
抗腫瘍活性を有するAKT/PKBの阻害剤
本発明は、Akt/PKB経路の阻害のための材料および方法に関する。1つの実施形態において、本発明の化合物は、Aktタンパク質のキナーゼ活性および/またはリン酸化レベルを阻害する。本発明は、式Iの化合物の有効量と細胞を接触させることを含む、Aktタンパク質の発現が上昇されるかまたは構成的に活性のある癌細胞または他の細胞を阻害または殺傷する方法にも関する。本発明は、人または動物に対して式Iの化合物の有効量を投与することを含む、人または動物中の癌または腫瘍を治療する方法にも関する。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
(関連出願の相互参照)
本出願は、2007年7月12日に出願された米国仮出願第60/949,365号の利益を主張し、任意の図、表、核酸配列、アミノ酸配列および図面を含むその全体は参照することにより本明細書に組み入れられる。
政府支援
【0002】
本出願の主題は、助成金番号R01CA107078、DAMD17−02−1−0671およびW81XWH−05−1−0021の下で、国立衛生研究所−国立癌研究所およびARMY/MRMCからの研究助成金によって支援された。従って、米国政府は本発明において一定の権利を有する。
【背景技術】
【0003】
プロテインキナーゼBとも呼ばれるAktは、セリン/スレオニンキナーゼのサブファミリーを示す。Aktは、v−akt癌遺伝子産物の細胞ホモログとして最初に記載され(Bellacosa et al. 1991)、それは3つのメンバーのAkt1/PKBα、Akt2/PKBβおよびAkt3/PKBγを有する(Cheng et al. 1992; Jones et al. 1991a; Jones et al. 1991b)。Aktの活性化は、膜輸送を仲介するプレクストリン相同性(PH)ドメインの完全性、ならびに活性化ループ中のThr308、およびSer473のリン酸化に依存する(Konishi et al. 1995)。PI3 Kによって生産されたフォスフォイノシチド(PtdIns−3,4−P2およびPtdIns−3,4,5−P3)は、AktのPHドメインへ直接結合し、分子中のコンフォメーション変化を引き起こし、それはAktの活性化ループをThr308でPDK1によってリン酸化することができる(Datta et al. 1999)。Aktの完全活性化もまた、C末端の疎水性モチーフ内のSer473のリン酸化に関連する(Datta et al. 1999)。Thr308のリン酸化におけるPDK1の役割は十分に確立されているが、Ser473のリン酸化の機構は論争の的になっている。この修飾に関与する多数の候補酵素が提案され、それらはインテグリン結合キナーゼ(Persad et al. 2001)、キナーゼPRK2の複合体中にある場合のPDK1(Wick et al. 2000)、自己リン酸化を介してAktそれ自体(Toker et al. 2000)、DNA依存性キナーゼ(Feng et al. 2004)、およびリクター(rictor)−mTOR複合体(Sarbassoy et al. 2005)を含む。Aktの活性は、ヒト悪性腫瘍中で頻繁に変異している腫瘍抑制因子PTENによって負に調節される(Vazquez et al. 2000)。PTENは、PtdIns−3,4,5−P3をPtdIns−4,5−P2に変換することによってPtdIns−3,4,5−P3の細胞内濃度を低下させ、その結果としてPI3K/Akt経路を阻害する、二重特異性タンパク質および脂質ホスファターゼをコードする(Stambolic et al. 1998)。
【0004】
Aktは多数の分子をリン酸化および/またはそれらと相互作用して、細胞の増殖、生存、移動および分化における役割を含む正常な細胞機能を発揮させる(Cheng et al. 2001)。多くの証拠から、Aktが腫瘍発生および進行において重大な役割を持つことが実証される。さらに、Akt経路の異常な過活性化はすべてのヒト腫瘍の最大50%で検出され(Sun et al. 2001; Cheng et al. 1997)、密接に化学耐性に関連する(West et al. 2002)。したがって、Aktは抗癌薬物開発のための魅力的な標的であった(West et al. 2002)。
【0005】
過去数年において、組み合わせ化学、ハイスループットおよび仮想スクリーニング、ならびに従来の医薬品化学を介して、Akt経路の何十もの阻害剤が同定された。Aktの脂質ベースの阻害剤は第1に開発されたものであり、ペリホシン(Kondapaka et al. 2003)、PX−316(Meuillet et al. 2004)およびホスファチジルイノシトールエーテル脂質類似体(Castillo et al. 2004)を含み、AktのPHドメインと相互作用するようにデザインされた。さらに、いくつかのAktアンタゴニストは、化学ライブラリーのハイスループットスクリーニングおよび合理的設計を使用して同定された。これらの阻害剤は、9−メトキシ−2−メチルエリプチシニウムアセタート(Jin et al. 2004)、インダゾール−ピリジンA−443654(Luo et al. 2005)、アイソフォーム特異的アロステリックキナーゼ阻害剤(Lindsley et al. 2005)、およびトリシリビン/TCNとも呼ばれるAkt/PKBシグナリング阻害剤−2(API−2)(Yang et al. 2004)を含む。API−2/TCNは、以前に行われた第I相および第II相の試験において抗腫瘍活性を示す三環式ヌクレオシドであるが、肝毒性、高血糖、血小板減少および高トリグリセリド血症を含む複数の毒性によりさらなる開発は妨げられた(Feun et al. 1993; Hoffman et al. 1996)。我々は、NCI多様性セットのスクリーニングによって、API−2がAktキナーゼ活性を阻害し、高Akt活性を示すヒト癌細胞の異種移植片のアポトーシスを刺激することを以前に示した(Yang et al. 2004)。この結果からこの薬物の研究に新たな関心が持たれ、より低用量でAktを阻害し、以前の関連した副作用なしに腫瘍細胞アポトーシスを誘導しうる可能性が提出される(Yang et al. 2004; Cheng et al. 2005)。
【発明の概要】
【0006】
本発明は、Akt/PKB経路の阻害のための化合物、組成物および方法に関する。1つの実施形態において、本発明の化合物は、Aktタンパク質のキナーゼ活性および/またはリン酸化レベルを阻害する。本発明の化合物は、式Iで示される一般的な構造を有する。特異的実施形態において、本発明の化合物(本明細書においてAPI−1と呼ばれる)は、構造:
【化1】

を有する。
【0007】
API−1などの本発明の化合物は、異常なAktを有するヒト腫瘍細胞におけるAktシグナリングを阻害し、細胞増殖の阻害およびアポトーシスの誘導を導く。異種移植ヌードマウスモデルにおいて、本発明の化合物は、過活性化されたAktを有する細胞における腫瘍増殖をかなり阻害するが、低レベルのAktを有する腫瘍においては阻害しない。
【0008】
本発明は、式Iの化合物の有効量と細胞を接触させることを含む、Aktタンパク質の発現が上昇されるかまたは構成的に活性のある癌細胞または他の細胞を阻害または殺傷する方法にも関する。
【0009】
本発明は、人または動物に対して式Iの化合物の有効量を投与することを含む、人または動物中の癌または腫瘍を治療する方法にも関する。
【0010】
セリン/スレオニンキナーゼAkt/PKB経路は、ヒト癌において頻繁に過活性化され、細胞外および細胞内の腫瘍形成シグナルの変換のための基本的な節点として機能し、したがってそれは分子治療のための標的を示す。国立癌研究所の多様性セットのスクリーニングによって、低分子のAkt経路阻害剤のAPI(Akt/PKBシグナリング阻害剤)−1が同定された。API−1は、Aktファミリーの3つのメンバーのキナーゼ活性およびリン酸化レベルを阻害する。しかしながら、それは上流活性化因子(PI3KおよびPDK1)の活性には効果がなかった。さらに、構成的に活性のあるAktのキナーゼ活性およびリン酸化レベルは、細胞培養において主としてAPI−1によって阻害されるが、それはAktキナーゼ活性にはインビトロで効果がなかった。API−1は、Aktについて高度に選択的であり、PKC、SGK、PKA、STAT3、Erk−1/2、またはJNKの活性化を阻害しない。API−1によるAktの阻害は、構成的に活性化されたAktを保有するヒト癌細胞において、細胞増殖停止およびアポトーシスの誘導をもたらした。重要なことには、API−1は、Aktが上昇したヒト癌細胞のヌードマウスにおいて腫瘍増殖を選択的に阻害したが、Aktが上昇しないこれらの癌細胞の増殖は阻害しない。これらのデーターは、API−1がインビトロおよびインビボで抗腫瘍活性を有するAkt経路阻害剤であり、過活性化Aktを発現する癌患者のための可能性のある抗癌剤でありえることを示唆する。
【0011】
Aktは、癌細胞の生存、増殖および腫瘍進行を調節する主要な経路である。上昇したレベルのAktキナーゼは、Bcr−Abl(グリーベック)、Her2/Neu(ハークプチン(Hercptin))、およびmTOR(ラパマイシン)の細胞毒性化学療法薬および低分子阻害剤を含む様々な癌治療法に対する耐性に寄与することがよく報告されていた。Aktのブロックは、過活性化Aktを有する癌細胞において腫瘍増殖を阻害し、化学療法および他の標的化された治療に対して癌細胞をより感受性にする。他の抗腫瘍薬物とのAPI−1の組合せはより強力な抗腫瘍効果を提供する。
【図面の簡単な説明】
【0012】
【図1A】NCI多様性セットからのAkt阻害剤の候補としてのAPI−1の同定の図示である。API−1の化学構造を示す。
【図1B】NCI多様性セットからのAktインヒビターの候補としてのAPI−1の同定の図示である。API−1がAktの3つのメンバーを阻害することを示す図示である。HEK293細胞をHA−Akt1、HA−AKT2およびHA−AKT3によりトランスフェクションし、EGF刺激前にAPI−1(10μM)により処理し、細胞を溶解し、抗HA抗体により免疫沈降した。免疫沈降物はインビトロキナーゼ分析を行なった(上部)。下部パネルは、トランスフェクションしたAkt1、AKT2およびAKT3の発現を示す、抗HA抗体により検出したウエスタンブロットである。
【図1C】NCI多様性セットからのAktインヒビターの候補としてのAPI−1の同定の図示である。API−1が、OVCAR3細胞(それらは過活性化Aktを発現する)においてAktのリン酸化レベルを阻害することを示す図示である。細胞を示された濃度でAPI−1により2時間処理し、抗リン酸化−Akt−S473抗体によるイムノブロット解析を行なった(上部パネル)。下部パネルは全Aktの発現を示す。
【図1D】NCI多様性セットからのAktインヒビターの候補としてのAPI−1の同定の図示である。API−1がインビトロでAktを阻害しないことを示した図示である。示された量のAPI−1を含むキナーゼバッファー中の構成的に活性のあるAkt組換えタンパク質のインビトロキナーゼ分析。化合物E(複数のキナーゼ阻害剤のATP競合物)を陽性対照として使用した。実験は3回反復した。
【図2A】API−1が、PI3K、PDK1、およびAGCキナーゼファミリーの非常によく関連したメンバーを阻害しないことを示す図示である。インビトロPI3Kキナーゼ分析を示す図示である。HEK293細胞は血清飢餓を行い、EGF刺激前に30分間API−1(10μM)またはワートマニン(1nM)により処理した。細胞を溶解し、抗p110α抗体により免疫沈降した。免疫沈降物は、基質としてPI−4−Pを使用する、インビトロキナーゼ分析を行なった。
【図2B】API−1が、PI3K、PDK1、およびAGCキナーゼファミリーの非常によく関連したメンバーを阻害しないことを示す図示である。PDK1活性化に対するAPI−1の効果を示す図示である。インビトロキナーゼ分析は、示された化合物の存在下で製造者説明書に従って、PDK1キナーゼキット(アップステート・バイオテクノロジー社 (Upstate Biotechnology Inc) )により実行された。
【図2C】API−1が、PI3K、PDK1、およびAGCキナーゼファミリーの非常によく関連したメンバーを阻害しないことを示す図示である。インビトロのPKAキナーゼ分析を示す図示である。組換えPKAを、示された阻害剤(API−1またはPKAI)および基質ケンプチドを含むADBバッファー(アップステート・バイオテクノロジー社)中でインキュベートした。キナーゼ活性を定量した。
【図2D】API−1が、PI3K、PDK1、およびAGCキナーゼファミリーの非常によく関連したメンバーを阻害しないことを示す図示である。生細胞における、PKA、PKCおよびPDKのキナーゼ活性に対するAPI−1の効果を示す図示である。OVCAR3細胞をAPI−1の示された濃度により1時間処理した。細胞を溶解し、示された抗体によりイムノブロットした。
【図2E】API−1が、PI3K、PDK1、およびAGCキナーゼファミリーの非常によく関連したメンバーを阻害しないことを示す図示である。インビトロSGKキナーゼ分析を示す図示である。組換えSGKタンパク質をAPI−1または化合物Eと共にインキュベートした。キナーゼ分析は、SGK基質ペプチドおよび[γ−32P]ATPの追加によって開始された。キナーゼ活性を定量した。
【図2F】API−1が、PI3K、PDK1、およびAGCキナーゼファミリーの非常によく関連したメンバーを阻害しないことを示す図示である。HEK293細胞をHA−SGKによりトランスフェクションし、EGF刺激前にAPI−1またはワートマニンにより処理した場合の結果を示す図示である。インビトロキナーゼは、基質としてヒストン−H2Bを使用して、HA−SGK免疫沈降物により実行された。
【図2G】API−1が、PI3K、PDK1、およびAGCキナーゼファミリーの非常によく関連したメンバーを阻害しないことを示す図示である。API−1が、Erk、p38、JNKおよびStat3のリン酸化を阻害しないことを示す図示である。OVCAR3細胞をAPI−1により3時間処理し、示された抗体によりイムノブロットした。
【図3A】API−1が、構成的に活性のあるAktおよびその下流の標的を阻害することを実証する図示である。API−1が構成的に活性のあるAktを阻害することを実証する図示である。HEK293細胞を、示されたHA−myr−Akt1、HA−Akt1−E40KおよびHA−myr−Akt2によりトランスフェクションした。API−1による1時間の処理に後続して、細胞を溶解して、抗HA抗体により免疫沈降させた。免疫沈降物に、インビトロキナーゼ分析(上部パネル)、および示された抗体によるイムノブロット(中央および下部のパネル)を行なった。
【図3B】API−1が、構成的に活性のあるAktおよびその下流の標的を阻害することを実証する図示である。API−1がAktの下流の標的のリン酸化を阻害することを実証する図示である。OVCAR3細胞をAPI−1(10μM)により示された時間処理し、示された抗体によりイムノブロットした。API−1は、Aktおよびその下流の標的(GSK3βおよびS6タンパク質)のリン酸化レベルを有意に低下させる。
【図3C】API−1が、構成的に活性のあるAktおよびその下流の標的を阻害することを実証する図示である。AKT−SI1は、Aktの下流の標的のリン酸化を阻害した。
【図3D】API−1が、構成的に活性のあるAktおよびその下流の標的を阻害することを実証する図示である。AKT−SI1は、mTORC1およびmTORC2複合体を妨害しなかった。
【図4A】API−1が、上昇したAktのヒト癌細胞においてAkt活性および細胞増殖を阻害し、アポトーシスを誘導することを示す図示である。API−1による処理に後続する結果を示すウエスタンブロットであり、Aktのリン酸化レベルおよびPARP切断は、示されたヒト癌細胞株において抗リン酸化−Akt−T308抗体および切断PARP抗体により検出された(上部および中央のパネル)。ブロットは抗アクチン抗体により再プロービングした(下部パネル)。
【図4B】API−1が、上昇したAktのヒト癌細胞においてAkt活性および細胞増殖を阻害し、アポトーシスを誘導することを示す図示である。示された細胞株を異なる用量のAPI−1により24時間処理し、次にMTT分析により解析した細胞増殖分析である。
【図4C】API−1が、上昇したAktのヒト癌細胞においてAkt活性および細胞増殖を阻害し、アポトーシスを誘導することを示す図示である。示された細胞株を異なる用量のAPI−1により24時間処理し、次にMTT分析により解析した細胞増殖分析である。
【図4D】API−1が、上昇したAktのヒト癌細胞においてAkt活性および細胞増殖を阻害し、アポトーシスを誘導することを示す図示である。細胞をAPI−1により処理し、アネキシンVおよびPIにより染色し、ファックスキャン(FACScan)によって解析したアポトーシス解析を示す。
【図4E】API−1が、上昇したAktのヒト癌細胞においてAkt活性および細胞増殖を阻害し、アポトーシスを誘導することを示す図示である。細胞をAPI−1により処理し、アネキシンVおよびPIにより染色し、ファックスキャンによって解析したアポトーシス解析を示す。
【図4F】API−1が、上昇したAktのヒト癌細胞においてAkt活性および細胞増殖を阻害し、アポトーシスを誘導することを示す図示である。細胞をAPI−1により処理し、アネキシンVおよびPIにより染色し、ファックスキャンによって解析したアポトーシス解析を示す。
【図4G】API−1が、上昇したAktのヒト癌細胞においてAkt活性および細胞増殖を阻害し、アポトーシスを誘導することを示す図示である。細胞をAPI−1により処理し、アネキシンVおよびPIにより染色し、ファックスキャンによって解析したアポトーシス解析を示す。
【図5A】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。対照群の写真(図5A)およびAPI−1−処理した群の写真(図5B)および関連したグラフ(図5C〜5F)である。右側に低レベルのAkt細胞および左側に上昇したレベルのAkt細胞で、腫瘍細胞をヌードマウスに皮下注射した。腫瘍が約100〜150mm3の平均サイズに達した時に、「材料および方法」において記述されているように、媒質または10mg/kg/日のAPI−1のいずれかにより動物を処理した。上昇したレベルのAktを発現するPANC1/OVCAR3(それぞれ図5Eおよび5C)、および低レベルのAktを示すCOLO357/OVCAR5(それぞれ図5Fおよび5D)を有するマウスの表示であり、異種移植はAPI−1または媒質(A)により処理された。パネルBは、10マウス/群による腫瘍成長曲線を示す。
【図5B】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。対照群の写真(図5A)およびAPI−1−処理した群の写真(図5B)および関連したグラフ(図5C〜5F)である。右側に低レベルのAkt細胞および左側に上昇したレベルのAkt細胞で、腫瘍細胞をヌードマウスに皮下注射した。腫瘍が約100〜150mm3の平均サイズに達した時に、「材料および方法」において記述されているように、媒質または10mg/kg/日のAPI−1のいずれかにより動物を処理した。上昇したレベルのAktを発現するPANC1/OVCAR3(それぞれ図5Eおよび5C)、および低レベルのAktを示すCOLO357/OVCAR5(それぞれ図5Fおよび5D)を有するマウスの表示であり、異種移植はAPI−1または媒質(A)により処理された。パネルBは、10マウス/群による腫瘍成長曲線を示す。
【図5C】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。対照群の写真(図5A)およびAPI−1−処理した群の写真(図5B)および関連したグラフ(図5C〜5F)である。右側に低レベルのAkt細胞および左側に上昇したレベルのAkt細胞で、腫瘍細胞をヌードマウスに皮下注射した。腫瘍が約100〜150mm3の平均サイズに達した時に、「材料および方法」において記述されているように、媒質または10mg/kg/日のAPI−1のいずれかにより動物を処理した。上昇したレベルのAktを発現するPANC1/OVCAR3(それぞれ図5Eおよび5C)、および低レベルのAktを示すCOLO357/OVCAR5(それぞれ図5Fおよび5D)を有するマウスの表示であり、異種移植はAPI−1または媒質(A)により処理された。パネルBは、10マウス/群による腫瘍成長曲線を示す。
【図5D】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。対照群の写真(図5A)およびAPI−1−処理した群の写真(図5B)および関連したグラフ(図5C〜5F)である。右側に低レベルのAkt細胞および左側に上昇したレベルのAkt細胞で、腫瘍細胞をヌードマウスに皮下注射した。腫瘍が約100〜150mm3の平均サイズに達した時に、「材料および方法」において記述されているように、媒質または10mg/kg/日のAPI−1のいずれかにより動物を処理した。上昇したレベルのAktを発現するPANC1/OVCAR3(それぞれ図5Eおよび5C)、および低レベルのAktを示すCOLO357/OVCAR5(それぞれ図5Fおよび5D)を有するマウスの表示であり、異種移植はAPI−1または媒質(A)により処理された。パネルBは、10マウス/群による腫瘍成長曲線を示す。
【図5E】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。対照群の写真(図5A)およびAPI−1−処理した群の写真(図5B)および関連したグラフ(図5C〜5F)である。右側に低レベルのAkt細胞および左側に上昇したレベルのAkt細胞で、腫瘍細胞をヌードマウスに皮下注射した。腫瘍が約100〜150mm3の平均サイズに達した時に、「材料および方法」において記述されているように、媒質または10mg/kg/日のAPI−1のいずれかにより動物を処理した。上昇したレベルのAktを発現するPANC1/OVCAR3(それぞれ図5Eおよび5C)、および低レベルのAktを示すCOLO357/OVCAR5(それぞれ図5Fおよび5D)を有するマウスの表示であり、異種移植はAPI−1または媒質(A)により処理された。パネルBは、10マウス/群による腫瘍成長曲線を示す。
【図5F】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。対照群の写真(図5A)およびAPI−1−処理した群の写真(図5B)および関連したグラフ(図5C〜5F)である。右側に低レベルのAkt細胞および左側に上昇したレベルのAkt細胞で、腫瘍細胞をヌードマウスに皮下注射した。腫瘍が約100〜150mm3の平均サイズに達した時に、「材料および方法」において記述されているように、媒質または10mg/kg/日のAPI−1のいずれかにより動物を処理した。上昇したレベルのAktを発現するPANC1/OVCAR3(それぞれ図5Eおよび5C)、および低レベルのAktを示すCOLO357/OVCAR5(それぞれ図5Fおよび5D)を有するマウスの表示であり、異種移植はAPI−1または媒質(A)により処理された。パネルBは、10マウス/群による腫瘍成長曲線を示す。
【図5G】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。実験の終了時の腫瘍サイズ(左側)および重量(右側)の例を示す。DMSO対照と比較して、API−1は、PANC1およびOVCAR3の異種移植の腫瘍重量を有意に減少させた(*P=0.02)。
【図5H】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。実験の終了時の腫瘍サイズ(左側)および重量(右側)の例を示す。DMSO対照と比較して、API−1は、PANC1およびOVCAR3の異種移植の腫瘍重量を有意に減少させた(*P=0.02)。
【図5I】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。実験の終了時の腫瘍サイズ(左側)および重量(右側)の例を示す。DMSO対照と比較して、API−1は、PANC1およびOVCAR3の異種移植の腫瘍重量を有意に減少させた(*P=0.02)。
【図5J】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。実験の終了時の腫瘍サイズ(左側)および重量(右側)の例を示す。DMSO対照と比較して、API−1は、PANC1およびOVCAR3の異種移植の腫瘍重量を有意に減少させた(*P=0.02)。
【図5K】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。API−1がインビボでAktリン酸化を阻害することを実証する。API−1処理された腫瘍試料および無処理の腫瘍試料を溶解し、示された抗体によりイムノブロットした。
【図5L】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。API−1がインビボでAktリン酸化を阻害することを実証する。API−1処理された腫瘍試料および無処理の腫瘍試料を溶解し、示された抗体によりイムノブロットした。
【図6A】AKT−SI1がIGF−1に誘導されるAktの細胞膜移行を阻害することを示した図である。HeLa細胞をカバーグラス上でMyc−AKT1によりトランスフェクションし、一晩血清飢餓を行い、次に30分間AKT−SI1あり(図6C)、またはAKT−SI1なしで(図6B)処理し、その後IGF1により15分間刺激した。固定に後続して、細胞を抗Mycモノクローナル抗体、続いてFITCコンジュゲート二次抗体により免疫染色して、サイトゾルまたは膜におけるエピトープのタグを付加したタンパク質の存在を示した(図6B)。AKT−SI1およびIGF1による処理のない細胞を対照として使用した(図6A)。
【図6B】AKT−SI1がIGF−1に誘導されるAktの細胞膜移行を阻害することを示した図である。HeLa細胞をカバーグラス上でMyc−AKT1によりトランスフェクションし、一晩血清飢餓を行い、次に30分間AKT−SI1あり(図6C)、またはAKT−SI1なしで(図6B)処理し、その後IGF1により15分間刺激した。固定に後続して、細胞を抗Mycモノクローナル抗体、続いてFITCコンジュゲート二次抗体により免疫染色して、サイトゾルまたは膜におけるエピトープのタグを付加したタンパク質の存在を示した(図6B)。AKT−SI1およびIGF1による処理のない細胞を対照として使用した(図6A)。
【図6C】AKT−SI1がIGF−1に誘導されるAktの細胞膜移行を阻害することを示した図である。HeLa細胞をカバーグラス上でMyc−AKT1によりトランスフェクションし、一晩血清飢餓を行い、次に30分間AKT−SI1あり(図6C)、またはAKT−SI1なしで(図6B)処理し、その後IGF1により15分間刺激した。固定に後続して、細胞を抗Mycモノクローナル抗体、続いてFITCコンジュゲート二次抗体により免疫染色して、サイトゾルまたは膜におけるエピトープのタグを付加したタンパク質の存在を示した(図6B)。AKT−SI1およびIGF1による処理のない細胞を対照として使用した(図6A)。
【図7A】AKT−SI1がAktキナーゼ活性、特に構成的に活性のあるAktの阻害においてAPI−2/TCNよりも強力であることを示した図である。AKT−SI1構造はAPI−2/TCNとリボース糖部分を共有しているが(図7A)、これらの2つの分子の残りの部分に化学的類似性はない。しかしながら、それらのAkt阻害能力を比較した。HEK293細胞を、野生型Myc−AKT1(図7Bおよび7C)および構成的に活性のあるMyc−AKT1−E17K(図7Dおよび7E)によりトランスフェクションした。36時間のインキュベーションに後続して、細胞に一晩飢餓血清を行なった。野生型Aktをトランスフェクションした細胞は、AKT−SI1(図7Bおよび7D)またはAPI−2/TCN(図7Cおよび7E)により30分間処理し、続いてEGFにより15分間刺激した。免疫沈降を抗Mycの抗体により実行し、免疫沈降物で基質としてヒストンH2Bを使用するインビトロキナーゼ分析を行なった(上部)。AKT−SI1およびAPI−2/TCNによるAktキナーゼ活性の阻害は、相対的活性として定量および計算した(中央)。ウエスタンブロット解析は免疫沈降したAKT1タンパク質を示す(下部パネル)。実験は3回反復した。
【図7B】AKT−SI1がAktキナーゼ活性、特に構成的に活性のあるAktの阻害においてAPI−2/TCNよりも強力であることを示した図である。AKT−SI1構造はAPI−2/TCNとリボース糖部分を共有しているが(図7A)、これらの2つの分子の残りの部分に化学的類似性はない。しかしながら、それらのAkt阻害能力を比較した。HEK293細胞を、野生型Myc−AKT1(図7Bおよび7C)および構成的に活性のあるMyc−AKT1−E17K(図7Dおよび7E)によりトランスフェクションした。36時間のインキュベーションに後続して、細胞に一晩飢餓血清を行なった。野生型Aktをトランスフェクションした細胞は、AKT−SI1(図7Bおよび7D)またはAPI−2/TCN(図7Cおよび7E)により30分間処理し、続いてEGFにより15分間刺激した。免疫沈降を抗Mycの抗体により実行し、免疫沈降物で基質としてヒストンH2Bを使用するインビトロキナーゼ分析を行なった(上部)。AKT−SI1およびAPI−2/TCNによるAktキナーゼ活性の阻害は、相対的活性として定量および計算した(中央)。ウエスタンブロット解析は免疫沈降したAKT1タンパク質を示す(下部パネル)。実験は3回反復した。
【図7C】AKT−SI1がAktキナーゼ活性、特に構成的に活性のあるAktの阻害においてAPI−2/TCNよりも強力であることを示した図である。AKT−SI1構造はAPI−2/TCNとリボース糖部分を共有しているが(図7A)、これらの2つの分子の残りの部分に化学的類似性はない。しかしながら、それらのAkt阻害能力を比較した。HEK293細胞を、野生型Myc−AKT1(図7Bおよび7C)および構成的に活性のあるMyc−AKT1−E17K(図7Dおよび7E)によりトランスフェクションした。36時間のインキュベーションに後続して、細胞に一晩飢餓血清を行なった。野生型Aktをトランスフェクションした細胞は、AKT−SI1(図7Bおよび7D)またはAPI−2/TCN(図7Cおよび7E)により30分間処理し、続いてEGFにより15分間刺激した。免疫沈降を抗Mycの抗体により実行し、免疫沈降物で基質としてヒストンH2Bを使用するインビトロキナーゼ分析を行なった(上部)。AKT−SI1およびAPI−2/TCNによるAktキナーゼ活性の阻害は、相対的活性として定量および計算した(中央)。ウエスタンブロット解析は免疫沈降したAKT1タンパク質を示す(下部パネル)。実験は3回反復した。
【図7D】AKT−SI1がAktキナーゼ活性、特に構成的に活性のあるAktの阻害においてAPI−2/TCNよりも強力であることを示した図である。AKT−SI1構造はAPI−2/TCNとリボース糖部分を共有しているが(図7A)、これらの2つの分子の残りの部分に化学的類似性はない。しかしながら、それらのAkt阻害能力を比較した。HEK293細胞を、野生型Myc−AKT1(図7Bおよび7C)および構成的に活性のあるMyc−AKT1−E17K(図7Dおよび7E)によりトランスフェクションした。36時間のインキュベーションに後続して、細胞に一晩飢餓血清を行なった。野生型Aktをトランスフェクションした細胞は、AKT−SI1(図7Bおよび7D)またはAPI−2/TCN(図7Cおよび7E)により30分間処理し、続いてEGFにより15分間刺激した。免疫沈降を抗Mycの抗体により実行し、免疫沈降物で基質としてヒストンH2Bを使用するインビトロキナーゼ分析を行なった(上部)。AKT−SI1およびAPI−2/TCNによるAktキナーゼ活性の阻害は、相対的活性として定量および計算した(中央)。ウエスタンブロット解析は免疫沈降したAKT1タンパク質を示す(下部パネル)。実験は3回反復した。
【図7E】AKT−SI1がAktキナーゼ活性、特に構成的に活性のあるAktの阻害においてAPI−2/TCNよりも強力であることを示した図である。AKT−SI1構造はAPI−2/TCNとリボース糖部分を共有しているが(図7A)、これらの2つの分子の残りの部分に化学的類似性はない。しかしながら、それらのAkt阻害能力を比較した。HEK293細胞を、野生型Myc−AKT1(図7Bおよび7C)および構成的に活性のあるMyc−AKT1−E17K(図7Dおよび7E)によりトランスフェクションした。36時間のインキュベーションに後続して、細胞に一晩飢餓血清を行なった。野生型Aktをトランスフェクションした細胞は、AKT−SI1(図7Bおよび7D)またはAPI−2/TCN(図7Cおよび7E)により30分間処理し、続いてEGFにより15分間刺激した。免疫沈降を抗Mycの抗体により実行し、免疫沈降物で基質としてヒストンH2Bを使用するインビトロキナーゼ分析を行なった(上部)。AKT−SI1およびAPI−2/TCNによるAktキナーゼ活性の阻害は、相対的活性として定量および計算した(中央)。ウエスタンブロット解析は免疫沈降したAKT1タンパク質を示す(下部パネル)。実験は3回反復した。
【図8A】AKT−SI1およびAPI−2/TCNがリン酸化Aktレベルを阻害することを示した図である。野生型Myc−AKT1および構成的に活性のあるMyc−AKT1−E17KによりトランスフェクションしたHEK293細胞を、示された試薬により処理し、抗リン酸化−Akt−S473抗体(上部)および抗Myc抗体(下部)によりイムノブロットした。
【図8B】AKT−SI1およびAPI−2/TCNがリン酸化Aktレベルを阻害することを示した図である。野生型Myc−AKT1および構成的に活性のあるMyc−AKT1−E17KによりトランスフェクションしたHEK293細胞を、示された試薬により処理し、抗リン酸化−Akt−S473抗体(上部)および抗Myc抗体(下部)によりイムノブロットした。
【発明を実施するための形態】
【0013】
本発明は、Akt/PKB経路を阻害する化合物および組成物に関する。1つの実施形態において、本発明の化合物は、Aktタンパク質のキナーゼ活性および/またはリン酸化レベルを阻害する。本発明の化合物は、式I:
【化2】
(I)
(式中、
Xは独立してO、NまたはSであり;
YはO、NまたはSであり;
R1は、独立して−H、−OH、−NH2、−NO2、ハロゲン、−OHにより任意に置換されるアルキル、または−OHにより任意に置換されるアルコキシであり;
および、R2は、−H、−OH、−NH2、−C(O)NH2、アルキル、またはアルコキシであり、それらのいずれかは、−OH、ハロゲン、アルキルまたはアルコキシにより任意に置換することができ;
R3は、−H、−OH、−NH2、−C(O)NH2、アルキルまたはアルコキシであり、それらのいずれかは、−OHまたはハロゲンまたはアルキルまたはアルコキシにより任意で置換することができる);または
その類似体もしくは薬学的に許容される塩で示される一般的な構造を有する。
【0014】
1つの実施形態において、各XはNである。例示された実施形態において、YはOである。別の例示された実施形態において、各R1は独立して−OHまたは−CH2OHである。なお別の実施形態において、R2は−OHにより任意に置換される−NH2である。さらに実施形態において、R2は−C(O)NH2である。
【0015】
具体的な実施形態において、本発明の化合物は、以下の構造(式II):
【化3】
(II)
を有する 。
【0016】
本発明の化合物は、PTEN変異などの上流レギュレーターの変化からの結果として生じる高度に活性化された野生型Aktだけでなく、myr−AKT1、Myr−AKT2およびE40K−AKT1を含む、構成的に活性のあるAkt変異体も阻害する。最近の研究は、脂質結合ポケット中のアミノ酸17でのグルタミン酸のリジン置換(E17K)をもたらす、ヒトの乳癌、結腸直腸癌および卵巣癌におけるAKT1のPHドメイン中の再発性体細胞変異を同定した(Carpten et al. 2007)。Lys17はポケットの静電的相互作用を変化させて、フォスフォイノシチドリガンドによる新しい水素結合を形成する。この変異は細胞膜への病的な局在を介してAKT1を活性化し、細胞を形質転換し、マウスにおける白血病を誘導する。さらに、E17K置換はアロステリックキナーゼ阻害剤への感受性を低下させる(Carpten et al. 2007)。E40K−AKT1変異体は細胞膜に局在できるE17Kに類似しているので(Bellacosa et al. 1998)、API−1はE17K−AKT1もまた阻害しうる。
【0017】
単離された化合物として本発明の化合物を投与することができるが、これらの化合物は、医薬組成物の一部としても投与することができる。本発明は、したがってさらに少なくとも1つの薬学的に許容される担体を伴う組成物を提供する。医薬組成物は、腸内投与、非経口投与、静脈内投与、筋肉内投与、局所投与、皮下投与などのような様々な投与経路に適合させることができる。当業者によって決定することができるように、継続的に、または明瞭なインターバルで投与することができる。
【0018】
本発明の化合物は、薬学的に有益な組成物を調製する公知の方法に従って製剤化することができる。製剤は、当業者に周知であり容易に利用可能な多数の出典中に記述されている。例えば、レミントンの製薬科学(Remington's Pharmaceutical Science)(Martin 1995)は、本発明に関連して使用できる製剤について記述する。投与に適切な製剤は、例えば、抗酸化剤、緩衝剤、静菌剤、および意図されるレシピエントの製剤を血液と等張にする溶質を含みうる水性の滅菌済み注射溶液;ならびに懸濁化剤および増粘剤を含みうる水性および非水性の滅菌済み懸濁物を含む。製剤は、単位用量またはマルチ用量の容器(例えば密封したアンプル、バイアル)中に提供され、使用前に、滅菌済み液体担体(例えば注射用の水)の条件のみを必要とする、冷凍乾燥(凍結乾燥)条件で保存することができる。即時注射溶液および懸濁物は、滅菌済みの粉末、顆粒、タブレットなどから調製することができる。特に上で言及された成分に加えて、本発明の組成物は、当該製剤のタイプを考慮して、当技術分野における従来の他の薬剤を含みうることが理解されるべきである。
【0019】
本明細書において使用されるように、アルキルは、直鎖または分岐鎖である、1〜20の炭素原子を有する飽和炭化水素基または一価不飽和炭化水素基もしくは多価不飽和炭化水素基を意味し、C1-Xアルキルは、1〜「X」の炭素原子数を含む直鎖または分岐鎖のアルキル基を意味する。例えば、C1-6アルキルは、1〜6の炭素原子を含む直鎖または分岐鎖のアルキル基を意味する。アルコキシは、アルキル基が以前に本明細書において記述されているようなものである、アルキル−O−基を意味する。
【0020】
用語「ハロゲン」は、フッ素、塩素、臭素、またはヨウ素などの周期表のハロゲンを意味する。
【0021】
用語「任意に置換される」は、任意の利用可能な1つまたは複数の位置で、1つまたは複数の有機基または無機基(例えばアルキル、アリール、ヘテロアリール、アシル、アルケニル、シクロアルキル、ヘテロシクロアルキル、シクロアルケニル、ヘテロシクロアルケニル、またはハロゲン)により任意に置換されることを意味する。
【0022】
飽和アルキル基の例は、メチル、エチル、N−プロピル、イソプロピル、N−ブチル、tert−ブチル、イソブチル、sec−ブチル、N−ペンチル、N−ヘキシル、N−ヘプチルおよびN−オクチルを含むが、これらに限定されない。不飽和アルキル基は、1つまたは複数の二重結合または三重結合を有するアルキル基である。不飽和アルキル基は、例えば、エテニル、プロペニル、ブテニル、ヘキセニル、ビニル、2−プロピニル、2−イソペンテニル、2−ブタジエニル、エチニル、1−プロピニル、3−プロピニルおよび3−ブチニルを含む。具体的には、「アルキル」は、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソ−ブチル、sec−ブチル、ペンチル、3−ペンチル、ヘキシル、ヘプチル、オクチル、ノニル、デシル、ウンデシル、ドデシル、トリデシル、テトラデシル、ペンタデシル、ビニル、1−プロペニル、2−プロペニル、1−ブテニル、2−ブテニル、3−ブテニル、1−ペンテニル、2−ペンテニル、3−ペンテニル、4−ペンテニル、1−ヘキセニル、2−ヘキセニル、3−ヘキセニル、4−ヘキセニル、5−ヘキセニル、1−ヘプテニル、2−ヘプテニル、3−ヘプテニル、4−ヘプテニル、5−ヘプテニル、1−ノネニル、2−ノネニル、3−ノネニル、4−ノネニル、5−ノネニル、6−ノネニル、7−ノネニル、8−ノネニル、1−デケニル、2−デケニル、3−デケニル、4−デケニル、5−デケニル、6−デケニル、7−デケニル、8−デケニル、9−デケニル;1−ウンデケニル、2−ウンデケニル、3−ウンデケニル、4−ウンデケニル、5−ウンデケニル、6−ウンデケニル、7−ウンデケニル、8−ウンデケニル、9−ウンデケニル、10−ウンデケニル、1−ドデケニル、2−ドデケニル、3−ドデケニル、4−ドデケニル、5−ドデケニル、6−ドデケニル、7−ドデケニル、8−ドデケニル、9−ドデケニル、10−ドデケニル、11−ドデケニル、1−トリデケニル、2−トリデケニル、3−トリデケニル、4−トリデケニル、5−トリデケニル、6−トリデケニル、7−トリデケニル、8−トリデケニル、9−トリデケニル、10−トリデケニル、11−トリデケニル、12−トリデケニル、1−テトラデケニル、2−テトラデケニル、3−テトラデケニル、4−テトラデケニル、5−テトラデケニル、6−テトラデケニル、7−テトラデケニル、8−テトラデケニル、9−テトラデケニル、10−テトラデケニル、11−テトラデケニル、12−テトラデケニル、13−テトラデケニル、1−ペンタデケニル、2−ペンタデケニル、3−ペンタデケニル、4−ペンタデケニル、5−ペンタデケニル、6−ペンタデケニル、7−ペンタデケニル、8−ペンタデケニル、9−ペンタデケニル、10−ペンタデケニル、11−ペンタデケニル、12−ペンタデケニル、13−ペンタデケニル、または14−ペンタデケニルを含むことができ;「アルコキシ」は、メトキシ、エトキシ、プロポキシ、イソプロポキシ、ブトキシ、イソ−ブトキシ、sec−ブトキシ、ペンタオキシ、3−ペンタオキシ、ヘキサオキシ、ヘプチロキシ、オクチロキシ、ノニロキシ、デシロキシ、ウンデシロキシ、ドデシロキシ、トリデシロキシ、テトラデシロキシ、またはペンタデシロキシを含むことができる。
【0023】
本発明の化合物は、当業者によって調製することができるすべて水和物および塩を含む。本発明の化合物が安定した無毒な酸性塩または塩基塩を形成するのに十分に塩基性または酸性の条件下で、塩として化合物を投与することは適切でありうる。薬学的に許容される塩の例は、生理学的な許容されるアニオンを形成する酸により形成された有機酸の付加塩(例えば、トシル酸塩、メタンスルホン酸塩、酢酸塩、クエン酸塩、マロン酸塩、酒石酸塩、コハク酸塩、安息香酸塩、アスコルビン酸塩、α−ケトグルタル酸塩およびα−グリセロリン酸塩)である。塩酸塩、硫酸塩、硝酸塩、重炭酸塩、および炭酸塩塩を含む、適切な無機塩類もまた形成することができる。
【0024】
化合物の薬学的に許容される塩は、当技術分野における周知の標準的手順を使用して、例えば、生理学的に許容されるアニオンを与える適切な酸とアミンなどの十分に塩基性の化合物を反応させることによって得ることができる。カルボン酸のアルカリ金属(例えばナトリウム、カリウムまたはリチウム)塩、またはアルカリ土類金属(例えばカルシウム)塩もまた作製することができる。
【0025】
本明細書において使用されるように、用語「類似体」は、実質的にもう一つの化合物と同種類のものであるが、例えば、側鎖基の追加、親構造の酸化または還元によって修飾されうる化合物を指す。本発明の化合物の類似体は、一般に公知の標準的反応を使用して容易に調製することができる。これらの標準的反応は、水素化反応、アルキル化反応、アセチル化反応および酸性化反応を含むが、これらに限定されない。化学的修飾は、分子中に存在するすべての官能基を保護すること、および所望の反応を行なった後にそれらを脱保護することによって、科学文献中で公知の標準的手順を使用して、当業者によって成し遂げることができる(Greene et al. 1999; Honda et al. 1997; Honda et al. 1998; Konoike et al. 1997; Honda et al. 2000;それらの各々はその全体を参照することにより本明細書に組み入れられる)。所望の生物学的活性(アポトーシスの誘導、細胞毒性、細胞静止、細胞周期停止の誘導、抗血管形成特性などのような)を示す類似体は、細胞分析または他のインビトロもしくはインビボの分析を使用して、同定または確認することができる。
【0026】
本発明の化合物は1つまたは複数の非対称的に置換された炭素原子(すなわち炭素中心)を含むことができることが理解される。本発明の化合物における1つまたは複数の不斉中心の存在は、立体異性体を生じさせることができ、各場合において、本発明は、鏡像異性体および偏左右異性体を含むそのようなすべての立体異性体、ならびにそのラセミ混合物を含む混合物に拡張するように理解されるべきである。
【0027】
本発明の化合物および組成物は、様々な非治療目的および治療目的に有用である。化合物および組成物は、動物およびヒトにおける異常な細胞増殖の減少のために使用できる。化合物のかかる抗増殖特性のために、それらはインビトロおよびインビボを含む様々な設定における望まれない細胞増殖を減少させるのに有用である。
【0028】
化合物およびそれらを含む組成物の治療適用は、現時点でまたは将来的に当業者に公知の任意の適切な治療方法および技術によって成し遂げることができる。さらに、本発明の化合物は、他の有用な化合物および組成物の調製のための出発材料または中間体としての用途を有する。
【0029】
本発明の化合物およびその組成物は、任意で不活性希釈剤などの薬学的に許容される担体と組み合わせて、望まれない細胞増殖の部位(腫瘍部位または良性の皮膚増殖など、例えば腫瘍または皮膚増殖に対して注射または局所的に適用される)または真菌感染の部位などの1つまたは複数の解剖学的な部位で局所的に投与することができる。本発明の化合物およびその組成物は、任意で不活性希釈剤など薬学的に許容される担体または経口送達のための吸収可能な食用担体と組み合わせて、全身的に(静脈内または経口的など)投与することができる。それらはハードシェルゼラチンカプセルまたはソフトシェルゼラチンカプセル中に封入できるか、タブレットに圧縮できるか、または患者の食餌の食品に直接組み入れることができる。経口の治療投与のために、活性化合物は1つまたは複数の賦形剤と組み合わせることができ、摂取可能なタブレット、バッカル錠、トローチ、カプセル、エリキシル、懸濁物、シロップ剤、ウエハー、エアゾールスプレーおよび同種のものの形態で使用できる。
【0030】
タブレット、トローチ、ピル、カプセルおよび同種のものは、以下のものもまた含むことができる。トラガカントゴム、アラビアゴム、トウモロコシデンプンまたはゼラチンなどの結合剤;リン酸カルシウムなどの賦形剤;トウモロコシデンプン、ジャガイモデンプン、アルギン酸および同種のものなどの崩壊剤;ステアリン酸マグネシウムなどの潤滑剤;およびスクロース、フルクトース、ラクトースもしくはアスパルテームなど甘味剤、またはペパーミント、ヒメコウジ油もしくはチェリーフレーバーなどの着香剤を追加できる。単位投与量形態がカプセルである場合、上述のタイプの材料に加えて、植物油またはポリエチレングリコールなどの液体担体を含むことができる。様々な他の材料は、被膜として、またはそうでなければ固体単位の投与量形態の物理的形態を修飾するように存在しうる。例えば、タブレット、ピルまたはカプセルは、ゼラチン、ワックス、セラックまたは糖および同種のものにより被覆できる。シロップまたはエリキシルは、活性化合物、甘味剤としてスクロースまたはフルクトース、防腐剤としてメチルパラベンおよびプロピルパラベン、色素、ならびにチェリーフレーバーまたはオレンジフレーバーなどの香味料を含むことができる。当然、任意の単位投与量形態の調製に使用される任意の材料は、使用する量で薬学的に許容され、実質的に非毒性であるべきである。さらに、活性化合物は徐放性の調製物および装置の中へ組み入れることができる。
【0031】
本発明の化合物および組成物(その薬学的に許容される塩または類似体を含む)は、注入または注射によって静脈内に、筋肉内に、または腹腔内に投与することができる。活性薬剤またはその塩の溶液は無毒な界面活性剤と任意で混合されて、水中で調製することができる。分散物は、グリセロール、液体ポリエチレングリコール、トリアセチンおよびその混合物、ならびに油中でも調製することができる。通常の保存および使用の条件下で、これらの調製物は、微生物の増殖を防止する防腐剤を含むことができる。
【0032】
注射または注入に適切な薬学的投与量形態は、任意でリポソーム中でカプセル化された滅菌済みの注射可能もしくは注入可能な溶液または分散物の即時調整に適合する活性成分を含む、滅菌済みの水性の溶液もしくは分散物または滅菌済みの粉末を含むことができる。最終的な投与量形態は、滅菌済みで流動的であり、製造および保存の条件下で安定しているべきである。液体担体または媒質は、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、液体ポリエチレングリコール、および同種のもの)、植物油、無毒なグリセリルエステル、および適切なそれらの混合物を含む、溶媒または液体分散媒でありえる。適切な流動性は、例えば、リポソームの形成によって、分散物の場合で必要とされる粒子サイズの維持によって、または界面活性剤の使用によって、維持することができる。任意で、微生物活動の防止は、様々な他の抗菌剤および抗真菌剤(例えば、パラベン、クロロブタノール、フェノール、ソルビン酸、チメロサール、および同種のもの)によって成し遂げることができる。多くの場合において、等張剤(例えば、糖、緩衝剤または塩化ナトリウム)を含むことは好ましいだろう。注射可能な組成物の持続的吸収は、吸収遅延剤(例えばモノステアリン酸アルミニウムおよびゼラチン)の含有によって成し遂げることができる。
【0033】
滅菌済みの注射可能な溶液は、上で列挙された様々な他の成分と共に適切な溶媒中に必要とされた量で本発明の化合物を組み入れ、必要に応じて続いて濾過滅菌することによって調製される。滅菌済みの注射可能な溶液の調製のための滅菌済み粉末の場合において、調製の好ましい方法は真空乾燥および凍結乾燥の技術であり、事前に滅菌済みの濾過溶液中に存在する任意の追加の所望成分を加えた活性成分の粉末をもたらす。
【0034】
局所投与のために、本発明の化合物は液体または固体として適用されうる。しかしながら、皮膚科学的に許容される担体(固体または液体でありうる)と組み合わせて、組成物として皮膚に局所的に投与することが一般的に望ましいだろう。本発明の化合物および組成物を被験体の皮膚に局所的に適用して、悪性腫瘍または良性腫瘍のサイズを減少させる(および完全な除去を含みうる)か、または感染部位を治療することができる。本発明の化合物は、増殖部位または感染部位に直接適用することができる。好ましくは、化合物は、軟膏、クリーム、ローション、溶液、チンキ、または同種のものなどの製剤で増殖部位または感染部位に適用される。皮膚の病変への薬理学的物質の送達のための薬物送達システムは、米国特許第5,167,649号中で記述されたようなものも使用することができる。
【0035】
有用な固体担体は、タルク、粘土、微結晶性セルロース、シリカ、アルミナ、および同種のものなどの微粉固体を含む。有用な液体担体は、水、アルコールもしくはグリコールまたは水−アルコール/グリコールブレンドを含み、化合物は任意で非毒性界面活性剤の補助によって、効果的なレベルで溶解または分散させることができる。香料および追加の抗生物質のような補助剤を追加して、規定の使用のための特性を最適化することができる。結果として生ずる液体組成物は、吸収性パッドから適用するか、救急絆および他の包帯を使用して浸透させるか、例えばポンプタイプ噴霧器またはエアゾール噴霧器を使用して罹患した領域上にスプレーすることができる。
【0036】
合成ポリマー、脂肪酸、脂肪酸塩およびエステルなどの増粘剤、脂肪アルコール、修飾セルロースまたは修飾鉱物材もまた、液体担体と共に使用して、使用者の皮膚への直接的な適用のために、塗布可能なペースト、ゲル、軟膏、石鹸、および同種のものを形成することができる。皮膚へ化合物を送達するために使用できる有用な皮膚科学的組成物の例は、米国特許第4,608,392号;米国特許第4,992,478号;米国特許第4,559,157号;および米国特許第4,820,508号で開示される。
【0037】
本発明の化合物および医薬組成物の有用な投与量は、インビトロの活性と動物モデルにおけるインビボの活性との比較によって決定することができる。マウス、および他の動物、ヒトに対する有効量の推定のための方法は、当技術分野で公知であり、例えば米国特許第4,938,949号を参照。
【0038】
本発明は、薬学的に許容される担体と組み合わせた本発明の化合物を含む医薬組成物にも関する。経口投与、局所投与または非経口投与に適合した、化合物の量を含む医薬組成物は、本発明の好ましい実施形態を構成する。本発明の文脈中で患者(特にヒト)に投与される用量は、適当な期間枠にわたって患者において治療効果を達成するのに十分であり、致死毒性がなく、許容されるレベル以上の副作用または病的状態を好ましくは引き起こさないものであるべきである。当業者は、病理学的状態の重症度およびステージに加えて、被験体の条件(健康)、被験体の体重、もしあれば併用療法の種類、処理の頻度、治療係数を含む様々な因子に、投与量が依存することを認識するだろう。
【0039】
本発明は、式Iの化合物、またはその塩もしくは類似体の有効量と細胞を接触させることを含む、Aktタンパク質の発現が上昇されるかまたは構成的に活性のある、癌細胞もしくは腫瘍細胞または他の細胞の生存または増殖を阻害するかまたは殺傷する方法にも関する。具体的な実施形態において、化合物は、式II、またはその塩もしくは類似体で示される構造を有する。1つの実施形態において、細胞はヒト細胞または他の哺乳類細胞である。本方法を使用して阻害または殺傷することができる癌細胞は、転移性の性質の細胞を含んでいる。したがって、癌細胞または腫瘍細胞の転移の阻害もまた本発明によって検討される。本方法はインビトロまたはインビボで実行することができる。
【0040】
本発明は、式Iの化合物、またはその塩もしくは類似体の有効量を人または動物に投与することを含む、人または動物における癌または腫瘍などの腫瘍学的障害を治療する方法にも関する。具体的な実施形態において、化合物は、式II、またはその塩もしくは類似体で示される式を有する。1つの実施形態において、本発明の化合物または組成物の有効量は、腫瘍学的障害を有し、その治療を必要とする患者に投与される。阻害剤は、化学療法、免疫療法および/または放射線療法の前か、それらの後か、またはそれらと併用して、投与することができる。本発明の方法は、腫瘍学的障害の治療を必要としているか、または必要としうる患者の同定を任意で含むことができる。本発明の方法を使用する治療を必要とする患者は、医学または獣医学の当業者に公知の標準的技術を必要に応じて使用して同定することができる。1つの実施形態において、患者は、霊長類(サル、チンパンジー、類人猿など)、イヌ、ネコ、ウシ、ブタもしくはウマ、または腫瘍学的障害がある他の動物などの、ヒトまたは他の哺乳類でありえる。患者への投与のための本発明の化合物の投与および製剤化のための手段は、当技術分野において公知であり、それらの実施例は本明細書において記述される。本発明の範囲内の腫瘍学的障害は、肛門、胆管、膀胱、骨、骨髄、腸管(大腸および直腸を含む)、乳房、目、胆嚢、腎臓、口、喉頭、食道、胃、精巣、頚管、頭部、首、卵巣、肺、中皮腫、神経内分泌、陰茎、皮膚、脊髄、甲状腺、膣、外陰部、子宮、肝臓、筋肉、膵臓、前立腺、血球(リンパ球および他の免疫系細胞を含む)および脳の癌および/または腫瘍を含むが、これらに限定されない。本発明による治療のために検討される具体的な癌は、癌腫、カポジ肉腫、黒色腫、中皮腫軟部組織肉腫、白血病(急性リンパ芽球性、急性骨髄性、慢性リンパ球性、慢性骨髄性、および他のもの)、およびリンパ腫(ホジキンおよび非ホジキン)、および多発性骨髄腫を含む。
【0041】
腫瘍学的障害の治療のために、本発明によって検討される化合物および組成物は、他の抗腫瘍性もしくは抗癌性の薬剤もしくは物質(例えば化学療法剤、免疫療法剤、放射線療法剤など)および/または放射線療法および/または腫瘍を除去する外科的治療の前か、それらの後か、またはそれらと組み合わせて、治療を必要とする患者に投与することができる。例えば、本発明の化合物および組成物は、タキソールまたはビンブラスチンなどの有糸分裂阻害剤、シクロホスアミドまたはイフォスファミドなどのアルキル化剤、5−フルオロウラシルまたはヒドロキシウレアなどの抗代謝剤、アドリアマイシンまたはブレオマイシンなどのDNAインターカレーター、エトポシドまたはカンプトテシンなどのトポイソメラーゼ阻害剤、アンジオスタチンなどの抗血管新生剤、タモキシフェンなどの抗エストロゲン、および/または例えばグリーベック(ノバルティスファーマ(Novartis Pharmaceuticals)社)およびハーセプチン(ジェネンテック(Genentech)社)などのそれぞれ他の抗癌薬物または抗体により、患者が、治療される予定か、または治療されているか、または治療されてきた、癌治療法において使用することができる。これらの他の物質または放射線治療は、本発明の化合物と同様の、または異なる回数で与えることができる。本発明の範囲内で検討される他の化学療法剤の例は、アルトレタミン、ブレオマイシン、ボルテゾミブ(ベルケイド)、ブスルファン、ホリナートカルシウム、カペシタビン、カルボプラチン、カルムスチン、クロラムブチル、シスプラチン、クラドリビン、クリサンタスパーゼ(crisantaspase)、シクロホスファミド、シタラビン、ダカルバジン、ダクチノマイシン、ダウノルビシン、ドセタキセル、ドキソルビシン、エピルビシン、エトポシド、フルダラビン、フルオロウラシル、ゲフィチニブ(イレッサ)、ゲムシタビン、ヒドロキシウレア、イダルビシン、イフォスファミド、イマチニブ(グリーベック)、イリノテカン、リポソームドキソルビシン、ロムスチン、メルファラン、メルカプトプリン、メトトレキサート、マイトマイシン、ミトキサントロン、オキサリプラチン、パクリタキセル、ペントスタチン、プロカルバジン、ラルチトレキセド、ストレプトゾシン、テガフール−ウラシル、テモゾロマイド、チオテパ、チオグアニン/チオグアニン(tioguanine/thioguanine)、トポテカン、トレオスルファン、ビンブラスチン、ビンクリスチン、ビンデシン、ビノレルビンを含むが、これらに限定されない。例示された実施形態において、化学療法剤はメルファランである。本発明の範囲内で検討された免疫療法剤の例は、アレムツズマブ、セツキシマブ(エルビタックス)、ゲムツズマブ、ヨウ素131トシツモマブ、リツキシマブ、トラスツザマブ(trastuzamab)(ハーセプチン)を含むが、これらに限定されない。本発明は、化学療法剤、免疫療法剤、放射線療法剤、または放射線療法の投与の前か、それらの後か、および/またはそれらと組み合わせて、本発明の化合物の有効量を投与することを含む、腫瘍学的障害を治療する方法にも関する。
【0042】
本発明は、本発明の化合物または組成物の有効量と細胞を接触させることによって、細胞中のAkt/PKB経路を阻害する方法にも関する。1つの実施形態において、化合物は、Akt1、AKT2および/またはAKT3タンパク質に結合し、それらの活性を阻害する。具体的な実施形態において、化合物は、式II、またはその塩もしくは類似体で示される構造を有する。1つの実施形態において、細胞はヒト細胞または哺乳類細胞であり、異常な増殖、生存、移動または分化を示す癌細胞もしくは腫瘍細胞または他の細胞になりえる。1つの実施形態において、細胞は、Akt1、AKT2、および/またはAKT3などのAktタンパク質を、構成的に発現するか、または上昇レベルもしくは異常レベルで発現する。
【0043】
本発明は、本発明の化合物または組成物の治療的有効量が人または動物に投与される、細胞中のAktタンパク質の構成的発現、異常な発現、または上昇した発現に関連する障害を有する人または動物を治療する方法にも関する。障害は、例えば、異常な細胞増殖、細胞生存、細胞移動、および/または細胞分化によって特徴づけられるものでありえる。1つの実施形態において、化合物は、Akt1、AKT2、および/またはAKT3タンパク質に結合し、それらの活性を阻害する。具体的な実施形態において、化合物は式II、またはその塩もしくは類似体で示される構造を有する。
【0044】
治療される障害または疾患の条件に依存して、適切な用量は標的細胞の増加または増殖を減少させる量でありえる。癌の情況において、適切な用量は、悪性腫瘍などの癌組織において、所望反応を達成することが公知である活性薬剤の濃度をもたらすものである。好ましい投与量は、管理し難い副作用なしに癌細胞増殖の最大の阻害をもたらす量である。当業者によって決定することができるように、化合物の投与は継続的、または異なるインターバルでありえる。
【0045】
いくつかの実施形態において、所望療法の治療のためのかかる投薬量の投与を提供するために、本発明の医薬組成物は、担体または希釈剤を含む全組成物の重量に基づいて、1つまたは複数の化合物の総重量で、約0.1%〜45%、特に1〜15%の間で含まれうる。例示的に、投与される活性のある成分の投与量レベルは、静脈内、0.01〜約20mg/kg;腹腔内、0.01〜約100mg/kg;皮下、0.01〜約100mg/kg;筋肉内、0.01〜約100mg/kg;経口的、0.01〜約200mg/kg、および好ましくは約1〜100mg/kg;鼻腔内滴下、0.01〜約20mg/kg;ならびにエアロゾル、0.01〜約20mg/kgの動物(身体)重量でありえる。
【0046】
本発明は、1つまたは複数の容器中に、本発明の1つまたは複数の化合物、または本発明の化合物を含む組成物、または前述のものの類似体もしくは塩を含むキットにも関する。1つの実施形態において、キットは、式IIの化合物、またはその薬学的に許容される塩もしくは類似体を含む。本発明のキットは、薬学的に許容される担体および/または希釈剤を任意で含むことができる。1つの実施形態において、本発明のキットは1つまたは複数の他の成分または添加物または本明細書において記述されるような補助剤を含む。他の実施形態において、キットは、本明細書において記述される薬剤などの1つまたは複数の抗癌剤を含む。1つの実施形態において、本発明のキットは、キットの化合物または組成物を投与する方法について記述する説明書または包装材料を含む。キットの容器は、任意の適切な材料(例えばガラス、プラスチック、金属など)、および任意の適切なサイズ、形状、または立体配置でありえる。1つの実施形態において、本発明の化合物は、タブレット、ピル、または粉末の形態などの固形としてキット中に提供される。他の実施形態において、本発明の化合物は液体または溶液としてキット中に提供される。1つの実施形態において、キットは、液体または溶液の形態で本発明の化合物を含むアンプルまたはシリンジを含む。
【0047】
開示された方法から利益を得る哺乳類種は、類人猿、チンパンジー、オランウータン、ヒト、サルなどの霊長類;イヌ、ネコ、モルモット、ハムスター、ベトナムポットベリーピッグ、ウサギ、およびフェレットなどの飼育された動物(例えばペット);ウシ、水牛、バイソン、ウマ、ロバ、ブタ、ヒツジ、およびヤギなどの飼育された家畜;クマ、ライオン、トラ、ヒョウ、ゾウ、カバ、サイ、キリン、アンテロープ、ナマケモノ、カモシカ、シマウマ、ウィルドビースト、プレーリードッグ、コアラ、カンガルー、コモリネズミ、アライグマ、パンダ、ハイエナ、アザラシ、アシカ、ゾウアザラシ、カワウソ、ネズミイルカ、イルカおよびクジラなどの典型的には動物園において見出される外来動物を含むが、これらに限定されない。開示された方法から利益を得てもよい他の種は魚類、両生類、鳥および爬虫類を含む。本明細書において使用されるように、「患者」、「被験体」という用語は同じ意味で使用され、かかるヒト種および非ヒト種を含むように意図される。同様に、本発明のインビトロの方法は、かかるヒト種および非ヒト種の細胞に対して実行することができる。
材料および方法
【0048】
細胞株およびNCI多様性セット。この研究において使用されるすべて細胞株は、ATCCから購入したか、または事前に記載されたものである。(Cheng et al. 1997; Jiang et al. 2000; Yang et al. 2004; Yang et al. 2005)。NCI構造多様性セットは、およそ140,000の化合物のNCI薬物寄託場所から選択された1,992の化合物のライブラリーである。これらの多様性セット化合物の選択、構造および活性に関する詳細なデーターは、NCIの開発的な治療プログラム(Developmental Therapeutics Program)ウェブサイトで見出すことができる(Jones et al. 1991)。
【0049】
Aktにより形質転換した細胞の増殖阻害についてのスクリーニング。AKT2により形質転換したNIH3T3細胞またはLXSNベクターによりトランスフェクションしたNIH3T3対照細胞(Cheng et al. 1997)を、96ウェル組織培養プレートにプレーティングした。5μMのNCI多様性セット化合物による処理に後続して、細胞増殖は、セルタイター96ワン・ソリューション・セル・プロリファレーション(CellTier 96 One Solution Cell Proliferation)キット(プロメガ(Promega)社)により検出された。AKT2により形質転換したNIH3T3細胞の増殖を阻害するが、LXSNによりトランスフェクションしたNIH3T3細胞は阻害しない化合物をAkt阻害剤の候補と見なし、詳しい解析を行なった。
【0050】
インビトロのプロテインキナーゼ、細胞生存およびアポトーシス分析。以前に記述されたように、インビトロのキナーゼを実行した(Jiang et al. 2000)。細胞生存はMTT(シグマ(Sigma)社)により分析した。アポトーシスは、アネキシンV(BDバイオサイエンス(BD Biosciences)社)により検出し、製造者の説明書に従って実行した。組換えのAktおよびPDK1は、アップステート・バイオテクノロジー(Upstate Biotechnology)社から購入した。
【0051】
ヌードマウス腫瘍異種移植モデルにおける抗腫瘍活性。腫瘍細胞を採取し、PBS中に再懸濁し、以前に報告されるように8週齢のメスヌードマウスの右脇腹および左脇腹(2×106細胞/脇腹)に皮下(s.c.)注射した(Sun et al. 1999)。腫瘍が約100〜150 mm3に達した時に、動物は無作為化され、媒質または薬物を毎日腹腔内(i.p.)投薬した。対照動物にジメチルスルホキシド(DMSO)(20%)媒質を投与し、処理動物に20%DMSO中のAPI−1(10mg/kg/日)を注射した。
【0052】
本明細書において参照または引用したすべての特許、特許出願、仮出願、および出版物は、すべて図および表を含むその全体を参照することにより、この明細書の明確な教示に矛盾しない程度まで組み入れられる。
【0053】
以下は本発明の実行のための手順を説明する実施例である。これらの実施例は限定として解釈されるべきでない。特に断りのない限り、すべてのパーセンテージは重量により、すべての溶媒混合比率は体積による。
【実施例1】
【0054】
NCI多様性セットのスクリーニングによる低分子Akt/PKB経路阻害剤−1(API−1)の同定
【0055】
Akt経路の異常な活性化がすべてのヒト悪性腫瘍のほとんど50%において起こり、Aktの阻害が細胞増殖停止およびアポトーシスを誘導するという事実のために、抗癌薬を見出だすために低分子Akt阻害剤を開発することは産業および学界からの関心を引いた(Cheng et al. 2005; Granville et al. 2006)。何十ものAkt阻害剤が報告されているが、それらの多くはインビボでの抗腫瘍活性を欠く。脂質ベースのAkt阻害剤(ペリホシン)は第I相および第II相の研究において報告された(Van Ummersen et al. 2004; Bailey et al. 2006)。しかしながら、どちらの研究においてもAktの修飾は査定されなかった。膵臓癌におけるペリホシンの最近の第II相研究は、第一ステージの間の許容できない有害事象を受けて終結した(Marsh et al. 2007)。それにもかかわらず、最小の副作用を備え他のキナーゼ活性の阻害を欠く強力な選択的Akt阻害剤を開発する必要がある。Aktの低分子阻害剤を同定するために、我々は、「材料および方法」中で記述されているように、AKT2により形質転換したNIH3T3細胞の増殖阻害が可能であるが、空ベクターLXSNによりトランスフェクションされたNIH3T3細胞の増殖阻害が可能でない薬剤について、NCIからの1,992化合物の化学的ライブラリー(NCI多様性セット)を評価した。三重実験は、32の化合物がAKT2により形質転換した細胞のみの増殖を阻害したことを示した。我々は、以前にそれらのうちの1つ(インビトロおよびインビボで抗腫瘍活性を有する全Akt阻害剤であり、現在第I相臨床試験中である、API−2/トリシリビンと命名された)の特性を評価した(Yang et al. 2004)。
【0056】
本研究において、我々は、Akt/PKB阻害剤−1(API−1)が生細胞におけるAktのキナーゼ活性およびリン酸化レベルを特異的に阻害することを示した。図1AはAPI−1(NSC177223)の化学構造を示し、それは化学名を有しておらず、NCI60細胞株において検査されていない(http://dtp.nci.nih.gov/)。API−1が、形質転換していない親細胞以上にAKT2形質転換細胞を選択的に阻害したので、API−1がAKT2キナーゼの阻害剤であるか、Aktの他の2つのメンバーも阻害するかどうかを最初に調べた。HAのタグを付加した野生型のAkt1、AKT2およびAKT3により、HEK293細胞をトランスフェクションした。一晩の血清飢餓に後続して、細胞はEGF刺激の前に60分間API−1により処理し、抗HA抗体により免疫沈降した。免疫沈降物はインビトロキナーゼ分析を行なった。図1Bは、API−1がAkt1、AKT2およびAKT3のインスリン誘導性キナーゼ活性を抑制したことを示す。API−1が生細胞においてAktを阻害するかどうかを次に調べる。OVCAR3細胞(リン酸化Aktを上昇したレベルで発現する)を、異なる用量のAPI−1により3時間処理した。抗リン酸化−Akt−S473抗体によるイムノブロット解析から、API−1がAktのリン酸化レベルおよびIC50を効率的に減少させるのは、およそ0.8μMであることが示された。しかしながら、全体のAktレベルは変化しなかった(図1C)。さらに、API−1が直接インビトロのAktキナーゼ活性を阻害するかどうかを調べた。構成的に活性のあるAkt組換えタンパク質を、異なる量のAPI−1、および陽性対照として化合物E(Aktを含む全キナーゼのATP競合阻害剤)を含むキナーゼバッファー中で、Akt/SGK基質ペプチド(アップステート社)と共にインキュベートした。三重実験から、API−1はAktキナーゼ活性に対する効果がないことが示され(図1D)、API−1が直接インビトロでAktを阻害せず、API−1がATPとしても、また基質競合物としても機能しないことが示唆された。
【実施例2】
【0057】
API−1はAktの上流の活性化因子を阻害しない
【0058】
Aktは、PTENにより負に調節されるPI3K依存的様式を介して、細胞外刺激および細胞内シグナル分子によって活性化される。PI3Kの活性化またはPTENの変異は、Aktキナーゼ活性の誘導を導くPDK1を活性化するだろう。したがって、AktのAPI−1阻害は、PI3KおよびPDK1などのAktの1つまたは複数の標的上流分子に由来しうる。この目的のために、API−1がPI3Kおよび/またはPDK1を阻害するかどうかを次に調べた。HEK293細胞は血清飢餓を行い、次にEGF刺激の前にAPI−1またはPI3K阻害剤(ワートマニン)により1時間処理した。PI3Kを抗p110α抗体により免疫沈降した。免疫沈降物は、基質としてPI−4−Pを使用してインビトロのPI3Kキナーゼ分析を行なった。図2A中で示されるように、EGFにより誘導されたPI3K活性はワートマニンによって阻害されたが、API−1によって阻害されなかった。API−1の効果を評価するために、リードアウト(アップステート・バイオテクノロジー社)としてSGKキナーゼを使用して、PDK1によりインビトロのPDK1キナーゼ分析を実行した。PDK1阻害剤UCN−01とは異なり(Sato et al. 2002)、API−1はインビトロのPDK1キナーゼ活性に効果がなかった(図2B)。生細胞におけるPDK1活性化に対するAPI−1の効果をさらに評価するために、OVCAR3細胞のAPI−1処理に後いて、PDK1−Ser241(自己リン酸化され、活性のために重要な残基(Casamayor et al. 1999))のリン酸化レベルを調べた。図2Dは、PDK1のリン酸化レベルがAPI−1によって阻害されなかったことを示す。
【実施例3】
【0059】
API−1は、AGCキナーゼメンバーのPKA、PKCおよびSGK、ならびに他のシグナリング分子のERK、JNK、p38およびSTAT3以上にAktに高度に選択的である。
【0060】
Aktは、AGC(PKA/PKG/PKC)キナーゼファミリーに属し、このファミリーは、PKA、PKC、血清およびグルココルチコイド誘導性キナーゼ(SGK)、p90リボソームS6キナーゼ、p70S6K、マイトジェンおよびストレス活性化プロテインキナーゼ、ならびにPKC関連キナーゼもまた含む。AGCキナーゼファミリーの中で、PKA、PKCおよびSGKのタンパク質構造は、他のメンバーよりもAktキナーゼに非常に近い。したがって、次にこれらの3つのキナーゼの酵素活性に対するAPI−1の効果を検討した。インビトロのPKAキナーゼ分析およびSGKキナーゼ分析は、キナーゼバッファー中のケンプチドまたはAkt/SGK基質ペプチドおよび[γ−32P]ATPの追加によるキナーゼ分析の開始の前の30分間、組換えPKAタンパク質または組換えSGKタンパク質との、示された用量のAPI−1または特異的阻害剤の前インキュベーションによって実行された。インビトロキナーゼ分析は、PKAおよびSGKのキナーゼ活性がPKAIおよび化合物Eによってそれぞれ阻害されるが、API−1にはそれらの活性に対する効果ないことを示した(図2Cおよび2E)。生細胞におけるPKAおよびPKCαの活性化に対するAPI−1の効果をさらに評価するために、OVCAR3細胞は示された用量のAPI−1またはPKAおよびPKCの特異的阻害剤により処理され、PKAおよびPKCのリン酸化レベルがAPI−1によって阻害されないことがイムノブロット解析により示された(図2D)。さらに、HEK293細胞をHAのタグを付加したSGKによりトランスフェクションした。インビトロキナーゼ分析から、EGFに誘導されたSGKキナーゼ活性はワートマニンによって減少されるが、API−1によって減少されないことが示された(図2F)。
【0061】
API−1が他の腫瘍形成性の生存経路に効果があるかどうか決定するために、OVCAR3細胞をAPI−1(10aM)により異なる時間処理し、市販で入手可能な抗リン酸化抗体によりイムノブロットした。API−1処理後には、Stat3、JNK、p38およびErk1/2のリン酸化レベルの検出可能な変化は観察されなかった(図2G)。これらのデーターは、API−1がAkt経路を特異的に阻害できることを示す。
【実施例4】
【0062】
API−1は構成的に活性のあるAktおよびその下流の標的を阻害する。
【0063】
API−1はAktをインビトロで直接阻害することができなかったが、PI3KおよびPDK1に対する影響なしに生細胞におけるAktのキナーゼ活性およびリン酸化を停止したので、この化合物がATP結合部位ではなくAktタンパク質と相互作用して、PDK1およびPDK2によるThr308およびSer473のリン酸化を防止することが推測された。これが真であるならば、構成的に活性のあるAktの活性化がまだThr308およびSer473のリン酸化を必要とするので、API−1は構成的に活性のあるAktの活性化もまた阻害すべきである(Sun et al. 2001)。この仮説を検証するために、HEK293細胞を、HAのタグを付加したMyr−Akt1、−Akt2および−Akt1E40Kによりトランスフェクションした。一晩の血清飢餓に後続して、細胞をAPI−1ありまたはAPIなしで処理した。Myr−Akt1、Myr−Akt2およびAkt1−E40Kを、抗HA抗体により免疫沈降した。免疫沈降物に、インビトロキナーゼ分析および抗リン酸化−Akt−T308抗体によるイムノブロット解析を行なった。図3Aは、Myr−Akt1、Myr−Akt2およびAkt1−E40Kのリン酸化レベルだけでなく、インビトロのAktキナーゼ活性もAPI−1によって阻害されたことを示し、API−1がAkt分子に結合して細胞内での活性化を阻害するという概念を支援する。
【0064】
Aktは多数のタンパク質のリン酸化を介してその細胞機能を発揮する(Datta et al. 1999)。次にAPI−1がAktの下流の標的を阻害するかどうかを調べた。GSK3βおよびmTORは主要なAkt標的のうちの2つであるので、GSKβおよびS6(p70S6Kの基質)のリン酸化レベルでAPI−1の影響を評価した。API−1によるOVCAR3の処理に続くイムノブロット解析から、API−1がそれらのリン酸化を非常に阻害することが明らかになった(図3B)。
【実施例5】
【0065】
API−1は細胞増殖を抑制し、Aktを過剰発現または活性化するヒト癌細胞株においてアポトーシスを誘導する
【0066】
Aktは主要な増殖促進および生存促進の経路である。上昇したAktキナーゼを有する癌細胞は化学療法薬に誘導された細胞増殖停止および細胞死に対してより高い耐性を示すが、Aktのノックダウンは異なるアポトーシス促進刺激によって誘導されるアポトーシスに感受性を持つ(Solit et al. 2003; Xu et al. 2003; Jetzt et al. 2003)。API−1が選択的にAkt経路を阻害する能力は、Aktの異常な発現/活性化を有するそれらの腫瘍細胞において、優先的に増殖を阻害することおよび/またはアポトーシスを誘導することを示唆する。これを検査するために、構成的に活性のあるAktを発現する細胞(Aktの過剰発現(OVCAR3、OVCAR8、MCF7およびPANC1)またはPTEN遺伝子の変異(MDA−MB−468PC−3およびLNCaP)によって引き起こされた)、および行なってない細胞(OVCAR5、MDA−MB435s、DU−145およびCOLO357)の処理にAPI−1を使用した。イムノブロット解析から、上昇したAktを発現する細胞においてAPI−1によりAktのリン酸化レベルが有意に阻害されるが、リン酸化Aktは低レベルのAktを示す細胞株においてもまたAPI−1により減少されることが示された(図4Aおよびデーター不掲載)。しかしながら、API−1はPARP切断を誘導し、低レベルのAktを有する細胞と比較して、Aktを過剰発現/活性化する細胞において、はるかに高い程度で細胞増殖を阻害する(図4A、4Bおよび4C)。API−1処理は、Aktを過剰発現/活性化する細胞株のOVCAR3、OVCA8、MDA−MB−468およびMCF7においておよそ50〜70%で細胞増殖を阻害したが、OVCAR5およびMDA−MB435s細胞において約10〜30%のみ阻害した(図4Bおよび4C)。さらに、API−1は、OVACAR3およびMDA−MB−468においてそれぞれ9倍および4.6倍でアポトーシスを誘導するが、API−1−処理したOVCAR5およびMDA−MB435s細胞においてはるかに少ないアポトーシスが観察された(図4D〜4G)。したがって、API−1は細胞増殖を阻害し、異常なAktを発現する細胞においてアポトーシスを優先的に誘導する。
【実施例6】
【0067】
API−1は、Aktを過剰発現するヌードマウスにおいて腫瘍の増殖を阻害する
【0068】
Aktの異常な活性化および過剰発現がヒトの卵巣癌および膵臓癌で頻繁に検出されること(Cheng et al. 1992)、およびAktのアンチセンスが癌細胞の増殖および浸潤を有意に阻害すること(Cheng et al. 1996)は、以前に示されている。さらに、PI3K、HSP70、Srcおよびファルネシルトランスフェラーゼの阻害剤によるAkt経路の阻害は、細胞増殖の停止およびアポトーシスの誘導をもたらした(Solit et al. 2003; Xu et al. 2003)。API−1は、上昇したAktレベルを有する癌細胞においてAktシグナリングを阻害しアポトーシスおよび細胞増殖停止を誘導するので(図4A〜4G)、ヌードマウスにおいて、上昇したAktレベルを有する腫瘍の増殖は、低レベルのAktを有する腫瘍よりも、API−1に対してより感受性であるべきだと推論された。この目的のために、Aktを過剰発現する細胞(OVCAR3およびPANC−1)をマウスの左脇腹に皮下移植し、低レベルのAktを発現する細胞株(OVCAR5およびCOLO357)を右脇腹に皮下移植した。腫瘍が約100〜150mm3の平均サイズに達した時に、動物を無作為化し、媒質またはAPI−1(10mg/kg/日)のいずれかにより腹腔内処理した。図5A〜5Jで示されるように、媒質対照により処理したOVCAR3およびPANC1の腫瘍は増殖し続けた。API−1は、OVCAR3およびPANC1腫瘍増殖をそれぞれ70%および50%阻害した(図5C〜5Fおよび5G〜5J)。これとは対照的に、API−1は、ヌードマウスにおけるOVCAR5およびCOLO357の細胞の増殖に対して効果がほとんどなかった(図5A〜5J)。API−1は、用量10mg/kg/日で、マウスの血糖値、体重、活動性および食物摂取量に対して影響はなかった(データー不掲載)。処理された腫瘍サンプルにおいて、Aktのリン酸化レベルはAPI−1によって約70%減少されたが、全Akt含有量の変化はなかった(図5K〜5L)。総合すれば、これらの結果は、API−1が上昇したAktレベルを有する腫瘍の増殖を選択的に阻害することを示す。
【0069】
本明細書において記述された実施例および実施形態が説明の目的のみのためのものあり、それらに照らして様々な修飾または変更が当業者に対して示唆され、本出願の趣旨および範囲、および添付された請求項の範囲内に含まれるべきであることが理解される。さらに、本明細書において開示された任意の発明またはその実施形態の任意の要素または限定は、任意のおよび/もしくは他のすべての要素もしくは限定(個別にまたは任意の組合せで)、または本明細書において開示された他の発明またはその実施形態と組み合わせることができ、すべてのかかる組合せは本発明の範囲でそれらに限定されずに検討される。
【0070】
参照文献
【発明の詳細な説明】
【0001】
(関連出願の相互参照)
本出願は、2007年7月12日に出願された米国仮出願第60/949,365号の利益を主張し、任意の図、表、核酸配列、アミノ酸配列および図面を含むその全体は参照することにより本明細書に組み入れられる。
政府支援
【0002】
本出願の主題は、助成金番号R01CA107078、DAMD17−02−1−0671およびW81XWH−05−1−0021の下で、国立衛生研究所−国立癌研究所およびARMY/MRMCからの研究助成金によって支援された。従って、米国政府は本発明において一定の権利を有する。
【背景技術】
【0003】
プロテインキナーゼBとも呼ばれるAktは、セリン/スレオニンキナーゼのサブファミリーを示す。Aktは、v−akt癌遺伝子産物の細胞ホモログとして最初に記載され(Bellacosa et al. 1991)、それは3つのメンバーのAkt1/PKBα、Akt2/PKBβおよびAkt3/PKBγを有する(Cheng et al. 1992; Jones et al. 1991a; Jones et al. 1991b)。Aktの活性化は、膜輸送を仲介するプレクストリン相同性(PH)ドメインの完全性、ならびに活性化ループ中のThr308、およびSer473のリン酸化に依存する(Konishi et al. 1995)。PI3 Kによって生産されたフォスフォイノシチド(PtdIns−3,4−P2およびPtdIns−3,4,5−P3)は、AktのPHドメインへ直接結合し、分子中のコンフォメーション変化を引き起こし、それはAktの活性化ループをThr308でPDK1によってリン酸化することができる(Datta et al. 1999)。Aktの完全活性化もまた、C末端の疎水性モチーフ内のSer473のリン酸化に関連する(Datta et al. 1999)。Thr308のリン酸化におけるPDK1の役割は十分に確立されているが、Ser473のリン酸化の機構は論争の的になっている。この修飾に関与する多数の候補酵素が提案され、それらはインテグリン結合キナーゼ(Persad et al. 2001)、キナーゼPRK2の複合体中にある場合のPDK1(Wick et al. 2000)、自己リン酸化を介してAktそれ自体(Toker et al. 2000)、DNA依存性キナーゼ(Feng et al. 2004)、およびリクター(rictor)−mTOR複合体(Sarbassoy et al. 2005)を含む。Aktの活性は、ヒト悪性腫瘍中で頻繁に変異している腫瘍抑制因子PTENによって負に調節される(Vazquez et al. 2000)。PTENは、PtdIns−3,4,5−P3をPtdIns−4,5−P2に変換することによってPtdIns−3,4,5−P3の細胞内濃度を低下させ、その結果としてPI3K/Akt経路を阻害する、二重特異性タンパク質および脂質ホスファターゼをコードする(Stambolic et al. 1998)。
【0004】
Aktは多数の分子をリン酸化および/またはそれらと相互作用して、細胞の増殖、生存、移動および分化における役割を含む正常な細胞機能を発揮させる(Cheng et al. 2001)。多くの証拠から、Aktが腫瘍発生および進行において重大な役割を持つことが実証される。さらに、Akt経路の異常な過活性化はすべてのヒト腫瘍の最大50%で検出され(Sun et al. 2001; Cheng et al. 1997)、密接に化学耐性に関連する(West et al. 2002)。したがって、Aktは抗癌薬物開発のための魅力的な標的であった(West et al. 2002)。
【0005】
過去数年において、組み合わせ化学、ハイスループットおよび仮想スクリーニング、ならびに従来の医薬品化学を介して、Akt経路の何十もの阻害剤が同定された。Aktの脂質ベースの阻害剤は第1に開発されたものであり、ペリホシン(Kondapaka et al. 2003)、PX−316(Meuillet et al. 2004)およびホスファチジルイノシトールエーテル脂質類似体(Castillo et al. 2004)を含み、AktのPHドメインと相互作用するようにデザインされた。さらに、いくつかのAktアンタゴニストは、化学ライブラリーのハイスループットスクリーニングおよび合理的設計を使用して同定された。これらの阻害剤は、9−メトキシ−2−メチルエリプチシニウムアセタート(Jin et al. 2004)、インダゾール−ピリジンA−443654(Luo et al. 2005)、アイソフォーム特異的アロステリックキナーゼ阻害剤(Lindsley et al. 2005)、およびトリシリビン/TCNとも呼ばれるAkt/PKBシグナリング阻害剤−2(API−2)(Yang et al. 2004)を含む。API−2/TCNは、以前に行われた第I相および第II相の試験において抗腫瘍活性を示す三環式ヌクレオシドであるが、肝毒性、高血糖、血小板減少および高トリグリセリド血症を含む複数の毒性によりさらなる開発は妨げられた(Feun et al. 1993; Hoffman et al. 1996)。我々は、NCI多様性セットのスクリーニングによって、API−2がAktキナーゼ活性を阻害し、高Akt活性を示すヒト癌細胞の異種移植片のアポトーシスを刺激することを以前に示した(Yang et al. 2004)。この結果からこの薬物の研究に新たな関心が持たれ、より低用量でAktを阻害し、以前の関連した副作用なしに腫瘍細胞アポトーシスを誘導しうる可能性が提出される(Yang et al. 2004; Cheng et al. 2005)。
【発明の概要】
【0006】
本発明は、Akt/PKB経路の阻害のための化合物、組成物および方法に関する。1つの実施形態において、本発明の化合物は、Aktタンパク質のキナーゼ活性および/またはリン酸化レベルを阻害する。本発明の化合物は、式Iで示される一般的な構造を有する。特異的実施形態において、本発明の化合物(本明細書においてAPI−1と呼ばれる)は、構造:
【化1】
を有する。
【0007】
API−1などの本発明の化合物は、異常なAktを有するヒト腫瘍細胞におけるAktシグナリングを阻害し、細胞増殖の阻害およびアポトーシスの誘導を導く。異種移植ヌードマウスモデルにおいて、本発明の化合物は、過活性化されたAktを有する細胞における腫瘍増殖をかなり阻害するが、低レベルのAktを有する腫瘍においては阻害しない。
【0008】
本発明は、式Iの化合物の有効量と細胞を接触させることを含む、Aktタンパク質の発現が上昇されるかまたは構成的に活性のある癌細胞または他の細胞を阻害または殺傷する方法にも関する。
【0009】
本発明は、人または動物に対して式Iの化合物の有効量を投与することを含む、人または動物中の癌または腫瘍を治療する方法にも関する。
【0010】
セリン/スレオニンキナーゼAkt/PKB経路は、ヒト癌において頻繁に過活性化され、細胞外および細胞内の腫瘍形成シグナルの変換のための基本的な節点として機能し、したがってそれは分子治療のための標的を示す。国立癌研究所の多様性セットのスクリーニングによって、低分子のAkt経路阻害剤のAPI(Akt/PKBシグナリング阻害剤)−1が同定された。API−1は、Aktファミリーの3つのメンバーのキナーゼ活性およびリン酸化レベルを阻害する。しかしながら、それは上流活性化因子(PI3KおよびPDK1)の活性には効果がなかった。さらに、構成的に活性のあるAktのキナーゼ活性およびリン酸化レベルは、細胞培養において主としてAPI−1によって阻害されるが、それはAktキナーゼ活性にはインビトロで効果がなかった。API−1は、Aktについて高度に選択的であり、PKC、SGK、PKA、STAT3、Erk−1/2、またはJNKの活性化を阻害しない。API−1によるAktの阻害は、構成的に活性化されたAktを保有するヒト癌細胞において、細胞増殖停止およびアポトーシスの誘導をもたらした。重要なことには、API−1は、Aktが上昇したヒト癌細胞のヌードマウスにおいて腫瘍増殖を選択的に阻害したが、Aktが上昇しないこれらの癌細胞の増殖は阻害しない。これらのデーターは、API−1がインビトロおよびインビボで抗腫瘍活性を有するAkt経路阻害剤であり、過活性化Aktを発現する癌患者のための可能性のある抗癌剤でありえることを示唆する。
【0011】
Aktは、癌細胞の生存、増殖および腫瘍進行を調節する主要な経路である。上昇したレベルのAktキナーゼは、Bcr−Abl(グリーベック)、Her2/Neu(ハークプチン(Hercptin))、およびmTOR(ラパマイシン)の細胞毒性化学療法薬および低分子阻害剤を含む様々な癌治療法に対する耐性に寄与することがよく報告されていた。Aktのブロックは、過活性化Aktを有する癌細胞において腫瘍増殖を阻害し、化学療法および他の標的化された治療に対して癌細胞をより感受性にする。他の抗腫瘍薬物とのAPI−1の組合せはより強力な抗腫瘍効果を提供する。
【図面の簡単な説明】
【0012】
【図1A】NCI多様性セットからのAkt阻害剤の候補としてのAPI−1の同定の図示である。API−1の化学構造を示す。
【図1B】NCI多様性セットからのAktインヒビターの候補としてのAPI−1の同定の図示である。API−1がAktの3つのメンバーを阻害することを示す図示である。HEK293細胞をHA−Akt1、HA−AKT2およびHA−AKT3によりトランスフェクションし、EGF刺激前にAPI−1(10μM)により処理し、細胞を溶解し、抗HA抗体により免疫沈降した。免疫沈降物はインビトロキナーゼ分析を行なった(上部)。下部パネルは、トランスフェクションしたAkt1、AKT2およびAKT3の発現を示す、抗HA抗体により検出したウエスタンブロットである。
【図1C】NCI多様性セットからのAktインヒビターの候補としてのAPI−1の同定の図示である。API−1が、OVCAR3細胞(それらは過活性化Aktを発現する)においてAktのリン酸化レベルを阻害することを示す図示である。細胞を示された濃度でAPI−1により2時間処理し、抗リン酸化−Akt−S473抗体によるイムノブロット解析を行なった(上部パネル)。下部パネルは全Aktの発現を示す。
【図1D】NCI多様性セットからのAktインヒビターの候補としてのAPI−1の同定の図示である。API−1がインビトロでAktを阻害しないことを示した図示である。示された量のAPI−1を含むキナーゼバッファー中の構成的に活性のあるAkt組換えタンパク質のインビトロキナーゼ分析。化合物E(複数のキナーゼ阻害剤のATP競合物)を陽性対照として使用した。実験は3回反復した。
【図2A】API−1が、PI3K、PDK1、およびAGCキナーゼファミリーの非常によく関連したメンバーを阻害しないことを示す図示である。インビトロPI3Kキナーゼ分析を示す図示である。HEK293細胞は血清飢餓を行い、EGF刺激前に30分間API−1(10μM)またはワートマニン(1nM)により処理した。細胞を溶解し、抗p110α抗体により免疫沈降した。免疫沈降物は、基質としてPI−4−Pを使用する、インビトロキナーゼ分析を行なった。
【図2B】API−1が、PI3K、PDK1、およびAGCキナーゼファミリーの非常によく関連したメンバーを阻害しないことを示す図示である。PDK1活性化に対するAPI−1の効果を示す図示である。インビトロキナーゼ分析は、示された化合物の存在下で製造者説明書に従って、PDK1キナーゼキット(アップステート・バイオテクノロジー社 (Upstate Biotechnology Inc) )により実行された。
【図2C】API−1が、PI3K、PDK1、およびAGCキナーゼファミリーの非常によく関連したメンバーを阻害しないことを示す図示である。インビトロのPKAキナーゼ分析を示す図示である。組換えPKAを、示された阻害剤(API−1またはPKAI)および基質ケンプチドを含むADBバッファー(アップステート・バイオテクノロジー社)中でインキュベートした。キナーゼ活性を定量した。
【図2D】API−1が、PI3K、PDK1、およびAGCキナーゼファミリーの非常によく関連したメンバーを阻害しないことを示す図示である。生細胞における、PKA、PKCおよびPDKのキナーゼ活性に対するAPI−1の効果を示す図示である。OVCAR3細胞をAPI−1の示された濃度により1時間処理した。細胞を溶解し、示された抗体によりイムノブロットした。
【図2E】API−1が、PI3K、PDK1、およびAGCキナーゼファミリーの非常によく関連したメンバーを阻害しないことを示す図示である。インビトロSGKキナーゼ分析を示す図示である。組換えSGKタンパク質をAPI−1または化合物Eと共にインキュベートした。キナーゼ分析は、SGK基質ペプチドおよび[γ−32P]ATPの追加によって開始された。キナーゼ活性を定量した。
【図2F】API−1が、PI3K、PDK1、およびAGCキナーゼファミリーの非常によく関連したメンバーを阻害しないことを示す図示である。HEK293細胞をHA−SGKによりトランスフェクションし、EGF刺激前にAPI−1またはワートマニンにより処理した場合の結果を示す図示である。インビトロキナーゼは、基質としてヒストン−H2Bを使用して、HA−SGK免疫沈降物により実行された。
【図2G】API−1が、PI3K、PDK1、およびAGCキナーゼファミリーの非常によく関連したメンバーを阻害しないことを示す図示である。API−1が、Erk、p38、JNKおよびStat3のリン酸化を阻害しないことを示す図示である。OVCAR3細胞をAPI−1により3時間処理し、示された抗体によりイムノブロットした。
【図3A】API−1が、構成的に活性のあるAktおよびその下流の標的を阻害することを実証する図示である。API−1が構成的に活性のあるAktを阻害することを実証する図示である。HEK293細胞を、示されたHA−myr−Akt1、HA−Akt1−E40KおよびHA−myr−Akt2によりトランスフェクションした。API−1による1時間の処理に後続して、細胞を溶解して、抗HA抗体により免疫沈降させた。免疫沈降物に、インビトロキナーゼ分析(上部パネル)、および示された抗体によるイムノブロット(中央および下部のパネル)を行なった。
【図3B】API−1が、構成的に活性のあるAktおよびその下流の標的を阻害することを実証する図示である。API−1がAktの下流の標的のリン酸化を阻害することを実証する図示である。OVCAR3細胞をAPI−1(10μM)により示された時間処理し、示された抗体によりイムノブロットした。API−1は、Aktおよびその下流の標的(GSK3βおよびS6タンパク質)のリン酸化レベルを有意に低下させる。
【図3C】API−1が、構成的に活性のあるAktおよびその下流の標的を阻害することを実証する図示である。AKT−SI1は、Aktの下流の標的のリン酸化を阻害した。
【図3D】API−1が、構成的に活性のあるAktおよびその下流の標的を阻害することを実証する図示である。AKT−SI1は、mTORC1およびmTORC2複合体を妨害しなかった。
【図4A】API−1が、上昇したAktのヒト癌細胞においてAkt活性および細胞増殖を阻害し、アポトーシスを誘導することを示す図示である。API−1による処理に後続する結果を示すウエスタンブロットであり、Aktのリン酸化レベルおよびPARP切断は、示されたヒト癌細胞株において抗リン酸化−Akt−T308抗体および切断PARP抗体により検出された(上部および中央のパネル)。ブロットは抗アクチン抗体により再プロービングした(下部パネル)。
【図4B】API−1が、上昇したAktのヒト癌細胞においてAkt活性および細胞増殖を阻害し、アポトーシスを誘導することを示す図示である。示された細胞株を異なる用量のAPI−1により24時間処理し、次にMTT分析により解析した細胞増殖分析である。
【図4C】API−1が、上昇したAktのヒト癌細胞においてAkt活性および細胞増殖を阻害し、アポトーシスを誘導することを示す図示である。示された細胞株を異なる用量のAPI−1により24時間処理し、次にMTT分析により解析した細胞増殖分析である。
【図4D】API−1が、上昇したAktのヒト癌細胞においてAkt活性および細胞増殖を阻害し、アポトーシスを誘導することを示す図示である。細胞をAPI−1により処理し、アネキシンVおよびPIにより染色し、ファックスキャン(FACScan)によって解析したアポトーシス解析を示す。
【図4E】API−1が、上昇したAktのヒト癌細胞においてAkt活性および細胞増殖を阻害し、アポトーシスを誘導することを示す図示である。細胞をAPI−1により処理し、アネキシンVおよびPIにより染色し、ファックスキャンによって解析したアポトーシス解析を示す。
【図4F】API−1が、上昇したAktのヒト癌細胞においてAkt活性および細胞増殖を阻害し、アポトーシスを誘導することを示す図示である。細胞をAPI−1により処理し、アネキシンVおよびPIにより染色し、ファックスキャンによって解析したアポトーシス解析を示す。
【図4G】API−1が、上昇したAktのヒト癌細胞においてAkt活性および細胞増殖を阻害し、アポトーシスを誘導することを示す図示である。細胞をAPI−1により処理し、アネキシンVおよびPIにより染色し、ファックスキャンによって解析したアポトーシス解析を示す。
【図5A】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。対照群の写真(図5A)およびAPI−1−処理した群の写真(図5B)および関連したグラフ(図5C〜5F)である。右側に低レベルのAkt細胞および左側に上昇したレベルのAkt細胞で、腫瘍細胞をヌードマウスに皮下注射した。腫瘍が約100〜150mm3の平均サイズに達した時に、「材料および方法」において記述されているように、媒質または10mg/kg/日のAPI−1のいずれかにより動物を処理した。上昇したレベルのAktを発現するPANC1/OVCAR3(それぞれ図5Eおよび5C)、および低レベルのAktを示すCOLO357/OVCAR5(それぞれ図5Fおよび5D)を有するマウスの表示であり、異種移植はAPI−1または媒質(A)により処理された。パネルBは、10マウス/群による腫瘍成長曲線を示す。
【図5B】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。対照群の写真(図5A)およびAPI−1−処理した群の写真(図5B)および関連したグラフ(図5C〜5F)である。右側に低レベルのAkt細胞および左側に上昇したレベルのAkt細胞で、腫瘍細胞をヌードマウスに皮下注射した。腫瘍が約100〜150mm3の平均サイズに達した時に、「材料および方法」において記述されているように、媒質または10mg/kg/日のAPI−1のいずれかにより動物を処理した。上昇したレベルのAktを発現するPANC1/OVCAR3(それぞれ図5Eおよび5C)、および低レベルのAktを示すCOLO357/OVCAR5(それぞれ図5Fおよび5D)を有するマウスの表示であり、異種移植はAPI−1または媒質(A)により処理された。パネルBは、10マウス/群による腫瘍成長曲線を示す。
【図5C】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。対照群の写真(図5A)およびAPI−1−処理した群の写真(図5B)および関連したグラフ(図5C〜5F)である。右側に低レベルのAkt細胞および左側に上昇したレベルのAkt細胞で、腫瘍細胞をヌードマウスに皮下注射した。腫瘍が約100〜150mm3の平均サイズに達した時に、「材料および方法」において記述されているように、媒質または10mg/kg/日のAPI−1のいずれかにより動物を処理した。上昇したレベルのAktを発現するPANC1/OVCAR3(それぞれ図5Eおよび5C)、および低レベルのAktを示すCOLO357/OVCAR5(それぞれ図5Fおよび5D)を有するマウスの表示であり、異種移植はAPI−1または媒質(A)により処理された。パネルBは、10マウス/群による腫瘍成長曲線を示す。
【図5D】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。対照群の写真(図5A)およびAPI−1−処理した群の写真(図5B)および関連したグラフ(図5C〜5F)である。右側に低レベルのAkt細胞および左側に上昇したレベルのAkt細胞で、腫瘍細胞をヌードマウスに皮下注射した。腫瘍が約100〜150mm3の平均サイズに達した時に、「材料および方法」において記述されているように、媒質または10mg/kg/日のAPI−1のいずれかにより動物を処理した。上昇したレベルのAktを発現するPANC1/OVCAR3(それぞれ図5Eおよび5C)、および低レベルのAktを示すCOLO357/OVCAR5(それぞれ図5Fおよび5D)を有するマウスの表示であり、異種移植はAPI−1または媒質(A)により処理された。パネルBは、10マウス/群による腫瘍成長曲線を示す。
【図5E】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。対照群の写真(図5A)およびAPI−1−処理した群の写真(図5B)および関連したグラフ(図5C〜5F)である。右側に低レベルのAkt細胞および左側に上昇したレベルのAkt細胞で、腫瘍細胞をヌードマウスに皮下注射した。腫瘍が約100〜150mm3の平均サイズに達した時に、「材料および方法」において記述されているように、媒質または10mg/kg/日のAPI−1のいずれかにより動物を処理した。上昇したレベルのAktを発現するPANC1/OVCAR3(それぞれ図5Eおよび5C)、および低レベルのAktを示すCOLO357/OVCAR5(それぞれ図5Fおよび5D)を有するマウスの表示であり、異種移植はAPI−1または媒質(A)により処理された。パネルBは、10マウス/群による腫瘍成長曲線を示す。
【図5F】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。対照群の写真(図5A)およびAPI−1−処理した群の写真(図5B)および関連したグラフ(図5C〜5F)である。右側に低レベルのAkt細胞および左側に上昇したレベルのAkt細胞で、腫瘍細胞をヌードマウスに皮下注射した。腫瘍が約100〜150mm3の平均サイズに達した時に、「材料および方法」において記述されているように、媒質または10mg/kg/日のAPI−1のいずれかにより動物を処理した。上昇したレベルのAktを発現するPANC1/OVCAR3(それぞれ図5Eおよび5C)、および低レベルのAktを示すCOLO357/OVCAR5(それぞれ図5Fおよび5D)を有するマウスの表示であり、異種移植はAPI−1または媒質(A)により処理された。パネルBは、10マウス/群による腫瘍成長曲線を示す。
【図5G】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。実験の終了時の腫瘍サイズ(左側)および重量(右側)の例を示す。DMSO対照と比較して、API−1は、PANC1およびOVCAR3の異種移植の腫瘍重量を有意に減少させた(*P=0.02)。
【図5H】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。実験の終了時の腫瘍サイズ(左側)および重量(右側)の例を示す。DMSO対照と比較して、API−1は、PANC1およびOVCAR3の異種移植の腫瘍重量を有意に減少させた(*P=0.02)。
【図5I】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。実験の終了時の腫瘍サイズ(左側)および重量(右側)の例を示す。DMSO対照と比較して、API−1は、PANC1およびOVCAR3の異種移植の腫瘍重量を有意に減少させた(*P=0.02)。
【図5J】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。実験の終了時の腫瘍サイズ(左側)および重量(右側)の例を示す。DMSO対照と比較して、API−1は、PANC1およびOVCAR3の異種移植の腫瘍重量を有意に減少させた(*P=0.02)。
【図5K】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。API−1がインビボでAktリン酸化を阻害することを実証する。API−1処理された腫瘍試料および無処理の腫瘍試料を溶解し、示された抗体によりイムノブロットした。
【図5L】マウス異種移植において、上昇したAktを有する癌細胞株中でAPI−1が抗腫瘍活性を示すことを実証する図である。API−1がインビボでAktリン酸化を阻害することを実証する。API−1処理された腫瘍試料および無処理の腫瘍試料を溶解し、示された抗体によりイムノブロットした。
【図6A】AKT−SI1がIGF−1に誘導されるAktの細胞膜移行を阻害することを示した図である。HeLa細胞をカバーグラス上でMyc−AKT1によりトランスフェクションし、一晩血清飢餓を行い、次に30分間AKT−SI1あり(図6C)、またはAKT−SI1なしで(図6B)処理し、その後IGF1により15分間刺激した。固定に後続して、細胞を抗Mycモノクローナル抗体、続いてFITCコンジュゲート二次抗体により免疫染色して、サイトゾルまたは膜におけるエピトープのタグを付加したタンパク質の存在を示した(図6B)。AKT−SI1およびIGF1による処理のない細胞を対照として使用した(図6A)。
【図6B】AKT−SI1がIGF−1に誘導されるAktの細胞膜移行を阻害することを示した図である。HeLa細胞をカバーグラス上でMyc−AKT1によりトランスフェクションし、一晩血清飢餓を行い、次に30分間AKT−SI1あり(図6C)、またはAKT−SI1なしで(図6B)処理し、その後IGF1により15分間刺激した。固定に後続して、細胞を抗Mycモノクローナル抗体、続いてFITCコンジュゲート二次抗体により免疫染色して、サイトゾルまたは膜におけるエピトープのタグを付加したタンパク質の存在を示した(図6B)。AKT−SI1およびIGF1による処理のない細胞を対照として使用した(図6A)。
【図6C】AKT−SI1がIGF−1に誘導されるAktの細胞膜移行を阻害することを示した図である。HeLa細胞をカバーグラス上でMyc−AKT1によりトランスフェクションし、一晩血清飢餓を行い、次に30分間AKT−SI1あり(図6C)、またはAKT−SI1なしで(図6B)処理し、その後IGF1により15分間刺激した。固定に後続して、細胞を抗Mycモノクローナル抗体、続いてFITCコンジュゲート二次抗体により免疫染色して、サイトゾルまたは膜におけるエピトープのタグを付加したタンパク質の存在を示した(図6B)。AKT−SI1およびIGF1による処理のない細胞を対照として使用した(図6A)。
【図7A】AKT−SI1がAktキナーゼ活性、特に構成的に活性のあるAktの阻害においてAPI−2/TCNよりも強力であることを示した図である。AKT−SI1構造はAPI−2/TCNとリボース糖部分を共有しているが(図7A)、これらの2つの分子の残りの部分に化学的類似性はない。しかしながら、それらのAkt阻害能力を比較した。HEK293細胞を、野生型Myc−AKT1(図7Bおよび7C)および構成的に活性のあるMyc−AKT1−E17K(図7Dおよび7E)によりトランスフェクションした。36時間のインキュベーションに後続して、細胞に一晩飢餓血清を行なった。野生型Aktをトランスフェクションした細胞は、AKT−SI1(図7Bおよび7D)またはAPI−2/TCN(図7Cおよび7E)により30分間処理し、続いてEGFにより15分間刺激した。免疫沈降を抗Mycの抗体により実行し、免疫沈降物で基質としてヒストンH2Bを使用するインビトロキナーゼ分析を行なった(上部)。AKT−SI1およびAPI−2/TCNによるAktキナーゼ活性の阻害は、相対的活性として定量および計算した(中央)。ウエスタンブロット解析は免疫沈降したAKT1タンパク質を示す(下部パネル)。実験は3回反復した。
【図7B】AKT−SI1がAktキナーゼ活性、特に構成的に活性のあるAktの阻害においてAPI−2/TCNよりも強力であることを示した図である。AKT−SI1構造はAPI−2/TCNとリボース糖部分を共有しているが(図7A)、これらの2つの分子の残りの部分に化学的類似性はない。しかしながら、それらのAkt阻害能力を比較した。HEK293細胞を、野生型Myc−AKT1(図7Bおよび7C)および構成的に活性のあるMyc−AKT1−E17K(図7Dおよび7E)によりトランスフェクションした。36時間のインキュベーションに後続して、細胞に一晩飢餓血清を行なった。野生型Aktをトランスフェクションした細胞は、AKT−SI1(図7Bおよび7D)またはAPI−2/TCN(図7Cおよび7E)により30分間処理し、続いてEGFにより15分間刺激した。免疫沈降を抗Mycの抗体により実行し、免疫沈降物で基質としてヒストンH2Bを使用するインビトロキナーゼ分析を行なった(上部)。AKT−SI1およびAPI−2/TCNによるAktキナーゼ活性の阻害は、相対的活性として定量および計算した(中央)。ウエスタンブロット解析は免疫沈降したAKT1タンパク質を示す(下部パネル)。実験は3回反復した。
【図7C】AKT−SI1がAktキナーゼ活性、特に構成的に活性のあるAktの阻害においてAPI−2/TCNよりも強力であることを示した図である。AKT−SI1構造はAPI−2/TCNとリボース糖部分を共有しているが(図7A)、これらの2つの分子の残りの部分に化学的類似性はない。しかしながら、それらのAkt阻害能力を比較した。HEK293細胞を、野生型Myc−AKT1(図7Bおよび7C)および構成的に活性のあるMyc−AKT1−E17K(図7Dおよび7E)によりトランスフェクションした。36時間のインキュベーションに後続して、細胞に一晩飢餓血清を行なった。野生型Aktをトランスフェクションした細胞は、AKT−SI1(図7Bおよび7D)またはAPI−2/TCN(図7Cおよび7E)により30分間処理し、続いてEGFにより15分間刺激した。免疫沈降を抗Mycの抗体により実行し、免疫沈降物で基質としてヒストンH2Bを使用するインビトロキナーゼ分析を行なった(上部)。AKT−SI1およびAPI−2/TCNによるAktキナーゼ活性の阻害は、相対的活性として定量および計算した(中央)。ウエスタンブロット解析は免疫沈降したAKT1タンパク質を示す(下部パネル)。実験は3回反復した。
【図7D】AKT−SI1がAktキナーゼ活性、特に構成的に活性のあるAktの阻害においてAPI−2/TCNよりも強力であることを示した図である。AKT−SI1構造はAPI−2/TCNとリボース糖部分を共有しているが(図7A)、これらの2つの分子の残りの部分に化学的類似性はない。しかしながら、それらのAkt阻害能力を比較した。HEK293細胞を、野生型Myc−AKT1(図7Bおよび7C)および構成的に活性のあるMyc−AKT1−E17K(図7Dおよび7E)によりトランスフェクションした。36時間のインキュベーションに後続して、細胞に一晩飢餓血清を行なった。野生型Aktをトランスフェクションした細胞は、AKT−SI1(図7Bおよび7D)またはAPI−2/TCN(図7Cおよび7E)により30分間処理し、続いてEGFにより15分間刺激した。免疫沈降を抗Mycの抗体により実行し、免疫沈降物で基質としてヒストンH2Bを使用するインビトロキナーゼ分析を行なった(上部)。AKT−SI1およびAPI−2/TCNによるAktキナーゼ活性の阻害は、相対的活性として定量および計算した(中央)。ウエスタンブロット解析は免疫沈降したAKT1タンパク質を示す(下部パネル)。実験は3回反復した。
【図7E】AKT−SI1がAktキナーゼ活性、特に構成的に活性のあるAktの阻害においてAPI−2/TCNよりも強力であることを示した図である。AKT−SI1構造はAPI−2/TCNとリボース糖部分を共有しているが(図7A)、これらの2つの分子の残りの部分に化学的類似性はない。しかしながら、それらのAkt阻害能力を比較した。HEK293細胞を、野生型Myc−AKT1(図7Bおよび7C)および構成的に活性のあるMyc−AKT1−E17K(図7Dおよび7E)によりトランスフェクションした。36時間のインキュベーションに後続して、細胞に一晩飢餓血清を行なった。野生型Aktをトランスフェクションした細胞は、AKT−SI1(図7Bおよび7D)またはAPI−2/TCN(図7Cおよび7E)により30分間処理し、続いてEGFにより15分間刺激した。免疫沈降を抗Mycの抗体により実行し、免疫沈降物で基質としてヒストンH2Bを使用するインビトロキナーゼ分析を行なった(上部)。AKT−SI1およびAPI−2/TCNによるAktキナーゼ活性の阻害は、相対的活性として定量および計算した(中央)。ウエスタンブロット解析は免疫沈降したAKT1タンパク質を示す(下部パネル)。実験は3回反復した。
【図8A】AKT−SI1およびAPI−2/TCNがリン酸化Aktレベルを阻害することを示した図である。野生型Myc−AKT1および構成的に活性のあるMyc−AKT1−E17KによりトランスフェクションしたHEK293細胞を、示された試薬により処理し、抗リン酸化−Akt−S473抗体(上部)および抗Myc抗体(下部)によりイムノブロットした。
【図8B】AKT−SI1およびAPI−2/TCNがリン酸化Aktレベルを阻害することを示した図である。野生型Myc−AKT1および構成的に活性のあるMyc−AKT1−E17KによりトランスフェクションしたHEK293細胞を、示された試薬により処理し、抗リン酸化−Akt−S473抗体(上部)および抗Myc抗体(下部)によりイムノブロットした。
【発明を実施するための形態】
【0013】
本発明は、Akt/PKB経路を阻害する化合物および組成物に関する。1つの実施形態において、本発明の化合物は、Aktタンパク質のキナーゼ活性および/またはリン酸化レベルを阻害する。本発明の化合物は、式I:
【化2】
(I)
(式中、
Xは独立してO、NまたはSであり;
YはO、NまたはSであり;
R1は、独立して−H、−OH、−NH2、−NO2、ハロゲン、−OHにより任意に置換されるアルキル、または−OHにより任意に置換されるアルコキシであり;
および、R2は、−H、−OH、−NH2、−C(O)NH2、アルキル、またはアルコキシであり、それらのいずれかは、−OH、ハロゲン、アルキルまたはアルコキシにより任意に置換することができ;
R3は、−H、−OH、−NH2、−C(O)NH2、アルキルまたはアルコキシであり、それらのいずれかは、−OHまたはハロゲンまたはアルキルまたはアルコキシにより任意で置換することができる);または
その類似体もしくは薬学的に許容される塩で示される一般的な構造を有する。
【0014】
1つの実施形態において、各XはNである。例示された実施形態において、YはOである。別の例示された実施形態において、各R1は独立して−OHまたは−CH2OHである。なお別の実施形態において、R2は−OHにより任意に置換される−NH2である。さらに実施形態において、R2は−C(O)NH2である。
【0015】
具体的な実施形態において、本発明の化合物は、以下の構造(式II):
【化3】
(II)
を有する 。
【0016】
本発明の化合物は、PTEN変異などの上流レギュレーターの変化からの結果として生じる高度に活性化された野生型Aktだけでなく、myr−AKT1、Myr−AKT2およびE40K−AKT1を含む、構成的に活性のあるAkt変異体も阻害する。最近の研究は、脂質結合ポケット中のアミノ酸17でのグルタミン酸のリジン置換(E17K)をもたらす、ヒトの乳癌、結腸直腸癌および卵巣癌におけるAKT1のPHドメイン中の再発性体細胞変異を同定した(Carpten et al. 2007)。Lys17はポケットの静電的相互作用を変化させて、フォスフォイノシチドリガンドによる新しい水素結合を形成する。この変異は細胞膜への病的な局在を介してAKT1を活性化し、細胞を形質転換し、マウスにおける白血病を誘導する。さらに、E17K置換はアロステリックキナーゼ阻害剤への感受性を低下させる(Carpten et al. 2007)。E40K−AKT1変異体は細胞膜に局在できるE17Kに類似しているので(Bellacosa et al. 1998)、API−1はE17K−AKT1もまた阻害しうる。
【0017】
単離された化合物として本発明の化合物を投与することができるが、これらの化合物は、医薬組成物の一部としても投与することができる。本発明は、したがってさらに少なくとも1つの薬学的に許容される担体を伴う組成物を提供する。医薬組成物は、腸内投与、非経口投与、静脈内投与、筋肉内投与、局所投与、皮下投与などのような様々な投与経路に適合させることができる。当業者によって決定することができるように、継続的に、または明瞭なインターバルで投与することができる。
【0018】
本発明の化合物は、薬学的に有益な組成物を調製する公知の方法に従って製剤化することができる。製剤は、当業者に周知であり容易に利用可能な多数の出典中に記述されている。例えば、レミントンの製薬科学(Remington's Pharmaceutical Science)(Martin 1995)は、本発明に関連して使用できる製剤について記述する。投与に適切な製剤は、例えば、抗酸化剤、緩衝剤、静菌剤、および意図されるレシピエントの製剤を血液と等張にする溶質を含みうる水性の滅菌済み注射溶液;ならびに懸濁化剤および増粘剤を含みうる水性および非水性の滅菌済み懸濁物を含む。製剤は、単位用量またはマルチ用量の容器(例えば密封したアンプル、バイアル)中に提供され、使用前に、滅菌済み液体担体(例えば注射用の水)の条件のみを必要とする、冷凍乾燥(凍結乾燥)条件で保存することができる。即時注射溶液および懸濁物は、滅菌済みの粉末、顆粒、タブレットなどから調製することができる。特に上で言及された成分に加えて、本発明の組成物は、当該製剤のタイプを考慮して、当技術分野における従来の他の薬剤を含みうることが理解されるべきである。
【0019】
本明細書において使用されるように、アルキルは、直鎖または分岐鎖である、1〜20の炭素原子を有する飽和炭化水素基または一価不飽和炭化水素基もしくは多価不飽和炭化水素基を意味し、C1-Xアルキルは、1〜「X」の炭素原子数を含む直鎖または分岐鎖のアルキル基を意味する。例えば、C1-6アルキルは、1〜6の炭素原子を含む直鎖または分岐鎖のアルキル基を意味する。アルコキシは、アルキル基が以前に本明細書において記述されているようなものである、アルキル−O−基を意味する。
【0020】
用語「ハロゲン」は、フッ素、塩素、臭素、またはヨウ素などの周期表のハロゲンを意味する。
【0021】
用語「任意に置換される」は、任意の利用可能な1つまたは複数の位置で、1つまたは複数の有機基または無機基(例えばアルキル、アリール、ヘテロアリール、アシル、アルケニル、シクロアルキル、ヘテロシクロアルキル、シクロアルケニル、ヘテロシクロアルケニル、またはハロゲン)により任意に置換されることを意味する。
【0022】
飽和アルキル基の例は、メチル、エチル、N−プロピル、イソプロピル、N−ブチル、tert−ブチル、イソブチル、sec−ブチル、N−ペンチル、N−ヘキシル、N−ヘプチルおよびN−オクチルを含むが、これらに限定されない。不飽和アルキル基は、1つまたは複数の二重結合または三重結合を有するアルキル基である。不飽和アルキル基は、例えば、エテニル、プロペニル、ブテニル、ヘキセニル、ビニル、2−プロピニル、2−イソペンテニル、2−ブタジエニル、エチニル、1−プロピニル、3−プロピニルおよび3−ブチニルを含む。具体的には、「アルキル」は、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソ−ブチル、sec−ブチル、ペンチル、3−ペンチル、ヘキシル、ヘプチル、オクチル、ノニル、デシル、ウンデシル、ドデシル、トリデシル、テトラデシル、ペンタデシル、ビニル、1−プロペニル、2−プロペニル、1−ブテニル、2−ブテニル、3−ブテニル、1−ペンテニル、2−ペンテニル、3−ペンテニル、4−ペンテニル、1−ヘキセニル、2−ヘキセニル、3−ヘキセニル、4−ヘキセニル、5−ヘキセニル、1−ヘプテニル、2−ヘプテニル、3−ヘプテニル、4−ヘプテニル、5−ヘプテニル、1−ノネニル、2−ノネニル、3−ノネニル、4−ノネニル、5−ノネニル、6−ノネニル、7−ノネニル、8−ノネニル、1−デケニル、2−デケニル、3−デケニル、4−デケニル、5−デケニル、6−デケニル、7−デケニル、8−デケニル、9−デケニル;1−ウンデケニル、2−ウンデケニル、3−ウンデケニル、4−ウンデケニル、5−ウンデケニル、6−ウンデケニル、7−ウンデケニル、8−ウンデケニル、9−ウンデケニル、10−ウンデケニル、1−ドデケニル、2−ドデケニル、3−ドデケニル、4−ドデケニル、5−ドデケニル、6−ドデケニル、7−ドデケニル、8−ドデケニル、9−ドデケニル、10−ドデケニル、11−ドデケニル、1−トリデケニル、2−トリデケニル、3−トリデケニル、4−トリデケニル、5−トリデケニル、6−トリデケニル、7−トリデケニル、8−トリデケニル、9−トリデケニル、10−トリデケニル、11−トリデケニル、12−トリデケニル、1−テトラデケニル、2−テトラデケニル、3−テトラデケニル、4−テトラデケニル、5−テトラデケニル、6−テトラデケニル、7−テトラデケニル、8−テトラデケニル、9−テトラデケニル、10−テトラデケニル、11−テトラデケニル、12−テトラデケニル、13−テトラデケニル、1−ペンタデケニル、2−ペンタデケニル、3−ペンタデケニル、4−ペンタデケニル、5−ペンタデケニル、6−ペンタデケニル、7−ペンタデケニル、8−ペンタデケニル、9−ペンタデケニル、10−ペンタデケニル、11−ペンタデケニル、12−ペンタデケニル、13−ペンタデケニル、または14−ペンタデケニルを含むことができ;「アルコキシ」は、メトキシ、エトキシ、プロポキシ、イソプロポキシ、ブトキシ、イソ−ブトキシ、sec−ブトキシ、ペンタオキシ、3−ペンタオキシ、ヘキサオキシ、ヘプチロキシ、オクチロキシ、ノニロキシ、デシロキシ、ウンデシロキシ、ドデシロキシ、トリデシロキシ、テトラデシロキシ、またはペンタデシロキシを含むことができる。
【0023】
本発明の化合物は、当業者によって調製することができるすべて水和物および塩を含む。本発明の化合物が安定した無毒な酸性塩または塩基塩を形成するのに十分に塩基性または酸性の条件下で、塩として化合物を投与することは適切でありうる。薬学的に許容される塩の例は、生理学的な許容されるアニオンを形成する酸により形成された有機酸の付加塩(例えば、トシル酸塩、メタンスルホン酸塩、酢酸塩、クエン酸塩、マロン酸塩、酒石酸塩、コハク酸塩、安息香酸塩、アスコルビン酸塩、α−ケトグルタル酸塩およびα−グリセロリン酸塩)である。塩酸塩、硫酸塩、硝酸塩、重炭酸塩、および炭酸塩塩を含む、適切な無機塩類もまた形成することができる。
【0024】
化合物の薬学的に許容される塩は、当技術分野における周知の標準的手順を使用して、例えば、生理学的に許容されるアニオンを与える適切な酸とアミンなどの十分に塩基性の化合物を反応させることによって得ることができる。カルボン酸のアルカリ金属(例えばナトリウム、カリウムまたはリチウム)塩、またはアルカリ土類金属(例えばカルシウム)塩もまた作製することができる。
【0025】
本明細書において使用されるように、用語「類似体」は、実質的にもう一つの化合物と同種類のものであるが、例えば、側鎖基の追加、親構造の酸化または還元によって修飾されうる化合物を指す。本発明の化合物の類似体は、一般に公知の標準的反応を使用して容易に調製することができる。これらの標準的反応は、水素化反応、アルキル化反応、アセチル化反応および酸性化反応を含むが、これらに限定されない。化学的修飾は、分子中に存在するすべての官能基を保護すること、および所望の反応を行なった後にそれらを脱保護することによって、科学文献中で公知の標準的手順を使用して、当業者によって成し遂げることができる(Greene et al. 1999; Honda et al. 1997; Honda et al. 1998; Konoike et al. 1997; Honda et al. 2000;それらの各々はその全体を参照することにより本明細書に組み入れられる)。所望の生物学的活性(アポトーシスの誘導、細胞毒性、細胞静止、細胞周期停止の誘導、抗血管形成特性などのような)を示す類似体は、細胞分析または他のインビトロもしくはインビボの分析を使用して、同定または確認することができる。
【0026】
本発明の化合物は1つまたは複数の非対称的に置換された炭素原子(すなわち炭素中心)を含むことができることが理解される。本発明の化合物における1つまたは複数の不斉中心の存在は、立体異性体を生じさせることができ、各場合において、本発明は、鏡像異性体および偏左右異性体を含むそのようなすべての立体異性体、ならびにそのラセミ混合物を含む混合物に拡張するように理解されるべきである。
【0027】
本発明の化合物および組成物は、様々な非治療目的および治療目的に有用である。化合物および組成物は、動物およびヒトにおける異常な細胞増殖の減少のために使用できる。化合物のかかる抗増殖特性のために、それらはインビトロおよびインビボを含む様々な設定における望まれない細胞増殖を減少させるのに有用である。
【0028】
化合物およびそれらを含む組成物の治療適用は、現時点でまたは将来的に当業者に公知の任意の適切な治療方法および技術によって成し遂げることができる。さらに、本発明の化合物は、他の有用な化合物および組成物の調製のための出発材料または中間体としての用途を有する。
【0029】
本発明の化合物およびその組成物は、任意で不活性希釈剤などの薬学的に許容される担体と組み合わせて、望まれない細胞増殖の部位(腫瘍部位または良性の皮膚増殖など、例えば腫瘍または皮膚増殖に対して注射または局所的に適用される)または真菌感染の部位などの1つまたは複数の解剖学的な部位で局所的に投与することができる。本発明の化合物およびその組成物は、任意で不活性希釈剤など薬学的に許容される担体または経口送達のための吸収可能な食用担体と組み合わせて、全身的に(静脈内または経口的など)投与することができる。それらはハードシェルゼラチンカプセルまたはソフトシェルゼラチンカプセル中に封入できるか、タブレットに圧縮できるか、または患者の食餌の食品に直接組み入れることができる。経口の治療投与のために、活性化合物は1つまたは複数の賦形剤と組み合わせることができ、摂取可能なタブレット、バッカル錠、トローチ、カプセル、エリキシル、懸濁物、シロップ剤、ウエハー、エアゾールスプレーおよび同種のものの形態で使用できる。
【0030】
タブレット、トローチ、ピル、カプセルおよび同種のものは、以下のものもまた含むことができる。トラガカントゴム、アラビアゴム、トウモロコシデンプンまたはゼラチンなどの結合剤;リン酸カルシウムなどの賦形剤;トウモロコシデンプン、ジャガイモデンプン、アルギン酸および同種のものなどの崩壊剤;ステアリン酸マグネシウムなどの潤滑剤;およびスクロース、フルクトース、ラクトースもしくはアスパルテームなど甘味剤、またはペパーミント、ヒメコウジ油もしくはチェリーフレーバーなどの着香剤を追加できる。単位投与量形態がカプセルである場合、上述のタイプの材料に加えて、植物油またはポリエチレングリコールなどの液体担体を含むことができる。様々な他の材料は、被膜として、またはそうでなければ固体単位の投与量形態の物理的形態を修飾するように存在しうる。例えば、タブレット、ピルまたはカプセルは、ゼラチン、ワックス、セラックまたは糖および同種のものにより被覆できる。シロップまたはエリキシルは、活性化合物、甘味剤としてスクロースまたはフルクトース、防腐剤としてメチルパラベンおよびプロピルパラベン、色素、ならびにチェリーフレーバーまたはオレンジフレーバーなどの香味料を含むことができる。当然、任意の単位投与量形態の調製に使用される任意の材料は、使用する量で薬学的に許容され、実質的に非毒性であるべきである。さらに、活性化合物は徐放性の調製物および装置の中へ組み入れることができる。
【0031】
本発明の化合物および組成物(その薬学的に許容される塩または類似体を含む)は、注入または注射によって静脈内に、筋肉内に、または腹腔内に投与することができる。活性薬剤またはその塩の溶液は無毒な界面活性剤と任意で混合されて、水中で調製することができる。分散物は、グリセロール、液体ポリエチレングリコール、トリアセチンおよびその混合物、ならびに油中でも調製することができる。通常の保存および使用の条件下で、これらの調製物は、微生物の増殖を防止する防腐剤を含むことができる。
【0032】
注射または注入に適切な薬学的投与量形態は、任意でリポソーム中でカプセル化された滅菌済みの注射可能もしくは注入可能な溶液または分散物の即時調整に適合する活性成分を含む、滅菌済みの水性の溶液もしくは分散物または滅菌済みの粉末を含むことができる。最終的な投与量形態は、滅菌済みで流動的であり、製造および保存の条件下で安定しているべきである。液体担体または媒質は、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、液体ポリエチレングリコール、および同種のもの)、植物油、無毒なグリセリルエステル、および適切なそれらの混合物を含む、溶媒または液体分散媒でありえる。適切な流動性は、例えば、リポソームの形成によって、分散物の場合で必要とされる粒子サイズの維持によって、または界面活性剤の使用によって、維持することができる。任意で、微生物活動の防止は、様々な他の抗菌剤および抗真菌剤(例えば、パラベン、クロロブタノール、フェノール、ソルビン酸、チメロサール、および同種のもの)によって成し遂げることができる。多くの場合において、等張剤(例えば、糖、緩衝剤または塩化ナトリウム)を含むことは好ましいだろう。注射可能な組成物の持続的吸収は、吸収遅延剤(例えばモノステアリン酸アルミニウムおよびゼラチン)の含有によって成し遂げることができる。
【0033】
滅菌済みの注射可能な溶液は、上で列挙された様々な他の成分と共に適切な溶媒中に必要とされた量で本発明の化合物を組み入れ、必要に応じて続いて濾過滅菌することによって調製される。滅菌済みの注射可能な溶液の調製のための滅菌済み粉末の場合において、調製の好ましい方法は真空乾燥および凍結乾燥の技術であり、事前に滅菌済みの濾過溶液中に存在する任意の追加の所望成分を加えた活性成分の粉末をもたらす。
【0034】
局所投与のために、本発明の化合物は液体または固体として適用されうる。しかしながら、皮膚科学的に許容される担体(固体または液体でありうる)と組み合わせて、組成物として皮膚に局所的に投与することが一般的に望ましいだろう。本発明の化合物および組成物を被験体の皮膚に局所的に適用して、悪性腫瘍または良性腫瘍のサイズを減少させる(および完全な除去を含みうる)か、または感染部位を治療することができる。本発明の化合物は、増殖部位または感染部位に直接適用することができる。好ましくは、化合物は、軟膏、クリーム、ローション、溶液、チンキ、または同種のものなどの製剤で増殖部位または感染部位に適用される。皮膚の病変への薬理学的物質の送達のための薬物送達システムは、米国特許第5,167,649号中で記述されたようなものも使用することができる。
【0035】
有用な固体担体は、タルク、粘土、微結晶性セルロース、シリカ、アルミナ、および同種のものなどの微粉固体を含む。有用な液体担体は、水、アルコールもしくはグリコールまたは水−アルコール/グリコールブレンドを含み、化合物は任意で非毒性界面活性剤の補助によって、効果的なレベルで溶解または分散させることができる。香料および追加の抗生物質のような補助剤を追加して、規定の使用のための特性を最適化することができる。結果として生ずる液体組成物は、吸収性パッドから適用するか、救急絆および他の包帯を使用して浸透させるか、例えばポンプタイプ噴霧器またはエアゾール噴霧器を使用して罹患した領域上にスプレーすることができる。
【0036】
合成ポリマー、脂肪酸、脂肪酸塩およびエステルなどの増粘剤、脂肪アルコール、修飾セルロースまたは修飾鉱物材もまた、液体担体と共に使用して、使用者の皮膚への直接的な適用のために、塗布可能なペースト、ゲル、軟膏、石鹸、および同種のものを形成することができる。皮膚へ化合物を送達するために使用できる有用な皮膚科学的組成物の例は、米国特許第4,608,392号;米国特許第4,992,478号;米国特許第4,559,157号;および米国特許第4,820,508号で開示される。
【0037】
本発明の化合物および医薬組成物の有用な投与量は、インビトロの活性と動物モデルにおけるインビボの活性との比較によって決定することができる。マウス、および他の動物、ヒトに対する有効量の推定のための方法は、当技術分野で公知であり、例えば米国特許第4,938,949号を参照。
【0038】
本発明は、薬学的に許容される担体と組み合わせた本発明の化合物を含む医薬組成物にも関する。経口投与、局所投与または非経口投与に適合した、化合物の量を含む医薬組成物は、本発明の好ましい実施形態を構成する。本発明の文脈中で患者(特にヒト)に投与される用量は、適当な期間枠にわたって患者において治療効果を達成するのに十分であり、致死毒性がなく、許容されるレベル以上の副作用または病的状態を好ましくは引き起こさないものであるべきである。当業者は、病理学的状態の重症度およびステージに加えて、被験体の条件(健康)、被験体の体重、もしあれば併用療法の種類、処理の頻度、治療係数を含む様々な因子に、投与量が依存することを認識するだろう。
【0039】
本発明は、式Iの化合物、またはその塩もしくは類似体の有効量と細胞を接触させることを含む、Aktタンパク質の発現が上昇されるかまたは構成的に活性のある、癌細胞もしくは腫瘍細胞または他の細胞の生存または増殖を阻害するかまたは殺傷する方法にも関する。具体的な実施形態において、化合物は、式II、またはその塩もしくは類似体で示される構造を有する。1つの実施形態において、細胞はヒト細胞または他の哺乳類細胞である。本方法を使用して阻害または殺傷することができる癌細胞は、転移性の性質の細胞を含んでいる。したがって、癌細胞または腫瘍細胞の転移の阻害もまた本発明によって検討される。本方法はインビトロまたはインビボで実行することができる。
【0040】
本発明は、式Iの化合物、またはその塩もしくは類似体の有効量を人または動物に投与することを含む、人または動物における癌または腫瘍などの腫瘍学的障害を治療する方法にも関する。具体的な実施形態において、化合物は、式II、またはその塩もしくは類似体で示される式を有する。1つの実施形態において、本発明の化合物または組成物の有効量は、腫瘍学的障害を有し、その治療を必要とする患者に投与される。阻害剤は、化学療法、免疫療法および/または放射線療法の前か、それらの後か、またはそれらと併用して、投与することができる。本発明の方法は、腫瘍学的障害の治療を必要としているか、または必要としうる患者の同定を任意で含むことができる。本発明の方法を使用する治療を必要とする患者は、医学または獣医学の当業者に公知の標準的技術を必要に応じて使用して同定することができる。1つの実施形態において、患者は、霊長類(サル、チンパンジー、類人猿など)、イヌ、ネコ、ウシ、ブタもしくはウマ、または腫瘍学的障害がある他の動物などの、ヒトまたは他の哺乳類でありえる。患者への投与のための本発明の化合物の投与および製剤化のための手段は、当技術分野において公知であり、それらの実施例は本明細書において記述される。本発明の範囲内の腫瘍学的障害は、肛門、胆管、膀胱、骨、骨髄、腸管(大腸および直腸を含む)、乳房、目、胆嚢、腎臓、口、喉頭、食道、胃、精巣、頚管、頭部、首、卵巣、肺、中皮腫、神経内分泌、陰茎、皮膚、脊髄、甲状腺、膣、外陰部、子宮、肝臓、筋肉、膵臓、前立腺、血球(リンパ球および他の免疫系細胞を含む)および脳の癌および/または腫瘍を含むが、これらに限定されない。本発明による治療のために検討される具体的な癌は、癌腫、カポジ肉腫、黒色腫、中皮腫軟部組織肉腫、白血病(急性リンパ芽球性、急性骨髄性、慢性リンパ球性、慢性骨髄性、および他のもの)、およびリンパ腫(ホジキンおよび非ホジキン)、および多発性骨髄腫を含む。
【0041】
腫瘍学的障害の治療のために、本発明によって検討される化合物および組成物は、他の抗腫瘍性もしくは抗癌性の薬剤もしくは物質(例えば化学療法剤、免疫療法剤、放射線療法剤など)および/または放射線療法および/または腫瘍を除去する外科的治療の前か、それらの後か、またはそれらと組み合わせて、治療を必要とする患者に投与することができる。例えば、本発明の化合物および組成物は、タキソールまたはビンブラスチンなどの有糸分裂阻害剤、シクロホスアミドまたはイフォスファミドなどのアルキル化剤、5−フルオロウラシルまたはヒドロキシウレアなどの抗代謝剤、アドリアマイシンまたはブレオマイシンなどのDNAインターカレーター、エトポシドまたはカンプトテシンなどのトポイソメラーゼ阻害剤、アンジオスタチンなどの抗血管新生剤、タモキシフェンなどの抗エストロゲン、および/または例えばグリーベック(ノバルティスファーマ(Novartis Pharmaceuticals)社)およびハーセプチン(ジェネンテック(Genentech)社)などのそれぞれ他の抗癌薬物または抗体により、患者が、治療される予定か、または治療されているか、または治療されてきた、癌治療法において使用することができる。これらの他の物質または放射線治療は、本発明の化合物と同様の、または異なる回数で与えることができる。本発明の範囲内で検討される他の化学療法剤の例は、アルトレタミン、ブレオマイシン、ボルテゾミブ(ベルケイド)、ブスルファン、ホリナートカルシウム、カペシタビン、カルボプラチン、カルムスチン、クロラムブチル、シスプラチン、クラドリビン、クリサンタスパーゼ(crisantaspase)、シクロホスファミド、シタラビン、ダカルバジン、ダクチノマイシン、ダウノルビシン、ドセタキセル、ドキソルビシン、エピルビシン、エトポシド、フルダラビン、フルオロウラシル、ゲフィチニブ(イレッサ)、ゲムシタビン、ヒドロキシウレア、イダルビシン、イフォスファミド、イマチニブ(グリーベック)、イリノテカン、リポソームドキソルビシン、ロムスチン、メルファラン、メルカプトプリン、メトトレキサート、マイトマイシン、ミトキサントロン、オキサリプラチン、パクリタキセル、ペントスタチン、プロカルバジン、ラルチトレキセド、ストレプトゾシン、テガフール−ウラシル、テモゾロマイド、チオテパ、チオグアニン/チオグアニン(tioguanine/thioguanine)、トポテカン、トレオスルファン、ビンブラスチン、ビンクリスチン、ビンデシン、ビノレルビンを含むが、これらに限定されない。例示された実施形態において、化学療法剤はメルファランである。本発明の範囲内で検討された免疫療法剤の例は、アレムツズマブ、セツキシマブ(エルビタックス)、ゲムツズマブ、ヨウ素131トシツモマブ、リツキシマブ、トラスツザマブ(trastuzamab)(ハーセプチン)を含むが、これらに限定されない。本発明は、化学療法剤、免疫療法剤、放射線療法剤、または放射線療法の投与の前か、それらの後か、および/またはそれらと組み合わせて、本発明の化合物の有効量を投与することを含む、腫瘍学的障害を治療する方法にも関する。
【0042】
本発明は、本発明の化合物または組成物の有効量と細胞を接触させることによって、細胞中のAkt/PKB経路を阻害する方法にも関する。1つの実施形態において、化合物は、Akt1、AKT2および/またはAKT3タンパク質に結合し、それらの活性を阻害する。具体的な実施形態において、化合物は、式II、またはその塩もしくは類似体で示される構造を有する。1つの実施形態において、細胞はヒト細胞または哺乳類細胞であり、異常な増殖、生存、移動または分化を示す癌細胞もしくは腫瘍細胞または他の細胞になりえる。1つの実施形態において、細胞は、Akt1、AKT2、および/またはAKT3などのAktタンパク質を、構成的に発現するか、または上昇レベルもしくは異常レベルで発現する。
【0043】
本発明は、本発明の化合物または組成物の治療的有効量が人または動物に投与される、細胞中のAktタンパク質の構成的発現、異常な発現、または上昇した発現に関連する障害を有する人または動物を治療する方法にも関する。障害は、例えば、異常な細胞増殖、細胞生存、細胞移動、および/または細胞分化によって特徴づけられるものでありえる。1つの実施形態において、化合物は、Akt1、AKT2、および/またはAKT3タンパク質に結合し、それらの活性を阻害する。具体的な実施形態において、化合物は式II、またはその塩もしくは類似体で示される構造を有する。
【0044】
治療される障害または疾患の条件に依存して、適切な用量は標的細胞の増加または増殖を減少させる量でありえる。癌の情況において、適切な用量は、悪性腫瘍などの癌組織において、所望反応を達成することが公知である活性薬剤の濃度をもたらすものである。好ましい投与量は、管理し難い副作用なしに癌細胞増殖の最大の阻害をもたらす量である。当業者によって決定することができるように、化合物の投与は継続的、または異なるインターバルでありえる。
【0045】
いくつかの実施形態において、所望療法の治療のためのかかる投薬量の投与を提供するために、本発明の医薬組成物は、担体または希釈剤を含む全組成物の重量に基づいて、1つまたは複数の化合物の総重量で、約0.1%〜45%、特に1〜15%の間で含まれうる。例示的に、投与される活性のある成分の投与量レベルは、静脈内、0.01〜約20mg/kg;腹腔内、0.01〜約100mg/kg;皮下、0.01〜約100mg/kg;筋肉内、0.01〜約100mg/kg;経口的、0.01〜約200mg/kg、および好ましくは約1〜100mg/kg;鼻腔内滴下、0.01〜約20mg/kg;ならびにエアロゾル、0.01〜約20mg/kgの動物(身体)重量でありえる。
【0046】
本発明は、1つまたは複数の容器中に、本発明の1つまたは複数の化合物、または本発明の化合物を含む組成物、または前述のものの類似体もしくは塩を含むキットにも関する。1つの実施形態において、キットは、式IIの化合物、またはその薬学的に許容される塩もしくは類似体を含む。本発明のキットは、薬学的に許容される担体および/または希釈剤を任意で含むことができる。1つの実施形態において、本発明のキットは1つまたは複数の他の成分または添加物または本明細書において記述されるような補助剤を含む。他の実施形態において、キットは、本明細書において記述される薬剤などの1つまたは複数の抗癌剤を含む。1つの実施形態において、本発明のキットは、キットの化合物または組成物を投与する方法について記述する説明書または包装材料を含む。キットの容器は、任意の適切な材料(例えばガラス、プラスチック、金属など)、および任意の適切なサイズ、形状、または立体配置でありえる。1つの実施形態において、本発明の化合物は、タブレット、ピル、または粉末の形態などの固形としてキット中に提供される。他の実施形態において、本発明の化合物は液体または溶液としてキット中に提供される。1つの実施形態において、キットは、液体または溶液の形態で本発明の化合物を含むアンプルまたはシリンジを含む。
【0047】
開示された方法から利益を得る哺乳類種は、類人猿、チンパンジー、オランウータン、ヒト、サルなどの霊長類;イヌ、ネコ、モルモット、ハムスター、ベトナムポットベリーピッグ、ウサギ、およびフェレットなどの飼育された動物(例えばペット);ウシ、水牛、バイソン、ウマ、ロバ、ブタ、ヒツジ、およびヤギなどの飼育された家畜;クマ、ライオン、トラ、ヒョウ、ゾウ、カバ、サイ、キリン、アンテロープ、ナマケモノ、カモシカ、シマウマ、ウィルドビースト、プレーリードッグ、コアラ、カンガルー、コモリネズミ、アライグマ、パンダ、ハイエナ、アザラシ、アシカ、ゾウアザラシ、カワウソ、ネズミイルカ、イルカおよびクジラなどの典型的には動物園において見出される外来動物を含むが、これらに限定されない。開示された方法から利益を得てもよい他の種は魚類、両生類、鳥および爬虫類を含む。本明細書において使用されるように、「患者」、「被験体」という用語は同じ意味で使用され、かかるヒト種および非ヒト種を含むように意図される。同様に、本発明のインビトロの方法は、かかるヒト種および非ヒト種の細胞に対して実行することができる。
材料および方法
【0048】
細胞株およびNCI多様性セット。この研究において使用されるすべて細胞株は、ATCCから購入したか、または事前に記載されたものである。(Cheng et al. 1997; Jiang et al. 2000; Yang et al. 2004; Yang et al. 2005)。NCI構造多様性セットは、およそ140,000の化合物のNCI薬物寄託場所から選択された1,992の化合物のライブラリーである。これらの多様性セット化合物の選択、構造および活性に関する詳細なデーターは、NCIの開発的な治療プログラム(Developmental Therapeutics Program)ウェブサイトで見出すことができる(Jones et al. 1991)。
【0049】
Aktにより形質転換した細胞の増殖阻害についてのスクリーニング。AKT2により形質転換したNIH3T3細胞またはLXSNベクターによりトランスフェクションしたNIH3T3対照細胞(Cheng et al. 1997)を、96ウェル組織培養プレートにプレーティングした。5μMのNCI多様性セット化合物による処理に後続して、細胞増殖は、セルタイター96ワン・ソリューション・セル・プロリファレーション(CellTier 96 One Solution Cell Proliferation)キット(プロメガ(Promega)社)により検出された。AKT2により形質転換したNIH3T3細胞の増殖を阻害するが、LXSNによりトランスフェクションしたNIH3T3細胞は阻害しない化合物をAkt阻害剤の候補と見なし、詳しい解析を行なった。
【0050】
インビトロのプロテインキナーゼ、細胞生存およびアポトーシス分析。以前に記述されたように、インビトロのキナーゼを実行した(Jiang et al. 2000)。細胞生存はMTT(シグマ(Sigma)社)により分析した。アポトーシスは、アネキシンV(BDバイオサイエンス(BD Biosciences)社)により検出し、製造者の説明書に従って実行した。組換えのAktおよびPDK1は、アップステート・バイオテクノロジー(Upstate Biotechnology)社から購入した。
【0051】
ヌードマウス腫瘍異種移植モデルにおける抗腫瘍活性。腫瘍細胞を採取し、PBS中に再懸濁し、以前に報告されるように8週齢のメスヌードマウスの右脇腹および左脇腹(2×106細胞/脇腹)に皮下(s.c.)注射した(Sun et al. 1999)。腫瘍が約100〜150 mm3に達した時に、動物は無作為化され、媒質または薬物を毎日腹腔内(i.p.)投薬した。対照動物にジメチルスルホキシド(DMSO)(20%)媒質を投与し、処理動物に20%DMSO中のAPI−1(10mg/kg/日)を注射した。
【0052】
本明細書において参照または引用したすべての特許、特許出願、仮出願、および出版物は、すべて図および表を含むその全体を参照することにより、この明細書の明確な教示に矛盾しない程度まで組み入れられる。
【0053】
以下は本発明の実行のための手順を説明する実施例である。これらの実施例は限定として解釈されるべきでない。特に断りのない限り、すべてのパーセンテージは重量により、すべての溶媒混合比率は体積による。
【実施例1】
【0054】
NCI多様性セットのスクリーニングによる低分子Akt/PKB経路阻害剤−1(API−1)の同定
【0055】
Akt経路の異常な活性化がすべてのヒト悪性腫瘍のほとんど50%において起こり、Aktの阻害が細胞増殖停止およびアポトーシスを誘導するという事実のために、抗癌薬を見出だすために低分子Akt阻害剤を開発することは産業および学界からの関心を引いた(Cheng et al. 2005; Granville et al. 2006)。何十ものAkt阻害剤が報告されているが、それらの多くはインビボでの抗腫瘍活性を欠く。脂質ベースのAkt阻害剤(ペリホシン)は第I相および第II相の研究において報告された(Van Ummersen et al. 2004; Bailey et al. 2006)。しかしながら、どちらの研究においてもAktの修飾は査定されなかった。膵臓癌におけるペリホシンの最近の第II相研究は、第一ステージの間の許容できない有害事象を受けて終結した(Marsh et al. 2007)。それにもかかわらず、最小の副作用を備え他のキナーゼ活性の阻害を欠く強力な選択的Akt阻害剤を開発する必要がある。Aktの低分子阻害剤を同定するために、我々は、「材料および方法」中で記述されているように、AKT2により形質転換したNIH3T3細胞の増殖阻害が可能であるが、空ベクターLXSNによりトランスフェクションされたNIH3T3細胞の増殖阻害が可能でない薬剤について、NCIからの1,992化合物の化学的ライブラリー(NCI多様性セット)を評価した。三重実験は、32の化合物がAKT2により形質転換した細胞のみの増殖を阻害したことを示した。我々は、以前にそれらのうちの1つ(インビトロおよびインビボで抗腫瘍活性を有する全Akt阻害剤であり、現在第I相臨床試験中である、API−2/トリシリビンと命名された)の特性を評価した(Yang et al. 2004)。
【0056】
本研究において、我々は、Akt/PKB阻害剤−1(API−1)が生細胞におけるAktのキナーゼ活性およびリン酸化レベルを特異的に阻害することを示した。図1AはAPI−1(NSC177223)の化学構造を示し、それは化学名を有しておらず、NCI60細胞株において検査されていない(http://dtp.nci.nih.gov/)。API−1が、形質転換していない親細胞以上にAKT2形質転換細胞を選択的に阻害したので、API−1がAKT2キナーゼの阻害剤であるか、Aktの他の2つのメンバーも阻害するかどうかを最初に調べた。HAのタグを付加した野生型のAkt1、AKT2およびAKT3により、HEK293細胞をトランスフェクションした。一晩の血清飢餓に後続して、細胞はEGF刺激の前に60分間API−1により処理し、抗HA抗体により免疫沈降した。免疫沈降物はインビトロキナーゼ分析を行なった。図1Bは、API−1がAkt1、AKT2およびAKT3のインスリン誘導性キナーゼ活性を抑制したことを示す。API−1が生細胞においてAktを阻害するかどうかを次に調べる。OVCAR3細胞(リン酸化Aktを上昇したレベルで発現する)を、異なる用量のAPI−1により3時間処理した。抗リン酸化−Akt−S473抗体によるイムノブロット解析から、API−1がAktのリン酸化レベルおよびIC50を効率的に減少させるのは、およそ0.8μMであることが示された。しかしながら、全体のAktレベルは変化しなかった(図1C)。さらに、API−1が直接インビトロのAktキナーゼ活性を阻害するかどうかを調べた。構成的に活性のあるAkt組換えタンパク質を、異なる量のAPI−1、および陽性対照として化合物E(Aktを含む全キナーゼのATP競合阻害剤)を含むキナーゼバッファー中で、Akt/SGK基質ペプチド(アップステート社)と共にインキュベートした。三重実験から、API−1はAktキナーゼ活性に対する効果がないことが示され(図1D)、API−1が直接インビトロでAktを阻害せず、API−1がATPとしても、また基質競合物としても機能しないことが示唆された。
【実施例2】
【0057】
API−1はAktの上流の活性化因子を阻害しない
【0058】
Aktは、PTENにより負に調節されるPI3K依存的様式を介して、細胞外刺激および細胞内シグナル分子によって活性化される。PI3Kの活性化またはPTENの変異は、Aktキナーゼ活性の誘導を導くPDK1を活性化するだろう。したがって、AktのAPI−1阻害は、PI3KおよびPDK1などのAktの1つまたは複数の標的上流分子に由来しうる。この目的のために、API−1がPI3Kおよび/またはPDK1を阻害するかどうかを次に調べた。HEK293細胞は血清飢餓を行い、次にEGF刺激の前にAPI−1またはPI3K阻害剤(ワートマニン)により1時間処理した。PI3Kを抗p110α抗体により免疫沈降した。免疫沈降物は、基質としてPI−4−Pを使用してインビトロのPI3Kキナーゼ分析を行なった。図2A中で示されるように、EGFにより誘導されたPI3K活性はワートマニンによって阻害されたが、API−1によって阻害されなかった。API−1の効果を評価するために、リードアウト(アップステート・バイオテクノロジー社)としてSGKキナーゼを使用して、PDK1によりインビトロのPDK1キナーゼ分析を実行した。PDK1阻害剤UCN−01とは異なり(Sato et al. 2002)、API−1はインビトロのPDK1キナーゼ活性に効果がなかった(図2B)。生細胞におけるPDK1活性化に対するAPI−1の効果をさらに評価するために、OVCAR3細胞のAPI−1処理に後いて、PDK1−Ser241(自己リン酸化され、活性のために重要な残基(Casamayor et al. 1999))のリン酸化レベルを調べた。図2Dは、PDK1のリン酸化レベルがAPI−1によって阻害されなかったことを示す。
【実施例3】
【0059】
API−1は、AGCキナーゼメンバーのPKA、PKCおよびSGK、ならびに他のシグナリング分子のERK、JNK、p38およびSTAT3以上にAktに高度に選択的である。
【0060】
Aktは、AGC(PKA/PKG/PKC)キナーゼファミリーに属し、このファミリーは、PKA、PKC、血清およびグルココルチコイド誘導性キナーゼ(SGK)、p90リボソームS6キナーゼ、p70S6K、マイトジェンおよびストレス活性化プロテインキナーゼ、ならびにPKC関連キナーゼもまた含む。AGCキナーゼファミリーの中で、PKA、PKCおよびSGKのタンパク質構造は、他のメンバーよりもAktキナーゼに非常に近い。したがって、次にこれらの3つのキナーゼの酵素活性に対するAPI−1の効果を検討した。インビトロのPKAキナーゼ分析およびSGKキナーゼ分析は、キナーゼバッファー中のケンプチドまたはAkt/SGK基質ペプチドおよび[γ−32P]ATPの追加によるキナーゼ分析の開始の前の30分間、組換えPKAタンパク質または組換えSGKタンパク質との、示された用量のAPI−1または特異的阻害剤の前インキュベーションによって実行された。インビトロキナーゼ分析は、PKAおよびSGKのキナーゼ活性がPKAIおよび化合物Eによってそれぞれ阻害されるが、API−1にはそれらの活性に対する効果ないことを示した(図2Cおよび2E)。生細胞におけるPKAおよびPKCαの活性化に対するAPI−1の効果をさらに評価するために、OVCAR3細胞は示された用量のAPI−1またはPKAおよびPKCの特異的阻害剤により処理され、PKAおよびPKCのリン酸化レベルがAPI−1によって阻害されないことがイムノブロット解析により示された(図2D)。さらに、HEK293細胞をHAのタグを付加したSGKによりトランスフェクションした。インビトロキナーゼ分析から、EGFに誘導されたSGKキナーゼ活性はワートマニンによって減少されるが、API−1によって減少されないことが示された(図2F)。
【0061】
API−1が他の腫瘍形成性の生存経路に効果があるかどうか決定するために、OVCAR3細胞をAPI−1(10aM)により異なる時間処理し、市販で入手可能な抗リン酸化抗体によりイムノブロットした。API−1処理後には、Stat3、JNK、p38およびErk1/2のリン酸化レベルの検出可能な変化は観察されなかった(図2G)。これらのデーターは、API−1がAkt経路を特異的に阻害できることを示す。
【実施例4】
【0062】
API−1は構成的に活性のあるAktおよびその下流の標的を阻害する。
【0063】
API−1はAktをインビトロで直接阻害することができなかったが、PI3KおよびPDK1に対する影響なしに生細胞におけるAktのキナーゼ活性およびリン酸化を停止したので、この化合物がATP結合部位ではなくAktタンパク質と相互作用して、PDK1およびPDK2によるThr308およびSer473のリン酸化を防止することが推測された。これが真であるならば、構成的に活性のあるAktの活性化がまだThr308およびSer473のリン酸化を必要とするので、API−1は構成的に活性のあるAktの活性化もまた阻害すべきである(Sun et al. 2001)。この仮説を検証するために、HEK293細胞を、HAのタグを付加したMyr−Akt1、−Akt2および−Akt1E40Kによりトランスフェクションした。一晩の血清飢餓に後続して、細胞をAPI−1ありまたはAPIなしで処理した。Myr−Akt1、Myr−Akt2およびAkt1−E40Kを、抗HA抗体により免疫沈降した。免疫沈降物に、インビトロキナーゼ分析および抗リン酸化−Akt−T308抗体によるイムノブロット解析を行なった。図3Aは、Myr−Akt1、Myr−Akt2およびAkt1−E40Kのリン酸化レベルだけでなく、インビトロのAktキナーゼ活性もAPI−1によって阻害されたことを示し、API−1がAkt分子に結合して細胞内での活性化を阻害するという概念を支援する。
【0064】
Aktは多数のタンパク質のリン酸化を介してその細胞機能を発揮する(Datta et al. 1999)。次にAPI−1がAktの下流の標的を阻害するかどうかを調べた。GSK3βおよびmTORは主要なAkt標的のうちの2つであるので、GSKβおよびS6(p70S6Kの基質)のリン酸化レベルでAPI−1の影響を評価した。API−1によるOVCAR3の処理に続くイムノブロット解析から、API−1がそれらのリン酸化を非常に阻害することが明らかになった(図3B)。
【実施例5】
【0065】
API−1は細胞増殖を抑制し、Aktを過剰発現または活性化するヒト癌細胞株においてアポトーシスを誘導する
【0066】
Aktは主要な増殖促進および生存促進の経路である。上昇したAktキナーゼを有する癌細胞は化学療法薬に誘導された細胞増殖停止および細胞死に対してより高い耐性を示すが、Aktのノックダウンは異なるアポトーシス促進刺激によって誘導されるアポトーシスに感受性を持つ(Solit et al. 2003; Xu et al. 2003; Jetzt et al. 2003)。API−1が選択的にAkt経路を阻害する能力は、Aktの異常な発現/活性化を有するそれらの腫瘍細胞において、優先的に増殖を阻害することおよび/またはアポトーシスを誘導することを示唆する。これを検査するために、構成的に活性のあるAktを発現する細胞(Aktの過剰発現(OVCAR3、OVCAR8、MCF7およびPANC1)またはPTEN遺伝子の変異(MDA−MB−468PC−3およびLNCaP)によって引き起こされた)、および行なってない細胞(OVCAR5、MDA−MB435s、DU−145およびCOLO357)の処理にAPI−1を使用した。イムノブロット解析から、上昇したAktを発現する細胞においてAPI−1によりAktのリン酸化レベルが有意に阻害されるが、リン酸化Aktは低レベルのAktを示す細胞株においてもまたAPI−1により減少されることが示された(図4Aおよびデーター不掲載)。しかしながら、API−1はPARP切断を誘導し、低レベルのAktを有する細胞と比較して、Aktを過剰発現/活性化する細胞において、はるかに高い程度で細胞増殖を阻害する(図4A、4Bおよび4C)。API−1処理は、Aktを過剰発現/活性化する細胞株のOVCAR3、OVCA8、MDA−MB−468およびMCF7においておよそ50〜70%で細胞増殖を阻害したが、OVCAR5およびMDA−MB435s細胞において約10〜30%のみ阻害した(図4Bおよび4C)。さらに、API−1は、OVACAR3およびMDA−MB−468においてそれぞれ9倍および4.6倍でアポトーシスを誘導するが、API−1−処理したOVCAR5およびMDA−MB435s細胞においてはるかに少ないアポトーシスが観察された(図4D〜4G)。したがって、API−1は細胞増殖を阻害し、異常なAktを発現する細胞においてアポトーシスを優先的に誘導する。
【実施例6】
【0067】
API−1は、Aktを過剰発現するヌードマウスにおいて腫瘍の増殖を阻害する
【0068】
Aktの異常な活性化および過剰発現がヒトの卵巣癌および膵臓癌で頻繁に検出されること(Cheng et al. 1992)、およびAktのアンチセンスが癌細胞の増殖および浸潤を有意に阻害すること(Cheng et al. 1996)は、以前に示されている。さらに、PI3K、HSP70、Srcおよびファルネシルトランスフェラーゼの阻害剤によるAkt経路の阻害は、細胞増殖の停止およびアポトーシスの誘導をもたらした(Solit et al. 2003; Xu et al. 2003)。API−1は、上昇したAktレベルを有する癌細胞においてAktシグナリングを阻害しアポトーシスおよび細胞増殖停止を誘導するので(図4A〜4G)、ヌードマウスにおいて、上昇したAktレベルを有する腫瘍の増殖は、低レベルのAktを有する腫瘍よりも、API−1に対してより感受性であるべきだと推論された。この目的のために、Aktを過剰発現する細胞(OVCAR3およびPANC−1)をマウスの左脇腹に皮下移植し、低レベルのAktを発現する細胞株(OVCAR5およびCOLO357)を右脇腹に皮下移植した。腫瘍が約100〜150mm3の平均サイズに達した時に、動物を無作為化し、媒質またはAPI−1(10mg/kg/日)のいずれかにより腹腔内処理した。図5A〜5Jで示されるように、媒質対照により処理したOVCAR3およびPANC1の腫瘍は増殖し続けた。API−1は、OVCAR3およびPANC1腫瘍増殖をそれぞれ70%および50%阻害した(図5C〜5Fおよび5G〜5J)。これとは対照的に、API−1は、ヌードマウスにおけるOVCAR5およびCOLO357の細胞の増殖に対して効果がほとんどなかった(図5A〜5J)。API−1は、用量10mg/kg/日で、マウスの血糖値、体重、活動性および食物摂取量に対して影響はなかった(データー不掲載)。処理された腫瘍サンプルにおいて、Aktのリン酸化レベルはAPI−1によって約70%減少されたが、全Akt含有量の変化はなかった(図5K〜5L)。総合すれば、これらの結果は、API−1が上昇したAktレベルを有する腫瘍の増殖を選択的に阻害することを示す。
【0069】
本明細書において記述された実施例および実施形態が説明の目的のみのためのものあり、それらに照らして様々な修飾または変更が当業者に対して示唆され、本出願の趣旨および範囲、および添付された請求項の範囲内に含まれるべきであることが理解される。さらに、本明細書において開示された任意の発明またはその実施形態の任意の要素または限定は、任意のおよび/もしくは他のすべての要素もしくは限定(個別にまたは任意の組合せで)、または本明細書において開示された他の発明またはその実施形態と組み合わせることができ、すべてのかかる組合せは本発明の範囲でそれらに限定されずに検討される。
【0070】
参照文献
【特許請求の範囲】
【請求項1】
式Iの化合物:
【化1】
(I)
(式中、
Xは独立してO、NまたはSであり;
YはO、NまたはSであり;
R1は、独立して−H、−OH、−NH2、−NO2、ハロゲン、−OHにより任意に置換されるアルキル、または−OHにより任意に置換されるアルコキシであり;
R2は、−H、−OH、−NH2、−C(O)NH2、アルキル、またはアルコキシであり、それらのいずれかは、−OH、ハロゲン、アルキルまたはアルコキシにより任意で置換することができ;
R3は、−H、−OH、−NH2、−C(O)NH2、アルキルまたはアルコキシであり、それらのいずれかは、−OH、ハロゲン、アルキルまたはアルコキシにより任意で置換することができる);または
その薬学的に許容される塩。
【請求項2】
各XがNであるか、またはYがOである、請求項1に記載の化合物。
【請求項3】
各R1が、独立して−OHまたは−CH2OHである、請求項1に記載の化合物。
【請求項4】
R2が、−OHにより任意に置換される−NH2である、請求項1に記載の化合物。
【請求項5】
R2が、−C(O)NH2である、請求項1に記載の化合物。
【請求項6】
前記化合物が、構造:
【化2】
を有する、請求項1に記載の化合物
【請求項7】
請求項1〜6のいずれか一項に記載の化合物を含む組成物。
【請求項8】
前記組成物が、薬学的に許容される担体または希釈剤を含む、請求項7に記載の組成物。
【請求項9】
前記組成物が、1つまたは複数の抗癌剤を含む、請求項7に記載の組成物。
【請求項10】
前記抗癌剤が、アルトレタミン、ブレオマイシン、ボルテゾミブ(ベルケイド)、ブスルファン、ホリナートカルシウム、カペシタビン、カルボプラチン、カルムスチン、クロラムブチル、シスプラチン、クラドリビン、クリサンタスパーゼ、シクロホスファミド、シタラビン、ダカルバジン、ダクチノマイシン、ダウノルビシン、ドセタキセル、ドキソルビシン、エピルビシン、エトポシド、フルダラビン、フルオロウラシル、ゲフィチニブ(イレッサ)、ゲムシタビン、ヒドロキシウレア、イダルビシン、イフォスファミド、イマチニブ(グリーベック)、イリノテカン、リポソームドキソルビシン、ロムスチン、メルファラン、メルカプトプリン、メトトレキサート、マイトマイシン、ミトキサントロン、オキサリプラチン、パクリタキセル、ペントスタチン、プロカルバジン、ラルチトレキセド、ストレプトゾシン、テガフール−ウラシル、テモゾロマイド、チオテパ、チオグアニン/チオグアニン、トポテカン、トレオスルファン、ビンブラスチン、ビンクリスチン、ビンデシン、ビノレルビン、メルファラン、アレムツズマブ、セツキシマブ(エルビタックス)、ゲムツズマブ、ヨウ素131トシツモマブ、リツキシマブ、またはトラスツザマブ(ハーセプチン)である、請求項9に記載の組成物。
【請求項11】
前記組成物が、1つまたは複数の有糸分裂阻害剤、アルキル化剤、抗代謝剤、DNAインターカレーター、トポイソメラーゼ阻害剤、抗血管新生剤、または抗エストロゲンを含む、請求項7に記載の組成物。
【請求項12】
Aktタンパク質の上昇した発現もしくは構成的に活性のある発現を有する細胞の生存もしくは増殖を阻害するかまたは細胞を殺傷する方法であって、請求項1〜6のいずれかに記載の化合物または請求項7〜11のいずれかに記載の組成物の有効量と該細胞を接触させることを含む該方法。
【請求項13】
前記細胞がヒト細胞である、請求項12に記載の方法。
【請求項14】
前記細胞が、癌または腫瘍細胞である、請求項12に記載の方法。
【請求項15】
前記癌細胞が、肛門、胆管、膀胱、骨、骨髄、腸管(大腸および直腸を含む)、乳房、目、胆嚢、腎臓、口、喉頭、食道、胃、精巣、頚管、頭部、首、卵巣、肺、中皮腫、神経内分泌、陰茎、皮膚、脊髄、甲状腺、膣、外陰部、子宮、肝臓、筋肉、膵臓、前立腺、血球(リンパ球および他の免疫系細胞を含む)、または脳の癌細胞である、請求項14に記載の方法。
【請求項16】
人または動物における腫瘍学的障害を治療する方法であって、請求項1〜6のいずれかに記載の化合物または請求項7〜11のいずれかに記載の組成物の治療上の有効量の投与を含む該方法。
【請求項17】
前記腫瘍学的障害が、肛門、胆管、膀胱、骨、骨髄、腸管(大腸および直腸を含む)、乳房、目、胆嚢、腎臓、口、喉頭、食道、胃、精巣、頚管、頭部、首、卵巣、肺、中皮腫、神経内分泌、陰茎、皮膚、脊髄、甲状腺、膣、外陰部、子宮、肝臓、筋肉、膵臓、前立腺、血球(リンパ球および他の免疫系細胞を含む)、または脳の癌および/または腫瘍である、請求項16に記載の方法。
【請求項18】
前記化合物が、抗腫瘍剤および/または抗癌剤、および/または放射線療法、および/または外科的治療の前に、それらの後に、またはそれらと組み合わせて投与される、請求項16に記載の方法。
【請求項19】
前記腫瘍学的障害が、Aktタンパク質の上昇した発現または構成的に活性のある発現を有する細胞に関連するかまたは特徴づけられる、請求項16に記載の方法。
【請求項20】
前記方法が、腫瘍学的障害を有しているかまたは治療を必要とする人または動物を同定することをさらに含む、請求項16に記載の方法。
【請求項21】
前記化合物の投与経路が、腸内投与、非経口投与、静脈内投与、筋肉内投与、経口投与、皮下投与、局所投与、または鼻腔内投与である、請求項16に記載の方法。
【請求項22】
1つまたは複数の容器中に、請求項1〜6のいずれか一項に記載の1つまたは複数の化合物または請求項7〜11のいずれか一項に記載の1つまたは複数の組成物を含むキット。
【請求項23】
前記キットが、薬学的に許容される担体および/または希釈剤をさらに含む、請求項22に記載のキット。
【請求項24】
前記キットが、1つまたは複数の抗癌剤をさらに含む、請求項22に記載のキット。
【請求項25】
前記抗癌剤が、アルトレタミン、ブレオマイシン、ボルテゾミブ(ベルケイド)、ブスルファン、ホリナートカルシウム、カペシタビン、カルボプラチン、カルムスチン、クロラムブチル、シスプラチン、クラドリビン、クリサンタスパーゼ、シクロホスファミド、シタラビン、ダカルバジン、ダクチノマイシン、ダウノルビシン、ドセタキセル、ドキソルビシン、エピルビシン、エトポシド、フルダラビン、フルオロウラシル、ゲフィチニブ(イレッサ)、ゲムシタビン、ヒドロキシウレア、イダルビシン、イフォスファミド、イマチニブ(グリーベック)、イリノテカン、リポソームドキソルビシン、ロムスチン、メルファラン、メルカプトプリン、メトトレキサート、マイトマイシン、ミトキサントロン、オキサリプラチン、パクリタキセル、ペントスタチン、プロカルバジン、ラルチトレキセド、ストレプトゾシン、テガフール−ウラシル、テモゾロマイド、チオテパ、チオグアニン/チオグアニン、トポテカン、トレオスルファン、ビンブラスチン、ビンクリスチン、ビンデシン、ビノレルビン、メルファラン、アレムツズマブ、セツキシマブ(エルビタックス)、ゲムツズマブ、ヨウ素131トシツモマブ、リツキシマブ、またはトラスツザマブ(ハーセプチン)である、請求項24に記載のキット
【請求項26】
前記キットが、1つまたは複数の有糸分裂阻害剤、アルキル化剤、抗代謝剤、DNAインターカレーター、トポイソメラーゼ阻害剤、抗血管新生剤、または抗エストロゲンをさらに含む、請求項22に記載のキット。
【請求項27】
細胞におけるAkt/PKB経路を阻害する方法であって、請求項1〜6のいずれかに記載の化合物または請求項7〜11のいずれかに記載の組成物の有効量と細胞を接触させることを含む該方法。
【請求項28】
前記化合物が、Akt1、AKT2、および/またはAKT3に結合し、それらのキナーゼ活性を阻害する、請求項27に記載の方法。
【請求項29】
前記細胞がAktタンパク質を構成的に発現するか、または前記細胞がAktタンパク質を上昇したレベルで発現する、請求項27に記載の方法。
【請求項30】
前記細胞が腫瘍または癌細胞である、請求項27に記載の方法。
【請求項31】
細胞増殖、細胞生存、細胞移動、および/または細胞分化が阻害される、請求項27に記載の方法。
【請求項32】
前記細胞がヒト細胞である、請求項27に記載の方法。
【請求項33】
前記障害が、細胞におけるAktタンパク質の構成的発現、異常な発現または上昇した発現に関連する、人または動物における障害を治療する方法であって、請求項1〜6のいずれかに記載の化合物または請求項7〜11のいずれかに記載の組成物の有効量の投与を含む該方法。
【請求項34】
前記化合物が、Akt1、AKT2、またはAKT3に結合し、それらのキナーゼ活性を阻害する、請求項33に記載の方法。
【請求項35】
前記障害が、異常な細胞増殖、細胞生存、細胞移動、および/または細胞分化によって特徴づけられる、請求項33に記載の方法。
【請求項1】
式Iの化合物:
【化1】
(I)
(式中、
Xは独立してO、NまたはSであり;
YはO、NまたはSであり;
R1は、独立して−H、−OH、−NH2、−NO2、ハロゲン、−OHにより任意に置換されるアルキル、または−OHにより任意に置換されるアルコキシであり;
R2は、−H、−OH、−NH2、−C(O)NH2、アルキル、またはアルコキシであり、それらのいずれかは、−OH、ハロゲン、アルキルまたはアルコキシにより任意で置換することができ;
R3は、−H、−OH、−NH2、−C(O)NH2、アルキルまたはアルコキシであり、それらのいずれかは、−OH、ハロゲン、アルキルまたはアルコキシにより任意で置換することができる);または
その薬学的に許容される塩。
【請求項2】
各XがNであるか、またはYがOである、請求項1に記載の化合物。
【請求項3】
各R1が、独立して−OHまたは−CH2OHである、請求項1に記載の化合物。
【請求項4】
R2が、−OHにより任意に置換される−NH2である、請求項1に記載の化合物。
【請求項5】
R2が、−C(O)NH2である、請求項1に記載の化合物。
【請求項6】
前記化合物が、構造:
【化2】
を有する、請求項1に記載の化合物
【請求項7】
請求項1〜6のいずれか一項に記載の化合物を含む組成物。
【請求項8】
前記組成物が、薬学的に許容される担体または希釈剤を含む、請求項7に記載の組成物。
【請求項9】
前記組成物が、1つまたは複数の抗癌剤を含む、請求項7に記載の組成物。
【請求項10】
前記抗癌剤が、アルトレタミン、ブレオマイシン、ボルテゾミブ(ベルケイド)、ブスルファン、ホリナートカルシウム、カペシタビン、カルボプラチン、カルムスチン、クロラムブチル、シスプラチン、クラドリビン、クリサンタスパーゼ、シクロホスファミド、シタラビン、ダカルバジン、ダクチノマイシン、ダウノルビシン、ドセタキセル、ドキソルビシン、エピルビシン、エトポシド、フルダラビン、フルオロウラシル、ゲフィチニブ(イレッサ)、ゲムシタビン、ヒドロキシウレア、イダルビシン、イフォスファミド、イマチニブ(グリーベック)、イリノテカン、リポソームドキソルビシン、ロムスチン、メルファラン、メルカプトプリン、メトトレキサート、マイトマイシン、ミトキサントロン、オキサリプラチン、パクリタキセル、ペントスタチン、プロカルバジン、ラルチトレキセド、ストレプトゾシン、テガフール−ウラシル、テモゾロマイド、チオテパ、チオグアニン/チオグアニン、トポテカン、トレオスルファン、ビンブラスチン、ビンクリスチン、ビンデシン、ビノレルビン、メルファラン、アレムツズマブ、セツキシマブ(エルビタックス)、ゲムツズマブ、ヨウ素131トシツモマブ、リツキシマブ、またはトラスツザマブ(ハーセプチン)である、請求項9に記載の組成物。
【請求項11】
前記組成物が、1つまたは複数の有糸分裂阻害剤、アルキル化剤、抗代謝剤、DNAインターカレーター、トポイソメラーゼ阻害剤、抗血管新生剤、または抗エストロゲンを含む、請求項7に記載の組成物。
【請求項12】
Aktタンパク質の上昇した発現もしくは構成的に活性のある発現を有する細胞の生存もしくは増殖を阻害するかまたは細胞を殺傷する方法であって、請求項1〜6のいずれかに記載の化合物または請求項7〜11のいずれかに記載の組成物の有効量と該細胞を接触させることを含む該方法。
【請求項13】
前記細胞がヒト細胞である、請求項12に記載の方法。
【請求項14】
前記細胞が、癌または腫瘍細胞である、請求項12に記載の方法。
【請求項15】
前記癌細胞が、肛門、胆管、膀胱、骨、骨髄、腸管(大腸および直腸を含む)、乳房、目、胆嚢、腎臓、口、喉頭、食道、胃、精巣、頚管、頭部、首、卵巣、肺、中皮腫、神経内分泌、陰茎、皮膚、脊髄、甲状腺、膣、外陰部、子宮、肝臓、筋肉、膵臓、前立腺、血球(リンパ球および他の免疫系細胞を含む)、または脳の癌細胞である、請求項14に記載の方法。
【請求項16】
人または動物における腫瘍学的障害を治療する方法であって、請求項1〜6のいずれかに記載の化合物または請求項7〜11のいずれかに記載の組成物の治療上の有効量の投与を含む該方法。
【請求項17】
前記腫瘍学的障害が、肛門、胆管、膀胱、骨、骨髄、腸管(大腸および直腸を含む)、乳房、目、胆嚢、腎臓、口、喉頭、食道、胃、精巣、頚管、頭部、首、卵巣、肺、中皮腫、神経内分泌、陰茎、皮膚、脊髄、甲状腺、膣、外陰部、子宮、肝臓、筋肉、膵臓、前立腺、血球(リンパ球および他の免疫系細胞を含む)、または脳の癌および/または腫瘍である、請求項16に記載の方法。
【請求項18】
前記化合物が、抗腫瘍剤および/または抗癌剤、および/または放射線療法、および/または外科的治療の前に、それらの後に、またはそれらと組み合わせて投与される、請求項16に記載の方法。
【請求項19】
前記腫瘍学的障害が、Aktタンパク質の上昇した発現または構成的に活性のある発現を有する細胞に関連するかまたは特徴づけられる、請求項16に記載の方法。
【請求項20】
前記方法が、腫瘍学的障害を有しているかまたは治療を必要とする人または動物を同定することをさらに含む、請求項16に記載の方法。
【請求項21】
前記化合物の投与経路が、腸内投与、非経口投与、静脈内投与、筋肉内投与、経口投与、皮下投与、局所投与、または鼻腔内投与である、請求項16に記載の方法。
【請求項22】
1つまたは複数の容器中に、請求項1〜6のいずれか一項に記載の1つまたは複数の化合物または請求項7〜11のいずれか一項に記載の1つまたは複数の組成物を含むキット。
【請求項23】
前記キットが、薬学的に許容される担体および/または希釈剤をさらに含む、請求項22に記載のキット。
【請求項24】
前記キットが、1つまたは複数の抗癌剤をさらに含む、請求項22に記載のキット。
【請求項25】
前記抗癌剤が、アルトレタミン、ブレオマイシン、ボルテゾミブ(ベルケイド)、ブスルファン、ホリナートカルシウム、カペシタビン、カルボプラチン、カルムスチン、クロラムブチル、シスプラチン、クラドリビン、クリサンタスパーゼ、シクロホスファミド、シタラビン、ダカルバジン、ダクチノマイシン、ダウノルビシン、ドセタキセル、ドキソルビシン、エピルビシン、エトポシド、フルダラビン、フルオロウラシル、ゲフィチニブ(イレッサ)、ゲムシタビン、ヒドロキシウレア、イダルビシン、イフォスファミド、イマチニブ(グリーベック)、イリノテカン、リポソームドキソルビシン、ロムスチン、メルファラン、メルカプトプリン、メトトレキサート、マイトマイシン、ミトキサントロン、オキサリプラチン、パクリタキセル、ペントスタチン、プロカルバジン、ラルチトレキセド、ストレプトゾシン、テガフール−ウラシル、テモゾロマイド、チオテパ、チオグアニン/チオグアニン、トポテカン、トレオスルファン、ビンブラスチン、ビンクリスチン、ビンデシン、ビノレルビン、メルファラン、アレムツズマブ、セツキシマブ(エルビタックス)、ゲムツズマブ、ヨウ素131トシツモマブ、リツキシマブ、またはトラスツザマブ(ハーセプチン)である、請求項24に記載のキット
【請求項26】
前記キットが、1つまたは複数の有糸分裂阻害剤、アルキル化剤、抗代謝剤、DNAインターカレーター、トポイソメラーゼ阻害剤、抗血管新生剤、または抗エストロゲンをさらに含む、請求項22に記載のキット。
【請求項27】
細胞におけるAkt/PKB経路を阻害する方法であって、請求項1〜6のいずれかに記載の化合物または請求項7〜11のいずれかに記載の組成物の有効量と細胞を接触させることを含む該方法。
【請求項28】
前記化合物が、Akt1、AKT2、および/またはAKT3に結合し、それらのキナーゼ活性を阻害する、請求項27に記載の方法。
【請求項29】
前記細胞がAktタンパク質を構成的に発現するか、または前記細胞がAktタンパク質を上昇したレベルで発現する、請求項27に記載の方法。
【請求項30】
前記細胞が腫瘍または癌細胞である、請求項27に記載の方法。
【請求項31】
細胞増殖、細胞生存、細胞移動、および/または細胞分化が阻害される、請求項27に記載の方法。
【請求項32】
前記細胞がヒト細胞である、請求項27に記載の方法。
【請求項33】
前記障害が、細胞におけるAktタンパク質の構成的発現、異常な発現または上昇した発現に関連する、人または動物における障害を治療する方法であって、請求項1〜6のいずれかに記載の化合物または請求項7〜11のいずれかに記載の組成物の有効量の投与を含む該方法。
【請求項34】
前記化合物が、Akt1、AKT2、またはAKT3に結合し、それらのキナーゼ活性を阻害する、請求項33に記載の方法。
【請求項35】
前記障害が、異常な細胞増殖、細胞生存、細胞移動、および/または細胞分化によって特徴づけられる、請求項33に記載の方法。
【図1A】

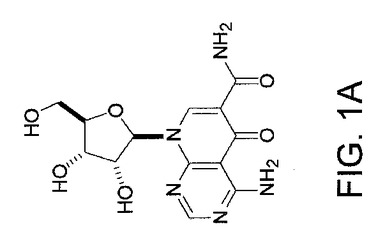
【図1B】
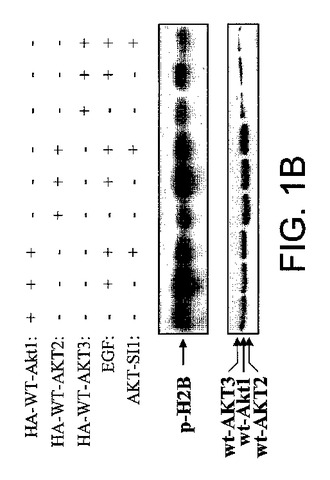
【図1C】
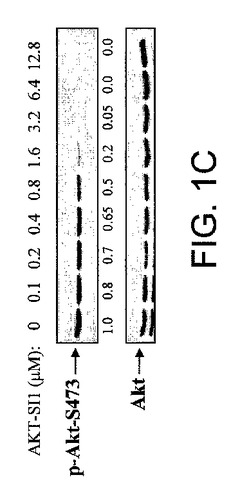
【図1D】
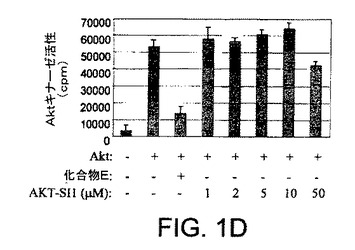
【図2A】
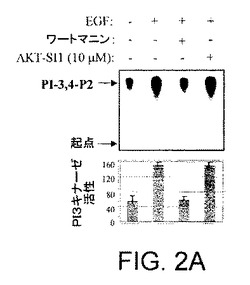
【図2B】
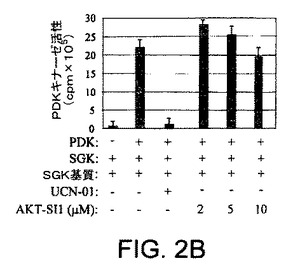
【図2C】
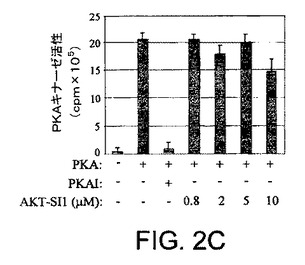
【図2D】
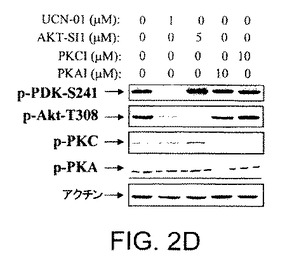
【図2E】
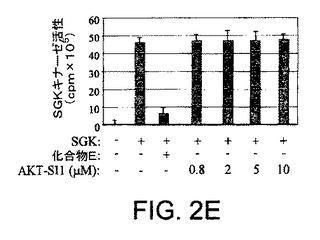
【図2F】
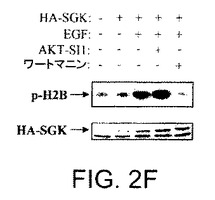
【図2G】
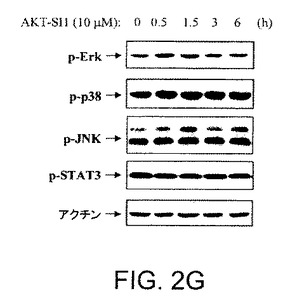
【図3A】
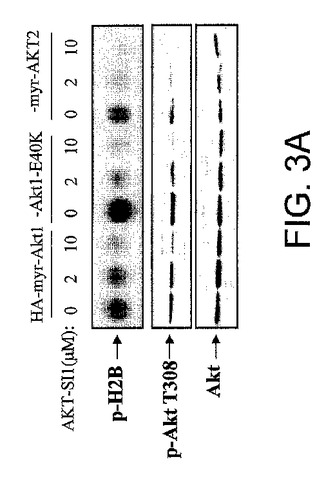
【図3B】
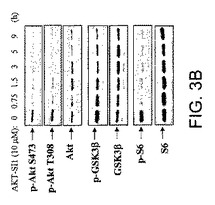
【図3C】
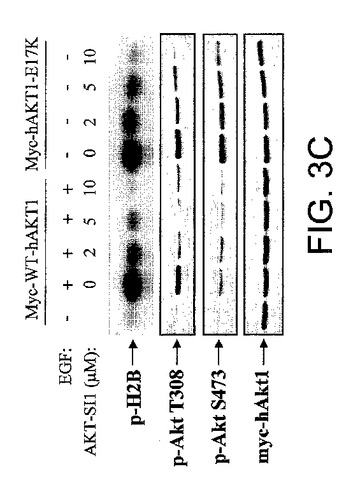
【図3D】
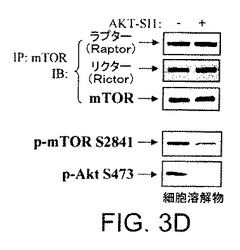
【図4A】
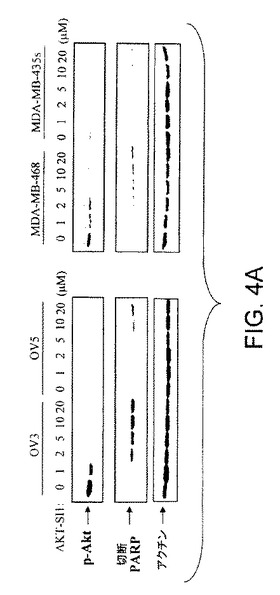
【図4B】
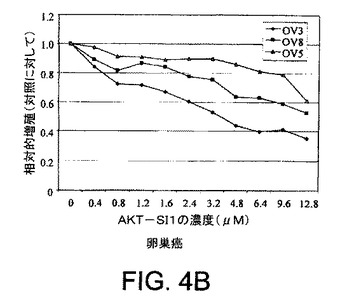
【図4C】
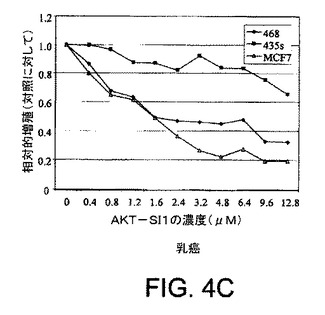
【図4D】
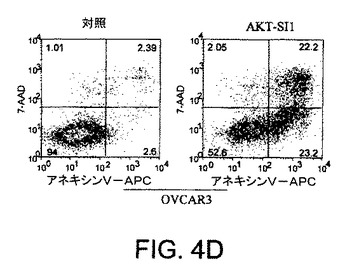
【図4E】
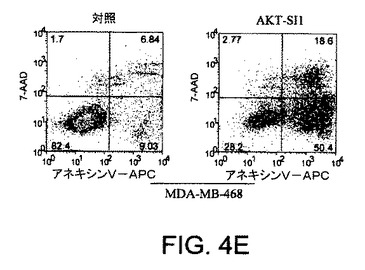
【図4F】
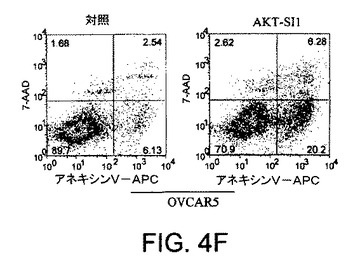
【図4G】
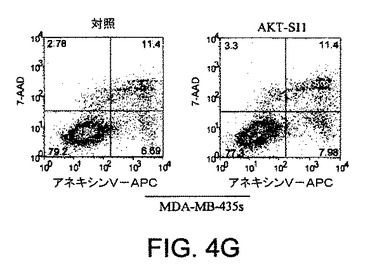
【図5A】

【図5B】
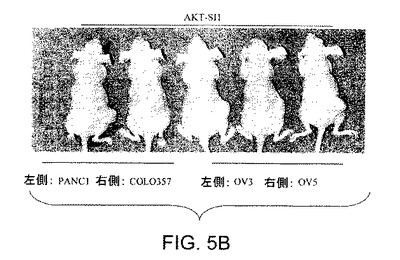
【図5C】
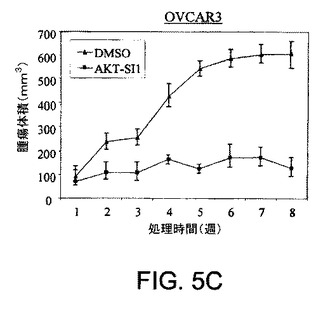
【図5D】
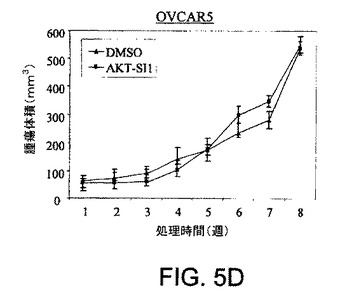
【図5E】
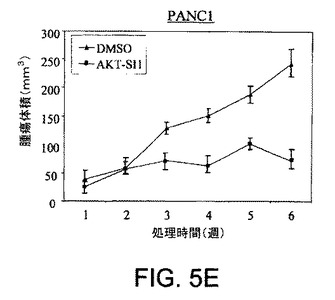
【図5F】
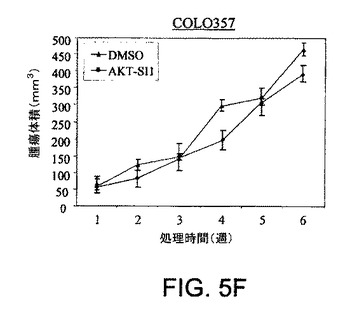
【図5G】

【図5H】
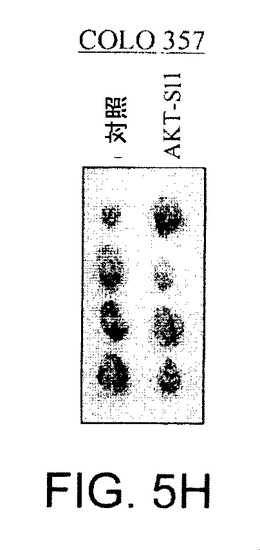
【図5I】
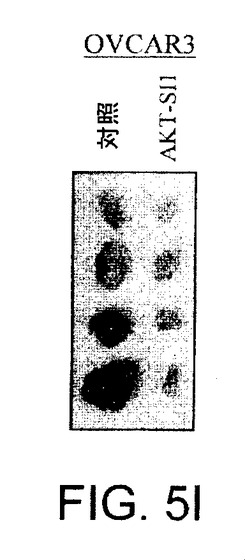
【図5J】

【図5K】
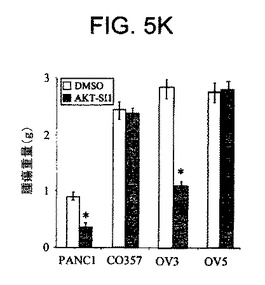
【図5L】
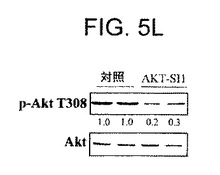
【図6】

【図7A】
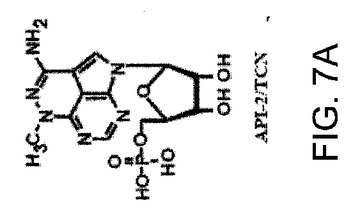
【図7B】
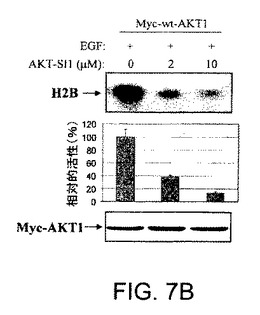
【図7C】
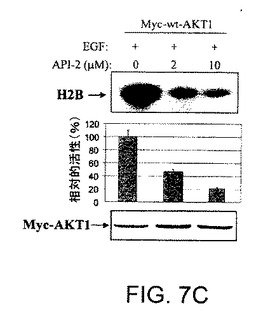
【図7D】
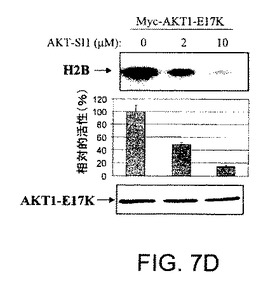
【図7E】
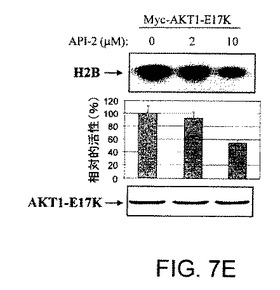
【図8A】
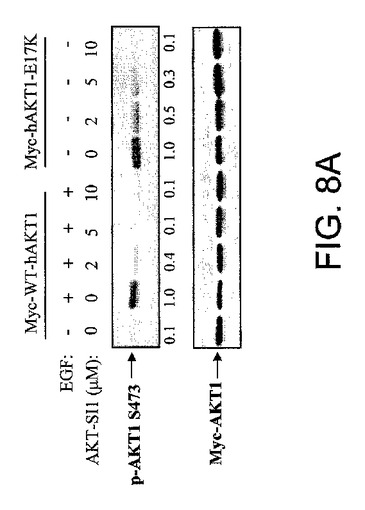
【図8B】
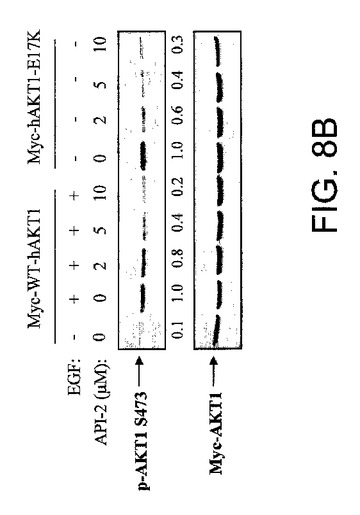
【図1B】
【図1C】
【図1D】
【図2A】
【図2B】
【図2C】
【図2D】
【図2E】
【図2F】
【図2G】
【図3A】
【図3B】
【図3C】
【図3D】
【図4A】
【図4B】
【図4C】
【図4D】
【図4E】
【図4F】
【図4G】
【図5A】
【図5B】
【図5C】
【図5D】
【図5E】
【図5F】
【図5G】
【図5H】
【図5I】
【図5J】
【図5K】
【図5L】
【図6】
【図7A】
【図7B】
【図7C】
【図7D】
【図7E】
【図8A】
【図8B】
【公表番号】特表2010−533206(P2010−533206A)
【公表日】平成22年10月21日(2010.10.21)
【国際特許分類】
【出願番号】特願2010−516297(P2010−516297)
【出願日】平成20年7月14日(2008.7.14)
【国際出願番号】PCT/US2008/069974
【国際公開番号】WO2009/009793
【国際公開日】平成21年1月15日(2009.1.15)
【出願人】(398014333)ユニヴァーシティ オブ サウス フロリダ (17)
【Fターム(参考)】
【公表日】平成22年10月21日(2010.10.21)
【国際特許分類】
【出願日】平成20年7月14日(2008.7.14)
【国際出願番号】PCT/US2008/069974
【国際公開番号】WO2009/009793
【国際公開日】平成21年1月15日(2009.1.15)
【出願人】(398014333)ユニヴァーシティ オブ サウス フロリダ (17)
【Fターム(参考)】
[ Back to top ]