
改変型放出鎮痛性懸濁液
非ステロイド性抗炎症薬、特に、イブプロフェンといったプロピオン酸誘導体を、フェニレフリンといったより短い治療上有効な血漿濃度持続時間を有する第2の活性成分と共に含む医薬品剤形、およびこれと同じものを投与する方法が提供される。この方法は、拡張された期間にわたって、改善された治療学的効果、特に、痛みの軽減と共に鬱血除去の軽減をもたらす。
【発明の詳細な説明】
【開示の内容】
【0001】
本発明は、少なくとも2つの活性成分の投与のための液体剤形に適している改変型放出医薬品調合物に関する。より詳細には、この剤形は、同様の期間にわたって血漿中に含まれる全ての活性成分の医薬品に好適な血漿濃度を提供する速度で、活性成分を放出する。
【0002】
〔発明の背景〕
痛み、炎症、および発熱を処置するための治療薬としては、鎮痛薬、抗炎症薬(anti-inflammatories)、および解熱薬を含む。非ステロイド性抗炎症性薬(Non-steroidal anti-inflammatory drug)(NSAID)は、そのような治療薬の一種である。NSAIDとしては、プロピオン酸誘導体、酢酸誘導体、フェナム酸誘導体(fenamic acid derivatives)、ビフェニルカルボン酸誘導体(biphenylcarbodylic acid derivatives)、オキシカム(oxicams)、およびシクロオキシゲナーゼ−2(COX−2)選択性NSAIDを含む。
【0003】
プロピオン酸誘導体としては、例えば、イブプロフェン、ナプロキセン、およびケトプロフェンを含む。イブプロフェンは特に、広く使用されていてよく知られている、鎮痛特性および解熱特性を持つNSAIDである。イブプロフェンは、何年もの間、多くの形態で大衆薬として商業的に入手可能となってきた。イブプロフェンは、2−(4−イソブチルフェニル)−プロピオン酸として化学的に知られている。
【0004】
速効性放出NSAIDは、典型的には、約4〜6時間毎に投与される。典型的には、NSAIDの一日量は、約50〜約2000ミリグラム、好ましくは約100〜1600ミリグラム、最も好ましくは約200〜約1200ミリグラムの範囲にわたる。
【0005】
多くの他の活性成分は、それらの比較的短い持続時間ゆえに、より頻繁に投与される。例えば、鬱血除去薬であるフェニレフリンの治療上有効な血漿濃度は、約2.5時間±.7時間であり、そのため、フェニレフリンは典型的には、2〜4時間毎に投与される。
【0006】
NSAIDと、持続時間がより短い、医薬品として好適な血漿濃度を有する他の活性成分とを含有する単一製品を投与するためには、後者の放出を制御する必要があるだろう。特には身体における薬のより長期間にわたる作用を提供するために、剤形から患者の胃腸(「g.i.」)液への薬または他の活性成分の放出の速度を減らすことがよく知られている。
【0007】
経口的に送達される薬が身体中のその作用部位に到達する速度は、g.i.粘膜を介した血中への薬の吸収の速度および程度を含めた多数の因子に依存する。しかしながら、薬が血中へと吸収されることができるより前に、薬はまずg.i.液中に溶解されなければならない。多くの薬にとって、g.i.膜を通る吸収は、g.i.液中へのそれらの溶解に比べて比較的速く、そのため、このことが薬の溶解を薬の吸収における速度制限ステップにしている。したがって、配合者は、薬の溶解速度を改変することにより血中への薬の吸収速度を効果的に制御することがある。
【0008】
薬の治療学的効力の開始および持続は広く変化し、それらの対応する吸収、分配、代謝、および排出もまた広く変化するので、異なる方法で異なる薬の放出を改変すること、あるいは、第1の薬が剤形から速効性で放出されるようにし、一方で第2の薬が「改変型」式、例えば遅延型または制御型のいずれかで放出されることが知られている。
【0009】
剤形が改変された速度で薬を送達することができるようにするよく知られたメカニズム(例えば、持続型、延長型、拡張型、または遅滞型の放出)としては、拡散、浸食、および浸透を含む。多くの場合、特定の活性成分に対して、特に望ましい改変型放出プロフィールを達成する上述のメカニズムの組み合わせを使用する剤形を設計することが実用的である。
【0010】
不都合なことに、多くの改変型放出施用は、最終的な大きいサイズおよび重さを有する固体投薬ユニットを用いる。そのような投薬ユニットの投与は、特に子供たちや高齢者などの嚥下が困難な患者たちに、問題を提示する。したがって、咀嚼可能か、もしくは経口的に崩壊可能な個体形態、または液体形態のいずれかの形態で改変型放出内服薬を提供することがさらに望ましい。多くの患者たちにとって、液体経口剤形は、さらなる咀嚼ステップ無しで嚥下されることができるため、より好ましいものである。
【0011】
経口液体形態は、速効性放出プロフィールで薬物を送達するために、長年、一般に使用されてきた。例えば、米国特許第5,374,659号;同第4,788,220号;同第4,975,465号;および同第5,183,829号を参照のこと。しかしながら、改変型放出薬物を液体剤形に組み込むことは、有意な調合物変化をもたらす。特に、コーティングされるか、または化学的に結合された粒子が典型的に、薬の改変型放出部分を保有するために用いられる。例えば、米国特許第5,980,882号は、薬の放出速度を遅くするために、キレート剤と共に薬−樹脂複合体を使用することを開示している。米国特許第4,847,077号は、延長型で連続的な薬の放出を提供するために、薬−樹脂複合体粒子上に設けられた水透過性拡散隔膜コーティングを使用することを開示している。
【0012】
そのような粒子の特性だけでなく、それらを懸濁するための液体媒体の特性も、粒子が均一に分散した状態に維持されることができるように、適合性でなければならない。特に難題は、患者による摂取の前の液体剤形の貯蔵寿命の間の懸濁された粒子から懸濁液の媒質中への薬の中途での放出を防ぐことである。加えて、液体剤形の保管寿命の間ずっと液体剤形の所望の溶解プロフィールと所望の用量均一性とを維持することが、経口用液体改変型放出懸濁液製品を処方するにあたって取り組まれるべき別の難題である。不都合なことに、多くの場合、例えば速効性放出のイブプロフェンと改変型放出の第2の活性成分(たとえば、フェニレフリン)とを含有する製品を処方するときに、イブプロフェンと本技術分野で既知の改変型放出コーティング剤との間の相互作用が原因で、これらの問題に遭遇する。
【0013】
米国特許出願第20060057205号は、フェニレフリンと鎮痛薬といった少なくとも第2の薬とを含む液体剤形を開示しており、この剤形は、イオン交換樹脂と共に、両薬の複合体の粒子を含んでおり、この粒子は、ポリメタクリレートといった改変型放出コーティングでコーティングされている。しかしながら、我々は、イブプロフェンといったコーティングされていないプロピオン酸誘導体は、半透性の改変型放出コーティング(たとえば、エチルセルロースおよびポリメタクリレートを含有するもの)と相互作用することがあることを発見した。有害なことに、この相互作用は多くの場合、コーティングされた薬の放出速度および意図された改変型放出特性を損なう。
【0014】
したがって、イブプロフェン粒子とフェニレフリンといった他の活性成分の改変型放出粒子とを含む改変型放出剤形であって、味が良いだけでなく、投与後の所望の放出プロフィールを保証する安定な形態にある、剤形を有することが望ましいであろう。特に、使用者に、速効性放出用量のイブプロフェンと持続型放出用量の第2の活性成分との両方を、イブプロフェンと持続型放出コーティングとの間の相互作用を起こすことなく、提供するような鎮痛薬製品を有することがさらに望ましいであろう。
【0015】
〔発明の概要〕
本発明は本書にて請求されているように、液体懸濁剤中のNSAIDSの投与のために好適な剤形のような医薬品剤形であって、かかる剤形は、
a)NSAIDといった第1の活性成分を含む第1の部分であって、この第1の活性成分は、剤形が溶解媒質に接触すると実質的に速効式に剤形から放出される、第1の部分と、
b)第2の部分であって、
i)第2の活性成分を有するイオン交換樹脂粒子であって、第2の活性成分は、第1の活性成分と同じでも、または異なっていてもよく、ある一つの実施形態ではフェニレフリンといったものであってもよく、イオン交換樹脂上に結合されていて薬−樹脂複合体粒子を形成している、イオン交換樹脂粒子、
ii)薬−樹脂複合体粒子のそれぞれを実質的に被覆している半透性コーティング層、および、
iii)ii)の粒子のそれぞれを実質的に被覆している保護コーティング層、
を含んでいる、第2の部分と、
を含む、からなる、および/または、から本質的になり、
第2の活性成分は、剤形が溶解媒質に接触すると改変型放出式に第2の部分から放出され、剤形の第2の部分から放出されたときの第2の活性成分の治療学的効果の持続時間は、第1の活性成分の治療学的効果の持続時間と実質的に同じである、医薬品剤形、ならびにその投与のための方法を提供する。
【0016】
〔発明の詳細な説明〕
本明細書中の説明に基づいて当業者が本発明をその十分な範囲まで利用することができると思料される。下記の詳細な実施形態は、例示としてのみ解釈されるべきものであって、開示のいずれもが、いかようにも限定的に解釈されるべきではない。
【0017】
他に定義されない限り、本明細書中で使用されている全ての技術用語および科学用語は、本発明が属する技術分野における当業者により一般に理解されているのと同じ意味を有する。また、本明細書中で言及されている全ての文献、特許出願、特許、およびその他の参照文献は、参照により組み込まれる。本明細書中で使用される全てのパーセンテージは、他に特定されない限り、重量%である。加えて、本明細書中で示されている全ての範囲は、2つの終点の間の値のあらゆる組み合わせをすべて含めたものを含むものと意味される。
【0018】
本明細書中で使用される用語「実質的に被覆する」または「実質的に連続的に」は、コーティングが概ね連続的で、コアまたは下層の全表面を概ね被覆していて、そのため、活性成分または下層のうちのわずかだけ露出するか、あるいは全く露出しないようになっていることを意味している。
【0019】
本明細書中で使用されている「ATDAIRD」は、有効な速効性放出用量」の特定の活性成分の作用の平均治療学的持続時間を意味するものとする。例えば、速効性放出用量のイブプロフェンまたはケトプロフェンの典型的な作用の持続時間、すなわち治療学的効果の期間は、約4〜約6時間である。したがって、イブプロフェンまたはケトプロフェンについてのATDAIRDは5時間である。速効性放出用量のナプロキセンの作用の典型的な持続時間は、約8〜約12時間である。したがって、ナプロキセンについてのATDAIRDは10時間である。速効性放出用量のフェニレフリンの作用の典型的な持続時間は、約2〜約4時間である。したがって、フェニレフリンについてのATDAIRDは3時間である。特定の活性成分の作用の治療学的持続時間は、特定の活性成分を含む速効性放出製品についてのラベルにおける投薬説明書から容易に決定されることができる。
【0020】
本明細書中で使用されている「改変型放出(modified release)」は、g.i.液といった溶解媒質中での活性成分の変更された放出または溶解に対して適用するものとする。改変型式で放出され得る活性成分または複数の成分は、例えば、剤形、コーティング、もしくは粒子、またはこれらのいずれかの部分、たとえば液体懸濁媒質の全体に分散した粒子といった部分の中に含まれていてもよい。改変型放出の種類としては、1)拡張型放出(extended release)、または2)遅延型放出(delayed release)を含む。全般的に、改変型放出剤形は、活性成分を、摂取後の時間の拡張された期間にわたって有用にするよう処方され、これにより、従来の剤形における同じ活性成分の投薬に比べて、投薬頻度を減らすことができる。改変型放出剤形は、活性成分の組み合わせを使用することも可能にし、このとき、ある一つの活性成分の持続時間は、他の活性成分の持続時間と異なってもよい。
【0021】
投与後、活性成分が実質的に連続的で調節された手法で剤形から放出され、剤形からの活性成分の完全な放出、すなわち枯渇のための時間が、同じものの速効性放出剤形に関連する時間よりも長いことが、「拡張型放出」により意味される。拡張型放出の種類としては、制御型、持続型、延長型、ゼロオーダーの放出などを含む。
【0022】
投与後、活性成分が剤形から放出されない時間の少なくとも1つの期間がある、すなわち、活性成分の放出が、経口投与後すぐではない時間に起こることが、「遅延型放出」により意味される。
【0023】
本明細書中で使用されている「溶解媒質」は、例えば、製品のテストのために使用されるインビトロの溶解媒質、または胃腸液といった、本発明の懸濁液剤形が溶解されることができるあらゆる好適な液体環境を意味するものとする。本発明の懸濁液剤形からの活性成分または複数の成分の溶解をテストするために使用される好適なインビトロの溶解媒質としては、本明細書中に参照により組み込まれるUSP23(1995)の第786頁に記述されているものを含む。
【0024】
本明細書中で使用されている「実質的にコーティングされた」は、粒子の表面積の約20%未満、例えば、約15%未満、または約1.0%未満が露出されている、例えば、所望のコーティングで被覆されていないことを意味するものとする。
【0025】
「腸溶性」は、約5.0を超えるか、約5.5を超えるか、または約6.0を超えるpH、あるいは腸管で見られるpHで溶解されることができることを意味するものとする。
【0026】
「液体剤形」は、非排他的に、懸濁液またはエリキシルを含んでもよく、この中で、活性成分の一つ以上が溶解されるか、部分的に溶解されるか、または、溶解されていない状態もしくは懸濁した状態にある。
【0027】
本明細書中で使用されている「薬−樹脂複合体」は、限定されるものではないが医薬品活性成分を含めた、いずれかの活性成分と、イオン交換樹脂との結合された形態を意味するものとする。薬−樹脂複合体はまた、本技術分野では「レジネート」とも称される。
【0028】
本明細書中で使用されている「速効性放出(immediate release)」は、少なくとも1つの活性成分の溶解特質がその活性成分を含有する速効性放出タブレットについてのUSP規格を満足することを意味する。速効性放出特性を有する活性成分は、活性成分の溶解を遅延または延長させることを意図することなく、胃腸内容物に溶解され得る。例えば、アセトアミノフェンタブレットの場合、USP24は、pH5.8リン酸緩衝液中、USP装置2(パドル)を50rpmで用いて、剤形に含まれるアセトアミノフェンの少なくとも80%が投薬後30分以内に剤形から放出されることを規定しており、イブプロフェンタブレットの場合には、USP24は、pH7.2のリン酸緩衝液中、USP装置2(パドル)を50rpmで用いて、剤形に含まれるイブプロフェンの少なくとも80%が投薬後60分以内に剤形から放出されることを規定している。USP 24, 2000 Version, 19〜20および856 (1999)を参照のこと。加えて、イブプロフェン懸濁液は、USP装置2(パドル)を50rpmで用いるpH5.6の酢酸緩衝液(acetate buffer)を用いた溶解について分析されることもあり、ここでは、剤形に含まれるイブプロフェンの少なくとも80%が、速効性放出用量について投薬後60分以内に剤形から放出される。
【0029】
本明細書中で使用されている薬の「放出速度」は、単位時間当たりの剤形から放出される薬の量、例えば、時間当たり放出される薬のミリグラム数(mg/hr)をさしている。薬の放出速度は、本技術分野で既知のインビトロの剤形溶解テスト条件の下、算出される。本明細書中で使用される、「投与後」に規定された時間で得られる薬の放出速度は、適切な溶解試験の開始後の規定された時間で得られるインビトロの薬の放出速度、例えば、USP24に示されているものをさしている。
【0030】
本明細書中で使用されている「治療学的効果」は、病気を診断、処置、治療、緩和、または防止するか、または身体の構造もしくはあらゆる機能に影響を及ぼすことが意図された活性成分のあらゆる効果または作用を意味するものとする。
【0031】
本明細書中で使用されている「半透性」は、膜が適切な溶解媒質、例えば、胃腸液またはインビトロの溶解媒質に接触しているときに、水がそのような膜を透過することができ、また、塩および本明細書中に記述されている活性成分を含めたその他の分子がそのような膜を通ってゆっくりと拡散させられることを意味するものとする。
【0032】
本明細書中で使用されている「水不溶性」は、U.S. Pharmacopeia第24版により定義されているように水中に、実質的に不溶性であるか、事実上不溶性であるか、または僅かにだけ可溶性である組成を意味している。これらの組成物は、完全な溶解のために、かかる組成物の部(part)当たり、少なくとも約100部の溶媒を必要とする。
【0033】
本明細書中で使用されている「侵食性(Erodible)」は、適切な溶解媒質に接触したときに、表面の侵食を介して組成物が溶解することを意味するものとする。
【0034】
本明細書中で使用されている「保護コーティング」は、乾燥媒体、例えば剤形のマトリックス中の、または、液体剤形実施形態においては液体媒体媒質中の、他の粒子または他の活性成分と反応しないコーティングを意味するものとする。
【0035】
本明細書中で使用されている用語「フェニレフリン」は、ベンザインメタノール,3−ヒドロキシ−α−[(メチルアミノ)メチル]を意味し、限定されるものではないが、医薬品に許容可能なそれらの塩、エステル、異性体、または誘導体を含む。
【0036】
本明細書中で使用されている「粒子」は、結晶、顆粒、凝集物、または溶解されていないあらゆる固体材料である。
【0037】
本発明のある一つの実施形態は、活性成分の投与のために好適な改変型放出医薬品剤形に向けられており、この剤形は、a)速効性放出部分、例えば、剤形から速効性で放出される少なくとも1つの活性成分を含む部分と、b)改変型放出部分、例えば、時間の改変された期間にわたって実質的に連続的に血流中に放出される少なくとも1つの活性成分を含む部分とを含む。
【0038】
ある一つの実施形態において、剤形の第2の部分から放出されたときの第2の活性成分の改変型放出の治療学的効果が、第1の活性成分の速効性放出の治療学的効果の持続時間と実質的に同じになるように、剤形が溶解媒質に接触すると活性成分が改変型放出式に第2の部分から放出される。「第1の活性成分の速効性治療学的効果の持続時間と実質的に同じ」とは、第2の活性成分の治療学的効果の持続時間が、第1の活性成分の持続時間と同じであるか、またはその持続時間の約1時間以内、すなわち、例えば約1/2時間以内、または約15分以内、または約10分以内であることを意味するものとする。他の実施形態において、剤形の第2の部分から放出されたときの第2の活性成分の改変型放出の治療学的効果は、例えば、剤形の最初の投与後、少なくとも約4時間〜約6時間、または約4時間〜約8時間、または約4時間〜約12時間であってもよい。
【0039】
速効性放出部分は、剤形内で、分子レベルで分散されている、例えば溶融または溶解されている1つ以上の活性成分を含んでもよく、あるいは、活性成分は、次にコーティングされてもよいし、またはコーティングされなくてもよい粒子の形態にあってもよい。活性成分が粒子の形態にある実施形態において、粒子は(コーティングされていようと、またはコーティングされてなかろうと)典型的には、約1ミクロン〜約2000ミクロンの平均粒径を有する。ある一つの実施形態において、そのような粒子は、約1ミクロン〜約300ミクロンの平均粒径を有する結晶の形態にある。他の実施形態において、粒子は、約25ミクロン〜約2000ミクロン、例えば、約25ミクロン〜約1000ミクロン、または約25ミクロン〜約400ミクロンの平均粒径を有する顆粒またはペレットの形態にある。
【0040】
改変型放出部分は、改変型放出特性を有する様々な粒子中に、少なくとも1つの活性成分を含む。ある一つの実施形態において、改変型放出部分中のこれらの粒子のコアは、改変型放出組成物で実質的にコーティングされた純粋な結晶形態にある活性成分から構成されてもよい。あるいは、粒子コアは、本技術分野で既知の結合剤、賦形剤などといったオプションの成分と、1つ以上の活性成分から構成された顆粒との混合物から構成されてもよく、そのような顆粒もまた、改変型放出組成物で実質的にコーティングされる。他の実施形態において、活性成分粒子は、改変型放出組成物で構成されたマトリックスの全体に分散されてもよい。
【0041】
さらなる他の実施形態において、1つ以上の活性成分は、樹脂、例えばイオン交換樹脂に、化学的に結合されるか、または「複合体化され(complexed)」て、薬−樹脂複合体粒子(または活性成分樹脂粒子)を形成してもよく、この複合体は、まず半透性コーティング層で実質的にコーティングされ、その後、保護コーティング層で実質的にコーティングされる。当業者は、過度の実験を行うことなく、本実施形態で使用するための特定のイオン交換樹脂が、例えば、活性成分のイオン電荷といったいくつかの因子に依存することを容易に認識するであろう。NSAID活性成分のために好適なイオン交換樹脂の例としては、限定されるものではないが、スチレン/ジビニルベンゼンコポリマー、およびRohm & Haasから「Duolite(登録商標)AP143」という商品名で商業的に入手可能なコレスチラミンを含む。フェニレフリンまたは偽性エフェドリンといった正の電荷を持つ活性成分のために好適なイオン交換樹脂の例としては、限定されるものではないが、スルホン化スチレン/ジビニルベンゼンコポリマーから誘導されたスルホン酸カチオン性イオン交換樹脂、たとえば、Rohm & Haasから一般商品名「Amberlite」で商業的に入手可能なもの、例えば、「Amberlite IRP69」、および、Dow Chemical Companyから商品名「Dowex」で商業的に入手可能なもの、例えば、「Dowex Marathon」、「Dowex Monosphere」、および「Dowex XYS-40010.00」を含む。Amberlite IRP 60およびDowex XYS-40010.00の製品は、約8%のジビニルベンゼンと架橋結合したポリスチレンから構成されたスルホン化ポリマーであり、乾燥樹脂(H+形態)の約4.5〜5.5meq./gのイオン交換能力を有する。Amberlite IRP-69製品は、約47μm〜約149μmの粒径範囲を備えた不規則な形になった粒子から構成されており、一方、Dow XYS-40010.00製品は、約45μm〜約150μmの粒径範囲を有する球状粒子から構成されている。他の好適なイオン交換樹脂である「Dow XYS-40013.00」は、約8%のジビニルベンゼンと架橋結合されたポリスチレンから構成されたポリマーであり、四級アンモニウム基で官能性を持たせてあり、その交換能力は、標準的には、乾燥樹脂の約3〜約4meq./gの範囲内にある。他の好適なイオン交換樹脂は、ポラクリリンカリウム(polacrilin potassium)を含み、これは、ジビニルベンゼンカリウム塩を含むメタクリル酸ポリマー(methacrylic acid polymers with divinylbenzene potassium salt)としても知られている。ポラクリリンカリウム(polacrin potassium)の好適な例としては、限定されるものではないが、Rohm & Haasから商品名「Amberlite IRP-64」で商業的に入手可能なものを含み、これは、メタクリル酸(methcrylic acid)とジビニルベンゼンとのコポリマーから誘導されたカチオン性イオン交換樹脂である。ポリマー樹脂との複合体形成のさらなる詳細は、本技術分野においてよく知られており、例えば、米国特許第4,221,778号;同第5,980,882号;同第4,847,077号;および同第6,001,392号に開示されている。
【0042】
イオン交換樹脂は、一般に、強酸カチオン、強塩基カチオン、弱酸カチオン、および弱塩基カチオンを含めた種々の種類に分類される。全般的に、薬は、好適な樹脂の水性懸濁液に混合され、その後、樹脂−薬複合体が洗浄され、乾燥される。樹脂への薬の結合は、洗浄物からの媒質溶離のpHを分析するか、または、洗浄物のナトリウム濃度における変化を測定することにより実証され得る。
【0043】
フェニレフリンを水性混合物を介してイオン交換樹脂に結合させることが望ましい実施形態においては、塩酸フェニレフリン(PHE−HCl)および樹脂が混合物になるように初めに配合されてもよい。その後、樹脂のナトリウムイオンが、フェニレフリンのプロトン化形態(PHE−H+)に交換されてもよい。典型的には、結果として生じる薬−樹脂複合体は、フェニレフリンと樹脂との重量比が約20:80〜約80:20、すなわち、例えば約30:70〜約70:30である。
【0044】
ある一つの特定の実施形態において、剤形の改変型放出部分は、イオン交換樹脂を実質的に含まない。イオン交換樹脂の量が、剤形中の全ての活性成分粒子の全重量に基づいて、約1パーセント未満、例えば、約0.5パーセント未満、または約0.1パーセント未満であることが、「イオン交換樹脂を実質的に含まない」により意味される。
【0045】
本発明に従うと、薬−樹脂複合体粒子は、半透性コーティングで実質的にコーティングされる。粒子表面の約80%、例えば、約85%、または約99%がコーティングされていることが、「実質的にコーティングされる」により意味される。
【0046】
好適な半透性コーティングの例としては、限定されるものではないが、セルロースアセテート、エチルセルロース、非腸溶性ポリメタクリレート、ならびに、これらのコポリマーおよび混合物といったポリマーを含む。例示的な非腸溶性ポリメタクリレートとしては、限定されるものではないが、Rohm Pharmaから商品名「EUDRAGIT NE」で商業的に入手可能なポリ(エチルアクリレート,メチルメタクリレート)[2:1];Rohm Pharmaから商品名「EUDRAGIT FS」で商業的に入手可能なポリ(メチルアクリレート,メチルメタクリレート,メタクリル酸)[7:3:1];Rohm Pharmaから商品名「EUDRAGIT RL」で商業的に入手可能なポリ(エチルアクリレート,メチルメタクリレート,トリエチルアンモニオエチルメタクリレートクロライド)[1:2:0.2](poly(ethyl acrylate, methyl methacrylate, triethylammonioethyl methacrylate chloride) 1:2:0.2);Rohm Pharmaから商品名「EUDRAGIT RS」で商業的に入手可能なポリ(エチルアクリレート,メチルメタクリレート,トリエチレンアンモニオエチルメタクリレートクロライド)[1:2:0.1](poly(ethyl acrylate, methyl methacrylate, triethyleammonioethyl methacrylate chloride 1:2:0.1)、ならびに、これらのコポリマーおよび混合物を含む。本技術分野ではアセチルセルロース、セルロースジアセテート、およびセルローストリアセテートという一般名でも既知であるセルロースアセテートは、Eastman Chemical Companyから商業的に入手可能である。本技術分野ではセルロースエチルエーテルとしても既知であるエチルセルロースは、Dow Corporationから商品名「ETHOCEL」で商業的に入手可能である。ある一つの実施形態において、半透性コーティングは、セルロースアセテート、エチルセルロース、およびこれらの混合物から選択されてもよい。
【0047】
コーティングされた薬−樹脂複合体粒子は、その後、保護コーティングで実質的にコーティングされる。コーティング粒子表面の約80%、例えば、約85%、または約99%がその後、保護コーティング層でコーティングされることが、「実質的にコーティングされる」により意味される。
【0048】
好適な保護コーティングの例としては、限定されるものではないが、ヒドロキシプロピルメチルセルロースフタレート(ヒプロメロースフタレート(hypromellose phthalate)としても知られている)、ヒドロキシプロピルメチルセルロースアセテートスクシネート、セルロースアセテートフタレート、ポリビニルアセテートフタレート、セラック、腸溶性ポリメタクリレート系ポリマー(enteric polymethacrylate-based polymers)、ならびに、これらのコポリマーおよび混合物を含む腸溶性ポリマーから構成されたものを含む。好適な腸溶性ポリメタクリレート系ポリマーの例としては、限定されるものではないが、Rohm Pharma GmbHから商品名「EUDRAGIT S」ポリマーで商業的に入手可能なポリ(メタクリル酸,メチルメタクリレート)[1:2];Rohm Pharma GmbHから商品名「EUDRAGIT L-100, L-30D, L 12.5 and L12.5 P」ポリマーで商業的に入手可能なポリ(メタクリル酸,メチルメタクリレート)[1:1];ならびに、Rohm Pharmaから商品名「EUDRAGIT L30-D 55 and L-100-55」、Eastman Chemicalから商品名「Eastacryl 30D」、およびColorcon Corporationから商品名「Acryl-EZE」、およびBASF Fine Chemicalsから商品名「Kollicoat MAE 30D」で商業的に入手可能なポリ(メタクリル酸,エチルアクリレート)[1:1]を含む。ある一つの実施形態において、腸溶性ポリマーは、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートスクシネート、セルロースアセテートフタレート、ポリビニルアセテートフタレート、セラック、ならびに、これらのコポリマーおよび混合物といった非アクリル酸塩化合物(non-acrylate compounds)から選択されてもよい。
【0049】
ある一つの実施形態において、保護コーティングは、脂肪酸エステル、ワックス、またはこれらの混合物といった脂質の形態で提供されてもよい。好適な脂肪酸エステルの例としては、限定されるものではないが、スクロース脂肪酸エステル;モノグリセリド、ジグリセリド、およびトリグリセリド;グリセリルベヘナート(glyceryl behenate);グリセリルパルミトステアレート(glyceryl palmitostearate);グリセリルトリステアレート(glyceryl tristearate);グリセリルトリラウリレート(glyceryl trilaurylate);グリセリルミリステート;GLYCOWAX-932;ラウロイルマクロゴール−32グリセリド;ステアロイルマクロゴール−32グリセリド;約C10〜C40の脂肪酸鎖長を有するものなどの脂肪酸エステル;およびこれらの混合物を含む。
【0050】
好適なワックスの例としては、限定されるものではないが、カルナウバろう、鯨ろう、蜜ろう、カンデリラろう、セラックろう、カルヌバろう(carnuba wax)、蜜ろう、微晶ろう、およびパラフィンろうなど、ならびにこれらの混合物を含む。
【0051】
オプションとして、半透性コーティングおよび/または保護コーティングは、可塑剤を含んでもよい。好適な可塑剤の例としては、限定されるものではないが、ポリエチレングリコール;プロピレングリコール;グリセリン;ソルビトール;トリエチルシトレート;トリブチルシトレート;ジブチルセバケート(dibutyl sebecate);ヒマシ油、菜種油、オリーブ油、およびゴマ油などの植物油;ポリソルベート、ラウリル硫酸ナトリウム、およびスルホコハク酸ジオクチルナトリウムなどの界面活性物質;グリセロールのモノアセテート;グリセロールのジアセテート;グリセロールのトリアセテート;天然ゴム(natural gum);トリアセチン;アセチルトリブチルシトレート(acetyltributyl citrate);ジエチルオキサレート(diethyloxalate);ジエチルマレート(diethylmalate);ジエチルフマレート;ジエチルマロネート;ジオクチルフタレート;ジブチルスクシネート;グリセロールトリブチレート;グリセロールモノステアレート;水素化されたヒマシ油;置換されたトリグリセリドおよびグリセリド;ならびに、これらの混合物を含む。
【0052】
ある一つの実施形態において、好適な可塑剤は、半透性コーティングの全乾燥重量に基づいて、約0.1%〜約40%、例えば、約1%〜約30%、または約5%〜約20%の量で使用され得る。
【0053】
ある一つの実施形態において、好適な可塑剤は、保護コーティングの全乾燥重量に基づいて、約0.1%〜約40%、すなわち例えば、約1%〜約30%、または約5%〜約20%の量で使用され得る。
【0054】
ある一つの実施形態において、改変型放出粒子における半透性コーティング層と保護腸溶性コーティング層との重量比は、約10:90〜約90:10、または約20:80〜約80:20である。
【0055】
ある一つの実施形態において、改変型放出粒子は、腸溶性ポリマーを実質的に含まず、すなわち例えば、改変型放出粒子は、改変型放出粒子の全重量に基づいて、約1パーセント未満、または約0.25パーセント未満の腸溶性ポリマーを含む。
【0056】
ある一つの実施形態において、薬−樹脂複合体は、溶媒和または含浸剤(solvating or impregnating agent)で処理されてもよく、これは、活性成分および樹脂が混合されている間か、または活性成分が樹脂に結合された後で加えられる。好適な含浸剤の例としては、限定されるものではないが、ソルビトール、ポリエチレングリコール、グリセロール、プロピレングリコール、マンニトール、ラクチトール(lactitol)、ラクトース、メチルセルロース、およびこれらの混合物を含む。含浸剤は、乾燥樹脂の重量当たり、約5部〜約50部の量で存在し得る。
【0057】
ある一つの実施形態において、薬−樹脂複合体の酸化を抑制することにより薬−樹脂複合体を安定化するために、キレート剤が剤形に加えられてもよい。好適なキレート剤としては、限定されるものではないが、エチレンジアミン四酢酸(EDTA)およびEDTAの塩を含み、EDTAの塩としては、限定されるものではないが、エデト酸カルシウム二ナトリウム、エデト酸三ナトリウム、エデト酸二ナトリウム、およびエデト酸ナトリウムを含む。キレート剤は、最終的な剤形の重量の、約0.005パーセント〜約10パーセントの量で存在し得る。
【0058】
第2の保護コーティング層の下にある第1の半透性コーティング層を含む活性成分−樹脂複合体粒子は、改変型放出組成物の全乾燥重量に基づいて、約1パーセント〜約99パーセント、例えば、約5パーセント〜約80パーセントの第1の半透性コーティング層、および約5パーセント〜約99パーセント、例えば約10パーセント〜約90パーセントの第2の保護コーティング層を含む改変型放出組成物を産生する。
【0059】
2つのコーティング層のそれぞれの厚みは、所望の改変型放出特性、選択される活性成分などに依存して変化してもよいが、典型的には、約0.01ミクロン〜約500ミクロン、例えば、約0.1ミクロン〜約100ミクロンの範囲にわたり得る。
【0060】
粒子上の第1のコーティング層の表面積当たりの乾燥重量は、約0.1mg/cm2〜約10mg/cm2、すなわち例えば、約0.5mg/cm2〜約5mg/cm2である。粒子上の第2のコーティング層の表面積当たりの乾燥重量は、約0.1mg/cm2〜約10mg/cm2、例えば、約0.5mg/cm2〜約8mg/cm2である。
【0061】
活性成分−樹脂複合体粒子の第1の半透性コーティング層の追加後の重量増加は、コーティングされていない活性成分−樹脂複合体粒子の乾燥重量に基づいて、約1パーセント〜約200パーセント、例えば、約20パーセント〜約150パーセントである。活性成分−樹脂複合体粒子の第2の保護コーティング層の追加後の重量増加は、第1の半透性コーティング層でコーティングされた活性成分−樹脂複合体粒子の乾燥重量に基づいて、約25パーセント〜約400パーセント、例えば、約40パーセント〜約400パーセントである。
【0062】
コーティングされた活性成分粒子は、本技術分野で既知のあらゆる好適な方法により形成されてもよい。好適な粒子形成およびコーティング方法としては、高速攪拌造粒(high sheer granulation)、流動層造粒(fluid bed granulation)、例えば、ローター造粒、流動層コーティング、ウルスターコーティング(wurster coating)、コアセルベーション(coaccervation)、吹き付け乾燥、吹き付け凝結などを含み、例えば、Pharmaceutical Dosage Forms: Tablets Volume 3, edited by Herbert A. Lieberman and Leon Lachman, Chapters 2, 3, and 4 (1982)に記述されている。
【0063】
活性成分をイオン交換樹脂に結合させて薬−樹脂複合体を形成することにより粒子が形成されるある一つの実施形態においては、結果として生じる粒子は、ウルスター流動層コーティングを用いて半透性層がまずコーティングされ、その後、腸溶性層がウルスター流動層コーティングを用いてコーティングされる。コーティング材料は、溶媒を含む溶液または分散系を介して粒子上に吹き付けられてもよく、この溶媒としては、限定されるものではないが、水、エタノール、メタノール、アセトン、ヘキサン、シクロヘキサン、メチレンクロライド、イソプロパノール、およびこれらの混合物を含む。例えば、米国特許第4,847,077号を参照のこと。
【0064】
ある一つの実施形態において、薬−樹脂複合体の平均直径は、約20ミクロン〜約400ミクロン、または約20ミクロン〜約300ミクロンである。ある一つの実施形態において、第1のコーティング層でコーティングされた薬−樹脂複合体粒子の平均直径は、約20ミクロン〜約800ミクロン、例えば、約50ミクロン〜約400ミクロンであり、第1および第2のコーティング層の両方でコーティングされた薬−樹脂複合体粒子の平均直径は、約50〜約1000ミクロン、例えば、約100ミクロン〜約400ミクロンである。
【0065】
本発明の剤形は、1つ以上の活性な薬剤または成分を含む。広く好適な活性成分としては、例えば、医薬品、鉱物、ビタミンおよびその他の機能性食品(nutraceutical)、オーラルケア剤、香味料(flavorant)、およびこれらの混合物を含む。好適な医薬品としては、鎮痛薬、抗炎症剤、抗関節炎薬、麻酔薬、抗ヒスタミン薬、鎮咳薬、抗生物質、抗感染剤(anti-infective agents)、抗ウイルス薬、抗凝固薬、抗うつ薬、抗糖尿病剤(antidiabetic agents)、制吐薬、抗膨満薬(antiflatulents)、抗真菌薬、鎮痙薬、食欲抑制剤、気管支拡張薬、心血管作用薬(cardiovascular agents)、中枢神経系作用薬、中枢神経系刺激薬、鬱血除去薬、経口避妊薬、利尿薬、去痰薬、胃腸作用薬、偏頭痛製剤、乗物酔い製品、ムコリチック物(mucolytics)、筋弛緩薬、骨粗鬆症製剤、ポリジメチルシロキサン、呼吸器作用薬、睡眠補助薬(sleep-aids)、尿路作用薬、およびこれらの混合物を含む。
【0066】
好適な香味料としては、メントール、ペパーミント、ミント香料、果物香料、チョコレート、バニラ、風船ガム香料、コーヒー香料、リキュール香料、およびこれらの組み合わせなどを含む。
【0067】
好適な胃腸作用薬の例としては、制酸薬、例えば、炭酸カルシウム、水酸化マグネシウム、酸化マグネシウム、炭酸マグネシウム、水酸化アルミニウム、炭酸水素ナトリウム、炭酸ジヒドロキシアルミニウムナトリウム(dihydroxialuminum sodium carbonate);刺激性緩下薬(stimulant laxatives)、例えば、ビサコジル、カスカラサグラダ、ダントロン、センナ、フェノールフタレイン、アロエ、ヒマシ油、リシノール酸、およびデヒドロコール酸、ならびにこれらの混合物;H2レセプター拮抗薬、例えば、ファモチジン(famotadine)、ラニチジン、シメチジン(cimetadine)、ニザチジン;プロトンポンプ阻害薬、例えば、オメプラゾール、またはランソプラゾール;胃腸細胞保護薬(gastrointestinal cytoprotectives)、例えば、スクラフレート(sucraflate)、およびミソプロストール;胃腸運動促進薬(gastrointestinal prokinetics)、例えば、プルカロプリド、H. pyloriに対する抗生物質(antibiotics for H. pylori)、例えば、クラリスロマイシン、アモキシシリン、テトラサイクリン、およびメトロニダゾール;止瀉薬、例えば、ジフェノキシラート、およびロペラミド;グリコピロレート;制吐薬、例えば、オンダンセトロン、鎮痛薬、例えば、メサラミンを含む。
【0068】
限定されるものではないが、ジメチコーン、およびシメチコンを含む、好適なポリジメチルシロキサンの例は、米国特許第4,906,478号、同第5,275,822号、および同第6,103,260号に開示されているものであり、各特許文献の内容は、参照により本明細書中に明白に組み込まれる。本明細書中で使用されている用語「シメチコン」は、限定されるものではないが、シメチコンおよびジメチコーンを含めたポリジメチルシロキサンのより広い分類をさしている。
【0069】
本発明のある一つの実施形態において、少なくとも1つの活性成分は、ビサコジル、アルブテロール、ファモチジン、ラニチジン、シメチジン、プルカロプリド、ジフェノキシラート、ロペラミド、ラクターゼ、メサラミン、ビスマス、制酸薬、ならびに、医薬品に許容可能なこれらの塩、エステル、異性体、および混合物から選択され得る。
【0070】
他の実施形態において、少なくとも1つの活性成分は、鎮痛薬、抗炎症薬、および解熱薬、例えば、以下のものを含めた非ステロイド性抗炎症薬(NSAID)、すなわち、a)プロピオン酸誘導体、例えば、イブプロフェン、ナプロキセン、ケトプロフェンなど、b)酢酸誘導体、例えば、インドメタシン、ジクロフェナク、スリンダク、トルメチンなど、c)フェナム酸誘導体、例えば、メフェナム酸、メクロフェナム酸、フルフェナム酸など、d)ビフェニルカルボン酸誘導体、例えば、ジフルニサル、フルフェニサル(flufenisal)など、e)オキシカム、例えば、ピロキシカム、スドキシカム、イソキシカム、メロキシカムなど、f)シクロオキシゲナーゼ−2(COX−2)選択性NSAID、g)アスピリン、およびh)上記のものの医薬品に許容可能な塩から選択される。
【0071】
ある一つの特定の実施形態において、少なくとも1つの活性成分は、プロピオン酸誘導体NSAIDから選択され、これは、医薬品に許容可能な鎮痛薬/非ステロイド性抗炎症薬であり、遊離−CH(CH3)COOHもしくは−CH2CH2COOH、または医薬品に許容可能な塩の基、例えば、−CH(CH3)COO−Na+もしくはCH2CH2COO−Na+を有し、これらは、典型的には、環構造、好ましくは芳香環構造に直接またはカルボニル官能基を介して結合される。
【0072】
有用なプロピオン酸誘導体の例としては、イブプロフェン、ナプロキセン、ベノキサプロフェン(benoxaprofen)、ナプロキセンナトリウム、フェンブフェン、フルルビプロフェン、フェノプロフェン、フェノプロフェンカルシウム、フルルビプロフェン、チアプロフェン酸塩(tiaprofenic)、オキサプロジン、フェンブプロフェン(fenbuprofen)、ケトプロフェン、インドプロフェン(indoprofen)、ピルプロフェン、カルプロフェン(carpofen)、オキサプロフェン(oxaprofen)、プラノプロフェン、ミクロプロフェン(microprofen)、チオキサプロフェン、スプロフェン、アルミノプロフェン、チアプロフェン酸、フルプロフェン(fluprofen)、ブクロキシ酸(bucloxic acid)、ならびに、医薬品に許容可能なこれらの塩、誘導体、および組み合わせを含む。
【0073】
本発明のある一つの実施形態においては、プロピオン酸誘導体は、イブプロフェン、ケトプロフェン、フルビプロフェン(flubiprofen)、ならびに、医薬品に許容可能なこれらの塩、および組み合わせから選択される。
【0074】
他の実施形態において、プロピオン酸誘導体は、イブプロフェン、2−(4−イソブチルフェニル)プロピオン酸、または医薬品に許容可能なこれらの塩、例えば、イブプロフェンのアルギニン、リジン、またはヒスチジンの塩である。その他の医薬品に許容可能なイブプロフェンの塩は、米国特許第4,279,926号、同第4,873,231号、同第5,424,075号、および同第5,510,385号に記述されており、これらの内容は参照として組み込まれる。
【0075】
本発明の他の特定の実施形態において、少なくとも1つの活性成分は、アセトアミノフェン、アセチルサリチル酸、イブプロフェン、ナプロキセン、ケトプロフェン、フルルビプロフェン、ジクロフェナク、シクロベンザプリン、メロキシカム、ロフェコキシブ(rofecoxib)、セレコキシブ(celecoxib)、ならびに、医薬品に許容可能なこれらの塩、エステル、異性体、および混合物から選択されてもよい。
【0076】
本発明の他の特定の実施形態において、少なくとも1つの活性成分は、偽性エフェドリン、フェニレフリン、フェニルプロパノラミン、クロルフェニラミン、デキストロメトルファン、ジフェンヒドラミン、クロフェジアノール(clofedianol)、アステミゾール、テルフェナジン、フェキソフェナジン(fexofenadine)、ロラタジン(loratadine)、デスロラタジン、セチリジン(cetirizine)、およびこれらの混合物、ならびに、医薬品に許容可能なこれらの塩、エステル、異性体、および混合物から選択されてもよい。
【0077】
特定の実施形態において、樹脂複合体に結合された活性成分は、フェニレフリン、偽性エフェドリン、デキストロメトルファン、ジフェンヒドラミン、クロルフェニラミン、およびこれらの混合物から選択される。ある一つの実施形態において、樹脂をベースとする粒子は、フェニレフリン、偽性エフェドリン、デキストロメトルファン、ジフェンヒドラミン、クロルフェニラミン、およびこれらの混合物の塩酸塩および臭化水素酸塩を用いて結合される。
【0078】
他の特定の実施形態において、少なくとも1つの活性成分は、NSAIDまたは医薬品に許容可能なその塩であり、薬樹脂複合体に結合されるその他の活性成分は、フェニレフリンおよび/または偽性エフェドリン(psuedoephedrine)である。
【0079】
ある一つの実施形態において、活性成分または複数の成分の治療上有効な量は、「単位用量容積(unit dose volume)」で示されてもよく、これは、粉末または水性懸濁液の形態の場合もある。本明細書中で使用されている「治療上有効な量」は、経口投与に対して所望の治療学的応答を生み出す活性成分の量である。当業者は、例えば、投与されている特定の活性成分;活性成分の生物学的利用能特質;所望の用量療法;患者の年齢および体重などといった因子を考慮することにより、所定の患者に対しての活性成分の「治療上有効な量」を用意に決定することができる。本明細書中で使用されている「単位用量容積」は、患者にある用量の所定の製品を経口的に投与するのに都合のよいあらゆる容積であり得る。
【0080】
この実施形態において、「単位用量容積」は、典型的には投薬指導が付随し、これは、例えば、患者の年齢または体重に依存する複数回の単位用量容積であってもよい活性成分の量を患者に服用するように指示する。典型的には、懸濁液の単位用量容積は、最小の患者に対して治療上有効である量の活性成分を含むであろう。例えば、好適な単位用量容積は、小さじ一杯(約5mL)、大さじ一杯(約15mL)、一滴(one dropper)、または1ミリリットルを含み得る。
【0081】
本発明に従うと、NSAIDを含む剤形は、特定の痛み軽減処置で、拡張された時間の期間、例えば約4時間または約6時間の期間にわたって血中に活性成分を放出する一回の投与で、処置が必要なほ乳類に提供されてもよい。ゼロ時点で、速効性放出用量部分中の活性成分を介して、最初の用量のNSAIDがほ乳類に提供される、すなわち、投与される。次に、第2の活性成分が、改変型放出用量部分中の活性成分を介して、活性成分を含む調合物の最初の投与から次の約4〜約6時間の間にわたって血中に放出される。言い換えると、調合物は、最初の投与から約4または約6時間後には、溶解されていない第2の活性成分をまだ保持している。
【0082】
本発明を実施するにあたって、剤形は、活性成分の全重量に基づいて、約0.01パーセント〜約40パーセントの第1の活性成分の速効性放出用量粒子部分と、約0.01パーセント〜約40パーセント、すなわち、約.01パーセント〜約10パーセントの二重コーティングされた第2の活性成分の改変型放出用量部分とから構成されてもよい。本明細書中で使用されている「部分(portion)」は、あらゆるオプションの構成成分と共に同定されている活性成分の総計を意味するが、液体媒体、あるいは、固体剤形の場合には速効性放出用量粒子が結合され得るマトリックスまたはその他の乾燥媒体を含まないものとする。速効性放出用量部分および改変型放出用量部分は、適切な媒体と結合されて、1)必要な場合には即席で懸濁されることができる乾燥混合物、2)エリキシル、または懸濁液のようにすぐ使用できる液体剤形、または3)固体または半固体剤形のうちのいずれかを形成してもよい。
【0083】
媒体の好適な構成物は、限定することなく、溶媒;構造化剤(structuring agents);膨潤剤;界面活性物質;糖;緩衝物質、例えば、クエン酸、およびクエン酸ナトリウム;グリシン、ならびに塩化水素酸、リン酸ナトリウム、およびリン酸カリウム;保存剤、および細菌発育阻止剤、例えばp−ヒドロキシ安息香酸のエステル;着色料;ならびに、医薬品中で一般に使用される種々の調味および甘味料を含むことができる。
【0084】
好適な甘味料の例としては、限定されるものではないが、あらゆる既知の甘味付け剤、例えば、糖、糖アルコール、高強度甘味料(high intensity sweeteners)、およびこれらの混合物を含む。好適な糖としては、限定されるものではないが、スクロース、デキストロース、高フルクトーストウモロコシシロップ(high fructose corn syrup)、およびマルトースを含む。好適な糖アルコールとしては、限定されるものではないが、ソルビトール、キシリトール、およびマンニトールを含む。好適な高強度甘味料としては、限定されるものではないが、スクラロース(sucralose)、アスパルテーム、サッカリン、およびアセスルファムK(acesulfame K)を含む。
【0085】
ある一つの実施形態において、有効量の緩衝剤が、液体懸濁液剤形の改変型放出部分中に含まれる少なくとも1つの活性成分のpKaを液体懸濁液剤形の全体のpHよりも大きくさせるために使用される。
【0086】
加えて、媒体は、水から構成されてもよいし、あるいは、水と、例えばグリコール、アルコール、およびグリセロールといった本技術分野で既知の医薬品に許容可能な水混和性共溶媒との混合物から構成されてもよい。
【0087】
ある実施形態において、剤形は、本技術分野において既知のあらゆる懸濁系、例えば、典型的には、1つ以上の構造化剤および/または1つ以上の膨潤剤を含むものを含んでもよい。ある一つの実施形態において、剤形は、液体懸濁液剤形の全重量に基づいて、約0.1パーセント〜約10パーセントの懸濁系を含む。好適な懸濁系は、例えば、米国特許第5,374,659号、同第5,621,005号、および同第5,409,907号で開示されているものを含み、これらの特許文献は全て、その全体が本明細書中に参照により組み込まれる。
【0088】
本発明での使用のために好適な構造化剤は、親水コロイドといった親水性ポリマーを含む。好適な親水コロイドの例としては、アルギナート、寒天、ガーゴム、イナゴマメ(locust bean)、カラゲナン、タラ(tara)、アラビアゴム、トラガカントゴム、ペクチン、キサンタン、ゲル化剤、マルトデキストリン(maltodextrin)、ガラクトマンナン、プスツラン(pusstulan)、ラミナリン、スクレログルカン(scleroglucan)、アラビアゴム、イヌリン、カラヤ、ウェラン(whelan)、ラムサン(rhamsan)、ズーグラン(zooglan)、メチラン(methylan)、キチン、シクロデキストリン、キトサン、およびこれらの組み合わせを含む。本発明のある実施形態において、キサンタンガムは、構造化剤である。
【0089】
キサンタンガムは、高分子量の天然炭水化物であり、より詳細には、多糖類である。本発明での使用のために好適なある一つのキサンタンガムは、Xanthomonas campestrisにより生産される高分子量の多糖類である。この多糖類を生み出すための技術および菌株は、米国特許第4,752,580号および同第3,485,719号に記述されており、その開示は参照として本明細書中に組み込まれる。ある一つの実施形態において、キサンタンガムは、LV model Brookfield Synchro-Lectric viscometer at 60 rpm, no. 3 spindleにより、25℃で測定されたときに、1パーセント塩溶液中、約1000〜約1700cP(mPa−sec)の粘度を有し得る。好適なキサンタンガムは、例えばCP Kelcoから商品名「Keltrol」、「Keltrol TF」、および「Keltrol 1000」で入手可能である。
【0090】
膨潤剤は、適切な水性環境に曝されると、ネットワーク系を形成することなく膨張する。アルファ化でんぷんは、特に優れた膨潤剤である。「インスタント化」でんぷんとしても知られるアルファ化でんぷんは、冷水に加えられると直ちに膨潤して肥厚し始めるように下ごしらえされている。ある一つの特に好適なアルファ化でんぷんは、改変され、安定化されていて、ろう様のトウモロコシ食品でんぷんから調製され、National Starch Companyから「INSTANT STARCH, ULTRASPERSE-M」として商業的に入手可能である。その他の好適な膨潤剤としては、限定されるものではないが、微結晶性セルロースおよび/またはヒドロキシプロピルメチルセルロースを含む。
【0091】
ある一つの実施形態において、懸濁系は、アルファ化でんぷん膨潤剤と共にキサンタンガム構造化剤から構成される。他の実施形態において、懸濁系は、液体懸濁液剤形の全重量に基づいて、約0.01パーセント〜約1パーセント、または約0.05パーセント〜約0.40パーセントのキサンタンガムと、約1パーセント〜約10パーセント、または約0.5パーセント〜約3.0パーセントのアルファ化でんぷん(例えば、National Starch Companyから商品名「INSTANT STARCH, ULTRASPERSE-M」で商業的に入手可能)とから構成される。
【0092】
剤形が液体形態、例えば懸濁液またはエリキシルである実施形態において、液体剤形のpHは、その中に含まれるコーティングされていない活性成分の溶解度を最小限にし、かつ化学的安定度を最大限にするよう最適化されるべきである。コーティングされていない活性成分が塩基性、例えば炭酸カルシウムである、ある一つの実施形態において、剤形のpHは、塩基性のコーティングされていない活性成分のpKaよりも2pH単位だけ上になるべく近づくようにされ得る。コーティングされていない活性成分が酸性、例えばイブプロフェンである実施形態においては、剤形のpHは、酸性のコーティングされていない活性成分のpKaの2pH単位だけ下になるべく近づくようにされ得る。コーティングされていない活性成分としてイブプロフェンを用いるある実施形態において、剤形のpHは、約1.0〜約5.0、例えば約1.0〜約4.0の範囲にされ得る。
【0093】
剤形は、既知のpH調節剤を用いて緩衝化され、所望のpH範囲内に懸濁液のpHを維持することができる。好適なpH−調節剤は、所望の程度のpH緩衝を提供するのに十分な量で剤形中に存在し得る。pH調節剤は、典型的には、剤形100mL当たり、約0〜約1グラムの範囲で存在するであろう。
【0094】
コーティングされていない活性薬剤としてイブプロフェンを含み、かつ例えばカルボキシメチルセルロースナトリウム(sodium carboxymethylcellulose)といったアルカリ性ポリマーを有する懸濁系を含む実施形態において、pH調節剤は、例えば、クエン酸、リンゴ酸、グルタミン酸など、味覚がマスクされた経口懸濁液での使用のために許容可能な味覚特質を有する弱有機酸から選択され得る。
【0095】
ある一つの実施形態において、剤形はオプションで、剤形の所望のpH範囲内で活性を有する抗菌性保存剤を含んでもよい。そのような医薬品懸濁液中で有用な保存剤は、限定されるものではないが、安息香酸ナトリウム、ソルビン酸カリウム、エデト酸の塩(salts of edetate)(エチレンジアミン四酢酸、すなわちEDTAとも知られていて、例えばエデト酸二ナトリウム)およびパラベン(parabens)(例えば、メチル、エチル、プロピル、およびブチルp−ヒドロキシ安息香酸エステル)を含む。上述で列挙されている保存剤は、例示的なものであるが、各保存剤は、保存剤の適合性および効力を保証するために、各調合物において、実験的な根拠により評価されなれければならない。医薬品調合物中の保存剤の効力を評価するための方法は、当業者に既知である。
【0096】
あるオプションの実施形態において、本発明の剤形は、ある疎水性活性薬剤の分散を補助するために湿潤剤として使用するための界面活性物質を利用してもよい。その他のある実施形態において、本発明の剤形は、界面活性物質を実質的に含まなくてもよい。ここで使用されている「界面活性物質を実質的に含まない」とは、懸濁液が、約0.1%未満、例えば約0.05%未満の界面活性物質を含むことを意味しているものとする。好適な界面活性物質の例としては、限定されるものではないが、ソルビタンオレイン酸エステル、例えば、polysorbate 80としても知られているポリオキシエチレンソルビタンモノオレイン酸塩(polyoxyethylene sorbitan monooleate)を含む。
【0097】
ある一つの実施形態において、剤形は、水性医薬品懸濁液組成物の形態にあり、水性医薬品懸濁液の容量当たり(w/vまたはg/100mL)の活性成分の全重量に基づいて、約0パーセントを超え約30パーセント以下、例えば、約0.05パーセント〜約20パーセント、または約0.5パーセント〜約10パーセント、または約0.5パーセント〜約5パーセントの第1の活性成分と、約0パーセントを超え約10パーセント以下、例えば、約0.01パーセント〜約10パーセント、または約0.03パーセント〜約5パーセントの第2の改変型放出活性成分とから構成される。
【0098】
ある実施形態においては、第1の活性成分はイブプロフェンであり、懸濁液剤形の速効性放出部分中の第1の活性成分の量は、水性懸濁液剤形の容量当たり(w/v)の第1の活性成分の全重量に基づいて、小さじ一杯の水性懸濁液剤形当たり、約25〜約400mg、例えば、約50mg〜約200mgであるか、または1mLの水性懸濁液剤形当たり約20mgの第1の活性成分であり、この量は、水性懸濁液剤形の容量当たり(w/v)の第1の活性成分の全重量に基づいて、約0.25パーセント〜約4パーセントに等しく、また、第2の活性成分は、フェニレフリンまたは偽性エフェドリンであり、懸濁液剤形の改変型放出部分中の第2の活性成分の量は、水性懸濁液剤形の容量当たり(w/v)の第2の活性成分の全重量に基づいて、小さじ一杯の水性懸濁液剤形当たり約1mg〜約20mg、例えば、約1mg〜約10mgであるか、または1mLの水性懸濁液剤形当たり、約1.5mgの第2の活性成分であり、この量は、水性懸濁液剤形の容量当たり(w/v)の第2の活性成分の全重量に基づいて、約0.01パーセント〜約0.3パーセントに等しい。
【0099】
本発明のある一つの実施形態は、液体の測定可能な懸濁液組成物に向けられており、この組成物は、懸濁液の全重量に基づいて、a)約0.05パーセント〜約40パーセントの第1の速効性放出活性成分と、b)約20パーセント〜約80パーセントの水と、c)約0.1パーセント〜約10パーセントの懸濁系と、d)約0パーセント〜約40パーセント、例えば約20パーセント〜約40パーセントの甘味付け剤と、e)約0パーセント〜約0.5パーセントの賦形剤と、約0.01パーセント〜約_10パーセントの改変型放出粒子の第2の部分とを含む。
【0100】
他の実施形態において、本発明の剤形は、剤形の全重量に基づいて、約0.1パーセント〜約10パーセント、例えば、約0.1〜約5パーセントの第1の速効性放出部分と、約0.05パーセント〜約10パーセント、例えば、約0.05パーセント〜約5パーセントの第2の改変型放出部分とを含む。ある一つの実施形態において、剤形の第2の改変型放出部分は、第2の部分の全乾燥重量に基づいて、約5パーセント〜約80パーセント、例えば、約5パーセント〜約70パーセントの第1の半透性コーティング層と、約10パーセント〜約90パーセント、例えば、約10パーセント〜約80パーセントの第2の保護コーティング層と、約1パーセント〜約50パーセント、例えば、約1パーセント〜約30パーセントの薬−樹脂複合体粒子とから構成される。
【0101】
本発明に従うと、剤形は、剤形の全重量に基づいて、約0.1パーセント〜約5パーセント、例えば、約0.5パーセント〜約3パーセントの第1の活性成分と、約0.005パーセント〜約1パーセント、例えば、約0.01パーセント〜約0.5パーセントの第2の活性成分とを含有する。
【0102】
ある実施形態において、本発明の懸濁液の粘度は、Brookfield DV-I+ Viscometerにより、# 31 spindleを用いて、12rpmの速度で、約25℃の温度条件下測定されたときに、約400cps〜約1500cpsの範囲にわたり得る。
【0103】
本発明の剤形は、1回の投与で、有効量の第1の活性成分、例えばNSAIDなどATDAIRDが約5である有効量の第1の活性成分と、同じ剤形中で、フェニレフリンまたは偽性エフェドリン(psuedoephedrine)などATDAIRDが約3である有効量の第2の活性成分とを送達するように意図されており、両方の活性成分は、より長いATDAIRD期間にわたって剤形から放出されることができる。
【0104】
「有効量」の鎮痛薬とは、患者における痛みからの軽減をもたらすものである。例えば、イブプロフェンの典型的な成人用量は、4〜6時間毎とすれば患者の体重kg当たり約2.9〜約12mgの範囲にわたり、典型的な一日量は約11.6〜約72mg/kg/日の範囲にわたり得る。従って、典型的な70kgの成人に対する有効量のイブプロフェンの投与は、例えば40mg/mLのイブプロフェンを含有する本発明の調合物約5mL〜約60mLを一日に1回または2回投与することを含み得る。典型的な小児科用量のイブプロフェンは、4〜6時間毎とすれば約5〜約10mg/kgの範囲にわたり、典型的な一日量は約20〜約60mg/kg/日の範囲にわたり得る。典型的な15kgの子供に対する有効量のイブプロフェンの投与は、例えば20mg/mLのイブプロフェンを含有する本発明の調合物約5mL〜約60mLを一日に1回または2回投与することを含み得る。
【0105】
「有効量」の鬱血除去薬とは、鬱滞の効果的な軽減を提供するもの、すなわち、腫脹を減らすことにより、鼻の通路および/または洞などの、鬱滞を解消する薬物である。例えば、フェニレフリンの典型的な成人用量は、6時間毎として患者の体重kg当たり約0.14〜約0.29mg、または、約0.60〜約1.0mg/kg/日の範囲にわたる典型的な一日量で、典型的な成人について6時間毎として約10〜約20mg、または、典型的な成人について、約0.86mg/kg/日もしくは1日当たり約60mgのフェニレフリンの範囲にわたり得る。したがって、典型的な70kgの成人に対する有効量のフェニレフリンの投与は、例えば3mg/mLのフェニレフリンを含有する、本発明の調合物約2.5mL〜約10mL、または約5mLを、一日に1〜4回投与することを含み得る。フェニレフリンの典型的な小児科用量は、2〜4時間毎として約0.25〜約0.75mg/kg、または、約1.0〜約2.7mg/kg/日の範囲にわたる典型的な一日量で、典型的な子供について6時間毎として約3.75〜約11.25mg、または、典型的な子供について、約2mg/kg/日もしくは1日当たり約30mgのフェニレフリンの範囲にわたり得る。典型的な15kgの子供に対する有効量のフェニレフリンの投与は、例えば1.5mg/mLのフェニレフリンを含有する、本発明の調合物約2.5mL〜約10mLを、一日に1〜4回投与することを含み得る。
【0106】
ある一つの実施形態において、本発明の剤形の経口投与は、剤形から第2の活性成分を放出し続け、これにより、その治療学的効果の持続時間が、第1の活性成分の持続時間に匹敵するようになる改変型放出用量で、NSAIDといった第1の活性成分を使用者に提供する。
【0107】
ある一つの実施形態において、液体剤形は、イブプロフェンおよびフェニレフリンの両方を含有する速効性放出部分と、追加量のフェニレフリンを含有する改変型放出部分とを含む。この実施形態において、イブプロフェンの速効性放出用量は、懸濁液の約25mg/5mL〜約200mg/5mL、例えば懸濁液の約50mg/5mL〜約200mg/5mLの範囲にわたり得、フェニレフリンの速効性放出用量は、懸濁液5mL当たり約2.5mg〜約15mgの速効性放出フェニレフリン、例えば懸濁液5mL当たり約2.5mg〜約10mgの速効性放出フェニレフリンの範囲にわたり得る。この実施形態において、フェニレフリンの改変型放出用量は、懸濁液5mL当たり約2.5mg〜約20mgの改変型放出フェニレフリン、例えば懸濁液5mL当たり約5〜約15mgの改変型放出フェニレフリンの範囲にわたり得る。
【0108】
本発明の他の実施形態は、偽性エフェドリンまたはフェニレフリン(phenylepherine)の経口投与により、鼻および呼吸器の鬱滞の緩和が必要な人物のそのような鬱滞を緩和すると共に、そのような人物に本発明の対象の剤形を投与することにより、頭痛、関節痛、病的に分泌物の多い鼻通路の状態(watery nasal passages)、流涙(weeping eyes)、洞の鬱滞および痛み、咳嗽、粘液の過剰な浸出、および気管支炎といった関連する状態をも回復させる方法に向けられている。
【0109】
有利なことに、我々は、剤形が、懸濁液のような液体剤形として設計されているか、または投与前に水で再構成されることができる乾燥剤形として設計されているかにかかわらず、製品の保管寿命の間、および処置の期間の間ずっと剤形の改変型放出部分の放出特質を効果的に安定化する方法を予想外にも見出した。特に、我々は、摂取前の製品中の粒子からの活性成分の放出を防止しながら、一方で、g.i.液中でのそれらの同じ粒子からの活性成分の改変型放出を可能にするという難題を克服した。
【0110】
我々はさらに、第1の活性薬剤と第2の活性薬剤上の半透性コーティングとの間の相互作用を克服することにより、第2のコーティングされた活性薬剤の治療学的効果の持続時間を、コーティングされていない第1の活性薬剤が保持するのに匹敵する持続時間まで延長させる方法を見出した。
【0111】
有利には、本発明の調合物は、例えば、(i)正確に測定可能な単一用量乾燥調合物、または液体懸濁液、(ii)必要に応じて再懸濁されるように異なる量の顆粒を測定することにより得ることができる有意な用量柔軟性を有する、複数用量の顆粒状の調合物、(iii)複数用量の液体懸濁液、および(iv)特に小児科施用で有用な、活性成分が懸濁した濃縮されたドロップを含めた、様々な形式で使用され得る。
【0112】
加えて、調合物は、投与し、嚥下するのに都合がよく、一日量の活性成分の数が減るので、全患者適応性(overall patient compliance)が達成される。嚥下および投与の容易性により、小児科での実施におけるさらなる利点が予測される。
【0113】
下記の実施例がさらに本発明を説明するが、本発明をいかなるようにも限定することを意味するものではない。
【0114】
<例1:フェニレフリン−樹脂複合体粒子の調製>
好適なステンレス鋼容器中で、2125.0gの塩酸フェニレフリンを、2000.0gの脱イオン水に溶解した。これに対して、Rohm & Haas Corporationから商業的に入手可能なAmberlite IRP-69イオン交換樹脂を2500.0g加え、実験室ミキサーを用いて100RPMで少なくとも12時間、攪拌した。結果として得られたスラリーを、減圧ろ過を用いてWattman #4ろ紙を通してろ過した。結果として得られた固形分を1〜2時間風乾し、湿った樹脂粒子を産生し、この樹脂粒子を次に、Glatt流動層GPCG-1ドライヤーで10分間、50℃で乾燥した。結果として得られた乾燥粒子は、約32.0%のフェニレフリンを含んでいた。残った結合していないフェニレフリンが、ろ過されたスラリー溶液中に残っていた。
【0115】
<例2:エチルセルロース半透性第1コーティング溶液の調製>
Dow Chemical Corporationから商品名「Ethocel 10 CPS」で商業的に入手可能なエチルセルロース690グラムと、アセチルクエン酸トリブチル(acetyltributyl citrate)(ATBC)150gと、ステアリン酸マグネシウムUSP20mgとを、周囲条件下で、溶媒の全重量に基づいて3780gのアセトンと3780gのイソプロピルアルコールとを含む溶媒(50:50混合物)中に分散させることにより、コーティング溶液を調製した。この溶液を、実験室ミキサーを用いて75RPMで少なくとも60分間、混合した。
【0116】
結果として得られたコーティング溶液は、湿った全コーティング溶液に基づいて、8.19%のエチルセルロースと、1.78%のアセチルクエン酸トリブチルと、0.24%のステアリン酸マグネシウムと、44.89%のアセトンと、44.89%のイソプロピルアルコールとを含んでいた。この溶液は、10%の固形分を含んでいた。固形分の相対量は、乾燥したコーティング溶液の全重量パーセントに基づいて、エチルセルロース80.23%、ATBC17.44%、およびステアリン酸マグネシウム2.33%であった。
【0117】
<例3:保護用(腸溶性)第2コーティング溶液の調製>
Rohm Pharmaから商業的に入手可能な、商品名「Eudragit L30D-55」で商業的に入手可能なメタクリレートコポリマー分散系(30%固形分)4193.1gを、精製水2396.0g中に分散させることによりコーティング溶液を調製し、周囲条件下で、5分間25RPMで混合した。これに対して、50.8gのモノステアリン酸グリセロールと、126.8gのクエン酸トリエチルとを、50RPMで少なくとも30分間混合しながら加えた。
【0118】
結果として得られたコーティング溶液は、湿った全コーティング溶液に基づいて、61.97%のEudragit L30D-55(Eudragit L30D-55の30%が固形分)、0.75%のモノステアリン酸グリセロール、1.87%のクエン酸トリエチル、および35.41%の精製水を含んでいた。
【0119】
固形分の相当量は、乾燥したコーティング溶液の全重量パーセントに基づいて、Eudragit L30D-55が87.63%、モノステアリン酸グリセロールが3.53%、クエン酸トリエチルが8.83%であった。
【0120】
<例4:単一の半透性エチルセルロース層でコーティングされた薬−樹脂フェニレフリン複合体の調製>
例1から得られた薬−樹脂複合体粒子1000.0グラムを、Glatt GPCG-1/3コーティングユニットに配置し、例2に従って調製されたエチルセルロースコーティング溶液を約37〜42℃の製品温度条件下で、約15.0g/分の速度で、約2バール(約200kPa)の霧化気圧で噴霧することにより、この溶液でコーティングした。結果として得られたコーティングされたフェニレフリン顆粒は、コーティングされたフェニレフリン顆粒の全乾燥重量に基づいて、約47.9%の半透性コーティングを含んだ。
【0121】
<例5:半透性層および外側腸溶性層でコーティングされた薬−樹脂フェニレフリン複合体の調製>
例4に従って調製されたコーティングされたフェニレフリン粒子750.0グラムを、Glatt GPCG-1/3コーティングユニットに配置し、例3に従って調製された腸溶性Eudragit L30Dコーティング溶液を、約54〜71℃の製品温度条件下で、約15.0g/分の速度で、約2バール(約200kPa)の霧化気圧で噴霧することにより、この溶液でコーティングした。結果として得られたコーティングされたフェニレフリン顆粒は、二重コーティングされたフェニレフリン顆粒の全乾燥重量に基づいて、約65.7%の外側保護用腸溶性コーティングを含んだ。最終的な乾燥した二重コーティングされた粒子中の成分の量を表1に示す。
【表1】

【0122】
<例6:速効性放出イブプロフェン用量を含む懸濁液ベースの調製>
【表2】
【0123】
上記の表2に示されているように、1300.0gの精製水USPをScott Turbon高速せん断ミキサー(Scott Turbon high shear mixer)を備えた混合用タンクに入れ、良好なボルテックスを生み出すために約500rpm〜約1000rpmで混合した。次に、この混合用タンクに、アルファ化でんぶんとキサンタンガムとを加え、20分間混合した。これに対して、次にグリセリンを加え、5分間混合した。これに対して、次にスクロースを加え、12分間混合した。ポリソルベート−80NF、クエン酸無水物USP、およびアセスルファムKを次々と加え、その後、結果として得られた混合物を10分間混合した。イブプロフェンUSPをバッチに加え、約500rpmおよび1000rpmで20分間混合した。これに対して次に、残りの精製水を加え、10分間混合した。
【0124】
<例7:速効性放出イブプロフェン用量および単一半透性層でコーティングされたフェニレフリン−樹脂複合体を含む懸濁液の調製>
例4に従って調製されたコーティングされたフェニレフリン粒子600mgを、好適な100mLメスフラスコに加え、例6に従って調製された懸濁液ベースで100.0mL容量まで希釈し、その後、結果として得られる懸濁液が視覚的に均一になるまで、手動でくるくると回転させて(end-over-end)混合した。結果として得られた懸濁液は、100mg/5mLの速効性放出イブプロフェン用量と、5mg/5mLのフェニレフリン用量とを含んだ。
【0125】
<例8:速効性放出イブプロフェン用量ならびに半透性層および外側保護用腸溶性層でコーティングされたフェニレフリン−樹脂複合体を含む懸濁液の調製>
例5に従って調製された、5.7%の活性フェニレフリンHClを含むコーティングされたフェニレフリン1724.0mgを、好適な100mLメスフラスコに加えた。次に、結果として得られた懸濁液を、例6に従って調製された懸濁液ベースで100.0mL容量まで希釈し、結果として得られる懸濁液が均一になるまで手動でくるくると回転させて混合した。結果として得られた最終的な懸濁液は、100mg/5mLの速効性放出イブプロフェン用量と、5mg/5mLの改変型放出フェニレフリン用量とを含んだ。
【0126】
<例9:例7および例8の懸濁液の溶解分析>
溶解媒質:
pH1(0.1N HCl)媒質:1000mLの0.1N HClをパドル付きのUSP Type II装置の3つの容器それぞれに配置した。次に、例7で生成された最終的な懸濁液のサンプル5.0mLを3つの容器のそれぞれに独立して加え、混合物が視覚的に均一になるまで50r.p.m.の速度で、37℃で混合した。
【0127】
例7から得られたサンプルを例8から得られたサンプルに置換して、この手順を繰り返した。
【0128】
例4から得られた粒子約20.5mg、および例5から得られた粒子約133.0mgでそれぞれ置換して、この手順をさらに2回繰り返した。これらは、約4.1mgの用量の遊離塩基(free base)としてのフェニレフリンに基づいて算出した。
【0129】
1時間pH変化媒質:750mLの0.1N HClを、パドル付きのUSP Type II装置の3つのさらなる容器のそれぞれに配置した。次に、例7で生成された最終的な懸濁液のサンプル5.0mLを3つの容器のそれぞれに独立して加え、混合物が均一になるまで50r.p.m.の速度で、37℃で混合した。1時間後、サンプル10mLを器から取り出し、次に、これに対して0.2MのNa3PO4を250mL加え、これにより、媒質組成を約6.8のpHで0.05Mのリン酸ナトリウム緩衝剤約990mLへと変化させた。
【0130】
例7から得られたサンプルを例8から得られたサンプルに置換して、この手順を繰り返した。
【0131】
次に、例4から得られた粒子約20.5mg、および例5から得られた粒子約133.0mgでそれぞれ置換して、この手順をさらに2回繰り返した。これらは、約4.1mgの用量の遊離塩基としてのフェニレフリンに基づいて算出した。
【0132】
pH7.2(リン酸緩衝)媒質:NaOHでpH7.2に調節された0.05MのKH2PO4緩衝剤1000mLを、パドル付きのUSP Type II装置の3つのさらなる容器にそれぞれ配置した。次に、例7で生成された最終的な懸濁液のサンプル5.0mLを3つの容器のそれぞれに独立して加え、混合物が視覚的に均一になるまで50r.p.m.の速度で、37℃で混合した。
【0133】
例7から得られたサンプルを例8から得られたサンプルに置換して、この手順を繰り返した。
【0134】
次に、例4から得られた粒子約20.5mg、および例5から得られた粒子約133.0mgでそれぞれ置換して、この手順をさらに2回繰り返した。これらは、約4.1mgの用量の遊離塩基としてのフェニレフリンに基づいて算出した。
【0135】
サンプリングおよび分析:
その後、それぞれ1、2、3、4、6、7、および8時間後、懸濁液/緩衝剤混合物のサンプル10mLをそれぞれの容器から独立して取り出した。
【0136】
1、2、3、4、6、7、および8時間それぞれでのフェニレフリンおよびイブプロフェンについての溶解曲線を導き出すために、波長270nmに設定された、Waters(登録商標)Alliance 2695分離モジュールおよびWaters(登録商標)2996 PDA検出器を備えた高圧液体クロマトグラフ(HPLC)を用いてイブプロフェンおよびフェニレフリンの含量について、各サンプル10mLを独立して分析した。溶解サンプルのそれぞれを、フェニレフリン(遊離塩基)を0.004mg/mLで含み、イブプロフェン(遊離酸)を0.10mg/mLで含む、フェニレフリン遊離塩基およびイブプロフェン遊離酸の100%放出に必要とされる理論濃度に相関した混合された標準と比較した。
【0137】
HPLCで使用された移動層は、0.2%H3PO4/メタノール/アセトニトリル(40/35/25)中の20mMのドデシル硫酸ナトリウム(SDS)を用いて調製した。注入量を50μLとし、運転時間を約10分間とし、ポンプ流量(pump flow)を1.0mL/分とした。分析のために用いたカラムは、Phenomenex Luna C8(2)(3μm、4.6x75mm)であった。
【0138】
HPLCの結果を図1〜4に示す。この図では、3つの器中に溶解されたフェニレフリンのパーセントの平均を表す点がそれぞれグラフ化されている。
【0139】
図1および図3の比較により、エチルセルロースでコーティングされた粒子がイブプロフェン含有懸濁液に配置されると、この粒子からのフェニレフリンの放出速度が実質的に増加したことが示された。対照的に、図2および図4の比較により、エチルセルロースの内側層と腸溶性ポリマーの外側層とでコーティングされた粒子がイブプロフェン含有懸濁液に配置されると、この粒子からのフェニレフリン放出速度は実質的にそのままであったことが示された。
【図面の簡単な説明】
【0140】
【図1】3つの異なる媒質中での時間に対する例4に従って調製されたエチルセルロース半透性コーティング層でコーティングされた粒子中のフェニレフリンの放出された(すなわち、溶解)パーセンテージを示している。
【図2】3つの異なる媒質中での時間に対する例5に従って調製されたエチルセルロース半透性コーティング層および外部腸溶性コーティング層でコーティングされた粒子中のフェニレフリンの放出された(すなわち、溶解)パーセンテージを示している。
【図3】時間に対する例7に従って調製された速効性放出イブプロフェンを含む懸濁液中のエチルセルロースコーティング層でコーティングされた粒子中のフェニレフリンの放出された(すなわち、溶解)パーセンテージを示している。
【図4】時間に対する例8に従って調製された速効性放出イブプロフェンを含む懸濁液中のエチルセルロースコーティング層および外部腸溶性コーティング層でコーティングされた粒子中のフェニレフリンの放出された(すなわち、溶解)パーセンテージを示している。
【開示の内容】
【0001】
本発明は、少なくとも2つの活性成分の投与のための液体剤形に適している改変型放出医薬品調合物に関する。より詳細には、この剤形は、同様の期間にわたって血漿中に含まれる全ての活性成分の医薬品に好適な血漿濃度を提供する速度で、活性成分を放出する。
【0002】
〔発明の背景〕
痛み、炎症、および発熱を処置するための治療薬としては、鎮痛薬、抗炎症薬(anti-inflammatories)、および解熱薬を含む。非ステロイド性抗炎症性薬(Non-steroidal anti-inflammatory drug)(NSAID)は、そのような治療薬の一種である。NSAIDとしては、プロピオン酸誘導体、酢酸誘導体、フェナム酸誘導体(fenamic acid derivatives)、ビフェニルカルボン酸誘導体(biphenylcarbodylic acid derivatives)、オキシカム(oxicams)、およびシクロオキシゲナーゼ−2(COX−2)選択性NSAIDを含む。
【0003】
プロピオン酸誘導体としては、例えば、イブプロフェン、ナプロキセン、およびケトプロフェンを含む。イブプロフェンは特に、広く使用されていてよく知られている、鎮痛特性および解熱特性を持つNSAIDである。イブプロフェンは、何年もの間、多くの形態で大衆薬として商業的に入手可能となってきた。イブプロフェンは、2−(4−イソブチルフェニル)−プロピオン酸として化学的に知られている。
【0004】
速効性放出NSAIDは、典型的には、約4〜6時間毎に投与される。典型的には、NSAIDの一日量は、約50〜約2000ミリグラム、好ましくは約100〜1600ミリグラム、最も好ましくは約200〜約1200ミリグラムの範囲にわたる。
【0005】
多くの他の活性成分は、それらの比較的短い持続時間ゆえに、より頻繁に投与される。例えば、鬱血除去薬であるフェニレフリンの治療上有効な血漿濃度は、約2.5時間±.7時間であり、そのため、フェニレフリンは典型的には、2〜4時間毎に投与される。
【0006】
NSAIDと、持続時間がより短い、医薬品として好適な血漿濃度を有する他の活性成分とを含有する単一製品を投与するためには、後者の放出を制御する必要があるだろう。特には身体における薬のより長期間にわたる作用を提供するために、剤形から患者の胃腸(「g.i.」)液への薬または他の活性成分の放出の速度を減らすことがよく知られている。
【0007】
経口的に送達される薬が身体中のその作用部位に到達する速度は、g.i.粘膜を介した血中への薬の吸収の速度および程度を含めた多数の因子に依存する。しかしながら、薬が血中へと吸収されることができるより前に、薬はまずg.i.液中に溶解されなければならない。多くの薬にとって、g.i.膜を通る吸収は、g.i.液中へのそれらの溶解に比べて比較的速く、そのため、このことが薬の溶解を薬の吸収における速度制限ステップにしている。したがって、配合者は、薬の溶解速度を改変することにより血中への薬の吸収速度を効果的に制御することがある。
【0008】
薬の治療学的効力の開始および持続は広く変化し、それらの対応する吸収、分配、代謝、および排出もまた広く変化するので、異なる方法で異なる薬の放出を改変すること、あるいは、第1の薬が剤形から速効性で放出されるようにし、一方で第2の薬が「改変型」式、例えば遅延型または制御型のいずれかで放出されることが知られている。
【0009】
剤形が改変された速度で薬を送達することができるようにするよく知られたメカニズム(例えば、持続型、延長型、拡張型、または遅滞型の放出)としては、拡散、浸食、および浸透を含む。多くの場合、特定の活性成分に対して、特に望ましい改変型放出プロフィールを達成する上述のメカニズムの組み合わせを使用する剤形を設計することが実用的である。
【0010】
不都合なことに、多くの改変型放出施用は、最終的な大きいサイズおよび重さを有する固体投薬ユニットを用いる。そのような投薬ユニットの投与は、特に子供たちや高齢者などの嚥下が困難な患者たちに、問題を提示する。したがって、咀嚼可能か、もしくは経口的に崩壊可能な個体形態、または液体形態のいずれかの形態で改変型放出内服薬を提供することがさらに望ましい。多くの患者たちにとって、液体経口剤形は、さらなる咀嚼ステップ無しで嚥下されることができるため、より好ましいものである。
【0011】
経口液体形態は、速効性放出プロフィールで薬物を送達するために、長年、一般に使用されてきた。例えば、米国特許第5,374,659号;同第4,788,220号;同第4,975,465号;および同第5,183,829号を参照のこと。しかしながら、改変型放出薬物を液体剤形に組み込むことは、有意な調合物変化をもたらす。特に、コーティングされるか、または化学的に結合された粒子が典型的に、薬の改変型放出部分を保有するために用いられる。例えば、米国特許第5,980,882号は、薬の放出速度を遅くするために、キレート剤と共に薬−樹脂複合体を使用することを開示している。米国特許第4,847,077号は、延長型で連続的な薬の放出を提供するために、薬−樹脂複合体粒子上に設けられた水透過性拡散隔膜コーティングを使用することを開示している。
【0012】
そのような粒子の特性だけでなく、それらを懸濁するための液体媒体の特性も、粒子が均一に分散した状態に維持されることができるように、適合性でなければならない。特に難題は、患者による摂取の前の液体剤形の貯蔵寿命の間の懸濁された粒子から懸濁液の媒質中への薬の中途での放出を防ぐことである。加えて、液体剤形の保管寿命の間ずっと液体剤形の所望の溶解プロフィールと所望の用量均一性とを維持することが、経口用液体改変型放出懸濁液製品を処方するにあたって取り組まれるべき別の難題である。不都合なことに、多くの場合、例えば速効性放出のイブプロフェンと改変型放出の第2の活性成分(たとえば、フェニレフリン)とを含有する製品を処方するときに、イブプロフェンと本技術分野で既知の改変型放出コーティング剤との間の相互作用が原因で、これらの問題に遭遇する。
【0013】
米国特許出願第20060057205号は、フェニレフリンと鎮痛薬といった少なくとも第2の薬とを含む液体剤形を開示しており、この剤形は、イオン交換樹脂と共に、両薬の複合体の粒子を含んでおり、この粒子は、ポリメタクリレートといった改変型放出コーティングでコーティングされている。しかしながら、我々は、イブプロフェンといったコーティングされていないプロピオン酸誘導体は、半透性の改変型放出コーティング(たとえば、エチルセルロースおよびポリメタクリレートを含有するもの)と相互作用することがあることを発見した。有害なことに、この相互作用は多くの場合、コーティングされた薬の放出速度および意図された改変型放出特性を損なう。
【0014】
したがって、イブプロフェン粒子とフェニレフリンといった他の活性成分の改変型放出粒子とを含む改変型放出剤形であって、味が良いだけでなく、投与後の所望の放出プロフィールを保証する安定な形態にある、剤形を有することが望ましいであろう。特に、使用者に、速効性放出用量のイブプロフェンと持続型放出用量の第2の活性成分との両方を、イブプロフェンと持続型放出コーティングとの間の相互作用を起こすことなく、提供するような鎮痛薬製品を有することがさらに望ましいであろう。
【0015】
〔発明の概要〕
本発明は本書にて請求されているように、液体懸濁剤中のNSAIDSの投与のために好適な剤形のような医薬品剤形であって、かかる剤形は、
a)NSAIDといった第1の活性成分を含む第1の部分であって、この第1の活性成分は、剤形が溶解媒質に接触すると実質的に速効式に剤形から放出される、第1の部分と、
b)第2の部分であって、
i)第2の活性成分を有するイオン交換樹脂粒子であって、第2の活性成分は、第1の活性成分と同じでも、または異なっていてもよく、ある一つの実施形態ではフェニレフリンといったものであってもよく、イオン交換樹脂上に結合されていて薬−樹脂複合体粒子を形成している、イオン交換樹脂粒子、
ii)薬−樹脂複合体粒子のそれぞれを実質的に被覆している半透性コーティング層、および、
iii)ii)の粒子のそれぞれを実質的に被覆している保護コーティング層、
を含んでいる、第2の部分と、
を含む、からなる、および/または、から本質的になり、
第2の活性成分は、剤形が溶解媒質に接触すると改変型放出式に第2の部分から放出され、剤形の第2の部分から放出されたときの第2の活性成分の治療学的効果の持続時間は、第1の活性成分の治療学的効果の持続時間と実質的に同じである、医薬品剤形、ならびにその投与のための方法を提供する。
【0016】
〔発明の詳細な説明〕
本明細書中の説明に基づいて当業者が本発明をその十分な範囲まで利用することができると思料される。下記の詳細な実施形態は、例示としてのみ解釈されるべきものであって、開示のいずれもが、いかようにも限定的に解釈されるべきではない。
【0017】
他に定義されない限り、本明細書中で使用されている全ての技術用語および科学用語は、本発明が属する技術分野における当業者により一般に理解されているのと同じ意味を有する。また、本明細書中で言及されている全ての文献、特許出願、特許、およびその他の参照文献は、参照により組み込まれる。本明細書中で使用される全てのパーセンテージは、他に特定されない限り、重量%である。加えて、本明細書中で示されている全ての範囲は、2つの終点の間の値のあらゆる組み合わせをすべて含めたものを含むものと意味される。
【0018】
本明細書中で使用される用語「実質的に被覆する」または「実質的に連続的に」は、コーティングが概ね連続的で、コアまたは下層の全表面を概ね被覆していて、そのため、活性成分または下層のうちのわずかだけ露出するか、あるいは全く露出しないようになっていることを意味している。
【0019】
本明細書中で使用されている「ATDAIRD」は、有効な速効性放出用量」の特定の活性成分の作用の平均治療学的持続時間を意味するものとする。例えば、速効性放出用量のイブプロフェンまたはケトプロフェンの典型的な作用の持続時間、すなわち治療学的効果の期間は、約4〜約6時間である。したがって、イブプロフェンまたはケトプロフェンについてのATDAIRDは5時間である。速効性放出用量のナプロキセンの作用の典型的な持続時間は、約8〜約12時間である。したがって、ナプロキセンについてのATDAIRDは10時間である。速効性放出用量のフェニレフリンの作用の典型的な持続時間は、約2〜約4時間である。したがって、フェニレフリンについてのATDAIRDは3時間である。特定の活性成分の作用の治療学的持続時間は、特定の活性成分を含む速効性放出製品についてのラベルにおける投薬説明書から容易に決定されることができる。
【0020】
本明細書中で使用されている「改変型放出(modified release)」は、g.i.液といった溶解媒質中での活性成分の変更された放出または溶解に対して適用するものとする。改変型式で放出され得る活性成分または複数の成分は、例えば、剤形、コーティング、もしくは粒子、またはこれらのいずれかの部分、たとえば液体懸濁媒質の全体に分散した粒子といった部分の中に含まれていてもよい。改変型放出の種類としては、1)拡張型放出(extended release)、または2)遅延型放出(delayed release)を含む。全般的に、改変型放出剤形は、活性成分を、摂取後の時間の拡張された期間にわたって有用にするよう処方され、これにより、従来の剤形における同じ活性成分の投薬に比べて、投薬頻度を減らすことができる。改変型放出剤形は、活性成分の組み合わせを使用することも可能にし、このとき、ある一つの活性成分の持続時間は、他の活性成分の持続時間と異なってもよい。
【0021】
投与後、活性成分が実質的に連続的で調節された手法で剤形から放出され、剤形からの活性成分の完全な放出、すなわち枯渇のための時間が、同じものの速効性放出剤形に関連する時間よりも長いことが、「拡張型放出」により意味される。拡張型放出の種類としては、制御型、持続型、延長型、ゼロオーダーの放出などを含む。
【0022】
投与後、活性成分が剤形から放出されない時間の少なくとも1つの期間がある、すなわち、活性成分の放出が、経口投与後すぐではない時間に起こることが、「遅延型放出」により意味される。
【0023】
本明細書中で使用されている「溶解媒質」は、例えば、製品のテストのために使用されるインビトロの溶解媒質、または胃腸液といった、本発明の懸濁液剤形が溶解されることができるあらゆる好適な液体環境を意味するものとする。本発明の懸濁液剤形からの活性成分または複数の成分の溶解をテストするために使用される好適なインビトロの溶解媒質としては、本明細書中に参照により組み込まれるUSP23(1995)の第786頁に記述されているものを含む。
【0024】
本明細書中で使用されている「実質的にコーティングされた」は、粒子の表面積の約20%未満、例えば、約15%未満、または約1.0%未満が露出されている、例えば、所望のコーティングで被覆されていないことを意味するものとする。
【0025】
「腸溶性」は、約5.0を超えるか、約5.5を超えるか、または約6.0を超えるpH、あるいは腸管で見られるpHで溶解されることができることを意味するものとする。
【0026】
「液体剤形」は、非排他的に、懸濁液またはエリキシルを含んでもよく、この中で、活性成分の一つ以上が溶解されるか、部分的に溶解されるか、または、溶解されていない状態もしくは懸濁した状態にある。
【0027】
本明細書中で使用されている「薬−樹脂複合体」は、限定されるものではないが医薬品活性成分を含めた、いずれかの活性成分と、イオン交換樹脂との結合された形態を意味するものとする。薬−樹脂複合体はまた、本技術分野では「レジネート」とも称される。
【0028】
本明細書中で使用されている「速効性放出(immediate release)」は、少なくとも1つの活性成分の溶解特質がその活性成分を含有する速効性放出タブレットについてのUSP規格を満足することを意味する。速効性放出特性を有する活性成分は、活性成分の溶解を遅延または延長させることを意図することなく、胃腸内容物に溶解され得る。例えば、アセトアミノフェンタブレットの場合、USP24は、pH5.8リン酸緩衝液中、USP装置2(パドル)を50rpmで用いて、剤形に含まれるアセトアミノフェンの少なくとも80%が投薬後30分以内に剤形から放出されることを規定しており、イブプロフェンタブレットの場合には、USP24は、pH7.2のリン酸緩衝液中、USP装置2(パドル)を50rpmで用いて、剤形に含まれるイブプロフェンの少なくとも80%が投薬後60分以内に剤形から放出されることを規定している。USP 24, 2000 Version, 19〜20および856 (1999)を参照のこと。加えて、イブプロフェン懸濁液は、USP装置2(パドル)を50rpmで用いるpH5.6の酢酸緩衝液(acetate buffer)を用いた溶解について分析されることもあり、ここでは、剤形に含まれるイブプロフェンの少なくとも80%が、速効性放出用量について投薬後60分以内に剤形から放出される。
【0029】
本明細書中で使用されている薬の「放出速度」は、単位時間当たりの剤形から放出される薬の量、例えば、時間当たり放出される薬のミリグラム数(mg/hr)をさしている。薬の放出速度は、本技術分野で既知のインビトロの剤形溶解テスト条件の下、算出される。本明細書中で使用される、「投与後」に規定された時間で得られる薬の放出速度は、適切な溶解試験の開始後の規定された時間で得られるインビトロの薬の放出速度、例えば、USP24に示されているものをさしている。
【0030】
本明細書中で使用されている「治療学的効果」は、病気を診断、処置、治療、緩和、または防止するか、または身体の構造もしくはあらゆる機能に影響を及ぼすことが意図された活性成分のあらゆる効果または作用を意味するものとする。
【0031】
本明細書中で使用されている「半透性」は、膜が適切な溶解媒質、例えば、胃腸液またはインビトロの溶解媒質に接触しているときに、水がそのような膜を透過することができ、また、塩および本明細書中に記述されている活性成分を含めたその他の分子がそのような膜を通ってゆっくりと拡散させられることを意味するものとする。
【0032】
本明細書中で使用されている「水不溶性」は、U.S. Pharmacopeia第24版により定義されているように水中に、実質的に不溶性であるか、事実上不溶性であるか、または僅かにだけ可溶性である組成を意味している。これらの組成物は、完全な溶解のために、かかる組成物の部(part)当たり、少なくとも約100部の溶媒を必要とする。
【0033】
本明細書中で使用されている「侵食性(Erodible)」は、適切な溶解媒質に接触したときに、表面の侵食を介して組成物が溶解することを意味するものとする。
【0034】
本明細書中で使用されている「保護コーティング」は、乾燥媒体、例えば剤形のマトリックス中の、または、液体剤形実施形態においては液体媒体媒質中の、他の粒子または他の活性成分と反応しないコーティングを意味するものとする。
【0035】
本明細書中で使用されている用語「フェニレフリン」は、ベンザインメタノール,3−ヒドロキシ−α−[(メチルアミノ)メチル]を意味し、限定されるものではないが、医薬品に許容可能なそれらの塩、エステル、異性体、または誘導体を含む。
【0036】
本明細書中で使用されている「粒子」は、結晶、顆粒、凝集物、または溶解されていないあらゆる固体材料である。
【0037】
本発明のある一つの実施形態は、活性成分の投与のために好適な改変型放出医薬品剤形に向けられており、この剤形は、a)速効性放出部分、例えば、剤形から速効性で放出される少なくとも1つの活性成分を含む部分と、b)改変型放出部分、例えば、時間の改変された期間にわたって実質的に連続的に血流中に放出される少なくとも1つの活性成分を含む部分とを含む。
【0038】
ある一つの実施形態において、剤形の第2の部分から放出されたときの第2の活性成分の改変型放出の治療学的効果が、第1の活性成分の速効性放出の治療学的効果の持続時間と実質的に同じになるように、剤形が溶解媒質に接触すると活性成分が改変型放出式に第2の部分から放出される。「第1の活性成分の速効性治療学的効果の持続時間と実質的に同じ」とは、第2の活性成分の治療学的効果の持続時間が、第1の活性成分の持続時間と同じであるか、またはその持続時間の約1時間以内、すなわち、例えば約1/2時間以内、または約15分以内、または約10分以内であることを意味するものとする。他の実施形態において、剤形の第2の部分から放出されたときの第2の活性成分の改変型放出の治療学的効果は、例えば、剤形の最初の投与後、少なくとも約4時間〜約6時間、または約4時間〜約8時間、または約4時間〜約12時間であってもよい。
【0039】
速効性放出部分は、剤形内で、分子レベルで分散されている、例えば溶融または溶解されている1つ以上の活性成分を含んでもよく、あるいは、活性成分は、次にコーティングされてもよいし、またはコーティングされなくてもよい粒子の形態にあってもよい。活性成分が粒子の形態にある実施形態において、粒子は(コーティングされていようと、またはコーティングされてなかろうと)典型的には、約1ミクロン〜約2000ミクロンの平均粒径を有する。ある一つの実施形態において、そのような粒子は、約1ミクロン〜約300ミクロンの平均粒径を有する結晶の形態にある。他の実施形態において、粒子は、約25ミクロン〜約2000ミクロン、例えば、約25ミクロン〜約1000ミクロン、または約25ミクロン〜約400ミクロンの平均粒径を有する顆粒またはペレットの形態にある。
【0040】
改変型放出部分は、改変型放出特性を有する様々な粒子中に、少なくとも1つの活性成分を含む。ある一つの実施形態において、改変型放出部分中のこれらの粒子のコアは、改変型放出組成物で実質的にコーティングされた純粋な結晶形態にある活性成分から構成されてもよい。あるいは、粒子コアは、本技術分野で既知の結合剤、賦形剤などといったオプションの成分と、1つ以上の活性成分から構成された顆粒との混合物から構成されてもよく、そのような顆粒もまた、改変型放出組成物で実質的にコーティングされる。他の実施形態において、活性成分粒子は、改変型放出組成物で構成されたマトリックスの全体に分散されてもよい。
【0041】
さらなる他の実施形態において、1つ以上の活性成分は、樹脂、例えばイオン交換樹脂に、化学的に結合されるか、または「複合体化され(complexed)」て、薬−樹脂複合体粒子(または活性成分樹脂粒子)を形成してもよく、この複合体は、まず半透性コーティング層で実質的にコーティングされ、その後、保護コーティング層で実質的にコーティングされる。当業者は、過度の実験を行うことなく、本実施形態で使用するための特定のイオン交換樹脂が、例えば、活性成分のイオン電荷といったいくつかの因子に依存することを容易に認識するであろう。NSAID活性成分のために好適なイオン交換樹脂の例としては、限定されるものではないが、スチレン/ジビニルベンゼンコポリマー、およびRohm & Haasから「Duolite(登録商標)AP143」という商品名で商業的に入手可能なコレスチラミンを含む。フェニレフリンまたは偽性エフェドリンといった正の電荷を持つ活性成分のために好適なイオン交換樹脂の例としては、限定されるものではないが、スルホン化スチレン/ジビニルベンゼンコポリマーから誘導されたスルホン酸カチオン性イオン交換樹脂、たとえば、Rohm & Haasから一般商品名「Amberlite」で商業的に入手可能なもの、例えば、「Amberlite IRP69」、および、Dow Chemical Companyから商品名「Dowex」で商業的に入手可能なもの、例えば、「Dowex Marathon」、「Dowex Monosphere」、および「Dowex XYS-40010.00」を含む。Amberlite IRP 60およびDowex XYS-40010.00の製品は、約8%のジビニルベンゼンと架橋結合したポリスチレンから構成されたスルホン化ポリマーであり、乾燥樹脂(H+形態)の約4.5〜5.5meq./gのイオン交換能力を有する。Amberlite IRP-69製品は、約47μm〜約149μmの粒径範囲を備えた不規則な形になった粒子から構成されており、一方、Dow XYS-40010.00製品は、約45μm〜約150μmの粒径範囲を有する球状粒子から構成されている。他の好適なイオン交換樹脂である「Dow XYS-40013.00」は、約8%のジビニルベンゼンと架橋結合されたポリスチレンから構成されたポリマーであり、四級アンモニウム基で官能性を持たせてあり、その交換能力は、標準的には、乾燥樹脂の約3〜約4meq./gの範囲内にある。他の好適なイオン交換樹脂は、ポラクリリンカリウム(polacrilin potassium)を含み、これは、ジビニルベンゼンカリウム塩を含むメタクリル酸ポリマー(methacrylic acid polymers with divinylbenzene potassium salt)としても知られている。ポラクリリンカリウム(polacrin potassium)の好適な例としては、限定されるものではないが、Rohm & Haasから商品名「Amberlite IRP-64」で商業的に入手可能なものを含み、これは、メタクリル酸(methcrylic acid)とジビニルベンゼンとのコポリマーから誘導されたカチオン性イオン交換樹脂である。ポリマー樹脂との複合体形成のさらなる詳細は、本技術分野においてよく知られており、例えば、米国特許第4,221,778号;同第5,980,882号;同第4,847,077号;および同第6,001,392号に開示されている。
【0042】
イオン交換樹脂は、一般に、強酸カチオン、強塩基カチオン、弱酸カチオン、および弱塩基カチオンを含めた種々の種類に分類される。全般的に、薬は、好適な樹脂の水性懸濁液に混合され、その後、樹脂−薬複合体が洗浄され、乾燥される。樹脂への薬の結合は、洗浄物からの媒質溶離のpHを分析するか、または、洗浄物のナトリウム濃度における変化を測定することにより実証され得る。
【0043】
フェニレフリンを水性混合物を介してイオン交換樹脂に結合させることが望ましい実施形態においては、塩酸フェニレフリン(PHE−HCl)および樹脂が混合物になるように初めに配合されてもよい。その後、樹脂のナトリウムイオンが、フェニレフリンのプロトン化形態(PHE−H+)に交換されてもよい。典型的には、結果として生じる薬−樹脂複合体は、フェニレフリンと樹脂との重量比が約20:80〜約80:20、すなわち、例えば約30:70〜約70:30である。
【0044】
ある一つの特定の実施形態において、剤形の改変型放出部分は、イオン交換樹脂を実質的に含まない。イオン交換樹脂の量が、剤形中の全ての活性成分粒子の全重量に基づいて、約1パーセント未満、例えば、約0.5パーセント未満、または約0.1パーセント未満であることが、「イオン交換樹脂を実質的に含まない」により意味される。
【0045】
本発明に従うと、薬−樹脂複合体粒子は、半透性コーティングで実質的にコーティングされる。粒子表面の約80%、例えば、約85%、または約99%がコーティングされていることが、「実質的にコーティングされる」により意味される。
【0046】
好適な半透性コーティングの例としては、限定されるものではないが、セルロースアセテート、エチルセルロース、非腸溶性ポリメタクリレート、ならびに、これらのコポリマーおよび混合物といったポリマーを含む。例示的な非腸溶性ポリメタクリレートとしては、限定されるものではないが、Rohm Pharmaから商品名「EUDRAGIT NE」で商業的に入手可能なポリ(エチルアクリレート,メチルメタクリレート)[2:1];Rohm Pharmaから商品名「EUDRAGIT FS」で商業的に入手可能なポリ(メチルアクリレート,メチルメタクリレート,メタクリル酸)[7:3:1];Rohm Pharmaから商品名「EUDRAGIT RL」で商業的に入手可能なポリ(エチルアクリレート,メチルメタクリレート,トリエチルアンモニオエチルメタクリレートクロライド)[1:2:0.2](poly(ethyl acrylate, methyl methacrylate, triethylammonioethyl methacrylate chloride) 1:2:0.2);Rohm Pharmaから商品名「EUDRAGIT RS」で商業的に入手可能なポリ(エチルアクリレート,メチルメタクリレート,トリエチレンアンモニオエチルメタクリレートクロライド)[1:2:0.1](poly(ethyl acrylate, methyl methacrylate, triethyleammonioethyl methacrylate chloride 1:2:0.1)、ならびに、これらのコポリマーおよび混合物を含む。本技術分野ではアセチルセルロース、セルロースジアセテート、およびセルローストリアセテートという一般名でも既知であるセルロースアセテートは、Eastman Chemical Companyから商業的に入手可能である。本技術分野ではセルロースエチルエーテルとしても既知であるエチルセルロースは、Dow Corporationから商品名「ETHOCEL」で商業的に入手可能である。ある一つの実施形態において、半透性コーティングは、セルロースアセテート、エチルセルロース、およびこれらの混合物から選択されてもよい。
【0047】
コーティングされた薬−樹脂複合体粒子は、その後、保護コーティングで実質的にコーティングされる。コーティング粒子表面の約80%、例えば、約85%、または約99%がその後、保護コーティング層でコーティングされることが、「実質的にコーティングされる」により意味される。
【0048】
好適な保護コーティングの例としては、限定されるものではないが、ヒドロキシプロピルメチルセルロースフタレート(ヒプロメロースフタレート(hypromellose phthalate)としても知られている)、ヒドロキシプロピルメチルセルロースアセテートスクシネート、セルロースアセテートフタレート、ポリビニルアセテートフタレート、セラック、腸溶性ポリメタクリレート系ポリマー(enteric polymethacrylate-based polymers)、ならびに、これらのコポリマーおよび混合物を含む腸溶性ポリマーから構成されたものを含む。好適な腸溶性ポリメタクリレート系ポリマーの例としては、限定されるものではないが、Rohm Pharma GmbHから商品名「EUDRAGIT S」ポリマーで商業的に入手可能なポリ(メタクリル酸,メチルメタクリレート)[1:2];Rohm Pharma GmbHから商品名「EUDRAGIT L-100, L-30D, L 12.5 and L12.5 P」ポリマーで商業的に入手可能なポリ(メタクリル酸,メチルメタクリレート)[1:1];ならびに、Rohm Pharmaから商品名「EUDRAGIT L30-D 55 and L-100-55」、Eastman Chemicalから商品名「Eastacryl 30D」、およびColorcon Corporationから商品名「Acryl-EZE」、およびBASF Fine Chemicalsから商品名「Kollicoat MAE 30D」で商業的に入手可能なポリ(メタクリル酸,エチルアクリレート)[1:1]を含む。ある一つの実施形態において、腸溶性ポリマーは、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートスクシネート、セルロースアセテートフタレート、ポリビニルアセテートフタレート、セラック、ならびに、これらのコポリマーおよび混合物といった非アクリル酸塩化合物(non-acrylate compounds)から選択されてもよい。
【0049】
ある一つの実施形態において、保護コーティングは、脂肪酸エステル、ワックス、またはこれらの混合物といった脂質の形態で提供されてもよい。好適な脂肪酸エステルの例としては、限定されるものではないが、スクロース脂肪酸エステル;モノグリセリド、ジグリセリド、およびトリグリセリド;グリセリルベヘナート(glyceryl behenate);グリセリルパルミトステアレート(glyceryl palmitostearate);グリセリルトリステアレート(glyceryl tristearate);グリセリルトリラウリレート(glyceryl trilaurylate);グリセリルミリステート;GLYCOWAX-932;ラウロイルマクロゴール−32グリセリド;ステアロイルマクロゴール−32グリセリド;約C10〜C40の脂肪酸鎖長を有するものなどの脂肪酸エステル;およびこれらの混合物を含む。
【0050】
好適なワックスの例としては、限定されるものではないが、カルナウバろう、鯨ろう、蜜ろう、カンデリラろう、セラックろう、カルヌバろう(carnuba wax)、蜜ろう、微晶ろう、およびパラフィンろうなど、ならびにこれらの混合物を含む。
【0051】
オプションとして、半透性コーティングおよび/または保護コーティングは、可塑剤を含んでもよい。好適な可塑剤の例としては、限定されるものではないが、ポリエチレングリコール;プロピレングリコール;グリセリン;ソルビトール;トリエチルシトレート;トリブチルシトレート;ジブチルセバケート(dibutyl sebecate);ヒマシ油、菜種油、オリーブ油、およびゴマ油などの植物油;ポリソルベート、ラウリル硫酸ナトリウム、およびスルホコハク酸ジオクチルナトリウムなどの界面活性物質;グリセロールのモノアセテート;グリセロールのジアセテート;グリセロールのトリアセテート;天然ゴム(natural gum);トリアセチン;アセチルトリブチルシトレート(acetyltributyl citrate);ジエチルオキサレート(diethyloxalate);ジエチルマレート(diethylmalate);ジエチルフマレート;ジエチルマロネート;ジオクチルフタレート;ジブチルスクシネート;グリセロールトリブチレート;グリセロールモノステアレート;水素化されたヒマシ油;置換されたトリグリセリドおよびグリセリド;ならびに、これらの混合物を含む。
【0052】
ある一つの実施形態において、好適な可塑剤は、半透性コーティングの全乾燥重量に基づいて、約0.1%〜約40%、例えば、約1%〜約30%、または約5%〜約20%の量で使用され得る。
【0053】
ある一つの実施形態において、好適な可塑剤は、保護コーティングの全乾燥重量に基づいて、約0.1%〜約40%、すなわち例えば、約1%〜約30%、または約5%〜約20%の量で使用され得る。
【0054】
ある一つの実施形態において、改変型放出粒子における半透性コーティング層と保護腸溶性コーティング層との重量比は、約10:90〜約90:10、または約20:80〜約80:20である。
【0055】
ある一つの実施形態において、改変型放出粒子は、腸溶性ポリマーを実質的に含まず、すなわち例えば、改変型放出粒子は、改変型放出粒子の全重量に基づいて、約1パーセント未満、または約0.25パーセント未満の腸溶性ポリマーを含む。
【0056】
ある一つの実施形態において、薬−樹脂複合体は、溶媒和または含浸剤(solvating or impregnating agent)で処理されてもよく、これは、活性成分および樹脂が混合されている間か、または活性成分が樹脂に結合された後で加えられる。好適な含浸剤の例としては、限定されるものではないが、ソルビトール、ポリエチレングリコール、グリセロール、プロピレングリコール、マンニトール、ラクチトール(lactitol)、ラクトース、メチルセルロース、およびこれらの混合物を含む。含浸剤は、乾燥樹脂の重量当たり、約5部〜約50部の量で存在し得る。
【0057】
ある一つの実施形態において、薬−樹脂複合体の酸化を抑制することにより薬−樹脂複合体を安定化するために、キレート剤が剤形に加えられてもよい。好適なキレート剤としては、限定されるものではないが、エチレンジアミン四酢酸(EDTA)およびEDTAの塩を含み、EDTAの塩としては、限定されるものではないが、エデト酸カルシウム二ナトリウム、エデト酸三ナトリウム、エデト酸二ナトリウム、およびエデト酸ナトリウムを含む。キレート剤は、最終的な剤形の重量の、約0.005パーセント〜約10パーセントの量で存在し得る。
【0058】
第2の保護コーティング層の下にある第1の半透性コーティング層を含む活性成分−樹脂複合体粒子は、改変型放出組成物の全乾燥重量に基づいて、約1パーセント〜約99パーセント、例えば、約5パーセント〜約80パーセントの第1の半透性コーティング層、および約5パーセント〜約99パーセント、例えば約10パーセント〜約90パーセントの第2の保護コーティング層を含む改変型放出組成物を産生する。
【0059】
2つのコーティング層のそれぞれの厚みは、所望の改変型放出特性、選択される活性成分などに依存して変化してもよいが、典型的には、約0.01ミクロン〜約500ミクロン、例えば、約0.1ミクロン〜約100ミクロンの範囲にわたり得る。
【0060】
粒子上の第1のコーティング層の表面積当たりの乾燥重量は、約0.1mg/cm2〜約10mg/cm2、すなわち例えば、約0.5mg/cm2〜約5mg/cm2である。粒子上の第2のコーティング層の表面積当たりの乾燥重量は、約0.1mg/cm2〜約10mg/cm2、例えば、約0.5mg/cm2〜約8mg/cm2である。
【0061】
活性成分−樹脂複合体粒子の第1の半透性コーティング層の追加後の重量増加は、コーティングされていない活性成分−樹脂複合体粒子の乾燥重量に基づいて、約1パーセント〜約200パーセント、例えば、約20パーセント〜約150パーセントである。活性成分−樹脂複合体粒子の第2の保護コーティング層の追加後の重量増加は、第1の半透性コーティング層でコーティングされた活性成分−樹脂複合体粒子の乾燥重量に基づいて、約25パーセント〜約400パーセント、例えば、約40パーセント〜約400パーセントである。
【0062】
コーティングされた活性成分粒子は、本技術分野で既知のあらゆる好適な方法により形成されてもよい。好適な粒子形成およびコーティング方法としては、高速攪拌造粒(high sheer granulation)、流動層造粒(fluid bed granulation)、例えば、ローター造粒、流動層コーティング、ウルスターコーティング(wurster coating)、コアセルベーション(coaccervation)、吹き付け乾燥、吹き付け凝結などを含み、例えば、Pharmaceutical Dosage Forms: Tablets Volume 3, edited by Herbert A. Lieberman and Leon Lachman, Chapters 2, 3, and 4 (1982)に記述されている。
【0063】
活性成分をイオン交換樹脂に結合させて薬−樹脂複合体を形成することにより粒子が形成されるある一つの実施形態においては、結果として生じる粒子は、ウルスター流動層コーティングを用いて半透性層がまずコーティングされ、その後、腸溶性層がウルスター流動層コーティングを用いてコーティングされる。コーティング材料は、溶媒を含む溶液または分散系を介して粒子上に吹き付けられてもよく、この溶媒としては、限定されるものではないが、水、エタノール、メタノール、アセトン、ヘキサン、シクロヘキサン、メチレンクロライド、イソプロパノール、およびこれらの混合物を含む。例えば、米国特許第4,847,077号を参照のこと。
【0064】
ある一つの実施形態において、薬−樹脂複合体の平均直径は、約20ミクロン〜約400ミクロン、または約20ミクロン〜約300ミクロンである。ある一つの実施形態において、第1のコーティング層でコーティングされた薬−樹脂複合体粒子の平均直径は、約20ミクロン〜約800ミクロン、例えば、約50ミクロン〜約400ミクロンであり、第1および第2のコーティング層の両方でコーティングされた薬−樹脂複合体粒子の平均直径は、約50〜約1000ミクロン、例えば、約100ミクロン〜約400ミクロンである。
【0065】
本発明の剤形は、1つ以上の活性な薬剤または成分を含む。広く好適な活性成分としては、例えば、医薬品、鉱物、ビタミンおよびその他の機能性食品(nutraceutical)、オーラルケア剤、香味料(flavorant)、およびこれらの混合物を含む。好適な医薬品としては、鎮痛薬、抗炎症剤、抗関節炎薬、麻酔薬、抗ヒスタミン薬、鎮咳薬、抗生物質、抗感染剤(anti-infective agents)、抗ウイルス薬、抗凝固薬、抗うつ薬、抗糖尿病剤(antidiabetic agents)、制吐薬、抗膨満薬(antiflatulents)、抗真菌薬、鎮痙薬、食欲抑制剤、気管支拡張薬、心血管作用薬(cardiovascular agents)、中枢神経系作用薬、中枢神経系刺激薬、鬱血除去薬、経口避妊薬、利尿薬、去痰薬、胃腸作用薬、偏頭痛製剤、乗物酔い製品、ムコリチック物(mucolytics)、筋弛緩薬、骨粗鬆症製剤、ポリジメチルシロキサン、呼吸器作用薬、睡眠補助薬(sleep-aids)、尿路作用薬、およびこれらの混合物を含む。
【0066】
好適な香味料としては、メントール、ペパーミント、ミント香料、果物香料、チョコレート、バニラ、風船ガム香料、コーヒー香料、リキュール香料、およびこれらの組み合わせなどを含む。
【0067】
好適な胃腸作用薬の例としては、制酸薬、例えば、炭酸カルシウム、水酸化マグネシウム、酸化マグネシウム、炭酸マグネシウム、水酸化アルミニウム、炭酸水素ナトリウム、炭酸ジヒドロキシアルミニウムナトリウム(dihydroxialuminum sodium carbonate);刺激性緩下薬(stimulant laxatives)、例えば、ビサコジル、カスカラサグラダ、ダントロン、センナ、フェノールフタレイン、アロエ、ヒマシ油、リシノール酸、およびデヒドロコール酸、ならびにこれらの混合物;H2レセプター拮抗薬、例えば、ファモチジン(famotadine)、ラニチジン、シメチジン(cimetadine)、ニザチジン;プロトンポンプ阻害薬、例えば、オメプラゾール、またはランソプラゾール;胃腸細胞保護薬(gastrointestinal cytoprotectives)、例えば、スクラフレート(sucraflate)、およびミソプロストール;胃腸運動促進薬(gastrointestinal prokinetics)、例えば、プルカロプリド、H. pyloriに対する抗生物質(antibiotics for H. pylori)、例えば、クラリスロマイシン、アモキシシリン、テトラサイクリン、およびメトロニダゾール;止瀉薬、例えば、ジフェノキシラート、およびロペラミド;グリコピロレート;制吐薬、例えば、オンダンセトロン、鎮痛薬、例えば、メサラミンを含む。
【0068】
限定されるものではないが、ジメチコーン、およびシメチコンを含む、好適なポリジメチルシロキサンの例は、米国特許第4,906,478号、同第5,275,822号、および同第6,103,260号に開示されているものであり、各特許文献の内容は、参照により本明細書中に明白に組み込まれる。本明細書中で使用されている用語「シメチコン」は、限定されるものではないが、シメチコンおよびジメチコーンを含めたポリジメチルシロキサンのより広い分類をさしている。
【0069】
本発明のある一つの実施形態において、少なくとも1つの活性成分は、ビサコジル、アルブテロール、ファモチジン、ラニチジン、シメチジン、プルカロプリド、ジフェノキシラート、ロペラミド、ラクターゼ、メサラミン、ビスマス、制酸薬、ならびに、医薬品に許容可能なこれらの塩、エステル、異性体、および混合物から選択され得る。
【0070】
他の実施形態において、少なくとも1つの活性成分は、鎮痛薬、抗炎症薬、および解熱薬、例えば、以下のものを含めた非ステロイド性抗炎症薬(NSAID)、すなわち、a)プロピオン酸誘導体、例えば、イブプロフェン、ナプロキセン、ケトプロフェンなど、b)酢酸誘導体、例えば、インドメタシン、ジクロフェナク、スリンダク、トルメチンなど、c)フェナム酸誘導体、例えば、メフェナム酸、メクロフェナム酸、フルフェナム酸など、d)ビフェニルカルボン酸誘導体、例えば、ジフルニサル、フルフェニサル(flufenisal)など、e)オキシカム、例えば、ピロキシカム、スドキシカム、イソキシカム、メロキシカムなど、f)シクロオキシゲナーゼ−2(COX−2)選択性NSAID、g)アスピリン、およびh)上記のものの医薬品に許容可能な塩から選択される。
【0071】
ある一つの特定の実施形態において、少なくとも1つの活性成分は、プロピオン酸誘導体NSAIDから選択され、これは、医薬品に許容可能な鎮痛薬/非ステロイド性抗炎症薬であり、遊離−CH(CH3)COOHもしくは−CH2CH2COOH、または医薬品に許容可能な塩の基、例えば、−CH(CH3)COO−Na+もしくはCH2CH2COO−Na+を有し、これらは、典型的には、環構造、好ましくは芳香環構造に直接またはカルボニル官能基を介して結合される。
【0072】
有用なプロピオン酸誘導体の例としては、イブプロフェン、ナプロキセン、ベノキサプロフェン(benoxaprofen)、ナプロキセンナトリウム、フェンブフェン、フルルビプロフェン、フェノプロフェン、フェノプロフェンカルシウム、フルルビプロフェン、チアプロフェン酸塩(tiaprofenic)、オキサプロジン、フェンブプロフェン(fenbuprofen)、ケトプロフェン、インドプロフェン(indoprofen)、ピルプロフェン、カルプロフェン(carpofen)、オキサプロフェン(oxaprofen)、プラノプロフェン、ミクロプロフェン(microprofen)、チオキサプロフェン、スプロフェン、アルミノプロフェン、チアプロフェン酸、フルプロフェン(fluprofen)、ブクロキシ酸(bucloxic acid)、ならびに、医薬品に許容可能なこれらの塩、誘導体、および組み合わせを含む。
【0073】
本発明のある一つの実施形態においては、プロピオン酸誘導体は、イブプロフェン、ケトプロフェン、フルビプロフェン(flubiprofen)、ならびに、医薬品に許容可能なこれらの塩、および組み合わせから選択される。
【0074】
他の実施形態において、プロピオン酸誘導体は、イブプロフェン、2−(4−イソブチルフェニル)プロピオン酸、または医薬品に許容可能なこれらの塩、例えば、イブプロフェンのアルギニン、リジン、またはヒスチジンの塩である。その他の医薬品に許容可能なイブプロフェンの塩は、米国特許第4,279,926号、同第4,873,231号、同第5,424,075号、および同第5,510,385号に記述されており、これらの内容は参照として組み込まれる。
【0075】
本発明の他の特定の実施形態において、少なくとも1つの活性成分は、アセトアミノフェン、アセチルサリチル酸、イブプロフェン、ナプロキセン、ケトプロフェン、フルルビプロフェン、ジクロフェナク、シクロベンザプリン、メロキシカム、ロフェコキシブ(rofecoxib)、セレコキシブ(celecoxib)、ならびに、医薬品に許容可能なこれらの塩、エステル、異性体、および混合物から選択されてもよい。
【0076】
本発明の他の特定の実施形態において、少なくとも1つの活性成分は、偽性エフェドリン、フェニレフリン、フェニルプロパノラミン、クロルフェニラミン、デキストロメトルファン、ジフェンヒドラミン、クロフェジアノール(clofedianol)、アステミゾール、テルフェナジン、フェキソフェナジン(fexofenadine)、ロラタジン(loratadine)、デスロラタジン、セチリジン(cetirizine)、およびこれらの混合物、ならびに、医薬品に許容可能なこれらの塩、エステル、異性体、および混合物から選択されてもよい。
【0077】
特定の実施形態において、樹脂複合体に結合された活性成分は、フェニレフリン、偽性エフェドリン、デキストロメトルファン、ジフェンヒドラミン、クロルフェニラミン、およびこれらの混合物から選択される。ある一つの実施形態において、樹脂をベースとする粒子は、フェニレフリン、偽性エフェドリン、デキストロメトルファン、ジフェンヒドラミン、クロルフェニラミン、およびこれらの混合物の塩酸塩および臭化水素酸塩を用いて結合される。
【0078】
他の特定の実施形態において、少なくとも1つの活性成分は、NSAIDまたは医薬品に許容可能なその塩であり、薬樹脂複合体に結合されるその他の活性成分は、フェニレフリンおよび/または偽性エフェドリン(psuedoephedrine)である。
【0079】
ある一つの実施形態において、活性成分または複数の成分の治療上有効な量は、「単位用量容積(unit dose volume)」で示されてもよく、これは、粉末または水性懸濁液の形態の場合もある。本明細書中で使用されている「治療上有効な量」は、経口投与に対して所望の治療学的応答を生み出す活性成分の量である。当業者は、例えば、投与されている特定の活性成分;活性成分の生物学的利用能特質;所望の用量療法;患者の年齢および体重などといった因子を考慮することにより、所定の患者に対しての活性成分の「治療上有効な量」を用意に決定することができる。本明細書中で使用されている「単位用量容積」は、患者にある用量の所定の製品を経口的に投与するのに都合のよいあらゆる容積であり得る。
【0080】
この実施形態において、「単位用量容積」は、典型的には投薬指導が付随し、これは、例えば、患者の年齢または体重に依存する複数回の単位用量容積であってもよい活性成分の量を患者に服用するように指示する。典型的には、懸濁液の単位用量容積は、最小の患者に対して治療上有効である量の活性成分を含むであろう。例えば、好適な単位用量容積は、小さじ一杯(約5mL)、大さじ一杯(約15mL)、一滴(one dropper)、または1ミリリットルを含み得る。
【0081】
本発明に従うと、NSAIDを含む剤形は、特定の痛み軽減処置で、拡張された時間の期間、例えば約4時間または約6時間の期間にわたって血中に活性成分を放出する一回の投与で、処置が必要なほ乳類に提供されてもよい。ゼロ時点で、速効性放出用量部分中の活性成分を介して、最初の用量のNSAIDがほ乳類に提供される、すなわち、投与される。次に、第2の活性成分が、改変型放出用量部分中の活性成分を介して、活性成分を含む調合物の最初の投与から次の約4〜約6時間の間にわたって血中に放出される。言い換えると、調合物は、最初の投与から約4または約6時間後には、溶解されていない第2の活性成分をまだ保持している。
【0082】
本発明を実施するにあたって、剤形は、活性成分の全重量に基づいて、約0.01パーセント〜約40パーセントの第1の活性成分の速効性放出用量粒子部分と、約0.01パーセント〜約40パーセント、すなわち、約.01パーセント〜約10パーセントの二重コーティングされた第2の活性成分の改変型放出用量部分とから構成されてもよい。本明細書中で使用されている「部分(portion)」は、あらゆるオプションの構成成分と共に同定されている活性成分の総計を意味するが、液体媒体、あるいは、固体剤形の場合には速効性放出用量粒子が結合され得るマトリックスまたはその他の乾燥媒体を含まないものとする。速効性放出用量部分および改変型放出用量部分は、適切な媒体と結合されて、1)必要な場合には即席で懸濁されることができる乾燥混合物、2)エリキシル、または懸濁液のようにすぐ使用できる液体剤形、または3)固体または半固体剤形のうちのいずれかを形成してもよい。
【0083】
媒体の好適な構成物は、限定することなく、溶媒;構造化剤(structuring agents);膨潤剤;界面活性物質;糖;緩衝物質、例えば、クエン酸、およびクエン酸ナトリウム;グリシン、ならびに塩化水素酸、リン酸ナトリウム、およびリン酸カリウム;保存剤、および細菌発育阻止剤、例えばp−ヒドロキシ安息香酸のエステル;着色料;ならびに、医薬品中で一般に使用される種々の調味および甘味料を含むことができる。
【0084】
好適な甘味料の例としては、限定されるものではないが、あらゆる既知の甘味付け剤、例えば、糖、糖アルコール、高強度甘味料(high intensity sweeteners)、およびこれらの混合物を含む。好適な糖としては、限定されるものではないが、スクロース、デキストロース、高フルクトーストウモロコシシロップ(high fructose corn syrup)、およびマルトースを含む。好適な糖アルコールとしては、限定されるものではないが、ソルビトール、キシリトール、およびマンニトールを含む。好適な高強度甘味料としては、限定されるものではないが、スクラロース(sucralose)、アスパルテーム、サッカリン、およびアセスルファムK(acesulfame K)を含む。
【0085】
ある一つの実施形態において、有効量の緩衝剤が、液体懸濁液剤形の改変型放出部分中に含まれる少なくとも1つの活性成分のpKaを液体懸濁液剤形の全体のpHよりも大きくさせるために使用される。
【0086】
加えて、媒体は、水から構成されてもよいし、あるいは、水と、例えばグリコール、アルコール、およびグリセロールといった本技術分野で既知の医薬品に許容可能な水混和性共溶媒との混合物から構成されてもよい。
【0087】
ある実施形態において、剤形は、本技術分野において既知のあらゆる懸濁系、例えば、典型的には、1つ以上の構造化剤および/または1つ以上の膨潤剤を含むものを含んでもよい。ある一つの実施形態において、剤形は、液体懸濁液剤形の全重量に基づいて、約0.1パーセント〜約10パーセントの懸濁系を含む。好適な懸濁系は、例えば、米国特許第5,374,659号、同第5,621,005号、および同第5,409,907号で開示されているものを含み、これらの特許文献は全て、その全体が本明細書中に参照により組み込まれる。
【0088】
本発明での使用のために好適な構造化剤は、親水コロイドといった親水性ポリマーを含む。好適な親水コロイドの例としては、アルギナート、寒天、ガーゴム、イナゴマメ(locust bean)、カラゲナン、タラ(tara)、アラビアゴム、トラガカントゴム、ペクチン、キサンタン、ゲル化剤、マルトデキストリン(maltodextrin)、ガラクトマンナン、プスツラン(pusstulan)、ラミナリン、スクレログルカン(scleroglucan)、アラビアゴム、イヌリン、カラヤ、ウェラン(whelan)、ラムサン(rhamsan)、ズーグラン(zooglan)、メチラン(methylan)、キチン、シクロデキストリン、キトサン、およびこれらの組み合わせを含む。本発明のある実施形態において、キサンタンガムは、構造化剤である。
【0089】
キサンタンガムは、高分子量の天然炭水化物であり、より詳細には、多糖類である。本発明での使用のために好適なある一つのキサンタンガムは、Xanthomonas campestrisにより生産される高分子量の多糖類である。この多糖類を生み出すための技術および菌株は、米国特許第4,752,580号および同第3,485,719号に記述されており、その開示は参照として本明細書中に組み込まれる。ある一つの実施形態において、キサンタンガムは、LV model Brookfield Synchro-Lectric viscometer at 60 rpm, no. 3 spindleにより、25℃で測定されたときに、1パーセント塩溶液中、約1000〜約1700cP(mPa−sec)の粘度を有し得る。好適なキサンタンガムは、例えばCP Kelcoから商品名「Keltrol」、「Keltrol TF」、および「Keltrol 1000」で入手可能である。
【0090】
膨潤剤は、適切な水性環境に曝されると、ネットワーク系を形成することなく膨張する。アルファ化でんぷんは、特に優れた膨潤剤である。「インスタント化」でんぷんとしても知られるアルファ化でんぷんは、冷水に加えられると直ちに膨潤して肥厚し始めるように下ごしらえされている。ある一つの特に好適なアルファ化でんぷんは、改変され、安定化されていて、ろう様のトウモロコシ食品でんぷんから調製され、National Starch Companyから「INSTANT STARCH, ULTRASPERSE-M」として商業的に入手可能である。その他の好適な膨潤剤としては、限定されるものではないが、微結晶性セルロースおよび/またはヒドロキシプロピルメチルセルロースを含む。
【0091】
ある一つの実施形態において、懸濁系は、アルファ化でんぷん膨潤剤と共にキサンタンガム構造化剤から構成される。他の実施形態において、懸濁系は、液体懸濁液剤形の全重量に基づいて、約0.01パーセント〜約1パーセント、または約0.05パーセント〜約0.40パーセントのキサンタンガムと、約1パーセント〜約10パーセント、または約0.5パーセント〜約3.0パーセントのアルファ化でんぷん(例えば、National Starch Companyから商品名「INSTANT STARCH, ULTRASPERSE-M」で商業的に入手可能)とから構成される。
【0092】
剤形が液体形態、例えば懸濁液またはエリキシルである実施形態において、液体剤形のpHは、その中に含まれるコーティングされていない活性成分の溶解度を最小限にし、かつ化学的安定度を最大限にするよう最適化されるべきである。コーティングされていない活性成分が塩基性、例えば炭酸カルシウムである、ある一つの実施形態において、剤形のpHは、塩基性のコーティングされていない活性成分のpKaよりも2pH単位だけ上になるべく近づくようにされ得る。コーティングされていない活性成分が酸性、例えばイブプロフェンである実施形態においては、剤形のpHは、酸性のコーティングされていない活性成分のpKaの2pH単位だけ下になるべく近づくようにされ得る。コーティングされていない活性成分としてイブプロフェンを用いるある実施形態において、剤形のpHは、約1.0〜約5.0、例えば約1.0〜約4.0の範囲にされ得る。
【0093】
剤形は、既知のpH調節剤を用いて緩衝化され、所望のpH範囲内に懸濁液のpHを維持することができる。好適なpH−調節剤は、所望の程度のpH緩衝を提供するのに十分な量で剤形中に存在し得る。pH調節剤は、典型的には、剤形100mL当たり、約0〜約1グラムの範囲で存在するであろう。
【0094】
コーティングされていない活性薬剤としてイブプロフェンを含み、かつ例えばカルボキシメチルセルロースナトリウム(sodium carboxymethylcellulose)といったアルカリ性ポリマーを有する懸濁系を含む実施形態において、pH調節剤は、例えば、クエン酸、リンゴ酸、グルタミン酸など、味覚がマスクされた経口懸濁液での使用のために許容可能な味覚特質を有する弱有機酸から選択され得る。
【0095】
ある一つの実施形態において、剤形はオプションで、剤形の所望のpH範囲内で活性を有する抗菌性保存剤を含んでもよい。そのような医薬品懸濁液中で有用な保存剤は、限定されるものではないが、安息香酸ナトリウム、ソルビン酸カリウム、エデト酸の塩(salts of edetate)(エチレンジアミン四酢酸、すなわちEDTAとも知られていて、例えばエデト酸二ナトリウム)およびパラベン(parabens)(例えば、メチル、エチル、プロピル、およびブチルp−ヒドロキシ安息香酸エステル)を含む。上述で列挙されている保存剤は、例示的なものであるが、各保存剤は、保存剤の適合性および効力を保証するために、各調合物において、実験的な根拠により評価されなれければならない。医薬品調合物中の保存剤の効力を評価するための方法は、当業者に既知である。
【0096】
あるオプションの実施形態において、本発明の剤形は、ある疎水性活性薬剤の分散を補助するために湿潤剤として使用するための界面活性物質を利用してもよい。その他のある実施形態において、本発明の剤形は、界面活性物質を実質的に含まなくてもよい。ここで使用されている「界面活性物質を実質的に含まない」とは、懸濁液が、約0.1%未満、例えば約0.05%未満の界面活性物質を含むことを意味しているものとする。好適な界面活性物質の例としては、限定されるものではないが、ソルビタンオレイン酸エステル、例えば、polysorbate 80としても知られているポリオキシエチレンソルビタンモノオレイン酸塩(polyoxyethylene sorbitan monooleate)を含む。
【0097】
ある一つの実施形態において、剤形は、水性医薬品懸濁液組成物の形態にあり、水性医薬品懸濁液の容量当たり(w/vまたはg/100mL)の活性成分の全重量に基づいて、約0パーセントを超え約30パーセント以下、例えば、約0.05パーセント〜約20パーセント、または約0.5パーセント〜約10パーセント、または約0.5パーセント〜約5パーセントの第1の活性成分と、約0パーセントを超え約10パーセント以下、例えば、約0.01パーセント〜約10パーセント、または約0.03パーセント〜約5パーセントの第2の改変型放出活性成分とから構成される。
【0098】
ある実施形態においては、第1の活性成分はイブプロフェンであり、懸濁液剤形の速効性放出部分中の第1の活性成分の量は、水性懸濁液剤形の容量当たり(w/v)の第1の活性成分の全重量に基づいて、小さじ一杯の水性懸濁液剤形当たり、約25〜約400mg、例えば、約50mg〜約200mgであるか、または1mLの水性懸濁液剤形当たり約20mgの第1の活性成分であり、この量は、水性懸濁液剤形の容量当たり(w/v)の第1の活性成分の全重量に基づいて、約0.25パーセント〜約4パーセントに等しく、また、第2の活性成分は、フェニレフリンまたは偽性エフェドリンであり、懸濁液剤形の改変型放出部分中の第2の活性成分の量は、水性懸濁液剤形の容量当たり(w/v)の第2の活性成分の全重量に基づいて、小さじ一杯の水性懸濁液剤形当たり約1mg〜約20mg、例えば、約1mg〜約10mgであるか、または1mLの水性懸濁液剤形当たり、約1.5mgの第2の活性成分であり、この量は、水性懸濁液剤形の容量当たり(w/v)の第2の活性成分の全重量に基づいて、約0.01パーセント〜約0.3パーセントに等しい。
【0099】
本発明のある一つの実施形態は、液体の測定可能な懸濁液組成物に向けられており、この組成物は、懸濁液の全重量に基づいて、a)約0.05パーセント〜約40パーセントの第1の速効性放出活性成分と、b)約20パーセント〜約80パーセントの水と、c)約0.1パーセント〜約10パーセントの懸濁系と、d)約0パーセント〜約40パーセント、例えば約20パーセント〜約40パーセントの甘味付け剤と、e)約0パーセント〜約0.5パーセントの賦形剤と、約0.01パーセント〜約_10パーセントの改変型放出粒子の第2の部分とを含む。
【0100】
他の実施形態において、本発明の剤形は、剤形の全重量に基づいて、約0.1パーセント〜約10パーセント、例えば、約0.1〜約5パーセントの第1の速効性放出部分と、約0.05パーセント〜約10パーセント、例えば、約0.05パーセント〜約5パーセントの第2の改変型放出部分とを含む。ある一つの実施形態において、剤形の第2の改変型放出部分は、第2の部分の全乾燥重量に基づいて、約5パーセント〜約80パーセント、例えば、約5パーセント〜約70パーセントの第1の半透性コーティング層と、約10パーセント〜約90パーセント、例えば、約10パーセント〜約80パーセントの第2の保護コーティング層と、約1パーセント〜約50パーセント、例えば、約1パーセント〜約30パーセントの薬−樹脂複合体粒子とから構成される。
【0101】
本発明に従うと、剤形は、剤形の全重量に基づいて、約0.1パーセント〜約5パーセント、例えば、約0.5パーセント〜約3パーセントの第1の活性成分と、約0.005パーセント〜約1パーセント、例えば、約0.01パーセント〜約0.5パーセントの第2の活性成分とを含有する。
【0102】
ある実施形態において、本発明の懸濁液の粘度は、Brookfield DV-I+ Viscometerにより、# 31 spindleを用いて、12rpmの速度で、約25℃の温度条件下測定されたときに、約400cps〜約1500cpsの範囲にわたり得る。
【0103】
本発明の剤形は、1回の投与で、有効量の第1の活性成分、例えばNSAIDなどATDAIRDが約5である有効量の第1の活性成分と、同じ剤形中で、フェニレフリンまたは偽性エフェドリン(psuedoephedrine)などATDAIRDが約3である有効量の第2の活性成分とを送達するように意図されており、両方の活性成分は、より長いATDAIRD期間にわたって剤形から放出されることができる。
【0104】
「有効量」の鎮痛薬とは、患者における痛みからの軽減をもたらすものである。例えば、イブプロフェンの典型的な成人用量は、4〜6時間毎とすれば患者の体重kg当たり約2.9〜約12mgの範囲にわたり、典型的な一日量は約11.6〜約72mg/kg/日の範囲にわたり得る。従って、典型的な70kgの成人に対する有効量のイブプロフェンの投与は、例えば40mg/mLのイブプロフェンを含有する本発明の調合物約5mL〜約60mLを一日に1回または2回投与することを含み得る。典型的な小児科用量のイブプロフェンは、4〜6時間毎とすれば約5〜約10mg/kgの範囲にわたり、典型的な一日量は約20〜約60mg/kg/日の範囲にわたり得る。典型的な15kgの子供に対する有効量のイブプロフェンの投与は、例えば20mg/mLのイブプロフェンを含有する本発明の調合物約5mL〜約60mLを一日に1回または2回投与することを含み得る。
【0105】
「有効量」の鬱血除去薬とは、鬱滞の効果的な軽減を提供するもの、すなわち、腫脹を減らすことにより、鼻の通路および/または洞などの、鬱滞を解消する薬物である。例えば、フェニレフリンの典型的な成人用量は、6時間毎として患者の体重kg当たり約0.14〜約0.29mg、または、約0.60〜約1.0mg/kg/日の範囲にわたる典型的な一日量で、典型的な成人について6時間毎として約10〜約20mg、または、典型的な成人について、約0.86mg/kg/日もしくは1日当たり約60mgのフェニレフリンの範囲にわたり得る。したがって、典型的な70kgの成人に対する有効量のフェニレフリンの投与は、例えば3mg/mLのフェニレフリンを含有する、本発明の調合物約2.5mL〜約10mL、または約5mLを、一日に1〜4回投与することを含み得る。フェニレフリンの典型的な小児科用量は、2〜4時間毎として約0.25〜約0.75mg/kg、または、約1.0〜約2.7mg/kg/日の範囲にわたる典型的な一日量で、典型的な子供について6時間毎として約3.75〜約11.25mg、または、典型的な子供について、約2mg/kg/日もしくは1日当たり約30mgのフェニレフリンの範囲にわたり得る。典型的な15kgの子供に対する有効量のフェニレフリンの投与は、例えば1.5mg/mLのフェニレフリンを含有する、本発明の調合物約2.5mL〜約10mLを、一日に1〜4回投与することを含み得る。
【0106】
ある一つの実施形態において、本発明の剤形の経口投与は、剤形から第2の活性成分を放出し続け、これにより、その治療学的効果の持続時間が、第1の活性成分の持続時間に匹敵するようになる改変型放出用量で、NSAIDといった第1の活性成分を使用者に提供する。
【0107】
ある一つの実施形態において、液体剤形は、イブプロフェンおよびフェニレフリンの両方を含有する速効性放出部分と、追加量のフェニレフリンを含有する改変型放出部分とを含む。この実施形態において、イブプロフェンの速効性放出用量は、懸濁液の約25mg/5mL〜約200mg/5mL、例えば懸濁液の約50mg/5mL〜約200mg/5mLの範囲にわたり得、フェニレフリンの速効性放出用量は、懸濁液5mL当たり約2.5mg〜約15mgの速効性放出フェニレフリン、例えば懸濁液5mL当たり約2.5mg〜約10mgの速効性放出フェニレフリンの範囲にわたり得る。この実施形態において、フェニレフリンの改変型放出用量は、懸濁液5mL当たり約2.5mg〜約20mgの改変型放出フェニレフリン、例えば懸濁液5mL当たり約5〜約15mgの改変型放出フェニレフリンの範囲にわたり得る。
【0108】
本発明の他の実施形態は、偽性エフェドリンまたはフェニレフリン(phenylepherine)の経口投与により、鼻および呼吸器の鬱滞の緩和が必要な人物のそのような鬱滞を緩和すると共に、そのような人物に本発明の対象の剤形を投与することにより、頭痛、関節痛、病的に分泌物の多い鼻通路の状態(watery nasal passages)、流涙(weeping eyes)、洞の鬱滞および痛み、咳嗽、粘液の過剰な浸出、および気管支炎といった関連する状態をも回復させる方法に向けられている。
【0109】
有利なことに、我々は、剤形が、懸濁液のような液体剤形として設計されているか、または投与前に水で再構成されることができる乾燥剤形として設計されているかにかかわらず、製品の保管寿命の間、および処置の期間の間ずっと剤形の改変型放出部分の放出特質を効果的に安定化する方法を予想外にも見出した。特に、我々は、摂取前の製品中の粒子からの活性成分の放出を防止しながら、一方で、g.i.液中でのそれらの同じ粒子からの活性成分の改変型放出を可能にするという難題を克服した。
【0110】
我々はさらに、第1の活性薬剤と第2の活性薬剤上の半透性コーティングとの間の相互作用を克服することにより、第2のコーティングされた活性薬剤の治療学的効果の持続時間を、コーティングされていない第1の活性薬剤が保持するのに匹敵する持続時間まで延長させる方法を見出した。
【0111】
有利には、本発明の調合物は、例えば、(i)正確に測定可能な単一用量乾燥調合物、または液体懸濁液、(ii)必要に応じて再懸濁されるように異なる量の顆粒を測定することにより得ることができる有意な用量柔軟性を有する、複数用量の顆粒状の調合物、(iii)複数用量の液体懸濁液、および(iv)特に小児科施用で有用な、活性成分が懸濁した濃縮されたドロップを含めた、様々な形式で使用され得る。
【0112】
加えて、調合物は、投与し、嚥下するのに都合がよく、一日量の活性成分の数が減るので、全患者適応性(overall patient compliance)が達成される。嚥下および投与の容易性により、小児科での実施におけるさらなる利点が予測される。
【0113】
下記の実施例がさらに本発明を説明するが、本発明をいかなるようにも限定することを意味するものではない。
【0114】
<例1:フェニレフリン−樹脂複合体粒子の調製>
好適なステンレス鋼容器中で、2125.0gの塩酸フェニレフリンを、2000.0gの脱イオン水に溶解した。これに対して、Rohm & Haas Corporationから商業的に入手可能なAmberlite IRP-69イオン交換樹脂を2500.0g加え、実験室ミキサーを用いて100RPMで少なくとも12時間、攪拌した。結果として得られたスラリーを、減圧ろ過を用いてWattman #4ろ紙を通してろ過した。結果として得られた固形分を1〜2時間風乾し、湿った樹脂粒子を産生し、この樹脂粒子を次に、Glatt流動層GPCG-1ドライヤーで10分間、50℃で乾燥した。結果として得られた乾燥粒子は、約32.0%のフェニレフリンを含んでいた。残った結合していないフェニレフリンが、ろ過されたスラリー溶液中に残っていた。
【0115】
<例2:エチルセルロース半透性第1コーティング溶液の調製>
Dow Chemical Corporationから商品名「Ethocel 10 CPS」で商業的に入手可能なエチルセルロース690グラムと、アセチルクエン酸トリブチル(acetyltributyl citrate)(ATBC)150gと、ステアリン酸マグネシウムUSP20mgとを、周囲条件下で、溶媒の全重量に基づいて3780gのアセトンと3780gのイソプロピルアルコールとを含む溶媒(50:50混合物)中に分散させることにより、コーティング溶液を調製した。この溶液を、実験室ミキサーを用いて75RPMで少なくとも60分間、混合した。
【0116】
結果として得られたコーティング溶液は、湿った全コーティング溶液に基づいて、8.19%のエチルセルロースと、1.78%のアセチルクエン酸トリブチルと、0.24%のステアリン酸マグネシウムと、44.89%のアセトンと、44.89%のイソプロピルアルコールとを含んでいた。この溶液は、10%の固形分を含んでいた。固形分の相対量は、乾燥したコーティング溶液の全重量パーセントに基づいて、エチルセルロース80.23%、ATBC17.44%、およびステアリン酸マグネシウム2.33%であった。
【0117】
<例3:保護用(腸溶性)第2コーティング溶液の調製>
Rohm Pharmaから商業的に入手可能な、商品名「Eudragit L30D-55」で商業的に入手可能なメタクリレートコポリマー分散系(30%固形分)4193.1gを、精製水2396.0g中に分散させることによりコーティング溶液を調製し、周囲条件下で、5分間25RPMで混合した。これに対して、50.8gのモノステアリン酸グリセロールと、126.8gのクエン酸トリエチルとを、50RPMで少なくとも30分間混合しながら加えた。
【0118】
結果として得られたコーティング溶液は、湿った全コーティング溶液に基づいて、61.97%のEudragit L30D-55(Eudragit L30D-55の30%が固形分)、0.75%のモノステアリン酸グリセロール、1.87%のクエン酸トリエチル、および35.41%の精製水を含んでいた。
【0119】
固形分の相当量は、乾燥したコーティング溶液の全重量パーセントに基づいて、Eudragit L30D-55が87.63%、モノステアリン酸グリセロールが3.53%、クエン酸トリエチルが8.83%であった。
【0120】
<例4:単一の半透性エチルセルロース層でコーティングされた薬−樹脂フェニレフリン複合体の調製>
例1から得られた薬−樹脂複合体粒子1000.0グラムを、Glatt GPCG-1/3コーティングユニットに配置し、例2に従って調製されたエチルセルロースコーティング溶液を約37〜42℃の製品温度条件下で、約15.0g/分の速度で、約2バール(約200kPa)の霧化気圧で噴霧することにより、この溶液でコーティングした。結果として得られたコーティングされたフェニレフリン顆粒は、コーティングされたフェニレフリン顆粒の全乾燥重量に基づいて、約47.9%の半透性コーティングを含んだ。
【0121】
<例5:半透性層および外側腸溶性層でコーティングされた薬−樹脂フェニレフリン複合体の調製>
例4に従って調製されたコーティングされたフェニレフリン粒子750.0グラムを、Glatt GPCG-1/3コーティングユニットに配置し、例3に従って調製された腸溶性Eudragit L30Dコーティング溶液を、約54〜71℃の製品温度条件下で、約15.0g/分の速度で、約2バール(約200kPa)の霧化気圧で噴霧することにより、この溶液でコーティングした。結果として得られたコーティングされたフェニレフリン顆粒は、二重コーティングされたフェニレフリン顆粒の全乾燥重量に基づいて、約65.7%の外側保護用腸溶性コーティングを含んだ。最終的な乾燥した二重コーティングされた粒子中の成分の量を表1に示す。
【表1】
【0122】
<例6:速効性放出イブプロフェン用量を含む懸濁液ベースの調製>
【表2】
【0123】
上記の表2に示されているように、1300.0gの精製水USPをScott Turbon高速せん断ミキサー(Scott Turbon high shear mixer)を備えた混合用タンクに入れ、良好なボルテックスを生み出すために約500rpm〜約1000rpmで混合した。次に、この混合用タンクに、アルファ化でんぶんとキサンタンガムとを加え、20分間混合した。これに対して、次にグリセリンを加え、5分間混合した。これに対して、次にスクロースを加え、12分間混合した。ポリソルベート−80NF、クエン酸無水物USP、およびアセスルファムKを次々と加え、その後、結果として得られた混合物を10分間混合した。イブプロフェンUSPをバッチに加え、約500rpmおよび1000rpmで20分間混合した。これに対して次に、残りの精製水を加え、10分間混合した。
【0124】
<例7:速効性放出イブプロフェン用量および単一半透性層でコーティングされたフェニレフリン−樹脂複合体を含む懸濁液の調製>
例4に従って調製されたコーティングされたフェニレフリン粒子600mgを、好適な100mLメスフラスコに加え、例6に従って調製された懸濁液ベースで100.0mL容量まで希釈し、その後、結果として得られる懸濁液が視覚的に均一になるまで、手動でくるくると回転させて(end-over-end)混合した。結果として得られた懸濁液は、100mg/5mLの速効性放出イブプロフェン用量と、5mg/5mLのフェニレフリン用量とを含んだ。
【0125】
<例8:速効性放出イブプロフェン用量ならびに半透性層および外側保護用腸溶性層でコーティングされたフェニレフリン−樹脂複合体を含む懸濁液の調製>
例5に従って調製された、5.7%の活性フェニレフリンHClを含むコーティングされたフェニレフリン1724.0mgを、好適な100mLメスフラスコに加えた。次に、結果として得られた懸濁液を、例6に従って調製された懸濁液ベースで100.0mL容量まで希釈し、結果として得られる懸濁液が均一になるまで手動でくるくると回転させて混合した。結果として得られた最終的な懸濁液は、100mg/5mLの速効性放出イブプロフェン用量と、5mg/5mLの改変型放出フェニレフリン用量とを含んだ。
【0126】
<例9:例7および例8の懸濁液の溶解分析>
溶解媒質:
pH1(0.1N HCl)媒質:1000mLの0.1N HClをパドル付きのUSP Type II装置の3つの容器それぞれに配置した。次に、例7で生成された最終的な懸濁液のサンプル5.0mLを3つの容器のそれぞれに独立して加え、混合物が視覚的に均一になるまで50r.p.m.の速度で、37℃で混合した。
【0127】
例7から得られたサンプルを例8から得られたサンプルに置換して、この手順を繰り返した。
【0128】
例4から得られた粒子約20.5mg、および例5から得られた粒子約133.0mgでそれぞれ置換して、この手順をさらに2回繰り返した。これらは、約4.1mgの用量の遊離塩基(free base)としてのフェニレフリンに基づいて算出した。
【0129】
1時間pH変化媒質:750mLの0.1N HClを、パドル付きのUSP Type II装置の3つのさらなる容器のそれぞれに配置した。次に、例7で生成された最終的な懸濁液のサンプル5.0mLを3つの容器のそれぞれに独立して加え、混合物が均一になるまで50r.p.m.の速度で、37℃で混合した。1時間後、サンプル10mLを器から取り出し、次に、これに対して0.2MのNa3PO4を250mL加え、これにより、媒質組成を約6.8のpHで0.05Mのリン酸ナトリウム緩衝剤約990mLへと変化させた。
【0130】
例7から得られたサンプルを例8から得られたサンプルに置換して、この手順を繰り返した。
【0131】
次に、例4から得られた粒子約20.5mg、および例5から得られた粒子約133.0mgでそれぞれ置換して、この手順をさらに2回繰り返した。これらは、約4.1mgの用量の遊離塩基としてのフェニレフリンに基づいて算出した。
【0132】
pH7.2(リン酸緩衝)媒質:NaOHでpH7.2に調節された0.05MのKH2PO4緩衝剤1000mLを、パドル付きのUSP Type II装置の3つのさらなる容器にそれぞれ配置した。次に、例7で生成された最終的な懸濁液のサンプル5.0mLを3つの容器のそれぞれに独立して加え、混合物が視覚的に均一になるまで50r.p.m.の速度で、37℃で混合した。
【0133】
例7から得られたサンプルを例8から得られたサンプルに置換して、この手順を繰り返した。
【0134】
次に、例4から得られた粒子約20.5mg、および例5から得られた粒子約133.0mgでそれぞれ置換して、この手順をさらに2回繰り返した。これらは、約4.1mgの用量の遊離塩基としてのフェニレフリンに基づいて算出した。
【0135】
サンプリングおよび分析:
その後、それぞれ1、2、3、4、6、7、および8時間後、懸濁液/緩衝剤混合物のサンプル10mLをそれぞれの容器から独立して取り出した。
【0136】
1、2、3、4、6、7、および8時間それぞれでのフェニレフリンおよびイブプロフェンについての溶解曲線を導き出すために、波長270nmに設定された、Waters(登録商標)Alliance 2695分離モジュールおよびWaters(登録商標)2996 PDA検出器を備えた高圧液体クロマトグラフ(HPLC)を用いてイブプロフェンおよびフェニレフリンの含量について、各サンプル10mLを独立して分析した。溶解サンプルのそれぞれを、フェニレフリン(遊離塩基)を0.004mg/mLで含み、イブプロフェン(遊離酸)を0.10mg/mLで含む、フェニレフリン遊離塩基およびイブプロフェン遊離酸の100%放出に必要とされる理論濃度に相関した混合された標準と比較した。
【0137】
HPLCで使用された移動層は、0.2%H3PO4/メタノール/アセトニトリル(40/35/25)中の20mMのドデシル硫酸ナトリウム(SDS)を用いて調製した。注入量を50μLとし、運転時間を約10分間とし、ポンプ流量(pump flow)を1.0mL/分とした。分析のために用いたカラムは、Phenomenex Luna C8(2)(3μm、4.6x75mm)であった。
【0138】
HPLCの結果を図1〜4に示す。この図では、3つの器中に溶解されたフェニレフリンのパーセントの平均を表す点がそれぞれグラフ化されている。
【0139】
図1および図3の比較により、エチルセルロースでコーティングされた粒子がイブプロフェン含有懸濁液に配置されると、この粒子からのフェニレフリンの放出速度が実質的に増加したことが示された。対照的に、図2および図4の比較により、エチルセルロースの内側層と腸溶性ポリマーの外側層とでコーティングされた粒子がイブプロフェン含有懸濁液に配置されると、この粒子からのフェニレフリン放出速度は実質的にそのままであったことが示された。
【図面の簡単な説明】
【0140】
【図1】3つの異なる媒質中での時間に対する例4に従って調製されたエチルセルロース半透性コーティング層でコーティングされた粒子中のフェニレフリンの放出された(すなわち、溶解)パーセンテージを示している。
【図2】3つの異なる媒質中での時間に対する例5に従って調製されたエチルセルロース半透性コーティング層および外部腸溶性コーティング層でコーティングされた粒子中のフェニレフリンの放出された(すなわち、溶解)パーセンテージを示している。
【図3】時間に対する例7に従って調製された速効性放出イブプロフェンを含む懸濁液中のエチルセルロースコーティング層でコーティングされた粒子中のフェニレフリンの放出された(すなわち、溶解)パーセンテージを示している。
【図4】時間に対する例8に従って調製された速効性放出イブプロフェンを含む懸濁液中のエチルセルロースコーティング層および外部腸溶性コーティング層でコーティングされた粒子中のフェニレフリンの放出された(すなわち、溶解)パーセンテージを示している。
【特許請求の範囲】
【請求項1】
液体医薬品剤形において、
a)NSAIDおよび/または医薬品に許容可能なその塩から構成される第1の活性成分を含む第1の部分であって、前記第1の活性成分は、前記剤形が溶解媒質に接触すると実質的に速効式に前記剤形から放出される、第1の部分と、
b)第2の部分であって、
i)第2の活性成分を有するイオン交換樹脂粒子であって、前記第2の活性成分は、前記イオン交換樹脂上に結合されていて薬−樹脂複合体粒子を形成している、イオン交換樹脂粒子、
ii)前記薬−樹脂複合体粒子を実質的に被覆してコーティングされた粒子を形成している半透性コーティング層、および、
iii)前記コーティングされた粒子を実質的に被覆している保護コーティング層、
を含んでいる、第2の部分と、
を含み、
前記第2の活性成分は、前記剤形が前記溶解媒質に接触すると改変型放出式に前記第2の部分から放出され、
前記剤形の前記第2の部分から放出されたときの前記第2の活性成分の治療学的効果の持続時間は、前記第1の活性成分の治療学的効果の持続時間と実質的に同じである、剤形。
【請求項2】
請求項1に記載の剤形において、
前記第1の部分および前記第2の部分の投与のための媒体、
をさらに含む、剤形。
【請求項3】
請求項2に記載の剤形において、
前記媒体は、溶媒、構造化剤、膨潤剤、懸濁系、界面活性物質、甘味料、緩衝剤、保存剤、細菌発育阻止剤、着色料、調味剤、およびこれらの混合物からなる群より選択される、剤形。
【請求項4】
請求項2に記載の剤形において、
前記媒体は、水から構成され、
前記剤形は、液体懸濁液の形態にある、剤形。
【請求項5】
請求項2に記載の剤形において、
前記媒体は、水、または、水と、グリコール、アルコール、グリセロール、およびこれらの混合物からなる群より選択される医薬品に許容可能な水混和性共溶媒との混合物からなる群より選択される、剤形。
【請求項6】
請求項1に記載の剤形において、
前記第2の部分の前記イオン交換樹脂粒子は、スチレン/ジビニルベンゼンコポリマー、コレスチラミン(cholestryamines)、ポラクリリンカリウム、およびこれらの混合物から構成される、剤形。
【請求項7】
請求項1に記載の剤形において、
前記半透性コーティング層は、セルロースアセテート、エチルセルロース、非腸溶性ポリメタクリレート、およびこれらの混合物からなる群より選択される1つ以上の薬剤から構成される、剤形。
【請求項8】
請求項1に記載の剤形において、
前記保護コーティング層は、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートスクシネート、セルロースアセテートフタレート、ポリビニルアセテートフタレート、セラック、腸溶性ポリメタクリレート系ポリマー、ならびに、これらのコポリマーおよび混合物からなる群より選択される腸溶性ポリマーから構成される、剤形。
【請求項9】
請求項1に記載の剤形において、
前記保護コーティング層は、脂質、ワックス、またはこれらの混合物から構成される、剤形。
【請求項10】
請求項1に記載の剤形において、
前記保護コーティング層は、スクロース脂肪酸エステル;モノグリセリド;ジグリセリド;トリグリセリド;グリセリルベヘナート;グリセリルパルミトステアレート;グリセリルトリステアレート;グリセリルトリラウリレート;グリセリルミリステート;GLYCOWAX-932;ラウロイルマクロゴール−32グリセリド;ステアロイルマクロゴール−32グリセリド;約C10〜約C40の脂肪酸鎖長を有する脂肪酸エステル;およびこれらの混合物からなる群より選択される、剤形。
【請求項11】
請求項1に記載の剤形において、
前記保護コーティング層は、カルナウバろう、鯨ろう、蜜ろう、カンデリラろう、セラックろう、カルヌバろう、蜜ろう、微晶ろう、およびパラフィンろう、ならびにこれらの混合物からなる群より選択されるワックスから構成される、剤形。
【請求項12】
請求項1に記載の剤形において、
前記半透性コーティング層は、前記半透性コーティング層の全乾燥重量に基づいて、約0.1%〜約40%の可塑剤をさらに含む、剤形。
【請求項13】
請求項1に記載の剤形において、
前記保護コーティングは、前記保護コーティングの全乾燥重量に基づいて、約0.1%〜約40%の可塑剤をさらに含む、剤形。
【請求項14】
請求項1に記載の剤形において、
前記半透性コーティング層に対する前記保護コーティング層の重量比は、約10:90〜約90:10である、剤形。
【請求項15】
請求項1に記載の剤形において、
前記第2の部分は、前記第2の部分の全乾燥重量に基づいて、
a)約5パーセント〜約80パーセントの前記第1のコーティング層と、
b)約10パーセント〜約90パーセントの前記第2のコーティング層と、
c)約1パーセント〜約50パーセントの前記薬−樹脂複合体粒子と、
から構成される、剤形。
【請求項16】
請求項1に記載の剤形において、
前記剤形の全重量に基づいて、約0.1%〜約5%の前記第1の部分と、約0.05%〜約5%の前記第2の部分とをさらに含む、剤形。
【請求項17】
請求項1に記載の剤形において、
前記剤形の全重量に基づいて、
a)約0.1%〜約5%の前記第1の活性成分と、
b)約0.005%〜約1%の前記第2の活性成分と、
をさらに含む、剤形。
【請求項18】
請求項1に記載の剤形において、
前記第1の活性成分は、プロピオン酸誘導体、または医薬品に許容可能なその塩であり、
前記第2の活性成分は、フェニレフリン、または医薬品に許容可能なその塩である、剤形。
【請求項19】
痛みの処置が必要なほ乳類のその痛みを処置する方法において、
前記剤形の投与後少なくとも約4時間〜約6時間の期間にわたって前記ほ乳類に痛みの軽減をもたらすのに有効な量の請求項1の剤形を投与すること、
を含む、方法。
【請求項20】
洞の鬱滞の処置が必要なほ乳類のその洞の鬱滞を処置する方法において、
前記剤形の投与後少なくとも約4時間〜約6時間の期間にわたって前記ほ乳類に痛みの軽減をもたらすのに有効な量の請求項1の剤形を投与すること、
を含む、方法。
【請求項21】
医薬品剤形中のNSAIDおよび鬱血除去薬をこれが必要なほ乳類に対して投与する方法において、
前記ほ乳類が、投与時に、速効性放出用量の前記NSAIDまたは医薬品に許容可能なその塩を受け取り、前記剤形の投与後、約4時間〜約6時間の期間にわたって、改変型放出用量の前記鬱血除去薬を受け取るように、ほ乳類に前記剤形を提供すること、
を含み、
さらなるNSAIDまたは鬱血除去薬が前記期間の間、提供されない、方法。
【請求項22】
液体懸濁液剤形において、
a)前記剤形が溶解媒質に接触すると実質的に速効式に前記剤形から放出されるNSAIDまたは医薬品に許容可能なその塩を含む第1の部分と、
b)鬱血除去薬を含む粒子の第2の部分であって、前記剤形が前記溶解媒質に接触すると改変型放出式に前記粒子から前記鬱血除去薬が放出される、第2の部分と、
c)水、または、水と、グリコール、アルコール、グリセロール、およびこれらの混合物からなる群より選択される医薬品に許容可能な水混和性共溶媒との混合物と、
を含み、
前記鬱血除去薬は、前記剤形が前記溶解媒質に接触する改変型放出式に前記第2の部分から放出され、
前記剤形の前記第2の部分から放出されたときの前記鬱血除去薬の治療学的効果の持続時間は、前記NSAIDの治療学的効果の持続時間と実質的に同じである、剤形。
【請求項23】
液体医薬品剤形において、
a)NSAIDおよび/または医薬品に許容可能なその塩から構成される第1の活性成分を含む第1の部分であって、前記第1の活性成分は、前記剤形が溶解媒質に接触すると実質的に速効式に前記剤形から放出される、第1の部分と、
b)第2の部分であって、
i)第2の活性成分を有するイオン交換樹脂粒子であって、前記第2の活性成分は、前記イオン交換樹脂上に結合されていて薬−樹脂複合体粒子を形成している、イオン交換樹脂粒子、
ii)前記薬−樹脂複合体粒子を実質的に被覆してコーティングされた粒子を形成している半透性コーティング層、および、
iii)前記コーティングされた粒子を実質的に被覆している保護コーティング層、
を含んでいる、第2の部分と、
を含み、
前記第2の活性成分は、前記剤形が前記溶解媒質に接触すると改変型放出式に前記第2の部分から放出され、
前記第2の活性成分は、前記第1の活性成分と同じであるか、または異なる、剤形。
【請求項24】
液体医薬品剤形において、
a)NSAIDまたは医薬品に許容可能なその塩から構成される第1の活性成分を含む第1の部分であって、前記第1の活性成分は、前記剤形が溶解媒質に接触すると実質的に速効式に前記剤形から放出される、第1の部分と、
b)第2の部分であって、
i)第2の活性成分を有するカチオン交換樹脂粒子であって、前記第2の活性成分は、前記カチオン交換樹脂上に結合されていて薬−樹脂複合体粒子を形成しており、前記第2の活性成分は、フェニレフリンまたは医薬品に許容可能なその塩から構成されており、前記カチオン交換樹脂粒子は、スチレンおよびジビニルベンゼンのスルホン化コポリマー、またはメタクリル酸およびジビニルベンゼンのコポリマーから構成されている、カチオン交換樹脂粒子、
ii)前記薬−樹脂複合体粒子を実質的に被覆してコーティングされた粒子を形成している半透性コーティング層であって、エチルセルロース、または非腸溶性ポリメタクリレートから構成されている、半透性コーティング層、および、
iii)前記コーティングされた粒子を実質的に被覆している保護コーティング層、
を含んでいる、第2の部分と、
を含み、
前記第2の活性成分は、前記剤形が前記溶解媒質に接触すると改変型放出式に前記第2の部分から放出され、
前記剤形の前記第2の部分から放出されたときの前記第2の活性成分の治療学的効果の持続時間は、約4時間〜約6時間であり、前記第1の活性成分の治療学的効果の持続時間は、約4時間〜約6時間である、剤形。
【請求項1】
液体医薬品剤形において、
a)NSAIDおよび/または医薬品に許容可能なその塩から構成される第1の活性成分を含む第1の部分であって、前記第1の活性成分は、前記剤形が溶解媒質に接触すると実質的に速効式に前記剤形から放出される、第1の部分と、
b)第2の部分であって、
i)第2の活性成分を有するイオン交換樹脂粒子であって、前記第2の活性成分は、前記イオン交換樹脂上に結合されていて薬−樹脂複合体粒子を形成している、イオン交換樹脂粒子、
ii)前記薬−樹脂複合体粒子を実質的に被覆してコーティングされた粒子を形成している半透性コーティング層、および、
iii)前記コーティングされた粒子を実質的に被覆している保護コーティング層、
を含んでいる、第2の部分と、
を含み、
前記第2の活性成分は、前記剤形が前記溶解媒質に接触すると改変型放出式に前記第2の部分から放出され、
前記剤形の前記第2の部分から放出されたときの前記第2の活性成分の治療学的効果の持続時間は、前記第1の活性成分の治療学的効果の持続時間と実質的に同じである、剤形。
【請求項2】
請求項1に記載の剤形において、
前記第1の部分および前記第2の部分の投与のための媒体、
をさらに含む、剤形。
【請求項3】
請求項2に記載の剤形において、
前記媒体は、溶媒、構造化剤、膨潤剤、懸濁系、界面活性物質、甘味料、緩衝剤、保存剤、細菌発育阻止剤、着色料、調味剤、およびこれらの混合物からなる群より選択される、剤形。
【請求項4】
請求項2に記載の剤形において、
前記媒体は、水から構成され、
前記剤形は、液体懸濁液の形態にある、剤形。
【請求項5】
請求項2に記載の剤形において、
前記媒体は、水、または、水と、グリコール、アルコール、グリセロール、およびこれらの混合物からなる群より選択される医薬品に許容可能な水混和性共溶媒との混合物からなる群より選択される、剤形。
【請求項6】
請求項1に記載の剤形において、
前記第2の部分の前記イオン交換樹脂粒子は、スチレン/ジビニルベンゼンコポリマー、コレスチラミン(cholestryamines)、ポラクリリンカリウム、およびこれらの混合物から構成される、剤形。
【請求項7】
請求項1に記載の剤形において、
前記半透性コーティング層は、セルロースアセテート、エチルセルロース、非腸溶性ポリメタクリレート、およびこれらの混合物からなる群より選択される1つ以上の薬剤から構成される、剤形。
【請求項8】
請求項1に記載の剤形において、
前記保護コーティング層は、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートスクシネート、セルロースアセテートフタレート、ポリビニルアセテートフタレート、セラック、腸溶性ポリメタクリレート系ポリマー、ならびに、これらのコポリマーおよび混合物からなる群より選択される腸溶性ポリマーから構成される、剤形。
【請求項9】
請求項1に記載の剤形において、
前記保護コーティング層は、脂質、ワックス、またはこれらの混合物から構成される、剤形。
【請求項10】
請求項1に記載の剤形において、
前記保護コーティング層は、スクロース脂肪酸エステル;モノグリセリド;ジグリセリド;トリグリセリド;グリセリルベヘナート;グリセリルパルミトステアレート;グリセリルトリステアレート;グリセリルトリラウリレート;グリセリルミリステート;GLYCOWAX-932;ラウロイルマクロゴール−32グリセリド;ステアロイルマクロゴール−32グリセリド;約C10〜約C40の脂肪酸鎖長を有する脂肪酸エステル;およびこれらの混合物からなる群より選択される、剤形。
【請求項11】
請求項1に記載の剤形において、
前記保護コーティング層は、カルナウバろう、鯨ろう、蜜ろう、カンデリラろう、セラックろう、カルヌバろう、蜜ろう、微晶ろう、およびパラフィンろう、ならびにこれらの混合物からなる群より選択されるワックスから構成される、剤形。
【請求項12】
請求項1に記載の剤形において、
前記半透性コーティング層は、前記半透性コーティング層の全乾燥重量に基づいて、約0.1%〜約40%の可塑剤をさらに含む、剤形。
【請求項13】
請求項1に記載の剤形において、
前記保護コーティングは、前記保護コーティングの全乾燥重量に基づいて、約0.1%〜約40%の可塑剤をさらに含む、剤形。
【請求項14】
請求項1に記載の剤形において、
前記半透性コーティング層に対する前記保護コーティング層の重量比は、約10:90〜約90:10である、剤形。
【請求項15】
請求項1に記載の剤形において、
前記第2の部分は、前記第2の部分の全乾燥重量に基づいて、
a)約5パーセント〜約80パーセントの前記第1のコーティング層と、
b)約10パーセント〜約90パーセントの前記第2のコーティング層と、
c)約1パーセント〜約50パーセントの前記薬−樹脂複合体粒子と、
から構成される、剤形。
【請求項16】
請求項1に記載の剤形において、
前記剤形の全重量に基づいて、約0.1%〜約5%の前記第1の部分と、約0.05%〜約5%の前記第2の部分とをさらに含む、剤形。
【請求項17】
請求項1に記載の剤形において、
前記剤形の全重量に基づいて、
a)約0.1%〜約5%の前記第1の活性成分と、
b)約0.005%〜約1%の前記第2の活性成分と、
をさらに含む、剤形。
【請求項18】
請求項1に記載の剤形において、
前記第1の活性成分は、プロピオン酸誘導体、または医薬品に許容可能なその塩であり、
前記第2の活性成分は、フェニレフリン、または医薬品に許容可能なその塩である、剤形。
【請求項19】
痛みの処置が必要なほ乳類のその痛みを処置する方法において、
前記剤形の投与後少なくとも約4時間〜約6時間の期間にわたって前記ほ乳類に痛みの軽減をもたらすのに有効な量の請求項1の剤形を投与すること、
を含む、方法。
【請求項20】
洞の鬱滞の処置が必要なほ乳類のその洞の鬱滞を処置する方法において、
前記剤形の投与後少なくとも約4時間〜約6時間の期間にわたって前記ほ乳類に痛みの軽減をもたらすのに有効な量の請求項1の剤形を投与すること、
を含む、方法。
【請求項21】
医薬品剤形中のNSAIDおよび鬱血除去薬をこれが必要なほ乳類に対して投与する方法において、
前記ほ乳類が、投与時に、速効性放出用量の前記NSAIDまたは医薬品に許容可能なその塩を受け取り、前記剤形の投与後、約4時間〜約6時間の期間にわたって、改変型放出用量の前記鬱血除去薬を受け取るように、ほ乳類に前記剤形を提供すること、
を含み、
さらなるNSAIDまたは鬱血除去薬が前記期間の間、提供されない、方法。
【請求項22】
液体懸濁液剤形において、
a)前記剤形が溶解媒質に接触すると実質的に速効式に前記剤形から放出されるNSAIDまたは医薬品に許容可能なその塩を含む第1の部分と、
b)鬱血除去薬を含む粒子の第2の部分であって、前記剤形が前記溶解媒質に接触すると改変型放出式に前記粒子から前記鬱血除去薬が放出される、第2の部分と、
c)水、または、水と、グリコール、アルコール、グリセロール、およびこれらの混合物からなる群より選択される医薬品に許容可能な水混和性共溶媒との混合物と、
を含み、
前記鬱血除去薬は、前記剤形が前記溶解媒質に接触する改変型放出式に前記第2の部分から放出され、
前記剤形の前記第2の部分から放出されたときの前記鬱血除去薬の治療学的効果の持続時間は、前記NSAIDの治療学的効果の持続時間と実質的に同じである、剤形。
【請求項23】
液体医薬品剤形において、
a)NSAIDおよび/または医薬品に許容可能なその塩から構成される第1の活性成分を含む第1の部分であって、前記第1の活性成分は、前記剤形が溶解媒質に接触すると実質的に速効式に前記剤形から放出される、第1の部分と、
b)第2の部分であって、
i)第2の活性成分を有するイオン交換樹脂粒子であって、前記第2の活性成分は、前記イオン交換樹脂上に結合されていて薬−樹脂複合体粒子を形成している、イオン交換樹脂粒子、
ii)前記薬−樹脂複合体粒子を実質的に被覆してコーティングされた粒子を形成している半透性コーティング層、および、
iii)前記コーティングされた粒子を実質的に被覆している保護コーティング層、
を含んでいる、第2の部分と、
を含み、
前記第2の活性成分は、前記剤形が前記溶解媒質に接触すると改変型放出式に前記第2の部分から放出され、
前記第2の活性成分は、前記第1の活性成分と同じであるか、または異なる、剤形。
【請求項24】
液体医薬品剤形において、
a)NSAIDまたは医薬品に許容可能なその塩から構成される第1の活性成分を含む第1の部分であって、前記第1の活性成分は、前記剤形が溶解媒質に接触すると実質的に速効式に前記剤形から放出される、第1の部分と、
b)第2の部分であって、
i)第2の活性成分を有するカチオン交換樹脂粒子であって、前記第2の活性成分は、前記カチオン交換樹脂上に結合されていて薬−樹脂複合体粒子を形成しており、前記第2の活性成分は、フェニレフリンまたは医薬品に許容可能なその塩から構成されており、前記カチオン交換樹脂粒子は、スチレンおよびジビニルベンゼンのスルホン化コポリマー、またはメタクリル酸およびジビニルベンゼンのコポリマーから構成されている、カチオン交換樹脂粒子、
ii)前記薬−樹脂複合体粒子を実質的に被覆してコーティングされた粒子を形成している半透性コーティング層であって、エチルセルロース、または非腸溶性ポリメタクリレートから構成されている、半透性コーティング層、および、
iii)前記コーティングされた粒子を実質的に被覆している保護コーティング層、
を含んでいる、第2の部分と、
を含み、
前記第2の活性成分は、前記剤形が前記溶解媒質に接触すると改変型放出式に前記第2の部分から放出され、
前記剤形の前記第2の部分から放出されたときの前記第2の活性成分の治療学的効果の持続時間は、約4時間〜約6時間であり、前記第1の活性成分の治療学的効果の持続時間は、約4時間〜約6時間である、剤形。
【図1】

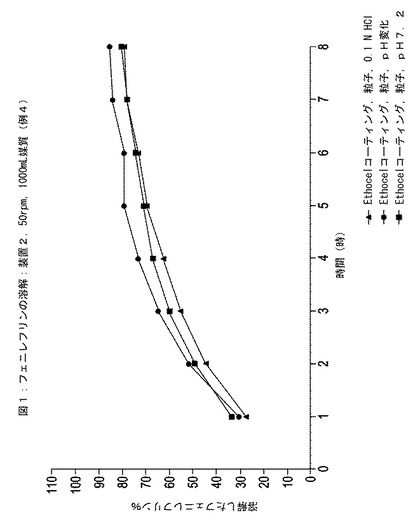
【図2】
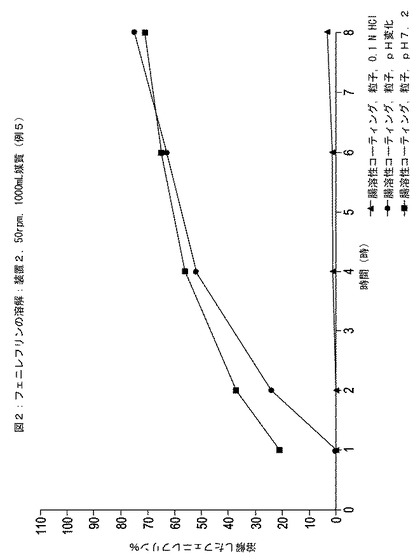
【図3】
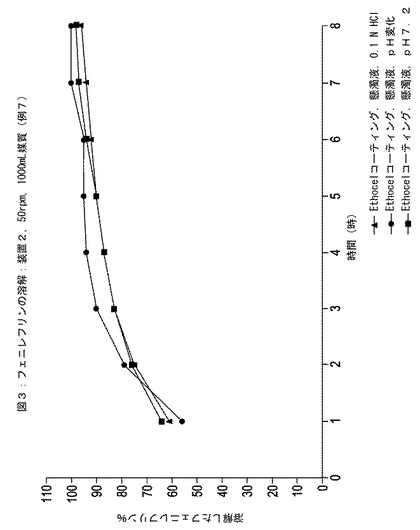
【図4】
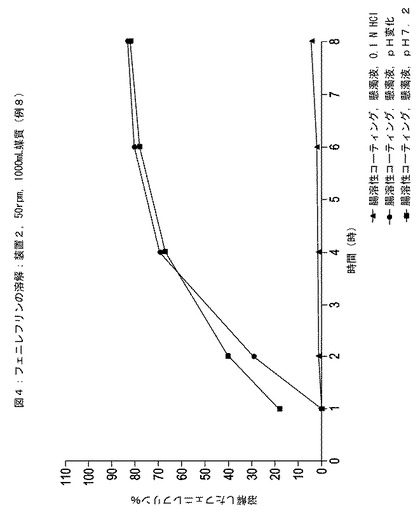
【図2】
【図3】
【図4】
【公表番号】特表2010−510320(P2010−510320A)
【公表日】平成22年4月2日(2010.4.2)
【国際特許分類】
【出願番号】特願2009−538480(P2009−538480)
【出願日】平成19年11月20日(2007.11.20)
【国際出願番号】PCT/US2007/085166
【国際公開番号】WO2008/064192
【国際公開日】平成20年5月29日(2008.5.29)
【出願人】(506105814)マクニール−ピーピーシー・インコーポレーテツド (69)
【Fターム(参考)】
【公表日】平成22年4月2日(2010.4.2)
【国際特許分類】
【出願日】平成19年11月20日(2007.11.20)
【国際出願番号】PCT/US2007/085166
【国際公開番号】WO2008/064192
【国際公開日】平成20年5月29日(2008.5.29)
【出願人】(506105814)マクニール−ピーピーシー・インコーポレーテツド (69)
【Fターム(参考)】
[ Back to top ]