
抗菌性ポリマー製品、その製造方法および使用方法
本発明は、抗菌性ポリマー製品であって、ポリマー全体に分散され、かつ、約200ナノメートル未満の粒子サイズを有する金属塩粒子を含有する、抗菌性ポリマー製品と、それらを製造する方法と、に関するものである。

【発明の詳細な説明】
【開示の内容】
【0001】
〔発明の分野〕
本発明は、抗菌性ポリマー製品、その製造方法および使用方法に関するものである。
【0002】
〔発明の背景〕
抗菌特性を有する材料は、多くの適用で使用されてきた。カテーテル、補綴具、インプラント、眼用装置等の医療装置の場合に、装置表面への細菌感染は、重篤感染症および装置故障をもたらすことがある。また、表面に集中した感染症は、食品の腐敗、食物が原因となる疾病の蔓延、および、材料の生物付着にも関係している。したがって、健康器具および生物医学装置、食品、および、個人衛生に関する各産業で利用する抗菌材料を進展させることに、重大な関心がある。
【0003】
銀塩類は、術後感染、歯科、創傷治療および医療装置の消毒剤として、ヒトのヘルスケアおよび医薬品に使用される長い歴史を有している。硝酸銀は、新生児眼炎(ophthalmic neonatorum)の予防に使用されてきた。コロイド銀は、1800年代に導入され、医療用の硝酸銀の代替物質として、1930年代以前に広く使用された。
【0004】
最近になって、銀化合物は、可溶性および不溶性の塩類、ポリマー類とゼオライトとを結合した錯体、金属銀および酸化銀等の種々の形態で、医療装置に添加されてきた。しかし、当該銀化合物の多くがポリマー組成物中に組み込まれている場合に、当該ポリマー組成物は、非常に漠然とした一貫性のない銀の充填、製造方法の複雑化、不都合な銀の急速放出、あるいは、有効性の欠如を含む複数の欠陥を被る。
【0005】
ポリマーマトリクス中への銀の導入に関する幾つかの方法が、開示されてきており、当該方法は、銀錯化合物の還元または合成等の化学的精密検査(chemical workups)、予備形成された銀粒子をポリマーに混合させること、あるいは、スパッタリング法およびプラズマ堆積法等の複雑な物理的方法を含む。これらの方法は、複雑であり、ポリマー材料中への銀化合物の一貫した充填を与えるとは限らない。銀塩等の微量金属塩類(oligodynamic metal salts)をコロイド金属塩粒子として医療装置に導入する方法が、開示されてきた。しかし、光重合法によって形成された装置への当該塩類の導入方法、および、還元剤を含む反応混合物中への当該塩類の導入方法は、開示されていなかった。
【0006】
1950年代以降、視力を改善するためのコンタクトレンズは、商業的に使用されるようになってきた。最初のコンタクトレンズは、硬質材料で形成された。当該コンタクトレンズは、起床中の数時間、患者によって使用され、かつ、洗浄のために取り外された。このコンタクトレンズの分野における現在の開発は、洗浄のために取り外さずに、数日以上の連続装用が可能なソフトコンタクトレンズを生み出した。多くの患者が当該ソフトコンタクトレンズを快適性の増大から好むが、当該ソフトコンタクトレンズはユーザーに多少の有害反応を生じさせることがある。当該ソフトコンタクトレンズの長期使用は、ソフトコンタクトレンズ表面上での細菌または他の微生物、特に緑膿菌(Pseudomonas aeruginosa)の増大を促進することになる。細菌および他の微生物の増大は、コンタクトレンズによる急性結膜充血(contact lens acute red eye)およびこの同類の症状等、有害な副作用を生じさせることになる。細菌および他の微生物の問題は、十中八九、ソフトコンタクトレンズの長期使用に関連しているが、細菌および他の微生物の増大は、ハードコンタクトレンズ装用者にも同様に生じる。
【0007】
したがって、依然として、眼用装置の表面上での細菌または他の微生物の増殖、および/あるいは、当該表面への細菌または他の微生物の付着を抑制するコンタクトレンズ等の眼用装置を製造する必要がある。さらに、コンタクトレンズの表面上での細菌または他の微生物の増殖、および/あるいは、当該表面への細菌または他の微生物の付着を促進しないコンタクトレンズ等の眼用装置を製造する必要がある。また、依然として、細菌または他の微生物の増殖に関係した有害反応を抑制するコンタクトレンズを製造する必要がある。
【0008】
〔発明の概要〕
一つの実施の形態において、本発明は、少なくとも一つのポリマーから形成された製品に関するものであり、当該ポリマーは、当該ポリマー全体に均質に分散され、かつ、約200ナノメートル未満の粒子サイズを有する抗菌性の金属塩粒子を含み、当該製品は、緑膿菌(Pseudomonas aeruginosa)および黄色ブドウ球菌(s. aureus)のうち、少なくとも一方において少なくとも約0.5の対数減少値を呈し、かつ、約70マイクロメートルの厚さで、CSIレンズと比較して約100%未満のヘイズ値(haze value)を呈するものである。
【0009】
他の実施の形態において、本発明は、
(a)少なくとも一つの塩前駆体を、任意に反応性ポリマー混合物の少なくとも一つの成分と共に、溶媒中に溶解させて、塩前駆体の混合物を形成するステップ;
(b)少なくとも一つの金属剤および少なくとも一つの分散剤を、任意に少なくとも一つの反応性成分と共に、溶媒中に溶解させることによって、分散剤・金属剤の錯体を形成して、金属剤混合物を形成するステップであって、当該複数の溶媒および成分は同一または異なるものであってもよい、ステップ;
(c)少なくとも一つの抗菌性金属塩:[Mq+]a[Xz-]bを含む粒子含有混合物を形成するための粒子形成条件下で、当該塩前駆体の混合物および当該金属剤混合物を混合するステップ;
(d)ステップ(a)および(b)に反応性成分が含まれていない場合において、このステップ(d)で少なくとも一つの反応性成分が添加されることを条件として、任意に、追加の反応性成分を当該粒子含有混合物に混合して、粒子含有反応性混合物を形成するステップ;ならびに、
ステップ(c)で抗菌性ポリマー製品中にMq+として添加された当該金属剤由来のMの少なくとも約90%を維持する上で十分な反応条件下で、当該ポリマー製品を形成するために、当該粒子含有反応性混合物を反応させるステップ、
を含む方法に関するものである。
【0010】
さらに他の実施の形態において、本発明は、約200ナノメートル以下の粒子サイズを有する安定化抗菌性金属塩粒子と少なくとも一つのフリーラジカル反応性成分とを含む反応性混合物を、当該金属塩粒子に応じて調整された臨界波長を超える波長の光、熱、あるいは、これらの組み合わせを用いて、硬化させて、抗菌性金属塩粒子を含む製品を形成することを含む方法に関するものである。
【0011】
〔発明の詳細な記述〕
本発明は、緑膿菌(Pseudomonas aeruginosa)、黄色ブドウ球菌(Staphyloccus aureus)のうち、少なくとも一方、あるいは、両方において少なくとも約0.5の対数減少値、および約100%未満のヘイズ値(haze value)を呈する抗菌性の製品であって、当該製品が作られる少なくとも一つのポリマー全体に、均質に分散した約200ナノメートル未満の粒子サイズを有する抗菌性金属塩粒子を含むか、当該粒子から本質的に成るか、あるいは、当該粒子で構成される抗菌性の製品から成るものである。一部の実施の形態において、粒子サイズは、約100ナノメートル未満であり、他の実施の形態においては、約50ナノメートル未満である。当該製品中の抗菌性金属塩粒子の粒子サイズは、走査型電子顕微鏡によって測定されてもよい。
【0012】
この明細書で使用されているように、用語「抗菌性」とは、製品が以下の特性:製品への細菌または他の微生物の付着の抑制、製品上での細菌または他の微生物の増殖の抑制、および、製品の表面上または製品の周辺領域内での細菌または他の微生物に対する殺菌のうち、一つ以上の特性を呈することを意味する。本発明のために、製品への細菌または他の微生物の付着、製品上での細菌または他の微生物の増殖、および、製品の表面上の細菌または他の微生物の存在は、全体的に「微生物のコロニー形成」と呼ばれる。好適には、本発明の製品は、生存している細菌または他の微生物において少なくとも約0.25の対数減少値を呈するものであり、一部の実施の形態においては少なくとも約0.5の対数減少値を呈するものであり、一部の実施の形態においては少なくとも約1.0の対数減少値を(90%以上の抑制効果)呈するものである。このような細菌または他の微生物は、緑膿菌(Pseudomonas aeruginosa)、アカントアメーバ属(Acanthamoeba species)、黄色ブドウ球菌(Staphyoccus aureus)、大腸菌(E. coli)、表皮ブドウ球菌(Staphyoccus epidermidis)、および、霊菌(Serratia marcesens)を含むが、これらに限定されるものではない。
【0013】
フリーラジカル反応性成分は、フリーラジカル開始反応によって重合され得る重合性成分を含む。限定されないフリーラジカル反応基の例は、(メタ)クリレート基((meth)acrylates)、スチリル基、ビニル基、ビニルエーテル基、C1-6アルキル(メタ)クリレート基、(メタ)クリルアミド基、C1-6アルキル(メタ)クリルアミド基、N-ビニルラクタム基、N-ビニルアミド基、C2-12アルケニル基、C2-12アルケニルフェニル基、C2-12アルケニルナフチル基、C2-6アルケニルフェニルC1-6アルキル基、O-ビニルカルバメート基、および、O-ビニルカーボネート基を含む。
【0014】
この明細書で使用されているように、用語「金属塩」とは、一般式:[Mq+]a[Xz-]bを有する、あらゆる分子を意味し、ここで、Xは、あらゆる負帯電イオンを含み、a、b、qおよびzは、それぞれ個別に1以上の整数であり、q(a)=z(b)である。Mは、限定するものではないが、以下のイオン:Al+3、Cr+2、Cr+3、Cd+1、Cd+2、Co+2、Co+3、Ca+2、Mg+2、Ni+2、Ti+2、Ti+3、Ti+4、V+2、V+3、V+5、Sr+2、Fe+2、Fe+3、Au+2、Au+3、Au+1、Ag+2、Ag+1、Pd+2、Pd+4、Pt+2、Pt+4、Cu+1、Cu+2、Mn+2、Mn+3、Mn+4、Zn+2、Se+4、Se+2、および、これらの混合物から選択された、あらゆる正帯電金属イオンであってもよい。他の実施の形態において、Mは、Al+3、Co+2、Co+3、Ca+2、Mg+2、Ni+2、Ti+2、Ti+3、Ti+4、V+2、V+3、V+5、Sr+2、Fe+2、Fe+3、Au+2、Au+3、Au+1、Ag+2、Ag+1、Pd+2、Pd+4、Pt+2、Pt+4、Cu+1、Cu+2、Mn+2、Mn+3、Mn+4、Se+4およびZn+2、および、これらの混合物から選択されてもよい。Xの例は、CO3-2、NO3-1、PO4-3、Cl-1、I-1、Br-1、S-1、O-2、酢酸塩、これらの混合物、および、これらの同類のものを含むが、これらに限定されるものではない。さらに、Xは、CO3-2、SO4-2、PO4-3、Cl-1、I-1、Br-1、S-1、O-2、酢酸塩、および、これらの同類のもの、例えばC1-5アルキルCO2-1などを含む負帯電イオンを含む。他の実施の形態において、Xは、CO3-2、SO4-2、Cl-1、I-1、Br-1、酢酸塩、および、これらの混合物を包含してもよい。この明細書で使用されているように、用語「金属塩」は、米国公開特許US-2003-0043341-A1号明細書に開示された金属塩のように、ゼオライトを含まない。一つの実施の形態において、aは、1、2または3である。一つの実施の形態において、bは、1、2または3である。一つの実施の形態において、金属イオンは、Mg+2、Zn+2、Cu+1、Cu+2、Au+2、Au+3、Au+1、Pd+2、Pd+4、Pt+2、Pt+4、Ag+2およびAg+1、および、これらの混合物から選択される。特に好適な金属イオンは、Ag+1である。適切な金属塩の例は、硫化マンガン、酸化亜鉛、炭酸亜鉛、硫酸カルシウム、硫化セレン、ヨウ化銅、硫化銅、および、リン酸銅を含むが、これらに限定されるものではない。銀塩の例は、炭酸銀、リン酸銀、硫化銀、塩化銀、臭化銀、ヨウ化銀、および、酸化銀を含むが、これらに限定されるものではない。一つの実施の形態において、金属塩は、ヨウ化銀、塩化銀、および、臭化銀等、少なくとも一つの銀塩を含む。
【0015】
本発明の一部の実施の形態における金属Mの少なくとも約90%、および、一部の実施の形態における当該金属Mの少なくとも約95%は、金属塩:[Mq+]a[Xz-]bの形態である。この割合は、イオン性金属および非イオン性金属(metal0)の測定値から算出されてもよい。例えば、製品がヒドロゲルコンタクトレンズであり、かつ、抗菌性金属塩がヨウ化銀である場合に、イオン性金属は、リン酸緩衝生理溶液(米国バージニア州ハーンドンのメディア・テック・インク(Media Tech, Inc.)から市販されたダルベッコ(Dulbecco)社製のリン酸緩衝生理食塩水10X)中で当該レンズを抽出し、他の塩が抽出溶液中に存在しなくなるまで、使用する米国薬局方(USP)AppVII中に記載された方法を用いることによって算出され得る。抽出後に、製品は、機器中性子放射化分析法(INAA)を用いることによって測定される。非イオン性銀(Ag0)が使用条件下で抽出可能でないときに、抽出後にレンズ中で測定された全銀は、非イオン性銀の酸化状態(Ag0 oxidation state)にある。
【0016】
製品が、血液、尿、涙あるいは唾液のような水混和性の体液に接触している医療装置であり、かつ、約12時間を超える抗菌効果が望ましい場合における実施の形態で、金属塩は、25℃の純水中での約2×10−10未満のKspを有している。一つの実施の形態において、金属塩は、約2.0×10−17モル/リットルを超えない溶解度積定数を有している。特定の実施の形態において、製品は、生物医学装置、眼用装置すなわちコンタクトレンズであってもよい。
【0017】
この明細書で使用されているように、用語「純(pure)」は、米国フロリダ州ボーカラートンのシーアールシー出版社(CRC Press)から1993年に発行された「化学および物理のシーアールシーハンドブック第74版」中で定義されているように、使用された水の質を云う。種々の塩に関して、25℃の純水中で測定された溶解度積定数(Ksp)は、米国フロリダ州ボーカラートンのシーアールシー出版社(CRC Press)から1993年に発行された「化学および物理のシーアールシーハンドブック74版」中で公開されている。例えば、仮に、金属塩が炭酸銀(Ag2CO3)である場合に、溶解度積定数(Ksp)は、以下の式によって示される。
Ag2CO3(複数のAg2CO3)→2Ag+(水性媒体中)+CO32-(水性媒体中)
溶解度積定数(Ksp)は、以下のように算出される。
Ksp=[Ag+]2[CO32]
炭酸銀が溶解するときに、溶液中には、二つの銀の陽イオンごとに一つの炭酸アニオンが存在する、すなわち、[CO32]=1/2[Ag+]の式が成立することから、溶解度積定数の等式は、溶解された銀濃度について解くために、以下のように再整理され得る。
Ksp=[Ag+]2(1/2[Ag+])=1/2[Ag+]3
[Ag+]=(2 Ksp)1/3
【0018】
25℃で測定された場合における溶解度積定数が約2×10−10を超えない金属塩を含む製品が、1日〜30日まで、あるいは、それ以上の期間にわたって、レンズから金属を連続的に放出するはずであることが見出された。一つの実施の形態において、適切な金属塩は、ヨウ化銀、塩化銀、臭化銀、および、これらの混合物を含む。他の実施の形態において、金属塩は、ヨウ化銀を含む。
【0019】
本発明の製品は、ポリマー類から形成されると共に、当該製品は、食品、薬剤および医療装置、生物医学装置、および、これらの同類のもの用の包装体を含む、包装体、保存容器およびラップにおける適用を見出すことができる。生物医学装置は、カテーテル、ステント、血液保存袋およびチューブ、補綴物、インプラント、ならびに、眼用装置を含み、この眼用装置は、眼用レンズ(レンズの詳細な説明は、以下のとおりである。)を含む。一つの実施の形態において、本発明の製品は、光重合性ポリマー、特に、可視光の被爆によって重合化される成分等のフリーラジカル反応性成分から形成される。他の実施の形態において、製品は、使用中に、可視光および紫外光に被爆される。このような製品は、包装体、保存容器、プラスチックラップ、および眼用装置を含む。一つの実施の形態において、本発明の製品は、眼用装置である。
【0020】
上述した製品は、この技術分野において既知であり、かつ、種々のポリマーから形成されてもよい。一部の実施の形態において、製品は、一つのポリマーから形成され、かつ、他のポリマーで被覆されてもよい。抗菌性ポリマーは、装置または当該装置の部品の形態になるように成形され、あるいは、被膜として使用されてもよい。
【0021】
上述した多くの実施の形態において、製品の透明度は、ユーザーにとって心配事である。例えば、限定されない一つの実施の形態において、製品がコンタクトレンズ等の眼用装置である場合に、本発明に使用される金属塩の非常に小さな粒子サイズは、その金属塩を特に適切なものにする。一部の実施の形態において、本発明は、約200ナノメートル未満、あるいは、約100ナノメートル未満の達成粒子サイズ(achieved particles sizes)を有すると共に、一部の実施の形態において、約50ナノメートル未満の達成粒子サイズを有する。この非常に小さな粒子サイズは、可視光の波長よりも小さいものであり、当該粒子サイズは、本発明の製品を、透明度が望まれる場合における適用に対して特に有用なものにする。このような実施の形態は、コンタクトレンズ、眼内レンズ、血液保存袋およびチューブ、ならびに、食品包装体を含むが、これらに限定されるものではない。ポリマーの光学的品質が要求されない場合の適用において、上記の粒子サイズの範囲よりも大きな粒子も使用されてもよい。
【0022】
一つの実施の形態において、金属塩粒子も、製品が形成される上述した少なくとも一つのポリマー全体に、均質に分散されている。この明細書で使用されているように、用語「均質に分散される」とは、粒子の凝集が形成されず、かつ、粒子が、抗菌性金属塩を含むポリマーの特定部分に実質的に集中していないことを意味する。一つの実施の形態において、「均質に分散される」とは、ポリマーの任意の二つの領域間における金属塩粒子の濃度(乾燥製品の重量に基づいて重量%として測定される)の違いが約20%未満であることを意味する。他の実施の形態において、ポリマーの任意の二つの領域間における金属塩粒子の濃度の違いが約10%未満であり、さらに他の実施の形態において、ポリマーの任意の二つの領域間における金属塩粒子の濃度の違いが約5%未満である。分散の均質性は、特性X線の放出を誘発する高エネルギー電子を使用する元素分析技術を用いて、最終製品中で測定されてもよい。この適用では、電子プローブ微量分析法(EPM)が使用された(カメラ(Cameca)SX100と、20キロ電子ボルト、50ナノアンペアおよび20マイクロメートルの分析条件を用いる四つの波長分光計を備えた自動電子マイクロプローブSX50)。
【0023】
一つの実施の形態において、本発明の製品には、視認できる霞み(visual haze)も、不都合な色もない。抗菌性製品の透明度は、以下に詳細に記述されるCSIレンズに対する、約70マイクロメートルの厚さを有するサンプルを用いて測定されたヘイズ(haze)の百分率によって評価された。約100%未満のヘイズ値、約50%未満のヘイズ値は、本発明を用いることで、容易に達成され得る。
【0024】
最終ポリマー製品の色は、分光光度計を用いて測定され、かつ、国際照明委員会(CIE)の1976年のL*a*b*表色系で記録され得る。本発明の製品は、約89を超えるL*を有してもよく、一部の実施の形態において、約90を超えるL*を有し、a*は、約2未満であり、一部の実施の形態において、a*は約1.4未満であってもよい。色測定は、紫外線吸収剤、染料(handling tints)、フォトクロミック化合物、および、これらの同類等、最終製品の色に影響を与えうるポリマー成分を含まないポリマー類に対して実施されることになる。
【0025】
ポリマー中の金属塩の量は、乾燥ポリマーの全重量に基づいて測定される。ポリマー中の金属塩の量は、製品の最終用途および使用要件によって決定される。例えば、製品がコンタクトレンズである場合の一つの実施の形態において、透明度および色は、重要である。製品がコンタクトレンズであり、かつ、金属塩がヨウ化銀である場合の複数の実施の形態において、ポリマー中の銀の量は、ポリマーの乾燥重量に基づいて、約100ppm〜約1000ppmであり、一部の実施の形態では、ポリマーの乾燥重量に基づいて、200ppm〜約1000ppmである。他の実施の形態において、ポリマー中の銀の量は、ポリマーの乾燥重量に基づいて、約0.00001重量%(0.1ppm)〜約10.0重量%、好ましくは約0.0001重量%(1ppm)〜約1.0重量%、最も好ましくは約0.0001重量%(1ppm)〜約0.1重量%であってもよい。金属塩の添加については、金属塩の分子量が、金属イオンの重量%の金属塩への変換率を決定し、この技術分野における当業者は、抗菌性金属の所望量を与える上で必要な塩の量を算出することができる。
【0026】
一つの実施の形態において、本発明の製品は、
(a)反応性ポリマー混合物の少なくとも一つの成分中に少なくとも一つの塩前駆体を溶解させて塩前駆体混合物を形成すること;
(b)当該反応性ポリマー混合物の少なくとも一つの成分中に少なくとも一つの金属剤および少なくとも一つの分散剤を溶解させることによって、金属剤・分散剤錯体を形成して、金属剤混合物を形成すること;
(c)粒子形成条件下で、当該塩前駆体混合物および当該金属剤混合物を混合して粒子含有反応性混合物を形成すること;
(d)任意に、追加の反応性ポリマー成分を当該粒子含有反応性混合物に混合すること;ならびに、
(e)当該粒子含有反応性混合物を反応させて、金属塩を含む抗菌性ポリマー製品または部品を形成することであって、抗菌性金属Mの少なくとも約90%が金属塩の形態で存在する、形成すること、
によって形成されてもよい。
【0027】
用語「金属塩」は、上述した意味を有している。用語「塩前駆体」とは、金属イオンで置換できる陽イオンを含有する、あらゆる化合物または組成物(水溶液を含む)を云う。この実施の形態において、当該塩前駆体が約1マイクログラム/ミリリットル以上でレンズ処方中に溶解できることが好ましい。当該用語は、「抗菌性コンタクトレンズおよび使用方法(Antimicrobial Contact Lenses and Methods of Use)」という名称の米国公開特許US2003/0043341号明細書に記述されているようにゼオライトを含まず、あるいは、「活性銀を含有する抗菌性コンタクトレンズおよび製造方法(Antimicrobial Contact Lenses Containing Activated Silver and Methods for Their Production)」という名称の国際公開WO02/062402号に記述されたような活性銀を含まない。塩前駆体は、少なくとも化学量論的な量で、反応性混合物中に添加され、一部の実施の形態において、プラスチック製の最終製品に望ましい抗菌性金属の量に関連してモル過剰で、反応性混合物中に添加される。例えば、20マイクログラムのヨウ化銀が金属塩として製品中に存在している場合の一つの実施の形態において、ヨウ化ナトリウムは、少なくとも12マイクログラムの量で、反応性混合物中に存在している。塩前駆体の例は、塩化ナトリウム、ヨウ化ナトリウム、臭化ナトリウム、塩化リチウム、硫化リチウム、硫化ナトリウム、硫化カリウム、四塩化銀ナトリウム(sodium tetrachloro argentate)、これらの混合物、および、これらの同類のものなど、無機分子を含むが、これらに限定されるものではない。有機分子の例は、テトラアルキル乳酸アンモニウム(tetra-alkyl ammonium lactate)、テトラアルキル硫酸アンモニウム(tetra-alkyl ammonium sulfate)、テトラアルキル酢酸ホスホニウム(tetra-alkyl phosphonium acetate)、テトラアルキル硫酸ホスホニウム(tetra-alkyl phosphonium sulfate)、テトラアルキル塩化アンモニウム(tetra-alkyl ammonium chloride)、テトラアルキル塩化ホスホニウム(tetra-alkyl phosphonium chloride)、臭化物、ヨウ化物等のハロゲン化四級アンモニウムまたはハロゲン化四級ホスホニウム(quaternary ammonium or phosphonium halides)、および、これらの同類の物質を含むが、これらに限定されるものではない。一つの実施の形態において、塩前駆体は、ヨウ化ナトリウムを含む。
【0028】
用語「金属剤」とは、金属イオンを含有する、あらゆる組成物(水溶液を含む)を云う。このような組成物の例は、硝酸銀、銀トリフレート(silver triflate)、酢酸銀、テトラフルオロホウ酸銀(silver tetrafluoroborate)、硝酸銅、硫酸銅、硫酸マグネシウム、硫酸亜鉛、これらの混合物、および、これらの同類の水溶液または有機性溶液を含むが、これらに限定されるものではない。溶液中での金属剤の適切な濃度は、最終製品に含まれることになる金属塩の所望量に基づいて算出され得る。例えば、一つの実施の形態において、金属剤の濃度は、最終製品中に、約0.00001重量%(0.1ppm)〜約10.0重量%、約0.0001重量%(1ppm)〜約1.0重量%の金属塩を与えるように、他の実施の形態において、約0.0001重量%(1ppm)〜約0.1重量%の金属塩を与えるように、選択される。
【0029】
一部の実施の形態において、安定した色が望ましい。例えば、プラスチック製品が眼用装置である場合に、当該眼用装置が反応性混合物と同一の色および透明度を有することが望ましいことがある。銀塩は、感光的であることが知られている。したがって、仮に、銀塩の形成、および当該銀塩を含有する製品の硬化を気にかけない場合に、所望の銀塩は、当該製品中に生成されない。例えば、ヨウ化銀は、約400ナノメートル未満の波長光に対して感光的であるので、仮に、当該感光性を気にかけない場合に、光開始によって硬化される反応性混合物が、銀塩が減少したことを示す、不都合な黄色または茶色のレンズを形成する場合がある。光還元は、選択された金属塩の結合エネルギーに相当する波長(臨界波長)より大きな光の波長で、金属塩を含む反応性混合物を硬化させることによって、最小限化されてもよい。例えば、ヨウ化銀は、60キロカロリー/モルの結合エネルギーを有している。この結合エネルギーに関連した波長は、電磁方程式:
EAgI=hc/(λNA)
を用いて算出されてもよい。ここで、hはプランク定数であり、cは光の速度であり、λは入射放射線の波長であり、NAはアボガドロ数(Avagadro’s number)である。
【0030】
ヨウ化銀では、λは477ナノメートルである。臨界波長への調節は、型材および包装材料および溶液によるエネルギーの吸収または反射の説明となるように行われてもよい。したがって、例えば、製品がヨウ化銀を含むコンタクトレンズであり、当該製品が、10%のエネルギー伝送損失を計上するプラスチック製モールドを用いる直接成型によって形成される場合に、調節された臨界波長は、
λ=(1-10%)×477ナノメートル
λ=429ナノメートル
【0031】
したがって、この実施の形態における硬化条件は、約429ナノメートルより大きな波長光を含む。代わりに、反応性混合物は、熱硬化等を含むが、これに限定されない、光を含まない条件を用いて、硬化され得る。
【0032】
また、光還元は、実質的にすべての金属剤が金属塩に変換されるように、金属剤と比較して、モル過剰の塩前駆体を用いて、最小限化され得る。約1.1:1以上である塩前駆体:金属剤のモル比は、許容される。このモル比は、最終製品中に含まれる抗菌性金属Mの少なくとも約90%が金属塩の形態であることを保証する。一部の実施の形態において、製品は、重合開始剤、および、紫外光以外の条件を用いて硬化される。
【0033】
金属剤混合物および塩前駆体混合物のうち、少なくとも一方は、少なくとも一つの分散剤をさらに含み、一つの実施の形態では、金属剤混合物が、少なくとも一つの分散剤をさらに含む。適切な分散剤は、孤立電子対を備えた官能基を含有するポリマー類を含む。分散剤の例は、ヒドロキシアルキルメチルセルロースポリマー類、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレンオキシド、澱粉、ペクチン、ゼラチン等の多糖類;ポリジメチルアクリルアミドを含むポリアクリルアミド、ポリアクリル酸、3−アミノプロピルトリエトキシシラン(3-aminopropyltriethoxysilane)(APS)、メチルトリエトキシシラン(methyl-triethoxysilane)(MTS)、フェニルトリメトキシシラン(phenyl-trimethoxysilane)(PTS)、ビニルトリエトキシシラン(vinyl-triethoxysilane)(VTS)および3−グリシドキシプロピルトリメトキシシラン(3-glycidoxypropyltrimethoxysilane)(GPS)等の有機アルコキシシラン類(organoalkoxysilanes)、ポリエチレングリコール、ポリプロピレングリコール、グリセリンのホウ酸エステル(BAGE)等のポリエーテル類、約10,000を超える分子量を有し、かつ、ヒドロキシル基およびウレタン基等を含むが、これらに限定されない水素結合基等、粘性を増大させる基を含有するシリコンマクロマー、ならびに、これらの混合物を含む。
【0034】
一つの実施の形態において、分散剤は、ヒドロキシアルキルメチルセルロースポリマー類、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレンオキシド、グリセリン、グリセリンのホウ酸エステル(BAGE)、ゼラチンおよびポリアクリル酸、および、これらの混合物からなる群より選択される。他の実施の形態において、分散剤は、ヒドロキシプロピルメチルセルロース、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレンオキシド、ゼラチン、グリセリン、グリセリンのホウ酸エステル(BAGE)、および、これらの混合物からなる群より選択される。さらに他の実施の形態において、分散剤は、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレンオキシド、および、これらの混合物からなる群より選択される。
【0035】
分散剤がポリマーである場合に、当該ポリマーは、ある範囲の分子量を有することができる。約1000から数百万以下の分子量が使用されてもよい。上限は、金属塩混合物、塩前駆体混合物および反応性混合物中への分散剤の溶解度のみによって境界される。ゼラチンおよびメチルセルロース等のグリコシド系ポリマー類では、分子量は、百万を超えてもよい。ポリビニルアルコール、ポリビニルピロリドンおよびポリアクリル酸のような非グリコシド系ポリマー類では、分子量は、約2,500ダルトン〜約2,000,000ダルトン(約4.151×10−21g〜約3.321×10−18g)の範囲であってもよく、一部の実施の形態では、約10,000ダルトン〜約1,800,000ダルトン(約1.661×10−20g〜約2.989×10−18g)の範囲であってもよく、他の実施の形態では、約20,000ダルトン〜約1,500,000ダルトン(約3.321×10−20g〜約2.491×10−18g)の範囲であってもよい。一部の実施の形態において、約50,000ダルトン(約8.303×10−20g)を超える分子量は、この範囲内で分散剤が一部のポリマー系内で、より安定化効果を与える場合に、使用されてもよい。
【0036】
あるいは、分散状態安定化ポリマー(dispersion-stabilizing polymers)の分子量は、ジョン・ワイリー・アンド・サンズ・インク(John Wiley & Sons Inc.)発行の「ポリマー科学および工学の百科事典第2版」第17巻、第198頁〜第257頁の「N−ビニルアミドポリマー」項(Encyclopedia of Polymer Science and Engineering, N-Vinyl Amide Polymers, Second edition, Vo1 17, pgs. 198-257)中に記述されているように、動粘測定法に基づくK値によっても表示され得る。この方法で表示される場合に、非グリコシド系分散剤ポリマー類は、約5〜約150のK値を有してもよく、一部の実施の形態では、約5〜約100のK値、約5〜約70のK値を有してもよく、他の実施の形態では、約5〜約50のK値を有してもよい。
【0037】
金属塩ナノ粒子が反応性ポリマー混合物中に直接、形成される場合に、分散剤は、当該反応性ポリマー混合物の全成分の重量%に基づいて、約0.001重量%〜約40重量%の間の量で存在してもよい。一部の実施の形態において、分散剤は、約0.01重量%〜約30重量%の間の量で存在してもよく、他の実施の形態では、約0.1重量%〜約30重量%の間の量で存在してもよい。一部の実施の形態において、分散剤は、ポリビニルアルコールを含むコンタクトレンズが製造される場合など、ポリマー製品を形成する上で使用される反応性成分でもある。これらの実施の形態において、使用される分散剤の量は、当該反応性ポリマー混合物の全成分の重量%に基づいて、約90重量%以下の量であってもよく、一部の実施の形態では、約100重量%以下の量であってもよい。
【0038】
一部の実施の形態において、分散剤は、結果的に得られたポリマーに追加の利点を与える。例えば、ポリビニルピロリドン(PVP)が粒子安定剤である場合に、当該ポリビニルピロリドンは、分散安定性をもたらすことに加えて、湿潤性、摩擦係数、含水率、離型性、および、同類のものを改善することができる。これらの実施の形態において、分散安定性を与える上で必要な量より多い量の分散剤を含む必要がある、あるいは、当該多い量の分散剤を含むことが望ましい場合がある。これらの実施の形態において、所望サイズの粒子が形成されることを保証するために、脱気ステップおよび熟成ステップ等、他の工程条件の均衡を保つことが望ましいはずである。
【0039】
塩前駆体混合物および金属剤混合物は、粒子形成条件下で混合される。この明細書で使用されているように、粒子形成条件は、約200ナノメートル未満の平均粒子サイズ、一部の実施の形態では約100ナノメートル未満の平均粒子サイズ、他の実施の形態では約50ナノメートル未満の平均粒子サイズを有し、かつ、反応性混合物全体に分散された金属塩粒子を形成する上で適した時間、温度およびpHを含む。
【0040】
混合温度は、反応性混合物中の反応性成分に依存して変更されてもよい。概ね、反応性混合物の凝固点より高く、約100℃以下の混合温度が使用されてもよい。一部の実施の形態において、約10℃〜約90℃の間の混合温度が使用されてもよく、他の実施の形態では約10℃〜約50℃の間の混合温度が有用である。
【0041】
塩前駆体混合物または金属剤混合物のいずれか一方あるいは両方は、反応性混合物と混合する前に脱気されてもよい。
【0042】
一つの実施の形態において、塩前駆体混合物または金属剤混合物のいずれか一方は、単独噴流等の流れによって導入されてもよく、あるいは、塩前駆体混合物および金属剤混合物の双方は、二重噴流によって同時に導入されてもよい。単独噴流法において、例えば、金属剤混合物の溶液は、制御速度で噴流によって流されて、塩前駆体混合物および分散剤を含有する撹拌溶液中に導入される。あるいは、二重噴流法は、分散剤を含有する撹拌溶液中に、二つの別の噴流によって塩前駆体混合物および金属剤混合物の双方を同時に添加するために使用されてもよい。一部の実施の形態において、更なる量の分散剤、塩前駆体混合物および/または金属剤混合物を添加することが望ましい場合がある。
【0043】
塩前駆体混合物および金属剤混合物は、約10分間未満の時間中、反応性混合物中に添加されてもよく、一部の実施の形態では、約10秒〜約5分間の付加時間中、添加されてもよい。
【0044】
得られる溶液が均質であり、かつ、安定した分散液が形成される限り、あらゆる混合時間が使用されてもよい。この明細書で使用されているように、安定した分散液は、少なくとも約12時間、実質的に変化しない。商業的に望ましい混合時間は、約1分間〜数日の範囲を含めてもよく、一部の実施の形態では、約10分間〜約12時間の範囲を含めてもよい。
【0045】
高剪断性の混合方法は、低分子量のポリマーと一緒に使用されてもよく、上述した範囲の下限値での混合時間を可能にすることができる。
【0046】
また、反応性混合物は、真空下で、あるいは、当該反応性混合物中のいずれの成分とも反応しないガスを用いて、脱気されてもよい。適切な不活性ガスは、窒素、アルゴン、これらを含む混合物、および、これらの同類のものを含む。脱気処理は、完全真空(例えば、10mbar(10ヘクトパスカル))以下の圧力、および、約60分間以内の時間、一部の実施の形態では約40分間以内の時間を用いて、実行されてもよい。所定の反応性混合物と一緒に使用される温度条件および圧力条件と同様に、脱気ステップの継続時間も、使用される溶媒の揮発度等の他の要素によって決められてもよい。
【0047】
上記処理方法は、脱気ステップ前に、粒子熟成ステップをさらに含めてもよい。非常に小さな粒子は、加熱中に、より大きな粒子よりも、容易に溶解する。したがって、透明度が重要である場合の適用(眼用装置等)において、さらなる処理(殺菌ステップ、溶融加工ステップ、焼鈍ステップ、焼結ステップ等)中または保存中に、過度に熟成しない程度に粒子を大きくすることを保証する粒子熟成ステップを含むことが望ましいことがある。当該粒子熟成ステップにおいて、反応性混合物は、微粉を低減するために、約5分間〜1時間の期間、約30℃〜約70℃の温度に加熱される。この粒子熟成ステップは、殺菌される医療装置にとって特に有用であることがある。例えば、プラスチック製品がレンズである場合に、当該レンズが形成されるときに、当該レンズは、視認できる霞みが存在しない状態である必要があり、かつ、処理(殺菌包装処理ステップを含む)、保存、および、使用の間中、視認できる霞みが存在しない状態を維持する必要がある。また、微粉の形成は、分散剤の量を低減させることによって、軽減されてもよい。
【0048】
一つの実施の形態において、反応性混合物中の少なくとも約90%の粒子は、約100ナノメートル未満の粒子サイズを有しており、他の実施の形態では、反応性混合物中の少なくとも約90%の粒子は、約80ナノメートル未満の粒子サイズを有しており、さらに他の実施の形態では、反応性混合物中の少なくとも約90%の粒子は、約60ナノメートル未満の粒子サイズを有している。反応性混合物中の粒子の粒子サイズは、以下の検査法の項で記述されているように、光散乱法(レーザー光散乱法または動的光散乱法のいずれか一方)によって測定されてもよい。
【0049】
本発明の粒子含有反応性混合物は、視認できる霞みも、不都合な色もない。不都合な色の欠如は、白色を背景にして主観的に、あるいは、以下に記述されるL*a*b*表色法を用いて、評価されてもよい。
【0050】
追加の成分は、混合ステップ中に、任意に添加されてもよい。追加のポリマー成分は、反応性のモノマー類、プレポリマー類、および、マクロマー類、重合開始剤、架橋剤、連鎖移動剤、紫外線吸収剤、湿潤剤、剥離補助剤(release aids)、フォトクロミック化合物、栄養補助化合物および医薬化合物、着色剤、染料、顔料、これらの組み合わせ、および、これらの同類の物質を含む。当該追加のポリマー成分は、モノマー類、オリゴマー類あるいはプレポリマー類を含む、いかなる形態でも添加されてもよい。
【0051】
仮に、反応性混合物のいずれかの成分が金属剤と反応して元素金属(elemental metal)を形成することができ、かつ、当該元素金属が望ましいものではない場合に、一つの実施の形態において、当該成分は、金属塩粒子が形成された後であるが、反応性混合物を硬化させてポリマー製品を形成する前に、当該反応性混合物中に添加される。例えば、硝酸銀(AgNO3)がN,N-ジメチルアクリルアミド(DMA)と反応して不都合な非イオン性銀(Ag0)を形成できることが見出されてきた。したがって、N,N-ジメチルアクリルアミド(DMA)を含む反応性混合物では、当該N,N-ジメチルアクリルアミド(DMA)は、一つの実施の形態(金属塩がヨウ化銀である場合)において、金属塩粒子(ヨウ化銀)が形成された後に、当該反応性混合物中に添加される。この技術分野における当業者は、ある成分が還元剤として作用するか否かについて、当該成分を溶媒中で金属剤と混合し、かつ、化学分析によって、あるいは、特定の場合に、当該混合物の外観の変化によって分析することで、容易に決定することができる。
【0052】
あるいは、ナノ粒子の金属塩は、反応性ポリマー混合物とは別に形成されてもよい。例えば、安定化金属塩粒子は、少なくとも一つの塩前駆体を含む塩前駆体溶液を形成すること;
約20重量%〜約50重量%の間の少なくとも一つの分散剤であって、少なくとも約1000の平均分子量を有する、分散剤と、少なくとも一つの金属剤とを含む金属剤溶液を形成すること;
添加中に溶液の透明度を維持し、かつ、約200ナノメートル未満の粒子サイズを有する安定化金属塩粒子を含む生成物溶液を形成する上で十分な速度で、一方の溶液を他方の溶液中に添加すること;および、
当該安定化金属塩粒子を乾燥させることによって形成されてもよい。安定化金属塩粒子は、約200ナノメートル未満の粒子サイズを有する金属塩粒子であり、かつ、少なくとも一つの分散剤と錯化されている。一部の実施の形態において、安定化金属塩粒子は、約100ナノメートル未満の粒子サイズを有しており、他の一部の実施の形態では約50ナノメートル未満の粒子サイズを有している。
【0053】
この実施の形態において、金属剤および塩前駆体の溶液は、(a)金属剤、塩前駆体および分散剤を溶解でき、(b)金属剤を金属に還元せず、かつ、(c)既知の方法によって容易に除去できる、任意の溶媒を用いて形成される。水、アルコール類、または、これらの混合物が使用されてもよい。金属剤および塩前駆体を可溶化できる適切なアルコール類が選択されてもよい。硝酸銀およびヨウ化銀が金属剤および塩前駆体として使用される場合に、三級アミルアルコール、トリプロピレングリコール・モノメチルエーテル等のアルコール類、これらの混合物、および、これらと水との混合物が使用されてもよい。また、水も単独で使用されてもよい。
【0054】
上述したいずれの分散剤も使用されてもよい。混合物は使用されてもよい。分散剤は、金属剤溶液または塩前駆体溶液のいずれか一方または両方に含まれてもよく、あるいは、金属剤溶液および塩前駆体溶液が添加される第三の溶液に含めることができる。一つの実施の形態において、金属剤溶液も、少なくとも一つの分散剤を含む。金属剤溶液および塩前駆体溶液の双方が少なくとも一つの分散剤を含む場合の実施の形態において、当該分散剤は、同一のものであってもよく、あるいは、異なったものであってもよい。
【0055】
分散剤は、約500ナノメートル未満の金属塩粒子サイズを与える上で十分な量(粒子サイズ安定化有効量)で含まれている。最終製品の透明度が重要である場合の実施の形態において、粒子サイズは約200ナノメートル未満であり、一部の実施の形態では約100ナノメートル未満であり、他の実施の形態では約50ナノメートル未満である。一つの実施の形態において、少なくとも約20重量%の分散剤は、所望の粒子サイズが達成されることを保証するために、少なくとも一つの溶液中で使用される。一部の実施の形態において、金属剤に対する分散剤単位のモル比は、少なくとも約1.5、あるいは、少なくとも約2であり、一部の実施の形態では少なくとも約4である。この明細書で使用されているように、分散剤単位は、分散剤中の反復単位である。一部の実施の形態において、両溶液中での分散剤の濃度が同一であることは便利なはずである。
【0056】
溶液中での分散剤の濃度の上限値は、選択された溶媒中での当該分散剤の溶解度、および、当該溶液の取扱い易さによって決定されてもよい。一つの実施の形態において、各溶液は、約50センチポアズ(cps)未満の粘度を有している。一つの実施の形態において、生成物溶液は、約50重量%以下の分散剤を有してもよい。上述したように、金属剤溶液および塩前駆体溶液は、同一または異なる濃度の分散剤を有してもよい。すべての重量%は、当該溶液中の全成分の全重量に基づいている。
【0057】
この実施の形態では、金属剤溶液および塩前駆体溶液中の金属剤および塩前駆体の各濃度は、選択された溶媒中での金属剤および塩前駆体の溶解度の限界までであって、少なくとも約1500ppmであることが望ましく、一部の実施の形態では約5000ppm〜溶解度の限界の間であることがのぞましく、一部の実施の形態では約5000ppm〜約50,000ppm(5重量%)の間であることが望ましく、また、他の実施の形態では約5000ppm〜約20,000ppm(2重量%)の間であることが望ましい。
【0058】
溶液の混合は、室温で実行されてもよく、撹拌法を含めることが有効となる。渦が形成される速度以上の撹拌速度が使用されてもよい。選択された撹拌速度は、泡立て、発泡、あるいは、混合容器からの溶液の損失を生じさせるものであってはならない。撹拌は、添加中、継続される。
【0059】
混合は、大気圧あるいは減圧下で実行されてもよい。一部の実施の形態において、混合は、溶液に泡立て、または、発泡を生じさせてもよい。発泡または泡立ては、高濃度の金属塩が入るポケットを形成させ、当該ポケットが所望の粒子サイズより大きなサイズの粒子をもたらすことがある場合に、望ましくない。これらの場合には、減圧条件が使用されてもよい。圧力は、大気圧と選択された溶媒の蒸気圧との間の任意の圧力とすることができる。一つの実施の形態において、水が溶媒である場合に、当該圧力は、大気圧と約40mbar(約40ヘクトパスカル)との間であってもよい。
【0060】
塩前駆体溶液および金属剤溶液の添加速度は、混合中に溶液の透明度を維持するように選択される。局所的な僅かな霞みは、当該溶液が撹拌中に透明である限り、許容され得る。当該溶液の透明度は、視覚的に観察されるか、あるいは、紫外・可視分光法(UV-VIS)を用いてモニターされてもよい。適切な添加速度は、所望の濃度を有する一連の溶液を調製し、かつ、異なる添加速度での溶液の透明度をモニターすることによって決定されてもよい。この方法は、実施例26〜31に例証されている。また、塩前駆体溶液への分散剤の導入は、添加速度を上げることができる。
【0061】
他の実施の形態において、速い添加速度が望ましい場合に、金属剤および分散剤は、塩前駆体溶液と混合する前に、錯体形成時間を含む錯体形成条件下で混合されることが許容される。金属剤溶液中の分散剤が金属剤を含む錯体を形成すると考えられる。この実施の形態において、金属剤溶液および塩前駆体溶液を混合する前に、金属剤が分散剤と完全に錯化することが望ましい。「完全に錯化される」とは、実質的に全部の金属イオンが少なくとも一つの分散剤と錯化されたことを意味する。「実質的に全部」とは、当該金属イオンの少なくとも約90%、一部の実施の形態では少なくとも約95%が少なくとも一つの分散剤と錯化されたことを意味する。
【0062】
錯体形成時間は、紫外・可視分光法(UV-VIS)あるいはフーリエ変換赤外分光法(FTIR)等の分光法により、溶液中でモニターされてもよい。分散剤を含まない金属剤溶液の複数のスペクトルは測定される。当該金属剤溶液の複数のスペクトルは、分散剤の添加後にモニターされ、かつ、スペクトル変化もモニターされる。錯体形成時間は、スペクトル変化が安定する時間である。
【0063】
あるいは、錯化時間は、同一濃度を有する一連の金属剤・分散剤溶液を形成し、各溶液を異なる時間(実施例1では、3時間、6時間、12時間、24時間、72時間および1週間)で混合し、各金属剤・分散剤溶液を塩前駆体溶液とバッチ式で混合することによって、経験的に測定されてもよい。錯体形成時間で混合された金属剤・分散剤溶液は、添加速度を制御せずに、金属剤溶液および塩前駆体溶液が一緒に直接注入される場合に、透明な溶液を形成することになる。例えば、20ミリリットルの金属剤溶液は、20ミリリットルの塩前駆体溶液に1秒間以内で添加されてもよい。
【0064】
錯化条件は、錯化時間(上述されている)、温度、金属剤に対する分散剤の比率、および、撹拌速度を含む。温度、金属剤に対する分散剤のモル比、および、撹拌速度の上昇は、錯化時間を短縮するはずである。この明細書に記載された教示に関連して、この技術分野における当業者は、この明細書に開示された錯化レベルを得るために、諸条件を変更することができるはずである。
【0065】
仮に、金属剤および分散剤が完全に錯化されていない場合に、混合条件は、錯化されていない金属塩の形成よりも多く、分散剤・金属剤錯体を形成するために、混合物中での反応を偏らせる(bias)ように選択されてもよい。この偏り(biasing)は、(a)塩前駆体中の分散剤の濃度、あるいは、塩前駆体溶液および金属剤溶液が添加される溶液中の分散剤の濃度、ならびに、(b)金属剤溶液および塩前駆体溶液の混合速度を制御することによって達成されてもよい。
【0066】
一旦、金属剤溶液および塩前駆体溶液が混合されたら、生成物溶液は乾燥されてもよい。凍結乾燥機、噴霧乾燥機およびこれらの同類等、従来のあらゆる乾燥設備が、使用されてもよい。乾燥設備および乾燥方法は、この技術分野において周知である。適切な噴霧乾燥機の例は、ジーイーエー・ニロ・インク(GEA Niro Inc.)から入手可能な乾燥機等、回転式噴霧乾燥機である。噴霧乾燥では、噴霧導入口の温度は、選択された溶媒の引火点を超えている。
【0067】
凍結乾燥機は、ジーイーエー・ニロ・インク(GEA Niro Inc.)を含む、多くの製造者から入手可能である。凍結乾燥温度および圧力は、この技術分野における当業者によって周知なように、溶媒を昇華させるように選択される。選択された乾燥方法について、従来の範囲内のあらゆる温度が使用されてもよい。
【0068】
生成物溶液は、得られる粉末が約10重量%未満の溶媒含有率を有するようになるまで、一部の実施の形態では約5重量%未満の溶媒含有率を有するようになるまで、また、他の一部の実施の形態では約2重量%未満の溶媒含有率を有するようになるまで、乾燥処理される。高濃度の溶媒は、安定化金属塩を形成するために使用された溶媒がポリマー製品を形成するために使用された反応性混合物と相溶性である場合に、適切であることがある。当該粉末は、この粉末を水中に分散させ、透過電子顕微鏡法、光子相関分光分析法、あるいは、動的光散乱法によって測定されるように、約100ナノメートル以下の粒子サイズ、約50ナノメートル以下の粒子サイズ、また、一部の実施の形態では約15ナノメートル以下の粒子サイズを有する安定化金属塩を含む。
【0069】
安定化金属塩粉末は、反応性混合物に直接、添加されてもよい。安定化金属塩粉末の添加量は、所望レベルの抗菌性金属イオンを与えるために、容易に算出されることができる。
【0070】
金属塩を含む反応性混合物は、抗菌性ポリマー製品を形成するために、反応させられる。反応条件は、当該反応性混合物中の成分に基づいて、この技術分野における当業者によって容易に選択されてもよい。例えば、抗菌性ポリマー製品がフリーラジカル反応性成分から形成されたコンタクトレンズである場合に、反応性混合物は、重合開始剤を含み、かつ、反応条件は、光硬化または熱硬化の条件を含めてもよい。抗菌性金属塩が、ヨウ化銀、塩化銀および臭化銀等の感光性である場合に、上述した臨界波長を下回る波長に対する当該金属塩の被爆は、銀イオン(Ag+)を非イオン性銀(Ag0)に変換することから、結果的に、当該塩が組み込まれた製品の色を暗くすることになる。したがって、一つの実施の形態において、フリーラジカル反応性成分が使用される場合に、硬化処理は、可視光に対する被爆によって実行される。他の実施の形態において、反応性混合物は、少なくとも一つの紫外線吸収化合物をさらに含み、かつ、当該反応性混合物は、可視光、熱、あるいは、これらの組み合わせを用いて硬化処理される。さらに他の実施の形態において、反応性混合物は、少なくとも一つの紫外線吸収化合物および可視光重合開始剤をさらに含み、かつ、当該反応性混合物は、可視光を用いて硬化処理される。
【0071】
金属塩は、種々のポリマー類中に形成されてもよく、あるいは、当該ポリマー類中に添加されてもよい。適切なポリマー類は、使用目的に基づいて選択されてもよい。例えば、食品包装用では、ポリエチレンテレフタレート、高密度ポリエチレンおよびポリプロピレン等のポリマー類は、食品および飲料の容器用に一般的に使用されており、低密度ポリエチレンは、プラスチック製ラップに一般的に使用されている。
【0072】
関節置換体等、種々の埋め込み型装置は、高次架橋された超高分子量ポリエチレン(UHMWPE)を用いて形成されており、当該ポリエチレンは、通常、少なくとも約400,000の分子量を有しており、一部の実施の形態では当該分子量が約1,000,000〜約10,000,000であり、当該ポリエチレンは、通常、本質的にゼロ(0)であるメルトインデックス(米国材料試験協会(ASTM) D-1238)、および、8を超える比重低下、一部の実施の形態では約25〜30の間の比重低下によって定義されている。
【0073】
縫合糸および創傷包帯を作る際のヤーンとしての使用に適した、適切な吸収性ポリマーの例は、ラクチド(乳酸、D-ラクチド、L-ラクチドおよびメソラクチドを含む)、グリコリド(グリコール酸を含む)、ε-カプロラクトン、p-ジオキサノン(1,4-ジオキサン-2-オン)、トリメチレン炭酸塩(trimethylene carbonate)(1,3-ジオキサン-2-オン)の各ホモポリマー類および各コポリマー類を含むが、これらに限定されない脂肪族ポリエステル類;トリメチレン炭酸塩、δ-バテロラクトン(δ-vaterolactone)、β-ブチロラクトン、γ-ブチロラクトン、ε-デカラクトン(ε-decalactone)、ヒドロキシ酪酸塩、ヒドロキシ吉草酸塩(hydroxyvalerate)、1,4-ジオキセパン-2-オン(1,4-dioxepan-2-one)(その二量体1,5,8,12-テトラオキサシクロテトラデカン-7,14-ジオン(1,5,8,12-tetraoxacyclotetradecane-7,14-dione)を含む)、1,5-ジオキセパン-2-オン、6,6-ジメチル-1,4-ジオキサン-2-オンの各アルキル誘導体;および、これらのポリマー混合物を含むが、これらに限定されるものではない。
【0074】
縫合糸は、ポリアミド類(ポリヘキサメチレンアジポアミド(polyhexamethylene adipamide)(ナイロン66)、ポリヘキサメチレンセバクアミド(polyhexamethylene sebacamide)(ナイロン610)、ポリカプロアミド(polycapramide)(ナイロン6)、ポリドデカンアミド(polydodecanamide)(ナイロン12)およびポリヘキサメチレンイソフタルアミド(polyhexamethylene isophthalamide)(ナイロン61)、これらのコポリマー類およびこれらの混合物)、ポリエステル類(例えば、ポリエチレンテレフタレート、ポリブチルテレフタレート、これらのコポリマー類およびこれらの混合物)、フルオロポリマー類(例えば、ポリテトラフルオロエチレンおよびポリフッ化ビニリデン)、ポリオレフィン類(例えば、アイソタクチックおよびシンジオタクチックなポリプロピレンおよびこれらの混合物の他、ヘテロタクチックなポリプロピレン(1985年12月10日に発行され、エシコン・インク(Ethicon, Inc.)に譲渡された米国特許第4,557,264号明細書に記述されたポリエチレン等であり、当該文献は参照によってこの明細書に組み込まれる)およびポリエチレン(1985年12月10日に発行され、エシコン・インク(Ethicon, Inc.)に譲渡された米国特許第4,557,264号明細書に記述されたポリエチレン等)と混合したアイソタクチックまたはシンジオタクチックなポリプロピレンを主成分とする混合物、ならびに、これらの混合物等であるが、これらに限定されない非吸収性ポリマー材料からも形成され得る。
【0075】
涙点プラグ(punctal plugs)の本体は、シリコーン、シリコーン混合物、例えば、pHEMA(ポリヒドロキシエチルメタクリレート)、ポリエチレングリコール、ポリビニルピロリドンおよびグリセロールの各親水性モノマー類等のシリコーンコポリマー類、例えば米国特許第5,962,548号明細書、米国特許第6,020,445号明細書、米国特許第6,099,852号明細書、米国特許第6,367,929号明細書および米国特許第6,822,016号明細書に記述されたポリマー類等のシリコーンヒドロゲルポリマーを含むが、これらに限定されない、任意の適切な生体適合性ポリマー類から形成されてもよい。他の適切な生体適合性ポリマー類は、例えば、ポリ(エチレングリコール);ポリ(エチレンオキシド);ポリ(プロピレングリコール);ポリ(ビニルアルコール);ポリ(ヒドロキシエチルメタクリレート);ポリ(ビニルピロリドン);ポリアクリル酸;ポリ(エチルオキサゾリン);ポリ(ジメチルアクリルアミド);例えばホスホリルコリン誘導体等のリン脂質;ポリスルホベタイン類(polysulfobetains);例えばヒアルロン酸、デキストラン、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、ジェランガム、グアーガム、ヘパラン硫酸、コンドロイチン硫酸(chondritin sulfate)、ヘパリンおよびアルギン酸塩等の多糖類および炭水化物;例えばゼラチン、コラーゲン、アルブミンおよびオボアルブミン等の蛋白質;ポリアミノ酸;例えばポリテトラフルオロエチレン(「PTFE」)、ポリフッ化ビニリデン(「PVDF」)およびテフロン等のフッ素化ポリマー類;ポリプロピレン;ポリエチレン;ナイロン;ならびに、エチレンビニルアルコール(「EVA」)を含む。
【0076】
超音波外科機器のポリマー部品は、ポリイミド類、フルオラエチレンプロペン(fluora ethylene propene)(FEPテフロン)、PTFEテフロン、シリコーンゴム、エチレン・プロピレンジエンモノマー(EPDM)ゴムから形成されてもよく、これらの材料のいずれかは、テフロンまたはグラファイト等の材料で充填されてもよく、あるいは充填されなくてもよい。このような例は、米国公開特許US20050192610号明細書および米国特許第6,458,142号明細書に開示されている。
【0077】
上述のポリマー類の製造方法は周知であり、かつ、安定化金属塩粒子は、溶融混練により、あるいは、重合工程中に、容易に組み込まれうる。各系に適切な分散剤は、分散剤および分散剤・金属剤錯体の熱安定性を考慮することによって容易に選択されることができる。
【0078】
一つの実施の形態において、抗菌性ポリマー製品はレンズである。この明細書で使用されているように、用語「レンズ」は、眼球内または眼球上に配する眼用装置を云う。これらの眼用装置は、光学補正、治療効果、美容効果、あるいは、これらの組み合わせを付与することができる。用語「レンズ」は、ソフトコンタクトレンズ、ハードコンタクトレンズ、眼内レンズ、被せレンズ(overlay lenses)、眼球用挿入体(ocular inserts)、および、涙点プラグ等であるが、これに限定されない光学挿入体(optical inserts)を含むが、これらに限定されるものではない。
【0079】
ソフトコンタクトレンズは、シリコーンヒドロゲルおよびフルオロヒドロゲルを含むが、これらに限定されない、シリコーンエラストマーまたはヒドロゲルから形成されてもよい。好適には、本発明のレンズは、エタフィルコンA(etafilcon A)から形成されたレンズ等のレンズに匹敵する光学的な透明度を呈する、光学的に透明なものである。
【0080】
本発明の金属塩は、米国特許第5,710,302号明細書、国際公開WO9421698号、欧州特許第406161号、日本特許出願JP2000016905号、米国特許第5,998,498号明細書、米国特許出願09/532,943号、米国特許第6,087,415号明細書、米国特許第5,760,100号明細書、米国特許第5,776,999号明細書、米国特許第5,789,461号明細書、米国特許第5,849,811号明細書および米国特許第5,965,631号明細書に記述されたソフトコンタクトレンズ処方中に添加されてもよい。さらに、本発明の金属塩は、市販のソフトコンタクトレンズ処方中に添加されてもよい。ソフトコンタクトレンズの処方例は、エタフィルコンA、ゲンフィルコンA(genfilcon A)、レネフィルコンA(lenefilcon A)、ポリマコン(polymacon)、アクアフィルコンA(acquafilcon A)、バラフィルコンA(balafilcon A)、ロトラフィルコンA(lotrafilcon A)、ロトラフィルコンB、ガリフィルコン(galyfilcon)、セノフィルコン(senofilcon)およびコムフィルコン(comfilcon)の各処方を含むが、これらに限定されるものではない。一つの実施の形態において、コンタクトレンズ処方は、エタフィルコンA、バラフィルコンA、アクアフィルコンA、ロトラフィルコンA、ロトラフィルコンB、セノフィルコン、ガリフィルコン、コムフィルコンであり、他の実施の形態では、コンタクトレンズ処方は、米国特許第5,998,498号明細書、米国特許出願09/532,943号、2000年8月30に出願された、米国特許出願09/532,943号の一部継続出願、国際公開WO03/022321号、米国特許第6,087,415号明細書、米国特許第5,760,100号明細書、米国特許第5,776,999号明細書、米国特許第5,789,461号明細書、米国特許第5,849,811号明細書および米国特許第5,965,631号明細書で調製されたエタフィルコンA、ガリフィルコン、コムフィルコンおよびシリコーンヒドロゲルである。これらの特許文献ばかりでなく、この段落で開示された他の全特許文献は、参照によって、この明細書にそっくりそのまま組み込まれる。一つの実施の形態において、本発明の金属塩は、米国特許出願11/757484号に記述されているように、少なくとも約41の親水率(hydrophilicity index)を有するレンズ材料中に添加されてもよい。一つの実施の形態において、製品は、ガリフィルコンから形成されたコンタクトレンズである。
【0081】
ハードコンタクトレンズは、ポリ(メチル)メタクリレートのポリマー類、シリコンアクリレート(silicon acrylates)、シリコーンアクリレート(silicone acrylates)、フルオロアクリレート、フルオロエーテル、ポリアセチレンおよびポリイミドを含むが、これらに限定されないポリマー類から形成され、ここで、代表例の調製は、米国特許第4,330,383号明細書に見出されることができる。本発明の眼内レンズは、既知の材料を用いて形成され得る。例えば、当該レンズは、ポリメチルメタクリレート、ポリスチレン、ポリカーボネートまたはこれらの同類の材料、および、これらの組み合わせを含むが、これらに限定されない硬質材料から形成されてもよい。さらに、ヒドロゲル、シリコーン材料、アクリル系材料、フルオロカーボン材料、および、これらの同類の材料、またはこれらの組み合わせを非限定的に含む可撓性材料が使用されてもよい。典型的な眼内レンズは、国際公開WO0026698号、国際公開WO0022460号、国際公開WO9929750号、国際公開WO9927978号および国際公開WO0022459号、米国特許第4,301,012号明細書、米国特許第4,872,876号明細書、米国特許第4,863,464号明細書、米国特許第4,725,277号明細書、米国特許第4,731,079号明細書に記述されている。金属塩は、上述したようなハードコンタクトレンズ処方および眼内レンズ処方中に添加されてもよい。
【0082】
眼用レンズを含む生物医学装置は、被膜が抗菌性金属の活性を抑制しない、すなわち、被膜が当該活性を不都合に低下させないことを条件として、生体組織との適合性を向上させるために被覆されてもよい。したがって、本発明の製品は、レンズを被覆するのに使用される多くの物質で被覆されてもよい。あるいは、安定化金属塩粒子は、あらゆる既知の被膜組成物に都合よく添加されてもよく、一つの実施の形態では、当該粒子は、本発明の教示にしたがって、浸漬被覆溶液等の溶液および反応性混合物、トランスファー成型被膜(mold transfer coatings)、反応性被膜およびこれらに同類の被膜から形成される溶液組成物中に添加されてもよい。適切な被膜例は、米国特許第6,087,415号明細書および米国公開特許US2001/0086160号明細書に開示されたような、結合剤または結合層を用いた被膜、米国特許第5,779,943号明細書に開示されたような潜在的な親水性被膜、米国特許第5,275,838号明細書に開示されたようなポリエチレンオキシド星状被膜、米国特許第4,973,493号明細書に開示されたような共有結合被膜、米国特許第5,135,297号明細書に記述されたように被覆される製品と接触した反応性モノマーの重合化および架橋化によって形成された被膜、米国特許第6,200,626号明細書に開示されたようなグラフト重合被膜、欧州特許第1,287,060号、米国特許第6,689,480号明細書および国際公開WO2004/060431号に開示されたような非反応性または錯体形成被膜、欧州特許第1252222号、米国特許第7,022,379号明細書、米国特許第6,896,926号明細書、米国公開特許US2004/0224098号明細書、米国公開特許US2005058844号明細書および米国特許第6,827,966号明細書に開示されたような「多層被膜」、国際公開WO03/011551A1号に開示されたようなトランスファー成型被膜、および、米国特許第5,760,100号明細書に開示された表面改質方法を含むが、これらに限定されるものではない。米国特許第6,193,369号明細書に開示されたようなケイ酸塩被膜、および、米国特許第6,213,604号明細書に開示されたようなプラズマ被膜は、抗菌性金属塩を含む眼用装置等の製品上に塗布されてもよい。これらの適用および特許文献は、手順、組成物および方法に関して参照されることによって、この明細書に組み込まれる。
【0083】
上述したレンズ処方の多くは、ユーザーが1日〜30日の範囲の連続期間、当該レンズを挿入することを可能にする。眼の中にレンズが長く存在すればするほど、細菌および他の微生物が当該レンズ表面上で増大することになる可能性が高くなることが知られている。本発明のレンズは、コンタクトレンズ等のポリマー製品上での細菌の増大抑制に役立つ。
【0084】
またさらに、本発明は、哺乳類の眼領域内に配されたレンズ上での微生物のコロニー形成に関連した有害事象を軽減する方法であって、当該方法は、少なくとも約14日間、哺乳類の眼上に、少なくとも一つの抗菌性金属塩を含む抗菌性レンズを配置することを含むか、配置することから成るか、あるいは、配置することから本質的に成る、方法を含み、当該抗菌性レンズは、当該14日の期間後に、少なくとも約0.5マイクログラムの抽出可能(extractible)な抗菌性金属を含むものである。他の実施の形態において、当該抗菌性レンズは、少なくとも約30日後に、少なくとも約0.5マイクログラムの当該抽出可能(extractible)な抗菌性金属を含むものである。この実施の形態において、当該抗菌性レンズは、連続的に装用されてもよく、あるいは、毎日装用するモード(就寝前に取り外し、かつ、起床時に再挿入するモード)で装用されてもよい。抗菌性金属塩の抽出は、上述した条件を用いて決定されうる。さらに他の実施の形態において、本発明の抗菌性レンズは、予定の装用期間中に、1日当たり0.5マイクログラムの抗菌性金属を放出する上で十分な抗菌性金属塩の初期濃度を含む。予定の装用期間は、レンズが、患者による装用に推奨される長さの期間である。
【0085】
用語「レンズ」、「抗菌性レンズ」および「金属塩」は、すべて、上述した意味および好適な範囲を有している。語句「微生物のコロニー形成に関連した有害事象」は、コンタクトレンズによる眼の炎症、コンタクトレンズに関連した辺縁性潰瘍、コンタクトレンズに関連した眼の充血、浸潤性角膜炎、細菌性角膜炎、および、これらの同類の症状を含むが、これらに限定されるものではない。用語「哺乳類」とは、あらゆる温血性の高等脊椎動物を意味し、好適な哺乳類は、ヒトである。
【0086】
以下の検査方法が、実施例において使用された。
【0087】
レンズのオートクレーブ処理後におけるレンズの銀の含有率は、機器中性子放射化分析法「INAA」によって決定された。この機器中性子放射化分析法(INAA)は、核反応における中性子で放射化されることによって特定放射性核種の人工的な誘導に基づく定性元素分析法および定量元素分析法である。サンプルの放射の後には、放射性核種の崩壊によって放出される特徴的ガンマ線の定量測定法が行われる。特定のエネルギーで検出されたガンマ線は、特定の放射性核種の存在の兆候であり、この兆候は高度の特異性を可能にする。ベッカー・ディー・エー(Becker, D. A.);グリーンバーグ・アール・アール(Greenberg, R. R.);ストーン・エス・エフ(Stone, S. F.)による1992年の放射分析核化学誌(J. Radioanal. Nucl. Chem.)第160(1)巻の第41頁〜第53頁;ベッカー・ディー・エー(Becker, D. A.);アンダーソン・ディー・エル(Anderson, D. L.);リンドストローム・アール・エム(Lindstrom, R. M.);グリーンバーグ・アール・アール(Greenberg, R. R.);ガリティ・ケー・エム(Garrity, K. M.);マッケー・イー・エー(Mackey, E. A.)による1994年の放射分析核科学誌(J. Radioanal. Nucl. Chem.)第179(1)巻の第149頁〜第154頁。コンタクトレンズ材料中での銀およびヨウ化物の含有量を定量化するために使用された当該機器中性子放射化分析法(INAA)では、以下の二つの核反応を使用する:
1.活性化反応において、原子炉で生成された放射性の中性子の捕獲後に、110Agは、安定した109Agから生成され、かつ、128Iは、安定した127Iから生成される。
2.崩壊反応において、110Ag(τ1/2=24.6秒)および128I(τ1/2=25分)は、当該放射性核種に特徴的なエネルギー(Agに関しては657.8キロ電子ボルト、および、Iに関しては443キロ電子ボルト)で初期濃度に比例する陰電子放出によって、崩壊する。
放射性標準品およびサンプルからの110Agおよび128Iの崩壊に特異的なガンマ線の放出は、ガンマ線分光法、定評のある波高分析法によって測定され、これにより、分析物の濃度の測定値を得ることができる。
【0088】
霞みは、室温で、平坦な黒色背景上に置かれた、20ミリメートル×40ミリメートル×10ミリメートルの透明なガラス製セル内のホウ酸緩衝食塩水中に検査用の水和レンズを配置し、下から、当該セルの法線に対して66°の角度で、光ファイバ灯(屈折力(power)4〜5.4に設定された口径0.5インチ(約1.27センチメートル)の光誘導装置を備えたチタン・ツール・サプライ社(Titan Tool Supply Co.)製の光ファイバ灯)からの光を照射し、上から、レンズセルに垂直に、レンズプラットホームより14ミリメートル上に配置されたビデオカメラ(ナビター(Navitar)社製ズームレンズTV Zoom 7000を備えたカメラDVC1300C:19130RGB)で当該レンズ画像を撮影することによって、測定される。背景の散乱は、EPIX XCAP V1.0ソフトウエアを用いて空白セルの画像を減じることによって、当該レンズの散乱から差し引かれる。差し引かれた散乱光の画像は、当該レンズの中央部分の10ミリメートル上を積分した後に、ヘイズ値100に任意に設定されるジオプター:−1.00のCSI Thin Lens(登録商標)を、ヘイズ値ゼロに設定された無レンズの状態と比較することによって定量分析される。五つのレンズが分析されると共に、これらの結果は、標準的なCSIレンズの百分率として、ヘイズ値を得るように平均化される。
【0089】
主観的な霞みの測定は、絞りを全開に設定する「暗視野」モードで、ニコン社製顕微鏡SMZ1500を用いて行われた。評価対象のレンズは、SSPSで満たされたガラス製のペトリ皿内に配置された後に、顕微鏡の検査台上に置かれた。この方法から得られる定量値は、以下のように、上記で測定されたヘイズ値の百分率と概ね一致する:
「濃い霞み」>〜100%
「薄い霞み」<〜70%
「非常に薄い霞み」<〜40%
【0090】
色は、以下のように測定された:サンプルは、室温で、ホウ酸緩衝硫酸ナトリウムの包装用溶液(SSPS)中において平衡化された。過剰な湿気は、レンズ表面から除去された。当該レンズは、顕微鏡用スライド板上に配置され、かつ、スポンジ状の綿棒を用いて、ならして平坦化された。一滴の包装用溶液が当該レンズに滴下され、第二の顕微鏡用スライド板で覆われ、当該レンズ上または下に気泡が存在しないことを確実にした。当該レンズの中央部分は、ソフトウエアQA Master 2000を搭載した比色計X-Rite Model SP64の開口上の白色背景前に位置決めされる。この比色計は、ワンデイ・アキュビュー(1 DAY ACUVUE)コンタクトレンズを用いて調整される。三つの示度が観測され、かつ、その平均値が記録される。上述した検査法を用いて、6回測定され、かつ、その平均化されたワンデイ・アキュビュー(1 DAY ACUVUE)コンタクトレンズに関するL*a*b*値は、以下のとおりである。
L*=72.33±0.04、a*=1.39±0、b*=0.38±0.01
【0091】
反応性混合物の紫外・可視(UV-VIS)スペクトルは、機器UVICAM UV300を用いて測定された。データは、1回の走査および1.5ナノメートルの帯域幅を用いて、200ナノメートル〜800ナノメートルから収集された。各実施例に掲げられた基線溶媒(baseline solvent)を使用した。未処理データは、グラフ作成および分析のために、エクセル(Excel)にエクスポートされた。複数のスペクトルは、比較目的でグラフ化された上記波長に対して標準化された。銀を含有するモノマー類では、銀含有成分の添加の24時間後の紫外・可視(UV-VIS)データが取得された。
【0092】
平衡化したパーキン・エルマー(Perkin Elmer)社製の走査型スペクトロメーター・ラムダ19 UV/VVIS(二重単色光分光器システム(double monochormator system))を用い、200ナノメートル〜800ナノメートルの範囲の全域を1ナノメートル間隔で走査し、以下の設定:幅4ナノメートルのスリット、960ナノメートル/分の走査速度、平面度(smooth)=2ナノメートル、近赤外線(NIR)感度=3、ランプ変更=319.2ナノメートルおよび検出器変更=860.8ナノメートルで、レンズの紫外・可視(UV-VIS)スペクトル(200ナノメートル〜800ナノメートルでの光透過率%)が取得された。レンズは、円形のサンプルホルダー上に平面になるように配置され、かつ、レンズの皺および伸びを最小限にするために挟みつけられる。当該レンズおよびホルダーは、包装用溶液で満たされたキュベット内に配置され、かつ、当該レンズの前面がサンプルビームに面するように配向される。複数のスペクトルは、当該機器に搭載されたソフトウエアを用い、かつ、等式:%Tave=S/Nを用いて算出されるが、ここで、Sは特定領域での%Tの合計であり、かつ、Nは波長の数である。
【0093】
プラスチック製品全体への金属塩の分布は、実施例23に記述されているように、電子プローブ微量分析法(EPM)を用いて測定された。
【0094】
粒子サイズは、レーザー光散乱法または動的光散乱法を用いて測定された。約500ナノメートルを超える粒子サイズ範囲を有するサンプルでは、レーザー回折粒子サイズ分析器Horiba-LA930が使用された。機器の点検は、%Tの空試験値に基づいて実行された。1ミリリットルのサンプル溶液は、媒体としての150ミリリットルの水を収容した循環浴中に導入された。1.7−0.1iの相対屈折率および循環速度5が使用された。サンプルは、超音波処理を用いる機器内での測定前に、2分間、超音波処理された。分析には、トリトン(Triton)(登録商標)X-100(ユニオンカーバイド(Union Carbide)社から市販されている)(0.1%)が界面活性剤として使用された。三通りの分析が実行され、かつ、当該分析結果は、互いに一致することを確認するために比較された。機器は、粒子サイズの平均値と共に粒子サイズ分布を示すグラフを含む報告を与えた。
【0095】
約500ナノメートル未満の粒子サイズ範囲を有するサンプルでは、動的光散乱装置Malvern 4700が使用された。機器の点検は、サンプル分析前に、NISTの追跡可能な標準サイズのポリスチレン粒子(NIST traceable standard size polystyrene particles)を用いて実行された。1ミリリットルのサンプルは、水で20ミリリットルに希釈され、かつ、当該サンプルは、ブランソン(Branson)社製の超音波プローブを用いて、1分間、超音波処理され、かつ、相対屈折率および粘度値の双方は、上記ソフトウエアに入力された。機器は、粒子サイズの平均値と共に粒子サイズ分布を示すグラフを含む報告を与えた。
【0096】
レンズは、以下の方法を用いて、黄色ブドウ球菌(S. aureus)に対する有効性に関して評価された。黄色ブドウ球菌(Staphylococcus aureus)の臨床分離株031(Clinical Isolate 031)の培養液は、トリプシン大豆培地(TSB)中で、一晩、増殖された。培養液は、リン酸緩衝食塩水(PBS、pH=7.4±0.2)中で三回、洗浄され、かつ、細菌ペレットは、10ミリリットルの2%トリプシン大豆培地・リン酸緩衝食塩水(TSB- PBS)中で再懸濁された。細菌接種材料は、最終濃度が約1×108コロニー形成単位/ミリリットル(cfu/mL)になるように調製された。連続希釈法は、約1×104cfu/ミリリットルの接種材料濃度を達成するように2%トリプシン大豆培地・リン酸緩衝食塩水(TSB- PBS)中で実行された。
【0097】
殺菌済みのコンタクトレンズは、残留溶液を除去するために、30ミリリットルのリン酸緩衝食塩水(PBS、pH=7.4±0.2)を三回交換しながら、水洗された。水洗された各コンタクトレンズは、500マイクロリットルの細菌接種材料と共に、殺菌済みの組織培養プレートの別個の検査穴内に配置された後に、当該プレートには、35±2℃で20±2時間、震盪機‐培養器(100回転/分)内で回転が与えられた。各レンズ、および対応する細胞懸濁液は、各検査穴から除去され、0.05重量/総容量(w/v)%のツイーン(Tween)(商標)80を含有する9.5ミリリットルのリン酸緩衝食塩水(PBS)(TPBS)中に配置された。
【0098】
その後、レンズ、および対応する細胞懸濁液は、1600回転/分で3分間、ボルテックスされ、残留する細菌のレンズに対する粘着をなくす遠心力を利用した。得られる上澄液の生菌数は、標準希釈液およびプレートカウント法を用いて算出された。レンズに関連して回収された生菌数の結果は平均化された。
【0099】
本発明を説明するために、以下の実施例が組み込まれている。これらの実施例は本発明を限定するものではない。当該実施例は、本発明を実施する方法を示唆するものにすぎない。コンタクトレンズならびに他の特殊製品に見識のある者は、本発明を実施する他の方法を見出せる。しかし、これらの方法は、本発明の範囲内に入るものであると見なされる。
【0100】
〔実施例〕
以下の略称が、実施例で使用された。
AHM:3-アリルオキシ-2-ヒドロキシプロピルメタクリレート(3-allyloxy-2-hydroxypropyl methacrylate)
AMBN:2,2'-アゾビス(2-メチルブチロニトリル)(2,2'-Azobis(2-Methylbutyronitrile))
BHT:ブチル化ヒドリキシトルエン
Blue HEMA:実施例4または米国特許第5,944,853号明細書に記述されるような反応性青色4号とヒドロキシエチルメタクリレート(HEMA)との反応生成物
CGI 1850:1-ヒドロキシシクロヘキシルフェニルケトン(1-hydroxycyclohexyl phenyl ketone)とビス(2,6-ジメチオキシベンゾイル)-2,4-4-トリメチルペンチルホスフィンオキシド(bis (2,6-dimethyoxybenzoyl)-2,4-4-trimethylpentyl phosphine oxide)との1:1(w/w)の混合物
CGI 819:ビス(2,4,6-トリメチルベンゾイル)-フェニルホスフィンオキシド(bis(2,4,6-trimethylbenzoyl)-phenylphosphineoxide)
DI水:脱イオン水
DMA:N,N-ジメチルアクリルアミド
DAROCUR 1173:2-ヒドロキシ-2-メチル-1-フェニル-プロパン-1-オン(2-hydroxy-2-methyl-1-phenyl-propan-1-one)
EGDMA:エチレングリコールジメタクリレート
HEMA:ヒドロキシエチルメタクリレート
BAGE:グリセリンのホウ酸エステル
IPA:イソプロピルアルコール
MAA:メタクリル酸
Macromer:実施例22で製造されるようなシリコーン含有マクロマー
mPDMS:モノメタクリロキシプロピル末端化ポリジメチルシロキサン(mono-methacryloxypropyl terminated polydimethylsiloxane)(分子量:800〜1000)
Norbloc:2-(2'-ヒドロキシ-5-メタクリロキシエチルフェニル)-2H-ベンゾトリアゾール(2-(2’-hydroxy-5-methacrylyloxyethylphenyl)-2H-benzotriazole)
HO-mPDMS:実施例21で調製されるようなモノ-(3-メタクリロキシ-2-ヒドロキシプロピロキシ)プロピル末端化、モノ-ブチル末端化ポリジメチルシロキサン(mono-(3-methacryloxy-2-hydroxypropyloxy)propyl terminated, mono-butyl terminated polydimethylsiloxane)(分子量:612)
AgI Particles:合成実施例3に従って形成されたヨウ化銀粒子
ppm:乾燥レンズの1グラム当たりのサンプルの百万分の一マイクログラム
PAA:ポリアクリル酸(分子量:2000)
PVP:ポリビニルピロリドン
PVA:ポリビニルアルコール
SiMMA:3-メタクリロキシ-2-ヒドロキシプロピロキシ)プロピルビス(トリメチルシロキシ)メチルシラン3-methacryloxy-2-hydroxypropyloxy)propylbis (trimethylsiloxy)methylsilane)
SSPS:以下に記載するように作られた、ホウ酸緩衝硫酸ナトリウムの包装用溶液
TAA:三級アミルアルコール
TBACB:テトラブチルアンモニウム3-クロロベンゾエート(tetrabutylammonium 3-chlorobenzoate)
THF:テトラヒドロフラン
TRIS:3-メタクリロキシプロピルトリス(トリメチルシロキシ)シラン(3-methacryloxypropyltris(trimethylsiloxy)silane)
w/w:重量/全重量
w/v:重量/全容量
v/v:容量/全容量
【0101】
使用される以下の組成物が調製された。
【0102】
涙様液(Tear-like fluid)(TLF)緩衝液:
涙様液緩衝液(TLF緩衝液)は、0.137グラムの重炭酸ナトリウム(シグマ(Sigma)社製S8875)および0.01グラムのD-グルコース(シグマ(Sigma)社製G5400)を、カルシウムおよびマグネシウムを含有するリン酸緩衝食塩水(PBS)(シグマ(Sigma)社製D8662)中に添加することによって、調製された。この涙様液緩衝液は、各成分が完全に溶解されるまで(約5分間)、室温で撹拌された。
【0103】
脂質保存溶液は、約60℃で、約1時間、十分に撹拌しながら、透明になるまで、以下の脂質を涙様液緩衝液中に混合することによって調製された。
コルステリルリノール酸(シグマ(Sigma)社製C0289) 24ミリグラム/ミリリットル
酢酸リナリル(シグマ(Sigma)社製L2807) 20ミリグラム/ミリリットル
トリオレイン(シグマ(Sigma)社製7140) 16ミリグラム/ミリリットル
オレイン酸プロピルエステル(シグマ(Sigma)社製O9625) 12ミリグラム/ミリリットル
ウンデシレン酸(シグマ(Sigma)社製U8502) 3ミリグラム/ミリリットル
コレステロール(シグマ(Sigma)社製C8667) 1.6ミリグラム/ミリリットル
【0104】
当該脂質保存溶液(0.1ミリリットル)は、0.015グラムのムチン(ウシ顎下腺由来のムチン類:(シグマ(Sigma)社製M3895, 型式1-S))と混合された。3つの1ミリリットル涙様液緩衝液が、脂質ムチン混合物中に添加された。当該溶液は、全成分が溶解状態になるまで(約1時間)、撹拌された。全量が100ミリリットルになるまで、適量の涙様液緩衝液が添加され、かつ、十分に混合された。
【0105】
以下の成分は、前記で調製された当該100ミリリットルの脂質ムチン混合物中に、一成分ずつ、以下に列挙された順序で、添加された。全添加時間は、約1時間であった。
ウシ血漿由来の酸性糖蛋白質(シグマ(Sigma)社製G3643) 0.05ミリグラム/ミリリットル
ウシ胎児血清(シグマ(Sigma)社製F2442) 0.1%
ウシ血漿由来のガンマ・グロブリン(シグマ(Sigma)社製G7516) 0.3ミリグラム/ミリリットル
ベータ・ラクトグロブリン(牛乳リポカリン(bovine milk lipocaline))(シグマ(Sigma)社製L3908) 1.3ミリグラム/ミリリットル
鶏卵白由来のリゾチーム(シグマ(Sigma)社製L7651) 2ミリグラム/ミリリットル
ウシ初乳由来のラクトフェリン(シグマ(Sigma)社製L4765) 2ミリグラム/ミリリットル
【0106】
得られた溶液は、一晩、4℃で放置した。当該溶液のpHは、1規定の塩酸で、7.4に調整した。当該溶液は、使用前に−20℃で、濾過され、かつ、保存された。
【0107】
〔ホウ酸緩衝硫酸ナトリウムの包装用溶液(SSPS)〕
包装用溶液は、脱イオン水中に以下の成分を含有している:
0.18重量%のホウ酸ナトリウム[1330-43-4]、マリンクロート(Mallinckrodt)社製
0.91重量%のホウ酸[10043-35-3]、マリンクロート(Mallinckrodt)社製
1.4重量%の硫酸ナトリウム[7757-82-6]、シグマ(Sigma)社製
0.005重量%のメチルエーテルセルロース[232-674-9]、フィッシャー・サイエンティフィック(Fisher Scientific)社製
【0108】
〔実施例1〕
12.6グラムの5%PVP(K値12)脱イオン水溶液が調製された。3.94グラムの1%硝酸銀溶液が添加され、室温で、磁石式の撹拌棒を用いて5分間、混合された。その後、3.47グラムの1%ヨウ化ナトリウム溶液が添加され、室温で、磁石式の撹拌棒を用いて5分間、混合された。透明なヨウ化銀ナノ分散液が得られた。
【0109】
〔比較例1〕
1.0グラムの1%硝酸銀溶液が、室温で、1.0グラムの1%ヨウ化ナトリウム溶液中に添加された。ヨウ化銀沈殿物を含む非常に濁った分散液が得られた。その後、5グラムの5%PVP(K値12)溶液が、磁石式の撹拌棒を用いて48時間、混合しながら、添加された。沈殿物は解消されなかった。
【0110】
〔実施例2〕
PVP(K値12)に代えて、当初の溶液が、98%加水分解されたポリビニルアルコール(セルボル(Celvol)09-523:米国テキサス州ダラスのセラニーズ・ケミカルズ(Celanese Chemicals)社製)で調製されたことを除き、実施例1が繰り返された。透明なナノ分散液が得られた。
【0111】
〔合成実施例1〕
撹拌装置、温度調節装置、および、冷却および加熱用のジャケットを備えた、5リットルのガラス製反応器には、以下の化合物の混合物が充填された。
【表1】
【0112】
反応器温度が71℃まで上げられ、かつ、2.11グラムのAMBNが添加された。AMBNが溶解され、かつ、当該反応器が遅い窒素流で覆われた。20時間、温度が71℃に保持された。
【0113】
ネジ付き蓋および磁石式の撹拌棒を備えた五つの1リットル瓶が用意され、未処理の生成物が各瓶内に600グラムずつ注入された。溶液は、磁石式の撹拌棒で絶えず撹拌しながら、水浴中で60℃に加熱された。その後、54グラムのヘプタン(9%)が添加され、かつ、当該溶液が60℃に再加熱された。撹拌操作が停止され、かつ、当該瓶が60℃の水浴中に置かれた。温度が、20時間かけて、24℃まで下げられた。ここで、上相は透明な液状になり、かつ、下相は半固形状になった。上相は、最も大きい部分を占めている(瓶の全量の約80%)が、固形状ポリマーの含有率は小さい(凡そ1.5%〜2.5%)。
【0114】
各瓶内の上相は廃棄され、かつ、下相は、固形分12%および水3%を特徴とする2125グラムのポリマー溶液を得るために、水性エタノール中に再溶解された。
【0115】
このポリマー溶液は、以下のパラメータで、不活性ループ(Inert Loop)、出口用フィルタ、および、高性能サイクロンを備えたミニ噴霧乾燥機(Mini Spray Dryer)B-290を用いて、噴霧乾燥された。
【表2】
【0116】
結果は、細かく白色でフワフワした250グラムの粉末をもたらし、この粉末は、約97%まで乾燥されていた。この粉末は、磁石式の撹拌棒を備えた、いくつかの1リットルフラスコ(約77グラムずつ)に移された。これらのフラスコは、当該フラスコ中の材料をさらに乾燥させるために、100℃〜130℃で一晩、30mbar(30ヘクトパスカル)未満の減圧下で、真空処理された。
【0117】
翌朝、真空状態は、乾燥アルゴン雰囲気により破られ、かつ、当該フラスコは、制御された乾燥窒素雰囲気が充填されたボックス内に移された。当該フラスコの総重量は、温度が下がった後に、確認された。各1リットルフラスコには、300グラムのNMP(無水N-メチルピロリドン:プルム(purum);無水物;分子篩済み(over molecular sieves)のフルカ(Fluka)社製)が添加されて、当該粉末を完全に溶解させると共に、当該複数のフラスコでは、その均質性に関する点検が行われた。MAH(メタクリル酸無水物:純度98%)は、50cc(50ミリリットル)の円筒ガラス容器内で秤量されると共に、移す前に、メタクリル酸無水物(MAH)を希釈するために、50グラムの無水N-メチルピロリドン(NMP)が添加された。当該ガラス容器内を水洗して完全に移したことを保証するために、他の50グラムの無水N-メチルピロリドン(NMP)が使用された。トリエチルアミン(フルカ(Fluka)社からのpuriss p.a.)がフィン付ピペットを用いて直接、添加された。蓋が堅く締め付けられ、テープで封止されて、窒素流が止められた。反応は、約40時間、続けられた。
【0118】
上述のように製造されたポリマーは、以下のように精製された。75グラムのポリマーは、400ミリリットルの無水N-メチルピロリドン(NMP)中で溶解された。二つの5リットルガラス製ビーカーには、それぞれ4リットルの脱イオン水、30ミリリットルの発煙HCl(塩酸)および磁石式の撹拌棒が入れられた。当該ビーカーには、それぞれ200ミリリットルずつ、約10ミリリットル/秒の速度で、先の反応で得られた官能性生成物が徐々に注入された。沈殿物が生じ、水相が除去された。膨潤した残留ポリマーは、300ミリリットルのエタノール中に再溶解された。
【0119】
さらに二つの5リットルガラス製ビーカーには、それぞれ4リットルの脱イオン水および磁石式の撹拌棒が入れられた。それぞれ4リットルの脱イオン水が充填された二つの5リットルガラス製ビーカーには、ポリマー/エタノール溶液が注入されて、沈殿物が再度、生じた。水相が除去されると共に、残留塩酸をさらに抽出するために、新しい脱イオン水が添加された。約12時間後に、水相が除去されると共に、膨潤したポリマー材料の重量が確認された(約120グラム)。
【0120】
膨潤したポリマー材料は、固形含有率を13±0.5%にするために、エタノール中に再溶解され、その後、当該溶液は、25ミリメートルGD/X0.45マイクロメートルワットマンフィルタ(25 mm GD/X 0.45μm Whatmann filter)により濾過された。当該溶液は、不活性ループ(Inert Loop)、出口用フィルタ、および、高性能サイクロンを備えたミニ噴霧乾燥機(Mini Spray Dryer)B-290を用いて、噴霧乾燥された。以下のパラメータが適用された。
【表3】
【0121】
結果は、細かく白色でフワフワした約155グラムの粉末をもたらした。
【0122】
〔実施例3〕
合成実施例1で調製されたコポリマー(3.49グラム)は、4.9グラムのマスターバッチ溶液(希釈剤としてのプロピレングリコール99.89%、光重合開始剤としてのジメトキシベンゾイルビス(アシル)ホスフィンオキシド(Dimethoxybenzoyl bis(acyl) phosphine oxide)1.10%、および、阻害剤としての4-メトキシフェノール0.011%を含む)と混合された。実施例1で調製された2グラムのナノ分散液が秤量されてから、コポリマー/マスターバッチ溶液と混合された。得られた混合物は、その内部に閉じ込められた空気を除去するために、2500回転/分で15分間、遠心分離処理された。透明なプレポリマーが得られた。
【0123】
プレポリマーは、窒素下で12時間、脱気された熱可塑性コンタクトレンズ用型(ポリスチレン製の前曲面および後曲面)に分注された。この型内のプレポリマーは、室温で30ミリワット/平方センチメートルの光量で、20℃で空中において30秒間、照射された。その後、レンズは、20℃の脱イオン水中で20分間、水和され、ホウ酸緩衝硫酸ナトリウムの包装用溶液(SSPS)中に収容され、121℃で18分間、殺菌処理された。レンズは、暗視野の顕微鏡下で測定されるように、非常に薄い霞みを呈していた。中性子活性化法を用いて測定された五つのレンズにおける銀の平均含有量は、9.72マイクログラムであり、標準偏差が0.16マイクログラム/レンズであることが分かった。
【0124】
〔実施例4〕
0.339グラムのPVP(K値12)粉末は、3.487グラムの1%ヨウ化ナトリウム溶液中に添加され、10分間、混合されて、塩前駆体溶液Aを形成した。PVP(K値12、0.266グラム)は、4.29グラムの1%硝酸銀溶液にゆっくり添加されて金属剤溶液Bを形成した。塩前駆体溶液A(0.379グラム)は、以下の表1に示された17.603グラムのモノマー混合物中に添加され、かつ、3分間、混合された。その後、金属剤溶液B(0.3963グラム)は、当該モノマー混合物中に添加され、かつ、10分間、撹拌された。
【0125】
当該モノマー混合物は、真空(29水銀柱インチ(約98.21キロパスカル))下で20分間、脱気処理された。モノマー混合物は、熱可塑性コンタクトレンズ用型(ポリスチレン製の前曲面および後曲面)に分注され、かつ、この型内のモノマー混合物は、室温で5ミリワット/平方センチメートルの光量で、窒素下で6分間、照射された。その後、レンズは、20℃の脱イオン水中で水和され、SSPS中に収容され、121℃で約20分間、高圧殺菌器中で殺菌処理された。レンズは、暗視野の顕微鏡下で測定されるように、非常に薄い霞みを呈していた。銀の含有量は、中性子活性化法を用いて測定され、当該銀の含有量は、4.7マイクログラムであり、標準偏差が0.11マイクログラム/レンズであった。
【表4】
【0126】
〔実施例5〕
PVP(K値12、0.946グラム)は、表1に挙げられた30.7グラムの反応性混合物にゆっくり添加され、25分間混合することにより溶解された。0.0177グラムの硝酸銀(固体)は、添加され、かつ、溶解されるまで混合された。その後、0.0300グラムのヨウ化ナトリウム(固体)は、添加され、その混合物は、室温で1時間、混合されて、粒子含有反応性混合物を形成した。粒子含有反応性混合物は、真空(29水銀柱インチ(約98.21キロパスカル))下で10分間、脱気処理された。粒子含有反応性混合物は、実施例4に記述されているように、コンタクトレンズ用型(ポリスチレン製の前曲面および後曲面)に分注され、硬化され、水和され、包装され、殺菌処理された。レンズは、暗視野の顕微鏡下で測定されるように、非常に薄い霞みを呈していた。銀の含有量は、中性子活性化法を用いて測定され、当該銀の含有量は、12.8マイクログラムであり、標準偏差が0.4マイクログラム/レンズであった。
【0127】
〔実施例6〜13〕
以下の各実施例において、二つの混合物が調製された。塩前駆体混合物(「SPM」)は、表1に挙げられた反応性混合物、表2に挙げられた量のPVP(K値12)およびヨウ化ナトリウムを混合することによって調製された。PVPの濃度(重量%)は、粒子含有反応性混合物中のPVPの重量%として挙げられている。金属剤混合物(「MAM」)は、表1に挙げられた反応性混合物、表2に挙げられた量の硝酸銀を混合することによって調製された。各混合物は、全成分が一体化されるまで混合されて透明な混合物が形成された(約5時間〜約19時間)。各実施例において、略同一容量の塩前駆体混合物(SPM)および金属剤混合物(MAM)は、混合されて、表3の第2欄に挙げられた、硝酸銀に対するヨウ化ナトリウムのモル比を有する反応性混合物を形成した。1時間混合した実施例8を除き、各反応性混合物が30分間以上、混合された。反応性混合物は、表3に挙げられた条件下で脱気処理された。脱気処理された各反応性混合物は、実施例4に記述されているように、分注され、硬化され、水和され、包装され、殺菌処理された。レンズの霞みは、暗視野の顕微鏡下で測定された。銀の含有量は、中性子活性化法を用いて測定された。全レンズ中における標的の銀取り込み量(targeted silver uptake)は、約10マイクログラムであった。結果は、以下の表3に記録された。
【表5】
【表6】
【0128】
モル過剰のヨウ化ナトリウムを含有した実施例6および7は、殺菌前において非常に薄い霞みを呈しており、殺菌後において薄い霞みおよび通常の色を呈していた。対照的に、同一条件を用いて調製されたが、過剰の硝酸銀を含有した実施例8、9および10は、それぞれ、黄色、薄茶色および茶色を呈していた。したがって、実施例6および7は、特に、金属剤が金属塩よりも感光性である場合に、金属剤の金属塩への変換を保証する処理条件が改善された色を呈する製品をもたらすことを示している。
【0129】
2.6%のPVPおよび50分間の脱気ステップを有した実施例12は、殺菌前において非常に薄い霞みを呈しているが、殺菌後において濃い霞みを呈しており、したがって、この実施例12は、殺菌中に、粒子の熟成が生じ得ることを示唆している。しかし、反応性混合物が硬化される前に、粒子の熟成ステップが追加された場合(実施例13:70℃で20分間)に、得られたレンズは、殺菌後において非常に薄い霞みを呈していた。
【0130】
〔実施例14〕
以下の表4中の反応性混合物が調製された。反応性成分は、全反応性成分(希釈剤を除く)の重量%として記録され、かつ、当該希釈剤は、最終反応性混合物の重量%である。固体の硝酸銀(0.040グラム)は、28.09グラムのモノマー混合物中に添加された。その後、ヨウ化ナトリウム(固体:0.0427グラム)は、当該混合物中に添加され、かつ、室温で1時間、混合された。混合後に、容器の底部に固形分が未だ残っていた。反応性混合物は、熱可塑性コンタクトレンズ用型(ゼオン(Zeon, Corp.)社から入手されたゼオノール(Zeonor:登録商標)から製造された前曲面および後曲面)に分注され、かつ、この型内の混合物には、室温で5ミリワット/平方センチメートルの光量で、窒素下で10分間、照射された。その後、レンズは、25℃の脱イオン水中で水和され、ホウ酸緩衝硫酸ナトリウムの包装用溶液中に収容され、121℃で20分間、高圧殺菌器中で殺菌処理された。レンズは、暗視野の顕微鏡下で測定されるように、非常に薄い霞みを呈していたが、僅かに淡い黒色を呈していた。銀の含有量は、中性子活性化法を用いて測定されたところでは、6.2マイクログラムであり、標準偏差が0.21マイクログラム/レンズであった。
【表7】
【0131】
〔実施例15〕
PVP K値12(9.29グラム)は、200.00グラムのTPME中に、撹拌しながら、ゆっくりと添加され、かつ、20分間、混合された。その後、0.7040グラムの固体の硝酸銀が当該溶液中に添加されて、金属剤溶液を形成した。この金属剤溶液は、磁石式の撹拌装置を用いて、6時間、撹拌された。
【0132】
ヨウ化ナトリウム(0.8880グラム)は、200.13グラムのTPME中に添加されて、塩前駆体溶液を形成した。この塩前駆体溶液は、磁石式の撹拌装置を用いて、6時間、撹拌された。金属剤溶液(170.89グラム)は、絶えず撹拌しながら、当該塩前駆体溶液(171.21グラム)中に混入された。透明なナノ分散液が得られた。溶液は、25分間、混合された。その後、全ナノ分散液は、以下の表5に挙げられた500.20グラムの反応性混合物中に混合された。
【表8】
【0133】
反応性混合物は、29水銀柱インチ(740mmHg)(約98.21キロパスカル)で15分間、脱気処理された。反応性混合物は、熱可塑性コンタクトレンズ用型(ゼオン(Zeon, Corp.)社から入手したゼオノール(Zeonor:登録商標)から製造された前曲面および後曲面)に分注され、室温で5ミリワット/平方センチメートルの光量で、窒素下で6分間、照射された。その後、レンズは、30分間、20℃の脱イオン水中で、その後、60分間、70%のIPA中で水和された後、1分間、脱イオン水で水洗され、2時間以上、脱イオン水で水洗が行われており、これらの操作はすべて室温で行われた。その後、レンズは、点検され、ホウ酸緩衝硫酸ナトリウムの包装用溶液(SSPS)中に収容され、121℃で18分間、高圧殺菌器中で殺菌処理された。
【0134】
レンズは、10.70マイクログラムの銀の平均取り込み量を有しており、標準偏差(5枚のレンズ)は0.2マイクログラム/レンズであった。レンズの霞みは、68%であり、標準偏差(5枚のレンズ)は8.9%であった。
【0135】
〔実施例16〕
419.5グラムの反応性混合物が、表6に挙げられた成分から調製された。
【0136】
HEMAは、TPMEに添加されて、HEMA/TPME(HEMA:TPME=5.1:10)溶液を形成し、かつ、清浄な琥珀色の瓶内で1時間、混合された。
【0137】
金属剤混合物は、清浄な琥珀色の瓶内で、7グラムのPVP(K値12)を70.0グラムの当該HEMA/TPME溶液中にゆっくりと添加し、かつ、磁石式の撹拌棒で撹拌することによって、形成された。金属剤混合物は、全PVP(K値12)が溶解されるまで、混合された。硝酸銀(0.49グラム)が添加され、かつ、全固形分が溶解されるまで、6時間、混合された。
【0138】
塩前駆体混合物は、清浄な琥珀色の瓶内で、0.42グラムのヨウ化ナトリウムを30グラムの当該HEMA/TPME溶液中に添加し、かつ、全固形分が溶解されるまで、6時間、磁石式の撹拌棒で混合することによって、形成された。
【0139】
金属剤(67.02グラム)混合物は、撹拌しながら、塩前駆体混合物中にゆっくりと注入され、かつ、1時間、混合された。金属塩のヨウ化銀を含有する透明な分散液が得られた。
【0140】
表6に挙げられた成分を有する反応性混合物が調製された。反応性成分(419.5グラム)および金属塩分散液(80.5グラム)は、琥珀色の瓶内で混合され、かつ、約24時間より長く混合された。その後、反応性混合物は、3マイクロメートルのフィルタで濾過され、かつ、29水銀柱インチ(約98.21キロパスカル)下で15分間、脱気処理された。
【表9】
【0141】
反応性混合物は、熱可塑性コンタクトレンズ用型(ゼオン(Zeon, Corp.)社から入手したゼオノール(Zeonor:登録商標)から製造された前曲面および後曲面)に分注され、室温で5ミリワット/平方センチメートルの光量で、窒素下で6分間、照射された。その後、レンズは、30分間、20℃の脱イオン水中で、その後、60分間、70%のIPA中で水和された後、1分間、脱イオン水で水洗され、2時間以上、脱イオン水で水洗が行われており、これらの操作はすべて室温で行われた。その後、レンズは、点検され、ホウ酸緩衝硫酸ナトリウムの包装用溶液中に収容され、121℃で18分間、高圧殺菌器中で殺菌処理された。
【0142】
レンズは、10.60マイクログラムの銀の平均取り込み量を有しており、標準偏差(5枚のレンズ)は0.2マイクログラム/レンズであった。レンズの霞みは、38.6%であり、標準偏差(5枚のレンズ)は4.3%であった。
【0143】
〔実施例17〕
0.0243グラムのPVP(K値12)は、10.0037グラムのTPME中に、ゆっくりと添加され、磁石式の撹拌棒を用いて、20分間、混合された。その後、0.0199グラムの硝酸銀が当該溶液中に添加され、かつ、当該溶液は、室温で4時間、混合されて、溶液Aを得た。0.054グラムの固体のヨウ化ナトリウムは、10.0326グラムのTPME中に添加され、かつ、室温で4時間、混合されて、溶液Bを得た。溶液Aは、溶液B中に注入され、かつ、20分間、混合されて、TPME中のヨウ化銀の透明なナノ分散液を得た。
【0144】
上述のように調製された4.20グラムのヨウ化銀ナノ分散液は、以下の表7に示されるような組成を有する5.13グラムのモノマー混合物中に添加され、かつ、12時間、混合された。その後、モノマーは、22水銀柱インチ(約74.50キロパスカル)の真空下で20分間、脱気処理された。反応性混合物は、熱可塑性コンタクトレンズ用型(ゼオン(Zeon, Corp.)社から入手したゼオノール(Zeonor:登録商標)から製造された前曲面および後曲面)に分注され、室温で5ミリワット/平方センチメートルの光量で、窒素下で6分間、照射された。その後、レンズは、30分間、20℃の脱イオン水中で、その後、60分間、70%のIPA中で水和された後、1分間、脱イオン水で水洗され、2時間以上、脱イオン水で水洗が行われており、これらの操作はすべて室温で行われた。その後、レンズは、点検され、ホウ酸緩衝硫酸ナトリウムの包装用溶液中に収容され、121℃で18分間、高圧殺菌器中で殺菌処理された。
【0145】
レンズは、12マイクログラムの銀の含有率を有しており、標準偏差(5枚のサンプル)は0.1マイクログラムであった。レンズの霞みは、84%であり、標準偏差(5枚のサンプル)は4であった。
【表10】
【0146】
〔比較例2〕
アキュビュー・アドバンス(ACUVUE ADVANCE:登録商標)ブランドのコンタクトレンズとしてビスタコン(Vistakon)社から入手可能な、ガリフィルコン系の硬化水和レンズは、ブリスターパック内の脱イオン水中に置かれた。過剰の脱イオン水が除去されると共に、0.8ミリリットルの塩前駆体混合物(脱イオン水中に1100ppmのヨウ化ナトリウムを含有する)が、当該レンズを収容しているブリスター中に添加され、かつ、室温で一晩、放置された。当該塩前駆体混合物が除去されると共に、0.8ミリリットルの金属剤混合物(脱イオン水中に700ppmの硝酸銀および5%PVP(K値90)を含有する)が添加された。3分後に、金属塩混合物が除去されると共に、脱イオン水(900マイクロリットル)が当該ブリスターに添加され、約5分間、放置され、最終的に除去された。この脱イオン水処理は、さらに二回反復されると共に、当該レンズは、SSPSを収容しているガラス製バイアル瓶に移された。このバイアル瓶は、封止されると共に、122℃で30分間、オートクレーブ処理された。レンズは、機器中性子放射化分析法(INAA)によって分析され、その結果、約16マイクログラムの銀を含有していた。
【0147】
〔実施例18および比較例2〕
実施例6および比較例2のレンズ中の銀の相対含有率は、電子プローブ微量分析法(EPM)を用いて測定され、コンタクトレンズ全体における含有銀の分布状態を確認した。
【0148】
複数のサンプルは、半分に切断され、かつ、試料を固定するための二つの小ネジ用に穴開け加工され雌ネジを切られた(tapped)、直径25ミリメートルのアルミニウム製ホルダー内に垂直にレンズ全体を取り付けることによって、プロファイル解析用に調製された。当該レンズは、材料の半分が当該ホルダー表面より上に出るように、固定された。その後、1枚刃の清浄なカミソリは、切断面を引き裂かないように一回の滑らかな行程で、レンズを半分に切断するために使用された。その後、これらのサンプルには、伝導性を確保するために、真空蒸発装置内で炭素被膜処理が施された。これらのサンプルの遠端には、伝導性をよりよくするためコロイド状炭素塗料で軽く塗装された。
【0149】
残りのレンズ半体の直径近くからの切片は、当該残りのレンズ半体から切り出されると共に、凹面を上にして、上面上の二つの両面炭素製「粘着タブ」を備えた直径25ミリメートルのホルダー上に注意深く置かれた。
【0150】
レンズの凸面は、二つの「粘着タブ」上に当該レンズ材料の凸面側の残りの弦を取り付けることによって、分析された。清浄なテフロン材料シート(厚さ0.032インチ(約0.0813センチメートル))は、コンタクトレンズが平坦になるように、炭素製「粘着タブ」にコンタクトレンズを押圧するために使用された。これらのサンプルには、炭素真空蒸発装置内で、20ナノメートル〜40ナノメートルの純正仕様(Spec-Pure)タイプのグラファイト被膜処理が施された。これらのサンプルの遠端には、伝導性をよりよくするためコロイド状炭素塗料で塗装された。
【0151】
当該複数のサンプルは、カメラSX50(1988年)、または、20キロ電子ボルト、50ナノアンペア、および、焦点ボケをしたビームサイズ:20マイクロメートルを含むレンズ表面を分析するための分析条件を用いる四つの波長分光計を備えた自動電子マイクロプローブSX100(2005年)のいずれかを用いて、分析された。当該ビームサイズは、プロファイル解析のために、5マイクロメートルに低減された。計数時間は、ピーク時で160秒であり、各オフピーク時で80秒であった。
【0152】
バックグラウンド位置は、スペクトル干渉が存在しないように選択された。バックグラウンド強度は、オフピーク位置間の線形補間によって算出された。また、強度は、検出器不感時間、ビームドリフトおよび標準強度ドリフトについて補正された。あらゆる分析では、有意なドリフトが指摘されなかった。銀に関する検出限界は、約40ppmであった。
【0153】
プロファイル解析の取得は、レンズ輪郭表面のうち、凸面(前曲面)を突き止め、かつ、その点から全横断を開始することによって、行われた。表面分析は、レンズ材料の切片の一側面で開始し、かつ、レンズ全体を横断する250マイクロメートルまたは500マイクロメートルの段差を用いることによって、実行された。この段差は、概ね、総距離(サンプル表面当たり25〜50データ点)が約8ミリメートル〜12ミリメートルであった。全データ点は、約4時間待った後でもサンプル表面を完全に平坦化されなかったサンプルについて分光光度計の焦点ボケが生じず、Zフォーカスに関して安定化することが確実になるように、手動で確認された。
【0154】
金属の銀は、銀の一次標準物質として使用された。標準物質および未確認物質には、20ナノメートルの純正仕様(Spec-Pure)タイプのグラファイト被膜処理が施され、かつ、標準物質および未確認物質については、標準物質の計数時間がピーク時で10秒であり、各オフピーク時で5秒間であることを除き、上述した条件下で実行された。
【0155】
図1は、実施例16および比較例1のレンズ中の銀の分布を編集して示すグラフであって、当該銀がレンズ形成後に、当該レンズ中に沈殿している状態を示すグラフである。図1から理解できるように、実施例21のレンズ中の金属塩の濃度(複数の四角形を結ぶ線によって示されている)は、レンズ全体で一貫している。比較例2のレンズでは、図1は、分析されたレンズが、当該レンズの前面および後面の20%以内の高濃度の銀を有しているが、レンズ中心部分では非常に少ない銀を有していることをも示している(複数のダイアモンド形を結ぶ線によって示されている)。
【0156】
〔実施例19〕
実施例16に従って形成されたレンズは、以下の方法を用いて、レンズから放出される銀について評価された。
【0157】
検査されるレンズは、過剰の液体を除去するために殺菌ガーゼを用いて拭き取られた後に、24穴を有し、かつ、各穴に1ミリリットルの涙様液(TLF)を収容する殺菌済みの細胞培養プレートの穴に、1レンズ/穴の割合で移された。当該プレートは、蒸発および脱水を防止するために被覆され、かつ、少なくとも100回転/分で撹拌しながら、35℃で培養された。24時間ごとに、当該レンズは、新鮮な1ミリリットル容量の涙様液(TLF)に移された。測定が実行される各時間間隔で、上記穴の中から、最低3枚のレンズが取り出され、かつ、100ミリリットルのリン酸緩衝食塩水(PBS)で三回〜五回、水洗された。これらのレンズは、過剰の液体を除去するためにペーパータオル上で拭き取られ、かつ、プロピレン・シンチレーションバイアル瓶(1レンズ/瓶)に移された。銀の含有率は、中性子活性化法によって分析された。
【0158】
比較例2のレンズも、上述したように検査された。両レンズの結果は、表3に示されている。図2において、ダイアモンド形状の点を結ぶ実線は実施例16のレンズからの結果であり、かつ、四角形状の点を結ぶ破線は比較例2のレンズの評価からの結果である。図2は、本発明のレンズが、比較例2のレンズよりも遅く、かつ、一貫して抗菌性金属を放出することを明らかに示している(ここで、レンズが形成された後に、銀塩がレンズ中に沈殿している)。
【0159】
〔実施例20〕
実施例16および比較例2の各レンズは、以下の方法を用いて、細菌に対する有効性に関して評価された。黄色ブドウ球菌(Staphylococcus aureus)ATCC#15442(米国メリーランド州ロックビルのアメリカン・タイプ・カルチャー・コレクション)の培養液は、トリプシン大豆培地中で、一晩、増殖された。培養液は、リン酸緩衝食塩水(PBS、pH=7.4±0.2)中で三回、洗浄され、かつ、細菌ペレットは、10ミリリットルの2%トリプシン大豆培地・リン酸緩衝食塩水(TSB- PBS)中で再懸濁された。細菌接種材料は、最終濃度が約1×108コロニー形成単位/ミリリットル(cfu/mL)になるように調製された。連続希釈法は、約1×104cfu/mLの接種材料濃度を達成するように2%トリプシン大豆培地・リン酸緩衝食塩水(TSB- PBS)中で実行された。
【0160】
殺菌済みのコンタクトレンズは、残留溶液を除去するために、30ミリリットルのリン酸緩衝食塩水(PBS、pH=7.4±0.2)を三回交換しながら水洗された。水洗された各コンタクトレンズは、500マイクロリットルの細菌接種材料と共に、殺菌済みの組織培養プレートの別個の検査穴内に配置された後に、当該プレートには、35±2℃で約20時間、震盪機‐培養器(100回転/分)内で回転が与えられた。各レンズは、ガラス製バイアル瓶から取り出され、三回交換したリン酸緩衝食塩水で五回、水洗されて、緩く結合した細胞群を除去した。培養後に、各タイプのレンズのうち、3枚のレンズは、初期の対細菌有効性(以下に記述される)を測定するために、取り出され、かつ、残りのレンズは、上述したように、500マイクロリットルの涙様液(TLF)を収容する、新たなマイクロタイタープレートの穴内に移された。
【0161】
当該残りのレンズは、1ミリリットル/レンズの涙様液(TLF)を収容する、個々の組織培養プレートの穴内で7日間および14日間、当該レンズが24時間ごとに新鮮な涙様液(TLF)溶液に移されながら、培養された。
【0162】
培養期間の末において(培養後、7日間および14日間)、測定対象のレンズは、穴から取り出され、緩く結合した細胞群を除去するために、三回交換したリン酸緩衝食塩水で五回、水洗され、0.05重量/総容量(w/v)%のツイーン(Tween(商標))80を含有する約10ミリリットルのリン酸緩衝食塩水(PBS)中に配置され、2000回転/分で3分間、ボルテックスされ、残留する細菌のレンズに対する粘着をなくす遠心力を利用した。得られる上澄液の生菌数は、流動式血球計算器RBD 3000を用いて算出され、かつ、3枚のレンズに付着した検出可能な生菌数の結果は平均化された。この結果は、図3に示されている。ビスタコン(Vistakon)社から入手可能な、ヒドラクリア(Hydraclear:商標)を備えたアキュビュー(登録商標)・アドバンス(商標)ブランドのコンタクトレンズは、対照として使用された。
【0163】
実施例16および比較例2のレンズに関する結果は、図3に示されている。ダイアモンド形状の点を結ぶ実線は実施例16のレンズからの結果であり、かつ、四角形状の点を結ぶ破線は比較例2のレンズの評価からの結果である。図3は、本発明のレンズが、14日間、一貫して細菌(緑膿菌(Pseudomonas aeruginosa))の対数減少値4を呈することを示している。本発明のレンズと異なり、比較例2のレンズは、最初の7日間で、対数減少値3を呈するものの、その後、評価期間の残存期間で、当該対数減少値が低下し、14日目で、約1の対数減少値を呈した。したがって、本発明のレンズは、比較例2のレンズよりも、細菌に対する有効性が高く、かつ、長期間に及ぶことを示した。
【0164】
〔実施例21〕
45.5キログラムの3-アリルオキシ-2-ヒドロキシプロピルメタクリレート(AHM)およびブチル化ヒドリキシトルエン(BHT)を含む撹拌溶液には、10ミリリットルのプラチナジビニルテトラメチルジシロキサン・キシレン溶液(Pt (0) divinyltetramethyldisiloxane solution in xylenes)(Pt濃度:2.25%)が添加された後に、44.9キログラムのn-ブチルポリジメチルシランが添加された。反応熱は、約20℃の反応温度を維持するために調節された。n-ブチルポリジメチルシランが完全に消費された後に、プラチナ(Pt)触媒は、6.9グラムのジエチルエチレンジアミンの添加により、失活処理された。未処理の反応性混合物は、ラフィネートの3-アリルオキシ-2-ヒドロキシプロピルメタクリレート(AHM)の残留量が0.1%未満になるまで、181キログラムのエチレングリコールで数回にわたって抽出された。10グラムのブチル化ヒドリキシトルエン(BHT)は、得られたラフィネートに添加され、溶解するまで撹拌された後に、残留エチレングリコールが除去され、64.5キログラムのOH-mPDMSを得た。得られた液体には、6.45グラムの4-メトキシフェノール(MeHQ)が添加され、撹拌され、濾過されて、最終的に、64.39キログラムのOH-mPDMSを無色油として得た。
【0165】
〔合成実施例2:マクロマーの調製〕
室温で、窒素下の乾燥ボックス内に収容された乾燥容器内には、30.0グラム(0.277モル)のビス(ジメチルアミノ)メチルシラン、TBACB)の1M溶液(1000ミリリットルの乾燥THF中に386.0グラムのTBACBを含有する)13.75ミリリットルと、61.39グラム(0.578モル)のp‐キシレンと、154.28グラム(1.541モル)のメチルメタクリレート(開始剤に対して1.4当量)と、1892.13グラム(9.352モル)の2-(トリメチルシロキシ)エチルメタクリレート(開始剤に対して8.5当量)と、4399.78グラム(61.01モル)のTHFとの溶液が添加された。熱電対およびコンデンサーを備え、かつ、すべての口が窒素源に接続された三つ口を備えた乾燥した丸底フラスコには、乾燥ボックス内で調製された上記混合物が添加された。
【0166】
反応性混合物は、撹拌して窒素を追い出しながら、15℃まで冷却された。当該溶液が15℃に達した後に、191.75グラム(1.100モル)の1-トリメチルシロキシ-1-メトキシ-2-メチルプロペン(1当量)が反応容器内に投入された。反応では、約62℃以下の発熱が許容され、その後、11ミリリットルの乾燥THF中に154.4グラムのTBACBを含有する0.4M溶液30ミリリットルが残余の反応中に、定量された。反応温度が30℃に達し、かつ、定量操作が開始された後に、467.56グラム(2.311モル)の2-(トリメチルシロキシ)エチルメタクリレート(開始剤に対して2.1当量)と3.812グラム(3.63モル)のn-ブチルモノメタクリロキシプロピル-ポリジメチルシロキサン(開始剤に対して3.3当量)と3673.84グラム(8.689モル)のTRIS(開始剤に対して7.9当量)と20.0グラムのビス(ジメチルアミノ)メチルシランとの溶液が添加された。
【0167】
混合物は、約38℃〜約42℃まで発熱された後に、30℃まで冷却された。その際、10.0グラム(0.076モル)のビス(ジメチルアミノ)メチルシランと154.26グラム(1.541モル)のメチルメタクリレート(開始剤に対して1.4当量)と1892.13グラム(9.352モル)の2-(トリメチルシロキシ)エチルメタクリレート(開始剤に対して8.5当量)との溶液が添加され、かつ、その混合物は、再び、約40℃まで発熱された。反応温度は、約30℃まで下げられ、かつ、2ガロン(7.6リットル)のTHFが添加されて粘性を低下させた。439.69グラムの水と740.6グラムのメタノールと8.8グラム(0.068モル)のジクロロ酢酸との溶液が添加され、その混合物は、HEMA上の保護基によるブロックを解除するために、4.5時間、還流処理された。その後、揮発性成分が除去されると共に、蒸気温度が110℃に達するまで、水分除去に役立つトルエンが添加された。
【0168】
反応用フラスコは、約110℃に維持され、かつ、443グラム(2.201モル)のTMIとビスマスK-KAT 348(5.94グラム)との溶液が添加された。混合物の反応は、イソシアネートのピークが赤外線によって現れるまで行われる。トルエンは、減圧下で蒸発されて、オフホワイトの無水で蝋質の反応性モノマーを得た。マクロマーは、アセトン:マクロマーの重量比が約2:1となるように、アセトン中に置かれた。24時間後に、マクロマーを沈積するために、水が添加されると共に、当該マクロマーは、濾過され、かつ、45℃〜60℃の間の温度で真空オーブンを用いて20時間〜30時間、乾燥された。
【0169】
〔合成実施例3:ヨウ化銀ナノ分散液の形成〕
金属剤溶液および塩前駆体溶液は、以下のように形成された。10,000ppmの硝酸銀は、200グラムの50重量/全重量(w/w)%PVP(K値12)脱イオン水溶液中に撹拌しながら、溶解された。ヨウ化ナトリウム(10,000ppm)は、200グラムの50重量/全重量(w/w)%PVP(K値12)脱イオン水溶液中に撹拌しながら、溶解された。硝酸銀を含有する金属塩溶液は、塩前駆体溶液中に、2013回転/分で撹拌しながら、200グラム/時の速度で添加された。金属塩溶液は、空気中で噴霧乾燥された。導入口温度は185℃であり、出口温度は90℃であり、供給速度は2.7キログラム/時であった。安定化ヨウ化銀ナノ粒子は、5重量%未満の含水率を有していた。
【0170】
安定化ヨウ化銀ナノ粒子粉末(0.32グラム)は、199.7グラムの脱イオン水中に溶解されて、溶液を調製した。ヨウ化銀ナノ粒子粉末は、ヨウ化銀として名目上6600ppmの銀濃度を有していた。最終溶液中における銀濃度は、算出されたところでは、11ppmであった。
【0171】
〔実施例22〕
米国公開特許US2005/0013842 A1号明細書の実施例10の方法は、以下のように実行された。
硝酸銀(0.127グラム)は、75ミリリットルの脱イオン水中に溶解されて、0.01M硝酸銀溶液を調製した。ポリアクリル酸(PAA、2グラム)は、48ミリリットルの脱イオン水中に溶解されて、4重量/全重量(w/w)%のポリアクリル酸溶液を調製した。200ミリリットルの脱イオン水中には、水素化ホウ素ナトリウム(0.008グラム)が添加されて、1mM溶液を調製した。
この1mM水素化ホウ素ナトリウム溶液(197ミリリットル)は、撹拌棒を備えたビーカー内に置かれた。このビーカーは、氷水浴中に沈められた。この設備は、撹拌プレート上に置かれた。0.01M硝酸銀溶液(2ミリリットル)は、4重量/全重量(w/w)%のポリアクリル酸溶液(1ミリリットル)と混合され、かつ、氷水浴中で冷却された。硝酸銀・ポリアクリル酸溶液の混合物は、急速撹拌しながら、冷却された1mM水素化ホウ素ナトリウム溶液中に急速添加された。これらの溶液の混合後に、茶色‐黄色への急激な変色(immediate brown-yellow discoloration)が観察された。溶液は、8時間混合された後に、保存用の琥珀色の透明広口瓶に移された。添加された硝酸銀の量に基づいて、最終溶液の銀濃度は、算出されたところでは、11ppmであった。
【0172】
この実施例22の銀含有溶液の紫外・可視(UV-VIS)スペクトルが測定されており、当該スペクトルは、合成実施例3で調製されたヨウ化銀/PVP水溶液の紫外・可視(UV-VIS)スペクトルと共に、図4に示されている。図4から理解できるように、この実施例22の溶液のスペクトルは、約420ナノメートルを中心とする広いピークを呈していた。対照的に、合成実施例3のヨウ化銀/PVP水性分散液の紫外・可視(UV-VIS)スペクトルの主ピークは、330ナノメートルを中心とするものであった。チャン・ゼット(Zang, Z)らによれば、このピークは、当該水溶液中に、PVPに結合したイオン形態(Ag+)で存在する銀の相互作用に起因することがある。図4における複数のスペクトル差は、本発明の反応性混合物および眼用装置中の銀がイオン形態で存在する可能性があるが、実施例23A、23Bおよび23Eの反応性混合物中に存在する銀が非イオン性銀(Ag0)として存在していることを示している。
【0173】
〔実施例23Aおよび23B(比較例)〕
表9に挙げられた複数のモノマー成分(光重合開始剤Darocur 1173以外の成分)は、表9に挙げられた量で、琥珀色のガラス製バイアル瓶内で混合され、かつ、混合用の瓶回転装置上で回転が与えられた。
【0174】
実施例23Aにおいて、硝酸銀溶液(フィッシャー(Fisher)社製の無水エタノール54ミリリットル中にフィッシャー(Fisher)社製の米国化学会(A.C.S.)品質の0.025グラムの硝酸銀が溶解された)は、硝酸銀の供給源として使用された。実施例23Bにおいて、硝酸銀溶液(フィッシャー(Fisher)社製の無水エタノール54ミリリットル中にフィッシャー(Fisher)社製の米国化学会(A.C.S.)品質の0.305グラムの硝酸銀が溶解された)は、硝酸銀の供給源として使用された。
【表11】
【0175】
実施例23Aおよび23Bで調製された5ミリリットルの各反応性混合物は、24時間、そのままにしておかれた。反応性混合物の色は、L*a*b*表色系および上述した方法を用いて、定量的に測定された。また、反応性混合物の色は、白色蛍光灯下で、主観的に評価された。これらの結果は、以下の表10に開示されている。
【0176】
実施例23Aおよび23Bの反応性混合物に関する紫外・可視(UV-VIS)スペクトルが測定されており、図5に示されている。
【0177】
光重合開始剤(Darocur 1173)が添加され、かつ、各処方は、660水銀柱ミリメートル(87.99キロパスカル)の真空下で5分間〜7分間、脱気処理された。その後、処方は、窒素グローブボックスに移された。コンタクトレンズは、少なくとも24時間、窒素グローブボックス中で脱酸素化されたゼオノール(Zeonor:登録商標)製の前曲面型およびポリプロピレン製の後曲面型を用いて調製された。レンズ収容空洞当たり100マイクロリットルの投与量が使用され、かつ、レンズ型を保持するフレームが水晶板の下に配置された。レンズは、紫外線照射灯(平行な四灯一組のフィリップス(Philips)社製TL09/20)下、室温で60分間、硬化処理された。
【0178】
硬化後に、レンズ型が手動で開けられると共に、70:30のIPA:脱イオン水の混合物を含む瓶内に、レンズが、1枚のレンズ当たり〜5ミリリットルの溶液を用いて放出された。少なくとも60分後に、レンズ型がピンセットで取り出され、溶液が静かに注入され、瓶には、新鮮なIPA:脱イオン水(70:30)混合物が充填された。レンズには、瓶回転装置上で回転が与えられ、少なくとも60分後に、溶液が静かに注入され、瓶には、新鮮な脱イオン水が充填された。レンズには、さらに、少なくとも60分間、瓶回転装置上で回転が与えられ、溶液が静かに注入され、瓶には、新鮮な脱イオン水が充填された。レンズは、ガラス製バイアル瓶内の5ミリリットルのリン酸緩衝包装用溶液中に収容され、当該バイアル瓶がシリコーン製封止材料およびアルミニウム固定キャップで封止され、122℃で30分間、オートクレーブ処理された。レンズ中の銀含有率は、機器中性子放射化分析法(INAA)を用いることによって測定され、表9に記録されている。
【0179】
〔実施例23Cおよび23D〕
硝酸銀/エタノール溶液に代えて、合成実施例3で調製された安定化AgI/PVP粉末が添加されたことを除き、実施例23Aおよび23Bが繰り返された。溶液の色は、実施例23Aおよび23Bに記述されているように、測定され、表10に記録されている。実施例23Cおよび23Dの反応性混合物に関する紫外・可視(UV-VIS)スペクトルが測定されており、そのスペクトルは図5に示されている。レンズは、実施例23Aおよび23Bに記述されているように形成され、かつ、ガラス製バイアル瓶内に入れられた、50ppmのメチルセルロースを含有する5ミリリットルのホウ酸緩衝硫酸ナトリウムの包装用溶液(SSPS)中に収容され、当該バイアル瓶がシリコーン製封止材料およびアルミニウム固定キャップで封止され、122℃で30分間、オートクレーブ処理された。レンズ中の銀含有率は、機器中性子放射化分析法(INAA)を用いることによって測定され、表9に記録されている。
【0180】
〔実施例23E〕
銀供給源が、硝酸銀/エタノール溶液に代えて、11.25グラムのDMA中に溶解された0.026グラムの硝酸銀および0.011グラムのポリアクリル酸(PAA)であることを除き、実施例23Aが繰り返された。溶液の色は、実施例23Aおよび23Bに記述されているように、測定されており、表10に記録されている。実施例23Eの反応性混合物に関する紫外・可視(UV-VIS)スペクトルが測定されており、そのスペクトルは図5に示されている。レンズは、実施例23Aおよび23Bに記述されているように形成された。レンズ中の銀含有率は、機器中性子放射化分析法(INAA)を用いることによって測定され、表9に記録されている。
【0181】
〔実施例23F〕
銀が全く添加されなかったことを除き、実施例23Aが繰り返された。
【表12】
【0182】
図5は、実施例23A〜23Fの反応性混合物に関する紫外・可視(UV-VIS)スペクトルの比較を示している。実施例23F(銀を含有しない対照の処方)は、グラフ化された領域内で、いかなるピークをも示さなかった。低濃度の銀を含有する反応性混合物(実施例23A)も、いかなる明確なピークをも示さなかった。しかし、実施例23Bは、435ナノメートルに明確なピークを示しており、この場合に、米国公開特許US2005/0013842号明細書によれば、非イオン性銀(Ag0)の存在が確認されている。
【0183】
実施例23Cに関するスペクトルでは、紫外・可視(UV-VIS)スペクトル中の417ナノメートルに明確な遷移が観察された。当該遷移は、実施例23Dの反応性混合物(約389ppmの標的銀濃度を有する)のスペクトルに存在するように現れたが、信号は雑音が多く、当該スペクトル領域内において飽和状態に近くなっていた。チャン・ゼット(Zang, Z)、チャオ・ビー(Zhao, B)およびフー・エル(Hu, L)は、1996年1月に発行された固体化学誌第121巻第1頁、第5頁、第105頁〜第110頁の「化学還元法により合成された銀超微粉末のPVP防御機構」(Journal of Solid State Chemistry January 1996, 121, Issue 1, 5, 105-110. PVP Protective Mechanism of Ultrafine Silver Powder Synthesized by Chemical Reduction Processes)において、彼らがコロイド状ヨウ化銀の紫外・可視(UV-VIS)スペクトルを分析したときに、サンプル23Cに非常に類似し、420ナノメートルに吸収段部を有するスペクトルプロファイルを得た。さらに、彼らは、水素化ホウ素ナトリウムを用いたコロイド状ヨウ化銀の非イオン性銀(Ag0)への還元時に、ピークの位置および形状が、サンプル23Bの紫外・可視(UV-VIS)スペクトルにおいて観察されるものと非常に類似していたことを発見した。科学文献中のデータに基づいて、実施例23Bの硝酸銀系のモノマー類と比較して、実施例23Cで観察されたピークの異なる形状および位置は、異なる酸化状態を有する銀粒子の存在を示すものと見なされる。
【0184】
〔実施例24A〜24F〕
実施例23A〜23Fで形成されたレンズは、上述した検査方法に記述された手順を用いて、黄色ブドウ球菌(Staphylococcus aureus)031に対する有効性に関して検査された。その結果は、以下の表11に記録されている。
【表13】
【0185】
実施例23Aおよび23Bは、それぞれ実施例23Cおよび23Dに類似する、レンズ中の銀濃度を呈している。しかし、抗菌活性データは、硝酸銀を含有するモノマー類(実施例23A、23Bおよび23E)から形成されたレンズが、実施例23Fで形成された対照レンズと比較されたときに、抗菌活性を呈しなかったことを示している。これに対して、実施例23Cおよび23Dに従って形成され、かつ、金属塩ナノ粒子を含有するレンズは、当該対照レンズと比較して、少なくとも1の対数減少値を呈していた。
【0186】
〔実施例25A〕
可視光重合開始剤CGI 819が使用され、かつ、レンズが、可視光照射灯(平行な四灯一組のフィリップス(Philips)社製TL03/20)下、室温で30分間、硬化処理されたことを除き、実施例23Dが繰り返された。硬化されたレンズは、実施例23Dに開示されているように、放出され、抽出され、水和処理され、包装され、かつ、オートクレーブ処理された。銀濃度、ヨウ化物濃度および明度は測定され、以下の表12に示されている。また、実施例23D(紫外線硬化法で形成されている、同一の処方)のレンズの銀濃度、ヨウ化物濃度および明度も測定され、以下の表12および表13に示されている。
【0187】
〔実施例25B〕
硬化前の処方に2重量%のNorblocが添加され、かつ、エタノール濃度が2%だけ下げられたことを除き、実施例25Aが繰り返された。銀濃度、ヨウ化物濃度および明度は、水和処理および殺菌処理後に測定され、以下の表12および表13に示されている。
【表14】
【0188】
実施例23Dに従って、紫外線硬化法を用いて形成されたレンズ中の銀:ヨウ化物のモル比(水和処理および殺菌処理後に測定された)は、観察されたところでは、約2であった。このデータは、レンズ中の銀含有量の約半分が、硬化処理中に、ヨウ化銀から異なる酸化状態の銀に変換されたことを示唆している。実施例23Dにおいては、紫外光がヨウ化銀(AgI)を非イオン性銀(Ag0)およびヨウ素分子(I2)に変換したものと考えられる。ヨウ素分子(I2)がIPA中に溶解可能であるので、当該ヨウ素分子(I2)は水和処理中に除去された。反応性混合物に添加されたヨウ化銀に基づいて予定される、レンズ中の銀:ヨウ化物のモル比は、約1であった。
【0189】
実施例25Aおよび25Bで形成されたレンズ中の銀:ヨウ化物のモル比は、可視光硬化法を用いると、約1であった。したがって、紫外光の範囲外の硬化条件の使用は、ヨウ化銀等の抗菌性金属塩を塩の形態で維持する上で、重要である。
【表15】
【0190】
表13中の比色分析データに基づくと、可視光硬化法を用いて調製された同程度の銀濃度を有する複数のレンズ(実施例25Aおよび25B)は、実施例23Dで、紫外線で硬化されて形成された複数のレンズよりもかなり薄い黄色(低b*値)を呈した。
【0191】
〔実施例26〜28〕
100,000ppmのPVP(K値12)溶液が脱イオン水中で調製された。この溶液(溶液A)は、ヨウ化ナトリウム(NaI)溶液および硝酸銀(AgNO3)溶液を調製するためのベースを与えた。約1500ppm、約5000ppm、および、約10000ppmのヨウ化ナトリウムおよび硝酸銀のそれぞれを含有する溶液が調製された。各溶液は、粒子が可視的に観察されなくなるまで、撹拌された。20ミリリットルのヨウ化ナトリウム溶液は清浄な瓶内に置かれ、かつ、磁石式の撹拌装置は当該瓶内に置かれた。撹拌装置の撹拌速度は300回転/分に設定され、かつ、20ミリリットルの硝酸銀が当該ヨウ化ナトリウム溶液中に、以下の表14に示された速度で添加された。すべての混合処理は、室温で実行された。溶液の霞みは、表14中に挙げられた添加時間の最後に、主観的に評価され、その結果は、以下の表14に記録されている。実施例は、表14に示された各濃度および添加速度ごとに繰り返された。
【表16】
【0192】
〔実施例29〜31〕
ヨウ化ナトリウム溶液が硝酸銀溶液に添加されたことを除き、実施例26〜28が繰り返された。これらの結果は、以下の表15に示されている。
【表17】
【0193】
〔実施例32〕
金属剤溶液および塩前駆体溶液が、室温で、〜5日間、瓶回転装置上で混合された後に、20ミリリットルの各溶液がバッチ式で混合された(約1秒で一緒に注入された)ことを除き、実施例31が繰り返された。結果は、PVP‐AgI錯体を含む透明な溶液であった。
【0194】
〔実施例33〜39〕
約10ミリリットルの700ppmの硝酸銀溶液は、表16に示されたPVP濃度(脱イオン水中でのPVP(K値12)の濃度:1%〜35%)で、PVP(K値12):脱イオン水溶液中で形成された。各硝酸銀溶液は、手動で震盪しながら、10ミリリットルの1100ppmのヨウ化ナトリウム/脱イオン水溶液(PVPを含まない)中に滴下されて、分散液を形成した。実施例33では乳白色であったが、残りの実施例では硝酸銀の添加中、透明のままであった。粒子サイズの測定は、得られたヨウ化銀分散液に対して、レーザー光散乱法(実施例33)および光子相関分光分析法(実施例35〜39)を用いて実行された。データは、粒子サイズ分布のz平均として記録されている。
【表18】
【0195】
表16のデータは、図5にグラフで示されている。表16のデータは、金属塩の形成中のPVPの存在が粒子サイズを実質的に(少なくとも2桁)低減させることを明確に示している。
【0196】
〔実施例40〜44〕
PVPに代えて、表17に挙げられた分散剤が、表17に挙げられた濃度で使用されたことを除き、実施例34が繰り返された。粒子サイズの測定は、得られたヨウ化銀分散液に対して、レーザー光散乱法(実施例40、41および43)および光子相関分光分析法(実施例42、44)を用いて実行された。データは、粒子サイズ分布のz平均として記録されている。
【表19】
【0197】
〔実施例45〕
以下の表18に示された各成分は、表18に挙げられた量で、琥珀色のガラス製バイアル瓶内に一緒に混合され、かつ、瓶回転装置上で回転が与えられた。混合物は、コンタクトレンズ用型(ゼオノール(Zeonor:登録商標)前曲面用型および後曲面用型)内へ分注され、かつ、以下の条件:酸素2.8%±0.5%、可視光硬化(フィリップス(Philips)社製TL03ランプ)、強度プロファイル:25℃での光量1±0.5ミリワット/平方センチメートル(10秒〜60秒)、80℃±5℃での光量5.5±0.5ミリワット/平方センチメートル(304秒〜600秒)で硬化処理された。レンズは、IPA/水混合物中で水和され、50ppmのメチルセルロースを含有する950マイクロリットルのホウ酸緩衝硫酸ナトリウムの包装用溶液(SSPS)中で個別のポリプロピレン製ブリスターパック内に収容され、かつ、124℃で18分間、オートクレーブ処理された。
【表20】
【0198】
この実施例45で形成された12枚のレンズは、上述した検査方法に記述された手順を用いて、黄色ブドウ球菌(Staphylococcus aureus)031に対する有効性に関して検査された。対照レンズは、実施例45の方法で形成されたが、ヨウ化銀ナノ粒子を含有していなかった。銀を含有するレンズの対数減少値(対照レンズと対比して)は、確認されたところでは、3.3±0.2(平均値±標準偏差)であった。
【0199】
〔実施例46〕
実施例45のレンズは、実施例45の対照レンズと共に、二重盲検対側性臨床試験(double masked, contralateral clinical trial)で、30人の患者(全員が現在、コンタクトレンズ装用者)によって装用された。これらの患者は、毎日装用方式で、14日間、レンズを装用し、オプティフリー・リプレニッシュ(OptiFree RepleniSH)を使用し、レンズの洗浄および消毒中に、当該レンズを擦るように指示された。実施例45のレンズは、ベースラインで約10マイクログラムの銀を含有していた。
【0200】
当該試験を終了した26人の患者からの装用済みのレンズは、14日間の装用期間の最後に収集され、機器中性子放射化分析法(INAA)によって銀含有率について検査された。機器中性子放射化分析法(INAA)のデータから、銀の放出の平均速度が算出されたところでは、0.5マイクログラム/日であった。また、レンズは、上述した検査方法の項に記述された方法を用いて、黄色ブドウ球菌(S. aureus)に対する活性に関して検査された。実施例45のレンズの対数減少値(装用済みの対照と対比して)は、確認されたところでは、3.4±1.2(平均値±標準偏差)であった。
【図面の簡単な説明】
【0201】
【図1】実施例16および比較例2の各レンズ中の銀濃度を、当該レンズ縁部からの距離の関数として示すグラフである。
【図2】銀の放出状態の比較を、実施例16および比較例2で形成されたコンタクトレンズに関する時間関数として示すグラフである。
【図3】緑膿菌(Pseudomonas aeruginosa)に対する有効性の比較を、実施例16および比較例2で形成されたコンタクトレンズに関する時間関数として示すグラフである。
【図4】実施例22および合成実施例3の各混合物に関する紫外・可視(UV-VIS)スペクトルを示す。
【図5】実施例23Aおよび23Bの反応性混合物に関する紫外・可視(UV-VIS)スペクトルを示す。
【開示の内容】
【0001】
〔発明の分野〕
本発明は、抗菌性ポリマー製品、その製造方法および使用方法に関するものである。
【0002】
〔発明の背景〕
抗菌特性を有する材料は、多くの適用で使用されてきた。カテーテル、補綴具、インプラント、眼用装置等の医療装置の場合に、装置表面への細菌感染は、重篤感染症および装置故障をもたらすことがある。また、表面に集中した感染症は、食品の腐敗、食物が原因となる疾病の蔓延、および、材料の生物付着にも関係している。したがって、健康器具および生物医学装置、食品、および、個人衛生に関する各産業で利用する抗菌材料を進展させることに、重大な関心がある。
【0003】
銀塩類は、術後感染、歯科、創傷治療および医療装置の消毒剤として、ヒトのヘルスケアおよび医薬品に使用される長い歴史を有している。硝酸銀は、新生児眼炎(ophthalmic neonatorum)の予防に使用されてきた。コロイド銀は、1800年代に導入され、医療用の硝酸銀の代替物質として、1930年代以前に広く使用された。
【0004】
最近になって、銀化合物は、可溶性および不溶性の塩類、ポリマー類とゼオライトとを結合した錯体、金属銀および酸化銀等の種々の形態で、医療装置に添加されてきた。しかし、当該銀化合物の多くがポリマー組成物中に組み込まれている場合に、当該ポリマー組成物は、非常に漠然とした一貫性のない銀の充填、製造方法の複雑化、不都合な銀の急速放出、あるいは、有効性の欠如を含む複数の欠陥を被る。
【0005】
ポリマーマトリクス中への銀の導入に関する幾つかの方法が、開示されてきており、当該方法は、銀錯化合物の還元または合成等の化学的精密検査(chemical workups)、予備形成された銀粒子をポリマーに混合させること、あるいは、スパッタリング法およびプラズマ堆積法等の複雑な物理的方法を含む。これらの方法は、複雑であり、ポリマー材料中への銀化合物の一貫した充填を与えるとは限らない。銀塩等の微量金属塩類(oligodynamic metal salts)をコロイド金属塩粒子として医療装置に導入する方法が、開示されてきた。しかし、光重合法によって形成された装置への当該塩類の導入方法、および、還元剤を含む反応混合物中への当該塩類の導入方法は、開示されていなかった。
【0006】
1950年代以降、視力を改善するためのコンタクトレンズは、商業的に使用されるようになってきた。最初のコンタクトレンズは、硬質材料で形成された。当該コンタクトレンズは、起床中の数時間、患者によって使用され、かつ、洗浄のために取り外された。このコンタクトレンズの分野における現在の開発は、洗浄のために取り外さずに、数日以上の連続装用が可能なソフトコンタクトレンズを生み出した。多くの患者が当該ソフトコンタクトレンズを快適性の増大から好むが、当該ソフトコンタクトレンズはユーザーに多少の有害反応を生じさせることがある。当該ソフトコンタクトレンズの長期使用は、ソフトコンタクトレンズ表面上での細菌または他の微生物、特に緑膿菌(Pseudomonas aeruginosa)の増大を促進することになる。細菌および他の微生物の増大は、コンタクトレンズによる急性結膜充血(contact lens acute red eye)およびこの同類の症状等、有害な副作用を生じさせることになる。細菌および他の微生物の問題は、十中八九、ソフトコンタクトレンズの長期使用に関連しているが、細菌および他の微生物の増大は、ハードコンタクトレンズ装用者にも同様に生じる。
【0007】
したがって、依然として、眼用装置の表面上での細菌または他の微生物の増殖、および/あるいは、当該表面への細菌または他の微生物の付着を抑制するコンタクトレンズ等の眼用装置を製造する必要がある。さらに、コンタクトレンズの表面上での細菌または他の微生物の増殖、および/あるいは、当該表面への細菌または他の微生物の付着を促進しないコンタクトレンズ等の眼用装置を製造する必要がある。また、依然として、細菌または他の微生物の増殖に関係した有害反応を抑制するコンタクトレンズを製造する必要がある。
【0008】
〔発明の概要〕
一つの実施の形態において、本発明は、少なくとも一つのポリマーから形成された製品に関するものであり、当該ポリマーは、当該ポリマー全体に均質に分散され、かつ、約200ナノメートル未満の粒子サイズを有する抗菌性の金属塩粒子を含み、当該製品は、緑膿菌(Pseudomonas aeruginosa)および黄色ブドウ球菌(s. aureus)のうち、少なくとも一方において少なくとも約0.5の対数減少値を呈し、かつ、約70マイクロメートルの厚さで、CSIレンズと比較して約100%未満のヘイズ値(haze value)を呈するものである。
【0009】
他の実施の形態において、本発明は、
(a)少なくとも一つの塩前駆体を、任意に反応性ポリマー混合物の少なくとも一つの成分と共に、溶媒中に溶解させて、塩前駆体の混合物を形成するステップ;
(b)少なくとも一つの金属剤および少なくとも一つの分散剤を、任意に少なくとも一つの反応性成分と共に、溶媒中に溶解させることによって、分散剤・金属剤の錯体を形成して、金属剤混合物を形成するステップであって、当該複数の溶媒および成分は同一または異なるものであってもよい、ステップ;
(c)少なくとも一つの抗菌性金属塩:[Mq+]a[Xz-]bを含む粒子含有混合物を形成するための粒子形成条件下で、当該塩前駆体の混合物および当該金属剤混合物を混合するステップ;
(d)ステップ(a)および(b)に反応性成分が含まれていない場合において、このステップ(d)で少なくとも一つの反応性成分が添加されることを条件として、任意に、追加の反応性成分を当該粒子含有混合物に混合して、粒子含有反応性混合物を形成するステップ;ならびに、
ステップ(c)で抗菌性ポリマー製品中にMq+として添加された当該金属剤由来のMの少なくとも約90%を維持する上で十分な反応条件下で、当該ポリマー製品を形成するために、当該粒子含有反応性混合物を反応させるステップ、
を含む方法に関するものである。
【0010】
さらに他の実施の形態において、本発明は、約200ナノメートル以下の粒子サイズを有する安定化抗菌性金属塩粒子と少なくとも一つのフリーラジカル反応性成分とを含む反応性混合物を、当該金属塩粒子に応じて調整された臨界波長を超える波長の光、熱、あるいは、これらの組み合わせを用いて、硬化させて、抗菌性金属塩粒子を含む製品を形成することを含む方法に関するものである。
【0011】
〔発明の詳細な記述〕
本発明は、緑膿菌(Pseudomonas aeruginosa)、黄色ブドウ球菌(Staphyloccus aureus)のうち、少なくとも一方、あるいは、両方において少なくとも約0.5の対数減少値、および約100%未満のヘイズ値(haze value)を呈する抗菌性の製品であって、当該製品が作られる少なくとも一つのポリマー全体に、均質に分散した約200ナノメートル未満の粒子サイズを有する抗菌性金属塩粒子を含むか、当該粒子から本質的に成るか、あるいは、当該粒子で構成される抗菌性の製品から成るものである。一部の実施の形態において、粒子サイズは、約100ナノメートル未満であり、他の実施の形態においては、約50ナノメートル未満である。当該製品中の抗菌性金属塩粒子の粒子サイズは、走査型電子顕微鏡によって測定されてもよい。
【0012】
この明細書で使用されているように、用語「抗菌性」とは、製品が以下の特性:製品への細菌または他の微生物の付着の抑制、製品上での細菌または他の微生物の増殖の抑制、および、製品の表面上または製品の周辺領域内での細菌または他の微生物に対する殺菌のうち、一つ以上の特性を呈することを意味する。本発明のために、製品への細菌または他の微生物の付着、製品上での細菌または他の微生物の増殖、および、製品の表面上の細菌または他の微生物の存在は、全体的に「微生物のコロニー形成」と呼ばれる。好適には、本発明の製品は、生存している細菌または他の微生物において少なくとも約0.25の対数減少値を呈するものであり、一部の実施の形態においては少なくとも約0.5の対数減少値を呈するものであり、一部の実施の形態においては少なくとも約1.0の対数減少値を(90%以上の抑制効果)呈するものである。このような細菌または他の微生物は、緑膿菌(Pseudomonas aeruginosa)、アカントアメーバ属(Acanthamoeba species)、黄色ブドウ球菌(Staphyoccus aureus)、大腸菌(E. coli)、表皮ブドウ球菌(Staphyoccus epidermidis)、および、霊菌(Serratia marcesens)を含むが、これらに限定されるものではない。
【0013】
フリーラジカル反応性成分は、フリーラジカル開始反応によって重合され得る重合性成分を含む。限定されないフリーラジカル反応基の例は、(メタ)クリレート基((meth)acrylates)、スチリル基、ビニル基、ビニルエーテル基、C1-6アルキル(メタ)クリレート基、(メタ)クリルアミド基、C1-6アルキル(メタ)クリルアミド基、N-ビニルラクタム基、N-ビニルアミド基、C2-12アルケニル基、C2-12アルケニルフェニル基、C2-12アルケニルナフチル基、C2-6アルケニルフェニルC1-6アルキル基、O-ビニルカルバメート基、および、O-ビニルカーボネート基を含む。
【0014】
この明細書で使用されているように、用語「金属塩」とは、一般式:[Mq+]a[Xz-]bを有する、あらゆる分子を意味し、ここで、Xは、あらゆる負帯電イオンを含み、a、b、qおよびzは、それぞれ個別に1以上の整数であり、q(a)=z(b)である。Mは、限定するものではないが、以下のイオン:Al+3、Cr+2、Cr+3、Cd+1、Cd+2、Co+2、Co+3、Ca+2、Mg+2、Ni+2、Ti+2、Ti+3、Ti+4、V+2、V+3、V+5、Sr+2、Fe+2、Fe+3、Au+2、Au+3、Au+1、Ag+2、Ag+1、Pd+2、Pd+4、Pt+2、Pt+4、Cu+1、Cu+2、Mn+2、Mn+3、Mn+4、Zn+2、Se+4、Se+2、および、これらの混合物から選択された、あらゆる正帯電金属イオンであってもよい。他の実施の形態において、Mは、Al+3、Co+2、Co+3、Ca+2、Mg+2、Ni+2、Ti+2、Ti+3、Ti+4、V+2、V+3、V+5、Sr+2、Fe+2、Fe+3、Au+2、Au+3、Au+1、Ag+2、Ag+1、Pd+2、Pd+4、Pt+2、Pt+4、Cu+1、Cu+2、Mn+2、Mn+3、Mn+4、Se+4およびZn+2、および、これらの混合物から選択されてもよい。Xの例は、CO3-2、NO3-1、PO4-3、Cl-1、I-1、Br-1、S-1、O-2、酢酸塩、これらの混合物、および、これらの同類のものを含むが、これらに限定されるものではない。さらに、Xは、CO3-2、SO4-2、PO4-3、Cl-1、I-1、Br-1、S-1、O-2、酢酸塩、および、これらの同類のもの、例えばC1-5アルキルCO2-1などを含む負帯電イオンを含む。他の実施の形態において、Xは、CO3-2、SO4-2、Cl-1、I-1、Br-1、酢酸塩、および、これらの混合物を包含してもよい。この明細書で使用されているように、用語「金属塩」は、米国公開特許US-2003-0043341-A1号明細書に開示された金属塩のように、ゼオライトを含まない。一つの実施の形態において、aは、1、2または3である。一つの実施の形態において、bは、1、2または3である。一つの実施の形態において、金属イオンは、Mg+2、Zn+2、Cu+1、Cu+2、Au+2、Au+3、Au+1、Pd+2、Pd+4、Pt+2、Pt+4、Ag+2およびAg+1、および、これらの混合物から選択される。特に好適な金属イオンは、Ag+1である。適切な金属塩の例は、硫化マンガン、酸化亜鉛、炭酸亜鉛、硫酸カルシウム、硫化セレン、ヨウ化銅、硫化銅、および、リン酸銅を含むが、これらに限定されるものではない。銀塩の例は、炭酸銀、リン酸銀、硫化銀、塩化銀、臭化銀、ヨウ化銀、および、酸化銀を含むが、これらに限定されるものではない。一つの実施の形態において、金属塩は、ヨウ化銀、塩化銀、および、臭化銀等、少なくとも一つの銀塩を含む。
【0015】
本発明の一部の実施の形態における金属Mの少なくとも約90%、および、一部の実施の形態における当該金属Mの少なくとも約95%は、金属塩:[Mq+]a[Xz-]bの形態である。この割合は、イオン性金属および非イオン性金属(metal0)の測定値から算出されてもよい。例えば、製品がヒドロゲルコンタクトレンズであり、かつ、抗菌性金属塩がヨウ化銀である場合に、イオン性金属は、リン酸緩衝生理溶液(米国バージニア州ハーンドンのメディア・テック・インク(Media Tech, Inc.)から市販されたダルベッコ(Dulbecco)社製のリン酸緩衝生理食塩水10X)中で当該レンズを抽出し、他の塩が抽出溶液中に存在しなくなるまで、使用する米国薬局方(USP)AppVII中に記載された方法を用いることによって算出され得る。抽出後に、製品は、機器中性子放射化分析法(INAA)を用いることによって測定される。非イオン性銀(Ag0)が使用条件下で抽出可能でないときに、抽出後にレンズ中で測定された全銀は、非イオン性銀の酸化状態(Ag0 oxidation state)にある。
【0016】
製品が、血液、尿、涙あるいは唾液のような水混和性の体液に接触している医療装置であり、かつ、約12時間を超える抗菌効果が望ましい場合における実施の形態で、金属塩は、25℃の純水中での約2×10−10未満のKspを有している。一つの実施の形態において、金属塩は、約2.0×10−17モル/リットルを超えない溶解度積定数を有している。特定の実施の形態において、製品は、生物医学装置、眼用装置すなわちコンタクトレンズであってもよい。
【0017】
この明細書で使用されているように、用語「純(pure)」は、米国フロリダ州ボーカラートンのシーアールシー出版社(CRC Press)から1993年に発行された「化学および物理のシーアールシーハンドブック第74版」中で定義されているように、使用された水の質を云う。種々の塩に関して、25℃の純水中で測定された溶解度積定数(Ksp)は、米国フロリダ州ボーカラートンのシーアールシー出版社(CRC Press)から1993年に発行された「化学および物理のシーアールシーハンドブック74版」中で公開されている。例えば、仮に、金属塩が炭酸銀(Ag2CO3)である場合に、溶解度積定数(Ksp)は、以下の式によって示される。
Ag2CO3(複数のAg2CO3)→2Ag+(水性媒体中)+CO32-(水性媒体中)
溶解度積定数(Ksp)は、以下のように算出される。
Ksp=[Ag+]2[CO32]
炭酸銀が溶解するときに、溶液中には、二つの銀の陽イオンごとに一つの炭酸アニオンが存在する、すなわち、[CO32]=1/2[Ag+]の式が成立することから、溶解度積定数の等式は、溶解された銀濃度について解くために、以下のように再整理され得る。
Ksp=[Ag+]2(1/2[Ag+])=1/2[Ag+]3
[Ag+]=(2 Ksp)1/3
【0018】
25℃で測定された場合における溶解度積定数が約2×10−10を超えない金属塩を含む製品が、1日〜30日まで、あるいは、それ以上の期間にわたって、レンズから金属を連続的に放出するはずであることが見出された。一つの実施の形態において、適切な金属塩は、ヨウ化銀、塩化銀、臭化銀、および、これらの混合物を含む。他の実施の形態において、金属塩は、ヨウ化銀を含む。
【0019】
本発明の製品は、ポリマー類から形成されると共に、当該製品は、食品、薬剤および医療装置、生物医学装置、および、これらの同類のもの用の包装体を含む、包装体、保存容器およびラップにおける適用を見出すことができる。生物医学装置は、カテーテル、ステント、血液保存袋およびチューブ、補綴物、インプラント、ならびに、眼用装置を含み、この眼用装置は、眼用レンズ(レンズの詳細な説明は、以下のとおりである。)を含む。一つの実施の形態において、本発明の製品は、光重合性ポリマー、特に、可視光の被爆によって重合化される成分等のフリーラジカル反応性成分から形成される。他の実施の形態において、製品は、使用中に、可視光および紫外光に被爆される。このような製品は、包装体、保存容器、プラスチックラップ、および眼用装置を含む。一つの実施の形態において、本発明の製品は、眼用装置である。
【0020】
上述した製品は、この技術分野において既知であり、かつ、種々のポリマーから形成されてもよい。一部の実施の形態において、製品は、一つのポリマーから形成され、かつ、他のポリマーで被覆されてもよい。抗菌性ポリマーは、装置または当該装置の部品の形態になるように成形され、あるいは、被膜として使用されてもよい。
【0021】
上述した多くの実施の形態において、製品の透明度は、ユーザーにとって心配事である。例えば、限定されない一つの実施の形態において、製品がコンタクトレンズ等の眼用装置である場合に、本発明に使用される金属塩の非常に小さな粒子サイズは、その金属塩を特に適切なものにする。一部の実施の形態において、本発明は、約200ナノメートル未満、あるいは、約100ナノメートル未満の達成粒子サイズ(achieved particles sizes)を有すると共に、一部の実施の形態において、約50ナノメートル未満の達成粒子サイズを有する。この非常に小さな粒子サイズは、可視光の波長よりも小さいものであり、当該粒子サイズは、本発明の製品を、透明度が望まれる場合における適用に対して特に有用なものにする。このような実施の形態は、コンタクトレンズ、眼内レンズ、血液保存袋およびチューブ、ならびに、食品包装体を含むが、これらに限定されるものではない。ポリマーの光学的品質が要求されない場合の適用において、上記の粒子サイズの範囲よりも大きな粒子も使用されてもよい。
【0022】
一つの実施の形態において、金属塩粒子も、製品が形成される上述した少なくとも一つのポリマー全体に、均質に分散されている。この明細書で使用されているように、用語「均質に分散される」とは、粒子の凝集が形成されず、かつ、粒子が、抗菌性金属塩を含むポリマーの特定部分に実質的に集中していないことを意味する。一つの実施の形態において、「均質に分散される」とは、ポリマーの任意の二つの領域間における金属塩粒子の濃度(乾燥製品の重量に基づいて重量%として測定される)の違いが約20%未満であることを意味する。他の実施の形態において、ポリマーの任意の二つの領域間における金属塩粒子の濃度の違いが約10%未満であり、さらに他の実施の形態において、ポリマーの任意の二つの領域間における金属塩粒子の濃度の違いが約5%未満である。分散の均質性は、特性X線の放出を誘発する高エネルギー電子を使用する元素分析技術を用いて、最終製品中で測定されてもよい。この適用では、電子プローブ微量分析法(EPM)が使用された(カメラ(Cameca)SX100と、20キロ電子ボルト、50ナノアンペアおよび20マイクロメートルの分析条件を用いる四つの波長分光計を備えた自動電子マイクロプローブSX50)。
【0023】
一つの実施の形態において、本発明の製品には、視認できる霞み(visual haze)も、不都合な色もない。抗菌性製品の透明度は、以下に詳細に記述されるCSIレンズに対する、約70マイクロメートルの厚さを有するサンプルを用いて測定されたヘイズ(haze)の百分率によって評価された。約100%未満のヘイズ値、約50%未満のヘイズ値は、本発明を用いることで、容易に達成され得る。
【0024】
最終ポリマー製品の色は、分光光度計を用いて測定され、かつ、国際照明委員会(CIE)の1976年のL*a*b*表色系で記録され得る。本発明の製品は、約89を超えるL*を有してもよく、一部の実施の形態において、約90を超えるL*を有し、a*は、約2未満であり、一部の実施の形態において、a*は約1.4未満であってもよい。色測定は、紫外線吸収剤、染料(handling tints)、フォトクロミック化合物、および、これらの同類等、最終製品の色に影響を与えうるポリマー成分を含まないポリマー類に対して実施されることになる。
【0025】
ポリマー中の金属塩の量は、乾燥ポリマーの全重量に基づいて測定される。ポリマー中の金属塩の量は、製品の最終用途および使用要件によって決定される。例えば、製品がコンタクトレンズである場合の一つの実施の形態において、透明度および色は、重要である。製品がコンタクトレンズであり、かつ、金属塩がヨウ化銀である場合の複数の実施の形態において、ポリマー中の銀の量は、ポリマーの乾燥重量に基づいて、約100ppm〜約1000ppmであり、一部の実施の形態では、ポリマーの乾燥重量に基づいて、200ppm〜約1000ppmである。他の実施の形態において、ポリマー中の銀の量は、ポリマーの乾燥重量に基づいて、約0.00001重量%(0.1ppm)〜約10.0重量%、好ましくは約0.0001重量%(1ppm)〜約1.0重量%、最も好ましくは約0.0001重量%(1ppm)〜約0.1重量%であってもよい。金属塩の添加については、金属塩の分子量が、金属イオンの重量%の金属塩への変換率を決定し、この技術分野における当業者は、抗菌性金属の所望量を与える上で必要な塩の量を算出することができる。
【0026】
一つの実施の形態において、本発明の製品は、
(a)反応性ポリマー混合物の少なくとも一つの成分中に少なくとも一つの塩前駆体を溶解させて塩前駆体混合物を形成すること;
(b)当該反応性ポリマー混合物の少なくとも一つの成分中に少なくとも一つの金属剤および少なくとも一つの分散剤を溶解させることによって、金属剤・分散剤錯体を形成して、金属剤混合物を形成すること;
(c)粒子形成条件下で、当該塩前駆体混合物および当該金属剤混合物を混合して粒子含有反応性混合物を形成すること;
(d)任意に、追加の反応性ポリマー成分を当該粒子含有反応性混合物に混合すること;ならびに、
(e)当該粒子含有反応性混合物を反応させて、金属塩を含む抗菌性ポリマー製品または部品を形成することであって、抗菌性金属Mの少なくとも約90%が金属塩の形態で存在する、形成すること、
によって形成されてもよい。
【0027】
用語「金属塩」は、上述した意味を有している。用語「塩前駆体」とは、金属イオンで置換できる陽イオンを含有する、あらゆる化合物または組成物(水溶液を含む)を云う。この実施の形態において、当該塩前駆体が約1マイクログラム/ミリリットル以上でレンズ処方中に溶解できることが好ましい。当該用語は、「抗菌性コンタクトレンズおよび使用方法(Antimicrobial Contact Lenses and Methods of Use)」という名称の米国公開特許US2003/0043341号明細書に記述されているようにゼオライトを含まず、あるいは、「活性銀を含有する抗菌性コンタクトレンズおよび製造方法(Antimicrobial Contact Lenses Containing Activated Silver and Methods for Their Production)」という名称の国際公開WO02/062402号に記述されたような活性銀を含まない。塩前駆体は、少なくとも化学量論的な量で、反応性混合物中に添加され、一部の実施の形態において、プラスチック製の最終製品に望ましい抗菌性金属の量に関連してモル過剰で、反応性混合物中に添加される。例えば、20マイクログラムのヨウ化銀が金属塩として製品中に存在している場合の一つの実施の形態において、ヨウ化ナトリウムは、少なくとも12マイクログラムの量で、反応性混合物中に存在している。塩前駆体の例は、塩化ナトリウム、ヨウ化ナトリウム、臭化ナトリウム、塩化リチウム、硫化リチウム、硫化ナトリウム、硫化カリウム、四塩化銀ナトリウム(sodium tetrachloro argentate)、これらの混合物、および、これらの同類のものなど、無機分子を含むが、これらに限定されるものではない。有機分子の例は、テトラアルキル乳酸アンモニウム(tetra-alkyl ammonium lactate)、テトラアルキル硫酸アンモニウム(tetra-alkyl ammonium sulfate)、テトラアルキル酢酸ホスホニウム(tetra-alkyl phosphonium acetate)、テトラアルキル硫酸ホスホニウム(tetra-alkyl phosphonium sulfate)、テトラアルキル塩化アンモニウム(tetra-alkyl ammonium chloride)、テトラアルキル塩化ホスホニウム(tetra-alkyl phosphonium chloride)、臭化物、ヨウ化物等のハロゲン化四級アンモニウムまたはハロゲン化四級ホスホニウム(quaternary ammonium or phosphonium halides)、および、これらの同類の物質を含むが、これらに限定されるものではない。一つの実施の形態において、塩前駆体は、ヨウ化ナトリウムを含む。
【0028】
用語「金属剤」とは、金属イオンを含有する、あらゆる組成物(水溶液を含む)を云う。このような組成物の例は、硝酸銀、銀トリフレート(silver triflate)、酢酸銀、テトラフルオロホウ酸銀(silver tetrafluoroborate)、硝酸銅、硫酸銅、硫酸マグネシウム、硫酸亜鉛、これらの混合物、および、これらの同類の水溶液または有機性溶液を含むが、これらに限定されるものではない。溶液中での金属剤の適切な濃度は、最終製品に含まれることになる金属塩の所望量に基づいて算出され得る。例えば、一つの実施の形態において、金属剤の濃度は、最終製品中に、約0.00001重量%(0.1ppm)〜約10.0重量%、約0.0001重量%(1ppm)〜約1.0重量%の金属塩を与えるように、他の実施の形態において、約0.0001重量%(1ppm)〜約0.1重量%の金属塩を与えるように、選択される。
【0029】
一部の実施の形態において、安定した色が望ましい。例えば、プラスチック製品が眼用装置である場合に、当該眼用装置が反応性混合物と同一の色および透明度を有することが望ましいことがある。銀塩は、感光的であることが知られている。したがって、仮に、銀塩の形成、および当該銀塩を含有する製品の硬化を気にかけない場合に、所望の銀塩は、当該製品中に生成されない。例えば、ヨウ化銀は、約400ナノメートル未満の波長光に対して感光的であるので、仮に、当該感光性を気にかけない場合に、光開始によって硬化される反応性混合物が、銀塩が減少したことを示す、不都合な黄色または茶色のレンズを形成する場合がある。光還元は、選択された金属塩の結合エネルギーに相当する波長(臨界波長)より大きな光の波長で、金属塩を含む反応性混合物を硬化させることによって、最小限化されてもよい。例えば、ヨウ化銀は、60キロカロリー/モルの結合エネルギーを有している。この結合エネルギーに関連した波長は、電磁方程式:
EAgI=hc/(λNA)
を用いて算出されてもよい。ここで、hはプランク定数であり、cは光の速度であり、λは入射放射線の波長であり、NAはアボガドロ数(Avagadro’s number)である。
【0030】
ヨウ化銀では、λは477ナノメートルである。臨界波長への調節は、型材および包装材料および溶液によるエネルギーの吸収または反射の説明となるように行われてもよい。したがって、例えば、製品がヨウ化銀を含むコンタクトレンズであり、当該製品が、10%のエネルギー伝送損失を計上するプラスチック製モールドを用いる直接成型によって形成される場合に、調節された臨界波長は、
λ=(1-10%)×477ナノメートル
λ=429ナノメートル
【0031】
したがって、この実施の形態における硬化条件は、約429ナノメートルより大きな波長光を含む。代わりに、反応性混合物は、熱硬化等を含むが、これに限定されない、光を含まない条件を用いて、硬化され得る。
【0032】
また、光還元は、実質的にすべての金属剤が金属塩に変換されるように、金属剤と比較して、モル過剰の塩前駆体を用いて、最小限化され得る。約1.1:1以上である塩前駆体:金属剤のモル比は、許容される。このモル比は、最終製品中に含まれる抗菌性金属Mの少なくとも約90%が金属塩の形態であることを保証する。一部の実施の形態において、製品は、重合開始剤、および、紫外光以外の条件を用いて硬化される。
【0033】
金属剤混合物および塩前駆体混合物のうち、少なくとも一方は、少なくとも一つの分散剤をさらに含み、一つの実施の形態では、金属剤混合物が、少なくとも一つの分散剤をさらに含む。適切な分散剤は、孤立電子対を備えた官能基を含有するポリマー類を含む。分散剤の例は、ヒドロキシアルキルメチルセルロースポリマー類、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレンオキシド、澱粉、ペクチン、ゼラチン等の多糖類;ポリジメチルアクリルアミドを含むポリアクリルアミド、ポリアクリル酸、3−アミノプロピルトリエトキシシラン(3-aminopropyltriethoxysilane)(APS)、メチルトリエトキシシラン(methyl-triethoxysilane)(MTS)、フェニルトリメトキシシラン(phenyl-trimethoxysilane)(PTS)、ビニルトリエトキシシラン(vinyl-triethoxysilane)(VTS)および3−グリシドキシプロピルトリメトキシシラン(3-glycidoxypropyltrimethoxysilane)(GPS)等の有機アルコキシシラン類(organoalkoxysilanes)、ポリエチレングリコール、ポリプロピレングリコール、グリセリンのホウ酸エステル(BAGE)等のポリエーテル類、約10,000を超える分子量を有し、かつ、ヒドロキシル基およびウレタン基等を含むが、これらに限定されない水素結合基等、粘性を増大させる基を含有するシリコンマクロマー、ならびに、これらの混合物を含む。
【0034】
一つの実施の形態において、分散剤は、ヒドロキシアルキルメチルセルロースポリマー類、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレンオキシド、グリセリン、グリセリンのホウ酸エステル(BAGE)、ゼラチンおよびポリアクリル酸、および、これらの混合物からなる群より選択される。他の実施の形態において、分散剤は、ヒドロキシプロピルメチルセルロース、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレンオキシド、ゼラチン、グリセリン、グリセリンのホウ酸エステル(BAGE)、および、これらの混合物からなる群より選択される。さらに他の実施の形態において、分散剤は、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレンオキシド、および、これらの混合物からなる群より選択される。
【0035】
分散剤がポリマーである場合に、当該ポリマーは、ある範囲の分子量を有することができる。約1000から数百万以下の分子量が使用されてもよい。上限は、金属塩混合物、塩前駆体混合物および反応性混合物中への分散剤の溶解度のみによって境界される。ゼラチンおよびメチルセルロース等のグリコシド系ポリマー類では、分子量は、百万を超えてもよい。ポリビニルアルコール、ポリビニルピロリドンおよびポリアクリル酸のような非グリコシド系ポリマー類では、分子量は、約2,500ダルトン〜約2,000,000ダルトン(約4.151×10−21g〜約3.321×10−18g)の範囲であってもよく、一部の実施の形態では、約10,000ダルトン〜約1,800,000ダルトン(約1.661×10−20g〜約2.989×10−18g)の範囲であってもよく、他の実施の形態では、約20,000ダルトン〜約1,500,000ダルトン(約3.321×10−20g〜約2.491×10−18g)の範囲であってもよい。一部の実施の形態において、約50,000ダルトン(約8.303×10−20g)を超える分子量は、この範囲内で分散剤が一部のポリマー系内で、より安定化効果を与える場合に、使用されてもよい。
【0036】
あるいは、分散状態安定化ポリマー(dispersion-stabilizing polymers)の分子量は、ジョン・ワイリー・アンド・サンズ・インク(John Wiley & Sons Inc.)発行の「ポリマー科学および工学の百科事典第2版」第17巻、第198頁〜第257頁の「N−ビニルアミドポリマー」項(Encyclopedia of Polymer Science and Engineering, N-Vinyl Amide Polymers, Second edition, Vo1 17, pgs. 198-257)中に記述されているように、動粘測定法に基づくK値によっても表示され得る。この方法で表示される場合に、非グリコシド系分散剤ポリマー類は、約5〜約150のK値を有してもよく、一部の実施の形態では、約5〜約100のK値、約5〜約70のK値を有してもよく、他の実施の形態では、約5〜約50のK値を有してもよい。
【0037】
金属塩ナノ粒子が反応性ポリマー混合物中に直接、形成される場合に、分散剤は、当該反応性ポリマー混合物の全成分の重量%に基づいて、約0.001重量%〜約40重量%の間の量で存在してもよい。一部の実施の形態において、分散剤は、約0.01重量%〜約30重量%の間の量で存在してもよく、他の実施の形態では、約0.1重量%〜約30重量%の間の量で存在してもよい。一部の実施の形態において、分散剤は、ポリビニルアルコールを含むコンタクトレンズが製造される場合など、ポリマー製品を形成する上で使用される反応性成分でもある。これらの実施の形態において、使用される分散剤の量は、当該反応性ポリマー混合物の全成分の重量%に基づいて、約90重量%以下の量であってもよく、一部の実施の形態では、約100重量%以下の量であってもよい。
【0038】
一部の実施の形態において、分散剤は、結果的に得られたポリマーに追加の利点を与える。例えば、ポリビニルピロリドン(PVP)が粒子安定剤である場合に、当該ポリビニルピロリドンは、分散安定性をもたらすことに加えて、湿潤性、摩擦係数、含水率、離型性、および、同類のものを改善することができる。これらの実施の形態において、分散安定性を与える上で必要な量より多い量の分散剤を含む必要がある、あるいは、当該多い量の分散剤を含むことが望ましい場合がある。これらの実施の形態において、所望サイズの粒子が形成されることを保証するために、脱気ステップおよび熟成ステップ等、他の工程条件の均衡を保つことが望ましいはずである。
【0039】
塩前駆体混合物および金属剤混合物は、粒子形成条件下で混合される。この明細書で使用されているように、粒子形成条件は、約200ナノメートル未満の平均粒子サイズ、一部の実施の形態では約100ナノメートル未満の平均粒子サイズ、他の実施の形態では約50ナノメートル未満の平均粒子サイズを有し、かつ、反応性混合物全体に分散された金属塩粒子を形成する上で適した時間、温度およびpHを含む。
【0040】
混合温度は、反応性混合物中の反応性成分に依存して変更されてもよい。概ね、反応性混合物の凝固点より高く、約100℃以下の混合温度が使用されてもよい。一部の実施の形態において、約10℃〜約90℃の間の混合温度が使用されてもよく、他の実施の形態では約10℃〜約50℃の間の混合温度が有用である。
【0041】
塩前駆体混合物または金属剤混合物のいずれか一方あるいは両方は、反応性混合物と混合する前に脱気されてもよい。
【0042】
一つの実施の形態において、塩前駆体混合物または金属剤混合物のいずれか一方は、単独噴流等の流れによって導入されてもよく、あるいは、塩前駆体混合物および金属剤混合物の双方は、二重噴流によって同時に導入されてもよい。単独噴流法において、例えば、金属剤混合物の溶液は、制御速度で噴流によって流されて、塩前駆体混合物および分散剤を含有する撹拌溶液中に導入される。あるいは、二重噴流法は、分散剤を含有する撹拌溶液中に、二つの別の噴流によって塩前駆体混合物および金属剤混合物の双方を同時に添加するために使用されてもよい。一部の実施の形態において、更なる量の分散剤、塩前駆体混合物および/または金属剤混合物を添加することが望ましい場合がある。
【0043】
塩前駆体混合物および金属剤混合物は、約10分間未満の時間中、反応性混合物中に添加されてもよく、一部の実施の形態では、約10秒〜約5分間の付加時間中、添加されてもよい。
【0044】
得られる溶液が均質であり、かつ、安定した分散液が形成される限り、あらゆる混合時間が使用されてもよい。この明細書で使用されているように、安定した分散液は、少なくとも約12時間、実質的に変化しない。商業的に望ましい混合時間は、約1分間〜数日の範囲を含めてもよく、一部の実施の形態では、約10分間〜約12時間の範囲を含めてもよい。
【0045】
高剪断性の混合方法は、低分子量のポリマーと一緒に使用されてもよく、上述した範囲の下限値での混合時間を可能にすることができる。
【0046】
また、反応性混合物は、真空下で、あるいは、当該反応性混合物中のいずれの成分とも反応しないガスを用いて、脱気されてもよい。適切な不活性ガスは、窒素、アルゴン、これらを含む混合物、および、これらの同類のものを含む。脱気処理は、完全真空(例えば、10mbar(10ヘクトパスカル))以下の圧力、および、約60分間以内の時間、一部の実施の形態では約40分間以内の時間を用いて、実行されてもよい。所定の反応性混合物と一緒に使用される温度条件および圧力条件と同様に、脱気ステップの継続時間も、使用される溶媒の揮発度等の他の要素によって決められてもよい。
【0047】
上記処理方法は、脱気ステップ前に、粒子熟成ステップをさらに含めてもよい。非常に小さな粒子は、加熱中に、より大きな粒子よりも、容易に溶解する。したがって、透明度が重要である場合の適用(眼用装置等)において、さらなる処理(殺菌ステップ、溶融加工ステップ、焼鈍ステップ、焼結ステップ等)中または保存中に、過度に熟成しない程度に粒子を大きくすることを保証する粒子熟成ステップを含むことが望ましいことがある。当該粒子熟成ステップにおいて、反応性混合物は、微粉を低減するために、約5分間〜1時間の期間、約30℃〜約70℃の温度に加熱される。この粒子熟成ステップは、殺菌される医療装置にとって特に有用であることがある。例えば、プラスチック製品がレンズである場合に、当該レンズが形成されるときに、当該レンズは、視認できる霞みが存在しない状態である必要があり、かつ、処理(殺菌包装処理ステップを含む)、保存、および、使用の間中、視認できる霞みが存在しない状態を維持する必要がある。また、微粉の形成は、分散剤の量を低減させることによって、軽減されてもよい。
【0048】
一つの実施の形態において、反応性混合物中の少なくとも約90%の粒子は、約100ナノメートル未満の粒子サイズを有しており、他の実施の形態では、反応性混合物中の少なくとも約90%の粒子は、約80ナノメートル未満の粒子サイズを有しており、さらに他の実施の形態では、反応性混合物中の少なくとも約90%の粒子は、約60ナノメートル未満の粒子サイズを有している。反応性混合物中の粒子の粒子サイズは、以下の検査法の項で記述されているように、光散乱法(レーザー光散乱法または動的光散乱法のいずれか一方)によって測定されてもよい。
【0049】
本発明の粒子含有反応性混合物は、視認できる霞みも、不都合な色もない。不都合な色の欠如は、白色を背景にして主観的に、あるいは、以下に記述されるL*a*b*表色法を用いて、評価されてもよい。
【0050】
追加の成分は、混合ステップ中に、任意に添加されてもよい。追加のポリマー成分は、反応性のモノマー類、プレポリマー類、および、マクロマー類、重合開始剤、架橋剤、連鎖移動剤、紫外線吸収剤、湿潤剤、剥離補助剤(release aids)、フォトクロミック化合物、栄養補助化合物および医薬化合物、着色剤、染料、顔料、これらの組み合わせ、および、これらの同類の物質を含む。当該追加のポリマー成分は、モノマー類、オリゴマー類あるいはプレポリマー類を含む、いかなる形態でも添加されてもよい。
【0051】
仮に、反応性混合物のいずれかの成分が金属剤と反応して元素金属(elemental metal)を形成することができ、かつ、当該元素金属が望ましいものではない場合に、一つの実施の形態において、当該成分は、金属塩粒子が形成された後であるが、反応性混合物を硬化させてポリマー製品を形成する前に、当該反応性混合物中に添加される。例えば、硝酸銀(AgNO3)がN,N-ジメチルアクリルアミド(DMA)と反応して不都合な非イオン性銀(Ag0)を形成できることが見出されてきた。したがって、N,N-ジメチルアクリルアミド(DMA)を含む反応性混合物では、当該N,N-ジメチルアクリルアミド(DMA)は、一つの実施の形態(金属塩がヨウ化銀である場合)において、金属塩粒子(ヨウ化銀)が形成された後に、当該反応性混合物中に添加される。この技術分野における当業者は、ある成分が還元剤として作用するか否かについて、当該成分を溶媒中で金属剤と混合し、かつ、化学分析によって、あるいは、特定の場合に、当該混合物の外観の変化によって分析することで、容易に決定することができる。
【0052】
あるいは、ナノ粒子の金属塩は、反応性ポリマー混合物とは別に形成されてもよい。例えば、安定化金属塩粒子は、少なくとも一つの塩前駆体を含む塩前駆体溶液を形成すること;
約20重量%〜約50重量%の間の少なくとも一つの分散剤であって、少なくとも約1000の平均分子量を有する、分散剤と、少なくとも一つの金属剤とを含む金属剤溶液を形成すること;
添加中に溶液の透明度を維持し、かつ、約200ナノメートル未満の粒子サイズを有する安定化金属塩粒子を含む生成物溶液を形成する上で十分な速度で、一方の溶液を他方の溶液中に添加すること;および、
当該安定化金属塩粒子を乾燥させることによって形成されてもよい。安定化金属塩粒子は、約200ナノメートル未満の粒子サイズを有する金属塩粒子であり、かつ、少なくとも一つの分散剤と錯化されている。一部の実施の形態において、安定化金属塩粒子は、約100ナノメートル未満の粒子サイズを有しており、他の一部の実施の形態では約50ナノメートル未満の粒子サイズを有している。
【0053】
この実施の形態において、金属剤および塩前駆体の溶液は、(a)金属剤、塩前駆体および分散剤を溶解でき、(b)金属剤を金属に還元せず、かつ、(c)既知の方法によって容易に除去できる、任意の溶媒を用いて形成される。水、アルコール類、または、これらの混合物が使用されてもよい。金属剤および塩前駆体を可溶化できる適切なアルコール類が選択されてもよい。硝酸銀およびヨウ化銀が金属剤および塩前駆体として使用される場合に、三級アミルアルコール、トリプロピレングリコール・モノメチルエーテル等のアルコール類、これらの混合物、および、これらと水との混合物が使用されてもよい。また、水も単独で使用されてもよい。
【0054】
上述したいずれの分散剤も使用されてもよい。混合物は使用されてもよい。分散剤は、金属剤溶液または塩前駆体溶液のいずれか一方または両方に含まれてもよく、あるいは、金属剤溶液および塩前駆体溶液が添加される第三の溶液に含めることができる。一つの実施の形態において、金属剤溶液も、少なくとも一つの分散剤を含む。金属剤溶液および塩前駆体溶液の双方が少なくとも一つの分散剤を含む場合の実施の形態において、当該分散剤は、同一のものであってもよく、あるいは、異なったものであってもよい。
【0055】
分散剤は、約500ナノメートル未満の金属塩粒子サイズを与える上で十分な量(粒子サイズ安定化有効量)で含まれている。最終製品の透明度が重要である場合の実施の形態において、粒子サイズは約200ナノメートル未満であり、一部の実施の形態では約100ナノメートル未満であり、他の実施の形態では約50ナノメートル未満である。一つの実施の形態において、少なくとも約20重量%の分散剤は、所望の粒子サイズが達成されることを保証するために、少なくとも一つの溶液中で使用される。一部の実施の形態において、金属剤に対する分散剤単位のモル比は、少なくとも約1.5、あるいは、少なくとも約2であり、一部の実施の形態では少なくとも約4である。この明細書で使用されているように、分散剤単位は、分散剤中の反復単位である。一部の実施の形態において、両溶液中での分散剤の濃度が同一であることは便利なはずである。
【0056】
溶液中での分散剤の濃度の上限値は、選択された溶媒中での当該分散剤の溶解度、および、当該溶液の取扱い易さによって決定されてもよい。一つの実施の形態において、各溶液は、約50センチポアズ(cps)未満の粘度を有している。一つの実施の形態において、生成物溶液は、約50重量%以下の分散剤を有してもよい。上述したように、金属剤溶液および塩前駆体溶液は、同一または異なる濃度の分散剤を有してもよい。すべての重量%は、当該溶液中の全成分の全重量に基づいている。
【0057】
この実施の形態では、金属剤溶液および塩前駆体溶液中の金属剤および塩前駆体の各濃度は、選択された溶媒中での金属剤および塩前駆体の溶解度の限界までであって、少なくとも約1500ppmであることが望ましく、一部の実施の形態では約5000ppm〜溶解度の限界の間であることがのぞましく、一部の実施の形態では約5000ppm〜約50,000ppm(5重量%)の間であることが望ましく、また、他の実施の形態では約5000ppm〜約20,000ppm(2重量%)の間であることが望ましい。
【0058】
溶液の混合は、室温で実行されてもよく、撹拌法を含めることが有効となる。渦が形成される速度以上の撹拌速度が使用されてもよい。選択された撹拌速度は、泡立て、発泡、あるいは、混合容器からの溶液の損失を生じさせるものであってはならない。撹拌は、添加中、継続される。
【0059】
混合は、大気圧あるいは減圧下で実行されてもよい。一部の実施の形態において、混合は、溶液に泡立て、または、発泡を生じさせてもよい。発泡または泡立ては、高濃度の金属塩が入るポケットを形成させ、当該ポケットが所望の粒子サイズより大きなサイズの粒子をもたらすことがある場合に、望ましくない。これらの場合には、減圧条件が使用されてもよい。圧力は、大気圧と選択された溶媒の蒸気圧との間の任意の圧力とすることができる。一つの実施の形態において、水が溶媒である場合に、当該圧力は、大気圧と約40mbar(約40ヘクトパスカル)との間であってもよい。
【0060】
塩前駆体溶液および金属剤溶液の添加速度は、混合中に溶液の透明度を維持するように選択される。局所的な僅かな霞みは、当該溶液が撹拌中に透明である限り、許容され得る。当該溶液の透明度は、視覚的に観察されるか、あるいは、紫外・可視分光法(UV-VIS)を用いてモニターされてもよい。適切な添加速度は、所望の濃度を有する一連の溶液を調製し、かつ、異なる添加速度での溶液の透明度をモニターすることによって決定されてもよい。この方法は、実施例26〜31に例証されている。また、塩前駆体溶液への分散剤の導入は、添加速度を上げることができる。
【0061】
他の実施の形態において、速い添加速度が望ましい場合に、金属剤および分散剤は、塩前駆体溶液と混合する前に、錯体形成時間を含む錯体形成条件下で混合されることが許容される。金属剤溶液中の分散剤が金属剤を含む錯体を形成すると考えられる。この実施の形態において、金属剤溶液および塩前駆体溶液を混合する前に、金属剤が分散剤と完全に錯化することが望ましい。「完全に錯化される」とは、実質的に全部の金属イオンが少なくとも一つの分散剤と錯化されたことを意味する。「実質的に全部」とは、当該金属イオンの少なくとも約90%、一部の実施の形態では少なくとも約95%が少なくとも一つの分散剤と錯化されたことを意味する。
【0062】
錯体形成時間は、紫外・可視分光法(UV-VIS)あるいはフーリエ変換赤外分光法(FTIR)等の分光法により、溶液中でモニターされてもよい。分散剤を含まない金属剤溶液の複数のスペクトルは測定される。当該金属剤溶液の複数のスペクトルは、分散剤の添加後にモニターされ、かつ、スペクトル変化もモニターされる。錯体形成時間は、スペクトル変化が安定する時間である。
【0063】
あるいは、錯化時間は、同一濃度を有する一連の金属剤・分散剤溶液を形成し、各溶液を異なる時間(実施例1では、3時間、6時間、12時間、24時間、72時間および1週間)で混合し、各金属剤・分散剤溶液を塩前駆体溶液とバッチ式で混合することによって、経験的に測定されてもよい。錯体形成時間で混合された金属剤・分散剤溶液は、添加速度を制御せずに、金属剤溶液および塩前駆体溶液が一緒に直接注入される場合に、透明な溶液を形成することになる。例えば、20ミリリットルの金属剤溶液は、20ミリリットルの塩前駆体溶液に1秒間以内で添加されてもよい。
【0064】
錯化条件は、錯化時間(上述されている)、温度、金属剤に対する分散剤の比率、および、撹拌速度を含む。温度、金属剤に対する分散剤のモル比、および、撹拌速度の上昇は、錯化時間を短縮するはずである。この明細書に記載された教示に関連して、この技術分野における当業者は、この明細書に開示された錯化レベルを得るために、諸条件を変更することができるはずである。
【0065】
仮に、金属剤および分散剤が完全に錯化されていない場合に、混合条件は、錯化されていない金属塩の形成よりも多く、分散剤・金属剤錯体を形成するために、混合物中での反応を偏らせる(bias)ように選択されてもよい。この偏り(biasing)は、(a)塩前駆体中の分散剤の濃度、あるいは、塩前駆体溶液および金属剤溶液が添加される溶液中の分散剤の濃度、ならびに、(b)金属剤溶液および塩前駆体溶液の混合速度を制御することによって達成されてもよい。
【0066】
一旦、金属剤溶液および塩前駆体溶液が混合されたら、生成物溶液は乾燥されてもよい。凍結乾燥機、噴霧乾燥機およびこれらの同類等、従来のあらゆる乾燥設備が、使用されてもよい。乾燥設備および乾燥方法は、この技術分野において周知である。適切な噴霧乾燥機の例は、ジーイーエー・ニロ・インク(GEA Niro Inc.)から入手可能な乾燥機等、回転式噴霧乾燥機である。噴霧乾燥では、噴霧導入口の温度は、選択された溶媒の引火点を超えている。
【0067】
凍結乾燥機は、ジーイーエー・ニロ・インク(GEA Niro Inc.)を含む、多くの製造者から入手可能である。凍結乾燥温度および圧力は、この技術分野における当業者によって周知なように、溶媒を昇華させるように選択される。選択された乾燥方法について、従来の範囲内のあらゆる温度が使用されてもよい。
【0068】
生成物溶液は、得られる粉末が約10重量%未満の溶媒含有率を有するようになるまで、一部の実施の形態では約5重量%未満の溶媒含有率を有するようになるまで、また、他の一部の実施の形態では約2重量%未満の溶媒含有率を有するようになるまで、乾燥処理される。高濃度の溶媒は、安定化金属塩を形成するために使用された溶媒がポリマー製品を形成するために使用された反応性混合物と相溶性である場合に、適切であることがある。当該粉末は、この粉末を水中に分散させ、透過電子顕微鏡法、光子相関分光分析法、あるいは、動的光散乱法によって測定されるように、約100ナノメートル以下の粒子サイズ、約50ナノメートル以下の粒子サイズ、また、一部の実施の形態では約15ナノメートル以下の粒子サイズを有する安定化金属塩を含む。
【0069】
安定化金属塩粉末は、反応性混合物に直接、添加されてもよい。安定化金属塩粉末の添加量は、所望レベルの抗菌性金属イオンを与えるために、容易に算出されることができる。
【0070】
金属塩を含む反応性混合物は、抗菌性ポリマー製品を形成するために、反応させられる。反応条件は、当該反応性混合物中の成分に基づいて、この技術分野における当業者によって容易に選択されてもよい。例えば、抗菌性ポリマー製品がフリーラジカル反応性成分から形成されたコンタクトレンズである場合に、反応性混合物は、重合開始剤を含み、かつ、反応条件は、光硬化または熱硬化の条件を含めてもよい。抗菌性金属塩が、ヨウ化銀、塩化銀および臭化銀等の感光性である場合に、上述した臨界波長を下回る波長に対する当該金属塩の被爆は、銀イオン(Ag+)を非イオン性銀(Ag0)に変換することから、結果的に、当該塩が組み込まれた製品の色を暗くすることになる。したがって、一つの実施の形態において、フリーラジカル反応性成分が使用される場合に、硬化処理は、可視光に対する被爆によって実行される。他の実施の形態において、反応性混合物は、少なくとも一つの紫外線吸収化合物をさらに含み、かつ、当該反応性混合物は、可視光、熱、あるいは、これらの組み合わせを用いて硬化処理される。さらに他の実施の形態において、反応性混合物は、少なくとも一つの紫外線吸収化合物および可視光重合開始剤をさらに含み、かつ、当該反応性混合物は、可視光を用いて硬化処理される。
【0071】
金属塩は、種々のポリマー類中に形成されてもよく、あるいは、当該ポリマー類中に添加されてもよい。適切なポリマー類は、使用目的に基づいて選択されてもよい。例えば、食品包装用では、ポリエチレンテレフタレート、高密度ポリエチレンおよびポリプロピレン等のポリマー類は、食品および飲料の容器用に一般的に使用されており、低密度ポリエチレンは、プラスチック製ラップに一般的に使用されている。
【0072】
関節置換体等、種々の埋め込み型装置は、高次架橋された超高分子量ポリエチレン(UHMWPE)を用いて形成されており、当該ポリエチレンは、通常、少なくとも約400,000の分子量を有しており、一部の実施の形態では当該分子量が約1,000,000〜約10,000,000であり、当該ポリエチレンは、通常、本質的にゼロ(0)であるメルトインデックス(米国材料試験協会(ASTM) D-1238)、および、8を超える比重低下、一部の実施の形態では約25〜30の間の比重低下によって定義されている。
【0073】
縫合糸および創傷包帯を作る際のヤーンとしての使用に適した、適切な吸収性ポリマーの例は、ラクチド(乳酸、D-ラクチド、L-ラクチドおよびメソラクチドを含む)、グリコリド(グリコール酸を含む)、ε-カプロラクトン、p-ジオキサノン(1,4-ジオキサン-2-オン)、トリメチレン炭酸塩(trimethylene carbonate)(1,3-ジオキサン-2-オン)の各ホモポリマー類および各コポリマー類を含むが、これらに限定されない脂肪族ポリエステル類;トリメチレン炭酸塩、δ-バテロラクトン(δ-vaterolactone)、β-ブチロラクトン、γ-ブチロラクトン、ε-デカラクトン(ε-decalactone)、ヒドロキシ酪酸塩、ヒドロキシ吉草酸塩(hydroxyvalerate)、1,4-ジオキセパン-2-オン(1,4-dioxepan-2-one)(その二量体1,5,8,12-テトラオキサシクロテトラデカン-7,14-ジオン(1,5,8,12-tetraoxacyclotetradecane-7,14-dione)を含む)、1,5-ジオキセパン-2-オン、6,6-ジメチル-1,4-ジオキサン-2-オンの各アルキル誘導体;および、これらのポリマー混合物を含むが、これらに限定されるものではない。
【0074】
縫合糸は、ポリアミド類(ポリヘキサメチレンアジポアミド(polyhexamethylene adipamide)(ナイロン66)、ポリヘキサメチレンセバクアミド(polyhexamethylene sebacamide)(ナイロン610)、ポリカプロアミド(polycapramide)(ナイロン6)、ポリドデカンアミド(polydodecanamide)(ナイロン12)およびポリヘキサメチレンイソフタルアミド(polyhexamethylene isophthalamide)(ナイロン61)、これらのコポリマー類およびこれらの混合物)、ポリエステル類(例えば、ポリエチレンテレフタレート、ポリブチルテレフタレート、これらのコポリマー類およびこれらの混合物)、フルオロポリマー類(例えば、ポリテトラフルオロエチレンおよびポリフッ化ビニリデン)、ポリオレフィン類(例えば、アイソタクチックおよびシンジオタクチックなポリプロピレンおよびこれらの混合物の他、ヘテロタクチックなポリプロピレン(1985年12月10日に発行され、エシコン・インク(Ethicon, Inc.)に譲渡された米国特許第4,557,264号明細書に記述されたポリエチレン等であり、当該文献は参照によってこの明細書に組み込まれる)およびポリエチレン(1985年12月10日に発行され、エシコン・インク(Ethicon, Inc.)に譲渡された米国特許第4,557,264号明細書に記述されたポリエチレン等)と混合したアイソタクチックまたはシンジオタクチックなポリプロピレンを主成分とする混合物、ならびに、これらの混合物等であるが、これらに限定されない非吸収性ポリマー材料からも形成され得る。
【0075】
涙点プラグ(punctal plugs)の本体は、シリコーン、シリコーン混合物、例えば、pHEMA(ポリヒドロキシエチルメタクリレート)、ポリエチレングリコール、ポリビニルピロリドンおよびグリセロールの各親水性モノマー類等のシリコーンコポリマー類、例えば米国特許第5,962,548号明細書、米国特許第6,020,445号明細書、米国特許第6,099,852号明細書、米国特許第6,367,929号明細書および米国特許第6,822,016号明細書に記述されたポリマー類等のシリコーンヒドロゲルポリマーを含むが、これらに限定されない、任意の適切な生体適合性ポリマー類から形成されてもよい。他の適切な生体適合性ポリマー類は、例えば、ポリ(エチレングリコール);ポリ(エチレンオキシド);ポリ(プロピレングリコール);ポリ(ビニルアルコール);ポリ(ヒドロキシエチルメタクリレート);ポリ(ビニルピロリドン);ポリアクリル酸;ポリ(エチルオキサゾリン);ポリ(ジメチルアクリルアミド);例えばホスホリルコリン誘導体等のリン脂質;ポリスルホベタイン類(polysulfobetains);例えばヒアルロン酸、デキストラン、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース、ジェランガム、グアーガム、ヘパラン硫酸、コンドロイチン硫酸(chondritin sulfate)、ヘパリンおよびアルギン酸塩等の多糖類および炭水化物;例えばゼラチン、コラーゲン、アルブミンおよびオボアルブミン等の蛋白質;ポリアミノ酸;例えばポリテトラフルオロエチレン(「PTFE」)、ポリフッ化ビニリデン(「PVDF」)およびテフロン等のフッ素化ポリマー類;ポリプロピレン;ポリエチレン;ナイロン;ならびに、エチレンビニルアルコール(「EVA」)を含む。
【0076】
超音波外科機器のポリマー部品は、ポリイミド類、フルオラエチレンプロペン(fluora ethylene propene)(FEPテフロン)、PTFEテフロン、シリコーンゴム、エチレン・プロピレンジエンモノマー(EPDM)ゴムから形成されてもよく、これらの材料のいずれかは、テフロンまたはグラファイト等の材料で充填されてもよく、あるいは充填されなくてもよい。このような例は、米国公開特許US20050192610号明細書および米国特許第6,458,142号明細書に開示されている。
【0077】
上述のポリマー類の製造方法は周知であり、かつ、安定化金属塩粒子は、溶融混練により、あるいは、重合工程中に、容易に組み込まれうる。各系に適切な分散剤は、分散剤および分散剤・金属剤錯体の熱安定性を考慮することによって容易に選択されることができる。
【0078】
一つの実施の形態において、抗菌性ポリマー製品はレンズである。この明細書で使用されているように、用語「レンズ」は、眼球内または眼球上に配する眼用装置を云う。これらの眼用装置は、光学補正、治療効果、美容効果、あるいは、これらの組み合わせを付与することができる。用語「レンズ」は、ソフトコンタクトレンズ、ハードコンタクトレンズ、眼内レンズ、被せレンズ(overlay lenses)、眼球用挿入体(ocular inserts)、および、涙点プラグ等であるが、これに限定されない光学挿入体(optical inserts)を含むが、これらに限定されるものではない。
【0079】
ソフトコンタクトレンズは、シリコーンヒドロゲルおよびフルオロヒドロゲルを含むが、これらに限定されない、シリコーンエラストマーまたはヒドロゲルから形成されてもよい。好適には、本発明のレンズは、エタフィルコンA(etafilcon A)から形成されたレンズ等のレンズに匹敵する光学的な透明度を呈する、光学的に透明なものである。
【0080】
本発明の金属塩は、米国特許第5,710,302号明細書、国際公開WO9421698号、欧州特許第406161号、日本特許出願JP2000016905号、米国特許第5,998,498号明細書、米国特許出願09/532,943号、米国特許第6,087,415号明細書、米国特許第5,760,100号明細書、米国特許第5,776,999号明細書、米国特許第5,789,461号明細書、米国特許第5,849,811号明細書および米国特許第5,965,631号明細書に記述されたソフトコンタクトレンズ処方中に添加されてもよい。さらに、本発明の金属塩は、市販のソフトコンタクトレンズ処方中に添加されてもよい。ソフトコンタクトレンズの処方例は、エタフィルコンA、ゲンフィルコンA(genfilcon A)、レネフィルコンA(lenefilcon A)、ポリマコン(polymacon)、アクアフィルコンA(acquafilcon A)、バラフィルコンA(balafilcon A)、ロトラフィルコンA(lotrafilcon A)、ロトラフィルコンB、ガリフィルコン(galyfilcon)、セノフィルコン(senofilcon)およびコムフィルコン(comfilcon)の各処方を含むが、これらに限定されるものではない。一つの実施の形態において、コンタクトレンズ処方は、エタフィルコンA、バラフィルコンA、アクアフィルコンA、ロトラフィルコンA、ロトラフィルコンB、セノフィルコン、ガリフィルコン、コムフィルコンであり、他の実施の形態では、コンタクトレンズ処方は、米国特許第5,998,498号明細書、米国特許出願09/532,943号、2000年8月30に出願された、米国特許出願09/532,943号の一部継続出願、国際公開WO03/022321号、米国特許第6,087,415号明細書、米国特許第5,760,100号明細書、米国特許第5,776,999号明細書、米国特許第5,789,461号明細書、米国特許第5,849,811号明細書および米国特許第5,965,631号明細書で調製されたエタフィルコンA、ガリフィルコン、コムフィルコンおよびシリコーンヒドロゲルである。これらの特許文献ばかりでなく、この段落で開示された他の全特許文献は、参照によって、この明細書にそっくりそのまま組み込まれる。一つの実施の形態において、本発明の金属塩は、米国特許出願11/757484号に記述されているように、少なくとも約41の親水率(hydrophilicity index)を有するレンズ材料中に添加されてもよい。一つの実施の形態において、製品は、ガリフィルコンから形成されたコンタクトレンズである。
【0081】
ハードコンタクトレンズは、ポリ(メチル)メタクリレートのポリマー類、シリコンアクリレート(silicon acrylates)、シリコーンアクリレート(silicone acrylates)、フルオロアクリレート、フルオロエーテル、ポリアセチレンおよびポリイミドを含むが、これらに限定されないポリマー類から形成され、ここで、代表例の調製は、米国特許第4,330,383号明細書に見出されることができる。本発明の眼内レンズは、既知の材料を用いて形成され得る。例えば、当該レンズは、ポリメチルメタクリレート、ポリスチレン、ポリカーボネートまたはこれらの同類の材料、および、これらの組み合わせを含むが、これらに限定されない硬質材料から形成されてもよい。さらに、ヒドロゲル、シリコーン材料、アクリル系材料、フルオロカーボン材料、および、これらの同類の材料、またはこれらの組み合わせを非限定的に含む可撓性材料が使用されてもよい。典型的な眼内レンズは、国際公開WO0026698号、国際公開WO0022460号、国際公開WO9929750号、国際公開WO9927978号および国際公開WO0022459号、米国特許第4,301,012号明細書、米国特許第4,872,876号明細書、米国特許第4,863,464号明細書、米国特許第4,725,277号明細書、米国特許第4,731,079号明細書に記述されている。金属塩は、上述したようなハードコンタクトレンズ処方および眼内レンズ処方中に添加されてもよい。
【0082】
眼用レンズを含む生物医学装置は、被膜が抗菌性金属の活性を抑制しない、すなわち、被膜が当該活性を不都合に低下させないことを条件として、生体組織との適合性を向上させるために被覆されてもよい。したがって、本発明の製品は、レンズを被覆するのに使用される多くの物質で被覆されてもよい。あるいは、安定化金属塩粒子は、あらゆる既知の被膜組成物に都合よく添加されてもよく、一つの実施の形態では、当該粒子は、本発明の教示にしたがって、浸漬被覆溶液等の溶液および反応性混合物、トランスファー成型被膜(mold transfer coatings)、反応性被膜およびこれらに同類の被膜から形成される溶液組成物中に添加されてもよい。適切な被膜例は、米国特許第6,087,415号明細書および米国公開特許US2001/0086160号明細書に開示されたような、結合剤または結合層を用いた被膜、米国特許第5,779,943号明細書に開示されたような潜在的な親水性被膜、米国特許第5,275,838号明細書に開示されたようなポリエチレンオキシド星状被膜、米国特許第4,973,493号明細書に開示されたような共有結合被膜、米国特許第5,135,297号明細書に記述されたように被覆される製品と接触した反応性モノマーの重合化および架橋化によって形成された被膜、米国特許第6,200,626号明細書に開示されたようなグラフト重合被膜、欧州特許第1,287,060号、米国特許第6,689,480号明細書および国際公開WO2004/060431号に開示されたような非反応性または錯体形成被膜、欧州特許第1252222号、米国特許第7,022,379号明細書、米国特許第6,896,926号明細書、米国公開特許US2004/0224098号明細書、米国公開特許US2005058844号明細書および米国特許第6,827,966号明細書に開示されたような「多層被膜」、国際公開WO03/011551A1号に開示されたようなトランスファー成型被膜、および、米国特許第5,760,100号明細書に開示された表面改質方法を含むが、これらに限定されるものではない。米国特許第6,193,369号明細書に開示されたようなケイ酸塩被膜、および、米国特許第6,213,604号明細書に開示されたようなプラズマ被膜は、抗菌性金属塩を含む眼用装置等の製品上に塗布されてもよい。これらの適用および特許文献は、手順、組成物および方法に関して参照されることによって、この明細書に組み込まれる。
【0083】
上述したレンズ処方の多くは、ユーザーが1日〜30日の範囲の連続期間、当該レンズを挿入することを可能にする。眼の中にレンズが長く存在すればするほど、細菌および他の微生物が当該レンズ表面上で増大することになる可能性が高くなることが知られている。本発明のレンズは、コンタクトレンズ等のポリマー製品上での細菌の増大抑制に役立つ。
【0084】
またさらに、本発明は、哺乳類の眼領域内に配されたレンズ上での微生物のコロニー形成に関連した有害事象を軽減する方法であって、当該方法は、少なくとも約14日間、哺乳類の眼上に、少なくとも一つの抗菌性金属塩を含む抗菌性レンズを配置することを含むか、配置することから成るか、あるいは、配置することから本質的に成る、方法を含み、当該抗菌性レンズは、当該14日の期間後に、少なくとも約0.5マイクログラムの抽出可能(extractible)な抗菌性金属を含むものである。他の実施の形態において、当該抗菌性レンズは、少なくとも約30日後に、少なくとも約0.5マイクログラムの当該抽出可能(extractible)な抗菌性金属を含むものである。この実施の形態において、当該抗菌性レンズは、連続的に装用されてもよく、あるいは、毎日装用するモード(就寝前に取り外し、かつ、起床時に再挿入するモード)で装用されてもよい。抗菌性金属塩の抽出は、上述した条件を用いて決定されうる。さらに他の実施の形態において、本発明の抗菌性レンズは、予定の装用期間中に、1日当たり0.5マイクログラムの抗菌性金属を放出する上で十分な抗菌性金属塩の初期濃度を含む。予定の装用期間は、レンズが、患者による装用に推奨される長さの期間である。
【0085】
用語「レンズ」、「抗菌性レンズ」および「金属塩」は、すべて、上述した意味および好適な範囲を有している。語句「微生物のコロニー形成に関連した有害事象」は、コンタクトレンズによる眼の炎症、コンタクトレンズに関連した辺縁性潰瘍、コンタクトレンズに関連した眼の充血、浸潤性角膜炎、細菌性角膜炎、および、これらの同類の症状を含むが、これらに限定されるものではない。用語「哺乳類」とは、あらゆる温血性の高等脊椎動物を意味し、好適な哺乳類は、ヒトである。
【0086】
以下の検査方法が、実施例において使用された。
【0087】
レンズのオートクレーブ処理後におけるレンズの銀の含有率は、機器中性子放射化分析法「INAA」によって決定された。この機器中性子放射化分析法(INAA)は、核反応における中性子で放射化されることによって特定放射性核種の人工的な誘導に基づく定性元素分析法および定量元素分析法である。サンプルの放射の後には、放射性核種の崩壊によって放出される特徴的ガンマ線の定量測定法が行われる。特定のエネルギーで検出されたガンマ線は、特定の放射性核種の存在の兆候であり、この兆候は高度の特異性を可能にする。ベッカー・ディー・エー(Becker, D. A.);グリーンバーグ・アール・アール(Greenberg, R. R.);ストーン・エス・エフ(Stone, S. F.)による1992年の放射分析核化学誌(J. Radioanal. Nucl. Chem.)第160(1)巻の第41頁〜第53頁;ベッカー・ディー・エー(Becker, D. A.);アンダーソン・ディー・エル(Anderson, D. L.);リンドストローム・アール・エム(Lindstrom, R. M.);グリーンバーグ・アール・アール(Greenberg, R. R.);ガリティ・ケー・エム(Garrity, K. M.);マッケー・イー・エー(Mackey, E. A.)による1994年の放射分析核科学誌(J. Radioanal. Nucl. Chem.)第179(1)巻の第149頁〜第154頁。コンタクトレンズ材料中での銀およびヨウ化物の含有量を定量化するために使用された当該機器中性子放射化分析法(INAA)では、以下の二つの核反応を使用する:
1.活性化反応において、原子炉で生成された放射性の中性子の捕獲後に、110Agは、安定した109Agから生成され、かつ、128Iは、安定した127Iから生成される。
2.崩壊反応において、110Ag(τ1/2=24.6秒)および128I(τ1/2=25分)は、当該放射性核種に特徴的なエネルギー(Agに関しては657.8キロ電子ボルト、および、Iに関しては443キロ電子ボルト)で初期濃度に比例する陰電子放出によって、崩壊する。
放射性標準品およびサンプルからの110Agおよび128Iの崩壊に特異的なガンマ線の放出は、ガンマ線分光法、定評のある波高分析法によって測定され、これにより、分析物の濃度の測定値を得ることができる。
【0088】
霞みは、室温で、平坦な黒色背景上に置かれた、20ミリメートル×40ミリメートル×10ミリメートルの透明なガラス製セル内のホウ酸緩衝食塩水中に検査用の水和レンズを配置し、下から、当該セルの法線に対して66°の角度で、光ファイバ灯(屈折力(power)4〜5.4に設定された口径0.5インチ(約1.27センチメートル)の光誘導装置を備えたチタン・ツール・サプライ社(Titan Tool Supply Co.)製の光ファイバ灯)からの光を照射し、上から、レンズセルに垂直に、レンズプラットホームより14ミリメートル上に配置されたビデオカメラ(ナビター(Navitar)社製ズームレンズTV Zoom 7000を備えたカメラDVC1300C:19130RGB)で当該レンズ画像を撮影することによって、測定される。背景の散乱は、EPIX XCAP V1.0ソフトウエアを用いて空白セルの画像を減じることによって、当該レンズの散乱から差し引かれる。差し引かれた散乱光の画像は、当該レンズの中央部分の10ミリメートル上を積分した後に、ヘイズ値100に任意に設定されるジオプター:−1.00のCSI Thin Lens(登録商標)を、ヘイズ値ゼロに設定された無レンズの状態と比較することによって定量分析される。五つのレンズが分析されると共に、これらの結果は、標準的なCSIレンズの百分率として、ヘイズ値を得るように平均化される。
【0089】
主観的な霞みの測定は、絞りを全開に設定する「暗視野」モードで、ニコン社製顕微鏡SMZ1500を用いて行われた。評価対象のレンズは、SSPSで満たされたガラス製のペトリ皿内に配置された後に、顕微鏡の検査台上に置かれた。この方法から得られる定量値は、以下のように、上記で測定されたヘイズ値の百分率と概ね一致する:
「濃い霞み」>〜100%
「薄い霞み」<〜70%
「非常に薄い霞み」<〜40%
【0090】
色は、以下のように測定された:サンプルは、室温で、ホウ酸緩衝硫酸ナトリウムの包装用溶液(SSPS)中において平衡化された。過剰な湿気は、レンズ表面から除去された。当該レンズは、顕微鏡用スライド板上に配置され、かつ、スポンジ状の綿棒を用いて、ならして平坦化された。一滴の包装用溶液が当該レンズに滴下され、第二の顕微鏡用スライド板で覆われ、当該レンズ上または下に気泡が存在しないことを確実にした。当該レンズの中央部分は、ソフトウエアQA Master 2000を搭載した比色計X-Rite Model SP64の開口上の白色背景前に位置決めされる。この比色計は、ワンデイ・アキュビュー(1 DAY ACUVUE)コンタクトレンズを用いて調整される。三つの示度が観測され、かつ、その平均値が記録される。上述した検査法を用いて、6回測定され、かつ、その平均化されたワンデイ・アキュビュー(1 DAY ACUVUE)コンタクトレンズに関するL*a*b*値は、以下のとおりである。
L*=72.33±0.04、a*=1.39±0、b*=0.38±0.01
【0091】
反応性混合物の紫外・可視(UV-VIS)スペクトルは、機器UVICAM UV300を用いて測定された。データは、1回の走査および1.5ナノメートルの帯域幅を用いて、200ナノメートル〜800ナノメートルから収集された。各実施例に掲げられた基線溶媒(baseline solvent)を使用した。未処理データは、グラフ作成および分析のために、エクセル(Excel)にエクスポートされた。複数のスペクトルは、比較目的でグラフ化された上記波長に対して標準化された。銀を含有するモノマー類では、銀含有成分の添加の24時間後の紫外・可視(UV-VIS)データが取得された。
【0092】
平衡化したパーキン・エルマー(Perkin Elmer)社製の走査型スペクトロメーター・ラムダ19 UV/VVIS(二重単色光分光器システム(double monochormator system))を用い、200ナノメートル〜800ナノメートルの範囲の全域を1ナノメートル間隔で走査し、以下の設定:幅4ナノメートルのスリット、960ナノメートル/分の走査速度、平面度(smooth)=2ナノメートル、近赤外線(NIR)感度=3、ランプ変更=319.2ナノメートルおよび検出器変更=860.8ナノメートルで、レンズの紫外・可視(UV-VIS)スペクトル(200ナノメートル〜800ナノメートルでの光透過率%)が取得された。レンズは、円形のサンプルホルダー上に平面になるように配置され、かつ、レンズの皺および伸びを最小限にするために挟みつけられる。当該レンズおよびホルダーは、包装用溶液で満たされたキュベット内に配置され、かつ、当該レンズの前面がサンプルビームに面するように配向される。複数のスペクトルは、当該機器に搭載されたソフトウエアを用い、かつ、等式:%Tave=S/Nを用いて算出されるが、ここで、Sは特定領域での%Tの合計であり、かつ、Nは波長の数である。
【0093】
プラスチック製品全体への金属塩の分布は、実施例23に記述されているように、電子プローブ微量分析法(EPM)を用いて測定された。
【0094】
粒子サイズは、レーザー光散乱法または動的光散乱法を用いて測定された。約500ナノメートルを超える粒子サイズ範囲を有するサンプルでは、レーザー回折粒子サイズ分析器Horiba-LA930が使用された。機器の点検は、%Tの空試験値に基づいて実行された。1ミリリットルのサンプル溶液は、媒体としての150ミリリットルの水を収容した循環浴中に導入された。1.7−0.1iの相対屈折率および循環速度5が使用された。サンプルは、超音波処理を用いる機器内での測定前に、2分間、超音波処理された。分析には、トリトン(Triton)(登録商標)X-100(ユニオンカーバイド(Union Carbide)社から市販されている)(0.1%)が界面活性剤として使用された。三通りの分析が実行され、かつ、当該分析結果は、互いに一致することを確認するために比較された。機器は、粒子サイズの平均値と共に粒子サイズ分布を示すグラフを含む報告を与えた。
【0095】
約500ナノメートル未満の粒子サイズ範囲を有するサンプルでは、動的光散乱装置Malvern 4700が使用された。機器の点検は、サンプル分析前に、NISTの追跡可能な標準サイズのポリスチレン粒子(NIST traceable standard size polystyrene particles)を用いて実行された。1ミリリットルのサンプルは、水で20ミリリットルに希釈され、かつ、当該サンプルは、ブランソン(Branson)社製の超音波プローブを用いて、1分間、超音波処理され、かつ、相対屈折率および粘度値の双方は、上記ソフトウエアに入力された。機器は、粒子サイズの平均値と共に粒子サイズ分布を示すグラフを含む報告を与えた。
【0096】
レンズは、以下の方法を用いて、黄色ブドウ球菌(S. aureus)に対する有効性に関して評価された。黄色ブドウ球菌(Staphylococcus aureus)の臨床分離株031(Clinical Isolate 031)の培養液は、トリプシン大豆培地(TSB)中で、一晩、増殖された。培養液は、リン酸緩衝食塩水(PBS、pH=7.4±0.2)中で三回、洗浄され、かつ、細菌ペレットは、10ミリリットルの2%トリプシン大豆培地・リン酸緩衝食塩水(TSB- PBS)中で再懸濁された。細菌接種材料は、最終濃度が約1×108コロニー形成単位/ミリリットル(cfu/mL)になるように調製された。連続希釈法は、約1×104cfu/ミリリットルの接種材料濃度を達成するように2%トリプシン大豆培地・リン酸緩衝食塩水(TSB- PBS)中で実行された。
【0097】
殺菌済みのコンタクトレンズは、残留溶液を除去するために、30ミリリットルのリン酸緩衝食塩水(PBS、pH=7.4±0.2)を三回交換しながら、水洗された。水洗された各コンタクトレンズは、500マイクロリットルの細菌接種材料と共に、殺菌済みの組織培養プレートの別個の検査穴内に配置された後に、当該プレートには、35±2℃で20±2時間、震盪機‐培養器(100回転/分)内で回転が与えられた。各レンズ、および対応する細胞懸濁液は、各検査穴から除去され、0.05重量/総容量(w/v)%のツイーン(Tween)(商標)80を含有する9.5ミリリットルのリン酸緩衝食塩水(PBS)(TPBS)中に配置された。
【0098】
その後、レンズ、および対応する細胞懸濁液は、1600回転/分で3分間、ボルテックスされ、残留する細菌のレンズに対する粘着をなくす遠心力を利用した。得られる上澄液の生菌数は、標準希釈液およびプレートカウント法を用いて算出された。レンズに関連して回収された生菌数の結果は平均化された。
【0099】
本発明を説明するために、以下の実施例が組み込まれている。これらの実施例は本発明を限定するものではない。当該実施例は、本発明を実施する方法を示唆するものにすぎない。コンタクトレンズならびに他の特殊製品に見識のある者は、本発明を実施する他の方法を見出せる。しかし、これらの方法は、本発明の範囲内に入るものであると見なされる。
【0100】
〔実施例〕
以下の略称が、実施例で使用された。
AHM:3-アリルオキシ-2-ヒドロキシプロピルメタクリレート(3-allyloxy-2-hydroxypropyl methacrylate)
AMBN:2,2'-アゾビス(2-メチルブチロニトリル)(2,2'-Azobis(2-Methylbutyronitrile))
BHT:ブチル化ヒドリキシトルエン
Blue HEMA:実施例4または米国特許第5,944,853号明細書に記述されるような反応性青色4号とヒドロキシエチルメタクリレート(HEMA)との反応生成物
CGI 1850:1-ヒドロキシシクロヘキシルフェニルケトン(1-hydroxycyclohexyl phenyl ketone)とビス(2,6-ジメチオキシベンゾイル)-2,4-4-トリメチルペンチルホスフィンオキシド(bis (2,6-dimethyoxybenzoyl)-2,4-4-trimethylpentyl phosphine oxide)との1:1(w/w)の混合物
CGI 819:ビス(2,4,6-トリメチルベンゾイル)-フェニルホスフィンオキシド(bis(2,4,6-trimethylbenzoyl)-phenylphosphineoxide)
DI水:脱イオン水
DMA:N,N-ジメチルアクリルアミド
DAROCUR 1173:2-ヒドロキシ-2-メチル-1-フェニル-プロパン-1-オン(2-hydroxy-2-methyl-1-phenyl-propan-1-one)
EGDMA:エチレングリコールジメタクリレート
HEMA:ヒドロキシエチルメタクリレート
BAGE:グリセリンのホウ酸エステル
IPA:イソプロピルアルコール
MAA:メタクリル酸
Macromer:実施例22で製造されるようなシリコーン含有マクロマー
mPDMS:モノメタクリロキシプロピル末端化ポリジメチルシロキサン(mono-methacryloxypropyl terminated polydimethylsiloxane)(分子量:800〜1000)
Norbloc:2-(2'-ヒドロキシ-5-メタクリロキシエチルフェニル)-2H-ベンゾトリアゾール(2-(2’-hydroxy-5-methacrylyloxyethylphenyl)-2H-benzotriazole)
HO-mPDMS:実施例21で調製されるようなモノ-(3-メタクリロキシ-2-ヒドロキシプロピロキシ)プロピル末端化、モノ-ブチル末端化ポリジメチルシロキサン(mono-(3-methacryloxy-2-hydroxypropyloxy)propyl terminated, mono-butyl terminated polydimethylsiloxane)(分子量:612)
AgI Particles:合成実施例3に従って形成されたヨウ化銀粒子
ppm:乾燥レンズの1グラム当たりのサンプルの百万分の一マイクログラム
PAA:ポリアクリル酸(分子量:2000)
PVP:ポリビニルピロリドン
PVA:ポリビニルアルコール
SiMMA:3-メタクリロキシ-2-ヒドロキシプロピロキシ)プロピルビス(トリメチルシロキシ)メチルシラン3-methacryloxy-2-hydroxypropyloxy)propylbis (trimethylsiloxy)methylsilane)
SSPS:以下に記載するように作られた、ホウ酸緩衝硫酸ナトリウムの包装用溶液
TAA:三級アミルアルコール
TBACB:テトラブチルアンモニウム3-クロロベンゾエート(tetrabutylammonium 3-chlorobenzoate)
THF:テトラヒドロフラン
TRIS:3-メタクリロキシプロピルトリス(トリメチルシロキシ)シラン(3-methacryloxypropyltris(trimethylsiloxy)silane)
w/w:重量/全重量
w/v:重量/全容量
v/v:容量/全容量
【0101】
使用される以下の組成物が調製された。
【0102】
涙様液(Tear-like fluid)(TLF)緩衝液:
涙様液緩衝液(TLF緩衝液)は、0.137グラムの重炭酸ナトリウム(シグマ(Sigma)社製S8875)および0.01グラムのD-グルコース(シグマ(Sigma)社製G5400)を、カルシウムおよびマグネシウムを含有するリン酸緩衝食塩水(PBS)(シグマ(Sigma)社製D8662)中に添加することによって、調製された。この涙様液緩衝液は、各成分が完全に溶解されるまで(約5分間)、室温で撹拌された。
【0103】
脂質保存溶液は、約60℃で、約1時間、十分に撹拌しながら、透明になるまで、以下の脂質を涙様液緩衝液中に混合することによって調製された。
コルステリルリノール酸(シグマ(Sigma)社製C0289) 24ミリグラム/ミリリットル
酢酸リナリル(シグマ(Sigma)社製L2807) 20ミリグラム/ミリリットル
トリオレイン(シグマ(Sigma)社製7140) 16ミリグラム/ミリリットル
オレイン酸プロピルエステル(シグマ(Sigma)社製O9625) 12ミリグラム/ミリリットル
ウンデシレン酸(シグマ(Sigma)社製U8502) 3ミリグラム/ミリリットル
コレステロール(シグマ(Sigma)社製C8667) 1.6ミリグラム/ミリリットル
【0104】
当該脂質保存溶液(0.1ミリリットル)は、0.015グラムのムチン(ウシ顎下腺由来のムチン類:(シグマ(Sigma)社製M3895, 型式1-S))と混合された。3つの1ミリリットル涙様液緩衝液が、脂質ムチン混合物中に添加された。当該溶液は、全成分が溶解状態になるまで(約1時間)、撹拌された。全量が100ミリリットルになるまで、適量の涙様液緩衝液が添加され、かつ、十分に混合された。
【0105】
以下の成分は、前記で調製された当該100ミリリットルの脂質ムチン混合物中に、一成分ずつ、以下に列挙された順序で、添加された。全添加時間は、約1時間であった。
ウシ血漿由来の酸性糖蛋白質(シグマ(Sigma)社製G3643) 0.05ミリグラム/ミリリットル
ウシ胎児血清(シグマ(Sigma)社製F2442) 0.1%
ウシ血漿由来のガンマ・グロブリン(シグマ(Sigma)社製G7516) 0.3ミリグラム/ミリリットル
ベータ・ラクトグロブリン(牛乳リポカリン(bovine milk lipocaline))(シグマ(Sigma)社製L3908) 1.3ミリグラム/ミリリットル
鶏卵白由来のリゾチーム(シグマ(Sigma)社製L7651) 2ミリグラム/ミリリットル
ウシ初乳由来のラクトフェリン(シグマ(Sigma)社製L4765) 2ミリグラム/ミリリットル
【0106】
得られた溶液は、一晩、4℃で放置した。当該溶液のpHは、1規定の塩酸で、7.4に調整した。当該溶液は、使用前に−20℃で、濾過され、かつ、保存された。
【0107】
〔ホウ酸緩衝硫酸ナトリウムの包装用溶液(SSPS)〕
包装用溶液は、脱イオン水中に以下の成分を含有している:
0.18重量%のホウ酸ナトリウム[1330-43-4]、マリンクロート(Mallinckrodt)社製
0.91重量%のホウ酸[10043-35-3]、マリンクロート(Mallinckrodt)社製
1.4重量%の硫酸ナトリウム[7757-82-6]、シグマ(Sigma)社製
0.005重量%のメチルエーテルセルロース[232-674-9]、フィッシャー・サイエンティフィック(Fisher Scientific)社製
【0108】
〔実施例1〕
12.6グラムの5%PVP(K値12)脱イオン水溶液が調製された。3.94グラムの1%硝酸銀溶液が添加され、室温で、磁石式の撹拌棒を用いて5分間、混合された。その後、3.47グラムの1%ヨウ化ナトリウム溶液が添加され、室温で、磁石式の撹拌棒を用いて5分間、混合された。透明なヨウ化銀ナノ分散液が得られた。
【0109】
〔比較例1〕
1.0グラムの1%硝酸銀溶液が、室温で、1.0グラムの1%ヨウ化ナトリウム溶液中に添加された。ヨウ化銀沈殿物を含む非常に濁った分散液が得られた。その後、5グラムの5%PVP(K値12)溶液が、磁石式の撹拌棒を用いて48時間、混合しながら、添加された。沈殿物は解消されなかった。
【0110】
〔実施例2〕
PVP(K値12)に代えて、当初の溶液が、98%加水分解されたポリビニルアルコール(セルボル(Celvol)09-523:米国テキサス州ダラスのセラニーズ・ケミカルズ(Celanese Chemicals)社製)で調製されたことを除き、実施例1が繰り返された。透明なナノ分散液が得られた。
【0111】
〔合成実施例1〕
撹拌装置、温度調節装置、および、冷却および加熱用のジャケットを備えた、5リットルのガラス製反応器には、以下の化合物の混合物が充填された。
【表1】
【0112】
反応器温度が71℃まで上げられ、かつ、2.11グラムのAMBNが添加された。AMBNが溶解され、かつ、当該反応器が遅い窒素流で覆われた。20時間、温度が71℃に保持された。
【0113】
ネジ付き蓋および磁石式の撹拌棒を備えた五つの1リットル瓶が用意され、未処理の生成物が各瓶内に600グラムずつ注入された。溶液は、磁石式の撹拌棒で絶えず撹拌しながら、水浴中で60℃に加熱された。その後、54グラムのヘプタン(9%)が添加され、かつ、当該溶液が60℃に再加熱された。撹拌操作が停止され、かつ、当該瓶が60℃の水浴中に置かれた。温度が、20時間かけて、24℃まで下げられた。ここで、上相は透明な液状になり、かつ、下相は半固形状になった。上相は、最も大きい部分を占めている(瓶の全量の約80%)が、固形状ポリマーの含有率は小さい(凡そ1.5%〜2.5%)。
【0114】
各瓶内の上相は廃棄され、かつ、下相は、固形分12%および水3%を特徴とする2125グラムのポリマー溶液を得るために、水性エタノール中に再溶解された。
【0115】
このポリマー溶液は、以下のパラメータで、不活性ループ(Inert Loop)、出口用フィルタ、および、高性能サイクロンを備えたミニ噴霧乾燥機(Mini Spray Dryer)B-290を用いて、噴霧乾燥された。
【表2】
【0116】
結果は、細かく白色でフワフワした250グラムの粉末をもたらし、この粉末は、約97%まで乾燥されていた。この粉末は、磁石式の撹拌棒を備えた、いくつかの1リットルフラスコ(約77グラムずつ)に移された。これらのフラスコは、当該フラスコ中の材料をさらに乾燥させるために、100℃〜130℃で一晩、30mbar(30ヘクトパスカル)未満の減圧下で、真空処理された。
【0117】
翌朝、真空状態は、乾燥アルゴン雰囲気により破られ、かつ、当該フラスコは、制御された乾燥窒素雰囲気が充填されたボックス内に移された。当該フラスコの総重量は、温度が下がった後に、確認された。各1リットルフラスコには、300グラムのNMP(無水N-メチルピロリドン:プルム(purum);無水物;分子篩済み(over molecular sieves)のフルカ(Fluka)社製)が添加されて、当該粉末を完全に溶解させると共に、当該複数のフラスコでは、その均質性に関する点検が行われた。MAH(メタクリル酸無水物:純度98%)は、50cc(50ミリリットル)の円筒ガラス容器内で秤量されると共に、移す前に、メタクリル酸無水物(MAH)を希釈するために、50グラムの無水N-メチルピロリドン(NMP)が添加された。当該ガラス容器内を水洗して完全に移したことを保証するために、他の50グラムの無水N-メチルピロリドン(NMP)が使用された。トリエチルアミン(フルカ(Fluka)社からのpuriss p.a.)がフィン付ピペットを用いて直接、添加された。蓋が堅く締め付けられ、テープで封止されて、窒素流が止められた。反応は、約40時間、続けられた。
【0118】
上述のように製造されたポリマーは、以下のように精製された。75グラムのポリマーは、400ミリリットルの無水N-メチルピロリドン(NMP)中で溶解された。二つの5リットルガラス製ビーカーには、それぞれ4リットルの脱イオン水、30ミリリットルの発煙HCl(塩酸)および磁石式の撹拌棒が入れられた。当該ビーカーには、それぞれ200ミリリットルずつ、約10ミリリットル/秒の速度で、先の反応で得られた官能性生成物が徐々に注入された。沈殿物が生じ、水相が除去された。膨潤した残留ポリマーは、300ミリリットルのエタノール中に再溶解された。
【0119】
さらに二つの5リットルガラス製ビーカーには、それぞれ4リットルの脱イオン水および磁石式の撹拌棒が入れられた。それぞれ4リットルの脱イオン水が充填された二つの5リットルガラス製ビーカーには、ポリマー/エタノール溶液が注入されて、沈殿物が再度、生じた。水相が除去されると共に、残留塩酸をさらに抽出するために、新しい脱イオン水が添加された。約12時間後に、水相が除去されると共に、膨潤したポリマー材料の重量が確認された(約120グラム)。
【0120】
膨潤したポリマー材料は、固形含有率を13±0.5%にするために、エタノール中に再溶解され、その後、当該溶液は、25ミリメートルGD/X0.45マイクロメートルワットマンフィルタ(25 mm GD/X 0.45μm Whatmann filter)により濾過された。当該溶液は、不活性ループ(Inert Loop)、出口用フィルタ、および、高性能サイクロンを備えたミニ噴霧乾燥機(Mini Spray Dryer)B-290を用いて、噴霧乾燥された。以下のパラメータが適用された。
【表3】
【0121】
結果は、細かく白色でフワフワした約155グラムの粉末をもたらした。
【0122】
〔実施例3〕
合成実施例1で調製されたコポリマー(3.49グラム)は、4.9グラムのマスターバッチ溶液(希釈剤としてのプロピレングリコール99.89%、光重合開始剤としてのジメトキシベンゾイルビス(アシル)ホスフィンオキシド(Dimethoxybenzoyl bis(acyl) phosphine oxide)1.10%、および、阻害剤としての4-メトキシフェノール0.011%を含む)と混合された。実施例1で調製された2グラムのナノ分散液が秤量されてから、コポリマー/マスターバッチ溶液と混合された。得られた混合物は、その内部に閉じ込められた空気を除去するために、2500回転/分で15分間、遠心分離処理された。透明なプレポリマーが得られた。
【0123】
プレポリマーは、窒素下で12時間、脱気された熱可塑性コンタクトレンズ用型(ポリスチレン製の前曲面および後曲面)に分注された。この型内のプレポリマーは、室温で30ミリワット/平方センチメートルの光量で、20℃で空中において30秒間、照射された。その後、レンズは、20℃の脱イオン水中で20分間、水和され、ホウ酸緩衝硫酸ナトリウムの包装用溶液(SSPS)中に収容され、121℃で18分間、殺菌処理された。レンズは、暗視野の顕微鏡下で測定されるように、非常に薄い霞みを呈していた。中性子活性化法を用いて測定された五つのレンズにおける銀の平均含有量は、9.72マイクログラムであり、標準偏差が0.16マイクログラム/レンズであることが分かった。
【0124】
〔実施例4〕
0.339グラムのPVP(K値12)粉末は、3.487グラムの1%ヨウ化ナトリウム溶液中に添加され、10分間、混合されて、塩前駆体溶液Aを形成した。PVP(K値12、0.266グラム)は、4.29グラムの1%硝酸銀溶液にゆっくり添加されて金属剤溶液Bを形成した。塩前駆体溶液A(0.379グラム)は、以下の表1に示された17.603グラムのモノマー混合物中に添加され、かつ、3分間、混合された。その後、金属剤溶液B(0.3963グラム)は、当該モノマー混合物中に添加され、かつ、10分間、撹拌された。
【0125】
当該モノマー混合物は、真空(29水銀柱インチ(約98.21キロパスカル))下で20分間、脱気処理された。モノマー混合物は、熱可塑性コンタクトレンズ用型(ポリスチレン製の前曲面および後曲面)に分注され、かつ、この型内のモノマー混合物は、室温で5ミリワット/平方センチメートルの光量で、窒素下で6分間、照射された。その後、レンズは、20℃の脱イオン水中で水和され、SSPS中に収容され、121℃で約20分間、高圧殺菌器中で殺菌処理された。レンズは、暗視野の顕微鏡下で測定されるように、非常に薄い霞みを呈していた。銀の含有量は、中性子活性化法を用いて測定され、当該銀の含有量は、4.7マイクログラムであり、標準偏差が0.11マイクログラム/レンズであった。
【表4】
【0126】
〔実施例5〕
PVP(K値12、0.946グラム)は、表1に挙げられた30.7グラムの反応性混合物にゆっくり添加され、25分間混合することにより溶解された。0.0177グラムの硝酸銀(固体)は、添加され、かつ、溶解されるまで混合された。その後、0.0300グラムのヨウ化ナトリウム(固体)は、添加され、その混合物は、室温で1時間、混合されて、粒子含有反応性混合物を形成した。粒子含有反応性混合物は、真空(29水銀柱インチ(約98.21キロパスカル))下で10分間、脱気処理された。粒子含有反応性混合物は、実施例4に記述されているように、コンタクトレンズ用型(ポリスチレン製の前曲面および後曲面)に分注され、硬化され、水和され、包装され、殺菌処理された。レンズは、暗視野の顕微鏡下で測定されるように、非常に薄い霞みを呈していた。銀の含有量は、中性子活性化法を用いて測定され、当該銀の含有量は、12.8マイクログラムであり、標準偏差が0.4マイクログラム/レンズであった。
【0127】
〔実施例6〜13〕
以下の各実施例において、二つの混合物が調製された。塩前駆体混合物(「SPM」)は、表1に挙げられた反応性混合物、表2に挙げられた量のPVP(K値12)およびヨウ化ナトリウムを混合することによって調製された。PVPの濃度(重量%)は、粒子含有反応性混合物中のPVPの重量%として挙げられている。金属剤混合物(「MAM」)は、表1に挙げられた反応性混合物、表2に挙げられた量の硝酸銀を混合することによって調製された。各混合物は、全成分が一体化されるまで混合されて透明な混合物が形成された(約5時間〜約19時間)。各実施例において、略同一容量の塩前駆体混合物(SPM)および金属剤混合物(MAM)は、混合されて、表3の第2欄に挙げられた、硝酸銀に対するヨウ化ナトリウムのモル比を有する反応性混合物を形成した。1時間混合した実施例8を除き、各反応性混合物が30分間以上、混合された。反応性混合物は、表3に挙げられた条件下で脱気処理された。脱気処理された各反応性混合物は、実施例4に記述されているように、分注され、硬化され、水和され、包装され、殺菌処理された。レンズの霞みは、暗視野の顕微鏡下で測定された。銀の含有量は、中性子活性化法を用いて測定された。全レンズ中における標的の銀取り込み量(targeted silver uptake)は、約10マイクログラムであった。結果は、以下の表3に記録された。
【表5】
【表6】
【0128】
モル過剰のヨウ化ナトリウムを含有した実施例6および7は、殺菌前において非常に薄い霞みを呈しており、殺菌後において薄い霞みおよび通常の色を呈していた。対照的に、同一条件を用いて調製されたが、過剰の硝酸銀を含有した実施例8、9および10は、それぞれ、黄色、薄茶色および茶色を呈していた。したがって、実施例6および7は、特に、金属剤が金属塩よりも感光性である場合に、金属剤の金属塩への変換を保証する処理条件が改善された色を呈する製品をもたらすことを示している。
【0129】
2.6%のPVPおよび50分間の脱気ステップを有した実施例12は、殺菌前において非常に薄い霞みを呈しているが、殺菌後において濃い霞みを呈しており、したがって、この実施例12は、殺菌中に、粒子の熟成が生じ得ることを示唆している。しかし、反応性混合物が硬化される前に、粒子の熟成ステップが追加された場合(実施例13:70℃で20分間)に、得られたレンズは、殺菌後において非常に薄い霞みを呈していた。
【0130】
〔実施例14〕
以下の表4中の反応性混合物が調製された。反応性成分は、全反応性成分(希釈剤を除く)の重量%として記録され、かつ、当該希釈剤は、最終反応性混合物の重量%である。固体の硝酸銀(0.040グラム)は、28.09グラムのモノマー混合物中に添加された。その後、ヨウ化ナトリウム(固体:0.0427グラム)は、当該混合物中に添加され、かつ、室温で1時間、混合された。混合後に、容器の底部に固形分が未だ残っていた。反応性混合物は、熱可塑性コンタクトレンズ用型(ゼオン(Zeon, Corp.)社から入手されたゼオノール(Zeonor:登録商標)から製造された前曲面および後曲面)に分注され、かつ、この型内の混合物には、室温で5ミリワット/平方センチメートルの光量で、窒素下で10分間、照射された。その後、レンズは、25℃の脱イオン水中で水和され、ホウ酸緩衝硫酸ナトリウムの包装用溶液中に収容され、121℃で20分間、高圧殺菌器中で殺菌処理された。レンズは、暗視野の顕微鏡下で測定されるように、非常に薄い霞みを呈していたが、僅かに淡い黒色を呈していた。銀の含有量は、中性子活性化法を用いて測定されたところでは、6.2マイクログラムであり、標準偏差が0.21マイクログラム/レンズであった。
【表7】
【0131】
〔実施例15〕
PVP K値12(9.29グラム)は、200.00グラムのTPME中に、撹拌しながら、ゆっくりと添加され、かつ、20分間、混合された。その後、0.7040グラムの固体の硝酸銀が当該溶液中に添加されて、金属剤溶液を形成した。この金属剤溶液は、磁石式の撹拌装置を用いて、6時間、撹拌された。
【0132】
ヨウ化ナトリウム(0.8880グラム)は、200.13グラムのTPME中に添加されて、塩前駆体溶液を形成した。この塩前駆体溶液は、磁石式の撹拌装置を用いて、6時間、撹拌された。金属剤溶液(170.89グラム)は、絶えず撹拌しながら、当該塩前駆体溶液(171.21グラム)中に混入された。透明なナノ分散液が得られた。溶液は、25分間、混合された。その後、全ナノ分散液は、以下の表5に挙げられた500.20グラムの反応性混合物中に混合された。
【表8】
【0133】
反応性混合物は、29水銀柱インチ(740mmHg)(約98.21キロパスカル)で15分間、脱気処理された。反応性混合物は、熱可塑性コンタクトレンズ用型(ゼオン(Zeon, Corp.)社から入手したゼオノール(Zeonor:登録商標)から製造された前曲面および後曲面)に分注され、室温で5ミリワット/平方センチメートルの光量で、窒素下で6分間、照射された。その後、レンズは、30分間、20℃の脱イオン水中で、その後、60分間、70%のIPA中で水和された後、1分間、脱イオン水で水洗され、2時間以上、脱イオン水で水洗が行われており、これらの操作はすべて室温で行われた。その後、レンズは、点検され、ホウ酸緩衝硫酸ナトリウムの包装用溶液(SSPS)中に収容され、121℃で18分間、高圧殺菌器中で殺菌処理された。
【0134】
レンズは、10.70マイクログラムの銀の平均取り込み量を有しており、標準偏差(5枚のレンズ)は0.2マイクログラム/レンズであった。レンズの霞みは、68%であり、標準偏差(5枚のレンズ)は8.9%であった。
【0135】
〔実施例16〕
419.5グラムの反応性混合物が、表6に挙げられた成分から調製された。
【0136】
HEMAは、TPMEに添加されて、HEMA/TPME(HEMA:TPME=5.1:10)溶液を形成し、かつ、清浄な琥珀色の瓶内で1時間、混合された。
【0137】
金属剤混合物は、清浄な琥珀色の瓶内で、7グラムのPVP(K値12)を70.0グラムの当該HEMA/TPME溶液中にゆっくりと添加し、かつ、磁石式の撹拌棒で撹拌することによって、形成された。金属剤混合物は、全PVP(K値12)が溶解されるまで、混合された。硝酸銀(0.49グラム)が添加され、かつ、全固形分が溶解されるまで、6時間、混合された。
【0138】
塩前駆体混合物は、清浄な琥珀色の瓶内で、0.42グラムのヨウ化ナトリウムを30グラムの当該HEMA/TPME溶液中に添加し、かつ、全固形分が溶解されるまで、6時間、磁石式の撹拌棒で混合することによって、形成された。
【0139】
金属剤(67.02グラム)混合物は、撹拌しながら、塩前駆体混合物中にゆっくりと注入され、かつ、1時間、混合された。金属塩のヨウ化銀を含有する透明な分散液が得られた。
【0140】
表6に挙げられた成分を有する反応性混合物が調製された。反応性成分(419.5グラム)および金属塩分散液(80.5グラム)は、琥珀色の瓶内で混合され、かつ、約24時間より長く混合された。その後、反応性混合物は、3マイクロメートルのフィルタで濾過され、かつ、29水銀柱インチ(約98.21キロパスカル)下で15分間、脱気処理された。
【表9】
【0141】
反応性混合物は、熱可塑性コンタクトレンズ用型(ゼオン(Zeon, Corp.)社から入手したゼオノール(Zeonor:登録商標)から製造された前曲面および後曲面)に分注され、室温で5ミリワット/平方センチメートルの光量で、窒素下で6分間、照射された。その後、レンズは、30分間、20℃の脱イオン水中で、その後、60分間、70%のIPA中で水和された後、1分間、脱イオン水で水洗され、2時間以上、脱イオン水で水洗が行われており、これらの操作はすべて室温で行われた。その後、レンズは、点検され、ホウ酸緩衝硫酸ナトリウムの包装用溶液中に収容され、121℃で18分間、高圧殺菌器中で殺菌処理された。
【0142】
レンズは、10.60マイクログラムの銀の平均取り込み量を有しており、標準偏差(5枚のレンズ)は0.2マイクログラム/レンズであった。レンズの霞みは、38.6%であり、標準偏差(5枚のレンズ)は4.3%であった。
【0143】
〔実施例17〕
0.0243グラムのPVP(K値12)は、10.0037グラムのTPME中に、ゆっくりと添加され、磁石式の撹拌棒を用いて、20分間、混合された。その後、0.0199グラムの硝酸銀が当該溶液中に添加され、かつ、当該溶液は、室温で4時間、混合されて、溶液Aを得た。0.054グラムの固体のヨウ化ナトリウムは、10.0326グラムのTPME中に添加され、かつ、室温で4時間、混合されて、溶液Bを得た。溶液Aは、溶液B中に注入され、かつ、20分間、混合されて、TPME中のヨウ化銀の透明なナノ分散液を得た。
【0144】
上述のように調製された4.20グラムのヨウ化銀ナノ分散液は、以下の表7に示されるような組成を有する5.13グラムのモノマー混合物中に添加され、かつ、12時間、混合された。その後、モノマーは、22水銀柱インチ(約74.50キロパスカル)の真空下で20分間、脱気処理された。反応性混合物は、熱可塑性コンタクトレンズ用型(ゼオン(Zeon, Corp.)社から入手したゼオノール(Zeonor:登録商標)から製造された前曲面および後曲面)に分注され、室温で5ミリワット/平方センチメートルの光量で、窒素下で6分間、照射された。その後、レンズは、30分間、20℃の脱イオン水中で、その後、60分間、70%のIPA中で水和された後、1分間、脱イオン水で水洗され、2時間以上、脱イオン水で水洗が行われており、これらの操作はすべて室温で行われた。その後、レンズは、点検され、ホウ酸緩衝硫酸ナトリウムの包装用溶液中に収容され、121℃で18分間、高圧殺菌器中で殺菌処理された。
【0145】
レンズは、12マイクログラムの銀の含有率を有しており、標準偏差(5枚のサンプル)は0.1マイクログラムであった。レンズの霞みは、84%であり、標準偏差(5枚のサンプル)は4であった。
【表10】
【0146】
〔比較例2〕
アキュビュー・アドバンス(ACUVUE ADVANCE:登録商標)ブランドのコンタクトレンズとしてビスタコン(Vistakon)社から入手可能な、ガリフィルコン系の硬化水和レンズは、ブリスターパック内の脱イオン水中に置かれた。過剰の脱イオン水が除去されると共に、0.8ミリリットルの塩前駆体混合物(脱イオン水中に1100ppmのヨウ化ナトリウムを含有する)が、当該レンズを収容しているブリスター中に添加され、かつ、室温で一晩、放置された。当該塩前駆体混合物が除去されると共に、0.8ミリリットルの金属剤混合物(脱イオン水中に700ppmの硝酸銀および5%PVP(K値90)を含有する)が添加された。3分後に、金属塩混合物が除去されると共に、脱イオン水(900マイクロリットル)が当該ブリスターに添加され、約5分間、放置され、最終的に除去された。この脱イオン水処理は、さらに二回反復されると共に、当該レンズは、SSPSを収容しているガラス製バイアル瓶に移された。このバイアル瓶は、封止されると共に、122℃で30分間、オートクレーブ処理された。レンズは、機器中性子放射化分析法(INAA)によって分析され、その結果、約16マイクログラムの銀を含有していた。
【0147】
〔実施例18および比較例2〕
実施例6および比較例2のレンズ中の銀の相対含有率は、電子プローブ微量分析法(EPM)を用いて測定され、コンタクトレンズ全体における含有銀の分布状態を確認した。
【0148】
複数のサンプルは、半分に切断され、かつ、試料を固定するための二つの小ネジ用に穴開け加工され雌ネジを切られた(tapped)、直径25ミリメートルのアルミニウム製ホルダー内に垂直にレンズ全体を取り付けることによって、プロファイル解析用に調製された。当該レンズは、材料の半分が当該ホルダー表面より上に出るように、固定された。その後、1枚刃の清浄なカミソリは、切断面を引き裂かないように一回の滑らかな行程で、レンズを半分に切断するために使用された。その後、これらのサンプルには、伝導性を確保するために、真空蒸発装置内で炭素被膜処理が施された。これらのサンプルの遠端には、伝導性をよりよくするためコロイド状炭素塗料で軽く塗装された。
【0149】
残りのレンズ半体の直径近くからの切片は、当該残りのレンズ半体から切り出されると共に、凹面を上にして、上面上の二つの両面炭素製「粘着タブ」を備えた直径25ミリメートルのホルダー上に注意深く置かれた。
【0150】
レンズの凸面は、二つの「粘着タブ」上に当該レンズ材料の凸面側の残りの弦を取り付けることによって、分析された。清浄なテフロン材料シート(厚さ0.032インチ(約0.0813センチメートル))は、コンタクトレンズが平坦になるように、炭素製「粘着タブ」にコンタクトレンズを押圧するために使用された。これらのサンプルには、炭素真空蒸発装置内で、20ナノメートル〜40ナノメートルの純正仕様(Spec-Pure)タイプのグラファイト被膜処理が施された。これらのサンプルの遠端には、伝導性をよりよくするためコロイド状炭素塗料で塗装された。
【0151】
当該複数のサンプルは、カメラSX50(1988年)、または、20キロ電子ボルト、50ナノアンペア、および、焦点ボケをしたビームサイズ:20マイクロメートルを含むレンズ表面を分析するための分析条件を用いる四つの波長分光計を備えた自動電子マイクロプローブSX100(2005年)のいずれかを用いて、分析された。当該ビームサイズは、プロファイル解析のために、5マイクロメートルに低減された。計数時間は、ピーク時で160秒であり、各オフピーク時で80秒であった。
【0152】
バックグラウンド位置は、スペクトル干渉が存在しないように選択された。バックグラウンド強度は、オフピーク位置間の線形補間によって算出された。また、強度は、検出器不感時間、ビームドリフトおよび標準強度ドリフトについて補正された。あらゆる分析では、有意なドリフトが指摘されなかった。銀に関する検出限界は、約40ppmであった。
【0153】
プロファイル解析の取得は、レンズ輪郭表面のうち、凸面(前曲面)を突き止め、かつ、その点から全横断を開始することによって、行われた。表面分析は、レンズ材料の切片の一側面で開始し、かつ、レンズ全体を横断する250マイクロメートルまたは500マイクロメートルの段差を用いることによって、実行された。この段差は、概ね、総距離(サンプル表面当たり25〜50データ点)が約8ミリメートル〜12ミリメートルであった。全データ点は、約4時間待った後でもサンプル表面を完全に平坦化されなかったサンプルについて分光光度計の焦点ボケが生じず、Zフォーカスに関して安定化することが確実になるように、手動で確認された。
【0154】
金属の銀は、銀の一次標準物質として使用された。標準物質および未確認物質には、20ナノメートルの純正仕様(Spec-Pure)タイプのグラファイト被膜処理が施され、かつ、標準物質および未確認物質については、標準物質の計数時間がピーク時で10秒であり、各オフピーク時で5秒間であることを除き、上述した条件下で実行された。
【0155】
図1は、実施例16および比較例1のレンズ中の銀の分布を編集して示すグラフであって、当該銀がレンズ形成後に、当該レンズ中に沈殿している状態を示すグラフである。図1から理解できるように、実施例21のレンズ中の金属塩の濃度(複数の四角形を結ぶ線によって示されている)は、レンズ全体で一貫している。比較例2のレンズでは、図1は、分析されたレンズが、当該レンズの前面および後面の20%以内の高濃度の銀を有しているが、レンズ中心部分では非常に少ない銀を有していることをも示している(複数のダイアモンド形を結ぶ線によって示されている)。
【0156】
〔実施例19〕
実施例16に従って形成されたレンズは、以下の方法を用いて、レンズから放出される銀について評価された。
【0157】
検査されるレンズは、過剰の液体を除去するために殺菌ガーゼを用いて拭き取られた後に、24穴を有し、かつ、各穴に1ミリリットルの涙様液(TLF)を収容する殺菌済みの細胞培養プレートの穴に、1レンズ/穴の割合で移された。当該プレートは、蒸発および脱水を防止するために被覆され、かつ、少なくとも100回転/分で撹拌しながら、35℃で培養された。24時間ごとに、当該レンズは、新鮮な1ミリリットル容量の涙様液(TLF)に移された。測定が実行される各時間間隔で、上記穴の中から、最低3枚のレンズが取り出され、かつ、100ミリリットルのリン酸緩衝食塩水(PBS)で三回〜五回、水洗された。これらのレンズは、過剰の液体を除去するためにペーパータオル上で拭き取られ、かつ、プロピレン・シンチレーションバイアル瓶(1レンズ/瓶)に移された。銀の含有率は、中性子活性化法によって分析された。
【0158】
比較例2のレンズも、上述したように検査された。両レンズの結果は、表3に示されている。図2において、ダイアモンド形状の点を結ぶ実線は実施例16のレンズからの結果であり、かつ、四角形状の点を結ぶ破線は比較例2のレンズの評価からの結果である。図2は、本発明のレンズが、比較例2のレンズよりも遅く、かつ、一貫して抗菌性金属を放出することを明らかに示している(ここで、レンズが形成された後に、銀塩がレンズ中に沈殿している)。
【0159】
〔実施例20〕
実施例16および比較例2の各レンズは、以下の方法を用いて、細菌に対する有効性に関して評価された。黄色ブドウ球菌(Staphylococcus aureus)ATCC#15442(米国メリーランド州ロックビルのアメリカン・タイプ・カルチャー・コレクション)の培養液は、トリプシン大豆培地中で、一晩、増殖された。培養液は、リン酸緩衝食塩水(PBS、pH=7.4±0.2)中で三回、洗浄され、かつ、細菌ペレットは、10ミリリットルの2%トリプシン大豆培地・リン酸緩衝食塩水(TSB- PBS)中で再懸濁された。細菌接種材料は、最終濃度が約1×108コロニー形成単位/ミリリットル(cfu/mL)になるように調製された。連続希釈法は、約1×104cfu/mLの接種材料濃度を達成するように2%トリプシン大豆培地・リン酸緩衝食塩水(TSB- PBS)中で実行された。
【0160】
殺菌済みのコンタクトレンズは、残留溶液を除去するために、30ミリリットルのリン酸緩衝食塩水(PBS、pH=7.4±0.2)を三回交換しながら水洗された。水洗された各コンタクトレンズは、500マイクロリットルの細菌接種材料と共に、殺菌済みの組織培養プレートの別個の検査穴内に配置された後に、当該プレートには、35±2℃で約20時間、震盪機‐培養器(100回転/分)内で回転が与えられた。各レンズは、ガラス製バイアル瓶から取り出され、三回交換したリン酸緩衝食塩水で五回、水洗されて、緩く結合した細胞群を除去した。培養後に、各タイプのレンズのうち、3枚のレンズは、初期の対細菌有効性(以下に記述される)を測定するために、取り出され、かつ、残りのレンズは、上述したように、500マイクロリットルの涙様液(TLF)を収容する、新たなマイクロタイタープレートの穴内に移された。
【0161】
当該残りのレンズは、1ミリリットル/レンズの涙様液(TLF)を収容する、個々の組織培養プレートの穴内で7日間および14日間、当該レンズが24時間ごとに新鮮な涙様液(TLF)溶液に移されながら、培養された。
【0162】
培養期間の末において(培養後、7日間および14日間)、測定対象のレンズは、穴から取り出され、緩く結合した細胞群を除去するために、三回交換したリン酸緩衝食塩水で五回、水洗され、0.05重量/総容量(w/v)%のツイーン(Tween(商標))80を含有する約10ミリリットルのリン酸緩衝食塩水(PBS)中に配置され、2000回転/分で3分間、ボルテックスされ、残留する細菌のレンズに対する粘着をなくす遠心力を利用した。得られる上澄液の生菌数は、流動式血球計算器RBD 3000を用いて算出され、かつ、3枚のレンズに付着した検出可能な生菌数の結果は平均化された。この結果は、図3に示されている。ビスタコン(Vistakon)社から入手可能な、ヒドラクリア(Hydraclear:商標)を備えたアキュビュー(登録商標)・アドバンス(商標)ブランドのコンタクトレンズは、対照として使用された。
【0163】
実施例16および比較例2のレンズに関する結果は、図3に示されている。ダイアモンド形状の点を結ぶ実線は実施例16のレンズからの結果であり、かつ、四角形状の点を結ぶ破線は比較例2のレンズの評価からの結果である。図3は、本発明のレンズが、14日間、一貫して細菌(緑膿菌(Pseudomonas aeruginosa))の対数減少値4を呈することを示している。本発明のレンズと異なり、比較例2のレンズは、最初の7日間で、対数減少値3を呈するものの、その後、評価期間の残存期間で、当該対数減少値が低下し、14日目で、約1の対数減少値を呈した。したがって、本発明のレンズは、比較例2のレンズよりも、細菌に対する有効性が高く、かつ、長期間に及ぶことを示した。
【0164】
〔実施例21〕
45.5キログラムの3-アリルオキシ-2-ヒドロキシプロピルメタクリレート(AHM)およびブチル化ヒドリキシトルエン(BHT)を含む撹拌溶液には、10ミリリットルのプラチナジビニルテトラメチルジシロキサン・キシレン溶液(Pt (0) divinyltetramethyldisiloxane solution in xylenes)(Pt濃度:2.25%)が添加された後に、44.9キログラムのn-ブチルポリジメチルシランが添加された。反応熱は、約20℃の反応温度を維持するために調節された。n-ブチルポリジメチルシランが完全に消費された後に、プラチナ(Pt)触媒は、6.9グラムのジエチルエチレンジアミンの添加により、失活処理された。未処理の反応性混合物は、ラフィネートの3-アリルオキシ-2-ヒドロキシプロピルメタクリレート(AHM)の残留量が0.1%未満になるまで、181キログラムのエチレングリコールで数回にわたって抽出された。10グラムのブチル化ヒドリキシトルエン(BHT)は、得られたラフィネートに添加され、溶解するまで撹拌された後に、残留エチレングリコールが除去され、64.5キログラムのOH-mPDMSを得た。得られた液体には、6.45グラムの4-メトキシフェノール(MeHQ)が添加され、撹拌され、濾過されて、最終的に、64.39キログラムのOH-mPDMSを無色油として得た。
【0165】
〔合成実施例2:マクロマーの調製〕
室温で、窒素下の乾燥ボックス内に収容された乾燥容器内には、30.0グラム(0.277モル)のビス(ジメチルアミノ)メチルシラン、TBACB)の1M溶液(1000ミリリットルの乾燥THF中に386.0グラムのTBACBを含有する)13.75ミリリットルと、61.39グラム(0.578モル)のp‐キシレンと、154.28グラム(1.541モル)のメチルメタクリレート(開始剤に対して1.4当量)と、1892.13グラム(9.352モル)の2-(トリメチルシロキシ)エチルメタクリレート(開始剤に対して8.5当量)と、4399.78グラム(61.01モル)のTHFとの溶液が添加された。熱電対およびコンデンサーを備え、かつ、すべての口が窒素源に接続された三つ口を備えた乾燥した丸底フラスコには、乾燥ボックス内で調製された上記混合物が添加された。
【0166】
反応性混合物は、撹拌して窒素を追い出しながら、15℃まで冷却された。当該溶液が15℃に達した後に、191.75グラム(1.100モル)の1-トリメチルシロキシ-1-メトキシ-2-メチルプロペン(1当量)が反応容器内に投入された。反応では、約62℃以下の発熱が許容され、その後、11ミリリットルの乾燥THF中に154.4グラムのTBACBを含有する0.4M溶液30ミリリットルが残余の反応中に、定量された。反応温度が30℃に達し、かつ、定量操作が開始された後に、467.56グラム(2.311モル)の2-(トリメチルシロキシ)エチルメタクリレート(開始剤に対して2.1当量)と3.812グラム(3.63モル)のn-ブチルモノメタクリロキシプロピル-ポリジメチルシロキサン(開始剤に対して3.3当量)と3673.84グラム(8.689モル)のTRIS(開始剤に対して7.9当量)と20.0グラムのビス(ジメチルアミノ)メチルシランとの溶液が添加された。
【0167】
混合物は、約38℃〜約42℃まで発熱された後に、30℃まで冷却された。その際、10.0グラム(0.076モル)のビス(ジメチルアミノ)メチルシランと154.26グラム(1.541モル)のメチルメタクリレート(開始剤に対して1.4当量)と1892.13グラム(9.352モル)の2-(トリメチルシロキシ)エチルメタクリレート(開始剤に対して8.5当量)との溶液が添加され、かつ、その混合物は、再び、約40℃まで発熱された。反応温度は、約30℃まで下げられ、かつ、2ガロン(7.6リットル)のTHFが添加されて粘性を低下させた。439.69グラムの水と740.6グラムのメタノールと8.8グラム(0.068モル)のジクロロ酢酸との溶液が添加され、その混合物は、HEMA上の保護基によるブロックを解除するために、4.5時間、還流処理された。その後、揮発性成分が除去されると共に、蒸気温度が110℃に達するまで、水分除去に役立つトルエンが添加された。
【0168】
反応用フラスコは、約110℃に維持され、かつ、443グラム(2.201モル)のTMIとビスマスK-KAT 348(5.94グラム)との溶液が添加された。混合物の反応は、イソシアネートのピークが赤外線によって現れるまで行われる。トルエンは、減圧下で蒸発されて、オフホワイトの無水で蝋質の反応性モノマーを得た。マクロマーは、アセトン:マクロマーの重量比が約2:1となるように、アセトン中に置かれた。24時間後に、マクロマーを沈積するために、水が添加されると共に、当該マクロマーは、濾過され、かつ、45℃〜60℃の間の温度で真空オーブンを用いて20時間〜30時間、乾燥された。
【0169】
〔合成実施例3:ヨウ化銀ナノ分散液の形成〕
金属剤溶液および塩前駆体溶液は、以下のように形成された。10,000ppmの硝酸銀は、200グラムの50重量/全重量(w/w)%PVP(K値12)脱イオン水溶液中に撹拌しながら、溶解された。ヨウ化ナトリウム(10,000ppm)は、200グラムの50重量/全重量(w/w)%PVP(K値12)脱イオン水溶液中に撹拌しながら、溶解された。硝酸銀を含有する金属塩溶液は、塩前駆体溶液中に、2013回転/分で撹拌しながら、200グラム/時の速度で添加された。金属塩溶液は、空気中で噴霧乾燥された。導入口温度は185℃であり、出口温度は90℃であり、供給速度は2.7キログラム/時であった。安定化ヨウ化銀ナノ粒子は、5重量%未満の含水率を有していた。
【0170】
安定化ヨウ化銀ナノ粒子粉末(0.32グラム)は、199.7グラムの脱イオン水中に溶解されて、溶液を調製した。ヨウ化銀ナノ粒子粉末は、ヨウ化銀として名目上6600ppmの銀濃度を有していた。最終溶液中における銀濃度は、算出されたところでは、11ppmであった。
【0171】
〔実施例22〕
米国公開特許US2005/0013842 A1号明細書の実施例10の方法は、以下のように実行された。
硝酸銀(0.127グラム)は、75ミリリットルの脱イオン水中に溶解されて、0.01M硝酸銀溶液を調製した。ポリアクリル酸(PAA、2グラム)は、48ミリリットルの脱イオン水中に溶解されて、4重量/全重量(w/w)%のポリアクリル酸溶液を調製した。200ミリリットルの脱イオン水中には、水素化ホウ素ナトリウム(0.008グラム)が添加されて、1mM溶液を調製した。
この1mM水素化ホウ素ナトリウム溶液(197ミリリットル)は、撹拌棒を備えたビーカー内に置かれた。このビーカーは、氷水浴中に沈められた。この設備は、撹拌プレート上に置かれた。0.01M硝酸銀溶液(2ミリリットル)は、4重量/全重量(w/w)%のポリアクリル酸溶液(1ミリリットル)と混合され、かつ、氷水浴中で冷却された。硝酸銀・ポリアクリル酸溶液の混合物は、急速撹拌しながら、冷却された1mM水素化ホウ素ナトリウム溶液中に急速添加された。これらの溶液の混合後に、茶色‐黄色への急激な変色(immediate brown-yellow discoloration)が観察された。溶液は、8時間混合された後に、保存用の琥珀色の透明広口瓶に移された。添加された硝酸銀の量に基づいて、最終溶液の銀濃度は、算出されたところでは、11ppmであった。
【0172】
この実施例22の銀含有溶液の紫外・可視(UV-VIS)スペクトルが測定されており、当該スペクトルは、合成実施例3で調製されたヨウ化銀/PVP水溶液の紫外・可視(UV-VIS)スペクトルと共に、図4に示されている。図4から理解できるように、この実施例22の溶液のスペクトルは、約420ナノメートルを中心とする広いピークを呈していた。対照的に、合成実施例3のヨウ化銀/PVP水性分散液の紫外・可視(UV-VIS)スペクトルの主ピークは、330ナノメートルを中心とするものであった。チャン・ゼット(Zang, Z)らによれば、このピークは、当該水溶液中に、PVPに結合したイオン形態(Ag+)で存在する銀の相互作用に起因することがある。図4における複数のスペクトル差は、本発明の反応性混合物および眼用装置中の銀がイオン形態で存在する可能性があるが、実施例23A、23Bおよび23Eの反応性混合物中に存在する銀が非イオン性銀(Ag0)として存在していることを示している。
【0173】
〔実施例23Aおよび23B(比較例)〕
表9に挙げられた複数のモノマー成分(光重合開始剤Darocur 1173以外の成分)は、表9に挙げられた量で、琥珀色のガラス製バイアル瓶内で混合され、かつ、混合用の瓶回転装置上で回転が与えられた。
【0174】
実施例23Aにおいて、硝酸銀溶液(フィッシャー(Fisher)社製の無水エタノール54ミリリットル中にフィッシャー(Fisher)社製の米国化学会(A.C.S.)品質の0.025グラムの硝酸銀が溶解された)は、硝酸銀の供給源として使用された。実施例23Bにおいて、硝酸銀溶液(フィッシャー(Fisher)社製の無水エタノール54ミリリットル中にフィッシャー(Fisher)社製の米国化学会(A.C.S.)品質の0.305グラムの硝酸銀が溶解された)は、硝酸銀の供給源として使用された。
【表11】
【0175】
実施例23Aおよび23Bで調製された5ミリリットルの各反応性混合物は、24時間、そのままにしておかれた。反応性混合物の色は、L*a*b*表色系および上述した方法を用いて、定量的に測定された。また、反応性混合物の色は、白色蛍光灯下で、主観的に評価された。これらの結果は、以下の表10に開示されている。
【0176】
実施例23Aおよび23Bの反応性混合物に関する紫外・可視(UV-VIS)スペクトルが測定されており、図5に示されている。
【0177】
光重合開始剤(Darocur 1173)が添加され、かつ、各処方は、660水銀柱ミリメートル(87.99キロパスカル)の真空下で5分間〜7分間、脱気処理された。その後、処方は、窒素グローブボックスに移された。コンタクトレンズは、少なくとも24時間、窒素グローブボックス中で脱酸素化されたゼオノール(Zeonor:登録商標)製の前曲面型およびポリプロピレン製の後曲面型を用いて調製された。レンズ収容空洞当たり100マイクロリットルの投与量が使用され、かつ、レンズ型を保持するフレームが水晶板の下に配置された。レンズは、紫外線照射灯(平行な四灯一組のフィリップス(Philips)社製TL09/20)下、室温で60分間、硬化処理された。
【0178】
硬化後に、レンズ型が手動で開けられると共に、70:30のIPA:脱イオン水の混合物を含む瓶内に、レンズが、1枚のレンズ当たり〜5ミリリットルの溶液を用いて放出された。少なくとも60分後に、レンズ型がピンセットで取り出され、溶液が静かに注入され、瓶には、新鮮なIPA:脱イオン水(70:30)混合物が充填された。レンズには、瓶回転装置上で回転が与えられ、少なくとも60分後に、溶液が静かに注入され、瓶には、新鮮な脱イオン水が充填された。レンズには、さらに、少なくとも60分間、瓶回転装置上で回転が与えられ、溶液が静かに注入され、瓶には、新鮮な脱イオン水が充填された。レンズは、ガラス製バイアル瓶内の5ミリリットルのリン酸緩衝包装用溶液中に収容され、当該バイアル瓶がシリコーン製封止材料およびアルミニウム固定キャップで封止され、122℃で30分間、オートクレーブ処理された。レンズ中の銀含有率は、機器中性子放射化分析法(INAA)を用いることによって測定され、表9に記録されている。
【0179】
〔実施例23Cおよび23D〕
硝酸銀/エタノール溶液に代えて、合成実施例3で調製された安定化AgI/PVP粉末が添加されたことを除き、実施例23Aおよび23Bが繰り返された。溶液の色は、実施例23Aおよび23Bに記述されているように、測定され、表10に記録されている。実施例23Cおよび23Dの反応性混合物に関する紫外・可視(UV-VIS)スペクトルが測定されており、そのスペクトルは図5に示されている。レンズは、実施例23Aおよび23Bに記述されているように形成され、かつ、ガラス製バイアル瓶内に入れられた、50ppmのメチルセルロースを含有する5ミリリットルのホウ酸緩衝硫酸ナトリウムの包装用溶液(SSPS)中に収容され、当該バイアル瓶がシリコーン製封止材料およびアルミニウム固定キャップで封止され、122℃で30分間、オートクレーブ処理された。レンズ中の銀含有率は、機器中性子放射化分析法(INAA)を用いることによって測定され、表9に記録されている。
【0180】
〔実施例23E〕
銀供給源が、硝酸銀/エタノール溶液に代えて、11.25グラムのDMA中に溶解された0.026グラムの硝酸銀および0.011グラムのポリアクリル酸(PAA)であることを除き、実施例23Aが繰り返された。溶液の色は、実施例23Aおよび23Bに記述されているように、測定されており、表10に記録されている。実施例23Eの反応性混合物に関する紫外・可視(UV-VIS)スペクトルが測定されており、そのスペクトルは図5に示されている。レンズは、実施例23Aおよび23Bに記述されているように形成された。レンズ中の銀含有率は、機器中性子放射化分析法(INAA)を用いることによって測定され、表9に記録されている。
【0181】
〔実施例23F〕
銀が全く添加されなかったことを除き、実施例23Aが繰り返された。
【表12】
【0182】
図5は、実施例23A〜23Fの反応性混合物に関する紫外・可視(UV-VIS)スペクトルの比較を示している。実施例23F(銀を含有しない対照の処方)は、グラフ化された領域内で、いかなるピークをも示さなかった。低濃度の銀を含有する反応性混合物(実施例23A)も、いかなる明確なピークをも示さなかった。しかし、実施例23Bは、435ナノメートルに明確なピークを示しており、この場合に、米国公開特許US2005/0013842号明細書によれば、非イオン性銀(Ag0)の存在が確認されている。
【0183】
実施例23Cに関するスペクトルでは、紫外・可視(UV-VIS)スペクトル中の417ナノメートルに明確な遷移が観察された。当該遷移は、実施例23Dの反応性混合物(約389ppmの標的銀濃度を有する)のスペクトルに存在するように現れたが、信号は雑音が多く、当該スペクトル領域内において飽和状態に近くなっていた。チャン・ゼット(Zang, Z)、チャオ・ビー(Zhao, B)およびフー・エル(Hu, L)は、1996年1月に発行された固体化学誌第121巻第1頁、第5頁、第105頁〜第110頁の「化学還元法により合成された銀超微粉末のPVP防御機構」(Journal of Solid State Chemistry January 1996, 121, Issue 1, 5, 105-110. PVP Protective Mechanism of Ultrafine Silver Powder Synthesized by Chemical Reduction Processes)において、彼らがコロイド状ヨウ化銀の紫外・可視(UV-VIS)スペクトルを分析したときに、サンプル23Cに非常に類似し、420ナノメートルに吸収段部を有するスペクトルプロファイルを得た。さらに、彼らは、水素化ホウ素ナトリウムを用いたコロイド状ヨウ化銀の非イオン性銀(Ag0)への還元時に、ピークの位置および形状が、サンプル23Bの紫外・可視(UV-VIS)スペクトルにおいて観察されるものと非常に類似していたことを発見した。科学文献中のデータに基づいて、実施例23Bの硝酸銀系のモノマー類と比較して、実施例23Cで観察されたピークの異なる形状および位置は、異なる酸化状態を有する銀粒子の存在を示すものと見なされる。
【0184】
〔実施例24A〜24F〕
実施例23A〜23Fで形成されたレンズは、上述した検査方法に記述された手順を用いて、黄色ブドウ球菌(Staphylococcus aureus)031に対する有効性に関して検査された。その結果は、以下の表11に記録されている。
【表13】
【0185】
実施例23Aおよび23Bは、それぞれ実施例23Cおよび23Dに類似する、レンズ中の銀濃度を呈している。しかし、抗菌活性データは、硝酸銀を含有するモノマー類(実施例23A、23Bおよび23E)から形成されたレンズが、実施例23Fで形成された対照レンズと比較されたときに、抗菌活性を呈しなかったことを示している。これに対して、実施例23Cおよび23Dに従って形成され、かつ、金属塩ナノ粒子を含有するレンズは、当該対照レンズと比較して、少なくとも1の対数減少値を呈していた。
【0186】
〔実施例25A〕
可視光重合開始剤CGI 819が使用され、かつ、レンズが、可視光照射灯(平行な四灯一組のフィリップス(Philips)社製TL03/20)下、室温で30分間、硬化処理されたことを除き、実施例23Dが繰り返された。硬化されたレンズは、実施例23Dに開示されているように、放出され、抽出され、水和処理され、包装され、かつ、オートクレーブ処理された。銀濃度、ヨウ化物濃度および明度は測定され、以下の表12に示されている。また、実施例23D(紫外線硬化法で形成されている、同一の処方)のレンズの銀濃度、ヨウ化物濃度および明度も測定され、以下の表12および表13に示されている。
【0187】
〔実施例25B〕
硬化前の処方に2重量%のNorblocが添加され、かつ、エタノール濃度が2%だけ下げられたことを除き、実施例25Aが繰り返された。銀濃度、ヨウ化物濃度および明度は、水和処理および殺菌処理後に測定され、以下の表12および表13に示されている。
【表14】
【0188】
実施例23Dに従って、紫外線硬化法を用いて形成されたレンズ中の銀:ヨウ化物のモル比(水和処理および殺菌処理後に測定された)は、観察されたところでは、約2であった。このデータは、レンズ中の銀含有量の約半分が、硬化処理中に、ヨウ化銀から異なる酸化状態の銀に変換されたことを示唆している。実施例23Dにおいては、紫外光がヨウ化銀(AgI)を非イオン性銀(Ag0)およびヨウ素分子(I2)に変換したものと考えられる。ヨウ素分子(I2)がIPA中に溶解可能であるので、当該ヨウ素分子(I2)は水和処理中に除去された。反応性混合物に添加されたヨウ化銀に基づいて予定される、レンズ中の銀:ヨウ化物のモル比は、約1であった。
【0189】
実施例25Aおよび25Bで形成されたレンズ中の銀:ヨウ化物のモル比は、可視光硬化法を用いると、約1であった。したがって、紫外光の範囲外の硬化条件の使用は、ヨウ化銀等の抗菌性金属塩を塩の形態で維持する上で、重要である。
【表15】
【0190】
表13中の比色分析データに基づくと、可視光硬化法を用いて調製された同程度の銀濃度を有する複数のレンズ(実施例25Aおよび25B)は、実施例23Dで、紫外線で硬化されて形成された複数のレンズよりもかなり薄い黄色(低b*値)を呈した。
【0191】
〔実施例26〜28〕
100,000ppmのPVP(K値12)溶液が脱イオン水中で調製された。この溶液(溶液A)は、ヨウ化ナトリウム(NaI)溶液および硝酸銀(AgNO3)溶液を調製するためのベースを与えた。約1500ppm、約5000ppm、および、約10000ppmのヨウ化ナトリウムおよび硝酸銀のそれぞれを含有する溶液が調製された。各溶液は、粒子が可視的に観察されなくなるまで、撹拌された。20ミリリットルのヨウ化ナトリウム溶液は清浄な瓶内に置かれ、かつ、磁石式の撹拌装置は当該瓶内に置かれた。撹拌装置の撹拌速度は300回転/分に設定され、かつ、20ミリリットルの硝酸銀が当該ヨウ化ナトリウム溶液中に、以下の表14に示された速度で添加された。すべての混合処理は、室温で実行された。溶液の霞みは、表14中に挙げられた添加時間の最後に、主観的に評価され、その結果は、以下の表14に記録されている。実施例は、表14に示された各濃度および添加速度ごとに繰り返された。
【表16】
【0192】
〔実施例29〜31〕
ヨウ化ナトリウム溶液が硝酸銀溶液に添加されたことを除き、実施例26〜28が繰り返された。これらの結果は、以下の表15に示されている。
【表17】
【0193】
〔実施例32〕
金属剤溶液および塩前駆体溶液が、室温で、〜5日間、瓶回転装置上で混合された後に、20ミリリットルの各溶液がバッチ式で混合された(約1秒で一緒に注入された)ことを除き、実施例31が繰り返された。結果は、PVP‐AgI錯体を含む透明な溶液であった。
【0194】
〔実施例33〜39〕
約10ミリリットルの700ppmの硝酸銀溶液は、表16に示されたPVP濃度(脱イオン水中でのPVP(K値12)の濃度:1%〜35%)で、PVP(K値12):脱イオン水溶液中で形成された。各硝酸銀溶液は、手動で震盪しながら、10ミリリットルの1100ppmのヨウ化ナトリウム/脱イオン水溶液(PVPを含まない)中に滴下されて、分散液を形成した。実施例33では乳白色であったが、残りの実施例では硝酸銀の添加中、透明のままであった。粒子サイズの測定は、得られたヨウ化銀分散液に対して、レーザー光散乱法(実施例33)および光子相関分光分析法(実施例35〜39)を用いて実行された。データは、粒子サイズ分布のz平均として記録されている。
【表18】
【0195】
表16のデータは、図5にグラフで示されている。表16のデータは、金属塩の形成中のPVPの存在が粒子サイズを実質的に(少なくとも2桁)低減させることを明確に示している。
【0196】
〔実施例40〜44〕
PVPに代えて、表17に挙げられた分散剤が、表17に挙げられた濃度で使用されたことを除き、実施例34が繰り返された。粒子サイズの測定は、得られたヨウ化銀分散液に対して、レーザー光散乱法(実施例40、41および43)および光子相関分光分析法(実施例42、44)を用いて実行された。データは、粒子サイズ分布のz平均として記録されている。
【表19】
【0197】
〔実施例45〕
以下の表18に示された各成分は、表18に挙げられた量で、琥珀色のガラス製バイアル瓶内に一緒に混合され、かつ、瓶回転装置上で回転が与えられた。混合物は、コンタクトレンズ用型(ゼオノール(Zeonor:登録商標)前曲面用型および後曲面用型)内へ分注され、かつ、以下の条件:酸素2.8%±0.5%、可視光硬化(フィリップス(Philips)社製TL03ランプ)、強度プロファイル:25℃での光量1±0.5ミリワット/平方センチメートル(10秒〜60秒)、80℃±5℃での光量5.5±0.5ミリワット/平方センチメートル(304秒〜600秒)で硬化処理された。レンズは、IPA/水混合物中で水和され、50ppmのメチルセルロースを含有する950マイクロリットルのホウ酸緩衝硫酸ナトリウムの包装用溶液(SSPS)中で個別のポリプロピレン製ブリスターパック内に収容され、かつ、124℃で18分間、オートクレーブ処理された。
【表20】
【0198】
この実施例45で形成された12枚のレンズは、上述した検査方法に記述された手順を用いて、黄色ブドウ球菌(Staphylococcus aureus)031に対する有効性に関して検査された。対照レンズは、実施例45の方法で形成されたが、ヨウ化銀ナノ粒子を含有していなかった。銀を含有するレンズの対数減少値(対照レンズと対比して)は、確認されたところでは、3.3±0.2(平均値±標準偏差)であった。
【0199】
〔実施例46〕
実施例45のレンズは、実施例45の対照レンズと共に、二重盲検対側性臨床試験(double masked, contralateral clinical trial)で、30人の患者(全員が現在、コンタクトレンズ装用者)によって装用された。これらの患者は、毎日装用方式で、14日間、レンズを装用し、オプティフリー・リプレニッシュ(OptiFree RepleniSH)を使用し、レンズの洗浄および消毒中に、当該レンズを擦るように指示された。実施例45のレンズは、ベースラインで約10マイクログラムの銀を含有していた。
【0200】
当該試験を終了した26人の患者からの装用済みのレンズは、14日間の装用期間の最後に収集され、機器中性子放射化分析法(INAA)によって銀含有率について検査された。機器中性子放射化分析法(INAA)のデータから、銀の放出の平均速度が算出されたところでは、0.5マイクログラム/日であった。また、レンズは、上述した検査方法の項に記述された方法を用いて、黄色ブドウ球菌(S. aureus)に対する活性に関して検査された。実施例45のレンズの対数減少値(装用済みの対照と対比して)は、確認されたところでは、3.4±1.2(平均値±標準偏差)であった。
【図面の簡単な説明】
【0201】
【図1】実施例16および比較例2の各レンズ中の銀濃度を、当該レンズ縁部からの距離の関数として示すグラフである。
【図2】銀の放出状態の比較を、実施例16および比較例2で形成されたコンタクトレンズに関する時間関数として示すグラフである。
【図3】緑膿菌(Pseudomonas aeruginosa)に対する有効性の比較を、実施例16および比較例2で形成されたコンタクトレンズに関する時間関数として示すグラフである。
【図4】実施例22および合成実施例3の各混合物に関する紫外・可視(UV-VIS)スペクトルを示す。
【図5】実施例23Aおよび23Bの反応性混合物に関する紫外・可視(UV-VIS)スペクトルを示す。
【特許請求の範囲】
【請求項1】
少なくとも一つのポリマーから形成された製品において、
前記ポリマーは、前記ポリマー全体に均質に分散され、かつ、約200ナノメートル未満の粒子サイズを有する抗菌性の金属塩粒子を含み、
前記製品は、緑膿菌および黄色ブドウ球菌のうち、少なくとも一方において少なくとも約0.5の対数減少値を呈し、かつ、約70マイクロメートルの厚さで、CSIレンズと比較して約100%未満のヘイズ値を呈する、製品。
【請求項2】
請求項1に記載の製品において、
前記製品は、医療装置である、製品。
【請求項3】
請求項1に記載の製品において、
前記製品は、眼用装置である、製品。
【請求項4】
請求項3に記載の眼用装置において、
前記眼用装置は、予定された装用期間後に、少なくとも約0.5マイクログラムの抗菌性金属を含む、装置。
【請求項5】
請求項1に記載の製品において、
前記抗菌性の金属塩粒子は、式:[Mq+]a[Xz-]bのものであり、ここで、Xは、あらゆる負帯電イオンであり、Mは、あらゆる正帯電金属であり、a、b、qおよびzは、それぞれ個別に1以上の整数であり、q(a)=z(b)である、製品。
【請求項6】
請求項5に記載の製品において、
Mは、Al+3、Co+2、Co+3、Ca+2、Mg+2、Ni+2、Ti+2、Ti+3、Ti+4、V+2、V+3、V+5、Sr+2、Fe+2、Fe+3、Au+2、Au+3、Au+1、Ag+2、Ag+1、Pd+2、Pd+4、Pt+2、Pt+4、Cu+1、Cu+2、Mn+2、Mn+3、Mn+4、Se+4、および、Zn+2からなる群より選択されている、製品。
【請求項7】
請求項1に記載の製品において、
Mは、Mg+2、Zn+2、Cu+1、Cu+2、Au+2、Au+3、Au+1、Pd+2、Pd+4、Pt+2、Pt+4、Ag+2、および、Ag+1からなる群より選択されている、製品。
【請求項8】
請求項5に記載の製品において、
Mは、Ag+1を含む、製品。
【請求項9】
請求項5に記載の製品において、
Xは、CO3-2、SO4-2、PO4-3、Cl-1、I-1、Br-1、S-1、O-2、および、酢酸塩を含むイオン類からなる群より選択されている、製品。
【請求項10】
請求項5に記載の製品において、
Xは、CO3-2、SO4-2、Cl-1、I-1、Br-1、および、酢酸塩からなる群より選択されている、製品。
【請求項11】
請求項1に記載の製品において、
前記金属塩粒子は、炭酸銀、リン酸銀、硫化銀、塩化銀、臭化銀、ヨウ化銀、および、酸化銀の各粒子からなる群より選択されている、製品。
【請求項12】
請求項1に記載の製品において、
前記金属塩粒子は、硫化マンガン、酸化亜鉛、炭酸亜鉛、硫酸カルシウム、硫化セレン、ヨウ化銅、硫化銅、および、リン酸銅からなる群より選択された少なくとも一つの塩を含む、製品。
【請求項13】
請求項5に記載の製品において、
前記金属塩は、約25℃の純水中で測定された、2×10−10以下の溶解度積定数を有している、製品。
【請求項14】
請求項1に記載の製品において、
前記金属塩粒子は、約100ナノメートル未満の粒子サイズを有している、製品。
【請求項15】
請求項1に記載の製品において、
前記ポリマーは、前記製品の乾燥重量に基づいて、約0.1ppm〜約10重量%の金属を含む、製品。
【請求項16】
請求項1に記載の製品において、
前記ポリマーは、前記製品の乾燥重量に基づいて、約1ppm〜約1重量%の金属を含む、製品。
【請求項17】
請求項1に記載の製品において、
前記ポリマーは、ヒドロゲルである、製品。
【請求項18】
請求項1に記載の製品において、
前記製品は、約4未満のb*値、少なくとも約89のL*値、あるいは、両方の値をさらに含む、製品。
【請求項19】
請求項1に記載の製品において、
前記ポリマーは、少なくとも一つの紫外線吸収化合物をさらに含む、製品。
【請求項20】
請求項1に記載の製品において、
前記少なくとも一つの紫外線吸収化合物は、紫外光の少なくとも約90%が前記装置を通過することを妨げる上で十分な量で存在している、製品。
【請求項21】
方法において、
(a)少なくとも一つの塩前駆体を、任意に反応性ポリマー混合物の少なくとも一つの成分と共に、溶媒中に溶解させて、塩前駆体の混合物を形成するステップと、
(b)少なくとも一つの金属剤および少なくとも一つの分散剤を、任意に少なくとも一つの反応性成分と共に、溶媒中に溶解させることによって、分散剤・金属剤の錯体を形成して、金属剤混合物を形成するステップであって、前記溶媒および成分は同一または異なるものであってもよい、ステップと、
(c)少なくとも一つの抗菌性金属塩:[Mq+]a[Xz-]bを含む粒子含有混合物を形成するための粒子形成条件下で、前記塩前駆体の混合物および前記金属剤混合物を混合するステップと、
(d)ステップ(a)および(b)に反応性成分が含まれていない場合において、このステップ(d)で少なくとも一つの反応性成分が添加されることを条件として、任意に、追加の反応性成分を前記粒子含有混合物に混合して、粒子含有反応性混合物を形成するステップと、
(e)ステップ(c)で抗菌性ポリマー製品中にMq+として添加された前記金属剤由来のMの少なくとも約90%を維持する上で十分な反応条件下で、前記ポリマー製品を形成するために、前記粒子含有反応性混合物を反応させるステップと、
を含む、方法。
【請求項22】
請求項21に記載の方法において、
ステップ(a)または(b)で任意に使用された前記少なくとも一つの反応性成分は、前記金属剤と反応しないものである、方法。
【請求項23】
請求項21に記載の方法において、
前記金属剤との反応性を有する反応性成分は、混合ステップ(d)で前記粒子含有反応性混合物に添加される、方法。
【請求項24】
請求項21に記載の方法において、
前記分散剤は、ヒドロキシアルキルメチルセルロースポリマー類、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレンオキシド、澱粉、ペクチン、ポリアクリルアミド、ゼラチン、ポリアクリル酸、有機アルコキシシラン類である3−アミノプロピルトリエトキシシラン、メチルトリエトキシシラン、フェニルトリメトキシシラン、ビニルトリエトキシシラン、および3−グリシドキシプロピルトリメトキシシラン、グリセリンのホウ酸エステル、および、これらの混合物からなる群より選択される、方法。
【請求項25】
請求項21に記載の方法において、
前記分散剤は、ヒドロキシアルキルメチルセルロースポリマー類、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレンオキシド、ゼラチンおよびポリアクリル酸、グリセリンのホウ酸エステル、および、これらの混合物からなる群より選択される、方法。
【請求項26】
請求項21に記載の方法において、
前記分散剤は、ヒドロキシプロピルメチルセルロース、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレンオキシド、ゼラチンおよびポリアクリル酸、および、これらの混合物からなる群より選択される、方法。
【請求項27】
請求項21に記載の方法において、
前記分散剤は、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレンオキシド、および、ポリアクリル酸、および、これらの混合物からなる群より選択される、方法。
【請求項28】
請求項27に記載の方法において、
前記分散剤は、約2,000,000未満の分子量を有する、方法。
【請求項29】
請求項27に記載の方法において、
前記分散剤は、約20,000〜約1,500,000の間の分子量を有する、方法。
【請求項30】
請求項21に記載の方法において、
前記金属剤混合物は、約40重量%以下の前記分散剤を含む、方法。
【請求項31】
請求項21に記載の方法において、
前記金属剤混合物は、0.01重量%〜約30重量%の間の前記分散剤を含む、方法。
【請求項32】
請求項21に記載の方法において、
前記塩前駆体混合物は、約10重量%以下の前記塩前駆体を含む、方法。
【請求項33】
請求項21に記載の方法において、
前記金属塩前駆体混合物は、前記金属剤に対してモル過剰で、前記塩前駆体を含む、方法。
【請求項34】
請求項21に記載の方法において、
前記金属剤混合物は、約10重量%以下の前記金属剤を含む、方法。
【請求項35】
請求項21に記載の方法において、
前記溶媒は、ステップ(d)前に、粒子含有混合物から除去される、方法。
【請求項36】
方法において、
約200ナノメートル以下の粒子サイズを有する安定化抗菌性金属塩粒子および少なくとも一つのフリーラジカル反応性成分を含む反応性混合物を、前記金属塩粒子に応じて調整された臨界波長を超える波長の光、熱、あるいは、これらの組み合わせを用いて、硬化させて、抗菌性金属塩粒子を含む製品を形成すること、
を含む、方法。
【請求項37】
請求項36に記載の方法において、
前記安定化抗菌性金属塩粒子は、少なくとも一つの銀金属塩を含み、
前記調整された臨界波長は、約430ナノメートルである、方法。
【請求項38】
請求項36に記載の方法において、
前記抗菌性金属は、式:[Mq+]a[Xz-]bを有し、前記ポリマー中のMの少なくとも約90%は、Mq+である、方法。
【請求項39】
請求項36に記載の方法において、
前記反応性混合物は、少なくとも一つの紫外線吸収化合物をさらに含む、方法。
【請求項1】
少なくとも一つのポリマーから形成された製品において、
前記ポリマーは、前記ポリマー全体に均質に分散され、かつ、約200ナノメートル未満の粒子サイズを有する抗菌性の金属塩粒子を含み、
前記製品は、緑膿菌および黄色ブドウ球菌のうち、少なくとも一方において少なくとも約0.5の対数減少値を呈し、かつ、約70マイクロメートルの厚さで、CSIレンズと比較して約100%未満のヘイズ値を呈する、製品。
【請求項2】
請求項1に記載の製品において、
前記製品は、医療装置である、製品。
【請求項3】
請求項1に記載の製品において、
前記製品は、眼用装置である、製品。
【請求項4】
請求項3に記載の眼用装置において、
前記眼用装置は、予定された装用期間後に、少なくとも約0.5マイクログラムの抗菌性金属を含む、装置。
【請求項5】
請求項1に記載の製品において、
前記抗菌性の金属塩粒子は、式:[Mq+]a[Xz-]bのものであり、ここで、Xは、あらゆる負帯電イオンであり、Mは、あらゆる正帯電金属であり、a、b、qおよびzは、それぞれ個別に1以上の整数であり、q(a)=z(b)である、製品。
【請求項6】
請求項5に記載の製品において、
Mは、Al+3、Co+2、Co+3、Ca+2、Mg+2、Ni+2、Ti+2、Ti+3、Ti+4、V+2、V+3、V+5、Sr+2、Fe+2、Fe+3、Au+2、Au+3、Au+1、Ag+2、Ag+1、Pd+2、Pd+4、Pt+2、Pt+4、Cu+1、Cu+2、Mn+2、Mn+3、Mn+4、Se+4、および、Zn+2からなる群より選択されている、製品。
【請求項7】
請求項1に記載の製品において、
Mは、Mg+2、Zn+2、Cu+1、Cu+2、Au+2、Au+3、Au+1、Pd+2、Pd+4、Pt+2、Pt+4、Ag+2、および、Ag+1からなる群より選択されている、製品。
【請求項8】
請求項5に記載の製品において、
Mは、Ag+1を含む、製品。
【請求項9】
請求項5に記載の製品において、
Xは、CO3-2、SO4-2、PO4-3、Cl-1、I-1、Br-1、S-1、O-2、および、酢酸塩を含むイオン類からなる群より選択されている、製品。
【請求項10】
請求項5に記載の製品において、
Xは、CO3-2、SO4-2、Cl-1、I-1、Br-1、および、酢酸塩からなる群より選択されている、製品。
【請求項11】
請求項1に記載の製品において、
前記金属塩粒子は、炭酸銀、リン酸銀、硫化銀、塩化銀、臭化銀、ヨウ化銀、および、酸化銀の各粒子からなる群より選択されている、製品。
【請求項12】
請求項1に記載の製品において、
前記金属塩粒子は、硫化マンガン、酸化亜鉛、炭酸亜鉛、硫酸カルシウム、硫化セレン、ヨウ化銅、硫化銅、および、リン酸銅からなる群より選択された少なくとも一つの塩を含む、製品。
【請求項13】
請求項5に記載の製品において、
前記金属塩は、約25℃の純水中で測定された、2×10−10以下の溶解度積定数を有している、製品。
【請求項14】
請求項1に記載の製品において、
前記金属塩粒子は、約100ナノメートル未満の粒子サイズを有している、製品。
【請求項15】
請求項1に記載の製品において、
前記ポリマーは、前記製品の乾燥重量に基づいて、約0.1ppm〜約10重量%の金属を含む、製品。
【請求項16】
請求項1に記載の製品において、
前記ポリマーは、前記製品の乾燥重量に基づいて、約1ppm〜約1重量%の金属を含む、製品。
【請求項17】
請求項1に記載の製品において、
前記ポリマーは、ヒドロゲルである、製品。
【請求項18】
請求項1に記載の製品において、
前記製品は、約4未満のb*値、少なくとも約89のL*値、あるいは、両方の値をさらに含む、製品。
【請求項19】
請求項1に記載の製品において、
前記ポリマーは、少なくとも一つの紫外線吸収化合物をさらに含む、製品。
【請求項20】
請求項1に記載の製品において、
前記少なくとも一つの紫外線吸収化合物は、紫外光の少なくとも約90%が前記装置を通過することを妨げる上で十分な量で存在している、製品。
【請求項21】
方法において、
(a)少なくとも一つの塩前駆体を、任意に反応性ポリマー混合物の少なくとも一つの成分と共に、溶媒中に溶解させて、塩前駆体の混合物を形成するステップと、
(b)少なくとも一つの金属剤および少なくとも一つの分散剤を、任意に少なくとも一つの反応性成分と共に、溶媒中に溶解させることによって、分散剤・金属剤の錯体を形成して、金属剤混合物を形成するステップであって、前記溶媒および成分は同一または異なるものであってもよい、ステップと、
(c)少なくとも一つの抗菌性金属塩:[Mq+]a[Xz-]bを含む粒子含有混合物を形成するための粒子形成条件下で、前記塩前駆体の混合物および前記金属剤混合物を混合するステップと、
(d)ステップ(a)および(b)に反応性成分が含まれていない場合において、このステップ(d)で少なくとも一つの反応性成分が添加されることを条件として、任意に、追加の反応性成分を前記粒子含有混合物に混合して、粒子含有反応性混合物を形成するステップと、
(e)ステップ(c)で抗菌性ポリマー製品中にMq+として添加された前記金属剤由来のMの少なくとも約90%を維持する上で十分な反応条件下で、前記ポリマー製品を形成するために、前記粒子含有反応性混合物を反応させるステップと、
を含む、方法。
【請求項22】
請求項21に記載の方法において、
ステップ(a)または(b)で任意に使用された前記少なくとも一つの反応性成分は、前記金属剤と反応しないものである、方法。
【請求項23】
請求項21に記載の方法において、
前記金属剤との反応性を有する反応性成分は、混合ステップ(d)で前記粒子含有反応性混合物に添加される、方法。
【請求項24】
請求項21に記載の方法において、
前記分散剤は、ヒドロキシアルキルメチルセルロースポリマー類、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレンオキシド、澱粉、ペクチン、ポリアクリルアミド、ゼラチン、ポリアクリル酸、有機アルコキシシラン類である3−アミノプロピルトリエトキシシラン、メチルトリエトキシシラン、フェニルトリメトキシシラン、ビニルトリエトキシシラン、および3−グリシドキシプロピルトリメトキシシラン、グリセリンのホウ酸エステル、および、これらの混合物からなる群より選択される、方法。
【請求項25】
請求項21に記載の方法において、
前記分散剤は、ヒドロキシアルキルメチルセルロースポリマー類、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレンオキシド、ゼラチンおよびポリアクリル酸、グリセリンのホウ酸エステル、および、これらの混合物からなる群より選択される、方法。
【請求項26】
請求項21に記載の方法において、
前記分散剤は、ヒドロキシプロピルメチルセルロース、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレンオキシド、ゼラチンおよびポリアクリル酸、および、これらの混合物からなる群より選択される、方法。
【請求項27】
請求項21に記載の方法において、
前記分散剤は、ポリビニルアルコール、ポリビニルピロリドン、ポリエチレンオキシド、および、ポリアクリル酸、および、これらの混合物からなる群より選択される、方法。
【請求項28】
請求項27に記載の方法において、
前記分散剤は、約2,000,000未満の分子量を有する、方法。
【請求項29】
請求項27に記載の方法において、
前記分散剤は、約20,000〜約1,500,000の間の分子量を有する、方法。
【請求項30】
請求項21に記載の方法において、
前記金属剤混合物は、約40重量%以下の前記分散剤を含む、方法。
【請求項31】
請求項21に記載の方法において、
前記金属剤混合物は、0.01重量%〜約30重量%の間の前記分散剤を含む、方法。
【請求項32】
請求項21に記載の方法において、
前記塩前駆体混合物は、約10重量%以下の前記塩前駆体を含む、方法。
【請求項33】
請求項21に記載の方法において、
前記金属塩前駆体混合物は、前記金属剤に対してモル過剰で、前記塩前駆体を含む、方法。
【請求項34】
請求項21に記載の方法において、
前記金属剤混合物は、約10重量%以下の前記金属剤を含む、方法。
【請求項35】
請求項21に記載の方法において、
前記溶媒は、ステップ(d)前に、粒子含有混合物から除去される、方法。
【請求項36】
方法において、
約200ナノメートル以下の粒子サイズを有する安定化抗菌性金属塩粒子および少なくとも一つのフリーラジカル反応性成分を含む反応性混合物を、前記金属塩粒子に応じて調整された臨界波長を超える波長の光、熱、あるいは、これらの組み合わせを用いて、硬化させて、抗菌性金属塩粒子を含む製品を形成すること、
を含む、方法。
【請求項37】
請求項36に記載の方法において、
前記安定化抗菌性金属塩粒子は、少なくとも一つの銀金属塩を含み、
前記調整された臨界波長は、約430ナノメートルである、方法。
【請求項38】
請求項36に記載の方法において、
前記抗菌性金属は、式:[Mq+]a[Xz-]bを有し、前記ポリマー中のMの少なくとも約90%は、Mq+である、方法。
【請求項39】
請求項36に記載の方法において、
前記反応性混合物は、少なくとも一つの紫外線吸収化合物をさらに含む、方法。
【図1】

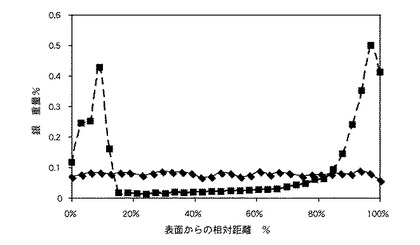
【図2】
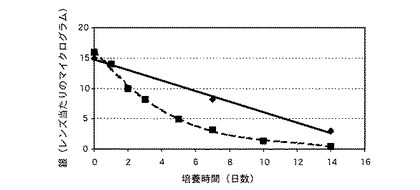
【図3】
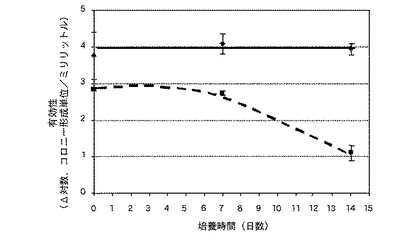
【図4】
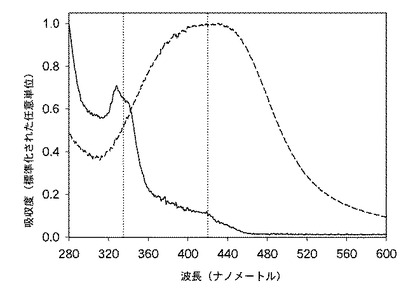
【図5】
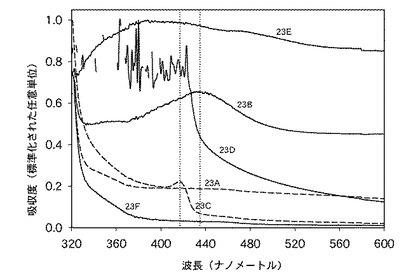
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2010−514463(P2010−514463A)
【公表日】平成22年5月6日(2010.5.6)
【国際特許分類】
【出願番号】特願2009−534627(P2009−534627)
【出願日】平成19年10月22日(2007.10.22)
【国際出願番号】PCT/US2007/022491
【国際公開番号】WO2008/127299
【国際公開日】平成20年10月23日(2008.10.23)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.テフロン
【出願人】(591175675)ジョンソン・アンド・ジョンソン・ビジョン・ケア・インコーポレイテッド (44)
【Fターム(参考)】
【公表日】平成22年5月6日(2010.5.6)
【国際特許分類】
【出願日】平成19年10月22日(2007.10.22)
【国際出願番号】PCT/US2007/022491
【国際公開番号】WO2008/127299
【国際公開日】平成20年10月23日(2008.10.23)
【公序良俗違反の表示】
(特許庁注:以下のものは登録商標)
1.テフロン
【出願人】(591175675)ジョンソン・アンド・ジョンソン・ビジョン・ケア・インコーポレイテッド (44)
【Fターム(参考)】
[ Back to top ]