
抗菌剤として有用な新規化合物
【課題】新たな抗生物質を提供すること。
【解決手段】下記式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステル。

【解決手段】下記式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステル。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、抗菌剤として有用な新規化合物に関する。
【背景技術】
【0002】
ペニシリン(Penicillin)の発見以来、セファロスポリンを含む数多くの合成β-ラクタム系抗生物質は多くの人々を様々な病気から救ってきた。しかし、現在、大量の抗生物質の使用により耐性菌が顕在化し、今まで有効であったβ-ラクタム系抗生物質が無効となる例が増加している。従来一つの抗生物質は20-30年くらい使用すると耐性菌が出現すると報告されている。このような問題を解決するために、新たな抗生物質の開発が望まれている。
【発明の開示】
【発明が解決しようとする課題】
【0003】
本発明は、新たな抗生物質を提供することを目的とする。
【課題を解決するための手段】
【0004】
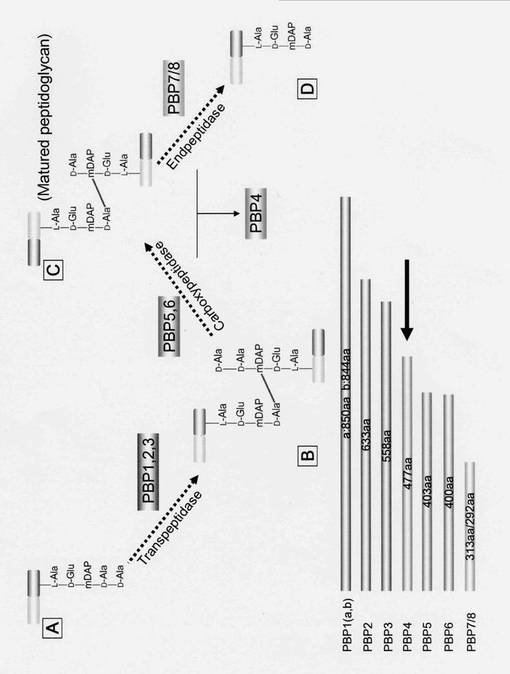
ペニシリン結合タンパク質(penicillin binding protein; PBP)は、ペニシリンに代表されるβ−ラクタム系抗生物質と特異的に結合する。12種類のPBPが大腸菌で知られているが、このうち、PBP1, 2, 3, 4は、細菌の細胞表面のペプチドグリカン層の合成・分解・再構成を担っている(図1)。PBPの活性がペニシリン等の抗生物質によって阻害されると、細菌は死に至る。現在、国内で市販されているセフェム系抗生物質として、塩野義製薬株式会社から、フロモックス(商品名)が販売されており、国内では抗生剤として40%の市場率を示している。このフロモックスの化合物は、塩野義製薬株式会社が開発を行った抗生物質であるが、本発明者とその共同研究者らによって、フロモックスの化合物と大腸菌由来ペニシリン結合タンパク質4との相互作用の機構の解明が行われた(Biochemistry. 2006 Jan 24;45(3):783-92.)。今回、本発明者は、インフルエンザ菌(Haemophilus influrenzae)由来のペニシリン結合タンパク質(PBP4)の立体構造を解明し、その結果に基づいて、新たな抗生物質の設計・合成を行ったところ、2つの化合物について、PBP4との相互作用及び抗菌活性が認められた。本発明は、これらの知見に基づいて完成されたものである。
【0005】
本発明の要旨は以下の通りである。
(1)下記式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステル。
【0006】
【化1】
(式中、Rは下記の式 (b)又は(c)で表される基である)
【0007】
【化2】
(2)(1)記載の化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルを含む医薬組成物。
(3)抗菌剤である(2)記載の医薬組成物。
(4)(1)記載の化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルとペニシリン結合タンパク質との複合体。
(5)ペニシリン結合タンパク質がペニシリン結合タンパク質4である(4)記載の複合体。
(6)ペニシリン結合タンパク質4が病原菌に由来する(5)記載の複合体。
(7)病原菌がインフルエンザ菌である(6)記載の複合体。
(8)(4)〜(7)のいずれかに記載の複合体の結晶。
(9)結晶の晶系が単斜結晶であり、空間群がP21である(8)記載の結晶。
(10)結晶の単位格子が、a=64.5±1Å、b=92.5±1Å、c=104.9±1Å、α=90°、β=107.7°、γ=90°である(9)記載の結晶。
【発明の効果】
【0008】
本発明により新規化合物が提供された。これらの化合物は、抗生物質として有用である。
【発明を実施するための最良の形態】
【0009】
以下、本発明の実施の形態についてより詳細に説明する。
【0010】
本発明は、下記式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルを提供する。
【0011】
【化1】
(式中、Rは下記の式(b)又は(c)で表される基である)
【0012】
【化2】
式(I)で表される化合物は、以下の2つの化合物である。
【0013】
3,3-Dimethyl-7-oxo-6-[2-(2-oxo-imidazolidin-1-ylamino)-2-phenyl-acetylamino]-4-thia-1-aza-bicyclo[3.2.0]heptane-2-carboxylic acid(化合物18)
【0014】
【化5】
;及びPotassium 6-(2-ethoxy-propionylamino)-3,3-dimethyl-7-oxo-4-thia-1-aza-bicyclo[3.2.0]heptane-2-carboxylate(化合物31)
【0015】
【化6】
これらの化合物は、下記の合成スキームで製造することができる。
【0016】
(化合物18の合成スキーム)
【0017】
【化3】
(化合物31の合成スキーム)
【0018】
【化4】
式(I)で表される化合物の医薬的に許容される塩としては、ナトリウム、カリウム、リチウム等のアルカリ金属塩、カルシウム、マグネシウム等のアルカリ土類金属塩、シクロヘキシルアミン、トリメチルアミン、ジエタノールアミン等の有機アミン塩、アルギニン、リジン等の塩基性アミノ酸塩、アンモニウム塩などを例示することができるが、これらに限定されない。
【0019】
式(I)で表される化合物の医薬的に許容されるエステルは、ペニシリン系、セファロスポリン系抗生物質について慣用されているエステルであるとよく、C2−6アルカノイルオキシC1−6アルキルエステル(具体的には、アセトキシメチルエステル、1−アセトキシエチルエステル、1−アセトキシブチルエステル、2−アセトキシエチルエステル、プロピオニルオキシメチルエステル、ピバロイルオキシメチルエステルなど)、1−(C1−6アルコキシ)C1−6アルキルエステル(具体的には、メトキシメチルエステル、エトキシメチルエステル、イソプロポキシメチルエステル、1−メトキシエチルエステル、1−エトキシエチルエステルなど)、1−(C1−6アルキルチオ)C1−6アルキルエステル(具体的には、メチルチオメチルエステル、エチルチオメチルエステルなど)などを例示することができるが、これらに限定されない。
【0020】
式(I)で表される化合物の分子内に不斉炭炭素が存在する場合は、ラセミ体および光学活性体も包含される。
【0021】
また、式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルは、様々な溶媒和物、例えば水、メタノール、エタノール、ジメチルホルムアミド、酢酸エチルなどとの溶媒和物としても存在でき、これらの溶媒和物も本発明に包含される。
【0022】
式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルは抗菌活性を有するので、病原性細菌により生じる種々の疾患(例えば、感染症など)の予防及び/又は治療のために使用できる。抗菌剤として式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルを使用する場合に対象となる菌は、式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルが抗菌活性を示す限り特に限定されず、グラム陽性菌又はグラム陰性菌のいずれであってもよいが、特に、Haemophilus Influenzae、病原性大腸菌などの菌に対して効果的である。式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルは、単独で使用してもよいし、他の抗菌剤と組合わせて使用してもよい。
【0023】
式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルは、常法により製剤化した医薬製剤(例えば、注射剤、カプセル剤、錠剤、散剤、顆粒剤など)として、ヒト又は動物に投与することができる。例えば、有効成分の量に換算して、1日あたり約0.1〜1000 mg/kg(体重)、好ましくは1日あたり約 1〜 500 mg/kg(体重)の投与量で、1回または数回に分けて経口投与するとよいが、その投与量や投与回数は、症状、年齢、投与方法などにより適宜変更しうる。注射剤に製剤化する場合には、蒸留水、生理食塩水などの担体を用いるとよく、カプセル剤、錠剤、散剤、顆粒剤に製剤化する場合には、デンプン、乳糖、白糖、炭酸カルシウムなどの賦形剤、デンプンのり液、アラビアゴム、ゼラチン、アルギン酸ナトリウム、カルボキシメチルセルロース、ヒドロキシプロピルセルロースなどの結合剤、ステアリン酸マグネシウム、タルクなどの滑沢剤など、デンプン、寒天、結晶セルロース、炭酸カルシウム、炭酸水素ナトリウム、アルギン酸ナトリウムなどの崩壊剤などを用いるとよい。製剤中の有効成分の含有率は、1〜99重量%の間で変動させることができる。例えば、錠剤、カプセル剤、顆粒剤、散剤などの形態をとる場合には、有効成分を5〜80重量%含有させるのが好ましく、注射剤の場合には、有効成分を1〜10重量%含有させるのが好ましい。
【0024】
本発明は、式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルとペニシリン結合タンパク質との複合体も提供する。複数種類のペニシリン結合タンパク質が知られており、例えば、大腸菌では12種類のペニシリン結合タンパク質が知られている(J. Bacteriol. 181, 3981-93 (1993))。ペニシリン結合タンパク質としては、ペニシリン結合タンパク質4(PBP4)が好ましい。ペニシリン結合タンパク質は、病原菌由来のものであるとよく、病原菌としては、インフルエンザ菌、肺炎球菌、病原性大腸菌などを例示することができる。Haemophilus influenzae 由来のPBP4のアミノ酸配列を配列番号2に示す。
【0025】
ペニシリン結合タンパク質は、例えば、Biochemistry, Vol. 45, No.3, pages 783-792に記載の方法により調製することができる。簡単に説明すると、細菌のゲノムDNAから成熟PBP4のコード領域をPCRでクローニングする。生成物を適当な制限酵素で消化し、プラスミドに連結する。このプラスミドを宿主細胞に導入する。形質転換細胞を増殖させた後、超音波で破砕し、細胞溶解物を遠心分離し、セファロースカラムにかけ、適当な塩勾配で溶出させる。結晶化するためには、さらに精製した後に濃縮する。結晶化は蒸気拡散法で行うとよい。結晶は、25%(v/v)グリセロール含有リザーバー溶液からなる抗凍結剤溶液に移し、液体窒素中でフラッシュ冷却してもよい。
【0026】
式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルとペニシリン結合タンパク質との複合体は、バッチ法、透析法、蒸気拡散法などの手法により結晶化することができる。例えば、PBPの結晶を、式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルで飽和した母液、0.1MHEPES pH 7.5、25%PEG6000、10% glycerolに2時間程度20℃で浸漬することにより、式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルとペニシリン結合タンパク質との複合体の結晶を作製することができる。
【0027】
式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルとペニシリン結合タンパク質との複合体の結晶は、晶系が単斜結晶であり、空間群がP21であるとよく、 結晶の単位格子が、a=64.5±1Å、b=92.5±1Å、c=104.9±1Å、α=90°、β=107.7°、γ=90°であるものが好ましい。
【0028】
本発明の複合体は、構造解析を行うことにより、抗生物質の反応機構の解明や新規抗生物質の設計に役立つ。
【実施例】
【0029】
以下、実施例に基づいて本発明を詳細に説明するが、本発明はこれらの実施例に限定されるものではない。
【0030】
〔製造例1〕
3,3-Dimethyl-7-oxo-6-[2-(2-oxo-imidazolidin-1-ylamino)-2-phenyl-acetylamino]-4-thia-1-aza-bicyclo[3.2.0]heptane-2-carboxylic acid(化合物18)の製造
【0031】
【化3】
工程1
【0032】
【化7】
2N硫酸3リットルに溶解した2-イミダゾリジノン126 gの溶液に、外部を冷やしながら13分かけて亜硝酸ナトリウム101 gを小分けにして添加し、内部温度を3℃-6℃に維持した。混合物を0℃で1.5時間攪拌した。1時間かけて20℃以下で亜鉛末(220 g)を添加した。過剰の亜鉛を濾過により除去し、pHが6に達するまで固体水酸化バリウムを濾液に添加した。白色固体を濾過により除去した。pHが7.9に達するまで水性(50%)水酸化ナトリウムを濾液に添加し、固形物を濾過により除去した。濾液を減圧で濃縮した。メタノールを添加し、沈殿塩を濾過により除去した。カラムクロマトグラフィーで精製した後、1-アミノ-2-イミダゾリジノン10 gを得た。
【0033】
工程2
【0034】
【化8】
アセトン(11 ml)に溶解した(+)-6-アミノペニシラン酸1.08 g(5 mmol)を冷却し(0℃)し、トリエチルアミン(0.7 ml)で処理した。30分間攪拌した後、混合物を0℃に維持しながら、アセトン(17.5 ml)に溶解したベンジルブロミド(0.6 ml)を分割して添加し、混合物を0℃で4時間以上攪拌した後、乾燥エーテル(150 ml)中に注いだ。沈殿物を濾過で除き、濾過物を炭酸水素ナトリウム、次いで水で洗浄した。p-トルエンスルホン酸(850 mg)を含有するアセトン(50 ml)で有機相を処理して、ベンジル6β-アミノペニシラナートトルエン-p-スルホナート塩の結晶沈殿物を得た。この塩を酢酸エチルでスラリー化し、st炭酸水素ナトリウム溶液とともに振盪した。有機相を分離し、乾燥し、蒸発させて、所望の化合物500 mgを油状物として得た。
【0035】
工程3
【0036】
【化9】
乾燥塩化メチレン20 mlに溶解したフェニールグリオキシル酸600 mgの溶液に、1-アミノ-2-イミダゾリジノン410 mgを添加し、室温で2時間攪拌し、混合物を0℃まで1時間冷却し、濾過後、ろ過ケーク700 mgを得た。これは次の工程に直接用いることができた。
【0037】
工程4
【0038】
【化10】
メタノール15 mlに溶解したHHPS060-1 700 mgの攪拌溶液に、シアノ水素化ほう素ナトリウム285 mg(1.5eq)を添加した。その後、溶液のpHを3-4に保ちながら、塩化水素で飽和したメタノールを0℃で滴下した。その後、溶液のpHを3-4に保ちながら、混合物を室温で2.5時間攪拌した。メタノールを蒸発させ、混合物を次の工程に直接用いた。
【0039】
工程5
【0040】
【化11】
15 mlのアセトンと3 mlの水に溶解したHHPS060-2の溶液に、K2CO3 616 mg(1.5 eq)を添加した。その後、0℃で、クロロ炭酸ベンジルエステル561 mg (1.1 eq)を滴下した。室温で4時間攪拌した。アセトンを蒸発させ、50 mlの酢酸エチルを添加し、1N HClでpH 2に酸性化した。有機相をNa2SO4で乾燥させ、カラムクロマトグラフィー(PE/EA=1/3)で精製した。化合物HHPS060-3 500 mgを得た。
【0041】
工程6
【0042】
【化12】
乾燥塩化メチレン15 mlに溶解したHHPS060-3 500 mgの溶液に、HATU 770 mg (1.5 eq)を添加し、室温で1時間攪拌した。さらに、HHPS060-SM2 415 mg (1eq)を添加した後、DIPEA 350 mg (2.0 eq)を滴下し、室温で4時間攪拌した。塩化メチレン35 mlを添加し、1N HCl (15 ml×3)、st NaHCO3 (15 ml×3)、st NaCl (15 ml×3)で洗浄した。有機相をNa2SO4で乾燥させ、カラムクロマトグラフィー(PE/EA=2/3)で精製した。化合物HHPS060-4 600 mgを得た。化合物HHPS060-4のLCMSとMNRをそれぞれ図5と6に示す。
【0043】
工程7
【0044】
【化13】
酢酸エチル20 mlに溶解したHHPS060-4 300 mgの溶液に、Pd/C 900 mgを添加し、室温で水和物ボール(hydrate ball)下にて7時間攪拌した。HPLCで精製した後、最終化合物HHPS060を得た。化合物HHPS060のLCMSとMNRをそれぞれ図7と8に示す。
【0045】
〔製造例2〕
Potassium 6-(2-ethoxy-propionylamino)-3,3-dimethyl-7-oxo-4-thia-1-aza-bicyclo[3.2.0]heptane
-2-carboxylate(化合物31)の製造
【0046】
【化4】
工程1
【0047】
【化14】
NaH (2.24 g, 94.3 mmol)を冷却THF (135 ml, 0℃)に添加した後、この溶液に1.5時間で乳酸エチル(10.42 g, 88.2 mmol)を滴下した。その後、EtI (16.50 g, 106 mmol)を添加し、反応混合物を室温まで温め、一晩攪拌した。飽和NH4Cl aq (150 ml)を添加することにより反応を停止させ、Et2O (75 ml×3)で抽出した。有機相を合わせ、鹹水で洗浄し、無水MgSO4で乾燥した。濾過及び溶媒除去の後、黄色の油状物が残った。この油状物を蒸留で精製して、所望の生成物6.9 gを得た(収率:53.6%)。
【0048】
工程2
【0049】
【化15】
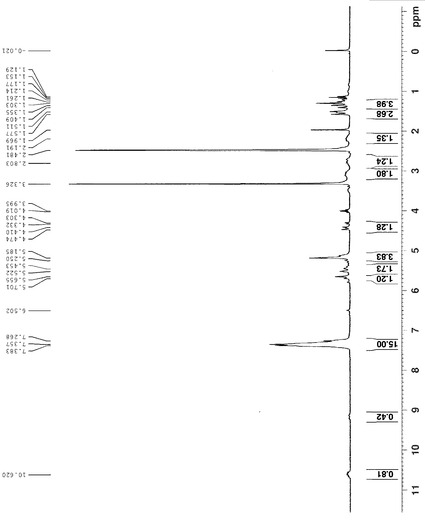
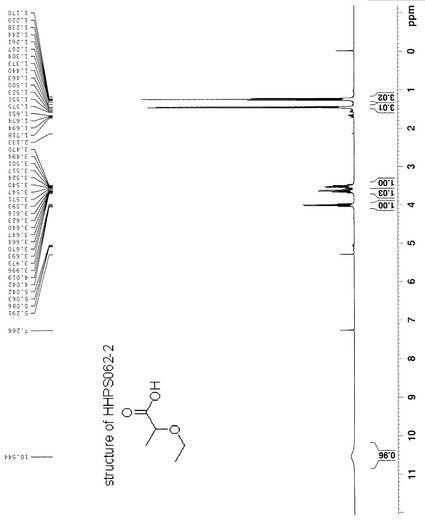
95%エタノール(50 ml)に溶解したHHPS062-1 (2.0 g, 13.7 mmol)とKOH (1.92 g, 34 mmol)を含有する溶液を5時間還流した。エタノールを減圧で除去し、黄色の油状物を得た。これを水(30 ml)中に入れ、EA (15 ml×3)で抽出した。水相を6N HCLでpH 1まで酸性化し、EA (15 ml×3)で抽出した。第2の抽出からの有機相を合わせ、鹹水で洗浄し、無水MgSO4で乾燥した。濾過及び溶媒除去の後、黄色の油状物が残った。これを蒸留で精製して、所望の生成物1.4 gを得た(収率:74%)。この化合物の構造を1H-NMRで確認した。化合物HHPS062-2の1H-NMRを図9に示す。
【0050】
工程3(J.C.S. Perkin 1975,562-567参照)
【0051】
【化16】
6-APA (6.0 g, 27.8 mmol)をアセトン30 mlに添加し、冷却し、TEA (4.2 g, 41.7 mmol)で30分間処理し、アセトン5 mlに溶解したBnBr (4.7 g, 27.8 mmol)の溶液を添加し、混合物を0℃で4時間攪拌した。その後、反応混合物を乾燥エーテルに注ぎ、得られた沈殿物を濾過し、濾液をNaHCO3 aqで洗浄した。TSOHを含有するアセトンで有機相を処理して、結晶沈殿物3.5 gを得た(収率:29.1%)。構造をLC-MSで確認した。
【0052】
工程4
【0053】
【化17】
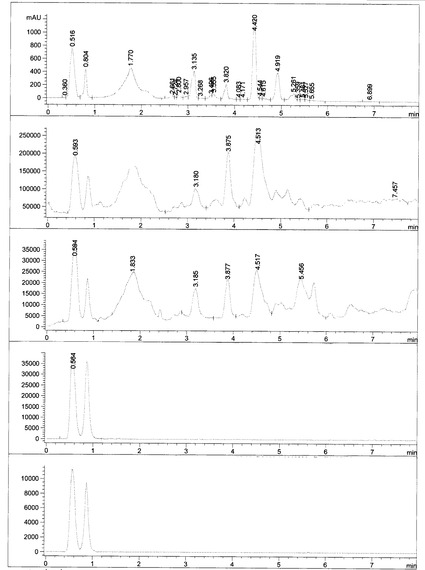
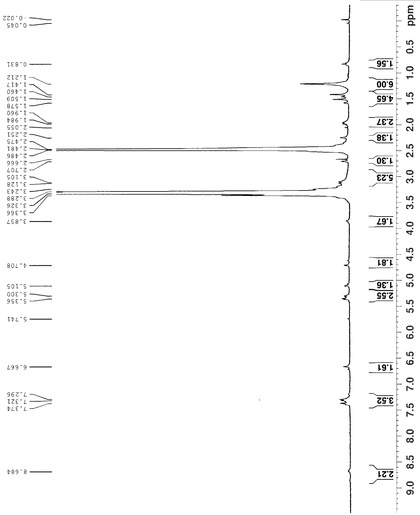
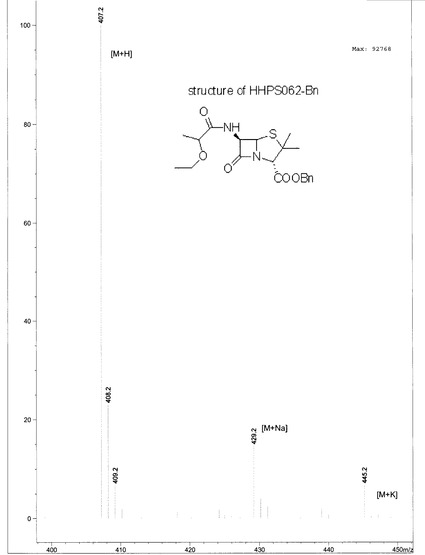
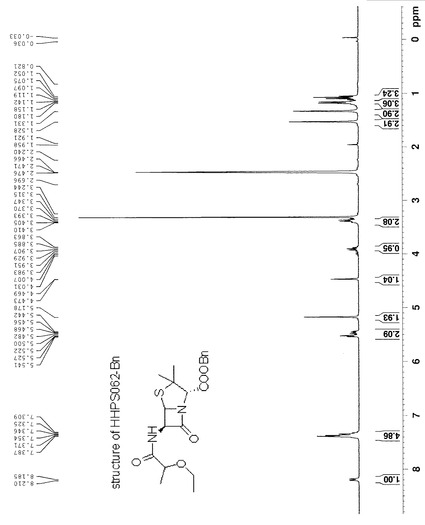
HHPS062-2 (118 mg, 1 mmol)をCH2Cl2 5mlに溶解した後、HATU (447 mg, 1.2 mmol)を添加し、懸濁液を得た。この懸濁液にDIPEA (430 mg, 3.3 mmol)を添加し、わずかに黄色の溶液を得た。同時に、6-APA-Bn (432 mg, 1mmol)をCH2Cl2 6mlに入れ、もう一つのDIPEA (430 mg, 3.3 mmol)を混合物に添加した後、透明な無色溶液になった。この溶液をHHPS062-2溶液にシリンジを通して添加し、反応溶液を3時間攪拌した。この溶液を最初に1N HCL (10 ml×3)、次に1N NaHCO3 (20 ml×2)で洗浄した後、有機相を無水MgSO4で乾燥し、溶媒を除去した後、残渣をシリカゲルカラムクロマトグラフィー(EA/PE=1/4)で精製し、生成物を無色油状物250 mgの形状で得た(収率:61.5%)。構造を1H-NMR及びLC-MSで確認した。化合物HHPS-062-BnのLS, MS及び1H-NMRをそれぞれ図10, 11及び12に示す。
【0054】
工程5(JOC 1990,55,5510-5517参照)
【0055】
【化18】
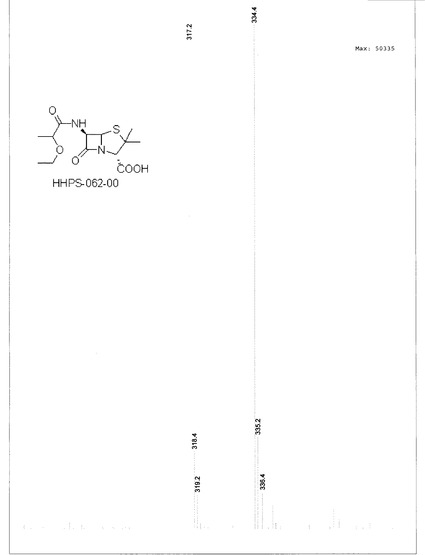
HHPS062-Bn (150 mg, 0.37 mmol)を精製EA 20mlに溶解した後、Pd/C (10%) 60 mgを添加し、反応フラスコにH2を充填し、混合物を周囲温度で6時間攪拌した。出発物質が完全に消費されたとき(TLCでモニター)、Pd/Cを濾過で除去し、溶媒を減圧留去で除去した。残渣をn-へキサン(10 ml×3)で洗浄し、得られた油状物をオイルポンプで乾燥し、所望の生成物を白色粉末70 mgの形状で得た(収率:61.8%)。構造を1H-NMR及びLC-MSで確認した(図13, 14, 15及び16)。
【0056】
〔実施例1〕抗菌活性の測定
Ampicillin、化合物18、化合物31及びAmoxillin(Amoxil, GlaxoSmithKline)の各化合物1mgを400ulの滅菌水に溶解した。大腸菌DH5αを撒いておいたLBプレートに、滅菌済み5mmろ紙を置き、そこに4ulの各化合物溶液をしみ込ませ、終夜37℃で培養した。結果を図3に示す。その結果、現在市販されている抗菌剤のAmpicillin、Amoxillin(Amoxil, GlaxoSmithKline)同様、化合物18及び化合物31においても、大腸菌DH5αに対する抗菌作用を確認する事が出来た。グラム陰性菌であるインフルエンザ菌への抗菌作用も期待できる。
【0057】
〔実施例2〕Haemophilus influenzae菌由来のペニシリン結合タンパク質4と化合物18又は31との複合体のX線構造解析
Haemophilus influenzae由来のPBP4をコードしている遺伝子領域(配列番号1)を発現プラスミド(pET28b ベクター(インビトロジェン))に導入し、このプラスミドをタンパク質発現宿主(BL21(DE3) / pLysS(インビトロジェン))に導入した。形質転換後の宿主細胞を37℃でO.D.600が0.5〜0.6に成るまで震盪培養し、1mM IPTGを添加し20℃終夜培養を行った。培養後、4000 rpmで集菌し、バッファー(0.1 M Tris pH8.5、0.1 M NaCl、1 mM PMSF)で懸濁し、超音波破砕をし、19,000 rpmで遠心分離を行い、上清を陰イオン交換カラムにかけ、塩濃度勾配で溶出させた。SDS-PAGEでPBP4を含むフラクションを確認し、それらをまとめ、陽イオン交換カラムかけ、塩濃度勾配で溶出した。
【0058】
SDS-PAGEでPBP4を含むフラクションを確認し、濃縮し、ゲル濾過カラムにかけた。最後に再度、陰イオン交換カラム(monoQ)にかけ、フロースルーを回収した。これを5mg/mlまで濃縮し、結晶化サンプルとした(10 mM Tris pH8.0、0.1 M NaCl)。
【0059】
結晶化方法:5mg/mlまで濃縮した結晶化サンプルと、母液(0.1 M HEPES pH7.5、15-20% (w/v) PEG600)を1:1の割合で混ぜ、ハンギングドロップ蒸気拡散法を用いて、一週間ほど20℃で静置した。
【0060】
複合体の調整法:上記の方法で作成した結晶のドロップに、結晶が出来たその母液で化合物を溶かし込んだ溶液を直接ドロップに0.1-0.3μl入れ、1時間ほど20℃で静置した。
【0061】
X線構造解析方法:高エネルギー加速器研究機構でイメージデータを、軸をたてて200枚ほど測定し、大腸菌のPBP4をテンプレートとしCCP4プログラムのMolrepを用いて分子置換法を行った。その結果、非対称単位内に2分子のHiPBP4を確認する事ができた。抗菌剤との複合体構造は、インフルエンザ菌由来PBP4のネイティブ構造をテンプレートとして分子置換を行った。
【0062】
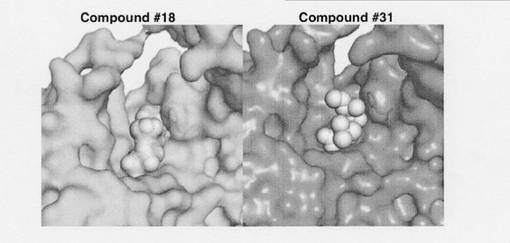
化合物とインフルエンザ菌由来PBP4との立体配座を図3に示す。インフルエンザ菌由来PBP4と抗菌剤との複合体形成時の位置関係を図4に示す。
【産業上の利用可能性】
【0063】
本発明の化合物は、抗菌剤として有用である。また、本発明の化合物とPBP4との複合体の結晶の構造解析を行うことにより、抗菌活性が高い化合物の設計が可能となる。
【図面の簡単な説明】
【0064】
【図1】ペニシリン結合タンパク質(PBP)の働きを示す模式図である。
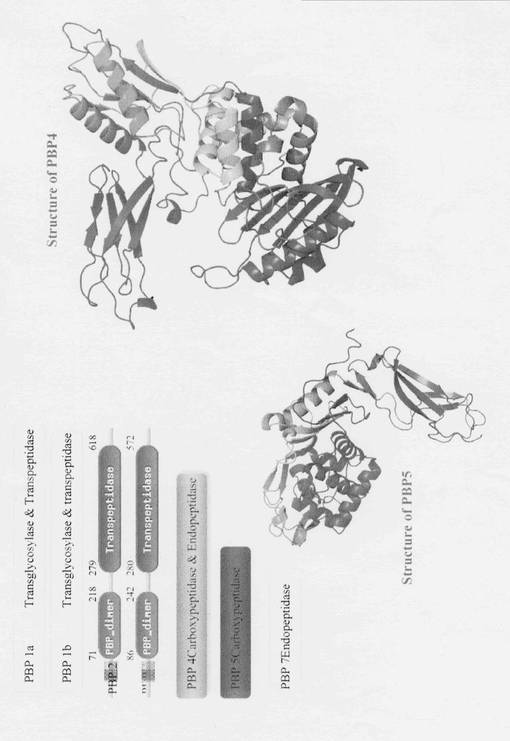
【図2】Haemophilus influenzae由来のPBPの機能とモデル構造を示す。
【図3】大腸菌DH5αを用いた、ディスク法(LBプレート)による抗菌作用テスト及び化合物とインフルエンザ菌由来PBP4との立体配座を示す。
【図4】インフルエンザ菌由来PBP4と抗菌剤との複合体形成時の位置関係を示す。
【図5】化合物HHPS060-4のLCMSを示す。
【図6】化合物HHPS060-4のMNRを示す。
【図7】化合物HHPS060のLCMS示す。
【図8】化合物HHPS060のMNRを示す。
【図9】化合物HHPS062-2の1H-NMRを示す。
【図10】化合物HHPS-062-BnのLSを示す。
【図11】化合物HHPS-062-BnのMSを示す。
【図12】化合物HHPS-062-Bnの1H-NMRを示す。
【図13】化合物HHPS-062-00のLSを示す。
【図14】化合物HHPS-062-00のMSを示す。
【図15】化合物HHPS-062-00のMSを示す。
【図16】化合物HHPS-062-00の1H-NMRを示す。
【配列表フリーテキスト】
【0065】
<配列番号1>
配列番号1は、Haemophilus influenzae 由来のPBP4をコードしている遺伝子領域のヌクレオチド配列を示す。
<配列番号2>
配列番号2は、Haemophilus influenzae 由来のPBP4のアミノ酸配列を示す。
【技術分野】
【0001】
本発明は、抗菌剤として有用な新規化合物に関する。
【背景技術】
【0002】
ペニシリン(Penicillin)の発見以来、セファロスポリンを含む数多くの合成β-ラクタム系抗生物質は多くの人々を様々な病気から救ってきた。しかし、現在、大量の抗生物質の使用により耐性菌が顕在化し、今まで有効であったβ-ラクタム系抗生物質が無効となる例が増加している。従来一つの抗生物質は20-30年くらい使用すると耐性菌が出現すると報告されている。このような問題を解決するために、新たな抗生物質の開発が望まれている。
【発明の開示】
【発明が解決しようとする課題】
【0003】
本発明は、新たな抗生物質を提供することを目的とする。
【課題を解決するための手段】
【0004】
ペニシリン結合タンパク質(penicillin binding protein; PBP)は、ペニシリンに代表されるβ−ラクタム系抗生物質と特異的に結合する。12種類のPBPが大腸菌で知られているが、このうち、PBP1, 2, 3, 4は、細菌の細胞表面のペプチドグリカン層の合成・分解・再構成を担っている(図1)。PBPの活性がペニシリン等の抗生物質によって阻害されると、細菌は死に至る。現在、国内で市販されているセフェム系抗生物質として、塩野義製薬株式会社から、フロモックス(商品名)が販売されており、国内では抗生剤として40%の市場率を示している。このフロモックスの化合物は、塩野義製薬株式会社が開発を行った抗生物質であるが、本発明者とその共同研究者らによって、フロモックスの化合物と大腸菌由来ペニシリン結合タンパク質4との相互作用の機構の解明が行われた(Biochemistry. 2006 Jan 24;45(3):783-92.)。今回、本発明者は、インフルエンザ菌(Haemophilus influrenzae)由来のペニシリン結合タンパク質(PBP4)の立体構造を解明し、その結果に基づいて、新たな抗生物質の設計・合成を行ったところ、2つの化合物について、PBP4との相互作用及び抗菌活性が認められた。本発明は、これらの知見に基づいて完成されたものである。
【0005】
本発明の要旨は以下の通りである。
(1)下記式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステル。
【0006】
【化1】
(式中、Rは下記の式 (b)又は(c)で表される基である)
【0007】
【化2】
(2)(1)記載の化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルを含む医薬組成物。
(3)抗菌剤である(2)記載の医薬組成物。
(4)(1)記載の化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルとペニシリン結合タンパク質との複合体。
(5)ペニシリン結合タンパク質がペニシリン結合タンパク質4である(4)記載の複合体。
(6)ペニシリン結合タンパク質4が病原菌に由来する(5)記載の複合体。
(7)病原菌がインフルエンザ菌である(6)記載の複合体。
(8)(4)〜(7)のいずれかに記載の複合体の結晶。
(9)結晶の晶系が単斜結晶であり、空間群がP21である(8)記載の結晶。
(10)結晶の単位格子が、a=64.5±1Å、b=92.5±1Å、c=104.9±1Å、α=90°、β=107.7°、γ=90°である(9)記載の結晶。
【発明の効果】
【0008】
本発明により新規化合物が提供された。これらの化合物は、抗生物質として有用である。
【発明を実施するための最良の形態】
【0009】
以下、本発明の実施の形態についてより詳細に説明する。
【0010】
本発明は、下記式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルを提供する。
【0011】
【化1】
(式中、Rは下記の式(b)又は(c)で表される基である)
【0012】
【化2】
式(I)で表される化合物は、以下の2つの化合物である。
【0013】
3,3-Dimethyl-7-oxo-6-[2-(2-oxo-imidazolidin-1-ylamino)-2-phenyl-acetylamino]-4-thia-1-aza-bicyclo[3.2.0]heptane-2-carboxylic acid(化合物18)
【0014】
【化5】
;及びPotassium 6-(2-ethoxy-propionylamino)-3,3-dimethyl-7-oxo-4-thia-1-aza-bicyclo[3.2.0]heptane-2-carboxylate(化合物31)
【0015】
【化6】
これらの化合物は、下記の合成スキームで製造することができる。
【0016】
(化合物18の合成スキーム)
【0017】
【化3】
(化合物31の合成スキーム)
【0018】
【化4】
式(I)で表される化合物の医薬的に許容される塩としては、ナトリウム、カリウム、リチウム等のアルカリ金属塩、カルシウム、マグネシウム等のアルカリ土類金属塩、シクロヘキシルアミン、トリメチルアミン、ジエタノールアミン等の有機アミン塩、アルギニン、リジン等の塩基性アミノ酸塩、アンモニウム塩などを例示することができるが、これらに限定されない。
【0019】
式(I)で表される化合物の医薬的に許容されるエステルは、ペニシリン系、セファロスポリン系抗生物質について慣用されているエステルであるとよく、C2−6アルカノイルオキシC1−6アルキルエステル(具体的には、アセトキシメチルエステル、1−アセトキシエチルエステル、1−アセトキシブチルエステル、2−アセトキシエチルエステル、プロピオニルオキシメチルエステル、ピバロイルオキシメチルエステルなど)、1−(C1−6アルコキシ)C1−6アルキルエステル(具体的には、メトキシメチルエステル、エトキシメチルエステル、イソプロポキシメチルエステル、1−メトキシエチルエステル、1−エトキシエチルエステルなど)、1−(C1−6アルキルチオ)C1−6アルキルエステル(具体的には、メチルチオメチルエステル、エチルチオメチルエステルなど)などを例示することができるが、これらに限定されない。
【0020】
式(I)で表される化合物の分子内に不斉炭炭素が存在する場合は、ラセミ体および光学活性体も包含される。
【0021】
また、式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルは、様々な溶媒和物、例えば水、メタノール、エタノール、ジメチルホルムアミド、酢酸エチルなどとの溶媒和物としても存在でき、これらの溶媒和物も本発明に包含される。
【0022】
式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルは抗菌活性を有するので、病原性細菌により生じる種々の疾患(例えば、感染症など)の予防及び/又は治療のために使用できる。抗菌剤として式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルを使用する場合に対象となる菌は、式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルが抗菌活性を示す限り特に限定されず、グラム陽性菌又はグラム陰性菌のいずれであってもよいが、特に、Haemophilus Influenzae、病原性大腸菌などの菌に対して効果的である。式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルは、単独で使用してもよいし、他の抗菌剤と組合わせて使用してもよい。
【0023】
式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルは、常法により製剤化した医薬製剤(例えば、注射剤、カプセル剤、錠剤、散剤、顆粒剤など)として、ヒト又は動物に投与することができる。例えば、有効成分の量に換算して、1日あたり約0.1〜1000 mg/kg(体重)、好ましくは1日あたり約 1〜 500 mg/kg(体重)の投与量で、1回または数回に分けて経口投与するとよいが、その投与量や投与回数は、症状、年齢、投与方法などにより適宜変更しうる。注射剤に製剤化する場合には、蒸留水、生理食塩水などの担体を用いるとよく、カプセル剤、錠剤、散剤、顆粒剤に製剤化する場合には、デンプン、乳糖、白糖、炭酸カルシウムなどの賦形剤、デンプンのり液、アラビアゴム、ゼラチン、アルギン酸ナトリウム、カルボキシメチルセルロース、ヒドロキシプロピルセルロースなどの結合剤、ステアリン酸マグネシウム、タルクなどの滑沢剤など、デンプン、寒天、結晶セルロース、炭酸カルシウム、炭酸水素ナトリウム、アルギン酸ナトリウムなどの崩壊剤などを用いるとよい。製剤中の有効成分の含有率は、1〜99重量%の間で変動させることができる。例えば、錠剤、カプセル剤、顆粒剤、散剤などの形態をとる場合には、有効成分を5〜80重量%含有させるのが好ましく、注射剤の場合には、有効成分を1〜10重量%含有させるのが好ましい。
【0024】
本発明は、式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルとペニシリン結合タンパク質との複合体も提供する。複数種類のペニシリン結合タンパク質が知られており、例えば、大腸菌では12種類のペニシリン結合タンパク質が知られている(J. Bacteriol. 181, 3981-93 (1993))。ペニシリン結合タンパク質としては、ペニシリン結合タンパク質4(PBP4)が好ましい。ペニシリン結合タンパク質は、病原菌由来のものであるとよく、病原菌としては、インフルエンザ菌、肺炎球菌、病原性大腸菌などを例示することができる。Haemophilus influenzae 由来のPBP4のアミノ酸配列を配列番号2に示す。
【0025】
ペニシリン結合タンパク質は、例えば、Biochemistry, Vol. 45, No.3, pages 783-792に記載の方法により調製することができる。簡単に説明すると、細菌のゲノムDNAから成熟PBP4のコード領域をPCRでクローニングする。生成物を適当な制限酵素で消化し、プラスミドに連結する。このプラスミドを宿主細胞に導入する。形質転換細胞を増殖させた後、超音波で破砕し、細胞溶解物を遠心分離し、セファロースカラムにかけ、適当な塩勾配で溶出させる。結晶化するためには、さらに精製した後に濃縮する。結晶化は蒸気拡散法で行うとよい。結晶は、25%(v/v)グリセロール含有リザーバー溶液からなる抗凍結剤溶液に移し、液体窒素中でフラッシュ冷却してもよい。
【0026】
式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルとペニシリン結合タンパク質との複合体は、バッチ法、透析法、蒸気拡散法などの手法により結晶化することができる。例えば、PBPの結晶を、式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルで飽和した母液、0.1MHEPES pH 7.5、25%PEG6000、10% glycerolに2時間程度20℃で浸漬することにより、式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルとペニシリン結合タンパク質との複合体の結晶を作製することができる。
【0027】
式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルとペニシリン結合タンパク質との複合体の結晶は、晶系が単斜結晶であり、空間群がP21であるとよく、 結晶の単位格子が、a=64.5±1Å、b=92.5±1Å、c=104.9±1Å、α=90°、β=107.7°、γ=90°であるものが好ましい。
【0028】
本発明の複合体は、構造解析を行うことにより、抗生物質の反応機構の解明や新規抗生物質の設計に役立つ。
【実施例】
【0029】
以下、実施例に基づいて本発明を詳細に説明するが、本発明はこれらの実施例に限定されるものではない。
【0030】
〔製造例1〕
3,3-Dimethyl-7-oxo-6-[2-(2-oxo-imidazolidin-1-ylamino)-2-phenyl-acetylamino]-4-thia-1-aza-bicyclo[3.2.0]heptane-2-carboxylic acid(化合物18)の製造
【0031】
【化3】
工程1
【0032】
【化7】
2N硫酸3リットルに溶解した2-イミダゾリジノン126 gの溶液に、外部を冷やしながら13分かけて亜硝酸ナトリウム101 gを小分けにして添加し、内部温度を3℃-6℃に維持した。混合物を0℃で1.5時間攪拌した。1時間かけて20℃以下で亜鉛末(220 g)を添加した。過剰の亜鉛を濾過により除去し、pHが6に達するまで固体水酸化バリウムを濾液に添加した。白色固体を濾過により除去した。pHが7.9に達するまで水性(50%)水酸化ナトリウムを濾液に添加し、固形物を濾過により除去した。濾液を減圧で濃縮した。メタノールを添加し、沈殿塩を濾過により除去した。カラムクロマトグラフィーで精製した後、1-アミノ-2-イミダゾリジノン10 gを得た。
【0033】
工程2
【0034】
【化8】
アセトン(11 ml)に溶解した(+)-6-アミノペニシラン酸1.08 g(5 mmol)を冷却し(0℃)し、トリエチルアミン(0.7 ml)で処理した。30分間攪拌した後、混合物を0℃に維持しながら、アセトン(17.5 ml)に溶解したベンジルブロミド(0.6 ml)を分割して添加し、混合物を0℃で4時間以上攪拌した後、乾燥エーテル(150 ml)中に注いだ。沈殿物を濾過で除き、濾過物を炭酸水素ナトリウム、次いで水で洗浄した。p-トルエンスルホン酸(850 mg)を含有するアセトン(50 ml)で有機相を処理して、ベンジル6β-アミノペニシラナートトルエン-p-スルホナート塩の結晶沈殿物を得た。この塩を酢酸エチルでスラリー化し、st炭酸水素ナトリウム溶液とともに振盪した。有機相を分離し、乾燥し、蒸発させて、所望の化合物500 mgを油状物として得た。
【0035】
工程3
【0036】
【化9】
乾燥塩化メチレン20 mlに溶解したフェニールグリオキシル酸600 mgの溶液に、1-アミノ-2-イミダゾリジノン410 mgを添加し、室温で2時間攪拌し、混合物を0℃まで1時間冷却し、濾過後、ろ過ケーク700 mgを得た。これは次の工程に直接用いることができた。
【0037】
工程4
【0038】
【化10】
メタノール15 mlに溶解したHHPS060-1 700 mgの攪拌溶液に、シアノ水素化ほう素ナトリウム285 mg(1.5eq)を添加した。その後、溶液のpHを3-4に保ちながら、塩化水素で飽和したメタノールを0℃で滴下した。その後、溶液のpHを3-4に保ちながら、混合物を室温で2.5時間攪拌した。メタノールを蒸発させ、混合物を次の工程に直接用いた。
【0039】
工程5
【0040】
【化11】
15 mlのアセトンと3 mlの水に溶解したHHPS060-2の溶液に、K2CO3 616 mg(1.5 eq)を添加した。その後、0℃で、クロロ炭酸ベンジルエステル561 mg (1.1 eq)を滴下した。室温で4時間攪拌した。アセトンを蒸発させ、50 mlの酢酸エチルを添加し、1N HClでpH 2に酸性化した。有機相をNa2SO4で乾燥させ、カラムクロマトグラフィー(PE/EA=1/3)で精製した。化合物HHPS060-3 500 mgを得た。
【0041】
工程6
【0042】
【化12】
乾燥塩化メチレン15 mlに溶解したHHPS060-3 500 mgの溶液に、HATU 770 mg (1.5 eq)を添加し、室温で1時間攪拌した。さらに、HHPS060-SM2 415 mg (1eq)を添加した後、DIPEA 350 mg (2.0 eq)を滴下し、室温で4時間攪拌した。塩化メチレン35 mlを添加し、1N HCl (15 ml×3)、st NaHCO3 (15 ml×3)、st NaCl (15 ml×3)で洗浄した。有機相をNa2SO4で乾燥させ、カラムクロマトグラフィー(PE/EA=2/3)で精製した。化合物HHPS060-4 600 mgを得た。化合物HHPS060-4のLCMSとMNRをそれぞれ図5と6に示す。
【0043】
工程7
【0044】
【化13】
酢酸エチル20 mlに溶解したHHPS060-4 300 mgの溶液に、Pd/C 900 mgを添加し、室温で水和物ボール(hydrate ball)下にて7時間攪拌した。HPLCで精製した後、最終化合物HHPS060を得た。化合物HHPS060のLCMSとMNRをそれぞれ図7と8に示す。
【0045】
〔製造例2〕
Potassium 6-(2-ethoxy-propionylamino)-3,3-dimethyl-7-oxo-4-thia-1-aza-bicyclo[3.2.0]heptane
-2-carboxylate(化合物31)の製造
【0046】
【化4】
工程1
【0047】
【化14】
NaH (2.24 g, 94.3 mmol)を冷却THF (135 ml, 0℃)に添加した後、この溶液に1.5時間で乳酸エチル(10.42 g, 88.2 mmol)を滴下した。その後、EtI (16.50 g, 106 mmol)を添加し、反応混合物を室温まで温め、一晩攪拌した。飽和NH4Cl aq (150 ml)を添加することにより反応を停止させ、Et2O (75 ml×3)で抽出した。有機相を合わせ、鹹水で洗浄し、無水MgSO4で乾燥した。濾過及び溶媒除去の後、黄色の油状物が残った。この油状物を蒸留で精製して、所望の生成物6.9 gを得た(収率:53.6%)。
【0048】
工程2
【0049】
【化15】
95%エタノール(50 ml)に溶解したHHPS062-1 (2.0 g, 13.7 mmol)とKOH (1.92 g, 34 mmol)を含有する溶液を5時間還流した。エタノールを減圧で除去し、黄色の油状物を得た。これを水(30 ml)中に入れ、EA (15 ml×3)で抽出した。水相を6N HCLでpH 1まで酸性化し、EA (15 ml×3)で抽出した。第2の抽出からの有機相を合わせ、鹹水で洗浄し、無水MgSO4で乾燥した。濾過及び溶媒除去の後、黄色の油状物が残った。これを蒸留で精製して、所望の生成物1.4 gを得た(収率:74%)。この化合物の構造を1H-NMRで確認した。化合物HHPS062-2の1H-NMRを図9に示す。
【0050】
工程3(J.C.S. Perkin 1975,562-567参照)
【0051】
【化16】
6-APA (6.0 g, 27.8 mmol)をアセトン30 mlに添加し、冷却し、TEA (4.2 g, 41.7 mmol)で30分間処理し、アセトン5 mlに溶解したBnBr (4.7 g, 27.8 mmol)の溶液を添加し、混合物を0℃で4時間攪拌した。その後、反応混合物を乾燥エーテルに注ぎ、得られた沈殿物を濾過し、濾液をNaHCO3 aqで洗浄した。TSOHを含有するアセトンで有機相を処理して、結晶沈殿物3.5 gを得た(収率:29.1%)。構造をLC-MSで確認した。
【0052】
工程4
【0053】
【化17】
HHPS062-2 (118 mg, 1 mmol)をCH2Cl2 5mlに溶解した後、HATU (447 mg, 1.2 mmol)を添加し、懸濁液を得た。この懸濁液にDIPEA (430 mg, 3.3 mmol)を添加し、わずかに黄色の溶液を得た。同時に、6-APA-Bn (432 mg, 1mmol)をCH2Cl2 6mlに入れ、もう一つのDIPEA (430 mg, 3.3 mmol)を混合物に添加した後、透明な無色溶液になった。この溶液をHHPS062-2溶液にシリンジを通して添加し、反応溶液を3時間攪拌した。この溶液を最初に1N HCL (10 ml×3)、次に1N NaHCO3 (20 ml×2)で洗浄した後、有機相を無水MgSO4で乾燥し、溶媒を除去した後、残渣をシリカゲルカラムクロマトグラフィー(EA/PE=1/4)で精製し、生成物を無色油状物250 mgの形状で得た(収率:61.5%)。構造を1H-NMR及びLC-MSで確認した。化合物HHPS-062-BnのLS, MS及び1H-NMRをそれぞれ図10, 11及び12に示す。
【0054】
工程5(JOC 1990,55,5510-5517参照)
【0055】
【化18】
HHPS062-Bn (150 mg, 0.37 mmol)を精製EA 20mlに溶解した後、Pd/C (10%) 60 mgを添加し、反応フラスコにH2を充填し、混合物を周囲温度で6時間攪拌した。出発物質が完全に消費されたとき(TLCでモニター)、Pd/Cを濾過で除去し、溶媒を減圧留去で除去した。残渣をn-へキサン(10 ml×3)で洗浄し、得られた油状物をオイルポンプで乾燥し、所望の生成物を白色粉末70 mgの形状で得た(収率:61.8%)。構造を1H-NMR及びLC-MSで確認した(図13, 14, 15及び16)。
【0056】
〔実施例1〕抗菌活性の測定
Ampicillin、化合物18、化合物31及びAmoxillin(Amoxil, GlaxoSmithKline)の各化合物1mgを400ulの滅菌水に溶解した。大腸菌DH5αを撒いておいたLBプレートに、滅菌済み5mmろ紙を置き、そこに4ulの各化合物溶液をしみ込ませ、終夜37℃で培養した。結果を図3に示す。その結果、現在市販されている抗菌剤のAmpicillin、Amoxillin(Amoxil, GlaxoSmithKline)同様、化合物18及び化合物31においても、大腸菌DH5αに対する抗菌作用を確認する事が出来た。グラム陰性菌であるインフルエンザ菌への抗菌作用も期待できる。
【0057】
〔実施例2〕Haemophilus influenzae菌由来のペニシリン結合タンパク質4と化合物18又は31との複合体のX線構造解析
Haemophilus influenzae由来のPBP4をコードしている遺伝子領域(配列番号1)を発現プラスミド(pET28b ベクター(インビトロジェン))に導入し、このプラスミドをタンパク質発現宿主(BL21(DE3) / pLysS(インビトロジェン))に導入した。形質転換後の宿主細胞を37℃でO.D.600が0.5〜0.6に成るまで震盪培養し、1mM IPTGを添加し20℃終夜培養を行った。培養後、4000 rpmで集菌し、バッファー(0.1 M Tris pH8.5、0.1 M NaCl、1 mM PMSF)で懸濁し、超音波破砕をし、19,000 rpmで遠心分離を行い、上清を陰イオン交換カラムにかけ、塩濃度勾配で溶出させた。SDS-PAGEでPBP4を含むフラクションを確認し、それらをまとめ、陽イオン交換カラムかけ、塩濃度勾配で溶出した。
【0058】
SDS-PAGEでPBP4を含むフラクションを確認し、濃縮し、ゲル濾過カラムにかけた。最後に再度、陰イオン交換カラム(monoQ)にかけ、フロースルーを回収した。これを5mg/mlまで濃縮し、結晶化サンプルとした(10 mM Tris pH8.0、0.1 M NaCl)。
【0059】
結晶化方法:5mg/mlまで濃縮した結晶化サンプルと、母液(0.1 M HEPES pH7.5、15-20% (w/v) PEG600)を1:1の割合で混ぜ、ハンギングドロップ蒸気拡散法を用いて、一週間ほど20℃で静置した。
【0060】
複合体の調整法:上記の方法で作成した結晶のドロップに、結晶が出来たその母液で化合物を溶かし込んだ溶液を直接ドロップに0.1-0.3μl入れ、1時間ほど20℃で静置した。
【0061】
X線構造解析方法:高エネルギー加速器研究機構でイメージデータを、軸をたてて200枚ほど測定し、大腸菌のPBP4をテンプレートとしCCP4プログラムのMolrepを用いて分子置換法を行った。その結果、非対称単位内に2分子のHiPBP4を確認する事ができた。抗菌剤との複合体構造は、インフルエンザ菌由来PBP4のネイティブ構造をテンプレートとして分子置換を行った。
【0062】
化合物とインフルエンザ菌由来PBP4との立体配座を図3に示す。インフルエンザ菌由来PBP4と抗菌剤との複合体形成時の位置関係を図4に示す。
【産業上の利用可能性】
【0063】
本発明の化合物は、抗菌剤として有用である。また、本発明の化合物とPBP4との複合体の結晶の構造解析を行うことにより、抗菌活性が高い化合物の設計が可能となる。
【図面の簡単な説明】
【0064】
【図1】ペニシリン結合タンパク質(PBP)の働きを示す模式図である。
【図2】Haemophilus influenzae由来のPBPの機能とモデル構造を示す。
【図3】大腸菌DH5αを用いた、ディスク法(LBプレート)による抗菌作用テスト及び化合物とインフルエンザ菌由来PBP4との立体配座を示す。
【図4】インフルエンザ菌由来PBP4と抗菌剤との複合体形成時の位置関係を示す。
【図5】化合物HHPS060-4のLCMSを示す。
【図6】化合物HHPS060-4のMNRを示す。
【図7】化合物HHPS060のLCMS示す。
【図8】化合物HHPS060のMNRを示す。
【図9】化合物HHPS062-2の1H-NMRを示す。
【図10】化合物HHPS-062-BnのLSを示す。
【図11】化合物HHPS-062-BnのMSを示す。
【図12】化合物HHPS-062-Bnの1H-NMRを示す。
【図13】化合物HHPS-062-00のLSを示す。
【図14】化合物HHPS-062-00のMSを示す。
【図15】化合物HHPS-062-00のMSを示す。
【図16】化合物HHPS-062-00の1H-NMRを示す。
【配列表フリーテキスト】
【0065】
<配列番号1>
配列番号1は、Haemophilus influenzae 由来のPBP4をコードしている遺伝子領域のヌクレオチド配列を示す。
<配列番号2>
配列番号2は、Haemophilus influenzae 由来のPBP4のアミノ酸配列を示す。
【特許請求の範囲】
【請求項1】
下記式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステル。
【化1】
(式中、Rは下記の式(b)又は(c)で表される基である)
【化2】
【請求項2】
請求項1記載の化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルを含む医薬組成物。
【請求項3】
抗菌剤である請求項2記載の医薬組成物。
【請求項4】
請求項1記載の化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルとペニシリン結合タンパク質との複合体。
【請求項5】
ペニシリン結合タンパク質がペニシリン結合タンパク質4である請求項4記載の複合体。
【請求項6】
ペニシリン結合タンパク質4が病原菌に由来する請求項5記載の複合体。
【請求項7】
病原菌がインフルエンザ菌である請求項6記載の複合体。
【請求項8】
請求項4〜7のいずれかに記載の複合体の結晶。
【請求項9】
結晶の晶系が単斜結晶であり、空間群がP21である請求項8記載の結晶。
【請求項10】
結晶の単位格子が、a=64.5オングストローム、b=92.5オングストローム、c=104.9オングストローム、α=90°、β=107.7°、γ=90°である請求項9記載の結晶。
【請求項1】
下記式(I)で表される化合物、その医薬的に許容される塩又はその医薬的に許容されるエステル。
【化1】
(式中、Rは下記の式(b)又は(c)で表される基である)
【化2】
【請求項2】
請求項1記載の化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルを含む医薬組成物。
【請求項3】
抗菌剤である請求項2記載の医薬組成物。
【請求項4】
請求項1記載の化合物、その医薬的に許容される塩又はその医薬的に許容されるエステルとペニシリン結合タンパク質との複合体。
【請求項5】
ペニシリン結合タンパク質がペニシリン結合タンパク質4である請求項4記載の複合体。
【請求項6】
ペニシリン結合タンパク質4が病原菌に由来する請求項5記載の複合体。
【請求項7】
病原菌がインフルエンザ菌である請求項6記載の複合体。
【請求項8】
請求項4〜7のいずれかに記載の複合体の結晶。
【請求項9】
結晶の晶系が単斜結晶であり、空間群がP21である請求項8記載の結晶。
【請求項10】
結晶の単位格子が、a=64.5オングストローム、b=92.5オングストローム、c=104.9オングストローム、α=90°、β=107.7°、γ=90°である請求項9記載の結晶。
【図5】

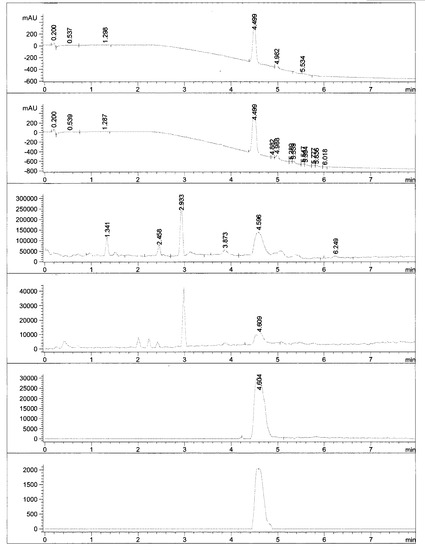
【図6】
【図7】
【図8】
【図9】
【図10】

【図11】
【図12】
【図13】

【図14】
【図15】
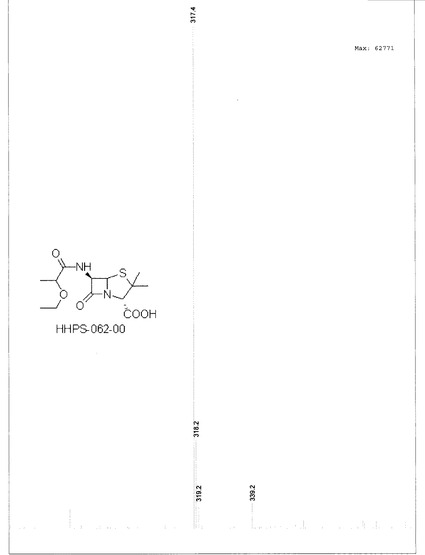
【図16】
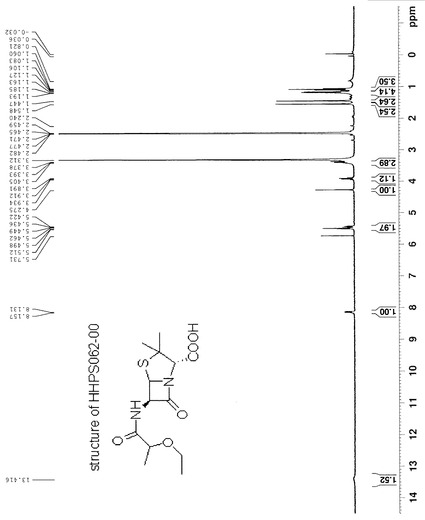
【図1】
【図2】
【図3】
【図4】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図1】
【図2】
【図3】
【図4】
【公開番号】特開2010−47520(P2010−47520A)
【公開日】平成22年3月4日(2010.3.4)
【国際特許分類】
【出願番号】特願2008−213424(P2008−213424)
【出願日】平成20年8月22日(2008.8.22)
【公序良俗違反の表示】
特許法第64条第2項第4号の規定により図面の一部または全部を不掲載とする。
【出願人】(505155528)公立大学法人横浜市立大学 (101)
【Fターム(参考)】
【公開日】平成22年3月4日(2010.3.4)
【国際特許分類】
【出願日】平成20年8月22日(2008.8.22)
【公序良俗違反の表示】
特許法第64条第2項第4号の規定により図面の一部または全部を不掲載とする。
【出願人】(505155528)公立大学法人横浜市立大学 (101)
【Fターム(参考)】
[ Back to top ]