
改善されたキメラメガヌクレアーゼ酵素及びその使用
本発明は、増強された切断活性及び変更された配列特異性を有する変異体I-Dmo I派生物をコードするポリペプチド、並びにこれらのポリペプチドの使用に関する。これらのポリペプチドは少なくとも第1I-DmoIドメインを含み、該ペプチド配列は残基15、19及び/又は20の少なくとも1つと前記第1I-DmoIドメインの27位、29位、33位、35位、37位、75位、76位、77位、81位の少なくとも1つの置換を含む。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、改善された活性及び変化されたDNA標的配列を有する改変I-DmoIドメインを含む、キメラメガヌクレアーゼ酵素に関する。特に、本発明は、I-CreI単量体に連結された改変I-DmoIドメインを含むキメラメガヌクレアーゼ酵素に関する。
【背景技術】
【0002】
所定の遺伝子座を設計する方策の中でも、メガヌクレアーゼのような稀に切断をするDNAエンドヌクレアーゼの使用は、稀切断性(rare-cutting)DNAエンドヌクレアーゼによるDNA二本鎖破断(DSB)及び該破断の部位での相同組み換え事象をもたらすことを介する遺伝子ターゲッティングの成功率を向上させるための有力なツールとして浮上してきた。
【0003】
メガヌクレアーゼは、大きい(12〜45 bp)DNA標的部位を認識するエンドヌクレアーゼである。天然においては、メガヌクレアーゼは、非常に稀に切断をするエンドヌクレアーゼのファミリーであるホーミングエンドヌクレアーゼを実質的に含む。このファミリーは、酵母サッカロミセス セレビシエ(Saccharomyces cerevisiae)のミトコンドリアグループIイントロンにより元来コードされるタンパク質I-SceI(オメガヌクレアーゼ)のインビボでの使用により最初に特徴付けられた。イントロンのORF、独立遺伝子又は介在配列(インテイン)によりコードされるホーミングエンドヌクレアーゼは、それらを、細菌系R/MIIから一般的に単離されてきた「古典的な」制限酵素から区別する、顕著な構造的及び機能的特性を示す。
【0004】
ホーミングエンドヌクレアーゼは、12〜40 bpに及ぶDNAの認識配列を有するのに対して、「古典的な」制限酵素はもっと短いDNAの範囲、3〜8 bpの範囲を認識する(いわゆるレアカッター(rare-cutter)については12 bpまで)。したがって、ホーミングエンドヌクレアーゼは、ヒトゲノムと同程度に大きく複雑なゲノム中でさえも切断の頻度が非常に低い。
【0005】
グループIイントロン又はインテインによりコードされるいくつかのホーミングエンドヌクレアーゼは、それらの各遺伝子要素の対立遺伝子のイントロンレス又はインテインレス部位に戻ること(homing)を促進することが示されている。部位特異的な二本鎖破断をイントロンレス又はインテインレスの対立遺伝子中に作ることにより、これらのヌクレアーゼは、コード配列を複製し、DNAレベルでのイントロン又は介在配列の挿入を導く遺伝子変換過程に関与する、組み換え生成末端(recombinogenic ends)を作り出す。
【0006】
ホーミングエンドヌクレアーゼは、保存されたアミノ酸モチーフに基づいて分類された4つの別々のファミリーに属する。レビューとして、Chevalier及びStoddard(Nucleic Acids Research, 2001, 29, 3757-3774)を参照されたい。
【0007】
これらのファミリーの1つと本発明の主題は、ホーミングエンドヌクレアーゼファミリーのうちで最も大きいLAGLIDADGファミリーである。このファミリーは、保存された三次元構造(下記参照)により特徴付けられるが、上記の触媒中心である短いペプチドを除いて、一次配列レベルでは非常に不十分な保存を示す。このファミリーは、このペプチドの共通配列からLAGLIDADGと称され、各LAGLIDADGタンパク質において1つまたは2つのコピーが見出された。
【0008】
1つのLAGLIDADG (L)を有するホーミングエンドヌクレアーゼは、分子量がおよそ20 kDaであり、ホモ二量体として作用する。2つのコピーを有するもの(LL)は、25 kDa (230アミノ酸)〜50 kDa (HO、545アミノ酸)の範囲であり、各モチーフの間に70〜150残基を有し、単量体として作用する。標的配列の切断は認識部位の内側で生じ、3'OHの突出を有する4ヌクレオチドの互い違いの(staggered)切り口を残す。
【0009】
I-Ceu IおよびI-Cre I (163アミノ酸)は、1つのLAGLIDADGモチーフ(モノ- LAGLIDADG)を有するホーミングエンドヌクレアーゼである。I-DmoI (194アミノ酸、SWISSPROTアクセッション番号P21505 (配列番号22))、I-SceI、PI-PfuI及びPI-SceIは、2つのLAGLIDADGモチーフを有するホーミングエンドヌクレアーゼである。
【0010】
本発明においては特に記載しない限り、残基の番号は、SWISSPROT番号P21505 (配列番号22)又は構造PDBコード1b24のI-DmoI配列のアミノ酸番号付けを適用する。
【0011】
X線結晶解析を用いる構造モデルにより、I-CreI (PDBコード1g9y)、I-Dmo I (PDBコード1b24)、PI-Sce I、PI-Pfu Iが作り出されている。そのDNA部位に結合したI-Cre I及びPI-Sce I (Moureら、Nat Struct Biol、2002、9: 764-70)の構造も明らかになっており、特定のタンパク質-DNA接触についての多くの予測が導かれる。
【0012】
I-CreI (配列番号24)のような単独のモチーフを有するLAGLIDADGタンパク質は、ホモ二量体を形成し、パリンドローム又は偽パリンドローム(pseudo-palidromic) DNA配列を切断するのに対して、I-SceIのようなより大きい2重モチーフタンパク質は単量体であり、非パリンドローム標的を切断する。いくつかの異なるLAGLIDADGタンパク質は結晶化されており、それらは、一次配列レベルでの類似性の欠如とは対照的に、コア構造の顕著な保存を示している(Juricaら、Mol Cell. 1998; 2:469-76、Chevalierら、Nat Struct Biol. 2001; 8:312-6、Chevalierら、J Mol Biol. 2003; 329:253-69、Moureら、J Mol Biol. 2003; 334:685-95、Moureら、Nat Struct Biol. 2002; 9:764-70、Ichiyanagiら、J Mol Biol. 2000; 300:889-901、Duanら、Cell. 1997; 89:555-64、Bolduら、Genes Dev. 2003; 17:2875-88、Silvaら、J Mol Biol. 1999; 286:1123-36)。
【0013】
このコア構造において、二量体LAGLIDADGタンパク質中の2つの単量体又は単量体LAGLIDADGタンパク質中の2つのドメインが貢献する2つの特徴的なαββαββα折り畳みが、2回転対称で互いに面している。DNA結合は、逆平行βシートに折り畳まれ、かつDNAヘリックス主溝上のサドルを形成している各ドメインからの4つのβ鎖に依存する。触媒コアは中央にあり、両方の対称的単量体/ドメインの寄与を有する。このコア構造に加えて、他のドメイン:例えばPI-SceIでは、タンパク質スプライシングドメインを有するインテインと、さらなるDNA結合ドメインとを見出すことができる(Moureら、Nat Struct Biol. 2002; 9:764-70、Grindlら、Nucleic Acids Res. 1998; 26:1857-62)。
【0014】
LAGLIDADGファミリーの個々のメンバーの間での配列の保存を明らかに欠如するにもかかわらず、構造比較はLAGLIDADGタンパク質が、それらはI-CreIのような二量体又はI-DmoIのような単量体のように切断するが、同様の活性化コンフォメーションをとることを示す。全ての構造において、LAGLIDADGモチーフは中心にあり、2回転(偽-)対称軸が2つの単量体または見かけのドメインを隔てる2つのパックされたα-ヘリックスを形成する。
【0015】
LAGLIDADGモチーフは、I-CreI中の残基13〜21、並びにI-DmoI中の14〜22位及び110〜118位に該当する。LAGLIDADG α-へリックスのどちら側においても、4つのβ-シートは、タンパク質と標的DNA配列のハーフサイト(half site)との相互作用を促進するDNA結合インターフェースを提供する。I-DmoIは、第1ドメイン(残基1〜95)と第2ドメイン(残基105〜194)がリンカー(残基96〜104)により隔てられることを除いて、I-CreI二量体に類似する(Epinatら、Nucleic Acids Res、2003、31: 2952-62)。
【0016】
I-SceIは、哺乳動物細胞のゲノム標的において相同組み換えを1000倍以上に刺激するために用いられた最初のホーミングエンドヌクレアーゼであった(Choulikaら、Mol Cell Biol. 1995; 15:1968-73、Cohen-Tannoudjiら、Mol Cell Biol. 1998; 18:1444-8、Donohoら、Mol Cell Biol. 1998; 18:4070-8、Alwinら、Mol Ther. 2005; 12:610-7、Porteus、Mol Ther. 2006; 13:438-46、Rouetら、Mol Cell Biol. 1994; 14:8096-106)。
【0017】
近年、I-SceIは、マウス肝臓における標的とされた組み換えをインビボで刺激するためにも用いられ、肝細胞の1%まで組み換えを観察することができた(Goubleら、J Gene Med. 2006; 8:616-22)。そのような方法論に固有の制限は、興味対象の遺伝子座(locus)に天然のI-SceI切断部位を予め導入する必要があることである。
この制限を回避するために、過去何年にもわたって、仕立てられた切断特異性を有するジンクフィンガーヌクレアーゼを作り出す甚大な努力がなされてきた(Porteus M Hら、Nat Biotechnol. 2005; 23:967-73、Ashworthら、Nature. 2006; 441:656-9、Urnovら、Nature. 2005; 435、646-651、Smithら、Nucleic Acids Res. 2006、2006; 34:e149)。
【0018】
それらの高いレベルの特異性が与えられたなら、ホーミングエンドヌクレアーゼは、仕立てられたエンドヌクレアーゼを設計するための理想的な折り畳みを表す。いくつかの研究により、LAGLIDADGタンパク質からのDNA結合ドメインが設計され得ることが示されている(Chevalierら、Nucleic Acids Res. 2001; 29:3757-74)。
【0019】
PI-SceI (Gimbleら、J Mol Biol. 2003; 334:993-1008)、I-CreI (Seligmanら、Nucleic Acids Res. 2002; 30:3870-9、Sussmanら、J Mol Biol. 2004; 342:31-41、Rosenら、Nucleic Acids Res. 2006; Arnouldら、J Mol Biol. 2006; 355:443-58)、I-SceI (Doyonら、J Am Chem Soc. 2006; 128:2477-84) 及びI-MsoI (Ashworthら、Nature. 2006; 441:656-9)を含む、いくつかのLAGLIDADGタンパク質が、新たな配列結合又は切断特異性を得るために、合理的又は半合理的な(semi-rational)突然変異生成及びスクリーニングにより改変された。
【0020】
近年、ハイスループットスクリーニング法で補助された半合理的設計により、本出願人らは、LAGLIDADGファミリーからのホモ二量体であるI-CreIから何千もの新規タンパク質を得ることを可能としている(Smithら、Nucleic Acids Res. 2006; 34: e149; Arnouldら、J Mol Biol. 2006; 355:443-58)。
本出願人らは、I-CreIのDNA結合サブドメインを以前に同定し、これらが変異体を組み合わせて組み立てること(combinatorial assembly)を許容するのに十分に独立していたことを示している(Smithら、Nucleic Acids Res. 2006; 34: e149)。これらの発見により、選択された標的を切断する、設計されたI-CreI派生物の第二世代の産生が可能となった。
【0021】
この組み合わせの方策は、ヒトRAG1及びXPC遺伝子内に位置する天然DNA標的配列を切断するメガヌクレアーゼの創出により説明されている(Smithら、Nucleic Acids Res. 2006; 34: e149; Arnouldら、J Mol Biol. 2007; 371:49-65)。
【0022】
しかしながら、4つまでのサブドメインを組み合わせる能力により、標的となり得るDNA配列の数は顕著に増加しているが、所定のサイズの天然の標的配列にあり得る配列の完全な範囲に作用できる一組の酵素を製造することは依然として難しい。
最も分かりにくい因子の一つは、I-CreI標的部位の4つの中心のヌクレオチドの影響である。この領域において塩基特異的なタンパク質-DNA相互作用がないにもかかわらず、無作為に突然変異させた部位のライブラリーからの切断可能なI-CreI標的のインビトロ選択により、これら4つの塩基対の切断活性への重要性が明らかとなった(Argastら、J Mol Biol. 1998; 280:345-53)。より一般的には、あり得る全ての22塩基対配列を切断する、設計されたメガヌクレアーゼがI-CreI足場(scaffold)のみに由来し得ることはありそうにない。
【0023】
別の方策は、別個のメガヌクレアーゼからのドメインを組み合わせることである。このアプローチは、I-CreIとI-DmoIとの間のドメイン交換による新しいメガヌクレアーゼの創出により説明されており、2つの半分の親標的配列の融合に相当するハイブリッド配列を切断するメガヌクレアーゼの創出へと導いている(Epinatら、Nucleic Acids Res. 2003; 31:2952-62, Chevalierら、Mol. Cell. 2002; 10:895-905)。
【0024】
I-DmoIは、超好熱性古細菌デサルフロコッカス・モビリス(Desulfurococcus mobilis)からの22 kDaのエンドヌクレアーゼである。それは、どちらもLAGLIDADGモチーフを有する、2つの類似のドメインを含む単量体タンパク質である。以下D1234(配列番号30)と称するDNA標的なしの該タンパク質単独の構造は解明されている(Silvaら、J Mol Biol. 1999; 286:1123-36)。
【0025】
Chevalierらの研究グループ(Mol Cell. 2002; 10:895-905)は、2つのエンドヌクレアーゼI-DmoI及びI-CreIに基づいて、E-DreI(設計された(Engineered) I-DmoI/I-CreI)と呼ばれるキメラタンパク質を組み立てた。E-DreIは、該酵素の元の(initial)足場を作り出すための柔軟なリンカーにより連結されたI-CreIホモ二量体の単一のサブユニットへのI-DmoIのN末端ドメインの融合体からなる。そして、Chevalierらは、予め作成したI-DmoI及びI-CreIモデルの要素を組み合わせることにより作成した3Dモデルから予測される、いずれの潜在的な立体障害(steric clashes)も緩和できるように、コンピュータを利用した界面アルゴリズムの予測に基づいて多数の残基改変物を作った。
上記のChevalierら、2002において、2つの構成分子が面している表面間の残基は同定された;特に、E-DreI足場の47位、51位、55位、108位、193位及び194位の残基は、潜在的に障害することが同定された。これらの残基はアラニンに置き換えられたが、そのような改変タンパク質は不溶性であることが見出された。
【0026】
残基番号は、天然のI-DmoIリンカーの長さを模倣する3アミノ酸NGNリンカーにより隔てられるI-CreIの最後の156残基に融合したI-DmoIのN末端ドメインから101残基(第1メチオニンで始まる)を含むE-DreIオープンリーディングフレームを参照する。
そして、残基47、51、55、108、193及び194、並びに残基12、13、17、19、52、105、109及び113のコンピュータを利用した再設計と;それに続く、これらの残基において改変されたE-DreI酵素の可溶性を決定するための、選択された配列に対するインビボでのタンパク質折り畳みアッセイを介して、界面が最適化された。最終足場が、I-DmoIのI19、H51及びH55並びにI-CreIのE8、L11、F16、K96及びL97 (E105、L108、F113、K193及びL194に該当する)の改変で設計された。
【0027】
キメラDNA標的dre3(本出願人の命名を用いるとC12D34 (配列番号31))との複合体中のE-DreI (Chevalierら、Mol Cell. 2002; 10:895-905)の構造は、本明細書の図2中に示されるように解明されている。E-DreIは、このハイブリッドC12D34 (配列番号31)標的のみを認識し切断できることが示された。この構造から、多数の残基が、E-DreIのその標的ハイブリッド部位への塩基特異的接触であることが予測され、これらの残基はE-DreI中のI-DmoIの25、29、31、33、34、35、37、70、75、76、77、79、81であり;及びE-DreI中のI-CreIの残基123、125、127、130、135、137、139、141、163、165、167、172であった。
【0028】
本出願人も、設計されたホーミングヌクレアーゼ酵素により切断されるDNA標的配列の範囲を広げようとするためにDmoCre足場についての実験をこれまでに行っている。DmoCreは、2つのホーミングエンドヌクレアーゼI-DmoI及びI-CreIから組み立てられたキメラ分子である。それは、I-CreI単量体に連結したI-DmoIからのN末端部分を含む。DmoCreは足場としてすばらしい利点を有し得る:I-DmoI部分における変異は、I-CreIドメインにおける変異と組み合わせることができ、かつ数千のそのような種々のI-CreI分子はすでに同定され、プロファイルされている (Smith Jら、Nucleic Acids Res. 2006;34(22):e149 , Arnould Sら、J Mol Biol. 2006; 355:443-58, Arnould Sら、J Mol Biol. 2007; 371:49-65)。
【0029】
DNA標的なしのI-DmoIタンパク質単独の構造(Silvaら、J Mol Biol. 1999; 286:1123-36)及びI-CreIとそのDNA標的C1234 (配列番号28)との複合体の構造(Juricaら、Mol Cell. 1998; 2:469-76、Chevalierら、J Mol Biol. 2003; 329:253-69)に基づいて、キメラDmoCreエンドヌクレアーゼが組み立てられている(Epinatら、Nucleic Acids Res, 2003, 31: 2952-62)。DmoCreは、残基F109までのI-DmoIとそれに続く残基L13からのI-CreIに相当する単量体タンパク質である。立体障害を避けるために、I107はロイシン残基に変異されている。さらに、I-CreIの残基96及び97に近いことが見出されたI-DmoIの残基47、51及び55は、それぞれアラニン、アラニン及びアスパラギン酸に変異された。
【0030】
DmoCreは、インビトロで活性であることが示されており(Epinatら、Nucleic Acids Res, 2003, 31: 2952-62)、C1234 (配列番号28)又はC1221 (配列番号29) (C1234に由来するパリンドローム標的)の左部分及びD1234 (配列番号30)の右部分から構成されるハイブリッド標的C12D34 (配列番号31)を切断できた(図1)。さらに、それらのDNA標的配列を37°Cでより効率的に切断できるI-DmoI及びDmoCre変異型が、酵母細胞における無作為突然変異生成及びスクリーニングにより同定された(WO 2005/105989; Prietoら、J. Biol Chem. 2007 Nov 12; [出版に先んじるEpub])。
【0031】
それゆえ、E-DreI及びDmoCreキメラ酵素のみが、ハイブリッド標的C12D34 (配列番号31)を認識して切断できる。また、E-DreI及びDmoCreの足場は、共通して残基47、51及び55の改変を有する。
【発明の概要】
【発明が解決しようとする課題】
【0032】
本発明者らは、より広い一連の標的配列を認識する新世代のキメラ酵素を作り出すことに興味があり、それゆえ彼らは、キメラI-DmoI酵素における構成要素又は2つの異なるヌクレアーゼからの触媒ドメインを含むキメラ酵素のいずれかとして用いるためのI-DmoI酵素の第1ドメインのさらなる増強を研究した。新しいDNA配列を標的にできるようにし、そしてDNA標的配列を含む興味対象の部位に二本鎖破断を生じさせ得るようにすることにより、本出願人は、DNA組み換え事象、DNA喪失または細胞死を引き起こすことができるツールを提供する。
【0033】
この二本鎖破断は、特定の配列を修復し、特定の配列を改変し、変異した遺伝子に代えて機能的遺伝子を回復し、興味対象の内因性遺伝子を弱めるか又は活性化し、興味対象の部位に変異を導入し、外因性遺伝子又はその一部を導入し、内因性遺伝子若しくはその一部を不活性化するか又は検出し、染色体腕を転移させ、或いは未修復及び分解したDNAを放出するために用いることができる。それゆえ、そのような改変されたメガヌクレアーゼ酵素は、ユーザーにそのような改変されたメガヌクレアーゼ酵素の治療、研究または他の生産用途におけるより広い様々な潜在的な選択肢を与える。
【0034】
それゆえ、本発明者らは、これらのキメラ酵素が認識し切断できるDNA標的の数を増やそうとすることにより、少なくとも1つのI-DmoIドメインを含むキメラメガヌクレアーゼ酵素を改善しようと努めた。
【課題を解決するための手段】
【0035】
したがって、本発明は、少なくとも第1I-DmoIドメインを含み、該第1I-DmoIドメインの残基15、19又は20の少なくとも1つの置換及び27位、29位、33位、35位、37位、75位、76位、77位又は81位の残基の少なくとも1つの置換を含むことを特徴とし、±2位、±3位、±4位、±5位、±6位、±7位、±8位、±9位、±10位の少なくとも1つにおいて配列番号30の野生型I-DmoI DNA標的ハーフサイトとは異なるI-DmoI DNA標的ハーフサイトを認識する、I-DmoIエンドヌクレアーゼ又はそれらのキメラ派生物の配列を含むポリペプチドに関する。
【図面の簡単な説明】
【0036】
【図1】異なる22 bpのDNA標的を示す配列比較である。
【図2】DNA標的と結合しているE-DreIの構造を示す。
【図3】(A)DNA標的と結合しているE-DreIの分子表面を示す。(B)DNAとの相互作用している残基29、33及び35を示す拡大図。
【図4】pCLS1055の概略の制限マップ。
【図5】pCLS0542の概略の制限マップ。
【図6】8 DC10NNN標的に対するDClib2ライブラリーからのDmoCre2変異体の1次スクリーニングの例を示す。
【図7】DmoCre2のヒットマップ及び64 DC10NNN標的に対するDClib2ライブラリーを示す。
【図8】(A)DNA標的と結合しているE-DreIの分子表面。(B)DNAとの相互作用している残基75、76及び77を示す拡大図である。
【図9】DmoCre4及び64 DC4NNN標的に対するDClib4ライブラリーのヒットマップ。
【図10】いくつかの異なる22 bpのDNA標的を示す。
【図11】5CAGD34標的(配列番号33)に対するDCSca2_5CAG変異体及びDCSca4_5CAG変異体の1次スクリーニングの例を表す。
【図12】異なる22 bpのDNA標的を示す。
【図13】RAG1.10.2D34標的(配列番号35)、RAG1.10.2 (配列番号34)、C12D34 (配列番号31)、D1234 (配列番号30)及びC1221 (配列番号29) DNA標的に対するDmoM2変異体の酵母切断アッセイ。
【図14】異なる22 bpのDNA標的を示す。
【図15】それぞれの各標的に対するRAG1.10.2D34カッター及びRAG1.10.3D34カッターの2次スクリーニングの例を表す。
【図16】それぞれの標的に対する精製されたRAG1.10.2D34カッター及びRAG1.10.3D34カッターの2次スクリーニング。
【図17】64 DC4NNN標的に対するD4Clib2Bライブラリーのヒットマップ。
【図18】(A)自身のDNA標的と結合しているE-DreIの分子表面を示す。(B)DNAとの相互作用している残基37及び81を示す拡大図である。(C)残基37の付近にある残基27を示す別の拡大図である。
【図19】64 DC7NNN標的に対するD7Clib2ライブラリーのヒットマップ。
【図20】組み合わされたDC10TGG4ACTに対するSeqDC10NNN4ACTライブラリーからのDmoCre2変異体の1次スクリーニングの例を表す。
【図21】RAG1.10.3DC4ACT標的(A)及びRAG1.10.3DC4TAT標的(B)に対するRAG1.10.3DC4NNNライブラリーからの変異体の1次スクリーニングの例を表す。
【発明を実施するための形態】
【0037】
この明細書を通じて、記載されるDmoCreキメラ酵素は、クローニング手段のために2位にバリンを含む。この付加的残基は、キメラ酵素配列内の残基の番号付けには含まれない。それゆえ、例えばキメラ酵素の19位の残基は、実際にはこのキメラ酵素の第20番目の残基である。
【0038】
本発明者らは、I-DmoIの第1ドメインと別の機能的エンドヌクレアーゼドメイン若しくは単量体との組み合わせを含むキメラ酵素のような、改善されたI-DmoIエンドヌクレアーゼ又はその派生物をコードするポリペプチドを提供する。このポリペプチドは、天然のI-DmoIタンパク質の残基1〜95に該当する第1I-DmoIドメインにおいて2つ以上のアミノ酸残基の変異(changes)を有する。特に、第1I-DmoIドメインはI-DmoIアミノ酸配列(配列番号22)の1位〜95位に該当し、I-DmoIリンカーは96位〜104位に該当し、第2I-DmoIドメインの開始部(beginning)は105位〜109位に該当し、これは本出願人が開発し、本明細書に記載した2つの新しいキメラメガヌクレアーゼ足場である、DmoCre2及びDmoCre4に用いられた完全断片である。好ましくは、完全な109残基の断片は、第1I-DmoIドメイン断片としてキメラ酵素中に用いられる
【0039】
残基15、19及び20の変異は、本発明者らによりDmoCre2と呼ばれるキメラタンパク質の活性が増加する結果になることが、本発明者らによる実験により示されている。残基29、33及び35の変異は、I-DmoI DNA標的ハーフサイト(配列番号30)の±8位〜±10位にて、I-DmoIのこの改変されたドメインにより認識される配列を変化させることが、本出願人により初めて示されている。残基75、76及び77の変異は、I-DmoI DNA標的ハーフサイト(配列番号30)の±2位〜±4位にて、I-DmoIのこの改変されたドメインにより認識される配列を変化させることが、本発明者らにより初めて示されている。残基27、37及び81の変更は、I-DmoI DNA標的ハーフサイト(配列番号30)の±5位〜±7位にて、I-DmoIのこの改変されたドメインにより認識される配列を変化させることが、本発明者らにより初めて示されている。したがって、本発明者らは、ハイブリッド配列C12D34とは異なる標的配列を認識できる改善された第1I-DmoIドメインを提供する。該I-DmoI DNA標的ハーフサイト(配列番号30)はAAGTTCCGGCGであり、+2〜+4及び+8〜+10の領域は太字で、+5〜+7の領域は斜字である。
【0040】
そのようなポリペプチドは、改変されたメガヌクレーゼを含み、DmoCre及びE-DreIにより認識され切断されるハイブリッド標的配列以外の、より広い範囲のDNA標的配列を認識し切断できるようにする。
【0041】
特に、15位、19位又は20位の残基の少なくとも1つが任意のアミノ酸に置換される。
特に、本発明によるポリペプチドは、グルタミンへ変異される15位のリジンの改変である、L15Q変異を含み得る。
特に、本発明によるポリペプチドは、アスパラギン酸へ変異される19位のイソロイシンの改変である、I19D変異を含み得る。残基19の改変により、I-DmoIドメインをより活性にすることが本出願人によって示されている。
特に、本発明によるポリペプチドは、セリン又はアラニンへ変異される20位のグリシンの改変である、G20S又はG20A変異を含み得る。
【0042】
特に、ポリペプチドは、107位において少なくとも1つの改変された残基も含み得る。
特に、本発明によるポリペプチドは、リジンへ変更される107位のイソロイシンの改変である、I107L改変を含み得る。残基107の改変は、I-DmoIドメインと、他の酵素、例えばI-CreIのドメインとの間の立体障害を防ぐだろう。
【0043】
特に、任意のアミノ酸による29位、33位又は35位の残基の少なくとも1つの置換は、±8位、±9位、±10位の少なくとも1つにおいて配列番号30の野生型I-DmoI DNA標的ハーフサイトとは異なるI-DmoI DNA標的ハーフサイトの該ポリペプチドの認識を変化させる。
【0044】
特に、任意のアミノ酸による75位、76位又は77位の残基の少なくとも1つの置換は、±2位、±3位、±4位の少なくとも1つにおいて配列番号30の野生型I-DmoI DNA標的ハーフサイトとは異なるI-DmoI DNA標的ハーフサイトの該ポリペプチドの認識を変化させる。
【0045】
特に、任意のアミノ酸による27位、37位又は81位の残基の少なくとも1つの置換は、±5位、±6位、±7位の少なくとも1つにおいて配列番号30の野生型I-DmoI DNA標的ハーフサイトとは異なるI-DmoI DNA標的ハーフサイトの該ポリペプチドの認識を変化させる。
【0046】
特に、該ポリペプチドは、配列番号1の配列に由来する。
本願において、に由来するとは、元の配列から作り出され、そして元の機能性を保持するように改変されるが、元の機能性を保持するかぎり、元の配列と比較して残基の変異及び/又は付加若しくは欠失を有する、任意の核酸又はタンパク質の配列を意味する。
配列番号1は、DmoCre2 (配列番号2)中のI-DmoIドメインとして本発明で用いられた残基15及び19にて改変されたI-DmoIドメインの配列である。このI-DmoIドメインも残基107への改変を含むが、Epinatら(Nucleic Acids Res、2003、31: 2952-62)のようなL47A、H51A及びL55Dへの改変は含まない。
【0047】
特に、該ポリペプチドは、配列番号27の配列に由来する。
配列番号27は、DmoCre4 (配列番号9)中のI-DmoIドメインとして本発明で用いられる残基19、20及び109にて改変されたI-DmoIドメインの配列である。このI-DmoIドメインは、Epinatら(Nucleic Acids Res、2003、31: 2952-62)のようなL47A、H51A及びL55Dへの改変は含まない。
【0048】
特に、該ポリペプチドは、二量体LAGLIDADGホーミングエンドヌクレアーゼの配列又は別の単量体LAGLIDADGホーミングエンドヌクレアーゼのドメインへの該第1I-Dmo Iドメインの融合体からなるキメラI-Dmo Iエンドヌクレアーゼである。
【0049】
本発明は、単一の単量体タンパク質における配列番号22の残基1〜95、或いは本発明により変化された2つのI-DmoIドメインの組み合わせを含む、改変された第1I-DmoIドメイン及び第2野生型I-DmoIドメインの両方を含む、改変されたI-DmoIエンドヌクレアーゼ酵素に関する。改変されたI-DmoIドメインが、I-Sce I、I-Chu I、I-Cre I、I-Csm I, PI-Sce I, PI-Tli I、PI-Mtu I、I-Ceu I、I-Sce II、I-Sce III、HO、PI-Civ I、PI-Ctr I、PI-Aae I、PI-Bsu I、PI-Dha I、 PI-Dra I、PI-Mav I、PI-Mch I、PI-Mfu I、PI-Mfl I、PI-Mga I、PI-Mgo I、PI-Min I、PI-Mka I、PI-Mle I、PI-Mma I、PI-Msh I、PI-Msm I、PI-Mth I、PI-Mtu I、PI-Mxe I、PI-Npu I、PI-Pfu I、PI-Rma I、PI-Spb I、PI-Ssp I、PI-Fac I、PI-Mja I、PI-Pho I、PI-Tag I、PI-Thy I、PI-Tko I、I-MsoI及びPI-Tsp I;好ましくは、I-Sce I、I-Chu I、I-Dmo I、I-Csm I、PI-Sce I、PI-Pfu I、PI-Tli I、PI-Mtu I、及びI-Ceu Iのような別のLAGLIDADGエンドヌクレアーゼのドメインと組み合わされ得ることも本発明の一面である。
【0050】
また、本発明は、I-DmoIの第1ドメインの配列が、(i) 上記の第1I-Dmo Iドメインの4位、49位、52位、92位、94位及び/若しくは95位の残基の1つ、並びに/或いは(ii) I-Dmo Iの第2ドメインのリンカー又は開始部の101位、102位及び/若しくは109位の残基の1つ
の群から選択される少なくとも1つのさらなる残基の置換も含むポリペプチドに関する。
【0051】
上記のポリペプチドの有利な実施形態によれば:
- 4位のアスパラギンがイソロイシンに変異され(N4I)、
- 49位のリジンがアルギニンに変異され(K49R)、
- 52位のイソロイシンンがフェニルアラニンに変異され(I52F)、
- 92位のアラニンがスレオニンに変異され(A92T)、
- 94位のメチオニンがリジンに変異され(M94K)、
- 95位のロイシンがグルタミンに変異され(L95Q)、
-(存在する場合)101位のフェニルアラニンがシステインに変異され(F101C)、
-(存在する場合)102位のアスパラギンがイソロイシンに変異され(N102I)、及び/又は
-(存在する場合)109位のフェニルアラニンがイソロイシンに変異される(F109I)。
【0052】
特に、該ポリペプチドの第1I-Dmo Iドメインは、上記のキメラ-DmoエンドヌクレアーゼのNH2末端にある。
特に、キメラ-Dmoエンドヌクレアーゼの一部を形成する二量体LAGLIDADGホーミングエンドヌクレアーゼは、I-CreIである。
【0053】
特に、キメラI-Dmo Iエンドヌクレアーゼは、配列番号2の配列に由来する。
配列番号2は、残基15、19及び107にて改変されたI-Dmo Iドメインを含む、本発明の好ましいDmoCre2キメラエンドヌクレアーゼのペプチド配列である。
【0054】
特に、本発明によるポリペプチドは、配列番号9の配列に由来する。
配列番号9は、残基19、20及び109にて改変されたI-Dmo Iドメインを含む、本発明の好ましいDmoCre4キメラエンドヌクレアーゼのペプチド配列である。
【0055】
特に、本発明のこの第一面によるポリペプチドは、検出可能なタグをNH2末端及び/又はCOOH末端に含み得る。
【0056】
本発明はポリヌクレオチドにも関し、このポリヌクレオチドは本発明によるポリペプチドをコードすることを特徴とする。
【0057】
本発明は、本発明によるポリヌクレオチドを含むことを特徴とする、ベクターにも関する。
本発明は、本発明によるポリヌクレオチド又はベクターにより改変されることを特徴とする、宿主細胞にも関する。
上記のポリヌクレオチドを含む組み換えベクターは、公知の組み換えDNA及び遺伝子工学的技術により得ることができ、宿主細胞に導入できる。
【0058】
本発明のポリペプチドは、前記ポリペプチドの発現に適切な条件下で、該ポリペプチドをコードするポリヌクレオチド配列を含む発現ベクターを有する宿主細胞を培養し、該宿主細胞培養物から該ポリペプチドを回収することにより得ることができる。
【0059】
本発明は、本発明によるポリヌクレオチド又ベクターにより、その構成細胞の全て又は一部が改変されることを特徴とする、非ヒトトランスジェニック動物にも関する。
本発明は、本発明によるポリヌクレオチド又ベクターにより、その構成細胞の全て又は一部が改変されることを特徴とする、トランスジェニック植物にも関する。
【0060】
本発明は、第1I-DmoIドメインの残基15、19、20の少なくとも1つの置換及び27位、29位、33位、35位、37位、75位、76位、77位又は81位の残基の少なくとも1つの置換からなる該第1I-DmoIドメインを少なくとも含み、I-CreI単量体の配列に融合され、該I-CreI単量体配列が前記I-CreI単量体の44位、68位、70位、75位又は77位の残基の少なくとも1つの改変を含む、ポリペプチドにも関する。
【0061】
I-CreI単量体における残基番号の参照は、参照I-CreI単量体配列の配列番号24に適用する。そのようなポリペプチドは、例えば5CAGD34 (配列番号33)標的を切断できる。5CAGD34 (配列番号33)は、I-DmoI標的DNA配列(配列番号30)の第2ハーフに融合された5CAG_P標的(配列番号32)の第1ハーフである。5CAG_P標的(配列番号32)は、±3位、±4位及び±5位にて配列CAGへ改変された野生型I-CreI標的DNA配列に当てはまる。
全ての標的配列は、22又は24 bpのパリンドローム配列である。それゆえ、それらは、改変されたヌクレオチドに続く接尾辞_Pによってのみ記載されるだろう。
【0062】
本発明は、第1I-DmoIドメインの残基15、19、20のなくとも1つの置換及び27位、29位、33位、35位、37位、75位、76位、77位又は81位の残基の少なくとも1つの置換を含む該第1I-DmoIドメインを少なくとも含み、I-CreI単量体の配列に融合され、該I-CreI単量体配列が該I-CreI単量体の28位、30位、32位、33位、38位又は40位の残基の少なくとも1つの改変を含む、I-DmoIエンドヌクレアーゼ又はそれらのキメラ派生物の配列を含むポリペプチドにも関する。
【0063】
そのようなポリペプチドは、例えばRAG1.10.2D34標的(配列番号35)又はRAG1.10.3D34 標的(配列番号39)を切断できる。RAG1.10.2D34は、I-DmoI標的DNA配列(配列番号30)の第2ハーフに融合されたRAG1.10.2 DNA標的(配列番号34)の第1ハーフである。RAG1.10.3D34は、I-DmoI標的DNA配列(配列番号30)の第2ハーフに融合されたRAG1.10.3 DNA標的(配列番号38)の第1ハーフである。
【0064】
本発明は、第1I-DmoIドメインの残基15、19、20のなくとも1つの置換及び27位、29位、33位、35位、37位、75位、76位、77位又は81位の残基の少なくとも1つの置換にある(consisting in)該第1I-DmoIドメインを少なくとも含み、I-CreI単量体の配列に融合され、該I-CreI単量体配列が該I-CreIドメインの37位、79位、81位の残基の少なくとも1つの改変を含む、I-DmoIエンドヌクレアーゼ又はそれらのキメラ派生物の配列を含むポリペプチドにも関する。
【0065】
27位、37位又は81位が改変される場合、そのようなポリペプチドは、C12D34 (配列番号31) DNA標的配列の+5〜+7であるDmoCreの7NNN部分が野生型ヌクレオチド配列標的CGAとは異なっている標的を切断できる。
【0066】
本発明のよりよい理解のために、またどのようにして同じく実施するのかを示すために、添付の図面を参照して、本発明による特定の実施形態、方法及びプロセスを一例としてのみ示される:
【0067】
図1:異なる22 bpのDNA標的を示す配列比較であって、C1234 (配列番号28)は野生型I-CreI標的であり、C1221 (配列番号29)はC1234 (配列番号28)の左部分に由来するパリンドローム配列であり、D1234 (配列番号30)は野生型I-DmoI標的であり、そしてC12D34 (配列番号31)はキメラDmoCreタンパク質のハイブリッド標的であり、またDC10NNN標的(配列番号8)は+8位、+9位及び+10にて縮重が導入されたC12D34 (配列番号31)からの派生物である。
【0068】
図2:DNA標的と結合しているE-DreIの構造を示す(PDBコード1MOW)。
図3A:DNA標的と結合しているE-DreIの分子表面を示す。
図3B:DNAとの相互作用している残基29、33及び35を示す拡大図。点線は水素結合を示す。
図4:pCLS1055の概略の制限マップ。
図5:pCLS0542の概略の制限マップ。
図6:8 DC10NNN標的に対するDClib2ライブラリーからのDmoCre2変異体の1次スクリーニングの例を示す。
【0069】
図7:DmoCre2のヒットマップ及び64 DC10NNN標的に対するDClib2ライブラリーを示す。ヒットマップ中に示される各標的は、C12D34 (配列番号31)の相補鎖である;例えばヒットマップ中のCGGは、以下の実施例2で定義されるDC10CCGに相当する。
【0070】
図8:DNA標的と結合しているE-DreIの分子表面(図8A)。無作為化のために選択された結合の領域(+2位、+3位、+4位の塩基対並びにタンパク質残基75、76及び77)は赤で強調されている。図8Bは、DNAとの相互作用している残基75、76及び77を示す拡大図である。点線は水素結合を示す。
【0071】
図9:DmoCre4及び64 DC4NNN標的(DC4TTCはない)からの63に対するDClib4ライブラリーのヒットマップ。D4Clib4ヒットマップにおいて、それぞれの切断された標的の下の数は、この標的を切断するクローンの数である。これらのクローンのいくつかは、重複した配列を有し得る。各標的において、灰色レベルは切断強度の平均に比例する。ヒットマップ中に示される各標的は、C12D34 (配列番号31)の相補鎖である;例えばヒットマップ中のAGGは、以下の実施例3で定義されるDC4CCTに相当する。
【0072】
図10:いくつかの異なる22 bpのDNA標的を示す。5CAG_P (配列番号32)パリンドローム標的は、灰色のボックスで強調される±5位、±4位、±3位において相違を有するC1221に由来する。I-CreI標的部分において、5CAGD34 (配列番号33)標的は、5CAG_P (配列番号32)がC1221とは異なるのと同様に、C12D34 (配列番号31)とは異なる。
【0073】
図11:この図は、5CAGD34標的(配列番号33)に対するDCSca2_5CAG変異体及びDCSca4_5CAG変異体の1次スクリーニングの例を表す。DCSca2_5CAGライブラリースクリーニングでは、我々は9ドットクラスタ形式を用いた。各9ドット酵母クラスタにおいて、異なる変異体が、左上のドットの5CAGD34 (配列番号33)標的に対して試験される。DCSca4_5CAGライブラリースクリーニングでは、我々は4ドットクラスタ形式を用いた。各4ドットクラスタにおいて、異なる変異体が、左の2ドットの5CAGD34 (配列番号33)標的に対して試験され、一方、右の2ドットは内部対照クラスタである。H10、H11及びH12クラスタは陽性対照及び陰性対照を含む。
【0074】
図12:異なる22 bpのDNA標的を示す。RAG1.10.2 DNA配列(配列番号34)は、C1221 (配列番号29)に由来するパリンドローム標的である。10GTT及び5CAGモジュールは灰色のボックスで強調されている。I-CreI標的部分では、RAG1.10.2D34標的(配列番号35)は、RAG1.10.2 (配列番号32)がC1222とは異なるのと同様に、C12D34 (配列番号31)とは異なる。
【0075】
図13:RAG1.10.2D34標的(配列番号35)、RAG1.10.2 (配列番号34)、C12D34 (配列番号31)、D1234 (配列番号30)及びC1221 (配列番号29) DNA標的に対するDmoM2変異体の酵母切断アッセイ。各4ドット酵母クラスタにおいて、左の2ドットは、表示された標的を有するDmoM2変異体の切断アッセイを示し、一方、右の2ドットは内部対照である。
【0076】
図14:異なる22 bpのDNA標的を示す。RAG1.10.3 DNA配列は、C1221に由来するパリンドローム標的である。10 TGG及び5 GAGモジュールは灰色のボックスで強調されている。I-CreI標的部分では、RAG1.10.3D34標的(配列番号35)は、RAG1.10.3がC1222とは異なるのと同様に、C12D34とは異なる。
【0077】
図15:この図は、それぞれの各標的に対するRAG1.10.2D34カッター及びRAG1.10.3D34カッターの2次スクリーニングの例を表す。RAG1.10.2D34標的では、異なる変異体が、左上の各酵母クラスタにおけるその標的に対して試験される。RAG1.10.3D34標的では、異なる変異体が、左下の各酵母クラスタにおけるその標的に対して試験される。各4ドットクラスタにおいて、右の2ドットは内部対照クラスタである。
【0078】
図16:それぞれの標的に対する精製されたRAG1.10.2D34カッター及びRAG1.10.3D34カッターの2次スクリーニング。試験設計が示されている。RAG1.10.2D34スクリーニングでは、最初の変異体はRG2D2であり、一方、RAG1.10.3D34スクリーニングでは、それはRG3D3である。精製されたRAG1.10.2D34カッターはA9、B3、C5に位置し、黒の円はそれぞれAmel1_RG2D、Amel2_RG2D、Amel3_RG2Dである。精製されたRAG1.10.3D34カッターはA8、B3、E3に位置し、黒の円はそれぞれAmel1_RG3D、Amel2_RG3D、Amel3_RG3Dである。
【0079】
図17:64 DC4NNN標的に対するD4Clib2Bライブラリーのヒットマップ。それぞれの切断された標的の下の数は、この標的を切断する異なる配列を有するDmoCre2変異体の数である。各標的において、灰色レベルは切断強度の平均に比例する。ヒットマップ中に示される各標的は、C12D34 (配列番号31)の相補鎖である;例えばヒットマップ中のCAGは、実施例3で定義されるDC4CTGに相当する。
【0080】
図18:DNA標的と結合しているE-DreIの分子表面を示す(図17A)。無作為化のために選択された結合の領域(+5位、+6位、+7位の塩基対ならびにタンパク質残基37及び81)は黒で強調されている。図16Bは、DNAとの相互作用している残基37及び81を示す拡大図である。点線は水素結合を示す。図16Cは、残基37の付近にある残基27を示す別の拡大図である。
【0081】
図19:64 DC7NNN標的に対するD7Clib2ライブラリーのヒットマップ。それぞれの切断された標的の下の数は、この標的を切断する異なる配列を有するDmoCre2変異体の数である。各標的において、灰色レベルは切断強度の平均に比例する。ヒットマップ中に示される各標的は、C12D34 (配列番号31)の相補鎖を参照する;例えばヒットマップ中のGGAは、実施例9で定義されるDC7TCCに相当する。
【0082】
図20:この図は、組み合わされたDC10TGG4ACT標的に対するSeqDC10NNN4ACTライブラリーからのDmoCre2変異体の1次スクリーニングの例を表す。各酵母クラスタにおいて、右の2ドットは実験の内部対照である。他の4ドットにおいて、1ドットはSeqDC10NNN4ACTライブラリーからの1つの変異体に相当する。3つの陽性クローンは黒丸である。
【0083】
図21:この図は、RAG1.10.3DC4ACT標的(A)及びRAG1.10.3DC4TAT標的(B)に対するRAG1.10.3DC4NNNライブラリーからの変異体の1次スクリーニングの例を表す。各酵母クラスタにおいて、右上のドットはAmel2_RG3D変異体に相当し、右下のドットは実験の内部対照である。他の4ドットにおいて、1ドットはRAG1.10.3DC4NNNライブラリーからの1つの変異体に相当する。いくつかの陽性クローンは黒丸である。
【0084】
ここに記載されるものは、発明者らにより意図される特定の形態の一例である。以下の記載において、多数の特定の詳説は完全な理解を提供するために記述される。しかしながら、本発明がこれらの特定の詳説に限定されることなく実施され得ることは当業者に明らかであろう。他の例では、記載を不必要にあいまいにしないようにするため、公知の方法及び構造は記載されない。
【0085】
定義
- ポリペプチド配列中のアミノ酸残基は、本明細書において、1文字コードに従って表し、例えばQはGln又はグルタミン残基を意味し、RはArg又はアルギニン残基を意味し、DはAsp又はアスパラギン酸残基を意味する。
- 疎水性アミノ酸は、ロイシン(L)、バリン(V)、イソロイシン(I)、アラニン(A)、メチオニン(M)、フェニルアラニン(F)、トリプトファン(W)及びチロシン(Y)のことである。
【0086】
- ヌクレオチドは、次のように表す:1文字コードは、ヌクレオシドの塩基を表すために用いる:aはアデニンであり、tはチミンであり、cはシトシンであり、gはグアニンである。縮重ヌクレオチドについて、rはg又はa (プリンヌクレオチド)を表し、kはg又はtを表し、sはg又はcを表し、wはa又はtを表し、mはa又はcを表し、yはt又はc (ピリミジンヌクレオチド)を表し、dはg、a又はtを表し、vはg、a又はcを表し、bはg、t又はcを表し、hはa、t又はcを表し、nはg、a、t又はcを表す。
【0087】
- 「メガヌクレアーゼ」により、12〜45 bpの二本鎖DNA標的配列を有するエンドヌクレアーゼを意図する。
- 「親のLAGLIDADGホーミングエンドヌクレアーゼ」により、野生型ホモ二量体LAGLIDADGホーミングエンドヌクレアーゼ、又はその機能的変異型を意図する。上記の親のLAGLIDADGホーミングエンドヌクレアーゼは、22〜24 bpの二本鎖DNA標的を切断できる機能的エンドヌクレアーゼに組み合わされる、単量体、2つのLAGLIDADGホーミングエンドヌクレアーゼのコアドメインを含む二量体(ホモ二量体又はヘテロ二量体)であり得る。
【0088】
- 「ホモ二量体LAGLIDADGホーミングエンドヌクレアーゼ」により、単独LAGLIDADGモチーフを有し、パリンドロームDNA標的配列を切断するI-CreI若しくはI-MsoIのような野生型ホモ二量体LAGLIDADGホーミングエンドヌクレアーゼ、又はその機能的変異型を意図する。
- 「LAGLIDADGホーミングエンドヌクレアーゼ変異型」又は「変異型」により、LAGLIDADGホーミングエンドヌクレアーゼ配列の少なくとも1つのアミノ酸を、異なるアミノ酸で置き換えることにより得られるタンパク質を意図する。
【0089】
- 「機能的変異型」により、DNA標的、好ましくは、野生型LAGLIDADGホーミングエンドヌクレアーゼにより切断されない新しいDNA標的を切断できるLAGLIDADGホーミングエンドヌクレアーゼ変異型を意図する。例えば、このような変異型は、DNA標的配列に接触するか、又は直接若しくは間接的に該DNA標的と相互作用する位置にてアミノ酸変異を有する。
【0090】
- 「新規な特異性を有するホーミングエンドヌクレアーゼ変異型」により、親のホーミングエンドヌクレアーゼのものとは異なる切断されるDNA標的のパターン(切断プロフィール)を有する変異型を意図する。変異型は、親のホーミングエンドヌクレアーゼより少ない(制限されたプロフィール)又はより多い標的を切断できる。好ましくは、変異型は、親のホーミングエンドヌクレアーゼにより切断されない少なくとも1つの標的を切断できる。
等価で、同様に用いられる用語「新規な特異性」「改変された特異性」「新規な切断特異性」「新規な基質特異性」は、DNA標的配列のヌクレオチドに対する変異型の特異性のことである。
【0091】
- 「I-CreI」により、配列SWISSPROT P05725又はpdbアクセッションコード1g9y (配列番号24)を有する野生型I-CreIを意図する。
- 「I-DmoI」により、配列SWISSPROT番号P21505 (配列番号22)又は構造PDBコード1b24を有する野生型I-DmoIを意図する。
【0092】
- 「ドメイン」又は「コアドメイン」により、約100アミノ酸残基の配列に相当する、LAGLIDADGファミリーのホーミングエンドヌクレアーゼの特徴的なαββαββα折り畳みである「LAGLIDADGホーミングエンドヌクレアーゼコアドメイン」を意図する。該ドメインは、DNA標的の一方の半分と相互作用する逆平行ベータシートに折り畳まれる4つのベータ鎖を含む。このドメインは、DNA標的の他方の半分と相互作用する別のLAGLIDADGホーミングエンドヌクレアーゼコアドメインと会合して、該DNA標的を切断できる機能エンドヌクレアーゼを形成できる。例えば、二量体ホーミングエンドヌクレアーゼI-CreI (163アミノ酸)の場合、LAGLIDADGホーミングエンドヌクレアーゼコアドメインは、残基6〜94に相当する。単量体ホーミングエンドヌクレアーゼの場合、2つのこのようなドメインが、エンドヌクレアーゼの配列中に見出される;例えば、I-DmoI (194アミノ酸)、第1ドメイン(少なくとも残基1〜95及び第2ドメイン(残基105〜194)がリンカー(残基96〜104)によって隔てられている。
【0093】
- 「サブドメイン」により、ホーミングエンドヌクレアーゼDNA標的ハーフサイトの独特の(distinct)部分と相互作用するLAGLIDADGホーミングエンドヌクレアーゼコアドメインの領域を意図する。
- 「ベータヘアピン」により、ループ又はターンにより接続されたLAGLIDADGホーミングエンドヌクレアーゼコアドメインの逆並行ベータシートの2つの連続するベータ鎖を意図する。
- 「C1221」により、パリンドローム「21」を形成するようにI-CreI標的部位「12」が逆に反復される第1のハーフのことであることを意図する。
【0094】
- 「切断活性」により、本発明の変異型の切断活性は、PCT出願WO 2004/067736; Epinatら、Nucleic Acids Res., 2003, 31, 2952-2962; Chamesら、Nucleic Acids Res., 2005, 33, e178及びArnouldら、J. Mol. Biol., 2006, 355, 443-458に記載されるように、レポーターベクターを用いる酵母又は哺乳動物細胞における直列反復組み換えアッセイにより測定され得る。レポーターベクターは、酵母又は哺乳動物発現ベクター中にクローニングされる、レポーター遺伝子の2つの短縮された非機能的コピー(直列反復)及びキメラDNA標的配列を、介在配列内に含む。DNA標的配列は、異なるヌクレオチドにより少なくとも1つのヌクレオチドが置換される親のホーミングエンドヌクレアーゼ切断部位に由来する。好ましくは、DNA切断部位の1つ以上の位置での4つの塩基(g、a、c、t)異なる組み合わせを表すパリンドローム又は非パリンドロームDNA標的のパネルが試験される(n個の変異される位置の4n パリンドローム標的)。変異型の発現は、DNA標的配列を切断できる機能的エンドヌクレアーゼとなる。この切断は、直接反復間の相同組み換えを誘導して、結果として機能的レポーター遺伝子を生じさせ、その発現は適切なアッセイによりモニターできる。
【0095】
- 「DNA標的」「DNA標的配列」「標的配列」「標的部位」「標的」「部位」「認識部位」「認識配列」「ホーミング認識部位」「ホーミング部位」「切断部位」により、LAGLIDADGホーミングエンドヌクレアーゼにより認識され且つ切断される20〜24 bpの二本鎖パリンドローム、部分的パリンドローム(偽パリンドローム)又は非パリンドロームのポリヌクレオチド配列を意図する。これらの用語は、そこでエンドヌクレアーゼにより二本鎖破断(切断)が誘導される独特のDNAの位置、好ましくはゲノムの位置のことをいう。DNA標的は、二本鎖ポリヌクレオチドの一方の鎖の5'から3'の配列により定義される。例えば、野生型I-CreIにより切断されるパリンドロームDNA標的配列は、5'- t-12c-11a-10a-9a-8a-7c-6g-5t-4c-3g-2t-1a+1c+2g+3a+4c+5g+6t+7t+8t+9t+10g+11a+12 (配列番号29)の配列により定義される。DNA標的の切断は、センス鎖及びアンチセンス鎖のそれぞれについて、+2位及び-2位のヌクレオチドで生じる。そうでないと記載しない限り、メガヌクレアーゼ変異型によるDNA標的の切断が生じる位置は、DNA標的のセンス鎖上の切断部位に相当する。
【0096】
- 「DNA標的ハーフサイト」「ハーフ切断部位」又は「ハーフサイト」により、各LAGLIDADGホーミングエンドヌクレアーゼコアドメインが結合するDNA標的の部分を意図する。
- 「DC10NNN」(配列番号8)により、これが配列の+8位、+9位及び+10位にて変異を有するDmoCreの標的配列であることを意図し、それゆえ10位から連続して逆向きに3つのヌクレオチドで変異可能な10位におけるDmoCreである。同様に、DC4NNN (配列番号36)は、配列の+2位、+3位及び+4位にて変異を有するDmoCreの標的配列のことをいい;またDC7NNN (配列番号37)は、配列の+5位、+6位及び+7位にて変異を有するDmoCreの標的配列のことをいう。
【0097】
- 「キメラDNA標的」又は「ハイブリッドDNA標的」により、2つの親のメガヌクレアーゼ標的配列の異なる半分の融合を意図する。さらに、該標的の少なくとも一方の半分は、別個のサブドメインが結合するヌクレオチドの組み合わせ(組み合わせたDNA標的)を含み得る。
- 「変異」により、核酸/アミノ酸配列における、1つ以上のヌクレオチド/アミノ酸の置換、欠失、及び/又は付加を意味する。
【0098】
- 「相同な」により、配列同士の間の相同組み換えを導くのに十分な別の配列との同一性を有する、より具体的には少なくとも95%の同一性、好ましくは97%の同一性、より好ましくは99%を有する配列を意図する。
- 「同一性」は、2つの核酸分子又はポリペプチド間の配列同一性のことをいう。同一性は、比較の目的のために整列させ得るそれぞれの配列中の位置の比較により決定できる。比較される配列中の位置が同じ塩基により占められる場合、その分子同士は、その位置において同一である。核酸又はアミノ酸配列間の類似性又は同一性の程度は、核酸配列により共有される位置での同一又は一致するヌクレオチドの数の関数である。種々のアラインメントアルゴリズム及び/又はプログラムを用いて2つの配列間の同一性を計算でき、例えば、GCG配列解析パッケージ(University of Wisconsin, Madison, Wis.)の一部分として利用可能であり、例えばデフォルト設定で用い得るFASTA又はBLASTを含む。
【0099】
- 「個体」は、哺乳動物、及びその他の脊椎動物(例えば鳥類、魚類及び爬虫類)を含む。用語「哺乳動物」及び「哺乳類」は、本明細書で用いる場合、その子に授乳し、生存する子を出産する(真獣類(eutharian)又は胎盤哺乳類(placental mammals))又は産卵する(後獣類(metatharian)又は無胎盤哺乳類(nonplacental mammals))単孔類、有袋類及び有胎盤類(placental)を含む、いずれの脊椎動物のことをいう。哺乳動物の種の例は、ヒト及びその他の霊長類(例えばサル、チンパンジー)、げっ歯類(例えばラット、マウス、モルモット)並びに反すう動物(例えばウシ、ブタ、ウマ)を含む。
【0100】
- 「遺伝病」は、部分的又は完全な、直接的又は間接的な1又は複数の遺伝子における異常による、任意の疾患のことをいう。該異常は、変異、挿入又は欠失であり得る。該変異は、点突然変異であり得る。上記の異常は、遺伝子のコード配列又はその調節配列に影響し得る。該異常は、ゲノム配列の構造又はコードされるmRNAの構造若しくは安定性に影響し得る。該遺伝病は、劣性又は優性であり得る。このような遺伝病は、限定されないが、嚢胞性繊維症, ハンチントン舞踏病、家族性高コレステロール血症(LDL受容体欠損)、肝芽腫、ウィルソン病、先天性肝性ポリフィリン病、肝臓代謝の遺伝性障害、レッシュナイハン症候群、鎌状赤血球貧血、サラセミア、色素性乾皮症、ファンコーニ貧血、色素性網膜炎、毛細管拡張性運動失調、ブルーム症候群、網膜芽腫、デュシェンヌ型筋ジストロフィー、及びテイ-サックス病であり得る。
【0101】
- 「RAG遺伝子」により、哺乳動物のRAG1又はRAG2遺伝子を意図する。例えば、ヒトRAG遺伝子は、アクセッション番号NC_000011.8のもとNCBIデータベースにおいて利用できる:RAG1 (遺伝子ID:5896)配列及びRAG2 (遺伝子ID:5897)配列は、それぞれ36546139 位〜36557877位及び36570071位〜36576362位(マイナス鎖)で位置が定められている。両遺伝子は、短い非翻訳領域及び長い非翻訳領域がそれぞれ5' 末端及び3'末端に連結したRAGタンパク質をコードするORFを含む、短い非翻訳エクソン1及びエクソン2を有している。
【0102】
- 「RAG1.10」は、ヒトRAG1遺伝子(アクセッション番号NC_000011.8、836546139位〜36557877位)の5270位、RAG1のコーディングエクソンから7 bp上流に位置する22 bpの(非パリンドロームの)標的である。
- 「RAG1.10.2」(配列番号34)は、RAG1.10標的の第1ハーフに由来するパリンドローム標的(tgttctcagg tacctgagaaca)である。
- 「RAG1.10.2D34」:「RAG1.10.2D34」(配列番号35)により、D34で表されるI-DmoI標的配列の第2ハーフに結合する、上で定義されるRAG1.10.2標的配列の第1部分を含む配列を意味する。この配列は「tgttctcagg taagttccggcg」である。
【0103】
- 「RAG1.10.3」(配列番号38)は、RAG1.10標的の第1ハーフに由来するパリンドローム標的(ctggctgaggtacctcagccag)である。
- 「RAG1.10.3D34」:「RAG1.10.3D34」(配列番号39)により、D34で表されるI-DmoI標的配列の第2ハーフに結合する、上で定義されるRAG1.10.3標的配列の第1部分を含む配列を意味する。この配列は「ttggctgaggtaagttccggcg」である。
【0104】
- 「ベクター」:本発明に用い得るベクターは、限定されないが、ウイルスベクター、プラスミド、RNAベクター、或いは染色体、非染色体、半合成(semi-synthetic)又は合成の核酸からなり得る直鎖状若しくは環状のDNA又はRNA分子を含む。好ましいベクターは、自律複製できるもの(エピソームベクター)及び/又は連結された核酸の発現を可能にするもの(発現ベクター)である。多数の適切なベクターが当業者に知られ、商業的に入手可能である。
【0105】
ウイルスベクターは、レトロウイルス、アデノウイルス、パルボウイルス(例えばアデノ随伴ウイルス)、コロナウイルス、マイナス鎖RNAウイルス、例えばオルトミクソウイルス(例えばインフルエンザウイルス)、ラブドウイルス(例えば狂犬病及び水疱性口内炎ウイルス)、パラミクソウイルス(例えば麻疹及びセンダイ)、プラス鎖RNAウイルス、例えばピコルナウイルス及びアルファウイルス、並びにアデノウイルス、ヘルペスウイルス(例えば単純ヘルペスウイルス1及び2型、エプスタイン-バーウイルス、サイトメガロウイルス)及びポックスウイルス(例えばワクシニア、鶏痘及びカナリア痘)を含む二本鎖DNAウイルスを含む。その他のウイルスは、例えばノーウォークウイルス、トガウイルス、フラビウイルス、レオウイルス、パポバウイルス、ヘパドナウイルス及び肝炎ウイルスを含む。レトロウイルスの例は、トリ白血病肉腫、哺乳類C型、B型ウイルス、D型ウイルス、HTLV-BLV群、レンチウイルス、スプマウイルスを含む(Coffin, J. M., Retroviridae: The viruses and their replication, In Fundamental Virology、第3版、B. N. Fieldsら編、Lippincott-Raven Publishers、フィラデルフィア、1996)。用語「ベクター」は、それが連結されている別の核酸を輸送できる核酸分子のことをいう。好ましいベクターのあるタイプは、エピソーム、すなわち染色体外の複製が可能な核酸である。好ましいベクターは、自律複製及び/又はそれらが連結されている核酸の発現が可能なものである。それらが作動可能に(operably)連結されている遺伝子の発現に指向できるベクターは、本明細書において「発現ベクター」という。本発明によるベクターは、限定されないが、YAC (酵母人工染色体)、BAC (細菌人工)、バキュロウイルスベクター、ファージ、ファージミド、コスミド、ウイルスベクター、プラスミド、RNAベクター、或いは染色体、非染色体、半合成又は合成のDNAからなり得る直鎖状若しくは環状DNA又はRNA分子を含む。一般に、組み換えDNA技術において実用性のある発現ベクターは、しばしば、一般に環状二本鎖DNAループをいう「プラスミド」の形態にあり、そのベクターの形態においてそれらは染色体に結合しない。適切なベクターの多数は、当業者に知られている。
【0106】
ベクターは、選択マーカー、例えば:真核細胞培養にはネオマイシンホスホトランスフェラーゼ、ヒスチジノールデヒドロゲナーゼ、ジヒドロ葉酸レダクターゼ、ハイグロマイシンホスホトランスフェラーゼ、単純ヘルペスウイルスチミジンキナーゼ、アデノシンデアミナーゼ、グルタミンシンセターゼ、及びヒポキサンチン-グアニンホスホリボシルトランスフェラーゼ;エス・セレビシエ(S. cerevisiae)にはTRP1;イー・コリ(E. coli)にはテトラサイクリン、リファンピシン又はアンピシリン耐性などを含み得る。しかしながら、本発明は、同等の機能を提供し、かつ本発明に続いて知られることとなる発現ベクターのようなその他の形態を含むことを意図する。
【0107】
好ましい上記のベクターは、本発明のポリペプチドをコードする配列が適切な転写及び翻訳調節エレメントの調節下に置かれ、該タンパク質の産生及び合成を可能にする発現ベクターである。したがって、上記のポリヌクレオチドは発現カセットに含まれる。より詳細には、該ベクターは、複製開始点、上記のコードするポリヌクレオチドが作動可能に連結されるプロモータ、リボソーム部位、RNAスプライシング部位(ゲノムDNAを用いる場合)、ポリアデニレーション部位及び転写終結部位を含む。それはエンハンサーも含み得る。プロモータの選択は、ポリペプチドを発現させる細胞に依存する。
【実施例】
【0108】
実施例1:活性が増大したDmoCreの改良
本発明者らは、現存するDmoCre足場を、この酵素の全体的な活性を増大させることにより改良することを計画した。特に、DmoCreのI-DmoI N-末端α-へリックスにI-DmoIの残基15、19及び20 (配列番号22)に相当する3つの変異を導入した。
G20S変異は、酵母においてより活性なDmoCreタンパク質を導くが、2つの変異L15Q及びI19Dは、CHO細胞において、以前に記載された染色体外SSA (一本鎖アニーリング)組み換えアッセイ(Arnouldら、Mol Biol. 2006 Jan 20;355(3):443〜58)により示されるようにタンパク質を活性にする。よって、この実施例において用いた最終的なDmoCre足場は、L15Q、I19D及びG20S変異を有し、これらは全てI-DmoI N-末端LAGLIDADG α-ヘリックスに局在する。上記の野生型I-DmoIドメインは、配列番号1に示す。
この足場をDmoCre2といい、以下の実験において用いた。DmoCre2のペプチド配列は、配列番号2に示す。
【0109】
実施例2:縮重DC10NNN_P標的を切断するDmoCre2由来変異体の作製
DmoCre2タンパク質についての新しい配列特異性を工学的に作製する可能性を研究するために、本発明者らは、C12D34 DNA標的の+8位〜+10位の3つの隣接するヌクレオチドについて調べた。図2に示す構造により、これらの3つの塩基対とDmoCre2タンパク質残基との間の接触を詳細に調べることが可能になった。
図3Aは、そのDNA標的と結合したハイブリッド酵素の分子表面を示す。無作為化のために選択した結合の領域(+8位、+9位、+10位の塩基対、及び配列番号22の残基29、33及び35に相当するタンパク質残基)を強調する。図3Bは、DNA標的と相互作用している残基29、33及び35を示す拡大図である。点線は水素結合を表す。よって、この分析を用いて、3つのDmoCre2残基が正確に示される:DNAと接触するR33及びE35、並びにDNAと近く、E35と相互作用しているようであるY29。
DmoCre2タンパク質についての新しい切断特異性を単離するために、29位、33位及び35位で変異させたDmoCre2変異体ライブラリー(DClib2)を構築し、酵母を形質転換し、酵母スクリーニングアッセイを用いて(以下を参照)、+8位〜+10位で縮重した64個の標的(DC10NNN (配列番号8)と称する)に対してスクリーニングした。DC10NNN標的は、5' CAAAACGTCGTAAGTTCCNNNC 3' (配列番号8)であり、ここでNNNは+8位〜+10位を表し、これらの位置におけるA、C、G及びTの全ての組み合わせが、64個の標的DC10NNN配列を構成する。
【0110】
材料及び方法
64個の標的ベクターの構築:
標的は、以下のようにしてクローニングした:ゲートウェイクローニング配列と接する64個の標的配列のそれぞれに対応するオリゴヌクレオチドを、Proligoに注文した:
5'TGGCATACAAGTTTTCNNNGGAACTTACGACGTTTTGACAATCGTCTGTCA 3' (配列番号3)。一本鎖オリゴヌクレオチドのPCR増幅により作製した二本鎖標的DNAを、ゲートウェイプロトコル(Invitrogen)を用いて酵母レポーターベクター(pCLS1055、図4)にクローニングした。酵母レポーターベクターでエス・セレビシエ(S. cerevisiae) FYBL2-7B株(MAT-α, ura3Δ851, trp1Δ63, leu2Δ1, lys2Δ202)を形質転換した。
【0111】
DmoCre2 DClib2変異体ライブラリーの構築:
29位、33位又は35位に変異を含むDmoCre2由来コード配列を作製するために、DmoCreコード配列の5'末端(aa 1位〜43位)又は3'末端(36位〜264位)を増幅する2回の別々のオーバーラップPCR反応を行った。3'末端について、PCR増幅は、ベクター(pCLS0542、図5)に特異的なプライマー(Gal10R 5'-ACAACCTTGATTGGAGACTTGACC-3' (配列番号4))及びアミノ酸36〜46についてのDmoCreコード配列に特異的なプライマー(Dmo10CreFor 5'- TATCGTGTTGTGATCACCCAGAAGTCTGAAAAC-3' (配列番号5))を用いて行う。5'末端について、PCR増幅は、ベクターpCLS0542に特異的なプライマー(Gal10F 5'-GCAACTTTAGTGCTGACACATACAGG-3' (配列番号6))及びアミノ酸23〜43についてのDmoCreコード配列に特異的なプライマー(Dmo10CreRev 5'-CTTCTGGGTGATCACAACACGATAMNNGCTMNNGTTACCTTTMNNTTTCAGCTTGTACAGGCC-3' (配列番号7))を用いて行う。
【0112】
29位、33位及び35位でのNNKコドンをもたらすオリゴヌクレオチド中のMNNコードは、これらの位置にて20個の可能なアミノ酸のうちでの縮重をもたらす。次いで、2つのオーバーラップPCRフラグメントのそれぞれ25 ngと、NcoI及びEagIでの消化により線状にした75 ngのベクターDNA (pCLS0542)とを用いて、酵母サッカロミセス・セレビシエ(Saccharomyces cerevisiae) FYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietzら, Methods Enzymol. 2002; 350:87〜96)。両方の群の変異を含むインタクトなコード配列を、酵母でのインビボ相同組み換えにより作製する。DClib2核酸多様性は323 = 32768であり、形質転換の後にライブラリー多様性の約7%である2232個のクローンを採集した。
【0113】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
スクリーニングは、以前に記載されたようにして行った(Arnouldら, J Mol Biol. 2006; 355:443〜58)。具体的には、交配を、コロニーグリッダーを用いて行った(QpixII, Genetix)。変異体を、YPDプレートを覆うナイロンフィルタ上に、低い格子密度(low gridding density) (約4スポット/cm2)を用いてグリッドした。2回目のグリッド手順を、同じフィルタ上に行って、それぞれの標的について異なるレポーターを有する酵母株からなる第2層をスポットした。メンブレンを、固体寒天YPDリッチ培地上に置き、30℃にて一晩インキュベートして交配を可能にした。次に、フィルタを、ロイシン及びトリプトファンを欠き、ガラクトース(2%)を炭素源として含む合成培地に移し、37℃にて5日間インキュベートして、発現及び標的ベクターを有する二倍体を選択した。5日後に、フィルタを、0.5 Mリン酸ナトリウムバッファー、pH 7.0中の0.02% X-Gal、0.1% SDS、6%ジメチルホルムアミド(DMF)、7mM β-メルカプトエタノール、1%アガロースを含む固体アガロース培地上に置き、37℃にてインキュベートして、β-ガラクトシダーゼ活性をモニターした。結果を走査により分析し、独自のソフトウェアを用いて定量を行った。
【0114】
変異体の配列決定
プラスミドを発現する変異体を回収するために、酵母DNAを標準的なプロトコルを用いて抽出し、大腸菌(E. coli)を形質転換するために用いた。変異ORFの配列決定を、次いで、プラスミドに対してMillegen SAにより行った。或いは、ORFを酵母DNAからPCRにより増幅し(Akadaら, Biotechniques. 2000; 28:668〜70, 672, 674)、配列決定を、PCR生成物に対して直接、Millegen SAにより行った。
【0115】
結果
上記の酵母スクリーニングアッセイを用いて、DmoCre2 DClib2ライブラリーを構成する2232個のクローンを、64個のDC10NNN標的に対してスクリーニングした。スクリーニングにより、少なくとも1つのDC10NNN標的(配列番号8)を切断できる519個の陽性クローンが得られ(図6)、配列決定の後に432個のユニークメガヌクレアーゼが得られた。元のDmoCre2タンパク質は、64個のDC10NNN標的のうち13個を切断できる。図7に示すDClib2ヒットマップは、DmoCreコード配列の29位、33位及び35位に変異を導入することにより、57個のDC10NNN標的がDmoCre2由来変異体により切断できるようになったことを示す。このスクリーニングアプローチにより、よって、DC10NNN標的についてのDmoCre2切断スペクトルを広げることができ、新しい切断特異性を単離することが可能になる。
【0116】
以下の表Iを参照して、本発明者らにより同定された種々のDClib2クローンを列挙し、これらのそれぞれにおける残基の変化を示すとともに、切断することが示されたDC10標的配列も示す。一番上の列は、+8位〜+10位の3つのヌクレオチドを示し、図は、挿入断片を欠く酵母からの陰性対照と比較した比色反応の強度を示す。具体的には、「0」の値は、このアッセイにおいてバックグラウンドノイズの試験されたレベルに等しい実験結果を表す。「_」の値は、この試料がこの特定のヌクレオチドの組み合わせについて試験されなかったことを示す。
【0117】
【表1−1】

【0118】
【表1−2】
【0119】
【表1−3】
【0120】
【表1−4】
【0121】
【表1−5】
【0122】
【表1−6】
【0123】
【表1−7】
【0124】
【表1−8】
【0125】
【表1−9】
【0126】
【表1−10】
【0127】
【表1−11】
【0128】
【表1−12】
【0129】
【表1−13】
【0130】
【表1−14】
【0131】
【表1−15】
【0132】
【表1−16】
【0133】
【表1−17】
【0134】
【表1−18】
【0135】
【表1−19】
【0136】
【表1−20】
【0137】
【表1−21】
【0138】
【表1−22】
【0139】
【表1−23】
【0140】
【表1−24】
【0141】
実施例3:縮重DC4NNN_P標的を切断するDmoCre由来変異体の作製
本出願人らは、酵母及びCHO細胞において活性な別のDmoCre足場も開発し、これもまた残基20でG20S置換にて改変されているこの足場を、配列番号22の残基19及び109に相当する残基にて改変し(I19D及びF109Y改変)、DmoCre4 (配列番号9)と命名した。
DmoCre4タンパク質(配列番号9)についての追加の特異性を見出す可能性を研究するために、C12D34 DNA標的の+2位〜+4位の3つの隣接するヌクレオチドについて調べた。図2に示す構造により、これらの3つの塩基対とタンパク質残基との間の接触を詳細に調べることが可能になった(図8)。配列番号22の残基D75、T76及びR77に相当する3つのDmoCre4残基が同定され、これらはDNA標的と接触する。DmoCre4タンパク質についての新しい切断特異性を単離するために、75位、76位又は77位で変異させたDmoCre4変異体ライブラリー(D4Clib4)を構築し、酵母を形質転換し、酵母スクリーニングアッセイを用いて、+2位〜+4位で縮重した64個の標的(DC4NNN (配列番号36)と称する)に対してスクリーニングした。このようなアプローチは、I-CreIタンパク質についてすでに詳細に記載されている(Smith Jら, Nucleic Acids Res. 2006; Arnould Sら, J Mol Biol. 2006; 355:443〜58, Arnould Sら, . J Mol Biol. 2007; 371:49〜65)。
【0142】
材料及び方法
64個の標的ベクターの構築:
標的は、以下のようにしてクローニングした:ゲートウェイクローニング配列と接する64個の標的配列のそれぞれに対応するオリゴヌクレオチドを、Proligoに注文した:
5'TGGCATACAAGTTTTCGCCGGANNNTACGACGTTTTGACAATCGTCTGTCA 3'(配列番号10)。一本鎖オリゴヌクレオチドのPCR増幅により作製した二本鎖標的DNAを、ゲートウェイプロトコル(Invitrogen)を用いて酵母レポーターベクター(pCLS1055、図4)にクローニングした。酵母レポーターベクターでエス・セレビシエFYBL2-7B株(MAT-α, ura3Δ851, trp1Δ63, leu2Δ1, lys2Δ202)を形質転換した。
【0143】
DmoCre4 D4Clib4変異体ライブラリーの構築:
75位、76位及び77位に変異を含むDmoCre4由来コード配列を作製するために、DmoCre4コード配列の5'末端 (aa 1位〜74位)又は3'末端(66位〜264位)を増幅する2つの別々のオーバーラップPCR反応を行った。3'末端について、PCR増幅は、ベクター(pCLS0542、図5)に特異的なプライマー(Gal10R 5'-ACAACCTTGATTGGAGACTTGACC-3') (配列番号11)及びアミノ酸66〜83についてのDmoCreコード配列に特異的なプライマー(DClib4For 5'-AAATCTAAAATCCAGATCGTTAAGGGTNNKNNKNNKTATGAGCTGCGTGTGAGC-3') (配列番号12)を用いて行う。75位〜77位でのNNKコドンは、これらの位置にて20個の可能なアミノ酸のうちでの縮重をもたらす。5'末端について、PCR増幅は、ベクターpCLS0542に特異的なプライマー(Gal10F 5'-GCAACTTTAGTGCTGACACATACAGG-3') (配列番号13)及びアミノ酸66〜74についてのDmoCreコード配列に特異的なプライマー(DClib4Rev 5'-ACCCTTAACGATCTGGATTTTAGATTT-3') (配列番号14)を用いて行う。2つのオーバーラップPCRフラグメントのそれぞれ25 ngと、NcoI及びEagIでの消化により線状にした75 ngのベクターDNA (pCLS0542)とを用いて、酵母サッカロミセス・セレビシエFYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietz R Dら, Methods Enzymol. 2002; 350:87〜96)。インタクトなDmoCreコード配列を、酵母でのインビボ相同組み換えにより作製する。D4Clib4核酸多様性は、323 = 32768である。形質転換の後に、ライブラリー 多様性の約14%である4464個のクローンを採集した。
【0144】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0145】
結果
上記の酵母スクリーニングアッセイを用いて、DmoCre4 D4Clib4 ライブラリーを構成する4464個のクローンを、DC4GAA標的以外の64個全てのDC4NNN標的に対してスクリーニングした。スクリーニングにより、少なくとも1つのDC4NNN標的(配列番号36)を切断できる1194個の陽性クローンが得られた。これらのクローンは、配列レベルでは特徴決定しなかった。元のDmoCre4タンパク質は、63個のDC4NNN標的のうち4個を切断できる。図9に示すD4Clib4ヒットマップは、DmoCre4コード配列の75位、76位及び77位に変異を導入することにより、21個のDC4NNN標的がDmoCre4由来変異体により切断できるようになったことを示す。我々のスクリーニングアプローチにより、よって、DC4NNN標的についてのDmoCre4切断スペクトルを広げることができ、新しい切断特異性を単離することが可能になる。
【0146】
実施例4:5CAGD34標的を切断するDmoCre由来変異体の作製
本発明者らは、C1221に由来し、±5位、±4位、±3位にて縮重するパリンドロームDNA標的に対するI-CreIタンパク質の特異性を改変できたことを以前に示した(Arnouldら, J Mol Biol. 2006; 355:443〜58)。44位、68位、70位、75位及び77位にてI-CreIコード配列に変異を導入することにより、5CAG_P標的(配列番号32)を切断するI-CreI由来変異体を得ることができた。
この実施例では、これらの同じ変異をDmoCre2又はDmoCre4コード配列に導入することにより、5CAGD34標的(配列番号33)を切断するDmoCre由来変異体を作製できることを示す(図10)。これらのDmoCre由来変異体を作製するために、5CAG_P標的(配列番号32)を切断できる36個のI-CreI変異体を採用した。I-CreI部分のコード配列を、DmoCre2又はDmoCre4タンパク質から、制限酵素消化により除去し、5CAG_Pカッターコード配列に置き換えた。DmoCre2及びDmoCre4タンパク質にそれぞれ基づく2つの変異体ライブラリーDCSca2_5CAG及びDCSca4_5CAGを構築し、5CAGD34標的(配列番号33)に対して、上記のような酵母スクリーニングアッセイを用いてスクリーニングした。
【0147】
材料及び方法
5CAGD34標的ベクターの構築
標的は、以下のようにしてクローニングした:ゲートウェイクローニング配列で挟まれた標的配列に対応するオリゴヌクレオチドを、Proligoに注文した:5' TGGCATACAAGTTTTCGCCGGAACTTACCTGGTTTTGACAATCGTCTGTCA 3' (配列番号15)。一本鎖オリゴヌクレオチドのPCR増幅により作製した二本鎖標的DNAを、ゲートウェイプロトコル(Invitrogen)を用いて酵母レポーターベクター(pCLS1055、図4)にクローニングした。酵母レポーターベクターでエス・セレビシエFYBL2-7B株(MAT-α, ura3Δ851, trp1Δ63, leu2Δ1, lys2Δ202)を形質転換した。
【0148】
DCsca2_5CAG変異体ライブラリーの構築:
5CAG_P標的切断を担うI-CreI部分の配列に変異を含むDmoCre2由来コード配列を作製するために、I-CreI由来5CAG_Pカッターのそれぞれについてのaa 13〜148の領域を増幅するPCR反応を行った。PCR増幅を、プライマーCreNgoLib (5' CGTGAGCAGCTGGCGTTCCTGGCCGGCTTTGTGGACGGTGAC-3' (配列番号16))及びCreMluLib (5'-ACGAACGGTTTCAGAAGTGGTTTTACGCGTCTTAG-3' (配列番号17))を用いて行った。
36個のPCRフラグメントを、次いで、プールした。DmoCre2タンパク質についての酵母発現ベクターを、次いで、NgoMIV及びMluIで消化して、DmoCre2タンパク質の残基111〜238をカバーするフラグメントを除去した。最後に、オーバーラップPCRプール25 ngと、75 ngの消化ベクターDNAとを用いて、酵母サッカロミセス・セレビシエFYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietz R Dら, Methods Enzymol. 2002; 350:87〜96)。5CAG_Pカッターに特徴的な変異を含むインタクトなDmoCreコード配列を、酵母でのインビボ相同組み換えにより作製した。形質転換の後に、ライブラリー多様性の約5倍の186個のクローンを採集した。
【0149】
DCSca4_5CAG変異体ライブラリーの構築
5CAG_P標的切断を担うI-CreI部分の配列に変異を有するDmoCre4由来コード配列を作製するために、I-CreI由来5CAG_Pカッターのそれぞれについてのaa 13〜148の領域を増幅するPCR反応を行った。PCR増幅を、プライマーCreMluLib及びCreNgoLibY (5' CGTGAGCAGCTGGCGTACCTGGCCGGCTTTGTGGACGGTGAC-3') (配列番号18)を用いて行ったが」、これは、DmoCre4タンパク質に特徴的なF109Y変異を考慮に入れている。36個のPCRフラグメントを、次いで、プールした。DmoCre4タンパク質についての酵母レポーターベクターを、次いで、制限酵素NgoMIV及びMluIで消化して、DmoCre4タンパク質の残基111〜238をカバーするフラグメントを除去した。最後に、PCRプール25 ngと、75 ngの消化ベクターDNAとを用いて、酵母サッカロミセス・セレビシエFYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietz R Dら, Methods Enzymol. 2002; 350:87〜96)。5CAG_Pカッターに特徴的な変異を含むインタクトなDmoCreコード配列を、酵母でのインビボ相同組み換えにより作製した。形質転換の後に、ライブラリー多様性の約5倍の186個のクローンを採集した。
【0150】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0151】
結果
実施例1に記載される酵母スクリーニングアッセイを用いて、DCSca2_5CAGライブラリーを構成する186個のクローンと、DCSca4_5CAG ライブラリーを構成する186個のクローンを、5CAGD34 (配列番号33)標的に対してスクリーニングした。最初のライブラリー及び次のライブラリーからそれぞれ、全体的により強い切断効率を有する32個及び40個の陽性クローンが得られた。陽性の例を図11に示す。5CAGD34標的(配列番号33)を切断するクローンは、5CAG_P標的(配列番号32)を切断しない(データ示さず)。よって、本発明者らは、特定のI-CreI変異をDmoCre足場に導入して、組み合わせ標的を効率的に切断できることを示した。
【0152】
実施例5:RAG1.10.2D34標的を切断するDmoCre由来変異体の作製
RAG1.10.2 DNAパリンドローム標的(配列番号34)は、I-CreI C1221標的(配列番号29)に由来する(図12)。本発明者らは、10GTT (例えばヌクレオチド8、9及び10がそれぞれG、T及びTである)及び5CAG I-CreI由来変異体を組み合わせ、次いで、最初に単離されたRAG1.10.2カッターに対してランダム突然変異誘発ステップを行うことにより、どのようにして、RAG1.10.2標的を非常に強く切断するI-CreI由来変異体を得ることができたかを以前に示した(WO2008/010009)。このM2と呼ばれる変異体は、野生型I-CreI配列と比較して以下の変異を有する:配列番号24のN30R、Y33N、Q44A、R68Y、R70S、I77R。これらの同じ変異をDmoCre2のI-CreI部分に導入し、DmoM2とよばれる得られたDmoCre変異体(配列番号71)のRAG1.10.2D34 (配列番号35)組み合わせ標的に対する活性を、酵母切断アッセイを用いて調べた。
【0153】
材料及び方法
RAG1.10.2D34標的ベクターの構築
標的は、以下のようにしてクローニングした:ゲートウェイクローニング配列に挟まれた標的配列に対応するオリゴヌクレオチドを、Proligoに注文した:5' TGGCATACAAGTTTTCGCCGGAACTTACCTGAGAACAACAATCGTCTGTCA 3' (配列番号19)。一本鎖オリゴヌクレオチドのPCR増幅により作製した二本鎖標的DNAを、ゲートウェイプロトコル(Invitrogen)を用いて酵母レポーターベクター(pCLS1055、図4)にクローニングした。酵母レポーターベクターでエス・セレビシエFYBL2-7B株(MAT-α, ura3Δ851, trp1Δ63, leu2Δ1, lys2Δ202)を形質転換した。
【0154】
DmoM2の構築
RAG1.10.2 M2変異体に特異的なI-CreI部分に変異を含むDmoCre2由来コード配列を作製するために、M2変異体のaa 9〜146の領域を増幅するPCR反応を行った。PCR増幅は、プライマーCreNgoFor (5' TTCCTGCTGTACCTGGCCGGCTTTGTGG-3' (配列番号20))及びCreMluRev (5'- TTCAGAAGTGGTTTTACGCGTCTTAG -3' (配列番号21))を用いて行う。次いで、PCRフラグメントを、DmoCre2タンパク質についてのORFを含む酵母発現ベクターと同様に、制限酵素NgoMIV及びMluIで消化した。ライゲーション反応を行い、大腸菌DH5αをライゲーション混合物で形質転換した。得られたDmoM2変異体を、次いで増幅して配列決定した。
【0155】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0156】
結果
この酵母切断アッセイを用いて、組み合わせRAG1.10.2D34標的(配列番号35)及びその他の異なる標的に対するDmoM2変異体の活性を調べた。図13は、DmoM2が、組み合わせRAG1.10.2D34標的(配列番号35)を特異的に切断することを示す。よって、本発明者らは、DmoCre足場のI-CreI部分に、標的配列についてのコンビナトリアル及び/又は最適化実験の間にI-CreI足場において以前に単離された変異を導入して、該標的配列の一部分を含む組み合わせDmoCre標的を効率的に切断できたことを示した。
【0157】
実施例6:RAG1.10.2D34又はRAG1.10.3D34標的を切断するDmoCre由来変異体の作製
RAG1.10.2及びRAG1.10.3 DNAパリンドローム標的(配列番号34及び38)は、I-CreI C1221標的(配列番号29)に由来する(図14)。本発明者らは、10GTT及び5CAG I-CreI由来変異体を組み合わせることにより、どのようにして、RAG1.10.2標的を非常に強く切断するI-CreI由来変異体を得ることができたか、又は10TGG及び5GAG I-CreI由来変異体を組み合わせることにより、どのようにして、RAG1.10.3標的を非常に強く切断するI-CreI由来変異体を得ることができたかを以前に示した(WO2008/010009及びWO2008/010093)。実施例4に記載したのと同じ方法論を用いて、I-CreI部分のコード配列を、DmoCre2タンパク質から、制限酵素消化により除去し、RAG1.10.2カッターコード配列又はRAG1.10.3カッターコード配列のいずれかで置き換えた。以下の表IIは、I-CreI部分における変異を、RAG1.10.2カッターコード配列について配列番号24のI-CreI 配列における残基の番号付けを参照にしてまとめ、以下の表IIIは、I-CreI部分における変異をRAG1.10.3カッターコード配列についてまとめる。DCSca2_RAG1.10.2及びDCSca2_RAG1.10.3変異体ライブラリーを作製するために、33個のRAG1.10.2カッター及び35個のRAG1.10.3カッターをそれぞれ用いた。次いで、2つの変異体ライブラリーを、上記の酵母スクリーニングアッセイを用いてそれぞれRAG1.10.2D34及びRAG1.10.3D34標的に対して、また、親の標的に対してもスクリーニングした。
【0158】
【表2】
【0159】
【表3】
【0160】
材料及び方法
RAG1.10.3D34標的ベクターの構築
標的は、以下のようにしてクローニングした:ゲートウェイクローニング配列に挟まれた標的配列に対応するオリゴヌクレオチドを、Proligoに注文した:5'TGGCATACAAGTTTTCGCCGGAACTTACCTCAGCCAGACAATCGTCTGTCA-3' (配列番号19)。一本鎖オリゴヌクレオチドのPCR増幅により作製した二本鎖標的DNAを、ゲートウェイプロトコル(Invitrogen)を用いて酵母レポーターベクター(pCLS1055、図4)にクローニングした。酵母レポーターベクターでエス・セレビシエFYBL2-7B株(MAT-α, ura3Δ851, trp1Δ63, leu2Δ1, lys2Δ202)を形質転換した。
【0161】
DCSca2_RAG1.10.2変異体ライブラリーの構築
RAG1.10.2標的切断を担うI-CreI部分の配列に変異を含むDmoCre2由来コード配列を作製するために、33個のI-CreI由来RAG1.10.2カッターのそれぞれについてのaa 13〜148の領域を増幅するPCR反応を行い、さらに、プライマーは、DmoCre2を含む発現ベクターの配列にいずれかの末端にて相同な部分も含む。PCR増幅は、プライマーCreNgoLib (5' CGTGAGCAGCTGGCGTTCCTGGCCGGCTTTGTGGACGGTGAC-3' (配列番号16))及びCreMluLib (5'-ACGAACGGTTTCAGAAGT GGTTTTACGCGTCTTAG-3' (配列番号17))を用いて行う。
33個のPCRフラグメントを、次いで、プールした。DmoCre2タンパク質についての酵母発現ベクターを、次いで、NgoMIV及びMluIで消化して、DmoCre2タンパク質の残基111〜238をカバーするフラグメントを除去した。最後に、25 ngのPCRプールと、75 ngの消化ベクターDNAとを用いて、酵母サッカロミセス・セレビシエFYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietz R Dら, Methods Enzymol. 2002; 350:87〜96)。RAG1.10.2カッターに特徴的な変異を含むインタクトなDmoCreコード配列を、酵母でのインビボ相同組み換えにより作製した。形質転換の後に、ライブラリー多様性の約5倍の186個のクローンを採集した。
【0162】
DCSca2_RAG1.10.3変異体ライブラリーの構築
方法論は、35個のRAG1.10.3カッターのプールを用いた以外は、DCSca2_RAG1.10.2変異体ライブラリーと同じであった。
【0163】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0164】
結果
実施例2に記載される酵母スクリーニングアッセイを用いて、DCSca2_RAG1.10.2変異体ライブラリーを構成する186個のクローンと、DCSca2_RAG1.10.3変異体ライブラリーを構成する186個のクローンを、それぞれRAG1.10.2D34及びRAG1.10.3D34標的に対してスクリーニングした。DCSca2_RAG1.10.2ライブラリーからは36個の陽性クローンが得られ、そのうち24個のクローンを再配置して、図15に示す2回目のスクリーニングに供した。24個のクローンは、8個のユニーク配列を示し(表IVを参照)、RAG1.10.2D34標的に特異的である。これらは、RAG1.10.2、C1221及びC12D34標的のいずれも切断しない。DCSca2_RAG1.10.3ライブラリーからは52個の陽性クローンが得られ、そのうち33個のクローンを再配置して、図15に示す2回目のスクリーニングに供した。33個のクローンは、6個のユニーク配列を示し(表IVを参照)、RAG1.10.3D34標的に特異的である。これらは、RAG1.10.3、C1221及びC12D34標的のいずれも切断しない。よって、本発明者らは、特定のI-CreI変異を、DmoCre足場に、ライブラリーアプローチを用いて導入して、組み合わせ標的を効率的かつ特異的に切断できることを示した。
【0165】
実施例7:ランダム突然変異誘発によるRAG1.10.2D34及びRAG1.10.3D34メガヌクレアーゼの改良
実施例6で同定したRAG1.10.2D34及びRAG1.10.3D34カッターの切断効率を改良するために、1回のランダム突然変異誘発を、実施例6で単離した、選択されたRAG1.10.2D34及びRAG1.10.3D34カッターに対して行った。それぞれの標的ついて、実施例6に記載したもののうち3つの変異体を選択した。表IVを参照。これらのDNAをプールし、PCR無作為化についての鋳型として用いた。変異体ライブラリーを酵母において構築し、適切な標的に対してスクリーニングした。
【0166】
材料及び方法
ランダム突然変異誘発によるライブラリーの構築
それぞれの変異体プールに対して、0.3 mMの濃度のMn2+を用いるPCRによるランダム突然変異誘発を行った。用いたプライマーは、preATGCreFor (5'GCATAAATTACTATACTTCTATAGACACGCAAACACAAATACACAGCGGCCTTGCCACC-3' (配列番号40))及びICreIpostRev (5'-GGCTCGAGGAGCTCGTCTAGAGGATCGCTCGAGTTATCAGTCGGCCGC-3' (配列番号41))である。
およそ25ngのPCR生成物と、NcoI及びEagIでの消化により線状にした75ngのベクターDNA (pCLS542、図5)を用いて、酵母サッカロミセス・セレビシエFYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietz及びWoods 2002)。DmoCre変異体についてのインタクトなコード配列を含む発現プラスミドを、PCR生成物のオーバーラップ部分と消化ベクターとの間の酵母でのインビボ相同組み換えにより作製した。
【0167】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0168】
結果
以下の表IVは、8個のRAG1.10.2D34カッターと、6個のRAG1.10.3D34カッターの配列を示す。これらのうち、カッターのそれぞれのクラスの最初の3つ(表IVにおいて下線を付す)を選択して、無作為化PCRを行った。これらの配列はDmoCre2タンパク質に由来し、126位、128位、130位、131位、136位、138位、142位、166位、168位、173位及び175位(配列番号2)の残基にて異なる。これらの位置は、I-CreIのそれぞれ28位、30位、32位、33位、38位、40位、44位、68位、70位、75位及び77位(配列番号24)に対応する。上記のように、これらのRAG1.10.2D34及びRAG1.10.3D34カッターは、DmoCre2中に存在する変異、すなわちL15Q、I19D及びG20Sの変異も含み、これらは全てI-DmoI N-末端LAGLIDADGアルファ-ヘリックス中にある。
【0169】
【表4】
【0170】
無作為化PCRから創出した変異体ライブラリーを、次いで、我々の酵母スクリーニングアッセイにおいて、それらのそれぞれの標的に対してスクリーニングした。RG2D2及びRG3D3変異体を対照として用いた。対照変異体と比較して活性が増大した変異体を選択し、図16に示す2回目のスクリーニングに供した。それぞれの標的について、切断活性が改善された3つの変異体を丸で囲む。これらの選択された変異体を単離し、配列決定したが、表Vはそれらの配列を示す。
【0171】
【表5】
【0172】
表Vは、RAG1.10.2D34標的についての切断活性の改善が、V105A、E80K及びY66Hの変異をI-CreI部分に導入したことによることを示す(位置の番号付けはI-CreI配列である配列番号24を参照する)。RAG1.10.3D34標的の場合、活性の増大は、追加の変異によっては得られなかったが、突然変異誘発を行うために用いた3つのRAG1.10.3D34カッター間の変異の交換により得られた。
【0173】
実施例8:縮重DC4NNN_P標的を切断する新しいDmoCre由来変異体の作製
DC4NNN標的(配列番号36)についての特異性を有するDmoCre足場を探索するために、DmoCre2タンパク質に基づく新しい変異体ライブラリーを酵母において作製した。実施例3で述べたように、配列番号22の3つの残基D75、T76及びR77は、C12D34標的の+2位〜+4位の3つの塩基と接触する。配列番号22の残基T41も含まれ、C12D34標的の+4位のチミンのメチル基とファンデルワールス接触を確立する。本発明者らは、この残基の変異が、DC4NNN標的に対するDmoCre2タンパク質についての新しい特異性を提供できるのではないかと考えた。よって、DmoCre2タンパク質についての新しい切断特異性を単離するために、配列番号22 (I-DmoI部分)の残基41、75又は77に対応する位置で変異されたDmoCre2変異体ライブラリー(D4Clib2Bis)を構築し、酵母を形質転換し、酵母スクリーニングアッセイを用いて、+2位〜+4位で縮重した64個の標的(DC4NNN 配列番号36)に対してスクリーニングした。
【0174】
材料及び方法
DmoCre2 D4Clib2Bis変異体ライブラリーの構築:
配列番号22 (I-DmoI部分)の41位、75位及び77位にて変異を含むDmoCre2由来コード配列を作製するために、異なるPCR反応を行った。1回目のPCR反応は、ベクターpCLS0542に特異的なプライマー(Gal10F 5'-GCAACTTTAGTGCTGACACATACAGG-3' (配列番号13))及びプライマーDCaa49-37Rev (5'- TTTAATCAGGTTTTCAGACTTCTGMNNGATCACAACACG-3' (配列番号42))を用い、これはDmoCre2コード配列の5'末端(aa 1位〜49位)を増幅する。3'末端増幅について、2回のPCR反応を行った。1回目の反応は、DmoCre2の残基42〜74の間の領域を、プライマーDCaa42-50For (5'-CAGAAGTCTGAAAACCTGATTAAACAA-3' (配列番号43))及びDCaa74-66Rev (5'-ACCCTTAACGATCTGGATTTTAGATTT-3' (配列番号44))を用いて増幅する。2回目の反応は、DmoCre2の3'末端(68位〜264位)を、プライマーDCaa68-81For (5'-AAAATCCAGATCGTTAAGGGTNNKACCNNKTATGAGCTGCGT-3' (配列番号45))及びベクター(pCLS0542、図5)に特異的なプライマー(Gal10R 5'-ACAACCTTGATTGGAGACTTGACC-3' (配列番号4))を用いて増幅する。
2つのPCRフラグメントを精製し、DCaa42-50For及びGal10Rのプライマーを用いて行うアセンブリPCRにおいて鋳型として用いた。
次いで、2つのオーバーラップPCRフラグメント(1位〜49位及び42位〜264位)のそれぞれ25ngと、NcoI及びEagIでの消化により線状にしたオーバーラップする75ngのベクターDNA (pCLS0542)とを用いて、酵母サッカロミセス・セレビシエFYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietz R Dら, Methods Enzymol. 2002; 350:87〜96)。インタクトなDmoCreコードを、酵母でのインビボ相同組み換えにより作製した。形質転換の後に、2232個のクローンを採集した。
【0175】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0176】
結果
上記の酵母スクリーニングアッセイを用いて、DmoCre2 D4Clib2Bisライブラリーを構成する2232個のクローンを、DC4TTC標的以外の64個全てのDC4NNN標的に対してスクリーニングした。スクリーニングにより、少なくとも1つのDC4NNN標的(配列番号36)を切断できる335個の陽性クローンを得た。これらのクローンを再配置し、配列決定し(221個のユニーク配列を単離した)、2回目のスクリーニングに供した。元のDmoCre2タンパク質は、63個のDC4NNN標的のうち4個を切断できる。図17に示すD4Clib2Bisヒットマップは、41位、75位及び77位の変異をDmoCre2コード配列に導入することにより、32個のDC4NNN標的をこれらのDmoCre2由来変異体により切断できるようになることを示す。
この数を、実施例3で記載した変異体ライブラリーにより切断された21個のDC4NNN標的と比較しなければならない。このスクリーニングアプローチにおいて41位を変異させることにより、DC4NNN標的についてのDmoCre2切断スペクトルを広げることができ、新しい切断特性を単離することが可能になる。
【0177】
実施例9:縮重DC7NNN_P標的を切断する新しいDmoCre由来変異体の作製
DmoCre2タンパク質についての新しい配列特異性を工学的に作製する可能性を研究するために、本出願人らは、C12D34 DNA標的の+5位〜+7位の3つの隣接するヌクレオチドについて調べた。図2に示す構造により、これらの3つの塩基対とDmoCre2タンパク質残基との間の接触を詳細に調べることが可能になった。
図18Aは、そのDNA標的と結合したハイブリッド酵素の分子表面を示す。無作為化のために選択した結合の領域(+5位、+6位、+7位の塩基対、及び37及び81のタンパク質残基)を強調する。+5位〜+7位で縮重した64個の標的をDC7NNN (配列番号37)と称する。DC7NNN標的は、5' CAAAACGTCGTAAGTNNNGGCG 3' (配列番号37)であり、ここでNNNは+5位〜+7位を表し、これらの位置におけるA、C、G及びTの全ての組み合わせが、64個の標的DC7NNN配列を構成する。図18Bは、DNAと相互作用している配列番号22の2つのアルギニン残基37及び81を示す拡大図である。点線は水素結合を表す。これらのアルギニン残基の1つ又は2つを変異させることにより、DC7NNN標的に対するDmoCre2の切断活性が急激に減少するか又は完全に喪失される。
構造をより詳細に検討することにより、アルギニン残基37が、配列番号22のロイシン残基27と疎水的に接触していることが示される(図18C)。よって、27位での変異が、アルギニン37での変異を補うことができるだろう。
DmoCre2タンパク質についての新しい切断特異性を単離するために、27位及び37位で変異させたDmoCre2変異体ライブラリー(D7Clib2)を構築し、酵母を形質転換し、以下の酵母スクリーニングアッセイを用いて、DC7GAC以外の64個全てのDC7NNN標的に対してスクリーニングした。
【0178】
材料及び方法
64個の標的ベクターの構築:
標的は、以下のようにしてクローニングした:ゲートウェイクローニング配列と接する64個の標的配列のそれぞれに対応するオリゴヌクレオチドを、Proligoに注文した:5'TGGCATACAAGTTTTCGCCNNNACTTACGACGTTTTGACAATCGTCTGTCA-3', (配列番号3)。一本鎖オリゴヌクレオチドのPCR増幅により作製した二本鎖標的DNAを、ゲートウェイプロトコル(Invitrogen)を用いて酵母レポーターベクター(pCLS1055、図4)にクローニングした。酵母レポーターベクターでエス・セレビシエFYBL2-7B株(MAT-α, ura3Δ851, trp1Δ63, leu2Δ1, lys2Δ202)を形質転換した。
【0179】
DmoCre2 DClib2変異体ライブラリーの構築:
27位及び37位に変異を含むDmoCre2由来コード配列を作製するために、DmoCreコード配列の5'末端(aa 1位〜43位)又は3'末端(38位〜264位)を増幅する2回の別々のオーバーラップPCR反応を行った。3'末端について、PCR増幅は、ベクター(pCLS0542、図5)に特異的なプライマー(Gal10R 5'-ACAACCTTGATTGGAGACTTGACC-3' (配列番号4))及びアミノ酸38〜46についてのDmoCreコード配列に特異的なプライマー(DC37For 5'GTTGTGATCACCCAGAAGTCTGAAAAC-3' (配列番号46))を用いて行う。5'末端について、PCR増幅は、ベクターpCLS0542に特異的なプライマー(Gal10F 5'-GCAACTTTAGTGCTGACACATACAGG-3' (配列番号6))及びアミノ酸23〜43についてのDmoCreコード配列に特異的なプライマー(DC3727ScanRev 5'-CTTCTGGGTGATCACAACMNNATATTCGCTACGGTTACCTTTATATTTMNNCTTGTACAGGCC-3' (配列番号47))を用いて行う。
【0180】
27位及び37位でのNNKコドンをもたらすオリゴヌクレオチド中のMNNコードは、これらの位置にて20個の可能なアミノ酸のうちでの縮重をもたらす。次いで、2つのオーバーラップPCRフラグメントのそれぞれ25 ngと、NcoI及びEagIでの消化により線状にした75 ngのオーバーラップベクターDNA (pCLS0542)とを用いて、酵母サッカロミセス・セレビシエFYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietzら, Methods Enzymol. 2002; 350:87〜96)。
両方の群の変異を含むインタクトなコード配列を、酵母でのインビボ相同組み換えにより作製した。D7Clib2の核酸多様性は322 = 1024であり、形質転換の後にほぼライブラリー全体の多様性を表す1116個のクローンを採集した。
【0181】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0182】
結果
上記の酵母スクリーニングアッセイを用いて、DmoCre2 D4Clib2Bisライブラリーを構成する1116個のクローンを、DC4GTC標的以外の64個全てのDC4NNN標的に対してスクリーニングした。スクリーニングにより、少なくとも1つのDC7NNN標的を切断できる174個の陽性クローンが得られた。これらのクローンを再配置し、配列決定し(75個のユニーク配列を単離した)、2回目のスクリーニングに供した。元のDmoCre2タンパク質は、63個のDC7NNN標的のうち、9個(DC7CCC, DC7TCC, DC7ACC, DC7GCC, DC7TTC, DC7ATC, DC7TCT, DC7ACT及びDC7TTT)を切断できた。図19に示すD7Clib2ヒットマップは、DmoCre2コード配列中に27位及び37位にて変異を導入することにより、19個のDC7NNN標的がDmoCre2由来変異体により切断できるようになったことを示す。我々のスクリーニングアプローチにより、よって、DC7NNN標的についてのDmoCre2切断スペクトル広げることができ、新しい切断特異性を単離することが可能になる。
【0183】
実施例10:2組の変異を組み合わせ、かつ組み合わせDC10TGG4ACT標的を切断する新しいDmoCre由来変異体の作製
DmoCre2タンパク質について以前に単離された異なる組の変異を組み合わせて、組み合わせ標的を切断する可能性を調べた。まず、野生型I-DmoI (配列番号22)中の75位、76位及び77位に相当する残基で変異させ、DC4ACT標的を切断できる8個のDmoCre2由来変異体を選択した。配列番号22中の75〜77位に相当する残基での配列については表VIを参照されたい。これらの変異体を用いて、配列番号22中の29位及び33位のアミノ酸に相当するDmoCre2残基で縮重した変異体ライブラリー(SeqDC10NNN4ACT)を創出した。得られたライブラリーを、最後に、酵母において、組み合わせDC10TGG4ACT標的に対してスクリーニングした。
【0184】
材料及び方法
DC10TGG4ACT標的ベクターの構築:
標的は、以下のようにしてクローニングした:ゲートウェイクローニング配列に接する標的配列に対応するオリゴヌクレオチドを、Proligoに注文した:5'TGGCATACAAGTTTTCCCAGGAAGTTACGACGTTTTGACAATCGTCTGTCA-3' 配列番号60。一本鎖オリゴヌクレオチドのPCR増幅により作製した二本鎖標的DNAを、ゲートウェイプロトコル(Invitrogen)を用いて酵母レポーターベクター(pCLS1055、図4)にクローニングした。酵母レポーターベクターでエス・セレビシエFYBL2-7B株(MAT-α, ura3Δ851, trp1Δ63, leu2Δ1, lys2Δ202)を形質転換した。
【0185】
DmoCre2 SeqDC10NNN4ACT変異体ライブラリーの構築:
まず、DC4ACT標的を切断できる8個のDmoCre2変異体についてのDNAコードをプールした。次いで、このDNAプールを、2回の別々のオーバーラップPCR反応のための鋳型として用いて、29位及び33位での変異を含むDmoCre2由来コード配列を作製した。1回目のPCR反応は、DmoCre2コード配列の5'末端(aa 1位〜40位)を、プライマーGal10F (5'-GCAACTTTAGTGCTGACACATACAGG-3' 配列番号6)及びD10CreRev2 (5'- GATCACAACACGATATTCGCTMNNGTTACCTTTMNNTTTCAGCTTGTA-3' 配列番号61)を用いて増幅し、2回目のPCR反応は、DmoCre2コード配列の3'末端(34位〜264位)を、特異的プライマーGal10R (5'-ACAACCTTGATTGGAGACTTGACC-3' 配列番号4)及びD10CreFor2 (5'-AGCGAATATCGTGTTGTGATCACCCAGAAGTCTG-3' 配列番号62)を用いて増幅する。
【0186】
29位及び33位でのNNKコドンをもたらすD10CreRev2オリゴヌクレオチド中のMNNコードは、これらの位置にて20個の可能なアミノ酸のうちでの縮重をもたらす。次いで、2つのオーバーラップPCRフラグメントのそれぞれ25 ngと、NcoI及びEagIでの消化により線状にした75 ngのオーバーラップベクターDNA (pCLS0542、図5) とを用いて、酵母サッカロミセス・セレビシエFYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietzら, Methods Enzymol. 2002; 350:87〜96)。インタクトなコード配列を、酵母でのインビボ相同組み換えにより作製した。形質転換の後に、SeqDC10NNN4ACTライブラリーの2232個のクローンを採集した。
【0187】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0188】
結果
DC4ACT標的を切断できる8個のDmoCre2由来変異体を選択した。これらの変異体は、配列番号22の75位、76位及び77位に対応する残基にて変異を有し、以下の表VIに列挙される。
【0189】
【表6】
【0190】
SeqDC10NNN4ACTライブラリーを、次いで、我々の酵母スクリーニングアッセイを用いて、組み合わせDC10TGG4ACT標的に対してスクリーニングした。スクリーニングアッセイにより11個の陽性クローンが得られ、スクリーニングの一部を図20に示し、ここでは3個の陽性クローンを黒丸で囲む。よって、我々はここで、C12D34標的の+8位〜+10位のヌクレオチドと相互作用する残基の変異を、C12D34標的の+2位〜+4位のヌクレオチドと相互作用する残基の変異と組み合わせて、組み合わせ標的を切断できることを示す。
【0191】
実施例11:RAG1.10.3DC4ACT及びRAG1.10.3DC4TAT標的を切断する新しいRAG1.10.3D34由来変異体の作製
以下の2つの標的の可能性のあるカッターを見出すために(図14): RAG1.10.3DC4ACT (5'-CTGGCTGAGGTAACTTCCGGCG-3' 配列番号72)及びRAG1.10.3DC4TAT (5'- CTGGCTGAGGTATATTCCGGCG-3' 配列番号73)、実施例7に記載される改良RAG1.10.3D34カッター(Amel2_RG3D変異体、配列番号58)を採用して、野生型I-DmoI (配列番号22)の75位、76位及び77位に対応するAmel2_RG3D (配列番号58)の残基を縮重する変異体ライブラリー(RAG1.10.3DC4NNN)を構築した。
【0192】
材料及び方法
RAG1.10.3DC4ACT及びRAG1.10.3DC4TAT標的ベクターの構築:
標的は、以下のようにしてクローニングした:ゲートウェイクローニング配列と接する上記の標的配列の相補鎖に相当するオリゴヌクレオチドを、Proligoに注文した: 5'TGGCATACAAGTTTTCGCCGGAAGTTACCTCAGCCAGACAATCGTCTGTCA-3' 配列番号74 (RAG1.10.3DC4ACT標的について)及び5'TGGCATACAAGTTTTCGCCGGAATATACCTCAGCCAGACAATCGTCTGTCA-3' 配列番号75 (RAG1.10.3DC4TAT標的について)。一本鎖オリゴヌクレオチドのPCR増幅により作製した二本鎖標的DNAを、ゲートウェイプロトコル(Invitrogen)を用いて酵母レポーターベクター(pCLS1055、図4)にクローニングした。酵母レポーターベクターでエス・セレビシエFYBL2-7B株(MAT-α, ura3Δ851, trp1Δ63, leu2Δ1, lys2Δ202)を形質転換した。
【0193】
RAG1.10.3DC4NNN変異体ライブラリーの構築
Amel2_RG3DのDNA (配列番号58)を鋳型として用いて、D4Clib4作製について実施例3に記載されるのと同じプロトコルを用いて、RAG1.10.3DC4NNN変異体ライブラリーを構築した。2232個のクローンを採集した。
【0194】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0195】
結果
RAG1.10.3DC4NNNライブラリーを構成する2232個のクローンを、2つの標的RAG1.10.3DC4ACT (配列番号72)及びRAG1.10.3DC4TAT (配列番号73)に対して、我々の酵母スクリーニングアッセイを用いてスクリーニングした。スクリーニングにより、RAG1.10.3DC4ACT標的に対して68個の陽性クローン(図21、A)、及びRAG1.10.3DC4TAT標的に対して26個の陽性クローン(図21、B)が得られた。Amel2_RG3D変異体(右上の点の対照)は、RAG1.10.3DC4ACT標的もRAG1.10.3DC4TAT標的も切断しなかった。それぞれの陽性クローンは、2つの標的のうちの1つだけを切断することが見出された。これらの結果は、得られた変異体の特異性を示す。このスクリーニングにより、よって、DmoCreタンパク質のCre部分に変異を導入した後に(Amel2_RG3D変異体)、I-DmoI部分にさらに変異を加えることによりタンパク質をさらに工学的に改変して、組み合わせ標的を特異的に切断することが可能であることが示される。
【技術分野】
【0001】
本発明は、改善された活性及び変化されたDNA標的配列を有する改変I-DmoIドメインを含む、キメラメガヌクレアーゼ酵素に関する。特に、本発明は、I-CreI単量体に連結された改変I-DmoIドメインを含むキメラメガヌクレアーゼ酵素に関する。
【背景技術】
【0002】
所定の遺伝子座を設計する方策の中でも、メガヌクレアーゼのような稀に切断をするDNAエンドヌクレアーゼの使用は、稀切断性(rare-cutting)DNAエンドヌクレアーゼによるDNA二本鎖破断(DSB)及び該破断の部位での相同組み換え事象をもたらすことを介する遺伝子ターゲッティングの成功率を向上させるための有力なツールとして浮上してきた。
【0003】
メガヌクレアーゼは、大きい(12〜45 bp)DNA標的部位を認識するエンドヌクレアーゼである。天然においては、メガヌクレアーゼは、非常に稀に切断をするエンドヌクレアーゼのファミリーであるホーミングエンドヌクレアーゼを実質的に含む。このファミリーは、酵母サッカロミセス セレビシエ(Saccharomyces cerevisiae)のミトコンドリアグループIイントロンにより元来コードされるタンパク質I-SceI(オメガヌクレアーゼ)のインビボでの使用により最初に特徴付けられた。イントロンのORF、独立遺伝子又は介在配列(インテイン)によりコードされるホーミングエンドヌクレアーゼは、それらを、細菌系R/MIIから一般的に単離されてきた「古典的な」制限酵素から区別する、顕著な構造的及び機能的特性を示す。
【0004】
ホーミングエンドヌクレアーゼは、12〜40 bpに及ぶDNAの認識配列を有するのに対して、「古典的な」制限酵素はもっと短いDNAの範囲、3〜8 bpの範囲を認識する(いわゆるレアカッター(rare-cutter)については12 bpまで)。したがって、ホーミングエンドヌクレアーゼは、ヒトゲノムと同程度に大きく複雑なゲノム中でさえも切断の頻度が非常に低い。
【0005】
グループIイントロン又はインテインによりコードされるいくつかのホーミングエンドヌクレアーゼは、それらの各遺伝子要素の対立遺伝子のイントロンレス又はインテインレス部位に戻ること(homing)を促進することが示されている。部位特異的な二本鎖破断をイントロンレス又はインテインレスの対立遺伝子中に作ることにより、これらのヌクレアーゼは、コード配列を複製し、DNAレベルでのイントロン又は介在配列の挿入を導く遺伝子変換過程に関与する、組み換え生成末端(recombinogenic ends)を作り出す。
【0006】
ホーミングエンドヌクレアーゼは、保存されたアミノ酸モチーフに基づいて分類された4つの別々のファミリーに属する。レビューとして、Chevalier及びStoddard(Nucleic Acids Research, 2001, 29, 3757-3774)を参照されたい。
【0007】
これらのファミリーの1つと本発明の主題は、ホーミングエンドヌクレアーゼファミリーのうちで最も大きいLAGLIDADGファミリーである。このファミリーは、保存された三次元構造(下記参照)により特徴付けられるが、上記の触媒中心である短いペプチドを除いて、一次配列レベルでは非常に不十分な保存を示す。このファミリーは、このペプチドの共通配列からLAGLIDADGと称され、各LAGLIDADGタンパク質において1つまたは2つのコピーが見出された。
【0008】
1つのLAGLIDADG (L)を有するホーミングエンドヌクレアーゼは、分子量がおよそ20 kDaであり、ホモ二量体として作用する。2つのコピーを有するもの(LL)は、25 kDa (230アミノ酸)〜50 kDa (HO、545アミノ酸)の範囲であり、各モチーフの間に70〜150残基を有し、単量体として作用する。標的配列の切断は認識部位の内側で生じ、3'OHの突出を有する4ヌクレオチドの互い違いの(staggered)切り口を残す。
【0009】
I-Ceu IおよびI-Cre I (163アミノ酸)は、1つのLAGLIDADGモチーフ(モノ- LAGLIDADG)を有するホーミングエンドヌクレアーゼである。I-DmoI (194アミノ酸、SWISSPROTアクセッション番号P21505 (配列番号22))、I-SceI、PI-PfuI及びPI-SceIは、2つのLAGLIDADGモチーフを有するホーミングエンドヌクレアーゼである。
【0010】
本発明においては特に記載しない限り、残基の番号は、SWISSPROT番号P21505 (配列番号22)又は構造PDBコード1b24のI-DmoI配列のアミノ酸番号付けを適用する。
【0011】
X線結晶解析を用いる構造モデルにより、I-CreI (PDBコード1g9y)、I-Dmo I (PDBコード1b24)、PI-Sce I、PI-Pfu Iが作り出されている。そのDNA部位に結合したI-Cre I及びPI-Sce I (Moureら、Nat Struct Biol、2002、9: 764-70)の構造も明らかになっており、特定のタンパク質-DNA接触についての多くの予測が導かれる。
【0012】
I-CreI (配列番号24)のような単独のモチーフを有するLAGLIDADGタンパク質は、ホモ二量体を形成し、パリンドローム又は偽パリンドローム(pseudo-palidromic) DNA配列を切断するのに対して、I-SceIのようなより大きい2重モチーフタンパク質は単量体であり、非パリンドローム標的を切断する。いくつかの異なるLAGLIDADGタンパク質は結晶化されており、それらは、一次配列レベルでの類似性の欠如とは対照的に、コア構造の顕著な保存を示している(Juricaら、Mol Cell. 1998; 2:469-76、Chevalierら、Nat Struct Biol. 2001; 8:312-6、Chevalierら、J Mol Biol. 2003; 329:253-69、Moureら、J Mol Biol. 2003; 334:685-95、Moureら、Nat Struct Biol. 2002; 9:764-70、Ichiyanagiら、J Mol Biol. 2000; 300:889-901、Duanら、Cell. 1997; 89:555-64、Bolduら、Genes Dev. 2003; 17:2875-88、Silvaら、J Mol Biol. 1999; 286:1123-36)。
【0013】
このコア構造において、二量体LAGLIDADGタンパク質中の2つの単量体又は単量体LAGLIDADGタンパク質中の2つのドメインが貢献する2つの特徴的なαββαββα折り畳みが、2回転対称で互いに面している。DNA結合は、逆平行βシートに折り畳まれ、かつDNAヘリックス主溝上のサドルを形成している各ドメインからの4つのβ鎖に依存する。触媒コアは中央にあり、両方の対称的単量体/ドメインの寄与を有する。このコア構造に加えて、他のドメイン:例えばPI-SceIでは、タンパク質スプライシングドメインを有するインテインと、さらなるDNA結合ドメインとを見出すことができる(Moureら、Nat Struct Biol. 2002; 9:764-70、Grindlら、Nucleic Acids Res. 1998; 26:1857-62)。
【0014】
LAGLIDADGファミリーの個々のメンバーの間での配列の保存を明らかに欠如するにもかかわらず、構造比較はLAGLIDADGタンパク質が、それらはI-CreIのような二量体又はI-DmoIのような単量体のように切断するが、同様の活性化コンフォメーションをとることを示す。全ての構造において、LAGLIDADGモチーフは中心にあり、2回転(偽-)対称軸が2つの単量体または見かけのドメインを隔てる2つのパックされたα-ヘリックスを形成する。
【0015】
LAGLIDADGモチーフは、I-CreI中の残基13〜21、並びにI-DmoI中の14〜22位及び110〜118位に該当する。LAGLIDADG α-へリックスのどちら側においても、4つのβ-シートは、タンパク質と標的DNA配列のハーフサイト(half site)との相互作用を促進するDNA結合インターフェースを提供する。I-DmoIは、第1ドメイン(残基1〜95)と第2ドメイン(残基105〜194)がリンカー(残基96〜104)により隔てられることを除いて、I-CreI二量体に類似する(Epinatら、Nucleic Acids Res、2003、31: 2952-62)。
【0016】
I-SceIは、哺乳動物細胞のゲノム標的において相同組み換えを1000倍以上に刺激するために用いられた最初のホーミングエンドヌクレアーゼであった(Choulikaら、Mol Cell Biol. 1995; 15:1968-73、Cohen-Tannoudjiら、Mol Cell Biol. 1998; 18:1444-8、Donohoら、Mol Cell Biol. 1998; 18:4070-8、Alwinら、Mol Ther. 2005; 12:610-7、Porteus、Mol Ther. 2006; 13:438-46、Rouetら、Mol Cell Biol. 1994; 14:8096-106)。
【0017】
近年、I-SceIは、マウス肝臓における標的とされた組み換えをインビボで刺激するためにも用いられ、肝細胞の1%まで組み換えを観察することができた(Goubleら、J Gene Med. 2006; 8:616-22)。そのような方法論に固有の制限は、興味対象の遺伝子座(locus)に天然のI-SceI切断部位を予め導入する必要があることである。
この制限を回避するために、過去何年にもわたって、仕立てられた切断特異性を有するジンクフィンガーヌクレアーゼを作り出す甚大な努力がなされてきた(Porteus M Hら、Nat Biotechnol. 2005; 23:967-73、Ashworthら、Nature. 2006; 441:656-9、Urnovら、Nature. 2005; 435、646-651、Smithら、Nucleic Acids Res. 2006、2006; 34:e149)。
【0018】
それらの高いレベルの特異性が与えられたなら、ホーミングエンドヌクレアーゼは、仕立てられたエンドヌクレアーゼを設計するための理想的な折り畳みを表す。いくつかの研究により、LAGLIDADGタンパク質からのDNA結合ドメインが設計され得ることが示されている(Chevalierら、Nucleic Acids Res. 2001; 29:3757-74)。
【0019】
PI-SceI (Gimbleら、J Mol Biol. 2003; 334:993-1008)、I-CreI (Seligmanら、Nucleic Acids Res. 2002; 30:3870-9、Sussmanら、J Mol Biol. 2004; 342:31-41、Rosenら、Nucleic Acids Res. 2006; Arnouldら、J Mol Biol. 2006; 355:443-58)、I-SceI (Doyonら、J Am Chem Soc. 2006; 128:2477-84) 及びI-MsoI (Ashworthら、Nature. 2006; 441:656-9)を含む、いくつかのLAGLIDADGタンパク質が、新たな配列結合又は切断特異性を得るために、合理的又は半合理的な(semi-rational)突然変異生成及びスクリーニングにより改変された。
【0020】
近年、ハイスループットスクリーニング法で補助された半合理的設計により、本出願人らは、LAGLIDADGファミリーからのホモ二量体であるI-CreIから何千もの新規タンパク質を得ることを可能としている(Smithら、Nucleic Acids Res. 2006; 34: e149; Arnouldら、J Mol Biol. 2006; 355:443-58)。
本出願人らは、I-CreIのDNA結合サブドメインを以前に同定し、これらが変異体を組み合わせて組み立てること(combinatorial assembly)を許容するのに十分に独立していたことを示している(Smithら、Nucleic Acids Res. 2006; 34: e149)。これらの発見により、選択された標的を切断する、設計されたI-CreI派生物の第二世代の産生が可能となった。
【0021】
この組み合わせの方策は、ヒトRAG1及びXPC遺伝子内に位置する天然DNA標的配列を切断するメガヌクレアーゼの創出により説明されている(Smithら、Nucleic Acids Res. 2006; 34: e149; Arnouldら、J Mol Biol. 2007; 371:49-65)。
【0022】
しかしながら、4つまでのサブドメインを組み合わせる能力により、標的となり得るDNA配列の数は顕著に増加しているが、所定のサイズの天然の標的配列にあり得る配列の完全な範囲に作用できる一組の酵素を製造することは依然として難しい。
最も分かりにくい因子の一つは、I-CreI標的部位の4つの中心のヌクレオチドの影響である。この領域において塩基特異的なタンパク質-DNA相互作用がないにもかかわらず、無作為に突然変異させた部位のライブラリーからの切断可能なI-CreI標的のインビトロ選択により、これら4つの塩基対の切断活性への重要性が明らかとなった(Argastら、J Mol Biol. 1998; 280:345-53)。より一般的には、あり得る全ての22塩基対配列を切断する、設計されたメガヌクレアーゼがI-CreI足場(scaffold)のみに由来し得ることはありそうにない。
【0023】
別の方策は、別個のメガヌクレアーゼからのドメインを組み合わせることである。このアプローチは、I-CreIとI-DmoIとの間のドメイン交換による新しいメガヌクレアーゼの創出により説明されており、2つの半分の親標的配列の融合に相当するハイブリッド配列を切断するメガヌクレアーゼの創出へと導いている(Epinatら、Nucleic Acids Res. 2003; 31:2952-62, Chevalierら、Mol. Cell. 2002; 10:895-905)。
【0024】
I-DmoIは、超好熱性古細菌デサルフロコッカス・モビリス(Desulfurococcus mobilis)からの22 kDaのエンドヌクレアーゼである。それは、どちらもLAGLIDADGモチーフを有する、2つの類似のドメインを含む単量体タンパク質である。以下D1234(配列番号30)と称するDNA標的なしの該タンパク質単独の構造は解明されている(Silvaら、J Mol Biol. 1999; 286:1123-36)。
【0025】
Chevalierらの研究グループ(Mol Cell. 2002; 10:895-905)は、2つのエンドヌクレアーゼI-DmoI及びI-CreIに基づいて、E-DreI(設計された(Engineered) I-DmoI/I-CreI)と呼ばれるキメラタンパク質を組み立てた。E-DreIは、該酵素の元の(initial)足場を作り出すための柔軟なリンカーにより連結されたI-CreIホモ二量体の単一のサブユニットへのI-DmoIのN末端ドメインの融合体からなる。そして、Chevalierらは、予め作成したI-DmoI及びI-CreIモデルの要素を組み合わせることにより作成した3Dモデルから予測される、いずれの潜在的な立体障害(steric clashes)も緩和できるように、コンピュータを利用した界面アルゴリズムの予測に基づいて多数の残基改変物を作った。
上記のChevalierら、2002において、2つの構成分子が面している表面間の残基は同定された;特に、E-DreI足場の47位、51位、55位、108位、193位及び194位の残基は、潜在的に障害することが同定された。これらの残基はアラニンに置き換えられたが、そのような改変タンパク質は不溶性であることが見出された。
【0026】
残基番号は、天然のI-DmoIリンカーの長さを模倣する3アミノ酸NGNリンカーにより隔てられるI-CreIの最後の156残基に融合したI-DmoIのN末端ドメインから101残基(第1メチオニンで始まる)を含むE-DreIオープンリーディングフレームを参照する。
そして、残基47、51、55、108、193及び194、並びに残基12、13、17、19、52、105、109及び113のコンピュータを利用した再設計と;それに続く、これらの残基において改変されたE-DreI酵素の可溶性を決定するための、選択された配列に対するインビボでのタンパク質折り畳みアッセイを介して、界面が最適化された。最終足場が、I-DmoIのI19、H51及びH55並びにI-CreIのE8、L11、F16、K96及びL97 (E105、L108、F113、K193及びL194に該当する)の改変で設計された。
【0027】
キメラDNA標的dre3(本出願人の命名を用いるとC12D34 (配列番号31))との複合体中のE-DreI (Chevalierら、Mol Cell. 2002; 10:895-905)の構造は、本明細書の図2中に示されるように解明されている。E-DreIは、このハイブリッドC12D34 (配列番号31)標的のみを認識し切断できることが示された。この構造から、多数の残基が、E-DreIのその標的ハイブリッド部位への塩基特異的接触であることが予測され、これらの残基はE-DreI中のI-DmoIの25、29、31、33、34、35、37、70、75、76、77、79、81であり;及びE-DreI中のI-CreIの残基123、125、127、130、135、137、139、141、163、165、167、172であった。
【0028】
本出願人も、設計されたホーミングヌクレアーゼ酵素により切断されるDNA標的配列の範囲を広げようとするためにDmoCre足場についての実験をこれまでに行っている。DmoCreは、2つのホーミングエンドヌクレアーゼI-DmoI及びI-CreIから組み立てられたキメラ分子である。それは、I-CreI単量体に連結したI-DmoIからのN末端部分を含む。DmoCreは足場としてすばらしい利点を有し得る:I-DmoI部分における変異は、I-CreIドメインにおける変異と組み合わせることができ、かつ数千のそのような種々のI-CreI分子はすでに同定され、プロファイルされている (Smith Jら、Nucleic Acids Res. 2006;34(22):e149 , Arnould Sら、J Mol Biol. 2006; 355:443-58, Arnould Sら、J Mol Biol. 2007; 371:49-65)。
【0029】
DNA標的なしのI-DmoIタンパク質単独の構造(Silvaら、J Mol Biol. 1999; 286:1123-36)及びI-CreIとそのDNA標的C1234 (配列番号28)との複合体の構造(Juricaら、Mol Cell. 1998; 2:469-76、Chevalierら、J Mol Biol. 2003; 329:253-69)に基づいて、キメラDmoCreエンドヌクレアーゼが組み立てられている(Epinatら、Nucleic Acids Res, 2003, 31: 2952-62)。DmoCreは、残基F109までのI-DmoIとそれに続く残基L13からのI-CreIに相当する単量体タンパク質である。立体障害を避けるために、I107はロイシン残基に変異されている。さらに、I-CreIの残基96及び97に近いことが見出されたI-DmoIの残基47、51及び55は、それぞれアラニン、アラニン及びアスパラギン酸に変異された。
【0030】
DmoCreは、インビトロで活性であることが示されており(Epinatら、Nucleic Acids Res, 2003, 31: 2952-62)、C1234 (配列番号28)又はC1221 (配列番号29) (C1234に由来するパリンドローム標的)の左部分及びD1234 (配列番号30)の右部分から構成されるハイブリッド標的C12D34 (配列番号31)を切断できた(図1)。さらに、それらのDNA標的配列を37°Cでより効率的に切断できるI-DmoI及びDmoCre変異型が、酵母細胞における無作為突然変異生成及びスクリーニングにより同定された(WO 2005/105989; Prietoら、J. Biol Chem. 2007 Nov 12; [出版に先んじるEpub])。
【0031】
それゆえ、E-DreI及びDmoCreキメラ酵素のみが、ハイブリッド標的C12D34 (配列番号31)を認識して切断できる。また、E-DreI及びDmoCreの足場は、共通して残基47、51及び55の改変を有する。
【発明の概要】
【発明が解決しようとする課題】
【0032】
本発明者らは、より広い一連の標的配列を認識する新世代のキメラ酵素を作り出すことに興味があり、それゆえ彼らは、キメラI-DmoI酵素における構成要素又は2つの異なるヌクレアーゼからの触媒ドメインを含むキメラ酵素のいずれかとして用いるためのI-DmoI酵素の第1ドメインのさらなる増強を研究した。新しいDNA配列を標的にできるようにし、そしてDNA標的配列を含む興味対象の部位に二本鎖破断を生じさせ得るようにすることにより、本出願人は、DNA組み換え事象、DNA喪失または細胞死を引き起こすことができるツールを提供する。
【0033】
この二本鎖破断は、特定の配列を修復し、特定の配列を改変し、変異した遺伝子に代えて機能的遺伝子を回復し、興味対象の内因性遺伝子を弱めるか又は活性化し、興味対象の部位に変異を導入し、外因性遺伝子又はその一部を導入し、内因性遺伝子若しくはその一部を不活性化するか又は検出し、染色体腕を転移させ、或いは未修復及び分解したDNAを放出するために用いることができる。それゆえ、そのような改変されたメガヌクレアーゼ酵素は、ユーザーにそのような改変されたメガヌクレアーゼ酵素の治療、研究または他の生産用途におけるより広い様々な潜在的な選択肢を与える。
【0034】
それゆえ、本発明者らは、これらのキメラ酵素が認識し切断できるDNA標的の数を増やそうとすることにより、少なくとも1つのI-DmoIドメインを含むキメラメガヌクレアーゼ酵素を改善しようと努めた。
【課題を解決するための手段】
【0035】
したがって、本発明は、少なくとも第1I-DmoIドメインを含み、該第1I-DmoIドメインの残基15、19又は20の少なくとも1つの置換及び27位、29位、33位、35位、37位、75位、76位、77位又は81位の残基の少なくとも1つの置換を含むことを特徴とし、±2位、±3位、±4位、±5位、±6位、±7位、±8位、±9位、±10位の少なくとも1つにおいて配列番号30の野生型I-DmoI DNA標的ハーフサイトとは異なるI-DmoI DNA標的ハーフサイトを認識する、I-DmoIエンドヌクレアーゼ又はそれらのキメラ派生物の配列を含むポリペプチドに関する。
【図面の簡単な説明】
【0036】
【図1】異なる22 bpのDNA標的を示す配列比較である。
【図2】DNA標的と結合しているE-DreIの構造を示す。
【図3】(A)DNA標的と結合しているE-DreIの分子表面を示す。(B)DNAとの相互作用している残基29、33及び35を示す拡大図。
【図4】pCLS1055の概略の制限マップ。
【図5】pCLS0542の概略の制限マップ。
【図6】8 DC10NNN標的に対するDClib2ライブラリーからのDmoCre2変異体の1次スクリーニングの例を示す。
【図7】DmoCre2のヒットマップ及び64 DC10NNN標的に対するDClib2ライブラリーを示す。
【図8】(A)DNA標的と結合しているE-DreIの分子表面。(B)DNAとの相互作用している残基75、76及び77を示す拡大図である。
【図9】DmoCre4及び64 DC4NNN標的に対するDClib4ライブラリーのヒットマップ。
【図10】いくつかの異なる22 bpのDNA標的を示す。
【図11】5CAGD34標的(配列番号33)に対するDCSca2_5CAG変異体及びDCSca4_5CAG変異体の1次スクリーニングの例を表す。
【図12】異なる22 bpのDNA標的を示す。
【図13】RAG1.10.2D34標的(配列番号35)、RAG1.10.2 (配列番号34)、C12D34 (配列番号31)、D1234 (配列番号30)及びC1221 (配列番号29) DNA標的に対するDmoM2変異体の酵母切断アッセイ。
【図14】異なる22 bpのDNA標的を示す。
【図15】それぞれの各標的に対するRAG1.10.2D34カッター及びRAG1.10.3D34カッターの2次スクリーニングの例を表す。
【図16】それぞれの標的に対する精製されたRAG1.10.2D34カッター及びRAG1.10.3D34カッターの2次スクリーニング。
【図17】64 DC4NNN標的に対するD4Clib2Bライブラリーのヒットマップ。
【図18】(A)自身のDNA標的と結合しているE-DreIの分子表面を示す。(B)DNAとの相互作用している残基37及び81を示す拡大図である。(C)残基37の付近にある残基27を示す別の拡大図である。
【図19】64 DC7NNN標的に対するD7Clib2ライブラリーのヒットマップ。
【図20】組み合わされたDC10TGG4ACTに対するSeqDC10NNN4ACTライブラリーからのDmoCre2変異体の1次スクリーニングの例を表す。
【図21】RAG1.10.3DC4ACT標的(A)及びRAG1.10.3DC4TAT標的(B)に対するRAG1.10.3DC4NNNライブラリーからの変異体の1次スクリーニングの例を表す。
【発明を実施するための形態】
【0037】
この明細書を通じて、記載されるDmoCreキメラ酵素は、クローニング手段のために2位にバリンを含む。この付加的残基は、キメラ酵素配列内の残基の番号付けには含まれない。それゆえ、例えばキメラ酵素の19位の残基は、実際にはこのキメラ酵素の第20番目の残基である。
【0038】
本発明者らは、I-DmoIの第1ドメインと別の機能的エンドヌクレアーゼドメイン若しくは単量体との組み合わせを含むキメラ酵素のような、改善されたI-DmoIエンドヌクレアーゼ又はその派生物をコードするポリペプチドを提供する。このポリペプチドは、天然のI-DmoIタンパク質の残基1〜95に該当する第1I-DmoIドメインにおいて2つ以上のアミノ酸残基の変異(changes)を有する。特に、第1I-DmoIドメインはI-DmoIアミノ酸配列(配列番号22)の1位〜95位に該当し、I-DmoIリンカーは96位〜104位に該当し、第2I-DmoIドメインの開始部(beginning)は105位〜109位に該当し、これは本出願人が開発し、本明細書に記載した2つの新しいキメラメガヌクレアーゼ足場である、DmoCre2及びDmoCre4に用いられた完全断片である。好ましくは、完全な109残基の断片は、第1I-DmoIドメイン断片としてキメラ酵素中に用いられる
【0039】
残基15、19及び20の変異は、本発明者らによりDmoCre2と呼ばれるキメラタンパク質の活性が増加する結果になることが、本発明者らによる実験により示されている。残基29、33及び35の変異は、I-DmoI DNA標的ハーフサイト(配列番号30)の±8位〜±10位にて、I-DmoIのこの改変されたドメインにより認識される配列を変化させることが、本出願人により初めて示されている。残基75、76及び77の変異は、I-DmoI DNA標的ハーフサイト(配列番号30)の±2位〜±4位にて、I-DmoIのこの改変されたドメインにより認識される配列を変化させることが、本発明者らにより初めて示されている。残基27、37及び81の変更は、I-DmoI DNA標的ハーフサイト(配列番号30)の±5位〜±7位にて、I-DmoIのこの改変されたドメインにより認識される配列を変化させることが、本発明者らにより初めて示されている。したがって、本発明者らは、ハイブリッド配列C12D34とは異なる標的配列を認識できる改善された第1I-DmoIドメインを提供する。該I-DmoI DNA標的ハーフサイト(配列番号30)はAAGTTCCGGCGであり、+2〜+4及び+8〜+10の領域は太字で、+5〜+7の領域は斜字である。
【0040】
そのようなポリペプチドは、改変されたメガヌクレーゼを含み、DmoCre及びE-DreIにより認識され切断されるハイブリッド標的配列以外の、より広い範囲のDNA標的配列を認識し切断できるようにする。
【0041】
特に、15位、19位又は20位の残基の少なくとも1つが任意のアミノ酸に置換される。
特に、本発明によるポリペプチドは、グルタミンへ変異される15位のリジンの改変である、L15Q変異を含み得る。
特に、本発明によるポリペプチドは、アスパラギン酸へ変異される19位のイソロイシンの改変である、I19D変異を含み得る。残基19の改変により、I-DmoIドメインをより活性にすることが本出願人によって示されている。
特に、本発明によるポリペプチドは、セリン又はアラニンへ変異される20位のグリシンの改変である、G20S又はG20A変異を含み得る。
【0042】
特に、ポリペプチドは、107位において少なくとも1つの改変された残基も含み得る。
特に、本発明によるポリペプチドは、リジンへ変更される107位のイソロイシンの改変である、I107L改変を含み得る。残基107の改変は、I-DmoIドメインと、他の酵素、例えばI-CreIのドメインとの間の立体障害を防ぐだろう。
【0043】
特に、任意のアミノ酸による29位、33位又は35位の残基の少なくとも1つの置換は、±8位、±9位、±10位の少なくとも1つにおいて配列番号30の野生型I-DmoI DNA標的ハーフサイトとは異なるI-DmoI DNA標的ハーフサイトの該ポリペプチドの認識を変化させる。
【0044】
特に、任意のアミノ酸による75位、76位又は77位の残基の少なくとも1つの置換は、±2位、±3位、±4位の少なくとも1つにおいて配列番号30の野生型I-DmoI DNA標的ハーフサイトとは異なるI-DmoI DNA標的ハーフサイトの該ポリペプチドの認識を変化させる。
【0045】
特に、任意のアミノ酸による27位、37位又は81位の残基の少なくとも1つの置換は、±5位、±6位、±7位の少なくとも1つにおいて配列番号30の野生型I-DmoI DNA標的ハーフサイトとは異なるI-DmoI DNA標的ハーフサイトの該ポリペプチドの認識を変化させる。
【0046】
特に、該ポリペプチドは、配列番号1の配列に由来する。
本願において、に由来するとは、元の配列から作り出され、そして元の機能性を保持するように改変されるが、元の機能性を保持するかぎり、元の配列と比較して残基の変異及び/又は付加若しくは欠失を有する、任意の核酸又はタンパク質の配列を意味する。
配列番号1は、DmoCre2 (配列番号2)中のI-DmoIドメインとして本発明で用いられた残基15及び19にて改変されたI-DmoIドメインの配列である。このI-DmoIドメインも残基107への改変を含むが、Epinatら(Nucleic Acids Res、2003、31: 2952-62)のようなL47A、H51A及びL55Dへの改変は含まない。
【0047】
特に、該ポリペプチドは、配列番号27の配列に由来する。
配列番号27は、DmoCre4 (配列番号9)中のI-DmoIドメインとして本発明で用いられる残基19、20及び109にて改変されたI-DmoIドメインの配列である。このI-DmoIドメインは、Epinatら(Nucleic Acids Res、2003、31: 2952-62)のようなL47A、H51A及びL55Dへの改変は含まない。
【0048】
特に、該ポリペプチドは、二量体LAGLIDADGホーミングエンドヌクレアーゼの配列又は別の単量体LAGLIDADGホーミングエンドヌクレアーゼのドメインへの該第1I-Dmo Iドメインの融合体からなるキメラI-Dmo Iエンドヌクレアーゼである。
【0049】
本発明は、単一の単量体タンパク質における配列番号22の残基1〜95、或いは本発明により変化された2つのI-DmoIドメインの組み合わせを含む、改変された第1I-DmoIドメイン及び第2野生型I-DmoIドメインの両方を含む、改変されたI-DmoIエンドヌクレアーゼ酵素に関する。改変されたI-DmoIドメインが、I-Sce I、I-Chu I、I-Cre I、I-Csm I, PI-Sce I, PI-Tli I、PI-Mtu I、I-Ceu I、I-Sce II、I-Sce III、HO、PI-Civ I、PI-Ctr I、PI-Aae I、PI-Bsu I、PI-Dha I、 PI-Dra I、PI-Mav I、PI-Mch I、PI-Mfu I、PI-Mfl I、PI-Mga I、PI-Mgo I、PI-Min I、PI-Mka I、PI-Mle I、PI-Mma I、PI-Msh I、PI-Msm I、PI-Mth I、PI-Mtu I、PI-Mxe I、PI-Npu I、PI-Pfu I、PI-Rma I、PI-Spb I、PI-Ssp I、PI-Fac I、PI-Mja I、PI-Pho I、PI-Tag I、PI-Thy I、PI-Tko I、I-MsoI及びPI-Tsp I;好ましくは、I-Sce I、I-Chu I、I-Dmo I、I-Csm I、PI-Sce I、PI-Pfu I、PI-Tli I、PI-Mtu I、及びI-Ceu Iのような別のLAGLIDADGエンドヌクレアーゼのドメインと組み合わされ得ることも本発明の一面である。
【0050】
また、本発明は、I-DmoIの第1ドメインの配列が、(i) 上記の第1I-Dmo Iドメインの4位、49位、52位、92位、94位及び/若しくは95位の残基の1つ、並びに/或いは(ii) I-Dmo Iの第2ドメインのリンカー又は開始部の101位、102位及び/若しくは109位の残基の1つ
の群から選択される少なくとも1つのさらなる残基の置換も含むポリペプチドに関する。
【0051】
上記のポリペプチドの有利な実施形態によれば:
- 4位のアスパラギンがイソロイシンに変異され(N4I)、
- 49位のリジンがアルギニンに変異され(K49R)、
- 52位のイソロイシンンがフェニルアラニンに変異され(I52F)、
- 92位のアラニンがスレオニンに変異され(A92T)、
- 94位のメチオニンがリジンに変異され(M94K)、
- 95位のロイシンがグルタミンに変異され(L95Q)、
-(存在する場合)101位のフェニルアラニンがシステインに変異され(F101C)、
-(存在する場合)102位のアスパラギンがイソロイシンに変異され(N102I)、及び/又は
-(存在する場合)109位のフェニルアラニンがイソロイシンに変異される(F109I)。
【0052】
特に、該ポリペプチドの第1I-Dmo Iドメインは、上記のキメラ-DmoエンドヌクレアーゼのNH2末端にある。
特に、キメラ-Dmoエンドヌクレアーゼの一部を形成する二量体LAGLIDADGホーミングエンドヌクレアーゼは、I-CreIである。
【0053】
特に、キメラI-Dmo Iエンドヌクレアーゼは、配列番号2の配列に由来する。
配列番号2は、残基15、19及び107にて改変されたI-Dmo Iドメインを含む、本発明の好ましいDmoCre2キメラエンドヌクレアーゼのペプチド配列である。
【0054】
特に、本発明によるポリペプチドは、配列番号9の配列に由来する。
配列番号9は、残基19、20及び109にて改変されたI-Dmo Iドメインを含む、本発明の好ましいDmoCre4キメラエンドヌクレアーゼのペプチド配列である。
【0055】
特に、本発明のこの第一面によるポリペプチドは、検出可能なタグをNH2末端及び/又はCOOH末端に含み得る。
【0056】
本発明はポリヌクレオチドにも関し、このポリヌクレオチドは本発明によるポリペプチドをコードすることを特徴とする。
【0057】
本発明は、本発明によるポリヌクレオチドを含むことを特徴とする、ベクターにも関する。
本発明は、本発明によるポリヌクレオチド又はベクターにより改変されることを特徴とする、宿主細胞にも関する。
上記のポリヌクレオチドを含む組み換えベクターは、公知の組み換えDNA及び遺伝子工学的技術により得ることができ、宿主細胞に導入できる。
【0058】
本発明のポリペプチドは、前記ポリペプチドの発現に適切な条件下で、該ポリペプチドをコードするポリヌクレオチド配列を含む発現ベクターを有する宿主細胞を培養し、該宿主細胞培養物から該ポリペプチドを回収することにより得ることができる。
【0059】
本発明は、本発明によるポリヌクレオチド又ベクターにより、その構成細胞の全て又は一部が改変されることを特徴とする、非ヒトトランスジェニック動物にも関する。
本発明は、本発明によるポリヌクレオチド又ベクターにより、その構成細胞の全て又は一部が改変されることを特徴とする、トランスジェニック植物にも関する。
【0060】
本発明は、第1I-DmoIドメインの残基15、19、20の少なくとも1つの置換及び27位、29位、33位、35位、37位、75位、76位、77位又は81位の残基の少なくとも1つの置換からなる該第1I-DmoIドメインを少なくとも含み、I-CreI単量体の配列に融合され、該I-CreI単量体配列が前記I-CreI単量体の44位、68位、70位、75位又は77位の残基の少なくとも1つの改変を含む、ポリペプチドにも関する。
【0061】
I-CreI単量体における残基番号の参照は、参照I-CreI単量体配列の配列番号24に適用する。そのようなポリペプチドは、例えば5CAGD34 (配列番号33)標的を切断できる。5CAGD34 (配列番号33)は、I-DmoI標的DNA配列(配列番号30)の第2ハーフに融合された5CAG_P標的(配列番号32)の第1ハーフである。5CAG_P標的(配列番号32)は、±3位、±4位及び±5位にて配列CAGへ改変された野生型I-CreI標的DNA配列に当てはまる。
全ての標的配列は、22又は24 bpのパリンドローム配列である。それゆえ、それらは、改変されたヌクレオチドに続く接尾辞_Pによってのみ記載されるだろう。
【0062】
本発明は、第1I-DmoIドメインの残基15、19、20のなくとも1つの置換及び27位、29位、33位、35位、37位、75位、76位、77位又は81位の残基の少なくとも1つの置換を含む該第1I-DmoIドメインを少なくとも含み、I-CreI単量体の配列に融合され、該I-CreI単量体配列が該I-CreI単量体の28位、30位、32位、33位、38位又は40位の残基の少なくとも1つの改変を含む、I-DmoIエンドヌクレアーゼ又はそれらのキメラ派生物の配列を含むポリペプチドにも関する。
【0063】
そのようなポリペプチドは、例えばRAG1.10.2D34標的(配列番号35)又はRAG1.10.3D34 標的(配列番号39)を切断できる。RAG1.10.2D34は、I-DmoI標的DNA配列(配列番号30)の第2ハーフに融合されたRAG1.10.2 DNA標的(配列番号34)の第1ハーフである。RAG1.10.3D34は、I-DmoI標的DNA配列(配列番号30)の第2ハーフに融合されたRAG1.10.3 DNA標的(配列番号38)の第1ハーフである。
【0064】
本発明は、第1I-DmoIドメインの残基15、19、20のなくとも1つの置換及び27位、29位、33位、35位、37位、75位、76位、77位又は81位の残基の少なくとも1つの置換にある(consisting in)該第1I-DmoIドメインを少なくとも含み、I-CreI単量体の配列に融合され、該I-CreI単量体配列が該I-CreIドメインの37位、79位、81位の残基の少なくとも1つの改変を含む、I-DmoIエンドヌクレアーゼ又はそれらのキメラ派生物の配列を含むポリペプチドにも関する。
【0065】
27位、37位又は81位が改変される場合、そのようなポリペプチドは、C12D34 (配列番号31) DNA標的配列の+5〜+7であるDmoCreの7NNN部分が野生型ヌクレオチド配列標的CGAとは異なっている標的を切断できる。
【0066】
本発明のよりよい理解のために、またどのようにして同じく実施するのかを示すために、添付の図面を参照して、本発明による特定の実施形態、方法及びプロセスを一例としてのみ示される:
【0067】
図1:異なる22 bpのDNA標的を示す配列比較であって、C1234 (配列番号28)は野生型I-CreI標的であり、C1221 (配列番号29)はC1234 (配列番号28)の左部分に由来するパリンドローム配列であり、D1234 (配列番号30)は野生型I-DmoI標的であり、そしてC12D34 (配列番号31)はキメラDmoCreタンパク質のハイブリッド標的であり、またDC10NNN標的(配列番号8)は+8位、+9位及び+10にて縮重が導入されたC12D34 (配列番号31)からの派生物である。
【0068】
図2:DNA標的と結合しているE-DreIの構造を示す(PDBコード1MOW)。
図3A:DNA標的と結合しているE-DreIの分子表面を示す。
図3B:DNAとの相互作用している残基29、33及び35を示す拡大図。点線は水素結合を示す。
図4:pCLS1055の概略の制限マップ。
図5:pCLS0542の概略の制限マップ。
図6:8 DC10NNN標的に対するDClib2ライブラリーからのDmoCre2変異体の1次スクリーニングの例を示す。
【0069】
図7:DmoCre2のヒットマップ及び64 DC10NNN標的に対するDClib2ライブラリーを示す。ヒットマップ中に示される各標的は、C12D34 (配列番号31)の相補鎖である;例えばヒットマップ中のCGGは、以下の実施例2で定義されるDC10CCGに相当する。
【0070】
図8:DNA標的と結合しているE-DreIの分子表面(図8A)。無作為化のために選択された結合の領域(+2位、+3位、+4位の塩基対並びにタンパク質残基75、76及び77)は赤で強調されている。図8Bは、DNAとの相互作用している残基75、76及び77を示す拡大図である。点線は水素結合を示す。
【0071】
図9:DmoCre4及び64 DC4NNN標的(DC4TTCはない)からの63に対するDClib4ライブラリーのヒットマップ。D4Clib4ヒットマップにおいて、それぞれの切断された標的の下の数は、この標的を切断するクローンの数である。これらのクローンのいくつかは、重複した配列を有し得る。各標的において、灰色レベルは切断強度の平均に比例する。ヒットマップ中に示される各標的は、C12D34 (配列番号31)の相補鎖である;例えばヒットマップ中のAGGは、以下の実施例3で定義されるDC4CCTに相当する。
【0072】
図10:いくつかの異なる22 bpのDNA標的を示す。5CAG_P (配列番号32)パリンドローム標的は、灰色のボックスで強調される±5位、±4位、±3位において相違を有するC1221に由来する。I-CreI標的部分において、5CAGD34 (配列番号33)標的は、5CAG_P (配列番号32)がC1221とは異なるのと同様に、C12D34 (配列番号31)とは異なる。
【0073】
図11:この図は、5CAGD34標的(配列番号33)に対するDCSca2_5CAG変異体及びDCSca4_5CAG変異体の1次スクリーニングの例を表す。DCSca2_5CAGライブラリースクリーニングでは、我々は9ドットクラスタ形式を用いた。各9ドット酵母クラスタにおいて、異なる変異体が、左上のドットの5CAGD34 (配列番号33)標的に対して試験される。DCSca4_5CAGライブラリースクリーニングでは、我々は4ドットクラスタ形式を用いた。各4ドットクラスタにおいて、異なる変異体が、左の2ドットの5CAGD34 (配列番号33)標的に対して試験され、一方、右の2ドットは内部対照クラスタである。H10、H11及びH12クラスタは陽性対照及び陰性対照を含む。
【0074】
図12:異なる22 bpのDNA標的を示す。RAG1.10.2 DNA配列(配列番号34)は、C1221 (配列番号29)に由来するパリンドローム標的である。10GTT及び5CAGモジュールは灰色のボックスで強調されている。I-CreI標的部分では、RAG1.10.2D34標的(配列番号35)は、RAG1.10.2 (配列番号32)がC1222とは異なるのと同様に、C12D34 (配列番号31)とは異なる。
【0075】
図13:RAG1.10.2D34標的(配列番号35)、RAG1.10.2 (配列番号34)、C12D34 (配列番号31)、D1234 (配列番号30)及びC1221 (配列番号29) DNA標的に対するDmoM2変異体の酵母切断アッセイ。各4ドット酵母クラスタにおいて、左の2ドットは、表示された標的を有するDmoM2変異体の切断アッセイを示し、一方、右の2ドットは内部対照である。
【0076】
図14:異なる22 bpのDNA標的を示す。RAG1.10.3 DNA配列は、C1221に由来するパリンドローム標的である。10 TGG及び5 GAGモジュールは灰色のボックスで強調されている。I-CreI標的部分では、RAG1.10.3D34標的(配列番号35)は、RAG1.10.3がC1222とは異なるのと同様に、C12D34とは異なる。
【0077】
図15:この図は、それぞれの各標的に対するRAG1.10.2D34カッター及びRAG1.10.3D34カッターの2次スクリーニングの例を表す。RAG1.10.2D34標的では、異なる変異体が、左上の各酵母クラスタにおけるその標的に対して試験される。RAG1.10.3D34標的では、異なる変異体が、左下の各酵母クラスタにおけるその標的に対して試験される。各4ドットクラスタにおいて、右の2ドットは内部対照クラスタである。
【0078】
図16:それぞれの標的に対する精製されたRAG1.10.2D34カッター及びRAG1.10.3D34カッターの2次スクリーニング。試験設計が示されている。RAG1.10.2D34スクリーニングでは、最初の変異体はRG2D2であり、一方、RAG1.10.3D34スクリーニングでは、それはRG3D3である。精製されたRAG1.10.2D34カッターはA9、B3、C5に位置し、黒の円はそれぞれAmel1_RG2D、Amel2_RG2D、Amel3_RG2Dである。精製されたRAG1.10.3D34カッターはA8、B3、E3に位置し、黒の円はそれぞれAmel1_RG3D、Amel2_RG3D、Amel3_RG3Dである。
【0079】
図17:64 DC4NNN標的に対するD4Clib2Bライブラリーのヒットマップ。それぞれの切断された標的の下の数は、この標的を切断する異なる配列を有するDmoCre2変異体の数である。各標的において、灰色レベルは切断強度の平均に比例する。ヒットマップ中に示される各標的は、C12D34 (配列番号31)の相補鎖である;例えばヒットマップ中のCAGは、実施例3で定義されるDC4CTGに相当する。
【0080】
図18:DNA標的と結合しているE-DreIの分子表面を示す(図17A)。無作為化のために選択された結合の領域(+5位、+6位、+7位の塩基対ならびにタンパク質残基37及び81)は黒で強調されている。図16Bは、DNAとの相互作用している残基37及び81を示す拡大図である。点線は水素結合を示す。図16Cは、残基37の付近にある残基27を示す別の拡大図である。
【0081】
図19:64 DC7NNN標的に対するD7Clib2ライブラリーのヒットマップ。それぞれの切断された標的の下の数は、この標的を切断する異なる配列を有するDmoCre2変異体の数である。各標的において、灰色レベルは切断強度の平均に比例する。ヒットマップ中に示される各標的は、C12D34 (配列番号31)の相補鎖を参照する;例えばヒットマップ中のGGAは、実施例9で定義されるDC7TCCに相当する。
【0082】
図20:この図は、組み合わされたDC10TGG4ACT標的に対するSeqDC10NNN4ACTライブラリーからのDmoCre2変異体の1次スクリーニングの例を表す。各酵母クラスタにおいて、右の2ドットは実験の内部対照である。他の4ドットにおいて、1ドットはSeqDC10NNN4ACTライブラリーからの1つの変異体に相当する。3つの陽性クローンは黒丸である。
【0083】
図21:この図は、RAG1.10.3DC4ACT標的(A)及びRAG1.10.3DC4TAT標的(B)に対するRAG1.10.3DC4NNNライブラリーからの変異体の1次スクリーニングの例を表す。各酵母クラスタにおいて、右上のドットはAmel2_RG3D変異体に相当し、右下のドットは実験の内部対照である。他の4ドットにおいて、1ドットはRAG1.10.3DC4NNNライブラリーからの1つの変異体に相当する。いくつかの陽性クローンは黒丸である。
【0084】
ここに記載されるものは、発明者らにより意図される特定の形態の一例である。以下の記載において、多数の特定の詳説は完全な理解を提供するために記述される。しかしながら、本発明がこれらの特定の詳説に限定されることなく実施され得ることは当業者に明らかであろう。他の例では、記載を不必要にあいまいにしないようにするため、公知の方法及び構造は記載されない。
【0085】
定義
- ポリペプチド配列中のアミノ酸残基は、本明細書において、1文字コードに従って表し、例えばQはGln又はグルタミン残基を意味し、RはArg又はアルギニン残基を意味し、DはAsp又はアスパラギン酸残基を意味する。
- 疎水性アミノ酸は、ロイシン(L)、バリン(V)、イソロイシン(I)、アラニン(A)、メチオニン(M)、フェニルアラニン(F)、トリプトファン(W)及びチロシン(Y)のことである。
【0086】
- ヌクレオチドは、次のように表す:1文字コードは、ヌクレオシドの塩基を表すために用いる:aはアデニンであり、tはチミンであり、cはシトシンであり、gはグアニンである。縮重ヌクレオチドについて、rはg又はa (プリンヌクレオチド)を表し、kはg又はtを表し、sはg又はcを表し、wはa又はtを表し、mはa又はcを表し、yはt又はc (ピリミジンヌクレオチド)を表し、dはg、a又はtを表し、vはg、a又はcを表し、bはg、t又はcを表し、hはa、t又はcを表し、nはg、a、t又はcを表す。
【0087】
- 「メガヌクレアーゼ」により、12〜45 bpの二本鎖DNA標的配列を有するエンドヌクレアーゼを意図する。
- 「親のLAGLIDADGホーミングエンドヌクレアーゼ」により、野生型ホモ二量体LAGLIDADGホーミングエンドヌクレアーゼ、又はその機能的変異型を意図する。上記の親のLAGLIDADGホーミングエンドヌクレアーゼは、22〜24 bpの二本鎖DNA標的を切断できる機能的エンドヌクレアーゼに組み合わされる、単量体、2つのLAGLIDADGホーミングエンドヌクレアーゼのコアドメインを含む二量体(ホモ二量体又はヘテロ二量体)であり得る。
【0088】
- 「ホモ二量体LAGLIDADGホーミングエンドヌクレアーゼ」により、単独LAGLIDADGモチーフを有し、パリンドロームDNA標的配列を切断するI-CreI若しくはI-MsoIのような野生型ホモ二量体LAGLIDADGホーミングエンドヌクレアーゼ、又はその機能的変異型を意図する。
- 「LAGLIDADGホーミングエンドヌクレアーゼ変異型」又は「変異型」により、LAGLIDADGホーミングエンドヌクレアーゼ配列の少なくとも1つのアミノ酸を、異なるアミノ酸で置き換えることにより得られるタンパク質を意図する。
【0089】
- 「機能的変異型」により、DNA標的、好ましくは、野生型LAGLIDADGホーミングエンドヌクレアーゼにより切断されない新しいDNA標的を切断できるLAGLIDADGホーミングエンドヌクレアーゼ変異型を意図する。例えば、このような変異型は、DNA標的配列に接触するか、又は直接若しくは間接的に該DNA標的と相互作用する位置にてアミノ酸変異を有する。
【0090】
- 「新規な特異性を有するホーミングエンドヌクレアーゼ変異型」により、親のホーミングエンドヌクレアーゼのものとは異なる切断されるDNA標的のパターン(切断プロフィール)を有する変異型を意図する。変異型は、親のホーミングエンドヌクレアーゼより少ない(制限されたプロフィール)又はより多い標的を切断できる。好ましくは、変異型は、親のホーミングエンドヌクレアーゼにより切断されない少なくとも1つの標的を切断できる。
等価で、同様に用いられる用語「新規な特異性」「改変された特異性」「新規な切断特異性」「新規な基質特異性」は、DNA標的配列のヌクレオチドに対する変異型の特異性のことである。
【0091】
- 「I-CreI」により、配列SWISSPROT P05725又はpdbアクセッションコード1g9y (配列番号24)を有する野生型I-CreIを意図する。
- 「I-DmoI」により、配列SWISSPROT番号P21505 (配列番号22)又は構造PDBコード1b24を有する野生型I-DmoIを意図する。
【0092】
- 「ドメイン」又は「コアドメイン」により、約100アミノ酸残基の配列に相当する、LAGLIDADGファミリーのホーミングエンドヌクレアーゼの特徴的なαββαββα折り畳みである「LAGLIDADGホーミングエンドヌクレアーゼコアドメイン」を意図する。該ドメインは、DNA標的の一方の半分と相互作用する逆平行ベータシートに折り畳まれる4つのベータ鎖を含む。このドメインは、DNA標的の他方の半分と相互作用する別のLAGLIDADGホーミングエンドヌクレアーゼコアドメインと会合して、該DNA標的を切断できる機能エンドヌクレアーゼを形成できる。例えば、二量体ホーミングエンドヌクレアーゼI-CreI (163アミノ酸)の場合、LAGLIDADGホーミングエンドヌクレアーゼコアドメインは、残基6〜94に相当する。単量体ホーミングエンドヌクレアーゼの場合、2つのこのようなドメインが、エンドヌクレアーゼの配列中に見出される;例えば、I-DmoI (194アミノ酸)、第1ドメイン(少なくとも残基1〜95及び第2ドメイン(残基105〜194)がリンカー(残基96〜104)によって隔てられている。
【0093】
- 「サブドメイン」により、ホーミングエンドヌクレアーゼDNA標的ハーフサイトの独特の(distinct)部分と相互作用するLAGLIDADGホーミングエンドヌクレアーゼコアドメインの領域を意図する。
- 「ベータヘアピン」により、ループ又はターンにより接続されたLAGLIDADGホーミングエンドヌクレアーゼコアドメインの逆並行ベータシートの2つの連続するベータ鎖を意図する。
- 「C1221」により、パリンドローム「21」を形成するようにI-CreI標的部位「12」が逆に反復される第1のハーフのことであることを意図する。
【0094】
- 「切断活性」により、本発明の変異型の切断活性は、PCT出願WO 2004/067736; Epinatら、Nucleic Acids Res., 2003, 31, 2952-2962; Chamesら、Nucleic Acids Res., 2005, 33, e178及びArnouldら、J. Mol. Biol., 2006, 355, 443-458に記載されるように、レポーターベクターを用いる酵母又は哺乳動物細胞における直列反復組み換えアッセイにより測定され得る。レポーターベクターは、酵母又は哺乳動物発現ベクター中にクローニングされる、レポーター遺伝子の2つの短縮された非機能的コピー(直列反復)及びキメラDNA標的配列を、介在配列内に含む。DNA標的配列は、異なるヌクレオチドにより少なくとも1つのヌクレオチドが置換される親のホーミングエンドヌクレアーゼ切断部位に由来する。好ましくは、DNA切断部位の1つ以上の位置での4つの塩基(g、a、c、t)異なる組み合わせを表すパリンドローム又は非パリンドロームDNA標的のパネルが試験される(n個の変異される位置の4n パリンドローム標的)。変異型の発現は、DNA標的配列を切断できる機能的エンドヌクレアーゼとなる。この切断は、直接反復間の相同組み換えを誘導して、結果として機能的レポーター遺伝子を生じさせ、その発現は適切なアッセイによりモニターできる。
【0095】
- 「DNA標的」「DNA標的配列」「標的配列」「標的部位」「標的」「部位」「認識部位」「認識配列」「ホーミング認識部位」「ホーミング部位」「切断部位」により、LAGLIDADGホーミングエンドヌクレアーゼにより認識され且つ切断される20〜24 bpの二本鎖パリンドローム、部分的パリンドローム(偽パリンドローム)又は非パリンドロームのポリヌクレオチド配列を意図する。これらの用語は、そこでエンドヌクレアーゼにより二本鎖破断(切断)が誘導される独特のDNAの位置、好ましくはゲノムの位置のことをいう。DNA標的は、二本鎖ポリヌクレオチドの一方の鎖の5'から3'の配列により定義される。例えば、野生型I-CreIにより切断されるパリンドロームDNA標的配列は、5'- t-12c-11a-10a-9a-8a-7c-6g-5t-4c-3g-2t-1a+1c+2g+3a+4c+5g+6t+7t+8t+9t+10g+11a+12 (配列番号29)の配列により定義される。DNA標的の切断は、センス鎖及びアンチセンス鎖のそれぞれについて、+2位及び-2位のヌクレオチドで生じる。そうでないと記載しない限り、メガヌクレアーゼ変異型によるDNA標的の切断が生じる位置は、DNA標的のセンス鎖上の切断部位に相当する。
【0096】
- 「DNA標的ハーフサイト」「ハーフ切断部位」又は「ハーフサイト」により、各LAGLIDADGホーミングエンドヌクレアーゼコアドメインが結合するDNA標的の部分を意図する。
- 「DC10NNN」(配列番号8)により、これが配列の+8位、+9位及び+10位にて変異を有するDmoCreの標的配列であることを意図し、それゆえ10位から連続して逆向きに3つのヌクレオチドで変異可能な10位におけるDmoCreである。同様に、DC4NNN (配列番号36)は、配列の+2位、+3位及び+4位にて変異を有するDmoCreの標的配列のことをいい;またDC7NNN (配列番号37)は、配列の+5位、+6位及び+7位にて変異を有するDmoCreの標的配列のことをいう。
【0097】
- 「キメラDNA標的」又は「ハイブリッドDNA標的」により、2つの親のメガヌクレアーゼ標的配列の異なる半分の融合を意図する。さらに、該標的の少なくとも一方の半分は、別個のサブドメインが結合するヌクレオチドの組み合わせ(組み合わせたDNA標的)を含み得る。
- 「変異」により、核酸/アミノ酸配列における、1つ以上のヌクレオチド/アミノ酸の置換、欠失、及び/又は付加を意味する。
【0098】
- 「相同な」により、配列同士の間の相同組み換えを導くのに十分な別の配列との同一性を有する、より具体的には少なくとも95%の同一性、好ましくは97%の同一性、より好ましくは99%を有する配列を意図する。
- 「同一性」は、2つの核酸分子又はポリペプチド間の配列同一性のことをいう。同一性は、比較の目的のために整列させ得るそれぞれの配列中の位置の比較により決定できる。比較される配列中の位置が同じ塩基により占められる場合、その分子同士は、その位置において同一である。核酸又はアミノ酸配列間の類似性又は同一性の程度は、核酸配列により共有される位置での同一又は一致するヌクレオチドの数の関数である。種々のアラインメントアルゴリズム及び/又はプログラムを用いて2つの配列間の同一性を計算でき、例えば、GCG配列解析パッケージ(University of Wisconsin, Madison, Wis.)の一部分として利用可能であり、例えばデフォルト設定で用い得るFASTA又はBLASTを含む。
【0099】
- 「個体」は、哺乳動物、及びその他の脊椎動物(例えば鳥類、魚類及び爬虫類)を含む。用語「哺乳動物」及び「哺乳類」は、本明細書で用いる場合、その子に授乳し、生存する子を出産する(真獣類(eutharian)又は胎盤哺乳類(placental mammals))又は産卵する(後獣類(metatharian)又は無胎盤哺乳類(nonplacental mammals))単孔類、有袋類及び有胎盤類(placental)を含む、いずれの脊椎動物のことをいう。哺乳動物の種の例は、ヒト及びその他の霊長類(例えばサル、チンパンジー)、げっ歯類(例えばラット、マウス、モルモット)並びに反すう動物(例えばウシ、ブタ、ウマ)を含む。
【0100】
- 「遺伝病」は、部分的又は完全な、直接的又は間接的な1又は複数の遺伝子における異常による、任意の疾患のことをいう。該異常は、変異、挿入又は欠失であり得る。該変異は、点突然変異であり得る。上記の異常は、遺伝子のコード配列又はその調節配列に影響し得る。該異常は、ゲノム配列の構造又はコードされるmRNAの構造若しくは安定性に影響し得る。該遺伝病は、劣性又は優性であり得る。このような遺伝病は、限定されないが、嚢胞性繊維症, ハンチントン舞踏病、家族性高コレステロール血症(LDL受容体欠損)、肝芽腫、ウィルソン病、先天性肝性ポリフィリン病、肝臓代謝の遺伝性障害、レッシュナイハン症候群、鎌状赤血球貧血、サラセミア、色素性乾皮症、ファンコーニ貧血、色素性網膜炎、毛細管拡張性運動失調、ブルーム症候群、網膜芽腫、デュシェンヌ型筋ジストロフィー、及びテイ-サックス病であり得る。
【0101】
- 「RAG遺伝子」により、哺乳動物のRAG1又はRAG2遺伝子を意図する。例えば、ヒトRAG遺伝子は、アクセッション番号NC_000011.8のもとNCBIデータベースにおいて利用できる:RAG1 (遺伝子ID:5896)配列及びRAG2 (遺伝子ID:5897)配列は、それぞれ36546139 位〜36557877位及び36570071位〜36576362位(マイナス鎖)で位置が定められている。両遺伝子は、短い非翻訳領域及び長い非翻訳領域がそれぞれ5' 末端及び3'末端に連結したRAGタンパク質をコードするORFを含む、短い非翻訳エクソン1及びエクソン2を有している。
【0102】
- 「RAG1.10」は、ヒトRAG1遺伝子(アクセッション番号NC_000011.8、836546139位〜36557877位)の5270位、RAG1のコーディングエクソンから7 bp上流に位置する22 bpの(非パリンドロームの)標的である。
- 「RAG1.10.2」(配列番号34)は、RAG1.10標的の第1ハーフに由来するパリンドローム標的(tgttctcagg tacctgagaaca)である。
- 「RAG1.10.2D34」:「RAG1.10.2D34」(配列番号35)により、D34で表されるI-DmoI標的配列の第2ハーフに結合する、上で定義されるRAG1.10.2標的配列の第1部分を含む配列を意味する。この配列は「tgttctcagg taagttccggcg」である。
【0103】
- 「RAG1.10.3」(配列番号38)は、RAG1.10標的の第1ハーフに由来するパリンドローム標的(ctggctgaggtacctcagccag)である。
- 「RAG1.10.3D34」:「RAG1.10.3D34」(配列番号39)により、D34で表されるI-DmoI標的配列の第2ハーフに結合する、上で定義されるRAG1.10.3標的配列の第1部分を含む配列を意味する。この配列は「ttggctgaggtaagttccggcg」である。
【0104】
- 「ベクター」:本発明に用い得るベクターは、限定されないが、ウイルスベクター、プラスミド、RNAベクター、或いは染色体、非染色体、半合成(semi-synthetic)又は合成の核酸からなり得る直鎖状若しくは環状のDNA又はRNA分子を含む。好ましいベクターは、自律複製できるもの(エピソームベクター)及び/又は連結された核酸の発現を可能にするもの(発現ベクター)である。多数の適切なベクターが当業者に知られ、商業的に入手可能である。
【0105】
ウイルスベクターは、レトロウイルス、アデノウイルス、パルボウイルス(例えばアデノ随伴ウイルス)、コロナウイルス、マイナス鎖RNAウイルス、例えばオルトミクソウイルス(例えばインフルエンザウイルス)、ラブドウイルス(例えば狂犬病及び水疱性口内炎ウイルス)、パラミクソウイルス(例えば麻疹及びセンダイ)、プラス鎖RNAウイルス、例えばピコルナウイルス及びアルファウイルス、並びにアデノウイルス、ヘルペスウイルス(例えば単純ヘルペスウイルス1及び2型、エプスタイン-バーウイルス、サイトメガロウイルス)及びポックスウイルス(例えばワクシニア、鶏痘及びカナリア痘)を含む二本鎖DNAウイルスを含む。その他のウイルスは、例えばノーウォークウイルス、トガウイルス、フラビウイルス、レオウイルス、パポバウイルス、ヘパドナウイルス及び肝炎ウイルスを含む。レトロウイルスの例は、トリ白血病肉腫、哺乳類C型、B型ウイルス、D型ウイルス、HTLV-BLV群、レンチウイルス、スプマウイルスを含む(Coffin, J. M., Retroviridae: The viruses and their replication, In Fundamental Virology、第3版、B. N. Fieldsら編、Lippincott-Raven Publishers、フィラデルフィア、1996)。用語「ベクター」は、それが連結されている別の核酸を輸送できる核酸分子のことをいう。好ましいベクターのあるタイプは、エピソーム、すなわち染色体外の複製が可能な核酸である。好ましいベクターは、自律複製及び/又はそれらが連結されている核酸の発現が可能なものである。それらが作動可能に(operably)連結されている遺伝子の発現に指向できるベクターは、本明細書において「発現ベクター」という。本発明によるベクターは、限定されないが、YAC (酵母人工染色体)、BAC (細菌人工)、バキュロウイルスベクター、ファージ、ファージミド、コスミド、ウイルスベクター、プラスミド、RNAベクター、或いは染色体、非染色体、半合成又は合成のDNAからなり得る直鎖状若しくは環状DNA又はRNA分子を含む。一般に、組み換えDNA技術において実用性のある発現ベクターは、しばしば、一般に環状二本鎖DNAループをいう「プラスミド」の形態にあり、そのベクターの形態においてそれらは染色体に結合しない。適切なベクターの多数は、当業者に知られている。
【0106】
ベクターは、選択マーカー、例えば:真核細胞培養にはネオマイシンホスホトランスフェラーゼ、ヒスチジノールデヒドロゲナーゼ、ジヒドロ葉酸レダクターゼ、ハイグロマイシンホスホトランスフェラーゼ、単純ヘルペスウイルスチミジンキナーゼ、アデノシンデアミナーゼ、グルタミンシンセターゼ、及びヒポキサンチン-グアニンホスホリボシルトランスフェラーゼ;エス・セレビシエ(S. cerevisiae)にはTRP1;イー・コリ(E. coli)にはテトラサイクリン、リファンピシン又はアンピシリン耐性などを含み得る。しかしながら、本発明は、同等の機能を提供し、かつ本発明に続いて知られることとなる発現ベクターのようなその他の形態を含むことを意図する。
【0107】
好ましい上記のベクターは、本発明のポリペプチドをコードする配列が適切な転写及び翻訳調節エレメントの調節下に置かれ、該タンパク質の産生及び合成を可能にする発現ベクターである。したがって、上記のポリヌクレオチドは発現カセットに含まれる。より詳細には、該ベクターは、複製開始点、上記のコードするポリヌクレオチドが作動可能に連結されるプロモータ、リボソーム部位、RNAスプライシング部位(ゲノムDNAを用いる場合)、ポリアデニレーション部位及び転写終結部位を含む。それはエンハンサーも含み得る。プロモータの選択は、ポリペプチドを発現させる細胞に依存する。
【実施例】
【0108】
実施例1:活性が増大したDmoCreの改良
本発明者らは、現存するDmoCre足場を、この酵素の全体的な活性を増大させることにより改良することを計画した。特に、DmoCreのI-DmoI N-末端α-へリックスにI-DmoIの残基15、19及び20 (配列番号22)に相当する3つの変異を導入した。
G20S変異は、酵母においてより活性なDmoCreタンパク質を導くが、2つの変異L15Q及びI19Dは、CHO細胞において、以前に記載された染色体外SSA (一本鎖アニーリング)組み換えアッセイ(Arnouldら、Mol Biol. 2006 Jan 20;355(3):443〜58)により示されるようにタンパク質を活性にする。よって、この実施例において用いた最終的なDmoCre足場は、L15Q、I19D及びG20S変異を有し、これらは全てI-DmoI N-末端LAGLIDADG α-ヘリックスに局在する。上記の野生型I-DmoIドメインは、配列番号1に示す。
この足場をDmoCre2といい、以下の実験において用いた。DmoCre2のペプチド配列は、配列番号2に示す。
【0109】
実施例2:縮重DC10NNN_P標的を切断するDmoCre2由来変異体の作製
DmoCre2タンパク質についての新しい配列特異性を工学的に作製する可能性を研究するために、本発明者らは、C12D34 DNA標的の+8位〜+10位の3つの隣接するヌクレオチドについて調べた。図2に示す構造により、これらの3つの塩基対とDmoCre2タンパク質残基との間の接触を詳細に調べることが可能になった。
図3Aは、そのDNA標的と結合したハイブリッド酵素の分子表面を示す。無作為化のために選択した結合の領域(+8位、+9位、+10位の塩基対、及び配列番号22の残基29、33及び35に相当するタンパク質残基)を強調する。図3Bは、DNA標的と相互作用している残基29、33及び35を示す拡大図である。点線は水素結合を表す。よって、この分析を用いて、3つのDmoCre2残基が正確に示される:DNAと接触するR33及びE35、並びにDNAと近く、E35と相互作用しているようであるY29。
DmoCre2タンパク質についての新しい切断特異性を単離するために、29位、33位及び35位で変異させたDmoCre2変異体ライブラリー(DClib2)を構築し、酵母を形質転換し、酵母スクリーニングアッセイを用いて(以下を参照)、+8位〜+10位で縮重した64個の標的(DC10NNN (配列番号8)と称する)に対してスクリーニングした。DC10NNN標的は、5' CAAAACGTCGTAAGTTCCNNNC 3' (配列番号8)であり、ここでNNNは+8位〜+10位を表し、これらの位置におけるA、C、G及びTの全ての組み合わせが、64個の標的DC10NNN配列を構成する。
【0110】
材料及び方法
64個の標的ベクターの構築:
標的は、以下のようにしてクローニングした:ゲートウェイクローニング配列と接する64個の標的配列のそれぞれに対応するオリゴヌクレオチドを、Proligoに注文した:
5'TGGCATACAAGTTTTCNNNGGAACTTACGACGTTTTGACAATCGTCTGTCA 3' (配列番号3)。一本鎖オリゴヌクレオチドのPCR増幅により作製した二本鎖標的DNAを、ゲートウェイプロトコル(Invitrogen)を用いて酵母レポーターベクター(pCLS1055、図4)にクローニングした。酵母レポーターベクターでエス・セレビシエ(S. cerevisiae) FYBL2-7B株(MAT-α, ura3Δ851, trp1Δ63, leu2Δ1, lys2Δ202)を形質転換した。
【0111】
DmoCre2 DClib2変異体ライブラリーの構築:
29位、33位又は35位に変異を含むDmoCre2由来コード配列を作製するために、DmoCreコード配列の5'末端(aa 1位〜43位)又は3'末端(36位〜264位)を増幅する2回の別々のオーバーラップPCR反応を行った。3'末端について、PCR増幅は、ベクター(pCLS0542、図5)に特異的なプライマー(Gal10R 5'-ACAACCTTGATTGGAGACTTGACC-3' (配列番号4))及びアミノ酸36〜46についてのDmoCreコード配列に特異的なプライマー(Dmo10CreFor 5'- TATCGTGTTGTGATCACCCAGAAGTCTGAAAAC-3' (配列番号5))を用いて行う。5'末端について、PCR増幅は、ベクターpCLS0542に特異的なプライマー(Gal10F 5'-GCAACTTTAGTGCTGACACATACAGG-3' (配列番号6))及びアミノ酸23〜43についてのDmoCreコード配列に特異的なプライマー(Dmo10CreRev 5'-CTTCTGGGTGATCACAACACGATAMNNGCTMNNGTTACCTTTMNNTTTCAGCTTGTACAGGCC-3' (配列番号7))を用いて行う。
【0112】
29位、33位及び35位でのNNKコドンをもたらすオリゴヌクレオチド中のMNNコードは、これらの位置にて20個の可能なアミノ酸のうちでの縮重をもたらす。次いで、2つのオーバーラップPCRフラグメントのそれぞれ25 ngと、NcoI及びEagIでの消化により線状にした75 ngのベクターDNA (pCLS0542)とを用いて、酵母サッカロミセス・セレビシエ(Saccharomyces cerevisiae) FYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietzら, Methods Enzymol. 2002; 350:87〜96)。両方の群の変異を含むインタクトなコード配列を、酵母でのインビボ相同組み換えにより作製する。DClib2核酸多様性は323 = 32768であり、形質転換の後にライブラリー多様性の約7%である2232個のクローンを採集した。
【0113】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
スクリーニングは、以前に記載されたようにして行った(Arnouldら, J Mol Biol. 2006; 355:443〜58)。具体的には、交配を、コロニーグリッダーを用いて行った(QpixII, Genetix)。変異体を、YPDプレートを覆うナイロンフィルタ上に、低い格子密度(low gridding density) (約4スポット/cm2)を用いてグリッドした。2回目のグリッド手順を、同じフィルタ上に行って、それぞれの標的について異なるレポーターを有する酵母株からなる第2層をスポットした。メンブレンを、固体寒天YPDリッチ培地上に置き、30℃にて一晩インキュベートして交配を可能にした。次に、フィルタを、ロイシン及びトリプトファンを欠き、ガラクトース(2%)を炭素源として含む合成培地に移し、37℃にて5日間インキュベートして、発現及び標的ベクターを有する二倍体を選択した。5日後に、フィルタを、0.5 Mリン酸ナトリウムバッファー、pH 7.0中の0.02% X-Gal、0.1% SDS、6%ジメチルホルムアミド(DMF)、7mM β-メルカプトエタノール、1%アガロースを含む固体アガロース培地上に置き、37℃にてインキュベートして、β-ガラクトシダーゼ活性をモニターした。結果を走査により分析し、独自のソフトウェアを用いて定量を行った。
【0114】
変異体の配列決定
プラスミドを発現する変異体を回収するために、酵母DNAを標準的なプロトコルを用いて抽出し、大腸菌(E. coli)を形質転換するために用いた。変異ORFの配列決定を、次いで、プラスミドに対してMillegen SAにより行った。或いは、ORFを酵母DNAからPCRにより増幅し(Akadaら, Biotechniques. 2000; 28:668〜70, 672, 674)、配列決定を、PCR生成物に対して直接、Millegen SAにより行った。
【0115】
結果
上記の酵母スクリーニングアッセイを用いて、DmoCre2 DClib2ライブラリーを構成する2232個のクローンを、64個のDC10NNN標的に対してスクリーニングした。スクリーニングにより、少なくとも1つのDC10NNN標的(配列番号8)を切断できる519個の陽性クローンが得られ(図6)、配列決定の後に432個のユニークメガヌクレアーゼが得られた。元のDmoCre2タンパク質は、64個のDC10NNN標的のうち13個を切断できる。図7に示すDClib2ヒットマップは、DmoCreコード配列の29位、33位及び35位に変異を導入することにより、57個のDC10NNN標的がDmoCre2由来変異体により切断できるようになったことを示す。このスクリーニングアプローチにより、よって、DC10NNN標的についてのDmoCre2切断スペクトルを広げることができ、新しい切断特異性を単離することが可能になる。
【0116】
以下の表Iを参照して、本発明者らにより同定された種々のDClib2クローンを列挙し、これらのそれぞれにおける残基の変化を示すとともに、切断することが示されたDC10標的配列も示す。一番上の列は、+8位〜+10位の3つのヌクレオチドを示し、図は、挿入断片を欠く酵母からの陰性対照と比較した比色反応の強度を示す。具体的には、「0」の値は、このアッセイにおいてバックグラウンドノイズの試験されたレベルに等しい実験結果を表す。「_」の値は、この試料がこの特定のヌクレオチドの組み合わせについて試験されなかったことを示す。
【0117】
【表1−1】
【0118】
【表1−2】
【0119】
【表1−3】
【0120】
【表1−4】
【0121】
【表1−5】
【0122】
【表1−6】
【0123】
【表1−7】
【0124】
【表1−8】
【0125】
【表1−9】
【0126】
【表1−10】
【0127】
【表1−11】
【0128】
【表1−12】
【0129】
【表1−13】
【0130】
【表1−14】
【0131】
【表1−15】
【0132】
【表1−16】
【0133】
【表1−17】
【0134】
【表1−18】
【0135】
【表1−19】
【0136】
【表1−20】
【0137】
【表1−21】
【0138】
【表1−22】
【0139】
【表1−23】
【0140】
【表1−24】
【0141】
実施例3:縮重DC4NNN_P標的を切断するDmoCre由来変異体の作製
本出願人らは、酵母及びCHO細胞において活性な別のDmoCre足場も開発し、これもまた残基20でG20S置換にて改変されているこの足場を、配列番号22の残基19及び109に相当する残基にて改変し(I19D及びF109Y改変)、DmoCre4 (配列番号9)と命名した。
DmoCre4タンパク質(配列番号9)についての追加の特異性を見出す可能性を研究するために、C12D34 DNA標的の+2位〜+4位の3つの隣接するヌクレオチドについて調べた。図2に示す構造により、これらの3つの塩基対とタンパク質残基との間の接触を詳細に調べることが可能になった(図8)。配列番号22の残基D75、T76及びR77に相当する3つのDmoCre4残基が同定され、これらはDNA標的と接触する。DmoCre4タンパク質についての新しい切断特異性を単離するために、75位、76位又は77位で変異させたDmoCre4変異体ライブラリー(D4Clib4)を構築し、酵母を形質転換し、酵母スクリーニングアッセイを用いて、+2位〜+4位で縮重した64個の標的(DC4NNN (配列番号36)と称する)に対してスクリーニングした。このようなアプローチは、I-CreIタンパク質についてすでに詳細に記載されている(Smith Jら, Nucleic Acids Res. 2006; Arnould Sら, J Mol Biol. 2006; 355:443〜58, Arnould Sら, . J Mol Biol. 2007; 371:49〜65)。
【0142】
材料及び方法
64個の標的ベクターの構築:
標的は、以下のようにしてクローニングした:ゲートウェイクローニング配列と接する64個の標的配列のそれぞれに対応するオリゴヌクレオチドを、Proligoに注文した:
5'TGGCATACAAGTTTTCGCCGGANNNTACGACGTTTTGACAATCGTCTGTCA 3'(配列番号10)。一本鎖オリゴヌクレオチドのPCR増幅により作製した二本鎖標的DNAを、ゲートウェイプロトコル(Invitrogen)を用いて酵母レポーターベクター(pCLS1055、図4)にクローニングした。酵母レポーターベクターでエス・セレビシエFYBL2-7B株(MAT-α, ura3Δ851, trp1Δ63, leu2Δ1, lys2Δ202)を形質転換した。
【0143】
DmoCre4 D4Clib4変異体ライブラリーの構築:
75位、76位及び77位に変異を含むDmoCre4由来コード配列を作製するために、DmoCre4コード配列の5'末端 (aa 1位〜74位)又は3'末端(66位〜264位)を増幅する2つの別々のオーバーラップPCR反応を行った。3'末端について、PCR増幅は、ベクター(pCLS0542、図5)に特異的なプライマー(Gal10R 5'-ACAACCTTGATTGGAGACTTGACC-3') (配列番号11)及びアミノ酸66〜83についてのDmoCreコード配列に特異的なプライマー(DClib4For 5'-AAATCTAAAATCCAGATCGTTAAGGGTNNKNNKNNKTATGAGCTGCGTGTGAGC-3') (配列番号12)を用いて行う。75位〜77位でのNNKコドンは、これらの位置にて20個の可能なアミノ酸のうちでの縮重をもたらす。5'末端について、PCR増幅は、ベクターpCLS0542に特異的なプライマー(Gal10F 5'-GCAACTTTAGTGCTGACACATACAGG-3') (配列番号13)及びアミノ酸66〜74についてのDmoCreコード配列に特異的なプライマー(DClib4Rev 5'-ACCCTTAACGATCTGGATTTTAGATTT-3') (配列番号14)を用いて行う。2つのオーバーラップPCRフラグメントのそれぞれ25 ngと、NcoI及びEagIでの消化により線状にした75 ngのベクターDNA (pCLS0542)とを用いて、酵母サッカロミセス・セレビシエFYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietz R Dら, Methods Enzymol. 2002; 350:87〜96)。インタクトなDmoCreコード配列を、酵母でのインビボ相同組み換えにより作製する。D4Clib4核酸多様性は、323 = 32768である。形質転換の後に、ライブラリー 多様性の約14%である4464個のクローンを採集した。
【0144】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0145】
結果
上記の酵母スクリーニングアッセイを用いて、DmoCre4 D4Clib4 ライブラリーを構成する4464個のクローンを、DC4GAA標的以外の64個全てのDC4NNN標的に対してスクリーニングした。スクリーニングにより、少なくとも1つのDC4NNN標的(配列番号36)を切断できる1194個の陽性クローンが得られた。これらのクローンは、配列レベルでは特徴決定しなかった。元のDmoCre4タンパク質は、63個のDC4NNN標的のうち4個を切断できる。図9に示すD4Clib4ヒットマップは、DmoCre4コード配列の75位、76位及び77位に変異を導入することにより、21個のDC4NNN標的がDmoCre4由来変異体により切断できるようになったことを示す。我々のスクリーニングアプローチにより、よって、DC4NNN標的についてのDmoCre4切断スペクトルを広げることができ、新しい切断特異性を単離することが可能になる。
【0146】
実施例4:5CAGD34標的を切断するDmoCre由来変異体の作製
本発明者らは、C1221に由来し、±5位、±4位、±3位にて縮重するパリンドロームDNA標的に対するI-CreIタンパク質の特異性を改変できたことを以前に示した(Arnouldら, J Mol Biol. 2006; 355:443〜58)。44位、68位、70位、75位及び77位にてI-CreIコード配列に変異を導入することにより、5CAG_P標的(配列番号32)を切断するI-CreI由来変異体を得ることができた。
この実施例では、これらの同じ変異をDmoCre2又はDmoCre4コード配列に導入することにより、5CAGD34標的(配列番号33)を切断するDmoCre由来変異体を作製できることを示す(図10)。これらのDmoCre由来変異体を作製するために、5CAG_P標的(配列番号32)を切断できる36個のI-CreI変異体を採用した。I-CreI部分のコード配列を、DmoCre2又はDmoCre4タンパク質から、制限酵素消化により除去し、5CAG_Pカッターコード配列に置き換えた。DmoCre2及びDmoCre4タンパク質にそれぞれ基づく2つの変異体ライブラリーDCSca2_5CAG及びDCSca4_5CAGを構築し、5CAGD34標的(配列番号33)に対して、上記のような酵母スクリーニングアッセイを用いてスクリーニングした。
【0147】
材料及び方法
5CAGD34標的ベクターの構築
標的は、以下のようにしてクローニングした:ゲートウェイクローニング配列で挟まれた標的配列に対応するオリゴヌクレオチドを、Proligoに注文した:5' TGGCATACAAGTTTTCGCCGGAACTTACCTGGTTTTGACAATCGTCTGTCA 3' (配列番号15)。一本鎖オリゴヌクレオチドのPCR増幅により作製した二本鎖標的DNAを、ゲートウェイプロトコル(Invitrogen)を用いて酵母レポーターベクター(pCLS1055、図4)にクローニングした。酵母レポーターベクターでエス・セレビシエFYBL2-7B株(MAT-α, ura3Δ851, trp1Δ63, leu2Δ1, lys2Δ202)を形質転換した。
【0148】
DCsca2_5CAG変異体ライブラリーの構築:
5CAG_P標的切断を担うI-CreI部分の配列に変異を含むDmoCre2由来コード配列を作製するために、I-CreI由来5CAG_Pカッターのそれぞれについてのaa 13〜148の領域を増幅するPCR反応を行った。PCR増幅を、プライマーCreNgoLib (5' CGTGAGCAGCTGGCGTTCCTGGCCGGCTTTGTGGACGGTGAC-3' (配列番号16))及びCreMluLib (5'-ACGAACGGTTTCAGAAGTGGTTTTACGCGTCTTAG-3' (配列番号17))を用いて行った。
36個のPCRフラグメントを、次いで、プールした。DmoCre2タンパク質についての酵母発現ベクターを、次いで、NgoMIV及びMluIで消化して、DmoCre2タンパク質の残基111〜238をカバーするフラグメントを除去した。最後に、オーバーラップPCRプール25 ngと、75 ngの消化ベクターDNAとを用いて、酵母サッカロミセス・セレビシエFYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietz R Dら, Methods Enzymol. 2002; 350:87〜96)。5CAG_Pカッターに特徴的な変異を含むインタクトなDmoCreコード配列を、酵母でのインビボ相同組み換えにより作製した。形質転換の後に、ライブラリー多様性の約5倍の186個のクローンを採集した。
【0149】
DCSca4_5CAG変異体ライブラリーの構築
5CAG_P標的切断を担うI-CreI部分の配列に変異を有するDmoCre4由来コード配列を作製するために、I-CreI由来5CAG_Pカッターのそれぞれについてのaa 13〜148の領域を増幅するPCR反応を行った。PCR増幅を、プライマーCreMluLib及びCreNgoLibY (5' CGTGAGCAGCTGGCGTACCTGGCCGGCTTTGTGGACGGTGAC-3') (配列番号18)を用いて行ったが」、これは、DmoCre4タンパク質に特徴的なF109Y変異を考慮に入れている。36個のPCRフラグメントを、次いで、プールした。DmoCre4タンパク質についての酵母レポーターベクターを、次いで、制限酵素NgoMIV及びMluIで消化して、DmoCre4タンパク質の残基111〜238をカバーするフラグメントを除去した。最後に、PCRプール25 ngと、75 ngの消化ベクターDNAとを用いて、酵母サッカロミセス・セレビシエFYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietz R Dら, Methods Enzymol. 2002; 350:87〜96)。5CAG_Pカッターに特徴的な変異を含むインタクトなDmoCreコード配列を、酵母でのインビボ相同組み換えにより作製した。形質転換の後に、ライブラリー多様性の約5倍の186個のクローンを採集した。
【0150】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0151】
結果
実施例1に記載される酵母スクリーニングアッセイを用いて、DCSca2_5CAGライブラリーを構成する186個のクローンと、DCSca4_5CAG ライブラリーを構成する186個のクローンを、5CAGD34 (配列番号33)標的に対してスクリーニングした。最初のライブラリー及び次のライブラリーからそれぞれ、全体的により強い切断効率を有する32個及び40個の陽性クローンが得られた。陽性の例を図11に示す。5CAGD34標的(配列番号33)を切断するクローンは、5CAG_P標的(配列番号32)を切断しない(データ示さず)。よって、本発明者らは、特定のI-CreI変異をDmoCre足場に導入して、組み合わせ標的を効率的に切断できることを示した。
【0152】
実施例5:RAG1.10.2D34標的を切断するDmoCre由来変異体の作製
RAG1.10.2 DNAパリンドローム標的(配列番号34)は、I-CreI C1221標的(配列番号29)に由来する(図12)。本発明者らは、10GTT (例えばヌクレオチド8、9及び10がそれぞれG、T及びTである)及び5CAG I-CreI由来変異体を組み合わせ、次いで、最初に単離されたRAG1.10.2カッターに対してランダム突然変異誘発ステップを行うことにより、どのようにして、RAG1.10.2標的を非常に強く切断するI-CreI由来変異体を得ることができたかを以前に示した(WO2008/010009)。このM2と呼ばれる変異体は、野生型I-CreI配列と比較して以下の変異を有する:配列番号24のN30R、Y33N、Q44A、R68Y、R70S、I77R。これらの同じ変異をDmoCre2のI-CreI部分に導入し、DmoM2とよばれる得られたDmoCre変異体(配列番号71)のRAG1.10.2D34 (配列番号35)組み合わせ標的に対する活性を、酵母切断アッセイを用いて調べた。
【0153】
材料及び方法
RAG1.10.2D34標的ベクターの構築
標的は、以下のようにしてクローニングした:ゲートウェイクローニング配列に挟まれた標的配列に対応するオリゴヌクレオチドを、Proligoに注文した:5' TGGCATACAAGTTTTCGCCGGAACTTACCTGAGAACAACAATCGTCTGTCA 3' (配列番号19)。一本鎖オリゴヌクレオチドのPCR増幅により作製した二本鎖標的DNAを、ゲートウェイプロトコル(Invitrogen)を用いて酵母レポーターベクター(pCLS1055、図4)にクローニングした。酵母レポーターベクターでエス・セレビシエFYBL2-7B株(MAT-α, ura3Δ851, trp1Δ63, leu2Δ1, lys2Δ202)を形質転換した。
【0154】
DmoM2の構築
RAG1.10.2 M2変異体に特異的なI-CreI部分に変異を含むDmoCre2由来コード配列を作製するために、M2変異体のaa 9〜146の領域を増幅するPCR反応を行った。PCR増幅は、プライマーCreNgoFor (5' TTCCTGCTGTACCTGGCCGGCTTTGTGG-3' (配列番号20))及びCreMluRev (5'- TTCAGAAGTGGTTTTACGCGTCTTAG -3' (配列番号21))を用いて行う。次いで、PCRフラグメントを、DmoCre2タンパク質についてのORFを含む酵母発現ベクターと同様に、制限酵素NgoMIV及びMluIで消化した。ライゲーション反応を行い、大腸菌DH5αをライゲーション混合物で形質転換した。得られたDmoM2変異体を、次いで増幅して配列決定した。
【0155】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0156】
結果
この酵母切断アッセイを用いて、組み合わせRAG1.10.2D34標的(配列番号35)及びその他の異なる標的に対するDmoM2変異体の活性を調べた。図13は、DmoM2が、組み合わせRAG1.10.2D34標的(配列番号35)を特異的に切断することを示す。よって、本発明者らは、DmoCre足場のI-CreI部分に、標的配列についてのコンビナトリアル及び/又は最適化実験の間にI-CreI足場において以前に単離された変異を導入して、該標的配列の一部分を含む組み合わせDmoCre標的を効率的に切断できたことを示した。
【0157】
実施例6:RAG1.10.2D34又はRAG1.10.3D34標的を切断するDmoCre由来変異体の作製
RAG1.10.2及びRAG1.10.3 DNAパリンドローム標的(配列番号34及び38)は、I-CreI C1221標的(配列番号29)に由来する(図14)。本発明者らは、10GTT及び5CAG I-CreI由来変異体を組み合わせることにより、どのようにして、RAG1.10.2標的を非常に強く切断するI-CreI由来変異体を得ることができたか、又は10TGG及び5GAG I-CreI由来変異体を組み合わせることにより、どのようにして、RAG1.10.3標的を非常に強く切断するI-CreI由来変異体を得ることができたかを以前に示した(WO2008/010009及びWO2008/010093)。実施例4に記載したのと同じ方法論を用いて、I-CreI部分のコード配列を、DmoCre2タンパク質から、制限酵素消化により除去し、RAG1.10.2カッターコード配列又はRAG1.10.3カッターコード配列のいずれかで置き換えた。以下の表IIは、I-CreI部分における変異を、RAG1.10.2カッターコード配列について配列番号24のI-CreI 配列における残基の番号付けを参照にしてまとめ、以下の表IIIは、I-CreI部分における変異をRAG1.10.3カッターコード配列についてまとめる。DCSca2_RAG1.10.2及びDCSca2_RAG1.10.3変異体ライブラリーを作製するために、33個のRAG1.10.2カッター及び35個のRAG1.10.3カッターをそれぞれ用いた。次いで、2つの変異体ライブラリーを、上記の酵母スクリーニングアッセイを用いてそれぞれRAG1.10.2D34及びRAG1.10.3D34標的に対して、また、親の標的に対してもスクリーニングした。
【0158】
【表2】
【0159】
【表3】
【0160】
材料及び方法
RAG1.10.3D34標的ベクターの構築
標的は、以下のようにしてクローニングした:ゲートウェイクローニング配列に挟まれた標的配列に対応するオリゴヌクレオチドを、Proligoに注文した:5'TGGCATACAAGTTTTCGCCGGAACTTACCTCAGCCAGACAATCGTCTGTCA-3' (配列番号19)。一本鎖オリゴヌクレオチドのPCR増幅により作製した二本鎖標的DNAを、ゲートウェイプロトコル(Invitrogen)を用いて酵母レポーターベクター(pCLS1055、図4)にクローニングした。酵母レポーターベクターでエス・セレビシエFYBL2-7B株(MAT-α, ura3Δ851, trp1Δ63, leu2Δ1, lys2Δ202)を形質転換した。
【0161】
DCSca2_RAG1.10.2変異体ライブラリーの構築
RAG1.10.2標的切断を担うI-CreI部分の配列に変異を含むDmoCre2由来コード配列を作製するために、33個のI-CreI由来RAG1.10.2カッターのそれぞれについてのaa 13〜148の領域を増幅するPCR反応を行い、さらに、プライマーは、DmoCre2を含む発現ベクターの配列にいずれかの末端にて相同な部分も含む。PCR増幅は、プライマーCreNgoLib (5' CGTGAGCAGCTGGCGTTCCTGGCCGGCTTTGTGGACGGTGAC-3' (配列番号16))及びCreMluLib (5'-ACGAACGGTTTCAGAAGT GGTTTTACGCGTCTTAG-3' (配列番号17))を用いて行う。
33個のPCRフラグメントを、次いで、プールした。DmoCre2タンパク質についての酵母発現ベクターを、次いで、NgoMIV及びMluIで消化して、DmoCre2タンパク質の残基111〜238をカバーするフラグメントを除去した。最後に、25 ngのPCRプールと、75 ngの消化ベクターDNAとを用いて、酵母サッカロミセス・セレビシエFYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietz R Dら, Methods Enzymol. 2002; 350:87〜96)。RAG1.10.2カッターに特徴的な変異を含むインタクトなDmoCreコード配列を、酵母でのインビボ相同組み換えにより作製した。形質転換の後に、ライブラリー多様性の約5倍の186個のクローンを採集した。
【0162】
DCSca2_RAG1.10.3変異体ライブラリーの構築
方法論は、35個のRAG1.10.3カッターのプールを用いた以外は、DCSca2_RAG1.10.2変異体ライブラリーと同じであった。
【0163】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0164】
結果
実施例2に記載される酵母スクリーニングアッセイを用いて、DCSca2_RAG1.10.2変異体ライブラリーを構成する186個のクローンと、DCSca2_RAG1.10.3変異体ライブラリーを構成する186個のクローンを、それぞれRAG1.10.2D34及びRAG1.10.3D34標的に対してスクリーニングした。DCSca2_RAG1.10.2ライブラリーからは36個の陽性クローンが得られ、そのうち24個のクローンを再配置して、図15に示す2回目のスクリーニングに供した。24個のクローンは、8個のユニーク配列を示し(表IVを参照)、RAG1.10.2D34標的に特異的である。これらは、RAG1.10.2、C1221及びC12D34標的のいずれも切断しない。DCSca2_RAG1.10.3ライブラリーからは52個の陽性クローンが得られ、そのうち33個のクローンを再配置して、図15に示す2回目のスクリーニングに供した。33個のクローンは、6個のユニーク配列を示し(表IVを参照)、RAG1.10.3D34標的に特異的である。これらは、RAG1.10.3、C1221及びC12D34標的のいずれも切断しない。よって、本発明者らは、特定のI-CreI変異を、DmoCre足場に、ライブラリーアプローチを用いて導入して、組み合わせ標的を効率的かつ特異的に切断できることを示した。
【0165】
実施例7:ランダム突然変異誘発によるRAG1.10.2D34及びRAG1.10.3D34メガヌクレアーゼの改良
実施例6で同定したRAG1.10.2D34及びRAG1.10.3D34カッターの切断効率を改良するために、1回のランダム突然変異誘発を、実施例6で単離した、選択されたRAG1.10.2D34及びRAG1.10.3D34カッターに対して行った。それぞれの標的ついて、実施例6に記載したもののうち3つの変異体を選択した。表IVを参照。これらのDNAをプールし、PCR無作為化についての鋳型として用いた。変異体ライブラリーを酵母において構築し、適切な標的に対してスクリーニングした。
【0166】
材料及び方法
ランダム突然変異誘発によるライブラリーの構築
それぞれの変異体プールに対して、0.3 mMの濃度のMn2+を用いるPCRによるランダム突然変異誘発を行った。用いたプライマーは、preATGCreFor (5'GCATAAATTACTATACTTCTATAGACACGCAAACACAAATACACAGCGGCCTTGCCACC-3' (配列番号40))及びICreIpostRev (5'-GGCTCGAGGAGCTCGTCTAGAGGATCGCTCGAGTTATCAGTCGGCCGC-3' (配列番号41))である。
およそ25ngのPCR生成物と、NcoI及びEagIでの消化により線状にした75ngのベクターDNA (pCLS542、図5)を用いて、酵母サッカロミセス・セレビシエFYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietz及びWoods 2002)。DmoCre変異体についてのインタクトなコード配列を含む発現プラスミドを、PCR生成物のオーバーラップ部分と消化ベクターとの間の酵母でのインビボ相同組み換えにより作製した。
【0167】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0168】
結果
以下の表IVは、8個のRAG1.10.2D34カッターと、6個のRAG1.10.3D34カッターの配列を示す。これらのうち、カッターのそれぞれのクラスの最初の3つ(表IVにおいて下線を付す)を選択して、無作為化PCRを行った。これらの配列はDmoCre2タンパク質に由来し、126位、128位、130位、131位、136位、138位、142位、166位、168位、173位及び175位(配列番号2)の残基にて異なる。これらの位置は、I-CreIのそれぞれ28位、30位、32位、33位、38位、40位、44位、68位、70位、75位及び77位(配列番号24)に対応する。上記のように、これらのRAG1.10.2D34及びRAG1.10.3D34カッターは、DmoCre2中に存在する変異、すなわちL15Q、I19D及びG20Sの変異も含み、これらは全てI-DmoI N-末端LAGLIDADGアルファ-ヘリックス中にある。
【0169】
【表4】
【0170】
無作為化PCRから創出した変異体ライブラリーを、次いで、我々の酵母スクリーニングアッセイにおいて、それらのそれぞれの標的に対してスクリーニングした。RG2D2及びRG3D3変異体を対照として用いた。対照変異体と比較して活性が増大した変異体を選択し、図16に示す2回目のスクリーニングに供した。それぞれの標的について、切断活性が改善された3つの変異体を丸で囲む。これらの選択された変異体を単離し、配列決定したが、表Vはそれらの配列を示す。
【0171】
【表5】
【0172】
表Vは、RAG1.10.2D34標的についての切断活性の改善が、V105A、E80K及びY66Hの変異をI-CreI部分に導入したことによることを示す(位置の番号付けはI-CreI配列である配列番号24を参照する)。RAG1.10.3D34標的の場合、活性の増大は、追加の変異によっては得られなかったが、突然変異誘発を行うために用いた3つのRAG1.10.3D34カッター間の変異の交換により得られた。
【0173】
実施例8:縮重DC4NNN_P標的を切断する新しいDmoCre由来変異体の作製
DC4NNN標的(配列番号36)についての特異性を有するDmoCre足場を探索するために、DmoCre2タンパク質に基づく新しい変異体ライブラリーを酵母において作製した。実施例3で述べたように、配列番号22の3つの残基D75、T76及びR77は、C12D34標的の+2位〜+4位の3つの塩基と接触する。配列番号22の残基T41も含まれ、C12D34標的の+4位のチミンのメチル基とファンデルワールス接触を確立する。本発明者らは、この残基の変異が、DC4NNN標的に対するDmoCre2タンパク質についての新しい特異性を提供できるのではないかと考えた。よって、DmoCre2タンパク質についての新しい切断特異性を単離するために、配列番号22 (I-DmoI部分)の残基41、75又は77に対応する位置で変異されたDmoCre2変異体ライブラリー(D4Clib2Bis)を構築し、酵母を形質転換し、酵母スクリーニングアッセイを用いて、+2位〜+4位で縮重した64個の標的(DC4NNN 配列番号36)に対してスクリーニングした。
【0174】
材料及び方法
DmoCre2 D4Clib2Bis変異体ライブラリーの構築:
配列番号22 (I-DmoI部分)の41位、75位及び77位にて変異を含むDmoCre2由来コード配列を作製するために、異なるPCR反応を行った。1回目のPCR反応は、ベクターpCLS0542に特異的なプライマー(Gal10F 5'-GCAACTTTAGTGCTGACACATACAGG-3' (配列番号13))及びプライマーDCaa49-37Rev (5'- TTTAATCAGGTTTTCAGACTTCTGMNNGATCACAACACG-3' (配列番号42))を用い、これはDmoCre2コード配列の5'末端(aa 1位〜49位)を増幅する。3'末端増幅について、2回のPCR反応を行った。1回目の反応は、DmoCre2の残基42〜74の間の領域を、プライマーDCaa42-50For (5'-CAGAAGTCTGAAAACCTGATTAAACAA-3' (配列番号43))及びDCaa74-66Rev (5'-ACCCTTAACGATCTGGATTTTAGATTT-3' (配列番号44))を用いて増幅する。2回目の反応は、DmoCre2の3'末端(68位〜264位)を、プライマーDCaa68-81For (5'-AAAATCCAGATCGTTAAGGGTNNKACCNNKTATGAGCTGCGT-3' (配列番号45))及びベクター(pCLS0542、図5)に特異的なプライマー(Gal10R 5'-ACAACCTTGATTGGAGACTTGACC-3' (配列番号4))を用いて増幅する。
2つのPCRフラグメントを精製し、DCaa42-50For及びGal10Rのプライマーを用いて行うアセンブリPCRにおいて鋳型として用いた。
次いで、2つのオーバーラップPCRフラグメント(1位〜49位及び42位〜264位)のそれぞれ25ngと、NcoI及びEagIでの消化により線状にしたオーバーラップする75ngのベクターDNA (pCLS0542)とを用いて、酵母サッカロミセス・セレビシエFYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietz R Dら, Methods Enzymol. 2002; 350:87〜96)。インタクトなDmoCreコードを、酵母でのインビボ相同組み換えにより作製した。形質転換の後に、2232個のクローンを採集した。
【0175】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0176】
結果
上記の酵母スクリーニングアッセイを用いて、DmoCre2 D4Clib2Bisライブラリーを構成する2232個のクローンを、DC4TTC標的以外の64個全てのDC4NNN標的に対してスクリーニングした。スクリーニングにより、少なくとも1つのDC4NNN標的(配列番号36)を切断できる335個の陽性クローンを得た。これらのクローンを再配置し、配列決定し(221個のユニーク配列を単離した)、2回目のスクリーニングに供した。元のDmoCre2タンパク質は、63個のDC4NNN標的のうち4個を切断できる。図17に示すD4Clib2Bisヒットマップは、41位、75位及び77位の変異をDmoCre2コード配列に導入することにより、32個のDC4NNN標的をこれらのDmoCre2由来変異体により切断できるようになることを示す。
この数を、実施例3で記載した変異体ライブラリーにより切断された21個のDC4NNN標的と比較しなければならない。このスクリーニングアプローチにおいて41位を変異させることにより、DC4NNN標的についてのDmoCre2切断スペクトルを広げることができ、新しい切断特性を単離することが可能になる。
【0177】
実施例9:縮重DC7NNN_P標的を切断する新しいDmoCre由来変異体の作製
DmoCre2タンパク質についての新しい配列特異性を工学的に作製する可能性を研究するために、本出願人らは、C12D34 DNA標的の+5位〜+7位の3つの隣接するヌクレオチドについて調べた。図2に示す構造により、これらの3つの塩基対とDmoCre2タンパク質残基との間の接触を詳細に調べることが可能になった。
図18Aは、そのDNA標的と結合したハイブリッド酵素の分子表面を示す。無作為化のために選択した結合の領域(+5位、+6位、+7位の塩基対、及び37及び81のタンパク質残基)を強調する。+5位〜+7位で縮重した64個の標的をDC7NNN (配列番号37)と称する。DC7NNN標的は、5' CAAAACGTCGTAAGTNNNGGCG 3' (配列番号37)であり、ここでNNNは+5位〜+7位を表し、これらの位置におけるA、C、G及びTの全ての組み合わせが、64個の標的DC7NNN配列を構成する。図18Bは、DNAと相互作用している配列番号22の2つのアルギニン残基37及び81を示す拡大図である。点線は水素結合を表す。これらのアルギニン残基の1つ又は2つを変異させることにより、DC7NNN標的に対するDmoCre2の切断活性が急激に減少するか又は完全に喪失される。
構造をより詳細に検討することにより、アルギニン残基37が、配列番号22のロイシン残基27と疎水的に接触していることが示される(図18C)。よって、27位での変異が、アルギニン37での変異を補うことができるだろう。
DmoCre2タンパク質についての新しい切断特異性を単離するために、27位及び37位で変異させたDmoCre2変異体ライブラリー(D7Clib2)を構築し、酵母を形質転換し、以下の酵母スクリーニングアッセイを用いて、DC7GAC以外の64個全てのDC7NNN標的に対してスクリーニングした。
【0178】
材料及び方法
64個の標的ベクターの構築:
標的は、以下のようにしてクローニングした:ゲートウェイクローニング配列と接する64個の標的配列のそれぞれに対応するオリゴヌクレオチドを、Proligoに注文した:5'TGGCATACAAGTTTTCGCCNNNACTTACGACGTTTTGACAATCGTCTGTCA-3', (配列番号3)。一本鎖オリゴヌクレオチドのPCR増幅により作製した二本鎖標的DNAを、ゲートウェイプロトコル(Invitrogen)を用いて酵母レポーターベクター(pCLS1055、図4)にクローニングした。酵母レポーターベクターでエス・セレビシエFYBL2-7B株(MAT-α, ura3Δ851, trp1Δ63, leu2Δ1, lys2Δ202)を形質転換した。
【0179】
DmoCre2 DClib2変異体ライブラリーの構築:
27位及び37位に変異を含むDmoCre2由来コード配列を作製するために、DmoCreコード配列の5'末端(aa 1位〜43位)又は3'末端(38位〜264位)を増幅する2回の別々のオーバーラップPCR反応を行った。3'末端について、PCR増幅は、ベクター(pCLS0542、図5)に特異的なプライマー(Gal10R 5'-ACAACCTTGATTGGAGACTTGACC-3' (配列番号4))及びアミノ酸38〜46についてのDmoCreコード配列に特異的なプライマー(DC37For 5'GTTGTGATCACCCAGAAGTCTGAAAAC-3' (配列番号46))を用いて行う。5'末端について、PCR増幅は、ベクターpCLS0542に特異的なプライマー(Gal10F 5'-GCAACTTTAGTGCTGACACATACAGG-3' (配列番号6))及びアミノ酸23〜43についてのDmoCreコード配列に特異的なプライマー(DC3727ScanRev 5'-CTTCTGGGTGATCACAACMNNATATTCGCTACGGTTACCTTTATATTTMNNCTTGTACAGGCC-3' (配列番号47))を用いて行う。
【0180】
27位及び37位でのNNKコドンをもたらすオリゴヌクレオチド中のMNNコードは、これらの位置にて20個の可能なアミノ酸のうちでの縮重をもたらす。次いで、2つのオーバーラップPCRフラグメントのそれぞれ25 ngと、NcoI及びEagIでの消化により線状にした75 ngのオーバーラップベクターDNA (pCLS0542)とを用いて、酵母サッカロミセス・セレビシエFYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietzら, Methods Enzymol. 2002; 350:87〜96)。
両方の群の変異を含むインタクトなコード配列を、酵母でのインビボ相同組み換えにより作製した。D7Clib2の核酸多様性は322 = 1024であり、形質転換の後にほぼライブラリー全体の多様性を表す1116個のクローンを採集した。
【0181】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0182】
結果
上記の酵母スクリーニングアッセイを用いて、DmoCre2 D4Clib2Bisライブラリーを構成する1116個のクローンを、DC4GTC標的以外の64個全てのDC4NNN標的に対してスクリーニングした。スクリーニングにより、少なくとも1つのDC7NNN標的を切断できる174個の陽性クローンが得られた。これらのクローンを再配置し、配列決定し(75個のユニーク配列を単離した)、2回目のスクリーニングに供した。元のDmoCre2タンパク質は、63個のDC7NNN標的のうち、9個(DC7CCC, DC7TCC, DC7ACC, DC7GCC, DC7TTC, DC7ATC, DC7TCT, DC7ACT及びDC7TTT)を切断できた。図19に示すD7Clib2ヒットマップは、DmoCre2コード配列中に27位及び37位にて変異を導入することにより、19個のDC7NNN標的がDmoCre2由来変異体により切断できるようになったことを示す。我々のスクリーニングアプローチにより、よって、DC7NNN標的についてのDmoCre2切断スペクトル広げることができ、新しい切断特異性を単離することが可能になる。
【0183】
実施例10:2組の変異を組み合わせ、かつ組み合わせDC10TGG4ACT標的を切断する新しいDmoCre由来変異体の作製
DmoCre2タンパク質について以前に単離された異なる組の変異を組み合わせて、組み合わせ標的を切断する可能性を調べた。まず、野生型I-DmoI (配列番号22)中の75位、76位及び77位に相当する残基で変異させ、DC4ACT標的を切断できる8個のDmoCre2由来変異体を選択した。配列番号22中の75〜77位に相当する残基での配列については表VIを参照されたい。これらの変異体を用いて、配列番号22中の29位及び33位のアミノ酸に相当するDmoCre2残基で縮重した変異体ライブラリー(SeqDC10NNN4ACT)を創出した。得られたライブラリーを、最後に、酵母において、組み合わせDC10TGG4ACT標的に対してスクリーニングした。
【0184】
材料及び方法
DC10TGG4ACT標的ベクターの構築:
標的は、以下のようにしてクローニングした:ゲートウェイクローニング配列に接する標的配列に対応するオリゴヌクレオチドを、Proligoに注文した:5'TGGCATACAAGTTTTCCCAGGAAGTTACGACGTTTTGACAATCGTCTGTCA-3' 配列番号60。一本鎖オリゴヌクレオチドのPCR増幅により作製した二本鎖標的DNAを、ゲートウェイプロトコル(Invitrogen)を用いて酵母レポーターベクター(pCLS1055、図4)にクローニングした。酵母レポーターベクターでエス・セレビシエFYBL2-7B株(MAT-α, ura3Δ851, trp1Δ63, leu2Δ1, lys2Δ202)を形質転換した。
【0185】
DmoCre2 SeqDC10NNN4ACT変異体ライブラリーの構築:
まず、DC4ACT標的を切断できる8個のDmoCre2変異体についてのDNAコードをプールした。次いで、このDNAプールを、2回の別々のオーバーラップPCR反応のための鋳型として用いて、29位及び33位での変異を含むDmoCre2由来コード配列を作製した。1回目のPCR反応は、DmoCre2コード配列の5'末端(aa 1位〜40位)を、プライマーGal10F (5'-GCAACTTTAGTGCTGACACATACAGG-3' 配列番号6)及びD10CreRev2 (5'- GATCACAACACGATATTCGCTMNNGTTACCTTTMNNTTTCAGCTTGTA-3' 配列番号61)を用いて増幅し、2回目のPCR反応は、DmoCre2コード配列の3'末端(34位〜264位)を、特異的プライマーGal10R (5'-ACAACCTTGATTGGAGACTTGACC-3' 配列番号4)及びD10CreFor2 (5'-AGCGAATATCGTGTTGTGATCACCCAGAAGTCTG-3' 配列番号62)を用いて増幅する。
【0186】
29位及び33位でのNNKコドンをもたらすD10CreRev2オリゴヌクレオチド中のMNNコードは、これらの位置にて20個の可能なアミノ酸のうちでの縮重をもたらす。次いで、2つのオーバーラップPCRフラグメントのそれぞれ25 ngと、NcoI及びEagIでの消化により線状にした75 ngのオーバーラップベクターDNA (pCLS0542、図5) とを用いて、酵母サッカロミセス・セレビシエFYC2-6A株(MAT-α, trp1Δ63, leu2Δ1, his3Δ200)を、高効率LiAc形質転換プロトコルを用いて形質転換した(Gietzら, Methods Enzymol. 2002; 350:87〜96)。インタクトなコード配列を、酵母でのインビボ相同組み換えにより作製した。形質転換の後に、SeqDC10NNN4ACTライブラリーの2232個のクローンを採集した。
【0187】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0188】
結果
DC4ACT標的を切断できる8個のDmoCre2由来変異体を選択した。これらの変異体は、配列番号22の75位、76位及び77位に対応する残基にて変異を有し、以下の表VIに列挙される。
【0189】
【表6】
【0190】
SeqDC10NNN4ACTライブラリーを、次いで、我々の酵母スクリーニングアッセイを用いて、組み合わせDC10TGG4ACT標的に対してスクリーニングした。スクリーニングアッセイにより11個の陽性クローンが得られ、スクリーニングの一部を図20に示し、ここでは3個の陽性クローンを黒丸で囲む。よって、我々はここで、C12D34標的の+8位〜+10位のヌクレオチドと相互作用する残基の変異を、C12D34標的の+2位〜+4位のヌクレオチドと相互作用する残基の変異と組み合わせて、組み合わせ標的を切断できることを示す。
【0191】
実施例11:RAG1.10.3DC4ACT及びRAG1.10.3DC4TAT標的を切断する新しいRAG1.10.3D34由来変異体の作製
以下の2つの標的の可能性のあるカッターを見出すために(図14): RAG1.10.3DC4ACT (5'-CTGGCTGAGGTAACTTCCGGCG-3' 配列番号72)及びRAG1.10.3DC4TAT (5'- CTGGCTGAGGTATATTCCGGCG-3' 配列番号73)、実施例7に記載される改良RAG1.10.3D34カッター(Amel2_RG3D変異体、配列番号58)を採用して、野生型I-DmoI (配列番号22)の75位、76位及び77位に対応するAmel2_RG3D (配列番号58)の残基を縮重する変異体ライブラリー(RAG1.10.3DC4NNN)を構築した。
【0192】
材料及び方法
RAG1.10.3DC4ACT及びRAG1.10.3DC4TAT標的ベクターの構築:
標的は、以下のようにしてクローニングした:ゲートウェイクローニング配列と接する上記の標的配列の相補鎖に相当するオリゴヌクレオチドを、Proligoに注文した: 5'TGGCATACAAGTTTTCGCCGGAAGTTACCTCAGCCAGACAATCGTCTGTCA-3' 配列番号74 (RAG1.10.3DC4ACT標的について)及び5'TGGCATACAAGTTTTCGCCGGAATATACCTCAGCCAGACAATCGTCTGTCA-3' 配列番号75 (RAG1.10.3DC4TAT標的について)。一本鎖オリゴヌクレオチドのPCR増幅により作製した二本鎖標的DNAを、ゲートウェイプロトコル(Invitrogen)を用いて酵母レポーターベクター(pCLS1055、図4)にクローニングした。酵母レポーターベクターでエス・セレビシエFYBL2-7B株(MAT-α, ura3Δ851, trp1Δ63, leu2Δ1, lys2Δ202)を形質転換した。
【0193】
RAG1.10.3DC4NNN変異体ライブラリーの構築
Amel2_RG3DのDNA (配列番号58)を鋳型として用いて、D4Clib4作製について実施例3に記載されるのと同じプロトコルを用いて、RAG1.10.3DC4NNN変異体ライブラリーを構築した。2232個のクローンを採集した。
【0194】
メガヌクレアーゼ発現クローンの交配及び酵母でのスクリーニング:
実験は、上記の実施例2と同様にして行った。
【0195】
結果
RAG1.10.3DC4NNNライブラリーを構成する2232個のクローンを、2つの標的RAG1.10.3DC4ACT (配列番号72)及びRAG1.10.3DC4TAT (配列番号73)に対して、我々の酵母スクリーニングアッセイを用いてスクリーニングした。スクリーニングにより、RAG1.10.3DC4ACT標的に対して68個の陽性クローン(図21、A)、及びRAG1.10.3DC4TAT標的に対して26個の陽性クローン(図21、B)が得られた。Amel2_RG3D変異体(右上の点の対照)は、RAG1.10.3DC4ACT標的もRAG1.10.3DC4TAT標的も切断しなかった。それぞれの陽性クローンは、2つの標的のうちの1つだけを切断することが見出された。これらの結果は、得られた変異体の特異性を示す。このスクリーニングにより、よって、DmoCreタンパク質のCre部分に変異を導入した後に(Amel2_RG3D変異体)、I-DmoI部分にさらに変異を加えることによりタンパク質をさらに工学的に改変して、組み合わせ標的を特異的に切断することが可能であることが示される。
【特許請求の範囲】
【請求項1】
少なくとも第1I-DmoIドメインを含み、前記第1I-DmoIドメインの15位、19位又は20位の残基の少なくとも1つの置換及び27位、29位、33位、35位、37位、75位、76位、77位又は81位の残基の少なくとも1つの置換を含むことを特徴とし、
±2位、±3位、±4位、±5位、±6位、±7位、±8位、±9位、±10位の少なくとも1つにおいて配列番号30の野生型I-DmoI DNA標的ハーフサイトとは異なるI-DmoI DNA標的ハーフサイトを認識する、
I-DmoIエンドヌクレアーゼ又はそれらのキメラ派生物の配列を含むポリペプチド。
【請求項2】
15位、19位又は20位の残基の少なくとも1つが任意のアミノ酸に置換される、請求項1に記載のポリペプチド。
【請求項3】
20位の残基がセリン又はアラニンに変異される(G20S又はG20A)、請求項1又は2に記載のポリペプチド。
【請求項4】
15位のリジンがグルタミンとなる(L15Q)、請求項1〜3のいずれか1項に記載のポリペプチド。
【請求項5】
19位のイソロイシンがアスパラギン酸となる(I19D)、請求項1〜4のいずれか1項に記載のポリペプチド。
【請求項6】
任意のアミノ酸による29位、33位又は35位の残基の少なくとも1つの置換が、±8位、±9位、±10位の少なくとも1つにおいて配列番号30の野生型I-DmoI DNA標的ハーフサイトとは異なるI-DmoI DNA標的ハーフサイトの前記ポリペプチドの認識を変化させる、請求項1〜5のいずれか1項に記載のポリペプチド。
【請求項7】
任意のアミノ酸による75位、76位又は77位の残基の少なくとも1つの置換が、±2位、±3位、±4位の少なくとも1つにおいて配列番号30の野生型I-DmoI DNA標的ハーフサイトとは異なるI-DmoI DNA標的ハーフサイトの前記ポリペプチドの認識を変化させる、請求項1〜6のいずれか1項に記載のポリペプチド。
【請求項8】
任意のアミノ酸による27位、37位又は81位の残基の少なくとも1つの置換が、±5位、±6位、±7位の少なくとも1つにおいて配列番号30の野生型I-DmoI DNA標的ハーフサイトとは異なるI-DmoI DNA標的ハーフサイトの前記ポリペプチドの認識を変化させる、請求項1〜7のいずれか1項に記載のポリペプチド。
【請求項9】
配列番号1の配列に由来する、請求項1に記載のポリペプチド。
【請求項10】
配列番号27の配列に由来する、請求項1に記載のポリペプチド。
【請求項11】
前記ポリペプチドが、二量体LAGLIDADGホーミングエンドヌクレアーゼの配列又は別の単量体LAGLIDADGホーミングエンドヌクレアーゼのドメインへの前記第1I-Dmo Iドメインの融合体からなるキメラ-Dmoエンドヌクレアーゼである、請求項1〜10のいずれか1項に記載のポリペプチド。
【請求項12】
前記第1I-Dmo Iドメインが、I-Sce I、I-Chu I、I-Cre I、I-Csm I, PI-Sce I, PI-Tli I、PI-Mtu I、I-Ceu I、I-Sce II、I-Sce III、HO、PI-Civ I、PI-Ctr I、PI-Aae I、PI-Bsu I、PI-Dha I、 PI-Dra I、PI-Mav I、PI-Mch I、PI-Mfu I、PI-Mfl I、PI-Mga I、PI-Mgo I、PI-Min I、PI-Mka I、PI-Mle I、PI-Mma I、PI-Msh I、PI-Msm I、PI-Mth I、PI-Mtu I、PI-Mxe I、PI-Npu I、PI-Pfu I、PI-Rma I、PI-Spb I、PI-Ssp I、PI-Fac I、PI-Mja I、PI-Pho I、PI-Tag I、PI-Thy I、PI-Tko I、PI-Tsp I、I-MsoIの群の酵素の1つから選択される第2ドメインに融合される、請求項1〜11のいずれか1項に記載のポリペプチド。
【請求項13】
前記配列が、(i) 前記第1I-Dmo Iドメインの4位、49位、52位、92位、94位及び/若しくは95位の残基の1つ、並びに/或いは(ii) 存在する場合、I-Dmo Iの第2ドメインのリンカー又は開始部の101位、102位及び/若しくは109位の残基の1つ
の群から選択される少なくとも1つのさらなる残基の置換を含む、請求項1〜12のいずれか1項に記載のポリペプチド。
【請求項14】
4位のアスパラギンがイソロイシンに変異され(N4I)、49位のリジンがアルギニンに変異され(K49R)、52位のイソロイシンンがフェニルアラニンに変異され(I52F)、92位のアラニンがスレオニンに変異され(A92T)、94位のメチオニンがリジンに変異され(M94K)、95位のロイシンがグルタミンに変異され(L95Q)、(存在する場合)101位のフェニルアラニンがシステインに変異され(F101C)、(存在する場合)102位のアスパラギンがイソロイシンに変異され(N102I)、及び/又は(存在する場合)109位のフェニルアラニンがイソロイシンに変異される(F109I)、請求項13に記載のポリペプチド。
【請求項15】
第1I-Dmo Iドメインが、前記キメラ-DmoエンドヌクレアーゼのNH2末端にある、請求項1〜14のいずれか1項に記載のポリペプチド。
【請求項16】
前記二量体LAGLIDADGホーミングエンドヌクレアーゼがI-CreIであることを特徴とする、請求項1〜15のいずれか1項に記載のポリペプチド。
【請求項17】
配列番号2の配列に由来することを特徴とする、請求項1〜16のいずれか1項に記載のポリペプチド。
【請求項18】
配列番号9の配列に由来することを特徴とする、請求項1〜17のいずれか1項に記載のポリペプチド。
【請求項19】
検出可能なタグをNH2末端及び/又はCOOH末端に含むことを特徴とする、請求項1〜18のいずれか1項に記載のポリペプチド。
【請求項20】
請求項1〜19のいずれか1項に記載のポリペプチドをコードすることを特徴とする、ポリヌクレオチド。
【請求項21】
請求項20に記載のポリヌクレオチドを含むことを特徴とする、ベクター。
【請求項22】
請求項20に記載のポリヌクレオチド又は請求項21に記載のベクターにより改変されることを特徴とする、宿主細胞。
【請求項23】
請求項20に記載のポリヌクレオチド又は請求項21に記載のベクターにより、その細胞の全て又は一部が改変されることを特徴とする、非ヒトトランスジェニック動物。
【請求項24】
請求項20に記載のポリヌクレオチド又は請求項21に記載のベクターにより、その細胞の全て又は一部が改変されることを特徴とする、トランスジェニック植物。
【請求項25】
請求項1〜19のいずれか1項に記載のポリペプチド、請求項20に記載のポリヌクレオチド、請求項21に記載のベクター、請求項22に記載の細胞、請求項23に記載の非ヒト動物若しくは請求項24に記載の植物の、新規なDNA標的特異性を有するメガヌクレアーゼの選択及び/又はスクリーニングのための使用。
【請求項26】
前記I-CreI単量体配列が、前記I-CreI単量体の44位、68位、70位、75位、77位の残基の少なくとも1つの改変を含む、請求項16〜18のいずれか1項に記載のポリペプチド。
【請求項27】
前記I-CreI単量体配列が、前記I-CreI単量体の28位、30位、32位、33位、38位、40位の残基の少なくとも1つの改変を含む、請求項16〜18又は26のいずれか1項に記載のポリペプチド。
【請求項28】
前記I-CreI単量体配列が、前記I-CreI単量体の37位、79位、81位の残基の少なくとも1つの改変を含む、請求項16〜18、26又は27のいずれか1項に記載のポリペプチド。
【請求項1】
少なくとも第1I-DmoIドメインを含み、前記第1I-DmoIドメインの15位、19位又は20位の残基の少なくとも1つの置換及び27位、29位、33位、35位、37位、75位、76位、77位又は81位の残基の少なくとも1つの置換を含むことを特徴とし、
±2位、±3位、±4位、±5位、±6位、±7位、±8位、±9位、±10位の少なくとも1つにおいて配列番号30の野生型I-DmoI DNA標的ハーフサイトとは異なるI-DmoI DNA標的ハーフサイトを認識する、
I-DmoIエンドヌクレアーゼ又はそれらのキメラ派生物の配列を含むポリペプチド。
【請求項2】
15位、19位又は20位の残基の少なくとも1つが任意のアミノ酸に置換される、請求項1に記載のポリペプチド。
【請求項3】
20位の残基がセリン又はアラニンに変異される(G20S又はG20A)、請求項1又は2に記載のポリペプチド。
【請求項4】
15位のリジンがグルタミンとなる(L15Q)、請求項1〜3のいずれか1項に記載のポリペプチド。
【請求項5】
19位のイソロイシンがアスパラギン酸となる(I19D)、請求項1〜4のいずれか1項に記載のポリペプチド。
【請求項6】
任意のアミノ酸による29位、33位又は35位の残基の少なくとも1つの置換が、±8位、±9位、±10位の少なくとも1つにおいて配列番号30の野生型I-DmoI DNA標的ハーフサイトとは異なるI-DmoI DNA標的ハーフサイトの前記ポリペプチドの認識を変化させる、請求項1〜5のいずれか1項に記載のポリペプチド。
【請求項7】
任意のアミノ酸による75位、76位又は77位の残基の少なくとも1つの置換が、±2位、±3位、±4位の少なくとも1つにおいて配列番号30の野生型I-DmoI DNA標的ハーフサイトとは異なるI-DmoI DNA標的ハーフサイトの前記ポリペプチドの認識を変化させる、請求項1〜6のいずれか1項に記載のポリペプチド。
【請求項8】
任意のアミノ酸による27位、37位又は81位の残基の少なくとも1つの置換が、±5位、±6位、±7位の少なくとも1つにおいて配列番号30の野生型I-DmoI DNA標的ハーフサイトとは異なるI-DmoI DNA標的ハーフサイトの前記ポリペプチドの認識を変化させる、請求項1〜7のいずれか1項に記載のポリペプチド。
【請求項9】
配列番号1の配列に由来する、請求項1に記載のポリペプチド。
【請求項10】
配列番号27の配列に由来する、請求項1に記載のポリペプチド。
【請求項11】
前記ポリペプチドが、二量体LAGLIDADGホーミングエンドヌクレアーゼの配列又は別の単量体LAGLIDADGホーミングエンドヌクレアーゼのドメインへの前記第1I-Dmo Iドメインの融合体からなるキメラ-Dmoエンドヌクレアーゼである、請求項1〜10のいずれか1項に記載のポリペプチド。
【請求項12】
前記第1I-Dmo Iドメインが、I-Sce I、I-Chu I、I-Cre I、I-Csm I, PI-Sce I, PI-Tli I、PI-Mtu I、I-Ceu I、I-Sce II、I-Sce III、HO、PI-Civ I、PI-Ctr I、PI-Aae I、PI-Bsu I、PI-Dha I、 PI-Dra I、PI-Mav I、PI-Mch I、PI-Mfu I、PI-Mfl I、PI-Mga I、PI-Mgo I、PI-Min I、PI-Mka I、PI-Mle I、PI-Mma I、PI-Msh I、PI-Msm I、PI-Mth I、PI-Mtu I、PI-Mxe I、PI-Npu I、PI-Pfu I、PI-Rma I、PI-Spb I、PI-Ssp I、PI-Fac I、PI-Mja I、PI-Pho I、PI-Tag I、PI-Thy I、PI-Tko I、PI-Tsp I、I-MsoIの群の酵素の1つから選択される第2ドメインに融合される、請求項1〜11のいずれか1項に記載のポリペプチド。
【請求項13】
前記配列が、(i) 前記第1I-Dmo Iドメインの4位、49位、52位、92位、94位及び/若しくは95位の残基の1つ、並びに/或いは(ii) 存在する場合、I-Dmo Iの第2ドメインのリンカー又は開始部の101位、102位及び/若しくは109位の残基の1つ
の群から選択される少なくとも1つのさらなる残基の置換を含む、請求項1〜12のいずれか1項に記載のポリペプチド。
【請求項14】
4位のアスパラギンがイソロイシンに変異され(N4I)、49位のリジンがアルギニンに変異され(K49R)、52位のイソロイシンンがフェニルアラニンに変異され(I52F)、92位のアラニンがスレオニンに変異され(A92T)、94位のメチオニンがリジンに変異され(M94K)、95位のロイシンがグルタミンに変異され(L95Q)、(存在する場合)101位のフェニルアラニンがシステインに変異され(F101C)、(存在する場合)102位のアスパラギンがイソロイシンに変異され(N102I)、及び/又は(存在する場合)109位のフェニルアラニンがイソロイシンに変異される(F109I)、請求項13に記載のポリペプチド。
【請求項15】
第1I-Dmo Iドメインが、前記キメラ-DmoエンドヌクレアーゼのNH2末端にある、請求項1〜14のいずれか1項に記載のポリペプチド。
【請求項16】
前記二量体LAGLIDADGホーミングエンドヌクレアーゼがI-CreIであることを特徴とする、請求項1〜15のいずれか1項に記載のポリペプチド。
【請求項17】
配列番号2の配列に由来することを特徴とする、請求項1〜16のいずれか1項に記載のポリペプチド。
【請求項18】
配列番号9の配列に由来することを特徴とする、請求項1〜17のいずれか1項に記載のポリペプチド。
【請求項19】
検出可能なタグをNH2末端及び/又はCOOH末端に含むことを特徴とする、請求項1〜18のいずれか1項に記載のポリペプチド。
【請求項20】
請求項1〜19のいずれか1項に記載のポリペプチドをコードすることを特徴とする、ポリヌクレオチド。
【請求項21】
請求項20に記載のポリヌクレオチドを含むことを特徴とする、ベクター。
【請求項22】
請求項20に記載のポリヌクレオチド又は請求項21に記載のベクターにより改変されることを特徴とする、宿主細胞。
【請求項23】
請求項20に記載のポリヌクレオチド又は請求項21に記載のベクターにより、その細胞の全て又は一部が改変されることを特徴とする、非ヒトトランスジェニック動物。
【請求項24】
請求項20に記載のポリヌクレオチド又は請求項21に記載のベクターにより、その細胞の全て又は一部が改変されることを特徴とする、トランスジェニック植物。
【請求項25】
請求項1〜19のいずれか1項に記載のポリペプチド、請求項20に記載のポリヌクレオチド、請求項21に記載のベクター、請求項22に記載の細胞、請求項23に記載の非ヒト動物若しくは請求項24に記載の植物の、新規なDNA標的特異性を有するメガヌクレアーゼの選択及び/又はスクリーニングのための使用。
【請求項26】
前記I-CreI単量体配列が、前記I-CreI単量体の44位、68位、70位、75位、77位の残基の少なくとも1つの改変を含む、請求項16〜18のいずれか1項に記載のポリペプチド。
【請求項27】
前記I-CreI単量体配列が、前記I-CreI単量体の28位、30位、32位、33位、38位、40位の残基の少なくとも1つの改変を含む、請求項16〜18又は26のいずれか1項に記載のポリペプチド。
【請求項28】
前記I-CreI単量体配列が、前記I-CreI単量体の37位、79位、81位の残基の少なくとも1つの改変を含む、請求項16〜18、26又は27のいずれか1項に記載のポリペプチド。
【図1】


【図2】
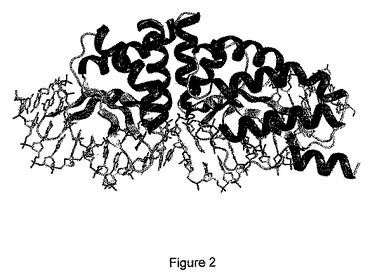
【図3A】
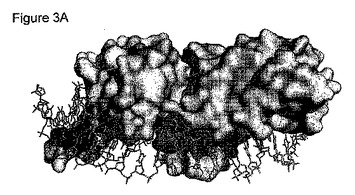
【図3B】
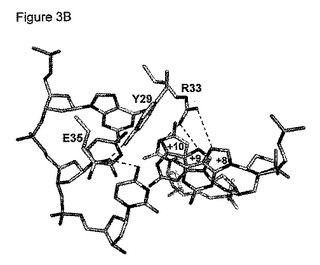
【図4】
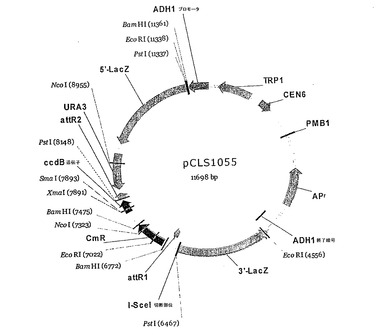
【図5】
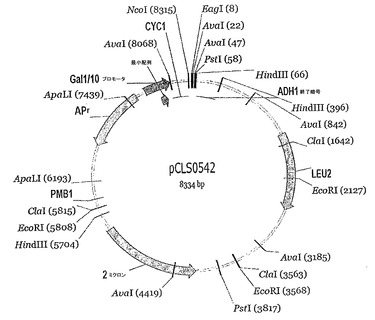
【図6】
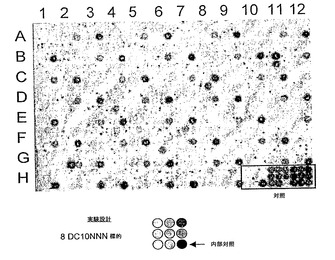
【図7】
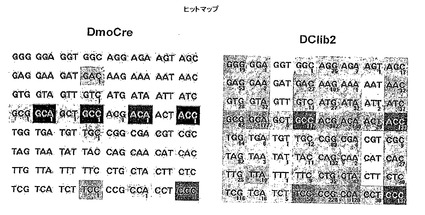
【図8A】
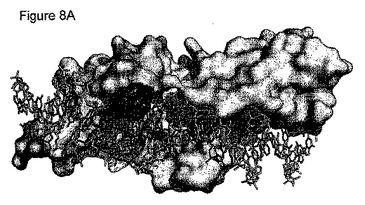
【図8B】

【図9】

【図10】

【図11】
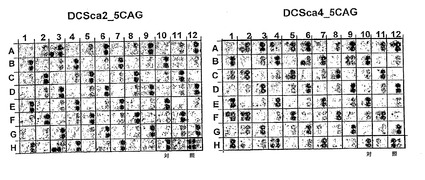
【図12】

【図13】

【図14】

【図15】
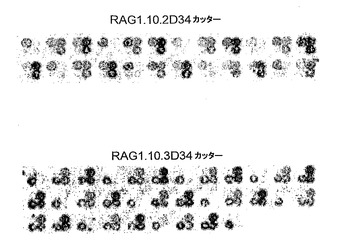
【図16】
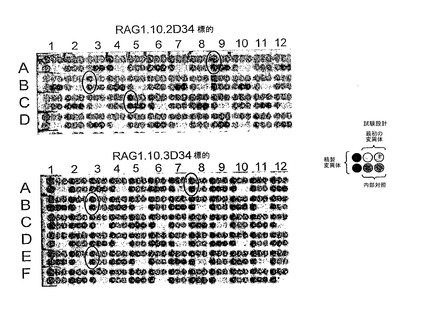
【図17】
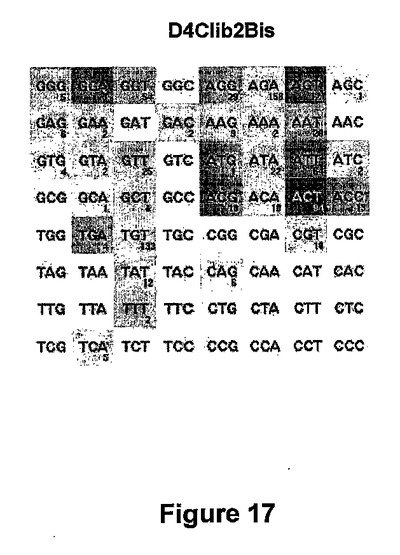
【図18A】

【図18B】
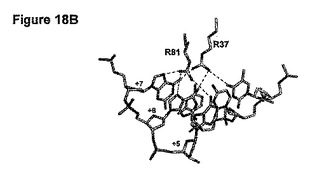
【図18C】
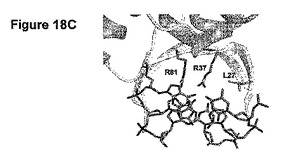
【図19】
【図20】

【図21】

【図2】
【図3A】
【図3B】
【図4】
【図5】
【図6】
【図7】
【図8A】
【図8B】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18A】
【図18B】
【図18C】
【図19】
【図20】
【図21】
【公表番号】特表2011−505824(P2011−505824A)
【公表日】平成23年3月3日(2011.3.3)
【国際特許分類】
【出願番号】特願2010−537538(P2010−537538)
【出願日】平成20年12月12日(2008.12.12)
【国際出願番号】PCT/IB2008/003744
【国際公開番号】WO2009/074873
【国際公開日】平成21年6月18日(2009.6.18)
【出願人】(508245231)
【氏名又は名称原語表記】CELLECTIS
【住所又は居所原語表記】102 avenue Gaston Roussel,F−93235 Romainville Cedex,France
【Fターム(参考)】
【公表日】平成23年3月3日(2011.3.3)
【国際特許分類】
【出願日】平成20年12月12日(2008.12.12)
【国際出願番号】PCT/IB2008/003744
【国際公開番号】WO2009/074873
【国際公開日】平成21年6月18日(2009.6.18)
【出願人】(508245231)
【氏名又は名称原語表記】CELLECTIS
【住所又は居所原語表記】102 avenue Gaston Roussel,F−93235 Romainville Cedex,France
【Fターム(参考)】
[ Back to top ]