
改善された反応速度を有するB12依存性デヒドラターゼ
改善された反応速度を有するB12依存性デヒドラターゼの配列を提供することで、グリセロールおよび1,3−プロパンジオール存在下における酵素の自殺不活性化の速度が低下する。酵素は、グリセロールデヒドラターゼのα−サブユニットをコードするDhaB1遺伝子を標的にした、エラープローンPCRおよびオリゴヌクレオチド−誘導変異誘発を使用して作り出される。改善された反応速度を有する変異株は、これもまたここで開示されるハイスループットアッセイを使用して迅速に同定される。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、1,3−プロパンジオールを生成するための分子生物学分野およびB12依存性デヒドラターゼの使用に関する。より具体的には、酵素不活性化速度が低下するように反応速度が改善されたB12依存性デヒドラターゼを作り出し、選択する方法について述べる。
【背景技術】
【0002】
1,3−プロパンジオールは、いくつかの用途において、ポリエステル、ポリエーテル、およびポリウレタンを製造するための出発原料をはじめとする有用性を有する。1,3−プロパンジオールを製造するための方法としては、伝統的化学経路および生物学的経路の双方が挙げられる。1,3−プロパンジオールを製造するための生物学的方法については、最近述べられている(非特許文献1)。1,3−プロパンジオールを生物学的に製造するのには、二段階逐次反応の基質としてのグリセロールが必要である。まずデヒドラターゼ(典型的に補酵素B12依存性デヒドラターゼ)が、グリセロールを中間体3−ヒドロキシプロピオンアルデヒド(3−HP)に変換する。次にNADH−(またはNADPH)−依存酸化還元酵素(式1および2参照)によって、3−HPが1,3−プロパンジオールに還元される。
グリセロール→3−HP+H2O(式1)
3−HP+NADH+H+→1,3−プロパンジオール+NAD+(式2)
【0003】
1,3−プロパンジオールはさらに代謝されることなく、その結果、培養液中に高濃度で蓄積する。
【0004】
典型的にグリセロールは、1,3−プロパンジオールを生物学的に生成するための出発原料として使用される。しかしグルコースおよびその他の炭水化物もまた、1,3−プロパンジオール生成のための適切な基質である。具体的にはラフェンド(Laffend)ら(特許文献1、特許文献2)は、デヒドラターゼ活性を含んでなる単一微生物を使用して、グリセロールまたはジヒドロキシアセトン以外の炭素基質(特に、例えばグルコース)から1,3−プロパンジオールを製造する方法を開示する。エンプテイジ(Emptage)ら(特許文献3)は、3−HPを1,3−プロパンジオールに変換する非特異的触媒活性(dhaTによってコードされる1,3−プロパンジオール酸化還元酵素とは区別される)によって得られる力価(リットル当たりの生成グラム数)の顕著な増大について述べる。ペイン(Payne)ら(特許文献4)は、1,3−プロパンジオールを生物学的に生成するのに有用な特定のベクターおよびプラスミドを開示する。セレヴァン(Cervin)ら(特許文献5)は、1,3−プロパンジオールの高収率生成のための改善された大腸菌(E.coli)株を開示する。特許文献1、特許文献3、特許文献4、および特許文献5は、参照によってその全体を本明細書に援用したものとする。
【0005】
グリセロールを3−HPに変換するのに関与する酵素は、主として補酵素B12依存性グリセロールデヒドラターゼ(E.C.4.2.1.30)および補酵素B12依存性ジオールデヒドラターゼ(E.C.4.2.1.28)として知られる補酵素B12依存性酵素である。これらの異なるが関係のある補酵素B12依存性酵素は、それらの分子および生化学的特性に関しては十分に研究されている。補酵素B12依存性デヒドラターゼの遺伝子は、例えば肺炎桿菌(Klebsiella pneumoniae)、シトロバクター・フロインディ(Citrobacter freundii)、クロストリジウム・パスツーリアナム(Clostridium pasteurianum)、ネズミチフス菌(Salmonella typhimurium)、クレブシエラ・オキシトカ(Klebsiella oxytoca)、およびラクトバシラス・コリノイデス(Lactobacillus collinoides)で同定されている(非特許文献2、非特許文献3、および非特許文献4)。
【0006】
文献中で使用される遺伝子命名法には幅広いバリエーションがあるが、各例で補酵素B12依存性デヒドラターゼは、大型または「α」サブユニット、中型または「β」サブユニット、および小型または「γ」サブユニットの3個のサブユニットから構成される。これらのサブユニットは、α2β2γ2構造体にアセンブルされてアポ酵素を形成する。補酵素B12(活性補助因子化学種)は、アポ酵素に結合して触媒的に活性のホロ酵素を形成する。補酵素B12は、それによって触媒作用が起きるラジカル機序に関与するので、触媒の活性のために必要とされる。
【0007】
生化学的に、補酵素B12依存性グリセロールおよび補酵素B12依存性ジオールデヒドラターゼの双方が、グリセロールおよびその他の基質による機序ベースの自殺不活性化を被ることが知られている(非特許文献3、非特許文献5)。さらに不活性化は、ホロ酵素と高濃度の1,3−プロパンジオールとの相互作用を通じて起きる。不活性化は補酵素B12補助因子のコバルト−炭素(Co−C)結合の開裂を伴い、5’−デオキシアデノシンの形成と不活性なコバラミン化学種をもたらす。不活性なコバラミン化学種はデヒドラターゼ密接に結合したままであり、補酵素B12依存性デヒドラターゼ再活性化因子(「デヒドラターゼ再活性化因子」)の介入なしには解離が起きない。この不活性化は3−HP形成に結びついた反応速度を顕著に低下でき、それによって間接的に1,3−プロパンジオール生成を減少させる。
【0008】
補酵素B12依存性デヒドラターゼ不活性化の影響は、部分的に克服できる。例えば不活性化は、デヒドラターゼ活性を再活性化するのに関与するタンパク質に依存して克服できる。デヒドラターゼ再活性化因子については、特許文献6、特許文献7、非特許文献3、非特許文献6、および非特許文献7で述べられる。再活性化は多段階プロセスで起きる。最初にATP−依存プロセスにおいて、不活性化された補酵素B12依存性デヒドラターゼとデヒドラターゼ再活性化因子との相互作用が、密接に結合した不活性なコバラミン化学種の放出を引き起こし、アポ酵素が生成する。引き続いてデヒドラターゼアポ酵素が補酵素B12と結合して、触媒的に活性なホロ酵素を再形成しても良く、(別のATP−依存プロセスで酵素作用によって)不活性なコバラミン化学種が補酵素B12に再生されても良い。しかしデヒドラターゼ再活性化および補酵素B12再生プロセスの双方がATPを必要とするので、デヒドラターゼ活性復元のためにデヒドラターゼ再活性化因子だけに依存することは、本質的に限定される。これらのATP−依存プロセスは、特に引き続く3−HPから1,3−プロパンジオールへの反応が存在して、1,3−プロパンジオール濃度が高い場合、グリセロールを3−HPに変換するプロセスに対する顕著なエネルギー負荷を意味する。
【0009】
代案としては、1,3−プロパンジオール生成中に培地に添加する補酵素B12の量を増大させる、あるいは培養液に(生体内で(in vivo)補酵素B12に変換される)ビタミンB12を補充する、のいずれかで微生物に追加的な補酵素B12を供給することが可能である。しかしどちらの場合もこれらの添加経費が、プロセスの経済性を顕著に妨げるかもしれない。
【0010】
クロウ(Croux)ら(特許文献8)は、補酵素B12非依存性デヒドラターゼを発現する組み換え微生物を使用して1,3−プロパンジオールを製造するプロセスを開発することで、補酵素B12依存性デヒドラターゼに関する問題に対処した。しかしこの解決法の有用性は、特定の好ましいプロセス条件下でB12非依存性デヒドラターが機能する能力によって限定されるかもしれない(例えば好気性条件下で(非特許文献8))。
【0011】
原理上は、自然発生微生物株から不活性化速度が低下した補酵素B12依存性デヒドラターゼを単離することが可能なはずである。低下した不活性化速度は、微生物宿主における補酵素B12依存性デヒドラターゼの代謝回転率(生成物mol/ホロ酵素mol)を増大させ、これによりデヒドラターゼおよび補酵素B12の要求量が低下する。このアプローチはそのレベルのデヒドラターゼおよび補酵素B12を維持するのに必要なエネルギーも低下させるであろう。しかし実際には、補酵素B12依存性デヒドラターゼの多様性は不活性化速度に関して限られていることが分かっている。
【0012】
【特許文献1】国際公開第96/35796号パンフレット
【特許文献2】米国特許第5,686,276号明細書
【特許文献3】国際公開第01/012833号パンフレット
【特許文献4】米国特許出願第60/374931号明細書
【特許文献5】米国特許出願第60/416192号明細書
【特許文献6】国際公開第98/21341号パンフレット
【特許文献7】米国特許第6,013,494号明細書
【特許文献8】国際公開第01/04324 A1号パンフレット
【非特許文献1】ゼング(Zeng)ら、Adv.Biochem.Eng.Biotechnol.、74:239〜259(2002)
【非特許文献2】トラヤ(Toraya)T.、アミノ酸残基および関連ラジカルに関与する金属結合酵素(Metalloenzymes Involving Amino Acid−Residue and Related Radicals)、シーゲル(Sigel)H.およびシーゲル(Sigel)A.編;生物学的システム中の金属イオン(Metal Ions in Biological Systems);マーセルデッカー(Marcel Dekker):ニューヨーク(New York)、1994;第30巻、pp217〜254
【非特許文献3】ダニエル(Daniel)ら、FEMS Microbilogy Reviews、22:553〜566(1999)
【非特許文献4】サウヴェージ(Sauvageot)ら、FEMS Microbiology Letters、209:69〜74(2002)
【非特許文献5】サイフェルト(Seifert)ら、Eur.J.Biochem.、268:2369〜2378(2001)
【非特許文献6】トラヤ(Toraya)およびモリ(Mori)、J.Biol.Chem.、274:3372(1999)
【非特許文献7】トビマツ(Tobimatsu)ら、J.Bacteriol.、181:4110(1999)
【非特許文献8】ハートマニス(Hartmanis)およびステードマン(Stadman)、Arch.Biochem.Biophys.、245:144〜52(1986)
【発明の開示】
【発明が解決しようとする課題】
【0013】
したがって解決すべき問題は、現在入手できる補酵素B12依存性デヒドラターゼ酵素が、工業化合物の生成のために工業用途で必要な反応速度を提供できないことである。
【課題を解決するための手段】
【0014】
出願人らは、
(a)B12依存性デヒドラターゼホロ酵素と、グリセロールおよび少なくとも25mMの1,3−プロパンジオールを含んでなる混合物とを接触させ、
(b)B12依存性デヒドラターゼホロ酵素をスクリーニングして、B12依存性デヒドラターゼの代謝回転率を推定する
ことを含んでなる、改善された反応速度を有するB12依存性デヒドラターゼのスクリーニング方法を提供する。
【0015】
方法のスクリーニングステップは、
(a)補酵素B12、デヒドラターゼ再活性化因子、または内在性B12依存性デヒドラターゼの供給源を有さない細胞をグリセロールを含まない発酵性炭素基質上に生育させ、
(b)細胞を透過化処理し、
(c)補酵素B12、グリセロール、および少なくとも25mMの1,3−プロパンジオールの混合物をステップ(b)の透過化処理された細胞添加し、
(d)ステップ(c)で生産された3−ヒドロキシプロピオンアルデヒドを定量化する
ことをさらに含んでなり、
定量化は、T1、T2、およびT(600)よりなる群から選択される測定である。
【0016】
本発明は、配列番号:40、44、48、52、56、60、64、68、72、76、80、84、88、92、96、100、104、108、112、116、120、124、128、132、136、140、143、147、151、155、159、163、167、171、175、179、182、186、189、192、195、198、201、204、208、212、215、218、221、225、229、233、237、241、245、249、253、257、261、265、269、273、277、281、285、289、293、297、301、304、307、310、313、316、319、322、325、328、331、334、および337よりなる群から選択される、B12依存性変異株デヒドラターゼをコードする核酸配列をさらに含む。より好ましいB12依存性変異株デヒドラターゼは、配列番号:40、44、48、52、56、60、64、68、72、76、80、84、88、92、96、100、104、108、112、116、120、124、128、132、136、140、143、147、151、155、159、163、167、171、175、179、182、186、189、192、195、198、201、204、208、212、215、218、221、225、229、233、237、241、245、249、253、257、261、265、269、273、277、281、285、289、293、297、301、304、307、310、313、316、319、322、325、328、331、334、および337よりなる群から選択される核酸配列によってコードされる。B12依存性変異株デヒドラターゼをコードするより好ましい核酸配列は、配列番号:140、179、186、189、192、195、198、201、212、215、218、301、304、307、310、313、316、319、322、325、328、331、334、および337よりなる群から選択されるα−βサブユニット融合を含んでなる。B12依存性変異株デヒドラターゼをコードする最も好ましい核酸配列は、配列番号313、322、および328よりなる群から選択されるα−βサブユニット融合を含んでなる。
【0017】
本発明はまた、α−βサブユニット融合のαおよびβサブユニット間にリンカー配列さらに含んでなる核酸配列を含み、リンカー配列は、配列番号18および配列番号19よりなる群から選択される。
【0018】
本発明はまた、
a)ホットスポット1またはホットスポット2を含んでなるB12依存性デヒドラターゼホロ酵素と変異原因物質とを接触させ、
b)ステップA)で生産された変異株を改善されたkcatおよび/または安定性についてスクリーニングし、そして
c)ステップa)およびb)を反復する
ことを含んでなる、改善された反応速度を有するB12依存性デヒドラターゼ変異株作り出す方法を含む。
【0019】
標準デヒドラターゼと比較して改善された反応速度を有するB12依存性デヒドラターゼを同定するための本発明のさらに別の方法は、
a)デヒドラターゼホロ酵素と5〜10mMのグリセロールおよび/または10〜300mMの1,3−プロパンジオールとを接触させ、
b)ハイスループットアッセイにおいて、kcatおよび総酵素代謝回転を推定するのに十分に隔たった少なくとも2個の時点で測定し、
c)標準デヒドラターゼと比較して改善された反応速度を有するB12依存性変異株を選択する
ことを含んでなる。
【0020】
標準デヒドラターゼと比較して改善された反応速度を有するB12依存性デヒドラターゼを同定するなおも別の本発明の方法は、
a)デヒドラターゼホロ酵素と5〜50mMグリセロールおよび>300mMの1,3−プロパンジオールとを接触させ、
b)ハイスループットアッセイにおいて、T0から十分に隔たった1つの時点で測定して、総酵素代謝回転数を推定し、そして
c)標準デヒドラターゼと比較して改善された反応速度を有するB12依存性変異株を選択する
ことを含んでなる。
【0021】
配列リスト
出願人らは、特許出願におけるヌクレオチドおよびアミノ酸配列開示を司る規則(EPO通達OJ 12/1992、追補No.2で公開されるEPO長官決定の付録IおよびII)、37 C.F.R.1.821〜1.825および付録AおよびB(ヌクレオチドおよび/またはアミノ酸配列を含む出願の開示に関する要件)で述べられる、世界知的所有権機関(WIPO)標準ST.25(1998)およびEPOおよびPCTの配列表要件(規則5.2および49.5(aの2)、および実施細則第208号および附属書C)に一致する346個の配列を提供した。配列説明は、参照によって本明細書に援用するNucleic Acids Research、13:3021〜3030(1985)およびBiochemical Journal、219(2):345〜373(1984)で述べられるIUPAC−IYUB標準に準拠して定義される、ヌクレオチド配列性状のための1文字コードおよびアミノ酸のための3文字コードを含む。
【0022】
配列番号1は、肺炎桿菌(Klebsiella pneumoniae)ATCC25955(エンプテイジ(Emptage)ら、国際公開第01/012833 A2号パンフレット)から単離された野生型GDHを含有する12.1kBのEcoRI−SalI断片である。野生型GDHは、α−サブユニット(bp 7044〜8711)、β−サブユニット(bp 8724〜9308)、およびγ−サブユニット(bp 9311〜9736)によってコードされる。GDHのα、β、およびγ−サブユニットのアミノ酸配列は、それぞれ配列番号2、配列番号3、および配列番号4として提供される。
【0023】
配列番号5および6は、プラスミドpGH20からのGDHのクローニングのために使用されるプライマーDHA−F1およびDHA−R1をそれぞれコードする。
【0024】
配列番号7は、GDHのα−サブユニット全体およびβ−サブユニットの一部のクローニングのために使用される逆転写プライマーDHA−R2をコードする。
【0025】
配列番号8〜14は、α−サブユニットの領域性無作為変異誘発のために使用されるプライマーpGD20RM−F1、pGD20RM−R1、TB4BF、TB4BR、pGD20RM−F2、pGD20RM−R2、およびGD−Cをそれぞれコードする。
【0026】
配列番号15〜17は、領域性無作為変異株ライブラリーの調製のために使用される変性プライマーpGD20RM−F3、TB4B−R1、およびpGD20RM−R4をそれぞれコードする。
【0027】
配列番号18は、融合タンパク質Sma3002のα−およびβ−サブユニット間のリンカーをコードする。配列番号19は、融合タンパク質Xba3009のα−およびβ−サブユニット間のリンカーをコードする。
【0028】
配列番号20〜25は、第二世代変異株GDHの合成のために使用されるプライマー2−F4−F1、2−F4−R1、12−B1−F1、12−B1−R1、16−H5−F1、および16−H5−R1をそれぞれコードする。
【0029】
配列番号26〜29は、純粋融合変異株1−E1および22−G7を作り出すために使用されるプライマー1−E1−F1、1−E1−R1、22−G7−F1、および22−G7−R1をそれぞれコードする。
【0030】
配列番号30〜39は、Sma3002−由来変異株の合成のために使用されるプライマー7A−C1−F1、7A−C1−R1、7C−A5−F1、7C−A5−R1、8−C9−F1、8−C9−R1、9−D7−F1、9−D7−R1、10−G6−F1、および10−G6−R1をそれぞれコードする。
【0031】
野生型GDHから誘導される変異酵素は、それぞれの核酸配列およびアミノ酸配列に従って、以下のSEQ識別番号を割り当てられる(表1)。
【0032】
【表1】
【0033】
【表2】
【0034】
【表3】
【0035】
配列番号340〜342は、プライマーT53−SM、L509−SM、およびV224−SMをそれぞれコードする。
【0036】
配列番号343および344は、プライマーGDHM−F1およびGDHM−R1をそれぞれのコードする。
【0037】
配列番号345および346は、プライマーGDHM−F2およびGDHM−Rをそれぞれのコードする。
【発明を実施するための最良の形態】
【0038】
出願人らは、工業用途で使用するための、具体的には3−ヒドロキシプロピオンアルデヒドおよび1,3−プロパンジオールを製造するための改善された反応速度を有する(すなわちグリセロールおよび1,3−プロパンジオール存在下で増大した総代謝回転数を提供する)人工的補酵素B12依存性デヒドラターゼを提供することで既述の問題を解決した。変異誘発技術によって作り出されたこれらのデヒドラターゼは、それらがそれから作り出された野生型酵素と比較して、増大したkcatおよび/または低下した酵素不活性化速度のいずれかを有することで特徴づけられる。
【0039】
本発明の人工的補酵素B12依存性デヒドラターゼは、触媒作用速度の不当な犠牲なしに不活性化速度を低下させる。デヒドラターゼ不活性化問題のこの解決法は、微生物宿主において補酵素B12依存性デヒドラターゼの代謝回転率(生成物mol/ホロ酵素mol)を増大させるので好ましい。増大した代謝回転率の効果は、デヒドラターゼ要求量、補酵素B12要求量、およびエネルギー消費を低下させて、そのデヒドラターゼおよび補酵素B12レベルを維持する。さらに補酵素B12依存性デヒドラターゼは、工業プロセス条件下での1,3−プロパンジオール生成のために効率的に作動することが実証されている。
【0040】
一連の変異株デヒドラターゼを提供するのに加えて、本発明は2個のハイスループットアッセイも提供して、変異株デヒドラターゼのスクリーニングを容易にする。どちらの方法も補酵素B12、デヒドラターゼ再活性化因子、またはB12依存性依存デヒドラターゼの供給源を有さない細胞における、アポ酵素形態のB12依存性デヒドラターゼの存在に依存する。したがって補酵素B12および基質グリセロールの添加に基づいて、デヒドラターゼ活性の開始を正確に制御することが可能である。
【0041】
具体的には、変異補酵素B12依存性デヒドラターゼ遺伝子の大きなライブラリーを作成して、遺伝子産物を発現させ、グリセロールおよび1,3−プロパンジオール混合物存在下での不活性化低下についてハイスループット様式でスクリーニングした。スクリーニング方法は、低下した不活性化速度および改善されたグリセロール触媒作用速度を有するこれらの人工的補酵素B12依存性デヒドラターゼの容易で迅速な同定を可能にする。引き続くデヒドラターゼエンジニアリングの工程は、なおさらに改善されたデヒドラターゼ変異体の生成および同定を可能にする。
【0042】
ここでの教示に基づいて、本発明で述べられるように、グリセロールから3−HPへの転換を触媒できるB12依存性デヒドラターゼ活性をコードする多様な遺伝子が、変異誘発およびスクリーニングのための標的として適することは、当業者には明らかである。したがって改善された反応速度(すなわちグリセロールおよび1,3−プロパンジオール存在下における増大した総代謝回転数)を有する多様な変異株デヒドラターゼが、この発明の手段によって同定できることが期待される。
【0043】
定義
明細書および請求項の解釈のために、以下の略語および定義が使用される。
【0044】
「ポリメラーゼ連鎖反応」は、PCRという形に省略する。
【0045】
「3−ヒドロキシプロピオンアルデヒド」は、3−HPという形に省略する。
【0046】
「デヒドラターゼ」という用語は、グリセロール分子を生成物3−ヒドロキシプロピオンアルデヒドに異性化または変換できるあらゆる酵素を指すために使用する。上述のようにいくつかのデヒドラターゼは、補助因子として補酵素B12を必要とする。「補酵素B12依存性デヒドラターゼ」および「B12依存性デヒドラターゼ」という用語は区別なく使用され、補酵素B12を必要とするデヒドラターゼを指す。補酵素B12依存性デヒドラターゼは、補酵素B12依存性デヒドラターゼ(E.C.4.2.1.30)および補酵素B12依存性デヒドラターゼ(E.C.4.2.1.28)を含んでなる。代案としては、デヒドラターゼは補酵素B12非依存性であっても良い。これらの酵素は補助因子として補酵素B12を必要とせず、「B12非依存性デヒドラターゼ」という用語で呼ばれる。本発明の目的で、「グリセロールデヒドラターゼ」と言う用語は、特に補酵素B12依存性デヒドラターゼ(E.C.4.2.1.30)を指すために使用され、「ジオールデヒドラターゼ」と言う用語は、特に補酵素B12依存性デヒドラターゼ(E.C.4.2.1.28)を指すために使用される(出願人らの命名法の意図的選択は、B12非依存性デヒドラターゼを「ジオールデヒドラターゼ」および「グリセロールデヒドラターゼ」とそれぞれ称したハートマニス(Hartmanis)およびステードマン(Stadman)、同上、およびクラウス(Croux)ら、同上、の選択と混同すべきでない)。
【0047】
「アポ酵素」と言う用語は、タンパク質から構成される酵素の部分を指す。アポ酵素は、酵素が機能するために必要とするかもしれない非タンパク質構造を含まないので、アポ酵素は触媒的に不活性であっても良い。「補助因子」と言う用語は、アポ酵素によって触媒の活性のために必要とされる非タンパク質構造を指す。「ホロ酵素」と言う用語は、触媒的に活性なタンパク質−補助因子複合体を指す。補酵素B12依存性デヒドラターゼアポ酵素は、ホロ酵素を形成するために補助因子補酵素B12を必要とする。
【0048】
「補酵素B12」および「アデノシルコバラミン」と言う用語は区別なく使用されて、5’−デオキシアデノシルコバラミンを意味する。
【0049】
「ビタミンB12」および「シアノコバラミン」と言う用語は区別なく使用されて、上軸5’−デオキシ−5’−アデノシルリガンドが、シアノ部分にによって置換される補酵素B12の誘導体を指す。
【0050】
「ヒドロキソコバラミン」は、上軸5’−デオキシアデノシルリガンドがヒドロキシ部分によって置換される補酵素B12の誘導体を指す。アクアコバラミンは、ヒドロキソコバラミンのプロトン化形態である。
【0051】
グリセロールまたは1,3−プロパンジオールによるB12非依存性デヒドラターゼホロ酵素の不活性化は、上軸5’−デオキシ−5’−アデノシルリガンドを失った不活性なコバラミン化学種の形成をもたらす。「補酵素B12前駆物質」と言う用語は、上軸5’−デオキシアデノシルリガンドが置換されている補酵素B12の誘導体を指す。
【0052】
ここでの用法では、「GDH」と言う用語は、特に肺炎桿菌(Klebsiella pneumoniae)ATCC25955から単離され、配列番号1のbp 7044〜8711、bp 8724〜9308、およびbp 9311〜9736によってコードされるB12依存性グリセロールデヒドラターゼを指す。「GDH」と言う用語は、α、β、およびγ−サブユニットのアセンブルされた複合体を指すのに使用され、アポ酵素またはホロ酵素を指しても良い。GDHの個々のサブユニットへの言及は、例えば「GDHのα−サブユニット」または「GDHα−サブユニット」などのサブユニットを特定する。同様にα−およびβ−およびγ−サブユニットを総称して、あるいはGDHの個々のサブユニットに言及して、GDHのアミノ酸配列に言及しても良い。GDHのα、β、およびγ−サブユニットのアミノ酸配列は、それぞれ配列番号2、配列番号3、および配列番号4として提供される。
【0053】
この発明の開示の目的では、GDHは、人工的(変異株)誘導体との比較でDNAおよびアミノ酸配列の標準として使用される。GDHはまた、本発明を使用して作り出された人工的誘導体の反応速度がそれに対して測定される、野生型反応速度の標準として使用される。ここではGDHが標準材料として使用されるが、あらゆる自然発生的補酵素B12−依存デヒドラターゼ(例えば表2に列挙されるもの)が本発明においてGDHと区別なく使用でき、それに対して人工的誘導体の反応速度が測定される。
【0054】
「遺伝的に変更される」と言う用語は、形質転換または変異によって遺伝的材料を変化させるプロセスを指す。
【0055】
ここでの用法では、「変異株」と言う用語は、変異プロセスによって生成したGDH酵素またはGDH配列を含有する細菌クローン、プラスミド、ライブラリーまたはベクターを指す。代案としては、変異株と言う用語は、変異プロセスによって生じたGDH酵素、GDHアミノ酸配列、またはGDH DNA配列を直接指す。したがって変異株GDHは(野生型)GDHとは異なる。
【0056】
「改善された反応速度」と言う用語は、グリセロールおよび/または1,3−プロパンジオール存在下で、野生型酵素に対して低下したデヒドラターゼ不活性化速度を指す。したがって改善された反応速度は、グリセロールおよび1,3−プロパンジオール存在下で、増大した総酵素代謝回転数に関係がある。これはkcatの増大および/または酵素不活性化速度の低下のどちらかによって達成できる。
【0057】
「触媒効率」と言う用語は、酵素のkcat/KMとして定義される。「触媒効率」は、基質に対する酵素の特異性を定量化するのに使用される。
【0058】
「kcat」、「KM」、および「Ki」と言う用語は当業者に知られており、(フェルスト(Ferst)著、「酵素構造および機序(Enzyme Structure and Mechanism)」第二版、W.H.フリーマン(Freeman)、ニューヨーク(New York)、1985、pp98〜120)で述べられている。
【0059】
「kcat」と言う用語は、「代謝回転数」と呼ばれることが多い。「kcat」と言う用語は、単位時間あたり活性部位あたりの生成物に変換される基質分子の最大数、または単位時間あたりに酵素が回転する数として定義される。kcat=V最大/[E]、(式中、[E]は酵素濃度である)(フェルスト(Ferst)著、「酵素構造および機序(Enzyme Structure and Mechanism)」第二版、W.H.フリーマン(Freeman)、ニューヨーク(New York)、1985、pp98〜120)。「総代謝回転」および「総代謝回転数」と言う用語は、ここでは反応開始(T0)とホロ酵素の完全な不活性化が起きる間に、B12依存性デヒドラターゼホロ酵素とグリセロールとの(場合により1,3−プロパンジオール存在下での)反応によって形成される生成物量を指すのに使用される。
【0060】
「Ki」と言う用語は、1,3−プロパンジオールの阻害定数を指す。
【0061】
「k観察された不活性」と言う用語は、B12依存性デヒドラターゼホロ酵素と過剰なグリセロールおよび/または過剰な1,3−プロパンジオールとの反応において観察される、不活性化の一次反応速度定数を指す。過剰なグリセロールのみの存在下で、k観察された不活性はk不活性グリセロールに等しい。過剰な1,3−プロパンジオールのみの存在下で、k観察された不活性はk不活性1,3−プロパンジオールに等しい。過剰なグリセロールおよび過剰な1,3−プロパンジオール単独双方存在下で、k観察された不活性は、k不活性グリセロールおよびk不活性1,3−プロパンジオール双方の関数に等しい。
【0062】
「T1」と言う用語は、10mMのグリセロールおよび50mMの1,3−プロパンジオール存在下で、反応開始30秒後に測定されるGDH酵素反応によって作られる生成物量を指す。T1はkcatに比例する。
【0063】
「T2」と言う用語は、10mMのグリセロールおよび50mMの1,3−プロパンジオール存在下で、反応開始40分後に測定されるGDH酵素反応によって作られる生成物量を指す。T2はkcat/k観察された不活性に比例し、総酵素総代謝回転数を反映する。
【0064】
「T2/T1比率」と言う用語は、T2とT1との比率を指す。T2/T1比率は、1/k観察された不活性に比例する。
【0065】
「T2(600)」と言う用語は、10mMのグリセロールおよび600mMの1,3−プロパンジオール存在下で、反応開始40分後に測定されるGDH酵素反応によって作られる生成物量を指す。T2(600)はkcat/k観察された不活性に比例し、600mMの1,3−プロパンジオール存在下における総酵素総代謝回転数を反映する。
【0066】
「T(600)」と言う用語は、10mMのグリセロールおよび600mMの1,3−プロパンジオール存在下で、反応開始70分後に測定されるGDH酵素反応によって作られる生成物量を指す。T(600)はkcat/k観察された不活性に比例する。T(600)値は、600mM1,3−プロパンジオール存在下においてT2(600)よりも正確な総酵素代謝回転数を与える。
【0067】
「ポリペプチド」および「タンパク質」と言う用語は、区別なく使用される。
【0068】
「宿主細胞」または「宿主生物体」と言う用語は、外来性または非相同的遺伝子を受け入れてこれらの遺伝子を発現し、活性遺伝子産物を生成できる微生物を指す。
【0069】
「単離された」と言う用語は、それが自然に関連する少なくとも1個の構成要素から取り除かれるタンパク質またはDNA配列を指す。
【0070】
「単離された核酸分子」は、場合により合成、非天然または修飾ヌクレオチド塩基を含有する、一本鎖または二本鎖であるRNAまたはDNAのポリマーである。DNAポリマーの形態の単離された核酸分子は、1個またはそれ以上のcDNA、ゲノムDNAまたは合成DNAの断片を含んでなっても良い。
【0071】
「遺伝子」とは、特定のタンパク質を発現する核酸断片を指し、場合によってはコード配列に先行する(5’非コード配列)およびそれに続く(3’非コード配列)制御配列を含んでも良い。「天然遺伝子」および「野生型遺伝子」は、自然界にそれ自体の制御配列と共に見られる遺伝子を指す。「キメラ遺伝子」とは、自然界に共に見られない制御およびコード配列を含んでなる天然遺伝子でないあらゆる遺伝子を指す。したがって、キメラ遺伝子は、異なる供給源から誘導される制御配列およびコード配列、あるいは同一供給源から誘導されるが、自然界に見られるのとは異なるやり方で配列される制御配列およびコード配列を含んでなっても良い。「内因性の遺伝子」とは、生物体ゲノムにおいてその天然位置にある天然遺伝子を指す。「外来性の」遺伝子とは、宿主生物体において常態では見られないが、代わりに遺伝子移入によって宿主生物体に導入される遺伝子を指す。外来性の遺伝子は、非天然生物体中に挿入された天然遺伝子、あるいはキメラ遺伝子を含んでなることができる。「導入遺伝子」とは、形質転換によってゲノム中に導入される遺伝子である。
【0072】
「コードする」および「コーディング」と言う用語は、それによって遺伝子が、転写および翻訳の機序を通じてアミノ酸配列を生成するプロセスを指す。特定のアミノ酸配列をコードするプロセスは、コードされるアミノ酸における変化を引き起こさない塩基置換を伴うかもしれないDNA配列を含み、あるいは1個またはそれ以上アミノ酸を変更するかもしれないが、DNA配列によってコードされるタンパク質の官能特性に影響しない塩基置換を伴うDNA配列を含むものと理解される。したがって本発明は、特定の例示的な配列以上のものを包含するものと理解される。
【0073】
「アミノ酸」と言う用語は、タンパク質またはポリペプチドの基本的化学構造単位を指す。アミノ酸は、参照によって本明細書に援用するNucleic Acids Research、13:3021〜3030(1985)およびBiochemical Journal、219(2):345〜373(1984)で述べられるIUPAC−IYUB標準に準拠して定義されるように、1文字コードまたはアミノ酸のための3文字コードのいずれかによって識別される。
【0074】
特定のタンパク質では、DNAコード領域内の点置換変異、および得られるアミノ酸変化は、以下の表記法の1つを使用して、標準DNAおよびアミノ酸配列を参照して特定される。例えばGDHにおける変異の場合、変異は下で述べる表記法の1つを使用して記述される。
1.詳細な表記法:最初に野生型コドンのヌクレオチド配列が提示され、続いて「α」、「β」または「γ」記号(GDHのα−、β−、またはγ−サブユニットの変異を区別するため)、3文字略語の特定の野生型アミノ酸、およびその位置が提示される。次に野生型情報の後に、参照される変異中に存在する特定のヌクレオチドおよびアミノ酸修飾が続く。この表記法の例はGGG(α−Gly63)からGGA(Gly)であり、ここではα−サブユニットの63番目のコドンが、変異株においてヌクレオチド配列が「GGG」から「GGA」に変更されるようにサイレント変異を被る。
2.「簡略」表記法:「α」、「β]、または「γ」の記号(GDHのα−、β−またはγ−サブユニットにおける変異と区別するため)に、野生型アミノ酸の1文字略語、コドン位置、および変異株アミノ酸の1文字略語が続く。この表記法の例はα−V224Lであり、変異株におけるα−サブユニット中のコドン224における野生型バリンからロイシンへの変異を表す。特定部位で化学的に同等のアミノ酸の生成をもたらす(しかしコードされるタンパク質官能特性に影響しない)遺伝子における変更が一般的であることは、技術分野で良く知られている。
【0075】
「実質的に同様の」とは、1個またはそれ以上のヌクレオチド塩基の変化が、1個またはそれ以上アミノ酸の置換をもたらすが、DNA配列によってコードされるタンパク質の官能特性に影響しない核酸分子を指す。「実質的に同様の」とはまた、1個またはそれ以上ヌクレオチド塩基の変化が、アンチセンスまたはコサプレッション技術によって、遺伝子発現の変更を媒介する核酸分子の能力に影響しない核酸分子を指す。「実質的に同様の」とはまた、アンチセンスまたはコサプレッション技術による遺伝子発現の変更を媒介する能力に対して、得られる転写物の官能特性、または得られるタンパク質分子の官能特性の変更に実質的に影響しない、本発明の核酸分子の修飾(1個またはそれ以上のヌクレオチド塩基の欠失または挿入などの)を指す。本発明は特定の例示的な配列以上のものを包含する。
【0076】
提案される各修飾は、十分に技術分野の日常的技術の範囲内であり、コードされる生成物の生物学的活性維持の判定も同様である。さらに当業者は、この発明によって実質的に包含される同様の配列が、ストリンジェントな条件(0.1×SSC、0.1%SDS、65℃および2×SSC、0.1%SDSでの洗浄とそれに続く0.1×SSC、0.1%SDS)下で、ここで例示する配列とハイブリッドするそれらの能力によっても定義されることを認識する。本発明の好ましい実質的に同様の核酸断片は、そのDNA配列が、ここで報告する核酸断片のDNA配列と少なくとも80%同一である核酸断片である。より好ましい核酸断片は、ここで報告する核酸断片のDNA配列と少なくとも90%同一である。最も好ましいのは、ここで報告する核酸断片のDNA配列と少なくとも95%同一である核酸断片である。
【0077】
核酸断片は、温度および溶液イオン強度の適切な条件下で、核酸断片の一本鎖形態がその他の核酸断片とアニールできる場合、cDNA、ゲノムDNA、またはRNAなどの別の核酸断片と「ハイブリッド形成可能」である。ハイブリッド形成および洗浄条件については良く知られており、サムブルック(Sambrook)J.、フリッチュ(Fritsch)E.F.およびマニアティス(Maniatis)T.、分子クローニング:実験室マニュアル(Molecular Cloning:A Laboratory Manual)、第二版、コールドスプリングハーバーラボラトリー(Cold Spring Harbor Laboratory):コールドスプリングハーバー(Cold Spring Harbor)、ニューヨーク(NY)(1989)の特に第11章およびその表11.1で例証される。
【0078】
「かなりの部分」とは、当業者による配列の手動評価によって、あるいはBLAST(「基礎的局在性整列化検索ツール(Basic Local Alignment Search Tool)」、アルトシュル(Altschul)ら、J.Mol.Biol.、215:403〜410(1993)、www.ncbi.nlm.nih.gov/BLAST/も参照されたい)などのアルゴリズムを使用したコンピュータ支援配列比較および同定によって、遺伝子またはポリペプチドの推定上の同定が得られるのに十分な、そのポリペプチドまたは遺伝子のヌクレオチド配列のアミノ酸配列を含んでなる、アミノ酸またはヌクレオチド配列を指す。推定的にポリペプチドまたは核酸配列が既知のタンパク質または遺伝子に相同的であると同定するためには、概して10個以上の隣接するアミノ酸または30個以上のヌクレオチド配列が必要である。さらにヌクレオチド配列に関して、20〜30個の隣接するヌクレオチドを含んでなる遺伝子特異的オリゴヌクレオチドプローブを配列依存遺伝子同定法(例えばサザンハイブリッド形成)および単離(例えば細菌コロニーまたはバクテリオファージ・プラークの原位置(in situ)ハイブリッド形成)において使用しても良い。さらにプライマーを含んでなる特定の核酸分子を得るために、塩基12〜15個の短いオリゴヌクレオチドをPCRで増幅プライマーとして使用しても良い。したがってヌクレオチド配列の「かなりの部分」は、配列を含んでなる核酸分子の特定の同定および/または単離ができるようにする十分な配列を含んでなる。本明細書は、1つまたは複数の特定タンパク質をコードする、部分的または完全なアミノ酸およびヌクレオチド配列を教示する。当業者はここで報告される配列の利益を享受して、開示された配列の全てまたはかなりの部分を当業者に知られている目的のために使用できる。したがって本発明は、添付の配列表で報告されるような完全な配列、ならびに上で定義されるようなこれらの配列のかなりの部分を含んでなる。
【0079】
「相補的」と言う用語は、二本鎖DNA間の関係について述べる。例えばDNAについて、アデノシンはチミンに相補的であり、シトシンはグアニンに相補的である。したがって本発明は添付の配列表で報告されるような完全な配列、ならびに実質的に同様の核酸配列に相補的な単離された核酸分子も含む。
【0080】
「パーセント同一性」と言う用語は、技術分野で知られているように、配列を比較して判定される2つ以上のポリペプチド配列または2つ以上のポリヌクレオチド配列の関係である。技術分野で「同一性」とは、場合によっては、このような配列のストリング間の整合によって判定されるような、ポリペプチドまたはポリヌクレオチド配列間の配列関連性の程度も意味する。「同一性」および「類似性」は、1.)「計算分子生物学(Computational Molecular Biology)」、レスク(Lesk)A.M.編、オックスフォードユニバーシティ(Oxford University)、ニューヨーク(New York)、1988、2.)「バイオコンピューティング:情報科学およびゲノムプロジェクト(Biocomputing:Informatics and Genome Projects)」、スミス(Smith)D.W.編、アカデミック(Academic)、ニューヨーク(New York)、1993、3.)「配列データのコンピュータ分析(Computer Analysis of Sequence Data)」、第一部、グリフィン(Griffin)A.M.、およびグリフィン(Griffin)H.G.編、Humana、ニュージャージー(New Jersey)、1994、4.)「分子生物学における配列分析(Sequence Analysis in Molecular Bioloy)」、フォン・ハインェ(von Heinje)G.編、アカデミック(Academic)、ニューヨーク(New York)、1987、5.)「配列分析入門(Sequence Analysis Primer)」、グリブスコフ(Gribskov)M.およびデュヴルー(Devereux)J.編、ストックトン(Stockton)、ニューヨーク(New York)、1991で述べられるものをはじめとするが、これに限定されるものではない、既知の方法によって容易に計算できる。同一性を判定する好ましい方法は、試験される配列間に最大の整合を与えるようにデザインされる。
【0081】
同一性および類似性を判定する方法は、一般に入手できるコンピュータプログラムで体系化される。2個の配列間の同一性および類似性を判定する好ましいコンピュータプログラム法としては、それらの標準デフォルト値がgap形成ペナルティ=12およびgap延長ペナルティ=4である、ニードルマン(Needleman)およびブンシュ(Wunsch)アルゴリズムを使用するGCGプログラムパッケージにあるGCGパイルアッププログラム(デュヴルー(Devereux)ら、Nucleic Acids Res.12:387〜395(1984))、BLASTP、BLASTN、およびFASTA、(ピアソン(Pearson)ら、Proc.Natl.Acad.Sci.USA、85:2444〜2448(1988))が挙げられるが、これに限定されるものではない。BLASTXプログラムは、NCBIおよびその他の供給元から一般に入手できる(「BLASTマニュアル(BLAST Manual)」、アルトシュル(Altschul)ら、Natl.Cent.Biotechnol.Inf.、Natl.Library Med.(NCBINLM)NIH、Bethesda、MD;アルトシュル(Altschul)ら、J.Mol.Biol.、215:403〜410(1990);アルトシュル(Altschul)ら、Nucleic Acids Res.、25:3389〜3402(1997))。同一性を判定する別の好ましい方法は、ヨッタン−ハイン(Jotun−Hein)アルゴリズムを使用するDNASTARタンパク質アライメントプロトコル法による(ハイン(Hein)ら、Methods Enzymol.、183:626−645 (1990))。アラインメントのためのヨッタン−ハイン(Jotun−Hein)法のデフォルト変数は、1.)複数アラインメントではgapペナルティ=11、gap長さペナルティ=3、および2.)対合アラインメントではktuple=6である。一例として、参照ヌクレオチド配列に対して少なくとも例えば95%の「同一性」を有するヌクレオチド配列を有するポリヌクレオチドとは、ポリヌクレオチド配列が参照ヌクレオチド配列の各100個のヌクレオチドあたり5点の変異を含んでも良いこと以外は、ポリヌクレオチドのヌクレオチド配列が、参照配列と同一であることを意味する。換言すると、参照ヌクレオチド配列と少なくとも95%同一のヌクレオチド配列を有するポリヌクレオチドを得るためには、参照配列中の5%までのヌクレオチドが、欠失しまたは別のヌクレオチドで置換されていても良く、あるいは参照配列中の全ヌクレオチドの5%までのヌクレオチドの数が、参照配列中に挿入されていても良い。参照配列のこれらの変異は、参照ヌクレオチド配列の5’または3’末端部位で生じても、あるいはこれの末端部位間のあらゆる位置で、参照配列中の、または参照配列中の1つまたは複数の隣接するグループ中のヌクレオチドに個別に挿入されて生じても良い。同じように参照アミノ酸配列に対して少なくとも例えば95%の同一性を有するアミノ酸配列を有するポリペプチドとは、ポリペプチド配列が参照アミノ酸の各100個のアミノ酸あたり5個までのアミノ酸の変化を含むこと以外は、ポリペプチドのアミノ酸配列が参照配列に同一であることを意味する。換言すると参照アミノ酸配列と少なくとも95%同一のアミノ酸配列を有するポリペプチドを得るためには、参照配列中の5%までのアミノ酸残基が欠失しまたは別のアミノ酸で置換されていても良く、あるいは参照配列中の全アミノ酸残基の5%までのアミノ酸の数が、参照配列中に挿入されていても良い。参照配列のこれらの変化は、参照アミノ酸配列のアミノまたはカルボキシ末端部位で生じても、あるいはこれらの末端部位間のあらゆる位置で参照配列中の、または参照配列中の1つまたは複数の隣接するグループ中の残基間に個別に挿入されて生じても良い。
【0082】
「相同的」と言う用語は、特定の宿主細胞において天然または自然発生的なタンパク質またはポリペプチドを指す。本発明は組換えDNA技術を通じて、相同的タンパク質を生成する微生物を含む。
【0083】
「パーセント相同性」と言う用語は、ポリペプチド間のアミノ酸配列同一性の程度を指す。第1のアミノ酸配列が第2のアミノ酸配列と同一である場合、第1および第2のアミノ酸配列は100%相同性を示す。あらゆる2個のポリペプチド間の相同性は、どちらかの配列の特定部位の整合アミノ酸総数の一次関数であり、例えば2個の配列のどちらかのアミノ酸総数の半分が同一ならば、2個の配列は50%の相同性を示すと言われる。
【0084】
「コドン縮重」とは、コードされるポリペプチドのアミノ酸配列に影響しないヌクレオチド配列の変動を可能にする遺伝子コード中の分岐を指す。当業者は、特定のアミノ酸を特定化するヌクレオチドコドンの使用における、特定の宿主細胞によって示される「コドン−バイアス」について良く知っている。
【0085】
結果として生じるタンパク質分子の機能特性に実質的に影響しない、沈黙変化を生じる配列中の欠失、挿入または置換などの配列の修飾も考察された。例えば遺伝子コードの縮重を反映する、あるいは特定部位で化学的に等価のアミノ酸の生成をもたらす遺伝子配列中の変化が考察された。場合によってはタンパク質の生物学的活性変化の効果を研究するために、配列の変異体を作り出すことが実際望ましいかもしれない。提案される各変更は、十分に技術分野のルーチン技術の範囲内であり、コードされる生成物中の生物学的活性保持の判定についても同様である。
【0086】
「発現」と言う用語は、遺伝子産物配列の遺伝子コードから遺伝子産物への転写および翻訳を指す。
【0087】
「プラスミド」、「ベクター」、および「カセット」と言う用語は、細胞の中心的代謝の一部ではない遺伝子を運ぶことが多く、通常環状二本鎖DNA分子の形態である染色体外の要素を指す。このような要素は、あらゆる供給源から誘導される一本鎖または二本鎖DNAまたはRNAの配列、ゲノム一体化配列、直鎖または環状のファージまたはヌクレオチド配列を自律的に複製するかもしれず、そこではいくつかのヌクレオチド配列が独自の構成に連結または組み換えされ、それは選択された遺伝子産物のために、適切な3’非翻訳配列と共に、プロモーター断片およびDNA配列を細胞中に導入することができる。「形質転換カセット」とは、外来性遺伝子を含有し、外来遺伝子に加えて特定の宿主細胞の形質転換を容易にする要素を有する特定のベクターを指す。「発現カセット」とは、外来性遺伝子を含有し、外来性遺伝子に加えて外来性宿主におけるその遺伝子の促進された発現を可能にする要素を有する特定のベクターを指す。
【0088】
「遺伝子組み換え」と言う用語は、交差または非依存性組み合わせのプロセスによって、親テンプレート分子中に存在しない遺伝的組み合わせがそれによって形成されるプロセスを指す。したがって遺伝子組み換えは、親テンプレート分子から得ることができる遺伝的配列の全ての組み合わせを含み(それによって新たに生成された「遺伝子組み換え産生物」の各ヌクレオチド位置が、その特定のヌクレオチド位置のあらゆる親テンプレートから誘導できる)、さらに遺伝子組み換えは、新たな変異(すなわち、欠失、置換、挿入)の導入を含む。
【0089】
「遺伝子組み換えされたポリペプチド」と言う用語は、遺伝子組み換えされた遺伝子またはDNAによってコードされたポリペプチドを意味する。遺伝子組み換えされたポリペプチドは、変更または増強された特性を有することが多い。
【0090】
ポリペプチドまたはタンパク質に適用される「変更された特性」と言う用語は、アッセイ法によって測定できる、ヌクレオチド配列によってコードされるタンパク質に付随する特性を指し、その特性は、天然配列に付随するものと比較して増強されまたは減少する。変更されても良い酵素の好ましい特性の例としては、酵素の活性、基質特異性、阻害物質に対する安定性、熱安定性、プロテアーゼ安定性、溶剤安定性、洗浄剤安定性、および折りたたみ特性が挙げられる。「増強された生物学的特性」とは、天然配列に付随するものを超える変更された特性を指す。「減少した生物学的特性」とは、天然配列に付随するものに満たない変更された特性を指す。
【0091】
「テンプレート」または「親テンプレート」と言う用語は、ワトソン−クリック塩基対形成規則に従って、DNAまたはRNAポリメラーゼによって複写されて、新しいDNAまたはRNA鎖を生成する核酸分子を指す。テンプレート分子から生成される第1のコピーは相補的配列を有するので、テンプレート(または「モデル」)中の配列情報は保存される。テンプレート分子は単一または二本鎖であっても良く、あらゆる供給源に由来しても良い。
【0092】
「複製」は、「テンプレート核酸」の核酸鎖の相補的コピーが、ポリメラーゼ酵素によって合成されるプロセスである。「プライマー誘導」複製では、このプロセスは、複製を開始するために、「二重の」「オリゴヌクレオチド」の末端ヌクレオチドの(デオキシ)リボース部分の3’位置にヒドロキシル基(OH)を必要とする。
【0093】
核酸の「5’領域」および「3’領域」は、遺伝子組み換えが起きることが望ましいヌクレオチド領域を参照して、相対的用語として使用される。これらの領域はテンプレート分子内またはテンプレート分子に付着する側方DNA配列内であっても良い。不対プライマーが、これらの5’および3’領域の部分にアニールする。
【0094】
「側方配列」または「側方DNA断片」とは、それに不対プライマーがアニールしても良いユニークなヌクレオチド配列(テンプレート分子に対して)を提供するために、テンプレート分子の5’または3’領域のいずれかに付着するDNAの短い断片を指す。
【0095】
「完全長伸張生成物」とは、親テンプレートの5’および3’領域の間に含まれるのと非常によく似た長さ(約100塩基以内)を有する、プライマー誘導複製によって生成されるヌクレオチド配列である。
【0096】
「増幅」は、いくつかの「テンプレート核酸」のコピーが、線形または対数いずれかの様式で増加するように複製がサイクル様式で反復されるプロセスである。
【0097】
「プライマー」と言う用語は、相補鎖の合成がポリメラーゼによって触媒される条件下に置くと、相補鎖に沿った核酸合成または複製の開始点として作用できる(合成または天然)オリゴヌクレオチドを指す。プライマーは、プライマーそれ自体と核酸の相補鎖との間の配列相補性に基づいて、相補鎖にアニールする能力を有さねばならないが、塩基のいくらかのミスマッチは許される。プライマーサイズ、塩基配列、相補性、および標的相互作用の要件については下でより詳細に考察する。「プライマー」と言う用語自体は、ここでは出願人によって一般的に使用され、核酸複製プロセスを開始するために機能する(すなわち合成をプライムできる)あらゆる配列−結合オリゴヌクレオチドを包含する。
【0098】
「フォーワードプライマー」と言う用語は、二本鎖テンプレート分子のセンス鎖の5’領域で合成をプライムでき、または一本鎖テンプレート分子の5’領域で合成をプライムできるプライマーを指す。
【0099】
「逆転写プライマー」と言う用語は、二本鎖テンプレート分子アンチセンス鎖の5’領域で合成をプライムでき、または二本鎖テンプレート分子のアンチセンス鎖である一本鎖テンプレート分子の5’領域で合成をプライムできるプライマーを指す。
【0100】
「対合プライマー」と言う用語は、単一テンプレート分子にアニールして、プライマー誘導核酸増幅プロセスによってテンプレートの正確なコピーの合成を可能にするようにデザインされる、フォーワードおよび逆転写プライマーからなるプライマーのペアを指す。二本鎖テンプレート分子の場合、フォーワードプライマーがアンチセンス鎖(テンプレートとして使用されるセンス鎖の相補的コピー)の正確なコピーを生成し、逆転写プライマーがセンス鎖(テンプレートとして使用されるアンチセンス鎖の相補的コピー)の正確なコピーを生成するので、フォーワードおよび逆転写プライマーは、二本鎖テンプレートの正確なコピーの合成を可能にする。対照的にテンプレート分子が一本鎖である場合、そのテンプレートの正確なコピーは、プライマー誘導核酸増幅プロセスを使用して生成される。
【0101】
「不対プライマー」と言う用語は、単一テンプレート分子にアニールして、プライマー誘導核酸増幅プロセスによってそのテンプレートの正確なコピーの合成を可能にするようにデザインされていない、フォーワードおよび逆転写プライマーからなるプライマーの対を指す。その代わりフォーワードプライマーは第1のテンプレート分子にアニールするが、第2のテンプレート分子にアニールできない。逆転写プライマーは、第1のテンプレート分子と配列が異なる第2のテンプレート分子にアニールするが、第1のテンプレート分子にアニールできない。このユニークな不対プライマーのデザインは、テンプレート切り替えを通じた複製中に遺伝子組み換えが起きない限り、一本鎖または二本鎖テンプレート分子がプライマー誘導核酸増幅プロセスによって増幅できないことを確実にする。
【0102】
「プライマー誘導伸張」と言う用語は、プライマーが使用されて、核酸分子の線形または対数増幅において核酸配列の複製を援助する、技術分野で知られているあらゆる方法を指す。例えばプライマー誘導伸張は、ポリメラーゼ連鎖反応(PCR)、リガーゼ鎖反応(LCR)、および鎖置換増幅(SDA)をはじめとするがこ、これに限定されるものではない、技術分野で知られているいくつかのスキームのいずれかによって実行しても良い。
【0103】
GDHおよびコード遺伝子
グリセロールから3−HPへの転換について、この反応の触媒に関与するデヒドラターゼは、B12依存性デヒドラターゼまたはB12非依存性デヒドラターゼであることができる。しかし本発明の目的では、デヒドラターゼはB12依存性デヒドラターゼである。このB12依存性デヒドラターゼは、GDH、グリセロールデヒドラターゼ(E.C.4.2.1.30)、またはジオールデヒドラターゼ(E.C.4.2.1.28)であることができる。これらの各酵素はα2β2γ2構造を有する。文献で使用される遺伝子命名法には幅広いバリエーションがあるので、明瞭さのために、肺炎桿菌(Klebsiella pneumoniae)、シトロバクター・フロインディ(Citrobacter freundii)、クロストリジウム・パスツーリアナム(Clostridium pasteurianum)、ネズミチフス菌(Salmonella typhimurium)、ラクトバシラス・コリノイデ(Lactobacillus collinoide)、およびクレブシエラ・オキシトカ(Klebsiella oxytoca)のデヒドラターゼ遺伝子の遺伝子名およびGenBank番号を示す比較チャートを表2に示し、同定を容易にする。また遺伝子については、例えばダニエル(Daniel)ら、(FEMS Microbiol.Rev.、22:553、(1999))およびトラヤ(Toraya)およびモリ(Mori)(J. Biol.Chem.、274:3372、(1999))で述べられる。
【0104】
【表4】
【0105】
本発明の目的では、肺炎桿菌(K.pneumoniae)(国際公開第01/012833 A2号パンフレット)のdhaB1、dhaB2、およびdhaB3(それぞれ配列番号1および配列番号2、3、および4)によってコードされるGDHが、変異誘発の標的として使用される。この酵素、グリセロールデヒドラターゼは好ましい基質がグリセロールであり、2個の63kDaのαサブユニット、2個の21kDaのβサブユニット、および2個の16kDaのγサブユニットから構成されることが知られている。しかし当業者はここで述べる技術において、シトロバクター・フロインディ(Citrobacter freundii)、クロストリジウム・パスツーリアナム(Clostridium pasteurianum)、またはその他の肺炎桿菌(K.pneumoniae)株のグリセロールデヒドラターゼ、あるいはネズミチフス菌(Salmonella typhimurium)、クレブシエラ・オキシトカまたは肺炎桿菌(K.pneumoniae)のジオールデヒドラターゼもまた適切であることを認識するであろう。同様にその活性がグリセロールの3−HPへの転換を触媒できるB12依存性デヒドラターゼ活性をコードするあらゆる遺伝子が、GDH酵素の機能を変更しないアミノ酸置換、欠失または付加を包含するあらゆるアミノ酸配列をはじめとする本発明の標的として適切なはずである。したがって当業者はその他の供給源から単離されるGDHをコードする遺伝子もまた、本発明で使用するのに適切であることを理解するであろう。
【0106】
グリセロールまたは1,3−プロパンジオール存在下におけるB12依存性デヒドラターゼ不活性化
B12依存性デヒドラターゼは、触媒作用中にグリセロールによって、または1,3−プロパンジオールとの相互作用によって可逆的不活性化を被る。不活性化は、補酵素B12補助因子のコバルト−炭素(Co−C)結合の開裂を伴い、5’−デオキシアデノシンおよび不活性なコバラミン化学種の形成をもたらす。不活性なコバラミン化学種はデヒドラターゼとの密接な結合を保ち、補酵素B12依存性デヒドラターゼ再活性化因子の介入なしには解離が起きない。
【0107】
図1は、GDH不活性化に付随する典型的な経時変化を示す。これらの酵素活性アッセイは、肺炎桿菌(K.pneumoniae)(国際公開第01/012833 A2号パンフレット)のdhaB1、dhaB2、およびdhaB3(それぞれ配列番号1および配列番号2、3、および4)によってコードされるGDHを使用して、実施される。図1上方の曲線は、基質としての10mMのグリセロール(Km約0.5mM)とのGDHの反応の経時変化を示す一方、下方の曲線は、アッセイに50mMの1,3−プロパンジオール(Ki約15mM)を含めることの影響を示す。明らかに3−HP生成物形成速度は、一次不活性化速度定数(k観察された不活性)に従って、不活性化が起きるに連れて時間と共に迅速に低下する。
【0108】
改善された活性を有する変異株GDH
観察されたGDH不活性化に基づいて、野生型GDHと比較して低下した不活性化速度を有する一連の変異株GDHを作り出した。典型的にアプローチは、グリセロールおよび1,3−プロパンジオール存在下で増大した総代謝回転数を有する変異酵素を作り出して、単離することを伴う。これは、kcatの増大および/または酵素不活性化速度の低下のいずれかによって達成できる。
【0109】
GDH活性を改善するプロセスは、GDH遺伝子を含んでなる発現ベクターの構築、GDHコード配列の変異誘発、そして最後に低下した不活性化速度を有する変異体の単離を伴う。引き続く変異誘発の工程によって、GDHコード配列の発達が可能になる。
【0110】
変異株B12依存性デヒドラターゼライブラリーは、変異誘発のための出発原料としてあらゆる野生型(または実質的に同様の)B12依存性デヒドラターゼを使用して調製できる。
【0111】
B12依存性デヒドラターゼの変異誘発の伝統的方法
B12依存性デヒドラターゼ変異誘発のために、多様なアプローチを使用しても良い。ここで使用される2つの適切なアプローチとしては、エラープローンPCR(ロング(Leung)ら、Techniques、1:11〜5(1989);ジョウ(Zhou)ら、Nucleic Acids Res.、19:6052〜6052(1991);およびスピー(Spee)ら、Nucleic Acids Res.、21:777〜778(1993))、および生体内(in vivo)変異誘発が挙げられる。
【0112】
エラープローンPCRの主要な利点は、この方法によって導入された全ての変異がB12依存性デヒドラターゼ遺伝子内であり、PCR条件を変化させることであらゆる変化が容易に制御できることである。代案としては、大腸菌(E.coli)XL1−Red菌株、およびカリフォルニア州ラ・ホーヤのストラタジーン(Stratagene(La Jolla、CA))からのエピキュリアン・コリ(Epicurian coli)XL1−Redミューテータ菌株(グリーナー(Greener)およびキャラハン(Callahan)、Strategies、7:32〜34(1994)も参照されたい)などの市販される材料を使用して、生体内(in vivo)変異誘発を用いても良い。この菌株では、一次DNA修復経路の3つ(mutS、mutD、およびmutT)が欠損しており、野生型よりも5000倍高い変異速度がもたらされる。生体内(in vivo)変異誘発は、(エラープローンPCRのように)連結効率に依存しないが、変異はベクターのあらゆる領域で生じても良く、変異速度は一般にはるかに低い。
【0113】
代案としては、「遺伝子シャッフリング」法(米国特許第5,605,793号明細書、同第5,811,238号明細書、同第5,830,72号明細書、および同第5,837,458号明細書)、または核酸間での遺伝子組み換え発生の活性を促進するあらゆる同様の手段を使用して、低下した不活性化速度を有する変異株B12依存性デヒドラターゼを構築しても良いことが考察される。遺伝子シャッフリング法は、その容易な実行と高速な変異誘発のために特に魅力的である。遺伝子シャッフリングのプロセスは、関心のある遺伝子と同様の(または異なる)追加的なDNA領域集団の存在下で、関心のある遺伝子を特定サイズの断片に限定することを伴う。次に断片のプールを変性させ再アニールして、変異遺伝子を作り出す。次に変異した遺伝子を変更した活性についてスクリーニングする。
【0114】
野生型B12依存性デヒドラターゼ配列を変異させて、この方法によって変更されまたは増強される活性についてスクリーニングしても良い。配列は二本鎖であるべきで、50bp〜10kB範囲の様々な長さであることができる。配列は技術分野で良く知られている制限エンドヌクレアーゼを使用して、約10bp〜1000bpの範囲の断片に無作為に消化されても良い(マニアティス(Maniatis)、同上)。完全長配列に加えて、配列の全部(または部分)とハイブリッド形成可能な断片集団を添加しても良い。同様に、野生型配列とハイブリッド形成可能でない断片集団を添加しても良い。典型的にこれらの追加的断片集団は、総核酸と比較して重量で約10倍〜20倍過剰に添加される。一般にこのプロセスは、混合物中に約100〜1000個の異なる特定の核酸断片の生成を可能にする。無作為核酸断片の混合集団は、変性されて一本鎖核酸断片を形成し、次に再アニールされる。その他の一本鎖核酸断片との相同性領域を有する一本鎖核酸断片のみが再アニールする。無作為核酸断片は、加熱によって変性されても良い。当業者は、二本鎖核酸を完全に変性するのに必要な条件を判定できる。好ましくは、温度は、約80℃〜100℃である。核酸断片を冷却によって再アニールしても良い。好ましくは、温度は約20℃〜75℃である。再生は、ポリエチレングリコール(「PEG」)または塩の添加によって加速できる。塩濃度は、好ましくは0mM〜200mMである。アニールされた核酸断片は、次に核酸ポリメラーゼおよびdNTP(すなわちdATP、dCTP、dGTPおよびdTTP)存在下でインキュベートされる。核酸ポリメラーゼは、クレノウ断片、Taqポリメラーゼまたは技術分野で知られているあらゆるその他のDNAポリメラーゼであっても良い。アニールに先だって、アニールと同時に、またはアニール後に、ポリメラーゼを無作為核酸断片に添加しても良い。変性、再生、およびポリメラーゼ存在下でのインキュベーションのサイクルは、所望の回数反復される。好ましくはサイクルは2〜50回反復され、より好ましくは、手順は10〜40回反復される。得られる核酸は約50bp〜約100kBのより大きな二本鎖ポリヌクレオチドであり、標準クローニングおよび発現プロトコルによって、発現および変更された活性についてスクリーニングしても良い(マニアティス(Maniatis)、同上)。
【0115】
上で例証した方法(B12依存性デヒドラターゼをコードする遺伝子を直接に変異誘発するようにデザインされる)に加えて、変異株を作り出す伝統的方法をここで述べられる目的のために使用できる。例えばB12依存性デヒドラターゼ活性を有する野生型細胞を放射線または化学変異原などの多様な作用物質に暴露して、次に所望の表現型についてスクリーニングしても良い。放射線によって変異を作り出す場合、紫外線(UV)または電離放射線のどちらを使用しても良い。遺伝的変異に適した短波長UVの波長は200nm〜300nmの範囲内であり、254nmが好ましい。この波長内のUV放射線は、主にグアニジンおよびシトシンからアデニンおよびチミジンへの核酸配列内変化を引き起こす。全ての細胞は、ほとんどのUV誘導される変異を修復するDNA修復機序を有するので、カフェインおよびその他の阻害物質などの作用物質を添加して修復プロセスを妨害し、いくつかの効果的な変異を最大化しても良い。300nm〜400nm範囲の光を使用する長波UV変異もまた可能であるが、この範囲は、一般に、DNAと相互作用する様々な賦活物質(ソラレン染料など)との組み合わせで使用しない限り、短波長UV光ほど効果的でない。同様に化学作用物質による変異誘発も変異株を生成するのに効果的であり、一般に使用される物質としては、複製しないDNAに影響する化学物質(HNO2およびNH2OHなど)、ならびに複製するDNAに影響する作用物質(フレームシフト変異を引き起こすことで注目すべきアクリジン染料など)が挙げられる。放射線または化学作用物質を使用して変異株を作り出す特定の方法については、技術分野において文書で十分に立証されている。例えばトーマスD.ブロック(Thomas D.Brock)著、生物工学:工業微生物学テキスト(Biotechnology:A Textbook of Industrial Microbilogy)、第二版、Sinauer Associates:マサチューセッツ州サンダーランド(Sunderland、MA)(1989)、またはデシュパンデ・ムクンド(Deshpande,Mukund)V.、Appl.Biochem.Biotechnol.、36:227(1992)を参照されたい。
【0116】
変異誘発方法にかかわらず、酵素がグリセロールおよび1,3−プロパンジオール存在下で増大した総代謝回転数を有するように、遺伝子を発達させても良い。これはkcatの増大および/または酵素不活性化速度の低下のどちらかによって達成できる。
【0117】
不対プライマーを使用した遺伝子組み換え発生伸張法を使用したB12依存性デヒドラターゼの変異誘発
図5は、2個の遺伝子の遺伝子組み換えに基づく、不対プライマーを使用した遺伝子組み換え発生伸張法の原理を示す。この方法は、親遺伝子がそれらの5’および3’末端に異なるDNA配列を有することを必要とする。親遺伝子が、同一の5’および3’配列を有する場合、短い側方DNA断片は、(図5のステップAで示されるように)標準PCRによって遺伝子の5’または3’末端に付着されていなくてはならない。
【0118】
次に熱サイクルのための2個の不対プライマーのデザインに続いて、PCR生成物を遺伝子組み換え発生伸張法のテンプレーとして使用できる。プライマー−1はテンプレート−1の5’末端とアニールするが、テンプレート−2の5’末端とは結合しない。プライマー−2はテンプレート−2の3’末端とアニールするが、テンプレート−1の3’末端とは結合しない(ステップB)。これによってどちらの親テンプレートも熱サイクル反応によって増幅されないことが確実になる。
【0119】
短いアニールおよび合成サイクルを実施することで、一連の短いDNA断片を作り出す(ステップC)。十分な相同性があれば、これらのDNA断片のいくつかは引き続くアニールサイクルで、異なるテンプレートとアニールする(すなわち「テンプレート切り替え」)(ステップD)。引き続いて図5で示されるように、組み換えDNA断片が作られる。最後にテンプレート−1の5’末端およびテンプレート−2の3’末端を有する組み換えDNA遺伝子が作り出される(ステップE)。
【0120】
この時点で、これまでの不対プライマー−1およびプライマー−2は、新たに作り出された組み換え遺伝子のための対合プライマーになる。さらなるアニールおよび合成サイクルによって、テンプレート−1の5’末端およびテンプレート−2の3’末端を有する組み換えDNA遺伝子のプールを増幅する(図5のステップF)。増幅ステップ中に、組み換えDNA遺伝子をさらに遺伝子組み換えすることができ、それによって組み換えDNA遺伝子の交差数が増加する。理論的には、全ての増幅された生成物が組み換えDNA生成物であるべきである。これらの組み換え生成物は親テンプレート分子から直接誘導でき、あるいは追加的変異(例えば挿入、欠失、および置換)が最終遺伝子組み換え産生物中に組み込まれても良い。最終反応混合物中への親テンプレートの混入はごくわずかである。
【0121】
この方法論についてさらに詳しくは、参照によって本明細書に援用する米国特許出願第0/374366号明細書で開示される。
【0122】
改善された速度性能を有するB12依存性デヒドラターゼ変異体の同定
改善された速度性能を有するB12依存性デヒドラターゼ変異体を(大集団から)同定するために、ハイスループットアッセイの開発は、スクリーニングを極めて容易にする。2回の測定に依存して、標準(例えば野生型GDH)と比較したkcatの改善および/またはデヒドラターゼ変異体の総代謝回転数の推定を提供する、単純なスクリーニング方法がここで開示される。具体的には、GDH変異遺伝子ライブラリーを標準方法によってGDH発現について大腸菌(E.coli)中にクローン化し、単離株を得てレノックス・ブロス中で培養し、透過化処理して、次にGDH活性についてアッセイした。スクリーニング方法はホールセルに含有されるGDHについて述べられるが、当業者は方法を容易に変更して、粗製または精製調製品中の酵素に適用できることを認識するであろう。
【0123】
ここで開発されたGDHアッセイは、補酵素B12、デヒドラターゼ再活性化因子、またはB12依存性デヒドラターゼ供給源を有さない細胞における、アポ酵素形態のGDH酵素の存在に依存する。触媒的に活性のGDHホロ酵素は、培養液への補酵素B12の添加によってのみ形成する。したがってGDHの反応は、補酵素B12および基質グリセロールの添加によって、高精度で効果的にスイッチ「オン」できる。これによってGDH活性の持続時間(タイミング)の正確な制御が可能になり、したがって正確なGDH活性の測定が可能になる。反応は0時(時間=t0)に開始され、反応の初期相中(t0の直後、および酵素不活性化が起きる前、時間=t1)の生成物形成が測定される。さらに生成物形成が、さらにより長時間測定される(t0の直後、および酵素不活性化完了後、時間=t2)。生成物(3−HP)形成の判定は、比色アルデヒド分析に基づく(ズーレック(Zurek)G.、およびU.カルスト(Karst)、Analytica Chimica Acta、351:247〜257(1997))。
【0124】
飽和濃度の補酵素B12およびグリセロール存在下のGDHでは、経時的に生成する3−HPの量(y)は、次によって推定される。
y=T0+AMP(1-exp(−k観察された不活性*時間))
式中、T0はt0時の3−HPの量(バックグラウンド)であり、AMPはt0からGDHが完全に不活性化されるまでの間に生成する3−HPの量(総代謝回転数)であり、k観察された不活性は、観察された一次不活性化速度定数である。1,3−プロパンジオールが存在しないので、k観察された不活性は、全てk不活性グリセロール(グリセロールに起因する一次不活性化速度定数)に起因する(図1上方の曲線を参照されたい)。AMPは、[GDH]*kcat/k観察された不活性に等しい。経時変化を使用して、固定濃度のGDH、補酵素B12、およびグリセロールのための適切な時間(t1およびt2)を確立する。T0について補正した後、t1で形成した生成物量(T1、酵素不活性化が起きる前)を使用して[GDH]*kcatを推定し、t2で形成した生成物(T2、酵素不活性化の完了後)を使用して、総代謝回転数および[GDH]*kcat/k観察された不活性を推定する。したがってT1値はkcatに正比例し、T2値はkcat/k観察された不活性に正比例し、T2/T1は1/k観察された不活性を与える。
【0125】
適切な時間(t1およびt2)を(1,3−プロパンジオール不在下で)固定濃度の野生型GDH、補酵素B12およびグリセロールについて判定した後、T1およびT2の値を同一アッセイ条件下で、しかし1,3−プロパンジオールを添加して再度測定した(図1下方の曲線)。グリセロールおよび1,3−プロパンジオール存在下では競合的阻害が起き、k観察された不活性はkgly(グリセロールに起因する一次不活性化速度定数)およびk1,3−プロパンジオール(1,3−プロパンジオールに起因する一次不活性化速度定数)の双方の関数である。適切な1,3−プロパンジオール濃度において、T1値は1,3−プロパンジオール不在下での値(1,3−プロパンジオールおよびグリセロールの競合的阻害を反映する)から測定可能に低下し、T2値は1,3−プロパンジオール不在下での値(k1,3−プロパンジオール>kglyと言う事実を反映する)から大幅に低下した。二点アッセイでは、グリセロールは、5〜50mM、好ましくは10mMの濃度で存在し、1,3−プロパンジオールは10〜300mM、好ましくは50mMの濃度で存在する。
【0126】
出願人らは、グリセロールおよび1,3−プロパンジオール存在下で、野生型GDH−対−変異体株を調べる条件を確立した。顕微鏡的レベルでは、GDHへの変異の導入は、グリセロールおよび/または1,3−プロパンジオールに対する酵素親和力kcat、ならびにそれぞれのk不活性値に影響するかもしれない。最初は親和性および速度定数が互いに非依存に変動するという保証がなかったが、これらのパラメーターのバリエーションが、10mMのグリセロールおよび50mMの1,3−プロパンジオール存在下で実施された二点アッセイ(T1およびT2)の結果にいかに影響できるかを考えることは有用であった。具体的には、緩慢な不活性化(k観察された不活性の低下)は、増大するT2/T1比率をもたらす。他方、T1またはT2いずれかのバリエーションは、酵素の固有の特性の変化に起因することができる。例えばグリセロールおよび1,3−プロパンジオールの相対親和性が一定で、kcatおよびk不活性値を比例して低下させると、T2/T1は増大するがT1は低くなる。他方、グリセロールとの正常な相互作用を有するが、1,3−プロパンジオールへの低下した相対親和力、または1,3−プロパンジオール不活性化のより低い速度定数を有する変異株は、ほぼ正常なT1、上昇したT2、およびより高いT2/T1比率を示すべきである。正常なグリセロールおよび1,3−プロパンジオール親和性を有するが、どちらかの化合物に対する低下したk不活性を有する変異株もまた、正常なT1を示し、増大するT2およびT2/T1比率を示すべきである。k不活性における、あるいは基質または1,3−プロパンジオール親和力における変化がないkcatの増大は、T1およびT2を増大させるが、T2/T1比率は増大させない。上述の多数のバリエーションにもかかわらず、ここでの研究の目的は、グリセロールおよび1,3−プロパンジオール存在下で、GDH酵素の総代謝回転数を増大させることである。これはkcatの増大および/または酵素不活性化速度の低下のどちらかによって達成できる。したがって野生型と比較して、より高いT1および/またはより高いT2および/またはより高いT2/T1値、またはこれらのパラメーターのあらゆる組み合わせより成る群から選択される少なくとも1つの改善を示す変異株は、改善された特性を有すると見なされる。
【0127】
1,3−プロパンジオール濃度と比べてグリセロール濃度が低い場合、(kcatの推定のための)T1の測定は可能でないので、1,3−プロパンジオールによる顕著な不活性化が起きる。1,3−プロパンジオール生成プロセスにおいては高い1,3−プロパンジオール濃度および低いグリセロール濃度が望ましいので、これらの条件は妥当である。問題のこの側面を認識して、グリセロールがxx〜yymM、好ましくは10mMの濃度で存在し、1,3−プロパンジオールが300mMを超え、好ましくは600mMの濃度で存在すること以外は、上述のようにして一点アッセイが提供される。この一点アッセイでは、時間がt終点によって示され、3−HPの生成量がT(XXX)によって示され、XXXは1,3−プロパンジオールの濃度(mM)である。
【0128】
補酵素B12不活性化に影響する重大なアミノ酸の同定
出願人らは、野生型遺伝子と比べて、低下した補酵素B12不活性化速度を有する多様な変異株GDH酵素を開示する。これらの変異株は、上述の変異誘発およびスクリーニング方法を使用して同定された。変異株、変更アミノ酸残基、1/k観察された不活性活性(T2/T1として測定される)、およびT2を表1にまとめる。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に掲載する。
【0129】
【表5】
【0130】
【表6】
【0131】
【表7】
【0132】
【表8】
【0133】
【表9】
【0134】
【表10】
【0135】
上述の変異誘発およびスクリーニング方法を使用して同定された追加的な変異株を表4にまとめる。この表は各変異株、変更アミノ酸残基、およびT(600)活性に関する情報を提示する。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に掲載する。
【0136】
【表11】
【0137】
【表12】
【0138】
低下した補酵素B12−不活性化速度を有する多様な変異株GDH酵素が上で開示される。本発明の好ましい変異株としては、配列番号:40、44、48、52、56、60、64、68、72、76、80、84、88、92、96、100、104、108、112、116、120、124、128、132、136、140、143、147、151、155、159、163、167、171、175、179、182、186、189、192、195、198、201、204、208、212、215、218、221、225、229、233、237、241、245、249、253、257、261、265、269、273、277、281、285、289、293、297、301、304、307、310、313、316、319、322、325、328、331、334、および337が挙げられる。本発明のより好ましい変異株は配列番号:140、179、186、189、192、195、198、201、212、215、218、301、304、307、310、313、316、319、322、325、328、331、334、および337である。本発明の最も好ましい変異株は、配列番号:313、322、および328である。
【0139】
上の変異株に加えて、それぞれが表2および3に列挙された変異のいくつかの組み合わせをさらに有する、改善された反応速度を有する変異株の大きなプールが合成できる。例えば複数点変異を有する変異株GDHにおける各変異を酵素の全体的反応速度の改善に向けた望ましい効果について評価できる。所望ならば、反応速度を高めなかったあらゆる変異は野生型配列に戻せる。同様に一点変異のみを有する上のリストのGDH変異株を上で開示される別の変異と組み合わせて、酵素の活性をさらに改善することができる。出願人らは、GDH酵素における以下の有用な変異を開示し、これらは多様を組み合わせで使用して野生型遺伝子と比較し、低下した補酵素B12不活性化速度を有する代案の変異株GDH酵素を生成しても良い。これらの変異としては、
1.α−サブユニット中:TCA(α−Ser41)からTCG(Ser)、GTG(α−Val44)からGCG(Ala)、GTG(α−Val44)からGAG(Glu)、GGT(α−Gly47)からGGC(Gly)、CAG(α−Gln59)からCGG(Arg)、ATG(α−Met62)からGTG(Val)、ATG(α−Met62)からACG(Thr)、ATG(α−Met62)からCTG(Leu)、ATC(α−Ile63)からGTC(Val)、GGG(α−Gly63)からGGA(Gly)、CGA(α−Arg65)からCAA(Gln)、ATC(α−Ile67)からGTC(Val)、TAC(α−Tyr70)からAAC(Asn)、GTT(α−Val74)からGTC(Val)、GTT(α−Val74)からATT(Ile)、ACG(α−Thr77)からGCG(Ala)、GTG(α−Val86)からGAG(Glu)、CAC(α−His96)からCAT(His)、ATC(α−Ile102)からACC(Thr)、ATC(α−Ile102)からGTC(Val)、ATC(α−Ile105)からATT(Ile)、GTC(α−Val115)からGCC(Ala)、GAG(α−Glu116)からGAA(Glu)、GCG(α−Ala119)からACG(Thr)、GTG(α−Val124)からGCG(Ala)、CGT(α−Arg134)からCGC(Arg)、CGG(α−Arg137)からAGG(Arg)、AAC(α−Asn141)からATC(Ile)、TGC(α−Cys143)からTGT(Cys)、CTC(α−Leu148)からCGC(Arg)、AAA(α−Lys149)からAGA(Arg)、AAA(α−Lys149)からCAA(Gln)、GAT(α−Asp150)からCAT(His)、GAT(α−Asp150)からGAC(Asp)、AAT(α−Asn151)からAAC(Asn)、CCG(α−Pro152)からCCC(Pro)、TCA(α−Ser168)からCCA(Pro)、TGC(α−Cys193)からAGC(Ser)、GAG(α−Glu209)からGAA(Glu)、ATG(α−Met214)からTTG(Leu)、GGC(α−Gly216)からGGG(Gly)、TTA(α−Leu217)からGTA(Val)、AGC(α−Ser219)からAAC(Asn)、GTG(α−Val224)からCTG(Leu)、GTG(α−Val224)からATG(Met)、GTG(α−Val224)からTTG(Leu)、GTC(α−Val226)からGCC(Ala)、GCG(α−Ala231)からACG(Thr)、TTT(α−Phe233)からCTT(Leu)、GGC(α−Gly236)からAGC(Ser)、GAG(α−Glu240)からGAA(Glu)、CAG(α−Gln242)からCAA(Gln)、ATG(α−Met257)からGTG(Val)、ATG(α−Met257)からACG(Thr)、CTG(α−Leu268)からCTA(Leu)、TAT(α−Tyr271)からTGT(Cys)、ACT(α−Asn288)からACC(Asn)、GTG(α−Val301)からGTA(Val)、ATG(α−Met306)からCTG(Leu)、ATG(α−Met306)からTTG(Leu)、GCT(α−Ala309)からGCC(Ala)、ATT(α−Ile314)からGTT(Val)、CTG(α−Leu318)からTTG(Leu)、CAG(α−Gln337)からCAA(Gln)、TTC(α−Phe339)からGTC(Val)、CGC(α−Arg346)からCGG(Arg)、ACC(α−Thr350)からGCC(Ala)、GCC(α−Ala376)からGCT(Ala)、GTT(α−Val423)からATT(Ile)、CGC(α−Arg425)からCGT(Arg)、CCG(α−Pro430)からTCG(Ser)、AAC(α−Asn447)からAAT(Asn)、CCG(α−Pro450)からCCA(Pro)、GCG(α−Ala460)からGCA(Ala)、GTG(α−Val461)からGGG(Gly)、GAA(α−Glu462)からGAG(Glu)、AAC(α−Asn468)からAAT(Asn)、ACC(α−Thr470)からGCC(Ala)、AGC(α−Ser481)からAGT(Ser)、AAT(α−Asn489)からAGT(Ser)、ACC(α−Thr499)からGCC(Ala)、TAC(α−Tyr502)からCAC(His)、CTC(α−Leu509)からTTC(Phe)、CTC(α−Leu509)からTTT(Phe)、TTC(α−Phe513)からCTC(Leu)、AAC(α−Asn520)からAGC(Ser)、CGC(α−Arg533)からGGC(Gly)、GTT(α−Val549)からGCT(Ala)、ACC(α−Thr553)からACG(Thr)、TAA(αの停止)からCAA(Gln)、TAA(αの停止)からGAA(Glu)、
2.β−サブユニット中:CAA(β−Gln2)からCGA(Arg);TTT(β−Phe11)からTTA(Leu)、TTT(β−Phe11)からTTC(Phe)、CTG(β−Leu13)からCCG(Pro)、AAAβ−Lys14)からAGA(Arg)、GGG(β−Gly19)からGAG(Glu)、GAT(β−Asp24からGGT(Gly)、GAT(β−Asp24)からGAA(Glu)、GAA(β−Glu25)からGAG(Glu)、GCC(β−Ala27)からTCC(Ser)、GAA(β−Glu29)からGAG(Glu)、ACT(β−Thr45)からGCT(Ala)、GCG(β−Ala53)からGTG(Val)、AAA(β−Lys56)からAGA(Arg)、CTG(β−Leu58)からCTT(Leu)、GAA(β−Glu64)からGAG(Glu)、CTT(β−Leu67)からCTC(Leu)、CGG(β−Arg70)からCGA(Arg)、GCC(β−Ala88)からGCT(Ala)、GAT(β−Asp111)からGAA(Glu)、CTG(β−Leu113)からCCG(Pro)、TCT(β−Ser122)からCCC(Pro)、GAG(β−Glu130)からGGG(Gly)、CCG(β−Pro152)からACG(Thr)、CCG(β−Pro152)からTCG(Ser)、AAC(β−Asn155)からAGC(Ser)、AAC(β−Asn155)からAAG(Lys)、AAA(β−Lys166)からAGA(Arg)、AAA(β−Lys173)からGAA(Glu)、GAC(β−Asp181)からGGC(Gly)、CCC(β−Pro184)からCCT(Pro)、および
3.γ−サブユニット中:AAA(γ−Lys4)からAAG(Lys)、ATC(γ−Ile21)からACC(Thr)、AAA(γ−Lys27)からAGA(Arg)、GAG(γ−Glu35)からAAG(Lys)、ATC(γ−Ile49)からACC(Thr)、ACC(γ−Thr53)からGCC(Ala)、ACC(γ−Thr53)からTCC(Ser)、ACC(γ−Thr53)からTGT(Cys)、CAT(γ−His67)からTAT(Tyr)、AAT(γ−Asn72)からAGT(Ser)、CAG(γ−Gln101)からCGG(Arg)、ACC(γ−Thr114)からTCC(Ser)、ACC(γ−Thr114)からGCC(Ala)、GCC(γ−Ala122)からGTC(Val)、GCG(γ−Ala128)からGTG(Val)、およびCTG(γ−Leu137)からCTA(Leu)が挙げられる。
【0140】
同様のヌクレオチドおよびアミノ酸変異は、野生型GDH以外のその他のB12依存性デヒドラターゼ中で作ることができる。関心のあるB12依存性デヒドラターゼとGDHを並べることで、変異を必要とする正確なヌクレオチド/塩基の位置が示される。
【0141】
本発明は、上述の特定の変異のみならず化学的に同等のアミノ酸の置換を許すものも包含する。したがって例えばアミノ酸が脂肪族非極性アミノ酸であるアラニンによって置換されると、同一部位が、化学的に同等のアミノ酸であるセリンによって置換されても良いことが期待される。
【0142】
デヒドラターゼのタンパク質工学技術
今や、遺伝的工学技術および分子グラフィックスの方法、すなわちタンパク質工学技術によって、三次元構造情報と古典的タンパク質化学を組み合わせることで、タンパク質の多くの特性の変性を試みることが可能である。変更された活性を有する酵素を得るこのアプローチは、最初にモデル分子の生成、または未知の構築と同様の配列を有する既知の構築の使用に依存する。
【0143】
ここでの目的では、基質フリー形態のB12依存性グリセロールデヒドラターゼの三次元結晶構造がX線結晶構造解析によってあらかじめ求められ(リャオ(Liao)ら、J.Inorganic Biochem.(近刊))、さらに1,2−プロパンジオールとの複合体中の酵素の構造も報告されている(ヤマニシ(Yamanishi)ら、Eur.J.Biochem.、269:4484〜4494(2002))。これらのB12依存性グリセロールデヒドラターゼの三次元モデル、および変異の「ホットスポット」が反応速度において改善された活性を示す(自殺不活性化速度が低下した)これらのデヒドラターゼ酵素内のどこに典型的に位置するのかという理解から、デヒドラターゼ構造内の領域を標的にすることができ、そこでは構造の代案の変更がタンパク質の特性に所望の変化をもたらすかもしれない。したがって例えば以下の野生型残基を標的とした領域的部位−誘導変異誘発からは、改善された触媒活性を有する追加的なデヒドラターゼ変異株が生じることが期待される。
1)残基62〜70(α−サブユニットのN−末端から2番目のαらせんを包含する)および/または
2)残基219〜236(活性部位近くの領域、TIMバレルの4番目のβ−鎖の部分と、続くα−サブユニットのループおよび短いらせんを包含する)。
【0144】
宿主細胞における組み換えデヒドラターゼの発現
デヒドラターゼをコードする遺伝子の発現による3−HPおよび1,3−プロパンジオールの組み換え生成のための適切な宿主細胞は、原核生物または真核生物のどちらであっても良く、活性酵素を発現するそれらの能力によってのみ制限される。好ましい宿主は、典型的にシトロバクター(Citrobacter)、エンテロバクター(Enterobacter)、クロストリジウム(Clostridium)、クレブシエラ(Klebsiella)、アエロバクター(Aerobacter)、ラクトバシラス(Lactobacillu)、アスペルギルス(Aspergillus)、サッカロミセス(Saccharomyces)、分裂酵母(Schizosaccharomyces)、チゴサッカロミセス(Zygosaccharomyces)、ピチア(Pichia)、クリヴェロミセス(Kluyveromyces)、カンジダ(Candida)、ハンゼヌラ(Hansenula)、デバリオミセス(Debaryomyces)、ケカビ(Mucor)、トルロプシス(Torulopsis)、メチロバクター(Methylobacter)、エシェリキア(Escherichia)、サルモネラ(Salmonella)、バシラス(Bacillus)、ストレプトマイセス(Streptomyces)およびシュードモナス(Pseudomonas)などの3−HPまたは1,3−プロパンジオールの生成に有用なものである。本発明でより好ましいのは、大腸菌(Escherichia coli)、E.ブラタエ(E.blattae)、クレブシエラ(Klebsiella)、シトロバクター(Citrobacter)、およびアエロバクター(Aerobacter)である。
【0145】
宿主細胞中に適切なデヒドラターゼをコードする遺伝子のクローニング、形質転換および発現に適したベクター、形質転換方法、および発現カセットは、当業者には良く知られている。適切なベクターは、宿主細胞として用いられる細菌と適合性のものである。したがって適切なベクターは、例えば細菌、ウイルス(例えばバクテリオファージT7またはM−13由来ファージなど)、コスミド、酵母または植物から誘導できる。このようなベクターを得て使用するプロトコルは、技術分野で知られている(サムブルック(Sambrook)ら、同上)。
【0146】
典型的にベクターまたはカセットは、関連性のある遺伝子の転写および翻訳を導く配列、選択可能マーカー、および自律的複製または染色体組み込みができるようにする配列を含有する。適切なベクターは、転写イニシエーション制御を含む遺伝子の5’領域、および転写終結を制御するDNA断片の3’領域を含んでなる。双方の制御領域が、形質転換宿主細胞に相同的な遺伝子から誘導されることが最も好ましいが、このような制御領域は、必ずしも産生宿主として選択された特定種に固有の遺伝子から誘導されなくて良いものと理解される。
【0147】
所望の宿主細胞中で本発明の当該遺伝子の発現を推進するのに有用なイニシエーション制御領域(またはプロモーター)は多数あり、当業者にはなじみが深い。CYC1、HIS3、GAL1、GAL10、ADH1、PGK、PHO5、GAPDH、ADC1、TRP1、URA3、LEU2、ENO、TPI(サッカロミセス(Saccharomyces)中での発現に有用)、AOX1(ピチア中での発現に有用)、およびlac、trp、λPL、λPR、T7、tac、およびtrc(大腸菌(E.coli)中での発現に有用)をはじめとするが、これに限定されるものではない、これらの遺伝子を推進できる実質的にあらゆるプロモーターが本発明のために適切である。
【0148】
終結制御領域はまた、好ましい宿主に固有の様々な遺伝子から誘導されても良い。場合により、終結部位は必要ないかもしれないが、含まれることが最も好ましい。
【0149】
ひとたび適切なカセットが構築されると、それらは適切な宿主細胞を形質転換するために使用される。変異株デヒドラターゼをコードする遺伝子を含有するカセットの宿主細胞内への導入は、形質転換(例えばカルシウム透過化処理細胞、電気穿孔法を使用する)によって、あるいは組換え型ファージウイルスを使用した形質移入などの既知の手順によって実行しても良い(サムブルック(Sambrook)ら、同上)。
【0150】
発酵を通じた工業生産
本発明で使用するための発酵培養液は、適切な炭素基質を含有しなくてはならない。適切な基質は発酵科学の当業者に良く知られている。好ましい炭素基質は、グリセロール、ジヒドロキシアセトン、単糖類、オリゴ糖、多糖類、および一炭素基質である。より好ましいのは、糖(例えばグルコース、フルクトース、スクロース)および一炭素基質(例えばメタノールおよび二酸化炭素)である。最も好ましいのはグルコースである。
【0151】
適切な炭素供給源に加えて、発酵培養液は、培養の生育と3−HPおよび1,3−プロパンジオール生成に必要な酵素的経路の促進に適した、当業者に知られている適切なミネラル、塩、補助因子、緩衝液、およびその他の構成要素を含有しなくてはならない。Co(II)塩および/またはビタミンB12またはそれらの前駆物質に特に注意が払われる。
【0152】
典型的に細胞は、30℃において適切な培養液中で培養された。本発明の好ましい生育培養液は、ルリア−ベルターニ(Luria Bertani)(LB)ブロス、サブロー・デキストロース(Sabouraud Dextrose)(SD)ブロス、またはイースト・モルト抽出物(YM)ブロスなどの一般的な商業的に調製された培養液である。その他の特定または合成培養液を使用しても良く、特定微生物の生育に適した培養液は、微生物または発酵科学の当業者には既知である。
【0153】
バッチ、流加バッチ、または連続プロセスを使用して本発明を実施すること、およびあらゆる既知の発酵様式が適することが考察された(ブロック(Brock)ら、同上によって多様な方法が詳述されている)。さらに細胞を基材上にホールセル触媒として固定し、3−HPまたは1,3−プロパンジオール生産の発酵条件に曝すことが考察された。
【0154】
改善されたB12依存性デヒドラターゼ変異株は、使用する炭素基質とは無関係に、多様なプロセスにおいて3−HPおよび1,3−プロパンジオールの生成のために有用であることが期待される(米国特許第5686276号明細書、グリセロールから3−HPAへ)。当該株の組み換えは、参照によって本明細書に援用する米国特許出願第10/420587号明細書(デュポン(DuPont)2002)のプラスミドで述べられるデヒドラターゼからスワップアウトできる。
【0155】
以下の実施例で本発明をさらに明らかにする。これらの実施例は、本発明の好ましい実施態様を示しながら、例証のみのために提供されるものと理解される。上記考察およびこれらの実施例から、当業者は本発明の本質的特質を把握でき、その範囲と精神を逸脱することなく、本発明の様々な変化と修正を行って、様々な利用法および条件に適合させることができる。
【実施例】
【0156】
一般方法:
PCR増幅、エンド−およびエキソヌクレアーゼによるDNA修飾(DNAクローニングおよび連結のための所望の末端を生成するため)、および細菌形質転換のために必要とされる手順は、技術分野で良く知られている。ここでは標準分子クローニング技術が使用され、サムブルック(Sambrook)J.、フリッチュ(Fritsch)E.F.、およびマニアティス(Maniatis)T.、「分子クローニング:実験室マニュアル(Molecular Cloning:A Laboratory Manual)」、第二版、コールドスプリングハーバーラボラトリー(Cold Spring Harbor Laboratory)、コールドスプリングハーバー(Cold Spring Harbor)、ニューヨーク(NY)、(1989)(以下マニアティス);シルハビー(Silhavy)T.J.、ベンナン(Bennan)M.L.およびエンクイスト(Enquist)L.W.、遺伝子融合実験(Experiments with Gene Fusions)、(コールドスプリングハーバーラボラトリー(Cold Spring Harbor Laboratory):コールドスプリング(Cold Spring)、ニューヨーク(NY)、1984);およびオースベル(Ausubel)ら、分子生物学現代プロトコル(Current Protocols in Molecular Biology)(Greene Publishing and Wiley−Interscience;1987)で述べられている。
【0157】
細菌培養の維持および生育に適した材料および方法は、技術分野で良く知られている。以下の実施例で使用するのに適した技術は、下に述べられる。
1.)「一般微生物学方法マニュアル(Manual of Methods for General Bacteriology)」、フィリップス・ゲアハルト(Phillipp Gerhardt)、R.G.E.マレー(R.G.E.Murray)、ラルフN.コスティロウ(Ralph N.Costilow)、ユージーンW.ネスター(Eugene W.Nester)、ウィリスA.ウッド(Willis A.Wood)、ノエルR.クリーグ(Noel R.Krieg)、およびG.ブリッグス・フィリップス(G.Briggs Phillips)編、米国微生物学会(American Society for Microbiology)、ワシントンDC(Washington,D.C.)(1994)または
2.)ブロック(Brock)T.D.著、バイオテクノロジー:工業的微生物学テキストブック(Biotechnology:A Textbook of Industrial Microbiology)、第二版、Sinauer Associates:マサチューセッツ州サンダーランド(Sunderland, MA)(1989)。
細菌細胞の生育および維持のために使用される全ての試薬、制限酵素および材料は、特に断りのない限り、ウィスコンシン州ミルウォーキーのアルドリッチ・ケミカルズ(Aldrich Chemicals(Milwaukee、WI))、ミシガン州デトロイトのディフコ・ラボラトリーズ(DIFCO Laboratories(Detroit、MI))、メリーランド州ゲーサーズバーグのギブコ/BRL(GIBCO/BRL(Gaithersburg、MD))、ミズーリ州セントルイスのシグマケミカル(Sigma Chemical Company(St.Louis、MO))またはウィスコンシン州マディソンのプロメガ(Promega(Madison、WI))から得た。PCR反応は、特に断りのない限りカリフォルニア州フォスターシティのPEアプライド・バイオシステムズ(PE Applied Biosystems(Foster City、CA))からのAmplitaqまたはAmplitaq金酵素を使用して、GeneAMP PCRシステム9700上で実施した。サイクル条件および反応は、ここで特に断りのない限り、製造元の説明書に従って標準化した。
【0158】
DNA配列決定反応は、特に断りのない限り、インディアナ州インディアナポリスのロシュ・アプライド・サイエンス(Roche Applied Science(Indianapolis、IN))からのエキスパンド高忠実度PCRシステム(Expand High Fidelity PCR System)を使用して、PEアプライド・バイオシステムズ(PE Applied Biosystems)からのABI 377自動化シーケンサーまたはマサチューセッツ州ウォルサムのMJリサーチ(MJ Research(Waltham、MA))からのPTC−200 DNAエンジン上で実施した。同様に、データはメリーランド州ベセズダのインフォマックス(InforMax,Inc.(Bethesda、MD))からのベクターNTIプログラム、またはウィスコンシン州マディソンのDNAスター(DNASTAR Inc.(Madison、WI))からのDNAスタープログラムを使用して管理した。
【0159】
略語の意味は次のとおり。「sec」は秒を意味し、「min」は分を意味し、「hr」は時間を意味し、「d」は日を意味し、「μL」はマイクロリットルを意味し、「mL」はミリリットルを意味し、「L」はリットルを意味し、「mm」はミリメートルを意味し、「μm」はマイクロメートルを意味し、「nm」はナノメートルを意味し、「mM」はミリモル(millimolar)を意味し、「μM」はマイクロモル(micromolar)を意味し、「nM」はナノモルを意味し、「M」はモル濃度を意味し、「mmol」はミリモル(millimole)を意味し、「μmol」はマイクロモル(micromole)を意味し、「ng」はナノグラムを意味し、「mg」はミリグラムを意味し、「g」はグラムを意味し、「kB」はキロベースを意味し、「mU」はミリ単位を意味し、「U」は単位を意味する。
【0160】
菌株、ベクターおよび培養条件
大腸菌(Escherichia coli)BL21(DE3)細胞を酵素過剰発現のために使用した(シュスターB.(Shuster,B.)およびレティJ.(Retey,J.)、FEBS Lett.349:252〜254(1994))。大腸菌(Escherichia coli)XL1−Blue細胞は、カリフォルニア州ラ・ホーヤのストラタジーン(Stratagene(La Jolla、CA))から購入した。野生型大腸菌(Escherichia coli)5Kは、最初にコネティカット州ニューヘーブンのイェール大学大腸菌遺伝子ストックセンター(Coli Genetic Stock Center)(CGSC#4510;エール大学(Yale University)(コネティカット州ニューヘーブン(New Haven、CT)))から得て、ラムダDE3(5K(DE3))で溶原化した。ベクターpBluescript II SK+はストラタジーン(Stratagene)から購入した。
【0161】
分子生物学的用途のための全てのキットは、特に断りのない限り製造元の説明書に従って使用した。
【0162】
実施例1
「Xba−ライブラリー」の構築:GDHのα−、β−およびγ−サブユニットを標的とする無作為変異誘発
エラープローンPCR増幅を使用して、肺炎桿菌(Klebsiella pneumoniae)dhaB1、dhaB2、およびdhaB3遺伝子を標的とする無作為変異株ライブラリーを作り出した。ライブラリーの代表的な配列分析は、kBあたりほぼ4.2点の変異があることを実証し、酵素活性測定は、ライブラリー中の約15〜25%の変異株が活性であると判定した。
【0163】
エラープローンPCR増幅
エンプタージェ(Emptage)ら(国際公開第01/12833号パンフレット)は、肺炎桿菌(Klebsiella pneumoniae)dhaB1、dhaB2、およびdhaB3遺伝子(配列番号1)を含んでなるプラスミドpDT2の構築について述べる。プラスミドpGD20は、GDH遺伝子の発現をT7プロモーターの制御下におくpBluescript II SK+(ストラタジーン(Stratagene))に、pDT2のHindIII/XbaI断片(dhaB1、dhaB2、およびdhaB3を含有する)を挿入して構築された。
【0164】
GDHの3個の遺伝子全てを標的とする無作為に生成された変異株ライブラリーを作り出した。最初に以下のプライマーを使用して、エラープローンPCRによって、dhaB1(1668bp)、dhaB2(585bp)およびdhaB3(426bp)を含んでなる配列をpGD20から増幅した。DHA−F1(配列番号5)およびDHA−R1(配列番号6)。カリフォルニア州パロアルトのクロンテック(Clontech Laboratories,Inc.(Palo Alto、CA))からのクロンテック(Clontech)変異誘発キットを使用して、エラープローンPCRを実施した。反応混合物は以下から構成された。38μLのPCR等級水、5μLの10×アドバンタック・プラス(AdvanTaq Plus)緩衝液、2μLのMnSO4(8mM)、1μLのdGTP(2mM)、1μLの50×ダイバーシファイdNTPミックス(Diversify dNTP Mix)、1μLプライマー混合物、1μLテンプレートDNA、および1μLのアドバンタック・プラス(AdvanTaq Plus)ポリメラーゼ。製造元の説明書に従って熱サイクル反応を実施した。2.7kBのPCR生成物をHind III/Xba Iで消化して、ライゲーションの準備をした。
【0165】
変異株ライブラリー構築
Hind IIIおよびXba I消化を使用してpGD20コンストラクトからGDHの3個の全遺伝子を含有する全挿入断片を除去できるが、挿入断片サイズはほぼ2.7kBであり、一方ベクターサイズは約2.9kBである。アガロースゲル上でのこれらの2つの断片の分離を容易にするために、Hind III、Xba IおよびPst Iを使用してpGD20を消化した。Pst Iはベクターを切断せず、代わりに挿入断片を3カ所のみで切断し、挿入断片から4個の小さな断片を生じる。したがって消化されたベクターは、アガロースゲル上でその他のDNA断片からの混入なしに、2.9kB周辺で移動する。次にHind III/Xba I/Pst I消化されたベクターとHind III/Xba I−消化されたエラープローンPCR生成物をライゲートする。エタノール沈殿後、ライゲーション混合物を形質転換する準備が整った。
【0166】
ライゲーション混合物の形質転換
T7プロモーターが変異株ライブラリーのために使用されたので、ラムダDE3で溶原化された大腸菌(E.coli)細胞を変異酵素発現のための宿主細胞として使用した。具体的には、5K(DE3)大腸菌(E.coli)株を変異株ライブラリー構築のために使用した。最初に電気穿孔適格性の5K(DE3)細胞を次のようにして作成した。2L滅菌フラスコ内の500mLのLBブロスに、一晩培養した2.5mLの細胞を添加した。OD600が0.5〜0.8に達するまで、培養を振盪機上で37℃でインキュベートした。次に細胞を氷の上で10分間インキュベートし、続いて4℃で10分間遠心分離した。細胞を500mLの氷冷水で1回洗浄した後、細胞を1〜2mLの10%氷冷グリセロールに再懸濁した。滅菌エッペンドルフ管内にアリコート(50μL)を作成し、即座にドライアイス内で凍結した。コンピテントな細胞を−80℃で保存した。
【0167】
形質転換のために、1μLのライゲーション混合物を40μLのコンピテントな細胞に添加し、サンプルを0.1cmのギャップを残して電気穿孔キュベット内に移した。1.7kv/cmの電圧を電気穿孔のために使用した。アンピシリン存在下で細胞をLBプレート上にのせ、37℃で一晩インキュベートした。
【0168】
「Xba」変異株ライブラリーのDNA配列分析
DNA配列決定分析のために、9個の変異株コロニーを無作為にピックアップした。配列決定後、生じた変異数、変異位置、および観察された特定タイプの変異を各変異株について分析した。分析からは、変異株中に全タイプの塩基置換が存在することが明らかになり、特定の変異タイプに対するバイアスの欠如が示唆された。さらに分析された9個の変異株クローン中にただ1個の欠失変異が観察され、塩基挿入変異は同定されなかった。これは変異株ライブラリー中の欠失および挿入の頻度が非常に低いことを示唆した。平均変異速度はkBあたり4.2点の変異であった。GDH活性測定は、ライブラリー中の約15〜25%の変異株が活性であることを示した。
【0169】
実施例2
「Sma−ライブラリー」の構築:GDHのα−サブユニットと、β−サブユニットの一部を標的とする無作為変異誘発
主に肺炎桿菌(Klebsiella pneumoniae)dhaB1遺伝子を標的とする第2の無作為変異株ライブラリーを作成した。ライブラリーの代表的な配列分析は、kBあたりほぼ4.5点の変異があることを実証した。酵素活性測定は、ライブラリー中の約15〜25%の変異株が活性であると判定した。
【0170】
エラープローンPCR増幅および変異株ライブラリー構築
以下のプライマーを使用し、pGD20をテンプレートとして使用したエラープローンPCRによって、dhaB1遺伝子全体およびdhaB2遺伝子のほぼ200bpの部分を増幅した。DHA−F1(配列番号5)およびDHA−R2(配列番号7)。実施例1で述べたようにして、クロンテック(Clontech)変異誘発キットを使用してエラープローンPCR反応を実施した。次に1.9kBのPCR生成物をHind IIIおよびSma Iで消化した。
【0171】
ベクターを調製するために、pGD20プラスミドをHind IIIおよびSma Iで消化して(野生型dhaB1遺伝子と、dhaB2遺伝子の一部を除去して)、次にアガロースゲルから精製した。Hind III/Sma I−消化されたエラープローンPCR生成物とHind III/Sma I−消化したpGD20ベクターをライゲートした。実施例1で変異株ライブラリーの作成について述べたのと同様にして、ライゲーション混合物を電気穿孔によって大腸菌(E.coli)5K(DE3)中に形質転換した。
【0172】
「Sma」変異株ライブラリーのDNA配列分析
10個の変異株コロニーをDNA配列決定分析のために無作為にピックアップし、ライブラリーの完全性を調べた。各変異株を生じた変異数、変異位置、および観察された特定タイプの変異について判定した。これらの結果に基づいて、Sma−ライブラリー中の平均変異速度は、kBあたり4.5点変異と判定された。あらゆる特定の変異タイプについての明らかなバイアスはなく、変異株ライブラリー中の欠失および挿入の頻度は非常に低かった。酵素測定は、ライブラリー中の約15〜25%の変異株が活性であることを明らかにした。
【0173】
実施例3
GDHのα−サブユニットのアミノ酸番号141〜152(「PpuMI−ライブラリー」)、219〜226(「4BR1−ライブラリー」)および330〜342(「RsrII−ライブラリー」)を標的にした領域的無作為変異誘発
GDHの結晶構造(リャオ(Liao)ら、J.Inorganic Biochem.、93(1−2):84〜91(2003);ヤマニシ(Yamanishi)ら、Eur.J.Biochem.269:4484〜4494(2002))に基づいて、GDHのα−サブユニットの以下の領域を領域性無作為変異誘発の標的とした。1)アミノ酸番号141〜152、2)アミノ酸番号219〜226、および3)アミノ酸番号330〜342。これらの各領域は長さがかなり短いので、オリゴ−誘導変異誘発アプローチを使用してこれらの3個の変異株ライブラリーを制作した。これは変異させる各領域の上流または下流のユニークな制限部位に対応するサイレント変異を最初に作り出して、クローニングを容易にする多段階プロセスを伴う。このようにして変性オリゴヌクレオチドプライマーを調製して、PCR反応において使用しα−サブユニットの標的領域で変異誘発した。次にこれらの変異誘発されたPCR断片を大腸菌(E.coli)中にクローン化して、「PpuMI−ライブラリー」、「4BR1−ライブラリー」、および「RsrII−ライブラリー」を作り出した。
【0174】
pGD20中へのサイレント変異の導入
プラスミドpGD20中にユニークな制限部位を作成するために、各標的領域の上流または下流に1個のサイレント変異を生成した。各領域で点変異を作成するために、以下のプライマー対を使用した。各プライマー中の太字の大文字で示すヌクレオチドは、特定の変異が導入される位置を示す。
【0175】
【表13】
【0176】
カリフォルニア州ラ・ホーヤのストラタジーン(Stratagene(La Jolla、CA))からのストラタジーン(Stratagene)クイックチェンジ(QuikChange)部位−誘導変異誘発キットを使用して、製造元の説明書に従って変異誘発実験を実施した。変異誘発に続いて、各変異株クローンからプラスミドを精製した。制限酵素消化と、それに続く直接的DNA配列分析から点変異を確認した。
【0177】
オリゴ−誘導変異誘発
領域性無作為変異株ライブラリーを製造するために、3個の変性オリゴヌクレオチドを合成した(下に示すpGD20RM−F3、TB4B−R1、およびpGD20RM−R4)。大文字で示すヌクレオチドのためのオリゴヌクレオチド合成のために、標準条件を使用した。対照的に太字の小文字で示すヌクレオチドは、合成中に各プライマーの下に示す変性ヌクレオチド混合物を使用した。したがって1〜2点変異は、プライマーの各「変性領域」をもたらすことが予測された。
【0178】
pGD20RM−F3(a.a.141〜a.a.152領域のため−「PpuMIライブラリー」):
5’−GCC CGC AGG ACC CCC TCC aac cag tgc cac gtc acc aat ctc aaa gat aat ccg GTG CAG ATT−3’(配列番号15)
a=2%G、2%C、および2%Tと混合される94%A、
g=2%A、2%C、および2%Tと混合される94%G、
c=2%G、2%A、および2%Tと混合される94%C、
t=2%G、2%C、および2%Aと混合される94%T。
【0179】
TB4B−R1(a.a.219〜a.a.226領域のため−「4BR1ライブラリー」):
5’−GCG TGG GTT AAC cag cta cgc cga gac ggt gtc ggt cta cGG CAC CGA AGC GGT ATT TAC C−3’(配列番号16)
a=2%G、2%C、および2%Tと混合される94%A、
g=2%A、2%C、および2%Tと混合される94%G、
c=2%A、2%T、および2%Gと混合される94%C、
t=2%A、2%G、および2%Cと混合される94%T。
【0180】
pGD20RM−R4(a.a.330〜a.a.342領域のため−「RsrIIライブラリー」):
5’−GGT GCG CGC GGT GCG GCG AAT ATC cga gtg gga gaa agt ctg gtc gtt ggc gga cgc cac ttc GAG GTC GAG−3’(配列番号17)
a=1.66%G、1.66%C、および1.66%Tと混合される95%A、
g=1.66%A、1.66%C、および1.66%Tと混合される95%G、
c=1.66%G、1.66%A、および1.66%Tと混合される95%C、
t=1.66%G、1.66%C、および1.66%Aと混合される95%T。
【0181】
次に高忠実度PCR反応を実施し、以下のプライマー対を使用して変異原性PCR断片を生成する。
●PpuMI−ライブラリー:pGD20RM−F3およびDHA−R2(配列番号15および7)、
●4BR−1ライブラリー:TB4B−R1およびGD−C(配列番号16および14)、
●RsrII−ライブラリー:DHA−F1およびpGD20RM−R4(配列番号5および17)。
【0182】
PCR断片をアガロースゲルから精製し、次にPpuM I/Xba I(PpuMI−ライブラリーのため)、Hpa I/Xba I(4BR−1ライブラリーのため)、およびHind III/Rsr II(RsrII−ライブラリーのため)でそれぞれ消化した。
【0183】
変異株ライブラリー構築
(部位−誘導変異誘発によって、PpuM I、Hpa IまたはRsr IIのユニークな制限酵素消化部位が導入されている)変異pGD20コンストラクトをPpuM I/Xba I、Hpa I/Xba IまたはHind III/Rsr IIでそれぞれ消化した。直線化ベクターをアガロースゲルから精製し、次に制限酵素−消化したPCR生成物にライゲートした。実施例1で述べたようにして、ライゲーション混合物を大腸菌(E.coli)株5K(DE3)中に電気穿孔し、変異株ライブラリーを調製した。
【0184】
領域性ライブラリーのための配列決定分析
各領域性ライブラリーからのDNA配列決定分析のために、9〜10個の変異株コロニーを無作為にピックアップした。PpuMI−ライブラリーでは、変異株あたりの平均変異速度は2.8個の変異(n=9)であった。4BR1−ライブラリー(n=8)では、変異株あたりの平均変異速度は2.0個の変異であった。最後にRsr IIライブラリー(n=10)からの配列決定データからは、変異株あたり2.2個の変異の平均変異速度が明らかになった。3個のライブラリーのいずれにおいても、挿入または欠失のいずれも観察されなかった。全てのタイプの塩基置換が検出されたが、あらゆる特定の変異タイプに対するバイアスの欠如が示唆された。
【0185】
実施例4
スクリーニング・アッセイ開発
B12補酵素および基質(グリセロール)の添加によって、B12依存性デヒドラターゼ反応が手動で「開始」できるスクリーニング・アッセイを開発した。比色分析アルデヒドアッセイによって、デヒドラターゼ反応生成物3−HPを検出する。このアッセイを使用して、グリセロールおよび1,3−プロパンジオール存在下での典型的なB12依存性デヒドラターゼ反応の経時変化に関わる予備的な分析により、反応初期速度v、および観察された酵素不活性化速度k観察された不活性を推定する迅速な理論的技術の開発が可能になった。
【0186】
スクリーニング・アッセイ原理
酵素の補助因子である補酵素B12の供給源を有さなければ、野生型B12依存性デヒドラターゼまたは変異株B12依存性デヒドラターゼのどちらかを発現する培養は、アポ−B12依存性デヒドラターゼを生成する。対照的にホロ酵素は、トルエン−および洗剤−透過化処理されたホールセルに補酵素が添加されれば自然発生的に形成する。したがってこのような細胞に補酵素と基質を添加することで、B12依存性デヒドラターゼ反応を「開始する」ことが可能である。反応生成物である3−HPは、試薬3−メチル−2−ベンゾチアゾリノン(MBTH)および酸化剤である塩化第二鉄を使用して、比色分析アルデヒドアッセイにより検出できる(ズーレック(Zurek)G.、およびカルスト(Karst)U.、Analytica Chimica Acta、351:247〜257(1997))。
【0187】
(1,3−プロパンジオール有りまたは無しで)典型的なGDH反応の経時変化を調べる予備的アッセイ
野生型GDHを生成する細胞を、15%レノックス・ブロス(メリーランド州ゲーサーズバーグのギブコ−BRL(Gibco−BRL(Rockville、MD))からの15容積のレノックス・ブロスを85容積の0.5%NaClで希釈し、続いて滅菌して作成した)中で、ニュージャージー州ニュー・ブランズウィックのニュー・ブランズウィック・サイエンティフィック(New Brunswick Scientific(New Brunswick、NJ))からのイノヴァ(Innova)4300恒温器内で振盪(250rpm)して、37℃で一晩生育させた。細胞を次のようにして透過化処理した。培養(0.3mL)のアリコートをカリフォルニア州フラートンのベックマン−コールター(Beckman−Coulter(Fullerton、CA))からのポリプロピレン深型ウェルプレートの方形ウェル内に分配し、2.5%(v/v)のトリトン(Triton)X−100洗浄剤を含有する10μLのトルエンを各ウェルに入れた。次にプレートをドイツ国シュタウフェンのIKA−ヴェルク(IKA−Werke Gmbh.(Staufen、Germany))からのIKA MTS4振盪機上で10分間、最大速度で振盪した。補酵素B12を保護するため赤色光の下で作業して、5容積の基質溶液を1容積の透過化処理した細胞懸濁液に添加して反応を開始した。基質溶液は、0.1MのK−Hepes(pH8)中に、12mMのグリセロールおよび24μMの補酵素B12を含有した。定刻のインターバルに、反応の12.5μlのアリコートを12.5μlの0.4Mグリシン−HCl(pH2.7)中の3mg/mL MBTHを含有する96−ウェルプレートのウェルに入れた。標本採取の少なくとも20分後に、125μLの10mM HCl中の5.5mM FeCl3をMBTH−含有サンプルのそれぞれに添加した。さらに20分以上してから、カリフォルニア州サニーベールのモレキュラー・デバイシス(Molecular Devices(Sunnyvale、CA))からのスペクトラマックス(Spectramax)160プレートリーダーを使用して、GDH活性に伴う青色を670nmでの吸光度として定量化した。基質溶液を50mMの1,3−プロパンジオールで補って、同様の実験を行った。
【0188】
アッセイ結果の理論的な分析
図1で示されるように経時変化反応からのデータを時間−対−OD670でプロットした。具体的には、図1上方の曲線が、基質とした10mMのグリセロール(Km約0.5mM)とのGDH(室温、0.1M K−HEPES中、pH8)の反応の経時変化を示す。時間と共に不活性化が起きるので、生成物形成速度は迅速に低下する。3個のパラメーターを以下の式に当てはめて、点を通過する理論的曲線が描かれる。
y=T0+amp(1−exp(−k観察された不活性*時間))
これらのパラメーターは、
1.)バックグラウンド(アッセイのt=0(T0))値)、
2.)アッセイ中に生じる制限総吸光度の振幅値(amp)、および
3.)曲率を制御する一次不活性化速度定数(k観察された不活性)である。
フィッティングパラメーターは次のように反応の速度特性に関連する。振幅は観察された酵素不活性化速度(k観察された不活性)によって除された反応初期速度(v)に等しい。初期速度は[GDH][gly]kcat/(Km+[gly])である。アッセイバックグラウンドを差し引くと、さらなる分析は以下を明らかにする。
1.反応中に採取された、vの推定のための初期点(T1)、および振幅のための末期点(T2)の2個のサンプルのみからvおよびk観察された不活性の双方が推定でき、
2.1/k観察された不活性はT2/T1として推定できる。
【0189】
図1下方の曲線は、50mMの1,3−プロパンジオール(Ki約15mM)をアッセイに含めることの影響を示す。反応初期速度は約20%だけ低下したが、不活性化は約3時間早かった。これは、競合的阻害が比較的小さいが、酵素−1,3−プロパンジオール複合体の不活性化の速度定数(k不活性1,3−プロパンジオール)が、酵素−グリセロール複合体のそれ(k不活性グリセロール)を超えるためである。
【0190】
双方の不活性化プロセスを組み込んだ速度スキームを下に示す。
【0191】
【化1】
【0192】
実施例5
二点ハイスループット・スクリーニング・アッセイ
実施例4で述べた方法論に基づいて、ハイスループット・スクリーニング・アッセイ・プロトコルを使用して、実施例1、2、および3で調製した変異株コロニーを調べた。このアッセイでは、アッセイ中に2つの時点で特にGDH反応生成物を測定した。T1はT0の30秒後に測定し、T2はT0の40分後に測定した。
【0193】
0.15mL/ウェルで15%レノックス・ブロスを含有する標準96−ウェルマイクロタイタープレートの94個のウェル内に、プレート培養したライブラリーからのコロニーをピックアップして入れた。残る2個のウェルには、野生型GDHを生成する細胞を接種し、細胞を静止恒温器内で4〜6時間37℃で培養した。代案としては、ピックアップした細胞をスクリーニング前に−80℃で保存することが望ましい場合、保存する前に、グリセロール(10%v/v)を含む培地およびピックアップした細胞を一晩培養した。スクリーニング前に、カリフォルニア州サンディエゴのV&Pサイエンティフィック(V&P Scientific(San Diego、CA))からの96−ピン接種器によって、あらかじめ凍結した細胞をグリセロールなしで15%レノックス・ブロスを含有する新鮮な96−ウェルプレートに移し、6〜16時間37℃で振盪せずに培養した。
【0194】
GDH変異株スクリーニングでは、細胞培養プロトコルは次のとおりであった。カリフォルニア州フラートンのベックマン−コールター(Beckman−Coulter(Fullerton、CA))からのポリプロピレン深型ウェル96−プレートのウェル内に、15%レノックス・ブロス培地を分配した(0.3mL/ウェル)。96ピン−長ピン接種機(V&Pサイエンティフィック(V&P Scientific))を使用して、浅型96−ウェルプレートから深型ウェルへ細胞を接種して、ミネソタ州セントポールの3Mヘルスケア(3M Health Care(St.Paul、MN))からの3インチ幅のミクロポア外科手術用テープによってプレートを覆った。アッセイが完了するまで浅型96−ウェルプレートを4℃で保存し、潜在的に改善されたと同定される系統をさらに調べるために、生細胞の供給源としてそれらを使用した。深型ウェルプレート中の細胞は、37℃で一晩(250rpm)振盪しながら培養した。ニュージャージー州ニュー・ブランズウィックのニュー・ブランズウィック・サイエンティフィック(New Brunswick Scientific(New Brunswick、NJ))からのイノヴァ(Innova)4300恒温器内の空気は、プラスチックトレー内の湿ったスポンジによって加湿した。
【0195】
2.5%(v/v)のトリトン(Triton)X−100洗浄剤を含有する10μLのトルエンを各ウェルに添加して、96−ウェルプレート内で細胞を透過化処理した。次にプレートをドイツ国シュタウフェンのIKA−ヴェルク(IKA−Werke Gmbh.(Staufen、Germany))からのIKA MTS4振盪機上で10分間、最大速度で振盪した。赤色プラスチックフィルムで覆って白色室内光から基質混合物を保護した、ペンシルベニア州フィラデルフィアのローム・アンド・ハース(Rohm&Haas(Philadelphia、PA))からのプレキシグラス(Plexiglas)(登録商標)閉鎖容器内にあるバイオメック(Biomek)2000ロボット(ベックマン−コールター(Beckman−Coulter)を使用して、アリコート(8μL)の透過化処理された細胞を96−ウェル反応プレートに移した。0.1Mカリウム−HEPES緩衝液中(pH8)に24μM補酵素B12、12mMグリセロール、および50mM 1,3−プロパンジオールを含有する40μLの基質混合物(赤色光の下で調製されて、必要になるまでホイルを巻いた容器内に−20℃で保存)をロボットによって細胞に添加して、室温での反応を開始した。基質添加の30秒後、12.5μLの反応のアリコート(T1サンプル)を0.4Mグリシン−HCl(pH2.7)中の12.5μL 3−メチル−2−ベンゾチアゾリノン(MBTH)をウェルが含有する第2のプレートに移した。基質添加のほぼ40分後、MBTHを含有する第3のプレートに、同様の反応のアリコート(T2サンプル)を移した。この移動の少なくとも20分後、125μLの10mM HCl中の5.5mM FeCl3溶液を第2および第3のプレート(MBTHを含有する)の各ウェルに添加した。さらに20〜60分後、カリフォルニア州サニーベールのモレキュラー・デバイシス(Molecular Devices(Sunnyvale、CA))からのスペクトラマックス(Spectramax)160プレートリーダーを使用して、GDH活性に伴う青色を670nmでの吸光度として定量化した。
【0196】
プレートリーダーからのデータをワシントン州レドモンドのマイクロソフト(Microsoft)(登録商標)(Redmond,WA)からの修正したエクセルコンピュータープログラムに移した。このプログラムは、各反応プレートからの第1(T1)および第2(T2)のサンプルをマッチさせ、アウトプットをプレート、プレート内の位置、および各反応におけるT1、T2、およびT2/T1比率を示す結果の表にするように組まれている。T1、T2、またはT2/T1比率において非常に高い値を示すサンプルについてデータを調べた。これは、これらのあらゆるパラメーターでデータをソートするプログラムの能力によって容易になった。最も有望な候補をさらに詳しく調べるために、保留した浅型96−ウェルプレートから画線培養した。
【0197】
実施例6
GDH変異株ライブラリーのスクリーニングおよびポジティブヒットの同定
実施例5で述べた自動化されたハイスループットアッセイを使用して、5個の変異株ライブラリーからのほぼ100,000個の変異株コロニーをスクリーニングした。これらのライブラリーは次を含んだ。
1.)実施例1で述べたように、α−、β−、およびγ−サブユニット(DhaB1、DhaB2、およびDhaB3)を標的にしたXbaライブラリー、
2.)実施例2で述べたように、α−と、β−サブユニットの小さな部分を標的にしたSmaライブラリー、
3.)実施例3で述べたように、α−サブユニットのa.a.141〜a.a.152を標的にしたPpuMIライブラリー、
4.)実施例3で述べたように、α−サブユニットのa.a.219〜a.a.226を標的にした4BR1ライブラリー、および
5.)実施例3で述べたように、α−サブユニットのa.a.330〜a.a.342を標的にしたRsrIIライブラリー。
【0198】
あらゆる個々の単離株は、上述の5個のライブラリーの1つから誘導された。ほとんどの個々の単離株は、ライブラリー供給源を示すラベルとそれに続く数(例えば「Xba3010」)によって一義的に同定された。第1のスクリーニングからの全ての推定上の「ヒット」は、フォローアップアッセイ中で確認した。ほとんどの変異効果は、下で述べるような速度パラメーターに基づいて、4個の大まかなカテゴリーに従って分類された。変異遺伝子の配列分析とそれに続く野生型遺伝子との比較によって、各変異株遺伝子中に存在する特定の点変異の同定が可能になった。
【0199】
変異株ライブラリーのスクリーニングおよびヒットの確認
ほぼ100,000個の変異株の一次スクリーニングに続いて、フォローアップ確認アッセイによって推定上のヒットが確認された。簡単に述べると、各推定上のヒットを8個のウェル中で再アッセイした。それぞれの個々のクローンからの結果を統計学的に分析して、T1(30秒で測定されるアルデヒド量)およびT2(40分で測定されるアルデヒド量)の平均値および標準偏差を得た。これらの結果を野生型酵素と比較した。
【0200】
図2は、y−軸上のOD670nmに対してx−軸上にアッセイ数をプロットする典型的なフォローアップアッセイ結果を示す。Xba3010および野生型クローンに関するT1およびT2でのそれぞれの個々のアッセイ(n=8)からの結果が提示される。平均値は横線として図示され、数的には括弧±標準偏差で提示される。一般に、標準偏差は約10〜15%であった。
【0201】
スクリーニング結果
表6は野生型を超えるT2/T1比率、または野生型を超えるT2のいずれか、またはその双方を有するヒットのフォローアップアッセイ結果をまとめる。T2/T1比率は、1,3−プロパンジオールおよびグリセロール存在下で起きる40分の反応での酵素安定性の指標を提供する。T2は、完全に不活性化される前の酵素の総代謝回転数を示唆する。T2/T1比率が高いほど酵素の安定性が良くなる。したがって改善された安定性を有する変異酵素は、野生型酵素と比較して、グリセロールおよび1,3−プロパンジオール存在下で低下した不活性化速度を有する。
【0202】
表6では、野生型の結果への標準化に続く、フォローアップアッセイからのT1およびT2の平均値が報告される。ほとんどの変異酵素は以下の定義に基づいて、タイプ−1、−2、−3、または-4変異株に分類される。
●タイプ1変異株:野生型と比べてT2/T1比率が改善されている一方、T2値の絶対値が減少している。
●タイプ−2変異株:T2/T1比率およびT2の双方が野生型よりも改善されている一方、T2改善の程度はT2/T1比率の改善に満たない。
●タイプ−3変異株:T2/T1比率およびT2の双方が野生型よりも改善されており、双方の改善程度が同様である。
●タイプ−4変異株:T2/T1比率が野生型に対して同等または低下しているが、T2が改善されている。
【0203】
表6はまた、各変異株において同定された特定の点変異をまとめる。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に掲載する。
【0204】
【表14】
【0205】
【表15】
【0206】
【表16】
【0207】
【表17】
【0208】
配列決定結果に見られるように、ほとんどの変異はアミノ酸置換として同定される(サイレント変異を除く)。しかし変異株の2つ(Sma3002[配列番号140]およびXba3009[配列番号179])は、融合タンパク質である。具体的には、α−サブユニット(dhaB1)をコードする遺伝子の停止コドン(TAA)は、Sma3002およびXba3009中でそれぞれCAA(Gln)またはGAA(Glu)に変化する。この停止コドンと、β−サブユニット(dhaB2)をコードする遺伝子の最初のコドンの間には15 bpがあるので、これらの融合タンパク質のどちらもβ−サブユニット中にフレームシフトを引き起こさなかった。これにより、双方の変異酵素は活性を維持できるようになった。β−サブユニットの最初のコドン(GTG)は、通常fMet−tRNAによって認識される。しかし融合変異株では、このコドンがVal−tRNAによって認識されなくてはならない。Sma3002融合タンパク質は、6個のアミノ酸残基Gln−Gly−Gly−Ile−Pro−Val(配列番号18)からなるリンカーを含有した。Xba3009融合タンパク質では、リンカーはGlu−Gly−Gly−Ile−Pro−Val(配列番号19)であった。
【0209】
実施例7
精製酵素を使用した変異株の生化学分析
各酵素の過剰発現および精製に続いて、5個の変異株および野生型GDHのより詳しい性質決定を実施した。
【0210】
過剰発現および精製
野生型酵素と共にさらに生化学分析するため、Xba3007(タイプ−1、配列番号40)、Sma3002(タイプ−2、配列番号140)、Xba3010(タイプ−3、配列番号175)、Xba3009(タイプ−4、配列番号179)および4BR1001(タイプ−2、配列番号171))を選択した。最初に5K(DE3)大腸菌(E.coli)宿主菌株からプラスミドを精製して、大腸菌(E.coli)BL21(DE3)中に形質転換した。1.0mMのIPTGを添加する前に、細胞をアンピシリン含有LB培地中で0.6〜1.0のOD600に培養した。37℃で3時間の誘導後、遠心分離によって細胞を採集し、20mMのHEPES−KOH緩衝液(pH8.0)で1回洗浄した。細胞ペレットを−80℃で保存した。
【0211】
酵素精製のため、細胞ペレットを最初にカリフォルニア州パロアルトのロシュ(Roche、Palo Alto、CA)からの完全なミニプロテアーゼ阻害剤カクテル錠剤および0.5mMのEDTAを含有する、20mMのHEPES−KOH緩衝液(pH8.0)に再懸濁した。細胞を超音波処理によって破壊し(ブランソン(Branson)モデル450;20%出力、50%パルス、氷浴中4分間)、続いて遠心分離した(40,000×g、30分間、4℃)。透明な上清を遠沈し(110,000×g、4℃、1時間)、(NH4)2SO4を氷上の上清中に緩慢に添加して、溶液を50%飽和とした。溶液を氷上で25分間撹拌し、続いて遠心分離した(40,000×g、30分間、4℃)。ペレットを2mLの泳動用緩衝液(100mMのHEPES−KOH(pH8.2)、100mMの1,2−プロパンジオールおよび1mMのDTT)中に再懸濁し、次に泳動用緩衝液で平衡化したニュージャージー州ピスカタウェイのファーマシア・バイオテック(Pharmacia Biotech(Piscataway、NJ))からの16/60高負荷スーパーデックス(Hiload Superdex)200粒径排除カラムに入れた。
【0212】
流速0.25mL/分の緩衝液によって酵素を溶出し、画分コレクター(3mL/画分)を使用して溶出液を収集した。実施例5で述べたアッセイを使用して画分を酵素活性についてアッセイし、次に活性画分をプールして、マサチューセッツベッドフォードのミリポア(Milipore(Bedford、MA))からのセントリコン(Centricon)YM100を使用して濃縮した。100mMのHEPES−KOH(pH8.2)および1mMのDTTからなる新鮮な泳動用緩衝液を使用して、濃縮された酵素を同一カラムにもう一度通過させた。精製した酵素は、10〜20%の勾配ゲルおよびクマシー・ブルー染色を使用したSDS−PAGE電気泳動による評定で、純度75〜95%であった。
【0213】
変異株GDH酵素の生化学的性質決定:
精製した野生型および変異株GDH酵素を使用した詳細な酵素速度分析を実施した。MBTH−比色分析アルデヒドアッセイ(ズーレック(Zurek)G.、およびカルスト(Karst)U.、Analytica Chimica Acta、351:247〜257(1997))を使用して生成物形成を測定し、酵素活性を求めて、KMおよびV最大はラインウェーバー−バーク・プロットから求めた。kcatをV最大から計算した。求めた酵素のKMおよびkcatを表7に示す。
【0214】
【表18】
【0215】
最後に、各変異酵素の不活性化特性の詳細な分析を実施した。実施例4で述べたようにして、グリセロール不活性化速度定数を測定した。空気不活性化速度定数のために、21μMの補酵素B12を含有する0.1M K−HEPES緩衝液(pH8)で酵素を27μg/mLに希釈し、次に空気中において室温でインキュベートした。総代謝回転数をインキュベーションの0.25、0.5、1、2、5、10、15、20および30分後に測定した。総代謝回転数を測定するために、200mMのグリセロール、24mMの補酵素B12、および0.1MのK−HEPES緩衝液(pH8)を含有する190μLの反応溶液に10μLの酵素溶液を添加して、反応混合物を室温で2時間インキュベートした。MBTH−比色分析アルデヒドアッセイ(ズーレック(Zurek)G.、およびU.(カルスト(Karst)U.、同上)を使用して3−HP濃度を測定することで、総代謝回転数を推定した。時間−対−総代謝回転数でデータをプロットし、A=A0exp(−k空気t)(式中、「A」は総代謝回転数、「A0」は0時の総代謝回転数、「k空気」は空気不活性化速度定数、および「t」は時間である)を使用して、曲線のあてはめにより総代謝回転数を推定した。
【0216】
1,3−プロパンジオール不活性化を測定するために、21μMの補酵素B12および様々な濃度の1,3−プロパンジオール(すなわち、1、20、100および300mM)を含有する0.1M K−HEPES緩衝液(pH8)中で、酵素を27μg/mLに希釈した。空気不活性化速度定数の測定のために使用した方法により、各1,3−プロパンジオール濃度について不活性化速度定数を推定した。不活性化速度定数を1,3−プロパンジオール濃度に対してプロットし、曲線から1,3−プロパンジオールによる最大不活性化速度定数を推定した。1,3−プロパンジオールの解離定数は、不活性化速度定数が最大不活性化速度定数の半分になる1,3−プロパンジオール濃度であった。
【0217】
この不活性化特性の分析結果を下の表8に示す。グリセロールまたは1,3−プロパンジオールまたは光いずれかの不在下、しかし空気(酸素)存在下では、B12依存性デヒドラターゼホロ酵素の不活性化が二相速度で起きる。kglyは、1,3−プロパンジオールなしに10mMのグリセロール存在下で測定される不活性化速度定数である。全ての変異株および野生型デヒドラターゼは二相の空気不活性化を示したので、k空気、Fは急速相のための空気不活性化速度定数であり、k空気、Sは緩慢相の空気不活性化速度定数であり、k1,3−プロパンジオールは1,3−プロパンジオールによる最大不活性化速度定数である。Kdは1,3−プロパンジオールの解離定数である。
【0218】
【表19】
【0219】
実施例8
選択した第一世代変異株の組み合わせによる2回目の変異誘発
1回目の変異誘発から得られた変異株をさらに改善するために、2回目の変異誘発を実施して、いくつかの第一世代変異株からの変異を組み合わせた。
【0220】
第2世代変異株を製造するために、最初に、複数の変異を含有するいくつかの第一世代変異株からのプラスミド(例えばSma3002またはXba3009)を宿主細胞から精製した。次にこれらのプラスミドに、Xba3007、Xba3029または4BR1001で見つかった一点変異を導入して、第二世代変異株2−F4、12−B1、13−B7、および16−H5を生成した。表9はこれらの各変異株、およびそれらを生成するのに使用したプライマーに関する詳細をまとめる。各プライマー中の太字の大文字で示すヌクレオチドは、特定の変異が導入される位置を示す。
【0221】
【表20】
【0222】
カリフォルニア州ラ・ホーヤのストラタジーン(Stratagene(La Jolla、CA))からのストラタジーン(Stratagene)クイックチェンジ(QuikChange)部位−誘導変異誘発キットを使用して、実施例3で述べたようにして変異誘発実験を実施した。
【0223】
実施例5で述べたようにして、各第二世代変異株のT2/T1比率およびT2値を求めた。これらの結果を下の表10に示す。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に掲載する。
【0224】
【表21】
【0225】
実施例9
純粋融合変異株の構築および分析
2個の融合変異株であるXba3009(配列番号179)およびSma3002(配列番号140)については、実施例6で述べた。どちらも融合それ自体に加えてその他の変異を含有した。α−およびβ−融合の効果を調査するために、2個の純粋融合変異株(1E1および20G7)を構築した。
【0226】
純粋融合変異株の構築
α−サブユニット(TAA)の停止コドンをCAA(1E1)またはGAA(22−G7)に変化させた。この修飾は、野生型GDHプラスミド中に一点変異を導入することで達成した。点変異を作成するために、以下の2対のプライマーを使用した。
1−E1変異株のため:
1−E1−F1:5’−gac acc att gaa Caa ggc ggt att cct−3’(配列番号26)
1−E1−R1:5’−agg aat acc gcc ttG ttc aat ggt gtc−3’(配列番号27)
22−G7変異株のため:
22−G7−F1:
5’−ccc gac acc att gaa Gaa ggc ggt att cct gtg−3’(配列番号28)
22−G7−R1:
5’−cac agg aat acc gcc ttC ttc aat ggt gtc ggg−3’(配列番号29)
【0227】
各プライマー中の太字の大文字で示すヌクレオチドは、特定の変異が導入される位置を示す。
【0228】
カリフォルニア州ラ・ホーヤのストラタジーン(Stratagene(La Jolla、CA))からのストラタジーン(Stratagene)クイックチェンジ(QuikChange)部位−誘導変異誘発キットを使用して、実施例3で述べたようにして変異誘発実験を実施した。実施例5で述べたようにして、これらの第二世代変異株のT2/T1比率およびT2値を求めた。これらの結果を下の表11に示す。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に掲載する。
【0229】
【表22】
【0230】
実施例10
変異株Sma3002に見つかった変異の分析
実施例6で同定された第一世代変異株であるSma3002(配列番号140)は、T2/T1比率およびT2値の双方に顕著な改善を示した。それは4個の点変異を含有し、その1つは実施例9で詳細に探求した融合変異を含んだ。これらの変異の影響を個別に調査するため、さらに2個の変異株(α−Y271Cおよびβ−Q2R)を構築した。
【0231】
前に従った方法論を使用して、下に示すようなユニークにデザインされたプライマーを使用して、野生型プラスミドに単一点変異を導入した。前の実施例と同じく、各プライマー中の太字の大文字で示すヌクレオチドは、特定の変異が導入される位置を示す。
【0232】
【表23】
【0233】
カリフォルニア州ラ・ホーヤのストラタジーン(Stratagene(La Jolla、CA))からのストラタジーン(Stratagene)クイックチェンジ(QuikChange)部位−誘導変異誘発キットを使用して、実施例3で述べたようにして変異誘発実験を実施した。実施例5で述べたようにして、これらの変異株のT2/T1比率およびT2値を測定した。表13に結果を示す。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に掲載する。
【0234】
【表24】
【0235】
α−Y271C変異はT2値を低下させるが、T2/T1比率を変化させることができなかった。したがってこの変異は、酵素のkcatを低下させる。別の変異β−Q2Rは、T2またはT2/T1比率のいずれにも顕著に影響しないようである。
【0236】
Sma3002(配列番号140)における3個の非融合変異のいずれかが協力して作用し、1,3−プロパンジオールおよびグリセロール存在下での酵素の安定性を増大させるかどうかを判定するために、3個の追加的な変異株を作成した。これらの各変異株では、野生型DNA配列を単一点変異として導入することで、Sma3002における3個の非融合変異(α−Y271C、α−Y502H、またはβ−Q2R)の1つを除去した。これからSma3002中に存在した元の非融合変異の内2つをそれぞれ含有する、3個のSma3002−由来変異株が得られた。ストラタジーン(Stratagene)クイックチェンジ(QuikChange)部位−誘導変異誘発キットを使用して、実施例3で述べたようにして、Sma3002プラスミド中に単一点変異を導入し、プライマーを下の表14に示す。
【0237】
【表25】
【0238】
実施例5で述べたようにして、これらの変異株のT2/T1比率およびT2値を測定した。表15はこの分析の結果を示す。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に掲載する。
【0239】
【表26】
【0240】
実施例11
いくつかの第一世代変異の第二世代変異株への添加による3回目の変異誘発
ここでT2として報告する値で測定されるGDH酵素の総代謝回転の改善に向けて、先行する実施例でかなりの改善がなされたが、3回目の変異誘発を施した。これらの反応では、T2値の最大の改善を示す2個の第二世代変異株(すなわち1−E1および8−C9)に、第一世代変異株から選択される点変異を導入した。これは最初に、宿主細胞から1−E1および8−C9変異株プラスミドを精製することで達成した。次に下の表16に示すプライマーと、カリフォルニア州ラ・ホーヤのストラタジーン(Stratagene(La Jolla、CA))からのストラタジーン(Stratagene)クイックチェンジ(QuikChange)部位−誘導変異誘発キットを使用して、実施例3で述べたようにして、これらのプラスミドにXba3007、Xba3029、または4BR1001に見つかった単一アミノ酸置換変異を導入した。
【0241】
【表27】
【0242】
これらの第三世代変異株のT2/T1比率およびT2値は、実施例5で述べたようにして測定した。表17は結果を示す。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に掲載する。
【0243】
【表28】
【0244】
全体的に第三世代変異株では、酵素安定性にかなりの改善があった。全ての変異株は、最良の第一世代変異株(すなわち、Sma3002(配列番号140))と比較して改善された安定性(T2/T1比率)および総代謝回転数(T2)を有した。実際、変異株20−B9および21−D10は、これまでに作成した全変異株を超えるT2値を有した。
【0245】
実施例12
いくつかの第二および第三世代変異株の生化学的性質決定
選択された第二世および第三世代変異株をさらに詳しく性質決定するために、実施例4で述べる方法を使用して、10mMのグリセロールおよび50mMの1,3−プロパンジオール存在下でこれらの変異株の不活性化速度定数を求めた。これらの結果を下に示す表18にまとめる。
【0246】
【表29】
【0247】
期待されたように、結果はT2/T1比率の測定と一致した。
【0248】
改善されたGDHの一用途は、1,3−プロパンジオール生物生産であるので、高濃度の1,3−プロパンジオール存在下における変異株の総代謝回転数を求めることが重要である。上述のアッセイは、わずか50mM存在下で実施した。1,3−プロパンジオール生物生産のための発酵の後期では、1,3−プロパンジオール濃度が1Mに達することができる。したがって第2の二点アッセイを実施した。具体的には、いくつかの有望な変異株について、10mMのグリセロールおよび600mMの1,3−プロパンジオール存在下で、T2値も測定した。表19に結果をまとめる。
【0249】
【表30】
【0250】
結果は、T2(50mM)について得られたものと幾分同様であったが、数個の変異株は、予期された以上に良好に機能した。Sma3002のT2(600mM)は野生型酵素のそれよりも3.3倍高かった。各第三世代変異株(15−E4、18−D7、20−B9、および21−D10)は、Sma3002と比較してT2(600mM)にさらなる改善を示した。結果は、より高いT2(600mM)を有するこれらの第三世代変異株が、より高濃度の1,3−プロパンジオール中でより耐性があることを示唆した。これらの変異株は、1,3−プロパンジオール生物生産のために非常に有用であることが期待される。
【0251】
実施例13
高濃度の1,3−プロパンジオール存在下で総酵素代謝回転数を測定するための一点ハイスループット・スクリーニング・アッセイ
1,3−プロパンジオール濃度が約300mMよりも高い場合、反応開始後ほとんど即座にGDHの不活性化が起きる。これらの条下ではT1が正確に測定できないので、実施例4および5で述べたハイスループット・スクリーニング・アッセイを使用して、変異株をスクリーニングすることができない。高濃度の1,3−プロパンジオール存在下での総酵素代謝回転数は、ここで改善することが望ましい重要な酵素速度パラメーターの1つである。この総酵素代謝回転数は、不活性化速度の低下またはkcatの増大のいずれかによって改善できる。高濃度の1,3−プロパンジオール存在下で、改善された総酵素代謝回転数を有する変異株についてスクリーニングするために、修正したハイスループット・スクリーニング・アッセイを開発した。
【0252】
簡単に述べると、実施例5で述べたようにして、変異株細胞を96−ウェルプレート内で培養し透過化処理した。カリフォルニア州フラートンのベックマン−コールター(Beckman−Coulter(Fullerton、CA))からのバイオメック(Biomek)2000ロボットを使用して、アリコート(8μL)の透過化処理された細胞を96−ウェル反応プレートに移した。英国ハンプシャー州ニューミルトンのジェネティクス(Genetix(New Milton、Hampshire、UK))からのQfill2を使用して、0.1Mカリウム−HEPES緩衝液(pH8)中の24μM補酵素B12、12mMグリセロール、および720mMの1,3−プロパンジオールを含有する40μLの基質を細胞に添加して、室温での反応を開始した。プレートを室温でほぼ70分間培養した後、バイオメック(Biomek)2000ロボット(ベックマン−コールター(Beckman−Coulter)を使用して、ウェルが12.5μLの0.4Mグリシン−HCl(pH2.7)中の3−メチル−2−ベンゾチアゾリノン(MBTH)を含有する第2のプレートに、反応の12.5μLのアリコートを移した。70分の反応時間によって総代謝回転数の正確な測定ができるようになる。実施例5で述べたようにして、生成物である3−HPの濃度を求めた。カリフォルニア州サニーベールのモレキュラー・デバイシス(Molecular Devices(Sunnyvale、CA))からのスペクトラマックス(Spectramax)160プレートリーダーを使用して670nmでの吸光度を測定し、総酵素代謝回転数(T(600))を推定した。
【0253】
Qfill2は、基質を15秒以内に1枚の96−ウェルプレートに添加できるので、この一点比色分析アッセイは実行するのがが簡単である。さらにアッセイは実施例4および5で述べたアッセイと比較して、より高いスループット能力を提供できる。野生型GDHおよび異なる酵素速度パラメーターを有するいくつかの変異株について、実施例5および本実施例で実施されたアッセイの比較結果を表20に示す。
【0254】
【表31】
【0255】
これらの結果は、各アッセイが異なる速度パラメーターについてスクリーニングすることを実証する。本実施例で述べられるスクリーニング・アッセイによって同定される変異株は、より高濃度の1,3−プロパンジオールに対してより抵抗性であり、一般に改善された安定性と妥当なkcat値を有する。
【0256】
実施例14
酵素構造と変異の間の相互関係
この実施例では、野生型GDHと比較して増大したT2または増大したT2/T1比率を有する変異の位置をGDHの三次元結晶構築に関して調べた。これによって変異が、改善された反応速度(不活性化速度が低下する)をもたらすことが多い変異「ホットスポット」と見なすことができるデヒドラターゼ内の領域の同定が可能になる。これらの領域における代案の配列変更は、おそらく反応速度に改善を有する追加的な変異株をもたらす。
【0257】
三次元構造に相関する変異位置
基質フリー形態のデヒドラターゼの三次元結晶構造は、X線結晶構造解析によって求められており(リャオ(Liao)ら、J.Inorganic Biochem.93(1−2):84〜91(2003))、さらに1,2−プロパンジオール複合体中の酵素の構造も報告されている(ヤマニシ(Yamanishi)ら、Eur.J.Biochem.269:4484〜4494(2002))。これらの構造に基づいて、各GDHサブユニットの模式図上に、T2/T1比率またはT2のいずれかに改善をもたらす変異(実施例6〜12からの)をマッピングした。より具体的には、図3はα−サブユニット上に単一点変異(野生型GDHと比較して)を含有する変異株の分布を示す(全ての単一点変異の88%に相当)。対照的に、図4はα−サブユニッ上に複数点変異(野生型GDHと比較して)を含有する変異株の分布を示す。このサブユニットは全ての複数点変異の58%を含有し、残りの変異は、β−(31%)およびγ−(11%)サブユニットにそれぞれ分布した。
【0258】
ポジティブヒットから同定されたホットスポット
単一点および複数点変異株の双方における変異は、GDHの全ての3個のサブユニットに位置して大型α−サブユニット全体に分布するが、単一点変異を含有する変異株および複数点変異を含有する変異株は、α−サブユニット中に2個の共通のホットスポットを示す。
【0259】
1つの変異ホットスポットは、α−サブユニットのN−末端から2番目のαらせん(残基62〜70)上に位置する。この領域では5個の単一点変異株が見つかり、内3つは残基62に異なるアミノ酸置換の変異を有し、1つは残基65に変異を有し、1つは残基70に変異を有する(図3)。複数点変異株からの6個の変異部位はこのらせん上にも位置し、残基62に3つ、残基63、67および70にそれぞれ1つある(図4)。
【0260】
第2のホットスポットは、4番目のβ−鎖の部分、TIMバレルとそれに続くα−サブユニットのループおよび短いらせん(残基224〜236)を含む領域である。このホットスポットは活性部位の近辺にある。この領域では5個の単一部位変異が見つかっている。残基224は2個の変異株において異なるアミノ酸置換を有した。残基226は、2個の同一変異株の変異部位である。1個の変異株は、残基233上にある。さらに複数部位変異株の3個の変異部位は、この領域(残基226、231、および236)に位置する。
【0261】
実施例15
GDH変異株の部位飽和変異誘発
実施例9で特徴づけられる変異株中に存在する3個の変異部位に、部位飽和変異誘発を実施した。具体的には、これらの部位はγ−Thr53(変異株Xba3009に見られる)、α−Leu509(変異株Xba3029に見られる)およびα−Val224(変異株4BR1001に見られる)であった。飽和変異誘発ライブラリーを調製するために、1−E1および8−C9変異株(それぞれ実施例9および10)をそれらの宿主細胞から精製し、表21に示す変性プライマーと共にテンプレートとして使用した。
【0262】
【表32】
【0263】
カリフォルニア州ラ・ホーヤのストラタジーン(Stratagene(La Jolla、CA))からのストラタジーン(Stratagene)クイックチェンジ(QuikChange)部位−誘導変異誘発キットを使用して、製造元の説明書に従って、GDH−SM1、GDH−SM2、GDH−SM3、およびGDH−SM4ライブラリーを調製した。プラスミドの大腸菌(E.coli)株5K(DE3)への電気穿孔に続いて(実施例1で述べたように)、一点GDHアッセイを使用して各ライブラリーからの88個の変異株コロニーをスクリーニングして、高濃度1,3−プロパンジオール存在下での総酵素代謝回転数を推定した。次に各スクリーニングからの最良のヒットをDNA配列分析にかけた。以下の表に結果を示す。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に提供する。
【0264】
【表33】
【0265】
全ての4個の飽和変異株(GDH−SM1−G11、GSH−SM2−B11、GDH−SM3−D2、およびGDH−SM4−H2[太字で示す])は、それらが由来した親変異遺伝子との比較で、T(600)のさらなる改善を示した。興味深いことに、変異株GDH−SM4−H2では、3個の塩基変化が同定された(すなわち、ACC(γ−Thr53)からTGT(Cys))。このタイプの変異は、エラープローンPCRを使用して生成することが極めて困難である。
【0266】
実施例16
不対プライマーを使用してGDH変異株を生成する遺伝子組み換え発生伸張法
無作為変異誘発、合理的デザインの変異誘発、および飽和変異誘発(実施例6、8−11、および15)を使用したグリセロールおよび1,3−プロパンジオール存在下でのGDH不活性化速度の顕著な改善にも関わらず、工業用途のためにはさらなる改善が望ましかった。したがって実施例6、9、10、および15からの24個のグリセロールデヒドラターゼ変異株を不対プライマー法を使用した単一遺伝子組み換え発生伸張反応の親テンプレートとして使用した(米国特許出願第60/360279号明細書)。
【0267】
短い側方DNA断片の親遺伝子5’または3’末端への付着
PCRによって短い側方DNA断片を親遺伝子の5’または3’末端に付着し、引き続いてそれを不対プライマーを使用した遺伝子組み換え発生伸張法のための結合部位として使用した。GDHを含有するプラスミドを宿主細胞から精製し、テンプレートとして使用した。
【0268】
具体的には、インディアナ州インディアナポリスのロシュ・アプライド・サイエンス(Roche Applied Science(Indianapolis、IN))からのエキスパンド高忠実度PCRシステム(Expand High Fidelity PCR System)を使用した標準高忠実度PCR反応において、フォーワードプライマーGDHM−F1(配列番号343)および逆転写プライマーGDHM−R1(配列番号344)を使用して、以下のテンプレート遺伝子を増幅した。1−E1(配列番号198)、野生型GDH(配列番号1)、Xba3023(配列番号182)、Xba3010(配列番号175)、8−C9(配列番号212)、4BR1001(配列番号171)、Xba3015(配列番号147)、Xba3008(配列番号151)、Sma3003(配列番号143)、Xba3016(配列番号155)、およびXba3020(配列番号159)。次に得られたPCR生成物を当モル比で共に混合し、「混合物−1」と称した。
【0269】
フォーワードプライマーGDHM−F2(配列番号345)および逆転写プライマーGDHM−R2(配列番号346)を使用して、以下の遺伝子を同様にして増幅した。Xba3007(配列番号40)、Xba3029(配列番号44)、Xba3025(配列番号52)、Sma3009(配列番号179)、Sma3010(配列番号175)、Sma3008(配列番号151)、RsrII001(配列番号136)、PpuMI002(配列番号128)、KG005(配列番号253)、GDH−SM1−G11(配列番号301)、GDH−SM2−B11(配列番号304)、GDH−SM3−D2(配列番号307)、およびGDH−SM4−H2(配列番号310)。次に得られたPCR生成物を当モル比で共に混合し、「混合物−2」と称した。
【0270】
カリフォルニア州バレンシアのキアゲン(Qiagen(Valencia、CA))からのキアゲン(Qiagen)DNA抽出キットを使用して、増幅された生成物をアガロースゲルから精製し、次に不対プライマーによる遺伝子組み換え発生伸張法のための親テンプレートとして使用した。
【0271】
不対プライマー法を使用した遺伝子組み換え発生変異生成物の作成
2個のプライマーPADH316F1(配列番号29)およびT7T(配列番号30)を使用して、上述の24個の親テンプレートから遺伝子組み換え産生物(すなわちGDH変異遺伝子)を作成した。配列番号343によって生成される短い5’DNA断片の添加により、PADH316F1は、「混合物−1」(すなわち、1−E1、野生型GDH、Xba3023、Xba3010、8−C9、4BR1001、Xba3015、Xba3008、Sma3003、Xba3016、およびXba3020)中のテンプレートの5’末端とアニールする。しかしプライマーPADH316F1は、「混合物−2」テンプレートの5’末端に結合しない。同様にプライマーT7Tは、「混合物−2」テンプレート(すなわち、Xba3007、Xba3029、Xba3025、Sma3009、Sma3010、Sma3008、RsrII001、PpuMI002、KG005、GDH−SM1−G11、GDH−SM2−B11、GDH−SM3−D2、およびGDH−SM4−H2)の3’末端にアニールするが、「混合物−1」テンプレートの3’末端には結合しない。
【0272】
反応のために以下の反応混合物をアセンブルした。10ng「混合物−1」、10ng「混合物−2」、各200μMのdNTP、1×PCR緩衝液(最終濃度、1.5mM MgCl2)、286nMのpADH316F1、286nMのT7T、カリフォルニア州バレンシアのキアゲン(Qiagen(Valencia、CA))からの0.625Uのホット・スター・タック(HotStarTaq)、dH2Oで25μLに調節。熱サイクル条件は次のとおりであった。95℃で2分間変性、続いて95℃で30秒の60回のサイクル、1秒+63〜69℃の間の勾配でサイクルあたり1秒、72℃で7分間の最終伸張、および4℃での保持。ニューヨーク州ウェストベリーのエッペンドルフ・サイエンティフィック(Eppendorf Scientific、Inc.(Westbury、NY))からのエッペンドルフ・マスターサイクラー・グラジェント(Eppendorf Mastercycler gradient)5331を熱サイクル反応のために使用した。
【0273】
反応で使用した2個のプライマーは、いかなるテンプレートの5’および3’末端とも同時にマッチしないので、いずれの親テンプレートも反応中に増幅できない。しかし5’および3’末端の双方を有するあらゆる組み換えDNA生成物が、引き続く熱サイクル中で増幅される。不対プライマー反応に続いて、反応混合物をアガロースゲルにロードした。約66〜67℃において、不対プライマー反応から大量の2.7kBのPCR生成物が得られた。テンプレートとして使用したオリジナルの親分子それ自身が約2.7kBだったので、このサイズの生成物は予期された。
【0274】
遺伝子組み換え発生GDH変異株ライブラリーの作成およびスクリーニング
組み換えGDH DNA生成物(ほぼ2.7kB)をキアゲン(Qiagen)DNA抽出キットを使用してゲルから精製し、Xba IおよびHind IIIで消化して、次にXbaI−HindIII−消化したpGD20ベクターとリゲートした(実施例1)。実施例1で述べたように、電気穿孔によるライゲーション混合物の大腸菌(E.coli)株5K(DE3)への形質転換によって、変異株ライブラリーを得た。ライブラリーサイズはライゲーション反応あたり30万個のコロニーを超えた。
【0275】
6,000〜7,000個の変異株コロニーをアガロースプレートからピックアップし、実施例13で述べたように一点ハイスループット・スクリーニング・アッセイを使用してスクリーニングした。一次スクリーニングに続いて、フォローアップ確認アッセイにより推定上のヒットが確認された。簡単に述べると、各推定上のヒットを8個のウェル内で再アッセイした。個々の各クローンからの結果を統計学的に分析して、T(600)の平均値および標準偏差を得た。これらの結果を野生型GDH酵素と比較した。
【0276】
さらに二点アッセイを使用していくつかのヒットをさらに詳しく調査し(実施例5)、これらのヒットの初期反応速度および不活性化の変化を大まかに推定した。
【0277】
表23はいくつかの遺伝子組み換え発生GDH変異株について、これらの様々なアッセイの結果をまとめる。
【0278】
【表34】
【0279】
異なる組み換えGDH変異株は(同様のT(600)値を有するにもかかわらず)異なる酵素速度を示したが、これは単に各アッセイ手段のパラメーターの違いを反映するだけである。例えば変異株SHGDH24およびSHGDH43は同一のT(600)値を有したが、SHGDH24が野生型と比較して軽度に改善されたT1値のみを有したのに対し、SHGDH43は大幅に低下したT1を有した。SHGDH24では酵素のkcatは低下しなかったが、対照的にSHGDH43は大幅に低下したkcatを有した。どちらの場合もSHGDH24およびSHGDH43双方のGDH酵素安定性が増大することで、総酵素代謝回転数の改善が可能になった。
【0280】
SHGDH51はこれらの変異株中で安定性について最大の改善を示したが、そのkcatは大幅に低下したので、この変異株は最高の総酵素代謝回転数を有さなかった。SHGDH37はこれらの変異株中でT(600)について最大の改善を示したが、そのT2値はSHGDH12のそれよりも低く、SHGDH37が高濃度の1,3−プロパンジオールに対して、SHGDH12よりも抵抗性であることが示唆された。
【0281】
しかしいずれにせよ結果は明らかに、不対プライマー法を使用した新しい変異株の作成により、GDH安定性におけるかなりの改善があったことを実証した。具体的には、無作為変異誘発を通じて得られた最良の変異株(すなわち変異株Sma3002;配列番号140)は3.3のT(600)を有した。無作為変異誘発およびスクリーニングを通じて同定された変異株の合理的な組み合わせ、または部位飽和変異誘発を使用したこれらの変異の半無作為組み合わせからは、T(600)が最大の4.3である変異株(すなわち変異株GDH−SM1−G11;配列番号301)が得られた。したがって不対プライマーを使用した遺伝子組み換え発生伸張法アプローチを使用した無作為遺伝子組み換えは、前述の合理的かつ半無作為アプローチよりも強力な技術であると結論される。
【0282】
変異遺伝子の配列分析
表23に列挙される組み換えGDH変異株からプラスミドDNAを精製し、それぞれのGDH遺伝子全体を配列分析した。変異株の分析と、それに続くオリジナル野生型GDH遺伝子配列との比較は、これらの組み換え変異遺伝子が表24に示す単一塩基置換変異(点変異)を含有することを示す。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に提供する。
【0283】
【表35】
【0284】
【表36】
【0285】
興味深いことに、全ての変異株(配列番号313、316、319、322、325、328、331、334、337)はα−β融合変異を含有し(元来Sma3002およびXba3009変異株に見られたように[実施例6])、この変異がT(600)値の改善にとって非常に重要であることが示唆された。SHGDH43は、4個の新たに作り出された変異に加えて、4個の異なる親遺伝子(すなわち4BR1001、1−E1、Xba3008、およびGDH−SM1−G11)からの変異を含有した。これは遺伝子組み換え発生PCR中に、4個の異なる親遺伝子間で少なくとも4つの交差が起きたことを示唆した。SHGDH25およびSHGDH51はいくつかの新たに作り出された変異に加えて、3個の親遺伝子(すなわち、SHGDH25ではSma3003、4BR1001、および1−E1;SHGDH51ではRsrII001、1−E1、およびXba3007)からの変異を含有した。
【図面の簡単な説明】
【0286】
【図1】グリセロール存在下で、またはグリセロールおよび1,3−プロパンジオール存在下のどちらかで実施された、典型的なGDH反応の経時変化を示す。
【図2】実施例6で述べられるような典型的なフォローアップアッセイの図示される結果である。
【図3】(野生型GDHと比較して)α−サブユニット上に一点変異を含有する変異株の分布を示す。
【図4】(野生型のGDHと比較して)α−サブユニット上に複数点変異を含有する変異株の分布を示す。
【図5】不対プライマーを使用した遺伝子組み換え発生伸張法の原理を示す。
【技術分野】
【0001】
本発明は、1,3−プロパンジオールを生成するための分子生物学分野およびB12依存性デヒドラターゼの使用に関する。より具体的には、酵素不活性化速度が低下するように反応速度が改善されたB12依存性デヒドラターゼを作り出し、選択する方法について述べる。
【背景技術】
【0002】
1,3−プロパンジオールは、いくつかの用途において、ポリエステル、ポリエーテル、およびポリウレタンを製造するための出発原料をはじめとする有用性を有する。1,3−プロパンジオールを製造するための方法としては、伝統的化学経路および生物学的経路の双方が挙げられる。1,3−プロパンジオールを製造するための生物学的方法については、最近述べられている(非特許文献1)。1,3−プロパンジオールを生物学的に製造するのには、二段階逐次反応の基質としてのグリセロールが必要である。まずデヒドラターゼ(典型的に補酵素B12依存性デヒドラターゼ)が、グリセロールを中間体3−ヒドロキシプロピオンアルデヒド(3−HP)に変換する。次にNADH−(またはNADPH)−依存酸化還元酵素(式1および2参照)によって、3−HPが1,3−プロパンジオールに還元される。
グリセロール→3−HP+H2O(式1)
3−HP+NADH+H+→1,3−プロパンジオール+NAD+(式2)
【0003】
1,3−プロパンジオールはさらに代謝されることなく、その結果、培養液中に高濃度で蓄積する。
【0004】
典型的にグリセロールは、1,3−プロパンジオールを生物学的に生成するための出発原料として使用される。しかしグルコースおよびその他の炭水化物もまた、1,3−プロパンジオール生成のための適切な基質である。具体的にはラフェンド(Laffend)ら(特許文献1、特許文献2)は、デヒドラターゼ活性を含んでなる単一微生物を使用して、グリセロールまたはジヒドロキシアセトン以外の炭素基質(特に、例えばグルコース)から1,3−プロパンジオールを製造する方法を開示する。エンプテイジ(Emptage)ら(特許文献3)は、3−HPを1,3−プロパンジオールに変換する非特異的触媒活性(dhaTによってコードされる1,3−プロパンジオール酸化還元酵素とは区別される)によって得られる力価(リットル当たりの生成グラム数)の顕著な増大について述べる。ペイン(Payne)ら(特許文献4)は、1,3−プロパンジオールを生物学的に生成するのに有用な特定のベクターおよびプラスミドを開示する。セレヴァン(Cervin)ら(特許文献5)は、1,3−プロパンジオールの高収率生成のための改善された大腸菌(E.coli)株を開示する。特許文献1、特許文献3、特許文献4、および特許文献5は、参照によってその全体を本明細書に援用したものとする。
【0005】
グリセロールを3−HPに変換するのに関与する酵素は、主として補酵素B12依存性グリセロールデヒドラターゼ(E.C.4.2.1.30)および補酵素B12依存性ジオールデヒドラターゼ(E.C.4.2.1.28)として知られる補酵素B12依存性酵素である。これらの異なるが関係のある補酵素B12依存性酵素は、それらの分子および生化学的特性に関しては十分に研究されている。補酵素B12依存性デヒドラターゼの遺伝子は、例えば肺炎桿菌(Klebsiella pneumoniae)、シトロバクター・フロインディ(Citrobacter freundii)、クロストリジウム・パスツーリアナム(Clostridium pasteurianum)、ネズミチフス菌(Salmonella typhimurium)、クレブシエラ・オキシトカ(Klebsiella oxytoca)、およびラクトバシラス・コリノイデス(Lactobacillus collinoides)で同定されている(非特許文献2、非特許文献3、および非特許文献4)。
【0006】
文献中で使用される遺伝子命名法には幅広いバリエーションがあるが、各例で補酵素B12依存性デヒドラターゼは、大型または「α」サブユニット、中型または「β」サブユニット、および小型または「γ」サブユニットの3個のサブユニットから構成される。これらのサブユニットは、α2β2γ2構造体にアセンブルされてアポ酵素を形成する。補酵素B12(活性補助因子化学種)は、アポ酵素に結合して触媒的に活性のホロ酵素を形成する。補酵素B12は、それによって触媒作用が起きるラジカル機序に関与するので、触媒の活性のために必要とされる。
【0007】
生化学的に、補酵素B12依存性グリセロールおよび補酵素B12依存性ジオールデヒドラターゼの双方が、グリセロールおよびその他の基質による機序ベースの自殺不活性化を被ることが知られている(非特許文献3、非特許文献5)。さらに不活性化は、ホロ酵素と高濃度の1,3−プロパンジオールとの相互作用を通じて起きる。不活性化は補酵素B12補助因子のコバルト−炭素(Co−C)結合の開裂を伴い、5’−デオキシアデノシンの形成と不活性なコバラミン化学種をもたらす。不活性なコバラミン化学種はデヒドラターゼ密接に結合したままであり、補酵素B12依存性デヒドラターゼ再活性化因子(「デヒドラターゼ再活性化因子」)の介入なしには解離が起きない。この不活性化は3−HP形成に結びついた反応速度を顕著に低下でき、それによって間接的に1,3−プロパンジオール生成を減少させる。
【0008】
補酵素B12依存性デヒドラターゼ不活性化の影響は、部分的に克服できる。例えば不活性化は、デヒドラターゼ活性を再活性化するのに関与するタンパク質に依存して克服できる。デヒドラターゼ再活性化因子については、特許文献6、特許文献7、非特許文献3、非特許文献6、および非特許文献7で述べられる。再活性化は多段階プロセスで起きる。最初にATP−依存プロセスにおいて、不活性化された補酵素B12依存性デヒドラターゼとデヒドラターゼ再活性化因子との相互作用が、密接に結合した不活性なコバラミン化学種の放出を引き起こし、アポ酵素が生成する。引き続いてデヒドラターゼアポ酵素が補酵素B12と結合して、触媒的に活性なホロ酵素を再形成しても良く、(別のATP−依存プロセスで酵素作用によって)不活性なコバラミン化学種が補酵素B12に再生されても良い。しかしデヒドラターゼ再活性化および補酵素B12再生プロセスの双方がATPを必要とするので、デヒドラターゼ活性復元のためにデヒドラターゼ再活性化因子だけに依存することは、本質的に限定される。これらのATP−依存プロセスは、特に引き続く3−HPから1,3−プロパンジオールへの反応が存在して、1,3−プロパンジオール濃度が高い場合、グリセロールを3−HPに変換するプロセスに対する顕著なエネルギー負荷を意味する。
【0009】
代案としては、1,3−プロパンジオール生成中に培地に添加する補酵素B12の量を増大させる、あるいは培養液に(生体内で(in vivo)補酵素B12に変換される)ビタミンB12を補充する、のいずれかで微生物に追加的な補酵素B12を供給することが可能である。しかしどちらの場合もこれらの添加経費が、プロセスの経済性を顕著に妨げるかもしれない。
【0010】
クロウ(Croux)ら(特許文献8)は、補酵素B12非依存性デヒドラターゼを発現する組み換え微生物を使用して1,3−プロパンジオールを製造するプロセスを開発することで、補酵素B12依存性デヒドラターゼに関する問題に対処した。しかしこの解決法の有用性は、特定の好ましいプロセス条件下でB12非依存性デヒドラターが機能する能力によって限定されるかもしれない(例えば好気性条件下で(非特許文献8))。
【0011】
原理上は、自然発生微生物株から不活性化速度が低下した補酵素B12依存性デヒドラターゼを単離することが可能なはずである。低下した不活性化速度は、微生物宿主における補酵素B12依存性デヒドラターゼの代謝回転率(生成物mol/ホロ酵素mol)を増大させ、これによりデヒドラターゼおよび補酵素B12の要求量が低下する。このアプローチはそのレベルのデヒドラターゼおよび補酵素B12を維持するのに必要なエネルギーも低下させるであろう。しかし実際には、補酵素B12依存性デヒドラターゼの多様性は不活性化速度に関して限られていることが分かっている。
【0012】
【特許文献1】国際公開第96/35796号パンフレット
【特許文献2】米国特許第5,686,276号明細書
【特許文献3】国際公開第01/012833号パンフレット
【特許文献4】米国特許出願第60/374931号明細書
【特許文献5】米国特許出願第60/416192号明細書
【特許文献6】国際公開第98/21341号パンフレット
【特許文献7】米国特許第6,013,494号明細書
【特許文献8】国際公開第01/04324 A1号パンフレット
【非特許文献1】ゼング(Zeng)ら、Adv.Biochem.Eng.Biotechnol.、74:239〜259(2002)
【非特許文献2】トラヤ(Toraya)T.、アミノ酸残基および関連ラジカルに関与する金属結合酵素(Metalloenzymes Involving Amino Acid−Residue and Related Radicals)、シーゲル(Sigel)H.およびシーゲル(Sigel)A.編;生物学的システム中の金属イオン(Metal Ions in Biological Systems);マーセルデッカー(Marcel Dekker):ニューヨーク(New York)、1994;第30巻、pp217〜254
【非特許文献3】ダニエル(Daniel)ら、FEMS Microbilogy Reviews、22:553〜566(1999)
【非特許文献4】サウヴェージ(Sauvageot)ら、FEMS Microbiology Letters、209:69〜74(2002)
【非特許文献5】サイフェルト(Seifert)ら、Eur.J.Biochem.、268:2369〜2378(2001)
【非特許文献6】トラヤ(Toraya)およびモリ(Mori)、J.Biol.Chem.、274:3372(1999)
【非特許文献7】トビマツ(Tobimatsu)ら、J.Bacteriol.、181:4110(1999)
【非特許文献8】ハートマニス(Hartmanis)およびステードマン(Stadman)、Arch.Biochem.Biophys.、245:144〜52(1986)
【発明の開示】
【発明が解決しようとする課題】
【0013】
したがって解決すべき問題は、現在入手できる補酵素B12依存性デヒドラターゼ酵素が、工業化合物の生成のために工業用途で必要な反応速度を提供できないことである。
【課題を解決するための手段】
【0014】
出願人らは、
(a)B12依存性デヒドラターゼホロ酵素と、グリセロールおよび少なくとも25mMの1,3−プロパンジオールを含んでなる混合物とを接触させ、
(b)B12依存性デヒドラターゼホロ酵素をスクリーニングして、B12依存性デヒドラターゼの代謝回転率を推定する
ことを含んでなる、改善された反応速度を有するB12依存性デヒドラターゼのスクリーニング方法を提供する。
【0015】
方法のスクリーニングステップは、
(a)補酵素B12、デヒドラターゼ再活性化因子、または内在性B12依存性デヒドラターゼの供給源を有さない細胞をグリセロールを含まない発酵性炭素基質上に生育させ、
(b)細胞を透過化処理し、
(c)補酵素B12、グリセロール、および少なくとも25mMの1,3−プロパンジオールの混合物をステップ(b)の透過化処理された細胞添加し、
(d)ステップ(c)で生産された3−ヒドロキシプロピオンアルデヒドを定量化する
ことをさらに含んでなり、
定量化は、T1、T2、およびT(600)よりなる群から選択される測定である。
【0016】
本発明は、配列番号:40、44、48、52、56、60、64、68、72、76、80、84、88、92、96、100、104、108、112、116、120、124、128、132、136、140、143、147、151、155、159、163、167、171、175、179、182、186、189、192、195、198、201、204、208、212、215、218、221、225、229、233、237、241、245、249、253、257、261、265、269、273、277、281、285、289、293、297、301、304、307、310、313、316、319、322、325、328、331、334、および337よりなる群から選択される、B12依存性変異株デヒドラターゼをコードする核酸配列をさらに含む。より好ましいB12依存性変異株デヒドラターゼは、配列番号:40、44、48、52、56、60、64、68、72、76、80、84、88、92、96、100、104、108、112、116、120、124、128、132、136、140、143、147、151、155、159、163、167、171、175、179、182、186、189、192、195、198、201、204、208、212、215、218、221、225、229、233、237、241、245、249、253、257、261、265、269、273、277、281、285、289、293、297、301、304、307、310、313、316、319、322、325、328、331、334、および337よりなる群から選択される核酸配列によってコードされる。B12依存性変異株デヒドラターゼをコードするより好ましい核酸配列は、配列番号:140、179、186、189、192、195、198、201、212、215、218、301、304、307、310、313、316、319、322、325、328、331、334、および337よりなる群から選択されるα−βサブユニット融合を含んでなる。B12依存性変異株デヒドラターゼをコードする最も好ましい核酸配列は、配列番号313、322、および328よりなる群から選択されるα−βサブユニット融合を含んでなる。
【0017】
本発明はまた、α−βサブユニット融合のαおよびβサブユニット間にリンカー配列さらに含んでなる核酸配列を含み、リンカー配列は、配列番号18および配列番号19よりなる群から選択される。
【0018】
本発明はまた、
a)ホットスポット1またはホットスポット2を含んでなるB12依存性デヒドラターゼホロ酵素と変異原因物質とを接触させ、
b)ステップA)で生産された変異株を改善されたkcatおよび/または安定性についてスクリーニングし、そして
c)ステップa)およびb)を反復する
ことを含んでなる、改善された反応速度を有するB12依存性デヒドラターゼ変異株作り出す方法を含む。
【0019】
標準デヒドラターゼと比較して改善された反応速度を有するB12依存性デヒドラターゼを同定するための本発明のさらに別の方法は、
a)デヒドラターゼホロ酵素と5〜10mMのグリセロールおよび/または10〜300mMの1,3−プロパンジオールとを接触させ、
b)ハイスループットアッセイにおいて、kcatおよび総酵素代謝回転を推定するのに十分に隔たった少なくとも2個の時点で測定し、
c)標準デヒドラターゼと比較して改善された反応速度を有するB12依存性変異株を選択する
ことを含んでなる。
【0020】
標準デヒドラターゼと比較して改善された反応速度を有するB12依存性デヒドラターゼを同定するなおも別の本発明の方法は、
a)デヒドラターゼホロ酵素と5〜50mMグリセロールおよび>300mMの1,3−プロパンジオールとを接触させ、
b)ハイスループットアッセイにおいて、T0から十分に隔たった1つの時点で測定して、総酵素代謝回転数を推定し、そして
c)標準デヒドラターゼと比較して改善された反応速度を有するB12依存性変異株を選択する
ことを含んでなる。
【0021】
配列リスト
出願人らは、特許出願におけるヌクレオチドおよびアミノ酸配列開示を司る規則(EPO通達OJ 12/1992、追補No.2で公開されるEPO長官決定の付録IおよびII)、37 C.F.R.1.821〜1.825および付録AおよびB(ヌクレオチドおよび/またはアミノ酸配列を含む出願の開示に関する要件)で述べられる、世界知的所有権機関(WIPO)標準ST.25(1998)およびEPOおよびPCTの配列表要件(規則5.2および49.5(aの2)、および実施細則第208号および附属書C)に一致する346個の配列を提供した。配列説明は、参照によって本明細書に援用するNucleic Acids Research、13:3021〜3030(1985)およびBiochemical Journal、219(2):345〜373(1984)で述べられるIUPAC−IYUB標準に準拠して定義される、ヌクレオチド配列性状のための1文字コードおよびアミノ酸のための3文字コードを含む。
【0022】
配列番号1は、肺炎桿菌(Klebsiella pneumoniae)ATCC25955(エンプテイジ(Emptage)ら、国際公開第01/012833 A2号パンフレット)から単離された野生型GDHを含有する12.1kBのEcoRI−SalI断片である。野生型GDHは、α−サブユニット(bp 7044〜8711)、β−サブユニット(bp 8724〜9308)、およびγ−サブユニット(bp 9311〜9736)によってコードされる。GDHのα、β、およびγ−サブユニットのアミノ酸配列は、それぞれ配列番号2、配列番号3、および配列番号4として提供される。
【0023】
配列番号5および6は、プラスミドpGH20からのGDHのクローニングのために使用されるプライマーDHA−F1およびDHA−R1をそれぞれコードする。
【0024】
配列番号7は、GDHのα−サブユニット全体およびβ−サブユニットの一部のクローニングのために使用される逆転写プライマーDHA−R2をコードする。
【0025】
配列番号8〜14は、α−サブユニットの領域性無作為変異誘発のために使用されるプライマーpGD20RM−F1、pGD20RM−R1、TB4BF、TB4BR、pGD20RM−F2、pGD20RM−R2、およびGD−Cをそれぞれコードする。
【0026】
配列番号15〜17は、領域性無作為変異株ライブラリーの調製のために使用される変性プライマーpGD20RM−F3、TB4B−R1、およびpGD20RM−R4をそれぞれコードする。
【0027】
配列番号18は、融合タンパク質Sma3002のα−およびβ−サブユニット間のリンカーをコードする。配列番号19は、融合タンパク質Xba3009のα−およびβ−サブユニット間のリンカーをコードする。
【0028】
配列番号20〜25は、第二世代変異株GDHの合成のために使用されるプライマー2−F4−F1、2−F4−R1、12−B1−F1、12−B1−R1、16−H5−F1、および16−H5−R1をそれぞれコードする。
【0029】
配列番号26〜29は、純粋融合変異株1−E1および22−G7を作り出すために使用されるプライマー1−E1−F1、1−E1−R1、22−G7−F1、および22−G7−R1をそれぞれコードする。
【0030】
配列番号30〜39は、Sma3002−由来変異株の合成のために使用されるプライマー7A−C1−F1、7A−C1−R1、7C−A5−F1、7C−A5−R1、8−C9−F1、8−C9−R1、9−D7−F1、9−D7−R1、10−G6−F1、および10−G6−R1をそれぞれコードする。
【0031】
野生型GDHから誘導される変異酵素は、それぞれの核酸配列およびアミノ酸配列に従って、以下のSEQ識別番号を割り当てられる(表1)。
【0032】
【表1】
【0033】
【表2】
【0034】
【表3】
【0035】
配列番号340〜342は、プライマーT53−SM、L509−SM、およびV224−SMをそれぞれコードする。
【0036】
配列番号343および344は、プライマーGDHM−F1およびGDHM−R1をそれぞれのコードする。
【0037】
配列番号345および346は、プライマーGDHM−F2およびGDHM−Rをそれぞれのコードする。
【発明を実施するための最良の形態】
【0038】
出願人らは、工業用途で使用するための、具体的には3−ヒドロキシプロピオンアルデヒドおよび1,3−プロパンジオールを製造するための改善された反応速度を有する(すなわちグリセロールおよび1,3−プロパンジオール存在下で増大した総代謝回転数を提供する)人工的補酵素B12依存性デヒドラターゼを提供することで既述の問題を解決した。変異誘発技術によって作り出されたこれらのデヒドラターゼは、それらがそれから作り出された野生型酵素と比較して、増大したkcatおよび/または低下した酵素不活性化速度のいずれかを有することで特徴づけられる。
【0039】
本発明の人工的補酵素B12依存性デヒドラターゼは、触媒作用速度の不当な犠牲なしに不活性化速度を低下させる。デヒドラターゼ不活性化問題のこの解決法は、微生物宿主において補酵素B12依存性デヒドラターゼの代謝回転率(生成物mol/ホロ酵素mol)を増大させるので好ましい。増大した代謝回転率の効果は、デヒドラターゼ要求量、補酵素B12要求量、およびエネルギー消費を低下させて、そのデヒドラターゼおよび補酵素B12レベルを維持する。さらに補酵素B12依存性デヒドラターゼは、工業プロセス条件下での1,3−プロパンジオール生成のために効率的に作動することが実証されている。
【0040】
一連の変異株デヒドラターゼを提供するのに加えて、本発明は2個のハイスループットアッセイも提供して、変異株デヒドラターゼのスクリーニングを容易にする。どちらの方法も補酵素B12、デヒドラターゼ再活性化因子、またはB12依存性依存デヒドラターゼの供給源を有さない細胞における、アポ酵素形態のB12依存性デヒドラターゼの存在に依存する。したがって補酵素B12および基質グリセロールの添加に基づいて、デヒドラターゼ活性の開始を正確に制御することが可能である。
【0041】
具体的には、変異補酵素B12依存性デヒドラターゼ遺伝子の大きなライブラリーを作成して、遺伝子産物を発現させ、グリセロールおよび1,3−プロパンジオール混合物存在下での不活性化低下についてハイスループット様式でスクリーニングした。スクリーニング方法は、低下した不活性化速度および改善されたグリセロール触媒作用速度を有するこれらの人工的補酵素B12依存性デヒドラターゼの容易で迅速な同定を可能にする。引き続くデヒドラターゼエンジニアリングの工程は、なおさらに改善されたデヒドラターゼ変異体の生成および同定を可能にする。
【0042】
ここでの教示に基づいて、本発明で述べられるように、グリセロールから3−HPへの転換を触媒できるB12依存性デヒドラターゼ活性をコードする多様な遺伝子が、変異誘発およびスクリーニングのための標的として適することは、当業者には明らかである。したがって改善された反応速度(すなわちグリセロールおよび1,3−プロパンジオール存在下における増大した総代謝回転数)を有する多様な変異株デヒドラターゼが、この発明の手段によって同定できることが期待される。
【0043】
定義
明細書および請求項の解釈のために、以下の略語および定義が使用される。
【0044】
「ポリメラーゼ連鎖反応」は、PCRという形に省略する。
【0045】
「3−ヒドロキシプロピオンアルデヒド」は、3−HPという形に省略する。
【0046】
「デヒドラターゼ」という用語は、グリセロール分子を生成物3−ヒドロキシプロピオンアルデヒドに異性化または変換できるあらゆる酵素を指すために使用する。上述のようにいくつかのデヒドラターゼは、補助因子として補酵素B12を必要とする。「補酵素B12依存性デヒドラターゼ」および「B12依存性デヒドラターゼ」という用語は区別なく使用され、補酵素B12を必要とするデヒドラターゼを指す。補酵素B12依存性デヒドラターゼは、補酵素B12依存性デヒドラターゼ(E.C.4.2.1.30)および補酵素B12依存性デヒドラターゼ(E.C.4.2.1.28)を含んでなる。代案としては、デヒドラターゼは補酵素B12非依存性であっても良い。これらの酵素は補助因子として補酵素B12を必要とせず、「B12非依存性デヒドラターゼ」という用語で呼ばれる。本発明の目的で、「グリセロールデヒドラターゼ」と言う用語は、特に補酵素B12依存性デヒドラターゼ(E.C.4.2.1.30)を指すために使用され、「ジオールデヒドラターゼ」と言う用語は、特に補酵素B12依存性デヒドラターゼ(E.C.4.2.1.28)を指すために使用される(出願人らの命名法の意図的選択は、B12非依存性デヒドラターゼを「ジオールデヒドラターゼ」および「グリセロールデヒドラターゼ」とそれぞれ称したハートマニス(Hartmanis)およびステードマン(Stadman)、同上、およびクラウス(Croux)ら、同上、の選択と混同すべきでない)。
【0047】
「アポ酵素」と言う用語は、タンパク質から構成される酵素の部分を指す。アポ酵素は、酵素が機能するために必要とするかもしれない非タンパク質構造を含まないので、アポ酵素は触媒的に不活性であっても良い。「補助因子」と言う用語は、アポ酵素によって触媒の活性のために必要とされる非タンパク質構造を指す。「ホロ酵素」と言う用語は、触媒的に活性なタンパク質−補助因子複合体を指す。補酵素B12依存性デヒドラターゼアポ酵素は、ホロ酵素を形成するために補助因子補酵素B12を必要とする。
【0048】
「補酵素B12」および「アデノシルコバラミン」と言う用語は区別なく使用されて、5’−デオキシアデノシルコバラミンを意味する。
【0049】
「ビタミンB12」および「シアノコバラミン」と言う用語は区別なく使用されて、上軸5’−デオキシ−5’−アデノシルリガンドが、シアノ部分にによって置換される補酵素B12の誘導体を指す。
【0050】
「ヒドロキソコバラミン」は、上軸5’−デオキシアデノシルリガンドがヒドロキシ部分によって置換される補酵素B12の誘導体を指す。アクアコバラミンは、ヒドロキソコバラミンのプロトン化形態である。
【0051】
グリセロールまたは1,3−プロパンジオールによるB12非依存性デヒドラターゼホロ酵素の不活性化は、上軸5’−デオキシ−5’−アデノシルリガンドを失った不活性なコバラミン化学種の形成をもたらす。「補酵素B12前駆物質」と言う用語は、上軸5’−デオキシアデノシルリガンドが置換されている補酵素B12の誘導体を指す。
【0052】
ここでの用法では、「GDH」と言う用語は、特に肺炎桿菌(Klebsiella pneumoniae)ATCC25955から単離され、配列番号1のbp 7044〜8711、bp 8724〜9308、およびbp 9311〜9736によってコードされるB12依存性グリセロールデヒドラターゼを指す。「GDH」と言う用語は、α、β、およびγ−サブユニットのアセンブルされた複合体を指すのに使用され、アポ酵素またはホロ酵素を指しても良い。GDHの個々のサブユニットへの言及は、例えば「GDHのα−サブユニット」または「GDHα−サブユニット」などのサブユニットを特定する。同様にα−およびβ−およびγ−サブユニットを総称して、あるいはGDHの個々のサブユニットに言及して、GDHのアミノ酸配列に言及しても良い。GDHのα、β、およびγ−サブユニットのアミノ酸配列は、それぞれ配列番号2、配列番号3、および配列番号4として提供される。
【0053】
この発明の開示の目的では、GDHは、人工的(変異株)誘導体との比較でDNAおよびアミノ酸配列の標準として使用される。GDHはまた、本発明を使用して作り出された人工的誘導体の反応速度がそれに対して測定される、野生型反応速度の標準として使用される。ここではGDHが標準材料として使用されるが、あらゆる自然発生的補酵素B12−依存デヒドラターゼ(例えば表2に列挙されるもの)が本発明においてGDHと区別なく使用でき、それに対して人工的誘導体の反応速度が測定される。
【0054】
「遺伝的に変更される」と言う用語は、形質転換または変異によって遺伝的材料を変化させるプロセスを指す。
【0055】
ここでの用法では、「変異株」と言う用語は、変異プロセスによって生成したGDH酵素またはGDH配列を含有する細菌クローン、プラスミド、ライブラリーまたはベクターを指す。代案としては、変異株と言う用語は、変異プロセスによって生じたGDH酵素、GDHアミノ酸配列、またはGDH DNA配列を直接指す。したがって変異株GDHは(野生型)GDHとは異なる。
【0056】
「改善された反応速度」と言う用語は、グリセロールおよび/または1,3−プロパンジオール存在下で、野生型酵素に対して低下したデヒドラターゼ不活性化速度を指す。したがって改善された反応速度は、グリセロールおよび1,3−プロパンジオール存在下で、増大した総酵素代謝回転数に関係がある。これはkcatの増大および/または酵素不活性化速度の低下のどちらかによって達成できる。
【0057】
「触媒効率」と言う用語は、酵素のkcat/KMとして定義される。「触媒効率」は、基質に対する酵素の特異性を定量化するのに使用される。
【0058】
「kcat」、「KM」、および「Ki」と言う用語は当業者に知られており、(フェルスト(Ferst)著、「酵素構造および機序(Enzyme Structure and Mechanism)」第二版、W.H.フリーマン(Freeman)、ニューヨーク(New York)、1985、pp98〜120)で述べられている。
【0059】
「kcat」と言う用語は、「代謝回転数」と呼ばれることが多い。「kcat」と言う用語は、単位時間あたり活性部位あたりの生成物に変換される基質分子の最大数、または単位時間あたりに酵素が回転する数として定義される。kcat=V最大/[E]、(式中、[E]は酵素濃度である)(フェルスト(Ferst)著、「酵素構造および機序(Enzyme Structure and Mechanism)」第二版、W.H.フリーマン(Freeman)、ニューヨーク(New York)、1985、pp98〜120)。「総代謝回転」および「総代謝回転数」と言う用語は、ここでは反応開始(T0)とホロ酵素の完全な不活性化が起きる間に、B12依存性デヒドラターゼホロ酵素とグリセロールとの(場合により1,3−プロパンジオール存在下での)反応によって形成される生成物量を指すのに使用される。
【0060】
「Ki」と言う用語は、1,3−プロパンジオールの阻害定数を指す。
【0061】
「k観察された不活性」と言う用語は、B12依存性デヒドラターゼホロ酵素と過剰なグリセロールおよび/または過剰な1,3−プロパンジオールとの反応において観察される、不活性化の一次反応速度定数を指す。過剰なグリセロールのみの存在下で、k観察された不活性はk不活性グリセロールに等しい。過剰な1,3−プロパンジオールのみの存在下で、k観察された不活性はk不活性1,3−プロパンジオールに等しい。過剰なグリセロールおよび過剰な1,3−プロパンジオール単独双方存在下で、k観察された不活性は、k不活性グリセロールおよびk不活性1,3−プロパンジオール双方の関数に等しい。
【0062】
「T1」と言う用語は、10mMのグリセロールおよび50mMの1,3−プロパンジオール存在下で、反応開始30秒後に測定されるGDH酵素反応によって作られる生成物量を指す。T1はkcatに比例する。
【0063】
「T2」と言う用語は、10mMのグリセロールおよび50mMの1,3−プロパンジオール存在下で、反応開始40分後に測定されるGDH酵素反応によって作られる生成物量を指す。T2はkcat/k観察された不活性に比例し、総酵素総代謝回転数を反映する。
【0064】
「T2/T1比率」と言う用語は、T2とT1との比率を指す。T2/T1比率は、1/k観察された不活性に比例する。
【0065】
「T2(600)」と言う用語は、10mMのグリセロールおよび600mMの1,3−プロパンジオール存在下で、反応開始40分後に測定されるGDH酵素反応によって作られる生成物量を指す。T2(600)はkcat/k観察された不活性に比例し、600mMの1,3−プロパンジオール存在下における総酵素総代謝回転数を反映する。
【0066】
「T(600)」と言う用語は、10mMのグリセロールおよび600mMの1,3−プロパンジオール存在下で、反応開始70分後に測定されるGDH酵素反応によって作られる生成物量を指す。T(600)はkcat/k観察された不活性に比例する。T(600)値は、600mM1,3−プロパンジオール存在下においてT2(600)よりも正確な総酵素代謝回転数を与える。
【0067】
「ポリペプチド」および「タンパク質」と言う用語は、区別なく使用される。
【0068】
「宿主細胞」または「宿主生物体」と言う用語は、外来性または非相同的遺伝子を受け入れてこれらの遺伝子を発現し、活性遺伝子産物を生成できる微生物を指す。
【0069】
「単離された」と言う用語は、それが自然に関連する少なくとも1個の構成要素から取り除かれるタンパク質またはDNA配列を指す。
【0070】
「単離された核酸分子」は、場合により合成、非天然または修飾ヌクレオチド塩基を含有する、一本鎖または二本鎖であるRNAまたはDNAのポリマーである。DNAポリマーの形態の単離された核酸分子は、1個またはそれ以上のcDNA、ゲノムDNAまたは合成DNAの断片を含んでなっても良い。
【0071】
「遺伝子」とは、特定のタンパク質を発現する核酸断片を指し、場合によってはコード配列に先行する(5’非コード配列)およびそれに続く(3’非コード配列)制御配列を含んでも良い。「天然遺伝子」および「野生型遺伝子」は、自然界にそれ自体の制御配列と共に見られる遺伝子を指す。「キメラ遺伝子」とは、自然界に共に見られない制御およびコード配列を含んでなる天然遺伝子でないあらゆる遺伝子を指す。したがって、キメラ遺伝子は、異なる供給源から誘導される制御配列およびコード配列、あるいは同一供給源から誘導されるが、自然界に見られるのとは異なるやり方で配列される制御配列およびコード配列を含んでなっても良い。「内因性の遺伝子」とは、生物体ゲノムにおいてその天然位置にある天然遺伝子を指す。「外来性の」遺伝子とは、宿主生物体において常態では見られないが、代わりに遺伝子移入によって宿主生物体に導入される遺伝子を指す。外来性の遺伝子は、非天然生物体中に挿入された天然遺伝子、あるいはキメラ遺伝子を含んでなることができる。「導入遺伝子」とは、形質転換によってゲノム中に導入される遺伝子である。
【0072】
「コードする」および「コーディング」と言う用語は、それによって遺伝子が、転写および翻訳の機序を通じてアミノ酸配列を生成するプロセスを指す。特定のアミノ酸配列をコードするプロセスは、コードされるアミノ酸における変化を引き起こさない塩基置換を伴うかもしれないDNA配列を含み、あるいは1個またはそれ以上アミノ酸を変更するかもしれないが、DNA配列によってコードされるタンパク質の官能特性に影響しない塩基置換を伴うDNA配列を含むものと理解される。したがって本発明は、特定の例示的な配列以上のものを包含するものと理解される。
【0073】
「アミノ酸」と言う用語は、タンパク質またはポリペプチドの基本的化学構造単位を指す。アミノ酸は、参照によって本明細書に援用するNucleic Acids Research、13:3021〜3030(1985)およびBiochemical Journal、219(2):345〜373(1984)で述べられるIUPAC−IYUB標準に準拠して定義されるように、1文字コードまたはアミノ酸のための3文字コードのいずれかによって識別される。
【0074】
特定のタンパク質では、DNAコード領域内の点置換変異、および得られるアミノ酸変化は、以下の表記法の1つを使用して、標準DNAおよびアミノ酸配列を参照して特定される。例えばGDHにおける変異の場合、変異は下で述べる表記法の1つを使用して記述される。
1.詳細な表記法:最初に野生型コドンのヌクレオチド配列が提示され、続いて「α」、「β」または「γ」記号(GDHのα−、β−、またはγ−サブユニットの変異を区別するため)、3文字略語の特定の野生型アミノ酸、およびその位置が提示される。次に野生型情報の後に、参照される変異中に存在する特定のヌクレオチドおよびアミノ酸修飾が続く。この表記法の例はGGG(α−Gly63)からGGA(Gly)であり、ここではα−サブユニットの63番目のコドンが、変異株においてヌクレオチド配列が「GGG」から「GGA」に変更されるようにサイレント変異を被る。
2.「簡略」表記法:「α」、「β]、または「γ」の記号(GDHのα−、β−またはγ−サブユニットにおける変異と区別するため)に、野生型アミノ酸の1文字略語、コドン位置、および変異株アミノ酸の1文字略語が続く。この表記法の例はα−V224Lであり、変異株におけるα−サブユニット中のコドン224における野生型バリンからロイシンへの変異を表す。特定部位で化学的に同等のアミノ酸の生成をもたらす(しかしコードされるタンパク質官能特性に影響しない)遺伝子における変更が一般的であることは、技術分野で良く知られている。
【0075】
「実質的に同様の」とは、1個またはそれ以上のヌクレオチド塩基の変化が、1個またはそれ以上アミノ酸の置換をもたらすが、DNA配列によってコードされるタンパク質の官能特性に影響しない核酸分子を指す。「実質的に同様の」とはまた、1個またはそれ以上ヌクレオチド塩基の変化が、アンチセンスまたはコサプレッション技術によって、遺伝子発現の変更を媒介する核酸分子の能力に影響しない核酸分子を指す。「実質的に同様の」とはまた、アンチセンスまたはコサプレッション技術による遺伝子発現の変更を媒介する能力に対して、得られる転写物の官能特性、または得られるタンパク質分子の官能特性の変更に実質的に影響しない、本発明の核酸分子の修飾(1個またはそれ以上のヌクレオチド塩基の欠失または挿入などの)を指す。本発明は特定の例示的な配列以上のものを包含する。
【0076】
提案される各修飾は、十分に技術分野の日常的技術の範囲内であり、コードされる生成物の生物学的活性維持の判定も同様である。さらに当業者は、この発明によって実質的に包含される同様の配列が、ストリンジェントな条件(0.1×SSC、0.1%SDS、65℃および2×SSC、0.1%SDSでの洗浄とそれに続く0.1×SSC、0.1%SDS)下で、ここで例示する配列とハイブリッドするそれらの能力によっても定義されることを認識する。本発明の好ましい実質的に同様の核酸断片は、そのDNA配列が、ここで報告する核酸断片のDNA配列と少なくとも80%同一である核酸断片である。より好ましい核酸断片は、ここで報告する核酸断片のDNA配列と少なくとも90%同一である。最も好ましいのは、ここで報告する核酸断片のDNA配列と少なくとも95%同一である核酸断片である。
【0077】
核酸断片は、温度および溶液イオン強度の適切な条件下で、核酸断片の一本鎖形態がその他の核酸断片とアニールできる場合、cDNA、ゲノムDNA、またはRNAなどの別の核酸断片と「ハイブリッド形成可能」である。ハイブリッド形成および洗浄条件については良く知られており、サムブルック(Sambrook)J.、フリッチュ(Fritsch)E.F.およびマニアティス(Maniatis)T.、分子クローニング:実験室マニュアル(Molecular Cloning:A Laboratory Manual)、第二版、コールドスプリングハーバーラボラトリー(Cold Spring Harbor Laboratory):コールドスプリングハーバー(Cold Spring Harbor)、ニューヨーク(NY)(1989)の特に第11章およびその表11.1で例証される。
【0078】
「かなりの部分」とは、当業者による配列の手動評価によって、あるいはBLAST(「基礎的局在性整列化検索ツール(Basic Local Alignment Search Tool)」、アルトシュル(Altschul)ら、J.Mol.Biol.、215:403〜410(1993)、www.ncbi.nlm.nih.gov/BLAST/も参照されたい)などのアルゴリズムを使用したコンピュータ支援配列比較および同定によって、遺伝子またはポリペプチドの推定上の同定が得られるのに十分な、そのポリペプチドまたは遺伝子のヌクレオチド配列のアミノ酸配列を含んでなる、アミノ酸またはヌクレオチド配列を指す。推定的にポリペプチドまたは核酸配列が既知のタンパク質または遺伝子に相同的であると同定するためには、概して10個以上の隣接するアミノ酸または30個以上のヌクレオチド配列が必要である。さらにヌクレオチド配列に関して、20〜30個の隣接するヌクレオチドを含んでなる遺伝子特異的オリゴヌクレオチドプローブを配列依存遺伝子同定法(例えばサザンハイブリッド形成)および単離(例えば細菌コロニーまたはバクテリオファージ・プラークの原位置(in situ)ハイブリッド形成)において使用しても良い。さらにプライマーを含んでなる特定の核酸分子を得るために、塩基12〜15個の短いオリゴヌクレオチドをPCRで増幅プライマーとして使用しても良い。したがってヌクレオチド配列の「かなりの部分」は、配列を含んでなる核酸分子の特定の同定および/または単離ができるようにする十分な配列を含んでなる。本明細書は、1つまたは複数の特定タンパク質をコードする、部分的または完全なアミノ酸およびヌクレオチド配列を教示する。当業者はここで報告される配列の利益を享受して、開示された配列の全てまたはかなりの部分を当業者に知られている目的のために使用できる。したがって本発明は、添付の配列表で報告されるような完全な配列、ならびに上で定義されるようなこれらの配列のかなりの部分を含んでなる。
【0079】
「相補的」と言う用語は、二本鎖DNA間の関係について述べる。例えばDNAについて、アデノシンはチミンに相補的であり、シトシンはグアニンに相補的である。したがって本発明は添付の配列表で報告されるような完全な配列、ならびに実質的に同様の核酸配列に相補的な単離された核酸分子も含む。
【0080】
「パーセント同一性」と言う用語は、技術分野で知られているように、配列を比較して判定される2つ以上のポリペプチド配列または2つ以上のポリヌクレオチド配列の関係である。技術分野で「同一性」とは、場合によっては、このような配列のストリング間の整合によって判定されるような、ポリペプチドまたはポリヌクレオチド配列間の配列関連性の程度も意味する。「同一性」および「類似性」は、1.)「計算分子生物学(Computational Molecular Biology)」、レスク(Lesk)A.M.編、オックスフォードユニバーシティ(Oxford University)、ニューヨーク(New York)、1988、2.)「バイオコンピューティング:情報科学およびゲノムプロジェクト(Biocomputing:Informatics and Genome Projects)」、スミス(Smith)D.W.編、アカデミック(Academic)、ニューヨーク(New York)、1993、3.)「配列データのコンピュータ分析(Computer Analysis of Sequence Data)」、第一部、グリフィン(Griffin)A.M.、およびグリフィン(Griffin)H.G.編、Humana、ニュージャージー(New Jersey)、1994、4.)「分子生物学における配列分析(Sequence Analysis in Molecular Bioloy)」、フォン・ハインェ(von Heinje)G.編、アカデミック(Academic)、ニューヨーク(New York)、1987、5.)「配列分析入門(Sequence Analysis Primer)」、グリブスコフ(Gribskov)M.およびデュヴルー(Devereux)J.編、ストックトン(Stockton)、ニューヨーク(New York)、1991で述べられるものをはじめとするが、これに限定されるものではない、既知の方法によって容易に計算できる。同一性を判定する好ましい方法は、試験される配列間に最大の整合を与えるようにデザインされる。
【0081】
同一性および類似性を判定する方法は、一般に入手できるコンピュータプログラムで体系化される。2個の配列間の同一性および類似性を判定する好ましいコンピュータプログラム法としては、それらの標準デフォルト値がgap形成ペナルティ=12およびgap延長ペナルティ=4である、ニードルマン(Needleman)およびブンシュ(Wunsch)アルゴリズムを使用するGCGプログラムパッケージにあるGCGパイルアッププログラム(デュヴルー(Devereux)ら、Nucleic Acids Res.12:387〜395(1984))、BLASTP、BLASTN、およびFASTA、(ピアソン(Pearson)ら、Proc.Natl.Acad.Sci.USA、85:2444〜2448(1988))が挙げられるが、これに限定されるものではない。BLASTXプログラムは、NCBIおよびその他の供給元から一般に入手できる(「BLASTマニュアル(BLAST Manual)」、アルトシュル(Altschul)ら、Natl.Cent.Biotechnol.Inf.、Natl.Library Med.(NCBINLM)NIH、Bethesda、MD;アルトシュル(Altschul)ら、J.Mol.Biol.、215:403〜410(1990);アルトシュル(Altschul)ら、Nucleic Acids Res.、25:3389〜3402(1997))。同一性を判定する別の好ましい方法は、ヨッタン−ハイン(Jotun−Hein)アルゴリズムを使用するDNASTARタンパク質アライメントプロトコル法による(ハイン(Hein)ら、Methods Enzymol.、183:626−645 (1990))。アラインメントのためのヨッタン−ハイン(Jotun−Hein)法のデフォルト変数は、1.)複数アラインメントではgapペナルティ=11、gap長さペナルティ=3、および2.)対合アラインメントではktuple=6である。一例として、参照ヌクレオチド配列に対して少なくとも例えば95%の「同一性」を有するヌクレオチド配列を有するポリヌクレオチドとは、ポリヌクレオチド配列が参照ヌクレオチド配列の各100個のヌクレオチドあたり5点の変異を含んでも良いこと以外は、ポリヌクレオチドのヌクレオチド配列が、参照配列と同一であることを意味する。換言すると、参照ヌクレオチド配列と少なくとも95%同一のヌクレオチド配列を有するポリヌクレオチドを得るためには、参照配列中の5%までのヌクレオチドが、欠失しまたは別のヌクレオチドで置換されていても良く、あるいは参照配列中の全ヌクレオチドの5%までのヌクレオチドの数が、参照配列中に挿入されていても良い。参照配列のこれらの変異は、参照ヌクレオチド配列の5’または3’末端部位で生じても、あるいはこれの末端部位間のあらゆる位置で、参照配列中の、または参照配列中の1つまたは複数の隣接するグループ中のヌクレオチドに個別に挿入されて生じても良い。同じように参照アミノ酸配列に対して少なくとも例えば95%の同一性を有するアミノ酸配列を有するポリペプチドとは、ポリペプチド配列が参照アミノ酸の各100個のアミノ酸あたり5個までのアミノ酸の変化を含むこと以外は、ポリペプチドのアミノ酸配列が参照配列に同一であることを意味する。換言すると参照アミノ酸配列と少なくとも95%同一のアミノ酸配列を有するポリペプチドを得るためには、参照配列中の5%までのアミノ酸残基が欠失しまたは別のアミノ酸で置換されていても良く、あるいは参照配列中の全アミノ酸残基の5%までのアミノ酸の数が、参照配列中に挿入されていても良い。参照配列のこれらの変化は、参照アミノ酸配列のアミノまたはカルボキシ末端部位で生じても、あるいはこれらの末端部位間のあらゆる位置で参照配列中の、または参照配列中の1つまたは複数の隣接するグループ中の残基間に個別に挿入されて生じても良い。
【0082】
「相同的」と言う用語は、特定の宿主細胞において天然または自然発生的なタンパク質またはポリペプチドを指す。本発明は組換えDNA技術を通じて、相同的タンパク質を生成する微生物を含む。
【0083】
「パーセント相同性」と言う用語は、ポリペプチド間のアミノ酸配列同一性の程度を指す。第1のアミノ酸配列が第2のアミノ酸配列と同一である場合、第1および第2のアミノ酸配列は100%相同性を示す。あらゆる2個のポリペプチド間の相同性は、どちらかの配列の特定部位の整合アミノ酸総数の一次関数であり、例えば2個の配列のどちらかのアミノ酸総数の半分が同一ならば、2個の配列は50%の相同性を示すと言われる。
【0084】
「コドン縮重」とは、コードされるポリペプチドのアミノ酸配列に影響しないヌクレオチド配列の変動を可能にする遺伝子コード中の分岐を指す。当業者は、特定のアミノ酸を特定化するヌクレオチドコドンの使用における、特定の宿主細胞によって示される「コドン−バイアス」について良く知っている。
【0085】
結果として生じるタンパク質分子の機能特性に実質的に影響しない、沈黙変化を生じる配列中の欠失、挿入または置換などの配列の修飾も考察された。例えば遺伝子コードの縮重を反映する、あるいは特定部位で化学的に等価のアミノ酸の生成をもたらす遺伝子配列中の変化が考察された。場合によってはタンパク質の生物学的活性変化の効果を研究するために、配列の変異体を作り出すことが実際望ましいかもしれない。提案される各変更は、十分に技術分野のルーチン技術の範囲内であり、コードされる生成物中の生物学的活性保持の判定についても同様である。
【0086】
「発現」と言う用語は、遺伝子産物配列の遺伝子コードから遺伝子産物への転写および翻訳を指す。
【0087】
「プラスミド」、「ベクター」、および「カセット」と言う用語は、細胞の中心的代謝の一部ではない遺伝子を運ぶことが多く、通常環状二本鎖DNA分子の形態である染色体外の要素を指す。このような要素は、あらゆる供給源から誘導される一本鎖または二本鎖DNAまたはRNAの配列、ゲノム一体化配列、直鎖または環状のファージまたはヌクレオチド配列を自律的に複製するかもしれず、そこではいくつかのヌクレオチド配列が独自の構成に連結または組み換えされ、それは選択された遺伝子産物のために、適切な3’非翻訳配列と共に、プロモーター断片およびDNA配列を細胞中に導入することができる。「形質転換カセット」とは、外来性遺伝子を含有し、外来遺伝子に加えて特定の宿主細胞の形質転換を容易にする要素を有する特定のベクターを指す。「発現カセット」とは、外来性遺伝子を含有し、外来性遺伝子に加えて外来性宿主におけるその遺伝子の促進された発現を可能にする要素を有する特定のベクターを指す。
【0088】
「遺伝子組み換え」と言う用語は、交差または非依存性組み合わせのプロセスによって、親テンプレート分子中に存在しない遺伝的組み合わせがそれによって形成されるプロセスを指す。したがって遺伝子組み換えは、親テンプレート分子から得ることができる遺伝的配列の全ての組み合わせを含み(それによって新たに生成された「遺伝子組み換え産生物」の各ヌクレオチド位置が、その特定のヌクレオチド位置のあらゆる親テンプレートから誘導できる)、さらに遺伝子組み換えは、新たな変異(すなわち、欠失、置換、挿入)の導入を含む。
【0089】
「遺伝子組み換えされたポリペプチド」と言う用語は、遺伝子組み換えされた遺伝子またはDNAによってコードされたポリペプチドを意味する。遺伝子組み換えされたポリペプチドは、変更または増強された特性を有することが多い。
【0090】
ポリペプチドまたはタンパク質に適用される「変更された特性」と言う用語は、アッセイ法によって測定できる、ヌクレオチド配列によってコードされるタンパク質に付随する特性を指し、その特性は、天然配列に付随するものと比較して増強されまたは減少する。変更されても良い酵素の好ましい特性の例としては、酵素の活性、基質特異性、阻害物質に対する安定性、熱安定性、プロテアーゼ安定性、溶剤安定性、洗浄剤安定性、および折りたたみ特性が挙げられる。「増強された生物学的特性」とは、天然配列に付随するものを超える変更された特性を指す。「減少した生物学的特性」とは、天然配列に付随するものに満たない変更された特性を指す。
【0091】
「テンプレート」または「親テンプレート」と言う用語は、ワトソン−クリック塩基対形成規則に従って、DNAまたはRNAポリメラーゼによって複写されて、新しいDNAまたはRNA鎖を生成する核酸分子を指す。テンプレート分子から生成される第1のコピーは相補的配列を有するので、テンプレート(または「モデル」)中の配列情報は保存される。テンプレート分子は単一または二本鎖であっても良く、あらゆる供給源に由来しても良い。
【0092】
「複製」は、「テンプレート核酸」の核酸鎖の相補的コピーが、ポリメラーゼ酵素によって合成されるプロセスである。「プライマー誘導」複製では、このプロセスは、複製を開始するために、「二重の」「オリゴヌクレオチド」の末端ヌクレオチドの(デオキシ)リボース部分の3’位置にヒドロキシル基(OH)を必要とする。
【0093】
核酸の「5’領域」および「3’領域」は、遺伝子組み換えが起きることが望ましいヌクレオチド領域を参照して、相対的用語として使用される。これらの領域はテンプレート分子内またはテンプレート分子に付着する側方DNA配列内であっても良い。不対プライマーが、これらの5’および3’領域の部分にアニールする。
【0094】
「側方配列」または「側方DNA断片」とは、それに不対プライマーがアニールしても良いユニークなヌクレオチド配列(テンプレート分子に対して)を提供するために、テンプレート分子の5’または3’領域のいずれかに付着するDNAの短い断片を指す。
【0095】
「完全長伸張生成物」とは、親テンプレートの5’および3’領域の間に含まれるのと非常によく似た長さ(約100塩基以内)を有する、プライマー誘導複製によって生成されるヌクレオチド配列である。
【0096】
「増幅」は、いくつかの「テンプレート核酸」のコピーが、線形または対数いずれかの様式で増加するように複製がサイクル様式で反復されるプロセスである。
【0097】
「プライマー」と言う用語は、相補鎖の合成がポリメラーゼによって触媒される条件下に置くと、相補鎖に沿った核酸合成または複製の開始点として作用できる(合成または天然)オリゴヌクレオチドを指す。プライマーは、プライマーそれ自体と核酸の相補鎖との間の配列相補性に基づいて、相補鎖にアニールする能力を有さねばならないが、塩基のいくらかのミスマッチは許される。プライマーサイズ、塩基配列、相補性、および標的相互作用の要件については下でより詳細に考察する。「プライマー」と言う用語自体は、ここでは出願人によって一般的に使用され、核酸複製プロセスを開始するために機能する(すなわち合成をプライムできる)あらゆる配列−結合オリゴヌクレオチドを包含する。
【0098】
「フォーワードプライマー」と言う用語は、二本鎖テンプレート分子のセンス鎖の5’領域で合成をプライムでき、または一本鎖テンプレート分子の5’領域で合成をプライムできるプライマーを指す。
【0099】
「逆転写プライマー」と言う用語は、二本鎖テンプレート分子アンチセンス鎖の5’領域で合成をプライムでき、または二本鎖テンプレート分子のアンチセンス鎖である一本鎖テンプレート分子の5’領域で合成をプライムできるプライマーを指す。
【0100】
「対合プライマー」と言う用語は、単一テンプレート分子にアニールして、プライマー誘導核酸増幅プロセスによってテンプレートの正確なコピーの合成を可能にするようにデザインされる、フォーワードおよび逆転写プライマーからなるプライマーのペアを指す。二本鎖テンプレート分子の場合、フォーワードプライマーがアンチセンス鎖(テンプレートとして使用されるセンス鎖の相補的コピー)の正確なコピーを生成し、逆転写プライマーがセンス鎖(テンプレートとして使用されるアンチセンス鎖の相補的コピー)の正確なコピーを生成するので、フォーワードおよび逆転写プライマーは、二本鎖テンプレートの正確なコピーの合成を可能にする。対照的にテンプレート分子が一本鎖である場合、そのテンプレートの正確なコピーは、プライマー誘導核酸増幅プロセスを使用して生成される。
【0101】
「不対プライマー」と言う用語は、単一テンプレート分子にアニールして、プライマー誘導核酸増幅プロセスによってそのテンプレートの正確なコピーの合成を可能にするようにデザインされていない、フォーワードおよび逆転写プライマーからなるプライマーの対を指す。その代わりフォーワードプライマーは第1のテンプレート分子にアニールするが、第2のテンプレート分子にアニールできない。逆転写プライマーは、第1のテンプレート分子と配列が異なる第2のテンプレート分子にアニールするが、第1のテンプレート分子にアニールできない。このユニークな不対プライマーのデザインは、テンプレート切り替えを通じた複製中に遺伝子組み換えが起きない限り、一本鎖または二本鎖テンプレート分子がプライマー誘導核酸増幅プロセスによって増幅できないことを確実にする。
【0102】
「プライマー誘導伸張」と言う用語は、プライマーが使用されて、核酸分子の線形または対数増幅において核酸配列の複製を援助する、技術分野で知られているあらゆる方法を指す。例えばプライマー誘導伸張は、ポリメラーゼ連鎖反応(PCR)、リガーゼ鎖反応(LCR)、および鎖置換増幅(SDA)をはじめとするがこ、これに限定されるものではない、技術分野で知られているいくつかのスキームのいずれかによって実行しても良い。
【0103】
GDHおよびコード遺伝子
グリセロールから3−HPへの転換について、この反応の触媒に関与するデヒドラターゼは、B12依存性デヒドラターゼまたはB12非依存性デヒドラターゼであることができる。しかし本発明の目的では、デヒドラターゼはB12依存性デヒドラターゼである。このB12依存性デヒドラターゼは、GDH、グリセロールデヒドラターゼ(E.C.4.2.1.30)、またはジオールデヒドラターゼ(E.C.4.2.1.28)であることができる。これらの各酵素はα2β2γ2構造を有する。文献で使用される遺伝子命名法には幅広いバリエーションがあるので、明瞭さのために、肺炎桿菌(Klebsiella pneumoniae)、シトロバクター・フロインディ(Citrobacter freundii)、クロストリジウム・パスツーリアナム(Clostridium pasteurianum)、ネズミチフス菌(Salmonella typhimurium)、ラクトバシラス・コリノイデ(Lactobacillus collinoide)、およびクレブシエラ・オキシトカ(Klebsiella oxytoca)のデヒドラターゼ遺伝子の遺伝子名およびGenBank番号を示す比較チャートを表2に示し、同定を容易にする。また遺伝子については、例えばダニエル(Daniel)ら、(FEMS Microbiol.Rev.、22:553、(1999))およびトラヤ(Toraya)およびモリ(Mori)(J. Biol.Chem.、274:3372、(1999))で述べられる。
【0104】
【表4】
【0105】
本発明の目的では、肺炎桿菌(K.pneumoniae)(国際公開第01/012833 A2号パンフレット)のdhaB1、dhaB2、およびdhaB3(それぞれ配列番号1および配列番号2、3、および4)によってコードされるGDHが、変異誘発の標的として使用される。この酵素、グリセロールデヒドラターゼは好ましい基質がグリセロールであり、2個の63kDaのαサブユニット、2個の21kDaのβサブユニット、および2個の16kDaのγサブユニットから構成されることが知られている。しかし当業者はここで述べる技術において、シトロバクター・フロインディ(Citrobacter freundii)、クロストリジウム・パスツーリアナム(Clostridium pasteurianum)、またはその他の肺炎桿菌(K.pneumoniae)株のグリセロールデヒドラターゼ、あるいはネズミチフス菌(Salmonella typhimurium)、クレブシエラ・オキシトカまたは肺炎桿菌(K.pneumoniae)のジオールデヒドラターゼもまた適切であることを認識するであろう。同様にその活性がグリセロールの3−HPへの転換を触媒できるB12依存性デヒドラターゼ活性をコードするあらゆる遺伝子が、GDH酵素の機能を変更しないアミノ酸置換、欠失または付加を包含するあらゆるアミノ酸配列をはじめとする本発明の標的として適切なはずである。したがって当業者はその他の供給源から単離されるGDHをコードする遺伝子もまた、本発明で使用するのに適切であることを理解するであろう。
【0106】
グリセロールまたは1,3−プロパンジオール存在下におけるB12依存性デヒドラターゼ不活性化
B12依存性デヒドラターゼは、触媒作用中にグリセロールによって、または1,3−プロパンジオールとの相互作用によって可逆的不活性化を被る。不活性化は、補酵素B12補助因子のコバルト−炭素(Co−C)結合の開裂を伴い、5’−デオキシアデノシンおよび不活性なコバラミン化学種の形成をもたらす。不活性なコバラミン化学種はデヒドラターゼとの密接な結合を保ち、補酵素B12依存性デヒドラターゼ再活性化因子の介入なしには解離が起きない。
【0107】
図1は、GDH不活性化に付随する典型的な経時変化を示す。これらの酵素活性アッセイは、肺炎桿菌(K.pneumoniae)(国際公開第01/012833 A2号パンフレット)のdhaB1、dhaB2、およびdhaB3(それぞれ配列番号1および配列番号2、3、および4)によってコードされるGDHを使用して、実施される。図1上方の曲線は、基質としての10mMのグリセロール(Km約0.5mM)とのGDHの反応の経時変化を示す一方、下方の曲線は、アッセイに50mMの1,3−プロパンジオール(Ki約15mM)を含めることの影響を示す。明らかに3−HP生成物形成速度は、一次不活性化速度定数(k観察された不活性)に従って、不活性化が起きるに連れて時間と共に迅速に低下する。
【0108】
改善された活性を有する変異株GDH
観察されたGDH不活性化に基づいて、野生型GDHと比較して低下した不活性化速度を有する一連の変異株GDHを作り出した。典型的にアプローチは、グリセロールおよび1,3−プロパンジオール存在下で増大した総代謝回転数を有する変異酵素を作り出して、単離することを伴う。これは、kcatの増大および/または酵素不活性化速度の低下のいずれかによって達成できる。
【0109】
GDH活性を改善するプロセスは、GDH遺伝子を含んでなる発現ベクターの構築、GDHコード配列の変異誘発、そして最後に低下した不活性化速度を有する変異体の単離を伴う。引き続く変異誘発の工程によって、GDHコード配列の発達が可能になる。
【0110】
変異株B12依存性デヒドラターゼライブラリーは、変異誘発のための出発原料としてあらゆる野生型(または実質的に同様の)B12依存性デヒドラターゼを使用して調製できる。
【0111】
B12依存性デヒドラターゼの変異誘発の伝統的方法
B12依存性デヒドラターゼ変異誘発のために、多様なアプローチを使用しても良い。ここで使用される2つの適切なアプローチとしては、エラープローンPCR(ロング(Leung)ら、Techniques、1:11〜5(1989);ジョウ(Zhou)ら、Nucleic Acids Res.、19:6052〜6052(1991);およびスピー(Spee)ら、Nucleic Acids Res.、21:777〜778(1993))、および生体内(in vivo)変異誘発が挙げられる。
【0112】
エラープローンPCRの主要な利点は、この方法によって導入された全ての変異がB12依存性デヒドラターゼ遺伝子内であり、PCR条件を変化させることであらゆる変化が容易に制御できることである。代案としては、大腸菌(E.coli)XL1−Red菌株、およびカリフォルニア州ラ・ホーヤのストラタジーン(Stratagene(La Jolla、CA))からのエピキュリアン・コリ(Epicurian coli)XL1−Redミューテータ菌株(グリーナー(Greener)およびキャラハン(Callahan)、Strategies、7:32〜34(1994)も参照されたい)などの市販される材料を使用して、生体内(in vivo)変異誘発を用いても良い。この菌株では、一次DNA修復経路の3つ(mutS、mutD、およびmutT)が欠損しており、野生型よりも5000倍高い変異速度がもたらされる。生体内(in vivo)変異誘発は、(エラープローンPCRのように)連結効率に依存しないが、変異はベクターのあらゆる領域で生じても良く、変異速度は一般にはるかに低い。
【0113】
代案としては、「遺伝子シャッフリング」法(米国特許第5,605,793号明細書、同第5,811,238号明細書、同第5,830,72号明細書、および同第5,837,458号明細書)、または核酸間での遺伝子組み換え発生の活性を促進するあらゆる同様の手段を使用して、低下した不活性化速度を有する変異株B12依存性デヒドラターゼを構築しても良いことが考察される。遺伝子シャッフリング法は、その容易な実行と高速な変異誘発のために特に魅力的である。遺伝子シャッフリングのプロセスは、関心のある遺伝子と同様の(または異なる)追加的なDNA領域集団の存在下で、関心のある遺伝子を特定サイズの断片に限定することを伴う。次に断片のプールを変性させ再アニールして、変異遺伝子を作り出す。次に変異した遺伝子を変更した活性についてスクリーニングする。
【0114】
野生型B12依存性デヒドラターゼ配列を変異させて、この方法によって変更されまたは増強される活性についてスクリーニングしても良い。配列は二本鎖であるべきで、50bp〜10kB範囲の様々な長さであることができる。配列は技術分野で良く知られている制限エンドヌクレアーゼを使用して、約10bp〜1000bpの範囲の断片に無作為に消化されても良い(マニアティス(Maniatis)、同上)。完全長配列に加えて、配列の全部(または部分)とハイブリッド形成可能な断片集団を添加しても良い。同様に、野生型配列とハイブリッド形成可能でない断片集団を添加しても良い。典型的にこれらの追加的断片集団は、総核酸と比較して重量で約10倍〜20倍過剰に添加される。一般にこのプロセスは、混合物中に約100〜1000個の異なる特定の核酸断片の生成を可能にする。無作為核酸断片の混合集団は、変性されて一本鎖核酸断片を形成し、次に再アニールされる。その他の一本鎖核酸断片との相同性領域を有する一本鎖核酸断片のみが再アニールする。無作為核酸断片は、加熱によって変性されても良い。当業者は、二本鎖核酸を完全に変性するのに必要な条件を判定できる。好ましくは、温度は、約80℃〜100℃である。核酸断片を冷却によって再アニールしても良い。好ましくは、温度は約20℃〜75℃である。再生は、ポリエチレングリコール(「PEG」)または塩の添加によって加速できる。塩濃度は、好ましくは0mM〜200mMである。アニールされた核酸断片は、次に核酸ポリメラーゼおよびdNTP(すなわちdATP、dCTP、dGTPおよびdTTP)存在下でインキュベートされる。核酸ポリメラーゼは、クレノウ断片、Taqポリメラーゼまたは技術分野で知られているあらゆるその他のDNAポリメラーゼであっても良い。アニールに先だって、アニールと同時に、またはアニール後に、ポリメラーゼを無作為核酸断片に添加しても良い。変性、再生、およびポリメラーゼ存在下でのインキュベーションのサイクルは、所望の回数反復される。好ましくはサイクルは2〜50回反復され、より好ましくは、手順は10〜40回反復される。得られる核酸は約50bp〜約100kBのより大きな二本鎖ポリヌクレオチドであり、標準クローニングおよび発現プロトコルによって、発現および変更された活性についてスクリーニングしても良い(マニアティス(Maniatis)、同上)。
【0115】
上で例証した方法(B12依存性デヒドラターゼをコードする遺伝子を直接に変異誘発するようにデザインされる)に加えて、変異株を作り出す伝統的方法をここで述べられる目的のために使用できる。例えばB12依存性デヒドラターゼ活性を有する野生型細胞を放射線または化学変異原などの多様な作用物質に暴露して、次に所望の表現型についてスクリーニングしても良い。放射線によって変異を作り出す場合、紫外線(UV)または電離放射線のどちらを使用しても良い。遺伝的変異に適した短波長UVの波長は200nm〜300nmの範囲内であり、254nmが好ましい。この波長内のUV放射線は、主にグアニジンおよびシトシンからアデニンおよびチミジンへの核酸配列内変化を引き起こす。全ての細胞は、ほとんどのUV誘導される変異を修復するDNA修復機序を有するので、カフェインおよびその他の阻害物質などの作用物質を添加して修復プロセスを妨害し、いくつかの効果的な変異を最大化しても良い。300nm〜400nm範囲の光を使用する長波UV変異もまた可能であるが、この範囲は、一般に、DNAと相互作用する様々な賦活物質(ソラレン染料など)との組み合わせで使用しない限り、短波長UV光ほど効果的でない。同様に化学作用物質による変異誘発も変異株を生成するのに効果的であり、一般に使用される物質としては、複製しないDNAに影響する化学物質(HNO2およびNH2OHなど)、ならびに複製するDNAに影響する作用物質(フレームシフト変異を引き起こすことで注目すべきアクリジン染料など)が挙げられる。放射線または化学作用物質を使用して変異株を作り出す特定の方法については、技術分野において文書で十分に立証されている。例えばトーマスD.ブロック(Thomas D.Brock)著、生物工学:工業微生物学テキスト(Biotechnology:A Textbook of Industrial Microbilogy)、第二版、Sinauer Associates:マサチューセッツ州サンダーランド(Sunderland、MA)(1989)、またはデシュパンデ・ムクンド(Deshpande,Mukund)V.、Appl.Biochem.Biotechnol.、36:227(1992)を参照されたい。
【0116】
変異誘発方法にかかわらず、酵素がグリセロールおよび1,3−プロパンジオール存在下で増大した総代謝回転数を有するように、遺伝子を発達させても良い。これはkcatの増大および/または酵素不活性化速度の低下のどちらかによって達成できる。
【0117】
不対プライマーを使用した遺伝子組み換え発生伸張法を使用したB12依存性デヒドラターゼの変異誘発
図5は、2個の遺伝子の遺伝子組み換えに基づく、不対プライマーを使用した遺伝子組み換え発生伸張法の原理を示す。この方法は、親遺伝子がそれらの5’および3’末端に異なるDNA配列を有することを必要とする。親遺伝子が、同一の5’および3’配列を有する場合、短い側方DNA断片は、(図5のステップAで示されるように)標準PCRによって遺伝子の5’または3’末端に付着されていなくてはならない。
【0118】
次に熱サイクルのための2個の不対プライマーのデザインに続いて、PCR生成物を遺伝子組み換え発生伸張法のテンプレーとして使用できる。プライマー−1はテンプレート−1の5’末端とアニールするが、テンプレート−2の5’末端とは結合しない。プライマー−2はテンプレート−2の3’末端とアニールするが、テンプレート−1の3’末端とは結合しない(ステップB)。これによってどちらの親テンプレートも熱サイクル反応によって増幅されないことが確実になる。
【0119】
短いアニールおよび合成サイクルを実施することで、一連の短いDNA断片を作り出す(ステップC)。十分な相同性があれば、これらのDNA断片のいくつかは引き続くアニールサイクルで、異なるテンプレートとアニールする(すなわち「テンプレート切り替え」)(ステップD)。引き続いて図5で示されるように、組み換えDNA断片が作られる。最後にテンプレート−1の5’末端およびテンプレート−2の3’末端を有する組み換えDNA遺伝子が作り出される(ステップE)。
【0120】
この時点で、これまでの不対プライマー−1およびプライマー−2は、新たに作り出された組み換え遺伝子のための対合プライマーになる。さらなるアニールおよび合成サイクルによって、テンプレート−1の5’末端およびテンプレート−2の3’末端を有する組み換えDNA遺伝子のプールを増幅する(図5のステップF)。増幅ステップ中に、組み換えDNA遺伝子をさらに遺伝子組み換えすることができ、それによって組み換えDNA遺伝子の交差数が増加する。理論的には、全ての増幅された生成物が組み換えDNA生成物であるべきである。これらの組み換え生成物は親テンプレート分子から直接誘導でき、あるいは追加的変異(例えば挿入、欠失、および置換)が最終遺伝子組み換え産生物中に組み込まれても良い。最終反応混合物中への親テンプレートの混入はごくわずかである。
【0121】
この方法論についてさらに詳しくは、参照によって本明細書に援用する米国特許出願第0/374366号明細書で開示される。
【0122】
改善された速度性能を有するB12依存性デヒドラターゼ変異体の同定
改善された速度性能を有するB12依存性デヒドラターゼ変異体を(大集団から)同定するために、ハイスループットアッセイの開発は、スクリーニングを極めて容易にする。2回の測定に依存して、標準(例えば野生型GDH)と比較したkcatの改善および/またはデヒドラターゼ変異体の総代謝回転数の推定を提供する、単純なスクリーニング方法がここで開示される。具体的には、GDH変異遺伝子ライブラリーを標準方法によってGDH発現について大腸菌(E.coli)中にクローン化し、単離株を得てレノックス・ブロス中で培養し、透過化処理して、次にGDH活性についてアッセイした。スクリーニング方法はホールセルに含有されるGDHについて述べられるが、当業者は方法を容易に変更して、粗製または精製調製品中の酵素に適用できることを認識するであろう。
【0123】
ここで開発されたGDHアッセイは、補酵素B12、デヒドラターゼ再活性化因子、またはB12依存性デヒドラターゼ供給源を有さない細胞における、アポ酵素形態のGDH酵素の存在に依存する。触媒的に活性のGDHホロ酵素は、培養液への補酵素B12の添加によってのみ形成する。したがってGDHの反応は、補酵素B12および基質グリセロールの添加によって、高精度で効果的にスイッチ「オン」できる。これによってGDH活性の持続時間(タイミング)の正確な制御が可能になり、したがって正確なGDH活性の測定が可能になる。反応は0時(時間=t0)に開始され、反応の初期相中(t0の直後、および酵素不活性化が起きる前、時間=t1)の生成物形成が測定される。さらに生成物形成が、さらにより長時間測定される(t0の直後、および酵素不活性化完了後、時間=t2)。生成物(3−HP)形成の判定は、比色アルデヒド分析に基づく(ズーレック(Zurek)G.、およびU.カルスト(Karst)、Analytica Chimica Acta、351:247〜257(1997))。
【0124】
飽和濃度の補酵素B12およびグリセロール存在下のGDHでは、経時的に生成する3−HPの量(y)は、次によって推定される。
y=T0+AMP(1-exp(−k観察された不活性*時間))
式中、T0はt0時の3−HPの量(バックグラウンド)であり、AMPはt0からGDHが完全に不活性化されるまでの間に生成する3−HPの量(総代謝回転数)であり、k観察された不活性は、観察された一次不活性化速度定数である。1,3−プロパンジオールが存在しないので、k観察された不活性は、全てk不活性グリセロール(グリセロールに起因する一次不活性化速度定数)に起因する(図1上方の曲線を参照されたい)。AMPは、[GDH]*kcat/k観察された不活性に等しい。経時変化を使用して、固定濃度のGDH、補酵素B12、およびグリセロールのための適切な時間(t1およびt2)を確立する。T0について補正した後、t1で形成した生成物量(T1、酵素不活性化が起きる前)を使用して[GDH]*kcatを推定し、t2で形成した生成物(T2、酵素不活性化の完了後)を使用して、総代謝回転数および[GDH]*kcat/k観察された不活性を推定する。したがってT1値はkcatに正比例し、T2値はkcat/k観察された不活性に正比例し、T2/T1は1/k観察された不活性を与える。
【0125】
適切な時間(t1およびt2)を(1,3−プロパンジオール不在下で)固定濃度の野生型GDH、補酵素B12およびグリセロールについて判定した後、T1およびT2の値を同一アッセイ条件下で、しかし1,3−プロパンジオールを添加して再度測定した(図1下方の曲線)。グリセロールおよび1,3−プロパンジオール存在下では競合的阻害が起き、k観察された不活性はkgly(グリセロールに起因する一次不活性化速度定数)およびk1,3−プロパンジオール(1,3−プロパンジオールに起因する一次不活性化速度定数)の双方の関数である。適切な1,3−プロパンジオール濃度において、T1値は1,3−プロパンジオール不在下での値(1,3−プロパンジオールおよびグリセロールの競合的阻害を反映する)から測定可能に低下し、T2値は1,3−プロパンジオール不在下での値(k1,3−プロパンジオール>kglyと言う事実を反映する)から大幅に低下した。二点アッセイでは、グリセロールは、5〜50mM、好ましくは10mMの濃度で存在し、1,3−プロパンジオールは10〜300mM、好ましくは50mMの濃度で存在する。
【0126】
出願人らは、グリセロールおよび1,3−プロパンジオール存在下で、野生型GDH−対−変異体株を調べる条件を確立した。顕微鏡的レベルでは、GDHへの変異の導入は、グリセロールおよび/または1,3−プロパンジオールに対する酵素親和力kcat、ならびにそれぞれのk不活性値に影響するかもしれない。最初は親和性および速度定数が互いに非依存に変動するという保証がなかったが、これらのパラメーターのバリエーションが、10mMのグリセロールおよび50mMの1,3−プロパンジオール存在下で実施された二点アッセイ(T1およびT2)の結果にいかに影響できるかを考えることは有用であった。具体的には、緩慢な不活性化(k観察された不活性の低下)は、増大するT2/T1比率をもたらす。他方、T1またはT2いずれかのバリエーションは、酵素の固有の特性の変化に起因することができる。例えばグリセロールおよび1,3−プロパンジオールの相対親和性が一定で、kcatおよびk不活性値を比例して低下させると、T2/T1は増大するがT1は低くなる。他方、グリセロールとの正常な相互作用を有するが、1,3−プロパンジオールへの低下した相対親和力、または1,3−プロパンジオール不活性化のより低い速度定数を有する変異株は、ほぼ正常なT1、上昇したT2、およびより高いT2/T1比率を示すべきである。正常なグリセロールおよび1,3−プロパンジオール親和性を有するが、どちらかの化合物に対する低下したk不活性を有する変異株もまた、正常なT1を示し、増大するT2およびT2/T1比率を示すべきである。k不活性における、あるいは基質または1,3−プロパンジオール親和力における変化がないkcatの増大は、T1およびT2を増大させるが、T2/T1比率は増大させない。上述の多数のバリエーションにもかかわらず、ここでの研究の目的は、グリセロールおよび1,3−プロパンジオール存在下で、GDH酵素の総代謝回転数を増大させることである。これはkcatの増大および/または酵素不活性化速度の低下のどちらかによって達成できる。したがって野生型と比較して、より高いT1および/またはより高いT2および/またはより高いT2/T1値、またはこれらのパラメーターのあらゆる組み合わせより成る群から選択される少なくとも1つの改善を示す変異株は、改善された特性を有すると見なされる。
【0127】
1,3−プロパンジオール濃度と比べてグリセロール濃度が低い場合、(kcatの推定のための)T1の測定は可能でないので、1,3−プロパンジオールによる顕著な不活性化が起きる。1,3−プロパンジオール生成プロセスにおいては高い1,3−プロパンジオール濃度および低いグリセロール濃度が望ましいので、これらの条件は妥当である。問題のこの側面を認識して、グリセロールがxx〜yymM、好ましくは10mMの濃度で存在し、1,3−プロパンジオールが300mMを超え、好ましくは600mMの濃度で存在すること以外は、上述のようにして一点アッセイが提供される。この一点アッセイでは、時間がt終点によって示され、3−HPの生成量がT(XXX)によって示され、XXXは1,3−プロパンジオールの濃度(mM)である。
【0128】
補酵素B12不活性化に影響する重大なアミノ酸の同定
出願人らは、野生型遺伝子と比べて、低下した補酵素B12不活性化速度を有する多様な変異株GDH酵素を開示する。これらの変異株は、上述の変異誘発およびスクリーニング方法を使用して同定された。変異株、変更アミノ酸残基、1/k観察された不活性活性(T2/T1として測定される)、およびT2を表1にまとめる。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に掲載する。
【0129】
【表5】
【0130】
【表6】
【0131】
【表7】
【0132】
【表8】
【0133】
【表9】
【0134】
【表10】
【0135】
上述の変異誘発およびスクリーニング方法を使用して同定された追加的な変異株を表4にまとめる。この表は各変異株、変更アミノ酸残基、およびT(600)活性に関する情報を提示する。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に掲載する。
【0136】
【表11】
【0137】
【表12】
【0138】
低下した補酵素B12−不活性化速度を有する多様な変異株GDH酵素が上で開示される。本発明の好ましい変異株としては、配列番号:40、44、48、52、56、60、64、68、72、76、80、84、88、92、96、100、104、108、112、116、120、124、128、132、136、140、143、147、151、155、159、163、167、171、175、179、182、186、189、192、195、198、201、204、208、212、215、218、221、225、229、233、237、241、245、249、253、257、261、265、269、273、277、281、285、289、293、297、301、304、307、310、313、316、319、322、325、328、331、334、および337が挙げられる。本発明のより好ましい変異株は配列番号:140、179、186、189、192、195、198、201、212、215、218、301、304、307、310、313、316、319、322、325、328、331、334、および337である。本発明の最も好ましい変異株は、配列番号:313、322、および328である。
【0139】
上の変異株に加えて、それぞれが表2および3に列挙された変異のいくつかの組み合わせをさらに有する、改善された反応速度を有する変異株の大きなプールが合成できる。例えば複数点変異を有する変異株GDHにおける各変異を酵素の全体的反応速度の改善に向けた望ましい効果について評価できる。所望ならば、反応速度を高めなかったあらゆる変異は野生型配列に戻せる。同様に一点変異のみを有する上のリストのGDH変異株を上で開示される別の変異と組み合わせて、酵素の活性をさらに改善することができる。出願人らは、GDH酵素における以下の有用な変異を開示し、これらは多様を組み合わせで使用して野生型遺伝子と比較し、低下した補酵素B12不活性化速度を有する代案の変異株GDH酵素を生成しても良い。これらの変異としては、
1.α−サブユニット中:TCA(α−Ser41)からTCG(Ser)、GTG(α−Val44)からGCG(Ala)、GTG(α−Val44)からGAG(Glu)、GGT(α−Gly47)からGGC(Gly)、CAG(α−Gln59)からCGG(Arg)、ATG(α−Met62)からGTG(Val)、ATG(α−Met62)からACG(Thr)、ATG(α−Met62)からCTG(Leu)、ATC(α−Ile63)からGTC(Val)、GGG(α−Gly63)からGGA(Gly)、CGA(α−Arg65)からCAA(Gln)、ATC(α−Ile67)からGTC(Val)、TAC(α−Tyr70)からAAC(Asn)、GTT(α−Val74)からGTC(Val)、GTT(α−Val74)からATT(Ile)、ACG(α−Thr77)からGCG(Ala)、GTG(α−Val86)からGAG(Glu)、CAC(α−His96)からCAT(His)、ATC(α−Ile102)からACC(Thr)、ATC(α−Ile102)からGTC(Val)、ATC(α−Ile105)からATT(Ile)、GTC(α−Val115)からGCC(Ala)、GAG(α−Glu116)からGAA(Glu)、GCG(α−Ala119)からACG(Thr)、GTG(α−Val124)からGCG(Ala)、CGT(α−Arg134)からCGC(Arg)、CGG(α−Arg137)からAGG(Arg)、AAC(α−Asn141)からATC(Ile)、TGC(α−Cys143)からTGT(Cys)、CTC(α−Leu148)からCGC(Arg)、AAA(α−Lys149)からAGA(Arg)、AAA(α−Lys149)からCAA(Gln)、GAT(α−Asp150)からCAT(His)、GAT(α−Asp150)からGAC(Asp)、AAT(α−Asn151)からAAC(Asn)、CCG(α−Pro152)からCCC(Pro)、TCA(α−Ser168)からCCA(Pro)、TGC(α−Cys193)からAGC(Ser)、GAG(α−Glu209)からGAA(Glu)、ATG(α−Met214)からTTG(Leu)、GGC(α−Gly216)からGGG(Gly)、TTA(α−Leu217)からGTA(Val)、AGC(α−Ser219)からAAC(Asn)、GTG(α−Val224)からCTG(Leu)、GTG(α−Val224)からATG(Met)、GTG(α−Val224)からTTG(Leu)、GTC(α−Val226)からGCC(Ala)、GCG(α−Ala231)からACG(Thr)、TTT(α−Phe233)からCTT(Leu)、GGC(α−Gly236)からAGC(Ser)、GAG(α−Glu240)からGAA(Glu)、CAG(α−Gln242)からCAA(Gln)、ATG(α−Met257)からGTG(Val)、ATG(α−Met257)からACG(Thr)、CTG(α−Leu268)からCTA(Leu)、TAT(α−Tyr271)からTGT(Cys)、ACT(α−Asn288)からACC(Asn)、GTG(α−Val301)からGTA(Val)、ATG(α−Met306)からCTG(Leu)、ATG(α−Met306)からTTG(Leu)、GCT(α−Ala309)からGCC(Ala)、ATT(α−Ile314)からGTT(Val)、CTG(α−Leu318)からTTG(Leu)、CAG(α−Gln337)からCAA(Gln)、TTC(α−Phe339)からGTC(Val)、CGC(α−Arg346)からCGG(Arg)、ACC(α−Thr350)からGCC(Ala)、GCC(α−Ala376)からGCT(Ala)、GTT(α−Val423)からATT(Ile)、CGC(α−Arg425)からCGT(Arg)、CCG(α−Pro430)からTCG(Ser)、AAC(α−Asn447)からAAT(Asn)、CCG(α−Pro450)からCCA(Pro)、GCG(α−Ala460)からGCA(Ala)、GTG(α−Val461)からGGG(Gly)、GAA(α−Glu462)からGAG(Glu)、AAC(α−Asn468)からAAT(Asn)、ACC(α−Thr470)からGCC(Ala)、AGC(α−Ser481)からAGT(Ser)、AAT(α−Asn489)からAGT(Ser)、ACC(α−Thr499)からGCC(Ala)、TAC(α−Tyr502)からCAC(His)、CTC(α−Leu509)からTTC(Phe)、CTC(α−Leu509)からTTT(Phe)、TTC(α−Phe513)からCTC(Leu)、AAC(α−Asn520)からAGC(Ser)、CGC(α−Arg533)からGGC(Gly)、GTT(α−Val549)からGCT(Ala)、ACC(α−Thr553)からACG(Thr)、TAA(αの停止)からCAA(Gln)、TAA(αの停止)からGAA(Glu)、
2.β−サブユニット中:CAA(β−Gln2)からCGA(Arg);TTT(β−Phe11)からTTA(Leu)、TTT(β−Phe11)からTTC(Phe)、CTG(β−Leu13)からCCG(Pro)、AAAβ−Lys14)からAGA(Arg)、GGG(β−Gly19)からGAG(Glu)、GAT(β−Asp24からGGT(Gly)、GAT(β−Asp24)からGAA(Glu)、GAA(β−Glu25)からGAG(Glu)、GCC(β−Ala27)からTCC(Ser)、GAA(β−Glu29)からGAG(Glu)、ACT(β−Thr45)からGCT(Ala)、GCG(β−Ala53)からGTG(Val)、AAA(β−Lys56)からAGA(Arg)、CTG(β−Leu58)からCTT(Leu)、GAA(β−Glu64)からGAG(Glu)、CTT(β−Leu67)からCTC(Leu)、CGG(β−Arg70)からCGA(Arg)、GCC(β−Ala88)からGCT(Ala)、GAT(β−Asp111)からGAA(Glu)、CTG(β−Leu113)からCCG(Pro)、TCT(β−Ser122)からCCC(Pro)、GAG(β−Glu130)からGGG(Gly)、CCG(β−Pro152)からACG(Thr)、CCG(β−Pro152)からTCG(Ser)、AAC(β−Asn155)からAGC(Ser)、AAC(β−Asn155)からAAG(Lys)、AAA(β−Lys166)からAGA(Arg)、AAA(β−Lys173)からGAA(Glu)、GAC(β−Asp181)からGGC(Gly)、CCC(β−Pro184)からCCT(Pro)、および
3.γ−サブユニット中:AAA(γ−Lys4)からAAG(Lys)、ATC(γ−Ile21)からACC(Thr)、AAA(γ−Lys27)からAGA(Arg)、GAG(γ−Glu35)からAAG(Lys)、ATC(γ−Ile49)からACC(Thr)、ACC(γ−Thr53)からGCC(Ala)、ACC(γ−Thr53)からTCC(Ser)、ACC(γ−Thr53)からTGT(Cys)、CAT(γ−His67)からTAT(Tyr)、AAT(γ−Asn72)からAGT(Ser)、CAG(γ−Gln101)からCGG(Arg)、ACC(γ−Thr114)からTCC(Ser)、ACC(γ−Thr114)からGCC(Ala)、GCC(γ−Ala122)からGTC(Val)、GCG(γ−Ala128)からGTG(Val)、およびCTG(γ−Leu137)からCTA(Leu)が挙げられる。
【0140】
同様のヌクレオチドおよびアミノ酸変異は、野生型GDH以外のその他のB12依存性デヒドラターゼ中で作ることができる。関心のあるB12依存性デヒドラターゼとGDHを並べることで、変異を必要とする正確なヌクレオチド/塩基の位置が示される。
【0141】
本発明は、上述の特定の変異のみならず化学的に同等のアミノ酸の置換を許すものも包含する。したがって例えばアミノ酸が脂肪族非極性アミノ酸であるアラニンによって置換されると、同一部位が、化学的に同等のアミノ酸であるセリンによって置換されても良いことが期待される。
【0142】
デヒドラターゼのタンパク質工学技術
今や、遺伝的工学技術および分子グラフィックスの方法、すなわちタンパク質工学技術によって、三次元構造情報と古典的タンパク質化学を組み合わせることで、タンパク質の多くの特性の変性を試みることが可能である。変更された活性を有する酵素を得るこのアプローチは、最初にモデル分子の生成、または未知の構築と同様の配列を有する既知の構築の使用に依存する。
【0143】
ここでの目的では、基質フリー形態のB12依存性グリセロールデヒドラターゼの三次元結晶構造がX線結晶構造解析によってあらかじめ求められ(リャオ(Liao)ら、J.Inorganic Biochem.(近刊))、さらに1,2−プロパンジオールとの複合体中の酵素の構造も報告されている(ヤマニシ(Yamanishi)ら、Eur.J.Biochem.、269:4484〜4494(2002))。これらのB12依存性グリセロールデヒドラターゼの三次元モデル、および変異の「ホットスポット」が反応速度において改善された活性を示す(自殺不活性化速度が低下した)これらのデヒドラターゼ酵素内のどこに典型的に位置するのかという理解から、デヒドラターゼ構造内の領域を標的にすることができ、そこでは構造の代案の変更がタンパク質の特性に所望の変化をもたらすかもしれない。したがって例えば以下の野生型残基を標的とした領域的部位−誘導変異誘発からは、改善された触媒活性を有する追加的なデヒドラターゼ変異株が生じることが期待される。
1)残基62〜70(α−サブユニットのN−末端から2番目のαらせんを包含する)および/または
2)残基219〜236(活性部位近くの領域、TIMバレルの4番目のβ−鎖の部分と、続くα−サブユニットのループおよび短いらせんを包含する)。
【0144】
宿主細胞における組み換えデヒドラターゼの発現
デヒドラターゼをコードする遺伝子の発現による3−HPおよび1,3−プロパンジオールの組み換え生成のための適切な宿主細胞は、原核生物または真核生物のどちらであっても良く、活性酵素を発現するそれらの能力によってのみ制限される。好ましい宿主は、典型的にシトロバクター(Citrobacter)、エンテロバクター(Enterobacter)、クロストリジウム(Clostridium)、クレブシエラ(Klebsiella)、アエロバクター(Aerobacter)、ラクトバシラス(Lactobacillu)、アスペルギルス(Aspergillus)、サッカロミセス(Saccharomyces)、分裂酵母(Schizosaccharomyces)、チゴサッカロミセス(Zygosaccharomyces)、ピチア(Pichia)、クリヴェロミセス(Kluyveromyces)、カンジダ(Candida)、ハンゼヌラ(Hansenula)、デバリオミセス(Debaryomyces)、ケカビ(Mucor)、トルロプシス(Torulopsis)、メチロバクター(Methylobacter)、エシェリキア(Escherichia)、サルモネラ(Salmonella)、バシラス(Bacillus)、ストレプトマイセス(Streptomyces)およびシュードモナス(Pseudomonas)などの3−HPまたは1,3−プロパンジオールの生成に有用なものである。本発明でより好ましいのは、大腸菌(Escherichia coli)、E.ブラタエ(E.blattae)、クレブシエラ(Klebsiella)、シトロバクター(Citrobacter)、およびアエロバクター(Aerobacter)である。
【0145】
宿主細胞中に適切なデヒドラターゼをコードする遺伝子のクローニング、形質転換および発現に適したベクター、形質転換方法、および発現カセットは、当業者には良く知られている。適切なベクターは、宿主細胞として用いられる細菌と適合性のものである。したがって適切なベクターは、例えば細菌、ウイルス(例えばバクテリオファージT7またはM−13由来ファージなど)、コスミド、酵母または植物から誘導できる。このようなベクターを得て使用するプロトコルは、技術分野で知られている(サムブルック(Sambrook)ら、同上)。
【0146】
典型的にベクターまたはカセットは、関連性のある遺伝子の転写および翻訳を導く配列、選択可能マーカー、および自律的複製または染色体組み込みができるようにする配列を含有する。適切なベクターは、転写イニシエーション制御を含む遺伝子の5’領域、および転写終結を制御するDNA断片の3’領域を含んでなる。双方の制御領域が、形質転換宿主細胞に相同的な遺伝子から誘導されることが最も好ましいが、このような制御領域は、必ずしも産生宿主として選択された特定種に固有の遺伝子から誘導されなくて良いものと理解される。
【0147】
所望の宿主細胞中で本発明の当該遺伝子の発現を推進するのに有用なイニシエーション制御領域(またはプロモーター)は多数あり、当業者にはなじみが深い。CYC1、HIS3、GAL1、GAL10、ADH1、PGK、PHO5、GAPDH、ADC1、TRP1、URA3、LEU2、ENO、TPI(サッカロミセス(Saccharomyces)中での発現に有用)、AOX1(ピチア中での発現に有用)、およびlac、trp、λPL、λPR、T7、tac、およびtrc(大腸菌(E.coli)中での発現に有用)をはじめとするが、これに限定されるものではない、これらの遺伝子を推進できる実質的にあらゆるプロモーターが本発明のために適切である。
【0148】
終結制御領域はまた、好ましい宿主に固有の様々な遺伝子から誘導されても良い。場合により、終結部位は必要ないかもしれないが、含まれることが最も好ましい。
【0149】
ひとたび適切なカセットが構築されると、それらは適切な宿主細胞を形質転換するために使用される。変異株デヒドラターゼをコードする遺伝子を含有するカセットの宿主細胞内への導入は、形質転換(例えばカルシウム透過化処理細胞、電気穿孔法を使用する)によって、あるいは組換え型ファージウイルスを使用した形質移入などの既知の手順によって実行しても良い(サムブルック(Sambrook)ら、同上)。
【0150】
発酵を通じた工業生産
本発明で使用するための発酵培養液は、適切な炭素基質を含有しなくてはならない。適切な基質は発酵科学の当業者に良く知られている。好ましい炭素基質は、グリセロール、ジヒドロキシアセトン、単糖類、オリゴ糖、多糖類、および一炭素基質である。より好ましいのは、糖(例えばグルコース、フルクトース、スクロース)および一炭素基質(例えばメタノールおよび二酸化炭素)である。最も好ましいのはグルコースである。
【0151】
適切な炭素供給源に加えて、発酵培養液は、培養の生育と3−HPおよび1,3−プロパンジオール生成に必要な酵素的経路の促進に適した、当業者に知られている適切なミネラル、塩、補助因子、緩衝液、およびその他の構成要素を含有しなくてはならない。Co(II)塩および/またはビタミンB12またはそれらの前駆物質に特に注意が払われる。
【0152】
典型的に細胞は、30℃において適切な培養液中で培養された。本発明の好ましい生育培養液は、ルリア−ベルターニ(Luria Bertani)(LB)ブロス、サブロー・デキストロース(Sabouraud Dextrose)(SD)ブロス、またはイースト・モルト抽出物(YM)ブロスなどの一般的な商業的に調製された培養液である。その他の特定または合成培養液を使用しても良く、特定微生物の生育に適した培養液は、微生物または発酵科学の当業者には既知である。
【0153】
バッチ、流加バッチ、または連続プロセスを使用して本発明を実施すること、およびあらゆる既知の発酵様式が適することが考察された(ブロック(Brock)ら、同上によって多様な方法が詳述されている)。さらに細胞を基材上にホールセル触媒として固定し、3−HPまたは1,3−プロパンジオール生産の発酵条件に曝すことが考察された。
【0154】
改善されたB12依存性デヒドラターゼ変異株は、使用する炭素基質とは無関係に、多様なプロセスにおいて3−HPおよび1,3−プロパンジオールの生成のために有用であることが期待される(米国特許第5686276号明細書、グリセロールから3−HPAへ)。当該株の組み換えは、参照によって本明細書に援用する米国特許出願第10/420587号明細書(デュポン(DuPont)2002)のプラスミドで述べられるデヒドラターゼからスワップアウトできる。
【0155】
以下の実施例で本発明をさらに明らかにする。これらの実施例は、本発明の好ましい実施態様を示しながら、例証のみのために提供されるものと理解される。上記考察およびこれらの実施例から、当業者は本発明の本質的特質を把握でき、その範囲と精神を逸脱することなく、本発明の様々な変化と修正を行って、様々な利用法および条件に適合させることができる。
【実施例】
【0156】
一般方法:
PCR増幅、エンド−およびエキソヌクレアーゼによるDNA修飾(DNAクローニングおよび連結のための所望の末端を生成するため)、および細菌形質転換のために必要とされる手順は、技術分野で良く知られている。ここでは標準分子クローニング技術が使用され、サムブルック(Sambrook)J.、フリッチュ(Fritsch)E.F.、およびマニアティス(Maniatis)T.、「分子クローニング:実験室マニュアル(Molecular Cloning:A Laboratory Manual)」、第二版、コールドスプリングハーバーラボラトリー(Cold Spring Harbor Laboratory)、コールドスプリングハーバー(Cold Spring Harbor)、ニューヨーク(NY)、(1989)(以下マニアティス);シルハビー(Silhavy)T.J.、ベンナン(Bennan)M.L.およびエンクイスト(Enquist)L.W.、遺伝子融合実験(Experiments with Gene Fusions)、(コールドスプリングハーバーラボラトリー(Cold Spring Harbor Laboratory):コールドスプリング(Cold Spring)、ニューヨーク(NY)、1984);およびオースベル(Ausubel)ら、分子生物学現代プロトコル(Current Protocols in Molecular Biology)(Greene Publishing and Wiley−Interscience;1987)で述べられている。
【0157】
細菌培養の維持および生育に適した材料および方法は、技術分野で良く知られている。以下の実施例で使用するのに適した技術は、下に述べられる。
1.)「一般微生物学方法マニュアル(Manual of Methods for General Bacteriology)」、フィリップス・ゲアハルト(Phillipp Gerhardt)、R.G.E.マレー(R.G.E.Murray)、ラルフN.コスティロウ(Ralph N.Costilow)、ユージーンW.ネスター(Eugene W.Nester)、ウィリスA.ウッド(Willis A.Wood)、ノエルR.クリーグ(Noel R.Krieg)、およびG.ブリッグス・フィリップス(G.Briggs Phillips)編、米国微生物学会(American Society for Microbiology)、ワシントンDC(Washington,D.C.)(1994)または
2.)ブロック(Brock)T.D.著、バイオテクノロジー:工業的微生物学テキストブック(Biotechnology:A Textbook of Industrial Microbiology)、第二版、Sinauer Associates:マサチューセッツ州サンダーランド(Sunderland, MA)(1989)。
細菌細胞の生育および維持のために使用される全ての試薬、制限酵素および材料は、特に断りのない限り、ウィスコンシン州ミルウォーキーのアルドリッチ・ケミカルズ(Aldrich Chemicals(Milwaukee、WI))、ミシガン州デトロイトのディフコ・ラボラトリーズ(DIFCO Laboratories(Detroit、MI))、メリーランド州ゲーサーズバーグのギブコ/BRL(GIBCO/BRL(Gaithersburg、MD))、ミズーリ州セントルイスのシグマケミカル(Sigma Chemical Company(St.Louis、MO))またはウィスコンシン州マディソンのプロメガ(Promega(Madison、WI))から得た。PCR反応は、特に断りのない限りカリフォルニア州フォスターシティのPEアプライド・バイオシステムズ(PE Applied Biosystems(Foster City、CA))からのAmplitaqまたはAmplitaq金酵素を使用して、GeneAMP PCRシステム9700上で実施した。サイクル条件および反応は、ここで特に断りのない限り、製造元の説明書に従って標準化した。
【0158】
DNA配列決定反応は、特に断りのない限り、インディアナ州インディアナポリスのロシュ・アプライド・サイエンス(Roche Applied Science(Indianapolis、IN))からのエキスパンド高忠実度PCRシステム(Expand High Fidelity PCR System)を使用して、PEアプライド・バイオシステムズ(PE Applied Biosystems)からのABI 377自動化シーケンサーまたはマサチューセッツ州ウォルサムのMJリサーチ(MJ Research(Waltham、MA))からのPTC−200 DNAエンジン上で実施した。同様に、データはメリーランド州ベセズダのインフォマックス(InforMax,Inc.(Bethesda、MD))からのベクターNTIプログラム、またはウィスコンシン州マディソンのDNAスター(DNASTAR Inc.(Madison、WI))からのDNAスタープログラムを使用して管理した。
【0159】
略語の意味は次のとおり。「sec」は秒を意味し、「min」は分を意味し、「hr」は時間を意味し、「d」は日を意味し、「μL」はマイクロリットルを意味し、「mL」はミリリットルを意味し、「L」はリットルを意味し、「mm」はミリメートルを意味し、「μm」はマイクロメートルを意味し、「nm」はナノメートルを意味し、「mM」はミリモル(millimolar)を意味し、「μM」はマイクロモル(micromolar)を意味し、「nM」はナノモルを意味し、「M」はモル濃度を意味し、「mmol」はミリモル(millimole)を意味し、「μmol」はマイクロモル(micromole)を意味し、「ng」はナノグラムを意味し、「mg」はミリグラムを意味し、「g」はグラムを意味し、「kB」はキロベースを意味し、「mU」はミリ単位を意味し、「U」は単位を意味する。
【0160】
菌株、ベクターおよび培養条件
大腸菌(Escherichia coli)BL21(DE3)細胞を酵素過剰発現のために使用した(シュスターB.(Shuster,B.)およびレティJ.(Retey,J.)、FEBS Lett.349:252〜254(1994))。大腸菌(Escherichia coli)XL1−Blue細胞は、カリフォルニア州ラ・ホーヤのストラタジーン(Stratagene(La Jolla、CA))から購入した。野生型大腸菌(Escherichia coli)5Kは、最初にコネティカット州ニューヘーブンのイェール大学大腸菌遺伝子ストックセンター(Coli Genetic Stock Center)(CGSC#4510;エール大学(Yale University)(コネティカット州ニューヘーブン(New Haven、CT)))から得て、ラムダDE3(5K(DE3))で溶原化した。ベクターpBluescript II SK+はストラタジーン(Stratagene)から購入した。
【0161】
分子生物学的用途のための全てのキットは、特に断りのない限り製造元の説明書に従って使用した。
【0162】
実施例1
「Xba−ライブラリー」の構築:GDHのα−、β−およびγ−サブユニットを標的とする無作為変異誘発
エラープローンPCR増幅を使用して、肺炎桿菌(Klebsiella pneumoniae)dhaB1、dhaB2、およびdhaB3遺伝子を標的とする無作為変異株ライブラリーを作り出した。ライブラリーの代表的な配列分析は、kBあたりほぼ4.2点の変異があることを実証し、酵素活性測定は、ライブラリー中の約15〜25%の変異株が活性であると判定した。
【0163】
エラープローンPCR増幅
エンプタージェ(Emptage)ら(国際公開第01/12833号パンフレット)は、肺炎桿菌(Klebsiella pneumoniae)dhaB1、dhaB2、およびdhaB3遺伝子(配列番号1)を含んでなるプラスミドpDT2の構築について述べる。プラスミドpGD20は、GDH遺伝子の発現をT7プロモーターの制御下におくpBluescript II SK+(ストラタジーン(Stratagene))に、pDT2のHindIII/XbaI断片(dhaB1、dhaB2、およびdhaB3を含有する)を挿入して構築された。
【0164】
GDHの3個の遺伝子全てを標的とする無作為に生成された変異株ライブラリーを作り出した。最初に以下のプライマーを使用して、エラープローンPCRによって、dhaB1(1668bp)、dhaB2(585bp)およびdhaB3(426bp)を含んでなる配列をpGD20から増幅した。DHA−F1(配列番号5)およびDHA−R1(配列番号6)。カリフォルニア州パロアルトのクロンテック(Clontech Laboratories,Inc.(Palo Alto、CA))からのクロンテック(Clontech)変異誘発キットを使用して、エラープローンPCRを実施した。反応混合物は以下から構成された。38μLのPCR等級水、5μLの10×アドバンタック・プラス(AdvanTaq Plus)緩衝液、2μLのMnSO4(8mM)、1μLのdGTP(2mM)、1μLの50×ダイバーシファイdNTPミックス(Diversify dNTP Mix)、1μLプライマー混合物、1μLテンプレートDNA、および1μLのアドバンタック・プラス(AdvanTaq Plus)ポリメラーゼ。製造元の説明書に従って熱サイクル反応を実施した。2.7kBのPCR生成物をHind III/Xba Iで消化して、ライゲーションの準備をした。
【0165】
変異株ライブラリー構築
Hind IIIおよびXba I消化を使用してpGD20コンストラクトからGDHの3個の全遺伝子を含有する全挿入断片を除去できるが、挿入断片サイズはほぼ2.7kBであり、一方ベクターサイズは約2.9kBである。アガロースゲル上でのこれらの2つの断片の分離を容易にするために、Hind III、Xba IおよびPst Iを使用してpGD20を消化した。Pst Iはベクターを切断せず、代わりに挿入断片を3カ所のみで切断し、挿入断片から4個の小さな断片を生じる。したがって消化されたベクターは、アガロースゲル上でその他のDNA断片からの混入なしに、2.9kB周辺で移動する。次にHind III/Xba I/Pst I消化されたベクターとHind III/Xba I−消化されたエラープローンPCR生成物をライゲートする。エタノール沈殿後、ライゲーション混合物を形質転換する準備が整った。
【0166】
ライゲーション混合物の形質転換
T7プロモーターが変異株ライブラリーのために使用されたので、ラムダDE3で溶原化された大腸菌(E.coli)細胞を変異酵素発現のための宿主細胞として使用した。具体的には、5K(DE3)大腸菌(E.coli)株を変異株ライブラリー構築のために使用した。最初に電気穿孔適格性の5K(DE3)細胞を次のようにして作成した。2L滅菌フラスコ内の500mLのLBブロスに、一晩培養した2.5mLの細胞を添加した。OD600が0.5〜0.8に達するまで、培養を振盪機上で37℃でインキュベートした。次に細胞を氷の上で10分間インキュベートし、続いて4℃で10分間遠心分離した。細胞を500mLの氷冷水で1回洗浄した後、細胞を1〜2mLの10%氷冷グリセロールに再懸濁した。滅菌エッペンドルフ管内にアリコート(50μL)を作成し、即座にドライアイス内で凍結した。コンピテントな細胞を−80℃で保存した。
【0167】
形質転換のために、1μLのライゲーション混合物を40μLのコンピテントな細胞に添加し、サンプルを0.1cmのギャップを残して電気穿孔キュベット内に移した。1.7kv/cmの電圧を電気穿孔のために使用した。アンピシリン存在下で細胞をLBプレート上にのせ、37℃で一晩インキュベートした。
【0168】
「Xba」変異株ライブラリーのDNA配列分析
DNA配列決定分析のために、9個の変異株コロニーを無作為にピックアップした。配列決定後、生じた変異数、変異位置、および観察された特定タイプの変異を各変異株について分析した。分析からは、変異株中に全タイプの塩基置換が存在することが明らかになり、特定の変異タイプに対するバイアスの欠如が示唆された。さらに分析された9個の変異株クローン中にただ1個の欠失変異が観察され、塩基挿入変異は同定されなかった。これは変異株ライブラリー中の欠失および挿入の頻度が非常に低いことを示唆した。平均変異速度はkBあたり4.2点の変異であった。GDH活性測定は、ライブラリー中の約15〜25%の変異株が活性であることを示した。
【0169】
実施例2
「Sma−ライブラリー」の構築:GDHのα−サブユニットと、β−サブユニットの一部を標的とする無作為変異誘発
主に肺炎桿菌(Klebsiella pneumoniae)dhaB1遺伝子を標的とする第2の無作為変異株ライブラリーを作成した。ライブラリーの代表的な配列分析は、kBあたりほぼ4.5点の変異があることを実証した。酵素活性測定は、ライブラリー中の約15〜25%の変異株が活性であると判定した。
【0170】
エラープローンPCR増幅および変異株ライブラリー構築
以下のプライマーを使用し、pGD20をテンプレートとして使用したエラープローンPCRによって、dhaB1遺伝子全体およびdhaB2遺伝子のほぼ200bpの部分を増幅した。DHA−F1(配列番号5)およびDHA−R2(配列番号7)。実施例1で述べたようにして、クロンテック(Clontech)変異誘発キットを使用してエラープローンPCR反応を実施した。次に1.9kBのPCR生成物をHind IIIおよびSma Iで消化した。
【0171】
ベクターを調製するために、pGD20プラスミドをHind IIIおよびSma Iで消化して(野生型dhaB1遺伝子と、dhaB2遺伝子の一部を除去して)、次にアガロースゲルから精製した。Hind III/Sma I−消化されたエラープローンPCR生成物とHind III/Sma I−消化したpGD20ベクターをライゲートした。実施例1で変異株ライブラリーの作成について述べたのと同様にして、ライゲーション混合物を電気穿孔によって大腸菌(E.coli)5K(DE3)中に形質転換した。
【0172】
「Sma」変異株ライブラリーのDNA配列分析
10個の変異株コロニーをDNA配列決定分析のために無作為にピックアップし、ライブラリーの完全性を調べた。各変異株を生じた変異数、変異位置、および観察された特定タイプの変異について判定した。これらの結果に基づいて、Sma−ライブラリー中の平均変異速度は、kBあたり4.5点変異と判定された。あらゆる特定の変異タイプについての明らかなバイアスはなく、変異株ライブラリー中の欠失および挿入の頻度は非常に低かった。酵素測定は、ライブラリー中の約15〜25%の変異株が活性であることを明らかにした。
【0173】
実施例3
GDHのα−サブユニットのアミノ酸番号141〜152(「PpuMI−ライブラリー」)、219〜226(「4BR1−ライブラリー」)および330〜342(「RsrII−ライブラリー」)を標的にした領域的無作為変異誘発
GDHの結晶構造(リャオ(Liao)ら、J.Inorganic Biochem.、93(1−2):84〜91(2003);ヤマニシ(Yamanishi)ら、Eur.J.Biochem.269:4484〜4494(2002))に基づいて、GDHのα−サブユニットの以下の領域を領域性無作為変異誘発の標的とした。1)アミノ酸番号141〜152、2)アミノ酸番号219〜226、および3)アミノ酸番号330〜342。これらの各領域は長さがかなり短いので、オリゴ−誘導変異誘発アプローチを使用してこれらの3個の変異株ライブラリーを制作した。これは変異させる各領域の上流または下流のユニークな制限部位に対応するサイレント変異を最初に作り出して、クローニングを容易にする多段階プロセスを伴う。このようにして変性オリゴヌクレオチドプライマーを調製して、PCR反応において使用しα−サブユニットの標的領域で変異誘発した。次にこれらの変異誘発されたPCR断片を大腸菌(E.coli)中にクローン化して、「PpuMI−ライブラリー」、「4BR1−ライブラリー」、および「RsrII−ライブラリー」を作り出した。
【0174】
pGD20中へのサイレント変異の導入
プラスミドpGD20中にユニークな制限部位を作成するために、各標的領域の上流または下流に1個のサイレント変異を生成した。各領域で点変異を作成するために、以下のプライマー対を使用した。各プライマー中の太字の大文字で示すヌクレオチドは、特定の変異が導入される位置を示す。
【0175】
【表13】
【0176】
カリフォルニア州ラ・ホーヤのストラタジーン(Stratagene(La Jolla、CA))からのストラタジーン(Stratagene)クイックチェンジ(QuikChange)部位−誘導変異誘発キットを使用して、製造元の説明書に従って変異誘発実験を実施した。変異誘発に続いて、各変異株クローンからプラスミドを精製した。制限酵素消化と、それに続く直接的DNA配列分析から点変異を確認した。
【0177】
オリゴ−誘導変異誘発
領域性無作為変異株ライブラリーを製造するために、3個の変性オリゴヌクレオチドを合成した(下に示すpGD20RM−F3、TB4B−R1、およびpGD20RM−R4)。大文字で示すヌクレオチドのためのオリゴヌクレオチド合成のために、標準条件を使用した。対照的に太字の小文字で示すヌクレオチドは、合成中に各プライマーの下に示す変性ヌクレオチド混合物を使用した。したがって1〜2点変異は、プライマーの各「変性領域」をもたらすことが予測された。
【0178】
pGD20RM−F3(a.a.141〜a.a.152領域のため−「PpuMIライブラリー」):
5’−GCC CGC AGG ACC CCC TCC aac cag tgc cac gtc acc aat ctc aaa gat aat ccg GTG CAG ATT−3’(配列番号15)
a=2%G、2%C、および2%Tと混合される94%A、
g=2%A、2%C、および2%Tと混合される94%G、
c=2%G、2%A、および2%Tと混合される94%C、
t=2%G、2%C、および2%Aと混合される94%T。
【0179】
TB4B−R1(a.a.219〜a.a.226領域のため−「4BR1ライブラリー」):
5’−GCG TGG GTT AAC cag cta cgc cga gac ggt gtc ggt cta cGG CAC CGA AGC GGT ATT TAC C−3’(配列番号16)
a=2%G、2%C、および2%Tと混合される94%A、
g=2%A、2%C、および2%Tと混合される94%G、
c=2%A、2%T、および2%Gと混合される94%C、
t=2%A、2%G、および2%Cと混合される94%T。
【0180】
pGD20RM−R4(a.a.330〜a.a.342領域のため−「RsrIIライブラリー」):
5’−GGT GCG CGC GGT GCG GCG AAT ATC cga gtg gga gaa agt ctg gtc gtt ggc gga cgc cac ttc GAG GTC GAG−3’(配列番号17)
a=1.66%G、1.66%C、および1.66%Tと混合される95%A、
g=1.66%A、1.66%C、および1.66%Tと混合される95%G、
c=1.66%G、1.66%A、および1.66%Tと混合される95%C、
t=1.66%G、1.66%C、および1.66%Aと混合される95%T。
【0181】
次に高忠実度PCR反応を実施し、以下のプライマー対を使用して変異原性PCR断片を生成する。
●PpuMI−ライブラリー:pGD20RM−F3およびDHA−R2(配列番号15および7)、
●4BR−1ライブラリー:TB4B−R1およびGD−C(配列番号16および14)、
●RsrII−ライブラリー:DHA−F1およびpGD20RM−R4(配列番号5および17)。
【0182】
PCR断片をアガロースゲルから精製し、次にPpuM I/Xba I(PpuMI−ライブラリーのため)、Hpa I/Xba I(4BR−1ライブラリーのため)、およびHind III/Rsr II(RsrII−ライブラリーのため)でそれぞれ消化した。
【0183】
変異株ライブラリー構築
(部位−誘導変異誘発によって、PpuM I、Hpa IまたはRsr IIのユニークな制限酵素消化部位が導入されている)変異pGD20コンストラクトをPpuM I/Xba I、Hpa I/Xba IまたはHind III/Rsr IIでそれぞれ消化した。直線化ベクターをアガロースゲルから精製し、次に制限酵素−消化したPCR生成物にライゲートした。実施例1で述べたようにして、ライゲーション混合物を大腸菌(E.coli)株5K(DE3)中に電気穿孔し、変異株ライブラリーを調製した。
【0184】
領域性ライブラリーのための配列決定分析
各領域性ライブラリーからのDNA配列決定分析のために、9〜10個の変異株コロニーを無作為にピックアップした。PpuMI−ライブラリーでは、変異株あたりの平均変異速度は2.8個の変異(n=9)であった。4BR1−ライブラリー(n=8)では、変異株あたりの平均変異速度は2.0個の変異であった。最後にRsr IIライブラリー(n=10)からの配列決定データからは、変異株あたり2.2個の変異の平均変異速度が明らかになった。3個のライブラリーのいずれにおいても、挿入または欠失のいずれも観察されなかった。全てのタイプの塩基置換が検出されたが、あらゆる特定の変異タイプに対するバイアスの欠如が示唆された。
【0185】
実施例4
スクリーニング・アッセイ開発
B12補酵素および基質(グリセロール)の添加によって、B12依存性デヒドラターゼ反応が手動で「開始」できるスクリーニング・アッセイを開発した。比色分析アルデヒドアッセイによって、デヒドラターゼ反応生成物3−HPを検出する。このアッセイを使用して、グリセロールおよび1,3−プロパンジオール存在下での典型的なB12依存性デヒドラターゼ反応の経時変化に関わる予備的な分析により、反応初期速度v、および観察された酵素不活性化速度k観察された不活性を推定する迅速な理論的技術の開発が可能になった。
【0186】
スクリーニング・アッセイ原理
酵素の補助因子である補酵素B12の供給源を有さなければ、野生型B12依存性デヒドラターゼまたは変異株B12依存性デヒドラターゼのどちらかを発現する培養は、アポ−B12依存性デヒドラターゼを生成する。対照的にホロ酵素は、トルエン−および洗剤−透過化処理されたホールセルに補酵素が添加されれば自然発生的に形成する。したがってこのような細胞に補酵素と基質を添加することで、B12依存性デヒドラターゼ反応を「開始する」ことが可能である。反応生成物である3−HPは、試薬3−メチル−2−ベンゾチアゾリノン(MBTH)および酸化剤である塩化第二鉄を使用して、比色分析アルデヒドアッセイにより検出できる(ズーレック(Zurek)G.、およびカルスト(Karst)U.、Analytica Chimica Acta、351:247〜257(1997))。
【0187】
(1,3−プロパンジオール有りまたは無しで)典型的なGDH反応の経時変化を調べる予備的アッセイ
野生型GDHを生成する細胞を、15%レノックス・ブロス(メリーランド州ゲーサーズバーグのギブコ−BRL(Gibco−BRL(Rockville、MD))からの15容積のレノックス・ブロスを85容積の0.5%NaClで希釈し、続いて滅菌して作成した)中で、ニュージャージー州ニュー・ブランズウィックのニュー・ブランズウィック・サイエンティフィック(New Brunswick Scientific(New Brunswick、NJ))からのイノヴァ(Innova)4300恒温器内で振盪(250rpm)して、37℃で一晩生育させた。細胞を次のようにして透過化処理した。培養(0.3mL)のアリコートをカリフォルニア州フラートンのベックマン−コールター(Beckman−Coulter(Fullerton、CA))からのポリプロピレン深型ウェルプレートの方形ウェル内に分配し、2.5%(v/v)のトリトン(Triton)X−100洗浄剤を含有する10μLのトルエンを各ウェルに入れた。次にプレートをドイツ国シュタウフェンのIKA−ヴェルク(IKA−Werke Gmbh.(Staufen、Germany))からのIKA MTS4振盪機上で10分間、最大速度で振盪した。補酵素B12を保護するため赤色光の下で作業して、5容積の基質溶液を1容積の透過化処理した細胞懸濁液に添加して反応を開始した。基質溶液は、0.1MのK−Hepes(pH8)中に、12mMのグリセロールおよび24μMの補酵素B12を含有した。定刻のインターバルに、反応の12.5μlのアリコートを12.5μlの0.4Mグリシン−HCl(pH2.7)中の3mg/mL MBTHを含有する96−ウェルプレートのウェルに入れた。標本採取の少なくとも20分後に、125μLの10mM HCl中の5.5mM FeCl3をMBTH−含有サンプルのそれぞれに添加した。さらに20分以上してから、カリフォルニア州サニーベールのモレキュラー・デバイシス(Molecular Devices(Sunnyvale、CA))からのスペクトラマックス(Spectramax)160プレートリーダーを使用して、GDH活性に伴う青色を670nmでの吸光度として定量化した。基質溶液を50mMの1,3−プロパンジオールで補って、同様の実験を行った。
【0188】
アッセイ結果の理論的な分析
図1で示されるように経時変化反応からのデータを時間−対−OD670でプロットした。具体的には、図1上方の曲線が、基質とした10mMのグリセロール(Km約0.5mM)とのGDH(室温、0.1M K−HEPES中、pH8)の反応の経時変化を示す。時間と共に不活性化が起きるので、生成物形成速度は迅速に低下する。3個のパラメーターを以下の式に当てはめて、点を通過する理論的曲線が描かれる。
y=T0+amp(1−exp(−k観察された不活性*時間))
これらのパラメーターは、
1.)バックグラウンド(アッセイのt=0(T0))値)、
2.)アッセイ中に生じる制限総吸光度の振幅値(amp)、および
3.)曲率を制御する一次不活性化速度定数(k観察された不活性)である。
フィッティングパラメーターは次のように反応の速度特性に関連する。振幅は観察された酵素不活性化速度(k観察された不活性)によって除された反応初期速度(v)に等しい。初期速度は[GDH][gly]kcat/(Km+[gly])である。アッセイバックグラウンドを差し引くと、さらなる分析は以下を明らかにする。
1.反応中に採取された、vの推定のための初期点(T1)、および振幅のための末期点(T2)の2個のサンプルのみからvおよびk観察された不活性の双方が推定でき、
2.1/k観察された不活性はT2/T1として推定できる。
【0189】
図1下方の曲線は、50mMの1,3−プロパンジオール(Ki約15mM)をアッセイに含めることの影響を示す。反応初期速度は約20%だけ低下したが、不活性化は約3時間早かった。これは、競合的阻害が比較的小さいが、酵素−1,3−プロパンジオール複合体の不活性化の速度定数(k不活性1,3−プロパンジオール)が、酵素−グリセロール複合体のそれ(k不活性グリセロール)を超えるためである。
【0190】
双方の不活性化プロセスを組み込んだ速度スキームを下に示す。
【0191】
【化1】
【0192】
実施例5
二点ハイスループット・スクリーニング・アッセイ
実施例4で述べた方法論に基づいて、ハイスループット・スクリーニング・アッセイ・プロトコルを使用して、実施例1、2、および3で調製した変異株コロニーを調べた。このアッセイでは、アッセイ中に2つの時点で特にGDH反応生成物を測定した。T1はT0の30秒後に測定し、T2はT0の40分後に測定した。
【0193】
0.15mL/ウェルで15%レノックス・ブロスを含有する標準96−ウェルマイクロタイタープレートの94個のウェル内に、プレート培養したライブラリーからのコロニーをピックアップして入れた。残る2個のウェルには、野生型GDHを生成する細胞を接種し、細胞を静止恒温器内で4〜6時間37℃で培養した。代案としては、ピックアップした細胞をスクリーニング前に−80℃で保存することが望ましい場合、保存する前に、グリセロール(10%v/v)を含む培地およびピックアップした細胞を一晩培養した。スクリーニング前に、カリフォルニア州サンディエゴのV&Pサイエンティフィック(V&P Scientific(San Diego、CA))からの96−ピン接種器によって、あらかじめ凍結した細胞をグリセロールなしで15%レノックス・ブロスを含有する新鮮な96−ウェルプレートに移し、6〜16時間37℃で振盪せずに培養した。
【0194】
GDH変異株スクリーニングでは、細胞培養プロトコルは次のとおりであった。カリフォルニア州フラートンのベックマン−コールター(Beckman−Coulter(Fullerton、CA))からのポリプロピレン深型ウェル96−プレートのウェル内に、15%レノックス・ブロス培地を分配した(0.3mL/ウェル)。96ピン−長ピン接種機(V&Pサイエンティフィック(V&P Scientific))を使用して、浅型96−ウェルプレートから深型ウェルへ細胞を接種して、ミネソタ州セントポールの3Mヘルスケア(3M Health Care(St.Paul、MN))からの3インチ幅のミクロポア外科手術用テープによってプレートを覆った。アッセイが完了するまで浅型96−ウェルプレートを4℃で保存し、潜在的に改善されたと同定される系統をさらに調べるために、生細胞の供給源としてそれらを使用した。深型ウェルプレート中の細胞は、37℃で一晩(250rpm)振盪しながら培養した。ニュージャージー州ニュー・ブランズウィックのニュー・ブランズウィック・サイエンティフィック(New Brunswick Scientific(New Brunswick、NJ))からのイノヴァ(Innova)4300恒温器内の空気は、プラスチックトレー内の湿ったスポンジによって加湿した。
【0195】
2.5%(v/v)のトリトン(Triton)X−100洗浄剤を含有する10μLのトルエンを各ウェルに添加して、96−ウェルプレート内で細胞を透過化処理した。次にプレートをドイツ国シュタウフェンのIKA−ヴェルク(IKA−Werke Gmbh.(Staufen、Germany))からのIKA MTS4振盪機上で10分間、最大速度で振盪した。赤色プラスチックフィルムで覆って白色室内光から基質混合物を保護した、ペンシルベニア州フィラデルフィアのローム・アンド・ハース(Rohm&Haas(Philadelphia、PA))からのプレキシグラス(Plexiglas)(登録商標)閉鎖容器内にあるバイオメック(Biomek)2000ロボット(ベックマン−コールター(Beckman−Coulter)を使用して、アリコート(8μL)の透過化処理された細胞を96−ウェル反応プレートに移した。0.1Mカリウム−HEPES緩衝液中(pH8)に24μM補酵素B12、12mMグリセロール、および50mM 1,3−プロパンジオールを含有する40μLの基質混合物(赤色光の下で調製されて、必要になるまでホイルを巻いた容器内に−20℃で保存)をロボットによって細胞に添加して、室温での反応を開始した。基質添加の30秒後、12.5μLの反応のアリコート(T1サンプル)を0.4Mグリシン−HCl(pH2.7)中の12.5μL 3−メチル−2−ベンゾチアゾリノン(MBTH)をウェルが含有する第2のプレートに移した。基質添加のほぼ40分後、MBTHを含有する第3のプレートに、同様の反応のアリコート(T2サンプル)を移した。この移動の少なくとも20分後、125μLの10mM HCl中の5.5mM FeCl3溶液を第2および第3のプレート(MBTHを含有する)の各ウェルに添加した。さらに20〜60分後、カリフォルニア州サニーベールのモレキュラー・デバイシス(Molecular Devices(Sunnyvale、CA))からのスペクトラマックス(Spectramax)160プレートリーダーを使用して、GDH活性に伴う青色を670nmでの吸光度として定量化した。
【0196】
プレートリーダーからのデータをワシントン州レドモンドのマイクロソフト(Microsoft)(登録商標)(Redmond,WA)からの修正したエクセルコンピュータープログラムに移した。このプログラムは、各反応プレートからの第1(T1)および第2(T2)のサンプルをマッチさせ、アウトプットをプレート、プレート内の位置、および各反応におけるT1、T2、およびT2/T1比率を示す結果の表にするように組まれている。T1、T2、またはT2/T1比率において非常に高い値を示すサンプルについてデータを調べた。これは、これらのあらゆるパラメーターでデータをソートするプログラムの能力によって容易になった。最も有望な候補をさらに詳しく調べるために、保留した浅型96−ウェルプレートから画線培養した。
【0197】
実施例6
GDH変異株ライブラリーのスクリーニングおよびポジティブヒットの同定
実施例5で述べた自動化されたハイスループットアッセイを使用して、5個の変異株ライブラリーからのほぼ100,000個の変異株コロニーをスクリーニングした。これらのライブラリーは次を含んだ。
1.)実施例1で述べたように、α−、β−、およびγ−サブユニット(DhaB1、DhaB2、およびDhaB3)を標的にしたXbaライブラリー、
2.)実施例2で述べたように、α−と、β−サブユニットの小さな部分を標的にしたSmaライブラリー、
3.)実施例3で述べたように、α−サブユニットのa.a.141〜a.a.152を標的にしたPpuMIライブラリー、
4.)実施例3で述べたように、α−サブユニットのa.a.219〜a.a.226を標的にした4BR1ライブラリー、および
5.)実施例3で述べたように、α−サブユニットのa.a.330〜a.a.342を標的にしたRsrIIライブラリー。
【0198】
あらゆる個々の単離株は、上述の5個のライブラリーの1つから誘導された。ほとんどの個々の単離株は、ライブラリー供給源を示すラベルとそれに続く数(例えば「Xba3010」)によって一義的に同定された。第1のスクリーニングからの全ての推定上の「ヒット」は、フォローアップアッセイ中で確認した。ほとんどの変異効果は、下で述べるような速度パラメーターに基づいて、4個の大まかなカテゴリーに従って分類された。変異遺伝子の配列分析とそれに続く野生型遺伝子との比較によって、各変異株遺伝子中に存在する特定の点変異の同定が可能になった。
【0199】
変異株ライブラリーのスクリーニングおよびヒットの確認
ほぼ100,000個の変異株の一次スクリーニングに続いて、フォローアップ確認アッセイによって推定上のヒットが確認された。簡単に述べると、各推定上のヒットを8個のウェル中で再アッセイした。それぞれの個々のクローンからの結果を統計学的に分析して、T1(30秒で測定されるアルデヒド量)およびT2(40分で測定されるアルデヒド量)の平均値および標準偏差を得た。これらの結果を野生型酵素と比較した。
【0200】
図2は、y−軸上のOD670nmに対してx−軸上にアッセイ数をプロットする典型的なフォローアップアッセイ結果を示す。Xba3010および野生型クローンに関するT1およびT2でのそれぞれの個々のアッセイ(n=8)からの結果が提示される。平均値は横線として図示され、数的には括弧±標準偏差で提示される。一般に、標準偏差は約10〜15%であった。
【0201】
スクリーニング結果
表6は野生型を超えるT2/T1比率、または野生型を超えるT2のいずれか、またはその双方を有するヒットのフォローアップアッセイ結果をまとめる。T2/T1比率は、1,3−プロパンジオールおよびグリセロール存在下で起きる40分の反応での酵素安定性の指標を提供する。T2は、完全に不活性化される前の酵素の総代謝回転数を示唆する。T2/T1比率が高いほど酵素の安定性が良くなる。したがって改善された安定性を有する変異酵素は、野生型酵素と比較して、グリセロールおよび1,3−プロパンジオール存在下で低下した不活性化速度を有する。
【0202】
表6では、野生型の結果への標準化に続く、フォローアップアッセイからのT1およびT2の平均値が報告される。ほとんどの変異酵素は以下の定義に基づいて、タイプ−1、−2、−3、または-4変異株に分類される。
●タイプ1変異株:野生型と比べてT2/T1比率が改善されている一方、T2値の絶対値が減少している。
●タイプ−2変異株:T2/T1比率およびT2の双方が野生型よりも改善されている一方、T2改善の程度はT2/T1比率の改善に満たない。
●タイプ−3変異株:T2/T1比率およびT2の双方が野生型よりも改善されており、双方の改善程度が同様である。
●タイプ−4変異株:T2/T1比率が野生型に対して同等または低下しているが、T2が改善されている。
【0203】
表6はまた、各変異株において同定された特定の点変異をまとめる。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に掲載する。
【0204】
【表14】
【0205】
【表15】
【0206】
【表16】
【0207】
【表17】
【0208】
配列決定結果に見られるように、ほとんどの変異はアミノ酸置換として同定される(サイレント変異を除く)。しかし変異株の2つ(Sma3002[配列番号140]およびXba3009[配列番号179])は、融合タンパク質である。具体的には、α−サブユニット(dhaB1)をコードする遺伝子の停止コドン(TAA)は、Sma3002およびXba3009中でそれぞれCAA(Gln)またはGAA(Glu)に変化する。この停止コドンと、β−サブユニット(dhaB2)をコードする遺伝子の最初のコドンの間には15 bpがあるので、これらの融合タンパク質のどちらもβ−サブユニット中にフレームシフトを引き起こさなかった。これにより、双方の変異酵素は活性を維持できるようになった。β−サブユニットの最初のコドン(GTG)は、通常fMet−tRNAによって認識される。しかし融合変異株では、このコドンがVal−tRNAによって認識されなくてはならない。Sma3002融合タンパク質は、6個のアミノ酸残基Gln−Gly−Gly−Ile−Pro−Val(配列番号18)からなるリンカーを含有した。Xba3009融合タンパク質では、リンカーはGlu−Gly−Gly−Ile−Pro−Val(配列番号19)であった。
【0209】
実施例7
精製酵素を使用した変異株の生化学分析
各酵素の過剰発現および精製に続いて、5個の変異株および野生型GDHのより詳しい性質決定を実施した。
【0210】
過剰発現および精製
野生型酵素と共にさらに生化学分析するため、Xba3007(タイプ−1、配列番号40)、Sma3002(タイプ−2、配列番号140)、Xba3010(タイプ−3、配列番号175)、Xba3009(タイプ−4、配列番号179)および4BR1001(タイプ−2、配列番号171))を選択した。最初に5K(DE3)大腸菌(E.coli)宿主菌株からプラスミドを精製して、大腸菌(E.coli)BL21(DE3)中に形質転換した。1.0mMのIPTGを添加する前に、細胞をアンピシリン含有LB培地中で0.6〜1.0のOD600に培養した。37℃で3時間の誘導後、遠心分離によって細胞を採集し、20mMのHEPES−KOH緩衝液(pH8.0)で1回洗浄した。細胞ペレットを−80℃で保存した。
【0211】
酵素精製のため、細胞ペレットを最初にカリフォルニア州パロアルトのロシュ(Roche、Palo Alto、CA)からの完全なミニプロテアーゼ阻害剤カクテル錠剤および0.5mMのEDTAを含有する、20mMのHEPES−KOH緩衝液(pH8.0)に再懸濁した。細胞を超音波処理によって破壊し(ブランソン(Branson)モデル450;20%出力、50%パルス、氷浴中4分間)、続いて遠心分離した(40,000×g、30分間、4℃)。透明な上清を遠沈し(110,000×g、4℃、1時間)、(NH4)2SO4を氷上の上清中に緩慢に添加して、溶液を50%飽和とした。溶液を氷上で25分間撹拌し、続いて遠心分離した(40,000×g、30分間、4℃)。ペレットを2mLの泳動用緩衝液(100mMのHEPES−KOH(pH8.2)、100mMの1,2−プロパンジオールおよび1mMのDTT)中に再懸濁し、次に泳動用緩衝液で平衡化したニュージャージー州ピスカタウェイのファーマシア・バイオテック(Pharmacia Biotech(Piscataway、NJ))からの16/60高負荷スーパーデックス(Hiload Superdex)200粒径排除カラムに入れた。
【0212】
流速0.25mL/分の緩衝液によって酵素を溶出し、画分コレクター(3mL/画分)を使用して溶出液を収集した。実施例5で述べたアッセイを使用して画分を酵素活性についてアッセイし、次に活性画分をプールして、マサチューセッツベッドフォードのミリポア(Milipore(Bedford、MA))からのセントリコン(Centricon)YM100を使用して濃縮した。100mMのHEPES−KOH(pH8.2)および1mMのDTTからなる新鮮な泳動用緩衝液を使用して、濃縮された酵素を同一カラムにもう一度通過させた。精製した酵素は、10〜20%の勾配ゲルおよびクマシー・ブルー染色を使用したSDS−PAGE電気泳動による評定で、純度75〜95%であった。
【0213】
変異株GDH酵素の生化学的性質決定:
精製した野生型および変異株GDH酵素を使用した詳細な酵素速度分析を実施した。MBTH−比色分析アルデヒドアッセイ(ズーレック(Zurek)G.、およびカルスト(Karst)U.、Analytica Chimica Acta、351:247〜257(1997))を使用して生成物形成を測定し、酵素活性を求めて、KMおよびV最大はラインウェーバー−バーク・プロットから求めた。kcatをV最大から計算した。求めた酵素のKMおよびkcatを表7に示す。
【0214】
【表18】
【0215】
最後に、各変異酵素の不活性化特性の詳細な分析を実施した。実施例4で述べたようにして、グリセロール不活性化速度定数を測定した。空気不活性化速度定数のために、21μMの補酵素B12を含有する0.1M K−HEPES緩衝液(pH8)で酵素を27μg/mLに希釈し、次に空気中において室温でインキュベートした。総代謝回転数をインキュベーションの0.25、0.5、1、2、5、10、15、20および30分後に測定した。総代謝回転数を測定するために、200mMのグリセロール、24mMの補酵素B12、および0.1MのK−HEPES緩衝液(pH8)を含有する190μLの反応溶液に10μLの酵素溶液を添加して、反応混合物を室温で2時間インキュベートした。MBTH−比色分析アルデヒドアッセイ(ズーレック(Zurek)G.、およびU.(カルスト(Karst)U.、同上)を使用して3−HP濃度を測定することで、総代謝回転数を推定した。時間−対−総代謝回転数でデータをプロットし、A=A0exp(−k空気t)(式中、「A」は総代謝回転数、「A0」は0時の総代謝回転数、「k空気」は空気不活性化速度定数、および「t」は時間である)を使用して、曲線のあてはめにより総代謝回転数を推定した。
【0216】
1,3−プロパンジオール不活性化を測定するために、21μMの補酵素B12および様々な濃度の1,3−プロパンジオール(すなわち、1、20、100および300mM)を含有する0.1M K−HEPES緩衝液(pH8)中で、酵素を27μg/mLに希釈した。空気不活性化速度定数の測定のために使用した方法により、各1,3−プロパンジオール濃度について不活性化速度定数を推定した。不活性化速度定数を1,3−プロパンジオール濃度に対してプロットし、曲線から1,3−プロパンジオールによる最大不活性化速度定数を推定した。1,3−プロパンジオールの解離定数は、不活性化速度定数が最大不活性化速度定数の半分になる1,3−プロパンジオール濃度であった。
【0217】
この不活性化特性の分析結果を下の表8に示す。グリセロールまたは1,3−プロパンジオールまたは光いずれかの不在下、しかし空気(酸素)存在下では、B12依存性デヒドラターゼホロ酵素の不活性化が二相速度で起きる。kglyは、1,3−プロパンジオールなしに10mMのグリセロール存在下で測定される不活性化速度定数である。全ての変異株および野生型デヒドラターゼは二相の空気不活性化を示したので、k空気、Fは急速相のための空気不活性化速度定数であり、k空気、Sは緩慢相の空気不活性化速度定数であり、k1,3−プロパンジオールは1,3−プロパンジオールによる最大不活性化速度定数である。Kdは1,3−プロパンジオールの解離定数である。
【0218】
【表19】
【0219】
実施例8
選択した第一世代変異株の組み合わせによる2回目の変異誘発
1回目の変異誘発から得られた変異株をさらに改善するために、2回目の変異誘発を実施して、いくつかの第一世代変異株からの変異を組み合わせた。
【0220】
第2世代変異株を製造するために、最初に、複数の変異を含有するいくつかの第一世代変異株からのプラスミド(例えばSma3002またはXba3009)を宿主細胞から精製した。次にこれらのプラスミドに、Xba3007、Xba3029または4BR1001で見つかった一点変異を導入して、第二世代変異株2−F4、12−B1、13−B7、および16−H5を生成した。表9はこれらの各変異株、およびそれらを生成するのに使用したプライマーに関する詳細をまとめる。各プライマー中の太字の大文字で示すヌクレオチドは、特定の変異が導入される位置を示す。
【0221】
【表20】
【0222】
カリフォルニア州ラ・ホーヤのストラタジーン(Stratagene(La Jolla、CA))からのストラタジーン(Stratagene)クイックチェンジ(QuikChange)部位−誘導変異誘発キットを使用して、実施例3で述べたようにして変異誘発実験を実施した。
【0223】
実施例5で述べたようにして、各第二世代変異株のT2/T1比率およびT2値を求めた。これらの結果を下の表10に示す。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に掲載する。
【0224】
【表21】
【0225】
実施例9
純粋融合変異株の構築および分析
2個の融合変異株であるXba3009(配列番号179)およびSma3002(配列番号140)については、実施例6で述べた。どちらも融合それ自体に加えてその他の変異を含有した。α−およびβ−融合の効果を調査するために、2個の純粋融合変異株(1E1および20G7)を構築した。
【0226】
純粋融合変異株の構築
α−サブユニット(TAA)の停止コドンをCAA(1E1)またはGAA(22−G7)に変化させた。この修飾は、野生型GDHプラスミド中に一点変異を導入することで達成した。点変異を作成するために、以下の2対のプライマーを使用した。
1−E1変異株のため:
1−E1−F1:5’−gac acc att gaa Caa ggc ggt att cct−3’(配列番号26)
1−E1−R1:5’−agg aat acc gcc ttG ttc aat ggt gtc−3’(配列番号27)
22−G7変異株のため:
22−G7−F1:
5’−ccc gac acc att gaa Gaa ggc ggt att cct gtg−3’(配列番号28)
22−G7−R1:
5’−cac agg aat acc gcc ttC ttc aat ggt gtc ggg−3’(配列番号29)
【0227】
各プライマー中の太字の大文字で示すヌクレオチドは、特定の変異が導入される位置を示す。
【0228】
カリフォルニア州ラ・ホーヤのストラタジーン(Stratagene(La Jolla、CA))からのストラタジーン(Stratagene)クイックチェンジ(QuikChange)部位−誘導変異誘発キットを使用して、実施例3で述べたようにして変異誘発実験を実施した。実施例5で述べたようにして、これらの第二世代変異株のT2/T1比率およびT2値を求めた。これらの結果を下の表11に示す。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に掲載する。
【0229】
【表22】
【0230】
実施例10
変異株Sma3002に見つかった変異の分析
実施例6で同定された第一世代変異株であるSma3002(配列番号140)は、T2/T1比率およびT2値の双方に顕著な改善を示した。それは4個の点変異を含有し、その1つは実施例9で詳細に探求した融合変異を含んだ。これらの変異の影響を個別に調査するため、さらに2個の変異株(α−Y271Cおよびβ−Q2R)を構築した。
【0231】
前に従った方法論を使用して、下に示すようなユニークにデザインされたプライマーを使用して、野生型プラスミドに単一点変異を導入した。前の実施例と同じく、各プライマー中の太字の大文字で示すヌクレオチドは、特定の変異が導入される位置を示す。
【0232】
【表23】
【0233】
カリフォルニア州ラ・ホーヤのストラタジーン(Stratagene(La Jolla、CA))からのストラタジーン(Stratagene)クイックチェンジ(QuikChange)部位−誘導変異誘発キットを使用して、実施例3で述べたようにして変異誘発実験を実施した。実施例5で述べたようにして、これらの変異株のT2/T1比率およびT2値を測定した。表13に結果を示す。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に掲載する。
【0234】
【表24】
【0235】
α−Y271C変異はT2値を低下させるが、T2/T1比率を変化させることができなかった。したがってこの変異は、酵素のkcatを低下させる。別の変異β−Q2Rは、T2またはT2/T1比率のいずれにも顕著に影響しないようである。
【0236】
Sma3002(配列番号140)における3個の非融合変異のいずれかが協力して作用し、1,3−プロパンジオールおよびグリセロール存在下での酵素の安定性を増大させるかどうかを判定するために、3個の追加的な変異株を作成した。これらの各変異株では、野生型DNA配列を単一点変異として導入することで、Sma3002における3個の非融合変異(α−Y271C、α−Y502H、またはβ−Q2R)の1つを除去した。これからSma3002中に存在した元の非融合変異の内2つをそれぞれ含有する、3個のSma3002−由来変異株が得られた。ストラタジーン(Stratagene)クイックチェンジ(QuikChange)部位−誘導変異誘発キットを使用して、実施例3で述べたようにして、Sma3002プラスミド中に単一点変異を導入し、プライマーを下の表14に示す。
【0237】
【表25】
【0238】
実施例5で述べたようにして、これらの変異株のT2/T1比率およびT2値を測定した。表15はこの分析の結果を示す。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に掲載する。
【0239】
【表26】
【0240】
実施例11
いくつかの第一世代変異の第二世代変異株への添加による3回目の変異誘発
ここでT2として報告する値で測定されるGDH酵素の総代謝回転の改善に向けて、先行する実施例でかなりの改善がなされたが、3回目の変異誘発を施した。これらの反応では、T2値の最大の改善を示す2個の第二世代変異株(すなわち1−E1および8−C9)に、第一世代変異株から選択される点変異を導入した。これは最初に、宿主細胞から1−E1および8−C9変異株プラスミドを精製することで達成した。次に下の表16に示すプライマーと、カリフォルニア州ラ・ホーヤのストラタジーン(Stratagene(La Jolla、CA))からのストラタジーン(Stratagene)クイックチェンジ(QuikChange)部位−誘導変異誘発キットを使用して、実施例3で述べたようにして、これらのプラスミドにXba3007、Xba3029、または4BR1001に見つかった単一アミノ酸置換変異を導入した。
【0241】
【表27】
【0242】
これらの第三世代変異株のT2/T1比率およびT2値は、実施例5で述べたようにして測定した。表17は結果を示す。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に掲載する。
【0243】
【表28】
【0244】
全体的に第三世代変異株では、酵素安定性にかなりの改善があった。全ての変異株は、最良の第一世代変異株(すなわち、Sma3002(配列番号140))と比較して改善された安定性(T2/T1比率)および総代謝回転数(T2)を有した。実際、変異株20−B9および21−D10は、これまでに作成した全変異株を超えるT2値を有した。
【0245】
実施例12
いくつかの第二および第三世代変異株の生化学的性質決定
選択された第二世および第三世代変異株をさらに詳しく性質決定するために、実施例4で述べる方法を使用して、10mMのグリセロールおよび50mMの1,3−プロパンジオール存在下でこれらの変異株の不活性化速度定数を求めた。これらの結果を下に示す表18にまとめる。
【0246】
【表29】
【0247】
期待されたように、結果はT2/T1比率の測定と一致した。
【0248】
改善されたGDHの一用途は、1,3−プロパンジオール生物生産であるので、高濃度の1,3−プロパンジオール存在下における変異株の総代謝回転数を求めることが重要である。上述のアッセイは、わずか50mM存在下で実施した。1,3−プロパンジオール生物生産のための発酵の後期では、1,3−プロパンジオール濃度が1Mに達することができる。したがって第2の二点アッセイを実施した。具体的には、いくつかの有望な変異株について、10mMのグリセロールおよび600mMの1,3−プロパンジオール存在下で、T2値も測定した。表19に結果をまとめる。
【0249】
【表30】
【0250】
結果は、T2(50mM)について得られたものと幾分同様であったが、数個の変異株は、予期された以上に良好に機能した。Sma3002のT2(600mM)は野生型酵素のそれよりも3.3倍高かった。各第三世代変異株(15−E4、18−D7、20−B9、および21−D10)は、Sma3002と比較してT2(600mM)にさらなる改善を示した。結果は、より高いT2(600mM)を有するこれらの第三世代変異株が、より高濃度の1,3−プロパンジオール中でより耐性があることを示唆した。これらの変異株は、1,3−プロパンジオール生物生産のために非常に有用であることが期待される。
【0251】
実施例13
高濃度の1,3−プロパンジオール存在下で総酵素代謝回転数を測定するための一点ハイスループット・スクリーニング・アッセイ
1,3−プロパンジオール濃度が約300mMよりも高い場合、反応開始後ほとんど即座にGDHの不活性化が起きる。これらの条下ではT1が正確に測定できないので、実施例4および5で述べたハイスループット・スクリーニング・アッセイを使用して、変異株をスクリーニングすることができない。高濃度の1,3−プロパンジオール存在下での総酵素代謝回転数は、ここで改善することが望ましい重要な酵素速度パラメーターの1つである。この総酵素代謝回転数は、不活性化速度の低下またはkcatの増大のいずれかによって改善できる。高濃度の1,3−プロパンジオール存在下で、改善された総酵素代謝回転数を有する変異株についてスクリーニングするために、修正したハイスループット・スクリーニング・アッセイを開発した。
【0252】
簡単に述べると、実施例5で述べたようにして、変異株細胞を96−ウェルプレート内で培養し透過化処理した。カリフォルニア州フラートンのベックマン−コールター(Beckman−Coulter(Fullerton、CA))からのバイオメック(Biomek)2000ロボットを使用して、アリコート(8μL)の透過化処理された細胞を96−ウェル反応プレートに移した。英国ハンプシャー州ニューミルトンのジェネティクス(Genetix(New Milton、Hampshire、UK))からのQfill2を使用して、0.1Mカリウム−HEPES緩衝液(pH8)中の24μM補酵素B12、12mMグリセロール、および720mMの1,3−プロパンジオールを含有する40μLの基質を細胞に添加して、室温での反応を開始した。プレートを室温でほぼ70分間培養した後、バイオメック(Biomek)2000ロボット(ベックマン−コールター(Beckman−Coulter)を使用して、ウェルが12.5μLの0.4Mグリシン−HCl(pH2.7)中の3−メチル−2−ベンゾチアゾリノン(MBTH)を含有する第2のプレートに、反応の12.5μLのアリコートを移した。70分の反応時間によって総代謝回転数の正確な測定ができるようになる。実施例5で述べたようにして、生成物である3−HPの濃度を求めた。カリフォルニア州サニーベールのモレキュラー・デバイシス(Molecular Devices(Sunnyvale、CA))からのスペクトラマックス(Spectramax)160プレートリーダーを使用して670nmでの吸光度を測定し、総酵素代謝回転数(T(600))を推定した。
【0253】
Qfill2は、基質を15秒以内に1枚の96−ウェルプレートに添加できるので、この一点比色分析アッセイは実行するのがが簡単である。さらにアッセイは実施例4および5で述べたアッセイと比較して、より高いスループット能力を提供できる。野生型GDHおよび異なる酵素速度パラメーターを有するいくつかの変異株について、実施例5および本実施例で実施されたアッセイの比較結果を表20に示す。
【0254】
【表31】
【0255】
これらの結果は、各アッセイが異なる速度パラメーターについてスクリーニングすることを実証する。本実施例で述べられるスクリーニング・アッセイによって同定される変異株は、より高濃度の1,3−プロパンジオールに対してより抵抗性であり、一般に改善された安定性と妥当なkcat値を有する。
【0256】
実施例14
酵素構造と変異の間の相互関係
この実施例では、野生型GDHと比較して増大したT2または増大したT2/T1比率を有する変異の位置をGDHの三次元結晶構築に関して調べた。これによって変異が、改善された反応速度(不活性化速度が低下する)をもたらすことが多い変異「ホットスポット」と見なすことができるデヒドラターゼ内の領域の同定が可能になる。これらの領域における代案の配列変更は、おそらく反応速度に改善を有する追加的な変異株をもたらす。
【0257】
三次元構造に相関する変異位置
基質フリー形態のデヒドラターゼの三次元結晶構造は、X線結晶構造解析によって求められており(リャオ(Liao)ら、J.Inorganic Biochem.93(1−2):84〜91(2003))、さらに1,2−プロパンジオール複合体中の酵素の構造も報告されている(ヤマニシ(Yamanishi)ら、Eur.J.Biochem.269:4484〜4494(2002))。これらの構造に基づいて、各GDHサブユニットの模式図上に、T2/T1比率またはT2のいずれかに改善をもたらす変異(実施例6〜12からの)をマッピングした。より具体的には、図3はα−サブユニット上に単一点変異(野生型GDHと比較して)を含有する変異株の分布を示す(全ての単一点変異の88%に相当)。対照的に、図4はα−サブユニッ上に複数点変異(野生型GDHと比較して)を含有する変異株の分布を示す。このサブユニットは全ての複数点変異の58%を含有し、残りの変異は、β−(31%)およびγ−(11%)サブユニットにそれぞれ分布した。
【0258】
ポジティブヒットから同定されたホットスポット
単一点および複数点変異株の双方における変異は、GDHの全ての3個のサブユニットに位置して大型α−サブユニット全体に分布するが、単一点変異を含有する変異株および複数点変異を含有する変異株は、α−サブユニット中に2個の共通のホットスポットを示す。
【0259】
1つの変異ホットスポットは、α−サブユニットのN−末端から2番目のαらせん(残基62〜70)上に位置する。この領域では5個の単一点変異株が見つかり、内3つは残基62に異なるアミノ酸置換の変異を有し、1つは残基65に変異を有し、1つは残基70に変異を有する(図3)。複数点変異株からの6個の変異部位はこのらせん上にも位置し、残基62に3つ、残基63、67および70にそれぞれ1つある(図4)。
【0260】
第2のホットスポットは、4番目のβ−鎖の部分、TIMバレルとそれに続くα−サブユニットのループおよび短いらせん(残基224〜236)を含む領域である。このホットスポットは活性部位の近辺にある。この領域では5個の単一部位変異が見つかっている。残基224は2個の変異株において異なるアミノ酸置換を有した。残基226は、2個の同一変異株の変異部位である。1個の変異株は、残基233上にある。さらに複数部位変異株の3個の変異部位は、この領域(残基226、231、および236)に位置する。
【0261】
実施例15
GDH変異株の部位飽和変異誘発
実施例9で特徴づけられる変異株中に存在する3個の変異部位に、部位飽和変異誘発を実施した。具体的には、これらの部位はγ−Thr53(変異株Xba3009に見られる)、α−Leu509(変異株Xba3029に見られる)およびα−Val224(変異株4BR1001に見られる)であった。飽和変異誘発ライブラリーを調製するために、1−E1および8−C9変異株(それぞれ実施例9および10)をそれらの宿主細胞から精製し、表21に示す変性プライマーと共にテンプレートとして使用した。
【0262】
【表32】
【0263】
カリフォルニア州ラ・ホーヤのストラタジーン(Stratagene(La Jolla、CA))からのストラタジーン(Stratagene)クイックチェンジ(QuikChange)部位−誘導変異誘発キットを使用して、製造元の説明書に従って、GDH−SM1、GDH−SM2、GDH−SM3、およびGDH−SM4ライブラリーを調製した。プラスミドの大腸菌(E.coli)株5K(DE3)への電気穿孔に続いて(実施例1で述べたように)、一点GDHアッセイを使用して各ライブラリーからの88個の変異株コロニーをスクリーニングして、高濃度1,3−プロパンジオール存在下での総酵素代謝回転数を推定した。次に各スクリーニングからの最良のヒットをDNA配列分析にかけた。以下の表に結果を示す。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に提供する。
【0264】
【表33】
【0265】
全ての4個の飽和変異株(GDH−SM1−G11、GSH−SM2−B11、GDH−SM3−D2、およびGDH−SM4−H2[太字で示す])は、それらが由来した親変異遺伝子との比較で、T(600)のさらなる改善を示した。興味深いことに、変異株GDH−SM4−H2では、3個の塩基変化が同定された(すなわち、ACC(γ−Thr53)からTGT(Cys))。このタイプの変異は、エラープローンPCRを使用して生成することが極めて困難である。
【0266】
実施例16
不対プライマーを使用してGDH変異株を生成する遺伝子組み換え発生伸張法
無作為変異誘発、合理的デザインの変異誘発、および飽和変異誘発(実施例6、8−11、および15)を使用したグリセロールおよび1,3−プロパンジオール存在下でのGDH不活性化速度の顕著な改善にも関わらず、工業用途のためにはさらなる改善が望ましかった。したがって実施例6、9、10、および15からの24個のグリセロールデヒドラターゼ変異株を不対プライマー法を使用した単一遺伝子組み換え発生伸張反応の親テンプレートとして使用した(米国特許出願第60/360279号明細書)。
【0267】
短い側方DNA断片の親遺伝子5’または3’末端への付着
PCRによって短い側方DNA断片を親遺伝子の5’または3’末端に付着し、引き続いてそれを不対プライマーを使用した遺伝子組み換え発生伸張法のための結合部位として使用した。GDHを含有するプラスミドを宿主細胞から精製し、テンプレートとして使用した。
【0268】
具体的には、インディアナ州インディアナポリスのロシュ・アプライド・サイエンス(Roche Applied Science(Indianapolis、IN))からのエキスパンド高忠実度PCRシステム(Expand High Fidelity PCR System)を使用した標準高忠実度PCR反応において、フォーワードプライマーGDHM−F1(配列番号343)および逆転写プライマーGDHM−R1(配列番号344)を使用して、以下のテンプレート遺伝子を増幅した。1−E1(配列番号198)、野生型GDH(配列番号1)、Xba3023(配列番号182)、Xba3010(配列番号175)、8−C9(配列番号212)、4BR1001(配列番号171)、Xba3015(配列番号147)、Xba3008(配列番号151)、Sma3003(配列番号143)、Xba3016(配列番号155)、およびXba3020(配列番号159)。次に得られたPCR生成物を当モル比で共に混合し、「混合物−1」と称した。
【0269】
フォーワードプライマーGDHM−F2(配列番号345)および逆転写プライマーGDHM−R2(配列番号346)を使用して、以下の遺伝子を同様にして増幅した。Xba3007(配列番号40)、Xba3029(配列番号44)、Xba3025(配列番号52)、Sma3009(配列番号179)、Sma3010(配列番号175)、Sma3008(配列番号151)、RsrII001(配列番号136)、PpuMI002(配列番号128)、KG005(配列番号253)、GDH−SM1−G11(配列番号301)、GDH−SM2−B11(配列番号304)、GDH−SM3−D2(配列番号307)、およびGDH−SM4−H2(配列番号310)。次に得られたPCR生成物を当モル比で共に混合し、「混合物−2」と称した。
【0270】
カリフォルニア州バレンシアのキアゲン(Qiagen(Valencia、CA))からのキアゲン(Qiagen)DNA抽出キットを使用して、増幅された生成物をアガロースゲルから精製し、次に不対プライマーによる遺伝子組み換え発生伸張法のための親テンプレートとして使用した。
【0271】
不対プライマー法を使用した遺伝子組み換え発生変異生成物の作成
2個のプライマーPADH316F1(配列番号29)およびT7T(配列番号30)を使用して、上述の24個の親テンプレートから遺伝子組み換え産生物(すなわちGDH変異遺伝子)を作成した。配列番号343によって生成される短い5’DNA断片の添加により、PADH316F1は、「混合物−1」(すなわち、1−E1、野生型GDH、Xba3023、Xba3010、8−C9、4BR1001、Xba3015、Xba3008、Sma3003、Xba3016、およびXba3020)中のテンプレートの5’末端とアニールする。しかしプライマーPADH316F1は、「混合物−2」テンプレートの5’末端に結合しない。同様にプライマーT7Tは、「混合物−2」テンプレート(すなわち、Xba3007、Xba3029、Xba3025、Sma3009、Sma3010、Sma3008、RsrII001、PpuMI002、KG005、GDH−SM1−G11、GDH−SM2−B11、GDH−SM3−D2、およびGDH−SM4−H2)の3’末端にアニールするが、「混合物−1」テンプレートの3’末端には結合しない。
【0272】
反応のために以下の反応混合物をアセンブルした。10ng「混合物−1」、10ng「混合物−2」、各200μMのdNTP、1×PCR緩衝液(最終濃度、1.5mM MgCl2)、286nMのpADH316F1、286nMのT7T、カリフォルニア州バレンシアのキアゲン(Qiagen(Valencia、CA))からの0.625Uのホット・スター・タック(HotStarTaq)、dH2Oで25μLに調節。熱サイクル条件は次のとおりであった。95℃で2分間変性、続いて95℃で30秒の60回のサイクル、1秒+63〜69℃の間の勾配でサイクルあたり1秒、72℃で7分間の最終伸張、および4℃での保持。ニューヨーク州ウェストベリーのエッペンドルフ・サイエンティフィック(Eppendorf Scientific、Inc.(Westbury、NY))からのエッペンドルフ・マスターサイクラー・グラジェント(Eppendorf Mastercycler gradient)5331を熱サイクル反応のために使用した。
【0273】
反応で使用した2個のプライマーは、いかなるテンプレートの5’および3’末端とも同時にマッチしないので、いずれの親テンプレートも反応中に増幅できない。しかし5’および3’末端の双方を有するあらゆる組み換えDNA生成物が、引き続く熱サイクル中で増幅される。不対プライマー反応に続いて、反応混合物をアガロースゲルにロードした。約66〜67℃において、不対プライマー反応から大量の2.7kBのPCR生成物が得られた。テンプレートとして使用したオリジナルの親分子それ自身が約2.7kBだったので、このサイズの生成物は予期された。
【0274】
遺伝子組み換え発生GDH変異株ライブラリーの作成およびスクリーニング
組み換えGDH DNA生成物(ほぼ2.7kB)をキアゲン(Qiagen)DNA抽出キットを使用してゲルから精製し、Xba IおよびHind IIIで消化して、次にXbaI−HindIII−消化したpGD20ベクターとリゲートした(実施例1)。実施例1で述べたように、電気穿孔によるライゲーション混合物の大腸菌(E.coli)株5K(DE3)への形質転換によって、変異株ライブラリーを得た。ライブラリーサイズはライゲーション反応あたり30万個のコロニーを超えた。
【0275】
6,000〜7,000個の変異株コロニーをアガロースプレートからピックアップし、実施例13で述べたように一点ハイスループット・スクリーニング・アッセイを使用してスクリーニングした。一次スクリーニングに続いて、フォローアップ確認アッセイにより推定上のヒットが確認された。簡単に述べると、各推定上のヒットを8個のウェル内で再アッセイした。個々の各クローンからの結果を統計学的に分析して、T(600)の平均値および標準偏差を得た。これらの結果を野生型GDH酵素と比較した。
【0276】
さらに二点アッセイを使用していくつかのヒットをさらに詳しく調査し(実施例5)、これらのヒットの初期反応速度および不活性化の変化を大まかに推定した。
【0277】
表23はいくつかの遺伝子組み換え発生GDH変異株について、これらの様々なアッセイの結果をまとめる。
【0278】
【表34】
【0279】
異なる組み換えGDH変異株は(同様のT(600)値を有するにもかかわらず)異なる酵素速度を示したが、これは単に各アッセイ手段のパラメーターの違いを反映するだけである。例えば変異株SHGDH24およびSHGDH43は同一のT(600)値を有したが、SHGDH24が野生型と比較して軽度に改善されたT1値のみを有したのに対し、SHGDH43は大幅に低下したT1を有した。SHGDH24では酵素のkcatは低下しなかったが、対照的にSHGDH43は大幅に低下したkcatを有した。どちらの場合もSHGDH24およびSHGDH43双方のGDH酵素安定性が増大することで、総酵素代謝回転数の改善が可能になった。
【0280】
SHGDH51はこれらの変異株中で安定性について最大の改善を示したが、そのkcatは大幅に低下したので、この変異株は最高の総酵素代謝回転数を有さなかった。SHGDH37はこれらの変異株中でT(600)について最大の改善を示したが、そのT2値はSHGDH12のそれよりも低く、SHGDH37が高濃度の1,3−プロパンジオールに対して、SHGDH12よりも抵抗性であることが示唆された。
【0281】
しかしいずれにせよ結果は明らかに、不対プライマー法を使用した新しい変異株の作成により、GDH安定性におけるかなりの改善があったことを実証した。具体的には、無作為変異誘発を通じて得られた最良の変異株(すなわち変異株Sma3002;配列番号140)は3.3のT(600)を有した。無作為変異誘発およびスクリーニングを通じて同定された変異株の合理的な組み合わせ、または部位飽和変異誘発を使用したこれらの変異の半無作為組み合わせからは、T(600)が最大の4.3である変異株(すなわち変異株GDH−SM1−G11;配列番号301)が得られた。したがって不対プライマーを使用した遺伝子組み換え発生伸張法アプローチを使用した無作為遺伝子組み換えは、前述の合理的かつ半無作為アプローチよりも強力な技術であると結論される。
【0282】
変異遺伝子の配列分析
表23に列挙される組み換えGDH変異株からプラスミドDNAを精製し、それぞれのGDH遺伝子全体を配列分析した。変異株の分析と、それに続くオリジナル野生型GDH遺伝子配列との比較は、これらの組み換え変異遺伝子が表24に示す単一塩基置換変異(点変異)を含有することを示す。酵素のDNA配列のSEQ識別番号は、表の一番目の欄に提供する。
【0283】
【表35】
【0284】
【表36】
【0285】
興味深いことに、全ての変異株(配列番号313、316、319、322、325、328、331、334、337)はα−β融合変異を含有し(元来Sma3002およびXba3009変異株に見られたように[実施例6])、この変異がT(600)値の改善にとって非常に重要であることが示唆された。SHGDH43は、4個の新たに作り出された変異に加えて、4個の異なる親遺伝子(すなわち4BR1001、1−E1、Xba3008、およびGDH−SM1−G11)からの変異を含有した。これは遺伝子組み換え発生PCR中に、4個の異なる親遺伝子間で少なくとも4つの交差が起きたことを示唆した。SHGDH25およびSHGDH51はいくつかの新たに作り出された変異に加えて、3個の親遺伝子(すなわち、SHGDH25ではSma3003、4BR1001、および1−E1;SHGDH51ではRsrII001、1−E1、およびXba3007)からの変異を含有した。
【図面の簡単な説明】
【0286】
【図1】グリセロール存在下で、またはグリセロールおよび1,3−プロパンジオール存在下のどちらかで実施された、典型的なGDH反応の経時変化を示す。
【図2】実施例6で述べられるような典型的なフォローアップアッセイの図示される結果である。
【図3】(野生型GDHと比較して)α−サブユニット上に一点変異を含有する変異株の分布を示す。
【図4】(野生型のGDHと比較して)α−サブユニット上に複数点変異を含有する変異株の分布を示す。
【図5】不対プライマーを使用した遺伝子組み換え発生伸張法の原理を示す。
【特許請求の範囲】
【請求項1】
(c)B12依存性デヒドラターゼホロ酵素と、グリセロールおよび少なくとも25mMの1,3−プロパンジオールを含んでなる混合物とを接触させ、
(d)B12依存性デヒドラターゼホロ酵素をスクリーニングして、B12依存性デヒドラターゼの代謝回転率を推定する
ことを含んでなる、改善された反応速度を有するB12依存性デヒドラターゼのスクリーニング方法。
【請求項2】
(b)のスクリーニングステップが、
e)補酵素B12、デヒドラターゼ再活性化因子、または内因性のB12依存性デヒドラターゼの供給源を有さない細胞をグリセロールを含まない発酵性炭素基質上に生育させ、
f)細胞を透過化処理し、
g)補酵素B12、グリセロール、および少なくとも25mMの1,3−プロパンジオールの混合物をステップ(b)の透過化処理された細胞に添加し、
h)ステップ(c)で生産された3−ヒドロキシプロピオンアルデヒドを定量化する
ことを含んでなり、
定量化が、T1、T2、およびT(600)よりなる群から選択される測定である請求項1に記載の方法。
【請求項3】
配列番号:40、44、48、52、56、60、64、68、72、76、80、84、88、92、96、100、104、108、112、116、120、124、128、132、136、140、143、147、151、155、159、163、167、171、175、179、182、186、189、192、195、198、201、204、208、212、215、218、221、225、229、233、237、241、245、249、253、257、261、265、269、273、277、281、285、289、293、297、301、304、307、310、313、316、319、322、325、328、331、334、および337よりなる群から選択される、B12依存性変異株デヒドラターゼをコードする核酸配列。
【請求項4】
配列番号:40、44、48、52、56、60、64、68、72、76、80、84、88、92、96、100、104、108、112、116、120、124、128、132、136、140、143、147、151、155、159、163、167、171、175、179、182、186、189、192、195、198、201、204、208、212、215、218、221、225、229、233、237、241、245、249、253、257、261、265、269、273、277、281、285、289、293、297、301、304、307、310、313、316、319、322、325、328、331、334、および337よりなる群から選択される核酸配列によってコードされるB12依存性変異株デヒドラターゼ。
【請求項5】
配列番号:140、179、186、189、192、195、198、201、212、215、218、301、304、307、310、313、316、319、322、325、328、331、334、および337よりなる群から選択されるα−βサブユニット融合を含んでなる、B12依存性変異株デヒドラターゼをコードする核酸配列。
【請求項6】
配列番号313、322、および328よりなる群から選択されるα−βサブユニット融合を含んでなる、B12依存性変異株デヒドラターゼをコードする核酸配列。
【請求項7】
α−βサブユニット融合のαおよびβサブユニット間のリンカー配列をさらに含んでなり、リンカー配列が、配列番号18および配列番号19よりなる群から選択される請求項5に記載の核酸配列。
【請求項8】
d)ホットスポット1またはホットスポット2を含んでなるB12依存性デヒドラターゼホロ酵素と変異原因物質とを接触させ、
e)ステップA)で生産された変異株を改善されたkcatおよび/または安定性についてスクリーニングし、そして
f)ステップa)およびb)を反復する
ことを含んでなる、改善された反応速度を有するB12依存性デヒドラターゼ変異株を作り出す方法。
【請求項9】
d)デヒドラターゼホロ酵素と5〜10mMのグリセロールおよび/または10〜300mMの1,3−プロパンジオールとを接触させ、
e)ハイスループットアッセイにおいて、kcatおよび総酵素代謝回転を推定するのに十分に隔たった少なくとも2つの時点で測定し、
f)標準デヒドラターゼと比較して改善された反応速度を有するB12依存性変異株を選択する
ことを含んでなる、標準デヒドラターゼと比較して改善された反応速度を有するB12依存性デヒドラターゼを同定する方法。
【請求項10】
d)デヒドラターゼホロ酵素と5〜50mMのグリセロールおよび>300mMの1,3−プロパンジオールとを接触させ、
e)ハイスループットアッセイにおいて、T0から十分に隔たった1つの時点で測定して、総酵素代謝回転数を推定し、そして
f)標準デヒドラターゼと比較して改善された反応速度を有するB12依存性変異株を選択する
ことを含んでなる、標準デヒドラターゼと比較して改善された反応速度を有するB12依存性デヒドラターゼを同定する方法。
【請求項1】
(c)B12依存性デヒドラターゼホロ酵素と、グリセロールおよび少なくとも25mMの1,3−プロパンジオールを含んでなる混合物とを接触させ、
(d)B12依存性デヒドラターゼホロ酵素をスクリーニングして、B12依存性デヒドラターゼの代謝回転率を推定する
ことを含んでなる、改善された反応速度を有するB12依存性デヒドラターゼのスクリーニング方法。
【請求項2】
(b)のスクリーニングステップが、
e)補酵素B12、デヒドラターゼ再活性化因子、または内因性のB12依存性デヒドラターゼの供給源を有さない細胞をグリセロールを含まない発酵性炭素基質上に生育させ、
f)細胞を透過化処理し、
g)補酵素B12、グリセロール、および少なくとも25mMの1,3−プロパンジオールの混合物をステップ(b)の透過化処理された細胞に添加し、
h)ステップ(c)で生産された3−ヒドロキシプロピオンアルデヒドを定量化する
ことを含んでなり、
定量化が、T1、T2、およびT(600)よりなる群から選択される測定である請求項1に記載の方法。
【請求項3】
配列番号:40、44、48、52、56、60、64、68、72、76、80、84、88、92、96、100、104、108、112、116、120、124、128、132、136、140、143、147、151、155、159、163、167、171、175、179、182、186、189、192、195、198、201、204、208、212、215、218、221、225、229、233、237、241、245、249、253、257、261、265、269、273、277、281、285、289、293、297、301、304、307、310、313、316、319、322、325、328、331、334、および337よりなる群から選択される、B12依存性変異株デヒドラターゼをコードする核酸配列。
【請求項4】
配列番号:40、44、48、52、56、60、64、68、72、76、80、84、88、92、96、100、104、108、112、116、120、124、128、132、136、140、143、147、151、155、159、163、167、171、175、179、182、186、189、192、195、198、201、204、208、212、215、218、221、225、229、233、237、241、245、249、253、257、261、265、269、273、277、281、285、289、293、297、301、304、307、310、313、316、319、322、325、328、331、334、および337よりなる群から選択される核酸配列によってコードされるB12依存性変異株デヒドラターゼ。
【請求項5】
配列番号:140、179、186、189、192、195、198、201、212、215、218、301、304、307、310、313、316、319、322、325、328、331、334、および337よりなる群から選択されるα−βサブユニット融合を含んでなる、B12依存性変異株デヒドラターゼをコードする核酸配列。
【請求項6】
配列番号313、322、および328よりなる群から選択されるα−βサブユニット融合を含んでなる、B12依存性変異株デヒドラターゼをコードする核酸配列。
【請求項7】
α−βサブユニット融合のαおよびβサブユニット間のリンカー配列をさらに含んでなり、リンカー配列が、配列番号18および配列番号19よりなる群から選択される請求項5に記載の核酸配列。
【請求項8】
d)ホットスポット1またはホットスポット2を含んでなるB12依存性デヒドラターゼホロ酵素と変異原因物質とを接触させ、
e)ステップA)で生産された変異株を改善されたkcatおよび/または安定性についてスクリーニングし、そして
f)ステップa)およびb)を反復する
ことを含んでなる、改善された反応速度を有するB12依存性デヒドラターゼ変異株を作り出す方法。
【請求項9】
d)デヒドラターゼホロ酵素と5〜10mMのグリセロールおよび/または10〜300mMの1,3−プロパンジオールとを接触させ、
e)ハイスループットアッセイにおいて、kcatおよび総酵素代謝回転を推定するのに十分に隔たった少なくとも2つの時点で測定し、
f)標準デヒドラターゼと比較して改善された反応速度を有するB12依存性変異株を選択する
ことを含んでなる、標準デヒドラターゼと比較して改善された反応速度を有するB12依存性デヒドラターゼを同定する方法。
【請求項10】
d)デヒドラターゼホロ酵素と5〜50mMのグリセロールおよび>300mMの1,3−プロパンジオールとを接触させ、
e)ハイスループットアッセイにおいて、T0から十分に隔たった1つの時点で測定して、総酵素代謝回転数を推定し、そして
f)標準デヒドラターゼと比較して改善された反応速度を有するB12依存性変異株を選択する
ことを含んでなる、標準デヒドラターゼと比較して改善された反応速度を有するB12依存性デヒドラターゼを同定する方法。
【図1】

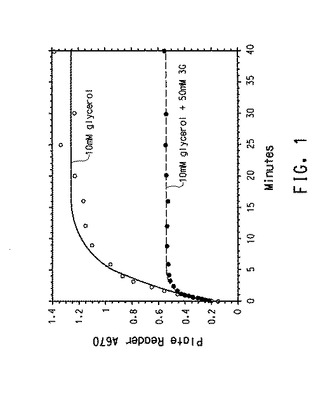
【図2】
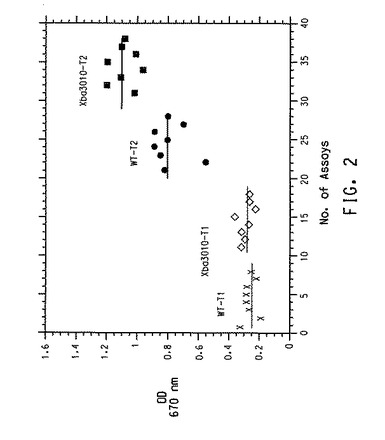
【図3】
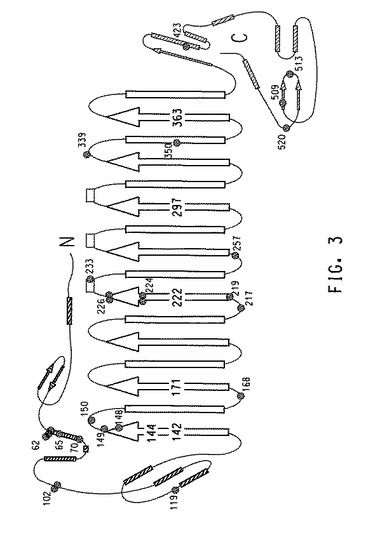
【図4】
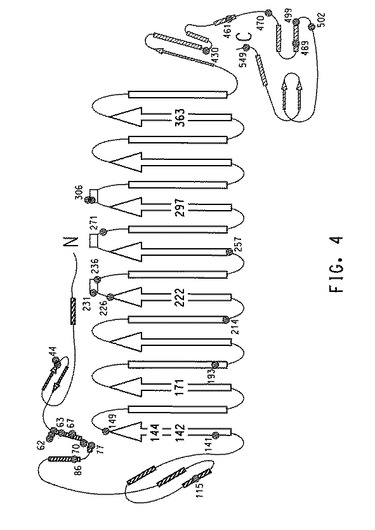
【図5】
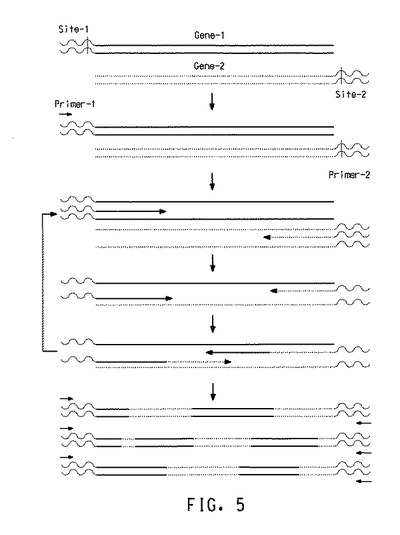
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2007−524342(P2007−524342A)
【公表日】平成19年8月30日(2007.8.30)
【国際特許分類】
【出願番号】特願2004−562258(P2004−562258)
【出願日】平成15年12月16日(2003.12.16)
【国際出願番号】PCT/US2003/040397
【国際公開番号】WO2004/056963
【国際公開日】平成16年7月8日(2004.7.8)
【出願人】(390023674)イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー (2,692)
【氏名又は名称原語表記】E.I.DU PONT DE NEMOURS AND COMPANY
【Fターム(参考)】
【公表日】平成19年8月30日(2007.8.30)
【国際特許分類】
【出願日】平成15年12月16日(2003.12.16)
【国際出願番号】PCT/US2003/040397
【国際公開番号】WO2004/056963
【国際公開日】平成16年7月8日(2004.7.8)
【出願人】(390023674)イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー (2,692)
【氏名又は名称原語表記】E.I.DU PONT DE NEMOURS AND COMPANY
【Fターム(参考)】
[ Back to top ]