
持続した薬物送達のための水不溶性複合体を含む薬学的処方物
【課題】ペプチド化合物(例えば、ペプチド、ポリペプチド、タンパク質、ペプチド擬似物(peptidomimetic)など)、好ましくは薬学的に活性なペプチド化合物、および複合体の投与の際にインビボでペプチド化合物の持続した送達を行い得るキャリア巨大分子からなる、安定な水不溶性複合体を含む薬学的組成物を提供すること。
【解決手段】薬学的に活性なペプチドとキャリア巨大分子との固体イオン性複合体を含む薬学的組成物であって、ここで該複合体のペプチド含有量が少なくとも57重量%である、組成物。
【解決手段】薬学的に活性なペプチドとキャリア巨大分子との固体イオン性複合体を含む薬学的組成物であって、ここで該複合体のペプチド含有量が少なくとも57重量%である、組成物。
【発明の詳細な説明】
【背景技術】
【0001】
発明の背景
種々の疾患および臨床的障害が、薬学的に活性なペプチドの投与により処置される。そのような例の1つに前立腺癌があるが、これは性ホルモン依存性癌であり、そして男性ホルモンの合成を調節する黄体ホルモン(LH)の産生を妨げる黄体形成ホルモン放出ホルモン(LHRH)アナログの投与により処置され得る。詳細には、LH産生を減少させるために、黄体形成ホルモン放出ホルモンレセプター(例えば、ロイプロリドおよびゴセレリン)のスーパーアゴニストとして作用するLHRHのペプチドアナログが使用されてきた。
【0002】
多くの場合、薬学的に活性なペプチドの治療的効果は、長期間に渡りインビボで継続的にそれが存在することに依存する。インビボでのペプチドの継続的な送達を達成するために、繰り返し投与を行う必要をさけるために持続した放出または持続した送達処方が望ましい。持続した薬物送達のための一つの試みは、マイクロカプセル化によるものである。この方法において、活性成分は微粒子を生成するポリマー膜内に封じ込められている。例えば、LHRHスーパーアゴニスト(例えば、ロイプロリドおよびゴセレリン)は、典型的には、数週間から数ヶ月にわたりスーパーアゴニストの持続した送達を提供する貯蔵注射(depot injection)に適した処方物を調製するために、ポリ−ラクチド/ポリ−グリコリド共重合体を含む微粒子内にカプセル化される(例えば、米国特許第4,675,189号;4,677,191号;5,480,656号および4,728,721号を参照のこと)。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】米国特許第4,675,189号明細書
【特許文献2】米国特許第4,677,191号明細書
【特許文献1】米国特許第5,480,656号明細書
【特許文献1】米国特許第4,728,721号明細書
【発明の概要】
【発明が解決しようとする課題】
【0004】
薬学的に活性なペプチドをインビボで長期間にわたり継続的に投与するためのさらに持続した送達処方物が必要である。
【課題を解決するための手段】
【0005】
発明の要旨
本発明は、ペプチド化合物(例えば、ペプチド、ポリペプチド、タンパク質、ペプチド擬似物(peptidomimetic)など)、好ましくは薬学的に活性なペプチド化合物、および複合体の投与の際にインビボでペプチド化合物の持続した送達を行い得るキャリア巨大分子からなる、安定な水不溶性複合体を含む薬学的組成物を提供する。従って、本発明の複合体は、長期間(例えば、1ヶ月)にわたって被験体への薬学的に活性なペプチド化合物の継続した送達を可能にし得る。さらに、ペプチド化合物とキャリア巨大分子との、強固で安定な複合体での会合は、処方物へのペプチド化合物の高濃度での添加(load)を可能にする。
【0006】
本発明の複合体は、実質的に水不溶性複合体が形成されるような条件下(例えば、ペプチド化合物およびキャリア巨大分子の水性溶液が、複合体が沈殿するまで混合される)で、ペプチド化合物とキャリア巨大分子とを組み合わせることにより形成される。複合体は、固体形態(例えば、ペースト、顆粒、粉末、または凍結乾燥体)であっても良く、または粉末形態の複合体が安定な液体懸濁物または半固体分散体を形成するのに十分に細かく粉末化され得る。
【0007】
好適な実施態様において、水不溶性複合体のペプチド化合物はLHRHアナログ、より好ましくはLHRHアンタゴニストであり、そしてキャリア巨大分子は、アニオン性ポリマー、好ましくはカルボキシメチルセルロースである。本発明の複合体は、インビボでの投与の前に、例えばγ線照射または電子線照射による滅菌化に適している。
【0008】
本発明のLHRHアナログ含有化合物を被験体へ投与することによる、LHRHアナログで処置可能な状態における被験体を処置するための方法もまた、提供される。好適な実施態様において、本発明の処置方法は、前立腺癌の処置に使用される。
【図面の簡単な説明】
【0009】
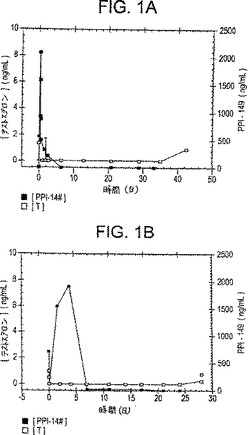
【図1】図1は、PPI-149およびカルボキシメチルセルロースの複合体の筋肉内注射に続く時間経過に伴う、ラット(左のグラフ)およびイヌ(右のグラフ)における、血漿テストステロンレベル(ng/ml;白四角)および血漿PPI-149レベル(ng/ml;黒四角)を描いたグラフを示す。
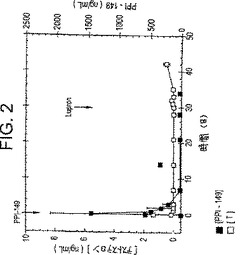
【図2】図2は、0日目におけるLHRHアンタゴニストPPI-149およびカフレボキシメチノレセルロースの複合体の筋肉内注射、および30日目におけるLHRHアゴニストLupronTMの注射に続く時間経過に伴う、ラットにおける血漿テストステロンレベル(ng/ml;白四角)および血漿PPI-149レベル(ng/ml;黒四角)を描いたグラフであり、PPI-149前処置によりLupronTM誘導テストステロンサージが抑制されることを示している。
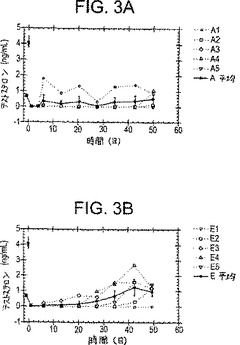
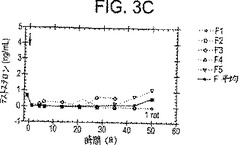
【図3−1】図3A〜3Cは、PPI-149-CMC(図3A)、PPI-258-CMC(図3B)、またはCetrorelixTM-CMC(図3C)の筋肉内注射に続く時間経過に伴う、雄性Sprague-Dawleyラットにおける血漿テストステロンレベル(ng/ml)を描いた一連のグラフである。
【図3−2】図3A〜3Cは、PPI-149-CMC(図3A)、PPI-258-CMC(図3B)、またはCetrorelixTM-CMC(図3C)の筋肉内注射に続く時間経過に伴う、雄性Sprague-Dawleyラットにおける血漿テストステロンレベル(ng/ml)を描いた一連のグラフである。
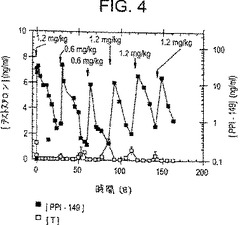
【図4】図4は、指示された用量で28日間隔におけるPPI-149-CMCの皮下注射に続く時間経過に伴う、イヌにおける血漿テストステロンレベル(ng/ml;白四角)および血漿PPI-149レベル(ng/ml;黒四角)を描いたグラフであり、血漿テストステロンレベルが長期にわたり抑制されることを示している。
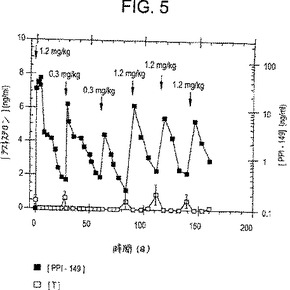
【図5】図5は、指示された用量で28日間隔におけるPPI-149-CMCの筋肉内注射に続く時間経過に伴う、イヌにおける血漿テストステロンレベル(ng/ml;白四角)および血漿PPI-149レベル(ng/ml;黒四角)を描いたグラフであり、血漿テストステロンレベルが長期にわたり抑制されることを示している。
【発明を実施するための形態】
【0010】
発明の詳細な説明
本発明は、ペプチド化合物(例えば、ペプチド、ポリペプチド、タンパク質、ペプチド擬似物など)およびキャリア巨大分子からなる、安定な水不溶性複合体を含む薬学的組成物、そのような組成物の作製法、およびそのような組成物の使用法に関する。本発明の薬学的組成物の利点は、薬学的に活性なペプチド化合物を、全身的または局所的のいずれかで、長期間(例えば、数週間、1ヶ月、または数ヶ月)にわたり送達し得ること、およびペプチド化合物を複合体へ高濃度で添加し得ること、を含む。
【0011】
本発明がより容易に理解され得るために、特定の用語をあらかじめ定義する。
【0012】
本明細書中で使用されるように、用語「ペプチド化合物」は、少なくとも部分的に、アミド結合(すなわちペプチド結合)により連結されたアミノ酸残基からなる化合物を示すことを意図する。用語「ペプチド化合物」は、ペプチド、ポリペプチド、およびタンパク質を包含することを意図する。代表的には、ペプチドは、約100よりも少ないアミノ酸からなり、より代表的には、約50よりも少ないアミノ酸残基からなり、さらにより代表的には、約25より少ないアミノ酸残基からなる。用語「ペプチド化合物」は、さらにペプチドアナログ、ペプチド誘導体、および天然に生じるアミノ酸からなるペプチドの化学構造を模倣したペプチド擬似物を包含することを意図する。ペプチドアナログの例は、1つ以上の非天然アミノ酸を含むペプチドを含む。ペプチド誘導体の例は、アミノ酸側鎖、ペプチド骨格、またはアミノ-もしくはカルボキシ-末端が誘導体化されているペプチドを含む(例えば、メチル化アミド連結したペプチド化合物)。ペプチド擬似物の例は、ペプチド骨格が1つ以上のベンゾジアゼピン分子で置換されているペプチド化合物(例えば、James、G.L.ら、(1993)Science 260:1937〜1942を参照のこと)、全てのL-アミノ酸が対応するD-アミノ酸で置換された「反転」(inverso)ペプチド、アミノ酸配列が逆転(「逆」)しており、かつ全てのL-アミノ酸が対応するD-アミノ酸で置換(「反転」)された「逆-反転」(retro-inverso)ペプチド(Sistoによる米国特許第4,522,752号を参照のこと)、およびペプチド骨格(すなわちアミド結合)擬似物のような他のアイソスター(isostere)(アミド窒素、α-炭素、アミドカルボニルの修飾、アミド結合の全置換、延長、欠失(deletion)、または骨格連結を含む)を含む。ψ[CH2S]、ψ[CH2NH]、ψ[CSNH2]、ψ[NHCO]、ψ[COCH2]、およびψ[(E)または(Z)CH=CH]を含む、いくつかのペプチド骨格改変が公知である。上記で使用される名称において、ψはアミド結合が存在しないことを意味する。アミド基と置換される構造は、角括弧(bracket)内に指定されている。他の可能な改変は、N-アルキル(またはアリール)置換(ψ[CONR])、骨格連結によるラクタムおよび他の環式構造の構築、並びにC-末端ヒドロキシメチル誘導体、O-修飾誘導体、およびN-末端修飾誘導体(アルキルアミドおよびヒドラジンのような置換アミドを含む)を含む他の誘導体を含む。
【0013】
本明細書中で使用されるように、用語「薬学的に活性なペプチド化合物」は、その存在する形態またはインビボでのプロセシングに際してのいずれかにおいて、薬理学的活性を示すペプチド化合物を示すことを意図する(すなわち、薬学的に活性なペプチド化合物は、構造的薬理学的活性であるペプチド化合物、および薬理学的活性を示すために投与に続いてインビボで何らかの方法で代謝もしくはプロセシングされなければならない「プロドラッグ」形態であるペプチド化合物を含む)。
【0014】
本明細書中で使用されるように、用語「多価カチオン性ペプチド化合物」および「多価アニオン性ペプチド化合物」は、それぞれ多重の正電荷または負電荷を含むペプチド化合物を示すことを意図する。「二価カチオン性」または「二価アニオン性」ペプチド化合物は、それぞれ2個の正電荷または負電荷を含むペプチド化合物を示すことを意図する。「三価カチオン性」または「三価アニオン性」ペプチド化合物は、それぞれ3個の正電荷または負電荷を含むペプチド化合物を示すことを意図する。
【0015】
本明細書中で使用されるように、用語「LHRHアナログ」は、黄体形成ホルモン放出ホルモンの構造を模倣するペプチド化合物を包含することを意図する。LHRHアナログは、LHRHアゴニストまたはLHRHアンタゴニストであり得る。
【0016】
本明細書中で使用されるように、「LHRHアゴニスト」は、黄体形成ホルモンの放出を刺激するように黄体形成ホルモン放出ホルモンレセプター(LHRH-R)を刺激する化合物を示すことを意図するか、または「LHRHアンタゴニスト」は、黄体形成ホルモンの放出を阻害するようにLHRH-Rを阻害する化合物を示す。LHRHアゴニストの例は、ロイプロリド(商標名:Lupronc(登録商標);Abbott/TAP)、ゴセレリン(商標名:Zoladex(登録商標);Zeneca)、ブセレリン(Hoechst)、トリプトレリン(Decapeptyl、D-Trp-6-LHRH、およびDebiopharm(登録商標)としても公知;Ipsen/Beaufour)、ナファレリン(商標名”Synarel(登録商標);Syntex)、ルトレリン(Wyeth)、シストレリン(cystorelin)(Hoechst)、ゴナドレリン(Ayerst)、およびヒストレリン(histrerlin)(Ortho)を含む。
【0017】
本明細書中で使用されるように、用語「LHRHアンタゴニスト」は、黄体形成ホルモンの放出を阻害するような黄体形成ホルモン放出ホルモンレセプターを阻害する化合物を示すことを意図する。LHRHアンタゴニストの例は、Antide、Cetrorelix、以下に記載される化合物:Folkersらの米国特許第5,470,947号;FolkersらによるPCT出願第WO89/01944:Havivの米国特許第5,413,990号;Havivの米国特許第5,300,492号;Koerberらの米国特許第5,371,070号;Hoegerらの米国特許第5,296,468号;Janakyらの米国特許第5,171,835号;Coyらの米国特許第5,003,011号;Coyの米国特許第4,431,635号;Deらの米国特許第4,992,421号;Roeskeの米国特許第4,851,385号;Nestor、Jr.らの米国特許第4,801,577号;およびRoeskeらの米国特許第4,689,396号、ならびに以下に開示される化合物:米国特許出願番号第08/480,494号、表題「LHRHアンタゴニストペプチド」、およびその対応するPCT出願(PCT出願番号第PCT/US96/09852)、同じく表題「LHRHアンタゴニストペプチド」を含む。これらの内容はいずれも全て特に本明細書中で参考として援用される。特に好適なLHRHアンタゴニストは、以下の構造を含む:Ac-D-Nal1、4-Cl-D-Phe2、D-Pal3、N-Me-Tyr5、D-Asn6、Lys(iPr)8、本明細書中でPPI-149として示されるD-Ala10-LHRH。
【0018】
本明細書中で使用されるように、用語「キャリア巨大分子」は、ペプチド化合物と複合化して水不溶性複合体を形成し得る巨大分子を示すことを意図する。ペプチド化合物と複合化する前は、代表的にはキャリア巨大分子は水溶性である。好ましくは、巨大分子は、少なくとも5kDa、より好ましくは10kDaの分子量を有する。用語「アニオン性キャリア巨大分子」は、アニオン性ポリマーのような負電荷を帯びた高分子量の分子を含むことを意図する。用語「カチオン性キャリア巨大分子」は、カチオン性ポリマーのような正電荷を帯びた高分子量の分子を含むことを意図する。
【0019】
本明細書中で使用されるように、用語「水不溶性複合体」は、本明細書中に記載される手順に従ってペプチド化合物およびキャリア巨大分子の適切な化合に際して形成される、物理的および科学的に安定な複合体を示すことを意図する。代表的には、この複合体は、ペプチド化合物およびキャリア巨大分子の水性調製物の化合において生成する沈殿の形態をとる。機構によって制限されることを意図しないが、本発明の好適な水不溶性複合体の形成は、ペプチド化合物がカチオン性であり、そしてキャリア分子がアニオン性であるか、またはその逆である状況に置けるイオン性相互作用を含む(すなわち、少なくとも部分的にそれにより媒介される)と考えられる。さらに、またはあるいは、本発明の水不溶性複合体の形成は、疎水性相互作用を含み得る(すなわち、少なくとも部分的にそれにより媒介される)。なおさらに、本発明の水不溶性複合体の形成は、共有相互作用を含み得る(すなわち、少なくとも部分的にそれにより媒介される)。「水不溶性」であるという複合体の記述は、それが水性溶液から沈殿することによって示されるように、複合体が実質的にまたは容易に水に溶解しないことを示すことを意図する。しかしながら、本発明の「水不溶性」複合体がインビトロでまたはインビボでの水性生理学的環境においてのいずれかで限られた水溶性(すなわち、部分的溶解性)を示し得ることが理解されるべきである。
【0020】
本明細書中で使用されるように、用語「持続した送達」は、投与に続く一定の期間、好ましくは少なくとも数日、1週間、または数週間に渡りインビボでの薬学的な薬剤の継続した送達を示すことを意図する。薬剤の持続した送達は、例えば、時間経過に伴う薬剤の継続した治療的効果により実証され得る(例えば、LHRHアナログについては、アナログの持続した送達は時間経過に伴うテストステロン合成の継続した抑制により実証され得る)。あるいは、薬剤の持続した送達は、時間経過に伴うインビボでの薬剤の存在の検出により実証され得る。
【0021】
本明細書中で使用されるように、用語「被験体」は、温血動物、好ましくは哺乳動物、より好ましくは霊長類、最も好ましくはヒトを含むことを意図する。
【0022】
本明細書中で使用されるように、用語「被験体への投与」は、被験体の所望の部位へ組成物を送達するための任意の適切な経路による、組成物(例えば薬学的処方物)の被験体への投薬、送達、または塗布を示すことを意図し、非経口もしくは経口経路、筋肉内注射、皮下/皮内注射、静脈注射、口内投与、直腸、結腸、膣、鼻内による経皮送達および投与、もしくは気道経路のいずれかによる送達を含む。
【0023】
本明細書中で使用されるように、用語「LHRHアナログで処置可能な状態」は、LHRHアゴニストまたはLHRHアンタゴニストの投与が所望の効果(例えば、治療的に有益な効果)をあげる疾患、障害、および他の状熊を含むことを意図する。LHRHアナログで処置可能な状態の例は、ホルモン依存性癌(前立腺癌、乳癌、卵巣癌、子宮癌、および精巣癌を含む)、良性前立腺肥大、性的早熟、子宮内膜症、子宮筋肺、不妊症(インビトロ受精を介して)および受精(すなわち、避妊的使用)を含む。
【0024】
本発明の1つの局面は、薬学的に活性なペプチド化合物とキャリア巨大分子との水不溶性複合体を含む薬学的組成物に関する。好適な実施態様において、水不溶性複合体の形成は、少なくとも部分的には薬学的に活性なペプチドとキャリア巨大分子との間のイオン性相互作用により媒介される。これらの実施態様において、薬学的に活性なペプチド化合物がカチオン性であり、キャリア巨大分子がアニオン性であるか、薬学的に活性なペプチド化合物がアニオン性であり、キャリア巨大分子がカチオン性であるかのいずれかである。別の実施態様において、水不溶性複合体の形成は、少なくとも部分的には薬学的に活性なペプチド化合物とキャリア巨大分子との間の疎水性相互作用により媒介される。好適な実施態様において、複合体に使用されるペプチド化合物は、二価または三価のカチオン性ペプチド化合物のような多価カチオン性ペプチド化合物であり、そしてキャリア巨大分子は、アニオン性巨大分子である。
【0025】
本発明の薬学的組成物は、被験体への組成物の投与後、インビボで被験体へのペプチド化合物の持続した送達を可能にし、ここで持続した送達の持続時間は、複合体を形成するために使用したペプチド化合物およびキャリア巨大分子の濃度に依存して変化し得る。例えば、1つの実施態様において、水不溶性複合体の1回の用量は、薬学的組成物が被験体に投与された後、少なくとも1週間被験体へペプチド化合物の持続した送達を提供する。別の実施態様において、水不溶性複合体の1回の用量は、薬学的組成物が被験体に投与された後、少なくとも2週間被験体へペプチド化合物の持続した送達を提供する。さらに別の1つの実施態様において、水不溶性複合体の1回の用量は、薬学的組成物が被験体に投与された後、少なくとも3週間被験体へペプチド化合物の持続した送達を提供する。さらに別の実施態様において、水不溶性複合体の1回の用量は、薬学的組成物が被験体に投与された後、少なくとも4週間被験体へペプチド化合物の持続した送達を提供する。さらに長期の、または短期の持続時間での持続した送達を提供する処方物(例えば、1日、1〜7日、1ヶ月、2ヶ月、3ヶ月などの継続した送達を提供する処方物)もまた、本発明に包含される。数ヶ月にわたるペプチド化合物の継続した送達は、例えば、毎月繰り返して服用することにより達成され得、各服用がほぼ1ヶ月の間ペプチド化合物の持続した送達を提供する(例えば、実施例14を参照のこと)。
【0026】
ペプチド化合物が、ペプチド化合物とキャリア巨大分子との結合によりキャリア巨大分子との水不溶性非共有結合複合体を形成し得る限り、任意の大きさのペプチド化合物が複合体への使用に適するものであり得る。しかしながら、特定の好適な実施態様において、ペプチド化合物は、約5〜約20アミノ酸長のペプチドであるか、約8〜約15アミノ酸長のペプチドであるか、または約8〜約12アミノ酸長のペプチドである。種々の薬学的に活性なペプチドが、処方物に使用され得、その非制限的な例は、LHRHアナログ(さらに以下で議論される)、ブラジキニンアナログ、副甲状腺ホルモン、副腎皮質刺激ホルモン(ACTH)、カルシトニン、およびバソプレッシンアナログ(例えば、1-デアミノ-8-D-アルギニンバソプレッシン(DDAVP))を含む。
【0027】
種々のキャリア巨大分子が本発明の水不溶性複合体の形成に適したものであり得るが、好ましい巨大分子はポリマーであり、好ましくは水溶性ポリマーである。好適な実施態様において、キャリア巨大分子は、アニオン性ポリアルコール誘導体のようなアニオン性ポリマー、またはそのフラグメント、およびその塩(例えば、ナトリウム塩)である。ポリアルコールが誘導体化され得るアニオン性部分は、例えば、カルボキシレート、ホスフェート、またはスルフェート基を含む。特に好適なアニオン性ポリマーは、アニオン性ポリサッカリド誘導体、またはそのフラグメント、およびその塩(例えば、ナトリウム塩)である。キャリア巨大分子は、単一の分子種(例えば、単一の型のポリマー)または2つ以上の異なる分子種(例えば、2つの型のポリマーの混合物)を含み得る。特定のアニオン性ポリマーの例は、カルボキシメチルセルロース、アルギン、アルギネート、アニオン性アセテートポリマー、アニオン性アクリル酸ポリマー、キサンタンガム、グリコール酸ナトリウムスターチ、およびそのフラグメント、誘導体、および薬学的に受容可能な塩、ならびにアニオン性カラギーナン誘導体、アニオン性ポリガラクツロン酸誘導体、ならびに硫酸化およびスルホン化ポリスチレン誘導体を含む。好適なアニオン性ポリマーは、カルボキシメチルセルロースナトリウム塩である。カチオン性ポリマーの例は、ポリ-L-リジンおよび他の塩基性アミノ酸ポリマーを含む。
【0028】
本発明の特定の好適な実施態様において、水不溶性複合体のペプチド化合物は、LHRHアナログであり、例えばLHRHアゴニスト、または、より好ましくはLHRHアンタゴニストである。代表的には、そのようなLHRHアナログは、10アミノ酸長である。好ましいLHRHアンタゴニストは、ペプチド化合物を含むLHRHアンタゴニストを含み、ここで天然の哺乳動物のLHRHの6位におけるアミノ酸に対応するペプチド化合物の残基はD-アスパラギン(D-Asn)構造を含む。本明細書中で使用されるように、用語「D-アスパラギン構造」は、D-Asn、ならびにD-Asnの機能的活性を保持するそのアナログ、誘導体、および擬似物を含むことを意図する。他の好ましいLHRHアンタゴニストは、以下の構造を含むペプチド化合物を含むLHRHアンタゴニストまたはその薬学的に受容可能な塩を含む:A-B-C-D-E-F-G-H-I-J
ここで
Aは、ピロ-Glu、Ac-D-Nal、Ac-D-QalAc-Sar、またはAc-D-Palであり
Bは、His、または4-Cl-D-Pheであり
Cは、Trp、D-Pal、D-Nal、L-Nal、D-Pal(N-0)、またはD-Trpであり
Dは、Serであり
Eは、N-Me-Ala、Tyr、N-Me-Tyr、Ser、Lys(iPr)、4-Cl-Phe、His、Asn、MeLAla、Arg、またはIleであり;
Fは、
【0029】
【化1】

ここで
RおよびXは、独立して、Hまたはアルキルであり;そして
Lは、小極性部分を含み;
Gは、Leu、またはTrpであり;
Hは、Lys(iPr)、Gln、Met、またはArgであり
Iは、Proであり;そして
Jは、Gly-NH2、またはD-Ala-NH2である。
【0030】
用語「小極性部分」は、小さな立体的かさ高さを有し、そして比較的極性である部分を示す。極性は、Pスケールによる親水性度として測定する。1-オクタノールと水との間の分配係数、Pが、化合物の親水性度の測定に対する標準として使用されている。親水性度は、logP、分配係数の対数として表され得る(Hanschら、Nature 194:178(1962);Fujitaら、J.Am.Chem.Soc.86:5175(1964))。多くの分子に対する親水性度の標準表、および多くの官能基に対する親油性度(疎水性度)置換基定数(πで表記)が、編集されている(例えば、HanschおよびLeo、「化学および生物学における相関分析に対する置換基定数」Wiley、New York.New York(1979)を参照のこと)。膨大な範囲の候補親水性部分の親水性度が、これらの表の助けによりかなり正確に予測され得る。例えば、ナフタレンのlogP(オクタノール/水)の実測値は、3.45である。-OHに対する置換基定数Pは、-0.67である。従って、β-ナフトールに対するlogPの予測値は、3.45+(-0.67)=2.78である。この値は、β-ナフトールに対するlogPの実測値、2.84と良く一致する。本明細書中で使用されるように、用語「小極性部分」は、-1と+2の間のlogP、およびTrpの立体的かさ高さよりも小さい立体的かさ高さを有する部分を示す。
【0031】
特定の実施態様において、Lは小極性部分を含むが、但しFはD-Cit、D-Hci、またはD-CitあるいはD-Hciの低級アルキル誘導体ではない。好ましくは、Fは、D-Asn、D-Gln、およびD-Thrからなる群より選択される。より好ましくは、FはD-Asnである。好ましくは、Eは、チロシン(Tyr)またはN-メチル-チロシン(N-Me-Tyr)である。特定の好適な実施熊様において、LHRHアンタゴニストは以下の構造式を有する:Ac-D-Nal1、4-Cl-D-Phe2、D-Pal3、N-Me-Tyr5、D-Asn6、Lys(iPr)8、D-Ala10-LHRH(本明細書中でPPI-149として示される)。本発明の特定の好適な複合体は、PPI-149およびカルボキシメチルセルロースを含む。
【0032】
水不溶性複合体に加えて、本発明の薬学的処方物はさらなる薬学的に受容可能なキャリアおよび/または賦形剤を含み得る。本明細書中で使用されるように、「薬学的に受容可能なキャリア」は、生理学的に適合性である、任意のそして全ての溶媒、分散媒体、コーティング、抗菌剤および抗真菌剤、等張液および吸収遅延剤などを含む。好ましくは、キャリアは静脈内、筋肉内、皮下または非経口投与(例えば、注射による)に適している。賦形剤は、薬学的に受容可能な安定剤および崩壊剤を含む。
【0033】
キャリア巨大分子と複合体化したLHRHアナログの薬学的処方物に加えて、本発明はさらに、そのような複合体およびそのような複合体を含有するシリンジを含むパッケージ化処方物を包含する。例えば、本発明はLHRHアナログ(好ましくはPPI-149)およびキャリア巨大分子(好ましくは、カルボキシメチルセルロース)の水不溶性複合体を含み、LHRHアナログで処置可能な状態の被験体を処置するための水不溶性複合体の使用説明書と合わせてパッケージ化された、LHRHアナログで処置可能な状態の被験体を処置するためのパッケージ化処方物を提供する。別の実施態様において、本発明は管腔を有するシリンジを提供し、ここでLHRHアナログ(好ましくはPPI-149)およびキャリア巨大分子(好ましくは、カルボキシメチルセルロース)の水不溶性複合体は、管腔中に含まれる。
【0034】
本発明の複合体は、ペプチド化合物およびキャリア巨大分子の水不溶性複合体が形成するような条件下で、ペプチド化合物およびキャリア巨大分子を結合させることにより調製される。従って、本発明の別の局面は、薬学的処方物の調製法に関する。1つの実施態様において、この方法は以下を含む:
ペプチド化合物およびキャリア巨大分子を提供する工程;
ペプチド化合物およびキャリア巨大分子の水不溶性複合体が形成するような条件下で、ペプチド化合物およびキャリア巨大分子を結合させる工程:および
水不溶性複合体を含む薬学的処方物を調製する工程。
【0035】
例えば、ペプチド化合物の溶液とキャリア巨大分子の溶液とを、ペプチド化合物およびキャリア巨大分子の水不溶性複合体が溶液から沈殿してくるまで合わせる。特定の実施態様において、ペプチド化合物およびキャリア巨大分子の溶液は水性溶液である。あるいは、ペプチド化合物またはキャリア分子(または両方)が2つの混合前に実質的に水溶性でない場合、ペプチド化合物および/またはキャリア巨大分子は、複合体の2つの成分を合わせる前に、アルコール(例えばエタノール)のような水混和性溶媒に溶解され得る。水不溶性複合体の調製方法の別の実施態様において、ペプチド化合物の溶液とキャリア巨大分子の溶液とを、ペプチド化合物およびキャリア巨大分子の水不溶性複合体が溶液から沈殿してくるまで合わせ、そして加熱する。水不溶性複合体を得るために必要なペプチド化合物およびキャリア巨大分子の量は、使用される特定のペプチド化合物およびキャリア巨大分子、使用される特定の溶媒、および/または複合体を得るために使用する手順に依存して変化し得る。しかしながら、代表的には、ペプチド化合物はモルを基準にして、キャリア巨大分子に対して過剰である。しばしば、ペプチド化合物はまた、実施例において示されるように、重量/重量を基準にして過剰である。特定の実施態様において、キャリア巨大分子(好ましくは、カルボキシメチルセルロースナトリウム)、およびペプチド化合物(好ましくは、PPI-149)は、0.2:1(w/w)のキャリア巨大分子:ペプチド化合物の比で合わされる。種々の他の実施態様において、キャリア巨大分子:ペプチド化合物の比(w/w)は、例えば、0.5:1、0.4:1、0.3:1、0.25:1、0.15:1または0.1:1であり得る。本発明の水不溶性複合体の調製のための条件および手順の非制限的な例が、実施例1〜5および8〜9でさらに記載される。
【0036】
ペプチド化合物/巨大分子複合体が、いったん溶液から沈殿すると、沈殿物は、濾過(例えば、0.45ミクロンナイロン膜を通して)、遠心分離などのような当該分野で公知の手段により溶液から取り出され得る。次いで、回収されたペーストが乾燥され得(例えば、真空でまたは70℃のオーブンで)、そして固体が当該分野で公知の手段により粉砕または摩砕されて粉末にされ得る(例えば、ハンマーあるいはゴア粉砕、または乳鉢および乳棒での摩砕)。粉砕または摩砕に続いて、粉末は網(好ましくは90ミクロンの網)でふるいにかけられ、均一な粒子分布が得られ得る。さらに、回収されたペーストは、冷凍され、凍結乾燥して乾燥され得る。複合体の粉末形態は、キャリア溶液に分散されて注射に適した液体懸濁物または半固体分散体を形成し得る。従って、種々の実施態様において、本発明の薬学的処方物は、乾燥固体、液体懸濁物、または半固体分散体である。液体懸濁物における使用に適した液体キャリアの例は、生理食塩水溶液、グリセリン溶液、およびレシチン溶液を含む。
【0037】
別の実施態様において、本発明の薬学的処方物は滅菌処方物である。例えば、水不溶性複合体の形成に続いて、この複合体は、最適にはγ線照射または電子線滅菌により滅菌され得る。従って、上記で記載される薬学的処方物を調製するための本発明の方法はさらに、水不溶性複合体のγ線照射または電子線照射による滅菌を含み得る。好ましくは、処方物は少なくとも15KGyのγ線線量を使用するγ線照射により滅菌される。他の実施熊様において、処方物は少なくとも19KGyまたは少なくとも24KGyのγ線線量を使用するγ線照射により滅菌される。実施例11で実証されるように、本発明の処方物はγ線照射に際して十分に安定なままである。
【0038】
あるいは、滅菌薬学的処方物を調製するために、水不溶性複合体は従来の滅菌技術を使用して単離され得る(例えば、滅菌された出発物質の使用、および無菌での製造プロセスの実行)。従って、上記で記載される薬学的処方物を調製するための方法の別の実施態様において、水不溶性複合体は無菌の手順を使用して形成される。
【0039】
本発明の水不溶性複合体の形成方法はさらに、実施例1〜5および8〜9に記載される。本発明の方法により調製される、粉末、液体懸濁物、半固体分散体、乾燥固体(例えば、凍結乾燥固体)、およびそれらの滅菌形態(例えば、γ線照射により)を含む、薬学的処方物がまた、本発明に包含される。
【0040】
本発明のさらに別の局面は、水不溶性複合体に含まれる薬学的に活性なペプチド化合物により処置可能な状態を患っている被験体を処置するための、本発明の薬学的処方物の使用法に関する。従って、好適な実施態様において、本発明は、LHRHアナログとキャリア巨大分子との水不溶性複合体を含む薬学的処方物を被験体に投与する工程を含む、LHRHアナログで処置可能な状態の被験体を処置するための方法を提供する。
【0041】
薬学的処方物が、所望の治療的結果を得るために適した任意の経路により被験体に投与され得るが、好ましい投与経路は非経口経路、特に筋肉内(i.m.)注射および皮下/皮内(s.c./i.d.)注射である。あるいは、処方物は経口で被験体に投与され得る。他の適した非経口経路は、静脈内注射、口腔投与、直腸、膣、鼻内または気道経路による経皮送達および投与を含む。i.m.またはs.c./i.d.経路により数週間から数ヶ月の持続した送達を提供する処方物が、代替的な経路により投与される場合、他の生理学的機構による薬剤のクリアランスにより同等の時間の間の薬剤の持続した送達が起こらないかもしれない(すなわち、投薬形態が送達部位から排泄され(clear)得るため、長期治療的効果がi.m.またはs.c./i.d.注射で観察されるものほど長い期間は観察されない)ことを注意するべきである。
【0042】
薬学的処方物は、治療的に有効な量のLHRHアナログを含有する。「治療的に有効な量」は、投薬において、そして必要な期間にわたり、所望の結果を得るために有効な量を示す。LHRHアナログの治療的に有効な量は、個体の疾患状態、年齢、および体重、ならびにLHRHアナログの個体において所望の応答を導き出す能力(単独または1つ以上の他の薬物との組み合わせにおいて)のような因子によって変化し得る。投薬レジメンは、最適の治療応答を提供するために調整され得る。治療的に有効な量はまた、治療的に有益な効果がアンタゴニストのどのような毒性または有害な影響にも勝る(outweighed)量である。LHRHアナログの治療的に有効な量に対する非制限的な範囲は、0.01〜10mg/kgである。28日間の血漿テストステロンレベルの持続した減少のためのLHRHアナログPPI-149の好適な投薬量は、ほぼ1mL以下の液体懸濁物の体積中、ほぼ0.1〜10mg/kg、より好ましくは0.3〜1.2mg/kg(遊離のペプチドとして表される)である。投薬量の値が緩和されるべき状熊の重篤度によって変化し得ることが注意されるべきである。任意の特定の被験体に対して、特定の投薬レジメンが、個々の必要性によっておよび組成物の投与または投与の監督を行う個人の専門的判断によって、経時的に調整されるべきであること、および本明細書中で記述される投薬範囲が例示に過ぎず、そして請求の組成物の範囲または実施を制限することを意図しないことが、さらに理解されるべきである。
【0043】
本発明の処置方法は、LHRHアナログの投与が所望の臨床的効果を有する種々の状態、疾患、および障害の処置に適用され得る。疾患および障害の例は、ホルモン依存性癌(例えば、前立腺癌、乳癌、卵巣癌、子宮癌、および精巣癌)、良性前立腺肥大、性的早熟、子宮内膜症および子宮筋腫を含む。従って、本発明は、本発明の薬学的処方物の投与によりこれらの疾患および障害を処置する方法を提供する。さらに、LHRHアナログは排卵を変更するために使用され得る。従って、本発明の方法はまた、インビトロでの妊娠および避妊目的に使用され得る。
【0044】
特定の好適な実施態様において、この方法は、前立腺癌の処置のために使用され得、処方物に使用されるLHRHアナログはLHRHアンタゴニスト、最も好ましくはPPI-149であり、そしてこの方法は筋肉内または皮下投与による投与後少なくとも4週間インビボでLHRHアナログの持続した送達を可能にする。本発明により処方されたLHRHアナログ、好ましくはPPI-149は、前立腺癌を患っている被験体にLHRHアナログを投与することにより、前立腺癌細胞の成長を阻害するために使用され得る。さらに、本発明により処方されたLHRHアンタゴニスト、好ましくはPPI-149は、LHRHアゴニスト治療を開始する前に前立腺癌を患っている被験体にLHRHアンタゴニスト、好ましくはPPI-149を前投与することにより、LHRHアゴニストの使用に伴うテストステロンの急増を阻害するために使用され得る。本発明の処方物が適用され得る、LHRHアゴニスト誘導テストステロン急増を阻害するための方法、およびLHRHアンタゴニストを使用する前立腺癌を処置するための他の方法は、米国特許出願番号第08/573,109号、表題「LHRHアンタゴニストを使用する前立腺を処置するための方法」(1995年12月15日出願)およびその一部継続特許出願番号第08/755,593号、同じく表題「LHRHアンタゴニストを使用する前立腺を処置するための方法」(1996年11月25日出願)においてさらに記載され、この両者の内容が公開PCT出願WO97/22357号に援用される。米国出願および公開PCT出願の全内容が、参考として本明細書中に特に援用される。
【0045】
薬学的に活性なペプチド化合物のキャリア巨大分子との複合体化のための特定のプロセスが、以下の実施例1〜5および8〜9に記述される。LHRHアンタゴニスト含有複合体が、インビボで薬学的に活性なペプチドの持続した送達を可能にし得ること(実施例6)およびLHRH-アゴニスト誘導テストステロン急増を阻害し得ること(実施例7)を実証する試験結果もまた、記載される。本発明をさらに例示する以下の実施例は、制限として解釈されるべきではない。本出願全体にわたり引用される全ての参考文献、特許、および公開特許出願の内容は、参考として本明細書中に援用される。
【実施例】
【0046】
実施例1:
LHRHアンタゴニストPPI-149 100ml溶液を、PPI-149 6.25mg/mlを水に溶解して調製した。等量の米国薬局方カルボキシメチルセルロースナトリウム(CMC)(低粘度グレード(grade)、Hercules Chemical Co.)サンプル(最小100ml)を、0.125%w/vで調製し、溶解するまで混合した。各等量のPPI-149およびCMC溶液を混合し(0.2:1(w/w)のCMC:ペプチド比を与える)、そして固体物質を得た。その固体物質を一晩撹拌し、その後0.45ミクロンのナイロンフィルターで濾過することで回収した。その溶液濾液のHPLC評価では、少なくとも95%のPPI-149化合物が固体複合体に変換したことを示した。溶液から取り除いた。回収した白色ペーストを水で2回すすぎ、そしてその後バイアルへ移し、減圧下で乾燥した。72時間の乾燥後、白色粉末を633mg得た。固体物質をその後、乳鉢および乳棒で粉末化した。元素分析は、複合体中に57%のペプチドを示した。
【0047】
実施例2:
PPI-149 25mgを1mlの水に溶解した。これに0.5%カルボキシメチルセルロース溶液を1ml加えた。混合により、混合物が絹状の白色固体を形成した。混合物を加熱して5分間環流し、そして羊毛状白色沈殿を形成した。遠心分離/デカンテーションでこの物質を分離した。その固体を水中で再懸濁し、遠心分離を繰り返して回収した。その溶液濾液のHPLC評価は、少なくとも90%のPPI-149化合物が固体複合体へ変換したことを示した。その白色沈殿を減圧下乾燥し、そして固体物質を乳鉢および乳棒で粉末化した。元素分析は、複合体中に77%のペプチドを示した。
【0048】
実施例3:
PPI-149 50mgを2mlの5%マンニトールに溶解し、そして2mlの0.5%カルボキシメチルセルロース(低粘度、米国薬局方、Spectrum Quality Chemicals)と混合した。混合物を撹拌し、そして即座に白色沈殿が生成した。懸濁液を凍結し、そして凍結乾燥により乾燥し、PPI-149持続送達(sustained delivery)複合体を得た。
【0049】
実施例4:
PPI-149 25mgを1mlの水に溶解した。これに1mlの0.5%アルギン酸ナトリウム、米国薬局方(Spectrum)を加えた。混合後、混合物は即座に白色沈殿を形成した。遠心分離/デカンテーションでこの物質を単離した。その固体を水中で再懸濁し、遠心分離を繰り返して回収した。その白色沈殿を減圧下乾燥した。元素分析を実施して、66%のペプチド含有量を得た。
【0050】
実施例5:
PPI-149 25mgを1mlの水に溶解した。pHを11.0に調整するために、アンモニアを加えた。これに1mlの0.5%アルギン酸、米国薬局方(Spectrum)を加えた。混合後、混合物は即座に白色沈殿を形成した。遠心分離/デカンテーションでこの物質を分離した。その固体を水中で再懸濁し、遠心分離を繰り返して回収した。その白色沈殿を減圧下乾燥した。元素分析を実施して、79%のペプチド含有量を得た。
【0051】
実施例6:
LHRHアンタゴニストPPI-149およびカルボキシメチルセルロースの水不溶性複合体を、前述の実施例に従って調製した。PPI-149/CMC複合体の懸濁液を調製し、1回投与量をラットおよびイヌの筋肉内に注射した。ラットへの投与量は50μg/kg/日×60日であり、そしてイヌへの投与量は40μg/kg/日×28日であった。血漿テストステロンレベル(ng/mlにおいて)を、動物におけるLHRHアンタゴニストの活性の尺度として種々の時点で決定した。図1のグラフに示した代表的な結果は、PPI-149/CMC複合体の筋肉内注射が、ラットにおいては少なくとも42日間、およびイヌにおいては少なくとも28日間、血漿テストステロンレベルの抑制の持続に導くことを実証し(図1中の白い四角で示す)、LHRHアンタゴニストの送達の持続を実証する。動物におけるPPI-149の血漿レベル(ng/mlにおいて)をまたモニターした(図1中の黒い四角で示す)。最初のPPI-149急上昇が最初の約8日間に観察され、その時以後、PPI-149は実質的に血漿中に検出不可能であった。約8日以降の血漿中のPPI-149検出が不可能であるにもかかわらず、テストステロンレベル結果は、PPI-149が、インビボにおいて実験の過程中、依然として治療上活性であったことを実証する。
【0052】
実施例7:
LHRHアンタゴニストPPI-149およびカルボキシメチルセルロースの水不溶性複合体を、前述の実施例に従って調製した。PPI-149/CMC複合体の懸濁液を調製し、1回投与量をラットの筋肉内に、0日に注射した。30日に、LHRHアゴニストLupronTM(ロイプロリド(leuprolide))をラットに注射した。血漿テストステロンレベル(ng/mlにおいて;図2中の白い四角で示す)を、動物におけるLHRHアンタゴニストの活性の尺度として種々の時点で決定した。動物におけるPPI-149の血漿レベル(ng/mlにおいて)をまたモニターした(図2中の黒い四角で示す)。図2のグラフに示した代表的な結果は、PPI-149/CMC複合体での前処置が、速やかに血漿テストステロンを去勢(castration)レベルに減少させ、そしてその上LHRHアゴニスト誘導テストステロンの急激な上昇をブロックすることを実証する。約8日を越えると血漿中にPPI-149が検出され得ないにもかかわらず、テスロステロンレベル結果は、PPI-149が、インビボにおいて実験の過程中、依然として治療上活性であったことを実証する。
【0053】
実施例8:
この実施例において、LHRHアナログPPI-258およびカルボキシメチルセルロース(CMC)との間に、不溶性複合体を生成した。PPI-258は、以下の構造を有する:アセチル-D-ナフチルアラニル-D-4-Cl-フェニルアラニル-D-ピリジルアラニル-L-セリル-L-チロシル-D-アスパラギニル-L-ロイシル-L-Ne-イソプロピル-リジル-L-プロピル-D-アラニル-アミド。PPI-258/CMCデポ剤の調製のために、174.8mg(148.6mg正味重量)のPPI-258を29.72mLの水に加え、そして材料を撹拌し、ペプチドを懸濁させそして溶解した。この撹拌溶液に、1.85mlの2%CMCナトリウム溶液(Hercules)を加えた。固体沈殿物が即座に認められた。加熱環流後、懸濁液が半透明になり、その後白色沈殿が現れる。5分間の環流後、反応物を冷却し、固体を遠心分離で分離した。固体を水ですすぎ、1晩減圧下乾燥した。乾燥粉末を乳鉢および乳棒で粉末化し、90ミクロンのステンレス綱のふるいを通してふるった。ふるった粉末(90ミクロンふるい)を回収してキャラクタリゼーションした。総収量は乾燥粉末で198.4mgであり、粉砕工程後、110.8mgのサイズ調整された粉末を得た。キャラクタリゼーションは、以下の構成の複合体組成を提供した。:ペプチドPPI-258-80%、CMC-18.8%、水-6.6%。
【0054】
実施例9:
この実施例において、LHRHアナログCetrorelixTM(SB-75としてまた公知である)およびカルボキシメチルセルロース(CMC)との間に、不溶性複合体を形成した。CetrorelixTMは、以下の構造を有する:アセチル-D-ナフチルアラニル-D-4-Cl-フェニルアラニル-D-ピリジルアラニル-L-セリル-L-チロシル-D-シトルリル-L-ロイシル-L-アルギニル-L-プロピル-D-アラニル-アミド。CetrorelixTM/CMCデポ剤の調製のために、102.8mg(87mg正味重量)のCetrorelixTMを17.4mLの水に加え、そして物質を撹拌して懸濁させ、そしてペプチドを溶解した。この撹拌溶液に、1.1mlの2%CMCナトリウム溶液(Hercules)を加えた。固まり状の白色沈殿物が即座に認められた。懸濁液を5分間加熱環流し、冷却して白色固体沈殿を得た。固体を遠心分離で単離し、水ですすぎ、1晩減圧下乾燥した。乾燥粉末を乳鉢および乳棒で粉末化し、90ミクロンのステンレス綱のふるいを通してふるった。その粉末を回収してキャラクタリゼーションした。総収量は乾燥粉末で95mgであり、粉砕工程後、60mgのサイズ調整(size)された粉末を得た。キャラクタリゼーションは、以下の構成の複合組成を提供した。:ペプチドCetrorelixTM-75%、CMC-20.7%、水-6.5%。
【0055】
実施例10:
この実施例において、前述の3実施例で述べられたように、CMCデポ剤処方物として調製された、3種の異なるLHRHアナログPPI-149、PPI-258およびCetrorelixTMの持続放出をインビボにおいて試験した。3種の異なる処方物ビヒクルを生理食塩水、グリセリン(15%グリセリン/4%デキストロース)およびレシチンでテストした。Sprague-Dawleyラット(オス25体、重量範囲300-325g)を使用し、そしてLHRHアナログの有効性を血漿テストステロンレベルの減少に基づいて決定した。
【0056】
投与の投与量および経路は以下のとおりであった:
【0057】
【表1】
ペプチドの実際の投与量は300μg/kg/日を30日間であり、これは1回200μLの筋肉内(IM)もしくは皮下(SC)注射として与え、2.7mg/ラットであった。5ラット/グループに注射するために必要な総体積は、13.5mg/mLの活性ペプチド濃度で1.3mLであった。以下に示すように、注射の体積を一定に保ち、そして粉体の重量を以下のように総ペプチド含量について調整した:
【0058】
【表2】
試験品目の1回の200μL筋肉内、もしくは皮下注射は、それぞれ左後肢の上部側面もしくは肩胛骨間の皮下に、麻酔下で0日に行った。
【0059】
血漿テストステロンレベルをテストするために、血液約0.4mLを後眼窩洞から投与後1日および3、7、14、21、28および35日に取り出した。標準的な方法でテストステロン血漿レベルを決定するために血液を処理して血漿にし、そしてドライアイス上で凍結した。
【0060】
図3A-3Cに示される代表的な結果は、オスSprague-Dawleyラットにおける血漿テストステロンレベルが少なくとも28日間、そして50日間ほど、CMCデポ剤処方物として調製されたLHRHアナログPPI-149、PPI-258およびCentrolixTMの持続放出に応答して減少し、そして低いレベルで保持されることを実証する(それぞれ、図3A、3Bおよび3Cに示す)。これらの結果は、3種の処方物全てが、インビボにおいて血漿テストステロンレベルの減少に有効であり、長時間にわたり減少した血漿テストステロンレベルを保持することを示す。
【0061】
実施例11:
この実施例においてPPI-149-CMC処方物を、殺菌の目的でガンマ線照射に曝露し、その後、照射処方物の物理的および化学的性質の両方から評価を行った。以下に述べたデータは、ガンマ線照射がPPI-149-CMCデポ剤の殺菌に実現性のある方法であることを示している。
【0062】
ペプチド安定性
各約40mgの、2個の分離されたPPI-149-CMCロットを多数のType Iのガラスバイアルに別々に詰め(空気頭部空隙下)、ゴムストッパーおよびアルミニウムシールでシールした。バイアルをその後種々のみかけの線量のガンマ線照射にさらした。2つのロットのそれぞれについて、各ガンマ照射曝露の各レベルにおいて、2個のバイアルを、ペプチド純度(%で表す)について分析した。その結果は、γ線照射線量24KGy以下において、PPI-149-CMCが一貫してペプチド純度において2%未満の減少を示した(HPLC不純物プロフィールで決定した)ことを示した。より高いガンマ曝露線量の利用の第2の研究を、追加的なPPI-149-CMCの実験室ロットで実施した。PPI-149-CMCは、高いガンマ線量に曝露した場合、著しく良好な化学的安定性を実証した。
【0063】
次の予備処方の研究は、PPI-149-CMCγ線照射後に得た分解プロフィールと、PPI-149注射可能溶液(1mg/mL)のオートクレーブ後に得た分解プロフィールとを比較するために実行した。2個のサンプルを調製した:a)19KGyのγ線照射に曝露したPPI-149-CMC;b)オートクレーブ(121℃/20分)に供されたPPI-149溶液(1mg/mL)。2個のサンプルのHPLCクロマトグラムは、2個のサンプルについての分解プロフィールが定性的に同様でありそうであることを実証した(同様の主要ピークの相対保持時間が与えられた)。
【0064】
ガンマ線照射後のストレス下安定性保存
ストレスを受けた保存予備処方物の研究が、ガンマ線照射後のバイアルにおいてまた実施された。2個のPPI-149-CMCの実験室ロットからの密封されたバイアルを19KGyのガンマ線照射に曝露し、1ヶ月まで25℃、37℃および50℃で保存した。これらの予備処方物研究における化学的安定性データは、投与量19KGyのγ照射後のストレスを受ける保存安定性が、高度にストレスを受ける保存状態下(例えば、50℃で1週間)でさえ、主たる化学的不安定性をもたらさなかったことを示した。データはγ照射線量19KGy以下を示し、28日まで50℃以下でのPPI-149-CMCの保存は一貫してペプチド純度において2%未満の減少を示した(HPLC不純物プロフィールで決定した)。研究した2個のロット間の初期水分含量の明らかな差異にもかかわらず、初期の予備処方安定サンプルもしくは1ヶ月まで保存したもののどちらにも、ペプチド純度における顕著な差が測定されなかった。
【0065】
PPI-149-CMC粒子サイズ分析
レーザー光散乱を用いた粒子サイズ法が発達しており、PPI-149-CMCのサイズ研究に適用できる。その方法の使用を例示するために、PPI-149-CMCの粒子サイズへのガンマ線照射の影響を調べるために実施した予備処方実験を示す。この実験は、アモルファス固体物質は保存により粒子凝集する傾向であり得るという以前の理解で考えた。PPI-149-CMC実験室ロットの2個の試料をtype Iのガラスバイアルに詰め、灰色のブチルゴムストッパーで閉め、アルミニウムシールで密封した。粒子評価を、投与量15.5KGyのγ線照射への曝露の前および後に実施した。粒子評価はレーザー光散乱(逆フーリエレンズを備えたMalvern Mastersizer STMを使用)で実施した。レーザー光散乱による粒子サイズ分析のための20mgのサンプルを、約0.5mLの脱イオン水へ激しく撹拌することにより分散し、その後周囲温度の浴で5分間超音波処理した。バックグラウンド計算を実行後、定性実験法を実施した。サンプル分散液を約20%の暗黒化(obscuration)が得られるまで、連続フィードリザーバー(feed reservoir)(公称体積 約60mL)へ滴下した。ミキサー回転速度は、実験(バックグラウンド確認も加えて)を通して2700rpmで保持された。この速度でボルテックス由来の泡は生じなかったが、十分に安定した分散液を保持した。8スキャンを行った。得られたデータの分析は、とった任意のデータポイントの極値(extreme)として0.03%以下の標準偏差を示した。サンプル分散液をリザーバーに15分間保持し、そしてその後再測定した場合、顕著な変化は得られず、実験の過程を通して粒子溶解が無かったことを示した。
【0066】
サンプルを、上記に挙げた実験パラメータを使用して分析した。8スキャンを実行し、そして平均粒子直径データを測定した。2つの異なるサイズ分布が見られ、そして全てが粒子サイズの高端で明らかなカットオフを有しており、粒子凝集の無いことを示した。PPI-149-CMCの1ロットは、ガンマ線照射前では、サンプルのガンマ線照射後よりも明らかに低い平均体積直径を有した。この予備処方研究は、殺菌プロセス中にいくらかの粒子凝集が起こったことを示すように考えられる。
【0067】
実施例12:
この実施例では、PPI-149-CMCの固体状態形態に対するγ線照射および温度/湿度の両方のストレスに対する有効性を研究するために、種々の予備処方実験を行った。
【0068】
X線粉末回折
最初の実験においては、2個の60mgのPPI-149-CMCサンプルを(エアーヘッドスペース下(air headspace)で)、I型ガラスバイアル中にパックし、灰色のブチルゴムストッパーで閉め、そしてアルミニウムシールでシールした。次いで、一つのサンプルを、19.0KGyのγ線照射線量に曝露した。次に、固体状態形態の2個の60mgのサンプルをX線粉末回折によって研究した。回析図を19.0Kgyのγ線照射線量に曝露する前および後で比較した。
【0069】
次の研究では、PPI-149-CMCの60mgサンプル(γ線照射後)をI型ガラスバイアル中に置き、そして予め平衡化した50℃/75%相対湿度の一定湿度インキュベーター中に5日間置いた。インキュベーターから回収した後すぐに、サンプル容器を灰色のブチルゴムのストッパーで閉め、そしてアルミニウムシールでシールした。次いで、このストレスにさらしたサンプルのX線粉末回折を、容器中にて室温に維持した同じロットの別のサンプルと比較した。グラファイトモノクロメーターおよび50kV、40mAで操作されているCu(λ=1.54Å)X線源を備えた、Siemens D500自動化Powder Diffractometerを用いて、これらのサンプルを分析した。2θ走査範囲は、4〜40°であり、0.05°/1.2秒段階の段階走査窓を用いる。ビームスリットはNo.(1)1°、(2)1°、(3)1°、(4)0.15°および(5)0.15°の幅にセットした。2θ校正をNBSマイカ標準(SRM675)を用いて行った。サンプルを、ゼロバックグラウンドサンプルプレートを用いて分析した。
【0070】
データはγ線照射の前には、PPI-149-CMCは、明らかな結晶構造、または擬結晶構造を有さないことを示した。実際、アモルファス固体に特徴的なX線粉末回折パターン(回析図中の2°〜20°20の間の有意なピークのない幅広のハンプ)を示した。照射後のPPI-149-CMCサンプルは、非照射サンプルと非常に類似の回析パターンを生じ、これはガンマ線照射処理(19KGyまでおよび19KGyを含む線量)が、明らかに物質中での固体状態の多型転移を誘導しないことを示す。類似の様式で、PPI-149-CMCの温度/湿度ストレスをかけたサンプルは、非照射サンプルおよび照射サンプルの両方と非常に類似の回析パターンを生成し、このことはppI-149-CMCが、物質内での固体状態多型転移を過度に誘導する傾向がないことを強く示唆する。
【0071】
吸湿性
PPI-149-CMC(照射後)での予備処方研究を、一定温度(25℃)で種々の相対湿度の条件下で平衡湿気取り込み(重量増加によって測定)を決定するためによって行った。相対湿度(RH%)の関数としての平衡湿気(水%)の分析は、湿気の含量が約80%相対湿度まで徐々に増加することを示した。高い相対湿度(95%RH)において、PPI-149-CMCは、有意な湿分吸収が可能であった。80%RH以下の相対湿度において、湿気からの保護という点で有意な予防策は、不必要であると考えられる;従って、特定の製造工程は周囲湿度条件下で行い得る(ただし、極端な湿度は避ける)。
【0072】
実施例13:
この実施例において、PPI-149-CMCの融解研究を行った。実験はシンクを用いる条件、およびシンクを用いない条件の両方を利用して行った。PPI-149-CMCは、25℃で、pH7.3の0.1Mリン酸緩衝化生理食塩水に約100μg/mlの溶解度(遊離ペプチドとして測定され表される)を有した。シンク条件下において(所定の温度での、この系での飽和溶解度の<10%として定義される)、撹拌しない場合でさえ、PPI-149-CMCは迅速に溶解した(遊離のペプチドとして測定、および示した)。類似の実験において、PPI-149-CMCの平衡溶解度を25℃でpH7.3の0.1Mリン酸緩衝化生理食塩水中で決定し、(遊離のペプチドとして測定、および示し)これには3個のサンプルを用いた:PPI-149-CMC単独、さらに10(重量)%のPPI-149(遊離のペプチドとして示したが、酢酸と結合したPPI-149として導入した)が存在する場合でのPPI-149-CMC、およびさらに50(重量)%米国薬局方カルボキシメチルセルロースナトリウムが存在する場合でのPPI-149-CMC。3つのサンプルは全て、表面上は類似のペプチド平衡溶解度を示した。選択された緩衝液系は生理学的条件下に近いので、PPI-149-CMC中に存在するさらに遊離のカルボキシメチルセルロースまたはペプチド種の存在は、溶解性に影響を与えそうにないと考えられる。
【0073】
実施例14:
この実施例において、PPI-149-CMCの反復した皮下(SC)および筋肉内(IM)用量での薬物動力学、薬力学、および安全性をイヌにおいて特徴付けた。
【0074】
3ヶ月間行った最初の研究において、種々の再構成ビヒクル中のPPI-149-CMCの月一回IM注射またはSC注射を、1.2mg/kg(1日目)、0.3mg/kgまたは0.6mg/kg(29日目)、および1.2mg/kg(57日目)で用いて、40匹の雄ビーグル犬を評価した。5匹のイヌの8グループを以下に示すように本研究に割り当てた:
【0075】
【表3】
a.再構成ビヒクルをPPI-149-CMCを粒子懸濁物として再構成するのに用いた。
これらは以下を含む(水中に):
1. グリセリン=15%グリセリン/5%デキストロース
2. PEG=4%ポリエチレングリコール-3350/4%マンニトール
3. レシチン=0.5%レシチン/5%マンニトール
b.記号:臨床研究に用いられる再構成ビヒクルは0.9%塩化ナトリウム米国薬局方
c.全用量はペプチド(PPI-149)含量に関して示す。
d.完全な解剖学的組織学および顕微鏡学的組織学用に3匹の動物を85日目に屠殺した。
【0076】
この研究は、異なるビヒクル中での初回用量でのPPI-149-CMCの効力が、処置の最初の1ヶ月の間に評価されるように設定した。研究中の2ヶ月目の間、この計画において、有効な「維持」用量を決定することを試みるために、イヌはより低い投与量のPPI-149-CMCを受けた。3ヶ月目は、PPI-149-CMCの長期安全性および効力特性を評価するために計画した。
【0077】
再構成ビヒクルの1つの中に処方されたPPI-149-CMCのIMもしくはSC用量、またはコントロール体のIM用量を、各投与日に右後ろ足の上部側面(IM)へ、または肩甲部の中央(SC)に投与した。物質を23gの短いベベル針を備えた1ccのツベルクリンシリンジに引き込んだ。投与の直前に注射部位をアルコール綿棒で拭く。注入する用量は、ペプチド/kg体重の特定用量に基づいていた。全ての用量は、投与されたPPI-149ペプチドの量をいうということに留意するべきである。
【0078】
実験の間はずっと、毒性または薬理学的効果および通常の行動および様子における変化の明らかな徴候について、各動物を1日少なくとも2回観察した。全ての臨床的に異常な所見を記録した。
【0079】
初回用量の投与の前、および投与後の種々の時点で、全血球算定(CBC)、血清化学分析、ならびに週に2回の放射イムノアッセイによるPPI-149濃度およびテストステロン濃度の決定のために、血液を回収した。
【0080】
研究の3ヶ月後、9匹の動物を屠殺し、そしてそれらの組織を、全体的な病理学的分析および組織病理学的分析のために回収した。屠殺のため、ビヒクルコントロールグループ、IM投与グループの1つ、およびSC投与グループの1つから、動物を選択した。3ヶ月目の屠殺物の、全体的な病理学および組織病理学のために回収した組織は:投与部位(SCまたはIM)、副腎、大動脈、骨、骨髄、脳、横隔膜、精巣上体、食道、視神経付きの眼、心臓、腎臓、大腸(盲腸、結腸)、胆嚢を有する肝臓、気管支付きの肺、リンパ節、膵臓、脳下垂体、尿道付きの前立腺、唾液腺、座骨神経、骨格筋、皮膚、小腸(十二指腸、空腸、回腸)、脊髄、脾臓、胃、精巣、胸腺、副甲状腺付きの甲状腺(thryoid gland)、舌、気管、膀胱(urinary bladdr)、および全体的な病変。
【0081】
実験の間、処置動物またはコントロール動物のいずれも、血液学または血液化学的にはベースラインから有意な変化はなかった。3ヶ月目の全体的なおよび組織学的評価において、屠殺物はこのLHRHアンタゴニストに期待されたような、精巣および前立腺中の変化を除いては、PPI-149-CMC処理イヌおよびコントロール(ビヒクル処置)動物との間の明らかな差を示さなかった。
【0082】
PPI-149-CMCの薬物力学に関して、種々の再構築ビヒクル中の再懸濁した1.2mg/kg PPI-149-CMCで処理し、およびIMまたはSCを投与した全てのイヌは、類似の血漿PPI-149の薬物動力学プロフィールを示した(血漿濃度は最初の2日間にピークを有し、次いで次の月の間ゆっくりと、指数関数的に減少する)。この研究に用いた3個の再構築したビヒクルの任意のものに再懸濁した場合、PPI-149-CMCは、PPI-149の類似の血漿分布を示した。
【0083】
PPI-149-CMCの内分泌効力に関して、全てのイヌにおいて、去勢レベル(<0.6ng/mL)のテストステロンが、PPI-149-CMC投与の開始24時間以内に観察され、そしてレベルは、一般に投与経路または再構築ビヒクルの選択に関わらず、最初の1ヶ月間を通じて去勢範囲を維持した。35匹のうち26匹のイヌ(75%)が、29日目のPPI-149-CMCの第2用量の投与直前に得た血液サンプル中のテストステロンの去勢レベルを有した。これらの結果は、イヌの1.2mg/kgの初回用量が首尾良く血漿テストステロンの迅速で長期間持続する抑制(>28日)を誘導することを示す。投与の2月目において、「維持」用量の効力(初回用量より低い用量)を研究した場合、結果はPPI-149-CMCの0.3mg/kgまたは0.6mg/kgの投与が、35匹中30匹のイヌの20日以上の間のテストステロンの去勢レベルの維持を示した。処置の2ヶ月目の最後(57日目)で、35匹中21匹のイヌ(60%)が去勢レベルを維持し、一方14動物が正常範囲(>0.6%ng/mL)のテストステロンを有した。3ヶ月目の初めに1.2mg/kgの用量を投与した。PPI-149の血漿濃度は、続く28日間維持され、一方テストステロンの血漿レベルは再び「去勢」であった。3ヶ月目の最後(85日目)までに、テストステロンの血漿レベルは35匹のPPI-149-CMC処置イヌの30匹が去勢範囲にあることが示された。
【0084】
要約すると、35匹のイヌが、種々の再構築ビヒクルで、IMまたはSC投与を用いて、1日目に1.2mg/kgのPPI-149-CMC、29日目に0.3mg/kgまたは0.6mg/kgのPPI-149-CMC、ならびに57日目に1.2mg/kgのPPI-149-CMCを受けた。これらの35匹のうち19動物(54%)が、治療の全経過を通じて去勢範囲を維持した血漿テストステロンレベルを有した。従って、28日の間隔でのPPI-149-CMCの投与は、血漿テストステロンの完全な抑制を生じることができた。これは迅速(全動物が24時間以内に去勢レベルを有した)であり、そして長期間持続する(投与の経過にわたって維持された)。
【0085】
上記と類似の研究を、PPI-149-CMCの長期的な安全性および効力の特徴をさらに評価するため、6ヶ月間イヌで行った。動物は、IMまたはSCのいずれかで1.2mg/kg PPI-149-CMCの初回用量を受け、そして28日の間隔で5回の続く用量(0.3・g/kg、0.6mg/kg、または1.2mg/kgのいずれかの濃度)を受けた。血漿テストステロンレベルおよびPPI-149レベルを、放射イムノアッセイによって一定間隔で評価した。代表的な結果を図4(SC処置について)および図5(IM処置について)に示す(これは、血漿テストステロンレベル(白四角)およびPPI-149レベル(黒四角)を例示する)。PPI-149-CMCの各投与に用いた特定の投薬量をグラフに示す。図4および図5に例示した結果は、28日間隔でのPPI-149-CMCの投与が、血漿テストステロンの完全な抑制を生じ得たことをさらに実証する。これは迅速でかつ長く持続し、6ヶ月もの長い間、減少した血漿テストステロンレベルを維持する。
【0086】
等価物
当業者は、本明細書に記載された本発明の特定の実施態様に対する多くの等価物を、日常的実験以外を用いることなく認識するか、または確認しうる。このような等価物は、以下の請求の範囲に包含されることが意図される。
【背景技術】
【0001】
発明の背景
種々の疾患および臨床的障害が、薬学的に活性なペプチドの投与により処置される。そのような例の1つに前立腺癌があるが、これは性ホルモン依存性癌であり、そして男性ホルモンの合成を調節する黄体ホルモン(LH)の産生を妨げる黄体形成ホルモン放出ホルモン(LHRH)アナログの投与により処置され得る。詳細には、LH産生を減少させるために、黄体形成ホルモン放出ホルモンレセプター(例えば、ロイプロリドおよびゴセレリン)のスーパーアゴニストとして作用するLHRHのペプチドアナログが使用されてきた。
【0002】
多くの場合、薬学的に活性なペプチドの治療的効果は、長期間に渡りインビボで継続的にそれが存在することに依存する。インビボでのペプチドの継続的な送達を達成するために、繰り返し投与を行う必要をさけるために持続した放出または持続した送達処方が望ましい。持続した薬物送達のための一つの試みは、マイクロカプセル化によるものである。この方法において、活性成分は微粒子を生成するポリマー膜内に封じ込められている。例えば、LHRHスーパーアゴニスト(例えば、ロイプロリドおよびゴセレリン)は、典型的には、数週間から数ヶ月にわたりスーパーアゴニストの持続した送達を提供する貯蔵注射(depot injection)に適した処方物を調製するために、ポリ−ラクチド/ポリ−グリコリド共重合体を含む微粒子内にカプセル化される(例えば、米国特許第4,675,189号;4,677,191号;5,480,656号および4,728,721号を参照のこと)。
【先行技術文献】
【特許文献】
【0003】
【特許文献1】米国特許第4,675,189号明細書
【特許文献2】米国特許第4,677,191号明細書
【特許文献1】米国特許第5,480,656号明細書
【特許文献1】米国特許第4,728,721号明細書
【発明の概要】
【発明が解決しようとする課題】
【0004】
薬学的に活性なペプチドをインビボで長期間にわたり継続的に投与するためのさらに持続した送達処方物が必要である。
【課題を解決するための手段】
【0005】
発明の要旨
本発明は、ペプチド化合物(例えば、ペプチド、ポリペプチド、タンパク質、ペプチド擬似物(peptidomimetic)など)、好ましくは薬学的に活性なペプチド化合物、および複合体の投与の際にインビボでペプチド化合物の持続した送達を行い得るキャリア巨大分子からなる、安定な水不溶性複合体を含む薬学的組成物を提供する。従って、本発明の複合体は、長期間(例えば、1ヶ月)にわたって被験体への薬学的に活性なペプチド化合物の継続した送達を可能にし得る。さらに、ペプチド化合物とキャリア巨大分子との、強固で安定な複合体での会合は、処方物へのペプチド化合物の高濃度での添加(load)を可能にする。
【0006】
本発明の複合体は、実質的に水不溶性複合体が形成されるような条件下(例えば、ペプチド化合物およびキャリア巨大分子の水性溶液が、複合体が沈殿するまで混合される)で、ペプチド化合物とキャリア巨大分子とを組み合わせることにより形成される。複合体は、固体形態(例えば、ペースト、顆粒、粉末、または凍結乾燥体)であっても良く、または粉末形態の複合体が安定な液体懸濁物または半固体分散体を形成するのに十分に細かく粉末化され得る。
【0007】
好適な実施態様において、水不溶性複合体のペプチド化合物はLHRHアナログ、より好ましくはLHRHアンタゴニストであり、そしてキャリア巨大分子は、アニオン性ポリマー、好ましくはカルボキシメチルセルロースである。本発明の複合体は、インビボでの投与の前に、例えばγ線照射または電子線照射による滅菌化に適している。
【0008】
本発明のLHRHアナログ含有化合物を被験体へ投与することによる、LHRHアナログで処置可能な状態における被験体を処置するための方法もまた、提供される。好適な実施態様において、本発明の処置方法は、前立腺癌の処置に使用される。
【図面の簡単な説明】
【0009】
【図1】図1は、PPI-149およびカルボキシメチルセルロースの複合体の筋肉内注射に続く時間経過に伴う、ラット(左のグラフ)およびイヌ(右のグラフ)における、血漿テストステロンレベル(ng/ml;白四角)および血漿PPI-149レベル(ng/ml;黒四角)を描いたグラフを示す。
【図2】図2は、0日目におけるLHRHアンタゴニストPPI-149およびカフレボキシメチノレセルロースの複合体の筋肉内注射、および30日目におけるLHRHアゴニストLupronTMの注射に続く時間経過に伴う、ラットにおける血漿テストステロンレベル(ng/ml;白四角)および血漿PPI-149レベル(ng/ml;黒四角)を描いたグラフであり、PPI-149前処置によりLupronTM誘導テストステロンサージが抑制されることを示している。
【図3−1】図3A〜3Cは、PPI-149-CMC(図3A)、PPI-258-CMC(図3B)、またはCetrorelixTM-CMC(図3C)の筋肉内注射に続く時間経過に伴う、雄性Sprague-Dawleyラットにおける血漿テストステロンレベル(ng/ml)を描いた一連のグラフである。
【図3−2】図3A〜3Cは、PPI-149-CMC(図3A)、PPI-258-CMC(図3B)、またはCetrorelixTM-CMC(図3C)の筋肉内注射に続く時間経過に伴う、雄性Sprague-Dawleyラットにおける血漿テストステロンレベル(ng/ml)を描いた一連のグラフである。
【図4】図4は、指示された用量で28日間隔におけるPPI-149-CMCの皮下注射に続く時間経過に伴う、イヌにおける血漿テストステロンレベル(ng/ml;白四角)および血漿PPI-149レベル(ng/ml;黒四角)を描いたグラフであり、血漿テストステロンレベルが長期にわたり抑制されることを示している。
【図5】図5は、指示された用量で28日間隔におけるPPI-149-CMCの筋肉内注射に続く時間経過に伴う、イヌにおける血漿テストステロンレベル(ng/ml;白四角)および血漿PPI-149レベル(ng/ml;黒四角)を描いたグラフであり、血漿テストステロンレベルが長期にわたり抑制されることを示している。
【発明を実施するための形態】
【0010】
発明の詳細な説明
本発明は、ペプチド化合物(例えば、ペプチド、ポリペプチド、タンパク質、ペプチド擬似物など)およびキャリア巨大分子からなる、安定な水不溶性複合体を含む薬学的組成物、そのような組成物の作製法、およびそのような組成物の使用法に関する。本発明の薬学的組成物の利点は、薬学的に活性なペプチド化合物を、全身的または局所的のいずれかで、長期間(例えば、数週間、1ヶ月、または数ヶ月)にわたり送達し得ること、およびペプチド化合物を複合体へ高濃度で添加し得ること、を含む。
【0011】
本発明がより容易に理解され得るために、特定の用語をあらかじめ定義する。
【0012】
本明細書中で使用されるように、用語「ペプチド化合物」は、少なくとも部分的に、アミド結合(すなわちペプチド結合)により連結されたアミノ酸残基からなる化合物を示すことを意図する。用語「ペプチド化合物」は、ペプチド、ポリペプチド、およびタンパク質を包含することを意図する。代表的には、ペプチドは、約100よりも少ないアミノ酸からなり、より代表的には、約50よりも少ないアミノ酸残基からなり、さらにより代表的には、約25より少ないアミノ酸残基からなる。用語「ペプチド化合物」は、さらにペプチドアナログ、ペプチド誘導体、および天然に生じるアミノ酸からなるペプチドの化学構造を模倣したペプチド擬似物を包含することを意図する。ペプチドアナログの例は、1つ以上の非天然アミノ酸を含むペプチドを含む。ペプチド誘導体の例は、アミノ酸側鎖、ペプチド骨格、またはアミノ-もしくはカルボキシ-末端が誘導体化されているペプチドを含む(例えば、メチル化アミド連結したペプチド化合物)。ペプチド擬似物の例は、ペプチド骨格が1つ以上のベンゾジアゼピン分子で置換されているペプチド化合物(例えば、James、G.L.ら、(1993)Science 260:1937〜1942を参照のこと)、全てのL-アミノ酸が対応するD-アミノ酸で置換された「反転」(inverso)ペプチド、アミノ酸配列が逆転(「逆」)しており、かつ全てのL-アミノ酸が対応するD-アミノ酸で置換(「反転」)された「逆-反転」(retro-inverso)ペプチド(Sistoによる米国特許第4,522,752号を参照のこと)、およびペプチド骨格(すなわちアミド結合)擬似物のような他のアイソスター(isostere)(アミド窒素、α-炭素、アミドカルボニルの修飾、アミド結合の全置換、延長、欠失(deletion)、または骨格連結を含む)を含む。ψ[CH2S]、ψ[CH2NH]、ψ[CSNH2]、ψ[NHCO]、ψ[COCH2]、およびψ[(E)または(Z)CH=CH]を含む、いくつかのペプチド骨格改変が公知である。上記で使用される名称において、ψはアミド結合が存在しないことを意味する。アミド基と置換される構造は、角括弧(bracket)内に指定されている。他の可能な改変は、N-アルキル(またはアリール)置換(ψ[CONR])、骨格連結によるラクタムおよび他の環式構造の構築、並びにC-末端ヒドロキシメチル誘導体、O-修飾誘導体、およびN-末端修飾誘導体(アルキルアミドおよびヒドラジンのような置換アミドを含む)を含む他の誘導体を含む。
【0013】
本明細書中で使用されるように、用語「薬学的に活性なペプチド化合物」は、その存在する形態またはインビボでのプロセシングに際してのいずれかにおいて、薬理学的活性を示すペプチド化合物を示すことを意図する(すなわち、薬学的に活性なペプチド化合物は、構造的薬理学的活性であるペプチド化合物、および薬理学的活性を示すために投与に続いてインビボで何らかの方法で代謝もしくはプロセシングされなければならない「プロドラッグ」形態であるペプチド化合物を含む)。
【0014】
本明細書中で使用されるように、用語「多価カチオン性ペプチド化合物」および「多価アニオン性ペプチド化合物」は、それぞれ多重の正電荷または負電荷を含むペプチド化合物を示すことを意図する。「二価カチオン性」または「二価アニオン性」ペプチド化合物は、それぞれ2個の正電荷または負電荷を含むペプチド化合物を示すことを意図する。「三価カチオン性」または「三価アニオン性」ペプチド化合物は、それぞれ3個の正電荷または負電荷を含むペプチド化合物を示すことを意図する。
【0015】
本明細書中で使用されるように、用語「LHRHアナログ」は、黄体形成ホルモン放出ホルモンの構造を模倣するペプチド化合物を包含することを意図する。LHRHアナログは、LHRHアゴニストまたはLHRHアンタゴニストであり得る。
【0016】
本明細書中で使用されるように、「LHRHアゴニスト」は、黄体形成ホルモンの放出を刺激するように黄体形成ホルモン放出ホルモンレセプター(LHRH-R)を刺激する化合物を示すことを意図するか、または「LHRHアンタゴニスト」は、黄体形成ホルモンの放出を阻害するようにLHRH-Rを阻害する化合物を示す。LHRHアゴニストの例は、ロイプロリド(商標名:Lupronc(登録商標);Abbott/TAP)、ゴセレリン(商標名:Zoladex(登録商標);Zeneca)、ブセレリン(Hoechst)、トリプトレリン(Decapeptyl、D-Trp-6-LHRH、およびDebiopharm(登録商標)としても公知;Ipsen/Beaufour)、ナファレリン(商標名”Synarel(登録商標);Syntex)、ルトレリン(Wyeth)、シストレリン(cystorelin)(Hoechst)、ゴナドレリン(Ayerst)、およびヒストレリン(histrerlin)(Ortho)を含む。
【0017】
本明細書中で使用されるように、用語「LHRHアンタゴニスト」は、黄体形成ホルモンの放出を阻害するような黄体形成ホルモン放出ホルモンレセプターを阻害する化合物を示すことを意図する。LHRHアンタゴニストの例は、Antide、Cetrorelix、以下に記載される化合物:Folkersらの米国特許第5,470,947号;FolkersらによるPCT出願第WO89/01944:Havivの米国特許第5,413,990号;Havivの米国特許第5,300,492号;Koerberらの米国特許第5,371,070号;Hoegerらの米国特許第5,296,468号;Janakyらの米国特許第5,171,835号;Coyらの米国特許第5,003,011号;Coyの米国特許第4,431,635号;Deらの米国特許第4,992,421号;Roeskeの米国特許第4,851,385号;Nestor、Jr.らの米国特許第4,801,577号;およびRoeskeらの米国特許第4,689,396号、ならびに以下に開示される化合物:米国特許出願番号第08/480,494号、表題「LHRHアンタゴニストペプチド」、およびその対応するPCT出願(PCT出願番号第PCT/US96/09852)、同じく表題「LHRHアンタゴニストペプチド」を含む。これらの内容はいずれも全て特に本明細書中で参考として援用される。特に好適なLHRHアンタゴニストは、以下の構造を含む:Ac-D-Nal1、4-Cl-D-Phe2、D-Pal3、N-Me-Tyr5、D-Asn6、Lys(iPr)8、本明細書中でPPI-149として示されるD-Ala10-LHRH。
【0018】
本明細書中で使用されるように、用語「キャリア巨大分子」は、ペプチド化合物と複合化して水不溶性複合体を形成し得る巨大分子を示すことを意図する。ペプチド化合物と複合化する前は、代表的にはキャリア巨大分子は水溶性である。好ましくは、巨大分子は、少なくとも5kDa、より好ましくは10kDaの分子量を有する。用語「アニオン性キャリア巨大分子」は、アニオン性ポリマーのような負電荷を帯びた高分子量の分子を含むことを意図する。用語「カチオン性キャリア巨大分子」は、カチオン性ポリマーのような正電荷を帯びた高分子量の分子を含むことを意図する。
【0019】
本明細書中で使用されるように、用語「水不溶性複合体」は、本明細書中に記載される手順に従ってペプチド化合物およびキャリア巨大分子の適切な化合に際して形成される、物理的および科学的に安定な複合体を示すことを意図する。代表的には、この複合体は、ペプチド化合物およびキャリア巨大分子の水性調製物の化合において生成する沈殿の形態をとる。機構によって制限されることを意図しないが、本発明の好適な水不溶性複合体の形成は、ペプチド化合物がカチオン性であり、そしてキャリア分子がアニオン性であるか、またはその逆である状況に置けるイオン性相互作用を含む(すなわち、少なくとも部分的にそれにより媒介される)と考えられる。さらに、またはあるいは、本発明の水不溶性複合体の形成は、疎水性相互作用を含み得る(すなわち、少なくとも部分的にそれにより媒介される)。なおさらに、本発明の水不溶性複合体の形成は、共有相互作用を含み得る(すなわち、少なくとも部分的にそれにより媒介される)。「水不溶性」であるという複合体の記述は、それが水性溶液から沈殿することによって示されるように、複合体が実質的にまたは容易に水に溶解しないことを示すことを意図する。しかしながら、本発明の「水不溶性」複合体がインビトロでまたはインビボでの水性生理学的環境においてのいずれかで限られた水溶性(すなわち、部分的溶解性)を示し得ることが理解されるべきである。
【0020】
本明細書中で使用されるように、用語「持続した送達」は、投与に続く一定の期間、好ましくは少なくとも数日、1週間、または数週間に渡りインビボでの薬学的な薬剤の継続した送達を示すことを意図する。薬剤の持続した送達は、例えば、時間経過に伴う薬剤の継続した治療的効果により実証され得る(例えば、LHRHアナログについては、アナログの持続した送達は時間経過に伴うテストステロン合成の継続した抑制により実証され得る)。あるいは、薬剤の持続した送達は、時間経過に伴うインビボでの薬剤の存在の検出により実証され得る。
【0021】
本明細書中で使用されるように、用語「被験体」は、温血動物、好ましくは哺乳動物、より好ましくは霊長類、最も好ましくはヒトを含むことを意図する。
【0022】
本明細書中で使用されるように、用語「被験体への投与」は、被験体の所望の部位へ組成物を送達するための任意の適切な経路による、組成物(例えば薬学的処方物)の被験体への投薬、送達、または塗布を示すことを意図し、非経口もしくは経口経路、筋肉内注射、皮下/皮内注射、静脈注射、口内投与、直腸、結腸、膣、鼻内による経皮送達および投与、もしくは気道経路のいずれかによる送達を含む。
【0023】
本明細書中で使用されるように、用語「LHRHアナログで処置可能な状態」は、LHRHアゴニストまたはLHRHアンタゴニストの投与が所望の効果(例えば、治療的に有益な効果)をあげる疾患、障害、および他の状熊を含むことを意図する。LHRHアナログで処置可能な状態の例は、ホルモン依存性癌(前立腺癌、乳癌、卵巣癌、子宮癌、および精巣癌を含む)、良性前立腺肥大、性的早熟、子宮内膜症、子宮筋肺、不妊症(インビトロ受精を介して)および受精(すなわち、避妊的使用)を含む。
【0024】
本発明の1つの局面は、薬学的に活性なペプチド化合物とキャリア巨大分子との水不溶性複合体を含む薬学的組成物に関する。好適な実施態様において、水不溶性複合体の形成は、少なくとも部分的には薬学的に活性なペプチドとキャリア巨大分子との間のイオン性相互作用により媒介される。これらの実施態様において、薬学的に活性なペプチド化合物がカチオン性であり、キャリア巨大分子がアニオン性であるか、薬学的に活性なペプチド化合物がアニオン性であり、キャリア巨大分子がカチオン性であるかのいずれかである。別の実施態様において、水不溶性複合体の形成は、少なくとも部分的には薬学的に活性なペプチド化合物とキャリア巨大分子との間の疎水性相互作用により媒介される。好適な実施態様において、複合体に使用されるペプチド化合物は、二価または三価のカチオン性ペプチド化合物のような多価カチオン性ペプチド化合物であり、そしてキャリア巨大分子は、アニオン性巨大分子である。
【0025】
本発明の薬学的組成物は、被験体への組成物の投与後、インビボで被験体へのペプチド化合物の持続した送達を可能にし、ここで持続した送達の持続時間は、複合体を形成するために使用したペプチド化合物およびキャリア巨大分子の濃度に依存して変化し得る。例えば、1つの実施態様において、水不溶性複合体の1回の用量は、薬学的組成物が被験体に投与された後、少なくとも1週間被験体へペプチド化合物の持続した送達を提供する。別の実施態様において、水不溶性複合体の1回の用量は、薬学的組成物が被験体に投与された後、少なくとも2週間被験体へペプチド化合物の持続した送達を提供する。さらに別の1つの実施態様において、水不溶性複合体の1回の用量は、薬学的組成物が被験体に投与された後、少なくとも3週間被験体へペプチド化合物の持続した送達を提供する。さらに別の実施態様において、水不溶性複合体の1回の用量は、薬学的組成物が被験体に投与された後、少なくとも4週間被験体へペプチド化合物の持続した送達を提供する。さらに長期の、または短期の持続時間での持続した送達を提供する処方物(例えば、1日、1〜7日、1ヶ月、2ヶ月、3ヶ月などの継続した送達を提供する処方物)もまた、本発明に包含される。数ヶ月にわたるペプチド化合物の継続した送達は、例えば、毎月繰り返して服用することにより達成され得、各服用がほぼ1ヶ月の間ペプチド化合物の持続した送達を提供する(例えば、実施例14を参照のこと)。
【0026】
ペプチド化合物が、ペプチド化合物とキャリア巨大分子との結合によりキャリア巨大分子との水不溶性非共有結合複合体を形成し得る限り、任意の大きさのペプチド化合物が複合体への使用に適するものであり得る。しかしながら、特定の好適な実施態様において、ペプチド化合物は、約5〜約20アミノ酸長のペプチドであるか、約8〜約15アミノ酸長のペプチドであるか、または約8〜約12アミノ酸長のペプチドである。種々の薬学的に活性なペプチドが、処方物に使用され得、その非制限的な例は、LHRHアナログ(さらに以下で議論される)、ブラジキニンアナログ、副甲状腺ホルモン、副腎皮質刺激ホルモン(ACTH)、カルシトニン、およびバソプレッシンアナログ(例えば、1-デアミノ-8-D-アルギニンバソプレッシン(DDAVP))を含む。
【0027】
種々のキャリア巨大分子が本発明の水不溶性複合体の形成に適したものであり得るが、好ましい巨大分子はポリマーであり、好ましくは水溶性ポリマーである。好適な実施態様において、キャリア巨大分子は、アニオン性ポリアルコール誘導体のようなアニオン性ポリマー、またはそのフラグメント、およびその塩(例えば、ナトリウム塩)である。ポリアルコールが誘導体化され得るアニオン性部分は、例えば、カルボキシレート、ホスフェート、またはスルフェート基を含む。特に好適なアニオン性ポリマーは、アニオン性ポリサッカリド誘導体、またはそのフラグメント、およびその塩(例えば、ナトリウム塩)である。キャリア巨大分子は、単一の分子種(例えば、単一の型のポリマー)または2つ以上の異なる分子種(例えば、2つの型のポリマーの混合物)を含み得る。特定のアニオン性ポリマーの例は、カルボキシメチルセルロース、アルギン、アルギネート、アニオン性アセテートポリマー、アニオン性アクリル酸ポリマー、キサンタンガム、グリコール酸ナトリウムスターチ、およびそのフラグメント、誘導体、および薬学的に受容可能な塩、ならびにアニオン性カラギーナン誘導体、アニオン性ポリガラクツロン酸誘導体、ならびに硫酸化およびスルホン化ポリスチレン誘導体を含む。好適なアニオン性ポリマーは、カルボキシメチルセルロースナトリウム塩である。カチオン性ポリマーの例は、ポリ-L-リジンおよび他の塩基性アミノ酸ポリマーを含む。
【0028】
本発明の特定の好適な実施態様において、水不溶性複合体のペプチド化合物は、LHRHアナログであり、例えばLHRHアゴニスト、または、より好ましくはLHRHアンタゴニストである。代表的には、そのようなLHRHアナログは、10アミノ酸長である。好ましいLHRHアンタゴニストは、ペプチド化合物を含むLHRHアンタゴニストを含み、ここで天然の哺乳動物のLHRHの6位におけるアミノ酸に対応するペプチド化合物の残基はD-アスパラギン(D-Asn)構造を含む。本明細書中で使用されるように、用語「D-アスパラギン構造」は、D-Asn、ならびにD-Asnの機能的活性を保持するそのアナログ、誘導体、および擬似物を含むことを意図する。他の好ましいLHRHアンタゴニストは、以下の構造を含むペプチド化合物を含むLHRHアンタゴニストまたはその薬学的に受容可能な塩を含む:A-B-C-D-E-F-G-H-I-J
ここで
Aは、ピロ-Glu、Ac-D-Nal、Ac-D-QalAc-Sar、またはAc-D-Palであり
Bは、His、または4-Cl-D-Pheであり
Cは、Trp、D-Pal、D-Nal、L-Nal、D-Pal(N-0)、またはD-Trpであり
Dは、Serであり
Eは、N-Me-Ala、Tyr、N-Me-Tyr、Ser、Lys(iPr)、4-Cl-Phe、His、Asn、MeLAla、Arg、またはIleであり;
Fは、
【0029】
【化1】
ここで
RおよびXは、独立して、Hまたはアルキルであり;そして
Lは、小極性部分を含み;
Gは、Leu、またはTrpであり;
Hは、Lys(iPr)、Gln、Met、またはArgであり
Iは、Proであり;そして
Jは、Gly-NH2、またはD-Ala-NH2である。
【0030】
用語「小極性部分」は、小さな立体的かさ高さを有し、そして比較的極性である部分を示す。極性は、Pスケールによる親水性度として測定する。1-オクタノールと水との間の分配係数、Pが、化合物の親水性度の測定に対する標準として使用されている。親水性度は、logP、分配係数の対数として表され得る(Hanschら、Nature 194:178(1962);Fujitaら、J.Am.Chem.Soc.86:5175(1964))。多くの分子に対する親水性度の標準表、および多くの官能基に対する親油性度(疎水性度)置換基定数(πで表記)が、編集されている(例えば、HanschおよびLeo、「化学および生物学における相関分析に対する置換基定数」Wiley、New York.New York(1979)を参照のこと)。膨大な範囲の候補親水性部分の親水性度が、これらの表の助けによりかなり正確に予測され得る。例えば、ナフタレンのlogP(オクタノール/水)の実測値は、3.45である。-OHに対する置換基定数Pは、-0.67である。従って、β-ナフトールに対するlogPの予測値は、3.45+(-0.67)=2.78である。この値は、β-ナフトールに対するlogPの実測値、2.84と良く一致する。本明細書中で使用されるように、用語「小極性部分」は、-1と+2の間のlogP、およびTrpの立体的かさ高さよりも小さい立体的かさ高さを有する部分を示す。
【0031】
特定の実施態様において、Lは小極性部分を含むが、但しFはD-Cit、D-Hci、またはD-CitあるいはD-Hciの低級アルキル誘導体ではない。好ましくは、Fは、D-Asn、D-Gln、およびD-Thrからなる群より選択される。より好ましくは、FはD-Asnである。好ましくは、Eは、チロシン(Tyr)またはN-メチル-チロシン(N-Me-Tyr)である。特定の好適な実施熊様において、LHRHアンタゴニストは以下の構造式を有する:Ac-D-Nal1、4-Cl-D-Phe2、D-Pal3、N-Me-Tyr5、D-Asn6、Lys(iPr)8、D-Ala10-LHRH(本明細書中でPPI-149として示される)。本発明の特定の好適な複合体は、PPI-149およびカルボキシメチルセルロースを含む。
【0032】
水不溶性複合体に加えて、本発明の薬学的処方物はさらなる薬学的に受容可能なキャリアおよび/または賦形剤を含み得る。本明細書中で使用されるように、「薬学的に受容可能なキャリア」は、生理学的に適合性である、任意のそして全ての溶媒、分散媒体、コーティング、抗菌剤および抗真菌剤、等張液および吸収遅延剤などを含む。好ましくは、キャリアは静脈内、筋肉内、皮下または非経口投与(例えば、注射による)に適している。賦形剤は、薬学的に受容可能な安定剤および崩壊剤を含む。
【0033】
キャリア巨大分子と複合体化したLHRHアナログの薬学的処方物に加えて、本発明はさらに、そのような複合体およびそのような複合体を含有するシリンジを含むパッケージ化処方物を包含する。例えば、本発明はLHRHアナログ(好ましくはPPI-149)およびキャリア巨大分子(好ましくは、カルボキシメチルセルロース)の水不溶性複合体を含み、LHRHアナログで処置可能な状態の被験体を処置するための水不溶性複合体の使用説明書と合わせてパッケージ化された、LHRHアナログで処置可能な状態の被験体を処置するためのパッケージ化処方物を提供する。別の実施態様において、本発明は管腔を有するシリンジを提供し、ここでLHRHアナログ(好ましくはPPI-149)およびキャリア巨大分子(好ましくは、カルボキシメチルセルロース)の水不溶性複合体は、管腔中に含まれる。
【0034】
本発明の複合体は、ペプチド化合物およびキャリア巨大分子の水不溶性複合体が形成するような条件下で、ペプチド化合物およびキャリア巨大分子を結合させることにより調製される。従って、本発明の別の局面は、薬学的処方物の調製法に関する。1つの実施態様において、この方法は以下を含む:
ペプチド化合物およびキャリア巨大分子を提供する工程;
ペプチド化合物およびキャリア巨大分子の水不溶性複合体が形成するような条件下で、ペプチド化合物およびキャリア巨大分子を結合させる工程:および
水不溶性複合体を含む薬学的処方物を調製する工程。
【0035】
例えば、ペプチド化合物の溶液とキャリア巨大分子の溶液とを、ペプチド化合物およびキャリア巨大分子の水不溶性複合体が溶液から沈殿してくるまで合わせる。特定の実施態様において、ペプチド化合物およびキャリア巨大分子の溶液は水性溶液である。あるいは、ペプチド化合物またはキャリア分子(または両方)が2つの混合前に実質的に水溶性でない場合、ペプチド化合物および/またはキャリア巨大分子は、複合体の2つの成分を合わせる前に、アルコール(例えばエタノール)のような水混和性溶媒に溶解され得る。水不溶性複合体の調製方法の別の実施態様において、ペプチド化合物の溶液とキャリア巨大分子の溶液とを、ペプチド化合物およびキャリア巨大分子の水不溶性複合体が溶液から沈殿してくるまで合わせ、そして加熱する。水不溶性複合体を得るために必要なペプチド化合物およびキャリア巨大分子の量は、使用される特定のペプチド化合物およびキャリア巨大分子、使用される特定の溶媒、および/または複合体を得るために使用する手順に依存して変化し得る。しかしながら、代表的には、ペプチド化合物はモルを基準にして、キャリア巨大分子に対して過剰である。しばしば、ペプチド化合物はまた、実施例において示されるように、重量/重量を基準にして過剰である。特定の実施態様において、キャリア巨大分子(好ましくは、カルボキシメチルセルロースナトリウム)、およびペプチド化合物(好ましくは、PPI-149)は、0.2:1(w/w)のキャリア巨大分子:ペプチド化合物の比で合わされる。種々の他の実施態様において、キャリア巨大分子:ペプチド化合物の比(w/w)は、例えば、0.5:1、0.4:1、0.3:1、0.25:1、0.15:1または0.1:1であり得る。本発明の水不溶性複合体の調製のための条件および手順の非制限的な例が、実施例1〜5および8〜9でさらに記載される。
【0036】
ペプチド化合物/巨大分子複合体が、いったん溶液から沈殿すると、沈殿物は、濾過(例えば、0.45ミクロンナイロン膜を通して)、遠心分離などのような当該分野で公知の手段により溶液から取り出され得る。次いで、回収されたペーストが乾燥され得(例えば、真空でまたは70℃のオーブンで)、そして固体が当該分野で公知の手段により粉砕または摩砕されて粉末にされ得る(例えば、ハンマーあるいはゴア粉砕、または乳鉢および乳棒での摩砕)。粉砕または摩砕に続いて、粉末は網(好ましくは90ミクロンの網)でふるいにかけられ、均一な粒子分布が得られ得る。さらに、回収されたペーストは、冷凍され、凍結乾燥して乾燥され得る。複合体の粉末形態は、キャリア溶液に分散されて注射に適した液体懸濁物または半固体分散体を形成し得る。従って、種々の実施態様において、本発明の薬学的処方物は、乾燥固体、液体懸濁物、または半固体分散体である。液体懸濁物における使用に適した液体キャリアの例は、生理食塩水溶液、グリセリン溶液、およびレシチン溶液を含む。
【0037】
別の実施態様において、本発明の薬学的処方物は滅菌処方物である。例えば、水不溶性複合体の形成に続いて、この複合体は、最適にはγ線照射または電子線滅菌により滅菌され得る。従って、上記で記載される薬学的処方物を調製するための本発明の方法はさらに、水不溶性複合体のγ線照射または電子線照射による滅菌を含み得る。好ましくは、処方物は少なくとも15KGyのγ線線量を使用するγ線照射により滅菌される。他の実施熊様において、処方物は少なくとも19KGyまたは少なくとも24KGyのγ線線量を使用するγ線照射により滅菌される。実施例11で実証されるように、本発明の処方物はγ線照射に際して十分に安定なままである。
【0038】
あるいは、滅菌薬学的処方物を調製するために、水不溶性複合体は従来の滅菌技術を使用して単離され得る(例えば、滅菌された出発物質の使用、および無菌での製造プロセスの実行)。従って、上記で記載される薬学的処方物を調製するための方法の別の実施態様において、水不溶性複合体は無菌の手順を使用して形成される。
【0039】
本発明の水不溶性複合体の形成方法はさらに、実施例1〜5および8〜9に記載される。本発明の方法により調製される、粉末、液体懸濁物、半固体分散体、乾燥固体(例えば、凍結乾燥固体)、およびそれらの滅菌形態(例えば、γ線照射により)を含む、薬学的処方物がまた、本発明に包含される。
【0040】
本発明のさらに別の局面は、水不溶性複合体に含まれる薬学的に活性なペプチド化合物により処置可能な状態を患っている被験体を処置するための、本発明の薬学的処方物の使用法に関する。従って、好適な実施態様において、本発明は、LHRHアナログとキャリア巨大分子との水不溶性複合体を含む薬学的処方物を被験体に投与する工程を含む、LHRHアナログで処置可能な状態の被験体を処置するための方法を提供する。
【0041】
薬学的処方物が、所望の治療的結果を得るために適した任意の経路により被験体に投与され得るが、好ましい投与経路は非経口経路、特に筋肉内(i.m.)注射および皮下/皮内(s.c./i.d.)注射である。あるいは、処方物は経口で被験体に投与され得る。他の適した非経口経路は、静脈内注射、口腔投与、直腸、膣、鼻内または気道経路による経皮送達および投与を含む。i.m.またはs.c./i.d.経路により数週間から数ヶ月の持続した送達を提供する処方物が、代替的な経路により投与される場合、他の生理学的機構による薬剤のクリアランスにより同等の時間の間の薬剤の持続した送達が起こらないかもしれない(すなわち、投薬形態が送達部位から排泄され(clear)得るため、長期治療的効果がi.m.またはs.c./i.d.注射で観察されるものほど長い期間は観察されない)ことを注意するべきである。
【0042】
薬学的処方物は、治療的に有効な量のLHRHアナログを含有する。「治療的に有効な量」は、投薬において、そして必要な期間にわたり、所望の結果を得るために有効な量を示す。LHRHアナログの治療的に有効な量は、個体の疾患状態、年齢、および体重、ならびにLHRHアナログの個体において所望の応答を導き出す能力(単独または1つ以上の他の薬物との組み合わせにおいて)のような因子によって変化し得る。投薬レジメンは、最適の治療応答を提供するために調整され得る。治療的に有効な量はまた、治療的に有益な効果がアンタゴニストのどのような毒性または有害な影響にも勝る(outweighed)量である。LHRHアナログの治療的に有効な量に対する非制限的な範囲は、0.01〜10mg/kgである。28日間の血漿テストステロンレベルの持続した減少のためのLHRHアナログPPI-149の好適な投薬量は、ほぼ1mL以下の液体懸濁物の体積中、ほぼ0.1〜10mg/kg、より好ましくは0.3〜1.2mg/kg(遊離のペプチドとして表される)である。投薬量の値が緩和されるべき状熊の重篤度によって変化し得ることが注意されるべきである。任意の特定の被験体に対して、特定の投薬レジメンが、個々の必要性によっておよび組成物の投与または投与の監督を行う個人の専門的判断によって、経時的に調整されるべきであること、および本明細書中で記述される投薬範囲が例示に過ぎず、そして請求の組成物の範囲または実施を制限することを意図しないことが、さらに理解されるべきである。
【0043】
本発明の処置方法は、LHRHアナログの投与が所望の臨床的効果を有する種々の状態、疾患、および障害の処置に適用され得る。疾患および障害の例は、ホルモン依存性癌(例えば、前立腺癌、乳癌、卵巣癌、子宮癌、および精巣癌)、良性前立腺肥大、性的早熟、子宮内膜症および子宮筋腫を含む。従って、本発明は、本発明の薬学的処方物の投与によりこれらの疾患および障害を処置する方法を提供する。さらに、LHRHアナログは排卵を変更するために使用され得る。従って、本発明の方法はまた、インビトロでの妊娠および避妊目的に使用され得る。
【0044】
特定の好適な実施態様において、この方法は、前立腺癌の処置のために使用され得、処方物に使用されるLHRHアナログはLHRHアンタゴニスト、最も好ましくはPPI-149であり、そしてこの方法は筋肉内または皮下投与による投与後少なくとも4週間インビボでLHRHアナログの持続した送達を可能にする。本発明により処方されたLHRHアナログ、好ましくはPPI-149は、前立腺癌を患っている被験体にLHRHアナログを投与することにより、前立腺癌細胞の成長を阻害するために使用され得る。さらに、本発明により処方されたLHRHアンタゴニスト、好ましくはPPI-149は、LHRHアゴニスト治療を開始する前に前立腺癌を患っている被験体にLHRHアンタゴニスト、好ましくはPPI-149を前投与することにより、LHRHアゴニストの使用に伴うテストステロンの急増を阻害するために使用され得る。本発明の処方物が適用され得る、LHRHアゴニスト誘導テストステロン急増を阻害するための方法、およびLHRHアンタゴニストを使用する前立腺癌を処置するための他の方法は、米国特許出願番号第08/573,109号、表題「LHRHアンタゴニストを使用する前立腺を処置するための方法」(1995年12月15日出願)およびその一部継続特許出願番号第08/755,593号、同じく表題「LHRHアンタゴニストを使用する前立腺を処置するための方法」(1996年11月25日出願)においてさらに記載され、この両者の内容が公開PCT出願WO97/22357号に援用される。米国出願および公開PCT出願の全内容が、参考として本明細書中に特に援用される。
【0045】
薬学的に活性なペプチド化合物のキャリア巨大分子との複合体化のための特定のプロセスが、以下の実施例1〜5および8〜9に記述される。LHRHアンタゴニスト含有複合体が、インビボで薬学的に活性なペプチドの持続した送達を可能にし得ること(実施例6)およびLHRH-アゴニスト誘導テストステロン急増を阻害し得ること(実施例7)を実証する試験結果もまた、記載される。本発明をさらに例示する以下の実施例は、制限として解釈されるべきではない。本出願全体にわたり引用される全ての参考文献、特許、および公開特許出願の内容は、参考として本明細書中に援用される。
【実施例】
【0046】
実施例1:
LHRHアンタゴニストPPI-149 100ml溶液を、PPI-149 6.25mg/mlを水に溶解して調製した。等量の米国薬局方カルボキシメチルセルロースナトリウム(CMC)(低粘度グレード(grade)、Hercules Chemical Co.)サンプル(最小100ml)を、0.125%w/vで調製し、溶解するまで混合した。各等量のPPI-149およびCMC溶液を混合し(0.2:1(w/w)のCMC:ペプチド比を与える)、そして固体物質を得た。その固体物質を一晩撹拌し、その後0.45ミクロンのナイロンフィルターで濾過することで回収した。その溶液濾液のHPLC評価では、少なくとも95%のPPI-149化合物が固体複合体に変換したことを示した。溶液から取り除いた。回収した白色ペーストを水で2回すすぎ、そしてその後バイアルへ移し、減圧下で乾燥した。72時間の乾燥後、白色粉末を633mg得た。固体物質をその後、乳鉢および乳棒で粉末化した。元素分析は、複合体中に57%のペプチドを示した。
【0047】
実施例2:
PPI-149 25mgを1mlの水に溶解した。これに0.5%カルボキシメチルセルロース溶液を1ml加えた。混合により、混合物が絹状の白色固体を形成した。混合物を加熱して5分間環流し、そして羊毛状白色沈殿を形成した。遠心分離/デカンテーションでこの物質を分離した。その固体を水中で再懸濁し、遠心分離を繰り返して回収した。その溶液濾液のHPLC評価は、少なくとも90%のPPI-149化合物が固体複合体へ変換したことを示した。その白色沈殿を減圧下乾燥し、そして固体物質を乳鉢および乳棒で粉末化した。元素分析は、複合体中に77%のペプチドを示した。
【0048】
実施例3:
PPI-149 50mgを2mlの5%マンニトールに溶解し、そして2mlの0.5%カルボキシメチルセルロース(低粘度、米国薬局方、Spectrum Quality Chemicals)と混合した。混合物を撹拌し、そして即座に白色沈殿が生成した。懸濁液を凍結し、そして凍結乾燥により乾燥し、PPI-149持続送達(sustained delivery)複合体を得た。
【0049】
実施例4:
PPI-149 25mgを1mlの水に溶解した。これに1mlの0.5%アルギン酸ナトリウム、米国薬局方(Spectrum)を加えた。混合後、混合物は即座に白色沈殿を形成した。遠心分離/デカンテーションでこの物質を単離した。その固体を水中で再懸濁し、遠心分離を繰り返して回収した。その白色沈殿を減圧下乾燥した。元素分析を実施して、66%のペプチド含有量を得た。
【0050】
実施例5:
PPI-149 25mgを1mlの水に溶解した。pHを11.0に調整するために、アンモニアを加えた。これに1mlの0.5%アルギン酸、米国薬局方(Spectrum)を加えた。混合後、混合物は即座に白色沈殿を形成した。遠心分離/デカンテーションでこの物質を分離した。その固体を水中で再懸濁し、遠心分離を繰り返して回収した。その白色沈殿を減圧下乾燥した。元素分析を実施して、79%のペプチド含有量を得た。
【0051】
実施例6:
LHRHアンタゴニストPPI-149およびカルボキシメチルセルロースの水不溶性複合体を、前述の実施例に従って調製した。PPI-149/CMC複合体の懸濁液を調製し、1回投与量をラットおよびイヌの筋肉内に注射した。ラットへの投与量は50μg/kg/日×60日であり、そしてイヌへの投与量は40μg/kg/日×28日であった。血漿テストステロンレベル(ng/mlにおいて)を、動物におけるLHRHアンタゴニストの活性の尺度として種々の時点で決定した。図1のグラフに示した代表的な結果は、PPI-149/CMC複合体の筋肉内注射が、ラットにおいては少なくとも42日間、およびイヌにおいては少なくとも28日間、血漿テストステロンレベルの抑制の持続に導くことを実証し(図1中の白い四角で示す)、LHRHアンタゴニストの送達の持続を実証する。動物におけるPPI-149の血漿レベル(ng/mlにおいて)をまたモニターした(図1中の黒い四角で示す)。最初のPPI-149急上昇が最初の約8日間に観察され、その時以後、PPI-149は実質的に血漿中に検出不可能であった。約8日以降の血漿中のPPI-149検出が不可能であるにもかかわらず、テストステロンレベル結果は、PPI-149が、インビボにおいて実験の過程中、依然として治療上活性であったことを実証する。
【0052】
実施例7:
LHRHアンタゴニストPPI-149およびカルボキシメチルセルロースの水不溶性複合体を、前述の実施例に従って調製した。PPI-149/CMC複合体の懸濁液を調製し、1回投与量をラットの筋肉内に、0日に注射した。30日に、LHRHアゴニストLupronTM(ロイプロリド(leuprolide))をラットに注射した。血漿テストステロンレベル(ng/mlにおいて;図2中の白い四角で示す)を、動物におけるLHRHアンタゴニストの活性の尺度として種々の時点で決定した。動物におけるPPI-149の血漿レベル(ng/mlにおいて)をまたモニターした(図2中の黒い四角で示す)。図2のグラフに示した代表的な結果は、PPI-149/CMC複合体での前処置が、速やかに血漿テストステロンを去勢(castration)レベルに減少させ、そしてその上LHRHアゴニスト誘導テストステロンの急激な上昇をブロックすることを実証する。約8日を越えると血漿中にPPI-149が検出され得ないにもかかわらず、テスロステロンレベル結果は、PPI-149が、インビボにおいて実験の過程中、依然として治療上活性であったことを実証する。
【0053】
実施例8:
この実施例において、LHRHアナログPPI-258およびカルボキシメチルセルロース(CMC)との間に、不溶性複合体を生成した。PPI-258は、以下の構造を有する:アセチル-D-ナフチルアラニル-D-4-Cl-フェニルアラニル-D-ピリジルアラニル-L-セリル-L-チロシル-D-アスパラギニル-L-ロイシル-L-Ne-イソプロピル-リジル-L-プロピル-D-アラニル-アミド。PPI-258/CMCデポ剤の調製のために、174.8mg(148.6mg正味重量)のPPI-258を29.72mLの水に加え、そして材料を撹拌し、ペプチドを懸濁させそして溶解した。この撹拌溶液に、1.85mlの2%CMCナトリウム溶液(Hercules)を加えた。固体沈殿物が即座に認められた。加熱環流後、懸濁液が半透明になり、その後白色沈殿が現れる。5分間の環流後、反応物を冷却し、固体を遠心分離で分離した。固体を水ですすぎ、1晩減圧下乾燥した。乾燥粉末を乳鉢および乳棒で粉末化し、90ミクロンのステンレス綱のふるいを通してふるった。ふるった粉末(90ミクロンふるい)を回収してキャラクタリゼーションした。総収量は乾燥粉末で198.4mgであり、粉砕工程後、110.8mgのサイズ調整された粉末を得た。キャラクタリゼーションは、以下の構成の複合体組成を提供した。:ペプチドPPI-258-80%、CMC-18.8%、水-6.6%。
【0054】
実施例9:
この実施例において、LHRHアナログCetrorelixTM(SB-75としてまた公知である)およびカルボキシメチルセルロース(CMC)との間に、不溶性複合体を形成した。CetrorelixTMは、以下の構造を有する:アセチル-D-ナフチルアラニル-D-4-Cl-フェニルアラニル-D-ピリジルアラニル-L-セリル-L-チロシル-D-シトルリル-L-ロイシル-L-アルギニル-L-プロピル-D-アラニル-アミド。CetrorelixTM/CMCデポ剤の調製のために、102.8mg(87mg正味重量)のCetrorelixTMを17.4mLの水に加え、そして物質を撹拌して懸濁させ、そしてペプチドを溶解した。この撹拌溶液に、1.1mlの2%CMCナトリウム溶液(Hercules)を加えた。固まり状の白色沈殿物が即座に認められた。懸濁液を5分間加熱環流し、冷却して白色固体沈殿を得た。固体を遠心分離で単離し、水ですすぎ、1晩減圧下乾燥した。乾燥粉末を乳鉢および乳棒で粉末化し、90ミクロンのステンレス綱のふるいを通してふるった。その粉末を回収してキャラクタリゼーションした。総収量は乾燥粉末で95mgであり、粉砕工程後、60mgのサイズ調整(size)された粉末を得た。キャラクタリゼーションは、以下の構成の複合組成を提供した。:ペプチドCetrorelixTM-75%、CMC-20.7%、水-6.5%。
【0055】
実施例10:
この実施例において、前述の3実施例で述べられたように、CMCデポ剤処方物として調製された、3種の異なるLHRHアナログPPI-149、PPI-258およびCetrorelixTMの持続放出をインビボにおいて試験した。3種の異なる処方物ビヒクルを生理食塩水、グリセリン(15%グリセリン/4%デキストロース)およびレシチンでテストした。Sprague-Dawleyラット(オス25体、重量範囲300-325g)を使用し、そしてLHRHアナログの有効性を血漿テストステロンレベルの減少に基づいて決定した。
【0056】
投与の投与量および経路は以下のとおりであった:
【0057】
【表1】
ペプチドの実際の投与量は300μg/kg/日を30日間であり、これは1回200μLの筋肉内(IM)もしくは皮下(SC)注射として与え、2.7mg/ラットであった。5ラット/グループに注射するために必要な総体積は、13.5mg/mLの活性ペプチド濃度で1.3mLであった。以下に示すように、注射の体積を一定に保ち、そして粉体の重量を以下のように総ペプチド含量について調整した:
【0058】
【表2】
試験品目の1回の200μL筋肉内、もしくは皮下注射は、それぞれ左後肢の上部側面もしくは肩胛骨間の皮下に、麻酔下で0日に行った。
【0059】
血漿テストステロンレベルをテストするために、血液約0.4mLを後眼窩洞から投与後1日および3、7、14、21、28および35日に取り出した。標準的な方法でテストステロン血漿レベルを決定するために血液を処理して血漿にし、そしてドライアイス上で凍結した。
【0060】
図3A-3Cに示される代表的な結果は、オスSprague-Dawleyラットにおける血漿テストステロンレベルが少なくとも28日間、そして50日間ほど、CMCデポ剤処方物として調製されたLHRHアナログPPI-149、PPI-258およびCentrolixTMの持続放出に応答して減少し、そして低いレベルで保持されることを実証する(それぞれ、図3A、3Bおよび3Cに示す)。これらの結果は、3種の処方物全てが、インビボにおいて血漿テストステロンレベルの減少に有効であり、長時間にわたり減少した血漿テストステロンレベルを保持することを示す。
【0061】
実施例11:
この実施例においてPPI-149-CMC処方物を、殺菌の目的でガンマ線照射に曝露し、その後、照射処方物の物理的および化学的性質の両方から評価を行った。以下に述べたデータは、ガンマ線照射がPPI-149-CMCデポ剤の殺菌に実現性のある方法であることを示している。
【0062】
ペプチド安定性
各約40mgの、2個の分離されたPPI-149-CMCロットを多数のType Iのガラスバイアルに別々に詰め(空気頭部空隙下)、ゴムストッパーおよびアルミニウムシールでシールした。バイアルをその後種々のみかけの線量のガンマ線照射にさらした。2つのロットのそれぞれについて、各ガンマ照射曝露の各レベルにおいて、2個のバイアルを、ペプチド純度(%で表す)について分析した。その結果は、γ線照射線量24KGy以下において、PPI-149-CMCが一貫してペプチド純度において2%未満の減少を示した(HPLC不純物プロフィールで決定した)ことを示した。より高いガンマ曝露線量の利用の第2の研究を、追加的なPPI-149-CMCの実験室ロットで実施した。PPI-149-CMCは、高いガンマ線量に曝露した場合、著しく良好な化学的安定性を実証した。
【0063】
次の予備処方の研究は、PPI-149-CMCγ線照射後に得た分解プロフィールと、PPI-149注射可能溶液(1mg/mL)のオートクレーブ後に得た分解プロフィールとを比較するために実行した。2個のサンプルを調製した:a)19KGyのγ線照射に曝露したPPI-149-CMC;b)オートクレーブ(121℃/20分)に供されたPPI-149溶液(1mg/mL)。2個のサンプルのHPLCクロマトグラムは、2個のサンプルについての分解プロフィールが定性的に同様でありそうであることを実証した(同様の主要ピークの相対保持時間が与えられた)。
【0064】
ガンマ線照射後のストレス下安定性保存
ストレスを受けた保存予備処方物の研究が、ガンマ線照射後のバイアルにおいてまた実施された。2個のPPI-149-CMCの実験室ロットからの密封されたバイアルを19KGyのガンマ線照射に曝露し、1ヶ月まで25℃、37℃および50℃で保存した。これらの予備処方物研究における化学的安定性データは、投与量19KGyのγ照射後のストレスを受ける保存安定性が、高度にストレスを受ける保存状態下(例えば、50℃で1週間)でさえ、主たる化学的不安定性をもたらさなかったことを示した。データはγ照射線量19KGy以下を示し、28日まで50℃以下でのPPI-149-CMCの保存は一貫してペプチド純度において2%未満の減少を示した(HPLC不純物プロフィールで決定した)。研究した2個のロット間の初期水分含量の明らかな差異にもかかわらず、初期の予備処方安定サンプルもしくは1ヶ月まで保存したもののどちらにも、ペプチド純度における顕著な差が測定されなかった。
【0065】
PPI-149-CMC粒子サイズ分析
レーザー光散乱を用いた粒子サイズ法が発達しており、PPI-149-CMCのサイズ研究に適用できる。その方法の使用を例示するために、PPI-149-CMCの粒子サイズへのガンマ線照射の影響を調べるために実施した予備処方実験を示す。この実験は、アモルファス固体物質は保存により粒子凝集する傾向であり得るという以前の理解で考えた。PPI-149-CMC実験室ロットの2個の試料をtype Iのガラスバイアルに詰め、灰色のブチルゴムストッパーで閉め、アルミニウムシールで密封した。粒子評価を、投与量15.5KGyのγ線照射への曝露の前および後に実施した。粒子評価はレーザー光散乱(逆フーリエレンズを備えたMalvern Mastersizer STMを使用)で実施した。レーザー光散乱による粒子サイズ分析のための20mgのサンプルを、約0.5mLの脱イオン水へ激しく撹拌することにより分散し、その後周囲温度の浴で5分間超音波処理した。バックグラウンド計算を実行後、定性実験法を実施した。サンプル分散液を約20%の暗黒化(obscuration)が得られるまで、連続フィードリザーバー(feed reservoir)(公称体積 約60mL)へ滴下した。ミキサー回転速度は、実験(バックグラウンド確認も加えて)を通して2700rpmで保持された。この速度でボルテックス由来の泡は生じなかったが、十分に安定した分散液を保持した。8スキャンを行った。得られたデータの分析は、とった任意のデータポイントの極値(extreme)として0.03%以下の標準偏差を示した。サンプル分散液をリザーバーに15分間保持し、そしてその後再測定した場合、顕著な変化は得られず、実験の過程を通して粒子溶解が無かったことを示した。
【0066】
サンプルを、上記に挙げた実験パラメータを使用して分析した。8スキャンを実行し、そして平均粒子直径データを測定した。2つの異なるサイズ分布が見られ、そして全てが粒子サイズの高端で明らかなカットオフを有しており、粒子凝集の無いことを示した。PPI-149-CMCの1ロットは、ガンマ線照射前では、サンプルのガンマ線照射後よりも明らかに低い平均体積直径を有した。この予備処方研究は、殺菌プロセス中にいくらかの粒子凝集が起こったことを示すように考えられる。
【0067】
実施例12:
この実施例では、PPI-149-CMCの固体状態形態に対するγ線照射および温度/湿度の両方のストレスに対する有効性を研究するために、種々の予備処方実験を行った。
【0068】
X線粉末回折
最初の実験においては、2個の60mgのPPI-149-CMCサンプルを(エアーヘッドスペース下(air headspace)で)、I型ガラスバイアル中にパックし、灰色のブチルゴムストッパーで閉め、そしてアルミニウムシールでシールした。次いで、一つのサンプルを、19.0KGyのγ線照射線量に曝露した。次に、固体状態形態の2個の60mgのサンプルをX線粉末回折によって研究した。回析図を19.0Kgyのγ線照射線量に曝露する前および後で比較した。
【0069】
次の研究では、PPI-149-CMCの60mgサンプル(γ線照射後)をI型ガラスバイアル中に置き、そして予め平衡化した50℃/75%相対湿度の一定湿度インキュベーター中に5日間置いた。インキュベーターから回収した後すぐに、サンプル容器を灰色のブチルゴムのストッパーで閉め、そしてアルミニウムシールでシールした。次いで、このストレスにさらしたサンプルのX線粉末回折を、容器中にて室温に維持した同じロットの別のサンプルと比較した。グラファイトモノクロメーターおよび50kV、40mAで操作されているCu(λ=1.54Å)X線源を備えた、Siemens D500自動化Powder Diffractometerを用いて、これらのサンプルを分析した。2θ走査範囲は、4〜40°であり、0.05°/1.2秒段階の段階走査窓を用いる。ビームスリットはNo.(1)1°、(2)1°、(3)1°、(4)0.15°および(5)0.15°の幅にセットした。2θ校正をNBSマイカ標準(SRM675)を用いて行った。サンプルを、ゼロバックグラウンドサンプルプレートを用いて分析した。
【0070】
データはγ線照射の前には、PPI-149-CMCは、明らかな結晶構造、または擬結晶構造を有さないことを示した。実際、アモルファス固体に特徴的なX線粉末回折パターン(回析図中の2°〜20°20の間の有意なピークのない幅広のハンプ)を示した。照射後のPPI-149-CMCサンプルは、非照射サンプルと非常に類似の回析パターンを生じ、これはガンマ線照射処理(19KGyまでおよび19KGyを含む線量)が、明らかに物質中での固体状態の多型転移を誘導しないことを示す。類似の様式で、PPI-149-CMCの温度/湿度ストレスをかけたサンプルは、非照射サンプルおよび照射サンプルの両方と非常に類似の回析パターンを生成し、このことはppI-149-CMCが、物質内での固体状態多型転移を過度に誘導する傾向がないことを強く示唆する。
【0071】
吸湿性
PPI-149-CMC(照射後)での予備処方研究を、一定温度(25℃)で種々の相対湿度の条件下で平衡湿気取り込み(重量増加によって測定)を決定するためによって行った。相対湿度(RH%)の関数としての平衡湿気(水%)の分析は、湿気の含量が約80%相対湿度まで徐々に増加することを示した。高い相対湿度(95%RH)において、PPI-149-CMCは、有意な湿分吸収が可能であった。80%RH以下の相対湿度において、湿気からの保護という点で有意な予防策は、不必要であると考えられる;従って、特定の製造工程は周囲湿度条件下で行い得る(ただし、極端な湿度は避ける)。
【0072】
実施例13:
この実施例において、PPI-149-CMCの融解研究を行った。実験はシンクを用いる条件、およびシンクを用いない条件の両方を利用して行った。PPI-149-CMCは、25℃で、pH7.3の0.1Mリン酸緩衝化生理食塩水に約100μg/mlの溶解度(遊離ペプチドとして測定され表される)を有した。シンク条件下において(所定の温度での、この系での飽和溶解度の<10%として定義される)、撹拌しない場合でさえ、PPI-149-CMCは迅速に溶解した(遊離のペプチドとして測定、および示した)。類似の実験において、PPI-149-CMCの平衡溶解度を25℃でpH7.3の0.1Mリン酸緩衝化生理食塩水中で決定し、(遊離のペプチドとして測定、および示し)これには3個のサンプルを用いた:PPI-149-CMC単独、さらに10(重量)%のPPI-149(遊離のペプチドとして示したが、酢酸と結合したPPI-149として導入した)が存在する場合でのPPI-149-CMC、およびさらに50(重量)%米国薬局方カルボキシメチルセルロースナトリウムが存在する場合でのPPI-149-CMC。3つのサンプルは全て、表面上は類似のペプチド平衡溶解度を示した。選択された緩衝液系は生理学的条件下に近いので、PPI-149-CMC中に存在するさらに遊離のカルボキシメチルセルロースまたはペプチド種の存在は、溶解性に影響を与えそうにないと考えられる。
【0073】
実施例14:
この実施例において、PPI-149-CMCの反復した皮下(SC)および筋肉内(IM)用量での薬物動力学、薬力学、および安全性をイヌにおいて特徴付けた。
【0074】
3ヶ月間行った最初の研究において、種々の再構成ビヒクル中のPPI-149-CMCの月一回IM注射またはSC注射を、1.2mg/kg(1日目)、0.3mg/kgまたは0.6mg/kg(29日目)、および1.2mg/kg(57日目)で用いて、40匹の雄ビーグル犬を評価した。5匹のイヌの8グループを以下に示すように本研究に割り当てた:
【0075】
【表3】
a.再構成ビヒクルをPPI-149-CMCを粒子懸濁物として再構成するのに用いた。
これらは以下を含む(水中に):
1. グリセリン=15%グリセリン/5%デキストロース
2. PEG=4%ポリエチレングリコール-3350/4%マンニトール
3. レシチン=0.5%レシチン/5%マンニトール
b.記号:臨床研究に用いられる再構成ビヒクルは0.9%塩化ナトリウム米国薬局方
c.全用量はペプチド(PPI-149)含量に関して示す。
d.完全な解剖学的組織学および顕微鏡学的組織学用に3匹の動物を85日目に屠殺した。
【0076】
この研究は、異なるビヒクル中での初回用量でのPPI-149-CMCの効力が、処置の最初の1ヶ月の間に評価されるように設定した。研究中の2ヶ月目の間、この計画において、有効な「維持」用量を決定することを試みるために、イヌはより低い投与量のPPI-149-CMCを受けた。3ヶ月目は、PPI-149-CMCの長期安全性および効力特性を評価するために計画した。
【0077】
再構成ビヒクルの1つの中に処方されたPPI-149-CMCのIMもしくはSC用量、またはコントロール体のIM用量を、各投与日に右後ろ足の上部側面(IM)へ、または肩甲部の中央(SC)に投与した。物質を23gの短いベベル針を備えた1ccのツベルクリンシリンジに引き込んだ。投与の直前に注射部位をアルコール綿棒で拭く。注入する用量は、ペプチド/kg体重の特定用量に基づいていた。全ての用量は、投与されたPPI-149ペプチドの量をいうということに留意するべきである。
【0078】
実験の間はずっと、毒性または薬理学的効果および通常の行動および様子における変化の明らかな徴候について、各動物を1日少なくとも2回観察した。全ての臨床的に異常な所見を記録した。
【0079】
初回用量の投与の前、および投与後の種々の時点で、全血球算定(CBC)、血清化学分析、ならびに週に2回の放射イムノアッセイによるPPI-149濃度およびテストステロン濃度の決定のために、血液を回収した。
【0080】
研究の3ヶ月後、9匹の動物を屠殺し、そしてそれらの組織を、全体的な病理学的分析および組織病理学的分析のために回収した。屠殺のため、ビヒクルコントロールグループ、IM投与グループの1つ、およびSC投与グループの1つから、動物を選択した。3ヶ月目の屠殺物の、全体的な病理学および組織病理学のために回収した組織は:投与部位(SCまたはIM)、副腎、大動脈、骨、骨髄、脳、横隔膜、精巣上体、食道、視神経付きの眼、心臓、腎臓、大腸(盲腸、結腸)、胆嚢を有する肝臓、気管支付きの肺、リンパ節、膵臓、脳下垂体、尿道付きの前立腺、唾液腺、座骨神経、骨格筋、皮膚、小腸(十二指腸、空腸、回腸)、脊髄、脾臓、胃、精巣、胸腺、副甲状腺付きの甲状腺(thryoid gland)、舌、気管、膀胱(urinary bladdr)、および全体的な病変。
【0081】
実験の間、処置動物またはコントロール動物のいずれも、血液学または血液化学的にはベースラインから有意な変化はなかった。3ヶ月目の全体的なおよび組織学的評価において、屠殺物はこのLHRHアンタゴニストに期待されたような、精巣および前立腺中の変化を除いては、PPI-149-CMC処理イヌおよびコントロール(ビヒクル処置)動物との間の明らかな差を示さなかった。
【0082】
PPI-149-CMCの薬物力学に関して、種々の再構築ビヒクル中の再懸濁した1.2mg/kg PPI-149-CMCで処理し、およびIMまたはSCを投与した全てのイヌは、類似の血漿PPI-149の薬物動力学プロフィールを示した(血漿濃度は最初の2日間にピークを有し、次いで次の月の間ゆっくりと、指数関数的に減少する)。この研究に用いた3個の再構築したビヒクルの任意のものに再懸濁した場合、PPI-149-CMCは、PPI-149の類似の血漿分布を示した。
【0083】
PPI-149-CMCの内分泌効力に関して、全てのイヌにおいて、去勢レベル(<0.6ng/mL)のテストステロンが、PPI-149-CMC投与の開始24時間以内に観察され、そしてレベルは、一般に投与経路または再構築ビヒクルの選択に関わらず、最初の1ヶ月間を通じて去勢範囲を維持した。35匹のうち26匹のイヌ(75%)が、29日目のPPI-149-CMCの第2用量の投与直前に得た血液サンプル中のテストステロンの去勢レベルを有した。これらの結果は、イヌの1.2mg/kgの初回用量が首尾良く血漿テストステロンの迅速で長期間持続する抑制(>28日)を誘導することを示す。投与の2月目において、「維持」用量の効力(初回用量より低い用量)を研究した場合、結果はPPI-149-CMCの0.3mg/kgまたは0.6mg/kgの投与が、35匹中30匹のイヌの20日以上の間のテストステロンの去勢レベルの維持を示した。処置の2ヶ月目の最後(57日目)で、35匹中21匹のイヌ(60%)が去勢レベルを維持し、一方14動物が正常範囲(>0.6%ng/mL)のテストステロンを有した。3ヶ月目の初めに1.2mg/kgの用量を投与した。PPI-149の血漿濃度は、続く28日間維持され、一方テストステロンの血漿レベルは再び「去勢」であった。3ヶ月目の最後(85日目)までに、テストステロンの血漿レベルは35匹のPPI-149-CMC処置イヌの30匹が去勢範囲にあることが示された。
【0084】
要約すると、35匹のイヌが、種々の再構築ビヒクルで、IMまたはSC投与を用いて、1日目に1.2mg/kgのPPI-149-CMC、29日目に0.3mg/kgまたは0.6mg/kgのPPI-149-CMC、ならびに57日目に1.2mg/kgのPPI-149-CMCを受けた。これらの35匹のうち19動物(54%)が、治療の全経過を通じて去勢範囲を維持した血漿テストステロンレベルを有した。従って、28日の間隔でのPPI-149-CMCの投与は、血漿テストステロンの完全な抑制を生じることができた。これは迅速(全動物が24時間以内に去勢レベルを有した)であり、そして長期間持続する(投与の経過にわたって維持された)。
【0085】
上記と類似の研究を、PPI-149-CMCの長期的な安全性および効力の特徴をさらに評価するため、6ヶ月間イヌで行った。動物は、IMまたはSCのいずれかで1.2mg/kg PPI-149-CMCの初回用量を受け、そして28日の間隔で5回の続く用量(0.3・g/kg、0.6mg/kg、または1.2mg/kgのいずれかの濃度)を受けた。血漿テストステロンレベルおよびPPI-149レベルを、放射イムノアッセイによって一定間隔で評価した。代表的な結果を図4(SC処置について)および図5(IM処置について)に示す(これは、血漿テストステロンレベル(白四角)およびPPI-149レベル(黒四角)を例示する)。PPI-149-CMCの各投与に用いた特定の投薬量をグラフに示す。図4および図5に例示した結果は、28日間隔でのPPI-149-CMCの投与が、血漿テストステロンの完全な抑制を生じ得たことをさらに実証する。これは迅速でかつ長く持続し、6ヶ月もの長い間、減少した血漿テストステロンレベルを維持する。
【0086】
等価物
当業者は、本明細書に記載された本発明の特定の実施態様に対する多くの等価物を、日常的実験以外を用いることなく認識するか、または確認しうる。このような等価物は、以下の請求の範囲に包含されることが意図される。
【特許請求の範囲】
【請求項1】
薬学的に活性なペプチド化合物とキャリア巨大分子との水不溶性複合体を含む、薬学的組成物。
【請求項2】
前記水不溶性複合体の処方物が、前記薬学的に活性なペプチド化合物と前記キャリア巨大分子との間のイオン性相互作用により、少なくとも部分的に媒介される、請求項1に記載の薬学的組成物。
【請求項3】
前記薬学的に活性なペプチド化合物がカチオン性であり、かつ前記キャリア巨大分子がアニオン性である、請求項2に記載の薬学的組成物。
【請求項4】
前記薬学的に活性なペプチド化合物がアニオン性であり、前記キャリア巨大分子がカチオン性である、請求項2に記載の薬学的組成物。
【請求項5】
前記水不溶性複合体の処方物が、前記薬学的に活性なペプチド化合物と前記キャリア巨大分子との間の疎水性相互作用により、少なくとも部分的に媒介される、請求項1に記載の薬学的組成物。
【請求項6】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも1週間、該被験体に前記薬学的に活性なペプチドの持続した送達を提供する、請求項1に記載の薬学的組成物。
【請求項7】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも2週間、該被験体に前記薬学的に活性なペプチドの持続した送達を提供する、請求項1に記載の薬学的組成物。
【請求項8】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも3週間、該被験体に前記薬学的に活性なペプチドの持続した送達を提供する、請求項1に記載の薬学的組成物。
【請求項9】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも4週間、該被験体に前記薬学的に活性なペプチドの持続した送達を提供する、請求項1に記載の薬学的組成物。
【請求項10】
前記薬学的に活性なペプチドが多価カチオン性またはアニオン性ペプチドである、請求項1に記載の薬学的組成物。
【請求項11】
前記ペプチドが5〜20アミノ酸長である、請求項1に記載の薬学的組成物。
【請求項12】
前記ペプチドが8〜15アミノ酸長である、請求項1に記載の薬学的組成物。
【請求項13】
前記ペプチドが8〜12アミノ酸長である、請求項1に記載の薬学的組成物。
【請求項14】
前記キャリア巨大分子が、アニオン性ポリマーである、請求項1に記載の薬学的組成物。
【請求項15】
前記キャリア巨大分子が、アニオン性ポリアルコール誘導体、もしくはそのフラグメント、またはそれらの薬学的に受容可能な塩である、請求項1に記載の薬学的組成物。
【請求項16】
前記キャリア巨大分子が、アニオン性ポリサッカリド誘導体、もしくはそのフラグメント、またはそれらの薬学的に受容可能な塩である、請求項1に記載の薬学的組成物。
【請求項17】
前記キャリア巨大分子が、カルボキシメチルセルロースまたはその薬学的に受容可能な塩である、請求項1に記載の薬学的組成物。
【請求項18】
前記キャリア巨大分子が、アルギン、アルギネート、アニオン性アセテートポリマー、アニオン性アクリル酸ポリマー、キサンタンガム、アニオン性カラギーナン誘導体、アニオン性ポリガラクツロン酸誘導体、グリコール酸ナトリウムスターチ、ならびにそのフラグメント、誘導体、および薬学的に受容可能な塩からなる群より選択される、請求項1に記載の薬学的組成物。
【請求項19】
乾燥した固体である、請求項1に記載の薬学的組成物。
【請求項20】
液体懸濁物または半固体分散体である、請求項1に記載の薬学的組成物。
【請求項21】
水不溶性複合体を含む薬学的組成物であって、ここで該水不溶性複合体が、薬学的に活性なペプチド化合物およびキャリア巨大分子から本質的になる、薬学的組成物。
【請求項22】
LHRHアナログとキャリア巨大分子との水不溶性複合体からなる、薬学的組成物。
【請求項23】
前記水不溶性複合体の処方物が、前記LHRHアナログと前記キャリア巨大分子との間のイオン性相互作用により、少なくとも部分的に媒介される、請求項22に記載の薬学的組成物。
【請求項24】
前記水不溶性複合体が、前記LHRHアナログと前記キャリア巨大分子との間の疎水性相互作用により、少なくとも部分的に媒介される、請求項22に記載の薬学的組成物。
【請求項25】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも1週間、該被験体に前記LHRHアナログの持続した送達を提供する、請求項22に記載の薬学的組成物。
【請求項26】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも2週間、該被験体に前記LHRHアナログの持続した送達を提供する、請求項22に記載の薬学的組成物。
【請求項27】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも3週間、該被験体に前記LHRHアナログの持続した送達を提供する、請求項22に記載の薬学的組成物。
【請求項28】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも4週間、該被験体に前記LHRHアナログの持続した送達を提供する、請求項22に記載の薬学的組成物。
【請求項29】
前記LHRHアナログがLHRHアンタゴニストである、請求項22に記載の薬学的組成物。
【請求項30】
前記LHRHアナログがペプチド化合物を含み、ここで天然の哺乳動物LHRHの6位のアミノ酸に対応する該ペプチド化合物の残基がD−アスパラギン構造を含む、請求項29に記載の薬学的組成物。
【請求項31】
前記LHRHアンタゴニストが、以下の構造:
A−B−C−D−E−F−G−H−I−J
を含むペプチド化合物またはその薬学的に受容可能な塩であって、ここで、
Aは、ピロ−Glu、Ac−D−Nal、Ac−D−Qal、Ac−Sar、またはAc−D−Palであり
Bは、His、または4−Cl−D−Pheであり
Cは、Trp、D−Pal、D−Nal、L−Nal、D−Pal(N−O)、またはD−Trpであり
Dは、Serであり
Eは、N−Me−Ala、Tyr、N−Me−Tyr、Ser、Lys(iPr)、4−Cl−Phe、His、Asn、Met、Ala、Arg、またはIleであり;
Fは、
【化1】
ここで
RおよびXは、独立してHまたはアルキルであり;そして
Lは小極性部分を含み;
Gは、Leu、またはTrpであり;
Hは、Lys(iPr)、Gln、Met、またはArgであり
Iは、Proであり;そして
Jは、Gly−NH2、またはD−Ala−NH2である、
請求項29に記載の薬学的組成物。
【請求項32】
Fが、D−Asn、D−GlnおよびD−Thrからなる群より選択される、請求項31に記載の薬学的組成物。
【請求項33】
FがD−Asnである、請求項31に記載の薬学的組成物。
【請求項34】
EがチロシンまたはN−メチルチロシンである、請求項31に記載の薬学的組成物。
【請求項35】
前記LHRHアンタゴニストが、以下の構造:Ac−D−Nal1、4−Cl−D−Phe2、D−Pal3、N−Me−Tyr5、D−Asn6、Lys(iPr)8、D−Ala10−LHRHを有する、請求項29に記載の薬学的組成物。
【請求項36】
前記キャリア巨大分子がアニオン性ポリマーである、請求項22に記載の薬学的組成物。
【請求項37】
前記キャリア巨大分子が、アニオン性ポリアルコール誘導体、もしくはそのフラグメント、またはそれらの薬学的に受容可能な塩である、請求項22に記載の薬学的組成物。
【請求項38】
前記キャリア巨大分子が、アニオン性ポリサッカリド誘導体、もしくはそのフラグメント、またはそれらの薬学的に受容可能な塩である、請求項22に記載の薬学的組成物。
【請求項39】
前記キャリア巨大分子が、カルボキシメチルセルロースまたはその薬学的に受容可能な塩である、請求項22に記載の薬学的組成物。
【請求項40】
前記キャリア巨大分子が、アルギン、アルギネート、アニオン性アセテートポリマー、アニオン性アクリル酸ポリマー、キサンタンガム、アニオン性カラギーナン誘導体、アニオン性ポリガラクツロン酸誘導体、グリコール酸ナトリウムスターチ、ならびにそのフラグメント、誘導体、および薬学的に受容可能な塩からなる群より選択される、請求項22に記載の薬学的組成物。
【請求項41】
乾燥した固体である、請求項22に記載の薬学的組成物。
【請求項42】
液体懸濁物または半固体分散体である、請求項22に記載の薬学的組成物。
【請求項43】
LHRHアナログで処置可能な状態の被験体を処置するためのパッケージ化処方物であって、以下:
LHRHアナログで処置可能な状態の被験体を処置するための水不溶性複合体の使用説明書と合わせてパッケージ化された、LHRHアナログとキャリア巨大分子との水不溶性複合体を含む、パッケージ化処方物。
【請求項44】
前記LHRHアンタゴニストが、以下の構造:Ac−D−Nal1、4−Cl−D−Phe2、D−Pal3、N−Me−Tyr5、D−Asn6、Lys(iPr)8、D−Ala10−LHRHを有し、前記キャリア巨大分子がカルボキシメチルセルロースまたはその薬学的に受容可能な塩である、請求項43に記載のパッケージ化処方物。
【請求項45】
管腔を有するシリンジにおいて、改良が、該管腔に、LHRHアナログとキャリア巨大分子との水不溶性複合体の液体懸濁液を含むことである、シリンジ。
【請求項46】
前記LHRHアナログが、以下の構造:Ac−D−Nal1、4−Cl−D−Phe2、D−Pal3、N−Me−Tyr5、D−Asn6、Lys(iPr)8、D−Ala10−LHRHを有し、前記キャリア巨大分子がカルボキシメチルセルロースまたはその薬学的に受容可能な塩である、請求項45に記載のシリンジ。
【請求項47】
薬学的処方物を調製する方法であって、該方法は、以下:
ペプチド化合物およびキャリア巨大分子を提供する工程;
該ペプチド化合物と該キャリア巨大分子とを、該ペプチド化合物と該キャリア巨大分子との水不溶性複合体が形成するような条件下で組み合わせる工程;および
該水不溶性複合体を含む薬学的処方物を調製する工程、
を包含する、方法。
【請求項48】
前記ペプチド化合物の溶液と前記キャリア巨大分子の溶液とが、該ペプチド化合物と該キャリア巨大分子との水不溶性複合体が沈殿するまで組み合わされる、請求項47に記載の方法。
【請求項49】
前記ペプチド化合物の前記溶液および前記キャリア巨大分子の前記溶液が、水溶液である、請求項48に記載の方法。
【請求項50】
前記ペプチド化合物の前記溶液と前記キャリア巨大分子の前記溶液とが、該ペプチド化合物と該キャリア巨大分子との水不溶性複合体が沈殿するまで、組み合わされ、そして加熱される、請求項48に記載の方法。
【請求項51】
前記水不溶性複合体を、γ線照射または電子線照射によって滅菌する工程をさらに包含する、請求項47に記載の方法。
【請求項52】
前記水不溶性複合体が、無菌の手順を使用して形成される、請求項47に記載の方法。
【請求項53】
前記ペプチド化合物がカチオン性であり、かつ前記キャリア巨大分子がアニオン性である、請求項47に記載の方法。
【請求項54】
前記ペプチド化合物がアニオン性であり、かつ前記キャリア巨大分子がカチオン性である、請求項47に記載の方法。
【請求項55】
前記ペプチド化合物が、多価カチオン性またはアニオン性ペプチドである、請求項47に記載の方法。
【請求項56】
前記ペプチド化合物が、LHRHアナログである、請求項47に記載の方法。
【請求項57】
前記LHRHアナログがLHRHアンタゴニストである、請求項56に記載の方法。
【請求項58】
前記LHRHアンタゴニストがペプチド化合物を含み、ここで天然の哺乳動物LHRHの6位のアミノ酸に対応する該ペプチド化合物の残基がD−アスパラギン構造を含む、請求項57に記載の方法。
【請求項59】
前記LHRHアンタゴニストが、以下の構造:
A−B−C−D−E−F−G−H−I−J
を含むペプチド化合物またはその薬学的に受容可能な塩であって、ここで、
Aは、ピロ−Glu、Ac−D−Nal、Ac−D−Qal、Ac−Sar、またはAc−D−Palであり
Bは、His、または4−Cl−D−Pheであり
Cは、Trp、D−Pal、D−Nal、L−Nal、D−Pal(N−O)、またはD−Trpであり
Dは、Serであり
Eは、N−Me−Ala、Tyr、N−Me−Tyr、Ser、Lys(iPr)、4−Cl−Phe、His、Asn、Met、Ala、Arg、またはIleであり;
Fは、
【化2】
ここで
RおよびXは、独立してHまたはアルキルであり;そして
Lは小極性部分を含み;
Gは、Leu、またはTrpであり;
Hは、Lys(iPr)、Gln、Met、またはArgであり
Iは、Proであり;そして
Jは、Gly−NH2、またはD−Ala−NH2である、
請求項57に記載の方法。
【請求項60】
Fが、D−Asn、D−GlnおよびD−Thrからなる群より選択される、請求項59に記載の方法。
【請求項61】
FがD−Asnである、請求項59に記載の方法。
【請求項62】
EがチロシンまたはN−メチルチロシンである、請求項59に記載の方法。
【請求項63】
前記LHRHアンタゴニストが、以下の構造:Ac−D−Nal1、4−Cl−D−Phe2、D−Pal3、N−Me−Tyr5、D−Asn6、Lys(iPr)8、D−Ala10−LHRHを有する、請求項57に記載の方法。
【請求項64】
前記キャリア巨大分子がアニオン性ポリマーである、請求項47に記載の方法。
【請求項65】
前記キャリア巨大分子が、アニオン性ポリアルコール誘導体、もしくはそのフラグメント、またはそれらの薬学的に受容可能な塩である、請求項47に記載の方法。
【請求項66】
前記キャリア巨大分子が、アニオン性ポリサッカリド誘導体、もしくはそのフラグメント、またはそれらの薬学的に受容可能な塩である、請求項47に記載の方法。
【請求項67】
前記キャリア巨大分子が、カルボキシメチルセルロースまたはその薬学的に受容可能な塩である、請求項47に記載の方法。
【請求項68】
前記キャリア巨大分子が、アルギン、アルギネート、アニオン性アセテートポリマー、アニオン性アクリル酸ポリマー、キサンタンガム、アニオン性カラギーナン誘導体、アニオン性ポリガラクツロン酸誘導体、グリコール酸ナトリウムスターチ、ならびにそのフラグメント、誘導体、および薬学的に受容可能な塩からなる群より選択される、請求項47に記載の方法。
【請求項69】
乾燥した固体である、請求項47に記載の方法。
【請求項70】
液体懸濁物または半固体分散体である、請求項47に記載の方法。
【請求項71】
請求項47〜70のいずれか1項に記載の方法に従って調製された、薬学的処方物。
【請求項72】
LHRHアナログで処置可能な状態について被験体を処置する方法であって、該方法は、該被験体に、LHRHアナログとキャリア巨大分子との水不溶性複合体を含む薬学的処方物を投与する工程、
を包含する、方法。
【請求項73】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも1週間、該被験体に前記LHRHアナログの持続した送達を提供する、請求項72に記載の方法。
【請求項74】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも2週間、該被験体に前記LHRHアナログの持続した送達を提供する、請求項72に記載の方法。
【請求項75】
前記水不溶性非共有結合複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも3週間、該被験体に前記LHRHアナログの持続した送達を提供する、請求項72に記載の方法。
【請求項76】
前記水不溶性非共有結合複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも4週間、該被験体に前記LHRHアナログの持続した送達を提供する、請求項72に記載の方法。
【請求項77】
前記LHRHアナログがLHRHアンタゴニストである、請求項72に記載の方法。
【請求項78】
前記LHRHアンタゴニストが、以下の構造:Ac−D−Nal1、4−Cl−D−Phe2、D−Pal3、N−Me−Tyr5、D−Asn6、Lys(iPr)8、D−Ala10−LHRHを有する、請求項77に記載の方法。
【請求項79】
前記キャリア巨大分子がアニオン性ポリマーである、請求項72に記載の方法。
【請求項80】
前記キャリア巨大分子が、アニオン性ポリアルコール誘導体、もしくはそのフラグメント、またはそれらの薬学的に受容可能な塩である、請求項72に記載の方法。
【請求項81】
前記キャリア巨大分子が、アニオン性ポリサッカリド誘導体、もしくはそのフラグメント、またはそれらの薬学的に受容可能な塩である、請求項72に記載の方法。
【請求項82】
前記キャリア巨大分子が、カルボキシメチルセルロースまたはその薬学的に受容可能な塩である、請求項72に記載の方法。
【請求項83】
前記キャリア巨大分子が、アルギン、アルギネート、アニオン性アセテートポリマー、アニオン性アクリル酸ポリマー、キサンタンガム、アニオン性カラギーナン誘導体、アニオン性ポリガラクツロン酸誘導体、グリコール酸ナトリウムスターチ、ならびにそのフラグメント、誘導体、および薬学的に受容可能な塩からなる群より選択される、請求項72に記載の方法。
【請求項84】
前記薬学的処方物が、前記被験体に非経口経路により投与される、請求項72に記載の方法。
【請求項85】
前記薬学的処方物が、前記被験体に経口的に投与される、請求項72に記載の方法。
【請求項86】
前記薬学的処方物が、筋肉内注射および皮下/皮内注射により投与される、請求項72に記載の方法。
【請求項87】
LHRHアナログで処置可能な前記状態が、ホルモン依存性癌である、請求項72に記載の方法。
【請求項88】
前記ホルモン依存性癌が前立腺癌である、請求項87に記載の方法。
【請求項89】
LHRHアナログで処置可能な前記状態が、良性前立腺肥大、性的早熟、子宮内膜症および子宮筋腫からなる群より選択される、請求項72に記載の方法。
【請求項90】
前記LHRHアナログが、体外受精または避妊目的で投与される、請求項72に記載の方法。
【請求項1】
薬学的に活性なペプチド化合物とキャリア巨大分子との水不溶性複合体を含む、薬学的組成物。
【請求項2】
前記水不溶性複合体の処方物が、前記薬学的に活性なペプチド化合物と前記キャリア巨大分子との間のイオン性相互作用により、少なくとも部分的に媒介される、請求項1に記載の薬学的組成物。
【請求項3】
前記薬学的に活性なペプチド化合物がカチオン性であり、かつ前記キャリア巨大分子がアニオン性である、請求項2に記載の薬学的組成物。
【請求項4】
前記薬学的に活性なペプチド化合物がアニオン性であり、前記キャリア巨大分子がカチオン性である、請求項2に記載の薬学的組成物。
【請求項5】
前記水不溶性複合体の処方物が、前記薬学的に活性なペプチド化合物と前記キャリア巨大分子との間の疎水性相互作用により、少なくとも部分的に媒介される、請求項1に記載の薬学的組成物。
【請求項6】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも1週間、該被験体に前記薬学的に活性なペプチドの持続した送達を提供する、請求項1に記載の薬学的組成物。
【請求項7】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも2週間、該被験体に前記薬学的に活性なペプチドの持続した送達を提供する、請求項1に記載の薬学的組成物。
【請求項8】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも3週間、該被験体に前記薬学的に活性なペプチドの持続した送達を提供する、請求項1に記載の薬学的組成物。
【請求項9】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも4週間、該被験体に前記薬学的に活性なペプチドの持続した送達を提供する、請求項1に記載の薬学的組成物。
【請求項10】
前記薬学的に活性なペプチドが多価カチオン性またはアニオン性ペプチドである、請求項1に記載の薬学的組成物。
【請求項11】
前記ペプチドが5〜20アミノ酸長である、請求項1に記載の薬学的組成物。
【請求項12】
前記ペプチドが8〜15アミノ酸長である、請求項1に記載の薬学的組成物。
【請求項13】
前記ペプチドが8〜12アミノ酸長である、請求項1に記載の薬学的組成物。
【請求項14】
前記キャリア巨大分子が、アニオン性ポリマーである、請求項1に記載の薬学的組成物。
【請求項15】
前記キャリア巨大分子が、アニオン性ポリアルコール誘導体、もしくはそのフラグメント、またはそれらの薬学的に受容可能な塩である、請求項1に記載の薬学的組成物。
【請求項16】
前記キャリア巨大分子が、アニオン性ポリサッカリド誘導体、もしくはそのフラグメント、またはそれらの薬学的に受容可能な塩である、請求項1に記載の薬学的組成物。
【請求項17】
前記キャリア巨大分子が、カルボキシメチルセルロースまたはその薬学的に受容可能な塩である、請求項1に記載の薬学的組成物。
【請求項18】
前記キャリア巨大分子が、アルギン、アルギネート、アニオン性アセテートポリマー、アニオン性アクリル酸ポリマー、キサンタンガム、アニオン性カラギーナン誘導体、アニオン性ポリガラクツロン酸誘導体、グリコール酸ナトリウムスターチ、ならびにそのフラグメント、誘導体、および薬学的に受容可能な塩からなる群より選択される、請求項1に記載の薬学的組成物。
【請求項19】
乾燥した固体である、請求項1に記載の薬学的組成物。
【請求項20】
液体懸濁物または半固体分散体である、請求項1に記載の薬学的組成物。
【請求項21】
水不溶性複合体を含む薬学的組成物であって、ここで該水不溶性複合体が、薬学的に活性なペプチド化合物およびキャリア巨大分子から本質的になる、薬学的組成物。
【請求項22】
LHRHアナログとキャリア巨大分子との水不溶性複合体からなる、薬学的組成物。
【請求項23】
前記水不溶性複合体の処方物が、前記LHRHアナログと前記キャリア巨大分子との間のイオン性相互作用により、少なくとも部分的に媒介される、請求項22に記載の薬学的組成物。
【請求項24】
前記水不溶性複合体が、前記LHRHアナログと前記キャリア巨大分子との間の疎水性相互作用により、少なくとも部分的に媒介される、請求項22に記載の薬学的組成物。
【請求項25】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも1週間、該被験体に前記LHRHアナログの持続した送達を提供する、請求項22に記載の薬学的組成物。
【請求項26】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも2週間、該被験体に前記LHRHアナログの持続した送達を提供する、請求項22に記載の薬学的組成物。
【請求項27】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも3週間、該被験体に前記LHRHアナログの持続した送達を提供する、請求項22に記載の薬学的組成物。
【請求項28】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも4週間、該被験体に前記LHRHアナログの持続した送達を提供する、請求項22に記載の薬学的組成物。
【請求項29】
前記LHRHアナログがLHRHアンタゴニストである、請求項22に記載の薬学的組成物。
【請求項30】
前記LHRHアナログがペプチド化合物を含み、ここで天然の哺乳動物LHRHの6位のアミノ酸に対応する該ペプチド化合物の残基がD−アスパラギン構造を含む、請求項29に記載の薬学的組成物。
【請求項31】
前記LHRHアンタゴニストが、以下の構造:
A−B−C−D−E−F−G−H−I−J
を含むペプチド化合物またはその薬学的に受容可能な塩であって、ここで、
Aは、ピロ−Glu、Ac−D−Nal、Ac−D−Qal、Ac−Sar、またはAc−D−Palであり
Bは、His、または4−Cl−D−Pheであり
Cは、Trp、D−Pal、D−Nal、L−Nal、D−Pal(N−O)、またはD−Trpであり
Dは、Serであり
Eは、N−Me−Ala、Tyr、N−Me−Tyr、Ser、Lys(iPr)、4−Cl−Phe、His、Asn、Met、Ala、Arg、またはIleであり;
Fは、
【化1】
ここで
RおよびXは、独立してHまたはアルキルであり;そして
Lは小極性部分を含み;
Gは、Leu、またはTrpであり;
Hは、Lys(iPr)、Gln、Met、またはArgであり
Iは、Proであり;そして
Jは、Gly−NH2、またはD−Ala−NH2である、
請求項29に記載の薬学的組成物。
【請求項32】
Fが、D−Asn、D−GlnおよびD−Thrからなる群より選択される、請求項31に記載の薬学的組成物。
【請求項33】
FがD−Asnである、請求項31に記載の薬学的組成物。
【請求項34】
EがチロシンまたはN−メチルチロシンである、請求項31に記載の薬学的組成物。
【請求項35】
前記LHRHアンタゴニストが、以下の構造:Ac−D−Nal1、4−Cl−D−Phe2、D−Pal3、N−Me−Tyr5、D−Asn6、Lys(iPr)8、D−Ala10−LHRHを有する、請求項29に記載の薬学的組成物。
【請求項36】
前記キャリア巨大分子がアニオン性ポリマーである、請求項22に記載の薬学的組成物。
【請求項37】
前記キャリア巨大分子が、アニオン性ポリアルコール誘導体、もしくはそのフラグメント、またはそれらの薬学的に受容可能な塩である、請求項22に記載の薬学的組成物。
【請求項38】
前記キャリア巨大分子が、アニオン性ポリサッカリド誘導体、もしくはそのフラグメント、またはそれらの薬学的に受容可能な塩である、請求項22に記載の薬学的組成物。
【請求項39】
前記キャリア巨大分子が、カルボキシメチルセルロースまたはその薬学的に受容可能な塩である、請求項22に記載の薬学的組成物。
【請求項40】
前記キャリア巨大分子が、アルギン、アルギネート、アニオン性アセテートポリマー、アニオン性アクリル酸ポリマー、キサンタンガム、アニオン性カラギーナン誘導体、アニオン性ポリガラクツロン酸誘導体、グリコール酸ナトリウムスターチ、ならびにそのフラグメント、誘導体、および薬学的に受容可能な塩からなる群より選択される、請求項22に記載の薬学的組成物。
【請求項41】
乾燥した固体である、請求項22に記載の薬学的組成物。
【請求項42】
液体懸濁物または半固体分散体である、請求項22に記載の薬学的組成物。
【請求項43】
LHRHアナログで処置可能な状態の被験体を処置するためのパッケージ化処方物であって、以下:
LHRHアナログで処置可能な状態の被験体を処置するための水不溶性複合体の使用説明書と合わせてパッケージ化された、LHRHアナログとキャリア巨大分子との水不溶性複合体を含む、パッケージ化処方物。
【請求項44】
前記LHRHアンタゴニストが、以下の構造:Ac−D−Nal1、4−Cl−D−Phe2、D−Pal3、N−Me−Tyr5、D−Asn6、Lys(iPr)8、D−Ala10−LHRHを有し、前記キャリア巨大分子がカルボキシメチルセルロースまたはその薬学的に受容可能な塩である、請求項43に記載のパッケージ化処方物。
【請求項45】
管腔を有するシリンジにおいて、改良が、該管腔に、LHRHアナログとキャリア巨大分子との水不溶性複合体の液体懸濁液を含むことである、シリンジ。
【請求項46】
前記LHRHアナログが、以下の構造:Ac−D−Nal1、4−Cl−D−Phe2、D−Pal3、N−Me−Tyr5、D−Asn6、Lys(iPr)8、D−Ala10−LHRHを有し、前記キャリア巨大分子がカルボキシメチルセルロースまたはその薬学的に受容可能な塩である、請求項45に記載のシリンジ。
【請求項47】
薬学的処方物を調製する方法であって、該方法は、以下:
ペプチド化合物およびキャリア巨大分子を提供する工程;
該ペプチド化合物と該キャリア巨大分子とを、該ペプチド化合物と該キャリア巨大分子との水不溶性複合体が形成するような条件下で組み合わせる工程;および
該水不溶性複合体を含む薬学的処方物を調製する工程、
を包含する、方法。
【請求項48】
前記ペプチド化合物の溶液と前記キャリア巨大分子の溶液とが、該ペプチド化合物と該キャリア巨大分子との水不溶性複合体が沈殿するまで組み合わされる、請求項47に記載の方法。
【請求項49】
前記ペプチド化合物の前記溶液および前記キャリア巨大分子の前記溶液が、水溶液である、請求項48に記載の方法。
【請求項50】
前記ペプチド化合物の前記溶液と前記キャリア巨大分子の前記溶液とが、該ペプチド化合物と該キャリア巨大分子との水不溶性複合体が沈殿するまで、組み合わされ、そして加熱される、請求項48に記載の方法。
【請求項51】
前記水不溶性複合体を、γ線照射または電子線照射によって滅菌する工程をさらに包含する、請求項47に記載の方法。
【請求項52】
前記水不溶性複合体が、無菌の手順を使用して形成される、請求項47に記載の方法。
【請求項53】
前記ペプチド化合物がカチオン性であり、かつ前記キャリア巨大分子がアニオン性である、請求項47に記載の方法。
【請求項54】
前記ペプチド化合物がアニオン性であり、かつ前記キャリア巨大分子がカチオン性である、請求項47に記載の方法。
【請求項55】
前記ペプチド化合物が、多価カチオン性またはアニオン性ペプチドである、請求項47に記載の方法。
【請求項56】
前記ペプチド化合物が、LHRHアナログである、請求項47に記載の方法。
【請求項57】
前記LHRHアナログがLHRHアンタゴニストである、請求項56に記載の方法。
【請求項58】
前記LHRHアンタゴニストがペプチド化合物を含み、ここで天然の哺乳動物LHRHの6位のアミノ酸に対応する該ペプチド化合物の残基がD−アスパラギン構造を含む、請求項57に記載の方法。
【請求項59】
前記LHRHアンタゴニストが、以下の構造:
A−B−C−D−E−F−G−H−I−J
を含むペプチド化合物またはその薬学的に受容可能な塩であって、ここで、
Aは、ピロ−Glu、Ac−D−Nal、Ac−D−Qal、Ac−Sar、またはAc−D−Palであり
Bは、His、または4−Cl−D−Pheであり
Cは、Trp、D−Pal、D−Nal、L−Nal、D−Pal(N−O)、またはD−Trpであり
Dは、Serであり
Eは、N−Me−Ala、Tyr、N−Me−Tyr、Ser、Lys(iPr)、4−Cl−Phe、His、Asn、Met、Ala、Arg、またはIleであり;
Fは、
【化2】
ここで
RおよびXは、独立してHまたはアルキルであり;そして
Lは小極性部分を含み;
Gは、Leu、またはTrpであり;
Hは、Lys(iPr)、Gln、Met、またはArgであり
Iは、Proであり;そして
Jは、Gly−NH2、またはD−Ala−NH2である、
請求項57に記載の方法。
【請求項60】
Fが、D−Asn、D−GlnおよびD−Thrからなる群より選択される、請求項59に記載の方法。
【請求項61】
FがD−Asnである、請求項59に記載の方法。
【請求項62】
EがチロシンまたはN−メチルチロシンである、請求項59に記載の方法。
【請求項63】
前記LHRHアンタゴニストが、以下の構造:Ac−D−Nal1、4−Cl−D−Phe2、D−Pal3、N−Me−Tyr5、D−Asn6、Lys(iPr)8、D−Ala10−LHRHを有する、請求項57に記載の方法。
【請求項64】
前記キャリア巨大分子がアニオン性ポリマーである、請求項47に記載の方法。
【請求項65】
前記キャリア巨大分子が、アニオン性ポリアルコール誘導体、もしくはそのフラグメント、またはそれらの薬学的に受容可能な塩である、請求項47に記載の方法。
【請求項66】
前記キャリア巨大分子が、アニオン性ポリサッカリド誘導体、もしくはそのフラグメント、またはそれらの薬学的に受容可能な塩である、請求項47に記載の方法。
【請求項67】
前記キャリア巨大分子が、カルボキシメチルセルロースまたはその薬学的に受容可能な塩である、請求項47に記載の方法。
【請求項68】
前記キャリア巨大分子が、アルギン、アルギネート、アニオン性アセテートポリマー、アニオン性アクリル酸ポリマー、キサンタンガム、アニオン性カラギーナン誘導体、アニオン性ポリガラクツロン酸誘導体、グリコール酸ナトリウムスターチ、ならびにそのフラグメント、誘導体、および薬学的に受容可能な塩からなる群より選択される、請求項47に記載の方法。
【請求項69】
乾燥した固体である、請求項47に記載の方法。
【請求項70】
液体懸濁物または半固体分散体である、請求項47に記載の方法。
【請求項71】
請求項47〜70のいずれか1項に記載の方法に従って調製された、薬学的処方物。
【請求項72】
LHRHアナログで処置可能な状態について被験体を処置する方法であって、該方法は、該被験体に、LHRHアナログとキャリア巨大分子との水不溶性複合体を含む薬学的処方物を投与する工程、
を包含する、方法。
【請求項73】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも1週間、該被験体に前記LHRHアナログの持続した送達を提供する、請求項72に記載の方法。
【請求項74】
前記水不溶性複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも2週間、該被験体に前記LHRHアナログの持続した送達を提供する、請求項72に記載の方法。
【請求項75】
前記水不溶性非共有結合複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも3週間、該被験体に前記LHRHアナログの持続した送達を提供する、請求項72に記載の方法。
【請求項76】
前記水不溶性非共有結合複合体の1回の用量が、前記薬学的組成物を被験体に投与した後少なくとも4週間、該被験体に前記LHRHアナログの持続した送達を提供する、請求項72に記載の方法。
【請求項77】
前記LHRHアナログがLHRHアンタゴニストである、請求項72に記載の方法。
【請求項78】
前記LHRHアンタゴニストが、以下の構造:Ac−D−Nal1、4−Cl−D−Phe2、D−Pal3、N−Me−Tyr5、D−Asn6、Lys(iPr)8、D−Ala10−LHRHを有する、請求項77に記載の方法。
【請求項79】
前記キャリア巨大分子がアニオン性ポリマーである、請求項72に記載の方法。
【請求項80】
前記キャリア巨大分子が、アニオン性ポリアルコール誘導体、もしくはそのフラグメント、またはそれらの薬学的に受容可能な塩である、請求項72に記載の方法。
【請求項81】
前記キャリア巨大分子が、アニオン性ポリサッカリド誘導体、もしくはそのフラグメント、またはそれらの薬学的に受容可能な塩である、請求項72に記載の方法。
【請求項82】
前記キャリア巨大分子が、カルボキシメチルセルロースまたはその薬学的に受容可能な塩である、請求項72に記載の方法。
【請求項83】
前記キャリア巨大分子が、アルギン、アルギネート、アニオン性アセテートポリマー、アニオン性アクリル酸ポリマー、キサンタンガム、アニオン性カラギーナン誘導体、アニオン性ポリガラクツロン酸誘導体、グリコール酸ナトリウムスターチ、ならびにそのフラグメント、誘導体、および薬学的に受容可能な塩からなる群より選択される、請求項72に記載の方法。
【請求項84】
前記薬学的処方物が、前記被験体に非経口経路により投与される、請求項72に記載の方法。
【請求項85】
前記薬学的処方物が、前記被験体に経口的に投与される、請求項72に記載の方法。
【請求項86】
前記薬学的処方物が、筋肉内注射および皮下/皮内注射により投与される、請求項72に記載の方法。
【請求項87】
LHRHアナログで処置可能な前記状態が、ホルモン依存性癌である、請求項72に記載の方法。
【請求項88】
前記ホルモン依存性癌が前立腺癌である、請求項87に記載の方法。
【請求項89】
LHRHアナログで処置可能な前記状態が、良性前立腺肥大、性的早熟、子宮内膜症および子宮筋腫からなる群より選択される、請求項72に記載の方法。
【請求項90】
前記LHRHアナログが、体外受精または避妊目的で投与される、請求項72に記載の方法。
【図1】

【図2】
【図3−1】
【図3−2】
【図4】
【図5】
【図2】
【図3−1】
【図3−2】
【図4】
【図5】
【公開番号】特開2011−105760(P2011−105760A)
【公開日】平成23年6月2日(2011.6.2)
【国際特許分類】
【外国語出願】
【出願番号】特願2011−24133(P2011−24133)
【出願日】平成23年2月7日(2011.2.7)
【分割の表示】特願2006−272340(P2006−272340)の分割
【原出願日】平成9年12月11日(1997.12.11)
【出願人】(506301003)プレイシス ファーマシューティカルズ インコーポレイテッド (2)
【Fターム(参考)】
【公開日】平成23年6月2日(2011.6.2)
【国際特許分類】
【出願番号】特願2011−24133(P2011−24133)
【出願日】平成23年2月7日(2011.2.7)
【分割の表示】特願2006−272340(P2006−272340)の分割
【原出願日】平成9年12月11日(1997.12.11)
【出願人】(506301003)プレイシス ファーマシューティカルズ インコーポレイテッド (2)
【Fターム(参考)】
[ Back to top ]