
抗IGF−I受容体抗体

【課題】インスリン様増殖因子-I受容体と特異的に結合しかつ阻害し、IGF-I、IGF-IIおよび血清の腫瘍細胞の増殖および生存に対する効果に拮抗し、かつ作動薬活性を実質的に欠く抗体、ヒト化抗体、再表面形成された抗体、抗体フラグメント、誘導体化抗体、および上記物質の細胞傷害薬とのコンジュゲートを提供する。
【解決手段】上記抗体およびそのフラグメントを利用した、増加したIGF-I受容体を発現する腫瘍、例えば乳癌、大腸癌、肺癌、前立腺癌、卵巣癌、滑膜癌、骨肉腫および膵癌の治療。上記の誘導体化した抗体を利用したIGF-I受容体の増強したレベルを発現する腫瘍の診断およびイメージング。
【解決手段】上記抗体およびそのフラグメントを利用した、増加したIGF-I受容体を発現する腫瘍、例えば乳癌、大腸癌、肺癌、前立腺癌、卵巣癌、滑膜癌、骨肉腫および膵癌の治療。上記の誘導体化した抗体を利用したIGF-I受容体の増強したレベルを発現する腫瘍の診断およびイメージング。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はヒトインスリン様増殖因子-I受容体(IGF-I受容体)と結合する抗体に関する。さらに特定すれば、本発明はIGF-I受容体の細胞機能を阻害する抗IGF-I受容体抗体に関する。なおさらに特定すれば、本発明はIGF-I、IGF-IIおよび血清の腫瘍細胞の増殖および生存に対する効果に拮抗しかつそれ自身は実質的に作動薬活性を欠く抗IGF-I受容体抗体に関する。本発明はまた、上記抗体のフラグメント、上記抗体のヒト化および再表面形成バージョン、上記抗体のコンジュゲート、抗体誘導体、ならびに上記の診断、研究および治療応用における利用にも関する。本発明はさらに、上記抗体およびそのフラグメントから作製される改良された抗体またはそのフラグメントに関する。他の態様においては、本発明は抗体またはそのフラグメントをコードするポリヌクレオチド、およびそのポリヌクレオチドを含むベクターに関する。
【背景技術】
【0002】
インスリン様増殖因子-I受容体(IGF-I受容体)は、膜貫通へテロテトラマータンパク質であり、細胞外の2つのα鎖および膜をまたがる2つのβ鎖をジスルフィド結合したβ-α-α-βコンフィギュレーションの形態を有する。リガンド、すなわちインスリン様増殖因子-I(IGF-I)およびインスリン様増殖因子-II(IGF-II)がIGF-I受容体の細胞外ドメインと結合すると、細胞内チロシンキナーゼドメインを活性化して受容体の自己リン酸化および基質リン酸化が起こる。IGF-I受容体はインスリン受容体と相同性があり、β鎖チロシンキナーゼドメインにおいて84%の高い配列類似性を有しかつα鎖細胞外システインリッチドメインにおいて48%の低い配列類似性を有する(Ulrich, A.ら, 1986, EMBO, 5,2503-2512;Fujita-Yamaguchi, Y.ら, 1986, J. Biol. Chem., 261, 16727-16731;LeRoith, D.ら, 1995, Endocrine Reviews, 16, 143-163)。IGF-I受容体およびそのリガンド(IGF-IおよびIGF-II)は多数の生理学的プロセスに重要な役割を果たし、その役割は胚形成時の成長および発生、成体における代謝、細胞増殖および細胞分化を含む(LeRoith, D., 2000, Endocrinology, 141,1287-1288;LeRoith, D., 1997, New England J. Med., 336, 633-640)。
【0003】
IGF-IとIGF-IIは両方とも、主にIGF結合タンパク質との複合体で存在する血中の内分泌ホルモンとして、および局所で産生されるパラ分泌および自己分泌増殖因子として機能する(Humbel, R.E., 1990, Eur. J. Biochem., 190, 445-462;Cohick, W.S.およびClemmons, D.R., 1993, Annu. Rev. Physiol. 55, 131-153)。
【0004】
IGF-I受容体は腫瘍細胞の増殖、形質転換および生存を促進することが示唆されている(Baserga, R.ら, 1997, Biochem. Biophys. Acta, 1332, F105-F126;Blakesley, V.A.ら, 1997, Journal of Endocrinology, 152, 339-344;Kaleko, M., Rutter, W.J.,およびMiller, A.D. 1990, Mol. Cell. Biol., 10, 464-473)。すなわち、腫瘍型のいくつかはIGF-I受容体の通常のレベルより高いレベルを発現することが公知であり、それらは乳癌、大腸癌、卵巣癌、滑膜肉腫、膵癌を含む(Khandwala, H.M.ら, 2000, Endocrine Reviews, 21, 215-244;Werner, H.およびLeRoith, D., 1996, Adv. Cancer Res., 68, 183-223;Happerfield, L.C.ら, 1997, J. Pathol., 183, 412-417;Frier, S.ら, 1999, Gut, 44, 704-708;van Dam, P.A.ら, 1994, J. Clin. Pathol., 47, 914-919;Xie, Y.ら, 1999, Cancer Res., 59, 3588-3591;Bergmann, U.ら, 1995, Cancer Res., 55, 2007-2011)。In vitroでIGF-IおよびIGF-IIは、複数のヒト腫瘍細胞系統、例えば肺癌、乳癌、大腸癌、骨肉腫および子宮頸癌に対する強力な分裂促進因子であることが示されている(Ankrapp, D.P.およびBevan, D.R., 1993, Cancer Res., 53, 3399-3404;Cullen, K.J., 1990, Cancer Res., 50, 48-53;Hermanto, U.ら, 2000, Cell Growth & Differentiation, 11, 655-664;Guo, Y. S.ら, 1995, J. Am. Coll. Surg., 181, 145-154;Kappel, C.C.ら, 1994, Cance
r Res., 54, 2803-2807;Steller, M.A.ら, 1996, Cancer Res., 56, 1761-1765)。これらの腫瘍および腫瘍細胞系統のいくつかはまた、高レベルのIGF-IまたはIGF-IIを発現して、自己分泌またパラ分泌の方法でそれら自身の増殖を刺激することができる(Quinn, K. A.ら, 1996, J. Biol. Chem., 271, 11477-11483)。
【0005】
疫学研究は、IGF-Iの血漿レベルの増加(およびIGF結合タンパク質-3のレベルの低下)は前立腺癌、大腸癌、肺癌および乳癌のリスクの増加と相関があることを示している(Chan, J. M.ら, 1998, Science, 279, 563-566;Wolk, A.ら, 1998, J. Natl. Cancer Inst., 90,911-915;Ma, J.ら, 1999, J. Natl. Cancer Inst., 91, 620-625;Yu, H.ら, 1999, J. Natl. Cancer Inst., 91, 151-156;Hankinson, S.E.ら, 1998, Lancet, 351, 1393-1396)。癌予防として、血漿のIGF-Iレベルを低下するかまたはIGF-I受容体の機能を抑制する手法が示唆されている(Wu, Y.ら, 2002, Cancer Res., 62, 1030-1035;Grimberg, A.およびCohen P., 2000, J. Cell. Physiol., 183, 1-9)。
【0006】
IGF-I受容体は、腫瘍細胞を増殖因子欠乏、足場非依存性、または細胞傷害薬処理により起こるアポトーシスから保護する(Navarro, M.およびBaserga, R., 2001, Endocrinology, 142, 1073-1081;Baserga, R.ら, 1997, Biochem. Biophys. Acta, 1332, F105-F126)。分裂促進、トランスフォーメーションおよび抗アポトーシス活性にとって重要であるIGF-I受容体のドメインは、突然変異分析により同定されている。
【0007】
例えば、IGF-I受容体のチロシン1251残基は、抗アポトーシスおよびトランスフォーメーション活性にとって極めて重要であるが分裂促進活性にとっては重要でないことが同定されている(O'Connor, R.ら, 1997, Mol. Cell. Biol., 17, 427-435;Miura, M.ら, 1995, J. Biol. Chem., 270, 22639-22644)。リガンドにより活性化されるIGF-I受容体の細胞内シグナル伝達経路はインスリン受容体基質(IRS-1およびIRS-2)のチロシン残基のリン酸化に関わり、これがホスファチジルイノシトール-3-キナーゼ(PI3キナーゼ)を膜へ補給する。膜結合したPI3キナーゼのリン脂質産物はセリン/トレオニンキナーゼAktを活性化し、その基質であるプロアポトーシスタンパク質BADがリン酸化されて不活性状態となる(Datta, S. R., Brunet, A. and Greenberg, M. E., 1999, Genes & Development, 13, 2905-2927;Kulik, G., Klippel, A.およびWeber, M. J., 1997, Mol. Cell. Biol. 17, 1595-1606)。MCF-7ヒト乳癌細胞のIGF-I受容体の分裂促進シグナル伝達はPI3キナーゼを必要とし、分裂促進因子活性化タンパク質キナーゼに依存しないが、分化ラットクロム親和細胞腫PC12細胞の生存シグナル伝達は、PI3キナーゼと分裂促進因子活性化タンパク質キナーゼの両方の経路を必要とする(Dufourny, B.ら, 1997, J. Biol. Chem., 272, 31163-31171;Parrizas, M., Saltiel, A. R. and LeRoith, D., 1997, J. Biol. Chem., 272, 154-161)。
【0008】
アンチセンス手法によるIGF-I受容体レベルの下方調節は、複数の腫瘍細胞系統、例えば黒色腫、肺癌、卵巣癌、グリア芽細胞腫、神経芽細胞腫および横紋筋肉腫のin vivoおよびin vitro腫瘍形成能を低下することが示されている(Resnicoff, M.ら, 1994, Cancer Res., 54, 4848-4850;Lee, C.-T.ら, 1996, Cancer Res., 56, 3038-3041;Muller, M.ら, 1998, Int. J. Cancer, 77, 567-571;Trojan, J.ら, 1993, Science, 259, 94-97;Liu, X.ら, 1998, Cancer Res., 58, 5432-5438;Shapiro, D. N.ら, 1994, J. Clin. Invest., 94, 1235-1242)。同様に、IGF-I受容体の優性ネガティブ突然変異は、IGF-I受容体を過剰発現する形質転換Rat-1細胞のin vivo腫瘍形成能、およびin vitro増殖を低下することが報じられている(Prager, D.ら, 1994, Proc. Natl. Acad. Sci. USA, 91, 2181-2185)。
【0009】
IGF-I受容体mRNAに対するアンチセンスを発現する腫瘍細胞は、生物拡散室内の動物に注射すると大量のアポトーシスを起こす。この観察によれば、腫瘍細胞はIGF-I受容体の阻害によりアポトーシスに対する感受性が正常細胞より高くなるという仮説に基づいて、IGF-I受容体は魅力的な治療標的であることを示す(Resnicoff, M.ら, 1995, Cancer Res., 55, 2463-2469;Baserga, R., 1995, Cancer Res., 55, 249-252)。
【0010】
腫瘍細胞中のIGF-I受容体の機能を阻害する他の手法として、IGF-I受容体の細胞外ドメインと結合してその活性化を阻害する抗IGF-I受容体抗体が利用されている。IGF-I受容体に対するマウスモノクローナル抗体を開発するいくつかの試みが報じられており、それらの中で2種の阻害抗体(IR3および1H7)は入手可能があり、それらの利用が複数のIGF-I受容体研究において報じられている。
【0011】
部分精製したインスリン受容体の胎盤調製物を用いてマウスを免疫感作すると、インスリン受容体と選択的に結合するIR1、およびIGF-I受容体(ソマトメジンC受容体)の優先的免疫沈降を示すがインスリン受容体の弱い免疫沈降も示す2種の抗体、IR2とIR3が得られるので、それを利用してIR3抗体を発生させた(Kull, F. C.ら, 1983, J. Biol. Chem., 258, 6561-6566)。
【0012】
精製したIGF-I受容体の胎盤調製物を利用して1H7抗体を発生させた。該胎盤調製物を用いてマウスを免疫感作すると、3種の刺激性抗体に加えて阻害性抗体1H7が得られる(Li, S.-L.ら, 1993, Biochem. Biophys. Res. Commun., 196, 92-98;Xiong, L.ら, 1992, Proc. Natl. Acad. Sci. USA, 89, 5356-5360)。
【0013】
他の報文では、IGF-I受容体の高レベルを発現するトランスフェクトされた3T3細胞を用いてマウスを免疫感作すると、ヒトIGF-I受容体に特異的なマウスモノクローナル抗体のパネルが得られるので、このパネルを結合競合によりおよびIGF-Iのトランスフェクトされた3T3細胞との結合の阻害または刺激により7グループに分類した(Soos, M. A.ら, 1992, J. Biol. Chem., 267, 12955-12963)。
【0014】
このように、IR3抗体はin vitroで最も汎用されるIGF-I受容体研究用の阻害性抗体であるが、これはIGF-I受容体を発現するトランスフェクトされた3T3およびCHO細胞に作動薬活性を示す欠点が悩みである(Kato, H.ら, 1993, J. Biol. Chem., 268, 2655-2661;Steele-Perkins, G.およびRoth, R. A., 1990, Biochem. Biophys. Res. Commun., 171, 1244-1251)。同様に、Soosらが開発した抗体のパネルの中で、最も阻害性抗体の24-57および24-60もトランスフェクトされた3T3細胞に作動薬活性を示した(Soos, M. A.ら, 1992, J. Biol. Chem., 267, 12955-12963)。IR3抗体は、IGF-Iの無傷の細胞におけるおよび可溶化後の発現された受容体との結合を阻害する(しかしIGF-IIを阻害しない)ことが報じられるが、細胞のDNA合成をin vitroで刺激するIGF-IとIGF-II両方の能力を阻害することが示されている(Steele-Perkins, G.およびRoth, R.A., 1990, Biochem. Biophys. Res. Commun., 171, 1244-1251)。IR3抗体の結合エピトープは、キメラのインスリンIGF-I受容体構築物からIGF-I受容体の223〜274領域であると推定されている(Gustafson, T. A.およびRutter, W. J., 1990, J. Biol. Chem., 265, 18663-18667;Soos, M. A.ら, 1992, J. Biol. Chem., 267, 12955-12963)。
【0015】
MCF-7ヒト乳癌細胞系統は、IGF-IおよびIGF-IIのin vitro増殖応答を実証するために利用される典型的なモデル細胞系統である(Dufourny, B.ら, 1997, J. Biol. Chem., 272, 31163-31171)。MCF-7細胞において、IR3抗体は、無血清状態で外因的に加えられたIGF-IおよびIGF-IIの刺激性効果を不完全にほぼ80%だけ遮断する。また、IR3抗体は10%血清中でMCF-7細胞の増殖を有意に阻害しない(25%未満)(Cullen, K. J.ら, 1990, Cancer Res., 50, 48-53)。MCF-7細胞の血清刺激増殖のIR3抗体によるin vitroでのこの弱い阻害は、IR3抗体処理がヌードマウスにおけるMCF-7異種移植片の増殖を有意に阻害しなかったin vivo研究の結果と関係がありうる(Arteaga, C. L.ら, 1989, J. Clin. Invest., 84, 1418-1423)。
【0016】
IR3および他の報じられた抗体の弱い作動薬活性、ならびにそれらがより生理学的な血清刺激の条件(無血清条件で外因的に加えたIGF-IまたはIGF-IIによる刺激の代わりに)のもとでMCF-7細胞などの腫瘍細胞の増殖を有意に阻害する能力が無いことの故に、腫瘍細胞の血清刺激増殖を有意に阻害ししかしそれ自身による有意な作動薬活性を示さない新しい抗IGF-I受容体抗体に対するニーズがある。
【発明の概要】
【発明が解決しようとする課題】
【0017】
従って、インスリン様増殖因子-I受容体と特異的に結合し、受容体に拮抗することにより受容体の細胞活性を阻害し、そしてまた受容体に対する作動薬活性を実質的に欠く抗体、抗体フラグメントおよび抗体誘導体を提供することが本発明の目的である。
【課題を解決するための手段】
【0018】
従って第1の実施形態においては、軽および重鎖可変域の両方のアミノ酸配列、軽および重鎖に対する遺伝子のcDNA配列、そのCDR(相補性決定領域)の同定、その表面アミノ酸の同定、ならびに組換え型においてそれを発現する方法に関して、本明細書で全て特徴付けられているマウス抗体EM164が提供される。
【0019】
第2の実施形態においては、抗体EM164の再表面形成されたまたはヒト化されたバージョンであって、抗体またはそのフラグメントの表面に曝される残基の軽および重鎖の両方が置換えられて公知のヒト抗体表面にさらによく似ている上記バージョンが提供される。そのようなヒト化された抗体は、マウスEM164と比較して、治療または診断薬としての有用性が一層増加しうる。抗体EM164のヒト化されたバージョンはまた、軽および重鎖可変域両方のそれぞれのアミノ酸配列、軽および重鎖に対する遺伝子のDNA配列、そのCDRの同定、その表面アミノ酸の同定、ならびに組換え型においてそれを発現する方法の開示に関して、本明細書において全て特徴づけられている。
【0020】
第3の実施形態においては、増殖刺激因子、例えば血清、インスリン様増殖因子-Iおよびインスリン様増殖因子-IIの存在のもとで、癌細胞の増殖をほぼ80%以上阻害することができる抗体が提供される。
【0021】
第4の実施形態においては、抗体または抗体フラグメントであって、
それぞれ配列番号1〜3:
SYWMH (配列番号1)、
EINPSNGRTNYNEKFKR (配列番号2)、
GRPDYYGSSKWYFDV (配列番号3)により表されるアミノ酸配列を有するCDRを含む重鎖を有しかつ
それぞれ配列番号4〜6:
RSSQSIVHSNVNTYLE (配列番号4)、
KVSNRFS (配列番号5)、
FQGSHVPPT (配列番号6)により表されるアミノ酸配列を有するCDRを含む軽鎖を有する上記抗体または抗体フラグメントが提供される。
【0022】
第5の実施形態においては、配列番号7:
QVQLQQSGAELVKPGASVKLSCKASGYTFTSYWMHWVKQRPGQGLEWIGEINPSNGRTNYNEKFKRKATLTVDKSSSTAYMQLSSLTSEDSAVYYFARGRPDYYGSSKWYFDVWGAGTTVTVSS(配列番号7)
により表されるアミノ酸配列と少なくとも90%配列同一性を共有するアミノ酸配列を持つ重鎖を有する抗体が提供される。
【0023】
同様に、配列番号8:
DVLMTQTPLSLPVSLGDQASISCRSSQSIVHSNVNTYLEWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGTDFTLRISRVEAEDLGIYYCFQGSHVPPTFGGGTKLEIKR(配列番号8)
により表されるアミノ酸配列と少なくとも90%配列同一性を共有するアミノ酸配列を持つ軽鎖を有する抗体が提供される。
【0024】
第6の実施形態においては、配列番号9〜12:
DVVMTQTPLSLPVSLGDPASISCRSSQSIVHSNVNTYLEWYLQKPGQSPRLLIYKVSNRFSGVPDRFSGSGAGTDFTLRISRVEAEDLGIYYCFQGSHVPPTFGGGTKLEIKR(配列番号9);
DVLMTQTPLSLPVSLGDPASISCRSSQSIVHSNVNTYLEWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGAGTDFTLRISRVEAEDLGIYYCFQGSHVPPTFGGGTKLEIKR(配列番号10);
DVLMTQTPLSLPVSLGDPASISCRSSQSIVHSNVNTYLEWYLQKPGQSPRLLIYKVSNRFSGVPDRFSGSGAGTDFTLRISRVEAEDLGIYYCFQGSHVPPTFGGGTKLEIKR(配列番号11);または
DVVMTQTPLSLPVSLGDPASISCRSSQSIVHSNVNTYLEWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGAGTDFTLRISRVEAEDLGIYYCFQGSHVPPTFGGGTKLEIKR(配列番号12)
のうちの1つに対応するアミノ酸配列を持つヒト化されたまたは再表面形成された軽鎖可変域を有する抗体が提供される。
【0025】
同様に、配列番号13:
QVQLVQSGAEVVKPGASVKLSCKASGYTFTSYWMHWVKQRPGQGLEWIGEINPSNGRTNYNQKFQGKATLTVDKSSSTAYMQLSSLTSEDSAVYYFARGRPDYYGSSKWYFDVWGQGTTVTVSS(配列番号13)
に対応するアミノ酸配列を持つヒト化されたまたは再表面形成された重鎖可変域を有する抗体が提供される。
【0026】
第7の実施形態においては、改良された特性を有する本発明の抗体または抗体フラグメントが提供される。例えば、改良されたIGF-I受容体に対する親和性を有する抗体または抗体フラグメントが本発明の抗体またはフラグメントの親和性成熟により調製される。
【0027】
本発明はさらに、上記抗体のコンジュゲートであって、直接にまたは切断可能もしくは非切断可能なリンカーを経由して細胞傷害薬が本発明の抗体もしくは抗体のエピトープ結合フラグメントと共有結合していることを特徴とする上記コンジュゲートを提供する。好ましい実施形態において、細胞傷害薬はタキソール、メイタンシノイド、CC-1065またはCC-1065類似体である。
【0028】
本発明はさらに、研究または診断応用に利用するためにさらに標識されている抗体またはそのフラグメントを提供する。好ましい実施形態において、標識は放射標識、蛍光体、発色体、イメージング剤または金属イオンである。
【0029】
上記抗体またはフラグメントを癌を有すると疑われる被験者に投与し、そして被験者の体内の標識の分布を測定またはモニターする診断方法も提供される。
【0030】
第8の実施形態においては、本発明は癌を有する被験者の治療方法であって、本発明の抗体、抗体フラグメントまたは抗体コンジュゲートを単独でまたは他の細胞傷害または治療薬と組合わせて投与することによる上記治療方法を提供する。癌は、1以上の、例えば乳癌、大腸癌、卵巣癌、骨肉腫、子宮頸癌、前立腺癌、肺癌、滑膜癌、膵癌、またはこれからIGF-I受容体レベルが高いことで確認される他の癌であってもよい。
【図面の簡単な説明】
【0031】
【図1】精製EM164抗体の、ヒトY1251F-IGF-I受容体またはヒトインスリン受容体を過剰発現する細胞との特異的結合の蛍光活性化細胞選別(フローサイトメトリー)(FACS)分析を示す。
【図2】EM164抗体のビオチン化ヒトIGF-I受容体との結合に対する結合滴定曲線を示す。
【図3】EM164抗体による、ビオチン化IGF-Iのヒト乳癌MCF-7細胞との結合の阻害を示す。
【図4】EM164抗体による、MCF-7細胞におけるIGF-I受容体のIGF-I刺激自己リン酸化の阻害を示す。
【図5】EM164抗体による、MCF-7細胞におけるIGF-I刺激IRS-1リン酸化の阻害を示す。
【図6】EM164抗体による、SaOS-2細胞におけるIGF-I刺激シグナル伝達の阻害を示す。
【図7】MTTアッセイにより評価した、様々な増殖条件下でのMCF-7細胞の増殖および生存に与えるEM164抗体の効果を示す。
【図8】様々な血清濃度のもとでの、MCF-7細胞の増殖と生存に与えるEM164抗体の効果を示す。
【図9】EM164抗体による、IGF-Iおよび血清に刺激されたNCI-H838細胞の増殖と生存の阻害を示す。
【図10】EM164抗体、タキソール、またはEM164抗体とタキソールの併用による処理が、マウスにおけるCalu-6肺癌異種移植片の増殖に与える効果を示す。
【図11】ヒト化EM164抗体(v.1.0)およびマウスEM164抗体の結合の間の競合を示す。
【図12】マウス抗IGF-I受容体抗体EM164の軽鎖リーダーおよび可変域の、cDNA(配列番号49)およびアミノ酸配列(配列番号50)を示す。矢印はフレームワークの出発点を示す。Kabatによる3種のCDR配列に下線を引いた。
【図13】マウス抗IGF-I受容体抗体EM164の重鎖リーダーおよび可変域の、cDNA(配列番号51)およびアミノ酸配列(配列番号52)を示す。矢印はフレームワークの出発点を示す。Kabatによる3種のCDR配列に下線を引いた。
【図14】Chothia正準クラス定義(Chothia canonical class definitions)から確認した抗体EM164の軽および重鎖CDRアミノ酸配列を示す。重鎖CDRに対するAbMモデルソフトウエア定義(AbM modeling software definitions)も示す。軽鎖のCDR1は配列番号4、CDR2は配列番号5、そしてCDR3は配列番号6である。重鎖のCDR1は配列番号1、CDR2は配列番号2、そしてCDR3は配列番号3である。AbM重鎖のCDR1は配列番号53、CDR2は配列番号54、そしてCDR3は配列番号55である。
【図15】抗IGF-I-受容体抗体EM164の軽鎖および重鎖アミノ酸配列とCr1(配列番号56)およびJ558.c(配列番号57)遺伝子の生殖系列配列とのアラインメントを示す。ダッシュ(-)は配列同一性を示す。
【図16】組換えキメラおよびヒト化EM164抗体を構築しかつ発現するために利用したプラスミドを示す。A)軽鎖クローニングプラスミド、B)重鎖クローニングプラスミド、C)哺乳類動物抗体発現プラスミド。
【図17】EM164の表面残基を予測するために用いた構造ファイルのセット中の抗体127種からスクリーニングした軽鎖のうちの最も相同的な10種のアミノ酸配列を示す。em164 LC(配列番号58)、2jel(配列番号59)、2pcp(配列番号60)、1nqb(配列番号61)、1kel(配列番号62)、1hyx(配列番号63)、1igf(配列番号64)、1tet(配列番号65)、1clz(配列番号66)、1bln(配列番号67)、1cly(配列番号68)、コンセンサス(配列番号69)。
【図18】EM164の表面残基を予測するために用いた構造ファイルのセット中の抗体127種からスクリーニングした重鎖のうちの最も相同的な10種のアミノ酸配列を示す。em164 HC(配列番号70)、1nqb(配列番号71)、1ngp(配列番号72)、1fbi(配列番号73)、1afv(配列番号74)、1yuh(配列番号75)、1plg(配列番号76)、1d5b(配列番号77)、1ae6(配列番号78)、1axs(配列番号79)、3hfl(配列番号80)、コンセンサス(配列番号81)。
【図19】10種の最も相同的な構造からの(A)軽鎖および(B)重鎖可変域残基のそれぞれに対する平均アクセシビリティを示す。数字はKabat抗体配列位置番号を表す。
【図20】マウスEM164(muEM164)およびヒト化EM164(huEM164)抗体に対する軽鎖可変域アミノ酸配列を示す。muEM164(配列番号82)、huEM164 V1.0(配列番号83)、huEM164 V1.1(配列番号84)、huEM164 V1.2(配列番号85)、huEM164 V1.3(配列番号86)。
【図21】マウス(muEM164、配列番号87)およびヒト化EM164抗体(huEM164、配列番号88)に対する重鎖可変域アミノ酸配列を示す。
【図22】huEM164 v1.0の可変域DNAおよびアミノ酸配列を、軽鎖(DNA配列番号89、アミノ酸配列番号90)と重鎖(DNA配列番号91、アミノ酸配列番号92)の両方に対して示す。
【図23A】ヒト化EM164 v1.1(DNA配列番号93;アミノ酸配列番号94)、v1.2(DNA配列番号95;アミノ酸配列番号96)およびv1.3(DNA配列番号97;アミノ酸配列番号98)の軽鎖可変域DNAおよびアミノ酸配列を示す。
【図23B】ヒト化EM164 v1.1(DNA配列番号93;アミノ酸配列番号94)、v1.2(DNA配列番号95;アミノ酸配列番号96)およびv1.3(DNA配列番号97;アミノ酸配列番号98)の軽鎖可変域DNAおよびアミノ酸配列を示す(図23Aの続き)。
【図24】ヒト化EM164 v1.0抗体およびマウスEM164抗体による、MCF-7細胞のIGF-I刺激増殖および生存の阻害を示す。
【図25】EM164がMCF-7細胞のIGF-I-刺激サイクリングを抑制することを示す。
【図26】EM164がIGF-Iおよび血清の抗アポトーシス効果を抑制することを示す。EM164を用いる処理はアポトーシス細胞死をもたらし、それが切断されたCK18タンパク質のレベル増加により実証される。
【図27】免疫不全マウスにおけるヒトBxPC-3膵癌異種移植片の増殖に対する、EM164抗体、ゲミシタビン、またはEM164抗体とゲミシタビンの併用による処理の効果を示す。
【発明を実施するための形態】
【0032】
本発明者らは、細胞表面上のヒトインスリン様増殖因子-I受容体(IGF-IR)と結合する新規の抗体を発見しかつそれを改良した。本抗体およびフラグメントは、受容体それ自身を活性化する能力がなくしかも受容体の細胞機能を阻害するというユニークな能力を有する。従って、IGF-IRと結合して阻害する従来公知の抗体はIGF-IRリガンドの不在時でも受容体を活性化するのに対して、本発明の抗体またはフラグメントはIGF-IRに拮抗するが実質的に作動薬活性を欠く。さらに本発明の抗体と抗体フラグメントは、血清の存在のもとでのヒト腫瘍細胞、例えばMCF-7細胞の増殖を80%以上阻害し、この値は従来公知の抗IGF-IR抗体を用いて得られるより高い阻害度である。
【0033】
本発明は、本明細書でEM164と呼び、軽および重鎖の両方のアミノ酸配列、そのCDRの同定、表面アミノ酸の同定、および組換え型におけるその発現に関して、全て特徴付けているマウス抗IGF-IR抗体から進める。
【0034】
生殖系列配列のEM164配列とのアラインメントを図15に示す。その比較によりEM164中の可能性のある体細胞突然変異が同定され、それらは軽鎖のCDR1および重鎖のCDR2にそれぞれ1つづつ含まれる。
【0035】
抗体EM164軽および重鎖、ならびにヒト化バージョンの1次アミノ酸およびDNA配列を本明細書に開示する。しかし、本発明の範囲はこれらの配列を含む抗体とフラグメントに限定されるものではない。それどころか、インスリン様増殖因子-I受容体と特異的に結合しかつ受容体の生物学的活性に拮抗し、しかし作動活性を実質的に欠く全ての抗体とフラグメントは本発明の範囲に包含される。従って、抗体と抗体フラグメントは、抗体EM164またはヒト化誘導体とその骨格、CDR、軽鎖および重鎖のアミノ酸配列が異なっていてもよく、それでもなお本発明の範囲内に包含されうる。
【0036】
抗体EM164のCDRはモデリングにより同定され、その分子構造は予測されている。再び、CDRはエピトープ認識にとって重要であるものの、それは本発明の抗体とフラグメントにとって必須ではない。すなわち、例えば本発明の抗体の親和性成熟により作られる、改良された特性を有する抗体とフラグメントが提供される。
【0037】
多様な抗体と抗体フラグメント、ならびに抗体模倣物を、特定のセットのCDRとフランキングする可変および定常域配列内の突然変異、欠失および/または挿入により作ることができる。このようにして、例えば、所与のセットのCDRに対して異なる重鎖の置換により異なるクラスのAbを得ることが可能であり、この方法により、例えば、IgG1〜4、IgM、IgA1〜2、IgD、IgE抗体型およびイソ型を作ることができる。同様に、本発明の範囲に含まれる人工抗体は、CDRの所与のセットを完全な合成フレームワーク内に埋め込むことにより作ることができる。本明細書において用語「可変」は、可変ドメインのある特定部分であって、抗体間で配列が異なりかつ各特定抗体のその抗原との結合および特異性に利用される上記部分を記載するために使用される。しかし、その可変性は通常、抗体の可変ドメイン全体に等しく分布するものではない。可変性は典型的には軽鎖および重鎖両方の可変ドメイン中の相補性決定領域(CDR)または超可変域と呼ばれる3セグメントに濃縮される。可変ドメインのより高度に保存された部分はフレームワーク(FR)と呼ばれる。重および軽鎖の可変ドメインはそれぞれ、主にβシートコンフィギュレーションである4つのフレームワーク領域を含み、それらのフレームワークは3つのCDRにより接続される。上記CDRは、接続する(場合によってはβシート構造の部分となる)ループを形成する。それぞれの鎖のCDRは、FR領域によって密接した近位に一緒に保持され、他の鎖からのCDRとともに抗体の抗原結合部位の形成に寄与する(E. A. Kabatら 「免疫学的に重要なタンパク質の配列(Sequences of Proteins of Immunological Interest)」, 第5版, 1991, NIH)。定常ドメインは抗体と抗原の結合に直接関わらないが、様々なエフェクター機能、例えば抗体の抗体依存性細胞毒性への関与を示す。
【0038】
ヒト化抗体、または他の哺乳類動物による非拒絶に適合させた抗体は、複数の技術、例えば再表面形成およびCDR移植術を用いて作ることができる。再表面形成技術においては、分子モデル、統計解析および突然変異誘発を組合わせ、可変域の非CDR表面を標的宿主の既知抗体の表面に似ているように調節する。抗体を再表面形成する計画と方法、および色々な宿主内の抗体の免疫原性を低下させる他の方法は、米国特許第5,639,641号に開示されていて、これは本明細書にその全てが参照により組み入れられる。CDR移植技術においては、マウス重および軽鎖CDRを全ヒトフレームワーク配列中に移植する。
【0039】
本発明はまた、本明細書に記載した抗体の機能的同等物も含む。機能的同等物はこれらの抗体と比較しうる結合特性を有し、例えば、キメラ、ヒト化および1本鎖抗体ならびにそれらのフラグメントを含む。このような機能的同等物を作るほう方法は、PCT出願WO93/21319、欧州特許出願第239,400号;PCT出願WO89/09622;欧州特許出願第338,745号;および欧州特許出願第332,424号に開示されていて、これらはそれぞれの全てが参照により組み入れられる。
【0040】
機能的同等物は、本発明の抗体の可変もしくは超可変域のアミノ酸配列と実質的に同じアミノ酸配列をもつポリペプチドを含む。アミノ酸配列に適用される「実質的に同じ」は、本明細書においては、PearsonおよびLipman, Proc. Natl. Acad. Sci. USA 85, 2444-2448 (1988)によるFASTAサーチ法により決定して、他のアミノ酸配列と少なくともほぼ90%、さらに好ましくは少なくともほぼ95%配列同一性をもつ配列として定義される。
【0041】
キメラ抗体は好ましくは、実質的にまたは専らヒト抗体定常域から誘導された定常域および実質的にまたは専らヒト以外の哺乳類動物の可変域配列から誘導された可変域を有する。抗体のヒト化型は、例えばマウス抗体の相補性決定領域をヒトフレームワークドメイン中に置換することにより作られる(例えばPCT公開第W092/22653号を参照)。ヒト化抗体は好ましくは、実質的にまたは専ら対応するヒト抗体領域から誘導された定常域と相補性決定領域(CDR)以外の可変域、および実質的にまたは専らヒト以外の哺乳類動物から誘導されたCDRを有する。
【0042】
機能性同等物はまた、1本鎖抗体(scFv)としても知られる1本鎖抗体フラグメントを含む。これらのフラグメントは、1以上の相互接続リンカーを用いてまたは用いないで、少なくとも1つの抗体可変軽鎖配列(VL)のフラグメントと繋留された少なくとも1つの抗体可変重鎖配列(VH)のフラグメントを含有する。そのようなリンカーは、短いフレキシブルなペプチドであって、(VL)と(VH)ドメインが連結されるとそれらの適当な3次元フォールディングが起こり、1本鎖抗体フラグメントが誘導された元の全抗体の標的分子結合特異性を維持することを確保するように選択することができる。一般的に(VL)または(VH)配列のカルボキシ末端を、このようなペプチドリンカーにより相補的(VL)および(VH)配列のアミノ酸末端と共有結合で連結することができる。1本鎖抗体フラグメントは、分子クローニング、抗体ファージディスプレイライブラリーまたは類似の技術により作製することができる。これらのタンパク質は、真核生物細胞または細菌を含む原核生物細胞のいずれで産生させてもよい。
【0043】
1本鎖抗体フラグメントは、本明細書に記載の全抗体の可変または相補性決定領域(CDR)の少なくとも1つを有するが、これらの抗体の定常ドメインのいくらかまたは全てを欠いている。これらの定常ドメインは抗原結合に必要でないが、全抗体構造の主要部を構成する。従って、1本鎖抗体フラグメントは、定常ドメインの一部または全てを含有する抗体の利用に関連するいくつかの問題を克服することができる。例えば、1本鎖抗体フラグメントは、生物学的分子と重鎖定常域との間の望ましくない相互作用、または他の欲しない生物学的活性を持たない傾向がある。さらに、1本鎖抗体フラグメントは全抗体よりかなり小さく従ってより大きい毛細管透過性を有するので、1本鎖抗体フラグメントが局在化して標的抗原結合部位とより効率的に結合することが可能になる。また、抗体フラグメントは真核生物細胞において比較的大規模に生産することができるので、その生産が容易になる。さらに、1本鎖抗体フラグメントは比較的サイズが小さいので、全抗体よりレシピエントの免疫応答を引起す可能性が低い。
【0044】
機能的同等物はさらに、全抗体のそれと同じかまたは比較しうる結合特性を有する抗体のフラグメントを含む。そのようなフラグメントはFabフラグメントの1つもしくは両方またはF(ab')2フラグメントを含有してもよい。好ましくは抗体フラグメントは全抗体の6つの相補性決定領域の全てを含有するが、全てより少ない数、例えば3つ、4つまたは5つのかかる領域を含有するフラグメントもまた機能的である。さらに、機能的同等物は、次の免疫グロブリンクラス:IgG、IgM、IgA、IgDまたはIgE、およびそれらのサブクラスのいずれか1つであってもまたはそのメンバーの組合わせであってもよい。
【0045】
本明細書に記載した抗IGF-I受容体抗体EM164およびそのヒト化変異体に対するアミノ酸および核酸配列の知識を利用して、同じようにヒトIGF-I受容体と結合してIGF-I受容体の細胞機能を阻害する他の抗体を開発することができる。複数の研究は、1以上のアミノ酸変化を抗体の配列の様々な位置へ導入することの抗体の特性、例えば結合および発現のレベル、に与える効果を1次抗体配列の知識に基づいて調査している(Yang, W. P.ら, 1995, J. Mol. Biol., 254, 392-403;Rader, C.ら, 1998, Proc. Natl. Acad. Sci. USA, 95, 8910-8915;Vaughan, T. J.ら, 1998, Nature Biotechnology, 16, 535-539)。
【0046】
これらの研究において、オリゴヌクレオチド媒介の位置指定突然変異誘発、カセット突然変異誘発、誤りがちな(error-prone)PCR、DNAシャッフリング、または大腸菌(E. coli)の変異誘発株などの技術を用いて、CDR1、CDR2、CDR3、またはフレームワーク領域の重および軽鎖遺伝子の配列を変えることにより、1次抗体の変異体が作製されている(Vaughan, T. J.ら, 1998, Nature Biotechnology, 16, 535-539;Adey, N. B.ら, 1996, 第16章, pp.277-291, in 「ペプチドとタンパク質のファージディスプレイ(Phage Display of Peptides and Proteins)」, Kay, B.K.ら編, Academic Press)。1次抗体の配列を変えるこれらの方法は、2次抗体の親和性の改良をもたらしている(Gram, H.ら, 1992, Proc. Natl. Acad. Sci. USA, 89, 3576-3580;Boder, E. T.ら, 2000, Proc. Natl. Acad. Sci. USA, 97, 10701-10705;Davies, J. and Riechmann, L., 1996, Immunotechnolgy, 2, 169-179;Thompson, J.ら, 1996, J. Mol. Biol., 256, 77-88;Short, M. K.ら, 2002, J. Biol. Chem., 277, 16365-16370;Furukawa, K.ら, 2001, J. Biol. Chem., 276, 27622-27628)。
【0047】
抗体の1以上のアミノ酸残基を変える類似の定方向(directed)手法により、本発明に記載した抗体配列を利用して改良された機能をもつ抗IGF-I受容体抗体を開発することができる。
【0048】
本発明のコンジュゲートは、細胞傷害薬に連結された本発明に記載の抗体、フラグメント、およびそれらの類似体を含む。好ましい細胞傷害薬はメイタンシノイド、タキサンおよびCC-1065の類似体である。コンジュゲートはin vitroの方法により調製することができる。細胞傷害薬を抗体と連結する目的で連結基を用いる。適当な連結基は当技術分野では周知であり、ジスルフィド基、チオエーテル基、酸に不安定な基、感光性基、ペプチダーゼ易分解性基およびエステラーゼ易分解性基を含む。好ましい連結基はジスルフィド基およびチオエーテル基である。例えば、コンジュゲートは、ジスルフィド交換反応を用いてまたは抗体と細胞傷害薬の間にチオエーテル基を形成することにより構築することができる。
【0049】
メイタンシノイドとメイタンシノイド類似体が好ましい細胞傷害薬として挙げられる。好適なメイタンシノイドとメイタンシノイド類似体の例は、米国特許第4,424,219号;第4,256,746号;第4,294,757号;第4,307,016号;第4,313,946号;第4,315,929号;第4,331,598号;第4,361,650号;第4,362,663号;第4,364,866号;第4,450,254号;第4,322,348号;第4,371,533号;第6,333,410号;第5,475,092号;第5,585,499号;および第5,846,545号に開示されている。
【0050】
タキサンも好ましい細胞傷害薬である。本発明に使用するのに好適なタキサンは米国特許第6,372,738号および第6,340,701号に開示されている。
【0051】
CC-1065とその類似体も本発明に使用するのに好ましい細胞傷害薬である。CC-1065とその類似体は米国特許第6,372,738号;第6,340,701号;第5,846,545号および第5,585,499号に開示されている。
【0052】
このような細胞傷害性コンジュゲートを調製するための魅力的な候補薬はCC-1065であり、これはストレプトマイセス・ゼレンシス(Streptomyces zelensis)の培養ブロスから単離された強力な抗腫瘍抗生物質である。CC-1065は通常使用される抗癌薬、例えばドキソルビシン、メトトレキセートおよびビンクリスチンよりもin vitroでほぼ1000倍以上強力である(B.K. Bhuyanら, Cancer Res., 42, 3532-3537 (1982))。
【0053】
メトトレキセート、ダウノルビシン、ドキソルビシン、ビンクリスチン、ビンブラスチン、メルファラン、マイトマイシンC、クロラムブシル、およびカリケアマイシンなどの細胞傷害薬も本発明のコンジュゲートを調製するのに好適であり、薬物分子を抗体分子と仲介担体分子、例えば血清アルブミンを介して連結することもできる。
【0054】
診断上の応用については、典型的には本発明の抗体を検出可能部分を用いて標識しうる。検出可能部分は、検出可能なシグナルを直接的にまたは間接的に作製しうるのであればいずれのものでもよい。例えば、検出可能部分は放射性同位体、例えば3H、14C、32P、35S、もしくは131I;蛍光もしくは化学発光化合物、例えばフルオレセインチオシアネート、ローダミン、もしくはルシフェリン;または酵素、例えばアルカリホスファターゼ、β-ガラクトシダーゼ、もしくは西洋わさびペルオキシダーゼであってもよい。
【0055】
抗体を検出可能な部分とコンジュゲートするために、当技術分野で公知のいずれの方法を採用してもよく、Hunter,ら, Nature 144:945 (1962);David,ら, Biochemistry 13:1014 (1974);Pain,ら, J. Immunol. Meth. 40:219 (1981);およびNygren, J. Histochem. and Cytochem. 30:407 (1982)により記載された方法を含む。
【0056】
本発明の抗体はいずれの公知のアッセイ方法、例えば競合結合アッセイ、直接および間接サンドイッチアッセイおよび免疫沈降アッセイにおいても使用することができる(Zola, 「モノクローナル抗体:技術のマニュアル(Monoclonal Antibodies: A Manual of Techniques)」, pp.147-158 (CRC Press, Inc., 1987))。
【0057】
本発明の抗体はまた、in vivoイメージングにも有用であり、ここでは検出可能部分、例えば放射不透明剤または放射性同位体を用いて標識した抗体を、被験者に好ましくはその血流中に投与し、そして標識した抗体の宿主中の位置をアッセイする。このイメージング技術は悪性腫瘍の段階判定および治療に有用である。抗体を、核磁気共鳴、放射線学または他の当技術分野で知られる検出手段のいずれかにより宿主中で検出可能であるいずれの部分を用いて標識してもよい。
【0058】
本発明の抗体はまた、アフィニティ精製剤としても有用である。このプロセスにおいては、抗体を好適な支持体、例えばセファデックス(Sephadex)樹脂または濾紙上に当技術分野で周知の方法を用いて固定する。
【0059】
本発明の抗体はまた、それらの細胞中のIGF-I受容体機能の阻害に基づく生物学的研究の試薬としても有用である。
【0060】
治療上の応用については、本発明の抗体またはコンジュゲートを、製薬上許容される剤形で被験者に投与する。それらを、ボーラスとしてまたはある期間にわたる連続輸液により静脈内に、筋肉内、皮下、関節内、滑膜内、くも膜下腔内、経口、局所または吸入経路により投与することができる。抗体はまた、腫瘍内、腫瘍周囲、病変部内、または病変部周囲経路により投与して、局所ならびに全身治療効果を与えてもよい。好適な製薬上許容される担体、希釈剤、および賦形剤は当業者に周知でありかつ当業者が臨床症状に基づいて決定することができる。好適な担体、希釈剤、および/または賦形剤の例としては、(1)約1mg/ml〜25mg/mlヒト血清アルブミンを含有するダルベッコ(Dulbecco)リン酸緩衝化生理食塩水、pH約7.4、(2)0.9%生理食塩水(0.9%w/v NaCl)、および(3)5%(w/v)デキスロースが挙げられる。本発明の方法はin vitro、in vivo、またはex vivoで実施することができる。
【0061】
他の治療処置においては、本発明の抗体、抗体フラグメントまたはコンジュゲートを1以上のさらなる治療薬とともに同時投与する。好適な治療薬は、限定されるものでないが、細胞傷害または細胞分裂停止薬を含む。タキソールは、細胞傷害薬でもある好ましい治療薬である。
【0062】
癌治療薬は、癌細胞を殺滅するかまたはその増殖を制限する一方、宿主への障害を最小化することを求める薬剤である。従って、そのような薬剤は、癌細胞の健康な宿主細胞との特性の相違(例えば代謝、血管新生または細胞表面抗原提示)を利用することができる。腫瘍形態学の相違は可能性のある介入部位となる:例えば、第2の治療薬は抗体、例えば固形腫瘍内部の血管新生を遅延させてそれにより増殖速度を遅くするのに有用である抗VEGF抗体であってもよい。他の治療薬としては、限定されるものでないが、補助薬、例えば塩酸グラニセトロン、男性ホルモンインヒビター、例えば酢酸ロイプロリド、抗生物質、例えばドキソルビシン、抗女性ホルモン剤、例えばタモキシフェン、代謝拮抗剤、例えばインターフェロンα-2a、細胞傷害薬、例えばタキソール、酵素阻害剤、例えばrasファルネシルトランスフェラーゼ阻害剤、免疫調節物質、例えばアルデスロイキン、および窒素マスターバッチ誘導体、例えば塩酸メルファランなどが挙げられる。
【0063】
水溶液剤形で存在するとき(凍結乾燥されているのでなく)、抗体は典型的には約0.1mg/ml〜100mg/mlの濃度で製剤されるが、この範囲外の広い変化も許容される。疾患の治療に適当な抗体またはコンジュゲートの投与量は、先に規定した治療する疾患の型、疾患の重篤度と経過、抗体を予防用にまたは治療用に投与するかどうか、先の治療の経過、患者の臨床歴と抗体に対する応答、および主治医の熟慮に依存しうる。抗体を、好適なように、患者に1回でまたは一連の治療全体にわたって投与する。
【0064】
疾患の型と重篤度に依存して、約0.015〜15mg抗体/kg患者体重が、例えば1回以上の分離投与によりまたは連続輸液により患者に投与する最初の候補投与量である。数日以上にわたる反復投与については、症状に依存して、所望の病状抑制が起こるまで治療を繰返す。しかし他の投与体制も有用でありうるのであって、それらを排除するものではない。
【実施例】
【0065】
本発明をこれから次の実施例を参照して記載するが、これは説明のためだけであって、本発明を限定することを意図しない。
【0066】
実施例1:マウスEM164抗体
この最初の実施例においては、本発明のマウス抗体の完全な1次アミノ酸構造およびcDNA配列を、その結合特性および組換え型におけるその発現方法とともに開示する。従って、本発明の抗体とその調製法の全てのかつ完全な開示が提供され、免疫学分野の業者であれば、過剰な実験をすることなく、上記抗体を調製することができよう。
【0067】
A.抗IGF-I受容体モノクローナル抗体ハイブリドーマの作製
Y1251F突然変異をもつヒトIGF-I受容体を発現する細胞系統は高い数(1細胞当たり〜107)のIGF-I受容体を発現するので、この細胞系統を免疫感作に用いた。IGF-I受容体の細胞質ドメインにおけるY1251F突然変異は、トランスフォーメーションおよび抗アポトーシスシグナル伝達の損失をもたらしたが、IGF-I結合およびIGF-I刺激による分裂促進シグナル伝達に影響を与えなかった(O'Connor, R.ら, 1997, Mol. Cell. Biol., 17, 427-435;Miura, M.ら, 1995, J. Biol. Chem., 270, 22639-22644)。突然変異は他の点で抗体作製に影響を与えなかった、何故なら、この実施例の抗体は、Y1251Fと野生型受容体の両方に対して同等であるIGF-I受容体の細胞外ドメインと結合するからである。
【0068】
Y1251F突然変異をもつヒトIGF-I受容体を発現する細胞系統はIGF-I-受容体-欠損マウスの3T3様細胞から、Y1251F-突然変異ヒトIGF-I-受容体遺伝子と一緒にピューロマイシン耐性遺伝子によるトランスフェクションにより作製し、そしてピューロマイシン(2.5μg/mL)を用いて、および高いIGF-I受容体発現についてFACSソーティングにより選択した(Miura, M.ら, 1995, J. Biol. Chem., 270, 22639-22644)。高レベルのIGF-I受容体発現を有する細胞系統をさらに、ほとんどの細胞にとって毒性がある高濃度、例えば25μg/mLのピューロマイシンを用いて選択した。生存コロニーを拾い、高レベルのIGF-I受容体発現を提示するものを選択した。
【0069】
6月齢のCAF1/J雌性マウスを、第0日にY1251F突然変異したヒトIGF-I-受容体を過剰発現する細胞(5x105細胞、0.2mL PBS中に懸濁)を用いて腹腔内に免疫感作した。動物を0.2mL細胞懸濁液を用いて次のように追加免疫した:第2日、1x106細胞;第5日、2x106細胞;第7、9、12、および23日、1x107細胞。第26日にマウスを犠牲にしてその脾臓を取除いた。
【0070】
脾臓を2つの艶消しガラススライド間で磨砕して単一細胞懸濁液を得て、これをペニシリンとストレプトマイシンを含有する無血清RPMI培地(SFM)を用いて洗浄した。脾臓細胞ペレットを0.83%(w/v)塩化アンモニウム水溶液10mL中に、10分間氷上で再懸濁し、赤血球を溶解し、次いで無血清培地(SFM)を用いて洗浄した。脾臓細胞(1.2x108)を非分泌マウス骨髄腫細胞系統P3X63Ag8.653(ATCC、Rockville、MD;Cat.#CRL1580)からの骨髄腫細胞(4x107)とともにチューブ中にプールし、そして無血清RPMI-1640培地(SFM)を用いて洗浄した。上清を除去して細胞ペレットを残りの培地中に再懸濁した。チューブを37℃の水の入ったビーカー内に置いて、チューブを静かに振りながら、1.5mLのポリエチレングリコール溶液(50%(w/v)PEG、平均分子量1500を含有する75mM HEPES、pH 8)をゆっくりと0.5mL/分の滴下速度で加えた。1分間待った後に、10mLのSFMを次のように加えた:最初の1分間にわたり1mL、次の1分間にわたり2mL、そして第3の1分間にわたり7mL。次いでさらに10mLをゆっくりと1分間にわたり加えた。細胞を遠心分離によりペレット化し、SFM中で洗浄し、5%胎児ウシ血清(FBS)、ヒポキサンチン/アミノプテリン/チミジン(HAT)、ペニシリン、ストレプトマイシン、および10%ハイブリドーマクローニングサプリメント(HCS)を補充したRPMI-1640増殖培地中に再懸濁した。細胞を96ウエル平底組織培養プレート中に、1ウエル当たり200μL中の2x105脾臓細胞の割合で播いた。5〜7日後に、1ウエル当たり100μLを取除いて、ヒポキサンチン/チミジン(HT)および5%FBSを補充した増殖培地と置換えた。免疫感作およびハイブリドーマ産生に用いた一般条件は、J. LangoneおよびH. Vunakis(編, 「酵素学の方法(Methods in Enzymology)」, Vol. 121, 「免疫化学技術、第I部(Immunochemical Techniques, Part I)」;1986;Academic Press, Florida)ならびにE. HarlowおよびD. Lane(「抗体:研究室マニュアル(Antibodies: A Laboratory Manual)」; 1988; Cold Spring Harbor Laboratory Press, New York)に記載された通りである。他の免疫感作およびハイブリドーマ産生の技術も利用することができ、当業者は周知している。
【0071】
ハイブリドーマクローンから得た培養物上清を、ELISAにより、精製ヒトIGF-I受容体との結合について、ELISAとFACSにより、ヒトIGF-I受容体を過剰発現する細胞との特異的結合についておよびヒトインスリン受容体を過剰発現する細胞との結合の欠如について、以下に記載の通りスクリーニングした。ヒトIGF-I受容体を過剰発現する細胞に対する結合親和性がヒトインスリン受容体を過剰発現する細胞に対する結合親和性より高いクローンを拡大してサブクローニングした。サブクローンの培養上清を上記結合アッセイによってさらにスクリーニングした。この方法で、サブクローン3F1-C8-D7(EM164)を選択し、重および軽鎖遺伝子をクローニングし、そして下記の通り配列決定した。
【0072】
ハイブリドーマクローンから得た上清をIGF-I受容体との結合についてスクリーニングするのに用いるため、以下の方法によりヒトIGF-I受容体を単離した。組換えIGF-Iをビオチン化試薬、例えばスルホ-NHS-LC-ビオチン、スルホ-NHS-SS-ビオチン、またはNHS-PEO4-ビオチンを用いて改変することにより、ビオチン化IGF-Iを調製した。ビオチン化IGF-Iをストレプトアビジン-アガロースビーズ上に吸収させ、ヒト野生型またはY1251F突然変異体IGFRを過剰発現した細胞から得たライセートとともにインキュベートした。ビーズを2〜4M尿素および界面活性剤、例えばトリトンX-100またはオクチル-β-グルコシドを含有するバッファーを用いて洗浄しかつ溶出した。溶出したIGF-I受容体をPBSに対して透析し、SDS-PAGEにより還元条件のもとで純度を分析し、その結果はIGF-I受容体のαおよびβ鎖バンドの分子量である約135kDaおよび95kDaをそれぞれ示した。
【0073】
ハイブリドーマ上清の精製IGF-I受容体との結合を確認するために、Immulon-4HB ELISAプレート(Dynatech)を、pH 9.5の50mM CHESバッファー中に希釈した(100μL;4℃、一夜)精製ヒトIGF-I受容体サンプル(アフィニティ精製したサンプルの尿素/オクチル-β-グルコシド溶出液から透析により調製した)を用いてコーティングした。ウエルをブロッキングバッファー(10mg/mL BSAを含む、50mMトリス、150mM NaCl、pH 7.5、および0.1% tween-20を含有するTBS-Tバッファー)200μLを用いてブロックし、ハイブリドーマクローン(100μL;ブロッキングバッファーに希釈された)からの上清とともに約1〜12時間インキュベートし、TBS-Tバッファーを用いて洗浄し、そしてヤギ抗マウスIgG-Fc抗体-西洋わさびペルオキシダーゼ(HRP)コンジュゲート(100μL;0.8μg/mLを含むブロッキングバッファー;Jackson ImmunoResearch Laboratories)とともにインキュベートし、次いで洗浄しそしてABTS/H2O2基質(0.5mg/mL ABTS、0.03% H2O2を含む0.1Mクエン酸バッファー、pH 4.2)を用いて405nmにて検出した。典型的な3F1ハイブリドーマ サブクローンからの上清は約1.2吸収単位のシグナルを3分以内の発現で生じ、他のハイブリドーマクローンからの上清で得た0.0と対照的であった。このELISAに対する一般条件は、E. HarlowおよびD. Lane(「抗体の利用:研究室マニュアル(Using Antibodies: A Laboratory Manual)」;1999, Cold Spring Harbor Laboratory Press, New York)に記載の抗体結合および検出に対する標準ELISA条件と類似し、この標準条件を利用することもできる。
【0074】
ハイブリドーマ上清のヒトIGF-I受容体との特異的結合およびヒトインスリン受容体との非結合に対するスクリーニングを、ELISAを用いて、ヒトY1251F-IGF-I受容体過剰発現細胞系統およびヒトインスリン受容体過剰発現細胞系統について実施した。両方の細胞系統をIGF-I受容体欠損マウスの3T3様細胞から作製した。IGF-I受容体過剰発現細胞およびインスリン受容体過剰発現細胞を別々に組織培養フラスコから迅速トリプシン/EDTA処理により収穫し、10%FBSを含有する増殖培地中に懸濁し、遠心分離によりペレット化し、そしてPBSを用いて洗浄した。洗浄した細胞(100μL、約1〜3x106細胞/mL)を、植物性血球凝集素(PHA)(100μL、20μg PHA/mL)を用いてコーティングしたImmulon-2HBプレートのウエルに加え、遠心分離してPHAコーティングウエルに10分間接着させた。細胞が接着したウエルを指ではじいてPBSを除去し、次いで一夜37℃で乾燥した。ウエルをPBS中の5mg/mL BSA溶液を用いて1時間37℃でブロックし、次いで静かにPBSを用いて洗浄した。次いでハイブリドーマクローン(100μL;ブロッキングバッファー中に希釈)からの上清のアリコートをIGF-I-受容体過剰発現細胞を含有するウエルおよびインスリン受容体過剰発現細胞を含有するウエルに加え、そして常温で1時間インキュベートした。ウエルをPBSを用いて洗浄し、ヤギ抗マウスIgG-Fc抗体-西洋わさびペルオキシダーゼコンジュゲート(100μL;ブロッキングバッファー中に0.8μg/mLを含む)とともに1時間インキュベートし、次いで洗浄し、そしてABTS/H2O2基質を用いて検出した。典型的な3F1ハイブリドーマサブクローンからの上清はIGF-I受容体を過剰発現する細胞とインキュベーションすると、12分以内の発現で0.88吸収単位のシグナルを生じ、ヒトインスリン受容体を過剰発現する細胞とのインキュベーションで得た0.22吸収単位の値とは対照的であった。
【0075】
ハイブリドーマをIntegra-CL350フラスコ(Integra Biosciences, Maryland)中で製造業者の明細書に従って増殖し、精製EM164抗体を得た。ほぼ0.5〜1mg/mL抗体の収率が、抗体標準を用いるELISAおよびSDS-PAGE/クーマシーブルー染色による定量に基づいて、Integraフラスコから収穫した上清から得られた。抗体をアフィニティクロマトグラフィにより、プロテインA-アガロースビーズカラム上に標準精製条件のもとで供給して精製し、3M NaClを含有する100mMトリスバッファー、pH 8.9で洗浄し、次いで150mM NaClを含有する100 mM酢酸溶液に溶出した。抗体を含有する溶出した画分を冷2M K2HPO4溶液により中和して、PBS中で4℃にて透析した。抗体の濃度は、280nmで測定することにより決定した(吸光係数=1.4mg-1mLcm-1)。精製抗体サンプルを還元条件下のSDS-PAGEおよびクーマシーブルー染色により分析し、その結果は、それぞれ約55kDaおよび25kDaである抗体の重および軽鎖のバンドだけを示した。精製抗体のイソ型はκ軽鎖をもつIgG1であった。
【0076】
B.EM164抗体の結合特性
精製EM164抗体の特異的結合を、蛍光活性化細胞選別(フローサイトメトリー)(FACS)によりIGF-I受容体を過剰発現する細胞を用いておよびヒトインスリン受容体を過剰発現する細胞を用いて実証した(図1)。100μL冷FACSバッファー(1mg/mL BSAを含むダルベッコ(Dulbecco's)MEM培地)中のEM164抗体(50-100nM)のインキュベーションを、IGF-I受容体を過剰発現する細胞を用いておよびインスリン受容体を過剰発現する細胞を用いて(2x105細胞/mL)、丸底96ウエルプレート中で1時間実施した。細胞を遠心分離によりペレット化し、冷FACSバッファーを用いて静かに指ではじくことにより洗浄し、次いでヤギ抗マウスIgG抗体-FITCコンジュゲート(100μL;10μg/mL FACSバッファー)とともに氷上で1時間インキュベートした。細胞をペレット化し、洗浄し、そしてPBS中の1%ホルムアルデヒド溶液120μLに再懸濁した。プレートをFACS Caliburリーダー(BD Biosciences)を用いて分析した。
【0077】
IGF-I受容体を過剰発現する細胞をEM164抗体とインキュベートすると強い蛍光シフトが得られ、インスリン受容体を過剰発現する細胞をEM164抗体とインキュベートしたときに得た非有意なシフトとは対照的であった(図1)。この結果はEM164抗体のIGF-I受容体との結合が選択的であって、インスリン受容体とは結合しないことを示した。対照抗体である、抗IGF-I受容体抗体1H7(Santa Cruz Biotechnology)および抗インスリン受容体α抗体(BD Pharmingen Laboratories)は、それぞれIGF-I受容体およびインスリン受容体を過剰発現する細胞とインキュベートすると蛍光シフトを生じた(図1)。強い蛍光シフトはまた、IGF-I受容体を発現するEM164抗体およびヒト乳癌MCF-7細胞(Dufourny, B.ら, 1997, J. Biol. Chem., 272, 31163-31171)を用いるFACSアッセイによっても観察され、この結果はEM164抗体がヒト腫瘍細胞の表面上のヒトIGF-I受容体と結合することを示した。
【0078】
EM164抗体のヒトIGF-I受容体との結合に対する解離定数(Kd)を、複数濃度における抗体の結合のELISA滴定により、直接コーティングしたIGF-I受容体(上記のとおり、ビオチン化IGF-Iを用いてアフィニティ精製された)または間接的に捕獲したビオチン化IGF-I受容体により決定した。ビオチン化IGF-I受容体を、IGF-I受容体を過剰発現する細胞からPEO-マレイミド-ビオチン試薬(Pierce、Molecular Biosciences)を用いて界面活性剤可溶化ライセートのビオチン化により調製し、これをNHS-アガロースビーズ上に固定した抗IGF-I受容体β鎖抗体を用いてアフィニティ精製し、NP-40界面活性剤を含有するバッファー中の2〜4M尿素溶液を用いて溶出し、そしてPBSに透析した。
【0079】
EM164抗体のビオチン化IGF-I受容体との結合に対するKdの測定は、Immulon-2HBプレートを、炭酸バッファー(150mM炭酸ナトリウム、350mM炭酸水素ナトリウム)中の1μg/mLストレプトアビジン溶液100μLを用いて4℃で一夜、コーティングして実施した。ストレプトアビジンでコーティングしたウエルを、ブロッキングバッファー(10mg/mL BSAを含むTBS-Tバッファー)200μLを用いてブロックし、TBS-Tバッファーを用いて洗浄し、そしてビオチン化IGF-I受容体(10〜100ng)とともに4時間常温でインキュベートした。間接的に捕獲したビオチン化IGF-I受容体を含有するウエルを次いで洗浄し、そしてEM164抗体とブロッキングバッファー中で複数の濃度(5.1x10-13M〜200nM)において2時間、常温でインキュベートし、次いで一夜4℃にてインキュベートした。ウエルを次にTBS-Tバッファーを用いて洗浄し、そしてヤギ抗マウスIgGH+L抗体-西洋わさびペルオキシダーゼコンジュゲート(100μL;0.5μg/mLを含むブロッキングバッファー)とインキュベートし、次いで洗浄し、そしてABTS/H2O2基質を用いて405nmにて検出した。Kd値を1つの部位との結合に対する非線形回帰により計算した。
【0080】
同様な結合滴定を、E. HarlowおよびD. Lane(「抗体の利用:研究室マニュアル(Using Antibodies: A Laboratory Manual)」;1999, Cold Spring Harbor Laboratory Press, New York)に記載のように、抗体のパパイン消化により調製したEM164抗体のFabフラグメントを用いて実施した。
【0081】
EM164抗体のビオチン化ヒトIGF-I受容体との結合に対する結合滴定曲線から、0.1nMのKd値を得た(図2)。EM164抗体のFabフラグメントもヒトIGF-I受容体と0.3nMのKd値で非常に強く結合し、この結果は、EM164抗体のIGF-I受容体に対するモノマー結合も非常に強いことを示した。
【0082】
EM164抗体によるIGF-I受容体の結合に対する解離定数のこの極めて低い値は一部は非常に遅いkoff速度によるものであって、これは1〜2日の長い洗浄後に観察される固定されたIGF-I受容体と結合した抗体の強い結合シグナルにより立証された。
【0083】
EM164抗体はIGF-I受容体の免疫沈降に使用することができ、これはヒト乳癌MCF-7細胞の界面活性剤可溶化ライセートのプロテインG-アガロースビーズ上に固定されたEM164抗体(Pierce Chemical Company)とのインキュベーションにより実証される。EM164抗体免疫沈降のウェスタンブロットを、ウサギポリクローナル抗IGF-I受容体β鎖(C-末端)抗体(Santa Cruz Biotechnology)およびヤギ抗ウサギIgG抗体西洋わさびペルオキシダーゼコンジュゲートを用いて処理し、次いで洗浄し、そして増強化学発光(ECL)検出により検出した。MCF-7細胞からのEM164免疫沈降物のウェスタンブロットは、IGF-I受容体のβ鎖に対応するバンドを約95kDaに、そしてプロIGF-I受容体に対応するバンドを約220kDaに表した。同様な免疫沈降をEM164抗体の結合の種特異性を調べるために他の細胞型に対して実施した。EM164抗体はcos-7細胞(アフリカミドリザル)からのIGF-I受容体とも結合したが、3T3細胞(マウス)、CHO細胞(チャイニーズハムスター)またはヤギ線維芽細胞(ヤギ)のIGF-I受容体と結合しなかった。EM164抗体はMCF-7細胞からのライセートのウェスタンブロット中のSDS変性ヒトIGF-I受容体を検出しなかった。この結果は、EM164抗体が自然の、非変性ヒトIGF-I受容体のコンフォメーションのエピトープと結合することを示した。
【0084】
EM164抗体の結合ドメインを、16量体C末端片(残基704〜719)と融合したL1およびL2ドメインにフランキングするシステインリッチドメイン(残基1〜468)を含みかつC末端エピトープタグで終わる末端切断α鎖構築物を用いてさらに特徴づけた。残基469〜703を欠如するこのより小さいIGF-I受容体は、IGF-Iと結合するが、未変性の全長IGF-I受容体と比較すると強さが劣ると報じられている(Molina, L.ら, 2000, FEBS Letters, 467, 226-230;Kristensen, C.ら, 1999, J. Biol. Chem., 274, 37251-37356)。そこで、残基704〜719であるC末端片と融合した残基1〜468を含み、かつC末端mycエピトープタグがフランキングする末端切断IGF-I受容体α鎖構築物を調製した。この構築物を発現する安定な細胞系統であって、かつまたこの構築物をヒト胚腎293T細胞において一過的に発現する細胞系統を構築した。EM164抗体のこの末端切断IGF-I受容体α鎖構築物との強い結合が観察された。試験した2つの抗体のうち、IR3(Calbiochem)もこの末端切断α鎖と結合したが、1H7抗体(Santa Cruz Biotechnology)は結合せず、これはEM164抗体のエピトープが明らかに1H7抗体のそれとは別であることを示した。
【0085】
C.EM164抗体による、IGF-IのMCF-7細胞との結合の阻害
IGF-Iのヒト乳癌MCF-7細胞との結合はEM164抗体により阻害された(図3)。MCF-7細胞を5μg/mL EM164抗体とともにまたはEM164抗体なしで2時間、無血清培地においてインキュベートし、次いで50ng/mLビオチン化IGF-Iとともに20分間、37℃にてインキュベートした。次いで細胞を2回、無血清培地を用いて洗浄して非結合ビオチン-IGF-Iを除去し、次いで1%NP-40およびプロテアーゼ阻害剤を含有する50mM HEPES、pH 7.4中に溶解した。Immulon-2HB ELISAプレートをマウスモノクローナル抗IGF-I受容体β鎖抗体を用いてコーティングし、これを用いてライセートからIGF-I受容体および結合したビオチン-IGF-Iを捕獲した。コーティングされた抗体のIGF-I受容体β鎖細胞質C末端ドメインとの結合は、ビオチン-IGF-IのIGF-I受容体細胞外ドメインとの結合を妨げなかった。ウエルを洗浄し、ストレプトアビジン-西洋わさびペルオキシダーゼコンジュゲートとともにインキュベートし、再び洗浄し、次いでABTS/H2O2基質を用いて検出した。5μg/mL EM164抗体によるMCF-7細胞に対するIGF-I結合の阻害は本質的に定量的であって、ビオチン-IGF-Iを欠く対照を用いて得たELISAバックグラウンドのそれとほとんど同等であった。
【0086】
EM164抗体によるIGF-IのMCF-7細胞との結合の阻害に対する上記アッセイに加えて、次のアッセイはEM164抗体がMCF-7細胞に結合したIGF-Iと極めて効果的に置換わることを実証し、これは生理学的条件のもとで拮抗性抗IGF-I受容体抗体が結合した内因生理学的リガンド(例えば、IGF-IまたはIGF-II)と置換わるために所望されることである。このIGF-I置換えアッセイにおいて、12ウエルプレート中で増殖したMCF-7細胞を血清飢餓させ、次いでビオチン化IGF-I(20〜50ng/mL)とともに無血清培地中で37℃(または4℃)にて1〜2時間インキュベートした。結合したビオチン化IGF-Iをもつ細胞を、次いでEM164抗体または対照抗体(10-100μg/mL)を用いて37℃(または4℃)で30分間〜4時間処理した。次いで細胞をPBSを用いて洗浄し、1% NP-40を含有する溶解バッファー中で4℃で溶解した。ELISAを上記の通り実施し、ライセートからIGF-I受容体を捕獲し、次いで受容体と結合したビオチン化IGF-Iをストレプトアビジン-西洋わさびペルオキシダーゼコンジュゲートを用いて検出した。このELISAは、EM164抗体が細胞に前結合したビオチン化IGF-Iと37℃にてほぼ完全に(30分間以内に90%および4時間以内に〜100%)かつ4℃にて2時間で約50%だけ置換わりうることを実証した。他の実験においては、NCI-H838肺癌細胞をビオチン-IGF-Iとともにインキュベートし、次いで洗浄し、そしてEM164抗体とともに4℃にて2時間インキュベートして、結合したビオチン-IGF-Iの80%減少を得た。従ってEM164抗体は、癌細胞から前結合したIGF-Iを追い出す点で極めて有効であり、これは結合した内因性生理学的リガンドを追い出すことによるIGF-I受容体の拮抗作用として治療上重要でありうる。
【0087】
抗IGF-I受容体β鎖抗体(Santa Cruz Biotechnology;sc-713)を用いるウェスタンブロット分析に基づくと、MCF-7細胞のEM164抗体との4℃で2時間(または37℃で30分間)のインキュベーションはIGF-I受容体の有意な下方調節を生じなかったが、EM164抗体との37℃で2時間のさらに長いインキュベーションはIGF-I受容体の25%下方調節を生じた。従って、これらの短時間実験における4℃および37℃にてのEM164抗体によるIGF-Iの結合の阻害および結合したIGF-Iの置換えは、EM164抗体の結合による受容体の下方調節によって説明することができない。IGF-IのIGF-I受容体との結合の強力な阻害およびEM164抗体による前結合したIGF-Iの置換えの機構は、結合部位の共有を介してまたは立体閉塞を介してまたはアロ立体効果を介しての結合に対する競合であると思われる。
【0088】
D.EM164抗体による、IGF-I受容体が媒介する細胞シグナル伝達の阻害
乳癌MCF-7細胞および骨肉腫SaOS-2細胞のEM164抗体による処理は細胞内IGF-I受容体シグナル伝達をほとんど完全に阻害した。これをIGF-I受容体自己リン酸化の阻害によりおよびその下流のエフェクター、例えばインスリン受容体基質-1(IRS-1)、AktおよびErk1/2(図4〜6)のリン酸化の阻害により示す。
【0089】
図4においては、MCF-7細胞を12ウエルプレート中で通常培地で3日間増殖し、次いで20μg/mL EM164抗体(または抗B4対照抗体)を用いて無血清培地で3時間処理し、次に50ng/mL IGF-Iを用いて20分間37℃で刺激した。次いで細胞を、プロテアーゼおよびホスファターゼインヒビターを含有する氷冷溶解バッファー(50mM HEPESバッファー、pH 7.4、1% NP-40、1mMオルトバナジン酸ナトリウム、100mMフッ化ナトリウム、10mMピロリン酸ナトリウム、2.5mM EDTA、10μMロイペプチン、5μMペプスタチン、1mM PMSF、5mMベンズアミジン、および5μg/mLアプロチニン)中に溶解した。ELISAプレートを抗IGF-I受容体β鎖C末端モノクローナル抗体TC123を用いてプレコートし、そしてライセートサンプルとともに5時間常温でインキュベートし、IGF-I受容体を捕獲した。次いで捕獲したIGF-I受容体を含有するウエルを洗浄し、ビオチン化抗ホスホチロシン抗体(PY20;0.25μg/mL;BD Transduction Laboratories)とともに30分間インキュベートし、次いで洗浄し、ストレプトアビジン-西洋わさびペルオキシダーゼコンジュゲート(0.8μg/mL)とともに30分間インキュベートした。ウエルを洗浄し、ABTS/H2O2基質を用いて検出した。対照抗B4抗体を使用すると、IGF-I受容体のIGF-I刺激自己リン酸化の無阻害を示した。対照的に、EM164抗体を用いて処理するとIGF-I受容体のIGF-I刺激自己リン酸化の完全な阻害を得た(図4)。
【0090】
インスリン受容体基質-1(IRS-1)のリン酸化の阻害を実証するため、固定した抗IRS-1抗体を使ってライセートからIRS-1を捕獲するELISAを用い、次いでホスホリル化IRS-1と結合するホスファチジルイノシトール-3-キナーゼ(PI3キナーゼ)に随伴するp85サブユニットを測定した(Jackson, J. G.ら, 1998, J. Biol. Chem., 273, 9994-10003)。図5において、MCF-7細胞を5μg/mL抗体(EM164またはIR3)とともに無血清培地において2時間処理し、次いで50ng/mL IGF-Iを用いて10分間37℃にて刺激した。抗IRS-1抗体(ウサギポリクローナル;Upstate Biotechnology)を間接的にELISAプレート上にコーティングしたヤギ抗ウサギIgG抗体とのインキュベーションにより捕獲し、次いでこれを利用して細胞ライセートからIRS-1を4℃の一夜インキュベーションにより捕獲した。次いでウエルをマウスモノクローナル抗p85-PI3キナーゼ抗体(Upstate Biotechnology)と4時間インキュベートし、次に、ヤギ抗マウス-IgG-抗体-HRPコンジュゲートにより30分間処理した。ウエルを次いで洗浄し、ABTS/H2O2基質を用いて検出した(図5)。図5に示すように、EM164抗体によるIGF-I-刺激IRS-1リン酸化の阻害はIR3抗体より効果的であり、かつEM164抗体はIGF-Iの不在の細胞とインキュベートしたとき、IRS-1リン酸化に対する作動活性を示さなかった。
【0091】
他の下流エフェクター、例えばAktおよびErk1/2の活性化も、SaOS-2細胞(図6)およびMCF-7細胞においてEM164抗体により用量に依存する様式で阻害され、これはライセートのウェスタンブロットおよびリン酸化特異的抗体(ウサギポリクローナル抗-ホスホ-Ser473Aktポリクローナルおよび抗ホスホ-ERK1/2抗体;Cell Signaling Technology)を用いて示した通りである。汎ERK抗体は全てのレーンで等しいタンパク質負荷を示した(図6)。EM164抗体によるSaOS-2細胞の処理は、Erk1/2のEGF-刺激リン酸化を阻害せず、従ってIGF-I受容体シグナル伝達経路のEM164抗体による阻害の特異性を実証した。
【0092】
E.EM164抗体による、ヒト腫瘍細胞のIGF-I-、IGF-II-および血清-刺激増殖および生存の阻害
複数のヒト腫瘍細胞系統を、無血清条件でIGF-Iに対するそれらの増殖および生存応答について試験した。これらの細胞系統をIGF-I、IGF-II、または血清の存在のもとでEM164抗体を用いて処理し、それらの増殖および生存応答をMTTアッセイを用いて2〜4日後に測定した。ほぼ1500細胞を96ウエルプレートで通常培地にまいて、翌日、それを無血清培地(トランスフェリンおよびBSAを補充した無血清RPMI培地、またはDufourny, B.ら, 1997, J. Biol. Chem., 272, 31163-31171が規定したフェノールレッドを含まない培地のいずれか)と置換えた。無血清培地における増殖の1日後に、細胞を約75μLの10μg/mL抗体とともに30分間〜3時間インキュベートし、次いで25μLのIGF-I(またはIGF-IIまたは血清)溶液を加えて、10ng/mL IGF-I、または20ng/mL IGF-II、または0.04〜10%血清の最終濃度を得た。複数の実験において、細胞を最初IGF-Iを用いて15分間刺激した後にEM164抗体を加えるか、またはIGF-IとEM164抗体の両方を一緒に加えた。次いで細胞をさらに2〜3日増殖させた。次いでMTT(3-(4,5)-ジメチルチアゾール-2-イル)-2,5-ジフェニルテトラゾリウムブロミド)のPBS中の5mg/mL溶液、25μLを加え、細胞をインキュベーターに戻して2〜3時間置いた。次いで培地を除去して、100μL DMSOと置き換え、混合し、そしてプレートの吸収を545nmにて測定した。複数のヒト腫瘍細胞系統はIGF-IまたはIGF-IIまたは血清を加えると増殖および生存の応答を示し、その応答はEM164抗体により有意に阻害され、抗体をIGF-Iの前に加えたか、またはIGF-Iを抗体の前に加えたか、またはIGF-Iと抗体の両方を一緒に加えたかどうかとは無関係であった(表1)。
【表1】

【0093】
EM164抗体は、乳癌MCF-7細胞(図7および8)のIGF-Iまたは血清刺激増殖および生存を強く阻害した。別の実験において、EM164抗体はMCF-7細胞のIGF-II刺激増殖および生存を強く阻害した。市販の抗体、例えばIR3抗体を用いる先の報告は、図7でIR3および1H7抗体に対して確認したように、MCF-7細胞の血清刺激増殖および生存に弱い阻害だけを示した(Cullen, K. J.ら, 1990, Cancer Res., 50, 48-53)。対照的に、EM164抗体はMCF-7細胞の血清またはIGF刺激増殖の強力な阻害剤であった。図8に示したように、EM164抗体は広範囲の血清濃度(0.04〜10%血清)にわたってMCF-7細胞の増殖および生存を等しく有効に阻害した。
【0094】
EM164抗体によるMCF-7細胞の増殖阻害を細胞数の計測により測定した。すなわち、12ウエルプレートにおいて、約7500の細胞を、10μg/mL EM164抗体の存在または不在のもとで10%FBSを含むRPMI培地中にまいた。増殖の5日後に、無処理の対照サンプルの細胞数は20.5 x 104細胞であり、EM164抗体処理サンプルのわずか1.7 x 104細胞の細胞数と対照的であった。EM164抗体による処理は、MCF-7細胞の増殖を5日間に約12倍だけ阻害した。EM164抗体によるこの阻害は、報じられたMCF-7細胞に対する6日間アッセイでのIR3抗体を用いる2.5倍の阻害(Rohlik, Q. T.ら, 1987, Biochem. Biophys. Res. Commun., 149, 276-281)より有意に大きかった。
【0095】
非小細胞肺癌系統NCI-H838のIGF-Iおよび血清刺激増殖および生存もまた、EM164抗体により、対照抗B4抗体と比較して強く阻害された(図9)。無血清培地中のEM164抗体による処理は、NCI-H838とMCF-7細胞の両方に対して、無処理サンプルより小さいシグナルを産生したが、それは恐らく、EM164抗体はこれらの細胞の自己分泌およびパラ分泌IGF-IおよびIGF-II刺激も阻害したからであろう(図7および9)。HT29大腸癌細胞のコロニーサイズも、EM164抗体により処理すると非常に減少した。
【0096】
従ってEM164抗体は、腫瘍細胞、例えばMCF-7細胞およびNCI-H838細胞の血清刺激増殖を80%以上阻害することでその有効性が公知の全ての抗IGF-I受容体抗体のなかでもユニークな存在である。
【0097】
EM164抗体は細胞周期のG0/G1期における増殖停止を引起し、IGF-Iの分裂促進効果を無効にした。細胞周期分析について、MCF-7細胞をIGF-I(20ng/mL)を用いてEM164(μg/mL)の存在または不在のもとで1日間処理し、次いでヨウ化プロピジウム染色とフローサイトメトリーにより分析した。図25に示すように、EM164不在のもとでIGF-I刺激に応答する細胞のサイクリング(細胞の41%がS期そして50%がG0/G1期)が、EM164処理細胞においては抑制された(細胞わずかに9%がS期そして77%がG0/G1期)。
【0098】
細胞増殖の阻害に加え、EM164抗体処理は細胞のアポトーシスをもたらした。アポトーシスの測定については、EM164の存在または不在のもとで1日間IGF-Iまたは血清とともにインキュベートしたNCI-H838肺癌細胞において、カスパーゼによるサイトケラチンCK18タンパク質の切断を測定した(図26)。EM164の不在のもとで、IGF-Iまたは血清を加えると、無IGF-I対照と比較してカスパーゼ切断されたCK18シグナルが低下し、IGF-Iおよび血清はカスパーゼの活性を阻止することを示した。EM164を用いて処理するとIGF-Iおよび血清の抗アポトーシス効果を抑制し、それはEM164存在時の切断されたCK18レベルがEM164不在時のそれより大きいことにより示された(図26)。
【0099】
F.他の細胞毒性および細胞分裂停止作用薬と併用したEM164抗体による、ヒト腫瘍細胞の増殖および生存の相乗的阻害
EM164抗体のタキソールとの併用投与は、非小細胞肺癌Calu6細胞の増殖および生存に対してタキソール単独より有意に阻害性があった。同様に、EM164抗体のカンプトセシンとの併用は、大腸癌HT29細胞の増殖および生存に対してカンプトセシン単独より有意に阻害性があった。EM164抗体単独では有機の化学毒性薬と同じような毒性は期待されないので、EM164抗体の主に細胞分裂停止効果と化学毒性薬の細胞傷害効果の間の相乗作用は、臨床環境における併用癌治療法に非常に効果的でありうる。
【0100】
EM164抗体の抗EGF受容体抗体(KS77)との併用効果は、複数の腫瘍細胞系統、例えばHT-3細胞、RD細胞、MCF-7細胞、およびA431細胞の増殖および生存に対して、EM164抗体またはKS77抗体単独より有意に高い阻害性があった。従って、2種の増殖因子受容体、例えばIGF-I受容体およびEGF受容体に対する中和化抗体を併用することの相乗効果はまた、臨床癌治療において有用でありうる。
【0101】
EM164抗体の細胞傷害薬とのコンジュゲートはまた、IGF-I受容体を過剰発現する腫瘍に標的を定めた細胞傷害薬の送達において貴重である。EM164抗体の放射標識または他の標識とのコンジュゲートは、IGF-I受容体を過剰発現する腫瘍の治療およびイメージングに利用することができる。
【0102】
G.単一作用薬としてまたは抗癌薬と併用する場合、免疫不全マウスのヒト癌異種移植片におけるEM164処理の効果
ヒト非小細胞肺癌Calu-6異種移植片を免疫不全マウス中に、1x107 Calu-6細胞の皮下注射により確立した。図10に示すように、確立された100mm3 Calu-6異種移植片を含有するこれらのマウスを、1処理グループ当たり5匹のマウスを用いて、EM164抗体単独(0.8mg/マウスの6注射、静脈内、毎週2回)またはタキソール単独(タキソールの5注射、腹腔内、毎週2回;15mg/kg)、またはタキソールとEM164抗体処理の併用、またはPBS単独(200μL/マウス、6注射、毎週2回、静脈)によって処理した。腫瘍の増殖は、EM164抗体処理により、PBS対照と比較して有意に遅くなった。EM164抗体は、マウスの体重測定に基づいて毒性が観察されなかった。タキソール単独処理は14日まで有効であったが、その後腫瘍は増殖を回復し始めた。しかしタキソールとEM164抗体の併用により処理したグループでは、タキソール単独で治療したグループと比較して腫瘍の増殖が有意に遅くなった。
【0103】
ヒト膵癌異種移植片を、5週齢雌性SCID/ICRマウス(Taconic)に107 BxPC-3細胞を含むPBSを皮下注射することにより確立した(第0日)。80mm3の確立された腫瘍を抱くマウスを、次いでEM164単独(0.8mg/マウスの13回注射;側方尾静脈の静脈内、第12、16、19、23、26、29、36、43、50、54、58、61および64日に)、ゲムシタビン単独(150mg/kg/マウスの2回注射;腹腔内、第12および19日に)、上記スケジュールによるゲムシタビンとEM164の併用、PBS単独、および対照抗体単独(EM164と同じスケジュール)によって、5処理グループのそれぞれに5匹のマウスを用いて、処理した。図27に示したように、EM164単独、またはゲムシタビンとの併用による処理は、最初に、EM164処理グループ中の5動物の4動物および併用処理グループの全動物に腫瘍異種移植片の全退行が生じた。1以上の動物の測定しうる腫瘍再増殖は、EM164グループにおいて第43日に、そして併用治療グループにおいて第68日に見られ、対照処理グループと比較して第74日に有意に小さい平均腫瘍容積を生じただけであった(それぞれP=0.029および0.002;ツー・テイルドT検定(two-tailed T-test);図27)。他の研究において、EM164抗体処理(単独または抗EGF受容体抗体と併用;腹腔内注射)は確立されたマウス中のBxPC-3異種移植片の増殖を阻害した。
【0104】
H.EM164抗体の軽および重鎖のクローニングと配列決定
EM164ハイブリドーマ細胞から得た全RNAを精製した。逆転写酵素反応を、4〜5μg全RNAおよびオリゴdTもしくは無作為ヘキサマープライマーのいずれかを用いて実施した。
【0105】
PCR反応をCoら(J. Immunol., 148, 1149-1154 (1992))が記載したRACE法およびWangら(J. Immunol. Methods, 233, 167-177 (2000))が記載した変性プライマーを用いて実施した。RACE PCR法は、第1鎖cDNAの3'末端にポリGテーリングを加える中間ステップを必要とした。RT反応物をQianeasy(Qiagen)カラムを用いて精製し、50μl 1 X NEBバッファー4で溶出した。dGテーリング反応はその溶出物に、0.25mM CoCl2、1mM dGTP、および5単位の末端トランスフェラーゼ(NEB)を含む1 X NEBバッファー4を用いて実施した。その混合物を37℃にて30分間インキュベートし、次いで反応物(10μl)の1/5を直接PCR反応に加えてテンプレートDNAとして利用した。
【0106】
RACEと変性PCR反応は、プライマーとテンプレートの相違を除くと、同じ反応であった。末端トランスフェラーゼ反応物をRACE PCRテンプレートに直接使用する一方、RT反応混合物を変性PCR反応に直接使用した。
【0107】
RACEおよび変性PCR反応において、同じ3'軽鎖プライマー:
HindKL - tatagagctcaagcttggatggtgggaagatggatacagttggtgc(配列番号14)、
および3’重鎖プライマー:
Bgl2IgG1 - ggaagatctatagacagatgggggtgtcgttttggc(配列番号15)
を使用した。
【0108】
RACE PCRにおいて、軽および重鎖の両方に対して、1つのポリC5'プライマー
:EcoPolyC - TATATCTAGAATTCCCCCCCCCCCCCCCCC(配列番号16)
を用いる一方、縮重5'末端PCRプライマーは、軽鎖に対して:
Sac1MK - GGGAGCTCGAYATTGTGMTSACMCARWCTMCA(配列番号17)、
および重鎖に対して:
EcoR1MH1 - CTTCCGGAATTCSARGTNMAGCTGSAGSAGTC(配列番号18)と
EcoR1MH2 - CTTCCGGAATTCSARGTNMAGCTGSAGSAGTCWGG(配列番号19)
の等しい混合物であった。
【0109】
上記プライマー配列において、混合塩基は次のとおり定義した:H=A+T+C、S=g+C、Y=C+T、K=G+T、M=A+C、R=A+g、W=A+T、V=A+C+G。
【0110】
PCR反応を、次のプログラムを用いて実施した:1)94℃3分間、2)94℃15秒間、3)45℃1分間、4)72℃2分間、5)ステップ2)に戻るサイクリング29回、および6)72℃10分間の最終伸長による完了。
【0111】
PCR産物を、PCRプライマーにより作製した制限酵素切断部位を利用してpBluescript II SK+(Stratagene)中にクローニングした。
【0112】
複数の個々の軽および重鎖クローンを、通常の方法により配列決定して同定し、起こりうるポリメラーゼが起こす配列誤差を回避した(図12および13)。Chotia正準分類定義を用いて、3つの軽鎖および重鎖CDRを同定した(図12〜14)。
【0113】
NCBI IgBlastデータベースは、抗IGF-I受容体抗体軽鎖可変域はマウスIgVk Cr1生殖系列遺伝子由来である一方、重鎖可変域は恐らくIgVh J558.c生殖系列遺伝子由来であることを示した(図15)。
【0114】
マウスEM164抗体のタンパク質配列決定を実施して図12および13に示した配列を確かめた。精製EM164抗体の軽および重鎖タンパク質をゲル(SDS-PAGE、還元条件)からPVDF膜に移し、PVDF膜から切除し、タンパク質配列決定により分析した。軽鎖のN末端配列をEdman配列決定によりDVLMTQTPLS(配列番号20)であると決定し、これはEM164ハイブリドーマから得たクローニングした軽鎖遺伝子のN末端配列と整合する。
【0115】
重鎖のN末端はEdmanタンパク質配列決定に対してブロックされていることがわかった。質量1129.5(M+H+、モノアイソトープ)の重鎖のトリプシン消化ペプチドフラグメントをPSD(post-source decay)を経由してフラグメント化し、その配列がGRPDYYGSSK(配列番号21)であると決定した。質量2664.2(M+H+、モノアイソトープ)の重鎖の他のトリプシン消化ペプチドフラグメントもPSD(post-source decay)を経由してフラグメント化し、その配列がSSSTAYMQLSSLTSEDSAVYYFAR(配列番号22)であると同定した。これらの配列の両方は、EM164ハイブリドーマから得たクローニングした重鎖遺伝子のCDR3およびフレームワーク3(FR3)の配列と完全に整合する。
【0116】
I.EM164抗体の組換え発現
軽および重鎖の一対の配列を単一の哺乳類動物発現ベクター中にクローニングした(図16)。ヒト可変配列用のPCRプライマーにより、pBluescriptIIクローニングベクター中にヒトシグナル配列との結合を可能にする制限酵素切断部位を作製しておく一方、可変配列を哺乳類動物発現プラスミド中に、EcoRIとBsiWIまたはHindIIIとApaI部位を軽鎖または重鎖用にそれぞれ用いてクローニングした(図16)。軽鎖可変配列をインフレームでヒトIgK定常域にクローニングし、そして重鎖可変配列をヒトIgγ1定常域配列中にクローニングした。最終発現プラスミドにおいて、ヒトCMVプロモーターは軽および重鎖両方のcDNA配列の発現を駆動した。組換えマウスEM164抗体の発現と精製は当技術分野で周知の方法によって進めた。
【0117】
実施例2:EM164抗体のヒト化バージョン
治療または診断薬として適当なヒト化バージョンを提供するためのEM164抗体の再表面形成は、全般的に米国特許第5,639,641号に開示された原理と方法に従い、次の通り進めた。
【0118】
A.表面予測
解析された構造をもつ1セットの抗体に対する可変域残基の溶媒アクセシビリティを用いてマウス抗IGF-I受容体抗体(EM164)可変域の表面残基を予測した。1セットの127種のユニークな抗体構造ファイル(表2)に対するアミノ酸溶媒アクセシビリティを、MCソフトウエアパッケージ(Pedersenら, 1994, J. Mol. Biol., 235, 959-973)を用いて計算した。127種の構造のセットから10種の最も類似した軽鎖および重鎖アミノ酸配列を配列アラインメントにより決定した。それぞれの可変域残基に対する平均溶媒アクセシビリティを計算し、そして30%を超える平均アクセシビリティをもつ位置を表面残基とみなした。25%〜35%の平均アクセシビリティをもつ位置をさらに、2つの同一フランキング残基をもつ構造だけに対して個々の残基アクセシビリティを計算することにより試験した。
【表2】
【0119】
B.分子モデル
マウスEM164の分子モデルを、Oxford Molecular社ソフトウエアパッケージAbMを用いて作製した。抗体フレームワークは、最も類似したアミノ酸配列をもつ抗体に対する構造ファイルから、すなわち軽鎖に対して2jelからおよび重鎖に対して1nqbから構築した。非正準CDRは、非重複の解析された構造を含有するC-α構造データベースを検索することにより構築した。CDRの5Å以内にある残基を決定した。
【0120】
C.ヒトAb選択
マウスEM164の表面位置を、Kabatデータベースにおけるヒト抗体配列の対応する位置と比較した(Johnson, G.およびWu, T. T. (2001) Nucleic Acids Research, 29: 205-206)。抗体データベース管理ソフトウエアSR(Searle 1998)を用いて、天然の重および軽鎖ヒト抗体対からの抗体表面残基を抽出してアラインメントを実施した。CDRの5Å以内にある位置を特に配慮し、最も同一の表面残基をもつヒト抗体表面を選んで、マウス抗IGF-I受容体抗体表面残基と置換えた。
【0121】
D.PCR突然変異誘発
マウスEM164cDNAクローン(上記)上でPCR突然変異誘発を実施して、再表面形成されたヒトEM164(本明細書ではhuEM164と呼ぶ)を構築した。プライマーセットは、全ての試験したhuEM164のバージョンに8個のアミノ酸変化が要求されるように設計し、そしてさらなるプライマーは、代わりに2つの5Å残基変化を作るように設計した(表3)。PCR反応を、次のプログラムを用いて実施した:1)94℃1分間、2)94℃15秒間、3)55℃1分間、4)72℃2分間、5)ステップ2)に戻るサイクリング29回、6)72℃4分間の最終伸長による完了。PCR産物をその対応する制限酵素によって消化し、それらを上記のようにpBluescriptクローニングベクター中にクローニングした。クローンを配列決定して所望のアミノ酸変化を確認した。
【表3】
【0122】
E.可変域表面残基
Pedersenら(J. Mol. Biol., 235, 959-973, 1994)およびRoguskaら(タンパク質 Eng., 9, 895-904, 1996)が記載した抗体再表面形成技術は、マウス抗体可変配列の表面残基を予想することから始める。表面残基は、その全表面積の少なくとも30%が水分子にアクセス可能であるアミノ酸として定義される。
【0123】
127種の抗体構造ファイルのセット中に10種の最も相同的な抗体を同定した(図17および18)。それぞれのKabat位置に対する溶媒アクセシビリティを、これらのアラインメントした配列に対して平均化し、それぞれの残基に対する相対的アクセシビリティの分布を図19に示した。軽および重鎖は両方とも、少なくとも30%の平均相対アクセシビリティをもつ26残基を有し(図19):これらの残基はEM164に対して予想された表面残基であった。複数の残基は25%〜35%の平均アクセシビリティを有したので、これらを、残基のどちらか1つの側にフランキングする2つの同一残基をもつ抗体だけを平均化することにより、さらに試験した(表4および5)。このさらなる分析の後、以上同定した表面残基の元来のセットは変らないまま残った。
【表4】
【表5】
【0124】
F.CDRの5Å以内にある残基を決定するための分子モデル
AbMソフトウエアパッケージを用いて作製した上記分子モデルを分析して、どのEM164表面残基がCDRの5Å以内にあるかを確認した。マウスEM164抗体を再表面形成するために、CDR外側の全ての表面残基をヒトの対応残基に変えなければならないが、CDRの5Å以内の残基は抗原特異性にも寄与しうるので、特別な注意を払って処理した。従って、これらの後者残基を同定してヒト化プロセスの間を通して注意深く配慮しなければならない。再表面形成に用いるCDR定義は重鎖CDR2に対するAbM定義と残る5つのCDRに対するKabat定義を組合わせる(図14)。表6はEM164モデルの軽または重鎖配列両方のいずれかのCDR残基の5Å以内にあった残基を示す。
【表6】
【0125】
G.最も相同的なヒト表面の同定
EM164を再表面形成するためのヒト抗体表面候補を、Kabat抗体配列データベース内で、規定した残基だけの抗体データベースに対する位置のサーチングを提供するSRソフトウエアを利用して同定した。自然対合を保存するために、軽および重鎖両方の表面残基を一緒に比較した。Kabatデータベースからの最も相同的なヒト表面を配列同一性の格付け順に整列した。トップ5の表面を表7に与える。ついでこれらの表面を比較して、CDRの5Å以内で必要となる変更が最小であるのはどれかを同定した。白血病B細胞抗体、CLL 1.69は最少数の表面残基変化(全部で10)を必要とし、そのうち2つだけがCDRの5Å以内にあった。
【0126】
EM164の全長可変域配列もKabatヒト抗体データベースとアラインメントして、再び、CLL 1.69を最も類似したヒト可変域配列として同定した。総合して、これらの配列比較はヒト白血病B細胞抗体CLL 1.69がEM164に対するヒト表面として好ましい選択であると同定した。
【表7】
【0127】
H. ヒト化EM164遺伝子の構築
EM164(表7)の表面残基変化を上記のPCR突然変異誘発技術を利用して作った。CLL 1.69に対する表面残基のうちの8残基はCDRの5Å以内になかったので、これらの残基はヒト化EM164の全てのバージョンにおいてマウスからヒトに変更した(表8および表9)。CDRの5Å以内にあった2つの軽鎖表面残基(Kabat位置3および45)はヒトに変更するかまたはマウスに保持した。まとめると、これらのオプションにより、4種のEM164ヒト化バージョンが構築された(図22および23)。
【0128】
4種のヒト化バージョンのうち、バージョン1.0は全ての10ヒト表面残基を有する。CDRの近傍の変化を最も保存するバージョンは、CDRの5Å以内にあるマウス表面の両方を保持したバージョン 1.1である。全ての4種のヒト化EM164抗体遺伝子を抗体発現プラスミド(図16)中にクローニングして、一過性および安定したトランスフェクションに用いた。
【表8】
【0129】
I.全長IGF-I受容体および末端切断IGF-I受容体α鎖との結合に対する、ヒト化EM164抗体バージョンの親和性とマウスEM164抗体の親和性との比較
ヒト化EM164抗体バージョン1.0-1.3の親和性をマウスEM164抗体の親和性と、上記ビオチン化全長ヒトIGF-I受容体またはmyc-エピトープ標識末端切断IGF-I受容体α鎖を用いる競合アッセイを通して比較した。ヒト化EM164抗体サンプルを、ヒト胚性腎293T細胞における適当な発現ベクターの一過性トランスフェクションにより得て、抗体濃度を精製ヒト化抗体標準を用いるELISAにより決定した。ELISA結合競合を測定するために、ヒト化抗体サンプルと様々な濃度のマウスEM164抗体の混合物を、間接的に捕獲したビオチン化全長IGF-I受容体またはmycエピトープ標識末端切断IGF-I受容体α鎖とともにインキュベートした。平衡後に、結合したヒト化抗体をヤギ抗ヒトFab'2抗体-西洋わさびペルオキシダーゼコンジュゲートを用いて検出した。([結合したマウスAb]/[結合したヒト化Ab])対([マウスAb]/[ヒト化Ab])のプロットは、理論的に(Kdヒト化Ab/KdマウスAb)の勾配をもつ直線を生じるので、これを用いてヒト化とマウス抗体の相対親和性を決定した。
【0130】
競合アッセイの例を図11に示す。Immulon-2HB ELISAプレートを、1ウエル当たり5μg/mLストレプトアビジンを含む炭酸バッファー100μLを用いて常温で7時間コーティングした。ストレプトアビジンをコーティングしたウエルをブロッキングバッファー(10mg/mL BSAを含むTBS-Tバッファー)200μLを用いて1時間ブロックし、TBS-Tバッファーを用いて洗浄し、そしてビオチン化IGF-I受容体(1ウエル当たり5ng)を用いて一夜4℃にてインキュベートした。次いで間接的に捕獲したビオチン化IGF-I受容体を含有するウエルを洗浄し、ヒト化EM164抗体(15.5ng)とマウス抗体(0ng、または16.35ng、または32.7ng、または65.4ng、または163.5ng)の混合物を含むブロッキングバッファー100μLを用いて2時間常温でインキュベートし、次いで一夜4℃にてインキュベートした。次いでウエルをTBS-Tバッファーを用いて洗浄し、ヤギ抗ヒトFab'2抗体-西洋わさびペルオキシダーゼコンジュゲートとともに1時間(100μL;1μg/mLを含むブロッキングバッファー)インキュベートし、その後洗浄し、そしてABTS/H2O2基質を用いて405nmにて検出した。
【0131】
([結合したマウスAb]/[結合したヒト化Ab])対([マウスAb]/[ヒト化Ab])のプロットは、0.52の勾配(=Kdヒト化Ab/KdマウスAb)をもつ直線(r2=0.996)を生じた。従って、ヒト化抗体バージョン1.0はIGF-I受容体と、マウスEM164抗体より強く結合した。勾配に対する約0.5〜0.8の類似した値を、ヒト化EM164抗体のバージョン1.0、1.1、1.2および1.3のマウスEM164抗体との全長IGF-I受容体または末端切断IGF-I受容体α鎖競合に対して得たので、これは、EM164抗体の全てのヒト化バージョンが親マウスEM164抗体のそれより優れた類似の親和性を有することを示した。重鎖に92F→Cの突然変異をもつEM164抗体のキメラバージョンは、マウスEM164抗体との類似の結合競合において約3の勾配を示し、これはEM164の92F→Cの突然変異はマウスEM164抗体がIGF-I受容体との結合に示したのより3倍低い親和性を有することを示した。ヒト化EM164 v1.0抗体は、マウスEM164抗体が示したのと類似のMCF-7細胞のIGF-I刺激増殖および生存の阻害を示した(図24)。ヒト化EM164 v1.0抗体によるMCF-7細胞の血清刺激増殖および阻害はマウスEM164抗体による阻害と類似した。
【表9】
【0132】
J.本明細書に記載のマウスおよびヒト化抗体配列から出発して、改良された抗IGF-I-受容体抗体を提供する方法
抗IGF-I受容体抗体EM164とそのヒト化変異体のアミノ酸および核酸配列を利用して、改良されたかつまた本発明の範囲内にある他の抗体を開発した。そのような改良された特性としては、IGF-I受容体に対する親和性の増加が挙げられる。複数の研究は、1以上のアミノ酸変化を抗体の配列の様々な位置に導入することのその特性、例えば結合および発現レベルに与える効果を、一次抗体配列の知識に基づいて、調査している(Yang, W.P.ら, 1995, J. Mol. Biol., 254, 392-403;Rader, C.ら, 1998, Proc. Natl. Acad. Sci. USA, 95, 8910-8915;Vaughan, T.J.ら, 1998, Nature Biotechnology, 16, 535-539)。
【0133】
これらの研究においては、オリゴヌクレオチド媒介位置指定突然変異誘発、カセット突然変異誘発、誤りがちの(error-prone)PCR、DNAシャッフリング、または大腸菌(E. coli)の変異誘発株などの方法を用いて、CDR1、CDR2、CDR3、またはフレームワーク領域中の軽および重鎖遺伝子の配列を変えることにより、1次抗体の変異体を作製している(Vaughan, T.J.ら, 1998, Nature Biotechnology, 16, 535-539;Adey, N.B.ら, 1996, 第16章, pp.277-291, in 「ペプチドとタンパク質のファージディスプレイ(Phage Display of Peptides and Proteins)」, Kay, B.K.ら編, Academic Press)。1次抗体の配列を変えるこれらの方法は、標準のスクリーニング技術の利用を通して、その2次抗体の親和性の改良をもたらしている(Gram, H.ら, 1992, Proc. Natl. Acad. Sci. USA, 89, 3576-3580;Boder, E.T.ら, 2000, Proc. Natl. Acad. Sci. USA, 97, 10701-10705;Davies, J.およびRiechmann, L., 1996, Immunotechnolgy, 2, 169-179;Thompson, J.ら, 1996, J. Mol. Biol., 256, 77-88;Short, M.K.ら, 2002, J. Biol. Chem., 277, 16365-16370;Furukawa, K.ら, 2001, J. Biol. Chem., 276, 27622-27628)。
【0134】
抗体の1以上のアミノ酸残基を変える類似の定方向手法(directed strategy)により、本発明に記載の抗体配列を用いて、改良された機能をもつ抗IGF-I受容体抗体、例えば治療薬の結合に利用する共有結合改変のための好都合な結合点に遊離アミノ基またはチオールなどの好適な基を有する抗体を開発することができる。
【0135】
K.マウス、キメラおよび他の抗IGF-I受容体抗体のための代わりの発現系
マウス抗IGF-I受容体抗体を、ヒト化抗体を発現するために利用したプラスミド(上記)と類似の哺乳類動物発現プラスミドからも発現した。軽鎖κおよび重鎖γ-1配列を含むマウス定常域を有する発現プラスミドは公知である(McLeanら, 2000, Mol Immunol., 37, 837-845)。これらのプラスミドを、いずれかの抗体可変域、例えばマウス抗IGF-I受容体抗体を単純な制限酵素消化とクローニングにより受容するように設計した。発現プラスミド中にこれらと適合しうる制限酵素切断部位を創出するために、抗IGF-1受容体抗体のさらなるPCRが通常必要であった。
【0136】
全マウス抗IGF-I受容体抗体を発現する代わりの手法は、キメラ抗IGF-I受容体抗体発現プラスミド中のヒト定常域を置換えることであった。キメラ発現プラスミド(図16)を可変域のためのおよび軽および重鎖定常域の両方のためのカセットを用いて構築した。抗体可変配列を制限酵素消化によりこの発現プラスミド中にクローニングしたように、別々の制限酵素消化を用いていずれかの定常域にクローニングした。抗IGF-1抗体可変域のクローニングについて本明細書に記載したRNAのように、κ軽鎖およびγ-1重鎖cDNAを、例えばマウスハイブリドーマRNAからクローニングした。同様に、好適なプライマーをKabatデータベース中の利用しうる配列から設計した(表10を参照)。例えば、RT-PCRを用いて定常域配列をクローニングしそしてこれらの断片をキメラ抗IGF-I受容体抗体発現プラスミド中にクローニングするために必要な制限酵素切断部位を創出した。このプラスミドを用いて次に標準哺乳類動物発現系、例えばCHO細胞系統において全マウス抗IGF-I受容体抗体を発現した。
【表10】
【0137】
寄託の表明
マウスEM164抗体を作るハイブリドーマはATCC(American Type Culture Collection, PO Box 1549, Manassas, VA 20108)に、2002年6月14日ブダペスト条約の条項のもとで、受託番号PTA-4457として寄託した。
【0138】
ある特定の特許および印刷出版物が本発明の開示に参照されていて、それらの教示は本明細書にそれらのそれぞれの全てが参照によりそれぞれ組込まれる。
【0139】
本発明は詳細にかつその特定の実施形態を参照して記載されているが、当業者であれば、本発明の精神と範囲から逸脱することなくそれに様々な変化と改変をなしうることは明らかであろう。
【技術分野】
【0001】
本発明はヒトインスリン様増殖因子-I受容体(IGF-I受容体)と結合する抗体に関する。さらに特定すれば、本発明はIGF-I受容体の細胞機能を阻害する抗IGF-I受容体抗体に関する。なおさらに特定すれば、本発明はIGF-I、IGF-IIおよび血清の腫瘍細胞の増殖および生存に対する効果に拮抗しかつそれ自身は実質的に作動薬活性を欠く抗IGF-I受容体抗体に関する。本発明はまた、上記抗体のフラグメント、上記抗体のヒト化および再表面形成バージョン、上記抗体のコンジュゲート、抗体誘導体、ならびに上記の診断、研究および治療応用における利用にも関する。本発明はさらに、上記抗体およびそのフラグメントから作製される改良された抗体またはそのフラグメントに関する。他の態様においては、本発明は抗体またはそのフラグメントをコードするポリヌクレオチド、およびそのポリヌクレオチドを含むベクターに関する。
【背景技術】
【0002】
インスリン様増殖因子-I受容体(IGF-I受容体)は、膜貫通へテロテトラマータンパク質であり、細胞外の2つのα鎖および膜をまたがる2つのβ鎖をジスルフィド結合したβ-α-α-βコンフィギュレーションの形態を有する。リガンド、すなわちインスリン様増殖因子-I(IGF-I)およびインスリン様増殖因子-II(IGF-II)がIGF-I受容体の細胞外ドメインと結合すると、細胞内チロシンキナーゼドメインを活性化して受容体の自己リン酸化および基質リン酸化が起こる。IGF-I受容体はインスリン受容体と相同性があり、β鎖チロシンキナーゼドメインにおいて84%の高い配列類似性を有しかつα鎖細胞外システインリッチドメインにおいて48%の低い配列類似性を有する(Ulrich, A.ら, 1986, EMBO, 5,2503-2512;Fujita-Yamaguchi, Y.ら, 1986, J. Biol. Chem., 261, 16727-16731;LeRoith, D.ら, 1995, Endocrine Reviews, 16, 143-163)。IGF-I受容体およびそのリガンド(IGF-IおよびIGF-II)は多数の生理学的プロセスに重要な役割を果たし、その役割は胚形成時の成長および発生、成体における代謝、細胞増殖および細胞分化を含む(LeRoith, D., 2000, Endocrinology, 141,1287-1288;LeRoith, D., 1997, New England J. Med., 336, 633-640)。
【0003】
IGF-IとIGF-IIは両方とも、主にIGF結合タンパク質との複合体で存在する血中の内分泌ホルモンとして、および局所で産生されるパラ分泌および自己分泌増殖因子として機能する(Humbel, R.E., 1990, Eur. J. Biochem., 190, 445-462;Cohick, W.S.およびClemmons, D.R., 1993, Annu. Rev. Physiol. 55, 131-153)。
【0004】
IGF-I受容体は腫瘍細胞の増殖、形質転換および生存を促進することが示唆されている(Baserga, R.ら, 1997, Biochem. Biophys. Acta, 1332, F105-F126;Blakesley, V.A.ら, 1997, Journal of Endocrinology, 152, 339-344;Kaleko, M., Rutter, W.J.,およびMiller, A.D. 1990, Mol. Cell. Biol., 10, 464-473)。すなわち、腫瘍型のいくつかはIGF-I受容体の通常のレベルより高いレベルを発現することが公知であり、それらは乳癌、大腸癌、卵巣癌、滑膜肉腫、膵癌を含む(Khandwala, H.M.ら, 2000, Endocrine Reviews, 21, 215-244;Werner, H.およびLeRoith, D., 1996, Adv. Cancer Res., 68, 183-223;Happerfield, L.C.ら, 1997, J. Pathol., 183, 412-417;Frier, S.ら, 1999, Gut, 44, 704-708;van Dam, P.A.ら, 1994, J. Clin. Pathol., 47, 914-919;Xie, Y.ら, 1999, Cancer Res., 59, 3588-3591;Bergmann, U.ら, 1995, Cancer Res., 55, 2007-2011)。In vitroでIGF-IおよびIGF-IIは、複数のヒト腫瘍細胞系統、例えば肺癌、乳癌、大腸癌、骨肉腫および子宮頸癌に対する強力な分裂促進因子であることが示されている(Ankrapp, D.P.およびBevan, D.R., 1993, Cancer Res., 53, 3399-3404;Cullen, K.J., 1990, Cancer Res., 50, 48-53;Hermanto, U.ら, 2000, Cell Growth & Differentiation, 11, 655-664;Guo, Y. S.ら, 1995, J. Am. Coll. Surg., 181, 145-154;Kappel, C.C.ら, 1994, Cance
r Res., 54, 2803-2807;Steller, M.A.ら, 1996, Cancer Res., 56, 1761-1765)。これらの腫瘍および腫瘍細胞系統のいくつかはまた、高レベルのIGF-IまたはIGF-IIを発現して、自己分泌またパラ分泌の方法でそれら自身の増殖を刺激することができる(Quinn, K. A.ら, 1996, J. Biol. Chem., 271, 11477-11483)。
【0005】
疫学研究は、IGF-Iの血漿レベルの増加(およびIGF結合タンパク質-3のレベルの低下)は前立腺癌、大腸癌、肺癌および乳癌のリスクの増加と相関があることを示している(Chan, J. M.ら, 1998, Science, 279, 563-566;Wolk, A.ら, 1998, J. Natl. Cancer Inst., 90,911-915;Ma, J.ら, 1999, J. Natl. Cancer Inst., 91, 620-625;Yu, H.ら, 1999, J. Natl. Cancer Inst., 91, 151-156;Hankinson, S.E.ら, 1998, Lancet, 351, 1393-1396)。癌予防として、血漿のIGF-Iレベルを低下するかまたはIGF-I受容体の機能を抑制する手法が示唆されている(Wu, Y.ら, 2002, Cancer Res., 62, 1030-1035;Grimberg, A.およびCohen P., 2000, J. Cell. Physiol., 183, 1-9)。
【0006】
IGF-I受容体は、腫瘍細胞を増殖因子欠乏、足場非依存性、または細胞傷害薬処理により起こるアポトーシスから保護する(Navarro, M.およびBaserga, R., 2001, Endocrinology, 142, 1073-1081;Baserga, R.ら, 1997, Biochem. Biophys. Acta, 1332, F105-F126)。分裂促進、トランスフォーメーションおよび抗アポトーシス活性にとって重要であるIGF-I受容体のドメインは、突然変異分析により同定されている。
【0007】
例えば、IGF-I受容体のチロシン1251残基は、抗アポトーシスおよびトランスフォーメーション活性にとって極めて重要であるが分裂促進活性にとっては重要でないことが同定されている(O'Connor, R.ら, 1997, Mol. Cell. Biol., 17, 427-435;Miura, M.ら, 1995, J. Biol. Chem., 270, 22639-22644)。リガンドにより活性化されるIGF-I受容体の細胞内シグナル伝達経路はインスリン受容体基質(IRS-1およびIRS-2)のチロシン残基のリン酸化に関わり、これがホスファチジルイノシトール-3-キナーゼ(PI3キナーゼ)を膜へ補給する。膜結合したPI3キナーゼのリン脂質産物はセリン/トレオニンキナーゼAktを活性化し、その基質であるプロアポトーシスタンパク質BADがリン酸化されて不活性状態となる(Datta, S. R., Brunet, A. and Greenberg, M. E., 1999, Genes & Development, 13, 2905-2927;Kulik, G., Klippel, A.およびWeber, M. J., 1997, Mol. Cell. Biol. 17, 1595-1606)。MCF-7ヒト乳癌細胞のIGF-I受容体の分裂促進シグナル伝達はPI3キナーゼを必要とし、分裂促進因子活性化タンパク質キナーゼに依存しないが、分化ラットクロム親和細胞腫PC12細胞の生存シグナル伝達は、PI3キナーゼと分裂促進因子活性化タンパク質キナーゼの両方の経路を必要とする(Dufourny, B.ら, 1997, J. Biol. Chem., 272, 31163-31171;Parrizas, M., Saltiel, A. R. and LeRoith, D., 1997, J. Biol. Chem., 272, 154-161)。
【0008】
アンチセンス手法によるIGF-I受容体レベルの下方調節は、複数の腫瘍細胞系統、例えば黒色腫、肺癌、卵巣癌、グリア芽細胞腫、神経芽細胞腫および横紋筋肉腫のin vivoおよびin vitro腫瘍形成能を低下することが示されている(Resnicoff, M.ら, 1994, Cancer Res., 54, 4848-4850;Lee, C.-T.ら, 1996, Cancer Res., 56, 3038-3041;Muller, M.ら, 1998, Int. J. Cancer, 77, 567-571;Trojan, J.ら, 1993, Science, 259, 94-97;Liu, X.ら, 1998, Cancer Res., 58, 5432-5438;Shapiro, D. N.ら, 1994, J. Clin. Invest., 94, 1235-1242)。同様に、IGF-I受容体の優性ネガティブ突然変異は、IGF-I受容体を過剰発現する形質転換Rat-1細胞のin vivo腫瘍形成能、およびin vitro増殖を低下することが報じられている(Prager, D.ら, 1994, Proc. Natl. Acad. Sci. USA, 91, 2181-2185)。
【0009】
IGF-I受容体mRNAに対するアンチセンスを発現する腫瘍細胞は、生物拡散室内の動物に注射すると大量のアポトーシスを起こす。この観察によれば、腫瘍細胞はIGF-I受容体の阻害によりアポトーシスに対する感受性が正常細胞より高くなるという仮説に基づいて、IGF-I受容体は魅力的な治療標的であることを示す(Resnicoff, M.ら, 1995, Cancer Res., 55, 2463-2469;Baserga, R., 1995, Cancer Res., 55, 249-252)。
【0010】
腫瘍細胞中のIGF-I受容体の機能を阻害する他の手法として、IGF-I受容体の細胞外ドメインと結合してその活性化を阻害する抗IGF-I受容体抗体が利用されている。IGF-I受容体に対するマウスモノクローナル抗体を開発するいくつかの試みが報じられており、それらの中で2種の阻害抗体(IR3および1H7)は入手可能があり、それらの利用が複数のIGF-I受容体研究において報じられている。
【0011】
部分精製したインスリン受容体の胎盤調製物を用いてマウスを免疫感作すると、インスリン受容体と選択的に結合するIR1、およびIGF-I受容体(ソマトメジンC受容体)の優先的免疫沈降を示すがインスリン受容体の弱い免疫沈降も示す2種の抗体、IR2とIR3が得られるので、それを利用してIR3抗体を発生させた(Kull, F. C.ら, 1983, J. Biol. Chem., 258, 6561-6566)。
【0012】
精製したIGF-I受容体の胎盤調製物を利用して1H7抗体を発生させた。該胎盤調製物を用いてマウスを免疫感作すると、3種の刺激性抗体に加えて阻害性抗体1H7が得られる(Li, S.-L.ら, 1993, Biochem. Biophys. Res. Commun., 196, 92-98;Xiong, L.ら, 1992, Proc. Natl. Acad. Sci. USA, 89, 5356-5360)。
【0013】
他の報文では、IGF-I受容体の高レベルを発現するトランスフェクトされた3T3細胞を用いてマウスを免疫感作すると、ヒトIGF-I受容体に特異的なマウスモノクローナル抗体のパネルが得られるので、このパネルを結合競合によりおよびIGF-Iのトランスフェクトされた3T3細胞との結合の阻害または刺激により7グループに分類した(Soos, M. A.ら, 1992, J. Biol. Chem., 267, 12955-12963)。
【0014】
このように、IR3抗体はin vitroで最も汎用されるIGF-I受容体研究用の阻害性抗体であるが、これはIGF-I受容体を発現するトランスフェクトされた3T3およびCHO細胞に作動薬活性を示す欠点が悩みである(Kato, H.ら, 1993, J. Biol. Chem., 268, 2655-2661;Steele-Perkins, G.およびRoth, R. A., 1990, Biochem. Biophys. Res. Commun., 171, 1244-1251)。同様に、Soosらが開発した抗体のパネルの中で、最も阻害性抗体の24-57および24-60もトランスフェクトされた3T3細胞に作動薬活性を示した(Soos, M. A.ら, 1992, J. Biol. Chem., 267, 12955-12963)。IR3抗体は、IGF-Iの無傷の細胞におけるおよび可溶化後の発現された受容体との結合を阻害する(しかしIGF-IIを阻害しない)ことが報じられるが、細胞のDNA合成をin vitroで刺激するIGF-IとIGF-II両方の能力を阻害することが示されている(Steele-Perkins, G.およびRoth, R.A., 1990, Biochem. Biophys. Res. Commun., 171, 1244-1251)。IR3抗体の結合エピトープは、キメラのインスリンIGF-I受容体構築物からIGF-I受容体の223〜274領域であると推定されている(Gustafson, T. A.およびRutter, W. J., 1990, J. Biol. Chem., 265, 18663-18667;Soos, M. A.ら, 1992, J. Biol. Chem., 267, 12955-12963)。
【0015】
MCF-7ヒト乳癌細胞系統は、IGF-IおよびIGF-IIのin vitro増殖応答を実証するために利用される典型的なモデル細胞系統である(Dufourny, B.ら, 1997, J. Biol. Chem., 272, 31163-31171)。MCF-7細胞において、IR3抗体は、無血清状態で外因的に加えられたIGF-IおよびIGF-IIの刺激性効果を不完全にほぼ80%だけ遮断する。また、IR3抗体は10%血清中でMCF-7細胞の増殖を有意に阻害しない(25%未満)(Cullen, K. J.ら, 1990, Cancer Res., 50, 48-53)。MCF-7細胞の血清刺激増殖のIR3抗体によるin vitroでのこの弱い阻害は、IR3抗体処理がヌードマウスにおけるMCF-7異種移植片の増殖を有意に阻害しなかったin vivo研究の結果と関係がありうる(Arteaga, C. L.ら, 1989, J. Clin. Invest., 84, 1418-1423)。
【0016】
IR3および他の報じられた抗体の弱い作動薬活性、ならびにそれらがより生理学的な血清刺激の条件(無血清条件で外因的に加えたIGF-IまたはIGF-IIによる刺激の代わりに)のもとでMCF-7細胞などの腫瘍細胞の増殖を有意に阻害する能力が無いことの故に、腫瘍細胞の血清刺激増殖を有意に阻害ししかしそれ自身による有意な作動薬活性を示さない新しい抗IGF-I受容体抗体に対するニーズがある。
【発明の概要】
【発明が解決しようとする課題】
【0017】
従って、インスリン様増殖因子-I受容体と特異的に結合し、受容体に拮抗することにより受容体の細胞活性を阻害し、そしてまた受容体に対する作動薬活性を実質的に欠く抗体、抗体フラグメントおよび抗体誘導体を提供することが本発明の目的である。
【課題を解決するための手段】
【0018】
従って第1の実施形態においては、軽および重鎖可変域の両方のアミノ酸配列、軽および重鎖に対する遺伝子のcDNA配列、そのCDR(相補性決定領域)の同定、その表面アミノ酸の同定、ならびに組換え型においてそれを発現する方法に関して、本明細書で全て特徴付けられているマウス抗体EM164が提供される。
【0019】
第2の実施形態においては、抗体EM164の再表面形成されたまたはヒト化されたバージョンであって、抗体またはそのフラグメントの表面に曝される残基の軽および重鎖の両方が置換えられて公知のヒト抗体表面にさらによく似ている上記バージョンが提供される。そのようなヒト化された抗体は、マウスEM164と比較して、治療または診断薬としての有用性が一層増加しうる。抗体EM164のヒト化されたバージョンはまた、軽および重鎖可変域両方のそれぞれのアミノ酸配列、軽および重鎖に対する遺伝子のDNA配列、そのCDRの同定、その表面アミノ酸の同定、ならびに組換え型においてそれを発現する方法の開示に関して、本明細書において全て特徴づけられている。
【0020】
第3の実施形態においては、増殖刺激因子、例えば血清、インスリン様増殖因子-Iおよびインスリン様増殖因子-IIの存在のもとで、癌細胞の増殖をほぼ80%以上阻害することができる抗体が提供される。
【0021】
第4の実施形態においては、抗体または抗体フラグメントであって、
それぞれ配列番号1〜3:
SYWMH (配列番号1)、
EINPSNGRTNYNEKFKR (配列番号2)、
GRPDYYGSSKWYFDV (配列番号3)により表されるアミノ酸配列を有するCDRを含む重鎖を有しかつ
それぞれ配列番号4〜6:
RSSQSIVHSNVNTYLE (配列番号4)、
KVSNRFS (配列番号5)、
FQGSHVPPT (配列番号6)により表されるアミノ酸配列を有するCDRを含む軽鎖を有する上記抗体または抗体フラグメントが提供される。
【0022】
第5の実施形態においては、配列番号7:
QVQLQQSGAELVKPGASVKLSCKASGYTFTSYWMHWVKQRPGQGLEWIGEINPSNGRTNYNEKFKRKATLTVDKSSSTAYMQLSSLTSEDSAVYYFARGRPDYYGSSKWYFDVWGAGTTVTVSS(配列番号7)
により表されるアミノ酸配列と少なくとも90%配列同一性を共有するアミノ酸配列を持つ重鎖を有する抗体が提供される。
【0023】
同様に、配列番号8:
DVLMTQTPLSLPVSLGDQASISCRSSQSIVHSNVNTYLEWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGTDFTLRISRVEAEDLGIYYCFQGSHVPPTFGGGTKLEIKR(配列番号8)
により表されるアミノ酸配列と少なくとも90%配列同一性を共有するアミノ酸配列を持つ軽鎖を有する抗体が提供される。
【0024】
第6の実施形態においては、配列番号9〜12:
DVVMTQTPLSLPVSLGDPASISCRSSQSIVHSNVNTYLEWYLQKPGQSPRLLIYKVSNRFSGVPDRFSGSGAGTDFTLRISRVEAEDLGIYYCFQGSHVPPTFGGGTKLEIKR(配列番号9);
DVLMTQTPLSLPVSLGDPASISCRSSQSIVHSNVNTYLEWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGAGTDFTLRISRVEAEDLGIYYCFQGSHVPPTFGGGTKLEIKR(配列番号10);
DVLMTQTPLSLPVSLGDPASISCRSSQSIVHSNVNTYLEWYLQKPGQSPRLLIYKVSNRFSGVPDRFSGSGAGTDFTLRISRVEAEDLGIYYCFQGSHVPPTFGGGTKLEIKR(配列番号11);または
DVVMTQTPLSLPVSLGDPASISCRSSQSIVHSNVNTYLEWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGAGTDFTLRISRVEAEDLGIYYCFQGSHVPPTFGGGTKLEIKR(配列番号12)
のうちの1つに対応するアミノ酸配列を持つヒト化されたまたは再表面形成された軽鎖可変域を有する抗体が提供される。
【0025】
同様に、配列番号13:
QVQLVQSGAEVVKPGASVKLSCKASGYTFTSYWMHWVKQRPGQGLEWIGEINPSNGRTNYNQKFQGKATLTVDKSSSTAYMQLSSLTSEDSAVYYFARGRPDYYGSSKWYFDVWGQGTTVTVSS(配列番号13)
に対応するアミノ酸配列を持つヒト化されたまたは再表面形成された重鎖可変域を有する抗体が提供される。
【0026】
第7の実施形態においては、改良された特性を有する本発明の抗体または抗体フラグメントが提供される。例えば、改良されたIGF-I受容体に対する親和性を有する抗体または抗体フラグメントが本発明の抗体またはフラグメントの親和性成熟により調製される。
【0027】
本発明はさらに、上記抗体のコンジュゲートであって、直接にまたは切断可能もしくは非切断可能なリンカーを経由して細胞傷害薬が本発明の抗体もしくは抗体のエピトープ結合フラグメントと共有結合していることを特徴とする上記コンジュゲートを提供する。好ましい実施形態において、細胞傷害薬はタキソール、メイタンシノイド、CC-1065またはCC-1065類似体である。
【0028】
本発明はさらに、研究または診断応用に利用するためにさらに標識されている抗体またはそのフラグメントを提供する。好ましい実施形態において、標識は放射標識、蛍光体、発色体、イメージング剤または金属イオンである。
【0029】
上記抗体またはフラグメントを癌を有すると疑われる被験者に投与し、そして被験者の体内の標識の分布を測定またはモニターする診断方法も提供される。
【0030】
第8の実施形態においては、本発明は癌を有する被験者の治療方法であって、本発明の抗体、抗体フラグメントまたは抗体コンジュゲートを単独でまたは他の細胞傷害または治療薬と組合わせて投与することによる上記治療方法を提供する。癌は、1以上の、例えば乳癌、大腸癌、卵巣癌、骨肉腫、子宮頸癌、前立腺癌、肺癌、滑膜癌、膵癌、またはこれからIGF-I受容体レベルが高いことで確認される他の癌であってもよい。
【図面の簡単な説明】
【0031】
【図1】精製EM164抗体の、ヒトY1251F-IGF-I受容体またはヒトインスリン受容体を過剰発現する細胞との特異的結合の蛍光活性化細胞選別(フローサイトメトリー)(FACS)分析を示す。
【図2】EM164抗体のビオチン化ヒトIGF-I受容体との結合に対する結合滴定曲線を示す。
【図3】EM164抗体による、ビオチン化IGF-Iのヒト乳癌MCF-7細胞との結合の阻害を示す。
【図4】EM164抗体による、MCF-7細胞におけるIGF-I受容体のIGF-I刺激自己リン酸化の阻害を示す。
【図5】EM164抗体による、MCF-7細胞におけるIGF-I刺激IRS-1リン酸化の阻害を示す。
【図6】EM164抗体による、SaOS-2細胞におけるIGF-I刺激シグナル伝達の阻害を示す。
【図7】MTTアッセイにより評価した、様々な増殖条件下でのMCF-7細胞の増殖および生存に与えるEM164抗体の効果を示す。
【図8】様々な血清濃度のもとでの、MCF-7細胞の増殖と生存に与えるEM164抗体の効果を示す。
【図9】EM164抗体による、IGF-Iおよび血清に刺激されたNCI-H838細胞の増殖と生存の阻害を示す。
【図10】EM164抗体、タキソール、またはEM164抗体とタキソールの併用による処理が、マウスにおけるCalu-6肺癌異種移植片の増殖に与える効果を示す。
【図11】ヒト化EM164抗体(v.1.0)およびマウスEM164抗体の結合の間の競合を示す。
【図12】マウス抗IGF-I受容体抗体EM164の軽鎖リーダーおよび可変域の、cDNA(配列番号49)およびアミノ酸配列(配列番号50)を示す。矢印はフレームワークの出発点を示す。Kabatによる3種のCDR配列に下線を引いた。
【図13】マウス抗IGF-I受容体抗体EM164の重鎖リーダーおよび可変域の、cDNA(配列番号51)およびアミノ酸配列(配列番号52)を示す。矢印はフレームワークの出発点を示す。Kabatによる3種のCDR配列に下線を引いた。
【図14】Chothia正準クラス定義(Chothia canonical class definitions)から確認した抗体EM164の軽および重鎖CDRアミノ酸配列を示す。重鎖CDRに対するAbMモデルソフトウエア定義(AbM modeling software definitions)も示す。軽鎖のCDR1は配列番号4、CDR2は配列番号5、そしてCDR3は配列番号6である。重鎖のCDR1は配列番号1、CDR2は配列番号2、そしてCDR3は配列番号3である。AbM重鎖のCDR1は配列番号53、CDR2は配列番号54、そしてCDR3は配列番号55である。
【図15】抗IGF-I-受容体抗体EM164の軽鎖および重鎖アミノ酸配列とCr1(配列番号56)およびJ558.c(配列番号57)遺伝子の生殖系列配列とのアラインメントを示す。ダッシュ(-)は配列同一性を示す。
【図16】組換えキメラおよびヒト化EM164抗体を構築しかつ発現するために利用したプラスミドを示す。A)軽鎖クローニングプラスミド、B)重鎖クローニングプラスミド、C)哺乳類動物抗体発現プラスミド。
【図17】EM164の表面残基を予測するために用いた構造ファイルのセット中の抗体127種からスクリーニングした軽鎖のうちの最も相同的な10種のアミノ酸配列を示す。em164 LC(配列番号58)、2jel(配列番号59)、2pcp(配列番号60)、1nqb(配列番号61)、1kel(配列番号62)、1hyx(配列番号63)、1igf(配列番号64)、1tet(配列番号65)、1clz(配列番号66)、1bln(配列番号67)、1cly(配列番号68)、コンセンサス(配列番号69)。
【図18】EM164の表面残基を予測するために用いた構造ファイルのセット中の抗体127種からスクリーニングした重鎖のうちの最も相同的な10種のアミノ酸配列を示す。em164 HC(配列番号70)、1nqb(配列番号71)、1ngp(配列番号72)、1fbi(配列番号73)、1afv(配列番号74)、1yuh(配列番号75)、1plg(配列番号76)、1d5b(配列番号77)、1ae6(配列番号78)、1axs(配列番号79)、3hfl(配列番号80)、コンセンサス(配列番号81)。
【図19】10種の最も相同的な構造からの(A)軽鎖および(B)重鎖可変域残基のそれぞれに対する平均アクセシビリティを示す。数字はKabat抗体配列位置番号を表す。
【図20】マウスEM164(muEM164)およびヒト化EM164(huEM164)抗体に対する軽鎖可変域アミノ酸配列を示す。muEM164(配列番号82)、huEM164 V1.0(配列番号83)、huEM164 V1.1(配列番号84)、huEM164 V1.2(配列番号85)、huEM164 V1.3(配列番号86)。
【図21】マウス(muEM164、配列番号87)およびヒト化EM164抗体(huEM164、配列番号88)に対する重鎖可変域アミノ酸配列を示す。
【図22】huEM164 v1.0の可変域DNAおよびアミノ酸配列を、軽鎖(DNA配列番号89、アミノ酸配列番号90)と重鎖(DNA配列番号91、アミノ酸配列番号92)の両方に対して示す。
【図23A】ヒト化EM164 v1.1(DNA配列番号93;アミノ酸配列番号94)、v1.2(DNA配列番号95;アミノ酸配列番号96)およびv1.3(DNA配列番号97;アミノ酸配列番号98)の軽鎖可変域DNAおよびアミノ酸配列を示す。
【図23B】ヒト化EM164 v1.1(DNA配列番号93;アミノ酸配列番号94)、v1.2(DNA配列番号95;アミノ酸配列番号96)およびv1.3(DNA配列番号97;アミノ酸配列番号98)の軽鎖可変域DNAおよびアミノ酸配列を示す(図23Aの続き)。
【図24】ヒト化EM164 v1.0抗体およびマウスEM164抗体による、MCF-7細胞のIGF-I刺激増殖および生存の阻害を示す。
【図25】EM164がMCF-7細胞のIGF-I-刺激サイクリングを抑制することを示す。
【図26】EM164がIGF-Iおよび血清の抗アポトーシス効果を抑制することを示す。EM164を用いる処理はアポトーシス細胞死をもたらし、それが切断されたCK18タンパク質のレベル増加により実証される。
【図27】免疫不全マウスにおけるヒトBxPC-3膵癌異種移植片の増殖に対する、EM164抗体、ゲミシタビン、またはEM164抗体とゲミシタビンの併用による処理の効果を示す。
【発明を実施するための形態】
【0032】
本発明者らは、細胞表面上のヒトインスリン様増殖因子-I受容体(IGF-IR)と結合する新規の抗体を発見しかつそれを改良した。本抗体およびフラグメントは、受容体それ自身を活性化する能力がなくしかも受容体の細胞機能を阻害するというユニークな能力を有する。従って、IGF-IRと結合して阻害する従来公知の抗体はIGF-IRリガンドの不在時でも受容体を活性化するのに対して、本発明の抗体またはフラグメントはIGF-IRに拮抗するが実質的に作動薬活性を欠く。さらに本発明の抗体と抗体フラグメントは、血清の存在のもとでのヒト腫瘍細胞、例えばMCF-7細胞の増殖を80%以上阻害し、この値は従来公知の抗IGF-IR抗体を用いて得られるより高い阻害度である。
【0033】
本発明は、本明細書でEM164と呼び、軽および重鎖の両方のアミノ酸配列、そのCDRの同定、表面アミノ酸の同定、および組換え型におけるその発現に関して、全て特徴付けているマウス抗IGF-IR抗体から進める。
【0034】
生殖系列配列のEM164配列とのアラインメントを図15に示す。その比較によりEM164中の可能性のある体細胞突然変異が同定され、それらは軽鎖のCDR1および重鎖のCDR2にそれぞれ1つづつ含まれる。
【0035】
抗体EM164軽および重鎖、ならびにヒト化バージョンの1次アミノ酸およびDNA配列を本明細書に開示する。しかし、本発明の範囲はこれらの配列を含む抗体とフラグメントに限定されるものではない。それどころか、インスリン様増殖因子-I受容体と特異的に結合しかつ受容体の生物学的活性に拮抗し、しかし作動活性を実質的に欠く全ての抗体とフラグメントは本発明の範囲に包含される。従って、抗体と抗体フラグメントは、抗体EM164またはヒト化誘導体とその骨格、CDR、軽鎖および重鎖のアミノ酸配列が異なっていてもよく、それでもなお本発明の範囲内に包含されうる。
【0036】
抗体EM164のCDRはモデリングにより同定され、その分子構造は予測されている。再び、CDRはエピトープ認識にとって重要であるものの、それは本発明の抗体とフラグメントにとって必須ではない。すなわち、例えば本発明の抗体の親和性成熟により作られる、改良された特性を有する抗体とフラグメントが提供される。
【0037】
多様な抗体と抗体フラグメント、ならびに抗体模倣物を、特定のセットのCDRとフランキングする可変および定常域配列内の突然変異、欠失および/または挿入により作ることができる。このようにして、例えば、所与のセットのCDRに対して異なる重鎖の置換により異なるクラスのAbを得ることが可能であり、この方法により、例えば、IgG1〜4、IgM、IgA1〜2、IgD、IgE抗体型およびイソ型を作ることができる。同様に、本発明の範囲に含まれる人工抗体は、CDRの所与のセットを完全な合成フレームワーク内に埋め込むことにより作ることができる。本明細書において用語「可変」は、可変ドメインのある特定部分であって、抗体間で配列が異なりかつ各特定抗体のその抗原との結合および特異性に利用される上記部分を記載するために使用される。しかし、その可変性は通常、抗体の可変ドメイン全体に等しく分布するものではない。可変性は典型的には軽鎖および重鎖両方の可変ドメイン中の相補性決定領域(CDR)または超可変域と呼ばれる3セグメントに濃縮される。可変ドメインのより高度に保存された部分はフレームワーク(FR)と呼ばれる。重および軽鎖の可変ドメインはそれぞれ、主にβシートコンフィギュレーションである4つのフレームワーク領域を含み、それらのフレームワークは3つのCDRにより接続される。上記CDRは、接続する(場合によってはβシート構造の部分となる)ループを形成する。それぞれの鎖のCDRは、FR領域によって密接した近位に一緒に保持され、他の鎖からのCDRとともに抗体の抗原結合部位の形成に寄与する(E. A. Kabatら 「免疫学的に重要なタンパク質の配列(Sequences of Proteins of Immunological Interest)」, 第5版, 1991, NIH)。定常ドメインは抗体と抗原の結合に直接関わらないが、様々なエフェクター機能、例えば抗体の抗体依存性細胞毒性への関与を示す。
【0038】
ヒト化抗体、または他の哺乳類動物による非拒絶に適合させた抗体は、複数の技術、例えば再表面形成およびCDR移植術を用いて作ることができる。再表面形成技術においては、分子モデル、統計解析および突然変異誘発を組合わせ、可変域の非CDR表面を標的宿主の既知抗体の表面に似ているように調節する。抗体を再表面形成する計画と方法、および色々な宿主内の抗体の免疫原性を低下させる他の方法は、米国特許第5,639,641号に開示されていて、これは本明細書にその全てが参照により組み入れられる。CDR移植技術においては、マウス重および軽鎖CDRを全ヒトフレームワーク配列中に移植する。
【0039】
本発明はまた、本明細書に記載した抗体の機能的同等物も含む。機能的同等物はこれらの抗体と比較しうる結合特性を有し、例えば、キメラ、ヒト化および1本鎖抗体ならびにそれらのフラグメントを含む。このような機能的同等物を作るほう方法は、PCT出願WO93/21319、欧州特許出願第239,400号;PCT出願WO89/09622;欧州特許出願第338,745号;および欧州特許出願第332,424号に開示されていて、これらはそれぞれの全てが参照により組み入れられる。
【0040】
機能的同等物は、本発明の抗体の可変もしくは超可変域のアミノ酸配列と実質的に同じアミノ酸配列をもつポリペプチドを含む。アミノ酸配列に適用される「実質的に同じ」は、本明細書においては、PearsonおよびLipman, Proc. Natl. Acad. Sci. USA 85, 2444-2448 (1988)によるFASTAサーチ法により決定して、他のアミノ酸配列と少なくともほぼ90%、さらに好ましくは少なくともほぼ95%配列同一性をもつ配列として定義される。
【0041】
キメラ抗体は好ましくは、実質的にまたは専らヒト抗体定常域から誘導された定常域および実質的にまたは専らヒト以外の哺乳類動物の可変域配列から誘導された可変域を有する。抗体のヒト化型は、例えばマウス抗体の相補性決定領域をヒトフレームワークドメイン中に置換することにより作られる(例えばPCT公開第W092/22653号を参照)。ヒト化抗体は好ましくは、実質的にまたは専ら対応するヒト抗体領域から誘導された定常域と相補性決定領域(CDR)以外の可変域、および実質的にまたは専らヒト以外の哺乳類動物から誘導されたCDRを有する。
【0042】
機能性同等物はまた、1本鎖抗体(scFv)としても知られる1本鎖抗体フラグメントを含む。これらのフラグメントは、1以上の相互接続リンカーを用いてまたは用いないで、少なくとも1つの抗体可変軽鎖配列(VL)のフラグメントと繋留された少なくとも1つの抗体可変重鎖配列(VH)のフラグメントを含有する。そのようなリンカーは、短いフレキシブルなペプチドであって、(VL)と(VH)ドメインが連結されるとそれらの適当な3次元フォールディングが起こり、1本鎖抗体フラグメントが誘導された元の全抗体の標的分子結合特異性を維持することを確保するように選択することができる。一般的に(VL)または(VH)配列のカルボキシ末端を、このようなペプチドリンカーにより相補的(VL)および(VH)配列のアミノ酸末端と共有結合で連結することができる。1本鎖抗体フラグメントは、分子クローニング、抗体ファージディスプレイライブラリーまたは類似の技術により作製することができる。これらのタンパク質は、真核生物細胞または細菌を含む原核生物細胞のいずれで産生させてもよい。
【0043】
1本鎖抗体フラグメントは、本明細書に記載の全抗体の可変または相補性決定領域(CDR)の少なくとも1つを有するが、これらの抗体の定常ドメインのいくらかまたは全てを欠いている。これらの定常ドメインは抗原結合に必要でないが、全抗体構造の主要部を構成する。従って、1本鎖抗体フラグメントは、定常ドメインの一部または全てを含有する抗体の利用に関連するいくつかの問題を克服することができる。例えば、1本鎖抗体フラグメントは、生物学的分子と重鎖定常域との間の望ましくない相互作用、または他の欲しない生物学的活性を持たない傾向がある。さらに、1本鎖抗体フラグメントは全抗体よりかなり小さく従ってより大きい毛細管透過性を有するので、1本鎖抗体フラグメントが局在化して標的抗原結合部位とより効率的に結合することが可能になる。また、抗体フラグメントは真核生物細胞において比較的大規模に生産することができるので、その生産が容易になる。さらに、1本鎖抗体フラグメントは比較的サイズが小さいので、全抗体よりレシピエントの免疫応答を引起す可能性が低い。
【0044】
機能的同等物はさらに、全抗体のそれと同じかまたは比較しうる結合特性を有する抗体のフラグメントを含む。そのようなフラグメントはFabフラグメントの1つもしくは両方またはF(ab')2フラグメントを含有してもよい。好ましくは抗体フラグメントは全抗体の6つの相補性決定領域の全てを含有するが、全てより少ない数、例えば3つ、4つまたは5つのかかる領域を含有するフラグメントもまた機能的である。さらに、機能的同等物は、次の免疫グロブリンクラス:IgG、IgM、IgA、IgDまたはIgE、およびそれらのサブクラスのいずれか1つであってもまたはそのメンバーの組合わせであってもよい。
【0045】
本明細書に記載した抗IGF-I受容体抗体EM164およびそのヒト化変異体に対するアミノ酸および核酸配列の知識を利用して、同じようにヒトIGF-I受容体と結合してIGF-I受容体の細胞機能を阻害する他の抗体を開発することができる。複数の研究は、1以上のアミノ酸変化を抗体の配列の様々な位置へ導入することの抗体の特性、例えば結合および発現のレベル、に与える効果を1次抗体配列の知識に基づいて調査している(Yang, W. P.ら, 1995, J. Mol. Biol., 254, 392-403;Rader, C.ら, 1998, Proc. Natl. Acad. Sci. USA, 95, 8910-8915;Vaughan, T. J.ら, 1998, Nature Biotechnology, 16, 535-539)。
【0046】
これらの研究において、オリゴヌクレオチド媒介の位置指定突然変異誘発、カセット突然変異誘発、誤りがちな(error-prone)PCR、DNAシャッフリング、または大腸菌(E. coli)の変異誘発株などの技術を用いて、CDR1、CDR2、CDR3、またはフレームワーク領域の重および軽鎖遺伝子の配列を変えることにより、1次抗体の変異体が作製されている(Vaughan, T. J.ら, 1998, Nature Biotechnology, 16, 535-539;Adey, N. B.ら, 1996, 第16章, pp.277-291, in 「ペプチドとタンパク質のファージディスプレイ(Phage Display of Peptides and Proteins)」, Kay, B.K.ら編, Academic Press)。1次抗体の配列を変えるこれらの方法は、2次抗体の親和性の改良をもたらしている(Gram, H.ら, 1992, Proc. Natl. Acad. Sci. USA, 89, 3576-3580;Boder, E. T.ら, 2000, Proc. Natl. Acad. Sci. USA, 97, 10701-10705;Davies, J. and Riechmann, L., 1996, Immunotechnolgy, 2, 169-179;Thompson, J.ら, 1996, J. Mol. Biol., 256, 77-88;Short, M. K.ら, 2002, J. Biol. Chem., 277, 16365-16370;Furukawa, K.ら, 2001, J. Biol. Chem., 276, 27622-27628)。
【0047】
抗体の1以上のアミノ酸残基を変える類似の定方向(directed)手法により、本発明に記載した抗体配列を利用して改良された機能をもつ抗IGF-I受容体抗体を開発することができる。
【0048】
本発明のコンジュゲートは、細胞傷害薬に連結された本発明に記載の抗体、フラグメント、およびそれらの類似体を含む。好ましい細胞傷害薬はメイタンシノイド、タキサンおよびCC-1065の類似体である。コンジュゲートはin vitroの方法により調製することができる。細胞傷害薬を抗体と連結する目的で連結基を用いる。適当な連結基は当技術分野では周知であり、ジスルフィド基、チオエーテル基、酸に不安定な基、感光性基、ペプチダーゼ易分解性基およびエステラーゼ易分解性基を含む。好ましい連結基はジスルフィド基およびチオエーテル基である。例えば、コンジュゲートは、ジスルフィド交換反応を用いてまたは抗体と細胞傷害薬の間にチオエーテル基を形成することにより構築することができる。
【0049】
メイタンシノイドとメイタンシノイド類似体が好ましい細胞傷害薬として挙げられる。好適なメイタンシノイドとメイタンシノイド類似体の例は、米国特許第4,424,219号;第4,256,746号;第4,294,757号;第4,307,016号;第4,313,946号;第4,315,929号;第4,331,598号;第4,361,650号;第4,362,663号;第4,364,866号;第4,450,254号;第4,322,348号;第4,371,533号;第6,333,410号;第5,475,092号;第5,585,499号;および第5,846,545号に開示されている。
【0050】
タキサンも好ましい細胞傷害薬である。本発明に使用するのに好適なタキサンは米国特許第6,372,738号および第6,340,701号に開示されている。
【0051】
CC-1065とその類似体も本発明に使用するのに好ましい細胞傷害薬である。CC-1065とその類似体は米国特許第6,372,738号;第6,340,701号;第5,846,545号および第5,585,499号に開示されている。
【0052】
このような細胞傷害性コンジュゲートを調製するための魅力的な候補薬はCC-1065であり、これはストレプトマイセス・ゼレンシス(Streptomyces zelensis)の培養ブロスから単離された強力な抗腫瘍抗生物質である。CC-1065は通常使用される抗癌薬、例えばドキソルビシン、メトトレキセートおよびビンクリスチンよりもin vitroでほぼ1000倍以上強力である(B.K. Bhuyanら, Cancer Res., 42, 3532-3537 (1982))。
【0053】
メトトレキセート、ダウノルビシン、ドキソルビシン、ビンクリスチン、ビンブラスチン、メルファラン、マイトマイシンC、クロラムブシル、およびカリケアマイシンなどの細胞傷害薬も本発明のコンジュゲートを調製するのに好適であり、薬物分子を抗体分子と仲介担体分子、例えば血清アルブミンを介して連結することもできる。
【0054】
診断上の応用については、典型的には本発明の抗体を検出可能部分を用いて標識しうる。検出可能部分は、検出可能なシグナルを直接的にまたは間接的に作製しうるのであればいずれのものでもよい。例えば、検出可能部分は放射性同位体、例えば3H、14C、32P、35S、もしくは131I;蛍光もしくは化学発光化合物、例えばフルオレセインチオシアネート、ローダミン、もしくはルシフェリン;または酵素、例えばアルカリホスファターゼ、β-ガラクトシダーゼ、もしくは西洋わさびペルオキシダーゼであってもよい。
【0055】
抗体を検出可能な部分とコンジュゲートするために、当技術分野で公知のいずれの方法を採用してもよく、Hunter,ら, Nature 144:945 (1962);David,ら, Biochemistry 13:1014 (1974);Pain,ら, J. Immunol. Meth. 40:219 (1981);およびNygren, J. Histochem. and Cytochem. 30:407 (1982)により記載された方法を含む。
【0056】
本発明の抗体はいずれの公知のアッセイ方法、例えば競合結合アッセイ、直接および間接サンドイッチアッセイおよび免疫沈降アッセイにおいても使用することができる(Zola, 「モノクローナル抗体:技術のマニュアル(Monoclonal Antibodies: A Manual of Techniques)」, pp.147-158 (CRC Press, Inc., 1987))。
【0057】
本発明の抗体はまた、in vivoイメージングにも有用であり、ここでは検出可能部分、例えば放射不透明剤または放射性同位体を用いて標識した抗体を、被験者に好ましくはその血流中に投与し、そして標識した抗体の宿主中の位置をアッセイする。このイメージング技術は悪性腫瘍の段階判定および治療に有用である。抗体を、核磁気共鳴、放射線学または他の当技術分野で知られる検出手段のいずれかにより宿主中で検出可能であるいずれの部分を用いて標識してもよい。
【0058】
本発明の抗体はまた、アフィニティ精製剤としても有用である。このプロセスにおいては、抗体を好適な支持体、例えばセファデックス(Sephadex)樹脂または濾紙上に当技術分野で周知の方法を用いて固定する。
【0059】
本発明の抗体はまた、それらの細胞中のIGF-I受容体機能の阻害に基づく生物学的研究の試薬としても有用である。
【0060】
治療上の応用については、本発明の抗体またはコンジュゲートを、製薬上許容される剤形で被験者に投与する。それらを、ボーラスとしてまたはある期間にわたる連続輸液により静脈内に、筋肉内、皮下、関節内、滑膜内、くも膜下腔内、経口、局所または吸入経路により投与することができる。抗体はまた、腫瘍内、腫瘍周囲、病変部内、または病変部周囲経路により投与して、局所ならびに全身治療効果を与えてもよい。好適な製薬上許容される担体、希釈剤、および賦形剤は当業者に周知でありかつ当業者が臨床症状に基づいて決定することができる。好適な担体、希釈剤、および/または賦形剤の例としては、(1)約1mg/ml〜25mg/mlヒト血清アルブミンを含有するダルベッコ(Dulbecco)リン酸緩衝化生理食塩水、pH約7.4、(2)0.9%生理食塩水(0.9%w/v NaCl)、および(3)5%(w/v)デキスロースが挙げられる。本発明の方法はin vitro、in vivo、またはex vivoで実施することができる。
【0061】
他の治療処置においては、本発明の抗体、抗体フラグメントまたはコンジュゲートを1以上のさらなる治療薬とともに同時投与する。好適な治療薬は、限定されるものでないが、細胞傷害または細胞分裂停止薬を含む。タキソールは、細胞傷害薬でもある好ましい治療薬である。
【0062】
癌治療薬は、癌細胞を殺滅するかまたはその増殖を制限する一方、宿主への障害を最小化することを求める薬剤である。従って、そのような薬剤は、癌細胞の健康な宿主細胞との特性の相違(例えば代謝、血管新生または細胞表面抗原提示)を利用することができる。腫瘍形態学の相違は可能性のある介入部位となる:例えば、第2の治療薬は抗体、例えば固形腫瘍内部の血管新生を遅延させてそれにより増殖速度を遅くするのに有用である抗VEGF抗体であってもよい。他の治療薬としては、限定されるものでないが、補助薬、例えば塩酸グラニセトロン、男性ホルモンインヒビター、例えば酢酸ロイプロリド、抗生物質、例えばドキソルビシン、抗女性ホルモン剤、例えばタモキシフェン、代謝拮抗剤、例えばインターフェロンα-2a、細胞傷害薬、例えばタキソール、酵素阻害剤、例えばrasファルネシルトランスフェラーゼ阻害剤、免疫調節物質、例えばアルデスロイキン、および窒素マスターバッチ誘導体、例えば塩酸メルファランなどが挙げられる。
【0063】
水溶液剤形で存在するとき(凍結乾燥されているのでなく)、抗体は典型的には約0.1mg/ml〜100mg/mlの濃度で製剤されるが、この範囲外の広い変化も許容される。疾患の治療に適当な抗体またはコンジュゲートの投与量は、先に規定した治療する疾患の型、疾患の重篤度と経過、抗体を予防用にまたは治療用に投与するかどうか、先の治療の経過、患者の臨床歴と抗体に対する応答、および主治医の熟慮に依存しうる。抗体を、好適なように、患者に1回でまたは一連の治療全体にわたって投与する。
【0064】
疾患の型と重篤度に依存して、約0.015〜15mg抗体/kg患者体重が、例えば1回以上の分離投与によりまたは連続輸液により患者に投与する最初の候補投与量である。数日以上にわたる反復投与については、症状に依存して、所望の病状抑制が起こるまで治療を繰返す。しかし他の投与体制も有用でありうるのであって、それらを排除するものではない。
【実施例】
【0065】
本発明をこれから次の実施例を参照して記載するが、これは説明のためだけであって、本発明を限定することを意図しない。
【0066】
実施例1:マウスEM164抗体
この最初の実施例においては、本発明のマウス抗体の完全な1次アミノ酸構造およびcDNA配列を、その結合特性および組換え型におけるその発現方法とともに開示する。従って、本発明の抗体とその調製法の全てのかつ完全な開示が提供され、免疫学分野の業者であれば、過剰な実験をすることなく、上記抗体を調製することができよう。
【0067】
A.抗IGF-I受容体モノクローナル抗体ハイブリドーマの作製
Y1251F突然変異をもつヒトIGF-I受容体を発現する細胞系統は高い数(1細胞当たり〜107)のIGF-I受容体を発現するので、この細胞系統を免疫感作に用いた。IGF-I受容体の細胞質ドメインにおけるY1251F突然変異は、トランスフォーメーションおよび抗アポトーシスシグナル伝達の損失をもたらしたが、IGF-I結合およびIGF-I刺激による分裂促進シグナル伝達に影響を与えなかった(O'Connor, R.ら, 1997, Mol. Cell. Biol., 17, 427-435;Miura, M.ら, 1995, J. Biol. Chem., 270, 22639-22644)。突然変異は他の点で抗体作製に影響を与えなかった、何故なら、この実施例の抗体は、Y1251Fと野生型受容体の両方に対して同等であるIGF-I受容体の細胞外ドメインと結合するからである。
【0068】
Y1251F突然変異をもつヒトIGF-I受容体を発現する細胞系統はIGF-I-受容体-欠損マウスの3T3様細胞から、Y1251F-突然変異ヒトIGF-I-受容体遺伝子と一緒にピューロマイシン耐性遺伝子によるトランスフェクションにより作製し、そしてピューロマイシン(2.5μg/mL)を用いて、および高いIGF-I受容体発現についてFACSソーティングにより選択した(Miura, M.ら, 1995, J. Biol. Chem., 270, 22639-22644)。高レベルのIGF-I受容体発現を有する細胞系統をさらに、ほとんどの細胞にとって毒性がある高濃度、例えば25μg/mLのピューロマイシンを用いて選択した。生存コロニーを拾い、高レベルのIGF-I受容体発現を提示するものを選択した。
【0069】
6月齢のCAF1/J雌性マウスを、第0日にY1251F突然変異したヒトIGF-I-受容体を過剰発現する細胞(5x105細胞、0.2mL PBS中に懸濁)を用いて腹腔内に免疫感作した。動物を0.2mL細胞懸濁液を用いて次のように追加免疫した:第2日、1x106細胞;第5日、2x106細胞;第7、9、12、および23日、1x107細胞。第26日にマウスを犠牲にしてその脾臓を取除いた。
【0070】
脾臓を2つの艶消しガラススライド間で磨砕して単一細胞懸濁液を得て、これをペニシリンとストレプトマイシンを含有する無血清RPMI培地(SFM)を用いて洗浄した。脾臓細胞ペレットを0.83%(w/v)塩化アンモニウム水溶液10mL中に、10分間氷上で再懸濁し、赤血球を溶解し、次いで無血清培地(SFM)を用いて洗浄した。脾臓細胞(1.2x108)を非分泌マウス骨髄腫細胞系統P3X63Ag8.653(ATCC、Rockville、MD;Cat.#CRL1580)からの骨髄腫細胞(4x107)とともにチューブ中にプールし、そして無血清RPMI-1640培地(SFM)を用いて洗浄した。上清を除去して細胞ペレットを残りの培地中に再懸濁した。チューブを37℃の水の入ったビーカー内に置いて、チューブを静かに振りながら、1.5mLのポリエチレングリコール溶液(50%(w/v)PEG、平均分子量1500を含有する75mM HEPES、pH 8)をゆっくりと0.5mL/分の滴下速度で加えた。1分間待った後に、10mLのSFMを次のように加えた:最初の1分間にわたり1mL、次の1分間にわたり2mL、そして第3の1分間にわたり7mL。次いでさらに10mLをゆっくりと1分間にわたり加えた。細胞を遠心分離によりペレット化し、SFM中で洗浄し、5%胎児ウシ血清(FBS)、ヒポキサンチン/アミノプテリン/チミジン(HAT)、ペニシリン、ストレプトマイシン、および10%ハイブリドーマクローニングサプリメント(HCS)を補充したRPMI-1640増殖培地中に再懸濁した。細胞を96ウエル平底組織培養プレート中に、1ウエル当たり200μL中の2x105脾臓細胞の割合で播いた。5〜7日後に、1ウエル当たり100μLを取除いて、ヒポキサンチン/チミジン(HT)および5%FBSを補充した増殖培地と置換えた。免疫感作およびハイブリドーマ産生に用いた一般条件は、J. LangoneおよびH. Vunakis(編, 「酵素学の方法(Methods in Enzymology)」, Vol. 121, 「免疫化学技術、第I部(Immunochemical Techniques, Part I)」;1986;Academic Press, Florida)ならびにE. HarlowおよびD. Lane(「抗体:研究室マニュアル(Antibodies: A Laboratory Manual)」; 1988; Cold Spring Harbor Laboratory Press, New York)に記載された通りである。他の免疫感作およびハイブリドーマ産生の技術も利用することができ、当業者は周知している。
【0071】
ハイブリドーマクローンから得た培養物上清を、ELISAにより、精製ヒトIGF-I受容体との結合について、ELISAとFACSにより、ヒトIGF-I受容体を過剰発現する細胞との特異的結合についておよびヒトインスリン受容体を過剰発現する細胞との結合の欠如について、以下に記載の通りスクリーニングした。ヒトIGF-I受容体を過剰発現する細胞に対する結合親和性がヒトインスリン受容体を過剰発現する細胞に対する結合親和性より高いクローンを拡大してサブクローニングした。サブクローンの培養上清を上記結合アッセイによってさらにスクリーニングした。この方法で、サブクローン3F1-C8-D7(EM164)を選択し、重および軽鎖遺伝子をクローニングし、そして下記の通り配列決定した。
【0072】
ハイブリドーマクローンから得た上清をIGF-I受容体との結合についてスクリーニングするのに用いるため、以下の方法によりヒトIGF-I受容体を単離した。組換えIGF-Iをビオチン化試薬、例えばスルホ-NHS-LC-ビオチン、スルホ-NHS-SS-ビオチン、またはNHS-PEO4-ビオチンを用いて改変することにより、ビオチン化IGF-Iを調製した。ビオチン化IGF-Iをストレプトアビジン-アガロースビーズ上に吸収させ、ヒト野生型またはY1251F突然変異体IGFRを過剰発現した細胞から得たライセートとともにインキュベートした。ビーズを2〜4M尿素および界面活性剤、例えばトリトンX-100またはオクチル-β-グルコシドを含有するバッファーを用いて洗浄しかつ溶出した。溶出したIGF-I受容体をPBSに対して透析し、SDS-PAGEにより還元条件のもとで純度を分析し、その結果はIGF-I受容体のαおよびβ鎖バンドの分子量である約135kDaおよび95kDaをそれぞれ示した。
【0073】
ハイブリドーマ上清の精製IGF-I受容体との結合を確認するために、Immulon-4HB ELISAプレート(Dynatech)を、pH 9.5の50mM CHESバッファー中に希釈した(100μL;4℃、一夜)精製ヒトIGF-I受容体サンプル(アフィニティ精製したサンプルの尿素/オクチル-β-グルコシド溶出液から透析により調製した)を用いてコーティングした。ウエルをブロッキングバッファー(10mg/mL BSAを含む、50mMトリス、150mM NaCl、pH 7.5、および0.1% tween-20を含有するTBS-Tバッファー)200μLを用いてブロックし、ハイブリドーマクローン(100μL;ブロッキングバッファーに希釈された)からの上清とともに約1〜12時間インキュベートし、TBS-Tバッファーを用いて洗浄し、そしてヤギ抗マウスIgG-Fc抗体-西洋わさびペルオキシダーゼ(HRP)コンジュゲート(100μL;0.8μg/mLを含むブロッキングバッファー;Jackson ImmunoResearch Laboratories)とともにインキュベートし、次いで洗浄しそしてABTS/H2O2基質(0.5mg/mL ABTS、0.03% H2O2を含む0.1Mクエン酸バッファー、pH 4.2)を用いて405nmにて検出した。典型的な3F1ハイブリドーマ サブクローンからの上清は約1.2吸収単位のシグナルを3分以内の発現で生じ、他のハイブリドーマクローンからの上清で得た0.0と対照的であった。このELISAに対する一般条件は、E. HarlowおよびD. Lane(「抗体の利用:研究室マニュアル(Using Antibodies: A Laboratory Manual)」;1999, Cold Spring Harbor Laboratory Press, New York)に記載の抗体結合および検出に対する標準ELISA条件と類似し、この標準条件を利用することもできる。
【0074】
ハイブリドーマ上清のヒトIGF-I受容体との特異的結合およびヒトインスリン受容体との非結合に対するスクリーニングを、ELISAを用いて、ヒトY1251F-IGF-I受容体過剰発現細胞系統およびヒトインスリン受容体過剰発現細胞系統について実施した。両方の細胞系統をIGF-I受容体欠損マウスの3T3様細胞から作製した。IGF-I受容体過剰発現細胞およびインスリン受容体過剰発現細胞を別々に組織培養フラスコから迅速トリプシン/EDTA処理により収穫し、10%FBSを含有する増殖培地中に懸濁し、遠心分離によりペレット化し、そしてPBSを用いて洗浄した。洗浄した細胞(100μL、約1〜3x106細胞/mL)を、植物性血球凝集素(PHA)(100μL、20μg PHA/mL)を用いてコーティングしたImmulon-2HBプレートのウエルに加え、遠心分離してPHAコーティングウエルに10分間接着させた。細胞が接着したウエルを指ではじいてPBSを除去し、次いで一夜37℃で乾燥した。ウエルをPBS中の5mg/mL BSA溶液を用いて1時間37℃でブロックし、次いで静かにPBSを用いて洗浄した。次いでハイブリドーマクローン(100μL;ブロッキングバッファー中に希釈)からの上清のアリコートをIGF-I-受容体過剰発現細胞を含有するウエルおよびインスリン受容体過剰発現細胞を含有するウエルに加え、そして常温で1時間インキュベートした。ウエルをPBSを用いて洗浄し、ヤギ抗マウスIgG-Fc抗体-西洋わさびペルオキシダーゼコンジュゲート(100μL;ブロッキングバッファー中に0.8μg/mLを含む)とともに1時間インキュベートし、次いで洗浄し、そしてABTS/H2O2基質を用いて検出した。典型的な3F1ハイブリドーマサブクローンからの上清はIGF-I受容体を過剰発現する細胞とインキュベーションすると、12分以内の発現で0.88吸収単位のシグナルを生じ、ヒトインスリン受容体を過剰発現する細胞とのインキュベーションで得た0.22吸収単位の値とは対照的であった。
【0075】
ハイブリドーマをIntegra-CL350フラスコ(Integra Biosciences, Maryland)中で製造業者の明細書に従って増殖し、精製EM164抗体を得た。ほぼ0.5〜1mg/mL抗体の収率が、抗体標準を用いるELISAおよびSDS-PAGE/クーマシーブルー染色による定量に基づいて、Integraフラスコから収穫した上清から得られた。抗体をアフィニティクロマトグラフィにより、プロテインA-アガロースビーズカラム上に標準精製条件のもとで供給して精製し、3M NaClを含有する100mMトリスバッファー、pH 8.9で洗浄し、次いで150mM NaClを含有する100 mM酢酸溶液に溶出した。抗体を含有する溶出した画分を冷2M K2HPO4溶液により中和して、PBS中で4℃にて透析した。抗体の濃度は、280nmで測定することにより決定した(吸光係数=1.4mg-1mLcm-1)。精製抗体サンプルを還元条件下のSDS-PAGEおよびクーマシーブルー染色により分析し、その結果は、それぞれ約55kDaおよび25kDaである抗体の重および軽鎖のバンドだけを示した。精製抗体のイソ型はκ軽鎖をもつIgG1であった。
【0076】
B.EM164抗体の結合特性
精製EM164抗体の特異的結合を、蛍光活性化細胞選別(フローサイトメトリー)(FACS)によりIGF-I受容体を過剰発現する細胞を用いておよびヒトインスリン受容体を過剰発現する細胞を用いて実証した(図1)。100μL冷FACSバッファー(1mg/mL BSAを含むダルベッコ(Dulbecco's)MEM培地)中のEM164抗体(50-100nM)のインキュベーションを、IGF-I受容体を過剰発現する細胞を用いておよびインスリン受容体を過剰発現する細胞を用いて(2x105細胞/mL)、丸底96ウエルプレート中で1時間実施した。細胞を遠心分離によりペレット化し、冷FACSバッファーを用いて静かに指ではじくことにより洗浄し、次いでヤギ抗マウスIgG抗体-FITCコンジュゲート(100μL;10μg/mL FACSバッファー)とともに氷上で1時間インキュベートした。細胞をペレット化し、洗浄し、そしてPBS中の1%ホルムアルデヒド溶液120μLに再懸濁した。プレートをFACS Caliburリーダー(BD Biosciences)を用いて分析した。
【0077】
IGF-I受容体を過剰発現する細胞をEM164抗体とインキュベートすると強い蛍光シフトが得られ、インスリン受容体を過剰発現する細胞をEM164抗体とインキュベートしたときに得た非有意なシフトとは対照的であった(図1)。この結果はEM164抗体のIGF-I受容体との結合が選択的であって、インスリン受容体とは結合しないことを示した。対照抗体である、抗IGF-I受容体抗体1H7(Santa Cruz Biotechnology)および抗インスリン受容体α抗体(BD Pharmingen Laboratories)は、それぞれIGF-I受容体およびインスリン受容体を過剰発現する細胞とインキュベートすると蛍光シフトを生じた(図1)。強い蛍光シフトはまた、IGF-I受容体を発現するEM164抗体およびヒト乳癌MCF-7細胞(Dufourny, B.ら, 1997, J. Biol. Chem., 272, 31163-31171)を用いるFACSアッセイによっても観察され、この結果はEM164抗体がヒト腫瘍細胞の表面上のヒトIGF-I受容体と結合することを示した。
【0078】
EM164抗体のヒトIGF-I受容体との結合に対する解離定数(Kd)を、複数濃度における抗体の結合のELISA滴定により、直接コーティングしたIGF-I受容体(上記のとおり、ビオチン化IGF-Iを用いてアフィニティ精製された)または間接的に捕獲したビオチン化IGF-I受容体により決定した。ビオチン化IGF-I受容体を、IGF-I受容体を過剰発現する細胞からPEO-マレイミド-ビオチン試薬(Pierce、Molecular Biosciences)を用いて界面活性剤可溶化ライセートのビオチン化により調製し、これをNHS-アガロースビーズ上に固定した抗IGF-I受容体β鎖抗体を用いてアフィニティ精製し、NP-40界面活性剤を含有するバッファー中の2〜4M尿素溶液を用いて溶出し、そしてPBSに透析した。
【0079】
EM164抗体のビオチン化IGF-I受容体との結合に対するKdの測定は、Immulon-2HBプレートを、炭酸バッファー(150mM炭酸ナトリウム、350mM炭酸水素ナトリウム)中の1μg/mLストレプトアビジン溶液100μLを用いて4℃で一夜、コーティングして実施した。ストレプトアビジンでコーティングしたウエルを、ブロッキングバッファー(10mg/mL BSAを含むTBS-Tバッファー)200μLを用いてブロックし、TBS-Tバッファーを用いて洗浄し、そしてビオチン化IGF-I受容体(10〜100ng)とともに4時間常温でインキュベートした。間接的に捕獲したビオチン化IGF-I受容体を含有するウエルを次いで洗浄し、そしてEM164抗体とブロッキングバッファー中で複数の濃度(5.1x10-13M〜200nM)において2時間、常温でインキュベートし、次いで一夜4℃にてインキュベートした。ウエルを次にTBS-Tバッファーを用いて洗浄し、そしてヤギ抗マウスIgGH+L抗体-西洋わさびペルオキシダーゼコンジュゲート(100μL;0.5μg/mLを含むブロッキングバッファー)とインキュベートし、次いで洗浄し、そしてABTS/H2O2基質を用いて405nmにて検出した。Kd値を1つの部位との結合に対する非線形回帰により計算した。
【0080】
同様な結合滴定を、E. HarlowおよびD. Lane(「抗体の利用:研究室マニュアル(Using Antibodies: A Laboratory Manual)」;1999, Cold Spring Harbor Laboratory Press, New York)に記載のように、抗体のパパイン消化により調製したEM164抗体のFabフラグメントを用いて実施した。
【0081】
EM164抗体のビオチン化ヒトIGF-I受容体との結合に対する結合滴定曲線から、0.1nMのKd値を得た(図2)。EM164抗体のFabフラグメントもヒトIGF-I受容体と0.3nMのKd値で非常に強く結合し、この結果は、EM164抗体のIGF-I受容体に対するモノマー結合も非常に強いことを示した。
【0082】
EM164抗体によるIGF-I受容体の結合に対する解離定数のこの極めて低い値は一部は非常に遅いkoff速度によるものであって、これは1〜2日の長い洗浄後に観察される固定されたIGF-I受容体と結合した抗体の強い結合シグナルにより立証された。
【0083】
EM164抗体はIGF-I受容体の免疫沈降に使用することができ、これはヒト乳癌MCF-7細胞の界面活性剤可溶化ライセートのプロテインG-アガロースビーズ上に固定されたEM164抗体(Pierce Chemical Company)とのインキュベーションにより実証される。EM164抗体免疫沈降のウェスタンブロットを、ウサギポリクローナル抗IGF-I受容体β鎖(C-末端)抗体(Santa Cruz Biotechnology)およびヤギ抗ウサギIgG抗体西洋わさびペルオキシダーゼコンジュゲートを用いて処理し、次いで洗浄し、そして増強化学発光(ECL)検出により検出した。MCF-7細胞からのEM164免疫沈降物のウェスタンブロットは、IGF-I受容体のβ鎖に対応するバンドを約95kDaに、そしてプロIGF-I受容体に対応するバンドを約220kDaに表した。同様な免疫沈降をEM164抗体の結合の種特異性を調べるために他の細胞型に対して実施した。EM164抗体はcos-7細胞(アフリカミドリザル)からのIGF-I受容体とも結合したが、3T3細胞(マウス)、CHO細胞(チャイニーズハムスター)またはヤギ線維芽細胞(ヤギ)のIGF-I受容体と結合しなかった。EM164抗体はMCF-7細胞からのライセートのウェスタンブロット中のSDS変性ヒトIGF-I受容体を検出しなかった。この結果は、EM164抗体が自然の、非変性ヒトIGF-I受容体のコンフォメーションのエピトープと結合することを示した。
【0084】
EM164抗体の結合ドメインを、16量体C末端片(残基704〜719)と融合したL1およびL2ドメインにフランキングするシステインリッチドメイン(残基1〜468)を含みかつC末端エピトープタグで終わる末端切断α鎖構築物を用いてさらに特徴づけた。残基469〜703を欠如するこのより小さいIGF-I受容体は、IGF-Iと結合するが、未変性の全長IGF-I受容体と比較すると強さが劣ると報じられている(Molina, L.ら, 2000, FEBS Letters, 467, 226-230;Kristensen, C.ら, 1999, J. Biol. Chem., 274, 37251-37356)。そこで、残基704〜719であるC末端片と融合した残基1〜468を含み、かつC末端mycエピトープタグがフランキングする末端切断IGF-I受容体α鎖構築物を調製した。この構築物を発現する安定な細胞系統であって、かつまたこの構築物をヒト胚腎293T細胞において一過的に発現する細胞系統を構築した。EM164抗体のこの末端切断IGF-I受容体α鎖構築物との強い結合が観察された。試験した2つの抗体のうち、IR3(Calbiochem)もこの末端切断α鎖と結合したが、1H7抗体(Santa Cruz Biotechnology)は結合せず、これはEM164抗体のエピトープが明らかに1H7抗体のそれとは別であることを示した。
【0085】
C.EM164抗体による、IGF-IのMCF-7細胞との結合の阻害
IGF-Iのヒト乳癌MCF-7細胞との結合はEM164抗体により阻害された(図3)。MCF-7細胞を5μg/mL EM164抗体とともにまたはEM164抗体なしで2時間、無血清培地においてインキュベートし、次いで50ng/mLビオチン化IGF-Iとともに20分間、37℃にてインキュベートした。次いで細胞を2回、無血清培地を用いて洗浄して非結合ビオチン-IGF-Iを除去し、次いで1%NP-40およびプロテアーゼ阻害剤を含有する50mM HEPES、pH 7.4中に溶解した。Immulon-2HB ELISAプレートをマウスモノクローナル抗IGF-I受容体β鎖抗体を用いてコーティングし、これを用いてライセートからIGF-I受容体および結合したビオチン-IGF-Iを捕獲した。コーティングされた抗体のIGF-I受容体β鎖細胞質C末端ドメインとの結合は、ビオチン-IGF-IのIGF-I受容体細胞外ドメインとの結合を妨げなかった。ウエルを洗浄し、ストレプトアビジン-西洋わさびペルオキシダーゼコンジュゲートとともにインキュベートし、再び洗浄し、次いでABTS/H2O2基質を用いて検出した。5μg/mL EM164抗体によるMCF-7細胞に対するIGF-I結合の阻害は本質的に定量的であって、ビオチン-IGF-Iを欠く対照を用いて得たELISAバックグラウンドのそれとほとんど同等であった。
【0086】
EM164抗体によるIGF-IのMCF-7細胞との結合の阻害に対する上記アッセイに加えて、次のアッセイはEM164抗体がMCF-7細胞に結合したIGF-Iと極めて効果的に置換わることを実証し、これは生理学的条件のもとで拮抗性抗IGF-I受容体抗体が結合した内因生理学的リガンド(例えば、IGF-IまたはIGF-II)と置換わるために所望されることである。このIGF-I置換えアッセイにおいて、12ウエルプレート中で増殖したMCF-7細胞を血清飢餓させ、次いでビオチン化IGF-I(20〜50ng/mL)とともに無血清培地中で37℃(または4℃)にて1〜2時間インキュベートした。結合したビオチン化IGF-Iをもつ細胞を、次いでEM164抗体または対照抗体(10-100μg/mL)を用いて37℃(または4℃)で30分間〜4時間処理した。次いで細胞をPBSを用いて洗浄し、1% NP-40を含有する溶解バッファー中で4℃で溶解した。ELISAを上記の通り実施し、ライセートからIGF-I受容体を捕獲し、次いで受容体と結合したビオチン化IGF-Iをストレプトアビジン-西洋わさびペルオキシダーゼコンジュゲートを用いて検出した。このELISAは、EM164抗体が細胞に前結合したビオチン化IGF-Iと37℃にてほぼ完全に(30分間以内に90%および4時間以内に〜100%)かつ4℃にて2時間で約50%だけ置換わりうることを実証した。他の実験においては、NCI-H838肺癌細胞をビオチン-IGF-Iとともにインキュベートし、次いで洗浄し、そしてEM164抗体とともに4℃にて2時間インキュベートして、結合したビオチン-IGF-Iの80%減少を得た。従ってEM164抗体は、癌細胞から前結合したIGF-Iを追い出す点で極めて有効であり、これは結合した内因性生理学的リガンドを追い出すことによるIGF-I受容体の拮抗作用として治療上重要でありうる。
【0087】
抗IGF-I受容体β鎖抗体(Santa Cruz Biotechnology;sc-713)を用いるウェスタンブロット分析に基づくと、MCF-7細胞のEM164抗体との4℃で2時間(または37℃で30分間)のインキュベーションはIGF-I受容体の有意な下方調節を生じなかったが、EM164抗体との37℃で2時間のさらに長いインキュベーションはIGF-I受容体の25%下方調節を生じた。従って、これらの短時間実験における4℃および37℃にてのEM164抗体によるIGF-Iの結合の阻害および結合したIGF-Iの置換えは、EM164抗体の結合による受容体の下方調節によって説明することができない。IGF-IのIGF-I受容体との結合の強力な阻害およびEM164抗体による前結合したIGF-Iの置換えの機構は、結合部位の共有を介してまたは立体閉塞を介してまたはアロ立体効果を介しての結合に対する競合であると思われる。
【0088】
D.EM164抗体による、IGF-I受容体が媒介する細胞シグナル伝達の阻害
乳癌MCF-7細胞および骨肉腫SaOS-2細胞のEM164抗体による処理は細胞内IGF-I受容体シグナル伝達をほとんど完全に阻害した。これをIGF-I受容体自己リン酸化の阻害によりおよびその下流のエフェクター、例えばインスリン受容体基質-1(IRS-1)、AktおよびErk1/2(図4〜6)のリン酸化の阻害により示す。
【0089】
図4においては、MCF-7細胞を12ウエルプレート中で通常培地で3日間増殖し、次いで20μg/mL EM164抗体(または抗B4対照抗体)を用いて無血清培地で3時間処理し、次に50ng/mL IGF-Iを用いて20分間37℃で刺激した。次いで細胞を、プロテアーゼおよびホスファターゼインヒビターを含有する氷冷溶解バッファー(50mM HEPESバッファー、pH 7.4、1% NP-40、1mMオルトバナジン酸ナトリウム、100mMフッ化ナトリウム、10mMピロリン酸ナトリウム、2.5mM EDTA、10μMロイペプチン、5μMペプスタチン、1mM PMSF、5mMベンズアミジン、および5μg/mLアプロチニン)中に溶解した。ELISAプレートを抗IGF-I受容体β鎖C末端モノクローナル抗体TC123を用いてプレコートし、そしてライセートサンプルとともに5時間常温でインキュベートし、IGF-I受容体を捕獲した。次いで捕獲したIGF-I受容体を含有するウエルを洗浄し、ビオチン化抗ホスホチロシン抗体(PY20;0.25μg/mL;BD Transduction Laboratories)とともに30分間インキュベートし、次いで洗浄し、ストレプトアビジン-西洋わさびペルオキシダーゼコンジュゲート(0.8μg/mL)とともに30分間インキュベートした。ウエルを洗浄し、ABTS/H2O2基質を用いて検出した。対照抗B4抗体を使用すると、IGF-I受容体のIGF-I刺激自己リン酸化の無阻害を示した。対照的に、EM164抗体を用いて処理するとIGF-I受容体のIGF-I刺激自己リン酸化の完全な阻害を得た(図4)。
【0090】
インスリン受容体基質-1(IRS-1)のリン酸化の阻害を実証するため、固定した抗IRS-1抗体を使ってライセートからIRS-1を捕獲するELISAを用い、次いでホスホリル化IRS-1と結合するホスファチジルイノシトール-3-キナーゼ(PI3キナーゼ)に随伴するp85サブユニットを測定した(Jackson, J. G.ら, 1998, J. Biol. Chem., 273, 9994-10003)。図5において、MCF-7細胞を5μg/mL抗体(EM164またはIR3)とともに無血清培地において2時間処理し、次いで50ng/mL IGF-Iを用いて10分間37℃にて刺激した。抗IRS-1抗体(ウサギポリクローナル;Upstate Biotechnology)を間接的にELISAプレート上にコーティングしたヤギ抗ウサギIgG抗体とのインキュベーションにより捕獲し、次いでこれを利用して細胞ライセートからIRS-1を4℃の一夜インキュベーションにより捕獲した。次いでウエルをマウスモノクローナル抗p85-PI3キナーゼ抗体(Upstate Biotechnology)と4時間インキュベートし、次に、ヤギ抗マウス-IgG-抗体-HRPコンジュゲートにより30分間処理した。ウエルを次いで洗浄し、ABTS/H2O2基質を用いて検出した(図5)。図5に示すように、EM164抗体によるIGF-I-刺激IRS-1リン酸化の阻害はIR3抗体より効果的であり、かつEM164抗体はIGF-Iの不在の細胞とインキュベートしたとき、IRS-1リン酸化に対する作動活性を示さなかった。
【0091】
他の下流エフェクター、例えばAktおよびErk1/2の活性化も、SaOS-2細胞(図6)およびMCF-7細胞においてEM164抗体により用量に依存する様式で阻害され、これはライセートのウェスタンブロットおよびリン酸化特異的抗体(ウサギポリクローナル抗-ホスホ-Ser473Aktポリクローナルおよび抗ホスホ-ERK1/2抗体;Cell Signaling Technology)を用いて示した通りである。汎ERK抗体は全てのレーンで等しいタンパク質負荷を示した(図6)。EM164抗体によるSaOS-2細胞の処理は、Erk1/2のEGF-刺激リン酸化を阻害せず、従ってIGF-I受容体シグナル伝達経路のEM164抗体による阻害の特異性を実証した。
【0092】
E.EM164抗体による、ヒト腫瘍細胞のIGF-I-、IGF-II-および血清-刺激増殖および生存の阻害
複数のヒト腫瘍細胞系統を、無血清条件でIGF-Iに対するそれらの増殖および生存応答について試験した。これらの細胞系統をIGF-I、IGF-II、または血清の存在のもとでEM164抗体を用いて処理し、それらの増殖および生存応答をMTTアッセイを用いて2〜4日後に測定した。ほぼ1500細胞を96ウエルプレートで通常培地にまいて、翌日、それを無血清培地(トランスフェリンおよびBSAを補充した無血清RPMI培地、またはDufourny, B.ら, 1997, J. Biol. Chem., 272, 31163-31171が規定したフェノールレッドを含まない培地のいずれか)と置換えた。無血清培地における増殖の1日後に、細胞を約75μLの10μg/mL抗体とともに30分間〜3時間インキュベートし、次いで25μLのIGF-I(またはIGF-IIまたは血清)溶液を加えて、10ng/mL IGF-I、または20ng/mL IGF-II、または0.04〜10%血清の最終濃度を得た。複数の実験において、細胞を最初IGF-Iを用いて15分間刺激した後にEM164抗体を加えるか、またはIGF-IとEM164抗体の両方を一緒に加えた。次いで細胞をさらに2〜3日増殖させた。次いでMTT(3-(4,5)-ジメチルチアゾール-2-イル)-2,5-ジフェニルテトラゾリウムブロミド)のPBS中の5mg/mL溶液、25μLを加え、細胞をインキュベーターに戻して2〜3時間置いた。次いで培地を除去して、100μL DMSOと置き換え、混合し、そしてプレートの吸収を545nmにて測定した。複数のヒト腫瘍細胞系統はIGF-IまたはIGF-IIまたは血清を加えると増殖および生存の応答を示し、その応答はEM164抗体により有意に阻害され、抗体をIGF-Iの前に加えたか、またはIGF-Iを抗体の前に加えたか、またはIGF-Iと抗体の両方を一緒に加えたかどうかとは無関係であった(表1)。
【表1】
【0093】
EM164抗体は、乳癌MCF-7細胞(図7および8)のIGF-Iまたは血清刺激増殖および生存を強く阻害した。別の実験において、EM164抗体はMCF-7細胞のIGF-II刺激増殖および生存を強く阻害した。市販の抗体、例えばIR3抗体を用いる先の報告は、図7でIR3および1H7抗体に対して確認したように、MCF-7細胞の血清刺激増殖および生存に弱い阻害だけを示した(Cullen, K. J.ら, 1990, Cancer Res., 50, 48-53)。対照的に、EM164抗体はMCF-7細胞の血清またはIGF刺激増殖の強力な阻害剤であった。図8に示したように、EM164抗体は広範囲の血清濃度(0.04〜10%血清)にわたってMCF-7細胞の増殖および生存を等しく有効に阻害した。
【0094】
EM164抗体によるMCF-7細胞の増殖阻害を細胞数の計測により測定した。すなわち、12ウエルプレートにおいて、約7500の細胞を、10μg/mL EM164抗体の存在または不在のもとで10%FBSを含むRPMI培地中にまいた。増殖の5日後に、無処理の対照サンプルの細胞数は20.5 x 104細胞であり、EM164抗体処理サンプルのわずか1.7 x 104細胞の細胞数と対照的であった。EM164抗体による処理は、MCF-7細胞の増殖を5日間に約12倍だけ阻害した。EM164抗体によるこの阻害は、報じられたMCF-7細胞に対する6日間アッセイでのIR3抗体を用いる2.5倍の阻害(Rohlik, Q. T.ら, 1987, Biochem. Biophys. Res. Commun., 149, 276-281)より有意に大きかった。
【0095】
非小細胞肺癌系統NCI-H838のIGF-Iおよび血清刺激増殖および生存もまた、EM164抗体により、対照抗B4抗体と比較して強く阻害された(図9)。無血清培地中のEM164抗体による処理は、NCI-H838とMCF-7細胞の両方に対して、無処理サンプルより小さいシグナルを産生したが、それは恐らく、EM164抗体はこれらの細胞の自己分泌およびパラ分泌IGF-IおよびIGF-II刺激も阻害したからであろう(図7および9)。HT29大腸癌細胞のコロニーサイズも、EM164抗体により処理すると非常に減少した。
【0096】
従ってEM164抗体は、腫瘍細胞、例えばMCF-7細胞およびNCI-H838細胞の血清刺激増殖を80%以上阻害することでその有効性が公知の全ての抗IGF-I受容体抗体のなかでもユニークな存在である。
【0097】
EM164抗体は細胞周期のG0/G1期における増殖停止を引起し、IGF-Iの分裂促進効果を無効にした。細胞周期分析について、MCF-7細胞をIGF-I(20ng/mL)を用いてEM164(μg/mL)の存在または不在のもとで1日間処理し、次いでヨウ化プロピジウム染色とフローサイトメトリーにより分析した。図25に示すように、EM164不在のもとでIGF-I刺激に応答する細胞のサイクリング(細胞の41%がS期そして50%がG0/G1期)が、EM164処理細胞においては抑制された(細胞わずかに9%がS期そして77%がG0/G1期)。
【0098】
細胞増殖の阻害に加え、EM164抗体処理は細胞のアポトーシスをもたらした。アポトーシスの測定については、EM164の存在または不在のもとで1日間IGF-Iまたは血清とともにインキュベートしたNCI-H838肺癌細胞において、カスパーゼによるサイトケラチンCK18タンパク質の切断を測定した(図26)。EM164の不在のもとで、IGF-Iまたは血清を加えると、無IGF-I対照と比較してカスパーゼ切断されたCK18シグナルが低下し、IGF-Iおよび血清はカスパーゼの活性を阻止することを示した。EM164を用いて処理するとIGF-Iおよび血清の抗アポトーシス効果を抑制し、それはEM164存在時の切断されたCK18レベルがEM164不在時のそれより大きいことにより示された(図26)。
【0099】
F.他の細胞毒性および細胞分裂停止作用薬と併用したEM164抗体による、ヒト腫瘍細胞の増殖および生存の相乗的阻害
EM164抗体のタキソールとの併用投与は、非小細胞肺癌Calu6細胞の増殖および生存に対してタキソール単独より有意に阻害性があった。同様に、EM164抗体のカンプトセシンとの併用は、大腸癌HT29細胞の増殖および生存に対してカンプトセシン単独より有意に阻害性があった。EM164抗体単独では有機の化学毒性薬と同じような毒性は期待されないので、EM164抗体の主に細胞分裂停止効果と化学毒性薬の細胞傷害効果の間の相乗作用は、臨床環境における併用癌治療法に非常に効果的でありうる。
【0100】
EM164抗体の抗EGF受容体抗体(KS77)との併用効果は、複数の腫瘍細胞系統、例えばHT-3細胞、RD細胞、MCF-7細胞、およびA431細胞の増殖および生存に対して、EM164抗体またはKS77抗体単独より有意に高い阻害性があった。従って、2種の増殖因子受容体、例えばIGF-I受容体およびEGF受容体に対する中和化抗体を併用することの相乗効果はまた、臨床癌治療において有用でありうる。
【0101】
EM164抗体の細胞傷害薬とのコンジュゲートはまた、IGF-I受容体を過剰発現する腫瘍に標的を定めた細胞傷害薬の送達において貴重である。EM164抗体の放射標識または他の標識とのコンジュゲートは、IGF-I受容体を過剰発現する腫瘍の治療およびイメージングに利用することができる。
【0102】
G.単一作用薬としてまたは抗癌薬と併用する場合、免疫不全マウスのヒト癌異種移植片におけるEM164処理の効果
ヒト非小細胞肺癌Calu-6異種移植片を免疫不全マウス中に、1x107 Calu-6細胞の皮下注射により確立した。図10に示すように、確立された100mm3 Calu-6異種移植片を含有するこれらのマウスを、1処理グループ当たり5匹のマウスを用いて、EM164抗体単独(0.8mg/マウスの6注射、静脈内、毎週2回)またはタキソール単独(タキソールの5注射、腹腔内、毎週2回;15mg/kg)、またはタキソールとEM164抗体処理の併用、またはPBS単独(200μL/マウス、6注射、毎週2回、静脈)によって処理した。腫瘍の増殖は、EM164抗体処理により、PBS対照と比較して有意に遅くなった。EM164抗体は、マウスの体重測定に基づいて毒性が観察されなかった。タキソール単独処理は14日まで有効であったが、その後腫瘍は増殖を回復し始めた。しかしタキソールとEM164抗体の併用により処理したグループでは、タキソール単独で治療したグループと比較して腫瘍の増殖が有意に遅くなった。
【0103】
ヒト膵癌異種移植片を、5週齢雌性SCID/ICRマウス(Taconic)に107 BxPC-3細胞を含むPBSを皮下注射することにより確立した(第0日)。80mm3の確立された腫瘍を抱くマウスを、次いでEM164単独(0.8mg/マウスの13回注射;側方尾静脈の静脈内、第12、16、19、23、26、29、36、43、50、54、58、61および64日に)、ゲムシタビン単独(150mg/kg/マウスの2回注射;腹腔内、第12および19日に)、上記スケジュールによるゲムシタビンとEM164の併用、PBS単独、および対照抗体単独(EM164と同じスケジュール)によって、5処理グループのそれぞれに5匹のマウスを用いて、処理した。図27に示したように、EM164単独、またはゲムシタビンとの併用による処理は、最初に、EM164処理グループ中の5動物の4動物および併用処理グループの全動物に腫瘍異種移植片の全退行が生じた。1以上の動物の測定しうる腫瘍再増殖は、EM164グループにおいて第43日に、そして併用治療グループにおいて第68日に見られ、対照処理グループと比較して第74日に有意に小さい平均腫瘍容積を生じただけであった(それぞれP=0.029および0.002;ツー・テイルドT検定(two-tailed T-test);図27)。他の研究において、EM164抗体処理(単独または抗EGF受容体抗体と併用;腹腔内注射)は確立されたマウス中のBxPC-3異種移植片の増殖を阻害した。
【0104】
H.EM164抗体の軽および重鎖のクローニングと配列決定
EM164ハイブリドーマ細胞から得た全RNAを精製した。逆転写酵素反応を、4〜5μg全RNAおよびオリゴdTもしくは無作為ヘキサマープライマーのいずれかを用いて実施した。
【0105】
PCR反応をCoら(J. Immunol., 148, 1149-1154 (1992))が記載したRACE法およびWangら(J. Immunol. Methods, 233, 167-177 (2000))が記載した変性プライマーを用いて実施した。RACE PCR法は、第1鎖cDNAの3'末端にポリGテーリングを加える中間ステップを必要とした。RT反応物をQianeasy(Qiagen)カラムを用いて精製し、50μl 1 X NEBバッファー4で溶出した。dGテーリング反応はその溶出物に、0.25mM CoCl2、1mM dGTP、および5単位の末端トランスフェラーゼ(NEB)を含む1 X NEBバッファー4を用いて実施した。その混合物を37℃にて30分間インキュベートし、次いで反応物(10μl)の1/5を直接PCR反応に加えてテンプレートDNAとして利用した。
【0106】
RACEと変性PCR反応は、プライマーとテンプレートの相違を除くと、同じ反応であった。末端トランスフェラーゼ反応物をRACE PCRテンプレートに直接使用する一方、RT反応混合物を変性PCR反応に直接使用した。
【0107】
RACEおよび変性PCR反応において、同じ3'軽鎖プライマー:
HindKL - tatagagctcaagcttggatggtgggaagatggatacagttggtgc(配列番号14)、
および3’重鎖プライマー:
Bgl2IgG1 - ggaagatctatagacagatgggggtgtcgttttggc(配列番号15)
を使用した。
【0108】
RACE PCRにおいて、軽および重鎖の両方に対して、1つのポリC5'プライマー
:EcoPolyC - TATATCTAGAATTCCCCCCCCCCCCCCCCC(配列番号16)
を用いる一方、縮重5'末端PCRプライマーは、軽鎖に対して:
Sac1MK - GGGAGCTCGAYATTGTGMTSACMCARWCTMCA(配列番号17)、
および重鎖に対して:
EcoR1MH1 - CTTCCGGAATTCSARGTNMAGCTGSAGSAGTC(配列番号18)と
EcoR1MH2 - CTTCCGGAATTCSARGTNMAGCTGSAGSAGTCWGG(配列番号19)
の等しい混合物であった。
【0109】
上記プライマー配列において、混合塩基は次のとおり定義した:H=A+T+C、S=g+C、Y=C+T、K=G+T、M=A+C、R=A+g、W=A+T、V=A+C+G。
【0110】
PCR反応を、次のプログラムを用いて実施した:1)94℃3分間、2)94℃15秒間、3)45℃1分間、4)72℃2分間、5)ステップ2)に戻るサイクリング29回、および6)72℃10分間の最終伸長による完了。
【0111】
PCR産物を、PCRプライマーにより作製した制限酵素切断部位を利用してpBluescript II SK+(Stratagene)中にクローニングした。
【0112】
複数の個々の軽および重鎖クローンを、通常の方法により配列決定して同定し、起こりうるポリメラーゼが起こす配列誤差を回避した(図12および13)。Chotia正準分類定義を用いて、3つの軽鎖および重鎖CDRを同定した(図12〜14)。
【0113】
NCBI IgBlastデータベースは、抗IGF-I受容体抗体軽鎖可変域はマウスIgVk Cr1生殖系列遺伝子由来である一方、重鎖可変域は恐らくIgVh J558.c生殖系列遺伝子由来であることを示した(図15)。
【0114】
マウスEM164抗体のタンパク質配列決定を実施して図12および13に示した配列を確かめた。精製EM164抗体の軽および重鎖タンパク質をゲル(SDS-PAGE、還元条件)からPVDF膜に移し、PVDF膜から切除し、タンパク質配列決定により分析した。軽鎖のN末端配列をEdman配列決定によりDVLMTQTPLS(配列番号20)であると決定し、これはEM164ハイブリドーマから得たクローニングした軽鎖遺伝子のN末端配列と整合する。
【0115】
重鎖のN末端はEdmanタンパク質配列決定に対してブロックされていることがわかった。質量1129.5(M+H+、モノアイソトープ)の重鎖のトリプシン消化ペプチドフラグメントをPSD(post-source decay)を経由してフラグメント化し、その配列がGRPDYYGSSK(配列番号21)であると決定した。質量2664.2(M+H+、モノアイソトープ)の重鎖の他のトリプシン消化ペプチドフラグメントもPSD(post-source decay)を経由してフラグメント化し、その配列がSSSTAYMQLSSLTSEDSAVYYFAR(配列番号22)であると同定した。これらの配列の両方は、EM164ハイブリドーマから得たクローニングした重鎖遺伝子のCDR3およびフレームワーク3(FR3)の配列と完全に整合する。
【0116】
I.EM164抗体の組換え発現
軽および重鎖の一対の配列を単一の哺乳類動物発現ベクター中にクローニングした(図16)。ヒト可変配列用のPCRプライマーにより、pBluescriptIIクローニングベクター中にヒトシグナル配列との結合を可能にする制限酵素切断部位を作製しておく一方、可変配列を哺乳類動物発現プラスミド中に、EcoRIとBsiWIまたはHindIIIとApaI部位を軽鎖または重鎖用にそれぞれ用いてクローニングした(図16)。軽鎖可変配列をインフレームでヒトIgK定常域にクローニングし、そして重鎖可変配列をヒトIgγ1定常域配列中にクローニングした。最終発現プラスミドにおいて、ヒトCMVプロモーターは軽および重鎖両方のcDNA配列の発現を駆動した。組換えマウスEM164抗体の発現と精製は当技術分野で周知の方法によって進めた。
【0117】
実施例2:EM164抗体のヒト化バージョン
治療または診断薬として適当なヒト化バージョンを提供するためのEM164抗体の再表面形成は、全般的に米国特許第5,639,641号に開示された原理と方法に従い、次の通り進めた。
【0118】
A.表面予測
解析された構造をもつ1セットの抗体に対する可変域残基の溶媒アクセシビリティを用いてマウス抗IGF-I受容体抗体(EM164)可変域の表面残基を予測した。1セットの127種のユニークな抗体構造ファイル(表2)に対するアミノ酸溶媒アクセシビリティを、MCソフトウエアパッケージ(Pedersenら, 1994, J. Mol. Biol., 235, 959-973)を用いて計算した。127種の構造のセットから10種の最も類似した軽鎖および重鎖アミノ酸配列を配列アラインメントにより決定した。それぞれの可変域残基に対する平均溶媒アクセシビリティを計算し、そして30%を超える平均アクセシビリティをもつ位置を表面残基とみなした。25%〜35%の平均アクセシビリティをもつ位置をさらに、2つの同一フランキング残基をもつ構造だけに対して個々の残基アクセシビリティを計算することにより試験した。
【表2】
【0119】
B.分子モデル
マウスEM164の分子モデルを、Oxford Molecular社ソフトウエアパッケージAbMを用いて作製した。抗体フレームワークは、最も類似したアミノ酸配列をもつ抗体に対する構造ファイルから、すなわち軽鎖に対して2jelからおよび重鎖に対して1nqbから構築した。非正準CDRは、非重複の解析された構造を含有するC-α構造データベースを検索することにより構築した。CDRの5Å以内にある残基を決定した。
【0120】
C.ヒトAb選択
マウスEM164の表面位置を、Kabatデータベースにおけるヒト抗体配列の対応する位置と比較した(Johnson, G.およびWu, T. T. (2001) Nucleic Acids Research, 29: 205-206)。抗体データベース管理ソフトウエアSR(Searle 1998)を用いて、天然の重および軽鎖ヒト抗体対からの抗体表面残基を抽出してアラインメントを実施した。CDRの5Å以内にある位置を特に配慮し、最も同一の表面残基をもつヒト抗体表面を選んで、マウス抗IGF-I受容体抗体表面残基と置換えた。
【0121】
D.PCR突然変異誘発
マウスEM164cDNAクローン(上記)上でPCR突然変異誘発を実施して、再表面形成されたヒトEM164(本明細書ではhuEM164と呼ぶ)を構築した。プライマーセットは、全ての試験したhuEM164のバージョンに8個のアミノ酸変化が要求されるように設計し、そしてさらなるプライマーは、代わりに2つの5Å残基変化を作るように設計した(表3)。PCR反応を、次のプログラムを用いて実施した:1)94℃1分間、2)94℃15秒間、3)55℃1分間、4)72℃2分間、5)ステップ2)に戻るサイクリング29回、6)72℃4分間の最終伸長による完了。PCR産物をその対応する制限酵素によって消化し、それらを上記のようにpBluescriptクローニングベクター中にクローニングした。クローンを配列決定して所望のアミノ酸変化を確認した。
【表3】
【0122】
E.可変域表面残基
Pedersenら(J. Mol. Biol., 235, 959-973, 1994)およびRoguskaら(タンパク質 Eng., 9, 895-904, 1996)が記載した抗体再表面形成技術は、マウス抗体可変配列の表面残基を予想することから始める。表面残基は、その全表面積の少なくとも30%が水分子にアクセス可能であるアミノ酸として定義される。
【0123】
127種の抗体構造ファイルのセット中に10種の最も相同的な抗体を同定した(図17および18)。それぞれのKabat位置に対する溶媒アクセシビリティを、これらのアラインメントした配列に対して平均化し、それぞれの残基に対する相対的アクセシビリティの分布を図19に示した。軽および重鎖は両方とも、少なくとも30%の平均相対アクセシビリティをもつ26残基を有し(図19):これらの残基はEM164に対して予想された表面残基であった。複数の残基は25%〜35%の平均アクセシビリティを有したので、これらを、残基のどちらか1つの側にフランキングする2つの同一残基をもつ抗体だけを平均化することにより、さらに試験した(表4および5)。このさらなる分析の後、以上同定した表面残基の元来のセットは変らないまま残った。
【表4】
【表5】
【0124】
F.CDRの5Å以内にある残基を決定するための分子モデル
AbMソフトウエアパッケージを用いて作製した上記分子モデルを分析して、どのEM164表面残基がCDRの5Å以内にあるかを確認した。マウスEM164抗体を再表面形成するために、CDR外側の全ての表面残基をヒトの対応残基に変えなければならないが、CDRの5Å以内の残基は抗原特異性にも寄与しうるので、特別な注意を払って処理した。従って、これらの後者残基を同定してヒト化プロセスの間を通して注意深く配慮しなければならない。再表面形成に用いるCDR定義は重鎖CDR2に対するAbM定義と残る5つのCDRに対するKabat定義を組合わせる(図14)。表6はEM164モデルの軽または重鎖配列両方のいずれかのCDR残基の5Å以内にあった残基を示す。
【表6】
【0125】
G.最も相同的なヒト表面の同定
EM164を再表面形成するためのヒト抗体表面候補を、Kabat抗体配列データベース内で、規定した残基だけの抗体データベースに対する位置のサーチングを提供するSRソフトウエアを利用して同定した。自然対合を保存するために、軽および重鎖両方の表面残基を一緒に比較した。Kabatデータベースからの最も相同的なヒト表面を配列同一性の格付け順に整列した。トップ5の表面を表7に与える。ついでこれらの表面を比較して、CDRの5Å以内で必要となる変更が最小であるのはどれかを同定した。白血病B細胞抗体、CLL 1.69は最少数の表面残基変化(全部で10)を必要とし、そのうち2つだけがCDRの5Å以内にあった。
【0126】
EM164の全長可変域配列もKabatヒト抗体データベースとアラインメントして、再び、CLL 1.69を最も類似したヒト可変域配列として同定した。総合して、これらの配列比較はヒト白血病B細胞抗体CLL 1.69がEM164に対するヒト表面として好ましい選択であると同定した。
【表7】
【0127】
H. ヒト化EM164遺伝子の構築
EM164(表7)の表面残基変化を上記のPCR突然変異誘発技術を利用して作った。CLL 1.69に対する表面残基のうちの8残基はCDRの5Å以内になかったので、これらの残基はヒト化EM164の全てのバージョンにおいてマウスからヒトに変更した(表8および表9)。CDRの5Å以内にあった2つの軽鎖表面残基(Kabat位置3および45)はヒトに変更するかまたはマウスに保持した。まとめると、これらのオプションにより、4種のEM164ヒト化バージョンが構築された(図22および23)。
【0128】
4種のヒト化バージョンのうち、バージョン1.0は全ての10ヒト表面残基を有する。CDRの近傍の変化を最も保存するバージョンは、CDRの5Å以内にあるマウス表面の両方を保持したバージョン 1.1である。全ての4種のヒト化EM164抗体遺伝子を抗体発現プラスミド(図16)中にクローニングして、一過性および安定したトランスフェクションに用いた。
【表8】
【0129】
I.全長IGF-I受容体および末端切断IGF-I受容体α鎖との結合に対する、ヒト化EM164抗体バージョンの親和性とマウスEM164抗体の親和性との比較
ヒト化EM164抗体バージョン1.0-1.3の親和性をマウスEM164抗体の親和性と、上記ビオチン化全長ヒトIGF-I受容体またはmyc-エピトープ標識末端切断IGF-I受容体α鎖を用いる競合アッセイを通して比較した。ヒト化EM164抗体サンプルを、ヒト胚性腎293T細胞における適当な発現ベクターの一過性トランスフェクションにより得て、抗体濃度を精製ヒト化抗体標準を用いるELISAにより決定した。ELISA結合競合を測定するために、ヒト化抗体サンプルと様々な濃度のマウスEM164抗体の混合物を、間接的に捕獲したビオチン化全長IGF-I受容体またはmycエピトープ標識末端切断IGF-I受容体α鎖とともにインキュベートした。平衡後に、結合したヒト化抗体をヤギ抗ヒトFab'2抗体-西洋わさびペルオキシダーゼコンジュゲートを用いて検出した。([結合したマウスAb]/[結合したヒト化Ab])対([マウスAb]/[ヒト化Ab])のプロットは、理論的に(Kdヒト化Ab/KdマウスAb)の勾配をもつ直線を生じるので、これを用いてヒト化とマウス抗体の相対親和性を決定した。
【0130】
競合アッセイの例を図11に示す。Immulon-2HB ELISAプレートを、1ウエル当たり5μg/mLストレプトアビジンを含む炭酸バッファー100μLを用いて常温で7時間コーティングした。ストレプトアビジンをコーティングしたウエルをブロッキングバッファー(10mg/mL BSAを含むTBS-Tバッファー)200μLを用いて1時間ブロックし、TBS-Tバッファーを用いて洗浄し、そしてビオチン化IGF-I受容体(1ウエル当たり5ng)を用いて一夜4℃にてインキュベートした。次いで間接的に捕獲したビオチン化IGF-I受容体を含有するウエルを洗浄し、ヒト化EM164抗体(15.5ng)とマウス抗体(0ng、または16.35ng、または32.7ng、または65.4ng、または163.5ng)の混合物を含むブロッキングバッファー100μLを用いて2時間常温でインキュベートし、次いで一夜4℃にてインキュベートした。次いでウエルをTBS-Tバッファーを用いて洗浄し、ヤギ抗ヒトFab'2抗体-西洋わさびペルオキシダーゼコンジュゲートとともに1時間(100μL;1μg/mLを含むブロッキングバッファー)インキュベートし、その後洗浄し、そしてABTS/H2O2基質を用いて405nmにて検出した。
【0131】
([結合したマウスAb]/[結合したヒト化Ab])対([マウスAb]/[ヒト化Ab])のプロットは、0.52の勾配(=Kdヒト化Ab/KdマウスAb)をもつ直線(r2=0.996)を生じた。従って、ヒト化抗体バージョン1.0はIGF-I受容体と、マウスEM164抗体より強く結合した。勾配に対する約0.5〜0.8の類似した値を、ヒト化EM164抗体のバージョン1.0、1.1、1.2および1.3のマウスEM164抗体との全長IGF-I受容体または末端切断IGF-I受容体α鎖競合に対して得たので、これは、EM164抗体の全てのヒト化バージョンが親マウスEM164抗体のそれより優れた類似の親和性を有することを示した。重鎖に92F→Cの突然変異をもつEM164抗体のキメラバージョンは、マウスEM164抗体との類似の結合競合において約3の勾配を示し、これはEM164の92F→Cの突然変異はマウスEM164抗体がIGF-I受容体との結合に示したのより3倍低い親和性を有することを示した。ヒト化EM164 v1.0抗体は、マウスEM164抗体が示したのと類似のMCF-7細胞のIGF-I刺激増殖および生存の阻害を示した(図24)。ヒト化EM164 v1.0抗体によるMCF-7細胞の血清刺激増殖および阻害はマウスEM164抗体による阻害と類似した。
【表9】
【0132】
J.本明細書に記載のマウスおよびヒト化抗体配列から出発して、改良された抗IGF-I-受容体抗体を提供する方法
抗IGF-I受容体抗体EM164とそのヒト化変異体のアミノ酸および核酸配列を利用して、改良されたかつまた本発明の範囲内にある他の抗体を開発した。そのような改良された特性としては、IGF-I受容体に対する親和性の増加が挙げられる。複数の研究は、1以上のアミノ酸変化を抗体の配列の様々な位置に導入することのその特性、例えば結合および発現レベルに与える効果を、一次抗体配列の知識に基づいて、調査している(Yang, W.P.ら, 1995, J. Mol. Biol., 254, 392-403;Rader, C.ら, 1998, Proc. Natl. Acad. Sci. USA, 95, 8910-8915;Vaughan, T.J.ら, 1998, Nature Biotechnology, 16, 535-539)。
【0133】
これらの研究においては、オリゴヌクレオチド媒介位置指定突然変異誘発、カセット突然変異誘発、誤りがちの(error-prone)PCR、DNAシャッフリング、または大腸菌(E. coli)の変異誘発株などの方法を用いて、CDR1、CDR2、CDR3、またはフレームワーク領域中の軽および重鎖遺伝子の配列を変えることにより、1次抗体の変異体を作製している(Vaughan, T.J.ら, 1998, Nature Biotechnology, 16, 535-539;Adey, N.B.ら, 1996, 第16章, pp.277-291, in 「ペプチドとタンパク質のファージディスプレイ(Phage Display of Peptides and Proteins)」, Kay, B.K.ら編, Academic Press)。1次抗体の配列を変えるこれらの方法は、標準のスクリーニング技術の利用を通して、その2次抗体の親和性の改良をもたらしている(Gram, H.ら, 1992, Proc. Natl. Acad. Sci. USA, 89, 3576-3580;Boder, E.T.ら, 2000, Proc. Natl. Acad. Sci. USA, 97, 10701-10705;Davies, J.およびRiechmann, L., 1996, Immunotechnolgy, 2, 169-179;Thompson, J.ら, 1996, J. Mol. Biol., 256, 77-88;Short, M.K.ら, 2002, J. Biol. Chem., 277, 16365-16370;Furukawa, K.ら, 2001, J. Biol. Chem., 276, 27622-27628)。
【0134】
抗体の1以上のアミノ酸残基を変える類似の定方向手法(directed strategy)により、本発明に記載の抗体配列を用いて、改良された機能をもつ抗IGF-I受容体抗体、例えば治療薬の結合に利用する共有結合改変のための好都合な結合点に遊離アミノ基またはチオールなどの好適な基を有する抗体を開発することができる。
【0135】
K.マウス、キメラおよび他の抗IGF-I受容体抗体のための代わりの発現系
マウス抗IGF-I受容体抗体を、ヒト化抗体を発現するために利用したプラスミド(上記)と類似の哺乳類動物発現プラスミドからも発現した。軽鎖κおよび重鎖γ-1配列を含むマウス定常域を有する発現プラスミドは公知である(McLeanら, 2000, Mol Immunol., 37, 837-845)。これらのプラスミドを、いずれかの抗体可変域、例えばマウス抗IGF-I受容体抗体を単純な制限酵素消化とクローニングにより受容するように設計した。発現プラスミド中にこれらと適合しうる制限酵素切断部位を創出するために、抗IGF-1受容体抗体のさらなるPCRが通常必要であった。
【0136】
全マウス抗IGF-I受容体抗体を発現する代わりの手法は、キメラ抗IGF-I受容体抗体発現プラスミド中のヒト定常域を置換えることであった。キメラ発現プラスミド(図16)を可変域のためのおよび軽および重鎖定常域の両方のためのカセットを用いて構築した。抗体可変配列を制限酵素消化によりこの発現プラスミド中にクローニングしたように、別々の制限酵素消化を用いていずれかの定常域にクローニングした。抗IGF-1抗体可変域のクローニングについて本明細書に記載したRNAのように、κ軽鎖およびγ-1重鎖cDNAを、例えばマウスハイブリドーマRNAからクローニングした。同様に、好適なプライマーをKabatデータベース中の利用しうる配列から設計した(表10を参照)。例えば、RT-PCRを用いて定常域配列をクローニングしそしてこれらの断片をキメラ抗IGF-I受容体抗体発現プラスミド中にクローニングするために必要な制限酵素切断部位を創出した。このプラスミドを用いて次に標準哺乳類動物発現系、例えばCHO細胞系統において全マウス抗IGF-I受容体抗体を発現した。
【表10】
【0137】
寄託の表明
マウスEM164抗体を作るハイブリドーマはATCC(American Type Culture Collection, PO Box 1549, Manassas, VA 20108)に、2002年6月14日ブダペスト条約の条項のもとで、受託番号PTA-4457として寄託した。
【0138】
ある特定の特許および印刷出版物が本発明の開示に参照されていて、それらの教示は本明細書にそれらのそれぞれの全てが参照によりそれぞれ組込まれる。
【0139】
本発明は詳細にかつその特定の実施形態を参照して記載されているが、当業者であれば、本発明の精神と範囲から逸脱することなくそれに様々な変化と改変をなしうることは明らかであろう。
【特許請求の範囲】
【請求項1】
インスリン様増殖因子-I受容体(IGF-IR)と特異的に結合する抗体またはエピトープ結合抗体フラグメントであって、上記受容体の拮抗薬でありかつ上記受容体に対する作動薬活性を欠くことを特徴とする上記抗体またはエピトープ結合抗体フラグメント。
【請求項2】
再表面形成されている、請求項1に記載の抗体または抗体フラグメント。
【請求項3】
ヒトのものであるかまたはヒト化されている、請求項1に記載の抗体または抗体フラグメント。
【請求項4】
インスリン様増殖因子-I受容体と特異的に結合する抗体またはエピトープ結合抗体フラグメントであって、上記受容体の拮抗薬でありかつ上記受容体に対する作動薬活性を欠くものであり、血清およびインスリン様増殖因子-Iからなる群から選択される増殖刺激因子の存在のもとで癌細胞の増殖を約80%より大きく阻害することができる上記抗体または抗体フラグメント。
【請求項5】
上記癌細胞がヒト癌細胞である、請求項4に記載の抗体または抗体フラグメント。
【請求項6】
少なくとも1つの相補性決定領域を含み、上記相補性決定領域が配列番号1〜6:
SYWMH (配列番号1)、
EINPSNGRTNYNEKFKR (配列番号2)、
GRPDYYGSSKWYFDV (配列番号3)、
RSSQSIVHSNVNTYLE (配列番号4)、
KVSNRFS (配列番号5)、
FQGSHVPPT (配列番号6)
からなる群から選択される1つのアミノ酸配列を有する抗体またはエピトープ結合抗体フラグメントであって、IGF-IRに特異的に結合するものである、上記抗体またはエピトープ結合抗体フラグメント。
【請求項7】
少なくとも1つの重鎖可変域および少なくとも1つの軽鎖可変域を含み、上記重鎖可変域が配列番号1〜3:
SYWMH (配列番号1)、
EINPSNGRTNYNEKFKR (配列番号2)、
GRPDYYGSSKWYFDV (配列番号3)
によりそれぞれ表される3つの連続的な相補性決定領域を含み、かつ
上記軽鎖可変域が配列番号4〜6:
RSSQSIVHSNVNTYLE (配列番号4)、
KVSNRFS (配列番号5)、
FQGSHVPPT (配列番号6)
によりそれぞれ表される3つの連続的な相補性決定領域を含むことを特徴とする、IGF-IRに特異的に結合する抗体またはエピトープ結合抗体フラグメント。
【請求項8】
上記重鎖可変域が配列番号7:
QVQLQQSGAELVKPGASVKLSCKASGYTFTSYWMHWVKQRPGQGLEWIGEINPSNGRTNYNEKFKRKATLTVDKSSSTAYMQLSSLTSEDSAVYYFARGRPDYYGSSKWYFDVWGAGTTVTVSS(配列番号7)
により表されるアミノ酸配列と少なくとも90%配列同一性を有し、該抗体または抗体フラグメントがIGF-IRに特異的に結合する、請求項7に記載の抗体または抗体フラグメント。
【請求項9】
上記重鎖可変域が配列番号7により表される上記アミノ酸配列と少なくとも95%配列同一性を有し、該抗体または抗体フラグメントがIGF-IRに特異的に結合する、請求項8に記載の抗体または抗体フラグメント。
【請求項10】
上記重鎖可変域が配列番号7により表されるアミノ酸配列を有し、該抗体または抗体フラグメントがIGF-IRに特異的に結合する、請求項8に記載の抗体または抗体フラグメント。
【請求項11】
上記軽鎖可変域が配列番号8:
DVLMTQTPLSLPVSLGDQASISCRSSQSIVHSNVNTYLEWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGTDFTLRISRVEAEDLGIYYCFQGSHVPPTFGGGTKLEIKR(配列番号8)
により表されるアミノ酸配列と少なくとも90%配列同一性を有し、該抗体または抗体フラグメントがIGF-IRに特異的に結合する、請求項7に記載の抗体または抗体フラグメント。
【請求項12】
上記軽鎖可変域が配列番号8により表される上記アミノ酸配列と少なくとも95%配列同一性を有し、該抗体または抗体フラグメントがIGF-IRに特異的に結合する、請求項11に記載の抗体または抗体フラグメント。
【請求項13】
上記軽鎖可変域が配列番号8により表されるアミノ酸配列を有し、該抗体または抗体フラグメントがIGF-IRに特異的に結合する、請求項11に記載の抗体または抗体フラグメント。
【請求項14】
請求項1〜13のいずれか1項に記載の抗体または抗体フラグメントおよび製薬上許容される担体を含む医薬組成物。
【請求項15】
細胞傷害薬に連結された、請求項1〜13のいずれか1項に記載の抗体または抗体フラグメントを含むコンジュゲート。
【請求項16】
上記細胞傷害薬がメイタンシノイド、低分子薬剤、プロドラッグ、タキソイド、CC-1065およびCC-1065類似体からなる群から選択される、請求項15に記載のコンジュゲート。
【請求項17】
請求項16に記載のコンジュゲートおよび製薬上許容される担体を含む医薬組成物。
【請求項18】
上記抗体または抗体フラグメントが標識されている、請求項14または請求項17に記載の組成物を含む診断試薬。
【請求項19】
上記標識が放射標識、蛍光体、発色体、イメージング剤および金属イオンからなる群から選択される、請求項18に記載の診断試薬。
【請求項20】
IGF-IRを発現する癌細胞の増殖をex vivoにおいて阻害する方法であって、採取された上記細胞を請求項1〜13のいずれか1項に記載の抗体または抗体フラグメントと接触させることを含む上記方法。
【請求項21】
IGF-IRを発現する癌細胞の増殖を阻害するための医薬の製造における、請求項1〜13のいずれか1項に記載の抗体または抗体フラグメントの使用。
【請求項22】
IGF-IRを発現する癌患者を治療するための医薬組成物であって、請求項1〜13のいずれか1項に記載の抗体または抗体フラグメントの有効量を含む上記医薬組成物。
【請求項23】
さらに治療薬を含む、請求項22に記載の医薬組成物。
【請求項24】
上記治療薬が細胞傷害薬である、請求項23に記載の医薬組成物。
【請求項25】
IGF-IRを発現する癌患者を治療するための医薬組成物であって、請求項15または16に記載のコンジュゲートの有効量を含む上記医薬組成物。
【請求項26】
上記癌が乳癌、大腸癌、卵巣癌、骨肉腫、子宮頸癌、前立腺癌、肺癌、滑膜癌および膵癌からなる群から選択される癌である、請求項22〜25のいずれか1項に記載の医薬組成物。
【請求項27】
癌の疑いがある被験者を診断するための医薬組成物であって、請求項18または19に記載の診断試薬を含む上記医薬組成物。
【請求項28】
上記癌が乳癌、大腸癌、卵巣癌、骨肉腫、子宮頸癌、前立腺癌、肺癌、滑膜癌および膵癌からなる群から選択される癌である、請求項27に記載の医薬組成物。
【請求項29】
インスリン様増殖因子-I受容体と特異的に結合する改良された抗体またはエピトープ結合抗体フラグメントのスクリーニング方法であって、
(a)配列番号1〜8:
SYWMH (配列番号1),
EINPSNGRTNYNEKFKR (配列番号2),
GRPDYYGSSKWYFDV (配列番号3),
RSSQSIVHSNVNTYLE (配列番号4),
KVSNRFS (配列番号5),
FQGSHVPPT (配列番号6),
QVQLQQSGAELVKPGASVKLSCKASGYTFTSYWMHWVKQRPGQGLEWIGEINPSNGRTNYNEKFKRKATLTVDKSSSTAYMQLSSLTSEDSAVYYFARGRPDYYGSSKWYFDVWGAGTTVTVSS (配列番号7),
DVLMTQTPLSLPVSLGDQASISCRSSQSIVHSNVNTYLEWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGTDFTLRISRVEAEDLGIYYCFQGSHVPPTFGGGTKLEIKR (配列番号8);
からなる群から選択される少なくとも1つの配列を含む抗体またはそのエピトープ結合抗体フラグメントをコードするDNAを提供するステップ;
(b)上記DNA中に少なくとも1つのヌクレオチド突然変異、欠失または挿入を導入して、上記DNAによりコードされる上記抗体または抗体フラグメントのアミノ酸配列を変えるステップ;
(c)上記抗体または抗体フラグメントを発現するステップ;
(d)上記発現された抗体または抗体フラグメントを上記改良についてスクリーニングしてそれにより上記改良された抗体またはその抗体フラグメントを調製するステップ、
を含む、上記スクリーニング方法。
【請求項30】
上記改良がIGF-I受容体との親和性の増加である、請求項29に記載の方法。
【請求項31】
上記少なくとも1つのヌクレオチド突然変異、欠失または挿入を、オリゴヌクレオチドが媒介する位置指定突然変異誘発、カセット変異誘発、誤りがちなPCR、DNAシャッフリング、または大腸菌(E. coli)の突然変異体株の使用からなる群から選択される方法により行うことを特徴とする、請求項29に記載の方法。
【請求項32】
請求項6〜13のいずれか1項に記載の抗体または抗体フラグメントをコードするポリヌクレオチド。
【請求項33】
請求項6〜13のいずれか1項に記載の抗体または抗体フラグメントの軽鎖または重鎖可変域をコードするポリヌクレオチド。
【請求項34】
請求項32または33のポリヌクレオチドを含むベクター。
【請求項35】
上記抗体または抗体フラグメントを発現することができる発現ベクターである、請求項34に記載のベクター。
【請求項36】
上記IGF-IRがヒトIGF-IRである、請求項1〜13のいずれか1項に記載の抗体または抗体フラグメント。
【請求項37】
IGF-IRを発現する癌細胞のアポトーシスを誘導する方法であって、上記細胞を請求項1〜13のいずれか1項に記載の抗体または抗体フラグメントと接触させることを含む上記方法。
【請求項38】
抗体または抗体フラグメントが、
a)IGF-IRを活性化することなくIGF-IRの細胞機能を阻害する特性;
b)血清の存在のもとで少なくとも80%だけ腫瘍細胞増殖を阻害する特性;
c)IGF-IRと0.3x10-9M以下のKDで結合する特性;
d)IGF-IのIGF-IRとの結合を阻害する特性;
e)IGF-Iが媒介するIGF-IRによる細胞シグナル伝達を阻害する特性;
f)IGF-Iが誘導するIGF-IRリン酸化を阻害する特性;
g)IGF-IおよびIGF-IIが媒介する細胞増殖と生存を阻害する特性;
h)アフリカミドリザルIGF-IRと結合するが、マウス、チャイニーズハムスターまたはヤギIGF-IRと結合しない特性;および
i)IRS-1、AktおよびErk1/2の活性化を阻害する特性
からなる群から選択される少なくとも1つの特性を有することを特徴とする、請求項1〜13のいずれか1項に記載の抗体または抗体フラグメント。
【請求項39】
請求項38に記載の特性の全てを有する、請求項38に記載の抗体または抗体フラグメント。
【請求項40】
上記抗体または抗体フラグメントが
a)免疫グロブリン(Ig)G1-4、IgM、IgA1-2、IgDもしくはIgE分子であるかまたはそれらから誘導されるか;または
b)1本鎖抗体、1本鎖抗体フラグメント(scFv)、Fabフラグメント、F(ab’)2フラグメント、または少なくとも3つのCDRを含有するフラグメントである、
請求項1〜13のいずれか1項に記載の抗体または抗体フラグメント。
【請求項41】
請求項34または35に記載のベクターを含む宿主細胞。
【請求項42】
抗IGF-IR抗体またはエピトープ結合抗体フラグメントを作る方法であって、請求項41に記載の宿主細胞を上記抗IGF-IR抗体または抗体フラグメントを発現するのに好適な条件のもとで培養するステップ、および上記抗IGF-IR抗体または抗体フラグメントを回収するステップを含む上記方法。
【請求項43】
配列番号1、54、3、4、5および6からなる群より選択されるアミノ酸配列を含む、IGF-IRに特異的に結合する抗体またはそのエピトープ結合フラグメント。
【請求項44】
配列番号1、54、3、4、5および6のアミノ酸配列を含む、IGF-IRに特異的に結合する抗体またはそのエピトープ結合フラグメント。
【請求項45】
配列番号1、54、3、4、5および6からなる群より選択されるアミノ酸配列を有する少なくとも1つの相補性決定領域を含む軽鎖または重鎖を含む、IGF-IRに特異的に結合する抗体またはそのエピトープ結合フラグメントであって、該重鎖は、配列番号1、54および3のアミノ酸配列を含む3つの相補性決定領域を含み、該軽鎖は、配列番号4、5および6のアミノ酸配列を含む3つの相補性決定領域を含む、上記抗体またはそのフラグメント。
【請求項46】
少なくとも1つの重鎖可変域および少なくとも1つの軽鎖可変域を含む、IGF-IRに特異的に結合する抗体またはそのエピトープ結合フラグメントであって、該重鎖可変域は、配列番号1、54および3のアミノ酸配列を含む3つの相補性決定領域を含み、該軽鎖可変域は、配列番号4、5および6のアミノ酸配列を含む3つの相補性決定領域を含む、上記抗体またはそのフラグメント。
【請求項47】
該重鎖可変域は、配列番号1、54および3のアミノ酸配列を含む3つの重鎖相補性決定領域を含み、該軽鎖可変域は、配列番号8のアミノ酸配列を含む、請求項45または46に記載の抗体またはそのフラグメント。
【請求項48】
該重鎖可変域は、配列番号1、54および3のアミノ酸配列を含む3つの相補性決定領域を含み、該軽鎖可変域は、配列番号4、5および6のアミノ酸配列を含む3つの相補性決定領域を含み、該重鎖可変域は配列番号7のアミノ酸配列を含む、請求項45または46に記載の抗体またはそのフラグメント。
【請求項1】
インスリン様増殖因子-I受容体(IGF-IR)と特異的に結合する抗体またはエピトープ結合抗体フラグメントであって、上記受容体の拮抗薬でありかつ上記受容体に対する作動薬活性を欠くことを特徴とする上記抗体またはエピトープ結合抗体フラグメント。
【請求項2】
再表面形成されている、請求項1に記載の抗体または抗体フラグメント。
【請求項3】
ヒトのものであるかまたはヒト化されている、請求項1に記載の抗体または抗体フラグメント。
【請求項4】
インスリン様増殖因子-I受容体と特異的に結合する抗体またはエピトープ結合抗体フラグメントであって、上記受容体の拮抗薬でありかつ上記受容体に対する作動薬活性を欠くものであり、血清およびインスリン様増殖因子-Iからなる群から選択される増殖刺激因子の存在のもとで癌細胞の増殖を約80%より大きく阻害することができる上記抗体または抗体フラグメント。
【請求項5】
上記癌細胞がヒト癌細胞である、請求項4に記載の抗体または抗体フラグメント。
【請求項6】
少なくとも1つの相補性決定領域を含み、上記相補性決定領域が配列番号1〜6:
SYWMH (配列番号1)、
EINPSNGRTNYNEKFKR (配列番号2)、
GRPDYYGSSKWYFDV (配列番号3)、
RSSQSIVHSNVNTYLE (配列番号4)、
KVSNRFS (配列番号5)、
FQGSHVPPT (配列番号6)
からなる群から選択される1つのアミノ酸配列を有する抗体またはエピトープ結合抗体フラグメントであって、IGF-IRに特異的に結合するものである、上記抗体またはエピトープ結合抗体フラグメント。
【請求項7】
少なくとも1つの重鎖可変域および少なくとも1つの軽鎖可変域を含み、上記重鎖可変域が配列番号1〜3:
SYWMH (配列番号1)、
EINPSNGRTNYNEKFKR (配列番号2)、
GRPDYYGSSKWYFDV (配列番号3)
によりそれぞれ表される3つの連続的な相補性決定領域を含み、かつ
上記軽鎖可変域が配列番号4〜6:
RSSQSIVHSNVNTYLE (配列番号4)、
KVSNRFS (配列番号5)、
FQGSHVPPT (配列番号6)
によりそれぞれ表される3つの連続的な相補性決定領域を含むことを特徴とする、IGF-IRに特異的に結合する抗体またはエピトープ結合抗体フラグメント。
【請求項8】
上記重鎖可変域が配列番号7:
QVQLQQSGAELVKPGASVKLSCKASGYTFTSYWMHWVKQRPGQGLEWIGEINPSNGRTNYNEKFKRKATLTVDKSSSTAYMQLSSLTSEDSAVYYFARGRPDYYGSSKWYFDVWGAGTTVTVSS(配列番号7)
により表されるアミノ酸配列と少なくとも90%配列同一性を有し、該抗体または抗体フラグメントがIGF-IRに特異的に結合する、請求項7に記載の抗体または抗体フラグメント。
【請求項9】
上記重鎖可変域が配列番号7により表される上記アミノ酸配列と少なくとも95%配列同一性を有し、該抗体または抗体フラグメントがIGF-IRに特異的に結合する、請求項8に記載の抗体または抗体フラグメント。
【請求項10】
上記重鎖可変域が配列番号7により表されるアミノ酸配列を有し、該抗体または抗体フラグメントがIGF-IRに特異的に結合する、請求項8に記載の抗体または抗体フラグメント。
【請求項11】
上記軽鎖可変域が配列番号8:
DVLMTQTPLSLPVSLGDQASISCRSSQSIVHSNVNTYLEWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGTDFTLRISRVEAEDLGIYYCFQGSHVPPTFGGGTKLEIKR(配列番号8)
により表されるアミノ酸配列と少なくとも90%配列同一性を有し、該抗体または抗体フラグメントがIGF-IRに特異的に結合する、請求項7に記載の抗体または抗体フラグメント。
【請求項12】
上記軽鎖可変域が配列番号8により表される上記アミノ酸配列と少なくとも95%配列同一性を有し、該抗体または抗体フラグメントがIGF-IRに特異的に結合する、請求項11に記載の抗体または抗体フラグメント。
【請求項13】
上記軽鎖可変域が配列番号8により表されるアミノ酸配列を有し、該抗体または抗体フラグメントがIGF-IRに特異的に結合する、請求項11に記載の抗体または抗体フラグメント。
【請求項14】
請求項1〜13のいずれか1項に記載の抗体または抗体フラグメントおよび製薬上許容される担体を含む医薬組成物。
【請求項15】
細胞傷害薬に連結された、請求項1〜13のいずれか1項に記載の抗体または抗体フラグメントを含むコンジュゲート。
【請求項16】
上記細胞傷害薬がメイタンシノイド、低分子薬剤、プロドラッグ、タキソイド、CC-1065およびCC-1065類似体からなる群から選択される、請求項15に記載のコンジュゲート。
【請求項17】
請求項16に記載のコンジュゲートおよび製薬上許容される担体を含む医薬組成物。
【請求項18】
上記抗体または抗体フラグメントが標識されている、請求項14または請求項17に記載の組成物を含む診断試薬。
【請求項19】
上記標識が放射標識、蛍光体、発色体、イメージング剤および金属イオンからなる群から選択される、請求項18に記載の診断試薬。
【請求項20】
IGF-IRを発現する癌細胞の増殖をex vivoにおいて阻害する方法であって、採取された上記細胞を請求項1〜13のいずれか1項に記載の抗体または抗体フラグメントと接触させることを含む上記方法。
【請求項21】
IGF-IRを発現する癌細胞の増殖を阻害するための医薬の製造における、請求項1〜13のいずれか1項に記載の抗体または抗体フラグメントの使用。
【請求項22】
IGF-IRを発現する癌患者を治療するための医薬組成物であって、請求項1〜13のいずれか1項に記載の抗体または抗体フラグメントの有効量を含む上記医薬組成物。
【請求項23】
さらに治療薬を含む、請求項22に記載の医薬組成物。
【請求項24】
上記治療薬が細胞傷害薬である、請求項23に記載の医薬組成物。
【請求項25】
IGF-IRを発現する癌患者を治療するための医薬組成物であって、請求項15または16に記載のコンジュゲートの有効量を含む上記医薬組成物。
【請求項26】
上記癌が乳癌、大腸癌、卵巣癌、骨肉腫、子宮頸癌、前立腺癌、肺癌、滑膜癌および膵癌からなる群から選択される癌である、請求項22〜25のいずれか1項に記載の医薬組成物。
【請求項27】
癌の疑いがある被験者を診断するための医薬組成物であって、請求項18または19に記載の診断試薬を含む上記医薬組成物。
【請求項28】
上記癌が乳癌、大腸癌、卵巣癌、骨肉腫、子宮頸癌、前立腺癌、肺癌、滑膜癌および膵癌からなる群から選択される癌である、請求項27に記載の医薬組成物。
【請求項29】
インスリン様増殖因子-I受容体と特異的に結合する改良された抗体またはエピトープ結合抗体フラグメントのスクリーニング方法であって、
(a)配列番号1〜8:
SYWMH (配列番号1),
EINPSNGRTNYNEKFKR (配列番号2),
GRPDYYGSSKWYFDV (配列番号3),
RSSQSIVHSNVNTYLE (配列番号4),
KVSNRFS (配列番号5),
FQGSHVPPT (配列番号6),
QVQLQQSGAELVKPGASVKLSCKASGYTFTSYWMHWVKQRPGQGLEWIGEINPSNGRTNYNEKFKRKATLTVDKSSSTAYMQLSSLTSEDSAVYYFARGRPDYYGSSKWYFDVWGAGTTVTVSS (配列番号7),
DVLMTQTPLSLPVSLGDQASISCRSSQSIVHSNVNTYLEWYLQKPGQSPKLLIYKVSNRFSGVPDRFSGSGSGTDFTLRISRVEAEDLGIYYCFQGSHVPPTFGGGTKLEIKR (配列番号8);
からなる群から選択される少なくとも1つの配列を含む抗体またはそのエピトープ結合抗体フラグメントをコードするDNAを提供するステップ;
(b)上記DNA中に少なくとも1つのヌクレオチド突然変異、欠失または挿入を導入して、上記DNAによりコードされる上記抗体または抗体フラグメントのアミノ酸配列を変えるステップ;
(c)上記抗体または抗体フラグメントを発現するステップ;
(d)上記発現された抗体または抗体フラグメントを上記改良についてスクリーニングしてそれにより上記改良された抗体またはその抗体フラグメントを調製するステップ、
を含む、上記スクリーニング方法。
【請求項30】
上記改良がIGF-I受容体との親和性の増加である、請求項29に記載の方法。
【請求項31】
上記少なくとも1つのヌクレオチド突然変異、欠失または挿入を、オリゴヌクレオチドが媒介する位置指定突然変異誘発、カセット変異誘発、誤りがちなPCR、DNAシャッフリング、または大腸菌(E. coli)の突然変異体株の使用からなる群から選択される方法により行うことを特徴とする、請求項29に記載の方法。
【請求項32】
請求項6〜13のいずれか1項に記載の抗体または抗体フラグメントをコードするポリヌクレオチド。
【請求項33】
請求項6〜13のいずれか1項に記載の抗体または抗体フラグメントの軽鎖または重鎖可変域をコードするポリヌクレオチド。
【請求項34】
請求項32または33のポリヌクレオチドを含むベクター。
【請求項35】
上記抗体または抗体フラグメントを発現することができる発現ベクターである、請求項34に記載のベクター。
【請求項36】
上記IGF-IRがヒトIGF-IRである、請求項1〜13のいずれか1項に記載の抗体または抗体フラグメント。
【請求項37】
IGF-IRを発現する癌細胞のアポトーシスを誘導する方法であって、上記細胞を請求項1〜13のいずれか1項に記載の抗体または抗体フラグメントと接触させることを含む上記方法。
【請求項38】
抗体または抗体フラグメントが、
a)IGF-IRを活性化することなくIGF-IRの細胞機能を阻害する特性;
b)血清の存在のもとで少なくとも80%だけ腫瘍細胞増殖を阻害する特性;
c)IGF-IRと0.3x10-9M以下のKDで結合する特性;
d)IGF-IのIGF-IRとの結合を阻害する特性;
e)IGF-Iが媒介するIGF-IRによる細胞シグナル伝達を阻害する特性;
f)IGF-Iが誘導するIGF-IRリン酸化を阻害する特性;
g)IGF-IおよびIGF-IIが媒介する細胞増殖と生存を阻害する特性;
h)アフリカミドリザルIGF-IRと結合するが、マウス、チャイニーズハムスターまたはヤギIGF-IRと結合しない特性;および
i)IRS-1、AktおよびErk1/2の活性化を阻害する特性
からなる群から選択される少なくとも1つの特性を有することを特徴とする、請求項1〜13のいずれか1項に記載の抗体または抗体フラグメント。
【請求項39】
請求項38に記載の特性の全てを有する、請求項38に記載の抗体または抗体フラグメント。
【請求項40】
上記抗体または抗体フラグメントが
a)免疫グロブリン(Ig)G1-4、IgM、IgA1-2、IgDもしくはIgE分子であるかまたはそれらから誘導されるか;または
b)1本鎖抗体、1本鎖抗体フラグメント(scFv)、Fabフラグメント、F(ab’)2フラグメント、または少なくとも3つのCDRを含有するフラグメントである、
請求項1〜13のいずれか1項に記載の抗体または抗体フラグメント。
【請求項41】
請求項34または35に記載のベクターを含む宿主細胞。
【請求項42】
抗IGF-IR抗体またはエピトープ結合抗体フラグメントを作る方法であって、請求項41に記載の宿主細胞を上記抗IGF-IR抗体または抗体フラグメントを発現するのに好適な条件のもとで培養するステップ、および上記抗IGF-IR抗体または抗体フラグメントを回収するステップを含む上記方法。
【請求項43】
配列番号1、54、3、4、5および6からなる群より選択されるアミノ酸配列を含む、IGF-IRに特異的に結合する抗体またはそのエピトープ結合フラグメント。
【請求項44】
配列番号1、54、3、4、5および6のアミノ酸配列を含む、IGF-IRに特異的に結合する抗体またはそのエピトープ結合フラグメント。
【請求項45】
配列番号1、54、3、4、5および6からなる群より選択されるアミノ酸配列を有する少なくとも1つの相補性決定領域を含む軽鎖または重鎖を含む、IGF-IRに特異的に結合する抗体またはそのエピトープ結合フラグメントであって、該重鎖は、配列番号1、54および3のアミノ酸配列を含む3つの相補性決定領域を含み、該軽鎖は、配列番号4、5および6のアミノ酸配列を含む3つの相補性決定領域を含む、上記抗体またはそのフラグメント。
【請求項46】
少なくとも1つの重鎖可変域および少なくとも1つの軽鎖可変域を含む、IGF-IRに特異的に結合する抗体またはそのエピトープ結合フラグメントであって、該重鎖可変域は、配列番号1、54および3のアミノ酸配列を含む3つの相補性決定領域を含み、該軽鎖可変域は、配列番号4、5および6のアミノ酸配列を含む3つの相補性決定領域を含む、上記抗体またはそのフラグメント。
【請求項47】
該重鎖可変域は、配列番号1、54および3のアミノ酸配列を含む3つの重鎖相補性決定領域を含み、該軽鎖可変域は、配列番号8のアミノ酸配列を含む、請求項45または46に記載の抗体またはそのフラグメント。
【請求項48】
該重鎖可変域は、配列番号1、54および3のアミノ酸配列を含む3つの相補性決定領域を含み、該軽鎖可変域は、配列番号4、5および6のアミノ酸配列を含む3つの相補性決定領域を含み、該重鎖可変域は配列番号7のアミノ酸配列を含む、請求項45または46に記載の抗体またはそのフラグメント。
【図1】


【図2】
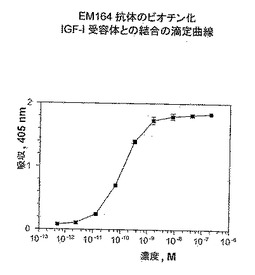
【図3】

【図4】
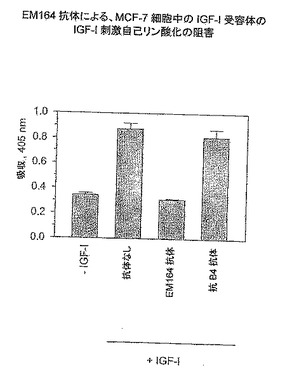
【図5】
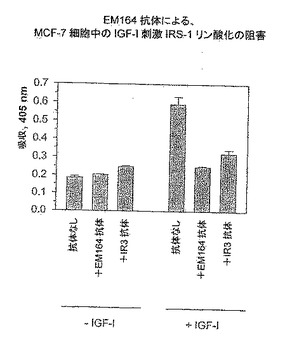
【図6】
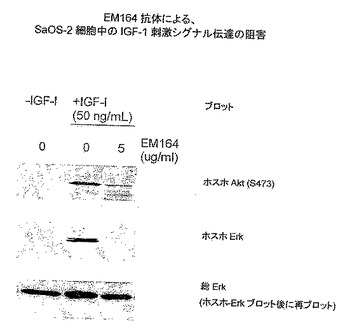
【図7】
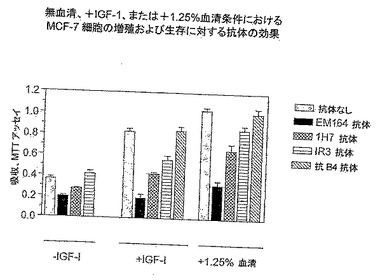
【図8】

【図9】
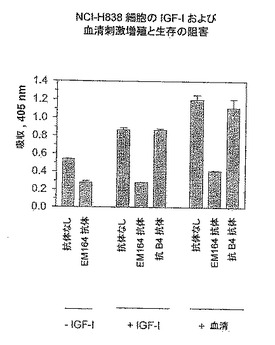
【図10】

【図11】
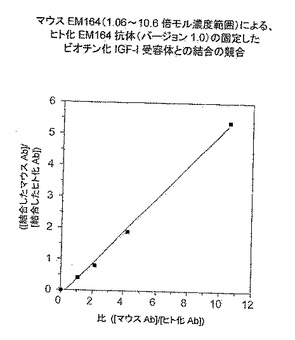
【図12】

【図13】

【図14】

【図15】

【図16】
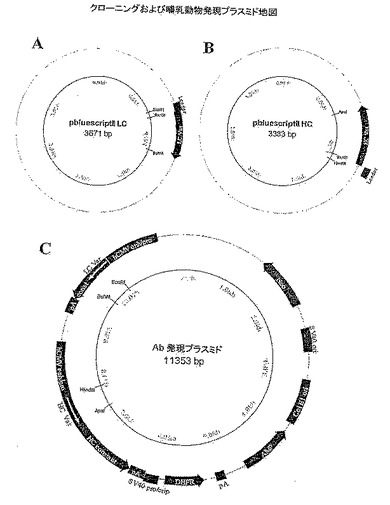
【図17】
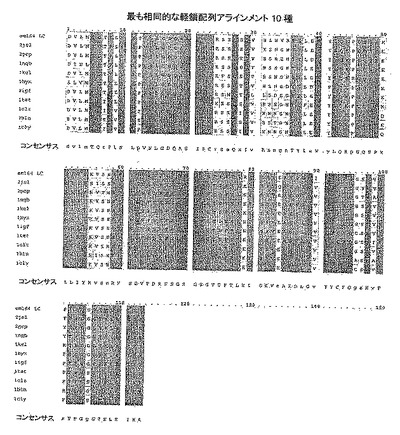
【図18】

【図19】

【図20】

【図21】

【図22】

【図23A】

【図23B】

【図24】
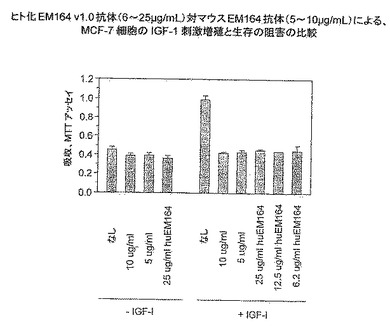
【図25】
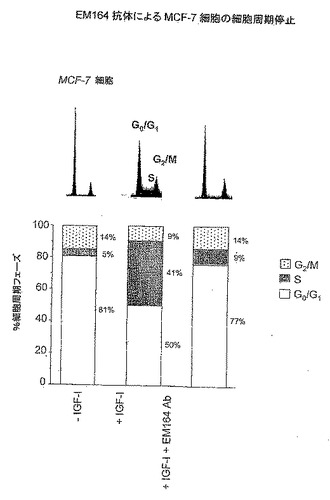
【図26】
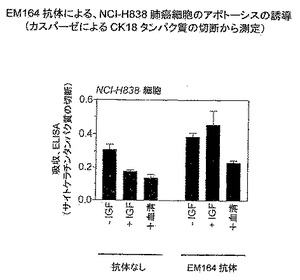
【図27】
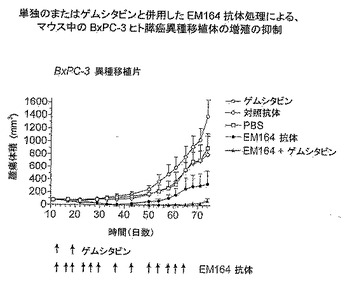
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23A】
【図23B】
【図24】
【図25】
【図26】
【図27】
【公開番号】特開2011−41564(P2011−41564A)
【公開日】平成23年3月3日(2011.3.3)
【国際特許分類】
【出願番号】特願2010−190561(P2010−190561)
【出願日】平成22年8月27日(2010.8.27)
【分割の表示】特願2004−513434(P2004−513434)の分割
【原出願日】平成15年6月12日(2003.6.12)
【出願人】(502103829)イムノージェン インコーポレーテッド (5)
【Fターム(参考)】
【公開日】平成23年3月3日(2011.3.3)
【国際特許分類】
【出願日】平成22年8月27日(2010.8.27)
【分割の表示】特願2004−513434(P2004−513434)の分割
【原出願日】平成15年6月12日(2003.6.12)
【出願人】(502103829)イムノージェン インコーポレーテッド (5)
【Fターム(参考)】
[ Back to top ]