
改変型熱ショックタンパク質−抗原性ペプチド複合体
【課題】 ワクチンまたは治療薬を商業生産する場合、大量のhsp-ペプチド複合体を定常的に供給することが有利である。従って、hsp-ペプチド複合体の信頼性のある供給源が必要である。
【解決手段】 本発明は、癌細胞または感染細胞の抗原性ペプチドに非共有結合で結合した改変型熱ショックタンパク質からなる、予防または治療に有効な免疫原性複合体を精製する方法に関する。本発明の方法は、分泌可能な改変型熱ショックタンパク質をコードするヌクレオチド配列を構築し、適切な宿主細胞内で該配列を発現させ、細胞培養物および細胞から免疫原性複合体を回収し、アフィニティークロマトグラフィーによって免疫原性複合体を精製することを含む。大量の免疫原性複合体が該遺伝子配列を含有する宿主細胞の大規模培養により得られる。前記複合体は、癌もしくは感染細胞に対して特異的な免疫応答を引き出すためのワクチンとして、または癌もしくは感染症を治療または予防するためのワクチンとして使用することができる。
【解決手段】 本発明は、癌細胞または感染細胞の抗原性ペプチドに非共有結合で結合した改変型熱ショックタンパク質からなる、予防または治療に有効な免疫原性複合体を精製する方法に関する。本発明の方法は、分泌可能な改変型熱ショックタンパク質をコードするヌクレオチド配列を構築し、適切な宿主細胞内で該配列を発現させ、細胞培養物および細胞から免疫原性複合体を回収し、アフィニティークロマトグラフィーによって免疫原性複合体を精製することを含む。大量の免疫原性複合体が該遺伝子配列を含有する宿主細胞の大規模培養により得られる。前記複合体は、癌もしくは感染細胞に対して特異的な免疫応答を引き出すためのワクチンとして、または癌もしくは感染症を治療または予防するためのワクチンとして使用することができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、米国立保健研究所(National Institutes of Health)により与えられた認可番号CA57904に基づいて政府の支援を得て行われた。政府は、本発明に関して一定の権利を有する。
【0002】
1. 緒言
本発明は、癌または感染症の予防および/または治療のためのワクチンとして有用な免疫原性物質を調製するための方法に関する。ワクチンには、改変型熱ショックタンパク質(hsp)(例えば、限定されるものではないが、hsp70、hsp9O、gp96、およびタンパク質ジスルフィドイソメラーゼ)と抗原性ペプチドとの非共有結合複合体が含まれている。ワクチンにより、特定のタイプの癌細胞または感染細胞に対する被験者の免疫応答を誘発または増大することができる。
【背景技術】
【0003】
2. 発明の背景
2.1. 癌の病理生物学
癌は、主に、特定の正常組織に由来する異常細胞の数の増大によって特性付けられる。また、疾患の過程には、これらの異常細胞による隣接組織の浸潤および循環系を介する局所リンパ節や遠隔部位へのこれらの異常細胞の拡散(転移)が関与する。臨床データおよび分子生物学的研究から示唆されるように、癌は、特定の条件下で新形成に発展する可能性のある新生物発生前の小さな変化から開始される多段階過程である。
【0004】
前癌性異常細胞増殖は、過形成、化生、または特に異形成によって例証されている(このような異常増殖条件に関するレビューについては、Robbins and Angell, 1976, Basic Pathology, 2d Ed., W.B. Saunders Co., Philadelphia, pp. 68-79を参照されたい)。過形成とは、構造または機能の有意な変化を伴わずに組織または器官の細胞数が増加する制御された細胞増殖の形態である。1例にすぎないが、子宮内膜過形成は、子宮内膜癌に先行して起こる場合が多い。化生とは、1つのタイプの成体細胞すなわち十分に分化した細胞が別のタイプの成体細胞と置き換わる制御された細胞増殖の形態である。化生は、上皮細胞または結合組織細胞で起こる可能性がある。非定型化生には、いくらか疾患性のある化生上皮が関与する。異形成は、多くの場合、癌の前兆であり、主に、上皮で見出される。また、異形成は、非新生物性細胞増殖の最も疾患性の強い形態であり、個々の細胞の均一性の消失や細胞の構造的配向の消失を伴う。異形成細胞は、多くの場合、濃く染色される異常に大きな核を有し、多形性を呈する。異形成は、特質上、慢性刺激または炎症が存在する場合に起こり、多くの場合、子宮頸管、気道、口腔、および胆嚢で見出される。
【0005】
新生物性病変は、クローン産生的に進化して、特に、新生物細胞が宿主の免疫学的監視から逃げる条件下で、浸潤、増殖、転移、および異質性に対する能力を増大させながら進行する可能性がある(Roitt, I., Brostoff, J. and Male, D., 1993, Immunology, 3rd ed., Mosby, St. Louis, pps. 17.1-17.12)。
【0006】
2.2. ワクチン接種
ワクチン接種により、世界中の多くの国々で、ポリオ、破傷風、水痘、麻疹などの特定の疾患が根絶された。この手法では、感染症を予防するために、免疫系の能力が利用された。タンパク質などの非生存物質を用いてこのようなワクチン接種を行うと、一般的には、抗体応答またはCD4+ヘルパーT細胞応答が誘発される(Raychaudhuri & Morrow, 1993, Immunology Today, 14:344-348)。一方、生存細胞や感染性ウイルスなどの生存物質を用いてワクチン接種または感染処理を行うと、一般的には、CD8+細胞傷害性Tリンパ球(CTL)応答が誘発される。CTL応答は、癌、感染性ウイルス、および細菌に対する防御を行う上で極めて重要である。これに起因して実用上の問題が生じる。なぜなら、CTL応答を達成する唯一の方法は、それ自体病原性である生存性薬剤を使用するものであるからである。この問題は、一般に、弱毒化されたウイルス株および細菌株を使用することによって、またはワクチン接種に使用することのできる細胞全体を死滅させることによって回避される。これらのストラテジーはうまく機能したが、弱毒化された株を使用する場合、弱毒化薬剤が遺伝子的に宿主DNAと組み換わって毒性株に変化する恐れがあるというリスクを常に伴う。したがって、タンパク質などの非生存物質を用いた特異的なワクチン接種を行うことによってCD8+CTL応答を誘発することのできる方法が必要である。
【0007】
腫瘍免疫学の時代は、PrehnおよびMainによる実験から始まった。彼らは、メチルコラントレン(MCA)誘発肉腫上の抗原を移植アッセイによりマウスの正常組織中で検出できなかったことから、これらの抗原が腫瘍特異的であることを明らかにした(Prehn et al., 1957, J. Natl. Cancer Inst. 18:769-778)。この考えは、MCA誘発腫瘍の発生元であるマウスにおいてMCA誘発腫瘍に対する腫瘍特異的耐性を誘発することができることを実証する更なる実験によって確かめられた(Klein et al., 1960, Cancer Res. 20:1561-1572)。
【0008】
その後の研究により、他の化学的もしくは物理的発癌物質で誘発された腫瘍または自然発生腫瘍についても、腫瘍特異的抗原が見出された(Kripke, 1974, J. Natl. Cancer Inst. 53:1333-1336; Vaage, 1968, Cancer Res. 28:2477- 2483; Carswell et al., 1970, J. Natl. Cancer Inst. 44:1281- 1288)。これらの研究では移植された腫瘍の増殖に対する防御免疫が腫瘍特異的抗原の判定基準として使用されたので、これらの抗原は、通常、「腫瘍特異的移植抗原」または「腫瘍特異的拒絶抗原」とも呼ばれる。いくつかの因子が腫瘍の免疫原性に大きく影響する可能性があり、こうした因子としては、例えば、関係する発癌物質の特異的タイプ、宿主の免疫適格性、および潜伏期間が挙げられる(Old et al., 1962, Ann. N.Y. Acad. Sci. 101:80-106; Bartlett, 1972, J. Natl. Cancer Inst. 49:493-504)。
【0009】
すべてというわけではないが、ほとんどの発癌物質は、突然変異を誘発して腫瘍特異的抗原を発現させる突然変異誘発物質である(Ames, 1979, Science 204:587-593; Weisburger et al., 1981, Science 214:401-407)。発癌物質の中には免疫抑制性のものもある(Malmgren et al., 1952, Proc. Soc. Exp. Biol. Med. 79:484-488)。実験的証拠から示唆されるように、腫瘍の免疫原性と潜伏期間(発癌物質への暴露から腫瘍出現までの時間)との間には、一定の逆相関関係がある(Old et al., 1962, Ann. N.Y. Acad. Sci. 101:80-106; および Bartlett, 1972, J. Natl. Cancer Inst. 49:493-504)。他の研究から、拒絶反応は起こさないが、それにもかかわらず、場合により特異的免疫応答を刺激する可能性のある腫瘍特異的抗原の存在が示唆された(Roitt I., Brostoff, J. and Male, D., 1993, Immunology, 3rd ed., Mosby, St. Louis, pp. 17.1-17.12)。
【0010】
2.3. 熱ショックタンパク質
熱ショックタンパク質(hsp)は、ストレスタンパク質とも呼ばれ、これらは同義的に用いられる。同定の対象となった最初のストレスタンパク質は、熱ショックに応答して細胞によって合成されるタンパク質であった。これまでに、hspの3つの主要なファミリーが分子量に基づいて同定された。これらのファミリーは、hsp60、hsp70、およびhsp90と呼ばれてきたものであり、これらの数字は、ストレスタンパク質のおよその分子量をキロダルトン単位で表している。その後、これらのファミリーの多くのメンバーが、栄養欠乏、代謝阻害、酸素ラジカル、細胞内病原体による感染などの他のストレス刺激に応答して誘導されることが明らかにされた。(Welch, May 1993, Scientific American 56-64; Young, 1990, Annu. Rev. Immunol. 8:401-420; Craig, 1993, Science 260:1902-1903; Gethirig et al., 1992, Nature 355:33-45; および Lindquist et al., 1988, Annu. Rev. Genetics 22:631-677を参照されたい。)
【0011】
主要なhspは、ストレスを受けた細胞中では非常に高レベルまで蓄積する可能性があるが、ストレスを受けなかった細胞中では低レベル〜中レベルで生成する。例えば、高誘導性哺乳類hsp7Oは、常温ではほとんど検出できないが、熱ショックを受けると細胞中で最も活発に合成されるタンパク質の1つになる(Welch et al., 1985, J. Cell. Biol. 101:1198-1211)。これとは対照的に、hsp90およびhsp60タンパク質は、常温において、すべてではないがほとんどの哺乳類細胞中に多量に存在し、しかも熱によって更に誘導される(Lai et al., 1984, Mol. Cell. Biol. 4:2802-2810; van Bergen en Henegouwen et al., 1987, Genes Dev. 1:525-531)。
【0012】
熱ショックタンパク質は、現存するタンパク質の中で最も高度に保存されているタンパク質に属する。例えば、E. coli由来のhsp70であるDnaKは、擦過傷由来のhsp70タンパク質に対して約50%のアミノ酸配列同一性を有する(Bardwell et al., 1984, Proc. Natl. Acad. Sci. 81:848-852)。また、hsp60ファミリーとhsp90ファミリーとの間にも、同じように高レベルのファミリー間の保存が見られる(Hickey et al., 1989, Mol. Cell. Biol. 9:2615-2626; Jindal, 1989, Mol. Cell. Biol. 9:2279-2283)。更に、hsp60、hsp70、およびhsp90ファミリーには、配列は例えば35%を超えるアミノ酸同一性を有するが発現レベルはストレスによって変化しないストレスタンパク質に関連付けられるタンパク質が含まれることが見出された。
【0013】
熱ショックおよび他の生理的ストレスに対する細胞応答に関する研究から、hspは、これらの有害な条件からの細胞保護に関与するだけではなく、ストレスを受けていない細胞中における本質的な生化学的および免疫学的過程にも関与することが実証された。hsp類は、様々な種類のシャペロン機能を発揮する。例えば、細胞質、核、ミトコンドリア、または小胞体に位置するhsp7O (Lindquist, S. et al., 1988, Ann. Rev. Genetics 22:631-677)は、免疫系の細胞への抗原の提示に関与するほか、正常細胞中におけるタンパク質の移動、折りたたみ、および集合にも関与する。
【0014】
シャペロン機能に関与すると考えられるいくつかのタンパク質は、小胞体(ER)内腔に存在するタンパク質であり、具体的には、例えば、タンパク質ジスルフィドイソメラーゼ(PDI; Gething et al.., 1992, Nature 355:33-45)、hsp90に関連付けられるGrp94またはERp99(Sorger & Pelham, 1987, J. Mol. Biol. 194:(2) 341-4)、およびhsp70に関連付けられるGrp78またはBiP(Munro et al., 1986, Cell 46:291-300; Haas & Webl, 1983, Nature 306:387-389)が挙げられる。これらのタンパク質は、突然変異体である折りたたまれていないグリコシル化の不完全なタンパク質に結合することが知られている(Machamer et al., 1990, J. Biol. Chem. 65:6879-6883; Gething et al., 1986, Cell 46:939-950)。ER中へのこれらのhspの局在化は、ER中に残留するのに必要なカルボキシル末端テトラペプチド(lys-Asp-Glu-Leu すなわち KDEL)によって媒介される(Munro & Pelham, 1987, Cell, 48:899-907)。一般に、この配列は、哺乳類細胞中ではKDELであり、Saccharcmyces cerevisiae中ではHDELであり、Schizosaccharomyces pombe中ではADELである(Pidoux & Armstrong, 1992, EMBO J. 11:1583-1591)。
【0015】
一般に、熱ショックタンパク質は、タンパク質またはペプチドに結合することができ、更に、結合したタンパク質またはペプチドをアデノシン三リン酸(ATP)の存在下で放出することができる。ATP加水分解はタンパク質集合の過程でhspが関与するときに起こると考えられている(Flynn et al., 1989, Science, 245:385-390)。
【0016】
2.4. 熱ショック/ストレスタンパク質hsp7O、hsp9O、およびgp96の免疫原性
Srivastavaらは、近交系マウスのメチルコラントレン誘発肉腫に対する免疫応答を実証した(1988, Immunol. Today 9:78-83)。これらの研究において、これらの腫瘍のそれぞれ異なった免疫原性に関与する分子は、96kDaの細胞表面糖タンパク質(gp96)および84〜86kDaの細胞内タンパク質として同定されることが分かった(Srivastava, P.K. et al., 1986, Proc. Natl. Acad. Sci. USA 83:3407-3411; Ullrich, S.J. et al., 1986, Proc. Natl. Acad. Sci. USA 83:3121-3125)。特定の腫瘍から単離されたgp96またはp84/86でマウスを免疫したところ、マウスは、この特定の腫瘍に対しては免疫されたが、抗原性の異なる腫瘍に対しては免疫されなかった。gp96およびp84/86をコードする遺伝子の単離および特性付けを行った結果、それらの間に顕著な相同性が存在することが判明し、更に、gp96およびp84/86はそれぞれ同じ熱ショックタンパク質に属する小胞体タンパク質および細胞質ゾルタンパク質であることが分かった(Srivastava, P.K. et al., 1988, Immunogenetics 28:205-207; Srivastava, P.K. et al., 1991, Curr. Top. Microbiol. Immunol. 167:109-123)。更に、hsp70は、それが単離された腫瘍に対する免疫は誘発するが、抗原的に異なる腫瘍に対する免疫は誘発しないことが分かった。しかしながら、いくつかのペプチドが欠乏したhsp70では、その免疫原性が失われることが判明した(Udono, M., and Srivastava, P.K., 1993, J. Exp. Med. 178:1391-1396)。これらの観測から示唆されるように、熱ショックタンパク質は、そのままでは免疫原性をもたないが、抗原性ペプチドと非共有結合複合体を形成し、この複合体は、抗原性ペプチドに対する特異的免疫を誘発することができる(Srivastava, P.K., 1993 Adv. Cancer Res. 62:153-177; Udono, H. et al., 1994, J. Immunol., 152:5398-5403; Suto, R. et al., 1995, Science, 269:1585-1588)。
【0017】
ストレスタンパク質とペプチドとの非共有結合複合体を癌細胞から精製し、これを癌の治療および予防のために使用することが、1996年4月11日付けのPCT公開WO 96/10411および1997年3月20日付けのPCT公開WO 97/10001に記載されている(SrivastavaおよびChandawarkarにより1997年2月7日に出願された同時係属米国特許出願第08/796,319号およびSrivastavaにより1997年2月7日に出願された同時係属米国特許出願第08/796,316号も参照されたい。それぞれの出願内容はすべて、参照により本明細書に組み入れる。)。
【0018】
また、ストレスタンパク質-ペプチド複合体を病原体感染細胞から単離して、ウイルスのような病原体、ならびに細菌、原生動物、真菌、寄生虫などの他の細胞内病原体によって引き起こされる感染症の治療および予防に使用することもできる。1995年9月21日付けのPCT公開WO 95/24923を参照されたい。
【0019】
また、ストレスタンパク質と抗原性ペプチドとをin vitroで複合体化することによって免疫原性ストレスタンパク質-ペプチド複合体を調製することもできる。このような複合体を癌および感染症の治療および予防に使用することが、1997年3月20日付けのPCT公開WO 97/10000に記載されている。更に、癌および感染症を治療するために熱ショックタンパク質と所定の抗原とを併用することが、1997年2月27日付けのPCT公開W097/06821に記載されている。
【0020】
養子免疫療法用に抗原提示細胞をin vitroで感作するためにストレスタンパク質-ペプチド複合体を使用することが、1997年3月20日付けのPCT公開WO97/10002に記載されている。
【0021】
免疫応答を刺激するために、ならびに感染症および癌を治療するために、真核細胞性熱ショックタンパク質をコードする発現可能なポリヌクレオチドを哺乳類細胞に投与することが、いずれも1997年2月27日付けのPCT公開WO 97/06685およびWO 97/06828に記載されている。
【0022】
細胞溶解物からストレスタンパク質-ペプチド複合体を精製することが既に報告されている。例えば、1995年9月21日付けのPCT公開WO 95/24923を参照されたい。
【先行技術文献】
【特許文献】
【0023】
【特許文献1】国際公開第96/10411号パンフレット
【特許文献2】国際公開第97/10001号パンフレット
【特許文献3】米国特許出願第08/796,319号明細書
【特許文献4】米国特許出願第08/796,316号明細書
【特許文献5】国際公開第95/24923号パンフレット
【特許文献6】国際公開第97/10000号パンフレット
【特許文献7】国際公開第97/06821号パンフレット
【特許文献8】国際公開第97/10002号パンフレット
【特許文献9】国際公開第97/06685号パンフレット
【特許文献10】国際公開第97/06828号パンフレット
【非特許文献】
【0024】
【非特許文献1】Robbins and Angell, 1976, Basic Pathology, 2d Ed., W.B. Saunders Co., Philadelphia, pp. 68-79
【非特許文献2】Roitt, I., Brostoff, J. and Male, D., 1993, Immunology, 3rd ed., Mosby, St. Louis, pps. 17.1-17.12
【非特許文献3】Raychaudhuri & Morrow, 1993, Immunology Today, 14:344-348
【非特許文献4】Prehn et al., 1957, J. Natl. Cancer Inst. 18:769-778
【非特許文献5】Klein et al., 1960, Cancer Res. 20:1561-1572
【非特許文献6】Kripke, 1974, J. Natl. Cancer Inst. 53:1333-1336
【非特許文献7】Vaage, 1968, Cancer Res. 28:2477- 2483
【非特許文献8】Carswell et al., 1970, J. Natl. Cancer Inst. 44:1281- 1288
【非特許文献9】Old et al., 1962, Ann. N.Y. Acad. Sci. 101:80-106
【非特許文献10】Bartlett, 1972, J. Natl. Cancer Inst. 49:493-504
【非特許文献11】Ames, 1979, Science 204:587-593
【非特許文献12】Weisburger et al., 1981, Science 214:401-407
【非特許文献13】Malmgren et al., 1952, Proc. Soc. Exp. Biol. Med. 79:484-488
【非特許文献14】Welch, May 1993, Scientific American 56-64
【非特許文献15】Young, 1990, Annu. Rev. Immunol. 8:401-420
【非特許文献16】Craig, 1993, Science 260:1902-1903
【非特許文献17】Gethirig et al., 1992, Nature 355:33-45
【非特許文献18】Lindquist et al., 1988, Annu. Rev. Genetics 22:631-677
【非特許文献19】Welch et al., 1985, J. Cell. Biol. 101:1198-1211
【非特許文献20】Lai et al., 1984, Mol. Cell. Biol. 4:2802-2810
【非特許文献21】van Bergen en Henegouwen et al., 1987, Genes Dev. 1:525-531
【非特許文献22】Bardwell et al., 1984, Proc. Natl. Acad. Sci. 81:848-852
【非特許文献23】Hickey et al., 1989, Mol. Cell. Biol. 9:2615-2626
【非特許文献24】Jindal, 1989, Mol. Cell. Biol. 9:2279-2283
【非特許文献25】Sorger & Pelham, 1987, J. Mol. Biol. 194:(2) 341-4
【非特許文献26】Machamer et al., 1990, J. Biol. Chem. 65:6879-6883
【非特許文献27】Gething et al., 1986, Cell 46:939-950
【非特許文献28】Munro & Pelham, 1987, Cell, 48:899-907
【非特許文献29】Pidoux & Armstrong, 1992, EMBO J. 11:1583-1591
【非特許文献30】Flynn et al., 1989, Science, 245:385-390
【非特許文献31】Srivastavaら, 1988, Immunol. Today 9:78-83
【非特許文献32】Srivastava, P.K. et al., 1986, Proc. Natl. Acad. Sci. USA 83:3407-3411
【非特許文献33】Ullrich, S.J. et al., 1986, Proc. Natl. Acad. Sci. USA 83:3121-3125
【非特許文献34】Srivastava, P.K. et al., 1988, Immunogenetics 28:205-207
【非特許文献35】Srivastava, P.K. et al., 1991, Curr. Top. Microbiol. Immunol. 167:109-123
【非特許文献36】Udono, M., and Srivastava, P.K., 1993, J. Exp. Med. 178:1391-1396
【非特許文献37】Srivastava, P.K., 1993 Adv. Cancer Res. 62:153-177
【非特許文献38】Udono, H. et al., 1994, J. Immunol., 152:5398-5403
【非特許文献39】Suto, R. et al., 1995, Science, 269:1585-1588
【発明の概要】
【発明が解決しようとする課題】
【0025】
癌に対するワクチンを調製する目的では、入手および使用可能な免疫原性物質の量は、出発癌細胞の量と直接関係している。被験者から得られる癌細胞の数は、特に、癌が初期段階にある場合、非常に少ないため、hsp-ペプチド複合体の生産に供しうる癌細胞の量はかなり制限されることが多い。ワクチンまたは治療薬を商業生産する場合、大量のhsp-ペプチド複合体を定常的に供給することが有利である。従って、hsp-ペプチド複合体の信頼性のある供給源が必要である。本発明の方法を用いると、患者からごく少量の組織しか得られない場合でさえも、便利にかつ迅速に治療用hsp-ペプチド複合体を提供することができる。
【課題を解決するための手段】
【0026】
3. 発明の概要
本発明は、癌または感染症の予防および治療に使用するための免疫原性組成物の調製方法に関する。
【0027】
本発明の方法によって調製される免疫原性組成物には、改変型熱ショックタンパク質(hsp)と抗原性(または免疫原性)ペプチドとの非共有結合分子複合体が含まれる。本発明の改変型熱ショックタンパク質は、それが発現される細胞によって分泌され、未改変の熱ショックタンパク質中に存在する小胞体残留配列を欠失しており、更に、ペプチドタグを含有する。細胞質中に天然に存在するhspの場合、それが発現される細胞によって分泌され、ペプチドタグを含有し、更に、未改変の熱ショックタンパク質中に存在しないリーダーペプチドを含有するように、hspを改変する。この改変は、ペプチドと改変型熱ショックタンパク質との非共有結合型の結合、および抗原提示に関与する非共有結合複合体の能力を、妨害することも低減することもない。改変型hspが組換え細胞中で発現されると、それらが分泌される。更に、この改変型hspは、アフィニティークロマトグラフィーを用いてペプチドタグにより精製することができる。
【0028】
一実施形態において、本発明は、改変型hspをコードするヌクレオチド配列(「改変型hsp遺伝子配列」)を含む核酸、ならびにこのような核酸を含むクローニングベクター、発現ベクター、および宿主細胞を提供する。一般に、本発明の方法には、改変型熱ショックタンパク質をコードするヌクレオチド配列を構築することと、発現ベクター中に改変型hsp遺伝子配列をクローン化することと、宿主細胞中に発現遺伝子構築物を導入することと、改変型hspが発現されるように宿主細胞を培養することと、改変型hspおよび/または改変型hsp-ペプチド複合体を精製することと、が含まれる。
一例として、本発明の改変型熱ショックタンパク質を調製する方法は、
(a) 熱ショックタンパク質の遺伝子配列が、(i)それを発現する細胞から分泌され、(ii)改変されていない熱ショックタンパク質中には存在する小胞体残留配列を欠失しており、かつ(iii)ペプチドタグを含む改変型熱ショックタンパク質をコードするように熱ショックタンパク質の遺伝子配列を改変すること、
(b) 該改変型熱ショックタンパク質の遺伝子配列が宿主細胞内で該改変型熱ショックタンパク質の発現を制御する少なくとも1つの調節領域に機能的に結合されるような発現構築物中に該改変型熱ショックタンパク質の遺伝子配列を連結すること、
(c) 宿主細胞に該改変型熱ショックタンパク質の遺伝子配列を導入すること、
(d) 該宿主細胞を培養して、該改変型熱ショックタンパク質の遺伝子配列によりコードされる該改変型熱ショックタンパク質を該宿主細胞において発現させること、
(e) 該改変型熱ショックタンパク質を細胞培養培地から回収すること、
を含んでなる。
【0029】
熱ショックタンパク質をコードするcDNAまたはゲノムDNAの入手および改変は、従来のDNAクローニングおよび突然変異誘発法により、DNA増幅法により、または合成法により行うことができる。一般に、hspをコードする配列は、発現前に、遺伝子の改変および複製を目的としてクローニングベクター中に挿入される。改変型hsp遺伝子配列は、好適な宿主細胞中においてin vitroでおよびin vivoでコード化改変型hspを発現するために、プロモーターなどの調節エレメント(1種または複数種)に機能的に連結された状態で、発現ベクターに挿入されるかまたは染色体内に組み込まれる。改変型hsp遺伝子配列を宿主細胞中に導入すると、宿主細胞によって改変型hsp遺伝子配列が発現される。これにより、改変型hspとペプチド(癌細胞または病原体によって特異的にコードされているペプチドを含む)との非共有結合複合体が、細胞内で産生される。
【0030】
従って、本発明は、抗原性細胞中における改変型hspと抗原性ペプチドとの免疫原性非共有結合複合体の産生および精製を行うための方法を提供する。この方法には、抗原性細胞中に改変型hsp遺伝子配列を導入することと、改変型hsp遺伝子配列を発現させるために組換え抗原性細胞を培養することと、組換え抗原性細胞から分泌された改変型hsp-抗原ペプチド複合体を細胞培養物の上清から回収および精製することとが含まれる。該複合体の抗原性ペプチドは、癌細胞や病原体感染細胞のような抗原性細胞中に見出される抗原性ペプチドを代表するものである。このような組換え抗原性細胞は、治療および予防に用いるためのワクチンとして有用である。組換え宿主細胞は、免疫原性複合体を生産するために、バッチでまたは連続的に、大量に培養することが可能である。改変型hsp配列を含む宿主細胞は、後で使用するために保存することができる(例えば凍結乾燥または冷凍により)。
【0031】
もう1つの実施形態において、本発明は、細胞中における改変型hspの産生および精製を行うための方法を提供する。この方法には、細胞中に改変型hsp遺伝子配列を導入することと、改変型hsp遺伝子配列を発現させるために組換え細胞を培養することと、組換え細胞から分泌された改変型hspを細胞培養物の上清から回収および精製することとが含まれる。好ましくは、改変型hspの精製は、ペプチドタグおよびアフィニティークロマトグラフィーを利用して行われる。本発明には更に、精製された改変型hspにin vitroで抗原性ペプチドを添加して治療および予防に用いるための免疫原性非共有結合複合体を形成できるようにすることが含まれる。
【0032】
更にもう1つの実施形態において、本発明は、免疫原性非共有結合複合体を産生および精製するための方法を提供する。この方法には、組換え細胞中で改変型hsp遺伝子配列と抗原性ペプチドをコードするヌクレオチド配列とを同時発現させることと、組換え細胞から分泌された改変型hsp-抗原性ペプチド複合体を細胞培養物の上清から回収および精製することとが含まれる。また、これらの組換え細胞は、治療および予防に用いるためのワクチンとして使用することができる。
【0033】
種々の実施形態において、改変型hspまたは改変型hsp-抗原性ペプチド複合体は、アフィニティークロマトグラフィーにより精製し、癌または感染症を予防および治療するためのワクチンとして使用することができる。
【0034】
改変型hsp-ペプチド複合体を含む免疫原性組成物、および改変型hsp-ペプチド複合体を分泌する組換え細胞は、本発明の方法に従って調製した場合、癌細胞または病原体に対する患者の免疫応答を誘発することができ、この免疫応答は、治療または予防に有効である。好ましくは、この患者は、改変型hspを発現するための癌細胞を提供した被験者である。このほか、癌細胞または感染細胞は、この患者以外の1人以上の被験者に由来するものであってもよいが、ただし、これらの被験者は、同じ組織タイプの癌(例えば、胃癌、乳癌、大腸癌、肺癌など)または同じタイプの病原体によって引き起こされた感染症を患っている者でなければならない。
【0035】
従って、本発明は、個体において抗原に対する免疫応答を誘発する方法を提供する。この方法には、抗原またはその断片に非共有結合で結合した改変型熱ショックタンパク質の免疫原性複合体および/またはこのような免疫原性複合体を分泌する組換え細胞を、個体に投与することが含まれる。本発明はまた、癌を患っているかまたは癌の予防が望まれる個体を対象に癌の治療または予防を行う方法を提供する。この方法には、癌に由来する抗原またはその断片に非共有結合で結合した改変型熱ショックタンパク質の免疫原性複合体および/またはこのような免疫原性複合体を分泌する組換え細胞を、個体に投与することが含まれる。更に、本発明は、感染症を患っているかまたは感染症の予防が望まれる個体を対象に感染症の治療または予防を行う方法を提供する。この方法には、感染細胞または病原体に由来する抗原またはその断片に非共有結合で結合した改変型熱ショックタンパク質の免疫原性複合体および/またはこのような免疫原性複合体を分泌する組換え細胞を、個体に投与することが含まれる。
【0036】
以下の節および項において、本発明の特定の組成物およびそれらの調製方法について説明する。
【図面の簡単な説明】
【0037】
4.図面の簡単な説明
【図1−1】gp96-Igの分泌および特性付け。図1a:gp96-Ig cDNAでトランスフェクトされたおよびトランスフェクトされていない小細胞肺癌系#7(SCLC)由来の上清のマウスIgGに対するELISA; 106/mlで細胞をプレートし、3日目および6日目に上清の試験を行った;標準として精製マウスIgG(500ng/ml)を使用した。図lb:プロテインAで精製されたgp96-IgのSDS PAGE。レーン1:クーマシーブルー染色(1μgタンパク質)、レーン2:モノクロナール抗gp96(抗Grp94、抗9G10)を用いたウェスタンブロット(100ngタンパク質)。
【図1−2】図lC:還元条件下および非還元条件下でマウスIgG(mIgG)およびCD3O-Ig融合タンパク質(CD3O-Ig)と比較した場合の精製改変型gp96-Ig融合タンパク質(gp96-Ig)のドデシル硫酸ナトリウム-ポリアクリルアミドゲル電気泳動(SDS-PAGE)分析。分子量マーカー(kDa単位)が、左側に示されている。
【図2−1】gp96-Igの分泌および細胞内局在化。図2a:黒丸:培養物上清中のgp96-Ig、白丸:細胞溶解物中のgp96-Ig。gp96-IgはELISAにより定量し、SCLC-gp96-Igは106/mlでプレートした。
【図2−2】図2b:透過性の付与されたSCLC-gp96-IgのFACS分析;破線、ヤギ抗ウサギIgG-FITC(陰性対照);実線、ヤギ抗マウスIgG-フィコエリトリン。図2c:透過性の付与されていないSCLCのFACS分析;トランスフェクトされていない。図2d:透過性の付与されていないSCLCのFACS分析;gp96-IgでトランスフェクトされたSCLC。両方のパネルにおいて、破線は、ヤギ抗ウサギIgG-FITCであり;実線は、ヤギ抗マウスIgG-FITCである。
【図3−1】擬似トランスフェクトされた細胞(三角形)およびトランスフェクトされていない細胞(白丸)と比較して、gp96-IgでトランスフェクトされたE.G7(図3a)およびLLC(図3b)(黒丸)では、腫瘍形成性が低下した。1パラメーターあたり1群6匹のマウスを使用した。
【図3−2】擬似トランスフェクトされた細胞(三角形)およびトランスフェクトされていない細胞(白丸)と比較して、gp96-IgでトランスフェクトされたE.G7(図3a)およびLLC(図3b)(黒丸)では、腫瘍形成性が低下した。1パラメーターあたり1群6匹のマウスを使用した。
【図4−1】分泌型gp96-Igは、腫瘍特異的記憶を引き起こす。gp96-IgでトランスフェクトされたE.G7(いずれのパネルにおいても黒丸)106個、放射線照射されたEG7(三角形)106個を用いて、2週間の時間間隔で2回、C57BL/6マウスを免役したか、または免疫しなかった(白丸)。2週間後、図に記載の数の腫瘍細胞でマウス(1群あたり6匹のマウス)をチャレンジした。図4a:E.G7; 図4b:EL4。
【図4−2】分泌型gp96-Igは、腫瘍特異的記憶を引き起こす。gp96-IgでトランスフェクトされたE.G7(いずれのパネルにおいても黒丸)106個、放射線照射されたEG7(三角形)106個を用いて、2週間の時間間隔で2回、C57BL/6マウスを免役したか、または免疫しなかった(白丸)。2週間後、図に記載の数の腫瘍細胞でマウス(1群あたり6匹のマウス)をチャレンジした。図4c:LLC; 図4d:LLC-ova。
【図5−1】E.G7-gp96-Ig 106個の拒絶反応に及ぼす免疫適格細胞の枯渇の影響。それぞれのマウスについて腫瘍増殖カーブが示されている。図5a〜5d(上側のパネル):皮下腫瘍接種の2日前の免疫適格細胞の枯渇。
【図5−2】E.G7-gp96-Ig 106個の拒絶反応に及ぼす免疫適格細胞の枯渇の影響。それぞれのマウスについて腫瘍増殖カーブが示されている。図5e〜5h(下側のパネル):皮下腫瘍接種の3日後の免疫適格細胞の枯渇。
【発明を実施するための形態】
【0038】
5.発明の詳細な説明
本発明は、抗原提示に関与する熱ショックタンパク質(hsp)を改変し、そして癌および感染症の予防ならびに治療に使用することができる免疫原性組成物を調製するための、組換えDNA技術の応用を意図する。
【0039】
本発明で使用する「抗原性細胞」は任意の抗原性細胞でよく、限定するわけではないが、癌細胞、前腫瘍細胞、細胞内病原体に感染した細胞、または病原体に感染した被験者から採取した細胞などである。
【0040】
癌細胞または病原体に感染した細胞中で、免疫原性hsp-ペプチド複合体が自然に産生される。こうしたhsp-ペプチド複合体を使用して、この複合体の宿主細胞中に同一の種類の癌細胞または感染細胞に対する特異的な免疫応答を誘発させてきた。したがって、これら複合体は癌または感染症の予防および治療に有用である。しかし、こうした免疫原性複合体は一般的に、こうした細胞から大量には分泌されない。さらに、多数の癌細胞または感染細胞(抗原性細胞)を取得するのは常に可能なまたは実現し得るものではないので、こうした細胞から取得し得るhsp-ペプチド複合体の量は時には非常に限定される。したがって、免疫原性hsp-ペプチド複合体の限定された供給の問題を克服するための方法を見出だすことが望ましい。この問題の一部分は、現在実用化されている、細胞溶解を必要とする、hsp-ペプチド複合体の精製方法によるものである。本発明は、抗原性細胞が免疫原性hsp-ペプチド複合体を培地中に分泌するように仕向けるための方法を提供する。本発明の抗原性細胞は所望の免疫原性複合体を継続的に産生および分泌し、これを培地から好都合に回収することができる。培地からのこうした免疫原性複合体を精製するための改良法も提供される。さらに、本発明は癌細胞および感染細胞の免疫原性を強化して、癌または感染症を予防および治療するためのワクチンとしてこれらの抗原性細胞を直接被験体に投与するすることができるようにするための方法を提供する。
【0041】
1実施形態において、本発明の方法によって調製される免疫原性組成物には、改変型熱ショックタンパク質(hsp)、および抗原性細胞中に存在するかまたは存在するタンパク質の一部分である抗原性ペプチドを含む非共有結合で結合した分子複合体が含まれる。本発明の改変型熱ショックタンパク質は、それを発現する細胞から分泌されるが、これは改変されていない熱ショックタンパク質中には存在する小胞体(ER)残留配列を欠失しており、またペプチドタグを含んでいる。この改変型hspが組換え細胞中で発現すると、それが分泌されて、アフィニティークロマトグラフィーを使用して、ペプチドタグによって精製することができる。
【0042】
別の実施形態において、本発明の改変型熱ショックタンパク質はそれを発現する細胞から分泌されるが、これはペプチドタグを含んでおり、また改変されていない熱ショックタンパク質中には存在しないリーダーペプチドを含んでいる。天然では細胞質中に存在するhspについて、アミノ末端に付加したリーダーペプチドがこれらのER中へのトランスロケーションを促進する。
【0043】
本発明の改変型hspは天然に存在するhspと同質の生物学的活性を示す。この改変は抗原性ペプチドに対して特異的におよび非共有結合で改変型hspに結合する能力、および関連する免疫細胞に対して抗原提示過程中にこの結合したペプチドを提示する能力に影響を与えない。従って、この改変型hspはin vitroおよび抗原性細胞中の両方で抗原性ペプチドと免疫原性非共有結合複合体を形成することができる。in vitroおよび抗原性細胞中で内因的に形成された改変型hsp-ペプチド複合体はいずれも、動物中でこの抗原性ペプチドが由来する抗原性細胞に対する特異的免疫応答を誘導することができる。
【0044】
特に、本発明は、天然には動物細胞の小胞体(ER)中に残留する熱ショックタンパク質(hsp)をコードするヌクレオチド配列の改変に関係する。本発明に従えば、ER中にhspの残留をシグナルするペプチドのストレッチをコードするhsp配列の一部の置換または欠失によって、hsp遺伝子配列、好ましくはcDNA配列を改変する。ER中にhspを残留させるシグナルは、カルボキシル末端に位置するXaa-Asp-Glu-Leu(XDEL、ここでXは任意のアミノ酸とすることができる)を含むペプチドのストレッチ中に存在することが知られている。hsp遺伝子配列はさらに、あるペプチドタグをコードするヌクレオチド配列の1つをhsp配列に付加することによって、改変される。得られる本発明の改変型hsp配列は、分泌されてER中に残留しない改変型hsp融合タンパク質をコードする。
【0045】
天然には細胞質中に存在するhspについては、ペプチドタグをコードするヌクレオチド配列ばかりでなく、リーダーペプチドをコードするヌクレオチド配列をも付加することによって、その遺伝子配列を改変する。この配列をhsp遺伝子配列のコード領域の5'末端に連結する。こうした疎水性リーダーペプチドを坦持するhspをER内腔内に取り込ませる。該リーダーペプチドはシグナル認識粒子によって認識され、これが、伸長していくhspペプチド鎖を粗面小胞体膜の細胞質ゾル側表面に誘導する。分泌可能なhspは膜を通過して完全に内腔中にトランスロケーションし、そこでリーダーペプチドがプロテアーゼによって消化される。
【0046】
本発明において、アフィニティークロマトグラフィーによる改変型hspまたは改変型hsp-ペプチド複合体の精製を容易にするために、種々の機能および親和性を持つ各種のペプチドタグを使用することができる。好ましいペプチドタグには免疫グロブリンの定常領域が含まれる。hspに融合させるペプチドタグに応じて、改変されたhspは二量体化などの新規性質を獲得することがあり、これを有利に活用して、改変型hspまたは改変型hsp-ペプチド複合体の機能を強化することもできる。ペプチドタグおよびリーダーペプチドをコードする配列、ならびにこうした配列をhsp配列に連結するための方法については、第5.1.4節に記載する。
【0047】
したがって、本発明は、改変型hspをコードするヌクレオチド配列(「改変型hsp遺伝子配列」)を含む核酸分子、ならびにこうした配列を含有するクローニングベクター、発現ベクターおよび組換え細胞を提供する。本発明は、改変型hsp遺伝子配列に相補的なヌクレオチド配列を含む核酸分子も包含する。改変型hsp遺伝子配列を改変型hsp-ペプチド複合体の産生に好適な宿主細胞中に導入する前に、中間細胞中のクローニングベクター中に該改変型hsp遺伝子配列をクローン化し、かつ/または複製によって増やすことができる。第5.2.1節に記載するような当技術分野で既知の任意の方法によって、改変型hsp遺伝子配列を含む発現構築物または発現ベクターを構築して、宿主細胞中に導入することができる。必要に応じて、改変型hspの発現のために、癌細胞、病原体に感染した細胞または正常細胞を含む、各種の細胞を使用することができる。
【0048】
本発明の発現遺伝子構築物には、改変型hspをコードするヌクレオチド配列、好ましくは改変型hspをコードする相補的DNA(cDNA)配列が含まれる。改変型hsp遺伝子配列は、適切な宿主細胞内で改変型hsp配列の発現を制御する少なくとも1つの調節領域(例えばプロモーター)に機能的に結合される。あるいは、宿主細胞内で相同組換えを促進する領域に、改変型hsp配列を隣接させて、染色体内のある部分に改変型hsp配列を挿入するようにし、それによって改変型hsp遺伝子配列を宿主細胞内で改変型hsp配列の発現を制御する少なくとも1つの調節領域に機能的に結合させることができる。改変型hsp遺伝子配列を含む両タイプの発現遺伝子構築物を発現可能な改変型hsp遺伝子配列とも称する。したがって、本発明は発現可能な改変型hsp遺伝子配列を含有する組換え細胞を提供する。
【0049】
hspをコードする配列およびペプチドタグをコードする配列、ならびにこうした配列を取得するための方法を第5.1.1節および第5.1.4節に詳細に記載する。ヌクレオチドの付加、欠失または置換によるhsp遺伝子配列の改変法を第5.1.2節に記載する。
【0050】
別の実施形態において、本発明は、改変型hsp遺伝子配列の発現を可能にする組換え細胞を培養し、そしてこの組換え細胞から分泌される改変型hspを回収および精製することを含む、細胞培養物から改変型hspを精製するための方法を提供する。一般的に、第5.3節に記載するような適切なアフィニティークロマトグラフィー法を使用する、分泌された改変型hspの培養上清からの精製は、改変型hsp上のペプチドタグによって容易になる。改良法では、細胞の溶解を必要とせず、これによって、継続的に増殖させて改変型hspまたは改変型hsp-ペプチド複合体を産生させることが可能になる。
【0051】
本発明の改変型hspは改変されていないhspと同様にペプチドに結合する能力があるので、組換え抗原性細胞中での改変型hsp遺伝子配列の発現時に、改変型hspが産生されて、これがER中でペプチドと結合し、非共有結合複合体を形成する。抗原性細胞の何らかのタンパク質が抗原性/免疫原性であるので、改変型hspと複合体化するペプチド/タンパク質が、これらが存在する抗原性細胞に特異的な免疫を宿主に与える。こうした非共有結合の免疫原性複合体は組換え細胞から分泌され、培地中に蓄積する。
【0052】
したがって、本発明は、改変型hspと細胞中の抗原性ペプチドの免疫原性非共有結合複合体を製造および精製するための方法も提供する。1実施形態において、本方法は、改変型hsp遺伝子配列を抗原性細胞中に導入し、この組換え抗原性細胞を培養して改変型hsp遺伝子配列を発現させ、組換え抗原性細胞から分泌される改変型hsp-抗原性ペプチド複合体を培地から回収し、精製することを含む。複合体の抗原性ペプチドは抗原性細胞の内側にある抗原性ペプチドの典型であるので、この抗原性ペプチドを使用して被験体にワクチン接種する前に、この抗原を単離および/または特性決定する必要はなく、またこれらの抗原の本質を知る必要すらない。第5.3節に記載するような、細胞培地からの改変型hspの精製に適用し得る方法は、その方法が改変型hspと抗原性ペプチドの非共有結合性結合を妨害しない限り、改変型hsp-抗原性ペプチド複合体の精製にも有用である。特定の1実施形態において、発現可能な改変型hsp遺伝子配列を含む組換え抗原性細胞を治療および予防用途のための免疫原性組成物に使用することができる。こうした組成物の免疫原性は当技術分野で既知の、また第5.5節および第6節に記載された方法によって試験することができる。hsp-ペプチド複合体のin vitroでの作製に使用するための改変型hspを調製する目的の場合は、分泌される改変型hspに不要の抗原性分子が負荷されないように、それ自体は抗原性でない宿主細胞を使用することが望ましい。
【0053】
組換え細胞を適度に大規模に連続式でまたはバッチ式で培養することによって、改変型hspまたは改変型hsp-ペプチド複合体を大量に製造することができる。組換え抗原性細胞の大規模連続またはバッチ培養の細胞培養培地から、改変型hspおよび組換え産生された抗原性タンパク質/ペプチドを含む所望の免疫原性複合体を精製することができる。改変型hsp-ペプチド複合体を分泌する永久細胞系は、有用な免疫原性組成物の一定で、再現性があり、また豊富な供給源となり得る。必要に応じて、改変型hsp遺伝子配列を含む組換え細胞をプールおよび/若しくは分取し、または増殖させ、または液体窒素下での凍結および保存によって保管し、組換え宿主細胞のバッチを将来何度も復元させて使用することができるようにする。
【0054】
抗原性タンパク質またはペプチドのコード配列が知られている、別の1実施形態においては、抗原性分子をコードするこの配列を発現遺伝子構築物中にクローン化し、発現可能な改変型hsp配列を含む組換え細胞中に導入することを意図する。改変型hspの発現についての記載(第5.2節参照)などの標準的技法によって、抗原性タンパク質またはペプチドの発現ベクター中へのクローニングを実施することができる。抗原性タンパク質またはペプチドは組換え細胞中で改変型hspと共発現する。これらの抗原性タンパク質またはペプチドは組換え細胞のER中で改変型hspと非共有結合複合体を形成する。こうした抗原性ペプチドは癌細胞中で発現される抗原性タンパク質の断片(例えば、腫瘍特異的抗原または腫瘍関連抗原の断片など)とすることができる。これらの細胞からの改変型hsp-抗原性ペプチド複合体は培養培地中に分泌され、第5.3節に記載と同様に回収し、アフィニティークロマトグラフィーによって精製することができる。発現可能な改変型hsp配列、および抗原をコードする発現遺伝子構築物の両方を含有する組換え細胞も、治療および予防用途のための免疫原性組成物として使用することができる。
【0055】
本発明はさらに、精製した改変型hspをin vitroで抗原性ペプチドと混合して免疫原性非共有結合複合体を形成させる方法を提供する。こうした複合体は癌または感染症の治療および予防において有用である。抗原性ペプチドは細胞源から精製するか、またはそのペプチドの配列が既知ならば、当技術分野で知られた方法によって、合成することができる。特定の1実施形態においては、ペプチドタグによって固相に可逆的に固定化された改変型hspとともに抗原性ペプチドをインキュベートして、固相上で抗原性ペプチドと改変型hspとの非共有結合複合体を形成させるようにする。
【0056】
したがって、本発明は改変型熱ショックタンパク質およびペプチドを複合体の形成のために十分な時間インキュベートすることを含む、ペプチドと非共有結合で結合した本発明の改変型熱ショックタンパク質の複合体をin vitroで調製するための方法を提供する。
【0057】
特許請求する方法によって調製された改変型hsp-ペプチド複合体および組換え抗原性細胞の両方を含む、本発明の免疫原性組成物は、個体の免疫担当能力を強化し、腫瘍細胞および病原体に感染した細胞の両方に対する特異的な免疫を誘発することができる。こうした免疫原性組成物は癌の進行を防止し、腫瘍細胞の増殖および進行を阻害し、そして病原体または病原体に感染した細胞の増殖を防止する能力もある。免疫原性組成物を使用して、腫瘍部位での炎症性反応を誘導し、治療する癌患者にある腫瘍負荷量を最終的に退縮させることができる。
【0058】
したがって、本発明は、個体中で抗原の1つに対する免疫応答を誘発する方法であって、この抗原若しくはその断片に非共有結合で結合した本発明の改変型熱ショックタンパク質の免疫原性複合体を該個体に投与することを含む方法を提供する。また本発明は、癌であるかまたは癌の予防が必要とされる個体において癌を治療または予防する方法であって、上記の抗原若しくはその断片に非共有結合で結合した本発明の改変型熱ショックタンパク質の免疫原性複合体を該個体に投与することを含む方法も提供する。また、感染症であるかまたは感染症の予防が必要とされる個体において感染症を治療または予防する方法であって、上記の抗原若しくはその断片に非共有結合で結合した本発明の改変型熱ショックタンパク質の免疫原性複合体をその個体に投与することを含む方法も本発明に包含される。
【0059】
癌細胞若しくは組織を採取した個体、または家族歴若しくは環境危険因子によって癌のリスクが増強した個体に自己由来の前記免疫原性組成物を投与することができる。同様に、病原体に感染した細胞若しくは抗原性細胞を取得した個体、または同一の病原体に感染するリスクがある個体に自己由来の前記免疫原性組成物を投与することができる。
【0060】
この癌の治療または予防方法は、改変型hspの発現のために癌細胞を提供しなかった他の個体に対しても、癌細胞の提供者と同一のタイプの癌であるならば、一般的に適用することができる。感染症の治療または予防についても、抗原性宿主細胞の提供者に感染した病原体と抗原性が同一の病原体に感染した個体ならば、前記方法を他の個体に適用することができるという、同一の原理が応用される。癌および感染症を治療または予防するための免疫原性組成物の使用については、第5.7節および第5.8節に記載する。
【0061】
5.1. 改変型hsp遺伝子配列の構築
ここに記載するのは、原核および真核細胞中で発現することができる改変型熱ショックタンパク質(hsp)をコードする遺伝子構築物の構築方法である。特定すると、改変型hspをコードするヌクレオチド配列の構築、その改変型hsp遺伝子配列の適切なクローニングベクター中へのインサート、および改変型hspおよび改変型hsp-ペプチド複合体の製造のための、この発現遺伝子構築物の適切な宿主細胞中への導入について、記載する。
【0062】
癌または感染症の治療および予防において有用な、本明細書中でストレスタンパク質と互換的に称する熱ショックタンパク質は、以下の基準のいずれか1つを満足する任意の細胞タンパク質の中から選択することができる:細胞がストレスの高い刺激に曝露されたときにその細胞内濃度が増加するタンパク質であること、その他のタンパク質またはペプチドに結合することができること、そしてアデノシン三リン酸(ATP)の存在下または低pHにおいて結合したタンパク質またはペプチドを放出する能力があること;あるいは、上記の特性のいずれかを有する任意の細胞タンパク質と少なくとも35%の相同性を示すタンパク質であること。本発明によって改変し、調製することができる複合体中のhspとして、限定するわけではないが、hsp70、hsp90、gp96、BiPおよびタンパク質ジスルフィドイソメラーゼが含まれる。好ましくは、hspはヒトhspである。好ましい複合体はタンパク質抗原の1つに非共有結合で結合した改変型ヒトhsp60、hsp70、hsp90、タンパク質ジスルフィドイソメラーゼ、またはBiPを含む。特定の実施形態において、複合体は通常は真核細胞の小胞体中に存在するヒトgp96の改変型を含む。
【0063】
hspの3つの主要なファミリー、すなわちhsp60、hsp70およびhsp90が現在のところ同定されている。さらに、タンパク質ジスルフィドイソメラーゼ(PDI)、ならびにチオレドキシン様ドメイン(群)を含有する小胞体中のその他のタンパク質、限定するわけではないがERp72およびERp61など、も包含される。これらのhspファミリーの全てのメンバーを本発明の実践によって改変し、調製することができるものと構想する。
【0064】
hsp60、hsp70、hsp90およびタンパク質ジスルフィドイソメラーゼファミリーは、配列においてストレスタンパク質に関係がある、例えばアミノ酸の同一性が35%を超えるが、発現レベルはストレスによって変更されないタンパク質で構成されることが発見された。したがって、本明細書で使用するストレスタンパク質または熱ショックタンパク質の定義は、細胞中での発現レベルがストレス性刺激に応答して増強されるこれらのファミリーのメンバーに少なくとも35%〜55%、好ましくは55%〜75%、最も好ましくは75%〜85%のアミノ酸同一性があるその他のタンパク質、それらの突然変異タンパク質、類似体および変異体を包含することを意図する。
【0065】
hsp遺伝子構築物を構築して改変する際に使用する常套的な分子生物学的反応を実施するために、基準となる論文、例えばMethods in Enzymology, 1987, volume 154, Academic Press;Sambrookら、1989, Molecular Cloning-A Laboratory Manual, 2nd Edition, Cold Spring Harbor Press, New York;およびAusubelら、Current Protocols in Molecular Biology, Greene Publishing Associates and Wiley Interscience, New York、に記載された操作法に従ってもよい。以下に詳細に記載する方法は単に説明のためのものであって、限定するつもりではない。市販の各種クローニングベクターおよび発現系を製造元の指示にしたがって使用してもよい。
【0066】
5.1.1. hsp遺伝子配列の単離
各種の実施形態において、本発明は改変型熱ショックタンパク質(hsp)、ならびに機能的に活性なその断片および誘導体のアミノ酸配列に関する。本明細書で使用する「機能的に活性な」改変型hspとは、抗原性ペプチドの結合、アデノシン三リン酸(ATP)の存在下または低pHにおける結合した抗原性ペプチドの放出、その他などの、未改変型hspに関係する1以上の公知の機能的活性を表出する改変型hspをいう。改変型hspおよび上記のようなその断片をコードする核酸、ならびにこれらの核酸に対して相補的かつこれらとハイブリダイズする能力がある核酸を提供する。
【0067】
天然に出現する熱ショックタンパク質のアミノ酸配列およびヌクレオチド配列はGenBankなどの配列データベースから一般的に入手可能である。Entrezなどのコンピュータプログラムを使用して、データベースを閲覧(browse)し、目的とする任意のアミノ酸配列および遺伝子配列データを受託番号によって検索することができる。類似の配列をアライメントスコアおよび統計によって分類する、FASTAおよびBLASTなどのプログラムを使用して、これらのデータベースを検索して、問題の配列に対して様々なの程度の類似性を有する配列を同定することができる。
【0068】
本発明の方法によって改変し、発現させることができるhspの限定するわけではない例としてのヌクレオチド配列が以下に公開されている。ヒトgp96:Genebank Accession No.X15187;Makiら、1990, Proc.Natl.Acad.Sci., 87:5658-5562、マウスgp96:Genebank Accession No.M16370;Srivastavaら、1987, Proc.Natl.Acad.Sci., 85:3807-3811、マウスBiP: Genebank Accession No.U16277;Haasら、1988, Proc.Natl.Acad.Sci. U.S.A., 85:2250-2254、ヒトBiP:Genebank Accession No.M19645;Tingら、1988, DNA 7:275-286、マウスhsp70:Genebank Accession No.M35021;Huntら、1990, Gene, 87:199-204、ヒトhsp70:Genbank Accession No.M24743;Huntら、1995, Proc.Natl.Acad.Sci. U.S.A., 82:6455-6489。遺伝コードの縮重性に起因して、用語「hsp遺伝子配列」は、天然に出現するヌクレオチド配列を称するばかりでなく、hspをコードする別の縮重DNA配列の全部をも包含する。
【0069】
hsp遺伝子のコード領域を取得するための核酸起源として、どのような真核細胞でも可能性としては機能し得る。hspをコードする核酸配列を脊椎動物、哺乳動物、ならびにヒトを含む霊長類起源から単離することができる。
【0070】
DNAは、クローン化DNA(例えばDNA「ライブラリー」の1つ)から当技術分野で公知の標準的操作法によるか、またはDNA増幅によって取得することができる。ゲノムDNAに由来するクローンには、コード領域の他に調節領域およびイントロンDNA領域が含有されることがあり、cDNAに由来するクローンはエクソン配列のみを含むことになる。いずれの起源でも、hsp遺伝子は該遺伝子の増殖のために好適なベクター中に分子クローン化される必要がある。
【0071】
ゲノムDNA由来のhsp遺伝子の分子クローニングにおいて、DNA断片を作製し、クローン化し、ゲノムライブラリーを形成する。関連するhspをコードする配列のいくつかが入手可能で、これを精製および標識することができるので、この標識したプローブへの核酸のハイブリダイゼーションによって、ゲノムDNAライブラリー中のクローン化DNA断片をスクリーニングすることができる(Benton,W.およびDavis,R., 1977, Science 196:180;Grunstein,M. および Hogness,D., 1975, Proc.Natl.Acad.Sci. U.S.A. 72:3961)。プローブに実質的に相同性があるDNA断片がハイブリダイズする。制限酵素消化(群)し、入手し得るならば公知の制限マップにしたがって予想されるサイズと断片のサイズを比較することによって、適切な断片を同定することも可能である。
【0072】
hspゲノムDNAを単離するための別法として、限定するわけではないが、公知の配列から遺伝子配列自体を化学的に合成するもの、またはhspをコードするmRNAに対するcDNAを作成するものが含まれる。例えば、hspを発現する細胞からhsp遺伝子のcDNAクローニングのためのRNAを単離することができる。当技術分野で公知の方法によってcDNAライブラリーを作製し、ゲノムDNAライブラリーをスクリーニングするための開示されているような方法によってスクリーニングすることができる。hspに対する抗体が入手し得るならば、hspを合成していると推定されるクローンに標識した抗体を結合させることによって、hspを同定することもできる。
【0073】
hspをコードするヌクレオチド配列のクローニングのためのその他の特定の実施形態を以下のように例示として提供するが、限定するものではない。
【0074】
特定の実施形態において、低ないし中程度ストリンジェンシーの条件下で、hspの1つをコードするヌクレオチド配列を含むプローブとハイブリダイズさせることによって、あるファミリー内の熱ショックタンパク質をコードするヌクレオチド配列を同定して取得することができる。
【0075】
限定するわけではない例示としての、こうした低ストリンジェンシー条件を使用する操作法は以下の通りである(Shilo および Weinberg, 1981, Proc.Natl.Acad.Sci. USA 78:6789-6792、も参照されたい)。35%ホルムアミド、5X SSC、50 mM Tris-HCl(pH 7.5)、5 mM EDTA、0.1% PVP、0.1% フィコール、1% BSA、および 500 μg/ml 変性サーモン精子 DNAを含有する溶液中、40℃で6時間、DNAを含有するフィルターを前処理する。以下の改変:0.02% PVP、0.02% フィコール、0.2% BSA、100 μg/ml サーモン精子 DNA、10%(wt/vol)硫酸デキストラン、をした同一の溶液中で、5〜20 X 106 cpm 32P標識プローブを使用して、ハイブリダイゼーションを実施する。フィルターをハイブリダイゼーション混合液中、40℃で18〜20時間インキュベートし、その後、2X SSC、25 mM Tris-HCl(pH 7.4)、5 mM EDTA、および 0.1% SDSを含有する溶液中、55℃で1.5時間洗浄する。洗浄液を新鮮な溶液と交換し、さらに60℃で1.5時間インキュベートする。フィルターをドライブロットし、オートラジオグラフィー用に曝露する。必要ならば、フィルターに65〜68℃で3回目の洗浄を行い、フィルムに再曝露する。使用することができる低ストリンジェンシーのその他の条件は当技術分野で公知である(例えば交差種ハイブリダイゼーションのために利用するもの)。
【0076】
別の実施形態において、選別の前に、ポリメラーゼ連鎖反応(PCR)を使用して、DNAクローンまたはゲノムライブラリー若しくはcDNAライブラリー中の所望の配列を増幅する。PCRは例えば熱サイクラーおよびTaqポリメラーゼ(Gene AmpTM)の使用によって実施することができる。増幅するDNAとして任意の種からのcDNAまたはゲノムDNAが含まれる。PCRのプライマーとして、関連するhspの公知の核酸配列に相当するオリゴヌクレオチドプライマーを使用することができる。好ましい実施形態において、オリゴヌクレオチドプライマーは少なくとも異種のhsp間で高度に保存されているhsp遺伝子の部分を表す。PCR反応で使用するために、いくつかの異なる縮重プライマーの合成を選択することもできる。PCR反応を開始させるために使用するハイブリダイゼーション条件のストリンジェンシーを変更することによって、公知のhspヌクレオチド配列と単離する核酸相同体間のヌクレオチド配列の類似性の程度をより大きくするかまたは小さくすることも可能である。交差種ハイブリダイゼーションのためには、低ストリンジェンシー条件が好ましい。同種ハイブリダイゼーションのためには、中程度のストリンジェント条件が好ましい。増幅が成功した後、hspをコードする配列をクローン化および配列決定することができる。増幅するhsp遺伝子のコード領域のサイズが1回のPCRで増幅するには大き過ぎる場合は、全長遺伝子を網羅する何回かのPCRを、好ましくは領域を重複させて、実施し、このPCR産物を連結して全長コード配列を形成することができる。あるいは、hsp遺伝子のセグメントが増幅されるならば、そのセグメントをクローン化して、完全cDNAまたはゲノムクローンを単離するためのプローブとして利用することができる。
【0077】
改変の前に、hsp遺伝子を適切なクローニングベクターの1つにインサートして宿主細胞中に導入し、この遺伝子配列の多数のコピーを作製させることができる。当技術分野で公知の多数のベクター−宿主系を使用することができる。例えば、限定するわけではないが、ラムダ誘導体などのバクテリオファージ、またはpBR322若しくはpUCプラスミド誘導体などのプラスミド、またはBluescriptベクター(Stratagene)などである。
【0078】
上記の方法はhspのクローンを取得または増量する方法を限定することを意味するものではない。本発明の改変型熱ショックタンパク質は、分泌されかつ細胞培養培地から容易に精製することができるように改変される。特に、本発明の改変型hspはhspを小胞体(ER)中に残留するシグナルとなるポリペプチドのセグメントを欠失している。残留シグナルは、このペプチドを欠失させるか、またはシグナルとして機能しないペプチドと置換することによって無能化される。その上、改変型hspには回収および精製を容易にするペプチドタグが含まれる。ペプチドタグは例えばカルボキシル末端など、抗原性ペプチドとの結合に関与しない、hspの任意の部分に融合させることができる。さらに、hspが本来小胞体中に残留する場合、分泌のためにER膜を通ってこれが移動するのを導くために、リーダーペプチドを添加する。好ましい実施形態において、通常はカルボキシル末端に位置しているhspの残留ペプチドをペプチドタグと置換する。
【0079】
5.1.2 熱ショックタンパク質遺伝子の改変
本発明の改変型熱ショックタンパク質は、その内部でこれを発現する細胞によって分泌されて、その細胞培養培地から容易に精製することができるように、改変される。特に、本発明の改変型hspはhspの小胞体(ER)中への残留シグナルとなるポリペプチドのセグメントを欠失している。こうしたペプチドは、限定するわけではないがgp96などのER中に残留するhsp中に存在する。残留シグナルはこのペプチドを欠失させるか、またはシグナルとして機能しないペプチドと置換することによって無能化される。さらに、改変型hspには回収および精製を容易にするペプチドタグが含まれる。ペプチドタグは抗原性ペプチドとの結合に関与しない、例えばカルボキシル末端などの任意のhspの部分に融合させることができる。好ましい実施形態において、通常はカルボキシル末端に位置しているhspの残留ペプチドをペプチドタグと置換する。さらに、hspが天然では細胞質中に残留する場合、分泌のためにER膜を通ってこれが移動するのを導くために、リーダーペプチドを添加する。
【0080】
本発明の改変型hsp中に存在する改変は当技術分野で公知の各種の方法によって生成させることができる。この改変生成をもたらす操作は遺伝子またはタンパク質レベルのいずれかで実施できるが、遺伝子レベルが好ましい。例えば、クローン化したhspのコード領域を当技術分野で公知の多数の組換えDNA法のいずれによっても改変することができる(Sambrookら、1990, Molecular Cloning,A Laboratory Manual, 2nd Edition, Cold Spring Harbor Laboratory, Cold Spring Harbor,New York;Ausubelら、Current Protocols in Molecular Biology, Chapter 8, Greene Publishing Associates および Wiley Interscience,New York)。以下の考察から、改変型hspをコードする最終的なヌクレオチド配列に到達するために、置換、欠失、インサートまたはこれらの任意の組み合わせが導入または組み合わされることが明らかであろう。
【0081】
別法として、改変型hspを化学的に合成することができる。例えば、ペプチドシンセサイザーの使用によって、所望の改変を含むhspの1部分に相当するペプチドを合成することができる。
【0082】
5.1.3 残留ペプチド
hspを小胞体(ER)中に残留させる原因となるペプチドは典型的にはカルボキシル末端に位置し、配列はXaa-Asp-Glu-Leu(またはXDEL)である(Munro & Pelham,1987,Cell,48:899-907)。用語「残留ペプチド」(retention peptide)は本明細書中ではこのテトラペプチド配列を意味するために使用するが、大部分の哺乳動物hsp中ではこれはKDEL(Lys-Asp-Glu-Leu)である。Saccharomyces cerevisiaeおよびSchizosaccharomyces pombe中の残留ペプチド配列はそれぞれHDEL(His-Asp-Glu-Leu)およびADEL(Ala-Asp-Glu-Leu)であることがわかっている(Pidoux & Armstrong,1992,EMBO J.11:1583-1591)。
【0083】
hspの残留ペプチドはその残留ペプチドを欠失させるか、または残留ペプチド中のアミノ酸を置換してシグナルを消滅させることによって、無能化することができる。一般論として、改変型hsp配列中に、改変型hspが細胞から分泌されるのを妨害する傾向がある何らかのシグナルが存在する場合は、その存在するシグナルを除去すべきである。個々のhspによるが、こうしたシグナルとして、膜貫通ドメインおよび細胞質ドメインが挙げられる。
【0084】
残留ペプチド配列またはその他のシグナルをコードするDNAのセグメントを除去するために、適切な部位が利用し得る場合は、hsp遺伝子配列をそうした部位で制限酵素(群)により切断して、残留ペプチドをコードするDNAの断片を放出させることができる。次に、hspをコードする領域の残留部を単離し、改変型hsp遺伝子配列を形成させるように連結する。
【0085】
あるいは、好都合な制限部位が利用可能でないならば、残留ペプチド配列をコードする領域に隣接する配列中に位置する制限部位を使用することによって、より大きなDNAの断片を放出させて、残留ペプチドをコードする配列を欠失している合成DNAの類似の断片と入れ替えることができる。適正な翻訳リーディングフレームが確実に維持されるように、注意しなければならない。
【0086】
所望ならば、当技術分野で公知の部位特異的突然変異誘発方法および/またはDNA増幅方法によって、適切な位置に制限部位を作製することができる。例えば、Shankarappaら、1992,PCR Method Appl.1:277-278、を参照されたい。目的のDNA中に所望の配列変更を導入するためには、ポリメラーゼ連鎖反応(PCR)が共通に使用される。プライマー配列中の任意の変更を容易にそのPCRのDNA産物中に組込むことができ、これが遺伝子配列中へのこの変更のその後の組込みを促す。例えば、所望の制限部位を組込んでいる合成オリゴヌクレオチドを適切な隣接配列プライマーとともに使用して、2つの隣接するDNA断片を増幅することができる。これらの増幅された断片のそれぞれが一方の末端に新しい制限部位を含有するようになる。この新しい部位と隣接部位の両方での酵素消化の後、増幅した断片を連結し、その後の操作用に準備したベクター中にサブクローン化する。コードされたタンパク質のアミノ酸配列が制限部位の導入によって変更されないことが必須である。
【0087】
その後の操作を促進するために、発現されたペプチド配列中のアミノ酸を置換(群)させる目的で、または制限部位を作製/欠失する目的で、DNA配列中の個々のヌクレオチドを改変するためには、当技術分野で公知の突然変異誘発のどのような技法でも使用することができる。こうした技法として、限定するわけではないが、化学的突然変異誘発、in vitro部位特異的突然変異誘発(Hutchinson,C. ら、1978,J.Biol.Chem.253:6551)、オリゴヌクレオチド特異的突然変異誘発(Smith,1985,Ann.Rev.Genet.19:423-463;Hillら、1987,Methods Enzymol.155:558-568)、PCRに基づく重複伸長(Hoら、1989,Gene 77:51-59)、PCRに基づくメガプライマー突然変異誘発(Sarkarら、1990,Biotechniques,8:404-407)その他が含まれる。二本鎖ジデオキシDNA配列決定によって、改変を確認することができる。
【0088】
上記の方法を応用して、テトラペプチド残留配列中の1以上のアミノ酸残基、特にAsp、GluおよびLeu残基を置換することができる。残留ペプチド配列内のあるアミノ酸に対する置換基はそのアミノ酸が属するクラスと異なるメンバーから選択することができる。非極性(疎水性)アミノ酸にはアラニン、ロイシン、イソロイシン、バリン、プロリン、フェニルアラニン、トリプトファンおよびメチオニンが含まれる。極性無電荷アミノ酸にはグリシン、セリン、トレオニン、システイン、チロシン、アスパラギンおよびグルタミンが含まれる。正に荷電した(塩基性)アミノ酸にはアルギニン、リシンおよびヒスチジンが含まれる。負に荷電した(酸性)アミノ酸にはアスパラギン酸およびグルタミン酸が含まれる。一般的に生化学的特性に最大の変化を産生することが期待される置換には以下のようなものがある:(a)親水性残基、例えばセリルまたはトレオニルを疎水性残基、例えばロイシル、イソロイシル、フェニルアラニル、バリルまたはアラニルの代わりに(またはこれらによって)置換する;(b)システインまたはプロリンを任意のその他の残基の代わりに(またはこれらによって)置換する;(c)正の電荷を有する側鎖を有する残基、例えばリシル、アルギニルまたはヒスチジルを負の電荷を有する残基、例えばグルタミルまたはアスパルチルの代わりに(またはこれらによって)置換する;あるいは、(d)嵩高な側鎖を有する残基、例えばフェニルアラニンを側鎖を持たないもの、例えばグリシンの代わりに(またはこれらによって)置換する。
【0089】
上記の方法は、hsp中の残留ペプチド配列および/またはその他のシグナルを欠失または消滅させることができる方法を限定することを意味するものではない。
【0090】
5.1.4 ペプチドタグおよび/またはリーダーペプチドの融合
本発明の改変型hspはまた、ペプチドタグを含んでなる融合タンパク質でもある。特定の具体例では、リーダーペプチドを同じく改変型hspと融合させて、それにより改変型hspを分泌のために小胞体(ER)へ輸送することを促進させ得る。
【0091】
種々の実施形態では、かかる融合タンパク質は、適当な読み取り枠においてhsp遺伝子配列と、ペプチドタグまたはリーダーペプチドをコードする配列とを連結することにより作製できる。ゲノム配列が用いられる場合は、確実に同じ翻訳読み取り枠内に改変型遺伝子が留まり、翻訳停止シグナルおよび/または疑似メッセンジャーRNAスプライシングシグナルによって妨げられないように注意しなければならない。
【0092】
好ましい実施形態では、このペプチドタグはそのアミノ末端でhspのカルボキシル末端と融合している。カルボキシル末端において融合が起こる厳密な部位は重要ではない。例えば、ペプチドタグは残留ペプチドと置き換わってもよい。最適な部位は通常の実験のより決定することができる。改変型hspの免疫原性は第5.5節に記載される方法により試験できる。
【0093】
限定されるものではないが、免疫グロブリン定常領域、ポリヒスチジン配列(Petty, 1996, Metal-chelate affinity chromatography, Current Protocols in Molecular Biology, 第2巻, Ausbelら編, Greene Publish, Assoc. & Wiley Interscience) 、グルタチオンS-トランスフェラーゼ(GST, Smith, 1993, Methods Mol. Cell Bio. 4:220-229)、大腸菌マルトース結合タンパク質(Guanら, 1987, Gene 67:21-30)および種々のセルロース結合ドメイン(米国特許第5,496,934号;第5,202,247号;第5,137,819号;Tommeら, 1994, Protein Eng. 7:117-123)など、当技術分野で公知の種々のペプチドタグがhspの改変に使用され得る。いくつかのペプチドタグは改変型hspに、多量体の形成能といった新規な構造特性を与え得る。結合したペプチドによる改変型hspの二量体化は、抗原提示過程でのhspとそのパートナーとの相互作用のアビディティ(avidity)を高めると考えられる。これらのペプチドタグは大抵、通常はホモポリマーとして存在するタンパク質に由来するものである。CD28(Leeら, 1990, J. Immunol. 145:344-352 )もしくはCD8(Shiueら, 1988, J. Exp. Med. 168:1993-2005)の細胞外ドメイン、または鎖内ジスルフィド結合部位を含有する免疫グロブリン分子部分などのペプチドタグは、多量体の形成をもたらし得る。他の可能性あるペプチドタグとしては、限定されるものではないが、以下の良く知られた例としてFLAGエピトープ、mycエピトープのアミノ酸408-439、インフルエンザウイルスヘマグルチニン(HA)エピトープなど、それに対してモノクローナル抗体が利用可能である短いアミノ酸配列がある。その他のペプチドタグは特異的結合パートナーにより認識され、従って、結合パートナーとの親和性結合によって単離が容易になるが、これらは固相支持体上にあるかまたは固相支持体上に固定化されていることが好ましい。当業者により認識されるように、前記のペプチドタグのコード領域を得るには、限定されるものではないが、DNAクローニング、DNA増幅および合成法をはじめとする多くの方法が使用できる。いくつかのペプチドタグ、ならびにそれらの検出および単離のための試薬は市販されている。
【0094】
好ましいペプチドタグは免疫グロブリン分子の不変部分である。典型的には、かかる部分は少なくとも免疫グロブリンH鎖の定常領域のCH2およびCH3ドメインを機能的に含んでなる。不変ドメインのFc部分のカルボキシル末端、またはH鎖もしくはL鎖のCH1に直に続くアミノ末端領域を用いて、融合が行われる。好適な免疫グロブリンを基礎とするペプチドタグは、IgG-1、-2、-3または-4サブタイプ、IgA、IgE、IgDまたはIgM、好ましくはIgG1から誘導し得る。好ましくは、改変型hspが、ヒトに対してin vivoで使用することを意図されている場合は、ヒト免疫グロブリンが用いられる。免疫グロブリンL鎖またはH鎖定常領域をコードする多くのDNAが公知であるか、あるいはcDNAライブラリーから容易に入手できる。例えば、Adamら, Biochemistry, 1980, 19:2711-2719; Goughら, 1980, Biochemistry, 19:2702-2710; Dolbyら, 1980, Proc. Natl. Acad. Sci. U.S.A., 77:6027-6031; Riceら, 1982, Proc. Natl. Acad. Sci. U.S.A., 79:7862-7865; Falknerら, 1982, Nature, 298-286-288; およびMorrisonら, 1984, Ann. Rev. Immunol, 2:239-256を参照。免疫グロブリンの検出には多くの免疫試薬および標識系が利用できるので、改変型hsp-Ig融合タンパク質(「改変型hsp-Ig」)は、酵素結合免疫吸着検定(ELISA)、免疫沈降、蛍光活性化細胞選別(FACS)など、当技術分野で公知の種々の免疫学的技術によって容易に検出および定量できる。同様に、ペプチドタグが容易に利用できる抗体を有するエピトープである場合、かかる試薬を前記の技術と併用して、ペプチドタグを含有する改変型hspを検出、定量および単離することができる。多くの例では、改変型hspに対する特異的抗体を開発する必要はない。
【0095】
特に好ましい実施形態は、残留ペプチドを欠く改変型hspと、ヒンジであるネズミ免疫グロブリンG-1(IgG-1)のCH2およびCH3ドメインとの融合である(Bowenら, J. Immunol. 156:442-9)。このペプチドは、Ig分子の他のシステインとのジスルフィド結合に通常関与する3つのシステイン残基を含む。いずれのシステインもタグとしての機能のためにはペプチドに必要とされないので、これらのシステイン残基の1以上を、例えばセリンなどの別のアミノ酸残基でさらに置換してもよい。第5.1.2節に記載されるもののような方法を適用してかかる置換をなすことができる。
【0096】
細菌および哺乳動物細胞由来の改変型hspの効率的な分泌のために、当技術分野で公知の種々のリーダー配列を使用できる(von Heijne, 1985, J. Mol. Biol. 184:99-105)。リーダーペプチドは意図される宿主細胞に基づいて選択され、細菌、酵母、ウイルス、動物、および哺乳動物配列が含まれる。例えば、ヘルペスウイルス糖タンパク質Dリーダーペプチドは種々の哺乳動物細胞における使用に好適である。哺乳動物細胞での使用に好ましいリーダーペプチドは、マウス免疫グロブリンκ鎖のV-J2-C領域から得られる(Bernerdら, 1981, Proc. Natl. Acad. Sci. 78:5812-5816)。
【0097】
公知であるか、またはライブラリーもしくは商業的供給者から容易に入手可能な所望のペプチドタグまたはリーダーペプチドをコードするDNA配列は本発明の実施に好適である。第5.1.1節に記載のhsp配列を得る方法もまた、ペプチドタグまたはリーダーペプチドをコードする配列を得るのに適用できる。
【0098】
5.2 改変型hspの産生
本発明の種々の実施形態では、改変型hspをコードする配列は、組換え細胞内での増幅および発現のための発現ベクターに挿入される。
【0099】
本明細書において発現構築物とは、適当な宿主細胞で改変型hspの発現を可能とする1以上の調節領域と機能的に結合された改変型hspをコードするヌクレオチド配列をいう。「機能的に結合される」とは、調節領域と発現される改変型hsp配列が、転写、および最終的には翻訳が可能なように結合され配置される連結をいう。
【0100】
改変型hspの転写に必要な調節領域は発現ベクターにより提供され得る。また、その同起源の開始コドンを欠く改変型hsp配列が発現される場合は、翻訳開始コドン(ATG)も提供され得る。適合する宿主-構築物系においては、宿主生物において改変型hsp配列の転写を果たすため、RNAポリメラーゼなどの細胞転写因子は、発現構築体の調節領域に結合するであろう。遺伝子発現に必要な調節領域の厳密な性質は宿主細胞によって異なると考えられる。一般に、RNAポリメラーゼと結合して機能的に結合された核酸配列の転写を誘導することができるプロモーターが必要とされる。かかる調節領域としては、TATAボックス、キャッピング配列」、CAAT配列など、転写および翻訳の開始に関与する5'非コード配列が挙げられる。非コード領域3'からコード配列にかけては、ターミネーターやポリアデニル化部位など、転写終結調節配列が含まれ得る。
【0101】
改変型hspの発現のためには、構成的調節領域および誘導調節領域の双方を使用し得る。組換え細胞の増殖に最適な条件と改変型hspの高レベルの発現のための条件とが異なる場合には、誘導プロモーターを使用することが望ましい。有用な調節領域の例は下記次節に示される。
【0102】
プロモーターなどの調節機能を有するDNA配列を改変型hsp遺伝子配列と結合させるため、あるいはベクターのクローニング部位に改変型hsp遺伝子配列を挿入するためには、当技術分野で良く知られた技術により、適合した制限部位を提供するリンカーまたはアダプターを、cDNAの末端に連結すればよい(Wuら, 1987, Methods in Enzymol 152:343-349)。制限酵素による切断の後、連結前に一本鎖DNA末端を消化するか、またはフィリングによって、平滑末端を作製することができる。あるいは、所望の制限酵素部位を含むプライマーを用いるPCRの使用によるDNAの増幅によって、DNA断片へ所望の制限酵素部位を導入することもできる。
【0103】
調節領域と機能的に結合された改変型hsp配列を含んでなる発現構築物は、さらにクローン化せずに、改変型hsp-ペプチド複合体の発現および産生のために適当な宿主細胞へ直接導入できる。例えば、米国特許第5,580,859号を参照。発現構築物はまた、例えば相同組換えによる改変型hsp配列の、宿主細胞のゲノムへの組み込みを助けるDNA配列を含んでもよい。この例においては、宿主細胞で改変型hspを増幅および発現するため、適当な宿主細胞に好適な複製起点を含んでなる発現ベクターを使用する必要はない。
【0104】
5.2.1 宿主-ベクター系
本明細書では改変型hspの発現に使用できるベクターおよび宿主細胞系が記載される。限定されるものではないが、プラスミド、コスミド、ファージ、ファージミドまたは改変型ウイルスを含む種々の発現ベクターが本発明で使用され得る。典型的にはかかる発現ベクターは、適当な宿主細胞おいてベクターを増幅するための機能的複製起点、改変型hsp遺伝子配列の挿入のための1以上の制限エンドヌクレアーゼ部位、および1以上の選択マーカーを含んでなる。発現ベクターは、限定されるものではないが、細菌、酵母、昆虫、哺乳動物およびヒトをはじめとする原核生物または真核生物に由来し得る適合する宿主細胞とともに使用しなければならない。
【0105】
発現構築物およびベクターは分泌される改変型hspの産生を目的とする宿主細胞へ導入される。in vitroで培養されたもの、または遺伝子操作されたものをはじめ、熱ショックタンパク質を産生でき、かつ、発現ベクターと適合する細胞種ならばいずれを使用してもよい。宿主細胞は、健康な人、癌患者、および感染症を有する患者を含む正常な被験者または罹病した被験者から、民間研究寄託機関、American Type Culture Collectionなどの好適な培養物収集機関、あるいは商業供給者から得られる。
【0106】
In vivoにおいて改変型hsp-抗原ペプチド複合体の産生および分泌を目的として改変型hsp遺伝子配列が導入できる細胞としては、限定されるものではないが、上皮細胞、内皮細胞、ケラチン生成細胞、繊維芽細胞、筋細胞、肝細胞;Tリンパ球、Bリンパ球、単球、マクロファージ、好中球、好酸球、巨核球、顆粒球などの血液細胞;種々の幹細胞または前駆細胞、特に造血幹細胞または前駆細胞、例えば骨髄、臍帯血、末梢血、胎児肝臓に由来するものなどが挙げられる。細胞種の選択は治療または予防される腫瘍または感染症の種類に依存し、当業者ならば決定できる。
【0107】
異なる宿主細胞は、タンパク質の翻訳後プロセッシングおよび改変型に関する特徴および特異的なメカニズムを持っている。受容者がhspをプロセッシングする過程と同様の特異的様式で発現された遺伝子産物を改変およびプロセッシングする宿主細胞を選択すればよい。多量のHspを産生するためには、本発明で用いる宿主細胞種は異種遺伝子の発現に使用されたものであり、大規模生産工程のために合理的に十分に特徴づけられ、開発されたものであることが好ましい。特殊な実施形態では、宿主細胞は、次に改変型hsp-ペプチド複合体または改変型hsp-ペプチド複合体を分泌する組換え細胞が投与される同一の宿主に由来するものであり、すなわち、改変型hspの発現および被験者への投与に用いられる細胞は被験者の自己由来のものである。
【0108】
特定の実施形態では、改変型hsp遺伝子配列を含んでなる発現構築物が抗原細胞へ導入される。本明細書において、抗原細胞とは、まだ腫瘍性ではないが、ウイルスなど癌を引き起こす感染性病原体に感染した前腫瘍性細胞;または例えばDNA傷害剤、放射線など突然変異誘発剤または発癌剤に曝された抗原細胞が挙げられる。使用可能な他の細胞としては、形態学的機能、生理学的機能、または生化学的機能により特徴づけられる、正常形態から腫瘍形態への遷移過程にある前腫瘍性細胞がある。
【0109】
好ましくは、本発明の方法で用いられる癌細胞および前腫瘍性細胞は哺乳動物起源である。本発明の本実施形態で意図される哺乳動物としては、ヒト、ペット動物(例えば、イヌやネコ)、家畜(例えば、ヒツジ、ウシ、ヤギ、ブタおよびウマ)、実験動物(例えば、マウス、ラットおよびウサギ)ならびに捕獲または捕獲されていない野生動物が挙げられる。
【0110】
種々の実施形態では、改変型hsp-ペプチド複合体を生産するため、またはワクチンとして使用するため本方法においていずれかの癌細胞、好ましくはヒト癌細胞を用いることができる。これらの癌細胞は、発現した改変型hspと非共有結合的に会合するようになる抗原ペプチドを提供する。本発明の方法によって製造される免疫組成物で治療または予防できる癌としては、限定されるものではないが、肉腫および癌腫などの腫瘍が挙げられる。本発明の方法に従う癌の例は、第5.7節に挙げられている。従って、複数の離れた部位に転移した癌をはじめ、前腫瘍性病変部や癌から単離された組織または細胞は、本方法で使用可能である。例えば、異常な増殖を示す組織に見られる細胞、循環中の白血病細胞、転移病変部、ならびに固形腫瘍組織が使用できる。
【0111】
もう1つの実施形態では、その細胞系の細胞が標的癌細胞の抗原と共通の少なくとも1以上の抗原決定基を有しているならば、前腫瘍性病変、癌組織または癌細胞由来の細胞系が使用できる。ヒト起源の癌組織、癌細胞、発癌性病原体に感染した細胞、その他の前腫瘍性細胞および細胞系が好ましい。最終的に複合体が投与される患者から切除した癌細胞が使用されること、すなわち、本発明の自己由来の実施形態が好ましいが、必ずしもすべての場合にそうであることが必要なわけではない(例えば、癌細胞は1以上の異なる個体に由来してもよい)。
【0112】
癌細胞または前腫瘍性細胞は当技術分野で公知のいずれの方法によっても同定できる。例えば、癌細胞は形態学、酵素アッセイ、増殖アッセイ、細胞遺伝学的特徴、DNAマッピング、DNA配列決定、発癌性ウイルスの有無、または突然変異誘発物質または発癌剤に曝された履歴、画像などによって同定できる。もう1つの例としては、癌細胞は外科技術、内視鏡検査またはその他の生検技術によって得ることもできる。癌細胞のいくつかの特有の特徴が分かっていれば、それらはまた、限定されるものではないが、アフィニティークロマトグラフィー、および蛍光活性化細胞選別(例えば、癌細胞により発現される抗原に対する蛍光タグ付き抗体による)など、当技術分野で公知の生化学的または免疫学的方法のいずれによっても取得または精製可能である。
【0113】
クローン化された、または均質な、または精製された癌細胞集団を用いる必要はない。癌組織、癌細胞、または細胞系は単一の個体から得てもよいし、あるいはいくつかの個体からプールしてもよい。改変型hspの発現のために使用される細胞に、標的癌細胞の少なくとも1以上の抗原決定基が存在する限り、in vivoの最終標的の細胞(例えば、意図される受容者の腫瘍由来の細胞)を用いる必要はない。さらには、離れた転移由来の細胞を用いて、原発性癌に対する免疫組成物を調製し得る。混合物中の実質的な数の細胞が癌細胞であり、かつ、標的癌細胞と少なくとも1つの抗原決定基を共有するならば、細胞混合物を用いることができる。特定の実施形態では、改変型hspの発現において使用される癌細胞は精製されている。
【0114】
大腸菌に基づくベクターは最も一般的であり、外来タンパク質の高レベルの発現に関して広い用途を持つ系である(Makrides, 1996, Microbiol Rev, 60:512-538)。大腸菌での発現に使用できる調節領域の例としては、限定されるものではないが、lac、trp、lpp、phoA、recA、tac、T3、T7およびλPL(Makrides, 1996, Microbiol Rev, 60:512-538)が挙げられる。原核生物発現ベクターの例としては、限定されるものではないが、λgt11などのλgtベクター系列(Huynhら, 1984, "DNA Cloning Techniques", 第I巻: A Practical Approach (D. Glover編), 49-78頁, IRL Press, Oxford)、およびpETベクター系列(Studierら, 1990, Methods Enzymol., 185:60-89)が挙げられる。しかしながら、原核生物宿主-ベクター系のあり得る欠点としては、哺乳動物細胞の翻訳後プロセッシングの多くを行うことができないことである。従って、真核生物宿主-ベクター系が好ましく、哺乳動物宿主-ベクター系がより好ましく、ヒト宿主-ベクター系が最も好ましい。
【0115】
哺乳動物宿主細胞における改変型hspの発現のためには、種々の調節領域が使用でき、例えばSV40初期および後期プロモーター、サイトメガロウイルス(CMV)前初期プロモーター、およびラウス肉腫ウイルスの長い末端反復(RSV-LTR)プロモーターがある。哺乳動物細胞で有用であり得る誘導プロモーターとしては、限定されるものではないが、メタロチオネインII遺伝子、マウス乳癌ウイルスグルココルチコイド応答性の長い末端反復(MMTV-LTR)、β-インターフェロン遺伝子およびhsp70遺伝子が挙げられる(Williamら, 1989, Cancer Res. 49:2735-42; Taylorら, 1990, Mol. Cell Biol., 10:165-75)。組換え宿主細胞において改変型hspの発現を駆動するには熱ショックプロモーターまたはストレスプロモーターを使用するのが有利であろう。
【0116】
特定の組織種の腫瘍細胞には、組織特異性を示し、かつ、トランスジェニック動物に利用されてきた以下の動物の調節領域もまた使用できる:膵腺房細胞において活性なエラスターゼI遺伝子制御領域(Swiftら, 1984, Cell 38:639-646; Ornitzら, 1986, Cold Spring Harbor Symp. Quant. Biol. 50:399-409; MacDonald, 1987, Hepatology 7:425-515);膵β細胞において活性なインスリン遺伝子制御領域(Hanahan, 1985, Nature 315:115-122)、リンパ系細胞において活性な免疫グロブリン遺伝子制御領域(Grosschedlら, 1984, Cell 38:647-658; Adamesら, 1985, Nature 318:533-538; Alexanderら, 1987, Mol. Cell. Biol. 7:1436-1444)、精巣、乳房、リンパ系およびマスト細胞において活性なマウス乳癌ウイルス制御領域(Lederら, 1986, Cell 45:485-495)、肝臓において活性なアルブミン遺伝子制御領域(Pinkertら, 1987, Genes and Devel. 1:268-276)、肝臓において活性なα-フェトプロテイン遺伝子制御領域(Krumlaufら, 1985, Mol. Cell. Biol. 5:1639-1648; Hammerら, 1987, Science 235:53-58)、肝臓において活性なα1-抗トリプシン遺伝子制御領域(Kelseyら, 1987, Genes and Devel. 1:161-171)、骨髄性細胞において活性なβ-グロビン遺伝子制御領域(Mogramら, 1985, Nature 315:338-340; Kolliasら, 1986, Cell 46:89-94)、脳の稀突起神経膠細胞において活性なミエリン塩基性タンパク質遺伝子制御領域(Readheadら, 1987, Cell 48:703-712)、骨格筋において活性なミオシンL鎖-2遺伝子制御領域(Sani, 1985, Nature 314:283-286)および視床下部において活性な性腺刺激ホルモン放出ホルモン遺伝子制御領域(Masonら, 1986, Science 234:1372-1378)。
【0117】
宿主細胞における改変型hspの発現効率は、発現ベクター中に適当な転写エンハンサーエレメント、例えばSV40ウイルス、B型肝炎ウイルス、サイトメガロウイルス、免疫グロブリン遺伝子、メタロチオネイン、β-アクチンで見られるものなどを包含させることにより増強され得る(Bittnerら, 1987, Mrthods in Enzymol. 153:516-544; Gorman, 1990, Curr. Op. In Biotechnol. 1:36-47を参照)。
【0118】
発現ベクターはまた、2種類以上の宿主細胞におけるベクターの維持および複製、または宿主の染色体へのベクターの組み込みを可能にする配列を含んでもよい。かかる配列としては、限定されるものではないが、複製起点、自律的複製配列(ARS)、セントロメアDNA、およびテロメアDNAが挙げられる。また、少なくとも2種の宿主細胞で複製および維持が可能なシャトルベクターを用いることも有利である。
【0119】
さらに、発現ベクターは、改変型hspをコードするDNAを含む宿主細胞を最初に単離、同定または追跡するための選択可能もしくはスクリーニング可能なマーカー遺伝子を含んでもよい。長期にわたって改変型hsp-ペプチド複合体を高収量で産生し、哺乳動物細胞内で安定な発現をすることが好ましい。哺乳動物細胞に関しては、限定されるものではないが、単純ヘルペスウイルスチミジンキナーゼ(Wiglerら, 1977, Cell 11:223)、ヒポキサンチン-グアニンホスホリボシルトランスフェラーゼ(SzybalskiおよびSzybalski, 1962, Proc. Natl. Acad. Sci. USA 48:2026)およびアデニンホスホリボシルトランスフェラーゼ(Lowyら, 1980, Cell 22:817)、それぞれtk-、hgprt-、またはaprt-細胞で使用できる遺伝子をはじめ、いくつかの選択系が使用され得る。また、メトトレキセート耐性を付与するジヒドロ葉酸レダクターゼ(dhfr)(Wiglerら, 1980, Proc. Natl. Acad. Sci. USA 77:3567; O'Hareら, 1981, Proc. Natl. Acad. Sci. USA 78:1527);ミコフェノール酸耐性を付与するgpt(MulliganおよびBerg, 1981, Proc. Natl. Acad. Sci. USA 78:2072);アミノグリコシドG-418耐性を付与するネオマイシンホスホトランスフェラーゼ(neo)(Colberre-Garapinら, 1981, J. Mol. Biol. 150:1);およびハイグロマイシン耐性を付与するハイグロマイシンホスホトランスフェラーゼ(hyg)(Santerreら, 1984, Gene 30:147)の選択に基づき、代謝拮抗物質耐性も使用できる。限定されるものではないが、ヒスチジノールおよびゼオシンTMなど、他の選択マーカーも使用できる。
【0120】
好ましい哺乳類宿主細胞としては、限定されるものではないが、ヒト、サルおよび齧歯類に由来するもの(例えば、Kriegler M. "Gene Transfer and Expression: A laboratory Manual", New York, freeman & Co. 1990を参照)、例えば、SV40で形質転換されたサル腎細胞系統(COS-7, ATCC CRL 1651);ヒト胎児腎系統(293、293-EBNAまたは懸濁培養での増殖のためにサブクローン化された293細胞、Grahamら, J. Gen. Virol., 36:59, 1977);ベビーハムスター腎細胞(BHK, ATCC CCL 10);チャイニーズハムスター卵巣細胞-DHFR(CHO, UrlaubおよびChasin, Proc. Natl. Acad. Sci. 77:4216, 1980);マウスセルトリ細胞(Mather, Biol. Reprod. 23:243-251, 1980);マウス繊維芽細胞(NIH-3T3)、サル腎細胞(CVI ATCC CCL 70);アフリカミドリザル腎細胞(VERO-76, ATCC CRL-1587);ヒト子宮頸癌細胞(HELA, ATCC CCL 2);イヌ腎細胞(MDCK, ATCC CCL 34);バッファローラット肝細胞(BRL 3A, ATCC CRL 1442);ヒト肺細胞(W138, ATCC CCL 75);ヒト肝細胞(Hep G2, HB 8065);およびマウス乳腺癌細胞(MMT 060562, ATCC CCL 51)が挙げられる。癌ワクチンとしての組換え細胞(改変型hsp-ペプチド複合体を産生する)の有用性を実証するために使用される癌細胞種の典型例は以下に示される:マウス繊維芽細胞系統NIH3T3、マウスルイス肺癌細胞系統LLC、肥満細胞腫細胞系統P815、マウスリンパ腫細胞系統EL4およびその卵白アルブミントランスフェクト体E.G7、マウス黒色腫細胞系統B16F10、マウス繊維肉腫細胞系統MC57、ならびにヒト小細胞肺癌細胞系統SCLC#2およびSCLC#7。
【0121】
多数あるウイルスに基づく発現系を哺乳類細胞とともに用いて、改変型hspを生産してもよい。DNAウイルス骨格を用いるベクターはシミアンウイルス40(SV40)(Hamerら, 1979, Cell 17:725)、アデノウイルス(Van Dorenら, 1984, Mol Cell Biol 4:1653)、アデノ随伴ウイルス(McLaughlinら, 1988, J Virol 62:1963)、およびウシパピローマウイルス(Zinnら, 1982, Proc Natl Acad Sci 79:4897)に由来するものである。発現ベクターとしてアデノウイルスを用いる場合には、供与DNA配列をアデノウイルス転写/翻訳制御複合体、例えば後期プロモーターおよび3つに分かれたリーダー配列に連結すればよい。このキメラ遺伝子を次いでin vivoまたはin vitro組換えによりアデノウイルスゲノムに挿入できる。ウイルスゲノムの非必須領域(例えばE1またはE3領域)に挿入すれば、生存可能であって感染した宿主で異種産物を発現することができる組換えウイルスが得られる。(例えば、LoganおよびShenk, 1984, Proc. Natl. Acad. Sci. (USA) 81:3655-3659を参照。)
【0122】
ウシパピローマウイルス(BPV)はヒトをはじめとする多くの高等脊椎動物に感染でき、そのDNAをエピソームとして複製する。組換え遺伝子の発現のために、哺乳類細胞において安定な多重コピー(20〜300コピー/細胞)の染色体外要素として存在する多くのシャトルベクターが開発されている。典型的には、これらのベクターはBPV DNAセグメント(全ゲノムまたは69%形質転換断片)、広い宿主範囲を有するプロモーター、ポリアデニル化シグナル、スプライス部位、選択マーカー、および大腸菌でベクターを増殖させる「非毒性」プラスミド配列を含む。細菌内で構築および増幅した後、発現遺伝子構築物は、例えばリン酸カルシウム共沈法により、哺乳類培養細胞中にトランスフェクトされる。形質転換された表現型を呈さない宿主細胞に関しては、ヒスチジノールおよびG418耐性などの優性の選択マーカーの使用により、形質転換体の選抜が行われる。第6節で記載されるように、改変型hsp遺伝子配列を、2種のBPVベクターpBCMGSNeoおよびpBCMGHis(Karasuyamaら, Eur. J. Immunol. 18:97-104; Oheら, Human Gene Therapy, 6:325-33)に挿入し、次いでこれを改変型hspの発現のため広範な細胞種にトランスフェクトした。
【0123】
別法として、ワクシニア7.5Kプロモーターを用いた。(例えば、Mackettら, 1982, Proc. Natl. Acad. Sci. (USA) 79:7415-7419; Mackettら, 1984, J. Virol. 49:857-864; Panicaliら, 1982, Proc. Natl. Acad. Sci. 79:4927-4931を参照。)ヒト宿主細胞を用いる場合には、エプスタインバーウイルス(EBV)複製起点(OriP)およびEBV核抗原1(EBNA-1;トランス作用複製因子)に基づくベクターが使用できる。かかるベクターは広範なヒト宿主細胞、例えば、EBO-pCD(Spickofskyら, 1990, DNA prot Eng Tech 2:14-18);pDR2およびλDR2(Clontech Laboratoriesから入手できる)とともに使用できる。
【0124】
改変型hspはまた、レトロウイルスに基づく発現系でも生産され得る。ウイルス遺伝子配列のほとんどは除去されて改変型hsp遺伝子で置換され、一方、除かれたウイルスの機能はトランスで補われるので、配列モロニーマウス白血病ウイルスなどのレトロウイルスが使用できる。トランスフェクションに対し、レトロウイルスは、例えば一次造血細胞をはじめとする広範な細胞種へ効果的に感染し、遺伝子を導入する。さらに、レトロウイルスベクターにより感染する宿主範囲は、ベクターのパッケージングに用いられるエンベロープの選択により操作できる。
【0125】
例えば、レトロウイルスベクターは5'長い末端反復配列(LTR)、3'LTR、パッケージングシグナル、細菌の複製起点、および選択マーカーを含んでなることができる。改変型hsp DNAは5'LTRと3'LTRの間の位置に挿入され、その結果、5'LTRプロモーターからの転写がクローン化されたDNAを転写することとなる。5'LTRは、限定されるものではないが、LTRプロモーター、R領域、U5領域、およびプライマー結合部位をこの順序で含むプロモーターを含んでなる。これらのLTRエレメントのヌクレオチド配列は当技術分野で十分公知である。感染細胞の選択を容易にするため、多剤選択マーカーだけでなく異種プロモーターもまた、発現ベクターに含めてよい。McLauchlinら, 1990, Prog Nucleic Acid Res and Molec Biol 38:91-135; Morgensternら, 1990, Nucleic Acid Res 18:3587-3596; Choulikaら, 1996, J Virol 70:1792-1798; Boesenら, 1994, Biotherapy 6:291-302; SalmonsおよびGunzberg, 1993, Human Gene Therapy 4:129-141;ならびにGrossmanおよびWilson, 1993, Curr. Opin. Genetics and Devel. 3:110-114を参照のこと。
【0126】
その他の有用な真核生物宿主-ベクター系としては、酵母および昆虫系がある。酵母では、構成または誘導プロモーターを含むいくつかのベクターが、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)(パン酵母)、シゾサッカロミセス・ポンペ(Schizosaccharomyces pombe)(分裂酵母)、ピヒア・パストリス(Pichia pastoris)およびハンセヌラ・ポリモルファ(Hansenula polymorpha)(メチロトローフ酵母)とともに使用され得る。概論としては、Current Protocols in Molecular Biology, 第2巻, 1988, Ausubelら編, Greene Publish. Assoc. & Wiley Interscience, 第13章; Grantら, 1987, Expression and Secretion Vectors for Yeast, Methods in Enzymology, WuおよびGrossman編, 1987, Acad. Press, N.Y., 第153巻, 516-544頁; Glover, 1986, DNA Cloning, 第II巻, IRL Press, Wash., D.C., 第3章,およびBitter, 1987, Heterologous Gene Expression in Yeast, Methods in Enzymology, BergerおよびKimmel編, Acad. Press, N.Y., 第152巻, 673-684頁;およびThe Molecular Biology of the Yeast Saccharomyces, 1982, Strathernら編, Cold Spring Harbor Press, 第IおよびII巻を参照のこと。
【0127】
昆虫系では、バキュロウイルスであるオートグラファ・カリフォルニカ(Autographa californica)核ポリヒドロシスウイルス(AcNPV)を、スポドプテラ・フルギペルダ(Spodoptera frugiperda)細胞にて改変型hspを発現するためのベクターとして使用できる。改変型hsp遺伝子配列は該ウイルスの非必須領域(例えば、ポリヘドリン遺伝子)内にクローン化し、AcNPVプロモーター(例えば、ポリヘドリンプロモーター)の制御下に置けばよい。これらの組換えウイルスは次いで、挿入されたDNAが発現する宿主細胞に感染させるのに用いる。(例えば、Smithら, 1983, J Virol 46:584; Smith, 米国特許第4,215,051号を参照。)。
【0128】
本明細書に記載のクローニングおよび発現ベクターはいずれも、当技術分野で十分公知の技術により、公知のDNA配列から合成および構築され得る。調節領域およびエンハンサーエレメントは、様々な天然および合成の両方の起源であり得る。いくつかのベクターおよび宿主細胞は商業的に入手できる。有用なベクターの例としては、限定されるものではないが、参照により本明細書に組み入れるCurrent Protocols in Molecular Biology, 1988, Ausubelら編, Greene Publish. Assoc. & Wiley Interscienceの付録5、およびClontech Laboratories, Stratagene Inc.およびInvitrogen, Inc.などの商業的供給者のカタログに記載されている。
【0129】
5.2.2 改変型hspの発現
改変型hspをコードするクローン化ヌクレオチド配列を含む発現構築物は、限定されるものではないが、原核細胞に関しては細菌形質転換(Hanahan, 1985, DNA Cloning, A Practical Approach, 1:109-136)、および真核細胞に関しては、リン酸カルシウム媒介トランスフェクション(Wiglerら, 1977, Cell 11:223-232)、リポソーム媒介トランスフェクション(Schaefer-Ridderら, 1982, Science 215:166-168)、エレクトロポレーション (Wolffら, 1987, Proc Natl Acad Sci 84:3344)、およびマイクロインジェクション(Cappechi, 1980, Cell 22:479-488)をはじめとする当技術分野で公知の種々の技術によって宿主細胞へ導入できる。改変型hspと抗原の同一宿主細胞中での同時発現も、実質的に同じ方法により達成できる。
【0130】
長期にわたって、正しくプロセッシングされた改変型hspまたは改変型hsp-ペプチド複合体を高収量で産生するように、哺乳類細胞内で安定した発現をすることが好ましい。改変型hspまたは改変型hsp-ペプチド複合体を安定して発現する細胞系統は、選択マーカーを含むベクターを用いることにより遺伝子工学的に作製してもよい。限定されるものではないが、例示すれば、発現構築物の導入後に、遺伝子操作された細胞を栄養強化培地中で1〜2日間増殖させ、次いで選択培地に切り替えればよい。発現構築物中の選択マーカーは選択に対する耐性を与え、最適には、細胞が、それらの染色体中へ該発現構築物を安定して組み込み、培養にて増殖し、さらには細胞系統にまで発達するようにさせる。かかる細胞は改変型hspを継続的に発現しつつ長期間培養可能である。
【0131】
組換え細胞は、標準的な条件の温度、インキュベーション時間、光学濃度および培地組成下で培養できる。あるいは、組換え抗原性細胞は、癌細胞または感染細胞の栄養要求性および生理的要求性を模倣した条件下で培養してもよい。しかしながら、組換え細胞の増殖のための条件は、改変型hspおよび抗原タンパク質の発現のための条件と異なっていてもよい。hsp-ペプチド複合体の産生を促進するために、改変された培養条件および培地を用いてもよい。例えば、改変型hspと同起源のプロモーターを保持する改変型hspを含む組換え細胞を、熱もしくは他の環境ストレス、または化学的ストレスに曝してもよい。改変型hspまたは改変型hsp-ペプチド複合体を産生するための至適条件を確立するために、当技術分野で公知のいずれの技術を適用してもよい。
【0132】
改変型hspを発現する組換え細胞がワクチンとして使用される、ある実施形態においては、得られた組換え細胞をin vivoで投与する前に改変型hsp遺伝子配列を細胞に導入する。かかる導入は、遺伝子治療技術を含む当技術分野において、限定されるものではないが、トランスフェクション、エレクトロポレーション、マイクロインジェクション、改変型hsp遺伝子配列を含有するウイルスまたはバクテリオファージベクターによる感染、細胞融合、染色体媒介遺伝子導入、微小細胞媒介遺伝子導入、スフェロプラスト融合などの公知のいずれの方法によっても行える。受容細胞の必要とされる発生学的および生理学的機能が破壊されない限り、外来遺伝子を細胞に導入するための当技術分野において公知の多くの技術 (例えば、LoefflerおよびBehr, 1993, Meth, Enzymol. 217:599-618; Cohenら, 1993, Meth. Enzymol. 217:618-644; Cline, 1985, Pharmac. Ther. 29:69-92を参照)を、本発明に従って使用してよい。この技術は、改変型hsp遺伝子配列の細胞への安定した導入を提供すべきであり、その結果、この配列が細胞により発現可能であり、さらにその子孫細胞により遺伝および発現可能であることが好ましい。
【0133】
得られた組換え細胞は、当技術分野で公知の種々の方法により患者に送達できる。好ましい実施形態では、上皮細胞を例えば皮下注射する。もう1つの実施形態では、組換え皮膚細胞を皮膚移植片として患者に適用してもよい。組換え血液細胞(例えば造血幹細胞または始原細胞)は、好ましくは静脈内投与される。使用が意図される細胞の量は、所望の効果、患者の状態などに依存し、当業者により決定できる。
【0134】
5.2.3. 改変型hspと抗原の同時発現
もう1つの実施形態では、タンパク質抗原またはそれらの一部をコードするヌクレオチド配列の発現型を、発現可能な改変型hsp遺伝子配列を含む組換え細胞中に導入でき、その結果としてこの抗原は改変型hspと同時発現される。抗原をコードするヌクレオチド配列を得る方法は、第5.4.5節に記載されている。限定されるものではないが、第5.2.2節に記載したような、抗原遺伝子配列の発現型を導入するためのいずれの技術を用いてもよい。タンパク質抗原またはそれらの一部は、組換え細胞のER中で改変型hspと非共有結合で結合し、得られた改変型hsp-抗原ペプチド複合体が分泌される。かかる複合体は、第5.3節に記載された方法、および当技術分野で公知の他の方法のいずれによっても、細胞培養培地から精製できる。精製された改変型hsp-抗原複合体は、癌もしくは感染症の治療または予防を目的として、被験者において抗原タンパク質に対する免疫反応を刺激するワクチンとして使用できる。
【0135】
さらに、改変型hsp遺伝子配列と抗原タンパク質をコードするヌクレオチド配列双方の発現型を含む組換え細胞を、被験者に注射するためのワクチンとして直接使用してもよい。前記のように、かかる細胞は、癌もしくは感染症の治療または予防を目的として、被験者において抗原タンパク質に対する免疫反応を刺激できる、改変型hsp-抗原複合体を分泌する。
【0136】
癌または感染症の治療および予防のための、かかる改変型hsp-ペプチド複合体と、改変型hspおよび抗原遺伝子配列の発現型を含む組換え細胞の用途は、第5.7節および5.8節に記載されている。
【0137】
5.3. 改変型hsp-ペプチド複合体の精製
一般に、本発明の改変型hspは、硫酸アンモニウム沈殿法、酸による抽出、陰イオンまたは陽イオン交換クロマトグラフィー、ホスホセルロースクロマトグラフフィー、イムノアフィニティークロマトグラフィー、ヒドロキシアパタイトクロマトグラフィー、およびレクチンクロマトグラフィーをはじめとする公知の方法により、組換え細胞培養物から回収および精製できる。
【0138】
細胞溶解物からのhsp70-ペプチド複合体の精製法はこれまでに記載されており、例えば、Udonoら, 1993, J. Exp. Med. 178:1391-1396を参照のこと。細胞溶解物からのhsp90-ペプチド複合体およびgp96-ペプチド複合体の精製は、例えば1995年9月21日付のWO 95/24923、および1997年3月20日付の WO 97/10000に記載されている。これらの方法を用いて本発明の改変型hspまたは改変型hsp-ペプチド複合体を組換え細胞から精製することができ、当技術分野に公知の重要でない改変により、改変型hspまたは改変型hsp-ペプチド複合体を細胞培養物から精製できる。
【0139】
しかしながら、本発明は改変型hsp上に存在するペプチドタグの特性に基づく、改変型hspの精製のための改良法を提供する。1つの方法は、タグとその結合相手の間の特異的な分子相互作用に基づくものである。もう1つの方法は、タグ上に存在するエピトープと抗体の免疫特異的結合に基づくものである。一般に当技術分野で十分公知のアフィニティークロマトグラフィーの原理をこれら双方の方法に適用できる。
【0140】
タグおよびその結合相手の特異的な分子相互作用に基づく、いくつかの方法を以下に記載する。
【0141】
免疫グロブリンの定常領域と融合された改変型hspの精製に一般的に適用可能な方法は、当技術分野で十分公知のプロテインAアフィニティークロマトグラフィー法である。ブドウ球菌のプロテインAは、42KDのポリペプチドであり、免疫グロブリンのH鎖の第2および第3の定常領域の間に位置する領域に特異的に結合する。異なるクラス、サブクラスおよび種の免疫グロブリンのFcドメインであるために、ヒトFc領域に対するプロテインAの親和性は強くても、他の種においては差異があるかもしれない。好ましくないサブクラスとしてはヒトIgG-3が挙げられ、ラットのサブクラスは最も好ましくない。特定のサブクラスに対しては、プロテインG(ブドウ球菌の)をプロテインAの代わりに精製に用いることができる。通常、抗体のアフィニティー精製用固相にはプロテイン-Aセファロース(PharmaciaまたはBiorad)を用い、免疫グロブリンのFcフラグメントと融合した改変型hspの精製にも、それを本質的に同様に使用できる。細胞上清に存在する、分泌された改変型hspは、固相上のプロテインAに特異的に結合するが、夾雑物は洗浄除去される。結合した改変型hspは、pHを段階的に下げた一連のクエン酸、酢酸およびグリシン-塩酸バッファーをはじめとする、当技術分野で公知の種々のバッファー系で溶出できる。組換え細胞が、改変型hspと同時に精製されるであろう抗体も産生する場合には、この方法は好ましくない。例えば、Langone, 1982, J. Immunol. Meth. 51:3; Wilchekら, 1982, Biochem. Intl. 4:629; Sjobringら, 1991, J, Biol. Chem. 26:399; 617-618頁, Antibodies A Laboratory Manual, HarlowおよびLane編集, Cold Spring Harbor laboratory, 1988)を参照のこと。
【0142】
あるいは、ポリヒスチジンタグを使用してもよく、その場合は改変型hspは金属キレートクロマトグラフィーで精製できる。通常、6個のヒスチジン配列であるこのポリヒスチジンタグは、ニトリロ三酢酸マトリックスのような固相に固定化できるニッケルイオン(Ni2+)のような2価の金属イオンに高い親和性を示す。ポリヒスチジンはNi2+-NTAアガロースに対してよく特徴付けられた親和性を有し、2つの穏やかな処理のいずれかにより溶出できる:イミダゾール(0.1-0.2M)は、効果的に該樹脂と結合部位を競合する;または、pHを6.0以下に下げることによりヒスチジンの側鎖をプロトン化し、結合を阻害する。この精製法は、細胞培養物の上清をNi2+-NTA-アガロースカラムにロードし、夾雑物を洗浄除去し、イミダゾールまたは弱酸により改変型hspを溶出させる。Ni2+-NTA-アガロースは、Sigma(St.Louis)およびQiagenのような商業的供給者から入手できる。改変型hspを検出し、定量するために用いられる、ポリヒスチジンタグを認識する抗体も利用できる。
【0143】
もう1つの使用可能なペプチドタグの例としては、元来、蠕虫である日本住血吸虫(Schistosoma japonicum)からクローン化されたグルタチオン-S-トランスフェラーゼ(GST)配列がある。一般に、大腸菌のような原核宿主細胞で発現される改変型hsp-GST融合物は、グルタチオンアガロースビーズによる吸着の後に、中性pHにて遊離還元グルタチオンの存在下で溶出させることにより、細胞培養物の上清から精製できる。変性条件は、精製中のいずれの過程においても必要とされないので、抗原ペプチドを保持する固定化された改変型hspのロードの際に用いることが望ましいであろう。さらに、GSTは一定条件下で二量体を形成することが知られているため、改変型hspの二量体が得られることもあり得る。Smith, 1993, Methods Mol. Cell Bio. 4:220-229を参照のこと。
【0144】
もう1つの有用な、使用可能なペプチドタグとしては、malE遺伝子にコードされる大腸菌のマルトース結合タンパク質(MBP)がある。細胞上清に存在する、分泌された改変型hsp-MBPはアミロース樹脂に結合するが、夾雑物は洗浄除去される。結合した改変型hsp-MBPはマルトースによりアミロース樹脂から溶出される。例えば、Guanら, 1987, Gene 67:21-30を参照のこと。
【0145】
改変型hspを精製するための第2の方法は、ポリクローナル抗体またはモノクローナル抗体が得られるようなエピトープを含むペプチドタグに対するものである。イムノアフィニティークロマトグラフィーおよび免疫沈降法のような、当技術分野で公知の、免疫特異的結合によるタンパク質の精製のための種々の方法が使用できる。例えば、Antibodies A Laboratory Manual, HarlowおよびLane編, 第13章、Cold Spring Harbor laboratory,1988; Current Protocols in Immunology, Coliganら編, 第8章, 第IおよびII節, John Wiley, 1991を参照のこと;なおその開示は両方とも、参照により本明細書に組み入れられる。
【0146】
前記の実施形態は、本発明の改変型hspを発現しているヒト細胞のような哺乳動物細胞の細胞培養培地から、分泌された改変型hsp-ペプチド複合体を回収および精製するために用いてよい。この方法は、改変型hspおよび/または改変型hsp-ペプチド複合体の中規模または大規模な精製に適用できる。改変型hsp-ペプチド複合体の精製のためには、pHを下げること、または変性条件を必要としない方法が最も好ましい。実施例では腫瘍細胞に関して記載したが、記載の方法は、例えば組織、単離細胞、または細胞内病原体に感染した真核細胞の不死化系統、または病原体に感染した被験者から得た細胞など、いずれの真核細胞からのhspの単離に使用してもよい。
【0147】
5.4. in vitroにおけるhsp-抗原性分子複合体の産生
改変型hspと組換え細胞中でそれらが内因的に関与するペプチドとの複合体が用いられない実施形態においては、改変型hspと抗原性分子の複合体をin vitroで調製する。当業者により認識されるように、以下に記載する方法による単離、化学的合成、または組換えによる産生のいずれかにより得られた抗原性ペプチドは、種々の改変型熱ショックタンパク質を用いてin vitro で再構成し非共有結合の改変型hsp-ペプチド免疫原性複合体を生成してもよい。かかる複合体は、免疫治療学的または予防のための本発明のワクチンに使用できる。改変型hsp-ペプチド複合体のin vitroにおける調製方法は、中規模または大規模で行うのに適用できる。
【0148】
改変型hspとの複合体形成のため、抗原性ペプチドとして使用するために、当技術分野で公知のものの中から、または、免疫測定法により抗体もしくはMHC分子と結合(抗原性)もしくは免疫応答できる(免疫原性)と決定されたものの中から、1以上の種の癌細胞、感染細胞もしくは感染性病原体に特異的な抗原またはそれらの抗原性部分を選択できる。
【0149】
5.4.1. 外因性抗原性分子
抗体との結合を検出することにより、問題の抗原の免疫原性または抗原性を決定するには、限定されるものではないが、ラジオイムノアッセイ、ELISA(酵素結合イムノソルベントアッセイ)、「サンドイッチ」イムノアッセイ、イムノラジオメトリックアッセイ、ゲル拡散沈降素反応、免疫拡散アッセイ、in vivoイムノアッセイ(例えば金コロイド、酵素または放射性同位元素標識を用いる)、ウエスタンブロット法、免疫沈降反応、凝集アッセイ(例えば、ゲル凝集検定法、血球凝集検定法)、補体固定アッセイ、免疫蛍光アッセイ、プロテインAアッセイ、および免疫電気泳動アッセイなどのような、競合および非競合アッセイ系を含む当技術分野に公知の種々の免疫検定法を用いることができる。1つの実施形態においては、一次抗体上の標識を検出することにより抗体結合を検出する。もう1つの実施形態においては、二次抗体または試薬と一次抗体との結合を検出することにより一次抗体を検出する。さらなる実施形態においては、二次抗体を標識する。当技術分野において、イムノアッセイで結合を検出するための多くの方法が公知であり、その使用が意図される。免疫原性を検出するための1つの実施形態においては、例えばin vitro細胞傷害性アッセイまたはin vivo遅延型過敏性アッセイなどの標準的な方法により、T細胞媒介応答を測定できる。
【0150】
抗原性分子としての使用のために、有用な可能性のある抗原またはそれらの誘導体も、病原体の感染性の中和における抗原の関与(そのような病原体による感染は治療または予防するのが望ましい)(Norrby, 1985, Summary, Vaccines 85, Lernerら(eds.), Cold Spring Harbor Laboratory, Cold Spring Harbor, New York, pp388-389)、タイプまたはグループ特異性、患者の抗血清または免疫細胞による認識、および/または抗原に特異的な抗血清もしくは免疫細胞の防御作用の実証などの、種々の基準により同定できる。さらに、病原体により引き起こされた疾病の治療または予防が望まれる場合には、抗原のコードされたエピトープが、時宜よく、または同じ病原体の種々の単離体間で、わずかな抗原性変化を示すか、あるいは全く変化を示さないのが好ましい。
【0151】
癌の治療または予防が望まれる場合、公知の腫瘍特異的抗原またはフラグメントもしくはそれらの誘導体が使用される。例えば、かかる腫瘍特異的または腫瘍関連抗原としては、限定されるものではないが、KS 1/4万能癌抗原(Perezおよび Walker, 1990, J. Immunol. 142:3662-3667; Bumal, 1998, Hybridoma 7(4):407-415);卵巣癌抗原(CA125)(Yuら, 1991, Cancer Res. 51(2):468-475);前立腺の酸リン酸塩(prostatic acid phosphate)(Tailerら, 1990, Nucl. Acids Res. 18(16):4928);前立腺特異的抗原(HenttuおよびVihko, 1989, Biochem. Biophys. Res. Comm. 160(2):903-910; Israeliら, 1993, Cancer Res. 53:227-230);黒色腫関連抗原p97(Estinら, 1989, J. Natl. Cancer Inst. 81(6):445-446); 黒色腫抗原gp75(Vijayasardahlら, 1990, J. Exp. Med. 171(4):1375-1380);高分子量黒色腫抗原(Nataliら, 1987, Cancer 59:55-63)および前立腺特異的膜抗原が挙げられる。
【0152】
特定の実施形態では、ある特定の腫瘍に特異的な抗原またはフラグメントもしくはそれらの誘導体を選択して改変型hspと複合体を形成し、次いでその腫瘍を有する患者に投与する。
【0153】
ウイルス性疾患の治療または予防が所望される場合は、既知のウイルスのエピトープを含む分子を用いる。例えば、かかる抗原性エピトープは、限定されるものではないが、A型肝炎、B型肝炎、C型肝炎、インフルエンザ、水痘、アデノウイルス、I型単純ヘルペス(HSV-I)、II型単純ヘルペス(HSV-II)、牛疫、ライノウイルス、ECHOウイルス、ロタウイルス、RSウイルス、パピローマウイルス、パポバウイルス、サイトメガロウイルス、エキノウイルス、アルボウイルス、ハンタウイルス、コクサッキーウイルス、ムンプスウイルス、麻疹ウイルス、風疹ウイルス、ポリオウイルス、I型ヒト免疫不全ウイルス(HIV-I)、およびII型ヒト免疫不全ウイルス(HIV-II)をはじめとするウイルスから製造してもよい。
【0154】
細菌感染の治療または予防が望ましい場合には、既知の細菌のエピトープを含む分子を用いるのが好ましい。例えば、限定されるものではないが、マイコバクテリア、リケッチア、マイコプラズマ、ナイセリアおよびレジオネラをはじめとする細菌から、かかる抗原性エピトープを製造してもよい。
【0155】
原生動物による感染の治療または予防が望ましい場合には、既知の原生動物のエピトープを含む分子を用いるのが好ましい。例えば、限定されるものではないが、リーシュマニア、コクジジオア(kokzidioa)、およびトリパノソーマをはじめとする原生動物から、かかる抗原性エピトープを製造してもよい。
【0156】
寄生虫による感染の治療または予防が望ましい場合には、既知の寄生虫のエピトープを含む分子を用いるのが好ましい。例えば、限定されるものではないが、クラミジアおよびリケッチアをはじめとする寄生虫から、かかる抗原エピトープを製造してもよい。
【0157】
5.4.2. hsp-ペプチド複合体からのペプチド
本発明の改変型hspとin vitroで複合体を形成するための抗原ペプチドはまた、ペプチドとhspの内因性複合体から得ることができる。hsp-ペプチド複合体からペプチドを溶離させるためには2つの方法を用いてよい。1つの試みは、ATP存在下でhsp-ペプチド複合体をインキュベートすることを含む。もう1つの試みは、低pHのバッファー中でこの複合体をインキュベートすることを含む。
【0158】
簡単に説明すると、目的の複合体をCentricon 10アッセンブリィー(Millipore)を通して遠心分離し、複合体と弱く結合している低分子量の物質を除去する。大分子量の画分は、SDS-PAGEにより除去および分析するが、低分子量画分は以下に記載するようにHPLCにより分析すればよい。ATPインキュベーションプロトコールにおいては、大分子量画分中のhsp-ペプチド複合体を10mMのATPとともに室温にて30分間インキュベートする。低pHのプロトコールにおいては、酢酸またはトリフルオロ酢酸(TFA)をhsp-ペプチド複合体に加え、最終濃度10%(vol/vol)とし、この混合物を室温または沸騰水浴中、またはその間のいずれかの温度において10分間インキュベートする(Van Bleakら, 1990, Nature 348:213-216;およびLiら, 1993, EMBO Journal 12:3143-3151参照)。
【0159】
得られたサンプルを前記に従いCentricon 10アッセンブリィーを通して遠心分離する。高分子量画分および低分子量画分を回収する。残りの大分子量のhsp-ペプチド複合体をATPとともに、または低pHにおいて再度インキュベートし、残存するペプチドを除去できる。
【0160】
得られた低分子量画分をプールし、蒸発濃縮して0.1%のTFAに溶解させる。次いで、例えば0.1%TFAで平衡化させたVYDAC C18逆相カラムを用いる逆相高速液体クロマトグラフィー(HPLC)により、溶解した物質を分画する。次いで、0.1%TFA中の0〜80%アセトニトリルの線形勾配を用いて流速約0.8ml/分でカラムを展開させ、結合した物質を溶出させる。ペプチドの溶出はOD210によりモニターでき、ペプチドを含む画分を回収する。
【0161】
5.4.3. MHC-ペプチド複合体からのペプチド
in vitroで本発明の改変型hspと複合体を形成させるには、MHC分子に結合するペプチドも使用できる。潜在的免疫原性ペプチドの、MHC分子からの単離法は当技術分野において公知であり、本明細書においては詳細に記載しないが(Falkら, 1990, Nature 348:248-251; Rotzscheら, 1990, Nature 348:252-254; Elliottら, 1990, Nature 348:191-197; Falkら, 1991, Nature 351:290-296; Demotzら, 1989, Nature 343:682-684; Rotzscheら, 1990, Science 249:283-287を参照)、その開示は参照により本明細書に組み入れられる。
【0162】
簡単に説明すると、従来のイムノアフィニティー法によりMHC-ペプチド複合体を単離してよい。次いで、アセトニトリル中の約0.1%TFAの存在下でこの複合体をインキュベートすることにより、ペプチドをMHC-ペプチドから溶離させればよい。従前のごとく逆相HPLCにより溶離させたペプチドを分画し、精製すればよい。
【0163】
5.4.4. 合成ペプチド
当技術分野に公知の手動または自動のいずれかのアミノ酸配列決定技術により、MHC分子から溶離させたペプチドまたはhspのアミノ酸配列を決定してもよい。潜在的な防御ペプチドのアミノ酸配列が決定された後、従来のペプチド合成法または当技術分野に公知の他の方法を用いて、所望の量のペプチドを合成してもよい。
【0164】
Merrifield, 1963, J. Am. Chem. Soc., 85:2149に記載されたものと同様の方法を用いて、前記で単離されたような同一のアミノ酸配列を有するペプチドを、固相合成法により合成してもよい。合成中、そのC末端に連結した、成長中のポリペプチド鎖、および不溶性高分子支持体、すなわちポリスチレンビーズに、側鎖が保護されたN-α-保護アミノ酸を段階的に加える。N-α-脱保護アミノ酸のアミノ基を、ジシクロヘキシルカルボジイミドのような試薬と反応させることにより活性化させたN-α-保護アミノ酸α-カルボキシル基に結合させることによりペプチドを合成する。遊離アミノ基を、活性化されたカルボキシル基に結合することにより、ペプチド結合の形成が起こる。最も一般的に用いられるN-α-保護基としては、酸に不安定なBocおよび塩基に不安定なFmocが挙げられる。適当な化学物質、樹脂、保護基、保護されたアミノ酸および試薬の詳細については、当技術分野において十分公知であり、本明細書においては詳しく論じない(Athertonら, 1989, Solid Phase Peptide Synthesis: A Practical Approach, IRL Press, およびBodansky, 1993, Peptide Chemistry, A Practical Textbook, 第2版, Springer-Verlagを参照)。
【0165】
得られたペプチドの精製は、ゲル浸透を用いる分取HPLC、分配および/またはイオン交換クロマトグラフィーのような従来の方法を用いて行える。適当なマトリックスおよびバッファーの選択は当技術分野において十分公知であり、本明細書では詳細に論じない。
【0166】
5.4.5 抗原をコードする遺伝子配列
本発明の特定の実施形態では、タンパク質抗原をコードするヌクレオチド配列またはその一部が、抗原の生産のために宿主細胞に導入できる。いずれの抗原タンパク質をコードするヌクレオチド配列でも得ることができ、実質的にhsp遺伝子配列のクローニングおよび発現に関して記載されている同様の方法により、発現のための発現ベクターにクローン化できる。この技術は第5.1−5.1.1節および第5.2−5.2.2節に記載されており、当技術分野で十分公知である。組換え抗原性タンパク質またはその一部はタンパク質に対して適当ないずれの方法によっても精製でき、次いで、これを用いて、第5.4.6節に記載されているようにin vitroで改変型hspと複合体を形成することができる。かかる改変型hsp-抗原複合体をワクチンとして用いて、癌または感染症の治療または予防の目的で、被験体において抗原性タンパク質に対する免疫応答を刺激することができる。
【0167】
5.4.6 in vitroにおける改変型hsp-ペプチド複合体の形成
改変型hspと抗原性分子とをin vitroで非共有結合的に複合体形成する好ましいプロトコールの例を以下に示す。ペプチドタグによって固相に可逆的に結合した改変型hspを用いて、複合体形成反応の前または後でバッファーの交換、洗浄および複合体の単離を容易にするのが有利でありうる。
【0168】
複合体形成に先立ち、改変型hspはATPまたは低pHで前処理して、注目されるhspと会合する可能性のあるいずれのペプチドも除去しておく。ATP法を用いる場合、Levyら, 1991, Cell 67:265-274により記載されたようにアピラナーゼの添加によって調製物から過剰なATPを除去する。低pH法を用いる場合、pH調整剤の添加により、バッファーを中性pHに再調整する。
【0169】
抗原性分子(1μg)と前処理した改変型hsp(9μg)を混合して抗原性分子:hspモル比をおよそ5:1とする。次いで、この混合物を20mMリン酸ナトリウムpH 7.2、350mM NaCl、3mM MgCl2、および1mMフェニルメチルスルホニルフルオリド(PMSF)を含有するものなど、好適な結合バッファー中、4℃〜45℃で15分〜3時間インキュベートする。この調製物をCentricon 10アッセンブリィー(Millipore)で遠心分離して結合していないいずれのペプチドも除去する。改変型hspが固相に結合している場合は、改変型hsp-ペプチド複合体を固相から溶離させる前に、形成した改変型hsp-ペプチド複合体を、未結合のペプチドを含まないよう洗浄することができる。ペプチドと改変型hspとの会合はSDS-PAGEでアッセイすることができる。これは、内在性hsp-ペプチド複合体から解離したペプチドのMHC-ペプチド複合体から単離されたペプチドをin vitroで複合体形成する好ましい方法である。
【0170】
本発明の別の実施形態では、タンパク質など内在性の抗原性分子に対して改変型hsp70の複合体を形成するために、5〜10μgの精製hspを、100μlの容量中、20mMリン酸ナトリウムバッファー pH 7.5、0.5M NaCl、3mM MgCl2および1mM ADPで等モル量の抗原性分子とともに37℃で1時間インキュベートすることが好ましい。このインキュベーション混合物を、リン酸緩衝食塩水で1mlにさらに希釈する。
【0171】
本発明のさらに別の実施形態では、ペプチドに対し改変型gp96の複合体を形成するために、アフィニティータグによって固相に固定化された5〜10μgの改変型gp96を、20mMリン酸ナトリウムバッファー pH 7.5、0.5M NaCl、3nM MgCl2を含有するものなど、好適なバッファー中、約50℃で約10分間、等モルまたは過剰量の抗原性ペプチドとともにインキュベートすることが好ましい。例えば、この方法のために、Igタグを含む改変型gp96はタンパク質A-セファロースに固定化できる。次いで、このインキュベーション混合物を室温で約30分間さらにインキュベートする。改変型hsp-ペプチド複合体が結合した固相を数回洗浄して、結合していないいずれのペプチドも除去する。次いで、改変型hsp-ペプチド複合体を適当な方法により固相から溶離させる。
【0172】
複合体形成の後、免疫原性hsp-抗原分子複合体は場合により、例えば以下に記載される混合リンパ球標的細胞アッセイ(MLTC)を用いてin vitroでアッセイできる。一度、免疫原性複合体が単離されれば、それらは場合により、以下に論じられる好ましい投与プロトコールおよび賦形剤を用いて動物モデルでさらに特性決定できる。
【0173】
5.5 hsp-ペプチド複合体の免疫原性の決定
任意の手順において、精製改変型hsp-ペプチド複合体は、当技術分野で十分公知の混合リンパ球標的培養アッセイ(MLTC)を用い、免疫原性に関してアッセイできる。
【0174】
限定されるものではないが例示すれば、以下の手法が使用できる。簡潔に言うと、マウスに候補となる改変型hsp-ペプチド複合体を皮下注射する。その他のマウスには正常な非組換え細胞由来の他のhsp-ペプチド複合体またはアッセイについて陽性対照として働く全感染細胞のいずれかを注射する。マウスには7〜10日おいて2回注射する。最後の免疫化の10日後、脾臓を摘出してリンパ球を遊離させる。遊離したリンパ球は、次ぎに、目的の複合体を発現した死細胞を添加することにより、in vitroで再び刺激すればよい。
【0175】
例えば、10%ウシ胎児血清を含有する3mlのRPMI培地中で、8x106個の免疫脾臓細胞を、4x104個のマイトマイシンCで処理したもしくはγ線照射した(5〜10,000ラッド)病原体感染細胞(すなわち、あり得る場合としては、感染性病原体の抗原をコードする遺伝子でトランスフェクトされた細胞)、または腫瘍細胞で刺激することができる。特定の場合には、T細胞増殖因子の供給源として培地中に33%の二次混合リンパ球培養上清またはインターロイキン2(IL-2)を含んでも良い(Glasebrookら, 1980, J. Exp. Med, 151:876を参照)。免疫後、細胞傷害性T細胞の一次応答を試験するためには、脾臓細胞を刺激せずに培養すればよい。いくつかの実験では、免疫化したマウスの脾臓細胞を、抗原的に異なる細胞で再刺激して、細胞傷害性T細胞の応答の特異性を決定することもできる。
【0176】
6日後、培養物を4時間の51Cr放出アッセイで細胞傷害性に関して試験した(Palladinoら, 1987, Cancer Res. 47:5074-5079およびBlachereら, 1993, J. Immunotherapy 14:352-356を参照)。このアッセイでは、混合リンパ球培養物を標的細胞懸濁物に加えて、種々のエフェクター:標的(E:T)比とする(通常は、1:1〜40:1)。標的細胞を、200mCi51Cr/mlを含有する培地中で37℃にて1時間、1x106個の標的細胞をインキュベートすることにより予め標識する。標識後、細胞を3回洗浄する。各アッセイ点(E:T比)は3回反復して行い、適当な対照を組み入れて自然51Cr放出(アッセイにリンパ球を加えない場合)と100%放出(界面活性剤で溶解した細胞)を測定する。細胞混合物を4時間インキュベートした後、200gで5分間の遠心分離によって細胞をペレットとする。上清中に放出された51Crの量をガンマカウンターにより測定する。細胞傷害性%は、(試験サンプル中のcpm−自然に放出されたcpm)/(界面活性剤で放出された全cpm−自然に放出されたcpm)にてcpmとして測定する。
【0177】
MHCクラスIカスケードをブロックするため、K-44ハイブリドーマ細胞(抗MHCクラスIハイブリドーマ)由来の濃縮ハイブリドーマ上清を、最終濃度が12.5%となるように試験サンプルに加える(Heikeら, 1994, J. Immunotherapy 15:165-174)。
【0178】
クロム放出アッセイの代替法としては、特異的な抗原で刺激した後のin vitroにおける細胞傷害性T細胞によるサイトカインの放出を測定するELISPOTアッセイがある。サイトカイン放出は、インターロイキン-2、腫瘍壊死因子αまたはインターロイキン-γなどの特定のサイトカインに特異的な抗体により検出する(例えば、Scheibenbogenら, 1997, Int. J. Cancer, 71:932-936を参照)。このアッセイは、T細胞によって分泌されるサイトカインを捕捉する、目的のサイトカインに特異的な抗体で予め被覆したマイクロタイタープレート中で行う。被覆したウェル内で24〜48時間T細胞をインキュベートした後、細胞傷害性T細胞を除去して、サイトカイン上の異なるエピトープを認識する標識二次抗体と置き換える。十分洗浄して結合していない抗体を除去した後、有色の反応生成物を生成する酵素基質をプレートに加える。サイトカイン産生細胞の数を顕微鏡下で数える。この方法はアッセイ時間が短く、かつ、多数の細胞傷害性T細胞を必要とせず鋭敏であるという利点を有する。
【0179】
5.6. 製剤
本発明の方法によって精製した改変型hspと抗原タンパク質またはペプチドの非共有結合複合体を、癌または感染症を治療または予防するために哺乳類に投与するための医薬製剤に製剤化することができる。治療剤の投与経路を選択する場合に考慮すべき要素は薬剤溶解性および吸収部位である。本発明の改変型hsp-抗原分子複合体を、限定されるものではないが、例えば、皮下、静脈内、または筋肉内などの任意の投与経路を使用して投与してもよいが、皮内または粘膜投与が好ましい。皮内または粘膜投与の利点はそれぞれ、より低用量であることと、吸収が迅速なことである。粘膜による投与経路としては、限定されるものではないが、経口、直腸、および鼻腔内投与が挙げられる。粘膜投与用の製剤には下記のような様々な製剤が適当である。投与経路は治療経過中に変更することができる。ペプチドと天然hspの複合体の好ましい用量、投与経路および治療計画については、WO96/10411およびWO97/10001として公開されたPCT国際特許出願に記載されており、これらは参照によりその全体を本明細書に組み入れるものとする。
【0180】
好ましい実施形態においては、ある量の改変型gp96-ペプチド複合体をヒトに投与する。すなわち約10〜600μg、好ましくは10〜100μg、最も好ましくは約25μgの範囲で、約4〜6週間、週に1回、種々の投与部位の皮内に逐次投与する。
【0181】
適合する医薬担体中に製剤化された非共有結合複合体を含んでなる組成物を調製、封入し、ヒト肉腫および癌腫、例えば、繊維肉腫、粘液肉腫、脂肪肉腫、軟骨肉腫、骨肉腫、脊椎腫、血管肉腫、内皮肉腫、リンパ管肉腫、リンパ管内皮肉腫、滑液腫、内皮腫、ユーイング肉腫、平滑筋肉腫、横紋筋肉腫、結腸癌、膵臓癌、乳癌、卵巣癌、前立腺癌、扁平上皮細胞癌、基底細胞癌、腺癌、汗腺癌、皮脂腺癌、乳頭癌、乳頭腺癌、包嚢腺癌、骨髄癌、気管支癌、腎細胞癌、肝癌、胆管癌、絨毛癌、精上皮腫、胚癌、ウィルムス腫、子宮頸癌、精巣癌、肺癌、小細胞肺癌、膀胱癌、上皮癌、神経膠腫、星状細胞腫、髄芽腫、頭蓋咽喉腫、上衣腫、松果体腫、血管芽腫、聴神経腫、乏突起膠細胞腫、髄膜腫、黒色腫、神経芽腫、網膜芽腫;白血病、例えば、急性リンパ球性白血病および急性骨髄性白血病(骨髄芽球性、前骨髄性、骨髄単球性、単球性、および赤白血病);慢性白血病(慢性骨髄性(顆粒球性)白血病および慢性リンパ球性白血病);および真性赤血球増加症、リンパ腫(ホジキン病および非ホジキン病)、多発性骨髄腫、ワルデンストレームマクログロブリン血症、およびH鎖病などのような記載した腫瘍の治療用に表示する。この複合体が水溶性である場合には、適当なバッファー中、例えば、リン酸緩衝生理食塩水または他の生理学上適合する溶液中で製剤化してもよい。また、得られた複合体が水性溶媒に難溶性である場合には、Tweenなどの非イオン性界面活性剤かポリエチレングリコールとともに製剤化してもよい。よって、非共有結合複合体およびその生理学上許容される溶媒和物を、吸入または通気(口または鼻のいずれかを通して)による投与、経口、口内、非経口、直腸投与、または固形腫瘍への直接注入による投与(腫瘍の場合)用に製剤化してもよい。
【0182】
経口投与のためには、医薬製剤は液体の形態、例えば、水溶液、シロップ、または懸濁液であってもよく、あるいは使用前に水または他の適当なビヒクルを加えて再構成する医薬品として提供してもよい。かかる液体製剤は懸濁剤(例えば、ソルビトールシロップ、セルロース誘導体または水素添加食用油);乳化剤(例えば、レシチンまたはアラビアゴム);非水性ビヒクル(例えば、アーモンド油、油性エステルまたは分留植物油);および防腐剤(例えば、p-ヒドロキシ安息香酸メチルもしくはプロピルまたはソルビン酸)のような製薬上許容される添加剤を加え、従来法によって調製すればよい。医薬組成物は例えば、結合剤(例えば、プレゲル化トウモロコシデンプン、ポリビニルピロリドンもしくはヒドロキシプロピルメチルセルロース);増量剤(例えば、ラクトース、微晶性セルロースもしくはリン酸水素カルシウム);滑沢剤(例えば、ステアリン酸マグネシウム、タルクもしくはシリカ);崩壊剤(例えば、ジャガイモデンプンもしくはグリコール酸ナトリウムデンプン);または湿潤剤(例えば、ラウリル硫酸ナトリウム)などの製薬上許容される賦形剤を加え、従来法によって調製された錠剤またはカプセル剤の形態をとってもよい。錠剤は当技術分野で公知の方法によって被覆してもよい。
【0183】
経口投与用の医薬製剤は、複合体の徐放性が得られるように適宜製剤化してもよい。かかる組成物は従来法で製剤化された錠剤またはトローチ剤の形態をとってもよい。
【0184】
吸入による投与のためには、複合体は適当な噴射剤、例えば、ジクロロジフルオロメタン、トリクロロジフルオロメタン、ジクロロテトラフルオロエタン、二酸化炭素または他の適当なガスを用いて加圧容器またはネブライザーから与えられるエアロゾルスプレー剤の形で便宜に送達され得る。加圧エアロゾル剤の場合では、用量単位は、定量を送達するためのバルブを提供することによって決定してもよい。吸入器または通気器で使用するために、複合体と、ラクトースまたはデンプンなどの適当な粉末基剤との粉末混合物を含む、例えば、ゼラチンのカプセル剤およびカートリッジを製剤化してもよい。
【0185】
複合体を注射、例えばボーラス注射または点滴による非経口投与用に製剤化することができる。注射用製剤は防腐剤を加えて単位剤形(例えば、アンプル)または複数回量容器で与えてもよい。組成物は懸濁液、溶液、または油性もしくは水性ビヒクル中のエマルションような形態をとってもよく、懸濁化剤、安定化剤および/または分散化剤などの配合剤を含んでもよい。また有効成分は使用前に適当なビヒクル、例えば、発熱物質フリーの滅菌水を加えて再構成するための粉末の形態であってもよい。
【0186】
また複合体は坐剤または滞留浣腸剤のような、例えばカカオ脂または他のグリセリドなどの通常の坐剤用基剤を含む直腸組成物に製剤化してもよい。
【0187】
前記の製剤に加え、複合体をデポー製剤として製剤化してもよい。かかる長期作用製剤は植え込み(皮下もしくは筋肉内)により、または筋肉内注射により投与してもよい。よって、例えば、複合体を適当なポリマー性もしくは疎水性物質(例えば、許容される油中のエマルションとして)またはイオン交換樹脂とともに、あるいは徐々に溶ける誘導体として、例えば、徐々に溶ける塩として製剤化してもよい。疎水性薬剤の送達ビヒクルまたは担体の例としては、リポソームおよびエマルションが公知である。
【0188】
必要なら、非共有結合複合体を含む1以上の単位剤形が含まれるパックまたはディスペンサー装置で複合体を提供してもよい。例えば、そのパックはブリスターパックのように、金属またはプラスチック箔を含んでなってもよい。パックまたはディスペンサー装置には投与についての説明書が添付される。
【0189】
また本発明では、本発明の治療計画を実施するためのキットも提供する。かかるキットは1以上の容器内に、治療上または予防上有効量の非共有結合改変型hsp-ペプチド複合体を、製薬上許容される形態で含んでなる。本発明のキットのバイアル中の改変型hsp-ペプチド複合体は、製薬上許容される溶液、例えば滅菌生理食塩水、ブドウ糖溶液、またはバッファー溶液の組み合わせ、あるいは他の製薬上許容される滅菌液の形態であってよい。また、複合体を凍結乾燥するか、または乾燥させてもよい;この場合、キットにはさらに容器内に、製薬上許容される溶液(例えば生理食塩水、ブドウ糖溶液など)、好ましくは滅菌されたものが含まれていてもよく、複合体を再構成して、注射用液を形成する。
【0190】
別の実施形態では、本発明のキットにはさらに、複合体を注入するための、好ましくは滅菌包装されたニードルもしくはシリンジ、および/または包装されたアルコールパッドが含まれている。場合により臨床医または患者による改変型hsp-ペプチド複合体の投与についての説明書が含まれている。
【0191】
5.7. 癌の予防および治療
なぜ、本発明により調製される非共有結合改変型hsp-ペプチド複合体または改変型hspを発現する組換え細胞によって提供される免疫治療を癌患者において使用することが望ましいかについては多くの理由がある。第1に、癌患者が免疫抑制される場合、ならびに麻酔による手術および後の化学療法によって免疫抑制を悪化させる場合、術前期間の適当な免疫治療によって、この免疫抑制を阻害するか、または逆転させることができる。このことによって感染合併症がより少なくなり、創傷治癒が加速され得る。第2に、術後、腫瘍の大きさは最小となり、この状況においては免疫治療が最も有効であるようである。第3の理由は、手術時に腫瘍細胞が循環系に流れ出る可能性があり、このときに適用した有効な免疫治療でこれらの細胞を排除し得るということである。
【0192】
特定の実施形態において、本発明の予防的および治療的有用性は術前か、または術後のいずれかの癌患者の免疫適格の向上、および癌細胞に対する腫瘍特異的な免疫性の誘導に向けられ、その目的は癌の抑制であり、最大の臨床上の目的は全面的な癌の退縮と根絶である。
【0193】
本発明によれば、好ましい癌の治療および予防法には1以上の個体、好ましくは治療が必要な個体から癌細胞を単離すること、およびかかる細胞中に、発現可能な改変型hsp遺伝子配列を、好ましくは発現遺伝子構築物として導入することが含まれる。前記の第5.1節の方法によって、改変型hsp遺伝子配列を、発現構築物の形態の、または染色体内に組み込まれた改変型hsp遺伝子配列が組換え細胞における改変型hspの発現に好適であるように遺伝子工学的に操作する。発現遺伝子構築物を含む組換え細胞を、発現遺伝子構築物によってコードされる改変型hspが組換え宿主細胞によって発現される条件下で培養する。癌細胞のペプチドと非共有結合した改変型熱ショックタンパク質複合体が分泌され、好ましくは第5.2節に記載の方法によって培地から精製する。投与経路に依存して、第5.4節の記載に従って改変型hsp-ペプチド複合体を製剤化し、個体に自己由来のものを(例えば、原発性の癌またはその転移を治療するために)、または同種組織タイプの癌の治療が必要な他の個体に、または家族の病歴もしくは環境危険因子が理由で癌の危険性が高い個体に投与する。hsp-ペプチド複合体の治療的および予防的使用法の例もPCT公開WO96/10411(1996年4月11日付)およびWO97/10001(1997年3月20日付)に記載されている。
【0194】
例えば、前記のように調製された改変型hsp-ペプチド複合体による治療は、術後のどの時点で開始してもよい。しかしながら、患者が化学療法を受けてきた場合には、通常4週間以上の間隔をおいた後に、hsp-抗原複合体を投与して免疫系を回復させる。治療計画には生理食塩水または他の生理学上適合する溶液に溶かした改変型hsp-抗原複合体の毎週の注入が含まれる。注入経路および部位は、例えば第1の注入は左腕に皮下投与し、第2の注入は右腕に、第3の注入は左腹部に、第4の注入は右腹部に、第5の注入は左腿部に、第6の注入は右腿部に、などとそれぞれの時期によって変える。同一部位には1回以上の注入間隔をおいた後に再び行う。さらに注入物を分配し、用量の半分をそれぞれ、同日に異なる部位へ投与する。全体的に見て、最初の4〜6回の注入を1週間間隔で与え、続いて2回の注入を2週間間隔で与え、その後は1ヶ月間隔で注入する計画が与えられる。
【0195】
あるいは、改変型hsp-ペプチド複合体を分泌する組換え腫瘍細胞を、患者へ注入するためのワクチンとして用いて、腫瘍細胞または腫瘍抗原を担持する細胞に対する免疫応答を刺激することができる。改変型hsp-ペプチド複合体を安定して発現し、分泌する自己組換え腫瘍細胞が好ましい。適当な用量を決定するため、組換え細胞によって分泌された改変型hsp-ペプチド複合体の量を定量し、in vivoでのばらつきのない分泌レベルを確実に達成することによって、ワクチン接種に使用する組換え細胞数を調整する。好ましい用量とは、24時間当たり約100ngの改変型gp96を分泌可能な組換え細胞数である。患者の安全性のため、患者への注入の直前に組換え腫瘍細胞を照射することが可能である(12000rad)。照射細胞は増殖せず、約7〜10日間、改変型hsp-ペプチド複合体を分泌し続けることができ、これは免疫応答を誘導するに十分である。
【0196】
本発明の方法によって調製された非共有結合hsp-ペプチド複合体を用いて、治療または予防できる癌には、限定されるものではないが、ヒト肉腫および癌腫、例えば、繊維肉腫、粘液肉腫、脂肪肉腫、軟骨肉腫、骨肉腫、脊椎腫、血管肉腫、内皮肉腫、リンパ管肉腫、リンパ管内皮肉腫、滑液腫、内皮腫、ユーイング肉腫、平滑筋肉腫、横紋筋肉腫、結腸癌、膵臓癌、乳癌、卵巣癌、前立腺癌、扁平上皮細胞癌、基底細胞癌、腺癌、汗腺癌、皮脂腺癌、乳頭癌、乳頭腺癌、包嚢腺癌、骨髄癌、気管支癌、腎細胞癌、肝癌、胆管癌、絨毛癌、精上皮腫、胚癌、ウィルムス腫、子宮頸癌、精巣癌、肺癌、小細胞肺癌、膀胱癌、上皮癌、神経膠腫、星状細胞腫、髄芽腫、頭蓋咽喉腫、上衣腫、松果体腫、血管芽腫、聴神経腫、乏突起膠細胞腫、髄膜腫、黒色腫、神経芽腫、網膜芽腫;白血病、例えば、急性リンパ球性白血病および急性骨髄性白血病(骨髄芽球性、前骨髄性、骨髄単球性、単球性、および赤白血病);慢性白血病(慢性骨髄性(顆粒球性)白血病および慢性リンパ球性白血病);および真性赤血球増加症、リンパ腫(ホジキン病および非ホジキン病)、多発性骨髄腫、ワルデンストレームマクログロブリン血症、およびH鎖病などが挙げられる。かかる癌の具体例を、下記の節で説明する。
【0197】
特定の実施形態において、癌は転移性のものである。別の特定の実施形態では、本発明のhsp-ペプチド分子複合体を投与する前に抗癌治療(例えば、放射線化学療法)を受けたという理由によって癌を有する患者が免疫抑制されている。別の特定の実施形態においては、癌は腫瘍である。
【0198】
悪性疾患の進行における改変型hsp-ペプチド複合体を用いた免疫治療の効果を、当業者に公知の任意の方法によってモニターすることができる。該方法としては、限定されるものではないが、a)細胞性免疫の評価として遅延型過敏性;b)in vitroでの細胞溶解性Tリンパ球の活性;c)腫瘍特異的抗原、例えば癌胎児性抗原(CEA)のレベル;d)コンピューター断層撮影スキャン(CT)のような手法を用いた腫瘍の形態変化;e)高い危険性のある個体の特定の癌に対する危険性についての推定バイオマーカーレベルの変化、およびf)ソノグラムを用いた腫瘍の形態変化、の測定が挙げられる。さらに使用できるその他の手法としては、シンチグラフィーおよび内視鏡検査法が挙げられる。
【0199】
また改変型hsp-ペプチド複合体を用いた免疫治療の予防効果を、特定の癌の危険性についての推定バイオマーカーレベルを測定することにより評価してもよい。例えば、前立腺癌の危険性の高まった個体においては、血清前立腺特異的抗原(PSA)をBrawerら, 1992, J. Urol. 147: 841-845, および Catalonaら, 1993, JAMA 270: 948-958に記載される手順によって測定する;または結腸直腸癌の危険性のある個体においては、CEAを当技術分野で公知の方法によって測定する;および乳癌の危険性の高まった個体においては、エストラジオールの16-α-ヒドロキシル化をSchneiderら, 1982, Proc. Natl. Acad. Sci. USA 79: 3047-3051に記載される手順によって測定する。前記に引用される参考文献は、参照によりその全体を本明細書に組み入れるものとする。
【0200】
5.8. 感染症の予防および治療
本発明の改変型hsp-ペプチド複合体は、細胞内病原体に感染した細胞、ならびに細胞内病原体によって形質転換された細胞から調製できることも明らかとなった。例えば、免疫原性hsp-ペプチド複合体をSV40のような形質転換ウイルスで形質転換した真核細胞から単離することができる。
【0201】
本発明の好ましい実施形態において、精製された改変型hsp-ペプチドワクチンは細胞内病原体によって引き起こされるヒト疾患の治療において特に有用であると考えられる。しかしながら、本明細書に記載される原理を用いて開発されたワクチンは同様に細胞内病原体によって引き起こされる他の哺乳類、例えばウシ、ウマ、ヒツジ、ヤギ、およびブタなどの飼育動物ならびにネコおよびイヌなどの家庭内ペットの疾患の治療に有用であることがわかる。
【0202】
本明細書に記載された方法に従って、限定されるものではないが、A型肝炎、B型肝炎、C型肝炎、インフルエンザ、水痘、アデノウイルス、HSV-I、HSV-II、牛痘、ライノウイルス、エコーウイルス、ロタウイルス、呼吸シンシチアルウイルス、パピローマウイルス、パポーバウイルス、サイトメガロウイルス、エキノウイルス、アルボウイルス、ハンタウイルス、コクサッキーウイルス、ムンプスウイルス、麻疹ウイルス、風疹ウイルス、ポリオウイルス、HSV-I、およびHSV-IIなどのウイルスに感染した細胞に対する免疫応答、特に細胞傷害性T細胞応答を刺激するワクチンを調製することができる。同様にまた、限定されるものではないが、マイコバクテリウム、リケッチア、マイコプラズマ、ナイセリア、およびレジオネラなどの細胞内細菌に感染した細胞に対する細胞傷害性T細胞応答を刺激するワクチンを調製することもできる。さらに、限定されるものではないが、リーシュマニア、kokzidioa、トリパノソーマなどの細胞内原生動物に感染した細胞に対する細胞傷害性T細胞応答を刺激するワクチンを調製することもできる。さらに、限定されるものではないが、クラミジアおよびリケッチアなどの細胞内寄生体に感染した細胞に対する細胞傷害性T細胞応答を刺激するワクチンを調製することができる。
【0203】
当業者には理解されるように、本明細書に記載されたプロトコールを用いてhsp-ペプチド複合体を、改変型hspを発現するよう遺伝子操作された任意の細胞、例えば細胞内病原体に感染した組織、単離された細胞または不死化真核細胞系から単離することができる。不死化動物細胞系を改変型hsp-ペプチド複合体の供給源として用いる場合、目的の病原体に感染し得る細胞系を用いることが重要である。さらに、ワクチン受容予定者と同一種から誘導した細胞を用いることが好ましい。改変型hsp遺伝子配列の発現可能形態をこれらの細胞系に導入する手法は、前記の第5.2.2節に記載されている。
【0204】
病原体が宿主細胞の溶解を引き起こすと予期される場合、細胞を病原体に感染させる前に、発現可能な改変型hsp遺伝子配列を宿主細胞に導入することが好ましい。例えば、HSV-Iに対して有効であると考えられる、ヒトへの投与用のhsp-ペプチド複合体を調製するためには、限定されるものではないが、発現可能な改変型hsp遺伝子配列ですでにトランスフェクトされたヒトCD4+T細胞、HepG2細胞、およびU937前単球細胞などのヒト細胞中でウイルスを増殖させることができる。同様に、例えばトランスフェクトされたヒト繊維芽細胞系およびMDCK細胞でインフルエンザウイルスを増殖させてもよく、マイコバクテリウムを、例えばトランスフェクトされたシュワン細胞中で培養してもよい。
【0205】
宿主細胞を溶解する直前に、分泌された改変型hsp-ペプチド複合体を含む細胞上清を採集してもよい。
【0206】
別の実施形態では、病原体の特定の抗原決定基をコードする遺伝子が同定された場合には、抗原をコードする遺伝子をトランスフェクトし、当技術分野で公知の手法を用いて改変型hsp遺伝子配列とともに、ヒトまたは哺乳類細胞系で同時発現させてもよい。改変型hsp-ペプチド複合体を分泌するかかる組換え抗原性細胞を、患者へ注入するためのワクチンとして用いて、感染細胞または抗原を担持する細胞に対する免疫応答を刺激することができる。改変型hsp-ペプチド複合体を安定して発現し、分泌する自己組換え抗原性細胞が好ましい。
【0207】
感染症の進行における改変型hsp-ペプチド複合体を用いた免疫治療の効果を、当業者に公知の任意の方法によってモニターすることができる。
【実施例】
【0208】
6. 実施例:哺乳動物細胞における改変型gp96-Igの産生
改変型hsp、gp96-Igについては、残留ペプチド(KDEL)のないgp96およびマウスIgGlの定常領域を含んでなるキメラタンパク質をコードするヌクレオチド配列を構築して、発現構築物の改変型gp96遺伝子配列をクローニングし、発現遺伝子構築物を種々の哺乳動物細胞へトランスフェクトして調製した。改変型hsp遺伝子配列を構築する方法、組換え細胞を作製する方法、改変型gp96を精製する方法については次の節で記載する。
【0209】
6.1 改変型gp96-Ig遺伝子配列の構築
ヒトgp96のコード領域は2,412塩基の長さであり、これはアミノ末端のシグナルペプチド(21アミノ酸残基)、疎水性残基が多く含まれる潜在的な膜貫通領域、およびカルボキシル末端の小胞体(ER)残留ペプチド配列(KDEL)をコードする。このタンパク質は804アミノ酸、推定分子量約96KDを有している。このコード領域は、マウスIgGlのヒンジ、CH2、およびCH3ドメインをコードする配列が含まれるが、KDELテトラペプチドをコードする配列なしに増幅した。免疫グロブリンを基にしたタグによってELISAによる検出、プロテインA-セファロースカラムでのアフィニティークロマトグラフィーによる精製、および蛍光活性化細胞選別解析による解析が容易になる。改変型gp96の分泌形態は、改変型hsp遺伝子配列でトランスフェクトした細胞によって産生される。改変型gp96は結合したペプチドを含んでいる。
【0210】
改変型gp96遺伝子配列を構築するために、全RNAをJurkat細胞(ヒト細胞系)から酸性イソチオシアン酸グアニジニウム-フェノール-クロロホルムで抽出した。二本鎖cDNAをGeneAmp RNA PCR キット(Perkin Elmer Cetus, Norwark, CT)を用いてRNAから調製し、ヒトgp96のコード領域をExpand TM Long Template PCR System (Boehringer Mannheim, Indianapolis, IN)のPwoおよびTaqポリメラーゼを用い、PCRによって増幅した。PCRサイクル条件は:鋳型DNAの変性、94℃で2分間;次いで94℃で10秒間、55℃で30秒間および68℃で2分間を10サイクル;さらに94℃で10秒間、55℃で30秒間および68℃で2分間を25サイクルと、各サイクルに20秒間の伸長サイクル、および最終伸長工程、68℃で7分間であった。用いたPCRプライマーはXhoI部位を含む5’-ATTACTCGAGGGCCGCACGCCATGAGGG-3’(順方向プライマー-1)および2)BamHI部位を含む5’-GCCCGGATCCTTCAGCTGTAGATTCCTTTGC-3’(逆方向プライマー-2)であった。2.4kb断片である増幅産物をアガロースゲル電気泳動により精製して、TAクローニング(Invitrogen, San Diego, CA)によってpCRIIベクターへクローン化し、次いでBluescript II SK(+/-)ファージミドへ再クローン化した。
【0211】
マウスIgGlのヒンジ、CH2、およびCH3ドメインをコードする配列(Igタグ)を、鋳型としてマウスIgGl cDNA、またはプラスミド、マウスIgGl-pCRIIを用い、PCRによって得た。IgGlのヒンジ部分にある3つのシステイン残基を標準手法によってセリン残基へと変異させた。PCRサイクル条件は:変性、95℃で2分間;次いで95℃で1分間、50℃で1分間および72℃で2分間を30サイクル、および最終伸長工程、72℃で10分間であった。PCRプライマーはBamHI部位を含む5’-GCGAGGATCCGTGCCCAGGGATTCTGGTTCTAAG-3’(順方向プライマー-3)およびNotI部位を含む5’-CTAAGCGGCCGCAAGGACACTGGGATCATTTACCAGG-3’(逆方向プライマー-4)であった。PCR産物は0.68kb断片であり、これをアガロースゲル電気泳動により精製して、TAクローニングによってpCRIIへクローン化し、Bluescript II SK(+/-)ファージミドへ挿入した。
【0212】
クローン化した改変型gp96をクローン化したIgタグと連結するために、gp96をコードする配列をpBluescriptのXhoIおよびBamHI部位へ挿入し、一方IgタグをpBluescriptのBamHIおよびNotI部位へ挿入した。融合タンパク質gp96-Igの発現をin vitro共役転写/翻訳 (Promega, Madison) によって確認した。次いで改変型gp96-Ig融合体をコードする配列をXhoIおよびNotI部位で切り離し、真核生物発現ベクター、pBCMGSNeoおよびpBCMGHisへ挿入した。これはCMVプロモーター下でgp96-Igを発現し、pBCMGHisはメタロチオネインプロモーター下でgp96-Igを発現するものである。改変型gp96-Ig配列のサイズは3.08kbであり、分泌されるキメラgp96の分子量は約121kDであると推定された。
【0213】
6.2 哺乳動物細胞における改変型gp96-Igの産生
マウス繊維芽細胞系NIH3T3、マウスルイス肺癌腫細胞系LLC、マウス肥満細胞腫細胞系P815、マウスリンパ腫細胞系EL4およびその卵白アルブミントランスフェクト化細胞E.G7、マウス黒色腫細胞系B16F10、マウス繊維肉腫細胞系MC57、およびヒト小細胞肺癌腫細胞系SCLC#2およびSCLC#7を改変型gp96-Ig遺伝子配列の発現に使用した。標準的な手法を用いてこれらの細胞へ発現遺伝子構築物を導入した。エピソームで維持される、ウシパピローマウイルス(BPV)に基づく、改変型配列(gp96-Ig-pBCMGSNeoおよびgp96-Ig-pBCMGHis)を含む発現ベクターpBCMGSNeoまたはpBCMGHisを使用して、リポフェクチン(GIBCO BRL, Gaithersburg, MD)によってLLC、B16F10、MC57、SCLC#2およびSCLC#7細胞を、リン酸カルシウムによってNIH3T3細胞を、エレクトロポレーションによってEL4、E.G7およびP815細胞をトランスフェクトした。NIH3T3を10%熱失活仔ウシ血清(GIBCO BRL)を補給したIMDM培地(GIBCO BRL, Grand Island, NY)で培養した。E.G7およびEL4を10%熱失活FCS(GIBCO BRL)および50μM β-メルカプトエタノール(Bio-Rad, CA)を補給したIMDM培地で培養した。他のすべての細胞系については10%熱失活FCSを補給したIMDM培地で培養した。NIH3T3、MC57、SCLC#2およびSCLC#7を単層培養物として維持し、0.05%トリプシンと、要すればEDTA(GIBCO BRL)で短時間トリプシン処理して継代した。トランスフェクトされた細胞を1mg/mlのG418(GIBCO)または2.5mMのL-ヒスチジノール(Sigma, St. Louis, MO)で2週間選抜し、希釈およびさらなる培養によって数回に分けて増殖させた。分泌されるgp96-Ig融合タンパク質を大量産生するクローンを制限希釈によって作製した。
【0214】
非トランスフェクト化細胞系ではマウスIgGが培養上清へ分泌されなかったが、改変型gp96-Ig遺伝子配列でトランスフェクトした細胞系では改変型gp96が分泌された。
【0215】
細胞を、10%FCSを補給したAIMVまたはIMDM培地にプレーティングして106/mlとし、異なる時点で培養上清を採取した。gp96-Igの細胞内発現を解析するために、細胞を同様にプレーティングし、採取し、PBSで洗浄し、3回の凍結解凍サイクルで溶解した。溶解液を600gで10分間遠心分離し、上清をさらに13,000gで60分間遠心分離した。最終上清を用いてgp96-Igの細胞内発現を定量した。
【0216】
細胞培養基中のgp96-Ig濃度を抗原としてIgタグを用いるELISAによって求めた。標識した抗マウス抗体によってIgタグを検出した。ELISAについては、平底96ウェルプレート(Becton Dickinson Labware, Oxnard, CA)をヤギ抗マウスIgG(5μg/ml)で4℃で一晩被覆し、PBS中1%ゼラチンで37℃で1時間ブロックした。ウェルを培養上清、または対照としてマウスIgG(ICN, Costa Mesa, CA)とともに37℃で1時間インキュベートし、ペルオキシダーゼ複合アフィニピュアF(ab’)2フラグメントヤギ抗マウスIgG(H+L)で37℃で1時間展開し、次いで2,2’-アジノ-ビス(3-エチルベンズチアゾリン-6-スルホン酸)(Sigma)とともにインキュベートした。吸光度をImmunoreader SLT-Labinstruments EAR 400AT(Austria)を用い、波長405nmで求めた。その吸光度をマウスIgG標準のものと比較してgp96-Ig濃度を求めた。
【0217】
【表1】

パピローマウイルスに基づくエピソーム発現ベクターのgp96-Igでトランスフェクトしたすべてのマウスおよびヒト細胞系では融合タンパク質が分泌された(図1aおよび表1)。擬似トランスフェクト化細胞ではgp96-Igは分泌されなかった。標準化条件下では、分泌される融合タンパク質のレベルはトランスフェクト化細胞によって様々であり、5ng/ml〜3300ng/mlであった。
【0218】
gp96-Ig分泌は時間に依存して、上清中に直線的に蓄積した(図2)。トランスフェクト化細胞の溶解液においては、低く、一定の定常状態レベルで細胞内gp96-Igが検出されたが、このことはそれが細胞内には蓄積しないことを示すものである。
【0219】
6.3 哺乳動物細胞によるgp96-Igの分泌
gp96-IgトランスフェクトSCLC細胞の膜を染色するために、ヤギ抗マウスIgG-FITCまたは対照としてヤギ抗ウサギIgG-FITCを用いて、4℃で15分間染色し、Becton Dickinson FACScanフローサイトメーターにより解析した。細胞内染色のために、4%パラホルムアルデヒドで細胞を固定し、1%サポニンにより浸透性を付与し、次いでヤギ抗マウスIgG-FITC(Boehringer Mannheim, Indianapolis, IN)、ヤギ抗マウスIgG-PE(Southern Biotechnology, Birmingham, AL)、ヤギ抗ウサギIgG-FITCまたはヤギ抗ゴールデンハムスターIgG-FITC(Jackson ImmunoResearch, West Grove, PA)で4℃で15分間染色し、フローサイトメーターにより解析した。
【0220】
膜の無傷なトランスフェクト化腫瘍細胞のFACS解析では、上記背景では抗マウスIgGによっては染色されないことが明らかになり、このことは融合タンパク質のIg部分が原形質膜の外片上には展示されないことを示している。これに対し、膜に透過性を付与した際は、gp96-Igは対照ヤギ抗ウサギIgG抗体ではなく、ヤギ抗マウスIgG抗体により細胞内で検出される。gp96の膜貫通ドメインによりgp96-Igの分泌は妨害されることなく、細胞内蓄積はなされない。これらのデータはこれまでの報告に一致し、このことは膜貫通ドメインは膜におけるgp96の固定には用いられず、gp96が内在性膜タンパク質ではないことを示唆するものである。
【0221】
6.4 改変型gp96-Igの精製
gp96-IgはプロテインAカラム(Bio-Rad, Hercules, CA)でのアフィニティークロマトグラフィーにより精製した。Ig融合タンパク質を精製するため、供給源としてgp96-Ig-トランスフェクト化SCLC#7(SCLC#7-gp96-Ig)の使用済み無血清培地を用いた。gp96-Ig-トランスフェクト化NIH-3T3細胞をAIMV培地にプレーティングして106/mlとし、6〜8日後に培養上清を採取した。遠心分離および濾過によって細胞屑を取り除いた後、上清の全タンパク質を硫酸アンモニウム沈殿(55%飽和)により濃縮し、PBSで透析した。サンプルを3.5M NaCl、1.6M グリシン、pH9.0(結合バッファー)で1:2に希釈し、プロテインAカラムに充填した。このカラムを結合バッファーで十分洗浄し、結合したタンパク質を0.1Mクエン酸、pH6.5で溶出した。タンパク質を含む画分をプールし、PBSで透析した。Micro BCAタンパク質アッセイ試薬キット(Pierce, Rockford, IL)によってgp96-Ig濃度を求めた。
【0222】
4〜15% Tris-グリシンゲル(Bio-Rad)上でドデシル硫酸ナトリウムポリアクリルアミドゲル電気泳動(SDS-PAGE)を行い、クーマシーブルーで染色した。ウエスタンブロット法では、SDS-PAGE上のタンパク質をニトロセルロースにブロットし、Grp94に特異的なラットモノクロナール抗体(9G10; StressGen, Victoria, Canada)、次いでペルオキシダーゼ複合アフィニピュアF(ab’)2フラグメントヤギ抗ラットIgG(H+L)(Jackson ImmunoResearch, West Grove, PA)でプローブした。
【0223】
プロテインAクロマトグラフィーにより精製したgp96-IgをSDS-PAGEにより解析した場合、推定分子量120kDをもつ主要なバンド、およびより高分子の2つの小バンドとして移動する物質についてはこれまでに改変されていないgp96と報告されたものである(図1b)。gp96に特異的なモノクロナール抗体についてのウエスタンブロッティングにより、融合タンパク質の正体が確認された。主要なバンドのみが染色されることから、小バンドが抗体により認識されないgp96のグリコシル化変異体であることが示唆される。
【0224】
また分泌される改変型gp96-Ig分子を、還元および非還元条件下でドデシル硫酸ナトリウム−ポリアクリルアミドゲル電気泳動(SDS-PAGE)によって解析した。図1Cでは、SCLC#2より分泌された精製gp96-Ig分子、マウスIgG、ならびにCD30とIgペプチドタグとの融合タンパク質の比較を示している。gp96(96kD)およびIgタグ(25kD)を含んでなるgp96-Ig融合タンパク質は適当なサイズの範囲にあると思われる。還元および非還元画分の双方において改変型gp96-Igに相当するトリプレットはこれまでにgp96に関して記載されている。
【0225】
7. 実施例:改変型gp96-Igを産生する腫瘍細胞の発癌性
本発明によれば、改変型hsp-ペプチド複合体を分泌する組換え腫瘍細胞はワクチンとして有用である。いずれの既存の腫瘍も持たない動物における発癌性を評価することで、かかる組換え細胞のin vivo免疫原性を試験した。
【0226】
組換え腫瘍細胞をマウスに注入し、かかる組換え細胞の発癌性が同一タイプの改変されていない(すなわち、発現遺伝子構築物のない)腫瘍細胞と比較して低下するかどうかを求めた。次の結果では、試験動物における腫瘍形成においてこれらの組換え腫瘍細胞がもとの腫瘍細胞よりも活性が低いことを示している。このことから、これらの動物の免疫系によって、もとの腫瘍細胞よりも免疫原性および/または抗原性の高い細胞に対して効力のある免疫応答が高められたことが示唆される。最初に組換え腫瘍細胞を注入した試験動物に存続している腫瘍細胞を単離し、培養に戻した。これらの腫瘍細胞によって改変型hsp-Ig分子が分泌されなくなったという観察から、これらの腫瘍細胞が選択された変異体であり、実際上免疫エフェクター機構によって標的化されなかったことが示唆される。
【0227】
7.1 E.G7およびE.G7-gp96-Igの相対的発癌性の比較
C57BL/6マウスの2群にそれぞれ、示された数のE.G7およびE.G7-gp96-Ig腫瘍生細胞を皮下注射した。結果は表2に示されている。
【0228】
【表2】
E.G7を注入したほとんどすべてのマウスが腫瘍を形成した。しかしながら、マウスのほとんどがE.G7-gp96-Ig腫瘍に拒絶反応を示した。このことより、E.G7-gp96-Igから分泌される改変型gp96はE.G7の腫瘍ペプチドを保有しており、E.G7に対する腫瘍免疫性が誘導され得ることが示唆される。E.G7-gp96-Ig腫瘍を有する2匹のマウスから腫瘍を摘出し、培地へ戻した。これらの腫瘍細胞では培地へ改変型gp96-Igが分泌されなかった。改変型gp96-Igを分泌する能力を失ったこれらの腫瘍変異体がin vivoで選択されたと考えられる。
【0229】
7.2 EL4およびEL4-gp96-Igの相対的発癌性の比較
C57BL/6マウスの2群にそれぞれ、示された数のEL4およびEL4-gp96-Ig腫瘍生細胞を皮下注射した。結果は表3に示されている。
【0230】
【表3】
EL4を注入したほとんどすべてのマウスが腫瘍を形成した。しかしながら、注入したマウスのほぼ50%がEL4-gp96-Ig腫瘍に拒絶反応を示した。この観察より、EL4-gp96-Igから分泌されるgp96はEL4の腫瘍ペプチドを保有しており、EL4細胞に対する腫瘍免疫性が誘導され得ることが示唆される。EL4-gp96-Ig腫瘍を有する3匹のマウスから腫瘍を摘出し、培地へ戻した。同様に、これらの腫瘍変異体によって改変型gp96-Igが分泌されなくなったことから、それらがin vivoでの免疫エフェクター機構を逃れたことが示唆される。
【0231】
7.3 MC57およびMC57-gp96-Igの相対的発癌性の比較
C57BL/6マウスに示された数の肝臓癌細胞を皮下注射した。MC57を注入したすべてのマウスは腫瘍を持たず生残した。しかしながら、MC57-gp96-Ig細胞を注入した1匹のマウスが腫瘍を形成した。この腫瘍を摘出し、培地へ戻した。摘出した腫瘍の培養上清における改変型gp96-Ig分子のレベルは、100μg/mlまで低下した。
【0232】
【表4】
7.4 B16F10およびB16F10-gp96-Igの相対的発癌性の比較
C57BL/6マウスの2群にそれぞれ、示された数のB16F10およびB16F10-gp96-Ig腫瘍生細胞を皮下注射した。B16F10-gp96-Ig細胞を注入したすべてのマウスは腫瘍に拒絶反応を示さなかった。B16F10細胞1x104個を注入した2匹のマウスは腫瘍に拒絶反応を示した。
【0233】
【表5】
7.5 E.G7-gp96-IgおよびLLC-gp96-Igの発癌性
さらなるin vivo研究のため、2種の細胞系を選択した。E.G7はEL4リンパ腫の卵白アルブミントランスフェクト化細胞である。E.G7はその関連する免疫原性にかかわらず、C57BL/6マウスに致死的な腫瘍を形成する。E.G7のgp96-Igトランスフェクションにより、E.G7-gp96-Ig免疫がEL4結合抗原および卵白アルブミン代用抗原のいずれか一方か、またはその双方に対して免疫するかどうかの決定がなされる。E.G7に対し、それは非造血性の低免疫原性腫瘍であるため、第2の腫瘍、gp96-Igまたは卵白アルブミンをコードする配列でトランスフェクトしたLLCを用いた。両細胞系は匹敵する量のgp96-Igを分泌する(表1参照)。
【0234】
PBS 200μl中の腫瘍生細胞をマウスの脇腹へ皮下注射してin vivoでの発癌性を求めた。少なくとも2ヶ月間、週に2回、腫瘍のサイズを2次元測定した。3週間後、平均腫瘍増殖が直径10mmを超えた場合、マウスは腫瘍陽性であるとして、犠牲にした。右脇腹に106個の生きたE.G7-gp96-Ig、または対照として照射したE.G7(PBS 200μl中)を皮下注射してマウスを免疫化した。2週間間隔で2回の免疫化を行った。
【0235】
C57BL/6マウスのE.G7の発癌性は、gp96-Ig分泌により擬似トランスフェクト化または非トランスフェクト化E.G7に比べ、100倍以上低下した。腫瘍細胞を分泌する107の熱ショックタンパク質の皮下接種によっては、接種したマウスの10%にしか腫瘍が生じなかった(図3)。EL4についても同様の発癌性の低下がgp96-Ig分泌により見られた。LLCによるgp96-Ig分泌はより穏やかな低下をもたらし、発癌性が約5倍低下した(図3)。これらの結果から、腫瘍の免疫原性を可能な限り高めることで、分泌されるgp96-Igによりそれらの発癌性が低下したことが示唆される。
【0236】
8. 実施例:改変型gp96-Igを発現する細胞のワクチン接種による防御効果
改変型gp96-Igを産生する組換え腫瘍細胞が防御免疫応答を刺激する能力を示すために、まずマウスに組換え腫瘍細胞をワクチン接種し、次いで改変型gp96-Ig遺伝子構築物を含まない腫瘍細胞の注入によってチャレンジした。
【0237】
右脇腹に1x106個の生きたE.G7-gp96-Igを皮下注射してマウス群を免疫化した。2週間間隔で2回の免疫化を行った。次いで最終免疫後2週間に左脇腹に示された数のE.G7またはEL4生細胞を皮下注射してマウスにチャレンジした。E.G7細胞は卵白アルブミン遺伝子構築物でトランスフェクトしたEL4細胞である。
【0238】
【表6】
表6の結果は、組換えE.G7-gp96-Ig細胞をワクチン接種した少数のマウスにE.G7およびEL4腫瘍が発生したことが示されている。よってかかる組換え腫瘍細胞を使用することで、予防措置として特異的抗原性関連腫瘍の発生を阻止または低減させ得ることを示している。
【0239】
8.1 E.G7-gp96-IgのE.G7およびLLC腫瘍細胞に対する防御効果
さらなるin vivo研究のため、細胞系E.G7およびLLCを選択した。前記のように、右脇腹にgp96-Igを分泌する非照射E.G7細胞を皮下注射してマウスを免疫化した。2週間間隔で2回の免疫化を行った。続いてそれらを、非トランスフェクト化または擬似トランスフェクト化E.G7で、EL4で、非トランスフェクト化LLCで、またLLC-ovaでチャレンジした。LLC-ovaを作製するために、発現ベクターpAc-NEO-OVAへクローン化したニワトリ卵白アルブミン遺伝子を使用し、リポフェクチン(Gibco BRL)によってLLCをトランスフェクトした。トランスフェクト化細胞を1mg/mlのG418(GIBCO BRL)で少なくとも2週間選抜し、それらの分泌レベルをELISAにより試験した。照射E.G7で免疫化したマウスおよびワクチン接種しないマウスをワクチン接種の対照とした。
【0240】
結果は図4に示されている。非免疫化マウスまたは照射細胞をワクチン接種したマウスより10倍高いE.G7のチャレンジ量に対し、E.G7-gp96-Ig免疫化マウスは防御された。EL4でチャレンジした場合には免疫化の効力がいっそうさらに示され、対照に比べ、チャレンジ量の50倍の増加が許容された。予期されたように、E.G7-gp96-Ig免疫化では非トランスフェクト化細胞またはベクターでトランスフェクトされたLLCによるチャレンジに対する防御は起こらないが、卵白アルブミントランスフェクト化LLCを使用する場合には適度の、約3倍増大した防御が観察された。EL4チャレンジに対するE.G7-gp96-Ig免疫化マウスの強い防御はE.G7およびEL4が共有する多数の腫瘍抗原によるものであろう。これに対し、LLC-ovaで得られた弱い防御は、卵白アルブミン由来の、H2b分子によって示される単一の、または限られた数のエピトープを認識するT細胞によるものである。
【0241】
9. 実施例:接種動物におけるコンピテント免疫細胞のin vivo枯渇および失活
E.G7-gp96-Igの拒絶反応における免疫機構の問題をコンピテント免疫細胞のin vivo枯渇および失活によってさらに試験した。
【0242】
CD4+およびCD8+細胞サブセットのin vivo枯渇に使用されるモノクロナール抗体はそれぞれ、GK1.5および2.43であった。それらをハイブリドーマ上清から精製し、次いでプロテインGアフィニティークロマトグラフィーに付した。生きた106個のE.G7-gp96-Ig(PBS 200μl中)を皮下注射する2日前、または3日後に、PBS 200μl中のGK1.5または2.43 100μgを腹腔内注射によって投与した。CD4およびCD8細胞の枯渇はFACS解析によって確認した。マクロファージの機能的阻害については、PBS 200μl中のカラゲーニン(II型; Sigma) 1mgを腹腔内注射によって投与した。
【0243】
gp96-Igを分泌する106個の腫瘍細胞、平均直径約8mmに増殖する腫瘍を定着させるに十分な用量を動物に接種した。後にこれは収縮して拒絶反応を起こす。抗CD8抗体2.43で処理したマウスでは腫瘍接種の2日前か、または3日後のいずれかに腫瘍拒絶反応が阻害される(図5)。たとえ抗CD4抗体GK1.5が14日を超える期間CD4細胞を完全に枯渇させたとしても、抗CD4抗体GK1.5では注入時間にかかわらず、腫瘍拒絶反応には何の効果もなかった。同様に、in vivoでマクロファージを失活させることが知られているカラゲーニンも、腫瘍拒絶反応には何ら効果を示さなかった。
【0244】
これらのデータは、分泌されるgp96-Igと結合するペプチドがクラスI MHCによって提示され、腫瘍特異的CD8+ CTL応答を刺激して腫瘍拒絶反応を誘導するという解釈に一致している。この応答はCD4の助けを受けないと考えられ、マクロファージを必要としない。本発明者らのモデル系では、gp96-Igは腫瘍生細胞によって分泌され、マウスを免疫化して、後に腫瘍拒絶反応を引き起こす。抗腫瘍免疫が起こった兆候として腫瘍増殖が6〜8日目までに減速し始め、次の週には腫瘍に拒絶反応を起こした。腫瘍接種の2日前か、または3日後、それらの枯渇時間にかかわらず、腫瘍拒絶反応にCD4細胞およびマクロファージは必要ではなかった。このことから、CD4細胞およびマクロファージは免疫の誘導に必須ではなく、この腫瘍系のエフェクター相にも必要でないことがわかる。他方、CD8細胞は分泌性gp96-Igに対する応答についてのすべての相で決定的に重要である。
【0245】
本発明は本明細書に記載される特定の実施形態により範囲を限定されるものではない。実際、本明細書に記載されるものに加え、本発明の様々な変形や変更が、これまでの説明および添付の図面から当業者には明らかであろう。かかる変形や変更は特許請求の範囲内にある。
【0246】
本明細書では様々な公報が引用されるが、その開示内容は参照によりその総てを本明細書に組み入れられる。
【技術分野】
【0001】
本発明は、米国立保健研究所(National Institutes of Health)により与えられた認可番号CA57904に基づいて政府の支援を得て行われた。政府は、本発明に関して一定の権利を有する。
【0002】
1. 緒言
本発明は、癌または感染症の予防および/または治療のためのワクチンとして有用な免疫原性物質を調製するための方法に関する。ワクチンには、改変型熱ショックタンパク質(hsp)(例えば、限定されるものではないが、hsp70、hsp9O、gp96、およびタンパク質ジスルフィドイソメラーゼ)と抗原性ペプチドとの非共有結合複合体が含まれている。ワクチンにより、特定のタイプの癌細胞または感染細胞に対する被験者の免疫応答を誘発または増大することができる。
【背景技術】
【0003】
2. 発明の背景
2.1. 癌の病理生物学
癌は、主に、特定の正常組織に由来する異常細胞の数の増大によって特性付けられる。また、疾患の過程には、これらの異常細胞による隣接組織の浸潤および循環系を介する局所リンパ節や遠隔部位へのこれらの異常細胞の拡散(転移)が関与する。臨床データおよび分子生物学的研究から示唆されるように、癌は、特定の条件下で新形成に発展する可能性のある新生物発生前の小さな変化から開始される多段階過程である。
【0004】
前癌性異常細胞増殖は、過形成、化生、または特に異形成によって例証されている(このような異常増殖条件に関するレビューについては、Robbins and Angell, 1976, Basic Pathology, 2d Ed., W.B. Saunders Co., Philadelphia, pp. 68-79を参照されたい)。過形成とは、構造または機能の有意な変化を伴わずに組織または器官の細胞数が増加する制御された細胞増殖の形態である。1例にすぎないが、子宮内膜過形成は、子宮内膜癌に先行して起こる場合が多い。化生とは、1つのタイプの成体細胞すなわち十分に分化した細胞が別のタイプの成体細胞と置き換わる制御された細胞増殖の形態である。化生は、上皮細胞または結合組織細胞で起こる可能性がある。非定型化生には、いくらか疾患性のある化生上皮が関与する。異形成は、多くの場合、癌の前兆であり、主に、上皮で見出される。また、異形成は、非新生物性細胞増殖の最も疾患性の強い形態であり、個々の細胞の均一性の消失や細胞の構造的配向の消失を伴う。異形成細胞は、多くの場合、濃く染色される異常に大きな核を有し、多形性を呈する。異形成は、特質上、慢性刺激または炎症が存在する場合に起こり、多くの場合、子宮頸管、気道、口腔、および胆嚢で見出される。
【0005】
新生物性病変は、クローン産生的に進化して、特に、新生物細胞が宿主の免疫学的監視から逃げる条件下で、浸潤、増殖、転移、および異質性に対する能力を増大させながら進行する可能性がある(Roitt, I., Brostoff, J. and Male, D., 1993, Immunology, 3rd ed., Mosby, St. Louis, pps. 17.1-17.12)。
【0006】
2.2. ワクチン接種
ワクチン接種により、世界中の多くの国々で、ポリオ、破傷風、水痘、麻疹などの特定の疾患が根絶された。この手法では、感染症を予防するために、免疫系の能力が利用された。タンパク質などの非生存物質を用いてこのようなワクチン接種を行うと、一般的には、抗体応答またはCD4+ヘルパーT細胞応答が誘発される(Raychaudhuri & Morrow, 1993, Immunology Today, 14:344-348)。一方、生存細胞や感染性ウイルスなどの生存物質を用いてワクチン接種または感染処理を行うと、一般的には、CD8+細胞傷害性Tリンパ球(CTL)応答が誘発される。CTL応答は、癌、感染性ウイルス、および細菌に対する防御を行う上で極めて重要である。これに起因して実用上の問題が生じる。なぜなら、CTL応答を達成する唯一の方法は、それ自体病原性である生存性薬剤を使用するものであるからである。この問題は、一般に、弱毒化されたウイルス株および細菌株を使用することによって、またはワクチン接種に使用することのできる細胞全体を死滅させることによって回避される。これらのストラテジーはうまく機能したが、弱毒化された株を使用する場合、弱毒化薬剤が遺伝子的に宿主DNAと組み換わって毒性株に変化する恐れがあるというリスクを常に伴う。したがって、タンパク質などの非生存物質を用いた特異的なワクチン接種を行うことによってCD8+CTL応答を誘発することのできる方法が必要である。
【0007】
腫瘍免疫学の時代は、PrehnおよびMainによる実験から始まった。彼らは、メチルコラントレン(MCA)誘発肉腫上の抗原を移植アッセイによりマウスの正常組織中で検出できなかったことから、これらの抗原が腫瘍特異的であることを明らかにした(Prehn et al., 1957, J. Natl. Cancer Inst. 18:769-778)。この考えは、MCA誘発腫瘍の発生元であるマウスにおいてMCA誘発腫瘍に対する腫瘍特異的耐性を誘発することができることを実証する更なる実験によって確かめられた(Klein et al., 1960, Cancer Res. 20:1561-1572)。
【0008】
その後の研究により、他の化学的もしくは物理的発癌物質で誘発された腫瘍または自然発生腫瘍についても、腫瘍特異的抗原が見出された(Kripke, 1974, J. Natl. Cancer Inst. 53:1333-1336; Vaage, 1968, Cancer Res. 28:2477- 2483; Carswell et al., 1970, J. Natl. Cancer Inst. 44:1281- 1288)。これらの研究では移植された腫瘍の増殖に対する防御免疫が腫瘍特異的抗原の判定基準として使用されたので、これらの抗原は、通常、「腫瘍特異的移植抗原」または「腫瘍特異的拒絶抗原」とも呼ばれる。いくつかの因子が腫瘍の免疫原性に大きく影響する可能性があり、こうした因子としては、例えば、関係する発癌物質の特異的タイプ、宿主の免疫適格性、および潜伏期間が挙げられる(Old et al., 1962, Ann. N.Y. Acad. Sci. 101:80-106; Bartlett, 1972, J. Natl. Cancer Inst. 49:493-504)。
【0009】
すべてというわけではないが、ほとんどの発癌物質は、突然変異を誘発して腫瘍特異的抗原を発現させる突然変異誘発物質である(Ames, 1979, Science 204:587-593; Weisburger et al., 1981, Science 214:401-407)。発癌物質の中には免疫抑制性のものもある(Malmgren et al., 1952, Proc. Soc. Exp. Biol. Med. 79:484-488)。実験的証拠から示唆されるように、腫瘍の免疫原性と潜伏期間(発癌物質への暴露から腫瘍出現までの時間)との間には、一定の逆相関関係がある(Old et al., 1962, Ann. N.Y. Acad. Sci. 101:80-106; および Bartlett, 1972, J. Natl. Cancer Inst. 49:493-504)。他の研究から、拒絶反応は起こさないが、それにもかかわらず、場合により特異的免疫応答を刺激する可能性のある腫瘍特異的抗原の存在が示唆された(Roitt I., Brostoff, J. and Male, D., 1993, Immunology, 3rd ed., Mosby, St. Louis, pp. 17.1-17.12)。
【0010】
2.3. 熱ショックタンパク質
熱ショックタンパク質(hsp)は、ストレスタンパク質とも呼ばれ、これらは同義的に用いられる。同定の対象となった最初のストレスタンパク質は、熱ショックに応答して細胞によって合成されるタンパク質であった。これまでに、hspの3つの主要なファミリーが分子量に基づいて同定された。これらのファミリーは、hsp60、hsp70、およびhsp90と呼ばれてきたものであり、これらの数字は、ストレスタンパク質のおよその分子量をキロダルトン単位で表している。その後、これらのファミリーの多くのメンバーが、栄養欠乏、代謝阻害、酸素ラジカル、細胞内病原体による感染などの他のストレス刺激に応答して誘導されることが明らかにされた。(Welch, May 1993, Scientific American 56-64; Young, 1990, Annu. Rev. Immunol. 8:401-420; Craig, 1993, Science 260:1902-1903; Gethirig et al., 1992, Nature 355:33-45; および Lindquist et al., 1988, Annu. Rev. Genetics 22:631-677を参照されたい。)
【0011】
主要なhspは、ストレスを受けた細胞中では非常に高レベルまで蓄積する可能性があるが、ストレスを受けなかった細胞中では低レベル〜中レベルで生成する。例えば、高誘導性哺乳類hsp7Oは、常温ではほとんど検出できないが、熱ショックを受けると細胞中で最も活発に合成されるタンパク質の1つになる(Welch et al., 1985, J. Cell. Biol. 101:1198-1211)。これとは対照的に、hsp90およびhsp60タンパク質は、常温において、すべてではないがほとんどの哺乳類細胞中に多量に存在し、しかも熱によって更に誘導される(Lai et al., 1984, Mol. Cell. Biol. 4:2802-2810; van Bergen en Henegouwen et al., 1987, Genes Dev. 1:525-531)。
【0012】
熱ショックタンパク質は、現存するタンパク質の中で最も高度に保存されているタンパク質に属する。例えば、E. coli由来のhsp70であるDnaKは、擦過傷由来のhsp70タンパク質に対して約50%のアミノ酸配列同一性を有する(Bardwell et al., 1984, Proc. Natl. Acad. Sci. 81:848-852)。また、hsp60ファミリーとhsp90ファミリーとの間にも、同じように高レベルのファミリー間の保存が見られる(Hickey et al., 1989, Mol. Cell. Biol. 9:2615-2626; Jindal, 1989, Mol. Cell. Biol. 9:2279-2283)。更に、hsp60、hsp70、およびhsp90ファミリーには、配列は例えば35%を超えるアミノ酸同一性を有するが発現レベルはストレスによって変化しないストレスタンパク質に関連付けられるタンパク質が含まれることが見出された。
【0013】
熱ショックおよび他の生理的ストレスに対する細胞応答に関する研究から、hspは、これらの有害な条件からの細胞保護に関与するだけではなく、ストレスを受けていない細胞中における本質的な生化学的および免疫学的過程にも関与することが実証された。hsp類は、様々な種類のシャペロン機能を発揮する。例えば、細胞質、核、ミトコンドリア、または小胞体に位置するhsp7O (Lindquist, S. et al., 1988, Ann. Rev. Genetics 22:631-677)は、免疫系の細胞への抗原の提示に関与するほか、正常細胞中におけるタンパク質の移動、折りたたみ、および集合にも関与する。
【0014】
シャペロン機能に関与すると考えられるいくつかのタンパク質は、小胞体(ER)内腔に存在するタンパク質であり、具体的には、例えば、タンパク質ジスルフィドイソメラーゼ(PDI; Gething et al.., 1992, Nature 355:33-45)、hsp90に関連付けられるGrp94またはERp99(Sorger & Pelham, 1987, J. Mol. Biol. 194:(2) 341-4)、およびhsp70に関連付けられるGrp78またはBiP(Munro et al., 1986, Cell 46:291-300; Haas & Webl, 1983, Nature 306:387-389)が挙げられる。これらのタンパク質は、突然変異体である折りたたまれていないグリコシル化の不完全なタンパク質に結合することが知られている(Machamer et al., 1990, J. Biol. Chem. 65:6879-6883; Gething et al., 1986, Cell 46:939-950)。ER中へのこれらのhspの局在化は、ER中に残留するのに必要なカルボキシル末端テトラペプチド(lys-Asp-Glu-Leu すなわち KDEL)によって媒介される(Munro & Pelham, 1987, Cell, 48:899-907)。一般に、この配列は、哺乳類細胞中ではKDELであり、Saccharcmyces cerevisiae中ではHDELであり、Schizosaccharomyces pombe中ではADELである(Pidoux & Armstrong, 1992, EMBO J. 11:1583-1591)。
【0015】
一般に、熱ショックタンパク質は、タンパク質またはペプチドに結合することができ、更に、結合したタンパク質またはペプチドをアデノシン三リン酸(ATP)の存在下で放出することができる。ATP加水分解はタンパク質集合の過程でhspが関与するときに起こると考えられている(Flynn et al., 1989, Science, 245:385-390)。
【0016】
2.4. 熱ショック/ストレスタンパク質hsp7O、hsp9O、およびgp96の免疫原性
Srivastavaらは、近交系マウスのメチルコラントレン誘発肉腫に対する免疫応答を実証した(1988, Immunol. Today 9:78-83)。これらの研究において、これらの腫瘍のそれぞれ異なった免疫原性に関与する分子は、96kDaの細胞表面糖タンパク質(gp96)および84〜86kDaの細胞内タンパク質として同定されることが分かった(Srivastava, P.K. et al., 1986, Proc. Natl. Acad. Sci. USA 83:3407-3411; Ullrich, S.J. et al., 1986, Proc. Natl. Acad. Sci. USA 83:3121-3125)。特定の腫瘍から単離されたgp96またはp84/86でマウスを免疫したところ、マウスは、この特定の腫瘍に対しては免疫されたが、抗原性の異なる腫瘍に対しては免疫されなかった。gp96およびp84/86をコードする遺伝子の単離および特性付けを行った結果、それらの間に顕著な相同性が存在することが判明し、更に、gp96およびp84/86はそれぞれ同じ熱ショックタンパク質に属する小胞体タンパク質および細胞質ゾルタンパク質であることが分かった(Srivastava, P.K. et al., 1988, Immunogenetics 28:205-207; Srivastava, P.K. et al., 1991, Curr. Top. Microbiol. Immunol. 167:109-123)。更に、hsp70は、それが単離された腫瘍に対する免疫は誘発するが、抗原的に異なる腫瘍に対する免疫は誘発しないことが分かった。しかしながら、いくつかのペプチドが欠乏したhsp70では、その免疫原性が失われることが判明した(Udono, M., and Srivastava, P.K., 1993, J. Exp. Med. 178:1391-1396)。これらの観測から示唆されるように、熱ショックタンパク質は、そのままでは免疫原性をもたないが、抗原性ペプチドと非共有結合複合体を形成し、この複合体は、抗原性ペプチドに対する特異的免疫を誘発することができる(Srivastava, P.K., 1993 Adv. Cancer Res. 62:153-177; Udono, H. et al., 1994, J. Immunol., 152:5398-5403; Suto, R. et al., 1995, Science, 269:1585-1588)。
【0017】
ストレスタンパク質とペプチドとの非共有結合複合体を癌細胞から精製し、これを癌の治療および予防のために使用することが、1996年4月11日付けのPCT公開WO 96/10411および1997年3月20日付けのPCT公開WO 97/10001に記載されている(SrivastavaおよびChandawarkarにより1997年2月7日に出願された同時係属米国特許出願第08/796,319号およびSrivastavaにより1997年2月7日に出願された同時係属米国特許出願第08/796,316号も参照されたい。それぞれの出願内容はすべて、参照により本明細書に組み入れる。)。
【0018】
また、ストレスタンパク質-ペプチド複合体を病原体感染細胞から単離して、ウイルスのような病原体、ならびに細菌、原生動物、真菌、寄生虫などの他の細胞内病原体によって引き起こされる感染症の治療および予防に使用することもできる。1995年9月21日付けのPCT公開WO 95/24923を参照されたい。
【0019】
また、ストレスタンパク質と抗原性ペプチドとをin vitroで複合体化することによって免疫原性ストレスタンパク質-ペプチド複合体を調製することもできる。このような複合体を癌および感染症の治療および予防に使用することが、1997年3月20日付けのPCT公開WO 97/10000に記載されている。更に、癌および感染症を治療するために熱ショックタンパク質と所定の抗原とを併用することが、1997年2月27日付けのPCT公開W097/06821に記載されている。
【0020】
養子免疫療法用に抗原提示細胞をin vitroで感作するためにストレスタンパク質-ペプチド複合体を使用することが、1997年3月20日付けのPCT公開WO97/10002に記載されている。
【0021】
免疫応答を刺激するために、ならびに感染症および癌を治療するために、真核細胞性熱ショックタンパク質をコードする発現可能なポリヌクレオチドを哺乳類細胞に投与することが、いずれも1997年2月27日付けのPCT公開WO 97/06685およびWO 97/06828に記載されている。
【0022】
細胞溶解物からストレスタンパク質-ペプチド複合体を精製することが既に報告されている。例えば、1995年9月21日付けのPCT公開WO 95/24923を参照されたい。
【先行技術文献】
【特許文献】
【0023】
【特許文献1】国際公開第96/10411号パンフレット
【特許文献2】国際公開第97/10001号パンフレット
【特許文献3】米国特許出願第08/796,319号明細書
【特許文献4】米国特許出願第08/796,316号明細書
【特許文献5】国際公開第95/24923号パンフレット
【特許文献6】国際公開第97/10000号パンフレット
【特許文献7】国際公開第97/06821号パンフレット
【特許文献8】国際公開第97/10002号パンフレット
【特許文献9】国際公開第97/06685号パンフレット
【特許文献10】国際公開第97/06828号パンフレット
【非特許文献】
【0024】
【非特許文献1】Robbins and Angell, 1976, Basic Pathology, 2d Ed., W.B. Saunders Co., Philadelphia, pp. 68-79
【非特許文献2】Roitt, I., Brostoff, J. and Male, D., 1993, Immunology, 3rd ed., Mosby, St. Louis, pps. 17.1-17.12
【非特許文献3】Raychaudhuri & Morrow, 1993, Immunology Today, 14:344-348
【非特許文献4】Prehn et al., 1957, J. Natl. Cancer Inst. 18:769-778
【非特許文献5】Klein et al., 1960, Cancer Res. 20:1561-1572
【非特許文献6】Kripke, 1974, J. Natl. Cancer Inst. 53:1333-1336
【非特許文献7】Vaage, 1968, Cancer Res. 28:2477- 2483
【非特許文献8】Carswell et al., 1970, J. Natl. Cancer Inst. 44:1281- 1288
【非特許文献9】Old et al., 1962, Ann. N.Y. Acad. Sci. 101:80-106
【非特許文献10】Bartlett, 1972, J. Natl. Cancer Inst. 49:493-504
【非特許文献11】Ames, 1979, Science 204:587-593
【非特許文献12】Weisburger et al., 1981, Science 214:401-407
【非特許文献13】Malmgren et al., 1952, Proc. Soc. Exp. Biol. Med. 79:484-488
【非特許文献14】Welch, May 1993, Scientific American 56-64
【非特許文献15】Young, 1990, Annu. Rev. Immunol. 8:401-420
【非特許文献16】Craig, 1993, Science 260:1902-1903
【非特許文献17】Gethirig et al., 1992, Nature 355:33-45
【非特許文献18】Lindquist et al., 1988, Annu. Rev. Genetics 22:631-677
【非特許文献19】Welch et al., 1985, J. Cell. Biol. 101:1198-1211
【非特許文献20】Lai et al., 1984, Mol. Cell. Biol. 4:2802-2810
【非特許文献21】van Bergen en Henegouwen et al., 1987, Genes Dev. 1:525-531
【非特許文献22】Bardwell et al., 1984, Proc. Natl. Acad. Sci. 81:848-852
【非特許文献23】Hickey et al., 1989, Mol. Cell. Biol. 9:2615-2626
【非特許文献24】Jindal, 1989, Mol. Cell. Biol. 9:2279-2283
【非特許文献25】Sorger & Pelham, 1987, J. Mol. Biol. 194:(2) 341-4
【非特許文献26】Machamer et al., 1990, J. Biol. Chem. 65:6879-6883
【非特許文献27】Gething et al., 1986, Cell 46:939-950
【非特許文献28】Munro & Pelham, 1987, Cell, 48:899-907
【非特許文献29】Pidoux & Armstrong, 1992, EMBO J. 11:1583-1591
【非特許文献30】Flynn et al., 1989, Science, 245:385-390
【非特許文献31】Srivastavaら, 1988, Immunol. Today 9:78-83
【非特許文献32】Srivastava, P.K. et al., 1986, Proc. Natl. Acad. Sci. USA 83:3407-3411
【非特許文献33】Ullrich, S.J. et al., 1986, Proc. Natl. Acad. Sci. USA 83:3121-3125
【非特許文献34】Srivastava, P.K. et al., 1988, Immunogenetics 28:205-207
【非特許文献35】Srivastava, P.K. et al., 1991, Curr. Top. Microbiol. Immunol. 167:109-123
【非特許文献36】Udono, M., and Srivastava, P.K., 1993, J. Exp. Med. 178:1391-1396
【非特許文献37】Srivastava, P.K., 1993 Adv. Cancer Res. 62:153-177
【非特許文献38】Udono, H. et al., 1994, J. Immunol., 152:5398-5403
【非特許文献39】Suto, R. et al., 1995, Science, 269:1585-1588
【発明の概要】
【発明が解決しようとする課題】
【0025】
癌に対するワクチンを調製する目的では、入手および使用可能な免疫原性物質の量は、出発癌細胞の量と直接関係している。被験者から得られる癌細胞の数は、特に、癌が初期段階にある場合、非常に少ないため、hsp-ペプチド複合体の生産に供しうる癌細胞の量はかなり制限されることが多い。ワクチンまたは治療薬を商業生産する場合、大量のhsp-ペプチド複合体を定常的に供給することが有利である。従って、hsp-ペプチド複合体の信頼性のある供給源が必要である。本発明の方法を用いると、患者からごく少量の組織しか得られない場合でさえも、便利にかつ迅速に治療用hsp-ペプチド複合体を提供することができる。
【課題を解決するための手段】
【0026】
3. 発明の概要
本発明は、癌または感染症の予防および治療に使用するための免疫原性組成物の調製方法に関する。
【0027】
本発明の方法によって調製される免疫原性組成物には、改変型熱ショックタンパク質(hsp)と抗原性(または免疫原性)ペプチドとの非共有結合分子複合体が含まれる。本発明の改変型熱ショックタンパク質は、それが発現される細胞によって分泌され、未改変の熱ショックタンパク質中に存在する小胞体残留配列を欠失しており、更に、ペプチドタグを含有する。細胞質中に天然に存在するhspの場合、それが発現される細胞によって分泌され、ペプチドタグを含有し、更に、未改変の熱ショックタンパク質中に存在しないリーダーペプチドを含有するように、hspを改変する。この改変は、ペプチドと改変型熱ショックタンパク質との非共有結合型の結合、および抗原提示に関与する非共有結合複合体の能力を、妨害することも低減することもない。改変型hspが組換え細胞中で発現されると、それらが分泌される。更に、この改変型hspは、アフィニティークロマトグラフィーを用いてペプチドタグにより精製することができる。
【0028】
一実施形態において、本発明は、改変型hspをコードするヌクレオチド配列(「改変型hsp遺伝子配列」)を含む核酸、ならびにこのような核酸を含むクローニングベクター、発現ベクター、および宿主細胞を提供する。一般に、本発明の方法には、改変型熱ショックタンパク質をコードするヌクレオチド配列を構築することと、発現ベクター中に改変型hsp遺伝子配列をクローン化することと、宿主細胞中に発現遺伝子構築物を導入することと、改変型hspが発現されるように宿主細胞を培養することと、改変型hspおよび/または改変型hsp-ペプチド複合体を精製することと、が含まれる。
一例として、本発明の改変型熱ショックタンパク質を調製する方法は、
(a) 熱ショックタンパク質の遺伝子配列が、(i)それを発現する細胞から分泌され、(ii)改変されていない熱ショックタンパク質中には存在する小胞体残留配列を欠失しており、かつ(iii)ペプチドタグを含む改変型熱ショックタンパク質をコードするように熱ショックタンパク質の遺伝子配列を改変すること、
(b) 該改変型熱ショックタンパク質の遺伝子配列が宿主細胞内で該改変型熱ショックタンパク質の発現を制御する少なくとも1つの調節領域に機能的に結合されるような発現構築物中に該改変型熱ショックタンパク質の遺伝子配列を連結すること、
(c) 宿主細胞に該改変型熱ショックタンパク質の遺伝子配列を導入すること、
(d) 該宿主細胞を培養して、該改変型熱ショックタンパク質の遺伝子配列によりコードされる該改変型熱ショックタンパク質を該宿主細胞において発現させること、
(e) 該改変型熱ショックタンパク質を細胞培養培地から回収すること、
を含んでなる。
【0029】
熱ショックタンパク質をコードするcDNAまたはゲノムDNAの入手および改変は、従来のDNAクローニングおよび突然変異誘発法により、DNA増幅法により、または合成法により行うことができる。一般に、hspをコードする配列は、発現前に、遺伝子の改変および複製を目的としてクローニングベクター中に挿入される。改変型hsp遺伝子配列は、好適な宿主細胞中においてin vitroでおよびin vivoでコード化改変型hspを発現するために、プロモーターなどの調節エレメント(1種または複数種)に機能的に連結された状態で、発現ベクターに挿入されるかまたは染色体内に組み込まれる。改変型hsp遺伝子配列を宿主細胞中に導入すると、宿主細胞によって改変型hsp遺伝子配列が発現される。これにより、改変型hspとペプチド(癌細胞または病原体によって特異的にコードされているペプチドを含む)との非共有結合複合体が、細胞内で産生される。
【0030】
従って、本発明は、抗原性細胞中における改変型hspと抗原性ペプチドとの免疫原性非共有結合複合体の産生および精製を行うための方法を提供する。この方法には、抗原性細胞中に改変型hsp遺伝子配列を導入することと、改変型hsp遺伝子配列を発現させるために組換え抗原性細胞を培養することと、組換え抗原性細胞から分泌された改変型hsp-抗原ペプチド複合体を細胞培養物の上清から回収および精製することとが含まれる。該複合体の抗原性ペプチドは、癌細胞や病原体感染細胞のような抗原性細胞中に見出される抗原性ペプチドを代表するものである。このような組換え抗原性細胞は、治療および予防に用いるためのワクチンとして有用である。組換え宿主細胞は、免疫原性複合体を生産するために、バッチでまたは連続的に、大量に培養することが可能である。改変型hsp配列を含む宿主細胞は、後で使用するために保存することができる(例えば凍結乾燥または冷凍により)。
【0031】
もう1つの実施形態において、本発明は、細胞中における改変型hspの産生および精製を行うための方法を提供する。この方法には、細胞中に改変型hsp遺伝子配列を導入することと、改変型hsp遺伝子配列を発現させるために組換え細胞を培養することと、組換え細胞から分泌された改変型hspを細胞培養物の上清から回収および精製することとが含まれる。好ましくは、改変型hspの精製は、ペプチドタグおよびアフィニティークロマトグラフィーを利用して行われる。本発明には更に、精製された改変型hspにin vitroで抗原性ペプチドを添加して治療および予防に用いるための免疫原性非共有結合複合体を形成できるようにすることが含まれる。
【0032】
更にもう1つの実施形態において、本発明は、免疫原性非共有結合複合体を産生および精製するための方法を提供する。この方法には、組換え細胞中で改変型hsp遺伝子配列と抗原性ペプチドをコードするヌクレオチド配列とを同時発現させることと、組換え細胞から分泌された改変型hsp-抗原性ペプチド複合体を細胞培養物の上清から回収および精製することとが含まれる。また、これらの組換え細胞は、治療および予防に用いるためのワクチンとして使用することができる。
【0033】
種々の実施形態において、改変型hspまたは改変型hsp-抗原性ペプチド複合体は、アフィニティークロマトグラフィーにより精製し、癌または感染症を予防および治療するためのワクチンとして使用することができる。
【0034】
改変型hsp-ペプチド複合体を含む免疫原性組成物、および改変型hsp-ペプチド複合体を分泌する組換え細胞は、本発明の方法に従って調製した場合、癌細胞または病原体に対する患者の免疫応答を誘発することができ、この免疫応答は、治療または予防に有効である。好ましくは、この患者は、改変型hspを発現するための癌細胞を提供した被験者である。このほか、癌細胞または感染細胞は、この患者以外の1人以上の被験者に由来するものであってもよいが、ただし、これらの被験者は、同じ組織タイプの癌(例えば、胃癌、乳癌、大腸癌、肺癌など)または同じタイプの病原体によって引き起こされた感染症を患っている者でなければならない。
【0035】
従って、本発明は、個体において抗原に対する免疫応答を誘発する方法を提供する。この方法には、抗原またはその断片に非共有結合で結合した改変型熱ショックタンパク質の免疫原性複合体および/またはこのような免疫原性複合体を分泌する組換え細胞を、個体に投与することが含まれる。本発明はまた、癌を患っているかまたは癌の予防が望まれる個体を対象に癌の治療または予防を行う方法を提供する。この方法には、癌に由来する抗原またはその断片に非共有結合で結合した改変型熱ショックタンパク質の免疫原性複合体および/またはこのような免疫原性複合体を分泌する組換え細胞を、個体に投与することが含まれる。更に、本発明は、感染症を患っているかまたは感染症の予防が望まれる個体を対象に感染症の治療または予防を行う方法を提供する。この方法には、感染細胞または病原体に由来する抗原またはその断片に非共有結合で結合した改変型熱ショックタンパク質の免疫原性複合体および/またはこのような免疫原性複合体を分泌する組換え細胞を、個体に投与することが含まれる。
【0036】
以下の節および項において、本発明の特定の組成物およびそれらの調製方法について説明する。
【図面の簡単な説明】
【0037】
4.図面の簡単な説明
【図1−1】gp96-Igの分泌および特性付け。図1a:gp96-Ig cDNAでトランスフェクトされたおよびトランスフェクトされていない小細胞肺癌系#7(SCLC)由来の上清のマウスIgGに対するELISA; 106/mlで細胞をプレートし、3日目および6日目に上清の試験を行った;標準として精製マウスIgG(500ng/ml)を使用した。図lb:プロテインAで精製されたgp96-IgのSDS PAGE。レーン1:クーマシーブルー染色(1μgタンパク質)、レーン2:モノクロナール抗gp96(抗Grp94、抗9G10)を用いたウェスタンブロット(100ngタンパク質)。
【図1−2】図lC:還元条件下および非還元条件下でマウスIgG(mIgG)およびCD3O-Ig融合タンパク質(CD3O-Ig)と比較した場合の精製改変型gp96-Ig融合タンパク質(gp96-Ig)のドデシル硫酸ナトリウム-ポリアクリルアミドゲル電気泳動(SDS-PAGE)分析。分子量マーカー(kDa単位)が、左側に示されている。
【図2−1】gp96-Igの分泌および細胞内局在化。図2a:黒丸:培養物上清中のgp96-Ig、白丸:細胞溶解物中のgp96-Ig。gp96-IgはELISAにより定量し、SCLC-gp96-Igは106/mlでプレートした。
【図2−2】図2b:透過性の付与されたSCLC-gp96-IgのFACS分析;破線、ヤギ抗ウサギIgG-FITC(陰性対照);実線、ヤギ抗マウスIgG-フィコエリトリン。図2c:透過性の付与されていないSCLCのFACS分析;トランスフェクトされていない。図2d:透過性の付与されていないSCLCのFACS分析;gp96-IgでトランスフェクトされたSCLC。両方のパネルにおいて、破線は、ヤギ抗ウサギIgG-FITCであり;実線は、ヤギ抗マウスIgG-FITCである。
【図3−1】擬似トランスフェクトされた細胞(三角形)およびトランスフェクトされていない細胞(白丸)と比較して、gp96-IgでトランスフェクトされたE.G7(図3a)およびLLC(図3b)(黒丸)では、腫瘍形成性が低下した。1パラメーターあたり1群6匹のマウスを使用した。
【図3−2】擬似トランスフェクトされた細胞(三角形)およびトランスフェクトされていない細胞(白丸)と比較して、gp96-IgでトランスフェクトされたE.G7(図3a)およびLLC(図3b)(黒丸)では、腫瘍形成性が低下した。1パラメーターあたり1群6匹のマウスを使用した。
【図4−1】分泌型gp96-Igは、腫瘍特異的記憶を引き起こす。gp96-IgでトランスフェクトされたE.G7(いずれのパネルにおいても黒丸)106個、放射線照射されたEG7(三角形)106個を用いて、2週間の時間間隔で2回、C57BL/6マウスを免役したか、または免疫しなかった(白丸)。2週間後、図に記載の数の腫瘍細胞でマウス(1群あたり6匹のマウス)をチャレンジした。図4a:E.G7; 図4b:EL4。
【図4−2】分泌型gp96-Igは、腫瘍特異的記憶を引き起こす。gp96-IgでトランスフェクトされたE.G7(いずれのパネルにおいても黒丸)106個、放射線照射されたEG7(三角形)106個を用いて、2週間の時間間隔で2回、C57BL/6マウスを免役したか、または免疫しなかった(白丸)。2週間後、図に記載の数の腫瘍細胞でマウス(1群あたり6匹のマウス)をチャレンジした。図4c:LLC; 図4d:LLC-ova。
【図5−1】E.G7-gp96-Ig 106個の拒絶反応に及ぼす免疫適格細胞の枯渇の影響。それぞれのマウスについて腫瘍増殖カーブが示されている。図5a〜5d(上側のパネル):皮下腫瘍接種の2日前の免疫適格細胞の枯渇。
【図5−2】E.G7-gp96-Ig 106個の拒絶反応に及ぼす免疫適格細胞の枯渇の影響。それぞれのマウスについて腫瘍増殖カーブが示されている。図5e〜5h(下側のパネル):皮下腫瘍接種の3日後の免疫適格細胞の枯渇。
【発明を実施するための形態】
【0038】
5.発明の詳細な説明
本発明は、抗原提示に関与する熱ショックタンパク質(hsp)を改変し、そして癌および感染症の予防ならびに治療に使用することができる免疫原性組成物を調製するための、組換えDNA技術の応用を意図する。
【0039】
本発明で使用する「抗原性細胞」は任意の抗原性細胞でよく、限定するわけではないが、癌細胞、前腫瘍細胞、細胞内病原体に感染した細胞、または病原体に感染した被験者から採取した細胞などである。
【0040】
癌細胞または病原体に感染した細胞中で、免疫原性hsp-ペプチド複合体が自然に産生される。こうしたhsp-ペプチド複合体を使用して、この複合体の宿主細胞中に同一の種類の癌細胞または感染細胞に対する特異的な免疫応答を誘発させてきた。したがって、これら複合体は癌または感染症の予防および治療に有用である。しかし、こうした免疫原性複合体は一般的に、こうした細胞から大量には分泌されない。さらに、多数の癌細胞または感染細胞(抗原性細胞)を取得するのは常に可能なまたは実現し得るものではないので、こうした細胞から取得し得るhsp-ペプチド複合体の量は時には非常に限定される。したがって、免疫原性hsp-ペプチド複合体の限定された供給の問題を克服するための方法を見出だすことが望ましい。この問題の一部分は、現在実用化されている、細胞溶解を必要とする、hsp-ペプチド複合体の精製方法によるものである。本発明は、抗原性細胞が免疫原性hsp-ペプチド複合体を培地中に分泌するように仕向けるための方法を提供する。本発明の抗原性細胞は所望の免疫原性複合体を継続的に産生および分泌し、これを培地から好都合に回収することができる。培地からのこうした免疫原性複合体を精製するための改良法も提供される。さらに、本発明は癌細胞および感染細胞の免疫原性を強化して、癌または感染症を予防および治療するためのワクチンとしてこれらの抗原性細胞を直接被験体に投与するすることができるようにするための方法を提供する。
【0041】
1実施形態において、本発明の方法によって調製される免疫原性組成物には、改変型熱ショックタンパク質(hsp)、および抗原性細胞中に存在するかまたは存在するタンパク質の一部分である抗原性ペプチドを含む非共有結合で結合した分子複合体が含まれる。本発明の改変型熱ショックタンパク質は、それを発現する細胞から分泌されるが、これは改変されていない熱ショックタンパク質中には存在する小胞体(ER)残留配列を欠失しており、またペプチドタグを含んでいる。この改変型hspが組換え細胞中で発現すると、それが分泌されて、アフィニティークロマトグラフィーを使用して、ペプチドタグによって精製することができる。
【0042】
別の実施形態において、本発明の改変型熱ショックタンパク質はそれを発現する細胞から分泌されるが、これはペプチドタグを含んでおり、また改変されていない熱ショックタンパク質中には存在しないリーダーペプチドを含んでいる。天然では細胞質中に存在するhspについて、アミノ末端に付加したリーダーペプチドがこれらのER中へのトランスロケーションを促進する。
【0043】
本発明の改変型hspは天然に存在するhspと同質の生物学的活性を示す。この改変は抗原性ペプチドに対して特異的におよび非共有結合で改変型hspに結合する能力、および関連する免疫細胞に対して抗原提示過程中にこの結合したペプチドを提示する能力に影響を与えない。従って、この改変型hspはin vitroおよび抗原性細胞中の両方で抗原性ペプチドと免疫原性非共有結合複合体を形成することができる。in vitroおよび抗原性細胞中で内因的に形成された改変型hsp-ペプチド複合体はいずれも、動物中でこの抗原性ペプチドが由来する抗原性細胞に対する特異的免疫応答を誘導することができる。
【0044】
特に、本発明は、天然には動物細胞の小胞体(ER)中に残留する熱ショックタンパク質(hsp)をコードするヌクレオチド配列の改変に関係する。本発明に従えば、ER中にhspの残留をシグナルするペプチドのストレッチをコードするhsp配列の一部の置換または欠失によって、hsp遺伝子配列、好ましくはcDNA配列を改変する。ER中にhspを残留させるシグナルは、カルボキシル末端に位置するXaa-Asp-Glu-Leu(XDEL、ここでXは任意のアミノ酸とすることができる)を含むペプチドのストレッチ中に存在することが知られている。hsp遺伝子配列はさらに、あるペプチドタグをコードするヌクレオチド配列の1つをhsp配列に付加することによって、改変される。得られる本発明の改変型hsp配列は、分泌されてER中に残留しない改変型hsp融合タンパク質をコードする。
【0045】
天然には細胞質中に存在するhspについては、ペプチドタグをコードするヌクレオチド配列ばかりでなく、リーダーペプチドをコードするヌクレオチド配列をも付加することによって、その遺伝子配列を改変する。この配列をhsp遺伝子配列のコード領域の5'末端に連結する。こうした疎水性リーダーペプチドを坦持するhspをER内腔内に取り込ませる。該リーダーペプチドはシグナル認識粒子によって認識され、これが、伸長していくhspペプチド鎖を粗面小胞体膜の細胞質ゾル側表面に誘導する。分泌可能なhspは膜を通過して完全に内腔中にトランスロケーションし、そこでリーダーペプチドがプロテアーゼによって消化される。
【0046】
本発明において、アフィニティークロマトグラフィーによる改変型hspまたは改変型hsp-ペプチド複合体の精製を容易にするために、種々の機能および親和性を持つ各種のペプチドタグを使用することができる。好ましいペプチドタグには免疫グロブリンの定常領域が含まれる。hspに融合させるペプチドタグに応じて、改変されたhspは二量体化などの新規性質を獲得することがあり、これを有利に活用して、改変型hspまたは改変型hsp-ペプチド複合体の機能を強化することもできる。ペプチドタグおよびリーダーペプチドをコードする配列、ならびにこうした配列をhsp配列に連結するための方法については、第5.1.4節に記載する。
【0047】
したがって、本発明は、改変型hspをコードするヌクレオチド配列(「改変型hsp遺伝子配列」)を含む核酸分子、ならびにこうした配列を含有するクローニングベクター、発現ベクターおよび組換え細胞を提供する。本発明は、改変型hsp遺伝子配列に相補的なヌクレオチド配列を含む核酸分子も包含する。改変型hsp遺伝子配列を改変型hsp-ペプチド複合体の産生に好適な宿主細胞中に導入する前に、中間細胞中のクローニングベクター中に該改変型hsp遺伝子配列をクローン化し、かつ/または複製によって増やすことができる。第5.2.1節に記載するような当技術分野で既知の任意の方法によって、改変型hsp遺伝子配列を含む発現構築物または発現ベクターを構築して、宿主細胞中に導入することができる。必要に応じて、改変型hspの発現のために、癌細胞、病原体に感染した細胞または正常細胞を含む、各種の細胞を使用することができる。
【0048】
本発明の発現遺伝子構築物には、改変型hspをコードするヌクレオチド配列、好ましくは改変型hspをコードする相補的DNA(cDNA)配列が含まれる。改変型hsp遺伝子配列は、適切な宿主細胞内で改変型hsp配列の発現を制御する少なくとも1つの調節領域(例えばプロモーター)に機能的に結合される。あるいは、宿主細胞内で相同組換えを促進する領域に、改変型hsp配列を隣接させて、染色体内のある部分に改変型hsp配列を挿入するようにし、それによって改変型hsp遺伝子配列を宿主細胞内で改変型hsp配列の発現を制御する少なくとも1つの調節領域に機能的に結合させることができる。改変型hsp遺伝子配列を含む両タイプの発現遺伝子構築物を発現可能な改変型hsp遺伝子配列とも称する。したがって、本発明は発現可能な改変型hsp遺伝子配列を含有する組換え細胞を提供する。
【0049】
hspをコードする配列およびペプチドタグをコードする配列、ならびにこうした配列を取得するための方法を第5.1.1節および第5.1.4節に詳細に記載する。ヌクレオチドの付加、欠失または置換によるhsp遺伝子配列の改変法を第5.1.2節に記載する。
【0050】
別の実施形態において、本発明は、改変型hsp遺伝子配列の発現を可能にする組換え細胞を培養し、そしてこの組換え細胞から分泌される改変型hspを回収および精製することを含む、細胞培養物から改変型hspを精製するための方法を提供する。一般的に、第5.3節に記載するような適切なアフィニティークロマトグラフィー法を使用する、分泌された改変型hspの培養上清からの精製は、改変型hsp上のペプチドタグによって容易になる。改良法では、細胞の溶解を必要とせず、これによって、継続的に増殖させて改変型hspまたは改変型hsp-ペプチド複合体を産生させることが可能になる。
【0051】
本発明の改変型hspは改変されていないhspと同様にペプチドに結合する能力があるので、組換え抗原性細胞中での改変型hsp遺伝子配列の発現時に、改変型hspが産生されて、これがER中でペプチドと結合し、非共有結合複合体を形成する。抗原性細胞の何らかのタンパク質が抗原性/免疫原性であるので、改変型hspと複合体化するペプチド/タンパク質が、これらが存在する抗原性細胞に特異的な免疫を宿主に与える。こうした非共有結合の免疫原性複合体は組換え細胞から分泌され、培地中に蓄積する。
【0052】
したがって、本発明は、改変型hspと細胞中の抗原性ペプチドの免疫原性非共有結合複合体を製造および精製するための方法も提供する。1実施形態において、本方法は、改変型hsp遺伝子配列を抗原性細胞中に導入し、この組換え抗原性細胞を培養して改変型hsp遺伝子配列を発現させ、組換え抗原性細胞から分泌される改変型hsp-抗原性ペプチド複合体を培地から回収し、精製することを含む。複合体の抗原性ペプチドは抗原性細胞の内側にある抗原性ペプチドの典型であるので、この抗原性ペプチドを使用して被験体にワクチン接種する前に、この抗原を単離および/または特性決定する必要はなく、またこれらの抗原の本質を知る必要すらない。第5.3節に記載するような、細胞培地からの改変型hspの精製に適用し得る方法は、その方法が改変型hspと抗原性ペプチドの非共有結合性結合を妨害しない限り、改変型hsp-抗原性ペプチド複合体の精製にも有用である。特定の1実施形態において、発現可能な改変型hsp遺伝子配列を含む組換え抗原性細胞を治療および予防用途のための免疫原性組成物に使用することができる。こうした組成物の免疫原性は当技術分野で既知の、また第5.5節および第6節に記載された方法によって試験することができる。hsp-ペプチド複合体のin vitroでの作製に使用するための改変型hspを調製する目的の場合は、分泌される改変型hspに不要の抗原性分子が負荷されないように、それ自体は抗原性でない宿主細胞を使用することが望ましい。
【0053】
組換え細胞を適度に大規模に連続式でまたはバッチ式で培養することによって、改変型hspまたは改変型hsp-ペプチド複合体を大量に製造することができる。組換え抗原性細胞の大規模連続またはバッチ培養の細胞培養培地から、改変型hspおよび組換え産生された抗原性タンパク質/ペプチドを含む所望の免疫原性複合体を精製することができる。改変型hsp-ペプチド複合体を分泌する永久細胞系は、有用な免疫原性組成物の一定で、再現性があり、また豊富な供給源となり得る。必要に応じて、改変型hsp遺伝子配列を含む組換え細胞をプールおよび/若しくは分取し、または増殖させ、または液体窒素下での凍結および保存によって保管し、組換え宿主細胞のバッチを将来何度も復元させて使用することができるようにする。
【0054】
抗原性タンパク質またはペプチドのコード配列が知られている、別の1実施形態においては、抗原性分子をコードするこの配列を発現遺伝子構築物中にクローン化し、発現可能な改変型hsp配列を含む組換え細胞中に導入することを意図する。改変型hspの発現についての記載(第5.2節参照)などの標準的技法によって、抗原性タンパク質またはペプチドの発現ベクター中へのクローニングを実施することができる。抗原性タンパク質またはペプチドは組換え細胞中で改変型hspと共発現する。これらの抗原性タンパク質またはペプチドは組換え細胞のER中で改変型hspと非共有結合複合体を形成する。こうした抗原性ペプチドは癌細胞中で発現される抗原性タンパク質の断片(例えば、腫瘍特異的抗原または腫瘍関連抗原の断片など)とすることができる。これらの細胞からの改変型hsp-抗原性ペプチド複合体は培養培地中に分泌され、第5.3節に記載と同様に回収し、アフィニティークロマトグラフィーによって精製することができる。発現可能な改変型hsp配列、および抗原をコードする発現遺伝子構築物の両方を含有する組換え細胞も、治療および予防用途のための免疫原性組成物として使用することができる。
【0055】
本発明はさらに、精製した改変型hspをin vitroで抗原性ペプチドと混合して免疫原性非共有結合複合体を形成させる方法を提供する。こうした複合体は癌または感染症の治療および予防において有用である。抗原性ペプチドは細胞源から精製するか、またはそのペプチドの配列が既知ならば、当技術分野で知られた方法によって、合成することができる。特定の1実施形態においては、ペプチドタグによって固相に可逆的に固定化された改変型hspとともに抗原性ペプチドをインキュベートして、固相上で抗原性ペプチドと改変型hspとの非共有結合複合体を形成させるようにする。
【0056】
したがって、本発明は改変型熱ショックタンパク質およびペプチドを複合体の形成のために十分な時間インキュベートすることを含む、ペプチドと非共有結合で結合した本発明の改変型熱ショックタンパク質の複合体をin vitroで調製するための方法を提供する。
【0057】
特許請求する方法によって調製された改変型hsp-ペプチド複合体および組換え抗原性細胞の両方を含む、本発明の免疫原性組成物は、個体の免疫担当能力を強化し、腫瘍細胞および病原体に感染した細胞の両方に対する特異的な免疫を誘発することができる。こうした免疫原性組成物は癌の進行を防止し、腫瘍細胞の増殖および進行を阻害し、そして病原体または病原体に感染した細胞の増殖を防止する能力もある。免疫原性組成物を使用して、腫瘍部位での炎症性反応を誘導し、治療する癌患者にある腫瘍負荷量を最終的に退縮させることができる。
【0058】
したがって、本発明は、個体中で抗原の1つに対する免疫応答を誘発する方法であって、この抗原若しくはその断片に非共有結合で結合した本発明の改変型熱ショックタンパク質の免疫原性複合体を該個体に投与することを含む方法を提供する。また本発明は、癌であるかまたは癌の予防が必要とされる個体において癌を治療または予防する方法であって、上記の抗原若しくはその断片に非共有結合で結合した本発明の改変型熱ショックタンパク質の免疫原性複合体を該個体に投与することを含む方法も提供する。また、感染症であるかまたは感染症の予防が必要とされる個体において感染症を治療または予防する方法であって、上記の抗原若しくはその断片に非共有結合で結合した本発明の改変型熱ショックタンパク質の免疫原性複合体をその個体に投与することを含む方法も本発明に包含される。
【0059】
癌細胞若しくは組織を採取した個体、または家族歴若しくは環境危険因子によって癌のリスクが増強した個体に自己由来の前記免疫原性組成物を投与することができる。同様に、病原体に感染した細胞若しくは抗原性細胞を取得した個体、または同一の病原体に感染するリスクがある個体に自己由来の前記免疫原性組成物を投与することができる。
【0060】
この癌の治療または予防方法は、改変型hspの発現のために癌細胞を提供しなかった他の個体に対しても、癌細胞の提供者と同一のタイプの癌であるならば、一般的に適用することができる。感染症の治療または予防についても、抗原性宿主細胞の提供者に感染した病原体と抗原性が同一の病原体に感染した個体ならば、前記方法を他の個体に適用することができるという、同一の原理が応用される。癌および感染症を治療または予防するための免疫原性組成物の使用については、第5.7節および第5.8節に記載する。
【0061】
5.1. 改変型hsp遺伝子配列の構築
ここに記載するのは、原核および真核細胞中で発現することができる改変型熱ショックタンパク質(hsp)をコードする遺伝子構築物の構築方法である。特定すると、改変型hspをコードするヌクレオチド配列の構築、その改変型hsp遺伝子配列の適切なクローニングベクター中へのインサート、および改変型hspおよび改変型hsp-ペプチド複合体の製造のための、この発現遺伝子構築物の適切な宿主細胞中への導入について、記載する。
【0062】
癌または感染症の治療および予防において有用な、本明細書中でストレスタンパク質と互換的に称する熱ショックタンパク質は、以下の基準のいずれか1つを満足する任意の細胞タンパク質の中から選択することができる:細胞がストレスの高い刺激に曝露されたときにその細胞内濃度が増加するタンパク質であること、その他のタンパク質またはペプチドに結合することができること、そしてアデノシン三リン酸(ATP)の存在下または低pHにおいて結合したタンパク質またはペプチドを放出する能力があること;あるいは、上記の特性のいずれかを有する任意の細胞タンパク質と少なくとも35%の相同性を示すタンパク質であること。本発明によって改変し、調製することができる複合体中のhspとして、限定するわけではないが、hsp70、hsp90、gp96、BiPおよびタンパク質ジスルフィドイソメラーゼが含まれる。好ましくは、hspはヒトhspである。好ましい複合体はタンパク質抗原の1つに非共有結合で結合した改変型ヒトhsp60、hsp70、hsp90、タンパク質ジスルフィドイソメラーゼ、またはBiPを含む。特定の実施形態において、複合体は通常は真核細胞の小胞体中に存在するヒトgp96の改変型を含む。
【0063】
hspの3つの主要なファミリー、すなわちhsp60、hsp70およびhsp90が現在のところ同定されている。さらに、タンパク質ジスルフィドイソメラーゼ(PDI)、ならびにチオレドキシン様ドメイン(群)を含有する小胞体中のその他のタンパク質、限定するわけではないがERp72およびERp61など、も包含される。これらのhspファミリーの全てのメンバーを本発明の実践によって改変し、調製することができるものと構想する。
【0064】
hsp60、hsp70、hsp90およびタンパク質ジスルフィドイソメラーゼファミリーは、配列においてストレスタンパク質に関係がある、例えばアミノ酸の同一性が35%を超えるが、発現レベルはストレスによって変更されないタンパク質で構成されることが発見された。したがって、本明細書で使用するストレスタンパク質または熱ショックタンパク質の定義は、細胞中での発現レベルがストレス性刺激に応答して増強されるこれらのファミリーのメンバーに少なくとも35%〜55%、好ましくは55%〜75%、最も好ましくは75%〜85%のアミノ酸同一性があるその他のタンパク質、それらの突然変異タンパク質、類似体および変異体を包含することを意図する。
【0065】
hsp遺伝子構築物を構築して改変する際に使用する常套的な分子生物学的反応を実施するために、基準となる論文、例えばMethods in Enzymology, 1987, volume 154, Academic Press;Sambrookら、1989, Molecular Cloning-A Laboratory Manual, 2nd Edition, Cold Spring Harbor Press, New York;およびAusubelら、Current Protocols in Molecular Biology, Greene Publishing Associates and Wiley Interscience, New York、に記載された操作法に従ってもよい。以下に詳細に記載する方法は単に説明のためのものであって、限定するつもりではない。市販の各種クローニングベクターおよび発現系を製造元の指示にしたがって使用してもよい。
【0066】
5.1.1. hsp遺伝子配列の単離
各種の実施形態において、本発明は改変型熱ショックタンパク質(hsp)、ならびに機能的に活性なその断片および誘導体のアミノ酸配列に関する。本明細書で使用する「機能的に活性な」改変型hspとは、抗原性ペプチドの結合、アデノシン三リン酸(ATP)の存在下または低pHにおける結合した抗原性ペプチドの放出、その他などの、未改変型hspに関係する1以上の公知の機能的活性を表出する改変型hspをいう。改変型hspおよび上記のようなその断片をコードする核酸、ならびにこれらの核酸に対して相補的かつこれらとハイブリダイズする能力がある核酸を提供する。
【0067】
天然に出現する熱ショックタンパク質のアミノ酸配列およびヌクレオチド配列はGenBankなどの配列データベースから一般的に入手可能である。Entrezなどのコンピュータプログラムを使用して、データベースを閲覧(browse)し、目的とする任意のアミノ酸配列および遺伝子配列データを受託番号によって検索することができる。類似の配列をアライメントスコアおよび統計によって分類する、FASTAおよびBLASTなどのプログラムを使用して、これらのデータベースを検索して、問題の配列に対して様々なの程度の類似性を有する配列を同定することができる。
【0068】
本発明の方法によって改変し、発現させることができるhspの限定するわけではない例としてのヌクレオチド配列が以下に公開されている。ヒトgp96:Genebank Accession No.X15187;Makiら、1990, Proc.Natl.Acad.Sci., 87:5658-5562、マウスgp96:Genebank Accession No.M16370;Srivastavaら、1987, Proc.Natl.Acad.Sci., 85:3807-3811、マウスBiP: Genebank Accession No.U16277;Haasら、1988, Proc.Natl.Acad.Sci. U.S.A., 85:2250-2254、ヒトBiP:Genebank Accession No.M19645;Tingら、1988, DNA 7:275-286、マウスhsp70:Genebank Accession No.M35021;Huntら、1990, Gene, 87:199-204、ヒトhsp70:Genbank Accession No.M24743;Huntら、1995, Proc.Natl.Acad.Sci. U.S.A., 82:6455-6489。遺伝コードの縮重性に起因して、用語「hsp遺伝子配列」は、天然に出現するヌクレオチド配列を称するばかりでなく、hspをコードする別の縮重DNA配列の全部をも包含する。
【0069】
hsp遺伝子のコード領域を取得するための核酸起源として、どのような真核細胞でも可能性としては機能し得る。hspをコードする核酸配列を脊椎動物、哺乳動物、ならびにヒトを含む霊長類起源から単離することができる。
【0070】
DNAは、クローン化DNA(例えばDNA「ライブラリー」の1つ)から当技術分野で公知の標準的操作法によるか、またはDNA増幅によって取得することができる。ゲノムDNAに由来するクローンには、コード領域の他に調節領域およびイントロンDNA領域が含有されることがあり、cDNAに由来するクローンはエクソン配列のみを含むことになる。いずれの起源でも、hsp遺伝子は該遺伝子の増殖のために好適なベクター中に分子クローン化される必要がある。
【0071】
ゲノムDNA由来のhsp遺伝子の分子クローニングにおいて、DNA断片を作製し、クローン化し、ゲノムライブラリーを形成する。関連するhspをコードする配列のいくつかが入手可能で、これを精製および標識することができるので、この標識したプローブへの核酸のハイブリダイゼーションによって、ゲノムDNAライブラリー中のクローン化DNA断片をスクリーニングすることができる(Benton,W.およびDavis,R., 1977, Science 196:180;Grunstein,M. および Hogness,D., 1975, Proc.Natl.Acad.Sci. U.S.A. 72:3961)。プローブに実質的に相同性があるDNA断片がハイブリダイズする。制限酵素消化(群)し、入手し得るならば公知の制限マップにしたがって予想されるサイズと断片のサイズを比較することによって、適切な断片を同定することも可能である。
【0072】
hspゲノムDNAを単離するための別法として、限定するわけではないが、公知の配列から遺伝子配列自体を化学的に合成するもの、またはhspをコードするmRNAに対するcDNAを作成するものが含まれる。例えば、hspを発現する細胞からhsp遺伝子のcDNAクローニングのためのRNAを単離することができる。当技術分野で公知の方法によってcDNAライブラリーを作製し、ゲノムDNAライブラリーをスクリーニングするための開示されているような方法によってスクリーニングすることができる。hspに対する抗体が入手し得るならば、hspを合成していると推定されるクローンに標識した抗体を結合させることによって、hspを同定することもできる。
【0073】
hspをコードするヌクレオチド配列のクローニングのためのその他の特定の実施形態を以下のように例示として提供するが、限定するものではない。
【0074】
特定の実施形態において、低ないし中程度ストリンジェンシーの条件下で、hspの1つをコードするヌクレオチド配列を含むプローブとハイブリダイズさせることによって、あるファミリー内の熱ショックタンパク質をコードするヌクレオチド配列を同定して取得することができる。
【0075】
限定するわけではない例示としての、こうした低ストリンジェンシー条件を使用する操作法は以下の通りである(Shilo および Weinberg, 1981, Proc.Natl.Acad.Sci. USA 78:6789-6792、も参照されたい)。35%ホルムアミド、5X SSC、50 mM Tris-HCl(pH 7.5)、5 mM EDTA、0.1% PVP、0.1% フィコール、1% BSA、および 500 μg/ml 変性サーモン精子 DNAを含有する溶液中、40℃で6時間、DNAを含有するフィルターを前処理する。以下の改変:0.02% PVP、0.02% フィコール、0.2% BSA、100 μg/ml サーモン精子 DNA、10%(wt/vol)硫酸デキストラン、をした同一の溶液中で、5〜20 X 106 cpm 32P標識プローブを使用して、ハイブリダイゼーションを実施する。フィルターをハイブリダイゼーション混合液中、40℃で18〜20時間インキュベートし、その後、2X SSC、25 mM Tris-HCl(pH 7.4)、5 mM EDTA、および 0.1% SDSを含有する溶液中、55℃で1.5時間洗浄する。洗浄液を新鮮な溶液と交換し、さらに60℃で1.5時間インキュベートする。フィルターをドライブロットし、オートラジオグラフィー用に曝露する。必要ならば、フィルターに65〜68℃で3回目の洗浄を行い、フィルムに再曝露する。使用することができる低ストリンジェンシーのその他の条件は当技術分野で公知である(例えば交差種ハイブリダイゼーションのために利用するもの)。
【0076】
別の実施形態において、選別の前に、ポリメラーゼ連鎖反応(PCR)を使用して、DNAクローンまたはゲノムライブラリー若しくはcDNAライブラリー中の所望の配列を増幅する。PCRは例えば熱サイクラーおよびTaqポリメラーゼ(Gene AmpTM)の使用によって実施することができる。増幅するDNAとして任意の種からのcDNAまたはゲノムDNAが含まれる。PCRのプライマーとして、関連するhspの公知の核酸配列に相当するオリゴヌクレオチドプライマーを使用することができる。好ましい実施形態において、オリゴヌクレオチドプライマーは少なくとも異種のhsp間で高度に保存されているhsp遺伝子の部分を表す。PCR反応で使用するために、いくつかの異なる縮重プライマーの合成を選択することもできる。PCR反応を開始させるために使用するハイブリダイゼーション条件のストリンジェンシーを変更することによって、公知のhspヌクレオチド配列と単離する核酸相同体間のヌクレオチド配列の類似性の程度をより大きくするかまたは小さくすることも可能である。交差種ハイブリダイゼーションのためには、低ストリンジェンシー条件が好ましい。同種ハイブリダイゼーションのためには、中程度のストリンジェント条件が好ましい。増幅が成功した後、hspをコードする配列をクローン化および配列決定することができる。増幅するhsp遺伝子のコード領域のサイズが1回のPCRで増幅するには大き過ぎる場合は、全長遺伝子を網羅する何回かのPCRを、好ましくは領域を重複させて、実施し、このPCR産物を連結して全長コード配列を形成することができる。あるいは、hsp遺伝子のセグメントが増幅されるならば、そのセグメントをクローン化して、完全cDNAまたはゲノムクローンを単離するためのプローブとして利用することができる。
【0077】
改変の前に、hsp遺伝子を適切なクローニングベクターの1つにインサートして宿主細胞中に導入し、この遺伝子配列の多数のコピーを作製させることができる。当技術分野で公知の多数のベクター−宿主系を使用することができる。例えば、限定するわけではないが、ラムダ誘導体などのバクテリオファージ、またはpBR322若しくはpUCプラスミド誘導体などのプラスミド、またはBluescriptベクター(Stratagene)などである。
【0078】
上記の方法はhspのクローンを取得または増量する方法を限定することを意味するものではない。本発明の改変型熱ショックタンパク質は、分泌されかつ細胞培養培地から容易に精製することができるように改変される。特に、本発明の改変型hspはhspを小胞体(ER)中に残留するシグナルとなるポリペプチドのセグメントを欠失している。残留シグナルは、このペプチドを欠失させるか、またはシグナルとして機能しないペプチドと置換することによって無能化される。その上、改変型hspには回収および精製を容易にするペプチドタグが含まれる。ペプチドタグは例えばカルボキシル末端など、抗原性ペプチドとの結合に関与しない、hspの任意の部分に融合させることができる。さらに、hspが本来小胞体中に残留する場合、分泌のためにER膜を通ってこれが移動するのを導くために、リーダーペプチドを添加する。好ましい実施形態において、通常はカルボキシル末端に位置しているhspの残留ペプチドをペプチドタグと置換する。
【0079】
5.1.2 熱ショックタンパク質遺伝子の改変
本発明の改変型熱ショックタンパク質は、その内部でこれを発現する細胞によって分泌されて、その細胞培養培地から容易に精製することができるように、改変される。特に、本発明の改変型hspはhspの小胞体(ER)中への残留シグナルとなるポリペプチドのセグメントを欠失している。こうしたペプチドは、限定するわけではないがgp96などのER中に残留するhsp中に存在する。残留シグナルはこのペプチドを欠失させるか、またはシグナルとして機能しないペプチドと置換することによって無能化される。さらに、改変型hspには回収および精製を容易にするペプチドタグが含まれる。ペプチドタグは抗原性ペプチドとの結合に関与しない、例えばカルボキシル末端などの任意のhspの部分に融合させることができる。好ましい実施形態において、通常はカルボキシル末端に位置しているhspの残留ペプチドをペプチドタグと置換する。さらに、hspが天然では細胞質中に残留する場合、分泌のためにER膜を通ってこれが移動するのを導くために、リーダーペプチドを添加する。
【0080】
本発明の改変型hsp中に存在する改変は当技術分野で公知の各種の方法によって生成させることができる。この改変生成をもたらす操作は遺伝子またはタンパク質レベルのいずれかで実施できるが、遺伝子レベルが好ましい。例えば、クローン化したhspのコード領域を当技術分野で公知の多数の組換えDNA法のいずれによっても改変することができる(Sambrookら、1990, Molecular Cloning,A Laboratory Manual, 2nd Edition, Cold Spring Harbor Laboratory, Cold Spring Harbor,New York;Ausubelら、Current Protocols in Molecular Biology, Chapter 8, Greene Publishing Associates および Wiley Interscience,New York)。以下の考察から、改変型hspをコードする最終的なヌクレオチド配列に到達するために、置換、欠失、インサートまたはこれらの任意の組み合わせが導入または組み合わされることが明らかであろう。
【0081】
別法として、改変型hspを化学的に合成することができる。例えば、ペプチドシンセサイザーの使用によって、所望の改変を含むhspの1部分に相当するペプチドを合成することができる。
【0082】
5.1.3 残留ペプチド
hspを小胞体(ER)中に残留させる原因となるペプチドは典型的にはカルボキシル末端に位置し、配列はXaa-Asp-Glu-Leu(またはXDEL)である(Munro & Pelham,1987,Cell,48:899-907)。用語「残留ペプチド」(retention peptide)は本明細書中ではこのテトラペプチド配列を意味するために使用するが、大部分の哺乳動物hsp中ではこれはKDEL(Lys-Asp-Glu-Leu)である。Saccharomyces cerevisiaeおよびSchizosaccharomyces pombe中の残留ペプチド配列はそれぞれHDEL(His-Asp-Glu-Leu)およびADEL(Ala-Asp-Glu-Leu)であることがわかっている(Pidoux & Armstrong,1992,EMBO J.11:1583-1591)。
【0083】
hspの残留ペプチドはその残留ペプチドを欠失させるか、または残留ペプチド中のアミノ酸を置換してシグナルを消滅させることによって、無能化することができる。一般論として、改変型hsp配列中に、改変型hspが細胞から分泌されるのを妨害する傾向がある何らかのシグナルが存在する場合は、その存在するシグナルを除去すべきである。個々のhspによるが、こうしたシグナルとして、膜貫通ドメインおよび細胞質ドメインが挙げられる。
【0084】
残留ペプチド配列またはその他のシグナルをコードするDNAのセグメントを除去するために、適切な部位が利用し得る場合は、hsp遺伝子配列をそうした部位で制限酵素(群)により切断して、残留ペプチドをコードするDNAの断片を放出させることができる。次に、hspをコードする領域の残留部を単離し、改変型hsp遺伝子配列を形成させるように連結する。
【0085】
あるいは、好都合な制限部位が利用可能でないならば、残留ペプチド配列をコードする領域に隣接する配列中に位置する制限部位を使用することによって、より大きなDNAの断片を放出させて、残留ペプチドをコードする配列を欠失している合成DNAの類似の断片と入れ替えることができる。適正な翻訳リーディングフレームが確実に維持されるように、注意しなければならない。
【0086】
所望ならば、当技術分野で公知の部位特異的突然変異誘発方法および/またはDNA増幅方法によって、適切な位置に制限部位を作製することができる。例えば、Shankarappaら、1992,PCR Method Appl.1:277-278、を参照されたい。目的のDNA中に所望の配列変更を導入するためには、ポリメラーゼ連鎖反応(PCR)が共通に使用される。プライマー配列中の任意の変更を容易にそのPCRのDNA産物中に組込むことができ、これが遺伝子配列中へのこの変更のその後の組込みを促す。例えば、所望の制限部位を組込んでいる合成オリゴヌクレオチドを適切な隣接配列プライマーとともに使用して、2つの隣接するDNA断片を増幅することができる。これらの増幅された断片のそれぞれが一方の末端に新しい制限部位を含有するようになる。この新しい部位と隣接部位の両方での酵素消化の後、増幅した断片を連結し、その後の操作用に準備したベクター中にサブクローン化する。コードされたタンパク質のアミノ酸配列が制限部位の導入によって変更されないことが必須である。
【0087】
その後の操作を促進するために、発現されたペプチド配列中のアミノ酸を置換(群)させる目的で、または制限部位を作製/欠失する目的で、DNA配列中の個々のヌクレオチドを改変するためには、当技術分野で公知の突然変異誘発のどのような技法でも使用することができる。こうした技法として、限定するわけではないが、化学的突然変異誘発、in vitro部位特異的突然変異誘発(Hutchinson,C. ら、1978,J.Biol.Chem.253:6551)、オリゴヌクレオチド特異的突然変異誘発(Smith,1985,Ann.Rev.Genet.19:423-463;Hillら、1987,Methods Enzymol.155:558-568)、PCRに基づく重複伸長(Hoら、1989,Gene 77:51-59)、PCRに基づくメガプライマー突然変異誘発(Sarkarら、1990,Biotechniques,8:404-407)その他が含まれる。二本鎖ジデオキシDNA配列決定によって、改変を確認することができる。
【0088】
上記の方法を応用して、テトラペプチド残留配列中の1以上のアミノ酸残基、特にAsp、GluおよびLeu残基を置換することができる。残留ペプチド配列内のあるアミノ酸に対する置換基はそのアミノ酸が属するクラスと異なるメンバーから選択することができる。非極性(疎水性)アミノ酸にはアラニン、ロイシン、イソロイシン、バリン、プロリン、フェニルアラニン、トリプトファンおよびメチオニンが含まれる。極性無電荷アミノ酸にはグリシン、セリン、トレオニン、システイン、チロシン、アスパラギンおよびグルタミンが含まれる。正に荷電した(塩基性)アミノ酸にはアルギニン、リシンおよびヒスチジンが含まれる。負に荷電した(酸性)アミノ酸にはアスパラギン酸およびグルタミン酸が含まれる。一般的に生化学的特性に最大の変化を産生することが期待される置換には以下のようなものがある:(a)親水性残基、例えばセリルまたはトレオニルを疎水性残基、例えばロイシル、イソロイシル、フェニルアラニル、バリルまたはアラニルの代わりに(またはこれらによって)置換する;(b)システインまたはプロリンを任意のその他の残基の代わりに(またはこれらによって)置換する;(c)正の電荷を有する側鎖を有する残基、例えばリシル、アルギニルまたはヒスチジルを負の電荷を有する残基、例えばグルタミルまたはアスパルチルの代わりに(またはこれらによって)置換する;あるいは、(d)嵩高な側鎖を有する残基、例えばフェニルアラニンを側鎖を持たないもの、例えばグリシンの代わりに(またはこれらによって)置換する。
【0089】
上記の方法は、hsp中の残留ペプチド配列および/またはその他のシグナルを欠失または消滅させることができる方法を限定することを意味するものではない。
【0090】
5.1.4 ペプチドタグおよび/またはリーダーペプチドの融合
本発明の改変型hspはまた、ペプチドタグを含んでなる融合タンパク質でもある。特定の具体例では、リーダーペプチドを同じく改変型hspと融合させて、それにより改変型hspを分泌のために小胞体(ER)へ輸送することを促進させ得る。
【0091】
種々の実施形態では、かかる融合タンパク質は、適当な読み取り枠においてhsp遺伝子配列と、ペプチドタグまたはリーダーペプチドをコードする配列とを連結することにより作製できる。ゲノム配列が用いられる場合は、確実に同じ翻訳読み取り枠内に改変型遺伝子が留まり、翻訳停止シグナルおよび/または疑似メッセンジャーRNAスプライシングシグナルによって妨げられないように注意しなければならない。
【0092】
好ましい実施形態では、このペプチドタグはそのアミノ末端でhspのカルボキシル末端と融合している。カルボキシル末端において融合が起こる厳密な部位は重要ではない。例えば、ペプチドタグは残留ペプチドと置き換わってもよい。最適な部位は通常の実験のより決定することができる。改変型hspの免疫原性は第5.5節に記載される方法により試験できる。
【0093】
限定されるものではないが、免疫グロブリン定常領域、ポリヒスチジン配列(Petty, 1996, Metal-chelate affinity chromatography, Current Protocols in Molecular Biology, 第2巻, Ausbelら編, Greene Publish, Assoc. & Wiley Interscience) 、グルタチオンS-トランスフェラーゼ(GST, Smith, 1993, Methods Mol. Cell Bio. 4:220-229)、大腸菌マルトース結合タンパク質(Guanら, 1987, Gene 67:21-30)および種々のセルロース結合ドメイン(米国特許第5,496,934号;第5,202,247号;第5,137,819号;Tommeら, 1994, Protein Eng. 7:117-123)など、当技術分野で公知の種々のペプチドタグがhspの改変に使用され得る。いくつかのペプチドタグは改変型hspに、多量体の形成能といった新規な構造特性を与え得る。結合したペプチドによる改変型hspの二量体化は、抗原提示過程でのhspとそのパートナーとの相互作用のアビディティ(avidity)を高めると考えられる。これらのペプチドタグは大抵、通常はホモポリマーとして存在するタンパク質に由来するものである。CD28(Leeら, 1990, J. Immunol. 145:344-352 )もしくはCD8(Shiueら, 1988, J. Exp. Med. 168:1993-2005)の細胞外ドメイン、または鎖内ジスルフィド結合部位を含有する免疫グロブリン分子部分などのペプチドタグは、多量体の形成をもたらし得る。他の可能性あるペプチドタグとしては、限定されるものではないが、以下の良く知られた例としてFLAGエピトープ、mycエピトープのアミノ酸408-439、インフルエンザウイルスヘマグルチニン(HA)エピトープなど、それに対してモノクローナル抗体が利用可能である短いアミノ酸配列がある。その他のペプチドタグは特異的結合パートナーにより認識され、従って、結合パートナーとの親和性結合によって単離が容易になるが、これらは固相支持体上にあるかまたは固相支持体上に固定化されていることが好ましい。当業者により認識されるように、前記のペプチドタグのコード領域を得るには、限定されるものではないが、DNAクローニング、DNA増幅および合成法をはじめとする多くの方法が使用できる。いくつかのペプチドタグ、ならびにそれらの検出および単離のための試薬は市販されている。
【0094】
好ましいペプチドタグは免疫グロブリン分子の不変部分である。典型的には、かかる部分は少なくとも免疫グロブリンH鎖の定常領域のCH2およびCH3ドメインを機能的に含んでなる。不変ドメインのFc部分のカルボキシル末端、またはH鎖もしくはL鎖のCH1に直に続くアミノ末端領域を用いて、融合が行われる。好適な免疫グロブリンを基礎とするペプチドタグは、IgG-1、-2、-3または-4サブタイプ、IgA、IgE、IgDまたはIgM、好ましくはIgG1から誘導し得る。好ましくは、改変型hspが、ヒトに対してin vivoで使用することを意図されている場合は、ヒト免疫グロブリンが用いられる。免疫グロブリンL鎖またはH鎖定常領域をコードする多くのDNAが公知であるか、あるいはcDNAライブラリーから容易に入手できる。例えば、Adamら, Biochemistry, 1980, 19:2711-2719; Goughら, 1980, Biochemistry, 19:2702-2710; Dolbyら, 1980, Proc. Natl. Acad. Sci. U.S.A., 77:6027-6031; Riceら, 1982, Proc. Natl. Acad. Sci. U.S.A., 79:7862-7865; Falknerら, 1982, Nature, 298-286-288; およびMorrisonら, 1984, Ann. Rev. Immunol, 2:239-256を参照。免疫グロブリンの検出には多くの免疫試薬および標識系が利用できるので、改変型hsp-Ig融合タンパク質(「改変型hsp-Ig」)は、酵素結合免疫吸着検定(ELISA)、免疫沈降、蛍光活性化細胞選別(FACS)など、当技術分野で公知の種々の免疫学的技術によって容易に検出および定量できる。同様に、ペプチドタグが容易に利用できる抗体を有するエピトープである場合、かかる試薬を前記の技術と併用して、ペプチドタグを含有する改変型hspを検出、定量および単離することができる。多くの例では、改変型hspに対する特異的抗体を開発する必要はない。
【0095】
特に好ましい実施形態は、残留ペプチドを欠く改変型hspと、ヒンジであるネズミ免疫グロブリンG-1(IgG-1)のCH2およびCH3ドメインとの融合である(Bowenら, J. Immunol. 156:442-9)。このペプチドは、Ig分子の他のシステインとのジスルフィド結合に通常関与する3つのシステイン残基を含む。いずれのシステインもタグとしての機能のためにはペプチドに必要とされないので、これらのシステイン残基の1以上を、例えばセリンなどの別のアミノ酸残基でさらに置換してもよい。第5.1.2節に記載されるもののような方法を適用してかかる置換をなすことができる。
【0096】
細菌および哺乳動物細胞由来の改変型hspの効率的な分泌のために、当技術分野で公知の種々のリーダー配列を使用できる(von Heijne, 1985, J. Mol. Biol. 184:99-105)。リーダーペプチドは意図される宿主細胞に基づいて選択され、細菌、酵母、ウイルス、動物、および哺乳動物配列が含まれる。例えば、ヘルペスウイルス糖タンパク質Dリーダーペプチドは種々の哺乳動物細胞における使用に好適である。哺乳動物細胞での使用に好ましいリーダーペプチドは、マウス免疫グロブリンκ鎖のV-J2-C領域から得られる(Bernerdら, 1981, Proc. Natl. Acad. Sci. 78:5812-5816)。
【0097】
公知であるか、またはライブラリーもしくは商業的供給者から容易に入手可能な所望のペプチドタグまたはリーダーペプチドをコードするDNA配列は本発明の実施に好適である。第5.1.1節に記載のhsp配列を得る方法もまた、ペプチドタグまたはリーダーペプチドをコードする配列を得るのに適用できる。
【0098】
5.2 改変型hspの産生
本発明の種々の実施形態では、改変型hspをコードする配列は、組換え細胞内での増幅および発現のための発現ベクターに挿入される。
【0099】
本明細書において発現構築物とは、適当な宿主細胞で改変型hspの発現を可能とする1以上の調節領域と機能的に結合された改変型hspをコードするヌクレオチド配列をいう。「機能的に結合される」とは、調節領域と発現される改変型hsp配列が、転写、および最終的には翻訳が可能なように結合され配置される連結をいう。
【0100】
改変型hspの転写に必要な調節領域は発現ベクターにより提供され得る。また、その同起源の開始コドンを欠く改変型hsp配列が発現される場合は、翻訳開始コドン(ATG)も提供され得る。適合する宿主-構築物系においては、宿主生物において改変型hsp配列の転写を果たすため、RNAポリメラーゼなどの細胞転写因子は、発現構築体の調節領域に結合するであろう。遺伝子発現に必要な調節領域の厳密な性質は宿主細胞によって異なると考えられる。一般に、RNAポリメラーゼと結合して機能的に結合された核酸配列の転写を誘導することができるプロモーターが必要とされる。かかる調節領域としては、TATAボックス、キャッピング配列」、CAAT配列など、転写および翻訳の開始に関与する5'非コード配列が挙げられる。非コード領域3'からコード配列にかけては、ターミネーターやポリアデニル化部位など、転写終結調節配列が含まれ得る。
【0101】
改変型hspの発現のためには、構成的調節領域および誘導調節領域の双方を使用し得る。組換え細胞の増殖に最適な条件と改変型hspの高レベルの発現のための条件とが異なる場合には、誘導プロモーターを使用することが望ましい。有用な調節領域の例は下記次節に示される。
【0102】
プロモーターなどの調節機能を有するDNA配列を改変型hsp遺伝子配列と結合させるため、あるいはベクターのクローニング部位に改変型hsp遺伝子配列を挿入するためには、当技術分野で良く知られた技術により、適合した制限部位を提供するリンカーまたはアダプターを、cDNAの末端に連結すればよい(Wuら, 1987, Methods in Enzymol 152:343-349)。制限酵素による切断の後、連結前に一本鎖DNA末端を消化するか、またはフィリングによって、平滑末端を作製することができる。あるいは、所望の制限酵素部位を含むプライマーを用いるPCRの使用によるDNAの増幅によって、DNA断片へ所望の制限酵素部位を導入することもできる。
【0103】
調節領域と機能的に結合された改変型hsp配列を含んでなる発現構築物は、さらにクローン化せずに、改変型hsp-ペプチド複合体の発現および産生のために適当な宿主細胞へ直接導入できる。例えば、米国特許第5,580,859号を参照。発現構築物はまた、例えば相同組換えによる改変型hsp配列の、宿主細胞のゲノムへの組み込みを助けるDNA配列を含んでもよい。この例においては、宿主細胞で改変型hspを増幅および発現するため、適当な宿主細胞に好適な複製起点を含んでなる発現ベクターを使用する必要はない。
【0104】
5.2.1 宿主-ベクター系
本明細書では改変型hspの発現に使用できるベクターおよび宿主細胞系が記載される。限定されるものではないが、プラスミド、コスミド、ファージ、ファージミドまたは改変型ウイルスを含む種々の発現ベクターが本発明で使用され得る。典型的にはかかる発現ベクターは、適当な宿主細胞おいてベクターを増幅するための機能的複製起点、改変型hsp遺伝子配列の挿入のための1以上の制限エンドヌクレアーゼ部位、および1以上の選択マーカーを含んでなる。発現ベクターは、限定されるものではないが、細菌、酵母、昆虫、哺乳動物およびヒトをはじめとする原核生物または真核生物に由来し得る適合する宿主細胞とともに使用しなければならない。
【0105】
発現構築物およびベクターは分泌される改変型hspの産生を目的とする宿主細胞へ導入される。in vitroで培養されたもの、または遺伝子操作されたものをはじめ、熱ショックタンパク質を産生でき、かつ、発現ベクターと適合する細胞種ならばいずれを使用してもよい。宿主細胞は、健康な人、癌患者、および感染症を有する患者を含む正常な被験者または罹病した被験者から、民間研究寄託機関、American Type Culture Collectionなどの好適な培養物収集機関、あるいは商業供給者から得られる。
【0106】
In vivoにおいて改変型hsp-抗原ペプチド複合体の産生および分泌を目的として改変型hsp遺伝子配列が導入できる細胞としては、限定されるものではないが、上皮細胞、内皮細胞、ケラチン生成細胞、繊維芽細胞、筋細胞、肝細胞;Tリンパ球、Bリンパ球、単球、マクロファージ、好中球、好酸球、巨核球、顆粒球などの血液細胞;種々の幹細胞または前駆細胞、特に造血幹細胞または前駆細胞、例えば骨髄、臍帯血、末梢血、胎児肝臓に由来するものなどが挙げられる。細胞種の選択は治療または予防される腫瘍または感染症の種類に依存し、当業者ならば決定できる。
【0107】
異なる宿主細胞は、タンパク質の翻訳後プロセッシングおよび改変型に関する特徴および特異的なメカニズムを持っている。受容者がhspをプロセッシングする過程と同様の特異的様式で発現された遺伝子産物を改変およびプロセッシングする宿主細胞を選択すればよい。多量のHspを産生するためには、本発明で用いる宿主細胞種は異種遺伝子の発現に使用されたものであり、大規模生産工程のために合理的に十分に特徴づけられ、開発されたものであることが好ましい。特殊な実施形態では、宿主細胞は、次に改変型hsp-ペプチド複合体または改変型hsp-ペプチド複合体を分泌する組換え細胞が投与される同一の宿主に由来するものであり、すなわち、改変型hspの発現および被験者への投与に用いられる細胞は被験者の自己由来のものである。
【0108】
特定の実施形態では、改変型hsp遺伝子配列を含んでなる発現構築物が抗原細胞へ導入される。本明細書において、抗原細胞とは、まだ腫瘍性ではないが、ウイルスなど癌を引き起こす感染性病原体に感染した前腫瘍性細胞;または例えばDNA傷害剤、放射線など突然変異誘発剤または発癌剤に曝された抗原細胞が挙げられる。使用可能な他の細胞としては、形態学的機能、生理学的機能、または生化学的機能により特徴づけられる、正常形態から腫瘍形態への遷移過程にある前腫瘍性細胞がある。
【0109】
好ましくは、本発明の方法で用いられる癌細胞および前腫瘍性細胞は哺乳動物起源である。本発明の本実施形態で意図される哺乳動物としては、ヒト、ペット動物(例えば、イヌやネコ)、家畜(例えば、ヒツジ、ウシ、ヤギ、ブタおよびウマ)、実験動物(例えば、マウス、ラットおよびウサギ)ならびに捕獲または捕獲されていない野生動物が挙げられる。
【0110】
種々の実施形態では、改変型hsp-ペプチド複合体を生産するため、またはワクチンとして使用するため本方法においていずれかの癌細胞、好ましくはヒト癌細胞を用いることができる。これらの癌細胞は、発現した改変型hspと非共有結合的に会合するようになる抗原ペプチドを提供する。本発明の方法によって製造される免疫組成物で治療または予防できる癌としては、限定されるものではないが、肉腫および癌腫などの腫瘍が挙げられる。本発明の方法に従う癌の例は、第5.7節に挙げられている。従って、複数の離れた部位に転移した癌をはじめ、前腫瘍性病変部や癌から単離された組織または細胞は、本方法で使用可能である。例えば、異常な増殖を示す組織に見られる細胞、循環中の白血病細胞、転移病変部、ならびに固形腫瘍組織が使用できる。
【0111】
もう1つの実施形態では、その細胞系の細胞が標的癌細胞の抗原と共通の少なくとも1以上の抗原決定基を有しているならば、前腫瘍性病変、癌組織または癌細胞由来の細胞系が使用できる。ヒト起源の癌組織、癌細胞、発癌性病原体に感染した細胞、その他の前腫瘍性細胞および細胞系が好ましい。最終的に複合体が投与される患者から切除した癌細胞が使用されること、すなわち、本発明の自己由来の実施形態が好ましいが、必ずしもすべての場合にそうであることが必要なわけではない(例えば、癌細胞は1以上の異なる個体に由来してもよい)。
【0112】
癌細胞または前腫瘍性細胞は当技術分野で公知のいずれの方法によっても同定できる。例えば、癌細胞は形態学、酵素アッセイ、増殖アッセイ、細胞遺伝学的特徴、DNAマッピング、DNA配列決定、発癌性ウイルスの有無、または突然変異誘発物質または発癌剤に曝された履歴、画像などによって同定できる。もう1つの例としては、癌細胞は外科技術、内視鏡検査またはその他の生検技術によって得ることもできる。癌細胞のいくつかの特有の特徴が分かっていれば、それらはまた、限定されるものではないが、アフィニティークロマトグラフィー、および蛍光活性化細胞選別(例えば、癌細胞により発現される抗原に対する蛍光タグ付き抗体による)など、当技術分野で公知の生化学的または免疫学的方法のいずれによっても取得または精製可能である。
【0113】
クローン化された、または均質な、または精製された癌細胞集団を用いる必要はない。癌組織、癌細胞、または細胞系は単一の個体から得てもよいし、あるいはいくつかの個体からプールしてもよい。改変型hspの発現のために使用される細胞に、標的癌細胞の少なくとも1以上の抗原決定基が存在する限り、in vivoの最終標的の細胞(例えば、意図される受容者の腫瘍由来の細胞)を用いる必要はない。さらには、離れた転移由来の細胞を用いて、原発性癌に対する免疫組成物を調製し得る。混合物中の実質的な数の細胞が癌細胞であり、かつ、標的癌細胞と少なくとも1つの抗原決定基を共有するならば、細胞混合物を用いることができる。特定の実施形態では、改変型hspの発現において使用される癌細胞は精製されている。
【0114】
大腸菌に基づくベクターは最も一般的であり、外来タンパク質の高レベルの発現に関して広い用途を持つ系である(Makrides, 1996, Microbiol Rev, 60:512-538)。大腸菌での発現に使用できる調節領域の例としては、限定されるものではないが、lac、trp、lpp、phoA、recA、tac、T3、T7およびλPL(Makrides, 1996, Microbiol Rev, 60:512-538)が挙げられる。原核生物発現ベクターの例としては、限定されるものではないが、λgt11などのλgtベクター系列(Huynhら, 1984, "DNA Cloning Techniques", 第I巻: A Practical Approach (D. Glover編), 49-78頁, IRL Press, Oxford)、およびpETベクター系列(Studierら, 1990, Methods Enzymol., 185:60-89)が挙げられる。しかしながら、原核生物宿主-ベクター系のあり得る欠点としては、哺乳動物細胞の翻訳後プロセッシングの多くを行うことができないことである。従って、真核生物宿主-ベクター系が好ましく、哺乳動物宿主-ベクター系がより好ましく、ヒト宿主-ベクター系が最も好ましい。
【0115】
哺乳動物宿主細胞における改変型hspの発現のためには、種々の調節領域が使用でき、例えばSV40初期および後期プロモーター、サイトメガロウイルス(CMV)前初期プロモーター、およびラウス肉腫ウイルスの長い末端反復(RSV-LTR)プロモーターがある。哺乳動物細胞で有用であり得る誘導プロモーターとしては、限定されるものではないが、メタロチオネインII遺伝子、マウス乳癌ウイルスグルココルチコイド応答性の長い末端反復(MMTV-LTR)、β-インターフェロン遺伝子およびhsp70遺伝子が挙げられる(Williamら, 1989, Cancer Res. 49:2735-42; Taylorら, 1990, Mol. Cell Biol., 10:165-75)。組換え宿主細胞において改変型hspの発現を駆動するには熱ショックプロモーターまたはストレスプロモーターを使用するのが有利であろう。
【0116】
特定の組織種の腫瘍細胞には、組織特異性を示し、かつ、トランスジェニック動物に利用されてきた以下の動物の調節領域もまた使用できる:膵腺房細胞において活性なエラスターゼI遺伝子制御領域(Swiftら, 1984, Cell 38:639-646; Ornitzら, 1986, Cold Spring Harbor Symp. Quant. Biol. 50:399-409; MacDonald, 1987, Hepatology 7:425-515);膵β細胞において活性なインスリン遺伝子制御領域(Hanahan, 1985, Nature 315:115-122)、リンパ系細胞において活性な免疫グロブリン遺伝子制御領域(Grosschedlら, 1984, Cell 38:647-658; Adamesら, 1985, Nature 318:533-538; Alexanderら, 1987, Mol. Cell. Biol. 7:1436-1444)、精巣、乳房、リンパ系およびマスト細胞において活性なマウス乳癌ウイルス制御領域(Lederら, 1986, Cell 45:485-495)、肝臓において活性なアルブミン遺伝子制御領域(Pinkertら, 1987, Genes and Devel. 1:268-276)、肝臓において活性なα-フェトプロテイン遺伝子制御領域(Krumlaufら, 1985, Mol. Cell. Biol. 5:1639-1648; Hammerら, 1987, Science 235:53-58)、肝臓において活性なα1-抗トリプシン遺伝子制御領域(Kelseyら, 1987, Genes and Devel. 1:161-171)、骨髄性細胞において活性なβ-グロビン遺伝子制御領域(Mogramら, 1985, Nature 315:338-340; Kolliasら, 1986, Cell 46:89-94)、脳の稀突起神経膠細胞において活性なミエリン塩基性タンパク質遺伝子制御領域(Readheadら, 1987, Cell 48:703-712)、骨格筋において活性なミオシンL鎖-2遺伝子制御領域(Sani, 1985, Nature 314:283-286)および視床下部において活性な性腺刺激ホルモン放出ホルモン遺伝子制御領域(Masonら, 1986, Science 234:1372-1378)。
【0117】
宿主細胞における改変型hspの発現効率は、発現ベクター中に適当な転写エンハンサーエレメント、例えばSV40ウイルス、B型肝炎ウイルス、サイトメガロウイルス、免疫グロブリン遺伝子、メタロチオネイン、β-アクチンで見られるものなどを包含させることにより増強され得る(Bittnerら, 1987, Mrthods in Enzymol. 153:516-544; Gorman, 1990, Curr. Op. In Biotechnol. 1:36-47を参照)。
【0118】
発現ベクターはまた、2種類以上の宿主細胞におけるベクターの維持および複製、または宿主の染色体へのベクターの組み込みを可能にする配列を含んでもよい。かかる配列としては、限定されるものではないが、複製起点、自律的複製配列(ARS)、セントロメアDNA、およびテロメアDNAが挙げられる。また、少なくとも2種の宿主細胞で複製および維持が可能なシャトルベクターを用いることも有利である。
【0119】
さらに、発現ベクターは、改変型hspをコードするDNAを含む宿主細胞を最初に単離、同定または追跡するための選択可能もしくはスクリーニング可能なマーカー遺伝子を含んでもよい。長期にわたって改変型hsp-ペプチド複合体を高収量で産生し、哺乳動物細胞内で安定な発現をすることが好ましい。哺乳動物細胞に関しては、限定されるものではないが、単純ヘルペスウイルスチミジンキナーゼ(Wiglerら, 1977, Cell 11:223)、ヒポキサンチン-グアニンホスホリボシルトランスフェラーゼ(SzybalskiおよびSzybalski, 1962, Proc. Natl. Acad. Sci. USA 48:2026)およびアデニンホスホリボシルトランスフェラーゼ(Lowyら, 1980, Cell 22:817)、それぞれtk-、hgprt-、またはaprt-細胞で使用できる遺伝子をはじめ、いくつかの選択系が使用され得る。また、メトトレキセート耐性を付与するジヒドロ葉酸レダクターゼ(dhfr)(Wiglerら, 1980, Proc. Natl. Acad. Sci. USA 77:3567; O'Hareら, 1981, Proc. Natl. Acad. Sci. USA 78:1527);ミコフェノール酸耐性を付与するgpt(MulliganおよびBerg, 1981, Proc. Natl. Acad. Sci. USA 78:2072);アミノグリコシドG-418耐性を付与するネオマイシンホスホトランスフェラーゼ(neo)(Colberre-Garapinら, 1981, J. Mol. Biol. 150:1);およびハイグロマイシン耐性を付与するハイグロマイシンホスホトランスフェラーゼ(hyg)(Santerreら, 1984, Gene 30:147)の選択に基づき、代謝拮抗物質耐性も使用できる。限定されるものではないが、ヒスチジノールおよびゼオシンTMなど、他の選択マーカーも使用できる。
【0120】
好ましい哺乳類宿主細胞としては、限定されるものではないが、ヒト、サルおよび齧歯類に由来するもの(例えば、Kriegler M. "Gene Transfer and Expression: A laboratory Manual", New York, freeman & Co. 1990を参照)、例えば、SV40で形質転換されたサル腎細胞系統(COS-7, ATCC CRL 1651);ヒト胎児腎系統(293、293-EBNAまたは懸濁培養での増殖のためにサブクローン化された293細胞、Grahamら, J. Gen. Virol., 36:59, 1977);ベビーハムスター腎細胞(BHK, ATCC CCL 10);チャイニーズハムスター卵巣細胞-DHFR(CHO, UrlaubおよびChasin, Proc. Natl. Acad. Sci. 77:4216, 1980);マウスセルトリ細胞(Mather, Biol. Reprod. 23:243-251, 1980);マウス繊維芽細胞(NIH-3T3)、サル腎細胞(CVI ATCC CCL 70);アフリカミドリザル腎細胞(VERO-76, ATCC CRL-1587);ヒト子宮頸癌細胞(HELA, ATCC CCL 2);イヌ腎細胞(MDCK, ATCC CCL 34);バッファローラット肝細胞(BRL 3A, ATCC CRL 1442);ヒト肺細胞(W138, ATCC CCL 75);ヒト肝細胞(Hep G2, HB 8065);およびマウス乳腺癌細胞(MMT 060562, ATCC CCL 51)が挙げられる。癌ワクチンとしての組換え細胞(改変型hsp-ペプチド複合体を産生する)の有用性を実証するために使用される癌細胞種の典型例は以下に示される:マウス繊維芽細胞系統NIH3T3、マウスルイス肺癌細胞系統LLC、肥満細胞腫細胞系統P815、マウスリンパ腫細胞系統EL4およびその卵白アルブミントランスフェクト体E.G7、マウス黒色腫細胞系統B16F10、マウス繊維肉腫細胞系統MC57、ならびにヒト小細胞肺癌細胞系統SCLC#2およびSCLC#7。
【0121】
多数あるウイルスに基づく発現系を哺乳類細胞とともに用いて、改変型hspを生産してもよい。DNAウイルス骨格を用いるベクターはシミアンウイルス40(SV40)(Hamerら, 1979, Cell 17:725)、アデノウイルス(Van Dorenら, 1984, Mol Cell Biol 4:1653)、アデノ随伴ウイルス(McLaughlinら, 1988, J Virol 62:1963)、およびウシパピローマウイルス(Zinnら, 1982, Proc Natl Acad Sci 79:4897)に由来するものである。発現ベクターとしてアデノウイルスを用いる場合には、供与DNA配列をアデノウイルス転写/翻訳制御複合体、例えば後期プロモーターおよび3つに分かれたリーダー配列に連結すればよい。このキメラ遺伝子を次いでin vivoまたはin vitro組換えによりアデノウイルスゲノムに挿入できる。ウイルスゲノムの非必須領域(例えばE1またはE3領域)に挿入すれば、生存可能であって感染した宿主で異種産物を発現することができる組換えウイルスが得られる。(例えば、LoganおよびShenk, 1984, Proc. Natl. Acad. Sci. (USA) 81:3655-3659を参照。)
【0122】
ウシパピローマウイルス(BPV)はヒトをはじめとする多くの高等脊椎動物に感染でき、そのDNAをエピソームとして複製する。組換え遺伝子の発現のために、哺乳類細胞において安定な多重コピー(20〜300コピー/細胞)の染色体外要素として存在する多くのシャトルベクターが開発されている。典型的には、これらのベクターはBPV DNAセグメント(全ゲノムまたは69%形質転換断片)、広い宿主範囲を有するプロモーター、ポリアデニル化シグナル、スプライス部位、選択マーカー、および大腸菌でベクターを増殖させる「非毒性」プラスミド配列を含む。細菌内で構築および増幅した後、発現遺伝子構築物は、例えばリン酸カルシウム共沈法により、哺乳類培養細胞中にトランスフェクトされる。形質転換された表現型を呈さない宿主細胞に関しては、ヒスチジノールおよびG418耐性などの優性の選択マーカーの使用により、形質転換体の選抜が行われる。第6節で記載されるように、改変型hsp遺伝子配列を、2種のBPVベクターpBCMGSNeoおよびpBCMGHis(Karasuyamaら, Eur. J. Immunol. 18:97-104; Oheら, Human Gene Therapy, 6:325-33)に挿入し、次いでこれを改変型hspの発現のため広範な細胞種にトランスフェクトした。
【0123】
別法として、ワクシニア7.5Kプロモーターを用いた。(例えば、Mackettら, 1982, Proc. Natl. Acad. Sci. (USA) 79:7415-7419; Mackettら, 1984, J. Virol. 49:857-864; Panicaliら, 1982, Proc. Natl. Acad. Sci. 79:4927-4931を参照。)ヒト宿主細胞を用いる場合には、エプスタインバーウイルス(EBV)複製起点(OriP)およびEBV核抗原1(EBNA-1;トランス作用複製因子)に基づくベクターが使用できる。かかるベクターは広範なヒト宿主細胞、例えば、EBO-pCD(Spickofskyら, 1990, DNA prot Eng Tech 2:14-18);pDR2およびλDR2(Clontech Laboratoriesから入手できる)とともに使用できる。
【0124】
改変型hspはまた、レトロウイルスに基づく発現系でも生産され得る。ウイルス遺伝子配列のほとんどは除去されて改変型hsp遺伝子で置換され、一方、除かれたウイルスの機能はトランスで補われるので、配列モロニーマウス白血病ウイルスなどのレトロウイルスが使用できる。トランスフェクションに対し、レトロウイルスは、例えば一次造血細胞をはじめとする広範な細胞種へ効果的に感染し、遺伝子を導入する。さらに、レトロウイルスベクターにより感染する宿主範囲は、ベクターのパッケージングに用いられるエンベロープの選択により操作できる。
【0125】
例えば、レトロウイルスベクターは5'長い末端反復配列(LTR)、3'LTR、パッケージングシグナル、細菌の複製起点、および選択マーカーを含んでなることができる。改変型hsp DNAは5'LTRと3'LTRの間の位置に挿入され、その結果、5'LTRプロモーターからの転写がクローン化されたDNAを転写することとなる。5'LTRは、限定されるものではないが、LTRプロモーター、R領域、U5領域、およびプライマー結合部位をこの順序で含むプロモーターを含んでなる。これらのLTRエレメントのヌクレオチド配列は当技術分野で十分公知である。感染細胞の選択を容易にするため、多剤選択マーカーだけでなく異種プロモーターもまた、発現ベクターに含めてよい。McLauchlinら, 1990, Prog Nucleic Acid Res and Molec Biol 38:91-135; Morgensternら, 1990, Nucleic Acid Res 18:3587-3596; Choulikaら, 1996, J Virol 70:1792-1798; Boesenら, 1994, Biotherapy 6:291-302; SalmonsおよびGunzberg, 1993, Human Gene Therapy 4:129-141;ならびにGrossmanおよびWilson, 1993, Curr. Opin. Genetics and Devel. 3:110-114を参照のこと。
【0126】
その他の有用な真核生物宿主-ベクター系としては、酵母および昆虫系がある。酵母では、構成または誘導プロモーターを含むいくつかのベクターが、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)(パン酵母)、シゾサッカロミセス・ポンペ(Schizosaccharomyces pombe)(分裂酵母)、ピヒア・パストリス(Pichia pastoris)およびハンセヌラ・ポリモルファ(Hansenula polymorpha)(メチロトローフ酵母)とともに使用され得る。概論としては、Current Protocols in Molecular Biology, 第2巻, 1988, Ausubelら編, Greene Publish. Assoc. & Wiley Interscience, 第13章; Grantら, 1987, Expression and Secretion Vectors for Yeast, Methods in Enzymology, WuおよびGrossman編, 1987, Acad. Press, N.Y., 第153巻, 516-544頁; Glover, 1986, DNA Cloning, 第II巻, IRL Press, Wash., D.C., 第3章,およびBitter, 1987, Heterologous Gene Expression in Yeast, Methods in Enzymology, BergerおよびKimmel編, Acad. Press, N.Y., 第152巻, 673-684頁;およびThe Molecular Biology of the Yeast Saccharomyces, 1982, Strathernら編, Cold Spring Harbor Press, 第IおよびII巻を参照のこと。
【0127】
昆虫系では、バキュロウイルスであるオートグラファ・カリフォルニカ(Autographa californica)核ポリヒドロシスウイルス(AcNPV)を、スポドプテラ・フルギペルダ(Spodoptera frugiperda)細胞にて改変型hspを発現するためのベクターとして使用できる。改変型hsp遺伝子配列は該ウイルスの非必須領域(例えば、ポリヘドリン遺伝子)内にクローン化し、AcNPVプロモーター(例えば、ポリヘドリンプロモーター)の制御下に置けばよい。これらの組換えウイルスは次いで、挿入されたDNAが発現する宿主細胞に感染させるのに用いる。(例えば、Smithら, 1983, J Virol 46:584; Smith, 米国特許第4,215,051号を参照。)。
【0128】
本明細書に記載のクローニングおよび発現ベクターはいずれも、当技術分野で十分公知の技術により、公知のDNA配列から合成および構築され得る。調節領域およびエンハンサーエレメントは、様々な天然および合成の両方の起源であり得る。いくつかのベクターおよび宿主細胞は商業的に入手できる。有用なベクターの例としては、限定されるものではないが、参照により本明細書に組み入れるCurrent Protocols in Molecular Biology, 1988, Ausubelら編, Greene Publish. Assoc. & Wiley Interscienceの付録5、およびClontech Laboratories, Stratagene Inc.およびInvitrogen, Inc.などの商業的供給者のカタログに記載されている。
【0129】
5.2.2 改変型hspの発現
改変型hspをコードするクローン化ヌクレオチド配列を含む発現構築物は、限定されるものではないが、原核細胞に関しては細菌形質転換(Hanahan, 1985, DNA Cloning, A Practical Approach, 1:109-136)、および真核細胞に関しては、リン酸カルシウム媒介トランスフェクション(Wiglerら, 1977, Cell 11:223-232)、リポソーム媒介トランスフェクション(Schaefer-Ridderら, 1982, Science 215:166-168)、エレクトロポレーション (Wolffら, 1987, Proc Natl Acad Sci 84:3344)、およびマイクロインジェクション(Cappechi, 1980, Cell 22:479-488)をはじめとする当技術分野で公知の種々の技術によって宿主細胞へ導入できる。改変型hspと抗原の同一宿主細胞中での同時発現も、実質的に同じ方法により達成できる。
【0130】
長期にわたって、正しくプロセッシングされた改変型hspまたは改変型hsp-ペプチド複合体を高収量で産生するように、哺乳類細胞内で安定した発現をすることが好ましい。改変型hspまたは改変型hsp-ペプチド複合体を安定して発現する細胞系統は、選択マーカーを含むベクターを用いることにより遺伝子工学的に作製してもよい。限定されるものではないが、例示すれば、発現構築物の導入後に、遺伝子操作された細胞を栄養強化培地中で1〜2日間増殖させ、次いで選択培地に切り替えればよい。発現構築物中の選択マーカーは選択に対する耐性を与え、最適には、細胞が、それらの染色体中へ該発現構築物を安定して組み込み、培養にて増殖し、さらには細胞系統にまで発達するようにさせる。かかる細胞は改変型hspを継続的に発現しつつ長期間培養可能である。
【0131】
組換え細胞は、標準的な条件の温度、インキュベーション時間、光学濃度および培地組成下で培養できる。あるいは、組換え抗原性細胞は、癌細胞または感染細胞の栄養要求性および生理的要求性を模倣した条件下で培養してもよい。しかしながら、組換え細胞の増殖のための条件は、改変型hspおよび抗原タンパク質の発現のための条件と異なっていてもよい。hsp-ペプチド複合体の産生を促進するために、改変された培養条件および培地を用いてもよい。例えば、改変型hspと同起源のプロモーターを保持する改変型hspを含む組換え細胞を、熱もしくは他の環境ストレス、または化学的ストレスに曝してもよい。改変型hspまたは改変型hsp-ペプチド複合体を産生するための至適条件を確立するために、当技術分野で公知のいずれの技術を適用してもよい。
【0132】
改変型hspを発現する組換え細胞がワクチンとして使用される、ある実施形態においては、得られた組換え細胞をin vivoで投与する前に改変型hsp遺伝子配列を細胞に導入する。かかる導入は、遺伝子治療技術を含む当技術分野において、限定されるものではないが、トランスフェクション、エレクトロポレーション、マイクロインジェクション、改変型hsp遺伝子配列を含有するウイルスまたはバクテリオファージベクターによる感染、細胞融合、染色体媒介遺伝子導入、微小細胞媒介遺伝子導入、スフェロプラスト融合などの公知のいずれの方法によっても行える。受容細胞の必要とされる発生学的および生理学的機能が破壊されない限り、外来遺伝子を細胞に導入するための当技術分野において公知の多くの技術 (例えば、LoefflerおよびBehr, 1993, Meth, Enzymol. 217:599-618; Cohenら, 1993, Meth. Enzymol. 217:618-644; Cline, 1985, Pharmac. Ther. 29:69-92を参照)を、本発明に従って使用してよい。この技術は、改変型hsp遺伝子配列の細胞への安定した導入を提供すべきであり、その結果、この配列が細胞により発現可能であり、さらにその子孫細胞により遺伝および発現可能であることが好ましい。
【0133】
得られた組換え細胞は、当技術分野で公知の種々の方法により患者に送達できる。好ましい実施形態では、上皮細胞を例えば皮下注射する。もう1つの実施形態では、組換え皮膚細胞を皮膚移植片として患者に適用してもよい。組換え血液細胞(例えば造血幹細胞または始原細胞)は、好ましくは静脈内投与される。使用が意図される細胞の量は、所望の効果、患者の状態などに依存し、当業者により決定できる。
【0134】
5.2.3. 改変型hspと抗原の同時発現
もう1つの実施形態では、タンパク質抗原またはそれらの一部をコードするヌクレオチド配列の発現型を、発現可能な改変型hsp遺伝子配列を含む組換え細胞中に導入でき、その結果としてこの抗原は改変型hspと同時発現される。抗原をコードするヌクレオチド配列を得る方法は、第5.4.5節に記載されている。限定されるものではないが、第5.2.2節に記載したような、抗原遺伝子配列の発現型を導入するためのいずれの技術を用いてもよい。タンパク質抗原またはそれらの一部は、組換え細胞のER中で改変型hspと非共有結合で結合し、得られた改変型hsp-抗原ペプチド複合体が分泌される。かかる複合体は、第5.3節に記載された方法、および当技術分野で公知の他の方法のいずれによっても、細胞培養培地から精製できる。精製された改変型hsp-抗原複合体は、癌もしくは感染症の治療または予防を目的として、被験者において抗原タンパク質に対する免疫反応を刺激するワクチンとして使用できる。
【0135】
さらに、改変型hsp遺伝子配列と抗原タンパク質をコードするヌクレオチド配列双方の発現型を含む組換え細胞を、被験者に注射するためのワクチンとして直接使用してもよい。前記のように、かかる細胞は、癌もしくは感染症の治療または予防を目的として、被験者において抗原タンパク質に対する免疫反応を刺激できる、改変型hsp-抗原複合体を分泌する。
【0136】
癌または感染症の治療および予防のための、かかる改変型hsp-ペプチド複合体と、改変型hspおよび抗原遺伝子配列の発現型を含む組換え細胞の用途は、第5.7節および5.8節に記載されている。
【0137】
5.3. 改変型hsp-ペプチド複合体の精製
一般に、本発明の改変型hspは、硫酸アンモニウム沈殿法、酸による抽出、陰イオンまたは陽イオン交換クロマトグラフィー、ホスホセルロースクロマトグラフフィー、イムノアフィニティークロマトグラフィー、ヒドロキシアパタイトクロマトグラフィー、およびレクチンクロマトグラフィーをはじめとする公知の方法により、組換え細胞培養物から回収および精製できる。
【0138】
細胞溶解物からのhsp70-ペプチド複合体の精製法はこれまでに記載されており、例えば、Udonoら, 1993, J. Exp. Med. 178:1391-1396を参照のこと。細胞溶解物からのhsp90-ペプチド複合体およびgp96-ペプチド複合体の精製は、例えば1995年9月21日付のWO 95/24923、および1997年3月20日付の WO 97/10000に記載されている。これらの方法を用いて本発明の改変型hspまたは改変型hsp-ペプチド複合体を組換え細胞から精製することができ、当技術分野に公知の重要でない改変により、改変型hspまたは改変型hsp-ペプチド複合体を細胞培養物から精製できる。
【0139】
しかしながら、本発明は改変型hsp上に存在するペプチドタグの特性に基づく、改変型hspの精製のための改良法を提供する。1つの方法は、タグとその結合相手の間の特異的な分子相互作用に基づくものである。もう1つの方法は、タグ上に存在するエピトープと抗体の免疫特異的結合に基づくものである。一般に当技術分野で十分公知のアフィニティークロマトグラフィーの原理をこれら双方の方法に適用できる。
【0140】
タグおよびその結合相手の特異的な分子相互作用に基づく、いくつかの方法を以下に記載する。
【0141】
免疫グロブリンの定常領域と融合された改変型hspの精製に一般的に適用可能な方法は、当技術分野で十分公知のプロテインAアフィニティークロマトグラフィー法である。ブドウ球菌のプロテインAは、42KDのポリペプチドであり、免疫グロブリンのH鎖の第2および第3の定常領域の間に位置する領域に特異的に結合する。異なるクラス、サブクラスおよび種の免疫グロブリンのFcドメインであるために、ヒトFc領域に対するプロテインAの親和性は強くても、他の種においては差異があるかもしれない。好ましくないサブクラスとしてはヒトIgG-3が挙げられ、ラットのサブクラスは最も好ましくない。特定のサブクラスに対しては、プロテインG(ブドウ球菌の)をプロテインAの代わりに精製に用いることができる。通常、抗体のアフィニティー精製用固相にはプロテイン-Aセファロース(PharmaciaまたはBiorad)を用い、免疫グロブリンのFcフラグメントと融合した改変型hspの精製にも、それを本質的に同様に使用できる。細胞上清に存在する、分泌された改変型hspは、固相上のプロテインAに特異的に結合するが、夾雑物は洗浄除去される。結合した改変型hspは、pHを段階的に下げた一連のクエン酸、酢酸およびグリシン-塩酸バッファーをはじめとする、当技術分野で公知の種々のバッファー系で溶出できる。組換え細胞が、改変型hspと同時に精製されるであろう抗体も産生する場合には、この方法は好ましくない。例えば、Langone, 1982, J. Immunol. Meth. 51:3; Wilchekら, 1982, Biochem. Intl. 4:629; Sjobringら, 1991, J, Biol. Chem. 26:399; 617-618頁, Antibodies A Laboratory Manual, HarlowおよびLane編集, Cold Spring Harbor laboratory, 1988)を参照のこと。
【0142】
あるいは、ポリヒスチジンタグを使用してもよく、その場合は改変型hspは金属キレートクロマトグラフィーで精製できる。通常、6個のヒスチジン配列であるこのポリヒスチジンタグは、ニトリロ三酢酸マトリックスのような固相に固定化できるニッケルイオン(Ni2+)のような2価の金属イオンに高い親和性を示す。ポリヒスチジンはNi2+-NTAアガロースに対してよく特徴付けられた親和性を有し、2つの穏やかな処理のいずれかにより溶出できる:イミダゾール(0.1-0.2M)は、効果的に該樹脂と結合部位を競合する;または、pHを6.0以下に下げることによりヒスチジンの側鎖をプロトン化し、結合を阻害する。この精製法は、細胞培養物の上清をNi2+-NTA-アガロースカラムにロードし、夾雑物を洗浄除去し、イミダゾールまたは弱酸により改変型hspを溶出させる。Ni2+-NTA-アガロースは、Sigma(St.Louis)およびQiagenのような商業的供給者から入手できる。改変型hspを検出し、定量するために用いられる、ポリヒスチジンタグを認識する抗体も利用できる。
【0143】
もう1つの使用可能なペプチドタグの例としては、元来、蠕虫である日本住血吸虫(Schistosoma japonicum)からクローン化されたグルタチオン-S-トランスフェラーゼ(GST)配列がある。一般に、大腸菌のような原核宿主細胞で発現される改変型hsp-GST融合物は、グルタチオンアガロースビーズによる吸着の後に、中性pHにて遊離還元グルタチオンの存在下で溶出させることにより、細胞培養物の上清から精製できる。変性条件は、精製中のいずれの過程においても必要とされないので、抗原ペプチドを保持する固定化された改変型hspのロードの際に用いることが望ましいであろう。さらに、GSTは一定条件下で二量体を形成することが知られているため、改変型hspの二量体が得られることもあり得る。Smith, 1993, Methods Mol. Cell Bio. 4:220-229を参照のこと。
【0144】
もう1つの有用な、使用可能なペプチドタグとしては、malE遺伝子にコードされる大腸菌のマルトース結合タンパク質(MBP)がある。細胞上清に存在する、分泌された改変型hsp-MBPはアミロース樹脂に結合するが、夾雑物は洗浄除去される。結合した改変型hsp-MBPはマルトースによりアミロース樹脂から溶出される。例えば、Guanら, 1987, Gene 67:21-30を参照のこと。
【0145】
改変型hspを精製するための第2の方法は、ポリクローナル抗体またはモノクローナル抗体が得られるようなエピトープを含むペプチドタグに対するものである。イムノアフィニティークロマトグラフィーおよび免疫沈降法のような、当技術分野で公知の、免疫特異的結合によるタンパク質の精製のための種々の方法が使用できる。例えば、Antibodies A Laboratory Manual, HarlowおよびLane編, 第13章、Cold Spring Harbor laboratory,1988; Current Protocols in Immunology, Coliganら編, 第8章, 第IおよびII節, John Wiley, 1991を参照のこと;なおその開示は両方とも、参照により本明細書に組み入れられる。
【0146】
前記の実施形態は、本発明の改変型hspを発現しているヒト細胞のような哺乳動物細胞の細胞培養培地から、分泌された改変型hsp-ペプチド複合体を回収および精製するために用いてよい。この方法は、改変型hspおよび/または改変型hsp-ペプチド複合体の中規模または大規模な精製に適用できる。改変型hsp-ペプチド複合体の精製のためには、pHを下げること、または変性条件を必要としない方法が最も好ましい。実施例では腫瘍細胞に関して記載したが、記載の方法は、例えば組織、単離細胞、または細胞内病原体に感染した真核細胞の不死化系統、または病原体に感染した被験者から得た細胞など、いずれの真核細胞からのhspの単離に使用してもよい。
【0147】
5.4. in vitroにおけるhsp-抗原性分子複合体の産生
改変型hspと組換え細胞中でそれらが内因的に関与するペプチドとの複合体が用いられない実施形態においては、改変型hspと抗原性分子の複合体をin vitroで調製する。当業者により認識されるように、以下に記載する方法による単離、化学的合成、または組換えによる産生のいずれかにより得られた抗原性ペプチドは、種々の改変型熱ショックタンパク質を用いてin vitro で再構成し非共有結合の改変型hsp-ペプチド免疫原性複合体を生成してもよい。かかる複合体は、免疫治療学的または予防のための本発明のワクチンに使用できる。改変型hsp-ペプチド複合体のin vitroにおける調製方法は、中規模または大規模で行うのに適用できる。
【0148】
改変型hspとの複合体形成のため、抗原性ペプチドとして使用するために、当技術分野で公知のものの中から、または、免疫測定法により抗体もしくはMHC分子と結合(抗原性)もしくは免疫応答できる(免疫原性)と決定されたものの中から、1以上の種の癌細胞、感染細胞もしくは感染性病原体に特異的な抗原またはそれらの抗原性部分を選択できる。
【0149】
5.4.1. 外因性抗原性分子
抗体との結合を検出することにより、問題の抗原の免疫原性または抗原性を決定するには、限定されるものではないが、ラジオイムノアッセイ、ELISA(酵素結合イムノソルベントアッセイ)、「サンドイッチ」イムノアッセイ、イムノラジオメトリックアッセイ、ゲル拡散沈降素反応、免疫拡散アッセイ、in vivoイムノアッセイ(例えば金コロイド、酵素または放射性同位元素標識を用いる)、ウエスタンブロット法、免疫沈降反応、凝集アッセイ(例えば、ゲル凝集検定法、血球凝集検定法)、補体固定アッセイ、免疫蛍光アッセイ、プロテインAアッセイ、および免疫電気泳動アッセイなどのような、競合および非競合アッセイ系を含む当技術分野に公知の種々の免疫検定法を用いることができる。1つの実施形態においては、一次抗体上の標識を検出することにより抗体結合を検出する。もう1つの実施形態においては、二次抗体または試薬と一次抗体との結合を検出することにより一次抗体を検出する。さらなる実施形態においては、二次抗体を標識する。当技術分野において、イムノアッセイで結合を検出するための多くの方法が公知であり、その使用が意図される。免疫原性を検出するための1つの実施形態においては、例えばin vitro細胞傷害性アッセイまたはin vivo遅延型過敏性アッセイなどの標準的な方法により、T細胞媒介応答を測定できる。
【0150】
抗原性分子としての使用のために、有用な可能性のある抗原またはそれらの誘導体も、病原体の感染性の中和における抗原の関与(そのような病原体による感染は治療または予防するのが望ましい)(Norrby, 1985, Summary, Vaccines 85, Lernerら(eds.), Cold Spring Harbor Laboratory, Cold Spring Harbor, New York, pp388-389)、タイプまたはグループ特異性、患者の抗血清または免疫細胞による認識、および/または抗原に特異的な抗血清もしくは免疫細胞の防御作用の実証などの、種々の基準により同定できる。さらに、病原体により引き起こされた疾病の治療または予防が望まれる場合には、抗原のコードされたエピトープが、時宜よく、または同じ病原体の種々の単離体間で、わずかな抗原性変化を示すか、あるいは全く変化を示さないのが好ましい。
【0151】
癌の治療または予防が望まれる場合、公知の腫瘍特異的抗原またはフラグメントもしくはそれらの誘導体が使用される。例えば、かかる腫瘍特異的または腫瘍関連抗原としては、限定されるものではないが、KS 1/4万能癌抗原(Perezおよび Walker, 1990, J. Immunol. 142:3662-3667; Bumal, 1998, Hybridoma 7(4):407-415);卵巣癌抗原(CA125)(Yuら, 1991, Cancer Res. 51(2):468-475);前立腺の酸リン酸塩(prostatic acid phosphate)(Tailerら, 1990, Nucl. Acids Res. 18(16):4928);前立腺特異的抗原(HenttuおよびVihko, 1989, Biochem. Biophys. Res. Comm. 160(2):903-910; Israeliら, 1993, Cancer Res. 53:227-230);黒色腫関連抗原p97(Estinら, 1989, J. Natl. Cancer Inst. 81(6):445-446); 黒色腫抗原gp75(Vijayasardahlら, 1990, J. Exp. Med. 171(4):1375-1380);高分子量黒色腫抗原(Nataliら, 1987, Cancer 59:55-63)および前立腺特異的膜抗原が挙げられる。
【0152】
特定の実施形態では、ある特定の腫瘍に特異的な抗原またはフラグメントもしくはそれらの誘導体を選択して改変型hspと複合体を形成し、次いでその腫瘍を有する患者に投与する。
【0153】
ウイルス性疾患の治療または予防が所望される場合は、既知のウイルスのエピトープを含む分子を用いる。例えば、かかる抗原性エピトープは、限定されるものではないが、A型肝炎、B型肝炎、C型肝炎、インフルエンザ、水痘、アデノウイルス、I型単純ヘルペス(HSV-I)、II型単純ヘルペス(HSV-II)、牛疫、ライノウイルス、ECHOウイルス、ロタウイルス、RSウイルス、パピローマウイルス、パポバウイルス、サイトメガロウイルス、エキノウイルス、アルボウイルス、ハンタウイルス、コクサッキーウイルス、ムンプスウイルス、麻疹ウイルス、風疹ウイルス、ポリオウイルス、I型ヒト免疫不全ウイルス(HIV-I)、およびII型ヒト免疫不全ウイルス(HIV-II)をはじめとするウイルスから製造してもよい。
【0154】
細菌感染の治療または予防が望ましい場合には、既知の細菌のエピトープを含む分子を用いるのが好ましい。例えば、限定されるものではないが、マイコバクテリア、リケッチア、マイコプラズマ、ナイセリアおよびレジオネラをはじめとする細菌から、かかる抗原性エピトープを製造してもよい。
【0155】
原生動物による感染の治療または予防が望ましい場合には、既知の原生動物のエピトープを含む分子を用いるのが好ましい。例えば、限定されるものではないが、リーシュマニア、コクジジオア(kokzidioa)、およびトリパノソーマをはじめとする原生動物から、かかる抗原性エピトープを製造してもよい。
【0156】
寄生虫による感染の治療または予防が望ましい場合には、既知の寄生虫のエピトープを含む分子を用いるのが好ましい。例えば、限定されるものではないが、クラミジアおよびリケッチアをはじめとする寄生虫から、かかる抗原エピトープを製造してもよい。
【0157】
5.4.2. hsp-ペプチド複合体からのペプチド
本発明の改変型hspとin vitroで複合体を形成するための抗原ペプチドはまた、ペプチドとhspの内因性複合体から得ることができる。hsp-ペプチド複合体からペプチドを溶離させるためには2つの方法を用いてよい。1つの試みは、ATP存在下でhsp-ペプチド複合体をインキュベートすることを含む。もう1つの試みは、低pHのバッファー中でこの複合体をインキュベートすることを含む。
【0158】
簡単に説明すると、目的の複合体をCentricon 10アッセンブリィー(Millipore)を通して遠心分離し、複合体と弱く結合している低分子量の物質を除去する。大分子量の画分は、SDS-PAGEにより除去および分析するが、低分子量画分は以下に記載するようにHPLCにより分析すればよい。ATPインキュベーションプロトコールにおいては、大分子量画分中のhsp-ペプチド複合体を10mMのATPとともに室温にて30分間インキュベートする。低pHのプロトコールにおいては、酢酸またはトリフルオロ酢酸(TFA)をhsp-ペプチド複合体に加え、最終濃度10%(vol/vol)とし、この混合物を室温または沸騰水浴中、またはその間のいずれかの温度において10分間インキュベートする(Van Bleakら, 1990, Nature 348:213-216;およびLiら, 1993, EMBO Journal 12:3143-3151参照)。
【0159】
得られたサンプルを前記に従いCentricon 10アッセンブリィーを通して遠心分離する。高分子量画分および低分子量画分を回収する。残りの大分子量のhsp-ペプチド複合体をATPとともに、または低pHにおいて再度インキュベートし、残存するペプチドを除去できる。
【0160】
得られた低分子量画分をプールし、蒸発濃縮して0.1%のTFAに溶解させる。次いで、例えば0.1%TFAで平衡化させたVYDAC C18逆相カラムを用いる逆相高速液体クロマトグラフィー(HPLC)により、溶解した物質を分画する。次いで、0.1%TFA中の0〜80%アセトニトリルの線形勾配を用いて流速約0.8ml/分でカラムを展開させ、結合した物質を溶出させる。ペプチドの溶出はOD210によりモニターでき、ペプチドを含む画分を回収する。
【0161】
5.4.3. MHC-ペプチド複合体からのペプチド
in vitroで本発明の改変型hspと複合体を形成させるには、MHC分子に結合するペプチドも使用できる。潜在的免疫原性ペプチドの、MHC分子からの単離法は当技術分野において公知であり、本明細書においては詳細に記載しないが(Falkら, 1990, Nature 348:248-251; Rotzscheら, 1990, Nature 348:252-254; Elliottら, 1990, Nature 348:191-197; Falkら, 1991, Nature 351:290-296; Demotzら, 1989, Nature 343:682-684; Rotzscheら, 1990, Science 249:283-287を参照)、その開示は参照により本明細書に組み入れられる。
【0162】
簡単に説明すると、従来のイムノアフィニティー法によりMHC-ペプチド複合体を単離してよい。次いで、アセトニトリル中の約0.1%TFAの存在下でこの複合体をインキュベートすることにより、ペプチドをMHC-ペプチドから溶離させればよい。従前のごとく逆相HPLCにより溶離させたペプチドを分画し、精製すればよい。
【0163】
5.4.4. 合成ペプチド
当技術分野に公知の手動または自動のいずれかのアミノ酸配列決定技術により、MHC分子から溶離させたペプチドまたはhspのアミノ酸配列を決定してもよい。潜在的な防御ペプチドのアミノ酸配列が決定された後、従来のペプチド合成法または当技術分野に公知の他の方法を用いて、所望の量のペプチドを合成してもよい。
【0164】
Merrifield, 1963, J. Am. Chem. Soc., 85:2149に記載されたものと同様の方法を用いて、前記で単離されたような同一のアミノ酸配列を有するペプチドを、固相合成法により合成してもよい。合成中、そのC末端に連結した、成長中のポリペプチド鎖、および不溶性高分子支持体、すなわちポリスチレンビーズに、側鎖が保護されたN-α-保護アミノ酸を段階的に加える。N-α-脱保護アミノ酸のアミノ基を、ジシクロヘキシルカルボジイミドのような試薬と反応させることにより活性化させたN-α-保護アミノ酸α-カルボキシル基に結合させることによりペプチドを合成する。遊離アミノ基を、活性化されたカルボキシル基に結合することにより、ペプチド結合の形成が起こる。最も一般的に用いられるN-α-保護基としては、酸に不安定なBocおよび塩基に不安定なFmocが挙げられる。適当な化学物質、樹脂、保護基、保護されたアミノ酸および試薬の詳細については、当技術分野において十分公知であり、本明細書においては詳しく論じない(Athertonら, 1989, Solid Phase Peptide Synthesis: A Practical Approach, IRL Press, およびBodansky, 1993, Peptide Chemistry, A Practical Textbook, 第2版, Springer-Verlagを参照)。
【0165】
得られたペプチドの精製は、ゲル浸透を用いる分取HPLC、分配および/またはイオン交換クロマトグラフィーのような従来の方法を用いて行える。適当なマトリックスおよびバッファーの選択は当技術分野において十分公知であり、本明細書では詳細に論じない。
【0166】
5.4.5 抗原をコードする遺伝子配列
本発明の特定の実施形態では、タンパク質抗原をコードするヌクレオチド配列またはその一部が、抗原の生産のために宿主細胞に導入できる。いずれの抗原タンパク質をコードするヌクレオチド配列でも得ることができ、実質的にhsp遺伝子配列のクローニングおよび発現に関して記載されている同様の方法により、発現のための発現ベクターにクローン化できる。この技術は第5.1−5.1.1節および第5.2−5.2.2節に記載されており、当技術分野で十分公知である。組換え抗原性タンパク質またはその一部はタンパク質に対して適当ないずれの方法によっても精製でき、次いで、これを用いて、第5.4.6節に記載されているようにin vitroで改変型hspと複合体を形成することができる。かかる改変型hsp-抗原複合体をワクチンとして用いて、癌または感染症の治療または予防の目的で、被験体において抗原性タンパク質に対する免疫応答を刺激することができる。
【0167】
5.4.6 in vitroにおける改変型hsp-ペプチド複合体の形成
改変型hspと抗原性分子とをin vitroで非共有結合的に複合体形成する好ましいプロトコールの例を以下に示す。ペプチドタグによって固相に可逆的に結合した改変型hspを用いて、複合体形成反応の前または後でバッファーの交換、洗浄および複合体の単離を容易にするのが有利でありうる。
【0168】
複合体形成に先立ち、改変型hspはATPまたは低pHで前処理して、注目されるhspと会合する可能性のあるいずれのペプチドも除去しておく。ATP法を用いる場合、Levyら, 1991, Cell 67:265-274により記載されたようにアピラナーゼの添加によって調製物から過剰なATPを除去する。低pH法を用いる場合、pH調整剤の添加により、バッファーを中性pHに再調整する。
【0169】
抗原性分子(1μg)と前処理した改変型hsp(9μg)を混合して抗原性分子:hspモル比をおよそ5:1とする。次いで、この混合物を20mMリン酸ナトリウムpH 7.2、350mM NaCl、3mM MgCl2、および1mMフェニルメチルスルホニルフルオリド(PMSF)を含有するものなど、好適な結合バッファー中、4℃〜45℃で15分〜3時間インキュベートする。この調製物をCentricon 10アッセンブリィー(Millipore)で遠心分離して結合していないいずれのペプチドも除去する。改変型hspが固相に結合している場合は、改変型hsp-ペプチド複合体を固相から溶離させる前に、形成した改変型hsp-ペプチド複合体を、未結合のペプチドを含まないよう洗浄することができる。ペプチドと改変型hspとの会合はSDS-PAGEでアッセイすることができる。これは、内在性hsp-ペプチド複合体から解離したペプチドのMHC-ペプチド複合体から単離されたペプチドをin vitroで複合体形成する好ましい方法である。
【0170】
本発明の別の実施形態では、タンパク質など内在性の抗原性分子に対して改変型hsp70の複合体を形成するために、5〜10μgの精製hspを、100μlの容量中、20mMリン酸ナトリウムバッファー pH 7.5、0.5M NaCl、3mM MgCl2および1mM ADPで等モル量の抗原性分子とともに37℃で1時間インキュベートすることが好ましい。このインキュベーション混合物を、リン酸緩衝食塩水で1mlにさらに希釈する。
【0171】
本発明のさらに別の実施形態では、ペプチドに対し改変型gp96の複合体を形成するために、アフィニティータグによって固相に固定化された5〜10μgの改変型gp96を、20mMリン酸ナトリウムバッファー pH 7.5、0.5M NaCl、3nM MgCl2を含有するものなど、好適なバッファー中、約50℃で約10分間、等モルまたは過剰量の抗原性ペプチドとともにインキュベートすることが好ましい。例えば、この方法のために、Igタグを含む改変型gp96はタンパク質A-セファロースに固定化できる。次いで、このインキュベーション混合物を室温で約30分間さらにインキュベートする。改変型hsp-ペプチド複合体が結合した固相を数回洗浄して、結合していないいずれのペプチドも除去する。次いで、改変型hsp-ペプチド複合体を適当な方法により固相から溶離させる。
【0172】
複合体形成の後、免疫原性hsp-抗原分子複合体は場合により、例えば以下に記載される混合リンパ球標的細胞アッセイ(MLTC)を用いてin vitroでアッセイできる。一度、免疫原性複合体が単離されれば、それらは場合により、以下に論じられる好ましい投与プロトコールおよび賦形剤を用いて動物モデルでさらに特性決定できる。
【0173】
5.5 hsp-ペプチド複合体の免疫原性の決定
任意の手順において、精製改変型hsp-ペプチド複合体は、当技術分野で十分公知の混合リンパ球標的培養アッセイ(MLTC)を用い、免疫原性に関してアッセイできる。
【0174】
限定されるものではないが例示すれば、以下の手法が使用できる。簡潔に言うと、マウスに候補となる改変型hsp-ペプチド複合体を皮下注射する。その他のマウスには正常な非組換え細胞由来の他のhsp-ペプチド複合体またはアッセイについて陽性対照として働く全感染細胞のいずれかを注射する。マウスには7〜10日おいて2回注射する。最後の免疫化の10日後、脾臓を摘出してリンパ球を遊離させる。遊離したリンパ球は、次ぎに、目的の複合体を発現した死細胞を添加することにより、in vitroで再び刺激すればよい。
【0175】
例えば、10%ウシ胎児血清を含有する3mlのRPMI培地中で、8x106個の免疫脾臓細胞を、4x104個のマイトマイシンCで処理したもしくはγ線照射した(5〜10,000ラッド)病原体感染細胞(すなわち、あり得る場合としては、感染性病原体の抗原をコードする遺伝子でトランスフェクトされた細胞)、または腫瘍細胞で刺激することができる。特定の場合には、T細胞増殖因子の供給源として培地中に33%の二次混合リンパ球培養上清またはインターロイキン2(IL-2)を含んでも良い(Glasebrookら, 1980, J. Exp. Med, 151:876を参照)。免疫後、細胞傷害性T細胞の一次応答を試験するためには、脾臓細胞を刺激せずに培養すればよい。いくつかの実験では、免疫化したマウスの脾臓細胞を、抗原的に異なる細胞で再刺激して、細胞傷害性T細胞の応答の特異性を決定することもできる。
【0176】
6日後、培養物を4時間の51Cr放出アッセイで細胞傷害性に関して試験した(Palladinoら, 1987, Cancer Res. 47:5074-5079およびBlachereら, 1993, J. Immunotherapy 14:352-356を参照)。このアッセイでは、混合リンパ球培養物を標的細胞懸濁物に加えて、種々のエフェクター:標的(E:T)比とする(通常は、1:1〜40:1)。標的細胞を、200mCi51Cr/mlを含有する培地中で37℃にて1時間、1x106個の標的細胞をインキュベートすることにより予め標識する。標識後、細胞を3回洗浄する。各アッセイ点(E:T比)は3回反復して行い、適当な対照を組み入れて自然51Cr放出(アッセイにリンパ球を加えない場合)と100%放出(界面活性剤で溶解した細胞)を測定する。細胞混合物を4時間インキュベートした後、200gで5分間の遠心分離によって細胞をペレットとする。上清中に放出された51Crの量をガンマカウンターにより測定する。細胞傷害性%は、(試験サンプル中のcpm−自然に放出されたcpm)/(界面活性剤で放出された全cpm−自然に放出されたcpm)にてcpmとして測定する。
【0177】
MHCクラスIカスケードをブロックするため、K-44ハイブリドーマ細胞(抗MHCクラスIハイブリドーマ)由来の濃縮ハイブリドーマ上清を、最終濃度が12.5%となるように試験サンプルに加える(Heikeら, 1994, J. Immunotherapy 15:165-174)。
【0178】
クロム放出アッセイの代替法としては、特異的な抗原で刺激した後のin vitroにおける細胞傷害性T細胞によるサイトカインの放出を測定するELISPOTアッセイがある。サイトカイン放出は、インターロイキン-2、腫瘍壊死因子αまたはインターロイキン-γなどの特定のサイトカインに特異的な抗体により検出する(例えば、Scheibenbogenら, 1997, Int. J. Cancer, 71:932-936を参照)。このアッセイは、T細胞によって分泌されるサイトカインを捕捉する、目的のサイトカインに特異的な抗体で予め被覆したマイクロタイタープレート中で行う。被覆したウェル内で24〜48時間T細胞をインキュベートした後、細胞傷害性T細胞を除去して、サイトカイン上の異なるエピトープを認識する標識二次抗体と置き換える。十分洗浄して結合していない抗体を除去した後、有色の反応生成物を生成する酵素基質をプレートに加える。サイトカイン産生細胞の数を顕微鏡下で数える。この方法はアッセイ時間が短く、かつ、多数の細胞傷害性T細胞を必要とせず鋭敏であるという利点を有する。
【0179】
5.6. 製剤
本発明の方法によって精製した改変型hspと抗原タンパク質またはペプチドの非共有結合複合体を、癌または感染症を治療または予防するために哺乳類に投与するための医薬製剤に製剤化することができる。治療剤の投与経路を選択する場合に考慮すべき要素は薬剤溶解性および吸収部位である。本発明の改変型hsp-抗原分子複合体を、限定されるものではないが、例えば、皮下、静脈内、または筋肉内などの任意の投与経路を使用して投与してもよいが、皮内または粘膜投与が好ましい。皮内または粘膜投与の利点はそれぞれ、より低用量であることと、吸収が迅速なことである。粘膜による投与経路としては、限定されるものではないが、経口、直腸、および鼻腔内投与が挙げられる。粘膜投与用の製剤には下記のような様々な製剤が適当である。投与経路は治療経過中に変更することができる。ペプチドと天然hspの複合体の好ましい用量、投与経路および治療計画については、WO96/10411およびWO97/10001として公開されたPCT国際特許出願に記載されており、これらは参照によりその全体を本明細書に組み入れるものとする。
【0180】
好ましい実施形態においては、ある量の改変型gp96-ペプチド複合体をヒトに投与する。すなわち約10〜600μg、好ましくは10〜100μg、最も好ましくは約25μgの範囲で、約4〜6週間、週に1回、種々の投与部位の皮内に逐次投与する。
【0181】
適合する医薬担体中に製剤化された非共有結合複合体を含んでなる組成物を調製、封入し、ヒト肉腫および癌腫、例えば、繊維肉腫、粘液肉腫、脂肪肉腫、軟骨肉腫、骨肉腫、脊椎腫、血管肉腫、内皮肉腫、リンパ管肉腫、リンパ管内皮肉腫、滑液腫、内皮腫、ユーイング肉腫、平滑筋肉腫、横紋筋肉腫、結腸癌、膵臓癌、乳癌、卵巣癌、前立腺癌、扁平上皮細胞癌、基底細胞癌、腺癌、汗腺癌、皮脂腺癌、乳頭癌、乳頭腺癌、包嚢腺癌、骨髄癌、気管支癌、腎細胞癌、肝癌、胆管癌、絨毛癌、精上皮腫、胚癌、ウィルムス腫、子宮頸癌、精巣癌、肺癌、小細胞肺癌、膀胱癌、上皮癌、神経膠腫、星状細胞腫、髄芽腫、頭蓋咽喉腫、上衣腫、松果体腫、血管芽腫、聴神経腫、乏突起膠細胞腫、髄膜腫、黒色腫、神経芽腫、網膜芽腫;白血病、例えば、急性リンパ球性白血病および急性骨髄性白血病(骨髄芽球性、前骨髄性、骨髄単球性、単球性、および赤白血病);慢性白血病(慢性骨髄性(顆粒球性)白血病および慢性リンパ球性白血病);および真性赤血球増加症、リンパ腫(ホジキン病および非ホジキン病)、多発性骨髄腫、ワルデンストレームマクログロブリン血症、およびH鎖病などのような記載した腫瘍の治療用に表示する。この複合体が水溶性である場合には、適当なバッファー中、例えば、リン酸緩衝生理食塩水または他の生理学上適合する溶液中で製剤化してもよい。また、得られた複合体が水性溶媒に難溶性である場合には、Tweenなどの非イオン性界面活性剤かポリエチレングリコールとともに製剤化してもよい。よって、非共有結合複合体およびその生理学上許容される溶媒和物を、吸入または通気(口または鼻のいずれかを通して)による投与、経口、口内、非経口、直腸投与、または固形腫瘍への直接注入による投与(腫瘍の場合)用に製剤化してもよい。
【0182】
経口投与のためには、医薬製剤は液体の形態、例えば、水溶液、シロップ、または懸濁液であってもよく、あるいは使用前に水または他の適当なビヒクルを加えて再構成する医薬品として提供してもよい。かかる液体製剤は懸濁剤(例えば、ソルビトールシロップ、セルロース誘導体または水素添加食用油);乳化剤(例えば、レシチンまたはアラビアゴム);非水性ビヒクル(例えば、アーモンド油、油性エステルまたは分留植物油);および防腐剤(例えば、p-ヒドロキシ安息香酸メチルもしくはプロピルまたはソルビン酸)のような製薬上許容される添加剤を加え、従来法によって調製すればよい。医薬組成物は例えば、結合剤(例えば、プレゲル化トウモロコシデンプン、ポリビニルピロリドンもしくはヒドロキシプロピルメチルセルロース);増量剤(例えば、ラクトース、微晶性セルロースもしくはリン酸水素カルシウム);滑沢剤(例えば、ステアリン酸マグネシウム、タルクもしくはシリカ);崩壊剤(例えば、ジャガイモデンプンもしくはグリコール酸ナトリウムデンプン);または湿潤剤(例えば、ラウリル硫酸ナトリウム)などの製薬上許容される賦形剤を加え、従来法によって調製された錠剤またはカプセル剤の形態をとってもよい。錠剤は当技術分野で公知の方法によって被覆してもよい。
【0183】
経口投与用の医薬製剤は、複合体の徐放性が得られるように適宜製剤化してもよい。かかる組成物は従来法で製剤化された錠剤またはトローチ剤の形態をとってもよい。
【0184】
吸入による投与のためには、複合体は適当な噴射剤、例えば、ジクロロジフルオロメタン、トリクロロジフルオロメタン、ジクロロテトラフルオロエタン、二酸化炭素または他の適当なガスを用いて加圧容器またはネブライザーから与えられるエアロゾルスプレー剤の形で便宜に送達され得る。加圧エアロゾル剤の場合では、用量単位は、定量を送達するためのバルブを提供することによって決定してもよい。吸入器または通気器で使用するために、複合体と、ラクトースまたはデンプンなどの適当な粉末基剤との粉末混合物を含む、例えば、ゼラチンのカプセル剤およびカートリッジを製剤化してもよい。
【0185】
複合体を注射、例えばボーラス注射または点滴による非経口投与用に製剤化することができる。注射用製剤は防腐剤を加えて単位剤形(例えば、アンプル)または複数回量容器で与えてもよい。組成物は懸濁液、溶液、または油性もしくは水性ビヒクル中のエマルションような形態をとってもよく、懸濁化剤、安定化剤および/または分散化剤などの配合剤を含んでもよい。また有効成分は使用前に適当なビヒクル、例えば、発熱物質フリーの滅菌水を加えて再構成するための粉末の形態であってもよい。
【0186】
また複合体は坐剤または滞留浣腸剤のような、例えばカカオ脂または他のグリセリドなどの通常の坐剤用基剤を含む直腸組成物に製剤化してもよい。
【0187】
前記の製剤に加え、複合体をデポー製剤として製剤化してもよい。かかる長期作用製剤は植え込み(皮下もしくは筋肉内)により、または筋肉内注射により投与してもよい。よって、例えば、複合体を適当なポリマー性もしくは疎水性物質(例えば、許容される油中のエマルションとして)またはイオン交換樹脂とともに、あるいは徐々に溶ける誘導体として、例えば、徐々に溶ける塩として製剤化してもよい。疎水性薬剤の送達ビヒクルまたは担体の例としては、リポソームおよびエマルションが公知である。
【0188】
必要なら、非共有結合複合体を含む1以上の単位剤形が含まれるパックまたはディスペンサー装置で複合体を提供してもよい。例えば、そのパックはブリスターパックのように、金属またはプラスチック箔を含んでなってもよい。パックまたはディスペンサー装置には投与についての説明書が添付される。
【0189】
また本発明では、本発明の治療計画を実施するためのキットも提供する。かかるキットは1以上の容器内に、治療上または予防上有効量の非共有結合改変型hsp-ペプチド複合体を、製薬上許容される形態で含んでなる。本発明のキットのバイアル中の改変型hsp-ペプチド複合体は、製薬上許容される溶液、例えば滅菌生理食塩水、ブドウ糖溶液、またはバッファー溶液の組み合わせ、あるいは他の製薬上許容される滅菌液の形態であってよい。また、複合体を凍結乾燥するか、または乾燥させてもよい;この場合、キットにはさらに容器内に、製薬上許容される溶液(例えば生理食塩水、ブドウ糖溶液など)、好ましくは滅菌されたものが含まれていてもよく、複合体を再構成して、注射用液を形成する。
【0190】
別の実施形態では、本発明のキットにはさらに、複合体を注入するための、好ましくは滅菌包装されたニードルもしくはシリンジ、および/または包装されたアルコールパッドが含まれている。場合により臨床医または患者による改変型hsp-ペプチド複合体の投与についての説明書が含まれている。
【0191】
5.7. 癌の予防および治療
なぜ、本発明により調製される非共有結合改変型hsp-ペプチド複合体または改変型hspを発現する組換え細胞によって提供される免疫治療を癌患者において使用することが望ましいかについては多くの理由がある。第1に、癌患者が免疫抑制される場合、ならびに麻酔による手術および後の化学療法によって免疫抑制を悪化させる場合、術前期間の適当な免疫治療によって、この免疫抑制を阻害するか、または逆転させることができる。このことによって感染合併症がより少なくなり、創傷治癒が加速され得る。第2に、術後、腫瘍の大きさは最小となり、この状況においては免疫治療が最も有効であるようである。第3の理由は、手術時に腫瘍細胞が循環系に流れ出る可能性があり、このときに適用した有効な免疫治療でこれらの細胞を排除し得るということである。
【0192】
特定の実施形態において、本発明の予防的および治療的有用性は術前か、または術後のいずれかの癌患者の免疫適格の向上、および癌細胞に対する腫瘍特異的な免疫性の誘導に向けられ、その目的は癌の抑制であり、最大の臨床上の目的は全面的な癌の退縮と根絶である。
【0193】
本発明によれば、好ましい癌の治療および予防法には1以上の個体、好ましくは治療が必要な個体から癌細胞を単離すること、およびかかる細胞中に、発現可能な改変型hsp遺伝子配列を、好ましくは発現遺伝子構築物として導入することが含まれる。前記の第5.1節の方法によって、改変型hsp遺伝子配列を、発現構築物の形態の、または染色体内に組み込まれた改変型hsp遺伝子配列が組換え細胞における改変型hspの発現に好適であるように遺伝子工学的に操作する。発現遺伝子構築物を含む組換え細胞を、発現遺伝子構築物によってコードされる改変型hspが組換え宿主細胞によって発現される条件下で培養する。癌細胞のペプチドと非共有結合した改変型熱ショックタンパク質複合体が分泌され、好ましくは第5.2節に記載の方法によって培地から精製する。投与経路に依存して、第5.4節の記載に従って改変型hsp-ペプチド複合体を製剤化し、個体に自己由来のものを(例えば、原発性の癌またはその転移を治療するために)、または同種組織タイプの癌の治療が必要な他の個体に、または家族の病歴もしくは環境危険因子が理由で癌の危険性が高い個体に投与する。hsp-ペプチド複合体の治療的および予防的使用法の例もPCT公開WO96/10411(1996年4月11日付)およびWO97/10001(1997年3月20日付)に記載されている。
【0194】
例えば、前記のように調製された改変型hsp-ペプチド複合体による治療は、術後のどの時点で開始してもよい。しかしながら、患者が化学療法を受けてきた場合には、通常4週間以上の間隔をおいた後に、hsp-抗原複合体を投与して免疫系を回復させる。治療計画には生理食塩水または他の生理学上適合する溶液に溶かした改変型hsp-抗原複合体の毎週の注入が含まれる。注入経路および部位は、例えば第1の注入は左腕に皮下投与し、第2の注入は右腕に、第3の注入は左腹部に、第4の注入は右腹部に、第5の注入は左腿部に、第6の注入は右腿部に、などとそれぞれの時期によって変える。同一部位には1回以上の注入間隔をおいた後に再び行う。さらに注入物を分配し、用量の半分をそれぞれ、同日に異なる部位へ投与する。全体的に見て、最初の4〜6回の注入を1週間間隔で与え、続いて2回の注入を2週間間隔で与え、その後は1ヶ月間隔で注入する計画が与えられる。
【0195】
あるいは、改変型hsp-ペプチド複合体を分泌する組換え腫瘍細胞を、患者へ注入するためのワクチンとして用いて、腫瘍細胞または腫瘍抗原を担持する細胞に対する免疫応答を刺激することができる。改変型hsp-ペプチド複合体を安定して発現し、分泌する自己組換え腫瘍細胞が好ましい。適当な用量を決定するため、組換え細胞によって分泌された改変型hsp-ペプチド複合体の量を定量し、in vivoでのばらつきのない分泌レベルを確実に達成することによって、ワクチン接種に使用する組換え細胞数を調整する。好ましい用量とは、24時間当たり約100ngの改変型gp96を分泌可能な組換え細胞数である。患者の安全性のため、患者への注入の直前に組換え腫瘍細胞を照射することが可能である(12000rad)。照射細胞は増殖せず、約7〜10日間、改変型hsp-ペプチド複合体を分泌し続けることができ、これは免疫応答を誘導するに十分である。
【0196】
本発明の方法によって調製された非共有結合hsp-ペプチド複合体を用いて、治療または予防できる癌には、限定されるものではないが、ヒト肉腫および癌腫、例えば、繊維肉腫、粘液肉腫、脂肪肉腫、軟骨肉腫、骨肉腫、脊椎腫、血管肉腫、内皮肉腫、リンパ管肉腫、リンパ管内皮肉腫、滑液腫、内皮腫、ユーイング肉腫、平滑筋肉腫、横紋筋肉腫、結腸癌、膵臓癌、乳癌、卵巣癌、前立腺癌、扁平上皮細胞癌、基底細胞癌、腺癌、汗腺癌、皮脂腺癌、乳頭癌、乳頭腺癌、包嚢腺癌、骨髄癌、気管支癌、腎細胞癌、肝癌、胆管癌、絨毛癌、精上皮腫、胚癌、ウィルムス腫、子宮頸癌、精巣癌、肺癌、小細胞肺癌、膀胱癌、上皮癌、神経膠腫、星状細胞腫、髄芽腫、頭蓋咽喉腫、上衣腫、松果体腫、血管芽腫、聴神経腫、乏突起膠細胞腫、髄膜腫、黒色腫、神経芽腫、網膜芽腫;白血病、例えば、急性リンパ球性白血病および急性骨髄性白血病(骨髄芽球性、前骨髄性、骨髄単球性、単球性、および赤白血病);慢性白血病(慢性骨髄性(顆粒球性)白血病および慢性リンパ球性白血病);および真性赤血球増加症、リンパ腫(ホジキン病および非ホジキン病)、多発性骨髄腫、ワルデンストレームマクログロブリン血症、およびH鎖病などが挙げられる。かかる癌の具体例を、下記の節で説明する。
【0197】
特定の実施形態において、癌は転移性のものである。別の特定の実施形態では、本発明のhsp-ペプチド分子複合体を投与する前に抗癌治療(例えば、放射線化学療法)を受けたという理由によって癌を有する患者が免疫抑制されている。別の特定の実施形態においては、癌は腫瘍である。
【0198】
悪性疾患の進行における改変型hsp-ペプチド複合体を用いた免疫治療の効果を、当業者に公知の任意の方法によってモニターすることができる。該方法としては、限定されるものではないが、a)細胞性免疫の評価として遅延型過敏性;b)in vitroでの細胞溶解性Tリンパ球の活性;c)腫瘍特異的抗原、例えば癌胎児性抗原(CEA)のレベル;d)コンピューター断層撮影スキャン(CT)のような手法を用いた腫瘍の形態変化;e)高い危険性のある個体の特定の癌に対する危険性についての推定バイオマーカーレベルの変化、およびf)ソノグラムを用いた腫瘍の形態変化、の測定が挙げられる。さらに使用できるその他の手法としては、シンチグラフィーおよび内視鏡検査法が挙げられる。
【0199】
また改変型hsp-ペプチド複合体を用いた免疫治療の予防効果を、特定の癌の危険性についての推定バイオマーカーレベルを測定することにより評価してもよい。例えば、前立腺癌の危険性の高まった個体においては、血清前立腺特異的抗原(PSA)をBrawerら, 1992, J. Urol. 147: 841-845, および Catalonaら, 1993, JAMA 270: 948-958に記載される手順によって測定する;または結腸直腸癌の危険性のある個体においては、CEAを当技術分野で公知の方法によって測定する;および乳癌の危険性の高まった個体においては、エストラジオールの16-α-ヒドロキシル化をSchneiderら, 1982, Proc. Natl. Acad. Sci. USA 79: 3047-3051に記載される手順によって測定する。前記に引用される参考文献は、参照によりその全体を本明細書に組み入れるものとする。
【0200】
5.8. 感染症の予防および治療
本発明の改変型hsp-ペプチド複合体は、細胞内病原体に感染した細胞、ならびに細胞内病原体によって形質転換された細胞から調製できることも明らかとなった。例えば、免疫原性hsp-ペプチド複合体をSV40のような形質転換ウイルスで形質転換した真核細胞から単離することができる。
【0201】
本発明の好ましい実施形態において、精製された改変型hsp-ペプチドワクチンは細胞内病原体によって引き起こされるヒト疾患の治療において特に有用であると考えられる。しかしながら、本明細書に記載される原理を用いて開発されたワクチンは同様に細胞内病原体によって引き起こされる他の哺乳類、例えばウシ、ウマ、ヒツジ、ヤギ、およびブタなどの飼育動物ならびにネコおよびイヌなどの家庭内ペットの疾患の治療に有用であることがわかる。
【0202】
本明細書に記載された方法に従って、限定されるものではないが、A型肝炎、B型肝炎、C型肝炎、インフルエンザ、水痘、アデノウイルス、HSV-I、HSV-II、牛痘、ライノウイルス、エコーウイルス、ロタウイルス、呼吸シンシチアルウイルス、パピローマウイルス、パポーバウイルス、サイトメガロウイルス、エキノウイルス、アルボウイルス、ハンタウイルス、コクサッキーウイルス、ムンプスウイルス、麻疹ウイルス、風疹ウイルス、ポリオウイルス、HSV-I、およびHSV-IIなどのウイルスに感染した細胞に対する免疫応答、特に細胞傷害性T細胞応答を刺激するワクチンを調製することができる。同様にまた、限定されるものではないが、マイコバクテリウム、リケッチア、マイコプラズマ、ナイセリア、およびレジオネラなどの細胞内細菌に感染した細胞に対する細胞傷害性T細胞応答を刺激するワクチンを調製することもできる。さらに、限定されるものではないが、リーシュマニア、kokzidioa、トリパノソーマなどの細胞内原生動物に感染した細胞に対する細胞傷害性T細胞応答を刺激するワクチンを調製することもできる。さらに、限定されるものではないが、クラミジアおよびリケッチアなどの細胞内寄生体に感染した細胞に対する細胞傷害性T細胞応答を刺激するワクチンを調製することができる。
【0203】
当業者には理解されるように、本明細書に記載されたプロトコールを用いてhsp-ペプチド複合体を、改変型hspを発現するよう遺伝子操作された任意の細胞、例えば細胞内病原体に感染した組織、単離された細胞または不死化真核細胞系から単離することができる。不死化動物細胞系を改変型hsp-ペプチド複合体の供給源として用いる場合、目的の病原体に感染し得る細胞系を用いることが重要である。さらに、ワクチン受容予定者と同一種から誘導した細胞を用いることが好ましい。改変型hsp遺伝子配列の発現可能形態をこれらの細胞系に導入する手法は、前記の第5.2.2節に記載されている。
【0204】
病原体が宿主細胞の溶解を引き起こすと予期される場合、細胞を病原体に感染させる前に、発現可能な改変型hsp遺伝子配列を宿主細胞に導入することが好ましい。例えば、HSV-Iに対して有効であると考えられる、ヒトへの投与用のhsp-ペプチド複合体を調製するためには、限定されるものではないが、発現可能な改変型hsp遺伝子配列ですでにトランスフェクトされたヒトCD4+T細胞、HepG2細胞、およびU937前単球細胞などのヒト細胞中でウイルスを増殖させることができる。同様に、例えばトランスフェクトされたヒト繊維芽細胞系およびMDCK細胞でインフルエンザウイルスを増殖させてもよく、マイコバクテリウムを、例えばトランスフェクトされたシュワン細胞中で培養してもよい。
【0205】
宿主細胞を溶解する直前に、分泌された改変型hsp-ペプチド複合体を含む細胞上清を採集してもよい。
【0206】
別の実施形態では、病原体の特定の抗原決定基をコードする遺伝子が同定された場合には、抗原をコードする遺伝子をトランスフェクトし、当技術分野で公知の手法を用いて改変型hsp遺伝子配列とともに、ヒトまたは哺乳類細胞系で同時発現させてもよい。改変型hsp-ペプチド複合体を分泌するかかる組換え抗原性細胞を、患者へ注入するためのワクチンとして用いて、感染細胞または抗原を担持する細胞に対する免疫応答を刺激することができる。改変型hsp-ペプチド複合体を安定して発現し、分泌する自己組換え抗原性細胞が好ましい。
【0207】
感染症の進行における改変型hsp-ペプチド複合体を用いた免疫治療の効果を、当業者に公知の任意の方法によってモニターすることができる。
【実施例】
【0208】
6. 実施例:哺乳動物細胞における改変型gp96-Igの産生
改変型hsp、gp96-Igについては、残留ペプチド(KDEL)のないgp96およびマウスIgGlの定常領域を含んでなるキメラタンパク質をコードするヌクレオチド配列を構築して、発現構築物の改変型gp96遺伝子配列をクローニングし、発現遺伝子構築物を種々の哺乳動物細胞へトランスフェクトして調製した。改変型hsp遺伝子配列を構築する方法、組換え細胞を作製する方法、改変型gp96を精製する方法については次の節で記載する。
【0209】
6.1 改変型gp96-Ig遺伝子配列の構築
ヒトgp96のコード領域は2,412塩基の長さであり、これはアミノ末端のシグナルペプチド(21アミノ酸残基)、疎水性残基が多く含まれる潜在的な膜貫通領域、およびカルボキシル末端の小胞体(ER)残留ペプチド配列(KDEL)をコードする。このタンパク質は804アミノ酸、推定分子量約96KDを有している。このコード領域は、マウスIgGlのヒンジ、CH2、およびCH3ドメインをコードする配列が含まれるが、KDELテトラペプチドをコードする配列なしに増幅した。免疫グロブリンを基にしたタグによってELISAによる検出、プロテインA-セファロースカラムでのアフィニティークロマトグラフィーによる精製、および蛍光活性化細胞選別解析による解析が容易になる。改変型gp96の分泌形態は、改変型hsp遺伝子配列でトランスフェクトした細胞によって産生される。改変型gp96は結合したペプチドを含んでいる。
【0210】
改変型gp96遺伝子配列を構築するために、全RNAをJurkat細胞(ヒト細胞系)から酸性イソチオシアン酸グアニジニウム-フェノール-クロロホルムで抽出した。二本鎖cDNAをGeneAmp RNA PCR キット(Perkin Elmer Cetus, Norwark, CT)を用いてRNAから調製し、ヒトgp96のコード領域をExpand TM Long Template PCR System (Boehringer Mannheim, Indianapolis, IN)のPwoおよびTaqポリメラーゼを用い、PCRによって増幅した。PCRサイクル条件は:鋳型DNAの変性、94℃で2分間;次いで94℃で10秒間、55℃で30秒間および68℃で2分間を10サイクル;さらに94℃で10秒間、55℃で30秒間および68℃で2分間を25サイクルと、各サイクルに20秒間の伸長サイクル、および最終伸長工程、68℃で7分間であった。用いたPCRプライマーはXhoI部位を含む5’-ATTACTCGAGGGCCGCACGCCATGAGGG-3’(順方向プライマー-1)および2)BamHI部位を含む5’-GCCCGGATCCTTCAGCTGTAGATTCCTTTGC-3’(逆方向プライマー-2)であった。2.4kb断片である増幅産物をアガロースゲル電気泳動により精製して、TAクローニング(Invitrogen, San Diego, CA)によってpCRIIベクターへクローン化し、次いでBluescript II SK(+/-)ファージミドへ再クローン化した。
【0211】
マウスIgGlのヒンジ、CH2、およびCH3ドメインをコードする配列(Igタグ)を、鋳型としてマウスIgGl cDNA、またはプラスミド、マウスIgGl-pCRIIを用い、PCRによって得た。IgGlのヒンジ部分にある3つのシステイン残基を標準手法によってセリン残基へと変異させた。PCRサイクル条件は:変性、95℃で2分間;次いで95℃で1分間、50℃で1分間および72℃で2分間を30サイクル、および最終伸長工程、72℃で10分間であった。PCRプライマーはBamHI部位を含む5’-GCGAGGATCCGTGCCCAGGGATTCTGGTTCTAAG-3’(順方向プライマー-3)およびNotI部位を含む5’-CTAAGCGGCCGCAAGGACACTGGGATCATTTACCAGG-3’(逆方向プライマー-4)であった。PCR産物は0.68kb断片であり、これをアガロースゲル電気泳動により精製して、TAクローニングによってpCRIIへクローン化し、Bluescript II SK(+/-)ファージミドへ挿入した。
【0212】
クローン化した改変型gp96をクローン化したIgタグと連結するために、gp96をコードする配列をpBluescriptのXhoIおよびBamHI部位へ挿入し、一方IgタグをpBluescriptのBamHIおよびNotI部位へ挿入した。融合タンパク質gp96-Igの発現をin vitro共役転写/翻訳 (Promega, Madison) によって確認した。次いで改変型gp96-Ig融合体をコードする配列をXhoIおよびNotI部位で切り離し、真核生物発現ベクター、pBCMGSNeoおよびpBCMGHisへ挿入した。これはCMVプロモーター下でgp96-Igを発現し、pBCMGHisはメタロチオネインプロモーター下でgp96-Igを発現するものである。改変型gp96-Ig配列のサイズは3.08kbであり、分泌されるキメラgp96の分子量は約121kDであると推定された。
【0213】
6.2 哺乳動物細胞における改変型gp96-Igの産生
マウス繊維芽細胞系NIH3T3、マウスルイス肺癌腫細胞系LLC、マウス肥満細胞腫細胞系P815、マウスリンパ腫細胞系EL4およびその卵白アルブミントランスフェクト化細胞E.G7、マウス黒色腫細胞系B16F10、マウス繊維肉腫細胞系MC57、およびヒト小細胞肺癌腫細胞系SCLC#2およびSCLC#7を改変型gp96-Ig遺伝子配列の発現に使用した。標準的な手法を用いてこれらの細胞へ発現遺伝子構築物を導入した。エピソームで維持される、ウシパピローマウイルス(BPV)に基づく、改変型配列(gp96-Ig-pBCMGSNeoおよびgp96-Ig-pBCMGHis)を含む発現ベクターpBCMGSNeoまたはpBCMGHisを使用して、リポフェクチン(GIBCO BRL, Gaithersburg, MD)によってLLC、B16F10、MC57、SCLC#2およびSCLC#7細胞を、リン酸カルシウムによってNIH3T3細胞を、エレクトロポレーションによってEL4、E.G7およびP815細胞をトランスフェクトした。NIH3T3を10%熱失活仔ウシ血清(GIBCO BRL)を補給したIMDM培地(GIBCO BRL, Grand Island, NY)で培養した。E.G7およびEL4を10%熱失活FCS(GIBCO BRL)および50μM β-メルカプトエタノール(Bio-Rad, CA)を補給したIMDM培地で培養した。他のすべての細胞系については10%熱失活FCSを補給したIMDM培地で培養した。NIH3T3、MC57、SCLC#2およびSCLC#7を単層培養物として維持し、0.05%トリプシンと、要すればEDTA(GIBCO BRL)で短時間トリプシン処理して継代した。トランスフェクトされた細胞を1mg/mlのG418(GIBCO)または2.5mMのL-ヒスチジノール(Sigma, St. Louis, MO)で2週間選抜し、希釈およびさらなる培養によって数回に分けて増殖させた。分泌されるgp96-Ig融合タンパク質を大量産生するクローンを制限希釈によって作製した。
【0214】
非トランスフェクト化細胞系ではマウスIgGが培養上清へ分泌されなかったが、改変型gp96-Ig遺伝子配列でトランスフェクトした細胞系では改変型gp96が分泌された。
【0215】
細胞を、10%FCSを補給したAIMVまたはIMDM培地にプレーティングして106/mlとし、異なる時点で培養上清を採取した。gp96-Igの細胞内発現を解析するために、細胞を同様にプレーティングし、採取し、PBSで洗浄し、3回の凍結解凍サイクルで溶解した。溶解液を600gで10分間遠心分離し、上清をさらに13,000gで60分間遠心分離した。最終上清を用いてgp96-Igの細胞内発現を定量した。
【0216】
細胞培養基中のgp96-Ig濃度を抗原としてIgタグを用いるELISAによって求めた。標識した抗マウス抗体によってIgタグを検出した。ELISAについては、平底96ウェルプレート(Becton Dickinson Labware, Oxnard, CA)をヤギ抗マウスIgG(5μg/ml)で4℃で一晩被覆し、PBS中1%ゼラチンで37℃で1時間ブロックした。ウェルを培養上清、または対照としてマウスIgG(ICN, Costa Mesa, CA)とともに37℃で1時間インキュベートし、ペルオキシダーゼ複合アフィニピュアF(ab’)2フラグメントヤギ抗マウスIgG(H+L)で37℃で1時間展開し、次いで2,2’-アジノ-ビス(3-エチルベンズチアゾリン-6-スルホン酸)(Sigma)とともにインキュベートした。吸光度をImmunoreader SLT-Labinstruments EAR 400AT(Austria)を用い、波長405nmで求めた。その吸光度をマウスIgG標準のものと比較してgp96-Ig濃度を求めた。
【0217】
【表1】
パピローマウイルスに基づくエピソーム発現ベクターのgp96-Igでトランスフェクトしたすべてのマウスおよびヒト細胞系では融合タンパク質が分泌された(図1aおよび表1)。擬似トランスフェクト化細胞ではgp96-Igは分泌されなかった。標準化条件下では、分泌される融合タンパク質のレベルはトランスフェクト化細胞によって様々であり、5ng/ml〜3300ng/mlであった。
【0218】
gp96-Ig分泌は時間に依存して、上清中に直線的に蓄積した(図2)。トランスフェクト化細胞の溶解液においては、低く、一定の定常状態レベルで細胞内gp96-Igが検出されたが、このことはそれが細胞内には蓄積しないことを示すものである。
【0219】
6.3 哺乳動物細胞によるgp96-Igの分泌
gp96-IgトランスフェクトSCLC細胞の膜を染色するために、ヤギ抗マウスIgG-FITCまたは対照としてヤギ抗ウサギIgG-FITCを用いて、4℃で15分間染色し、Becton Dickinson FACScanフローサイトメーターにより解析した。細胞内染色のために、4%パラホルムアルデヒドで細胞を固定し、1%サポニンにより浸透性を付与し、次いでヤギ抗マウスIgG-FITC(Boehringer Mannheim, Indianapolis, IN)、ヤギ抗マウスIgG-PE(Southern Biotechnology, Birmingham, AL)、ヤギ抗ウサギIgG-FITCまたはヤギ抗ゴールデンハムスターIgG-FITC(Jackson ImmunoResearch, West Grove, PA)で4℃で15分間染色し、フローサイトメーターにより解析した。
【0220】
膜の無傷なトランスフェクト化腫瘍細胞のFACS解析では、上記背景では抗マウスIgGによっては染色されないことが明らかになり、このことは融合タンパク質のIg部分が原形質膜の外片上には展示されないことを示している。これに対し、膜に透過性を付与した際は、gp96-Igは対照ヤギ抗ウサギIgG抗体ではなく、ヤギ抗マウスIgG抗体により細胞内で検出される。gp96の膜貫通ドメインによりgp96-Igの分泌は妨害されることなく、細胞内蓄積はなされない。これらのデータはこれまでの報告に一致し、このことは膜貫通ドメインは膜におけるgp96の固定には用いられず、gp96が内在性膜タンパク質ではないことを示唆するものである。
【0221】
6.4 改変型gp96-Igの精製
gp96-IgはプロテインAカラム(Bio-Rad, Hercules, CA)でのアフィニティークロマトグラフィーにより精製した。Ig融合タンパク質を精製するため、供給源としてgp96-Ig-トランスフェクト化SCLC#7(SCLC#7-gp96-Ig)の使用済み無血清培地を用いた。gp96-Ig-トランスフェクト化NIH-3T3細胞をAIMV培地にプレーティングして106/mlとし、6〜8日後に培養上清を採取した。遠心分離および濾過によって細胞屑を取り除いた後、上清の全タンパク質を硫酸アンモニウム沈殿(55%飽和)により濃縮し、PBSで透析した。サンプルを3.5M NaCl、1.6M グリシン、pH9.0(結合バッファー)で1:2に希釈し、プロテインAカラムに充填した。このカラムを結合バッファーで十分洗浄し、結合したタンパク質を0.1Mクエン酸、pH6.5で溶出した。タンパク質を含む画分をプールし、PBSで透析した。Micro BCAタンパク質アッセイ試薬キット(Pierce, Rockford, IL)によってgp96-Ig濃度を求めた。
【0222】
4〜15% Tris-グリシンゲル(Bio-Rad)上でドデシル硫酸ナトリウムポリアクリルアミドゲル電気泳動(SDS-PAGE)を行い、クーマシーブルーで染色した。ウエスタンブロット法では、SDS-PAGE上のタンパク質をニトロセルロースにブロットし、Grp94に特異的なラットモノクロナール抗体(9G10; StressGen, Victoria, Canada)、次いでペルオキシダーゼ複合アフィニピュアF(ab’)2フラグメントヤギ抗ラットIgG(H+L)(Jackson ImmunoResearch, West Grove, PA)でプローブした。
【0223】
プロテインAクロマトグラフィーにより精製したgp96-IgをSDS-PAGEにより解析した場合、推定分子量120kDをもつ主要なバンド、およびより高分子の2つの小バンドとして移動する物質についてはこれまでに改変されていないgp96と報告されたものである(図1b)。gp96に特異的なモノクロナール抗体についてのウエスタンブロッティングにより、融合タンパク質の正体が確認された。主要なバンドのみが染色されることから、小バンドが抗体により認識されないgp96のグリコシル化変異体であることが示唆される。
【0224】
また分泌される改変型gp96-Ig分子を、還元および非還元条件下でドデシル硫酸ナトリウム−ポリアクリルアミドゲル電気泳動(SDS-PAGE)によって解析した。図1Cでは、SCLC#2より分泌された精製gp96-Ig分子、マウスIgG、ならびにCD30とIgペプチドタグとの融合タンパク質の比較を示している。gp96(96kD)およびIgタグ(25kD)を含んでなるgp96-Ig融合タンパク質は適当なサイズの範囲にあると思われる。還元および非還元画分の双方において改変型gp96-Igに相当するトリプレットはこれまでにgp96に関して記載されている。
【0225】
7. 実施例:改変型gp96-Igを産生する腫瘍細胞の発癌性
本発明によれば、改変型hsp-ペプチド複合体を分泌する組換え腫瘍細胞はワクチンとして有用である。いずれの既存の腫瘍も持たない動物における発癌性を評価することで、かかる組換え細胞のin vivo免疫原性を試験した。
【0226】
組換え腫瘍細胞をマウスに注入し、かかる組換え細胞の発癌性が同一タイプの改変されていない(すなわち、発現遺伝子構築物のない)腫瘍細胞と比較して低下するかどうかを求めた。次の結果では、試験動物における腫瘍形成においてこれらの組換え腫瘍細胞がもとの腫瘍細胞よりも活性が低いことを示している。このことから、これらの動物の免疫系によって、もとの腫瘍細胞よりも免疫原性および/または抗原性の高い細胞に対して効力のある免疫応答が高められたことが示唆される。最初に組換え腫瘍細胞を注入した試験動物に存続している腫瘍細胞を単離し、培養に戻した。これらの腫瘍細胞によって改変型hsp-Ig分子が分泌されなくなったという観察から、これらの腫瘍細胞が選択された変異体であり、実際上免疫エフェクター機構によって標的化されなかったことが示唆される。
【0227】
7.1 E.G7およびE.G7-gp96-Igの相対的発癌性の比較
C57BL/6マウスの2群にそれぞれ、示された数のE.G7およびE.G7-gp96-Ig腫瘍生細胞を皮下注射した。結果は表2に示されている。
【0228】
【表2】
E.G7を注入したほとんどすべてのマウスが腫瘍を形成した。しかしながら、マウスのほとんどがE.G7-gp96-Ig腫瘍に拒絶反応を示した。このことより、E.G7-gp96-Igから分泌される改変型gp96はE.G7の腫瘍ペプチドを保有しており、E.G7に対する腫瘍免疫性が誘導され得ることが示唆される。E.G7-gp96-Ig腫瘍を有する2匹のマウスから腫瘍を摘出し、培地へ戻した。これらの腫瘍細胞では培地へ改変型gp96-Igが分泌されなかった。改変型gp96-Igを分泌する能力を失ったこれらの腫瘍変異体がin vivoで選択されたと考えられる。
【0229】
7.2 EL4およびEL4-gp96-Igの相対的発癌性の比較
C57BL/6マウスの2群にそれぞれ、示された数のEL4およびEL4-gp96-Ig腫瘍生細胞を皮下注射した。結果は表3に示されている。
【0230】
【表3】
EL4を注入したほとんどすべてのマウスが腫瘍を形成した。しかしながら、注入したマウスのほぼ50%がEL4-gp96-Ig腫瘍に拒絶反応を示した。この観察より、EL4-gp96-Igから分泌されるgp96はEL4の腫瘍ペプチドを保有しており、EL4細胞に対する腫瘍免疫性が誘導され得ることが示唆される。EL4-gp96-Ig腫瘍を有する3匹のマウスから腫瘍を摘出し、培地へ戻した。同様に、これらの腫瘍変異体によって改変型gp96-Igが分泌されなくなったことから、それらがin vivoでの免疫エフェクター機構を逃れたことが示唆される。
【0231】
7.3 MC57およびMC57-gp96-Igの相対的発癌性の比較
C57BL/6マウスに示された数の肝臓癌細胞を皮下注射した。MC57を注入したすべてのマウスは腫瘍を持たず生残した。しかしながら、MC57-gp96-Ig細胞を注入した1匹のマウスが腫瘍を形成した。この腫瘍を摘出し、培地へ戻した。摘出した腫瘍の培養上清における改変型gp96-Ig分子のレベルは、100μg/mlまで低下した。
【0232】
【表4】
7.4 B16F10およびB16F10-gp96-Igの相対的発癌性の比較
C57BL/6マウスの2群にそれぞれ、示された数のB16F10およびB16F10-gp96-Ig腫瘍生細胞を皮下注射した。B16F10-gp96-Ig細胞を注入したすべてのマウスは腫瘍に拒絶反応を示さなかった。B16F10細胞1x104個を注入した2匹のマウスは腫瘍に拒絶反応を示した。
【0233】
【表5】
7.5 E.G7-gp96-IgおよびLLC-gp96-Igの発癌性
さらなるin vivo研究のため、2種の細胞系を選択した。E.G7はEL4リンパ腫の卵白アルブミントランスフェクト化細胞である。E.G7はその関連する免疫原性にかかわらず、C57BL/6マウスに致死的な腫瘍を形成する。E.G7のgp96-Igトランスフェクションにより、E.G7-gp96-Ig免疫がEL4結合抗原および卵白アルブミン代用抗原のいずれか一方か、またはその双方に対して免疫するかどうかの決定がなされる。E.G7に対し、それは非造血性の低免疫原性腫瘍であるため、第2の腫瘍、gp96-Igまたは卵白アルブミンをコードする配列でトランスフェクトしたLLCを用いた。両細胞系は匹敵する量のgp96-Igを分泌する(表1参照)。
【0234】
PBS 200μl中の腫瘍生細胞をマウスの脇腹へ皮下注射してin vivoでの発癌性を求めた。少なくとも2ヶ月間、週に2回、腫瘍のサイズを2次元測定した。3週間後、平均腫瘍増殖が直径10mmを超えた場合、マウスは腫瘍陽性であるとして、犠牲にした。右脇腹に106個の生きたE.G7-gp96-Ig、または対照として照射したE.G7(PBS 200μl中)を皮下注射してマウスを免疫化した。2週間間隔で2回の免疫化を行った。
【0235】
C57BL/6マウスのE.G7の発癌性は、gp96-Ig分泌により擬似トランスフェクト化または非トランスフェクト化E.G7に比べ、100倍以上低下した。腫瘍細胞を分泌する107の熱ショックタンパク質の皮下接種によっては、接種したマウスの10%にしか腫瘍が生じなかった(図3)。EL4についても同様の発癌性の低下がgp96-Ig分泌により見られた。LLCによるgp96-Ig分泌はより穏やかな低下をもたらし、発癌性が約5倍低下した(図3)。これらの結果から、腫瘍の免疫原性を可能な限り高めることで、分泌されるgp96-Igによりそれらの発癌性が低下したことが示唆される。
【0236】
8. 実施例:改変型gp96-Igを発現する細胞のワクチン接種による防御効果
改変型gp96-Igを産生する組換え腫瘍細胞が防御免疫応答を刺激する能力を示すために、まずマウスに組換え腫瘍細胞をワクチン接種し、次いで改変型gp96-Ig遺伝子構築物を含まない腫瘍細胞の注入によってチャレンジした。
【0237】
右脇腹に1x106個の生きたE.G7-gp96-Igを皮下注射してマウス群を免疫化した。2週間間隔で2回の免疫化を行った。次いで最終免疫後2週間に左脇腹に示された数のE.G7またはEL4生細胞を皮下注射してマウスにチャレンジした。E.G7細胞は卵白アルブミン遺伝子構築物でトランスフェクトしたEL4細胞である。
【0238】
【表6】
表6の結果は、組換えE.G7-gp96-Ig細胞をワクチン接種した少数のマウスにE.G7およびEL4腫瘍が発生したことが示されている。よってかかる組換え腫瘍細胞を使用することで、予防措置として特異的抗原性関連腫瘍の発生を阻止または低減させ得ることを示している。
【0239】
8.1 E.G7-gp96-IgのE.G7およびLLC腫瘍細胞に対する防御効果
さらなるin vivo研究のため、細胞系E.G7およびLLCを選択した。前記のように、右脇腹にgp96-Igを分泌する非照射E.G7細胞を皮下注射してマウスを免疫化した。2週間間隔で2回の免疫化を行った。続いてそれらを、非トランスフェクト化または擬似トランスフェクト化E.G7で、EL4で、非トランスフェクト化LLCで、またLLC-ovaでチャレンジした。LLC-ovaを作製するために、発現ベクターpAc-NEO-OVAへクローン化したニワトリ卵白アルブミン遺伝子を使用し、リポフェクチン(Gibco BRL)によってLLCをトランスフェクトした。トランスフェクト化細胞を1mg/mlのG418(GIBCO BRL)で少なくとも2週間選抜し、それらの分泌レベルをELISAにより試験した。照射E.G7で免疫化したマウスおよびワクチン接種しないマウスをワクチン接種の対照とした。
【0240】
結果は図4に示されている。非免疫化マウスまたは照射細胞をワクチン接種したマウスより10倍高いE.G7のチャレンジ量に対し、E.G7-gp96-Ig免疫化マウスは防御された。EL4でチャレンジした場合には免疫化の効力がいっそうさらに示され、対照に比べ、チャレンジ量の50倍の増加が許容された。予期されたように、E.G7-gp96-Ig免疫化では非トランスフェクト化細胞またはベクターでトランスフェクトされたLLCによるチャレンジに対する防御は起こらないが、卵白アルブミントランスフェクト化LLCを使用する場合には適度の、約3倍増大した防御が観察された。EL4チャレンジに対するE.G7-gp96-Ig免疫化マウスの強い防御はE.G7およびEL4が共有する多数の腫瘍抗原によるものであろう。これに対し、LLC-ovaで得られた弱い防御は、卵白アルブミン由来の、H2b分子によって示される単一の、または限られた数のエピトープを認識するT細胞によるものである。
【0241】
9. 実施例:接種動物におけるコンピテント免疫細胞のin vivo枯渇および失活
E.G7-gp96-Igの拒絶反応における免疫機構の問題をコンピテント免疫細胞のin vivo枯渇および失活によってさらに試験した。
【0242】
CD4+およびCD8+細胞サブセットのin vivo枯渇に使用されるモノクロナール抗体はそれぞれ、GK1.5および2.43であった。それらをハイブリドーマ上清から精製し、次いでプロテインGアフィニティークロマトグラフィーに付した。生きた106個のE.G7-gp96-Ig(PBS 200μl中)を皮下注射する2日前、または3日後に、PBS 200μl中のGK1.5または2.43 100μgを腹腔内注射によって投与した。CD4およびCD8細胞の枯渇はFACS解析によって確認した。マクロファージの機能的阻害については、PBS 200μl中のカラゲーニン(II型; Sigma) 1mgを腹腔内注射によって投与した。
【0243】
gp96-Igを分泌する106個の腫瘍細胞、平均直径約8mmに増殖する腫瘍を定着させるに十分な用量を動物に接種した。後にこれは収縮して拒絶反応を起こす。抗CD8抗体2.43で処理したマウスでは腫瘍接種の2日前か、または3日後のいずれかに腫瘍拒絶反応が阻害される(図5)。たとえ抗CD4抗体GK1.5が14日を超える期間CD4細胞を完全に枯渇させたとしても、抗CD4抗体GK1.5では注入時間にかかわらず、腫瘍拒絶反応には何の効果もなかった。同様に、in vivoでマクロファージを失活させることが知られているカラゲーニンも、腫瘍拒絶反応には何ら効果を示さなかった。
【0244】
これらのデータは、分泌されるgp96-Igと結合するペプチドがクラスI MHCによって提示され、腫瘍特異的CD8+ CTL応答を刺激して腫瘍拒絶反応を誘導するという解釈に一致している。この応答はCD4の助けを受けないと考えられ、マクロファージを必要としない。本発明者らのモデル系では、gp96-Igは腫瘍生細胞によって分泌され、マウスを免疫化して、後に腫瘍拒絶反応を引き起こす。抗腫瘍免疫が起こった兆候として腫瘍増殖が6〜8日目までに減速し始め、次の週には腫瘍に拒絶反応を起こした。腫瘍接種の2日前か、または3日後、それらの枯渇時間にかかわらず、腫瘍拒絶反応にCD4細胞およびマクロファージは必要ではなかった。このことから、CD4細胞およびマクロファージは免疫の誘導に必須ではなく、この腫瘍系のエフェクター相にも必要でないことがわかる。他方、CD8細胞は分泌性gp96-Igに対する応答についてのすべての相で決定的に重要である。
【0245】
本発明は本明細書に記載される特定の実施形態により範囲を限定されるものではない。実際、本明細書に記載されるものに加え、本発明の様々な変形や変更が、これまでの説明および添付の図面から当業者には明らかであろう。かかる変形や変更は特許請求の範囲内にある。
【0246】
本明細書では様々な公報が引用されるが、その開示内容は参照によりその総てを本明細書に組み入れられる。
【特許請求の範囲】
【請求項1】
注射可能な用量のワクチンであって、被験者に投与されたときに、抗原に対してCD4非依存性のCD8+ Tリンパ球応答の活性化を生じるのに十分な量で改変型gp96熱ショックタンパク質と該抗原との複合体を分泌するように遺伝子操作された非増殖性組換え細胞を含んでなる前記ワクチン用量。
【請求項2】
組換え細胞がヒト細胞である、請求項1に記載のワクチン用量。
【請求項3】
組換え細胞が癌細胞である、請求項1に記載のワクチン用量。
【請求項4】
癌細胞が、肺癌細胞、卵巣癌細胞、膵臓癌細胞、または膀胱癌細胞である、請求項1に記載のワクチン用量。
【請求項5】
組換え細胞が、少なくとも2種の異なる種類の癌細胞を含み、ここで該異なる種類の癌細胞はそれぞれ異なるドナー由来である、請求項3に記載のワクチン用量。
【請求項6】
前記少なくとも2種の異なる種類の癌細胞が、少なくとも2種の肺癌細胞、少なくとも2種の卵巣癌細胞、少なくとも2種の膵臓癌細胞、及び少なくとも2種の膀胱癌細胞からなる群より選択される、請求項1に記載のワクチン用量。
【請求項7】
組換え細胞が、照射されたものである、請求項1に記載のワクチン用量。
【請求項8】
細胞の用量が24時間のin vitro培養で、少なくとも100ngの該複合体を分泌する、請求項1に記載のワクチン用量。
【請求項9】
抗原が前記細胞により発現される腫瘍関連抗原である、請求項1に記載のワクチン用量。
【請求項10】
抗原がウイルス性または細菌性の抗原である、請求項1に記載のワクチン用量。
【請求項11】
抗原がHIV抗原である、請求項1に記載のワクチン用量。
【請求項12】
組換え細胞が抗原をコードするベクターを含有する、請求項1に記載のワクチン用量。
【請求項13】
前記改変型熱ショックタンパク質が、小胞体残留配列を欠失している、請求項1に記載のワクチン用量。
【請求項14】
組換え細胞が凍結され、複数のアリコートに含有されている、請求項1に記載のワクチン用量。
【請求項15】
請求項1〜14のいずれか1項記載の組換え細胞の用量を、活性成分として含む、医薬。
【請求項16】
感染症または癌を治療または予防するための医薬の製造のための、請求項1〜14のいずれか1項記載の組換え細胞の用量を含む組成物の使用。
【請求項17】
以下の工程、
(a)改変型熱ショックタンパク質と抗原との複合体を分泌するように遺伝子操作された組換え細胞の培養物を用意すること、
(b)前記培養された組換え細胞による前記複合体の分泌速度を測定すること、
(c)工程(b)において測定された速度に基づき組換え細胞の用量を用意すること、
(d)前記組換え細胞の用量を、前記改変された熱ショックタンパク質−抗原複合体を分泌する能力が保持され、かつ該細胞の増殖能力が消失するよう照射すること、および
(e)照射した組換え細胞を複数のアリコートとして凍結すること、
を含む前記方法。
【請求項1】
注射可能な用量のワクチンであって、被験者に投与されたときに、抗原に対してCD4非依存性のCD8+ Tリンパ球応答の活性化を生じるのに十分な量で改変型gp96熱ショックタンパク質と該抗原との複合体を分泌するように遺伝子操作された非増殖性組換え細胞を含んでなる前記ワクチン用量。
【請求項2】
組換え細胞がヒト細胞である、請求項1に記載のワクチン用量。
【請求項3】
組換え細胞が癌細胞である、請求項1に記載のワクチン用量。
【請求項4】
癌細胞が、肺癌細胞、卵巣癌細胞、膵臓癌細胞、または膀胱癌細胞である、請求項1に記載のワクチン用量。
【請求項5】
組換え細胞が、少なくとも2種の異なる種類の癌細胞を含み、ここで該異なる種類の癌細胞はそれぞれ異なるドナー由来である、請求項3に記載のワクチン用量。
【請求項6】
前記少なくとも2種の異なる種類の癌細胞が、少なくとも2種の肺癌細胞、少なくとも2種の卵巣癌細胞、少なくとも2種の膵臓癌細胞、及び少なくとも2種の膀胱癌細胞からなる群より選択される、請求項1に記載のワクチン用量。
【請求項7】
組換え細胞が、照射されたものである、請求項1に記載のワクチン用量。
【請求項8】
細胞の用量が24時間のin vitro培養で、少なくとも100ngの該複合体を分泌する、請求項1に記載のワクチン用量。
【請求項9】
抗原が前記細胞により発現される腫瘍関連抗原である、請求項1に記載のワクチン用量。
【請求項10】
抗原がウイルス性または細菌性の抗原である、請求項1に記載のワクチン用量。
【請求項11】
抗原がHIV抗原である、請求項1に記載のワクチン用量。
【請求項12】
組換え細胞が抗原をコードするベクターを含有する、請求項1に記載のワクチン用量。
【請求項13】
前記改変型熱ショックタンパク質が、小胞体残留配列を欠失している、請求項1に記載のワクチン用量。
【請求項14】
組換え細胞が凍結され、複数のアリコートに含有されている、請求項1に記載のワクチン用量。
【請求項15】
請求項1〜14のいずれか1項記載の組換え細胞の用量を、活性成分として含む、医薬。
【請求項16】
感染症または癌を治療または予防するための医薬の製造のための、請求項1〜14のいずれか1項記載の組換え細胞の用量を含む組成物の使用。
【請求項17】
以下の工程、
(a)改変型熱ショックタンパク質と抗原との複合体を分泌するように遺伝子操作された組換え細胞の培養物を用意すること、
(b)前記培養された組換え細胞による前記複合体の分泌速度を測定すること、
(c)工程(b)において測定された速度に基づき組換え細胞の用量を用意すること、
(d)前記組換え細胞の用量を、前記改変された熱ショックタンパク質−抗原複合体を分泌する能力が保持され、かつ該細胞の増殖能力が消失するよう照射すること、および
(e)照射した組換え細胞を複数のアリコートとして凍結すること、
を含む前記方法。
【図1−1】

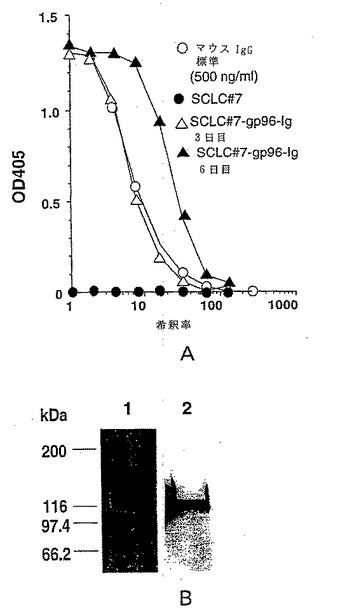
【図1−2】
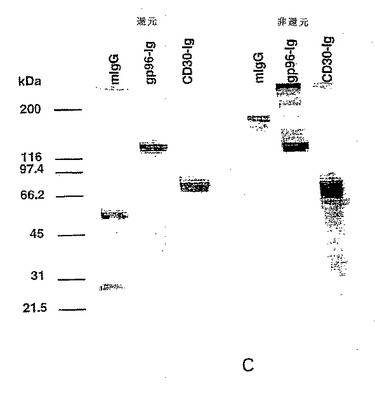
【図2−1】
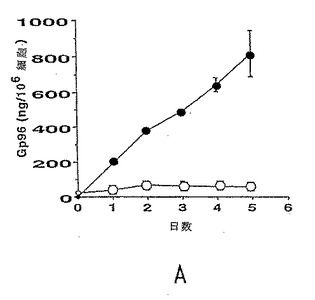
【図2−2】
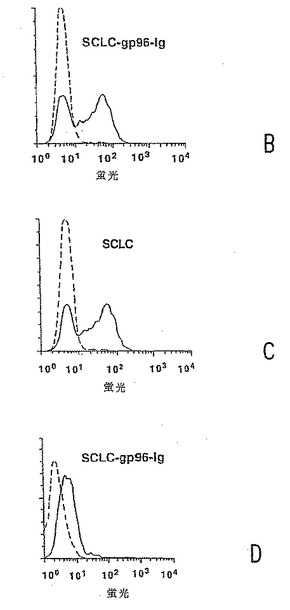
【図3−1】
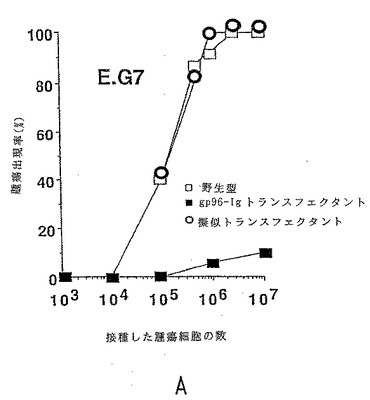
【図3−2】
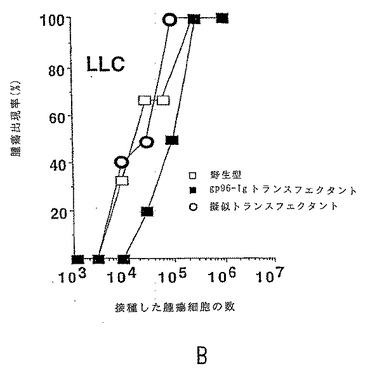
【図4−1】
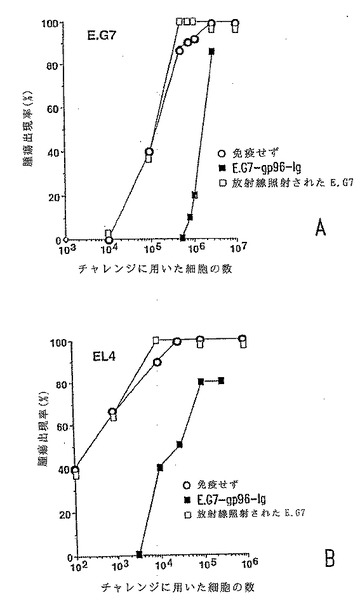
【図4−2】
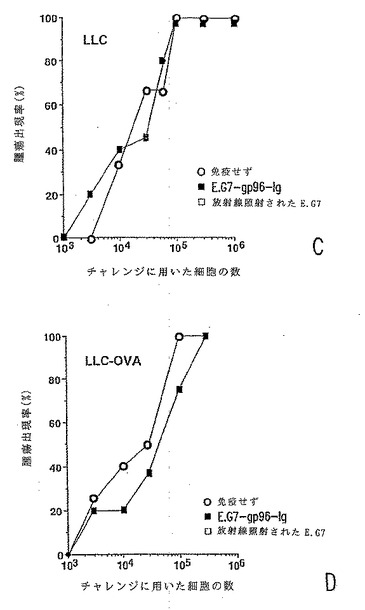
【図5−1】
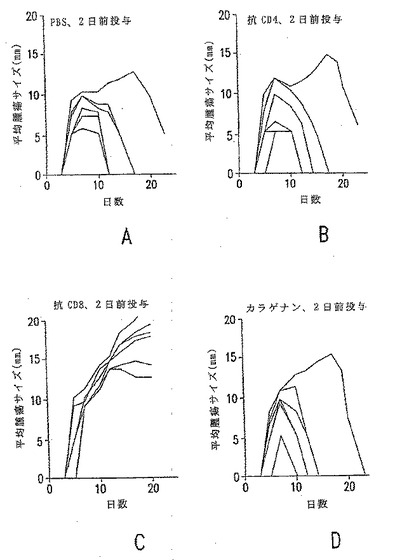
【図5−2】
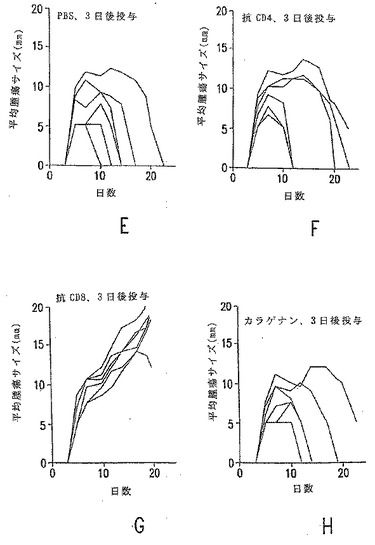
【図1−2】
【図2−1】
【図2−2】
【図3−1】
【図3−2】
【図4−1】
【図4−2】
【図5−1】
【図5−2】
【公開番号】特開2011−1378(P2011−1378A)
【公開日】平成23年1月6日(2011.1.6)
【国際特許分類】
【出願番号】特願2010−212423(P2010−212423)
【出願日】平成22年9月22日(2010.9.22)
【分割の表示】特願2000−532135(P2000−532135)の分割
【原出願日】平成11年2月19日(1999.2.19)
【出願人】(500389748)ユニバーシティー オブ マイアミ (3)
【Fターム(参考)】
【公開日】平成23年1月6日(2011.1.6)
【国際特許分類】
【出願日】平成22年9月22日(2010.9.22)
【分割の表示】特願2000−532135(P2000−532135)の分割
【原出願日】平成11年2月19日(1999.2.19)
【出願人】(500389748)ユニバーシティー オブ マイアミ (3)
【Fターム(参考)】
[ Back to top ]