
抗HER2抗体−薬剤コンジュゲートと化学療法剤の併用及び使用方法
立体異性体、幾何異性体、互変異性体、溶媒和化合物、代謝産物およびこれらの薬学的に許容可能な塩類を含む、抗体−薬剤コンジュゲートトラスツズマブ−MCC−DM1と化学療法剤との組合せは、腫瘍細胞増殖を阻害するために、そして、HER2およびKDR(VEGFRレセプター1)によって媒介される癌などの疾患を治療するために有用である。哺乳動物細胞における前記疾患や関連の病的状態のインビトロ、インサイツ及びインビボの診断、予防又は治療のための前記組合せの使用方法が開示される。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願の説明)
本非仮出願は、米国特許法規則1.53(b)の下で出願し、米国特許法119条(e)に基づき、2008年3月18日に出願の米国仮出願番号第61/037410号の優先権を主張する。この仮出願は出典明記により全体が援用される。
(技術分野)
本発明は、概して、癌などの過剰増殖性疾患に対して活性を有する化合物の製薬的併用に関する。また、本発明は、哺乳動物細胞又は関連の病的状態の、インビトロ、インサイツ及びインビボの診断又は治療のための化合物の併用の使用方法に関する。
【背景技術】
【0002】
HER2(ErbB2)レセプターチロシンは、膜貫通型レセプターの上皮性増殖因子レセプター(EGFR)ファミリのメンバーである。HER2の過剰発現は、およそ20%のヒトの乳癌において観察され、これらの腫瘍と関係する悪性の増殖および臨床結果不良に関係する(Slamon et al (1987) Science 235:177-182)。
トラスツズマブ(CAS180288-69-1、HERCEPTIN(登録商標)、huMAb4D5-8、rhuMAb HER2、Genentech)は、ヒト上皮細胞増殖因子レセプター2タンパク質であるHER2(ErbB2)の細胞外ドメインに対して、細胞ベースのアッセイにおいて高い親和性(Kd=5nM)で選択的に結合する、マウスHER2抗体の組み換えDNA由来のヒト化された、IgG1κ、モノクローナル抗体バージョンである(米国特許第5677171号;米国特許第5821337号;米国特許第6054297号;米国特許第6165464号;米国特許第6339142号;米国特許第6407213号;米国特許第6639055号;米国特許第6719971号;米国特許第6800738号;米国特許第7074404号;Coussens et al (1985) Science 230:1132-9;Slamon et al (1989) Science 244:707-12;Slamon et al (2001) New Engl. J. Med. 344: 783-792)。トラスツズマブは、HER2に結合するマウス抗体(4D5)の相補性決定領域と共にヒトフレームワーク領域を含有する。トラスツズマブは、HER2抗原と結合して、それにより癌細胞の増殖を阻害する。トラスツズマブは、インビトロアッセイ及び動物内において、HER2を過剰発現するヒト腫瘍細胞の増殖を阻害することが示された(Hudziak et al (1989) Mol Cell Biol 9:1165-72;Lewis et al (1993) Cancer Immunol Immunother; 37:255-63;Baselga et al (1998) Cancer Res. 58: 2825-2831)。トラスツズマブは抗体依存性細胞障害作用、ADCCのメディエーターである(Lewis et al (1993) Cancer Immunol Immunother 37(4): 255-263;Hotaling et al (1996) [abstract]. Proc. Annual Meeting Am Assoc Cancer Res; 37:471;Pegram MD, et al (1997) [abstract]. Proc Am Assoc Cancer Res; 38:602;Sliwkowski et al (1999) Seminars in Oncology 26(4), Suppl 12:60-70;Yarden Y. and Sliwkowski, M. (2001) Nature Reviews: Molecular Cell Biology, Macmillan Magazines, Ltd., Vol. 2: 127-137)。
【0003】
HERCEPTIN(登録商標)は、多くの従来の抗癌療法を受けたErbB2を過剰発現させている転移性乳癌患者の治療のために1998年に承認され(Baselga et al, (1996) J. Clin. Oncol. 14:737-744)、その後300000人以上の患者において使用されている(Slamon DJ, et al. N Engl J Med 2001;344:783 92;Vogel CL, et al. J Clin Oncol 2002;20:719 26;Marty M, et al. J Clin Oncol 2005;23:4265 74;Romond EH, et al. T N Engl J Med 2005;353:1673 84;Piccart-Gebhart MJ, et al. N Engl J Med 2005;353:1659 72;Slamon D, et al. [abstract]. Breast Cancer Res Treat 2006, 100 (Suppl 1): 52)。2006年に、FDAは、HER2陽性、リンパ節陽性の乳癌患者の補助治療のためのドキソルビシン、シクロホスファミドおよびパクリタキセルを含む治療投薬計画の一部として、HERCEPTIN(登録商標)(トラスツズマブ、Genentech Inc.)を承認した。HERCEPTIN(登録商標)が開発されたのでHER2陽性腫瘍患者は、化学療法単独よりも著明に良好な結果を得たが、実際にはすべてのHER2陽性、転移性乳癌(MBC)患者が有用な治療法を最終的に進歩させるであろう。MBC患者の結果を向上させる機会はまだある。トラスツズマブの多様な作用機構にもかかわらず、トラスツズマブにより治療される多くの患者は、治療が有効な期間の経過後に応答を示さないか又は応答が止まる。HER2+(HER2陽性)腫瘍にはHERCEPTIN(登録商標)に応答しないものもあり、応答する腫瘍を有する患者の多くは最終的に向上する。HERCEPTIN治療に対して応答しないか又は応答不良である、HER2を過剰発現する腫瘍患者又はHER2発現と関係する他の疾患患者のために、HER2に誘導された癌の治療法をさらに開発するという重要な臨床上の必要性がある。
【0004】
抗体を標的化した治療法に対する他のアプローチは、抗原を発現する癌細胞への細胞障害性剤の特異的運搬のために抗体を利用することである。細胞分裂阻害剤メイタンシンの誘導体であるメイタンシノイドは、ビンカアルカロイド薬剤と類似の方法で微小管に結合する(Issell BF et al (1978) Cancer Treat. Rev. 5: 199-207;Cabanillas F et al. (1979) Cancer Treat Rep, 63:507-9。トラスツズマブに連結したメイタンシノイドDM1からなる抗体−薬剤コンジュゲート(ADC)は、HER2を過剰発現するトラスツズマブ感受性かつトラスツズマブ耐性の腫瘍細胞株およびヒト乳癌の異種移植片モデルにおいて強力な抗腫瘍活性を示す。MCCリンカーを介して抗HER2マウス乳癌抗体TA.1に連結したメイタンシノイドのコンジュゲートは、ジスルフィドリンカーによる対応コンジュゲートの200分の1の強さであって(Chari et al (1992) Cancer Res. 127-133)。トラスツズマブに連結した、メイタンシノイドDM)からなる抗体−薬剤コンジュゲート(ADC)は、HER2を過剰発現するトラスツズマブ感受性かつトラスツズマブ耐性の腫瘍細胞株およびヒト癌の異種移植片モデルにおいて強力な抗腫瘍活性を示す。トラスツズマブ−MCC−DM1(T−DM1)は現在、HER2に対する治療法に抵抗性である疾患を有する患者において第II相臨床試験の評価が行われている(Beeram et al (2007) "A phase I study of trastuzumab-MCC-DM1 (T-DM1), a first-in-class HER2 antibody-drug conjugate (ADC), in patients (pts) with HER2+ metastatic breast cancer (BC)", American Society of Clinical Oncology 43rd: June 02 (Abs 1042;Krop et al, European Cancer Conference ECCO, Poster 2118, September 23-27, 2007, Barcelona;米国特許第7097840号;米国公開特許2005/0276812;米国公開特許2005/0166993)。
【0005】
数回の投薬計画又は投与様式において2以上の薬剤が共に用いられる併用療法は一般に、(i)最小の交差耐性を有する薬剤を組み合わせることによって耐性獲得が生じる頻度を低減する、(ii)僅かな副作用で有効性を達成する、すなわち治療指標を上げるために、毒性がオーバーラップしておらず類似の治療上の特性を有する薬剤の用量を減らす、(iii)細胞-周期段階又は生育特性を変えるなどといった、他の薬剤の使用によるある薬剤の作用に対して細胞を感作させる、そして(iv)2つの薬剤の生物活性の相加効果又は相加以上の効果を利用することによって有効性を上げるといった一又は複数の目的を有する(Pegram, M., et al (1999) Oncogene 18:2241-2251;Konecny, G., et al (2001) Breast Cancer Res. and Treatment 67:223-233;Pegram, M., et al (2004) J. of the Nat. Cancer Inst. 96(10): 739-749;Fitzgerald et al (2006) Nature Chem. Biol. 2(9): 458-466;Borisy et al (2003) Proc. Natl. Acad. Sci 100(13): 7977-7982)。
【0006】
ロエベ相加効果(Chou, T.C. and Talalay, P. (1977) J. Biol. Chem. 252:6438-6442;Chou, T.C. and Talalay, P. (1984) Adv. Enzyme Regul. 22:27-55;Berenbaum, M.C. (1989) Pharmacol. Rev. 41: 93-141)及びブリス非依存性/相乗効果(Bliss, C.I. (1956) Bacteriol. Rev. 20: 243-258;Greco et al (1995) Pharmacol. Rev. 47: 331-385)は、標的を50%阻害するために必要とされ、単純な場合のKiに等しい薬剤の用量であるIC50などのパラメーターに基づいて、単一療法と比較して併用療法について予想される用量−応答相関を算出するために用いられる方法である。
HER2二量体化阻害薬抗体およびEGFR阻害薬は、癌に対する併用療法について報告されている(米国公開特許2007/0020261)。トラスツズマブ−MCC−DM1(T−DM1)及びパーツズマブはMBC患者における活性が示され、パーツズマブとトラスツズマブの組合せはHER陽性MBC患者において活性であることが示されている(Baselga J, et al. "A Phase II trial of trastuzumab and pertuzumab in patients with HER2-positive metastatic breast cancer that had progressed during trastuzumab therapy: full response data", European Society of Medical Oncology, Stockholm, Sweden, September 12?16, 2008)。
【発明の概要】
【0007】
本発明は概して、癌細胞の増殖を阻害するための、一又は複数の化学療法剤と組み合わせて投与される、抗HER2 抗体−薬剤コンジュゲート、トラスツズマブ−MCC−DM1に関する。トラスツズマブ−MCC−DM1と化学療法剤のある組合せは、インビトロ及びインビボでの癌細胞の増殖を阻害する際に相乗効果を示す。本発明の組合せおよび方法は、癌などの過剰増殖性疾患の治療において有用でありうる。この組合せは、哺乳動物における腫瘍増殖を阻害し得、ヒトの癌患者を治療するために有用でありうる。
一態様では、本発明は、併用製剤として又は交互により哺乳動物に治療上の組合せを投与することを含む、過剰増殖性疾患の治療方法であって、このとき治療上の組合せにはトラスツズマブ−MCC−DM1の治療上有効量と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される化学療法剤の治療上有効量が含まれる方法を包含する。
トラスツズマブ−MCC−DM1の治療上有効量と化学療法剤の治療上有効量は、併用製剤として又は交互により投与されてよい。
【0008】
また、本発明は、哺乳動物細胞、生物体又は関連する病的状態の、インビトロ、インサイツ及びインビボの診断又は治療のための組成物の使用に関する。
また、本発明は、治療的併用の投与により相乗効果が生じる方法にも関する。
本発明の他の態様は、化学療法剤がHER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される、トラスツズマブ−MCC−DM1と、一又は複数の薬学的に許容可能な担体、流動促進剤、希釈液又は賦形剤とを含む薬学的組成物である。
本発明の他の態様は、治療を必要とする哺乳動物にトラスツズマブ−MCC−DM1と化学療法剤の有効量を投与することを含む、過剰増殖性疾患または疾病の治療方法を提供する。トラスツズマブ-MCC-DM1と化学療法剤は薬学的製剤として組み合わせて投与するために共に製剤化されも、又は治療的併用として交互に(交代して、連続的な投与)別々に投与されてもよい。一実施態様では、T−DM1は注入によって供給され、化学療法剤は経口的に運搬される。
【0009】
本発明の他の態様は、組合せにトラスツズマブ−MCC−DM1および抗癌性の標準的な治療を有する化学療法剤を含む場合のインビボ有効性についての有効な薬剤組合せを予測するための方法を提供する。インビトロ細胞増殖およびインビボ腫瘍異種移植片実験からの有効性データは、定性的及び定量的に分析される。定量的分析法は、相乗効果、拮抗作用又は相加効果を示すための併用指標(CI)値を生成するアイソボログラム及びChou & Talalay中央有効値、又はブリス非依存性リボン(Bliss Independence ribbon)グラフ屈折に基づいてよい。
本発明の他の態様は、哺乳動物のHER2又はKDR9(VEGFレセプター1)によって媒介されるものを含む、癌などの疾患又は疾病を治療するための、本発明の治療上の併用の使用方法である。
本発明の他の態様は、哺乳動物のHER2又はKDR9(VEGFレセプター1)によって媒介されるものを含む、癌などの疾患又は疾病を治療するための医薬の調製における、本発明の治療上の併用の使用である。
本発明の他の態様には、トラスツズマブ−MCC−DM1、化学療法剤、容器および場合によって治療を示すパッケージ挿入物又はラベルを具備する製造品又はキットが含まれる。
本発明の他の態様は、癌の治療のために併用して用いられる化合物の決定方法であって、a)トラスツズマブ−MCC−DM1と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される化学療法剤とをインビトロ腫瘍細胞株に投与することと、b)相乗効果又は非相乗効果を測定することを含む方法を包含する。
【0010】
本発明の更なる利点および新しい特徴は、後述する説明にある程度示され、以下の明細書の実施例により当業者にある程度明らかになるか、又は本発明の実施によってわかるであろう。本発明の利点は、特に添付の請求の範囲において示される手段、組合せ、組成物および方法によって理解され、達成されてよい。
【図面の簡単な説明】
【0011】
【図1】トラスツズマブ、トラスツズマブ−MCC−DM1(T-DM1)およびトラスツズマブとT−DM1の併用のIC50倍数濃度に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。
【図2】トラスツズマブ、トラスツズマブ−MCC−DM1(T-DM1)およびトラスツズマブとT−DM1の併用のIC50倍数濃度に対する、3日のBT−474EEIインビトロ細胞生存度のプロット線を示す。
【図3】パーツズマブ、トラスツズマブ−MCC−DM1(T-DM1)およびパーツズマブとT−DM1の併用のIC50倍数濃度に対する、5日のMDA−MB−175インビトロ細胞生存度のプロット線を示す。
【図3a】パーツズマブ、トラスツズマブ−MCC−DM1(T-DM1)およびパーツズマブとT−DM1の併用のIC50倍数濃度に対する、5日のMDA−MB−175インビトロ細胞生存度のプロット線を示す。
【図4】用量反応のトラスツズマブ−MCC−DM1(T-DM1)と併用した様々な一定用量のパーツズマブ、および様々な用量のT−DM1単独に対する、5日のBT-474インビトロ細胞生存度のプロット線を示す。
【図5】用量反応のパーツズマブと併用した様々な一定用量のトラスツズマブ−MCC−DM1(T-DM1)、および様々な用量のパーツズマブ単独に対する、5日のBT-474インビトロ細胞生存度のプロット線を示す。
【図6】パーツズマブ、トラスツズマブ−MCC−DM1(T-DM1)およびパーツズマブとT−DM1の併用のIC50倍数濃度に対する、5日のBT−474インビトロ細胞生存度のプロット線を示す。
【図7】一定用量のラパチニブと併用した変更用量のT−DM1(4.5nM、14nM、41nM、123nM)、及び変更用量のT−DM1単独(0〜1000ng/ml)に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。
【図7a】T−DM1、ラパチニブおよび一定用量比のT−DM1とラパチニブの併用に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。
【図8a】T−DM1、ラパチニブおよび一定用量比のT−DM1とラパチニブの併用に対する、3日のBT−474インビトロ細胞生存度のプロット線を示す。
【図8】一定用量のラパチニブと併用した変更用量のT−DM1(1.5nM、4.5nM、14nM、41nM、123nM)、及び変更用量のT−DM1単独(0〜1000ng/ml)に対する、3日のBT−474インビトロ細胞生存度のプロット線を示す。
【図9】一定用量のラパチニブと併用した変更用量のT−DM1(14nM、41nM、123nM、370nM、1111nM)、及び変更用量のT−DM1単独(0〜1000ng/ml)に対する、3日のBT−474−EEIインビトロ細胞生存度のプロット線を示す。
【図10】以下を投与した後のSCIDベージュマウスの乳房脂肪パッドに接種したKPL-4腫瘍(1マウスにつきマトリゲル中3000000細胞)における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ADCバッファ、(2) パーツズマブ15mg/kg、(3) T−DM1 0.3mg/kg、(4) T−DM1 1mg/kg、(5) T−DM1 3mg/kg、(6) パーツズマブ15mg/kg+T−DM1 0.3mg、(7) パーツズマブ15mg/kg+T−DM1 1mg/kg、(8)パーツズマブ15mg/kg+T−DM1 3mg/kg。ADCバッファおよびT−DM1は0日目に1度投与した。パーツズマブは、0、7および14日目に1度投与した。
【図11】以下を投与した後のSCIDベージュマウスの乳房脂肪パッドに接種したKPL-4腫瘍(1マウスにつきマトリゲル中3000000細胞)における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ADCバッファ、(2) 5−FU 100mg/kg、(3) パーツズマブ40mg/kg、第一パーツズマブ用量(5、7および9群)は2倍の負荷投与量とした、(4) B20-4.1、5mg/kg、(5) T−DM1、5mg/kg、(6) 5−FU、100mg/kg+T−DM1、5mg、(7) パーツズマブ40mg/kg+T−DM1、5mg/kg、(8) B20-4.1、5mg/kg+T−DM1、5mg/kg、(9) B20-4.1、5mg/kg+パーツズマブ、40mg/kg。ADCバッファおよびT−DM1は単回静注により0日目に1度投与した。パーツズマブは0、7、14、21日目に投与した(qwk ×4)。5−FUは0、7および14日目に投与した(qwk ×3)。B20-4.1は、0、3、7、10、14、17、21および24日目に投与した(2×/wk ×8合計)。
【図12】以下を投与した後のCRLnu/nuマウスの乳房脂肪パッドに接種したMMTV-HER2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル(ADCバッファ)、(2) B20-4.1、5mg/kg、(3) T−DM1、3mg/kg、(4) T−DM1、5mg/kg、(5) T−DM1、10mg/kg、(6) B20-4.1、5mg/kg+T−DM1 3mg/kg、(7) B20-4.1、5mg/kg+T−DM1 5mg/kg、(8) B20-4.1、5mg/kg+T−DM1、10mg/kg。ADCバッファおよびT−DM1は0および21日目に投与した。B20-4.1は、0、3、7、10、14、17、21および24日目に投与した(2×/wk ×4、合計8)。
【図13】以下を投与した後のCRLnu/nuマウスの乳房脂肪パッドに接種したMMTV-HER2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル(ADCバッファ)、(2) T−DM1 10mg/kg、(3) 5−FU 100mg/kg、(4) ゲムシタビン120mg/kg、(5)カルボプラチン100mg/kg、(6) 5−FU 100mg/kg+T−DM1 10mg/kg、(7) ゲムシタビン120mg/kg+T−DM1 10mg/kg、(8) カルボプラチン100mg/kg+T−DM1 10mg/kg。ADCバッファ、T−DM1およびカルボプラチンは、0日目に単回注射した。単一の注射。5−FUは0、7および14日目に投与した(qwk ×3)。ゲムシタビンは0、3、6および9日目に投与した(q3d ×4)。
【図14】以下を投与した後のハーラン胸腺欠損ヌードマウスの乳房脂肪パッドに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル(PBSバッファ) iv、qwk ×4、(2) ラパチニブ101mg/kg、po、bid ×21、(3) パーツズマブ40mg/kg、iv、qwk ×4、(4) B20-4.1 5mg/kg、ip、2×/wk ×4、(5) T−DM1 15mg/kg、iv、最後までq3wk、(6) ラパチニブ101mg/kg、po、bid ×21+T−DM1 15mg/kg、iv、最後までq3wk、(7) パーツズマブ40mg/kg、iv、qwk ×4+T−DM1 15mg/kg、iv、最後までq3wk、(8) B20-4.1 5mg/kg、ip、2×/wk×4+T−DM1 15mg/kg、iv、最後までq3wk。
【図15】以下を投与した後のハーラン胸腺欠損ヌードマウスの乳房脂肪パッドに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル(PBSバッファ) po、bid ×21、(2) T−DM1、7.5mg/kg、iv、qd ×1、(3) T−DM1、15mg/kg、iv、qd ×1、(4) ABT−869、5mg/kg、po、bid ×21、(5) ABT−869、15mg/kg、po、bid ×21、(6) T−DM1、7.5mg/kg、iv、qd ×1+ABT−869、5mg/kg、po、bid ×21、(7) T−DM1 7.5mg/kg、iv、qd ×1+ABT−869、15mg/kg、po、bid ×21、(8) T−DM1、15mg/kg、iv、qd ×1+ABT−869、5mg/kg、po、bid ×21、(9) T−DM1、15mg/kg、iv、qd ×1+ABT−869、15mg/kg、po、bid ×21。
【図16】以下を投与した後のハーラン胸腺欠損ヌードマウスの乳房脂肪パッドに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル、iv、qwk ×3、(2) T−DM1、7.5mg/kg、iv、q3wk ×2、(3) T−DM1、15mg/kg、iv、q3wk ×2、(4)ドセタキセル、30mg/kg、iv、qwk ×3、(5) T−DM1、7.5mg/kg、iv、q3wk ×2+ドセタキセル、30mg/kg、iv、qwk ×3、(6) T−DM1、15mg/kg、iv、q3wk ×2+ドセタキセル、30mg/kg、iv、qwk ×3。
【図17】以下を投与した後のハーラン胸腺欠損ヌードマウスの乳房脂肪パッドに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル、po、qd×21、(2) T−DM1、7.5mg/kg、iv、q3wk ×2、(3) T−DM1、15mg/kg、iv、q3wk ×2、(4) ラパチニブ、100mg/kg、po、bid ×21、(5) T−DM1、7.5mg/kg、iv、q3wk ×2+ラパチニブ、100mg/kg、po、bid ×21、(6) T−DM1、15mg/kg、iv、q3wk ×2+ラパチニブ、100mg/kg、po、bid ×21。
【図18】5−FU、トラスツズマブ−MCC−DM1(T-DM1)および5−FUとT−DM1の一定用量比組合せのIC50倍数の濃度に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。
【図19】5−FU、トラスツズマブ−MCC−DM1(T-DM1)および5−FUとT−DM1の一定用量比組合せのIC50倍数濃度に対する、3日のBT−474インビトロ細胞生存度のプロット線を示す。
【図20】ゲムシタビン、トラスツズマブ−MCC−DM1(T-DM1)およびゲムシタビンとT−DM1の一定用量比組合せのIC50倍数濃度に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。
【図21】ゲムシタビン、トラスツズマブ−MCC−DM1(T-DM1)およびゲムシタビンとT−DM1の一定用量比組合せのIC50倍数濃度に対する、3日のMDA−MD−361インビトロ細胞生存度のプロット線を示す。
【図22】0.25×〜4×のIC50倍数濃度でのT−DM1、GDC−0941、およびT−DM1とGDC−0941(62.5nM〜1μM)の1:10一定用量比の組合せによる処置後3日の、KPL4インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。
【図23】0.0625×〜16×のIC50倍数濃度でのT−DM1、GDC−0941、およびT−DM1(1.25〜80ng/ml)とGDC−0941(31.25nM〜2μM)の1:25一定用量比の組合せによる処置後3日の、KPL4インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。
【図24】0から16×のIC50倍数濃度での、T−DM1、PI103、GDC−0941、およびT−DM1+PI103及びT−DM1+GDC−0941の一定用量比の組合せによる処置後3日の、Her2増幅、HERCEPTIN(登録商標)耐性、PIK3CA(H1047R)変異のKPL-4細胞インビトロ細胞生存度(増殖)のプロット線を示す。
【図25】T−DM1、GDC−0941、および160ng/ml以下のT−DM1濃度の一定用量比のT−DM1とGDC−0941組合せによる処置後24時間の、KPL4カスパーゼ3/7インビトロ細胞生存度(増殖)のプロット線を示す。
【図26】T−DM1、GDC−0941、および0〜200ng/mlのT−DM1濃度の一定用量比のT−DM1とGDC−0941組合せによる処置後3日の、KPL4インビトロ細胞生存度(増殖)のプロット線を示す。
【図27】0.125×〜8×のIC50倍数濃度での、T−DM1、GDC−0941、およびT−DM1(3.125〜50ng/ml)とGDC−0941(62.5nM〜1μM)の1:20一定用量比の組合せによる処置後3日の、MDA−0MB−361インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。
【図28】0.125×〜8×のIC50倍数濃度での、T−DM1、GDC−0941、およびT−DM1(3.125〜100ng/ml)とGDC−0941(62.5nM〜2μM)の1:20一定用量比の組合せによる処置後3日の、MDA−0MB−361インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。
【図29】0.125×〜4×のIC50倍数濃度での、T−DM1、GDC−0941、およびT−DM1(3.125〜100ng/ml)とGDC−0941(31.25nM〜1μM)の1:10一定用量比の組合せによる処置後3日の、BT−474インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。
【図30】0.25×〜4×のIC50倍数濃度での、T−DM1、GDC−0941、およびT−DM1(6.25〜100ng/ml)とGDC−0941(62.5nM〜1μM)の1:10一定用量比の組合せによる処置後3日の、BT-474インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。
【図31】0〜16×のIC50倍数濃度での、T−DM1、PI103、GDC−0941、およびT−DM1+PI103及びT−DM1+GDC−0941の一定用量比の組合せによる処置後3日の、Her2増幅、非PI3K変異のAU565細胞インビトロ細胞生存度(増殖)のプロット線を示す。
【図32】0〜16×のIC50倍数濃度での、T−DM1、PI103、GDC−0941、およびT−DM1+PI103及びT−DM1+GDC−0941の一定用量比の組合せによる処置後3日の、Her2増幅、PIK3CA(C420R)変異のEFM192A細胞インビトロ細胞生存度(増殖)のプロット線を示す。
【図33】0〜16×のIC50倍数濃度での、T−DM1、PI103、GDC−0941、およびT−DM1+PI103及びT−DM1+GDC−0941の一定用量比の組合せによる処置後の、Her2増幅、ハーセプチン(登録商標)耐性、PIK3CA(H1047R)変異のHCC1954細胞インビトロ細胞生存度(増殖)のプロット線を示す。
【図34】以下を投与した後のCRLnu/nuマウスに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル、po、qd×21、(2) T−DM1、10mg/kg、iv、q3wk、(3) 5−FU、100mg/kg、po、qwk ×2、(4) T−DM1、5mg/kg、iv、q3wk+5−FU、100mg/kg、po、qwk ×2。
【図35】以下を投与した後のCRLnu/nuマウスに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル、po、qd ×21、(2) T−DM1、5mg/kg、iv、qd ×1、(3) GDC−0941、100mg/kg、po、qd ×21、(4) GDC−0152、50mg/kg、po、qwk ×3、(5) T−DM1、5mg/kg、iv、qd ×1+GDC−0941、100mg/kg、po、qd ×21、(6) T−DM1、5mg/kg、iv、qd ×1+GDC−0152、50mg/kg、po、qwk ×3。
【図36】以下を投与した後のCRLnu/nuマウスに接種したMDA−MB−361.1乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル、po、qd ×21、(2) GDC−0941、25mg/kg、po、qd ×21、(3) GDC−0941、50mg/kg、po、qd ×21、(4) GDC−0941、100mg/kg、po、qd ×21、(5) T−DM1、3mg/kg、iv、qd ×1、(6) T−DM1、10mg/kg、iv、qd ×1、(7) GDC−0941、25mg/kg、po、qd ×21+T−DM1、3mg/kg、iv、qd ×1、(8) GDC−0941、50mg/kg、po、qd ×21+T−DM1、3mg/kg、iv、qd ×1、(9) GDC−0941、100mg/kg、po、qd ×21+T−DM1、3mg/kg、iv、qd ×1、(10) GDC−0941、25mg/kg、po、qd ×21+T−DM1、10mg/kg、iv、qd ×1、(11) GDC−0941、50mg/kg、po、qd ×21+T−DM1、10mg/kg、iv、qd ×1、(12) GDC−0941、100mg/kg、po、qd ×21+T−DM1、10mg/kg、iv、qd ×1。
【図37】以下を投与した後のCRLnu/nuマウスに接種したMDA−MB−361.1乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル[MCT(0.5%メチルセルロース/0.2%TWEEN80(登録商標))+琥珀酸塩バッファ(100mMの琥珀酸ナトリウム、100mg/mlのトレハロース、0.1%のTWEEN80、pH5.0)]、po+IV、qd ×21及びqd、(2) GNE-390、1.0mg/kg、po、qd ×21、(3) GNE−390、2.5mg/kg、po、qd ×21、(4) T−DM1、3mg/kg、iv、qd、(5) GNE−390、1.0mg/kg、po、qd ×21+T−DM1、3mg/kg、iv、qd、(6) GNE−390、2.5mg/kg、po、qd×21+T−DM1、3mg/kg、iv、qd。
【発明を実施するための形態】
【0012】
(例示的実施態様の詳細な説明)
以下、その例が添付の構造及び式で示される本発明の所定の実施態様が詳細に参照される。本発明は、列挙された実施態様に関連して記載されるが、それらは本発明をこれら実施形態に限定することを意図するものではないことが理解される。逆に、本発明は、特許請求の範囲に記載の本発明の範囲内に含まれ得るあらゆる代替物、改変物、及び均等物を包含するものである。当業者であれば、本発明の実施に使用され得、ここに記載されたものと類似か等価である多くの方法及び材料を認識できるであろう。本発明は、記載された方法及び材料には決して限定されない。援用される文献、特許、及び類似の資料のうちの一又は複数が、限定しないが、定義された用語、用語の使用法、記載された技術などを含む本願と異なるか又は矛盾する場合、本願が優先する。
【0013】
(定義)
本明細書中及び特許請求の範囲において用いられる「含む」、「含んでいる」、「包含する(include)」、「包含している」及び「包含する(includes)」なる用語は、述べられた形質、整数、成分又は工程の存在を特定することを意図するが、一又は複数の他の形質、整数、成分、工程、又はその群の存在又は付加を妨げない。
「処置する」および「処置」なる用語は、治療的処置と予防的処置ないし防御的処置との両方をいい、その目的は、望ましくない生理学的変化または障害、例えば、癌などの過剰増殖性状態の増殖、発生または広がりを予防するかまたは遅くする(減らす)ことである。本発明の目的で、有利または望ましい臨床結果としては、検出可能であれ検出不可能であれ、症状の軽減、疾患の程度の減少、疾患の安定化された(すなわち、悪化しない)状態、疾患の進行の遅延または減速、疾患状態の軽減または緩和、および寛解(部分的であれ全体的であれ)が含まれる。また、「処置」は、処置を受けない場合に予測される生存と比較される場合に、生存を延長することを意味し得る。処置を必要とするものとしては、その状態または障害をすでに有するもの、並びにその状態または障害を有しやすいもの、あるいはその状態または障害が予防されるべきであるものが含まれる。
【0014】
「治療上の有効量」なる表現は、(i)本明細書中に記載される特定の疾患、状態又は障害を処置するか、(ii)特定の疾患、状態または障害の一又は複数の症状を寛解、軽減または排除するか、あるいは(iii)特定の疾患、状態、または障害の一又は複数の症状の発生を予防または遅延させる、本発明の化合物の量を意味する。癌の場合、薬物の治療上の有効量は、癌細胞の数を減少させ得;腫瘍のサイズを減少させ得;周辺器官への癌細胞の浸潤を阻害し得(すなわち、ある程度まで遅くし得、そして好ましくは停止させ得);腫瘍の転移を阻害し得(すなわち、ある程度まで遅くし得、そして好ましくは停止させ得);腫瘍の増殖をある程度まで阻害し得;そして/または癌に関連する症状のうちの一又は複数をある程度まで緩和し得る。薬物が、存在する癌細胞の増殖を防止し得、そして/または殺傷し得る場合、この薬物は、細胞増殖抑制性および/または細胞障害性であり得る。癌治療のためには、効力は、例えば、疾患の進行までの時間(TTP)を評価すること、および/または応答速度(RR)を決定することによって測定され得る。
【0015】
「過剰増殖性疾患」は、前悪性及び非腫瘍性のステージを含む腫瘍、癌及び腫瘍性組織を示し、乾癬、子宮内膜症、ポリープおよび線維腺腫も含む。
「癌」及び「癌性」なる用語は、典型的に制御されない細胞増殖によって特徴付けられる哺乳動物の生理的な状態を指すか又は表す。「腫瘍」は一又は複数の癌性細胞を含む。癌の例には、癌腫、リンパ腫、芽細胞腫、肉腫、および白血病またはリンパ悪性疾患が挙げられるが、これらに限定されない。このような癌のより具体的な例としては、扁平上皮癌(例えば、上皮扁平上皮癌)、小細胞肺癌、非小細胞肺癌(「NSCLC」)、肺の腺癌および肺の鱗状癌腫を含む肺癌、腹膜の癌、肝細胞癌、胃腸癌を含む胃癌(gastric or stomach)、膵臓癌、神経膠芽細胞腫、子宮頚癌、卵巣癌、肝臓癌、膀胱癌、肝細胞種、乳癌、結腸癌、直腸癌、結腸直腸癌、子宮内膜または子宮の癌腫、唾液腺癌腫、腎臓癌または腎性癌、前立腺癌、外陰部癌、甲状腺癌、肝性癌腫、肛門癌腫、陰茎癌腫ならびに頭頸部癌が含まれる。
【0016】
作用機構に関係なく、「化学療法剤」は癌の治療において有用な化学化合物である。化学療法剤の種類には、限定するものではないが、アルキル化剤(alkyating agent)、アンチメタボライ、紡錘体阻害剤植物アルカロイド、細胞障害性/抗腫瘍抗生物質、トポイソメラーゼ阻害薬、抗体、光増感剤及びキナーゼ阻害薬が含まれる。化学療法剤は、「ターゲティング療法」と従来の化学療法とに用いられる化合物を含む。化学療法剤の例には、エルロチニブ(TARCEVA(登録商標), Genentech/OSI Pharm.)、ドセタキセル(TAXOTERE(登録商標), Sanofi-Aventis)、5−FU(フルオロウラシル、5‐フルオロウラシル、CAS番号51-21-8)、ゲムシタビン(GEMZAR(登録商標), Lilly)、PD-0325901(CAS番号391210-10-9, Pfizer)、シスプラチン(シス-ジアミン、ジクロロ白金(II)、CAS番号15663-27-1)、カルボプラチン(CAS番号41575-94-4)、パクリタキセル(TAXOL(登録商標), Bristol-Myers Squibb Oncology, Princeton, N.J.)、トラスツズマブ(HERCEPTIN(登録商標), Genentech)、テモゾロミド(4-メチル-5-オキソ-2,3,4,6,8-ペントアザ二環式[4.3.0]ノナ-2,7,9-トリエン-9-カルボキシアミド、CAS番号85622-93-1, TEMODAR(登録商標), TEMODAL(登録商標), Schering Plough)、タモキシフェン((Z)-2-[4-(1,2-ジフェニルブト-1-エニル)フェノキシ]-N,N-ジメチル-エタンアミド, NOLVADEX(登録商標), ISTUBAL(登録商標), VALODEX(登録商標))、及びドキソルビシン(ADRIAMYCIN(登録商標))、Akti-1/2、HPPD及びラパマイシンが含まれる。
【0017】
化学療法剤の他の例には、オキサリプラチン(ELOXATIN(登録商標), Sanofi)、ボルテゾミブ(VELCADE(登録商標), Millennium Pharm.)、スーテント(SUNITINIB(登録商標), SU11248, Pfizer)、レトロゾール(FEMARA(登録商標), Novartis)、メシル酸イマチニブ(GLEEVEC(登録商標), Novartis)、XL-518 (Mekインヒビター, Exelixis, 国際公開2007/044515)、ARRY-886 (Mekインヒビター, AZD6244, Array BioPharma, Astra Zeneca)、SF-1126(PI3Kインヒビター, Semafore Pharmaceuticals)、BEZ-235(PI3Kインヒビター, Novartis)、XL-147(PI3Kインヒビター, Exelixis)、PTK787/ZK 222584 (Novartis)、フルベストラント(FASLODEX(登録商標), AstraZeneca)、リューコボリン(フォリン酸)、ラパマイシン(シロリムス, RAPAMUNE(登録商標), Wyeth)、ラパチニブ(TYKERB(登録商標), GSK572016, Glaxo Smith Kline)、ロナファーニブ(SARASARTM, SCH 66336, Schering Plough)、ソラフェニブ(NEXAVAR(登録商標), BAY43-9006, Bayer Labs)、ゲフィチニブ(IRESSA(登録商標), AstraZeneca)、イリノテカン(CAMPTOSAR(登録商標), CPT-11, Pfizer)、ティピファニブ(ZARNESTRATM, Johnson & Johnson)、ABRAXANETM (Cremophor-free), パクリタキセルのアルブミン-変更ナノ粒子製剤(American Pharmaceutical Partners, Schaumberg, Il)、バンデタニブ(rINN, ZD6474, ZACTIMA(登録商標), AstraZeneca)、クロラムブシル、AG1478、AG1571(SU5271; Sugen)、テムシロリムス(TORISEL(登録商標), Wyeth)、パゾパニブ(GlaxoSmithKline)、カンフォスファミド(TELCYTA(登録商標), Telik)、チオテパ、及びシクロフォスファミド(CYTOXAN(登録商標)、NEOSAR(登録商標));ブスルファン、インプロスルファン及びピポスルファンのようなスルホン酸アルキル類;ベンゾドーパ(benzodopa)、カルボコン、メツレドーパ(meturedopa)、及びウレドーパ(uredopa)のようなアジリジン類;アルトレートアミン(altretamine)、トリエチレンメラミン、トリエチレンホスホラミド、トリエチレンチオホスホラミド(triethylenethiophosphaoramide)及びトリメチローロメラミン(trimethylolomelamine)を含むエチレンイミン類及びメチラメラミン類;アセトゲニン(acetogenins)(特にブラタシン(bullatacin)及びブラタシノン(bullatacinone));カンプトセシン(合成類似体トポテカン(topotecan)を含む);ブリオスタチン;カリスタチン(callystatin);CC-1065(そのアドゼレシン(adozelesin)、カルゼレシン(carzelesin)及びバイゼレシン(bizelesin)合成類似体を含む);クリプトフィシン(cryptophycin)(特にクリプトフィシン1及びクリプトフィシン8);ドラスタチン(dolastatin);デュオカルマイシン(duocarmycin )(合成類似体、KW−2189及びCBI−TM1を含む); エレトロビン(eleutherobin);パンクラチスタチン(pancratistatin);サルコディクチン(sarcodictyin);スポンジスタチン(spongistatin);クロランブシル、クロルナファジン(chlornaphazine)、チョロホスファミド(cholophosphamide)、エストラムスチン、イホスファミド、メクロレタミン、メクロレタミンオキシドヒドロクロリド、メルファラン、ノベンビチン(novembichin)、フェネステリン(phenesterine)、プレドニムスチン(prednimustine)、トロフォスファミド(trofosfamide)、ウラシルマスタード等のナイトロジェンマスタード;ニトロスレアス(nitrosureas)、例えばカルムスチン(carmustine)、クロロゾトシン(chlorozotocin)、フォテムスチン(fotemustine)、ロムスチン(lomustine)、ニムスチン、ラニムスチン;エネジイン(enediyne) 抗生物質等の抗生物質(例えば、カリケアマイシン(calicheamicin)、カリケアマイシンガンマ1I及びカリケアマイシンオメガI1、例えば、Agnew Chem Intl. Ed. Engl., 33:183-186(1994)を参照のこと;ダイネミシン(dynemicin)、ダイネミシンA(dynemicinA);クロドロネート(clodronate)などのビスホスホネート(bisphosphonates);エスペラマイシン(esperamicin); 同様にネオカルチノスタチン発光団及び関連色素蛋白エネジイン(enediyne) 抗生物質発光団)、アクラシノマイシン(aclacinomysins)、アクチノマイシン、オースラマイシン(authramycin)、アザセリン、ブレオマイシン(bleomycins)、カクチノマイシン(cactinomycin)、カラビシン(carabicin)、カルミノマイシン(carminomycin)、カルジノフィリン(carzinophilin)、クロモマイシン、ダクチノマイシン、ダウノルビシン、デトルビシン(detorubicin)、6-ジアゾ-5-オキソ-L-ノルロイシン、モルフォリノ−ドキソルビシン、シアノモルフォリノ-ドキソルビシン、2-ピロリノ-ドキソルビシン及びデオキシドキソルビシンを含む)、エピルビシン、エソルビシン(esorubicin)、イダルビシン、マセロマイシン(marcellomycin)、マイトマイシンCなどのマイトマイシン(mitomycins)、マイコフェノール酸(mycophenolic acid)、ノガラマイシン(nogalamycin)、オリボマイシン(olivomycins)、ペプロマイシン、ポトフィロマイシン(potfiromycin)、ピューロマイシン、クエラマイシン(quelamycin)、ロドルビシン(rodorubicin)、ストレプトニグリン、ストレプトゾシン、ツベルシジン(tubercidin)、ウベニメクス、ジノスタチン(zinostatin)、ゾルビシン(zorubicin);メトトレキセート及び5-フルオロウラシル(5-FU)のような抗-代謝産物;デノプテリン(denopterin)、メトトレキセート、プテロプテリン(pteropterin)、トリメトレキセート(trimetrexate)のような葉酸類似体;フルダラビン(fludarabine)、6-メルカプトプリン、チアミプリン、チオグアニンのようなプリン類似体;アンシタビン、アザシチジン(azacitidine)、6-アザウリジン(azauridine)、カルモフール、シタラビン、ジデオキシウリジン、ドキシフルリジン、エノシタビン(enocitabine)、フロキシウリジン(floxuridine)のようなピリミジン類似体;カルステロン(calusterone)、プロピオン酸ドロモスタノロン、エピチオスタノール、メピチオスタン、テストラクトン(testolactone)のようなアンドロゲン類;アミノグルテチミド、ミトタン、トリロスタンのような抗副腎剤;フロリン酸(frolinic acid)のような葉酸リプレニッシャー(replenisher);アセグラトン;アルドホスファミドグリコシド;アミノレブリン酸;エニルウラシル(eniluracil);アムサクリン(amsacrine);ベストラブシル(bestrabucil);ビサントレン(bisantrene);エダトラキセート(edatraxate);デフォファミン(defofamine);デメコルシン(demecolcine);ジアジコン(diaziquone);エルフォルニチン(elfornithine);酢酸エリプチニウム(elliptinium acetate);エポチロン(epothilone);エトグルシド(etoglucid);硝酸ガリウム;ヒドロキシ尿素;レンチナン;ロニダミン(lonidamine);メイタンシン(maytansine)及びアンサマイトシン(ansamitocin)のようなメイタンシノイド(maytansinoid);ミトグアゾン(mitoguazone);ミトキサントロン;モピダモール(mopidamol);ニトラクリン(nitracrine);ペントスタチン;フェナメット(phenamet);ピラルビシン;ポドフィリン酸(podophyllinic acid);2-エチルヒドラジド;プロカルバジン;PSK(登録商標)多糖類複合体(JHS Natural Products, Eugene, OR);ラゾキサン(razoxane);リゾキシン(rhizoxin);シゾフィラン;スピロゲルマニウム(spirogermanium);テニュアゾン酸(tenuazonic acid);トリアジコン(triaziquone);2,2',2''-トリクロロトリエチルアミン;トリコテセン(trichothecenes)(T-2トキシン、ベラキュリンA(verracurin A)、ロリデンA(roridin A)及びアングイデン(anguidine));ウレタン;ビンデシン;ダカルバジン;マンノムスチン(mannomustine);ミトブロニトール;ミトラクトール(mitolactol);ピポブロマン(pipobroman);ガシトシン(gacytosine);アラビノシド(「Ara-C」);シクロホスファミド;チオテパ;6-チオグアニン;メルカプトプリン;メトトレキセート;シスプラチン及びカルボプラチンのようなプラチナ類似体;ビンブラスチン;エトポシド(VP-16);ミトキサントン;ビンクリスチン;ビノレルビン(NAVELBINE(登録商標));ナベルビン(Navelbine);ノバントロン(novantrone);テニポシド;エダトレキセート;ダウノマイシン;アミノプテリン;カペシタビン(XELODA(登録商標), Roche);イバンドロナート(ibandronate);CPT-11;トポイソメラーゼインヒビターRFS2000;ジフルオロメチロールニチン(DMFO);レチノイン酸などのレチノイド類;、並びに上述したものの製薬的に許容可能な塩類、酸類及び誘導体が含まれる。
【0018】
また、「化学療法剤」の定義には、(i) 腫瘍に対するホルモン作用を調節又は阻害するように働く抗ホルモン剤、抗エストロゲン及び選択的エストロゲンレセプターモジュレーター(SERM)など、例えばタモキシフェン(NOLVADEX(登録商標)タモキシフェンクエン酸塩)、ラロキシフェン(raloxifene)、ドロロキシフェン、4-ヒドロキシタモキシフェン、トリオキシフェン(trioxifene)、ケオキシフェン(keoxifene)、LY117018、オナプリストーン(onapristone)、及びFARESTON(登録商標)(クエン酸トレミフェン);(ii) アロマターゼ酵素を阻害するアロマターゼ阻害物質、それらは副腎でのエストロゲン産生を調節するものであり、例えば4(5)-イミダゾール類、アミノグルテチミド、MEGASE(登録商標)(メゲストロールアセテート)、AROMASIN(登録商標)(エキセメスタン, Pfizer)、ホルメスタイン(formestanie)、ファドロゾール、RIVISOR(登録商標)(ゾロゾール(vorozole))、FEMARA(登録商標)(レトロゾール, Novartis)及びARIMIDEX(登録商標)(アナストロゾール, AstraZeneca);(iii) 抗アンドロゲン、例えばフルタミド(flutamide)、ニルタミド(nilutamide)、ビカルタミド、ロイプロリド、及びゴセレリン;並びにトロキサシタビン(troxacitabine)(1,3-ジオキソランヌクレオシドシトシン類似体);(iv) プロテインキナーゼインヒビター、例えばMEKインヒビター(国際公開2007/044515);(v) 脂質キナーゼインヒビター;(vi) アンチセンスオリゴヌクレオチド、特に不粘着性細胞増殖に関係するシグナル伝達経路の遺伝子の発現を抑制するもの、例えばPKC−α、Raf及びH−Ras、オブリメルセン(GENASENSE(登録商標), Genta Inc.);(vii) リボザイム、例えばVEGF発現インヒビター(例えばANGIOZYME(登録商標))及びHER2発現インヒビター;(viii) 遺伝子治療ワクチン、例えばALLOVECTIN(登録商標)、LEUVECTIN(登録商標)及びVAXID(登録商標)などのワクチン;PROLEUKIN(登録商標)rIL−2;LURTOTECAN(登録商標)などのトポイソメラーゼ1インヒビター;ABARELIX(登録商標)rmRH;(ix) ベバシズマブ(AVASTIN(登録商標), Genentech)などの抗血管形成剤;並びに上記のものの製薬的に許容可能な塩類、酸類又は誘導体が含まれる。
また、「化学療法剤」の定義には、治療的抗体、例として、アレムツズマブ(Campath)、ベバシズマブ(アバスチン(登録商標), Genentech);セツキシマブ(ERBITUX(登録商標), Imclone);パニツムマブ(VECTIBIX(登録商標), Amgen)、リツキシマブ(RITUXAN(登録商標), Genentech/Biogen Idec)、ペルツズマブ(OMNITARGTM, 2C4, Genentech)、トラスツズマブ(HERCEPTIN(登録商標), Genentech)、トシツモマブ(Bexxar, Corixia)、及び抗体薬剤コンジュゲート、ゲムツズマブオゾガマイシン(MYLOTARG(登録商標), Wyeth)が含まれる。
【0019】
トラスツズマブ−MCC−DM1と組み合わされる化学療法剤としての治療的能力を有するヒト化モノクローナル抗体には、アレムツズマブ、アポリズマブ、アセリズマブ、アトリズマブ、バピネオズマブ、ベバシズマブ、ビバツズマブ・メルタンシン、カンツズマブ・メルタンシン、セデリズマブ、セルトリズマブ・ペゴール、シドフシツズマブ、シドツズマブ、ダクリズマブ、エクリズマブ、エファリズマブ、エピラツズマブ、エルリズマブ、フェルビズマブ、フォントリズマブ、ゲムツズマブ・オゾガマイシン、イノツズマブ・オゾガマイシン、イピリムマブ、ラベツズマブ、リンツズマブ、マツズマブ、メポリズマブ、モタビズマブ、モトビズマブ、ナタリズマブ、ニモツズマブ、ノロビズマブ、ヌマビズマブ、オクレリズマブ、オマリズマブ、パリビズマブ、パスコリズマブ、ペクフシツズマブ、ペクツズマブ、パーツズマブ、パキセリズマブ、ラリビズマブ、ラニビズマブ、レスリビズマブ、レスリズマブ、レシビズマブ、ロベリズマブ、ルプリズマブ、シブロツズマブ、シプリズマブ、ソンツズマブ、タカツズマブ・テトラキセタン、タドシズマブ、タリズマブ、テフィバズマブ、トシリズマブ、トラリズマブ、トラスツズマブ、ツコツズマブ・セルモロイキン、ツクシツズマブ、ウマビズマブ、ウルトキサズマブおよびビシリズマブが含まれる。
【0020】
「代謝産物」は、特定の化合物又は塩の体内の代謝によって生産される生成物である。化合物の代謝産物は当分野で公知の慣例技術を使用して同定されてもよく、それらの活性は本明細書中に記載のものなどの試験を用いて決定されてもよい。このような生成物は、例えば、投与される化合物の酸化、還元、加水分解、アミド化、アミド分解、エステル化、エステル分解、酵素の切断などから生じうる。したがって、本発明は、代謝産物が得られるために十分な時間、本発明の化合物を哺乳動物と接触させることを含む方法によって産生される化合物を含む、本発明の化合物の代謝産物を含む。
【0021】
「パッケージ挿入物」という用語は、効能、用途、服用量、投与、配合禁忌及び/又はその治療薬の用途に関する警告についての情報を含む、治療薬の商業的包装を慣習的に含めた指示書を指すために用いられる。
【0022】
本明細書中で用いる「薬学的に許容可能な塩」なる表現は、本発明の化合物の薬学的に許容可能な有機塩又は無機塩を指す。例示的な塩には、限定するものではないが、硫酸塩、クエン酸塩、酢酸塩、シュウ酸塩、塩化物、臭化物、ヨウ素、硝酸塩、重硫酸塩、リン酸塩、酸性リン酸塩、イソニコチン酸塩、乳酸塩、サリチル酸塩、酸性クエン酸塩、酒石酸塩、オレイン酸塩、タンニン酸塩、パントテン酸塩、酒石酸水素塩、アスコルビン酸塩、琥珀酸塩、マレイン酸塩、gentisinate、フマル酸塩、グルコン酸塩、グロクロン酸塩、糖酸塩、蟻酸塩、安息香酸塩、グルタミン酸塩、メタンスルホン酸塩「メシレート」、エタンスルホン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩、及びパモ酸塩(すなわち、1,1'-メチレンビス(2-ヒドロキシ-3-ナフトエート))が含まれる。薬学的に許容可能な塩は、他の分子、例えば酢酸イオン、琥珀酸イオン又は他の対イオンの包含を伴ってもよい。対イオンは、親化合物上の荷電を安定化する任意の有機又は無機の部分でもよい。さらに、薬学的に許容可能な塩は、その構造内に複数の荷電原子を有してもよい。複数の荷電原子が薬学的に許容可能な塩の一部である場合は複数の対イオンを有しうる。したがって、薬学的に許容可能な塩は、一又は複数の荷電原子及び/又は一又は複数の対イオンを有してもよい。
【0023】
本発明の化合物が塩基性である場合、当分野で有用な任意の好適な方法、例えば塩酸、臭化水素酸、硫酸、硝酸、メタンスルホン酸、リン酸などといった無機酸による遊離塩基の処理、又は酢酸、マレイン酸、コハク酸、マンデル酸、フマル酸、マロン酸、ピルビン酸、シュウ酸、グリコール酸、サリチル酸、ピラノシジル酸、例としてグルクロン酸又はガラクツロン酸、αヒドロキシ酸、例えばクエン酸又は酒石酸、アミノ酸、例えばアスパラギン酸又はグルタミン酸、芳香族酸、例えば安息香酸又はニッケイ酸、スルホン酸、例えばp-トルエンスルホン酸又はエタンスルホン酸などといった有機酸による遊離塩基の処理によって調製されてもよい。一般に塩基性薬学化合物からの薬学的に有用又は許容可能な塩の形成に適すると考えられる酸は、例えばP. Stahl et al, Camille G. (eds.) Handbook of Pharmaceutical Salts. Properties, Selection and Use. (2002) Zurich: Wiley-VCH;S. Berge et al, Journal of Pharmaceutical Sciences (1977) 66(1) 1 19;P. Gould, International J. of Pharmaceutics (1986) 33 201 217;Anderson et al, The Practice of Medicinal Chemistry (1996), Academic Press, New York; Remington’s Pharmaceutical Sciences, 18th ed., (1995) Mack Publishing Co., Easton PA;及び、The Orange Book (Food & Drug Administration, Washington, D.C. ウェブサイト上)で論じられている。これらの開示内容は出典明記によって本明細書中に援用される。
【0024】
本発明の化合物が酸である場合、所望の薬学的に許容可能な塩は、任意の好適な方法、例えば、アミン(一次、二次又は三次)、アルカリ金属水酸化物又はアルカリ土類金属水酸化物などといった無機塩基又は有機塩基による遊離酸の処理によって調製されてもよい。好適な塩の例示的な例には、限定するものではないが、アミノ酸、例えばグリシン及びアルギニン、アンモニア、一次、二次、三次のアミン、及び環状アミン、例えばピペリジン、モルホリン及びピペラジンから得られる有機塩、並びにナトリウム、カルシウム、カリウム、マグネシウム、マンガン、鉄、銅、亜鉛、アルミニウム及びリチウムから得られる無機塩が含まれる。
【0025】
「薬学的に許容可能な」という表現は、物質又は組成物が製剤を含む他の成分と、及び/又は処置される哺乳動物と化学的及び/又は毒物学的に適合性がなければならないことを表す。
「溶媒和化合物」は、一又は複数の溶媒分子と本発明の化合物との物理的結合又は複合体を指す。本発明の化合物は溶媒和されていない状態及び溶媒和された形態で存在しうる。溶媒和化合物を形成する溶媒の例には、限定するものではないが、水、イソプロパノール、エタノール、メタノール、DMSO、酢酸エチル、酢酸及びエタノールアミンが含まれる。「水和物」なる用語は溶媒分子が水である場合の複合体を指す。この物理的な会合は、水素結合を含む、様々な程度のイオン的及び共有結合的結合を伴う。場合によって、例えば一又は複数の溶媒分子が結晶質固体の結晶格子に取り込まれている場合、溶媒和化合物は単離可能であろう。溶媒和化合物の調製は、例えばM. Caira et al, J. Pharmaceutical Sci., 93(3), 601 611 (2004)のように、一般に公知である。溶媒和化合物、ヘミ溶媒和化合物、水和物などの類似の調製は、E. C. van Tonder et al, AAPS PharmSciTech., 5(1), article 12 (2004);及び、A. L. Bingham et al, Chem. Commun., 603 604 (2001)によって記述される。 典型的な限定的でない方法は、室温より高い温度の所望の量の所望の溶媒(有機物又は水又はその混合物)に本発明の化合物を溶解することと、後に標準的な方法によって単離される結晶を形成するために十分な速度で溶液を冷やすことを伴う。例えばI.R.分光法といった分析技術は、溶媒和化合物(または水和物)としての結晶中の溶媒(または水)の存在を示す。
【0026】
本明細書中で用いる「相乗効果」なる用語は、2以上の単一の薬剤の相加効果よりも効果が高い治療的併用を指す。トラスツズマブ−MCC−DM1と一又は複数の化学療法剤との間の相乗作用の決定は、本明細書において記述されるアッセイから得られる結果に基づいてよい。これらのアッセイの結果は、併用インデックス「CI」を得るために、CalcuSynソフトウェアによる用量−効果分析とChou&Talalay組合せ法を用いて分析される(Chou and Talalay (1984) Adv. Enzyme Regul. 22:27-55)。本発明によって提供される組合せは、いくつかのアッセイシステムで評価され、データは、抗癌薬間の相乗作用、相加作用および拮抗作用を定量化するために、標準的なプログラムを利用して分析されうる。好適に利用されるプログラムは、"New Avenues in Developmental Cancer Chemotherapy," Academic Press, 1987, Chapter 2のChou and Talalayによって記述されるものである。0.8未満の併用インデックス(CI)値は相乗効果を示し、1.2を超える値は拮抗作用を示し、0.8〜1.2の値は相加効果を示す。併用療法は「相乗効果」を示してよく、「相乗的」である、つまり、共に用いる活性成分が別々の化合物を用いて得られる効果の合計よりも大きい場合に得られる効果である。相乗効果は、活性成分が:(1)同時に処方され、併用された単位投薬製剤として同時に投与又は送達されるか、(2)別個の製剤として交互に送達されるか、又は(3)ある種の他の計画による場合に、達成することができる。交互療法で送達される場合、相乗効果は、化合物が、例えば別々のシリンジで異なった注射によって逐次的に投与されるか又は送達される場合に達成されうる。一般に、交互療法の間、各活性成分の有効用量が逐次的、つまり連続的に投与される。
【0027】
(トラスツズマブ−MCC−DM1)
本発明は、以下の構造を有する、トラスツズマブ−MCC−DM1(T−DM1)、抗体−薬剤コンジュゲート(CAS Reg. 番号139504-50-0)を含有する治療的併用を含む。

ここで、Trは、リンカー部分MCCによってメイタンシノイド薬剤部分DM1に連結したトラスツズマブ(米国特許第5208020号;米国特許第6441163号)である。抗体に対する薬剤比率又は薬剤負荷は、トラスツズマブ−MCC−DM1の上記の構造のpによって表され、1からおよそ8の整数値である。薬剤負荷値pは1〜8である。トラスツズマブ-MCC-DM1は、1、2、3、4、5、6、7及び8の薬剤部分が抗体トラスツズマブに共有結合して付着している様々な負荷及び付着の抗体−薬剤コンジュゲートの混合すべてを含む(米国特許第7097840号;米国公開特許2005/0276812;米国公開特許2005/0166993)。トラスツズマブ-MCC-DM1は実施例1に従って調製されてよい。
【0028】
トラスツズマブは、哺乳動物細胞(チャイニーズハムスター卵巣、CHO)懸濁培養物によって生産される。HER2(又はc-erbB2)プロトオンコジーンは185kDaの膜貫通レセプタータンパク質をコードし、構造的に上皮性増殖因子レセプターに関連がある。HER2タンパク質過剰発現は、原発性乳癌の25%〜30%に観察され、固定腫瘍塊の免疫組織化学ベースの評価を使用して決定されてよい(Press MF, et al (1993) Cancer Res 53:4960-70。トラスツズマブは、マウス4D5抗体の、又はマウス4D5抗体由来の抗原結合残基を有する抗体である(ATCC CRL 10463、ブダペスト条約の下、1990年5月24日に米国培養細胞系統保存機関、12301 Parklawn Drive, Rockville, Md. 20852に寄託されている)。例示的なヒト化4D5抗体には、huMAb4D5-1、huMAb4D5-2、huMAb4D5-3、huMAb4D5-4、huMAb4D5-5、huMAb4D5-6、huMAb4D5-7、及び米国特許第5821337号に記載のhuMAb4D5-8(HERCEPTIN(登録商標))が含まれる。
第I相試験では、3週ごとのIV注入によって投与されるT−DM1の最大耐量(MTD)は、3.6mg/kgであった。DLT(用量制限毒性)は、4.8mg/kgで処置された3人の患者のうちの2人にグレード4の血小板減少からなった。3.6mg/kgでの2以上の関連グレードの有害事象は、まれであり、対処可能であった。この治療計画は十分に許容されるものであり、前記のような有意な臨床活性と関係していた。第II相試験は、3週間ごとに投与される3.6mg/kgの服用レベルで類似の耐容性を有し、用量の減少を必要とする患者の割合は極僅かであった(112人の患者のうちの3人)。ゆえに、1)3週間ごとの3.6mg/kgのT−DM1に示される有効性及び安全性、及び2)この患者集団に対する3週間投薬計画の利便性に基づいて、この試験で試験するために3週間ごとに投与される3.6mg/kgのT−DM1用量を選択した。
【0029】
(化学療法剤)
特定の化学療法剤は、インビトロ及びインビボの細胞増殖の阻害において、トラスツズマブ-MCC-DM1と組み合わせると、驚くべき予想外の性質を示した。このような化学療法剤には、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941、及びGNE-390が含まれる。
パーツズマブ(CA Reg.番号380610-27-5、OMNITARG(登録商標)、2C4、Genentech)は、HER2の二量体化を阻害する組換えヒト化モノクローナル抗体である(米国特許第6054297号;米国特許第6407213号;米国特許第6800738号;米国特許第6627196号、米国特許第6949245号;米国特許第7041292号)。パーツズマブおよびトラスツズマブは、HER-2チロシンキナーゼレセプターの異なる細胞外領域を標的とする(Nahta et al (2004) Cancer Res. 64: 2343-2346)。2C4(パーツズマブ)を発現するハイブリドーマ細胞株は、1999年4月8日に、ATCC HB-12697として、米国培養細胞系統保存機関(ATCC)、10801 University Boulevard, Manassas, Va. 20110-2209, USAに寄託された。パーツズマブは、他のHERレセプターファミリ、すなわちHER1/EGFR、HER3およびHER4と協働するHER2レセプターの能力を遮断する(Agus et al (2002) Cancer Cell 2:127-37;Jackson et al (2004) Cancer Res 64:2601-9;Takai et al (2005) Cancer 104:2701-8;米国特許第6949245号)。癌細胞において、他のHERファミリレセプターと協働するHER2の能力が干渉されると、細胞シグナル伝達が遮断され、最後には癌細胞増殖の阻害および癌細胞の死に至りうる。HDIは、その作用様式が特有であるために、HER2を過剰発現しないものを含む様々な腫瘍において作用する能力を有する(Mullen et al (2007) Molecular Cancer Therapeutics 6:93-100)。
【0030】
パーツズマブは、ヒトIgG1(κ)フレームワーク配列に基づく。これは2つの重鎖と2つの軽鎖からなる。トラスツズマブのように、パーツズマブはHER2の細胞外ドメインに対するものである。しかしながら、パーツズマブは、トラスツズマブとは軽鎖および重鎖のエピトープ結合領域が異なる。結果として、パーツズマブは、HER2のサブドメイン2として知られるものの中のエピトープに結合するのに対して、トラスツズマブのエピトープは、サブドメイン4に位置する(Cho et al. 2003; Franklin et al. 2004)。パーツズマブは、HER1(上皮性増殖因子レセプター;EGFR)、HER3およびHER4を含む他のHERファミリーメンバーとHER2との会合を遮断することによって作用する。この会合は、MAPキナーゼおよびPI3-キナーゼを経てリガンド存在下でのシグナル伝達に必要である。その結果、パーツズマブは、リガンドが惹起する細胞内シグナル伝達を阻害する。これらのシグナル伝達経路の阻害の結果、増殖休止およびアポトーシスがそれぞれ生じうる(Hanahan and Weinberg 2000)。パーツズマブおよびトラスツズマブがHER2レセプター上の異なるエピトープで結合するので、リガンドにより活性化される下流のシグナル伝達はトラスツズマブによって遮断されるが、パーツズマブによっては遮断されない。したがって、パーツズマブは、抗腫瘍剤としてその活性を発揮するために、HER2過剰発現を必要としない。さらに、それらが相補的に作用するために、パーツズマブとT−DM1の組合せは、HER2を過剰発現する疾患において役割を有する可能性がある。
【0031】
パーツズマブは、HER2を低レベルで発現するMBC、非小細胞肺癌、ホルモン抵抗性前立腺癌及び卵巣癌を含む様々な癌種において実施された5つの第II相試験において単一薬剤として評価された。第II相試験は、正常なHER2発現である転移性乳癌(MBC)患者の第二ライン又は第三ライン治療において、単一薬剤としてパーツズマブを評価した(Cortes et al. (2005) J. Clin. Oncol. 23:3068)。パーツズマブは、トラスツズマブと併用した2つの第II相試験において評価された(Baselga J, et al. "A Phase II trial of trastuzumab and pertuzumab in patients with HER2-positive metastatic breast cancer that had progressed during trastuzumab therapy: full response data", European Society of Medical Oncology, Stockholm, Sweden, September 12?16, 2008; Gelmon et al (2008) J. Clin. Oncol. 26:1026)。第一試験には、最大3つの従来のトラスツズマブを含む投薬計画を既に受けていたHER2陽性MBCを有する11人の患者が参加した(Portera et al. 2007)。
【0032】
ベバシズマブ(CAS Reg.番号216974-75-3、アバスチン(登録商標)、Genentech)は、癌の治療に用いられる、血管内皮性増殖因子に対する抗VEGFモノクローナル抗体であり(米国特許第7227004号;米国特許第6884879号;米国特許第7060269号;米国特許第7169901号;米国特許第7297334号)、新たな血管の形成を遮断することによって腫瘍増殖を阻害する。ベバシズマブは、転移性大腸癌及びほとんどの形式の転移性非小細胞肺癌の治療において標準的な化学療法と組み合わせて使用するために2004年FDAによって承認された、米国においては第一に臨床上有用な脈管形成阻害薬であった。アジュバント/非転移性大腸癌、転移性乳癌、転移性腎臓細胞カルチノーマ、転移性多形性神経膠芽腫、転移性卵巣癌、転移性ホルモン抵抗性前立腺癌、及び転移の転移性又は切除不能な局所進行性膵癌を有する患者のための安全性及び有効性を決定するために、いくつかの後期臨床試験が行われている。
【0033】
抗VEGF抗体は通常、VEGF-B又はVEGF-Cといった他のVEGFホモログにも、PlGF、PDGF又はbFGFといった他の増殖因子とも結合しない。好ましい抗VEGF抗体には、ハイブリドーマATCC HB 10709によって産生されるモノクローナル抗-VEGF抗体A4.6.1と同じエピトープに結合するモノクローナル抗体;ベバシズマブを含むがこれに限らず、Presta et al. (1997) Cancer Res. 57: 4593-4599に従って生成される組み換えヒト化抗VEGFモノクローナル抗体が含まれる。ベバシズマブは、ヒトVEGFとそのレセプターの結合を遮断するマウスの抗hVEGFモノクローナル抗体A.4.6.1由来の、変異したヒトIgG1フレームワーク領域および抗原結合性相補性決定領域を含む。大部分のフレームワーク領域を含むベバシズマブのアミノ酸配列のおよそ93%はヒトIgG1由来であり、配列のおよそ7%はマウス抗体A4.6.1由来である。ベバシズマブは、およそ149000ダルトンの分子量を有し、グリコシル化されている。ベバシズマブおよび他のヒト化抗VEGF抗体は、米国特許第6884879号においてさらに記述される。その他の抗VEGF抗体には、国際公開2005/012359の図27−29の何れかに記載される、G6又はB20系列抗体(例えばG6-31、B20-4.1)が含まれる。一実施態様では、B20系列抗体は、残基F17、M18、D19、Y21、Y25、Q89、I91、K101、E103およびC104を含むヒトVEGF上の機能的エピトープに結合する。
A4.6.1(ATCC HB 10709)およびB2.6.2(ATCC HB 10710)抗VEGF発現ハイブリドーマ細胞株は、米国培養細胞系統保存機関(ATCC)、10801 University Boulevard, Manassas, VA 20110-2209 USAに寄託され、維持されている。DNA29101-1276として同定されたATCC寄託物のヌクレオチド配列挿入物によってコードされるVEGF-Eポリペプチドを発現するクローン(米国特許第6391311号)は1998年3月5日に寄託され、ATCC、10801 University Boulevard, Manassas, Va. 20110-2209, USAにてATCC209653として維持された。
【0034】
5−FU(フルオロウラシル、5‐フルオロウラシル、CAS Reg.番号51-21-8)は、チミジル酸合成酵素阻害薬であって、結腸直腸癌及び膵臓癌を含む癌の治療において数十年の間の使われている(米国特許第2802005号、米国特許第2885396号;Barton et al (1972) Jour. Org. Chem. 37:329;Hansen, R.M. (1991) Cancer Invest. 9:637-642)。5−FUは5−フルオロ−1H−ピリミジン−2,4−ジオンと命名され、以下の構造を有する:
【0035】
カルボプラチン(CAS Reg.番号41575-94-4)は、卵巣カルチノーマ、肺、頭頸部癌(米国特許第4140707号)に対して使用される化学療法剤である。カルボプラチンは、アザニド;シクロブタン-1,1-ジカルボン酸プラチナと命名され、以下の構造を有する:
【0036】
ラパチニブ(CAS Reg.番号388082-78-8、TYKERB(登録商標)、GW572016、Glaxo SmithKline)は、腫瘍がHER2(ErbB2)を過剰発現する進行性又は転移性の乳癌を有し、アンスラサイクリン、タキサンおよびトラスツズマブを含む従来の治療法を受けていた患者の治療のために、カペシタビン(XELODA(登録商標)、Roche)と併用した使用について承認されている。ラパチニブは、EGFR/HER2プロテインキナーゼドメインのATP-結合ポケットに結合することによってレセプターの自己リン酸化および活性化を阻害するATP競合的上皮性増殖因子(EGFR)であり、HER2/neu(ErbB-2)二重チロシンキナーゼ阻害因子である(米国特許第6727256号;米国特許第6713485号;米国特許第7109333号;米国特許第6933299号;米国特許第7084147号;米国特許第7157466号;米国特許第7141576号)。ラパチニブは、N-(3−クロロ−4−(3−フルオロベンジルオキシ)フェニル)-6-(5-((2-(メチルスルホニル)エチルアミノ)メチル)フラン-2-イル)キナゾリン-4-アミンとして命名され、以下の構造を有する:
【0037】
ABT-869(Abbott and Genentech)は、癌の経口治療候補のための、VEGFおよびPDGFファミリレセプターチロシンキナーゼの複数標的阻害因子である(米国特許第7297709号;米国公開特許2004/235892;米国公開特許2007/104780)。非小細胞肺癌(NSCLC)、肝細胞癌(HCC)および腎臓細胞カルチノーマ(RCC)を治療する臨床試験が開始された。ABT-869は、1-(4-(3-アミノ-1H-インダゾール-4-イル)フェニル)-3-(2-フルオロ-5-メチルフェニル)ウレア(CAS番号796967-16-3)と命名され、以下の構造を有する:
【0038】
ドセタキセル(タキソテール(登録商標)、Sanofi-Aventis)は、乳癌、卵巣癌及びNSCLC癌を治療するために用いられる(米国特許第4814470号;米国特許第5438072号;米国特許第5698582号;米国特許第5714512号;米国特許第5750561号)。ドセタキセルは、5, 20-エポキシ-1, 2, 4, 7, 10, 13-ヘキサヒドロキシタックス-11-en-9-one 4-アセテート 2-ベンゾエート, トリヒドレートを有する(2R,3S)-N-カルボキシ-3-フェニルイソセリン, N-tert-ブチルエステル, 13-エステル(米国特許第4814470号;欧州特許第253738号;CAS Reg.番号114977-28-5)と命名され、以下の構造を有する:
【0039】
GDC-0941(Genentech Inc.)、薬物動態的及び薬学的な性質が期待される、PI3Kの選択的、経口的な生物利用能があるチエノピリミジン阻害因子である(Folkes et al (2008) Jour. of Med. Chem. 51(18): 5522-5532;米国公開特許2008/0076768;米国公開特許2008/0207611;Belvin et al, American Association for Cancer Research Annual Meeting 2008, 99th: April 15, Abstract 4004;Folkes et al, American Association for Cancer Research Annual Meeting 2008, 99th: April 14, Abstract LB-146;Friedman et al, American Association for Cancer Research Annual Meeting 2008, 99th: April 14, Abstract LB-110)。GDC-0941は、固形腫瘍細胞株に対する特定の化学療法剤と組み合わせたときにインビトロ及びインビボで相乗的な活性を示す(米国特許出願番号第12/208227号、Belvin et al "Combinations Of Phosphoinositide 3-Kinase Inhibitor Compounds And Chemotherapeutic Agents, And Methods Of Use", filed 10 Sept 2008)。GDC-0941は、4-(2-(1H-インダゾール-4-イル)-6-((4-(メチルスルホニル)ピペラジン-1-イル)メチル)チエノ[3,2-d]ピリミジン-4-イル)モルホリン(CAS Reg.番号957054‐30‐7)として命名され、以下の構造を有する:
【0040】
GNE-390(Genentech Inc.)は、薬物動態的及び薬学的な性質が期待される、PI3Kの選択的、経口的な生物利用能があるチエノピリミジン阻害因子である(米国公開特許2008/0242665;国際公開2008/070740)。GNE-390は、固形腫瘍細胞株に対する特定の化学療法剤と組み合わせたときにインビトロ及びインビボで相乗的な活性を示す(米国特許出願番号第12/208227号、Belvin et al "Combinations Of Phosphoinositide 3-Kinase Inhibitor Compounds And Chemotherapeutic Agents, And Methods Of Use", filed 10 Sept 2008)。GNE-390は、(S)-1-(4-((2-(2-アミノピリミジン-5-イル)-7-メチル-4-モルホリノチエノ[3,2-d]ピリミジン-6-イル)メチル)ピペラジン-1-イル)-2-ヒドロキシプロパン-1-オンと命名され、以下の構造を有する:
【0041】
(生物学的評価)
異なる化学療法剤又は生物学的な標的化剤と組み合わせてトラスツズマブ-MCC DM1 T−DM1を用いたインビトロ細胞培養試験を、多くのHER2増幅細胞株において実行した。データはChou & Talalay法を用いて分析し、各薬剤について倍数のIC50を設定して、各組合せについての併用インデックス(CI)値を決定した。0.7未満のCI値は相乗効果を意味し、0.7〜1.3のCI値は相加効果を意味し、そして、1.3を超えるCI値は拮抗作用を意味する。化学療法剤との組合せでは、T−DM1をドセタキセル又は5−FUと併用すると相加的又は相乗的な増殖抑制作用が生じたのに対して、ゲムシタビン又はカルボプラチンいずれかとの組合せは全く効果がないか、T-DM1と拮抗的であった。マウス異種移植片試験は、T−DM1をドセタキセル又は5−FUと併用すると個々の薬剤による治療と比べて目覚ましく腫瘍抑制作用が亢進されたという同じ結果を示した。T-DM1をカルボプラチンと併用するといずれかの薬剤単独の場合と比較して効果が亢進したのに対して、T−DM1とゲムシタビンとの組合せはT−DM1単独の場合より効果が低かった。細胞培養試験において、T-DM1をパーツズマブ、ラパチニブ又はGDC−0941のいずれかと組み合わせると相加的又は相乗的な増殖抑制作用が生じ、個々の薬剤によって治療した場合と比較してインビボでの腫瘍抑制作用が著しく亢進した。対照的に、コンジュゲートしていないトラスツズマブは、HER2上の同じエピトープの結合によってT−DM1の活性を中和した。抗体B20-4.1又は小分子阻害因子ABT−869のような抗血管形成剤とT−DM1との組合せを用いたインビボ試験により、B20-4.1と共に投与した最高用量のT−DM1(10又は15mg/kg)を除いて、試験したすべての組合せにより腫瘍抑制作用が亢進した。
【0042】
多数の抗癌剤とトラスツズマブ-MCC-DM1(T-DM1)との組合せは、HER2を過剰発現する胸部腫瘍細胞におけるインビトロ増殖抑制作用および乳癌異種移植片モデルにおけるインビボ腫瘍抑制有効性を測定することによって調べた。これらの試験において、トラスツズマブ−MCC−DM1を、細胞障害性化学療法剤、抗体又は小分子キナーゼ阻害因子のいずれかに加えた。
乳癌マウス異種移植片モデルにおける抗VEGFマウス抗体B20-4.1(Liang et al (2006) Jour. Biol. Chem. 281:951-961)、ベバシズマブ代用物とトラスツズマブ−MCC−DM1との組合せにより、B20-4.1単独より大きな腫瘍抑制作用が生じた。これらの試験結果は、HER2陽性乳癌における異なる抗腫瘍療法と組み合わせたトラスツズマブ−MCC−DM1を含む治療投薬計画の将来の臨床評価に、相乗効果および論理的根拠を予測する理由となる。
【0043】
トラスツズマブ-DM1とラパチニブ、又はHER2抗体パーツズマブ(HER2二量体化阻害薬)と組み合わせたトラスツズマブ-DM1といったHER2標的化剤の組合せにより、相乗的薬剤効果が観察された。
カルボプラチン又は5−FUと併用したトラスツズマブ-MCC-DM1は、それぞれの薬剤単独による処置と比較して活性の亢進を示したのに対して、ゲムシタビンによる併用処置では腫瘍抑制作用の増加は生じなかった。
p110アイソフォームの小分子キナーゼパン阻害因子であるGDC−0941によってPI3キナーゼ経路を遮断すると、トラスツズマブ-DM1の活性が亢進した。
T-DM1をPI3K阻害因子GDC−0941と併用すると、変異したPI3Kを有するHER2増幅乳癌株:BT-474(K111N)、MDA−361.1(E545K)およびKPL4(H1047R)において抗腫瘍活性が亢進した。インビトロでの併用処置により細胞増殖の相加的又は相乗的な阻害、並びにアポトーシスの増加が生じた。同様に、MDA−MB−361.1およびFo5 HER2増幅異種移植片モデルでは、単独薬剤活性と比較して併用薬剤処置によりインビボ有効性が増加した。各薬剤のためのバイオマーカーの生化学的分析により、T−DM1およびGDC−0941によってホスホ-Aktおよびホスホ-ERKを阻害すると、GDC−0941によるRbおよびPRAS40のリン酸化が低減し、T−DM1による処置後の有糸分裂マーカーホスホ-ヒストンH3およびサイクリンB1のレベルが増加したことが示された。さらに、T−DM1処置によりこれら乳癌モデルでのアポトーシスが生じ、これは、23kDaのPARP-切断断片の出現、Bcl-XLレベルの減少、並びにカスパーゼ3及び7の活性化によって決定される。T−DM1へのGDC−0941の添加によりアポトーシス誘導がさらに亢進した。これらの試験により、HER2増幅乳癌での薬剤併用使用の合理性が裏付けられ、そして、トラスツズマブ又はラパチニブベースの治療法を進ませる疾患を有する患者のための新たな治療手段が提供される。
【0044】
(インビトロ細胞増殖アッセイ)
トラスツズマブ−MCC−DM1の化学療法剤との組合せのインビトロ有効性は、実施例2の細胞増殖アッセイ;Promega Corp., Madison, WIから市販されている発光細胞生存アッセイであるCellTiter-Glo(登録商標)によって測定した。この均質なアッセイ方法は、甲虫類ルシフェラーゼの組換え発現に基づき(米国特許第5583024号;米国特許第5674713号;米国特許第5700670号)、代謝的に活性な細胞の指標である、存在するATPの定量化に基づいて、培養物中の生細胞の数を決定する(Crouch et al (1993) J. Immunol. Meth. 160:81-88;米国特許第6602677号)。CellTiter-Glo(登録商標)アッセイは、96か384のウェルフォーマットにて実行し、自動化ハイスループットスクリーニング(HTS)に準じた(Cree et al (1995) AntiCancer Drugs 6:398-404)。均一なアッセイ手順は、血清添加培地中で培養した細胞に単一試薬(CellTiter-Glo(登録商標)試薬)を直接添加することを含む。細胞洗浄、培地の除去および複数のピペッティング工程は必要ない。システムは、試薬の添加及び混合の10分後に384-ウェルフォーマット中僅か15細胞/ウェルを検出した。
【0045】
均質な「添加混合測定(add-mix-measure)」フォーマットにより細胞の溶解と存在するATPの量と比例した発光シグナルの生成が生じる。ATPの量は培養物中に存在する細胞の数に正比例する。CellTiter-Glo(登録商標)アッセイは、用いた細胞の種類及び培地に応じて、ルシフェラーゼ反応によって生成される「増殖タイプ(glow-type)」の発光シグナルを生成する。このシグナルは一般に5時間を超える半減期を有する。生細胞は相対的な発光単位(RLU)に反映される。基質であるカブトムシルシフェリンは、組み換えホタルルシフェラーゼによって酸化的に脱炭酸され、ATPがAMPに付随して転換され、光子が生成する。半減期の延長により試薬の注入を行う必要がなくなり、複数のプレートの連続的な又はバッチモード処理に柔軟性が生じる。この細胞増殖アッセイは、様々なマルチウェルフォーマット、例えば96又は384のウェルフォーマットと共に用いられる。データは、ルミノメーター又はCCDカメラ撮像装置によって記録されてよい。発光出力は、時間と共に測定される相対的な光単位(RLU)として表される。
【0046】
トラスツズマブ−MCC−DM1および化学療法剤との組合せの増殖抑制作用は、図1〜9および18〜33の腫瘍細胞株に対するCellTiter-Glo(登録商標)アッセイ(実施例2)によって測定した。
例示的実施態様には、癌の治療のために組み合わせて用いられる化合物の同定方法であって、a) インビトロ腫瘍細胞株にトラスツズマブ−MCC−DM1(T-DM1)および化学療法剤の治療的組合せを投与すること、及びb) 相乗的又は非相乗的な効果を測定することを含む方法が含まれる。1.3を超える併用指標(CI)値は拮抗作用を意味し、0.7〜1.3のCI値は加算効果を意味し、0.7未満のCI値は相乗的薬剤相互作用を意味する。
【0047】
図1は、トラスツズマブに感受性があるSK−BR−3細胞における倍数の個々のIC50値(表1)での様々な濃度のトラスツズマブ−MCC−DM1(T-DM1)と併用したトラスツズマブの拮抗作用を示す。生存可能な細胞の数をIC50倍数値に対してプロットする。各組合せについてIC10からIC90に対する併用指標(CI)は2を超えており、これはインビトロでの拮抗作用を示す。しかしながら、インビボのT−DM1+トラスツズマブの組合せは拮抗作用を示さない。
表1 SK-BR-3増殖−3日間
【0048】
図2は、トラスツズマブ耐性であるBT-474EEI細胞における倍数の個々のIC50値(表2)での様々な濃度のトラスツズマブ−MCC−DM1(T-DM1)と併用したトラスツズマブの拮抗作用を示す。生存可能な細胞の数をIC50倍数値に対してプロットする。各組合せについてIC10からIC90に対する併用指標(CI)は2を超えており、これは拮抗作用を示す。
表2 BT-474-EEI増殖−3日間
【0049】
図3は、MDA−MB−175細胞における倍数の個々のIC50値(表3)での様々な濃度のトラスツズマブ−MCC−DM1(T-DM1)と併用したパーツズマブの拮抗作用を示す。生存可能な細胞の数をIC50倍数値に対してプロットする。各組合せについてIC10からIC90に対する併用指標(CI)は1未満であり、平均CI=0.387であり、これは相乗作用を示す(表3)。
表3 MDA-MB-175増殖−5日間
【0050】
図3aは、パーツズマブ、トラスツズマブ−MCC−DM1(T-DM1)およびパーツズマブとT−DM1の併用のIC50倍数濃度に対する、5日のMDA−MB−175インビトロ細胞生存度のプロット線を示す。生存可能な細胞の数をIC50倍数値に対してプロットする。各組合せについてIC10からIC90に対する併用指標(CI)は1未満であり、平均CI=0.096であり、これは相乗作用を示す(表3a)。
表3a MDA-MB-175増殖−5日間
【0051】
図4は、用量反応のトラスツズマブ−MCC−DM1(T-DM1)と併用した様々な一定用量のパーツズマブ、および様々な用量のT−DM1単独に対する、5日のBT-474インビトロ細胞生存度のプロット線を示す。図4は、様々な用量のパーツズマブと組み合わせた一定用量のT−DM1の効果を示す。T−DM1にパーツズマブを加えると、T−DM1単独よりも僅かに大きな増殖抑制活性が生じる。
図5は、用量反応のパーツズマブと併用した様々な一定用量のトラスツズマブ−MCC−DM1(T-DM1)、および様々な用量のパーツズマブ単独に対する、5日のBT-474インビトロ細胞生存度のプロット線を示す。図5は、BT−474細胞増殖に対する様々な用量のT−DM1と併用した一定用量のパーツズマブの効果を示す。パーツズマブにT−DM1を添加するとパーツズマブ単独のの効果が亢進される。
【0052】
図6は、BT-474細胞における倍数の個々のIC50値(表4)での様々な濃度のトラスツズマブ−MCC−DM1(T-DM1)と併用したパーツズマブの相乗効果を示す。生存可能な細胞の数をIC50倍数値に対してプロットする。IC10からIC90の併用指標(CI)値は0.198から1.328である。この範囲の平均CIは0.658であり相乗効果を示す。
表4 BT-474増殖−5日間
【0053】
図7は、一定用量のラパチニブと併用した変更用量のT−DM1(4.5nM、14nM、41nM、123nM)、及び変更用量のT−DM1単独(0〜1000ng/ml)に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。T−DM1にラパチニブを添加すると、T−DM1単独よりも僅かに大きな増殖抑制作用が生じる。
図7aは、表7aに示すように、T−DM1、ラパチニブ、およびT−DM1とラパチニブの一定用量比併用に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。IC10からIC90の平均CI値は0.793であり、これは相加作用を示す。
表7a SK-BR-3増殖−3日間
【0054】
図8aは、表8aに示すように、T−DM1、ラパチニブおよびT−DM1とラパチニブの一定用量比併用に対する、3日のBT-474インビトロ細胞生存度のプロット線を示す。IC10からIC90の平均CI値は0.403であり、これは相乗作用を示す。
表8a BT−474増殖−3日間
【0055】
図8は、一定用量のラパチニブと併用した変更用量のT−DM1(1.5nM、4.5nM、14nM、41nM、123nM)、及び変更用量のT−DM1単独(0〜1000ng/ml)に対する、3日のBT−474インビトロ細胞生存度のプロット線を示す。T−DM1へラパチニブを添加すると、いずれかの薬剤単独と比較して大きな増殖抑制作用が生じる。
図9は、一定用量のラパチニブと併用した変更用量のT−DM1(14nM、41nM、123nM、370nM、1111nM)、及び変更用量のT−DM1単独(0〜1000ng/ml)に対する、3日のBT−474−EEIインビトロ細胞生存度のプロット線を示す。T−DM1へラパチニブを添加すると、いずれかの薬剤単独と比較して大きな増殖抑制作用が生じる。
【0056】
図18は、5−FU、トラスツズマブ−MCC−DM1(T-DM1)および5−FUとT−DM1の一定用量比組合せ(表18)のIC50倍数の濃度に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。5−FUとT−DM1の併用はSK−BR−3細胞に対して相加作用を有し、IC10からIC90の平均CIは0.952である。
表18 5−FU+T−DM1:SK-BR-3増殖−3日間
【0057】
図19は、5−FU、トラスツズマブ−MCC−DM1(T-DM1)および5−FUとT−DM1の一定用量比組合せ(表19)のIC50倍数濃度に対する、3日のBT−474インビトロ細胞生存度のプロット線を示す。5−FUとT−DM1の併用はBT-474細胞に対して相乗作用を有し、平均CI値は0.623である。
表19 5−FU+T−DM1:BT-474増殖−3日間
【0058】
図20は、ゲムシタビン、トラスツズマブ−MCC−DM1(T-DM1)およびゲムシタビンとT−DM1の一定用量比組合せ(表20)のIC50倍数濃度に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。ゲムシタビンをT−DM1と併用すると拮抗的な薬剤相互作用が生じ、試験したすべての組合せでCI値は1.3を上回る。
表20 ゲムシタビン(GEM)+T−DM1:SK-BR-3増殖−3日間
【0059】
図21は、ゲムシタビン、トラスツズマブ−MCC−DM1(T-DM1)およびゲムシタビンとT−DM1の一定用量比組合せ(表21)のIC50倍数濃度に対する、3日のMDA−MD−361インビトロ細胞生存度のプロット線を示す。薬剤併用により平均CI=1.706の拮抗作用が生じる。
表21 ゲムシタビン(GEM)+T−DM1:MDA-MD-361増殖−3日間
【0060】
図22は、0.25×〜4×のIC50倍数濃度でのT−DM1、GDC−0941、およびT−DM1(6.25〜100ng/ml)とGDC−0941(62.5nM〜1μM)の一定用量比の組合せによる処置後3日の、KPL4インビトロ細胞生存度(増殖)のプロット線を示す。表22は、算出したCI値と1.111の平均CIと共に、10〜90%の阻害範囲における効果を示す。
相加作用のブリス予測は図22において点線で表す。ブリス独立プロット線は、2つの単一化合物の組合せから算出した相加反応を示す。
表22 GDC−0941+T−DM1:KPL4増殖−3日間
【0061】
図23は、0.0625×〜16×のIC50倍数濃度でのT−DM1、GDC−0941、およびT−DM1(1.25〜80ng/ml)とGDC−0941(31.25nM〜2μM)の一定用量比の組合せによる処置後3日の、KPL4インビトロ細胞生存度(増殖)のプロット線を示す。相加作用のブリス予測を点線でプロットする。表23は、算出したCI値と0.802の平均CIと共に、10〜90%の阻害範囲における効果を示す。T−DM1とGDC−0941の併用はKPL4細胞株において相加作用を有する。表23 GDC-0941+T−DM1:KPL4増殖−3日間
【0062】
図24は、0から16×のIC50倍数濃度での、T−DM1、PI103、GDC−0941、およびT−DM1+PI103及びT−DM1+GDC−0941の一定用量比の組合せによる処置後、Her2増幅、HERCEPTIN(登録商標)耐性、PIK3CA(H1047R)変異のKPL-4細胞インビトロ細胞生存度(増殖)のプロット線を示す。表24は併用指標値を示す。この結果から、CI値が0.5から1の間であることからT−DM1とGDC−0941との間には中程度のインビトロ相乗作用が、そしてCI値が1に近かったことからT−DM−1とPI103との間には相加作用が示唆される。
表24 併用:KPL4増殖
【0063】
PI3K選択阻害因子であるPI103(Hayakawa et al (2007) Bioorg. Med. Chem. Lett. 17:2438-2442;Raynaud et al (2007) Cancer Res. 67: 5840-5850;Fan et al (2006) Cancer Cell 9:341-349;米国特許第6608053号)は以下の構造を有する:
【0064】
図25は、T−DM1、GDC−0941、および一定用量比のT−DM1とGDC−0941の組合せによる処置後24時間の、KPL4カスパーゼ3/7インビトロ細胞アポトーシス(プログラム細胞死)のプロット線を示す。T−DM1とGDC−0941の組合せにより、いずれかの薬剤単独と比較してアポトーシスが大きく亢進する。
図26は、T−DM1、GDC−0941、および一定用量比のT−DM1とGDC−0941の組合せによる処置後3日の、KPL4インビトロ細胞アポトーシス(プログラム細胞死)のプロット線を示す。T−DM1とGDC−0941の組合せにより、いずれかの薬剤単独と比較してアポトーシスが大きく亢進する。
【0065】
図27は、0.125×〜8×のIC50倍数濃度での、T−DM1、GDC−0941、およびT−DM1(3.125〜50ng/ml)とGDC−0941(62.5nM〜1μM)の一定用量比の組合せによる処置後3日の、MDA−MB−361インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。表27は、算出したCI値と0.888の平均CIと共に10〜90%の阻害範囲における効果を示す。T-DM1をGDC−0941と併用することにより、MDA−MB−361細胞において相加的な増殖抑制作用が生じ、平均CIは0.889である。
表27 GDC−0941+T−DM1:MDA-MB-361増殖−3日間
【0066】
図28は、0.125×〜8×のIC50倍数濃度での、T−DM1、GDC−0941、およびT−DM1(3.125〜100ng/ml)とGDC−0941(62.5nM〜2μM)の一定用量比の組合せによる処置後3日の、MDA−MB−361インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。表28は、算出したCI値と0.813の平均CIと共に10〜90%の阻害範囲における効果を示す。T-DM1をGDC−0941と併用することにより、MDA−MB−361細胞において相加的な増殖抑制作用が生じ、平均CIは0.813である。
表28 GDC-0941+T−DM1:MDA-MB-361増殖−3日間
【0067】
図29は、0.125×〜4×のIC50倍数濃度での、T−DM1、GDC−0941、およびT−DM1(3.125〜100ng/ml)とGDC−0941(31.25nM〜1μM)の一定用量比の組合せによる処置後3日の、BT−474インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。表29は、算出したCI値と1.2122の平均CIと共に10〜90%の阻害範囲における効果を示す。GDC-0941とT-DM1は、これらの用量比ではBT-474に対する併用効果を有しない。
表29 GDC-0941+T−DM1:BT-474増殖−3日間
【0068】
図30は、0.25×〜4×のIC50倍数濃度での、T−DM1、GDC−0941、およびT−DM1(6.25〜100ng/ml)とGDC−0941(62.5nM〜1μM)の一定用量比の組合せによる処置後3日の、BT-474インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。表30は、算出されたCI値及び0.997の平均CIと共に、10〜90%の阻害範囲における効果を示し、これは相加効果を示す。
表30 GDC-0941+T−DM1:BT-474増殖−3日間
【0069】
図31は、0〜16×のIC50倍数濃度での、T−DM1、PI103、GDC−0941、およびT−DM1+PI103及びT−DM1+GDC−0941の一定用量比の組合せによる処置後3日の、Her2増幅、非PI3K変異のAU565細胞のインビトロ細胞生存度(増殖)のプロット線を示す。表31は併用指標値を示す。これらの結果から、CI値は1を超えるのでT−DM−1とGDC−0941との間はインビトロ拮抗作用、そしてCI値が1に近いか又は僅かに1より大きいのでT−DM−1とPI103との間は相加作用又は僅かに拮抗作用が示唆される。
表31 併用:AU565増殖
【0070】
図32は、0〜16×のIC50倍数濃度での、T−DM1、PI103、GDC−0941、およびT−DM1+PI103及びT−DM1+GDC−0941の一定用量比の組合せによる処置後3日の、Her2増幅、PIK3CA(C420R)変異のEFM192A細胞のインビトロ細胞生存度(増殖)のプロット線を示す。表32は併用指標値を示す。これらの結果から、CI値が0.5〜1であるのでT−DM−1とGDC−0941との間には中程度のインビトロ相乗作用が、そして、CI値が0.5に近いのでT−DM−1とPI103との間には相乗作用が示唆される。
表32 併用:EFM192A増殖
【0071】
図33は、0〜16×のIC50倍数濃度での、T−DM1、PI103、GDC−0941、およびT−DM1+PI103及びT−DM1+GDC−0941の一定用量比の組合せによる処置後3日の、Her2増幅、ハーセプチン(登録商標)耐性、PIK3CA(H1047R)変異のHCC1954細胞インビトロ細胞生存度(増殖)のプロット線を示す。表33は併用指標値を示す。これらの結果から、CI値が1に近いのでT−DM−1とGDC−0941との間には相加作用又は僅かなインビトロ相乗作用が、そして、CI値が1未満であるのでT−DM−1とPI103との間には僅かな相乗作用が示唆される。
表33 併用:HCC1954増殖
【0072】
(インビボ腫瘍異種移植片有効性)
本発明の併用の有効性は、齧歯動物の癌細胞の同種異系移植片又は異種移植片を着床させて、腫瘍を併用にて処置することによってインビボで測定されてよい。細胞株、腫瘍細胞中の特定の突然変異の有無、トラスツズマブ−MCC−DM1と化学療法剤の投与、投薬計画及び他の要因に応じて、結果が変わりうることは予測される。被検体マウスを、薬剤(一又は複数)又はコントロール(ベヒクル)にて処置し、数週間以上モニターし、腫瘍倍加までの時間、ログ細胞障害及び腫瘍抑制を測定した(実施例3)。図10〜17および34〜37は、マウスにおける異種移植片腫瘍阻害によって、化学療法剤と組み合わせたトラスツズマブ−MCC−DM1の有効性を示す。
【0073】
図10は、以下を投与した後のSCIDベージュマウスの乳房脂肪パッドに接種したKPL-4腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ADCバッファ、(2) パーツズマブ15mg/kg、(3) T−DM1 0.3mg/kg、(4) T−DM1 1mg/kg、(5) T−DM1 3mg/kg、(6) パーツズマブ15mg/kg+T−DM1 0.3mg、(7) パーツズマブ15mg/kg+T−DM1 1mg/kg、(8)パーツズマブ15mg/kg+T−DM1 3mg/kg。ADCバッファを投与した動物(1)は0のPRおよび0のCRであった。15mg/kgでパーツズマブを投与した動物(2)は0のPRおよび0のCRであった。0.3mg/kgのT−DM1のみを投与した動物(3)は0のPRおよび0のCRであった。1mg/kgのT−DM1のみを投与した動物(4)は1のPRおよび0のCRであった。3mg/kgのT−DM1のみを投与した動物(5)は7のPRおよび0のCRであった。15mg/kgのパーツズマブと0.3mg/kgのT−DM1を投与した動物(6)は、5のPRおよび0のCRであった。15mg/kgのパーツズマブと1mg/kgのT−DM1を投与した動物(7)は、8のPRおよび0のCRであった。15mg/kgのパーツズマブと3mg/kgのT−DM1を投与した動物(8)は、8のPRおよび0のCRであった。パーツズマブとT−DM1の併用により、KPL4異種移植片においていずれかの薬剤単独よりも大きな抗腫瘍活性が生じる。
【0074】
図11は、以下を投与した後のSCIDベージュマウスの乳房脂肪パッドに接種したKPL-4腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ADCバッファ、(2) 5−FU 100mg/kg、(3) パーツズマブ40mg/kg、(4) B20-4.1、5mg/kg、(5) T−DM1、5mg/kg、(6) 5−FU、100mg/kg+T−DM1、5mg、(7) パーツズマブ40mg/kg+T−DM1、5mg/kg、(8) B20-4.1、5mg/kg+T−DM1、5mg/kg、(9) B20-4.1、5mg/kg+パーツズマブ、40mg/kg。試験終了時には、50mm3体積未満のすべて残存する腫瘍を組織学的に評価し、単一薬剤(5) T−DM1、5mg/kgの8試料、併用群(6) 5−FU、100mg/kg+T−DM1、5mgの5試料、および併用群(7) パーツズマブ、40mg/kg+T−DM1、5mg/kgの8試料は生きている腫瘍細胞の所見がなかったことが決定された。
【0075】
図12は、以下を投与した後のCRLnu/nuマウスの乳房脂肪パッドに接種したMMTV-HER2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル(ADCバッファ)、(2) B20-4.1、5mg/kg、(3) T−DM1、3mg/kg、(4) T−DM1、5mg/kg、(5) T−DM1、10mg/kg、(6) B20-4.1、5mg/kg+T−DM1 3mg/kg、(7) B20-4.1、5mg/kg+T−DM1 5mg/kg、(8) B20-4.1、5mg/kg+T−DM1、10mg/kg。T−DM1とB20-4.1との併用により、10mg/kgでなく、3および5mg/kgのT−DM1により腫瘍増殖阻害が亢進する。
【0076】
図13は、以下を投与した後のCRLnu/nuマウスの乳房脂肪パッドに接種したMMTV-HER2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル(ADCバッファ)、(2) T−DM1 10mg/kg、(3) 5−FU 100mg/kg、(4) ゲムシタビン、120mg/kg、(5)カルボプラチン、100mg/kg、(6) 5−FU、100mg/kg+T−DM1、10mg/kg、(7) ゲムシタビン、120mg/kg+T−DM1、10mg/kg、(8) カルボプラチン、100mg/kg+T−DM1、10mg/kg。T-DM1を、5−FU、カルボプラチン又はゲムシタビンのいずれかと併用すると、単一薬剤処置と比較して腫瘍抑制効果が亢進する。
【0077】
図14は、以下を投与した後のハーラン胸腺欠損ヌードマウスの乳房脂肪パッドに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍異種移植片における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル(PBSバッファ) iv、qwk ×4、(2) ラパチニブ101mg/kg、po、bid ×21、(3) パーツズマブ40mg/kg、iv、qwk ×4、(4) B20-4.1 5mg/kg、ip、2×/wk ×4、(5) T−DM1 15mg/kg、iv、最後までq3wk、(6) ラパチニブ101mg/kg、po、bid ×21+T−DM1 15mg/kg、iv、最後までq3wk、(7) パーツズマブ40mg/kg、iv、qwk ×4+T−DM1 15mg/kg、iv、最後までq3wk、(8) B20-4.1 5mg/kg、ip、2×/wk×4+T−DM1 15mg/kg、iv、最後までq3wk。
15mg/kg用量の単一薬剤T−DM1(5)は、15mg/kgのT−DM1と5mg/kgのB20-4.1の併用(8)とは有意には異ならない。この試験においてラパチニブとパーツズマブはベヒクルと異ならなかった。B20-4.1はベヒクルと比較して効果が増加する傾向を示した。T-DM1は単一薬剤としても効果的であった(p<0.01)。T−DM1のラパチニブとの併用は、ラパチニブ単独より有意に良好であったが(p<0.01)、T−DM1単独とは異ならなかった。T−DM1のパーツズマブとの併用は、パーツズマブ単独より有意に良好であったが(p<0.01)、T−DM1単独とは異ならなかった。T−DM1のB20-4.1との併用は、B20-4.1単独より有意に良好であったが(p<0.01)、T−DM1単独とは異ならなかった。
【0078】
図15は、以下を投与した後のハーラン胸腺欠損ヌードマウスの乳房脂肪パッドに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍異種移植片における経時的な平均腫瘍体積変化によるインビボ有効性のプロット線を示す:(1) ベヒクル(PBSバッファ) po、bid ×21、(2) T−DM1、7.5mg/kg、iv、qd ×1、(3) T−DM1、15mg/kg、iv、qd ×1、(4) ABT−869、5mg/kg、po、bid ×21、(5) ABT−869、15mg/kg、po、bid ×21、(6) T−DM1、7.5mg/kg、iv、qd ×1+ABT−869、5mg/kg、po、bid ×21、(7) T−DM1 7.5mg/kg、iv、qd ×1+ABT−869、15mg/kg、po、bid ×21、(8) T−DM1、15mg/kg、iv、qd ×1+ABT−869、5mg/kg、po、bid ×21、(9) T−DM1、15mg/kg、iv、qd ×1+ABT−869、15mg/kg、po、bid ×21。
T−DM1とABT−869、5mg/kgとの併用は 2の部分的応答を示したが(8)、単一薬剤ABT−869、5mg/kg(4)よりかなり効果的ではない。T−DM1とABT−869、15mg/kgとの併用(9)は、単一薬剤ABT−869、15mg/kg(5)よりわずかに効果的である。5mg/kgで投与したABT-869は、エンドポイントまでの時間までベヒクルより有意に良好であったが(p<0.01)、腫瘍倍加までの時間はベヒクルとは異ならなかった。15mg/kgで投与したABT-869と7.5又は15mg/kgで投与したT-DM1は、腫瘍倍加および腫瘍エンドポイントいずれの時間までもベヒクルよりも有意に良好であった(p<0.01)。7.5mg/kgのT−DM1と5mg/kgのABT−869の併用は、7.5mg/kgのT−DM1の単一薬剤と異ならなかった。5mg/kgのABT−869単一薬剤と比較して、7.5mg/kgのT−DM1+5mg/kgのABT−869の併用は、腫瘍倍加までの時間は有意に良好であったが(p<0.01)、エンドポイントまでの時間は異ならなかった。7.5mg/kgのT−DM1と15mg/kgのABT−869の併用は、いずれかの単一薬剤よりも有意に良好であった(p<0.01)。15mg/kgのT−DM1+5mg/kgのABT−869の併用は15mg/kgのT−DM1単一薬剤と異ならなかった。5mg/kgのABT-869単一薬剤と比較して、15mg/kgのT−DM1と5mg/kgのABT−869の併用は、エンドポイントまでの時間は異ならなかったが、腫瘍倍加までの時間は有意に異なった(p<0.01)。15mg/kgのT−DM1+15mg/kgのABT−869の併用は、15mg/kgのABT−869単独より有意に良好であり、腫瘍倍加までの時間は15mg/kgのT−DM1単独より良好であった(p<0.01)。15mg/kgのT−DM1と15mg/kgのT−DM1+15mg/kgのABT−869のエンドポイントまでの時間は異ならなかった。
【0079】
図16は、以下を投与した後のハーラン胸腺欠損ヌードマウスの乳房脂肪パッドに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍異種移植片における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル、iv、qwk ×3、(2) T−DM1、7.5mg/kg、iv、q3wk ×2、(3) T−DM1、15mg/kg、iv、q3wk ×2、(4)ドセタキセル、30mg/kg、iv、qwk ×3、(5) T−DM1、7.5mg/kg、iv、q3wk ×2+ドセタキセル、30mg/kg、iv、qwk ×3、(6) T−DM1、15mg/kg、iv、q3wk ×2+ドセタキセル、30mg/kg、iv、qwk ×3。
15mg/kgのT−DM1を投与した動物(3)は、6の部分応答(PR)および1の完全寛解(CR)となった。30mg/kgのドセタキセル単独を投与した動物(4)は2のPRであった。7.5mg/kgのT−DM1と30mg/kgのドセタキセルの併用を投与した動物(5)は10のPRであった。15mg/kgのT−DM1と30mg/kgのドセタキセルの併用を投与した動物(6)は、7のPRおよび3のCRの用量応答を示した。すべての単一薬剤群はベヒクル群とは有意に異なっていた(p<0.01)。7.5mg/kgのT−DM1+ドセタキセルの併用は、腫瘍倍加までの時間およびエンドポイントまでの時間のいずれも、いずれか単一薬剤よりも有意に良好であった(p<0.01)。7.5mg/kgのT−DM1群では客観的な応答はなく、ドセタキセル単一薬剤群では2の部分応答(PR)であった。7.5mg/kgのT−DM1とドセタキセルの併用により、9のPRおよび1の完全寛解(CR)が生じた。15mg/kgのT−DM1+ドセタキセルの併用は、腫瘍倍加までの時間およびエンドポイントまでの時間はいずれの単一薬剤よりも有意に良好であった(p<0.01)。単一薬剤15mg/kgのT−DM1処置により、5のPRおよび2のCRが生じた。15mg/kgのT−DM1+ドセタキセルの併用は、客観的な応答速度が7のPRおよび3のCRに増えた。この併用群のすべてのマウスは処置に対して客観的な応答を有した。
【0080】
図17は、以下を投与した後のハーラン胸腺欠損ヌードマウスの乳房脂肪パッドに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍異種移植片における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル、po、qd×21、(2) T−DM1、7.5mg/kg、iv、q3wk ×2、(3) T−DM1、15mg/kg、iv、q3wk ×2、(4) ラパチニブ、100mg/kg、po、bid ×21、(5) T−DM1、7.5mg/kg、iv、q3wk ×2+ラパチニブ、100mg/kg、po、bid ×21、(6) T−DM1、15mg/kg、iv、q3wk ×2+ラパチニブ、100mg/kg、po、bid ×21。
15mg/kgのT−DM1を投与した動物(3)は、6の部分応答(PR)および3の完全寛解(CR)であった。7.5mg/kgのT−DM1と100mg/kgのラパチニブの併用を投与した動物(5)は、4のPRおよび5のCRであった。15mg/kgのT−DM1と100mg/kgのラパチニブの併用を投与した動物(6)は、8のCRの用量応答を示した。すべての単一薬剤群は、腫瘍倍加までの時間およびエンドポイントまでの時間は共にベヒクルと有意に異なっていた(p<0.01)。ラパチニブと併用した7.5mg/kgで投与したT-DM1は、単一薬剤の7.5mg/kgのT−DM1又はラパチニブよりも有意に良好であった(p<0.01)。ラパチニブと併用して15mg/kgで投与したT-DM1は、ラパチニブ単一薬剤よりも有意に良好であった(p<0.01)。この併用は、単一薬剤の15mg/kgのT−DM1とは異ならなかった。
腫瘍倍加までの時間はカプラン-マイヤー統計学的分析によって2×Voとして測定した。腫瘍倍加までの時間と生存率分析は、ログ−ランク−p値によって定量化した。進行時間は、腫瘍体積が1000mm3に達する経過時間として測定し、腫瘍体積が1000mm3に達しない場合は生存時間として測定した。T-DM1をラパチニブと併用すると、単一薬剤処置と比較して腫瘍抑制作用が大きく亢進した。
【0081】
図34は、以下を投与した後のCRLnu/nuマウスに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル、po、qd×21、(2) T−DM1、10mg/kg、iv、q3wk、(3) 5−FU、100mg/kg、po、qwk ×2、(4) (5) T−DM1、5mg/kg、iv、q3wk+5−FU、100mg/kg、po、qwk ×2。ベヒクルを投与した動物は、0の部分応答(PR)および0の完全寛解(CR)であった。T-DM1を投与した動物は1のPRおよび0のCRであった。5−FUを投与した動物は0のPRおよび0のCRであった。T−DM1と5−FUの併用を投与した動物は、42日目の時点で、3のPRおよび0のCRであった。T−DM1と5−FUによる処置により、いずれかの薬剤単独と比較して腫瘍抑制作用が亢進した。
【0082】
図35は、以下を投与した後のCRLnu/nuマウスに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル、po、qd ×21、(2) T−DM1、5mg/kg、iv、q3wk、(3) GDC−0941、100mg/kg、po、bid×21、(4) GDC−0152、50mg/kg、po、qwk ×2、(5) T−DM1、5mg/kg、iv、q3wk+GDC−0941、100mg/kg、po、bid ×21、(6) T−DM1、5mg/kg、iv、q3wk+GDC−0152、50mg/kg、po、qwk ×2。T−DM1とGDC−0941による処置により単一薬剤処置と比較して腫瘍抑制作用が亢進するのに対して、T−DM1とGDC−0152の併用はT−DM1単独より効果が低かった。
GDC-0152はアポトーシスタンパク質の阻害因子であるカスパーゼの阻害因子である(Call et al (2008) The Lancet Oncology, 9(10): 1002-1011;Deveraux et al (1999) J Clin Immunol 19:388-398)。
【0083】
図36は、以下を投与した後のCRLnu/nuマウスに接種したMDA−MB−361.1乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル、po、qd ×21、(2) GDC−0941、25mg/kg、po、qd ×21、(3) GDC−0941、50mg/kg、po、qd ×21、(4) GDC−0941、100mg/kg、po、qd ×21、(5) T−DM1、3mg/kg、iv、q3wk、(6) T−DM1、10mg/kg、iv、q3wk、(7) GDC−0941、25mg/kg、po、qd ×21+T−DM1、3mg/kg、iv、q3wk、(8) GDC−0941、50mg/kg、po、qd ×21+T−DM1、3mg/kg、iv、q3wk、(9) GDC−0941、100mg/kg、po、qd ×21+T−DM1、3mg/kg、iv、q3wk、(10) GDC−0941、25mg/kg、po、qd ×21+T−DM1、10mg/kg、iv、q3wk、(11) GDC−0941、50mg/kg、po、qd ×21+T−DM1、10mg/kg、iv、q3wk、(12) GDC−0941、100mg/kg、po、qd ×21+T−DM1、10mg/kg、iv、q3wk。
ベヒクルを投与した動物(1)は、0の部分応答(PR)および0の完全寛解(CR)であった。25mg/kgのGDC−0941を投与した動物(2)は、0のPRおよび0のCRであった。50mg/kgのGDC−0941を投与した動物(3)は、1のPRおよび0のCRであった。100mg/kgのGDC−0941を投与した動物(4)は、0のPRおよび0のCRであった。3mg/kgのT−DM1のみを投与した動物(5)は、1(PR)および1(CR)であった。10mg/kgのT−DM1のみを投与した動物(6)は、8(PR)および1(CR)であった。3mg/kgのT−DM1と25mg/kgのGDC−0941の併用を投与した動物(7)は、5のPRおよび0のCRであった。3mg/kgのT−DM1と50mg/kgのGDC−0941の併用を投与した動物(8)は、3のPRおよび0のCRであった。3mg/kgのT−DM1と100mg/kgのGDC−0941の併用を投与した動物(9)は、3のPRおよび1のCRであった。10mg/kgのT−DM1と50mg/kgのGDC−0941の併用を投与した動物(10)は、9のPRおよび0のCRであった。10mg/kgのT−DM1と50mg/kgのGDC−0941の併用を投与した動物(11)は、7のPRおよび2のCRであった。10mg/kgのT−DM1と100mg/kgのGDC−0941の併用を投与した動物(12)は、9のPRおよび1のCRであった。
【0084】
図37は、以下を投与した後のCRLnu/nuマウスに接種したMDA−MB−361.1乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル[MCT(0.5%メチルセルロース/0.2%TWEEN80)+琥珀酸塩バッファ(100mMの琥珀酸ナトリウム、100mg/mlのトレハロース、0.1%のTWEEN80、pH5.0)]、po+IV、qd ×21及びqd、(2) GNE-390、1.0mg/kg、po、qd ×21、(3) GNE−390、2.5mg/kg、po、qd ×21、(4) T−DM1、3mg/kg、iv、qd、(5) GNE−390、1.0mg/kg、po、qd ×21+T−DM1、3mg/kg、iv、qd、(6) GNE−390、2.5mg/kg、po、qd×21+T−DM1、3mg/kg、iv、qd。
ベヒクルを投与した動物(1)は、0の部分応答(PR)および0の完全寛解(CR)であった。1.0mg/kgのGNE−390のみを投与した動物(2)は、0のPRおよび0のCRであった。2.5mg/kgのGNE−390のみを投与した動物(3)は、1のPRおよび0のCRであった。3mg/kgのT−DM1のみを投与した動物(5)は、1(PR)および1(CR)であった。3mg/kgのT−DM1のみを投与した動物(4)は、0のPRおよび0のCRであった。3mg/kgのT−DM1と25mg/kgのGNE−390の併用を投与した動物(5)は、3のPRおよび0のCRであった。3mg/kgのT−DM1と2.5mg/kgのGNE−390の併用を投与した動物(6)は、5のPRおよび1のCRであった。MDA−MB−361.1乳癌異種移植片モデルでは、GNE−390をT−DM1と併用すると、GNE−390又はT−DM1単独と比較して、部分応答及び完全寛解の腫瘍抑制応答数が有意に増加した。
【0085】
(薬学的組成物)
本発明の薬学的組成物又は製剤には、トラスツズマブ−MCC−DM1、化学療法剤、及び一又は複数の薬学的に許容可能な担体、流動促進剤、希釈剤又は賦形剤の組合せが含まれる。
本発明のトラスツズマブ−MCC−DM1と化学療法剤は、非溶媒和状態並びに水、エタノール等のような薬学的に許容可能な溶媒との溶媒和形態で存在し得、本発明は溶媒和形態と非溶媒和形態の双方を包含するものである。
【0086】
また、本発明のトラスツズマブ−MCC−DM1と化学療法剤は、異なった互変異性体形態で存在し得、そのようなあらゆる形態が本発明の範囲に包含される。「互変異性体」又は「互変異性体形態」なる用語は、低エネルギー障壁を介して相互転換可能な異なったエネルギーの構造異性体を意味する。例えば、プロトン互変異性体(屈光性互変異性体としても知られている)は、ケト-エノール及びイミン-エナミン異性化のような、プロトンの移動を介した相互転換を含む。原子価互変異性体は結合電子の幾らかの再編成による相互転換を含む。
【0087】
薬学的組成物は、トラスツズマブ−MCC−DM1及び、本明細書中に記載される他の薬剤の一覧から選択される化学療法剤を含む2以上(例えば2)の薬学的な活性剤、並びに任意の薬学的に不活性な賦形剤、希釈剤、担体又は流動促進剤からなる個々の用量単位とバルク組成物を包含する。バルク組成物と各個々の用量単位は、一定量の前述の薬学的な活性剤を含みうる。バルク組成物は、個々の用量単位に形成されていない材料である。例示的な用量単位は、錠剤、丸剤、カプセルなどといった経口用量単位である。同様に、本明細書中に記載される、本発明の薬学的組成物を投与することによる患者の治療方法もまた、バルク組成物及び個々の用量単位の投与を含むことが意図される。
【0088】
また、薬学的組成物は、一又は複数の原子が通常自然に見出される原子質量又は質量数とは異なる原子質量又は質量数を有する原子によって置き換えられるという点を別にすると、ここに記載されたものと同一である本発明の同位体標識化合物を包含する。任意の特定の原子又は特定された元素のあらゆる同位体が本発明の化合物とその使用の範囲内であると考えられる。本発明の化合物に導入されうる例示的な同位体は、水素、炭素、窒素、酸素、リン、硫黄、フッ素、塩素及びヨードの同位体、例えば2H、3H、11C、13C、14C、13N、15N、15O、17O、18O、32P、33P、35S、18F、36Cl、123I及び125Iを含む。本発明の同位体標識化合物(例えば3H及び14Cで標識されたもの)は化合物及び/又は基質組織分布アッセイにおいて有用である。トリチウム標識(3H)及び炭素-14(14C)同位体は、調製及び検出性の容易さのために有用である。更に、重水素(2H)のような重い同位体での置換により、より大なる代謝安定性から生じるある種の治療的利点(例えばインビボ半減期の増加又は投薬量要求の減少)が得られ、よってある状況下では好ましい場合がある。ポジトロン放出同位体、例えば15O、13N、11C及び18Fは基質レセプター占有性を試験するポジトロン放出断層撮影(PET)に有用である。本発明の同位体標識化合物は一般に以下のここでのスキーム及び/又は実施例に開示されたものと類似した手順に従って、非同位体標識試薬に同位体標識試薬を置き換えて調製することができる。
【0089】
ヒトを含む哺乳動物における過剰増殖性疾患の治療的処置(予防的処置を含む)のために治療的併用を使用するために、トラスツズマブ−MCC−DM1と化学療法剤を、標準的な製薬的実務に従って製剤化されてよい。本発明により、一又は複数の薬学的に許容可能な担体、流動促進剤、希釈剤又は賦形剤と共にトラスツズマブ−MCC−DM1を含有する薬学的組成物が提供される。
適切な担体、希釈剤及び賦形剤は当業者によく知られており、炭水化物、ワックス、水溶性及び/又は膨潤性ポリマー、親水性又は疎水性物質、ゼラチン、油、溶媒、水等のような材料が含まれる。使用される特定の担体、希釈剤又は賦形剤は、本発明の化合物が適用されている手段及び目的に依存する。溶媒は一般的には哺乳動物への投与が安全である(GRAS)と当業者により認識される溶媒に基づいて選択される。一般に、安全な溶媒は、非毒性水性溶媒、例えば水、及び水に可溶性又は混和性である他の非毒性溶媒である。適切な水性溶媒には、水、エタノール、プロピレングリコール、ポリエチレングリコール(例えば,PEG400,PEG300)等とその混合物が含まれる。製剤はまた一又は複数のバッファー、安定剤、界面活性剤、湿潤剤、潤滑剤、乳化剤、懸濁剤、保存料、抗酸化剤、不透明化剤、流動促進剤、加工助剤、着色料、甘味料、香料剤、香味料及び薬物(つまり、本発明の化合物又はその薬学的組成物)を上品に提供するためあるいは薬学的製品(つまり医薬)の製造を補助するための他の既知の添加剤を含みうる。
【0090】
製剤は一般的な溶解及び混合手順を使用して調製されうる。例えば、原体薬剤物質(つまり、本発明の化合物又は該化合物の安定化形態(例えばシクロデキストリン誘導体又は他の既知の複合体形成剤との複合体)を、上述の賦形剤の一又は複数の存在下で適切な溶媒に溶解させる。本発明の化合物は典型的には容易に制御できる薬剤の用量を提供し処方されたレジメンでの患者の服薬遵守を可能にするように薬学的投薬形態で製剤化される。
投与される薬学的組成物(又は製剤)は薬剤を投与するために使用される方法に応じて様々な形に包装されうる。一般に、流通品は適切な形で薬学的製剤をそこに入れる容器を含む。適切な容器は当業者にはよく知られており、瓶(プラスチック又はガラス)、サッシェ(sachets)、アンプル、プラスチック袋、金属シリンダー等のような材料を含む。容器はまた包装の内容物への軽率なアクセスを防止するために不正開封防止体を含みうる。また、容器には容器の内容物を記述するラベルが付着される。ラベルは適切な注意書きをまた含みうる。
【0091】
本発明の化合物の薬学的製剤は、薬学的に許容可能な希釈剤、担体、賦形剤又は安定剤により凍結乾燥製剤、粉砕粉剤、又は水溶液の形態に、様々な経路及び投与タイプ用に調製されうる(Remington's PharmaceutIcal Sciences(1995)18版, Mack Publ. Co., Easton, PA)。製剤化は、生理学的に許容可能な担体、つまり用いられる用量及び濃度でレシピエントに非毒性である担体と、適切なpH、雰囲気温度、所望の純度で混合することによってなされうる。製剤のpHは特定の用途及び化合物の濃度に主に依存するが、約3から約8の範囲でありうる。
薬学的製剤は好ましくは無菌である。特にインビボ投与に使用される製剤は無菌でなければならない。かかる滅菌は滅菌濾過膜を通した濾過によって直ぐに達成される。
通常は、薬学的製剤は固形組成物、凍結乾燥製剤又は水溶液として保存されうる。
【0092】
本発明の薬学的製剤は良好な医療行為に一致した形、つまり量、濃度、スケジュール、過程、ビヒクル及び投与経路で、用量が決められ、投与されうる。この点で考慮すべき要因には、治療中の特定の疾患、治療中の特定の哺乳動物、個々の患者の臨床状態、疾患の原因、薬剤の送達部位、投与方法、投与スケジュール、並びに医師に知られている他の因子が含まれる。投与される化合物の「治療的に有効な量」はそのような考慮によって左右され、凝固因子媒介疾患を防止し、軽減し又は治療するのに必要な最少の量である。そのような量は好ましくはホストに毒性であるか又はホストを出血に対してより高い感受性にする量以下である。
一般的な事項として、投与されるトラスツズマブ−MCC−DM1の用量当たりの最初の薬学的に有効な量は、一日当たり約0.01〜100mg/kg患者体重の範囲、すなわち一日当たり約0.1から20mg/kg患者体重で、使用される化合物の典型的な最初の範囲は0.3から15mg/kg/日である。
【0093】
許容される希釈剤、担体、賦形剤及び安定化剤は、用いられる用量及び濃度でレシピエントに非毒性であり、リン酸塩、クエン酸塩、及び他の有機酸などの緩衝液;アスコルビン酸及びメチオニンを含む抗酸化剤;保存料(塩化オクタデシルジメチルベンジルアンモニウム;塩化ヘキサメトニウム;塩化ベンザルコニウム;塩化ベンゼトニウム;フェノール;ブチル、エタノール又はベンジルアルコール;メチル又はプロピルパラベン等のアルキルパラベン;カテコール;レゾルシノール;シクロヘキサノール;3-ペンタノール;及びm-クレゾールなど);低分子量(約10残基未満)ポリペプチド;血清アルブミン、ゼラチン、又は免疫グロブリン等のタンパク質;ポリビニルピロリドン等の親水性ポリマー;グリシン、グルタミン、アスパラギン、ヒスチジン、アルギニン、又はリジン等のアミノ酸;グルコース、マンノース、又はデキストリンを含む単糖類、二糖類、及び他の炭水化物;EDTA等のキレート剤;スクロース、マンニトール、トレハロース又はソルビトールなどの糖;ナトリウムなどの塩形成対イオン;金属錯体(例えば、Zn-タンパク質錯体)及び/又はトゥイーン80を含むTWEENTM、プルロニクス(PLURONICSTM)又はPEG400を含むポリエチレングリコール(PEG)等の非イオン性界面活性剤を含む。また、活性な薬学的成分は、例えばコアセルベーション技術により又は界面重合により調製されたマイクロカプセル、例えば、各々ヒドロキシメチルセルロース又はゼラチン-マイクロカプセル及びポリ(メタクリル酸メチル)マイクロカプセル中、コロイド状薬物送達系(例えば、リポソーム、アルブミンミクロスフィア、マイクロエマルション、ナノ粒子及びナノカプセル)中、又はマイクロエマルション中に包括されていてもよい。このような技術は、Remington's Pharmaceutical Science 18版, (1995) Mack Publ. Co., Easton, PAに開示されている。
【0094】
薬学的製剤はここに詳細が記載される投与経路に適したものを含む。製剤は簡便には単位投薬形態で提供され得、薬学の分野でよく知られた方法の何れかによって調製されうる。技術及び製剤化は一般にRemington's Pharmaceutical Sciences 18版. (1995) Mack Publishing Co., Easton, PAに見出される。かかる方法は、活性成分を、一又は複数の副成分を構成する担体と組み合わせる工程を含む。一般に、製剤は、活性成分を、液体担体又は微細に粉砕した固形担体又は双方と均一かつ密に組み合わせ、ついで必要ならば生成物を成形することにより調製される。
【0095】
経口投与に適した化学療法剤の製剤は、予め定まった量のトラスツズマブ−MCC−DM1の化合物及び/又は化学療法剤をそれぞれ含む、丸薬、硬カプセルないしは軟カプセル剤、例えばゼラチンカプセル剤(capsules)、カプセル(cachet)、トローチ剤、ロゼンジ剤、水性ないしは油性懸濁液、分散可能パウダーないしは顆粒、エマルション、シロップ又はエリキシル剤のような別々の単位として調製されうる。このような製剤は薬学的組成物の製造のために当該分野で知られている任意の方法に従って調製することができ、そのような組成物は、口に合う製剤を提供するために、甘味料、香味料、着色剤及び保存剤を含む一又は複数の薬剤を含みうる。圧縮錠は、場合によってはバインダー、潤滑剤、不活性希釈剤、保存料、界面活性剤又は分散剤と混合せしめて粉末又は顆粒のような自由に流動する形態で活性成分を適切な機械で圧縮することによって調製することができる。成形錠剤は不活性な液体希釈剤で湿潤させた粉末化活性成分の混合物を適切な機械で成形することによって製造することができる。錠剤は場合によっては被覆し又は切り込み線を入れ、場合によっては活性成分の遅延又は制御放出をもたらすように製剤化される。
【0096】
本発明の薬学的製剤の錠剤賦形剤には、錠剤を形成する粉剤のバルク体積を増やすための充填材(または希釈剤);摂取され、薬剤の迅速溶解および吸収を促進する場合に、錠剤を小さな断片、理想的には個々の薬剤粒子に破壊する崩壊剤;顆粒および錠剤が必須の機械的強度によって形成され、圧縮された後に合わさって錠剤となり、包装、輸送および慣例的な処理の間にその成分粉に壊れるのを防ぐ結合剤;製造の間に錠剤を形成する粉の流動性を向上させるための流動促進剤;製造の間に錠剤を圧縮するために用いる機材に錠剤化粉末が付着しないようにするための滑沢剤が含まれる。これらは、圧縮による粉混合物の流量を向上させ、完結した錠剤が機材から放出されるように、摩擦および破損を最小化する。流動促進剤と同様の機能を有する抗粘着剤は、錠剤を形成する粉末と製造中に錠剤の形状を打ち抜くために用いられる機械との間の接着を低減する。錠剤に配合される香料は、好ましい味覚を与えるか又は不快な味覚をマスキングし、着色料は認識及び患者のコンプライアンスを得るためである。
【0097】
錠剤の製造に適した非毒性の薬学的に許容可能な賦形剤と混合せしめられて活性成分を含む錠剤が許容可能である。これらの賦形剤は、例えば不活性な希釈剤、例えば炭酸カルシウム又はナトリウム、ラクトース、リン酸カルシウム又はナトリウム;顆粒化及び崩壊剤、例えばトウモロコシデンプン、又はアルギン酸;結合剤、例えばデンプン、ゼラチン又はアカシア;及び潤滑剤、例えばステアリン酸マグネシウム、ステアリン酸又はタルクでありうる。錠剤は非被覆でも、又は胃腸管中での崩壊と吸着を遅延させるマイクロカプセル化を含む既知の方法によって被覆してもよく、それによって長時間にわたる持続作用をもたらす。例えば、時間遅延物質、例えばモノステアリン酸グリセリル又はジステアリン酸グリセリルを単独で又はロウと共に用いることができる。
【0098】
眼又は他の外部組織、例えば口及び皮膚の治療には、製剤は、好ましくは、例えば0.075から20%w/wの量で活性成分を含む局所用軟膏又はクリームとして適用される。軟膏に製剤される場合、活性成分はパラフィン系又は水混和性軟膏基剤と共に用いることができる。あるいは、活性成分は水中油クリーム基剤を用いてクリームに製剤化することができる。
所望される場合、クリーム基剤の水性相は多価アルコール、すなわち、例えばプロピレングリコール、ブタン1,3-ジオール、マンニトール、ソルビトール、グリセロール及びポリエチレングリコール(PEG400を含む)及びその混合物のような二又はそれ以上のヒドロキシル基を有するアルコールを含みうる。局所用製剤は、好ましくは、皮膚又は他の患部領域を通しての活性成分の吸収又は浸透を向上させる化合物を含みうる。そのような皮膚浸透向上剤の例はジメチルスルホキシド及び関連類似体を含む。
【0099】
本発明のエマルションの油性相は、脂肪又は油との、あるいは脂肪と油の双方との少なくとも一の乳化剤の混合物を含む、公知の方法で既知の成分から構成することができる。好ましくは、親水性乳化剤が、安定化剤として作用する親油性乳化剤と共に含有せしめられる。併せて、安定化剤と共に又は安定化剤を伴わない乳化剤は乳化ロウを構成し、油及び脂肪と共にロウはクリーム製剤の油性分散相を形成する乳化軟膏基剤を含む。本発明の製剤に使用するのに適した乳化剤及びエマルション安定化剤には、トゥイーン(Tween(登録商標))60、スパン(Span(登録商標))80、セトステアリルアルコール、ベンジルアルコール、ミリスチルアルコール、モノ-ステアリン酸グリセリル及びラウリル硫酸ナトリウムが含まれる。
本発明の薬学的組成物の水性懸濁液は水性懸濁液の製造に適した賦形剤と混合せしめられて活性物質を含む。そのような賦形剤には、懸濁剤、例えばナトリウムカルボキシメチルセルロース、クロスカルメローゼ、ポビドン、メチルセルロース、ヒドロキシプロピルメチルセルロース、アルギン酸ナトリウム、ポリビニルピロリドン、トラガカントガム及びアカシアガム、及び分散又は湿潤剤、例えば天然に生じるホスファチド(例えばレシチン)、脂肪酸とのアルキレンオキシドの縮合産物(例えばポリオキシエチレンステアレート)、長鎖脂肪族アルコールとのエチレンオキシドの縮合産物(例えばヘプタデカエチレンオキシセタノール)、脂肪酸とヘキシトール無水物から誘導された部分エステルとのエチレンオキシドの縮合産物(例えばポリオキシエチレンソルビタンモノオレアート)が含まれる。水性懸濁液はまた一又は複数の保存料、例えばエチル又はn-プロピルp-ヒドロキシ-ベンゾエート、一又は複数の着色剤、一又は複数の香味剤及び一又は複数の甘味料、例えばスクロース又はサッカリンを含みうる。
【0100】
薬学的組成物は滅菌された注射用製剤の形態、例えば滅菌注射用水性又は油性懸濁液であってもよい。この懸濁液は上に述べた好適な分散又は湿潤剤及び懸濁剤を用いて公知技術に従って製剤化することができる。滅菌された注射用製剤はまた1,3-ブタン-ジオール溶液又は凍結乾燥粉末から調製されたもののように、非毒性の非経口的に許容可能な希釈剤又は溶媒中の溶液又は懸濁液であってもよい。用いることができる許容可能なビヒクル及び溶媒は水、リンガー液及び等張塩化ナトリウム溶液である。また、滅菌固定化油を溶媒又は懸濁媒質として便宜的に用いることができる。この目的に対して、合成のモノ-又はジグリセリドを含む任意のブランドの固定化油を用いることができる。また、オレイン酸のような脂肪酸も同様に注射剤の調製に使用することができる。
単回投薬形態をつくるために担体物質と混合されうる活性成分の量は治療されるホストと特定の投与形式に応じて変わる。例えば、ヒトへの経口投与のための時間放出製剤は、全組成物の約5から約95%(重量:重量)と変わりうる適切で簡便な量の担体物質と共に配合されておよそ1から1000mgの活性物質を含みうる。薬学的組成物は投与のために容易に測定可能な量をもたらすように調製することができる。例えば、静脈点滴のための水溶液は、約30mL/hrの割合で適した体積の点滴が生じうるようにするために溶液1ミリリットル当たり約3から500μgの活性成分を含みうる。
【0101】
非経口投与に適した製剤には、抗酸化剤、バッファー、静菌剤及び意図したレシピエントの血液と製剤を等張にする溶質を含みうる水性及び非水性滅菌注射用溶液;及び懸濁剤及び増粘剤を含みうる水性及び非水性滅菌懸濁液が含まれる。
眼への局所投与に適した製剤には、好適な担体、特に活性成分のための水性溶媒に活性成分が溶解又は懸濁させられた点眼液がまた含まれる。活性成分はそのような製剤中に好ましくは0.5から20%、例えば0.5から10%、例えば約1.5%w/wの濃度で存在する。
口への局所投与に適した製剤には、香味基剤、通常はスクロース及びアカシア又はトラガカント中に活性成分を含むロゼンジ;ゼラチン及びグリセリン、又はスクロース及びアカシアのような不活性基剤に活性成分を含むパスティユ;及び適切な液体担体に活性成分を含むうがい薬が含まれる。
直腸投与のための製剤は、例えばココアバター又はサリチラートを含む好適な基剤を用いて座薬として提供することができる。
【0102】
肺内又は経鼻投与に適した製剤は、例えば0.1から500ミクロン(例えば0.5、1、30ミクロン、35ミクロン等々のような増分ミクロンで0.1から500ミクロンの範囲の粒子径を含む)の範囲の粒子径を有し、これが鼻経路を通る迅速な吸入又は肺胞嚢に達するように口からの吸入によって投与される。好適な製剤には、活性成分の水性又は油性溶液が含まれる。エアゾール又は乾燥粉末投与に適した製剤は常法によって調製することができ、以下に記載されるような疾患の治療又は予防にこれまで使用されている化合物のような他の治療剤と共に送達されうる。
膣投与に適した製剤は、活性成分に加えて、当該分野で適切であることが知られているような担体を含むペッサリー、タンポン、クリーム、ゲル、ペースト、フォーム又はスプレー製剤として提供することができる。
【0103】
製剤は、単位投薬又は複数投薬容器、例えば密封されたアンプル及びバイアルに包装することができ、使用直前に注射用の滅菌液体担体、例えば水の添加のみを必要とするフリーズドライ(凍結乾燥)条件で保存することができる。即時混合注射溶液及び懸濁液は既に記載された種類の滅菌粉末、顆粒及び錠剤から調製される。好適な単位投薬製剤は、活性成分の、上に記載されたような毎日の投薬又は毎日の部分用量単位、又はその適切なフラクションを含むものである。
本発明は、獣医学的担体と共に上述の少なくとも一の活性成分を含有する動物用医薬組成物を更に提供する。獣医学的担体は該組成物を投与する目的に有用な物質であり、不活性であるか獣医学分野で許容可能であり活性成分と相容性のある固形、液体又はガス状物質でありうる。これらの動物用医薬組成物は非経口的、経口的又は任意の他の所望の経路で投与されうる。
【0104】
(併用療法)
トラスツズマブ−MCC−DM1は、前悪性及び非腫瘍又は非悪性の過剰増殖性疾患とともに、腫瘍、癌及び腫瘍性組織を含む、過剰増殖性疾患又は障害の治療のために、他の化学療法剤と組み合わされて用いられてよい。ある実施態様では、トラスツズマブ−MCC−DM1は、薬学的併用製剤又は併用療法としての投薬計画において、抗過剰増殖性特性を有するか又は過剰増殖性疾患を治療するために有用である第二の化合物と組み合わされる。薬学的併用製剤又は投薬計画の第二化合物は好ましくは、トラスツズマブ−MCC−DM1に対して相補的な活性を有し、互いに悪影響を与えない。このような化合物は、意図する目的のために有効である量で適切に併用される。一実施態様では、本発明の組成物は、本明細書において記述されるような化学療法剤と組み合わせたトラスツズマブ−MCC−DM1を含む。実施例4および5はそれぞれ、T−DM1+パーツズマブおよびT−DM1+GDC−0941の臨床プロトコールである。
【0105】
本発明の治療的併用には、過剰増殖性疾患の治療での別々、同時又は連続した使用のための併用用製剤としての、トラスツズマブ−MCC−DM1と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869およびドセタキセルから選択される化学療法剤との投与を含む、製剤化、投薬計画又は治療の他の工程が含まれる。
併用療法剤は同時の又は逐次の投与計画として投与されうる。逐次に投与される場合、併用剤は、二以上の投与で投与されうる。併用投与は、別個の製剤又は単一の薬学的製剤を使用する同時投与と、好ましくは両方の(又は全ての)活性薬剤がその生物学的活性を同時に作用させる期間が存在している、何れかの順の連続投与を含む。
上記の同時投与される薬剤の何れかに対して適した投薬量は現在使用されるものであり、新たに同定された薬剤と他の化学療法剤又は治療との組み合わせた作用(相乗作用)のために低くされうる。
抗癌療法の具体的な実施態様では、トラスツズマブ−MCC−DM1は、本明細書中に記載されるホルモン剤又は抗体医薬を含む化学療法剤、並びに外科的治療法および放射線療法と組み合わされうる。トラスツズマブ−MCC−DM1および他の薬学的に活性な化学療法剤(一又は複数)の量と投与の相対的タイミングは、所望の併用治療効果を達成するために選択されであろう。
【0106】
(薬学的組成物の投与)
本発明の化合物は、治療される状態に適切な任意の経路によって投与されてよい。適切な経路には、経口、非経口(皮下、筋肉内、静脈内、動脈内、吸入、皮内、髄膜下、硬膜外および注入技術を含む)、経皮、直腸、経鼻、局所的(頬側および舌下を含む)、経膣、腹膜内、肺内及び鼻腔内が含まれる。また、局所投与は、経皮パッチ又はイオン泳動法装置といった経皮投与の使用を伴いうる。薬剤の製剤は、Remington's Pharmaceutical Sciences, 18th Ed., (1995) Mack Publishing Co., Easton, PAにおいて述べられている。薬剤製剤の他の例は、Liberman, H. A. and Lachman, L., Eds., Pharmaceutical Dosage Forms, Marcel Decker, Vol 3, 2nd Ed., New York, NYにおいて見られる。局所の免疫抑制性治療のために、化合物は、灌流を含む病巣内投与によって、あるいは移植の前に移植片を阻害剤と接触させることによって投与されてよい。周知のことであるが、好ましい経路は、例えばレシピエントの状態によって異なってよい。化合物が経口投与される場合、丸薬、カプセル、錠剤などとして、薬学的に許容可能な担体、流動促進剤又は賦形剤にて調製されてよい。化合物が非経口的に投与される場合、後述されるように、薬学的に許容可能な非経口ベヒクル又は希釈剤によって、そして、単位用量注射用形態に調製されてよい。
【0107】
ヒト患者を治療するためのトラスツズマブ−MCC−DM1の用量は、およそ100mgからおよそ500mgであってよい。吸収、分布、代謝および排出を含む薬物動態学的(PK)及び薬力学的(PD)性質に応じて、トラスツズマブ−MCC−DM1の用量は、6週間に1回、3週間に1回、毎週又はもっと頻繁に投与されてよい。トラスツズマブ−MCC−DM1と組み合わされて用いられる化学療法剤の用量は、およそ10mgからおよそ1000mgまでであってよい。化学療法剤は、6週間に1回、3週間に1回、毎週、又は1日に1回ないし2回などもっと頻繁に投与されてよい。加えて、毒性因子は用量および投薬計画に影響しうる。経口投与される場合、丸薬、カプセル又は錠剤は、特定の期間に、毎日またはもっと少ない頻度で摂取されてよい。投薬計画は、多くの治療サイクルのために繰り返されてよい。
【0108】
(治療方法)
(1)トラスツズマブ−MCC−DM1と(2)化学療法剤との治療的組合せは、限定するものではないがHER2経路の活性化に特徴があるものを含む疾患、状態および/または障害を治療するために有用である。したがって、本発明の他の態様は、HER2又はVEGFRレセプター1を標的化することによって治療されうる疾患又は状態の治療方法を含む。(1)トラスツズマブ−MCC−DM1と(2)化学療法剤との治療的組合せは、前悪性及び非腫瘍性又は非悪性の過剰増殖性疾患とともに、腫瘍、癌及び腫瘍性組織を含む、過剰増殖性疾患又は障害の治療のために、用いられてよい。
本発明の方法に従って治療されうる癌には、限定するものではないが、胸部、卵巣、子宮頸管、前立腺、精巣、泌尿生殖器、食道、喉頭、神経膠芽腫、神経芽細胞腫、胃、皮膚、角化棘細胞腫、肺、類表皮癌、大細胞カルチノーマ、非小細胞肺カルチノーマ(NSCLC)、小細胞カルチノーマ、肺腺癌、骨、大腸、腺腫、膵臓、腺癌、甲状腺、濾胞性カルチノーマ、未分化癌、乳頭カルチノーマ、精上皮腫、メラノーマ、肉腫、膀胱カルチノーマ、肝臓カルチノーマ及び胆汁路、腎臓カルチノーマ、骨髄疾患、リンパ腫、線毛細胞、頬側(口腔)及び咽頭、唇、舌、咽頭、小腸、大腸-直腸、大腸、直腸、脳及び中枢神経系、ホジキン、及び白血病が含まれる。
本発明の他の態様は、本明細書に記載の疾患又は状態に罹患している哺乳動物、例えばヒトの該疾患又は状態の治療に用いるための、薬学的組成物又は治療上の組合せを提供する。また、本明細書中に記載の疾患及び症状に罹っている哺乳動物などの温血動物の該障害の治療のための医薬の調製における薬学的組成物の使用を提供する。
【0109】
(製造品)
本発明の他の実施態様では、上述の疾患及び障害の処置に有用なトラスツズマブ−MCC−DM1を具備する製造品又は「キット」が提供される。一実施態様では、キットは、トラスツズマブ−MCC−DM1を含む容器を具備する。キットは更に容器にあるか付随させられるラベル又はパッケージ挿入物を具備する。「パッケージ挿入物」なる用語は、効能、使用法、用量、投与法、禁忌についての情報及び/又はかかる治療用製品の使用に関する注意書きを含む治療用製品の商業的パッケージに常套的に含まれる指示書を意味する。適切な容器は、例えば、瓶、バイアル、シリンジ、ブリスター包装等を含む。該容器はガラス又はプラスチック等の様々な材料から形成されうる。該容器は、症状を処置するのに効果的であるトラスツズマブ−MCC−DM1又はその製剤を収容し、無菌アクセスポートを有しうる(例えば、該容器は静脈内溶液バッグ又は皮下注射針により突き刺されうるストッパーを有しているバイアルでありうる)。組成物中の少なくとも一の活性剤はトラスツズマブ−MCC−DM1である。ラベル又はパッケージ挿入物は、組成物が例えば癌のような選択された症状を治療するために使用されることを示している。一実施態様では、ラベル又はパッケージ挿入物は、トラスツズマブ−MCC−DM1を含有する組成物が異常な細胞増殖から生じる疾患を治療するために使用できることをまた示しているであろう。ラベル又はパッケージ挿入物は、組成物を他の疾患の治療に使用できることもまた示している場合がある。あるいは、又は付加的に、製造品は、薬学的に許容可能なバッファー、例えば注射用静菌水(BWFI)、リン酸緩衝生理食塩水、リンガー液及びデキストロース溶液を含む第三容器を更に含みうる。それは、他のバッファー、希釈剤、フィルター、針、及びシリンジを含む商業的及び使用者の観点から望まれる他の材料を更に含みうる。
【0110】
キットは、トラスツズマブ−MCC−DM1と、もし存在するならば第二薬学的製剤の投与のための指示書を更に具備しうる。例えば、キットがトラスツズマブ−MCC−DM1を含有する第一組成物と第二薬学的製剤を含むならば、キットは、第一及び第二薬学的組成物を、それを必要とする患者に同時に、連続的に又は別個に投与するための指示書を更に具備しうる。
他の実施態様では、キットは、例えば錠剤又はカプセル剤のような、トラスツズマブ−MCC−DM1の固形経口形態を送達させるのに適している。かかるキットは好ましくは多くの単位投薬形態を含む。かかるキットは、その意図された使用順に配向された投薬量を持つカードを含みうる。かかるキットの例は「ブリスター包装」である。ブリスター包装は包装産業ではよく知られており、薬学的単位投薬形態を包装するために広く使用されている。所望される場合、記憶補助品が、例えば投薬量が投与されうる日を治療スケジュール中に示す数字、文字、又は他の印又はカレンダー挿入物の形態で提供されうる。
【0111】
一実施態様によれば、キットは、(a)トラスツズマブ−MCC−DM1が含まれる第一容器と;場合によっては(b)第二薬学的製剤がそこに含まれる第二容器とを具備し得、ここで、第二薬学的製剤は抗過剰増殖性活性を持つ第二化合物を含む。あるいは、又は付加的に、キットは薬学的に許容可能なバッファー、例えば注射用静菌水(BWFI)、リン酸緩衝生理食塩水、リンガー液及びデキストロース溶液を含む第三容器を更に具備しうる。それは、他のバッファー、希釈剤、フィルター、針、及びシリンジを含む商業的及び使用者の観点から望まれる他の材料を更に含みうる。
キットがトラスツズマブ−MCC−DM1の組成物と第二治療剤、すなわち化学療法剤を含む場合、キットは、別個の組成物を収容するための容器、例えば分割瓶又は分割フォイル小包を具備しうるが、別個の組成物は単一の分割されていない容器内に収容されてもよい。典型的には、キットは別個の組成物の投与のための指示書を含む。別個の成分が好ましくは異なった投薬形態(例えば経口と非経口)で投与され、異なった投薬間隔で投与される場合、又は併用される個々の成分の用量設定が処方医師によるのが望ましい場合に特に有利である。
【実施例】
【0112】
本発明を例示するために、以下の実施例が含まれる。しかしながら、これらの実施例は本発明を制限するものではなく、本発明を実施する方法を提案するためだけのものであると理解すべきである。
【0113】
実施例1 トラスツズマブ−MCC−DM1の調製
トラスツズマブは、50mM リン酸カリウム/50mM 塩化ナトリウム/2mM EDTA、pH6.5中に20mg/mlでバッファ-交換によってHERCEPTIN(登録商標)から精製し、スクシンイミジル 4(N-マレイミドメチル)シクロヘキサン-1-カルボキシレート(SMCC、Pierce Biotechnology, Inc)の7.5〜10モル等価物、DMSO又はDMA(ジメチルアセトアミド)中20mM、6.7mg/mlにて処理した(米国公開特許2005/0169933;米国公開特許2005/0276812)。室温のアルゴン下で2〜4時間撹拌した後、反応混合物は、50mM リン酸カリウム/50mM 塩化ナトリウム/2mM EDTA、pH6.5にて平衡化したセファデックスG25カラムに濾過した。あるいは、反応混合物は、pH6の30mM クエン酸塩および150mM 塩化ナトリウムにてゲル濾過した。抗体を含む分画を貯蔵し、分析した。トラスツズマブ−SMCCは88%回収した。
上記の薬剤−リンカー中間生成物であるトラスツズマブ−MCCは、50mM リン酸カリウム/50mM 塩化ナトリウム/2mM EDTA、pH6.5にて10mg/mlの終濃度に希釈し、ジメチルアセトアミド中DM1の10mM溶液(5 SMCC/トラスツズマブ、7.37mg/mlと予測される1.7等価物)と反応させた。DM1は、アンサミトシン発酵産物(米国特許第6790954号;米国特許第7432088号)から調製され、結合のために誘導体化されてよい(米国特許第6333410号;RE39151)。反応物は、アルゴン下の室温で4〜およそ16時間撹拌した。コンジュゲート反応混合物は、pH6.5の1×PBSにてセファデックスG25ゲル濾過カラム(1.5×4.9cm)に濾過した。あるいは、反応混合物は、pH5の10mM 琥珀酸塩および150mM 塩化ナトリウムにてゲル濾過した。252nm及び280nmの吸光度によって測定すると、DM1/トラスツズマブ比率(p)は3.1であった。また、抗体に対する薬剤の比率(p)は質量分析によって測定されてよい。また、コンジュゲートは、SDSポリアクリルアミドゲル電気泳動によってモニターされてよい。凝集はレーザー光散乱分析によって評価されてよい。
【0114】
あるいは、トラスツズマブ−MCC−DM1は、MCC−DM1リンカー−薬剤試薬を形成し、次いでトラスツズマブと反応させることによって調製されてよい。
一般的に、DM1とのトラスツズマブ−MCCのコンジュゲート反応により、付着しコンジュゲートしたDM1薬剤の数が異なる、すなわちpが1から約8の分布である薬剤負荷である抗体を含む不均質混合物となる。トラスツズマブ上の多くの異なる求核基、例えば終末リジンアミノ基がSMCCと反応しうる場合、トラスツズマブへのSMCCの異なる付着部位により、更なる次元の不均一性が存在する。ゆえに、トラスツズマブ−MCC−DM1は、単離され、精製された種間分子、並びに1から8の平均薬剤負荷の混合物を含み、このMCC−DM1はトラスツズマブ抗体の何れかの部位により付着している。
コンジュゲート反応からのトラスツズマブ−MCC−DM1の調製におけるトラスツズマブ抗体当たりのDM1薬剤部分の平均数は、質量分析、ELISAアッセイ、電気泳動法およびHPLCといった従来の手段によって特徴付けられうる。また、pに関するトラスツズマブ−MCC−DM1の定量的分布も決定されてよい。ELISAによって、ADCの特定の調製物におけるpの平均値が決定されてよい(Hamblett et al (2004) Clinical Cancer Res. 10: 7063-7070;Sanderson et al (2005) Clinical Cancer Res. 11: 843-852)。しかしながら、p(薬剤)値の分布は、抗体−抗原結合とELISAの検出限界によって識別されない。また、抗体−薬剤コンジュゲートの検出のためのELISAアッセイは、薬剤部分が抗体のどこに、例えば重鎖又は軽鎖の断片や特定のアミノ酸残基に付着しているかを決定しない。場合によって、pが他の薬剤負荷を有するトラスツズマブ−MCC−DM1からの特定の値である場合の均質なトラスツズマブ−MCC−DM1の分離、精製および特徴づけは、逆相HPLC又は電気泳動法といった手段によって達成されうる。
【0115】
実施例2 インビトロ細胞増殖アッセイ
本発明の組合せの有効性は、以下のプロトコールを使用した細胞増殖アッセイによって測定した(Promega Corp. Technical Bulletin TB288; Mendoza et al (2002) Cancer Res. 62: 5485-5488)。Cell-Titer Gloアッセイ試薬およびプロトコールは市販されている(Promega)。アッセイは、細胞に入り、細胞増殖に作用する化合物の能力を評価する。アッセイ原理は、細胞のATPを定量化することによって存在する生細胞の数を決定することである。Cell-Titer Gloはこの定量化のために使用する試薬である。それは、Cell-Titer Gloの添加により細胞溶解とルシフェラーゼ反応による発光シグナルの生成が生じる同種のアッセイである。発光シグナルは、存在するATPの量と比例している。
DMSOおよび培地プレート:ヌンクの96ウェル円錐形の底のポリプロピレンプレート(カタログ番号249946)。
細胞プレート:ファルコンの384-ウェル黒色、透明底(マイクロクリアー)、蓋付きのTCプレート(353962)。
細胞培養培地:RPMI又はDMEM高グルコース;ハムのF-12(50:50)、10%胎仔ウシ血清、2mM L−グルタミン。
Cell Titer-Glo:Promega(カタログ番号G7572)
【0116】
手順:
1日目−細胞プレートへの播種。細胞を回収し、3日間のアッセイのために384ウェルの細胞プレートに1ウェルにつき54μlにつき1000〜2000細胞で細胞を播種する。37℃、5%CO2下で、一昼夜(約16時間)インキュベートする。
2日目−細胞への薬剤添加。化合物希釈物、DMSOプレート(9ポイントについて1:2段階希釈)。96ウェルプレートの2列目に20μlの化合物(小分子薬剤では10mMの貯蔵液)を加える。高精度培地プレート(1:50希釈)を用いて、合計9ポイントについてプレート(10μl+10μl 100%DMSO)全体にわたって1:2段階希釈を行った。異なる96ウェル培地プレートのすべてのウェルに147μlの培地を添加する。Rapidplateを用いて、DMSOプレートの各ウェルからのDMSO+化合物の3μlを培地プレート上の各々対応するウェルに移す。2薬剤併用試験のために、Rapidplateを用いて、DMSOプレートの各ウェルからのDMSO+化合物の1薬剤1.5μlを培地プレート上の各々対応するウェルに移す。次いで、もう1つの薬剤1.5ulを培地プレートへ移す。
細胞への薬剤添加、細胞プレート(1:10希釈)、6μlの培地+化合物を細胞に直接添加する(すでに細胞上に54μlの培地)。頻繁に開けられないインキュベーター中で37℃、5%CO2で3日間インキュベートする。
5日目−プレートを反応させる、Cell Titer Gloバッファを室温で解凍する。37℃および室温に平衡な温度からおよそ30分間細胞プレートを取り除く。Cell Titer GloバッファをCell Titer Glo基質に添加する(瓶から瓶へ)。30μlのCell Titer Glo試薬を各ウェルの細胞に加える。およそ30分間プレートシェーカ上に置く。PerkinElmer Envision(1ウェルにつき0.1秒)又はAnalyst HTプレート読み取り機(1ウェルにつき0.5秒)にて発光を読み取る。
【0117】
細胞生存率アッセイおよび併用アッセイ:細胞は、384ウェルプレートの1ウェルにつき1000〜2000細胞を播いて16時間置いた。2日目に、96ウェルプレートに、DMSOにて9つの1:2段階希釈化合物希釈物を作製した。Rapidplateロボット(Zymark Corp., Hopkinton, MA)を用いて、化合物を更に増殖培地にて希釈した。次いで、希釈された化合物を384ウェルの細胞プレートの4通りのウェルに加え、37℃および5%CO2でインキュベートした。4日後に、生細胞の相対数を、製造業者の指示に従ってCell-Titer Glo(Promega)を用いて発光によって測定し、Envision又はWallac Multilabel読み取り機(PerkinElmer, Foster City)にて読み取った。EC50値は、Kaleidagraph4.0(Synergy Software)又はPrism4.0ソフトウェア(GraphPad, San Diego)を用いて算出した。併用アッセイの薬剤は、8×EC50濃度で始めて投与した。薬剤のEC50が2.5μMを超える症例では、最も高い濃度である10μMを用いた。すべてのアッセイにおいて、トラスツズマブ−MCC−DM1と化学療法剤は、同時又は4時間空けて(一方から他方までの間)投与した。
【0118】
他の例示的なインビトロ細胞増殖アッセイは以下の工程を含む。
1. 培地中におよそ104細胞(細胞株及び腫瘍種類については図1を参照)を含む100μlの細胞培養物の一定分量を、384ウェルの不透明な壁のプレートの各ウェルに置いた。
2. 培地を含むが細胞を含まないコントロールウェルを調製した。
3. 化合物を試験ウェルに加え、3〜5日間インキュベートした。
4. プレートを室温と平衡にしておよそ30分間置いた。
5. 各ウェルに存在する細胞培養培地の体積と等しい量のCellTiter-Glo試薬を添加した。
6. 内容物を軌道シェーカ上で2分間混合し、細胞溶解を誘導した。
7. プレートを室温で10分間インキュベートし、発光シグナルを安定させた。
8. 発光は、RLU=相対的発光単位として記録し、グラフに記した。
あるいは、96ウェルのプレート中に細胞を最適密度で播き、試験化合物の存在下で4日間インキュベートした。その後、アラマーブルーTMをアッセイ培地に加え、細胞を6時間インキュベートした後、544nmの励起、590nmの発光で読み取った。EC50値は、S字状の用量応答曲線フィットを用いて算出した。
【0119】
実施例3 インビボ腫瘍異種移植片
トランスジェニック実験に適する動物は、通常の市販元から入手することができる。雌CB−17SCIDベージュマウス(Charles River Laboratory)の各群の乳房脂肪パットに、3000000のKPL-4(Her2過剰発現)乳癌細胞をマトリゲルとともに移植した。雌胸腺欠損ヌードマウス(Charles River Laboratory or Harlan)の各群の乳房脂肪パッドに、2×2mm3断片のMMTV-Her2 Fo5トランスジェニック胸部腫瘍を移植した。0日目に、各腫瘍モデルに特化した計画に従って、マウス異種移植片に、薬剤、薬剤併用又はベヒクルを投与した。5−FU、ゲムシタビン、カルボプラチンおよびB20-4.1を腹膜内投与し、パーツズマブは示したように静脈内又は腹膜内投与し、トラスツズマブ−MCC−DM1とドセタキセルは静脈内投与し、ラパチニブ、GDC−0941およびABT−869は経管栄養により経口的に投与した。試験期間中、週に2回腫瘍サイズを記録した。マウスの体重も週に2回記録し、マウスを定期的に観察した。ウルトラCal IVカリパス(Model 54-10-111; Fred V. Fowler Co., Inc.; Newton, MA)を用いて腫瘍体積を二次元(長さおよび幅)で測定し、エクセルv.11.2(Microsoft Corporation; Redmond, WA)を用いて分析した。腫瘍阻害グラフは、KaleidaGraphバージョン3.6(Synergy Software; Reading, PA)を用いてプロットした。腫瘍体積は以下の式によって算出した:腫瘍サイズ(mm3)=(長い方の測定値×短い方の測定値2)×0.5。
【0120】
Adventurera Pro AV812スケール(Ohaus Corporation; Pine Brook, NJ)を用いて動物の体重を測定した。KaleidaGraphバージョン3.6を使用してグラフを作成した。重量変化の割合は以下の式を使用して算出した:群重量変化の割合=(1−(最初の重量/新たな重量))×100。
腫瘍体積が2000mm3を上回わるか又は体重喪失が開始時の重量の20%以上であるマウスを、規定のガイダンスに従ってすぐに安楽死させた。
試験の終了時(EOS)の腫瘍増殖遅延の割合(%TGD)は、以下の式を用いて算出した:%TGD=100×(処置群のエンドポイントまでの中位時間−コントロール群のエンドポイントまでの中位時間)/コントロール群のエンドポイントの中位時間。
腫瘍発生(TI)は、試験終了時に各群に残っている測定可能な腫瘍の数を基に決定した。部分的な応答(PR)は、開始時の腫瘍体積と比較して腫瘍体積の50%超かつ100%未満の減少として定義し、3つの連続した測定値について観察した。完全寛解(CR)は、開始時の腫瘍体積と比較して腫瘍体積の100%の減少として定義し、3つの連続した測定値について観察した。データを分析し、p値はJMP統計ソフトウェア、バージョン5.1.2(SAS Institute; Cary, NC)によるダネットのt検定を使用して決定した。試験終了時の個々の腫瘍体積と平均腫瘍体積±SEM値は、JMP統計ソフトウェア、バージョン5.1.2を用いて算出した。体重データは、開始時の体重±SEMからの平均の変化の割合に基づいてグラフ化した。
【0121】
実施例4 パーツズマブと併用したトラスツズマブ−MCC−DM1(T-DM1)の臨床研究
従来の治療を受けていながら進行したHER2陽性の局所進行性又は転移性の乳癌を有する患者に静脈内投与したパーツズマブと併用したトラスツズマブ−MCC−DM1(T−DM1)の、安定性、耐容性及び有効性の第1b/II相、非盲検試験を設定し、併用の安全性と耐容性を特徴付けた。何れかの治療ラインにおいてトラスツズマブを既に摂取したか、又は進行した疾患のためにHER2標的治療と組み合わせて化学療法剤を摂取したか、又は最も最新の治療を受けたが進行した、HER2陽性の局所進行性又は転移性の乳癌を有する患者に、併用を3週間ごとに投与した。他の目的は、この計画においてT−DM1とパーツズマブの併用が投与される場合のT−DM1の薬物動態学を評価することである。他の目的は、この計画において投与されるT−DM1とパーツズマブの併用の有効性の事前評価をすることであり、これは、変更した固形腫瘍の治療効果判定基準(RECIST)、バージョン1.0を用いた調査者評価に基づいた客観的な応答速度によって測定される。この試験の二次目的は以下の通りである:(1) この計画において投与されるT−DM1とパーツズマブの併用を摂取した患者の無進行生存率(PFS)を推定すること;(2) この計画において投与されるT−DM1とパーツズマブの併用の応答の継続期間を評価すること;そして、(3) T−DM1に対する抗治療的抗体の開発を評価すること。
【0122】
トラスツズマブを既に摂取しており、その最後の治療後ないしは治療の間に進行したHER2陽性の局所進行性又は転移性の乳癌を有する患者において、T−DM1は、パーツズマブと組み合わされて静脈内(IV)注入によって投与されるであろう。患者は、最低3週間の間隔の反復サイクルで、T-DM1とパーツズマブの併用を摂取する。
所定の用量レベルの患者は、DLT観察期間(T−DM1の第一用量の時間から21日間とする)中、試験薬剤の第一用量を摂取した後で比較的高い用量レベルでの患者の治療の前に、DLT(用量制限毒性)について観察される。DLT観察期間中にこれらの患者においてDLTが観察されなければ、次の用量レベルへの用量段階的拡大を進めてよい。
【0123】
DLTは、DLT観察期間内に生じる次の何れかの治療関連毒性として定義される。(1) グレード3以上 疾患進行又は他の明らかに認識できる原因によるものでない非血液学的な有害事象、何れかのグレードの脱毛症を除く;(2) グレード3 標準的な治療法に応答する下痢;(3) グレード3 標準的な治療法に応答する前投薬がない場合の嘔気又は嘔吐;(4) グレード3以上 肝臓又は骨転移の結果としての、72時間続く、血清ビリルビン、肝トランスアミナーゼ(ALT又はAST)又はアルカリホスファターゼ(ALP)の上昇、グレード2の肝トランスアミナーゼ又は基準値のALPレベルを有する患者は除く(正常の上限値[ULN]5以下)。肝トランスアミナーゼ又はALPレベルが10ULN以上であれば、DLTとみなされるであろう。(5) グレード4以上 24時間続く血小板減少;(6) グレード4以上 4日間続くか又は熱(経口又は経鼓膜の温度100.4°F又は38℃)を伴う好中球減少症(好中球の絶対数500/細胞/mm3未満);(7) 調査者が何れかの試験化合物と関連していると感じる、主観的な過度の毒性;(8) 治療の第2サイクルの開始を妨げる、治療関連の毒性。
【0124】
次の最も高い用量レベルに進むことが決定された後は、患者内の用量段階的拡大も可能となると考える。試験に参加する患者は、まず最初に完全用量のパーツズマブとともに用量を減らしたT−DM1(3.0mg/kg)を摂取する。いったんコホートがDLT観察期間を終えると、これらの患者は、次のサイクルのために両薬剤の全用量へ増やすことが可能であると考えられる。しかしながら、3.6mg/kgの服用レベルの安全性はDLTの評価に基づく。患者(試験の用量−段階的拡大段階の間に試験に参加した人々を含む)は、初めの経過観察腫瘍評価により試験中である場合、有効性についての評価が可能であるとみなす。超音波心臓診断図(ECHO)又はマルチゲート取得(MUGA)スキャンは、サイクル1の終わりに、次いで試験期間を通して3サイクルごとに実施すべきである。
【0125】
T-DM1製剤
T-DM1は、20mmのフルオロ樹脂の積層ストッパーと反転式の灰色のプラスチック製キャップによるアルミニウム密閉を具備した20mlのI型USP/ヨーロッパ薬局方ガラスバイアルの使い捨ての凍結乾燥製剤として提供されてよい。8.0mLの注射用滅菌水(SWFI)にて再構成した後に、結果として生じる生成物は、10mM 琥珀酸ナトリウム、pH5.0、6%(w/v)スクロースおよび0.02%(w/v)ポリソルベート20中20mg/mLのT−DM1を含有する。各20mLのバイアルはおよそ172mgのT−DM1を含有し、160mgのT−DM1の運搬を可能とする。示された体積のT−DM1溶液をバイアル(一又は複数)から取り除き、IVバッグに加える。再構成されたT−DM1は、0.45%又は0.9%の塩化ナトリウム注射(最低250mLの体積)を含有するPVC又は無天然ゴム・無PVCのポリオレフィンバッグ(PO)に希釈される。0.45%塩化ナトリウムを含有するPVC又はPOバッグの使用が好まれる。0.9%の塩化ナトリウムを含有するPVC又はPOバッグが使われる場合、0.22μmインラインフィルターの使用が推奨される。バッグはゆっくりと逆さにして、溶液を混合する。0.9%又は0.45%の塩化ナトリウム注射、USPを含有するポリ塩化ビニル(PVC)又は無天然ゴム無PVCのポリオレフィン(PO)バッグ中で希釈される注入用T−DM1溶液は、短期間の間、2℃〜8℃(36°F〜46°F)で保存されてよい。
【0126】
パーツズマブ製剤
パーツズマブは、20mm L−ヒスチジン(pH6.0)、120mM スクロースおよび0.02%ポリソルベート20において調製される30mg/mlのパーツズマブを含有する使い捨ての製剤として提供される。各20ccのバイアルは、およそ420mgのパーツズマブ(14.0mL/バイアル)を含有する。示される体積のパーツズマブ溶液をバイアルから取り出し、注射用の0.9%塩化ナトリウム溶液の250ccのIVバッグに加える。バッグをゆっくりと逆さにして溶液を混合し、投与前に粒子及び変色について視覚的に調べる。0.9%塩化ナトリウムを含有するポリエチレン又は非PVCのポリオレフィンバッグ中で希釈される注入用パーツズマブの溶液は、短期間の間、2℃〜8℃(36°F〜46°F)で保存されてよい。
【0127】
安全性結果測定
T−DM1およびパーツズマブの安全性および耐容性は、以下の一次安全性結果測定を使用して評価される:(1) 有害事象の発症、性質および重症度;(2) T−DM1および/またはパーツズマブの用量変更、用量遅延又は中止が生じるような、試験薬物投与中及び該投与後の有害現象又は物理的な所見および臨床検査室結果の変化;及び、(3) ECHO又はMUGAスキャンを含む心機能の変化(すなわち左室駆出分画率[LVEF]、分節性壁異常)。
【0128】
薬物動態学的及び薬力学的結果測定
以下のT−DM1およびパーツズマブの薬物動態学的パラメーターは、適切なときに、データが得るように、いずれかの非区画および/または集団方法を使用する試験治療を受けるすべての患者において決定する:(1) T−DM1(コンジュゲート)、総トラスツズマブ(DM1フリー及びDM1コンジュゲート)の血清濃度;(2) DM1フリーの血漿濃度;(3) 総曝露(濃度−時間曲線下の領域[AUC]);(4) 最大血清中濃度(Cmax);(5) 最小濃度(Cmin);(6) クリアランス;(7) 分布の体積;(8) 最終的な半減期;(9) T−DM1に対する抗治療的抗体。
【0129】
有効性結果測定
変更RECIST, v1.0を用いた客観的な応答速度を有効性結果測定として評価する。この研究の二次有効性結果測定は以下の通りである:(1) PFS、試験治療開始から任意の原因による試験中(試験治療の最終投薬から30日以内)の疾患の進行又は死亡の最初の発生までの時間として定義され、これは調査者によって変更RECIST, v1.0を使用して腫瘍評価の概要が決定される;そして、(2) 応答の持続時間、任意の原因による試験中(試験治療の最終投薬から30日以内)の、調査者によって変更RECIST(v1.0)を使用して腫瘍評価の概要が決定される疾患進行の時間まで、又は死亡までの、文書化された客観的応答の最初の発生として定義される。
【0130】
試験治療
T−DM1は、2.4、3.0又は3.6mg/kgのIV用量で、3週間ごとを超えない頻度で投与される。いずれの患者も、2.4mg/kg程度の低いT−DM1用量に段階的に漸減してよい。3.0mg/kgで治療を始める患者のコホートにおいて生じる毒性に応じて、そして、3.0mg/kgのT−DM1が許容できることが確認される場合、患者は、その後のサイクルにおいて3週間ごとに3.6mg/kgのIVの用量に漸増されてよい。パーツズマブは、1サイクルの1日目に840mgIVの負荷用量で、その後のサイクルでは3週間ごとに420mgのIVで投与される。
【0131】
統計的方法
この試験の一次有効性エンドポイントは調査者評価の客観的な応答であり、4週間隔以上の2つの連続的な事象において決定される完全もしくは部分的な応答として定められる。推定する客観的応答速度とともに対応する95%信頼区間も算出する。客観的な応答のために、妥当に基準値を超えない腫瘍評価を有する患者を不応答者として計数する。応答の持続時間およびPFSのために、経過観察を行わない患者のデータを、患者が進行していないことがわかっていた最後の日に打ち切って処理する。治療後腫瘍評価又は死のない患者のデータは、治療開始+1日の日に打ち切る。
【0132】
実施例5 GDC−0941と併用するトラスツズマブ−MCC−DM1(T-DM1)の臨床試験
従来のトラスツズマブベースの治療により進行したHER2陽性転移性の乳癌を有する患者への静脈内投与したT−DM1と経口投与したGDC−0941の併用の第Ib相、非盲検試験を設定し、併用の安全性、耐容性、薬物動態及び活性を特徴付けた。この試験の一次目的は以下の通りである:T−DM1を投与したときのGDC−0941の安全性および耐容性を評価すること;T−DM1を投与したときのGDC−0941のMTDを推定すること;T−DM1と併用して投与されるGDC−0941の推奨される第II相用量を同定すること;そして、T−DM1と併用して投与されるときのGDC−0941の観察される抗腫瘍活性を特徴付けること。薬物動態学的目的は以下の通りである:T−DM1の有無の下でのGDC−0941の薬物動態学の特徴を表すこと;そして、GDC−0941の相対的有無の下でのT−DM1の薬物動態学の特徴を表すこと。
【0133】
GDC-0941製剤
GDC-0941はPO投与を目的とする乾燥粉末である。調製された薬剤品は、サイズ0の殻に封入され、色によって区別される2つの強度(15および50mg)の硬質ゼラチンカプセルで提供される。カプセル製剤に含まれる賦形剤は、微結晶性セルロースNF/EP、ラウリル硫酸ナトリウムNF/DP(50mgの強度のみ)、クエン酸無水のUSP/EP、クロスカルメロースナトリウムNF/EP、コロイド性二酸化ケイ素NF/EPおよびステアリン酸マグネシウム(ウシのものでない)NF/EPである。GDC-0941カプセルは、36 Fから46 F(2℃から8℃)の間の冷蔵温度で保存される。患者は、36 Fから46 F(2℃から8℃)の間の冷蔵温度で試験薬剤を保存するように指示される。
【0134】
結果測定
安全性、薬物動態、薬力学および有効性の結果測定は、実施例4にあるように統計的方法を含む方法によって決定され、評価される。
【0135】
試験治療
試験治療は3週サイクルで投与される。試験治療から臨床的利益を得る患者は、開発状況、薬剤有効性および他の因子に応じて、別の試験で生じうる複数のサイクルの治療の可能性を有する。
試験の用量漸増期では、参加している患者は、空腹時にサイクル1の第1日目にGDC−0941の一回量を摂取し、GDC−0941 PK投薬前及び投薬後の試料を採取し、そして、患者内の多様性を観察する。GDC−0941の開始用量は60mgのqdであり、これは第I相試験において用量制限毒性がなく、単一の薬剤として安全であることが決定されている用量である。サイクル1の1日目に、完全用量のT−DM1は、負荷用量なしで3.6mg/kgの90分にわたるIVにより投与される。この後にGDC−0941の投薬を行う。患者は、初めのT−DM1注入後の90分間モニターされる。次いで、GDC-0941は1日1回、合計14用量の後、第1サイクルのために1週間空ける。
その後の患者におけるGDC−0941の用量漸増は、進行又は不耐容が生じるまで続ける。続く試験治療サイクルは3週間の長さであり、各サイクルの1日目の最初に3.6mg/kgのT−DM1の30分のIV投与と、T−DM1注入の後に投与されるGDC−0941によるものであり、合計2週間続け、1週間休む。投薬は、進行又は不耐容が生じるまで続ける。T−DM1は30〜90分(±10)のIV注入として投与され、これは親の試験においてどれほどのT−DM1が許容されたかに依存する。90分の注入が十分に許容される場合、続く注入は30(±10)分にわたって供給されてよい。
【0136】
先の記載は本発明の原理を単に例示するものとして考慮される。更に、当業者には多くの修正及び変更が直ぐに明らかであるので、発明を前記した正確な構成及び方法に限定することは望まれない。したがって、あらゆる適切な修正及び均等物が次の特許請求の範囲によって定まる発明の範囲に入ると考えられうる。
【技術分野】
【0001】
(関連出願の説明)
本非仮出願は、米国特許法規則1.53(b)の下で出願し、米国特許法119条(e)に基づき、2008年3月18日に出願の米国仮出願番号第61/037410号の優先権を主張する。この仮出願は出典明記により全体が援用される。
(技術分野)
本発明は、概して、癌などの過剰増殖性疾患に対して活性を有する化合物の製薬的併用に関する。また、本発明は、哺乳動物細胞又は関連の病的状態の、インビトロ、インサイツ及びインビボの診断又は治療のための化合物の併用の使用方法に関する。
【背景技術】
【0002】
HER2(ErbB2)レセプターチロシンは、膜貫通型レセプターの上皮性増殖因子レセプター(EGFR)ファミリのメンバーである。HER2の過剰発現は、およそ20%のヒトの乳癌において観察され、これらの腫瘍と関係する悪性の増殖および臨床結果不良に関係する(Slamon et al (1987) Science 235:177-182)。
トラスツズマブ(CAS180288-69-1、HERCEPTIN(登録商標)、huMAb4D5-8、rhuMAb HER2、Genentech)は、ヒト上皮細胞増殖因子レセプター2タンパク質であるHER2(ErbB2)の細胞外ドメインに対して、細胞ベースのアッセイにおいて高い親和性(Kd=5nM)で選択的に結合する、マウスHER2抗体の組み換えDNA由来のヒト化された、IgG1κ、モノクローナル抗体バージョンである(米国特許第5677171号;米国特許第5821337号;米国特許第6054297号;米国特許第6165464号;米国特許第6339142号;米国特許第6407213号;米国特許第6639055号;米国特許第6719971号;米国特許第6800738号;米国特許第7074404号;Coussens et al (1985) Science 230:1132-9;Slamon et al (1989) Science 244:707-12;Slamon et al (2001) New Engl. J. Med. 344: 783-792)。トラスツズマブは、HER2に結合するマウス抗体(4D5)の相補性決定領域と共にヒトフレームワーク領域を含有する。トラスツズマブは、HER2抗原と結合して、それにより癌細胞の増殖を阻害する。トラスツズマブは、インビトロアッセイ及び動物内において、HER2を過剰発現するヒト腫瘍細胞の増殖を阻害することが示された(Hudziak et al (1989) Mol Cell Biol 9:1165-72;Lewis et al (1993) Cancer Immunol Immunother; 37:255-63;Baselga et al (1998) Cancer Res. 58: 2825-2831)。トラスツズマブは抗体依存性細胞障害作用、ADCCのメディエーターである(Lewis et al (1993) Cancer Immunol Immunother 37(4): 255-263;Hotaling et al (1996) [abstract]. Proc. Annual Meeting Am Assoc Cancer Res; 37:471;Pegram MD, et al (1997) [abstract]. Proc Am Assoc Cancer Res; 38:602;Sliwkowski et al (1999) Seminars in Oncology 26(4), Suppl 12:60-70;Yarden Y. and Sliwkowski, M. (2001) Nature Reviews: Molecular Cell Biology, Macmillan Magazines, Ltd., Vol. 2: 127-137)。
【0003】
HERCEPTIN(登録商標)は、多くの従来の抗癌療法を受けたErbB2を過剰発現させている転移性乳癌患者の治療のために1998年に承認され(Baselga et al, (1996) J. Clin. Oncol. 14:737-744)、その後300000人以上の患者において使用されている(Slamon DJ, et al. N Engl J Med 2001;344:783 92;Vogel CL, et al. J Clin Oncol 2002;20:719 26;Marty M, et al. J Clin Oncol 2005;23:4265 74;Romond EH, et al. T N Engl J Med 2005;353:1673 84;Piccart-Gebhart MJ, et al. N Engl J Med 2005;353:1659 72;Slamon D, et al. [abstract]. Breast Cancer Res Treat 2006, 100 (Suppl 1): 52)。2006年に、FDAは、HER2陽性、リンパ節陽性の乳癌患者の補助治療のためのドキソルビシン、シクロホスファミドおよびパクリタキセルを含む治療投薬計画の一部として、HERCEPTIN(登録商標)(トラスツズマブ、Genentech Inc.)を承認した。HERCEPTIN(登録商標)が開発されたのでHER2陽性腫瘍患者は、化学療法単独よりも著明に良好な結果を得たが、実際にはすべてのHER2陽性、転移性乳癌(MBC)患者が有用な治療法を最終的に進歩させるであろう。MBC患者の結果を向上させる機会はまだある。トラスツズマブの多様な作用機構にもかかわらず、トラスツズマブにより治療される多くの患者は、治療が有効な期間の経過後に応答を示さないか又は応答が止まる。HER2+(HER2陽性)腫瘍にはHERCEPTIN(登録商標)に応答しないものもあり、応答する腫瘍を有する患者の多くは最終的に向上する。HERCEPTIN治療に対して応答しないか又は応答不良である、HER2を過剰発現する腫瘍患者又はHER2発現と関係する他の疾患患者のために、HER2に誘導された癌の治療法をさらに開発するという重要な臨床上の必要性がある。
【0004】
抗体を標的化した治療法に対する他のアプローチは、抗原を発現する癌細胞への細胞障害性剤の特異的運搬のために抗体を利用することである。細胞分裂阻害剤メイタンシンの誘導体であるメイタンシノイドは、ビンカアルカロイド薬剤と類似の方法で微小管に結合する(Issell BF et al (1978) Cancer Treat. Rev. 5: 199-207;Cabanillas F et al. (1979) Cancer Treat Rep, 63:507-9。トラスツズマブに連結したメイタンシノイドDM1からなる抗体−薬剤コンジュゲート(ADC)は、HER2を過剰発現するトラスツズマブ感受性かつトラスツズマブ耐性の腫瘍細胞株およびヒト乳癌の異種移植片モデルにおいて強力な抗腫瘍活性を示す。MCCリンカーを介して抗HER2マウス乳癌抗体TA.1に連結したメイタンシノイドのコンジュゲートは、ジスルフィドリンカーによる対応コンジュゲートの200分の1の強さであって(Chari et al (1992) Cancer Res. 127-133)。トラスツズマブに連結した、メイタンシノイドDM)からなる抗体−薬剤コンジュゲート(ADC)は、HER2を過剰発現するトラスツズマブ感受性かつトラスツズマブ耐性の腫瘍細胞株およびヒト癌の異種移植片モデルにおいて強力な抗腫瘍活性を示す。トラスツズマブ−MCC−DM1(T−DM1)は現在、HER2に対する治療法に抵抗性である疾患を有する患者において第II相臨床試験の評価が行われている(Beeram et al (2007) "A phase I study of trastuzumab-MCC-DM1 (T-DM1), a first-in-class HER2 antibody-drug conjugate (ADC), in patients (pts) with HER2+ metastatic breast cancer (BC)", American Society of Clinical Oncology 43rd: June 02 (Abs 1042;Krop et al, European Cancer Conference ECCO, Poster 2118, September 23-27, 2007, Barcelona;米国特許第7097840号;米国公開特許2005/0276812;米国公開特許2005/0166993)。
【0005】
数回の投薬計画又は投与様式において2以上の薬剤が共に用いられる併用療法は一般に、(i)最小の交差耐性を有する薬剤を組み合わせることによって耐性獲得が生じる頻度を低減する、(ii)僅かな副作用で有効性を達成する、すなわち治療指標を上げるために、毒性がオーバーラップしておらず類似の治療上の特性を有する薬剤の用量を減らす、(iii)細胞-周期段階又は生育特性を変えるなどといった、他の薬剤の使用によるある薬剤の作用に対して細胞を感作させる、そして(iv)2つの薬剤の生物活性の相加効果又は相加以上の効果を利用することによって有効性を上げるといった一又は複数の目的を有する(Pegram, M., et al (1999) Oncogene 18:2241-2251;Konecny, G., et al (2001) Breast Cancer Res. and Treatment 67:223-233;Pegram, M., et al (2004) J. of the Nat. Cancer Inst. 96(10): 739-749;Fitzgerald et al (2006) Nature Chem. Biol. 2(9): 458-466;Borisy et al (2003) Proc. Natl. Acad. Sci 100(13): 7977-7982)。
【0006】
ロエベ相加効果(Chou, T.C. and Talalay, P. (1977) J. Biol. Chem. 252:6438-6442;Chou, T.C. and Talalay, P. (1984) Adv. Enzyme Regul. 22:27-55;Berenbaum, M.C. (1989) Pharmacol. Rev. 41: 93-141)及びブリス非依存性/相乗効果(Bliss, C.I. (1956) Bacteriol. Rev. 20: 243-258;Greco et al (1995) Pharmacol. Rev. 47: 331-385)は、標的を50%阻害するために必要とされ、単純な場合のKiに等しい薬剤の用量であるIC50などのパラメーターに基づいて、単一療法と比較して併用療法について予想される用量−応答相関を算出するために用いられる方法である。
HER2二量体化阻害薬抗体およびEGFR阻害薬は、癌に対する併用療法について報告されている(米国公開特許2007/0020261)。トラスツズマブ−MCC−DM1(T−DM1)及びパーツズマブはMBC患者における活性が示され、パーツズマブとトラスツズマブの組合せはHER陽性MBC患者において活性であることが示されている(Baselga J, et al. "A Phase II trial of trastuzumab and pertuzumab in patients with HER2-positive metastatic breast cancer that had progressed during trastuzumab therapy: full response data", European Society of Medical Oncology, Stockholm, Sweden, September 12?16, 2008)。
【発明の概要】
【0007】
本発明は概して、癌細胞の増殖を阻害するための、一又は複数の化学療法剤と組み合わせて投与される、抗HER2 抗体−薬剤コンジュゲート、トラスツズマブ−MCC−DM1に関する。トラスツズマブ−MCC−DM1と化学療法剤のある組合せは、インビトロ及びインビボでの癌細胞の増殖を阻害する際に相乗効果を示す。本発明の組合せおよび方法は、癌などの過剰増殖性疾患の治療において有用でありうる。この組合せは、哺乳動物における腫瘍増殖を阻害し得、ヒトの癌患者を治療するために有用でありうる。
一態様では、本発明は、併用製剤として又は交互により哺乳動物に治療上の組合せを投与することを含む、過剰増殖性疾患の治療方法であって、このとき治療上の組合せにはトラスツズマブ−MCC−DM1の治療上有効量と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される化学療法剤の治療上有効量が含まれる方法を包含する。
トラスツズマブ−MCC−DM1の治療上有効量と化学療法剤の治療上有効量は、併用製剤として又は交互により投与されてよい。
【0008】
また、本発明は、哺乳動物細胞、生物体又は関連する病的状態の、インビトロ、インサイツ及びインビボの診断又は治療のための組成物の使用に関する。
また、本発明は、治療的併用の投与により相乗効果が生じる方法にも関する。
本発明の他の態様は、化学療法剤がHER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される、トラスツズマブ−MCC−DM1と、一又は複数の薬学的に許容可能な担体、流動促進剤、希釈液又は賦形剤とを含む薬学的組成物である。
本発明の他の態様は、治療を必要とする哺乳動物にトラスツズマブ−MCC−DM1と化学療法剤の有効量を投与することを含む、過剰増殖性疾患または疾病の治療方法を提供する。トラスツズマブ-MCC-DM1と化学療法剤は薬学的製剤として組み合わせて投与するために共に製剤化されも、又は治療的併用として交互に(交代して、連続的な投与)別々に投与されてもよい。一実施態様では、T−DM1は注入によって供給され、化学療法剤は経口的に運搬される。
【0009】
本発明の他の態様は、組合せにトラスツズマブ−MCC−DM1および抗癌性の標準的な治療を有する化学療法剤を含む場合のインビボ有効性についての有効な薬剤組合せを予測するための方法を提供する。インビトロ細胞増殖およびインビボ腫瘍異種移植片実験からの有効性データは、定性的及び定量的に分析される。定量的分析法は、相乗効果、拮抗作用又は相加効果を示すための併用指標(CI)値を生成するアイソボログラム及びChou & Talalay中央有効値、又はブリス非依存性リボン(Bliss Independence ribbon)グラフ屈折に基づいてよい。
本発明の他の態様は、哺乳動物のHER2又はKDR9(VEGFレセプター1)によって媒介されるものを含む、癌などの疾患又は疾病を治療するための、本発明の治療上の併用の使用方法である。
本発明の他の態様は、哺乳動物のHER2又はKDR9(VEGFレセプター1)によって媒介されるものを含む、癌などの疾患又は疾病を治療するための医薬の調製における、本発明の治療上の併用の使用である。
本発明の他の態様には、トラスツズマブ−MCC−DM1、化学療法剤、容器および場合によって治療を示すパッケージ挿入物又はラベルを具備する製造品又はキットが含まれる。
本発明の他の態様は、癌の治療のために併用して用いられる化合物の決定方法であって、a)トラスツズマブ−MCC−DM1と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される化学療法剤とをインビトロ腫瘍細胞株に投与することと、b)相乗効果又は非相乗効果を測定することを含む方法を包含する。
【0010】
本発明の更なる利点および新しい特徴は、後述する説明にある程度示され、以下の明細書の実施例により当業者にある程度明らかになるか、又は本発明の実施によってわかるであろう。本発明の利点は、特に添付の請求の範囲において示される手段、組合せ、組成物および方法によって理解され、達成されてよい。
【図面の簡単な説明】
【0011】
【図1】トラスツズマブ、トラスツズマブ−MCC−DM1(T-DM1)およびトラスツズマブとT−DM1の併用のIC50倍数濃度に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。
【図2】トラスツズマブ、トラスツズマブ−MCC−DM1(T-DM1)およびトラスツズマブとT−DM1の併用のIC50倍数濃度に対する、3日のBT−474EEIインビトロ細胞生存度のプロット線を示す。
【図3】パーツズマブ、トラスツズマブ−MCC−DM1(T-DM1)およびパーツズマブとT−DM1の併用のIC50倍数濃度に対する、5日のMDA−MB−175インビトロ細胞生存度のプロット線を示す。
【図3a】パーツズマブ、トラスツズマブ−MCC−DM1(T-DM1)およびパーツズマブとT−DM1の併用のIC50倍数濃度に対する、5日のMDA−MB−175インビトロ細胞生存度のプロット線を示す。
【図4】用量反応のトラスツズマブ−MCC−DM1(T-DM1)と併用した様々な一定用量のパーツズマブ、および様々な用量のT−DM1単独に対する、5日のBT-474インビトロ細胞生存度のプロット線を示す。
【図5】用量反応のパーツズマブと併用した様々な一定用量のトラスツズマブ−MCC−DM1(T-DM1)、および様々な用量のパーツズマブ単独に対する、5日のBT-474インビトロ細胞生存度のプロット線を示す。
【図6】パーツズマブ、トラスツズマブ−MCC−DM1(T-DM1)およびパーツズマブとT−DM1の併用のIC50倍数濃度に対する、5日のBT−474インビトロ細胞生存度のプロット線を示す。
【図7】一定用量のラパチニブと併用した変更用量のT−DM1(4.5nM、14nM、41nM、123nM)、及び変更用量のT−DM1単独(0〜1000ng/ml)に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。
【図7a】T−DM1、ラパチニブおよび一定用量比のT−DM1とラパチニブの併用に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。
【図8a】T−DM1、ラパチニブおよび一定用量比のT−DM1とラパチニブの併用に対する、3日のBT−474インビトロ細胞生存度のプロット線を示す。
【図8】一定用量のラパチニブと併用した変更用量のT−DM1(1.5nM、4.5nM、14nM、41nM、123nM)、及び変更用量のT−DM1単独(0〜1000ng/ml)に対する、3日のBT−474インビトロ細胞生存度のプロット線を示す。
【図9】一定用量のラパチニブと併用した変更用量のT−DM1(14nM、41nM、123nM、370nM、1111nM)、及び変更用量のT−DM1単独(0〜1000ng/ml)に対する、3日のBT−474−EEIインビトロ細胞生存度のプロット線を示す。
【図10】以下を投与した後のSCIDベージュマウスの乳房脂肪パッドに接種したKPL-4腫瘍(1マウスにつきマトリゲル中3000000細胞)における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ADCバッファ、(2) パーツズマブ15mg/kg、(3) T−DM1 0.3mg/kg、(4) T−DM1 1mg/kg、(5) T−DM1 3mg/kg、(6) パーツズマブ15mg/kg+T−DM1 0.3mg、(7) パーツズマブ15mg/kg+T−DM1 1mg/kg、(8)パーツズマブ15mg/kg+T−DM1 3mg/kg。ADCバッファおよびT−DM1は0日目に1度投与した。パーツズマブは、0、7および14日目に1度投与した。
【図11】以下を投与した後のSCIDベージュマウスの乳房脂肪パッドに接種したKPL-4腫瘍(1マウスにつきマトリゲル中3000000細胞)における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ADCバッファ、(2) 5−FU 100mg/kg、(3) パーツズマブ40mg/kg、第一パーツズマブ用量(5、7および9群)は2倍の負荷投与量とした、(4) B20-4.1、5mg/kg、(5) T−DM1、5mg/kg、(6) 5−FU、100mg/kg+T−DM1、5mg、(7) パーツズマブ40mg/kg+T−DM1、5mg/kg、(8) B20-4.1、5mg/kg+T−DM1、5mg/kg、(9) B20-4.1、5mg/kg+パーツズマブ、40mg/kg。ADCバッファおよびT−DM1は単回静注により0日目に1度投与した。パーツズマブは0、7、14、21日目に投与した(qwk ×4)。5−FUは0、7および14日目に投与した(qwk ×3)。B20-4.1は、0、3、7、10、14、17、21および24日目に投与した(2×/wk ×8合計)。
【図12】以下を投与した後のCRLnu/nuマウスの乳房脂肪パッドに接種したMMTV-HER2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル(ADCバッファ)、(2) B20-4.1、5mg/kg、(3) T−DM1、3mg/kg、(4) T−DM1、5mg/kg、(5) T−DM1、10mg/kg、(6) B20-4.1、5mg/kg+T−DM1 3mg/kg、(7) B20-4.1、5mg/kg+T−DM1 5mg/kg、(8) B20-4.1、5mg/kg+T−DM1、10mg/kg。ADCバッファおよびT−DM1は0および21日目に投与した。B20-4.1は、0、3、7、10、14、17、21および24日目に投与した(2×/wk ×4、合計8)。
【図13】以下を投与した後のCRLnu/nuマウスの乳房脂肪パッドに接種したMMTV-HER2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル(ADCバッファ)、(2) T−DM1 10mg/kg、(3) 5−FU 100mg/kg、(4) ゲムシタビン120mg/kg、(5)カルボプラチン100mg/kg、(6) 5−FU 100mg/kg+T−DM1 10mg/kg、(7) ゲムシタビン120mg/kg+T−DM1 10mg/kg、(8) カルボプラチン100mg/kg+T−DM1 10mg/kg。ADCバッファ、T−DM1およびカルボプラチンは、0日目に単回注射した。単一の注射。5−FUは0、7および14日目に投与した(qwk ×3)。ゲムシタビンは0、3、6および9日目に投与した(q3d ×4)。
【図14】以下を投与した後のハーラン胸腺欠損ヌードマウスの乳房脂肪パッドに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル(PBSバッファ) iv、qwk ×4、(2) ラパチニブ101mg/kg、po、bid ×21、(3) パーツズマブ40mg/kg、iv、qwk ×4、(4) B20-4.1 5mg/kg、ip、2×/wk ×4、(5) T−DM1 15mg/kg、iv、最後までq3wk、(6) ラパチニブ101mg/kg、po、bid ×21+T−DM1 15mg/kg、iv、最後までq3wk、(7) パーツズマブ40mg/kg、iv、qwk ×4+T−DM1 15mg/kg、iv、最後までq3wk、(8) B20-4.1 5mg/kg、ip、2×/wk×4+T−DM1 15mg/kg、iv、最後までq3wk。
【図15】以下を投与した後のハーラン胸腺欠損ヌードマウスの乳房脂肪パッドに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル(PBSバッファ) po、bid ×21、(2) T−DM1、7.5mg/kg、iv、qd ×1、(3) T−DM1、15mg/kg、iv、qd ×1、(4) ABT−869、5mg/kg、po、bid ×21、(5) ABT−869、15mg/kg、po、bid ×21、(6) T−DM1、7.5mg/kg、iv、qd ×1+ABT−869、5mg/kg、po、bid ×21、(7) T−DM1 7.5mg/kg、iv、qd ×1+ABT−869、15mg/kg、po、bid ×21、(8) T−DM1、15mg/kg、iv、qd ×1+ABT−869、5mg/kg、po、bid ×21、(9) T−DM1、15mg/kg、iv、qd ×1+ABT−869、15mg/kg、po、bid ×21。
【図16】以下を投与した後のハーラン胸腺欠損ヌードマウスの乳房脂肪パッドに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル、iv、qwk ×3、(2) T−DM1、7.5mg/kg、iv、q3wk ×2、(3) T−DM1、15mg/kg、iv、q3wk ×2、(4)ドセタキセル、30mg/kg、iv、qwk ×3、(5) T−DM1、7.5mg/kg、iv、q3wk ×2+ドセタキセル、30mg/kg、iv、qwk ×3、(6) T−DM1、15mg/kg、iv、q3wk ×2+ドセタキセル、30mg/kg、iv、qwk ×3。
【図17】以下を投与した後のハーラン胸腺欠損ヌードマウスの乳房脂肪パッドに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル、po、qd×21、(2) T−DM1、7.5mg/kg、iv、q3wk ×2、(3) T−DM1、15mg/kg、iv、q3wk ×2、(4) ラパチニブ、100mg/kg、po、bid ×21、(5) T−DM1、7.5mg/kg、iv、q3wk ×2+ラパチニブ、100mg/kg、po、bid ×21、(6) T−DM1、15mg/kg、iv、q3wk ×2+ラパチニブ、100mg/kg、po、bid ×21。
【図18】5−FU、トラスツズマブ−MCC−DM1(T-DM1)および5−FUとT−DM1の一定用量比組合せのIC50倍数の濃度に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。
【図19】5−FU、トラスツズマブ−MCC−DM1(T-DM1)および5−FUとT−DM1の一定用量比組合せのIC50倍数濃度に対する、3日のBT−474インビトロ細胞生存度のプロット線を示す。
【図20】ゲムシタビン、トラスツズマブ−MCC−DM1(T-DM1)およびゲムシタビンとT−DM1の一定用量比組合せのIC50倍数濃度に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。
【図21】ゲムシタビン、トラスツズマブ−MCC−DM1(T-DM1)およびゲムシタビンとT−DM1の一定用量比組合せのIC50倍数濃度に対する、3日のMDA−MD−361インビトロ細胞生存度のプロット線を示す。
【図22】0.25×〜4×のIC50倍数濃度でのT−DM1、GDC−0941、およびT−DM1とGDC−0941(62.5nM〜1μM)の1:10一定用量比の組合せによる処置後3日の、KPL4インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。
【図23】0.0625×〜16×のIC50倍数濃度でのT−DM1、GDC−0941、およびT−DM1(1.25〜80ng/ml)とGDC−0941(31.25nM〜2μM)の1:25一定用量比の組合せによる処置後3日の、KPL4インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。
【図24】0から16×のIC50倍数濃度での、T−DM1、PI103、GDC−0941、およびT−DM1+PI103及びT−DM1+GDC−0941の一定用量比の組合せによる処置後3日の、Her2増幅、HERCEPTIN(登録商標)耐性、PIK3CA(H1047R)変異のKPL-4細胞インビトロ細胞生存度(増殖)のプロット線を示す。
【図25】T−DM1、GDC−0941、および160ng/ml以下のT−DM1濃度の一定用量比のT−DM1とGDC−0941組合せによる処置後24時間の、KPL4カスパーゼ3/7インビトロ細胞生存度(増殖)のプロット線を示す。
【図26】T−DM1、GDC−0941、および0〜200ng/mlのT−DM1濃度の一定用量比のT−DM1とGDC−0941組合せによる処置後3日の、KPL4インビトロ細胞生存度(増殖)のプロット線を示す。
【図27】0.125×〜8×のIC50倍数濃度での、T−DM1、GDC−0941、およびT−DM1(3.125〜50ng/ml)とGDC−0941(62.5nM〜1μM)の1:20一定用量比の組合せによる処置後3日の、MDA−0MB−361インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。
【図28】0.125×〜8×のIC50倍数濃度での、T−DM1、GDC−0941、およびT−DM1(3.125〜100ng/ml)とGDC−0941(62.5nM〜2μM)の1:20一定用量比の組合せによる処置後3日の、MDA−0MB−361インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。
【図29】0.125×〜4×のIC50倍数濃度での、T−DM1、GDC−0941、およびT−DM1(3.125〜100ng/ml)とGDC−0941(31.25nM〜1μM)の1:10一定用量比の組合せによる処置後3日の、BT−474インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。
【図30】0.25×〜4×のIC50倍数濃度での、T−DM1、GDC−0941、およびT−DM1(6.25〜100ng/ml)とGDC−0941(62.5nM〜1μM)の1:10一定用量比の組合せによる処置後3日の、BT-474インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。
【図31】0〜16×のIC50倍数濃度での、T−DM1、PI103、GDC−0941、およびT−DM1+PI103及びT−DM1+GDC−0941の一定用量比の組合せによる処置後3日の、Her2増幅、非PI3K変異のAU565細胞インビトロ細胞生存度(増殖)のプロット線を示す。
【図32】0〜16×のIC50倍数濃度での、T−DM1、PI103、GDC−0941、およびT−DM1+PI103及びT−DM1+GDC−0941の一定用量比の組合せによる処置後3日の、Her2増幅、PIK3CA(C420R)変異のEFM192A細胞インビトロ細胞生存度(増殖)のプロット線を示す。
【図33】0〜16×のIC50倍数濃度での、T−DM1、PI103、GDC−0941、およびT−DM1+PI103及びT−DM1+GDC−0941の一定用量比の組合せによる処置後の、Her2増幅、ハーセプチン(登録商標)耐性、PIK3CA(H1047R)変異のHCC1954細胞インビトロ細胞生存度(増殖)のプロット線を示す。
【図34】以下を投与した後のCRLnu/nuマウスに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル、po、qd×21、(2) T−DM1、10mg/kg、iv、q3wk、(3) 5−FU、100mg/kg、po、qwk ×2、(4) T−DM1、5mg/kg、iv、q3wk+5−FU、100mg/kg、po、qwk ×2。
【図35】以下を投与した後のCRLnu/nuマウスに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル、po、qd ×21、(2) T−DM1、5mg/kg、iv、qd ×1、(3) GDC−0941、100mg/kg、po、qd ×21、(4) GDC−0152、50mg/kg、po、qwk ×3、(5) T−DM1、5mg/kg、iv、qd ×1+GDC−0941、100mg/kg、po、qd ×21、(6) T−DM1、5mg/kg、iv、qd ×1+GDC−0152、50mg/kg、po、qwk ×3。
【図36】以下を投与した後のCRLnu/nuマウスに接種したMDA−MB−361.1乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル、po、qd ×21、(2) GDC−0941、25mg/kg、po、qd ×21、(3) GDC−0941、50mg/kg、po、qd ×21、(4) GDC−0941、100mg/kg、po、qd ×21、(5) T−DM1、3mg/kg、iv、qd ×1、(6) T−DM1、10mg/kg、iv、qd ×1、(7) GDC−0941、25mg/kg、po、qd ×21+T−DM1、3mg/kg、iv、qd ×1、(8) GDC−0941、50mg/kg、po、qd ×21+T−DM1、3mg/kg、iv、qd ×1、(9) GDC−0941、100mg/kg、po、qd ×21+T−DM1、3mg/kg、iv、qd ×1、(10) GDC−0941、25mg/kg、po、qd ×21+T−DM1、10mg/kg、iv、qd ×1、(11) GDC−0941、50mg/kg、po、qd ×21+T−DM1、10mg/kg、iv、qd ×1、(12) GDC−0941、100mg/kg、po、qd ×21+T−DM1、10mg/kg、iv、qd ×1。
【図37】以下を投与した後のCRLnu/nuマウスに接種したMDA−MB−361.1乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す。(1) ベヒクル[MCT(0.5%メチルセルロース/0.2%TWEEN80(登録商標))+琥珀酸塩バッファ(100mMの琥珀酸ナトリウム、100mg/mlのトレハロース、0.1%のTWEEN80、pH5.0)]、po+IV、qd ×21及びqd、(2) GNE-390、1.0mg/kg、po、qd ×21、(3) GNE−390、2.5mg/kg、po、qd ×21、(4) T−DM1、3mg/kg、iv、qd、(5) GNE−390、1.0mg/kg、po、qd ×21+T−DM1、3mg/kg、iv、qd、(6) GNE−390、2.5mg/kg、po、qd×21+T−DM1、3mg/kg、iv、qd。
【発明を実施するための形態】
【0012】
(例示的実施態様の詳細な説明)
以下、その例が添付の構造及び式で示される本発明の所定の実施態様が詳細に参照される。本発明は、列挙された実施態様に関連して記載されるが、それらは本発明をこれら実施形態に限定することを意図するものではないことが理解される。逆に、本発明は、特許請求の範囲に記載の本発明の範囲内に含まれ得るあらゆる代替物、改変物、及び均等物を包含するものである。当業者であれば、本発明の実施に使用され得、ここに記載されたものと類似か等価である多くの方法及び材料を認識できるであろう。本発明は、記載された方法及び材料には決して限定されない。援用される文献、特許、及び類似の資料のうちの一又は複数が、限定しないが、定義された用語、用語の使用法、記載された技術などを含む本願と異なるか又は矛盾する場合、本願が優先する。
【0013】
(定義)
本明細書中及び特許請求の範囲において用いられる「含む」、「含んでいる」、「包含する(include)」、「包含している」及び「包含する(includes)」なる用語は、述べられた形質、整数、成分又は工程の存在を特定することを意図するが、一又は複数の他の形質、整数、成分、工程、又はその群の存在又は付加を妨げない。
「処置する」および「処置」なる用語は、治療的処置と予防的処置ないし防御的処置との両方をいい、その目的は、望ましくない生理学的変化または障害、例えば、癌などの過剰増殖性状態の増殖、発生または広がりを予防するかまたは遅くする(減らす)ことである。本発明の目的で、有利または望ましい臨床結果としては、検出可能であれ検出不可能であれ、症状の軽減、疾患の程度の減少、疾患の安定化された(すなわち、悪化しない)状態、疾患の進行の遅延または減速、疾患状態の軽減または緩和、および寛解(部分的であれ全体的であれ)が含まれる。また、「処置」は、処置を受けない場合に予測される生存と比較される場合に、生存を延長することを意味し得る。処置を必要とするものとしては、その状態または障害をすでに有するもの、並びにその状態または障害を有しやすいもの、あるいはその状態または障害が予防されるべきであるものが含まれる。
【0014】
「治療上の有効量」なる表現は、(i)本明細書中に記載される特定の疾患、状態又は障害を処置するか、(ii)特定の疾患、状態または障害の一又は複数の症状を寛解、軽減または排除するか、あるいは(iii)特定の疾患、状態、または障害の一又は複数の症状の発生を予防または遅延させる、本発明の化合物の量を意味する。癌の場合、薬物の治療上の有効量は、癌細胞の数を減少させ得;腫瘍のサイズを減少させ得;周辺器官への癌細胞の浸潤を阻害し得(すなわち、ある程度まで遅くし得、そして好ましくは停止させ得);腫瘍の転移を阻害し得(すなわち、ある程度まで遅くし得、そして好ましくは停止させ得);腫瘍の増殖をある程度まで阻害し得;そして/または癌に関連する症状のうちの一又は複数をある程度まで緩和し得る。薬物が、存在する癌細胞の増殖を防止し得、そして/または殺傷し得る場合、この薬物は、細胞増殖抑制性および/または細胞障害性であり得る。癌治療のためには、効力は、例えば、疾患の進行までの時間(TTP)を評価すること、および/または応答速度(RR)を決定することによって測定され得る。
【0015】
「過剰増殖性疾患」は、前悪性及び非腫瘍性のステージを含む腫瘍、癌及び腫瘍性組織を示し、乾癬、子宮内膜症、ポリープおよび線維腺腫も含む。
「癌」及び「癌性」なる用語は、典型的に制御されない細胞増殖によって特徴付けられる哺乳動物の生理的な状態を指すか又は表す。「腫瘍」は一又は複数の癌性細胞を含む。癌の例には、癌腫、リンパ腫、芽細胞腫、肉腫、および白血病またはリンパ悪性疾患が挙げられるが、これらに限定されない。このような癌のより具体的な例としては、扁平上皮癌(例えば、上皮扁平上皮癌)、小細胞肺癌、非小細胞肺癌(「NSCLC」)、肺の腺癌および肺の鱗状癌腫を含む肺癌、腹膜の癌、肝細胞癌、胃腸癌を含む胃癌(gastric or stomach)、膵臓癌、神経膠芽細胞腫、子宮頚癌、卵巣癌、肝臓癌、膀胱癌、肝細胞種、乳癌、結腸癌、直腸癌、結腸直腸癌、子宮内膜または子宮の癌腫、唾液腺癌腫、腎臓癌または腎性癌、前立腺癌、外陰部癌、甲状腺癌、肝性癌腫、肛門癌腫、陰茎癌腫ならびに頭頸部癌が含まれる。
【0016】
作用機構に関係なく、「化学療法剤」は癌の治療において有用な化学化合物である。化学療法剤の種類には、限定するものではないが、アルキル化剤(alkyating agent)、アンチメタボライ、紡錘体阻害剤植物アルカロイド、細胞障害性/抗腫瘍抗生物質、トポイソメラーゼ阻害薬、抗体、光増感剤及びキナーゼ阻害薬が含まれる。化学療法剤は、「ターゲティング療法」と従来の化学療法とに用いられる化合物を含む。化学療法剤の例には、エルロチニブ(TARCEVA(登録商標), Genentech/OSI Pharm.)、ドセタキセル(TAXOTERE(登録商標), Sanofi-Aventis)、5−FU(フルオロウラシル、5‐フルオロウラシル、CAS番号51-21-8)、ゲムシタビン(GEMZAR(登録商標), Lilly)、PD-0325901(CAS番号391210-10-9, Pfizer)、シスプラチン(シス-ジアミン、ジクロロ白金(II)、CAS番号15663-27-1)、カルボプラチン(CAS番号41575-94-4)、パクリタキセル(TAXOL(登録商標), Bristol-Myers Squibb Oncology, Princeton, N.J.)、トラスツズマブ(HERCEPTIN(登録商標), Genentech)、テモゾロミド(4-メチル-5-オキソ-2,3,4,6,8-ペントアザ二環式[4.3.0]ノナ-2,7,9-トリエン-9-カルボキシアミド、CAS番号85622-93-1, TEMODAR(登録商標), TEMODAL(登録商標), Schering Plough)、タモキシフェン((Z)-2-[4-(1,2-ジフェニルブト-1-エニル)フェノキシ]-N,N-ジメチル-エタンアミド, NOLVADEX(登録商標), ISTUBAL(登録商標), VALODEX(登録商標))、及びドキソルビシン(ADRIAMYCIN(登録商標))、Akti-1/2、HPPD及びラパマイシンが含まれる。
【0017】
化学療法剤の他の例には、オキサリプラチン(ELOXATIN(登録商標), Sanofi)、ボルテゾミブ(VELCADE(登録商標), Millennium Pharm.)、スーテント(SUNITINIB(登録商標), SU11248, Pfizer)、レトロゾール(FEMARA(登録商標), Novartis)、メシル酸イマチニブ(GLEEVEC(登録商標), Novartis)、XL-518 (Mekインヒビター, Exelixis, 国際公開2007/044515)、ARRY-886 (Mekインヒビター, AZD6244, Array BioPharma, Astra Zeneca)、SF-1126(PI3Kインヒビター, Semafore Pharmaceuticals)、BEZ-235(PI3Kインヒビター, Novartis)、XL-147(PI3Kインヒビター, Exelixis)、PTK787/ZK 222584 (Novartis)、フルベストラント(FASLODEX(登録商標), AstraZeneca)、リューコボリン(フォリン酸)、ラパマイシン(シロリムス, RAPAMUNE(登録商標), Wyeth)、ラパチニブ(TYKERB(登録商標), GSK572016, Glaxo Smith Kline)、ロナファーニブ(SARASARTM, SCH 66336, Schering Plough)、ソラフェニブ(NEXAVAR(登録商標), BAY43-9006, Bayer Labs)、ゲフィチニブ(IRESSA(登録商標), AstraZeneca)、イリノテカン(CAMPTOSAR(登録商標), CPT-11, Pfizer)、ティピファニブ(ZARNESTRATM, Johnson & Johnson)、ABRAXANETM (Cremophor-free), パクリタキセルのアルブミン-変更ナノ粒子製剤(American Pharmaceutical Partners, Schaumberg, Il)、バンデタニブ(rINN, ZD6474, ZACTIMA(登録商標), AstraZeneca)、クロラムブシル、AG1478、AG1571(SU5271; Sugen)、テムシロリムス(TORISEL(登録商標), Wyeth)、パゾパニブ(GlaxoSmithKline)、カンフォスファミド(TELCYTA(登録商標), Telik)、チオテパ、及びシクロフォスファミド(CYTOXAN(登録商標)、NEOSAR(登録商標));ブスルファン、インプロスルファン及びピポスルファンのようなスルホン酸アルキル類;ベンゾドーパ(benzodopa)、カルボコン、メツレドーパ(meturedopa)、及びウレドーパ(uredopa)のようなアジリジン類;アルトレートアミン(altretamine)、トリエチレンメラミン、トリエチレンホスホラミド、トリエチレンチオホスホラミド(triethylenethiophosphaoramide)及びトリメチローロメラミン(trimethylolomelamine)を含むエチレンイミン類及びメチラメラミン類;アセトゲニン(acetogenins)(特にブラタシン(bullatacin)及びブラタシノン(bullatacinone));カンプトセシン(合成類似体トポテカン(topotecan)を含む);ブリオスタチン;カリスタチン(callystatin);CC-1065(そのアドゼレシン(adozelesin)、カルゼレシン(carzelesin)及びバイゼレシン(bizelesin)合成類似体を含む);クリプトフィシン(cryptophycin)(特にクリプトフィシン1及びクリプトフィシン8);ドラスタチン(dolastatin);デュオカルマイシン(duocarmycin )(合成類似体、KW−2189及びCBI−TM1を含む); エレトロビン(eleutherobin);パンクラチスタチン(pancratistatin);サルコディクチン(sarcodictyin);スポンジスタチン(spongistatin);クロランブシル、クロルナファジン(chlornaphazine)、チョロホスファミド(cholophosphamide)、エストラムスチン、イホスファミド、メクロレタミン、メクロレタミンオキシドヒドロクロリド、メルファラン、ノベンビチン(novembichin)、フェネステリン(phenesterine)、プレドニムスチン(prednimustine)、トロフォスファミド(trofosfamide)、ウラシルマスタード等のナイトロジェンマスタード;ニトロスレアス(nitrosureas)、例えばカルムスチン(carmustine)、クロロゾトシン(chlorozotocin)、フォテムスチン(fotemustine)、ロムスチン(lomustine)、ニムスチン、ラニムスチン;エネジイン(enediyne) 抗生物質等の抗生物質(例えば、カリケアマイシン(calicheamicin)、カリケアマイシンガンマ1I及びカリケアマイシンオメガI1、例えば、Agnew Chem Intl. Ed. Engl., 33:183-186(1994)を参照のこと;ダイネミシン(dynemicin)、ダイネミシンA(dynemicinA);クロドロネート(clodronate)などのビスホスホネート(bisphosphonates);エスペラマイシン(esperamicin); 同様にネオカルチノスタチン発光団及び関連色素蛋白エネジイン(enediyne) 抗生物質発光団)、アクラシノマイシン(aclacinomysins)、アクチノマイシン、オースラマイシン(authramycin)、アザセリン、ブレオマイシン(bleomycins)、カクチノマイシン(cactinomycin)、カラビシン(carabicin)、カルミノマイシン(carminomycin)、カルジノフィリン(carzinophilin)、クロモマイシン、ダクチノマイシン、ダウノルビシン、デトルビシン(detorubicin)、6-ジアゾ-5-オキソ-L-ノルロイシン、モルフォリノ−ドキソルビシン、シアノモルフォリノ-ドキソルビシン、2-ピロリノ-ドキソルビシン及びデオキシドキソルビシンを含む)、エピルビシン、エソルビシン(esorubicin)、イダルビシン、マセロマイシン(marcellomycin)、マイトマイシンCなどのマイトマイシン(mitomycins)、マイコフェノール酸(mycophenolic acid)、ノガラマイシン(nogalamycin)、オリボマイシン(olivomycins)、ペプロマイシン、ポトフィロマイシン(potfiromycin)、ピューロマイシン、クエラマイシン(quelamycin)、ロドルビシン(rodorubicin)、ストレプトニグリン、ストレプトゾシン、ツベルシジン(tubercidin)、ウベニメクス、ジノスタチン(zinostatin)、ゾルビシン(zorubicin);メトトレキセート及び5-フルオロウラシル(5-FU)のような抗-代謝産物;デノプテリン(denopterin)、メトトレキセート、プテロプテリン(pteropterin)、トリメトレキセート(trimetrexate)のような葉酸類似体;フルダラビン(fludarabine)、6-メルカプトプリン、チアミプリン、チオグアニンのようなプリン類似体;アンシタビン、アザシチジン(azacitidine)、6-アザウリジン(azauridine)、カルモフール、シタラビン、ジデオキシウリジン、ドキシフルリジン、エノシタビン(enocitabine)、フロキシウリジン(floxuridine)のようなピリミジン類似体;カルステロン(calusterone)、プロピオン酸ドロモスタノロン、エピチオスタノール、メピチオスタン、テストラクトン(testolactone)のようなアンドロゲン類;アミノグルテチミド、ミトタン、トリロスタンのような抗副腎剤;フロリン酸(frolinic acid)のような葉酸リプレニッシャー(replenisher);アセグラトン;アルドホスファミドグリコシド;アミノレブリン酸;エニルウラシル(eniluracil);アムサクリン(amsacrine);ベストラブシル(bestrabucil);ビサントレン(bisantrene);エダトラキセート(edatraxate);デフォファミン(defofamine);デメコルシン(demecolcine);ジアジコン(diaziquone);エルフォルニチン(elfornithine);酢酸エリプチニウム(elliptinium acetate);エポチロン(epothilone);エトグルシド(etoglucid);硝酸ガリウム;ヒドロキシ尿素;レンチナン;ロニダミン(lonidamine);メイタンシン(maytansine)及びアンサマイトシン(ansamitocin)のようなメイタンシノイド(maytansinoid);ミトグアゾン(mitoguazone);ミトキサントロン;モピダモール(mopidamol);ニトラクリン(nitracrine);ペントスタチン;フェナメット(phenamet);ピラルビシン;ポドフィリン酸(podophyllinic acid);2-エチルヒドラジド;プロカルバジン;PSK(登録商標)多糖類複合体(JHS Natural Products, Eugene, OR);ラゾキサン(razoxane);リゾキシン(rhizoxin);シゾフィラン;スピロゲルマニウム(spirogermanium);テニュアゾン酸(tenuazonic acid);トリアジコン(triaziquone);2,2',2''-トリクロロトリエチルアミン;トリコテセン(trichothecenes)(T-2トキシン、ベラキュリンA(verracurin A)、ロリデンA(roridin A)及びアングイデン(anguidine));ウレタン;ビンデシン;ダカルバジン;マンノムスチン(mannomustine);ミトブロニトール;ミトラクトール(mitolactol);ピポブロマン(pipobroman);ガシトシン(gacytosine);アラビノシド(「Ara-C」);シクロホスファミド;チオテパ;6-チオグアニン;メルカプトプリン;メトトレキセート;シスプラチン及びカルボプラチンのようなプラチナ類似体;ビンブラスチン;エトポシド(VP-16);ミトキサントン;ビンクリスチン;ビノレルビン(NAVELBINE(登録商標));ナベルビン(Navelbine);ノバントロン(novantrone);テニポシド;エダトレキセート;ダウノマイシン;アミノプテリン;カペシタビン(XELODA(登録商標), Roche);イバンドロナート(ibandronate);CPT-11;トポイソメラーゼインヒビターRFS2000;ジフルオロメチロールニチン(DMFO);レチノイン酸などのレチノイド類;、並びに上述したものの製薬的に許容可能な塩類、酸類及び誘導体が含まれる。
【0018】
また、「化学療法剤」の定義には、(i) 腫瘍に対するホルモン作用を調節又は阻害するように働く抗ホルモン剤、抗エストロゲン及び選択的エストロゲンレセプターモジュレーター(SERM)など、例えばタモキシフェン(NOLVADEX(登録商標)タモキシフェンクエン酸塩)、ラロキシフェン(raloxifene)、ドロロキシフェン、4-ヒドロキシタモキシフェン、トリオキシフェン(trioxifene)、ケオキシフェン(keoxifene)、LY117018、オナプリストーン(onapristone)、及びFARESTON(登録商標)(クエン酸トレミフェン);(ii) アロマターゼ酵素を阻害するアロマターゼ阻害物質、それらは副腎でのエストロゲン産生を調節するものであり、例えば4(5)-イミダゾール類、アミノグルテチミド、MEGASE(登録商標)(メゲストロールアセテート)、AROMASIN(登録商標)(エキセメスタン, Pfizer)、ホルメスタイン(formestanie)、ファドロゾール、RIVISOR(登録商標)(ゾロゾール(vorozole))、FEMARA(登録商標)(レトロゾール, Novartis)及びARIMIDEX(登録商標)(アナストロゾール, AstraZeneca);(iii) 抗アンドロゲン、例えばフルタミド(flutamide)、ニルタミド(nilutamide)、ビカルタミド、ロイプロリド、及びゴセレリン;並びにトロキサシタビン(troxacitabine)(1,3-ジオキソランヌクレオシドシトシン類似体);(iv) プロテインキナーゼインヒビター、例えばMEKインヒビター(国際公開2007/044515);(v) 脂質キナーゼインヒビター;(vi) アンチセンスオリゴヌクレオチド、特に不粘着性細胞増殖に関係するシグナル伝達経路の遺伝子の発現を抑制するもの、例えばPKC−α、Raf及びH−Ras、オブリメルセン(GENASENSE(登録商標), Genta Inc.);(vii) リボザイム、例えばVEGF発現インヒビター(例えばANGIOZYME(登録商標))及びHER2発現インヒビター;(viii) 遺伝子治療ワクチン、例えばALLOVECTIN(登録商標)、LEUVECTIN(登録商標)及びVAXID(登録商標)などのワクチン;PROLEUKIN(登録商標)rIL−2;LURTOTECAN(登録商標)などのトポイソメラーゼ1インヒビター;ABARELIX(登録商標)rmRH;(ix) ベバシズマブ(AVASTIN(登録商標), Genentech)などの抗血管形成剤;並びに上記のものの製薬的に許容可能な塩類、酸類又は誘導体が含まれる。
また、「化学療法剤」の定義には、治療的抗体、例として、アレムツズマブ(Campath)、ベバシズマブ(アバスチン(登録商標), Genentech);セツキシマブ(ERBITUX(登録商標), Imclone);パニツムマブ(VECTIBIX(登録商標), Amgen)、リツキシマブ(RITUXAN(登録商標), Genentech/Biogen Idec)、ペルツズマブ(OMNITARGTM, 2C4, Genentech)、トラスツズマブ(HERCEPTIN(登録商標), Genentech)、トシツモマブ(Bexxar, Corixia)、及び抗体薬剤コンジュゲート、ゲムツズマブオゾガマイシン(MYLOTARG(登録商標), Wyeth)が含まれる。
【0019】
トラスツズマブ−MCC−DM1と組み合わされる化学療法剤としての治療的能力を有するヒト化モノクローナル抗体には、アレムツズマブ、アポリズマブ、アセリズマブ、アトリズマブ、バピネオズマブ、ベバシズマブ、ビバツズマブ・メルタンシン、カンツズマブ・メルタンシン、セデリズマブ、セルトリズマブ・ペゴール、シドフシツズマブ、シドツズマブ、ダクリズマブ、エクリズマブ、エファリズマブ、エピラツズマブ、エルリズマブ、フェルビズマブ、フォントリズマブ、ゲムツズマブ・オゾガマイシン、イノツズマブ・オゾガマイシン、イピリムマブ、ラベツズマブ、リンツズマブ、マツズマブ、メポリズマブ、モタビズマブ、モトビズマブ、ナタリズマブ、ニモツズマブ、ノロビズマブ、ヌマビズマブ、オクレリズマブ、オマリズマブ、パリビズマブ、パスコリズマブ、ペクフシツズマブ、ペクツズマブ、パーツズマブ、パキセリズマブ、ラリビズマブ、ラニビズマブ、レスリビズマブ、レスリズマブ、レシビズマブ、ロベリズマブ、ルプリズマブ、シブロツズマブ、シプリズマブ、ソンツズマブ、タカツズマブ・テトラキセタン、タドシズマブ、タリズマブ、テフィバズマブ、トシリズマブ、トラリズマブ、トラスツズマブ、ツコツズマブ・セルモロイキン、ツクシツズマブ、ウマビズマブ、ウルトキサズマブおよびビシリズマブが含まれる。
【0020】
「代謝産物」は、特定の化合物又は塩の体内の代謝によって生産される生成物である。化合物の代謝産物は当分野で公知の慣例技術を使用して同定されてもよく、それらの活性は本明細書中に記載のものなどの試験を用いて決定されてもよい。このような生成物は、例えば、投与される化合物の酸化、還元、加水分解、アミド化、アミド分解、エステル化、エステル分解、酵素の切断などから生じうる。したがって、本発明は、代謝産物が得られるために十分な時間、本発明の化合物を哺乳動物と接触させることを含む方法によって産生される化合物を含む、本発明の化合物の代謝産物を含む。
【0021】
「パッケージ挿入物」という用語は、効能、用途、服用量、投与、配合禁忌及び/又はその治療薬の用途に関する警告についての情報を含む、治療薬の商業的包装を慣習的に含めた指示書を指すために用いられる。
【0022】
本明細書中で用いる「薬学的に許容可能な塩」なる表現は、本発明の化合物の薬学的に許容可能な有機塩又は無機塩を指す。例示的な塩には、限定するものではないが、硫酸塩、クエン酸塩、酢酸塩、シュウ酸塩、塩化物、臭化物、ヨウ素、硝酸塩、重硫酸塩、リン酸塩、酸性リン酸塩、イソニコチン酸塩、乳酸塩、サリチル酸塩、酸性クエン酸塩、酒石酸塩、オレイン酸塩、タンニン酸塩、パントテン酸塩、酒石酸水素塩、アスコルビン酸塩、琥珀酸塩、マレイン酸塩、gentisinate、フマル酸塩、グルコン酸塩、グロクロン酸塩、糖酸塩、蟻酸塩、安息香酸塩、グルタミン酸塩、メタンスルホン酸塩「メシレート」、エタンスルホン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩、及びパモ酸塩(すなわち、1,1'-メチレンビス(2-ヒドロキシ-3-ナフトエート))が含まれる。薬学的に許容可能な塩は、他の分子、例えば酢酸イオン、琥珀酸イオン又は他の対イオンの包含を伴ってもよい。対イオンは、親化合物上の荷電を安定化する任意の有機又は無機の部分でもよい。さらに、薬学的に許容可能な塩は、その構造内に複数の荷電原子を有してもよい。複数の荷電原子が薬学的に許容可能な塩の一部である場合は複数の対イオンを有しうる。したがって、薬学的に許容可能な塩は、一又は複数の荷電原子及び/又は一又は複数の対イオンを有してもよい。
【0023】
本発明の化合物が塩基性である場合、当分野で有用な任意の好適な方法、例えば塩酸、臭化水素酸、硫酸、硝酸、メタンスルホン酸、リン酸などといった無機酸による遊離塩基の処理、又は酢酸、マレイン酸、コハク酸、マンデル酸、フマル酸、マロン酸、ピルビン酸、シュウ酸、グリコール酸、サリチル酸、ピラノシジル酸、例としてグルクロン酸又はガラクツロン酸、αヒドロキシ酸、例えばクエン酸又は酒石酸、アミノ酸、例えばアスパラギン酸又はグルタミン酸、芳香族酸、例えば安息香酸又はニッケイ酸、スルホン酸、例えばp-トルエンスルホン酸又はエタンスルホン酸などといった有機酸による遊離塩基の処理によって調製されてもよい。一般に塩基性薬学化合物からの薬学的に有用又は許容可能な塩の形成に適すると考えられる酸は、例えばP. Stahl et al, Camille G. (eds.) Handbook of Pharmaceutical Salts. Properties, Selection and Use. (2002) Zurich: Wiley-VCH;S. Berge et al, Journal of Pharmaceutical Sciences (1977) 66(1) 1 19;P. Gould, International J. of Pharmaceutics (1986) 33 201 217;Anderson et al, The Practice of Medicinal Chemistry (1996), Academic Press, New York; Remington’s Pharmaceutical Sciences, 18th ed., (1995) Mack Publishing Co., Easton PA;及び、The Orange Book (Food & Drug Administration, Washington, D.C. ウェブサイト上)で論じられている。これらの開示内容は出典明記によって本明細書中に援用される。
【0024】
本発明の化合物が酸である場合、所望の薬学的に許容可能な塩は、任意の好適な方法、例えば、アミン(一次、二次又は三次)、アルカリ金属水酸化物又はアルカリ土類金属水酸化物などといった無機塩基又は有機塩基による遊離酸の処理によって調製されてもよい。好適な塩の例示的な例には、限定するものではないが、アミノ酸、例えばグリシン及びアルギニン、アンモニア、一次、二次、三次のアミン、及び環状アミン、例えばピペリジン、モルホリン及びピペラジンから得られる有機塩、並びにナトリウム、カルシウム、カリウム、マグネシウム、マンガン、鉄、銅、亜鉛、アルミニウム及びリチウムから得られる無機塩が含まれる。
【0025】
「薬学的に許容可能な」という表現は、物質又は組成物が製剤を含む他の成分と、及び/又は処置される哺乳動物と化学的及び/又は毒物学的に適合性がなければならないことを表す。
「溶媒和化合物」は、一又は複数の溶媒分子と本発明の化合物との物理的結合又は複合体を指す。本発明の化合物は溶媒和されていない状態及び溶媒和された形態で存在しうる。溶媒和化合物を形成する溶媒の例には、限定するものではないが、水、イソプロパノール、エタノール、メタノール、DMSO、酢酸エチル、酢酸及びエタノールアミンが含まれる。「水和物」なる用語は溶媒分子が水である場合の複合体を指す。この物理的な会合は、水素結合を含む、様々な程度のイオン的及び共有結合的結合を伴う。場合によって、例えば一又は複数の溶媒分子が結晶質固体の結晶格子に取り込まれている場合、溶媒和化合物は単離可能であろう。溶媒和化合物の調製は、例えばM. Caira et al, J. Pharmaceutical Sci., 93(3), 601 611 (2004)のように、一般に公知である。溶媒和化合物、ヘミ溶媒和化合物、水和物などの類似の調製は、E. C. van Tonder et al, AAPS PharmSciTech., 5(1), article 12 (2004);及び、A. L. Bingham et al, Chem. Commun., 603 604 (2001)によって記述される。 典型的な限定的でない方法は、室温より高い温度の所望の量の所望の溶媒(有機物又は水又はその混合物)に本発明の化合物を溶解することと、後に標準的な方法によって単離される結晶を形成するために十分な速度で溶液を冷やすことを伴う。例えばI.R.分光法といった分析技術は、溶媒和化合物(または水和物)としての結晶中の溶媒(または水)の存在を示す。
【0026】
本明細書中で用いる「相乗効果」なる用語は、2以上の単一の薬剤の相加効果よりも効果が高い治療的併用を指す。トラスツズマブ−MCC−DM1と一又は複数の化学療法剤との間の相乗作用の決定は、本明細書において記述されるアッセイから得られる結果に基づいてよい。これらのアッセイの結果は、併用インデックス「CI」を得るために、CalcuSynソフトウェアによる用量−効果分析とChou&Talalay組合せ法を用いて分析される(Chou and Talalay (1984) Adv. Enzyme Regul. 22:27-55)。本発明によって提供される組合せは、いくつかのアッセイシステムで評価され、データは、抗癌薬間の相乗作用、相加作用および拮抗作用を定量化するために、標準的なプログラムを利用して分析されうる。好適に利用されるプログラムは、"New Avenues in Developmental Cancer Chemotherapy," Academic Press, 1987, Chapter 2のChou and Talalayによって記述されるものである。0.8未満の併用インデックス(CI)値は相乗効果を示し、1.2を超える値は拮抗作用を示し、0.8〜1.2の値は相加効果を示す。併用療法は「相乗効果」を示してよく、「相乗的」である、つまり、共に用いる活性成分が別々の化合物を用いて得られる効果の合計よりも大きい場合に得られる効果である。相乗効果は、活性成分が:(1)同時に処方され、併用された単位投薬製剤として同時に投与又は送達されるか、(2)別個の製剤として交互に送達されるか、又は(3)ある種の他の計画による場合に、達成することができる。交互療法で送達される場合、相乗効果は、化合物が、例えば別々のシリンジで異なった注射によって逐次的に投与されるか又は送達される場合に達成されうる。一般に、交互療法の間、各活性成分の有効用量が逐次的、つまり連続的に投与される。
【0027】
(トラスツズマブ−MCC−DM1)
本発明は、以下の構造を有する、トラスツズマブ−MCC−DM1(T−DM1)、抗体−薬剤コンジュゲート(CAS Reg. 番号139504-50-0)を含有する治療的併用を含む。
ここで、Trは、リンカー部分MCCによってメイタンシノイド薬剤部分DM1に連結したトラスツズマブ(米国特許第5208020号;米国特許第6441163号)である。抗体に対する薬剤比率又は薬剤負荷は、トラスツズマブ−MCC−DM1の上記の構造のpによって表され、1からおよそ8の整数値である。薬剤負荷値pは1〜8である。トラスツズマブ-MCC-DM1は、1、2、3、4、5、6、7及び8の薬剤部分が抗体トラスツズマブに共有結合して付着している様々な負荷及び付着の抗体−薬剤コンジュゲートの混合すべてを含む(米国特許第7097840号;米国公開特許2005/0276812;米国公開特許2005/0166993)。トラスツズマブ-MCC-DM1は実施例1に従って調製されてよい。
【0028】
トラスツズマブは、哺乳動物細胞(チャイニーズハムスター卵巣、CHO)懸濁培養物によって生産される。HER2(又はc-erbB2)プロトオンコジーンは185kDaの膜貫通レセプタータンパク質をコードし、構造的に上皮性増殖因子レセプターに関連がある。HER2タンパク質過剰発現は、原発性乳癌の25%〜30%に観察され、固定腫瘍塊の免疫組織化学ベースの評価を使用して決定されてよい(Press MF, et al (1993) Cancer Res 53:4960-70。トラスツズマブは、マウス4D5抗体の、又はマウス4D5抗体由来の抗原結合残基を有する抗体である(ATCC CRL 10463、ブダペスト条約の下、1990年5月24日に米国培養細胞系統保存機関、12301 Parklawn Drive, Rockville, Md. 20852に寄託されている)。例示的なヒト化4D5抗体には、huMAb4D5-1、huMAb4D5-2、huMAb4D5-3、huMAb4D5-4、huMAb4D5-5、huMAb4D5-6、huMAb4D5-7、及び米国特許第5821337号に記載のhuMAb4D5-8(HERCEPTIN(登録商標))が含まれる。
第I相試験では、3週ごとのIV注入によって投与されるT−DM1の最大耐量(MTD)は、3.6mg/kgであった。DLT(用量制限毒性)は、4.8mg/kgで処置された3人の患者のうちの2人にグレード4の血小板減少からなった。3.6mg/kgでの2以上の関連グレードの有害事象は、まれであり、対処可能であった。この治療計画は十分に許容されるものであり、前記のような有意な臨床活性と関係していた。第II相試験は、3週間ごとに投与される3.6mg/kgの服用レベルで類似の耐容性を有し、用量の減少を必要とする患者の割合は極僅かであった(112人の患者のうちの3人)。ゆえに、1)3週間ごとの3.6mg/kgのT−DM1に示される有効性及び安全性、及び2)この患者集団に対する3週間投薬計画の利便性に基づいて、この試験で試験するために3週間ごとに投与される3.6mg/kgのT−DM1用量を選択した。
【0029】
(化学療法剤)
特定の化学療法剤は、インビトロ及びインビボの細胞増殖の阻害において、トラスツズマブ-MCC-DM1と組み合わせると、驚くべき予想外の性質を示した。このような化学療法剤には、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941、及びGNE-390が含まれる。
パーツズマブ(CA Reg.番号380610-27-5、OMNITARG(登録商標)、2C4、Genentech)は、HER2の二量体化を阻害する組換えヒト化モノクローナル抗体である(米国特許第6054297号;米国特許第6407213号;米国特許第6800738号;米国特許第6627196号、米国特許第6949245号;米国特許第7041292号)。パーツズマブおよびトラスツズマブは、HER-2チロシンキナーゼレセプターの異なる細胞外領域を標的とする(Nahta et al (2004) Cancer Res. 64: 2343-2346)。2C4(パーツズマブ)を発現するハイブリドーマ細胞株は、1999年4月8日に、ATCC HB-12697として、米国培養細胞系統保存機関(ATCC)、10801 University Boulevard, Manassas, Va. 20110-2209, USAに寄託された。パーツズマブは、他のHERレセプターファミリ、すなわちHER1/EGFR、HER3およびHER4と協働するHER2レセプターの能力を遮断する(Agus et al (2002) Cancer Cell 2:127-37;Jackson et al (2004) Cancer Res 64:2601-9;Takai et al (2005) Cancer 104:2701-8;米国特許第6949245号)。癌細胞において、他のHERファミリレセプターと協働するHER2の能力が干渉されると、細胞シグナル伝達が遮断され、最後には癌細胞増殖の阻害および癌細胞の死に至りうる。HDIは、その作用様式が特有であるために、HER2を過剰発現しないものを含む様々な腫瘍において作用する能力を有する(Mullen et al (2007) Molecular Cancer Therapeutics 6:93-100)。
【0030】
パーツズマブは、ヒトIgG1(κ)フレームワーク配列に基づく。これは2つの重鎖と2つの軽鎖からなる。トラスツズマブのように、パーツズマブはHER2の細胞外ドメインに対するものである。しかしながら、パーツズマブは、トラスツズマブとは軽鎖および重鎖のエピトープ結合領域が異なる。結果として、パーツズマブは、HER2のサブドメイン2として知られるものの中のエピトープに結合するのに対して、トラスツズマブのエピトープは、サブドメイン4に位置する(Cho et al. 2003; Franklin et al. 2004)。パーツズマブは、HER1(上皮性増殖因子レセプター;EGFR)、HER3およびHER4を含む他のHERファミリーメンバーとHER2との会合を遮断することによって作用する。この会合は、MAPキナーゼおよびPI3-キナーゼを経てリガンド存在下でのシグナル伝達に必要である。その結果、パーツズマブは、リガンドが惹起する細胞内シグナル伝達を阻害する。これらのシグナル伝達経路の阻害の結果、増殖休止およびアポトーシスがそれぞれ生じうる(Hanahan and Weinberg 2000)。パーツズマブおよびトラスツズマブがHER2レセプター上の異なるエピトープで結合するので、リガンドにより活性化される下流のシグナル伝達はトラスツズマブによって遮断されるが、パーツズマブによっては遮断されない。したがって、パーツズマブは、抗腫瘍剤としてその活性を発揮するために、HER2過剰発現を必要としない。さらに、それらが相補的に作用するために、パーツズマブとT−DM1の組合せは、HER2を過剰発現する疾患において役割を有する可能性がある。
【0031】
パーツズマブは、HER2を低レベルで発現するMBC、非小細胞肺癌、ホルモン抵抗性前立腺癌及び卵巣癌を含む様々な癌種において実施された5つの第II相試験において単一薬剤として評価された。第II相試験は、正常なHER2発現である転移性乳癌(MBC)患者の第二ライン又は第三ライン治療において、単一薬剤としてパーツズマブを評価した(Cortes et al. (2005) J. Clin. Oncol. 23:3068)。パーツズマブは、トラスツズマブと併用した2つの第II相試験において評価された(Baselga J, et al. "A Phase II trial of trastuzumab and pertuzumab in patients with HER2-positive metastatic breast cancer that had progressed during trastuzumab therapy: full response data", European Society of Medical Oncology, Stockholm, Sweden, September 12?16, 2008; Gelmon et al (2008) J. Clin. Oncol. 26:1026)。第一試験には、最大3つの従来のトラスツズマブを含む投薬計画を既に受けていたHER2陽性MBCを有する11人の患者が参加した(Portera et al. 2007)。
【0032】
ベバシズマブ(CAS Reg.番号216974-75-3、アバスチン(登録商標)、Genentech)は、癌の治療に用いられる、血管内皮性増殖因子に対する抗VEGFモノクローナル抗体であり(米国特許第7227004号;米国特許第6884879号;米国特許第7060269号;米国特許第7169901号;米国特許第7297334号)、新たな血管の形成を遮断することによって腫瘍増殖を阻害する。ベバシズマブは、転移性大腸癌及びほとんどの形式の転移性非小細胞肺癌の治療において標準的な化学療法と組み合わせて使用するために2004年FDAによって承認された、米国においては第一に臨床上有用な脈管形成阻害薬であった。アジュバント/非転移性大腸癌、転移性乳癌、転移性腎臓細胞カルチノーマ、転移性多形性神経膠芽腫、転移性卵巣癌、転移性ホルモン抵抗性前立腺癌、及び転移の転移性又は切除不能な局所進行性膵癌を有する患者のための安全性及び有効性を決定するために、いくつかの後期臨床試験が行われている。
【0033】
抗VEGF抗体は通常、VEGF-B又はVEGF-Cといった他のVEGFホモログにも、PlGF、PDGF又はbFGFといった他の増殖因子とも結合しない。好ましい抗VEGF抗体には、ハイブリドーマATCC HB 10709によって産生されるモノクローナル抗-VEGF抗体A4.6.1と同じエピトープに結合するモノクローナル抗体;ベバシズマブを含むがこれに限らず、Presta et al. (1997) Cancer Res. 57: 4593-4599に従って生成される組み換えヒト化抗VEGFモノクローナル抗体が含まれる。ベバシズマブは、ヒトVEGFとそのレセプターの結合を遮断するマウスの抗hVEGFモノクローナル抗体A.4.6.1由来の、変異したヒトIgG1フレームワーク領域および抗原結合性相補性決定領域を含む。大部分のフレームワーク領域を含むベバシズマブのアミノ酸配列のおよそ93%はヒトIgG1由来であり、配列のおよそ7%はマウス抗体A4.6.1由来である。ベバシズマブは、およそ149000ダルトンの分子量を有し、グリコシル化されている。ベバシズマブおよび他のヒト化抗VEGF抗体は、米国特許第6884879号においてさらに記述される。その他の抗VEGF抗体には、国際公開2005/012359の図27−29の何れかに記載される、G6又はB20系列抗体(例えばG6-31、B20-4.1)が含まれる。一実施態様では、B20系列抗体は、残基F17、M18、D19、Y21、Y25、Q89、I91、K101、E103およびC104を含むヒトVEGF上の機能的エピトープに結合する。
A4.6.1(ATCC HB 10709)およびB2.6.2(ATCC HB 10710)抗VEGF発現ハイブリドーマ細胞株は、米国培養細胞系統保存機関(ATCC)、10801 University Boulevard, Manassas, VA 20110-2209 USAに寄託され、維持されている。DNA29101-1276として同定されたATCC寄託物のヌクレオチド配列挿入物によってコードされるVEGF-Eポリペプチドを発現するクローン(米国特許第6391311号)は1998年3月5日に寄託され、ATCC、10801 University Boulevard, Manassas, Va. 20110-2209, USAにてATCC209653として維持された。
【0034】
5−FU(フルオロウラシル、5‐フルオロウラシル、CAS Reg.番号51-21-8)は、チミジル酸合成酵素阻害薬であって、結腸直腸癌及び膵臓癌を含む癌の治療において数十年の間の使われている(米国特許第2802005号、米国特許第2885396号;Barton et al (1972) Jour. Org. Chem. 37:329;Hansen, R.M. (1991) Cancer Invest. 9:637-642)。5−FUは5−フルオロ−1H−ピリミジン−2,4−ジオンと命名され、以下の構造を有する:
【0035】
カルボプラチン(CAS Reg.番号41575-94-4)は、卵巣カルチノーマ、肺、頭頸部癌(米国特許第4140707号)に対して使用される化学療法剤である。カルボプラチンは、アザニド;シクロブタン-1,1-ジカルボン酸プラチナと命名され、以下の構造を有する:
【0036】
ラパチニブ(CAS Reg.番号388082-78-8、TYKERB(登録商標)、GW572016、Glaxo SmithKline)は、腫瘍がHER2(ErbB2)を過剰発現する進行性又は転移性の乳癌を有し、アンスラサイクリン、タキサンおよびトラスツズマブを含む従来の治療法を受けていた患者の治療のために、カペシタビン(XELODA(登録商標)、Roche)と併用した使用について承認されている。ラパチニブは、EGFR/HER2プロテインキナーゼドメインのATP-結合ポケットに結合することによってレセプターの自己リン酸化および活性化を阻害するATP競合的上皮性増殖因子(EGFR)であり、HER2/neu(ErbB-2)二重チロシンキナーゼ阻害因子である(米国特許第6727256号;米国特許第6713485号;米国特許第7109333号;米国特許第6933299号;米国特許第7084147号;米国特許第7157466号;米国特許第7141576号)。ラパチニブは、N-(3−クロロ−4−(3−フルオロベンジルオキシ)フェニル)-6-(5-((2-(メチルスルホニル)エチルアミノ)メチル)フラン-2-イル)キナゾリン-4-アミンとして命名され、以下の構造を有する:
【0037】
ABT-869(Abbott and Genentech)は、癌の経口治療候補のための、VEGFおよびPDGFファミリレセプターチロシンキナーゼの複数標的阻害因子である(米国特許第7297709号;米国公開特許2004/235892;米国公開特許2007/104780)。非小細胞肺癌(NSCLC)、肝細胞癌(HCC)および腎臓細胞カルチノーマ(RCC)を治療する臨床試験が開始された。ABT-869は、1-(4-(3-アミノ-1H-インダゾール-4-イル)フェニル)-3-(2-フルオロ-5-メチルフェニル)ウレア(CAS番号796967-16-3)と命名され、以下の構造を有する:
【0038】
ドセタキセル(タキソテール(登録商標)、Sanofi-Aventis)は、乳癌、卵巣癌及びNSCLC癌を治療するために用いられる(米国特許第4814470号;米国特許第5438072号;米国特許第5698582号;米国特許第5714512号;米国特許第5750561号)。ドセタキセルは、5, 20-エポキシ-1, 2, 4, 7, 10, 13-ヘキサヒドロキシタックス-11-en-9-one 4-アセテート 2-ベンゾエート, トリヒドレートを有する(2R,3S)-N-カルボキシ-3-フェニルイソセリン, N-tert-ブチルエステル, 13-エステル(米国特許第4814470号;欧州特許第253738号;CAS Reg.番号114977-28-5)と命名され、以下の構造を有する:
【0039】
GDC-0941(Genentech Inc.)、薬物動態的及び薬学的な性質が期待される、PI3Kの選択的、経口的な生物利用能があるチエノピリミジン阻害因子である(Folkes et al (2008) Jour. of Med. Chem. 51(18): 5522-5532;米国公開特許2008/0076768;米国公開特許2008/0207611;Belvin et al, American Association for Cancer Research Annual Meeting 2008, 99th: April 15, Abstract 4004;Folkes et al, American Association for Cancer Research Annual Meeting 2008, 99th: April 14, Abstract LB-146;Friedman et al, American Association for Cancer Research Annual Meeting 2008, 99th: April 14, Abstract LB-110)。GDC-0941は、固形腫瘍細胞株に対する特定の化学療法剤と組み合わせたときにインビトロ及びインビボで相乗的な活性を示す(米国特許出願番号第12/208227号、Belvin et al "Combinations Of Phosphoinositide 3-Kinase Inhibitor Compounds And Chemotherapeutic Agents, And Methods Of Use", filed 10 Sept 2008)。GDC-0941は、4-(2-(1H-インダゾール-4-イル)-6-((4-(メチルスルホニル)ピペラジン-1-イル)メチル)チエノ[3,2-d]ピリミジン-4-イル)モルホリン(CAS Reg.番号957054‐30‐7)として命名され、以下の構造を有する:
【0040】
GNE-390(Genentech Inc.)は、薬物動態的及び薬学的な性質が期待される、PI3Kの選択的、経口的な生物利用能があるチエノピリミジン阻害因子である(米国公開特許2008/0242665;国際公開2008/070740)。GNE-390は、固形腫瘍細胞株に対する特定の化学療法剤と組み合わせたときにインビトロ及びインビボで相乗的な活性を示す(米国特許出願番号第12/208227号、Belvin et al "Combinations Of Phosphoinositide 3-Kinase Inhibitor Compounds And Chemotherapeutic Agents, And Methods Of Use", filed 10 Sept 2008)。GNE-390は、(S)-1-(4-((2-(2-アミノピリミジン-5-イル)-7-メチル-4-モルホリノチエノ[3,2-d]ピリミジン-6-イル)メチル)ピペラジン-1-イル)-2-ヒドロキシプロパン-1-オンと命名され、以下の構造を有する:
【0041】
(生物学的評価)
異なる化学療法剤又は生物学的な標的化剤と組み合わせてトラスツズマブ-MCC DM1 T−DM1を用いたインビトロ細胞培養試験を、多くのHER2増幅細胞株において実行した。データはChou & Talalay法を用いて分析し、各薬剤について倍数のIC50を設定して、各組合せについての併用インデックス(CI)値を決定した。0.7未満のCI値は相乗効果を意味し、0.7〜1.3のCI値は相加効果を意味し、そして、1.3を超えるCI値は拮抗作用を意味する。化学療法剤との組合せでは、T−DM1をドセタキセル又は5−FUと併用すると相加的又は相乗的な増殖抑制作用が生じたのに対して、ゲムシタビン又はカルボプラチンいずれかとの組合せは全く効果がないか、T-DM1と拮抗的であった。マウス異種移植片試験は、T−DM1をドセタキセル又は5−FUと併用すると個々の薬剤による治療と比べて目覚ましく腫瘍抑制作用が亢進されたという同じ結果を示した。T-DM1をカルボプラチンと併用するといずれかの薬剤単独の場合と比較して効果が亢進したのに対して、T−DM1とゲムシタビンとの組合せはT−DM1単独の場合より効果が低かった。細胞培養試験において、T-DM1をパーツズマブ、ラパチニブ又はGDC−0941のいずれかと組み合わせると相加的又は相乗的な増殖抑制作用が生じ、個々の薬剤によって治療した場合と比較してインビボでの腫瘍抑制作用が著しく亢進した。対照的に、コンジュゲートしていないトラスツズマブは、HER2上の同じエピトープの結合によってT−DM1の活性を中和した。抗体B20-4.1又は小分子阻害因子ABT−869のような抗血管形成剤とT−DM1との組合せを用いたインビボ試験により、B20-4.1と共に投与した最高用量のT−DM1(10又は15mg/kg)を除いて、試験したすべての組合せにより腫瘍抑制作用が亢進した。
【0042】
多数の抗癌剤とトラスツズマブ-MCC-DM1(T-DM1)との組合せは、HER2を過剰発現する胸部腫瘍細胞におけるインビトロ増殖抑制作用および乳癌異種移植片モデルにおけるインビボ腫瘍抑制有効性を測定することによって調べた。これらの試験において、トラスツズマブ−MCC−DM1を、細胞障害性化学療法剤、抗体又は小分子キナーゼ阻害因子のいずれかに加えた。
乳癌マウス異種移植片モデルにおける抗VEGFマウス抗体B20-4.1(Liang et al (2006) Jour. Biol. Chem. 281:951-961)、ベバシズマブ代用物とトラスツズマブ−MCC−DM1との組合せにより、B20-4.1単独より大きな腫瘍抑制作用が生じた。これらの試験結果は、HER2陽性乳癌における異なる抗腫瘍療法と組み合わせたトラスツズマブ−MCC−DM1を含む治療投薬計画の将来の臨床評価に、相乗効果および論理的根拠を予測する理由となる。
【0043】
トラスツズマブ-DM1とラパチニブ、又はHER2抗体パーツズマブ(HER2二量体化阻害薬)と組み合わせたトラスツズマブ-DM1といったHER2標的化剤の組合せにより、相乗的薬剤効果が観察された。
カルボプラチン又は5−FUと併用したトラスツズマブ-MCC-DM1は、それぞれの薬剤単独による処置と比較して活性の亢進を示したのに対して、ゲムシタビンによる併用処置では腫瘍抑制作用の増加は生じなかった。
p110アイソフォームの小分子キナーゼパン阻害因子であるGDC−0941によってPI3キナーゼ経路を遮断すると、トラスツズマブ-DM1の活性が亢進した。
T-DM1をPI3K阻害因子GDC−0941と併用すると、変異したPI3Kを有するHER2増幅乳癌株:BT-474(K111N)、MDA−361.1(E545K)およびKPL4(H1047R)において抗腫瘍活性が亢進した。インビトロでの併用処置により細胞増殖の相加的又は相乗的な阻害、並びにアポトーシスの増加が生じた。同様に、MDA−MB−361.1およびFo5 HER2増幅異種移植片モデルでは、単独薬剤活性と比較して併用薬剤処置によりインビボ有効性が増加した。各薬剤のためのバイオマーカーの生化学的分析により、T−DM1およびGDC−0941によってホスホ-Aktおよびホスホ-ERKを阻害すると、GDC−0941によるRbおよびPRAS40のリン酸化が低減し、T−DM1による処置後の有糸分裂マーカーホスホ-ヒストンH3およびサイクリンB1のレベルが増加したことが示された。さらに、T−DM1処置によりこれら乳癌モデルでのアポトーシスが生じ、これは、23kDaのPARP-切断断片の出現、Bcl-XLレベルの減少、並びにカスパーゼ3及び7の活性化によって決定される。T−DM1へのGDC−0941の添加によりアポトーシス誘導がさらに亢進した。これらの試験により、HER2増幅乳癌での薬剤併用使用の合理性が裏付けられ、そして、トラスツズマブ又はラパチニブベースの治療法を進ませる疾患を有する患者のための新たな治療手段が提供される。
【0044】
(インビトロ細胞増殖アッセイ)
トラスツズマブ−MCC−DM1の化学療法剤との組合せのインビトロ有効性は、実施例2の細胞増殖アッセイ;Promega Corp., Madison, WIから市販されている発光細胞生存アッセイであるCellTiter-Glo(登録商標)によって測定した。この均質なアッセイ方法は、甲虫類ルシフェラーゼの組換え発現に基づき(米国特許第5583024号;米国特許第5674713号;米国特許第5700670号)、代謝的に活性な細胞の指標である、存在するATPの定量化に基づいて、培養物中の生細胞の数を決定する(Crouch et al (1993) J. Immunol. Meth. 160:81-88;米国特許第6602677号)。CellTiter-Glo(登録商標)アッセイは、96か384のウェルフォーマットにて実行し、自動化ハイスループットスクリーニング(HTS)に準じた(Cree et al (1995) AntiCancer Drugs 6:398-404)。均一なアッセイ手順は、血清添加培地中で培養した細胞に単一試薬(CellTiter-Glo(登録商標)試薬)を直接添加することを含む。細胞洗浄、培地の除去および複数のピペッティング工程は必要ない。システムは、試薬の添加及び混合の10分後に384-ウェルフォーマット中僅か15細胞/ウェルを検出した。
【0045】
均質な「添加混合測定(add-mix-measure)」フォーマットにより細胞の溶解と存在するATPの量と比例した発光シグナルの生成が生じる。ATPの量は培養物中に存在する細胞の数に正比例する。CellTiter-Glo(登録商標)アッセイは、用いた細胞の種類及び培地に応じて、ルシフェラーゼ反応によって生成される「増殖タイプ(glow-type)」の発光シグナルを生成する。このシグナルは一般に5時間を超える半減期を有する。生細胞は相対的な発光単位(RLU)に反映される。基質であるカブトムシルシフェリンは、組み換えホタルルシフェラーゼによって酸化的に脱炭酸され、ATPがAMPに付随して転換され、光子が生成する。半減期の延長により試薬の注入を行う必要がなくなり、複数のプレートの連続的な又はバッチモード処理に柔軟性が生じる。この細胞増殖アッセイは、様々なマルチウェルフォーマット、例えば96又は384のウェルフォーマットと共に用いられる。データは、ルミノメーター又はCCDカメラ撮像装置によって記録されてよい。発光出力は、時間と共に測定される相対的な光単位(RLU)として表される。
【0046】
トラスツズマブ−MCC−DM1および化学療法剤との組合せの増殖抑制作用は、図1〜9および18〜33の腫瘍細胞株に対するCellTiter-Glo(登録商標)アッセイ(実施例2)によって測定した。
例示的実施態様には、癌の治療のために組み合わせて用いられる化合物の同定方法であって、a) インビトロ腫瘍細胞株にトラスツズマブ−MCC−DM1(T-DM1)および化学療法剤の治療的組合せを投与すること、及びb) 相乗的又は非相乗的な効果を測定することを含む方法が含まれる。1.3を超える併用指標(CI)値は拮抗作用を意味し、0.7〜1.3のCI値は加算効果を意味し、0.7未満のCI値は相乗的薬剤相互作用を意味する。
【0047】
図1は、トラスツズマブに感受性があるSK−BR−3細胞における倍数の個々のIC50値(表1)での様々な濃度のトラスツズマブ−MCC−DM1(T-DM1)と併用したトラスツズマブの拮抗作用を示す。生存可能な細胞の数をIC50倍数値に対してプロットする。各組合せについてIC10からIC90に対する併用指標(CI)は2を超えており、これはインビトロでの拮抗作用を示す。しかしながら、インビボのT−DM1+トラスツズマブの組合せは拮抗作用を示さない。
表1 SK-BR-3増殖−3日間
【0048】
図2は、トラスツズマブ耐性であるBT-474EEI細胞における倍数の個々のIC50値(表2)での様々な濃度のトラスツズマブ−MCC−DM1(T-DM1)と併用したトラスツズマブの拮抗作用を示す。生存可能な細胞の数をIC50倍数値に対してプロットする。各組合せについてIC10からIC90に対する併用指標(CI)は2を超えており、これは拮抗作用を示す。
表2 BT-474-EEI増殖−3日間
【0049】
図3は、MDA−MB−175細胞における倍数の個々のIC50値(表3)での様々な濃度のトラスツズマブ−MCC−DM1(T-DM1)と併用したパーツズマブの拮抗作用を示す。生存可能な細胞の数をIC50倍数値に対してプロットする。各組合せについてIC10からIC90に対する併用指標(CI)は1未満であり、平均CI=0.387であり、これは相乗作用を示す(表3)。
表3 MDA-MB-175増殖−5日間
【0050】
図3aは、パーツズマブ、トラスツズマブ−MCC−DM1(T-DM1)およびパーツズマブとT−DM1の併用のIC50倍数濃度に対する、5日のMDA−MB−175インビトロ細胞生存度のプロット線を示す。生存可能な細胞の数をIC50倍数値に対してプロットする。各組合せについてIC10からIC90に対する併用指標(CI)は1未満であり、平均CI=0.096であり、これは相乗作用を示す(表3a)。
表3a MDA-MB-175増殖−5日間
【0051】
図4は、用量反応のトラスツズマブ−MCC−DM1(T-DM1)と併用した様々な一定用量のパーツズマブ、および様々な用量のT−DM1単独に対する、5日のBT-474インビトロ細胞生存度のプロット線を示す。図4は、様々な用量のパーツズマブと組み合わせた一定用量のT−DM1の効果を示す。T−DM1にパーツズマブを加えると、T−DM1単独よりも僅かに大きな増殖抑制活性が生じる。
図5は、用量反応のパーツズマブと併用した様々な一定用量のトラスツズマブ−MCC−DM1(T-DM1)、および様々な用量のパーツズマブ単独に対する、5日のBT-474インビトロ細胞生存度のプロット線を示す。図5は、BT−474細胞増殖に対する様々な用量のT−DM1と併用した一定用量のパーツズマブの効果を示す。パーツズマブにT−DM1を添加するとパーツズマブ単独のの効果が亢進される。
【0052】
図6は、BT-474細胞における倍数の個々のIC50値(表4)での様々な濃度のトラスツズマブ−MCC−DM1(T-DM1)と併用したパーツズマブの相乗効果を示す。生存可能な細胞の数をIC50倍数値に対してプロットする。IC10からIC90の併用指標(CI)値は0.198から1.328である。この範囲の平均CIは0.658であり相乗効果を示す。
表4 BT-474増殖−5日間
【0053】
図7は、一定用量のラパチニブと併用した変更用量のT−DM1(4.5nM、14nM、41nM、123nM)、及び変更用量のT−DM1単独(0〜1000ng/ml)に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。T−DM1にラパチニブを添加すると、T−DM1単独よりも僅かに大きな増殖抑制作用が生じる。
図7aは、表7aに示すように、T−DM1、ラパチニブ、およびT−DM1とラパチニブの一定用量比併用に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。IC10からIC90の平均CI値は0.793であり、これは相加作用を示す。
表7a SK-BR-3増殖−3日間
【0054】
図8aは、表8aに示すように、T−DM1、ラパチニブおよびT−DM1とラパチニブの一定用量比併用に対する、3日のBT-474インビトロ細胞生存度のプロット線を示す。IC10からIC90の平均CI値は0.403であり、これは相乗作用を示す。
表8a BT−474増殖−3日間
【0055】
図8は、一定用量のラパチニブと併用した変更用量のT−DM1(1.5nM、4.5nM、14nM、41nM、123nM)、及び変更用量のT−DM1単独(0〜1000ng/ml)に対する、3日のBT−474インビトロ細胞生存度のプロット線を示す。T−DM1へラパチニブを添加すると、いずれかの薬剤単独と比較して大きな増殖抑制作用が生じる。
図9は、一定用量のラパチニブと併用した変更用量のT−DM1(14nM、41nM、123nM、370nM、1111nM)、及び変更用量のT−DM1単独(0〜1000ng/ml)に対する、3日のBT−474−EEIインビトロ細胞生存度のプロット線を示す。T−DM1へラパチニブを添加すると、いずれかの薬剤単独と比較して大きな増殖抑制作用が生じる。
【0056】
図18は、5−FU、トラスツズマブ−MCC−DM1(T-DM1)および5−FUとT−DM1の一定用量比組合せ(表18)のIC50倍数の濃度に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。5−FUとT−DM1の併用はSK−BR−3細胞に対して相加作用を有し、IC10からIC90の平均CIは0.952である。
表18 5−FU+T−DM1:SK-BR-3増殖−3日間
【0057】
図19は、5−FU、トラスツズマブ−MCC−DM1(T-DM1)および5−FUとT−DM1の一定用量比組合せ(表19)のIC50倍数濃度に対する、3日のBT−474インビトロ細胞生存度のプロット線を示す。5−FUとT−DM1の併用はBT-474細胞に対して相乗作用を有し、平均CI値は0.623である。
表19 5−FU+T−DM1:BT-474増殖−3日間
【0058】
図20は、ゲムシタビン、トラスツズマブ−MCC−DM1(T-DM1)およびゲムシタビンとT−DM1の一定用量比組合せ(表20)のIC50倍数濃度に対する、3日のSK−BR−3インビトロ細胞生存度のプロット線を示す。ゲムシタビンをT−DM1と併用すると拮抗的な薬剤相互作用が生じ、試験したすべての組合せでCI値は1.3を上回る。
表20 ゲムシタビン(GEM)+T−DM1:SK-BR-3増殖−3日間
【0059】
図21は、ゲムシタビン、トラスツズマブ−MCC−DM1(T-DM1)およびゲムシタビンとT−DM1の一定用量比組合せ(表21)のIC50倍数濃度に対する、3日のMDA−MD−361インビトロ細胞生存度のプロット線を示す。薬剤併用により平均CI=1.706の拮抗作用が生じる。
表21 ゲムシタビン(GEM)+T−DM1:MDA-MD-361増殖−3日間
【0060】
図22は、0.25×〜4×のIC50倍数濃度でのT−DM1、GDC−0941、およびT−DM1(6.25〜100ng/ml)とGDC−0941(62.5nM〜1μM)の一定用量比の組合せによる処置後3日の、KPL4インビトロ細胞生存度(増殖)のプロット線を示す。表22は、算出したCI値と1.111の平均CIと共に、10〜90%の阻害範囲における効果を示す。
相加作用のブリス予測は図22において点線で表す。ブリス独立プロット線は、2つの単一化合物の組合せから算出した相加反応を示す。
表22 GDC−0941+T−DM1:KPL4増殖−3日間
【0061】
図23は、0.0625×〜16×のIC50倍数濃度でのT−DM1、GDC−0941、およびT−DM1(1.25〜80ng/ml)とGDC−0941(31.25nM〜2μM)の一定用量比の組合せによる処置後3日の、KPL4インビトロ細胞生存度(増殖)のプロット線を示す。相加作用のブリス予測を点線でプロットする。表23は、算出したCI値と0.802の平均CIと共に、10〜90%の阻害範囲における効果を示す。T−DM1とGDC−0941の併用はKPL4細胞株において相加作用を有する。表23 GDC-0941+T−DM1:KPL4増殖−3日間
【0062】
図24は、0から16×のIC50倍数濃度での、T−DM1、PI103、GDC−0941、およびT−DM1+PI103及びT−DM1+GDC−0941の一定用量比の組合せによる処置後、Her2増幅、HERCEPTIN(登録商標)耐性、PIK3CA(H1047R)変異のKPL-4細胞インビトロ細胞生存度(増殖)のプロット線を示す。表24は併用指標値を示す。この結果から、CI値が0.5から1の間であることからT−DM1とGDC−0941との間には中程度のインビトロ相乗作用が、そしてCI値が1に近かったことからT−DM−1とPI103との間には相加作用が示唆される。
表24 併用:KPL4増殖
【0063】
PI3K選択阻害因子であるPI103(Hayakawa et al (2007) Bioorg. Med. Chem. Lett. 17:2438-2442;Raynaud et al (2007) Cancer Res. 67: 5840-5850;Fan et al (2006) Cancer Cell 9:341-349;米国特許第6608053号)は以下の構造を有する:
【0064】
図25は、T−DM1、GDC−0941、および一定用量比のT−DM1とGDC−0941の組合せによる処置後24時間の、KPL4カスパーゼ3/7インビトロ細胞アポトーシス(プログラム細胞死)のプロット線を示す。T−DM1とGDC−0941の組合せにより、いずれかの薬剤単独と比較してアポトーシスが大きく亢進する。
図26は、T−DM1、GDC−0941、および一定用量比のT−DM1とGDC−0941の組合せによる処置後3日の、KPL4インビトロ細胞アポトーシス(プログラム細胞死)のプロット線を示す。T−DM1とGDC−0941の組合せにより、いずれかの薬剤単独と比較してアポトーシスが大きく亢進する。
【0065】
図27は、0.125×〜8×のIC50倍数濃度での、T−DM1、GDC−0941、およびT−DM1(3.125〜50ng/ml)とGDC−0941(62.5nM〜1μM)の一定用量比の組合せによる処置後3日の、MDA−MB−361インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。表27は、算出したCI値と0.888の平均CIと共に10〜90%の阻害範囲における効果を示す。T-DM1をGDC−0941と併用することにより、MDA−MB−361細胞において相加的な増殖抑制作用が生じ、平均CIは0.889である。
表27 GDC−0941+T−DM1:MDA-MB-361増殖−3日間
【0066】
図28は、0.125×〜8×のIC50倍数濃度での、T−DM1、GDC−0941、およびT−DM1(3.125〜100ng/ml)とGDC−0941(62.5nM〜2μM)の一定用量比の組合せによる処置後3日の、MDA−MB−361インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。表28は、算出したCI値と0.813の平均CIと共に10〜90%の阻害範囲における効果を示す。T-DM1をGDC−0941と併用することにより、MDA−MB−361細胞において相加的な増殖抑制作用が生じ、平均CIは0.813である。
表28 GDC-0941+T−DM1:MDA-MB-361増殖−3日間
【0067】
図29は、0.125×〜4×のIC50倍数濃度での、T−DM1、GDC−0941、およびT−DM1(3.125〜100ng/ml)とGDC−0941(31.25nM〜1μM)の一定用量比の組合せによる処置後3日の、BT−474インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。表29は、算出したCI値と1.2122の平均CIと共に10〜90%の阻害範囲における効果を示す。GDC-0941とT-DM1は、これらの用量比ではBT-474に対する併用効果を有しない。
表29 GDC-0941+T−DM1:BT-474増殖−3日間
【0068】
図30は、0.25×〜4×のIC50倍数濃度での、T−DM1、GDC−0941、およびT−DM1(6.25〜100ng/ml)とGDC−0941(62.5nM〜1μM)の一定用量比の組合せによる処置後3日の、BT-474インビトロ細胞生存度(増殖)のプロット線を示す。相加効果のブリス予測を点線でプロットする。表30は、算出されたCI値及び0.997の平均CIと共に、10〜90%の阻害範囲における効果を示し、これは相加効果を示す。
表30 GDC-0941+T−DM1:BT-474増殖−3日間
【0069】
図31は、0〜16×のIC50倍数濃度での、T−DM1、PI103、GDC−0941、およびT−DM1+PI103及びT−DM1+GDC−0941の一定用量比の組合せによる処置後3日の、Her2増幅、非PI3K変異のAU565細胞のインビトロ細胞生存度(増殖)のプロット線を示す。表31は併用指標値を示す。これらの結果から、CI値は1を超えるのでT−DM−1とGDC−0941との間はインビトロ拮抗作用、そしてCI値が1に近いか又は僅かに1より大きいのでT−DM−1とPI103との間は相加作用又は僅かに拮抗作用が示唆される。
表31 併用:AU565増殖
【0070】
図32は、0〜16×のIC50倍数濃度での、T−DM1、PI103、GDC−0941、およびT−DM1+PI103及びT−DM1+GDC−0941の一定用量比の組合せによる処置後3日の、Her2増幅、PIK3CA(C420R)変異のEFM192A細胞のインビトロ細胞生存度(増殖)のプロット線を示す。表32は併用指標値を示す。これらの結果から、CI値が0.5〜1であるのでT−DM−1とGDC−0941との間には中程度のインビトロ相乗作用が、そして、CI値が0.5に近いのでT−DM−1とPI103との間には相乗作用が示唆される。
表32 併用:EFM192A増殖
【0071】
図33は、0〜16×のIC50倍数濃度での、T−DM1、PI103、GDC−0941、およびT−DM1+PI103及びT−DM1+GDC−0941の一定用量比の組合せによる処置後3日の、Her2増幅、ハーセプチン(登録商標)耐性、PIK3CA(H1047R)変異のHCC1954細胞インビトロ細胞生存度(増殖)のプロット線を示す。表33は併用指標値を示す。これらの結果から、CI値が1に近いのでT−DM−1とGDC−0941との間には相加作用又は僅かなインビトロ相乗作用が、そして、CI値が1未満であるのでT−DM−1とPI103との間には僅かな相乗作用が示唆される。
表33 併用:HCC1954増殖
【0072】
(インビボ腫瘍異種移植片有効性)
本発明の併用の有効性は、齧歯動物の癌細胞の同種異系移植片又は異種移植片を着床させて、腫瘍を併用にて処置することによってインビボで測定されてよい。細胞株、腫瘍細胞中の特定の突然変異の有無、トラスツズマブ−MCC−DM1と化学療法剤の投与、投薬計画及び他の要因に応じて、結果が変わりうることは予測される。被検体マウスを、薬剤(一又は複数)又はコントロール(ベヒクル)にて処置し、数週間以上モニターし、腫瘍倍加までの時間、ログ細胞障害及び腫瘍抑制を測定した(実施例3)。図10〜17および34〜37は、マウスにおける異種移植片腫瘍阻害によって、化学療法剤と組み合わせたトラスツズマブ−MCC−DM1の有効性を示す。
【0073】
図10は、以下を投与した後のSCIDベージュマウスの乳房脂肪パッドに接種したKPL-4腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ADCバッファ、(2) パーツズマブ15mg/kg、(3) T−DM1 0.3mg/kg、(4) T−DM1 1mg/kg、(5) T−DM1 3mg/kg、(6) パーツズマブ15mg/kg+T−DM1 0.3mg、(7) パーツズマブ15mg/kg+T−DM1 1mg/kg、(8)パーツズマブ15mg/kg+T−DM1 3mg/kg。ADCバッファを投与した動物(1)は0のPRおよび0のCRであった。15mg/kgでパーツズマブを投与した動物(2)は0のPRおよび0のCRであった。0.3mg/kgのT−DM1のみを投与した動物(3)は0のPRおよび0のCRであった。1mg/kgのT−DM1のみを投与した動物(4)は1のPRおよび0のCRであった。3mg/kgのT−DM1のみを投与した動物(5)は7のPRおよび0のCRであった。15mg/kgのパーツズマブと0.3mg/kgのT−DM1を投与した動物(6)は、5のPRおよび0のCRであった。15mg/kgのパーツズマブと1mg/kgのT−DM1を投与した動物(7)は、8のPRおよび0のCRであった。15mg/kgのパーツズマブと3mg/kgのT−DM1を投与した動物(8)は、8のPRおよび0のCRであった。パーツズマブとT−DM1の併用により、KPL4異種移植片においていずれかの薬剤単独よりも大きな抗腫瘍活性が生じる。
【0074】
図11は、以下を投与した後のSCIDベージュマウスの乳房脂肪パッドに接種したKPL-4腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ADCバッファ、(2) 5−FU 100mg/kg、(3) パーツズマブ40mg/kg、(4) B20-4.1、5mg/kg、(5) T−DM1、5mg/kg、(6) 5−FU、100mg/kg+T−DM1、5mg、(7) パーツズマブ40mg/kg+T−DM1、5mg/kg、(8) B20-4.1、5mg/kg+T−DM1、5mg/kg、(9) B20-4.1、5mg/kg+パーツズマブ、40mg/kg。試験終了時には、50mm3体積未満のすべて残存する腫瘍を組織学的に評価し、単一薬剤(5) T−DM1、5mg/kgの8試料、併用群(6) 5−FU、100mg/kg+T−DM1、5mgの5試料、および併用群(7) パーツズマブ、40mg/kg+T−DM1、5mg/kgの8試料は生きている腫瘍細胞の所見がなかったことが決定された。
【0075】
図12は、以下を投与した後のCRLnu/nuマウスの乳房脂肪パッドに接種したMMTV-HER2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル(ADCバッファ)、(2) B20-4.1、5mg/kg、(3) T−DM1、3mg/kg、(4) T−DM1、5mg/kg、(5) T−DM1、10mg/kg、(6) B20-4.1、5mg/kg+T−DM1 3mg/kg、(7) B20-4.1、5mg/kg+T−DM1 5mg/kg、(8) B20-4.1、5mg/kg+T−DM1、10mg/kg。T−DM1とB20-4.1との併用により、10mg/kgでなく、3および5mg/kgのT−DM1により腫瘍増殖阻害が亢進する。
【0076】
図13は、以下を投与した後のCRLnu/nuマウスの乳房脂肪パッドに接種したMMTV-HER2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル(ADCバッファ)、(2) T−DM1 10mg/kg、(3) 5−FU 100mg/kg、(4) ゲムシタビン、120mg/kg、(5)カルボプラチン、100mg/kg、(6) 5−FU、100mg/kg+T−DM1、10mg/kg、(7) ゲムシタビン、120mg/kg+T−DM1、10mg/kg、(8) カルボプラチン、100mg/kg+T−DM1、10mg/kg。T-DM1を、5−FU、カルボプラチン又はゲムシタビンのいずれかと併用すると、単一薬剤処置と比較して腫瘍抑制効果が亢進する。
【0077】
図14は、以下を投与した後のハーラン胸腺欠損ヌードマウスの乳房脂肪パッドに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍異種移植片における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル(PBSバッファ) iv、qwk ×4、(2) ラパチニブ101mg/kg、po、bid ×21、(3) パーツズマブ40mg/kg、iv、qwk ×4、(4) B20-4.1 5mg/kg、ip、2×/wk ×4、(5) T−DM1 15mg/kg、iv、最後までq3wk、(6) ラパチニブ101mg/kg、po、bid ×21+T−DM1 15mg/kg、iv、最後までq3wk、(7) パーツズマブ40mg/kg、iv、qwk ×4+T−DM1 15mg/kg、iv、最後までq3wk、(8) B20-4.1 5mg/kg、ip、2×/wk×4+T−DM1 15mg/kg、iv、最後までq3wk。
15mg/kg用量の単一薬剤T−DM1(5)は、15mg/kgのT−DM1と5mg/kgのB20-4.1の併用(8)とは有意には異ならない。この試験においてラパチニブとパーツズマブはベヒクルと異ならなかった。B20-4.1はベヒクルと比較して効果が増加する傾向を示した。T-DM1は単一薬剤としても効果的であった(p<0.01)。T−DM1のラパチニブとの併用は、ラパチニブ単独より有意に良好であったが(p<0.01)、T−DM1単独とは異ならなかった。T−DM1のパーツズマブとの併用は、パーツズマブ単独より有意に良好であったが(p<0.01)、T−DM1単独とは異ならなかった。T−DM1のB20-4.1との併用は、B20-4.1単独より有意に良好であったが(p<0.01)、T−DM1単独とは異ならなかった。
【0078】
図15は、以下を投与した後のハーラン胸腺欠損ヌードマウスの乳房脂肪パッドに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍異種移植片における経時的な平均腫瘍体積変化によるインビボ有効性のプロット線を示す:(1) ベヒクル(PBSバッファ) po、bid ×21、(2) T−DM1、7.5mg/kg、iv、qd ×1、(3) T−DM1、15mg/kg、iv、qd ×1、(4) ABT−869、5mg/kg、po、bid ×21、(5) ABT−869、15mg/kg、po、bid ×21、(6) T−DM1、7.5mg/kg、iv、qd ×1+ABT−869、5mg/kg、po、bid ×21、(7) T−DM1 7.5mg/kg、iv、qd ×1+ABT−869、15mg/kg、po、bid ×21、(8) T−DM1、15mg/kg、iv、qd ×1+ABT−869、5mg/kg、po、bid ×21、(9) T−DM1、15mg/kg、iv、qd ×1+ABT−869、15mg/kg、po、bid ×21。
T−DM1とABT−869、5mg/kgとの併用は 2の部分的応答を示したが(8)、単一薬剤ABT−869、5mg/kg(4)よりかなり効果的ではない。T−DM1とABT−869、15mg/kgとの併用(9)は、単一薬剤ABT−869、15mg/kg(5)よりわずかに効果的である。5mg/kgで投与したABT-869は、エンドポイントまでの時間までベヒクルより有意に良好であったが(p<0.01)、腫瘍倍加までの時間はベヒクルとは異ならなかった。15mg/kgで投与したABT-869と7.5又は15mg/kgで投与したT-DM1は、腫瘍倍加および腫瘍エンドポイントいずれの時間までもベヒクルよりも有意に良好であった(p<0.01)。7.5mg/kgのT−DM1と5mg/kgのABT−869の併用は、7.5mg/kgのT−DM1の単一薬剤と異ならなかった。5mg/kgのABT−869単一薬剤と比較して、7.5mg/kgのT−DM1+5mg/kgのABT−869の併用は、腫瘍倍加までの時間は有意に良好であったが(p<0.01)、エンドポイントまでの時間は異ならなかった。7.5mg/kgのT−DM1と15mg/kgのABT−869の併用は、いずれかの単一薬剤よりも有意に良好であった(p<0.01)。15mg/kgのT−DM1+5mg/kgのABT−869の併用は15mg/kgのT−DM1単一薬剤と異ならなかった。5mg/kgのABT-869単一薬剤と比較して、15mg/kgのT−DM1と5mg/kgのABT−869の併用は、エンドポイントまでの時間は異ならなかったが、腫瘍倍加までの時間は有意に異なった(p<0.01)。15mg/kgのT−DM1+15mg/kgのABT−869の併用は、15mg/kgのABT−869単独より有意に良好であり、腫瘍倍加までの時間は15mg/kgのT−DM1単独より良好であった(p<0.01)。15mg/kgのT−DM1と15mg/kgのT−DM1+15mg/kgのABT−869のエンドポイントまでの時間は異ならなかった。
【0079】
図16は、以下を投与した後のハーラン胸腺欠損ヌードマウスの乳房脂肪パッドに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍異種移植片における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル、iv、qwk ×3、(2) T−DM1、7.5mg/kg、iv、q3wk ×2、(3) T−DM1、15mg/kg、iv、q3wk ×2、(4)ドセタキセル、30mg/kg、iv、qwk ×3、(5) T−DM1、7.5mg/kg、iv、q3wk ×2+ドセタキセル、30mg/kg、iv、qwk ×3、(6) T−DM1、15mg/kg、iv、q3wk ×2+ドセタキセル、30mg/kg、iv、qwk ×3。
15mg/kgのT−DM1を投与した動物(3)は、6の部分応答(PR)および1の完全寛解(CR)となった。30mg/kgのドセタキセル単独を投与した動物(4)は2のPRであった。7.5mg/kgのT−DM1と30mg/kgのドセタキセルの併用を投与した動物(5)は10のPRであった。15mg/kgのT−DM1と30mg/kgのドセタキセルの併用を投与した動物(6)は、7のPRおよび3のCRの用量応答を示した。すべての単一薬剤群はベヒクル群とは有意に異なっていた(p<0.01)。7.5mg/kgのT−DM1+ドセタキセルの併用は、腫瘍倍加までの時間およびエンドポイントまでの時間のいずれも、いずれか単一薬剤よりも有意に良好であった(p<0.01)。7.5mg/kgのT−DM1群では客観的な応答はなく、ドセタキセル単一薬剤群では2の部分応答(PR)であった。7.5mg/kgのT−DM1とドセタキセルの併用により、9のPRおよび1の完全寛解(CR)が生じた。15mg/kgのT−DM1+ドセタキセルの併用は、腫瘍倍加までの時間およびエンドポイントまでの時間はいずれの単一薬剤よりも有意に良好であった(p<0.01)。単一薬剤15mg/kgのT−DM1処置により、5のPRおよび2のCRが生じた。15mg/kgのT−DM1+ドセタキセルの併用は、客観的な応答速度が7のPRおよび3のCRに増えた。この併用群のすべてのマウスは処置に対して客観的な応答を有した。
【0080】
図17は、以下を投与した後のハーラン胸腺欠損ヌードマウスの乳房脂肪パッドに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍異種移植片における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル、po、qd×21、(2) T−DM1、7.5mg/kg、iv、q3wk ×2、(3) T−DM1、15mg/kg、iv、q3wk ×2、(4) ラパチニブ、100mg/kg、po、bid ×21、(5) T−DM1、7.5mg/kg、iv、q3wk ×2+ラパチニブ、100mg/kg、po、bid ×21、(6) T−DM1、15mg/kg、iv、q3wk ×2+ラパチニブ、100mg/kg、po、bid ×21。
15mg/kgのT−DM1を投与した動物(3)は、6の部分応答(PR)および3の完全寛解(CR)であった。7.5mg/kgのT−DM1と100mg/kgのラパチニブの併用を投与した動物(5)は、4のPRおよび5のCRであった。15mg/kgのT−DM1と100mg/kgのラパチニブの併用を投与した動物(6)は、8のCRの用量応答を示した。すべての単一薬剤群は、腫瘍倍加までの時間およびエンドポイントまでの時間は共にベヒクルと有意に異なっていた(p<0.01)。ラパチニブと併用した7.5mg/kgで投与したT-DM1は、単一薬剤の7.5mg/kgのT−DM1又はラパチニブよりも有意に良好であった(p<0.01)。ラパチニブと併用して15mg/kgで投与したT-DM1は、ラパチニブ単一薬剤よりも有意に良好であった(p<0.01)。この併用は、単一薬剤の15mg/kgのT−DM1とは異ならなかった。
腫瘍倍加までの時間はカプラン-マイヤー統計学的分析によって2×Voとして測定した。腫瘍倍加までの時間と生存率分析は、ログ−ランク−p値によって定量化した。進行時間は、腫瘍体積が1000mm3に達する経過時間として測定し、腫瘍体積が1000mm3に達しない場合は生存時間として測定した。T-DM1をラパチニブと併用すると、単一薬剤処置と比較して腫瘍抑制作用が大きく亢進した。
【0081】
図34は、以下を投与した後のCRLnu/nuマウスに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル、po、qd×21、(2) T−DM1、10mg/kg、iv、q3wk、(3) 5−FU、100mg/kg、po、qwk ×2、(4) (5) T−DM1、5mg/kg、iv、q3wk+5−FU、100mg/kg、po、qwk ×2。ベヒクルを投与した動物は、0の部分応答(PR)および0の完全寛解(CR)であった。T-DM1を投与した動物は1のPRおよび0のCRであった。5−FUを投与した動物は0のPRおよび0のCRであった。T−DM1と5−FUの併用を投与した動物は、42日目の時点で、3のPRおよび0のCRであった。T−DM1と5−FUによる処置により、いずれかの薬剤単独と比較して腫瘍抑制作用が亢進した。
【0082】
図35は、以下を投与した後のCRLnu/nuマウスに接種したMMTV-Her2 Fo5トランスジェニック乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル、po、qd ×21、(2) T−DM1、5mg/kg、iv、q3wk、(3) GDC−0941、100mg/kg、po、bid×21、(4) GDC−0152、50mg/kg、po、qwk ×2、(5) T−DM1、5mg/kg、iv、q3wk+GDC−0941、100mg/kg、po、bid ×21、(6) T−DM1、5mg/kg、iv、q3wk+GDC−0152、50mg/kg、po、qwk ×2。T−DM1とGDC−0941による処置により単一薬剤処置と比較して腫瘍抑制作用が亢進するのに対して、T−DM1とGDC−0152の併用はT−DM1単独より効果が低かった。
GDC-0152はアポトーシスタンパク質の阻害因子であるカスパーゼの阻害因子である(Call et al (2008) The Lancet Oncology, 9(10): 1002-1011;Deveraux et al (1999) J Clin Immunol 19:388-398)。
【0083】
図36は、以下を投与した後のCRLnu/nuマウスに接種したMDA−MB−361.1乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル、po、qd ×21、(2) GDC−0941、25mg/kg、po、qd ×21、(3) GDC−0941、50mg/kg、po、qd ×21、(4) GDC−0941、100mg/kg、po、qd ×21、(5) T−DM1、3mg/kg、iv、q3wk、(6) T−DM1、10mg/kg、iv、q3wk、(7) GDC−0941、25mg/kg、po、qd ×21+T−DM1、3mg/kg、iv、q3wk、(8) GDC−0941、50mg/kg、po、qd ×21+T−DM1、3mg/kg、iv、q3wk、(9) GDC−0941、100mg/kg、po、qd ×21+T−DM1、3mg/kg、iv、q3wk、(10) GDC−0941、25mg/kg、po、qd ×21+T−DM1、10mg/kg、iv、q3wk、(11) GDC−0941、50mg/kg、po、qd ×21+T−DM1、10mg/kg、iv、q3wk、(12) GDC−0941、100mg/kg、po、qd ×21+T−DM1、10mg/kg、iv、q3wk。
ベヒクルを投与した動物(1)は、0の部分応答(PR)および0の完全寛解(CR)であった。25mg/kgのGDC−0941を投与した動物(2)は、0のPRおよび0のCRであった。50mg/kgのGDC−0941を投与した動物(3)は、1のPRおよび0のCRであった。100mg/kgのGDC−0941を投与した動物(4)は、0のPRおよび0のCRであった。3mg/kgのT−DM1のみを投与した動物(5)は、1(PR)および1(CR)であった。10mg/kgのT−DM1のみを投与した動物(6)は、8(PR)および1(CR)であった。3mg/kgのT−DM1と25mg/kgのGDC−0941の併用を投与した動物(7)は、5のPRおよび0のCRであった。3mg/kgのT−DM1と50mg/kgのGDC−0941の併用を投与した動物(8)は、3のPRおよび0のCRであった。3mg/kgのT−DM1と100mg/kgのGDC−0941の併用を投与した動物(9)は、3のPRおよび1のCRであった。10mg/kgのT−DM1と50mg/kgのGDC−0941の併用を投与した動物(10)は、9のPRおよび0のCRであった。10mg/kgのT−DM1と50mg/kgのGDC−0941の併用を投与した動物(11)は、7のPRおよび2のCRであった。10mg/kgのT−DM1と100mg/kgのGDC−0941の併用を投与した動物(12)は、9のPRおよび1のCRであった。
【0084】
図37は、以下を投与した後のCRLnu/nuマウスに接種したMDA−MB−361.1乳房腫瘍における経時的なインビボ平均腫瘍体積変化のプロット線を示す:(1) ベヒクル[MCT(0.5%メチルセルロース/0.2%TWEEN80)+琥珀酸塩バッファ(100mMの琥珀酸ナトリウム、100mg/mlのトレハロース、0.1%のTWEEN80、pH5.0)]、po+IV、qd ×21及びqd、(2) GNE-390、1.0mg/kg、po、qd ×21、(3) GNE−390、2.5mg/kg、po、qd ×21、(4) T−DM1、3mg/kg、iv、qd、(5) GNE−390、1.0mg/kg、po、qd ×21+T−DM1、3mg/kg、iv、qd、(6) GNE−390、2.5mg/kg、po、qd×21+T−DM1、3mg/kg、iv、qd。
ベヒクルを投与した動物(1)は、0の部分応答(PR)および0の完全寛解(CR)であった。1.0mg/kgのGNE−390のみを投与した動物(2)は、0のPRおよび0のCRであった。2.5mg/kgのGNE−390のみを投与した動物(3)は、1のPRおよび0のCRであった。3mg/kgのT−DM1のみを投与した動物(5)は、1(PR)および1(CR)であった。3mg/kgのT−DM1のみを投与した動物(4)は、0のPRおよび0のCRであった。3mg/kgのT−DM1と25mg/kgのGNE−390の併用を投与した動物(5)は、3のPRおよび0のCRであった。3mg/kgのT−DM1と2.5mg/kgのGNE−390の併用を投与した動物(6)は、5のPRおよび1のCRであった。MDA−MB−361.1乳癌異種移植片モデルでは、GNE−390をT−DM1と併用すると、GNE−390又はT−DM1単独と比較して、部分応答及び完全寛解の腫瘍抑制応答数が有意に増加した。
【0085】
(薬学的組成物)
本発明の薬学的組成物又は製剤には、トラスツズマブ−MCC−DM1、化学療法剤、及び一又は複数の薬学的に許容可能な担体、流動促進剤、希釈剤又は賦形剤の組合せが含まれる。
本発明のトラスツズマブ−MCC−DM1と化学療法剤は、非溶媒和状態並びに水、エタノール等のような薬学的に許容可能な溶媒との溶媒和形態で存在し得、本発明は溶媒和形態と非溶媒和形態の双方を包含するものである。
【0086】
また、本発明のトラスツズマブ−MCC−DM1と化学療法剤は、異なった互変異性体形態で存在し得、そのようなあらゆる形態が本発明の範囲に包含される。「互変異性体」又は「互変異性体形態」なる用語は、低エネルギー障壁を介して相互転換可能な異なったエネルギーの構造異性体を意味する。例えば、プロトン互変異性体(屈光性互変異性体としても知られている)は、ケト-エノール及びイミン-エナミン異性化のような、プロトンの移動を介した相互転換を含む。原子価互変異性体は結合電子の幾らかの再編成による相互転換を含む。
【0087】
薬学的組成物は、トラスツズマブ−MCC−DM1及び、本明細書中に記載される他の薬剤の一覧から選択される化学療法剤を含む2以上(例えば2)の薬学的な活性剤、並びに任意の薬学的に不活性な賦形剤、希釈剤、担体又は流動促進剤からなる個々の用量単位とバルク組成物を包含する。バルク組成物と各個々の用量単位は、一定量の前述の薬学的な活性剤を含みうる。バルク組成物は、個々の用量単位に形成されていない材料である。例示的な用量単位は、錠剤、丸剤、カプセルなどといった経口用量単位である。同様に、本明細書中に記載される、本発明の薬学的組成物を投与することによる患者の治療方法もまた、バルク組成物及び個々の用量単位の投与を含むことが意図される。
【0088】
また、薬学的組成物は、一又は複数の原子が通常自然に見出される原子質量又は質量数とは異なる原子質量又は質量数を有する原子によって置き換えられるという点を別にすると、ここに記載されたものと同一である本発明の同位体標識化合物を包含する。任意の特定の原子又は特定された元素のあらゆる同位体が本発明の化合物とその使用の範囲内であると考えられる。本発明の化合物に導入されうる例示的な同位体は、水素、炭素、窒素、酸素、リン、硫黄、フッ素、塩素及びヨードの同位体、例えば2H、3H、11C、13C、14C、13N、15N、15O、17O、18O、32P、33P、35S、18F、36Cl、123I及び125Iを含む。本発明の同位体標識化合物(例えば3H及び14Cで標識されたもの)は化合物及び/又は基質組織分布アッセイにおいて有用である。トリチウム標識(3H)及び炭素-14(14C)同位体は、調製及び検出性の容易さのために有用である。更に、重水素(2H)のような重い同位体での置換により、より大なる代謝安定性から生じるある種の治療的利点(例えばインビボ半減期の増加又は投薬量要求の減少)が得られ、よってある状況下では好ましい場合がある。ポジトロン放出同位体、例えば15O、13N、11C及び18Fは基質レセプター占有性を試験するポジトロン放出断層撮影(PET)に有用である。本発明の同位体標識化合物は一般に以下のここでのスキーム及び/又は実施例に開示されたものと類似した手順に従って、非同位体標識試薬に同位体標識試薬を置き換えて調製することができる。
【0089】
ヒトを含む哺乳動物における過剰増殖性疾患の治療的処置(予防的処置を含む)のために治療的併用を使用するために、トラスツズマブ−MCC−DM1と化学療法剤を、標準的な製薬的実務に従って製剤化されてよい。本発明により、一又は複数の薬学的に許容可能な担体、流動促進剤、希釈剤又は賦形剤と共にトラスツズマブ−MCC−DM1を含有する薬学的組成物が提供される。
適切な担体、希釈剤及び賦形剤は当業者によく知られており、炭水化物、ワックス、水溶性及び/又は膨潤性ポリマー、親水性又は疎水性物質、ゼラチン、油、溶媒、水等のような材料が含まれる。使用される特定の担体、希釈剤又は賦形剤は、本発明の化合物が適用されている手段及び目的に依存する。溶媒は一般的には哺乳動物への投与が安全である(GRAS)と当業者により認識される溶媒に基づいて選択される。一般に、安全な溶媒は、非毒性水性溶媒、例えば水、及び水に可溶性又は混和性である他の非毒性溶媒である。適切な水性溶媒には、水、エタノール、プロピレングリコール、ポリエチレングリコール(例えば,PEG400,PEG300)等とその混合物が含まれる。製剤はまた一又は複数のバッファー、安定剤、界面活性剤、湿潤剤、潤滑剤、乳化剤、懸濁剤、保存料、抗酸化剤、不透明化剤、流動促進剤、加工助剤、着色料、甘味料、香料剤、香味料及び薬物(つまり、本発明の化合物又はその薬学的組成物)を上品に提供するためあるいは薬学的製品(つまり医薬)の製造を補助するための他の既知の添加剤を含みうる。
【0090】
製剤は一般的な溶解及び混合手順を使用して調製されうる。例えば、原体薬剤物質(つまり、本発明の化合物又は該化合物の安定化形態(例えばシクロデキストリン誘導体又は他の既知の複合体形成剤との複合体)を、上述の賦形剤の一又は複数の存在下で適切な溶媒に溶解させる。本発明の化合物は典型的には容易に制御できる薬剤の用量を提供し処方されたレジメンでの患者の服薬遵守を可能にするように薬学的投薬形態で製剤化される。
投与される薬学的組成物(又は製剤)は薬剤を投与するために使用される方法に応じて様々な形に包装されうる。一般に、流通品は適切な形で薬学的製剤をそこに入れる容器を含む。適切な容器は当業者にはよく知られており、瓶(プラスチック又はガラス)、サッシェ(sachets)、アンプル、プラスチック袋、金属シリンダー等のような材料を含む。容器はまた包装の内容物への軽率なアクセスを防止するために不正開封防止体を含みうる。また、容器には容器の内容物を記述するラベルが付着される。ラベルは適切な注意書きをまた含みうる。
【0091】
本発明の化合物の薬学的製剤は、薬学的に許容可能な希釈剤、担体、賦形剤又は安定剤により凍結乾燥製剤、粉砕粉剤、又は水溶液の形態に、様々な経路及び投与タイプ用に調製されうる(Remington's PharmaceutIcal Sciences(1995)18版, Mack Publ. Co., Easton, PA)。製剤化は、生理学的に許容可能な担体、つまり用いられる用量及び濃度でレシピエントに非毒性である担体と、適切なpH、雰囲気温度、所望の純度で混合することによってなされうる。製剤のpHは特定の用途及び化合物の濃度に主に依存するが、約3から約8の範囲でありうる。
薬学的製剤は好ましくは無菌である。特にインビボ投与に使用される製剤は無菌でなければならない。かかる滅菌は滅菌濾過膜を通した濾過によって直ぐに達成される。
通常は、薬学的製剤は固形組成物、凍結乾燥製剤又は水溶液として保存されうる。
【0092】
本発明の薬学的製剤は良好な医療行為に一致した形、つまり量、濃度、スケジュール、過程、ビヒクル及び投与経路で、用量が決められ、投与されうる。この点で考慮すべき要因には、治療中の特定の疾患、治療中の特定の哺乳動物、個々の患者の臨床状態、疾患の原因、薬剤の送達部位、投与方法、投与スケジュール、並びに医師に知られている他の因子が含まれる。投与される化合物の「治療的に有効な量」はそのような考慮によって左右され、凝固因子媒介疾患を防止し、軽減し又は治療するのに必要な最少の量である。そのような量は好ましくはホストに毒性であるか又はホストを出血に対してより高い感受性にする量以下である。
一般的な事項として、投与されるトラスツズマブ−MCC−DM1の用量当たりの最初の薬学的に有効な量は、一日当たり約0.01〜100mg/kg患者体重の範囲、すなわち一日当たり約0.1から20mg/kg患者体重で、使用される化合物の典型的な最初の範囲は0.3から15mg/kg/日である。
【0093】
許容される希釈剤、担体、賦形剤及び安定化剤は、用いられる用量及び濃度でレシピエントに非毒性であり、リン酸塩、クエン酸塩、及び他の有機酸などの緩衝液;アスコルビン酸及びメチオニンを含む抗酸化剤;保存料(塩化オクタデシルジメチルベンジルアンモニウム;塩化ヘキサメトニウム;塩化ベンザルコニウム;塩化ベンゼトニウム;フェノール;ブチル、エタノール又はベンジルアルコール;メチル又はプロピルパラベン等のアルキルパラベン;カテコール;レゾルシノール;シクロヘキサノール;3-ペンタノール;及びm-クレゾールなど);低分子量(約10残基未満)ポリペプチド;血清アルブミン、ゼラチン、又は免疫グロブリン等のタンパク質;ポリビニルピロリドン等の親水性ポリマー;グリシン、グルタミン、アスパラギン、ヒスチジン、アルギニン、又はリジン等のアミノ酸;グルコース、マンノース、又はデキストリンを含む単糖類、二糖類、及び他の炭水化物;EDTA等のキレート剤;スクロース、マンニトール、トレハロース又はソルビトールなどの糖;ナトリウムなどの塩形成対イオン;金属錯体(例えば、Zn-タンパク質錯体)及び/又はトゥイーン80を含むTWEENTM、プルロニクス(PLURONICSTM)又はPEG400を含むポリエチレングリコール(PEG)等の非イオン性界面活性剤を含む。また、活性な薬学的成分は、例えばコアセルベーション技術により又は界面重合により調製されたマイクロカプセル、例えば、各々ヒドロキシメチルセルロース又はゼラチン-マイクロカプセル及びポリ(メタクリル酸メチル)マイクロカプセル中、コロイド状薬物送達系(例えば、リポソーム、アルブミンミクロスフィア、マイクロエマルション、ナノ粒子及びナノカプセル)中、又はマイクロエマルション中に包括されていてもよい。このような技術は、Remington's Pharmaceutical Science 18版, (1995) Mack Publ. Co., Easton, PAに開示されている。
【0094】
薬学的製剤はここに詳細が記載される投与経路に適したものを含む。製剤は簡便には単位投薬形態で提供され得、薬学の分野でよく知られた方法の何れかによって調製されうる。技術及び製剤化は一般にRemington's Pharmaceutical Sciences 18版. (1995) Mack Publishing Co., Easton, PAに見出される。かかる方法は、活性成分を、一又は複数の副成分を構成する担体と組み合わせる工程を含む。一般に、製剤は、活性成分を、液体担体又は微細に粉砕した固形担体又は双方と均一かつ密に組み合わせ、ついで必要ならば生成物を成形することにより調製される。
【0095】
経口投与に適した化学療法剤の製剤は、予め定まった量のトラスツズマブ−MCC−DM1の化合物及び/又は化学療法剤をそれぞれ含む、丸薬、硬カプセルないしは軟カプセル剤、例えばゼラチンカプセル剤(capsules)、カプセル(cachet)、トローチ剤、ロゼンジ剤、水性ないしは油性懸濁液、分散可能パウダーないしは顆粒、エマルション、シロップ又はエリキシル剤のような別々の単位として調製されうる。このような製剤は薬学的組成物の製造のために当該分野で知られている任意の方法に従って調製することができ、そのような組成物は、口に合う製剤を提供するために、甘味料、香味料、着色剤及び保存剤を含む一又は複数の薬剤を含みうる。圧縮錠は、場合によってはバインダー、潤滑剤、不活性希釈剤、保存料、界面活性剤又は分散剤と混合せしめて粉末又は顆粒のような自由に流動する形態で活性成分を適切な機械で圧縮することによって調製することができる。成形錠剤は不活性な液体希釈剤で湿潤させた粉末化活性成分の混合物を適切な機械で成形することによって製造することができる。錠剤は場合によっては被覆し又は切り込み線を入れ、場合によっては活性成分の遅延又は制御放出をもたらすように製剤化される。
【0096】
本発明の薬学的製剤の錠剤賦形剤には、錠剤を形成する粉剤のバルク体積を増やすための充填材(または希釈剤);摂取され、薬剤の迅速溶解および吸収を促進する場合に、錠剤を小さな断片、理想的には個々の薬剤粒子に破壊する崩壊剤;顆粒および錠剤が必須の機械的強度によって形成され、圧縮された後に合わさって錠剤となり、包装、輸送および慣例的な処理の間にその成分粉に壊れるのを防ぐ結合剤;製造の間に錠剤を形成する粉の流動性を向上させるための流動促進剤;製造の間に錠剤を圧縮するために用いる機材に錠剤化粉末が付着しないようにするための滑沢剤が含まれる。これらは、圧縮による粉混合物の流量を向上させ、完結した錠剤が機材から放出されるように、摩擦および破損を最小化する。流動促進剤と同様の機能を有する抗粘着剤は、錠剤を形成する粉末と製造中に錠剤の形状を打ち抜くために用いられる機械との間の接着を低減する。錠剤に配合される香料は、好ましい味覚を与えるか又は不快な味覚をマスキングし、着色料は認識及び患者のコンプライアンスを得るためである。
【0097】
錠剤の製造に適した非毒性の薬学的に許容可能な賦形剤と混合せしめられて活性成分を含む錠剤が許容可能である。これらの賦形剤は、例えば不活性な希釈剤、例えば炭酸カルシウム又はナトリウム、ラクトース、リン酸カルシウム又はナトリウム;顆粒化及び崩壊剤、例えばトウモロコシデンプン、又はアルギン酸;結合剤、例えばデンプン、ゼラチン又はアカシア;及び潤滑剤、例えばステアリン酸マグネシウム、ステアリン酸又はタルクでありうる。錠剤は非被覆でも、又は胃腸管中での崩壊と吸着を遅延させるマイクロカプセル化を含む既知の方法によって被覆してもよく、それによって長時間にわたる持続作用をもたらす。例えば、時間遅延物質、例えばモノステアリン酸グリセリル又はジステアリン酸グリセリルを単独で又はロウと共に用いることができる。
【0098】
眼又は他の外部組織、例えば口及び皮膚の治療には、製剤は、好ましくは、例えば0.075から20%w/wの量で活性成分を含む局所用軟膏又はクリームとして適用される。軟膏に製剤される場合、活性成分はパラフィン系又は水混和性軟膏基剤と共に用いることができる。あるいは、活性成分は水中油クリーム基剤を用いてクリームに製剤化することができる。
所望される場合、クリーム基剤の水性相は多価アルコール、すなわち、例えばプロピレングリコール、ブタン1,3-ジオール、マンニトール、ソルビトール、グリセロール及びポリエチレングリコール(PEG400を含む)及びその混合物のような二又はそれ以上のヒドロキシル基を有するアルコールを含みうる。局所用製剤は、好ましくは、皮膚又は他の患部領域を通しての活性成分の吸収又は浸透を向上させる化合物を含みうる。そのような皮膚浸透向上剤の例はジメチルスルホキシド及び関連類似体を含む。
【0099】
本発明のエマルションの油性相は、脂肪又は油との、あるいは脂肪と油の双方との少なくとも一の乳化剤の混合物を含む、公知の方法で既知の成分から構成することができる。好ましくは、親水性乳化剤が、安定化剤として作用する親油性乳化剤と共に含有せしめられる。併せて、安定化剤と共に又は安定化剤を伴わない乳化剤は乳化ロウを構成し、油及び脂肪と共にロウはクリーム製剤の油性分散相を形成する乳化軟膏基剤を含む。本発明の製剤に使用するのに適した乳化剤及びエマルション安定化剤には、トゥイーン(Tween(登録商標))60、スパン(Span(登録商標))80、セトステアリルアルコール、ベンジルアルコール、ミリスチルアルコール、モノ-ステアリン酸グリセリル及びラウリル硫酸ナトリウムが含まれる。
本発明の薬学的組成物の水性懸濁液は水性懸濁液の製造に適した賦形剤と混合せしめられて活性物質を含む。そのような賦形剤には、懸濁剤、例えばナトリウムカルボキシメチルセルロース、クロスカルメローゼ、ポビドン、メチルセルロース、ヒドロキシプロピルメチルセルロース、アルギン酸ナトリウム、ポリビニルピロリドン、トラガカントガム及びアカシアガム、及び分散又は湿潤剤、例えば天然に生じるホスファチド(例えばレシチン)、脂肪酸とのアルキレンオキシドの縮合産物(例えばポリオキシエチレンステアレート)、長鎖脂肪族アルコールとのエチレンオキシドの縮合産物(例えばヘプタデカエチレンオキシセタノール)、脂肪酸とヘキシトール無水物から誘導された部分エステルとのエチレンオキシドの縮合産物(例えばポリオキシエチレンソルビタンモノオレアート)が含まれる。水性懸濁液はまた一又は複数の保存料、例えばエチル又はn-プロピルp-ヒドロキシ-ベンゾエート、一又は複数の着色剤、一又は複数の香味剤及び一又は複数の甘味料、例えばスクロース又はサッカリンを含みうる。
【0100】
薬学的組成物は滅菌された注射用製剤の形態、例えば滅菌注射用水性又は油性懸濁液であってもよい。この懸濁液は上に述べた好適な分散又は湿潤剤及び懸濁剤を用いて公知技術に従って製剤化することができる。滅菌された注射用製剤はまた1,3-ブタン-ジオール溶液又は凍結乾燥粉末から調製されたもののように、非毒性の非経口的に許容可能な希釈剤又は溶媒中の溶液又は懸濁液であってもよい。用いることができる許容可能なビヒクル及び溶媒は水、リンガー液及び等張塩化ナトリウム溶液である。また、滅菌固定化油を溶媒又は懸濁媒質として便宜的に用いることができる。この目的に対して、合成のモノ-又はジグリセリドを含む任意のブランドの固定化油を用いることができる。また、オレイン酸のような脂肪酸も同様に注射剤の調製に使用することができる。
単回投薬形態をつくるために担体物質と混合されうる活性成分の量は治療されるホストと特定の投与形式に応じて変わる。例えば、ヒトへの経口投与のための時間放出製剤は、全組成物の約5から約95%(重量:重量)と変わりうる適切で簡便な量の担体物質と共に配合されておよそ1から1000mgの活性物質を含みうる。薬学的組成物は投与のために容易に測定可能な量をもたらすように調製することができる。例えば、静脈点滴のための水溶液は、約30mL/hrの割合で適した体積の点滴が生じうるようにするために溶液1ミリリットル当たり約3から500μgの活性成分を含みうる。
【0101】
非経口投与に適した製剤には、抗酸化剤、バッファー、静菌剤及び意図したレシピエントの血液と製剤を等張にする溶質を含みうる水性及び非水性滅菌注射用溶液;及び懸濁剤及び増粘剤を含みうる水性及び非水性滅菌懸濁液が含まれる。
眼への局所投与に適した製剤には、好適な担体、特に活性成分のための水性溶媒に活性成分が溶解又は懸濁させられた点眼液がまた含まれる。活性成分はそのような製剤中に好ましくは0.5から20%、例えば0.5から10%、例えば約1.5%w/wの濃度で存在する。
口への局所投与に適した製剤には、香味基剤、通常はスクロース及びアカシア又はトラガカント中に活性成分を含むロゼンジ;ゼラチン及びグリセリン、又はスクロース及びアカシアのような不活性基剤に活性成分を含むパスティユ;及び適切な液体担体に活性成分を含むうがい薬が含まれる。
直腸投与のための製剤は、例えばココアバター又はサリチラートを含む好適な基剤を用いて座薬として提供することができる。
【0102】
肺内又は経鼻投与に適した製剤は、例えば0.1から500ミクロン(例えば0.5、1、30ミクロン、35ミクロン等々のような増分ミクロンで0.1から500ミクロンの範囲の粒子径を含む)の範囲の粒子径を有し、これが鼻経路を通る迅速な吸入又は肺胞嚢に達するように口からの吸入によって投与される。好適な製剤には、活性成分の水性又は油性溶液が含まれる。エアゾール又は乾燥粉末投与に適した製剤は常法によって調製することができ、以下に記載されるような疾患の治療又は予防にこれまで使用されている化合物のような他の治療剤と共に送達されうる。
膣投与に適した製剤は、活性成分に加えて、当該分野で適切であることが知られているような担体を含むペッサリー、タンポン、クリーム、ゲル、ペースト、フォーム又はスプレー製剤として提供することができる。
【0103】
製剤は、単位投薬又は複数投薬容器、例えば密封されたアンプル及びバイアルに包装することができ、使用直前に注射用の滅菌液体担体、例えば水の添加のみを必要とするフリーズドライ(凍結乾燥)条件で保存することができる。即時混合注射溶液及び懸濁液は既に記載された種類の滅菌粉末、顆粒及び錠剤から調製される。好適な単位投薬製剤は、活性成分の、上に記載されたような毎日の投薬又は毎日の部分用量単位、又はその適切なフラクションを含むものである。
本発明は、獣医学的担体と共に上述の少なくとも一の活性成分を含有する動物用医薬組成物を更に提供する。獣医学的担体は該組成物を投与する目的に有用な物質であり、不活性であるか獣医学分野で許容可能であり活性成分と相容性のある固形、液体又はガス状物質でありうる。これらの動物用医薬組成物は非経口的、経口的又は任意の他の所望の経路で投与されうる。
【0104】
(併用療法)
トラスツズマブ−MCC−DM1は、前悪性及び非腫瘍又は非悪性の過剰増殖性疾患とともに、腫瘍、癌及び腫瘍性組織を含む、過剰増殖性疾患又は障害の治療のために、他の化学療法剤と組み合わされて用いられてよい。ある実施態様では、トラスツズマブ−MCC−DM1は、薬学的併用製剤又は併用療法としての投薬計画において、抗過剰増殖性特性を有するか又は過剰増殖性疾患を治療するために有用である第二の化合物と組み合わされる。薬学的併用製剤又は投薬計画の第二化合物は好ましくは、トラスツズマブ−MCC−DM1に対して相補的な活性を有し、互いに悪影響を与えない。このような化合物は、意図する目的のために有効である量で適切に併用される。一実施態様では、本発明の組成物は、本明細書において記述されるような化学療法剤と組み合わせたトラスツズマブ−MCC−DM1を含む。実施例4および5はそれぞれ、T−DM1+パーツズマブおよびT−DM1+GDC−0941の臨床プロトコールである。
【0105】
本発明の治療的併用には、過剰増殖性疾患の治療での別々、同時又は連続した使用のための併用用製剤としての、トラスツズマブ−MCC−DM1と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869およびドセタキセルから選択される化学療法剤との投与を含む、製剤化、投薬計画又は治療の他の工程が含まれる。
併用療法剤は同時の又は逐次の投与計画として投与されうる。逐次に投与される場合、併用剤は、二以上の投与で投与されうる。併用投与は、別個の製剤又は単一の薬学的製剤を使用する同時投与と、好ましくは両方の(又は全ての)活性薬剤がその生物学的活性を同時に作用させる期間が存在している、何れかの順の連続投与を含む。
上記の同時投与される薬剤の何れかに対して適した投薬量は現在使用されるものであり、新たに同定された薬剤と他の化学療法剤又は治療との組み合わせた作用(相乗作用)のために低くされうる。
抗癌療法の具体的な実施態様では、トラスツズマブ−MCC−DM1は、本明細書中に記載されるホルモン剤又は抗体医薬を含む化学療法剤、並びに外科的治療法および放射線療法と組み合わされうる。トラスツズマブ−MCC−DM1および他の薬学的に活性な化学療法剤(一又は複数)の量と投与の相対的タイミングは、所望の併用治療効果を達成するために選択されであろう。
【0106】
(薬学的組成物の投与)
本発明の化合物は、治療される状態に適切な任意の経路によって投与されてよい。適切な経路には、経口、非経口(皮下、筋肉内、静脈内、動脈内、吸入、皮内、髄膜下、硬膜外および注入技術を含む)、経皮、直腸、経鼻、局所的(頬側および舌下を含む)、経膣、腹膜内、肺内及び鼻腔内が含まれる。また、局所投与は、経皮パッチ又はイオン泳動法装置といった経皮投与の使用を伴いうる。薬剤の製剤は、Remington's Pharmaceutical Sciences, 18th Ed., (1995) Mack Publishing Co., Easton, PAにおいて述べられている。薬剤製剤の他の例は、Liberman, H. A. and Lachman, L., Eds., Pharmaceutical Dosage Forms, Marcel Decker, Vol 3, 2nd Ed., New York, NYにおいて見られる。局所の免疫抑制性治療のために、化合物は、灌流を含む病巣内投与によって、あるいは移植の前に移植片を阻害剤と接触させることによって投与されてよい。周知のことであるが、好ましい経路は、例えばレシピエントの状態によって異なってよい。化合物が経口投与される場合、丸薬、カプセル、錠剤などとして、薬学的に許容可能な担体、流動促進剤又は賦形剤にて調製されてよい。化合物が非経口的に投与される場合、後述されるように、薬学的に許容可能な非経口ベヒクル又は希釈剤によって、そして、単位用量注射用形態に調製されてよい。
【0107】
ヒト患者を治療するためのトラスツズマブ−MCC−DM1の用量は、およそ100mgからおよそ500mgであってよい。吸収、分布、代謝および排出を含む薬物動態学的(PK)及び薬力学的(PD)性質に応じて、トラスツズマブ−MCC−DM1の用量は、6週間に1回、3週間に1回、毎週又はもっと頻繁に投与されてよい。トラスツズマブ−MCC−DM1と組み合わされて用いられる化学療法剤の用量は、およそ10mgからおよそ1000mgまでであってよい。化学療法剤は、6週間に1回、3週間に1回、毎週、又は1日に1回ないし2回などもっと頻繁に投与されてよい。加えて、毒性因子は用量および投薬計画に影響しうる。経口投与される場合、丸薬、カプセル又は錠剤は、特定の期間に、毎日またはもっと少ない頻度で摂取されてよい。投薬計画は、多くの治療サイクルのために繰り返されてよい。
【0108】
(治療方法)
(1)トラスツズマブ−MCC−DM1と(2)化学療法剤との治療的組合せは、限定するものではないがHER2経路の活性化に特徴があるものを含む疾患、状態および/または障害を治療するために有用である。したがって、本発明の他の態様は、HER2又はVEGFRレセプター1を標的化することによって治療されうる疾患又は状態の治療方法を含む。(1)トラスツズマブ−MCC−DM1と(2)化学療法剤との治療的組合せは、前悪性及び非腫瘍性又は非悪性の過剰増殖性疾患とともに、腫瘍、癌及び腫瘍性組織を含む、過剰増殖性疾患又は障害の治療のために、用いられてよい。
本発明の方法に従って治療されうる癌には、限定するものではないが、胸部、卵巣、子宮頸管、前立腺、精巣、泌尿生殖器、食道、喉頭、神経膠芽腫、神経芽細胞腫、胃、皮膚、角化棘細胞腫、肺、類表皮癌、大細胞カルチノーマ、非小細胞肺カルチノーマ(NSCLC)、小細胞カルチノーマ、肺腺癌、骨、大腸、腺腫、膵臓、腺癌、甲状腺、濾胞性カルチノーマ、未分化癌、乳頭カルチノーマ、精上皮腫、メラノーマ、肉腫、膀胱カルチノーマ、肝臓カルチノーマ及び胆汁路、腎臓カルチノーマ、骨髄疾患、リンパ腫、線毛細胞、頬側(口腔)及び咽頭、唇、舌、咽頭、小腸、大腸-直腸、大腸、直腸、脳及び中枢神経系、ホジキン、及び白血病が含まれる。
本発明の他の態様は、本明細書に記載の疾患又は状態に罹患している哺乳動物、例えばヒトの該疾患又は状態の治療に用いるための、薬学的組成物又は治療上の組合せを提供する。また、本明細書中に記載の疾患及び症状に罹っている哺乳動物などの温血動物の該障害の治療のための医薬の調製における薬学的組成物の使用を提供する。
【0109】
(製造品)
本発明の他の実施態様では、上述の疾患及び障害の処置に有用なトラスツズマブ−MCC−DM1を具備する製造品又は「キット」が提供される。一実施態様では、キットは、トラスツズマブ−MCC−DM1を含む容器を具備する。キットは更に容器にあるか付随させられるラベル又はパッケージ挿入物を具備する。「パッケージ挿入物」なる用語は、効能、使用法、用量、投与法、禁忌についての情報及び/又はかかる治療用製品の使用に関する注意書きを含む治療用製品の商業的パッケージに常套的に含まれる指示書を意味する。適切な容器は、例えば、瓶、バイアル、シリンジ、ブリスター包装等を含む。該容器はガラス又はプラスチック等の様々な材料から形成されうる。該容器は、症状を処置するのに効果的であるトラスツズマブ−MCC−DM1又はその製剤を収容し、無菌アクセスポートを有しうる(例えば、該容器は静脈内溶液バッグ又は皮下注射針により突き刺されうるストッパーを有しているバイアルでありうる)。組成物中の少なくとも一の活性剤はトラスツズマブ−MCC−DM1である。ラベル又はパッケージ挿入物は、組成物が例えば癌のような選択された症状を治療するために使用されることを示している。一実施態様では、ラベル又はパッケージ挿入物は、トラスツズマブ−MCC−DM1を含有する組成物が異常な細胞増殖から生じる疾患を治療するために使用できることをまた示しているであろう。ラベル又はパッケージ挿入物は、組成物を他の疾患の治療に使用できることもまた示している場合がある。あるいは、又は付加的に、製造品は、薬学的に許容可能なバッファー、例えば注射用静菌水(BWFI)、リン酸緩衝生理食塩水、リンガー液及びデキストロース溶液を含む第三容器を更に含みうる。それは、他のバッファー、希釈剤、フィルター、針、及びシリンジを含む商業的及び使用者の観点から望まれる他の材料を更に含みうる。
【0110】
キットは、トラスツズマブ−MCC−DM1と、もし存在するならば第二薬学的製剤の投与のための指示書を更に具備しうる。例えば、キットがトラスツズマブ−MCC−DM1を含有する第一組成物と第二薬学的製剤を含むならば、キットは、第一及び第二薬学的組成物を、それを必要とする患者に同時に、連続的に又は別個に投与するための指示書を更に具備しうる。
他の実施態様では、キットは、例えば錠剤又はカプセル剤のような、トラスツズマブ−MCC−DM1の固形経口形態を送達させるのに適している。かかるキットは好ましくは多くの単位投薬形態を含む。かかるキットは、その意図された使用順に配向された投薬量を持つカードを含みうる。かかるキットの例は「ブリスター包装」である。ブリスター包装は包装産業ではよく知られており、薬学的単位投薬形態を包装するために広く使用されている。所望される場合、記憶補助品が、例えば投薬量が投与されうる日を治療スケジュール中に示す数字、文字、又は他の印又はカレンダー挿入物の形態で提供されうる。
【0111】
一実施態様によれば、キットは、(a)トラスツズマブ−MCC−DM1が含まれる第一容器と;場合によっては(b)第二薬学的製剤がそこに含まれる第二容器とを具備し得、ここで、第二薬学的製剤は抗過剰増殖性活性を持つ第二化合物を含む。あるいは、又は付加的に、キットは薬学的に許容可能なバッファー、例えば注射用静菌水(BWFI)、リン酸緩衝生理食塩水、リンガー液及びデキストロース溶液を含む第三容器を更に具備しうる。それは、他のバッファー、希釈剤、フィルター、針、及びシリンジを含む商業的及び使用者の観点から望まれる他の材料を更に含みうる。
キットがトラスツズマブ−MCC−DM1の組成物と第二治療剤、すなわち化学療法剤を含む場合、キットは、別個の組成物を収容するための容器、例えば分割瓶又は分割フォイル小包を具備しうるが、別個の組成物は単一の分割されていない容器内に収容されてもよい。典型的には、キットは別個の組成物の投与のための指示書を含む。別個の成分が好ましくは異なった投薬形態(例えば経口と非経口)で投与され、異なった投薬間隔で投与される場合、又は併用される個々の成分の用量設定が処方医師によるのが望ましい場合に特に有利である。
【実施例】
【0112】
本発明を例示するために、以下の実施例が含まれる。しかしながら、これらの実施例は本発明を制限するものではなく、本発明を実施する方法を提案するためだけのものであると理解すべきである。
【0113】
実施例1 トラスツズマブ−MCC−DM1の調製
トラスツズマブは、50mM リン酸カリウム/50mM 塩化ナトリウム/2mM EDTA、pH6.5中に20mg/mlでバッファ-交換によってHERCEPTIN(登録商標)から精製し、スクシンイミジル 4(N-マレイミドメチル)シクロヘキサン-1-カルボキシレート(SMCC、Pierce Biotechnology, Inc)の7.5〜10モル等価物、DMSO又はDMA(ジメチルアセトアミド)中20mM、6.7mg/mlにて処理した(米国公開特許2005/0169933;米国公開特許2005/0276812)。室温のアルゴン下で2〜4時間撹拌した後、反応混合物は、50mM リン酸カリウム/50mM 塩化ナトリウム/2mM EDTA、pH6.5にて平衡化したセファデックスG25カラムに濾過した。あるいは、反応混合物は、pH6の30mM クエン酸塩および150mM 塩化ナトリウムにてゲル濾過した。抗体を含む分画を貯蔵し、分析した。トラスツズマブ−SMCCは88%回収した。
上記の薬剤−リンカー中間生成物であるトラスツズマブ−MCCは、50mM リン酸カリウム/50mM 塩化ナトリウム/2mM EDTA、pH6.5にて10mg/mlの終濃度に希釈し、ジメチルアセトアミド中DM1の10mM溶液(5 SMCC/トラスツズマブ、7.37mg/mlと予測される1.7等価物)と反応させた。DM1は、アンサミトシン発酵産物(米国特許第6790954号;米国特許第7432088号)から調製され、結合のために誘導体化されてよい(米国特許第6333410号;RE39151)。反応物は、アルゴン下の室温で4〜およそ16時間撹拌した。コンジュゲート反応混合物は、pH6.5の1×PBSにてセファデックスG25ゲル濾過カラム(1.5×4.9cm)に濾過した。あるいは、反応混合物は、pH5の10mM 琥珀酸塩および150mM 塩化ナトリウムにてゲル濾過した。252nm及び280nmの吸光度によって測定すると、DM1/トラスツズマブ比率(p)は3.1であった。また、抗体に対する薬剤の比率(p)は質量分析によって測定されてよい。また、コンジュゲートは、SDSポリアクリルアミドゲル電気泳動によってモニターされてよい。凝集はレーザー光散乱分析によって評価されてよい。
【0114】
あるいは、トラスツズマブ−MCC−DM1は、MCC−DM1リンカー−薬剤試薬を形成し、次いでトラスツズマブと反応させることによって調製されてよい。
一般的に、DM1とのトラスツズマブ−MCCのコンジュゲート反応により、付着しコンジュゲートしたDM1薬剤の数が異なる、すなわちpが1から約8の分布である薬剤負荷である抗体を含む不均質混合物となる。トラスツズマブ上の多くの異なる求核基、例えば終末リジンアミノ基がSMCCと反応しうる場合、トラスツズマブへのSMCCの異なる付着部位により、更なる次元の不均一性が存在する。ゆえに、トラスツズマブ−MCC−DM1は、単離され、精製された種間分子、並びに1から8の平均薬剤負荷の混合物を含み、このMCC−DM1はトラスツズマブ抗体の何れかの部位により付着している。
コンジュゲート反応からのトラスツズマブ−MCC−DM1の調製におけるトラスツズマブ抗体当たりのDM1薬剤部分の平均数は、質量分析、ELISAアッセイ、電気泳動法およびHPLCといった従来の手段によって特徴付けられうる。また、pに関するトラスツズマブ−MCC−DM1の定量的分布も決定されてよい。ELISAによって、ADCの特定の調製物におけるpの平均値が決定されてよい(Hamblett et al (2004) Clinical Cancer Res. 10: 7063-7070;Sanderson et al (2005) Clinical Cancer Res. 11: 843-852)。しかしながら、p(薬剤)値の分布は、抗体−抗原結合とELISAの検出限界によって識別されない。また、抗体−薬剤コンジュゲートの検出のためのELISAアッセイは、薬剤部分が抗体のどこに、例えば重鎖又は軽鎖の断片や特定のアミノ酸残基に付着しているかを決定しない。場合によって、pが他の薬剤負荷を有するトラスツズマブ−MCC−DM1からの特定の値である場合の均質なトラスツズマブ−MCC−DM1の分離、精製および特徴づけは、逆相HPLC又は電気泳動法といった手段によって達成されうる。
【0115】
実施例2 インビトロ細胞増殖アッセイ
本発明の組合せの有効性は、以下のプロトコールを使用した細胞増殖アッセイによって測定した(Promega Corp. Technical Bulletin TB288; Mendoza et al (2002) Cancer Res. 62: 5485-5488)。Cell-Titer Gloアッセイ試薬およびプロトコールは市販されている(Promega)。アッセイは、細胞に入り、細胞増殖に作用する化合物の能力を評価する。アッセイ原理は、細胞のATPを定量化することによって存在する生細胞の数を決定することである。Cell-Titer Gloはこの定量化のために使用する試薬である。それは、Cell-Titer Gloの添加により細胞溶解とルシフェラーゼ反応による発光シグナルの生成が生じる同種のアッセイである。発光シグナルは、存在するATPの量と比例している。
DMSOおよび培地プレート:ヌンクの96ウェル円錐形の底のポリプロピレンプレート(カタログ番号249946)。
細胞プレート:ファルコンの384-ウェル黒色、透明底(マイクロクリアー)、蓋付きのTCプレート(353962)。
細胞培養培地:RPMI又はDMEM高グルコース;ハムのF-12(50:50)、10%胎仔ウシ血清、2mM L−グルタミン。
Cell Titer-Glo:Promega(カタログ番号G7572)
【0116】
手順:
1日目−細胞プレートへの播種。細胞を回収し、3日間のアッセイのために384ウェルの細胞プレートに1ウェルにつき54μlにつき1000〜2000細胞で細胞を播種する。37℃、5%CO2下で、一昼夜(約16時間)インキュベートする。
2日目−細胞への薬剤添加。化合物希釈物、DMSOプレート(9ポイントについて1:2段階希釈)。96ウェルプレートの2列目に20μlの化合物(小分子薬剤では10mMの貯蔵液)を加える。高精度培地プレート(1:50希釈)を用いて、合計9ポイントについてプレート(10μl+10μl 100%DMSO)全体にわたって1:2段階希釈を行った。異なる96ウェル培地プレートのすべてのウェルに147μlの培地を添加する。Rapidplateを用いて、DMSOプレートの各ウェルからのDMSO+化合物の3μlを培地プレート上の各々対応するウェルに移す。2薬剤併用試験のために、Rapidplateを用いて、DMSOプレートの各ウェルからのDMSO+化合物の1薬剤1.5μlを培地プレート上の各々対応するウェルに移す。次いで、もう1つの薬剤1.5ulを培地プレートへ移す。
細胞への薬剤添加、細胞プレート(1:10希釈)、6μlの培地+化合物を細胞に直接添加する(すでに細胞上に54μlの培地)。頻繁に開けられないインキュベーター中で37℃、5%CO2で3日間インキュベートする。
5日目−プレートを反応させる、Cell Titer Gloバッファを室温で解凍する。37℃および室温に平衡な温度からおよそ30分間細胞プレートを取り除く。Cell Titer GloバッファをCell Titer Glo基質に添加する(瓶から瓶へ)。30μlのCell Titer Glo試薬を各ウェルの細胞に加える。およそ30分間プレートシェーカ上に置く。PerkinElmer Envision(1ウェルにつき0.1秒)又はAnalyst HTプレート読み取り機(1ウェルにつき0.5秒)にて発光を読み取る。
【0117】
細胞生存率アッセイおよび併用アッセイ:細胞は、384ウェルプレートの1ウェルにつき1000〜2000細胞を播いて16時間置いた。2日目に、96ウェルプレートに、DMSOにて9つの1:2段階希釈化合物希釈物を作製した。Rapidplateロボット(Zymark Corp., Hopkinton, MA)を用いて、化合物を更に増殖培地にて希釈した。次いで、希釈された化合物を384ウェルの細胞プレートの4通りのウェルに加え、37℃および5%CO2でインキュベートした。4日後に、生細胞の相対数を、製造業者の指示に従ってCell-Titer Glo(Promega)を用いて発光によって測定し、Envision又はWallac Multilabel読み取り機(PerkinElmer, Foster City)にて読み取った。EC50値は、Kaleidagraph4.0(Synergy Software)又はPrism4.0ソフトウェア(GraphPad, San Diego)を用いて算出した。併用アッセイの薬剤は、8×EC50濃度で始めて投与した。薬剤のEC50が2.5μMを超える症例では、最も高い濃度である10μMを用いた。すべてのアッセイにおいて、トラスツズマブ−MCC−DM1と化学療法剤は、同時又は4時間空けて(一方から他方までの間)投与した。
【0118】
他の例示的なインビトロ細胞増殖アッセイは以下の工程を含む。
1. 培地中におよそ104細胞(細胞株及び腫瘍種類については図1を参照)を含む100μlの細胞培養物の一定分量を、384ウェルの不透明な壁のプレートの各ウェルに置いた。
2. 培地を含むが細胞を含まないコントロールウェルを調製した。
3. 化合物を試験ウェルに加え、3〜5日間インキュベートした。
4. プレートを室温と平衡にしておよそ30分間置いた。
5. 各ウェルに存在する細胞培養培地の体積と等しい量のCellTiter-Glo試薬を添加した。
6. 内容物を軌道シェーカ上で2分間混合し、細胞溶解を誘導した。
7. プレートを室温で10分間インキュベートし、発光シグナルを安定させた。
8. 発光は、RLU=相対的発光単位として記録し、グラフに記した。
あるいは、96ウェルのプレート中に細胞を最適密度で播き、試験化合物の存在下で4日間インキュベートした。その後、アラマーブルーTMをアッセイ培地に加え、細胞を6時間インキュベートした後、544nmの励起、590nmの発光で読み取った。EC50値は、S字状の用量応答曲線フィットを用いて算出した。
【0119】
実施例3 インビボ腫瘍異種移植片
トランスジェニック実験に適する動物は、通常の市販元から入手することができる。雌CB−17SCIDベージュマウス(Charles River Laboratory)の各群の乳房脂肪パットに、3000000のKPL-4(Her2過剰発現)乳癌細胞をマトリゲルとともに移植した。雌胸腺欠損ヌードマウス(Charles River Laboratory or Harlan)の各群の乳房脂肪パッドに、2×2mm3断片のMMTV-Her2 Fo5トランスジェニック胸部腫瘍を移植した。0日目に、各腫瘍モデルに特化した計画に従って、マウス異種移植片に、薬剤、薬剤併用又はベヒクルを投与した。5−FU、ゲムシタビン、カルボプラチンおよびB20-4.1を腹膜内投与し、パーツズマブは示したように静脈内又は腹膜内投与し、トラスツズマブ−MCC−DM1とドセタキセルは静脈内投与し、ラパチニブ、GDC−0941およびABT−869は経管栄養により経口的に投与した。試験期間中、週に2回腫瘍サイズを記録した。マウスの体重も週に2回記録し、マウスを定期的に観察した。ウルトラCal IVカリパス(Model 54-10-111; Fred V. Fowler Co., Inc.; Newton, MA)を用いて腫瘍体積を二次元(長さおよび幅)で測定し、エクセルv.11.2(Microsoft Corporation; Redmond, WA)を用いて分析した。腫瘍阻害グラフは、KaleidaGraphバージョン3.6(Synergy Software; Reading, PA)を用いてプロットした。腫瘍体積は以下の式によって算出した:腫瘍サイズ(mm3)=(長い方の測定値×短い方の測定値2)×0.5。
【0120】
Adventurera Pro AV812スケール(Ohaus Corporation; Pine Brook, NJ)を用いて動物の体重を測定した。KaleidaGraphバージョン3.6を使用してグラフを作成した。重量変化の割合は以下の式を使用して算出した:群重量変化の割合=(1−(最初の重量/新たな重量))×100。
腫瘍体積が2000mm3を上回わるか又は体重喪失が開始時の重量の20%以上であるマウスを、規定のガイダンスに従ってすぐに安楽死させた。
試験の終了時(EOS)の腫瘍増殖遅延の割合(%TGD)は、以下の式を用いて算出した:%TGD=100×(処置群のエンドポイントまでの中位時間−コントロール群のエンドポイントまでの中位時間)/コントロール群のエンドポイントの中位時間。
腫瘍発生(TI)は、試験終了時に各群に残っている測定可能な腫瘍の数を基に決定した。部分的な応答(PR)は、開始時の腫瘍体積と比較して腫瘍体積の50%超かつ100%未満の減少として定義し、3つの連続した測定値について観察した。完全寛解(CR)は、開始時の腫瘍体積と比較して腫瘍体積の100%の減少として定義し、3つの連続した測定値について観察した。データを分析し、p値はJMP統計ソフトウェア、バージョン5.1.2(SAS Institute; Cary, NC)によるダネットのt検定を使用して決定した。試験終了時の個々の腫瘍体積と平均腫瘍体積±SEM値は、JMP統計ソフトウェア、バージョン5.1.2を用いて算出した。体重データは、開始時の体重±SEMからの平均の変化の割合に基づいてグラフ化した。
【0121】
実施例4 パーツズマブと併用したトラスツズマブ−MCC−DM1(T-DM1)の臨床研究
従来の治療を受けていながら進行したHER2陽性の局所進行性又は転移性の乳癌を有する患者に静脈内投与したパーツズマブと併用したトラスツズマブ−MCC−DM1(T−DM1)の、安定性、耐容性及び有効性の第1b/II相、非盲検試験を設定し、併用の安全性と耐容性を特徴付けた。何れかの治療ラインにおいてトラスツズマブを既に摂取したか、又は進行した疾患のためにHER2標的治療と組み合わせて化学療法剤を摂取したか、又は最も最新の治療を受けたが進行した、HER2陽性の局所進行性又は転移性の乳癌を有する患者に、併用を3週間ごとに投与した。他の目的は、この計画においてT−DM1とパーツズマブの併用が投与される場合のT−DM1の薬物動態学を評価することである。他の目的は、この計画において投与されるT−DM1とパーツズマブの併用の有効性の事前評価をすることであり、これは、変更した固形腫瘍の治療効果判定基準(RECIST)、バージョン1.0を用いた調査者評価に基づいた客観的な応答速度によって測定される。この試験の二次目的は以下の通りである:(1) この計画において投与されるT−DM1とパーツズマブの併用を摂取した患者の無進行生存率(PFS)を推定すること;(2) この計画において投与されるT−DM1とパーツズマブの併用の応答の継続期間を評価すること;そして、(3) T−DM1に対する抗治療的抗体の開発を評価すること。
【0122】
トラスツズマブを既に摂取しており、その最後の治療後ないしは治療の間に進行したHER2陽性の局所進行性又は転移性の乳癌を有する患者において、T−DM1は、パーツズマブと組み合わされて静脈内(IV)注入によって投与されるであろう。患者は、最低3週間の間隔の反復サイクルで、T-DM1とパーツズマブの併用を摂取する。
所定の用量レベルの患者は、DLT観察期間(T−DM1の第一用量の時間から21日間とする)中、試験薬剤の第一用量を摂取した後で比較的高い用量レベルでの患者の治療の前に、DLT(用量制限毒性)について観察される。DLT観察期間中にこれらの患者においてDLTが観察されなければ、次の用量レベルへの用量段階的拡大を進めてよい。
【0123】
DLTは、DLT観察期間内に生じる次の何れかの治療関連毒性として定義される。(1) グレード3以上 疾患進行又は他の明らかに認識できる原因によるものでない非血液学的な有害事象、何れかのグレードの脱毛症を除く;(2) グレード3 標準的な治療法に応答する下痢;(3) グレード3 標準的な治療法に応答する前投薬がない場合の嘔気又は嘔吐;(4) グレード3以上 肝臓又は骨転移の結果としての、72時間続く、血清ビリルビン、肝トランスアミナーゼ(ALT又はAST)又はアルカリホスファターゼ(ALP)の上昇、グレード2の肝トランスアミナーゼ又は基準値のALPレベルを有する患者は除く(正常の上限値[ULN]5以下)。肝トランスアミナーゼ又はALPレベルが10ULN以上であれば、DLTとみなされるであろう。(5) グレード4以上 24時間続く血小板減少;(6) グレード4以上 4日間続くか又は熱(経口又は経鼓膜の温度100.4°F又は38℃)を伴う好中球減少症(好中球の絶対数500/細胞/mm3未満);(7) 調査者が何れかの試験化合物と関連していると感じる、主観的な過度の毒性;(8) 治療の第2サイクルの開始を妨げる、治療関連の毒性。
【0124】
次の最も高い用量レベルに進むことが決定された後は、患者内の用量段階的拡大も可能となると考える。試験に参加する患者は、まず最初に完全用量のパーツズマブとともに用量を減らしたT−DM1(3.0mg/kg)を摂取する。いったんコホートがDLT観察期間を終えると、これらの患者は、次のサイクルのために両薬剤の全用量へ増やすことが可能であると考えられる。しかしながら、3.6mg/kgの服用レベルの安全性はDLTの評価に基づく。患者(試験の用量−段階的拡大段階の間に試験に参加した人々を含む)は、初めの経過観察腫瘍評価により試験中である場合、有効性についての評価が可能であるとみなす。超音波心臓診断図(ECHO)又はマルチゲート取得(MUGA)スキャンは、サイクル1の終わりに、次いで試験期間を通して3サイクルごとに実施すべきである。
【0125】
T-DM1製剤
T-DM1は、20mmのフルオロ樹脂の積層ストッパーと反転式の灰色のプラスチック製キャップによるアルミニウム密閉を具備した20mlのI型USP/ヨーロッパ薬局方ガラスバイアルの使い捨ての凍結乾燥製剤として提供されてよい。8.0mLの注射用滅菌水(SWFI)にて再構成した後に、結果として生じる生成物は、10mM 琥珀酸ナトリウム、pH5.0、6%(w/v)スクロースおよび0.02%(w/v)ポリソルベート20中20mg/mLのT−DM1を含有する。各20mLのバイアルはおよそ172mgのT−DM1を含有し、160mgのT−DM1の運搬を可能とする。示された体積のT−DM1溶液をバイアル(一又は複数)から取り除き、IVバッグに加える。再構成されたT−DM1は、0.45%又は0.9%の塩化ナトリウム注射(最低250mLの体積)を含有するPVC又は無天然ゴム・無PVCのポリオレフィンバッグ(PO)に希釈される。0.45%塩化ナトリウムを含有するPVC又はPOバッグの使用が好まれる。0.9%の塩化ナトリウムを含有するPVC又はPOバッグが使われる場合、0.22μmインラインフィルターの使用が推奨される。バッグはゆっくりと逆さにして、溶液を混合する。0.9%又は0.45%の塩化ナトリウム注射、USPを含有するポリ塩化ビニル(PVC)又は無天然ゴム無PVCのポリオレフィン(PO)バッグ中で希釈される注入用T−DM1溶液は、短期間の間、2℃〜8℃(36°F〜46°F)で保存されてよい。
【0126】
パーツズマブ製剤
パーツズマブは、20mm L−ヒスチジン(pH6.0)、120mM スクロースおよび0.02%ポリソルベート20において調製される30mg/mlのパーツズマブを含有する使い捨ての製剤として提供される。各20ccのバイアルは、およそ420mgのパーツズマブ(14.0mL/バイアル)を含有する。示される体積のパーツズマブ溶液をバイアルから取り出し、注射用の0.9%塩化ナトリウム溶液の250ccのIVバッグに加える。バッグをゆっくりと逆さにして溶液を混合し、投与前に粒子及び変色について視覚的に調べる。0.9%塩化ナトリウムを含有するポリエチレン又は非PVCのポリオレフィンバッグ中で希釈される注入用パーツズマブの溶液は、短期間の間、2℃〜8℃(36°F〜46°F)で保存されてよい。
【0127】
安全性結果測定
T−DM1およびパーツズマブの安全性および耐容性は、以下の一次安全性結果測定を使用して評価される:(1) 有害事象の発症、性質および重症度;(2) T−DM1および/またはパーツズマブの用量変更、用量遅延又は中止が生じるような、試験薬物投与中及び該投与後の有害現象又は物理的な所見および臨床検査室結果の変化;及び、(3) ECHO又はMUGAスキャンを含む心機能の変化(すなわち左室駆出分画率[LVEF]、分節性壁異常)。
【0128】
薬物動態学的及び薬力学的結果測定
以下のT−DM1およびパーツズマブの薬物動態学的パラメーターは、適切なときに、データが得るように、いずれかの非区画および/または集団方法を使用する試験治療を受けるすべての患者において決定する:(1) T−DM1(コンジュゲート)、総トラスツズマブ(DM1フリー及びDM1コンジュゲート)の血清濃度;(2) DM1フリーの血漿濃度;(3) 総曝露(濃度−時間曲線下の領域[AUC]);(4) 最大血清中濃度(Cmax);(5) 最小濃度(Cmin);(6) クリアランス;(7) 分布の体積;(8) 最終的な半減期;(9) T−DM1に対する抗治療的抗体。
【0129】
有効性結果測定
変更RECIST, v1.0を用いた客観的な応答速度を有効性結果測定として評価する。この研究の二次有効性結果測定は以下の通りである:(1) PFS、試験治療開始から任意の原因による試験中(試験治療の最終投薬から30日以内)の疾患の進行又は死亡の最初の発生までの時間として定義され、これは調査者によって変更RECIST, v1.0を使用して腫瘍評価の概要が決定される;そして、(2) 応答の持続時間、任意の原因による試験中(試験治療の最終投薬から30日以内)の、調査者によって変更RECIST(v1.0)を使用して腫瘍評価の概要が決定される疾患進行の時間まで、又は死亡までの、文書化された客観的応答の最初の発生として定義される。
【0130】
試験治療
T−DM1は、2.4、3.0又は3.6mg/kgのIV用量で、3週間ごとを超えない頻度で投与される。いずれの患者も、2.4mg/kg程度の低いT−DM1用量に段階的に漸減してよい。3.0mg/kgで治療を始める患者のコホートにおいて生じる毒性に応じて、そして、3.0mg/kgのT−DM1が許容できることが確認される場合、患者は、その後のサイクルにおいて3週間ごとに3.6mg/kgのIVの用量に漸増されてよい。パーツズマブは、1サイクルの1日目に840mgIVの負荷用量で、その後のサイクルでは3週間ごとに420mgのIVで投与される。
【0131】
統計的方法
この試験の一次有効性エンドポイントは調査者評価の客観的な応答であり、4週間隔以上の2つの連続的な事象において決定される完全もしくは部分的な応答として定められる。推定する客観的応答速度とともに対応する95%信頼区間も算出する。客観的な応答のために、妥当に基準値を超えない腫瘍評価を有する患者を不応答者として計数する。応答の持続時間およびPFSのために、経過観察を行わない患者のデータを、患者が進行していないことがわかっていた最後の日に打ち切って処理する。治療後腫瘍評価又は死のない患者のデータは、治療開始+1日の日に打ち切る。
【0132】
実施例5 GDC−0941と併用するトラスツズマブ−MCC−DM1(T-DM1)の臨床試験
従来のトラスツズマブベースの治療により進行したHER2陽性転移性の乳癌を有する患者への静脈内投与したT−DM1と経口投与したGDC−0941の併用の第Ib相、非盲検試験を設定し、併用の安全性、耐容性、薬物動態及び活性を特徴付けた。この試験の一次目的は以下の通りである:T−DM1を投与したときのGDC−0941の安全性および耐容性を評価すること;T−DM1を投与したときのGDC−0941のMTDを推定すること;T−DM1と併用して投与されるGDC−0941の推奨される第II相用量を同定すること;そして、T−DM1と併用して投与されるときのGDC−0941の観察される抗腫瘍活性を特徴付けること。薬物動態学的目的は以下の通りである:T−DM1の有無の下でのGDC−0941の薬物動態学の特徴を表すこと;そして、GDC−0941の相対的有無の下でのT−DM1の薬物動態学の特徴を表すこと。
【0133】
GDC-0941製剤
GDC-0941はPO投与を目的とする乾燥粉末である。調製された薬剤品は、サイズ0の殻に封入され、色によって区別される2つの強度(15および50mg)の硬質ゼラチンカプセルで提供される。カプセル製剤に含まれる賦形剤は、微結晶性セルロースNF/EP、ラウリル硫酸ナトリウムNF/DP(50mgの強度のみ)、クエン酸無水のUSP/EP、クロスカルメロースナトリウムNF/EP、コロイド性二酸化ケイ素NF/EPおよびステアリン酸マグネシウム(ウシのものでない)NF/EPである。GDC-0941カプセルは、36 Fから46 F(2℃から8℃)の間の冷蔵温度で保存される。患者は、36 Fから46 F(2℃から8℃)の間の冷蔵温度で試験薬剤を保存するように指示される。
【0134】
結果測定
安全性、薬物動態、薬力学および有効性の結果測定は、実施例4にあるように統計的方法を含む方法によって決定され、評価される。
【0135】
試験治療
試験治療は3週サイクルで投与される。試験治療から臨床的利益を得る患者は、開発状況、薬剤有効性および他の因子に応じて、別の試験で生じうる複数のサイクルの治療の可能性を有する。
試験の用量漸増期では、参加している患者は、空腹時にサイクル1の第1日目にGDC−0941の一回量を摂取し、GDC−0941 PK投薬前及び投薬後の試料を採取し、そして、患者内の多様性を観察する。GDC−0941の開始用量は60mgのqdであり、これは第I相試験において用量制限毒性がなく、単一の薬剤として安全であることが決定されている用量である。サイクル1の1日目に、完全用量のT−DM1は、負荷用量なしで3.6mg/kgの90分にわたるIVにより投与される。この後にGDC−0941の投薬を行う。患者は、初めのT−DM1注入後の90分間モニターされる。次いで、GDC-0941は1日1回、合計14用量の後、第1サイクルのために1週間空ける。
その後の患者におけるGDC−0941の用量漸増は、進行又は不耐容が生じるまで続ける。続く試験治療サイクルは3週間の長さであり、各サイクルの1日目の最初に3.6mg/kgのT−DM1の30分のIV投与と、T−DM1注入の後に投与されるGDC−0941によるものであり、合計2週間続け、1週間休む。投薬は、進行又は不耐容が生じるまで続ける。T−DM1は30〜90分(±10)のIV注入として投与され、これは親の試験においてどれほどのT−DM1が許容されたかに依存する。90分の注入が十分に許容される場合、続く注入は30(±10)分にわたって供給されてよい。
【0136】
先の記載は本発明の原理を単に例示するものとして考慮される。更に、当業者には多くの修正及び変更が直ぐに明らかであるので、発明を前記した正確な構成及び方法に限定することは望まれない。したがって、あらゆる適切な修正及び均等物が次の特許請求の範囲によって定まる発明の範囲に入ると考えられうる。
【特許請求の範囲】
【請求項1】
併用製剤としてかあるいは交代に治療的併用を哺乳動物に投与することを含む、過剰増殖性疾患の治療方法であって、このとき該併用製剤がトラスツズマブ−MCC−DM1の治療上有効量と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される化学療法剤の治療上有効量とを含むものである方法。
【請求項2】
HER2二量体化阻害性抗体がパーツズマブである、請求項1に記載の方法。
【請求項3】
抗VEGF抗体がベバシズマブである、請求項1に記載の方法。
【請求項4】
化学療法剤が5−FUである、請求項1に記載の方法。
【請求項5】
化学療法剤がカルボプラチンである、請求項1に記載の方法。
【請求項6】
化学療法剤がラパチニブである、請求項1に記載の方法。
【請求項7】
化学療法剤がABT−869である、請求項1に記載の方法。
【請求項8】
化学療法剤がドセタキセルである、請求項1に記載の方法。
【請求項9】
化学療法剤がGDC−0941である、請求項1に記載の方法。
【請求項10】
化学療法剤がGNE−390である、請求項1に記載の方法。
【請求項11】
トラスツズマブ−MCC−DM1の治療上有効量と化学療法剤の治療上有効量が併用製剤として投与される、請求項1に記載の方法。
【請求項12】
トラスツズマブ−MCC−DM1の治療上有効量と化学療法剤の治療上有効量が交代に投与される、請求項1に記載の方法。
【請求項13】
哺乳動物が、化学療法剤を投与され、続いて、トラスツズマブ−MCC−DM1を投与される、請求項12に記載の方法。
【請求項14】
治療的併用が、過剰増殖性疾患を有するヒトにおよそ3週間隔で投与される、請求項12に記載の方法。
【請求項15】
トラスツズマブ−MCC−DM1が、過剰増殖性疾患を有するヒトにおよそ1週から3週の間隔で投与される、請求項12に記載の方法。
【請求項16】
治療的併用の投与により相乗効果が生じる、請求項1に記載の方法。
【請求項17】
過剰増殖性疾患が癌である、請求項1に記載の方法。
【請求項18】
過剰増殖性疾患がErbB2を発現している癌である、請求項17に記載の方法。
【請求項19】
癌が、胸部、卵巣、子宮頸管、前立腺、精巣、泌尿生殖器、食道、喉頭、神経膠芽腫、神経芽細胞腫、胃、皮膚、角化棘細胞腫、肺、類表皮癌、大細胞カルチノーマ、非小細胞肺カルチノーマ(NSCLC)、小細胞カルチノーマ、肺腺癌、骨、大腸、腺腫、膵臓、腺癌、甲状腺、濾胞性カルチノーマ、未分化癌、乳頭カルチノーマ、精上皮腫、メラノーマ、肉腫、膀胱カルチノーマ、肝臓カルチノーマ、及び胆汁路、腎臓カルチノーマ、膵臓、骨髄疾患、リンパ腫、線毛細胞、頬側口腔、鼻咽頭、咽頭、唇、舌、口、小腸、大腸-直腸、大腸、直腸、脳及び中枢神経系、ホジキン、又は、白血病である、請求項17に記載の方法。
【請求項20】
トラスツズマブ−MCC−DM1の量および化学療法剤の量がそれぞれおよそ1mgからおよそ1000mgである、請求項1に記載の方法。
【請求項21】
トラスツズマブ−MCC−DM1の量および化学療法剤の量がおよそ1:10重量からおよそ10:1重量の比率である、請求項1に記載の方法。
【請求項22】
哺乳動物がHER2陽性患者である、請求項1に記載の方法。
【請求項23】
HER2陽性患者がトラスツズマブ又はラパチニブ療法を受けた、請求項1に記載の方法。
【請求項24】
トラスツズマブ−MCC−DM1と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される化学療法剤と、一又は複数の薬学的に許容可能な担体、流動促進剤、希釈液又は賦形剤とを含有してなる薬学的組成物。
【請求項25】
二酸化ケイ素、粉末セルロース、微結晶性セルロース、金属ステアリン酸塩類、アルミノケイ酸ナトリウム、安息香酸ナトリウム、炭酸カルシウム、ケイ酸カルシウム、トウモロコシ澱粉、炭酸マグネシウム、アスベストを含まないタルク、ステアロワットC、澱粉、澱粉1500、マグネシウムラウリル硫酸塩、酸化マグネシウムおよびこれらの組合せから選択される薬学的に許容可能な流動促進剤を含んでなる、請求項24に記載の薬学的組成物。
【請求項26】
トラスツズマブ−MCC−DM1の量および化学療法剤の量がそれぞれおよそ1mgからおよそ1000mgで存在する、請求項24に記載の薬学的組成物。
【請求項27】
トラスツズマブ−MCC−DM1の量および化学療法剤の量がおよそ1:10重量からおよそ10:1重量の比率で存在する、請求項24に記載の薬学的組成物。
【請求項28】
胸部、卵巣、子宮頸管、前立腺、精巣、泌尿生殖器、食道、喉頭、神経膠芽腫、神経芽細胞腫、胃、皮膚、角化棘細胞腫、肺、類表皮癌、大細胞カルチノーマ、非小細胞肺カルチノーマ(NSCLC)、小細胞カルチノーマ、肺腺癌、骨、大腸、腺腫、膵臓、腺癌、甲状腺、濾胞性カルチノーマ、未分化癌、乳頭カルチノーマ、精上皮腫、メラノーマ、肉腫、膀胱カルチノーマ、肝臓カルチノーマ、及び胆汁路、腎臓カルチノーマ、膵臓、骨髄疾患、リンパ腫、線毛細胞、頬側口腔、鼻咽頭、咽頭、唇、舌、口、小腸、大腸-直腸、大腸、直腸、脳及び中枢神経系、ホジキン、又は、白血病から選択される癌の治療のための医薬の製造における治療的併用の使用であって、
このとき、治療的併用は、併用製剤として、または、交代に哺乳動物に投与され、トラスツズマブ−MCC−DM1の治療上有効量と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される化学療法剤の治療上有効量とを含んでなるものである、治療的併用の使用。
【請求項29】
過剰増殖性疾患を治療するための製造品であって、
a) 併用製剤として、又は、交代に哺乳動物に投与され、トラスツズマブ−MCC−DM1の治療上有効量と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される化学療法剤の治療上有効量とを含有してなる治療的併用と、b) 使用のための指示書とを具備する製造品。
【請求項30】
癌の治療のための併用に使用される化合物の決定方法であって、
a) トラスツズマブ−MCC−DM1と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される化学療法剤との治療的併用をHER2増幅乳癌細胞に投与し、
b) 細胞増殖の阻害を測定して、非悪性と悪性の乳房細胞が細胞生存度によって識別される
ことを含む方法。
【請求項31】
HER2増幅乳癌細胞がBT-474である、請求項30に記載の方法。
【請求項32】
癌の治療のために使用される治療的併用の決定方法であって、
a) インビトロ腫瘍細胞株を、トラスツズマブ−MCC−DM1と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される化学療法剤との治療的併用にて処置し、
b) 相乗効果又は非相乗効果を測定することを含み、
これによって、癌の治療のための相乗的治療的併用が決定される方法。
【請求項1】
併用製剤としてかあるいは交代に治療的併用を哺乳動物に投与することを含む、過剰増殖性疾患の治療方法であって、このとき該併用製剤がトラスツズマブ−MCC−DM1の治療上有効量と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される化学療法剤の治療上有効量とを含むものである方法。
【請求項2】
HER2二量体化阻害性抗体がパーツズマブである、請求項1に記載の方法。
【請求項3】
抗VEGF抗体がベバシズマブである、請求項1に記載の方法。
【請求項4】
化学療法剤が5−FUである、請求項1に記載の方法。
【請求項5】
化学療法剤がカルボプラチンである、請求項1に記載の方法。
【請求項6】
化学療法剤がラパチニブである、請求項1に記載の方法。
【請求項7】
化学療法剤がABT−869である、請求項1に記載の方法。
【請求項8】
化学療法剤がドセタキセルである、請求項1に記載の方法。
【請求項9】
化学療法剤がGDC−0941である、請求項1に記載の方法。
【請求項10】
化学療法剤がGNE−390である、請求項1に記載の方法。
【請求項11】
トラスツズマブ−MCC−DM1の治療上有効量と化学療法剤の治療上有効量が併用製剤として投与される、請求項1に記載の方法。
【請求項12】
トラスツズマブ−MCC−DM1の治療上有効量と化学療法剤の治療上有効量が交代に投与される、請求項1に記載の方法。
【請求項13】
哺乳動物が、化学療法剤を投与され、続いて、トラスツズマブ−MCC−DM1を投与される、請求項12に記載の方法。
【請求項14】
治療的併用が、過剰増殖性疾患を有するヒトにおよそ3週間隔で投与される、請求項12に記載の方法。
【請求項15】
トラスツズマブ−MCC−DM1が、過剰増殖性疾患を有するヒトにおよそ1週から3週の間隔で投与される、請求項12に記載の方法。
【請求項16】
治療的併用の投与により相乗効果が生じる、請求項1に記載の方法。
【請求項17】
過剰増殖性疾患が癌である、請求項1に記載の方法。
【請求項18】
過剰増殖性疾患がErbB2を発現している癌である、請求項17に記載の方法。
【請求項19】
癌が、胸部、卵巣、子宮頸管、前立腺、精巣、泌尿生殖器、食道、喉頭、神経膠芽腫、神経芽細胞腫、胃、皮膚、角化棘細胞腫、肺、類表皮癌、大細胞カルチノーマ、非小細胞肺カルチノーマ(NSCLC)、小細胞カルチノーマ、肺腺癌、骨、大腸、腺腫、膵臓、腺癌、甲状腺、濾胞性カルチノーマ、未分化癌、乳頭カルチノーマ、精上皮腫、メラノーマ、肉腫、膀胱カルチノーマ、肝臓カルチノーマ、及び胆汁路、腎臓カルチノーマ、膵臓、骨髄疾患、リンパ腫、線毛細胞、頬側口腔、鼻咽頭、咽頭、唇、舌、口、小腸、大腸-直腸、大腸、直腸、脳及び中枢神経系、ホジキン、又は、白血病である、請求項17に記載の方法。
【請求項20】
トラスツズマブ−MCC−DM1の量および化学療法剤の量がそれぞれおよそ1mgからおよそ1000mgである、請求項1に記載の方法。
【請求項21】
トラスツズマブ−MCC−DM1の量および化学療法剤の量がおよそ1:10重量からおよそ10:1重量の比率である、請求項1に記載の方法。
【請求項22】
哺乳動物がHER2陽性患者である、請求項1に記載の方法。
【請求項23】
HER2陽性患者がトラスツズマブ又はラパチニブ療法を受けた、請求項1に記載の方法。
【請求項24】
トラスツズマブ−MCC−DM1と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される化学療法剤と、一又は複数の薬学的に許容可能な担体、流動促進剤、希釈液又は賦形剤とを含有してなる薬学的組成物。
【請求項25】
二酸化ケイ素、粉末セルロース、微結晶性セルロース、金属ステアリン酸塩類、アルミノケイ酸ナトリウム、安息香酸ナトリウム、炭酸カルシウム、ケイ酸カルシウム、トウモロコシ澱粉、炭酸マグネシウム、アスベストを含まないタルク、ステアロワットC、澱粉、澱粉1500、マグネシウムラウリル硫酸塩、酸化マグネシウムおよびこれらの組合せから選択される薬学的に許容可能な流動促進剤を含んでなる、請求項24に記載の薬学的組成物。
【請求項26】
トラスツズマブ−MCC−DM1の量および化学療法剤の量がそれぞれおよそ1mgからおよそ1000mgで存在する、請求項24に記載の薬学的組成物。
【請求項27】
トラスツズマブ−MCC−DM1の量および化学療法剤の量がおよそ1:10重量からおよそ10:1重量の比率で存在する、請求項24に記載の薬学的組成物。
【請求項28】
胸部、卵巣、子宮頸管、前立腺、精巣、泌尿生殖器、食道、喉頭、神経膠芽腫、神経芽細胞腫、胃、皮膚、角化棘細胞腫、肺、類表皮癌、大細胞カルチノーマ、非小細胞肺カルチノーマ(NSCLC)、小細胞カルチノーマ、肺腺癌、骨、大腸、腺腫、膵臓、腺癌、甲状腺、濾胞性カルチノーマ、未分化癌、乳頭カルチノーマ、精上皮腫、メラノーマ、肉腫、膀胱カルチノーマ、肝臓カルチノーマ、及び胆汁路、腎臓カルチノーマ、膵臓、骨髄疾患、リンパ腫、線毛細胞、頬側口腔、鼻咽頭、咽頭、唇、舌、口、小腸、大腸-直腸、大腸、直腸、脳及び中枢神経系、ホジキン、又は、白血病から選択される癌の治療のための医薬の製造における治療的併用の使用であって、
このとき、治療的併用は、併用製剤として、または、交代に哺乳動物に投与され、トラスツズマブ−MCC−DM1の治療上有効量と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される化学療法剤の治療上有効量とを含んでなるものである、治療的併用の使用。
【請求項29】
過剰増殖性疾患を治療するための製造品であって、
a) 併用製剤として、又は、交代に哺乳動物に投与され、トラスツズマブ−MCC−DM1の治療上有効量と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される化学療法剤の治療上有効量とを含有してなる治療的併用と、b) 使用のための指示書とを具備する製造品。
【請求項30】
癌の治療のための併用に使用される化合物の決定方法であって、
a) トラスツズマブ−MCC−DM1と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される化学療法剤との治療的併用をHER2増幅乳癌細胞に投与し、
b) 細胞増殖の阻害を測定して、非悪性と悪性の乳房細胞が細胞生存度によって識別される
ことを含む方法。
【請求項31】
HER2増幅乳癌細胞がBT-474である、請求項30に記載の方法。
【請求項32】
癌の治療のために使用される治療的併用の決定方法であって、
a) インビトロ腫瘍細胞株を、トラスツズマブ−MCC−DM1と、HER2二量体化阻害性抗体、抗VEGF抗体、5−FU、カルボプラチン、ラパチニブ、ABT−869、ドセタキセル、GDC−0941およびGNE−390から選択される化学療法剤との治療的併用にて処置し、
b) 相乗効果又は非相乗効果を測定することを含み、
これによって、癌の治療のための相乗的治療的併用が決定される方法。
【図1】

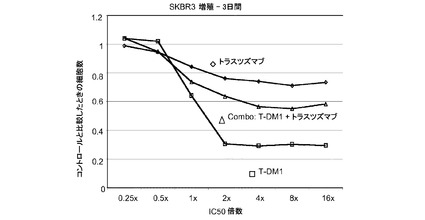
【図2】
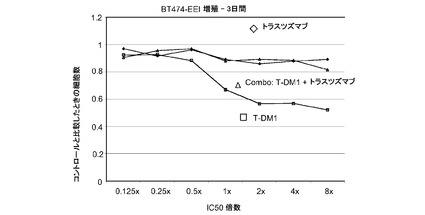
【図3】
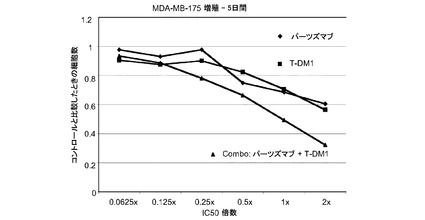
【図3a】
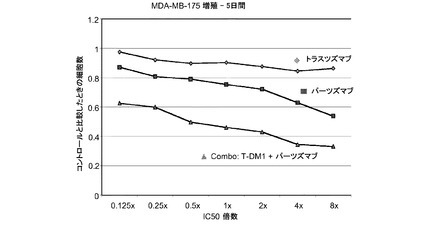
【図4】
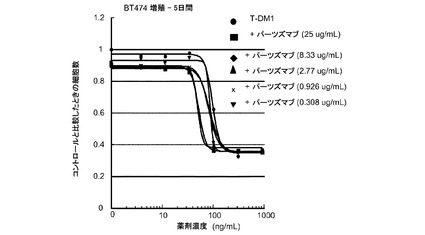
【図5】
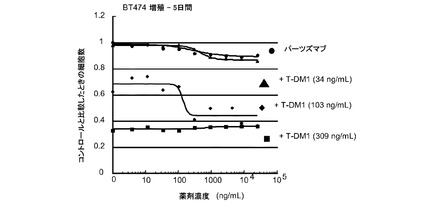
【図6】
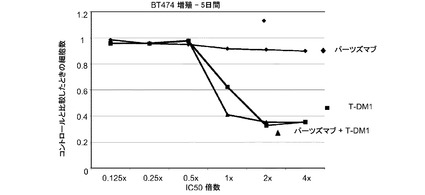
【図7】
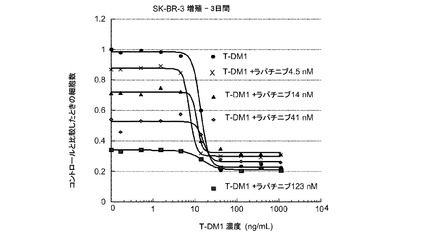
【図7a】
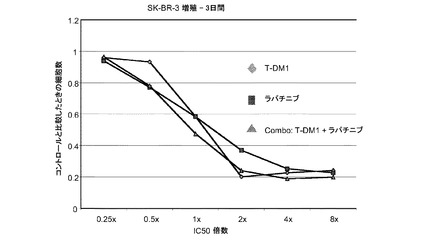
【図8】
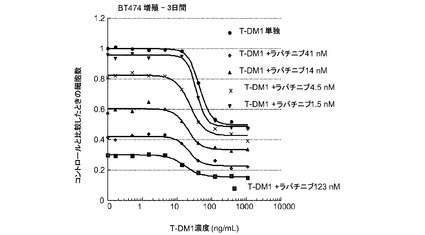
【図8a】
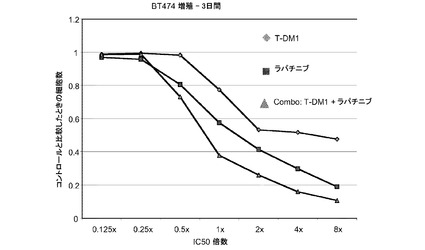
【図9】
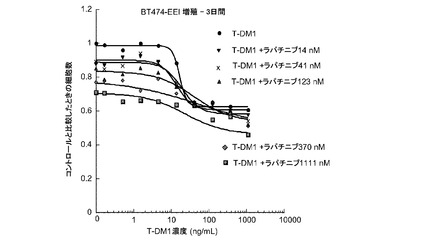
【図10】
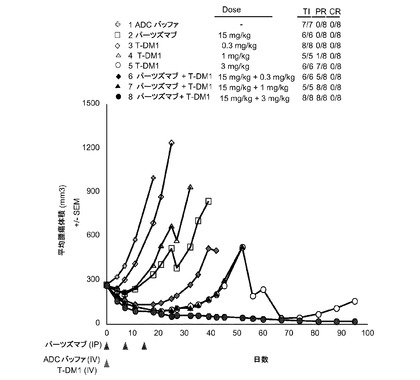
【図11】
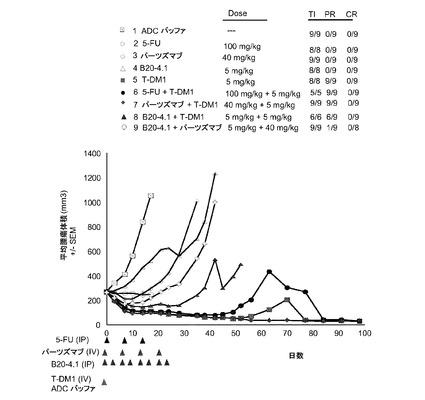
【図12】
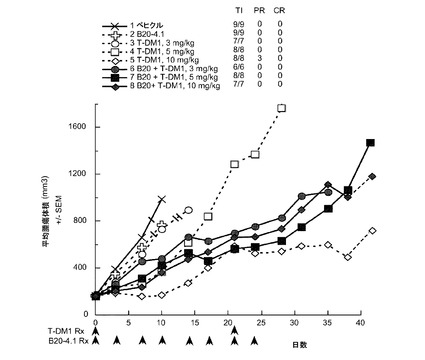
【図13】
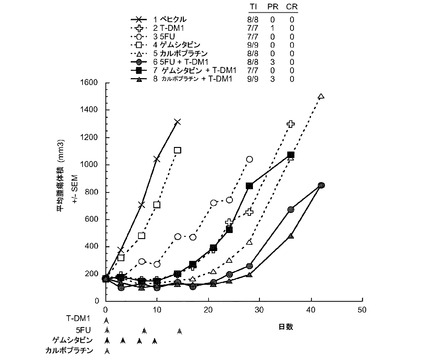
【図14】
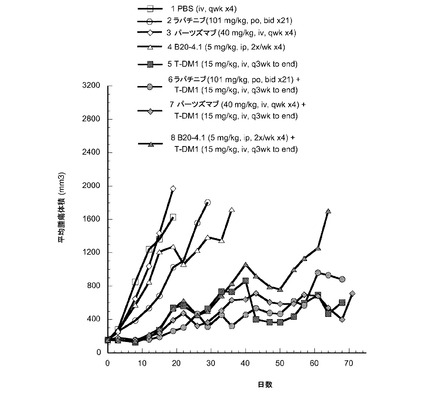
【図15】
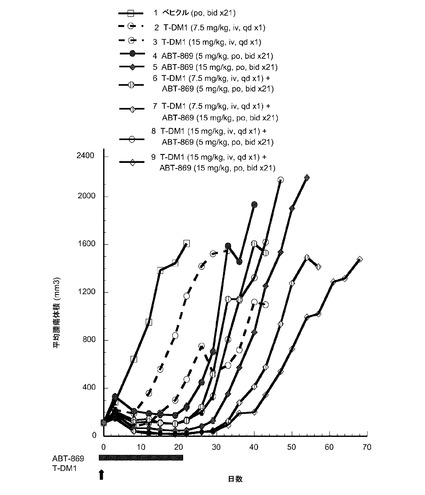
【図16】
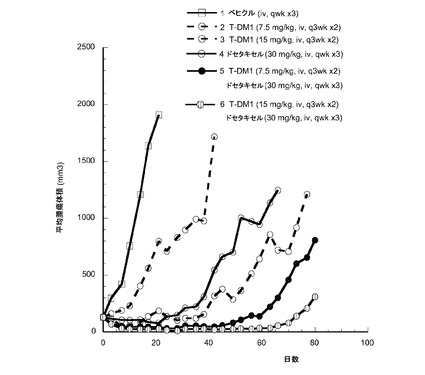
【図17】
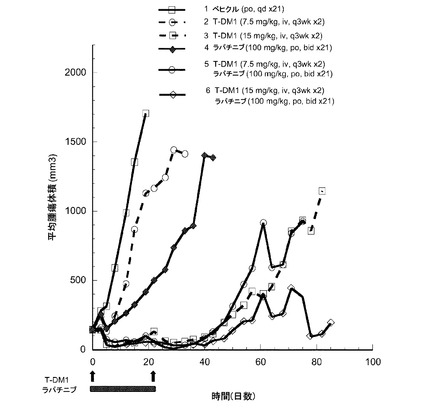
【図18】
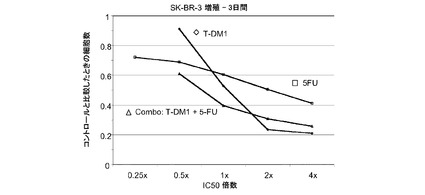
【図19】
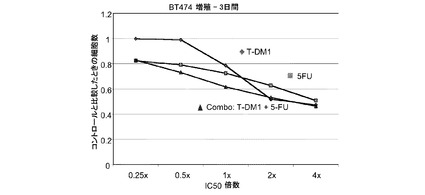
【図20】
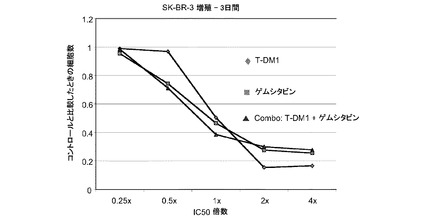
【図21】
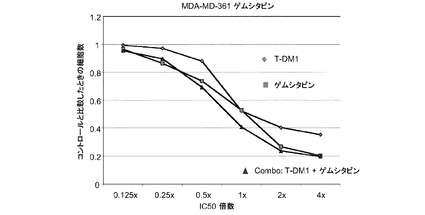
【図22】
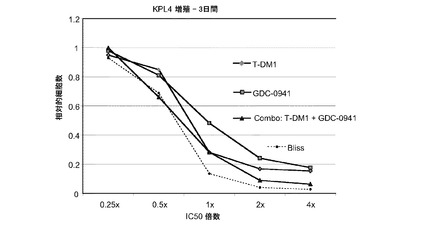
【図23】
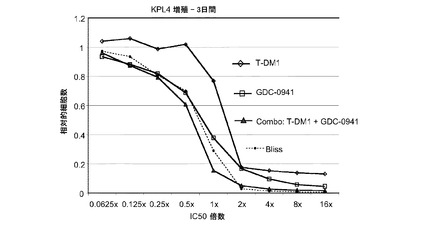
【図24】
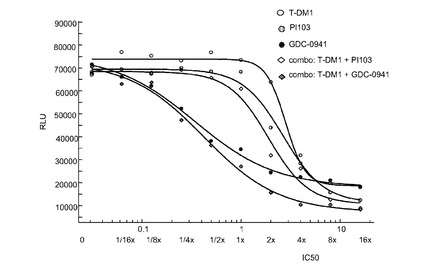
【図25】
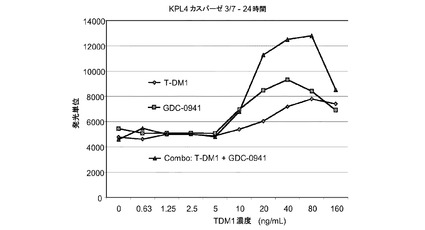
【図26】
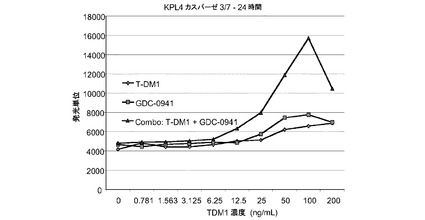
【図27】
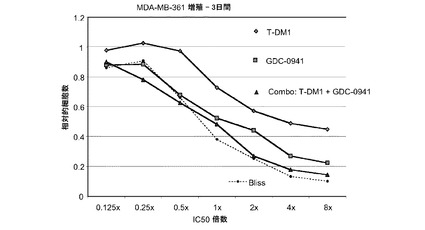
【図28】
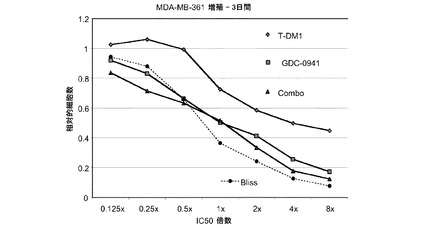
【図29】
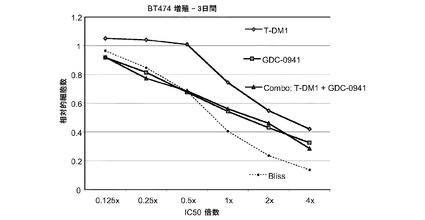
【図30】
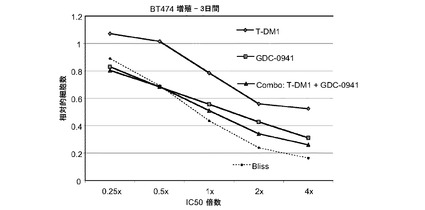
【図31】
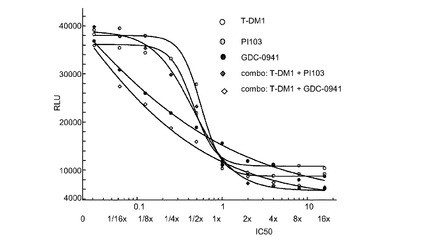
【図32】
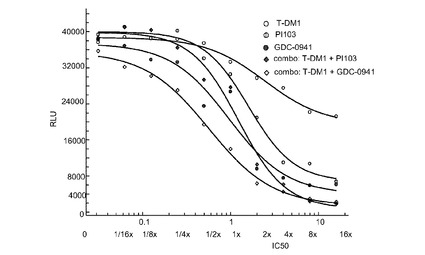
【図33】
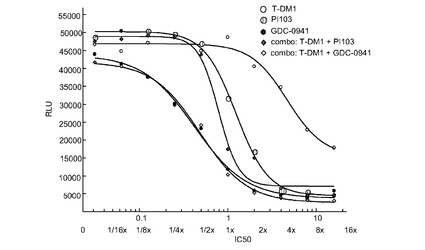
【図34】
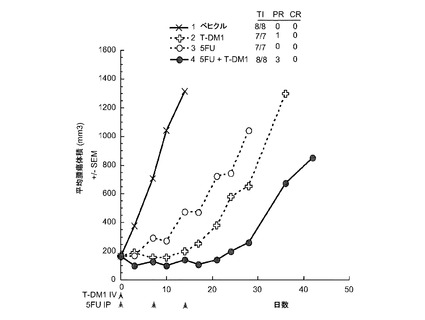
【図35】
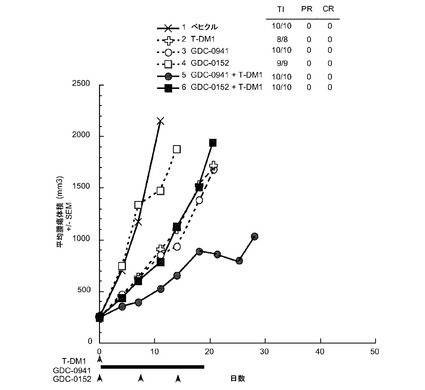
【図36】
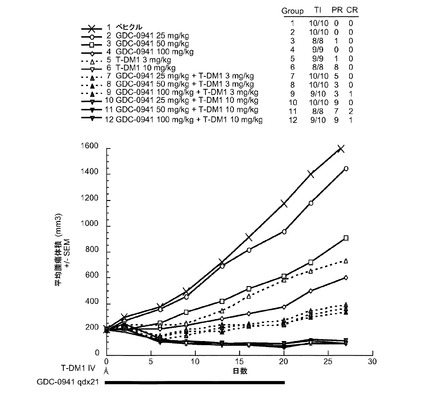
【図37】
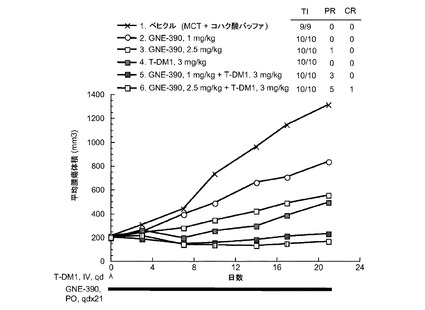
【図2】
【図3】
【図3a】
【図4】
【図5】
【図6】
【図7】
【図7a】
【図8】
【図8a】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【図31】
【図32】
【図33】
【図34】
【図35】
【図36】
【図37】
【公表番号】特表2012−500180(P2012−500180A)
【公表日】平成24年1月5日(2012.1.5)
【国際特許分類】
【出願番号】特願2011−500865(P2011−500865)
【出願日】平成21年3月10日(2009.3.10)
【国際出願番号】PCT/US2009/036608
【国際公開番号】WO2009/117277
【国際公開日】平成21年9月24日(2009.9.24)
【出願人】(509012625)ジェネンテック, インコーポレイテッド (357)
【Fターム(参考)】
【公表日】平成24年1月5日(2012.1.5)
【国際特許分類】
【出願日】平成21年3月10日(2009.3.10)
【国際出願番号】PCT/US2009/036608
【国際公開番号】WO2009/117277
【国際公開日】平成21年9月24日(2009.9.24)
【出願人】(509012625)ジェネンテック, インコーポレイテッド (357)
【Fターム(参考)】
[ Back to top ]