc-L-アラニン(1.5g、0.008mol)を60mlの酢酸エチルに溶解し、これに2.4gの2-エトキシカルボニル-1,2-ジヒドロキノリン(EEDQ)(0.010mol、1.2当量)を添加した。溶液を5分間撹拌し、その後D-フェニルグリシンメチルエステルHCl(1.5g、0.003mol)を添加した。撹拌を24時間続け、その後に溶液を3×25mL 10%(w/w)KHSO4水溶液、25mL飽和NaCl溶液、3×25飽和重炭酸ナトリウム溶液、および25mL飽和NaCl溶液で洗浄した。有機層を硫酸マグネシウム上で乾燥させ、蒸発させ透明な油を得た。油を20mlの蟻酸に溶解し、2時間室温で撹拌した。蒸発により酸を除去し、油を50mLの2-ブタノールおよび25mLのトルエンの混合物中に懸濁させた。混合物を24時間還流させ、撹拌しながら2時間にわたって冷却し、溶媒を真空下で減量しオリジナルの体積の約4分の1とした。固体を再結晶化させた。シクロ-D-フェニルグリシン-L-アラニンを融点範囲が260〜265℃の白色固体(1.1g、0.005mol、収率68%)として得た。
【0219】
実施例7
選択した化合物を0.9% NaClに溶解し、または30%ポリエチレングリコール400および70% 水の混合物に懸濁させ、動物モデルにおいて試験した。簡単に説明すると、化合物をカースワースファームズ(carsworth Farms)#1マウス(体重1gあたり0.01mgの容積)またはSDラット(体重1gあたり0.004mlの容積)に腹膜内または経口投与した。抗痙攣薬試薬を投与する前にピーク効果およびピーク神経障害の時間を決定した。
【0220】
最大電気ショック発作試験(MES)、電解質溶液(0.9% NaCl)滴を注入した角膜電極を動物の目に適用し、電気刺激(マウスでは50mA、ラットでは150mA;60Hz)を試験化合物のピーク効果時に0.2秒間加えた。動物を手で保持し、刺激時に解放し、発作を観察した。後脚緊張性伸筋成分の廃止(後脚緊張性伸展は身体面に対し90°を超えない)は、MESにより誘発される発作の広がりが化合物により阻害されることを示した。
【0221】
皮下ペンチレンテトラゾールしきい値試験(scMet)では、痙攣を起こさせる用量(CD97)のペンチレンテトラゾール(ラットでは85mg/kg)を試験化合物のピーク効果時に注射した。動物を隔離し、30分間観察して発作が起こるかどうかを確認した。少なくとも5秒間続く間代性の痙攣がないと、ペンチレンテトラゾールにより誘発される発作のしきい値を化合物により引き上げることができることが示された。
【0222】
実験動物における急性抗痙攣薬により誘発される毒性は通常、幾つかの種類の神経異常により特徴づけられる。マウスでは、これらの異常はロトロッド失調試験により検出することができる。この試験はラットでは幾分有益ではない。ロトロッド失調試験では、神経障害は、6rpmで回転するギザギザのついた棒上で少なくとも1分間、動物が平衡を維持することができないことにより示される。ラットは、位置感知試験により試験した。1つの後足をテーブルの縁を越えて徐々に下に下げると、正常な動物は正常な位置まで足を引き上げて戻す。足を正常な位置まで戻すことができないと神経障害が示される。
【0223】
実施例8
ジオキサピペラジン化合物の試験を12匹のマウスで、30、100、300mg/kg(各4匹のマウス)の用量で、試験化合物投与後30分および4時間に実施した。結果を表4に示す。
【0224】
(表4) 選択されたジオキサピペラジン化合物および試験データ
c=シクロ
Peg=フェニルグリシン
活性は0(不活性)から4までの段階に分けて示す
【0225】
表4に示されるように、c/D-フェニルグリシン-L-アラニンおよびc/D-フェニルグリシン-(S-Me)-L-システインは、この動物モデルシステムでは強い抗痙攣活性を示したが、幾つかの他のジオキサピペラジンはより弱い抗痙攣活性を示した。
【0226】
ある一定の他のジオキサピペラジンも合成し、試験した。これらの化合物のうち、c/L-アラニン-D-ロイシンが活性であることがわかった。
【0227】
実施例9:ビアリール抗癲癇誘発作用物質
さらに他の態様では、被験者において癲癇誘発および/または発作発生を阻害するための方法は有効量の化合物を被験者に投与し、癲癇誘発を阻害する段階を含み、ここでその化合物は以下の通りである。
【化64】
【0228】
より特定すると、好ましい化合物は以下の化学式を有する:
【化65】
(式中、各Xはそれぞれ、ハロゲン(クロロが好ましい)、ニトロ、シアノおよび置換または非置換のアルキルおよびアルコキシ基(トリフルオロメチルおよびメチルが好ましい)からなる群から選択され;nは0から5の整数であり(n=1が好ましい);およびYRおよびYSのうちの1つは水素であり、もう一方は置換または非置換のアミンであり、薬学的に許容されるその塩も含まれる)。
【0229】
(表5) 例示的なビアリールエーテル化合物
a.ピロカルピンを使用すると、化合物はラットでは100mg/kgで活性であるか、または不活性である。
【0230】
または、ビアリールエーテルはパラ置換されてもよい:
【化066】
例えば、前記、表2の化合物B8P79を参照のこと。
【0231】
生物学的データが示すように、RまたはS絶対立体化学のいずれかの鏡像異性体の方が、ラセミ化合物または他の立体異性体よりも生物学的に活性であるかもしれない。このような場合、その単一の立体異性体が好ましく、本発明による薬学的組成物は好ましくは、実質的にその立体異性体のみを含む。そのような立体化学異性体は、キラル開始材料からの不斉合成(例えば、桂皮酸エステルへのキラルアミンのマイケル付加後加水分解)、または下記で例示するように、ラセミ合成の分割のいずれかにより調製されてもよい。
【0232】
3-(3-トリフルオロメチルフェノキシ)-trans-桂皮酸メチル
3-[3-(トリフルオロメチル)フェノキシ]ベンズアルデヒド(8.05g、30mmol)および酢酸メチルトリフェニルホスホラニリデン(15.13g、45mmol)のTHF(200mL)溶液を24時間還流させて撹拌し、その後、室温まで冷却し、濃縮した。ヘキサンに溶解した0〜10%のEtOAc溶離液を用い、シリカゲル上でクロマトグラフィーにより残渣を精製すると、9.3g(96%)が得られた。
【0233】
3-(4-メチルフェノキシ)-trans-桂皮酸メチル
3-(4-メチルフェノキシ)ベンズアルデヒド(8.04g、37.9mmol)および酢酸メチルトリフェニルホスホラニリデン(19g、57mmol)のTHF(200mL)溶液を24時間還流させて撹拌し、その後、室温まで冷却し、濃縮した。ヘキサンに溶解した0〜10%のEtOAc溶離液を用い、シリカゲル上でクロマトグラフィーにより残渣を精製すると、9.6g(94.5%)が得られた。
【0234】
3-フェノキシ-trans-桂皮酸メチル
3-フェノキシベンズアルデヒド(8.03g、40.5mmol)および酢酸メチルトリフェニルホスホラニリデン(20g、60mmol)のTHF(200mL)溶液を24時間還流させて撹拌し、その後、室温まで冷却し、濃縮した。ヘキサンに溶解した0〜10%のEtOAc溶離液を用い、シリカゲル上でクロマトグラフィーにより残渣を精製すると、10.2g(99%)が得られた。
【0235】
(3R)-[(S)-(-)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチル
ブチルリチウム(ヘキサン中2.5M、9.9mL、24.75mmol)を0℃で、THF(200mL)に溶解した(S)-(-)-N-ベンジル-α-メチルベンジルアミン(5.3mL、25mmol)に添加した。赤色溶液を0℃で20分間撹拌し、-78℃まで冷却した。THF(20mL)に溶解した3-(3-トリフルオロメチルフェノキシ)-trans-桂皮酸メチル(4g、12.4mmol)を一滴ずつ添加した。混合物を2時間-78℃で撹拌し、飽和塩化アンモニウム(100mL)で急冷し、その後温め、飽和塩化ナトリウム水溶液(100mL)に注ぎ入れた。水層をEtOAc(2×100mL)で抽出、乾燥(Na2SO4)、濾過および蒸発させると残渣が得られ、これをヘキサンに溶解した0〜8%のEtOAc溶離液を用い、シリカゲル上でクロマトグラフィーにより精製した。収集した画分を蒸発させると3.2g(47%)が得られた。
【0236】
(3S)-[(R)-(+)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチル(4.1g)を、R-(+)-N-ベンジル-α-メチルベンジルアミンから同じ手順で調製し、収率は62%であった。
【0237】
(3R)-[(S)-(-)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチル
ブチルリチウム(ヘキサン中2.5M、12mL、30mmol)を0℃で、THF(200mL)に溶解した(S)-(-)-N-ベンジル-α-メチルベンジルアミン(6.3mL、30mmol)に添加した。赤色溶液を0℃で20分間撹拌し、-78℃まで冷却した。THF(20mL)に溶解した3-(3-トリフルオロメチルフェノキシ)-trans-桂皮酸メチル(4g、14.9mmol)を一滴ずつ添加した。混合物を2時間-78℃で撹拌し、飽和塩化アンモニウム(100mL)で急冷し、その後温め、飽和塩化ナトリウム水溶液(100mL)に注ぎ入れた。水層をEtOAc(2×100mL)抽出、乾燥(Na2SO4)、濾過および蒸発させると残渣が得られ、これをヘキサンに溶解した0〜8%のEtOAc溶離液を用い、シリカゲル上でクロマトグラフィーにより精製した。収集した画分を蒸発させると3.3g(46%)が得られた。
【0238】
(3S)-[(R)-(+)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチル(4.4g)を、R-(+)-N-ベンジル-α-メチルベンジルアミンから同じ手順で調製し、収率は62%であった。
【0239】
(3R)-[(S)-(-)-N-ベンジル-α-メチルベンジル]アミノ-(3-フェノキシフェニル)プロパン酸メチル
ブチルリチウム(ヘキサン中2.5M、13mL、32.5mmol)を0℃で、THF(200mL)に溶解した(S)-(-)-N-ベンジル-α-メチルベンジルアミン(6.6mL、31.6mmol)に添加した。赤色溶液を0℃で20分間撹拌し、-78℃まで冷却した。THF(20mL)に溶解した3-(4-メチルフェノキシ)-trans-桂皮酸メチル(4g、15.7mmol)を一滴ずつ添加した。混合物を-78℃で2時間撹拌し、飽和塩化アンモニウム(100mL)で急冷し、その後温め、飽和塩化ナトリウム水溶液(100mL)に注ぎ入れた。水層をEtOAc(2×100mL)抽出、乾燥(Na2SO4)、濾過および蒸発させると残渣が得られ、これをヘキサンに溶解した0〜8%のEtOAc溶離液を用い、シリカゲル上でクロマトグラフィーにより精製した。収集した画分を蒸発させると4.8g(66%)が得られた。
【0240】
(3S)-[(R)-(+)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチルを、R-(+)-N-ベンジル-α-メチルベンジルアミンから同じ手順で調製し、収率は51%であった。
【0241】
(3R)-アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチル
水素(1気圧)下、炭(700mg)上水酸化パラジウムの存在下、(3R)-[(S)-(-)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチル(3.2g、5.8mmol)のMeOH(60mL)、H2O(6mL)および酢酸(1.5mL)溶液を室温で36時間撹拌した。濾過、蒸発させ生成物を得た。生成物は精製せずに次の反応に使用した。
【0242】
(3S)-アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチルを、(3R)-[R-(+)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパノエート(3.9g、7.1mmol)から同じ手順で調製した。
【0243】
(3R)-アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロパン酸メチル
水素(1気圧)下、炭(530mg)上水酸化パラジウムの存在下、(3R)-[(S)-(-)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロパン酸メチル(3.3g、6.7mmol)のMeOH(60mL)、H2O(6mL)および酢酸(1.5mL)溶液を室温で36時間撹拌した。濾過、蒸発させ生成物を得た。生成物は精製せずに次の反応に使用した。
【0244】
(3S)-アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチルを、(3R)-[R-(+)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロパノエート(4.2g、8.5mmol)から同じ手順で調製した。
【0245】
(3R)-アミノ-3-(3-フェノキシフェニル)プロパン酸メチル
水素(1気圧)下、炭(700mg)上水酸化パラジウムの存在下、(3R)-[(S)-(-)-N-ベンジル-α-メチルベンジル]アミノ-3-(3-フェノキシフェニル)プロパン酸メチル(4.4g、9.1mmol)のMeOH(60mL)、H2O(6mL)および酢酸(1.5mL)溶液を室温で36時間撹拌した。濾過、蒸発させ生成物を得た。生成物は精製せずに次の反応に使用した。
【0246】
(3S)-アミノ-3-(3-フェノキシフェニル)プロパン酸メチルを、(3S)-[R-(+)-N-ベンジル-α-メチルベンジル]アミノ-3-(3-フェノキシフェニル)プロパノエート(3.7g、7.7mmol)から同じ手順で調製した。
【0247】
(3R)-アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロピオン酸塩酸塩(C1)
(3R)-アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチルを2N HCl(40mL)に溶解し、一晩中還流し、室温まで冷却し濃縮した。残渣を2N HCl(100mL)およびジエチルエーテル(30mL)に溶解した。水層と有機層との間に形成された油層を分離し、蒸発させ、一晩中ポンプで乾燥させ、白色粉末1.8gを得た:[α]20D-0.49°(c2.26、CH3OH)。
【0248】
(3S)-アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロピオン酸塩酸塩(C2)を、(3S)-アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチルから同じ手順により調製し、収率は74%であった:[α]20D+0.63°(c2.38、CH3OH)。
【0249】
(3R)-アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロピオン酸塩酸塩(C3)
(3R)-アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロパン酸メチルを2N HCl(40mL)に溶解し、一晩中還流し、室温まで冷却し濃縮した。残渣を2N HCl(100mL)に溶解し、濃縮した。白色沈澱を濾過し、ジエチルエーテル(10mL)で洗浄し、一晩中ポンプで乾燥させ、1.6gを得た:[α]20D-1.36°(c2.06、CH3OH)。
【0250】
(3S)-アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロピオン酸塩酸塩(C4)を、(3S)-アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロパン酸メチルから同じ手順により調製し、収率は65%であった:[α]20D+1.46°(c2.26、CH3OH)。
【0251】
(3R)-アミノ-3-(3-フェノキシフェニル)プロピオン酸塩酸塩(C5)
(3R)-アミノ-3-(3-フェノキシ)フェニルプロパン酸メチルを2N HCl(40mL)に溶解し、一晩中還流し、室温まで冷却し濃縮した。残渣を2N HCl(100mL)に溶解し、ジエチルエーテル(2×30mL)で洗浄した。水溶液を蒸発させ、一晩中ポンプで乾燥させ、白色粉末2.4gを得た:[α]20D-1.40°(c2.79、CH3OH)。
【0252】
(3S)-アミノ-3-(3-フェノキシフェニル)プロピオン酸塩酸塩(C7)(1.96g)を、(3S)-アミノ-3-(3-フェノキシフェニル)プロパン酸メチルから同じ手順により調製し、収率は87%であった:[α]20D+1.43°(c2.25、CH3OH)。
【0253】
(D)-(+)-3-アミノ-3-[3-(4-クロロフェノキシ)フェニル]プロピオン酸塩酸塩(C8)および(L)-(-)-3-アミノ-3-[3-(4-クロロフェノキシ)フェニル]プロピオン酸塩酸塩(C9)を、BOC-保護ラセミ3-アミノ-3-[3-(4-クロロフェノキシ)フェニル]プロピオン酸のEtOAcに溶解した(1R,2S)-(-)-エフェドリンによるジアステレオ異性体選択的再結晶化、それに続く当技術分野において認知された技術を用いたBOC基の酸性除去により製造した。これらの化合物の比旋光度は+1.07°および-1.04°であった(MeOH中でc=0.0118)。1Hおよび13C NMRは構造と一致した。
【0254】
(L)-(-)-3-アミノ-3-[3-(3,4-ジクロロフェノキシ)フェニル]プロピオン酸塩酸塩(C10)および(D)-(+)-3-アミノ-3-[3-(3,4-ジクロロフェノキシ)フェニル]プロピオン酸塩酸塩(C11)を酵素分割により調製した。塩化フェニルアセチルおよび3-アミノ-3-[3-(3,4-ジクロロフェノキシ)フェニル]プロピオン酸の反応から調製したラセミ3-[3-(3,4-ジクロロフェノキシ)フェニル]-3-フェニルアセチルアミノ-プロピオン酸(2.01g、4.5mmol)を30mLのEtOAcに溶解した。この溶液に30mLの1M リン酸緩衝液(pH=7.6)およびEupergitに固定された200mg(10% w/w)のペニシリンGアミダーゼ(PGA)を添加した。24時間後に反応を停止させ、濾過により酵素を除去した。アミンおよびアセチムアミド生成物をEtOAcと酸水溶液との間での分割により分離し、溶媒を減圧下で除去し、凍結乾燥により198mg(24%)の濃縮(L)-(-)-化合物を得た([α]D=-0.39°、MeOH中でc=0.0058)。1Hおよび13C NMRは構造と一致した。反応を24時間後に停止した。さらに、アセトアミドを6M HClで加水分解すると970mg(79%)の濃縮(D)-(+)-化合物が得られた([α]D=+0.13°、MeOH中でc=0.0173)。1Hおよび13C NMRは構造と一致した。
【0255】
等価物
当業者であれば、本明細書で記述した特定の手順に対する多くの等価物を認識し、またはせいぜい通常の実験を用いて確認することができる。そのような等価物は本発明の範囲内にあり、特許請求の範囲に含まれる。この出願全体において引用した他の参考文献、発行された特許および公開された特許出願全ての内容が、参照として明細書に組み込まれる。本発明およびその態様に関して、それらの特許、出願、および他の文書の適当な構成要素、手順、および方法を選択してもよい。
【図面の簡単な説明】
【0256】
【図1】本発明の方法に有用であるピリミジンおよびジヒドロピリミジン化合物の例を示す。
【図2】本発明のピリミジンおよびジヒドロピリミジン化合物を調製する合成スキームの例を示す。
【図3】本発明のβ-アミノ酸の合成の一つの態様を示す。
【図4】β-アミノ酸の精製スキームを示すフローチャートである。
【技術分野】
【0001】
関連出願
本出願は、2001年3月13日に提出された米国仮特許出願第60/275,618号の優先権を主張し;本出願は、さらに、1998年3月11日に提出された米国特許出願第09/041,371号(現在は、その全内容が参照として本明細書に組み入れられる米国特許第6,306,909号)に関し、その内容を開示する。
【背景技術】
【0002】
発明の背景
癲癇は、世界中の何十万人もの人が罹患している、発作を伴う重度の神経学的病態である。臨床的には、発作は、脳においてニューロンの集合体から突然電気が放電されることから生じる。結果として生じる神経細胞活性は、制御不可能な動きなどの症状となって顕在化する。
【0003】
発作は、「発作発生(ictogenesis)」と称される過程を通してニューロン集合体からの過剰な電気放電により引き起こされる単一の独立した臨床事象である。従って、発作は、単なる癲癇の症状である。癲癇は、正常な脳が変化し、「癲癇誘発」と称される過程を通じて反復性発作に感受性となる、根底にある一連の病的変換を特徴とする動的でしばしば進行的な過程である。発作発生および癲癇誘発は共通の生化学的経路を有すると考えられているが、二つの過程は同一ではない。発作発生(時間的および空間的な発作の開始および伝播)は、数秒間または数分間におよび起こる急速で明確な電気的/化学的事象である。癲癇誘発(正常な脳が、「癲癇誘発焦点」の開始および成熟により、自発的で偶発的で時間制限的な反復性発作に感受性の状態に転換される漸進的な過程)は、一般的に数ヶ月から数年間かけて生じる、緩徐な生化学的過程および/または組織学的過程である。癲癇誘発は二相過程である。第1相癲癇誘発は、最初の発作前の癲癇誘発過程の開始であり、発作、疾病(例えば髄膜炎)、または頭部への事故的打撃または脳に行われた外科手術などの外傷の結果であることが多い。第2相癲癇誘発は、発作にすでに感受性である脳が、依然として、頻度および/または重度の増加中の発作にさらに一層感受性となる過程を意味する。癲癇誘発に関与する過程は明確に確定されていないが、N-メチル-D-アスパルテート(NMDA)受容体により媒介されるニューロン間の興奮連関のアップレギュレーションが関与すると考える研究者もいる。また、γ-アミノ-酪酸(GABA)受容体により媒介されるニューロン間の抑制連関のダウンレギュレートを示唆する研究者もいる。
【0004】
癲癇発作が致死的であることは稀であるが、多数の患者が、発作の破壊的で危険性のある結果を避けるための薬物療法を必要としている。多くの場合、薬物療法は長期間必要とされ、生涯、処方薬を服用し続けなければならない場合もある。さらに、癲癇の管理に使用される薬物には、長期使用に伴う副作用があり、薬価がかなり高い場合もある。
【0005】
古い抗痙攣薬(例えばフェニトイン、バルプロ酸塩、およびカルバマゼピン)(イオンチャネル遮断剤)ならびに新しい薬剤(例えばフェルバメート、ガバペンチン、およびチアガビン)を含む、多種多様の薬物が癲癇発作の管理に使用できる。β-アラニンは、抗痙攣活性ならびにNMDA抑制活性およびGABA作動性刺激活性を示すと報告されているが、臨床的には使用されていない。現在入手可能な承認されている癲癇薬は抗痙攣剤であり、本明細書において「抗痙攣剤」という用語は、「抗発作」または「抗発作発生」と同義語であり;これらの薬物は、発作発生を遮断することにより発作を抑止できるが、癲癇誘発を遮断しないことから、癲癇には影響を及ぼさないと考えられている。従って、癲癇の治療に使用できる薬物は多数あるにも関わらず(すなわち、癲癇発作に伴う痙攣抑止により)、癲癇誘発を特徴とする病的変化の治療に一般的に承認されている薬物は全くない。癲癇誘発過程を抑制する一般的に承認されている方法は全くなく、抗癲癇誘発剤として認識されている一般的に承認されている薬物は全くない。
【発明の開示】
【0006】
発明の概要
本発明は、癲癇を含む痙攣疾患の治療および/または予防に有用である、方法および化合物、例えば抗発作発生化合物および/または抗癲癇誘発化合物に関する。
【0007】
一つの局面において、本発明は、被験者における癲癇誘発を抑制する方法を提供する。本方法は、それを必要とする被験者に、被験者における癲癇誘発が抑制されるように癲癇誘発に関連した経路中の一過程を変調する薬剤の有効量を投与する段階を含む。
【0008】
別の局面において、被験者における癲癇誘発抑制法を提供する。NMDA受容体に拮抗し、内因性GABA抑制を増強する薬剤の有効量を、被験者における癲癇誘発が抑制されるように、それを必要とする被験者に投与する。好ましい態様において、該薬剤は、NMDA受容体のグリシン結合部位に結合することにより、NMDA受容体に拮抗する。好ましい態様において、該薬剤は、グリア細胞のGABA取り込みを減少させることにより、GABA抑制を増強する。ある好ましい態様において、該薬剤は、NMDA受容体に拮抗し且つ内因性GABA抑制を増強するファルマコフォアを含む。該薬剤は経口投与でき、ある態様において、経口投与段階後に、該薬剤は、能動輸送シャトル機序により被験者の神経系に輸送され得る。好ましい態様において、抗癲癇誘発剤は、β-アミノアニオン化合物であり、アニオン部分は、カルボキシレート、サルフェート、スルホネート、スルフィネート、スルファメート、テトラゾリル、ホスフェート、ホスホネート、ホスフィネート、およびホスホロチオエートからなる群より選択される。ある態様において、該薬剤はβ-アミノ酸であるが、好ましくはβ-アラニンではない。
【0009】
別の局面において、本発明は、被験者における癲癇誘発を抑制する方法を提供する。該方法は、それを必要とする被験者に、癲癇誘発が抑制されるように、下記式
【化1】
(式中、Aは、生理的pHにおいて陰性基であり;R1は、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、またはアミノカルボニルであり;R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、もしくはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、環の原子数が3〜7である非置換もしくは置換の複素環を形成している)
で示される化合物;
またはその薬学的に許容される塩もしくはエステルの有効量を投与する段階を含む。
【0010】
別の局面において、本発明は、被験者における癲癇誘発を抑制する方法を提供する。該方法は、それを必要とする被験者に、癲癇誘発が抑制されるように、下記式
【化2】
(式中、点線は、選択的な単結合/二重結合(EまたはZ立体配置の)を示し;Aは、生理的pHにおいて陰性基であり;R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、もしくはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、環の原子数が3〜7である非置換または置換の複素環を形成しており;R4およびR5は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、アルコキシ、アリールオキシ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニルであるか;または、R4およびR5は、一緒に、環の原子数が5〜15である置換または非置換の炭素環または複素環を形成している)
で示される化合物;またはその薬学的に許容される塩もしくはエステルの有効量を投与する段階を含む。
【0011】
別の局面において、本発明は、被験者における痙攣疾患を抑制する方法を提供する。該方法は、痙攣疾患が抑制されるよう、それを必要とする被験者に、β-アミノアニオン化合物はβ-アラニンまたはタウリンではないという条件で、有効量のβ-アミノアニオン化合物を投与する段階を含む。
【0012】
別の局面において、本発明は、下記式:
【化3】
(式中、Aは、生理的pHにおいて陰性基であり;R1は、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、ニトロ、チオール、チオアルキル、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、またはアミノカルボニルであり;R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、もしくはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、環の原子数が3〜7である非置換または置換の複素環を形成している)
で示される抗癲癇誘発化合物;またはその薬学的に許容される塩もしくはエステルを提供し、抗癲癇誘発化合物は抗癲癇誘発活性を有する。好ましい態様において、Aはカルボキシレートを示す。
【0013】
ある好ましい態様において、該化合物は、α-シクロヘキシル-β-アラニン、α-(4-tert-ブチルシクロヘキシル)-β-アラニン、α-(4-フェニルシクロヘキシル)-β-アラニン、α-シクロドデシル-β-アラニン、β-(p-メトキシフェネチル)-β-アラニン、およびβ-(p-メチルフェネチル)-β-アラニン、およびその薬学的に許容される塩からなる群より選択されるか;または、該化合物は、β-(4-トリフルオロメチルフェニル)-β-アラニンおよびβ-[2-(4-ヒドロキシ-3-メトキシフェニル)エチル]-β-アラニン、およびその薬学的に許容される塩からなる群より選択されるか;または、該化合物は、β-(3-ペンチル)-β-アラニンおよびβ-(4-メチルシクロヘキシル)-β-アラニン、およびその薬学的に許容される塩からなる群より選択される。
【0014】
さらに別の局面において、本発明は、下記式
【化4】
(式中、Arは、非置換または置換のアリール基を示し;R6およびR6*は、各々独立的に、水素、アルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yは水素またはチアゾリル、トリアゾリル、およびイミダゾリルからなる群より選択される複素環式部分であり、ただし、Arが非置換フェニル基である場合、R7は水素、メチル、またはフェニルではない)
で示されるジオキサピペラジン化合物;またはその薬学的に許容される塩を提供する。
【0015】
被験者における痙攣疾患を抑制する方法も開示する。有効量の薬剤を、被験者における癲癇誘発および発作発生が抑制されるように、それを必要とする被験者に投与する。該薬剤は、ナトリウムまたはカルシウムイオンチャネルを遮断するか、あるいは、カリウムまたは塩化物イオンチャネルを開口し;少なくとも一つの活性、例えばNMDA受容体拮抗作用、内因性GABA抑制増強、カルシウム結合、鉄結合、亜鉛結合、NOシンターゼ阻害、および抗酸化活性を有する。望ましい態様において、該薬剤は、NMDA受容体に結合することにより、例えばNMDA受容体のグリシン結合部位に結合することによりNMDA受容体に拮抗する、および/または、グリア細胞のGABA取り込みを減少することによりGABA抑制を増強する。
【0016】
別の局面において、本発明は、痙攣疾患を抑制する方法を提供する。該方法は、それを必要とする被験者に、痙攣疾患が抑制されるように、下記式:
【化5】
(式中、Arは、非置換または置換のアリール基を示し;R6およびR6*は、各々独立的に、水素、アルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yは水素または複素環式部分、例えばチアゾリル、トリアゾリル、およびイミダゾリルであり、ただし、Arが非置換フェニル基である場合、R7は水素、メチル、またはフェニルではない)
で示される化合物;またはその薬学的に許容される塩もしくはエステルの有効量を投与する段階を含む。
【0017】
別の局面において、本発明は、下記式
【化6】
(式中、Arは、非置換または置換のアリール基を示し;R6は、水素またはアルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R6*は、抗酸化部分、NMDAアンタゴニスト、NOシンターゼ阻害剤、鉄キレート部分、Ca(II)キレート部分、またはZn(II)キレート部分であってよく;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yは複素環式部分、例えばチアゾリル、トリアゾリル、またはイミダゾリルである)
で示される化合物;またはその薬学的に許容される塩を提供する。好ましい態様において、R6*は、D-α-アミノアジピルである、および/またはR7はメルカプトメチルである。
【0018】
別の局面において、本発明は、それを必要とする被験者に、癲癇誘発が抑制されるように、下記式
【化7】
(式中、Arは、非置換または置換のアリール基を示し;R6は、水素またはアルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R6*は、抗酸化部分、NMDAアンタゴニスト、NOシンターゼ阻害剤、鉄キレート部分、Ca(II)キレート部分、またはZn(II)キレート部分であり得;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yは、チアゾリル、トリアゾリル、およびイミダゾリルからなる群より選択された複素環式部分である)
で示される化合物;またはその薬学的に許容される塩の有効量を投与する段階を含む、癲癇誘発および発作発生を同時に抑制する方法を提供する。
【0019】
別の局面において、本発明は、それを必要とする被験者に、NMDA受容体拮抗作用に関連した疾患が治療されるように、下記式
【化8】
(式中、Arは、非置換または置換のアリール基を示し;R6は、水素またはアルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R6*はNMDAアンタゴニスト部分であり;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yは、チアゾリル、トリアゾリル、およびイミダゾリルからなる群より選択された複素環式部分である)
で示される化合物;またはその薬学的に許容される塩の有効量を投与する段階を含む、NMDA受容体拮抗作用に関連した疾患を治療する方法を提供する。
【0020】
別の局面において、本発明は、下記式
【化9】
(式中、点線は、選択的な単結合/二重結合(EまたはZ立体配置の)を示し;R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、複素環の原子数が3〜7である非置換または置換の複素環を形成しており;R4およびR5は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、カルボキシル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R4およびR5は一緒に環の原子数が5〜15である置換または非置換の炭素環または複素環を形成し;R8は、水素、アルキル、アリール、または有機塩もしくは有機塩形成カチオンである)
で示されるβ-アミノカルボキシル化合物を調製する方法を提供する。該方法は、下記式
【化10】
(式中、点線は、各々、選択的な単結合を示し;Xは、ニトロ、アジド、またはNR2R3であり、ここでのR2およびR3は前記に定義した通りであり;Wは、-CNまたは-COORであり;R4およびR5は前記に定義した通りであり;R8は、水素、アルキル、アリール、または有機塩もしくは無機塩形成カチオンである)
で示される化合物を、還元的脱硫条件下で反応させて、β-アミノカルボキシル化合物を形成する段階を含む。
【0021】
別の局面において、本発明は、下記式
【化11】
(式中、R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、複素環の原子数が3〜7である非置換または置換の複素環を形成しており;R4およびR5は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、アルコキシ、アリールオキシ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニルであるか;または、R4およびR5は、一緒に、環の原子数が5〜15である置換または非置換の炭素環または複素環を形成しており;R8は、水素、アルキル、アリール、または有機塩もしくは無機塩形成カチオンである)
で示されるβ-アミノカルボキシル化合物を調製する方法を提供する。該方法は、下記式
【化12】
(式中、点線は、各々、選択的な単結合/二重結合を示し;Xは、ニトロ、アジド、またはNR2R3であり、R2およびR3は前記に定義した通りであり;Wは-CNまたは-COOR8であり;R8は、水素、アルキル、アリール、または有機塩もしくは無機塩形成カチオンであり;R4およびR5は前記に定義した通りである)
で示される化合物を、還元的脱硫条件下で反応させ、前記式のβ-アミノカルボキシル化合物を形成する段階を含み、ただし、Wが-CNである場合、該方法はさらに酸性化段階を含む。
【0022】
本発明はまた、必要とする被験者に、被験者における癲癇誘発が抑制されるように、式A-Bにより示される有効量の薬剤を投与することを含む、被験者における癲癇誘発および発作発生を抑制する方法を提供し、ここでのAは、ナトリウムまたはカルシウムイオンチャネル遮断活性を有するドメインであるか、あるいは、Aは、カリウムまたは塩化物チャネル開口活性を有し;Bは、少なくとも一つの活性、例えばNMDA受容体拮抗作用;内因性GABA抑制増強、カルシウム結合、鉄結合、亜鉛結合、NOシンターゼ阻害、および抗酸化活性を有するドメインである。好ましい態様において、薬剤のドメインAおよびBは共有結合している。好ましい態様において、Aはジオキサピペラジン部分である。
【0023】
さらに別の局面において、本発明は、それを必要とする被験者に、癲癇誘発が抑制されるように、下記式
【化13】
(式中、Aは生理的pHにおいて陰性基であり;R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、複素環の原子数が3〜7である非置換または置換の複素環を形成しており;R4およびR5は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、アルコキシ、アリールオキシ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニルであるか;または、R4およびR5は、一緒に、環の原子数が5〜15である置換または非置換の炭素環または複素環を形成している)
で示される化合物;またはその薬学的に許容される塩もしくはエステルの有効量を投与する段階を含む、癲癇誘発を抑制する方法を提供する。
【0024】
被験者における神経学的状態を抑制する方法には、それを必要とする被験者に、被験者における神経学的状態が抑制されるように、NMDA受容体に拮抗し、内因性GABA抑制を増強する薬剤の有効量を投与する段階が含まれる。神経学的状態は、例えば、卒中、アルツハイマー病、癌、および神経変性性疾患であり得る。
【0025】
β-アリール-β-アラニン化合物の調製法を提示し、これには、アリールアルデヒドをマロネート化合物およびアンモニウム化合物と、β-アリール-β-アラニン化合物が形成されるような条件下で反応させる段階が含まれる。
【0026】
癲癇誘発を抑制する他の方法には、それを必要とする被験者に、癲癇誘発が抑制されるように、下記式
【化14】
(式中、R9およびR10は、各々独立的に、水素、アルキル、アルケニル、アルキニル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、チオール、アルキルチオール、ニトロ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、およびアミノカルボニルであり得るか;または、R9およびR10は、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成し;R11は、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R10およびR11は、それらがそれぞれ結合している炭素原子および窒素原子と共に連結して、環の員数が4〜8である複素環を形成し;R12は、水素、アルキル、アリール、および炭水化物からなる群より選択される)
で示される化合物;またはその薬学的に許容される塩もしくはエステルの有効量を投与する段階を含む。
【0027】
別の局面において、癲癇誘発を抑制する方法には、それを必要とする被験者に、癲癇誘発が抑制されるように、下記式:
【化15】
(式中、R9a、R9b、R10a、R10bは、各々独立的に、水素、アルキル、アルケニル、アルキニル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、チオール、アルキルチオール、ニトロ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、およびアミノカルボニルであり得るか;または、R9aおよびR9bは、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成しているか;または、R10aおよびR10bは、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成しているか;または、R9aおよびR9bの一方は、R10aおよびR10bの一方と、それらが結合している2炭素単位と共に連結して、環の員数が4〜8である炭素環または複素環を形成し;R11は、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R10aおよびR10bの一方が、R11と、それらがそれぞれ結合している炭素原子および窒素原子と共に連結して、環の員数が4〜8である複素環を形成し;R12は、水素、アルキル、アリール、および炭水化物(例えば、糖、例えばリボースまたはデオキシリボース)からなる群より選択される)
で示される化合物;またはその薬学的に許容される塩もしくはエステルの有効量を投与する段階が含まれる。
【0028】
被験者における癲癇誘発を予防および/または抑制できる化合物を同定するためのファルマコフォアモデリング法は、本発明の一部であり、癲癇誘発に関与するタンパク質または分子に対して直接的または間接的薬理作用を引き起こすことが知られる二つ以上の化合物の構造の検証を特色とする。癲癇誘発に関与するこれらのタンパク質および分子には、細胞表面受容体分子(例えばNMDA受容体)または神経伝達物質輸送に関与する分子(例えばGABA輸送体)が含まれる。好ましくは、これらの化合物の構造には各々、化合物の薬理作用の少なくともいくつかを示すことのできる、一つまたは複数のファルマコフォアが含まれる。本発明の方法にはまた、二つ以上の化合物のファルマコフォア構造に基づいた平均的なファルマコフォア構造(例えば炭素骨格構造および/または3次元空間充填構造)の決定が含まれる。一つまたは複数の平均的なファルマコフォア構造を有する新規化合物を、実施例1に示したようなこれらの方法を使用して選択できる。
【0029】
関連した態様において、これらの方法は、癲癇誘発に関与する二つ以上のタンパク質または分子に対して直接的または間接的な薬理作用を引き起こすことが知られる、二つ以上の化合物の構造の検証を特徴とする。選択する新規化合物は、好ましくは、癲癇誘発に関与する異なるタンパク質または分子に対して活性である、一つ以上のファルマコフォアを有する。
【0030】
好ましい態様において、本発明の方法により選択される(例えば設計される)新規化合物は、被験者における癲癇誘発を抑制する。本発明のさらに他の目的は、卒中、アルツハイマー病、および神経変性性疾患の治療用の化合物および方法を提供することである。本発明のさらに他の目的は、新規の抗痙攣剤を提供することである。本発明のさらに他の目的は、卒中および疼痛を治療する化合物および方法を提供することである。本発明のこれらおよびその他の目的、特徴、および利点は、以下の記載および特許請求の範囲から明らかである。
【0031】
発明の詳細な説明
本発明は、癲癇および痙攣疾患の治療、癲癇誘発の抑制、および発作発生の抑制に有用な方法および薬剤;ならびに、本発明の抗痙攣剤および抗癲癇誘発剤の調製法に関する。本発明はさらに、痙攣疾患治療のための薬学的組成物、および本発明の抗痙攣化合物を含むキットにも関する。
【0032】
定義
便宜上、明細書、実施例および添付の特許請求の範囲に使用される特定の用語をここに集める。
【0033】
「癲癇誘発に関連した経路中の一過程」という用語には、第1相または第2相の癲癇誘発中に起こり、組織、すなわち中枢神経系(CNS)、例えば脳の組織における癲癇誘発変化をもたらす、生化学的過程または事象が含まれる。癲癇誘発に関連した経路中の過程の例は、下記に、より詳細に考察する。
【0034】
「NMDA受容体拮抗作用に関連した疾患」という用語には、NMDA受容体の異常(例えば過剰)活性を、NMDA受容体の拮抗作用により治療できる、被験者の疾患が含まれる。癲癇は、過剰なNMDA系の活性に関連した疾患である。過剰なNMDA系の活性に関連した疾患のその他の非制限的な例には、疼痛、卒中、不安、精神分裂病、その他の精神病、脳虚血、ハンチントン舞踏病、運動神経疾患、アルツハイマー病、AIDS痴呆、ならびに、過剰なNMDA受容体活性が疾患の少なくとも原因の一部であるその他の疾患(ヒトおよび動物における)が含まれる。例えば、Schoeppら、Eur J.Pharmacol.203:237-243(1991);Leesonら、J.Med.Chem 34:1243-1252(1991);Kulagowskiら、J.Med.Chem.37:1402-1405(1994);Mallamoら、J.Med.Chem.37:4438-4448(1994);およびそれに引用されている文献を参照されたい。
【0035】
「痙攣疾患」という用語には、被験者が、痙攣、例えば癲癇発作に起因する痙攣に患っている、疾患が含まれる。痙攣疾患には、癲癇および非癲癇性痙攣、例えば、被験者への痙攣剤の投与に起因する痙攣が含まれるがこれに限定されない。
【0036】
「癲癇誘発抑制」という用語には、癲癇誘発過程の予防、遅延、停止、または回復が含まれる。
【0037】
「抗癲癇誘発剤」という用語には、薬剤を被験者に投与した場合に、癲癇誘発を抑制できる薬剤が含まれる。
【0038】
「抗痙攣剤」という用語には、薬剤を被験者に投与した場合に、発作発生を抑制(例えば予防、遅延、停止、または回復)できる薬剤が含まれる。
【0039】
「ファルマコフォア」という用語は当技術分野において既知であり、これには、選択した生化学的作用、例えば酵素の阻害、受容体への結合、イオンのキレート形成などを果たすことのできる分子部分が含まれる。選択したファルマコフォアは、二つ以上の生化学的作用を有することができ、例えば、ある酵素の阻害剤であり、同時に第二酵素のアゴニストであることができる。治療剤は、同じまたは異なる生化学活性を有することのできる、一つ以上のファルマコフォアを含むことができる。当業者は、類似の構造および/または特性(例えば生物学的作用)を有する多くのファルマコフォアを合わせて、最適化または「平均ファルマコフォア」構造を予測または設計し得ることを認識していると考えられる。このような平均的なファルマコフォア構造は、個々のファルマコフォアを使用して平均的な構造を作製するよりも望ましいレベルの生物学的作用を与え得る。
【0040】
「陰性基」は、生理的pHにおいて負に荷電した基を意味する。好ましい陰性基には、カルボキシレート、サルフェート、スルホネート、スルフィネート、スルファメート、テトラゾリル、ホスフェート、ホスホネート、ホスフィネート、またはホスホロチオエートもしくはその機能的等価体が含まれる。陰性基の「機能的等価体」という用語には、生物学的等価体、例えばカルボキシレート基の生物学的等価体が含まれる。生物学的等価体には、古典的な生物学的に等価な同等体および非古典的な生物学的に等価な同等体の両方が含まれる。古典的および非古典的な生物学的等価体は当技術分野において既知である。例えば、Silverman,R.B. The Organic Chemistry of Drug Design and Drug Action、アカデミックプレス社:CA州サンディエゴ所在、1992、19〜23頁を参照されたい。特に好ましい陰性基はカルボキシレートである。
【0041】
「β-アミノアニオン化合物」という用語には、2炭素スペーサー単位により陰性基から離れている、例えば-NRaRb(RaおよびRbは、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、もしくはアリールオキシカルボニルであり得るか、または、RaおよびRbは、それらが結合している窒素原子と共に、環の原子数が3〜8である環式部分を形成している)などの、アミノ基を有する化合物が含まれる。従って、例えば、β-アミノアニオン化合物は、亜構造式A-C-C-NRaRb(Aは陰性基である)により示すことができる。好ましいβ-アミノアニオン化合物には、β-アミノ酸およびその類似体が含まれる。ある好ましい態様において、β-アミノアニオン化合物はβ-アラニンまたはタウリンではない。
【0042】
「還元的脱硫」という用語は当技術分野において既知であり、化合物から硫黄を還元的に除去する過程を意味する。還元的脱硫条件は当技術分野において既知であり、例えば、TiCl4/LiAlH4またはラネーニッケル/H2による処理が含まれる。一般に、Kharash,NおよびMeyers,C.Y.「The Chemistry of Organic Sulfur Compounds」Pergamon Press、ニューヨーク(1996)第2巻を参照されたい。
【0043】
「被験者」という用語は当技術分野において既知であり、恒温動物、より好ましくは、類人猿、サルおよびヒトの他に、ヒト以外の動物(例えば、ラット、マウス、ネコ、イヌ、ヒツジ、ウマ、ウシ)を含む哺乳動物を意味する。好ましい態様において、被験者はヒトである。
【0044】
特記しない限り、本発明の化学基は、置換されていても非置換でもよい。さらに、特記しない限り、この化学的置換基も置換されていても非置換でもよい。さらに、複数の置換基が、化学基または置換基上に存在してもよい。置換基の例には、アルケニル、アルキニル、ハロゲン、ヒドロキシル、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、カルボキシル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルアミノカルボニル、ジアルキルアミノカルボニル、アルキルチオカルボニル、アルコキシル、ホルミル、トリメチルシリル、ホスフェート、ホスホナート、ホスフィナート、シアノ、アミノ(アルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、およびアルキルアリールアミノを含む)、アシルアミノ(アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイルおよびウレイドを含む)、アミド、イミノ、スルフヒドリル、アルキルチオ、アリールチオ、チオカルボキシレート、サルフェート、アルキルスルフィニル、スルホナート、スルファモイル、スルホンアミド、ニトロ、トリフルオロメチル、シアノ、アジド、ヘテロサイクリル、アルキルアリール、および芳香族部分もしくはへテロ芳香族部分が含まれる。
【0045】
「アルキル」という用語は、直鎖アルキル基、分岐鎖アルキル基、シクロアルキル、ヘテロサイクリル、シクロアルキル(脂環式)基、アルキルで置換されたシクロアルキル基、およびシクロアルキルで置換されたアルキル基を含む、飽和脂肪族基を意味する。好ましい態様において、直鎖または分岐鎖アルキルは、その骨格に30以下の炭素原子を有し(例えば直鎖ではC1〜C30、分岐鎖ではC3〜C30)、より好ましくは骨格に20以下の炭素原子を有する。同様に、好ましいシクロアルキルは、その環構造に4〜10個の炭素原子、より好ましくは環構造に5、6または7個の炭素を有する。
【0046】
さらに、アルキル(例えばメチル、エチル、プロピル、ブチル、ペンチル、ヘキシルなど)は、「非置換アルキル」および「置換アルキル」の両方を含み、後者は、炭化水素骨格の一つ以上の炭素上の水素が置換基で置換されているアルキル部分を意味する。このような置換基には、例えば、ハロゲン、ヒドロキシル、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、カルボキシレート、アルキルカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルチオカルボニル、アルコキシル、ホスフェート、ホスホナート、ホスフィナート、シアノ、アミノ(アルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、およびアルキルアリールアミノを含む)、アシルアミノ(アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイル、およびウレイドを含む)、アミジノ、イミノ、スルフヒドリル、アルキルチオ、アリールチオ、チオカルボキシレート、サルフェート、スルホナート、スルファモイル、スルホンアミド、ニトロ、トリフルオロメチル、シアノ、アジド、ヘテロサイクリル、または芳香族部分もしくはヘテロ芳香族部分が含まれ得る。炭化水素鎖上の部分は、それ自体、適宜、置換されていてもよいことが当業者には理解される。シクロアルキルはさらに、例えば、前記した置換基で置換されていてもよい。「アラルキル」部分は、アリールで置換されたアルキルである(例えばフェニルメチル(すなわちベンジル))。
【0047】
「アリール」という用語は、0〜4個のヘテロ原子を含み得る5員環および6員環の単環芳香族基、例えば、ベンゼン、ピロール、フラン、チオフェン、イミダゾール、オキサゾール、チアゾール、トリアゾール、ピラゾール、ピリジン、ピラジン、ピリダジン、およびピリミジンなどが含まれる。アリール基にはまた、多環縮合芳香族基、例えばナフチル、キノリル、インドリルなどが含まれる。環構造にヘテロ原子を有するこのようなアリール基はまた、「アリール複素環」、「ヘテロアリール」、または「ヘテロ芳香族」とも称し得る。芳香族環(例えばフェニル、インドール、チオフェン)は、前記したような置換基(例えば、ハロゲン、ヒドロキシル、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、カルボキシレート、アルキルカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルチオカルボニル、アルコキシル、ホスフェート、ホスホナート、ホスフィナート、シアノ、アミノ(アルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、およびアルキルアリールアミノを含む)、アシルアミノ(アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイル、およびウレイドを含む)、アミジノ、イミノ、スルフヒドリル、アルキルチオ、アリールチオ、チオカルボキシレート、サルフェート、スルホナート、スルファモイル、スルホンアミド、ニトロ、トリフルオロメチル、シアノ、アジド、ヘテロサイクリル、または芳香族部分もしくはヘテロ芳香族部分)により、一つ以上の環位置において置換されていてもよい。アリール基はまた、脂環式または芳香族でない複素環式環と縮合または架橋して、テトラリンなどの多環を形成できる。
【0048】
「アルケニル」および「アルキニル」という用語には、前記したアルキルに対して、長さおよびあり得る置換基の点から、不飽和脂肪族基の類似体が含まれるが、それぞれ少なくとも一つの二重結合または三重結合および少なくとも二つの隣接炭素原子が含まれる。
【0049】
明細書および図面に使用したような「選択的な単結合/二重結合」は、点線を伴う実線で示され、適宜EまたはZ立体配置の単結合または二重結合であり得る二つの炭素原子の間の共有結合を意味する。例えば、下記構造はシクロヘキサンまたはシクロヘキセンを示すことができる:
【化16】
【0050】
炭素数を特記しない限り、「低級アルキル」とは、前記したようなアルキル基を意味するが、1〜10個の炭素原子、より好ましくは1〜6個の炭素原子をその骨格構造に有する。同様に、「低級アルケニル」および「低級アルキニル」は、類似した鎖長を有する。好ましいアルキル基は低級アルキルである。
【0051】
「ヘテロサイクリル」または「複素環式基」という用語は、3〜10員環、より好ましくは4〜7員環構造を意味し、環構造には、一つ以上のヘテロ原子、例えば2、3、または4個のヘテロ原子が含まれる。複素環式基は、ピロリジン、オキソラン、チオラン、ピペリジン、ピペラジン、モルホリン、ラクトン、ラクタム、例えばアゼチジノンおよびピロリジノン、スルタム、スルトンなどが含まれる。複素環式環は、一つ以上の位置において、前記したような置換基(ハロゲン、ヒドロキシル、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、カルボキシレート、アルキルカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルチオカルボニル、アルコキシル、ホスフェート、ホスホナート、ホスフィナート、シアノ、アミノ(アルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、およびアルキルアリールアミノを含む)、アシルアミノ(アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイル、およびウレイドを含む)、アミジノ、イミノ、スルフヒドリル、アルキルチオ、アリールチオ、チオカルボキシレート、サルフェート、スルホナート、スルファモイル、スルホンアミド、ニトロ、トリフルオロメチル、シアノ、アジド、ヘテロサイクリル、または芳香族もしくはヘテロ芳香族部分を含む)で置換されていてもよい。
【0052】
「ポリサイクリル」または「多環式基」という用語は、二つ以上の炭素が二つの隣接している環に共通している、例えば環が「縮合環」である、二つ以上の環式環(例えばシクロアルキル、シクロアルケニル、シクロアルキニル、アリールおよび/または複素環)を意味する。隣接していない原子により連結している環は、「架橋」環と称される。多環の各々の環は、前記したような置換基、例えば、ハロゲン、ヒドロキシル、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、カルボキシレート、アルキルカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルチオカルボニル、アルコキシル、ホスフェート、ホスホナート、ホスフィナート、シアノ、アミノ(アルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、およびアルキルアリールアミノを含む)、アシルアミノ(アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイル、およびウレイドを含む)、アミジノ、イミノ、スルフヒドリル、アルキルチオ、アリールチオ、チオカルボキシレート、サルフェート、スルホナート、スルファモイル、スルホンアミド、ニトロ、トリフルオロメチル、シアノ、アジド、ヘテロサイクリル、または芳香族部分もしくはヘテロ芳香族部分)により置換されていてもよい。
【0053】
本明細書に使用したような「ヘテロ原子」という用語は、炭素または水素以外のあらゆる元素の原子を意味する。好ましいヘテロ原子は、窒素、酸素、硫黄およびリンである。
【0054】
本明細書に使用したような「アリールアルデヒド」という用語は、式Ar-C(O)Hにより示される化合物を意味し、ここでのArは、アリール部分(前記したような)であり、-C(O)Hはホルミル基またはアルデヒド基である。好ましい態様において、アリールアルデヒドは、(置換または非置換)ベンズアルデヒドである。多種多様なアリールアルデヒドが商業的に入手可能であるか、または、市販の前駆体から慣用的な手順により調製することもできる。アリールアルデヒドの調製手順には、Vilsmeier-Haack反応(例えばJutz.Adv.Org.Chem.9、pt.1、225-342(1976)を参照されたい)、Gatterman反応(Truce、Org.React.9、37-72(1957))、Gatterman-Koch反応(Crounse、Org.React.5、290-300(1949))、およびReimer-Tieman反応(WynbergおよびMeijer、Org.React.28、1-36(1982))が含まれる。
【0055】
本発明のいくつかの化合物の構造は、不斉炭素原子を含むことに留意されたい。従って、このような不斉から生じる異性体(例えば全ての鏡像異性体およびジアステレオマー)が、特記しない限り本発明の範囲内に含まれると理解する。すなわち、特記しない限り、あらゆるキラル炭素中心は(R)または(S)立体化学であり得る。このような異性体は、実質的に純粋な形で、古典的な分離技術により、および立体化学的に制御された合成により得ることができる。さらに、アルケンは、適宜、E幾何またはZ幾何を含むことができる。
【0056】
I.痙攣疾患を治療する方法
一つの局面において、本発明は、痙攣疾患(癲癇を含む)を治療する方法を提供する。
【0057】
一つの局面において、本発明は、被験者における癲癇誘発を抑制する方法を提供する。該方法には、それを必要とする被験者に、被験者における癲癇誘発が抑制されるように、癲癇誘発に関連した経路中の一過程を変調する薬剤の有効量を投与する段階が含まれる。
【0058】
前記したように、N-メチル-D-アスパルテート(NMDA)受容体により媒介されるニューロン間の興奮連関のアップレギュレーション、および、γ-アミノ-酪酸(GABA)受容体により媒介されるニューロン間の抑制連関のダウンレギュレートは両方共、癲癇誘発に関与している。癲癇誘発に関連した経路中の他の過程には、癲癇誘発に関与する神経伝達物質である一酸化窒素(NO)遊離;過剰に遊離された場合にニューロンへの傷害を媒介し得るカルシウム(Ca2+)の遊離;過剰な亜鉛(Zn2+)に起因する神経毒性;過剰な鉄(Fe2+)に起因する神経毒性;および酸化的細胞傷害に起因する神経毒性が含まれる。従って、好ましい態様において、癲癇誘発を抑制するために被験者に投与できる薬剤は、好ましくは、癲癇誘発に関連した少なくとも一つの経路中の一つ以上の過程を抑制できる。例えば、癲癇誘発の抑制に有用な薬剤は、脳組織中のNOの遊離を減少できるか、NOの癲癇誘発作用を減弱でき;NMDA受容体に拮抗でき;内因性GABA抑制を増大でき;電位開口型イオンチャネルを遮断でき;Ca2+、Zn2+、またはFe2+を含むカチオンの遊離を減少できるか、該カチオンの遊離濃度を減少できるか(例えばキレート形成により)、またはその他の方法で該カチオンの癲癇誘発作用を低減できる。ある好ましい態様において、癲癇誘発を抑制するために被験者に投与できる薬剤は、癲癇誘発に関連した少なくとも一つの経路中の少なくとも二つの過程を抑制できる。
【0059】
癲癇誘発に関連した経路中の過程を変調できるファルマコフォアの非制限的な例には以下が含まれる:
・NOシンターゼ阻害剤、例えばL-アルギニンおよびそのアルキル化誘導体
・NMDA受容体のアンタゴニスト、例えば(R)-α-アミノ酸。例えば、NMDA受容体阻害剤の一般的総説に関する、Leeson,PおよびIverson,L.L.、J.Med.Chem.(1994)37:4053-4067を参照されたい;
・内因性GABA抑制増強剤、例えばGABAアミノトランスフェラーゼ失活剤、例えばγ-ビニル-GABA。例えば、GABA受容体アゴニストおよびアンタゴニストの総説に関する、Krogsgaard-Larsen,Pら、J.Med.Chem.(1994)37:2489-2505を参照されたい;
・Ca2+、Zn2+、またはFe2+のキレート化剤、例えばEDTA、EGTA、TNTA、2,2-ビピリジン-4,4-ジカルボキシレート、エンテロバクチン、ポルフィリン、クラウンエーテル、アザクラウンエーテルなど;
・抗酸化剤、例えばビタミンCおよびE、カロテノイド、例えばβ-カロテン、ブチル化フェノール、トロロクス(トコフェロール類似体)、セレニウムおよびグルタチオン。
【0060】
一つの好ましい態様において、該薬剤はNMDA受容体に拮抗し、内因性GABA抑制を増強する。ある好ましい態様において、該薬剤は経口投与する。好ましくは、経口投与後に、該薬剤は、能動輸送シャトル機序により被験者の神経系に輸送される。能動輸送シャトルの非制限的な例は、アミノ酸を血液脳関門(BBB)を通過して輸送できる、大きな中性アミノ酸輸送体である。
【0061】
別の態様において、本発明は癲癇誘発を抑制する方法を提供する。該方法には、それを必要とする被験者に、癲癇誘発が抑制されるように、式(式I):
【化17】
(式中、Aは、生理的pHにおいて陰性基であり;R1は、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、またはアミノカルボニルであり;R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、複素環の原子数が3〜7である非置換または置換の複素環を形成している)
で示される化合物;またはその薬学的に許容される塩もしくはエステルの有効量を投与する段階が含まれる。
【0062】
ある態様において、式Iの化合物は、癲癇誘発が抑制されるような、式(式II):
【化18】
(式中、点線は、選択的な単結合を示し;R4およびR5は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、アルコキシ、アリールオキシ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、複素環であるか;または、R4およびR5は、一緒に、環の原子数が5〜15(より好ましくは原子数5〜8)である置換または非置換の炭素環または複素環を形成しており;A、R2およびR3は前記に定義した通りである)
またはその薬学的に許容される塩もしくはエステルにより示すことができる。
【0063】
別の態様において、本発明は、癲癇誘発を抑制する方法を提供する。該方法には、それを必要とする被験者に、癲癇誘発が抑制されるように、式(式III)
【化19】
(式中、A、R2、R3、R4およびR5は前記に定義した通りである)
で示される化合物;またはその薬学的に許容される塩もしくはエステルの有効量を投与する段階が含まれる。好ましい態様において、Aはカルボキシレートである。特に好ましい態様において、Aはカルボキシレートであり、R4は水素であり、R5は(置換または非置換)アリール基である。別の好ましい態様において、R4およびR5は一緒に6員環、例えば2-、3-、または4-アミノ安息香酸、特にアントラニル酸を形成する。
【0064】
別の態様において、本発明は、癲癇誘発を抑制する方法を提供する。該方法には、それを必要とする被験者に、癲癇誘発が抑制されるように、α,α-二置換β-アラニン、α,β-二置換β-アラニン、β,β-二置換β-アラニン、α,β,α-三置換β-アラニン、α,β,β-三置換β-アラニン、α,α,N-三置換β-アラニン、α,β,N-三置換β-アラニン、β,β,N-三置換β-アラニン、α,α,N,N-四置換β-アラニン、α,β,N,N-四置換β-アラニン、β,β,N,N-四置換β-アラニン、α,α,β,β-四置換β-アラニン、α,α,β,N-四置換β-アラニン、α,β,β,N-四置換β-アラニン、α,α,β,N,N-五置換β-アラニン、α,β,β,N,N-五置換β-アラニン、α,α,β,β,N-五置換β-アラニン、α,α,β,β,N,N-六置換β-アラニン(全ての立体異性体を含む);またはその薬学的に許容される塩もしくはエステルを投与する段階が含まれる。
【0065】
被験者に投与する段階には、本発明の抗痙攣化合物および/または抗癲癇誘発化合物へと代謝される化合物の被験者への投与が含まれ得る。例えば、本発明の方法には、インビボで本発明の治療化合物へと変換されるプロドラッグの使用が含まれる。例えば、前記で引用したSilverman、第8章を参照されたい。このようなプロドラッグを使用すると、一般的には血液脳関門を通過しない化合物が血液脳関門を通過できるようになるように体内分布を変化させたり、または治療化合物の薬物動態を変化させることができる。例えば、陰性基(例えばカルボキシレート基)は、エチル基または脂肪基でエステル化して、カルボン酸エステルを生成できる。カルボン酸エステルを被験者に投与する場合、エステルは、酵素的または非酵素的に切断され、陰性基を顕現できる。
【0066】
別の例示的態様において、本発明の方法には、被験者に、ウラシルまたはその類似体(置換ピリミジン、UMPおよびウリジン、またはその類似体を含む)を投与する段階が含まれる。ウラシル化合物またはその代謝物(例えばジヒドロウラシルまたはβ-ウレイドプロピオネート)の投与により、本発明の活性化合物をインビボで形成することができる。従って、好ましい態様において、本発明の方法には、それを必要とする被験者に、例えば、ウラシル、ジヒドロウラシルまたはβ-ウレイドプロピオネート化合物を痙攣疾患を治療または予防するのに有効であるβ-アミノ酸化合物へとインビボで変換することにより、痙攣疾患を治療するおよび/または癲癇誘発を抑制するに有効な量の、有効量の置換または非置換のウラシル、ジヒドロウラシルまたはβ-ウレイドプロピオネート化合物、またはその誘導体もしくは類似体(またはその薬学的に許容される塩もしくはエステル)を投与する段階が含まれ得る。
【0067】
従って、ある態様において、被験者への投与に好ましい化合物には、ピリミジン、例えば置換ウラシル(インビボでβ-アミノアニオン化合物に変換され得る)が含まれる。好ましい態様において、該化合物は、式(式V):
【化20】
(式中、R9およびR10は、各々独立的に、水素、アルキル(シクロアルキル、ヘテロサイクリル、およびアラルキルを含む)、アルケニル、アルキニル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ(非置換および置換のアミノを含む)、ヒドロキシ、チオール、アルキルチオール、ニトロ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、またはアミノカルボニルであり得るか;または、R9およびR10は、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成し;R11は、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R10およびR11は、それらがそれぞれ結合している炭素原子および窒素原子と共に連結して、環の員数が4〜8である複素環を形成し;R12は、水素、アルキル、アリール、および炭水化物(例えば糖、例えばリボースまたはデオキシリボース)からなる群より選択される)
またはその薬学的に許容される塩もしくはエステルにより示すことができる。別の態様において、該化合物は、式(式Va):
【化21】
(式中、R9a、R9b、R10a、R10bは、各々独立的に、水素、アルキル(シクロアルキル、ヘテロサイクリル、およびアラルキルを含む)、アルケニル、アルキニル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ(非置換および置換のアミノを含む)、ヒドロキシ、チオール、アルキルチオール、ニトロ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、およびアミノカルボニルであり得るか;または、R9aおよびR9bは、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成しているか;または、R10aおよびR10bは、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成しているか;または、R9aおよびR9bの一方は、R10aおよびR10bの一方と、それらが結合している2炭素単位と共に連結して、環の員数が4〜8である炭素環または複素環を形成し;R11は、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R10aおよびR10bの一方が、R11と、それらがそれぞれ結合している炭素原子および窒素原子と共に連結して、環の員数が4〜8である複素環を形成し;R12は、水素、アルキル、アリール、および炭水化物(例えば、糖、例えばリボースまたはデオキシリボース)からなる群より選択される)
またはその薬学的に許容される塩もしくはエステルにより示すことができる。
【0068】
ピリミジン化合物、例えば5-フルオロウラシル(5FU)は、抗新生物剤として使用されてきた。5FUおよび類似化合物の抗癌活性は、5FUがチミジル酸シンターゼ(DNA合成に重要な酵素)を阻害する「自殺基質」機序に起因すると考えられている。好ましい態様において、痙攣疾患の治療(癲癇誘発の抑制)のために本発明に従って投与したピリミジン化合物およびジヒドロピリミジン化合物は、チミジル酸シンターゼを有意に阻害しない。理論により限定したくはないが、ピリミジン化合物によるチミジル酸シンターゼ阻害は、ピリミジン環の5位(すなわち式VaのR9)における電気陰性基の存在により増加すると考えられ、それ故、ピリミジン環の5位(すなわち式VaのR9)における非電気陰性基を該化合物に与えることにより減少させることができる。ピリミジン化合物のチミジル酸シンターゼへの結合能を減少させるために、十分な立体的なかさを有する置換基を与えることにより、チミジル酸シンターゼ阻害を減少させることができると考えられている。従って、好ましい態様において、本発明に従って投与する式Vの化合物において、R9は非電気陰性(すなわち中性または電気陽性)基(例えばアルキル、アリールまたはその他)である。好ましい態様において、式VのR9およびR10の少なくとも一方は立体的にかさ高い基(例えば長鎖または分岐アルキル、置換アリール、またはその他)であるか、または、R9およびR10は連結して、炭素環または複素環式環を形成している。
【0069】
本発明に従って使用するピリミジン化合物およびジヒドロピリミジン化合物の非制限的な例を、その活性代謝物の例と共に、図1に示す。
【0070】
置換または非置換のウラシル、またはその誘導体もしくは類似体の使用は、ラットの抗発作モデルで試験した場合に、あるウラシル化合物が抗発作発生特性(のみ)を有することが判明したので、特に有益であり得る。例えば、Medicinal Chemistry、Volume V;W.J.Close、L.Doub、M.A.Spielman;W.H.Hartung著;John Wiley and Sons 1961を参照されたい。従って、プロドラッグ型の化合物(ウラシル)は、抗発作活性を示すことができるが、代謝により生成されたβ-アミノアニオン化合物は抗癲癇誘発活性および/または抗痙攣活性を示すことができる。これらの活性は、独立的におよび組合せて、哺乳動物(ヒトを含む)の痙攣疾患の効果的療法を提供できる。
【0071】
ある態様において、本発明の活性剤は、NMDA受容体のグリシン結合部位に結合することによりNMDA受容体に拮抗する。ある好ましい態様において、該薬剤は、グリア細胞のGABA取り込みを減少させることによりGABA抑制を増強する。ある他の態様において、該薬剤は経口投与する。さらに他の態様において、該方法にはさらに、薬学的に許容されるビヒクル中の薬剤の投与が含まれる。
【0072】
さらに別の態様において、本発明は、痙攣疾患を抑制する方法を提供する。該方法には、それを必要とする被験者に、痙攣疾患が抑制されるように、有効量のβ-アミノアニオン化合物を投与する段階が含まれ;ただし、β-アミノアニオン化合物はβ-アラニンまたはタウリンではない。
【0073】
別の態様において、本発明は、被験者における痙攣疾患および癲癇誘発の両方を抑制する方法を提供する。該方法には、それを必要とする被験者に、被験者における癲癇誘発が抑制されるように、ナトリウムもしくはカルシウムイオンチャネルを遮断するか、または、カリウムもしくは塩化物イオンチャネルを開口し;NMDA受容体拮抗作用、内因性GABA抑制増強、カルシウム結合、鉄結合、NOシンターゼ阻害、および抗酸化活性からなる群より選択される少なくとも一つの活性を有する薬剤の有効量を投与する段階が含まれる。
【0074】
ナトリウムおよび/またはカルシウムイオンチャネル活性の遮断剤は当技術分野において公知であり、本発明の化合物および方法のA部分として使用できる。同様に、カリウムまたは塩化物チャネルを開口するあらゆる化合物を、本発明の化合物および方法におけるA部分として使用できる。NMDA受容体のアンタゴニストおよび内因性GABA抑制増強剤は当業者には公知であり、本発明の方法および化合物に使用できる。例えば、2,3-キノキサリジノンが、NMDA受容体アンタゴニスト活性を有すると報告されている(例えば、米国特許第5,721,234号を参照されたい)。カルシウムおよび亜鉛キレート剤の例には、前記したものの他に、エチレンジアミンテトラ酢酸(EDTA)、エチレングリコールビス(β-アミノエチルエーテル)-N,N,N',N'-テトラ酢酸などの二価カチオンのキレート形成に関して当技術分野において既知である部分が含まれる。鉄キレート剤の例には、エンテロバクチン、ピリドキサールイソニコチニルヒドラゾン、N,N'-ビス(2-ヒドロキシベンゾイル)-エチレンジアミン-N,N'-ジ酢酸(HBED)、および1-置換-2-アルキル-3-ヒドロキシ-4-ピリドン(1-(2’-カルボキシエチル)-2-メチル-3-ヒドロキシ-4-ピリドンを含む)、および鉄にキレートすることが当技術分野において既知である他の部分が含まれる。NOシンターゼ活性を阻害する化合物は当技術分野において既知であり、例えば、Nγ-置換アルギニン類似体、特にL立体配置のもの(L-Nγ-ニトロ-アルギニン(脳NOシンターゼの特異的阻害剤)、L-Nγ-アミノ-アルギニン、およびL-Nγ-アルキル-アルギニン);またはそのエステル、好ましくはメチルエステルが含まれる。抗酸化剤の例には、アスコルビン酸、トコフェロール(α-トコフェロールを含む)などが含まれる。
【0075】
別の態様において、本発明は、痙攣疾患を抑制する方法を提供する。該方法には、それを必要とする被験者に、痙攣疾患が抑制されるように、式(式IV):
【化22】
(式中、Arは、非置換または置換のアリール基を示し;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yはチアゾリル、トリアゾリル、およびイミダゾリルからなる群より選択される複素環式部分であり;R6およびR6*は、各々独立的に、水素、アルキル、アルキルカルボニル、またはアリールカルボニルである)
で示されるジオキサピペラジン(ジケトピペラジンとしても知られる)化合物;またはその薬学的に許容される塩の有効量を投与する段階が含まれる。好ましい態様において、R7は水素、メチルまたはフェニルではない。好ましい態様において、該化合物は、シクロ-D-フェニルグリシル-(S-Me)-L-システインである。ジオキサピペラジンの合成については、例えば、Kopple,K.D.ら、J.Org.Chem.33:862(1968);Slater,G.P.Chem.Ind.(ロンドン)32:1092(1969);Grahl-Nielsen,O. Tetrahedron Lett.1969:2827(1969)を参照されたい。選択したジオキサピペラジン化合物の合成は下記の実施例に記載する。
【0076】
別の態様において、本発明は、癲癇誘発および発作発生を同時に抑制する方法を提供し、該方法には、それを必要とする被験者に、癲癇誘発が抑制されるように、下記式:
【化23】
(式中、Arは、非置換または置換のアリール基を示し;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yはチアゾリル、トリアゾリル、およびイミダゾリルからなる群より選択される複素環式部分であり;R6は水素またはアルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R6*は、抗酸化部分、NMDAアンタゴニスト、NOシンターゼ阻害剤、鉄キレート部分、Ca(II)キレート部分、Zn(II)キレート部分、および抗酸化部分からなる群より選択される)
で示される化合物;またはその薬学的に許容される塩の有効量を投与する段階が含まれる。ある態様において、R7は、水素、メチル、またはフェニルではない。
【0077】
別の態様において、本発明は、NMDA受容体アンタゴニスト作用に関連した疾患を治療する方法を提供する。該方法には、それを必要とする被験者に、NMDA受容体アンタゴニスト作用に関連した疾患が治療されるように、下記式:
【化24】
(式中、Arは、非置換または置換のアリール基を示し;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yはチアゾリル、トリアゾリル、およびイミダゾリルからなる群より選択される複素環式部分であり;R6は水素またはアルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R6*は、NMDAアンタゴニスト部分である)
で示される化合物;またはその薬学的に許容される塩を投与する段階が含まれる。ある態様において、R7は、水素、メチル、またはフェニルである。
【0078】
さらに別の態様において、本発明は、被験者における発作発生および癲癇誘発を抑制する方法を提供する。該方法には、それを必要とする被験者に、被験者における癲癇誘発が抑制されるように、式A-Bにより示される薬剤の有効量を投与する段階が含まれ、ここでのAは、ナトリウムイオンチャネル遮断活性を有するドメインであり;Bは、NMDA受容体拮抗作用、GABA抑制増強、カルシウム結合、鉄結合、亜鉛結合、NOシンターゼ阻害、および抗酸化活性からなる群より選択される少なくとも一つの活性を有するドメインである。ある好ましい態様において、薬剤のドメインAおよびB(例えばファルマコフォア)は共有結合している。ある好ましい態様において、Aはジオキサピペラジン部分、フェニトイン部分、またはカルバマゼピン部分である。
【0079】
別の態様において、本発明は、被験者における発作発生および癲癇誘発を抑制する方法を提供する。該方法には、それを必要とする被験者に、被験者における癲癇誘発が抑制されるように、式A-Bにより示される薬剤の有効量を投与する段階が含まれ、ここでのAは、抗発作発生活性を有するドメインであり;Bは、NMDA受容体拮抗作用、GABA抑制増強;カルシウム結合;鉄結合;亜鉛結合;NOシンターゼ阻害;および抗酸化活性からなる群より選択された少なくとも一つの活性を有するドメインである。ある好ましい態様において、薬剤のドメインAおよびB(例えばファルマコフォア)は共有結合している。ある好ましい態様において、Aはジオキサピペラジン部分、フェニトイン部分、またはカルバマゼピン部分である。
【0080】
本発明に記載のハイブリッド薬物は、抗発作発生部分と、抗癲癇誘発部分を、好ましくはアミド結合またはエステル結合などの共有結合を介して接続することによる、二機能性分子であり得る。該連鎖は、選択的に、インビボで切断され得る。該連鎖は、A部分とB部分の間に屈曲性または十分な空間を与えるリンカー部分またはスペーサー部分を含んでいてもよく、これによりAおよびBが結合するまたはAおよびBが相互作用するそれぞれの部分との相互作用が可能となる。リンカーの例には、アミノ基含有AおよびB部分を連結するための二酸(例えばアジピン酸);または例えばカルボキシル基含有AおよびB部分を連結するためのジアミン(例えば1,6-ヘキサンジアミン);または、アミノ官能基化B部分をカルボキシ官能基化A部分に連結(またはその逆)するためのアミノ酸が含まれる。リンカーは、当業者に公知の考慮に従って所望の特性を与えるように選択できる。よって、二機能性分子は発作発生および癲癇誘発の両方を標的化する。当業者は、ハイブリッド薬物が、一つ以上の所望の平均的なファルマコフォアを含み得ることを認識していると考えられる。
【0081】
別の態様において、被験者における癲癇誘発および/または発作発生を抑制する方法には、癲癇誘発が抑制されるように、有効量の化合物を被験者に投与することが含まれ、ここでの化合物は、式A:
【化25】
(式中、R1は、水素、アルキル、アルケニル、アルキニル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R2はアルキル、アルケニル、アルキニル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;Aは生理的pHにおいて陰性基である)
およびその薬学的に許容される塩もしくはエステルである。
【0082】
式Aの好ましい態様において、Aはカルボン酸またはエステルである。式Aの別の好ましい態様において、R1は水素である。式Aのさらに別の好ましい態様において、R2はアルキル、例えばアリールアルキル、例えばフェニルアルキルである。
【0083】
式Aの化合物の例には、
【化26】
【化27】
【化28】
【化29】
【化30】
【化31】
およびその薬学的に許容される塩もしくはエステルが含まれる。
【0084】
別の態様において、被験者における癲癇誘発および/または発作発生を抑制する方法には、被験者に、癲癇誘発が抑制されるように、有効量の化合物を投与する段階が含まれ、ここでの該化合物は、式B:
【化32】
(式中、Aは生理的pHにおいて陰性基であり;Bはフェノキシで置換されたフェニル基である)
およびその薬学的に許容される塩もしくはエステルである。
【0085】
式Bの好ましい態様において、Aはカルボキシル基である。式Bの好ましい態様において、Bはアルキルフェノキシで置換されたフェニル基、例えばメチルフェノキシで置換されたフェニル基、またはハロフェノキシで置換されたフェニル基、例えばクロロフェノキシで置換されたフェニル基である。式Bの好ましい化合物は、以下に例示したような、単一の立体異性体である。
【0086】
式Bの化合物の例には
【化33】
【化34】
【化35】
およびその薬学的に許容される塩もしくはエステルが含まれる。
【0087】
式Bの化合物のさらに他の好ましい態様を、表5および以下に提示する:
【化36】
【化37】
【0088】
別の態様において、被験者における癲癇誘発および/または発作発生を抑制する方法には、被験者に、癲癇誘発が抑制されるように、有効量の化合物を投与することが含まれ、ここでの化合物は、式C:
【化38】
(式中、Aは生理的pHにおいて陰性基であり;Dは、二つ以上のアルコキシ部分またはアリールオキシ部分で置換されたアリール基である)
およびその薬学的に許容される塩もしくはエステルである。
【0089】
式Cの好ましい態様において、Aはカルボキシル基である。式Cの別の好ましい態様において、Dは、二つ以上のアルコキシ部分またはアリールオキシ部分で置換されたフェニル基である。式Cの別の好ましい態様において、Dは、二つ以上のアルコキシ(例えばメトキシ)基で置換されたフェニル基である。
【0090】
式Cの化合物の例には
【化39】
【化40】
【化41】
およびその薬学的に許容される塩が含まれる。
【0091】
別の態様において、被験者における癲癇誘発および/または発作発生を抑制する方法には、被験者に、癲癇誘発が抑制されるように、有効量の化合物を投与する段階が含まれ、ここでの化合物は、式D
【化42】
(式中、Aは生理的pHにおいて陰性基であり;mおよびnは1〜3であり;Eは、置換または非置換のフェニルである)
およびその薬学的に許容される塩もしくはエステルである。
【0092】
式Dの好ましい態様において、Aはカルボキシル基である。式Dの別の好ましい態様において、nは1であり、Eはジフェニルで置換されたメチルである。
【0093】
式Dの化合物の例には
【化43】
【化44】
【化45】
【化46】
およびその薬学的に許容される塩もしくはエステルが含まれる。
【0094】
さらに別の態様において、被験者における癲癇誘発および/または発作発生を抑制する方法には、癲癇誘発が抑制されるように、有効量の化合物を被験者に投与する段階が含まれ、ここでの化合物は、式E
【化47】
(式中、R13は、水素、アルキル、アリール、または有機塩もしくは無機塩形成カチオンであり;nは1〜5であり;tは1〜2(好ましい)であり;各Xは、独立的に、ハロゲン、ニトロ、シアノ、および置換もしくは非置換のアルキル基およびアルコキシ基からなる群より選択される)
およびその薬学的に許容される塩もしくはエステルである。
【0095】
式Eの好ましい態様において、R13は水素であり、tは2である。
【0096】
式Eの好ましい化合物の例には、以下が含まれる:
3-アミノ-3-(4-ニトロフェニル)プロピオン酸
【化48】
3-アミノ-3-(4-メチルフェニル)-2-カルボキシプロピオン酸
【化49】
3-アミノ-3-(4-メトキシフェニル)-2-カルボキシプロピオン酸
【化50】
3-アミノ-3-(4-ニトロフェニル)-2-カルボキシプロピオン酸
【化51】
【0097】
本発明の治療法に有用である化合物は、慣用的なスクリーニングアッセイ法により決定できる。例えば、下記の実施例2に記載した第1相癲癇誘発動物モデルを使用して、特定の化合物が、第1相癲癇誘発に対して抗癲癇誘発活性を示すかどうかを決定できる。慢性的な癲癇誘発は、ラットにおいて、Silverら(Ann.Neurol.(1991)29:356)により記載されたキンドリングアッセイ法(およびこれでスクリーニングした候補化合物)を用いてモデリングできる。同様に、抗痙攣剤として有用な化合物を、従来の動物モデル、例えばHorton,R.W.ら、Eur.J.Pharmacol.(1979)59:75-83に記載のマウスモデルでスクリーニングできる。例えば受容体または酵素の結合または阻害に有用な化合物またはファルマコフォアは、当業者に既知の従来の方法に従ってスクリーニングできる。例えば、GABA取り込み受容体への結合は、Schlewer(Schlewer,J.ら、J.Med.Chem.(1991)34:2547)により改変されたRamseyらの方法により定量できる。NMDA受容体上のグリシン部位への結合は、例えば、Kemp,Aら、Proc.Natl.Acad.Sci.USA(1988)85:6547に記載の方法に従って定量できる。電位開口型Na+チャネルに対する作用は、ラット海馬片における電位固定アッセイ法で評価できる。
【0098】
マウスまたはラットにおける抗痙攣活性および/または抗癲癇誘発活性について候補化合物をスクリーニングするのに適したアッセイ法を、下記の実施例4および5に記載する。
【0099】
II.化合物および化合物を同定するための方法
別の局面において、本発明は、癲癇および痙攣疾患を治療するのに有用な化合物を提供する。
【0100】
一つの態様において、本発明は、式(式I)
【化52】
(式中、Aは、生理的pHにおいて陰性基であり;R1は、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、またはアミノカルボニルであり;R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、複素環の原子数が3〜7である非置換または置換の複素環を形成している)
で示される抗癲癇誘発化合物(ここでの抗癲癇誘発化合物は、抗癲癇誘発活性を示す);またはその薬学的に許容される塩もしくはエステルを提供する。
【0101】
ある好ましい態様において、Aはカルボキシレートを示す。ある好ましい態様において、該化合物は、α-シクロヘキシル-β-アラニン、α-(4-tert-ブチルシクロヘキシル)-β-アラニン、α-(4-フェニルシクロヘキシル)-β-アラニン、α-シクロドデシル-β-アラニン、β-(p-メトキシフェネチル)-β-アラニン、β-(p-メチルフェネチル)-β-アラニン、およびその薬学的に許容される塩からなる群より選択される。他の好ましい態様において、該化合物は、β-(4-トリフルオロメチルフェニル)-β-アラニンおよびβ-[2-(4-ヒドロキシ-3-メトキシフェニル)エチル]-β-アラニンおよびその薬学的に許容される塩からなる群より選択される。さらに他の態様において、該化合物は、β-(3-ペンチル)-β-アラニンおよびβ-(4-メチルシクロヘキシル)-β-アラニンおよびその薬学的に許容される塩からなる群より選択される。
【0102】
別の態様において、本発明は、式(式IV)
【化53】
(式中、Arは、非置換または置換のアリール基を示し;R7は、水素、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yは水素またはチアゾリル、トリアゾリル、およびイミダゾリルからなる群より選択される複素環式部分であり;R6およびR6*は、各々独立的に、水素、アルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルからなる群より選択される)
で示されるジオキサピペラジン化合物またはその薬学的に許容される塩を提供する。ある好ましい態様において、Ar基が結合している炭素原子は「D」または「R」立体化学配置を有する。ある態様において、Arは非置換または置換のフェニル基である。ある態様において、Yは水素である。ある好ましい態様において、R6およびR6*の少なくとも一方は、抗酸化部分、NMDAアンタゴニスト、NOシンターゼ阻害剤、鉄キレート部分、Ca(II)キレート部分、およびZn(II)キレート部分からなる群より選択される。ある好ましい態様において、R7はメチルまたはメルカプトメチルである。
【0103】
ある好ましい態様において、R6およびR6*は、両方共に水素である。ある特に好ましい態様において、該化合物は、シクロフェニルグリシル-2-(アミノ-3-メルカプトブタン酸)、より好ましくはシクロ-D-フェニルグリシル-L-[2-(アミノ-3-メルカプトブタン酸)]である。関連した態様において、該化合物は、シクロ-D-フェニルグリシル-(S-Me)-L-システインである。いくつかの好ましい態様において、Arは非置換フェニル基である。ある態様において、R7は、水素、メチル、またはフェニルではない。
【0104】
別の態様において、本発明は、式(式IV)
【化54】
(式中、Arは、非置換または置換のアリール基を示し;R7は、アルキル、メルカプトアルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、シアノ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、または-(CH2)n-Yであり、ここでのnは1〜4の整数であり、Yは水素またはチアゾリル、トリアゾリル、およびイミダゾリルからなる群より選択される複素環式部分であり;R6は、水素またはアルキル、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;R6*は、抗酸化部分、NMDAアンタゴニスト、NOシンターゼ阻害剤、鉄キレート部分、Ca(II)キレート部分、およびZn(II)キレート部分からなる群より選択されるか;または、R6およびR6*の両方が、抗酸化部分、NMDAアンタゴニスト、NOシンターゼ阻害剤、鉄キレート部分、Ca(II)キレート部分、およびZn(II)キレート部分からなる群より選択される)
で示される化合物;またはその薬学的に許容される塩を提供する。ある好ましい態様において、R6*はD-α-アミノアジピルである。ある好ましい態様において、R7はメルカプトメチルである。ある態様において、R7は、水素、メチルまたはフェニルではない。ある好ましい態様において、R6*はさらに、切断可能な連鎖を含む。一つの態様において、該化合物はシクロ-D-フェニルグリシル-L-アラニンを含む。
【0105】
当業者には理解されるように、本発明の化合物には、単一のファルマコフォア(例えばジオキサピペラジン、ここでのジオキサピペラジン部分は単独のファルマコフォアである);または、β-アミノアニオン部分(ここでのβ-アミノアニオン部分は化合物の生化学的活性に関与している)を有し得る化合物が含まれる。本発明のある化合物には、二つの別個のファルマコフォアが含まれ、A-Bにより示される構造を有し、ここでのAおよびBは、生化学活性を有する各ドメインまたはファルマコフォア(例えば、独立した抗酸化部分を有する抗痙攣ジオキサピペラジン部分、例えばR6*)(本明細書ではハイブリッド」薬物とも称する)である。二つのファルマコフォアを含む化合物は、二つ以上の別個の受容体と相互作用できる。本発明の化合物が二つ以上のファルマコフォアを含む場合、ファルマコフォアは、当業者に既知の多種多様な技術により互いに連結させることができる。例えば、R6*により示されるファルマコフォアは、ジオキサピペラジン環の窒素へのアミド連鎖を介して、ジオキサピペラジン部分に共有結合できる。二つのファルマコフォア間の連鎖は、二つのファルマコフォアが互いにインビボで切断されるように(すなわち、インビボで不安定である連鎖の選択により)選択できる。このような生物学的に不安定な連鎖の例は、当技術分野において公知である。例えば、前記に引用したSilvermanを参照されたい。有利には、このような「ハイブリッド」の二つのファルマコフォア薬物は、体内に輸送され、脳などの部位または臓器に到達するように設計でき、そこで一つ以上のファルマコフォア部分が生物学的作用を示し、この部位でハイブリッド薬物は切断されて二つの活性薬物部分を与え得る。ハイブリッド薬物のいくつかの例は前記している。
【0106】
本発明は、さらに、本発明の治療化合物にインビボで変換されるプロドラッグの使用に関する。このようなプロドラッグを使用して、治療化合物の体内分布を変化(例えば、典型的には血液脳関門を通らない化合物が血液脳関門を通過できるようにするために)または薬物動態を変化させることができる。例えば、陰性基、例えばカルボキシレートまたはスルホネートは、例えばメチル基またはフェニル基でエステル化すると、カルボン酸エステルまたはスルホン酸エステルを生成することができる。カルボン酸エステルまたはスルホン酸エステルを被験者に投与した場合、エステルが酵素的または非酵素的に切断され、陰性基が出現する。エステルは環式(例えばラクトンまたはスルトン)でもよく、または、二つ以上のアニオン部分を連結基を通してエステル化してもよい。陰性基を部分(例えばアシルオキシメチルエステル)を用いてエステル化し、これは切断されると中間化合物が出現し、これはその後分解されると、活性化合物を生じることができる。別法として、アニオン部分は、インビボで能動的に輸送される基か、または標的臓器により選択的に取り込まれる基へとエステル化できる。エステルは、治療部分を特定の臓器へと特異的に標的化できるように選択できる。別の態様において、プロドラッグは、インビボで酸化されて治療化合物となる、還元形の陰性基、例えばカルボキシレートまたはスルホネート、例えばアルコールまたはチオールである。
【0107】
従って、前記したように、好ましい化合物には、インビボでβ-アミノアニオン化合物へと変換されることのできる、ピリミジン、例えば置換ウラシルが含まれる。好ましい態様において、該化合物は、式(式V):
【化55】
(式中、R9およびR10は、各々独立的に、水素、アルキル(シクロアルキル、ヘテロサイクリル、およびアラルキルを含む)、アルケニル、アルキニル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ(非置換および置換のアミノを含む)、ヒドロキシ、チオール、アルキルチオール、ニトロ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、またはアミノカルボニルからなる群より選択されるか;または、R9およびR10は、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成し;R11は、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R10およびR11は、それらがそれぞれ結合している炭素原子および窒素原子と共に連結して、環の員数が4〜8である複素環を形成し;R12は、水素、アルキル、アリール、および炭水化物(例えば糖、例えばリボースまたはデオキシリボース)からなる群より選択される)
またはその薬学的に許容される塩もしくはエステルにより示すことができる。別の態様において、該化合物は、式(式Va):
【化56】
(式中、R9a、R9b、R10a、R10bは、各々独立的に、水素、アルキル(シクロアルキル、ヘテロサイクリル、およびアラルキルを含む)、アルケニル、アルキニル、アリール、アルコキシ、アリールオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ(置換および非置換のアミノを含む)、ヒドロキシ、チオール、アルキルチオール、ニトロ、シアノ、ハロゲン、カルボキシル、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、またはアミノカルボニルからなる群より選択されるか;または、R9aおよびR9bは、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成しているか;または、R10aおよびR10bは、それらが結合している2炭素単位と共に、連結して環の員数が4〜8である炭素環または複素環を形成しているか;または、R9aおよびR9bの一方は、R10aおよびR10bの一方と、それらが結合している2炭素単位と共に連結して、環の員数が4〜8である炭素環または複素環を形成し;R11は、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R10aおよびR10bの一方が、R11と、それらがそれぞれ結合している炭素原子および窒素原子と共に連結して、環の員数が4〜8である複素環を形成し;R12は、水素、アルキル、アリール、および炭水化物(例えば、糖、例えばリボースまたはデオキシリボース)からなる群より選択される)
またはその薬学的に許容される塩もしくはエステルにより示すことができる。
【0108】
式VおよびVaの化合物は、種々の合成技術(そのいくつかは当技術分野において既知である)に従って調製できる。シンターゼの例を図2に示す。例えば、図2に示したように、バルビツール酸化合物を改変(例えばメシルクロリドおよびアミン塩基を用いてのメシル化により)して化合物を得、これはさらに官能基化できるか(例えば適切な求核試薬のミカエル付加により);または還元的に脱スルホン化して、適切な求ジエン体を用いてのその後のディールス・アルダー環化付加用の求ジエン体を得ることができる。
【0109】
本発明に有用な化合物はまた、治療化合物を標的臓器または臓器群に選択的に送達することを可能とする、担体または標的化部分も含み得る。例えば、治療化合物の脳への送達が望まれる場合、該化合物には、該化合物を脳に、能動輸送または受動輸送により標的化できる部分(「標的化部分」)が含まれ得る。例として、担体分子には、米国特許第4,450,564号および第5,389,623号に記載のような、酸化還元部分が含まれ得る。これらの特許は、脳に進入でき、そこで荷電したピリジニウム種へと酸化され、脳内に補足される、ジヒドロピリジン部分に連結した薬物を開示する。従って、薬物は脳に蓄積する。他の担体部分には、インビボで能動的または受動的に輸送され得る、アミノ酸またはチロキシンなどの化合物が含まれる。このような担体部分は、インビボで代謝的に除去されるか、または、活性化合物の一部として無傷で留まることができる。多くの標的化部分は既知であり、例えば、アシアログリコタンパク質(例えば、米国特許第5,166,320号を参照されたい)、および、受容体により媒介されるエンドサイトーシスを介して細胞に輸送される他のリガンドが含まれる。
【0110】
前記した標的化戦略およびプロドラッグ戦略を合わせて、プロドラッグとして所望の作用部位に輸送でき、その後、アンマスクされると、活性化合物が出現する化合物を作成することができる。
【0111】
別の局面において、本発明は、被験者における癲癇誘発を抑制できる化合物を同定するためのファルマコフォアモデリング法を提供する。これらの方法は、癲癇誘発に関与するタンパク質または分子に対して直接的または間接的薬理作用を引き起こすことが知られる二つ以上の化合物の構造の検証を特徴とする。癲癇誘発に関与するこれらのタンパク質および分子は、細胞表面受容体分子(例えばNMDA受容体)または神経伝達物質の輸送に関与する分子(例えばGABA輸送体)を含むと考えられている。好ましくは、これらの化合物の構造は各々、化合物の薬理作用の少なくとも一部を示す一つ以上のファルマコフォアを含む。本発明の方法にはまた、二つ以上の化合物のファルマコフォア構造に基づいた、平均的なファルマコフォア構造(例えば、炭素骨格構造および/または3次元空間充填構造)の決定が含まれる。一つ以上の平均的なファルマコフォア構造を有する新規化合物を、これらの方法を使用して選択できる。
【0112】
関連した態様において、これらの方法は、癲癇誘発に関与する二つ以上のタンパク質または分子に対して直接的または間接的薬理作用を引き起こすことが知られる二つ以上の化合物の構造の検証を特色とする。このような態様において、当業者は、選択した新規化合物が、好ましくは、癲癇誘発に関与する異なるタンパク質または分子に対して活性である一つ以上のファルマコフォアを有することを理解していると考えられる。
【0113】
好ましい態様において、本発明のこれらの方法により選択した(例えば設計した)新規化合物は、被験者における癲癇誘発を抑制する。
【0114】
化合物の同定法はさらに、癲癇誘発に関与するタンパク質または分子の少なくとも一部(例えば「偽受容体」)を刺激する他の相補的モデルの作成に依拠し得る。このような刺激を使用してさらに、一つ以上の平均的なファルマコフォアを含む新規候補化合物を評価できる。相補モデルは、癲癇誘発に関与するタンパク質分子と相互作用するファルマコフォアまたは化合物全体の構造に依拠した、アルゴリズムおよび/または方法を使用して作成できる。このような刺激の作成におけるアルゴリズムは当業者に公知であり、MM2分子力学力場が含まれる(例えば、Allinger(1977)J.Am.Chem.Soc.99:8127-8134、Allingerら(1988)J.Comp Chem.9:591-595、Liiら(1989)J.Comp.Chem.10:503-513、Cornellら(1995)J.Am.Chem.Soc.117:5179-5197、Wienerら(1986)J.Comp.Chem.7:230-252を参照されたい)。
【0115】
本発明はさらに、本発明の化合物の容器、および、治療有効量の化合物をそれを必要とする被験者に、被験者における痙攣疾患(例えば癲癇誘発)が抑制されるように使用するための説明書を含む、キットを提供する。本発明のキットは、本発明の化合物を使用、例えば投与するための便利な手段を提供する。特に好ましい態様において、キットは、治療有効量の化合物を、好ましくは単位投与形で含む。
【0116】
本発明はまた、検出マーカーで標識した本発明の化合物(例えば後記した化合物1〜14およびA1〜A32)を被験者に投与し;該被験者の脳のニューロンのNMDA受容体への該化合物の結合の増加を測定し、これにより該被験者における癲癇誘発容態を診断する段階を含む、被験者における癲癇誘発容態を診断する方法を提供する。
【0117】
本発明はさらに、検出マーカーで標識した本発明の化合物(例えば後記した化合物1〜14およびA1〜A32)を被験者に投与し;該被験者の脳のニューロンのGABA受容体への該化合物の結合の減少を測定し、これにより該被験者における癲癇誘発容態を診断する段階を含む、被験者における癲癇誘発容態を診断する方法を提供する。
【0118】
本明細書に使用したような「検出マーカーで標識した化合物」には、検出手段により標識した化合物が含まれ、放射能、蛍光、化学発光、および/または生物発光で標識した抗体が含まれる。
【0119】
標識として使用できる酵素の例には、リンゴ酸デヒドロゲナーゼ、スタフィロコッカスヌクレアーゼ、デルタ-V-ステロイドイソメラーゼ、酵母アルコールデヒドロゲナーゼ、α-グリセロリン酸デヒドロゲナーゼ、トリオースリン酸イソメラーゼ、セイヨウワサビペルオキシダーゼ、アルカリホスファターゼ、アスパラギナーゼ、グルコースオキシダーゼ、β-ガラクトシダーゼ、リボヌクレアーゼ、ウレアーゼ、カタラーゼ、グルコース-VI-リン酸デヒドロゲナーゼ、グルコアミラーゼ、およびアセチルコリンエステラーゼが含まれる。
【0120】
放射性標識の例には、3H、125I、131I、35S、14C、および好ましくは125Iが含まれる。蛍光標識の例には、フルオレセインイソチオシアネート、ローダミン、フィコエリトリン、フィコシアニン、アロフィコシアニン、o-フタルアルデヒド、およびフルオレサミンが含まれる。化学発光標識の例には、ルミノール、ルシフェリン、イソルミノール、理論量のアクリジニウムエステル、イミダゾール、アクリジニウム塩、およびそのシュウ酸エステルが含まれる。生物発光標識の例には、ルシフェリン、ルシフェラーゼ、およびエクオリンが含まれる。
【0121】
III.β-アミノアニオン化合物の調製法
本発明はさらに、β-アミノアニオン化合物の調製法を提供する。
【0122】
一つの態様において、本発明は、式(式VI):
【化57】
(式中、点線は、選択的な単結合/二重結合(EまたはZ立体配置の)を示し;R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、複素環の原子数が3〜7である非置換または置換の複素環を形成しており;R4およびR5は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、アルコキシ、アリールオキシ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、ヘテロサイクリルであるか;または、R4およびR5は、共に、環の原子数が5〜15(より好ましくは5〜8)である置換または非置換の炭素環または複素環を形成している)
で示されるβ-アミノカルボキシル化合物の調製法を提供する。該方法には、式VI
【化58】
(式中、点線は、選択的な単結合/二重結合を示し;Xは、ニトロ、アジド、またはNR2R3であり、ここでのR2およびR3は前記に定義した通りであり;Wは、-CNまたは-COOR8であり;R8は、水素、アルキル、アリール、または有機塩もしくは無機塩形成カチオンであり;R4およびR5は前記に定義した通りである)
で示される化合物を、β-アミノカルボキシルまたはβ-アミノ二トリル化合物が形成されるような、還元的脱硫条件下で反応させる段階が含まれる。ある好ましい態様において、R7は、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり、R3は水素である。
【0123】
式VIIの化合物は、当技術分野で公知の方法に従って調製できる。例えば、アミノチオフェンカルボキシレート(すなわち、式VIの化合物、ここでのWは-COOR8であり、R8はカチオンであり、Xはアミノ基であり、各点線は単結合である)の合成が、いくつかの方法により報告されている。例えば、Beck、J.Org.Chem(1972)37:3224;Meth-Cohn、J.Chem.Res.(1977)(S)294、(M)3262を参照されたい。還元的脱硫条件下におけるアミノチオフェンカルボキシレート(またはアミノチオフェンニトリル)の還元は、現在、良好な収率でβ-アミノ酸を生成することが判明している(アミノチオフェンニトリルにはニトリル基の加水分解も必要であり、これは公知の方法に従って達成できる、例えばLarock、Comprehensive Organic Transformations、VCH Publishers(1989)およびそれに引用されている文献を参照されたい)。好ましい態様において、還元的脱硫条件には、アミノチオフェンカルボキシレートが脱硫するように、アミノチオフェンカルボキシレートをラネーニッケルと反応させる段階が含まれる。
【0124】
別の態様において、本発明は、式VIII:
【化59】
(式中、R2およびR3は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであるか;または、R2およびR3は、それらが結合している窒素と共に、複素環の原子数が3〜7である、非置換または置換の複素環を形成しており;R4およびR5は、各々独立的に、水素、アルキル、アルケニル、アルキニル、シクロアルキル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アリールオキシカルボニル、アミノ、ヒドロキシ、シアノ、アルコキシ、アリールオキシ、カルボキシル、アルコキシカルボニル、アリールオキシカルボニル、ヘテロサイクリルであるか;または、R4およびR5は、一緒に、環の原子数が5〜15(より好ましくは5〜8)である置換または非置換の炭素環または複素環を形成しており;R8は、水素、アルキル、アリール、または有機塩もしくは有機塩形成カチオンである)
により示されるβ-アミノカルボキシル化合物の調製法を提供する。該方法には、式IX
【化60】
(式中、点線は、各々、選択的な単結合を示し;Xは、ニトロ、アジド、またはNR2R3であり、ここでのR2およびR3は前記に定義した通りであり;Wは、-CNまたは-COOR8であり、R8は、水素、アルキル、アリール、または有機塩もしくは無機塩形成カチオンであり;R4およびR5は、前記に定義した通りである)
で示される化合物を、式VIII(ここでのW=-CNである、カルボキシレートは還元的脱硫化および酸性化の後に形成される)で示されるβ-アミノカルボキシル化合物が形成されるような、還元的脱硫条件下で反応させる段階が含まれる。ある好ましい態様において、R2はアルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり、R3は水素である。
【0125】
式IXの化合物(またはそのエステル、これを既知の方法に従って加水分解すると、式IXの化合物を得ることができる)は、当技術分野において既知の方法に従って調製できる。例えば、米国特許第4,029,647号;HenriksenおよびAutrup、Acta Chem.Scand.26:3342(1972);またはHartkeおよびPeshkar、Pharm Zentralhalle 107:348(1968)を参照されたい。
【0126】
本発明の合成法は、以前に報告されたβ-アミノ酸の合成よりも優れた利点を提供する。例えば、本発明の方法により、2炭素骨格のどちらかの炭素または両方の炭素の置換された多種多様なβ-アミノ酸が得られ;生成される具体的なβ-アミノ酸は、出発物質のアミノチオフェンカルボキシレート(多種多様な置換基を用いて調製できる)により決定される。実施例1に下記したように、本発明の方法は、温和な条件下で、市販の試薬からほんの少数の段階で良好な収率でβ-アミノ酸を提供する。この方法により調製された化合物の例を実施例1に提示する。従って、本発明の方法は、一般的で迅速で簡単で高収率のβ-アミノ酸への経路を提供する。
【0127】
別の態様において、本発明は、β-アリール-β-アラニン化合物の調製法を提供する。この態様において、本発明は、しばしば容易に入手できる前駆体を使用して、多種多様の置換および非置換のβ-アリール-β-アラニン化合物を製造できる簡単でワンポットの反応を提供する。本明細書に使用した方法は、β-アラニン類似体を製造するための改法である。該方法には、アリールアルデヒドをマロネート化合物およびアンモニウム化合物と、β-アリール-β-アラニン化合物が形成される条件下で反応させる段階が含まれる。好ましい態様において、アリールアルデヒドは置換または非置換のベンズアルデヒドである。好ましい態様において、マロネート化合物はマロン酸である。好ましい態様において、アンモニウム化合物は、アンモニア、一級アミン、および二級アミンからなる群より選択される化合物のアンモニウム塩である。特に好ましいアンモニウム化合物は、アンモニアの塩、最も好ましくは酢酸アンモニウムである。好ましい態様において、溶媒は、エタノールなどの極性溶媒である。本発明に記載の合成例は、実施例3に記載する。
【0128】
β-アミノ酸は、本明細書に記載した抗癲癇誘発特性の他に、例えば合成中間体として、および商品化学物質としてのその他の用途を有することが理解される。例えば、β-ラクタム構造は、例えば、ペニシリン、カルバペネム、ノルカルジン(norcardins)、モノバクタムなどを含む、多くの商業的に価値ある抗生物質に存在する。β-アミノ酸をβ-ラクタムに変換する多種多様な方法が報告されている。例えば、Wang,W.-BおよびRoskamp,E.J.、J.Am.Chem.Soc.(1993)115:9417-9420およびそれに引用されている文献を参照されたい。従って、本発明は、さらに、β-ラクタムの合成法を提供する。該方法には、式VII(または式IX)の化合物を還元的脱硫条件にかけ、式VI(またはIもしくはVIII)の化合物を生成し、その後、式VI(またはIもしくはVIII)の化合物を環化してβ-ラクタムを形成する段階を含む。さらに、β-アミノ酸は、特定の癌患者の容態を改善することが示されている(例えば、Rougereau,Aら、Ann.Gastroenterol.Hepatol.(パリ)29(2):99-102(1993)を参照されたい)。従って、本発明は、癌の治療に有用な化合物の調製法を提供する。
【0129】
IV.ライブラリー
別の局面において、本発明は、式IV、式VI、または式VIIIの化合物のライブラリー、およびこのようなライブラリーの調製法を提供する。
【0130】
コンビナトリアルライブラリーの合成は当技術分野において公知であり、総説されている(例えば、E.M.Gordonら、J.Med.Chem.37:1385-1401(1994)を参照されたい)。従って、本発明は、式IV、式VI、または式VIIIの化合物のコンビナトリアルライブラリーの合成法を含む。このようなライブラリーは、多種多様な方法に従って合成できる。例えば、「スプリット・プール(split-pool)」戦略を実行して、化合物ライブラリーを作成できる。その後、固定された化合物ライブラリーを洗浄して不純物を除去できる。特定の態様において、固定された化合物を固相支持体から切断して、式IV、VIまたはVIIIの化合物を得ることができる。
【0131】
別のコンビナトリアル合成法の例において、「ディバーソマー(diversomer)ライブラリー」を、Hobbs,De Wittら(Proc.Natl.Acad.Sci.U.S.A.90:6909(1993))の方法により創造する。化合物ライブラリーの創製後、精製および後処理により、式IV、VIまたはVIIIで示される可溶性の置換化合物のライブラリーが得られる。
【0132】
Houghtenら、Nature 354:84-86(1991)の「ティーバッグ」技術を含む他の合成法を使用しても、該当発明に従って化合物のライブラリーを合成できる。
【0133】
コンビナトリアルライブラリーをスクリーニングして、ライブラリーのどのメンバーが所望の活性を有するかを決定でき、活性を有する場合には、活性種を同定できる。コンビナトリアルライブラリーのスクリーニング法が記載されている(例えば、Gordonら、J.Med.Chem.手順引用を参照されたい)。可溶性の化合物ライブラリーは、適切な受容体を用いての親和性アフィニティークロマトグラフィーによりスクリーニングして、受容体に対するリガンドを単離でき、その後、従来の技術(例えば質量分析法、NMRなど)により単離リガンドを同定できる。固定された化合物は、該化合物を可溶性受容体と接触させることによりスクリーニングでき;好ましくは、可溶性受容体は、リガンド結合を検出し指示できる標識(例えばフルオロフォア、比色定量酵素、放射性同位体、発光化合物など)にコンジュゲートしている。別法として、固定された化合物を選択的に遊離させ、膜中を拡散させ、受容体と相互作用させることができる。本発明のライブラリーのスクリーニングに有用なアッセイ法の例は当技術分野で既知である(例えば、E.M.Gordonら、J.Med.Chem.37:1385-1401(1994)を参照されたい)。
【0134】
化合物のコンビナトリアルライブラリーも、ライブラリーの各メンバーの実体をコードする「タグ」を用いて合成できる。例えば、米国特許第5,565,324号およびPCT公開公報WO94/08051号を参照されたい。一般に、この方法は、固相支持体または化合物に結合した、不活性であるが、容易に検出可能なタグの使用を特色とする。活性化合物が、前記した技術の一つにより検出される場合に、化合物の実体は、独特な随伴するタグの同定により決定される。このタギング法により、大きな化合物ライブラリーの合成が可能となり、これは非常に低いレベルで同定できる。
【0135】
好ましい態様において、本発明の化合物ライブラリーは、少なくとも30個の化合物、より好ましくは少なくとも100個の化合物、さらにより好ましくは少なくとも500個の化合物を含む。好ましい態様において、本発明の化合物ライブラリーは、109個より少ない化合物、より好ましくは108個より少ない化合物、さらにより好ましくは107個より少ない化合物を含む。
【0136】
化合物ライブラリーは、好ましくは、実質的に純粋である、すなわち、目的の生成物、例えばライブラリーのメンバー以外の化合物は実質的に含まない。好ましい態様において、本発明の方法に従って作成したライブラリーの純度は、少なくとも約50%、より好ましくは少なくとも約70%、さらにより好ましくは少なくとも約90%、最も好ましくは少なくとも約95%である。
【0137】
本発明のライブラリーは、本明細書に記載したように調製できる。一般に、本発明のライブラリーの合成に使用した少なくとも一つの出発物質は、雑種個体群として提供される。本明細書に使用したような「雑種個体群」という用語は、少なくとも二つの異なる化学実体、例えば異なる化学構造の化学実体を含む、個体群を意味する。例えば、式VIIの化合物の「雑種個体群」は、式VIIの少なくとも二つの異なる化合物を含む。化合物を固相支持体に固定するためのリンカーの雑種個体群の使用により、リンカー切断時に多種多様な化合物を作成できる。
【0138】
本発明のライブラリーは、とりわけ、薬物発見に有用である。例えば、本発明のライブラリーをスクリーニングして、ライブラリーが、予め選択された活性、例えば抗癲癇誘発活性または抗痙攣活性を有する化合物を含むかどうかを決定できる。
【0139】
V.薬学的組成物
別の局面において、本発明は、一つ以上の薬学的に許容される担体(添加剤)および/または希釈剤と共に製剤化された、治療有効量の一つ以上の前記した化合物を含む、薬学的に許容される組成物を提供する。本発明の薬学的組成物は、以下に適合したものを含む、固体型または液体型で投与するために特に製剤化され得る:(1)経口投与、例えば、舌に施薬するための水薬(水性または非水性の溶液または懸濁液)、錠剤、ボーラス、粉末、顆粒、ペースト;(2)非経口投与、例えば、滅菌溶液または懸濁液として、皮下、筋肉内または静脈内注射により;(3)局所適用、例えば、皮膚に塗るクリーム、軟膏、またはスプレーとして;または(4)膣内または直腸内、例えば膣座薬、クリーム、気泡として。好ましい態様において、治療化合物は経口投与する。本発明の化合物は、ヒトを含む被験者に、例えば哺乳動物に投与するための薬学的組成物として製剤化できる。
【0140】
本発明の化合物は、インビボでの医薬投与に適した生物学的に適合性の形態で被験者に投与する。「インビボで投与するに適した生物学的に適合性の形態」は、抗体の治療作用が毒性作用を上回る、投与化合物を意味する。被験者という用語は、免疫応答が誘発され得る生物、例えば哺乳動物を含むものとする。被験者の例には、ヒト、イヌ、ネコ、齧歯類(例えばマウスまたはラット)、およびそのトランスジェニック種が含まれる。治療有効量の本発明の治療組成物の投与は、所望の結果を達成するに必要な用量および投与期間において効果的な量として定義する。例えば、治療有効量の本発明の化合物は、個体の疾病状態、年齢、性別、および体重、ならびに、個体における所望の応答を誘発する抗体の能力などの因子に応じて変化し得る。投与方式は、最適な治療応答が得られるように調整し得る。例えば、数回に分割した用量を毎日投与しても、または、治療状況の緊急性に応じて比例的に減少させてもよい。
【0141】
活性化合物は、注射(皮下、静脈内など)、経口投与、吸入、経皮塗布、または直腸投与などの従来の様式で投与し得る。投与経路に応じて、活性化合物を材料でコーティングして、酵素、酸、および化合物を失活させ得る他の天然条件の作用から化合物を防御し得る。
【0142】
本発明の化合物は、酵素阻害剤と同時投与する適切な担体または希釈剤に含めて、またはリポソームなどの適切な担体に含めて被験者に投与できる。本明細書に使用したような「薬学的に許容される担体」は、生理食塩水または緩衝水溶液などの希釈剤を含むものとする。本発明の化合物を非経口投与以外で投与するために、その失活を防ぐ材料で抗体をコーティングするか、またはその材料と該化合物を同時投与する必要があり得る。リポソームには、水/油/水型乳剤ならびに従来のリポソームが含まれる(Strejanら、(1984)J.Neuroimmunol 7:27)。活性化合物はまた非経口的にまたは腹腔内に投与し得る。分散液は、グリセロール、液体ポリエチレングリコール、およびその混合物中で、および油中で調製できる。通常の貯蔵および使用の条件下で、これらの調製物は、微生物の増殖を防ぐための保存剤を含み得る。
【0143】
注射に使用するのに適した薬学的組成物には、滅菌水溶液(水溶性である場合)または分散液、ならびに、無菌注射溶液または分散液の即時調製用の滅菌粉末が含まれる。全ての場合において、組成物は無菌でなければならず、シリンジが容易に扱えるほど十分に流動性でなければならない。製造および貯蔵の条件下で安定でなければならず、細菌および真菌などの微生物の汚染作用に対して防御されていなければならない。薬学的に許容される担体は、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、および液体ポリエチレングリコールなど)およびその適切な混合物を含む、溶媒または分散媒体であり得る。適切な流動性が、例えば、レシチンなどのコーティングの使用により、分散液の場合には必要な粒子サイズの維持により、および界面活性剤の使用により維持できる。微生物の作用の防御は、種々の抗細菌剤および抗真菌剤、例えば、パラベン、クロロブタノール、フェノール、アスコルビン酸、チメロサールなどにより行なうことができる。多くの場合、等張剤、例えば、糖、ポリアルコール、例えばマンニトール、ソルビトール、塩化ナトリウムが組成物に含まれることが好ましい。注射用組成物の吸収延長は、組成物中に、吸収を遅延する薬剤、例えばモノステアリン酸アルミニウムおよびゼラチンを含めることにより行なうことができる。
【0144】
無菌注射溶液は、必要量の活性化合物を適切な溶媒中に、必要である場合には、前記に列挙した成分の一つまたは組合せて共に取り込み、その後、滅菌濾過することにより調製できる。一般に、分散液は、活性化合物を、基本分散媒体および前記に列挙したものからの他の必要な成分を含む、無菌ビヒクルに取り込むことにより調製する。無菌注射溶液の調製用の無菌粉末の場合、好ましい調製法は、真空乾燥法および凍結乾燥法であり、これにより活性成分の粉末と、以前に滅菌濾過したその溶液からの任意のその他の所望の成分が得られる。
【0145】
活性化合物を、前記したように適切に保護した場合、組成物は、例えば、不活性希釈剤または同化可能な食用担体と共に経口投与し得る。本明細書に使用したような「薬学的に許容される担体」には、任意および全ての溶媒、分散媒体、コーティング、抗細菌剤および抗真菌剤、等張剤および吸収遅延剤などが含まれる。薬学的に活性な物質のためにこのような媒体および薬剤を使用することは、当技術分野において公知である。任意の従来の媒体または薬剤が活性化合物と不適合性でない限り、治療組成物におけるその使用が考えられる。補助的活性化合物も組成物に取り込むことができる。
【0146】
投与を容易にし、用量を均一にするために、投与単位形で非経口組成物を製剤化することが特に有利である。本明細書に使用したような投与単位形は、治療したい哺乳動物被験者の単位量として適した物理的に独立した単位を意味し;各単位は、必要な薬学的担体と合わせて所望の治療効果が得られるように計算された、予め決定された量の活性化合物を含む。本発明の投与単位形の詳細は、活性化合物の独特な特徴および達成したい具体的な治療効果、および個体の治療的処置における活性化合物などの配合の分野に固有の限界により影響されこれに直接依存する。
【0147】
実施例
実施例1:ファルマコフォアモデルに基づく化合物の同定
2つの異なるクラスの化合物:(1)GABA摂取受容体の阻害物質および(2)NMDA受容体の共作用物質(co-agonist)の構造パラメータおよび特徴を組み入れたファルマコフォアモデルを開発した。
【0148】
以前のモデル(Murali Dharら、(1994) J. Med. Hem.37:2234、FalchおよびKrogsgaard-Larson (1991) Eur.J Med.Chem.26:69、N'Goka(1991)J. Med. Chem 34:2457)では、GABA摂取阻害物質が以下のものを含むことが示唆されている:
i)アミン基(好ましくは二級アミン)
ii)カルボキシル基
iii)親油基、好ましくは芳香族基
iv)アミンと親油基との間に配置された電子に富む官能基(二重結合または酸素)
v)アミン基と二重結合または酸素原子との間の2炭素鎖長。
【0149】
NMDA受容体錯体のグリシン共作用物質部位の拮抗物質に焦点が合わされた他の以前のモデル(例えば、LeesonおよびIverson(1994)J.Med.Chem 37:4053)では、NMDA受容体の共作用物質は望ましくは以下のものを含むことが示唆されている:
i)アミン基(好ましくは二級アミン)
ii)カルボキシル基
iii)2つの小さな親油基
iv)大きな親油基。
【0150】
この情報に基づき、クラスとしてはβ-アミノ酸およびその類似体であると考えられる平均のファルマコフォアモデル化合物を調製した。これらの化合物の重要なパラメータは以下のものを含む:
i)アミン基
ii)カルボキシル基
iii)β-アラニン骨格
iv)フレキシブルな脂肪親和性部分。
【0151】
所望の化合物のプロファイルをさらに精密化するために、一連の分枝モデル化計算(MM2分子機構力場)を使用して「平均的な受容体部位」の3次元視覚化を構成した。最初に、NMDA受容体上のグリシンサブサイトに結合することが知られている様々なプローブ分子を使用して、相補モデリングアプローチにより「擬似受容体」モデルを作成した。これを達成するために、周知のNMDA受容体サイトペプチドの断片を幾つかのプローブ分子(例えば、受容体に結合することが知られている化合物)付近に機械的に配置し、受容体をシミュレートした。すなわち、プローブ分子を鋳型として使用しその周りに受容体モデルを構成した。例えば、グルタミン酸塩の側鎖を使用してプローブ分子内の塩基性アンモニウム基に「合体」させた。脂肪親和性ポケットはフェニルアラニンの側鎖を用いてシミュレートした。そうすることにより、NMDA受容体上のグリシンサブサイトの「受容体」を数学的にモデル化した。次に同じ手順をグリアGABA摂取受容体に対し実施した。2つのモデル受容体をオーバーラップさせ、モデルハイブリッド受容体(平均的な受容体サイト)を設計した。このモデルハイブリッド受容体サイトは3つの「ポケット」を含んだ。アニオンポケットはカチオンポケットから7.7オングストローム離れて位置し、それぞれアンモニウム基およびカルボキシル基と相互作用することができる。移動性の脂肪親和性ポケットをアニオンポケットから5.2から8.1オングストロームの範囲の可変位置に配置した。上記条件を含むβ-アミノ酸類似体をモデルハイブリッド受容体中に挿入した。短い(2〜3炭素)フレキシブルアーム上に芳香族環を有するβ-置換β-アミノ酸を用いると最適適合が得られた。フレキシブルアームにより移動性の脂肪親和性ポケットとの相互作用が可能になると考えた。
【0152】
これらの方法により同定された候補化合物のリストを以下に示す。
【化61】
【0153】
多くのβ-アリールβ-アミノ酸化合物を、容易な「ワンポット」合成法によりさらに製造した。簡単に説明すると、置換ベンズアルデヒドの無水エチルアルコール溶液にマロン酸および過剰の酢酸アンモニウムを添加し、反応混合物を加熱して還流させた。反応混合物を冷却しβ-アリールβ-アラニンおよび(一定の場合)桂皮酸誘導体の混合物を得た。桂皮酸は(存在すれば)混合物の酸/塩基抽出により除去し、β-アリールβ-アラニンを得た。しばしば中程度から良好な収率であった。この方法により得られた候補化合物のリストを以下に列挙する。
【化62】
【化63】
【0154】
実施例2 候補化合物の癲癇誘発阻害に対する薬理学的有用性のインビボ評価
2つの群の候補類似体のインビボ試験を抗発作活性および神経毒性の両方に対し実施した。1つの発作モデルを、カナダ動物実験評議会のガイドラインに従い、クィーンズ大学動物倫理委員会の監督下、雄の成体SDラットを用いて実施した。この試験手順は、Turskiら(1984)Brain Res.321:237による以前の研究から採用されている。腹腔内(i.p.)注射により100mg/kgで試験化合物を投与した。塩酸ピロカルピンの腹腔内投与(350mg/kg)により20分後に発作が誘発された。防護は、ピロカルピン投与後30分の観察期間にわたり、慢性痙攣が起こらないものとして規定した。化合物1、2、3、5、8、10、11、13、A1、A4、A5、A11、A13、A14、A15、A16、A21、A26、A28、A29およびA31はこのアッセイ法で有意な抗発作活性を示した。抗発作活性を示す化合物のクラスとしては、N置換β-アミノ酸酸類似体(化合物1、2、3および10);β-置換β-アミノ酸類似体(化合物5、11、A1、A4、A5、A11、A13、A14、A15、A16、A21、A26、A28、A29およびA31);α-置換β-アミノ酸類似体(すなわち、化合物8および13)が挙げられる。
【0155】
候補化合物の抗発作および神経毒性を試験するための更なるアッセイ法には、最大電気ショック発作(MES)モデル、皮下ペンチレンテトラゾール(PTZ)-誘発発作モデル、およびロトロッド(rotorod)神経毒性試験が含まれた。全てのアッセイ法はNIHの癲癇部門における抗痙攣薬開発(ADD)プログラムにより実施した(例えば、StablesおよびKupferberg(1997)、NIH抗痙攣薬開発(ADD)プログラム:前臨床抗痙攣スクリーニングプロジェクト(The NIH anticonvulsant Drug Development (ADD) Program:Preclinical Anticonvulsant Screening Project、Libby & Sonsを参照のこと)。全ての化合物は雄のCarworth Farms #1マウスまたは雄のSDラットのいずれかを用いて試験した。各試験化合物は300、100および30mg/kgの腹腔内注射により投与した。
【0156】
MES-誘発発作モデルでは、試験化合物の抗発作活性は30分の観察期間にわたる後脚緊張性伸展の完全停止として規定した(例えば、「抗癲癇薬のための分子および細胞標的」G.Avanziniら(1997) John Libbey&Company Ltd.,pp191-198;James P. StablesおよびHarvey J. Kupferbergによる第16章「NIH抗痙攣薬開発(ADD)プログラム:前臨床抗痙攣スクリーニングプロジェクト」を参照のこと)。化合物9、10およびA3はこのアッセイ法で有意な抗発作活性を示した。
【0157】
PTZ-誘発発作モデルでは、発作は典型的にはPTZの腹腔内注射(マウスでは85mg/kgおよびラットでは70mg/kg)による試験化合物投与後0.5および4時間で誘発された。防護は、30分の観察期間にわたり慢性痙攣が阻害されることとして規定した。化合物9、10、A3、A7、A17、A22、A23、A24およびA25はこのアッセイ法では有意な抗発作活性を示した。
【0158】
ロトロッド神経毒性試験では、試験化合物の投与後、マウスを6rpmの速度で回転する直径1インチのぎざぎざのついたプラスチックロッド上に置いた。神経毒性は、1分の観察期間にわたりマウスが平衡を維持することができないものとして規定した。化合物1、2、4〜9、11、12、14、A3、A4、A6、A8、A9、A10、A17、A21、A22、A23、A26、A27、A28、A29、A30、A31およびA32はこのアッセイ法では神経毒性を示さなかった。しかしながら、いくらかの神経毒性を示した残りの化合物の毒性レベルは、カルバマジンおよびバルプロ酸などの抗発作薬に比べ低かった。
【0159】
実施例3:β-アミノ酸の合成:方法A
一般手順
無水酢酸によるN-アセチル保護
アセトアミドチオフェンカルボン酸エステルを、無水AcOH中で1時間、対応するアミノ化合物を過剰のAc2O(4当量)と還流させることにより調製した。混合物を冷水中に注ぎ入れ、生成物を濾過により単離し、水で洗浄し、EtOHから再結晶化した。
【0160】
レーニーニッケル触媒の合成
水(1.2 L)に溶解したNaOH(320.0g、8mol)溶液を2.0Lフラスコ中で機械的に撹拌した。氷浴で10℃まで冷却した後、ニッケルアルミニウム合金(250g)を少量ずつ90分にわたり添加した。得られた懸濁液を室温で1時間、50℃でさらに8時間撹拌した。懸濁液を目盛付きシリンダに移し、水性の上清のデカンテーションを行った。得られたスラリーを2.5MのNaOH水溶液(200mL)と共に振り混ぜ、その後にデカンテーションを行った。ニッケル触媒を水(150ml)中に懸濁させることにより30回洗浄し、その後にデカンテーションを行った。無水EtOH(100mL)を用いて洗浄を3度繰り返し、得られたレーニーニッケルを無水EtOH下で保存した。
【0161】
レーニーニッケル還元脱硫
アセトアミドチオフェンカルボン酸アルキル(20mmol)および新たに調製したレーニーニッケル(8当量)をEtOH(75mL)中で激しく撹拌しながら16時間還流させた。高温混合物を珪藻土(セライト)を通して濾過し、ニッケル残渣を熱EtOH(50mL)で洗浄した。濾液を濃縮すると、純粋なN-アセチル-β-アラニンアルキルエステルが透明油、ゴムまたは白色結晶として得られた。
【0162】
酸分解を介するN-アセチルおよびアルキルエステル脱保護
二重に保護したα-またはβ-置換β-アラニンを6M HCl中で5時間還流させた。溶液を蒸発させ(H2O、HCl、MeOHおよびAcOHを除去し)、残渣を蒸留H2O中に2度溶解し、濃縮した(残留HClを除去)。生成物をEtOHから再結晶化させ、塩酸塩を白色結晶として得た。または、粗生成物を最小量の熱H2Oに溶解し、NH4OHを滴下し遊離β-アミノ酸を沈澱させた。二体積のEtOHまたはMeOHを添加し生成物の分離を支援させ、凝集を防止させた。混合物を24時間冷却し(4℃)、さらに沈澱させ、その後濾過した。生成物を氷冷H2OおよびEtOHで洗浄し、その後MeOHまたはEtOHから再結晶化させ、純粋な置換β-アラニンを白色結晶として得た。
【0163】
TLC解析
下記の実験手順では、薄層クロマトグラフィー分析のために使用する溶媒は以下のように略す。
溶媒B:塩化メチレン:アセトン:酢酸100:100:0.5
溶媒I:酢酸エチル:メタノール9:1
溶媒J:クロロホルム:アセトン:水88:12:15
溶媒K:メタノール:酢酸5:1
溶媒L:エタノール:酢酸50:1
【0164】
アセトアミドチオフェンカルボン酸アルキルの合成
3-アセトアミドベンゾ[b]チオフェン-2-カルボン酸メチル
上記手順を用いて、3-アミドベンゾ[b]チオフェン-2-カルボン酸メチル(1.8596g、8.97mmol)をアセチル化し、EtOH再結晶化により精製し、微細な白色結晶として純粋生成物を得た(1.4723g、5.91mmol、65.9%);融点:178〜180℃;TLC:Rf=0.63(溶媒I)、0.55(溶媒J)、0.80(溶媒L);IR(cm-1):3271(NH)、3021(CH)、1716(エステルC=O)、1670(アミドC=O)、746(=CH)。
【0165】
3-アセトアミド-6-(トリフルオロメチル)ベンゾ[b]チオフェン-2-カルボン酸メチル
3-アミノ-6-(トリフルオロメチル)ベンゾ[b]チオフェン-2-カルボン酸メチル(1.4944g、5.43mmol)をアセチル化し、EtOH再結晶化により精製し、綿毛状淡黄色結晶として純粋生成物を得た(1.5261g、4.81mmol、88.6%);融点:204〜205℃;TLC:Rf=0.72(溶媒I)、0.78(溶媒L);IR(cm-1):3274(NH)、3069(CH芳香族)、2962(CH脂肪族)、1720(エステルC=O)、1676(アミノC=O)。
【0166】
2-アセトアミド-4,5,6,7-テトラヒドロベンゾ[b]チオフェン-3-カルボン酸メチル
2-アミノ-4,5,6,7-テトラヒドロベンゾ[b]チオフェン-3-カルボン酸メチル(3.0004g、14.20mmol)を、上述したようにアセチル化し、EtOH再結晶化により精製し、淡茶色結晶として純粋生成物を得た(3.3823g、13.35mmol、94.0%);融点:103〜106℃;TLC:Rf=0.68(溶媒I)、0.66(溶媒J)、0.76(溶媒L);IR(cm-1):3248(NH)、2932(CH)、1698(エステルC=O)、1668(アミドC=O)。
【0167】
2-アセトアミド-6-tert-ブチル-4,5,6,7-テトラヒドロベンゾ[b]チオフェン-3-カルボン酸メチル
2-アミノ-6-tert-ブチル-4,5,6,7-テトラヒドロベンゾ[b]チオフェン-3-カルボン酸メチル(1.3693g、5.12mmol)を、上述したようにアセチル化し、EtOH再結晶化により精製し、白色微細結晶として純粋生成物を得た(0.9312g、3.01mmol、58.8%);融点:117〜118℃;TLC:Rf=0.74(溶媒I)、0.70(溶媒J);IR(cm-1):3271(NH)、2953(CH)、1674(C=O)。
【0168】
2-アセトアミドシクロドデカ[b]チオフェン-3-カルボン酸エチル
2-アミノシクロドデカ[b]チオフェン-3-カルボン酸エチル(4.9236g、15.91mmol)を、上述したようにアセチル化し、EtOH再結晶化により精製し、淡茶色結晶として純粋生成物を得た(4.6058g、13.10mmol、82.3%);融点:54〜74℃;TLC:Rf=0.73(溶媒I);IR(cm-1):3358(NH)、2929(CH)、1710(エステルC=O)、1678(アミドC=O)。
【0169】
2-アセトアミド-4,5,6,7-テトラヒドロ-6-フェニルベンゾ[b]チオフェン-3-カルボン酸メチル
2-アミノ-4,5,6,7-テトラヒドロ-6-フェニルベンゾ[b]チオフェン-3-カルボン酸メチル(2.5046g、8.71mmol)を、上述したようにアセチル化し、EtOH再結晶化により精製し、オフホワイト微細粉末として純粋生成物を得た(2.3763g、7.21mmol、82.8%);融点:116〜117℃;TLC:Rf=0.79(溶媒I)、0.78(溶媒J);IR(cm-1):3255(NH)、3029(CH)、2925(CH)、1686(エステルC=O)、1668(アミドC=O)、703(=CH)。
【0170】
3-アセトアミド-5-フェニルチオフェン-2-カルボン酸メチル
3-アミノ-5-フェニルチオフェン-2-カルボン酸メチル(2.5031g、10.73mmol)を、上述したようにアセチル化し、EtOH再結晶化により精製し、白色結晶として純粋生成物を得た(2.7726g、10.07mmol、93.8%);融点:115℃;TLC:Rf=0.70(溶媒I)、0.70(溶媒J);IR(cm-1):3319(NH)、3122(CH)、2950(CH)、1715(エステルC=O)、1680(アミドC=O)、765(=CH)。
【0171】
3-アセトアミド-5-(4-メトキシフェニル)チオフェン-2-カルボン酸メチル
3-アミノ-5-(4-メトキシフェニル)チオフェン-2-カルボン酸メチル(2.5004g、9.50mmol)を、アセチル化し、EtOH再結晶化により精製し、白色微細結晶として純粋生成物を得た(2.7173g、8.90mmol、93.7%);融点:148〜149℃;TLC:Rf=0.68(溶媒I)、0.65(溶媒J);IR(cm-1):3303(NH)、3143(CH)、2943(CH)、1705(エステルC=O)、1663(アミドC=O)、817(=CH)。
【0172】
3-アセトアミド-5-(4-メチルフェニル)チオフェン-2-カルボン酸メチル
3-アミノ-5-(4-メチルフェニル)チオフェン-2-カルボン酸メチル(1.5098g、6.10mmol)を、上述したようにアセチル化し、EtOH再結晶化により精製し、綿毛状白色結晶として純粋生成物を得た(1.6694g、5.77mmol、94.6%);融点:127〜129℃;TLC:Rf=0.70(溶媒I)、0.64(溶媒J)、0.75(溶媒K);IR(cm-1):3316(NH)、2953(CH)、1710(エステルC=O)、1675(アミドC=O)、812(=CH)。
【0173】
3-アセトアミド-5-[3-メトキシ-4-(4-ニトロベンジルオキシ)フェニル]チオフェン-2-カルボン酸メチル
3-アミノ-5-[3-メトキシ-4-(4-ニトロベンジルオキシ)フェニル]チオフェン-2-カルボン酸メチル(1.5174g、3.66mmol)を、上述したようにアセチル化し、EtOH再結晶化により精製し、黄色結晶として純粋生成物を得た(1.5487g、3.39mmol、92.6%);融点:193〜194℃;TLC:Rf=0.68(溶媒I)、0.65(溶媒J);IR(cm-1):3326(NH)、3072(CH)、2944(CH)、1705(エステルC=O)、1671(アミドC=O)、836(=CH)。
【0174】
N-アセチル-α-置換-β-アラニンアルキルエステルの合成
N-アセチル-α-シクロヘキシル-β-アラニンメチルおよびエチルエステル
2-アセトアミド-4,5,6,7-テトラヒドロベンゾ[b]チオフェン-3-カルボン酸メチル(0.8125g、3.37mmol)を、レーニーニッケルを用いて還元脱硫し、淡黄色油として標題化合物を得た(0.6051g、2.81mmol、83.4%);TLC: Rf=0.80(溶媒I)、0.81(溶媒L);IR(cm-1):2894(CH脂肪族)、1738(エステルC=O)、1674(アミドC=O)。
【0175】
N-アセチル-α-シクロドデシル-β-アラニンエチルエステル
2-アセトアミドシクロドデカ[b]チオフェン-3-カルボン酸エチル(2.3366g、6.65mmol)を、レーニーニッケルを用いて還元脱硫し、黄色油として標題化合物を得た(2.1314g、6.55mmol、98.5%);TLC: Rf=0.75(溶媒I)、0.46(溶媒J);IR(cm-1):3316(NH)、2903(CH脂肪族)、1725(エステルC=O)、1661(アミドC=O)。
【0176】
N-アセチル-α-(4-tert-ブチルシクロヘキシル)-β-アラニンメチルエステル
2-アセトアミド-6-tert-ブチル-4,5,6,7-テトラヒドロベンゾ[b]チオフェン-3-カルボン酸メチル(0.8286g、2.68mmol)を、レーニーニッケルを用いて還元脱硫し、粘着性白色固体として標題化合物を得た(0.7466g、2.63mmol、98.3%);融点:73〜75℃;TLC: Rf=0.70(溶媒I)、0.33(溶媒J);IR(cm-1):3261(NH)、2943(CH脂肪族)、1735(エステルC=O)、1648(アミドC=O)。
【0177】
N-アセチル-α-(4-フェニルシクロヘキシル)-β-アラニンメチルエステル
2-アセトアミド-4,5,6,7-テトラヒドロ-6-フェニルベンゾ[b]チオフェン-3-カルボン酸メチル(2.0292g、6.16mmol)を、レーニーニッケルを用いて還元脱硫し、白色固体として標題化合物を得た(1.7908g、5.90mmol、95.8%);融点:75〜80℃;TLC: Rf=0.58(溶媒J)、0.79(溶媒L);IR(cm-1):3259(NH)、3079(=CH)、2929(CH脂肪族)、1730(エステルC=O)、1647(アミドC=O)、698(=CH)。
【0178】
N-アセチル-β-置換-β-アラニンメチルエステルの合成
N-アセチル-β-フェニル-β-アラニンメチルエステル
3-アセトアミドベンゾ[b]チオフェン-2-カルボン酸メチル(1.3742g、5.51mmol)を、レーニーニッケルを用いて還元脱硫し、淡黄茶色固体として標題化合物を得た(1.1876g、5.37mmol、97.4%);融点:58〜61℃;TLC: Rf=0.42(溶媒I)、0.24(溶媒J);IR(cm-1):3322(NH)、3061(CH芳香族)、2955(CH脂肪族)、1741(エステルC=O)、1649(アミドC=O)。
【0179】
N-アセチル-β-(4-トリフルオロメチルフェニル)-β-アラニンメチルエステル
3-アセトアミド-6-(トリフルオロメチル)ベンゾ[b]チオフェン-2-カルボン酸メチル(0.7014g、2.21mmol)を、レーニーニッケルを用いて還元脱硫し、透明油として標題化合物を得た(0.5961g、2.05mmol、92.6%);TLC: Rf=0.52(溶媒I)、0.86(溶媒L);IR(cm-1):3340(NH)、1736(エステルC=O)、1654(アミドC=O)。
【0180】
N-アセチル-β-フェネチル-β-アラニンメチルエステル
3-アセトアミド-5-フェニルチオフェン-2-カルボン酸メチル(2.3660g、8.59mmol)を、レーニーニッケルを用いて還元脱硫し、オフホワイトゴムとして標題化合物を得た(2.1108g、8.47mmol、98.6%);TLC: Rf=0.68(溶媒I)、0.65(溶媒J);IR(cm-1):3475(NH)、2893(CH脂肪族)、1735(エステルC=O)、1654(アミドC=O)。
【0181】
N-アセチル-β-(p-メトキシフェネチル)-β-アラニンメチルエステル
3-アセトアミド-5-(4-メトキシフェニル)チオフェン-2-カルボン酸メチル(1.8100g、5.93mmol)を、レーニーニッケルを用いて還元脱硫し、黄色油として標題化合物を得た(1.5544g、5.56mmol、93.8%);TLC: Rf=0.54(溶媒I)、0.25(溶媒J);IR(cm-1):3285(NH)、2944(CH)、1735(エステルC=O)、1651(アミドC=O)、728(=CH)。
【0182】
N-アセチル-β-[2-(4-メチルフェニル)エチル]-β-アラニンメチルエステル
3-アセトアミド-5-(4-メチルフェニル)チオフェン-2-カルボン酸メチル(1.4905g、5.15mmol)を、レーニーニッケルを用いて還元脱硫し、白色ゴムとして標題化合物を得た(1.3434g、5.10mmol、99.1%);融点:50〜51℃;TLC: Rf=0.63(溶媒I)、0.85(溶媒L);IR(cm-1):3288(NH)、2906(CH脂肪族)、1731(エステルC=O)、1639(アミドC=O)、807(=CH)。
【0183】
N-アセチル-β-[2-(3-メトキシ-4-ヒドロキシフェニル)エチル]-β-アラニンメチルエステル
3-アセトアミド-5-[3-メトキシ-4-(4-ニトロベンジルオキシ)フェニル]チオフェン-2-カルボン酸メチル(1.4481g、3.17mmol)を、レーニーニッケルを用いて還元脱硫した。濾過した溶液を熱EtOAcに溶解し、その後0.5N HCl(2×30mL)およびH2Oで洗浄した。有機層を乾燥させ(MgSO4)、濾過、濃縮し、黄色油として標題化合物を得た(0.5620g、1.90mmol、60.0%);TLC: Rf=0.80(溶媒L);IR(cm-1):3498(OH)、2905(CH脂肪族)、1743(エステルC=O)、1663(アミドC=O)、726(=CH)。
【0184】
α-置換-β-アラニンの合成
α-シクロヘキシル-β-アラニン
N-アセチル-α-シクロヘキシル-β-アラニンエチルおよびメチルエステル(2.4499g、10.77mmol)を脱保護し、白色微細結晶として標題化合物を得た(0.9573g、5.59mmol、51.9%);融点:238〜240℃;TLC: Rf=0.75(溶媒I);IR(cm-1):3300〜2700(OH)、2207、1635(カルボン酸C=O)。
【0185】
α-シクロドデシル-β-アラニン塩酸塩
N-アセチル-α-シクロドデシル-β-アラニンエチルエステル(2.1268g、6.83mmol)を脱保護し、白色結晶として標題化合物を得た(0.7322g、2.51mmol、36.7%);融点:201〜204℃;TLC: Rf=0.79(溶媒I)、0.80(溶媒L);IR(cm-1):3400〜2700(OH)、1722(カルボン酸C=O)。
【0186】
α-(4-tert-ブチルシクロヘキシル)-β-アラニン塩酸塩
N-アセチル-α-(4-tert-ブチルシクロヘキシル)-β-アラニンメチルエステル(0.7463g、2.63mmol)を脱保護し、白色微細結晶として標題化合物を得た(0.4347g、1.65mmol、62.7%);融点:230℃(dec);TLC: Rf=0.91(溶媒K);IR(cm-1):3400〜2700(OH)、1732(カルボン酸C=O)。
【0187】
α-(4-フェニルシクロヘキシル)-β-アラニン塩酸塩
N-アセチル-α-(4-フェニルシクロヘキシル)-β-アラニンメチルエステル(1.6699g、5.50mmol)を脱保護し、白色微細結晶として標題化合物を得た(0.5235g、1.84mmol、33.5%);融点:268℃(dec);TLC: Rf=0.74(溶媒I)、0.64(溶媒K);IR(cm-1):3300〜2500(OH)、1701(カルボン酸C=O)。
【0188】
β-置換-β-アラニンの合成
β-フェニル-β-アラニン
N-アセチル-β-フェニル-β-アラニンメチルエステル(1.1561g、5.23mmol)を脱保護し、白色微細結晶として標題化合物を得た(0.5275g、3.19mmol、61.1%);融点:220〜221℃;TLC: Rf=0.75(溶媒I);IR(cm-1):3305(鋭い:OH、H結合せず)、2195、1627(カルボン酸C=O)。
【0189】
β-(4-トリフルオロメチルフェニル)-β-アラニン塩酸塩
N-アセチル-β-(4-トリフルオロメチルフェニル)-β-アラニンメチルエステル(0.5850g、2.01mmol)を脱保護し、白色粉末として標題化合物を得た(0.5076g、1.87mmol、93.0%);融点:203℃(dec);TLC: Rf=0.60(溶媒H);IR(cm-1):3500〜2900(OH)、1715(カルボン酸C=O)。
【0190】
β-フェネチル-β-アラニン
N-アセチル-β-2-フェネチル-β-アラニンメチルエステル(1.5322g、6.15mmol)を脱保護し、白色結晶として標題化合物を得た(0.4709g、2.44mmol、39.6%);融点:211〜214℃;TLC: Rf=0.37(溶媒I)、0.74(溶媒L);IR(cm-1):3496、3310(鋭い:OH、H結合せず)、3028(CH)、2932(CH)、2162、1663(カルボン酸C=O)、702(=CH)。
【0191】
β-(p-メトキシフェネチル)-β-アラニン
N-アセチル-β-(p-メトキシフェネチル)-β-アラニンメチルエステル(1.1244g、4.03mmol)を脱保護し、MeOHから再結晶化し、オフホワイト結晶として標題化合物を得た(0.2761g、1.25mmol、31.0%);融点:180〜184℃;TLC: Rf=0.34(溶媒I)、0.74(溶媒K);IR(cm-1):3400〜2500(OH)、2171、1632(カルボン酸C=O)。
【0192】
β-(p-メチルフェネチル)-β-アラニン
N-アセチル-β-[2-(4-メチルフェニル)エチル]-β-アラニンメチルエステル(1.2884g、4.89mmol)を脱保護し、綿毛状白色結晶として標題化合物を得た(0.6779g、3.27mmol、66.9%);融点:206〜207℃;TLC: Rf=0.89(溶媒K);IR(cm-1):3530、3280(鋭い:OH、H結合せず)、3017(CH)、2166、1706(カルボン酸C=O)、810(=CH)。
【0193】
β-[2-(4-ヒドロキシ-3-メトキシフェニル)エチル]-β-アラニン塩酸塩
N-アセチル-β-[2-(4-ヒドロキシ-3-メトキシフェニル)エチル]-β-アラニンメチルエステル(0.5281g、1.79mmol)を脱保護し黄色油として標題化合物を得た(0.4852g、1.76mmol、98.4%);TLC: Rf=0.32(溶媒I);IR(cm-1):3447(OH)、1718(カルボン酸C=O)。
【0194】
2-アゼチジノンの合成
N-置換β-アミノ酸からのN-置換2-アゼチジノンの調製
CCl4(1.0mL、10mmol)およびトリエチルアミン(TEA)(1.7mL、12mmol)を、N-置換β-アミノ酸(10mmol)および(C6H5)3P(1.56g、1.2mmol)のMeCN(100mL)撹拌溶液に添加した。反応混合物を1.5時間還流させ、その後真空下で濃縮した。残渣をCH2Cl2(100mL)に溶解し、水および塩類溶液で洗浄した。有機層を乾燥させ(MgSO4)、蒸発乾燥させた。生成物を溶離液としてEtOAC/ヘキサン(1:2)を用いてシリカゲルフラッシュクロマトグラフィにより単離した。
【0195】
N-非置換β-アミノ酸からのN-シリル2-アゼチジノンの調製
N-ブロモスクシンイミド(2.14g、12mmol)およびTEA(1.7mL、12mmol)を、N-非置換β-アミノ酸(10mmol)および(C6H5)3P(1.56g、1.2mmol)のMeCN(100mL)撹拌溶液に添加した。反応混合物を室温で10時間撹拌し、その後真空下で濃縮した。残渣をCH2Cl2(60mL)に溶解し、t-ブチルジメチルシリルクロリド(2.25g、15mmol)およびジイソプロピルアミン(2.8mL、15mmol)で処理し、室温で5時間撹拌した。その後溶液をCH2Cl2(100mL)で希釈し、水および塩類溶液で洗浄した。有機層を乾燥させ(MgSO4)、蒸発乾燥させた。生成物を溶離液としてEtOAC/ヘキサン(1:7)を用いてシリカゲルフラッシュクロマトグラフィにより単離した。
【0196】
実施例4:β-アリールβ-アラニンの合成
β-アリールβ-アラニンをワンポット反応で調製した。簡単に説明すると、置換ベンズアルデヒドの無水エタノール溶液に、マロン酸および過剰の酢酸アンモニウムを添加し、反応混合物を加熱し還流させた。反応混合物を冷却しβ-アリールβ-アラニンおよび(一定の場合には)桂皮酸誘導体の混合物を得た。桂皮酸は(存在すれば)混合物の酸/塩基抽出により除去し、β-アリールβ-アラニンを得た。収率はしばしば中程度か良好であった。過程を図3に示した。ある一定のβ-アリールβ-アラニン化合物を合成するための実験手順の更なる詳細は以下に示す。化合物を精製するための代表的な精製スキームを図4に示す。本明細書で説明したように調製したある一定の化合物を以下の表1に示す。収率データは2列で示してあり、第2番目は以下の表2のものと同一である。
【0197】
(表1) ベンズアルデヒドから調製したβ-アリールβ-アラニンの平均収率
(反応条件は最適化していない)
【0198】
本方法により合成し選択した化合物を表1に示す。本発明のある一定のこれらの化合物および他の化合物の代表的な合成を以下で示す。
【0199】
対応するベンズアルデヒド誘導体を過剰の酢酸アンモニウム(〜2当量)およびマロン酸(1当量)と共に無水エタノール中で還流させ、反応を完了させる(TLCおよびNMRにより決定)ことによりβ-置換-β-アミノ酸を調製した。桂皮酸誘導体が副産物として生成した。反応混合物をその後、例えば図4で説明するように標準手順により処理した。
【0200】
β-3(3,4-ジクロロフェノキシ)フェニル-β-アラニン塩酸塩
上記手順を用いて、3-(3,4-ジクロロフェノキシ)ベンズアルデヒド(10g、37.4mmol)、酢酸アンモニウム(3.8437g、49.8mmol)およびマロン酸(3.8923g、37.4mmol)を無水エタノール(30mL)中で5時間(ゆっくりと)還流させた。白色固体のβ-3(3,4-ジクロロフェノキシ)フェニル-β-アラニンを濾過し、10mLの無水エタノールで2度洗浄した。続いて、このβ-3(3,4-ジクロロフェノキシ)フェニル-β-アラニンに10mLの3N HClを添加し、β-3(3,4-ジクロロフェノキシ)フェニル-β-アラニン塩酸塩(4.44g、12.2mmol、32.6%)を得た;融点164〜165℃;IR(KBr):3193、1609cm-1、Rf=0.55(溶媒24)、0.72(溶媒25)。
【0201】
β-4-ブロモフェニル-β-アラニン
4-ブロモベンズアルデヒド(10g、54mmol)、酢酸アンモニウム(8.663g、112.4mmol)およびマロン酸(5.6762g、54.5mmol)を無水エタノール(45mL)中で150時間(ゆっくりと)還流させた。白色固体を濾過し、50mLのNa2CO3および50mLのH2Oの温かい溶液(70℃)に溶解した。その後、この溶液を100mLのジエチルエーテルで3度抽出した。水層をさらに酸性化してpH7とし、白色固体β-4-ブロモフェニル-β-アラニン(4.5140g、18.49mmol、34.2%)を得た;融点:234℃;IR(KBr):3061、1594cm-1;TLC:Rf=0.35(溶媒24)、0.32(溶媒25)。
【0202】
β-4-フルオロフェニル-β-アラニン
4-フルオロベンズアルデヒド(10g、80mmol)、酢酸アンモニウム(8.2487g、107mmol)およびマロン酸(8.3285g、80mmol)を無水エタノール(60mL)中で48時間(ゆっくりと)還流させた。白色固体を濾過し、エタノール再結晶化により精製し、β-4-フルオロフェニル-β-アラニン(10.04g、54.8mmol、68.5%)を得た;融点:216〜217℃;IR(KBr):3160、1606cm-1;TLC:Rf=0.41(溶媒24)、0.42(溶媒25)。
【0203】
β-2,5-ジメトキシフェニル-β-アラニン
2,5-ジメトキシベンズアルデヒド(4.1437g、25mmol)、酢酸アンモニウム(3.1200g、40.47mmol)およびマロン酸(3.1244g、30.02mmol)を無水エタノール(60mL)中で6時間(ゆっくりと)還流させた。白色固体を濾過し、メタノール再結晶化により精製し、β-2,5-ジメトキシフェニル-β-アラニン(1.239g、5.5mmol、22.0%)を得た;融点:206〜208℃;IR(KBr):2944、1630cm-1;TLC:Rf=0.29(溶媒21)、0.66(溶媒23)。
【0204】
β-3-ブロモ-4-メトキシフェニル-β-アラニン
3-ブロモ-4-メトキシベンズアルデヒド(9.9835g、46.42mmol)、酢酸アンモニウム(7.2984g、94.69mmol)およびマロン酸(4.9124g、47.21mmol)を無水エタノール(110mL)中で281時間(ゆっくりと)還流させた。白色固体を濾過し、50mLのNa2CO3および50mLのH2Oの温かい溶液(70℃)に溶解した。その後、この溶液を100mLのジエチルエーテルで3度抽出した。水層をさらに酸性化してpH1とし、100mLの酢酸エチルで2度抽出した。続いて、水層を蒸発乾燥させ、その後白色残渣に30mLの無水エタノールを添加し、15分間撹拌し、濾過した。その後、同じ手順を2度繰り返した。最終混合物を濾過し、濾液を蒸発乾燥した。プロピレンオキシド(9.75mL、139.3mmol)をエタノール部分に添加した。溶液を撹拌し、50℃まで加熱し、β-3-ブロモ-4-メトキシフェニル-β-アラニン(3.0284g、11.05mmol、23.8%)を得た;融点:213℃;IR(KBr):2945、1604cm-1;TLC:Rf=0.26(溶媒24)、0.28(溶媒25)。
【0205】
前の段落に従い一般に合成される他の化合物およびそれらの分析データを以下の表2に示す。
【0206】
(表2) ベンズアルデヒドから調製されたβ-アリール-β-アラニン
【0207】
TLC解析
上記実験手順では、薄層クロマトグラフィー分析用に使用する溶媒は以下のように略記される:
溶媒21:アセトニトリル:酢酸:水 8:1:1
溶媒23:メタノール:酢酸 7:1
溶媒24:n-ブタノール:酢酸:水 4:1:1
溶媒25:メタノール:クロロホルム:酢酸 7:7:1
【0208】
β-アリール-β-アラニン、β-フェネチル-β-アラニン、α-シクロヘキシル-β-アラニン、およびα-置換-β-アラニン(およびそれらのある一定のエステルおよびアミド)、ならびに4'-置換N-アセチル-α-ピペリジニル-β-アラニンに対する他の分析データおよび生物学的データを表3-1から3-3において示す。
【0209】
(表3−1) 解析および生物活性データ
A. β-アリール-β-アラニンおよび前駆体
a.再結晶化のためにEtOH、H2Oまたは混合物を使用した。
b.ピロカルピンを使用すると、化合物はラットでは100mg/kgで活性であるか、または不活性である。
B. アリール置換β-フェネチル-β-アラニンおよび前駆体
a.可能であれば、再結晶化のためにEtOH、H2O、または混合物を使用した。
b.ピロカルピンを使用すると、化合物はラットでは100mg/kgで活性であるか、または不活性である。
【0210】
(表3−2) 解析および生物活性データ
C. 4'-置換α-シクロヘキシル-β-アラニンおよび前駆体
a.再結晶化のためにEtOH、H2Oまたは混合物を使用した。
b.ピロカルピンを使用すると、化合物はラットでは100mg/kgで活性であるか、または不活性である。
D. 4'-置換N-アセチル-α-ピペリジニル-β-アラニンメチルエステル
【0211】
(表3−3) 解析および生物活性データ
E. N-アセチル-α-置換-β-アラニンメチルエステルおよびα-置換-β-アラニン
a.最終合成段階の収率
b.ピロカルピンを使用すると、化合物はラットでは100mg/kgで活性であるか、または不活性である。
【0212】
実施例5
癲癇の「自発反復性発作」(SRS)モデルを使用して、I相癲癇誘発に対するモデルにおいて候補化合物を評価した(例えば、Mello,E.ら、Epilepsia(1993)34:985;Cavalheiro,J.ら、Epilepsia(1991)32:778を参照のこと)。SRSモデルでは、成体の雄SDラット(c.260g)に注射によりピロカルピンを投与する(380mg/kg、腹腔内)。25分以内に、動物は癲癇重積持続状態になり、これが典型的には15〜20時間続く(この段階で動物の約10%が死亡する)。ラットは自発的に回復させ、食物と水を自由に与え、16時間/8時間の明/暗サイクルで維持する。ラットは通常4つの群で研究する。約13〜15日に始めると、ラットは自発反復性発作を発症する。これは1週間につき約4〜5の割合で起こる。ラットは1日につき16時間ビデオテープ録画し、重要な発作挙動(点頭、前肢クロヌスおよび立ち上がり)に対しビデオテープを再検討する。動物は3ヶ月観察し、十分な数の発作の評価を可能とする。評価用の実験化合物は2つの時のうちのいずれかで投与することができる:時間1、1日目、癲癇重積持続状態の停止後SRSの発症前;または時間2、30日目、ラットは約2週間の間SRSを経験した時。時間Iに候補化合物を投与すると、抗癲癇誘発特性(発作の発症の阻害能力)を評価することができ;時間2に化合物を投与すると、定着した発作を抑制する能力を有する抗発作発生としての薬物評価が可能となる。
【0213】
参照として、時間1または時間2のいずれかで、標準抗痙攣薬フェニトインを投与した(20mg/kg/日、静脈内、10日間)。予測通り、フェニトインは時間1で投与した場合発作の発症を阻害する効果が無かったが、時間2で投与すると発作頻度を50%以上減少させるのに75%の効果を示した。
【0214】
対照的に、β-アラニンおよび類似(α-(4-tert-ブチルシクロヘキシル)-アラニン(実施例3を参照のこと)を、上記で概要を示した同じプロトコルを用い時間1または時間2のいずれかで相当する用量(20mg/kg/日、静脈内、10日間)で投与した。時間1では、これらの化合物のそれぞれが、少なくとも50%発作を減少させるのに75%の効果を示し、時間2では、各化合物は少なくとも50%発作を減少させるのに50%の効果を示した。
【0215】
前の表2および3で列挙した本発明の化合物について、実施例7に従う生物活性試験を行った。以下の化合物は少なくとも弱い活性を有することがわかった:塩酸β-p-メチルフェニル-β-アラニン、β-2-ヒドロキシ-3-メトキシフェニル-β-アラニン、β-3-メチル-4-メトキシフェニル-β-アラニン(わずか)、塩酸β-3-(3,4-ジクロロフェノキシ)フェニル-β-アラニン(中程度)、β-2,5-ジメチル-4-メトキシフェニル-β-アラニン、β-p-(トリフルオロメトキシ)フェニル-β-アラニン、およびβ-2-フルオロ-3-(トリフルオロメチル)フェニル-β-アラニン(中程度)。
【0216】
このように、β-アミノ酸は抗癲癇誘発化合物および抗発作発生化合物の両方としての活性を示す。
【0217】
実施例6
標準方法に従いジオキサピペラジン化合物を合成し、NMR、FAB-MS、融点およびHPLCによりキャラクタリゼーションを行った。幾つかの化合物の結晶構造を決定した。
【0218】
例示的な手順は以下の通りである。
Boc-L-アラニン(1.5g、0.008mol)を60mlの酢酸エチルに溶解し、これに2.4gの2-エトキシカルボニル-1,2-ジヒドロキノリン(EEDQ)(0.010mol、1.2当量)を添加した。溶液を5分間撹拌し、その後D-フェニルグリシンメチルエステルHCl(1.5g、0.003mol)を添加した。撹拌を24時間続け、その後に溶液を3×25mL 10%(w/w)KHSO4水溶液、25mL飽和NaCl溶液、3×25飽和重炭酸ナトリウム溶液、および25mL飽和NaCl溶液で洗浄した。有機層を硫酸マグネシウム上で乾燥させ、蒸発させ透明な油を得た。油を20mlの蟻酸に溶解し、2時間室温で撹拌した。蒸発により酸を除去し、油を50mLの2-ブタノールおよび25mLのトルエンの混合物中に懸濁させた。混合物を24時間還流させ、撹拌しながら2時間にわたって冷却し、溶媒を真空下で減量しオリジナルの体積の約4分の1とした。固体を再結晶化させた。シクロ-D-フェニルグリシン-L-アラニンを融点範囲が260〜265℃の白色固体(1.1g、0.005mol、収率68%)として得た。
【0219】
実施例7
選択した化合物を0.9% NaClに溶解し、または30%ポリエチレングリコール400および70% 水の混合物に懸濁させ、動物モデルにおいて試験した。簡単に説明すると、化合物をカースワースファームズ(carsworth Farms)#1マウス(体重1gあたり0.01mgの容積)またはSDラット(体重1gあたり0.004mlの容積)に腹膜内または経口投与した。抗痙攣薬試薬を投与する前にピーク効果およびピーク神経障害の時間を決定した。
【0220】
最大電気ショック発作試験(MES)、電解質溶液(0.9% NaCl)滴を注入した角膜電極を動物の目に適用し、電気刺激(マウスでは50mA、ラットでは150mA;60Hz)を試験化合物のピーク効果時に0.2秒間加えた。動物を手で保持し、刺激時に解放し、発作を観察した。後脚緊張性伸筋成分の廃止(後脚緊張性伸展は身体面に対し90°を超えない)は、MESにより誘発される発作の広がりが化合物により阻害されることを示した。
【0221】
皮下ペンチレンテトラゾールしきい値試験(scMet)では、痙攣を起こさせる用量(CD97)のペンチレンテトラゾール(ラットでは85mg/kg)を試験化合物のピーク効果時に注射した。動物を隔離し、30分間観察して発作が起こるかどうかを確認した。少なくとも5秒間続く間代性の痙攣がないと、ペンチレンテトラゾールにより誘発される発作のしきい値を化合物により引き上げることができることが示された。
【0222】
実験動物における急性抗痙攣薬により誘発される毒性は通常、幾つかの種類の神経異常により特徴づけられる。マウスでは、これらの異常はロトロッド失調試験により検出することができる。この試験はラットでは幾分有益ではない。ロトロッド失調試験では、神経障害は、6rpmで回転するギザギザのついた棒上で少なくとも1分間、動物が平衡を維持することができないことにより示される。ラットは、位置感知試験により試験した。1つの後足をテーブルの縁を越えて徐々に下に下げると、正常な動物は正常な位置まで足を引き上げて戻す。足を正常な位置まで戻すことができないと神経障害が示される。
【0223】
実施例8
ジオキサピペラジン化合物の試験を12匹のマウスで、30、100、300mg/kg(各4匹のマウス)の用量で、試験化合物投与後30分および4時間に実施した。結果を表4に示す。
【0224】
(表4) 選択されたジオキサピペラジン化合物および試験データ
c=シクロ
Peg=フェニルグリシン
活性は0(不活性)から4までの段階に分けて示す
【0225】
表4に示されるように、c/D-フェニルグリシン-L-アラニンおよびc/D-フェニルグリシン-(S-Me)-L-システインは、この動物モデルシステムでは強い抗痙攣活性を示したが、幾つかの他のジオキサピペラジンはより弱い抗痙攣活性を示した。
【0226】
ある一定の他のジオキサピペラジンも合成し、試験した。これらの化合物のうち、c/L-アラニン-D-ロイシンが活性であることがわかった。
【0227】
実施例9:ビアリール抗癲癇誘発作用物質
さらに他の態様では、被験者において癲癇誘発および/または発作発生を阻害するための方法は有効量の化合物を被験者に投与し、癲癇誘発を阻害する段階を含み、ここでその化合物は以下の通りである。
【化64】
【0228】
より特定すると、好ましい化合物は以下の化学式を有する:
【化65】
(式中、各Xはそれぞれ、ハロゲン(クロロが好ましい)、ニトロ、シアノおよび置換または非置換のアルキルおよびアルコキシ基(トリフルオロメチルおよびメチルが好ましい)からなる群から選択され;nは0から5の整数であり(n=1が好ましい);およびYRおよびYSのうちの1つは水素であり、もう一方は置換または非置換のアミンであり、薬学的に許容されるその塩も含まれる)。
【0229】
(表5) 例示的なビアリールエーテル化合物
a.ピロカルピンを使用すると、化合物はラットでは100mg/kgで活性であるか、または不活性である。
【0230】
または、ビアリールエーテルはパラ置換されてもよい:
【化066】
例えば、前記、表2の化合物B8P79を参照のこと。
【0231】
生物学的データが示すように、RまたはS絶対立体化学のいずれかの鏡像異性体の方が、ラセミ化合物または他の立体異性体よりも生物学的に活性であるかもしれない。このような場合、その単一の立体異性体が好ましく、本発明による薬学的組成物は好ましくは、実質的にその立体異性体のみを含む。そのような立体化学異性体は、キラル開始材料からの不斉合成(例えば、桂皮酸エステルへのキラルアミンのマイケル付加後加水分解)、または下記で例示するように、ラセミ合成の分割のいずれかにより調製されてもよい。
【0232】
3-(3-トリフルオロメチルフェノキシ)-trans-桂皮酸メチル
3-[3-(トリフルオロメチル)フェノキシ]ベンズアルデヒド(8.05g、30mmol)および酢酸メチルトリフェニルホスホラニリデン(15.13g、45mmol)のTHF(200mL)溶液を24時間還流させて撹拌し、その後、室温まで冷却し、濃縮した。ヘキサンに溶解した0〜10%のEtOAc溶離液を用い、シリカゲル上でクロマトグラフィーにより残渣を精製すると、9.3g(96%)が得られた。
【0233】
3-(4-メチルフェノキシ)-trans-桂皮酸メチル
3-(4-メチルフェノキシ)ベンズアルデヒド(8.04g、37.9mmol)および酢酸メチルトリフェニルホスホラニリデン(19g、57mmol)のTHF(200mL)溶液を24時間還流させて撹拌し、その後、室温まで冷却し、濃縮した。ヘキサンに溶解した0〜10%のEtOAc溶離液を用い、シリカゲル上でクロマトグラフィーにより残渣を精製すると、9.6g(94.5%)が得られた。
【0234】
3-フェノキシ-trans-桂皮酸メチル
3-フェノキシベンズアルデヒド(8.03g、40.5mmol)および酢酸メチルトリフェニルホスホラニリデン(20g、60mmol)のTHF(200mL)溶液を24時間還流させて撹拌し、その後、室温まで冷却し、濃縮した。ヘキサンに溶解した0〜10%のEtOAc溶離液を用い、シリカゲル上でクロマトグラフィーにより残渣を精製すると、10.2g(99%)が得られた。
【0235】
(3R)-[(S)-(-)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチル
ブチルリチウム(ヘキサン中2.5M、9.9mL、24.75mmol)を0℃で、THF(200mL)に溶解した(S)-(-)-N-ベンジル-α-メチルベンジルアミン(5.3mL、25mmol)に添加した。赤色溶液を0℃で20分間撹拌し、-78℃まで冷却した。THF(20mL)に溶解した3-(3-トリフルオロメチルフェノキシ)-trans-桂皮酸メチル(4g、12.4mmol)を一滴ずつ添加した。混合物を2時間-78℃で撹拌し、飽和塩化アンモニウム(100mL)で急冷し、その後温め、飽和塩化ナトリウム水溶液(100mL)に注ぎ入れた。水層をEtOAc(2×100mL)で抽出、乾燥(Na2SO4)、濾過および蒸発させると残渣が得られ、これをヘキサンに溶解した0〜8%のEtOAc溶離液を用い、シリカゲル上でクロマトグラフィーにより精製した。収集した画分を蒸発させると3.2g(47%)が得られた。
【0236】
(3S)-[(R)-(+)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチル(4.1g)を、R-(+)-N-ベンジル-α-メチルベンジルアミンから同じ手順で調製し、収率は62%であった。
【0237】
(3R)-[(S)-(-)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチル
ブチルリチウム(ヘキサン中2.5M、12mL、30mmol)を0℃で、THF(200mL)に溶解した(S)-(-)-N-ベンジル-α-メチルベンジルアミン(6.3mL、30mmol)に添加した。赤色溶液を0℃で20分間撹拌し、-78℃まで冷却した。THF(20mL)に溶解した3-(3-トリフルオロメチルフェノキシ)-trans-桂皮酸メチル(4g、14.9mmol)を一滴ずつ添加した。混合物を2時間-78℃で撹拌し、飽和塩化アンモニウム(100mL)で急冷し、その後温め、飽和塩化ナトリウム水溶液(100mL)に注ぎ入れた。水層をEtOAc(2×100mL)抽出、乾燥(Na2SO4)、濾過および蒸発させると残渣が得られ、これをヘキサンに溶解した0〜8%のEtOAc溶離液を用い、シリカゲル上でクロマトグラフィーにより精製した。収集した画分を蒸発させると3.3g(46%)が得られた。
【0238】
(3S)-[(R)-(+)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチル(4.4g)を、R-(+)-N-ベンジル-α-メチルベンジルアミンから同じ手順で調製し、収率は62%であった。
【0239】
(3R)-[(S)-(-)-N-ベンジル-α-メチルベンジル]アミノ-(3-フェノキシフェニル)プロパン酸メチル
ブチルリチウム(ヘキサン中2.5M、13mL、32.5mmol)を0℃で、THF(200mL)に溶解した(S)-(-)-N-ベンジル-α-メチルベンジルアミン(6.6mL、31.6mmol)に添加した。赤色溶液を0℃で20分間撹拌し、-78℃まで冷却した。THF(20mL)に溶解した3-(4-メチルフェノキシ)-trans-桂皮酸メチル(4g、15.7mmol)を一滴ずつ添加した。混合物を-78℃で2時間撹拌し、飽和塩化アンモニウム(100mL)で急冷し、その後温め、飽和塩化ナトリウム水溶液(100mL)に注ぎ入れた。水層をEtOAc(2×100mL)抽出、乾燥(Na2SO4)、濾過および蒸発させると残渣が得られ、これをヘキサンに溶解した0〜8%のEtOAc溶離液を用い、シリカゲル上でクロマトグラフィーにより精製した。収集した画分を蒸発させると4.8g(66%)が得られた。
【0240】
(3S)-[(R)-(+)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチルを、R-(+)-N-ベンジル-α-メチルベンジルアミンから同じ手順で調製し、収率は51%であった。
【0241】
(3R)-アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチル
水素(1気圧)下、炭(700mg)上水酸化パラジウムの存在下、(3R)-[(S)-(-)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチル(3.2g、5.8mmol)のMeOH(60mL)、H2O(6mL)および酢酸(1.5mL)溶液を室温で36時間撹拌した。濾過、蒸発させ生成物を得た。生成物は精製せずに次の反応に使用した。
【0242】
(3S)-アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチルを、(3R)-[R-(+)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパノエート(3.9g、7.1mmol)から同じ手順で調製した。
【0243】
(3R)-アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロパン酸メチル
水素(1気圧)下、炭(530mg)上水酸化パラジウムの存在下、(3R)-[(S)-(-)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロパン酸メチル(3.3g、6.7mmol)のMeOH(60mL)、H2O(6mL)および酢酸(1.5mL)溶液を室温で36時間撹拌した。濾過、蒸発させ生成物を得た。生成物は精製せずに次の反応に使用した。
【0244】
(3S)-アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチルを、(3R)-[R-(+)-N-ベンジル-α-メチルベンジル]アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロパノエート(4.2g、8.5mmol)から同じ手順で調製した。
【0245】
(3R)-アミノ-3-(3-フェノキシフェニル)プロパン酸メチル
水素(1気圧)下、炭(700mg)上水酸化パラジウムの存在下、(3R)-[(S)-(-)-N-ベンジル-α-メチルベンジル]アミノ-3-(3-フェノキシフェニル)プロパン酸メチル(4.4g、9.1mmol)のMeOH(60mL)、H2O(6mL)および酢酸(1.5mL)溶液を室温で36時間撹拌した。濾過、蒸発させ生成物を得た。生成物は精製せずに次の反応に使用した。
【0246】
(3S)-アミノ-3-(3-フェノキシフェニル)プロパン酸メチルを、(3S)-[R-(+)-N-ベンジル-α-メチルベンジル]アミノ-3-(3-フェノキシフェニル)プロパノエート(3.7g、7.7mmol)から同じ手順で調製した。
【0247】
(3R)-アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロピオン酸塩酸塩(C1)
(3R)-アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチルを2N HCl(40mL)に溶解し、一晩中還流し、室温まで冷却し濃縮した。残渣を2N HCl(100mL)およびジエチルエーテル(30mL)に溶解した。水層と有機層との間に形成された油層を分離し、蒸発させ、一晩中ポンプで乾燥させ、白色粉末1.8gを得た:[α]20D-0.49°(c2.26、CH3OH)。
【0248】
(3S)-アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロピオン酸塩酸塩(C2)を、(3S)-アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロパン酸メチルから同じ手順により調製し、収率は74%であった:[α]20D+0.63°(c2.38、CH3OH)。
【0249】
(3R)-アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロピオン酸塩酸塩(C3)
(3R)-アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロパン酸メチルを2N HCl(40mL)に溶解し、一晩中還流し、室温まで冷却し濃縮した。残渣を2N HCl(100mL)に溶解し、濃縮した。白色沈澱を濾過し、ジエチルエーテル(10mL)で洗浄し、一晩中ポンプで乾燥させ、1.6gを得た:[α]20D-1.36°(c2.06、CH3OH)。
【0250】
(3S)-アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロピオン酸塩酸塩(C4)を、(3S)-アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロパン酸メチルから同じ手順により調製し、収率は65%であった:[α]20D+1.46°(c2.26、CH3OH)。
【0251】
(3R)-アミノ-3-(3-フェノキシフェニル)プロピオン酸塩酸塩(C5)
(3R)-アミノ-3-(3-フェノキシ)フェニルプロパン酸メチルを2N HCl(40mL)に溶解し、一晩中還流し、室温まで冷却し濃縮した。残渣を2N HCl(100mL)に溶解し、ジエチルエーテル(2×30mL)で洗浄した。水溶液を蒸発させ、一晩中ポンプで乾燥させ、白色粉末2.4gを得た:[α]20D-1.40°(c2.79、CH3OH)。
【0252】
(3S)-アミノ-3-(3-フェノキシフェニル)プロピオン酸塩酸塩(C7)(1.96g)を、(3S)-アミノ-3-(3-フェノキシフェニル)プロパン酸メチルから同じ手順により調製し、収率は87%であった:[α]20D+1.43°(c2.25、CH3OH)。
【0253】
(D)-(+)-3-アミノ-3-[3-(4-クロロフェノキシ)フェニル]プロピオン酸塩酸塩(C8)および(L)-(-)-3-アミノ-3-[3-(4-クロロフェノキシ)フェニル]プロピオン酸塩酸塩(C9)を、BOC-保護ラセミ3-アミノ-3-[3-(4-クロロフェノキシ)フェニル]プロピオン酸のEtOAcに溶解した(1R,2S)-(-)-エフェドリンによるジアステレオ異性体選択的再結晶化、それに続く当技術分野において認知された技術を用いたBOC基の酸性除去により製造した。これらの化合物の比旋光度は+1.07°および-1.04°であった(MeOH中でc=0.0118)。1Hおよび13C NMRは構造と一致した。
【0254】
(L)-(-)-3-アミノ-3-[3-(3,4-ジクロロフェノキシ)フェニル]プロピオン酸塩酸塩(C10)および(D)-(+)-3-アミノ-3-[3-(3,4-ジクロロフェノキシ)フェニル]プロピオン酸塩酸塩(C11)を酵素分割により調製した。塩化フェニルアセチルおよび3-アミノ-3-[3-(3,4-ジクロロフェノキシ)フェニル]プロピオン酸の反応から調製したラセミ3-[3-(3,4-ジクロロフェノキシ)フェニル]-3-フェニルアセチルアミノ-プロピオン酸(2.01g、4.5mmol)を30mLのEtOAcに溶解した。この溶液に30mLの1M リン酸緩衝液(pH=7.6)およびEupergitに固定された200mg(10% w/w)のペニシリンGアミダーゼ(PGA)を添加した。24時間後に反応を停止させ、濾過により酵素を除去した。アミンおよびアセチムアミド生成物をEtOAcと酸水溶液との間での分割により分離し、溶媒を減圧下で除去し、凍結乾燥により198mg(24%)の濃縮(L)-(-)-化合物を得た([α]D=-0.39°、MeOH中でc=0.0058)。1Hおよび13C NMRは構造と一致した。反応を24時間後に停止した。さらに、アセトアミドを6M HClで加水分解すると970mg(79%)の濃縮(D)-(+)-化合物が得られた([α]D=+0.13°、MeOH中でc=0.0173)。1Hおよび13C NMRは構造と一致した。
【0255】
等価物
当業者であれば、本明細書で記述した特定の手順に対する多くの等価物を認識し、またはせいぜい通常の実験を用いて確認することができる。そのような等価物は本発明の範囲内にあり、特許請求の範囲に含まれる。この出願全体において引用した他の参考文献、発行された特許および公開された特許出願全ての内容が、参照として明細書に組み込まれる。本発明およびその態様に関して、それらの特許、出願、および他の文書の適当な構成要素、手順、および方法を選択してもよい。
【図面の簡単な説明】
【0256】
【図1】本発明の方法に有用であるピリミジンおよびジヒドロピリミジン化合物の例を示す。
【図2】本発明のピリミジンおよびジヒドロピリミジン化合物を調製する合成スキームの例を示す。
【図3】本発明のβ-アミノ酸の合成の一つの態様を示す。
【図4】β-アミノ酸の精製スキームを示すフローチャートである。
【特許請求の範囲】
【請求項1】
i) 各々が
a) 癲癇誘発に関与するポリペプチドに対して直接的または間接的な薬理作用を引き起こす能力、および
b) 該薬理作用の少なくとも一部を示すことが決定されているファルマコフォア
を有する二つ以上の化合物の構造を得る段階、
ii) 該二つ以上の化合物のファルマコフォアの構造に基づいた平均的なファルマコフォア構造を決定する段階、ならびに
iii) 平均的なファルマコフォアを含む新規化合物を選択する段階を含む、
被験者における癲癇誘発を抑制する化合物を同定する方法。
【請求項2】
i) 各々が
a) 癲癇誘発に関与するポリペプチドに対して直接的または間接的な薬理作用を引き起こす能力、および
b) 該薬理作用の少なくとも一部を示すことが決定されているファルマコフォア
を有する二つ以上の化合物の構造を得る段階、
ii) 該二つ以上の化合物のファルマコフォア構造に基づいた平均的なファルマコフォア構造を決定する段階、
iii) 癲癇誘発に関与する異なるポリペプチドについて、段階(i)および(ii)を少なくとも1回繰り返す段階、
iv) 前記の段階で決定した一つ以上の平均的なファルマコフォアを含む新規化合物を選択する段階
を含む、被験者における癲癇誘発を抑制する化合物を同定する方法。
【請求項3】
癲癇誘発に関与するポリペプチドに対する薬理活性が、阻害、アゴニスト作用、アンタゴニスト作用、キレート化、および結合からなる群より選択される、請求項1記載の方法。
【請求項4】
構造が炭素骨格構造である、請求項1記載の方法。
【請求項5】
構造が3次元空間充填構造である、請求項1記載の方法。
【請求項6】
癲癇誘発に関与するポリペプチドが細胞表面受容体である、請求項1記載の方法。
【請求項7】
癲癇誘発に関与するポリペプチドがNMDA受容体である、請求項6記載の方法。
【請求項8】
癲癇誘発に関与するポリペプチドが神経伝達物質の輸送に関与する、請求項1記載の方法。
【請求項9】
癲癇誘発に関与するポリペプチドがGABA輸送体である、請求項8記載の方法。
【請求項10】
癲癇誘発を抑制する請求項1記載の方法で同定された化合物の有効量を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法。
【請求項11】
癲癇誘発が抑制されるように有効量の化合物を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法であって、
該化合物が、下記式Aの化合物
【化1】
(式中
i) R1は、水素、アルキル、アルケニル、アルキニル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;
ii) R2は、アルキル、アルケニル、アルキニル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;
iii) Aは生理的pHにおいて陰性基である)
およびその薬学的に許容される塩もしくはエステルである、方法。
【請求項12】
Aがカルボキシルである、請求項11記載の方法。
【請求項13】
R1が水素である、請求項12記載の方法。
【請求項14】
R2がアルキルである、請求項13記載の方法。
【請求項15】
R2がアリールアルキルである、請求項14記載の方法。
【請求項16】
R2がフェニルアルキルである、請求項15記載の方法。
【請求項17】
化合物が、下記式の化合物
【化2】
【化3】
【化4】
【化5】
【化6】
【化7】
およびその薬学的に許容される塩もしくはエステルからなる群より選択される、請求項11記載の方法。
【請求項18】
癲癇誘発が抑制されるように有効量の化合物を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法であって、
該化合物が、下記式Bの化合物
【化8】
(式中、
i) Aは生理的pHにおいて陰性基であり;
ii) Bはフェノキシで置換されたフェニル基である)
およびその薬学的に許容される塩もしくはエステルである、方法。
【請求項19】
Aがカルボキシル基である、請求項18記載の方法。
【請求項20】
Bがアルキルフェノキシで置換されたフェニル基である、請求項19記載の方法。
【請求項21】
Bがメチルフェノキシで置換されたフェニル基である、請求項20記載の方法。
【請求項22】
Bがハロフェノキシで置換されたフェニル基である、請求項19記載の方法。
【請求項23】
Bがクロロフェノキシで置換されたフェニル基である、請求項22記載の方法。
【請求項24】
化合物が、下記式の化合物
【化9】
【化10】
【化11】
およびその薬学的に許容される塩もしくはエステルからなる群より選択される、請求項18記載の方法。
【請求項25】
癲癇誘発が抑制されるように有効量の化合物を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法であって、
該化合物が、下記式Cの化合物
【化12】
(式中、
i) Aは生理的pHにおいて陰性基であり;
ii) Dが、アルコキシおよびアリールオキシからなる群より選択される二つ以上の部分で置換されたアリール基である)
およびその薬学的に許容される塩である、方法。
【請求項26】
Aがカルボキシル基である、請求項25記載の方法。
【請求項27】
Dが、アルコキシおよびアリールオキシからなる群より選択される二つ以上の部分で置換されたフェニル基である、請求項26記載の方法。
【請求項28】
Dが、二つ以上のアルコキシ基で置換されたフェニル基である、請求項27記載の方法。
【請求項29】
アルコキシ基がメトキシ基である、請求項28記載の方法。
【請求項30】
化合物が、下記式の化合物
【化13】
【化14】
【化15】
およびその薬学的に許容される塩からなる群より選択される、請求項25記載の方法。
【請求項31】
癲癇誘発が抑制されるように有効量の化合物を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法であって、
該化合物は、下記式Dの化合物
【化16】
(式中、
i) Aは生理的pHにおいて陰性基であり;
ii) mおよびnは独立的に1、2、または3であり;
iii) Eは置換または非置換のフェニルである)
およびその薬学的に許容される塩である、方法。
【請求項32】
Aがカルボキシル基である、請求項31記載の方法。
【請求項33】
nが2である、請求項32記載の方法。
【請求項34】
nが1である、請求項32記載の方法。
【請求項35】
Eがジフェニルで置換されたメチルである、請求項34記載の方法。
【請求項36】
化合物が、下記式の化合物
【化17】
【化18】
【化19】
【化20】
およびその薬学的に許容される塩もしくはエステルからなる群より選択される、請求項31記載の方法。
【請求項37】
癲癇誘発が被験者において抑制されるように有効量の化合物を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法であって、該化合物が、下記式の化合物
【化21】
【化22】
【化23】
【化24】
およびその薬学的に許容される塩からなる群より選択され、その結果、癲癇誘発が被験者において抑制される方法。
【請求項38】
癲癇誘発が被験者において抑制されるように有効量の化合物を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法であって、該化合物が、下記式の化合物
【化25】
【化26】
【化27】
【化28】
【化29】
【化30】
【化31】
【化32】
【化33】
【化34】
【化35】
【化36】
【化37】
【化38】
およびその薬学的に許容される塩もしくはエステルからなる群より選択される、方法。
【請求項39】
被験者において癲癇誘発が抑制されるように、α,α-二置換β-アラニン、α,β-二置換β-アラニン、β,β-二置換β-アラニン、α,β,α-三置換β-アラニン、α,β,β-三置換β-アラニン、α,α,N-三置換β-アラニン、α,β,N-三置換β-アラニン、β,β,N-三置換β-アラニン、α,α,N,N-四置換β-アラニン、α,β,N,N-四置換β-アラニン、β,β,N,N-四置換β-アラニン、α,α,β,β-四置換β-アラニン、α,α,β,N-四置換β-アラニン、α,β,β,N-四置換β-アラニン、α,α,β,N,N-五置換β-アラニン、α,β,β,N,N-五置換β-アラニン、α,α,β,β,N-五置換β-アラニン、α,α,β,β,N,N-六置換β-アラニン、およびその薬学的に許容される塩もしくはエステルからなる群より選択される化合物の有効量を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法。
【請求項40】
被験者における癲癇誘発状態を診断する方法であって、検出可能なマーカーで標識されている下記式の化合物
【化39】
【化40】
【化41】
【化42】
からなる群より選択される化合物を該被験者に投与する段階;
該被験者の脳のニューロンのNMDA受容体への該化合物の結合の増加を測定し、これにより、該被験者における癲癇誘発状態を診断する段階を含む、方法。
【請求項41】
被験者における癲癇誘発状態を診断する方法であって、検出可能なマーカーで標識されている下記式の化合物
【化43】
【化44】
【化45】
【化46】
からなる群より選択された化合物を該被験者に投与する段階;
該被験者の脳のニューロンのGABA受容体への該化合物の結合の減少を測定し、これにより、該被験者における癲癇誘発状態を診断する段階を含む、方法。
【請求項42】
下記式の化合物
【化47】
または
【化48】
(式中、各Xは、独立的に、ハロゲン、ニトロ、シアノ、ならびに置換もしくは非置換のアルキル基およびアルコキシ基からなる群より選択され;nは0〜5の整数であり;YRおよびYSの一方が水素であり、他方が置換もしくは非置換のアミンである)、
およびその薬学的に許容される塩からなる群より選択される化合物を投与する段階を含む、被験者における癲癇誘発状態を診断する方法。
【請求項43】
被験者における癲癇誘発が抑制されるように、有効量の化合物を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法であって、
該化合物が、下記式の化合物
【化49】
または
【化50】
(式中、各Xは、独立的に、ハロゲン、ニトロ、シアノ、ならびに置換もしくは非置換のアルキル基およびアルコキシ基からなる群より選択され;nは0〜5の整数であり;YRおよびYSの一方が水素であり、他方が置換もしくは非置換のアミンである)、
およびその薬学的に許容される塩からなる群より選択される、方法。
【請求項44】
化合物が、(R)-3-アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロピオン酸、(S)-3-アミノ-3-[3-(トリフルオロメチルフェノキシ)フェニル]プロピオン酸、(R)-3-アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロピオン酸、(S)-3-アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロピオン酸、(R)-3-アミノ-3-[3-(フェノキシ)フェニル]プロピオン酸、(S)-3-アミノ-3-[3-(フェノキシ)フェニル]プロピオン酸、(D)-(+)-3-アミノ-3-[3-(4-クロロフェノキシ)フェニル]プロピオン酸、(L)-(-)-3-アミノ-3-[3-(4-クロロフェノキシ)フェニル]プロピオン酸、(L)-(-)-3-アミノ-3-[3-(3,4-ジクロロフェノキシ)フェニル]プロピオン酸、(D)-(+)-3-アミノ-3-[3-(3,4-ジクロロフェノキシ)フェニル]プロピオン酸、3-アミノ-3-(3-フェノキシ)フェニルプロピオン酸、およびその薬学的に許容される塩もしくはエステルからなる群より選択される、請求項42または43のいずれか一項に記載の方法。
【請求項45】
下記式の化合物
【化51】
(式中、R13は、水素、アルキル、アリール、または有機塩もしくは無機塩形成カチオンであり;nは1〜5であり;tは1〜2であり;各Xは、独立的に、ハロゲン、ニトロ、シアノ、ならびに置換もしくは非置換のアルキル基およびアルコキシ基からなる群より選択される)、
およびその薬学的に許容される塩もしくはエステルからなる群より選択される化合物を投与する段階を含む、被験者における癲癇誘発状態を診断する方法。
【請求項46】
被験者における癲癇誘発が抑制されるように、有効量の化合物を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法であって、
該化合物が、下記式の化合物
【化52】
(式中、R13は、水素、アルキル、アリールまたは有機塩もしくは無機塩形成カチオンであり;nは1〜5であり;tは1〜2であり;各Xは、独立的に、ハロゲン、ニトロ、シアノならびに置換もしくは非置換のアルキル基およびアルコキシ基からなる群より選択される)、
およびその薬学的に許容される塩もしくはエステルからなる群より選択される、方法。
【請求項47】
化合物が、3-アミノ-3-(4-ニトロフェニル)プロピオン酸、3-アミノ-3-(4-メチルフェニル)-2-カルボキシプロピオン酸、3-アミノ-3-(4-メトキシフェニル)-2-カルボキシプロピオン酸、3-アミノ-3-(4-ニトロフェニル)-2-カルボキシプロピオン酸、およびその薬学的に許容される塩もしくはエステルからなる群より選択される、請求項45または46のいずれか一項に記載の方法。
【請求項48】
アントラニル酸またはその薬学的に許容される塩を投与する段階を含む、被験者における癲癇誘発状態を診断する方法。
【請求項49】
被験者における癲癇誘発が抑制されるように、有効量のアントラニル酸を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法。
【請求項1】
i) 各々が
a) 癲癇誘発に関与するポリペプチドに対して直接的または間接的な薬理作用を引き起こす能力、および
b) 該薬理作用の少なくとも一部を示すことが決定されているファルマコフォア
を有する二つ以上の化合物の構造を得る段階、
ii) 該二つ以上の化合物のファルマコフォアの構造に基づいた平均的なファルマコフォア構造を決定する段階、ならびに
iii) 平均的なファルマコフォアを含む新規化合物を選択する段階を含む、
被験者における癲癇誘発を抑制する化合物を同定する方法。
【請求項2】
i) 各々が
a) 癲癇誘発に関与するポリペプチドに対して直接的または間接的な薬理作用を引き起こす能力、および
b) 該薬理作用の少なくとも一部を示すことが決定されているファルマコフォア
を有する二つ以上の化合物の構造を得る段階、
ii) 該二つ以上の化合物のファルマコフォア構造に基づいた平均的なファルマコフォア構造を決定する段階、
iii) 癲癇誘発に関与する異なるポリペプチドについて、段階(i)および(ii)を少なくとも1回繰り返す段階、
iv) 前記の段階で決定した一つ以上の平均的なファルマコフォアを含む新規化合物を選択する段階
を含む、被験者における癲癇誘発を抑制する化合物を同定する方法。
【請求項3】
癲癇誘発に関与するポリペプチドに対する薬理活性が、阻害、アゴニスト作用、アンタゴニスト作用、キレート化、および結合からなる群より選択される、請求項1記載の方法。
【請求項4】
構造が炭素骨格構造である、請求項1記載の方法。
【請求項5】
構造が3次元空間充填構造である、請求項1記載の方法。
【請求項6】
癲癇誘発に関与するポリペプチドが細胞表面受容体である、請求項1記載の方法。
【請求項7】
癲癇誘発に関与するポリペプチドがNMDA受容体である、請求項6記載の方法。
【請求項8】
癲癇誘発に関与するポリペプチドが神経伝達物質の輸送に関与する、請求項1記載の方法。
【請求項9】
癲癇誘発に関与するポリペプチドがGABA輸送体である、請求項8記載の方法。
【請求項10】
癲癇誘発を抑制する請求項1記載の方法で同定された化合物の有効量を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法。
【請求項11】
癲癇誘発が抑制されるように有効量の化合物を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法であって、
該化合物が、下記式Aの化合物
【化1】
(式中
i) R1は、水素、アルキル、アルケニル、アルキニル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;
ii) R2は、アルキル、アルケニル、アルキニル、アリール、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、またはアリールオキシカルボニルであり;
iii) Aは生理的pHにおいて陰性基である)
およびその薬学的に許容される塩もしくはエステルである、方法。
【請求項12】
Aがカルボキシルである、請求項11記載の方法。
【請求項13】
R1が水素である、請求項12記載の方法。
【請求項14】
R2がアルキルである、請求項13記載の方法。
【請求項15】
R2がアリールアルキルである、請求項14記載の方法。
【請求項16】
R2がフェニルアルキルである、請求項15記載の方法。
【請求項17】
化合物が、下記式の化合物
【化2】
【化3】
【化4】
【化5】
【化6】
【化7】
およびその薬学的に許容される塩もしくはエステルからなる群より選択される、請求項11記載の方法。
【請求項18】
癲癇誘発が抑制されるように有効量の化合物を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法であって、
該化合物が、下記式Bの化合物
【化8】
(式中、
i) Aは生理的pHにおいて陰性基であり;
ii) Bはフェノキシで置換されたフェニル基である)
およびその薬学的に許容される塩もしくはエステルである、方法。
【請求項19】
Aがカルボキシル基である、請求項18記載の方法。
【請求項20】
Bがアルキルフェノキシで置換されたフェニル基である、請求項19記載の方法。
【請求項21】
Bがメチルフェノキシで置換されたフェニル基である、請求項20記載の方法。
【請求項22】
Bがハロフェノキシで置換されたフェニル基である、請求項19記載の方法。
【請求項23】
Bがクロロフェノキシで置換されたフェニル基である、請求項22記載の方法。
【請求項24】
化合物が、下記式の化合物
【化9】
【化10】
【化11】
およびその薬学的に許容される塩もしくはエステルからなる群より選択される、請求項18記載の方法。
【請求項25】
癲癇誘発が抑制されるように有効量の化合物を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法であって、
該化合物が、下記式Cの化合物
【化12】
(式中、
i) Aは生理的pHにおいて陰性基であり;
ii) Dが、アルコキシおよびアリールオキシからなる群より選択される二つ以上の部分で置換されたアリール基である)
およびその薬学的に許容される塩である、方法。
【請求項26】
Aがカルボキシル基である、請求項25記載の方法。
【請求項27】
Dが、アルコキシおよびアリールオキシからなる群より選択される二つ以上の部分で置換されたフェニル基である、請求項26記載の方法。
【請求項28】
Dが、二つ以上のアルコキシ基で置換されたフェニル基である、請求項27記載の方法。
【請求項29】
アルコキシ基がメトキシ基である、請求項28記載の方法。
【請求項30】
化合物が、下記式の化合物
【化13】
【化14】
【化15】
およびその薬学的に許容される塩からなる群より選択される、請求項25記載の方法。
【請求項31】
癲癇誘発が抑制されるように有効量の化合物を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法であって、
該化合物は、下記式Dの化合物
【化16】
(式中、
i) Aは生理的pHにおいて陰性基であり;
ii) mおよびnは独立的に1、2、または3であり;
iii) Eは置換または非置換のフェニルである)
およびその薬学的に許容される塩である、方法。
【請求項32】
Aがカルボキシル基である、請求項31記載の方法。
【請求項33】
nが2である、請求項32記載の方法。
【請求項34】
nが1である、請求項32記載の方法。
【請求項35】
Eがジフェニルで置換されたメチルである、請求項34記載の方法。
【請求項36】
化合物が、下記式の化合物
【化17】
【化18】
【化19】
【化20】
およびその薬学的に許容される塩もしくはエステルからなる群より選択される、請求項31記載の方法。
【請求項37】
癲癇誘発が被験者において抑制されるように有効量の化合物を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法であって、該化合物が、下記式の化合物
【化21】
【化22】
【化23】
【化24】
およびその薬学的に許容される塩からなる群より選択され、その結果、癲癇誘発が被験者において抑制される方法。
【請求項38】
癲癇誘発が被験者において抑制されるように有効量の化合物を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法であって、該化合物が、下記式の化合物
【化25】
【化26】
【化27】
【化28】
【化29】
【化30】
【化31】
【化32】
【化33】
【化34】
【化35】
【化36】
【化37】
【化38】
およびその薬学的に許容される塩もしくはエステルからなる群より選択される、方法。
【請求項39】
被験者において癲癇誘発が抑制されるように、α,α-二置換β-アラニン、α,β-二置換β-アラニン、β,β-二置換β-アラニン、α,β,α-三置換β-アラニン、α,β,β-三置換β-アラニン、α,α,N-三置換β-アラニン、α,β,N-三置換β-アラニン、β,β,N-三置換β-アラニン、α,α,N,N-四置換β-アラニン、α,β,N,N-四置換β-アラニン、β,β,N,N-四置換β-アラニン、α,α,β,β-四置換β-アラニン、α,α,β,N-四置換β-アラニン、α,β,β,N-四置換β-アラニン、α,α,β,N,N-五置換β-アラニン、α,β,β,N,N-五置換β-アラニン、α,α,β,β,N-五置換β-アラニン、α,α,β,β,N,N-六置換β-アラニン、およびその薬学的に許容される塩もしくはエステルからなる群より選択される化合物の有効量を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法。
【請求項40】
被験者における癲癇誘発状態を診断する方法であって、検出可能なマーカーで標識されている下記式の化合物
【化39】
【化40】
【化41】
【化42】
からなる群より選択される化合物を該被験者に投与する段階;
該被験者の脳のニューロンのNMDA受容体への該化合物の結合の増加を測定し、これにより、該被験者における癲癇誘発状態を診断する段階を含む、方法。
【請求項41】
被験者における癲癇誘発状態を診断する方法であって、検出可能なマーカーで標識されている下記式の化合物
【化43】
【化44】
【化45】
【化46】
からなる群より選択された化合物を該被験者に投与する段階;
該被験者の脳のニューロンのGABA受容体への該化合物の結合の減少を測定し、これにより、該被験者における癲癇誘発状態を診断する段階を含む、方法。
【請求項42】
下記式の化合物
【化47】
または
【化48】
(式中、各Xは、独立的に、ハロゲン、ニトロ、シアノ、ならびに置換もしくは非置換のアルキル基およびアルコキシ基からなる群より選択され;nは0〜5の整数であり;YRおよびYSの一方が水素であり、他方が置換もしくは非置換のアミンである)、
およびその薬学的に許容される塩からなる群より選択される化合物を投与する段階を含む、被験者における癲癇誘発状態を診断する方法。
【請求項43】
被験者における癲癇誘発が抑制されるように、有効量の化合物を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法であって、
該化合物が、下記式の化合物
【化49】
または
【化50】
(式中、各Xは、独立的に、ハロゲン、ニトロ、シアノ、ならびに置換もしくは非置換のアルキル基およびアルコキシ基からなる群より選択され;nは0〜5の整数であり;YRおよびYSの一方が水素であり、他方が置換もしくは非置換のアミンである)、
およびその薬学的に許容される塩からなる群より選択される、方法。
【請求項44】
化合物が、(R)-3-アミノ-3-[3-(3-トリフルオロメチルフェノキシ)フェニル]プロピオン酸、(S)-3-アミノ-3-[3-(トリフルオロメチルフェノキシ)フェニル]プロピオン酸、(R)-3-アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロピオン酸、(S)-3-アミノ-3-[3-(4-メチルフェノキシ)フェニル]プロピオン酸、(R)-3-アミノ-3-[3-(フェノキシ)フェニル]プロピオン酸、(S)-3-アミノ-3-[3-(フェノキシ)フェニル]プロピオン酸、(D)-(+)-3-アミノ-3-[3-(4-クロロフェノキシ)フェニル]プロピオン酸、(L)-(-)-3-アミノ-3-[3-(4-クロロフェノキシ)フェニル]プロピオン酸、(L)-(-)-3-アミノ-3-[3-(3,4-ジクロロフェノキシ)フェニル]プロピオン酸、(D)-(+)-3-アミノ-3-[3-(3,4-ジクロロフェノキシ)フェニル]プロピオン酸、3-アミノ-3-(3-フェノキシ)フェニルプロピオン酸、およびその薬学的に許容される塩もしくはエステルからなる群より選択される、請求項42または43のいずれか一項に記載の方法。
【請求項45】
下記式の化合物
【化51】
(式中、R13は、水素、アルキル、アリール、または有機塩もしくは無機塩形成カチオンであり;nは1〜5であり;tは1〜2であり;各Xは、独立的に、ハロゲン、ニトロ、シアノ、ならびに置換もしくは非置換のアルキル基およびアルコキシ基からなる群より選択される)、
およびその薬学的に許容される塩もしくはエステルからなる群より選択される化合物を投与する段階を含む、被験者における癲癇誘発状態を診断する方法。
【請求項46】
被験者における癲癇誘発が抑制されるように、有効量の化合物を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法であって、
該化合物が、下記式の化合物
【化52】
(式中、R13は、水素、アルキル、アリールまたは有機塩もしくは無機塩形成カチオンであり;nは1〜5であり;tは1〜2であり;各Xは、独立的に、ハロゲン、ニトロ、シアノならびに置換もしくは非置換のアルキル基およびアルコキシ基からなる群より選択される)、
およびその薬学的に許容される塩もしくはエステルからなる群より選択される、方法。
【請求項47】
化合物が、3-アミノ-3-(4-ニトロフェニル)プロピオン酸、3-アミノ-3-(4-メチルフェニル)-2-カルボキシプロピオン酸、3-アミノ-3-(4-メトキシフェニル)-2-カルボキシプロピオン酸、3-アミノ-3-(4-ニトロフェニル)-2-カルボキシプロピオン酸、およびその薬学的に許容される塩もしくはエステルからなる群より選択される、請求項45または46のいずれか一項に記載の方法。
【請求項48】
アントラニル酸またはその薬学的に許容される塩を投与する段階を含む、被験者における癲癇誘発状態を診断する方法。
【請求項49】
被験者における癲癇誘発が抑制されるように、有効量のアントラニル酸を被験者に投与する段階を含む、被験者における癲癇誘発を抑制する方法。
【図1】

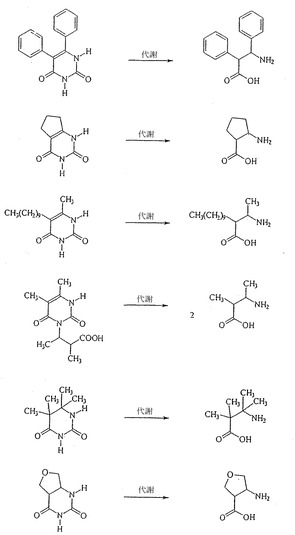
【図2】
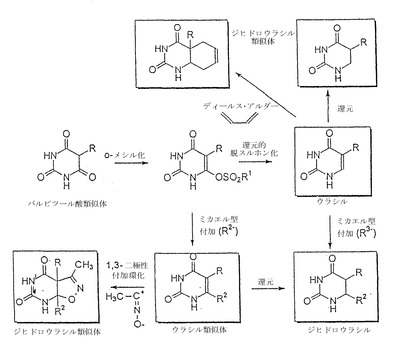
【図3】
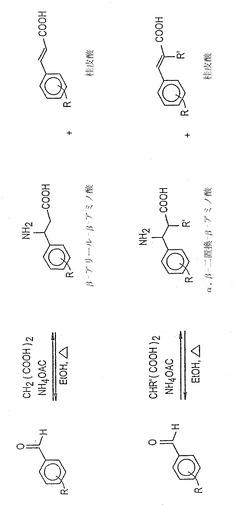
【図4】
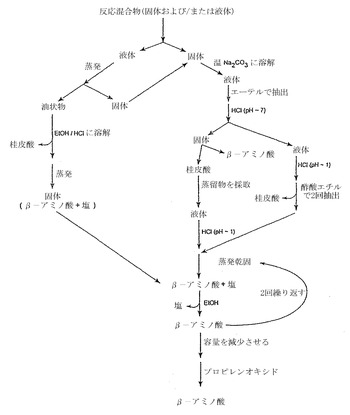
【図2】
【図3】
【図4】
【公開番号】特開2007−302678(P2007−302678A)
【公開日】平成19年11月22日(2007.11.22)
【国際特許分類】
【出願番号】特願2007−150044(P2007−150044)
【出願日】平成19年6月6日(2007.6.6)
【分割の表示】特願2002−572418(P2002−572418)の分割
【原出願日】平成14年3月13日(2002.3.13)
【出願人】(391018835)クイーンズ ユニバーシティ アット キングストン (9)
【氏名又は名称原語表記】QUEEN’S UNIVERSITY AT KINGSTON
【出願人】(304048517)ニューロケム (インターナショナル) リミテッド (1)
【Fターム(参考)】
【公開日】平成19年11月22日(2007.11.22)
【国際特許分類】
【出願日】平成19年6月6日(2007.6.6)
【分割の表示】特願2002−572418(P2002−572418)の分割
【原出願日】平成14年3月13日(2002.3.13)
【出願人】(391018835)クイーンズ ユニバーシティ アット キングストン (9)
【氏名又は名称原語表記】QUEEN’S UNIVERSITY AT KINGSTON
【出願人】(304048517)ニューロケム (インターナショナル) リミテッド (1)
【Fターム(参考)】
[ Back to top ]