
抗腫瘍ジヒドロピラン−2−オン化合物
ラスパイリイダ(Raspailiidae)科リトプロカミア(Lithoplocamia)属リチストイデス(lithistoides)種の海綿動物から得られる抗腫瘍化合物、及びその誘導体が提供される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規の抗腫瘍化合物、それらを含有する医薬組成物及び抗腫瘍剤としてのそれらの使用に関する。
【背景技術】
【0002】
1990年、グナセケラ(Gunasekera)SPらは、カリブ海の深層水海綿体であるディスコデルミア・ディソリュータ(Discodermia dissoluta)から新規なポリヒドロキシル化ラクトンである(+)−ディスコデルモリドの単離を報告した(グナセケラSPら、有機化学ジャーナル(J. Org. Chem.)1990年、55、4912〜4915及び有機化学ジャーナル1991年、56、1346)。
【0003】
【化1】

【0004】
この化合物は、臨床的に実証されている抗癌剤であるパクリタセキル(シフ(Schiff)ら、ネイチャー(Nature)1979年、277、665〜667)と同様の作用様式を有する、強力な有糸分裂阻害剤であることが明らかにされている(ハング(Hung)DTら、生物化学(Chem. Biol.)1996年、3、287〜293及びテル・ハール(ter Haar)Eら、生化学(Biochemistry)1996年、35、243〜250)。天然生成物は、両方とも、M期で細胞周期を停止させ、微小管形成を促進し、乳癌癌腫に対して同様の阻害効果を有する(それぞれ、2.4nM及び2.1nMのIC50)
【0005】
一方、N−アシルエンアミド官能基を含有する幾つかの特異な直鎖ジペプチドが、コンドロマイセス(Chondromyces)属に属する粘液細菌から単離されている(クンツ(Kunze)Bら、抗生物質ジャーナル(J. Antibiot.)1994年、47、881〜886及びヤンセン(Jansen)Rら、有機化学ジャーナル(J. Org. Chem.)1999年、1085〜1089)。特に、これらの化合物は、クロカシンA、B、C及びDであり、電子伝達インヒビターの群である。
【0006】
【化2】
【0007】
クロカシンA〜Dは、少数のグラム陽性細菌の増殖を適度に阻害し、動物細胞培養、並びに幾つかの酵母菌及び真菌の強力なインヒビターである。最も活性なものは、真菌のサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)に対して1.4ng/mLのMICを示し、L929マウス線維芽細胞培養に対して強力な毒性(0.06mg/LのIC50)を示した、クロカシンDである。
【0008】
癌は、動物及びヒトにおける死亡の主な原因である。癌に罹患している患者への投与に活性であり安全である抗腫瘍剤を得るために、多大な努力が今まで払われており、依然として払われている。本発明により解決される問題は、癌の治療に有用な化合物を提供することである。
【発明の概要】
【課題を解決するための手段】
【0009】
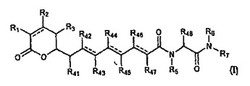
一つの態様において、本発明は、一般式Iの化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体を対象とし、
【0010】
【化3】
【0011】
式中、
R1は、水素、ORa、OCORa、OCOORa、NRaRb、NRaCORb及びNRaC(NRa)NRaRbから選択され;
R2及びR3は、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル及び置換又は非置換C2〜C12アルキニルから選択され;
R41、R42、R43、R44、R45、R46、R47及びR48は、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル及び置換又は非置換C2〜C12アルキニルから選択され;
R5、R6及びR7は、それぞれ独立して、水素、CORa、COORa、置換若しくは非置換C1〜C12アルキル、置換若しくは非置換C2〜C12アルケニル及び置換若しくは非置換C2〜C12アルキニルから選択されるか、又はR5及びR48は、それらが結合している対応するN原子及びC原子と一緒になって、置換若しくは非置換複素環式基を形成してもよく;
Ra及びRbは、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル、置換又は非置換アリール及び置換又は非置換複素環式基から選択され;そして
点線は、それぞれ任意の追加的な結合を表す。
【0012】
別の態様において、本発明は、薬剤として、特に癌を治療するための薬剤として使用される、式Iの化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体を対象とする。
【0013】
更なる態様において、本発明は、また、癌の治療における又は好ましくは癌の治療のための薬剤の調製における、式Iの化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体の使用を対象とする。本発明の別の態様は、治療方法及びこれらの方法において使用される化合物である。したがって、本発明は、更に、上記で定義された化合物の治療有効量を罹患している個体に投与することを含む癌に罹患している任意の哺乳動物、特にヒトを治療する方法を提供する。
【0014】
別の態様において、本発明は、式Iの化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体を薬学的に許容される担体又は希釈剤と一緒に含む、医薬組成物を対象とする。
【0015】
本発明は、また、ラスパイリイダ(Raspailiidae)科リトプロカミア(Lithoplocamia)属リチストイデス(lithistoides)種の海綿動物からの式Iの化合物の単離、それらを得るための方法、及びこれらの化合物からの誘導体の形成に関する。
【発明を実施するための形態】
【0016】
本発明は、上記で定義された一般式Iの化合物に関する。
【0017】
これらの化合物において、置換基を以下の指針に従って選択することができる。
【0018】
アルキル基は、分岐鎖又は非分岐鎖であることができ、好ましくは1〜約12個の炭素原子を有することができる。一つのより好ましい部類のアルキル基は、1〜約6個の炭素原子を有する。さらにより好ましいものは、1、2、3又は4個の炭素原子を有するアルキル基である。メチル、エチル、プロピル、イソプロピル、並びにtert−ブチル、sec−ブチル及びイソブチルを含むブチルが、本発明の化合物において特に好ましいアルキル基である。別の好ましい部類のアルキル基は、6〜約10個の炭素原子;さらにより好ましくは、7、8又は9個の炭素原子を有する。ヘプチル、オクチル及びノニルが、この部類の最も好ましいアルキル基である。
【0019】
本発明の化合物において好ましいアルケニル及びアルキニル基は、分岐鎖又は非分岐鎖であることができ、1つ以上の不飽和架橋及び2〜約12個の炭素原子を有することができる。一つのより好ましい部類のアルケニル及びアルキニル基は、2〜約6個の炭素原子を有する。さらにより好ましいものは、2、3又は4個の炭素原子を有するアルケニル及びアルキニル基である。別の好ましい部類のアルケニル及びアルキニル基は、4〜約10個の炭素原子、なおより好ましくは6〜約10個の炭素原子、さらにより好ましくは7、8又は9個の炭素原子を有する。
【0020】
本発明の化合物において適切なアリール基には、単環化合物、並び別々の及び/又は縮合したアリール基を含有する多環化合物を含む多環化合物が含まれる。典型的なアリール基は、1〜3個の分離又は縮合環及び6〜約18個の炭素環原子を含有する。好ましくは、アリール基は、6〜約10個の炭素環原子を含有する。特に好ましいアリール基には、置換又は非置換フェニル、置換又は非置換ナフチル、置換又は非置換ビフェニル、置換又は非置換フェナントリル及び置換又は非置換アントリルが含まれる。
【0021】
適切な複素環式基には、1〜3個の分離又は縮合環及び5〜約18個の環原子を含有する複素環式芳香族基及び複素脂環式基が含まれる。好ましくは、複素環式芳香族基及び複素脂環式基は、5〜約10個の環原子を含有する。本発明の化合物において適切な複素環式芳香族基は、N、O又はS原子から選択される1、2又は3個のヘテロ原子を含有し、例えば、8−クマリニルを含むクマリニル、8−キノリルを含むキノリル、イソキノリル、ピリジル、ピラジニル、ピラゾリル、ピリミジニル、フリル、ピロリル、チエニル、チアゾリル、イソチアゾリル、トリアゾリル、テトラゾリル、イソオキサゾリル、オキサゾリル、イミダゾリル、インドリル、イソインドリル、インダゾリル、インドリジニル、フタラジニル、プテリジニル、プリニル、オキサジアゾリル、チアジアゾリル、フラザニル、ピリダジニル、トリアジニル、シンノリニル、ベンゾイミダゾリル、ベンゾフラニル、ベンゾフラザニル、ベンゾチオフェニル、ベンゾチアゾリル、ベンゾオキサゾリル、キナゾリニル、キノキサリニル、ナフチリジニル及びフロピリジルが含まれる。本発明の化合物において適切な複素脂環式基は、N、O又はS原子から選択される1、2又は3個のヘテロ原子を含有し、例えば、ピロリジニル、テトラヒドロフラニル、ジヒドロフラニル、テトラヒドロチエニル、テトラヒドロチオピラニル、ピペリジル、モルホリニル、チオモルホリニル、チオキサニル、ピペラジニル、アゼチジニル、オキセタニル、チエタニル、ホモピペリジル、オキセパニル、チエパニル、オキサゼピニル、ジアゼピニル、チアゼピニル、1,2,3,6−テトラヒドロピリジル、2−ピロリニル、3−ピロリニル、インドリニル、2H−ピラニル、4H−ピラニル、ジオキサニル、1,3−ジオキソラニル、ピラゾリニル、ジチアニル、ジチオラニル、ジヒドロピラニル、ジヒドロチエニル、ジヒドロフラニル、ピラゾリジニル、イミダゾリニル、イミダゾリジニル、3−アザビシクロ〔3.1.0〕ヘキシル、3−アザビシクロ〔4.1.0〕ヘプチル、3H−インドリル及びキノリジニルが含まれる。
【0022】
上記に記述された基は、OR′、=O、SR′、SOR′、SO2R′、NO2、NHR′、N(R′)2、=N−R′、NHCOR′、N(COR′)2、NHSO2R′、NR′C(=NR′)NR′R′、CN、ハロゲン、COR′、COOR′、OCOR′、OCONHR′、OCON(R′)2、保護OH、置換又は非置換アリール及び置換又は非置換複素環式基のような1つ以上の適切な基により1つ以上の利用可能な位置において置換されていることができ、ここでR′基は、それぞれ独立して、水素、OH、NO2、NH2、SH、CN、ハロゲン、COH、COアルキル、CO2H、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル、置換又は非置換アリール及び置換又は非置換複素環式基からなる群より選択される。上記基がそれ自体置換される場合、置換基は、前記のリストから選択することができる。
【0023】
本発明の化合物において適切なハロゲン置換基には、F、Cl、Br及びIが含まれる。
【0024】
OHに対する適切な保護基は当業者によく知られている。有機化学の保護基についての一般的な概観が、有機合成における保護基(Protecting Groups in Organic Synthesis)、第4版、ウィリー・インターサイエンス(Wiley-Interscience)においてウッツ(Wuts)、PGM及びグリーン(Greene)TWにより、及び保護基(Protecting Groups)、第3版、ゲオルグシームフェアラーグ(Georg Thieme Verlag)においてコシエンスキー(Kocienski)PJにより提供されている。これらの参考文献は、OHの保護基について章を提供している。これらの参考文献は、全て、その全体が参照として組み込まれる。上記保護OHの例には、エーテル、シリルエーテル、エステル、スルホネート、スルフェネート及びスルフィネート、カーボネート、並びにカルバメートが挙げられる。
エーテルの場合では、OHの保護基は、メチル、メトキシメチル、メチルチオメチル、(フェニルジメチルシリル)メトキシメチル、ベンジルオキシメチル、p−メトキシベンジルオキシメチル、〔(3,4−ジメトキシベンジル)オキシ〕メチル、p−ニトロベンジルオキシメチル、o−ニトロベンジルオキシメチル、〔(R)−1−(2−ニトロフェニル)エトキシ〕メチル、(4−メトキシフェノキシ)メチル、グアイアコールメチル、〔(p−フェニルフェニル)オキシ〕メチル、t−ブトキシメチル、4−ペンテニルオキシメチル、シロキシメチル、2−メトキシエトキシメチル、2−シアノエトキシメチル、ビス(2−クロロエトキシ)メチル、2,2,2−トリクロロエトキシメチル、2−(トリメチルシリル)エトキシメチル、メントキシメチル、o−ビス(2−アセトキシエトキシ)メチル、テトラヒドロピラニル、フルオラステトラヒドロピラニル、3−ブロモテトラヒドロピラニル、テトラヒドロチオピラニル、1−メトキシシクロヘキシル、4−メトキシテトラヒドロピラニル、4−メトキシテトラヒドロチオピラニル、4−メトキシテトラヒドロチオピラニルS,S−ジオキシド、1−〔(2−クロロ−4−メチル)フェニル〕−4−メトキシピペリジン−4−イル、1−(2−フルオロフェニル)−4−メトキシピペリジン−4−イル、1−(4−クロロフェニル)−4−メトキシピペリジン−4−イル、1,4−ジオキサン−2−イル、テトラヒドロフラニル、テトラヒドロチオフラニル、2,3,3a,4,5,6,7,7a−オクタヒドロ−7,8,8−トリメチル−4,7−メタノベンゾフラン−2−イル、1−エトキシエチル、1−(2−クロロエトキシ)エチル、2−ヒドロキシエチル、2−ブロモエチル、1−〔2−(トリメチルシリル)エトキシ〕エチル、1−メチル−1−メトキシエチル、1−メチル−1−ベンジルオキシエチル、1−メチル−1−ベンジルオキシ−2−フルオロエチル、1−メチル−1−フェノキシエチル、2,2,2−トリクロロエチル、1,1−ジアニシル−2,2,2−トリクロロエチル、1,1,1,3,3,3−ヘキサフルオロ−2−フェニルイソプロピル、1−(2−シアノエトキシ)エチル、2−トリメチルシリルエチル、2−(ベンジルチオ)エチル、2−(フェニルセレニル)エチル、t−ブチル、シクロヘキシル、1−メチル−1′−シクロプロピルメチル、アリル、プレニル、シンナミル、2−フェンアリル、プロパルギル、p−クロロフェニル、p−メトキシフェニル、p−ニトロフェニル、2,4−ジニトロフェニル、2,3,5,6−テトラフルオロ−4−(トリフルオロメチル)フェニル、ベンジル、p−メトキシベンジル、3,4−ジメトキシベンジル、2,6−ジメトキシベンジル、o−ニトロベンジル、p−ニトロベンジル、ペンタジエニルニトロベンジル、ペンタジエニルニトロピペロニル、ハロベンジル、2,6−ジクロロベンジル、2,4−ジクロロベンジル、2,6−ジフルオロベンジル、p−シアノベンジル、フルオラスベンジル、4−フルオラスアルコキシベンジル、トリメチルシリルキシリル、p−フェニルベンジル、2−フェニル−2−プロピル、p−アシルアミノベンジル、p−アジドベンジル、4−アジド−3−クロロベンジル、2−トリフルオロメチルベンジル、4−トリフルオロメチルベンジル、p−(メチルスルフィニル)ベンジル、p−シレタニルベンジル、4−アセトキシベンジル、4−(2−トリメチルシリル)エトキシメトキシベンジル、2−ナフチルメチル、2−ピコリル、4−ピコリル、3−メチル−2−ピコリルN−オキシド、2−キノリニルメチル、6−メトキシ−2−(4−メチルフェニル−4−キノリンメチル)、1−ピレニルメチル、ジフェニルメチル、4−メトキシジフェニルメチル、4−フェニルジフェニルメチル、p,p′−ジニトロベンズヒドリル、5−ジベンゾスベリル、トリフェニルメチル、トリス(4−t−ブチルフェニル)メチル、α−ナフチルジフェニルメチル、p−メトキシフェニルジフェニルメチル、ジ(p−メトキシフェニル)フェニルメチル、トリ(p−メトキシフェニル)メチル、4−(4′−ブロモフェナシルオキシ)フェニルジフェニルメチル、4,4′,4″−トリス(4,5−ジクロロフタルイミドフェニル)メチル、4,4′,4″−トリス(レブリノイルオキシフェニル)メチル、4,4′,4″−トリス(ベンゾイルオキシフェニル)メチル、4,4′−ジメトキシ−3″−〔N−(イミダゾリルメチル)〕トリチル、4,4′−ジメトキシ−3″−〔N−(イミダゾリルエチル)カルバモイル〕トリチル、ビス(4−メトキシフェニル)−1′−ピレニルメチル、4−(17−テトラベンゾ〔a,c,g,i〕フルオレニルメチル)−4,4″−ジメトキシトリチル、9−アントリル、9−(9−フェニル)キサンテニル、9−フェニルチオキサンチル、9−(9−フェニル−10−オキソ)アントリル、1,3−ベンゾジチオラン−2−イル及び4,5−ビス(エトキシカルボニル)−〔1,3〕−ジオキソラン−2−イル、ベンズイソチアゾリルS,S−ジオキシドから選択することができる。シリルエーテルの場合では、OHの保護基は、トリメチルシリル、トリエチルシリル、トリイソプロピルシリル、ジメチルイソプロピルシリル、ジエチルイソプロピルシリル、ジメチルヘキシルシリル、2−ノルボルニルジメチルシリル、t−ブチルジメチルシリル、t−ブチルジフェニルシリル、トリベンジルシリル、トリ−p−キシリルシリル、トリフェニルシリル、ジフェニルメチルシリル、ジ−t−ブチルメチルシリル、ビス(t−ブチル)−1−ピレニルメトキシシリル、トリス(トリメチルシリル)シリル、(2−ヒドロキシスチリル)ジメチルシリル、(2−ヒドロキシスチリル)ジイソプロピルシリル、t−ブチルメトキシフェニルシリル、t−ブトキシジフェニルシリル、1,1,3,3−テトライソプロピル−3−〔2−(トリフェニルメトキシ)エトキシ〕ジシロキサン−1−イル及びフルオラスシリルから選択することができる。エステルの場合では、OHの保護基は、ホルメート、ベンゾイルホルメート、アセテート、クロロアセテート、ジクロロアセテート、トリクロロアセテート、トリクロロアセトアミデート、トリフルオロアセテート、メトキシアセテート、トリフェニルメトキシアセテート、フェノキシアセテート、p−クロロフェノキシアセテート、フェニルアセテート、ジフェニルアセテート、3−フェニルプロピオネート、ビスフルオラス鎖型プロパノイル、4−ペンテノエート、4−オキソペンタノエート、4,4−(エチレンジチオ)ペンタノエート、5〔3−ビス(4−メトキシフェニル)ヒドロキシメチルフェノキシ〕レブリネート、ピバロエート、1−アダマントエート、クロトネート、4−メトキシクロトネート、ベンゾエート、p−フェニルベンゾエート、2,4,6−トリメチルベンゾエート、4−ブロモベンゾエート、2,5−ジフルオロベンゾエート、p−ニトロベンゾエート、ピコリネート、ニコチネート、2−(アジドメチル)ベンゾエート、4−アジドブチレート、(2−アジドメチル)フェニルアセテート、2−{〔(トリチルチオ)オキシ〕メチル}ベンゾエート、2−{〔(4−メトキシトリチルチオ)オキシ〕メチル}ベンゾエート、2−{〔メチル(トリチルチオ)アミノ〕メチル}ベンゾエート、2−{{〔(4−メトキシトリチル)チオ〕メチルアミノ}−メチル}ベンゾエート、2−(アリルオキシ)フェニルアセテート、2−(プレニルオキシメチル)ベンゾエート、6−(レブリニルオキシメチル)−3−メトキシ−2−ニトロベンゾエート、6−(レブリニルオキシメチル)−3−メトキシ−4−ニトロベンゾエート、4−ベンジルオキシブチレート、4−トリアルキルシリルオキシブチレート、4−アセトキシ−2,2−ジメチルブチレート、2,2−ジメチル−4−ペンテノエート、2−ヨードベンゾエート、4−ニトロ−4−メチルペンタノエート、o−(ジブロモメチル)ベンゾエート、2−ホルミルベンゼンスルホネート、4−(メチルチオメトキシ)ブチレート、2−(メチルチオメトキシメチル)ベンゾエート、2−(クロロアセトキシメチル)ベンゾエート、2−〔(2−クロロアセトキシ)エチル〕ベンゾエート、2−〔2−(ベンジルオキシ)エチル〕ベンゾエート、2−〔2−(4−メトキシベンジルオキシ)エチル〕ベンゾエート、2,6−ジクロロ−4−メチルフェノキシアセテート、2,6−ジクロロ−4−(1,1,3,3−テトラメチルブチル)フェノキシアセテート、2,4−ビス(1,1−ジメチルプロピル)フェノキシアセテート、クロロジフェニルアセテート、イソブチレート、モノスクシノエート、(E)−2−メチル−2−ブテノエート、o−(メトキシカルボニル)ベンゾエート、α−ナフトエート、ニトレート、アルキルN,N,N′,N′−テトラメチルホスホロジアミデート及び2−クロロベンゾエートから選択することができる。スルホネート、スルフェネート及びスルフィネートの場合では、OHの保護基は、スルフェート、アリルスルホネート、メタンスルホネート、ベンジルスルホネート、トシレート、2−〔(4−ニトロフェニル)エチル〕スルホネート、2−トリフルオロメチルベンゼンスルホネート、4−モノメトキシトリチルスルフェネート、アルキル2,4−ジニトロフェニルスルフェネート、2,2,5,5−テトラメチルピロリジン−3−オン(one)−1−スルフィネート、ボレート及びジメチルホスフィノチオリルから選択することができる。カーボネートの場合では、OHの保護基は、メチルカーボネート、メトキシメチルカーボネート、9−フルオレニルメチルカーボネート、エチルカーボネート、ブロモエチルカーボネート、2−(メチルチオメトキシ)エチルカーボネート、2,2,2−トリクロロエチルカーボネート、1,1−ジメチル−2,2,2−トリクロロエチルカーボネート、2−(トリメチルシリル)エチルカーボネート、2−〔ジメチル(2−ナフチルメチル)シリル〕エチルカーボネート、2−(フェニルスルホニル)エチルカーボネート、2−(トリフェニルホスホニオ)エチルカーボネート、シス−〔4−〔〔(メトキシトリチル)スルフェニル〕オキシ〕テトラヒドロフラン−3−イル〕オキシカーボネート、イソブチルカーボネート、t−ブチルカーボネート、ビニルカーボネート、アリルカーボネート、シンナミルカーボネート、プロパルギルカーボネート、p−クロロフェニルカーボネート、p−ニトロフェニルカーボネート、4−エトキシ−1−ナフチルカーボネート、6−ブロモ−7−ヒドロキシクマリン−4−イルメチルカーボネート、ベンジルカーボネート、o−ニトロベンジルカーボネート、p−ニトロベンジルカーボネート、p−メトキシベンジルカーボネート、3,4−ジメトキシベンジルカーボネート、アントラキノン−2−イルメチルカーボネート、2−ダンシルエチルカーボネート、2−(4−ニトロフェニル)エチルカーボネート、2−(2,4−ジニトロフェニル)エチルカーボネート、2−(2−ニトロフェニル)プロピルカーボネート、アルキル2−(3,4−メチレンジオキシ−6−ニトロフェニル)プロピルカーボネート、2−シアノ−1−フェニルエチルカーボネート、2−(2−ピリジル)アミノ−1−フェニルエチルカーボネート、2−〔N−メチル−N−(2−ピリジル)〕アミノ−1−フェニルエチルカーボネート、フェナシルカーボネート、3′,5′−ジメトキシベンゾインカーボネート、メチルジチオカーボネート及びS−ベンジルチオカーボネートから選択することができる。そして、カルバメートの場合では、OHの保護基は、ジメチルチオカルバメート、N−フェニルカルバメート、N−メチル−N−(o−ニトロフェニル)カルバメートから選択することができる。これらの基の記述は、これらがOHの保護基の単なる例示として記述されているに過ぎないので、本発明の範囲の制限として解釈されるべきではなく、前記機能を有する更なる基が当業者によって知られている場合があり、それらは本発明に包含されると理解されるべきである。
【0025】
用語「薬学的に許容される塩、誘導体、プロドラッグ」は、患者に投与されると、本明細書に記載されている化合物を(直接的に又は間接的に)提供することができる任意の薬学的に許容される塩、エステル、溶媒和物、水和物又は任意のその他の化合物を意味する。しかし、薬学的に許容される塩ではないものも、薬学的に許容される塩の調製に有用でありうるので、本発明の範囲内であることが理解される。塩、プロドラッグ及び誘導体の調製は、当業界において既知の方法により実施することができる。
【0026】
例えば、本明細書において提供される化合物の薬学的に許容される塩は、塩基性又は酸性部分を含有する親化合物から、従来の化学方法によって合成される。一般に、上記塩は、例えば、これらの化合物の遊離酸又は塩基形態を、水中又は有機溶媒中又は2つの混合物中の適切な塩基又は酸の理論量と反応させることにより調製される。一般に、エーテル、酢酸エチル、エタノール、イソプロパノール又はアセトニトリルのような非水性媒質が好ましい。酸付加塩の例には、例えば塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、硫酸塩、硝酸塩、リン酸塩のような鉱酸付加塩、並びに、例えば酢酸塩、トリフルオロ酢酸塩、マレイン酸塩、フマル酸塩、クエン酸塩、シュウ酸塩、コハク酸塩、酒石酸塩、リンゴ酸塩、マンデル酸塩、メタンスルホン酸塩及びp−トルエンスルホン酸塩のような有機酸付加塩が挙げられる。アルカリ付加塩の例には、例えばナトリウム、カリウム、カルシウム及びアンモニウム塩のような無機塩、並びに、例えばエチレンジアミン、エタノールアミン、N,N−ジアルキレンエタノールアミン、トリエタノールアミン及び塩基性アミノ酸塩のような有機アルカリ塩が挙げられる。
【0027】
本発明の化合物は遊離化合物又は溶媒和物(例えば、水和物)のいずれかの結晶形態であることができ、両方の形態は本発明の範囲内であることが意図される。溶媒化の方法は、当業界において一般に知られている。
【0028】
式Iの化合物のプロドラッグである任意の化合物が、本発明の範囲及び意図の内である。用語「プロドラッグ」は、最も広範囲な意味において使用され、インビボ(in vivo)で本発明の化合物に変換される誘導体を包含する。上記誘導体は、当業者に容易に想起され、例えば、遊離ヒドロキシ基がエステル誘導体に変換される化合物が含まれる。
【0029】
本明細書において言及される任意の化合物は、上記特定の化合物、並びに特定の変形又は形態を表すことが意図される。特に、本明細書において言及される化合物は、不斉中心を有することができ、したがって、異なる鏡像異性形態で存在することができる。本明細書において言及される化合物の全ての光学異性体及び立体異性体、並びにこられの混合物は、本発明の範囲内であると考慮される。したがって、本明細書において言及される任意の所定の化合物は、ラセミ体、1つ以上の鏡像異性形態、1つ以上のジアステレオマー形態、1つ以上のアトロプ異性形態及びこれらの混合物のうちのいずれか1つを表すことが意図される。特に、上記の式Iにより表される本発明の化合物は、それらの非対称性に応じて鏡像異性体を又はジアステレオマーを含むことができる。二重結合に関する立体異性体も可能であり、したがって、幾つかの場合では、分子は(E)−異性体又は(Z)−異性体として存在することができる。分子が幾つかの二重結合を含有する場合、それぞれの二重結合は、分子の他の二重結合の立体異性体と同一又は異なることができる、それ自体の立体異性体を有し得る。単一の異性体又は異性体の混合物は本発明の範囲内である。
【0030】
更に、本明細書において言及される化合物は、幾何異性体(すなわち、シス及びトランス異性体)、互変異性体又はアトロプ異性体として存在することができる。特に、互変異性体という用語は、平衡で存在し、一つの異性体形態から別のものに容易に変換される、化合物の2つ以上の構造異性体のうちの1つを意味する。一般的な互変異性体対は、アミン−イミン、アミド−イミド、ケト−エノール、ラクタム−ラクチムなどである。加えて、本明細書において言及されるあらゆる化合物は、媒質に上記形態が存在する場合、水和物、溶媒和物及び多形体、並びにこれらの混合物を表すことが意図される。加えて、本明細書において言及される化合物は、同位体標識形態で存在することができる。本明細書において言及される化合物の全ての幾何異性体、互変異性体、アトロプ異性体、水和物、溶媒和物、多形体及び同位体標識形態、並びにこられの混合物は、本発明の範囲内であると考慮される。
【0031】
より簡潔な記載を提供するために、本明細書に提示されている幾つかの定量表現は、用語「約」により制限されない。用語「約」が明示的に使用されているか否かに関わらず、本明細書に提示されているあらゆる量は、実際の所定の値を意味することが意図され、上記所定の値の実験及び/又は測定条件に起因する等価値及び近似値を含む、上記所定値に対して当業界における通常の技術に基づいて合理的に推測される近似値を意味することも意図されることが理解される。
【0032】
一般式Iの化合物において、特に好ましいR1は、水素、ORa及びOCORaであり、ここでRaは、水素及び置換又は非置換C1〜C12アルキルから選択される。特に好ましいRaは、水素及び置換又は非置換C1〜C6アルキルであり、さらにより好ましいものは、水素、メチル、エチル、プロピル、イソプロピル及びtert−ブチルを含むブチルである。水素、OH及びメトキシが、最も好ましいR1基である。
【0033】
特に好ましいR2及びR3は、水素及び置換又は非置換C1〜C12アルキルである。より好ましいR2及びR3は、水素及び置換又は非置換C1〜C6アルキルであり、さらにより好ましいものは、水素である。
【0034】
特に好ましいR41、R42、R43、R44、R45、R46、R47及びR48は、水素及び置換又は非置換C1〜C12アルキルである。より好ましいR41、R42、R43、R44、R45、R46、R47及びR48は、水素及び置換又は非置換C1〜C6アルキルであり、より好ましいものは、水素、置換又は非置換メチル、置換又は非置換エチル、置換又は非置換プロピル、置換又は非置換イソプロピル、並びに置換又は非置換tert−ブチル、置換又は非置換イソブチル及び置換又は非置換sec−ブチルを含む置換又は非置換ブチルである。前記基の好ましい置換基は、OR′、=O、SR′、SOR′、SO2R′、NO2、NHR′、N(R′)2、=N−R′、NHCOR′、N(COR′)2、NHSO2R′、NR′C(=NR′)NR′R′、CN、ハロゲン、COR′、COOR′、OCOR′、OCONHR′、OCON(R′)2、保護OH、置換又は非置換アリール及び置換又は非置換複素環式基であり、ここでR′基は、それぞれ独立して、水素、OH、NO2、NH2、SH、CN、ハロゲン、COH、COアルキル、COOH、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル、置換又は非置換アリール及び置換又は非置換複素環式基からなる群より選択される。上記基がそれ自体置換される場合、置換基は、前記のリストから選択することができる。上記基のさらにより好ましい置換基は、OH、SCH3、SH、NH2、NHC(=NH)NH2、CONH2、COOH、フェニル、p−、m−又はo−ヒドロキシフェニル、1−、2−及び3−インドリルを含むインドリル、並びに4−及び5−イミダゾリルを含むイミダゾリルである。水素、メチル、イソプロピル、tert−ブチル及びベンジルが、最も好ましいR41、R42、R43、R44、R45、R46、R47及びR48基である。とりわけ、特に好ましいR42、R44、R45、R46及びR47は、水素である。特に好ましいR41及びR43は、メチルである。そして特に好ましいR48は、イソプロピル、tert−ブチル又はベンジルである。
【0035】
特に好ましいR5及びR6は、水素及び置換又は非置換C1〜C12アルキルである。より好ましいR5及びR6は、水素及び置換又は非置換C1〜C6アルキルであり、さらにより好ましいものは、水素、メチル、エチル、プロピル、イソプロピル及びtert−ブチルを含むブチルである。水素が特に好ましい。
【0036】
本発明の別の実施態様において、R5及びR48は、それらが結合している対応するN原子及びC原子と一緒になって、置換又は非置換複素環式基を形成することも好ましい。好ましい複素環式基は、1−、2−及び3−ピロリジニルを含むピロリジニルである。
【0037】
特に好ましいR7は、水素、置換又は非置換C1〜C12アルキル及び置換又は非置換C2〜C12アルケニルであり、より好ましいものは、水素、置換C1〜C12アルキル及び置換C2〜C12アルケニルである。好ましい置換アルキル及び置換アルケニルは、1つのみならず、2つ以上の置換基を表すことができる。より好ましいアルキル基は、6〜約10個の炭素原子;さらにより好ましくは、7、8又は9個の炭素原子を有するものである。ヘプチル、オクチル及びノニルが、最も好ましいアルキル基である。一方、より好ましいアルケニル基は、6〜約10個の炭素原子;さらにより好ましくは、7、8又は9個の炭素原子を有するものである。オクタ−1,6−ジエニル、オクタ−1,5−ジエニル、オクタ−1,4−ジエニル、オクタ−1,3−ジエニル、ノナ−1,7−ジエニル、ノナ−1,6−ジエニル、ノナ−1,5−ジエニル、ノナ−1,4−ジエニル、ノナ−1,3−ジエニル、ヘプタ−1,5−ジエニル、ヘプタ−1,4−ジエニル、ヘプタ−1,3−ジエニルが、最も好ましいアルケニル基である。前記アルキル及びアルケニル基の好ましい置換基は、OR′、=O、SR′、SOR′、SO2R′、NO2、NHR′、N(R′)2、=N−R′、NHCOR′、N(COR′)2、NHSO2R′、NR′C(=NR′)NR′R′、CN、ハロゲン、COR′、COOR′、OCOR′、OCONHR′、OCON(R′)2、保護OH、置換又は非置換アリール及び置換又は非置換複素環式基であり、ここでR′基は、それぞれ独立して、水素、OH、NO2、NH2、SH、CN、ハロゲン、COH、COアルキル、COOH、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル、置換又は非置換アリール及び置換又は非置換複素環式基からなる群より選択される。上記基がそれ自体置換される場合、置換基は、前記のリストから選択することができる。上記のアルキル及びアルケニル基のより好ましい置換基は、ハロゲン、OR′、=O、OCOR′、OCONHR′、OCON(R′)2及び保護OHであり、ここでR′基は、それぞれ、好ましくは水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル及び置換又は非置換アリールから選択される。これらのアルキル及びアルケニル基のさらにより好ましい置換基は、ハロゲン、OR′、=O、OCONHR′、OCON(R′)2及び保護OHであり、ここでOHの保護基は、好ましくはトリメチルシリル、トリエチルシリル、トリイソプロピルシリル、ジメチルイソプロピルシリル、ジエチルイソプロピルシリル、ジメチルヘキシルシリル、2−ノルボルニルジメチルシリル、t−ブチルジメチルシリル、t−ブチルジフェニルシリル、トリベンジルシリル、トリ−p−キシリルシリル、トリフェニルシリル、ジフェニルメチルシリル、ジ−t−ブチルメチルシリル、ビス(t−ブチル)−1−ピレニルメトキシシリル、トリス(トリメチルシリル)シリル、(2−ヒドロキシスチリル)ジメチルシリル、(2−ヒドロキシスチリル)ジイソプロピルシリル、t−ブチルメトキシフェニルシリル、t−ブトキシジフェニルシリル、1,1,3,3−テトライソプロピル−3−〔2−(トリフェニルメトキシ)エトキシ〕ジシロキサン−1−イル及びフルオラスシリルから選択され、R′基は、それぞれ、より好ましくは水素、非置換C1〜C6アルキル及び置換又は非置換アリールから選択される。Cl、OH、=O、OCONH2、OCONHフェニル、並びにOHの保護基が、好ましくはトリメチルシリル、トリエチルシリル、トリイソプロピルシリル、ジメチルイソプロピルシリル、ジエチルイソプロピルシリル、ジメチルヘキシルシリル、2−ノルボルニルジメチルシリル、t−ブチルジメチルシリル、t−ブチルジフェニルシリル、トリベンジルシリル、トリ−p−キシリルシリル、トリフェニルシリル、ジフェニルメチルシリル、ジ−t−ブチルメチルシリル、ビス(t−ブチル)−1−ピレニルメトキシシリル、トリス(トリメチルシリル)シリル、(2−ヒドロキシスチリル)ジメチルシリル、(2−ヒドロキシスチリル)ジイソプロピルシリル、t−ブチルメトキシフェニルシリル、t−ブトキシジフェニルシリル、1,1,3,3−テトライソプロピル−3−〔2−(トリフェニルメトキシ)エトキシ〕ジシロキサン−1−イル及びフルオラスシリルから選択される、保護OHは、これらのアルキル及びアルケニル基の最も好ましい置換基である。
【0038】
特に好ましいものは、点線で示された場所における1つ以上の追加的な結合の存在である。より好ましいものは、点線で示された全ての場所における追加的な結合の存在である。加えて、それぞれの二重結合の立体化学が(E)又は(Z)として存在することができる。単一の異性体又は異性体の混合物が本発明の範囲内である。
【0039】
より好ましくは、本発明は、一般式IIの化合物、又は薬学的に許容されるそれらの塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体を提供し、
【0040】
【化4】
【0041】
式中、
R1は、水素、ORa、OCORa、OCOORa、NRaRb、NRaCORb及びNRaC(NRa)NRaRbから選択され;
R41、R43及びR48は、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル及び置換又は非置換C2〜C12アルキニルから選択され;
R5、R6及びR7は、それぞれ独立して、水素、CORa、COORa、置換若しくは非置換C1〜C12アルキル、置換若しくは非置換C2〜C12アルケニル及び置換若しくは非置換C2〜C12アルキニルから選択されるか、又はR5及びR48は、それらが結合している対応するN原子及びC原子と一緒になって、置換若しくは非置換複素環式基を形成してもよく;
Ra及びRbは、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル、置換又は非置換アリール及び置換又は非置換複素環式基から選択され;そして
点線は、それぞれ任意の追加的な結合を表す。
【0042】
一般式IIの化合物において、特に好ましいR1は、水素、ORa及びOCORaであり、ここでRaは、水素及び置換又は非置換C1〜C12アルキルから選択される。特に好ましいRaは、水素及び置換又は非置換C1〜C6アルキルであり;さらにより好ましいものは、水素、メチル、エチル、プロピル、イソプロピル及びtert−ブチルを含むブチルである。水素、OH及びメトキシが、最も好ましいR1基である。
【0043】
特に好ましいR41、R43及びR48は、水素及び置換又は非置換C1〜C12アルキルである。より好ましいR41、R43及びR48は、水素及び置換又は非置換C1〜C6アルキルであり、さらにより好ましいものは、水素、置換又は非置換メチル、置換又は非置換エチル、置換又は非置換プロピル、置換又は非置換イソプロピル、並びに置換又は非置換tert−ブチル、置換又は非置換イソブチル及び置換又は非置換sec−ブチルを含む置換又は非置換ブチルである。前記基の好ましい置換基は、OR′、=O、SR′、SOR′、SO2R′、NO2、NHR′、N(R′)2、=N−R′、NHCOR′、N(COR′)2、NHSO2R′、NR′C(=NR′)NR′R′、CN、ハロゲン、COR′、COOR′、OCOR′、OCONHR′、OCON(R′)2、保護OH、置換又は非置換アリール及び置換又は非置換複素環式基であり、ここでR′基は、それぞれ独立して、水素、OH、NO2、NH2、SH、CN、ハロゲン、COH、COアルキル、COOH、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル、置換又は非置換アリール及び置換又は非置換複素環式基からなる群より選択される。上記基がそれ自体置換される場合、置換基は、前記のリストから選択することができる。上記基のさらにより好ましい置換基は、OH、SCH3、SH、NH2、NHC(=NH)NH2、CONH2、COOH、フェニル、p−、m−又はo−ヒドロキシフェニル、1−、2−及び3−インドリルを含むインドリル、並びに4−及び5−イミダゾリルを含むイミダゾリルである。水素、メチル、イソプロピル、tert−ブチル及びベンジルが、最も好ましいR41、R43及びR48基である。とりわけ、特に好ましいR41及びR43は、メチルであり、特に好ましいR48は、イソプロピル、tert−ブチル又はベンジルである。
【0044】
特に好ましいR5及びR6は、水素及び置換又は非置換C1〜C12アルキルである。より好ましいR5及びR6は、水素及び置換又は非置換C1〜C6アルキルであり;さらにより好ましいものは、水素、メチル、エチル、プロピル、イソプロピル及びtert−ブチルを含むブチルである。水素が最も好ましい。
【0045】
本発明の別の実施態様において、R5及びR48は、それらが結合している対応するN原子及びC原子と一緒になって、置換又は非置換複素環式基を形成することも好ましい。好ましい複素環式基は、1−、2−及び3−ピロリジニルを含むピロリジニルである。
【0046】
特に好ましいR7は、水素、置換又は非置換C1〜C12アルキル及び置換又は非置換C2〜C12アルケニルであり、より好ましいものは、水素、置換C1〜C12アルキル及び置換C2〜C12アルケニルである。より好ましいアルキル基は、6〜約10個の炭素原子;さらにより好ましくは、7、8又は9個の炭素原子を有するものである。ヘプチル、オクチル及びノニルが、最も好ましいアルキル基である。一方、より好ましいアルケニル基は、6〜約10個の炭素原子;さらにより好ましくは、7、8又は9個の炭素原子を有するものである。オクタ−1,6−ジエニル、オクタ−1,5−ジエニル、オクタ−1,4−ジエニル、オクタ−1,3−ジエニル、ノナ−1,7−ジエニル、ノナ−1,6−ジエニル、ノナ−1,5−ジエニル、ノナ−1,4−ジエニル、ノナ−1,3−ジエニル、ヘプタ−1,5−ジエニル、ヘプタ−1,4−ジエニル、ヘプタ−1,3−ジエニルが、最も好ましいアルケニル基である。前記アルキル及びアルケニル基の好ましい置換基は、OR′、=O、SR′、SOR′、SO2R′、NO2、NHR′、N(R′)2、=N−R′、NHCOR′、N(COR′)2、NHSO2R′、NR′C(=NR′)NR′R′、CN、ハロゲン、COR′、COOR′、OCOR′、OCONHR′、OCON(R′)2、保護OH、置換又は非置換アリール及び置換又は非置換複素環式基であり、ここでR′基は、それぞれ独立して、水素、OH、NO2、NH2、SH、CN、ハロゲン、COH、COアルキル、COOH、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル、置換又は非置換アリール及び置換又は非置換複素環式基からなる群より選択される。上記基がそれ自体置換される場合、置換基は、前記のリストから選択することができる。上記のアルキル及びアルケニル基のより好ましい置換基は、ハロゲン、OR′、=O、OCOR′、OCONHR′、OCON(R′)2及び保護OHであり、ここでR′基は、それぞれ、好ましくは水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル及び置換又は非置換アリールから選択される。これらのアルキル及びアルケニル基のさらにより好ましい置換基は、ハロゲン、OR′、=O、OCONHR′、OCON(R′)2及び保護OHであり、ここでOHの保護基は、好ましくはトリメチルシリル、トリエチルシリル、トリイソプロピルシリル、ジメチルイソプロピルシリル、ジエチルイソプロピルシリル、ジメチルヘキシルシリル、2−ノルボルニルジメチルシリル、t−ブチルジメチルシリル、t−ブチルジフェニルシリル、トリベンジルシリル、トリ−p−キシリルシリル、トリフェニルシリル、ジフェニルメチルシリル、ジ−t−ブチルメチルシリル、ビス(t−ブチル)−1−ピレニルメトキシシリル、トリス(トリメチルシリル)シリル、(2−ヒドロキシスチリル)ジメチルシリル、(2−ヒドロキシスチリル)ジイソプロピルシリル、t−ブチルメトキシフェニルシリル、t−ブトキシジフェニルシリル、1,1,3,3−テトライソプロピル−3−〔2−(トリフェニルメトキシ)エトキシ〕ジシロキサン−1−イル及びフルオラスシリルから選択され、R′基は、それぞれ、より好ましくは水素、非置換C1〜C6アルキル及び置換又は非置換アリールから選択される。Cl、OH、=O、OCONH2、OCONHフェニル、並びにOHの保護基が、好ましくはトリメチルシリル、トリエチルシリル、トリイソプロピルシリル、ジメチルイソプロピルシリル、ジエチルイソプロピルシリル、ジメチルヘキシルシリル、2−ノルボルニルジメチルシリル、t−ブチルジメチルシリル、t−ブチルジフェニルシリル、トリベンジルシリル、トリ−p−キシリルシリル、トリフェニルシリル、ジフェニルメチルシリル、ジ−t−ブチルメチルシリル、ビス(t−ブチル)−1−ピレニルメトキシシリル、トリス(トリメチルシリル)シリル、(2−ヒドロキシスチリル)ジメチルシリル、(2−ヒドロキシスチリル)ジイソプロピルシリル、t−ブチルメトキシフェニルシリル、t−ブトキシジフェニルシリル、 1,1,3,3−テトライソプロピル−3−〔2−(トリフェニルメトキシ)エトキシ〕ジシロキサン−1−イル及びフルオラスシリルから選択される、保護OHは、これらのアルキル及びアルケニル基の最も好ましい置換基である。
【0047】
特に好ましいものは、点線で示された場所における1つ以上の追加的な結合の存在である。より好ましいものは、点線で示された全ての場所における追加的な結合の存在である。なお、それぞれの二重結合の立体化学が(E)又は(Z)として存在することができる。単一の異性体又は異性体の混合物が本発明の範囲内である。
【0048】
本発明の特に好ましい化合物は、以下である:
【0049】
【化5】
【0050】
化合物1〜8は、ラスパイリイダ(Raspailiidae)科リトプロカミア(Lithoplocamia)属リチストイデス(lithistoides)種の海綿動物から単離された。
【0051】
リトプロカミア・リチストイデス(Lithoplocamia lithistoides )の試料は、メキシコのマサトラン(Mazatlan)にあるメキシコ国立自治大学(Universidad Nacional Autonoma de Mexico)の「海洋及び陸水学の科学研究所」(“Instituto de Ciencias del Mar y Limnologia”)に、参照コードLEB−ICML−UNAM−11−2004で寄託された。この海綿体は、マダガスカル(南17度06.071分/東49度51.385分)において、6〜20mの範囲の深さでスキューバダイビングにより手動で収集され、その記載は以下である。
【0052】
ラスパイリイダ(Raspailiidae)科:ラスパイリイダ・ヘンチェル(Raspailiidae Hentschel)、1923は、皮殻で覆われた、巨大な葉状の扇形又は分岐した成長形態を有し、通常は極めて剛毛な表面を有する海綿体である。個々の長厚突起又はオキセア(oxeas)を取り囲んでいる小型の薄い突起(フーパー&ウィーデンマイヤー(Hooper & Wiedenmayer)1994:図17)又はオキセア(フーパー&ウィーデンマイヤー1994:図5)のブラシからなる特殊な体外骨格が、典型的に存在する。襟体骨格は、圧縮軸骨格から羽毛網状の又は網状のみの構造まで多様である。海綿質繊維は、通常、核心交接刺(襟体突起、オキセア又は両方)を完全に包み込む。特別な分類の棘条突起(フーパー&ウィーデンマイヤー1994:図22)又は突起の変態(例えば、図22〜25、28)、有棘繊維が、繊維から直角に突き出している。微小骨片は通常不在であるが、単一の束晶(フーパー&ウィーデンマイヤー1994:図109)又は束(トリコドラグメータ(trichodragmata);フーパー&ウィーデンマイヤー1994:図110)が、幾つかの属において生じうる。ラスパイリイダ(Raspailiid)は、浅海から深さが少なくとも2460mまでの範囲に広く分布している(ハートマン(Hartman)1982)。
【0053】
リトプロカミア(Lithoplocamia)属リチストイデス(lithistoides )種は、皮殻で覆われた、巨大な成長形態であり、襟体骨格は、棘状棍棒体の高密度の等網糸又は不規則な半網糸の網状組織であり、1又は2つの大きさの分類において、軸性圧縮がなく、有棘の棘状突起がなく、平滑突起の軸外放射状管を有し、典型的には特殊なラスパイリイダ(raspailiid)体外骨格を有さず(しかし、存在する場合は、体外交接刺は、細長いオキセアである)、微小骨片が存在しない。
【0054】
リトプロカミア・リチストイデス(Lithoplocamia lithistoides)の試料は、また、ケニヤ(南04度40分5.5秒/東39度26分4.3秒、及び南3度38分36.5秒/東39度53分53.8秒)並びにタンザニア(南08度55分31.7秒/東39度34分53.5秒、及び南05度24.200分/東39度47.730分)において、30〜40mの範囲の深さで収集された。
【0055】
さらに、本発明の化合物は、合成により得ることができる。例えば、化合物1は、スキーム1に示されているように、異なるフラグメントを結合することによって作製できる。
【0056】
【化6】
【0057】
ここで、R、RI、RII、RIII、RIV、RV、RVI、RVII及びRVIIIは、所望の基又は必要とされる適切な保護基である。
【0058】
この方法は、以下の主要な工程を含むことができる:
a)標準的な文献の手順(コザワ(Kozawa)Yら、テトラヒドロン・レター(Tetrahedron Lett.)2002年、43、111)に従った、フラグメントCによるヨードアルケニル誘導体(フラグメントD)のアミド化によって、対応するエンアミド(フラグメントCD)を得ること。
b)有機合成において既知の手順(スコット(Scott)WJら、米国化学会ジャーナル(J. Am. Chem. Soc.)1984年、106、4630;ラバディ(Labadie)JWら、有機化学ジャーナル(J. Org. Chem.)1983年、48、4634〜4642;ファリーナ(Farina)Vら、有機反応(Organic Reactions)1998年、ウィリー(Wiley))に従った、フラグメントAとフラグメントBのスチル(Stille)型カップリング反応によって、直鎖ポリエン(フラグメントAB)を得ること。
c)フラグメントAB及びCDを、標準的な手順(ボダンスキー(Bodanszky)M及びボダンスキーA、ペプチド合成の実践(The Practice of Peptide Synthesis)、シュプリンガー・フェアラーグ、1993年)に従ってカップリングして、化合物1の炭素骨格を得ることができる。
d)アルコールORIIの脱保護、続くラクトン化は、有機合成において既知の手順(グリーン(Greene)及びウッツ(Wuts)、有機合成における保護基(Protective Groups in Organic Synthesis)、第3版、ウィリー・インターサイエンス(Wiley-Interscience);バーク(Burke)及びダンハイザー(Danheiser)、有機合成のための試薬ハンドブック:酸化及び還元剤(Handbook of Reagents for Organic Synthesis: Oxidizing and Reducing Agents)、ウィリー(Wiley);プラ(Pla)Dら、有機化学ジャーナル(J. Org. Chem.)2005年、70、8231)に従って達成することができる。
e)最後に、アルコールORIIIの脱保護、続くカルバメートの形成は、標準的な文献の手順(ラブ(Love)Bら、有機合成(Organic Syntheses)集、第5巻、162頁;第48巻、32頁;ミュラー(Muller)Eら、有機化学法(Methoden der Organischen Chemie)(フーベン・ワイル)(Houben-Weyl)、第4版、第8巻、G.シーム(Thieme)、シュットガルト、1952年、137頁)により達成して、化合物1を得ることができる。
【0059】
工程の順序を取り替えて最終化合物を得ることができる。例えば、フラグメントBCを第1工程で調製し、次にフラグメントA及びBと連続カップリングさせて、化合物1の炭素骨格を得ることができる。同様に、化合物1は、フラグメントA、B、C及びDを任意の順番で連続カップリングして調製することができる。別の選択肢は、任意の他のフラグメントとのカップリングの前に、フラグメントAにおけるラクトン部分の形成を実施することである。
【0060】
化合物1の類似体は、それぞれの場合において中間体化合物の適切な置換基を選択して、化合物1について記載したものと同等の方法により合成することができる。
【0061】
必要な場合、置換基に対して適切な保護基を使用して、反応性基が影響を受けないことを確実にすることができる。合成は、適切な工程で所望の置換基に変換することができる前駆体置換基を用いるように設計することができる。環構造における飽和又は不飽和を、合成の一部として導入又は除去することができる。出発材料及び試薬を、望ましいように変更して、意図される化合物の合成を確実にすることができる。加えて、類似体を、当業者に既知の合成有機化学における通常の手順によって、化合物1から合成することもできる。
【0062】
上記の合成経路を、望ましいように変更して、立体特異的化合物、並びに立体異性体の混合物を与えることができる。立体特異的試薬の使用又は合成の際にキラル中心を化合物に導入することを含む多様な方法により、特定の立体異性体又は特定の混合物を合成することが可能である。合成の際に1つ以上の立体中心を導入すること、また、存在する立体中心を反転することが可能である。加えて、化合物が当業者に既知の標準的な溶解技術により合成されると、立体異性体を分離することが可能である。
【0063】
式I及びIIの上記の化合物の重要な特徴は、それらの生体活性、特にそれらの細胞毒性及び抗有糸分裂活性である。
【0064】
本発明によって、細胞毒性及び抗有糸分裂活性を有する一般式I及びIIの化合物の新規医薬組成物、並びに抗腫瘍剤としてのそれらの使用が提供される。したがって、本発明は、更に、本発明の化合物、薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ又は立体異性体と薬学的に許容される担体とを含む、医薬組成物を提供する。
【0065】
医薬組成物の例には、経口、局所又は非経口投与用の任意の固体(錠剤、丸剤、カプセル剤、顆粒剤など)又は液体(液剤、懸濁剤又は乳剤)組成物が挙げられる。
【0066】
本発明の化合物又は組成物の投与は、静脈内注入、経口調合剤、並びに腹腔内及び静脈内投与のような任意の適切な方法によることができる。24時間までの注入時間を使用することが好ましく、より好ましくは1〜12時間であり、1〜6時間が最も好ましい。治療を病院に一晩入院することなく実施することを可能にする短い注入時間が、特に望ましい。しかし、注入は、必要であれば12〜24時間又はそれ以上であることができる。注入は、例えば1〜4週間の適切な間隔で実施することができる。本発明の化合物を含有する医薬組成物は、リポソーム又はナノ球体封入により、持続放出製剤において、又は他の標準的な送達手段により送達することができる。
【0067】
化合物の正確な投与量は、特定の製剤、適用様式、並びに特定の位置、宿主及び治療される腫瘍に応じて変わり得る。年齢、体重、性別、食事、投与時間、排出速度、宿主の状態、薬剤の組み合わせ、反応感受性及び疾患の重篤度のような他の要因が考慮され得る。投与は、最大耐量の範囲内で継続的に又は周期的に実施することができる。
【0068】
本発明の化合物及び組成物を他の薬剤と共に使用して、併用療法を提供することができる。他の薬剤は、同じ組成物の一部分を形成することができるか、又は同時若しくは異なる時に投与される別個の組成物として提供されうる。
【0069】
これらの化合物の抗腫瘍活性には、肺癌、結腸癌、乳癌及び子宮頸癌が含まれるが、これらに限定されない。
【0070】
実施例
実施例1:海洋生物及び収集の側面の記載
リトプロカミア・リチストイデス(Lithoplocamia lithistoides)は、マダガスカル(南17度06.071分/東49度51.385分)において、6〜20mの範囲の深さでのスキューバダイビングを用いて手動で収集された。動物材料は、ホセ・ルイス・カルバジョ(Jose Luis Carballo)(メキシコ自治大学(Universidad Autonoma de Mejico))により同定された。検体試料は、メキシコのマサトラン(Mazatlan)にあるメキシコ国立自治大学(Universidad Nacional Autonoma de Mexico)の「海洋及び陸水学の科学研究所」(“Instituto de Ciencias del Mar y Limnologia”)に、参照コードLEB−ICML−UNAM−11−2004で寄託された。
【0071】
実施例2:化合物1の単離
実施例1の冷凍検体(61g)を角切りし、H2O(3×200mL)、続いてMeOH:ジクロロメタンの混合物(1:1、3×200mL)により室温で抽出した。合わせた有機抽出物を濃縮して、粗物質1.11gを得た。この物質を、H2OからMeOHへの段階勾配によるLichroprep RP-18のVLCに付した。
【0072】
化合物1(1.6mg)を、半分取逆相HPLC(SymmetryPrep C18 7μm、7.8×150mm、MeCNが35〜100%の勾配H2O:MeCNを30分、UV検出、流速2.5mL/分、保持時間14.4分)により、MeOHを用いて溶離してフラクションから単離した。
化合物1:無定形の白色固体。(+)HRESIMS m/z606.2940[M+H]+(C31H4535ClN3O7で計算 606.2946);1H(500MHz)及び13C NMR(125MHz)表1を参照すること。
【0073】
【表1】
【化7】
【0074】
実施例3:化合物2、3、4、5、6及び7の単離
リトプロカミア・リチストイデス(Lithoplocamia lithistoides)の試料の第2群(7.66kg)を粉砕し、MeOH:ジクロロメタンの混合物(1:1、14L、2×5L、4L)で、徹底的に抽出した。溶媒を真空下で除去し、残留水溶液をEtOAc(12L、3×8L)で抽出した。有機層を蒸発させて、粗物質21.71gを得た。
【0075】
この物質を、H2O:MeOH(4:6)からMeOHの段階勾配を用いるRP-18カラムクロマトグラフィーに付した。H2O:MeOH(2:8、430mg)で溶離したフラクションをプールし、分取HPLC(Atlantis dC18、OBD、5μm、19×150mm、定組成H2O:MeOH(39:61)、流速:20mL/分、UV検出)に付して、純粋な化合物1(160.8mg)、2(13.2mg)及び7(1.89mg)、並びに3と4の混合物(11.4mg)及び5と6の混合物(10.0mg)を得た。純粋な化合物3(5.1mg)及び4(2.6mg)は、半分取HPLC(X-Terra Prep RP-18、10μm、10×150mm、MeOHが50〜70%の勾配H2O:MeOHを70分、流速:2.5mL/分、UV検出)による混合物の最終精製の後で得た。化合物5(3.6mg)及び6(1.0mg)は、半分取HPLC(X-Terra Prep RP-18、10μm、10×150mm、定組成H2O:MeOH(45:55)、流速:2.5mL/分、UV検出)により、同様の方法で分離した。
化合物2:無定形の白色固体。MS(ES)m/z606.3[M+H]+,628.3[M+Na]+;1H(500MHz)及び13C NMR(125MHz)表2を参照すること。
化合物3:無定形の白色固体。(+)HRESIMS m/z628.2774[M+Na]+(C31H4435ClN3O7Naで計算 628.2760);1H(500MHz)及び13C NMR(125MHz)表3を参照すること。
化合物4:無定形の白色固体。(+)HRESIMS m/z594.3152[M+Na]+(C31H45N3O7Naで計算 594.3150);1H(500MHz)及び13C NMR(125MHz)表4を参照すること。
化合物5:無定形の白色固体。MS(ES)m/z592.3[M+H]+,614.3[M+Na]+;1H(500MHz)及び13C NMR(125MHz)表5を参照すること。
化合物6:無定形の白色固体。MS(ES)m/z592.3[M+H]+,614.3[M+Na]+;1H(500MHz)及び13C NMR(125MHz)表6を参照すること。
化合物7:無定形の白色固体。(+)HRESIMS m/z427.2207[M+Na]+(C22H32N2O5Naで計算 427.2203);1H(500MHz)及び13C NMR(125MHz)表7を参照すること。
【0076】
【表2】
【化8】
【表3】
【化9】
【表4】
【化10】
【表5】
【化11】
【表6】
【化12】
【表7】
【化13】
【0077】
実施例4:化合物8の単離
実施例3に開示された抽出手順からもたらされた化合物1(61.6mg)含有フラクションを、半分取HPLC(Symmetryprep C-18、7μm、7.8×150mm、定組成H2O:CH3CN(55:45)、流速:2.3mL/分、UV検出)により更に精製して、0.9mgの化合物8を純粋な形態で得た。
化合物8:無定形の白色固体。MS(ES)m/z606.2[M+H]+,628.3[M+Na]+;1H(500MHz)及び13C NMR(125MHz)表8を参照すること。
【0078】
【表8】
【化14】
【0079】
実施例5:フラグメントAの合成
スキーム2は、スキーム1において提供された命名法に従って、フラグメントAの合成の幾つかの例を提供する。
【0080】
【化15】
【0081】
中間体9の合成
【0082】
【化16】
【0083】
ジクロロメタン/DMSO(331mL/149mL)の混合物中の(2S,3S)−3,5−ビス{〔(tert−ブチル)ジメチルシリル〕オキシ}−4−メチルペンタン−1−オール(P.プーカン(Phukan)、S.サスマール(Sasmal)及びM.E.マイヤー(Maier)有機化学ヨーロピアンジャーナル(Eur. J. Org. Chem.)2003年、1733〜1740)(50g、0.14mol)の0℃の溶液に、Et3N(96.1mL、0.69mol)を、添加漏斗により加えた。10分後、SO3・Pyr(54.8g、0.34mol)を少量ずつ加え、溶液を0℃で更に2時間撹拌した。次に、それをジクロロメタン(800ml)で希釈し、HCl(0.5N、800mL)で停止させた。有機層をデカントし、MgSO4で乾燥し、真空下で濃縮した。カラムクロマトグラフィー(ヘキサン/EtOAc 100:0〜10:1)による精製によって、45g(収率:90%)のアルデヒド9を得た。
1H-RMN (CDCl3, 300 MHz) δ: 9.79 (s, 1H), 4.30 (m, 1H), 3.65 (m, 2H), 2.51 (m, 1H), 1.69 (m, 2H), 1.04 (d, 3H, J = 6.9Hz), 0.85-0.88 (m, 18H), 0.03-0.07 (m, 12H).
13C-RMN (CDCl3, 75 MHz) δ: 205.4, 69.4, 59.6, 51.7, 37.5, 26.1, 26.0, 18.4, 18.2, 8.0, -4.3, -4.5, -5.2.
【0084】
中間体10の合成
【0085】
【化17】
【0086】
トルエン(625mL)中のアルデヒド9(45g、0.2mol)の溶液に、カルボエトキシエチリデン−トリフェニルホスホラン(113g、0.31mol)を加え、混合物を60℃で17時間加熱した。次に、溶媒を減圧下で除去し、得られた油状物をカラムクロマトグラフィー(ヘキサン/EtOAc 100:0〜10:1)により精製して、53.3g(収率:96%)のエステル化合物10を得た。
1H-RMN (CDCl3, 300 MHz) δ: 6.71 (dd, 1H, J = 1.5, 10.2 Hz), 4.19 (m, 2H), 3.77 (m, 1H), 3.66 (m, 2H), 2.61 (m, 1H), 1.85 (d, 3H, J = 1.5 Hz), 1.68 (m, 2H), 1.30 (t, 3H, J = 7.2 Hz), 0.98 (d, 3H, 6.9 Hz), 0.90 (m, 18H), 0.05 (m, 12H).
13C-RMN (CDCl3, 75 MHz) δ: 168.3, 145.4, 126.7, 72.2, 60.4, 59.7, 38.4, 38.0, 25.9, 18.2, 18.1, 14.3, 14.3, 12.6, -4.4, -4.6, -5.4.
【0087】
中間体11の合成
【0088】
【化18】
【0089】
無水THF(525mL)中のエステル10(46.7g、0.105mol)の−78℃に冷却した溶液に、アルゴン雰囲気下、トルエン(231mL、0.231mol)中の1M水素化ジイソブチルアルミニウム(DIBAL)を10分間かけて加え、混合物を−78℃で撹拌した。4時間後、反応をMeOH(10mL)で停止させ、酒石酸ナトリウムカリウムの飽和溶液(800mL)を加え、EtOAc(1000mL)で希釈した。この混合物を2時間撹拌し、次に有機層をデカントした。水性残渣を追加のEtOAc(2×400mL)で抽出し、合わせた有機層を乾燥し(Na2SO4)、溶媒を蒸発させた。得られた油状物をカラムクロマトグラフィー(ヘキサン/EtOAc 20:1〜10:1)により精製して、32.5g(収率:77%)のアルコール11を得た。
1H-RMN (CDCl3, 300 MHz) δ: 5.31 (d, 1H, J = 9.6 Hz), 3.98 (m, 2H), 3.66 (m, 3H), 2.49 (m, 1H), 1.67 (s, 3H), 1.70-1.62 (m, 2H), 0.91 (d, 3H, J = 6.9 Hz), 0.88 (m, 18H), 0.03 (m, 12H).
13C-RMN (CDCl3, 75 MHz) δ: 133.9, 129.8, 73.1, 69.1, 59.9, 37.8, 37.5, 25.9, 18.3, 18.1, 15.9, 13.9, -4.4, -4.4, -5.3.
【0090】
中間体12の合成
【0091】
【化19】
【0092】
エチルエステル(387mL)中のアルコール11(31.2g、77.5mmol)の溶液に、アルゴン雰囲気下、MnO2(101g、1.16mol)を加え、混合物を室温で2時間撹拌した。この混合物を、EtOAc(3L)により溶離するシリカゲルカラムで濾過し、得られた溶液を減圧下で乾燥して、29.1g(収率:94%)のアルデヒド12を得た。
1H-RMN (CDCl3, 300 MHz) δ: 9.37 (s, 1H), 6.44 (d, 1H, J = 9.6 Hz), 3.82 (dd, 1H, J = 6.3, 10.8 Hz), 3.65 (m, 2H), 2.82 (m, 1H), 1.74 (s, 3H), 1.67 (m, 2H), 1.02 (d, 3H, J = 6.9 Hz), 0.86 (s, 18H), 0.04-0.01 (m, 12H).
13C-RMN (CDCl3, 75 MHz) δ: 195.4, 157.8, 138.3, 134.5, 72.0, 59.5, 36.7, 37.5, 25.8, 18.2, 18.1, 14.3, 9.4, -4.4, -4.5, -5.4.
【0093】
中間体13の合成
【0094】
【化20】
【0095】
THF(727mL)中のヨウ化ヨードメチルトリフェニルホスホニウム(ギルバート・ストーク(Gilbert Stork),KZ.テトラへドロン・レターズ(Tetrahedron letters)1989年、30(17)、2173)(96.3g、181.7mmol)の懸濁液に、0℃で、ヘキサメチルジシラザンナトリウム(NaHMDS)の1M溶液(181.7mL、181.7mmol)を、添加漏斗により10分間かけてゆっくりと加えた。更に5分間撹拌した後、溶液を−78℃に冷却し、次に1,3−ジメチル−3,4,5,6−テトラヒドロ−2(1H)−ピリミジノン(DMPU)(43.9mL、363.4mmol)をカニューレにより加え、続いてTHF(727mL)に溶解したアルデヒド12(29.1g、72.7mmol)を加えた。温度を−78℃に保持し、その間、反応混合物を2時間撹拌した。ヘキサン(1L)を加え、得れたスラリーをセライトで濾過し、追加のヘキサン(3L)で洗浄した。濾液を減圧下で蒸発させ、得られた油状物をカラムクロマトグラフィー(ヘキサン/EtOAc 100:0〜20:1)により精製して、32g(収率:84%)のヨウ化物13を得た。
1H-RMN (CDCl3, 300 MHz) δ: 6.73 (d, 1H, J = 8.4 Hz), 6.09 (dd, 1H, J = 8.4, 1.2 Hz), 5.57 (dd, 1H, J = 9.6, 1.2 Hz), 3.63-3.71 (m, 3H), 2.58 (m, 1H), 1.90 (s, 3H), 1.70 (m, 2H), 0.96 (dd, 3H, J = 6.6, 1.2 Hz), 0.88 (s, 18H), 0.04 (m, 12H).
13C-RMN (CDCl3, 75 MHz) δ: 142.3, 138.1, 131.8, 74.6, 72.9, 59.8, 38.1, 37.9, 26.0, 18.3, 18.2, 15.7, 15.7, -4.4, -5.2, -5.2.
【0096】
中間体14の合成
【0097】
【化21】
【0098】
EtOH(114mL)中のヨウ化物13(12g、22.9mmol)の溶液に、ピリジニウムp−トルエンスルホネート(PPTS)(2.01g、8.0mmol)を加え、反応混合物を室温で25時間撹拌した。次に、溶媒を減圧下で除去し、得られた油状物をカラムクロマトグラフィー(ヘキサン/EtOAc 10:1)により精製して、8.7g(収率:93%)のアルコール14を得た。
1H-RMN (CDCl3, 300 MHz) δ: 6.69 (d, 1H, J = 8.4 Hz), 6.12 (d, 1H, J = 8.4 Hz), 5.47 (d, 1H, J = 9.9 Hz), 3.67-3.87 (m, 4H), 2.71 (m, 1H), 1.89 (s, 3H), 1.73-1.86 (m, 2H), 1.01 (d, 3H, J = 6.9 Hz), 0.91 (s, 9H), 0.087-0.115 (m, 6H).
13C-RMN (CDCl3, 75 MHz) δ: 142.4, 136.4, 132.6, 75.8, 75.2, 60.0, 38.1, 36.4, 26.1, 18.2, 17.1, 16.0, -4.1, -4.2.
【0099】
中間体15の合成
【0100】
【化22】
【0101】
ジクロロメタン/DMSO(50.9mL/22.9mL)の混合物中のアルコール14(8.7g、21.2mmol)の0℃の溶液に、Et3N(14.8mL、106mmol)を添加漏斗により加えた。10分後、SO3・Pyr(8.43g、53.0mol)を少量ずつ加え、溶液を0℃で更に2時間撹拌した。次に、それをジクロロメタン(800ml)で希釈し、HCl(0.5N、50mL)で停止させた。有機層をデカントし、MgSO4で乾燥し、真空下で濃縮した。カラムクロマトグラフィー(ヘキサン/EtOAc 10:1)による精製によって、6.9g(収率:80%)のアルデヒド15を得た。
1H-RMN (CDCl3, 300 MHz) δ: 9.89 (t, 1H, J = 1.5 Hz), 6.67 (d, 1H, J = 8.4 Hz), 6.13 (d, 1H, J = 8.4 Hz), 5.43 (d, 1H, J = 10.2 Hz), 3.98 (m, 1H), 2.59-2.69 (m, 3H), 1.85 (s, 3H), 1.01 (d, 3H, J = 6.6 Hz), 0.86 (s, 9H), 0.06 (s, 3H), 0.03 (s, 3H).
13C-RMN (CDCl3, 75 MHz) δ: 201.8, 141.9, 135.2, 133.3, 76.3, 71.9, 49.3, 39.3, 25.8, 18.0, 16.7, 15.9, -4.4, -4.5.
【0102】
中間体16aの合成
【0103】
【化23】
【0104】
アルゴン雰囲気下、−78℃で撹拌した、無水THF(390mL)中のジエチル(メトキシ〔メトキシカルボニル〕メチル)ホスホネート(5.51g、14.45mmol)及び18−クラウン−6(11.5g、43.34mmol)の溶液に、0.5Mカリウムビス(トリメチルシリル)アミド溶液(KHMDS)(43.34mL、21.67mmol)を滴加した。15分後、無水THF中のアルデヒド15(5.9g、14.45mmol)を30分間かけて滴加し、−78℃で90分間撹拌した。次に、反応を飽和NH4Cl溶液(200mL)で停止させ、室温に温め、ジクロロメタン(1000mL)で希釈した。有機層を乾燥し(Na2SO4)、減圧下で蒸発させた。カラムクロマトグラフィー(ヘキサン/Et2O 20:1)による精製によって、純粋な4.2g(59%)の(E)−16aを得た。
1H-RMN (CDCl3, 300 MHz) δ: 6.70 (d, 1H, J = 8.4 Hz), 6.08 (d, 1H, J = 8.4 Hz), 5.47 (d, 1H, J = 9.9 Hz), 5.37 (t, 1H, J = 7.2 Hz), 3.78 (s, 3H), 3.60 (s, 3H), 3.60 (m, 1H), 2.79 (m, 1H), 2.52-2.67 (m, 2H), 1.83 (s, 3H), 0.99 (d, 3H, J = 6.6 Hz), 0.89 (s, 9H), 0.05 (s, 3H), 0.04 (s, 3H).
13C-RMN (CDCl3, 75 MHz) δ: 163.7, 145.9, 142.1, 137.3, 132.1, 110.4, 75.4, 74.8, 55.4, 51.9, 38.1, 32.3, 25.9, 18.1, 16.5, 15.7, -4.3, -4.5.
【0105】
中間体16bの合成
【0106】
【化24】
【0107】
アルゴン雰囲気下、0℃で撹拌した、無水THF(2.4mL)中の〔ビス(2,2,2−トリフルオロエトキシ)ホスフィニル〕酢酸エチル(0.16mL、0.66mmol)及び18−クラウン−6(350mg、1.32mmol)の溶液に、KHMDS(1.23mL、0.62mmol)を滴加した。30分後、無水THF中のアルデヒド15(180mg、0.44mmol)を滴加し、−78℃で60分間撹拌した。次に、反応を飽和NH4Cl溶液で停止させ、室温に温め、EtOAcで希釈した。有機層を乾燥し(Na2SO4)、減圧下で蒸発させた。カラムクロマトグラフィー(ヘキサン/EtOAc 100:1〜15:1)による精製によって、172mg(収率:82%)の(Z)−16bを得た。
1H-RMN (CDCl3, 300 MHz) δ: 6.70 (d, 1H, J = 8.7 Hz), 6.44-6.36 (m, 1H), 6.09 (d, 1H, J = 8.7 Hz), 5.86-5.81 (m, 1H), 5.47 (d, 1H, J = 9.9 Hz), 4.14 (q, 2H, J = 7.2 Hz), 3.69-3.64 (m, 1H), 3.06-3.00 (m, 1H), 2.85-2.75 (m, 1H), 2.59-2.51 (m, 1H), 1.84 (s, 3H), 1.28 (t, 3H, J = 7.2 Hz), 1.00 (d, 3H, J = 6.6 Hz), 0.89 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H).
MS (ES) m/z 501.0 [M+Na]+
【0108】
中間体17aの合成
【0109】
【化25】
【0110】
MeOH(125mL)中のエステル16a(4.15g、8.39mmol)の溶液に、室温で、37%のHCl(1.04mL)を加え、反応混合物を6時間撹拌した。次に、混合物をNaHCO3の飽和溶液で中和し(pH7〜8)、有機溶媒を減圧下で蒸発させた。得られた懸濁液をジクロロメタン(3×200mL)で抽出し、乾燥し、蒸発させた。カラムクロマトグラフィー(ヘキサン/EtOAc 10:1〜2:1)による濾過によって、2.76g(収率:94%)のラクトン17aを得た。
1H-RMN (500 MHz, CDCl3) δ: 6.68 (d, 1H, J = 9.0 Hz), 6.20 (d, 1H, J = 8.5 Hz), 5.63 (dd, 1H, J = 2.5, 6.5 Hz), 5.43 (d, 1H, J = 10.0 Hz), 4.19 (m, 1H), 3.65 (s, 3H), 2.84 (m, 1H), 2.55 (m, 1H), 2.43 (dc, J = 1H, 3.0, 12.0, 15.0, 18.0 Hz), 1.87 (s, 3H), 1.16 (d, 3H, J = 6.5 Hz).
13C-RMN (125 MHz, CDCl3) δ: 161.6, 145.2, 141.8, 134.4, 132.7, 108.3, 81.7, 77.4, 55.4, 37.1, 26.6, 16.5, 16.1.
【0111】
中間体17bの合成
【0112】
【化26】
【0113】
MeOH(4.5mL)中のエステル16b(172mg、0.36mmol)の溶液に、室温で、37%のHCl(0.03mL)を加え、反応混合物を3時間撹拌した。次に、混合物をNaHCO3の飽和溶液で中和し(pH7〜8)、有機溶媒を減圧下で蒸発させた。得られた懸濁液をジクロロメタンで抽出し、乾燥し、蒸発させた。カラムクロマトグラフィー(ヘキサン/EtOAc 10:1〜5:1)による濾過によって、70mg(収率:61%)のラクトン17bを得た。
1H-RMN (CDCl3, 300 MHz) δ: 6.91-6.85 (m, 1H), 6.68 (d, 1H, J = 8.4 Hz), 6.62 (d, 1H, J = 8.4 Hz), 6.02 (dd, 1H, J = 2.7, 9.6 Hz), 5.45 (d, 1H, J = 9.9 Hz), 4.19 (m, 1H), 3.65 (s, 3H), 4.26-4.18 (m, 1H), 2.92-2.79 (m, 1H), 2.57-2.48 (m, 1H), 2.39-2.28 (m, 1H), 1.88 (s, 3H), 1.17 (d, 3H, J = 6.6 Hz).
【0114】
実施例6:フラグメントDの合成
【0115】
スキーム3は、スキーム1において提供された命名法に従って、フラグメントDの合成の幾つかの例を提供する。
【0116】
【化27】
【0117】
中間体19の合成
【0118】
【化28】
【0119】
ジクロロメタン(DCM)(918mL)中の中間体18(72.3g)の溶液に、室温で、3−クロロ過安息香酸(m−CPBA)(100g、0.58mol)を少量ずつ加え、混合物を室温で18時間撹拌した。白色の沈殿を、NaHCO3の飽和溶液で停止させ、DCM(3×250mL)で抽出し、再びNaHCO3の飽和溶液(3×250mL)で洗浄した。有機層を合わせ、Na2SO4で乾燥し、真空下で濃縮した。得られた油状物をシリカゲル(ヘキサン−AcOEt;15:1)で精製して、エポキシドを無色の油状物(64.5g、82%)として得た。無水THF(7.5mL)中のラセミエポキシド(30g)の溶液に、(R,R)Co(II)錯体(448mg、0.74mmol)を加え、続いてAcOH(0.14mL)を加えた。溶液を0℃に冷却し、水(1.2mL)を滴加した。反応混合物を室温に温め、18時間撹拌した。その時間の後、揮発性物質を真空下で濃縮し、粗物質をシリカゲルカラムに直接装填した。溶離剤としてヘキサン/EtOAc(15:1〜12:1)を使用するフラッシュクロマトグラフィーによって、キラルエポキシド(+)−19(13.6g、収率:46%)を無色の油状物として得た。
[α]D = +14.1 (c= 1, CHCl3).
1H NMR (CDCl3, 300 MHz) δ: 3.74 (t, 2H, J = 6.3 Hz), 3.01 (m, 1H), 2.74 (t, 1H, J = 4.6 Hz), 2.48 (dd, 1H, J = 5.1, 3.1 Hz), 1.70 (m, 2H), 0.87 (s, 9H), 0.04 (s, 6H).
13C RMN (CDCl3, 75 MHz) δ: 60.2, 50.2, 47.3, 36.1, 26.1, 18.4, -5.2.
【0120】
中間体20の合成
【0121】
【化29】
【0122】
プロピンを−78℃で濃縮し、無水THF(165mL)に溶解した。n−ブチルリチウムをAr下で30分間かけて滴加し、得られた白色の懸濁液を−78℃で更に30分間撹拌した。次に、無水THF(125mL)中の(+)(R)−2−〔2−(tert−ブチルジメチルシリルオキシ)エチル〕オキシラン19(23.7g)の溶液を滴加し、続いてBF3OEt2を加えた。混合物を−78℃で1時間、0℃で更に1時間撹拌した。反応をNH4Clの飽和水溶液(150mL)で停止させ、Et2O(3×150mL)で抽出した。合わせた有機層をNa2SO4で乾燥し、濾過し、濃縮した。フラッシュクロマトグラフィー(ヘキサン/EtOAc 10:1〜1:1)によって、22.7g(収率:80%)のアルコール20を無色の油状物として得た。
[α]D = +5.6 (c= 0.1, CHCl3).
1H-RMN (500 MHz, CDCl3) δ: 3.75-3.90 (m, 3H), 3.47 (d, 1H, J = 2.7 Hz, OH), 2.34 (m, 2H), 1.79, (t, 3H, J = 2.4 Hz), 1.75 (m, 2H), 0.89 (s, 9H), 0.07 (s, 6H).
13C-RMN (125 MHz, CDCl3) δ: 77.8, 75.8, 70.7, 62.4, 37.6, 27.6, 26.1, 18.3, 3.7, -5.3, -5.4.
MS (ES) m/z 243.2 [M+H]+, 265.2 [M+Na]+
【0123】
中間体21aの合成
【0124】
【化30】
【0125】
DCM中の中間体20(22.7g)及びp−メトキシベンジルトリクロロアセトイミデート(PMBTCA)の溶液を、Sc(OTf)3で処理した。混合物を室温で2時間撹拌し(TLCで検査)、反応を真空下で濃縮し、カラムクロマトグラフィー(ヘキサン/EtOAc 50:1〜15:1)により精製して、21aを黄色の油状物(18.3g;収率:55%)として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.25 (d, 2H, J = 8.7 Hz), 6.90 (d, 2H, J = 8.7 Hz), 4.45 (m, 2H), 3.80 (s, 3H), 3.65 (m, 3H), 2.40 (m, 2H), 1.82 (m, 2H), 1.79 (t, 3H, J = 2.4 Hz), 0.92 (s, 9H), 0.05 (s, 6H).
【0126】
中間体21bの合成
【0127】
【化31】
【0128】
N,N−ジメチルホルムアミド(DMF)(14mL)中のアルコール20(2.88g、11.9mmol)、tert−ブチルジフェニルシリルクロリド(4.39mL、16.89mmol)及び4−(ジメチルアミノ)ピリジン(43.6mg)の溶液を、室温で一晩撹拌した。混合物を水で希釈し、Et2Oで抽出し、有機相をブラインで洗浄し、Na2SO4で乾燥し、濾過し、濃縮した。フラッシュクロマトグラフィー(ヘキサン/EtOAc、95:1)によって、シリルエーテル21b(5.3g、収率:93%)を無色の液体として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.70-7.66 (m, 4H), 7.40-7.34 (m, 6H), 3.99-3.95 (m, 1H), 3.70-3.62 (m, 2H), 2.23-2.22 (m, 2H), 1.84-1.81 (m, 2H), 1.69 (t, 3H, J = 2.7 Hz), 1.05 (s, 9H), 0.84 (s, 9H), 0.01 (s, 6H).
13C-RMN (CDCl3, 75 MHz) δ: 136.1; 134.6; 129.7; 127.8; 77.8; 76.2; 69.9; 60.1; 39.6; 27.5; 27.2; 26.2; 19.6; 18.5; 3.7; -5.1.
【0129】
中間体22aの合成
【0130】
【化32】
【0131】
無水トルエン中の21aの溶液に、Ar下及び0℃で、シュワルツ(Schwartz)試薬(ビス(シクロペンタジエニル)ジルコニウム(IV)クロリドヒドリド、Cp2ZrHCl)を加え、反応を室温で5分間撹拌した。反応温度を20分間かけて50℃に増加し、50℃で2.30時間撹拌した。この時間の間に反応溶液は橙色になった。反応を0℃に冷却し、N−クロロスクシンイミドを一度に加えた。撹拌を室温で30分間続け、反応をヘキサン/EtOAc(95:5;500mL)で希釈した。固体を濾過により除去し、揮発物を蒸発させて、22aを黄色の油状物として得て、それを更に精製することなく使用した(15.1g;収率:86%)。
[α]D = +20.5 (c= 1, CHCl3).
1H NMR (CDCl3, 300 MHz) δ: 7.25 (d, 2H, J = 8.7 Hz), 6.87 (d, 2H, J = 8.7 Hz), 5.64 (td, 1H, J = 7.8, 0.9 Hz), 4.45 (q, 2H, J = 11.1 Hz), 3.80 (s, 3H), 3.70 (m, 2H), 3.62 (m, 1H), 2.27 (t, 2H, J = 6.9 Hz), 2.03 (s, 3H), 1.70 (m, 2H), 0.89 (s, 9H), 0.05 (s, 6H).
13C RMN (75 MHz, CDCl3) δ: 159.4, 130.9, 130.7, 129.6, 124.2, 114.0, 75.2, 71.4, 59.8, 55.5, 37.7, 33.8, 26.1, 21.2, 18.5, -5.1.
【0132】
中間体22bの合成
【0133】
【化33】
【0134】
酢酸エチル中の21b(4.73g、9.85mmol)、キノリン(0.582mL、4.92mmol)及びリンドラー触媒(2.18g)の混合液を含有するフラスコを、排気し、H2でフラッシュした。反応混合物を、H2下(1atm)、室温で2時間撹拌し、次にセライトプラグで濾過した。プラグを酢酸エチルですすぎ、合わせた濾液を0.1%のHClで洗浄した。有機層をNa2SO4で乾燥し、濾過し、濃縮して、中間体22b(4.27g、収率:90%)を無色の油状物として得て、それを更に精製することなく使用した。
1H NMR (CDCl3, 300 MHz) δ: 7.70-7.67 (m, 4H), 7.44-7.36 (m, 6H), 5.48 (m, 1H), 5.36-5.27 (m, 1H), 3.95-3.87 (m, 1H), 3.71-3.55 (m, 2H), 2.16 (dd, 2H, J = 6.9, 6.3 Hz), 1.73-1.66 (m, 2H), 1.41 (dd, 3H, J = 6.6, 1.2 Hz), 1.05 (s, 9H), 0.84 (s, 9H), -0.02 (s, 6H).
13C-RMN (CDCl3, 75 MHz) δ: 136.2; 134.8; 129.8; 127.8; 126.4; 125.8; 70.9; 60.4; 39.6; 34.8; 27.3; 26.2; 19.7; 18.5; 13.1; -5.1.
【0135】
中間体23aの合成
【0136】
【化34】
【0137】
無水THF中の22a(23g)の溶液に、Ar下及び0℃で、フッ化テトラブチルアンモニウム(TBAF)の溶液を20分間かけて滴加した(溶液は赤色になった)。反応混合物を室温で2時間撹拌し、次にNH4Clの飽和水溶液(200ml)で停止させた。合わせた層を分離し、水相をEtOAc(3×150ml)で十分に抽出した。合わせた有機層をNaSO4で乾燥し、濾過し、濃縮した。フラッシュクロマトグラフィー(ヘキサン/EtOAc 4:1〜1:1)によって、23aを無色の油状物(11.9g;収率:73%)として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.25 (d, 2H, J = 8.7 Hz), 6.86 (d, 2H, J = 8.7 Hz), 5.62 (t, 1H, J = 7.8 Hz), 4.45 (m, 2H), 3.80 (s, 3H), 3.70 (m, 3H), 2.35 (m, 2H), 2.03 (s, 3H), 1.75 (m, 2H).
【0138】
中間体23bの合成
【0139】
【化35】
【0140】
PPTS(837.7mg、3.33mmol)を、エタノール(80mL)中の22b(4g、8.33mmol)の溶液に、一度に加えた。反応混合物を室温で7時間撹拌し、次に濃縮した。残渣をDCMで希釈し、NaHCO3の飽和溶液で洗浄した。有機層を抽出し、Na2SO4で乾燥し、濾過し、濃縮した。フラッシュクロマトグラフィー(ヘキサン/EtOAc、95:1)によって、シリルエーテル23b(2.12g、収率:69%)を無色の油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.73-7.69 (m, 4H), 7.44-7.36 (m, 6H), 5.44-5.38 (m, 1H), 5.21-5.17 (m, 1H), 4.01-3.94 (m, 1H), 3.84-3.76 (m, 1H), 3.69-3.64 (m, 1H), 2.32-2.14 (m, 2H), 1.89-1.78 (m, 1H), 1.70-1.60 (m, 1H), 1.37 (d, 3H, J = 6.9 Hz), 1.07 (s, 9H).
13C-RMN (CDCl3, 75 MHz) δ: 136.2; 134.1; 130.0; 127.8; 126.3; 125.9; 72.3; 60.1; 37.7; 34.3; 27.2; 19.5; 13.0.
【0141】
中間体24aの合成
【0142】
【化36】
【0143】
(ジアセトキシヨード)ベンゼン(BAIB)(11.5g、35.7mmol)を、ジクロロメタン無水物(92mL)中のアルコール23a(9.2g、32.4mmol)及び2,2,6,6−テトラメチルピペリジン 1−オキシル(TEMPO)(515mg、3.3mmol)の溶液に加えた。反応混合物を、アルコールが検出されなくなるまで(TLC)、室温で20時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、DCM(3×100mL)で抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 4:1〜1:1)により精製して、24aを無色の油状物(6.3g;収率:70%)として得た。
1H NMR (CDCl3, 300 MHz) δ: 9.78 (s, 1H), 7.25 (d, 2H, J = 8.7 Hz), 6.85 (d, 2H, J = 8.7 Hz), 5.64 (t, 1H, J = 7.8 Hz), 4.45 (q, 2H, J = 11.1 Hz), 4.02 (m, 1H), 3.80 (s, 3H), 2.60 (m, 2H), 2.35 (m, 2H), 2.03 (s, 3H).
13C RMN (CDCl3, 75 MHz) δ: 201, 159.6, 132.1, 130.1, 129.7, 122.8, 114.1, 73.3, 71.5, 55.5, 48.3, 33.5, 21.3.
【0144】
中間体24bの合成
【0145】
【化37】
【0146】
BAIB(1.97g、6.11mmol)を、25mLのDCM中のアルコール23b(2.05g、5.56mmol)及びTEMPO(86.87mg、0.56mmol)の溶液に加えた。反応混合物を、アルコールが検出されなくなるまで(TLC)、室温で16〜18時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、DMCで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/DCM 5:1〜1:2)により精製して、24b(1.733mg、収率:79%)を無色の油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 9.72 (t, 1H, J = 2.7 Hz), 7.74-7.67 (m, 4H), 7.48-7.37 (m, 6H), 5.56-5.45 (m, 1H), 5.32-5.23 (m, 1H), 4.29-4.20 (m, 1H), 2.51-2.48 (m, 2H), 2.31-2.27 (m, 2H), 1.43 (dd, 3H, J = 6.9, 1.5 Hz), 1.06 (s, 9H).
13C-RMN (CDCl3, 75 MHz) δ: 202.3; 136.1; 134.0; 130.1; 127.9; 127.4; 125.1; 69.4; 50.1; 35.1; 27.2; 19.5; 13.1.
【0147】
中間体25aの合成
【0148】
【化38】
【0149】
無水THF(126mL)中のヨウ化ヨードメチルトリフェニルホスホニウム(16.6g;31mmol)の懸濁液に、室温で、THF(31.27mL)中のNaHMDSの1M溶液をゆっくりと加えた。2分間撹拌した後、黄色の混合物を−78℃に冷却し、次にTHF(82mL)中の24a(6.3g、22mmol)の溶液を加えた。反応混合物を−78℃で2時間、室温で5分間撹拌し、ヘキサンで希釈し、セライトプラグで濾過した。プラグをヘキサンですすぎ、合わせた濾液を減圧下で蒸発させ、得られた油状物をカラムクロマトグラフィー(ヘキサン/EtOAc 12:1〜8:1)により精製して、25aを黄色の油状物(5.6g;収率:62%)として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.25 (d, 2H, J = 8.7 Hz), 6.85 (d, 2H, J = 8.7 Hz), 6.25 (m, 2H) 5.64 (t, 1H, J = 7.8 Hz), 4.42 (m, 2H), 3.80 (s, 3H), 3.55(m, 1H), 2.40 (m, 2H), 2.25 (m, 2H), 2.03 (s, 3H).
【0150】
中間体25bの合成
【0151】
【化39】
【0152】
THF(60mL)中のヨードメチルトリフェニルホスホラン(3.32g、6.38mmol)の懸濁液に、室温で、THF中のNaHMDS(6.38mmol)の1M溶液6.83mLをゆっくりと加えた。2分間撹拌した後、黄色の混合物を−78℃に冷却し、次にTHF(40mL)中の24b(1.67g、4.56mmol)の溶液を加えた。反応混合物を−78℃で90分間、次に室温で5分間撹拌し、ヘキサンで希釈し、セライト/SiO2のプラグで濾過した。プラグをヘキサン/EtOAc(10:1〜5:1)によりすすいで、化合物25b(2g、収率:89%)を無色の油状物として得て、それを更に精製することなく使用した。
1H NMR (CDCl3, 300 MHz) δ: 7.70-7.66 (m, 4H), 7.45-7.34 (m, 6H), 6.21-6.31 (m, 2H), 5.49-5.43 (m, 1H), 5.35-5.27 (m, 1H), 3.94-3.75 (m, 1H), 2.30-2.27 (m, 2H), 2.24-2.04 (m, 2H), 1.43 (d, 3H, J = 6.6 Hz), 1.06 (s, 9H).
13C-RMN (CDCl3, 75 MHz) δ: 138.2; 136.2; 134.3; 129.9; 127.8; 126.4; 126.0; 84.1; 71.9; 41.6; 34.5; 27.2; 19.6; 13.2.
【0153】
中間体25cの合成
【0154】
【化40】
【0155】
2,3−ジクロロ−5,6−ジシアノ−p−ベンゾキノン(DDQ)(3.6g、16mmol)を、DCM−H2O(20:1)中の25a(5g;12mmol)の溶液に、Ar雰囲気下、室温で加えた。1:30h後(ヘキサン/EtOAc 4:1のTLCでは出発物質を示さなかった)、反応を、Et2O(200mL)に注ぐことにより停止させ、1M NaOH(3×50ml)及びブライン(50mL)で洗浄した。有機相をNa2SO4で乾燥し、濾過し、濃縮した。p−メトキシベンズアルデヒドのクロマトグラフ分離を、p−メトキシベンジルアルコールへの還元により促進した。この終点に向かって、Ar雰囲気下でNaBH4を有するMeOHにおいて得られた残渣の溶液を、室温で1時間保持した。次に反応混合物を、Et2O(100mL)に注ぐことによって停止させ、1M HCl(40mL)及びブライン(40mL)で洗浄した。有機相をNa2SO4で乾燥し、濾過し、濃縮した。得られた油状物をシリカゲル(ヘキサン/EtOAc 10:1〜4:1)で精製して、第二級アルコールを無色の油状物として得た(2.8g;収率:80%)。
【0156】
無水DCM中の第二級アルコール(2.8g;10mmol)の溶液に、Ar下及び0℃で、2,6−ルチジンを滴加し、続いてtert−ブチルジメチルシリルトリフルオロメタンスルホネート(TBSOTf)を加えた(ヘキサン/EtOAc 4:1のTLCでは出発物質を示さなかった)。この時点で、粗混合物を0.5M HCl(25mL)で停止させ、DCM(2×25mL)で抽出した。合わせた有機層をNaHCO3の飽和水溶液及びブラインで洗浄した。有機相をNaSO4で乾燥し、濾過し、濃縮した。フラッシュクロマトグラフィー(ヘキサン/EtOAc 100:1〜20:1)によって、25cを無色の油状物(3.14g;収率:80%)として得た。
1H NMR (CDCl3, 300 MHz) δ: 6.25 (m, 2H) 5.64 (t, 1H, J = 7.8 Hz), 3.82 (m, 1H), 2.38 (t, 2H, J = 6.0 Hz), 2.20 (t, 2H, J = 6.3 Hz), 2.03 (s, 3H), 0.86 (s, 9H), 0.05 (s, 6H).
13C RMN (CDCl3, 75 MHz) δ: 137.7, 130.9, 124.3, 84.6, 70.6, 42.5, 36.6, 25.9, 21.3, 18.2, -4.4.
【0157】
実施例7:フラグメントBCDの合成
スキーム4は、スキーム1において提供された命名法に従って、フラグメントBCDの合成の幾つかの例を提供する。
【0158】
【化41】
【0159】
中間体26aの合成
【0160】
【化42】
【0161】
再密封可能なシュレンク管に、ヨウ化銅(I)(148mg、0.78mmol)、炭酸カリウム(1.076g、7.78mmol)及びBoc−tert−LeuCONH2(ポズネフ(Pozdnev)V.F.、テトラヘドロン・レターズ(Tetrahedron Letters)1995年、36、7115〜7118に記載された手順に従って調製)(0.96g、4.15mmol)を投入し、排気し、アルゴンを充填した。N,N′−ジメチルエチレンジアミン(DMEDA)(0.166mL、1.55mmol)、ヨウ化ビニル25c(1.04g、2.59mmol)及び無水DMF(15mL)をアルゴン下で加えた。シュレンク管を密閉し、90℃で16〜18時間加熱し、室温に冷却した。得られた混合物をEtOAcで希釈し、水で停止させた。有機層を、水で洗浄し、Na2SO4で乾燥した。溶媒を減圧下で除去し、残渣をシリカゲルのフラッシュクロマトグラフィー(ヘキサン/EtOAc、20:1〜15:1)により精製した。中間体26a(670mg、収率、53%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.72 (d, 1H, J = 9.9 Hz), 6.70 (t, 1H, J = 9.6 Hz), 5.54 (t, 1H, J = 7.8 Hz), 5.35 (d, 1H, J = 9.0 Hz), 4.76 (q, 1H, J = 7.8 Hz), 3.89 (d, 1H, J = 9.0 Hz), 3.73-3.68 (m, 1H), 2.12 (m, 4H), 1.98 (s, 3H), 0.971 (s, 9H), 0.84 (s, 9H), 0.02 (s, 3H), 0.01 (s, 3H).
13C NMR (CDCl3, 75 MHz) δ: 168.9, 156.0 131.1, 123.9, 122.6, 108.2, 79.9, 71.6, 62.5, 36.5, 34.8, 33.8, 28.1, 26.7, 25.9, 21.2, 18.3, -4.3, -4.4.
【0162】
中間体26bの合成
【0163】
【化43】
【0164】
再密封可能なシュレンク管に、ヨウ化銅(I)(232.4mg、1.22mmol)、炭酸カリウム(1.688g、12.23mmol)及びBoc−tert−LeuCONH2(2.474g、6.12mmol)を投入し、排気し、アルゴンを充填した。N,N′−ジメチルエチレンジアミン(0.26mL、2.45mmol)、ヨウ化ビニル25b(2g、4.08mmol)及び無水DMF(35mL)をアルゴン下で加えた。シュレンク管を密閉し、90℃で16〜18時間加熱し、室温に冷却した。得られた混合物をEtOAcで希釈し、水で停止させた。有機層を、水で洗浄し、Na2SO4で乾燥した。溶媒を減圧下で除去し、残渣をシリカゲルのフラッシュクロマトグラフィー(ヘキサン/EtOAc、20:1〜15:1)により精製した。中間体26b(1.06g、収率:44%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.70-7.67 (m, 4H), 7.43-7.35 (m, 6H), 7.13 (d, 1H, J = 10.5 Hz), 6.67 (dd, 1H, J = 10.2, 9.6 Hz), 5.56-5.45 (m, 1H), 5.36-5.28 (m, 2H), 4.86-4.78 (m, 2H), 3.88-3.77 (m, 1H), 2.26-2.04 (m, 4H), 1.44 (d, 3H, J = 6.9 Hz), 1.43 (s, 9H), 1.06 (s, 9H), 0.96 (s, 9H).
【0165】
中間体26cの合成
【0166】
【化44】
【0167】
再密封可能なシュレンク管に、ヨウ化銅(I)(40.4mg、0.213mmol)、炭酸カリウム(294mg、2.13mmol)及びBoc−Val−CONH2(ポズネフ(Pozdnev)V.F.、テトラヘドロン・レターズ(Tetrahedron Letters)1995年、36、7115〜7118に記載された手順に従って調製)(230mg、1.06mmol)を投入し、排気し、アルゴンを充填した。N,N′−ジメチルエチレンジアミン(45μL、0.426mmol)、ヨウ化ビニル25c(283mg、0.71mmol)及び無水DMF(35mL)をアルゴン下で加えた。シュレンク管を密閉し、90℃で16〜18時間加熱し、室温に冷却した。得られた混合物をEtOAcで希釈し、水で停止させた。有機層を、水で洗浄し、Na2SO4で乾燥した。溶媒を減圧下で除去し、残渣をシリカゲルのフラッシュクロマトグラフィー(ヘキサン/EtOAc、7:1〜3:1)により精製した。中間体26c(270g、収率:77%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.80 (d, 1H, J = 9.3), 6.79-6.73 (m, 1H), 5.58 (t, 1H, J = 7.5 Hz), 5.02 (br s, 1H), 4.85-4.76 (m, 1H), 3.93 (dd, 1H, J = 8.4, 6.0 Hz), 3.80-3.73 (m, 1H), 2.12-2.22 (m, 5H), 2.02 (s, 3H), 1.45 (s, 9H), 0.98 (d, 3H, J = 6.9 Hz), 0.93 (d, 3H, J = 6.9 Hz), 0.89 (s, 9H), 0.07 (s, 3H), 0.06 (s, 3H).
13C NMR (CDCl3, 75 MHz) δ: 169.3, 131.1, 124.0, 122.7, 108.9, 71.6, 36.5, 33.8, 30.6, 28.5, 26.1, 21.3, 19.6, 18.3, 17.9, -4.3, -4.4.
【0168】
中間体26dの合成
【0169】
【化45】
【0170】
再密封可能なシュレンク管に、ヨウ化銅(I)(14.2mg、0.075mmol)、炭酸カリウム(104mg、0.75mmol)及びFmoc−Phe−CONH2(ポズネフ(Pozdnev)V.F.、テトラヘドロン・レターズ(Tetrahedron Letters)1995年、36、7115〜7118に記載された手順に従って調製)(145mg、0.375mmol)を投入し、排気し、アルゴンを充填した。N,N′−ジメチルエチレンジアミン(16μL、0.15mmol)、ヨウ化ビニル25c(100mg、0.25mmol)及び無水DMF(2.5mL)をアルゴン下で加えた。シュレンク管を密閉し、90℃で16〜18時間加熱し、室温に冷却した。得られた混合物をEtOAcで希釈し、水で停止させた。有機層を、水で洗浄し、Na2SO4で乾燥した。溶媒を減圧下で除去し、残渣をシリカゲルのフラッシュクロマトグラフィー(ヘキサン/EtOAc、4:1〜1:1)により精製した。中間体26d(46mg、収率:42%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 9.19 (d, 1H, J = 11.1 Hz), 7.36-7.21 (m, 5H), 6.77 (ddd, 1H, J = 10.2, 9.3, 0.9), 5.60 (br t, 1H, J = 7.8 Hz), 4.82-4.78 m, 1H), 3.79-3.71 (m, 1H), 3.67 (dd, 1H, J = 9.6, 3.9 Hz), 3.32 (dd, 1H, J = 13.8, 3.9 Hz), 2.69 (dd, 1H, J = 13.8, 9.6 Hz), 2.20-2.11 (m, 4H), 1.99 (s, 3H), 0.89 (s, 9H), 0.05 (s, 3H), 0.04 (s, 3H).
13C NMR (CDCl3, 75 MHz) δ: 171.9, 137.9, 130.9, 129.5, 129.1, 127.2, 124.1, 122.5, 107.9, 71.4, 56.6, 40.9, 36.3, 33.6, 26.1, 21.3, 18.3, -4.4, -4.5.
MS (ES) m/z 437.1 [M+H]+, 459.0 [M+Na]+
【0171】
中間体27aの合成
【0172】
【化46】
【0173】
エチレングリコール(30mL)中のアミノ保護誘導体26a(670mg、1.33mmol)の溶液を、200℃で10〜20分間加熱した。次に反応混合物を室温に冷却し、DCMで希釈し、ブラインで停止させ、水に注いだ。数滴の3M NaOHを、溶液がpH14に達するまで加え、次にDCMで十分に抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、真空下で濃縮して、第一級アミン27a(510mg、収率:95%)を黄色の油状物として得て、それを更に精製することなく使用した。
1H NMR (CDCl3, 300 MHz) δ: 8.77 (d, 1H, J = 9.9 Hz), 6.71 (t, 1H, J = 9.6 Hz), 5.56 (t, 1H, J = 7.8 Hz), 4.71 (m, 1H), 3.72 (m, 1H), 3.14 (s, 1H), 2.14 (m, 4H), 1.97 (s, 3H), 0.97 (s, 9H), 0.84 (s, 9H), 0.02 (s, 6H).
13C RMN (CDCl3, 75 MHz) δ: 171.2, 131.0, 124.1, 122.5, 107.1, 71.5, 64.3, 36.2, 34.5, 33.8, 26.5, 26.0, 21.2, 18.2, -4.4, -4.5.
【0174】
中間体27bの合成
【0175】
【化47】
【0176】
エチレングリコール(50mL)中のアミノ保護誘導体26b(847mg、1.43mmol)の溶液を、200℃で10〜20分間加熱した。次に反応混合物を室温に冷却し、DCMで希釈し、ブラインで停止させ、水に注いだ。数滴の3M NaOHを、溶液がpH14に達するまで加え、次にDCMで十分に抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、真空下で濃縮して、第一級アミン27b(435mg、62%)を、フラッシュクロマトグラフィー(ヘキサン/EtOAc 10:1〜1:2)による精製の後で、白色の泡状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 8.50 (d, 1H, J = 10.8 Hz), 7.70-7.66 (m, 4H), 7.45-7.33 (m, 6H), 6.67 (dd, 1H, J = 11.1, 9.3 Hz), 5.48-5.40 (m, 1H), 5.36-5.28 (m, 1H), 4.79 (dd, 1H, J = 16.2, 7.5 Hz), 3.87-3.79 (m, 1H), 3.08 (s, 1H), 2.22-2.14 (m, 4H), 1.43 (d, 3H, J = 6.9 Hz), 1.05 (s, 9H), 0.97 (s, 9H).
13C-RMN (CDCl3, 75 MHz) δ: 171.0; 136.1; 134.5; 129.8; 127.8; 126.3; 126.2; 122.1; 107.6; 72.6; 64.4; 34.0; 34.4; 32.8; 27.2; 26.9; 19.6; 13.2.
【0177】
中間体27cの合成
【0178】
【化48】
【0179】
エチレングリコール(15mL)中のアミノ保護誘導体26c(255mg、0.52mmol)の溶液を、200℃で10〜20分間加熱した。次に反応混合物を室温に冷却し、DCMで希釈し、ブラインで停止させ、水に注いだ。数滴の3M NaOHを、溶液がpH14に達するまで加え、次にDCMで十分に抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、真空下で濃縮して、第一級アミン27c(170mg、85%)を黄色の油状物として得て、それを更に精製することなく使用した。
1H NMR (CDCl3, 300 MHz) δ: 9.27 (d, 1H, J = 10.2), 6.76 (dd, 1H, J = 11.1, 9.6 Hz), 5.61 (t, 1H, J = 7.8 Hz), 4.80-4.72 (m, 1H), 3.81-3.73 (m, 1H), 3.31 (d, 1H, J = 3.6 Hz) 2.44-2.33 (m, 1H), 2.20-2.16 (m, 4H), 2.03 (s, 3H), 1.59 (br s, 2H), 1.00 (d, 3H, J = 6.9 Hz), 0.89 (s, 9H), 0.82 (d, 3H, J = 6.9 Hz), 0.05 (s, 6H).
13C NMR (CDCl3, 75 MHz) δ: 172.1, 131.1, 124.1, 122.5, 107.4, 71.5, 36.5, 33.7, 30.8, 26.0, 21.3, 20.0, 16.1, -4.3, -4.4.
【0180】
中間体28aの合成
【0181】
【化49】
【0182】
DCM/DMF(10:1、39.6mL)中のアミン27a(918mg、2.27mmol)の溶液に、無水DCM中の(Z)−3−トリブチルスタンニルプロペン酸(1028mg、2.84mmol)の溶液を、アルゴン雰囲気下で加え、次に0℃に冷却した。ジイソプロピルエチルアミン(DIPEA)(0.6mL、3.4mmol)、1−ヒドロキシ−7−アザベンゾトリアゾール(HOAt)(310mg、2.27mmol)及びN,N,N′,N′−テトラメチル−O−(7−アザベンゾトリアゾール−1−イル)ウロニウムヘキサフルオロホスフェート(HATU)(860mg、2.27mmol)を溶液に加え、30分後、冷浴を取り外した。反応混合物を室温で2時間撹拌し、NH4Clの飽和水溶液で停止させ、水に注ぎ、DCMで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 20:1〜15:1)により精製して、アミド28a(1110mg;収率:66%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.63 (d, 1H, J = 10.5 Hz), 6.97 (d, 1H, J = 12.3 Hz), 6.75 (d, 1H, J = 12.3 Hz), 6.72 (t, 1H, J = 9.5 Hz), 6.50 (d, 1H, J = 9.0 Hz), 5.56 (t, 1H, J = 6.6 Hz), 4.83 (q, 1H, J = 9.0 Hz), 4.41 (d, 1H, J = 9.6 Hz) 3.76 (m, 1H), 2.17 (m, 4H), 2.01 (s, 3H), 1.45 (m, 6H), 1.25 (m, 8H), 1.0 (s, 9H), 0.88 (s, 9H), 0.84 (m, 13H), 0.06 (s, 6H).
【0183】
中間体28bの合成
【0184】
【化50】
【0185】
DCM/DMF(4:1、12.5mL)中のアミン27b(575mg、1.17mmol)の溶液に、無水DCM中の(Z)−3−トリブチルスタンニルプロペン酸(505.6mg、1.4mmol)の溶液を、アルゴン雰囲気下で加え、次に0℃に冷却した。DIPEA(0.243mL、1.76mol)、7−ヒドロキシベンゾトリアゾール(HOBt)(189.2mg、1.4mmol)及びHATU(532.28mg、1.4mmol)を溶液に加え、30分後、冷浴を取り外した。反応混合物を室温で2時間撹拌し、NH4Clの飽和水溶液で停止させ、水に注ぎ、DCMで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 20:1〜15:1)により精製して、アミド28b(780.4mg;収率:77%)を白色の泡状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.70-7.68 (m, 4H), 7.43-7.36 (m, 6H), 7.02 (d, 1H, J = 12.3 Hz), 7.00 (d, 1H, J = 10.8 Hz), 6.75 (d, 1H, J = 12.3 Hz), 6.66 (t, 1H, J = 9.3 Hz), 6.26 (d, 1H, J = 9.6 Hz), 5.57-5.34 (m, 1H), 5.38-5.28 (m, 1H), 4.83 (dd, 1H, J = 16.5, 7.8 Hz), 4.31 (d, 1H, J = 9.6 Hz), 3.89-3.82 (m, 1H), 2.26-2.02 (m, 4H), 1.50-1.42 (m, 6H), 1.43 (d, 3H, J = 6.9 Hz), 1.33-1.20 (m, 6H), 1.06 (s, 9H), 0.96 (s, 9H), 0.95-0.83 (m, 15H).
13C-RMN (CDCl3, 75 MHz) δ: 168.0; 166.2; 153.8; 136.3; 136.1; 134.3; 130.0; 127.8; 126.7; 126.0; 121.6; 109.0; 72.6; 60.7; 35.7; 34.0; 32.7; 29..5; 27.7; 27.2; 26.7; 19.5; 14.0; 13.2; 11.8.
【0186】
中間体28cの合成
【0187】
【化51】
【0188】
DCM/DMF(10:1、7.7mL)中のアミン27c(170mg、0.437mmol)の溶液に、無水DCM中の(Z)−3−トリブチルスタンニルプロペン酸(197.2mg、0.546mmol)の溶液を、アルゴン雰囲気下で加え、次に0℃に冷却した。DIPEA(0.11mL、0.655mmol)、HOAt(59.4mg、0.437mmol)及びHATU(166mg、0.437mmol)を溶液に加え、30分後、冷浴を取り外した。反応混合物を室温で2時間撹拌し、NH4Clの飽和水溶液で停止させ、水に注ぎ、DCMで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 20:1〜15:1)により精製して、アミド28c(250mg、収率:78%)を白色の泡状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.94 (d, 1H, J = 10.8 Hz), 7.00 (d, 1H, J = 12.3 Hz), 6.75 (d, 1H, J = 12.3 Hz), 6.72 (t, 1H, J = 9.5 Hz), 6.50 (d, 1H, J = 9.0 Hz), 5.56 (t, J = 6.6 Hz, 1H), 4.83 (q, 1H, J = 9.0 Hz), 4.41 (t, 1H, J = 9.0 Hz), 3.76 (m, 1H), 2.17 (m, 4H), 2.01 (s, 3H), 1.45 (m, 7H), 1.25 (m, 8H), 0.88 (s, 9H), 0.84 (m, 19H), 0.06 (s, 6H).
13C-RMN (CDCl3, 75 MHz) δ: 169.2, 166.8, 153.8, 136.2, 131.1, 123.9, 122.6, 108.7, 71.6, 59.2, 36.5, 33.7, 31.4, 29.5, 29.4, 27.6, 26.1, 21.3, 19.5, 18.5, 18.3, 14.0, 11.8, -4.3, -4.4.
【0189】
中間体28dの合成
【0190】
【化52】
【0191】
DCM/DMF(10:1、1.3mL)中のアミン26d(44mg、0.1mmol)の溶液に、無水DCM中の(Z)−3−トリブチルスタンニルプロペン酸(45mg、0.125mmol)の溶液を、アルゴン雰囲気下で加え、次に0℃に冷却した。DIPEA(26μL、0.15mmol)、HOAt(13.6mg、0.1mmol)及びHATU(38mg、0.1mmol)を溶液に加え、30分後、冷浴を取り外した。反応混合物を室温で2時間撹拌し、NH4Clの飽和水溶液で停止させ、水に注ぎ、DCMで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 20:1〜15:1)により精製して、アミド28d(60mg、収率:80%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.43 (d, 1H, J = 10.8 Hz), 7.34-7.22 (m, 5H), 7.02 (d, 1H, J = 12.3 Hz), 6.70 (d, 1H, J = 12.3 Hz), 6.66 (dd, 1H, J = 9.9, 9.3 Hz), 6.34 (d, 1H, J = 7.8 Hz), 5.51 (dd, 1H, J = 8.1, 7.5 Hz), 4.81-4.71 (m, 2H), 3.68-3.59 (m, 1H), 3.18 (dd, 1H, J = 13.5, 6 Hz), 2.69 (dd, 1H, J = 13.5, 8.4 Hz), 2.11-2.04 (m, 2H), 2.01 (s, 3H), 1.96-1.87 (m, 1H), 1.80-1.70 (m, 1H), 1.53-1.43 (m, 8H), 1.31-1.24 (m, 10H), 0.89-0.85 (m, 9H), 0.88 (s, 9H), 0.04 (s, 3H), 0.01 (s, 3H).
13C NMR (CDCl3, 75 MHz) δ: 168.5, 166.5, 154.4, 136.7, 135.9, 131.0, 129.5, 129.1, 127.4, 124.0, 122.3, 108.8, 71.5, 55.1, 38.8, 36.6, 33.3, 29.5, 29.4, 27.6, 26.0, 21.3, 18.2, 14.0, 11.8, -4.3, -4.5.
MS (ES) m/z 781.2 [M+H]+, 803.2 [M+Na]+
【0192】
中間体28eの合成
【0193】
【化53】
【0194】
DCM/DMF(10:1、1mL)中のアミン27a(30mg、0.075mmol)の溶液に、無水DCM中の(E)−3−トリブチルスタンニルプロペン酸(33.5mg、0.095mmol)の溶液を、アルゴン雰囲気下で加え、次に0℃に冷却した。DIPEA(19μL、0.11mol)、HOAt(10mg、0.075mmol)及びHATU(27.5mg、0.075mmol)を溶液に加え、30分後、冷浴を取り外した。反応混合物を室温で2時間撹拌し、NH4Clの飽和水溶液で停止させ、水に注ぎ、DCMで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 6:1)により精製して、アミド28e(25mg、収率:45%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.68 (d, 1H, J = 9.2 Hz), 7.52 (d, 1H, J = 18.9 Hz), 6.73 (t, 1H, J = 9.2 Hz), 6.28 (d, 1H, J = 10.8 Hz), 6.25 (d, 1H, J = 18.9 Hz), 5.60 (t, 1H, J = 7.2 Hz), 4.83 (q, 1H, J = 9.2 Hz), 4.40 (d, 1H, J = 9.6 Hz), 3.77 (m, 1H), 2.17 (m, 4H), 2.01 (s, 3H), 1.45 (m, 6H), 1.25 (m, 8H), 1.0 (s, 9H), 0.88 (s, 9H), 0.84 (m, 13H), 0.06 (s, 6H).
【0195】
実施例8
スキーム5は、本発明の幾つかの化合物の合成を提供する。
【0196】
【化54】
【0197】
化合物29aの合成
【0198】
【化55】
【0199】
1−メチル−2−ピロリジノン(NMP)(14.7mL)中のアルケニルスタンナン28a(1.1g、1.47mmol)及び17a(0.62g、1.77mmol)の溶液に、0℃で、チオフェンカルボン酸銅(CuTC)(422mg、2.2mmol)を加えた。反応を、0℃で45分間、室温で20分間撹拌した。次に、粗混合物を中性アルミナのプラグで濾過し、EtOAc/エーテル 50:50で洗浄し、合わせた濾液を0.5N HCl(3×15mL)で洗浄した。有機溶液を乾燥し、蒸発させて粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 5:1〜1:1)により精製して、トリエン29a(0.66g、収率:66%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.89 (d, 1H, J = 10.8 Hz), 7.22 (dd, 1H, J = 12.3, 11.4 Hz), 6.86 (dd, 1H, J = 11.7, 11.4 Hz), 6.70 (dd, 1H, J = 9.9, 9.3 Hz), 6.35 (d, 1H, J = 9.3 Hz), 6.13 (d, 1H, J = 11.4 Hz), 5.66 (d, 1H, J = 11.4 Hz), 5.60 (dd, 1H, J = 5.4, 3.9 Hz), 5.55 (br t, 1H, J = 7.8 Hz), 5.26 (d, 1H, J = 10.2 Hz), 4.84-4.76 (m, 1H), 4.3 (d, 1H, J = 9.3 Hz), 4.20-4.16 (m, 1H), 3.77-3.69 (m, 1H), 3.63 (s, 3H), 2.89-2.77 (m, 1H), 2.41-2.33 (m, 2H), 2.19-2.13 (m, 4H), 2.00 (s, 3H), 1.82 (s, 3H), 1.13 (d, 3H, J = 6.9 Hz), 1.02 (s, 9H), 0.86 (s, 9H), 0.4 (s, 3H), 0.03 (s, 3H).
13C-RMN (CDCl3, 75 MHz) δ: 168.5; 166.4; 161.8; 145.4; 140.3, 137.3; 134.4; 134.3; 131.0, 124.3; 124.1, 122.4; 121.2; 108.7; 108.4; 82.0; 71.6; 60.6; 55.6; 37.5; 36.5, 35.1; 33.8; 26.5; 26.0; 21.3, 18.3, 17.4, 16.9, -4.3, -4.4.
【0200】
化合物29bの合成
【0201】
【化56】
【0202】
NMP(9mL)中のアルケニルスタンナン28b(780.4mg、0.904mmol)及び17a(377.4mg、1.085mmol)の溶液に、0℃で、チオフェンカルボン酸銅(258.5mg、1.36mmol)を加えた。反応を、0℃で45分間、室温で20分間撹拌した。次に、粗混合物を中性アルミナのプラグで濾過し、EtOAc/エーテル 50:50で洗浄し、合わせた濾液を0.5N HCl(3×10mL)で洗浄した。有機溶液を乾燥し、蒸発させて粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 5:1〜1:1)により精製して、トリエン29b(459.7mg、収率:66%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.66-7.64 (m, 4H), 7.43-7.32 (m, 7H), 7.23 (t, 1H, J = 11.7 Hz), 6.85 (t, 1H, J = 11.7 Hz), 6.62 (dd, 1H, J = 10.5, 9.3 Hz), 6.41 (d, 1H, J = 9.3 Hz), 6.11 (d, 1H, J = 11.7 Hz), 5.66 (d, 1H, J = 11.4 Hz), 5.60 (dd, 1H, J = 5.7, 5.1 Hz), 5.49-5.41 (m, 1H), 5.32-5.27 (m, 1H), 5.25 (d, 1H, J = 9.9 Hz), 4.83-4.75 (m, 1H), 4.32 (d, 1H, J = 9.3 Hz), 4.22-4.15 (m, 1H), 3.83-3.78 (m, 1H), 3.62 (s, 3H), 2.86-2.78 (m, 1H), 2.40-2.35 (m, 2H), 2.20-2.04 (m, 4H), 1.81 (s, 3H), 1.40 (d, 3H, J = 6.9 Hz), 1.13 (d, 3H, J = 6.9 Hz), 1.03 (s, 9H), 0.97 (s, 9H).
13C-RMN (CDCl3, 75 MHz) δ: 168.3; 166.3; 161.8; 145.4; 140.2, 137.3; 136.1; 134.8; 134.4; 134.3; 129.9; 127.8;126.4; 126.1; 124.4; 121.7; 121.2; 108.4; 109.1; 82.0; 72.6; 60.6; 55.6; 37.5; 35.2; 32.7; 31.1; 27.2; 26.8, 26.5; 19.5; 17.4; 16.9; 13.1.
【0203】
化合物29cの合成
【0204】
【化57】
【0205】
NMP(2.5mL)中のアルケニルスタンナン28c(250mg、0.34mmol)及び17a(142mg、0.409mmol)の溶液に、0℃で、チオフェンカルボン酸銅(97mg、0.51mmol)を加えた。反応を、0℃で45分間、室温で20分間撹拌した。次に、粗混合物を中性アルミナのプラグで濾過し、EtOAc/エーテル 50:50で洗浄し、合わせた濾液を0.5N HCl(3×10mL)で洗浄した。有機溶液を乾燥し、蒸発させて粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 10:1〜6:1)により精製して、トリエン29c(150mg、収率:67%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 8.21 (d, 1H, J = 10.8 Hz), 7.28 (t, 1H, J = 11.7 Hz), 6.88 (dd, 1H, J = 11.7, 11.4 Hz), 6.72 (dd, 1H, J = 10.2, 9.3 Hz), 6.42 (d, 1H, J = 8.4 Hz), 6.15 (d, 1H, J = 11.7 Hz), 5.66 (d, 1H, J = 11.4 Hz), 5.61 (dd, 1H, J = 5.7, 3.6 Hz), 5.56 (br t, 1H, J = 8.1 Hz), 5.27 (d, 1H, J = 9.9 Hz), 4.85-4.77 (m, 1H), 4.30 (dd, 1H, J = 8.1, 7.5 Hz), 4.24-4.16 (m, 1H), 3.79-3.72 (m, 1H), 3.66 (s, 3H), 2.88-2.80 (m, 1H), 2.42-2.37 (m, 2H), 2.18-2.14 (m, 5H), 2.00 (s, 3H), 1.83 (s, 3H), 1.14 (d, 3H J = 6.9 Hz), 0.97 (d, 3H, J = 6.6 Hz), 0.96 (d, 3H, J = 6.6 Hz), 0.86 (s, 9H), 0.4 (s, 6H).
13C-RMN (CDCl3, 75 MHz) δ: 169.2 166.8; 161.8; 145.4; 140.5, 137.7; 134.6; 134.3; 131.0, 124.3; 124.2, 122.6; 121.2; 108.6; 108.4; 82.0; 71.5; 58.9; 55.6; 37.5; 36.4; 33.8; 30.8, 26.5; 26.1; 21.3, 19.6, 18.5, 18.3, 17.4, 16.9, -4.3, -4.4.
【0206】
化合物29dの合成
【0207】
【化58】
【0208】
NMP(1mL)中のアルケニルスタンナン28d(60mg、0.08mmol)及び17a(32.4mg、0.09mmol)の溶液に、0℃で、チオフェンカルボン酸銅(22mg、0.12mmol)を加えた。反応を、0℃で45分間、室温で20分間撹拌した。次に、粗混合物を中性アルミナのプラグで濾過し、EtOAc/エーテル 50:50で洗浄し、合わせた濾液を0.5N HCl(3×10mL)で洗浄した。有機溶液を乾燥し、蒸発させて粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 4:1〜1:1)により精製して、トリエン29d(13mg、収率:25%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.59 (d, 1H, J = 11.1 Hz), 7.33-7.24 (m, 5H), 7.23 (t, 1H, J = 11.7 Hz), 6.90 (dd, 1H, J = 11.7, 11.4 Hz), 6.66 (dd, 1H, J = 10.5, 9 Hz), 6.24 (d, 1H, J = 7.2 Hz), 6.17 (d, 1H, J = 12.0 Hz), 5.63-5.58 (m, 2H), 5.51 (td, 1H, J = 7.8, 1.2 Hz), 5.28 (d, 1H, J = 10.8 Hz), 4.79-4.67 (m, 2H), 4.24-4.17 (m, 1H), 3.66 (s, 3H), 3.65-3.62 (m, 1H), 3.22 (dd, 1H, J = 13.5, 6.3 Hz), 3.04 (dd, 1H, J = 13.8, 8.4 Hz), 2.89-2.81 (m, 1H), 2.43-2.37 (m, 2H), 2.11-2.04 (m, 2H), 2.00 (s, 3H), 1.84 (s, 3H), 1.93-1.72 (m, 2H), 1.16 (d, 3H, J = 6.9 Hz), 0.86 (s, 9H), 0.03 (s, 3H), 0.01 (s, 3H).
13C NMR (CDCl3, 75 MHz) δ: 168.4, 166.5, 161.7, 145.5, 140.8, 138.1, 136.8, 134.5, 134.3, 131.0, 129.5, 129.1, 127.4, 124.2, 124.1, 122.3, 120.4, 108.7, 108.3, 82.0, 71.4, 55.7, 54.9, 38.3, 37.5, 36.6, 33.4, 26.5, 26.0, 21.3, 18.2, 17.4, 16.9, -4.3, -4.4.
MS (ES) m/z 711.2 [M+H]+
【0209】
化合物29eの合成
【0210】
【化59】
【0211】
NMP(1mL)中のアルケニルスタンナン28e(50mg、0.067mmol)及び17a(28mg、0.08mmol)の溶液に、0℃で、チオフェンカルボン酸銅(19.1mg、0.10mmol)を加えた。反応を、0℃で45分間、室温で20分間撹拌した。次に、粗混合物を中性アルミナのプラグで濾過し、EtOAc/エーテル 50:50で洗浄し、合わせた濾液を0.5N HCl(3×10mL)で洗浄した。有機溶液を乾燥し、蒸発させて粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 5:1〜1:1)により精製して、トリエン29e(33mg、収率:50%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.73 (d, 1H, J = 11.4 Hz), 7.70 (dd, 1H, J = 14.1, 11.7 Hz), 6.71 (dd, 1H J = 9.9, 9.7 Hz), 6.30 (d, 1H, J = 9.3 Hz), 6.13 (d, 1H, J = 12.9 Hz), 6.04 (dd, 1H, J = 11.7, 11.4 Hz), 5.93 (d, 1H, J = 15.0 Hz), 5.63 (br t, 1H, J = 4.5 Hz), 5.58-5.53 (m, 1H), 5.34 (d, 1H, J = 9.9 Hz), 4.85-4.78 (m, 1H), 4.41 (d, 1H, J = 9.3), 4.24-4.16 (m, 1H), 3.77-3.72 (m, 1H), 3.64 (s, 3H), 2.90-2.78 (m, 1H), 2.45-2.41 (m, 2H), 2.19-2.12 (m, 4H), 2.01 (s, 3H), 1.91 (s, 3H), 1.16 (d, 3H, J = 6.6 Hz), 1.02 (s, 9H), 0.87 (s, 9H), 0.06 (s, 3H), 0.04 (s, 3H).
13C NMR (CDCl3, 75 MHz) δ: 168.5, 166.1, 161.8, 145.4, 140.7, 138.0, 135.3, 134.9, 131.1, 125.8, 124.9, 124.0, 122.4, 108.7, 108.5, 81.9, 71.6, 60.9, 55.6, 37.6, 36.5, 35.2, 33.8, 29.9, 26.8, 26.1, 21.3, 18.3, 17.3, 16.9, -4.3, -4.4.
【0212】
化合物29fの合成
【0213】
【化60】
【0214】
NMP(0.9mL)中のアルケニルスタンナン28a(60mg、0.083mmol)及び17b(29mg、0.09mmol)の溶液に、0℃で、チオフェンカルボン酸銅(24mg、0.12mmol)を加えた。反応を、0℃で45分間、室温で20分間撹拌した。次に、粗混合物を中性アルミナのプラグで濾過し、EtOAc/エーテル 50:50で洗浄し、合わせた濾液を0.5N HCl(3×10mL)で洗浄した。有機溶液を乾燥し、蒸発させて粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 5:1〜1:1)により精製して、アミド29f(27mg、収率:50%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.62 (d, 1H, J = 10.5 Hz), 7.25 (dd, 1H, J = 12.6, 11.4 Hz), 6.94-6.84 (m, 2H), 6.73 (dd, 1H, J = 10.5, 9.0 Hz), 6.23 (d, 1H, J = 9.3 Hz), 6.17 (d, 1H, J = 11.4 Hz), 6.06-6.01 (m, 1H), 5.66 (d, 1H, J = 11.4 Hz), 5.60-5.55 (m, 1H), 5.29 (d, 1H, J = 9.9 Hz), 4.88-4.80 (m, 1H), 4.34 (d, 1H, J = 9.3 Hz), 4.27-4.19 (m, 1H), 3.79-3.72 (m, 1H), 2.90-2.81 (m, 1H), 2.36-2.30 (m, 2H), 2.21-2.13 (m, 4H), 2.03 (s, 3H), 1.85 (s, 3H), 1.17 (d, 3H, J = 6.6 Hz), 1.03 (s, 9H), 0.89 (s, 9H), 0.08 (s, 3H), 0.06 (s, 3H).
【0215】
化合物30aの合成
【0216】
【化61】
【0217】
THF(6mL)中の29a(275mg、0.41mmol)の溶液に、N2下及び室温で、THF(0.82mL、0.82mmol)中の1M TBAFを加えた。反応を室温で18時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、EtOAcで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製して、アルコール30a(175mg;収率:76%)を白色の固体として得た。
1H NMR (CDCl3, 300 MHz) δ: 9.00 (d, 1H, J = 10.2 Hz), 7.25 (dd, 1H, J = 12.0, 11.4 Hz), 6.86 (dd, 1H, J = 11.7, 11.4 Hz), 6.72 (dd, 1H, J = 9.6, 8.7 Hz), 6.68 (d, 1H, J = 8.7 Hz), 6.13 (d, 1H, J = 11.7 Hz), 5.68 (d, 1H, J = 11.4 Hz), 5.63-5.58 (m, 2H), 5.27 (d, 1H, J = 10.2 Hz), 4.85-4.76 (m, 1H), 4.42 (d, 1H, J = 9.3Hz), 4.25-4.17 (m, 1H), 3.70-3.69 (m, 1H), 3.63 (s, 3H), 3.48 (br s, 1H), 2.89-2.75 (m, 1H), 2.42-2.36 (m, 2H), 2.22-2.11 (m, 4H), 2.04 (s, 3H), 1.82 (s, 3H), 1.14 (d, 3H, J = 6.6 Hz), 1.03 (s, 9H).
【0218】
化合物30aの異性体(21S)−は、合成方法をラセミフラグメントDから出発して実施したときに得た。異性体〔(21S)−化合物30a及び(21R)−化合物30a〕の最終混合物を、半分取逆相HPLC(SymmetryPrep C18 7μm、7.8×150mm、MeCNが50〜60%の勾配H2O:MeCNを30分、UV検出、流速2.5mL/分、〔保持時間((21S−)30a):15.4分、保持時間((21R)−30a):14.7分〕)により分離し、(21S)−化合物30aを純粋な形態で得た。
【0219】
【化62】
【0220】
1H NMR (CDCl3, 300 MHz) δ: 8.62 (d, 1H, J = 10.2 Hz), 7.28-22 (m, 1H), 6.93-6.86 (m, 1H), 6.81-6.75 (m, 1H), 6.32 (d, 1H, J = 9.0 Hz), 6.17 (d, 1H, J = 11.7 Hz), 5.68-5.58 (m, 3H), 5.28 (d, 1H, J = 10.2 Hz), 4.93-4.84 (m, 1H), 4.32 (d, 1H, J = 9.3Hz), 4.25-4.17 (m, 1H), 3.78-3.67 (m, 1H), 3.66 (s, 3H), 2.89-2.81 (m, 1H), 2.43-2.38 (m, 2H), 2.28-2.20 (m, 4H), 2.08 (s, 3H), 1.84 (s, 3H), 1.16 (d, 3H J = 6.9 Hz), 1.02 (s, 9H).
【0221】
化合物30bの合成
【0222】
【化63】
【0223】
THF(7.5mL)中の29b(586mg、0.76mmol)の溶液に、N2下及び室温で、THF(1.53mL、2mmol)中の1M TBAFを加えた。反応を室温で18時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、EtOAcで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製して、アルコール30b(320mg、収率:80%)を白色の固体として得た。
1H NMR (CDCl3, 300 MHz) δ: 8.95 (d, 1H, J = 10.2 Hz), 7.25 (t, 1H, J = 12.0 Hz), 6.85 (t, 1H, J = 11.7 Hz), 6.73 (t, 1H, J = 9.6 Hz), 6.57 (d, 1H, J = 8.7 Hz), 6.12 (d, 1H, J = 11.4 Hz), 5.67 (d, 1H, J = 11.4 Hz), 5.61 (dd, 1H, J = 5.4, 3.9 Hz), 5.63-5.58 (m, 1H), 5.44-5.35 (m, 1H), 5.26 (d, 1H, J = 9.9 Hz), 4.86 (q, 1H, J = 8.1 Hz), 4.38 (d, 1H, J = 9.3 Hz), 4.24-4.16 (m, 1H), 3.81-3.71 (m, 1H), 3.64 (s, 3H), 2.96-2.92 (m, 1H), 2.86-2.79 (m, 1H), 2.41-2.37 (m, 2H), 2.28-2.14 (m, 4H), 1.82 (s, 3H), 1.61 (d, 3H, J = 6.6 Hz), 1.14 (d, 3H, J = 6.6 Hz), 1.02 (s, 9H).
13C-RMN (CDCl3, 75 MHz) δ: 168.7; 166.6; 161.8; 145.4; 140.3; 137.5; 134.4; 134.3; 127.7; 126.0; 124.4; 123.7; 121.1; 108.9; 108.4; 82.0; 72.1; 60.9; 55.7; 37.6; 35.0; 34.8; 33.2; 26.9; 26.5; 17.4; 16.9; 13.3.
【0224】
化合物30cの合成
【0225】
【化64】
【0226】
THF(4.8mL)中の29c(150mg、0.23mmol)の溶液に、N2下及び室温で、THF(0.45mL、0.45mmol)中の1M TBAFを加えた。反応を室温で18時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、EtOAcで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製して、アルコール30c(90mg、収率:73%)を白色の固体として得た。
1H NMR (CDCl3, 300 MHz) δ: 9.16 (d, 1H, J = 10.2 Hz), 7.26 (dd, 1H, J = 12.0, 11.1 Hz), 6.87 (dd, 1H, J = 11.7, 11.4 Hz), 6.79-6.70 (m, 2H), 6.14 (d, 1H, J = 11.7 Hz), 5.68 (d, 1H, J = 11.7 Hz), 5.63-5.58 (m, 2H), 5.27 (d, 1H, J = 9.6 Hz), 4.85-4.76 (m, 1H), 4.35 (dd, 1H, J = 8.4, 7.5 Hz), 4.24-4.17 (m, 1H), 3.70-3.69 (m, 1H), 3.63 (s, 3H), 3.43 (br s, 1H), 2.89-2.76 (m, 1H), 2.42-2.36 (m, 2H), 2.21-2.14 (m, 4H), 2.03 (s, 3H), 1.82 (s, 3H), 1.13 (d, 3H J = 6.9 Hz), 0.96 (d, 6H, J = 6.6 Hz).
13C-RMN (CDCl3, 75 MHz) δ: 169.7, 167.1, 161.8, 145.4, 140.5, 137.8, 134.6, 134.2, 131.6, 124.4, 123.9, 123.8, 120.7, 108.5, 108.4, 82.0, 59.1, 55.7, 37.5, 36.4, 33.5, 31.0, 26.5, 21.3, 19.5, 18.7, 17.4, 16.8.
MS (ES) m/z 549.0 [M+H]+, 571.1 [M+Na]+
【0227】
化合物30dの合成
【0228】
【化65】
【0229】
THF(0.32mL)中の29d(11mg、0.02mmol)の溶液に、N2下及び室温で、THF(0.03mL、0.03mmol)中の1M TBAFを加えた。反応を室温で18時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、EtOAcで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製して、アルコール30d(6mg、収率:65%)を白色の固体として得た。
1H NMR (CDCl3, 300 MHz) δ: 8.54 (d, 1H, J = 9.9 Hz), 7.34-7.23 (m, 5H), 7.20 (t, 1H, J = 11.7 Hz), 6.89 (dd, 1H, J = 11.7, 11.4 Hz), 6.72 (dd, 1H, J = 9.9, 9.3 Hz), 6.27 (d, 1H, J = 8.1 Hz), 6.17 (d, 1H, J = 11.7 Hz), 5.63-5.53 (m, 3H), 5.28 (d, 1H, J = 10.2 Hz), 4.84-4.76 (m, 1H), 4.75-4.68(m, 1H), 4.25-4.17 (m, 1H), 3.66 (s, 3H), 3.67-3.65 (m, 1H), 3.18 (dd, 1H, J = 13.8, 6.3 Hz), 3.06 (dd, 1H, J = 13.8, 8.1 Hz), 2.89-2.81 (m, 1H), 2.43-2.38 (m, 2H), 2.15-2.10 (m, 3H), 2.06 (s, 3H), 1.92-1.87 (m, 1H), 1.84 (s, 3H), 1.16 (d, 3H, J = 6.6 Hz).
13C NMR (CDCl3, 75 MHz) δ: 168.6, 166.6, 160.8, 145.8, 140.8, 138.1, 136.8, 134.6, 134.2 ,129.6, 129.0, 127.2, 124.1, 124.0, 123.4, 120.4, 108.3, 108.2, 82.0, 71.6, 55.7, 55.0, 38.4, 37.5, 36.4, 33.0, 26.5, 21.3, 17.4, 16.9.
MS (ES) m/z 597.2 [M+H]+.
【0230】
化合物30eの合成
【0231】
【化66】
【0232】
THF(1mL)中の29e(32mg、0.047mmol)の溶液に、N2下及び室温で、THF(0.094mL、0.094mmol)中の1M TBAFを加えた。反応を室温で18時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、EtOAcで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製して、アルコール30e(14mg、収率:55%)を白色の泡状物として得た。
1H NMR (CDCl3, 500 MHz) δ: 8.97 (d, 1H, J = 10.2 Hz), 7.71 (dd, 1H J = 14.7, 11.7 Hz), 6.74 (dd, 1H J = 9.3, 9.9 Hz), 6.57 (d, 1H, J = 9.0 Hz), 6.15 (d, 1H, J = 11.7 Hz), 6.03 (dd, 1H, J = 11.7, 11.4 Hz), 5.95 (d, 1H, J = 14.7 Hz), 5.65-5.58 (m, 2H), 5.35 (d, 1H, J = 9.9 Hz), 4.87-4.78 (m, 1H), 4.42 (d, 1H, J = 9.3), 4.25-4.18 (m, 1H), 3.72-3.68 (m, 1H), 3.65 (s, 3H), 3.25 (br s, 1H), 2.87-2.79 (m, 1H), 2.45-2.40 (m, 2H), 2.23-2.12 (m, 4H), 2.04 (s, 3H), 1.89 (s, 3H), 1.15 (d, 3H, J = 6.6 Hz).1.03 (s, 9H).
13C NMR (CDCl3, 75 MHz) δ: 168.8, 166.5, 161.68, 145.3, 140.9, 138.2, 135.4, 134.7, 132.0, 125.68, 124.6, 123.9, 123.6, 108.6, 108.4, 81.9, 71.7, 61.3, 55.7, 37.5, 36.5, 36.3, 34.9, 33.3, 26.9, 26.7, 21.3, 17.0, 16.7.
MS (ES) m/z 563.3 [M+H]+, 585.2 [M+Na]+
【0233】
化合物30fの合成
【0234】
【化67】
【0235】
THF(1mL)中の29f(28mg、0.04mmol)の溶液に、N2下及び室温で、THF(0.09mL、0.09mmol)中の1M TBAFを加えた。反応を室温で18時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、EtOAcで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製して、アルコール30f(17mg;収率:75%)を白色の固体として得た。
1H NMR (CDCl3, 300 MHz) δ: 9.05 (d, 1H, J = 10.2 Hz), 7.35 (m, 2H), 7.0 (dd, 1H, J = 11.7, 11.4 Hz), 6.73 (dd, 1H, J = 9.6, 8.7 Hz), 6.56 (d, 1H, J = 8.7 Hz), 6.05 (m, 3H), 5.63-5.58 (m, 2H), 5.30 (d, 1H, J = 10.2 Hz), 4.78 (m, 1H), 4.50 (d, 1H, J = 9.3Hz), 3.68 (m, 1H), 3.48 (br s, 1H), 2.45 (m, 1H), 2.42-2.36 (m, 2H), 2.22-2.11 (m, 4H), 2.04 (s, 3H), 1.82 (s, 3H), 1.14 (d, 3H J = 6.6 Hz), 1.03 (s, 9H).
【0236】
化合物1の合成
【0237】
【化68】
【0238】
ジクロロメタン(7.5mL)中の30a(300mg、0.53mmol)の溶液に、0℃で、トリクロロアセチルイソシアネート(TCAI)(76μl、0.64mmol)を加えた。反応を0℃で30分間撹拌し、次に中性酸化アルミニウムを加えた。混合物を5〜30分間撹拌し、次に酸化アルミニウムパッドに浸漬させた。生成物を、DCM/MeOH 50:1の混合物を使用して洗浄した。濾液を真空下で蒸発させて、粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 2:1〜1:2)により精製した。化合物1(0.26g、収率:81%)を白色の固体として得て、実施例2に報告されているものと同等の物理的特性及び分光特性(1H、13C RMN及びMS)を示した。
【0239】
化合物1の異性体(21S)−を、この2つの方法のいずれかにより得た:
A.−異性体(21R)−及び(21S)−化合物30aの混合物から出発し、半分取逆相HPLC(SymmetryPrep C18 7μm、7.8×150mm、MeOHが50〜100%の勾配H2O:MeOHを30分、UV検出、流速2.5mL/分、〔保持時間((21S)−1):19.2分、保持時間 ((21R)−1):19.8分〕)による(21S)−化合物1の最終分離と同じ手順に従う。
B.−化合物1について開示されたものと同じであるが、純粋な(21S)−化合物30aから出発する手順に従う。
【0240】
【化69】
【0241】
1H NMR (CDCl3, 500 MHz) δ: 8.69 (d, 1H, J = 10.5 Hz), 7.30 (t, 1H, J = 11.5 Hz), 6.90 (t, 1H, J = 11.5 Hz), 6.86-6.82 (m, 1H), 6.34 (d, 1H, J = 9.0 Hz), 6.17 (d, 1H, J = 11.5 Hz), 5.66 (d, 1H, J = 11.5 Hz), 5.64-5.62 (m, 1H), 5.59-5.56 (m, 1H), 5.29 (d, 1H, J = 9.5 Hz), 4.81-4.77 (m, 1H), 4.50-4.45 (m, 1H), 4.42 (d, 1H, J = 9.5 Hz), 4.25-4.20 (m, 1H), 3.66 (s, 3H), 2.89-2.81 (m, 1H), 2.44-2.31 (m, 5H), 2.24-2.17 (m, 1H), 2.06 (s, 3H), 1.84 (s, 3H), 1.16 (d, 3H, J = 6.5 Hz), 1.04 (s, 9H).
13C-RMN (125 MHz, CDCl3) δ: 168.4, 166.1, 157.2, 148.3, 145.2, 140.2, 137.4, 134.1, 134.0, 132.0, 124.7, 124.2, 122.4, 120.7, 108.1, 104.7, 81.8, 75.0, 60.8, 55.4, 37.2, 34.8, 32.5, 30.3, 26.7, 26.2, 21.0, 17.1, 16.6.
【0242】
化合物4の合成
【0243】
【化70】
【0244】
ジクロロメタン(1mL)中の30b(56mg、0.105mmol)の溶液に、0℃で、トリクロロアセチルイソシアネート(15μL、0.126mmol)を加えた。反応を0℃で30分間撹拌し、次に中性酸化アルミニウムを加えた。混合物を5〜30分間撹拌し、次に酸化アルミニウムパッドに浸漬させた。生成物を、DCM/MeOH 50:1の混合物を使用して洗浄した。濾液を真空下で蒸発させて、粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製した。化合物4(57.6mg、収率:96%)を白色の泡状物として得て、実施例3に報告されているものと同等の物理的特性及び分光特性(1H、13C RMN及びMS)を示した。
【0245】
化合物5の合成
【0246】
【化71】
【0247】
ジクロロメタン(2mL)中の30c(115mg、0.21mmol)の溶液に、0℃で、トリクロロアセチルイソシアネート(27μl、0.23mmol)を加えた。反応を0℃で30分間撹拌し、次に中性酸化アルミニウムを加えた。混合物を5〜30分間撹拌し、次に酸化アルミニウムパッドに浸漬させた。生成物を、DCM/MeOH 50:1の混合物を使用して洗浄した。濾液を真空下で蒸発させて、粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製した。化合物5(71mg、収率:57%)を白色の泡状物として得て、実施例3に報告されているものと同等の物理的特性及び分光特性(1H、13C RMN及びMS)を示した。
【0248】
異性体の最終混合物(15mg)を、半分取逆相HPLC(SymmetryPrep C18 7μm、7.8×150mm、MeOHが50〜70%の勾配H2O:MeOHを75分、UV検出、流速2.5mL/分、〔保持時間((15R)−7):18.15分、保持時間((15S)−7):19.62分〕)により分離し、3.1mgの純粋な(15R)−化合物7及び2.9mgの純粋な(15S)−化合物7を得た。
【0249】
化合物5の異性体(21S)−は、5を半逆相HPLC(SymmetryPrep C8、MeCNが45〜50%の勾配H2O:MeCNを30分、UV検出、流速4.7mL/分〔保持時間((21−S)−5):21.6分、保持時間((21R)−5):23.6分〕)により精製したときに単離した。両方の異性体を含有する試料(50mg)から出発して、上記の分離の後、39mgの純粋な(21R)−化合物5及び6.1mgの純粋な(21S)−化合物5を得た。
【0250】
【化72】
【0251】
1H NMR (CDCl3, 500 MHz) δ: 8.74 (d, 1H, J = 10.5 Hz), 7.29 (dd, 1H, J = 11.7, 11.4 Hz), 6.94 (dd, 1H, J = 11.7, 11.4 Hz), 6.84 (dd, 1H, J = 10.5, 9.3 Hz), 6.22 (m, 2H), 5.68 (d, 1H, J = 11.5 Hz), 5.63 (m, 2H), 5.42 (d, 1H, J = 9.3 Hz), 4.81 (m, 1H), 4.52 (m, 1H), 4.41 (m, 1H), 4.23 (m,1H), 3.66 (s, 3H), 2.91 (m, 1H), 2.49-2.38 (m, 3H), 2.35-2.31(m, 2H), 2.24-2.17 (m, 2H), 2.05 (s, 3H), 1.82 (s, 3H), 1.15 (d, 3H J = 6.6 Hz), 0.99 (d, 3H, J = 6.9 Hz) 0.96 (d, 3H, J = 6.9 Hz).
MS (ES) m/z 592.3 [M+H]+
【0252】
化合物31の合成
【0253】
【化73】
【0254】
ジクロロメタン(0.7mL)中の30d(5mg、0.008mmol)の溶液に、0℃で、トリクロロアセチルイソシアネート(1.1μl、0.009mmol)を加えた。反応を0℃で30分間撹拌し、次に中性酸化アルミニウムを加えた。混合物を5〜30分間撹拌し、次に酸化アルミニウムパッドに浸漬させた。生成物を、DCM/MeOH 50:1の混合物を使用して洗浄した。濾液を真空下で蒸発させて、粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製した。化合物31(3.5mg、収率:66%)を白色の固体として得た。
1H NMR (CDCl3, 300 MHz) δ: 8.43 (d, 1H, J = 10.5 Hz), 7.29-7.19 (m, 6H,), 6.91 (dd, 1H, J = 11.7, 11.4 Hz), 6.77 (br dd, 1H, J = 10.2, 9.6 Hz), 6.51 (d, 1H, J = 8.1 Hz), 6.16 (d, 1H, J = 11.4 Hz), 5.69-5.63 (m, 2H), 5.59-5.54 (m, 1H), 5.31 (d, 1H, J = 9.3 Hz), 5.12 (br s, 1H,), 4.91-4.84 (m, 1H), 4.76-4.70 (m, 1H), 4.31-4.13 (m, 2H, CH-5), 3.66 (s, 3H), 3.20 (dd, 1H, J = 13.5, 6.9 Hz), 3.09 (dd, 1H, J = 13.5, 6.6 Hz), 2.89-2.81 (m, 1H), 2.48-2.35 (m, 2H), 2.28-2.23 (m, 3H), 2.05 (s, 3H), 2.01-1.90 (m, 1H), 1.81 (s, 3H), 1.15 (d, 3H, J = 6.6 Hz).
13C NMR (CDCl3, 75 MHz) δ: 168.9, 166.5, 161.9, 157.1, 145.4, 140.6, 137.9, 136.4, 134.4, 133.9, 132.1, 129.7, 128.8, 127.2, 124.6, 124.6, 122.7, 120.7, 108.5, 105.6, 82.1, 74.8, 55.7, 54.6, 38.7, 37.2, 33.1, 30.5, 26.2, 21.2, 17.3, 16.4.
【0255】
化合物8の合成
【0256】
【化74】
【0257】
ジクロロメタン(1.7mL)中の30e(13mg、0.023mmol)の溶液に、0℃で、トリクロロアセチルイソシアネート(3μl、0.025mmol)を加えた。反応を0℃で30分間撹拌し、次に中性酸化アルミニウムを加えた。混合物を5〜30分間撹拌し、次に酸化アルミニウムパッドに浸漬させた。生成物を、DCM/MeOH 50:1の混合物を使用して洗浄した。濾液を真空下で蒸発させて、粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製した。化合物8(14.3mg、収率:100%)を白色の固体として得て、実施例4に報告されているものと同等の物理的特性及び分光特性(1H、13C RMN及びMS)を示した。
【0258】
実施例9
スキーム6は、本発明の幾つかの化合物の合成プロセスを示す。
【0259】
【化75】
【0260】
中間体32の合成
【0261】
【化76】
【0262】
アルゴン雰囲気、−78℃で撹拌した、無水THF(59mL)中の4−ホスホノクロトン酸トリエチル(3.7g、14.66mmol)及び18−クラウン−6(6.2g、23.46mmol)の溶液に、KHMDS(28.1ml、14.07mmol)を滴加した。15分後、アルデヒド12(2.35g、5.86mmol)を滴加し、室温で20時間撹拌した。次に、反応を飽和NH4Cl溶液(200mL)で停止させ、EtOAcで希釈した。有機相を乾燥し(Na2SO4)、減圧下で蒸発させた。カラムクロマトグラフィー(ヘキサン/EtOAc 20:1〜10:1)による精製によって、2.7g(収率:93%)のトリエン32を得た。
1H-RMN (300 MHz, CDCl3) δ: 7.31 (dd, 1H, J = 11.2, 15.3 Hz), 6.53 (d, 1H, J = 15.0 Hz), 6.21 (dd, 1H, J = 11.7, 13.8 Hz), 5.84 (d, 1H, J = 15.1 Hz), 5.61 (d, 1H, J = 9.6 Hz), 4.17 (m, 2H), 3.72 (m, 1H), 3.63 (m, 2H), 2.61 (m, 1H), 1.78 (s, 3H), 1.67 (m, 2H), 1.26 (m, 3H), 0.94 (d, 3H, J = 6.7 Hz), 0.87 (s, 18H), 0.01 (m, 12H).
【0263】
中間体33の合成
【0264】
【化77】
【0265】
EtOH(38mL)中の32(3.75g、7.54mmol)の溶液に、p-トルエンスルホン酸ピリジニウム(663mg、2.64mmol)を加えた。反応混合物を室温で17時間撹拌した。次に、溶媒を減圧下で除去し、得られた油状物をカラムクロマトグラフィー(ヘキサン/EtOAc 4:1〜1:1)により精製して、2.11g(収率:73%)のアルコール33を得た。
1H-RMN (300 MHz, CDCl3) δ: 7.31 (dd, 1H, J = 10.8, 15.0 Hz), 6.52 (d, 1H, J = 15.3 Hz), 6.23 (dd, 1H, J = 11.1, 15.0 Hz), 5.86 (d, 1H, J = 15.3 Hz), 5.52 (d, 1H, J = 9.9 Hz), 4.18 (q, 2H, J = 7.5 Hz), 3.72 (m, 3H), 2.73 (m, 1H), 1.82 (s, 3H), 1.68 (m, 2H), 1.28 (t, 3H, J = 7.2 Hz), 0.98 (d, 3H, J = 6.6 Hz), 0.88 (s, 9H), 0.08 (m, 6H).
【0266】
中間体34の合成
【0267】
【化78】
【0268】
アルコール33(130mg、0.34mmol)をDCM(3.4mL)中、不活性雰囲気下、室温で撹拌し、ペルヨージナン(DMP)(288.5mg、0.68mmol)を一度に加えた。反応を、完了するまで(TLC、約1時間)撹拌し、次に、NaHCO3(飽和溶液)で停止させ、DCMで抽出し、ブラインで洗浄し、硫酸マグネシウムで乾燥し、濾過し、真空下で濃縮した。生成物を、EtOAc/ヘキサン 1:4を用いて溶離するカラムクロマトグラフィーにより精製して、約125mg(収率:96%)のアルデヒド34を無色の油状物として得た。
1H-RMN (300 MHz, CDCl3) δ: 9.79 (s, 1H), 7.31 (dd, 1H, J = 11.1, 15.3 Hz), 6.52 (d, 1H, J = 15.3 Hz), 6.25 (dd, 1H, J = 11.1, 15.3 Hz), 5.87 (d, 1H, J = 15.3 Hz), 5.48 (d, 1H, J = 10.5 Hz), 4.19 (q, 2H, J = 7.2 Hz), 4.03 (m, 1H), 2.69 (m, 1H), 2.54 (m, 2H), 1.80 (s, 3H), 1.29 (t, 3H, J = 6.9 Hz), 1.01 (d, 3H, J = 6.9 Hz), 0.88 (s, 9H), 0.06 (m, 6H).
【0269】
中間体35の合成
【0270】
【化79】
【0271】
アルゴン雰囲気、−78℃で撹拌した、無水THF(10mL)中のホスホネート(170mg、0.67mmol)及び18−クラウン−6(357mg、1.35mmol)の溶液に、KHMDS(1.34ml、0.67mmol)を滴加した。15分後、無水THF(8.5mL)中のアルデヒド34(170mg、0.45mmol)の溶液を30分間かけて滴加し、−78℃で90分間撹拌した。次に、反応を飽和NH4Cl溶液で停止させ、室温に温め、ジクロロメタンで希釈した。有機相を乾燥し(Na2SO4)、減圧下で蒸発させた。カラムクロマトグラフィー(ヘキサン/EtOAc 20:1〜10:2)による精製によって、170mg(収率:82%)の(E)−35を得た。
1H-RMN (300 MHz, CDCl3) δ: 7.29 (dd, 1H, J = 10.8, 15.3 Hz), 6.50 (d, 1H, J = 15.3 Hz), 6.19 (dd, 1H, J = 10.8, 15.0 Hz), 5.83 (d, 1H, J = 15.3 Hz), 5.48 (d, 1H, J = 10.2 Hz), 5.33 (t, 1H, J = 7.2 Hz), 4.17 (m, 2H), 3.71 (s, 3H), 3.61 (m, 1H), 3.58 (s, 3H), 2.73 (m, 1H), 2.57 (m, 2H), 1.71 (s, 3H), 1.25 (m, 3H), 0.97 (d, 3H, J = 6.7 Hz), 0.88 (s, 9H), 0.03 (m, 6H).
【0272】
中間体36の合成
【0273】
【化80】
【0274】
LiOH(15.8mg、0.66mmol)を、20%水/ジオキサン(7mL)中のエステル35(140mg、0.30mmol)の溶液に加え、混合物を60℃で4時間撹拌した。混合物を冷却し、DCMで希釈し、HCl(0.5N、10mL)で洗浄した。水相をDCMで繰り返し抽出し、合わせた有機層をNa2SO4で乾燥し、濾過し、蒸発させて、粗二酸を得て、それを更に精製することなく次の工程に使用した。
濃HCl(43μL)を、MeOH(5.2mL)中の粗物質の溶液に加え、得られた混合物を室温で1時間撹拌した。次に、溶媒を減圧下で除去し、得られた油状物をカラムクロマトグラフィー(ヘキサン/EtOAc 1:4からEtOAc/MeOH 5:1)により精製して、72mg(収率:70%)の酸36を無色の油状物として得た。
1H-RMN (300 MHz, CDCl3) δ: 7.40 (dd, 1H, J = 10.8, 15.0 Hz), 6.57 (d, 1H, J = 15.0 Hz), 6.31 (dd, 1H, J = 11.4, 15.3 Hz), 5.89 (d, 1H, J = 15.0 Hz), 5.60 (m, 1H), 5.52 (d, 1H, J = 10.2 Hz), 4.22 (m, 1H), 3.64 (s, 3H), 2.90 (m, 1H), 2.38 (m, 2H), 1.84 (s, 3H), 1.15 (d, 3H, J = 6.9 Hz).
【0275】
化合物37の合成
【0276】
【化81】
【0277】
DCM/DMF(10:1、1.3mL)中のアミン27a(37.6mg、0.093mmol)の溶液に、無水DCM中の酸36(30mg、0.103mmol)の溶液を、アルゴン雰囲気下で加え、次に0℃に冷却した。DIPEA(26μL、0.14mmol)、HOAt(12.7mg、0.093mmol)及びHATU(35.4mg、0.093mmol)を溶液に加え、30分後、冷浴を取り外した。反応混合物を室温で3時間撹拌し、NH4Clの飽和水溶液で停止させ、水に注ぎ、DCMで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 2:1〜1:4)により精製して、アミド37(34.7mg、収率:55%)を油状物として得た。
1H NMR (CDCl3, 500 MHz) δ: 7.88 (d, 1H J = 10.5 Hz), 7.26 (dd, 1H, J = 14.4, 11.4 Hz), 6.72 (dd, 1H, J = 9.9, 9.6 Hz), 6.50 (d, 1H, J = 15.3 Hz), 6.31-6.22 (m, 2H), 5.94 (d, 1H, J = 14.7 Hz), 5.61-5.54 (m, 2H), 5.44 (d, 1H, J = 9.9 Hz), 4.87-4.79 (m, 1H), 4.45 (d, 1H, J = 9.3), 4.24-4.16 (m, 1H), 3.77-3.44 (m, 1H), 3.64 (s, 3H), 2.96-2.2.81 (m, 1H), 2.39-2.35 (m, 2H), 2.16-2.15 (m, 4H), 2.01 (s, 3H), 1.82 (s, 3H), 1.14 (d, 3H, J = 6.6 Hz).1.02 (s, 9H), 0.87 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H).
【0278】
化合物38の合成
【0279】
【化82】
【0280】
THF(0.6mL)中の37(28mg、0.04mmol)の溶液に、N2下及び室温で、THF(83μL、0.08mmol)中の1M TBAFを加えた。反応を室温で18時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、EtOAcで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製して、アルコール38(22mg、収率:96%)を白色の固体として得た。
1H NMR (CDCl3, 500 MHz) δ: 9.06 (d, 1H, J = 9.9 Hz), 7.24 (dd, 1H, J = 14.7, 10.5 Hz), 6.76 (d, 1H, J = 9.6 Hz), 6.74 (dd, 1H, J = 9.6, 9.6 Hz), 6.50 (d, 1H, J = 15.3 Hz), 6.25 (dd, 1H, J = 15.3, 11.1 Hz), 5.99 (d, 1H, J = 14.7 Hz), 5.65-5.60 (m, 2H), 5.45 (d, 1H, J = 9.9 Hz), 4.87-4.81 (m, 1H), 4.45 (d, 1H, J = 9.3), 4.24-4.16 (m, 1H), 3.74-3.64 (m, 1H), 3.64 (s, 3H), 3.22-3.17 (m, 1H), 2.95-2.2.82 (m, 1H), 2.40-2.35 (m, 2H), 2.23-2.16 (m, 4H), 2.05 (s, 3H), 1.81 (s, 3H), 1.13 (d, 3H, J = 6.6 Hz), 1.03 (s, 9H).
【0281】
化合物3の合成
【0282】
【化83】
【0283】
ジクロロメタン(0.35ml)中の化合物38(20mg、0.04mmol)の溶液に、0℃で、トリクロロアセチルイソシアネート(5.1μL、0.04mmol)を加えた。反応を0℃で30分間撹拌し、次に中性酸化アルミニウムを加えた。混合物を5〜30分間撹拌し、次に酸化アルミニウムパッドに浸漬させた。生成物を、DCM/MeOH 50:1の混合物を使用して洗浄した。濾液を真空下で蒸発させて、粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 1:1〜1:3)により精製した。化合物3(13.6mg、収率:63%)を白色の泡状物として得て、実施例3に報告されているものと同等の物理的特性及び分光特性(1H、13C RMN及びMS)を示した。
【0284】
実施例10
スキーム7は化合物7の合成プロセスを示す。
【0285】
【化84】
【0286】
中間体40の合成
【0287】
【化85】
【0288】
MeOH中のBoc−tert−LeuCONH2 39(ポズネフ(Pozdnev)V.F.、テトラヘドロン・レターズ(Tetrahedron Letters)1995年、36、7115〜7118に記載された手順に従って調製)(1.9g、8.26mmol)の溶液に、MeOH中1.25MのHClの溶液66mLを加えた。反応混合物を室温で2時間撹拌し、次に減圧下で濃縮した。次に固体をMeOHに懸濁し、濃水酸化アンモニウムで中和した。溶媒をロータリー蒸発で除去し、粗物質をジクロロメタンに溶解した。溶液をNa2SO4で乾燥し、濾過し、濃縮して、730mg(収率:68%)の中間体40を白色の固体として得た。
1H NMR (CDCl3, 300 MHz) δ: 6.65-6.55 (br s, 1H), 5.65-5.55 (br s, 1H), 3.18 (s, 1H), 1.65-1.55 (br s, 2H), 1.05 (s, 9H).
【0289】
中間体41の合成
【0290】
【化86】
【0291】
DCM/DMF(10:1、7.7mL)中のアミン40(100mg、0.77mmol)の溶液に、無水DCM中の(Z)−3−トリブチルスタンニルプロペン酸(306mg、0.85mmol)の溶液を、アルゴン雰囲気下で加え、次に0℃に冷却した。DIPEA(0.27mL、1.54mmol)、HOAt(115.6mg、0.85mmol)及びHATU(323mg、0.85mmol)を溶液に加え、30分後、冷浴を取り外した。反応混合物を室温で2時間撹拌し、NH4Clの飽和水溶液で停止させ、水に注ぎ、DCMで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 10:1〜1:1)により精製して、アミド41(228mg、収率:63%)を白色の泡状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 6.99 (d, 1H, J = 12.3 Hz), 6.77 (d, 1H, J = 12.3 Hz), 6.47 (d, 1H, J = 9.6 Hz), 6.38 (br s, 1H), 6.12 (br s, 1H), 4.46 (d, 1H, J = 9.9 Hz), 1.48-1.40 (m, 6H), 1.31-1.19 (m, 12H), 1.00 (s, 9H), 0.89-0.83 (m, 9H).
13C NMR (CDCl3, 75 MHz) δ: 173.4, 166.6, 153.4, 136.6, 60.0, 35.0, 29.6, 27.6, 26.8, 14.0, 11.7.
【0292】
化合物7の合成
【0293】
【化87】
【0294】
NMP(5mL)中のアルケニルスタンナン41(227.6mg、0.48mmol)及び17a(200.9mg、0.58mmol)の溶液に、0℃で、チオフェンカルボン酸銅(19.1mg、0.10mmol)を加えた。反応を、0℃で45分間、室温で20分間撹拌した。次に、粗混合物を中性アルミナのプラグで濾過し、EtOAc/エーテル 50:50で洗浄し、合わせた濾液を0.1%のHClで洗浄した。有機溶液を乾燥し、蒸発させて粗生成物を得て、それをカラムクロマトグラフィー(ジクロロメタン/MeOH 100:1〜10:1)により精製して、化合物7(65.7mg、収率:34%)を白色の泡状物として得た。この合成生成物は、実施例3に報告されているものと同等の物理的特性及び分光特性(1H、13C RMN及びMS)を示した。
【0295】
化合物7の異性体(15R)−は、アミノ酸(Boc−tert−LeuCONH2)のラセミ混合物を使用してこれらの反応を実施したときに得た。異性体の最終混合物(15mg)を、半分取逆相HPLC(SymmetryPrep C18 7μm、7.8×150mm、MeOHが50〜70%の勾配H2O:MeOHを75分、UV検出、流速2.5mL/分、〔保持時間((15R)−7):18.15分、保持時間((15S)−7):19.62分〕)により分離し、3.1mgの純粋な(15R)−化合物7及び2.9mgの純粋な(15S)−化合物7を得た。
【0296】
【化88】
【0297】
1H NMR (MeOD, 500 MHz) δ: 7.23 (dd, 1H, J = 12.5, 11.5 Hz), 6.94 (dd, 1H, J = 12.5, 11.5 Hz), 6.18 (d, 1H, J = 12.0 Hz), 5.91-5.88 (m, 1H), 5.86 (t, 1H, J = 4.5 Hz), 5.34 (d, 1H, J = 10.0 Hz), 4.33-4.28 (m, 1H), 3.64 (s, 3H), 2.92-2.88 (m, 1H),2.47-2.44 (m, 2H), 1.85 (s, 3H), 1.17 (d, 3H, J = 6.6 Hz ), 1.02 (s, 9H).
13C NMR (MeOD, 125 MHz) δ: 175.3, 168.6, 164.0, 146.0, 140.8, 138.2, 135.4, 135.4, 125.5, 121.9, 111.1, 83.5, 61.8, 55.9, 38.4, 35.0, 27.2, 27.1, 17.3, 16.8.
【0298】
実施例11
スキーム8は、化合物42及び43を得る方法を示す。
【0299】
【化89】
【0300】
化合物42の合成
【0301】
【化90】
【0302】
ピリジン(0.45mL)中のアルコール30a(5mg、8.8μmol)の溶液に、フェニルイソシアネート(29mL、0.27mmol)を加えた。反応を室温で20時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、EtOAcで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 20:1〜1:1)により精製して、化合物42(2.7mg、収率:44%)を白色の固体として得た。
1H NMR (CDCl3, 300 MHz) δ: 8.81-8.77 (m, 1H), 7.46-7.43 (m, 2H), 7.34-7.23 (m,3H), 7.11-7.06 (m, 2H), 6.88-6.77 (m, 2H), 6.39 (d, 1H, J = 9.9 Hz), 6.10 (d, 1H, J = 11.7 Hz), 5.67-5.57 (m, 3H), 5.27 (d, 1H, J = 9.9 Hz), 4.85-4.78 (m, 1H), 4.62-4.56 (m, 1H), 4.54 (d, 1H, J = 9.3 Hz), 4.25-4.17 (m, 1H), 3.66 (s, 3H), 2.87-2.80 (m, 1H), 2.41-2.38 (m, 5H), 2.21-2.13 (m, 1H), 2.08 (s, 3H), 1.82 (s, 3H), 1.16 (d, 3H J = 6.6 Hz), 1.07 (s, 9H).
【0303】
化合物43の合成
【0304】
【化91】
【0305】
アルコール30a(5mg、8.88μmol)をDCM(0.1mL)中、不活性雰囲気下、室温で撹拌し、ペルヨージナン(7.5mg、0.018mmol)を一度に加えた。反応を、完了するまで(TLC、約1時間)撹拌し、次に、NaHCO3(飽和溶液)で停止させ、DCMで抽出し、ブラインで洗浄し、硫酸マグネシウムで乾燥し、濾過し、真空下で濃縮した。生成物を、EtOAc/ヘキサン 1:1を用いて溶離するカラムクロマトグラフィーにより精製して、約4.5mg(収率:90%)のケトン43を無色の油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 8.32 (d, 1H, J = 9.9 Hz), 7.31-7.23 (m, 1H), 6.91 (dd, 1H, J = 11.7, 11.4 Hz), 6.84 (dd, 1H, J = 9.9, 8.7 Hz), 6.26 (d, 1H, J = 8.7 Hz), 6.18 (d, 1H, J = 11.4 Hz), 5.78-5.73 (m, 1H), 5.68 (d, 1H, J = 11.4 Hz), 5.64-5.61 (m, 1H), 5-30-5.27 (m, 1H), 4.94-4.86 (m, 1H), 4.41 (d, 1H, J = 9.0 Hz), 4.25-4.17 (m, 1H), 3.66 (s, 3H), 3.18 (dd, 4H, J = 18.3, 7.2 Hz), 2.89-2.75 (m, 1H), 2.44-2.38 (m, 2H), 2.04 (s, 3H), 1.84 (s,5 3H), 1.16 (d, 3H J = 6.9 Hz), 1.05 (s, 9H).
【0306】
実施例12:抗腫瘍活性の検出のためのバイオアッセイ
このアッセイの目的は、試験する試料のインビトロでの細胞増殖抑制性(腫瘍細胞増殖を遅延又は阻止する能力)又は細胞毒性(腫瘍細胞を死滅させる能力)の活性を評価することである。
【0307】
細胞株
【0308】
【表9】
【0309】
SBR比色アッセイを使用する細胞毒性活性の評価
スルホローダミンB(SRB)反応を使用する比色型のアッセイは、細胞増殖及び生存度の定量的測定のために採用されてきた(スケハン(Skehan)Pら、国立癌研究所ジャーナル(J. Natl. Cancer Inst.)1990年、82、1107〜1112に記載されている教示に従う)。
【0310】
この形態のアッセイは、SBS標準96ウエル細胞培養マイクロプレートを用いる(フェアクロス(Faircloth)ら、細胞科学における方法(Methods in cell science)、1988年、11(4)、201〜205;モスマン(Mosmann)ら、免疫学方法ジャーナル(Journal of. Immunological. Methods)1983年、65(1〜2)、55〜63)。異なる型のヒト癌に由来する、この研究に使用した全ての細胞株は、アメリカン・タイプ・カルチャー・コレクション(ATCC)から得た。
【0311】
細胞を、10%ウシ胎児血清(FBS)、2mMのL−グルタミン、100U/mLのペニシリン及び100U/mLのストレプトマイシンを補充したダルベッコー修飾イーグル培地(DMEM)に、37℃、5%CO2及び98%湿度で維持した。実験のために、細胞を、トリプシン処理を使用してサブコンフルエント培養から採取し、カウント及び平板培養する前に、新たな培地に再懸濁した。
【0312】
細胞を、150μLのアリコートにより1ウエルあたり5×103細胞で95ウエルマイクロタイタープレートに接種し、薬剤無含有培地において、18時間かけてプレート表面に結合させた。それぞれの細胞株に1つの対照(未処理)プレートを固定し(下記に記載するように)、0時間基準値として使用した。その後、試験試料を、50μLのアリコートにより、10〜0.00262μg/mLの範囲で10個の段階希釈で培養に加えた。48時間の暴露後、抗腫瘍効果をSRB法により推定した:簡潔には、細胞をPBSで2回洗浄し、1%グルタルアルデヒド溶液で15分間固定し、PBSで2回すすぎ、0.4%SRB溶液により室温で30分間染色した。次に細胞を1%酢酸溶液で数回すすぎ、風乾した。次にSRBを10mMトリズマ塩基溶液で抽出し、自動分光光度プレート読取機により490nmで吸光度を測定した。細胞生存は、対照細胞増殖の百分率として表した。試験した試料の最終効果は、NCIアルゴリズム(ボイド(Boyd)MR及びポール(Paull)KD、薬剤開発研究(Drug Dev. Res.)1995年、34,91〜104)を適用することにより評価した。
【0313】
三重培養の平均±SDを使用して、用量反応曲線を非線形回帰分析を使用して自動的に生成した。3つの基準パラメーターを自動補間法により計算した(NCIアルゴリズム):GI50=50%の増殖阻害を生じる濃度;TGI=総増殖阻害(細胞増殖抑制効果)及びLC50=50%の純細胞死滅(細胞毒性効果)を生じる濃度。
【0314】
抗有糸分裂アッセイプロトコール
細胞培養の有糸分裂率(有糸分裂が阻止された細胞の百分率)を、特異的有糸分裂マーカーを定量的に検出する特異的96ウエルマイクロプレート免疫アッセイを使用して評価した。HeLa細胞(h−子宮頸癌、ATCC番号CCL−2)を、試験する試料の存在下又は不在下で18時間インキュベートした。その後、細胞をPBSで洗浄し、75μLの新たに調製した溶解緩衝剤(1mMのEGTA(pH7.5)、0.5mMのPMSF及び1mMのNaVO3)中の氷により30分間溶解した。細胞抽出物のアリコート(60μL)を、高結合表面ELISAプレートに移し、speed-vacにより室温で2時間乾燥した。次にプレートを、100μLのPBS−1%BSAにより30℃で30分間ブロックし、抗MPM2一次マウスモノクローナル抗体(アップステート・バイオテクノロジー(Upstate Biotechnology)、カタログ番号05−368)により4℃で18時間及び適切なペルオキシダーゼ結合二次抗体により30℃で1時間連続的にインキュベートした。0.02%のツイーン20による集中的な洗浄の後、ペルオキシダーゼ反応を、30μLのTMB(3,3′,5,5′−テトラメチル−ベンジジン)を使用して30℃で30分間実施した。反応を、30μLの4%H2SO4溶液を加えることにより停止させた。アッセイを、マイクロプレート分光光度計により450nmでO.D.を測定することにより定量化した。結果を、未処理培養の対照と比較して、処理細胞培養において50%の有糸分裂阻止を生じた試料濃度をIC50として表した。
【0315】
表9及び10は、本発明の化合物の生物学的活性についてのデータを例示する。
【0316】
【表10】
【表11】
【技術分野】
【0001】
本発明は、新規の抗腫瘍化合物、それらを含有する医薬組成物及び抗腫瘍剤としてのそれらの使用に関する。
【背景技術】
【0002】
1990年、グナセケラ(Gunasekera)SPらは、カリブ海の深層水海綿体であるディスコデルミア・ディソリュータ(Discodermia dissoluta)から新規なポリヒドロキシル化ラクトンである(+)−ディスコデルモリドの単離を報告した(グナセケラSPら、有機化学ジャーナル(J. Org. Chem.)1990年、55、4912〜4915及び有機化学ジャーナル1991年、56、1346)。
【0003】
【化1】
【0004】
この化合物は、臨床的に実証されている抗癌剤であるパクリタセキル(シフ(Schiff)ら、ネイチャー(Nature)1979年、277、665〜667)と同様の作用様式を有する、強力な有糸分裂阻害剤であることが明らかにされている(ハング(Hung)DTら、生物化学(Chem. Biol.)1996年、3、287〜293及びテル・ハール(ter Haar)Eら、生化学(Biochemistry)1996年、35、243〜250)。天然生成物は、両方とも、M期で細胞周期を停止させ、微小管形成を促進し、乳癌癌腫に対して同様の阻害効果を有する(それぞれ、2.4nM及び2.1nMのIC50)
【0005】
一方、N−アシルエンアミド官能基を含有する幾つかの特異な直鎖ジペプチドが、コンドロマイセス(Chondromyces)属に属する粘液細菌から単離されている(クンツ(Kunze)Bら、抗生物質ジャーナル(J. Antibiot.)1994年、47、881〜886及びヤンセン(Jansen)Rら、有機化学ジャーナル(J. Org. Chem.)1999年、1085〜1089)。特に、これらの化合物は、クロカシンA、B、C及びDであり、電子伝達インヒビターの群である。
【0006】
【化2】
【0007】
クロカシンA〜Dは、少数のグラム陽性細菌の増殖を適度に阻害し、動物細胞培養、並びに幾つかの酵母菌及び真菌の強力なインヒビターである。最も活性なものは、真菌のサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)に対して1.4ng/mLのMICを示し、L929マウス線維芽細胞培養に対して強力な毒性(0.06mg/LのIC50)を示した、クロカシンDである。
【0008】
癌は、動物及びヒトにおける死亡の主な原因である。癌に罹患している患者への投与に活性であり安全である抗腫瘍剤を得るために、多大な努力が今まで払われており、依然として払われている。本発明により解決される問題は、癌の治療に有用な化合物を提供することである。
【発明の概要】
【課題を解決するための手段】
【0009】
一つの態様において、本発明は、一般式Iの化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体を対象とし、
【0010】
【化3】
【0011】
式中、
R1は、水素、ORa、OCORa、OCOORa、NRaRb、NRaCORb及びNRaC(NRa)NRaRbから選択され;
R2及びR3は、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル及び置換又は非置換C2〜C12アルキニルから選択され;
R41、R42、R43、R44、R45、R46、R47及びR48は、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル及び置換又は非置換C2〜C12アルキニルから選択され;
R5、R6及びR7は、それぞれ独立して、水素、CORa、COORa、置換若しくは非置換C1〜C12アルキル、置換若しくは非置換C2〜C12アルケニル及び置換若しくは非置換C2〜C12アルキニルから選択されるか、又はR5及びR48は、それらが結合している対応するN原子及びC原子と一緒になって、置換若しくは非置換複素環式基を形成してもよく;
Ra及びRbは、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル、置換又は非置換アリール及び置換又は非置換複素環式基から選択され;そして
点線は、それぞれ任意の追加的な結合を表す。
【0012】
別の態様において、本発明は、薬剤として、特に癌を治療するための薬剤として使用される、式Iの化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体を対象とする。
【0013】
更なる態様において、本発明は、また、癌の治療における又は好ましくは癌の治療のための薬剤の調製における、式Iの化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体の使用を対象とする。本発明の別の態様は、治療方法及びこれらの方法において使用される化合物である。したがって、本発明は、更に、上記で定義された化合物の治療有効量を罹患している個体に投与することを含む癌に罹患している任意の哺乳動物、特にヒトを治療する方法を提供する。
【0014】
別の態様において、本発明は、式Iの化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体を薬学的に許容される担体又は希釈剤と一緒に含む、医薬組成物を対象とする。
【0015】
本発明は、また、ラスパイリイダ(Raspailiidae)科リトプロカミア(Lithoplocamia)属リチストイデス(lithistoides)種の海綿動物からの式Iの化合物の単離、それらを得るための方法、及びこれらの化合物からの誘導体の形成に関する。
【発明を実施するための形態】
【0016】
本発明は、上記で定義された一般式Iの化合物に関する。
【0017】
これらの化合物において、置換基を以下の指針に従って選択することができる。
【0018】
アルキル基は、分岐鎖又は非分岐鎖であることができ、好ましくは1〜約12個の炭素原子を有することができる。一つのより好ましい部類のアルキル基は、1〜約6個の炭素原子を有する。さらにより好ましいものは、1、2、3又は4個の炭素原子を有するアルキル基である。メチル、エチル、プロピル、イソプロピル、並びにtert−ブチル、sec−ブチル及びイソブチルを含むブチルが、本発明の化合物において特に好ましいアルキル基である。別の好ましい部類のアルキル基は、6〜約10個の炭素原子;さらにより好ましくは、7、8又は9個の炭素原子を有する。ヘプチル、オクチル及びノニルが、この部類の最も好ましいアルキル基である。
【0019】
本発明の化合物において好ましいアルケニル及びアルキニル基は、分岐鎖又は非分岐鎖であることができ、1つ以上の不飽和架橋及び2〜約12個の炭素原子を有することができる。一つのより好ましい部類のアルケニル及びアルキニル基は、2〜約6個の炭素原子を有する。さらにより好ましいものは、2、3又は4個の炭素原子を有するアルケニル及びアルキニル基である。別の好ましい部類のアルケニル及びアルキニル基は、4〜約10個の炭素原子、なおより好ましくは6〜約10個の炭素原子、さらにより好ましくは7、8又は9個の炭素原子を有する。
【0020】
本発明の化合物において適切なアリール基には、単環化合物、並び別々の及び/又は縮合したアリール基を含有する多環化合物を含む多環化合物が含まれる。典型的なアリール基は、1〜3個の分離又は縮合環及び6〜約18個の炭素環原子を含有する。好ましくは、アリール基は、6〜約10個の炭素環原子を含有する。特に好ましいアリール基には、置換又は非置換フェニル、置換又は非置換ナフチル、置換又は非置換ビフェニル、置換又は非置換フェナントリル及び置換又は非置換アントリルが含まれる。
【0021】
適切な複素環式基には、1〜3個の分離又は縮合環及び5〜約18個の環原子を含有する複素環式芳香族基及び複素脂環式基が含まれる。好ましくは、複素環式芳香族基及び複素脂環式基は、5〜約10個の環原子を含有する。本発明の化合物において適切な複素環式芳香族基は、N、O又はS原子から選択される1、2又は3個のヘテロ原子を含有し、例えば、8−クマリニルを含むクマリニル、8−キノリルを含むキノリル、イソキノリル、ピリジル、ピラジニル、ピラゾリル、ピリミジニル、フリル、ピロリル、チエニル、チアゾリル、イソチアゾリル、トリアゾリル、テトラゾリル、イソオキサゾリル、オキサゾリル、イミダゾリル、インドリル、イソインドリル、インダゾリル、インドリジニル、フタラジニル、プテリジニル、プリニル、オキサジアゾリル、チアジアゾリル、フラザニル、ピリダジニル、トリアジニル、シンノリニル、ベンゾイミダゾリル、ベンゾフラニル、ベンゾフラザニル、ベンゾチオフェニル、ベンゾチアゾリル、ベンゾオキサゾリル、キナゾリニル、キノキサリニル、ナフチリジニル及びフロピリジルが含まれる。本発明の化合物において適切な複素脂環式基は、N、O又はS原子から選択される1、2又は3個のヘテロ原子を含有し、例えば、ピロリジニル、テトラヒドロフラニル、ジヒドロフラニル、テトラヒドロチエニル、テトラヒドロチオピラニル、ピペリジル、モルホリニル、チオモルホリニル、チオキサニル、ピペラジニル、アゼチジニル、オキセタニル、チエタニル、ホモピペリジル、オキセパニル、チエパニル、オキサゼピニル、ジアゼピニル、チアゼピニル、1,2,3,6−テトラヒドロピリジル、2−ピロリニル、3−ピロリニル、インドリニル、2H−ピラニル、4H−ピラニル、ジオキサニル、1,3−ジオキソラニル、ピラゾリニル、ジチアニル、ジチオラニル、ジヒドロピラニル、ジヒドロチエニル、ジヒドロフラニル、ピラゾリジニル、イミダゾリニル、イミダゾリジニル、3−アザビシクロ〔3.1.0〕ヘキシル、3−アザビシクロ〔4.1.0〕ヘプチル、3H−インドリル及びキノリジニルが含まれる。
【0022】
上記に記述された基は、OR′、=O、SR′、SOR′、SO2R′、NO2、NHR′、N(R′)2、=N−R′、NHCOR′、N(COR′)2、NHSO2R′、NR′C(=NR′)NR′R′、CN、ハロゲン、COR′、COOR′、OCOR′、OCONHR′、OCON(R′)2、保護OH、置換又は非置換アリール及び置換又は非置換複素環式基のような1つ以上の適切な基により1つ以上の利用可能な位置において置換されていることができ、ここでR′基は、それぞれ独立して、水素、OH、NO2、NH2、SH、CN、ハロゲン、COH、COアルキル、CO2H、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル、置換又は非置換アリール及び置換又は非置換複素環式基からなる群より選択される。上記基がそれ自体置換される場合、置換基は、前記のリストから選択することができる。
【0023】
本発明の化合物において適切なハロゲン置換基には、F、Cl、Br及びIが含まれる。
【0024】
OHに対する適切な保護基は当業者によく知られている。有機化学の保護基についての一般的な概観が、有機合成における保護基(Protecting Groups in Organic Synthesis)、第4版、ウィリー・インターサイエンス(Wiley-Interscience)においてウッツ(Wuts)、PGM及びグリーン(Greene)TWにより、及び保護基(Protecting Groups)、第3版、ゲオルグシームフェアラーグ(Georg Thieme Verlag)においてコシエンスキー(Kocienski)PJにより提供されている。これらの参考文献は、OHの保護基について章を提供している。これらの参考文献は、全て、その全体が参照として組み込まれる。上記保護OHの例には、エーテル、シリルエーテル、エステル、スルホネート、スルフェネート及びスルフィネート、カーボネート、並びにカルバメートが挙げられる。
エーテルの場合では、OHの保護基は、メチル、メトキシメチル、メチルチオメチル、(フェニルジメチルシリル)メトキシメチル、ベンジルオキシメチル、p−メトキシベンジルオキシメチル、〔(3,4−ジメトキシベンジル)オキシ〕メチル、p−ニトロベンジルオキシメチル、o−ニトロベンジルオキシメチル、〔(R)−1−(2−ニトロフェニル)エトキシ〕メチル、(4−メトキシフェノキシ)メチル、グアイアコールメチル、〔(p−フェニルフェニル)オキシ〕メチル、t−ブトキシメチル、4−ペンテニルオキシメチル、シロキシメチル、2−メトキシエトキシメチル、2−シアノエトキシメチル、ビス(2−クロロエトキシ)メチル、2,2,2−トリクロロエトキシメチル、2−(トリメチルシリル)エトキシメチル、メントキシメチル、o−ビス(2−アセトキシエトキシ)メチル、テトラヒドロピラニル、フルオラステトラヒドロピラニル、3−ブロモテトラヒドロピラニル、テトラヒドロチオピラニル、1−メトキシシクロヘキシル、4−メトキシテトラヒドロピラニル、4−メトキシテトラヒドロチオピラニル、4−メトキシテトラヒドロチオピラニルS,S−ジオキシド、1−〔(2−クロロ−4−メチル)フェニル〕−4−メトキシピペリジン−4−イル、1−(2−フルオロフェニル)−4−メトキシピペリジン−4−イル、1−(4−クロロフェニル)−4−メトキシピペリジン−4−イル、1,4−ジオキサン−2−イル、テトラヒドロフラニル、テトラヒドロチオフラニル、2,3,3a,4,5,6,7,7a−オクタヒドロ−7,8,8−トリメチル−4,7−メタノベンゾフラン−2−イル、1−エトキシエチル、1−(2−クロロエトキシ)エチル、2−ヒドロキシエチル、2−ブロモエチル、1−〔2−(トリメチルシリル)エトキシ〕エチル、1−メチル−1−メトキシエチル、1−メチル−1−ベンジルオキシエチル、1−メチル−1−ベンジルオキシ−2−フルオロエチル、1−メチル−1−フェノキシエチル、2,2,2−トリクロロエチル、1,1−ジアニシル−2,2,2−トリクロロエチル、1,1,1,3,3,3−ヘキサフルオロ−2−フェニルイソプロピル、1−(2−シアノエトキシ)エチル、2−トリメチルシリルエチル、2−(ベンジルチオ)エチル、2−(フェニルセレニル)エチル、t−ブチル、シクロヘキシル、1−メチル−1′−シクロプロピルメチル、アリル、プレニル、シンナミル、2−フェンアリル、プロパルギル、p−クロロフェニル、p−メトキシフェニル、p−ニトロフェニル、2,4−ジニトロフェニル、2,3,5,6−テトラフルオロ−4−(トリフルオロメチル)フェニル、ベンジル、p−メトキシベンジル、3,4−ジメトキシベンジル、2,6−ジメトキシベンジル、o−ニトロベンジル、p−ニトロベンジル、ペンタジエニルニトロベンジル、ペンタジエニルニトロピペロニル、ハロベンジル、2,6−ジクロロベンジル、2,4−ジクロロベンジル、2,6−ジフルオロベンジル、p−シアノベンジル、フルオラスベンジル、4−フルオラスアルコキシベンジル、トリメチルシリルキシリル、p−フェニルベンジル、2−フェニル−2−プロピル、p−アシルアミノベンジル、p−アジドベンジル、4−アジド−3−クロロベンジル、2−トリフルオロメチルベンジル、4−トリフルオロメチルベンジル、p−(メチルスルフィニル)ベンジル、p−シレタニルベンジル、4−アセトキシベンジル、4−(2−トリメチルシリル)エトキシメトキシベンジル、2−ナフチルメチル、2−ピコリル、4−ピコリル、3−メチル−2−ピコリルN−オキシド、2−キノリニルメチル、6−メトキシ−2−(4−メチルフェニル−4−キノリンメチル)、1−ピレニルメチル、ジフェニルメチル、4−メトキシジフェニルメチル、4−フェニルジフェニルメチル、p,p′−ジニトロベンズヒドリル、5−ジベンゾスベリル、トリフェニルメチル、トリス(4−t−ブチルフェニル)メチル、α−ナフチルジフェニルメチル、p−メトキシフェニルジフェニルメチル、ジ(p−メトキシフェニル)フェニルメチル、トリ(p−メトキシフェニル)メチル、4−(4′−ブロモフェナシルオキシ)フェニルジフェニルメチル、4,4′,4″−トリス(4,5−ジクロロフタルイミドフェニル)メチル、4,4′,4″−トリス(レブリノイルオキシフェニル)メチル、4,4′,4″−トリス(ベンゾイルオキシフェニル)メチル、4,4′−ジメトキシ−3″−〔N−(イミダゾリルメチル)〕トリチル、4,4′−ジメトキシ−3″−〔N−(イミダゾリルエチル)カルバモイル〕トリチル、ビス(4−メトキシフェニル)−1′−ピレニルメチル、4−(17−テトラベンゾ〔a,c,g,i〕フルオレニルメチル)−4,4″−ジメトキシトリチル、9−アントリル、9−(9−フェニル)キサンテニル、9−フェニルチオキサンチル、9−(9−フェニル−10−オキソ)アントリル、1,3−ベンゾジチオラン−2−イル及び4,5−ビス(エトキシカルボニル)−〔1,3〕−ジオキソラン−2−イル、ベンズイソチアゾリルS,S−ジオキシドから選択することができる。シリルエーテルの場合では、OHの保護基は、トリメチルシリル、トリエチルシリル、トリイソプロピルシリル、ジメチルイソプロピルシリル、ジエチルイソプロピルシリル、ジメチルヘキシルシリル、2−ノルボルニルジメチルシリル、t−ブチルジメチルシリル、t−ブチルジフェニルシリル、トリベンジルシリル、トリ−p−キシリルシリル、トリフェニルシリル、ジフェニルメチルシリル、ジ−t−ブチルメチルシリル、ビス(t−ブチル)−1−ピレニルメトキシシリル、トリス(トリメチルシリル)シリル、(2−ヒドロキシスチリル)ジメチルシリル、(2−ヒドロキシスチリル)ジイソプロピルシリル、t−ブチルメトキシフェニルシリル、t−ブトキシジフェニルシリル、1,1,3,3−テトライソプロピル−3−〔2−(トリフェニルメトキシ)エトキシ〕ジシロキサン−1−イル及びフルオラスシリルから選択することができる。エステルの場合では、OHの保護基は、ホルメート、ベンゾイルホルメート、アセテート、クロロアセテート、ジクロロアセテート、トリクロロアセテート、トリクロロアセトアミデート、トリフルオロアセテート、メトキシアセテート、トリフェニルメトキシアセテート、フェノキシアセテート、p−クロロフェノキシアセテート、フェニルアセテート、ジフェニルアセテート、3−フェニルプロピオネート、ビスフルオラス鎖型プロパノイル、4−ペンテノエート、4−オキソペンタノエート、4,4−(エチレンジチオ)ペンタノエート、5〔3−ビス(4−メトキシフェニル)ヒドロキシメチルフェノキシ〕レブリネート、ピバロエート、1−アダマントエート、クロトネート、4−メトキシクロトネート、ベンゾエート、p−フェニルベンゾエート、2,4,6−トリメチルベンゾエート、4−ブロモベンゾエート、2,5−ジフルオロベンゾエート、p−ニトロベンゾエート、ピコリネート、ニコチネート、2−(アジドメチル)ベンゾエート、4−アジドブチレート、(2−アジドメチル)フェニルアセテート、2−{〔(トリチルチオ)オキシ〕メチル}ベンゾエート、2−{〔(4−メトキシトリチルチオ)オキシ〕メチル}ベンゾエート、2−{〔メチル(トリチルチオ)アミノ〕メチル}ベンゾエート、2−{{〔(4−メトキシトリチル)チオ〕メチルアミノ}−メチル}ベンゾエート、2−(アリルオキシ)フェニルアセテート、2−(プレニルオキシメチル)ベンゾエート、6−(レブリニルオキシメチル)−3−メトキシ−2−ニトロベンゾエート、6−(レブリニルオキシメチル)−3−メトキシ−4−ニトロベンゾエート、4−ベンジルオキシブチレート、4−トリアルキルシリルオキシブチレート、4−アセトキシ−2,2−ジメチルブチレート、2,2−ジメチル−4−ペンテノエート、2−ヨードベンゾエート、4−ニトロ−4−メチルペンタノエート、o−(ジブロモメチル)ベンゾエート、2−ホルミルベンゼンスルホネート、4−(メチルチオメトキシ)ブチレート、2−(メチルチオメトキシメチル)ベンゾエート、2−(クロロアセトキシメチル)ベンゾエート、2−〔(2−クロロアセトキシ)エチル〕ベンゾエート、2−〔2−(ベンジルオキシ)エチル〕ベンゾエート、2−〔2−(4−メトキシベンジルオキシ)エチル〕ベンゾエート、2,6−ジクロロ−4−メチルフェノキシアセテート、2,6−ジクロロ−4−(1,1,3,3−テトラメチルブチル)フェノキシアセテート、2,4−ビス(1,1−ジメチルプロピル)フェノキシアセテート、クロロジフェニルアセテート、イソブチレート、モノスクシノエート、(E)−2−メチル−2−ブテノエート、o−(メトキシカルボニル)ベンゾエート、α−ナフトエート、ニトレート、アルキルN,N,N′,N′−テトラメチルホスホロジアミデート及び2−クロロベンゾエートから選択することができる。スルホネート、スルフェネート及びスルフィネートの場合では、OHの保護基は、スルフェート、アリルスルホネート、メタンスルホネート、ベンジルスルホネート、トシレート、2−〔(4−ニトロフェニル)エチル〕スルホネート、2−トリフルオロメチルベンゼンスルホネート、4−モノメトキシトリチルスルフェネート、アルキル2,4−ジニトロフェニルスルフェネート、2,2,5,5−テトラメチルピロリジン−3−オン(one)−1−スルフィネート、ボレート及びジメチルホスフィノチオリルから選択することができる。カーボネートの場合では、OHの保護基は、メチルカーボネート、メトキシメチルカーボネート、9−フルオレニルメチルカーボネート、エチルカーボネート、ブロモエチルカーボネート、2−(メチルチオメトキシ)エチルカーボネート、2,2,2−トリクロロエチルカーボネート、1,1−ジメチル−2,2,2−トリクロロエチルカーボネート、2−(トリメチルシリル)エチルカーボネート、2−〔ジメチル(2−ナフチルメチル)シリル〕エチルカーボネート、2−(フェニルスルホニル)エチルカーボネート、2−(トリフェニルホスホニオ)エチルカーボネート、シス−〔4−〔〔(メトキシトリチル)スルフェニル〕オキシ〕テトラヒドロフラン−3−イル〕オキシカーボネート、イソブチルカーボネート、t−ブチルカーボネート、ビニルカーボネート、アリルカーボネート、シンナミルカーボネート、プロパルギルカーボネート、p−クロロフェニルカーボネート、p−ニトロフェニルカーボネート、4−エトキシ−1−ナフチルカーボネート、6−ブロモ−7−ヒドロキシクマリン−4−イルメチルカーボネート、ベンジルカーボネート、o−ニトロベンジルカーボネート、p−ニトロベンジルカーボネート、p−メトキシベンジルカーボネート、3,4−ジメトキシベンジルカーボネート、アントラキノン−2−イルメチルカーボネート、2−ダンシルエチルカーボネート、2−(4−ニトロフェニル)エチルカーボネート、2−(2,4−ジニトロフェニル)エチルカーボネート、2−(2−ニトロフェニル)プロピルカーボネート、アルキル2−(3,4−メチレンジオキシ−6−ニトロフェニル)プロピルカーボネート、2−シアノ−1−フェニルエチルカーボネート、2−(2−ピリジル)アミノ−1−フェニルエチルカーボネート、2−〔N−メチル−N−(2−ピリジル)〕アミノ−1−フェニルエチルカーボネート、フェナシルカーボネート、3′,5′−ジメトキシベンゾインカーボネート、メチルジチオカーボネート及びS−ベンジルチオカーボネートから選択することができる。そして、カルバメートの場合では、OHの保護基は、ジメチルチオカルバメート、N−フェニルカルバメート、N−メチル−N−(o−ニトロフェニル)カルバメートから選択することができる。これらの基の記述は、これらがOHの保護基の単なる例示として記述されているに過ぎないので、本発明の範囲の制限として解釈されるべきではなく、前記機能を有する更なる基が当業者によって知られている場合があり、それらは本発明に包含されると理解されるべきである。
【0025】
用語「薬学的に許容される塩、誘導体、プロドラッグ」は、患者に投与されると、本明細書に記載されている化合物を(直接的に又は間接的に)提供することができる任意の薬学的に許容される塩、エステル、溶媒和物、水和物又は任意のその他の化合物を意味する。しかし、薬学的に許容される塩ではないものも、薬学的に許容される塩の調製に有用でありうるので、本発明の範囲内であることが理解される。塩、プロドラッグ及び誘導体の調製は、当業界において既知の方法により実施することができる。
【0026】
例えば、本明細書において提供される化合物の薬学的に許容される塩は、塩基性又は酸性部分を含有する親化合物から、従来の化学方法によって合成される。一般に、上記塩は、例えば、これらの化合物の遊離酸又は塩基形態を、水中又は有機溶媒中又は2つの混合物中の適切な塩基又は酸の理論量と反応させることにより調製される。一般に、エーテル、酢酸エチル、エタノール、イソプロパノール又はアセトニトリルのような非水性媒質が好ましい。酸付加塩の例には、例えば塩酸塩、臭化水素酸塩、ヨウ化水素酸塩、硫酸塩、硝酸塩、リン酸塩のような鉱酸付加塩、並びに、例えば酢酸塩、トリフルオロ酢酸塩、マレイン酸塩、フマル酸塩、クエン酸塩、シュウ酸塩、コハク酸塩、酒石酸塩、リンゴ酸塩、マンデル酸塩、メタンスルホン酸塩及びp−トルエンスルホン酸塩のような有機酸付加塩が挙げられる。アルカリ付加塩の例には、例えばナトリウム、カリウム、カルシウム及びアンモニウム塩のような無機塩、並びに、例えばエチレンジアミン、エタノールアミン、N,N−ジアルキレンエタノールアミン、トリエタノールアミン及び塩基性アミノ酸塩のような有機アルカリ塩が挙げられる。
【0027】
本発明の化合物は遊離化合物又は溶媒和物(例えば、水和物)のいずれかの結晶形態であることができ、両方の形態は本発明の範囲内であることが意図される。溶媒化の方法は、当業界において一般に知られている。
【0028】
式Iの化合物のプロドラッグである任意の化合物が、本発明の範囲及び意図の内である。用語「プロドラッグ」は、最も広範囲な意味において使用され、インビボ(in vivo)で本発明の化合物に変換される誘導体を包含する。上記誘導体は、当業者に容易に想起され、例えば、遊離ヒドロキシ基がエステル誘導体に変換される化合物が含まれる。
【0029】
本明細書において言及される任意の化合物は、上記特定の化合物、並びに特定の変形又は形態を表すことが意図される。特に、本明細書において言及される化合物は、不斉中心を有することができ、したがって、異なる鏡像異性形態で存在することができる。本明細書において言及される化合物の全ての光学異性体及び立体異性体、並びにこられの混合物は、本発明の範囲内であると考慮される。したがって、本明細書において言及される任意の所定の化合物は、ラセミ体、1つ以上の鏡像異性形態、1つ以上のジアステレオマー形態、1つ以上のアトロプ異性形態及びこれらの混合物のうちのいずれか1つを表すことが意図される。特に、上記の式Iにより表される本発明の化合物は、それらの非対称性に応じて鏡像異性体を又はジアステレオマーを含むことができる。二重結合に関する立体異性体も可能であり、したがって、幾つかの場合では、分子は(E)−異性体又は(Z)−異性体として存在することができる。分子が幾つかの二重結合を含有する場合、それぞれの二重結合は、分子の他の二重結合の立体異性体と同一又は異なることができる、それ自体の立体異性体を有し得る。単一の異性体又は異性体の混合物は本発明の範囲内である。
【0030】
更に、本明細書において言及される化合物は、幾何異性体(すなわち、シス及びトランス異性体)、互変異性体又はアトロプ異性体として存在することができる。特に、互変異性体という用語は、平衡で存在し、一つの異性体形態から別のものに容易に変換される、化合物の2つ以上の構造異性体のうちの1つを意味する。一般的な互変異性体対は、アミン−イミン、アミド−イミド、ケト−エノール、ラクタム−ラクチムなどである。加えて、本明細書において言及されるあらゆる化合物は、媒質に上記形態が存在する場合、水和物、溶媒和物及び多形体、並びにこれらの混合物を表すことが意図される。加えて、本明細書において言及される化合物は、同位体標識形態で存在することができる。本明細書において言及される化合物の全ての幾何異性体、互変異性体、アトロプ異性体、水和物、溶媒和物、多形体及び同位体標識形態、並びにこられの混合物は、本発明の範囲内であると考慮される。
【0031】
より簡潔な記載を提供するために、本明細書に提示されている幾つかの定量表現は、用語「約」により制限されない。用語「約」が明示的に使用されているか否かに関わらず、本明細書に提示されているあらゆる量は、実際の所定の値を意味することが意図され、上記所定の値の実験及び/又は測定条件に起因する等価値及び近似値を含む、上記所定値に対して当業界における通常の技術に基づいて合理的に推測される近似値を意味することも意図されることが理解される。
【0032】
一般式Iの化合物において、特に好ましいR1は、水素、ORa及びOCORaであり、ここでRaは、水素及び置換又は非置換C1〜C12アルキルから選択される。特に好ましいRaは、水素及び置換又は非置換C1〜C6アルキルであり、さらにより好ましいものは、水素、メチル、エチル、プロピル、イソプロピル及びtert−ブチルを含むブチルである。水素、OH及びメトキシが、最も好ましいR1基である。
【0033】
特に好ましいR2及びR3は、水素及び置換又は非置換C1〜C12アルキルである。より好ましいR2及びR3は、水素及び置換又は非置換C1〜C6アルキルであり、さらにより好ましいものは、水素である。
【0034】
特に好ましいR41、R42、R43、R44、R45、R46、R47及びR48は、水素及び置換又は非置換C1〜C12アルキルである。より好ましいR41、R42、R43、R44、R45、R46、R47及びR48は、水素及び置換又は非置換C1〜C6アルキルであり、より好ましいものは、水素、置換又は非置換メチル、置換又は非置換エチル、置換又は非置換プロピル、置換又は非置換イソプロピル、並びに置換又は非置換tert−ブチル、置換又は非置換イソブチル及び置換又は非置換sec−ブチルを含む置換又は非置換ブチルである。前記基の好ましい置換基は、OR′、=O、SR′、SOR′、SO2R′、NO2、NHR′、N(R′)2、=N−R′、NHCOR′、N(COR′)2、NHSO2R′、NR′C(=NR′)NR′R′、CN、ハロゲン、COR′、COOR′、OCOR′、OCONHR′、OCON(R′)2、保護OH、置換又は非置換アリール及び置換又は非置換複素環式基であり、ここでR′基は、それぞれ独立して、水素、OH、NO2、NH2、SH、CN、ハロゲン、COH、COアルキル、COOH、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル、置換又は非置換アリール及び置換又は非置換複素環式基からなる群より選択される。上記基がそれ自体置換される場合、置換基は、前記のリストから選択することができる。上記基のさらにより好ましい置換基は、OH、SCH3、SH、NH2、NHC(=NH)NH2、CONH2、COOH、フェニル、p−、m−又はo−ヒドロキシフェニル、1−、2−及び3−インドリルを含むインドリル、並びに4−及び5−イミダゾリルを含むイミダゾリルである。水素、メチル、イソプロピル、tert−ブチル及びベンジルが、最も好ましいR41、R42、R43、R44、R45、R46、R47及びR48基である。とりわけ、特に好ましいR42、R44、R45、R46及びR47は、水素である。特に好ましいR41及びR43は、メチルである。そして特に好ましいR48は、イソプロピル、tert−ブチル又はベンジルである。
【0035】
特に好ましいR5及びR6は、水素及び置換又は非置換C1〜C12アルキルである。より好ましいR5及びR6は、水素及び置換又は非置換C1〜C6アルキルであり、さらにより好ましいものは、水素、メチル、エチル、プロピル、イソプロピル及びtert−ブチルを含むブチルである。水素が特に好ましい。
【0036】
本発明の別の実施態様において、R5及びR48は、それらが結合している対応するN原子及びC原子と一緒になって、置換又は非置換複素環式基を形成することも好ましい。好ましい複素環式基は、1−、2−及び3−ピロリジニルを含むピロリジニルである。
【0037】
特に好ましいR7は、水素、置換又は非置換C1〜C12アルキル及び置換又は非置換C2〜C12アルケニルであり、より好ましいものは、水素、置換C1〜C12アルキル及び置換C2〜C12アルケニルである。好ましい置換アルキル及び置換アルケニルは、1つのみならず、2つ以上の置換基を表すことができる。より好ましいアルキル基は、6〜約10個の炭素原子;さらにより好ましくは、7、8又は9個の炭素原子を有するものである。ヘプチル、オクチル及びノニルが、最も好ましいアルキル基である。一方、より好ましいアルケニル基は、6〜約10個の炭素原子;さらにより好ましくは、7、8又は9個の炭素原子を有するものである。オクタ−1,6−ジエニル、オクタ−1,5−ジエニル、オクタ−1,4−ジエニル、オクタ−1,3−ジエニル、ノナ−1,7−ジエニル、ノナ−1,6−ジエニル、ノナ−1,5−ジエニル、ノナ−1,4−ジエニル、ノナ−1,3−ジエニル、ヘプタ−1,5−ジエニル、ヘプタ−1,4−ジエニル、ヘプタ−1,3−ジエニルが、最も好ましいアルケニル基である。前記アルキル及びアルケニル基の好ましい置換基は、OR′、=O、SR′、SOR′、SO2R′、NO2、NHR′、N(R′)2、=N−R′、NHCOR′、N(COR′)2、NHSO2R′、NR′C(=NR′)NR′R′、CN、ハロゲン、COR′、COOR′、OCOR′、OCONHR′、OCON(R′)2、保護OH、置換又は非置換アリール及び置換又は非置換複素環式基であり、ここでR′基は、それぞれ独立して、水素、OH、NO2、NH2、SH、CN、ハロゲン、COH、COアルキル、COOH、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル、置換又は非置換アリール及び置換又は非置換複素環式基からなる群より選択される。上記基がそれ自体置換される場合、置換基は、前記のリストから選択することができる。上記のアルキル及びアルケニル基のより好ましい置換基は、ハロゲン、OR′、=O、OCOR′、OCONHR′、OCON(R′)2及び保護OHであり、ここでR′基は、それぞれ、好ましくは水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル及び置換又は非置換アリールから選択される。これらのアルキル及びアルケニル基のさらにより好ましい置換基は、ハロゲン、OR′、=O、OCONHR′、OCON(R′)2及び保護OHであり、ここでOHの保護基は、好ましくはトリメチルシリル、トリエチルシリル、トリイソプロピルシリル、ジメチルイソプロピルシリル、ジエチルイソプロピルシリル、ジメチルヘキシルシリル、2−ノルボルニルジメチルシリル、t−ブチルジメチルシリル、t−ブチルジフェニルシリル、トリベンジルシリル、トリ−p−キシリルシリル、トリフェニルシリル、ジフェニルメチルシリル、ジ−t−ブチルメチルシリル、ビス(t−ブチル)−1−ピレニルメトキシシリル、トリス(トリメチルシリル)シリル、(2−ヒドロキシスチリル)ジメチルシリル、(2−ヒドロキシスチリル)ジイソプロピルシリル、t−ブチルメトキシフェニルシリル、t−ブトキシジフェニルシリル、1,1,3,3−テトライソプロピル−3−〔2−(トリフェニルメトキシ)エトキシ〕ジシロキサン−1−イル及びフルオラスシリルから選択され、R′基は、それぞれ、より好ましくは水素、非置換C1〜C6アルキル及び置換又は非置換アリールから選択される。Cl、OH、=O、OCONH2、OCONHフェニル、並びにOHの保護基が、好ましくはトリメチルシリル、トリエチルシリル、トリイソプロピルシリル、ジメチルイソプロピルシリル、ジエチルイソプロピルシリル、ジメチルヘキシルシリル、2−ノルボルニルジメチルシリル、t−ブチルジメチルシリル、t−ブチルジフェニルシリル、トリベンジルシリル、トリ−p−キシリルシリル、トリフェニルシリル、ジフェニルメチルシリル、ジ−t−ブチルメチルシリル、ビス(t−ブチル)−1−ピレニルメトキシシリル、トリス(トリメチルシリル)シリル、(2−ヒドロキシスチリル)ジメチルシリル、(2−ヒドロキシスチリル)ジイソプロピルシリル、t−ブチルメトキシフェニルシリル、t−ブトキシジフェニルシリル、1,1,3,3−テトライソプロピル−3−〔2−(トリフェニルメトキシ)エトキシ〕ジシロキサン−1−イル及びフルオラスシリルから選択される、保護OHは、これらのアルキル及びアルケニル基の最も好ましい置換基である。
【0038】
特に好ましいものは、点線で示された場所における1つ以上の追加的な結合の存在である。より好ましいものは、点線で示された全ての場所における追加的な結合の存在である。加えて、それぞれの二重結合の立体化学が(E)又は(Z)として存在することができる。単一の異性体又は異性体の混合物が本発明の範囲内である。
【0039】
より好ましくは、本発明は、一般式IIの化合物、又は薬学的に許容されるそれらの塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体を提供し、
【0040】
【化4】
【0041】
式中、
R1は、水素、ORa、OCORa、OCOORa、NRaRb、NRaCORb及びNRaC(NRa)NRaRbから選択され;
R41、R43及びR48は、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル及び置換又は非置換C2〜C12アルキニルから選択され;
R5、R6及びR7は、それぞれ独立して、水素、CORa、COORa、置換若しくは非置換C1〜C12アルキル、置換若しくは非置換C2〜C12アルケニル及び置換若しくは非置換C2〜C12アルキニルから選択されるか、又はR5及びR48は、それらが結合している対応するN原子及びC原子と一緒になって、置換若しくは非置換複素環式基を形成してもよく;
Ra及びRbは、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル、置換又は非置換アリール及び置換又は非置換複素環式基から選択され;そして
点線は、それぞれ任意の追加的な結合を表す。
【0042】
一般式IIの化合物において、特に好ましいR1は、水素、ORa及びOCORaであり、ここでRaは、水素及び置換又は非置換C1〜C12アルキルから選択される。特に好ましいRaは、水素及び置換又は非置換C1〜C6アルキルであり;さらにより好ましいものは、水素、メチル、エチル、プロピル、イソプロピル及びtert−ブチルを含むブチルである。水素、OH及びメトキシが、最も好ましいR1基である。
【0043】
特に好ましいR41、R43及びR48は、水素及び置換又は非置換C1〜C12アルキルである。より好ましいR41、R43及びR48は、水素及び置換又は非置換C1〜C6アルキルであり、さらにより好ましいものは、水素、置換又は非置換メチル、置換又は非置換エチル、置換又は非置換プロピル、置換又は非置換イソプロピル、並びに置換又は非置換tert−ブチル、置換又は非置換イソブチル及び置換又は非置換sec−ブチルを含む置換又は非置換ブチルである。前記基の好ましい置換基は、OR′、=O、SR′、SOR′、SO2R′、NO2、NHR′、N(R′)2、=N−R′、NHCOR′、N(COR′)2、NHSO2R′、NR′C(=NR′)NR′R′、CN、ハロゲン、COR′、COOR′、OCOR′、OCONHR′、OCON(R′)2、保護OH、置換又は非置換アリール及び置換又は非置換複素環式基であり、ここでR′基は、それぞれ独立して、水素、OH、NO2、NH2、SH、CN、ハロゲン、COH、COアルキル、COOH、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル、置換又は非置換アリール及び置換又は非置換複素環式基からなる群より選択される。上記基がそれ自体置換される場合、置換基は、前記のリストから選択することができる。上記基のさらにより好ましい置換基は、OH、SCH3、SH、NH2、NHC(=NH)NH2、CONH2、COOH、フェニル、p−、m−又はo−ヒドロキシフェニル、1−、2−及び3−インドリルを含むインドリル、並びに4−及び5−イミダゾリルを含むイミダゾリルである。水素、メチル、イソプロピル、tert−ブチル及びベンジルが、最も好ましいR41、R43及びR48基である。とりわけ、特に好ましいR41及びR43は、メチルであり、特に好ましいR48は、イソプロピル、tert−ブチル又はベンジルである。
【0044】
特に好ましいR5及びR6は、水素及び置換又は非置換C1〜C12アルキルである。より好ましいR5及びR6は、水素及び置換又は非置換C1〜C6アルキルであり;さらにより好ましいものは、水素、メチル、エチル、プロピル、イソプロピル及びtert−ブチルを含むブチルである。水素が最も好ましい。
【0045】
本発明の別の実施態様において、R5及びR48は、それらが結合している対応するN原子及びC原子と一緒になって、置換又は非置換複素環式基を形成することも好ましい。好ましい複素環式基は、1−、2−及び3−ピロリジニルを含むピロリジニルである。
【0046】
特に好ましいR7は、水素、置換又は非置換C1〜C12アルキル及び置換又は非置換C2〜C12アルケニルであり、より好ましいものは、水素、置換C1〜C12アルキル及び置換C2〜C12アルケニルである。より好ましいアルキル基は、6〜約10個の炭素原子;さらにより好ましくは、7、8又は9個の炭素原子を有するものである。ヘプチル、オクチル及びノニルが、最も好ましいアルキル基である。一方、より好ましいアルケニル基は、6〜約10個の炭素原子;さらにより好ましくは、7、8又は9個の炭素原子を有するものである。オクタ−1,6−ジエニル、オクタ−1,5−ジエニル、オクタ−1,4−ジエニル、オクタ−1,3−ジエニル、ノナ−1,7−ジエニル、ノナ−1,6−ジエニル、ノナ−1,5−ジエニル、ノナ−1,4−ジエニル、ノナ−1,3−ジエニル、ヘプタ−1,5−ジエニル、ヘプタ−1,4−ジエニル、ヘプタ−1,3−ジエニルが、最も好ましいアルケニル基である。前記アルキル及びアルケニル基の好ましい置換基は、OR′、=O、SR′、SOR′、SO2R′、NO2、NHR′、N(R′)2、=N−R′、NHCOR′、N(COR′)2、NHSO2R′、NR′C(=NR′)NR′R′、CN、ハロゲン、COR′、COOR′、OCOR′、OCONHR′、OCON(R′)2、保護OH、置換又は非置換アリール及び置換又は非置換複素環式基であり、ここでR′基は、それぞれ独立して、水素、OH、NO2、NH2、SH、CN、ハロゲン、COH、COアルキル、COOH、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル、置換又は非置換アリール及び置換又は非置換複素環式基からなる群より選択される。上記基がそれ自体置換される場合、置換基は、前記のリストから選択することができる。上記のアルキル及びアルケニル基のより好ましい置換基は、ハロゲン、OR′、=O、OCOR′、OCONHR′、OCON(R′)2及び保護OHであり、ここでR′基は、それぞれ、好ましくは水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル及び置換又は非置換アリールから選択される。これらのアルキル及びアルケニル基のさらにより好ましい置換基は、ハロゲン、OR′、=O、OCONHR′、OCON(R′)2及び保護OHであり、ここでOHの保護基は、好ましくはトリメチルシリル、トリエチルシリル、トリイソプロピルシリル、ジメチルイソプロピルシリル、ジエチルイソプロピルシリル、ジメチルヘキシルシリル、2−ノルボルニルジメチルシリル、t−ブチルジメチルシリル、t−ブチルジフェニルシリル、トリベンジルシリル、トリ−p−キシリルシリル、トリフェニルシリル、ジフェニルメチルシリル、ジ−t−ブチルメチルシリル、ビス(t−ブチル)−1−ピレニルメトキシシリル、トリス(トリメチルシリル)シリル、(2−ヒドロキシスチリル)ジメチルシリル、(2−ヒドロキシスチリル)ジイソプロピルシリル、t−ブチルメトキシフェニルシリル、t−ブトキシジフェニルシリル、1,1,3,3−テトライソプロピル−3−〔2−(トリフェニルメトキシ)エトキシ〕ジシロキサン−1−イル及びフルオラスシリルから選択され、R′基は、それぞれ、より好ましくは水素、非置換C1〜C6アルキル及び置換又は非置換アリールから選択される。Cl、OH、=O、OCONH2、OCONHフェニル、並びにOHの保護基が、好ましくはトリメチルシリル、トリエチルシリル、トリイソプロピルシリル、ジメチルイソプロピルシリル、ジエチルイソプロピルシリル、ジメチルヘキシルシリル、2−ノルボルニルジメチルシリル、t−ブチルジメチルシリル、t−ブチルジフェニルシリル、トリベンジルシリル、トリ−p−キシリルシリル、トリフェニルシリル、ジフェニルメチルシリル、ジ−t−ブチルメチルシリル、ビス(t−ブチル)−1−ピレニルメトキシシリル、トリス(トリメチルシリル)シリル、(2−ヒドロキシスチリル)ジメチルシリル、(2−ヒドロキシスチリル)ジイソプロピルシリル、t−ブチルメトキシフェニルシリル、t−ブトキシジフェニルシリル、 1,1,3,3−テトライソプロピル−3−〔2−(トリフェニルメトキシ)エトキシ〕ジシロキサン−1−イル及びフルオラスシリルから選択される、保護OHは、これらのアルキル及びアルケニル基の最も好ましい置換基である。
【0047】
特に好ましいものは、点線で示された場所における1つ以上の追加的な結合の存在である。より好ましいものは、点線で示された全ての場所における追加的な結合の存在である。なお、それぞれの二重結合の立体化学が(E)又は(Z)として存在することができる。単一の異性体又は異性体の混合物が本発明の範囲内である。
【0048】
本発明の特に好ましい化合物は、以下である:
【0049】
【化5】
【0050】
化合物1〜8は、ラスパイリイダ(Raspailiidae)科リトプロカミア(Lithoplocamia)属リチストイデス(lithistoides)種の海綿動物から単離された。
【0051】
リトプロカミア・リチストイデス(Lithoplocamia lithistoides )の試料は、メキシコのマサトラン(Mazatlan)にあるメキシコ国立自治大学(Universidad Nacional Autonoma de Mexico)の「海洋及び陸水学の科学研究所」(“Instituto de Ciencias del Mar y Limnologia”)に、参照コードLEB−ICML−UNAM−11−2004で寄託された。この海綿体は、マダガスカル(南17度06.071分/東49度51.385分)において、6〜20mの範囲の深さでスキューバダイビングにより手動で収集され、その記載は以下である。
【0052】
ラスパイリイダ(Raspailiidae)科:ラスパイリイダ・ヘンチェル(Raspailiidae Hentschel)、1923は、皮殻で覆われた、巨大な葉状の扇形又は分岐した成長形態を有し、通常は極めて剛毛な表面を有する海綿体である。個々の長厚突起又はオキセア(oxeas)を取り囲んでいる小型の薄い突起(フーパー&ウィーデンマイヤー(Hooper & Wiedenmayer)1994:図17)又はオキセア(フーパー&ウィーデンマイヤー1994:図5)のブラシからなる特殊な体外骨格が、典型的に存在する。襟体骨格は、圧縮軸骨格から羽毛網状の又は網状のみの構造まで多様である。海綿質繊維は、通常、核心交接刺(襟体突起、オキセア又は両方)を完全に包み込む。特別な分類の棘条突起(フーパー&ウィーデンマイヤー1994:図22)又は突起の変態(例えば、図22〜25、28)、有棘繊維が、繊維から直角に突き出している。微小骨片は通常不在であるが、単一の束晶(フーパー&ウィーデンマイヤー1994:図109)又は束(トリコドラグメータ(trichodragmata);フーパー&ウィーデンマイヤー1994:図110)が、幾つかの属において生じうる。ラスパイリイダ(Raspailiid)は、浅海から深さが少なくとも2460mまでの範囲に広く分布している(ハートマン(Hartman)1982)。
【0053】
リトプロカミア(Lithoplocamia)属リチストイデス(lithistoides )種は、皮殻で覆われた、巨大な成長形態であり、襟体骨格は、棘状棍棒体の高密度の等網糸又は不規則な半網糸の網状組織であり、1又は2つの大きさの分類において、軸性圧縮がなく、有棘の棘状突起がなく、平滑突起の軸外放射状管を有し、典型的には特殊なラスパイリイダ(raspailiid)体外骨格を有さず(しかし、存在する場合は、体外交接刺は、細長いオキセアである)、微小骨片が存在しない。
【0054】
リトプロカミア・リチストイデス(Lithoplocamia lithistoides)の試料は、また、ケニヤ(南04度40分5.5秒/東39度26分4.3秒、及び南3度38分36.5秒/東39度53分53.8秒)並びにタンザニア(南08度55分31.7秒/東39度34分53.5秒、及び南05度24.200分/東39度47.730分)において、30〜40mの範囲の深さで収集された。
【0055】
さらに、本発明の化合物は、合成により得ることができる。例えば、化合物1は、スキーム1に示されているように、異なるフラグメントを結合することによって作製できる。
【0056】
【化6】
【0057】
ここで、R、RI、RII、RIII、RIV、RV、RVI、RVII及びRVIIIは、所望の基又は必要とされる適切な保護基である。
【0058】
この方法は、以下の主要な工程を含むことができる:
a)標準的な文献の手順(コザワ(Kozawa)Yら、テトラヒドロン・レター(Tetrahedron Lett.)2002年、43、111)に従った、フラグメントCによるヨードアルケニル誘導体(フラグメントD)のアミド化によって、対応するエンアミド(フラグメントCD)を得ること。
b)有機合成において既知の手順(スコット(Scott)WJら、米国化学会ジャーナル(J. Am. Chem. Soc.)1984年、106、4630;ラバディ(Labadie)JWら、有機化学ジャーナル(J. Org. Chem.)1983年、48、4634〜4642;ファリーナ(Farina)Vら、有機反応(Organic Reactions)1998年、ウィリー(Wiley))に従った、フラグメントAとフラグメントBのスチル(Stille)型カップリング反応によって、直鎖ポリエン(フラグメントAB)を得ること。
c)フラグメントAB及びCDを、標準的な手順(ボダンスキー(Bodanszky)M及びボダンスキーA、ペプチド合成の実践(The Practice of Peptide Synthesis)、シュプリンガー・フェアラーグ、1993年)に従ってカップリングして、化合物1の炭素骨格を得ることができる。
d)アルコールORIIの脱保護、続くラクトン化は、有機合成において既知の手順(グリーン(Greene)及びウッツ(Wuts)、有機合成における保護基(Protective Groups in Organic Synthesis)、第3版、ウィリー・インターサイエンス(Wiley-Interscience);バーク(Burke)及びダンハイザー(Danheiser)、有機合成のための試薬ハンドブック:酸化及び還元剤(Handbook of Reagents for Organic Synthesis: Oxidizing and Reducing Agents)、ウィリー(Wiley);プラ(Pla)Dら、有機化学ジャーナル(J. Org. Chem.)2005年、70、8231)に従って達成することができる。
e)最後に、アルコールORIIIの脱保護、続くカルバメートの形成は、標準的な文献の手順(ラブ(Love)Bら、有機合成(Organic Syntheses)集、第5巻、162頁;第48巻、32頁;ミュラー(Muller)Eら、有機化学法(Methoden der Organischen Chemie)(フーベン・ワイル)(Houben-Weyl)、第4版、第8巻、G.シーム(Thieme)、シュットガルト、1952年、137頁)により達成して、化合物1を得ることができる。
【0059】
工程の順序を取り替えて最終化合物を得ることができる。例えば、フラグメントBCを第1工程で調製し、次にフラグメントA及びBと連続カップリングさせて、化合物1の炭素骨格を得ることができる。同様に、化合物1は、フラグメントA、B、C及びDを任意の順番で連続カップリングして調製することができる。別の選択肢は、任意の他のフラグメントとのカップリングの前に、フラグメントAにおけるラクトン部分の形成を実施することである。
【0060】
化合物1の類似体は、それぞれの場合において中間体化合物の適切な置換基を選択して、化合物1について記載したものと同等の方法により合成することができる。
【0061】
必要な場合、置換基に対して適切な保護基を使用して、反応性基が影響を受けないことを確実にすることができる。合成は、適切な工程で所望の置換基に変換することができる前駆体置換基を用いるように設計することができる。環構造における飽和又は不飽和を、合成の一部として導入又は除去することができる。出発材料及び試薬を、望ましいように変更して、意図される化合物の合成を確実にすることができる。加えて、類似体を、当業者に既知の合成有機化学における通常の手順によって、化合物1から合成することもできる。
【0062】
上記の合成経路を、望ましいように変更して、立体特異的化合物、並びに立体異性体の混合物を与えることができる。立体特異的試薬の使用又は合成の際にキラル中心を化合物に導入することを含む多様な方法により、特定の立体異性体又は特定の混合物を合成することが可能である。合成の際に1つ以上の立体中心を導入すること、また、存在する立体中心を反転することが可能である。加えて、化合物が当業者に既知の標準的な溶解技術により合成されると、立体異性体を分離することが可能である。
【0063】
式I及びIIの上記の化合物の重要な特徴は、それらの生体活性、特にそれらの細胞毒性及び抗有糸分裂活性である。
【0064】
本発明によって、細胞毒性及び抗有糸分裂活性を有する一般式I及びIIの化合物の新規医薬組成物、並びに抗腫瘍剤としてのそれらの使用が提供される。したがって、本発明は、更に、本発明の化合物、薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ又は立体異性体と薬学的に許容される担体とを含む、医薬組成物を提供する。
【0065】
医薬組成物の例には、経口、局所又は非経口投与用の任意の固体(錠剤、丸剤、カプセル剤、顆粒剤など)又は液体(液剤、懸濁剤又は乳剤)組成物が挙げられる。
【0066】
本発明の化合物又は組成物の投与は、静脈内注入、経口調合剤、並びに腹腔内及び静脈内投与のような任意の適切な方法によることができる。24時間までの注入時間を使用することが好ましく、より好ましくは1〜12時間であり、1〜6時間が最も好ましい。治療を病院に一晩入院することなく実施することを可能にする短い注入時間が、特に望ましい。しかし、注入は、必要であれば12〜24時間又はそれ以上であることができる。注入は、例えば1〜4週間の適切な間隔で実施することができる。本発明の化合物を含有する医薬組成物は、リポソーム又はナノ球体封入により、持続放出製剤において、又は他の標準的な送達手段により送達することができる。
【0067】
化合物の正確な投与量は、特定の製剤、適用様式、並びに特定の位置、宿主及び治療される腫瘍に応じて変わり得る。年齢、体重、性別、食事、投与時間、排出速度、宿主の状態、薬剤の組み合わせ、反応感受性及び疾患の重篤度のような他の要因が考慮され得る。投与は、最大耐量の範囲内で継続的に又は周期的に実施することができる。
【0068】
本発明の化合物及び組成物を他の薬剤と共に使用して、併用療法を提供することができる。他の薬剤は、同じ組成物の一部分を形成することができるか、又は同時若しくは異なる時に投与される別個の組成物として提供されうる。
【0069】
これらの化合物の抗腫瘍活性には、肺癌、結腸癌、乳癌及び子宮頸癌が含まれるが、これらに限定されない。
【0070】
実施例
実施例1:海洋生物及び収集の側面の記載
リトプロカミア・リチストイデス(Lithoplocamia lithistoides)は、マダガスカル(南17度06.071分/東49度51.385分)において、6〜20mの範囲の深さでのスキューバダイビングを用いて手動で収集された。動物材料は、ホセ・ルイス・カルバジョ(Jose Luis Carballo)(メキシコ自治大学(Universidad Autonoma de Mejico))により同定された。検体試料は、メキシコのマサトラン(Mazatlan)にあるメキシコ国立自治大学(Universidad Nacional Autonoma de Mexico)の「海洋及び陸水学の科学研究所」(“Instituto de Ciencias del Mar y Limnologia”)に、参照コードLEB−ICML−UNAM−11−2004で寄託された。
【0071】
実施例2:化合物1の単離
実施例1の冷凍検体(61g)を角切りし、H2O(3×200mL)、続いてMeOH:ジクロロメタンの混合物(1:1、3×200mL)により室温で抽出した。合わせた有機抽出物を濃縮して、粗物質1.11gを得た。この物質を、H2OからMeOHへの段階勾配によるLichroprep RP-18のVLCに付した。
【0072】
化合物1(1.6mg)を、半分取逆相HPLC(SymmetryPrep C18 7μm、7.8×150mm、MeCNが35〜100%の勾配H2O:MeCNを30分、UV検出、流速2.5mL/分、保持時間14.4分)により、MeOHを用いて溶離してフラクションから単離した。
化合物1:無定形の白色固体。(+)HRESIMS m/z606.2940[M+H]+(C31H4535ClN3O7で計算 606.2946);1H(500MHz)及び13C NMR(125MHz)表1を参照すること。
【0073】
【表1】
【化7】
【0074】
実施例3:化合物2、3、4、5、6及び7の単離
リトプロカミア・リチストイデス(Lithoplocamia lithistoides)の試料の第2群(7.66kg)を粉砕し、MeOH:ジクロロメタンの混合物(1:1、14L、2×5L、4L)で、徹底的に抽出した。溶媒を真空下で除去し、残留水溶液をEtOAc(12L、3×8L)で抽出した。有機層を蒸発させて、粗物質21.71gを得た。
【0075】
この物質を、H2O:MeOH(4:6)からMeOHの段階勾配を用いるRP-18カラムクロマトグラフィーに付した。H2O:MeOH(2:8、430mg)で溶離したフラクションをプールし、分取HPLC(Atlantis dC18、OBD、5μm、19×150mm、定組成H2O:MeOH(39:61)、流速:20mL/分、UV検出)に付して、純粋な化合物1(160.8mg)、2(13.2mg)及び7(1.89mg)、並びに3と4の混合物(11.4mg)及び5と6の混合物(10.0mg)を得た。純粋な化合物3(5.1mg)及び4(2.6mg)は、半分取HPLC(X-Terra Prep RP-18、10μm、10×150mm、MeOHが50〜70%の勾配H2O:MeOHを70分、流速:2.5mL/分、UV検出)による混合物の最終精製の後で得た。化合物5(3.6mg)及び6(1.0mg)は、半分取HPLC(X-Terra Prep RP-18、10μm、10×150mm、定組成H2O:MeOH(45:55)、流速:2.5mL/分、UV検出)により、同様の方法で分離した。
化合物2:無定形の白色固体。MS(ES)m/z606.3[M+H]+,628.3[M+Na]+;1H(500MHz)及び13C NMR(125MHz)表2を参照すること。
化合物3:無定形の白色固体。(+)HRESIMS m/z628.2774[M+Na]+(C31H4435ClN3O7Naで計算 628.2760);1H(500MHz)及び13C NMR(125MHz)表3を参照すること。
化合物4:無定形の白色固体。(+)HRESIMS m/z594.3152[M+Na]+(C31H45N3O7Naで計算 594.3150);1H(500MHz)及び13C NMR(125MHz)表4を参照すること。
化合物5:無定形の白色固体。MS(ES)m/z592.3[M+H]+,614.3[M+Na]+;1H(500MHz)及び13C NMR(125MHz)表5を参照すること。
化合物6:無定形の白色固体。MS(ES)m/z592.3[M+H]+,614.3[M+Na]+;1H(500MHz)及び13C NMR(125MHz)表6を参照すること。
化合物7:無定形の白色固体。(+)HRESIMS m/z427.2207[M+Na]+(C22H32N2O5Naで計算 427.2203);1H(500MHz)及び13C NMR(125MHz)表7を参照すること。
【0076】
【表2】
【化8】
【表3】
【化9】
【表4】
【化10】
【表5】
【化11】
【表6】
【化12】
【表7】
【化13】
【0077】
実施例4:化合物8の単離
実施例3に開示された抽出手順からもたらされた化合物1(61.6mg)含有フラクションを、半分取HPLC(Symmetryprep C-18、7μm、7.8×150mm、定組成H2O:CH3CN(55:45)、流速:2.3mL/分、UV検出)により更に精製して、0.9mgの化合物8を純粋な形態で得た。
化合物8:無定形の白色固体。MS(ES)m/z606.2[M+H]+,628.3[M+Na]+;1H(500MHz)及び13C NMR(125MHz)表8を参照すること。
【0078】
【表8】
【化14】
【0079】
実施例5:フラグメントAの合成
スキーム2は、スキーム1において提供された命名法に従って、フラグメントAの合成の幾つかの例を提供する。
【0080】
【化15】
【0081】
中間体9の合成
【0082】
【化16】
【0083】
ジクロロメタン/DMSO(331mL/149mL)の混合物中の(2S,3S)−3,5−ビス{〔(tert−ブチル)ジメチルシリル〕オキシ}−4−メチルペンタン−1−オール(P.プーカン(Phukan)、S.サスマール(Sasmal)及びM.E.マイヤー(Maier)有機化学ヨーロピアンジャーナル(Eur. J. Org. Chem.)2003年、1733〜1740)(50g、0.14mol)の0℃の溶液に、Et3N(96.1mL、0.69mol)を、添加漏斗により加えた。10分後、SO3・Pyr(54.8g、0.34mol)を少量ずつ加え、溶液を0℃で更に2時間撹拌した。次に、それをジクロロメタン(800ml)で希釈し、HCl(0.5N、800mL)で停止させた。有機層をデカントし、MgSO4で乾燥し、真空下で濃縮した。カラムクロマトグラフィー(ヘキサン/EtOAc 100:0〜10:1)による精製によって、45g(収率:90%)のアルデヒド9を得た。
1H-RMN (CDCl3, 300 MHz) δ: 9.79 (s, 1H), 4.30 (m, 1H), 3.65 (m, 2H), 2.51 (m, 1H), 1.69 (m, 2H), 1.04 (d, 3H, J = 6.9Hz), 0.85-0.88 (m, 18H), 0.03-0.07 (m, 12H).
13C-RMN (CDCl3, 75 MHz) δ: 205.4, 69.4, 59.6, 51.7, 37.5, 26.1, 26.0, 18.4, 18.2, 8.0, -4.3, -4.5, -5.2.
【0084】
中間体10の合成
【0085】
【化17】
【0086】
トルエン(625mL)中のアルデヒド9(45g、0.2mol)の溶液に、カルボエトキシエチリデン−トリフェニルホスホラン(113g、0.31mol)を加え、混合物を60℃で17時間加熱した。次に、溶媒を減圧下で除去し、得られた油状物をカラムクロマトグラフィー(ヘキサン/EtOAc 100:0〜10:1)により精製して、53.3g(収率:96%)のエステル化合物10を得た。
1H-RMN (CDCl3, 300 MHz) δ: 6.71 (dd, 1H, J = 1.5, 10.2 Hz), 4.19 (m, 2H), 3.77 (m, 1H), 3.66 (m, 2H), 2.61 (m, 1H), 1.85 (d, 3H, J = 1.5 Hz), 1.68 (m, 2H), 1.30 (t, 3H, J = 7.2 Hz), 0.98 (d, 3H, 6.9 Hz), 0.90 (m, 18H), 0.05 (m, 12H).
13C-RMN (CDCl3, 75 MHz) δ: 168.3, 145.4, 126.7, 72.2, 60.4, 59.7, 38.4, 38.0, 25.9, 18.2, 18.1, 14.3, 14.3, 12.6, -4.4, -4.6, -5.4.
【0087】
中間体11の合成
【0088】
【化18】
【0089】
無水THF(525mL)中のエステル10(46.7g、0.105mol)の−78℃に冷却した溶液に、アルゴン雰囲気下、トルエン(231mL、0.231mol)中の1M水素化ジイソブチルアルミニウム(DIBAL)を10分間かけて加え、混合物を−78℃で撹拌した。4時間後、反応をMeOH(10mL)で停止させ、酒石酸ナトリウムカリウムの飽和溶液(800mL)を加え、EtOAc(1000mL)で希釈した。この混合物を2時間撹拌し、次に有機層をデカントした。水性残渣を追加のEtOAc(2×400mL)で抽出し、合わせた有機層を乾燥し(Na2SO4)、溶媒を蒸発させた。得られた油状物をカラムクロマトグラフィー(ヘキサン/EtOAc 20:1〜10:1)により精製して、32.5g(収率:77%)のアルコール11を得た。
1H-RMN (CDCl3, 300 MHz) δ: 5.31 (d, 1H, J = 9.6 Hz), 3.98 (m, 2H), 3.66 (m, 3H), 2.49 (m, 1H), 1.67 (s, 3H), 1.70-1.62 (m, 2H), 0.91 (d, 3H, J = 6.9 Hz), 0.88 (m, 18H), 0.03 (m, 12H).
13C-RMN (CDCl3, 75 MHz) δ: 133.9, 129.8, 73.1, 69.1, 59.9, 37.8, 37.5, 25.9, 18.3, 18.1, 15.9, 13.9, -4.4, -4.4, -5.3.
【0090】
中間体12の合成
【0091】
【化19】
【0092】
エチルエステル(387mL)中のアルコール11(31.2g、77.5mmol)の溶液に、アルゴン雰囲気下、MnO2(101g、1.16mol)を加え、混合物を室温で2時間撹拌した。この混合物を、EtOAc(3L)により溶離するシリカゲルカラムで濾過し、得られた溶液を減圧下で乾燥して、29.1g(収率:94%)のアルデヒド12を得た。
1H-RMN (CDCl3, 300 MHz) δ: 9.37 (s, 1H), 6.44 (d, 1H, J = 9.6 Hz), 3.82 (dd, 1H, J = 6.3, 10.8 Hz), 3.65 (m, 2H), 2.82 (m, 1H), 1.74 (s, 3H), 1.67 (m, 2H), 1.02 (d, 3H, J = 6.9 Hz), 0.86 (s, 18H), 0.04-0.01 (m, 12H).
13C-RMN (CDCl3, 75 MHz) δ: 195.4, 157.8, 138.3, 134.5, 72.0, 59.5, 36.7, 37.5, 25.8, 18.2, 18.1, 14.3, 9.4, -4.4, -4.5, -5.4.
【0093】
中間体13の合成
【0094】
【化20】
【0095】
THF(727mL)中のヨウ化ヨードメチルトリフェニルホスホニウム(ギルバート・ストーク(Gilbert Stork),KZ.テトラへドロン・レターズ(Tetrahedron letters)1989年、30(17)、2173)(96.3g、181.7mmol)の懸濁液に、0℃で、ヘキサメチルジシラザンナトリウム(NaHMDS)の1M溶液(181.7mL、181.7mmol)を、添加漏斗により10分間かけてゆっくりと加えた。更に5分間撹拌した後、溶液を−78℃に冷却し、次に1,3−ジメチル−3,4,5,6−テトラヒドロ−2(1H)−ピリミジノン(DMPU)(43.9mL、363.4mmol)をカニューレにより加え、続いてTHF(727mL)に溶解したアルデヒド12(29.1g、72.7mmol)を加えた。温度を−78℃に保持し、その間、反応混合物を2時間撹拌した。ヘキサン(1L)を加え、得れたスラリーをセライトで濾過し、追加のヘキサン(3L)で洗浄した。濾液を減圧下で蒸発させ、得られた油状物をカラムクロマトグラフィー(ヘキサン/EtOAc 100:0〜20:1)により精製して、32g(収率:84%)のヨウ化物13を得た。
1H-RMN (CDCl3, 300 MHz) δ: 6.73 (d, 1H, J = 8.4 Hz), 6.09 (dd, 1H, J = 8.4, 1.2 Hz), 5.57 (dd, 1H, J = 9.6, 1.2 Hz), 3.63-3.71 (m, 3H), 2.58 (m, 1H), 1.90 (s, 3H), 1.70 (m, 2H), 0.96 (dd, 3H, J = 6.6, 1.2 Hz), 0.88 (s, 18H), 0.04 (m, 12H).
13C-RMN (CDCl3, 75 MHz) δ: 142.3, 138.1, 131.8, 74.6, 72.9, 59.8, 38.1, 37.9, 26.0, 18.3, 18.2, 15.7, 15.7, -4.4, -5.2, -5.2.
【0096】
中間体14の合成
【0097】
【化21】
【0098】
EtOH(114mL)中のヨウ化物13(12g、22.9mmol)の溶液に、ピリジニウムp−トルエンスルホネート(PPTS)(2.01g、8.0mmol)を加え、反応混合物を室温で25時間撹拌した。次に、溶媒を減圧下で除去し、得られた油状物をカラムクロマトグラフィー(ヘキサン/EtOAc 10:1)により精製して、8.7g(収率:93%)のアルコール14を得た。
1H-RMN (CDCl3, 300 MHz) δ: 6.69 (d, 1H, J = 8.4 Hz), 6.12 (d, 1H, J = 8.4 Hz), 5.47 (d, 1H, J = 9.9 Hz), 3.67-3.87 (m, 4H), 2.71 (m, 1H), 1.89 (s, 3H), 1.73-1.86 (m, 2H), 1.01 (d, 3H, J = 6.9 Hz), 0.91 (s, 9H), 0.087-0.115 (m, 6H).
13C-RMN (CDCl3, 75 MHz) δ: 142.4, 136.4, 132.6, 75.8, 75.2, 60.0, 38.1, 36.4, 26.1, 18.2, 17.1, 16.0, -4.1, -4.2.
【0099】
中間体15の合成
【0100】
【化22】
【0101】
ジクロロメタン/DMSO(50.9mL/22.9mL)の混合物中のアルコール14(8.7g、21.2mmol)の0℃の溶液に、Et3N(14.8mL、106mmol)を添加漏斗により加えた。10分後、SO3・Pyr(8.43g、53.0mol)を少量ずつ加え、溶液を0℃で更に2時間撹拌した。次に、それをジクロロメタン(800ml)で希釈し、HCl(0.5N、50mL)で停止させた。有機層をデカントし、MgSO4で乾燥し、真空下で濃縮した。カラムクロマトグラフィー(ヘキサン/EtOAc 10:1)による精製によって、6.9g(収率:80%)のアルデヒド15を得た。
1H-RMN (CDCl3, 300 MHz) δ: 9.89 (t, 1H, J = 1.5 Hz), 6.67 (d, 1H, J = 8.4 Hz), 6.13 (d, 1H, J = 8.4 Hz), 5.43 (d, 1H, J = 10.2 Hz), 3.98 (m, 1H), 2.59-2.69 (m, 3H), 1.85 (s, 3H), 1.01 (d, 3H, J = 6.6 Hz), 0.86 (s, 9H), 0.06 (s, 3H), 0.03 (s, 3H).
13C-RMN (CDCl3, 75 MHz) δ: 201.8, 141.9, 135.2, 133.3, 76.3, 71.9, 49.3, 39.3, 25.8, 18.0, 16.7, 15.9, -4.4, -4.5.
【0102】
中間体16aの合成
【0103】
【化23】
【0104】
アルゴン雰囲気下、−78℃で撹拌した、無水THF(390mL)中のジエチル(メトキシ〔メトキシカルボニル〕メチル)ホスホネート(5.51g、14.45mmol)及び18−クラウン−6(11.5g、43.34mmol)の溶液に、0.5Mカリウムビス(トリメチルシリル)アミド溶液(KHMDS)(43.34mL、21.67mmol)を滴加した。15分後、無水THF中のアルデヒド15(5.9g、14.45mmol)を30分間かけて滴加し、−78℃で90分間撹拌した。次に、反応を飽和NH4Cl溶液(200mL)で停止させ、室温に温め、ジクロロメタン(1000mL)で希釈した。有機層を乾燥し(Na2SO4)、減圧下で蒸発させた。カラムクロマトグラフィー(ヘキサン/Et2O 20:1)による精製によって、純粋な4.2g(59%)の(E)−16aを得た。
1H-RMN (CDCl3, 300 MHz) δ: 6.70 (d, 1H, J = 8.4 Hz), 6.08 (d, 1H, J = 8.4 Hz), 5.47 (d, 1H, J = 9.9 Hz), 5.37 (t, 1H, J = 7.2 Hz), 3.78 (s, 3H), 3.60 (s, 3H), 3.60 (m, 1H), 2.79 (m, 1H), 2.52-2.67 (m, 2H), 1.83 (s, 3H), 0.99 (d, 3H, J = 6.6 Hz), 0.89 (s, 9H), 0.05 (s, 3H), 0.04 (s, 3H).
13C-RMN (CDCl3, 75 MHz) δ: 163.7, 145.9, 142.1, 137.3, 132.1, 110.4, 75.4, 74.8, 55.4, 51.9, 38.1, 32.3, 25.9, 18.1, 16.5, 15.7, -4.3, -4.5.
【0105】
中間体16bの合成
【0106】
【化24】
【0107】
アルゴン雰囲気下、0℃で撹拌した、無水THF(2.4mL)中の〔ビス(2,2,2−トリフルオロエトキシ)ホスフィニル〕酢酸エチル(0.16mL、0.66mmol)及び18−クラウン−6(350mg、1.32mmol)の溶液に、KHMDS(1.23mL、0.62mmol)を滴加した。30分後、無水THF中のアルデヒド15(180mg、0.44mmol)を滴加し、−78℃で60分間撹拌した。次に、反応を飽和NH4Cl溶液で停止させ、室温に温め、EtOAcで希釈した。有機層を乾燥し(Na2SO4)、減圧下で蒸発させた。カラムクロマトグラフィー(ヘキサン/EtOAc 100:1〜15:1)による精製によって、172mg(収率:82%)の(Z)−16bを得た。
1H-RMN (CDCl3, 300 MHz) δ: 6.70 (d, 1H, J = 8.7 Hz), 6.44-6.36 (m, 1H), 6.09 (d, 1H, J = 8.7 Hz), 5.86-5.81 (m, 1H), 5.47 (d, 1H, J = 9.9 Hz), 4.14 (q, 2H, J = 7.2 Hz), 3.69-3.64 (m, 1H), 3.06-3.00 (m, 1H), 2.85-2.75 (m, 1H), 2.59-2.51 (m, 1H), 1.84 (s, 3H), 1.28 (t, 3H, J = 7.2 Hz), 1.00 (d, 3H, J = 6.6 Hz), 0.89 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H).
MS (ES) m/z 501.0 [M+Na]+
【0108】
中間体17aの合成
【0109】
【化25】
【0110】
MeOH(125mL)中のエステル16a(4.15g、8.39mmol)の溶液に、室温で、37%のHCl(1.04mL)を加え、反応混合物を6時間撹拌した。次に、混合物をNaHCO3の飽和溶液で中和し(pH7〜8)、有機溶媒を減圧下で蒸発させた。得られた懸濁液をジクロロメタン(3×200mL)で抽出し、乾燥し、蒸発させた。カラムクロマトグラフィー(ヘキサン/EtOAc 10:1〜2:1)による濾過によって、2.76g(収率:94%)のラクトン17aを得た。
1H-RMN (500 MHz, CDCl3) δ: 6.68 (d, 1H, J = 9.0 Hz), 6.20 (d, 1H, J = 8.5 Hz), 5.63 (dd, 1H, J = 2.5, 6.5 Hz), 5.43 (d, 1H, J = 10.0 Hz), 4.19 (m, 1H), 3.65 (s, 3H), 2.84 (m, 1H), 2.55 (m, 1H), 2.43 (dc, J = 1H, 3.0, 12.0, 15.0, 18.0 Hz), 1.87 (s, 3H), 1.16 (d, 3H, J = 6.5 Hz).
13C-RMN (125 MHz, CDCl3) δ: 161.6, 145.2, 141.8, 134.4, 132.7, 108.3, 81.7, 77.4, 55.4, 37.1, 26.6, 16.5, 16.1.
【0111】
中間体17bの合成
【0112】
【化26】
【0113】
MeOH(4.5mL)中のエステル16b(172mg、0.36mmol)の溶液に、室温で、37%のHCl(0.03mL)を加え、反応混合物を3時間撹拌した。次に、混合物をNaHCO3の飽和溶液で中和し(pH7〜8)、有機溶媒を減圧下で蒸発させた。得られた懸濁液をジクロロメタンで抽出し、乾燥し、蒸発させた。カラムクロマトグラフィー(ヘキサン/EtOAc 10:1〜5:1)による濾過によって、70mg(収率:61%)のラクトン17bを得た。
1H-RMN (CDCl3, 300 MHz) δ: 6.91-6.85 (m, 1H), 6.68 (d, 1H, J = 8.4 Hz), 6.62 (d, 1H, J = 8.4 Hz), 6.02 (dd, 1H, J = 2.7, 9.6 Hz), 5.45 (d, 1H, J = 9.9 Hz), 4.19 (m, 1H), 3.65 (s, 3H), 4.26-4.18 (m, 1H), 2.92-2.79 (m, 1H), 2.57-2.48 (m, 1H), 2.39-2.28 (m, 1H), 1.88 (s, 3H), 1.17 (d, 3H, J = 6.6 Hz).
【0114】
実施例6:フラグメントDの合成
【0115】
スキーム3は、スキーム1において提供された命名法に従って、フラグメントDの合成の幾つかの例を提供する。
【0116】
【化27】
【0117】
中間体19の合成
【0118】
【化28】
【0119】
ジクロロメタン(DCM)(918mL)中の中間体18(72.3g)の溶液に、室温で、3−クロロ過安息香酸(m−CPBA)(100g、0.58mol)を少量ずつ加え、混合物を室温で18時間撹拌した。白色の沈殿を、NaHCO3の飽和溶液で停止させ、DCM(3×250mL)で抽出し、再びNaHCO3の飽和溶液(3×250mL)で洗浄した。有機層を合わせ、Na2SO4で乾燥し、真空下で濃縮した。得られた油状物をシリカゲル(ヘキサン−AcOEt;15:1)で精製して、エポキシドを無色の油状物(64.5g、82%)として得た。無水THF(7.5mL)中のラセミエポキシド(30g)の溶液に、(R,R)Co(II)錯体(448mg、0.74mmol)を加え、続いてAcOH(0.14mL)を加えた。溶液を0℃に冷却し、水(1.2mL)を滴加した。反応混合物を室温に温め、18時間撹拌した。その時間の後、揮発性物質を真空下で濃縮し、粗物質をシリカゲルカラムに直接装填した。溶離剤としてヘキサン/EtOAc(15:1〜12:1)を使用するフラッシュクロマトグラフィーによって、キラルエポキシド(+)−19(13.6g、収率:46%)を無色の油状物として得た。
[α]D = +14.1 (c= 1, CHCl3).
1H NMR (CDCl3, 300 MHz) δ: 3.74 (t, 2H, J = 6.3 Hz), 3.01 (m, 1H), 2.74 (t, 1H, J = 4.6 Hz), 2.48 (dd, 1H, J = 5.1, 3.1 Hz), 1.70 (m, 2H), 0.87 (s, 9H), 0.04 (s, 6H).
13C RMN (CDCl3, 75 MHz) δ: 60.2, 50.2, 47.3, 36.1, 26.1, 18.4, -5.2.
【0120】
中間体20の合成
【0121】
【化29】
【0122】
プロピンを−78℃で濃縮し、無水THF(165mL)に溶解した。n−ブチルリチウムをAr下で30分間かけて滴加し、得られた白色の懸濁液を−78℃で更に30分間撹拌した。次に、無水THF(125mL)中の(+)(R)−2−〔2−(tert−ブチルジメチルシリルオキシ)エチル〕オキシラン19(23.7g)の溶液を滴加し、続いてBF3OEt2を加えた。混合物を−78℃で1時間、0℃で更に1時間撹拌した。反応をNH4Clの飽和水溶液(150mL)で停止させ、Et2O(3×150mL)で抽出した。合わせた有機層をNa2SO4で乾燥し、濾過し、濃縮した。フラッシュクロマトグラフィー(ヘキサン/EtOAc 10:1〜1:1)によって、22.7g(収率:80%)のアルコール20を無色の油状物として得た。
[α]D = +5.6 (c= 0.1, CHCl3).
1H-RMN (500 MHz, CDCl3) δ: 3.75-3.90 (m, 3H), 3.47 (d, 1H, J = 2.7 Hz, OH), 2.34 (m, 2H), 1.79, (t, 3H, J = 2.4 Hz), 1.75 (m, 2H), 0.89 (s, 9H), 0.07 (s, 6H).
13C-RMN (125 MHz, CDCl3) δ: 77.8, 75.8, 70.7, 62.4, 37.6, 27.6, 26.1, 18.3, 3.7, -5.3, -5.4.
MS (ES) m/z 243.2 [M+H]+, 265.2 [M+Na]+
【0123】
中間体21aの合成
【0124】
【化30】
【0125】
DCM中の中間体20(22.7g)及びp−メトキシベンジルトリクロロアセトイミデート(PMBTCA)の溶液を、Sc(OTf)3で処理した。混合物を室温で2時間撹拌し(TLCで検査)、反応を真空下で濃縮し、カラムクロマトグラフィー(ヘキサン/EtOAc 50:1〜15:1)により精製して、21aを黄色の油状物(18.3g;収率:55%)として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.25 (d, 2H, J = 8.7 Hz), 6.90 (d, 2H, J = 8.7 Hz), 4.45 (m, 2H), 3.80 (s, 3H), 3.65 (m, 3H), 2.40 (m, 2H), 1.82 (m, 2H), 1.79 (t, 3H, J = 2.4 Hz), 0.92 (s, 9H), 0.05 (s, 6H).
【0126】
中間体21bの合成
【0127】
【化31】
【0128】
N,N−ジメチルホルムアミド(DMF)(14mL)中のアルコール20(2.88g、11.9mmol)、tert−ブチルジフェニルシリルクロリド(4.39mL、16.89mmol)及び4−(ジメチルアミノ)ピリジン(43.6mg)の溶液を、室温で一晩撹拌した。混合物を水で希釈し、Et2Oで抽出し、有機相をブラインで洗浄し、Na2SO4で乾燥し、濾過し、濃縮した。フラッシュクロマトグラフィー(ヘキサン/EtOAc、95:1)によって、シリルエーテル21b(5.3g、収率:93%)を無色の液体として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.70-7.66 (m, 4H), 7.40-7.34 (m, 6H), 3.99-3.95 (m, 1H), 3.70-3.62 (m, 2H), 2.23-2.22 (m, 2H), 1.84-1.81 (m, 2H), 1.69 (t, 3H, J = 2.7 Hz), 1.05 (s, 9H), 0.84 (s, 9H), 0.01 (s, 6H).
13C-RMN (CDCl3, 75 MHz) δ: 136.1; 134.6; 129.7; 127.8; 77.8; 76.2; 69.9; 60.1; 39.6; 27.5; 27.2; 26.2; 19.6; 18.5; 3.7; -5.1.
【0129】
中間体22aの合成
【0130】
【化32】
【0131】
無水トルエン中の21aの溶液に、Ar下及び0℃で、シュワルツ(Schwartz)試薬(ビス(シクロペンタジエニル)ジルコニウム(IV)クロリドヒドリド、Cp2ZrHCl)を加え、反応を室温で5分間撹拌した。反応温度を20分間かけて50℃に増加し、50℃で2.30時間撹拌した。この時間の間に反応溶液は橙色になった。反応を0℃に冷却し、N−クロロスクシンイミドを一度に加えた。撹拌を室温で30分間続け、反応をヘキサン/EtOAc(95:5;500mL)で希釈した。固体を濾過により除去し、揮発物を蒸発させて、22aを黄色の油状物として得て、それを更に精製することなく使用した(15.1g;収率:86%)。
[α]D = +20.5 (c= 1, CHCl3).
1H NMR (CDCl3, 300 MHz) δ: 7.25 (d, 2H, J = 8.7 Hz), 6.87 (d, 2H, J = 8.7 Hz), 5.64 (td, 1H, J = 7.8, 0.9 Hz), 4.45 (q, 2H, J = 11.1 Hz), 3.80 (s, 3H), 3.70 (m, 2H), 3.62 (m, 1H), 2.27 (t, 2H, J = 6.9 Hz), 2.03 (s, 3H), 1.70 (m, 2H), 0.89 (s, 9H), 0.05 (s, 6H).
13C RMN (75 MHz, CDCl3) δ: 159.4, 130.9, 130.7, 129.6, 124.2, 114.0, 75.2, 71.4, 59.8, 55.5, 37.7, 33.8, 26.1, 21.2, 18.5, -5.1.
【0132】
中間体22bの合成
【0133】
【化33】
【0134】
酢酸エチル中の21b(4.73g、9.85mmol)、キノリン(0.582mL、4.92mmol)及びリンドラー触媒(2.18g)の混合液を含有するフラスコを、排気し、H2でフラッシュした。反応混合物を、H2下(1atm)、室温で2時間撹拌し、次にセライトプラグで濾過した。プラグを酢酸エチルですすぎ、合わせた濾液を0.1%のHClで洗浄した。有機層をNa2SO4で乾燥し、濾過し、濃縮して、中間体22b(4.27g、収率:90%)を無色の油状物として得て、それを更に精製することなく使用した。
1H NMR (CDCl3, 300 MHz) δ: 7.70-7.67 (m, 4H), 7.44-7.36 (m, 6H), 5.48 (m, 1H), 5.36-5.27 (m, 1H), 3.95-3.87 (m, 1H), 3.71-3.55 (m, 2H), 2.16 (dd, 2H, J = 6.9, 6.3 Hz), 1.73-1.66 (m, 2H), 1.41 (dd, 3H, J = 6.6, 1.2 Hz), 1.05 (s, 9H), 0.84 (s, 9H), -0.02 (s, 6H).
13C-RMN (CDCl3, 75 MHz) δ: 136.2; 134.8; 129.8; 127.8; 126.4; 125.8; 70.9; 60.4; 39.6; 34.8; 27.3; 26.2; 19.7; 18.5; 13.1; -5.1.
【0135】
中間体23aの合成
【0136】
【化34】
【0137】
無水THF中の22a(23g)の溶液に、Ar下及び0℃で、フッ化テトラブチルアンモニウム(TBAF)の溶液を20分間かけて滴加した(溶液は赤色になった)。反応混合物を室温で2時間撹拌し、次にNH4Clの飽和水溶液(200ml)で停止させた。合わせた層を分離し、水相をEtOAc(3×150ml)で十分に抽出した。合わせた有機層をNaSO4で乾燥し、濾過し、濃縮した。フラッシュクロマトグラフィー(ヘキサン/EtOAc 4:1〜1:1)によって、23aを無色の油状物(11.9g;収率:73%)として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.25 (d, 2H, J = 8.7 Hz), 6.86 (d, 2H, J = 8.7 Hz), 5.62 (t, 1H, J = 7.8 Hz), 4.45 (m, 2H), 3.80 (s, 3H), 3.70 (m, 3H), 2.35 (m, 2H), 2.03 (s, 3H), 1.75 (m, 2H).
【0138】
中間体23bの合成
【0139】
【化35】
【0140】
PPTS(837.7mg、3.33mmol)を、エタノール(80mL)中の22b(4g、8.33mmol)の溶液に、一度に加えた。反応混合物を室温で7時間撹拌し、次に濃縮した。残渣をDCMで希釈し、NaHCO3の飽和溶液で洗浄した。有機層を抽出し、Na2SO4で乾燥し、濾過し、濃縮した。フラッシュクロマトグラフィー(ヘキサン/EtOAc、95:1)によって、シリルエーテル23b(2.12g、収率:69%)を無色の油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.73-7.69 (m, 4H), 7.44-7.36 (m, 6H), 5.44-5.38 (m, 1H), 5.21-5.17 (m, 1H), 4.01-3.94 (m, 1H), 3.84-3.76 (m, 1H), 3.69-3.64 (m, 1H), 2.32-2.14 (m, 2H), 1.89-1.78 (m, 1H), 1.70-1.60 (m, 1H), 1.37 (d, 3H, J = 6.9 Hz), 1.07 (s, 9H).
13C-RMN (CDCl3, 75 MHz) δ: 136.2; 134.1; 130.0; 127.8; 126.3; 125.9; 72.3; 60.1; 37.7; 34.3; 27.2; 19.5; 13.0.
【0141】
中間体24aの合成
【0142】
【化36】
【0143】
(ジアセトキシヨード)ベンゼン(BAIB)(11.5g、35.7mmol)を、ジクロロメタン無水物(92mL)中のアルコール23a(9.2g、32.4mmol)及び2,2,6,6−テトラメチルピペリジン 1−オキシル(TEMPO)(515mg、3.3mmol)の溶液に加えた。反応混合物を、アルコールが検出されなくなるまで(TLC)、室温で20時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、DCM(3×100mL)で抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 4:1〜1:1)により精製して、24aを無色の油状物(6.3g;収率:70%)として得た。
1H NMR (CDCl3, 300 MHz) δ: 9.78 (s, 1H), 7.25 (d, 2H, J = 8.7 Hz), 6.85 (d, 2H, J = 8.7 Hz), 5.64 (t, 1H, J = 7.8 Hz), 4.45 (q, 2H, J = 11.1 Hz), 4.02 (m, 1H), 3.80 (s, 3H), 2.60 (m, 2H), 2.35 (m, 2H), 2.03 (s, 3H).
13C RMN (CDCl3, 75 MHz) δ: 201, 159.6, 132.1, 130.1, 129.7, 122.8, 114.1, 73.3, 71.5, 55.5, 48.3, 33.5, 21.3.
【0144】
中間体24bの合成
【0145】
【化37】
【0146】
BAIB(1.97g、6.11mmol)を、25mLのDCM中のアルコール23b(2.05g、5.56mmol)及びTEMPO(86.87mg、0.56mmol)の溶液に加えた。反応混合物を、アルコールが検出されなくなるまで(TLC)、室温で16〜18時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、DMCで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/DCM 5:1〜1:2)により精製して、24b(1.733mg、収率:79%)を無色の油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 9.72 (t, 1H, J = 2.7 Hz), 7.74-7.67 (m, 4H), 7.48-7.37 (m, 6H), 5.56-5.45 (m, 1H), 5.32-5.23 (m, 1H), 4.29-4.20 (m, 1H), 2.51-2.48 (m, 2H), 2.31-2.27 (m, 2H), 1.43 (dd, 3H, J = 6.9, 1.5 Hz), 1.06 (s, 9H).
13C-RMN (CDCl3, 75 MHz) δ: 202.3; 136.1; 134.0; 130.1; 127.9; 127.4; 125.1; 69.4; 50.1; 35.1; 27.2; 19.5; 13.1.
【0147】
中間体25aの合成
【0148】
【化38】
【0149】
無水THF(126mL)中のヨウ化ヨードメチルトリフェニルホスホニウム(16.6g;31mmol)の懸濁液に、室温で、THF(31.27mL)中のNaHMDSの1M溶液をゆっくりと加えた。2分間撹拌した後、黄色の混合物を−78℃に冷却し、次にTHF(82mL)中の24a(6.3g、22mmol)の溶液を加えた。反応混合物を−78℃で2時間、室温で5分間撹拌し、ヘキサンで希釈し、セライトプラグで濾過した。プラグをヘキサンですすぎ、合わせた濾液を減圧下で蒸発させ、得られた油状物をカラムクロマトグラフィー(ヘキサン/EtOAc 12:1〜8:1)により精製して、25aを黄色の油状物(5.6g;収率:62%)として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.25 (d, 2H, J = 8.7 Hz), 6.85 (d, 2H, J = 8.7 Hz), 6.25 (m, 2H) 5.64 (t, 1H, J = 7.8 Hz), 4.42 (m, 2H), 3.80 (s, 3H), 3.55(m, 1H), 2.40 (m, 2H), 2.25 (m, 2H), 2.03 (s, 3H).
【0150】
中間体25bの合成
【0151】
【化39】
【0152】
THF(60mL)中のヨードメチルトリフェニルホスホラン(3.32g、6.38mmol)の懸濁液に、室温で、THF中のNaHMDS(6.38mmol)の1M溶液6.83mLをゆっくりと加えた。2分間撹拌した後、黄色の混合物を−78℃に冷却し、次にTHF(40mL)中の24b(1.67g、4.56mmol)の溶液を加えた。反応混合物を−78℃で90分間、次に室温で5分間撹拌し、ヘキサンで希釈し、セライト/SiO2のプラグで濾過した。プラグをヘキサン/EtOAc(10:1〜5:1)によりすすいで、化合物25b(2g、収率:89%)を無色の油状物として得て、それを更に精製することなく使用した。
1H NMR (CDCl3, 300 MHz) δ: 7.70-7.66 (m, 4H), 7.45-7.34 (m, 6H), 6.21-6.31 (m, 2H), 5.49-5.43 (m, 1H), 5.35-5.27 (m, 1H), 3.94-3.75 (m, 1H), 2.30-2.27 (m, 2H), 2.24-2.04 (m, 2H), 1.43 (d, 3H, J = 6.6 Hz), 1.06 (s, 9H).
13C-RMN (CDCl3, 75 MHz) δ: 138.2; 136.2; 134.3; 129.9; 127.8; 126.4; 126.0; 84.1; 71.9; 41.6; 34.5; 27.2; 19.6; 13.2.
【0153】
中間体25cの合成
【0154】
【化40】
【0155】
2,3−ジクロロ−5,6−ジシアノ−p−ベンゾキノン(DDQ)(3.6g、16mmol)を、DCM−H2O(20:1)中の25a(5g;12mmol)の溶液に、Ar雰囲気下、室温で加えた。1:30h後(ヘキサン/EtOAc 4:1のTLCでは出発物質を示さなかった)、反応を、Et2O(200mL)に注ぐことにより停止させ、1M NaOH(3×50ml)及びブライン(50mL)で洗浄した。有機相をNa2SO4で乾燥し、濾過し、濃縮した。p−メトキシベンズアルデヒドのクロマトグラフ分離を、p−メトキシベンジルアルコールへの還元により促進した。この終点に向かって、Ar雰囲気下でNaBH4を有するMeOHにおいて得られた残渣の溶液を、室温で1時間保持した。次に反応混合物を、Et2O(100mL)に注ぐことによって停止させ、1M HCl(40mL)及びブライン(40mL)で洗浄した。有機相をNa2SO4で乾燥し、濾過し、濃縮した。得られた油状物をシリカゲル(ヘキサン/EtOAc 10:1〜4:1)で精製して、第二級アルコールを無色の油状物として得た(2.8g;収率:80%)。
【0156】
無水DCM中の第二級アルコール(2.8g;10mmol)の溶液に、Ar下及び0℃で、2,6−ルチジンを滴加し、続いてtert−ブチルジメチルシリルトリフルオロメタンスルホネート(TBSOTf)を加えた(ヘキサン/EtOAc 4:1のTLCでは出発物質を示さなかった)。この時点で、粗混合物を0.5M HCl(25mL)で停止させ、DCM(2×25mL)で抽出した。合わせた有機層をNaHCO3の飽和水溶液及びブラインで洗浄した。有機相をNaSO4で乾燥し、濾過し、濃縮した。フラッシュクロマトグラフィー(ヘキサン/EtOAc 100:1〜20:1)によって、25cを無色の油状物(3.14g;収率:80%)として得た。
1H NMR (CDCl3, 300 MHz) δ: 6.25 (m, 2H) 5.64 (t, 1H, J = 7.8 Hz), 3.82 (m, 1H), 2.38 (t, 2H, J = 6.0 Hz), 2.20 (t, 2H, J = 6.3 Hz), 2.03 (s, 3H), 0.86 (s, 9H), 0.05 (s, 6H).
13C RMN (CDCl3, 75 MHz) δ: 137.7, 130.9, 124.3, 84.6, 70.6, 42.5, 36.6, 25.9, 21.3, 18.2, -4.4.
【0157】
実施例7:フラグメントBCDの合成
スキーム4は、スキーム1において提供された命名法に従って、フラグメントBCDの合成の幾つかの例を提供する。
【0158】
【化41】
【0159】
中間体26aの合成
【0160】
【化42】
【0161】
再密封可能なシュレンク管に、ヨウ化銅(I)(148mg、0.78mmol)、炭酸カリウム(1.076g、7.78mmol)及びBoc−tert−LeuCONH2(ポズネフ(Pozdnev)V.F.、テトラヘドロン・レターズ(Tetrahedron Letters)1995年、36、7115〜7118に記載された手順に従って調製)(0.96g、4.15mmol)を投入し、排気し、アルゴンを充填した。N,N′−ジメチルエチレンジアミン(DMEDA)(0.166mL、1.55mmol)、ヨウ化ビニル25c(1.04g、2.59mmol)及び無水DMF(15mL)をアルゴン下で加えた。シュレンク管を密閉し、90℃で16〜18時間加熱し、室温に冷却した。得られた混合物をEtOAcで希釈し、水で停止させた。有機層を、水で洗浄し、Na2SO4で乾燥した。溶媒を減圧下で除去し、残渣をシリカゲルのフラッシュクロマトグラフィー(ヘキサン/EtOAc、20:1〜15:1)により精製した。中間体26a(670mg、収率、53%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.72 (d, 1H, J = 9.9 Hz), 6.70 (t, 1H, J = 9.6 Hz), 5.54 (t, 1H, J = 7.8 Hz), 5.35 (d, 1H, J = 9.0 Hz), 4.76 (q, 1H, J = 7.8 Hz), 3.89 (d, 1H, J = 9.0 Hz), 3.73-3.68 (m, 1H), 2.12 (m, 4H), 1.98 (s, 3H), 0.971 (s, 9H), 0.84 (s, 9H), 0.02 (s, 3H), 0.01 (s, 3H).
13C NMR (CDCl3, 75 MHz) δ: 168.9, 156.0 131.1, 123.9, 122.6, 108.2, 79.9, 71.6, 62.5, 36.5, 34.8, 33.8, 28.1, 26.7, 25.9, 21.2, 18.3, -4.3, -4.4.
【0162】
中間体26bの合成
【0163】
【化43】
【0164】
再密封可能なシュレンク管に、ヨウ化銅(I)(232.4mg、1.22mmol)、炭酸カリウム(1.688g、12.23mmol)及びBoc−tert−LeuCONH2(2.474g、6.12mmol)を投入し、排気し、アルゴンを充填した。N,N′−ジメチルエチレンジアミン(0.26mL、2.45mmol)、ヨウ化ビニル25b(2g、4.08mmol)及び無水DMF(35mL)をアルゴン下で加えた。シュレンク管を密閉し、90℃で16〜18時間加熱し、室温に冷却した。得られた混合物をEtOAcで希釈し、水で停止させた。有機層を、水で洗浄し、Na2SO4で乾燥した。溶媒を減圧下で除去し、残渣をシリカゲルのフラッシュクロマトグラフィー(ヘキサン/EtOAc、20:1〜15:1)により精製した。中間体26b(1.06g、収率:44%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.70-7.67 (m, 4H), 7.43-7.35 (m, 6H), 7.13 (d, 1H, J = 10.5 Hz), 6.67 (dd, 1H, J = 10.2, 9.6 Hz), 5.56-5.45 (m, 1H), 5.36-5.28 (m, 2H), 4.86-4.78 (m, 2H), 3.88-3.77 (m, 1H), 2.26-2.04 (m, 4H), 1.44 (d, 3H, J = 6.9 Hz), 1.43 (s, 9H), 1.06 (s, 9H), 0.96 (s, 9H).
【0165】
中間体26cの合成
【0166】
【化44】
【0167】
再密封可能なシュレンク管に、ヨウ化銅(I)(40.4mg、0.213mmol)、炭酸カリウム(294mg、2.13mmol)及びBoc−Val−CONH2(ポズネフ(Pozdnev)V.F.、テトラヘドロン・レターズ(Tetrahedron Letters)1995年、36、7115〜7118に記載された手順に従って調製)(230mg、1.06mmol)を投入し、排気し、アルゴンを充填した。N,N′−ジメチルエチレンジアミン(45μL、0.426mmol)、ヨウ化ビニル25c(283mg、0.71mmol)及び無水DMF(35mL)をアルゴン下で加えた。シュレンク管を密閉し、90℃で16〜18時間加熱し、室温に冷却した。得られた混合物をEtOAcで希釈し、水で停止させた。有機層を、水で洗浄し、Na2SO4で乾燥した。溶媒を減圧下で除去し、残渣をシリカゲルのフラッシュクロマトグラフィー(ヘキサン/EtOAc、7:1〜3:1)により精製した。中間体26c(270g、収率:77%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.80 (d, 1H, J = 9.3), 6.79-6.73 (m, 1H), 5.58 (t, 1H, J = 7.5 Hz), 5.02 (br s, 1H), 4.85-4.76 (m, 1H), 3.93 (dd, 1H, J = 8.4, 6.0 Hz), 3.80-3.73 (m, 1H), 2.12-2.22 (m, 5H), 2.02 (s, 3H), 1.45 (s, 9H), 0.98 (d, 3H, J = 6.9 Hz), 0.93 (d, 3H, J = 6.9 Hz), 0.89 (s, 9H), 0.07 (s, 3H), 0.06 (s, 3H).
13C NMR (CDCl3, 75 MHz) δ: 169.3, 131.1, 124.0, 122.7, 108.9, 71.6, 36.5, 33.8, 30.6, 28.5, 26.1, 21.3, 19.6, 18.3, 17.9, -4.3, -4.4.
【0168】
中間体26dの合成
【0169】
【化45】
【0170】
再密封可能なシュレンク管に、ヨウ化銅(I)(14.2mg、0.075mmol)、炭酸カリウム(104mg、0.75mmol)及びFmoc−Phe−CONH2(ポズネフ(Pozdnev)V.F.、テトラヘドロン・レターズ(Tetrahedron Letters)1995年、36、7115〜7118に記載された手順に従って調製)(145mg、0.375mmol)を投入し、排気し、アルゴンを充填した。N,N′−ジメチルエチレンジアミン(16μL、0.15mmol)、ヨウ化ビニル25c(100mg、0.25mmol)及び無水DMF(2.5mL)をアルゴン下で加えた。シュレンク管を密閉し、90℃で16〜18時間加熱し、室温に冷却した。得られた混合物をEtOAcで希釈し、水で停止させた。有機層を、水で洗浄し、Na2SO4で乾燥した。溶媒を減圧下で除去し、残渣をシリカゲルのフラッシュクロマトグラフィー(ヘキサン/EtOAc、4:1〜1:1)により精製した。中間体26d(46mg、収率:42%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 9.19 (d, 1H, J = 11.1 Hz), 7.36-7.21 (m, 5H), 6.77 (ddd, 1H, J = 10.2, 9.3, 0.9), 5.60 (br t, 1H, J = 7.8 Hz), 4.82-4.78 m, 1H), 3.79-3.71 (m, 1H), 3.67 (dd, 1H, J = 9.6, 3.9 Hz), 3.32 (dd, 1H, J = 13.8, 3.9 Hz), 2.69 (dd, 1H, J = 13.8, 9.6 Hz), 2.20-2.11 (m, 4H), 1.99 (s, 3H), 0.89 (s, 9H), 0.05 (s, 3H), 0.04 (s, 3H).
13C NMR (CDCl3, 75 MHz) δ: 171.9, 137.9, 130.9, 129.5, 129.1, 127.2, 124.1, 122.5, 107.9, 71.4, 56.6, 40.9, 36.3, 33.6, 26.1, 21.3, 18.3, -4.4, -4.5.
MS (ES) m/z 437.1 [M+H]+, 459.0 [M+Na]+
【0171】
中間体27aの合成
【0172】
【化46】
【0173】
エチレングリコール(30mL)中のアミノ保護誘導体26a(670mg、1.33mmol)の溶液を、200℃で10〜20分間加熱した。次に反応混合物を室温に冷却し、DCMで希釈し、ブラインで停止させ、水に注いだ。数滴の3M NaOHを、溶液がpH14に達するまで加え、次にDCMで十分に抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、真空下で濃縮して、第一級アミン27a(510mg、収率:95%)を黄色の油状物として得て、それを更に精製することなく使用した。
1H NMR (CDCl3, 300 MHz) δ: 8.77 (d, 1H, J = 9.9 Hz), 6.71 (t, 1H, J = 9.6 Hz), 5.56 (t, 1H, J = 7.8 Hz), 4.71 (m, 1H), 3.72 (m, 1H), 3.14 (s, 1H), 2.14 (m, 4H), 1.97 (s, 3H), 0.97 (s, 9H), 0.84 (s, 9H), 0.02 (s, 6H).
13C RMN (CDCl3, 75 MHz) δ: 171.2, 131.0, 124.1, 122.5, 107.1, 71.5, 64.3, 36.2, 34.5, 33.8, 26.5, 26.0, 21.2, 18.2, -4.4, -4.5.
【0174】
中間体27bの合成
【0175】
【化47】
【0176】
エチレングリコール(50mL)中のアミノ保護誘導体26b(847mg、1.43mmol)の溶液を、200℃で10〜20分間加熱した。次に反応混合物を室温に冷却し、DCMで希釈し、ブラインで停止させ、水に注いだ。数滴の3M NaOHを、溶液がpH14に達するまで加え、次にDCMで十分に抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、真空下で濃縮して、第一級アミン27b(435mg、62%)を、フラッシュクロマトグラフィー(ヘキサン/EtOAc 10:1〜1:2)による精製の後で、白色の泡状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 8.50 (d, 1H, J = 10.8 Hz), 7.70-7.66 (m, 4H), 7.45-7.33 (m, 6H), 6.67 (dd, 1H, J = 11.1, 9.3 Hz), 5.48-5.40 (m, 1H), 5.36-5.28 (m, 1H), 4.79 (dd, 1H, J = 16.2, 7.5 Hz), 3.87-3.79 (m, 1H), 3.08 (s, 1H), 2.22-2.14 (m, 4H), 1.43 (d, 3H, J = 6.9 Hz), 1.05 (s, 9H), 0.97 (s, 9H).
13C-RMN (CDCl3, 75 MHz) δ: 171.0; 136.1; 134.5; 129.8; 127.8; 126.3; 126.2; 122.1; 107.6; 72.6; 64.4; 34.0; 34.4; 32.8; 27.2; 26.9; 19.6; 13.2.
【0177】
中間体27cの合成
【0178】
【化48】
【0179】
エチレングリコール(15mL)中のアミノ保護誘導体26c(255mg、0.52mmol)の溶液を、200℃で10〜20分間加熱した。次に反応混合物を室温に冷却し、DCMで希釈し、ブラインで停止させ、水に注いだ。数滴の3M NaOHを、溶液がpH14に達するまで加え、次にDCMで十分に抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、真空下で濃縮して、第一級アミン27c(170mg、85%)を黄色の油状物として得て、それを更に精製することなく使用した。
1H NMR (CDCl3, 300 MHz) δ: 9.27 (d, 1H, J = 10.2), 6.76 (dd, 1H, J = 11.1, 9.6 Hz), 5.61 (t, 1H, J = 7.8 Hz), 4.80-4.72 (m, 1H), 3.81-3.73 (m, 1H), 3.31 (d, 1H, J = 3.6 Hz) 2.44-2.33 (m, 1H), 2.20-2.16 (m, 4H), 2.03 (s, 3H), 1.59 (br s, 2H), 1.00 (d, 3H, J = 6.9 Hz), 0.89 (s, 9H), 0.82 (d, 3H, J = 6.9 Hz), 0.05 (s, 6H).
13C NMR (CDCl3, 75 MHz) δ: 172.1, 131.1, 124.1, 122.5, 107.4, 71.5, 36.5, 33.7, 30.8, 26.0, 21.3, 20.0, 16.1, -4.3, -4.4.
【0180】
中間体28aの合成
【0181】
【化49】
【0182】
DCM/DMF(10:1、39.6mL)中のアミン27a(918mg、2.27mmol)の溶液に、無水DCM中の(Z)−3−トリブチルスタンニルプロペン酸(1028mg、2.84mmol)の溶液を、アルゴン雰囲気下で加え、次に0℃に冷却した。ジイソプロピルエチルアミン(DIPEA)(0.6mL、3.4mmol)、1−ヒドロキシ−7−アザベンゾトリアゾール(HOAt)(310mg、2.27mmol)及びN,N,N′,N′−テトラメチル−O−(7−アザベンゾトリアゾール−1−イル)ウロニウムヘキサフルオロホスフェート(HATU)(860mg、2.27mmol)を溶液に加え、30分後、冷浴を取り外した。反応混合物を室温で2時間撹拌し、NH4Clの飽和水溶液で停止させ、水に注ぎ、DCMで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 20:1〜15:1)により精製して、アミド28a(1110mg;収率:66%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.63 (d, 1H, J = 10.5 Hz), 6.97 (d, 1H, J = 12.3 Hz), 6.75 (d, 1H, J = 12.3 Hz), 6.72 (t, 1H, J = 9.5 Hz), 6.50 (d, 1H, J = 9.0 Hz), 5.56 (t, 1H, J = 6.6 Hz), 4.83 (q, 1H, J = 9.0 Hz), 4.41 (d, 1H, J = 9.6 Hz) 3.76 (m, 1H), 2.17 (m, 4H), 2.01 (s, 3H), 1.45 (m, 6H), 1.25 (m, 8H), 1.0 (s, 9H), 0.88 (s, 9H), 0.84 (m, 13H), 0.06 (s, 6H).
【0183】
中間体28bの合成
【0184】
【化50】
【0185】
DCM/DMF(4:1、12.5mL)中のアミン27b(575mg、1.17mmol)の溶液に、無水DCM中の(Z)−3−トリブチルスタンニルプロペン酸(505.6mg、1.4mmol)の溶液を、アルゴン雰囲気下で加え、次に0℃に冷却した。DIPEA(0.243mL、1.76mol)、7−ヒドロキシベンゾトリアゾール(HOBt)(189.2mg、1.4mmol)及びHATU(532.28mg、1.4mmol)を溶液に加え、30分後、冷浴を取り外した。反応混合物を室温で2時間撹拌し、NH4Clの飽和水溶液で停止させ、水に注ぎ、DCMで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 20:1〜15:1)により精製して、アミド28b(780.4mg;収率:77%)を白色の泡状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.70-7.68 (m, 4H), 7.43-7.36 (m, 6H), 7.02 (d, 1H, J = 12.3 Hz), 7.00 (d, 1H, J = 10.8 Hz), 6.75 (d, 1H, J = 12.3 Hz), 6.66 (t, 1H, J = 9.3 Hz), 6.26 (d, 1H, J = 9.6 Hz), 5.57-5.34 (m, 1H), 5.38-5.28 (m, 1H), 4.83 (dd, 1H, J = 16.5, 7.8 Hz), 4.31 (d, 1H, J = 9.6 Hz), 3.89-3.82 (m, 1H), 2.26-2.02 (m, 4H), 1.50-1.42 (m, 6H), 1.43 (d, 3H, J = 6.9 Hz), 1.33-1.20 (m, 6H), 1.06 (s, 9H), 0.96 (s, 9H), 0.95-0.83 (m, 15H).
13C-RMN (CDCl3, 75 MHz) δ: 168.0; 166.2; 153.8; 136.3; 136.1; 134.3; 130.0; 127.8; 126.7; 126.0; 121.6; 109.0; 72.6; 60.7; 35.7; 34.0; 32.7; 29..5; 27.7; 27.2; 26.7; 19.5; 14.0; 13.2; 11.8.
【0186】
中間体28cの合成
【0187】
【化51】
【0188】
DCM/DMF(10:1、7.7mL)中のアミン27c(170mg、0.437mmol)の溶液に、無水DCM中の(Z)−3−トリブチルスタンニルプロペン酸(197.2mg、0.546mmol)の溶液を、アルゴン雰囲気下で加え、次に0℃に冷却した。DIPEA(0.11mL、0.655mmol)、HOAt(59.4mg、0.437mmol)及びHATU(166mg、0.437mmol)を溶液に加え、30分後、冷浴を取り外した。反応混合物を室温で2時間撹拌し、NH4Clの飽和水溶液で停止させ、水に注ぎ、DCMで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 20:1〜15:1)により精製して、アミド28c(250mg、収率:78%)を白色の泡状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.94 (d, 1H, J = 10.8 Hz), 7.00 (d, 1H, J = 12.3 Hz), 6.75 (d, 1H, J = 12.3 Hz), 6.72 (t, 1H, J = 9.5 Hz), 6.50 (d, 1H, J = 9.0 Hz), 5.56 (t, J = 6.6 Hz, 1H), 4.83 (q, 1H, J = 9.0 Hz), 4.41 (t, 1H, J = 9.0 Hz), 3.76 (m, 1H), 2.17 (m, 4H), 2.01 (s, 3H), 1.45 (m, 7H), 1.25 (m, 8H), 0.88 (s, 9H), 0.84 (m, 19H), 0.06 (s, 6H).
13C-RMN (CDCl3, 75 MHz) δ: 169.2, 166.8, 153.8, 136.2, 131.1, 123.9, 122.6, 108.7, 71.6, 59.2, 36.5, 33.7, 31.4, 29.5, 29.4, 27.6, 26.1, 21.3, 19.5, 18.5, 18.3, 14.0, 11.8, -4.3, -4.4.
【0189】
中間体28dの合成
【0190】
【化52】
【0191】
DCM/DMF(10:1、1.3mL)中のアミン26d(44mg、0.1mmol)の溶液に、無水DCM中の(Z)−3−トリブチルスタンニルプロペン酸(45mg、0.125mmol)の溶液を、アルゴン雰囲気下で加え、次に0℃に冷却した。DIPEA(26μL、0.15mmol)、HOAt(13.6mg、0.1mmol)及びHATU(38mg、0.1mmol)を溶液に加え、30分後、冷浴を取り外した。反応混合物を室温で2時間撹拌し、NH4Clの飽和水溶液で停止させ、水に注ぎ、DCMで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 20:1〜15:1)により精製して、アミド28d(60mg、収率:80%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.43 (d, 1H, J = 10.8 Hz), 7.34-7.22 (m, 5H), 7.02 (d, 1H, J = 12.3 Hz), 6.70 (d, 1H, J = 12.3 Hz), 6.66 (dd, 1H, J = 9.9, 9.3 Hz), 6.34 (d, 1H, J = 7.8 Hz), 5.51 (dd, 1H, J = 8.1, 7.5 Hz), 4.81-4.71 (m, 2H), 3.68-3.59 (m, 1H), 3.18 (dd, 1H, J = 13.5, 6 Hz), 2.69 (dd, 1H, J = 13.5, 8.4 Hz), 2.11-2.04 (m, 2H), 2.01 (s, 3H), 1.96-1.87 (m, 1H), 1.80-1.70 (m, 1H), 1.53-1.43 (m, 8H), 1.31-1.24 (m, 10H), 0.89-0.85 (m, 9H), 0.88 (s, 9H), 0.04 (s, 3H), 0.01 (s, 3H).
13C NMR (CDCl3, 75 MHz) δ: 168.5, 166.5, 154.4, 136.7, 135.9, 131.0, 129.5, 129.1, 127.4, 124.0, 122.3, 108.8, 71.5, 55.1, 38.8, 36.6, 33.3, 29.5, 29.4, 27.6, 26.0, 21.3, 18.2, 14.0, 11.8, -4.3, -4.5.
MS (ES) m/z 781.2 [M+H]+, 803.2 [M+Na]+
【0192】
中間体28eの合成
【0193】
【化53】
【0194】
DCM/DMF(10:1、1mL)中のアミン27a(30mg、0.075mmol)の溶液に、無水DCM中の(E)−3−トリブチルスタンニルプロペン酸(33.5mg、0.095mmol)の溶液を、アルゴン雰囲気下で加え、次に0℃に冷却した。DIPEA(19μL、0.11mol)、HOAt(10mg、0.075mmol)及びHATU(27.5mg、0.075mmol)を溶液に加え、30分後、冷浴を取り外した。反応混合物を室温で2時間撹拌し、NH4Clの飽和水溶液で停止させ、水に注ぎ、DCMで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 6:1)により精製して、アミド28e(25mg、収率:45%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.68 (d, 1H, J = 9.2 Hz), 7.52 (d, 1H, J = 18.9 Hz), 6.73 (t, 1H, J = 9.2 Hz), 6.28 (d, 1H, J = 10.8 Hz), 6.25 (d, 1H, J = 18.9 Hz), 5.60 (t, 1H, J = 7.2 Hz), 4.83 (q, 1H, J = 9.2 Hz), 4.40 (d, 1H, J = 9.6 Hz), 3.77 (m, 1H), 2.17 (m, 4H), 2.01 (s, 3H), 1.45 (m, 6H), 1.25 (m, 8H), 1.0 (s, 9H), 0.88 (s, 9H), 0.84 (m, 13H), 0.06 (s, 6H).
【0195】
実施例8
スキーム5は、本発明の幾つかの化合物の合成を提供する。
【0196】
【化54】
【0197】
化合物29aの合成
【0198】
【化55】
【0199】
1−メチル−2−ピロリジノン(NMP)(14.7mL)中のアルケニルスタンナン28a(1.1g、1.47mmol)及び17a(0.62g、1.77mmol)の溶液に、0℃で、チオフェンカルボン酸銅(CuTC)(422mg、2.2mmol)を加えた。反応を、0℃で45分間、室温で20分間撹拌した。次に、粗混合物を中性アルミナのプラグで濾過し、EtOAc/エーテル 50:50で洗浄し、合わせた濾液を0.5N HCl(3×15mL)で洗浄した。有機溶液を乾燥し、蒸発させて粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 5:1〜1:1)により精製して、トリエン29a(0.66g、収率:66%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.89 (d, 1H, J = 10.8 Hz), 7.22 (dd, 1H, J = 12.3, 11.4 Hz), 6.86 (dd, 1H, J = 11.7, 11.4 Hz), 6.70 (dd, 1H, J = 9.9, 9.3 Hz), 6.35 (d, 1H, J = 9.3 Hz), 6.13 (d, 1H, J = 11.4 Hz), 5.66 (d, 1H, J = 11.4 Hz), 5.60 (dd, 1H, J = 5.4, 3.9 Hz), 5.55 (br t, 1H, J = 7.8 Hz), 5.26 (d, 1H, J = 10.2 Hz), 4.84-4.76 (m, 1H), 4.3 (d, 1H, J = 9.3 Hz), 4.20-4.16 (m, 1H), 3.77-3.69 (m, 1H), 3.63 (s, 3H), 2.89-2.77 (m, 1H), 2.41-2.33 (m, 2H), 2.19-2.13 (m, 4H), 2.00 (s, 3H), 1.82 (s, 3H), 1.13 (d, 3H, J = 6.9 Hz), 1.02 (s, 9H), 0.86 (s, 9H), 0.4 (s, 3H), 0.03 (s, 3H).
13C-RMN (CDCl3, 75 MHz) δ: 168.5; 166.4; 161.8; 145.4; 140.3, 137.3; 134.4; 134.3; 131.0, 124.3; 124.1, 122.4; 121.2; 108.7; 108.4; 82.0; 71.6; 60.6; 55.6; 37.5; 36.5, 35.1; 33.8; 26.5; 26.0; 21.3, 18.3, 17.4, 16.9, -4.3, -4.4.
【0200】
化合物29bの合成
【0201】
【化56】
【0202】
NMP(9mL)中のアルケニルスタンナン28b(780.4mg、0.904mmol)及び17a(377.4mg、1.085mmol)の溶液に、0℃で、チオフェンカルボン酸銅(258.5mg、1.36mmol)を加えた。反応を、0℃で45分間、室温で20分間撹拌した。次に、粗混合物を中性アルミナのプラグで濾過し、EtOAc/エーテル 50:50で洗浄し、合わせた濾液を0.5N HCl(3×10mL)で洗浄した。有機溶液を乾燥し、蒸発させて粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 5:1〜1:1)により精製して、トリエン29b(459.7mg、収率:66%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.66-7.64 (m, 4H), 7.43-7.32 (m, 7H), 7.23 (t, 1H, J = 11.7 Hz), 6.85 (t, 1H, J = 11.7 Hz), 6.62 (dd, 1H, J = 10.5, 9.3 Hz), 6.41 (d, 1H, J = 9.3 Hz), 6.11 (d, 1H, J = 11.7 Hz), 5.66 (d, 1H, J = 11.4 Hz), 5.60 (dd, 1H, J = 5.7, 5.1 Hz), 5.49-5.41 (m, 1H), 5.32-5.27 (m, 1H), 5.25 (d, 1H, J = 9.9 Hz), 4.83-4.75 (m, 1H), 4.32 (d, 1H, J = 9.3 Hz), 4.22-4.15 (m, 1H), 3.83-3.78 (m, 1H), 3.62 (s, 3H), 2.86-2.78 (m, 1H), 2.40-2.35 (m, 2H), 2.20-2.04 (m, 4H), 1.81 (s, 3H), 1.40 (d, 3H, J = 6.9 Hz), 1.13 (d, 3H, J = 6.9 Hz), 1.03 (s, 9H), 0.97 (s, 9H).
13C-RMN (CDCl3, 75 MHz) δ: 168.3; 166.3; 161.8; 145.4; 140.2, 137.3; 136.1; 134.8; 134.4; 134.3; 129.9; 127.8;126.4; 126.1; 124.4; 121.7; 121.2; 108.4; 109.1; 82.0; 72.6; 60.6; 55.6; 37.5; 35.2; 32.7; 31.1; 27.2; 26.8, 26.5; 19.5; 17.4; 16.9; 13.1.
【0203】
化合物29cの合成
【0204】
【化57】
【0205】
NMP(2.5mL)中のアルケニルスタンナン28c(250mg、0.34mmol)及び17a(142mg、0.409mmol)の溶液に、0℃で、チオフェンカルボン酸銅(97mg、0.51mmol)を加えた。反応を、0℃で45分間、室温で20分間撹拌した。次に、粗混合物を中性アルミナのプラグで濾過し、EtOAc/エーテル 50:50で洗浄し、合わせた濾液を0.5N HCl(3×10mL)で洗浄した。有機溶液を乾燥し、蒸発させて粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 10:1〜6:1)により精製して、トリエン29c(150mg、収率:67%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 8.21 (d, 1H, J = 10.8 Hz), 7.28 (t, 1H, J = 11.7 Hz), 6.88 (dd, 1H, J = 11.7, 11.4 Hz), 6.72 (dd, 1H, J = 10.2, 9.3 Hz), 6.42 (d, 1H, J = 8.4 Hz), 6.15 (d, 1H, J = 11.7 Hz), 5.66 (d, 1H, J = 11.4 Hz), 5.61 (dd, 1H, J = 5.7, 3.6 Hz), 5.56 (br t, 1H, J = 8.1 Hz), 5.27 (d, 1H, J = 9.9 Hz), 4.85-4.77 (m, 1H), 4.30 (dd, 1H, J = 8.1, 7.5 Hz), 4.24-4.16 (m, 1H), 3.79-3.72 (m, 1H), 3.66 (s, 3H), 2.88-2.80 (m, 1H), 2.42-2.37 (m, 2H), 2.18-2.14 (m, 5H), 2.00 (s, 3H), 1.83 (s, 3H), 1.14 (d, 3H J = 6.9 Hz), 0.97 (d, 3H, J = 6.6 Hz), 0.96 (d, 3H, J = 6.6 Hz), 0.86 (s, 9H), 0.4 (s, 6H).
13C-RMN (CDCl3, 75 MHz) δ: 169.2 166.8; 161.8; 145.4; 140.5, 137.7; 134.6; 134.3; 131.0, 124.3; 124.2, 122.6; 121.2; 108.6; 108.4; 82.0; 71.5; 58.9; 55.6; 37.5; 36.4; 33.8; 30.8, 26.5; 26.1; 21.3, 19.6, 18.5, 18.3, 17.4, 16.9, -4.3, -4.4.
【0206】
化合物29dの合成
【0207】
【化58】
【0208】
NMP(1mL)中のアルケニルスタンナン28d(60mg、0.08mmol)及び17a(32.4mg、0.09mmol)の溶液に、0℃で、チオフェンカルボン酸銅(22mg、0.12mmol)を加えた。反応を、0℃で45分間、室温で20分間撹拌した。次に、粗混合物を中性アルミナのプラグで濾過し、EtOAc/エーテル 50:50で洗浄し、合わせた濾液を0.5N HCl(3×10mL)で洗浄した。有機溶液を乾燥し、蒸発させて粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 4:1〜1:1)により精製して、トリエン29d(13mg、収率:25%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.59 (d, 1H, J = 11.1 Hz), 7.33-7.24 (m, 5H), 7.23 (t, 1H, J = 11.7 Hz), 6.90 (dd, 1H, J = 11.7, 11.4 Hz), 6.66 (dd, 1H, J = 10.5, 9 Hz), 6.24 (d, 1H, J = 7.2 Hz), 6.17 (d, 1H, J = 12.0 Hz), 5.63-5.58 (m, 2H), 5.51 (td, 1H, J = 7.8, 1.2 Hz), 5.28 (d, 1H, J = 10.8 Hz), 4.79-4.67 (m, 2H), 4.24-4.17 (m, 1H), 3.66 (s, 3H), 3.65-3.62 (m, 1H), 3.22 (dd, 1H, J = 13.5, 6.3 Hz), 3.04 (dd, 1H, J = 13.8, 8.4 Hz), 2.89-2.81 (m, 1H), 2.43-2.37 (m, 2H), 2.11-2.04 (m, 2H), 2.00 (s, 3H), 1.84 (s, 3H), 1.93-1.72 (m, 2H), 1.16 (d, 3H, J = 6.9 Hz), 0.86 (s, 9H), 0.03 (s, 3H), 0.01 (s, 3H).
13C NMR (CDCl3, 75 MHz) δ: 168.4, 166.5, 161.7, 145.5, 140.8, 138.1, 136.8, 134.5, 134.3, 131.0, 129.5, 129.1, 127.4, 124.2, 124.1, 122.3, 120.4, 108.7, 108.3, 82.0, 71.4, 55.7, 54.9, 38.3, 37.5, 36.6, 33.4, 26.5, 26.0, 21.3, 18.2, 17.4, 16.9, -4.3, -4.4.
MS (ES) m/z 711.2 [M+H]+
【0209】
化合物29eの合成
【0210】
【化59】
【0211】
NMP(1mL)中のアルケニルスタンナン28e(50mg、0.067mmol)及び17a(28mg、0.08mmol)の溶液に、0℃で、チオフェンカルボン酸銅(19.1mg、0.10mmol)を加えた。反応を、0℃で45分間、室温で20分間撹拌した。次に、粗混合物を中性アルミナのプラグで濾過し、EtOAc/エーテル 50:50で洗浄し、合わせた濾液を0.5N HCl(3×10mL)で洗浄した。有機溶液を乾燥し、蒸発させて粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 5:1〜1:1)により精製して、トリエン29e(33mg、収率:50%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.73 (d, 1H, J = 11.4 Hz), 7.70 (dd, 1H, J = 14.1, 11.7 Hz), 6.71 (dd, 1H J = 9.9, 9.7 Hz), 6.30 (d, 1H, J = 9.3 Hz), 6.13 (d, 1H, J = 12.9 Hz), 6.04 (dd, 1H, J = 11.7, 11.4 Hz), 5.93 (d, 1H, J = 15.0 Hz), 5.63 (br t, 1H, J = 4.5 Hz), 5.58-5.53 (m, 1H), 5.34 (d, 1H, J = 9.9 Hz), 4.85-4.78 (m, 1H), 4.41 (d, 1H, J = 9.3), 4.24-4.16 (m, 1H), 3.77-3.72 (m, 1H), 3.64 (s, 3H), 2.90-2.78 (m, 1H), 2.45-2.41 (m, 2H), 2.19-2.12 (m, 4H), 2.01 (s, 3H), 1.91 (s, 3H), 1.16 (d, 3H, J = 6.6 Hz), 1.02 (s, 9H), 0.87 (s, 9H), 0.06 (s, 3H), 0.04 (s, 3H).
13C NMR (CDCl3, 75 MHz) δ: 168.5, 166.1, 161.8, 145.4, 140.7, 138.0, 135.3, 134.9, 131.1, 125.8, 124.9, 124.0, 122.4, 108.7, 108.5, 81.9, 71.6, 60.9, 55.6, 37.6, 36.5, 35.2, 33.8, 29.9, 26.8, 26.1, 21.3, 18.3, 17.3, 16.9, -4.3, -4.4.
【0212】
化合物29fの合成
【0213】
【化60】
【0214】
NMP(0.9mL)中のアルケニルスタンナン28a(60mg、0.083mmol)及び17b(29mg、0.09mmol)の溶液に、0℃で、チオフェンカルボン酸銅(24mg、0.12mmol)を加えた。反応を、0℃で45分間、室温で20分間撹拌した。次に、粗混合物を中性アルミナのプラグで濾過し、EtOAc/エーテル 50:50で洗浄し、合わせた濾液を0.5N HCl(3×10mL)で洗浄した。有機溶液を乾燥し、蒸発させて粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 5:1〜1:1)により精製して、アミド29f(27mg、収率:50%)を油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 7.62 (d, 1H, J = 10.5 Hz), 7.25 (dd, 1H, J = 12.6, 11.4 Hz), 6.94-6.84 (m, 2H), 6.73 (dd, 1H, J = 10.5, 9.0 Hz), 6.23 (d, 1H, J = 9.3 Hz), 6.17 (d, 1H, J = 11.4 Hz), 6.06-6.01 (m, 1H), 5.66 (d, 1H, J = 11.4 Hz), 5.60-5.55 (m, 1H), 5.29 (d, 1H, J = 9.9 Hz), 4.88-4.80 (m, 1H), 4.34 (d, 1H, J = 9.3 Hz), 4.27-4.19 (m, 1H), 3.79-3.72 (m, 1H), 2.90-2.81 (m, 1H), 2.36-2.30 (m, 2H), 2.21-2.13 (m, 4H), 2.03 (s, 3H), 1.85 (s, 3H), 1.17 (d, 3H, J = 6.6 Hz), 1.03 (s, 9H), 0.89 (s, 9H), 0.08 (s, 3H), 0.06 (s, 3H).
【0215】
化合物30aの合成
【0216】
【化61】
【0217】
THF(6mL)中の29a(275mg、0.41mmol)の溶液に、N2下及び室温で、THF(0.82mL、0.82mmol)中の1M TBAFを加えた。反応を室温で18時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、EtOAcで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製して、アルコール30a(175mg;収率:76%)を白色の固体として得た。
1H NMR (CDCl3, 300 MHz) δ: 9.00 (d, 1H, J = 10.2 Hz), 7.25 (dd, 1H, J = 12.0, 11.4 Hz), 6.86 (dd, 1H, J = 11.7, 11.4 Hz), 6.72 (dd, 1H, J = 9.6, 8.7 Hz), 6.68 (d, 1H, J = 8.7 Hz), 6.13 (d, 1H, J = 11.7 Hz), 5.68 (d, 1H, J = 11.4 Hz), 5.63-5.58 (m, 2H), 5.27 (d, 1H, J = 10.2 Hz), 4.85-4.76 (m, 1H), 4.42 (d, 1H, J = 9.3Hz), 4.25-4.17 (m, 1H), 3.70-3.69 (m, 1H), 3.63 (s, 3H), 3.48 (br s, 1H), 2.89-2.75 (m, 1H), 2.42-2.36 (m, 2H), 2.22-2.11 (m, 4H), 2.04 (s, 3H), 1.82 (s, 3H), 1.14 (d, 3H, J = 6.6 Hz), 1.03 (s, 9H).
【0218】
化合物30aの異性体(21S)−は、合成方法をラセミフラグメントDから出発して実施したときに得た。異性体〔(21S)−化合物30a及び(21R)−化合物30a〕の最終混合物を、半分取逆相HPLC(SymmetryPrep C18 7μm、7.8×150mm、MeCNが50〜60%の勾配H2O:MeCNを30分、UV検出、流速2.5mL/分、〔保持時間((21S−)30a):15.4分、保持時間((21R)−30a):14.7分〕)により分離し、(21S)−化合物30aを純粋な形態で得た。
【0219】
【化62】
【0220】
1H NMR (CDCl3, 300 MHz) δ: 8.62 (d, 1H, J = 10.2 Hz), 7.28-22 (m, 1H), 6.93-6.86 (m, 1H), 6.81-6.75 (m, 1H), 6.32 (d, 1H, J = 9.0 Hz), 6.17 (d, 1H, J = 11.7 Hz), 5.68-5.58 (m, 3H), 5.28 (d, 1H, J = 10.2 Hz), 4.93-4.84 (m, 1H), 4.32 (d, 1H, J = 9.3Hz), 4.25-4.17 (m, 1H), 3.78-3.67 (m, 1H), 3.66 (s, 3H), 2.89-2.81 (m, 1H), 2.43-2.38 (m, 2H), 2.28-2.20 (m, 4H), 2.08 (s, 3H), 1.84 (s, 3H), 1.16 (d, 3H J = 6.9 Hz), 1.02 (s, 9H).
【0221】
化合物30bの合成
【0222】
【化63】
【0223】
THF(7.5mL)中の29b(586mg、0.76mmol)の溶液に、N2下及び室温で、THF(1.53mL、2mmol)中の1M TBAFを加えた。反応を室温で18時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、EtOAcで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製して、アルコール30b(320mg、収率:80%)を白色の固体として得た。
1H NMR (CDCl3, 300 MHz) δ: 8.95 (d, 1H, J = 10.2 Hz), 7.25 (t, 1H, J = 12.0 Hz), 6.85 (t, 1H, J = 11.7 Hz), 6.73 (t, 1H, J = 9.6 Hz), 6.57 (d, 1H, J = 8.7 Hz), 6.12 (d, 1H, J = 11.4 Hz), 5.67 (d, 1H, J = 11.4 Hz), 5.61 (dd, 1H, J = 5.4, 3.9 Hz), 5.63-5.58 (m, 1H), 5.44-5.35 (m, 1H), 5.26 (d, 1H, J = 9.9 Hz), 4.86 (q, 1H, J = 8.1 Hz), 4.38 (d, 1H, J = 9.3 Hz), 4.24-4.16 (m, 1H), 3.81-3.71 (m, 1H), 3.64 (s, 3H), 2.96-2.92 (m, 1H), 2.86-2.79 (m, 1H), 2.41-2.37 (m, 2H), 2.28-2.14 (m, 4H), 1.82 (s, 3H), 1.61 (d, 3H, J = 6.6 Hz), 1.14 (d, 3H, J = 6.6 Hz), 1.02 (s, 9H).
13C-RMN (CDCl3, 75 MHz) δ: 168.7; 166.6; 161.8; 145.4; 140.3; 137.5; 134.4; 134.3; 127.7; 126.0; 124.4; 123.7; 121.1; 108.9; 108.4; 82.0; 72.1; 60.9; 55.7; 37.6; 35.0; 34.8; 33.2; 26.9; 26.5; 17.4; 16.9; 13.3.
【0224】
化合物30cの合成
【0225】
【化64】
【0226】
THF(4.8mL)中の29c(150mg、0.23mmol)の溶液に、N2下及び室温で、THF(0.45mL、0.45mmol)中の1M TBAFを加えた。反応を室温で18時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、EtOAcで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製して、アルコール30c(90mg、収率:73%)を白色の固体として得た。
1H NMR (CDCl3, 300 MHz) δ: 9.16 (d, 1H, J = 10.2 Hz), 7.26 (dd, 1H, J = 12.0, 11.1 Hz), 6.87 (dd, 1H, J = 11.7, 11.4 Hz), 6.79-6.70 (m, 2H), 6.14 (d, 1H, J = 11.7 Hz), 5.68 (d, 1H, J = 11.7 Hz), 5.63-5.58 (m, 2H), 5.27 (d, 1H, J = 9.6 Hz), 4.85-4.76 (m, 1H), 4.35 (dd, 1H, J = 8.4, 7.5 Hz), 4.24-4.17 (m, 1H), 3.70-3.69 (m, 1H), 3.63 (s, 3H), 3.43 (br s, 1H), 2.89-2.76 (m, 1H), 2.42-2.36 (m, 2H), 2.21-2.14 (m, 4H), 2.03 (s, 3H), 1.82 (s, 3H), 1.13 (d, 3H J = 6.9 Hz), 0.96 (d, 6H, J = 6.6 Hz).
13C-RMN (CDCl3, 75 MHz) δ: 169.7, 167.1, 161.8, 145.4, 140.5, 137.8, 134.6, 134.2, 131.6, 124.4, 123.9, 123.8, 120.7, 108.5, 108.4, 82.0, 59.1, 55.7, 37.5, 36.4, 33.5, 31.0, 26.5, 21.3, 19.5, 18.7, 17.4, 16.8.
MS (ES) m/z 549.0 [M+H]+, 571.1 [M+Na]+
【0227】
化合物30dの合成
【0228】
【化65】
【0229】
THF(0.32mL)中の29d(11mg、0.02mmol)の溶液に、N2下及び室温で、THF(0.03mL、0.03mmol)中の1M TBAFを加えた。反応を室温で18時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、EtOAcで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製して、アルコール30d(6mg、収率:65%)を白色の固体として得た。
1H NMR (CDCl3, 300 MHz) δ: 8.54 (d, 1H, J = 9.9 Hz), 7.34-7.23 (m, 5H), 7.20 (t, 1H, J = 11.7 Hz), 6.89 (dd, 1H, J = 11.7, 11.4 Hz), 6.72 (dd, 1H, J = 9.9, 9.3 Hz), 6.27 (d, 1H, J = 8.1 Hz), 6.17 (d, 1H, J = 11.7 Hz), 5.63-5.53 (m, 3H), 5.28 (d, 1H, J = 10.2 Hz), 4.84-4.76 (m, 1H), 4.75-4.68(m, 1H), 4.25-4.17 (m, 1H), 3.66 (s, 3H), 3.67-3.65 (m, 1H), 3.18 (dd, 1H, J = 13.8, 6.3 Hz), 3.06 (dd, 1H, J = 13.8, 8.1 Hz), 2.89-2.81 (m, 1H), 2.43-2.38 (m, 2H), 2.15-2.10 (m, 3H), 2.06 (s, 3H), 1.92-1.87 (m, 1H), 1.84 (s, 3H), 1.16 (d, 3H, J = 6.6 Hz).
13C NMR (CDCl3, 75 MHz) δ: 168.6, 166.6, 160.8, 145.8, 140.8, 138.1, 136.8, 134.6, 134.2 ,129.6, 129.0, 127.2, 124.1, 124.0, 123.4, 120.4, 108.3, 108.2, 82.0, 71.6, 55.7, 55.0, 38.4, 37.5, 36.4, 33.0, 26.5, 21.3, 17.4, 16.9.
MS (ES) m/z 597.2 [M+H]+.
【0230】
化合物30eの合成
【0231】
【化66】
【0232】
THF(1mL)中の29e(32mg、0.047mmol)の溶液に、N2下及び室温で、THF(0.094mL、0.094mmol)中の1M TBAFを加えた。反応を室温で18時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、EtOAcで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製して、アルコール30e(14mg、収率:55%)を白色の泡状物として得た。
1H NMR (CDCl3, 500 MHz) δ: 8.97 (d, 1H, J = 10.2 Hz), 7.71 (dd, 1H J = 14.7, 11.7 Hz), 6.74 (dd, 1H J = 9.3, 9.9 Hz), 6.57 (d, 1H, J = 9.0 Hz), 6.15 (d, 1H, J = 11.7 Hz), 6.03 (dd, 1H, J = 11.7, 11.4 Hz), 5.95 (d, 1H, J = 14.7 Hz), 5.65-5.58 (m, 2H), 5.35 (d, 1H, J = 9.9 Hz), 4.87-4.78 (m, 1H), 4.42 (d, 1H, J = 9.3), 4.25-4.18 (m, 1H), 3.72-3.68 (m, 1H), 3.65 (s, 3H), 3.25 (br s, 1H), 2.87-2.79 (m, 1H), 2.45-2.40 (m, 2H), 2.23-2.12 (m, 4H), 2.04 (s, 3H), 1.89 (s, 3H), 1.15 (d, 3H, J = 6.6 Hz).1.03 (s, 9H).
13C NMR (CDCl3, 75 MHz) δ: 168.8, 166.5, 161.68, 145.3, 140.9, 138.2, 135.4, 134.7, 132.0, 125.68, 124.6, 123.9, 123.6, 108.6, 108.4, 81.9, 71.7, 61.3, 55.7, 37.5, 36.5, 36.3, 34.9, 33.3, 26.9, 26.7, 21.3, 17.0, 16.7.
MS (ES) m/z 563.3 [M+H]+, 585.2 [M+Na]+
【0233】
化合物30fの合成
【0234】
【化67】
【0235】
THF(1mL)中の29f(28mg、0.04mmol)の溶液に、N2下及び室温で、THF(0.09mL、0.09mmol)中の1M TBAFを加えた。反応を室温で18時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、EtOAcで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製して、アルコール30f(17mg;収率:75%)を白色の固体として得た。
1H NMR (CDCl3, 300 MHz) δ: 9.05 (d, 1H, J = 10.2 Hz), 7.35 (m, 2H), 7.0 (dd, 1H, J = 11.7, 11.4 Hz), 6.73 (dd, 1H, J = 9.6, 8.7 Hz), 6.56 (d, 1H, J = 8.7 Hz), 6.05 (m, 3H), 5.63-5.58 (m, 2H), 5.30 (d, 1H, J = 10.2 Hz), 4.78 (m, 1H), 4.50 (d, 1H, J = 9.3Hz), 3.68 (m, 1H), 3.48 (br s, 1H), 2.45 (m, 1H), 2.42-2.36 (m, 2H), 2.22-2.11 (m, 4H), 2.04 (s, 3H), 1.82 (s, 3H), 1.14 (d, 3H J = 6.6 Hz), 1.03 (s, 9H).
【0236】
化合物1の合成
【0237】
【化68】
【0238】
ジクロロメタン(7.5mL)中の30a(300mg、0.53mmol)の溶液に、0℃で、トリクロロアセチルイソシアネート(TCAI)(76μl、0.64mmol)を加えた。反応を0℃で30分間撹拌し、次に中性酸化アルミニウムを加えた。混合物を5〜30分間撹拌し、次に酸化アルミニウムパッドに浸漬させた。生成物を、DCM/MeOH 50:1の混合物を使用して洗浄した。濾液を真空下で蒸発させて、粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 2:1〜1:2)により精製した。化合物1(0.26g、収率:81%)を白色の固体として得て、実施例2に報告されているものと同等の物理的特性及び分光特性(1H、13C RMN及びMS)を示した。
【0239】
化合物1の異性体(21S)−を、この2つの方法のいずれかにより得た:
A.−異性体(21R)−及び(21S)−化合物30aの混合物から出発し、半分取逆相HPLC(SymmetryPrep C18 7μm、7.8×150mm、MeOHが50〜100%の勾配H2O:MeOHを30分、UV検出、流速2.5mL/分、〔保持時間((21S)−1):19.2分、保持時間 ((21R)−1):19.8分〕)による(21S)−化合物1の最終分離と同じ手順に従う。
B.−化合物1について開示されたものと同じであるが、純粋な(21S)−化合物30aから出発する手順に従う。
【0240】
【化69】
【0241】
1H NMR (CDCl3, 500 MHz) δ: 8.69 (d, 1H, J = 10.5 Hz), 7.30 (t, 1H, J = 11.5 Hz), 6.90 (t, 1H, J = 11.5 Hz), 6.86-6.82 (m, 1H), 6.34 (d, 1H, J = 9.0 Hz), 6.17 (d, 1H, J = 11.5 Hz), 5.66 (d, 1H, J = 11.5 Hz), 5.64-5.62 (m, 1H), 5.59-5.56 (m, 1H), 5.29 (d, 1H, J = 9.5 Hz), 4.81-4.77 (m, 1H), 4.50-4.45 (m, 1H), 4.42 (d, 1H, J = 9.5 Hz), 4.25-4.20 (m, 1H), 3.66 (s, 3H), 2.89-2.81 (m, 1H), 2.44-2.31 (m, 5H), 2.24-2.17 (m, 1H), 2.06 (s, 3H), 1.84 (s, 3H), 1.16 (d, 3H, J = 6.5 Hz), 1.04 (s, 9H).
13C-RMN (125 MHz, CDCl3) δ: 168.4, 166.1, 157.2, 148.3, 145.2, 140.2, 137.4, 134.1, 134.0, 132.0, 124.7, 124.2, 122.4, 120.7, 108.1, 104.7, 81.8, 75.0, 60.8, 55.4, 37.2, 34.8, 32.5, 30.3, 26.7, 26.2, 21.0, 17.1, 16.6.
【0242】
化合物4の合成
【0243】
【化70】
【0244】
ジクロロメタン(1mL)中の30b(56mg、0.105mmol)の溶液に、0℃で、トリクロロアセチルイソシアネート(15μL、0.126mmol)を加えた。反応を0℃で30分間撹拌し、次に中性酸化アルミニウムを加えた。混合物を5〜30分間撹拌し、次に酸化アルミニウムパッドに浸漬させた。生成物を、DCM/MeOH 50:1の混合物を使用して洗浄した。濾液を真空下で蒸発させて、粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製した。化合物4(57.6mg、収率:96%)を白色の泡状物として得て、実施例3に報告されているものと同等の物理的特性及び分光特性(1H、13C RMN及びMS)を示した。
【0245】
化合物5の合成
【0246】
【化71】
【0247】
ジクロロメタン(2mL)中の30c(115mg、0.21mmol)の溶液に、0℃で、トリクロロアセチルイソシアネート(27μl、0.23mmol)を加えた。反応を0℃で30分間撹拌し、次に中性酸化アルミニウムを加えた。混合物を5〜30分間撹拌し、次に酸化アルミニウムパッドに浸漬させた。生成物を、DCM/MeOH 50:1の混合物を使用して洗浄した。濾液を真空下で蒸発させて、粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製した。化合物5(71mg、収率:57%)を白色の泡状物として得て、実施例3に報告されているものと同等の物理的特性及び分光特性(1H、13C RMN及びMS)を示した。
【0248】
異性体の最終混合物(15mg)を、半分取逆相HPLC(SymmetryPrep C18 7μm、7.8×150mm、MeOHが50〜70%の勾配H2O:MeOHを75分、UV検出、流速2.5mL/分、〔保持時間((15R)−7):18.15分、保持時間((15S)−7):19.62分〕)により分離し、3.1mgの純粋な(15R)−化合物7及び2.9mgの純粋な(15S)−化合物7を得た。
【0249】
化合物5の異性体(21S)−は、5を半逆相HPLC(SymmetryPrep C8、MeCNが45〜50%の勾配H2O:MeCNを30分、UV検出、流速4.7mL/分〔保持時間((21−S)−5):21.6分、保持時間((21R)−5):23.6分〕)により精製したときに単離した。両方の異性体を含有する試料(50mg)から出発して、上記の分離の後、39mgの純粋な(21R)−化合物5及び6.1mgの純粋な(21S)−化合物5を得た。
【0250】
【化72】
【0251】
1H NMR (CDCl3, 500 MHz) δ: 8.74 (d, 1H, J = 10.5 Hz), 7.29 (dd, 1H, J = 11.7, 11.4 Hz), 6.94 (dd, 1H, J = 11.7, 11.4 Hz), 6.84 (dd, 1H, J = 10.5, 9.3 Hz), 6.22 (m, 2H), 5.68 (d, 1H, J = 11.5 Hz), 5.63 (m, 2H), 5.42 (d, 1H, J = 9.3 Hz), 4.81 (m, 1H), 4.52 (m, 1H), 4.41 (m, 1H), 4.23 (m,1H), 3.66 (s, 3H), 2.91 (m, 1H), 2.49-2.38 (m, 3H), 2.35-2.31(m, 2H), 2.24-2.17 (m, 2H), 2.05 (s, 3H), 1.82 (s, 3H), 1.15 (d, 3H J = 6.6 Hz), 0.99 (d, 3H, J = 6.9 Hz) 0.96 (d, 3H, J = 6.9 Hz).
MS (ES) m/z 592.3 [M+H]+
【0252】
化合物31の合成
【0253】
【化73】
【0254】
ジクロロメタン(0.7mL)中の30d(5mg、0.008mmol)の溶液に、0℃で、トリクロロアセチルイソシアネート(1.1μl、0.009mmol)を加えた。反応を0℃で30分間撹拌し、次に中性酸化アルミニウムを加えた。混合物を5〜30分間撹拌し、次に酸化アルミニウムパッドに浸漬させた。生成物を、DCM/MeOH 50:1の混合物を使用して洗浄した。濾液を真空下で蒸発させて、粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製した。化合物31(3.5mg、収率:66%)を白色の固体として得た。
1H NMR (CDCl3, 300 MHz) δ: 8.43 (d, 1H, J = 10.5 Hz), 7.29-7.19 (m, 6H,), 6.91 (dd, 1H, J = 11.7, 11.4 Hz), 6.77 (br dd, 1H, J = 10.2, 9.6 Hz), 6.51 (d, 1H, J = 8.1 Hz), 6.16 (d, 1H, J = 11.4 Hz), 5.69-5.63 (m, 2H), 5.59-5.54 (m, 1H), 5.31 (d, 1H, J = 9.3 Hz), 5.12 (br s, 1H,), 4.91-4.84 (m, 1H), 4.76-4.70 (m, 1H), 4.31-4.13 (m, 2H, CH-5), 3.66 (s, 3H), 3.20 (dd, 1H, J = 13.5, 6.9 Hz), 3.09 (dd, 1H, J = 13.5, 6.6 Hz), 2.89-2.81 (m, 1H), 2.48-2.35 (m, 2H), 2.28-2.23 (m, 3H), 2.05 (s, 3H), 2.01-1.90 (m, 1H), 1.81 (s, 3H), 1.15 (d, 3H, J = 6.6 Hz).
13C NMR (CDCl3, 75 MHz) δ: 168.9, 166.5, 161.9, 157.1, 145.4, 140.6, 137.9, 136.4, 134.4, 133.9, 132.1, 129.7, 128.8, 127.2, 124.6, 124.6, 122.7, 120.7, 108.5, 105.6, 82.1, 74.8, 55.7, 54.6, 38.7, 37.2, 33.1, 30.5, 26.2, 21.2, 17.3, 16.4.
【0255】
化合物8の合成
【0256】
【化74】
【0257】
ジクロロメタン(1.7mL)中の30e(13mg、0.023mmol)の溶液に、0℃で、トリクロロアセチルイソシアネート(3μl、0.025mmol)を加えた。反応を0℃で30分間撹拌し、次に中性酸化アルミニウムを加えた。混合物を5〜30分間撹拌し、次に酸化アルミニウムパッドに浸漬させた。生成物を、DCM/MeOH 50:1の混合物を使用して洗浄した。濾液を真空下で蒸発させて、粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製した。化合物8(14.3mg、収率:100%)を白色の固体として得て、実施例4に報告されているものと同等の物理的特性及び分光特性(1H、13C RMN及びMS)を示した。
【0258】
実施例9
スキーム6は、本発明の幾つかの化合物の合成プロセスを示す。
【0259】
【化75】
【0260】
中間体32の合成
【0261】
【化76】
【0262】
アルゴン雰囲気、−78℃で撹拌した、無水THF(59mL)中の4−ホスホノクロトン酸トリエチル(3.7g、14.66mmol)及び18−クラウン−6(6.2g、23.46mmol)の溶液に、KHMDS(28.1ml、14.07mmol)を滴加した。15分後、アルデヒド12(2.35g、5.86mmol)を滴加し、室温で20時間撹拌した。次に、反応を飽和NH4Cl溶液(200mL)で停止させ、EtOAcで希釈した。有機相を乾燥し(Na2SO4)、減圧下で蒸発させた。カラムクロマトグラフィー(ヘキサン/EtOAc 20:1〜10:1)による精製によって、2.7g(収率:93%)のトリエン32を得た。
1H-RMN (300 MHz, CDCl3) δ: 7.31 (dd, 1H, J = 11.2, 15.3 Hz), 6.53 (d, 1H, J = 15.0 Hz), 6.21 (dd, 1H, J = 11.7, 13.8 Hz), 5.84 (d, 1H, J = 15.1 Hz), 5.61 (d, 1H, J = 9.6 Hz), 4.17 (m, 2H), 3.72 (m, 1H), 3.63 (m, 2H), 2.61 (m, 1H), 1.78 (s, 3H), 1.67 (m, 2H), 1.26 (m, 3H), 0.94 (d, 3H, J = 6.7 Hz), 0.87 (s, 18H), 0.01 (m, 12H).
【0263】
中間体33の合成
【0264】
【化77】
【0265】
EtOH(38mL)中の32(3.75g、7.54mmol)の溶液に、p-トルエンスルホン酸ピリジニウム(663mg、2.64mmol)を加えた。反応混合物を室温で17時間撹拌した。次に、溶媒を減圧下で除去し、得られた油状物をカラムクロマトグラフィー(ヘキサン/EtOAc 4:1〜1:1)により精製して、2.11g(収率:73%)のアルコール33を得た。
1H-RMN (300 MHz, CDCl3) δ: 7.31 (dd, 1H, J = 10.8, 15.0 Hz), 6.52 (d, 1H, J = 15.3 Hz), 6.23 (dd, 1H, J = 11.1, 15.0 Hz), 5.86 (d, 1H, J = 15.3 Hz), 5.52 (d, 1H, J = 9.9 Hz), 4.18 (q, 2H, J = 7.5 Hz), 3.72 (m, 3H), 2.73 (m, 1H), 1.82 (s, 3H), 1.68 (m, 2H), 1.28 (t, 3H, J = 7.2 Hz), 0.98 (d, 3H, J = 6.6 Hz), 0.88 (s, 9H), 0.08 (m, 6H).
【0266】
中間体34の合成
【0267】
【化78】
【0268】
アルコール33(130mg、0.34mmol)をDCM(3.4mL)中、不活性雰囲気下、室温で撹拌し、ペルヨージナン(DMP)(288.5mg、0.68mmol)を一度に加えた。反応を、完了するまで(TLC、約1時間)撹拌し、次に、NaHCO3(飽和溶液)で停止させ、DCMで抽出し、ブラインで洗浄し、硫酸マグネシウムで乾燥し、濾過し、真空下で濃縮した。生成物を、EtOAc/ヘキサン 1:4を用いて溶離するカラムクロマトグラフィーにより精製して、約125mg(収率:96%)のアルデヒド34を無色の油状物として得た。
1H-RMN (300 MHz, CDCl3) δ: 9.79 (s, 1H), 7.31 (dd, 1H, J = 11.1, 15.3 Hz), 6.52 (d, 1H, J = 15.3 Hz), 6.25 (dd, 1H, J = 11.1, 15.3 Hz), 5.87 (d, 1H, J = 15.3 Hz), 5.48 (d, 1H, J = 10.5 Hz), 4.19 (q, 2H, J = 7.2 Hz), 4.03 (m, 1H), 2.69 (m, 1H), 2.54 (m, 2H), 1.80 (s, 3H), 1.29 (t, 3H, J = 6.9 Hz), 1.01 (d, 3H, J = 6.9 Hz), 0.88 (s, 9H), 0.06 (m, 6H).
【0269】
中間体35の合成
【0270】
【化79】
【0271】
アルゴン雰囲気、−78℃で撹拌した、無水THF(10mL)中のホスホネート(170mg、0.67mmol)及び18−クラウン−6(357mg、1.35mmol)の溶液に、KHMDS(1.34ml、0.67mmol)を滴加した。15分後、無水THF(8.5mL)中のアルデヒド34(170mg、0.45mmol)の溶液を30分間かけて滴加し、−78℃で90分間撹拌した。次に、反応を飽和NH4Cl溶液で停止させ、室温に温め、ジクロロメタンで希釈した。有機相を乾燥し(Na2SO4)、減圧下で蒸発させた。カラムクロマトグラフィー(ヘキサン/EtOAc 20:1〜10:2)による精製によって、170mg(収率:82%)の(E)−35を得た。
1H-RMN (300 MHz, CDCl3) δ: 7.29 (dd, 1H, J = 10.8, 15.3 Hz), 6.50 (d, 1H, J = 15.3 Hz), 6.19 (dd, 1H, J = 10.8, 15.0 Hz), 5.83 (d, 1H, J = 15.3 Hz), 5.48 (d, 1H, J = 10.2 Hz), 5.33 (t, 1H, J = 7.2 Hz), 4.17 (m, 2H), 3.71 (s, 3H), 3.61 (m, 1H), 3.58 (s, 3H), 2.73 (m, 1H), 2.57 (m, 2H), 1.71 (s, 3H), 1.25 (m, 3H), 0.97 (d, 3H, J = 6.7 Hz), 0.88 (s, 9H), 0.03 (m, 6H).
【0272】
中間体36の合成
【0273】
【化80】
【0274】
LiOH(15.8mg、0.66mmol)を、20%水/ジオキサン(7mL)中のエステル35(140mg、0.30mmol)の溶液に加え、混合物を60℃で4時間撹拌した。混合物を冷却し、DCMで希釈し、HCl(0.5N、10mL)で洗浄した。水相をDCMで繰り返し抽出し、合わせた有機層をNa2SO4で乾燥し、濾過し、蒸発させて、粗二酸を得て、それを更に精製することなく次の工程に使用した。
濃HCl(43μL)を、MeOH(5.2mL)中の粗物質の溶液に加え、得られた混合物を室温で1時間撹拌した。次に、溶媒を減圧下で除去し、得られた油状物をカラムクロマトグラフィー(ヘキサン/EtOAc 1:4からEtOAc/MeOH 5:1)により精製して、72mg(収率:70%)の酸36を無色の油状物として得た。
1H-RMN (300 MHz, CDCl3) δ: 7.40 (dd, 1H, J = 10.8, 15.0 Hz), 6.57 (d, 1H, J = 15.0 Hz), 6.31 (dd, 1H, J = 11.4, 15.3 Hz), 5.89 (d, 1H, J = 15.0 Hz), 5.60 (m, 1H), 5.52 (d, 1H, J = 10.2 Hz), 4.22 (m, 1H), 3.64 (s, 3H), 2.90 (m, 1H), 2.38 (m, 2H), 1.84 (s, 3H), 1.15 (d, 3H, J = 6.9 Hz).
【0275】
化合物37の合成
【0276】
【化81】
【0277】
DCM/DMF(10:1、1.3mL)中のアミン27a(37.6mg、0.093mmol)の溶液に、無水DCM中の酸36(30mg、0.103mmol)の溶液を、アルゴン雰囲気下で加え、次に0℃に冷却した。DIPEA(26μL、0.14mmol)、HOAt(12.7mg、0.093mmol)及びHATU(35.4mg、0.093mmol)を溶液に加え、30分後、冷浴を取り外した。反応混合物を室温で3時間撹拌し、NH4Clの飽和水溶液で停止させ、水に注ぎ、DCMで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 2:1〜1:4)により精製して、アミド37(34.7mg、収率:55%)を油状物として得た。
1H NMR (CDCl3, 500 MHz) δ: 7.88 (d, 1H J = 10.5 Hz), 7.26 (dd, 1H, J = 14.4, 11.4 Hz), 6.72 (dd, 1H, J = 9.9, 9.6 Hz), 6.50 (d, 1H, J = 15.3 Hz), 6.31-6.22 (m, 2H), 5.94 (d, 1H, J = 14.7 Hz), 5.61-5.54 (m, 2H), 5.44 (d, 1H, J = 9.9 Hz), 4.87-4.79 (m, 1H), 4.45 (d, 1H, J = 9.3), 4.24-4.16 (m, 1H), 3.77-3.44 (m, 1H), 3.64 (s, 3H), 2.96-2.2.81 (m, 1H), 2.39-2.35 (m, 2H), 2.16-2.15 (m, 4H), 2.01 (s, 3H), 1.82 (s, 3H), 1.14 (d, 3H, J = 6.6 Hz).1.02 (s, 9H), 0.87 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H).
【0278】
化合物38の合成
【0279】
【化82】
【0280】
THF(0.6mL)中の37(28mg、0.04mmol)の溶液に、N2下及び室温で、THF(83μL、0.08mmol)中の1M TBAFを加えた。反応を室温で18時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、EtOAcで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 3:1〜1:2)により精製して、アルコール38(22mg、収率:96%)を白色の固体として得た。
1H NMR (CDCl3, 500 MHz) δ: 9.06 (d, 1H, J = 9.9 Hz), 7.24 (dd, 1H, J = 14.7, 10.5 Hz), 6.76 (d, 1H, J = 9.6 Hz), 6.74 (dd, 1H, J = 9.6, 9.6 Hz), 6.50 (d, 1H, J = 15.3 Hz), 6.25 (dd, 1H, J = 15.3, 11.1 Hz), 5.99 (d, 1H, J = 14.7 Hz), 5.65-5.60 (m, 2H), 5.45 (d, 1H, J = 9.9 Hz), 4.87-4.81 (m, 1H), 4.45 (d, 1H, J = 9.3), 4.24-4.16 (m, 1H), 3.74-3.64 (m, 1H), 3.64 (s, 3H), 3.22-3.17 (m, 1H), 2.95-2.2.82 (m, 1H), 2.40-2.35 (m, 2H), 2.23-2.16 (m, 4H), 2.05 (s, 3H), 1.81 (s, 3H), 1.13 (d, 3H, J = 6.6 Hz), 1.03 (s, 9H).
【0281】
化合物3の合成
【0282】
【化83】
【0283】
ジクロロメタン(0.35ml)中の化合物38(20mg、0.04mmol)の溶液に、0℃で、トリクロロアセチルイソシアネート(5.1μL、0.04mmol)を加えた。反応を0℃で30分間撹拌し、次に中性酸化アルミニウムを加えた。混合物を5〜30分間撹拌し、次に酸化アルミニウムパッドに浸漬させた。生成物を、DCM/MeOH 50:1の混合物を使用して洗浄した。濾液を真空下で蒸発させて、粗生成物を得て、それをカラムクロマトグラフィー(ヘキサン/EtOAc 1:1〜1:3)により精製した。化合物3(13.6mg、収率:63%)を白色の泡状物として得て、実施例3に報告されているものと同等の物理的特性及び分光特性(1H、13C RMN及びMS)を示した。
【0284】
実施例10
スキーム7は化合物7の合成プロセスを示す。
【0285】
【化84】
【0286】
中間体40の合成
【0287】
【化85】
【0288】
MeOH中のBoc−tert−LeuCONH2 39(ポズネフ(Pozdnev)V.F.、テトラヘドロン・レターズ(Tetrahedron Letters)1995年、36、7115〜7118に記載された手順に従って調製)(1.9g、8.26mmol)の溶液に、MeOH中1.25MのHClの溶液66mLを加えた。反応混合物を室温で2時間撹拌し、次に減圧下で濃縮した。次に固体をMeOHに懸濁し、濃水酸化アンモニウムで中和した。溶媒をロータリー蒸発で除去し、粗物質をジクロロメタンに溶解した。溶液をNa2SO4で乾燥し、濾過し、濃縮して、730mg(収率:68%)の中間体40を白色の固体として得た。
1H NMR (CDCl3, 300 MHz) δ: 6.65-6.55 (br s, 1H), 5.65-5.55 (br s, 1H), 3.18 (s, 1H), 1.65-1.55 (br s, 2H), 1.05 (s, 9H).
【0289】
中間体41の合成
【0290】
【化86】
【0291】
DCM/DMF(10:1、7.7mL)中のアミン40(100mg、0.77mmol)の溶液に、無水DCM中の(Z)−3−トリブチルスタンニルプロペン酸(306mg、0.85mmol)の溶液を、アルゴン雰囲気下で加え、次に0℃に冷却した。DIPEA(0.27mL、1.54mmol)、HOAt(115.6mg、0.85mmol)及びHATU(323mg、0.85mmol)を溶液に加え、30分後、冷浴を取り外した。反応混合物を室温で2時間撹拌し、NH4Clの飽和水溶液で停止させ、水に注ぎ、DCMで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 10:1〜1:1)により精製して、アミド41(228mg、収率:63%)を白色の泡状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 6.99 (d, 1H, J = 12.3 Hz), 6.77 (d, 1H, J = 12.3 Hz), 6.47 (d, 1H, J = 9.6 Hz), 6.38 (br s, 1H), 6.12 (br s, 1H), 4.46 (d, 1H, J = 9.9 Hz), 1.48-1.40 (m, 6H), 1.31-1.19 (m, 12H), 1.00 (s, 9H), 0.89-0.83 (m, 9H).
13C NMR (CDCl3, 75 MHz) δ: 173.4, 166.6, 153.4, 136.6, 60.0, 35.0, 29.6, 27.6, 26.8, 14.0, 11.7.
【0292】
化合物7の合成
【0293】
【化87】
【0294】
NMP(5mL)中のアルケニルスタンナン41(227.6mg、0.48mmol)及び17a(200.9mg、0.58mmol)の溶液に、0℃で、チオフェンカルボン酸銅(19.1mg、0.10mmol)を加えた。反応を、0℃で45分間、室温で20分間撹拌した。次に、粗混合物を中性アルミナのプラグで濾過し、EtOAc/エーテル 50:50で洗浄し、合わせた濾液を0.1%のHClで洗浄した。有機溶液を乾燥し、蒸発させて粗生成物を得て、それをカラムクロマトグラフィー(ジクロロメタン/MeOH 100:1〜10:1)により精製して、化合物7(65.7mg、収率:34%)を白色の泡状物として得た。この合成生成物は、実施例3に報告されているものと同等の物理的特性及び分光特性(1H、13C RMN及びMS)を示した。
【0295】
化合物7の異性体(15R)−は、アミノ酸(Boc−tert−LeuCONH2)のラセミ混合物を使用してこれらの反応を実施したときに得た。異性体の最終混合物(15mg)を、半分取逆相HPLC(SymmetryPrep C18 7μm、7.8×150mm、MeOHが50〜70%の勾配H2O:MeOHを75分、UV検出、流速2.5mL/分、〔保持時間((15R)−7):18.15分、保持時間((15S)−7):19.62分〕)により分離し、3.1mgの純粋な(15R)−化合物7及び2.9mgの純粋な(15S)−化合物7を得た。
【0296】
【化88】
【0297】
1H NMR (MeOD, 500 MHz) δ: 7.23 (dd, 1H, J = 12.5, 11.5 Hz), 6.94 (dd, 1H, J = 12.5, 11.5 Hz), 6.18 (d, 1H, J = 12.0 Hz), 5.91-5.88 (m, 1H), 5.86 (t, 1H, J = 4.5 Hz), 5.34 (d, 1H, J = 10.0 Hz), 4.33-4.28 (m, 1H), 3.64 (s, 3H), 2.92-2.88 (m, 1H),2.47-2.44 (m, 2H), 1.85 (s, 3H), 1.17 (d, 3H, J = 6.6 Hz ), 1.02 (s, 9H).
13C NMR (MeOD, 125 MHz) δ: 175.3, 168.6, 164.0, 146.0, 140.8, 138.2, 135.4, 135.4, 125.5, 121.9, 111.1, 83.5, 61.8, 55.9, 38.4, 35.0, 27.2, 27.1, 17.3, 16.8.
【0298】
実施例11
スキーム8は、化合物42及び43を得る方法を示す。
【0299】
【化89】
【0300】
化合物42の合成
【0301】
【化90】
【0302】
ピリジン(0.45mL)中のアルコール30a(5mg、8.8μmol)の溶液に、フェニルイソシアネート(29mL、0.27mmol)を加えた。反応を室温で20時間撹拌し、次に、NH4Clの飽和水溶液で停止させ、EtOAcで抽出した。合わせた有機相をNa2SO4で乾燥し、濾過し、濃縮した。残渣をフラッシュクロマトグラフィー(ヘキサン/EtOAc 20:1〜1:1)により精製して、化合物42(2.7mg、収率:44%)を白色の固体として得た。
1H NMR (CDCl3, 300 MHz) δ: 8.81-8.77 (m, 1H), 7.46-7.43 (m, 2H), 7.34-7.23 (m,3H), 7.11-7.06 (m, 2H), 6.88-6.77 (m, 2H), 6.39 (d, 1H, J = 9.9 Hz), 6.10 (d, 1H, J = 11.7 Hz), 5.67-5.57 (m, 3H), 5.27 (d, 1H, J = 9.9 Hz), 4.85-4.78 (m, 1H), 4.62-4.56 (m, 1H), 4.54 (d, 1H, J = 9.3 Hz), 4.25-4.17 (m, 1H), 3.66 (s, 3H), 2.87-2.80 (m, 1H), 2.41-2.38 (m, 5H), 2.21-2.13 (m, 1H), 2.08 (s, 3H), 1.82 (s, 3H), 1.16 (d, 3H J = 6.6 Hz), 1.07 (s, 9H).
【0303】
化合物43の合成
【0304】
【化91】
【0305】
アルコール30a(5mg、8.88μmol)をDCM(0.1mL)中、不活性雰囲気下、室温で撹拌し、ペルヨージナン(7.5mg、0.018mmol)を一度に加えた。反応を、完了するまで(TLC、約1時間)撹拌し、次に、NaHCO3(飽和溶液)で停止させ、DCMで抽出し、ブラインで洗浄し、硫酸マグネシウムで乾燥し、濾過し、真空下で濃縮した。生成物を、EtOAc/ヘキサン 1:1を用いて溶離するカラムクロマトグラフィーにより精製して、約4.5mg(収率:90%)のケトン43を無色の油状物として得た。
1H NMR (CDCl3, 300 MHz) δ: 8.32 (d, 1H, J = 9.9 Hz), 7.31-7.23 (m, 1H), 6.91 (dd, 1H, J = 11.7, 11.4 Hz), 6.84 (dd, 1H, J = 9.9, 8.7 Hz), 6.26 (d, 1H, J = 8.7 Hz), 6.18 (d, 1H, J = 11.4 Hz), 5.78-5.73 (m, 1H), 5.68 (d, 1H, J = 11.4 Hz), 5.64-5.61 (m, 1H), 5-30-5.27 (m, 1H), 4.94-4.86 (m, 1H), 4.41 (d, 1H, J = 9.0 Hz), 4.25-4.17 (m, 1H), 3.66 (s, 3H), 3.18 (dd, 4H, J = 18.3, 7.2 Hz), 2.89-2.75 (m, 1H), 2.44-2.38 (m, 2H), 2.04 (s, 3H), 1.84 (s,5 3H), 1.16 (d, 3H J = 6.9 Hz), 1.05 (s, 9H).
【0306】
実施例12:抗腫瘍活性の検出のためのバイオアッセイ
このアッセイの目的は、試験する試料のインビトロでの細胞増殖抑制性(腫瘍細胞増殖を遅延又は阻止する能力)又は細胞毒性(腫瘍細胞を死滅させる能力)の活性を評価することである。
【0307】
細胞株
【0308】
【表9】
【0309】
SBR比色アッセイを使用する細胞毒性活性の評価
スルホローダミンB(SRB)反応を使用する比色型のアッセイは、細胞増殖及び生存度の定量的測定のために採用されてきた(スケハン(Skehan)Pら、国立癌研究所ジャーナル(J. Natl. Cancer Inst.)1990年、82、1107〜1112に記載されている教示に従う)。
【0310】
この形態のアッセイは、SBS標準96ウエル細胞培養マイクロプレートを用いる(フェアクロス(Faircloth)ら、細胞科学における方法(Methods in cell science)、1988年、11(4)、201〜205;モスマン(Mosmann)ら、免疫学方法ジャーナル(Journal of. Immunological. Methods)1983年、65(1〜2)、55〜63)。異なる型のヒト癌に由来する、この研究に使用した全ての細胞株は、アメリカン・タイプ・カルチャー・コレクション(ATCC)から得た。
【0311】
細胞を、10%ウシ胎児血清(FBS)、2mMのL−グルタミン、100U/mLのペニシリン及び100U/mLのストレプトマイシンを補充したダルベッコー修飾イーグル培地(DMEM)に、37℃、5%CO2及び98%湿度で維持した。実験のために、細胞を、トリプシン処理を使用してサブコンフルエント培養から採取し、カウント及び平板培養する前に、新たな培地に再懸濁した。
【0312】
細胞を、150μLのアリコートにより1ウエルあたり5×103細胞で95ウエルマイクロタイタープレートに接種し、薬剤無含有培地において、18時間かけてプレート表面に結合させた。それぞれの細胞株に1つの対照(未処理)プレートを固定し(下記に記載するように)、0時間基準値として使用した。その後、試験試料を、50μLのアリコートにより、10〜0.00262μg/mLの範囲で10個の段階希釈で培養に加えた。48時間の暴露後、抗腫瘍効果をSRB法により推定した:簡潔には、細胞をPBSで2回洗浄し、1%グルタルアルデヒド溶液で15分間固定し、PBSで2回すすぎ、0.4%SRB溶液により室温で30分間染色した。次に細胞を1%酢酸溶液で数回すすぎ、風乾した。次にSRBを10mMトリズマ塩基溶液で抽出し、自動分光光度プレート読取機により490nmで吸光度を測定した。細胞生存は、対照細胞増殖の百分率として表した。試験した試料の最終効果は、NCIアルゴリズム(ボイド(Boyd)MR及びポール(Paull)KD、薬剤開発研究(Drug Dev. Res.)1995年、34,91〜104)を適用することにより評価した。
【0313】
三重培養の平均±SDを使用して、用量反応曲線を非線形回帰分析を使用して自動的に生成した。3つの基準パラメーターを自動補間法により計算した(NCIアルゴリズム):GI50=50%の増殖阻害を生じる濃度;TGI=総増殖阻害(細胞増殖抑制効果)及びLC50=50%の純細胞死滅(細胞毒性効果)を生じる濃度。
【0314】
抗有糸分裂アッセイプロトコール
細胞培養の有糸分裂率(有糸分裂が阻止された細胞の百分率)を、特異的有糸分裂マーカーを定量的に検出する特異的96ウエルマイクロプレート免疫アッセイを使用して評価した。HeLa細胞(h−子宮頸癌、ATCC番号CCL−2)を、試験する試料の存在下又は不在下で18時間インキュベートした。その後、細胞をPBSで洗浄し、75μLの新たに調製した溶解緩衝剤(1mMのEGTA(pH7.5)、0.5mMのPMSF及び1mMのNaVO3)中の氷により30分間溶解した。細胞抽出物のアリコート(60μL)を、高結合表面ELISAプレートに移し、speed-vacにより室温で2時間乾燥した。次にプレートを、100μLのPBS−1%BSAにより30℃で30分間ブロックし、抗MPM2一次マウスモノクローナル抗体(アップステート・バイオテクノロジー(Upstate Biotechnology)、カタログ番号05−368)により4℃で18時間及び適切なペルオキシダーゼ結合二次抗体により30℃で1時間連続的にインキュベートした。0.02%のツイーン20による集中的な洗浄の後、ペルオキシダーゼ反応を、30μLのTMB(3,3′,5,5′−テトラメチル−ベンジジン)を使用して30℃で30分間実施した。反応を、30μLの4%H2SO4溶液を加えることにより停止させた。アッセイを、マイクロプレート分光光度計により450nmでO.D.を測定することにより定量化した。結果を、未処理培養の対照と比較して、処理細胞培養において50%の有糸分裂阻止を生じた試料濃度をIC50として表した。
【0315】
表9及び10は、本発明の化合物の生物学的活性についてのデータを例示する。
【0316】
【表10】
【表11】
【特許請求の範囲】
【請求項1】
一般式I:
【化92】
〔式中、R1は、水素、ORa、OCORa、OCOORa、NRaRb、NRaCORb及びNRaC(NRa)NRaRbから選択され;
R2及びR3は、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル及び置換又は非置換C2〜C12アルキニルから選択され;
R41、R42、R43、R44、R45、R46、R47及びR48は、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル及び置換又は非置換C2〜C12アルキニルから選択され;
R5、R6及びR7は、それぞれ独立して、水素、CORa、COORa、置換若しくは非置換C1〜C12アルキル、置換若しくは非置換C2〜C12アルケニル及び置換若しくは非置換C2〜C12アルキニルから選択されるか、又はR5及びR48は、それらが結合している対応するN原子及びC原子と一緒になって、置換若しくは非置換複素環式基を形成してもよく;
Ra及びRbは、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル、置換又は非置換アリール及び置換又は非置換複素環式基から選択され;並びに
点線は、それぞれ任意の追加的な結合を表す〕
の化合物、又は
薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項2】
以下の式II:
【化93】
〔式中、R1、R41、R43及びR48、R5、R6及びR7は、請求項1で定義されたとおりである〕を有する、請求項1に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項3】
R2及びR3が、それぞれ独立して、水素及び置換又は非置換C1〜C6アルキルから選択される、請求項1に記載の化合物、又はその薬学的に許容される塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項4】
R2及びR3が水素である、請求項3に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項5】
R1が、水素、ORa及びOCORaから選択され、ここでRaが、水素及び置換又は非置換C1〜C6アルキルから選択される、前記請求項のいずれか1項に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項6】
R1が、水素又はメトキシである、請求項5に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項7】
R42、R44、R45、R46及びR47基が、独立して、水素及び置換又は非置換C1〜C6アルキルから選択される、請求項1及び3〜6のいずれか1項記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項8】
R42、R44、R45、R46、R47基が、独立して、水素、置換又は非置換メチル、置換又は非置換イソプロピル及び置換又は非置換tert−ブチルから選択される、請求項7に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項9】
R42、R44、R45、R46及びR47が水素である、請求項8に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項10】
R41、R43及びR48基が、独立して、水素及び置換又は非置換C1〜C6アルキルから選択される、前記請求項のいずれか1項に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項11】
R41、R43及びR48基が、独立して、水素、置換又は非置換メチル、置換又は非置換イソプロピル及び置換又は非置換tert−ブチルから選択される、請求項10に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項12】
R41及びR43がメチルである、請求項11に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項13】
R48が、イソプロピル、tert−ブチル及びベンジルから選択される、請求項12に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項14】
R5及びR6が、それぞれ独立して、水素及び置換又は非置換C1〜C6アルキルからなる群から選択される、前記請求項のいずれか1項に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項15】
R5及びR6が水素である、請求項14に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項16】
R7が、水素、置換又は非置換C1〜C12アルキル及び置換又は非置換C2〜C12アルケニルから選択される、前記請求項のいずれか1項に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項17】
R7が、水素及び置換C2〜C12アルケニルから選択される、請求項16に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項18】
R7が、1つ以上の位置において、ハロゲン、OR′、=O、OCOR′、OCONHR′、OCON(R′)2及び保護OHで置換されているアルケニル基であり、ここでR′基が、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル及び置換又は非置換アリールから選択される、請求項17に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項19】
追加的な結合が点線で示された全ての場所において存在する、前記請求項のいずれか1項に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項20】
以下の式:
【化94】
を有する、請求項1若しくは2に記載の化合物、又は
薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ又は立体異性体。
【請求項21】
以下の式:
【化95】
を有する、請求項1若しくは2に記載の化合物、又は
薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ又は立体異性体。
【請求項22】
リトプロカミア・リチストイデス(Lithoplocamia lithistoides)からの抽出及び単離工程を実施することを含む、前記請求項のいずれか1項で定義された化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体を得るための方法。
【請求項23】
請求項1〜21のいずれか1項に記載の化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体及び薬学的に許容される希釈剤又は担体を含む医薬組成物。
【請求項24】
薬剤としての使用のための、請求項1〜21のいずれか1項に記載の化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項25】
癌を治療するための薬剤としての使用のための、請求項1〜21のいずれか1項に記載の化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項26】
薬剤の調製における、請求項1〜21のいずれか1項に記載の化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体の使用。
【請求項27】
薬剤が癌の治療において使用される、請求項26に記載の使用。
【請求項1】
一般式I:
【化92】
〔式中、R1は、水素、ORa、OCORa、OCOORa、NRaRb、NRaCORb及びNRaC(NRa)NRaRbから選択され;
R2及びR3は、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル及び置換又は非置換C2〜C12アルキニルから選択され;
R41、R42、R43、R44、R45、R46、R47及びR48は、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル及び置換又は非置換C2〜C12アルキニルから選択され;
R5、R6及びR7は、それぞれ独立して、水素、CORa、COORa、置換若しくは非置換C1〜C12アルキル、置換若しくは非置換C2〜C12アルケニル及び置換若しくは非置換C2〜C12アルキニルから選択されるか、又はR5及びR48は、それらが結合している対応するN原子及びC原子と一緒になって、置換若しくは非置換複素環式基を形成してもよく;
Ra及びRbは、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル、置換又は非置換アリール及び置換又は非置換複素環式基から選択され;並びに
点線は、それぞれ任意の追加的な結合を表す〕
の化合物、又は
薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項2】
以下の式II:
【化93】
〔式中、R1、R41、R43及びR48、R5、R6及びR7は、請求項1で定義されたとおりである〕を有する、請求項1に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項3】
R2及びR3が、それぞれ独立して、水素及び置換又は非置換C1〜C6アルキルから選択される、請求項1に記載の化合物、又はその薬学的に許容される塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項4】
R2及びR3が水素である、請求項3に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項5】
R1が、水素、ORa及びOCORaから選択され、ここでRaが、水素及び置換又は非置換C1〜C6アルキルから選択される、前記請求項のいずれか1項に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項6】
R1が、水素又はメトキシである、請求項5に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項7】
R42、R44、R45、R46及びR47基が、独立して、水素及び置換又は非置換C1〜C6アルキルから選択される、請求項1及び3〜6のいずれか1項記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項8】
R42、R44、R45、R46、R47基が、独立して、水素、置換又は非置換メチル、置換又は非置換イソプロピル及び置換又は非置換tert−ブチルから選択される、請求項7に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項9】
R42、R44、R45、R46及びR47が水素である、請求項8に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項10】
R41、R43及びR48基が、独立して、水素及び置換又は非置換C1〜C6アルキルから選択される、前記請求項のいずれか1項に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項11】
R41、R43及びR48基が、独立して、水素、置換又は非置換メチル、置換又は非置換イソプロピル及び置換又は非置換tert−ブチルから選択される、請求項10に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項12】
R41及びR43がメチルである、請求項11に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項13】
R48が、イソプロピル、tert−ブチル及びベンジルから選択される、請求項12に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項14】
R5及びR6が、それぞれ独立して、水素及び置換又は非置換C1〜C6アルキルからなる群から選択される、前記請求項のいずれか1項に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項15】
R5及びR6が水素である、請求項14に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項16】
R7が、水素、置換又は非置換C1〜C12アルキル及び置換又は非置換C2〜C12アルケニルから選択される、前記請求項のいずれか1項に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項17】
R7が、水素及び置換C2〜C12アルケニルから選択される、請求項16に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項18】
R7が、1つ以上の位置において、ハロゲン、OR′、=O、OCOR′、OCONHR′、OCON(R′)2及び保護OHで置換されているアルケニル基であり、ここでR′基が、それぞれ独立して、水素、置換又は非置換C1〜C12アルキル、置換又は非置換C2〜C12アルケニル、置換又は非置換C2〜C12アルキニル及び置換又は非置換アリールから選択される、請求項17に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項19】
追加的な結合が点線で示された全ての場所において存在する、前記請求項のいずれか1項に記載の化合物、又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項20】
以下の式:
【化94】
を有する、請求項1若しくは2に記載の化合物、又は
薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ又は立体異性体。
【請求項21】
以下の式:
【化95】
を有する、請求項1若しくは2に記載の化合物、又は
薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ又は立体異性体。
【請求項22】
リトプロカミア・リチストイデス(Lithoplocamia lithistoides)からの抽出及び単離工程を実施することを含む、前記請求項のいずれか1項で定義された化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体を得るための方法。
【請求項23】
請求項1〜21のいずれか1項に記載の化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体及び薬学的に許容される希釈剤又は担体を含む医薬組成物。
【請求項24】
薬剤としての使用のための、請求項1〜21のいずれか1項に記載の化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項25】
癌を治療するための薬剤としての使用のための、請求項1〜21のいずれか1項に記載の化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体。
【請求項26】
薬剤の調製における、請求項1〜21のいずれか1項に記載の化合物又は薬学的に許容されるその塩、誘導体、互変異性体、プロドラッグ若しくは立体異性体の使用。
【請求項27】
薬剤が癌の治療において使用される、請求項26に記載の使用。
【公表番号】特表2009−539943(P2009−539943A)
【公表日】平成21年11月19日(2009.11.19)
【国際特許分類】
【出願番号】特願2009−514811(P2009−514811)
【出願日】平成19年6月15日(2007.6.15)
【国際出願番号】PCT/EP2007/055959
【国際公開番号】WO2007/144423
【国際公開日】平成19年12月21日(2007.12.21)
【出願人】(508367692)ファルマ マル ソシエダッド アノニマ (1)
【Fターム(参考)】
【公表日】平成21年11月19日(2009.11.19)
【国際特許分類】
【出願日】平成19年6月15日(2007.6.15)
【国際出願番号】PCT/EP2007/055959
【国際公開番号】WO2007/144423
【国際公開日】平成19年12月21日(2007.12.21)
【出願人】(508367692)ファルマ マル ソシエダッド アノニマ (1)
【Fターム(参考)】
[ Back to top ]