
抗HIV薬

【課題】プロウイルスDNAからRNAへの転写過程を阻害する新たな抗HIV薬の提供。
【解決手段】式I

[式中、R1はHであり、且つR2は炭素数1〜3個のアルコキシ基であり、又はR1及びR2が一緒になって、−O−(CH2)2−O−の環を形成し、R3はH又は炭素数1〜3個のアルキル基である]で表される化合物を含有する抗HIV薬。
【解決手段】式I
[式中、R1はHであり、且つR2は炭素数1〜3個のアルコキシ基であり、又はR1及びR2が一緒になって、−O−(CH2)2−O−の環を形成し、R3はH又は炭素数1〜3個のアルキル基である]で表される化合物を含有する抗HIV薬。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、抗ヒト免疫不全ウイルス(HIV)薬に関する。
【背景技術】
【0002】
AIDS患者の予後は、逆転写酵素阻害薬やプロテアーゼ阻害薬の開発により大幅に改善したが、慢性毒性や薬剤耐性ウイルスの出現等の問題点が生じており、新たな作用機序を有する抗HIV薬の開発が必要とされている。例えば(特許文献1)には、ペルオキシソーム増殖因子活性化レセプター(PPAR)類に対して活性な下記の化合物が開示されており、対象となる疾患としてHIV感染症が挙げられている。
【化1】
【0003】
また、(特許文献2)には、HIV感染症のようなレトロウイルス感染症の治療用医薬の製造に使用できる化合物として、下記の(3S)テトラヒドロ−3−フラニル(1S,2R)−3−[[(4−アミノフェニル)スルホニル](イソブチル)アミノ]−1−ベンジル−2−(ホスホノオキシ)プロピルカルバミン酸カルシウムが開示されている。
【化2】
【0004】
一方、プロウイルスDNAからRNAへの転写過程は、HIVの複製に必須であるが、この段階を阻害する薬剤はまだ開発されていない。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特表2009−507079号公報
【特許文献2】特表2003−521447号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
そこで本発明は、プロウイルスDNAからRNAへの転写過程を阻害する新たな抗HIV薬を提供することを目的とする。
【課題を解決するための手段】
【0007】
HIV−1の転写過程は、宿主細胞因子のcyclin T1とCDK9、そしてHIV−1由来のTat及びTAR RNAにより形成される複合体によって開始される。そこで本発明者らは、これらの相互作用領域を標的とする低分子化合物を、3,000,000化合物のデータベースの中から、コンピューターを用いたin silicoスクリーニング(docking simulation)により選択した上で、in vitroにおける抗HIV−1アッセイにより同定することに成功し、本発明を完成させるに至った。すなわち、本発明の要旨は以下の通りである。
【0008】
(1)式I
【化3】
[式中、R1はHであり、且つR2は炭素数1〜3個のアルコキシ基であり、又はR1及びR2が一緒になって、−O−(CH2)2−O−の環を形成し、R3はH又は炭素数1〜3個のアルキル基である]
で表される化合物を含有する抗HIV薬。
【0009】
(2)式Iで表される化合物が、式II
【化4】
で表される化合物である前記(1)に記載の抗HIV薬。
【0010】
(3)式Iで表される化合物が、式III
【化5】
で表される化合物である前記(1)に記載の抗HIV薬。
【0011】
(4)式Iで表される化合物が、式IV
【化6】
で表される化合物である前記(1)に記載の抗HIV薬。
【発明の効果】
【0012】
本発明における化合物は、高い抗HIV活性を有し、抗HIV薬として好適に用いることができる。
【図面の簡単な説明】
【0013】
【図1】本発明の化合物についての抗HIV−1活性及び細胞毒性の試験結果を示すグラフである。
【図2】本発明の化合物についての抗HIV−1活性及び細胞毒性の試験結果を示すグラフである。
【図3】本発明の化合物についての抗HIV−1活性及び細胞毒性の試験結果を示すグラフである。
【発明を実施するための形態】
【0014】
以下、本発明を詳細に説明する。
本発明の抗HIV薬は、式Iで表される化合物を含有する。
【化7】
【0015】
ここで、式I中、R1はHであり、且つR2は炭素数1〜3個のアルコキシ基であり、又はR1及びR2が一緒になって、−O−(CH2)2−O−の環を形成し、R3はH又は炭素数1〜3個のアルキル基である。
【0016】
炭素数1〜3個のアルコキシ基は、直鎖状又は分岐状のいずれであってもよく、例えば、メトキシ基、エトキシ基、プロピルオキシ基、i−プロピルオキシ基等を挙げることができる。
【0017】
また、炭素数1〜3個のアルキル基としては、直鎖状又は分岐状のいずれであってもよく、例えば、メチル基、エチル基、n−プロピル基、イソプロピル基等を挙げることができる。
【0018】
上記式Iで表される化合物の中でも、抗HIV活性及び細胞毒性の観点から、特に、式IIで表されるN−[(6−メトキシ−2−オキソ−1H−キノリン−3−イル)メチル]−N−(ピリジン−3−イルメチル)−2,1,3−ベンゾチアジアゾール−4−スルホンアミドが好ましい。
【化8】
【0019】
また、別の好ましい化合物として、式IIIで表されるN−[(7−オキソ−3,6−ジヒドロ−2H−[1,4]ジオキシノ[2,3−g]キノリン−8−イル)メチル]−N−(ピリジン−3−イルメチル)−2,1,3−ベンゾチアジアゾール−4−スルホンアミドが挙げられる。
【化9】
【0020】
さらに、別の好ましい化合物として、式IVで表される5−メチル−N−[(7−オキソ−3,6−ジヒドロ−2H−[1,4]ジオキシノ[2,3−g]キノリン−8−イル)メチル]−N−(ピリジン−3−イルメチル)−2,1,3−ベンゾチアジアゾール−4−スルホンアミドが挙げられる。
【化10】
【0021】
上記の各化合物は、慣用の有機合成法により得ることができる。また、Asinex社(Moscow, Russia, http://www.asinex.com/library-gold.html)から入手可能である。
【0022】
上記の化合物は、抗HIV薬として、慣用の製剤担体と組み合わせて製剤化することができる。投与形態としては、特に限定はなく、必要に応じ適宜選択して使用され、錠剤、カプセル剤、顆粒剤、細粒剤、散剤、徐放性製剤、液剤、懸濁剤、エマルジョン剤、シロップ剤、エリキシル剤等の経口剤、注射剤、坐剤等の非経口剤が挙げられる。
【0023】
経口剤は、例えばデンプン、乳糖、白糖、マンニット、カルボキシメチルセルロース、コーンスターチ、無機塩類等を用いて常法に従って製造される。また、これらに加えて、結合剤、崩壊剤、界面活性剤、滑沢剤、流動性促進剤、矯味剤、着色剤、香料等を適宜添加することができる。
【0024】
結合剤としては、例えばデンプン、デキストリン、アラビアゴム、ゼラチン、ヒドロキシプロピルスターチ、メチルセルロース、カルボキシメチルセルロースナトリウム、ヒドロキシプロピルセルロース、結晶セルロース、エチルセルロース、ポリビニルピロリドン、マクロゴール等が挙げられる。
【0025】
崩壊剤としては、例えばデンプン、ヒドロキシプロピルスターチ、カルボキシメチルセルロースナトリウム、カルボキシメチルセルロースカルシウム、カルボキシメチルセルロース、低置換ヒドロキシプロピルセルロース等が挙げられる。
【0026】
界面活性剤としては、例えばラウリル硫酸ナトリウム、大豆レシチン、ショ糖脂肪酸エステル、ポリソルベート80等が挙げられる。
【0027】
滑沢剤としては、例えばタルク、ロウ類、水素添加植物油、ショ糖脂肪酸エステル、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸アルミニウム、ポリエチレングリコール等が挙げられる。
【0028】
流動性促進剤としては、例えば軽質無水ケイ酸、乾燥水酸化アルミニウムゲル、合成ケイ酸アルミニウム、ケイ酸マグネシウム等が挙げられる。
【0029】
注射剤は、常法に従って製造され、希釈剤として一般に注射用蒸留水、生理食塩水、ブドウ糖水溶液、オリーブ油、ゴマ油、ラッカセイ油、ダイズ油、トウモロコシ油、プロピレングリコール、ポリエチレングリコール等を用いることができる。さらに必要に応じて、殺菌剤、防腐剤、安定剤、等張化剤、無痛化剤等を加えてもよい。また、注射剤は、安定性の観点から、バイアル等に充填後冷凍し、通常の凍結乾燥技術により水分を除去し、使用直前に凍結乾燥物から液剤を再調製することもできる。式I〜式IVの化合物の注射剤中における割合は、5〜50重量%の間で変動させ得るが、これに限定されるものではない。
【0030】
その他の非経口剤としては、直腸内投与のための坐剤等が挙げられ、常法に従って製造される。
【0031】
製剤化した抗HIV薬は、剤形、投与経路等により異なるが、例えば、1日1〜4回を1週間から3ヶ月の期間、投与することが可能である。
【0032】
経口剤として所期の効果を発揮するためには、患者の年令、体重、疾患の程度により異なるが、通常成人の場合、式I〜式IVの化合物の重量として、例えば0.1〜1000mg、好ましくは1〜500mgを、1日数回に分けて服用することが適当である。
【0033】
非経口剤として所期の効果を発揮するためには、患者の年令、体重、疾患の程度により異なるが、通常成人の場合、式I〜式IVの化合物の重量として、例えば0.1〜1000mg、好ましくは1〜500mgを、静注、点滴静注、皮下注射、筋肉注射により投与することが適当である。
【実施例】
【0034】
以下、実施例に基づき本発明をさらに詳細に説明するが、これに限定されるものではない。
(In silicoスクリーニング)
全てのIn silico実験は、コンピュータソフトMOE(Chemical Computing Group Inc., Quebec, Canada)を用いて行った。In silicoドッキングの標的として用いられたヒトcyclin T1のモデル構造は、ウマcyclin T1とequine infectious anemia virus(EIAV)TatとEIAV TAR RNAの複合体構造(Protein Data Bank ID: 2w2h)を基にホモロジー・モデリングにより構築した(Anand K. et al., Nat. Struct. Mol. Biol. 2008 Dec; 15(12): 1287-92)。3,000,000化合物のデータベース(Namiki shoji, Tokyo, Japan)の中から薬剤としての性質を有する化合物をスクリーニングするために、分子量が350から600 Da、水素結合のドナー/アクセプター数が13個より少なく、回転可能な結合が7個より少なく、logP値が0から6の間をとる化合物を選別した。このようにして選別した化合物のドッキングシミュレーションを、ヒトcyclin T1におけるHIV−1 TatとTAR RNAが結合する部位に対して行った。その結果、最適なスコア値を示した254の化合物を合成し、in vitroにおける抗HIV−1活性を評価した。なお、合成した化合物は、Asinex社(Moscow, Russia, http://www.asinex.com/library-gold.html)から市販されているものを購入して使用した。
【0035】
(抗HIV−1試験)
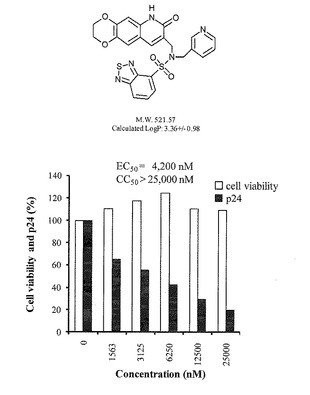
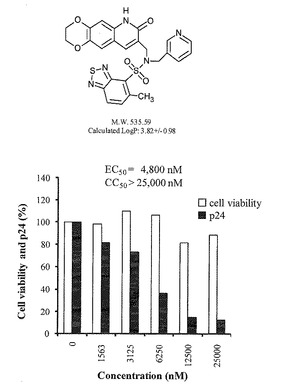
抗HIV−1試験は、HIV−1慢性感染細胞株であるOM10.1細胞を用いた。100μlの段階希釈された薬剤を含む96穴プレートに50μlのOM10.1細胞(4×105cells/ml)を加え、37℃、5%CO2で24時間培養した。その後、50μlのTNF−α(4ng/ml、Roche Diagnostics、Mannheim、Germany)を加え、37℃、5%CO2でさらに48時間培養した。100μlの培養上清を回収し、ELISA(ZeptoMetrix、Buffalo、NY)によりHIV−1 p24の産生量を定量して、産生を抑制する濃度(EC50)を測定した。また、化合物の細胞毒性(CC50)は、MTT法(Sigma-Aldrich、St. Louis、MO)により定量した。化合物A〜Jについての測定結果を下表及び図1〜3に示す。
【0036】
【表1】
【表2】
【表3】
【0037】
表及び図1〜3に示すように、本発明の化合物A〜Cは、それ以外の化合物に比べて、高い抗HIV−1活性を有し、細胞毒性が低いため、抗HIV−1薬として優れた化合物である。また、HIV−2においても、HIV−1と同じメカニズムによりウイルス遺伝子の転写が起こるため、上記実験結果より、本発明の化合物は抗HIV−2薬としても優れた活性を有し得るものである。
【技術分野】
【0001】
本発明は、抗ヒト免疫不全ウイルス(HIV)薬に関する。
【背景技術】
【0002】
AIDS患者の予後は、逆転写酵素阻害薬やプロテアーゼ阻害薬の開発により大幅に改善したが、慢性毒性や薬剤耐性ウイルスの出現等の問題点が生じており、新たな作用機序を有する抗HIV薬の開発が必要とされている。例えば(特許文献1)には、ペルオキシソーム増殖因子活性化レセプター(PPAR)類に対して活性な下記の化合物が開示されており、対象となる疾患としてHIV感染症が挙げられている。
【化1】
【0003】
また、(特許文献2)には、HIV感染症のようなレトロウイルス感染症の治療用医薬の製造に使用できる化合物として、下記の(3S)テトラヒドロ−3−フラニル(1S,2R)−3−[[(4−アミノフェニル)スルホニル](イソブチル)アミノ]−1−ベンジル−2−(ホスホノオキシ)プロピルカルバミン酸カルシウムが開示されている。
【化2】
【0004】
一方、プロウイルスDNAからRNAへの転写過程は、HIVの複製に必須であるが、この段階を阻害する薬剤はまだ開発されていない。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特表2009−507079号公報
【特許文献2】特表2003−521447号公報
【発明の概要】
【発明が解決しようとする課題】
【0006】
そこで本発明は、プロウイルスDNAからRNAへの転写過程を阻害する新たな抗HIV薬を提供することを目的とする。
【課題を解決するための手段】
【0007】
HIV−1の転写過程は、宿主細胞因子のcyclin T1とCDK9、そしてHIV−1由来のTat及びTAR RNAにより形成される複合体によって開始される。そこで本発明者らは、これらの相互作用領域を標的とする低分子化合物を、3,000,000化合物のデータベースの中から、コンピューターを用いたin silicoスクリーニング(docking simulation)により選択した上で、in vitroにおける抗HIV−1アッセイにより同定することに成功し、本発明を完成させるに至った。すなわち、本発明の要旨は以下の通りである。
【0008】
(1)式I
【化3】
[式中、R1はHであり、且つR2は炭素数1〜3個のアルコキシ基であり、又はR1及びR2が一緒になって、−O−(CH2)2−O−の環を形成し、R3はH又は炭素数1〜3個のアルキル基である]
で表される化合物を含有する抗HIV薬。
【0009】
(2)式Iで表される化合物が、式II
【化4】
で表される化合物である前記(1)に記載の抗HIV薬。
【0010】
(3)式Iで表される化合物が、式III
【化5】
で表される化合物である前記(1)に記載の抗HIV薬。
【0011】
(4)式Iで表される化合物が、式IV
【化6】
で表される化合物である前記(1)に記載の抗HIV薬。
【発明の効果】
【0012】
本発明における化合物は、高い抗HIV活性を有し、抗HIV薬として好適に用いることができる。
【図面の簡単な説明】
【0013】
【図1】本発明の化合物についての抗HIV−1活性及び細胞毒性の試験結果を示すグラフである。
【図2】本発明の化合物についての抗HIV−1活性及び細胞毒性の試験結果を示すグラフである。
【図3】本発明の化合物についての抗HIV−1活性及び細胞毒性の試験結果を示すグラフである。
【発明を実施するための形態】
【0014】
以下、本発明を詳細に説明する。
本発明の抗HIV薬は、式Iで表される化合物を含有する。
【化7】
【0015】
ここで、式I中、R1はHであり、且つR2は炭素数1〜3個のアルコキシ基であり、又はR1及びR2が一緒になって、−O−(CH2)2−O−の環を形成し、R3はH又は炭素数1〜3個のアルキル基である。
【0016】
炭素数1〜3個のアルコキシ基は、直鎖状又は分岐状のいずれであってもよく、例えば、メトキシ基、エトキシ基、プロピルオキシ基、i−プロピルオキシ基等を挙げることができる。
【0017】
また、炭素数1〜3個のアルキル基としては、直鎖状又は分岐状のいずれであってもよく、例えば、メチル基、エチル基、n−プロピル基、イソプロピル基等を挙げることができる。
【0018】
上記式Iで表される化合物の中でも、抗HIV活性及び細胞毒性の観点から、特に、式IIで表されるN−[(6−メトキシ−2−オキソ−1H−キノリン−3−イル)メチル]−N−(ピリジン−3−イルメチル)−2,1,3−ベンゾチアジアゾール−4−スルホンアミドが好ましい。
【化8】
【0019】
また、別の好ましい化合物として、式IIIで表されるN−[(7−オキソ−3,6−ジヒドロ−2H−[1,4]ジオキシノ[2,3−g]キノリン−8−イル)メチル]−N−(ピリジン−3−イルメチル)−2,1,3−ベンゾチアジアゾール−4−スルホンアミドが挙げられる。
【化9】
【0020】
さらに、別の好ましい化合物として、式IVで表される5−メチル−N−[(7−オキソ−3,6−ジヒドロ−2H−[1,4]ジオキシノ[2,3−g]キノリン−8−イル)メチル]−N−(ピリジン−3−イルメチル)−2,1,3−ベンゾチアジアゾール−4−スルホンアミドが挙げられる。
【化10】
【0021】
上記の各化合物は、慣用の有機合成法により得ることができる。また、Asinex社(Moscow, Russia, http://www.asinex.com/library-gold.html)から入手可能である。
【0022】
上記の化合物は、抗HIV薬として、慣用の製剤担体と組み合わせて製剤化することができる。投与形態としては、特に限定はなく、必要に応じ適宜選択して使用され、錠剤、カプセル剤、顆粒剤、細粒剤、散剤、徐放性製剤、液剤、懸濁剤、エマルジョン剤、シロップ剤、エリキシル剤等の経口剤、注射剤、坐剤等の非経口剤が挙げられる。
【0023】
経口剤は、例えばデンプン、乳糖、白糖、マンニット、カルボキシメチルセルロース、コーンスターチ、無機塩類等を用いて常法に従って製造される。また、これらに加えて、結合剤、崩壊剤、界面活性剤、滑沢剤、流動性促進剤、矯味剤、着色剤、香料等を適宜添加することができる。
【0024】
結合剤としては、例えばデンプン、デキストリン、アラビアゴム、ゼラチン、ヒドロキシプロピルスターチ、メチルセルロース、カルボキシメチルセルロースナトリウム、ヒドロキシプロピルセルロース、結晶セルロース、エチルセルロース、ポリビニルピロリドン、マクロゴール等が挙げられる。
【0025】
崩壊剤としては、例えばデンプン、ヒドロキシプロピルスターチ、カルボキシメチルセルロースナトリウム、カルボキシメチルセルロースカルシウム、カルボキシメチルセルロース、低置換ヒドロキシプロピルセルロース等が挙げられる。
【0026】
界面活性剤としては、例えばラウリル硫酸ナトリウム、大豆レシチン、ショ糖脂肪酸エステル、ポリソルベート80等が挙げられる。
【0027】
滑沢剤としては、例えばタルク、ロウ類、水素添加植物油、ショ糖脂肪酸エステル、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸アルミニウム、ポリエチレングリコール等が挙げられる。
【0028】
流動性促進剤としては、例えば軽質無水ケイ酸、乾燥水酸化アルミニウムゲル、合成ケイ酸アルミニウム、ケイ酸マグネシウム等が挙げられる。
【0029】
注射剤は、常法に従って製造され、希釈剤として一般に注射用蒸留水、生理食塩水、ブドウ糖水溶液、オリーブ油、ゴマ油、ラッカセイ油、ダイズ油、トウモロコシ油、プロピレングリコール、ポリエチレングリコール等を用いることができる。さらに必要に応じて、殺菌剤、防腐剤、安定剤、等張化剤、無痛化剤等を加えてもよい。また、注射剤は、安定性の観点から、バイアル等に充填後冷凍し、通常の凍結乾燥技術により水分を除去し、使用直前に凍結乾燥物から液剤を再調製することもできる。式I〜式IVの化合物の注射剤中における割合は、5〜50重量%の間で変動させ得るが、これに限定されるものではない。
【0030】
その他の非経口剤としては、直腸内投与のための坐剤等が挙げられ、常法に従って製造される。
【0031】
製剤化した抗HIV薬は、剤形、投与経路等により異なるが、例えば、1日1〜4回を1週間から3ヶ月の期間、投与することが可能である。
【0032】
経口剤として所期の効果を発揮するためには、患者の年令、体重、疾患の程度により異なるが、通常成人の場合、式I〜式IVの化合物の重量として、例えば0.1〜1000mg、好ましくは1〜500mgを、1日数回に分けて服用することが適当である。
【0033】
非経口剤として所期の効果を発揮するためには、患者の年令、体重、疾患の程度により異なるが、通常成人の場合、式I〜式IVの化合物の重量として、例えば0.1〜1000mg、好ましくは1〜500mgを、静注、点滴静注、皮下注射、筋肉注射により投与することが適当である。
【実施例】
【0034】
以下、実施例に基づき本発明をさらに詳細に説明するが、これに限定されるものではない。
(In silicoスクリーニング)
全てのIn silico実験は、コンピュータソフトMOE(Chemical Computing Group Inc., Quebec, Canada)を用いて行った。In silicoドッキングの標的として用いられたヒトcyclin T1のモデル構造は、ウマcyclin T1とequine infectious anemia virus(EIAV)TatとEIAV TAR RNAの複合体構造(Protein Data Bank ID: 2w2h)を基にホモロジー・モデリングにより構築した(Anand K. et al., Nat. Struct. Mol. Biol. 2008 Dec; 15(12): 1287-92)。3,000,000化合物のデータベース(Namiki shoji, Tokyo, Japan)の中から薬剤としての性質を有する化合物をスクリーニングするために、分子量が350から600 Da、水素結合のドナー/アクセプター数が13個より少なく、回転可能な結合が7個より少なく、logP値が0から6の間をとる化合物を選別した。このようにして選別した化合物のドッキングシミュレーションを、ヒトcyclin T1におけるHIV−1 TatとTAR RNAが結合する部位に対して行った。その結果、最適なスコア値を示した254の化合物を合成し、in vitroにおける抗HIV−1活性を評価した。なお、合成した化合物は、Asinex社(Moscow, Russia, http://www.asinex.com/library-gold.html)から市販されているものを購入して使用した。
【0035】
(抗HIV−1試験)
抗HIV−1試験は、HIV−1慢性感染細胞株であるOM10.1細胞を用いた。100μlの段階希釈された薬剤を含む96穴プレートに50μlのOM10.1細胞(4×105cells/ml)を加え、37℃、5%CO2で24時間培養した。その後、50μlのTNF−α(4ng/ml、Roche Diagnostics、Mannheim、Germany)を加え、37℃、5%CO2でさらに48時間培養した。100μlの培養上清を回収し、ELISA(ZeptoMetrix、Buffalo、NY)によりHIV−1 p24の産生量を定量して、産生を抑制する濃度(EC50)を測定した。また、化合物の細胞毒性(CC50)は、MTT法(Sigma-Aldrich、St. Louis、MO)により定量した。化合物A〜Jについての測定結果を下表及び図1〜3に示す。
【0036】
【表1】
【表2】
【表3】
【0037】
表及び図1〜3に示すように、本発明の化合物A〜Cは、それ以外の化合物に比べて、高い抗HIV−1活性を有し、細胞毒性が低いため、抗HIV−1薬として優れた化合物である。また、HIV−2においても、HIV−1と同じメカニズムによりウイルス遺伝子の転写が起こるため、上記実験結果より、本発明の化合物は抗HIV−2薬としても優れた活性を有し得るものである。
【特許請求の範囲】
【請求項1】
式I
【化1】
[式中、R1はHであり、且つR2は炭素数1〜3個のアルコキシ基であり、又はR1及びR2が一緒になって、−O−(CH2)2−O−の環を形成し、R3はH又は炭素数1〜3個のアルキル基である]
で表される化合物を含有する抗HIV薬。
【請求項2】
式Iで表される化合物が、式II
【化2】
で表される化合物である請求項1に記載の抗HIV薬。
【請求項3】
式Iで表される化合物が、式III
【化3】
で表される化合物である請求項1に記載の抗HIV薬。
【請求項4】
式Iで表される化合物が、式IV
【化4】
で表される化合物である請求項1に記載の抗HIV薬。
【請求項1】
式I
【化1】
[式中、R1はHであり、且つR2は炭素数1〜3個のアルコキシ基であり、又はR1及びR2が一緒になって、−O−(CH2)2−O−の環を形成し、R3はH又は炭素数1〜3個のアルキル基である]
で表される化合物を含有する抗HIV薬。
【請求項2】
式Iで表される化合物が、式II
【化2】
で表される化合物である請求項1に記載の抗HIV薬。
【請求項3】
式Iで表される化合物が、式III
【化3】
で表される化合物である請求項1に記載の抗HIV薬。
【請求項4】
式Iで表される化合物が、式IV
【化4】
で表される化合物である請求項1に記載の抗HIV薬。
【図1】


【図2】
【図3】
【図2】
【図3】
【公開番号】特開2012−201674(P2012−201674A)
【公開日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願番号】特願2011−70843(P2011−70843)
【出願日】平成23年3月28日(2011.3.28)
【出願人】(504258527)国立大学法人 鹿児島大学 (284)
【Fターム(参考)】
【公開日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願日】平成23年3月28日(2011.3.28)
【出願人】(504258527)国立大学法人 鹿児島大学 (284)
【Fターム(参考)】
[ Back to top ]