
改善された非沈静α2アゴニストを同定するための新規方法
交感神経増強状態、神経学的状態、眼症状および慢性疼痛を鎮静を伴うことなく予防または軽減する方法であって、α−2A/α1−A選択的アゴニストの有効量を対象に末梢投与し、それにより該状態または慢性疼痛を鎮静を伴うことなく予防または軽減し、前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有するまたはブリモニジンと比較して高いα−1A/α−2A効力比を有する、方法を提供する。

【発明の詳細な説明】
【発明の詳細な説明】
【0001】
(発明の分野)
本発明は、一般的には、痛みの処置および慢性の痛みの長期間にわたる緩和に関し、詳細には、α−アドレナリン作動性アゴニスト、およびα−2Aアドレナリン作動性受容体の選択的アンタゴニストに関する。
【0002】
(発明の背景)
臨床での痛みには、侵害受容性の痛みおよび神経障害性の痛みが含まれる。それぞれのタイプの痛みは、損傷部位および隣接する正常な組織における感覚過敏によって特徴づけられる。侵害受容性の痛みは、通常、継続時間が限定され、利用可能なオピオイド治療によく応答するが、神経障害性の痛みは、例えば、切断術の後で生じることが多い「ゴースト痛み」において明らかであるように、開始事象が治癒した後も長く持続し得る。慢性の痛み症候群(例えば、慢性の神経障害性の痛みなど)は、手術、圧迫傷害もしくは外傷、感染性因子、毒性薬物、炎症性障害、または代謝性疾患(例えば、糖尿病または虚血など)を含む様々な傷害のいずれかによって引き起こされる。
【0003】
残念なことではあるが、慢性の痛み(例えば、慢性の神経障害性の痛みなど)は、一般に、利用可能な薬物治療に対して抵抗性である。さらに、現在の治療は、認識変化、鎮静作用、悪心、そして麻酔薬の場合には耽溺などの重大な副作用を有している。神経障害性の痛みおよび他の慢性の痛みに苦しんでいる多くの患者は、年配者であるか、または、利用可能な鎮痛治療に関連する副作用に対するその寛容性を制限する医学的状態を有する。耐えることのできない副作用を生じさせることなく神経障害性の痛みを緩和することにおいて現在の治療が不十分であることは、多くの場合、慢性の痛みで苦しんでいる人々のうつ病傾向および自殺傾向において明らかである。
【0004】
(発明の要旨)
α−2アドレナリン作動性アゴニストは、呼吸抑制作用および潜在的耽溺性を有しないため、現在の鎮痛剤に対する代替として開発されつつある。そのような薬物は、脊椎投与されたとき、有用な鎮痛剤である。しかしながら、α−アドレナリン作動性アゴニストの望ましくない薬理学的性質、特に、鎮静作用および血圧低下により、これらの薬物の有用性が、経口投与または他の末梢経路によって投与されたときには制限されている。従って、経口経路または他の末梢経路によって投与することができ、かつ、鎮静作用および血圧低下などの望ましくない副作用を有しない効果的な鎮痛剤が求められている。本発明はこの要求を満たし、関連した利点もまた提供する。
【0005】
本発明はまた、現在までその痛みを抑えるために生涯にわたる毎日の薬物療法に直面している、慢性の痛みで苦しんでいる人々に対する新しい治療方法を提供する。残念ながら、慢性の神経障害性の痛みに対する利用可能な処置、例えば、三環系抗うつ剤、抗てんかん薬および局所麻酔注射などは、症状を一時的および様々な程度に緩和するにすぎない。利用できる処置はどれも、過敏になった痛み状態を逆戻りさせないか、または、神経障害性の痛みなどの痛みを治療しない。例えば、1ヶ月に1回または数回投与することができ、かつ、数週間または数ヶ月にわたって鎮痛活性を維持する効果的な薬物を、現在、利用することができない。従って、慢性の痛みからの長期間にわたる緩和をもたらす新規な方法が求められている。本発明はこの要求を満たし、関連した利点もまた提供する。
【0006】
本発明は、有効量のα−アドレナリン作動性アゴニストを含む医薬組成物と、有効量の選択的α−2Aアンタゴニストを含む医薬組成物とを対象に投与することによって対象において痛みを緩和する方法を提供する。本発明の方法は、神経障害性の痛み、例えば、糖尿病性神経障害に由来する痛みなど;内臓痛;手術後の痛み;ガンまたはガン処置に由来する痛み;炎症性の痛み、例えば、関節炎または過敏性腸症候群に由来する痛み;頭痛の痛みおよび筋肉痛(これらに限定されない)を含む様々なタイプの痛みを緩和するために有用である。
【0007】
様々なα−アドレナリン作動性アゴニストが本発明において有用であり、これらには、汎α−2アゴニストおよび汎α−1汎α−2アゴニストが含まれる。本発明の方法に従って痛みを緩和することにおいて有用なα−アドレナリン作動性アゴニストには、クロニジン、ブリモニジン、チザニジン、デキセメデトミジン、ノルエピネフリン、化合物1および化合物2、ならびに、それらのすべての医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が含まれるが、これらに限定されない。
【0008】
様々な選択的α−2Aアンタゴニストもまた、本発明において有用である。そのような選択的α−2Aアンタゴニストには、4-イミダゾール系化合物、例えば、化合物13ならびにその医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物、そして、BRL48962またはその医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が含まれるが、これらに限定されない。1つの実施形態において、本発明は、末梢限定的な選択的α−2Aアンタゴニストを用いて実施される。
【0009】
様々な投与経路が、本発明の方法に従って痛みを緩和するために有用である。1つの実施形態において、α−アドレナリン作動性アゴニストおよび選択的α−2Aアンタゴニストの両方が末梢投与される。他の実施形態において、α−アドレナリン作動性アゴニストが経口投与または皮下ミニポンプにより投与される。さらなる実施形態において、α−アドレナリン作動性アゴニストが経口投与または皮下ミニポンプにより投与され、選択的α−2Aアンタゴニストが任意の末梢経路によって投与される。さらに他の実施形態において、選択的α−2Aアンタゴニストが経口投与または皮下ミニポンプにより投与される。所望の場合には、選択的α−2Aアンタゴニストを経口投与または皮下ミニポンプにより投与することができ、α−アドレナリン作動性アゴニストを、任意の末梢経路によって、例えば、経口投与または皮下ミニポンプなどにより投与することができる。
【0010】
(発明の詳細な記載)
1つの実施形態において、本発明は、有効量のα−アドレナリン作動性アゴニストを含む医薬組成物と、有効量の選択的α−2Aアンタゴニストを含む医薬組成物とを対象に投与することによって対象において痛みを緩和する方法であって、α−アドレナリン作動性アゴニストおよび選択的α−2Aアンタゴニストがそれぞれ、少なくとも3日間の期間にわたって反復的または継続的に投与される方法を提供する。そのような方法では、痛みの緩和が、例えば、対象においてα−アドレナリン作動性アゴニストの著しいレベルが存在しないもとで継続し得る。
【0011】
本発明はさらに、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストで、同時での鎮静作用を伴わずに末梢痛覚消失を生じさせる能力を有するα−アドレナリン作動性アゴニストを含有する鎮痛剤組成物を提供する。1つの実施形態において、鎮痛剤組成物は、同時での鎮静作用を伴うことなく、また、血圧降下作用の実質的不存在下で末梢痛覚消失を生じさせる。別の実施形態において、本発明は、同時での鎮静作用を伴うことなく、痛みを少なくとも50%軽減するために十分である鎮痛剤組成物を提供する。さらなる実施形態において、痛みを少なくとも50%軽減させる鎮痛剤組成物の用量よりも少なくとも10倍、または100倍、または1000倍大きい用量の鎮痛剤組成物が、運動活動または筋肉活動における20%の減少を生じさせるために要求される。さらなる実施形態において、本発明は、同時での鎮静作用を伴うことなく、また、血圧降下作用の実質的不存在下で痛みを少なくとも50%軽減させるために十分な末梢痛覚消失を生じさせる鎮痛剤組成物を提供する。別の実施形態において、本発明は、同時での鎮静作用を伴わずに末梢痛覚消失を生じさせる能力を有する、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを含有する鎮痛剤組成物で、アゴニストがチオウレア系化合物またはその誘導体ではない鎮痛剤組成物を提供する。さらなる実施形態において、本発明は、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる能力を有する、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを含有する鎮痛剤組成物で、アゴニストがチオウレア系化合物または4-イミダゾール系化合物またはその誘導体ではない鎮痛剤組成物を提供する。
【0012】
本発明によりさらに、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストの有効量を含む医薬組成物を対象に末梢投与し、それにより、同時での鎮静作用を伴わない末梢痛覚消失を生じさせることによって対象において痛みを緩和する方法が提供される。そのような末梢痛覚消失は、同時での鎮静作用を伴うことなく、例えば、少なくとも50%、痛みを軽減させるために十分であり得る。別の実施形態において、末梢痛覚消失は血圧降下作用の実質的不存在下で生じる。1つの実施形態において、この方法は、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストで、チオウレア系化合物またはその誘導体ではないα−アドレナリン作動性アゴニストを使用して実施される。別の実施形態において、この方法は、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストで、チオウレア系化合物または4-イミダゾール系化合物またはその誘導体ではないα−アドレナリン作動性アゴニストを使用して実施される。様々なタイプおよび病因の痛みを本発明の方法に従って緩和することができる。非限定的な例として、本発明の方法は、神経障害性の痛み、例えば、糖尿病性神経障害に由来する痛みなど;内臓痛;手術後の痛み;ガンまたはガン処置に由来する痛み;炎症性の痛み、例えば、関節炎痛または過敏性腸症候群の痛みなど;頭痛の痛みおよび筋肉痛を緩和することにおいて有用であり得る。
【0013】
最小限のα−2Aアゴニスト活性を有する様々なα−アドレナリン作動性アゴニストが本発明の方法において有用であり得る。1つの実施形態において、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストである。そのようなアゴニストは、例えば、チオン系化合物であり、例えば、化合物3または化合物11またはその医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物などであり得る。1つの実施形態において、本発明の方法は、化合物3の(-)-エナンチオマーまたはその医薬的に許容され得る塩もしくはエステルである、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストを用いて実施される。
【0014】
本発明において有用な、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストには、イミダゾロン系化合物がさらに含まれるが、これに限定されない。最小限のα−2Aアゴニスト活性を有する有用なイミダゾロン系のα−2Bアゴニストは、例えば、化合物4またはその医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物であり得る。1つの実施形態において、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは化合物4の(+)-エナンチオマーまたはその医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物である。さらなる実施形態において、本発明の方法は、下記のいずれかの、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストを使用して実施される:化合物5、化合物6、化合物7、化合物8、化合物9または化合物14、あるいは、その塩、エステル、アミド、立体異性体またはラセミ混合物。最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、経口投与および皮下ミニポンプによる投与(これらに限定されない)を含む様々な経路のいずれかによって末梢投与される。
【0015】
本発明はまた、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングする方法で、α−2A受容体を、鎮痛活性を有するα−アドレナリン作動性アゴニストと接触させること、および、そのアゴニストがα−2Aアゴニスト活性を有するかどうかを明らかにすることによる方法を提供する。この方法では、α−2Aアゴニスト活性の不存在により、鎮痛活性を有するα−アドレナリン作動性アゴニストが、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
本発明ではさらに、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤をスクリーニングする方法であって、α−2A受容体を薬剤と接触させること、薬剤がα−2Aアゴニスト活性を有するかどうかを明らかにすること、α−2B受容体を薬剤と接触させること、および、薬剤がα−2Bアゴニスト活性を有するかどうかを明らかにすることによる方法が提供される。この方法では、α−2Aアゴニスト活性の不存在およびα−2Bアゴニスト活性の存在により、薬剤が、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0016】
本発明はまた、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングする方法であって、野生型レベルのα−2A受容体活性を少なくとも有するコントロール動物にα−アドレナリン作動性アゴニストを末梢投与すること、コントロール動物における痛覚消失についてアッセイすること、低下したレベルのα−2A受容体の発現または活性を有する対応する動物に、コントロール動物に投与された量と類似する量またはそれよりも大きい量のα−アドレナリン作動性アゴニストを末梢投与すること、および、低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失についてアッセイすることによる方法を提供する。この方法では、コントロール動物における痛覚消失の不存在、および低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失の存在により、α−アドレナリン作動性アゴニストが過度なα−2Aアゴニスト活性を有することが示され、かつ、コントロール動物における痛覚消失の存在、および低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失の存在により、α−アドレナリン作動性アゴニストが、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0017】
本発明のそのような方法において、コントロール動物は、例えば、両方のα−2A受容体遺伝子座において野生型であり得る。1つの実施形態において、コントロール動物は野生型動物であり、例えば、野生型マウスなどである。様々な対応する動物が本発明のスクリーニング方法において有用である。1つの実施形態において、本発明は、α−2A受容体遺伝子座においてホモ接合の点変異を有する対応する動物を用いて実施される。別の実施形態において、本発明は、α−2A受容体コード配列の内部に点変異を有する対応する動物を用いて実施される。そのような点変異は、例えば、Asp79と類似の残基で生じ得るし、例えば、Asp79からAsnへの変異であり得る。さらなる実施形態において、本発明は、ホモ接合のα−2Aノックアウト変異を有する対応する動物を用いて実施される。様々な方法論が、スルプロストン感作後に痛覚消失についてアッセイすること(これに限定されない)を含む本発明の方法で痛覚消失についてアッセイするために使用され得ることが理解される。
【0018】
所望の場合には、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングするための本発明の方法は、(a)野生型レベルのα−2A受容体活性およびα−2B受容体活性を少なくとも有するコントロール動物にα−アドレナリン作動性アゴニストを末梢投与すること、(b)コントロール動物における痛覚消失についてアッセイすること、(c)低下したレベルのα−2A受容体の発現または活性を有する対応する動物に、コントロール動物に投与された量と類似する量またはそれよりも大きい量のα−アドレナリン作動性アゴニストを末梢投与すること、(d)低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失についてアッセイすること、(e)低下したレベルのα−2B受容体の発現または活性を有する対応する動物にα−アドレナリン作動性アゴニストを末梢投与すること、および、(f)低下したレベルのα−2B受容体の発現または活性を有する対応する動物における痛覚消失についてアッセイすることによって実施することができ、この方法では、コントロール動物における痛覚消失の不存在、および低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失の存在により、α−アドレナリン作動性アゴニストが過度なα−2Aアゴニスト活性を有することが示され、かつ、コントロール動物における痛覚消失の存在、低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失の存在、および低下したレベルのα−2B受容体の発現または活性を有する対応する動物における痛覚消失の不存在により、α−アドレナリン作動性アゴニストが、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0019】
本発明はさらに、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングする方法であって、野生型レベルのα−2B受容体活性を少なくとも有するコントロール動物にα−アドレナリン作動性アゴニストを末梢投与すること、コントロール動物における痛覚消失についてアッセイすること、低下したレベルのα−2B受容体の発現または活性を有する対応する動物にα−アドレナリン作動性アゴニストを末梢投与すること、および、低下したレベルのα−2B受容体の発現または活性を有する対応する動物における痛覚消失についてアッセイすることによる方法を提供する。この方法では、コントロール動物における痛覚消失の存在、および低下したレベルのα−2B受容体の発現または活性を有する対応する動物における痛覚消失の不存在により、α−アドレナリン作動性アゴニストが、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0020】
本発明のそのような方法は、様々なコントロール動物、例えば、両方のα−2B受容体遺伝子座において野生型であるコントロール動物などを用いて実施することができる。1つの実施形態において、コントロール動物は野生型動物である。さらなる実施形態において、コントロール動物は野生型マウスである。同様に、様々な対応する動物が本発明のスクリーニング方法において有用であり、これらには、ヘテロ接合のα−2Bノックアウト変異またはホモ接合のα−2Bノックアウト変異を有する対応する動物が含まれる。痛覚消失は、様々な方法論のいずれかを使用してアッセイすることができる。1つの実施形態において、痛覚消失がスルプロストン感作後にアッセイされる。
【0021】
本発明はさらに、対象において慢性の痛みを長期間にわたって緩和するための方法を提供する。この方法は、少なくとも3日間の期間にわたってα−2A受容体活性化の不存在下で鎮痛性α−アドレナリン作動性受容体を対象において活性化し、その結果、慢性の痛みの緩和が、継続した受容体活性化の存在下で維持されるようにすることによって実施される。1つの実施形態において、本発明の方法は、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストの有効量を含有する医薬組成物を少なくとも3日間の期間にわたって対象に投与し、その結果、慢性の痛みの緩和が対象において著しいアゴニストレベルの不存在下で維持されるようにすることによって実施される。慢性の痛みの緩和は、例えば、対象において著しいアゴニストレベルの不存在下で少なくとも3週間にわたって維持され得る。本発明の方法は、任意のタイプの慢性の痛みを長期間にわたって緩和するために使用され得ることが理解される。非限定的な例として、そのような方法は、神経障害性の痛み;内臓痛;手術後の痛み;ガンまたはガン処置に由来する痛み;または炎症性の痛みを長期間にわたって緩和するために使用することができる。
【0022】
慢性の痛みの長期間にわたる緩和は、最小限のα−2Aアゴニスト活性を有する様々なα−アドレナリン作動性アゴニストのいずれかを用いて本発明の方法に従って達成することができる。例えば、慢性の痛みの長期間にわたる緩和を、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストを使用して達成することができる。最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストの例には、チオン系化合物、例えば、化合物3または化合物11またはその医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物などが含まれるが、これらに限定されない。1つの実施形態において、そのようなチオン系の、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、化合物3の(-)-エナンチオマーまたはその医薬的に許容され得る塩もしくはエステルである。最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストの例にはさらに、イミダゾロン系化合物、例えば、化合物4またはその医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物などが含まれるが、これらに限定されない。1つの実施形態において、そのようなイミダゾロン系の、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、化合物4の(+)-エナンチオマーまたはその医薬的に許容され得る塩もしくはエステルである。最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストの例にはまた、化合物5、化合物6、化合物7、化合物8および化合物9などの化合物、ならびに、それらの医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物が含まれるが、これらに限定されない。最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、すべての末梢投与経路(例えば、経口投与、または皮下ミニポンプによる投与)(これらに限定されない)を含む様々な経路のいずれかによって投与することができる。
【0023】
さらなる実施形態において、本発明の方法は、有効量のα−アドレナリン作動性アゴニストを含有する医薬組成物と、有効量の選択的α−2Aアンタゴニストを含有する医薬組成物とを少なくとも3日間の期間にわたって対象に投与し、その結果、慢性の痛みの緩和が、対象において著しいアゴニストレベルの不存在下で維持されるようにすることによって実施される。慢性の痛みの緩和は、対象において著しいアゴニストレベルの不存在下で、例えば、少なくとも3週間にわたって維持され得る。様々なα−アドレナリン作動性アゴニストが本発明において有用であり、これらには、クロニジン、ブリモニジン、チザニジン、デキセメデトミジン、ノルエピネフリン、ならびに、他の汎α−2アゴニストおよび汎α−1汎α−2アゴニスト、そして同様に、化合物1または化合物2ならびにそれらの医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が含まれる。同様に、様々な選択的α−2Aアンタゴニストが、慢性の痛みを長期間にわたって緩和することにおいて有用であり、これらには、化合物13ならびにその医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物、そして、末梢限定的な選択的α−2Aアンタゴニストが含まれるが、これらに限定されない。
【0024】
様々な投与経路が、慢性の痛みの長期間にわたる緩和のための医薬組成物を送達するために有用であり得る。そのような投与経路には、末梢投与、例えば、経口投与、または皮下ミニポンプによる投与が含まれるが、これらに限定されない。1つの実施形態において、α−アドレナリン作動性アゴニストおよび選択的α−2Aアンタゴニストの両方が末梢投与される。他の実施形態において、α−アドレナリン作動性アゴニストが、経口投与、または皮下ミニポンプによる投与によって投与され、選択的α−2Aアンタゴニストが任意の末梢経路によって投与される。さらにさらなる実施形態において、選択的α−2Aアンタゴニストが、経口投与、または皮下ミニポンプによる投与によって投与され、α−アドレナリン作動性アゴニストが、経口経路または皮下ミニポンプによる経路(これらに限定されない)を含む末梢経路によって投与される。
【0025】
アドレナリン作動性アゴニストは、カテコールアミン系化合物のノルエピネフリンおよびエピネフリンに対する様々な生理学的応答を媒介し、7個の膜貫通ドメインを有するGタンパク質共役型受容体スーパーファミリーのメンバーである。本明細書において「ブリモニジン」は次式
【化1】
を有する化合物を意味する。α−1アドレナリン作動性受容体が一般にはエフェクター器官における応答を媒介し、その一方で、α−2アドレナリン作動性受容体がシナプス後ならびにシナプス前に局在化しており、そこで神経伝達物質の放出を調節している。α−2アドレナリン作動性受容体の様々なアゴニストが、現在、高血圧、緑内障、痙性および注意欠陥障害の処置において、アヘン禁断症状の抑制において、また、全身麻酔に対する補助剤として臨床的に使用されている。
【0026】
α−2アドレナリン作動性受容体は、現在、その薬理学的特徴および分子的特徴に基づいて3つのサブタイプに分類されている:α−2A/D(ヒトにおけるα−2Aおよびラットにおけるα−2D)、α−2B、およびα−2C(Bylundら、Pharmacol. Rev.、46:121〜136 (1994);HeinおよびKobilka、Neuropharmacol.、34:357〜366 (1995))。α−2Aサブタイプおよびα−2Bサブタイプは一部の血管床における動脈収縮を調整することができ、α−2Aサブタイプおよびα−2Cサブタイプは交感神経終末からのノルエピネフリン放出のフィードバック阻害を媒介する。α−2Aサブタイプはまた、α−2アドレナリン作動性アゴニストの中枢作用の多くを媒介する(CalzadaおよびArtinano、Pharmacol. Res.、44:195〜208 (2001);Heinら、Ann. NY Acad. Science、881:265〜271 (1999);Karger(編)、α−Adrenoreceptors: Molecular Biology, Biochemistry and Pharmacology (1991))。
【0027】
α−2Aアゴニスト活性を有する非選択的α−アドレナリン作動性アゴニスト、例えば、クロニジンおよびデクセメジトミジンなどが、様々なタイプの痛みを処置するために使用されているが、そのような薬物は、鎮静作用から分離可能な痛覚消失を達成するために脊椎投与されなければならない。そのような汎アゴニストの中枢鎮痛作用は、脊椎内の後角において発現するα−2A受容体によって媒介される。本発明は、脊椎におけるα−2A受容体の前鎮痛性機能とは対照的に、末梢α−2A受容体が痛みを媒介するという驚くべき発見に基づいている。本発明はさらに、活性化されたとき、末梢痛覚消失を生じさせることができる、末梢で発現するアドレナリン作動性アゴニスト(α−2B受容体)の同定に基づいている。
【0028】
本明細書中に開示されるように、α−2A活性を有するα−アドレナリン作動性アゴニスト(例えば、汎アゴニストのクロニジンまたはブリモニジンなど)は、野生型動物に末梢投与されたとき、鎮静作用から分離可能な著しい痛覚消失を生じさせない。しかしながら、鎮静作用から分離可能な痛覚消失が、これらの薬物で末梢処置されたα−2Aノックアウトマウスにおいて観測されていた。さらに、鎮静作用から分離可能な鎮痛活性はまた、血液脳関門を容易に横断しないα−アドレナリン作動性アゴニストを末梢投与した後のα−2Aノックアウトマウスでも観測されていた(例えば、化合物1またはパラ-アミノ-クロニジン)(PAC;実施例IIBおよび図1Dを参照のこと)。この末梢鎮痛活性は野生型動物では観測されなかった(図1C)。このことは、α−アドレナリン作動性アゴニストの新規な鎮痛活性が、末梢α−2A受容体の活性化を妨げることによって現れることを示している。
【0029】
さらに、本明細書中、実施例IIIにおいて示されるように、Chungラット(これは末梢神経障害の広く受け入れられているモデルである)に、α−2汎アゴニスト(例えば、クロニジンなど)が、表1に示される選択的α−2Aアンタゴニスト(化合物13)と一緒に投与された。クロニジンが単独で末梢投与されたときに得られた結果とは対照的に、選択的α−2Aアンタゴニストと一緒のクロニジンの末梢への同時投与は、鎮静作用から分離可能な鎮痛作用を生じさせた。このことから、α−2A受容体の遮断は、α−アドレナリン作動性アゴニストの末梢鎮痛作用を遺伝子的に変化していない動物において生じさせ得ることが確認された。本明細書中、実施例IIIにおいてさらに開示されるように、多様な構造クラスの、最小限のα−2A活性を有するα−アドレナリン作動性アゴニスト、例えば、化合物3、化合物4、化合物5、化合物6、化合物7および化合物14などをChungモデルラットに腹腔内注射によって投与したとき、同時での鎮静作用を伴わない痛覚消失を生じさせた(下記の表2を参照のこと)。これらの結果は、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストの鎮痛活性がα−アドレナリン作動剤の以前に記載された鎮痛活性とは異なることを明らかにしており、また、最小限のα−2Aアゴニスト活性を有することによって特徴づけられるα−アドレナリン作動性アゴニストは有用な末梢鎮痛薬であり得ることをさらに示している。
【0030】
本明細書中、実施例IVにおいて開示される結果は、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストが、数日の薬物投与の後の少なくとも6週間にわたって持続する長期間にわたる痛み緩和を生じさせ得ることをさらに明らかにしている。本明細書中に開示されるように、Chungモデル動物が、皮下浸透圧ミニポンプを使用して3日間〜7日間、化合物8、化合物9、化合物3または化合物4(これらは、最小限のα−2Aアゴニスト活性を有する構造的に異なるα−アドレナリン作動性アゴニストである)で処置された。痛みの緩和が薬物処置の期間中にわたって観測された。例えば、化合物8は異疼痛を90〜100%緩和し、化合物9は異疼痛を60〜80%緩和した。さらに、試験されたアゴニストのそれぞれの鎮痛作用は、処置が終わった後の1ヶ月以上にわたって継続した。この長期間にわたる痛み緩和は、様々な試験された他の鎮痛剤の性質ではなかった。
【0031】
本明細書中にさらに開示されるように、様々な時点での化合物8の血漿中濃度のサンプリングから、非常に低い薬物レベルが、薬物処置を開始した後、10日目まで存在し、血漿中の薬物が14日目までに検出できなくなった(図5Bを参照のこと)が、痛みはこのときにはほとんど緩和していたことが明らかにされた。さらに、α−2アンタゴニストのラウオルシンが、経口胃管法または浸透圧ミニポンプによる数日間の投薬の後の様々な時点で、化合物8の鎮痛活性を阻害する能力についてアッセイされた。注目すべきことに、ラウオルシンは、処置の3日目に化合物8の鎮痛作用を阻害したが、4日目には阻害しなかった(図6を参照のこと)。まとめると、これらの結果は、鎮痛活性の持続期間が、薬物が血液中に残存している時間を超えていること、および、長期間にわたる投薬の後では、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、継続した受容体活性化または血漿中薬物レベルの不存在下でさえも、長期間にわたる痛み緩和をもたらし得ることを示している。
【0032】
本明細書中に開示されるさらなる結果は、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストはまた、Bennett部分座骨神経結紮モデルにおいて、また、過敏性腸症候群の動物モデルにおいて長期間にわたる痛み緩和を生じさせることを示している(実施例IV、図7Bおよび図8を参照のこと)。このことは、観測された長期間の鎮痛作用が神経障害性の痛みに対して特異的でないことを明らかにしている。これらの結果は、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストが、神経障害性の痛み、内臓痛、炎症性の痛み、手術後の痛みおよびガンの痛み(これらに限定されない)を含む様々なタイプの急性の痛みおよび慢性の痛みを処置するために使用され得ることを示している。
【0033】
これらの知見に基づいて、本発明は、α−2A受容体以外の末梢α−アドレナリン作動性受容体を作動させることによって対象において痛みを緩和する方法を提供する。1つの実施形態において、本発明は、末梢α−2B受容体を作動させることによって対象において痛みを緩和する方法を提供する。本発明はまた、有効量のα−アドレナリン作動性アゴニストを含有する医薬組成物と、有効量の選択的α−2Aアンタゴニストを含有する組成物とを対象に投与することによって対象において痛みを緩和する方法を提供する。そのような方法は、神経障害性の痛み、例えば、糖尿病性神経障害から生じる痛みなど;内臓痛;手術後の痛み;ガンまたはガン処置から生じる痛み;炎症性の痛み、例えば、関節炎または過敏性腸症候群から生じる痛み;頭痛の痛みおよび筋肉痛(これらに限定されない)を含む様々なタイプの痛みを緩和するために有用である。
【0034】
様々なα−アドレナリン作動性アゴニストが本発明において有用であり、これらには、汎α−2アゴニストおよび汎α−1汎α−2アゴニストならびにα−2Bアゴニストが含まれる。本発明の方法に従って痛みを緩和することにおいて有用なα−アドレナリン作動性アゴニストには、クロニジン、ブリモニジン、チザニジン、デキセメデトミジン、ノルエピネフリン、化合物1および化合物2、ならびに、すべてのそれらの医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が含まれるが、これらに限定されない。様々な選択的α−2Aアンタゴニストもまた本発明において有用である。そのような選択的α−2Aアンタゴニストには、化合物13、ならびにその医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物、BRL48962およびBRL44408、ならびに、それらの医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が含まれるが、これらに限定されない(Young他、Eur. J. Pharmacol.、168:381〜386 (1989))。1つの実施形態において、本発明は、末梢限定の選択的α−2Aアンタゴニストを用いて実施される。
【0035】
様々な投与経路が、本発明の方法に従って痛みを緩和するために有用であり得る。1つの実施形態において、α−アドレナリン作動性アゴニストおよび選択的α−2Aアンタゴニストの両方が末梢投与される。他の実施形態において、α−アドレナリン作動性アゴニストが経口投与または皮下ミニポンプにより投与される。さらなる実施形態において、α−アドレナリン作動性アゴニストが経口投与または皮下ミニポンプにより投与され、選択的α−2Aアンタゴニストが任意の末梢経路によって投与される。さらに他の実施形態において、選択的α−2Aアンタゴニストが経口投与または皮下ミニポンプにより投与される。所望される場合、選択的α−2Aアンタゴニストを経口投与または皮下ミニポンプにより投与することができ、一方で、α−アドレナリン作動性アゴニストが、例えば、経口投与または皮下ミニポンプにより末梢投与される。
【0036】
本発明はまた、有効量のα−アドレナリン作動性アゴニストを含有する医薬組成物と、有効量の選択的α−2Aアンタゴニストを含有する組成物とを対象に投与することによって対象において痛みを緩和する方法で、α−アドレナリン作動性アゴニストおよび選択的α−2Aアンタゴニストが少なくとも3日間の期間にわたって反復的または継続的に投与される方法を提供する。そのような方法において、痛み緩和は、例えば、対象において著しいα−アドレナリン作動性アゴニストレベルの不存在下で継続し得る。本発明は、1つまたは複数の医薬組成物を対象に投与することに依拠する方法を提供する。本明細書中で使用される用語「対象」は、痛みを経験することができる任意の動物を示し、例えば、ヒトまたは他の哺乳動物(例えば、霊長類、ウマ、ウシ、イヌまたはネコなど)を示す。
【0037】
本発明の方法は、急性の痛みおよび慢性の痛みの両方を処置するために、そして、非限定的な例として、起源が神経障害性、内臓性または炎症性である痛みを処置するために使用される。特定の実施形態において、本発明の方法は、神経障害性の痛み、内臓痛、手術後の痛み、ガンまたはガン処置から生じる痛み、および炎症性の痛みを処置するために使用される。
【0038】
急性の痛みおよび慢性の痛みの両方を本発明の方法によって処置することができ、用語「痛み」は急性の痛みおよび慢性の痛みの両方を包含する。本明細書中で使用される用語「急性の痛み」は、傷害(例えば、切り傷、挫傷、火傷など)によって、または、化学的刺激(例えば、カプサイシン(唐辛子の活性成分)にさらされたときに経験する痛みなど)によってもたらされる即時型の、一般には高い閾値の痛みを意味する。本明細書中で使用される用語「慢性の痛み」は、急性の痛みでない痛みを意味し、これには、神経障害性の痛み、内臓痛、炎症性の痛み、頭痛の痛み、筋肉痛および関連痛(これらに限定されない)が含まれる。慢性の痛みは、比較的長い持続期間(例えば、数年)の痛みであり、継続的または間断的であり得ることが理解される。
【0039】
1つの実施形態において、本発明の方法は、「神経障害性の痛み」を処置するために使用される。この場合、本明細書中で使用される「神経障害性の痛み」は、神経に対する傷害から生じる痛みを意味する用語である。神経障害性の痛みは、筋肉または結合組織における小さい皮膚神経または小さい神経が関係する急性の組織傷害によって引き起こされる痛みである侵害受容性の痛みから区別される。神経障害性の痛みとは対照的に、侵害受容性機構が関係する痛みは、通常、継続期間が組織修復の期間に限定され、一般には、利用可能な鎮痛剤またはオピオイドによって緩和される(Myers、Regional Anesthesia、20:173〜184 (1995))。神経障害性の痛みは典型的には、長く持続するか、または慢性的であり、最初の急性組織傷害の数日後または数ヶ月後に現れ得る。神経障害性の痛みは、持続的かつ無意識な痛み、ならびに、通常の場合には痛くない刺激に対する痛い応答である異疼痛、または、痛覚過敏(通常的にはありふれた痛い刺激(例えば、ピン刺激など)に対する強調された応答)を伴う。神経障害性の痛みは、一般には、オピオイド治療に対して抵抗性である(Myers、上掲、(1995))。
【0040】
本発明の方法は、末梢神経、後根神経節、脊髄、脳幹、視床または皮質の外傷、傷害または疾患(これらに限定されない)から生じる神経障害性の痛みを緩和することにおいて有用である。本発明の方法によって処置され得る神経障害性の痛みの例には、神経痛(例えば、帯状疱疹後神経痛など)、求心路遮断の痛み、および糖尿病性神経障害が含まれる。本発明の方法は、痛みの病因にかかわらず、神経障害性の痛みを緩和することにおいて有用であることが理解される。例として、本発明の方法は、末梢神経障害(例えば、神経腫など)から生じる神経障害性の痛み、神経圧迫から生じる神経障害性の痛み、神経の挫傷もしくは伸張または不完全な神経切断(transsection)から生じる神経障害性の痛み、あるいは単神経障害または多発神経障害から生じる神経障害性の痛みを緩和するために使用することができる。さらなる例として、本発明の方法は、後根神経節圧迫などの障害;脊髄の炎症;脊髄の打撲、腫瘍または半側切断;脳幹、視床または皮質の腫瘍または外傷から生じる神経障害性の痛みを緩和することにおいて有用である。
【0041】
上記で示されたように、本発明の方法は、単神経障害または多発神経障害から生じる神経障害性の痛みを緩和することができる。神経障害は、末梢神経系における機能的な乱れまたは病理学的な変化であり、臨床的には感覚ニューロンまたは運動ニューロンの異常によって特徴づけられる。単神経障害の用語は、1つだけの末梢神経が冒されていることを示し、一方、多発神経障害の用語は、いくつかの末梢神経が冒されていることを示す。神経障害の病因は既知または未知であり得る。既知の病因には、神経障害を生じさせる最も一般的な代謝性障害である糖尿病などの疾患状態または毒性状態の合併症、あるいは、刺激、虚血または血管炎が含まれる。本発明の方法によって処置され得る多発神経障害は、ポリオ後症候群、糖尿病、アルコール、アミロイド、毒素、HIV、甲状腺機能低下症、尿毒症、ビタミン欠乏症、化学療法、ddCまたはファブリー病(これらに限定されない)から生じ得る。本発明の方法は、病因が既知または未知であるこれらの神経障害または他の神経障害の痛みを緩和するために使用され得ることが理解される。
【0042】
さらなる非限定的な例として、本発明の方法は、クローン病、潰瘍性大腸炎、胃炎、過敏性腸症候群を含む慢性的な胃腸の炎症;および慢性的な内臓痛、例えば、ガンにより引き起こされる痛み、またはガンの処置に随伴する痛み(例えば、化学療法もしくは放射線療法に随伴する痛み)などを処置するために使用することができる。同様に、本発明の方法は、慢性の炎症性の痛み、例えば、関節炎(例えば、リウマチ様関節炎、痛風性関節炎または変形性関節症など);脊椎炎;または自己免疫疾患(例えば、エリテマトーデスなど)から生じる痛みを処置するために使用することができる。本発明の方法はさらに、頭痛の痛み;筋肉痛;ならびに、物質の乱用または中断に関連する痛み、および、病因が既知または未知である他のタイプの痛みを処置するために使用することができる。
【0043】
本発明の方法のいくつかは、部分的には「α−アドレナリン作動性アゴニスト」に依拠している。本明細書中で使用される「α−アドレナリン作動性アゴニスト」は、1つまたは複数のα−2アドレナリン作動性受容体において、ブリモニジンと比べて25%を超える効力を有する化合物、あるいは、1つまたは複数のα−1アドレナリン作動性受容体において、フェニルエフリンと比べて25%を超える効力を有する化合物を意味する用語である。そのような化合物は、1つまたは複数のα−アドレナリン作動性受容体に対して選択的であり得るか、または非選択的であり得る。従って、アドレナリン作動性アゴニストの用語は、すべてのα−1受容体およびα−2受容体においてアゴニスト活性を有する「汎α−1汎α−2アゴニスト」(例えば、ノルエピネフリンなど);汎α−2アゴニスト;α−2選択的アゴニスト;α−2Bアゴニスト;および、1つだけのα−アドレナリン作動性受容体に対して特異的であるアゴニストを包含するが、これらに限定されない。特定の実施形態において、本発明の方法では、1つまたは複数のα−2アドレナリン作動性受容体において、ブリモニジンと比べて30%を超える効力、40%を超える効力、50%を超える効力、60%を超える効力、70%を超える効力、80%を超える効力、90%を超える効力、100%を超える効力、または200%を超える効力を有するα−アドレナリン作動性アゴニストが利用される。さらなる実施形態において、本発明の方法では、1つまたは複数のα−1アドレナリン作動性受容体において、フェニルエフリンと比べて30%を超える効力、40%を超える効力、50%を超える効力、60%を超える効力、70%を超える効力、80%を超える効力、90%を超える効力、100%を超える効力、または200%を超える効力を有するα−アドレナリン作動性アゴニストが利用される。別の実施形態において、本発明は、顕著なα−2Aアンタゴニスト活性を有しないα−アドレナリン作動性アゴニストに依拠する。
【0044】
効力は、内因的活性としてもまた知られているが、化合物によって達成される最大受容体活性化の尺度であり、α−アドレナリン作動性受容体活性化の任意の受け入れられるアッセイを使用して、例えば、本明細書で後に記載するcAMPアッセイまたは受容体選択増幅技術(RSAT)アッセイを使用して決定することができる。効力は、それぞれの受容体サブタイプについて標準アゴニストの最大作用に対する薬物の最大作用の比率または百分率として表される。α−2受容体の効力が定義される場合、ブリモニジン(UK14304)が、α−2A受容体、α−2B受容体およびα−2C受容体に対する標準アゴニストとして一般に使用され、本発明において標準物として使用される。α−1受容体の効力が定義される場合、フェニルエフリンが、α−1A受容体、α−1B受容体およびα−1D受容体に対する受け入れられる標準アゴニストであり、本発明において標準物として使用される(Messierら、上掲、1995;Conklinら、上掲、1993)。
【0045】
本明細書中に開示されるように、α−2Bアゴニストは、痛みを緩和することにおいて、または、慢性の痛みを期間にわたって緩和するために有用であり得る。本明細書中で使用される用語「α−2Bアゴニスト」は、α−2Bアドレナリン作動性受容体において、ブリモニジンと比べて25%を超える効力を有する化合物を意味する。この用語は、他のα−アドレナリン作動性受容体と比較した場合、α−2B受容体に対して選択的または非選択的のいずれかであるアゴニストを包含することが理解される。従って、用語「α−2Bアゴニスト」は、下記においてさらに記載されるように、汎α−2アゴニストおよび汎α−1汎α−2アゴニスト、ならびに、α−2B受容体に対して選択的または特異的であるアゴニストを包含する。特定の実施形態において、本発明の方法では、α−2Bアドレナリン作動性受容体において、ブリモニジンと比べて30%を超える効力、40%を超える効力、50%を超える効力、60%を超える効力、70%を超える効力、80%を超える効力、90%を超える効力、100%を超える効力、または200%を超える効力を有するα−2Bアゴニストが利用される。例示的なα−2Bアゴニストには、クロニジン、ブリモニジン、化合物1および化合物2、化合物3〜12および14、ならびに、これらの化合物のすべての医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が含まれる。さらなる実施形態において、本発明の方法は、顕著なα−2Aアンタゴニスト活性を有しないα−2Bアゴニストに依拠する。
【0046】
本明細書中で使用される用語「汎α−2アゴニスト」は、α−2Aアドレナリン作動性受容体、α−2Bアドレナリン作動性受容体およびα−2Cアドレナリン作動性受容体のそれぞれにおいて、ブリモニジンと比べて25%を超える効力を有する化合物を意味し、汎α−1汎α−2アゴニストを包含する。様々な汎α−2アゴニストがこの分野では知られており、これらに、クロニジン、ブリモニジン、チザニジン、デキセメデトミジンおよびノルエピネフリンが含まれる。汎α−2アゴニストは、α−2A受容体、α−2B受容体およびα−2C受容体のそれぞれにおいて、ブリモニジンと比べて25%を超える効力を少なくとも有する。特定の実施形態において、本発明の方法は、α−2Aアドレナリン作動性受容体、α−2Bアドレナリン作動性受容体およびα−2Cアドレナリン作動性受容体において、ブリモニジンと比べて30%を超える効力、40%を超える効力、50%を超える効力、60%を超える効力、70%を超える効力、80%を超える効力、90%を超える効力、100%を超える効力、または200%を超える効力を有する汎α−2アゴニストを用いて実施される。汎α−2アゴニストの効力は様々なα−2受容体において異なり得ることが理解される。一例として、汎α−2アゴニストは、α−2A受容体において25%を超える効力、α−2B受容体において80%を超える効力、および、α−2C受容体において40%を超える効力を有し得る。
【0047】
本明細書中で使用される用語「汎α−1汎α−2アゴニスト」は、すべてのα−1受容体において、フェニルエフリンと比べて25%を超える効力を有し、かつ、すべてのα−2アドレナリン作動性受容体において、ブリモニジンと比べて25%を超える効力を有する化合物を意味する。特定の実施形態において、本発明の方法は、フェニルエフリンまたはブリモニジンに対して、それぞれ、すべてのα−1受容体およびα−2受容体において、30%を超える効力、40%を超える効力、50%を超える効力、60%を超える効力、70%を超える効力、80%を超える効力、90%を超える効力、100%を超える効力、または200%を超える効力を有する汎α−1汎α−2アゴニストに依拠する。本明細書中に開示されるように、選択的α−2Aアンタゴニストが、痛みを緩和するために、または、慢性の痛みを長期間にわたって緩和するために、α−アドレナリン作動性アゴニストと一緒に投与される。本明細書中で使用される用語「選択的α−2Aアンタゴニスト」は、(1)α−2Aにおいてブリモニジンと比べて25%未満の効力、(2)α−2Aにおいて100nM未満のKiを有し、さらに、(3)α−2Aよりもα−2Bにおいて少なくとも10倍大きいKiを有するか、または、α−2Bにおいてブリモニジンと比べて25%を超える効力を有する化合物を意味する。この定義から、非選択的なアンタゴニスト(例えば、ラウオルシンなど)がこの用語の範囲に含まれないことは当業者には明らかである。例示的な選択的α−2Aアンタゴニストが、化合物13、BRL48962およびBRL44408として本明細書中に提供される。これらの化合物の医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物もまた本発明において有用である。選択的α−2Aアンタゴニストは末梢限定の化合物であり得る。そのような化合物は、血液脳関門を容易に横断せず、従って、末梢投与されたとき、中枢神経系に入らない。
【0048】
α−2Aアンタゴニスト活性に加えて、「選択的α−2Aアンタゴニスト」もまた、化合物が上記に示された3つの基準を満たすならば、1つまたは複数のアドレナリン作動性受容体または他の受容体においてアゴニスト活性またはアンタゴニスト活性を有し得る。一例として、α−2Cアンタゴニスト活性を有する化合物で、α−2Cにおいて100nM未満のKi、および、α−2Cにおいてブリモニジンに対して25%未満の効力によって特徴づけられ、さらには、(19)α−2Aにおいてブリモニジンに対して25%未満の効力、α−2Aにおいて100nM未満のKi、および(3)α−2Aよりもα−2Bにおいて少なくとも10倍大きいKiを有する化合物が、本明細書中で定義される用語「選択的α−2Aアンタゴニスト」に包含される。同様に、α−2Aアンタゴニスト活性に加えて、α−2Bアゴニスト活性または他のアゴニスト活性を示す化合物もまた、用語「選択的α−2Aアンタゴニスト」に包含される。一例として、化合物13は、α−2Bにおいてブリモニジンに対して約40%の効力によって特徴づけられ、従って、基準(3)を満たし、さらには、(1)α−2Aにおいてブリモニジンと比べて5%の効力、および(2)α−2Aにおいて約0.08nMのKiを有しており、従って、本明細書中で使用される用語「選択的α−2Aアンタゴニスト」の定義の範囲に含まれる。特定の実施形態において、本発明の方法は、Kiが80nM未満、60nM未満、40nM未満、20nM未満、10nM未満、1nM未満または0.1nM未満である選択的α−2Aアンタゴニストを用いて実施される。さらなる実施形態において、本発明の方法は、α−2Aよりもα−2Bにおいて少なくとも20倍大きいKi、少なくとも30倍大きいKi、少なくとも40倍大きいKi、少なくとも50倍大きいKi、少なくとも100倍大きいKi、少なくとも200倍大きいKi、少なくとも500倍大きいKi、または少なくとも1000倍大きいKiを有する選択的α−2Aアンタゴニストを用いて実施される。さらにさらなる実施形態において、本発明の方法は、α−2Bにおいてブリモニジンと比べて30%を超える効力、40%を超える効力、50%を超える効力、60%を超える効力、70%を超える効力、80%を超える効力、90%を超える効力、100%を超える効力、または200%を超える効力を有する選択的α−2Aアンタゴニストを用いて実施される。
【0049】
1つの実施形態において、本発明は、最小限のα−2Bアンタゴニスト活性を有する選択的α−2Aアンタゴニストに依拠する。本明細書中で使用される用語「最小限のα−2Bアンタゴニスト活性を有する選択的α−2Aアンタゴニスト」は、α−2Bにおいて100nMを超えるKiをさらに有する本明細書中上記で定義されるような選択的α−2Aアンタゴニストを意味する。非限定的な例として、そのようなアンタゴニストは、α−2Bにおいて、200nMを超えるKi、300nMを超えるKi、400nMを超えるKi、500nMを超えるKi、1000nMを超えるKi、2000nMを超えるKi、3000nMを超えるKi、4000nMを超えるKi、または5000nMを超えるKiを有し得る。
【0050】
特異的α−2Aアンタゴニストはまた、本発明の方法においてα−2A受容体の活性化を防止するために使用することができる。本明細書中で使用される用語「特異的α−2Aアンタゴニスト」は、(1)α−2Aにおいてブリモニジンに対して25%未満の効力、(2)α−2Aにおいて100nM未満のKiを有し、さらには、(3)α−2A以外のα−アドレナリン作動性受容体のそれぞれにおいて、α−2Aにおけるよりも少なくとも10倍大きいKi、または、ブリモニジンもしくはフェニルエフリンと比べて25%を超える効力のいずれかを有する化合物を意味する。
【0051】
本発明はまた、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストに依拠する組成物および方法を提供する。具体的には、本発明は、同時での鎮静作用を伴わない末梢痛覚消失を生じさせることができる、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを含有する鎮痛剤組成物を提供する。1つの実施形態において、鎮痛剤組成物は、同時での鎮静作用を伴うことなく、また、血圧低下作用の実質的な不存在下で末梢痛覚消失を生じさせる。別の実施形態において、本発明は、同時での鎮静作用を伴うことなく、痛みを少なくとも50%軽減させるために十分な末梢痛覚消失を生じさせる鎮痛剤組成物を提供する。さらなる実施形態において、痛みを少なくとも50%軽減させるために要求される鎮痛剤組成物の用量よりも少なくとも10倍大きい、少なくとも100倍大きい、または少なくとも1000倍大きい用量の鎮痛剤組成物が、運動活動または筋肉活動における20%の軽減を生じさせるために要求される。さらに、さらなる実施形態において、本発明は、同時での鎮静作用を伴うことなく、また、血圧低下作用の実質的な不存在下で、痛みを少なくとも50%軽減させるために十分な末梢痛覚消失を生じさせる鎮痛剤組成物を提供する。別の実施形態において、本発明は、同時での鎮静作用を伴わない末梢痛覚消失を生じさせることができる、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを含有する鎮痛剤組成物で、アゴニストがチオウレア系化合物またはその誘導体でない鎮痛剤組成物を提供する。さらなる実施形態において、本発明は、同時での鎮静作用を伴わない末梢痛覚消失を生じさせることができる、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを含有する鎮痛剤組成物で、アゴニストがチオウレア系化合物または4-イミダゾール系化合物またはその誘導体でない鎮痛剤組成物を提供する。
【0052】
本発明はまた、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストの有効量を含有する医薬組成物を対象に投与し、それにより、同時での鎮静作用を伴わない末梢痛覚消失を生じさせることによって、対象において痛みを緩和する方法を提供する。そのような末梢痛覚消失は、同時での鎮静作用を伴うことなく、例えば、少なくとも50%、痛みを軽減させるために十分であり得る。別の実施形態において、末梢痛覚消失は血圧低下作用の実質的な不存在下で生じる。さらなる実施形態において、この方法は、チオウレア系化合物またはその誘導体でない、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを使用して実施される。また、さらに、さらなる実施形態において、この方法は、チオウレア系化合物または4-イミダゾール系化合物またはその誘導体でない、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを使用して実施される。様々なタイプおよび病因の痛みを本発明の方法に従って緩和することができる。非限定的な例として、本発明の方法は、神経障害性の痛み、例えば、糖尿病性神経障害から生じる痛みなど;内臓痛;手術後の痛み;ガンまたはガン処置から生じる痛み;炎症性の痛み、例えば、関節炎痛または過敏性腸症候群の痛みなど;頭痛の痛みおよび筋肉痛を緩和することにおいて有用であり得る。
【0053】
最小限のα−2Aアゴニスト活性を有する様々なα−アドレナリン作動性アゴニストが本発明の方法において有用であり得る。1つの実施形態において、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストである。そのようなアゴニストは、例えば、チオン系化合物であり、例えば、化合物3または化合物11またはその医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物などであり得る。1つの実施形態において、本発明の方法は、化合物3の(-)-エナンチオマーまたはその医薬的に許容され得る塩もしくはエステルである、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストを用いて実施される。
【0054】
本発明において有用な、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストにはさらに、イミダゾロン系化合物が含まれるが、これらに限定されない。有用なイミダゾロン系の、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、例えば、化合物4またはその医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物であり得る。1つの実施形態において、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、化合物4の(+)-エナンチオマー、またはその医薬的に許容され得る塩、エステルもしくはアミドである。さらなる実施形態において、本発明の方法は、下記のいずれかの、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストを使用して実施される:化合物5、化合物6、化合物7、化合物8、化合物9、化合物14、またはそれらの医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物。最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、経口投与および皮下ミニポンプによる投与(これらに限定されない)を含む様々な経路のいずれかによって末梢投与することができる。
【0055】
本明細書中で使用される用語「末梢痛覚消失」は、末梢投与の後で得られる痛みの軽減を意味する。下記においてさらに説明されるように、末梢投与は、中枢神経系の外側における対象への薬剤の導入を意味し、脊椎または脳に対する直接的な投与でない任意の投与経路を包含する。末梢痛覚消失は、痛みを、例えば、少なくとも30%、40%、50%、60%、70%、80%、90%または100%軽減させるために十分であり得る。本発明の組成物は、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる。末梢鎮静作用失は、本明細書中で使用される場合、運動活動または筋肉活動の減少を意味する用語である。本明細書中で使用される用語「同時での鎮静作用を伴わない」は、運動活動または筋肉活動の減少が1回以上の投薬において末梢痛覚消失に比較的ほとんど随伴しないことを意味する。具体的には、末梢投与されたとき、運動活動または筋肉活動における20%の減少を生じさせるために要求される用量が、痛みを少なくとも50%軽減させるために要求される用量よりも少なくとも3倍大きいならば、薬物は、「同時での鎮静作用を伴わない末梢痛覚消失」を生じさせる。特定の実施形態において、運動活動または筋肉活動における20%の減少を生じさせるために要求される用量は、痛みを少なくとも50%軽減させるために要求される用量よりも少なくとも4倍大きく、または5倍大きく、または6倍大きく、または7倍大きく、または8倍大きく、または9倍大きく、または10倍大きく、または25倍大きく、または50倍大きく、または100倍大きく、または200倍大きく、または500倍大きく、または1000倍大きく、または2000倍大きく、または5000倍大きい。末梢投与後の痛み軽減の程度および鎮静作用の程度を明らかにする様々な方法がこの分野では広く知られており、本明細書で後に記載する。
【0056】
本明細書中で使用される用語「最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニスト」は、(1)α−2Aアドレナリン作動性受容体においてブリモニジンに対して25%未満の効力を有すること、および(2)遺伝子的に変化していない動物において同時での鎮静作用を伴わない末梢痛覚消失を生じさせることができることによってさらに特徴づけられる、上記で定義されるようなα−アドレナリン作動性アゴニストを意味する。効力は、アゴニスト活性の任意の標準的なアッセイを使用して、例えば、本明細書中下記に記載されるcAMPアッセイまたはRSATアッセイなどを使用して測定されることが理解される。化合物3、化合物4、化合物5、化合物6、化合物7、化合物8、化合物9、化合物10、化合物11および化合物12、ならびに、それらの医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストの例として本明細書中に提供される。そのような化合物は、上記の表1に示されるように多様な構造クラスに属する。本発明において有用な、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、下記においてさらに記載されるように、1つまたは複数のα−1受容体において活性を有するα−アドレナリン作動性アゴニスト、最小限のα−2Aアゴニスト活性を有するα−2B/Cアゴニスト、最小限のα−2Aアゴニスト活性を有する特異的α−2Bアゴニスト、および、最小限のα−2Aアゴニスト活性を有する特異的α−2Cアゴニストを包含する。
【0057】
最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、α−アドレナリン作動性アゴニストをスクリーニングすることによって、例えば、様々なインビトロアッセイまたはインビボアッセイで、機能的活性について、α−2Aにおいて25%未満の効力を示すα−アドレナリン作動性アゴニストをスクリーニングすることによって容易に同定することができる。具体的には、そのようなアゴニストは、例えば、スルプロストン感作された痛みの広く受け入れられているモデルを使用して、野生型マウスおよびα−2Aノックアウトマウスの両方において、同時での鎮静作用を伴わない末梢痛覚消失についてアッセイすることができる。最小限よりも大きいα−2Aアゴニスト活性を有するアゴニストは、野生型のスルプロストン感作動物において同時での鎮静作用を伴わない末梢痛覚消失を生じさせないそのような化合物として排除することができ、だが、下記においてさらに記載されるように、同時での鎮静作用を伴わない末梢痛覚消失がα−2Aノックアウトマウスにおいて観測される。特定の実施形態において、本発明の方法は、同時での鎮静作用を伴うことなく、遺伝子的に変化していない動物において、痛みを少なくとも50%軽減させるために、または、少なくとも60%、70%、80%、90%もしくは100%軽減させるために十分な末梢痛覚消失を生じさせる、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを使用して実施される。
【0058】
本明細書中で使用される用語「最小限のα−2Aアゴニスト活性を有するα−2Bアゴニスト」は、(1)α−2B受容体においてブリモニジンに対して25%を超える効力、(2)α−2Bにおいて1000nM未満の効能、または、α−2Aよりもα−2Bにおいて少なくとも100倍大きい効能、(3)α−2A受容体においてブリモニジンに対して25%未満の効力、および(4)遺伝子的に変化していない動物において同時での鎮静作用を伴わない末梢痛覚消失を生じさせる能力を有することによって特徴づけられる化合物を意味する。最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストの例として、本明細書中には、化合物3、化合物4、化合物5、化合物6、化合物7、化合物8、化合物9、化合物10、化合物11、化合物12および化合物14、ならびに、これらの化合物の医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が提供される。本明細書中に例示されるように、これらの化合物は、部分的には、遺伝子的に変化していない動物において同時での鎮静作用を伴わない末梢痛覚消失を生じさせることができることによって特徴づけられる。特定の実施形態において、本発明において有用な、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、α−2Bにおいてブリモニジンと比べて30%を超える効力、40%を超える効力、50%を超える効力、60%を超える効力、70%を超える効力、80%を超える効力、90%を超える効力、100%を超える効力、または200%を超える効力を有する。さらなる実施形態において、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、α−2Aにおいてブリモニジンと比べて20%未満の効力、15%未満の効力、10%未満の効力、5%未満の効力、2%未満の効力または1%未満の効力を有する。さらなる実施形態において、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、α−2Bにおいて900nM未満の効能、800nM未満の効能、700nM未満の効能、600nM未満の効能、500nM未満の効能、400nM未満の効能、300nM未満の効能、200nM未満の効能、100nM未満の効能、50nM未満の効能、10nM未満の効能、または1nM未満の効能を有する。さらにさらなる実施形態において、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、α−2Aよりもα−2Bにおいて少なくとも200倍大きい効能、300倍大きい効能、少なくとも400倍大きい効能、500倍大きい効能、1000倍大きい効能、または10,000倍大きい効能を有する。さらなる実施形態において、本発明の方法は、同時での鎮静作用を伴うことなく、遺伝子的に変化していない動物において、痛みを少なくとも50%軽減させるために、または、少なくとも60%、70%、80%、90%もしくは100%軽減させるために十分な末梢痛覚消失を生じさせることができる、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストに依拠する。
【0059】
本明細書中で使用される用語「最小限のα−2Aアゴニスト活性を有するα−2B/Cアゴニスト」は、(1)α−2B受容体またはα−2C受容体または両者において、ブリモニジンと比べて25%を超える効力、(2)α−2B受容体またはα−2C受容体または両者において1000nM未満の効能、あるいは、α−2B受容体またはα−2C受容体または両者においてα−2A受容体に対して少なくとも100倍大きい効能、(3)α−2A受容体において、ブリモニジンと比べて25%未満の効力、および(4)遺伝子的に変化していない動物において同時での鎮静作用を伴わない末梢痛覚消失を生じさせる能力を有することによって特徴づけられる化合物を意味する。
【0060】
本明細書中で使用される用語「最小限のα−2Aアゴニスト活性を有する特異的α−2Bアゴニスト」は、(1)α−2Bにおいてブリモニジンと比べて25%を超える効力、(2)α−2Bにおいて1000nM未満の効能、または、α−2Aよりもα−2Bにおいて少なくとも100倍大きい効能、(3)α−2Aおよびα−2Cにおいてブリモニジンと比べて25%未満の効力または1/50の効能、(4)すべてのα−1受容体においてフェニルエフリンに対して25%未満の効力または1/50の効能、そして(5)遺伝子的に変化していない動物において同時での鎮静作用を伴わない末梢痛覚消失を生じさせる能力を有することによって特徴づけられる化合物を意味する。
【0061】
同様に、本明細書中で使用される用語「最小限のα−2Aアゴニスト活性を有する特異的α−2Cアゴニスト」は、(1)α−2Cにおいてブリモニジンと比べて25%を超える効力、(2)α−2Cにおいて1000nM未満の効能、または、α−2Aよりもα−2Cにおいて少なくとも100倍大きい効能、(3)α−2Aおよびα−2Bにおいてブリモニジンと比べて25%未満の効力または1/50の効能、(4)すべてのα−1受容体においてフェニルエフリンに対して25%未満の効力または1/50の効能、そして(5)遺伝子的に変化していない動物において同時での鎮静作用を伴わない末梢痛覚消失を生じさせる能力を有することによって特徴づけられる化合物を意味する。
【0062】
アゴニスト活性およびアンタゴニスト活性は、選択性および特異性を含めて、様々な日常的アッセイのいずれかを使用して特徴づけることができ、そのようなアッセイには、受容体選択および増幅技術(RSAT)アッセイ(Messierら、Pharmacol. Toxicol.、76:308〜11 (1995);Conklinら、Nature、363:274〜6 (1993));環状AMPアッセイ(Shimizuら、J. Neurochem.、16:1609〜1619 (1969));およびサイトセンサー微量生理機能測定アッセイ(Neveら、J. Biol. Chem.、267:25748〜25753 (1992))が含まれるが、これらに限定されない。そのようなアッセイは、一般には、1つだけのα−アドレナリン作動性受容体サブタイプのみを自然の状態で発現する細胞を使用して、または、1つだけの組換えα−アドレナリン作動性受容体サブタイプを発現するトランスフェクションされた細胞を使用して行われる。アドレナリン作動性受容体は、ヒト受容体、または、類似する薬理学を有するそのホモログであり得る。本明細書中に開示されるように、RSATアッセイが、ヒトα−2A(c10遺伝子)、ラットα−2B(RNG遺伝子)、ヒトα−2C(c4遺伝子)、ウシα−1A、ハムスターα−1Bおよびラットα−1Dで一過性にトランスフェクションされた細胞を使用して行われた。
【0063】
RSATアッセイでは、コンフルエンス細胞の混合集団における受容体含有細胞の選択的な増殖をもたらす接触阻害の受容体媒介による喪失が測定される。細胞数の増大が、所望される場合にはハイスループットアッセイ形式またはウルトラハイスループットアッセイ形式で、β-ガラクトシダーゼなどの適切な検出可能なマーカー遺伝子を用いて評価される。Gタンパク質(Gq)を活性化する受容体により、増殖応答が誘発される。α−アドレナリン作動性受容体は、通常の場合にはGiに共役しているが、Gi受容体認識ドメインを含有するハイブリッドGqタンパク質(これはGq/i5と呼ばれる)と同時発現させたときにはRSAT応答を活性化する。
【0064】
一例として、RSATアッセイを本質的には下記のように行うことができる。NIH-3T3細胞が15cmディッシュに2x106細胞の密度で置床され、10%子ウシ血清が補充されたダルベッコ改変イーグル培地で維持される。1日後、細胞が、p-SV-β-ガラクトシダーゼをコードする哺乳動物発現プラスミド(5〜10μg)、受容体をコードする哺乳動物発現プラスミド(1〜2μg)、およびGタンパク質をコードする哺乳動物発現プラスミド(1〜2μg)でリン酸カルシウム沈殿によって同時トランスフェクションされる。キャリアDNA(例えば、40μgのサケ精子DNA)もまた、トランスフェクション効率を増大させるために含めることができる。新鮮な培地が翌日に加えられる。1日〜2日後に細胞が集められ、50アッセイ分の量で凍結される。トランスフェクションされた細胞を解凍し、100μlの細胞が、例えば、96ウエルプレートにおいて三連でアッセイされる様々な濃度で、試験される化合物の100μl量に加えられる。インキュベーションが37℃で72時間〜96時間にわたって続けられる。リン酸塩緩衝化生理的食塩水で洗浄した後、β-ガラクトシダーゼ活性が、200μlの発色性基質(リン酸塩緩衝化生理的食塩水における3.5mMのO-ニトロフェニル-β-D-ガラクトピラノシド/0.5%NP-40)を加え、30℃で一晩インキュベーションして、420nmにおける光学密度を測定することによって測定される。吸光度は酵素活性の尺度であり、これは細胞数に依存し、受容体媒介による細胞増殖を反映する。それぞれの受容体における各薬物のEC50および最大作用(効力)が決定される。
【0065】
本発明はさらに、対象において慢性の痛みを長期間にわたって緩和するための方法を提供する。この方法は、慢性の痛みの緩和が、継続した受容体活性化の不存在下で維持されるように、少なくとも3日間の期間にわたって、α−2A受容体活性化の不存在下で鎮痛性α−アドレナリン作動性受容体を対象において活性化することによって実施される。1つの実施形態において、本発明の方法は、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストの有効量を含有する医薬組成物を少なくとも3日間の期間にわたって対象に投与し、その結果、慢性の痛みの緩和が対象において著しいアゴニストレベルの不存在下で維持されるようにすることによって実施される。慢性の痛みの緩和は、対象における著しいアゴニストレベルの不存在下で、例えば、少なくとも3週間にわたって維持され得る。本発明の方法は、任意のタイプの慢性の痛みを長期間にわたって緩和するために使用され得ることが理解される。非限定的な例として、そのような方法は、神経障害性の痛み;内臓痛;手術後の痛み;ガンまたはガン処置から生じる痛み;および炎症性の痛みを長期間にわたって緩和するために使用することができる。
【0066】
慢性の痛みの長期間にわたる緩和は、最小限のα−2Aアゴニスト活性を有する様々なα−アドレナリン作動性アゴニストのいずれかを用いて本発明の方法に従って達成され得る。慢性の痛みの長期間にわたる緩和を、例えば、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストを使用して達成することができる。最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストの例には、チオン系化合物、例えば、化合物3および化合物11、ならびに、それらの医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物などが含まれるが、これらに限定されない。1つの実施形態において、そのようなチオン系の、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、化合物3の(-)-エナンチオマーまたはその医薬的に許容され得る塩もしくはエステルである。最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストの例にはさらに、イミダゾロン系化合物、例えば、化合物4またはその医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物などが含まれるが、これらに限定されない。1つの実施形態において、そのようなイミダゾロン系の、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、化合物4の(+)-エナンチオマーまたはその医薬的に許容され得る塩もしくはエステルである。最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストの例にはまた、化合物5、化合物6、化合物7、化合物8および化合物9、ならびに、それらの医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物が含まれるが、これらに限定されない。本発明の方法において、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、末梢投与経路(例えば、経口投与、または皮下ミニポンプによる投与など)(これらに限定されない)を含む様々な経路のいずれかによって投与することができる。
【0067】
本発明はさらに、対象において慢性の痛みを長期間にわたって緩和するための方法で、有効量のα−アドレナリン作動性アゴニストを含有する医薬組成物と、有効量の選択的α−2Aアンタゴニストを含有する医薬組成物とを少なくとも3日間の期間にわたって対象に投与し、その結果、慢性の痛みの緩和が対象において著しいアゴニストレベルの不存在下で維持されるようにすることによる方法を提供する。慢性の痛みの緩和は、対象において著しいアゴニストレベルの不存在下で、例えば、少なくとも3週間にわたって維持され得る。様々なα−アドレナリン作動性アゴニストが本発明において有用であり、これらには、クロニジン、ブリモニジン、チザニジン、デキセメデトミジン、ノルエピネフリン、ならびに、他の汎α−2アゴニストおよび汎α−1汎α−2アゴニスト、そして同様に、化合物1または化合物2、ならびにそれらの医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が含まれる。同様に、様々な選択的α−2Aアンタゴニストが、慢性の痛みの長期間にわたる緩和において有用であり、これらには、化合物13ならびにその医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が含まれるが、これらに限定されない。様々な投与経路が、下記においてさらに議論されるように、慢性の痛みを長期間にわたって緩和するための医薬組成物を送達するために有用であることが理解される。そのような投与経路には、末梢投与、例えば、経口投与、または皮下ミニポンプによる投与などが包含されるが、これらに限定されない。
【0068】
本発明の方法は慢性の痛みの長期間にわたる緩和をもたらす。本明細書中で使用される用語「長期間にわたる緩和(長期間にわたって緩和する)」は、医薬組成物を最後に投与した後の少なくとも10日間にわたって持続する、痛みの著しい軽減を意味する。特定の実施形態において、緩和は、医薬組成物を最後に投与した後、少なくとも14日間、または少なくとも21日間、または少なくとも28日間、または少なくとも60日間、または少なくとも90日間にわたって持続する。さらなる実施形態において、本発明は、慢性の痛みを長期間にわたって緩和するための方法で、医薬組成物を最後に投与した後、少なくとも10日間、または少なくとも14日間、または少なくとも21日間、または少なくとも28日間、または少なくとも60日間、または少なくとも90日間にわたって痛みを少なくとも50%軽減させる方法を提供する。さらなる実施形態において、本発明は、慢性の痛みを長期間にわたって緩和するための方法で、医薬組成物を最後に投与した後、少なくとも10日間、または少なくとも14日間、または少なくとも21日間、または少なくとも28日間、または少なくとも60日間、または少なくとも90日間にわたって痛みを少なくとも80%軽減させる方法を提供する。他の実施形態において、本発明は、慢性の痛みを長期間にわたって緩和するための方法で、医薬組成物を最後に投与した後、少なくとも10日間、または少なくとも14日間、または少なくとも21日間、または少なくとも28日間、または少なくとも60日間、または少なくとも90日間にわたって痛みを少なくとも90%軽減させる方法を提供する。
【0069】
慢性の痛みを長期間にわたって緩和するための方法は、最小限のα−2A活性を有するα−アドレナリン作動性アゴニストを少なくとも3日間の期間にわたって投与することによって実施される。アゴニストは、少なくとも3日間の期間にわたって、例えば、3日間、4日間、5日間、6日間、7日間、8日間、9日間または10日間にわたって、反復した投薬または継続した投薬によって投与され得る。非限定的な例として、最小限のα−2A活性を有するα−アドレナリン作動性アゴニストを、3日間にわたって1日に3回、または、4日間にわたって1日に3回投与することができ、例えば、3日間にわたって1日に3回の経口投与、または、4日間にわたって1日に3回の経口投与により投与することができる。さらなる例として、最小限のα−2A活性を有するα−アドレナリン作動性アゴニストを継続的に投与することができ、例えば、埋め込まれた注入ミニポンプを介して静脈内投与することができ、または、3日間、4日間、5日間、6日間もしくは7日間にわたる持続放出配合物を使用して投与することができる。
【0070】
徐放性配合物が、慢性の痛みを長期間にわたって緩和するための本発明の方法において有用であり得ることが理解される。反復した投与が使用される場合、投与頻度は、部分的には、アゴニストの半減期に依存することがさらに理解される。所望される場合、本発明の方法は、長い半減期(例えば、少なくとも24時間、36時間、48時間または72時間の半減期)を有するアゴニストを1回だけ、または、ちょうど2回もしくは3回、投薬することによって実施することができる。
【0071】
種々の薬物送達手段が本発明の方法において組み合わせられ得ることが理解される。一例として、1日目における連続した静脈内投与を、少なくとも3日間の期間にわたってα−2A受容体活性化の不存在下で鎮痛性のα−アドレナリン作動性受容体を活性化するために、2日目および3日目での反復した経口投薬と組み合わせることができ、その結果、慢性の痛みの緩和が前記対象において著しいアゴニストレベルの不存在下で維持されるようになる。本明細書中下記においてさらに詳述されるように、投薬の頻度および継続期間は、部分的には、所望される緩和およびアゴニストの半減期に依存すること、そして、様々な投与経路が本発明の方法において有用であることが理解される。
【0072】
本発明によりまた、指定されたアゴニストまたはアンタゴニストを表す式に由来する医薬的に許容され得る塩、エステルおよびアミドが包含される。本発明において有用なアゴニストおよびアンタゴニストの好適な医薬的に許容され得る塩には、酸付加塩(これは、例えば、アゴニストまたはアンタゴニストの溶液を適切な酸(例えば、塩酸、硫酸、フマル酸、マレイン酸、コハク酸、酢酸、安息香酸、クエン酸、酒石酸、炭酸またはリン酸など)の溶液と混合することによって形成させることができる)が含まれるが、これらに限定されない。アゴニストまたはアンタゴニストが酸性成分を有する場合、その好適な医薬的に許容され得る塩には、アルカリ塩(例えば、ナトリウム塩またはカリウム塩など)、アルカリ土類塩(例えば、カルシウム塩またはマグネシウム塩など)、および、好適な有機リガンドと形成される塩(例えば、第四級アンモニウム塩)が含まれ得る。代表的な医薬的に許容され得る塩には、酢酸塩、ベンゼンスルホン酸塩、安息香酸塩、重炭酸塩、重硫酸塩、重酒石酸塩、ホウ酸塩、臭化物、カルシウムエデタート、カムシラート、炭酸塩、塩化物、クラブラン酸塩、クエン酸塩、二塩酸塩、エデト酸塩、エジシラート、エストラート、エシラート、フマル酸塩、グルセプタート、グルコン酸塩、グルタミン酸、グリコリルアルサニル酸塩、ヘキシルレゾルシン酸塩、ヒドラバミン、臭化水素酸塩、塩酸塩、ヒドロキシナフトエ酸塩、ヨウ化物、イソチオン酸塩、乳酸塩、ラクトビオン酸塩、ラウリン酸塩、リンゴ酸塩、マレイン酸塩、マンデル酸塩、メシラート、メチルブロミド、メチルニトラート、メチル硫酸塩、ムチン酸塩、ナプシラート、硝酸塩、N-メチルグルカミンアンモニウム塩、オレイン酸塩、パモ酸塩(エムボナート)、パルミチン酸塩、パントテン酸塩、リン酸塩/二リン酸塩、ポリガラクツロン酸塩、サリチル酸塩、ステアリン酸塩、硫酸塩、塩基性酢酸塩、コハク酸塩、タンニン酸塩、酒石酸塩、テオクラート、トシラート、トリエチオジドおよび吉草酸塩が含まれるが、これらに限定されない。
【0073】
本発明において有用なアゴニストおよびアンタゴニストの官能基を、化合物の薬理学的利用性を増強するために修飾することができる。そのような修飾は十分に化学の当業者の知識の範囲内であり、それらには、示されたアゴニストまたはアンタゴニストのエステル、アミド、エーテル、N-オキシドおよびプロドラッグが含まれるが、これらに限定されない。アゴニストまたはアンタゴニストの活性を増強することができる修飾の例には、例えば、エステル化、例えば、C1〜C6アルキルエステル、好ましくはC1〜C4アルキルエステルの形成が含まれ、この場合、アルキル基は直鎖または分枝鎖である。他の許容され得るエステルには、例えば、C5〜C7シクロアルキルエステルおよびアリールアルキルエステル(例えば、ベンジルエステルなど)が含まれる。そのようなエステルは、有機化学の技術分野で広く知られている従来の様々な方法を使用して、本明細書中に記載される化合物から調製することができる。
【0074】
他の医薬的に許容され得る修飾には、アミドの形成が含まれる。有用なアミド修飾には、例えば、アンモニア;第一級のC1〜C6ジアルキルアミン(この場合、アルキル基は直鎖または分枝鎖である);および、様々な置換を有するアリールアミンに由来するアミドが含まれる。第二級アミンの場合、アミンはまた5員環または6員環の形態であり得る。これらのアミドおよび他のアミドを調製するための様々な方法がこの分野では広く知られている。
【0075】
本発明において有用なアゴニストまたはアンタゴニストが少なくとも1つのキラル中心を有する場合、そのアゴニストまたはアンタゴニストは化学的に異なるエナンチオマーとして存在し得ることが理解される。さらに、アゴニストまたはアンタゴニストが2つ以上のキラル中心を有する場合、その化合物はジアステレオマーとして存在する。そのような異性体およびその混合物はすべてが、示されたアゴニストまたはアンタゴニストの範囲内に包含される。同様に、アゴニストまたはアンタゴニストが、その構造が互変異性体として存在することを可能にする構造的配置を有する場合、そのような互変異性体は、示されたアゴニストまたはアンタゴニストの範囲内に包含される。さらに、結晶形態において、アゴニストまたはアンタゴニストが多型体として存在する場合がある。溶媒が存在する場合、アゴニストは、例えば、水または一般的な有機溶媒との溶媒和物を形成する場合がある。そのような多型体、水和物および他の溶媒和物もまた、本明細書中で定義されるような示されたアゴニストまたはアンタゴニストの範囲内に包含される。
【0076】
本発明において有用なアゴニストまたはアンタゴニストは一般には医薬組成物で投与される。所望される場合、組成物は、場合により、同じ医薬組成物または異なる医薬組成物において、また、同じ投与経路または異なる投与経路によって、1つまたは複数の他の治療剤または鎮痛性物質と一緒に投与することができる。一例として、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストをガバペンチンなどの鎮痛剤と一緒に投与することができる。別の例として、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを、静脈内カクテルで、1つまたは複数のガン化学療法剤と一緒に投与することができる。
【0077】
本発明において有用な医薬組成物は、活性なアゴニストまたはアンタゴニストを含み、さらには、所望される場合、対象に投与されたとき、長期間または永続的な有害作用を実質的に有しない任意のキャリアまたは希釈剤である医薬的に許容され得るキャリアまたは希釈剤などの賦形剤を含むことができる。そのような賦形剤は、一般には、活性な化合物と混合されるか、または、活性な化合物を希釈もしくは包むことができる。キャリアは、活性な化合物に対する賦形剤またはビヒクルとして作用する固体または半固体または液体の薬剤であり得る。医薬的に許容され得るキャリアおよび希釈剤の例には、水、例えば、蒸留水または脱イオン水など;生理的食塩水;および他の水性媒体が含まれるが、これらに限定されない。有効成分は、所望するキャリアまたは希釈剤において可溶性であり得るか、または、所望するキャリアまたは希釈剤における懸濁物として送達され得ることが理解される。
【0078】
医薬組成物はさらに、所望する場合には、乳化剤、湿潤化剤、甘味剤もしくは矯味矯臭剤、張性調節剤、保存剤、緩衝剤または抗酸化剤などの1つまたは複数の剤を含むことができる。医薬組成物において有用な張性調節剤には、塩(例えば、塩化ナトリウム、塩化カリウムなど)、マンニトールまたはグリセリン、および他の医薬的に許容され得る張性調節剤が含まれる。本発明の医薬組成物において有用な保存剤には、塩化ベンザルコニウム、クロロブタノール、チメロサール、酢酸フェニル水銀および硝酸フェニル水銀が含まれるが、これらに限定されない。pHを調節するための様々な緩衝剤および手段を、医薬組成物を調製するために使用することができ、これらには、酢酸塩緩衝剤、クエン酸塩緩衝剤、リン酸塩緩衝剤およびホウ酸塩緩衝剤が含まれるが、これらに限定されない。同様に、本発明の医薬組成物において有用な様々な抗酸化剤がこの分野では広く知られており、これらには、例えば、メタ重亜硫酸ナトリウム、チオ硫酸ナトリウム、アセチルシステイン、ブチル化ヒドロキシアニソールおよびブチル化ヒドロキシトルエンが含まれる。薬理学の分野で知られているこれらの物質および他の物質が本発明において有用な医薬組成物に含まれ得ることが理解される。
【0079】
本発明のアゴニストおよびアンタゴニストは有効量で投与される。そのような有効量は、一般には、所望される治療効果を達成するために必要な最少用量であり、これは、例えば、大ざっぱには、痛みにより引き超される不快感を許容可能なレベルに軽減させるために必要なそのような量であり得る。そのような用量は、一般には、0.1mg/日〜1000mg/日の範囲であり、例えば、0.1mg/日〜500mg/日、0.5mg/日〜500mg/日、0.5mg/日〜100mg/日、0.5mg/日〜50mg/日、0.5mg/日〜20mg/日、0.5mg/日〜10mg/日または0.5mg/日〜5mg/日の範囲であり得るが、投与される実際の量は、痛みの重篤度、患者の年齢および体重、患者の全身的な身体状態、痛みの原因、および投与経路を含む関連した状況を考慮に入れて医師によって決定される。坐薬および持続放出配合物は本発明において有用であり得るし、これらには、例えば、皮膚パッチ、皮膚表面または皮膚下に置くための配合物、および筋肉内注射用配合物が含まれる。
【0080】
本発明の方法において有用な医薬組成物は、例えば、処置される痛みのタイプ、組成物に含められるアゴニストまたはアンタゴニスト、ならびに、対象の病歴、危険性因子および症状に依存して、様々な手段によって対象に投与することができる。本発明の方法のために好適な投与経路には、全身投与および局所投与の両方が含まれる。非限定的な例として、痛みを緩和するために、または、慢性の痛みを長期間にわたって緩和するために有用な医薬組成物は、経口投与もしくは皮下ミニポンプによって、皮膚パッチによって、静脈内注射、皮下注射もしくは筋肉内注射によって、局所的な滴剤、クリーム、ゲルもしくは軟膏によって、移植もしくは注入された持続放出配合物として、皮下ミニポンプもしくは埋め込まれたデバイスによって、クモ膜下ポンプもしくはクモ膜下注射によって、または硬膜外注射によって投与することができる。
【0081】
特定の実施形態において、本発明の方法は、アゴニストを含有する医薬組成物、アンタゴニストを含有する医薬組成物、または両方を末梢投与することによって実施される。一例として、α−アドレナリン作動性アゴニストを含有する医薬組成物、および選択的α−2Aアンタゴニストを含有する医薬組成物の一方または両方を末梢投与することができる。本明細書中で使用される用語「末梢投与」または用語「末梢投与される」は、薬剤を中枢神経系の外側で対象に導入することを意味する。末梢投与は、脊椎または脳への直接的な投与ではない任意の投与経路を包含する。そのため、クモ膜下投与および硬膜外投与、ならびに頭蓋注射または頭蓋埋め込みは用語「末梢投与」または用語「末梢投与される」の範囲に含まれないことは明らかである。さらに、いくつかの鎮痛剤は血液脳関門を横断することができ、従って、末梢投与後、中枢神経系および末梢神経系の全体に分布することは明らかである。
【0082】
末梢投与は局所的または全身的であり得る。局所投与では、局所投与された部位およびその周りに対して、投与部位から遠位の領域よりも著しく多い量の医薬組成物が送達される。全身投与では、本質的には対象の末梢神経系全体に対する医薬組成物の送達がもたらされ、そしてまた、組成物の性質に依存して中枢神経系への送達がもたらされることがある。
【0083】
本発明の方法において有用な末梢投与経路には、経口投与、局所投与、静脈内注射または他の注射、および埋め込まれたミニポンプまたは他の持続放出デバイスまたは配合物が包含されるが、これらに限定されない。本発明において有用な医薬組成物は、例えば、錠剤、液剤、カプセル剤または粉末剤などでの任意の許容され得る形態で経口投与によって;静脈内注射、腹腔内注射、筋肉内注射、皮下注射または非経口注射によって;経皮拡散またはエレクトロホレシスによって;滴剤、クリーム、ゲルまたは軟膏などでの任意の許容され得る形態で局所的に;また、ミニポンプまたは他の埋め込まれた持続放出デバイスまたは配合物によって末梢投与することができる。
【0084】
いくつかの実施形態では、本発明は、有効量のα−アドレナリン作動性アゴニストを含有する医薬組成物と、有効量の選択的α−2Aアンタゴニストを含有する医薬組成物との両方を対象に投与することによって実施される。そのような「混合治療」では、アゴニストおよびアンタゴニストが、同じ医薬組成物または異なる医薬組成物で、また、同じ投与経路または異なる投与経路によって独立して投与され得るか、または同時に送達され得ることが理解される。一例として、アゴニストおよびアンタゴニストの両方を経口投与することができ、この場合、アゴニストが1日に2回与えられ、アンタゴニストが1日に1回与えられる。別の例として、アゴニストを硬膜外に投与することができ、その一方で、アンタゴニストが経口投与される。さらなる例として、アゴニストおよびアンタゴニストを静脈内「カクテル」において一緒に投与することができる。典型的な局所眼科用組成物の成分を以下の表1に例示するがこれに限定されない:
【表1】
【0085】
本発明はまた、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングする方法で、α−2A受容体を、鎮痛活性を有するα−アドレナリン作動性アゴニストと接触させること;および、アゴニストがα−2Aアゴニスト活性を有するかどうかを明らかにすることによって行われる方法を提供する。この場合、α−2Aアゴニスト活性の不存在により、鎮痛活性を有するα−アドレナリン作動性アゴニストが、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0086】
本発明ではまた、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングする方法で、α−2A受容体を薬剤と接触させること;薬剤がα−2Aアゴニスト活性を有するかどうかを明らかにすること;α−2B受容体を薬剤と接触させること;および、薬剤がα−2Bアゴニスト活性を有するかどうかを明らかにすることによって行われる方法を提供する。この場合、α−2Aアゴニスト活性の不存在およびα−2Bアゴニスト活性の存在により、薬剤が、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0087】
本発明はまた、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングする方法で、野生型レベルのα−2A受容体活性を少なくとも有するコントロール動物にα−アドレナリン作動性アゴニストを末梢投与すること;コントロール動物における痛覚消失についてアッセイすること;低下したレベルのα−2A受容体の発現または活性を有する対応する動物に、コントロール動物に投与された量と類似する量またはそれよりも大きい量のα−アドレナリン作動性アゴニストを末梢投与すること;および、低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失についてアッセイすることによって行われる方法を提供する。この場合、コントロール動物における痛覚消失の不存在、および低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失の存在により、α−アドレナリン作動性アゴニストが過度なα−2Aアゴニスト活性を有することが示され、かつ、コントロール動物における痛覚消失の存在、および低下したレベルのα−2A受容体の発現または活性を有する前記対応する動物における痛覚消失の存在により、α−アドレナリン作動性アゴニストが、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0088】
本発明のそのような方法において、コントロール動物は、例えば、両方のα−2A受容体遺伝子座において野生型であり得る。1つの実施形態において、コントロール動物は、野生型マウスなどの野生型動物である。様々な対応する動物が本発明のスクリーニング方法において有用である。1つの実施形態において、本発明は、α−2A受容体遺伝子座においてホモ接合の点変異を有する対応する動物を用いて実施される。別の実施形態において、本発明は、α−2A受容体コード配列の内部に点変異を有する対応する動物を用いて実施される。そのような点変異は、例えば、Asp79と類似の残基で生じることがあり、例えば、Asp79からAsnへの変異であり得る。さらなる実施形態において、本発明は、ホモ接合のα−2Aノックアウト変異を有する対応する動物を用いて実施される。様々な方法論が、本発明の方法において痛覚消失についてアッセイするために使用され得ることが理解され、そのような方法論には、スルプロストン感作化後の痛覚消失についてスクリーニングすることが含まれるが、これに限定されない。
【0089】
所望される場合には、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングするための本発明の方法は、(a)野生型レベルのα−2A受容体活性およびα−2B受容体活性を少なくとも有するコントロール動物にα−アドレナリン作動性アゴニストを末梢投与すること;(b)コントロール動物における痛覚消失についてアッセイすること;(c)低下したレベルのα−2A受容体の発現または活性を有する対応する動物に、コントロール動物に投与された量と類似する量またはそれよりも大きい量のα−アドレナリン作動性アゴニストを末梢投与すること;(d)低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失についてアッセイすること;(e)低下したレベルのα−2B受容体の発現または活性を有する対応する動物にα−アドレナリン作動性アゴニストを末梢投与すること;および(f)低下したレベルのα−2B受容体の発現または活性を有する対応する動物における痛覚消失についてアッセイすることによって実施することができ、この場合、コントロール動物における痛覚消失の不存在、および低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失の存在により、α−アドレナリン作動性アゴニストが過度なα−2Aアゴニスト活性を有することが示され、かつ、コントロール動物における痛覚消失の存在、低下したレベルのα−2A受容体の発現または活性を有する前記対応する動物における痛覚消失の存在、および低下したレベルのα−2B受容体の発現または活性を有する対応する動物における痛覚消失の不存在により、α−アドレナリン作動性アゴニストが、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0090】
本発明はさらに、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングする方法で、野生型レベルのα−2B受容体活性を少なくとも有するコントロール動物にα−アドレナリン作動性アゴニストを末梢投与すること;コントロール動物における痛覚消失についてアッセイすること;低下したレベルのα−2B受容体の発現または活性を有する対応する動物にα−アドレナリン作動性アゴニストを末梢投与すること;および、低下したレベルのα−2B受容体の発現または活性を有する対応する動物における痛覚消失についてアッセイすることによる方法を提供する。この場合、コントロール動物における痛覚消失の存在、および低下したレベルのα−2B受容体の発現または活性を有する対応する動物における痛覚消失の不存在により、α−アドレナリン作動性アゴニストが、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0091】
本発明のそのような方法は、様々なコントロール動物を用いて、例えば、両方のα−2B受容体遺伝子座において野生型であるコントロール動物を用いて実施することができる。1つの実施形態において、コントロール動物は野生型動物である。さらなる実施形態において、コントロール動物は野生型マウスである。同様に、様々な対応する動物が本発明のスクリーニング方法において有用であり、対応する動物には、ヘテロ接合のα−2Bノックアウト変異またはホモ接合のα−2Bノックアウト変異を有する対応する動物が含まれる。痛覚消失は、様々な方法論のいずれかを使用してアッセイすることができる。1つの実施形態において、痛覚消失はスルプロストン感作化後にアッセイされる。
【0092】
本明細書中で使用される用語「コントロール動物」は、痛みを経験することができる任意の動物を意味する。コントロール動物は、例えば、内因性α−2A受容体の野生型レベルを発現することができるか、または、内因性α−2Aに加えてα−2A受容体の導入遺伝子を発現することができる。コントロール動物が両方のα−2A受容体遺伝子座において「野生型」である場合、コントロール動物は、自然界で通常的に見出されるレベルで天然に存在し、かつ発現するα−2A受容体配列を有する。同様に、コントロール動物が両方のα−2B受容体遺伝子座において「野生型」である場合、コントロール動物は、自然界で通常的に見出されるレベルで天然に存在し、かつ発現するα−2B受容体配列を有する。
【0093】
本明細書中で使用される用語「野生型動物」は、分子遺伝学により変化していない、マウスまたはラットなどの動物を意味する。「野生型動物」は、自然界で見出される野性型動物であり得るか、または、十分に特徴づけられた遺伝学的システムを有する同系交配動物もしくは他の実験室系統であり得る。本明細書中で使用される用語「対応する動物」は、低下したレベルのα−2A受容体の発現もしくは活性を有するか、または、低下したレベルのα−2B受容体の発現もしくは活性を有する、コントロール動物と同じ種の動物を意味する。対応する動物は、一般には、1つもしくは複数のα−2A受容体遺伝子座または1つもしくは複数のα−2B受容体遺伝子座を除いて、コントロール動物と遺伝子的に同一である。非限定的な例として、対応する動物は、α−2A遺伝子の一方もしくは両方またはα−2B遺伝子の一方もしくは両方を完全に有しないことがあり、あるいは、α−2A受容体コード配列もしくはα−2B受容体コード配列における欠失、挿入もしくは点変異、または、α−2Aもしくはα−2Bの発現を低下させるか、もしくは除去する、5'調節配列もしくは3'調節配列における欠失、挿入もしくは点変異を有することがあり、あるいは、低下した活性を有するα−2A受容体もしくはα−2B受容体(例えば、Asp79からAsnへの変異(これは約80%の受容体活性をなくす)を有するネズミα−2A受容体など)の野生型レベルを発現することがある。従って、1つの実施形態において、本発明のスクリーニング方法は、Asp79と類似の残基におけるホモ接合の点変異による低下したレベルのα−2A受容体の発現または活性を有する対応する動物を用いて実施される。そのような「類似の」残基は、ネズミα−2A受容体のホモログにおいて同じ相対的な位置に存在するそのようなアルギニン残基である。α−2Aノックアウトマウスおよびα−2Bノックアウトマウスの様々な系統、ならびに、低下したレベルのα−2A受容体の発現または活性をもたらすホモ接合の点変異を有する様々な系統がこの分野では広く知られているか、または、標準的な方法によって調製することができる(米国特許第5869079および5443505)。
【0094】
以下の実施例は説明を意図するものであり、本発明を限定するものではない。
【0095】
実施例1
感覚過敏の種々のメカニズムを有するマウスモデル
本実施例は、α−2Aおよびα−2Cノックアウトマウスの交感神経系の増大した緊張が、α−1A受容体活性化による触覚過敏の誘導を増大することを示すものである。
【0096】
A.スルプロストンーフェニレフィン誘発性触覚過敏の種々のメカニズム
野生型マウスおよびα−2Aノックアウトマウスにおける異疼痛に対するアッセイが行われた。簡単に記載すると、マウスが5匹〜6匹の動物群に分けられた。コントロールマウスには5μlのDMSOが投与され、一方、処置マウスには、様々な用量の示された薬剤を含有する5μlのDMSOが注射された。クモ膜下注射の後、各マウスは、観察のために、木材チップが底に敷かれた個々の13x8.5x13cmのプレキシガラスの囲いの中に入れられた。異疼痛が50分間の期間にわたって5分毎に1回評価され、応答が15分〜50分の時間枠で8回記録された。異疼痛は、小さいはけでマウスの側腹を軽くなでることによって評価され、下記のようにランク付けされた:0、反応なし;1、接触プローブから逃避しようとして軽く鳴くこと;2、激しい鳴き声が接触プローブによって誘発され、プローブに噛み付き、逃避しようと激しく努力すること。各動物に対する8回のスコアが合計され、群についての平均値が、平均総スコアを得るために求められた。
【0097】
非選択的α−2アンタゴニストのラウオルシンが、α−2Aノックアウトマウスにおいて、パラ-アミノ-クロニジン(PAC)によりもたらされる末梢痛覚消失に影響を及ぼすその能力についてアッセイされた。スルプロストンで感作されたα−2Aノックアウトマウスが、腹腔内注射によって100μg/kgで送達されたPACで処置された。上記に記載されるように、PACはα−2Aノックアウトマウスにおいて著しい痛覚消失を誘導した。しかしながら、図3に示されるように、PACの鎮痛作用はラウオルシン(0.3μg/kg)の腹腔内注射によって阻止された。これらの結果は、末梢のα−2受容体がα−2Aノックアウトマウスにおいて痛覚消失を媒介することを示している。
【0098】
鎮静作用が、下記のように、暗くしたチャンバーにおける探索行動を評価することによって分析された。マウスの体重を測定し、試験化合物を、5μlの体積でのクモ膜下注射によって、または1ml/kgの体積での腹腔内注射によって、示された用量で投与した。注射後5分〜30分での痛覚消失の測定に対応する所定の時点で、動物の活動が、ジギコムアナライザーチャンバー(Omnitech Electronic;Columbus、OH)にマウスを入れることによって自動的に測定された。ジギコムアナライザーチャンバーは、箱を十字に交差し、動物が動き回るときに遮断される光電池ビームを含む;チャンバーは、床の高さを上げることによってマウス用に変更された。全体的な動物の活動のコンピューター分析が5分間の期間にわたって行われた。学習した行動はデータに影響を及ぼし得るので、所与の動物はいずれもこのプロトコルのために多くても2回使用された。すべての動物には、研究の間に少なくとも2週間の休止期間が与えられた。
【0099】
ラット神経結紮モデルは、末梢の神経障害性の痛みの十分に受け入れられているモデルである。Chungモデルにおいて、左脊髄神経のL-5およびL-6の部分的結紮は、影響を受ける左足に、軽い接触に対する長く持続する過敏性をもたらす。この過敏性は、灼熱痛の神経障害性状態のヒトが経験する痛みと類似している(KimおよびChung、Pain、50:355-363(1992))。
【0100】
Chungラットにクモ膜下注射によって投与されたとき、汎α−2アゴニストのクロニジンは、鎮静作用から分離可能な著しい痛覚消失をもたらし、一方、腹腔内投与は、鎮静作用の不存在下で目立たない痛覚消失を生じさせたにすぎなかった。血液脳関門を容易に横断しない化合物である化合物1およびPACもまた、Chungラットモデルにおいて活性についてアッセイされた。予想されたように、いずれの化合物も、腹腔内投与されたとき、鎮静をもたらさない用量で著しい鎮痛作用を有しなかった。
【表2】
【0101】
選択的α−2Aアンタゴニストの化合物13とのクロニジンの同時投与は、腹腔内投与されたクロニジンの鎮痛作用の用量応答を左側に移動させた;0.3μg/kgの化合物13と組み合わせられたとき、30μg/kgが、劇的な異疼痛の逆転を得るために要求されただけであった(図4Aを参照のこと)。クロニジンのこの濃度は活動を減少させるが、化合物13の不存在下で異疼痛を逆転させる100μg/kgの用量ほどの鎮痛作用を全く有していなかった。
α−2のアウトマウスはDixon他(Ann. Rev. Pharmacol. Toxicol.、20:441-462(1980))の方法を使用して決定された。薬物後の閾値が薬物前の閾値と比較され、触覚感受性の逆転率(%)が15.1グラムの正常な閾値に基づいて計算された。この結果は、異疼痛の逆転%として表され、これは正常ラット(100%)に対する痛み閾値の逆転率を反映している。EP1受容体アンタゴニスト
【化2】
は米国特許第5843942に記載されているとおりに合成する。メマンチンは、最小限のα−2Aアゴニスト活性を有する、構造クラスが異なるα−アドレナリン作動性アゴニストが末梢投与され、Chungラットモデルにおいて痛みを緩和させる能力についてアッセイされた。表2には、チオン系化合物3、イミダゾロン系化合物4、チアゾール系化合物5、オキサゾール系化合物6、チオウレア系化合物7、および4-イミダゾール系化合物14を用いて得られた結果が示される。これらの化合物はそれぞれが、構造的に異なっているが、最小限のα−2Aアゴニスト活性を有するα2-B/C選択的α−アドレナリン作動性アゴニストである。一例として、化合物14は、最小限のα−2Aアゴニスト活性を有し、それにもかかわらず、著しいα−1アゴニスト活性を有するα−2B選択的アゴニストである。
【0102】
クロニジンおよび化合物3に対する鎮静作用および鎮痛作用の用量応答曲線の全面的な比較を、クロニジンについては20μg/kg〜100μg/kgの単回腹腔内用量、化合物3については1μg/kg〜1000μg/kgの単回腹腔内用量を使用して行った。異疼痛逆転の割合および全体的な活動の減少が上記のように決定された。図4Cに示されるように、クロニジンの鎮静作用が、痛覚消失を生じさせる用量よりも低い用量で生じた。具体的には、クロニジンは、著しい痛覚消失を生じさせる100μg/kgの用量で極めて大きい鎮静作用を有した。これらの結果は、クロニジンの末梢投薬からもたらされる痛覚消失が鎮静作用から分離可能でないことを明らかにしている。対照的に、図4Dに示される結果は、化合物3が、鎮静作用を生じさせることなく著しい痛覚消失を生じさせたことを明らかにしている。具体的には、鎮静作用が、異疼痛の強固な逆転を生じさせる用量よりも100倍大きい用量で生じなかった。これらの結果は、最小限のα−2Aアゴニスト活性を有するα−2Bアドレナリン作動性アゴニストが、同時での鎮静作用を伴うことなく80%の異疼痛の逆転を生じさせ得ることを明らかにしている。
【0103】
まとめると、これらの結果は、全身投薬の後で目立たない鎮痛活性を伴う非選択的α−アドレナリン作動性アゴニストが、α−2A受容体の活性化を妨げる薬剤と組み合わせられたとき、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる非常に効果的な薬剤に変わり得ることを明らかにしている。これらの結果はまた、最小限のα−2A受容体アゴニスト活性のみを有する選択的α−2アゴニストの鎮痛作用が、以前に記載されたα−アドレナリン作動性薬剤の鎮痛作用とは異なることを明らかにしている。
【0104】
構造が異なるα−アドレナリン作動性アゴニストが、長期間にわたる投薬の後のChungモデルラットにおける慢性の痛みの長期間にわたる緩和を生じさせる能力についてアッセイされた。具体的には、Chungモデル動物に、ビヒクルコントロール、または最小限のα2-A活性を有する下記のα−アドレナリン作動性アゴニストの0.1mg/kg/時間が、7日間、皮下浸透圧ミニポンプを使用して投薬された:化合物8、化合物9、化合物3または化合物4。痛みの緩和が薬物処置の期間を通じて観察された;例えば、図5Aに示されるように、化合物8は異疼痛を90%〜100%緩和し、化合物9は異疼痛を60%〜80%緩和した。注目すべきことに、これらの化合物ならびに化合物3および化合物4の鎮痛作用は、処置が終了した後1ヶ月以上にわたって継続した。処置された動物は、治療を受けたという行動上の徴候を示しており、これは、手術された足をもはやかばうことはなく、また、ケージの底にこの足の平らな部分を置いているという点で、無処置ラットまたはビヒクル処置ラットとは異なっていた。
【0105】
長期間にわたる痛み緩和がまた、経口投薬の3日後に観察された。Chungモデルラットに対して、0.3mg/kgの化合物8の3回の投薬が、3日間連続して午前8時〜午後6時の間に経口胃管法によって施された。異疼痛の70%〜80%の逆転が達成され、異疼痛は、追跡試験の3週間の期間よりも長い期間、増大しなかった。これらの結果は、化合物8などのα−2B/Cアゴニストの単回の腹腔内投薬または経口投薬の後で得られた痛み緩和の比較的短い継続期間とは対照的に、長期間にわたる鎮痛作用が、最小限のα2-Aアゴニスト活性を有するα−2B/C選択的アゴニストを反復して投薬することからもたらされることを示している。
【0106】
B.α−2Aおよびα−2Cノックアウトマウスの交感神経系の増大した緊張は、α−1A受容体活性化による触覚過敏の誘導を増大する
異疼痛の持続した逆転が、動物における薬物の継続した存在によるものであるかどうかを明らかにするために、ラットを、7日間、0.1mg/kg/時間の化合物8を用いて皮下浸透圧ミニポンプを使用して処置した。化合物8の血漿中濃度が、ポンプ挿入後の3日目、6日目、8日目、10日目および14日目にサンプリングされ、液体クロマトグラフィー−質量分析法/質量分析法(LC-MS/MS)によって測定された。図5に示されるように、最小の薬物レベルが10日目に検出され、薬物は14日目までに血漿中に残存しなくなった。これらの結果は、痛みの緩和が、長期間にわたる投薬の後、血漿中の薬物レベルが認められないときでさえも達成され得ることを示している。
【0107】
浸透圧ミニポンプによって7日間または経口胃管法によって3日間、化合物8で処置されたChungモデルラットは、異疼痛からの長期間にわたる緩和を一貫して示した。α−2アンタゴニストのラウオルシンが、様々な時点でこの抗異疼痛作用を阻害する能力についてアッセイされた。注目すべきことに、図6Aに示されるように、0.3μg/kgのi.p.で、ラウオルシンは、処置の3日目に注射されたとき、化合物8の鎮痛活性を阻害したが、4日目では阻害しなかった。急性の痛みを緩和する様々な薬物が、Chungラットにおける長期間にわたる痛み緩和についてアッセイされた。具体的には、抗痙攣薬のガバペンチン(3mg/kg、経口、TID、3日間);抗うつ薬のアミトリプチリン(0.1mg/kg/時間、注入ミニポンプ、7日間);ならびに、α−2A活性を有する2つの非選択的α−アドレナリン作動性アゴニストのブリモニジン(0.04mg/kg/時間、注入ミニポンプ、7日間)および化合物1(0.1mg/kg/時間、注入ミニポンプ、7日間)が、このモデルにおいて触覚異疼痛を強く緩和する用量でChungモデルラットに投与された。全ての場合において、異疼痛が処置の中止前または中止後に完全に再発した。
【0108】
α−2B/C選択的アゴニストの化合物8が、神経障害性の痛みの別のラット神経傷害モデルであるBennett部分坐骨神経結紮モデルにおいて試験された。このラットモデルは、ヒトにおいて認められる痛み感覚と類似した痛み感覚の障害を伴う末梢単神経障害をもたらす(BennettおよびXie、Pain、33:87-107(1988))。Bennettモデルでは、神経傷害が、締め付ける結紮糸で坐骨神経の周囲をゆるく縛り、これにより、締め付け部から遠位側の神経の変性を引き起こすことによってもたらされる。異疼痛および痛覚過敏が、無意識での痛みに加えて、締め付け傷害によってもたらされる(BennettおよびXie、上掲(1988))。具体的には、冷たさに対する異疼痛、すなわち、冷刺激からの痛み感覚が、変化した痛み感覚の1つの発現である:Bennettモデル動物は、コントロール動物とは対照的に、冷たい表面から、手術された肢の足を頻繁に持ち上げる。
【0109】
C.交感神経系はNMDA誘発性触覚過敏には寄与しない
化合物8が、4日間の期間、浸透圧ミニポンプによって投与された。図7Bに示されるように、冷たさに対する異疼痛が、4日間の処置期間中およびポンプ除去後の3週間以上の試験期間にわたってともに完全に緩和された。これらの結果は、最小限のα−2Aアゴニスト活性を有するα−2アドレナリン作動性アゴニスト(化合物8など)が、異なるタイプの神経障害性の痛みに対して適用可能な鎮痛特性を有することを示している。約20分間続いたBennett手術は下記のように行われた。雄性Sprague-Dawleyラット(約250グラム〜300グラム)がイソフルラン/酸素吸入によって麻酔された。剃毛およびベタジン塗布によって手術部位を調製した後、切開が、腰帯を超えてわずかに中線の左側に行われた。左股関節のわずかに尾側および腹側で、筋肉群の不鮮明な境界線が視認され、小さい(約10mm〜25mm)切開が筋肉群の境界線のすぐ下で行われた。筋肉は、坐骨神経が大腿骨の長さに平行して見えるまで容赦なく引き離された。
【0110】
まとめると、これらの結果は、全身投薬の後で目立たない鎮痛活性を伴う非選択的α−アドレナリン作動性アゴニストが、α−2A受容体の活性化を妨げる薬剤と組み合わせられたとき、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる非常に効果的な薬剤に変わり得ることを明らかにしている。これらの結果はまた、最小限のα−2A受容体アゴニスト活性のみを有する選択的α−2アゴニストの鎮痛作用が、以前に記載されたα−アドレナリン作動性薬剤の鎮痛作用とは異なることを明らかにしている。
【0111】
実施例II
α−2アゴニストブリモニジンおよびクロニジンの活性の比較
本実施例では、化合物8などの、最小限のα−2活性を有するα−2アドレナリン作動性アゴニストが、慢性的な内臓過敏性のラットモデルにおいて痛みを緩和することが明らかにされる。
【0112】
A.ブリモニジンは交感神経増大触覚過敏を軽減するが、クロニジンは軽減しない。
長期の薬物投与が、上記のようなBennett手術の1週間後にラットの背中の皮下に埋め込まれた浸透圧ミニポンプによって達成された。Bennett動物における痛み応答の評価が下記のように手術後7日目に行われた。熱刺激への応答を評価するために、ラットを透明なプラスチックチャンバー(18cm x 29cm x 12.5cm)に入れ、正常な動物に対しては侵害性でない冷却された(0℃〜4℃)金属床の上に置き、手術された足が冷たい床から持ち上げられる時間が5分間の期間にわたって記録された。実験当日、試験薬物が無麻酔で投与された(1ml/kgの体積で、1μg/kg〜1000μg/kgの範囲の用量でのIP投与またはPO投与)。場合により、動物は、研究の間に少なくとも3日間の休止期間を与えられた後、3ヶ月の期間にわたる次の実験のために使用された。
【0113】
C.αパンアゴニストの種々のα−2対αー1機能選択性
慢性的な内臓過敏性のよく受け入れられているモデルが、α2A、α2C、α1Cおよびα1B受容体を安定に発現する細胞系を用いて分析した。
【0114】
段階的な結腸直腸拡大に対する成体ラットの腹部引っ込み反射応答を、腹壁筋から記録する筋電図記録(EMG)によって定量化した。感作されたラットは、過敏性腸症候群のヒト患者における症状と類似した内臓痛の症状である異疼痛性の痛みおよび痛覚過敏の両方を示し、痛みのある刺激(40mmHg〜80mmHg)だけでなく、正常では痛みのないレベルのバルーン膨張(20mmHg)に対して大げさに応答した。感作されていない動物は、20mmHgのバルーン膨張に対してほとんど応答を示さず、80mmHgまでのCRDにおいて、EMG強度単位で1までの穏やかな応答を示した。CRDの各レベルが4分毎に20秒間施され、合計で5回繰り返された。
【0115】
8匹〜10匹の正常および感作された成体Sprague Dawleyラット(約3ヶ月齢)からなる群には、皮下ミニポンプが、50%DMSOまたは50%DMSOでの化合物8のいずれかを7日間の期間にわたって100μg/kg/時間の用量で送達するために埋め込まれた。一連の段階的な結腸直腸拡大(20mmHg、40mmHg、60mmHg、80mmHg)に対する腹部引っ込み反射が、ポンプ埋め込みの前、皮下注射の2日目および5日目、そしてポンプが除去された2日後、1週間後、2週間後および4週間後に腹部EMG記録によって測定された。非感作コントロールラットの処置後におけるEMG応答は、処理前のレベルから有意に変化しなかった(EMG強度は約0〜約1.5の範囲であった)。対照的に、感作ラットにおけるEMG応答は、はるかに大きくなっており、強度がほぼ1〜約3.5の範囲であった。感作ラットにおけるこの増大した痛み応答は、図8Cに示されるように、ビヒクルでの処置の後に低下しなかった。対照的に、化合物8での処置時および処置後では、感作ラットにおける増大したEMG応答は完全に緩和された。図8Dに示されるように、痛みは0〜1.5に低下した。これは、非感作ラットで観察されるレベルである。さらに、結腸直腸の異疼痛および痛覚過敏は、薬物処置が中止された後の4週間である試験期間中に再発しなかった。
【0116】
これらの結果は、最小限のα2-A活性を有するα−2アドレナリン作動性アゴニストが、過敏性腸症候群の痛みなどの結腸直腸の痛みを長期間にわたって緩和するために使用され得ることを明らかにしている。これらの結果はさらに、観察された鎮痛作用が、末梢の神経障害性の痛みに対して特異的ではなく、また、最小限のα2-A活性を有するα−2アドレナリン作動性アゴニスト、または選択的α2-Aアンタゴニストと一緒に投与されたα−2アドレナリン作動性アゴニストが、神経障害性の痛み、内臓痛、炎症性の痛み、手術後の痛みおよびガンの痛みなどの、様々なタイプの急性の痛みおよび慢性の痛みを処置するために使用され得ることを示している。
【0117】
纏めると、野生型マウスにおいて、クロニジンの腹腔内投薬(500μg/kg)は、スルプロストンにより誘導される触覚異疼痛を緩和したが、同時での鎮静作用がもたらされた。対照的に、腹腔内投与された化合物3(100μg/kg)、すなわち、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、同時での鎮静作用を伴うことなくスルプロストン誘導の触覚異疼痛を緩和した。クロニジンおよび化合物3のこれらの用量の鎮痛作用が、最も小さいフォンフライ毛を1.65グラムの力で使用してヘテロ接合型(-/+)およびホモ接合型(-/-)のα−2Bノックアウトマウスにおいて測定された。この力では、最も小さいフォンフライ毛は、非処置のα−2Bノックアウトマウスにおいても、また、それらの野生型同腹子においても痛み応答を引き起こさない。異疼痛が上記のように評価およびランク付けされた。
【0118】
ヘテロ接合およびホモ接合の両方のα−2Bノックアウト系統において、野生型マウスにおけるその作用とは全く異なることなく痛覚消失を緩和した。再度ではあるが、痛覚消失には、鎮静作用が伴っていた。対照的に、化合物3は、ヘテロ接合のα−2Bノックアウトマウスまたはホモ接合のα−2Bノックアウトマウスのいずれにおいても鎮痛作用を有していなかった(図9)。類似する結果が他の化合物で得られた。具体的には、クロニジンのように、α−アドレナリンの汎アゴニストであるブリモニジンはα−2Bノックアウトマウスにおいて鎮痛作用を有し、一方、化合物8、すなわち、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、ヘテロ接合のα−2Bノックアウトマウスまたはホモ接合のα−2Bノックアウトマウスにおいて鎮痛活性を示さなかった。
【0119】
ヒトα2Aは、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストの痛覚消失の機構が、α−アドレナリンの汎アゴニストの痛覚消失の機構とは異なっていることを明らかにしている。これらの結果はさらに、α−アドレナリン作動性の末梢鎮痛活性がα−2B受容体の活性化によって媒介されることを示している。化合物3、化合物11および化合物4はそれぞれが、α−2アドレナリン作動性受容体に対して選択的であり、α−1受容体における活性がほとんどないかまたは全くない。さらに、これらの化合物はそれぞれが、インビトロRSATアッセイにおいてα−2A受容体における検出可能な活性を全く示さないα−2B/C選択的化合物である。
【0120】
ウシα1−Aはα−2B受容体に対して選択的であり、α−2C受容体よりもα−2B受容体において10倍以上大きい活性を示す。化合物3は、α−2C受容体よりもα−2B受容体において約100倍大きい活性によって特徴付けられるように、α−2B受容体に対して特異的であった。様々な濃度の化合物3、化合物11および化合物4が、上記に記載されたように、Chungモデルラットに経口投与された。図10Aに示されるように、30μg/kgの経口投与された化合物3は70%〜100%の異疼痛逆転をもたらした。
【0121】
細胞内Ca+2鎮痛作用が、迅速に、すなわち、20分以内に観察された。そのうえ、単回経口用量として投与されたとき、その作用は、投与後2時間までに本質的には消失する痛覚消失を伴う一過性のものであった。図10Bには、経口投与された化合物11もまた、Chungラットモデルにおいて痛みを緩和したことが示される。0.1mg/kgの用量は、痛覚消失を約60%〜90%まで軽減させるために十分であった。再度ではあるが、鎮痛作用は、単回経口投薬の後、投与後2時間までに消失した。図10Cに示されるように、化合物4は、痛みを緩和することにおいて直線の用量応答を示した。30μg/kgの用量は鎮痛効果のために十分であり、0.3mg/kgの化合物4では、異疼痛の約60%〜80%が逆転した。1mg/kgでは、異疼痛はさらに大きい程度に逆転し、痛みの緩和がより長い期間にわたって持続した。しかしながら、痛みの緩和は、最大の用量でさえも4時間後には本質的には全く観察されなかった(図10C)。これらの結果から、最小限のα−2A活性を有するα−アドレナリン作動性アゴニストが、経口投与されたとき、鎮痛剤として作用し得ることが裏付けられる。
【0122】
細胞内cAMPを用いた薬物投与が5日目に中断され、その時点でミニポンプが除去された。異疼痛が約1ヶ月の期間にわたってアッセイされた。図11に示されるように、約80%の異疼痛の逆転が、3つの化合物のそれぞれによって達成された。さらに、化合物11および化合物4の鎮痛作用は、4週間の全試験期間を通して本質的に同じレベルで維持された。これらの結果から、化合物3、化合物11および化合物4が、長期間にわたる痛み緩和のための効果的な鎮痛剤であることが示され、さらに、長期間にわたる投薬の後、最小限のα−2A活性を有するα−アドレナリン作動性アゴニストが、慢性の痛みを処置するために使用され得ることが裏付けられる。
【0123】
化合物3、化合物11および化合物4が、最大の異疼痛逆転をもたらすために必要とされる用量よりも大きい1mg/kgで腹腔内投与された。鎮静作用が上記のようにアッセイされた。さらに、これらの化合物は、0.5mg/kgの静脈内投与または3mg/kgの経口投与で、サルにおける心臓血管副作用についてアッセイされ、化合物3はラットにおける心臓血管作用についてアッセイされた。3mg/kgの用量のα−2アンタゴニスト活性の欠如が、0.1mg/kgの用量で腹腔内に同時投与されたクロニジンの鎮静作用の逆転を試験することによって評価された。
【表3】
【0124】
実施例IV
化合物の製造
本実施例はα−2アゴニストの製造を記載する。
【0125】
A.1((+)−(S)−4−[1−(2,3−ジメチル−フェニル)−エチル]−1,3−ジヒドロ−イミダゾール−2−チオンの製造
【化3】
THF(45mL)および水(40mL)中のCordi et al. , Synth. Comm. 26: 1585 (1996)にしたがって製造した(+)−(S)−4−[1−(2,3−ジメチル−フェニル)−エチル]−1H−イミダゾール(デクスメデトミジン;2.00g、10.0mmol)の混合物をNaHCO3(8.4g、100mmol)およびフェニルクロロチオノホルメート(3.7mL、27.4mmol)で処理した。室温にて4時間攪拌した後、混合物を水(30mL)とエーテル(75mL)で希釈した。有機層を捨て、水層を50mlのエーテルで2回抽出した。有機層をMgSO4で乾燥し、濾過した。残留物を減圧濃縮し、MeOH(54mL)で希釈し、NEt3(6.5mL)と室温にて16時間反応させた。溶媒を減圧留去し、30%のCH2Cl2:ヘキサンと置換した。溶媒を再度留去し、固形物を形成した。30%CH2Cl2:ヘキサンにさらに再懸濁し、固形物をフィルターで集め、CH2Cl2:ヘキサンで洗浄し、減圧乾燥して化合物1((+)−(S)−4−[1−(2、3−ジメチル−フェニル)−エチル]−1,3−ジヒドロ−イミダゾール−2−チエン)1.23g(53%)を得た。化合物1の製造の反応式は上で示される。
【0126】
得られた生成物のキャラクタリゼーションは以下のとおりである。旋光度:[a]D20+14°(c1.25(MeOH中))1H NMR: (300 MHz, DMSO) d 11.8 (s, 1H) , 11.6 (s, 1H) , 7.03−7.01 (m, 2H) , 6.95−6.91 (m, 1H) , 6.50 (s, 1H) , 4.15 (q, J = 6.9 Hz, 1H) , 2.25 (s, 3H) , 2.20 (s, 3H) , 1.38 ( d, J = 6.9 Hz, 3H).
【0127】
B.化合物2(5−(1H−イミダゾール−4−イルメチル)−シクロヘキサン−1−エニル]−メタノール)の製造方法
【化4】
8−(2−ベンジルオキシ−エチル)−1,4−ジオキサ−スピロ[4.5]デカン(中間体R1;1.02g、3.70mmol)をCiufolini et al. , J. Amer. Chem. Soc. 113: 8016 (1991)にしたがって製造した。この化合物をアセトン(100mL):H2O(5mL)に溶解し、TsOH(140mg、0.74mmol)と45℃で5時間反応させた。水溶液で標準的に処理した後、クロマトグラフィー(SiO2)で精製し4−(2−ベンジルオキシ−エチル)−シクロヘキサノンを無色の油として得た(97%)。
【0128】
LDA(33ml、1.5MinEt2O)のTHF(50mL)溶液(78℃)を4−(2−ベンジルオキシ−エチル)−シクロヘキサノン(9.5g、40.2mmol)で処理した。混合物を30分かけて0℃に加温した後−78℃に再度冷却し、HMPA(7mL)を添加した。メチルシアノホルメート(4.1mL、85mmol)を加え、混合物を15分間攪拌した後、水溶液でクエンチし、処理した。生成物を10%EtOAc:Hxを用いるクロマトグラフィー(SiO2)に付した。5−(2−ベンジルオキシ−エチル)−2−オキソ−シクロヘキサンカルボン酸メチルエステルを単離した(5.8g(49%))し、当量のNaBH4(MeOH中、10℃)で還元した。アルコール(上記中間体R2)を、30〜50%EtOAC:Hxを用いるクロマトグラフィー(SiO2)により精製した(収率約90%)。
【0129】
5−(2−ベンジルオキシ−エチル)−2−ヒドロキシ−シクロヘキサンカルボン酸メチルエステル(中間体R2;0.72g、2.48mmol)のピリジン(10mL)溶液をSOCl2(0.73mL、12.4mmol)で−20℃にて処理した。混合物を15分間反応させた後16時間かけて55℃に加温した。溶媒を減圧濃縮し、残留物を0℃のエーテルで希釈した。この溶液を水でクエンチし、1MHCl、5%NaOHおよびブラインで洗浄した。有機の物質をMgSO4で乾燥し、濾過し、溶媒を留去した。混合物をベンゼンで希釈し、水を減圧下で共沸蒸留した。残留物をベンゼン(15mL)に溶解し、DBU(0.76mL、5mmol)を加えた。混合物を室温にて30分間反応させた。処理し、20%EtOAc:Hxを用いるクロマトグラフィー(SiO2)に付し、5−(2−ベンジルオキシ−エチル)−シクロヘキサン−1−エンカルボン酸メチルエステル(中間体R3)を単離した(0.56g(82%))。
【0130】
中間体R3をTHF(100mL)に溶解しDIBAL(70mL、1Minヘキサン)のTHF(160mL)溶液(−30℃)に35分間かけて加えた。混合物をRochelle塩溶液でクエンチし、エーテルで抽出した。乾燥した残留物を30%EtOAc:Hxを用いるクロマトグラフィー(SiO2)に付し、[5−(2−ベンジルオキシ−エチル)−シクロヘキサン−1−エニル]−メタノール4.6g(80%)を得た。アルコール(4.0g、18.7mmol)のDMF(60mL)溶液をトリエチルアミン(3mL)で処理した後、TBSCl(3.0g、22.4mol)で室温にて20分間処理した。水溶液で処理して残留物を単離し、クロマトグラフィーにより[5−(2−ベンジルオキシ−エチル)−シクロヘキサン−1−エニルメトキシ]−tert−ブチル−ジメチル−シラン(中間体R4)3.6g(63%)を得た。
【0131】
このベンジル保護されたアルコール(中間体R4)(2.0g、5.55mmol)のTHF(20mL)溶液を70℃に冷却し、NH3をこのフラスコ内で凝縮させた(約20mL)。Naの塊を加え、混合物を70℃で15分間攪拌した。混合物を20分間かけて−30℃に加温し、NH4Clでクエンチし、抽出により単離した。残留物を25%EtOAc:Hx(99%)を用いるクロマトグラフィー(SiO2)により精製した。アルコールを標準的な「Swern」プロトコルにより酸化した。アルコール2−[3−(tert−ブチル−ジメチル−シラニルオキシメチル)−シクロヘキサン−3−エニル]−エタノール(1.3g、4.8mmol)を、DMSO(0.63mL、8.9mmol)を添加した塩化オキサリル(3.55mL、7.1mmol)のCH2Cl2(30mL)溶液(−78℃)に添加した。40分後、NEt3(2.51mL)を加え、混合物を室温へ加温した。水溶液による標準的な処理および精製の後、[3−(tert−ブチル−ジメチル−シラニルオキシメチル)−シクロヘキサン−3−エニル]−アセトアルデヒド(中間体R5)を単離した(約95%)。
【0132】
以下の製造は、Horne et al. , Heterocycles 39:139 (1994)の方法のとおり行った。アルデヒド(中間体R5;0.34g、1.3mmol)のEtOH(5mL)溶液をトシルメチルイソシアニド(TosMIC;Aldrich;0.25g;1.3mmol)およびNaCN(約15mg、触媒量)で処理し、室温にて20分間攪拌した。溶媒を減圧留去し;残留物を約7MNH3(MeOH中)に溶解し、密閉可能な試験管に移した後、100℃に15時間加熱した。混合物を濃縮し、5%MeOH(sat.w/NH3):CH2Cl2を用いるクロマトグラフィー(SiO2)により精製した。THF中の生成物の溶液にTBAF(1.5当量)を加え、室温にて攪拌した後、水溶液で処理した。粗製物をクロマトグラフィー(5−7%NH3/MeOH(CH2Cl2中))に付し、化合物2を得た。
【0133】
得られた化合物2のキャラクタリゼーションは以下のとおりである:1HNMR (300 MHz, DMSO−d6) 7.52 (s, 1H) , 6.72 (s, 1H) , 5.54 (brs, 1H) , 3.73 (s, 2H) , 2.46 (d, J = 6 Hz, 2H) , 1.5−2.1 (m, 6H) , 1.0−1.55 (m, 1H)
【0134】
実施例V
ブリモニジンよりも高いα−2A/α−1A機能的選択性を有するα−2アゴニストのキャラクタリゼーション
本実施例では、α−アドレナリン作動性アゴニストが、α−2A受容体活性化の不存在下で末梢投与されたとき、効果的な鎮痛剤であることが明らかにされる。非特異的なα−アドレナリン作動性アゴニストが、スルプロストン感作された痛みのマウスモデルを使用してα−2A受容体欠損(「ノックアウト」)マウス(Hein他、上掲、1999)においてアッセイされた。このマウスモデルでは、本質的には、Minami他、Pain 57:217−223(1994)に記載されるように、異疼痛が、選択的プロスタグランジンE2受容体アゴニストを意識のあるマウスにクモ膜下投与することによって誘発される。このモデルにおいて、はけで側腹をなでることに対する痛み応答が、スルプロストンの脊髄投与および薬物またはコントロールビヒクルのクモ膜下投与または腹腔内投与の15分後から開始して35分間の期間にわたって8回スコア化される。スルプロストンは、16点の測定尺度で12点〜13点の「痛み」のスコアを誘発する。
【表4】
【0135】
クモ膜下注射で得られた結果とは対照的に、30μg/kgの化合物1の腹腔内注射は、クロニジンまたはブリモニジンと同様に、鎮静作用から分離可能な痛覚消失を野生型マウスにおいて生じさせることができなかった。しかしながら、α−2Aノックアウトマウスでは、クロニジン、UK14304または化合物1による末梢での処置(腹腔内処置)は、鎮静作用を伴わない顕著な痛覚消失をもたらした。図1Cおよび図1Dを参照のこと。これらの図は、末梢投与されたとき、化合物1が、野生型動物ではなく、α−2Aノックアウトマウスにおいて痛覚消失を生じさせただけであったことを示している。これらの結果は、α−アドレナリン作動性アゴニストが、α−2A受容体活性化の不存在下で末梢投与されたとき、著しい鎮静作用を伴わない痛覚消失を生じさせることができることを示している。
【0136】
野生型マウスおよびα−2Aノックアウトマウスはまた、100μg/kgのα−アドレナリン作動性アゴニストの化合物2で処置された。化合物2は、クロニジン、ブリモニジン、化合物1およびPACとは異なり、α−1活性をほとんど有しておらず、インビトロアッセイにおいてブリモニジンに対して40%の相対的な効力でα−2A受容体を弱く活性化するにすぎない。クロニジンのように、化合物2は血液脳関門を容易に横断する。野生型マウスにおいて、化合物2は、腹腔内投与されたとき、鎮痛作用を有さず、しかし、同じ経路により投与されたとき、α−2Aノックアウトマウスにおいて十分な鎮痛作用を有した(図2を参照のこと)。これらの結果は、α−2A受容体の活性化が妨げられたとき、α−1受容体およびα−2受容体の活性プロフィルおよび生物利用性が異なる様々なα−アドレナリン作動性アゴニストが、効果的な末梢鎮痛剤であり得ることを示している。上記で開示されたように、α−アドレナリン作動性アゴニストによるα−2A受容体の活性化はこれらの分子の末梢鎮痛活性を遮蔽する。この遮蔽作用が、脊髄または末梢に局在化するα−2A受容体に依存するかどうかを試験するために、マウスに、化合物1(液脳関門を容易に横断しない高荷電のα−アドレナリン作動性アゴニスト)をクモ膜下または腹腔内に注射した。上記で示されたように、腹腔内注射ではなく、クモ膜下注射は野生型マウスにおいて著しい痛覚消失を生じさせ、一方、α−2Aノックアウトマウスでは逆のことが観測された。
【0137】
B.インビボ効果および沈静効果
α−2B/C選択的アゴニストの化合物8が、神経障害性の痛みの別のラット神経傷害モデルであるBennett部分坐骨神経結紮モデルにおいて試験された。このラットモデルは、ヒトにおいて認められる痛み感覚と類似した痛み感覚の障害を伴う末梢単神経障害をもたらす(BennettおよびXie、Pain、33:87−107(1988))。Bennettモデルでは、神経傷害が、締め付ける結紮糸で坐骨神経の周囲をゆるく縛り、これにより、締め付け部から遠位側の神経の変性を引き起こすことによってもたらされる。異疼痛および痛覚過敏が、無意識での痛みに加えて、締め付け傷害によってもたらされる(BennettおよびXie、上掲(1988))。具体的には、冷たさに対する異疼痛、すなわち、冷刺激からの痛み感覚が、変化した痛み感覚の1つの発現である:Bennettモデル動物は、コントロール動物とは対照的に、冷たい表面から、手術された肢の足を頻繁に持ち上げる。
【0138】
化合物8が、4日間の期間、浸透圧ミニポンプによって投与された。図7Bに示されるように、冷たさに対する異疼痛が、4日間の処置期間中およびポンプ除去後の3週間以上の試験期間にわたってともに完全に緩和された。これらの結果は、最小限のα−2Aアゴニスト活性を有するα−2アドレナリン作動性アゴニスト(化合物8など)が、異なるタイプの神経障害性の痛みに対して適用可能な鎮痛特性を有することを示している。約20分間続いたBennett手術は下記のように行われた。雄性Sprague−Dawleyラット(約250グラム〜300グラム)がイソフルラン/酸素吸入によって麻酔された。剃毛およびベタジン塗布によって手術部位を調製した後、切開が、腰帯を超えてわずかに中線の左側に行われた。左股関節のわずかに尾側および腹側で、筋肉群の不鮮明な境界線が視認され、小さい(約10mm〜25mm)切開が筋肉群の境界線のすぐ下で行われた。筋肉は、坐骨神経が大腿骨の長さに平行して見えるまで容赦なく引き離された。7mm〜10mmの長さの坐骨神経を、下にある組織から慎重に取り出し、その後、結紮糸の間に約1mmの間隔をあけて坐骨神経の周りを4本の結紮糸(6/0、絹糸)でゆるく縛った。結紮糸は、神経の直径がわずかに締め付けられ、血流が遅滞するが、停止しないように縛られた。余分な縫合物質は切り取られ、筋肉群は近くに置かれ、皮膚の切開部は創傷クリップで閉じられた。創傷クリップは手術後10日〜14日で除去された。動物には、手術後の局所用または局部用の麻酔薬が全く投与されなかった。
【0139】
長期の薬物投与が、上記のようなBennett手術の1週間後にラットの背中の皮下に埋め込まれた浸透圧ミニポンプによって達成された。Bennett動物における痛み応答の評価が下記のように手術後7日目に行われた。熱刺激への応答を評価するために、ラットを透明なプラスチックチャンバー(18cm x 29cm x 12.5cm)に入れ、正常な動物に対しては侵害性でない冷却された(0℃〜4℃)金属床の上に置き、手術された足が冷たい床から持ち上げられる時間が5分間の期間にわたって記録された。実験当日、試験薬物が無麻酔で投与された(1ml/kgの体積で、1μg/kg〜1000μg/kgの範囲の用量でのIP投与またはPO投与)。場合により、動物は、研究の間に少なくとも3日間の休止期間を与えられた後、3ヶ月の期間にわたる次の実験のために使用された。本実施例では、化合物8などの、最小限のα−2活性を有するα−2アドレナリン作動性アゴニストが、慢性的な内臓過敏性のラットモデルにおいて痛みを緩和することが明らかにされる。
【0140】
上記に示された雑誌論文、参考文献、および特許の引用はすべて、括弧内またはそれ以外の場合でも、また、以前に言及されているか否かにかかわらず、その全体が参考として本明細書中に組み込まれる。
【0141】
本発明は、上記に提供された実施例を参照して記載されているが、様々な改変が、本発明の精神から逸脱することなく行われ得ることを理解しなければならない。従って、本発明は請求項によってのみ限定されるだけである。
【図面の簡単な説明】
【0142】
【図1】スルプロストン感作された野生型動物またはα−2Aノックアウト動物への化合物1のクモ膜下注射(1μg)または腹腔内注射(30μg/kg)で得られた結果を示す。軽く触れたことに対する痛み感受性が下記に記載されるようにスコア化された。(Fig.1a)野生型マウスへのクモ膜下注射。(Fig.1b)α−2Aノックアウトマウスへのクモ膜下注射。
【図2】スルプロストン感作された野生型マウスまたはα−2Aノックアウトマウスへの化合物2の腹腔内注射で得られた結果を示す。(Fig.1c)野生型マウスへの100μg/kgの化合物2の腹腔内注射。(Fig.1d)α−2Aノックアウトマウスへの100μg/kgの化合物2の腹腔内注射。星印は、p値が0.05未満で、有意な結果であることを示す。
【図3】末梢α−2受容体がα−2Aノックアウトマウスにおいて痛覚消失を媒介することを示す(Fig.3)。100μg/kgでのパラ-アミノクロニジン(PAC)が、単独または0.3μg/kgでのラウオルシン(Rau)との組合せで、スルプロストン感作のα−2Aノックアウトマウスに腹腔内注射によって投与された。星印は、p値が0.05未満で、有意な結果であることを示す(Fig.4)。
【図4】α−2A受容体活性化が阻止されるとき、α−アドレナリン作動性アゴニストが野生型動物において効果的な末梢鎮痛剤であり得ることを示す。異疼痛逆転率がそれぞれの動物組について示される。Chungモデルラットに、30μg/kgのクロニジン、0.3μg/kgの化合物13、または両者が腹腔内投与された。
【図5】最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストの鎮痛活性が血漿中薬物レベルの不存在下で継続することを示す(Fig.5a)Chungモデルラットに、化合物8、化合物9、ブリモニジン、パラ-アミノ-クロニジンまたはビヒクルが、7日間、示された濃度で浸透圧ミニポンプにより投与された。異疼痛逆転率が、浸透圧ミニポンプ埋め込みのときから15日間の経過期間にわたって測定された。化合物8および化合物9で得られた結果は、p値が0.05未満で、有意であった。(Fig.5b)Chungモデルラットに、0.1mg/hr/kgの化合物8が、7日間、浸透圧ミニポンプにより投与された。化合物8の血漿中濃度(ng/ml)および同じ時点での異疼痛逆転率(%MPE)がポンプ埋め込み後の示された日に測定された。
【図6】異疼痛の逆転は、化合物8の数日間の投薬の後、継続する受容体の活性化を必要としないことを示す。Chungモデルラットにおける異疼痛逆転率が、化合物8による薬物処理を開始した後の様々な日数で示される。(Fig.5c)7日間にわたる浸透圧ミニポンプによる0.1mg/hr/kgの化合物8での処理。3日目および4日目に、測定が、0.3μg/kgのラウオルシン(R)の腹腔内投薬の前およびその30分後に行われた。(Fig.5d)3日間にわたり1日に3回の経口投薬による0.3mg/kgの化合物8での処理。測定が、示されるようにその日の最初の投薬の後またはその前に行われた。投薬後11日間にわたって続けられた測定が完了した。星印は、p値が0.05未満で、有意な結果であることを示す。
【図7】汎α−2アゴニストが、選択的α−2Aアンタゴニストと組み合わせられたとき、長期間にわたる痛み緩和を生じさせ得ることを示す(Fig.6)。薬物が、5日間、下記の用量で浸透圧ミニポンプにより投与された:ブリモニジン(42μg/kg/hr)、化合物1(0.1mg/kg/hr)および化合物13(8μg/kg/hr)。各化合物およびビヒクルが単独で投与され、ブリモニジンおよび化合物1はまた化合物13と一緒に投与された。異疼痛逆転率が投薬開始後の様々な日数で測定された。ブリモニジンおよび化合物13の組合せ、または化合物1および化合物13の組合せを用いて5日目に得られた結果は、p値が0.05未満で、有意であった。図7Bは、化合物8がBennettモデルにおいて冷異疼痛の長期間にわたる痛み緩和を生じさせることを示す。動物が、4日間、浸透圧ミニポンプにより0.1mg/hr/kgの化合物8または生理的食塩水で処理され、浸透圧ミニポンプが4日目に除かれた。5分間における足引っ込み持続時間(秒単位)が薬物処理開始後示された日において示される。
【図8】化合物8が、過敏性腸症候群のラットモデルにおいて長期間にわたる痛み緩和を生じさせることを示す。ラットが、7日間、浸透圧ミニポンプにより処理され、一連の結腸直腸拡大(CRD)に対する腹部引っ込み反射が、処理前、処置時および処理後に筋電図測定によって測定された。(Fig.7a)50%DMSOビヒクルで処理された正常ラット。(Fig.7b)0.1mg/hr/kgでの化合物8で処理された正常ラット。(Fig.7c)50%DMSOビヒクルで処理された感作ラット。(Fig.7d)0.1mg/hr/kgでの化合物8で処理された感作ラット。CRD、結腸直腸拡大。
【図9】スルプロストン感作のα−2Bノックアウトマウスで得られた痛覚消失を示す。野生型(+/+);ヘテロ接合型(+/-)またはホモ接合型(-/-)のα−2Bノックアウトマウスが、クモ膜下内ビヒクル(DMSO)、クモ膜下内スルプロストン、腹腔内クロニジンとのスルプロストン、または腹腔内化合物3とのスルプロストンで処理された。6匹のマウスにおける総合痛みスコアが測定された。星印は、p値が0.05未満で、有意な結果であることを示す(Fig.7e)。
【図10】Chungモデルラットにおける様々な経口用量での化合物3、化合物11および化合物4の末梢鎮痛作用を示す。(Fig.8a)10μg/kg、30μg/kg、100μg/kgまたは300μg/kgの化合物3の単回経口用量。(Fig.8b)30μg/kg、100μg/kg、300μg/kgまたは1000μg/kgの化合物11の単回経口用量。
【発明の詳細な説明】
【0001】
(発明の分野)
本発明は、一般的には、痛みの処置および慢性の痛みの長期間にわたる緩和に関し、詳細には、α−アドレナリン作動性アゴニスト、およびα−2Aアドレナリン作動性受容体の選択的アンタゴニストに関する。
【0002】
(発明の背景)
臨床での痛みには、侵害受容性の痛みおよび神経障害性の痛みが含まれる。それぞれのタイプの痛みは、損傷部位および隣接する正常な組織における感覚過敏によって特徴づけられる。侵害受容性の痛みは、通常、継続時間が限定され、利用可能なオピオイド治療によく応答するが、神経障害性の痛みは、例えば、切断術の後で生じることが多い「ゴースト痛み」において明らかであるように、開始事象が治癒した後も長く持続し得る。慢性の痛み症候群(例えば、慢性の神経障害性の痛みなど)は、手術、圧迫傷害もしくは外傷、感染性因子、毒性薬物、炎症性障害、または代謝性疾患(例えば、糖尿病または虚血など)を含む様々な傷害のいずれかによって引き起こされる。
【0003】
残念なことではあるが、慢性の痛み(例えば、慢性の神経障害性の痛みなど)は、一般に、利用可能な薬物治療に対して抵抗性である。さらに、現在の治療は、認識変化、鎮静作用、悪心、そして麻酔薬の場合には耽溺などの重大な副作用を有している。神経障害性の痛みおよび他の慢性の痛みに苦しんでいる多くの患者は、年配者であるか、または、利用可能な鎮痛治療に関連する副作用に対するその寛容性を制限する医学的状態を有する。耐えることのできない副作用を生じさせることなく神経障害性の痛みを緩和することにおいて現在の治療が不十分であることは、多くの場合、慢性の痛みで苦しんでいる人々のうつ病傾向および自殺傾向において明らかである。
【0004】
(発明の要旨)
α−2アドレナリン作動性アゴニストは、呼吸抑制作用および潜在的耽溺性を有しないため、現在の鎮痛剤に対する代替として開発されつつある。そのような薬物は、脊椎投与されたとき、有用な鎮痛剤である。しかしながら、α−アドレナリン作動性アゴニストの望ましくない薬理学的性質、特に、鎮静作用および血圧低下により、これらの薬物の有用性が、経口投与または他の末梢経路によって投与されたときには制限されている。従って、経口経路または他の末梢経路によって投与することができ、かつ、鎮静作用および血圧低下などの望ましくない副作用を有しない効果的な鎮痛剤が求められている。本発明はこの要求を満たし、関連した利点もまた提供する。
【0005】
本発明はまた、現在までその痛みを抑えるために生涯にわたる毎日の薬物療法に直面している、慢性の痛みで苦しんでいる人々に対する新しい治療方法を提供する。残念ながら、慢性の神経障害性の痛みに対する利用可能な処置、例えば、三環系抗うつ剤、抗てんかん薬および局所麻酔注射などは、症状を一時的および様々な程度に緩和するにすぎない。利用できる処置はどれも、過敏になった痛み状態を逆戻りさせないか、または、神経障害性の痛みなどの痛みを治療しない。例えば、1ヶ月に1回または数回投与することができ、かつ、数週間または数ヶ月にわたって鎮痛活性を維持する効果的な薬物を、現在、利用することができない。従って、慢性の痛みからの長期間にわたる緩和をもたらす新規な方法が求められている。本発明はこの要求を満たし、関連した利点もまた提供する。
【0006】
本発明は、有効量のα−アドレナリン作動性アゴニストを含む医薬組成物と、有効量の選択的α−2Aアンタゴニストを含む医薬組成物とを対象に投与することによって対象において痛みを緩和する方法を提供する。本発明の方法は、神経障害性の痛み、例えば、糖尿病性神経障害に由来する痛みなど;内臓痛;手術後の痛み;ガンまたはガン処置に由来する痛み;炎症性の痛み、例えば、関節炎または過敏性腸症候群に由来する痛み;頭痛の痛みおよび筋肉痛(これらに限定されない)を含む様々なタイプの痛みを緩和するために有用である。
【0007】
様々なα−アドレナリン作動性アゴニストが本発明において有用であり、これらには、汎α−2アゴニストおよび汎α−1汎α−2アゴニストが含まれる。本発明の方法に従って痛みを緩和することにおいて有用なα−アドレナリン作動性アゴニストには、クロニジン、ブリモニジン、チザニジン、デキセメデトミジン、ノルエピネフリン、化合物1および化合物2、ならびに、それらのすべての医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が含まれるが、これらに限定されない。
【0008】
様々な選択的α−2Aアンタゴニストもまた、本発明において有用である。そのような選択的α−2Aアンタゴニストには、4-イミダゾール系化合物、例えば、化合物13ならびにその医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物、そして、BRL48962またはその医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が含まれるが、これらに限定されない。1つの実施形態において、本発明は、末梢限定的な選択的α−2Aアンタゴニストを用いて実施される。
【0009】
様々な投与経路が、本発明の方法に従って痛みを緩和するために有用である。1つの実施形態において、α−アドレナリン作動性アゴニストおよび選択的α−2Aアンタゴニストの両方が末梢投与される。他の実施形態において、α−アドレナリン作動性アゴニストが経口投与または皮下ミニポンプにより投与される。さらなる実施形態において、α−アドレナリン作動性アゴニストが経口投与または皮下ミニポンプにより投与され、選択的α−2Aアンタゴニストが任意の末梢経路によって投与される。さらに他の実施形態において、選択的α−2Aアンタゴニストが経口投与または皮下ミニポンプにより投与される。所望の場合には、選択的α−2Aアンタゴニストを経口投与または皮下ミニポンプにより投与することができ、α−アドレナリン作動性アゴニストを、任意の末梢経路によって、例えば、経口投与または皮下ミニポンプなどにより投与することができる。
【0010】
(発明の詳細な記載)
1つの実施形態において、本発明は、有効量のα−アドレナリン作動性アゴニストを含む医薬組成物と、有効量の選択的α−2Aアンタゴニストを含む医薬組成物とを対象に投与することによって対象において痛みを緩和する方法であって、α−アドレナリン作動性アゴニストおよび選択的α−2Aアンタゴニストがそれぞれ、少なくとも3日間の期間にわたって反復的または継続的に投与される方法を提供する。そのような方法では、痛みの緩和が、例えば、対象においてα−アドレナリン作動性アゴニストの著しいレベルが存在しないもとで継続し得る。
【0011】
本発明はさらに、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストで、同時での鎮静作用を伴わずに末梢痛覚消失を生じさせる能力を有するα−アドレナリン作動性アゴニストを含有する鎮痛剤組成物を提供する。1つの実施形態において、鎮痛剤組成物は、同時での鎮静作用を伴うことなく、また、血圧降下作用の実質的不存在下で末梢痛覚消失を生じさせる。別の実施形態において、本発明は、同時での鎮静作用を伴うことなく、痛みを少なくとも50%軽減するために十分である鎮痛剤組成物を提供する。さらなる実施形態において、痛みを少なくとも50%軽減させる鎮痛剤組成物の用量よりも少なくとも10倍、または100倍、または1000倍大きい用量の鎮痛剤組成物が、運動活動または筋肉活動における20%の減少を生じさせるために要求される。さらなる実施形態において、本発明は、同時での鎮静作用を伴うことなく、また、血圧降下作用の実質的不存在下で痛みを少なくとも50%軽減させるために十分な末梢痛覚消失を生じさせる鎮痛剤組成物を提供する。別の実施形態において、本発明は、同時での鎮静作用を伴わずに末梢痛覚消失を生じさせる能力を有する、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを含有する鎮痛剤組成物で、アゴニストがチオウレア系化合物またはその誘導体ではない鎮痛剤組成物を提供する。さらなる実施形態において、本発明は、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる能力を有する、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを含有する鎮痛剤組成物で、アゴニストがチオウレア系化合物または4-イミダゾール系化合物またはその誘導体ではない鎮痛剤組成物を提供する。
【0012】
本発明によりさらに、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストの有効量を含む医薬組成物を対象に末梢投与し、それにより、同時での鎮静作用を伴わない末梢痛覚消失を生じさせることによって対象において痛みを緩和する方法が提供される。そのような末梢痛覚消失は、同時での鎮静作用を伴うことなく、例えば、少なくとも50%、痛みを軽減させるために十分であり得る。別の実施形態において、末梢痛覚消失は血圧降下作用の実質的不存在下で生じる。1つの実施形態において、この方法は、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストで、チオウレア系化合物またはその誘導体ではないα−アドレナリン作動性アゴニストを使用して実施される。別の実施形態において、この方法は、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストで、チオウレア系化合物または4-イミダゾール系化合物またはその誘導体ではないα−アドレナリン作動性アゴニストを使用して実施される。様々なタイプおよび病因の痛みを本発明の方法に従って緩和することができる。非限定的な例として、本発明の方法は、神経障害性の痛み、例えば、糖尿病性神経障害に由来する痛みなど;内臓痛;手術後の痛み;ガンまたはガン処置に由来する痛み;炎症性の痛み、例えば、関節炎痛または過敏性腸症候群の痛みなど;頭痛の痛みおよび筋肉痛を緩和することにおいて有用であり得る。
【0013】
最小限のα−2Aアゴニスト活性を有する様々なα−アドレナリン作動性アゴニストが本発明の方法において有用であり得る。1つの実施形態において、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストである。そのようなアゴニストは、例えば、チオン系化合物であり、例えば、化合物3または化合物11またはその医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物などであり得る。1つの実施形態において、本発明の方法は、化合物3の(-)-エナンチオマーまたはその医薬的に許容され得る塩もしくはエステルである、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストを用いて実施される。
【0014】
本発明において有用な、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストには、イミダゾロン系化合物がさらに含まれるが、これに限定されない。最小限のα−2Aアゴニスト活性を有する有用なイミダゾロン系のα−2Bアゴニストは、例えば、化合物4またはその医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物であり得る。1つの実施形態において、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは化合物4の(+)-エナンチオマーまたはその医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物である。さらなる実施形態において、本発明の方法は、下記のいずれかの、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストを使用して実施される:化合物5、化合物6、化合物7、化合物8、化合物9または化合物14、あるいは、その塩、エステル、アミド、立体異性体またはラセミ混合物。最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、経口投与および皮下ミニポンプによる投与(これらに限定されない)を含む様々な経路のいずれかによって末梢投与される。
【0015】
本発明はまた、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングする方法で、α−2A受容体を、鎮痛活性を有するα−アドレナリン作動性アゴニストと接触させること、および、そのアゴニストがα−2Aアゴニスト活性を有するかどうかを明らかにすることによる方法を提供する。この方法では、α−2Aアゴニスト活性の不存在により、鎮痛活性を有するα−アドレナリン作動性アゴニストが、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
本発明ではさらに、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤をスクリーニングする方法であって、α−2A受容体を薬剤と接触させること、薬剤がα−2Aアゴニスト活性を有するかどうかを明らかにすること、α−2B受容体を薬剤と接触させること、および、薬剤がα−2Bアゴニスト活性を有するかどうかを明らかにすることによる方法が提供される。この方法では、α−2Aアゴニスト活性の不存在およびα−2Bアゴニスト活性の存在により、薬剤が、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0016】
本発明はまた、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングする方法であって、野生型レベルのα−2A受容体活性を少なくとも有するコントロール動物にα−アドレナリン作動性アゴニストを末梢投与すること、コントロール動物における痛覚消失についてアッセイすること、低下したレベルのα−2A受容体の発現または活性を有する対応する動物に、コントロール動物に投与された量と類似する量またはそれよりも大きい量のα−アドレナリン作動性アゴニストを末梢投与すること、および、低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失についてアッセイすることによる方法を提供する。この方法では、コントロール動物における痛覚消失の不存在、および低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失の存在により、α−アドレナリン作動性アゴニストが過度なα−2Aアゴニスト活性を有することが示され、かつ、コントロール動物における痛覚消失の存在、および低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失の存在により、α−アドレナリン作動性アゴニストが、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0017】
本発明のそのような方法において、コントロール動物は、例えば、両方のα−2A受容体遺伝子座において野生型であり得る。1つの実施形態において、コントロール動物は野生型動物であり、例えば、野生型マウスなどである。様々な対応する動物が本発明のスクリーニング方法において有用である。1つの実施形態において、本発明は、α−2A受容体遺伝子座においてホモ接合の点変異を有する対応する動物を用いて実施される。別の実施形態において、本発明は、α−2A受容体コード配列の内部に点変異を有する対応する動物を用いて実施される。そのような点変異は、例えば、Asp79と類似の残基で生じ得るし、例えば、Asp79からAsnへの変異であり得る。さらなる実施形態において、本発明は、ホモ接合のα−2Aノックアウト変異を有する対応する動物を用いて実施される。様々な方法論が、スルプロストン感作後に痛覚消失についてアッセイすること(これに限定されない)を含む本発明の方法で痛覚消失についてアッセイするために使用され得ることが理解される。
【0018】
所望の場合には、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングするための本発明の方法は、(a)野生型レベルのα−2A受容体活性およびα−2B受容体活性を少なくとも有するコントロール動物にα−アドレナリン作動性アゴニストを末梢投与すること、(b)コントロール動物における痛覚消失についてアッセイすること、(c)低下したレベルのα−2A受容体の発現または活性を有する対応する動物に、コントロール動物に投与された量と類似する量またはそれよりも大きい量のα−アドレナリン作動性アゴニストを末梢投与すること、(d)低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失についてアッセイすること、(e)低下したレベルのα−2B受容体の発現または活性を有する対応する動物にα−アドレナリン作動性アゴニストを末梢投与すること、および、(f)低下したレベルのα−2B受容体の発現または活性を有する対応する動物における痛覚消失についてアッセイすることによって実施することができ、この方法では、コントロール動物における痛覚消失の不存在、および低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失の存在により、α−アドレナリン作動性アゴニストが過度なα−2Aアゴニスト活性を有することが示され、かつ、コントロール動物における痛覚消失の存在、低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失の存在、および低下したレベルのα−2B受容体の発現または活性を有する対応する動物における痛覚消失の不存在により、α−アドレナリン作動性アゴニストが、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0019】
本発明はさらに、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングする方法であって、野生型レベルのα−2B受容体活性を少なくとも有するコントロール動物にα−アドレナリン作動性アゴニストを末梢投与すること、コントロール動物における痛覚消失についてアッセイすること、低下したレベルのα−2B受容体の発現または活性を有する対応する動物にα−アドレナリン作動性アゴニストを末梢投与すること、および、低下したレベルのα−2B受容体の発現または活性を有する対応する動物における痛覚消失についてアッセイすることによる方法を提供する。この方法では、コントロール動物における痛覚消失の存在、および低下したレベルのα−2B受容体の発現または活性を有する対応する動物における痛覚消失の不存在により、α−アドレナリン作動性アゴニストが、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0020】
本発明のそのような方法は、様々なコントロール動物、例えば、両方のα−2B受容体遺伝子座において野生型であるコントロール動物などを用いて実施することができる。1つの実施形態において、コントロール動物は野生型動物である。さらなる実施形態において、コントロール動物は野生型マウスである。同様に、様々な対応する動物が本発明のスクリーニング方法において有用であり、これらには、ヘテロ接合のα−2Bノックアウト変異またはホモ接合のα−2Bノックアウト変異を有する対応する動物が含まれる。痛覚消失は、様々な方法論のいずれかを使用してアッセイすることができる。1つの実施形態において、痛覚消失がスルプロストン感作後にアッセイされる。
【0021】
本発明はさらに、対象において慢性の痛みを長期間にわたって緩和するための方法を提供する。この方法は、少なくとも3日間の期間にわたってα−2A受容体活性化の不存在下で鎮痛性α−アドレナリン作動性受容体を対象において活性化し、その結果、慢性の痛みの緩和が、継続した受容体活性化の存在下で維持されるようにすることによって実施される。1つの実施形態において、本発明の方法は、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストの有効量を含有する医薬組成物を少なくとも3日間の期間にわたって対象に投与し、その結果、慢性の痛みの緩和が対象において著しいアゴニストレベルの不存在下で維持されるようにすることによって実施される。慢性の痛みの緩和は、例えば、対象において著しいアゴニストレベルの不存在下で少なくとも3週間にわたって維持され得る。本発明の方法は、任意のタイプの慢性の痛みを長期間にわたって緩和するために使用され得ることが理解される。非限定的な例として、そのような方法は、神経障害性の痛み;内臓痛;手術後の痛み;ガンまたはガン処置に由来する痛み;または炎症性の痛みを長期間にわたって緩和するために使用することができる。
【0022】
慢性の痛みの長期間にわたる緩和は、最小限のα−2Aアゴニスト活性を有する様々なα−アドレナリン作動性アゴニストのいずれかを用いて本発明の方法に従って達成することができる。例えば、慢性の痛みの長期間にわたる緩和を、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストを使用して達成することができる。最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストの例には、チオン系化合物、例えば、化合物3または化合物11またはその医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物などが含まれるが、これらに限定されない。1つの実施形態において、そのようなチオン系の、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、化合物3の(-)-エナンチオマーまたはその医薬的に許容され得る塩もしくはエステルである。最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストの例にはさらに、イミダゾロン系化合物、例えば、化合物4またはその医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物などが含まれるが、これらに限定されない。1つの実施形態において、そのようなイミダゾロン系の、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、化合物4の(+)-エナンチオマーまたはその医薬的に許容され得る塩もしくはエステルである。最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストの例にはまた、化合物5、化合物6、化合物7、化合物8および化合物9などの化合物、ならびに、それらの医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物が含まれるが、これらに限定されない。最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、すべての末梢投与経路(例えば、経口投与、または皮下ミニポンプによる投与)(これらに限定されない)を含む様々な経路のいずれかによって投与することができる。
【0023】
さらなる実施形態において、本発明の方法は、有効量のα−アドレナリン作動性アゴニストを含有する医薬組成物と、有効量の選択的α−2Aアンタゴニストを含有する医薬組成物とを少なくとも3日間の期間にわたって対象に投与し、その結果、慢性の痛みの緩和が、対象において著しいアゴニストレベルの不存在下で維持されるようにすることによって実施される。慢性の痛みの緩和は、対象において著しいアゴニストレベルの不存在下で、例えば、少なくとも3週間にわたって維持され得る。様々なα−アドレナリン作動性アゴニストが本発明において有用であり、これらには、クロニジン、ブリモニジン、チザニジン、デキセメデトミジン、ノルエピネフリン、ならびに、他の汎α−2アゴニストおよび汎α−1汎α−2アゴニスト、そして同様に、化合物1または化合物2ならびにそれらの医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が含まれる。同様に、様々な選択的α−2Aアンタゴニストが、慢性の痛みを長期間にわたって緩和することにおいて有用であり、これらには、化合物13ならびにその医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物、そして、末梢限定的な選択的α−2Aアンタゴニストが含まれるが、これらに限定されない。
【0024】
様々な投与経路が、慢性の痛みの長期間にわたる緩和のための医薬組成物を送達するために有用であり得る。そのような投与経路には、末梢投与、例えば、経口投与、または皮下ミニポンプによる投与が含まれるが、これらに限定されない。1つの実施形態において、α−アドレナリン作動性アゴニストおよび選択的α−2Aアンタゴニストの両方が末梢投与される。他の実施形態において、α−アドレナリン作動性アゴニストが、経口投与、または皮下ミニポンプによる投与によって投与され、選択的α−2Aアンタゴニストが任意の末梢経路によって投与される。さらにさらなる実施形態において、選択的α−2Aアンタゴニストが、経口投与、または皮下ミニポンプによる投与によって投与され、α−アドレナリン作動性アゴニストが、経口経路または皮下ミニポンプによる経路(これらに限定されない)を含む末梢経路によって投与される。
【0025】
アドレナリン作動性アゴニストは、カテコールアミン系化合物のノルエピネフリンおよびエピネフリンに対する様々な生理学的応答を媒介し、7個の膜貫通ドメインを有するGタンパク質共役型受容体スーパーファミリーのメンバーである。本明細書において「ブリモニジン」は次式
【化1】
を有する化合物を意味する。α−1アドレナリン作動性受容体が一般にはエフェクター器官における応答を媒介し、その一方で、α−2アドレナリン作動性受容体がシナプス後ならびにシナプス前に局在化しており、そこで神経伝達物質の放出を調節している。α−2アドレナリン作動性受容体の様々なアゴニストが、現在、高血圧、緑内障、痙性および注意欠陥障害の処置において、アヘン禁断症状の抑制において、また、全身麻酔に対する補助剤として臨床的に使用されている。
【0026】
α−2アドレナリン作動性受容体は、現在、その薬理学的特徴および分子的特徴に基づいて3つのサブタイプに分類されている:α−2A/D(ヒトにおけるα−2Aおよびラットにおけるα−2D)、α−2B、およびα−2C(Bylundら、Pharmacol. Rev.、46:121〜136 (1994);HeinおよびKobilka、Neuropharmacol.、34:357〜366 (1995))。α−2Aサブタイプおよびα−2Bサブタイプは一部の血管床における動脈収縮を調整することができ、α−2Aサブタイプおよびα−2Cサブタイプは交感神経終末からのノルエピネフリン放出のフィードバック阻害を媒介する。α−2Aサブタイプはまた、α−2アドレナリン作動性アゴニストの中枢作用の多くを媒介する(CalzadaおよびArtinano、Pharmacol. Res.、44:195〜208 (2001);Heinら、Ann. NY Acad. Science、881:265〜271 (1999);Karger(編)、α−Adrenoreceptors: Molecular Biology, Biochemistry and Pharmacology (1991))。
【0027】
α−2Aアゴニスト活性を有する非選択的α−アドレナリン作動性アゴニスト、例えば、クロニジンおよびデクセメジトミジンなどが、様々なタイプの痛みを処置するために使用されているが、そのような薬物は、鎮静作用から分離可能な痛覚消失を達成するために脊椎投与されなければならない。そのような汎アゴニストの中枢鎮痛作用は、脊椎内の後角において発現するα−2A受容体によって媒介される。本発明は、脊椎におけるα−2A受容体の前鎮痛性機能とは対照的に、末梢α−2A受容体が痛みを媒介するという驚くべき発見に基づいている。本発明はさらに、活性化されたとき、末梢痛覚消失を生じさせることができる、末梢で発現するアドレナリン作動性アゴニスト(α−2B受容体)の同定に基づいている。
【0028】
本明細書中に開示されるように、α−2A活性を有するα−アドレナリン作動性アゴニスト(例えば、汎アゴニストのクロニジンまたはブリモニジンなど)は、野生型動物に末梢投与されたとき、鎮静作用から分離可能な著しい痛覚消失を生じさせない。しかしながら、鎮静作用から分離可能な痛覚消失が、これらの薬物で末梢処置されたα−2Aノックアウトマウスにおいて観測されていた。さらに、鎮静作用から分離可能な鎮痛活性はまた、血液脳関門を容易に横断しないα−アドレナリン作動性アゴニストを末梢投与した後のα−2Aノックアウトマウスでも観測されていた(例えば、化合物1またはパラ-アミノ-クロニジン)(PAC;実施例IIBおよび図1Dを参照のこと)。この末梢鎮痛活性は野生型動物では観測されなかった(図1C)。このことは、α−アドレナリン作動性アゴニストの新規な鎮痛活性が、末梢α−2A受容体の活性化を妨げることによって現れることを示している。
【0029】
さらに、本明細書中、実施例IIIにおいて示されるように、Chungラット(これは末梢神経障害の広く受け入れられているモデルである)に、α−2汎アゴニスト(例えば、クロニジンなど)が、表1に示される選択的α−2Aアンタゴニスト(化合物13)と一緒に投与された。クロニジンが単独で末梢投与されたときに得られた結果とは対照的に、選択的α−2Aアンタゴニストと一緒のクロニジンの末梢への同時投与は、鎮静作用から分離可能な鎮痛作用を生じさせた。このことから、α−2A受容体の遮断は、α−アドレナリン作動性アゴニストの末梢鎮痛作用を遺伝子的に変化していない動物において生じさせ得ることが確認された。本明細書中、実施例IIIにおいてさらに開示されるように、多様な構造クラスの、最小限のα−2A活性を有するα−アドレナリン作動性アゴニスト、例えば、化合物3、化合物4、化合物5、化合物6、化合物7および化合物14などをChungモデルラットに腹腔内注射によって投与したとき、同時での鎮静作用を伴わない痛覚消失を生じさせた(下記の表2を参照のこと)。これらの結果は、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストの鎮痛活性がα−アドレナリン作動剤の以前に記載された鎮痛活性とは異なることを明らかにしており、また、最小限のα−2Aアゴニスト活性を有することによって特徴づけられるα−アドレナリン作動性アゴニストは有用な末梢鎮痛薬であり得ることをさらに示している。
【0030】
本明細書中、実施例IVにおいて開示される結果は、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストが、数日の薬物投与の後の少なくとも6週間にわたって持続する長期間にわたる痛み緩和を生じさせ得ることをさらに明らかにしている。本明細書中に開示されるように、Chungモデル動物が、皮下浸透圧ミニポンプを使用して3日間〜7日間、化合物8、化合物9、化合物3または化合物4(これらは、最小限のα−2Aアゴニスト活性を有する構造的に異なるα−アドレナリン作動性アゴニストである)で処置された。痛みの緩和が薬物処置の期間中にわたって観測された。例えば、化合物8は異疼痛を90〜100%緩和し、化合物9は異疼痛を60〜80%緩和した。さらに、試験されたアゴニストのそれぞれの鎮痛作用は、処置が終わった後の1ヶ月以上にわたって継続した。この長期間にわたる痛み緩和は、様々な試験された他の鎮痛剤の性質ではなかった。
【0031】
本明細書中にさらに開示されるように、様々な時点での化合物8の血漿中濃度のサンプリングから、非常に低い薬物レベルが、薬物処置を開始した後、10日目まで存在し、血漿中の薬物が14日目までに検出できなくなった(図5Bを参照のこと)が、痛みはこのときにはほとんど緩和していたことが明らかにされた。さらに、α−2アンタゴニストのラウオルシンが、経口胃管法または浸透圧ミニポンプによる数日間の投薬の後の様々な時点で、化合物8の鎮痛活性を阻害する能力についてアッセイされた。注目すべきことに、ラウオルシンは、処置の3日目に化合物8の鎮痛作用を阻害したが、4日目には阻害しなかった(図6を参照のこと)。まとめると、これらの結果は、鎮痛活性の持続期間が、薬物が血液中に残存している時間を超えていること、および、長期間にわたる投薬の後では、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、継続した受容体活性化または血漿中薬物レベルの不存在下でさえも、長期間にわたる痛み緩和をもたらし得ることを示している。
【0032】
本明細書中に開示されるさらなる結果は、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストはまた、Bennett部分座骨神経結紮モデルにおいて、また、過敏性腸症候群の動物モデルにおいて長期間にわたる痛み緩和を生じさせることを示している(実施例IV、図7Bおよび図8を参照のこと)。このことは、観測された長期間の鎮痛作用が神経障害性の痛みに対して特異的でないことを明らかにしている。これらの結果は、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストが、神経障害性の痛み、内臓痛、炎症性の痛み、手術後の痛みおよびガンの痛み(これらに限定されない)を含む様々なタイプの急性の痛みおよび慢性の痛みを処置するために使用され得ることを示している。
【0033】
これらの知見に基づいて、本発明は、α−2A受容体以外の末梢α−アドレナリン作動性受容体を作動させることによって対象において痛みを緩和する方法を提供する。1つの実施形態において、本発明は、末梢α−2B受容体を作動させることによって対象において痛みを緩和する方法を提供する。本発明はまた、有効量のα−アドレナリン作動性アゴニストを含有する医薬組成物と、有効量の選択的α−2Aアンタゴニストを含有する組成物とを対象に投与することによって対象において痛みを緩和する方法を提供する。そのような方法は、神経障害性の痛み、例えば、糖尿病性神経障害から生じる痛みなど;内臓痛;手術後の痛み;ガンまたはガン処置から生じる痛み;炎症性の痛み、例えば、関節炎または過敏性腸症候群から生じる痛み;頭痛の痛みおよび筋肉痛(これらに限定されない)を含む様々なタイプの痛みを緩和するために有用である。
【0034】
様々なα−アドレナリン作動性アゴニストが本発明において有用であり、これらには、汎α−2アゴニストおよび汎α−1汎α−2アゴニストならびにα−2Bアゴニストが含まれる。本発明の方法に従って痛みを緩和することにおいて有用なα−アドレナリン作動性アゴニストには、クロニジン、ブリモニジン、チザニジン、デキセメデトミジン、ノルエピネフリン、化合物1および化合物2、ならびに、すべてのそれらの医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が含まれるが、これらに限定されない。様々な選択的α−2Aアンタゴニストもまた本発明において有用である。そのような選択的α−2Aアンタゴニストには、化合物13、ならびにその医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物、BRL48962およびBRL44408、ならびに、それらの医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が含まれるが、これらに限定されない(Young他、Eur. J. Pharmacol.、168:381〜386 (1989))。1つの実施形態において、本発明は、末梢限定の選択的α−2Aアンタゴニストを用いて実施される。
【0035】
様々な投与経路が、本発明の方法に従って痛みを緩和するために有用であり得る。1つの実施形態において、α−アドレナリン作動性アゴニストおよび選択的α−2Aアンタゴニストの両方が末梢投与される。他の実施形態において、α−アドレナリン作動性アゴニストが経口投与または皮下ミニポンプにより投与される。さらなる実施形態において、α−アドレナリン作動性アゴニストが経口投与または皮下ミニポンプにより投与され、選択的α−2Aアンタゴニストが任意の末梢経路によって投与される。さらに他の実施形態において、選択的α−2Aアンタゴニストが経口投与または皮下ミニポンプにより投与される。所望される場合、選択的α−2Aアンタゴニストを経口投与または皮下ミニポンプにより投与することができ、一方で、α−アドレナリン作動性アゴニストが、例えば、経口投与または皮下ミニポンプにより末梢投与される。
【0036】
本発明はまた、有効量のα−アドレナリン作動性アゴニストを含有する医薬組成物と、有効量の選択的α−2Aアンタゴニストを含有する組成物とを対象に投与することによって対象において痛みを緩和する方法で、α−アドレナリン作動性アゴニストおよび選択的α−2Aアンタゴニストが少なくとも3日間の期間にわたって反復的または継続的に投与される方法を提供する。そのような方法において、痛み緩和は、例えば、対象において著しいα−アドレナリン作動性アゴニストレベルの不存在下で継続し得る。本発明は、1つまたは複数の医薬組成物を対象に投与することに依拠する方法を提供する。本明細書中で使用される用語「対象」は、痛みを経験することができる任意の動物を示し、例えば、ヒトまたは他の哺乳動物(例えば、霊長類、ウマ、ウシ、イヌまたはネコなど)を示す。
【0037】
本発明の方法は、急性の痛みおよび慢性の痛みの両方を処置するために、そして、非限定的な例として、起源が神経障害性、内臓性または炎症性である痛みを処置するために使用される。特定の実施形態において、本発明の方法は、神経障害性の痛み、内臓痛、手術後の痛み、ガンまたはガン処置から生じる痛み、および炎症性の痛みを処置するために使用される。
【0038】
急性の痛みおよび慢性の痛みの両方を本発明の方法によって処置することができ、用語「痛み」は急性の痛みおよび慢性の痛みの両方を包含する。本明細書中で使用される用語「急性の痛み」は、傷害(例えば、切り傷、挫傷、火傷など)によって、または、化学的刺激(例えば、カプサイシン(唐辛子の活性成分)にさらされたときに経験する痛みなど)によってもたらされる即時型の、一般には高い閾値の痛みを意味する。本明細書中で使用される用語「慢性の痛み」は、急性の痛みでない痛みを意味し、これには、神経障害性の痛み、内臓痛、炎症性の痛み、頭痛の痛み、筋肉痛および関連痛(これらに限定されない)が含まれる。慢性の痛みは、比較的長い持続期間(例えば、数年)の痛みであり、継続的または間断的であり得ることが理解される。
【0039】
1つの実施形態において、本発明の方法は、「神経障害性の痛み」を処置するために使用される。この場合、本明細書中で使用される「神経障害性の痛み」は、神経に対する傷害から生じる痛みを意味する用語である。神経障害性の痛みは、筋肉または結合組織における小さい皮膚神経または小さい神経が関係する急性の組織傷害によって引き起こされる痛みである侵害受容性の痛みから区別される。神経障害性の痛みとは対照的に、侵害受容性機構が関係する痛みは、通常、継続期間が組織修復の期間に限定され、一般には、利用可能な鎮痛剤またはオピオイドによって緩和される(Myers、Regional Anesthesia、20:173〜184 (1995))。神経障害性の痛みは典型的には、長く持続するか、または慢性的であり、最初の急性組織傷害の数日後または数ヶ月後に現れ得る。神経障害性の痛みは、持続的かつ無意識な痛み、ならびに、通常の場合には痛くない刺激に対する痛い応答である異疼痛、または、痛覚過敏(通常的にはありふれた痛い刺激(例えば、ピン刺激など)に対する強調された応答)を伴う。神経障害性の痛みは、一般には、オピオイド治療に対して抵抗性である(Myers、上掲、(1995))。
【0040】
本発明の方法は、末梢神経、後根神経節、脊髄、脳幹、視床または皮質の外傷、傷害または疾患(これらに限定されない)から生じる神経障害性の痛みを緩和することにおいて有用である。本発明の方法によって処置され得る神経障害性の痛みの例には、神経痛(例えば、帯状疱疹後神経痛など)、求心路遮断の痛み、および糖尿病性神経障害が含まれる。本発明の方法は、痛みの病因にかかわらず、神経障害性の痛みを緩和することにおいて有用であることが理解される。例として、本発明の方法は、末梢神経障害(例えば、神経腫など)から生じる神経障害性の痛み、神経圧迫から生じる神経障害性の痛み、神経の挫傷もしくは伸張または不完全な神経切断(transsection)から生じる神経障害性の痛み、あるいは単神経障害または多発神経障害から生じる神経障害性の痛みを緩和するために使用することができる。さらなる例として、本発明の方法は、後根神経節圧迫などの障害;脊髄の炎症;脊髄の打撲、腫瘍または半側切断;脳幹、視床または皮質の腫瘍または外傷から生じる神経障害性の痛みを緩和することにおいて有用である。
【0041】
上記で示されたように、本発明の方法は、単神経障害または多発神経障害から生じる神経障害性の痛みを緩和することができる。神経障害は、末梢神経系における機能的な乱れまたは病理学的な変化であり、臨床的には感覚ニューロンまたは運動ニューロンの異常によって特徴づけられる。単神経障害の用語は、1つだけの末梢神経が冒されていることを示し、一方、多発神経障害の用語は、いくつかの末梢神経が冒されていることを示す。神経障害の病因は既知または未知であり得る。既知の病因には、神経障害を生じさせる最も一般的な代謝性障害である糖尿病などの疾患状態または毒性状態の合併症、あるいは、刺激、虚血または血管炎が含まれる。本発明の方法によって処置され得る多発神経障害は、ポリオ後症候群、糖尿病、アルコール、アミロイド、毒素、HIV、甲状腺機能低下症、尿毒症、ビタミン欠乏症、化学療法、ddCまたはファブリー病(これらに限定されない)から生じ得る。本発明の方法は、病因が既知または未知であるこれらの神経障害または他の神経障害の痛みを緩和するために使用され得ることが理解される。
【0042】
さらなる非限定的な例として、本発明の方法は、クローン病、潰瘍性大腸炎、胃炎、過敏性腸症候群を含む慢性的な胃腸の炎症;および慢性的な内臓痛、例えば、ガンにより引き起こされる痛み、またはガンの処置に随伴する痛み(例えば、化学療法もしくは放射線療法に随伴する痛み)などを処置するために使用することができる。同様に、本発明の方法は、慢性の炎症性の痛み、例えば、関節炎(例えば、リウマチ様関節炎、痛風性関節炎または変形性関節症など);脊椎炎;または自己免疫疾患(例えば、エリテマトーデスなど)から生じる痛みを処置するために使用することができる。本発明の方法はさらに、頭痛の痛み;筋肉痛;ならびに、物質の乱用または中断に関連する痛み、および、病因が既知または未知である他のタイプの痛みを処置するために使用することができる。
【0043】
本発明の方法のいくつかは、部分的には「α−アドレナリン作動性アゴニスト」に依拠している。本明細書中で使用される「α−アドレナリン作動性アゴニスト」は、1つまたは複数のα−2アドレナリン作動性受容体において、ブリモニジンと比べて25%を超える効力を有する化合物、あるいは、1つまたは複数のα−1アドレナリン作動性受容体において、フェニルエフリンと比べて25%を超える効力を有する化合物を意味する用語である。そのような化合物は、1つまたは複数のα−アドレナリン作動性受容体に対して選択的であり得るか、または非選択的であり得る。従って、アドレナリン作動性アゴニストの用語は、すべてのα−1受容体およびα−2受容体においてアゴニスト活性を有する「汎α−1汎α−2アゴニスト」(例えば、ノルエピネフリンなど);汎α−2アゴニスト;α−2選択的アゴニスト;α−2Bアゴニスト;および、1つだけのα−アドレナリン作動性受容体に対して特異的であるアゴニストを包含するが、これらに限定されない。特定の実施形態において、本発明の方法では、1つまたは複数のα−2アドレナリン作動性受容体において、ブリモニジンと比べて30%を超える効力、40%を超える効力、50%を超える効力、60%を超える効力、70%を超える効力、80%を超える効力、90%を超える効力、100%を超える効力、または200%を超える効力を有するα−アドレナリン作動性アゴニストが利用される。さらなる実施形態において、本発明の方法では、1つまたは複数のα−1アドレナリン作動性受容体において、フェニルエフリンと比べて30%を超える効力、40%を超える効力、50%を超える効力、60%を超える効力、70%を超える効力、80%を超える効力、90%を超える効力、100%を超える効力、または200%を超える効力を有するα−アドレナリン作動性アゴニストが利用される。別の実施形態において、本発明は、顕著なα−2Aアンタゴニスト活性を有しないα−アドレナリン作動性アゴニストに依拠する。
【0044】
効力は、内因的活性としてもまた知られているが、化合物によって達成される最大受容体活性化の尺度であり、α−アドレナリン作動性受容体活性化の任意の受け入れられるアッセイを使用して、例えば、本明細書で後に記載するcAMPアッセイまたは受容体選択増幅技術(RSAT)アッセイを使用して決定することができる。効力は、それぞれの受容体サブタイプについて標準アゴニストの最大作用に対する薬物の最大作用の比率または百分率として表される。α−2受容体の効力が定義される場合、ブリモニジン(UK14304)が、α−2A受容体、α−2B受容体およびα−2C受容体に対する標準アゴニストとして一般に使用され、本発明において標準物として使用される。α−1受容体の効力が定義される場合、フェニルエフリンが、α−1A受容体、α−1B受容体およびα−1D受容体に対する受け入れられる標準アゴニストであり、本発明において標準物として使用される(Messierら、上掲、1995;Conklinら、上掲、1993)。
【0045】
本明細書中に開示されるように、α−2Bアゴニストは、痛みを緩和することにおいて、または、慢性の痛みを期間にわたって緩和するために有用であり得る。本明細書中で使用される用語「α−2Bアゴニスト」は、α−2Bアドレナリン作動性受容体において、ブリモニジンと比べて25%を超える効力を有する化合物を意味する。この用語は、他のα−アドレナリン作動性受容体と比較した場合、α−2B受容体に対して選択的または非選択的のいずれかであるアゴニストを包含することが理解される。従って、用語「α−2Bアゴニスト」は、下記においてさらに記載されるように、汎α−2アゴニストおよび汎α−1汎α−2アゴニスト、ならびに、α−2B受容体に対して選択的または特異的であるアゴニストを包含する。特定の実施形態において、本発明の方法では、α−2Bアドレナリン作動性受容体において、ブリモニジンと比べて30%を超える効力、40%を超える効力、50%を超える効力、60%を超える効力、70%を超える効力、80%を超える効力、90%を超える効力、100%を超える効力、または200%を超える効力を有するα−2Bアゴニストが利用される。例示的なα−2Bアゴニストには、クロニジン、ブリモニジン、化合物1および化合物2、化合物3〜12および14、ならびに、これらの化合物のすべての医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が含まれる。さらなる実施形態において、本発明の方法は、顕著なα−2Aアンタゴニスト活性を有しないα−2Bアゴニストに依拠する。
【0046】
本明細書中で使用される用語「汎α−2アゴニスト」は、α−2Aアドレナリン作動性受容体、α−2Bアドレナリン作動性受容体およびα−2Cアドレナリン作動性受容体のそれぞれにおいて、ブリモニジンと比べて25%を超える効力を有する化合物を意味し、汎α−1汎α−2アゴニストを包含する。様々な汎α−2アゴニストがこの分野では知られており、これらに、クロニジン、ブリモニジン、チザニジン、デキセメデトミジンおよびノルエピネフリンが含まれる。汎α−2アゴニストは、α−2A受容体、α−2B受容体およびα−2C受容体のそれぞれにおいて、ブリモニジンと比べて25%を超える効力を少なくとも有する。特定の実施形態において、本発明の方法は、α−2Aアドレナリン作動性受容体、α−2Bアドレナリン作動性受容体およびα−2Cアドレナリン作動性受容体において、ブリモニジンと比べて30%を超える効力、40%を超える効力、50%を超える効力、60%を超える効力、70%を超える効力、80%を超える効力、90%を超える効力、100%を超える効力、または200%を超える効力を有する汎α−2アゴニストを用いて実施される。汎α−2アゴニストの効力は様々なα−2受容体において異なり得ることが理解される。一例として、汎α−2アゴニストは、α−2A受容体において25%を超える効力、α−2B受容体において80%を超える効力、および、α−2C受容体において40%を超える効力を有し得る。
【0047】
本明細書中で使用される用語「汎α−1汎α−2アゴニスト」は、すべてのα−1受容体において、フェニルエフリンと比べて25%を超える効力を有し、かつ、すべてのα−2アドレナリン作動性受容体において、ブリモニジンと比べて25%を超える効力を有する化合物を意味する。特定の実施形態において、本発明の方法は、フェニルエフリンまたはブリモニジンに対して、それぞれ、すべてのα−1受容体およびα−2受容体において、30%を超える効力、40%を超える効力、50%を超える効力、60%を超える効力、70%を超える効力、80%を超える効力、90%を超える効力、100%を超える効力、または200%を超える効力を有する汎α−1汎α−2アゴニストに依拠する。本明細書中に開示されるように、選択的α−2Aアンタゴニストが、痛みを緩和するために、または、慢性の痛みを長期間にわたって緩和するために、α−アドレナリン作動性アゴニストと一緒に投与される。本明細書中で使用される用語「選択的α−2Aアンタゴニスト」は、(1)α−2Aにおいてブリモニジンと比べて25%未満の効力、(2)α−2Aにおいて100nM未満のKiを有し、さらに、(3)α−2Aよりもα−2Bにおいて少なくとも10倍大きいKiを有するか、または、α−2Bにおいてブリモニジンと比べて25%を超える効力を有する化合物を意味する。この定義から、非選択的なアンタゴニスト(例えば、ラウオルシンなど)がこの用語の範囲に含まれないことは当業者には明らかである。例示的な選択的α−2Aアンタゴニストが、化合物13、BRL48962およびBRL44408として本明細書中に提供される。これらの化合物の医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物もまた本発明において有用である。選択的α−2Aアンタゴニストは末梢限定の化合物であり得る。そのような化合物は、血液脳関門を容易に横断せず、従って、末梢投与されたとき、中枢神経系に入らない。
【0048】
α−2Aアンタゴニスト活性に加えて、「選択的α−2Aアンタゴニスト」もまた、化合物が上記に示された3つの基準を満たすならば、1つまたは複数のアドレナリン作動性受容体または他の受容体においてアゴニスト活性またはアンタゴニスト活性を有し得る。一例として、α−2Cアンタゴニスト活性を有する化合物で、α−2Cにおいて100nM未満のKi、および、α−2Cにおいてブリモニジンに対して25%未満の効力によって特徴づけられ、さらには、(19)α−2Aにおいてブリモニジンに対して25%未満の効力、α−2Aにおいて100nM未満のKi、および(3)α−2Aよりもα−2Bにおいて少なくとも10倍大きいKiを有する化合物が、本明細書中で定義される用語「選択的α−2Aアンタゴニスト」に包含される。同様に、α−2Aアンタゴニスト活性に加えて、α−2Bアゴニスト活性または他のアゴニスト活性を示す化合物もまた、用語「選択的α−2Aアンタゴニスト」に包含される。一例として、化合物13は、α−2Bにおいてブリモニジンに対して約40%の効力によって特徴づけられ、従って、基準(3)を満たし、さらには、(1)α−2Aにおいてブリモニジンと比べて5%の効力、および(2)α−2Aにおいて約0.08nMのKiを有しており、従って、本明細書中で使用される用語「選択的α−2Aアンタゴニスト」の定義の範囲に含まれる。特定の実施形態において、本発明の方法は、Kiが80nM未満、60nM未満、40nM未満、20nM未満、10nM未満、1nM未満または0.1nM未満である選択的α−2Aアンタゴニストを用いて実施される。さらなる実施形態において、本発明の方法は、α−2Aよりもα−2Bにおいて少なくとも20倍大きいKi、少なくとも30倍大きいKi、少なくとも40倍大きいKi、少なくとも50倍大きいKi、少なくとも100倍大きいKi、少なくとも200倍大きいKi、少なくとも500倍大きいKi、または少なくとも1000倍大きいKiを有する選択的α−2Aアンタゴニストを用いて実施される。さらにさらなる実施形態において、本発明の方法は、α−2Bにおいてブリモニジンと比べて30%を超える効力、40%を超える効力、50%を超える効力、60%を超える効力、70%を超える効力、80%を超える効力、90%を超える効力、100%を超える効力、または200%を超える効力を有する選択的α−2Aアンタゴニストを用いて実施される。
【0049】
1つの実施形態において、本発明は、最小限のα−2Bアンタゴニスト活性を有する選択的α−2Aアンタゴニストに依拠する。本明細書中で使用される用語「最小限のα−2Bアンタゴニスト活性を有する選択的α−2Aアンタゴニスト」は、α−2Bにおいて100nMを超えるKiをさらに有する本明細書中上記で定義されるような選択的α−2Aアンタゴニストを意味する。非限定的な例として、そのようなアンタゴニストは、α−2Bにおいて、200nMを超えるKi、300nMを超えるKi、400nMを超えるKi、500nMを超えるKi、1000nMを超えるKi、2000nMを超えるKi、3000nMを超えるKi、4000nMを超えるKi、または5000nMを超えるKiを有し得る。
【0050】
特異的α−2Aアンタゴニストはまた、本発明の方法においてα−2A受容体の活性化を防止するために使用することができる。本明細書中で使用される用語「特異的α−2Aアンタゴニスト」は、(1)α−2Aにおいてブリモニジンに対して25%未満の効力、(2)α−2Aにおいて100nM未満のKiを有し、さらには、(3)α−2A以外のα−アドレナリン作動性受容体のそれぞれにおいて、α−2Aにおけるよりも少なくとも10倍大きいKi、または、ブリモニジンもしくはフェニルエフリンと比べて25%を超える効力のいずれかを有する化合物を意味する。
【0051】
本発明はまた、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストに依拠する組成物および方法を提供する。具体的には、本発明は、同時での鎮静作用を伴わない末梢痛覚消失を生じさせることができる、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを含有する鎮痛剤組成物を提供する。1つの実施形態において、鎮痛剤組成物は、同時での鎮静作用を伴うことなく、また、血圧低下作用の実質的な不存在下で末梢痛覚消失を生じさせる。別の実施形態において、本発明は、同時での鎮静作用を伴うことなく、痛みを少なくとも50%軽減させるために十分な末梢痛覚消失を生じさせる鎮痛剤組成物を提供する。さらなる実施形態において、痛みを少なくとも50%軽減させるために要求される鎮痛剤組成物の用量よりも少なくとも10倍大きい、少なくとも100倍大きい、または少なくとも1000倍大きい用量の鎮痛剤組成物が、運動活動または筋肉活動における20%の軽減を生じさせるために要求される。さらに、さらなる実施形態において、本発明は、同時での鎮静作用を伴うことなく、また、血圧低下作用の実質的な不存在下で、痛みを少なくとも50%軽減させるために十分な末梢痛覚消失を生じさせる鎮痛剤組成物を提供する。別の実施形態において、本発明は、同時での鎮静作用を伴わない末梢痛覚消失を生じさせることができる、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを含有する鎮痛剤組成物で、アゴニストがチオウレア系化合物またはその誘導体でない鎮痛剤組成物を提供する。さらなる実施形態において、本発明は、同時での鎮静作用を伴わない末梢痛覚消失を生じさせることができる、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを含有する鎮痛剤組成物で、アゴニストがチオウレア系化合物または4-イミダゾール系化合物またはその誘導体でない鎮痛剤組成物を提供する。
【0052】
本発明はまた、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストの有効量を含有する医薬組成物を対象に投与し、それにより、同時での鎮静作用を伴わない末梢痛覚消失を生じさせることによって、対象において痛みを緩和する方法を提供する。そのような末梢痛覚消失は、同時での鎮静作用を伴うことなく、例えば、少なくとも50%、痛みを軽減させるために十分であり得る。別の実施形態において、末梢痛覚消失は血圧低下作用の実質的な不存在下で生じる。さらなる実施形態において、この方法は、チオウレア系化合物またはその誘導体でない、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを使用して実施される。また、さらに、さらなる実施形態において、この方法は、チオウレア系化合物または4-イミダゾール系化合物またはその誘導体でない、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを使用して実施される。様々なタイプおよび病因の痛みを本発明の方法に従って緩和することができる。非限定的な例として、本発明の方法は、神経障害性の痛み、例えば、糖尿病性神経障害から生じる痛みなど;内臓痛;手術後の痛み;ガンまたはガン処置から生じる痛み;炎症性の痛み、例えば、関節炎痛または過敏性腸症候群の痛みなど;頭痛の痛みおよび筋肉痛を緩和することにおいて有用であり得る。
【0053】
最小限のα−2Aアゴニスト活性を有する様々なα−アドレナリン作動性アゴニストが本発明の方法において有用であり得る。1つの実施形態において、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストである。そのようなアゴニストは、例えば、チオン系化合物であり、例えば、化合物3または化合物11またはその医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物などであり得る。1つの実施形態において、本発明の方法は、化合物3の(-)-エナンチオマーまたはその医薬的に許容され得る塩もしくはエステルである、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストを用いて実施される。
【0054】
本発明において有用な、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストにはさらに、イミダゾロン系化合物が含まれるが、これらに限定されない。有用なイミダゾロン系の、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、例えば、化合物4またはその医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物であり得る。1つの実施形態において、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、化合物4の(+)-エナンチオマー、またはその医薬的に許容され得る塩、エステルもしくはアミドである。さらなる実施形態において、本発明の方法は、下記のいずれかの、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストを使用して実施される:化合物5、化合物6、化合物7、化合物8、化合物9、化合物14、またはそれらの医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物。最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、経口投与および皮下ミニポンプによる投与(これらに限定されない)を含む様々な経路のいずれかによって末梢投与することができる。
【0055】
本明細書中で使用される用語「末梢痛覚消失」は、末梢投与の後で得られる痛みの軽減を意味する。下記においてさらに説明されるように、末梢投与は、中枢神経系の外側における対象への薬剤の導入を意味し、脊椎または脳に対する直接的な投与でない任意の投与経路を包含する。末梢痛覚消失は、痛みを、例えば、少なくとも30%、40%、50%、60%、70%、80%、90%または100%軽減させるために十分であり得る。本発明の組成物は、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる。末梢鎮静作用失は、本明細書中で使用される場合、運動活動または筋肉活動の減少を意味する用語である。本明細書中で使用される用語「同時での鎮静作用を伴わない」は、運動活動または筋肉活動の減少が1回以上の投薬において末梢痛覚消失に比較的ほとんど随伴しないことを意味する。具体的には、末梢投与されたとき、運動活動または筋肉活動における20%の減少を生じさせるために要求される用量が、痛みを少なくとも50%軽減させるために要求される用量よりも少なくとも3倍大きいならば、薬物は、「同時での鎮静作用を伴わない末梢痛覚消失」を生じさせる。特定の実施形態において、運動活動または筋肉活動における20%の減少を生じさせるために要求される用量は、痛みを少なくとも50%軽減させるために要求される用量よりも少なくとも4倍大きく、または5倍大きく、または6倍大きく、または7倍大きく、または8倍大きく、または9倍大きく、または10倍大きく、または25倍大きく、または50倍大きく、または100倍大きく、または200倍大きく、または500倍大きく、または1000倍大きく、または2000倍大きく、または5000倍大きい。末梢投与後の痛み軽減の程度および鎮静作用の程度を明らかにする様々な方法がこの分野では広く知られており、本明細書で後に記載する。
【0056】
本明細書中で使用される用語「最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニスト」は、(1)α−2Aアドレナリン作動性受容体においてブリモニジンに対して25%未満の効力を有すること、および(2)遺伝子的に変化していない動物において同時での鎮静作用を伴わない末梢痛覚消失を生じさせることができることによってさらに特徴づけられる、上記で定義されるようなα−アドレナリン作動性アゴニストを意味する。効力は、アゴニスト活性の任意の標準的なアッセイを使用して、例えば、本明細書中下記に記載されるcAMPアッセイまたはRSATアッセイなどを使用して測定されることが理解される。化合物3、化合物4、化合物5、化合物6、化合物7、化合物8、化合物9、化合物10、化合物11および化合物12、ならびに、それらの医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストの例として本明細書中に提供される。そのような化合物は、上記の表1に示されるように多様な構造クラスに属する。本発明において有用な、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、下記においてさらに記載されるように、1つまたは複数のα−1受容体において活性を有するα−アドレナリン作動性アゴニスト、最小限のα−2Aアゴニスト活性を有するα−2B/Cアゴニスト、最小限のα−2Aアゴニスト活性を有する特異的α−2Bアゴニスト、および、最小限のα−2Aアゴニスト活性を有する特異的α−2Cアゴニストを包含する。
【0057】
最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、α−アドレナリン作動性アゴニストをスクリーニングすることによって、例えば、様々なインビトロアッセイまたはインビボアッセイで、機能的活性について、α−2Aにおいて25%未満の効力を示すα−アドレナリン作動性アゴニストをスクリーニングすることによって容易に同定することができる。具体的には、そのようなアゴニストは、例えば、スルプロストン感作された痛みの広く受け入れられているモデルを使用して、野生型マウスおよびα−2Aノックアウトマウスの両方において、同時での鎮静作用を伴わない末梢痛覚消失についてアッセイすることができる。最小限よりも大きいα−2Aアゴニスト活性を有するアゴニストは、野生型のスルプロストン感作動物において同時での鎮静作用を伴わない末梢痛覚消失を生じさせないそのような化合物として排除することができ、だが、下記においてさらに記載されるように、同時での鎮静作用を伴わない末梢痛覚消失がα−2Aノックアウトマウスにおいて観測される。特定の実施形態において、本発明の方法は、同時での鎮静作用を伴うことなく、遺伝子的に変化していない動物において、痛みを少なくとも50%軽減させるために、または、少なくとも60%、70%、80%、90%もしくは100%軽減させるために十分な末梢痛覚消失を生じさせる、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを使用して実施される。
【0058】
本明細書中で使用される用語「最小限のα−2Aアゴニスト活性を有するα−2Bアゴニスト」は、(1)α−2B受容体においてブリモニジンに対して25%を超える効力、(2)α−2Bにおいて1000nM未満の効能、または、α−2Aよりもα−2Bにおいて少なくとも100倍大きい効能、(3)α−2A受容体においてブリモニジンに対して25%未満の効力、および(4)遺伝子的に変化していない動物において同時での鎮静作用を伴わない末梢痛覚消失を生じさせる能力を有することによって特徴づけられる化合物を意味する。最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストの例として、本明細書中には、化合物3、化合物4、化合物5、化合物6、化合物7、化合物8、化合物9、化合物10、化合物11、化合物12および化合物14、ならびに、これらの化合物の医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が提供される。本明細書中に例示されるように、これらの化合物は、部分的には、遺伝子的に変化していない動物において同時での鎮静作用を伴わない末梢痛覚消失を生じさせることができることによって特徴づけられる。特定の実施形態において、本発明において有用な、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、α−2Bにおいてブリモニジンと比べて30%を超える効力、40%を超える効力、50%を超える効力、60%を超える効力、70%を超える効力、80%を超える効力、90%を超える効力、100%を超える効力、または200%を超える効力を有する。さらなる実施形態において、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、α−2Aにおいてブリモニジンと比べて20%未満の効力、15%未満の効力、10%未満の効力、5%未満の効力、2%未満の効力または1%未満の効力を有する。さらなる実施形態において、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、α−2Bにおいて900nM未満の効能、800nM未満の効能、700nM未満の効能、600nM未満の効能、500nM未満の効能、400nM未満の効能、300nM未満の効能、200nM未満の効能、100nM未満の効能、50nM未満の効能、10nM未満の効能、または1nM未満の効能を有する。さらにさらなる実施形態において、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、α−2Aよりもα−2Bにおいて少なくとも200倍大きい効能、300倍大きい効能、少なくとも400倍大きい効能、500倍大きい効能、1000倍大きい効能、または10,000倍大きい効能を有する。さらなる実施形態において、本発明の方法は、同時での鎮静作用を伴うことなく、遺伝子的に変化していない動物において、痛みを少なくとも50%軽減させるために、または、少なくとも60%、70%、80%、90%もしくは100%軽減させるために十分な末梢痛覚消失を生じさせることができる、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストに依拠する。
【0059】
本明細書中で使用される用語「最小限のα−2Aアゴニスト活性を有するα−2B/Cアゴニスト」は、(1)α−2B受容体またはα−2C受容体または両者において、ブリモニジンと比べて25%を超える効力、(2)α−2B受容体またはα−2C受容体または両者において1000nM未満の効能、あるいは、α−2B受容体またはα−2C受容体または両者においてα−2A受容体に対して少なくとも100倍大きい効能、(3)α−2A受容体において、ブリモニジンと比べて25%未満の効力、および(4)遺伝子的に変化していない動物において同時での鎮静作用を伴わない末梢痛覚消失を生じさせる能力を有することによって特徴づけられる化合物を意味する。
【0060】
本明細書中で使用される用語「最小限のα−2Aアゴニスト活性を有する特異的α−2Bアゴニスト」は、(1)α−2Bにおいてブリモニジンと比べて25%を超える効力、(2)α−2Bにおいて1000nM未満の効能、または、α−2Aよりもα−2Bにおいて少なくとも100倍大きい効能、(3)α−2Aおよびα−2Cにおいてブリモニジンと比べて25%未満の効力または1/50の効能、(4)すべてのα−1受容体においてフェニルエフリンに対して25%未満の効力または1/50の効能、そして(5)遺伝子的に変化していない動物において同時での鎮静作用を伴わない末梢痛覚消失を生じさせる能力を有することによって特徴づけられる化合物を意味する。
【0061】
同様に、本明細書中で使用される用語「最小限のα−2Aアゴニスト活性を有する特異的α−2Cアゴニスト」は、(1)α−2Cにおいてブリモニジンと比べて25%を超える効力、(2)α−2Cにおいて1000nM未満の効能、または、α−2Aよりもα−2Cにおいて少なくとも100倍大きい効能、(3)α−2Aおよびα−2Bにおいてブリモニジンと比べて25%未満の効力または1/50の効能、(4)すべてのα−1受容体においてフェニルエフリンに対して25%未満の効力または1/50の効能、そして(5)遺伝子的に変化していない動物において同時での鎮静作用を伴わない末梢痛覚消失を生じさせる能力を有することによって特徴づけられる化合物を意味する。
【0062】
アゴニスト活性およびアンタゴニスト活性は、選択性および特異性を含めて、様々な日常的アッセイのいずれかを使用して特徴づけることができ、そのようなアッセイには、受容体選択および増幅技術(RSAT)アッセイ(Messierら、Pharmacol. Toxicol.、76:308〜11 (1995);Conklinら、Nature、363:274〜6 (1993));環状AMPアッセイ(Shimizuら、J. Neurochem.、16:1609〜1619 (1969));およびサイトセンサー微量生理機能測定アッセイ(Neveら、J. Biol. Chem.、267:25748〜25753 (1992))が含まれるが、これらに限定されない。そのようなアッセイは、一般には、1つだけのα−アドレナリン作動性受容体サブタイプのみを自然の状態で発現する細胞を使用して、または、1つだけの組換えα−アドレナリン作動性受容体サブタイプを発現するトランスフェクションされた細胞を使用して行われる。アドレナリン作動性受容体は、ヒト受容体、または、類似する薬理学を有するそのホモログであり得る。本明細書中に開示されるように、RSATアッセイが、ヒトα−2A(c10遺伝子)、ラットα−2B(RNG遺伝子)、ヒトα−2C(c4遺伝子)、ウシα−1A、ハムスターα−1Bおよびラットα−1Dで一過性にトランスフェクションされた細胞を使用して行われた。
【0063】
RSATアッセイでは、コンフルエンス細胞の混合集団における受容体含有細胞の選択的な増殖をもたらす接触阻害の受容体媒介による喪失が測定される。細胞数の増大が、所望される場合にはハイスループットアッセイ形式またはウルトラハイスループットアッセイ形式で、β-ガラクトシダーゼなどの適切な検出可能なマーカー遺伝子を用いて評価される。Gタンパク質(Gq)を活性化する受容体により、増殖応答が誘発される。α−アドレナリン作動性受容体は、通常の場合にはGiに共役しているが、Gi受容体認識ドメインを含有するハイブリッドGqタンパク質(これはGq/i5と呼ばれる)と同時発現させたときにはRSAT応答を活性化する。
【0064】
一例として、RSATアッセイを本質的には下記のように行うことができる。NIH-3T3細胞が15cmディッシュに2x106細胞の密度で置床され、10%子ウシ血清が補充されたダルベッコ改変イーグル培地で維持される。1日後、細胞が、p-SV-β-ガラクトシダーゼをコードする哺乳動物発現プラスミド(5〜10μg)、受容体をコードする哺乳動物発現プラスミド(1〜2μg)、およびGタンパク質をコードする哺乳動物発現プラスミド(1〜2μg)でリン酸カルシウム沈殿によって同時トランスフェクションされる。キャリアDNA(例えば、40μgのサケ精子DNA)もまた、トランスフェクション効率を増大させるために含めることができる。新鮮な培地が翌日に加えられる。1日〜2日後に細胞が集められ、50アッセイ分の量で凍結される。トランスフェクションされた細胞を解凍し、100μlの細胞が、例えば、96ウエルプレートにおいて三連でアッセイされる様々な濃度で、試験される化合物の100μl量に加えられる。インキュベーションが37℃で72時間〜96時間にわたって続けられる。リン酸塩緩衝化生理的食塩水で洗浄した後、β-ガラクトシダーゼ活性が、200μlの発色性基質(リン酸塩緩衝化生理的食塩水における3.5mMのO-ニトロフェニル-β-D-ガラクトピラノシド/0.5%NP-40)を加え、30℃で一晩インキュベーションして、420nmにおける光学密度を測定することによって測定される。吸光度は酵素活性の尺度であり、これは細胞数に依存し、受容体媒介による細胞増殖を反映する。それぞれの受容体における各薬物のEC50および最大作用(効力)が決定される。
【0065】
本発明はさらに、対象において慢性の痛みを長期間にわたって緩和するための方法を提供する。この方法は、慢性の痛みの緩和が、継続した受容体活性化の不存在下で維持されるように、少なくとも3日間の期間にわたって、α−2A受容体活性化の不存在下で鎮痛性α−アドレナリン作動性受容体を対象において活性化することによって実施される。1つの実施形態において、本発明の方法は、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストの有効量を含有する医薬組成物を少なくとも3日間の期間にわたって対象に投与し、その結果、慢性の痛みの緩和が対象において著しいアゴニストレベルの不存在下で維持されるようにすることによって実施される。慢性の痛みの緩和は、対象における著しいアゴニストレベルの不存在下で、例えば、少なくとも3週間にわたって維持され得る。本発明の方法は、任意のタイプの慢性の痛みを長期間にわたって緩和するために使用され得ることが理解される。非限定的な例として、そのような方法は、神経障害性の痛み;内臓痛;手術後の痛み;ガンまたはガン処置から生じる痛み;および炎症性の痛みを長期間にわたって緩和するために使用することができる。
【0066】
慢性の痛みの長期間にわたる緩和は、最小限のα−2Aアゴニスト活性を有する様々なα−アドレナリン作動性アゴニストのいずれかを用いて本発明の方法に従って達成され得る。慢性の痛みの長期間にわたる緩和を、例えば、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストを使用して達成することができる。最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストの例には、チオン系化合物、例えば、化合物3および化合物11、ならびに、それらの医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物などが含まれるが、これらに限定されない。1つの実施形態において、そのようなチオン系の、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、化合物3の(-)-エナンチオマーまたはその医薬的に許容され得る塩もしくはエステルである。最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストの例にはさらに、イミダゾロン系化合物、例えば、化合物4またはその医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物などが含まれるが、これらに限定されない。1つの実施形態において、そのようなイミダゾロン系の、最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストは、化合物4の(+)-エナンチオマーまたはその医薬的に許容され得る塩もしくはエステルである。最小限のα−2Aアゴニスト活性を有するα−2Bアゴニストの例にはまた、化合物5、化合物6、化合物7、化合物8および化合物9、ならびに、それらの医薬的に許容され得る塩、エステル、アミド、立体異性体もしくはラセミ混合物が含まれるが、これらに限定されない。本発明の方法において、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、末梢投与経路(例えば、経口投与、または皮下ミニポンプによる投与など)(これらに限定されない)を含む様々な経路のいずれかによって投与することができる。
【0067】
本発明はさらに、対象において慢性の痛みを長期間にわたって緩和するための方法で、有効量のα−アドレナリン作動性アゴニストを含有する医薬組成物と、有効量の選択的α−2Aアンタゴニストを含有する医薬組成物とを少なくとも3日間の期間にわたって対象に投与し、その結果、慢性の痛みの緩和が対象において著しいアゴニストレベルの不存在下で維持されるようにすることによる方法を提供する。慢性の痛みの緩和は、対象において著しいアゴニストレベルの不存在下で、例えば、少なくとも3週間にわたって維持され得る。様々なα−アドレナリン作動性アゴニストが本発明において有用であり、これらには、クロニジン、ブリモニジン、チザニジン、デキセメデトミジン、ノルエピネフリン、ならびに、他の汎α−2アゴニストおよび汎α−1汎α−2アゴニスト、そして同様に、化合物1または化合物2、ならびにそれらの医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が含まれる。同様に、様々な選択的α−2Aアンタゴニストが、慢性の痛みの長期間にわたる緩和において有用であり、これらには、化合物13ならびにその医薬的に許容され得る塩、エステル、アミド、立体異性体およびラセミ混合物が含まれるが、これらに限定されない。様々な投与経路が、下記においてさらに議論されるように、慢性の痛みを長期間にわたって緩和するための医薬組成物を送達するために有用であることが理解される。そのような投与経路には、末梢投与、例えば、経口投与、または皮下ミニポンプによる投与などが包含されるが、これらに限定されない。
【0068】
本発明の方法は慢性の痛みの長期間にわたる緩和をもたらす。本明細書中で使用される用語「長期間にわたる緩和(長期間にわたって緩和する)」は、医薬組成物を最後に投与した後の少なくとも10日間にわたって持続する、痛みの著しい軽減を意味する。特定の実施形態において、緩和は、医薬組成物を最後に投与した後、少なくとも14日間、または少なくとも21日間、または少なくとも28日間、または少なくとも60日間、または少なくとも90日間にわたって持続する。さらなる実施形態において、本発明は、慢性の痛みを長期間にわたって緩和するための方法で、医薬組成物を最後に投与した後、少なくとも10日間、または少なくとも14日間、または少なくとも21日間、または少なくとも28日間、または少なくとも60日間、または少なくとも90日間にわたって痛みを少なくとも50%軽減させる方法を提供する。さらなる実施形態において、本発明は、慢性の痛みを長期間にわたって緩和するための方法で、医薬組成物を最後に投与した後、少なくとも10日間、または少なくとも14日間、または少なくとも21日間、または少なくとも28日間、または少なくとも60日間、または少なくとも90日間にわたって痛みを少なくとも80%軽減させる方法を提供する。他の実施形態において、本発明は、慢性の痛みを長期間にわたって緩和するための方法で、医薬組成物を最後に投与した後、少なくとも10日間、または少なくとも14日間、または少なくとも21日間、または少なくとも28日間、または少なくとも60日間、または少なくとも90日間にわたって痛みを少なくとも90%軽減させる方法を提供する。
【0069】
慢性の痛みを長期間にわたって緩和するための方法は、最小限のα−2A活性を有するα−アドレナリン作動性アゴニストを少なくとも3日間の期間にわたって投与することによって実施される。アゴニストは、少なくとも3日間の期間にわたって、例えば、3日間、4日間、5日間、6日間、7日間、8日間、9日間または10日間にわたって、反復した投薬または継続した投薬によって投与され得る。非限定的な例として、最小限のα−2A活性を有するα−アドレナリン作動性アゴニストを、3日間にわたって1日に3回、または、4日間にわたって1日に3回投与することができ、例えば、3日間にわたって1日に3回の経口投与、または、4日間にわたって1日に3回の経口投与により投与することができる。さらなる例として、最小限のα−2A活性を有するα−アドレナリン作動性アゴニストを継続的に投与することができ、例えば、埋め込まれた注入ミニポンプを介して静脈内投与することができ、または、3日間、4日間、5日間、6日間もしくは7日間にわたる持続放出配合物を使用して投与することができる。
【0070】
徐放性配合物が、慢性の痛みを長期間にわたって緩和するための本発明の方法において有用であり得ることが理解される。反復した投与が使用される場合、投与頻度は、部分的には、アゴニストの半減期に依存することがさらに理解される。所望される場合、本発明の方法は、長い半減期(例えば、少なくとも24時間、36時間、48時間または72時間の半減期)を有するアゴニストを1回だけ、または、ちょうど2回もしくは3回、投薬することによって実施することができる。
【0071】
種々の薬物送達手段が本発明の方法において組み合わせられ得ることが理解される。一例として、1日目における連続した静脈内投与を、少なくとも3日間の期間にわたってα−2A受容体活性化の不存在下で鎮痛性のα−アドレナリン作動性受容体を活性化するために、2日目および3日目での反復した経口投薬と組み合わせることができ、その結果、慢性の痛みの緩和が前記対象において著しいアゴニストレベルの不存在下で維持されるようになる。本明細書中下記においてさらに詳述されるように、投薬の頻度および継続期間は、部分的には、所望される緩和およびアゴニストの半減期に依存すること、そして、様々な投与経路が本発明の方法において有用であることが理解される。
【0072】
本発明によりまた、指定されたアゴニストまたはアンタゴニストを表す式に由来する医薬的に許容され得る塩、エステルおよびアミドが包含される。本発明において有用なアゴニストおよびアンタゴニストの好適な医薬的に許容され得る塩には、酸付加塩(これは、例えば、アゴニストまたはアンタゴニストの溶液を適切な酸(例えば、塩酸、硫酸、フマル酸、マレイン酸、コハク酸、酢酸、安息香酸、クエン酸、酒石酸、炭酸またはリン酸など)の溶液と混合することによって形成させることができる)が含まれるが、これらに限定されない。アゴニストまたはアンタゴニストが酸性成分を有する場合、その好適な医薬的に許容され得る塩には、アルカリ塩(例えば、ナトリウム塩またはカリウム塩など)、アルカリ土類塩(例えば、カルシウム塩またはマグネシウム塩など)、および、好適な有機リガンドと形成される塩(例えば、第四級アンモニウム塩)が含まれ得る。代表的な医薬的に許容され得る塩には、酢酸塩、ベンゼンスルホン酸塩、安息香酸塩、重炭酸塩、重硫酸塩、重酒石酸塩、ホウ酸塩、臭化物、カルシウムエデタート、カムシラート、炭酸塩、塩化物、クラブラン酸塩、クエン酸塩、二塩酸塩、エデト酸塩、エジシラート、エストラート、エシラート、フマル酸塩、グルセプタート、グルコン酸塩、グルタミン酸、グリコリルアルサニル酸塩、ヘキシルレゾルシン酸塩、ヒドラバミン、臭化水素酸塩、塩酸塩、ヒドロキシナフトエ酸塩、ヨウ化物、イソチオン酸塩、乳酸塩、ラクトビオン酸塩、ラウリン酸塩、リンゴ酸塩、マレイン酸塩、マンデル酸塩、メシラート、メチルブロミド、メチルニトラート、メチル硫酸塩、ムチン酸塩、ナプシラート、硝酸塩、N-メチルグルカミンアンモニウム塩、オレイン酸塩、パモ酸塩(エムボナート)、パルミチン酸塩、パントテン酸塩、リン酸塩/二リン酸塩、ポリガラクツロン酸塩、サリチル酸塩、ステアリン酸塩、硫酸塩、塩基性酢酸塩、コハク酸塩、タンニン酸塩、酒石酸塩、テオクラート、トシラート、トリエチオジドおよび吉草酸塩が含まれるが、これらに限定されない。
【0073】
本発明において有用なアゴニストおよびアンタゴニストの官能基を、化合物の薬理学的利用性を増強するために修飾することができる。そのような修飾は十分に化学の当業者の知識の範囲内であり、それらには、示されたアゴニストまたはアンタゴニストのエステル、アミド、エーテル、N-オキシドおよびプロドラッグが含まれるが、これらに限定されない。アゴニストまたはアンタゴニストの活性を増強することができる修飾の例には、例えば、エステル化、例えば、C1〜C6アルキルエステル、好ましくはC1〜C4アルキルエステルの形成が含まれ、この場合、アルキル基は直鎖または分枝鎖である。他の許容され得るエステルには、例えば、C5〜C7シクロアルキルエステルおよびアリールアルキルエステル(例えば、ベンジルエステルなど)が含まれる。そのようなエステルは、有機化学の技術分野で広く知られている従来の様々な方法を使用して、本明細書中に記載される化合物から調製することができる。
【0074】
他の医薬的に許容され得る修飾には、アミドの形成が含まれる。有用なアミド修飾には、例えば、アンモニア;第一級のC1〜C6ジアルキルアミン(この場合、アルキル基は直鎖または分枝鎖である);および、様々な置換を有するアリールアミンに由来するアミドが含まれる。第二級アミンの場合、アミンはまた5員環または6員環の形態であり得る。これらのアミドおよび他のアミドを調製するための様々な方法がこの分野では広く知られている。
【0075】
本発明において有用なアゴニストまたはアンタゴニストが少なくとも1つのキラル中心を有する場合、そのアゴニストまたはアンタゴニストは化学的に異なるエナンチオマーとして存在し得ることが理解される。さらに、アゴニストまたはアンタゴニストが2つ以上のキラル中心を有する場合、その化合物はジアステレオマーとして存在する。そのような異性体およびその混合物はすべてが、示されたアゴニストまたはアンタゴニストの範囲内に包含される。同様に、アゴニストまたはアンタゴニストが、その構造が互変異性体として存在することを可能にする構造的配置を有する場合、そのような互変異性体は、示されたアゴニストまたはアンタゴニストの範囲内に包含される。さらに、結晶形態において、アゴニストまたはアンタゴニストが多型体として存在する場合がある。溶媒が存在する場合、アゴニストは、例えば、水または一般的な有機溶媒との溶媒和物を形成する場合がある。そのような多型体、水和物および他の溶媒和物もまた、本明細書中で定義されるような示されたアゴニストまたはアンタゴニストの範囲内に包含される。
【0076】
本発明において有用なアゴニストまたはアンタゴニストは一般には医薬組成物で投与される。所望される場合、組成物は、場合により、同じ医薬組成物または異なる医薬組成物において、また、同じ投与経路または異なる投与経路によって、1つまたは複数の他の治療剤または鎮痛性物質と一緒に投与することができる。一例として、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストをガバペンチンなどの鎮痛剤と一緒に投与することができる。別の例として、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストを、静脈内カクテルで、1つまたは複数のガン化学療法剤と一緒に投与することができる。
【0077】
本発明において有用な医薬組成物は、活性なアゴニストまたはアンタゴニストを含み、さらには、所望される場合、対象に投与されたとき、長期間または永続的な有害作用を実質的に有しない任意のキャリアまたは希釈剤である医薬的に許容され得るキャリアまたは希釈剤などの賦形剤を含むことができる。そのような賦形剤は、一般には、活性な化合物と混合されるか、または、活性な化合物を希釈もしくは包むことができる。キャリアは、活性な化合物に対する賦形剤またはビヒクルとして作用する固体または半固体または液体の薬剤であり得る。医薬的に許容され得るキャリアおよび希釈剤の例には、水、例えば、蒸留水または脱イオン水など;生理的食塩水;および他の水性媒体が含まれるが、これらに限定されない。有効成分は、所望するキャリアまたは希釈剤において可溶性であり得るか、または、所望するキャリアまたは希釈剤における懸濁物として送達され得ることが理解される。
【0078】
医薬組成物はさらに、所望する場合には、乳化剤、湿潤化剤、甘味剤もしくは矯味矯臭剤、張性調節剤、保存剤、緩衝剤または抗酸化剤などの1つまたは複数の剤を含むことができる。医薬組成物において有用な張性調節剤には、塩(例えば、塩化ナトリウム、塩化カリウムなど)、マンニトールまたはグリセリン、および他の医薬的に許容され得る張性調節剤が含まれる。本発明の医薬組成物において有用な保存剤には、塩化ベンザルコニウム、クロロブタノール、チメロサール、酢酸フェニル水銀および硝酸フェニル水銀が含まれるが、これらに限定されない。pHを調節するための様々な緩衝剤および手段を、医薬組成物を調製するために使用することができ、これらには、酢酸塩緩衝剤、クエン酸塩緩衝剤、リン酸塩緩衝剤およびホウ酸塩緩衝剤が含まれるが、これらに限定されない。同様に、本発明の医薬組成物において有用な様々な抗酸化剤がこの分野では広く知られており、これらには、例えば、メタ重亜硫酸ナトリウム、チオ硫酸ナトリウム、アセチルシステイン、ブチル化ヒドロキシアニソールおよびブチル化ヒドロキシトルエンが含まれる。薬理学の分野で知られているこれらの物質および他の物質が本発明において有用な医薬組成物に含まれ得ることが理解される。
【0079】
本発明のアゴニストおよびアンタゴニストは有効量で投与される。そのような有効量は、一般には、所望される治療効果を達成するために必要な最少用量であり、これは、例えば、大ざっぱには、痛みにより引き超される不快感を許容可能なレベルに軽減させるために必要なそのような量であり得る。そのような用量は、一般には、0.1mg/日〜1000mg/日の範囲であり、例えば、0.1mg/日〜500mg/日、0.5mg/日〜500mg/日、0.5mg/日〜100mg/日、0.5mg/日〜50mg/日、0.5mg/日〜20mg/日、0.5mg/日〜10mg/日または0.5mg/日〜5mg/日の範囲であり得るが、投与される実際の量は、痛みの重篤度、患者の年齢および体重、患者の全身的な身体状態、痛みの原因、および投与経路を含む関連した状況を考慮に入れて医師によって決定される。坐薬および持続放出配合物は本発明において有用であり得るし、これらには、例えば、皮膚パッチ、皮膚表面または皮膚下に置くための配合物、および筋肉内注射用配合物が含まれる。
【0080】
本発明の方法において有用な医薬組成物は、例えば、処置される痛みのタイプ、組成物に含められるアゴニストまたはアンタゴニスト、ならびに、対象の病歴、危険性因子および症状に依存して、様々な手段によって対象に投与することができる。本発明の方法のために好適な投与経路には、全身投与および局所投与の両方が含まれる。非限定的な例として、痛みを緩和するために、または、慢性の痛みを長期間にわたって緩和するために有用な医薬組成物は、経口投与もしくは皮下ミニポンプによって、皮膚パッチによって、静脈内注射、皮下注射もしくは筋肉内注射によって、局所的な滴剤、クリーム、ゲルもしくは軟膏によって、移植もしくは注入された持続放出配合物として、皮下ミニポンプもしくは埋め込まれたデバイスによって、クモ膜下ポンプもしくはクモ膜下注射によって、または硬膜外注射によって投与することができる。
【0081】
特定の実施形態において、本発明の方法は、アゴニストを含有する医薬組成物、アンタゴニストを含有する医薬組成物、または両方を末梢投与することによって実施される。一例として、α−アドレナリン作動性アゴニストを含有する医薬組成物、および選択的α−2Aアンタゴニストを含有する医薬組成物の一方または両方を末梢投与することができる。本明細書中で使用される用語「末梢投与」または用語「末梢投与される」は、薬剤を中枢神経系の外側で対象に導入することを意味する。末梢投与は、脊椎または脳への直接的な投与ではない任意の投与経路を包含する。そのため、クモ膜下投与および硬膜外投与、ならびに頭蓋注射または頭蓋埋め込みは用語「末梢投与」または用語「末梢投与される」の範囲に含まれないことは明らかである。さらに、いくつかの鎮痛剤は血液脳関門を横断することができ、従って、末梢投与後、中枢神経系および末梢神経系の全体に分布することは明らかである。
【0082】
末梢投与は局所的または全身的であり得る。局所投与では、局所投与された部位およびその周りに対して、投与部位から遠位の領域よりも著しく多い量の医薬組成物が送達される。全身投与では、本質的には対象の末梢神経系全体に対する医薬組成物の送達がもたらされ、そしてまた、組成物の性質に依存して中枢神経系への送達がもたらされることがある。
【0083】
本発明の方法において有用な末梢投与経路には、経口投与、局所投与、静脈内注射または他の注射、および埋め込まれたミニポンプまたは他の持続放出デバイスまたは配合物が包含されるが、これらに限定されない。本発明において有用な医薬組成物は、例えば、錠剤、液剤、カプセル剤または粉末剤などでの任意の許容され得る形態で経口投与によって;静脈内注射、腹腔内注射、筋肉内注射、皮下注射または非経口注射によって;経皮拡散またはエレクトロホレシスによって;滴剤、クリーム、ゲルまたは軟膏などでの任意の許容され得る形態で局所的に;また、ミニポンプまたは他の埋め込まれた持続放出デバイスまたは配合物によって末梢投与することができる。
【0084】
いくつかの実施形態では、本発明は、有効量のα−アドレナリン作動性アゴニストを含有する医薬組成物と、有効量の選択的α−2Aアンタゴニストを含有する医薬組成物との両方を対象に投与することによって実施される。そのような「混合治療」では、アゴニストおよびアンタゴニストが、同じ医薬組成物または異なる医薬組成物で、また、同じ投与経路または異なる投与経路によって独立して投与され得るか、または同時に送達され得ることが理解される。一例として、アゴニストおよびアンタゴニストの両方を経口投与することができ、この場合、アゴニストが1日に2回与えられ、アンタゴニストが1日に1回与えられる。別の例として、アゴニストを硬膜外に投与することができ、その一方で、アンタゴニストが経口投与される。さらなる例として、アゴニストおよびアンタゴニストを静脈内「カクテル」において一緒に投与することができる。典型的な局所眼科用組成物の成分を以下の表1に例示するがこれに限定されない:
【表1】
【0085】
本発明はまた、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングする方法で、α−2A受容体を、鎮痛活性を有するα−アドレナリン作動性アゴニストと接触させること;および、アゴニストがα−2Aアゴニスト活性を有するかどうかを明らかにすることによって行われる方法を提供する。この場合、α−2Aアゴニスト活性の不存在により、鎮痛活性を有するα−アドレナリン作動性アゴニストが、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0086】
本発明ではまた、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングする方法で、α−2A受容体を薬剤と接触させること;薬剤がα−2Aアゴニスト活性を有するかどうかを明らかにすること;α−2B受容体を薬剤と接触させること;および、薬剤がα−2Bアゴニスト活性を有するかどうかを明らかにすることによって行われる方法を提供する。この場合、α−2Aアゴニスト活性の不存在およびα−2Bアゴニスト活性の存在により、薬剤が、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0087】
本発明はまた、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングする方法で、野生型レベルのα−2A受容体活性を少なくとも有するコントロール動物にα−アドレナリン作動性アゴニストを末梢投与すること;コントロール動物における痛覚消失についてアッセイすること;低下したレベルのα−2A受容体の発現または活性を有する対応する動物に、コントロール動物に投与された量と類似する量またはそれよりも大きい量のα−アドレナリン作動性アゴニストを末梢投与すること;および、低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失についてアッセイすることによって行われる方法を提供する。この場合、コントロール動物における痛覚消失の不存在、および低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失の存在により、α−アドレナリン作動性アゴニストが過度なα−2Aアゴニスト活性を有することが示され、かつ、コントロール動物における痛覚消失の存在、および低下したレベルのα−2A受容体の発現または活性を有する前記対応する動物における痛覚消失の存在により、α−アドレナリン作動性アゴニストが、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0088】
本発明のそのような方法において、コントロール動物は、例えば、両方のα−2A受容体遺伝子座において野生型であり得る。1つの実施形態において、コントロール動物は、野生型マウスなどの野生型動物である。様々な対応する動物が本発明のスクリーニング方法において有用である。1つの実施形態において、本発明は、α−2A受容体遺伝子座においてホモ接合の点変異を有する対応する動物を用いて実施される。別の実施形態において、本発明は、α−2A受容体コード配列の内部に点変異を有する対応する動物を用いて実施される。そのような点変異は、例えば、Asp79と類似の残基で生じることがあり、例えば、Asp79からAsnへの変異であり得る。さらなる実施形態において、本発明は、ホモ接合のα−2Aノックアウト変異を有する対応する動物を用いて実施される。様々な方法論が、本発明の方法において痛覚消失についてアッセイするために使用され得ることが理解され、そのような方法論には、スルプロストン感作化後の痛覚消失についてスクリーニングすることが含まれるが、これに限定されない。
【0089】
所望される場合には、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングするための本発明の方法は、(a)野生型レベルのα−2A受容体活性およびα−2B受容体活性を少なくとも有するコントロール動物にα−アドレナリン作動性アゴニストを末梢投与すること;(b)コントロール動物における痛覚消失についてアッセイすること;(c)低下したレベルのα−2A受容体の発現または活性を有する対応する動物に、コントロール動物に投与された量と類似する量またはそれよりも大きい量のα−アドレナリン作動性アゴニストを末梢投与すること;(d)低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失についてアッセイすること;(e)低下したレベルのα−2B受容体の発現または活性を有する対応する動物にα−アドレナリン作動性アゴニストを末梢投与すること;および(f)低下したレベルのα−2B受容体の発現または活性を有する対応する動物における痛覚消失についてアッセイすることによって実施することができ、この場合、コントロール動物における痛覚消失の不存在、および低下したレベルのα−2A受容体の発現または活性を有する対応する動物における痛覚消失の存在により、α−アドレナリン作動性アゴニストが過度なα−2Aアゴニスト活性を有することが示され、かつ、コントロール動物における痛覚消失の存在、低下したレベルのα−2A受容体の発現または活性を有する前記対応する動物における痛覚消失の存在、および低下したレベルのα−2B受容体の発現または活性を有する対応する動物における痛覚消失の不存在により、α−アドレナリン作動性アゴニストが、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0090】
本発明はさらに、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤についてスクリーニングする方法で、野生型レベルのα−2B受容体活性を少なくとも有するコントロール動物にα−アドレナリン作動性アゴニストを末梢投与すること;コントロール動物における痛覚消失についてアッセイすること;低下したレベルのα−2B受容体の発現または活性を有する対応する動物にα−アドレナリン作動性アゴニストを末梢投与すること;および、低下したレベルのα−2B受容体の発現または活性を有する対応する動物における痛覚消失についてアッセイすることによる方法を提供する。この場合、コントロール動物における痛覚消失の存在、および低下したレベルのα−2B受容体の発現または活性を有する対応する動物における痛覚消失の不存在により、α−アドレナリン作動性アゴニストが、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる効果的な薬剤であることが示される。
【0091】
本発明のそのような方法は、様々なコントロール動物を用いて、例えば、両方のα−2B受容体遺伝子座において野生型であるコントロール動物を用いて実施することができる。1つの実施形態において、コントロール動物は野生型動物である。さらなる実施形態において、コントロール動物は野生型マウスである。同様に、様々な対応する動物が本発明のスクリーニング方法において有用であり、対応する動物には、ヘテロ接合のα−2Bノックアウト変異またはホモ接合のα−2Bノックアウト変異を有する対応する動物が含まれる。痛覚消失は、様々な方法論のいずれかを使用してアッセイすることができる。1つの実施形態において、痛覚消失はスルプロストン感作化後にアッセイされる。
【0092】
本明細書中で使用される用語「コントロール動物」は、痛みを経験することができる任意の動物を意味する。コントロール動物は、例えば、内因性α−2A受容体の野生型レベルを発現することができるか、または、内因性α−2Aに加えてα−2A受容体の導入遺伝子を発現することができる。コントロール動物が両方のα−2A受容体遺伝子座において「野生型」である場合、コントロール動物は、自然界で通常的に見出されるレベルで天然に存在し、かつ発現するα−2A受容体配列を有する。同様に、コントロール動物が両方のα−2B受容体遺伝子座において「野生型」である場合、コントロール動物は、自然界で通常的に見出されるレベルで天然に存在し、かつ発現するα−2B受容体配列を有する。
【0093】
本明細書中で使用される用語「野生型動物」は、分子遺伝学により変化していない、マウスまたはラットなどの動物を意味する。「野生型動物」は、自然界で見出される野性型動物であり得るか、または、十分に特徴づけられた遺伝学的システムを有する同系交配動物もしくは他の実験室系統であり得る。本明細書中で使用される用語「対応する動物」は、低下したレベルのα−2A受容体の発現もしくは活性を有するか、または、低下したレベルのα−2B受容体の発現もしくは活性を有する、コントロール動物と同じ種の動物を意味する。対応する動物は、一般には、1つもしくは複数のα−2A受容体遺伝子座または1つもしくは複数のα−2B受容体遺伝子座を除いて、コントロール動物と遺伝子的に同一である。非限定的な例として、対応する動物は、α−2A遺伝子の一方もしくは両方またはα−2B遺伝子の一方もしくは両方を完全に有しないことがあり、あるいは、α−2A受容体コード配列もしくはα−2B受容体コード配列における欠失、挿入もしくは点変異、または、α−2Aもしくはα−2Bの発現を低下させるか、もしくは除去する、5'調節配列もしくは3'調節配列における欠失、挿入もしくは点変異を有することがあり、あるいは、低下した活性を有するα−2A受容体もしくはα−2B受容体(例えば、Asp79からAsnへの変異(これは約80%の受容体活性をなくす)を有するネズミα−2A受容体など)の野生型レベルを発現することがある。従って、1つの実施形態において、本発明のスクリーニング方法は、Asp79と類似の残基におけるホモ接合の点変異による低下したレベルのα−2A受容体の発現または活性を有する対応する動物を用いて実施される。そのような「類似の」残基は、ネズミα−2A受容体のホモログにおいて同じ相対的な位置に存在するそのようなアルギニン残基である。α−2Aノックアウトマウスおよびα−2Bノックアウトマウスの様々な系統、ならびに、低下したレベルのα−2A受容体の発現または活性をもたらすホモ接合の点変異を有する様々な系統がこの分野では広く知られているか、または、標準的な方法によって調製することができる(米国特許第5869079および5443505)。
【0094】
以下の実施例は説明を意図するものであり、本発明を限定するものではない。
【0095】
実施例1
感覚過敏の種々のメカニズムを有するマウスモデル
本実施例は、α−2Aおよびα−2Cノックアウトマウスの交感神経系の増大した緊張が、α−1A受容体活性化による触覚過敏の誘導を増大することを示すものである。
【0096】
A.スルプロストンーフェニレフィン誘発性触覚過敏の種々のメカニズム
野生型マウスおよびα−2Aノックアウトマウスにおける異疼痛に対するアッセイが行われた。簡単に記載すると、マウスが5匹〜6匹の動物群に分けられた。コントロールマウスには5μlのDMSOが投与され、一方、処置マウスには、様々な用量の示された薬剤を含有する5μlのDMSOが注射された。クモ膜下注射の後、各マウスは、観察のために、木材チップが底に敷かれた個々の13x8.5x13cmのプレキシガラスの囲いの中に入れられた。異疼痛が50分間の期間にわたって5分毎に1回評価され、応答が15分〜50分の時間枠で8回記録された。異疼痛は、小さいはけでマウスの側腹を軽くなでることによって評価され、下記のようにランク付けされた:0、反応なし;1、接触プローブから逃避しようとして軽く鳴くこと;2、激しい鳴き声が接触プローブによって誘発され、プローブに噛み付き、逃避しようと激しく努力すること。各動物に対する8回のスコアが合計され、群についての平均値が、平均総スコアを得るために求められた。
【0097】
非選択的α−2アンタゴニストのラウオルシンが、α−2Aノックアウトマウスにおいて、パラ-アミノ-クロニジン(PAC)によりもたらされる末梢痛覚消失に影響を及ぼすその能力についてアッセイされた。スルプロストンで感作されたα−2Aノックアウトマウスが、腹腔内注射によって100μg/kgで送達されたPACで処置された。上記に記載されるように、PACはα−2Aノックアウトマウスにおいて著しい痛覚消失を誘導した。しかしながら、図3に示されるように、PACの鎮痛作用はラウオルシン(0.3μg/kg)の腹腔内注射によって阻止された。これらの結果は、末梢のα−2受容体がα−2Aノックアウトマウスにおいて痛覚消失を媒介することを示している。
【0098】
鎮静作用が、下記のように、暗くしたチャンバーにおける探索行動を評価することによって分析された。マウスの体重を測定し、試験化合物を、5μlの体積でのクモ膜下注射によって、または1ml/kgの体積での腹腔内注射によって、示された用量で投与した。注射後5分〜30分での痛覚消失の測定に対応する所定の時点で、動物の活動が、ジギコムアナライザーチャンバー(Omnitech Electronic;Columbus、OH)にマウスを入れることによって自動的に測定された。ジギコムアナライザーチャンバーは、箱を十字に交差し、動物が動き回るときに遮断される光電池ビームを含む;チャンバーは、床の高さを上げることによってマウス用に変更された。全体的な動物の活動のコンピューター分析が5分間の期間にわたって行われた。学習した行動はデータに影響を及ぼし得るので、所与の動物はいずれもこのプロトコルのために多くても2回使用された。すべての動物には、研究の間に少なくとも2週間の休止期間が与えられた。
【0099】
ラット神経結紮モデルは、末梢の神経障害性の痛みの十分に受け入れられているモデルである。Chungモデルにおいて、左脊髄神経のL-5およびL-6の部分的結紮は、影響を受ける左足に、軽い接触に対する長く持続する過敏性をもたらす。この過敏性は、灼熱痛の神経障害性状態のヒトが経験する痛みと類似している(KimおよびChung、Pain、50:355-363(1992))。
【0100】
Chungラットにクモ膜下注射によって投与されたとき、汎α−2アゴニストのクロニジンは、鎮静作用から分離可能な著しい痛覚消失をもたらし、一方、腹腔内投与は、鎮静作用の不存在下で目立たない痛覚消失を生じさせたにすぎなかった。血液脳関門を容易に横断しない化合物である化合物1およびPACもまた、Chungラットモデルにおいて活性についてアッセイされた。予想されたように、いずれの化合物も、腹腔内投与されたとき、鎮静をもたらさない用量で著しい鎮痛作用を有しなかった。
【表2】
【0101】
選択的α−2Aアンタゴニストの化合物13とのクロニジンの同時投与は、腹腔内投与されたクロニジンの鎮痛作用の用量応答を左側に移動させた;0.3μg/kgの化合物13と組み合わせられたとき、30μg/kgが、劇的な異疼痛の逆転を得るために要求されただけであった(図4Aを参照のこと)。クロニジンのこの濃度は活動を減少させるが、化合物13の不存在下で異疼痛を逆転させる100μg/kgの用量ほどの鎮痛作用を全く有していなかった。
α−2のアウトマウスはDixon他(Ann. Rev. Pharmacol. Toxicol.、20:441-462(1980))の方法を使用して決定された。薬物後の閾値が薬物前の閾値と比較され、触覚感受性の逆転率(%)が15.1グラムの正常な閾値に基づいて計算された。この結果は、異疼痛の逆転%として表され、これは正常ラット(100%)に対する痛み閾値の逆転率を反映している。EP1受容体アンタゴニスト
【化2】
は米国特許第5843942に記載されているとおりに合成する。メマンチンは、最小限のα−2Aアゴニスト活性を有する、構造クラスが異なるα−アドレナリン作動性アゴニストが末梢投与され、Chungラットモデルにおいて痛みを緩和させる能力についてアッセイされた。表2には、チオン系化合物3、イミダゾロン系化合物4、チアゾール系化合物5、オキサゾール系化合物6、チオウレア系化合物7、および4-イミダゾール系化合物14を用いて得られた結果が示される。これらの化合物はそれぞれが、構造的に異なっているが、最小限のα−2Aアゴニスト活性を有するα2-B/C選択的α−アドレナリン作動性アゴニストである。一例として、化合物14は、最小限のα−2Aアゴニスト活性を有し、それにもかかわらず、著しいα−1アゴニスト活性を有するα−2B選択的アゴニストである。
【0102】
クロニジンおよび化合物3に対する鎮静作用および鎮痛作用の用量応答曲線の全面的な比較を、クロニジンについては20μg/kg〜100μg/kgの単回腹腔内用量、化合物3については1μg/kg〜1000μg/kgの単回腹腔内用量を使用して行った。異疼痛逆転の割合および全体的な活動の減少が上記のように決定された。図4Cに示されるように、クロニジンの鎮静作用が、痛覚消失を生じさせる用量よりも低い用量で生じた。具体的には、クロニジンは、著しい痛覚消失を生じさせる100μg/kgの用量で極めて大きい鎮静作用を有した。これらの結果は、クロニジンの末梢投薬からもたらされる痛覚消失が鎮静作用から分離可能でないことを明らかにしている。対照的に、図4Dに示される結果は、化合物3が、鎮静作用を生じさせることなく著しい痛覚消失を生じさせたことを明らかにしている。具体的には、鎮静作用が、異疼痛の強固な逆転を生じさせる用量よりも100倍大きい用量で生じなかった。これらの結果は、最小限のα−2Aアゴニスト活性を有するα−2Bアドレナリン作動性アゴニストが、同時での鎮静作用を伴うことなく80%の異疼痛の逆転を生じさせ得ることを明らかにしている。
【0103】
まとめると、これらの結果は、全身投薬の後で目立たない鎮痛活性を伴う非選択的α−アドレナリン作動性アゴニストが、α−2A受容体の活性化を妨げる薬剤と組み合わせられたとき、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる非常に効果的な薬剤に変わり得ることを明らかにしている。これらの結果はまた、最小限のα−2A受容体アゴニスト活性のみを有する選択的α−2アゴニストの鎮痛作用が、以前に記載されたα−アドレナリン作動性薬剤の鎮痛作用とは異なることを明らかにしている。
【0104】
構造が異なるα−アドレナリン作動性アゴニストが、長期間にわたる投薬の後のChungモデルラットにおける慢性の痛みの長期間にわたる緩和を生じさせる能力についてアッセイされた。具体的には、Chungモデル動物に、ビヒクルコントロール、または最小限のα2-A活性を有する下記のα−アドレナリン作動性アゴニストの0.1mg/kg/時間が、7日間、皮下浸透圧ミニポンプを使用して投薬された:化合物8、化合物9、化合物3または化合物4。痛みの緩和が薬物処置の期間を通じて観察された;例えば、図5Aに示されるように、化合物8は異疼痛を90%〜100%緩和し、化合物9は異疼痛を60%〜80%緩和した。注目すべきことに、これらの化合物ならびに化合物3および化合物4の鎮痛作用は、処置が終了した後1ヶ月以上にわたって継続した。処置された動物は、治療を受けたという行動上の徴候を示しており、これは、手術された足をもはやかばうことはなく、また、ケージの底にこの足の平らな部分を置いているという点で、無処置ラットまたはビヒクル処置ラットとは異なっていた。
【0105】
長期間にわたる痛み緩和がまた、経口投薬の3日後に観察された。Chungモデルラットに対して、0.3mg/kgの化合物8の3回の投薬が、3日間連続して午前8時〜午後6時の間に経口胃管法によって施された。異疼痛の70%〜80%の逆転が達成され、異疼痛は、追跡試験の3週間の期間よりも長い期間、増大しなかった。これらの結果は、化合物8などのα−2B/Cアゴニストの単回の腹腔内投薬または経口投薬の後で得られた痛み緩和の比較的短い継続期間とは対照的に、長期間にわたる鎮痛作用が、最小限のα2-Aアゴニスト活性を有するα−2B/C選択的アゴニストを反復して投薬することからもたらされることを示している。
【0106】
B.α−2Aおよびα−2Cノックアウトマウスの交感神経系の増大した緊張は、α−1A受容体活性化による触覚過敏の誘導を増大する
異疼痛の持続した逆転が、動物における薬物の継続した存在によるものであるかどうかを明らかにするために、ラットを、7日間、0.1mg/kg/時間の化合物8を用いて皮下浸透圧ミニポンプを使用して処置した。化合物8の血漿中濃度が、ポンプ挿入後の3日目、6日目、8日目、10日目および14日目にサンプリングされ、液体クロマトグラフィー−質量分析法/質量分析法(LC-MS/MS)によって測定された。図5に示されるように、最小の薬物レベルが10日目に検出され、薬物は14日目までに血漿中に残存しなくなった。これらの結果は、痛みの緩和が、長期間にわたる投薬の後、血漿中の薬物レベルが認められないときでさえも達成され得ることを示している。
【0107】
浸透圧ミニポンプによって7日間または経口胃管法によって3日間、化合物8で処置されたChungモデルラットは、異疼痛からの長期間にわたる緩和を一貫して示した。α−2アンタゴニストのラウオルシンが、様々な時点でこの抗異疼痛作用を阻害する能力についてアッセイされた。注目すべきことに、図6Aに示されるように、0.3μg/kgのi.p.で、ラウオルシンは、処置の3日目に注射されたとき、化合物8の鎮痛活性を阻害したが、4日目では阻害しなかった。急性の痛みを緩和する様々な薬物が、Chungラットにおける長期間にわたる痛み緩和についてアッセイされた。具体的には、抗痙攣薬のガバペンチン(3mg/kg、経口、TID、3日間);抗うつ薬のアミトリプチリン(0.1mg/kg/時間、注入ミニポンプ、7日間);ならびに、α−2A活性を有する2つの非選択的α−アドレナリン作動性アゴニストのブリモニジン(0.04mg/kg/時間、注入ミニポンプ、7日間)および化合物1(0.1mg/kg/時間、注入ミニポンプ、7日間)が、このモデルにおいて触覚異疼痛を強く緩和する用量でChungモデルラットに投与された。全ての場合において、異疼痛が処置の中止前または中止後に完全に再発した。
【0108】
α−2B/C選択的アゴニストの化合物8が、神経障害性の痛みの別のラット神経傷害モデルであるBennett部分坐骨神経結紮モデルにおいて試験された。このラットモデルは、ヒトにおいて認められる痛み感覚と類似した痛み感覚の障害を伴う末梢単神経障害をもたらす(BennettおよびXie、Pain、33:87-107(1988))。Bennettモデルでは、神経傷害が、締め付ける結紮糸で坐骨神経の周囲をゆるく縛り、これにより、締め付け部から遠位側の神経の変性を引き起こすことによってもたらされる。異疼痛および痛覚過敏が、無意識での痛みに加えて、締め付け傷害によってもたらされる(BennettおよびXie、上掲(1988))。具体的には、冷たさに対する異疼痛、すなわち、冷刺激からの痛み感覚が、変化した痛み感覚の1つの発現である:Bennettモデル動物は、コントロール動物とは対照的に、冷たい表面から、手術された肢の足を頻繁に持ち上げる。
【0109】
C.交感神経系はNMDA誘発性触覚過敏には寄与しない
化合物8が、4日間の期間、浸透圧ミニポンプによって投与された。図7Bに示されるように、冷たさに対する異疼痛が、4日間の処置期間中およびポンプ除去後の3週間以上の試験期間にわたってともに完全に緩和された。これらの結果は、最小限のα−2Aアゴニスト活性を有するα−2アドレナリン作動性アゴニスト(化合物8など)が、異なるタイプの神経障害性の痛みに対して適用可能な鎮痛特性を有することを示している。約20分間続いたBennett手術は下記のように行われた。雄性Sprague-Dawleyラット(約250グラム〜300グラム)がイソフルラン/酸素吸入によって麻酔された。剃毛およびベタジン塗布によって手術部位を調製した後、切開が、腰帯を超えてわずかに中線の左側に行われた。左股関節のわずかに尾側および腹側で、筋肉群の不鮮明な境界線が視認され、小さい(約10mm〜25mm)切開が筋肉群の境界線のすぐ下で行われた。筋肉は、坐骨神経が大腿骨の長さに平行して見えるまで容赦なく引き離された。
【0110】
まとめると、これらの結果は、全身投薬の後で目立たない鎮痛活性を伴う非選択的α−アドレナリン作動性アゴニストが、α−2A受容体の活性化を妨げる薬剤と組み合わせられたとき、同時での鎮静作用を伴わない末梢痛覚消失を生じさせる非常に効果的な薬剤に変わり得ることを明らかにしている。これらの結果はまた、最小限のα−2A受容体アゴニスト活性のみを有する選択的α−2アゴニストの鎮痛作用が、以前に記載されたα−アドレナリン作動性薬剤の鎮痛作用とは異なることを明らかにしている。
【0111】
実施例II
α−2アゴニストブリモニジンおよびクロニジンの活性の比較
本実施例では、化合物8などの、最小限のα−2活性を有するα−2アドレナリン作動性アゴニストが、慢性的な内臓過敏性のラットモデルにおいて痛みを緩和することが明らかにされる。
【0112】
A.ブリモニジンは交感神経増大触覚過敏を軽減するが、クロニジンは軽減しない。
長期の薬物投与が、上記のようなBennett手術の1週間後にラットの背中の皮下に埋め込まれた浸透圧ミニポンプによって達成された。Bennett動物における痛み応答の評価が下記のように手術後7日目に行われた。熱刺激への応答を評価するために、ラットを透明なプラスチックチャンバー(18cm x 29cm x 12.5cm)に入れ、正常な動物に対しては侵害性でない冷却された(0℃〜4℃)金属床の上に置き、手術された足が冷たい床から持ち上げられる時間が5分間の期間にわたって記録された。実験当日、試験薬物が無麻酔で投与された(1ml/kgの体積で、1μg/kg〜1000μg/kgの範囲の用量でのIP投与またはPO投与)。場合により、動物は、研究の間に少なくとも3日間の休止期間を与えられた後、3ヶ月の期間にわたる次の実験のために使用された。
【0113】
C.αパンアゴニストの種々のα−2対αー1機能選択性
慢性的な内臓過敏性のよく受け入れられているモデルが、α2A、α2C、α1Cおよびα1B受容体を安定に発現する細胞系を用いて分析した。
【0114】
段階的な結腸直腸拡大に対する成体ラットの腹部引っ込み反射応答を、腹壁筋から記録する筋電図記録(EMG)によって定量化した。感作されたラットは、過敏性腸症候群のヒト患者における症状と類似した内臓痛の症状である異疼痛性の痛みおよび痛覚過敏の両方を示し、痛みのある刺激(40mmHg〜80mmHg)だけでなく、正常では痛みのないレベルのバルーン膨張(20mmHg)に対して大げさに応答した。感作されていない動物は、20mmHgのバルーン膨張に対してほとんど応答を示さず、80mmHgまでのCRDにおいて、EMG強度単位で1までの穏やかな応答を示した。CRDの各レベルが4分毎に20秒間施され、合計で5回繰り返された。
【0115】
8匹〜10匹の正常および感作された成体Sprague Dawleyラット(約3ヶ月齢)からなる群には、皮下ミニポンプが、50%DMSOまたは50%DMSOでの化合物8のいずれかを7日間の期間にわたって100μg/kg/時間の用量で送達するために埋め込まれた。一連の段階的な結腸直腸拡大(20mmHg、40mmHg、60mmHg、80mmHg)に対する腹部引っ込み反射が、ポンプ埋め込みの前、皮下注射の2日目および5日目、そしてポンプが除去された2日後、1週間後、2週間後および4週間後に腹部EMG記録によって測定された。非感作コントロールラットの処置後におけるEMG応答は、処理前のレベルから有意に変化しなかった(EMG強度は約0〜約1.5の範囲であった)。対照的に、感作ラットにおけるEMG応答は、はるかに大きくなっており、強度がほぼ1〜約3.5の範囲であった。感作ラットにおけるこの増大した痛み応答は、図8Cに示されるように、ビヒクルでの処置の後に低下しなかった。対照的に、化合物8での処置時および処置後では、感作ラットにおける増大したEMG応答は完全に緩和された。図8Dに示されるように、痛みは0〜1.5に低下した。これは、非感作ラットで観察されるレベルである。さらに、結腸直腸の異疼痛および痛覚過敏は、薬物処置が中止された後の4週間である試験期間中に再発しなかった。
【0116】
これらの結果は、最小限のα2-A活性を有するα−2アドレナリン作動性アゴニストが、過敏性腸症候群の痛みなどの結腸直腸の痛みを長期間にわたって緩和するために使用され得ることを明らかにしている。これらの結果はさらに、観察された鎮痛作用が、末梢の神経障害性の痛みに対して特異的ではなく、また、最小限のα2-A活性を有するα−2アドレナリン作動性アゴニスト、または選択的α2-Aアンタゴニストと一緒に投与されたα−2アドレナリン作動性アゴニストが、神経障害性の痛み、内臓痛、炎症性の痛み、手術後の痛みおよびガンの痛みなどの、様々なタイプの急性の痛みおよび慢性の痛みを処置するために使用され得ることを示している。
【0117】
纏めると、野生型マウスにおいて、クロニジンの腹腔内投薬(500μg/kg)は、スルプロストンにより誘導される触覚異疼痛を緩和したが、同時での鎮静作用がもたらされた。対照的に、腹腔内投与された化合物3(100μg/kg)、すなわち、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、同時での鎮静作用を伴うことなくスルプロストン誘導の触覚異疼痛を緩和した。クロニジンおよび化合物3のこれらの用量の鎮痛作用が、最も小さいフォンフライ毛を1.65グラムの力で使用してヘテロ接合型(-/+)およびホモ接合型(-/-)のα−2Bノックアウトマウスにおいて測定された。この力では、最も小さいフォンフライ毛は、非処置のα−2Bノックアウトマウスにおいても、また、それらの野生型同腹子においても痛み応答を引き起こさない。異疼痛が上記のように評価およびランク付けされた。
【0118】
ヘテロ接合およびホモ接合の両方のα−2Bノックアウト系統において、野生型マウスにおけるその作用とは全く異なることなく痛覚消失を緩和した。再度ではあるが、痛覚消失には、鎮静作用が伴っていた。対照的に、化合物3は、ヘテロ接合のα−2Bノックアウトマウスまたはホモ接合のα−2Bノックアウトマウスのいずれにおいても鎮痛作用を有していなかった(図9)。類似する結果が他の化合物で得られた。具体的には、クロニジンのように、α−アドレナリンの汎アゴニストであるブリモニジンはα−2Bノックアウトマウスにおいて鎮痛作用を有し、一方、化合物8、すなわち、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストは、ヘテロ接合のα−2Bノックアウトマウスまたはホモ接合のα−2Bノックアウトマウスにおいて鎮痛活性を示さなかった。
【0119】
ヒトα2Aは、最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストの痛覚消失の機構が、α−アドレナリンの汎アゴニストの痛覚消失の機構とは異なっていることを明らかにしている。これらの結果はさらに、α−アドレナリン作動性の末梢鎮痛活性がα−2B受容体の活性化によって媒介されることを示している。化合物3、化合物11および化合物4はそれぞれが、α−2アドレナリン作動性受容体に対して選択的であり、α−1受容体における活性がほとんどないかまたは全くない。さらに、これらの化合物はそれぞれが、インビトロRSATアッセイにおいてα−2A受容体における検出可能な活性を全く示さないα−2B/C選択的化合物である。
【0120】
ウシα1−Aはα−2B受容体に対して選択的であり、α−2C受容体よりもα−2B受容体において10倍以上大きい活性を示す。化合物3は、α−2C受容体よりもα−2B受容体において約100倍大きい活性によって特徴付けられるように、α−2B受容体に対して特異的であった。様々な濃度の化合物3、化合物11および化合物4が、上記に記載されたように、Chungモデルラットに経口投与された。図10Aに示されるように、30μg/kgの経口投与された化合物3は70%〜100%の異疼痛逆転をもたらした。
【0121】
細胞内Ca+2鎮痛作用が、迅速に、すなわち、20分以内に観察された。そのうえ、単回経口用量として投与されたとき、その作用は、投与後2時間までに本質的には消失する痛覚消失を伴う一過性のものであった。図10Bには、経口投与された化合物11もまた、Chungラットモデルにおいて痛みを緩和したことが示される。0.1mg/kgの用量は、痛覚消失を約60%〜90%まで軽減させるために十分であった。再度ではあるが、鎮痛作用は、単回経口投薬の後、投与後2時間までに消失した。図10Cに示されるように、化合物4は、痛みを緩和することにおいて直線の用量応答を示した。30μg/kgの用量は鎮痛効果のために十分であり、0.3mg/kgの化合物4では、異疼痛の約60%〜80%が逆転した。1mg/kgでは、異疼痛はさらに大きい程度に逆転し、痛みの緩和がより長い期間にわたって持続した。しかしながら、痛みの緩和は、最大の用量でさえも4時間後には本質的には全く観察されなかった(図10C)。これらの結果から、最小限のα−2A活性を有するα−アドレナリン作動性アゴニストが、経口投与されたとき、鎮痛剤として作用し得ることが裏付けられる。
【0122】
細胞内cAMPを用いた薬物投与が5日目に中断され、その時点でミニポンプが除去された。異疼痛が約1ヶ月の期間にわたってアッセイされた。図11に示されるように、約80%の異疼痛の逆転が、3つの化合物のそれぞれによって達成された。さらに、化合物11および化合物4の鎮痛作用は、4週間の全試験期間を通して本質的に同じレベルで維持された。これらの結果から、化合物3、化合物11および化合物4が、長期間にわたる痛み緩和のための効果的な鎮痛剤であることが示され、さらに、長期間にわたる投薬の後、最小限のα−2A活性を有するα−アドレナリン作動性アゴニストが、慢性の痛みを処置するために使用され得ることが裏付けられる。
【0123】
化合物3、化合物11および化合物4が、最大の異疼痛逆転をもたらすために必要とされる用量よりも大きい1mg/kgで腹腔内投与された。鎮静作用が上記のようにアッセイされた。さらに、これらの化合物は、0.5mg/kgの静脈内投与または3mg/kgの経口投与で、サルにおける心臓血管副作用についてアッセイされ、化合物3はラットにおける心臓血管作用についてアッセイされた。3mg/kgの用量のα−2アンタゴニスト活性の欠如が、0.1mg/kgの用量で腹腔内に同時投与されたクロニジンの鎮静作用の逆転を試験することによって評価された。
【表3】
【0124】
実施例IV
化合物の製造
本実施例はα−2アゴニストの製造を記載する。
【0125】
A.1((+)−(S)−4−[1−(2,3−ジメチル−フェニル)−エチル]−1,3−ジヒドロ−イミダゾール−2−チオンの製造
【化3】
THF(45mL)および水(40mL)中のCordi et al. , Synth. Comm. 26: 1585 (1996)にしたがって製造した(+)−(S)−4−[1−(2,3−ジメチル−フェニル)−エチル]−1H−イミダゾール(デクスメデトミジン;2.00g、10.0mmol)の混合物をNaHCO3(8.4g、100mmol)およびフェニルクロロチオノホルメート(3.7mL、27.4mmol)で処理した。室温にて4時間攪拌した後、混合物を水(30mL)とエーテル(75mL)で希釈した。有機層を捨て、水層を50mlのエーテルで2回抽出した。有機層をMgSO4で乾燥し、濾過した。残留物を減圧濃縮し、MeOH(54mL)で希釈し、NEt3(6.5mL)と室温にて16時間反応させた。溶媒を減圧留去し、30%のCH2Cl2:ヘキサンと置換した。溶媒を再度留去し、固形物を形成した。30%CH2Cl2:ヘキサンにさらに再懸濁し、固形物をフィルターで集め、CH2Cl2:ヘキサンで洗浄し、減圧乾燥して化合物1((+)−(S)−4−[1−(2、3−ジメチル−フェニル)−エチル]−1,3−ジヒドロ−イミダゾール−2−チエン)1.23g(53%)を得た。化合物1の製造の反応式は上で示される。
【0126】
得られた生成物のキャラクタリゼーションは以下のとおりである。旋光度:[a]D20+14°(c1.25(MeOH中))1H NMR: (300 MHz, DMSO) d 11.8 (s, 1H) , 11.6 (s, 1H) , 7.03−7.01 (m, 2H) , 6.95−6.91 (m, 1H) , 6.50 (s, 1H) , 4.15 (q, J = 6.9 Hz, 1H) , 2.25 (s, 3H) , 2.20 (s, 3H) , 1.38 ( d, J = 6.9 Hz, 3H).
【0127】
B.化合物2(5−(1H−イミダゾール−4−イルメチル)−シクロヘキサン−1−エニル]−メタノール)の製造方法
【化4】
8−(2−ベンジルオキシ−エチル)−1,4−ジオキサ−スピロ[4.5]デカン(中間体R1;1.02g、3.70mmol)をCiufolini et al. , J. Amer. Chem. Soc. 113: 8016 (1991)にしたがって製造した。この化合物をアセトン(100mL):H2O(5mL)に溶解し、TsOH(140mg、0.74mmol)と45℃で5時間反応させた。水溶液で標準的に処理した後、クロマトグラフィー(SiO2)で精製し4−(2−ベンジルオキシ−エチル)−シクロヘキサノンを無色の油として得た(97%)。
【0128】
LDA(33ml、1.5MinEt2O)のTHF(50mL)溶液(78℃)を4−(2−ベンジルオキシ−エチル)−シクロヘキサノン(9.5g、40.2mmol)で処理した。混合物を30分かけて0℃に加温した後−78℃に再度冷却し、HMPA(7mL)を添加した。メチルシアノホルメート(4.1mL、85mmol)を加え、混合物を15分間攪拌した後、水溶液でクエンチし、処理した。生成物を10%EtOAc:Hxを用いるクロマトグラフィー(SiO2)に付した。5−(2−ベンジルオキシ−エチル)−2−オキソ−シクロヘキサンカルボン酸メチルエステルを単離した(5.8g(49%))し、当量のNaBH4(MeOH中、10℃)で還元した。アルコール(上記中間体R2)を、30〜50%EtOAC:Hxを用いるクロマトグラフィー(SiO2)により精製した(収率約90%)。
【0129】
5−(2−ベンジルオキシ−エチル)−2−ヒドロキシ−シクロヘキサンカルボン酸メチルエステル(中間体R2;0.72g、2.48mmol)のピリジン(10mL)溶液をSOCl2(0.73mL、12.4mmol)で−20℃にて処理した。混合物を15分間反応させた後16時間かけて55℃に加温した。溶媒を減圧濃縮し、残留物を0℃のエーテルで希釈した。この溶液を水でクエンチし、1MHCl、5%NaOHおよびブラインで洗浄した。有機の物質をMgSO4で乾燥し、濾過し、溶媒を留去した。混合物をベンゼンで希釈し、水を減圧下で共沸蒸留した。残留物をベンゼン(15mL)に溶解し、DBU(0.76mL、5mmol)を加えた。混合物を室温にて30分間反応させた。処理し、20%EtOAc:Hxを用いるクロマトグラフィー(SiO2)に付し、5−(2−ベンジルオキシ−エチル)−シクロヘキサン−1−エンカルボン酸メチルエステル(中間体R3)を単離した(0.56g(82%))。
【0130】
中間体R3をTHF(100mL)に溶解しDIBAL(70mL、1Minヘキサン)のTHF(160mL)溶液(−30℃)に35分間かけて加えた。混合物をRochelle塩溶液でクエンチし、エーテルで抽出した。乾燥した残留物を30%EtOAc:Hxを用いるクロマトグラフィー(SiO2)に付し、[5−(2−ベンジルオキシ−エチル)−シクロヘキサン−1−エニル]−メタノール4.6g(80%)を得た。アルコール(4.0g、18.7mmol)のDMF(60mL)溶液をトリエチルアミン(3mL)で処理した後、TBSCl(3.0g、22.4mol)で室温にて20分間処理した。水溶液で処理して残留物を単離し、クロマトグラフィーにより[5−(2−ベンジルオキシ−エチル)−シクロヘキサン−1−エニルメトキシ]−tert−ブチル−ジメチル−シラン(中間体R4)3.6g(63%)を得た。
【0131】
このベンジル保護されたアルコール(中間体R4)(2.0g、5.55mmol)のTHF(20mL)溶液を70℃に冷却し、NH3をこのフラスコ内で凝縮させた(約20mL)。Naの塊を加え、混合物を70℃で15分間攪拌した。混合物を20分間かけて−30℃に加温し、NH4Clでクエンチし、抽出により単離した。残留物を25%EtOAc:Hx(99%)を用いるクロマトグラフィー(SiO2)により精製した。アルコールを標準的な「Swern」プロトコルにより酸化した。アルコール2−[3−(tert−ブチル−ジメチル−シラニルオキシメチル)−シクロヘキサン−3−エニル]−エタノール(1.3g、4.8mmol)を、DMSO(0.63mL、8.9mmol)を添加した塩化オキサリル(3.55mL、7.1mmol)のCH2Cl2(30mL)溶液(−78℃)に添加した。40分後、NEt3(2.51mL)を加え、混合物を室温へ加温した。水溶液による標準的な処理および精製の後、[3−(tert−ブチル−ジメチル−シラニルオキシメチル)−シクロヘキサン−3−エニル]−アセトアルデヒド(中間体R5)を単離した(約95%)。
【0132】
以下の製造は、Horne et al. , Heterocycles 39:139 (1994)の方法のとおり行った。アルデヒド(中間体R5;0.34g、1.3mmol)のEtOH(5mL)溶液をトシルメチルイソシアニド(TosMIC;Aldrich;0.25g;1.3mmol)およびNaCN(約15mg、触媒量)で処理し、室温にて20分間攪拌した。溶媒を減圧留去し;残留物を約7MNH3(MeOH中)に溶解し、密閉可能な試験管に移した後、100℃に15時間加熱した。混合物を濃縮し、5%MeOH(sat.w/NH3):CH2Cl2を用いるクロマトグラフィー(SiO2)により精製した。THF中の生成物の溶液にTBAF(1.5当量)を加え、室温にて攪拌した後、水溶液で処理した。粗製物をクロマトグラフィー(5−7%NH3/MeOH(CH2Cl2中))に付し、化合物2を得た。
【0133】
得られた化合物2のキャラクタリゼーションは以下のとおりである:1HNMR (300 MHz, DMSO−d6) 7.52 (s, 1H) , 6.72 (s, 1H) , 5.54 (brs, 1H) , 3.73 (s, 2H) , 2.46 (d, J = 6 Hz, 2H) , 1.5−2.1 (m, 6H) , 1.0−1.55 (m, 1H)
【0134】
実施例V
ブリモニジンよりも高いα−2A/α−1A機能的選択性を有するα−2アゴニストのキャラクタリゼーション
本実施例では、α−アドレナリン作動性アゴニストが、α−2A受容体活性化の不存在下で末梢投与されたとき、効果的な鎮痛剤であることが明らかにされる。非特異的なα−アドレナリン作動性アゴニストが、スルプロストン感作された痛みのマウスモデルを使用してα−2A受容体欠損(「ノックアウト」)マウス(Hein他、上掲、1999)においてアッセイされた。このマウスモデルでは、本質的には、Minami他、Pain 57:217−223(1994)に記載されるように、異疼痛が、選択的プロスタグランジンE2受容体アゴニストを意識のあるマウスにクモ膜下投与することによって誘発される。このモデルにおいて、はけで側腹をなでることに対する痛み応答が、スルプロストンの脊髄投与および薬物またはコントロールビヒクルのクモ膜下投与または腹腔内投与の15分後から開始して35分間の期間にわたって8回スコア化される。スルプロストンは、16点の測定尺度で12点〜13点の「痛み」のスコアを誘発する。
【表4】
【0135】
クモ膜下注射で得られた結果とは対照的に、30μg/kgの化合物1の腹腔内注射は、クロニジンまたはブリモニジンと同様に、鎮静作用から分離可能な痛覚消失を野生型マウスにおいて生じさせることができなかった。しかしながら、α−2Aノックアウトマウスでは、クロニジン、UK14304または化合物1による末梢での処置(腹腔内処置)は、鎮静作用を伴わない顕著な痛覚消失をもたらした。図1Cおよび図1Dを参照のこと。これらの図は、末梢投与されたとき、化合物1が、野生型動物ではなく、α−2Aノックアウトマウスにおいて痛覚消失を生じさせただけであったことを示している。これらの結果は、α−アドレナリン作動性アゴニストが、α−2A受容体活性化の不存在下で末梢投与されたとき、著しい鎮静作用を伴わない痛覚消失を生じさせることができることを示している。
【0136】
野生型マウスおよびα−2Aノックアウトマウスはまた、100μg/kgのα−アドレナリン作動性アゴニストの化合物2で処置された。化合物2は、クロニジン、ブリモニジン、化合物1およびPACとは異なり、α−1活性をほとんど有しておらず、インビトロアッセイにおいてブリモニジンに対して40%の相対的な効力でα−2A受容体を弱く活性化するにすぎない。クロニジンのように、化合物2は血液脳関門を容易に横断する。野生型マウスにおいて、化合物2は、腹腔内投与されたとき、鎮痛作用を有さず、しかし、同じ経路により投与されたとき、α−2Aノックアウトマウスにおいて十分な鎮痛作用を有した(図2を参照のこと)。これらの結果は、α−2A受容体の活性化が妨げられたとき、α−1受容体およびα−2受容体の活性プロフィルおよび生物利用性が異なる様々なα−アドレナリン作動性アゴニストが、効果的な末梢鎮痛剤であり得ることを示している。上記で開示されたように、α−アドレナリン作動性アゴニストによるα−2A受容体の活性化はこれらの分子の末梢鎮痛活性を遮蔽する。この遮蔽作用が、脊髄または末梢に局在化するα−2A受容体に依存するかどうかを試験するために、マウスに、化合物1(液脳関門を容易に横断しない高荷電のα−アドレナリン作動性アゴニスト)をクモ膜下または腹腔内に注射した。上記で示されたように、腹腔内注射ではなく、クモ膜下注射は野生型マウスにおいて著しい痛覚消失を生じさせ、一方、α−2Aノックアウトマウスでは逆のことが観測された。
【0137】
B.インビボ効果および沈静効果
α−2B/C選択的アゴニストの化合物8が、神経障害性の痛みの別のラット神経傷害モデルであるBennett部分坐骨神経結紮モデルにおいて試験された。このラットモデルは、ヒトにおいて認められる痛み感覚と類似した痛み感覚の障害を伴う末梢単神経障害をもたらす(BennettおよびXie、Pain、33:87−107(1988))。Bennettモデルでは、神経傷害が、締め付ける結紮糸で坐骨神経の周囲をゆるく縛り、これにより、締め付け部から遠位側の神経の変性を引き起こすことによってもたらされる。異疼痛および痛覚過敏が、無意識での痛みに加えて、締め付け傷害によってもたらされる(BennettおよびXie、上掲(1988))。具体的には、冷たさに対する異疼痛、すなわち、冷刺激からの痛み感覚が、変化した痛み感覚の1つの発現である:Bennettモデル動物は、コントロール動物とは対照的に、冷たい表面から、手術された肢の足を頻繁に持ち上げる。
【0138】
化合物8が、4日間の期間、浸透圧ミニポンプによって投与された。図7Bに示されるように、冷たさに対する異疼痛が、4日間の処置期間中およびポンプ除去後の3週間以上の試験期間にわたってともに完全に緩和された。これらの結果は、最小限のα−2Aアゴニスト活性を有するα−2アドレナリン作動性アゴニスト(化合物8など)が、異なるタイプの神経障害性の痛みに対して適用可能な鎮痛特性を有することを示している。約20分間続いたBennett手術は下記のように行われた。雄性Sprague−Dawleyラット(約250グラム〜300グラム)がイソフルラン/酸素吸入によって麻酔された。剃毛およびベタジン塗布によって手術部位を調製した後、切開が、腰帯を超えてわずかに中線の左側に行われた。左股関節のわずかに尾側および腹側で、筋肉群の不鮮明な境界線が視認され、小さい(約10mm〜25mm)切開が筋肉群の境界線のすぐ下で行われた。筋肉は、坐骨神経が大腿骨の長さに平行して見えるまで容赦なく引き離された。7mm〜10mmの長さの坐骨神経を、下にある組織から慎重に取り出し、その後、結紮糸の間に約1mmの間隔をあけて坐骨神経の周りを4本の結紮糸(6/0、絹糸)でゆるく縛った。結紮糸は、神経の直径がわずかに締め付けられ、血流が遅滞するが、停止しないように縛られた。余分な縫合物質は切り取られ、筋肉群は近くに置かれ、皮膚の切開部は創傷クリップで閉じられた。創傷クリップは手術後10日〜14日で除去された。動物には、手術後の局所用または局部用の麻酔薬が全く投与されなかった。
【0139】
長期の薬物投与が、上記のようなBennett手術の1週間後にラットの背中の皮下に埋め込まれた浸透圧ミニポンプによって達成された。Bennett動物における痛み応答の評価が下記のように手術後7日目に行われた。熱刺激への応答を評価するために、ラットを透明なプラスチックチャンバー(18cm x 29cm x 12.5cm)に入れ、正常な動物に対しては侵害性でない冷却された(0℃〜4℃)金属床の上に置き、手術された足が冷たい床から持ち上げられる時間が5分間の期間にわたって記録された。実験当日、試験薬物が無麻酔で投与された(1ml/kgの体積で、1μg/kg〜1000μg/kgの範囲の用量でのIP投与またはPO投与)。場合により、動物は、研究の間に少なくとも3日間の休止期間を与えられた後、3ヶ月の期間にわたる次の実験のために使用された。本実施例では、化合物8などの、最小限のα−2活性を有するα−2アドレナリン作動性アゴニストが、慢性的な内臓過敏性のラットモデルにおいて痛みを緩和することが明らかにされる。
【0140】
上記に示された雑誌論文、参考文献、および特許の引用はすべて、括弧内またはそれ以外の場合でも、また、以前に言及されているか否かにかかわらず、その全体が参考として本明細書中に組み込まれる。
【0141】
本発明は、上記に提供された実施例を参照して記載されているが、様々な改変が、本発明の精神から逸脱することなく行われ得ることを理解しなければならない。従って、本発明は請求項によってのみ限定されるだけである。
【図面の簡単な説明】
【0142】
【図1】スルプロストン感作された野生型動物またはα−2Aノックアウト動物への化合物1のクモ膜下注射(1μg)または腹腔内注射(30μg/kg)で得られた結果を示す。軽く触れたことに対する痛み感受性が下記に記載されるようにスコア化された。(Fig.1a)野生型マウスへのクモ膜下注射。(Fig.1b)α−2Aノックアウトマウスへのクモ膜下注射。
【図2】スルプロストン感作された野生型マウスまたはα−2Aノックアウトマウスへの化合物2の腹腔内注射で得られた結果を示す。(Fig.1c)野生型マウスへの100μg/kgの化合物2の腹腔内注射。(Fig.1d)α−2Aノックアウトマウスへの100μg/kgの化合物2の腹腔内注射。星印は、p値が0.05未満で、有意な結果であることを示す。
【図3】末梢α−2受容体がα−2Aノックアウトマウスにおいて痛覚消失を媒介することを示す(Fig.3)。100μg/kgでのパラ-アミノクロニジン(PAC)が、単独または0.3μg/kgでのラウオルシン(Rau)との組合せで、スルプロストン感作のα−2Aノックアウトマウスに腹腔内注射によって投与された。星印は、p値が0.05未満で、有意な結果であることを示す(Fig.4)。
【図4】α−2A受容体活性化が阻止されるとき、α−アドレナリン作動性アゴニストが野生型動物において効果的な末梢鎮痛剤であり得ることを示す。異疼痛逆転率がそれぞれの動物組について示される。Chungモデルラットに、30μg/kgのクロニジン、0.3μg/kgの化合物13、または両者が腹腔内投与された。
【図5】最小限のα−2Aアゴニスト活性を有するα−アドレナリン作動性アゴニストの鎮痛活性が血漿中薬物レベルの不存在下で継続することを示す(Fig.5a)Chungモデルラットに、化合物8、化合物9、ブリモニジン、パラ-アミノ-クロニジンまたはビヒクルが、7日間、示された濃度で浸透圧ミニポンプにより投与された。異疼痛逆転率が、浸透圧ミニポンプ埋め込みのときから15日間の経過期間にわたって測定された。化合物8および化合物9で得られた結果は、p値が0.05未満で、有意であった。(Fig.5b)Chungモデルラットに、0.1mg/hr/kgの化合物8が、7日間、浸透圧ミニポンプにより投与された。化合物8の血漿中濃度(ng/ml)および同じ時点での異疼痛逆転率(%MPE)がポンプ埋め込み後の示された日に測定された。
【図6】異疼痛の逆転は、化合物8の数日間の投薬の後、継続する受容体の活性化を必要としないことを示す。Chungモデルラットにおける異疼痛逆転率が、化合物8による薬物処理を開始した後の様々な日数で示される。(Fig.5c)7日間にわたる浸透圧ミニポンプによる0.1mg/hr/kgの化合物8での処理。3日目および4日目に、測定が、0.3μg/kgのラウオルシン(R)の腹腔内投薬の前およびその30分後に行われた。(Fig.5d)3日間にわたり1日に3回の経口投薬による0.3mg/kgの化合物8での処理。測定が、示されるようにその日の最初の投薬の後またはその前に行われた。投薬後11日間にわたって続けられた測定が完了した。星印は、p値が0.05未満で、有意な結果であることを示す。
【図7】汎α−2アゴニストが、選択的α−2Aアンタゴニストと組み合わせられたとき、長期間にわたる痛み緩和を生じさせ得ることを示す(Fig.6)。薬物が、5日間、下記の用量で浸透圧ミニポンプにより投与された:ブリモニジン(42μg/kg/hr)、化合物1(0.1mg/kg/hr)および化合物13(8μg/kg/hr)。各化合物およびビヒクルが単独で投与され、ブリモニジンおよび化合物1はまた化合物13と一緒に投与された。異疼痛逆転率が投薬開始後の様々な日数で測定された。ブリモニジンおよび化合物13の組合せ、または化合物1および化合物13の組合せを用いて5日目に得られた結果は、p値が0.05未満で、有意であった。図7Bは、化合物8がBennettモデルにおいて冷異疼痛の長期間にわたる痛み緩和を生じさせることを示す。動物が、4日間、浸透圧ミニポンプにより0.1mg/hr/kgの化合物8または生理的食塩水で処理され、浸透圧ミニポンプが4日目に除かれた。5分間における足引っ込み持続時間(秒単位)が薬物処理開始後示された日において示される。
【図8】化合物8が、過敏性腸症候群のラットモデルにおいて長期間にわたる痛み緩和を生じさせることを示す。ラットが、7日間、浸透圧ミニポンプにより処理され、一連の結腸直腸拡大(CRD)に対する腹部引っ込み反射が、処理前、処置時および処理後に筋電図測定によって測定された。(Fig.7a)50%DMSOビヒクルで処理された正常ラット。(Fig.7b)0.1mg/hr/kgでの化合物8で処理された正常ラット。(Fig.7c)50%DMSOビヒクルで処理された感作ラット。(Fig.7d)0.1mg/hr/kgでの化合物8で処理された感作ラット。CRD、結腸直腸拡大。
【図9】スルプロストン感作のα−2Bノックアウトマウスで得られた痛覚消失を示す。野生型(+/+);ヘテロ接合型(+/-)またはホモ接合型(-/-)のα−2Bノックアウトマウスが、クモ膜下内ビヒクル(DMSO)、クモ膜下内スルプロストン、腹腔内クロニジンとのスルプロストン、または腹腔内化合物3とのスルプロストンで処理された。6匹のマウスにおける総合痛みスコアが測定された。星印は、p値が0.05未満で、有意な結果であることを示す(Fig.7e)。
【図10】Chungモデルラットにおける様々な経口用量での化合物3、化合物11および化合物4の末梢鎮痛作用を示す。(Fig.8a)10μg/kg、30μg/kg、100μg/kgまたは300μg/kgの化合物3の単回経口用量。(Fig.8b)30μg/kg、100μg/kg、300μg/kgまたは1000μg/kgの化合物11の単回経口用量。
【特許請求の範囲】
【請求項1】
交感神経増強状態を鎮静を伴うことなく予防または軽減する方法であって、α−2A/α1−A選択的アゴニストの有効量を対象に末梢投与し、それにより該交感神経増強状態を鎮静を伴うことなく予防または軽減し、前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有するまたはブリモニジンと比較して高いα−1A/α−2A効力比を有する、方法。
【請求項2】
前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有する請求項1記載の方法。
【請求項3】
前記選択的アゴニストがブリモニジンと比較して少なくとも30%高いα−1A/α−2AEC50比を有する請求項1記載の方法。
【請求項4】
前記有効成分がブリモニジンと比較して少なくとも2倍のα−1A/α−2AEC50比を有する請求項1記載の方法。
【請求項5】
前記有効成分がブリモニジンと比較して少なくとも10倍のα−1A/α−2AEC50比を有する請求項1記載の方法。
【請求項6】
前記状態が感覚過敏である請求項1記載の方法。
【請求項7】
前記状態が頭痛を伴う感覚過敏である請求項6記載の方法。
【請求項8】
前記状態が片頭痛を伴う感覚過敏である請求項7記載の方法。
【請求項9】
前記状態が胃腸疾患である請求項1記載の方法。
【請求項10】
前記状態が過敏性腸症候群である請求項9記載の方法。
【請求項11】
前記胃腸疾患が消化不良である請求項9記載の方法。
【請求項12】
前記状態が皮膚病変である請求項1記載の方法。
【請求項13】
前記皮膚病変が乾癬である請求項12記載の方法。
【請求項14】
前記状態が心疾患である請求項1記載の方法。
【請求項15】
前記状態が頻脈である請求項14記載の方法。
【請求項16】
前記状態が末梢血管収縮である請求項1記載の方法。
【請求項17】
前記状態がパニック発作である請求項1記載の方法。
【請求項18】
前記状態が代謝障害である請求項1記載の方法。
【請求項19】
前記代謝障害がII型糖尿病である請求項18記載の方法。
【請求項20】
前記代謝障害がインスリン−耐性である請求項18記載の方法。
【請求項21】
前記代謝障害が肥満症である請求項18記載の方法。
【請求項22】
前記状態が筋肉収縮の障害である請求項1記載の方法。
【請求項23】
前記筋肉収縮の障害が骨格筋収縮の障害である請求項22記載の方法。
【請求項24】
前記筋肉収縮の障害が平滑筋収縮の障害である請求項22記載の方法。
【請求項25】
前記筋肉収縮の障害が痙縮である請求項22記載の方法。
【請求項26】
前記筋肉収縮の障害が緊張性頭痛を伴う請求項22記載の方法。
【請求項27】
前記状態が行動障害である請求項1記載の方法。
【請求項28】
前記有効量を経口投与する請求項1記載の方法。
【請求項29】
前記有効量を局所投与する請求項1記載の方法。
【請求項30】
前記有効量をパッチ経由で投与する請求項1記載の方法。
【請求項31】
慢性疼痛を鎮静を伴うことなく予防または軽減する方法であって、α−2A/α1−A選択的アゴニストの有効量を対象に末梢投与し、それにより慢性疼痛を鎮静を伴うことなく予防または軽減し、前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有するまたはブリモニジンと比較して高いα−1A/α−2A効力比を有する、方法。
【請求項32】
前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有する請求項31記載の方法。
【請求項33】
前記選択的アゴニストがブリモニジンと比較して少なくとも30%高いα−1A/α−2AEC50比を有する請求項31記載の方法。
【請求項34】
前記選択的アゴニストがブリモニジンと比較して少なくとも2倍のα−1A/α−2AEC50比を有する請求項31記載の方法。
【請求項35】
前記選択的アゴニストがブリモニジンと比較して少なくとも10倍のα−1A/α−2AEC50比を有する請求項31記載の方法。
【請求項36】
前記慢性疼痛が神経障害性疼痛である請求項31記載の方法。
【請求項37】
前記神経障害性疼痛が糖尿病性ニューロパシーに伴う請求項36記載の方法。
【請求項38】
前記神経障害性疼痛がヘルペス後神経痛に伴う請求項36記載の方法。
【請求項39】
前記慢性疼痛が癌に伴う請求項31記載の方法。
【請求項40】
前記慢性疼痛が術後疼痛である請求項31記載の方法。
【請求項41】
前記慢性疼痛がアロディニア痛である請求項41記載の方法。
【請求項42】
前記アロディニア痛が線維筋痛である請求項41記載の方法。
【請求項43】
前記慢性疼痛が複合性局所疼痛症候群に関連する(CRPS)請求項31記載の方法。
【請求項44】
前記慢性疼痛が内臓痛である請求項31記載の方法。
【請求項45】
前記内臓痛が過敏性腸症候群に伴う請求項44記載の方法。
【請求項46】
前記内臓痛が月経困難症に伴う請求項44記載の方法。
【請求項47】
前記慢性疼痛が頭痛に関連する請求項31記載の方法。
【請求項48】
前記頭痛が片頭痛である請求項47記載の方法。
【請求項49】
前記頭痛が非血管性である請求項47記載の方法。
【請求項50】
前記頭痛が群発性頭痛または毎日の緊張性頭痛である請求項47記載の方法。
【請求項51】
前記慢性疼痛が筋肉痛である請求項31記載の方法。
【請求項52】
前記筋肉痛が背中の痙攣に関連する請求項51記載の方法。
【請求項53】
前記有効量を経口投与する請求項31記載の方法。
【請求項54】
前記有効量を局所投与する請求項31記載の方法。
【請求項55】
前記有効量をパッチ経由で投与する請求項31記載の方法。
【請求項56】
神経学的状態を鎮静を伴うことなく予防または軽減する方法であって、α−2A/α1−A選択的アゴニストの有効量を対象に末梢投与し、それにより該神経学的状態を鎮静を伴うことなく予防または軽減し、前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有するまたはブリモニジンと比較して高いα−1A/α−2A効力比を有する、方法。
【請求項57】
前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有する請求項56記載の方法。
【請求項58】
前記選択的アゴニストがブリモニジンと比較して少なくとも30%高いα−1A/α−2AEC50比を有する請求項56記載の方法。
【請求項59】
前記有効成分がブリモニジンと比較して少なくとも2倍のα−1A/α−2AEC50比を有する請求項56記載の方法。
【請求項60】
前記有効成分がブリモニジンと比較して少なくとも10倍のα−1A/α−2AEC50比を有する請求項56記載の方法。
【請求項61】
前記神経学的状態が急性神経学的状態である請求項56記載の方法。
【請求項62】
前記急性神経学的状態が卒中である請求項61記載の方法。
【請求項63】
前記急性神経学的状態が頭部または脊椎外傷である請求項61記載の方法。
【請求項64】
前記急性神経学的状態が発作である請求項61記載の方法。
【請求項65】
前記神経学的状態が慢性神経学的状態である請求項56記載の方法。
【請求項66】
前記慢性神経学的状態が神経変性疾患である請求項56記載の方法。
【請求項67】
前記神経変性疾患がであるアルツハイマー病である請求項66記載の方法。
【請求項68】
前記神経変性疾患がであるパーキンソン病である請求項66記載の方法。
【請求項69】
前記神経変性疾患がであるハンチントン病である請求項66記載の方法。
【請求項70】
前記神経変性疾患が筋萎縮性側索硬化症または多発性硬化症である請求項66記載の方法。
【請求項71】
前記神経変性疾患がHIV関連認知症またはHIV関連ニューロパシーである請求項66記載の方法。
【請求項72】
前記神経変性疾患が眼疾患である請求項66記載の方法。
【請求項73】
前記眼疾患が緑内障である請求項72記載の方法。
【請求項74】
前記眼疾患が糖尿病性ニューロパシーである請求項72記載の方法。
【請求項75】
前記眼疾患が加齢性黄斑変性である請求項72記載の方法。
【請求項76】
前記慢性神経学的状態が、統合失調症、麻薬常用、麻薬離脱、麻薬依存、鬱および不安から選択される請求項65記載の方法。
【請求項77】
前記有効量を経口投与する請求項56記載の方法。
【請求項78】
前記有効量を局所投与する請求項56記載の方法。
【請求項79】
前記有効量をパッチ経由で投与する請求項56記載の方法。
【請求項80】
眼症状を鎮静を伴うことなく予防または軽減する方法であって、α−2A/α1−A選択的アゴニストの有効量を対象に末梢投与し、それにより該眼症状を鎮静を伴うことなく予防または軽減し、前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有するまたはブリモニジンと比較して高いα−1A/α−2A効力比を有する、方法。
【請求項81】
前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有する請求項80記載の方法。
【請求項82】
前記選択的アゴニストがブリモニジンと比較して少なくとも30%高いα−1A/α−2AEC50比を有する請求項80記載の方法。
【請求項83】
前記有効成分がブリモニジンと比較して少なくとも2倍のα−1A/α−2AEC50比を有する請求項80記載の方法。
【請求項84】
前記有効成分がブリモニジンと比較して少なくとも10倍のα−1A/α−2AEC50比を有する請求項80記載の方法。
【請求項85】
前記眼疾患が緑内障である請求項80記載の方法。
【請求項86】
前記眼疾患が黄斑変性である請求項80記載の方法。
【請求項87】
前記眼疾患が網膜症である請求項80記載の方法。
【請求項88】
前記網膜症が糖尿病性網膜症である請求項87記載の方法。
【請求項89】
前記有効量を経口投与する請求項80記載の方法。
【請求項90】
前記有効量を局所投与する請求項80記載の方法。
【請求項91】
前記有効量をパッチ経由で投与する請求項80記載の方法。
【請求項92】
末梢投与した場合に交感神経増強状態を鎮静を伴うことなく予防または軽減するα−2A/α1−A選択的アゴニストのスクリーニング方法であって、α−1A受容体と比較したα2−Aレセプターを活性化する試薬の機能的選択性を測定することを含んでなり、
α−1A受容体と比較してα2−Aレセプターを活性化する選択性が高い試薬が末梢投与した場合に交感神経増強状態を鎮静を伴うことなく予防または軽減するα−2A/α1−A選択的アゴニストである、方法。
【請求項93】
工程(a)α−2A受容体に対する前記試薬の効力、活性またはEC50を測定すること;工程(b)α−1A受容体に対する該試薬の効力、活性またはEC50を測定することを含んでなり、ブリモニジンと比較して高いα−1A効力を有するまたはブリモニジンと比較して高いα−1A/α−2A効力を有する試薬が交感神経増強状態を鎮静を伴うことなく予防または軽減するα−2A/α1−A選択的アゴニストである、請求項92記載の方法。
【請求項94】
前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有する請求項92記載の方法。
【請求項95】
前記選択的アゴニストがブリモニジンと比較して少なくとも30%高いα−1A/α−2AEC50比を有する請求項92記載の方法。
【請求項96】
前記有効成分がブリモニジンと比較して少なくとも2倍のα−1A/α−2AEC50比を有する請求項92記載の方法。
【請求項97】
前記有効成分がブリモニジンと比較して少なくとも10倍のα−1A/α−2AEC50比を有する請求項92記載の方法。
【請求項98】
前記工程(a)がアデニル酸サイクラーゼ活性の阻害の分析を含む請求項93記載の方法。
【請求項99】
前記アデニル酸サイクラーゼ活性の阻害の分析が、α−2Aを安定に発現するPC12細胞におけるものである請求項98記載の方法。
【請求項100】
前記PC12細胞がヒトα−2Aを安定に発現する請求項99記載の方法。
【請求項101】
前記工程(b)が細胞内カルシウムに関する分析を含む請求項93記載の方法。
【請求項102】
前記細胞内カルシウムをα1−Aを安定に発現するHEK293細胞において分析する請求項101記載の方法。
【請求項103】
前記HEK293細胞がウシα1−Aを安定に発現する請求項102記載の方法。
【請求項1】
交感神経増強状態を鎮静を伴うことなく予防または軽減する方法であって、α−2A/α1−A選択的アゴニストの有効量を対象に末梢投与し、それにより該交感神経増強状態を鎮静を伴うことなく予防または軽減し、前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有するまたはブリモニジンと比較して高いα−1A/α−2A効力比を有する、方法。
【請求項2】
前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有する請求項1記載の方法。
【請求項3】
前記選択的アゴニストがブリモニジンと比較して少なくとも30%高いα−1A/α−2AEC50比を有する請求項1記載の方法。
【請求項4】
前記有効成分がブリモニジンと比較して少なくとも2倍のα−1A/α−2AEC50比を有する請求項1記載の方法。
【請求項5】
前記有効成分がブリモニジンと比較して少なくとも10倍のα−1A/α−2AEC50比を有する請求項1記載の方法。
【請求項6】
前記状態が感覚過敏である請求項1記載の方法。
【請求項7】
前記状態が頭痛を伴う感覚過敏である請求項6記載の方法。
【請求項8】
前記状態が片頭痛を伴う感覚過敏である請求項7記載の方法。
【請求項9】
前記状態が胃腸疾患である請求項1記載の方法。
【請求項10】
前記状態が過敏性腸症候群である請求項9記載の方法。
【請求項11】
前記胃腸疾患が消化不良である請求項9記載の方法。
【請求項12】
前記状態が皮膚病変である請求項1記載の方法。
【請求項13】
前記皮膚病変が乾癬である請求項12記載の方法。
【請求項14】
前記状態が心疾患である請求項1記載の方法。
【請求項15】
前記状態が頻脈である請求項14記載の方法。
【請求項16】
前記状態が末梢血管収縮である請求項1記載の方法。
【請求項17】
前記状態がパニック発作である請求項1記載の方法。
【請求項18】
前記状態が代謝障害である請求項1記載の方法。
【請求項19】
前記代謝障害がII型糖尿病である請求項18記載の方法。
【請求項20】
前記代謝障害がインスリン−耐性である請求項18記載の方法。
【請求項21】
前記代謝障害が肥満症である請求項18記載の方法。
【請求項22】
前記状態が筋肉収縮の障害である請求項1記載の方法。
【請求項23】
前記筋肉収縮の障害が骨格筋収縮の障害である請求項22記載の方法。
【請求項24】
前記筋肉収縮の障害が平滑筋収縮の障害である請求項22記載の方法。
【請求項25】
前記筋肉収縮の障害が痙縮である請求項22記載の方法。
【請求項26】
前記筋肉収縮の障害が緊張性頭痛を伴う請求項22記載の方法。
【請求項27】
前記状態が行動障害である請求項1記載の方法。
【請求項28】
前記有効量を経口投与する請求項1記載の方法。
【請求項29】
前記有効量を局所投与する請求項1記載の方法。
【請求項30】
前記有効量をパッチ経由で投与する請求項1記載の方法。
【請求項31】
慢性疼痛を鎮静を伴うことなく予防または軽減する方法であって、α−2A/α1−A選択的アゴニストの有効量を対象に末梢投与し、それにより慢性疼痛を鎮静を伴うことなく予防または軽減し、前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有するまたはブリモニジンと比較して高いα−1A/α−2A効力比を有する、方法。
【請求項32】
前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有する請求項31記載の方法。
【請求項33】
前記選択的アゴニストがブリモニジンと比較して少なくとも30%高いα−1A/α−2AEC50比を有する請求項31記載の方法。
【請求項34】
前記選択的アゴニストがブリモニジンと比較して少なくとも2倍のα−1A/α−2AEC50比を有する請求項31記載の方法。
【請求項35】
前記選択的アゴニストがブリモニジンと比較して少なくとも10倍のα−1A/α−2AEC50比を有する請求項31記載の方法。
【請求項36】
前記慢性疼痛が神経障害性疼痛である請求項31記載の方法。
【請求項37】
前記神経障害性疼痛が糖尿病性ニューロパシーに伴う請求項36記載の方法。
【請求項38】
前記神経障害性疼痛がヘルペス後神経痛に伴う請求項36記載の方法。
【請求項39】
前記慢性疼痛が癌に伴う請求項31記載の方法。
【請求項40】
前記慢性疼痛が術後疼痛である請求項31記載の方法。
【請求項41】
前記慢性疼痛がアロディニア痛である請求項41記載の方法。
【請求項42】
前記アロディニア痛が線維筋痛である請求項41記載の方法。
【請求項43】
前記慢性疼痛が複合性局所疼痛症候群に関連する(CRPS)請求項31記載の方法。
【請求項44】
前記慢性疼痛が内臓痛である請求項31記載の方法。
【請求項45】
前記内臓痛が過敏性腸症候群に伴う請求項44記載の方法。
【請求項46】
前記内臓痛が月経困難症に伴う請求項44記載の方法。
【請求項47】
前記慢性疼痛が頭痛に関連する請求項31記載の方法。
【請求項48】
前記頭痛が片頭痛である請求項47記載の方法。
【請求項49】
前記頭痛が非血管性である請求項47記載の方法。
【請求項50】
前記頭痛が群発性頭痛または毎日の緊張性頭痛である請求項47記載の方法。
【請求項51】
前記慢性疼痛が筋肉痛である請求項31記載の方法。
【請求項52】
前記筋肉痛が背中の痙攣に関連する請求項51記載の方法。
【請求項53】
前記有効量を経口投与する請求項31記載の方法。
【請求項54】
前記有効量を局所投与する請求項31記載の方法。
【請求項55】
前記有効量をパッチ経由で投与する請求項31記載の方法。
【請求項56】
神経学的状態を鎮静を伴うことなく予防または軽減する方法であって、α−2A/α1−A選択的アゴニストの有効量を対象に末梢投与し、それにより該神経学的状態を鎮静を伴うことなく予防または軽減し、前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有するまたはブリモニジンと比較して高いα−1A/α−2A効力比を有する、方法。
【請求項57】
前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有する請求項56記載の方法。
【請求項58】
前記選択的アゴニストがブリモニジンと比較して少なくとも30%高いα−1A/α−2AEC50比を有する請求項56記載の方法。
【請求項59】
前記有効成分がブリモニジンと比較して少なくとも2倍のα−1A/α−2AEC50比を有する請求項56記載の方法。
【請求項60】
前記有効成分がブリモニジンと比較して少なくとも10倍のα−1A/α−2AEC50比を有する請求項56記載の方法。
【請求項61】
前記神経学的状態が急性神経学的状態である請求項56記載の方法。
【請求項62】
前記急性神経学的状態が卒中である請求項61記載の方法。
【請求項63】
前記急性神経学的状態が頭部または脊椎外傷である請求項61記載の方法。
【請求項64】
前記急性神経学的状態が発作である請求項61記載の方法。
【請求項65】
前記神経学的状態が慢性神経学的状態である請求項56記載の方法。
【請求項66】
前記慢性神経学的状態が神経変性疾患である請求項56記載の方法。
【請求項67】
前記神経変性疾患がであるアルツハイマー病である請求項66記載の方法。
【請求項68】
前記神経変性疾患がであるパーキンソン病である請求項66記載の方法。
【請求項69】
前記神経変性疾患がであるハンチントン病である請求項66記載の方法。
【請求項70】
前記神経変性疾患が筋萎縮性側索硬化症または多発性硬化症である請求項66記載の方法。
【請求項71】
前記神経変性疾患がHIV関連認知症またはHIV関連ニューロパシーである請求項66記載の方法。
【請求項72】
前記神経変性疾患が眼疾患である請求項66記載の方法。
【請求項73】
前記眼疾患が緑内障である請求項72記載の方法。
【請求項74】
前記眼疾患が糖尿病性ニューロパシーである請求項72記載の方法。
【請求項75】
前記眼疾患が加齢性黄斑変性である請求項72記載の方法。
【請求項76】
前記慢性神経学的状態が、統合失調症、麻薬常用、麻薬離脱、麻薬依存、鬱および不安から選択される請求項65記載の方法。
【請求項77】
前記有効量を経口投与する請求項56記載の方法。
【請求項78】
前記有効量を局所投与する請求項56記載の方法。
【請求項79】
前記有効量をパッチ経由で投与する請求項56記載の方法。
【請求項80】
眼症状を鎮静を伴うことなく予防または軽減する方法であって、α−2A/α1−A選択的アゴニストの有効量を対象に末梢投与し、それにより該眼症状を鎮静を伴うことなく予防または軽減し、前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有するまたはブリモニジンと比較して高いα−1A/α−2A効力比を有する、方法。
【請求項81】
前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有する請求項80記載の方法。
【請求項82】
前記選択的アゴニストがブリモニジンと比較して少なくとも30%高いα−1A/α−2AEC50比を有する請求項80記載の方法。
【請求項83】
前記有効成分がブリモニジンと比較して少なくとも2倍のα−1A/α−2AEC50比を有する請求項80記載の方法。
【請求項84】
前記有効成分がブリモニジンと比較して少なくとも10倍のα−1A/α−2AEC50比を有する請求項80記載の方法。
【請求項85】
前記眼疾患が緑内障である請求項80記載の方法。
【請求項86】
前記眼疾患が黄斑変性である請求項80記載の方法。
【請求項87】
前記眼疾患が網膜症である請求項80記載の方法。
【請求項88】
前記網膜症が糖尿病性網膜症である請求項87記載の方法。
【請求項89】
前記有効量を経口投与する請求項80記載の方法。
【請求項90】
前記有効量を局所投与する請求項80記載の方法。
【請求項91】
前記有効量をパッチ経由で投与する請求項80記載の方法。
【請求項92】
末梢投与した場合に交感神経増強状態を鎮静を伴うことなく予防または軽減するα−2A/α1−A選択的アゴニストのスクリーニング方法であって、α−1A受容体と比較したα2−Aレセプターを活性化する試薬の機能的選択性を測定することを含んでなり、
α−1A受容体と比較してα2−Aレセプターを活性化する選択性が高い試薬が末梢投与した場合に交感神経増強状態を鎮静を伴うことなく予防または軽減するα−2A/α1−A選択的アゴニストである、方法。
【請求項93】
工程(a)α−2A受容体に対する前記試薬の効力、活性またはEC50を測定すること;工程(b)α−1A受容体に対する該試薬の効力、活性またはEC50を測定することを含んでなり、ブリモニジンと比較して高いα−1A効力を有するまたはブリモニジンと比較して高いα−1A/α−2A効力を有する試薬が交感神経増強状態を鎮静を伴うことなく予防または軽減するα−2A/α1−A選択的アゴニストである、請求項92記載の方法。
【請求項94】
前記選択的アゴニストがブリモニジンと比較して低いα−1A作用を有する請求項92記載の方法。
【請求項95】
前記選択的アゴニストがブリモニジンと比較して少なくとも30%高いα−1A/α−2AEC50比を有する請求項92記載の方法。
【請求項96】
前記有効成分がブリモニジンと比較して少なくとも2倍のα−1A/α−2AEC50比を有する請求項92記載の方法。
【請求項97】
前記有効成分がブリモニジンと比較して少なくとも10倍のα−1A/α−2AEC50比を有する請求項92記載の方法。
【請求項98】
前記工程(a)がアデニル酸サイクラーゼ活性の阻害の分析を含む請求項93記載の方法。
【請求項99】
前記アデニル酸サイクラーゼ活性の阻害の分析が、α−2Aを安定に発現するPC12細胞におけるものである請求項98記載の方法。
【請求項100】
前記PC12細胞がヒトα−2Aを安定に発現する請求項99記載の方法。
【請求項101】
前記工程(b)が細胞内カルシウムに関する分析を含む請求項93記載の方法。
【請求項102】
前記細胞内カルシウムをα1−Aを安定に発現するHEK293細胞において分析する請求項101記載の方法。
【請求項103】
前記HEK293細胞がウシα1−Aを安定に発現する請求項102記載の方法。
【図1】

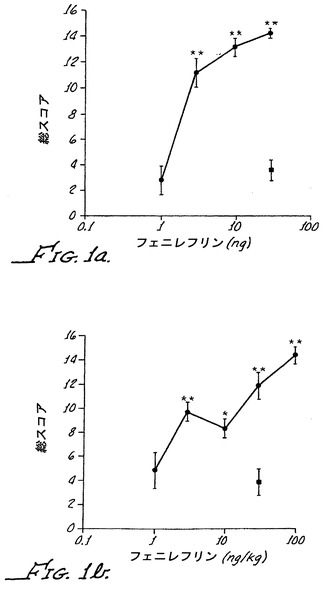
【図2】
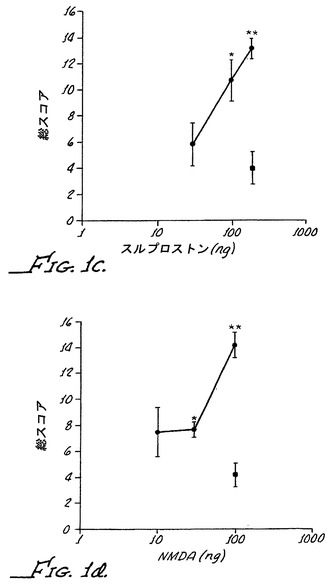
【図3】
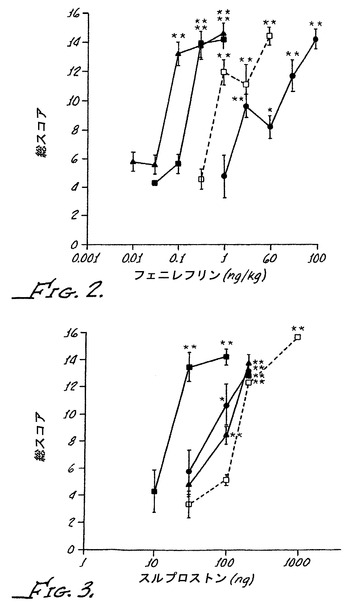
【図4】
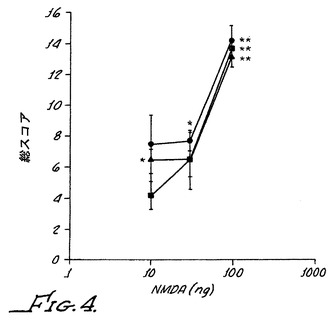
【図5】
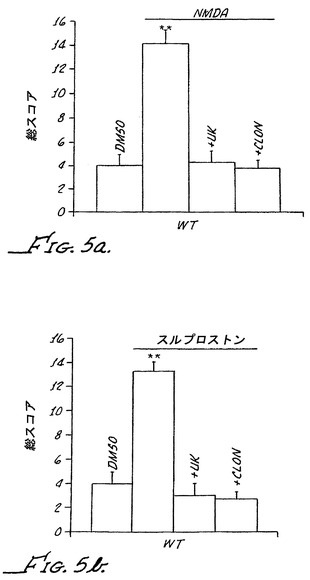
【図6】
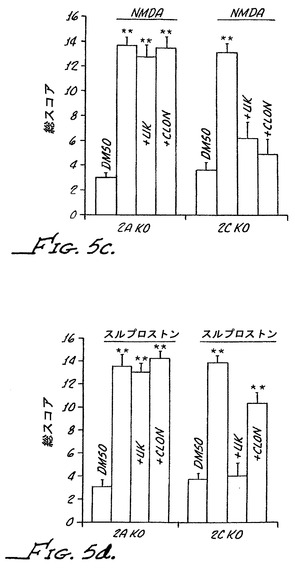
【図7】
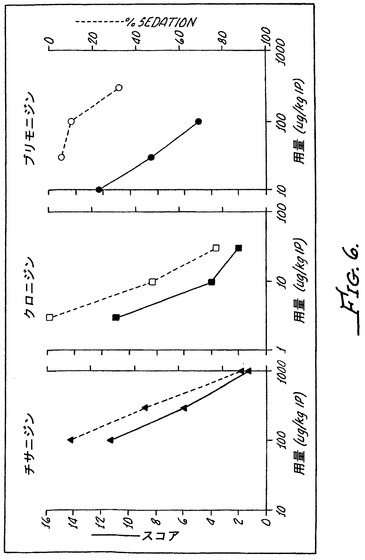
【図8】
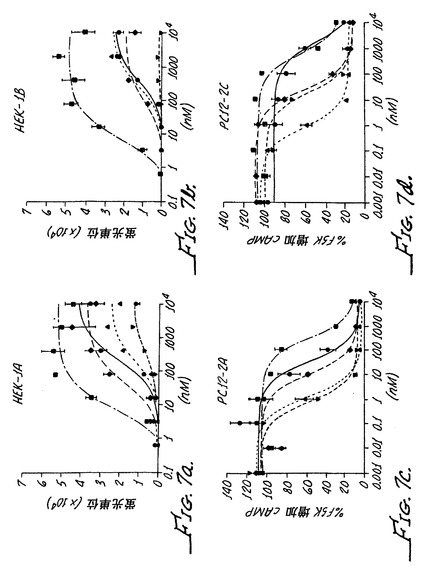
【図9】
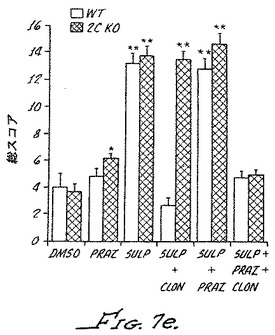
【図10】
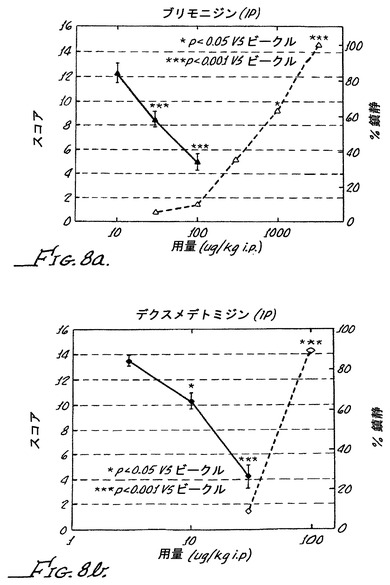
【図8c】
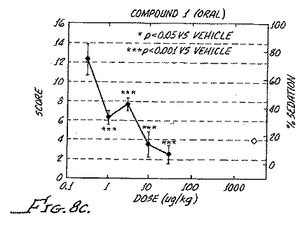
【図8d】
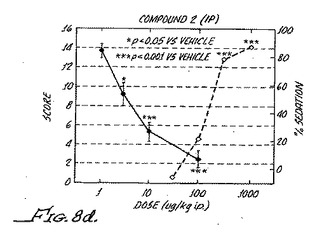
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図8c】
【図8d】
【公表番号】特表2007−505112(P2007−505112A)
【公表日】平成19年3月8日(2007.3.8)
【国際特許分類】
【出願番号】特願2006−526109(P2006−526109)
【出願日】平成16年8月19日(2004.8.19)
【国際出願番号】PCT/US2004/027170
【国際公開番号】WO2005/034849
【国際公開日】平成17年4月21日(2005.4.21)
【出願人】(591018268)アラーガン、インコーポレイテッド (293)
【氏名又は名称原語表記】ALLERGAN,INCORPORATED
【Fターム(参考)】
【公表日】平成19年3月8日(2007.3.8)
【国際特許分類】
【出願日】平成16年8月19日(2004.8.19)
【国際出願番号】PCT/US2004/027170
【国際公開番号】WO2005/034849
【国際公開日】平成17年4月21日(2005.4.21)
【出願人】(591018268)アラーガン、インコーポレイテッド (293)
【氏名又は名称原語表記】ALLERGAN,INCORPORATED
【Fターム(参考)】
[ Back to top ]