
抗糖尿病化合物の新規な結晶形態
フェノキシアルキルカルボン酸基に結合された2つのインドール化合物の新規な結晶性形態は、2型糖尿病、高血糖症、肥満症、異脂肪血症、または代謝症候群の治療に有用な選択的PPARガンマ部分作動薬である。新規な結晶形態は、第1の化合物の結晶性遊離酸二水和物および結晶性遊離酸無水物と、第2の化合物の遊離酸およびナトリウム塩のいくつかの結晶性形態を含む。本発明はまた、これらの新規な結晶形態を含む医薬組成物、この結晶形態およびこの医薬組成物を調製するための方法、ならびに2型糖尿病および他のPPARガンマ調節された疾患の治療における結晶形態の使用を含む医薬組成物に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、2型糖尿病、ならびに高血糖症、肥満症、異脂肪血症および代謝病を含む、PPARガンマ作動薬により調節される他の疾患の治療用の医薬有効成分として有用な化合物の新規な結晶形態および塩形態に関する。該化合物は選択的PPARガンマ部分作動薬(またSPPARgMまたはSPPARMとして知られている)である。
【背景技術】
【0002】
2型糖尿病は、依然として深刻な医療問題である。より有効で、副作用の少ない新しい治療が継続して求められている。
【発明の開示】
【課題を解決するための手段】
【0003】
本発明は、SPPARMまたはSPPARgMとしても知られるPPARガンマ部分作動薬として活性な化合物のクラスに属する化合物の新規な結晶形態、水和物、無水物および塩に関する。関連する化合物が、公開PCT出願04/020409に最初に開示されている。2つの構造的に類似する化合物の結晶形態、水和物、無水物、および/または塩が開示されている。(1)第1の化合物は、(2R)−2−{3−[3−(4−メトキシベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]フェノキシ}ブタン酸(化合物I)であり、結晶性二水和物および結晶性無水物として開示されている。ここに開示されている結晶性二水和物および結晶性無水物の形態は、精製の容易さ、処理の容易さおよび組成物の安定性など、医薬組成物の調製においての利点である特性を有し、先に開示された(2R)−2−{3−[3−(4−メトキシベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]フェノキシ}ブタン酸の非結晶形態に対して利点を有する。(2)第2の化合物は、(2R)−2−{3−[[3−(4−クロロベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]メチル]フェノキシ}酪酸(化合物II)であり、少なくとも5つの新規な結晶性多形形態を有するナトリウム塩および少なくとも3つの新規な結晶性多形形態を有する遊離酸としてここに開示されている。これらの多形形態の一部は、精製の容易さ、処理の容易さおよび組成物の安定性など、医薬組成物の調製においての利点である特性を有し、先に開示された化合物IIの固体形態に対して利点を有する。
【0004】
本発明はまた、新規な結晶性多形を含む医薬組成物、これらの多形形態およびこれらの医薬組成物を調製するための方法、2型糖尿病、高血糖症、肥満症、異脂肪血症および代謝病の治療用にこれらを使用するための方法に関する。
【発明を実施するための最良の形態】
【0005】
本発明は、構造式I(化合物I)
【0006】
【化3】

を有する(2R)−2−{3−[3−(4−メトキシベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]フェノキシ}ブタン酸の新規な結晶性二水和物および無水物の多形形態を提供する。
【0007】
上記構造は、公開PCT出願04/020409における実施例3として最初に開示された。’409特許公開における合成方法を使用して単離された化合物は、非結晶の固体である。発明者らはここに、非結晶化合物Iの結晶性二水和物および結晶性無水物を見い出した。これらの結晶化合物は、医薬組成物の調製においてきわめて容易に使用される。結晶性二水和物は、乾燥窒素流中で加温することによって結晶性無水物に容易に転換され、結晶性無水物は、湿窒素気流(RH>70%)中に放置することによって容易に結晶性二水和物に転換して戻る。結晶性二水和物は、非吸湿性である。
【0008】
本発明のさらなる実施形態は、検出可能な量の化合物Iの結晶性無水物形態または結晶性二水和物形態を含む原薬を提供する。「原薬(drug substance)」という用語は、活性医薬成分(API)を意味する。原薬中の結晶性無水物形態または結晶性二水和物形態の量は、X線粉末回折(XRPD)法、固体フッ素−19マジック角回転(MAS)核磁気共鳴分光法、固体炭素−13交差偏光マジック角回転(CPMAS)核磁気共鳴分光法、固体フーリエ変換赤外分光法およびラマン分光法などの物理的方法の使用によって定量することができる。この実施形態の第1のクラスにおいて、結晶性無水物形態または結晶性二水和物形態の約5重量%から約100重量%が原薬中に存在する。この実施形態の第2のクラスにおいて、結晶性無水物形態または結晶性二水和物形態の約10重量%から約100重量%が原薬中に存在する。この実施形態の第3のクラスにおいて、結晶性無水物形態または結晶性二水和物形態の約25重量%から約100重量%が原薬中に存在する。この実施形態の第4のクラスにおいて、結晶性無水物形態または結晶性二水和物形態の約50重量%から約100重量%が原薬中に存在する。この実施形態の第5のクラスにおいて、結晶性無水物形態または結晶性二水和物形態の約75重量%から約100重量%が原薬中に存在する。この実施形態の第6のクラスにおいて、実質的にすべての化合物I原薬は、結晶性無水物形態または結晶性二水和物形態であり、すなわち化合物I原薬は、実質的に純相結晶性無水物形態または純相結晶性二水和物形態である。
【0009】
本発明の別の態様は、PPARガンマ作動薬が適応される臨床状態の治療もしくは管理のための方法であり、このような治療もしくは管理を必要としている患者に化合物Iの結晶性無水物形態もしくは結晶性二水和物形態の治療有効量、または化合物Iの結晶性無水物形態または結晶性二水和物形態の治療有効量を含む医薬組成物を投与することを含む方法を提供する。このような臨床状態としては、2型糖尿病、高血糖症、肥満症、異脂肪血症および代謝症候群が挙げられる。「患者」は、ヒトを含む哺乳類である。患者はヒトの患者が最も多い。
【0010】
本発明はまた、PPARガンマ作動薬が適応される1以上の臨床状態の患者における治療または管理のための医薬の製造において本発明の結晶性無水物形態もしくは結晶性二水和物形態の使用を提供する。1つの実施形態において、臨床状態は2型糖尿病である。
【0011】
本発明の別の態様は、PPARガンマ作動薬が適用される1以上の臨床状態の患者の治療または管理において使用する結晶性無水物形態または結晶性二水和物形態を提供する。この態様の1つの実施形態では、臨床状態は2型糖尿病である。
【0012】
本発明はまた、1以上の医薬上許容される担体または賦形剤と共に結晶性無水物形態もしくは結晶性二水和物形態を含む医薬組成物を提供する。1つの実施形態において、医薬組成物は、活性医薬成分(API)が本発明の結晶性無水物形態もしくは結晶性二水和物形態の検出可能量を含む、医薬上許容される賦形剤と混合したAPIを含む。第2の実施形態において、医薬組成物は、APIが本発明の結晶性無水物形態または結晶性二水和物形態の約5重量%から約100重量%を含む、医薬上許容される賦形剤と混合したAPIを含む。この第2の実施形態の1つのクラスにおいて、このような組成物中のAPIは、結晶性無水物形態または結晶性二水和物形態の約10重量%から約100重量%を含む。この実施形態の第2のクラスでは、このような組成物中のAPIは、結晶性無水物形態または結晶性二水和物形態の約25重量%から約100重量%を含む。この実施形態の第3のクラスでは、このような組成物中のAPIは、結晶性無水物形態または結晶性二水和物形態の約50重量%から約100重量%を含む。この実施形態の第4のクラスにおいて、このような組成物中のAPIは、結晶性無水物形態または結晶性二水和物形態の約75重量%から約100重量%を含む。この実施形態の第5のクラスにおいて、実質的にすべてのAPIは、化合物Iの結晶性無水物形態または結晶性二水和物形態であり、すなわちAPIは、結晶性無水物形態における実質的に純相化合物Iであるかまたは結晶性二水和物形態における実質的に純相化合物Iである。
【0013】
本発明による組成物は、好適には、錠剤、丸薬、カプセル、粉末、顆粒、滅菌溶液もしくは懸濁液、定量エアロゾルもしくは液体スプレイ、ドロップ、アンプル、自動注入デバイスもしくは座薬などの単位投与形態にある。この組成物は、経口、非経口、経鼻、舌下、もしくは直腸投与、または吸入もしくは吹送による投与を対象としたものである。本発明による組成物の処方は、例えばRemington’s Pharmaceutical Science、第17版、1995年に記載の通り、当該技術分野に知られた方法によって都合良くもたらされる。
【0014】
投薬計画は、患者のタイプ、種、年齢、体重、性および患者の医学的状態;治療する症状の重症度;投与経路;ならびに患者の腎臓および肝臓の機能を含むさまざまな要因によって選択される。通常の技術を有する医者、獣医、または臨床医であれば、この状態の進行を防ぐ、対抗する、もしくは止める、または状態を治療もしくは管理するために必要とされる薬剤の有効量を容易に決定し、処方することができる。
【0015】
示された効果のために使用される場合、本発明の経口投与量は、1日当たり体重1kg当たり約0.01mg(mg/kg/day)から約100mg/kg/day、好ましくは0.01〜10mg/kg/dayの範囲である。経口投与のために、組成物は、例えば、遊離酸の形態で測定してAPI0.05、0.1、0.5、1.0、2.5、5.0、10.0、15.0、25.0、50.0、100、200、400、600、800、または1,000mgを含む錠剤もしくはカプセル、または治療結果を達成するために治療される患者にとって適切であり得るような他の投与形態で提供され得る。医薬は、通常API約0.5mgから約500mg、好ましくはAPI約1mgから約200mgを含む。経静脈的に、最も好ましい投与量は、定速輸液の間、約0.1から約10mg/kg/分の範囲である。有利には、本発明の結晶性無水物形態および結晶性二水和物形態は、1日1回投与する1日当たりの総量を1日2回、3回または4回の分割投与量で投与することができる。さらに、本発明の結晶性無水物形態および結晶性二水和物形態は、好適な経鼻投与の媒体の局所の使用を介して鼻孔内形態において、または通常の当業者によく知られた経皮皮膚用パッチ剤のそれらの形態を使用する経皮経路によって投与し得る。経皮送達系の形態において投与するために、投薬量投与は、当然、投与計画を通して間欠的ではなく連続的である。
【0016】
本発明の方法において、本明細書に詳細に述べる化合物Iの結晶性無水物形態および結晶性二水和物形態はAPIを形成することができ、投与の意図した形態、すなわち、経口用の錠剤、カプセル、エリキシル剤、シロップなどに関して好適に選択され従来の医薬慣行と一致している好適な薬剤希釈剤、賦形剤または担体(本明細書では、集合的に「担体」材料という)と混合して通常投与される。
【0017】
例えば、錠剤もしくはカプセルの形態での経口投与に対して、活性薬剤成分は、ラクトース、デンプン、サッカロース、グルコース、メチルセルロース、ステアリン酸マグネシウム、リン酸二カルシウム、硫酸カルシウム、マンニトール、ソルビトールなどの経口、非毒性の医薬上許容される不活性担体と合わせることができる;液体形態における経口投与として、経口APIは、エタノール、グリセロール、水などの任意の経口、非毒性、医薬上許容される不活性担体と合わせることができる。さらに、所望もしくは必要な場合、好適な結合剤、潤滑剤、崩壊剤および着色剤も、混合物に含むことができる。好適な結合剤としては、デンプン、ゼラチン、グルコースまたはベータ−ラクトースなどの天然砂糖、コーン甘味剤、アカシア、トラガカントまたはアルギン酸ナトリウム、カルボキシメチルセルロース、ポリエチレングリコール、ワックスなど天然ゴムおよび合成ゴムが挙げられる。これら投与形態で使用される潤滑剤としては、限定されることなく、オレイン酸ナトリウム、ステアリン酸ナトリウム、ステアリン酸マグネシウムおよびステアリルフマル酸ナトリウムが挙げられる。崩壊剤は、限定されることなく、デンプン、メチルセルロース、寒天、ベントナイト、キサンタンガムなどが挙げられる。使用され得る他の通常使用される添加物としては、安息香酸ナトリウム、酢酸ナトリウム、塩化ナトリムなどが挙げられる。
【0018】
本発明は同様に、下記式II
【0019】
【化4】
を有し、本明細書において化合物IIと称される(2R)−2−{3−[[3−(4−クロロベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]メチル]フェノキシ}酪酸の新規な多形形態を提供する。
【0020】
さまざまなレベルの安定性と調製の容易さを有する化合物IIの広範な結晶形態がある。V型ナトリウム塩およびC型遊離酸は再現性をもって調製される。C型酸は、熱力学的に安定であり、医薬製剤の調製に特に好適である。これは非水和物結晶性化合物である。
【0021】
これらの結晶形態は、化合物Iについて上記で提供されたものと類似の実施形態を有する。
【0022】
以下の非限定的な実施例は本発明を説明するためのものであり、本発明の範囲または精神を限定するものとして考えられるべきではない。
【0023】
本明細書における化合物IおよびIIは、ケト−エノールなどの互変異性体などの互変異性体として存在し得る。個々の互変異性体ならびにその混合物は、構造式Iの化合物に包含される。
【実施例】
【0024】
(実施例1)
(2R)−2−{3−[3−(4−メトキシベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]フェノキシ}ブタン酸(化合物I)の合成
化合物Iは下記に示す多段プロセスによって作られる。
【0025】
ステップ1:ニトロ化
【0026】
【化5】
【0027】
容器に酢酸(8.0L)を入れ、引き続き添加漏斗によって4−(トリフルオロメトキシ)アニリン(8.00Kg)を攪拌しながら20〜35℃で15〜20分かけて入れた。次いで、無水酢酸(18.08kg)を20〜35℃で15〜20分かけて添加した。この反応混合物を20〜25℃で20〜30分間攪拌した。96%硫酸(320ml)を添加漏斗によって25〜35℃で5分かけて、引き続き90%硝酸(2.08L)を攪拌しながら別の添加漏斗によって25〜35℃で60分かけて入れた。添加後、反応混合物を20〜25℃で0.5〜1時間攪拌した。水(60L)を冷却しながら15〜25℃で40〜50分かけて添加した。3〜4Lの水を添加後、固体生成物が徐々に結晶化した。このスラリーを室温で1時間攪拌し、黄色の固体生成物をろ過によって単離した。湿ったケーキを水(3×20L)で洗浄し、窒素気流を使用して乾燥し、乾燥生成物3を得た。別法として、反応混合物を冷水に注ぐことによって上述したクエンチを行うことができる。
【0028】
ステップ2:加水分解
【0029】
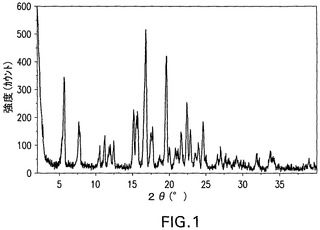
【化6】
【0030】
攪拌機、温度計プローブ、水冷浴および添加漏斗を備えた容器に、4−トリフルオロメトキシ−2−ニトロアセトアニリド(11.0Kg)およびメタノール(42L)を攪拌しながら室温で入れた。5NNaOH(10.41L)をスラリーに水浴を用いて20〜30℃で5〜10分かけて添加し、均一な濃い琥珀色の溶液を得た。この反応混合物を25〜30℃で1時間かけて攪拌した。反応が終了した後、水(63L)を氷水浴を用いて15〜25℃で40〜50分かけて添加した。水を添加後、このバッチを攪拌機を備えた200L容器に移した。このスラリーをろ過前に室温で2〜3時間さらに攪拌した。黄色の固体生成物をろ過によって単離し、湿ったケーキを3:2水/メタノール(2×12.5L)で洗浄し、真空吸引を用いて窒素下で乾燥し、2−ニトロ−4−トリフルオロメトキシアニリン4を得た。
【0031】
ステップ3:メーヤワインアリール化
【0032】
【化7】
【0033】
容器に酢酸イソプロペニル(30L)を入れ、次いで塩化銅(II)(2.93Kg)を攪拌しながら1回で18〜25℃で入れた。この溶液の温度を40〜45℃に調整した。酢酸イソプロペニル(5L)中アニリン4(4.05Kg)の溶液を、この反応混合物に添加漏斗によって1〜2時間かけて添加し、同時に亜硝酸イソブチル(2.84L)を別の添加漏斗で、反応温度を40〜50℃に保ちながら同じ時間をかけて添加した。
【0034】
反応混合物を18〜22℃に冷却し、トルエン(21.5L)で希釈し、1.0NHCl(21.5L)を添加して反応を停止した。反応混合物を15分間攪拌した。有機層を水(21.5L)、炭酸水素ナトリウム水溶液(21.5L)、および水(2.5L)で洗浄した。この有機層を部分真空(最高温度20℃)下で濃縮し、溶媒をトルエンに切り換えた。容量を10.75Lに調整し、均一な琥珀色の溶液を5〜10℃に冷却した。ヘプタン(3.6L)を添加し、混合物を1時間エージングした。ヘプタン(37L)を−5℃〜−10℃の温度を保ちながらゆっくり添加した。このスラリーを−25〜−20℃に冷却し、−25〜−20℃で1時間エージングし、ろ過した。濃い黄色のケーキをトルエン/ヘプタン(1:6v/v、11L)を用いて−25〜−20℃で洗浄し、さらにヘプタン(4L)を用いて−25〜20℃で洗浄し、室温、部分真空下、窒素気流で乾燥し、ケトン5を得た。
【0035】
ステップ4:インドール
【0036】
【化8】
【0037】
メタノール(7.6L)中基質5(2.0Kg)および5%Pd/C(484g)のスラリーを15℃、60psiの水素雰囲気下で1〜2時間水素添加し、次いで6時間で65℃に加熱した。反応混合物をソルカフロック(solca−floc)でろ過して触媒を除去し、MeOHで洗浄した。このバッチを濃縮し、溶媒をトルエンに切り換えた。6のトルエン溶液をカップリングステップにあるように使用した。
【0038】
ステップ5:3−ベンジルオキシヨードベンゼン
【0039】
【化9】
【0040】
容器に3−ヨードフェノール(6.8Kg)、粉末K2CO3(5.12Kg)、DMF(40.8L)および塩化ベンジル(4.30Kg)を入れた。得られたスラリーを68〜70℃に加熱し、6〜7時間攪拌した。反応混合物を35〜40℃に冷却し、水(40.8L)を同温で攪拌しながら1時間かけて添加した。水を添加後、得られたスラリーを室温で2〜3時間攪拌した。このバッチをろ過し、湿ったケーキを水(3×35L)で洗浄し、窒素下、真空吸引を12〜16時間して乾燥し、白色の結晶性固体として生成物7を得た。
【0041】
ステップ6:N−アリールインドール
【0042】
【化10】
【0043】
トルエン(9.6L)中インドール6(3.20Kg)の溶液に3−ベンジルオキシヨードベンゼン7(4.57Kg)、CuI(421g)、N,N−ジメチルエチレンジアミン(401g)、および炭酸セシウム(9.4Kg)を添加した。トルエン(6.5L)を添加した。このスラリーを激しく攪拌しながら105〜110℃で12時間エージングし、室温に冷却した。ソルカフロック(1.5Kg)、次いでトルエン(30L)を18〜25℃で添加した。次いで、水(30L)をよく攪拌しながら30〜60分かけて滴下した。この混合物を18〜25℃で1〜2時間エージングし、ろ過した。このケーキをトルエン(10L)ですすぎ洗いした。トルエンろ液を濃縮し、8の粗溶液を次のステップでそのまま使用した。この反応はまた、固体を懸濁したままに保つようによく混合し(反応器の底から、攪拌または再循環ループ)、まだ残留水分を有する前ステップからのベンジルオキシヨードベンゼンを使用して、2モル%の低い濃度で触媒としてCuCl2を使用して有利に行うことができる。空気の排除もまた、有利である。
【0044】
ステップ7:アシル化/脱ベンジル化
【0045】
【化11】
【0046】
トルエン(2.58L)中ベンジルオキシアリールインドール8(2.58Kg)の溶液を0℃に冷却し、ヘプタン(1.3L)中4−メトキシベンゾイルクロライド(1.58L)の溶液を10〜20分かけて添加した。ジエチルアルミニウムクロライド(1.8M、10.8L)の溶液を0〜5℃で30分かけて添加した。添加を終了すると直ちに、その反応物を室温まで加温した。水(2.12L)中6MHClを30分かけてゆっくり添加し、反応を止めた。得られたスラリーを室温まで温め、ヘプタン(49L)を添加し、混合物を室温で1〜2時間エージングし、次いでろ過した。反応物を冷水とヘプタンとの混合物に添加する逆クエンチを別に使用し得る。一夜窒素気流下で収集した固体を乾燥後、この物質をエタノール(17.3L)に溶解し、このスラリーを還流するまで加熱し、水(7.4L)中3M塩酸を添加した。この溶液を室温に冷却し、得られたスラリーをろ過し、エタノール中30%の3M塩酸で洗浄し、次いで窒素気流下で乾燥し、結晶性アシル化生成物9を得た。生成物をさらに、トルエンから1回の再結晶によって精製する。
【0047】
ステップ8A:
【0048】
【化12】
【0049】
ステップ8a:(S)2−ヒドロキシ酪酸イソブチル
容器にリン酸二水素カリウム(1.40Kg)および水(70.2L)を入れ、濃硫酸を添加することによってpHを7.5に調整した。ギ酸ナトリウム(1.54Kg)、および水(8L)中2−ケト酪酸カリウム塩(2Kg)の溶液を添加し、次いでβ−ニコチンアミドアデニンジヌクレオチド(96.3g)およびギ酸脱水素酵素(35.5g)を添加した。混合物を31〜37℃で攪拌し、水酸化ナトリウムを添加することによって溶液のpHを7.4に調整した。ニワトリ心臓酵素溶液(150mg)由来のL乳酸脱水素酵素VIII型を添加し、反応物を攪拌しながら33〜38℃で18時間エージングし、硫酸溶液を使用してpHを7.4〜7.6に調節した。別に、この反応をギ酸脱水素酵素ではなくグルコース脱水素酵素を使用して行い、再生されるように化学量論的な還元剤の還元を行うこともできる。
【0050】
転化が終了した後、イソブチルアルコール(40L)および塩化カリウム(5Kg)、次いで硫酸を添加し、pHを2に調整した。水層をイソブチルアルコール(3×40L)で抽出した。混合抽出物を約20%容量まで濃縮し、濃硫酸(5モル%)を添加し、この溶液を12時間エージングしてエステル化を完了した。溶液を10%重炭酸カリウム水溶液(10L)で洗浄した。この後処理においてN−メチルモルホリンを10%重炭酸カリウムの代りに使用することができる。層を分離し、有機層を水(10L)で洗浄した。分別蒸留カラムを使用した濃縮、および減圧下で溶媒のトルエンへの切り換えによって、トルエン中25%溶液として粗(S)2−ヒドロキシ酪酸イソブチルを得た。これをさらなる精製をすることなしに使用した。
【0051】
ステップ8B:(S)イソブチル2−トシルオキシ酪酸
酢酸エチル(14L)中(S)2−ヒドロキシ酪酸イソブチル(2.72Kg)およびp−トルエンスルホニルクロライド(3.6Kg)に20℃で、酢酸エチル(10L)中DABCO(286g)およびトリエチルアミン(4.7L)の溶液を2時間かけて添加した。この反応物を20〜25℃で4時間エージングした。この混合物を水(10L、2L、および2L)で3回洗浄し、有機層を濃縮し、溶媒をトルエンに切り換えた。(S)2−イソブチルトシル酪酸の濃縮トルエン溶液をカップリング反応におけるように使用した。別に、アセトニトリルを酢酸エチルの代りに使用することができ、この場合は反応はより速く進む。
【0052】
ステップ9:アシル化
【0053】
【化13】
【0054】
攪拌機、温度計プローブ、窒素注入口および添加漏斗を備えた50Lの容器に、DMSO(6L)、(S)トシルオキシ酪酸2−イソブチル(1.70Kg)およびインドールフェノール9(2.04Kg)を窒素下、20℃で入れた。粉末炭酸カリウム(1.88Kg)を添加した。DMSO(2L)を使用し、これらの物質を容器中にすすぎ洗いした。得られたスラリーを28〜30℃で24時間攪拌した。反応が完了したら、MTBE(10L)を添加し、スラリーを10℃に冷却した。水(15L)を添加し、層を室温(20〜25℃)でよく混合した。下側の水層を除去した。有機層を0.2M炭酸水素カリウム(2×5L)で洗浄した。粗生成物10を加水分解ステップにおいて精製することなく使用した。
【0055】
ステップ10:加水分解
【0056】
【化14】
【0057】
MTBE中エステル10(8.98Kg)の溶液をメタノール(27L)で希釈した。1M水酸化ナトリウム(30.8L)を室温で添加した。二相性反応混合物を40℃で2時間激しく攪拌した。(S)トシルオキシ酪酸2−イソブチルとのカップリング反応(ステップ9)由来の残留トルエン量に依存して、比較的長い反応時間(40℃で〜16時間)が必要とされる。反応混合物を室温に冷却し、MTBE(22L)で希釈した。5M塩酸(26.8L)を激しく攪拌しながら添加した。有機層を水(3×20L)で洗浄した。有機層をその最初の容量の約1/3に濃縮し、トルエンで洗い流して残存MTBEを取り除き、約40Lの全バッチ容量までトルエンで希釈した。この溶液に水(1L)を激しく攪拌しながら添加した。二相性混合物を室温(18〜23℃)で数分間エージングし、化合物I二水和物の種を用いて結晶種を入れた(実施例4)。このスラリーを室温(18〜23℃)で5時間激しく攪拌した。ヘプタン(51.5L)を攪拌しながら室温で2〜4時間かけて添加した。このバッチをろ過し、湿ったケーキを1:1トルエン/ヘプタン(15L)、引き続きヘプタン(15L)で洗浄し、乾燥窒素下で40〜50℃で乾燥し、白色の結晶性固体として化合物Iの遊離酸無水物を得た。上記で得られた固体無水物を湿った窒素(相対湿度>70%、不凝縮)の気流下に置くことによって、結晶性二水和物に水和する。
【0058】
(実施例2)
化合物Iの結晶性無水物および二水和物の調製
化合物Iの水和物と無水物は容易に相互転換する。実施例1由来の化合物I二水和物、または遊離酸としての他の供給源由来の粗もしくは純化合物I二水和物は、結晶性無水物にすることができるとともに、以下のように結晶性二水和物に転換して戻すことができる。化合物I(7.57Kgアッセイ、14.4モル)を十分に乾燥したトルエンに溶解し、30.3Lの全容量(4.0Lトルエン溶液/化合物IのKg)を有する溶液とする。
【0059】
水(1.03L;1.03Kg;57.4モル;4.00当量)をこの溶液に激しく攪拌しながら添加する。この二相性混合物を室温(18〜23℃)で数分間エージングし、次いで、化合物I二水和物の種(67g、0.12モル、0.0083当量)を用いて結晶種を入れる。このバッチは結晶種を入れた後、濁る。固体が徐々に析出し、濃いスラリーを得る。このスラリーを、上清の平衡(トルエン中60〜80mg/mlの化合物I)に達するまで室温(18〜23℃)で激しく攪拌する。
【0060】
次いで、ヘプタン(51.5L;35.2kg)を攪拌しながら室温で2〜4時間かけて添加する。得られた混合物を、上清の平衡(通常7mg/ml未満の化合物I)に達するまで室温で攪拌する。
【0061】
このバッチを室温でろ過する。湿ったケーキを1:1トルエン/ヘプタン洗浄液(15.1L)、次いでヘプタン洗浄液(15.1L)で洗浄し、次いで乾燥窒素下で約40〜50℃で乾燥し、白色の結晶性固体として化合物Iの遊離酸無水物を得る。
【0062】
この固体を湿った窒素気流(相対湿度>70%、不凝縮)下で結晶性二水和物に再水和する。この手順による固体化合物I二水和物の通常の収量は7.9Kg(14.0モル、97%収率)で、>99アッセイ%化学純度および>99.8%ee.を有する。
【0063】
(実施例2A)
化合物II二水和物を精製するための代りの手順を以下に示す。スケールは、120gmの二水和物である。
【0064】
二水和物(120g)およびイソプロピルアルコール(IPA;540mL)を晶析装置に入れる。この混合物を38〜40℃に加熱し、二水和物を溶解する。脱イオン水(273mL)を約10分かけて添加し、このバッチを33.6v%水とする。バッチ温度を38〜40℃に調整する。次いで、120gの、60/40v/v脱イオン水/IPA媒体ミルド種スラリー(50mg二水和物/gスラリー)を添加する。これにより6gの二水和物(5%)、74.8mLの脱イオン水、および49.9mLのIPAとなり、バッチは37.1v%水を含む。
【0065】
次いで、30gの、60/40v/v脱イオン水/IPAを次のフラッシュとして添加する。バッチは、現在37.9v%水を含む。種晶床を38〜40℃で1時間エージングする。次いで、バッチの温度を38〜40℃に維持しながら、脱イオン水(537mL)を2回に等分にして、第1回分を6時間かけて、第2回分を9時間かけて添加する。これによりバッチは、60/40v/v脱イオン水/IPAとなる。
【0066】
このバッチを15〜20℃に1.5時間かけて徐々に冷却し、次いで、これをエージングし、ろ過する。ケーキを480mLの脱イオン水で2回洗浄する。次いで、湿ったろ過ケーキを40℃のジャケット温度で湿った空気中で乾燥する。
【0067】
(実施例3)
化合物Iの結晶性無水物の調製
結晶性無水物を、実施例1および2に記載のように乾燥窒素下で、結晶性二水和物を40〜50℃に加温することによって作製する。
【0068】
(実施例4)
化合物I二水和物の種結晶の調製
化合物I(7.6g)の粗酸形態をトルエン(30mL)に溶解し、水(1.0mL)で処理する。得られた2相混合物を室温で2時間激しく攪拌する。この混合物を5℃で2日間放置する。界面にある少量の結晶をろ過し、より大きなバッチの化合物I二水和物のための種結晶として使用する。
【0069】
無水物および二水和物のキャラクタライゼイション
X線粉末回折調査は、分子構造、結晶化度および多形を特徴付けするために広範に使用されている。この二水和物および無水物のX線粉末回折パターンは、PW3040/60コンソールを備えたフィリップス分析用X’PertPRO X線回折システムで作成された。線源として、PW3373/00セラミックCuLFF X線管K−Alpha放射を使用した。X線粉末回折スペクトルを室温で記録した(CuKα放射、2°〜40°(2θ)、0.0167°のステップ、ステップ当たり5.08秒)。
【0070】
図1は、化合物Iの二水和物の特徴的X線粉末回折パターンを示す。二水和物は、16.8°、19.6°および5.7°2θのピークによって特徴付けられる。二水和物はさらに、22.4°、7.7°および15.2°2θのピークで特徴付けられることができる。これは、さらに、24.6°、11.2°および22.8°2θのピークで特徴付けられることができる。
【0071】
図6は、化合物Iの無水物の特徴的X線粉末回折パターンを示す。この無水物は、18.1°、11.9°および9.1°2θのピークによって特徴付けられる。無水物はさらに、14.3°、16.4°および23.7°2θのピークによって特徴付けられることができる。これはさらに、5.9°、13.8°および20.5°2θのピークによって特徴付けられることができる。
【0072】
結晶形態はまた、パーキンエルマーモデルTGA7装置を使用してTGA曲線を作成する熱重量分析によって特徴付けた。実験は、窒素気流下で、10℃/分の加熱速度で行った。重量/温度データを装置によって自動的に収集した。結果の分析を装置ソフトウェア内のデルタY関数を選択するとともに、減量がその間で計算される温度を選択することによって行った。減量を分解/蒸発の開始まで報告する。
【0073】
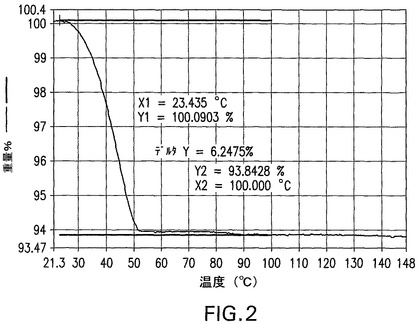
図2は、窒素気流下、10℃/分の加熱速度での化合物I二水和物の特徴的熱重量分析(TGA)曲線を示す。21℃と55℃との間で、6.25%の減量が生じる。この減量は、化合物Iの二水和物から2モルの水が失われるとして予測される重量変化と一致する(理論的減量は6.4%)。
【0074】
二水和物の水含量をまた、カールフィッシャー滴定によって測定した。二水和物の水含量は、6.4%の理論的水含量に対して、6.6%として測定された。
【0075】
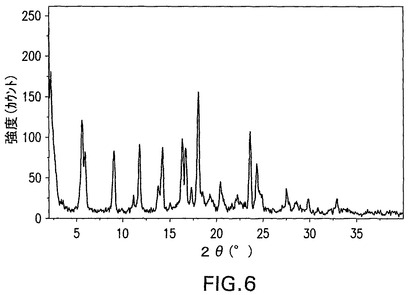
図7は、窒素気流下、10℃/分の加熱速度での化合物I無水物の特徴的熱重量分析(TGA)曲線を示す。20.9℃〜100.1℃の範囲の0.3%の減量は、化合物が無水であることを示す。
【0076】
図3は、化合物Iの無水物の特徴的示差走査熱量測定(DSC)曲線を示す。これはTAインスツルメントDSC2910示差走査熱量計を使用して得た。試料を窒素気流下、10℃/分の加熱速度で密閉パンの中で加熱した。データはこのシステムソフトウェアに含まれるDSC分析プログラムを使用して分析した。溶融吸熱は、吸熱が観察される範囲の温度の上方と下方のベースライン温度点間を積分した。記録されたデータは、開始温度、ピーク温度およびエンタルピーである。
【0077】
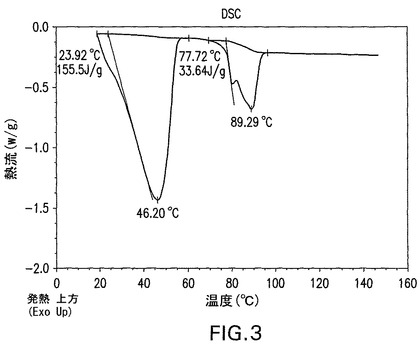
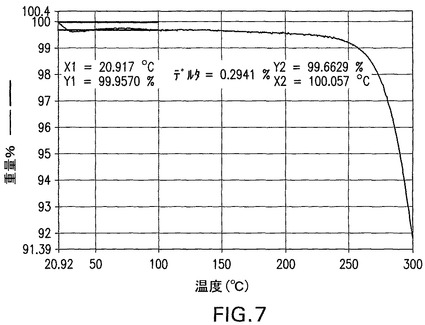
図8は、化合物Iの無水物の特徴的示差走査熱量測定曲線を示す。DSC曲線は、以下の条件下でTAインスツルメントQ1000示差走査熱量計を使用して得た。(1)試料を最初3℃/分で60℃まで加熱した。(2)試料を10℃/分で5℃まで冷却した後、60℃で30分間等温的に保持した。(3)次いで、調整運転を60秒ごとに1℃の変調および窒素気流下3℃/分の線形加熱速度で120℃まで行った。試料は密閉パンの中で加熱した。
【0078】
上記のX線粉末回折パターンに加えて、化合物Iの結晶性二水和物形態をさらに、固体炭素−13および固体フッ素−19核磁気共鳴(NMR)スペクトルによって特徴付けた。固体炭素−13NMRスペクトルは、ブルーカー(Bruker)4mm二重共鳴CPMASプローブを使用するブルーカー(Bruker)DSX400WB NMRシステムで得た。固体炭素−13NMRスペクトルは、可変増幅交差分極を備えた(プロトン/炭素−13)交差分極マジック角回転を利用した。試料を15.0kHzで回転し、全1,777スキャンを7秒のリサイクル遅れで収集した。FTを行う前に、20Hzの線幅拡大をスペクトルに対して適用した。化学シフトをグリシンのカルボニル炭素(176.03p.p.m.)を二次基準として使用するTMSスケールで記録する。
【0079】
固体フッ素−19NMRスペクトルは、ブルーカー(Bruker)4mm CRAMPSプローブを使用するブルーカー(Bruker)DSX400WB NMRシステムで得た。NMRスペクトルは、単純パルス獲得パルスプログラムを利用した。試料を15.0kHzで回転し、2秒のリサイクル遅れで全128スキャンを収集した。ベスペルエンドキャップ(vespel endcap)を、フッ素のバックグラウンドを最少化するために利用した。FTを行う前に、50Hzの線幅拡大をスペクトルに対して適用した。化学シフトを、−122p.p.m.の化学シフトに割り当てられる外部二次基準としてポリ(テトラフルオロエチレン)(テフロン(登録商標))を使用して記録する。
【0080】
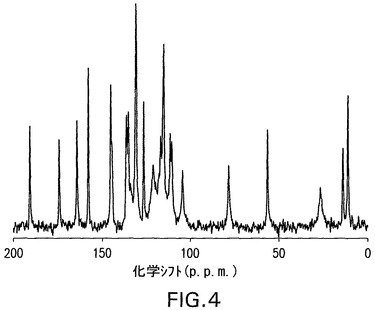
図4は、化合物Iの二水和物形態のための固体炭素−13CPMAS NMRスペクトルを示す。二水和物形態は、11.1、56.2、190.7p.p.m.の化学シフト値を有する特徴的なシグナルを示した。二水和物形態のさらなる特徴は、13.9、78.3、および174.2p.p.m.の化学シフト値を有するシグナルである。二水和物形態は、さらにもっと164.1、および157.6p.p.m.の化学シフト値を有するシグナルによって特徴付けられる。
【0081】
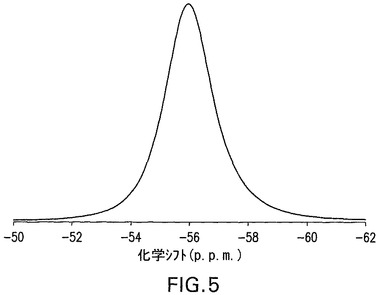
図5は、化合物Iの二水和物形態の固体フッ素−19MAS NMRスペクトルを示す。二水和物形態は、−56.0p.p.m.の化学シフト値を有する特徴的シグナルを示した。
【0082】
図9は、化合物Iの無水物形態の固体炭素−13CPMAS NMRスペクトルを示す。無水物形態は、10.3、54.1、および192.2p.p.m.の化学シフト値を有する特徴的シグナルを示した。無水物形態のさらなる特徴は、11.9、76.3、171.2p.p.m.の化学シフト値を有するシグナルであった。無水物形態はさらにもっと、163.0および159.3p.p.m.の化学シフト値を有するシグナルによって特徴付けられる。
【0083】
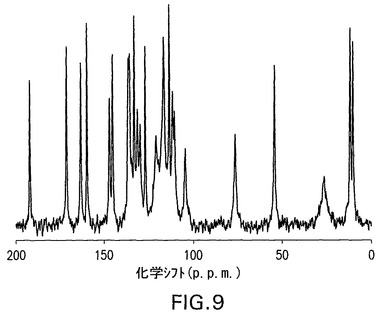
図10は、化合物Iの無水物形態の固体フッ素−19MAS NMRスペクトルを示す。無水物形態は、−55.6p.p.m.の化学シフト値を有する特徴的シグナルを示した。
【0084】
本発明の結晶性化合物Iの二水和物形態および結晶性化合物Iの無水物形態はそれぞれ、上記X線粉末回折、フッ素−19MAS NMR、炭素−13CPMAS NMR、およびDSC物理特性を有する、二水和物または無水物の少なくとも約5%の純相を有する。1つの実施形態において、純相は、上記固体の物理特性を有する、二水和物または無水物の少なくとも約10%である。第2の実施形態において、純相は、上記固体の物理特性を有する、二水和物または無水物の少なくとも約25%である。第3の実施形態において、純相は、上記固体の物理特性を有する、二水和物または無水物の少なくとも約50%である。第4の実施形態において、純相は、上記固体の物理特性を有する、二水和物または無水物の少なくとも約75%である。第5の実施形態において、純相は、上記固体の物理特性を有する、二水和物または無水物の少なくとも約90%である。第6の実施形態において、結晶性化合物Iは、上記固体の物理特性を有する、実質的に純相の二水和物または無水物である。「純相」の用語によって、本発明に記載された固体物理方法によって測定された化合物Iの別の特徴ある結晶性多形もしくは非結晶形態に関する化合物I無水物形態もしくは二水和物形態の固体純度を意味する。
【0085】
(実施例5)
(2R)−2−{3−[[3−(4−クロロベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]メチル]フェノキシ}酪酸(化合物II)の合成
ステップ1〜4は、実施例1のステップ1〜4と同じである。
【0086】
ステップ5
【0087】
【化15】
【0088】
ヘプタン(9.0L)中のクロトン酸メチル(3.00Kg)の溶液を窒素下、25〜35℃で20〜30分かけて臭素(1.55L)で処理した。反応混合物を30℃で1時間攪拌した。アセトニトリル(18L)を添加し、引き続き粉末炭酸カリウム(6.08Kg)を1回で添加した。反応物スラリーを60℃で1時間加温するとともに攪拌し、次いで、還流(74℃のバッチ温度)まで加熱した。ヘプタン/アセトニトリルアゼオトロープ(沸点69℃)を、溶媒15Lの除去をもって、蒸留除去した。アセトニトリル(ACN、6L)を常圧蒸留中に添加した。蒸留が完了したら、スラリーを80℃で3〜4時間エージングし、脱臭化水素反応を完了した。反応が完了したら、スラリーを20℃に冷却し、ろ過した。ケーキをアセトニトリル(2×6L)で洗浄した。生成物5〜7の溶液を、アルキル化反応においてさらに精製することなく使用した。
【0089】
ステップ6
【0090】
【化16】
【0091】
アセトニトリル中のメチル2−ブロモブタ−2−エノエート5−7(20.9Kg)および3−ヒドロキシベンジルアルコール(3.00Kg)の溶液を、粉末炭酸カリウム(6.54Kg)で処理し、このスラリーを80〜82℃に1時間かけて加温した。スラリーを80〜82℃で2時間エージングした。アセトニトリル(10L)を、常圧で2時間かけて蒸留除去した。反応混合物を50℃に冷却し、トルエン(20L)を添加した。このスラリーを減圧下、30〜40℃で濃縮し、全25Lの留出物を除去した。トルエン(10L)を添加し、スラリーを15℃に冷却した。水(20L)を添加し、相をよく混合した。水(7.5L)および50%w/v水酸化カリウム水溶液(5.31L)を添加した。2相混合物を攪拌し、35℃に5〜10時間加温した。混合物を20℃に冷却し、層を分離させた。下層(水層)を50Lの反応容器に入れ、15℃に冷却した。濃塩酸(2.25L)を添加し、pHを8〜9に調整した。IPAC(25L)を添加し、pHを濃塩酸(2.25L)で1.5(+/−0.2)に調整した。下側の水層を除去した。有機相を水(2×3.0L)で洗浄した。
【0092】
不飽和酸のIPAC溶液を減圧下、20〜30℃で濃縮し、溶媒をトルエンに切り換えた。容量を約30Lに調整した。スラリーを20℃で30分間エージングし、ろ過した。ケーキをトルエン(20L)で洗浄し、窒素気流下、ろ過漏斗上で一夜乾燥し、結晶性生成物5−8を得た。
【0093】
ステップ7
【0094】
【化17】
【0095】
不飽和酸5−8(3.922Kg)、トリエチルアミン(2.63L)およびメタノール(13.4L)を10ガロンのオートクレーブに入れた。メタノール(1L)を使用して移入を完了した。メタノール(800mL)中(R−BINAP)RuCl2(76.4g)のスラリーを、窒素下、1Lのステンレス鋼のボンベによって添加した。このバッチを100psi水素、20℃で20時間水素添加した。バッチを5ミクロンのインラインフィルターを通してろ過し、濃縮し、溶媒をトルエンに切り換えた。水(8L)および5MNaOH水溶液(4L)を添加し、相を分離させ、有機相を水(4L)で洗浄した。混合した水層をDarco G−60炭素(400g)を用いて60℃で1時間処理した。濃塩酸水溶液(300mL)を添加して混合物のpHをpH7に調整し、混合物を一夜(16時間)22℃まで冷却した。ソルカフロック(200g)を添加し、混合物をソルカフロックを介してろ過し、水(4L)で洗浄した。
【0096】
ろ液を10℃に冷却し、冷却によって13〜18℃の温度を維持しながら濃塩酸(3.7L)でpH=1に酸性化した。この混合物をIPAc(20L)で抽出した。このIPAc抽出物を塩水(brine)(2×4L)で洗浄し、生成物をヘプタン−IPAcの3:1混合物から結晶化した。結晶性生成物5−9を窒素気流下で乾燥した。
【0097】
ステップ8
【0098】
【化18】
【0099】
飽和酸(3.265Kg)、TsOH−H2O(58g)およびメタノール(45.7L)を60℃に14〜18時間加熱した。トリエチルアミン(86mL)を添加し、混合物を約10Lに濃縮した。この混合物の溶媒を酢酸イソプロピルに切り換え、生成物5−10を次のステップにおいて精製することなく使用した。
【0100】
ステップ9
【0101】
【化19】
【0102】
IPAC(10L)中メチルエステル(3.433Kg)の粗溶液にDMF(7.65L)を添加し、混合物を−15℃に冷却した。塩化チオニル(2.0Kg)を均一な淡い琥珀色の混合物に45分かけて添加した。混合物を20℃に30分かけて加温し、20℃で90分間エージングした。混合物を0℃に冷却し、n−ヘプタン(7.65L)を添加した。水(15.3L)を攪拌しながら5分かけて添加した。水相を除き、有機相を水(2×15L)で洗浄した。有機相を約4Lに濃縮し、生成物5−11溶液をさらに精製することなく使用した。
【0103】
ステップ10
【0104】
【化20】
【0105】
0℃のトルエン(9L)中ステップ4、実施例1のインドール生成物6(1.98Kg)の溶液にトルエン(6.1L)中1.8MEt2AlClを1〜2時間かけて添加した。得られた溶液を0℃で60分間攪拌した。n−ヘプタン(3L)中の溶液としてp−クロロベンゾイルクロライド(1.93Kg)を1〜2時間かけて添加し、反応物を12時間攪拌した。混合物を0℃に冷却し、メタノール(1.8L)を1時間かけて添加した。この反応物スラリーを1時間エージングした。n−ヘプタン(17L)を添加し、得られたスラリーを1時間エージングし、ろ過し、収集した沈殿物を追加のn−ヘプタン(22L)で洗浄した。
【0106】
赤色のケーキをすべての固体が溶解するまで還流下でメタノール(61L)中でスラリー化した。水(15L)を生成物の結晶化を誘導するために滴下漏斗によって1時間かけて添加した。得られたスラリーを室温に冷却し、生成物5−12をろ過によって単離し、窒素気流下で乾燥した。
【0107】
ステップ11
【0108】
【化21】
【0109】
DMF(5.70L)中アシルインドール5−12(2.52Kg)、ベンジルクロライド5−11(1.76Kg)、粉末炭酸カリウム(1.92Kg)、およびnBu4NI(263g)のスラリーを35℃で22時間攪拌した。この混合物を20℃に冷却し、次いでMTBE(4L)および酢酸(428g)を添加した。次いで、MTBE(17.4L)および水(14L)を添加し、水相を除去し、廃棄した。有機相を水(21L)で洗浄した。MTBE中生成物13の溶液を精製することなく使用した。
【0110】
ステップ12
【0111】
【化22】
【0112】
メタノール(11.2L)をMTBE中インドールメチルエステル5−13(3.71Kg)の粗溶液に添加した。1.0NNaOH(13.25L)を室温で添加した。得られた反応混合物を窒素下、40℃で4時間攪拌した。この反応混合物を室温に冷却し、5.0NHCl(2.91L)を激しく攪拌しながら5〜10分かけて添加した。有機層を30〜35℃で水(3×9L)を用いて洗浄した。有機層を減圧下で濃縮し、溶媒をトルエン(約24L)に切り換えた。ヘプタン(24L)を30分かけて添加し、得られたスラリーを室温で12〜14時間かけて攪拌した。生成物をろ過によって単離し、湿ったケーキを1:2トルエン/ヘプタン(12L)、次いでヘプタン(8L)で洗浄した。結晶性生成物化合物IIを窒素下、室温で乾燥した。
【0113】
(実施例6)
化合物IIの遊離酸(4g)をIPA(60ml)に溶解する。5N水酸化ナトリウム(1.46mL)を添加し、このスラリーを55℃に加温し、均一な溶液を得る。溶液を50℃に冷却し、1時間エージングして薄いスラリーを得る。スラリーを室温で一夜冷却する。スラリーをろ過し、真空中、40℃で一夜乾燥し、細かい結晶性針状物として化合物IIのナトリウム塩(3.6g)を得る。
【0114】
化合物IIの結晶形態
遊離酸としての化合物IIは、少なくとも3つの多形結晶形態を有し、A型、B型およびC型という。ナトリウム塩は、少なくとも5つの多形結晶形態を有し、I型、II型、III型、IV型およびV型という。実施例5に記載の合成はC型の遊離酸を生じ、これは好ましい遊離酸結晶塩形態である。実施例6に記載の合成はV型のナトリウム塩を生じ、これは好ましいナトリウム塩形態である。
【0115】
A型をIPA中の化合物IIのナトリウム塩の酸性化によって調製する。V型以外の化合物IIのナトリウム塩は、さまざまな溶媒にナトリウム塩を懸濁させることによって得る:IPA中の懸濁液からのI型およびIII型;α,α,α−トリフルオロトルエン中の懸濁液からのII型;およびTHF、AcOEtおよびACNからのIV型。V型のナトリウム塩を最も容易に得る。C型の遊離酸は、最も熱力学的に安定な酸形態であるようにみえる。
【0116】
(実施例7)
B型およびC型の遊離酸
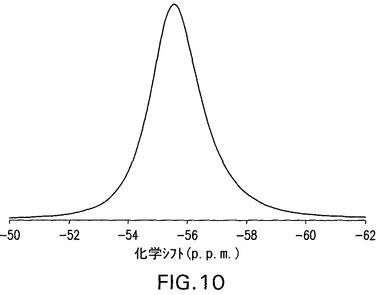
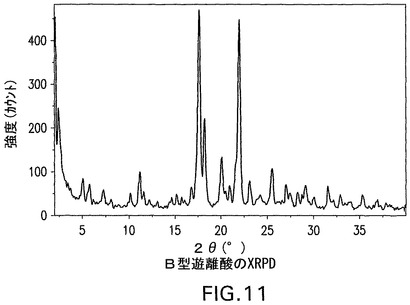
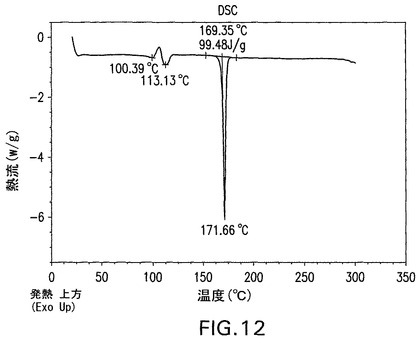
B型遊離酸は、100.4℃および113.1℃で発熱を示すDSC曲線(図12)を有する結晶性(図11)であり、脱水およびC型への転換に起因し得、引き続きC型の溶融が生じる(図15にみられる)。遊離酸(図3)の1モルにとって1.5モルの水に相当する4.7%の減量(図13)がある。図11は、結晶性B型の特徴的X線粉末回折パターンを示す。結晶性B型は、さらに17.7°、22.1°および18.3°2θのピークによって特徴付けられる。結晶性B型はさらに、11.2°、20.1°および5°2θのピークによって特徴付けられることができる。これはさらに、5.8°、10.2°および25.5°2θのピークによって特徴付けられることができる。
【0117】
C型遊離酸は、図14のXRPDによって示されるように結晶性である。C型遊離酸は、図15のDSCにおいて示されるように171.8℃の溶融開始温度、172.8℃でのピーク、および107.2J/gの融解熱を有する。C型遊離酸のTGAは、図16の曲線によって示されるように0.1%未満の最少の減量を示す。図14は、結晶性C型遊離酸の特徴的X線粉末回折パターンを示す。結晶性C型遊離酸は、21.6°、17°および16.3°2θのピークによって特徴付けられる。結晶性C型遊離酸はさらに、13.6°、5.5°、7.9°2θのピークによって特徴付けられることができる。結晶性C型遊離酸はさらに、18.4°、20.8°および20.1°2θのピークによって特徴付けられることができる。
【0118】
(実施例8)
V型ナトリウム塩
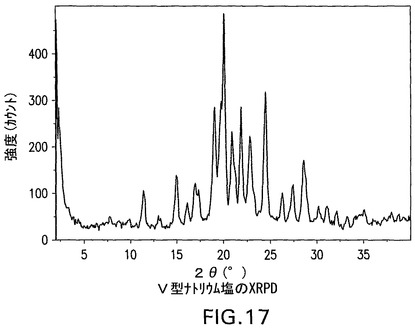
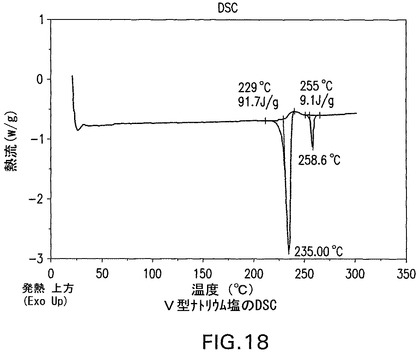
V型ナトリウム塩をXRPD、DSCおよびTGによって分析した。これは結晶性である(図17)。DSCは229℃の開始および235℃のピークを有する融点、ならびに255℃の開始および258.6℃のピークを有する二次吸熱を示す(図18)。TGA(図19)は、254℃まで0.2%の最少の減量を示す。図17は、V型結晶性ナトリウム塩の特徴的X線粉末回折パターンを示す。V型結晶性ナトリウム塩は、20°、19.7°および24.4°2θのピークによって特徴付けられる。V型結晶性ナトリウム塩はさらに、19°、21.8°および20.9°2θのピークによって特徴付けられることができる。V型結晶性ナトリム塩はさらに、11.4°、15°および16.1°2θのピークによって特徴付けられることができる。
【0119】
結晶形態の比較
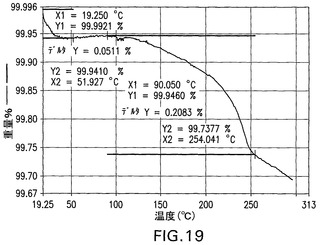
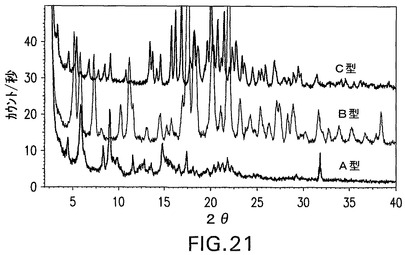
図20は、同じスケールで化合物IIのナトリウム塩の全5つの結晶形態(I、II、III、IV、およびV)のXRPDパターンを示す。
【0120】
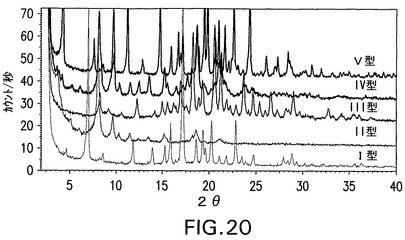
図21は、同じスケールで化合物IIの遊離酸の全3つの結晶形状(A、B、およびC)のXRPDパターンを示す。
【0121】
両方の図において、化合物IIのナトリウム塩と遊離酸との結晶形態は、独特のXRPDパターンを有することがわかる。
【0122】
(実施例9)
実施例1の合成手順の化合物4の化合物6への転換はまた、以下の手順によって行うことができる。
【0123】
【化23】
【0124】
メーヤワイン反応およびケタール化
酢酸イソプロペニル(22mL)中ニトロアニリン4(22.21g)の溶液およびiBuONO(94%、13.71g)の溶液を40〜50℃に維持し、酢酸イソプロペニル200mL中のCuCl2(2.01g)の懸濁液にそれぞれ別の添加漏斗からほぼ同じ速度(±10%)で1時間かけて添加する。この混合物を40〜50℃で1時間エージングし、25℃に冷却する。トルエン(100mL)を添加し、混合物を1MHCl(2×15mL)で洗浄する;大分部の銅は、この2回の少量の水性洗浄中に濃縮され、次いで水(2×100mL)で洗浄する。有機相を50mLに濃縮し、一定容量で200Torrで100mLのトルエンを使用して溶媒をトルエンに切り換える。次いで、混合物を、一定容量で200Torrで150mLのエタノールを使用して溶媒をエタノールに切り換える。50mLエタノール中ニトロケトン5を含む溶液をエチレングリコール(9.31g)、(EtO)3CH(21.03g)、およびp−TsOH(0.50g)と混合し、溶液を45℃に16時間または80℃に1〜2時間加熱し、次いで25℃に冷却した。ニトロケトン5の転化をHPLCにより監視した。
【0125】
水素添加
トリエチルアミン(0.28g)を前ステップの粗ニトロアリールケタールの溶液に添加し(pHは、pH紙を使用して測定して、<2から>5に変化した)、得られた混合物を耐圧瓶の中に置いた。ラネーニッケル(22.2gの湿ったスラリー)を添加し、混合物を40psigおよび25〜70℃(発熱性)で2時間水素添加した。混合物をDarco G60(1.1g)で25〜40℃で20分間処理し、次いでソルカフロックを介してろ過し、パッドをエタノール(50mL)で洗浄した。
【0126】
加水分解/環化
水25.9mL中クエン酸(27.3g)の溶液を前ステップからの粗生成物の溶液に添加した。この混合物を6時間エージングして、80℃で約95%の転化を達成した。次いで混合物を20℃に冷却し、pHが5になるまで十分量の50%NaOHを入れた。このバッチを200Torrで約50mlに濃縮し、次いで溶媒をトルエンに切り換え、留出物からエタノールを除去した。トルエン溶液を水(2×50mL)で洗浄した。次いで溶媒を常圧で水に切り換えた。所望のインドール生成物6は揮発性であり、トルエンが除去される際、留出物中に出始める。インドールは水に不溶であり、約83℃の融点を有する。蒸留の継続時、蒸気はトルエン/水混合物中に散布され、トルエン層中のインドールを補足するかまたは固体インドール(融点約83℃)がコンデンサーを詰まらせないように約85℃で凝縮させる。トルエンが除去された後、大部分のインドールは依然として蒸留容器中に残存し、蒸気蒸留(スチームストリッピング)を行うように水で蒸留して除き続けた。全てのインドールが留出物受けのトルエン層中に収集されるまで、水を蒸留物からリサイクして蒸留容器に戻す。水約65mlをインドール1g当たり蒸留する。
【0127】
(医薬組成物の実施例)
化合物Iの結晶性二水和物形態は、以下のように製剤することができる。化合物Iの100mg投与形態は、結晶性二水和物とアビセル(Avicel)(フィラー)、ラクトース(フィラー)、クロスカルメロースナトリウム(錠剤崩壊成分)、ラウリル硫酸ナトリム(界面活性剤)、およびステアリン酸マグネシウム(潤滑剤)とをローラーで固めて作製される顆粒形状の化合物I二水和物107mgから構成される。次いで、顆粒をゼラチンカプセルの中へ充填するかまたは錠剤に成形する。好ましくは、錠剤を濃色の不透明のフィルムでコートする。
【0128】
化合物IIの100mg投与形態は、上記と同じ手順を使用してC型結晶の100mgを処方することによって作製する。
【図面の簡単な説明】
【0129】
【図1】化合物Iの結晶性二水和物の特徴的X線粉末回折パターンを示す図である。
【図2】化合物Iの結晶性二水和物の特徴的熱重量分析(TGA)曲線を示す図である。
【図3】化合物Iの結晶性二水和物の特徴的示差走査熱量測定(DSC)曲線を示す図である。
【図4】化合物Iの結晶性二水和物形態の特徴的固体炭素−13CPMAS NMRスペクトルを示す図である。
【図5】化合物Iの結晶性二水和物形態の特徴的固体フッ素−19MAS NMRスペクトルを示す図である。
【図6】化合物Iの結晶性無水物の特徴的X線粉末回折パターンを示す図である。
【図7】化合物Iの結晶性無水物の特徴的熱重量分析(TGA)曲線を示す図である。
【図8】化合物Iの結晶性無水物の特徴的示差走査熱量測定(DSC)曲線を示す図である。
【図9】化合物Iの結晶性無水物形態の特徴的固体炭素−13CPMAS NMRスペクトルを示す図である。
【図10】化合物Iの結晶性無水物形態の特徴的固体フッ素−19MAS NMRスペクトルを示す図である。
【図11】化合物IIのB型遊離酸の特徴的X線粉末回折パターンを示す図である。
【図12】化合物IIのB型遊離酸の特徴的示差走査熱量測定(DSC)曲線を示す図である。
【図13】化合物IIのB型遊離酸の特徴的熱重量分析(TGA)曲線を示す図である。
【図14】化合物IIのC型遊離酸の特徴的X線粉末回折パターンを示す図である。
【図15】化合物IIのC型遊離酸の特徴的示差走査熱量測定(DSC)曲線を示す図である。
【図16】化合物IIのC型遊離酸の特徴的熱重量分析(TGA)曲線を示す図である。
【図17】化合物IIのV型ナトリウム塩の特徴的X線粉末回折パターンを示す図である。
【図18】化合物IIのV型ナトリウム塩の特徴的示差走査熱量測定(DSC)曲線を示す図である。
【図19】化合物IIのV型ナトリウム塩の特徴的熱重量分析(TGA)曲線を示す図である。
【図20】同じスケールの化合物IIのナトリウム塩の全5つの結晶形態のX線粉末回折パターンであり、それらが異なり、明確に区別できることを示す図である。
【図21】同じスケールの化合物IIの遊離酸の全3つの結晶形態のX線粉末回折パターンであり、それらが異なり、明確に区別できることを示す図である。
【技術分野】
【0001】
本発明は、2型糖尿病、ならびに高血糖症、肥満症、異脂肪血症および代謝病を含む、PPARガンマ作動薬により調節される他の疾患の治療用の医薬有効成分として有用な化合物の新規な結晶形態および塩形態に関する。該化合物は選択的PPARガンマ部分作動薬(またSPPARgMまたはSPPARMとして知られている)である。
【背景技術】
【0002】
2型糖尿病は、依然として深刻な医療問題である。より有効で、副作用の少ない新しい治療が継続して求められている。
【発明の開示】
【課題を解決するための手段】
【0003】
本発明は、SPPARMまたはSPPARgMとしても知られるPPARガンマ部分作動薬として活性な化合物のクラスに属する化合物の新規な結晶形態、水和物、無水物および塩に関する。関連する化合物が、公開PCT出願04/020409に最初に開示されている。2つの構造的に類似する化合物の結晶形態、水和物、無水物、および/または塩が開示されている。(1)第1の化合物は、(2R)−2−{3−[3−(4−メトキシベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]フェノキシ}ブタン酸(化合物I)であり、結晶性二水和物および結晶性無水物として開示されている。ここに開示されている結晶性二水和物および結晶性無水物の形態は、精製の容易さ、処理の容易さおよび組成物の安定性など、医薬組成物の調製においての利点である特性を有し、先に開示された(2R)−2−{3−[3−(4−メトキシベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]フェノキシ}ブタン酸の非結晶形態に対して利点を有する。(2)第2の化合物は、(2R)−2−{3−[[3−(4−クロロベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]メチル]フェノキシ}酪酸(化合物II)であり、少なくとも5つの新規な結晶性多形形態を有するナトリウム塩および少なくとも3つの新規な結晶性多形形態を有する遊離酸としてここに開示されている。これらの多形形態の一部は、精製の容易さ、処理の容易さおよび組成物の安定性など、医薬組成物の調製においての利点である特性を有し、先に開示された化合物IIの固体形態に対して利点を有する。
【0004】
本発明はまた、新規な結晶性多形を含む医薬組成物、これらの多形形態およびこれらの医薬組成物を調製するための方法、2型糖尿病、高血糖症、肥満症、異脂肪血症および代謝病の治療用にこれらを使用するための方法に関する。
【発明を実施するための最良の形態】
【0005】
本発明は、構造式I(化合物I)
【0006】
【化3】
を有する(2R)−2−{3−[3−(4−メトキシベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]フェノキシ}ブタン酸の新規な結晶性二水和物および無水物の多形形態を提供する。
【0007】
上記構造は、公開PCT出願04/020409における実施例3として最初に開示された。’409特許公開における合成方法を使用して単離された化合物は、非結晶の固体である。発明者らはここに、非結晶化合物Iの結晶性二水和物および結晶性無水物を見い出した。これらの結晶化合物は、医薬組成物の調製においてきわめて容易に使用される。結晶性二水和物は、乾燥窒素流中で加温することによって結晶性無水物に容易に転換され、結晶性無水物は、湿窒素気流(RH>70%)中に放置することによって容易に結晶性二水和物に転換して戻る。結晶性二水和物は、非吸湿性である。
【0008】
本発明のさらなる実施形態は、検出可能な量の化合物Iの結晶性無水物形態または結晶性二水和物形態を含む原薬を提供する。「原薬(drug substance)」という用語は、活性医薬成分(API)を意味する。原薬中の結晶性無水物形態または結晶性二水和物形態の量は、X線粉末回折(XRPD)法、固体フッ素−19マジック角回転(MAS)核磁気共鳴分光法、固体炭素−13交差偏光マジック角回転(CPMAS)核磁気共鳴分光法、固体フーリエ変換赤外分光法およびラマン分光法などの物理的方法の使用によって定量することができる。この実施形態の第1のクラスにおいて、結晶性無水物形態または結晶性二水和物形態の約5重量%から約100重量%が原薬中に存在する。この実施形態の第2のクラスにおいて、結晶性無水物形態または結晶性二水和物形態の約10重量%から約100重量%が原薬中に存在する。この実施形態の第3のクラスにおいて、結晶性無水物形態または結晶性二水和物形態の約25重量%から約100重量%が原薬中に存在する。この実施形態の第4のクラスにおいて、結晶性無水物形態または結晶性二水和物形態の約50重量%から約100重量%が原薬中に存在する。この実施形態の第5のクラスにおいて、結晶性無水物形態または結晶性二水和物形態の約75重量%から約100重量%が原薬中に存在する。この実施形態の第6のクラスにおいて、実質的にすべての化合物I原薬は、結晶性無水物形態または結晶性二水和物形態であり、すなわち化合物I原薬は、実質的に純相結晶性無水物形態または純相結晶性二水和物形態である。
【0009】
本発明の別の態様は、PPARガンマ作動薬が適応される臨床状態の治療もしくは管理のための方法であり、このような治療もしくは管理を必要としている患者に化合物Iの結晶性無水物形態もしくは結晶性二水和物形態の治療有効量、または化合物Iの結晶性無水物形態または結晶性二水和物形態の治療有効量を含む医薬組成物を投与することを含む方法を提供する。このような臨床状態としては、2型糖尿病、高血糖症、肥満症、異脂肪血症および代謝症候群が挙げられる。「患者」は、ヒトを含む哺乳類である。患者はヒトの患者が最も多い。
【0010】
本発明はまた、PPARガンマ作動薬が適応される1以上の臨床状態の患者における治療または管理のための医薬の製造において本発明の結晶性無水物形態もしくは結晶性二水和物形態の使用を提供する。1つの実施形態において、臨床状態は2型糖尿病である。
【0011】
本発明の別の態様は、PPARガンマ作動薬が適用される1以上の臨床状態の患者の治療または管理において使用する結晶性無水物形態または結晶性二水和物形態を提供する。この態様の1つの実施形態では、臨床状態は2型糖尿病である。
【0012】
本発明はまた、1以上の医薬上許容される担体または賦形剤と共に結晶性無水物形態もしくは結晶性二水和物形態を含む医薬組成物を提供する。1つの実施形態において、医薬組成物は、活性医薬成分(API)が本発明の結晶性無水物形態もしくは結晶性二水和物形態の検出可能量を含む、医薬上許容される賦形剤と混合したAPIを含む。第2の実施形態において、医薬組成物は、APIが本発明の結晶性無水物形態または結晶性二水和物形態の約5重量%から約100重量%を含む、医薬上許容される賦形剤と混合したAPIを含む。この第2の実施形態の1つのクラスにおいて、このような組成物中のAPIは、結晶性無水物形態または結晶性二水和物形態の約10重量%から約100重量%を含む。この実施形態の第2のクラスでは、このような組成物中のAPIは、結晶性無水物形態または結晶性二水和物形態の約25重量%から約100重量%を含む。この実施形態の第3のクラスでは、このような組成物中のAPIは、結晶性無水物形態または結晶性二水和物形態の約50重量%から約100重量%を含む。この実施形態の第4のクラスにおいて、このような組成物中のAPIは、結晶性無水物形態または結晶性二水和物形態の約75重量%から約100重量%を含む。この実施形態の第5のクラスにおいて、実質的にすべてのAPIは、化合物Iの結晶性無水物形態または結晶性二水和物形態であり、すなわちAPIは、結晶性無水物形態における実質的に純相化合物Iであるかまたは結晶性二水和物形態における実質的に純相化合物Iである。
【0013】
本発明による組成物は、好適には、錠剤、丸薬、カプセル、粉末、顆粒、滅菌溶液もしくは懸濁液、定量エアロゾルもしくは液体スプレイ、ドロップ、アンプル、自動注入デバイスもしくは座薬などの単位投与形態にある。この組成物は、経口、非経口、経鼻、舌下、もしくは直腸投与、または吸入もしくは吹送による投与を対象としたものである。本発明による組成物の処方は、例えばRemington’s Pharmaceutical Science、第17版、1995年に記載の通り、当該技術分野に知られた方法によって都合良くもたらされる。
【0014】
投薬計画は、患者のタイプ、種、年齢、体重、性および患者の医学的状態;治療する症状の重症度;投与経路;ならびに患者の腎臓および肝臓の機能を含むさまざまな要因によって選択される。通常の技術を有する医者、獣医、または臨床医であれば、この状態の進行を防ぐ、対抗する、もしくは止める、または状態を治療もしくは管理するために必要とされる薬剤の有効量を容易に決定し、処方することができる。
【0015】
示された効果のために使用される場合、本発明の経口投与量は、1日当たり体重1kg当たり約0.01mg(mg/kg/day)から約100mg/kg/day、好ましくは0.01〜10mg/kg/dayの範囲である。経口投与のために、組成物は、例えば、遊離酸の形態で測定してAPI0.05、0.1、0.5、1.0、2.5、5.0、10.0、15.0、25.0、50.0、100、200、400、600、800、または1,000mgを含む錠剤もしくはカプセル、または治療結果を達成するために治療される患者にとって適切であり得るような他の投与形態で提供され得る。医薬は、通常API約0.5mgから約500mg、好ましくはAPI約1mgから約200mgを含む。経静脈的に、最も好ましい投与量は、定速輸液の間、約0.1から約10mg/kg/分の範囲である。有利には、本発明の結晶性無水物形態および結晶性二水和物形態は、1日1回投与する1日当たりの総量を1日2回、3回または4回の分割投与量で投与することができる。さらに、本発明の結晶性無水物形態および結晶性二水和物形態は、好適な経鼻投与の媒体の局所の使用を介して鼻孔内形態において、または通常の当業者によく知られた経皮皮膚用パッチ剤のそれらの形態を使用する経皮経路によって投与し得る。経皮送達系の形態において投与するために、投薬量投与は、当然、投与計画を通して間欠的ではなく連続的である。
【0016】
本発明の方法において、本明細書に詳細に述べる化合物Iの結晶性無水物形態および結晶性二水和物形態はAPIを形成することができ、投与の意図した形態、すなわち、経口用の錠剤、カプセル、エリキシル剤、シロップなどに関して好適に選択され従来の医薬慣行と一致している好適な薬剤希釈剤、賦形剤または担体(本明細書では、集合的に「担体」材料という)と混合して通常投与される。
【0017】
例えば、錠剤もしくはカプセルの形態での経口投与に対して、活性薬剤成分は、ラクトース、デンプン、サッカロース、グルコース、メチルセルロース、ステアリン酸マグネシウム、リン酸二カルシウム、硫酸カルシウム、マンニトール、ソルビトールなどの経口、非毒性の医薬上許容される不活性担体と合わせることができる;液体形態における経口投与として、経口APIは、エタノール、グリセロール、水などの任意の経口、非毒性、医薬上許容される不活性担体と合わせることができる。さらに、所望もしくは必要な場合、好適な結合剤、潤滑剤、崩壊剤および着色剤も、混合物に含むことができる。好適な結合剤としては、デンプン、ゼラチン、グルコースまたはベータ−ラクトースなどの天然砂糖、コーン甘味剤、アカシア、トラガカントまたはアルギン酸ナトリウム、カルボキシメチルセルロース、ポリエチレングリコール、ワックスなど天然ゴムおよび合成ゴムが挙げられる。これら投与形態で使用される潤滑剤としては、限定されることなく、オレイン酸ナトリウム、ステアリン酸ナトリウム、ステアリン酸マグネシウムおよびステアリルフマル酸ナトリウムが挙げられる。崩壊剤は、限定されることなく、デンプン、メチルセルロース、寒天、ベントナイト、キサンタンガムなどが挙げられる。使用され得る他の通常使用される添加物としては、安息香酸ナトリウム、酢酸ナトリウム、塩化ナトリムなどが挙げられる。
【0018】
本発明は同様に、下記式II
【0019】
【化4】
を有し、本明細書において化合物IIと称される(2R)−2−{3−[[3−(4−クロロベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]メチル]フェノキシ}酪酸の新規な多形形態を提供する。
【0020】
さまざまなレベルの安定性と調製の容易さを有する化合物IIの広範な結晶形態がある。V型ナトリウム塩およびC型遊離酸は再現性をもって調製される。C型酸は、熱力学的に安定であり、医薬製剤の調製に特に好適である。これは非水和物結晶性化合物である。
【0021】
これらの結晶形態は、化合物Iについて上記で提供されたものと類似の実施形態を有する。
【0022】
以下の非限定的な実施例は本発明を説明するためのものであり、本発明の範囲または精神を限定するものとして考えられるべきではない。
【0023】
本明細書における化合物IおよびIIは、ケト−エノールなどの互変異性体などの互変異性体として存在し得る。個々の互変異性体ならびにその混合物は、構造式Iの化合物に包含される。
【実施例】
【0024】
(実施例1)
(2R)−2−{3−[3−(4−メトキシベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]フェノキシ}ブタン酸(化合物I)の合成
化合物Iは下記に示す多段プロセスによって作られる。
【0025】
ステップ1:ニトロ化
【0026】
【化5】
【0027】
容器に酢酸(8.0L)を入れ、引き続き添加漏斗によって4−(トリフルオロメトキシ)アニリン(8.00Kg)を攪拌しながら20〜35℃で15〜20分かけて入れた。次いで、無水酢酸(18.08kg)を20〜35℃で15〜20分かけて添加した。この反応混合物を20〜25℃で20〜30分間攪拌した。96%硫酸(320ml)を添加漏斗によって25〜35℃で5分かけて、引き続き90%硝酸(2.08L)を攪拌しながら別の添加漏斗によって25〜35℃で60分かけて入れた。添加後、反応混合物を20〜25℃で0.5〜1時間攪拌した。水(60L)を冷却しながら15〜25℃で40〜50分かけて添加した。3〜4Lの水を添加後、固体生成物が徐々に結晶化した。このスラリーを室温で1時間攪拌し、黄色の固体生成物をろ過によって単離した。湿ったケーキを水(3×20L)で洗浄し、窒素気流を使用して乾燥し、乾燥生成物3を得た。別法として、反応混合物を冷水に注ぐことによって上述したクエンチを行うことができる。
【0028】
ステップ2:加水分解
【0029】
【化6】
【0030】
攪拌機、温度計プローブ、水冷浴および添加漏斗を備えた容器に、4−トリフルオロメトキシ−2−ニトロアセトアニリド(11.0Kg)およびメタノール(42L)を攪拌しながら室温で入れた。5NNaOH(10.41L)をスラリーに水浴を用いて20〜30℃で5〜10分かけて添加し、均一な濃い琥珀色の溶液を得た。この反応混合物を25〜30℃で1時間かけて攪拌した。反応が終了した後、水(63L)を氷水浴を用いて15〜25℃で40〜50分かけて添加した。水を添加後、このバッチを攪拌機を備えた200L容器に移した。このスラリーをろ過前に室温で2〜3時間さらに攪拌した。黄色の固体生成物をろ過によって単離し、湿ったケーキを3:2水/メタノール(2×12.5L)で洗浄し、真空吸引を用いて窒素下で乾燥し、2−ニトロ−4−トリフルオロメトキシアニリン4を得た。
【0031】
ステップ3:メーヤワインアリール化
【0032】
【化7】
【0033】
容器に酢酸イソプロペニル(30L)を入れ、次いで塩化銅(II)(2.93Kg)を攪拌しながら1回で18〜25℃で入れた。この溶液の温度を40〜45℃に調整した。酢酸イソプロペニル(5L)中アニリン4(4.05Kg)の溶液を、この反応混合物に添加漏斗によって1〜2時間かけて添加し、同時に亜硝酸イソブチル(2.84L)を別の添加漏斗で、反応温度を40〜50℃に保ちながら同じ時間をかけて添加した。
【0034】
反応混合物を18〜22℃に冷却し、トルエン(21.5L)で希釈し、1.0NHCl(21.5L)を添加して反応を停止した。反応混合物を15分間攪拌した。有機層を水(21.5L)、炭酸水素ナトリウム水溶液(21.5L)、および水(2.5L)で洗浄した。この有機層を部分真空(最高温度20℃)下で濃縮し、溶媒をトルエンに切り換えた。容量を10.75Lに調整し、均一な琥珀色の溶液を5〜10℃に冷却した。ヘプタン(3.6L)を添加し、混合物を1時間エージングした。ヘプタン(37L)を−5℃〜−10℃の温度を保ちながらゆっくり添加した。このスラリーを−25〜−20℃に冷却し、−25〜−20℃で1時間エージングし、ろ過した。濃い黄色のケーキをトルエン/ヘプタン(1:6v/v、11L)を用いて−25〜−20℃で洗浄し、さらにヘプタン(4L)を用いて−25〜20℃で洗浄し、室温、部分真空下、窒素気流で乾燥し、ケトン5を得た。
【0035】
ステップ4:インドール
【0036】
【化8】
【0037】
メタノール(7.6L)中基質5(2.0Kg)および5%Pd/C(484g)のスラリーを15℃、60psiの水素雰囲気下で1〜2時間水素添加し、次いで6時間で65℃に加熱した。反応混合物をソルカフロック(solca−floc)でろ過して触媒を除去し、MeOHで洗浄した。このバッチを濃縮し、溶媒をトルエンに切り換えた。6のトルエン溶液をカップリングステップにあるように使用した。
【0038】
ステップ5:3−ベンジルオキシヨードベンゼン
【0039】
【化9】
【0040】
容器に3−ヨードフェノール(6.8Kg)、粉末K2CO3(5.12Kg)、DMF(40.8L)および塩化ベンジル(4.30Kg)を入れた。得られたスラリーを68〜70℃に加熱し、6〜7時間攪拌した。反応混合物を35〜40℃に冷却し、水(40.8L)を同温で攪拌しながら1時間かけて添加した。水を添加後、得られたスラリーを室温で2〜3時間攪拌した。このバッチをろ過し、湿ったケーキを水(3×35L)で洗浄し、窒素下、真空吸引を12〜16時間して乾燥し、白色の結晶性固体として生成物7を得た。
【0041】
ステップ6:N−アリールインドール
【0042】
【化10】
【0043】
トルエン(9.6L)中インドール6(3.20Kg)の溶液に3−ベンジルオキシヨードベンゼン7(4.57Kg)、CuI(421g)、N,N−ジメチルエチレンジアミン(401g)、および炭酸セシウム(9.4Kg)を添加した。トルエン(6.5L)を添加した。このスラリーを激しく攪拌しながら105〜110℃で12時間エージングし、室温に冷却した。ソルカフロック(1.5Kg)、次いでトルエン(30L)を18〜25℃で添加した。次いで、水(30L)をよく攪拌しながら30〜60分かけて滴下した。この混合物を18〜25℃で1〜2時間エージングし、ろ過した。このケーキをトルエン(10L)ですすぎ洗いした。トルエンろ液を濃縮し、8の粗溶液を次のステップでそのまま使用した。この反応はまた、固体を懸濁したままに保つようによく混合し(反応器の底から、攪拌または再循環ループ)、まだ残留水分を有する前ステップからのベンジルオキシヨードベンゼンを使用して、2モル%の低い濃度で触媒としてCuCl2を使用して有利に行うことができる。空気の排除もまた、有利である。
【0044】
ステップ7:アシル化/脱ベンジル化
【0045】
【化11】
【0046】
トルエン(2.58L)中ベンジルオキシアリールインドール8(2.58Kg)の溶液を0℃に冷却し、ヘプタン(1.3L)中4−メトキシベンゾイルクロライド(1.58L)の溶液を10〜20分かけて添加した。ジエチルアルミニウムクロライド(1.8M、10.8L)の溶液を0〜5℃で30分かけて添加した。添加を終了すると直ちに、その反応物を室温まで加温した。水(2.12L)中6MHClを30分かけてゆっくり添加し、反応を止めた。得られたスラリーを室温まで温め、ヘプタン(49L)を添加し、混合物を室温で1〜2時間エージングし、次いでろ過した。反応物を冷水とヘプタンとの混合物に添加する逆クエンチを別に使用し得る。一夜窒素気流下で収集した固体を乾燥後、この物質をエタノール(17.3L)に溶解し、このスラリーを還流するまで加熱し、水(7.4L)中3M塩酸を添加した。この溶液を室温に冷却し、得られたスラリーをろ過し、エタノール中30%の3M塩酸で洗浄し、次いで窒素気流下で乾燥し、結晶性アシル化生成物9を得た。生成物をさらに、トルエンから1回の再結晶によって精製する。
【0047】
ステップ8A:
【0048】
【化12】
【0049】
ステップ8a:(S)2−ヒドロキシ酪酸イソブチル
容器にリン酸二水素カリウム(1.40Kg)および水(70.2L)を入れ、濃硫酸を添加することによってpHを7.5に調整した。ギ酸ナトリウム(1.54Kg)、および水(8L)中2−ケト酪酸カリウム塩(2Kg)の溶液を添加し、次いでβ−ニコチンアミドアデニンジヌクレオチド(96.3g)およびギ酸脱水素酵素(35.5g)を添加した。混合物を31〜37℃で攪拌し、水酸化ナトリウムを添加することによって溶液のpHを7.4に調整した。ニワトリ心臓酵素溶液(150mg)由来のL乳酸脱水素酵素VIII型を添加し、反応物を攪拌しながら33〜38℃で18時間エージングし、硫酸溶液を使用してpHを7.4〜7.6に調節した。別に、この反応をギ酸脱水素酵素ではなくグルコース脱水素酵素を使用して行い、再生されるように化学量論的な還元剤の還元を行うこともできる。
【0050】
転化が終了した後、イソブチルアルコール(40L)および塩化カリウム(5Kg)、次いで硫酸を添加し、pHを2に調整した。水層をイソブチルアルコール(3×40L)で抽出した。混合抽出物を約20%容量まで濃縮し、濃硫酸(5モル%)を添加し、この溶液を12時間エージングしてエステル化を完了した。溶液を10%重炭酸カリウム水溶液(10L)で洗浄した。この後処理においてN−メチルモルホリンを10%重炭酸カリウムの代りに使用することができる。層を分離し、有機層を水(10L)で洗浄した。分別蒸留カラムを使用した濃縮、および減圧下で溶媒のトルエンへの切り換えによって、トルエン中25%溶液として粗(S)2−ヒドロキシ酪酸イソブチルを得た。これをさらなる精製をすることなしに使用した。
【0051】
ステップ8B:(S)イソブチル2−トシルオキシ酪酸
酢酸エチル(14L)中(S)2−ヒドロキシ酪酸イソブチル(2.72Kg)およびp−トルエンスルホニルクロライド(3.6Kg)に20℃で、酢酸エチル(10L)中DABCO(286g)およびトリエチルアミン(4.7L)の溶液を2時間かけて添加した。この反応物を20〜25℃で4時間エージングした。この混合物を水(10L、2L、および2L)で3回洗浄し、有機層を濃縮し、溶媒をトルエンに切り換えた。(S)2−イソブチルトシル酪酸の濃縮トルエン溶液をカップリング反応におけるように使用した。別に、アセトニトリルを酢酸エチルの代りに使用することができ、この場合は反応はより速く進む。
【0052】
ステップ9:アシル化
【0053】
【化13】
【0054】
攪拌機、温度計プローブ、窒素注入口および添加漏斗を備えた50Lの容器に、DMSO(6L)、(S)トシルオキシ酪酸2−イソブチル(1.70Kg)およびインドールフェノール9(2.04Kg)を窒素下、20℃で入れた。粉末炭酸カリウム(1.88Kg)を添加した。DMSO(2L)を使用し、これらの物質を容器中にすすぎ洗いした。得られたスラリーを28〜30℃で24時間攪拌した。反応が完了したら、MTBE(10L)を添加し、スラリーを10℃に冷却した。水(15L)を添加し、層を室温(20〜25℃)でよく混合した。下側の水層を除去した。有機層を0.2M炭酸水素カリウム(2×5L)で洗浄した。粗生成物10を加水分解ステップにおいて精製することなく使用した。
【0055】
ステップ10:加水分解
【0056】
【化14】
【0057】
MTBE中エステル10(8.98Kg)の溶液をメタノール(27L)で希釈した。1M水酸化ナトリウム(30.8L)を室温で添加した。二相性反応混合物を40℃で2時間激しく攪拌した。(S)トシルオキシ酪酸2−イソブチルとのカップリング反応(ステップ9)由来の残留トルエン量に依存して、比較的長い反応時間(40℃で〜16時間)が必要とされる。反応混合物を室温に冷却し、MTBE(22L)で希釈した。5M塩酸(26.8L)を激しく攪拌しながら添加した。有機層を水(3×20L)で洗浄した。有機層をその最初の容量の約1/3に濃縮し、トルエンで洗い流して残存MTBEを取り除き、約40Lの全バッチ容量までトルエンで希釈した。この溶液に水(1L)を激しく攪拌しながら添加した。二相性混合物を室温(18〜23℃)で数分間エージングし、化合物I二水和物の種を用いて結晶種を入れた(実施例4)。このスラリーを室温(18〜23℃)で5時間激しく攪拌した。ヘプタン(51.5L)を攪拌しながら室温で2〜4時間かけて添加した。このバッチをろ過し、湿ったケーキを1:1トルエン/ヘプタン(15L)、引き続きヘプタン(15L)で洗浄し、乾燥窒素下で40〜50℃で乾燥し、白色の結晶性固体として化合物Iの遊離酸無水物を得た。上記で得られた固体無水物を湿った窒素(相対湿度>70%、不凝縮)の気流下に置くことによって、結晶性二水和物に水和する。
【0058】
(実施例2)
化合物Iの結晶性無水物および二水和物の調製
化合物Iの水和物と無水物は容易に相互転換する。実施例1由来の化合物I二水和物、または遊離酸としての他の供給源由来の粗もしくは純化合物I二水和物は、結晶性無水物にすることができるとともに、以下のように結晶性二水和物に転換して戻すことができる。化合物I(7.57Kgアッセイ、14.4モル)を十分に乾燥したトルエンに溶解し、30.3Lの全容量(4.0Lトルエン溶液/化合物IのKg)を有する溶液とする。
【0059】
水(1.03L;1.03Kg;57.4モル;4.00当量)をこの溶液に激しく攪拌しながら添加する。この二相性混合物を室温(18〜23℃)で数分間エージングし、次いで、化合物I二水和物の種(67g、0.12モル、0.0083当量)を用いて結晶種を入れる。このバッチは結晶種を入れた後、濁る。固体が徐々に析出し、濃いスラリーを得る。このスラリーを、上清の平衡(トルエン中60〜80mg/mlの化合物I)に達するまで室温(18〜23℃)で激しく攪拌する。
【0060】
次いで、ヘプタン(51.5L;35.2kg)を攪拌しながら室温で2〜4時間かけて添加する。得られた混合物を、上清の平衡(通常7mg/ml未満の化合物I)に達するまで室温で攪拌する。
【0061】
このバッチを室温でろ過する。湿ったケーキを1:1トルエン/ヘプタン洗浄液(15.1L)、次いでヘプタン洗浄液(15.1L)で洗浄し、次いで乾燥窒素下で約40〜50℃で乾燥し、白色の結晶性固体として化合物Iの遊離酸無水物を得る。
【0062】
この固体を湿った窒素気流(相対湿度>70%、不凝縮)下で結晶性二水和物に再水和する。この手順による固体化合物I二水和物の通常の収量は7.9Kg(14.0モル、97%収率)で、>99アッセイ%化学純度および>99.8%ee.を有する。
【0063】
(実施例2A)
化合物II二水和物を精製するための代りの手順を以下に示す。スケールは、120gmの二水和物である。
【0064】
二水和物(120g)およびイソプロピルアルコール(IPA;540mL)を晶析装置に入れる。この混合物を38〜40℃に加熱し、二水和物を溶解する。脱イオン水(273mL)を約10分かけて添加し、このバッチを33.6v%水とする。バッチ温度を38〜40℃に調整する。次いで、120gの、60/40v/v脱イオン水/IPA媒体ミルド種スラリー(50mg二水和物/gスラリー)を添加する。これにより6gの二水和物(5%)、74.8mLの脱イオン水、および49.9mLのIPAとなり、バッチは37.1v%水を含む。
【0065】
次いで、30gの、60/40v/v脱イオン水/IPAを次のフラッシュとして添加する。バッチは、現在37.9v%水を含む。種晶床を38〜40℃で1時間エージングする。次いで、バッチの温度を38〜40℃に維持しながら、脱イオン水(537mL)を2回に等分にして、第1回分を6時間かけて、第2回分を9時間かけて添加する。これによりバッチは、60/40v/v脱イオン水/IPAとなる。
【0066】
このバッチを15〜20℃に1.5時間かけて徐々に冷却し、次いで、これをエージングし、ろ過する。ケーキを480mLの脱イオン水で2回洗浄する。次いで、湿ったろ過ケーキを40℃のジャケット温度で湿った空気中で乾燥する。
【0067】
(実施例3)
化合物Iの結晶性無水物の調製
結晶性無水物を、実施例1および2に記載のように乾燥窒素下で、結晶性二水和物を40〜50℃に加温することによって作製する。
【0068】
(実施例4)
化合物I二水和物の種結晶の調製
化合物I(7.6g)の粗酸形態をトルエン(30mL)に溶解し、水(1.0mL)で処理する。得られた2相混合物を室温で2時間激しく攪拌する。この混合物を5℃で2日間放置する。界面にある少量の結晶をろ過し、より大きなバッチの化合物I二水和物のための種結晶として使用する。
【0069】
無水物および二水和物のキャラクタライゼイション
X線粉末回折調査は、分子構造、結晶化度および多形を特徴付けするために広範に使用されている。この二水和物および無水物のX線粉末回折パターンは、PW3040/60コンソールを備えたフィリップス分析用X’PertPRO X線回折システムで作成された。線源として、PW3373/00セラミックCuLFF X線管K−Alpha放射を使用した。X線粉末回折スペクトルを室温で記録した(CuKα放射、2°〜40°(2θ)、0.0167°のステップ、ステップ当たり5.08秒)。
【0070】
図1は、化合物Iの二水和物の特徴的X線粉末回折パターンを示す。二水和物は、16.8°、19.6°および5.7°2θのピークによって特徴付けられる。二水和物はさらに、22.4°、7.7°および15.2°2θのピークで特徴付けられることができる。これは、さらに、24.6°、11.2°および22.8°2θのピークで特徴付けられることができる。
【0071】
図6は、化合物Iの無水物の特徴的X線粉末回折パターンを示す。この無水物は、18.1°、11.9°および9.1°2θのピークによって特徴付けられる。無水物はさらに、14.3°、16.4°および23.7°2θのピークによって特徴付けられることができる。これはさらに、5.9°、13.8°および20.5°2θのピークによって特徴付けられることができる。
【0072】
結晶形態はまた、パーキンエルマーモデルTGA7装置を使用してTGA曲線を作成する熱重量分析によって特徴付けた。実験は、窒素気流下で、10℃/分の加熱速度で行った。重量/温度データを装置によって自動的に収集した。結果の分析を装置ソフトウェア内のデルタY関数を選択するとともに、減量がその間で計算される温度を選択することによって行った。減量を分解/蒸発の開始まで報告する。
【0073】
図2は、窒素気流下、10℃/分の加熱速度での化合物I二水和物の特徴的熱重量分析(TGA)曲線を示す。21℃と55℃との間で、6.25%の減量が生じる。この減量は、化合物Iの二水和物から2モルの水が失われるとして予測される重量変化と一致する(理論的減量は6.4%)。
【0074】
二水和物の水含量をまた、カールフィッシャー滴定によって測定した。二水和物の水含量は、6.4%の理論的水含量に対して、6.6%として測定された。
【0075】
図7は、窒素気流下、10℃/分の加熱速度での化合物I無水物の特徴的熱重量分析(TGA)曲線を示す。20.9℃〜100.1℃の範囲の0.3%の減量は、化合物が無水であることを示す。
【0076】
図3は、化合物Iの無水物の特徴的示差走査熱量測定(DSC)曲線を示す。これはTAインスツルメントDSC2910示差走査熱量計を使用して得た。試料を窒素気流下、10℃/分の加熱速度で密閉パンの中で加熱した。データはこのシステムソフトウェアに含まれるDSC分析プログラムを使用して分析した。溶融吸熱は、吸熱が観察される範囲の温度の上方と下方のベースライン温度点間を積分した。記録されたデータは、開始温度、ピーク温度およびエンタルピーである。
【0077】
図8は、化合物Iの無水物の特徴的示差走査熱量測定曲線を示す。DSC曲線は、以下の条件下でTAインスツルメントQ1000示差走査熱量計を使用して得た。(1)試料を最初3℃/分で60℃まで加熱した。(2)試料を10℃/分で5℃まで冷却した後、60℃で30分間等温的に保持した。(3)次いで、調整運転を60秒ごとに1℃の変調および窒素気流下3℃/分の線形加熱速度で120℃まで行った。試料は密閉パンの中で加熱した。
【0078】
上記のX線粉末回折パターンに加えて、化合物Iの結晶性二水和物形態をさらに、固体炭素−13および固体フッ素−19核磁気共鳴(NMR)スペクトルによって特徴付けた。固体炭素−13NMRスペクトルは、ブルーカー(Bruker)4mm二重共鳴CPMASプローブを使用するブルーカー(Bruker)DSX400WB NMRシステムで得た。固体炭素−13NMRスペクトルは、可変増幅交差分極を備えた(プロトン/炭素−13)交差分極マジック角回転を利用した。試料を15.0kHzで回転し、全1,777スキャンを7秒のリサイクル遅れで収集した。FTを行う前に、20Hzの線幅拡大をスペクトルに対して適用した。化学シフトをグリシンのカルボニル炭素(176.03p.p.m.)を二次基準として使用するTMSスケールで記録する。
【0079】
固体フッ素−19NMRスペクトルは、ブルーカー(Bruker)4mm CRAMPSプローブを使用するブルーカー(Bruker)DSX400WB NMRシステムで得た。NMRスペクトルは、単純パルス獲得パルスプログラムを利用した。試料を15.0kHzで回転し、2秒のリサイクル遅れで全128スキャンを収集した。ベスペルエンドキャップ(vespel endcap)を、フッ素のバックグラウンドを最少化するために利用した。FTを行う前に、50Hzの線幅拡大をスペクトルに対して適用した。化学シフトを、−122p.p.m.の化学シフトに割り当てられる外部二次基準としてポリ(テトラフルオロエチレン)(テフロン(登録商標))を使用して記録する。
【0080】
図4は、化合物Iの二水和物形態のための固体炭素−13CPMAS NMRスペクトルを示す。二水和物形態は、11.1、56.2、190.7p.p.m.の化学シフト値を有する特徴的なシグナルを示した。二水和物形態のさらなる特徴は、13.9、78.3、および174.2p.p.m.の化学シフト値を有するシグナルである。二水和物形態は、さらにもっと164.1、および157.6p.p.m.の化学シフト値を有するシグナルによって特徴付けられる。
【0081】
図5は、化合物Iの二水和物形態の固体フッ素−19MAS NMRスペクトルを示す。二水和物形態は、−56.0p.p.m.の化学シフト値を有する特徴的シグナルを示した。
【0082】
図9は、化合物Iの無水物形態の固体炭素−13CPMAS NMRスペクトルを示す。無水物形態は、10.3、54.1、および192.2p.p.m.の化学シフト値を有する特徴的シグナルを示した。無水物形態のさらなる特徴は、11.9、76.3、171.2p.p.m.の化学シフト値を有するシグナルであった。無水物形態はさらにもっと、163.0および159.3p.p.m.の化学シフト値を有するシグナルによって特徴付けられる。
【0083】
図10は、化合物Iの無水物形態の固体フッ素−19MAS NMRスペクトルを示す。無水物形態は、−55.6p.p.m.の化学シフト値を有する特徴的シグナルを示した。
【0084】
本発明の結晶性化合物Iの二水和物形態および結晶性化合物Iの無水物形態はそれぞれ、上記X線粉末回折、フッ素−19MAS NMR、炭素−13CPMAS NMR、およびDSC物理特性を有する、二水和物または無水物の少なくとも約5%の純相を有する。1つの実施形態において、純相は、上記固体の物理特性を有する、二水和物または無水物の少なくとも約10%である。第2の実施形態において、純相は、上記固体の物理特性を有する、二水和物または無水物の少なくとも約25%である。第3の実施形態において、純相は、上記固体の物理特性を有する、二水和物または無水物の少なくとも約50%である。第4の実施形態において、純相は、上記固体の物理特性を有する、二水和物または無水物の少なくとも約75%である。第5の実施形態において、純相は、上記固体の物理特性を有する、二水和物または無水物の少なくとも約90%である。第6の実施形態において、結晶性化合物Iは、上記固体の物理特性を有する、実質的に純相の二水和物または無水物である。「純相」の用語によって、本発明に記載された固体物理方法によって測定された化合物Iの別の特徴ある結晶性多形もしくは非結晶形態に関する化合物I無水物形態もしくは二水和物形態の固体純度を意味する。
【0085】
(実施例5)
(2R)−2−{3−[[3−(4−クロロベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]メチル]フェノキシ}酪酸(化合物II)の合成
ステップ1〜4は、実施例1のステップ1〜4と同じである。
【0086】
ステップ5
【0087】
【化15】
【0088】
ヘプタン(9.0L)中のクロトン酸メチル(3.00Kg)の溶液を窒素下、25〜35℃で20〜30分かけて臭素(1.55L)で処理した。反応混合物を30℃で1時間攪拌した。アセトニトリル(18L)を添加し、引き続き粉末炭酸カリウム(6.08Kg)を1回で添加した。反応物スラリーを60℃で1時間加温するとともに攪拌し、次いで、還流(74℃のバッチ温度)まで加熱した。ヘプタン/アセトニトリルアゼオトロープ(沸点69℃)を、溶媒15Lの除去をもって、蒸留除去した。アセトニトリル(ACN、6L)を常圧蒸留中に添加した。蒸留が完了したら、スラリーを80℃で3〜4時間エージングし、脱臭化水素反応を完了した。反応が完了したら、スラリーを20℃に冷却し、ろ過した。ケーキをアセトニトリル(2×6L)で洗浄した。生成物5〜7の溶液を、アルキル化反応においてさらに精製することなく使用した。
【0089】
ステップ6
【0090】
【化16】
【0091】
アセトニトリル中のメチル2−ブロモブタ−2−エノエート5−7(20.9Kg)および3−ヒドロキシベンジルアルコール(3.00Kg)の溶液を、粉末炭酸カリウム(6.54Kg)で処理し、このスラリーを80〜82℃に1時間かけて加温した。スラリーを80〜82℃で2時間エージングした。アセトニトリル(10L)を、常圧で2時間かけて蒸留除去した。反応混合物を50℃に冷却し、トルエン(20L)を添加した。このスラリーを減圧下、30〜40℃で濃縮し、全25Lの留出物を除去した。トルエン(10L)を添加し、スラリーを15℃に冷却した。水(20L)を添加し、相をよく混合した。水(7.5L)および50%w/v水酸化カリウム水溶液(5.31L)を添加した。2相混合物を攪拌し、35℃に5〜10時間加温した。混合物を20℃に冷却し、層を分離させた。下層(水層)を50Lの反応容器に入れ、15℃に冷却した。濃塩酸(2.25L)を添加し、pHを8〜9に調整した。IPAC(25L)を添加し、pHを濃塩酸(2.25L)で1.5(+/−0.2)に調整した。下側の水層を除去した。有機相を水(2×3.0L)で洗浄した。
【0092】
不飽和酸のIPAC溶液を減圧下、20〜30℃で濃縮し、溶媒をトルエンに切り換えた。容量を約30Lに調整した。スラリーを20℃で30分間エージングし、ろ過した。ケーキをトルエン(20L)で洗浄し、窒素気流下、ろ過漏斗上で一夜乾燥し、結晶性生成物5−8を得た。
【0093】
ステップ7
【0094】
【化17】
【0095】
不飽和酸5−8(3.922Kg)、トリエチルアミン(2.63L)およびメタノール(13.4L)を10ガロンのオートクレーブに入れた。メタノール(1L)を使用して移入を完了した。メタノール(800mL)中(R−BINAP)RuCl2(76.4g)のスラリーを、窒素下、1Lのステンレス鋼のボンベによって添加した。このバッチを100psi水素、20℃で20時間水素添加した。バッチを5ミクロンのインラインフィルターを通してろ過し、濃縮し、溶媒をトルエンに切り換えた。水(8L)および5MNaOH水溶液(4L)を添加し、相を分離させ、有機相を水(4L)で洗浄した。混合した水層をDarco G−60炭素(400g)を用いて60℃で1時間処理した。濃塩酸水溶液(300mL)を添加して混合物のpHをpH7に調整し、混合物を一夜(16時間)22℃まで冷却した。ソルカフロック(200g)を添加し、混合物をソルカフロックを介してろ過し、水(4L)で洗浄した。
【0096】
ろ液を10℃に冷却し、冷却によって13〜18℃の温度を維持しながら濃塩酸(3.7L)でpH=1に酸性化した。この混合物をIPAc(20L)で抽出した。このIPAc抽出物を塩水(brine)(2×4L)で洗浄し、生成物をヘプタン−IPAcの3:1混合物から結晶化した。結晶性生成物5−9を窒素気流下で乾燥した。
【0097】
ステップ8
【0098】
【化18】
【0099】
飽和酸(3.265Kg)、TsOH−H2O(58g)およびメタノール(45.7L)を60℃に14〜18時間加熱した。トリエチルアミン(86mL)を添加し、混合物を約10Lに濃縮した。この混合物の溶媒を酢酸イソプロピルに切り換え、生成物5−10を次のステップにおいて精製することなく使用した。
【0100】
ステップ9
【0101】
【化19】
【0102】
IPAC(10L)中メチルエステル(3.433Kg)の粗溶液にDMF(7.65L)を添加し、混合物を−15℃に冷却した。塩化チオニル(2.0Kg)を均一な淡い琥珀色の混合物に45分かけて添加した。混合物を20℃に30分かけて加温し、20℃で90分間エージングした。混合物を0℃に冷却し、n−ヘプタン(7.65L)を添加した。水(15.3L)を攪拌しながら5分かけて添加した。水相を除き、有機相を水(2×15L)で洗浄した。有機相を約4Lに濃縮し、生成物5−11溶液をさらに精製することなく使用した。
【0103】
ステップ10
【0104】
【化20】
【0105】
0℃のトルエン(9L)中ステップ4、実施例1のインドール生成物6(1.98Kg)の溶液にトルエン(6.1L)中1.8MEt2AlClを1〜2時間かけて添加した。得られた溶液を0℃で60分間攪拌した。n−ヘプタン(3L)中の溶液としてp−クロロベンゾイルクロライド(1.93Kg)を1〜2時間かけて添加し、反応物を12時間攪拌した。混合物を0℃に冷却し、メタノール(1.8L)を1時間かけて添加した。この反応物スラリーを1時間エージングした。n−ヘプタン(17L)を添加し、得られたスラリーを1時間エージングし、ろ過し、収集した沈殿物を追加のn−ヘプタン(22L)で洗浄した。
【0106】
赤色のケーキをすべての固体が溶解するまで還流下でメタノール(61L)中でスラリー化した。水(15L)を生成物の結晶化を誘導するために滴下漏斗によって1時間かけて添加した。得られたスラリーを室温に冷却し、生成物5−12をろ過によって単離し、窒素気流下で乾燥した。
【0107】
ステップ11
【0108】
【化21】
【0109】
DMF(5.70L)中アシルインドール5−12(2.52Kg)、ベンジルクロライド5−11(1.76Kg)、粉末炭酸カリウム(1.92Kg)、およびnBu4NI(263g)のスラリーを35℃で22時間攪拌した。この混合物を20℃に冷却し、次いでMTBE(4L)および酢酸(428g)を添加した。次いで、MTBE(17.4L)および水(14L)を添加し、水相を除去し、廃棄した。有機相を水(21L)で洗浄した。MTBE中生成物13の溶液を精製することなく使用した。
【0110】
ステップ12
【0111】
【化22】
【0112】
メタノール(11.2L)をMTBE中インドールメチルエステル5−13(3.71Kg)の粗溶液に添加した。1.0NNaOH(13.25L)を室温で添加した。得られた反応混合物を窒素下、40℃で4時間攪拌した。この反応混合物を室温に冷却し、5.0NHCl(2.91L)を激しく攪拌しながら5〜10分かけて添加した。有機層を30〜35℃で水(3×9L)を用いて洗浄した。有機層を減圧下で濃縮し、溶媒をトルエン(約24L)に切り換えた。ヘプタン(24L)を30分かけて添加し、得られたスラリーを室温で12〜14時間かけて攪拌した。生成物をろ過によって単離し、湿ったケーキを1:2トルエン/ヘプタン(12L)、次いでヘプタン(8L)で洗浄した。結晶性生成物化合物IIを窒素下、室温で乾燥した。
【0113】
(実施例6)
化合物IIの遊離酸(4g)をIPA(60ml)に溶解する。5N水酸化ナトリウム(1.46mL)を添加し、このスラリーを55℃に加温し、均一な溶液を得る。溶液を50℃に冷却し、1時間エージングして薄いスラリーを得る。スラリーを室温で一夜冷却する。スラリーをろ過し、真空中、40℃で一夜乾燥し、細かい結晶性針状物として化合物IIのナトリウム塩(3.6g)を得る。
【0114】
化合物IIの結晶形態
遊離酸としての化合物IIは、少なくとも3つの多形結晶形態を有し、A型、B型およびC型という。ナトリウム塩は、少なくとも5つの多形結晶形態を有し、I型、II型、III型、IV型およびV型という。実施例5に記載の合成はC型の遊離酸を生じ、これは好ましい遊離酸結晶塩形態である。実施例6に記載の合成はV型のナトリウム塩を生じ、これは好ましいナトリウム塩形態である。
【0115】
A型をIPA中の化合物IIのナトリウム塩の酸性化によって調製する。V型以外の化合物IIのナトリウム塩は、さまざまな溶媒にナトリウム塩を懸濁させることによって得る:IPA中の懸濁液からのI型およびIII型;α,α,α−トリフルオロトルエン中の懸濁液からのII型;およびTHF、AcOEtおよびACNからのIV型。V型のナトリウム塩を最も容易に得る。C型の遊離酸は、最も熱力学的に安定な酸形態であるようにみえる。
【0116】
(実施例7)
B型およびC型の遊離酸
B型遊離酸は、100.4℃および113.1℃で発熱を示すDSC曲線(図12)を有する結晶性(図11)であり、脱水およびC型への転換に起因し得、引き続きC型の溶融が生じる(図15にみられる)。遊離酸(図3)の1モルにとって1.5モルの水に相当する4.7%の減量(図13)がある。図11は、結晶性B型の特徴的X線粉末回折パターンを示す。結晶性B型は、さらに17.7°、22.1°および18.3°2θのピークによって特徴付けられる。結晶性B型はさらに、11.2°、20.1°および5°2θのピークによって特徴付けられることができる。これはさらに、5.8°、10.2°および25.5°2θのピークによって特徴付けられることができる。
【0117】
C型遊離酸は、図14のXRPDによって示されるように結晶性である。C型遊離酸は、図15のDSCにおいて示されるように171.8℃の溶融開始温度、172.8℃でのピーク、および107.2J/gの融解熱を有する。C型遊離酸のTGAは、図16の曲線によって示されるように0.1%未満の最少の減量を示す。図14は、結晶性C型遊離酸の特徴的X線粉末回折パターンを示す。結晶性C型遊離酸は、21.6°、17°および16.3°2θのピークによって特徴付けられる。結晶性C型遊離酸はさらに、13.6°、5.5°、7.9°2θのピークによって特徴付けられることができる。結晶性C型遊離酸はさらに、18.4°、20.8°および20.1°2θのピークによって特徴付けられることができる。
【0118】
(実施例8)
V型ナトリウム塩
V型ナトリウム塩をXRPD、DSCおよびTGによって分析した。これは結晶性である(図17)。DSCは229℃の開始および235℃のピークを有する融点、ならびに255℃の開始および258.6℃のピークを有する二次吸熱を示す(図18)。TGA(図19)は、254℃まで0.2%の最少の減量を示す。図17は、V型結晶性ナトリウム塩の特徴的X線粉末回折パターンを示す。V型結晶性ナトリウム塩は、20°、19.7°および24.4°2θのピークによって特徴付けられる。V型結晶性ナトリウム塩はさらに、19°、21.8°および20.9°2θのピークによって特徴付けられることができる。V型結晶性ナトリム塩はさらに、11.4°、15°および16.1°2θのピークによって特徴付けられることができる。
【0119】
結晶形態の比較
図20は、同じスケールで化合物IIのナトリウム塩の全5つの結晶形態(I、II、III、IV、およびV)のXRPDパターンを示す。
【0120】
図21は、同じスケールで化合物IIの遊離酸の全3つの結晶形状(A、B、およびC)のXRPDパターンを示す。
【0121】
両方の図において、化合物IIのナトリウム塩と遊離酸との結晶形態は、独特のXRPDパターンを有することがわかる。
【0122】
(実施例9)
実施例1の合成手順の化合物4の化合物6への転換はまた、以下の手順によって行うことができる。
【0123】
【化23】
【0124】
メーヤワイン反応およびケタール化
酢酸イソプロペニル(22mL)中ニトロアニリン4(22.21g)の溶液およびiBuONO(94%、13.71g)の溶液を40〜50℃に維持し、酢酸イソプロペニル200mL中のCuCl2(2.01g)の懸濁液にそれぞれ別の添加漏斗からほぼ同じ速度(±10%)で1時間かけて添加する。この混合物を40〜50℃で1時間エージングし、25℃に冷却する。トルエン(100mL)を添加し、混合物を1MHCl(2×15mL)で洗浄する;大分部の銅は、この2回の少量の水性洗浄中に濃縮され、次いで水(2×100mL)で洗浄する。有機相を50mLに濃縮し、一定容量で200Torrで100mLのトルエンを使用して溶媒をトルエンに切り換える。次いで、混合物を、一定容量で200Torrで150mLのエタノールを使用して溶媒をエタノールに切り換える。50mLエタノール中ニトロケトン5を含む溶液をエチレングリコール(9.31g)、(EtO)3CH(21.03g)、およびp−TsOH(0.50g)と混合し、溶液を45℃に16時間または80℃に1〜2時間加熱し、次いで25℃に冷却した。ニトロケトン5の転化をHPLCにより監視した。
【0125】
水素添加
トリエチルアミン(0.28g)を前ステップの粗ニトロアリールケタールの溶液に添加し(pHは、pH紙を使用して測定して、<2から>5に変化した)、得られた混合物を耐圧瓶の中に置いた。ラネーニッケル(22.2gの湿ったスラリー)を添加し、混合物を40psigおよび25〜70℃(発熱性)で2時間水素添加した。混合物をDarco G60(1.1g)で25〜40℃で20分間処理し、次いでソルカフロックを介してろ過し、パッドをエタノール(50mL)で洗浄した。
【0126】
加水分解/環化
水25.9mL中クエン酸(27.3g)の溶液を前ステップからの粗生成物の溶液に添加した。この混合物を6時間エージングして、80℃で約95%の転化を達成した。次いで混合物を20℃に冷却し、pHが5になるまで十分量の50%NaOHを入れた。このバッチを200Torrで約50mlに濃縮し、次いで溶媒をトルエンに切り換え、留出物からエタノールを除去した。トルエン溶液を水(2×50mL)で洗浄した。次いで溶媒を常圧で水に切り換えた。所望のインドール生成物6は揮発性であり、トルエンが除去される際、留出物中に出始める。インドールは水に不溶であり、約83℃の融点を有する。蒸留の継続時、蒸気はトルエン/水混合物中に散布され、トルエン層中のインドールを補足するかまたは固体インドール(融点約83℃)がコンデンサーを詰まらせないように約85℃で凝縮させる。トルエンが除去された後、大部分のインドールは依然として蒸留容器中に残存し、蒸気蒸留(スチームストリッピング)を行うように水で蒸留して除き続けた。全てのインドールが留出物受けのトルエン層中に収集されるまで、水を蒸留物からリサイクして蒸留容器に戻す。水約65mlをインドール1g当たり蒸留する。
【0127】
(医薬組成物の実施例)
化合物Iの結晶性二水和物形態は、以下のように製剤することができる。化合物Iの100mg投与形態は、結晶性二水和物とアビセル(Avicel)(フィラー)、ラクトース(フィラー)、クロスカルメロースナトリウム(錠剤崩壊成分)、ラウリル硫酸ナトリム(界面活性剤)、およびステアリン酸マグネシウム(潤滑剤)とをローラーで固めて作製される顆粒形状の化合物I二水和物107mgから構成される。次いで、顆粒をゼラチンカプセルの中へ充填するかまたは錠剤に成形する。好ましくは、錠剤を濃色の不透明のフィルムでコートする。
【0128】
化合物IIの100mg投与形態は、上記と同じ手順を使用してC型結晶の100mgを処方することによって作製する。
【図面の簡単な説明】
【0129】
【図1】化合物Iの結晶性二水和物の特徴的X線粉末回折パターンを示す図である。
【図2】化合物Iの結晶性二水和物の特徴的熱重量分析(TGA)曲線を示す図である。
【図3】化合物Iの結晶性二水和物の特徴的示差走査熱量測定(DSC)曲線を示す図である。
【図4】化合物Iの結晶性二水和物形態の特徴的固体炭素−13CPMAS NMRスペクトルを示す図である。
【図5】化合物Iの結晶性二水和物形態の特徴的固体フッ素−19MAS NMRスペクトルを示す図である。
【図6】化合物Iの結晶性無水物の特徴的X線粉末回折パターンを示す図である。
【図7】化合物Iの結晶性無水物の特徴的熱重量分析(TGA)曲線を示す図である。
【図8】化合物Iの結晶性無水物の特徴的示差走査熱量測定(DSC)曲線を示す図である。
【図9】化合物Iの結晶性無水物形態の特徴的固体炭素−13CPMAS NMRスペクトルを示す図である。
【図10】化合物Iの結晶性無水物形態の特徴的固体フッ素−19MAS NMRスペクトルを示す図である。
【図11】化合物IIのB型遊離酸の特徴的X線粉末回折パターンを示す図である。
【図12】化合物IIのB型遊離酸の特徴的示差走査熱量測定(DSC)曲線を示す図である。
【図13】化合物IIのB型遊離酸の特徴的熱重量分析(TGA)曲線を示す図である。
【図14】化合物IIのC型遊離酸の特徴的X線粉末回折パターンを示す図である。
【図15】化合物IIのC型遊離酸の特徴的示差走査熱量測定(DSC)曲線を示す図である。
【図16】化合物IIのC型遊離酸の特徴的熱重量分析(TGA)曲線を示す図である。
【図17】化合物IIのV型ナトリウム塩の特徴的X線粉末回折パターンを示す図である。
【図18】化合物IIのV型ナトリウム塩の特徴的示差走査熱量測定(DSC)曲線を示す図である。
【図19】化合物IIのV型ナトリウム塩の特徴的熱重量分析(TGA)曲線を示す図である。
【図20】同じスケールの化合物IIのナトリウム塩の全5つの結晶形態のX線粉末回折パターンであり、それらが異なり、明確に区別できることを示す図である。
【図21】同じスケールの化合物IIの遊離酸の全3つの結晶形態のX線粉末回折パターンであり、それらが異なり、明確に区別できることを示す図である。
【特許請求の範囲】
【請求項1】
化合物Iは、構造式I
【化1】
を有する(2R)−2−{3−[3−(4−メトキシベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]フェノキシ}ブタン酸であり、結晶性遊離酸二水和物または結晶性遊離酸無水物であることを特徴とし、化合物IIは構造式II
【化2】
を有する(2R)−2−{3−[[3−(4−クロロベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]メチル]フェノキシ}酪酸であり、結晶性遊離酸または結晶性ナトリウム塩であることを特徴とする、化合物Iおよび化合物IIからなる群から選択される化合物。
【請求項2】
結晶性遊離酸二水和物または結晶性遊離酸無水物であることを特徴とする請求項1の化合物I。
【請求項3】
結晶性遊離酸二水和物であることを特徴とする請求項2の化合物I。
【請求項4】
結晶性遊離酸無水物であることを特徴とする請求項2の化合物I。
【請求項5】
X線粉末回折パターン、固体フッ素−19MAS核磁気共鳴スペクトル、固体炭素−13CPMAS核磁気共鳴スペクトル、示差走査熱量測定(DSC)曲線、および熱重量分析(TGA)曲線から選択される1以上のスペクトル特性を有する結晶性遊離酸二水和物であることを特徴とする請求項3の化合物I。
【請求項6】
X線粉末回折パターン、固体フッ素−19MAS核磁気共鳴スペクトル、固体炭素−13CPMAS核磁気共鳴スペクトル、示差走査熱量測定(DSC)曲線、および熱重量分析(TGA)曲線から選択される1以上のスペクトル特性を有する結晶性遊離酸無水物であることを特徴とする請求項4の化合物I。
【請求項7】
16.8°、19.6°、5.7°、22.4°、7.7°、15.2°、24.6°、11.2°および22.8°2θのXRPDピークを特徴とする請求項3の化合物I。
【請求項8】
11.1、56.2、190.7、13.9、78.3、174.2、164.1および157.6p.p.m.の化学シフト値を有する固体炭素−13CPMAS NMRスペクトルにおけるピークを特徴とする請求項3の化合物I。
【請求項9】
18.1°、11.9°、9.1°、14.3°、16.4°、23.7°、5.9°、13.8°、20.5°2θのXRPDピークを特徴とする請求項4の化合物I。
【請求項10】
10.3、54.1、192.2、11.9、76.3、171.2、163.0および159.3p.p.m.の化学シフト値を有する固体状態炭素−13CPMAS NMRスペクトルにおけるピークを特徴とする請求項4の化合物I。
【請求項11】
結晶性遊離酸または結晶性ナトリウム塩であることを特徴とする請求項1の化合物II。
【請求項12】
結晶性遊離酸であることを特徴とする請求項11の化合物II。
【請求項13】
結晶性ナトリウム塩であることを特徴とする請求項11の化合物II。
【請求項14】
X線粉末回折パターン、示差走査熱量測定(DSC)曲線、および熱重量分析(TGA)曲線から選択される1以上のスペクトル特性を有する結晶性遊離酸であることを特徴とする請求項12の化合物II。
【請求項15】
X線粉末回折パターン、示差走査熱量測定(DSC)曲線、および熱重量分析(TGA)曲線から選択される1以上のスペクトル特性を有する結晶性ナトリウム塩であることを特徴とする請求項13の化合物II。
【請求項16】
17.7°、22.1°、18.3°、11.2°、20.1°、5°、5.8°、10.2°および25.5°2θのXRPDピークによるB型結晶性遊離酸であることを特徴とする請求項14の化合物II。
【請求項17】
21.6°、17°、16.3°、13.6°、5.5°、7.9°、18.4°、20.8°および20.1°2θのXRPDピークによるC型結晶性遊離酸であることを特徴とする請求項14の化合物II。
【請求項18】
20°、19.7°、24.4°、19°、21.8°、20.9°、11.4°、15°および16.1°2θのXRPDピークによるV型結晶性ナトリウム塩であることを特徴とする請求項15の化合物II。
【請求項19】
図21に示されるX線粉末回折パターンを有することを特徴とし、A型、B型およびC型、またはその混合物から選択される結晶性遊離酸であることを特徴とする請求項12の化合物II。
【請求項20】
図20に示されるX線粉末回折パターンを有することを特徴とし、I型、II型、III型、IV型およびV型、またはその混合物から選択される結晶性ナトリウム塩であることを特徴とする請求項13の化合物II。
【請求項21】
化合物Iが結晶性遊離酸二水和物または結晶性遊離酸無水物であることを特徴とし、および化合物IIが結晶性遊離酸または結晶性ナトリウム塩であることを特徴とする、1種以上の医薬上許容される担体または賦形剤と共に請求項1の化合物Iまたは化合物IIを含む医薬組成物。
【請求項22】
化合物Iが、結晶性遊離酸二水和物であることを特徴とする化合物Iを含む請求項21の医薬組成物。
【請求項23】
化合物Iが、結晶性遊離酸無水物であることを特徴とする化合物Iを含む請求項21の医薬組成物。
【請求項24】
化合物IIが、C型結晶性遊離酸であることを特徴とする化合物IIを含む請求項21の医薬組成物。
【請求項25】
化合物IIが、V型結晶性ナトリウム塩であることを特徴とする化合物IIを含む請求項21の医薬組成物。
【請求項26】
治療を必要とする患者の2型糖尿病、高血糖症、肥満症、異脂肪血症、または代謝症候群を治療する方法であり、請求項1の結晶性化合物Iまたは結晶性化合物IIの治療有効量を患者に投与することを含み、化合物Iが、結晶性遊離酸二水和物または結晶性遊離酸無水物であることを特徴とし、および化合物IIが、結晶性遊離酸または結晶性ナトリウム塩であることを特徴とする、方法。
【請求項27】
ヒトまたは哺乳類患者の2型糖尿病、高血糖症、肥満症、異脂肪血症、または代謝症候群の治療用の医薬の製造における活性成分としての請求項1の結晶性化合物Iまたは結晶性化合物IIの使用であり、結晶性化合物Iが、結晶性遊離酸二水和物または結晶性遊離酸無水物であることを特徴とし、および化合物IIが、結晶性遊離酸または結晶性ナトリウム塩であることを特徴とする、使用。
【請求項1】
化合物Iは、構造式I
【化1】
を有する(2R)−2−{3−[3−(4−メトキシベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]フェノキシ}ブタン酸であり、結晶性遊離酸二水和物または結晶性遊離酸無水物であることを特徴とし、化合物IIは構造式II
【化2】
を有する(2R)−2−{3−[[3−(4−クロロベンゾイル)−2−メチル−6−(トリフルオロメトキシ)−1H−インドール−1−イル]メチル]フェノキシ}酪酸であり、結晶性遊離酸または結晶性ナトリウム塩であることを特徴とする、化合物Iおよび化合物IIからなる群から選択される化合物。
【請求項2】
結晶性遊離酸二水和物または結晶性遊離酸無水物であることを特徴とする請求項1の化合物I。
【請求項3】
結晶性遊離酸二水和物であることを特徴とする請求項2の化合物I。
【請求項4】
結晶性遊離酸無水物であることを特徴とする請求項2の化合物I。
【請求項5】
X線粉末回折パターン、固体フッ素−19MAS核磁気共鳴スペクトル、固体炭素−13CPMAS核磁気共鳴スペクトル、示差走査熱量測定(DSC)曲線、および熱重量分析(TGA)曲線から選択される1以上のスペクトル特性を有する結晶性遊離酸二水和物であることを特徴とする請求項3の化合物I。
【請求項6】
X線粉末回折パターン、固体フッ素−19MAS核磁気共鳴スペクトル、固体炭素−13CPMAS核磁気共鳴スペクトル、示差走査熱量測定(DSC)曲線、および熱重量分析(TGA)曲線から選択される1以上のスペクトル特性を有する結晶性遊離酸無水物であることを特徴とする請求項4の化合物I。
【請求項7】
16.8°、19.6°、5.7°、22.4°、7.7°、15.2°、24.6°、11.2°および22.8°2θのXRPDピークを特徴とする請求項3の化合物I。
【請求項8】
11.1、56.2、190.7、13.9、78.3、174.2、164.1および157.6p.p.m.の化学シフト値を有する固体炭素−13CPMAS NMRスペクトルにおけるピークを特徴とする請求項3の化合物I。
【請求項9】
18.1°、11.9°、9.1°、14.3°、16.4°、23.7°、5.9°、13.8°、20.5°2θのXRPDピークを特徴とする請求項4の化合物I。
【請求項10】
10.3、54.1、192.2、11.9、76.3、171.2、163.0および159.3p.p.m.の化学シフト値を有する固体状態炭素−13CPMAS NMRスペクトルにおけるピークを特徴とする請求項4の化合物I。
【請求項11】
結晶性遊離酸または結晶性ナトリウム塩であることを特徴とする請求項1の化合物II。
【請求項12】
結晶性遊離酸であることを特徴とする請求項11の化合物II。
【請求項13】
結晶性ナトリウム塩であることを特徴とする請求項11の化合物II。
【請求項14】
X線粉末回折パターン、示差走査熱量測定(DSC)曲線、および熱重量分析(TGA)曲線から選択される1以上のスペクトル特性を有する結晶性遊離酸であることを特徴とする請求項12の化合物II。
【請求項15】
X線粉末回折パターン、示差走査熱量測定(DSC)曲線、および熱重量分析(TGA)曲線から選択される1以上のスペクトル特性を有する結晶性ナトリウム塩であることを特徴とする請求項13の化合物II。
【請求項16】
17.7°、22.1°、18.3°、11.2°、20.1°、5°、5.8°、10.2°および25.5°2θのXRPDピークによるB型結晶性遊離酸であることを特徴とする請求項14の化合物II。
【請求項17】
21.6°、17°、16.3°、13.6°、5.5°、7.9°、18.4°、20.8°および20.1°2θのXRPDピークによるC型結晶性遊離酸であることを特徴とする請求項14の化合物II。
【請求項18】
20°、19.7°、24.4°、19°、21.8°、20.9°、11.4°、15°および16.1°2θのXRPDピークによるV型結晶性ナトリウム塩であることを特徴とする請求項15の化合物II。
【請求項19】
図21に示されるX線粉末回折パターンを有することを特徴とし、A型、B型およびC型、またはその混合物から選択される結晶性遊離酸であることを特徴とする請求項12の化合物II。
【請求項20】
図20に示されるX線粉末回折パターンを有することを特徴とし、I型、II型、III型、IV型およびV型、またはその混合物から選択される結晶性ナトリウム塩であることを特徴とする請求項13の化合物II。
【請求項21】
化合物Iが結晶性遊離酸二水和物または結晶性遊離酸無水物であることを特徴とし、および化合物IIが結晶性遊離酸または結晶性ナトリウム塩であることを特徴とする、1種以上の医薬上許容される担体または賦形剤と共に請求項1の化合物Iまたは化合物IIを含む医薬組成物。
【請求項22】
化合物Iが、結晶性遊離酸二水和物であることを特徴とする化合物Iを含む請求項21の医薬組成物。
【請求項23】
化合物Iが、結晶性遊離酸無水物であることを特徴とする化合物Iを含む請求項21の医薬組成物。
【請求項24】
化合物IIが、C型結晶性遊離酸であることを特徴とする化合物IIを含む請求項21の医薬組成物。
【請求項25】
化合物IIが、V型結晶性ナトリウム塩であることを特徴とする化合物IIを含む請求項21の医薬組成物。
【請求項26】
治療を必要とする患者の2型糖尿病、高血糖症、肥満症、異脂肪血症、または代謝症候群を治療する方法であり、請求項1の結晶性化合物Iまたは結晶性化合物IIの治療有効量を患者に投与することを含み、化合物Iが、結晶性遊離酸二水和物または結晶性遊離酸無水物であることを特徴とし、および化合物IIが、結晶性遊離酸または結晶性ナトリウム塩であることを特徴とする、方法。
【請求項27】
ヒトまたは哺乳類患者の2型糖尿病、高血糖症、肥満症、異脂肪血症、または代謝症候群の治療用の医薬の製造における活性成分としての請求項1の結晶性化合物Iまたは結晶性化合物IIの使用であり、結晶性化合物Iが、結晶性遊離酸二水和物または結晶性遊離酸無水物であることを特徴とし、および化合物IIが、結晶性遊離酸または結晶性ナトリウム塩であることを特徴とする、使用。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
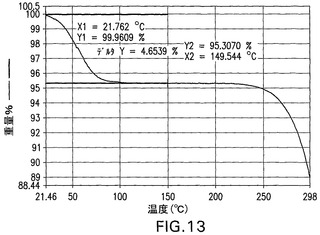
【図14】
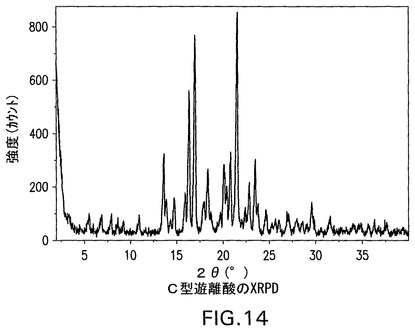
【図15】
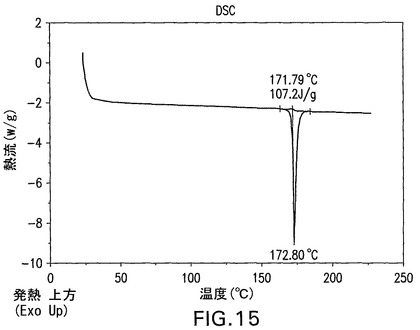
【図16】
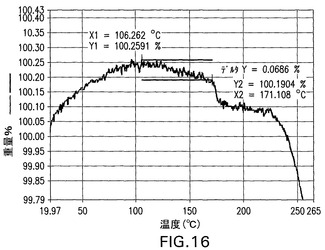
【図17】
【図18】
【図19】
【図20】
【図21】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【公表番号】特表2008−533033(P2008−533033A)
【公表日】平成20年8月21日(2008.8.21)
【国際特許分類】
【出願番号】特願2008−500934(P2008−500934)
【出願日】平成18年3月10日(2006.3.10)
【国際出願番号】PCT/US2006/008476
【国際公開番号】WO2006/099077
【国際公開日】平成18年9月21日(2006.9.21)
【出願人】(390023526)メルク エンド カムパニー インコーポレーテッド (924)
【氏名又は名称原語表記】MERCK & COMPANY INCOPORATED
【Fターム(参考)】
【公表日】平成20年8月21日(2008.8.21)
【国際特許分類】
【出願日】平成18年3月10日(2006.3.10)
【国際出願番号】PCT/US2006/008476
【国際公開番号】WO2006/099077
【国際公開日】平成18年9月21日(2006.9.21)
【出願人】(390023526)メルク エンド カムパニー インコーポレーテッド (924)
【氏名又は名称原語表記】MERCK & COMPANY INCOPORATED
【Fターム(参考)】
[ Back to top ]