
改良された抗体分子
【課題】持続的な治療効果を発揮できるよう抗体の血漿中半減期を長くすることで投与タンパク量を小さくし、高い安定性を付与することによって、長い投与間隔での皮下投与を可能にし、低コスト且つ利便性の高い第2世代の抗体医薬を提供する。
【解決手段】ヒト化抗IL-6レセプターIgG1抗体であるTOCILIZUMABの可変領域および定常領域のアミノ酸配列を改変することで、抗原中和能を増強させつつ、薬物動態を向上させることで投与頻度を少なくし持続的に治療効果を発揮し、且つ、免疫原性、安全性、物性(安定性および均一性)を改善させた、TOCILIZUMABより優れた第2世代の分子からなる医薬組成物、並びに、それらの医薬組成物の製造方法。
【解決手段】ヒト化抗IL-6レセプターIgG1抗体であるTOCILIZUMABの可変領域および定常領域のアミノ酸配列を改変することで、抗原中和能を増強させつつ、薬物動態を向上させることで投与頻度を少なくし持続的に治療効果を発揮し、且つ、免疫原性、安全性、物性(安定性および均一性)を改善させた、TOCILIZUMABより優れた第2世代の分子からなる医薬組成物、並びに、それらの医薬組成物の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、抗IL-6レセプター抗体を有効成分として含有する医薬組成物、および、その製造法等に関する。
【背景技術】
【0002】
抗体は血漿中での安定性が高く、副作用も少ないことから医薬品として注目されている。中でもIgG型の抗体医薬は多数上市されており、現在も数多くの抗体医薬が開発されている(非特許文献1、非特許文献2)。IL-6は様々な自己免疫疾患・炎症性疾患・悪性腫瘍等に関与するサイトカインであり(非特許文献3)、ヒト化抗IL-6レセプターIgG1抗体であるTOCILIZUMAB(トシリズマブ)はIL-6レセプターに特異的に結合する。TOCILIZUMABはIL-6の生物学的作用を中和することで、関節リウマチ等のIL-6が関連する疾患の治療薬として利用可能であると考えられており(特許文献1、2、3、非特許文献4)、日本においてはキャッスルマン病および関節リウマチの治療薬として承認されている(非特許文献5)。
【0003】
TOCILIZUMABのようなヒト化抗体は第1世代の抗体医薬であり、第1世代の抗体医薬を改良して薬効・利便性・コストを改善させた第2世代の抗体医薬が現在開発されている。第2世代の抗体医薬に適用可能な技術として様々な技術が開発されており、エフェクター機能、抗原結合能、薬物動態、安定性を向上させる、あるいは、免疫原性リスクを低減させる技術等が報告されている。薬効を増強させる、あるいは、投与量を低減させる方法として、IgG抗体のFc領域のアミノ酸置換により抗体依存性細胞障害活性(ADCC活性)や補体依存性細胞障害活性(CDC活性)を増強させる技術が報告されている(非特許文献6)。また、抗原結合能、抗原中和能を増強させる技術として、アフィニティーマチュレーション技術(非特許文献7)が報告されており、可変領域の相補性決定領域(CDR; complementarity determining region)などのアミノ酸に変異を導入することで抗原への結合活性を増強することが可能である。抗原結合能の増強によりin vitroの生物活性を向上させる、あるいは投与量を低減することが可能であり、さらにin vivoでの薬効を向上させることも可能である(非特許文献8)。現在、抗RSV抗体の第一世代薬であるPalivizumabより優れた効果を発揮するとされるMotavizumab(アフィニティーマチュレーションにより作製)の臨床試験が行われている(非特許文献9)。抗IL-6レセプター抗体においては、TOCILIZUMABよりアフィニティーが強いとされるアフィニティーが約0.05 nMの抗体が報告されているが(特許文献4)、0.05 nMより強いアフィニティーを有するヒト抗体あるいはヒト化抗体あるいはキメラ抗体の報告は無い。
【0004】
現在の抗体医薬が抱える問題として、投与タンパク量が非常に多いことによる高い製造コストが挙げられる。ヒト化抗IL-6レセプターIgG1抗体であるTOCILIZUMABにおいても投与量は8 mg/kg/month程度の静脈内注射が想定されている(非特許文献4)。また投与形態については、慢性的な自己免疫疾患の場合は皮下投与製剤が望ましいとされている。一般的に皮下投与製剤は高濃度製剤であることが必要であり、IgGタイプの抗体製剤の場合、安定性等の点から一般的には100 mg/mL程度の製剤が限度であると考えられる(非特許文献10)。持続的な治療効果を発揮できるよう抗体の血漿中半減期を長くすることで投与タンパク量を小さくし、高い安定性を付与することによって、長い投与間隔での皮下投与を可能にし、低コスト且つ利便性の高い第2世代の抗体医薬を提供することが可能である。
【0005】
抗体の薬物動態にはFcRnが大きく関与しており、抗体のアイソタイプ間の血漿中半減期の違いに関しては、IgG1およびIgG2が最も血漿中半減期に優れ、IgG3およびIgG4がそれより劣ることが知られている(非特許文献11)。血漿中半減期に優れるIgG1およびIgG2の抗体の血漿中半減期をさらに向上させる方法として、FcRnへの結合を増強する定常領域のアミノ酸置換が報告されている(非特許文献12、13)。また、免疫原性の観点からは定常領域よりも可変領域のアミノ酸置換により血漿中半減期をさらに向上させるほうが望ましいが(特許文献5)、これまで可変領域を改変することによりIL-6レセプター抗体の血漿中半減期を向上させた報告は無い。
【0006】
バイオ医薬品を開発するにあたってもうひとつの重要な問題は免疫原性である。一般的にマウス抗体はヒト化することによって免疫原性が低減される。ヒト化する際のテンプレートフレームワークにジャームライン配列を用いることにより、免疫原性のリスクはさらに軽減できるとされている(非特許文献14)。しかしながら、完全ヒト抗TNF抗体であるAdalimumabであっても13〜17%と高頻度で免疫原性が出現し、免疫原性が出現した患者においては治療効果の低減が見られている(非特許文献15、16)。ヒト抗体であってもCDRにT-cellエピトープが存在する可能性があり、CDR上のT-cellエピトープが免疫原性の原因となっている可能性がある。T-cellエピトープをin silicoあるいはin vitroで予測する方法が報告されているが(非特許文献17、18)、これらの方法を用いて予測されるT-cellエピトープを除去することで、免疫原性リスクを低減することが可能であると考えられる(非特許文献19)。
【0007】
ヒト化抗IL-6レセプターIgG1抗体であるTOCILIZUMABは、マウスPM1抗体をヒト化したIgG1抗体である。H鎖、L鎖それぞれNEW、REIのヒト配列をテンプレートフレームワークとして用いてCDRグラフティングが行われているが、活性保持に重要なアミノ酸として5アミノ酸がマウス配列としてフレームワークに残存している(非特許文献20)。これまでヒト化抗体であるTOCILIZUMABのフレームワークに残存するマウス配列の活性を低下させること無く完全ヒト化した報告は無い。また、TOCILIZUMABのCDR配列はマウス配列であり、Adalimumab同様、CDRにT-cellエピトープが存在する可能性があり、免疫原性のリスクは否定できない。TOCILIZUMABの臨床試験において、薬効用量である8 mg/kgにおいて抗TOCILIZUMAB抗体の出現は認められていないが、4 mg/kgおよび2 mg/kgにおいては該抗体が認められている(特許文献6)。これらのことから、TOCILIZUMABの免疫原性については改善の余地があると考えられる。しかしながら、これまでにアミノ酸置換により免疫原性リスクを改善させたTOCILIZUMABに関する報告は無い。
【0008】
TOCILIZUMABのアイソタイプはIgG1であるが、アイソタイプの違いはすなわち定常領域の配列の違いであり、定常領域の配列はエフェクター機能、薬物動態、物性等に大きく影響を与えると考えられていることから、抗体医薬の開発にとって定常領域の配列の選択は極めて重要である(非特許文献11)。近年、抗体医薬の安全性が非常に重要視されており、TGN1412のPhaseI臨床試験で見られた重大な副作用の原因の一つとして、抗体のFc部分とFcγレセプターの相互作用(エフェクター機能)が考えられている(非特許文献21)。抗原の生物学的作用を中和することが目的の抗体医薬においては、ADCC等のエフェクター機能に重要なFcγレセプターへの結合は不必要であり、副作用の点から考えるとFcγレセプターへの結合はむしろ好ましくない可能性も考えられる。Fcγレセプターへの結合を低下させる方法としては、IgG抗体のアイソタイプをIgG1からIgG2あるいはIgG4に変える方法があるが(非特許文献22)、FcγレセプターIへの結合および薬物動態の観点からはIgG4よりはIgG2が望ましいと考えられる(非特許文献11)。TOCILIZUMABのアイソタイプはIgG1であり、IL-6レセプター中和抗体であることから、ADCC等のエフェクター機能は必要とせず副作用の可能性を考えた場合、アイソタイプはIgG2が望ましい可能性も考えられる。
【0009】
一方、抗体を医薬品として開発するにあたり、そのタンパク質の物性、中でも均一性と安定性は極めて重要であり、IgG2アイソタイプは、ヒンジ領域のジスルフィド結合に由来するヘテロジェニティーが極めて多いことが報告されている(非特許文献23)。これに由来する目的物質/関連物質のヘテロジェニティーの製造間差を維持しつつ医薬品として大量に製造することは容易ではなくコスト増につながり、可能な限り単一物質であることが望まれる。さらに抗体のH鎖C末端配列のヘテロジェニティーとして、C末端アミノ酸のリジン残基の欠損、および、C末端の2アミノ酸のグリシン、リジンの欠損によるC末端カルボキシル基のアミド化が報告されており(非特許文献24)、IgG2アイソタイプの抗体を医薬品として開発する上には高い安定性を維持しつつこれらのヘテロジェニティーが低減されていることが望ましい。利便性に優れた安定な高濃度の皮下投与製剤を作製するためには、安定性が高いだけでなく、血漿中半減期に関してもTOCILIZUMABのアイソタイプであるIgG1よりも優れていることが望ましい。しかしながら、これまでにIgG2アイソタイプの定常領域の抗体に関するヘテロジェニティーが低減され、高い安定性を有し、IgG1アイソタイプの定常領域の抗体よりも優れた血漿中半減期を有する定常領域の配列の報告はない。
【0010】
なお、本発明の先行技術文献を以下に示す。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】WO 92/19759
【特許文献2】WO 96/11020
【特許文献3】WO 96/12503
【特許文献4】WO 2007/143168
【特許文献5】WO 2007/114319
【特許文献6】WO 2004/096273
【非特許文献】
【0012】
【非特許文献1】Janice M Reichert, Clark J Rosensweig, Laura B Faden & Matthew C Dewitz, Monoclonal antibody successes in the clinic, Nature Biotechnology 23, 1073 - 1078 (2005)
【非特許文献2】Pavlou AK, Belsey MJ., The therapeutic antibodies market to 2008., Eur J Pharm Biopharm. 2005 Apr;59(3):389-96.
【非特許文献3】Nishimoto N, Kishimoto T., Interleukin 6: from bench to bedside., Nat Clin Pract Rheumatol. 2006 Nov;2(11):619-26.
【非特許文献4】Maini RN, Taylor PC, Szechinski J, Pavelka K, Broll J, Balint G, Emery P, Raemen F, Petersen J, Smolen J, Thomson D, Kishimoto T; CHARISMA Study Group., Double-blind randomized controlled clinical trial of the interleukin-6 receptor antagonist, Tocilizumab, in European patients with rheumatoid arthritis who had an incomplete response to methotrexate., Arthritis Rheum. 2006 Sep;54(9):2817-29
【非特許文献5】Nishimoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S, Nakano N, Ikeda Y, Sasaki T, Nishioka K, Hara M, Taguchi H, Kimura Y, Kato Y, Asaoku H, Kumagai S, Kodama F, Nakahara H, Hagihara K, Yoshizaki K, Kishimoto T. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005 Oct 15;106(8):2627-32.
【非特許文献6】Kim SJ, Park Y, Hong HJ., Antibody engineering for the development of therapeutic antibodies., Mol Cells. 2005 Aug 31;20(1):17-29. Review.
【非特許文献7】Rothe A, Hosse RJ, Power BE. Ribosome display for improved biotherapeutic molecules. Expert Opin Biol Ther. 2006 Feb;6(2):177-87.
【非特許文献8】Rajpal A, Beyaz N, Haber L, Cappuccilli G, Yee H, Bhatt RR, Takeuchi T, Lerner RA, Crea R., A general method for greatly improving the affinity of antibodies by using combinatorial libraries., Proc Natl Acad Sci U S A. 2005 Jun 14;102(24):8466-71. Epub 2005 Jun 6.
【非特許文献9】Wu H, Pfarr DS, Johnson S, Brewah YA, Woods RM, Patel NK, White WI, Young JF, Kiener PA. Development of Motavizumab, an Ultra-potent Antibody for the Prevention of Respiratory Syncytial Virus Infection in the Upper and Lower Respiratory Tract. J Mol Biol. 2007, 368, 652-665
【非特許文献10】Shire SJ, Shahrokh Z, Liu J. Challenges in the development of high protein concentration formulations. J Pharm Sci. 2004 Jun;93(6):1390-402.
【非特許文献11】Salfeld JG. Isotype selection in antibody engineering.Nat Biotechnol. 2007 Dec;25(12):1369-72.
【非特許文献12】Hinton PR, Xiong JM, Johlfs MG, Tang MT, Keller S, Tsurushita N., An engineered human IgG1 antibody with longer serum half-life., J Immunol. 2006 Jan 1;176(1):346-56
【非特許文献13】Ghetie V, Popov S, Borvak J, Radu C, Matesoi D, Medesan C, Ober RJ, Ward ES., Increasing the serum persistence of an IgG fragment by random mutagenesis., Nat Biotechnol. 1997 Jul;15(7):637-40
【非特許文献14】Hwang WY, Almagro JC, Buss TN, Tan P, Foote J. Use of human germline genes in a CDR homology-based approach to antibody humanization.Methods. 2005 May;36(1):35-42
【非特許文献15】Bartelds GM, Wijbrandts CA, Nurmohamed MT, Stapel S, Lems WF, Aarden L, Dijkmans BA, Tak P, Wolbink GJ. Clinical response to adalimumab: The relationship with anti-adalimumab antibodies and serum adalimumab concentrations in rheumatoid arthritis. Ann Rheum Dis. 2007 Mar 9; [Epub ahead of print]
【非特許文献16】Bender NK, Heilig CE, Droll B, Wohlgemuth J, Armbruster FP, Heilig B. Immunogenicity, efficacy and adverse events of adalimumab in RA patients. Rheumatol Int. 2007 Jan;27(3):269-74.
【非特許文献17】Van Walle I, Gansemans Y, Parren PW, Stas P, Lasters I. Immunogenicity screening in protein drug development.Expert Opin Biol Ther. 2007 Mar;7(3):405-18.
【非特許文献18】Jones TD, Phillips WJ, Smith BJ, Bamford CA, Nayee PD, Baglin TP, Gaston JS, Baker MP. Identification and removal of a promiscuous CD4+ T cell epitope from the C1 domain of factor VIII. J Thromb Haemost. 2005 May;3(5):991-1000.
【非特許文献19】Chirino AJ, Ary ML, Marshall SA. Minimizing the immunogenicity of protein therapeutics. Drug Discov Today. 2004 Jan 15;9(2):82-90.
【非特許文献20】Sato K, Tsuchiya M, Saldanha J, Koishihara Y, Ohsugi Y, Kishimoto T, Bendig MM. Reshaping a human antibody to inhibit the interleukin 6-dependent tumor cell growth. Cancer Res. 1993 Feb 15;53(4):851-6.
【非特許文献21】Strand V, Kimberly R, Isaacs JD. Biologic therapies in rheumatology: lessons learned future directions. Nat Rev Drug Discov. 2007 Jan;6(1):75-92.
【非特許文献22】Gessner JE, Heiken H, Tamm A, Schmidt RE. The IgG Fc receptor family. Ann Hematol. 1998 Jun;76(6):231-48.
【非特許文献23】Dillon TM, Ricci MS, Vezina C, Flynn GC, Liu YD, Rehder DS, Plant M, Henkle B, Li Y, Deechongkit S, Varnum B, Wypych J, Balland A, Bondarenko PV. Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. J Biol Chem. 2008 Jun 6;283(23):16206-15.
【非特許文献24】Johnson KA, Paisley-Flango K, Tangarone BS, Porter TJ, Rouse JC. Cation exchange-HPLC and mass spectrometry reveal C-terminal amidation of an IgG1 heavy chain. Anal Biochem. 2007 Jan 1;360(1):75-83.
【発明の概要】
【発明が解決しようとする課題】
【0013】
本発明はこのような状況に鑑みて為されたものであり、その目的は、ヒト化抗IL-6レセプターIgG1抗体であるTOCILIZUMABの可変領域および定常領域のアミノ酸配列を改変することで、抗原中和能を増強させつつ、薬物動態を向上させることで投与頻度を少なくし持続的に治療効果を発揮し、且つ、免疫原性、安全性、物性(安定性および均一性)を改善させ、TOCILIZUMABより優れた第2世代の分子からなる医薬組成物(以下本明細書では「薬剤」あるいは「製剤」と記載する場合もある)、並びに、それらの医薬組成物の製造方法を提供することにある。
【課題を解決するための手段】
【0014】
本発明者らは、第1世代のヒト化抗IL-6レセプターIgG1抗体であるTOCILIZUMABの可変領域および定常領域のアミノ酸配列を改変することで、薬効を増強させつつ、薬物動態を向上させることで投与頻度を少なくし持続的に治療効果を発揮し、且つ、免疫原性、安全性、物性(安定性および均一性)を改善させ、TOCILIZUMABより優れた第2世代の分子の創製に向けて、鋭意研究を行った。その結果、本発明者らは、TOCILIZUMABの可変領域において、抗原への結合能(アフィニティー)を向上させるCDR変異を複数見出し、その組み合わせにより大幅にアフィニティーを向上させることに成功した。また本発明者らは、可変領域配列の等電点を低下させる改変を導入することで薬物動態を向上させることに成功した。また本発明者らは、抗原であるIL-6レセプターへの結合にpH依存性を付与することで1分子の抗体で複数回抗原を中和することを可能にし、薬物動態を向上させることに成功した。また本発明者らは、TOCILIZUMABのフレームワークに残存するマウス由来の配列を完全ヒト化し、可変領域においてin silicoで予測されたT-cellエピトープペプチドの数を低減させることで、免疫原性リスクを低減させることに成功した。さらに本発明者らは、TOCILIZUMABの定常領域において、安全性を高めるためにFcγレセプターへの結合をIgG1よりも低下させ、IgG1よりも薬物動態を改善させ、さらに安定性を低下させることなくIgG2のヒンジ領域のジスルフィド結合に由来するヘテロジェニティーおよびH鎖C末端に由来するヘテロジェニティーを低減させた新規な定常領域配列を見出すことに成功した。これらCDR領域アミノ酸配列の改変、可変領域アミノ酸配列の改変、定常領域アミノ酸配列の改変を適切に組み合わせることでTOCILIZUMABより優れた第2世代の分子の創製に成功した。
【0015】
本発明は、ヒト化抗IL-6レセプターIgG1抗体であるTOCILIZUMABの可変領域および定常領域のアミノ酸配列の改変により、より優れた抗原(IL-6レセプター)への結合能を有し、より優れた薬物動態を有し、より優れた安全性・免疫原性リスク・物性(安定性、均一性)を有するヒト化抗IL-6レセプターIgG抗体からなる医薬組成物、並びに、それらの医薬組成物の製造方法に関する。より具体的には、下記〔1〕〜〔11〕を提供するものである。
【0016】
〔1〕 以下の(a)〜(f)いずれかに記載のポリペプチド;
(a) 配列番号:1(VH4-M73のCDR1)の配列を有するCDR1、配列番号:2(VH4-M73のCDR2)の配列を有するCDR2、および配列番号:3(VH4-M73のCDR3)の配列を有するCDR3を含むポリペプチド、
(b) 配列番号:4(VH3-M73のCDR1)の配列を有するCDR1、配列番号:5(VH3-M73のCDR2)の配列を有するCDR2、および配列番号:6(VH3-M73のCDR3)の配列を有するCDR3を含むポリペプチド、
(c) 配列番号:7(VH5-M83のCDR1)の配列を有するCDR1、配列番号:8(VH5-M83のCDR2)の配列を有するCDR2、および配列番号:9(VH5-M83のCDR3)の配列を有するCDR3を含むポリペプチド、
(d) 配列番号:10(VL1のCDR1)の配列を有するCDR1、配列番号:11(VL1のCDR2)の配列を有するCDR2、および配列番号:12(VL1のCDR3)の配列を有するCDR3を含むポリペプチド、
(e) 配列番号:13(VL3のCDR1)の配列を有するCDR1、配列番号:14(VL3のCDR2)の配列を有するCDR2、および配列番号:15(VL3のCDR3)の配列を有するCDR3を含むポリペプチド、
(f) 配列番号:16(VL5のCDR1)の配列を有するCDR1、配列番号:17(VL5のCDR2)の配列を有するCDR2、および配列番号:18(VL5のCDR3)の配列を有するCDR3を含むポリペプチド。
〔2〕 以下の(a)〜(c)いずれかに記載の抗体;
(a) 配列番号:1(VH4-M73のCDR1)の配列を有するCDR1、配列番号:2(VH4-M73のCDR2)の配列を有するCDR2、および配列番号:3(VH4-M73のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに
配列番号:10(VL1のCDR1)の配列を有するCDR1、配列番号:11(VL1のCDR2)の配列を有するCDR2、および配列番号:12(VL1のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体、
(b) 配列番号:4(VH3-M73のCDR1)の配列を有するCDR1、配列番号:5(VH3-M73のCDR2)の配列を有するCDR2、および配列番号:6(VH3-M73のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに
配列番号:13(VL3のCDR1)の配列を有するCDR1、配列番号:14(VL3のCDR2)の配列を有するCDR2、および配列番号:15(VL3のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体、
(c) 配列番号:7(VH5-M83のCDR1)の配列を有するCDR1、配列番号:8(VH5-M83のCDR2)の配列を有するCDR2、および配列番号:9(VH5-M83のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに
配列番号:16(VL5のCDR1)の配列を有するCDR1、配列番号:17(VL5のCDR2)の配列を有するCDR2、および配列番号:18(VL5のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体。
〔3〕 以下の(a)〜(f)いずれかに記載の可変領域;
(a) 配列番号:19(VH4-M73の可変領域)の配列を有する重鎖可変領域、
(b) 配列番号:20(VH3-M73の可変領域)の配列を有する重鎖可変領域、
(c) 配列番号:21(VH5-M83の可変領域)の配列を有する重鎖可変領域、
(d) 配列番号:22(VL1の可変領域)の配列を有する軽鎖可変領域、
(e) 配列番号:23(VL3の可変領域)の配列を有する軽鎖可変領域、
(f) 配列番号:24(VL5の可変領域)の配列を有する軽鎖可変領域。
〔4〕 以下の(a)〜(c)いずれかに記載の抗体;
(a) 配列番号:19(VH4-M73の可変領域)の配列を有する重鎖可変領域および配列番号:22(VL1の可変領域)の配列を有する軽鎖可変領域を含む抗体、
(b) 配列番号:20(VH3-M73の可変領域)の配列を有する重鎖可変領域および配列番号:23(VL3の可変領域)の配列を有する軽鎖可変領域を含む抗体、
(c) 配列番号:21(VH5-M83の可変領域)の配列を有する重鎖可変領域および配列番号:24(VL5の可変領域)の配列を有する軽鎖可変領域を含む抗体。
〔5〕 以下の(a)〜(f)いずれかに記載の重鎖又は軽鎖;
(a) 配列番号:25(VH4-M73)の配列を有する重鎖、
(b) 配列番号:26(VH3-M73)の配列を有する重鎖、
(c) 配列番号:27(VH5-M83)の配列を有する重鎖、
(d) 配列番号:28(VL1)の配列を有する軽鎖、
(e) 配列番号:29(VL3)の配列を有する軽鎖、
(f) 配列番号:30(VL5)の配列を有する軽鎖。
〔6〕 以下の(a)〜(c)いずれかに記載の抗体;
(a) 配列番号:25(VH4-M73)の配列を有する重鎖および配列番号:28(VL1)の配列を有する軽鎖を含む抗体、
(b) 配列番号:26(VH3-M73)の配列を有する重鎖および配列番号:29(VL3)の配列を有する軽鎖を含む抗体、
(c) 配列番号:27(VH5-M83)の配列を有する重鎖および配列番号:30(VL5)の配列を有する軽鎖を含む抗体。
〔7〕 〔1〕〜〔6〕いずれかに記載のポリペプチドをコードする遺伝子。
〔8〕 〔7〕に記載の遺伝子を含むベクター。
〔9〕 〔8〕に記載のベクターを保持する宿主細胞。
〔10〕 〔9〕の宿主細胞を培養することにより、〔1〕〜〔6〕いずれかに記載のポリペプチドを製造する方法。
〔11〕 〔1〕〜〔6〕いずれかに記載のポリペプチド、または〔10〕に記載の方法によって製造されるポリペプチドを含む医薬組成物。
【発明の効果】
【0017】
本発明によって得られたヒト化抗IL-6レセプターIgG抗体は、薬効が増強し、薬物動態が向上したことで投与頻度を少なくすることができ、持続的に治療効果を発揮することができる。
【図面の簡単な説明】
【0018】
【図1】TOCILIZUMABのIL-6レセプターへの親和性を向上させる変異箇所をまとめたリストを示す図である。HCDR2のTOCILIZUMAB配列を配列番号:81、HCDR2の変異後配列(上段)を配列番号:82、HCDR2の変異後配列(下段)を配列番号:83、HCDR3のTOCILIZUMAB配列を配列番号:84、HCDR3の変異後配列(上段)を配列番号:85、HCDR3の変異後配列(下段)を配列番号:86、LCDR1のTOCILIZUMAB配列を配列番号:87、LCDR1の変異後配列(上段)を配列番号:88、LCDR1の変異後配列(下段)を配列番号:89、LCDR3のTOCILIZUMAB配列を配列番号:90、LCDR3の変異後配列(上段)を配列番号:91、LCDR3の変異後配列(下段)を配列番号:92に示す。
【図2】TOCILIZUMABとRDC-23のBaF/gp130における中和活性を示す図である。
【図3】TOCILIZUMABのIL-6レセプターへのIL-6レセプターへの結合を大きく低下させることなく可変領域の等電点を低下することできる変異箇所をまとめたリストを示す図である。図中の星印はヒト配列にするために等電点には影響しないが変異した箇所である。HFR1のTOCILIZUMAB配列を配列番号:93、HFR1の変異後配列を配列番号:94、HCDR1のTOCILIZUMAB配列を配列番号:95、HCDR1の変異後配列を配列番号:96、HFR2のTOCILIZUMAB配列を配列番号:97、HFR2の変異後配列を配列番号:98、HCDR2のTOCILIZUMAB配列を配列番号:81、HCDR2の変異後配列を配列番号:99、HFR4のTOCILIZUMAB配列を配列番号:100、HFR4の変異後配列を配列番号:101、LFR1のTOCILIZUMAB配列を配列番号:102、LFR1の変異後配列を配列番号:103、LCDR1のTOCILIZUMAB配列を配列番号:87、LCDR1の変異後配列を配列番号:104、LFR2のTOCILIZUMAB配列を配列番号:105、LFR2の変異後配列を配列番号:106、LCDR2のTOCILIZUMAB配列を配列番号:107、LCDR2の変異後配列を配列番号:108、109、LFR3のTOCILIZUMAB配列を配列番号:110、LFR3の変異後配列を配列番号:111、LFR4のTOCILIZUMAB配列を配列番号:112、LFR4の変異後配列を配列番号:113に示す。
【図4】TOCILIZUMABとH53/L28のBaF/gp130における中和活性を示す図である。
【図5】TOCILIZUMABとH53/L28をマウスに静脈内投与後の血漿中濃度推移を示したグラフである。
【図6】TOCILIZUMABとH53/L28をマウスに皮下投与後の血漿中濃度推移を示したグラフである。
【図7】IgG分子がエンドソーム内で膜型抗原から解離することで再度新たな抗原に結合することを示す模式図である。
【図8】TOCILIZUMABのIL-6レセプターへの結合にpH依存性(pH7.4で結合し、pH5.8で解離する)を付与することのできる変異箇所をまとめたリストを示す図である。HFR1のTOCILIZUMAB配列を配列番号:93、HFR1の変異後配列を配列番号:114、HCDR1のTOCILIZUMAB配列を配列番号:95、HCDR1の変異後配列を配列番号:115、LCDR1のTOCILIZUMAB配列を配列番号:87、LCDR1の変異後配列を配列番号:116、LCDR2のTOCILIZUMAB配列を配列番号:107、LCDR2の変異後配列を配列番号:117に示す。
【図9】TOCILIZUMABとH3pI/L73のBaF/gp130における中和活性を示す図である。
【図10】TOCILIZUMABとH3pI/L73をカニクイザルに静脈内投与後の血漿中濃度推移を示したグラフである。
【図11】TOCILIZUMABとH3pI/L73をヒトIL-6レセプタートランスジェニックマウスに静脈内投与後の血漿中濃度推移を示したグラフである。
【図12】TOCILIZUMAB、TOCILIZUMABΔK、およびTOCILIZUMABΔGKを陽イオン交換クロマトグラフィーによりC末端に由来するヘテロジェニティーの評価を行った結果を示した図である。
【図13】TOCILIZUMAB-IgG1、TOCILIZUMAB-IgG2、およびTOCILIZUMAB-SKSCを陽イオン交換クロマトグラフィーによりジスルフィド結合に由来するヘテロジェニティーの評価を行った結果を示した図である。
【図14】TOCILIZUMAB-IgG1、TOCILIZUMAB-IgG2、およびTOCILIZUMAB-SKSCを示差走査型熱量測定(DSC)により測定した変性曲線および各FabドメインのTm値を示した図である。
【図15】TOCILIZUMAB-IgG1、TOCILIZUMAB-M44、TOCILIZUMAB-M58およびTOCILIZUMAB-M73をヒトFcRnトランスジェニックマウスに静脈内投与後の血漿中濃度推移を示したグラフである。
【図16】TOCILIZUMAB、コントロールおよびFv5-M83のBaF/gp130における中和活性を示す図である。
【図17】TOCILIZUMAB、Fv3-M73およびFv4-M73のBaF/gp130における中和活性を示す図である。
【図18】TOCILIZUMAB、コントロール、Fv3-M73、Fv4-M73、およびFv5-M83をカニクイザルに静脈内投与後の血漿中濃度推移を示したグラフである。
【図19】TOCILIZUMAB、コントロール、Fv3-M73、Fv4-M73、およびFv5-M83をカニクイザルに静脈内投与後のCRP濃度推移を示したグラフである。
【図20】TOCILIZUMAB、コントロール、Fv3-M73、Fv4-M73、およびFv5-M83をカニクイザルに静脈内投与後の非結合型可溶型IL-6レセプター率の推移を示したグラフである。
【図21】TOCILIZUMABとFv4-M73によるヒトRA患者由来滑膜細胞からのMCP-1産生抑制作用を示す図である。
【図22】TOCILIZUMABとFv4-M73によるヒトRA患者由来滑膜細胞からのVEGF産生抑制作用を示す図である。
【発明を実施するための形態】
【0019】
本発明は以下の(a)〜(f)いずれかに記載のポリペプチドを提供する。
(a) 配列番号:1(VH4-M73のCDR1)の配列を有するCDR1、配列番号:2(VH4-M73のCDR2)の配列を有するCDR2、および配列番号:3(VH4-M73のCDR3)の配列を有するCDR3を含むポリペプチド、
(b) 配列番号:4(VH3-M73のCDR1)の配列を有するCDR1、配列番号:5(VH3-M73のCDR2)の配列を有するCDR2、および配列番号:6(VH3-M73のCDR3)の配列を有するCDR3を含むポリペプチド、
(c) 配列番号:7(VH5-M83のCDR1)の配列を有するCDR1、配列番号:8(VH5-M83のCDR2)の配列を有するCDR2、および配列番号:9(VH5-M83のCDR3)の配列を有するCDR3を含むポリペプチド、
(d) 配列番号:10(VL1のCDR1)の配列を有するCDR1、配列番号:11(VL1のCDR2)の配列を有するCDR2、および配列番号:12(VL1のCDR3)の配列を有するCDR3を含むポリペプチド、
(e) 配列番号:13(VL3のCDR1)の配列を有するCDR1、配列番号:14(VL3のCDR2)の配列を有するCDR2、および配列番号:15(VL3のCDR3)の配列を有するCDR3を含むポリペプチド、
(f) 配列番号:16(VL5のCDR1)の配列を有するCDR1、配列番号:17(VL5のCDR2)の配列を有するCDR2、および配列番号:18(VL5のCDR3)の配列を有するCDR3を含むポリペプチド。
【0020】
本発明においてポリペプチドは特に限定されないが、ヒトIL-6レセプターへの結合活性を有する抗原結合物質であることが好ましい。抗原結合物質の好ましい例として、抗体の重鎖可変領域(VH)、抗体の軽鎖可変領域(VL)、抗体の重鎖、抗体の軽鎖、抗体などを挙げることができる。
【0021】
上述の(a)〜(f)のポリペプチドにおいて、(a)〜(c)のポリペプチドの好ましい例として抗体の重鎖可変領域を挙げることができ、(d)〜(f)のポリペプチドの好ましい例として抗体の軽鎖可変領域を挙げることができる。
【0022】
これらの可変領域は抗ヒトIL-6レセプター抗体の一部として使用され得る。これらの可変領域が用いられた抗ヒトIL-6レセプター抗体は優れた結合活性、優れた薬物動態、優れた安全性・免疫原性、および/または優れた物性を有する。本発明において、優れた薬物動態、あるいは、薬物動態の向上とは、抗体を生体内に投与した際の血漿中濃度推移から算出される薬物動態パラメーターの一つである"クリアランス(Clearance、CL)"の減少、"濃度曲線下面積(Area Under Curve、AUC)"の増大、"平均滞留時間(Mean Residence Time)"の増大、"血漿中半減期(t1/2)"の増大、のいずれかを意味する。本発明において、優れた物性あるいは物性の向上とは、特に限定されないが、安定性の向上、ヘテロジェニティーの低減などを意味する。
【0023】
CDRと連結されるヒト抗体のフレームワーク領域(framework region;FR)は、CDRが良好な抗原結合部位を形成するものが選択される。本発明の可変領域に用いられるFRは特に限定されず、如何なるFRが用いられていてもよいが、ヒト由来のFRが用いられることが好ましい。ヒト由来のFRは天然配列を有するFRが用いられてもよいし、必要に応じ、CDRが適切な抗原結合部位を形成するように、天然配列を有するフレームワーク領域の1又は複数のアミノ酸を置換、欠失、付加および/または挿入等してもよい。たとえば、アミノ酸を置換したFRを用いた抗体の抗原への結合活性を測定し評価することによって所望の性質を有する変異FR配列が選択できる(Sato, K. et al., Cancer Res.(1993)53, 851-856)。
【0024】
又、上述のCDR配列において1又は複数のアミノ酸を置換、欠失、付加および/または挿入等してもよい。1又は複数のアミノ酸の置換、欠失、付加および/または挿入後のCDR配列は、結合活性、中和活性、安定性、免疫原性および/または薬物動態において改変前のCDR配列と同等の活性を有していることが好ましい。置換、欠失、付加および/または挿入されるアミノ酸の数は特に限定されないが、好ましくは1つのCDRにつき3アミノ酸以内、さらに好ましくは2アミノ酸以内、より好ましくは1アミノ酸である。
【0025】
1又は複数のアミノ酸残基を目的の他のアミノ酸に置換する方法としては、例えば、部位特異的変異誘発法(Hashimoto-Gotoh, T, Mizuno, T, Ogasahara, Y, and Nakagawa, M. (1995) An oligodeoxyribonucleotide-directed dual amber method for site-directed mutagenesis. Gene 152, 271-275、Zoller, MJ, and Smith, M.(1983) Oligonucleotide-directed mutagenesis of DNA fragments cloned into M13 vectors.Methods Enzymol. 100, 468-500、Kramer,W, Drutsa,V, Jansen,HW, Kramer,B, Pflugfelder,M, and Fritz,HJ(1984) The gapped duplex DNA approach to oligonucleotide-directed mutation construction. Nucleic Acids Res. 12, 9441-9456、Kramer W, and Fritz HJ(1987) Oligonucleotide-directed construction of mutations via gapped duplex DNA Methods. Enzymol. 154, 350-367、Kunkel,TA(1985) Rapid and efficient site-specific mutagenesis without phenotypic selection.Proc Natl Acad Sci U S A. 82, 488-492)が挙げられる。該方法を用いて、抗体の所望のアミノ酸を目的の他のアミノ酸に置換することができる。又、フレームワークシャッフリング(Mol Immunol. 2007 Apr;44(11):3049-60)およびCDR repair(US2006/0122377)等のライブラリー技術を用いることにより、フレームワークおよびCDRにおけるアミノ酸を適切な他のアミノ酸置換することも可能である。
【0026】
さらに、本発明は以下の(a)〜(c)いずれかに記載の抗体を提供する。
(a) 配列番号:1(VH4-M73のCDR1)の配列を有するCDR1、配列番号:2(VH4-M73のCDR2)の配列を有するCDR2、および配列番号:3(VH4-M73のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに配列番号:10(VL1のCDR1)の配列を有するCDR1、配列番号:11(VL1のCDR2)の配列を有するCDR2、および配列番号:12(VL1のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体、
(b) 配列番号:4(VH3-M73のCDR1)の配列を有するCDR1、配列番号:5(VH3-M73のCDR2)の配列を有するCDR2、および配列番号:6(VH3-M73のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに配列番号:13(VL3のCDR1)の配列を有するCDR1、配列番号:14(VL3のCDR2)の配列を有するCDR2、および配列番号:15(VL3のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体、
(c) 配列番号:7(VH5-M83のCDR1)の配列を有するCDR1、配列番号:8(VH5-M83のCDR2)の配列を有するCDR2、および配列番号:9(VH5-M83のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに配列番号:16(VL5のCDR1)の配列を有するCDR1、配列番号:17(VL5のCDR2)の配列を有するCDR2、および配列番号:18(VL5のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体。
【0027】
上述の抗体は優れた結合活性、優れた薬物動態、優れた安全性・免疫原性、および/または優れた物性を有する抗ヒトIL-6レセプター抗体として使用され得る。
【0028】
本発明のCDRと連結されるヒト抗体のフレームワーク領域は、CDRが良好な抗原結合部位を形成するものが選択される。本発明の可変領域に用いられるFRは特に限定されず、如何なるFRが用いられていてもよいが、ヒト由来のFRが用いられることが好ましい。ヒト由来のFRは天然配列を有するFRが用いられてもよいし、必要に応じ、CDRが適切な抗原結合部位を形成するように、天然配列を有するフレームワーク領域の1又は複数のアミノ酸を置換、欠失、付加および/または挿入等してもよい。たとえば、アミノ酸を置換したFRを用いた抗体の抗原への結合活性を測定し評価することによって所望の性質を有する変異FR配列が選択できる(Sato, K. et al., Cancer Res.(1993)53, 851-856)。
【0029】
又、本発明の抗体で用いられる定常領域は特に限定されず、如何なる定常領域が用いられてもよい。本発明の抗体で用いられる定常領域の好ましい例としては、ヒト由来の定常領域(IgG1、IgG2、IgG3、IgG4、Cκ、Cλ由来の定常領域など)を挙げることができる。ヒト由来の定常領域は1又は複数のアミノ酸が置換、欠失、付加および/または挿入されていてもよい。例えば、ヒト由来の定常領域の好ましい例として、重鎖定常領域の場合には配列番号:31(VH4-M73の定常領域)のアミノ酸配列を有する定常領域、配列番号:32(VH3-M73の定常領域)のアミノ酸配列を有する定常領域、配列番号:33(VH5-M83の定常領域)のアミノ酸配列を有する定常領域を挙げることができ、軽鎖定常領域の場合には配列番号:34(VL1)のアミノ酸配列を有する定常領域、配列番号:35(VL3)のアミノ酸配列を有する定常領域、配列番号:36(VL5)のアミノ酸配列を有する定常領域を挙げることができる。
【0030】
又、上述のCDR配列において1又は複数のアミノ酸を置換、欠失、付加および/または挿入等してもよい。1又は複数のアミノ酸の置換、欠失、付加および/または挿入後のCDR配列は、結合活性、中和活性、安定性、免疫原性および/または薬物動態において改変前のCDR配列と同等の活性を有していることが好ましい。置換、欠失、付加および/または挿入されるアミノ酸の数は特に限定されないが、好ましくは1つのCDRにつき3アミノ酸以内、さらに好ましくは2アミノ酸以内、より好ましくは1アミノ酸である。
【0031】
アミノ酸の置換、欠失、付加および/または挿入などは上述の方法により行うことも可能である。
【0032】
さらに本発明は以下の(a)〜(f)いずれかに記載の可変領域を提供する。
(a) 配列番号:19(VH4-M73の可変領域)の配列を有する重鎖可変領域、
(b) 配列番号:20(VH3-M73の可変領域)の配列を有する重鎖可変領域、
(c) 配列番号:21(VH5-M83の可変領域)の配列を有する重鎖可変領域、
(d) 配列番号:22(VL1の可変領域)の配列を有する軽鎖可変領域、
(e) 配列番号:23(VL3の可変領域)の配列を有する軽鎖可変領域、
(f) 配列番号:24(VL5の可変領域)の配列を有する軽鎖可変領域。
【0033】
上述の可変領域は抗ヒトIL-6レセプター抗体の一部として使用され得る。これらの可変領域が用いられた抗ヒトIL-6レセプター抗体は優れた結合活性、優れた薬物動態、優れた安全性・免疫原性、および/または優れた物性を有する。
【0034】
上述の可変領域は1又は複数(例えば5アミノ酸以内、好ましくは3アミノ酸以内)のアミノ酸が置換、欠失、付加および/または挿入していてもよい。1又は複数のアミノ酸残基を目的の他のアミノ酸に置換する方法としては、例えば、上述の方法を挙げることができる。
【0035】
また本発明は、上述の可変領域を含むポリペプチドを提供する。
【0036】
さらに本発明は以下の(a)〜(c)いずれかに記載の抗体を提供する。
(a) 配列番号:19(VH4-M73の可変領域)の配列を有する重鎖可変領域および配列番号:22(VL1の可変領域)の配列を有する軽鎖可変領域を含む抗体、
(b) 配列番号:20(VH3-M73の可変領域)の配列を有する重鎖可変領域および配列番号:23(VL3の可変領域)の配列を有する軽鎖可変領域を含む抗体、
(c) 配列番号:21(VH5-M83の可変領域)の配列を有する重鎖可変領域および配列番号:24(VL5の可変領域)の配列を有する軽鎖可変領域を含む抗体。
【0037】
上述の可変領域は抗ヒトIL-6レセプター抗体の一部として使用され得る。これらの可変領域が用いられた抗ヒトIL-6レセプター抗体は優れた結合活性、優れた薬物動態、優れた安全性・免疫原性、および/または優れた物性を有する。
【0038】
上述の可変領域は1又は複数(例えば5アミノ酸以内、好ましくは3アミノ酸以内)のアミノ酸が置換、欠失、付加および/または挿入していてもよい。1又は複数のアミノ酸残基を目的の他のアミノ酸に置換する方法としては、例えば、上述の方法を挙げることができる。
【0039】
又、本発明の抗体で用いられる定常領域は特に限定されず、如何なる定常領域が用いられてもよい。本発明の抗体で用いられる定常領域の好ましい例としては、ヒト由来の定常領域(IgG1、IgG2、IgG3、IgG4、κ鎖、λ鎖由来の定常領域など)を挙げることができる。ヒト由来の定常領域は1又は複数のアミノ酸が置換、欠失、付加および/または挿入されていてもよい。例えば、ヒト由来の定常領域の好ましい例として、重鎖定常領域の場合には配列番号:31(VH4-M73の定常領域)のアミノ酸配列を有する定常領域、配列番号:32(VH3-M73の定常領域)のアミノ酸配列を有する定常領域、配列番号:33(VH5-M83の定常領域)のアミノ酸配列を有する定常領域を挙げることができ、軽鎖定常領域の場合には配列番号:34(VL1)のアミノ酸配列を有する定常領域、配列番号:35(VL3)のアミノ酸配列を有する定常領域、配列番号:36(VL5)のアミノ酸配列を有する定常領域を挙げることができる。
【0040】
さらに本発明は以下の(a)〜(f)いずれかに記載の重鎖又は軽鎖を提供する。
(a) 配列番号:25(VH4-M73)の配列を有する重鎖、
(b) 配列番号:26(VH3-M73)の配列を有する重鎖、
(c) 配列番号:27(VH5-M83)の配列を有する重鎖、
(d) 配列番号:28(VL1)の配列を有する軽鎖、
(e) 配列番号:29(VL3)の配列を有する軽鎖、
(f) 配列番号:30(VL5)の配列を有する軽鎖。
【0041】
上述の重鎖又は軽鎖は抗ヒトIL-6レセプター抗体の一部として使用され得る。これらの重鎖又は軽鎖が用いられた抗ヒトIL-6レセプター抗体は、優れた結合活性、優れた薬物動態、優れた安全性・免疫原性、および/または優れた物性を有する。
【0042】
上述の重鎖又は軽鎖は、1又は複数(例えば10アミノ酸以内、好ましくは5アミノ酸以内、さらに好ましくは3アミノ酸以内)のアミノ酸が置換、欠失、付加および/または挿入していてもよい。1又は複数のアミノ酸残基を目的の他のアミノ酸に置換する方法としては、例えば、上述の方法を用いることが可能である。
【0043】
1又は複数のアミノ酸の置換、欠失、付加および/または挿入は可変領域において行われてもよいし、定常領域において行われてもよいし、又は可変領域と定常領域の両方において行われてもよい。
【0044】
さらに本発明は以下の(a)〜(c)いずれかに記載の抗体を提供する。
(a) 配列番号:25(VH4-M73)の配列を有する重鎖および配列番号:28(VL1)の配列を有する軽鎖を含む抗体、
(b) 配列番号:26(VH3-M73)の配列を有する重鎖および配列番号:29(VL3)の配列を有する軽鎖を含む抗体、
(c) 配列番号:27(VH5-M83)の配列を有する重鎖および配列番号:30(VL5)の配列を有する軽鎖を含む抗体。
【0045】
上述の抗体は優れた結合活性、優れた薬物動態、優れた安全性・免疫原性、および/または優れた物性を有する抗ヒトIL-6レセプター抗体である。
【0046】
上述の抗体は、1又は複数(例えば20アミノ酸以内、好ましくは10アミノ酸以内、さらに好ましくは5アミノ酸以内)のアミノ酸が置換、欠失、付加および/または挿入していてもよい。1又は複数のアミノ酸残基を目的の他のアミノ酸に置換する方法としては、例えば、上述の方法をあげることができる。
【0047】
1又は複数のアミノ酸の置換、欠失、付加および/または挿入は可変領域において行われてもよいし、定常領域において行われてもよいし、又は可変領域と定常領域の両方において行われてもよい。
【0048】
本発明の抗体は、ヒト化(humanized)抗体であることが好ましい。
【0049】
ヒト化抗体は、再構成(reshaped)ヒト抗体とも称され、これは、ヒト以外の哺乳動物由来のCDRをヒト抗体のCDRへ移植したものであり、その一般的な遺伝子組換え手法も知られている(欧州特許出願公開番号EP 125023号公報、WO 96/02576 号公報参照)。
【0050】
具体的には、例えば目的のCDRと目的のフレームワーク領域(FR)とを連結するように設計したDNA配列を、CDRおよびFR両方の末端領域にオーバーラップする部分を有するように作製した数個のオリゴヌクレオチドをプライマーとして用いてPCR法により合成する(WO98/13388号公報に記載の方法を参照)。得られたDNAをヒト抗体定常領域もしくはヒト抗体定常領域改変体をコードするDNAと連結し、次いで発現ベクターに組み込んで、これを宿主に導入し産生させることにより得られる(欧州特許出願公開番号EP 239400、国際特許出願公開番号WO 96/02576参照)。
【0051】
CDRと連結されるヒト抗体のフレームワーク領域は、CDRが良好な抗原結合部位を形成するものが選択される。必要に応じ、抗体の可変領域におけるフレームワーク領域のアミノ酸を置換、欠失、付加および/または挿入等してもよい。
【0052】
ヒト化抗体の定常領域には、ヒト抗体定常領域又はヒト抗体定常領域において1又は複数のアミノ酸が置換、欠失、付加および/または挿入したヒト抗体定常領域改変体を用いることができる。
【0053】
例えばH鎖では、Cγ1、Cγ2、Cγ3、Cγ4、Cμ、Cδ、Cα1、Cα2、Cεを、L鎖ではCκ、Cλを使用することができる。Cκのアミノ酸配列を配列番号:38に、当該アミノ酸配列をコードする塩基配列を配列番号:37に示す。Cγ1のアミノ酸配列を配列番号:40に、当該アミノ酸配列をコードする塩基配列を配列番号:39に示す。Cγ2のアミノ酸配列を配列番号:42に、当該アミノ酸配列をコードする塩基配列を配列番号:41に示す。Cγ4のアミノ酸配列を配列番号:44に、当該アミノ酸配列をコードする塩基配列を配列番号:43に示す。
【0054】
また、抗体またはその産生の安定性を改善するために、ヒト抗体C領域を修飾してもよい。ヒト化の際に用いられるヒト抗体は、IgG、IgM、IgA、IgE、IgDなど如何なるアイソタイプのヒト抗体でもよいが、本発明においてはIgGを用いることが好ましい。IgGとしては、IgG1、IgG2、IgG3、IgG4などを用いることが可能である。
【0055】
なお、ヒト化抗体を作製した後に、可変領域(例えば、CDR、FR)や定常領域中のアミノ酸を他のアミノ酸で置換、欠失、付加および/または挿入等してもよく、本発明のヒト化抗体には、そのようなアミノ酸置換等されたヒト化抗体も含まれる。
【0056】
本発明の抗体には、IL-6レセプターへの結合活性および/または中和活性を有する限り、IgGに代表される二価抗体だけでなく、一価抗体、若しくはIgMに代表される多価抗体も含まれる。本発明の多価抗体には、全て同じ抗原結合部位を有する多価抗体、または、一部もしくは全て異なる抗原結合部位を有する多価抗体が含まれる。本発明の抗体は、抗体の全長分子に限らず、IL-6レセプタータンパク質に結合する限り、低分子化抗体またはその修飾物であってもよい。
【0057】
低分子化抗体は、全長抗体(whole antibody、例えばwhole IgG等)の一部分が欠損している抗体断片を含む抗体であり、IL-6レセプターへの結合活性および/または中和活性を有する限り特に限定されない。本発明において低分子化抗体は、全長抗体の一部分を含む限り特に限定されないが、VH又はVLを含んでいることが好ましく、特に好ましくはVHとVLの両方を含む低分子化抗体である。又、本発明の低分子化抗体の他の好ましい例として、抗体のCDRを含む低分子化抗体を挙げることができる。低分子化抗体に含まれるCDRは抗体の6つのCDR全てが含まれいてもよいし、一部のCDRが含まれていてもよい。
【0058】
本発明における低分子化抗体は、全長抗体よりも分子量が小さくなることが好ましいが、例えば、ダイマー、トリマー、テトラマーなどの多量体を形成すること等もあり、全長抗体よりも分子量が大きくなることもある。
【0059】
抗体断片の具体例としては、例えば、Fab、Fab'、F(ab')2、Fvなどを挙げることができる。また、低分子化抗体の具体例としては、例えば、Fab、Fab'、F(ab')2、Fv、scFv(single chain Fv)、Diabody、sc(Fv)2(single chain (Fv)2)などを挙げることができる。これら抗体の多量体(例えば、ダイマー、トリマー、テトラマー、ポリマー)も、本発明の低分子化抗体に含まれる。
【0060】
抗体断片は、例えば、抗体を酵素で処理して抗体断片を生成させることによって得ることができる。抗体断片を生成する酵素として、例えばパパイン、ペプシン、あるいはプラスミンなどが公知である。あるいは、これら抗体断片をコードする遺伝子を構築し、これを発現ベクターに導入した後、適当な宿主細胞で発現させることができる(例えば、Co, M.S. et al., J. Immunol.(1994)152, 2968-2976、Better, M. & Horwitz, A. H. Methods in Enzymology(1989)178, 476-496、Pluckthun, A. & Skerra, A. Methods in Enzymology(1989)178, 476-496、Lamoyi, E., Methods in Enzymology(1989)121, 652-663、Rousseaux, J. et al., Methods in Enzymology(1989)121, 663-669、Bird, R. E. et al., TIBTECH(1991)9, 132-137参照)。
【0061】
消化酵素は、抗体断片の特定の位置を切断し、次のような特定の構造の抗体断片を与える。このような酵素的に得られた抗体断片に対して、遺伝子工学的手法を利用すると、抗体の任意の部分を欠失させることができる。
【0062】
上述の消化酵素を用いた場合に得られる抗体断片は以下のとおりである。
パパイン消化:F(ab)2またはFab
ペプシン消化:F(ab')2またはFab'
プラスミン消化:Facb
【0063】
本発明における低分子化抗体は、IL-6レセプターへの結合活性および/または中和活性を有する限り、任意の領域を欠失した抗体断片を含むことができる。
【0064】
ダイアボディーは、遺伝子融合により構築された二価(bivalent)の抗体断片を指す(Holliger P et al., Proc.Natl.Acad.Sci.USA 90: 6444-6448 (1993)、EP404,097号、WO93/11161号等)。ダイアボディーは、2本のポリペプチド鎖から構成されるダイマーである。通常、ダイマーを構成するポリペプチド鎖は、各々、同じ鎖中でVL及びVHがリンカーにより結合されている。ダイアボディーにおけるリンカーは、一般に、VLとVHが互いに結合できない位に短い。具体的には、リンカーを構成するアミノ酸残基は、例えば、5残基程度である。そのため、同一ポリペプチド鎖上にコードされるVLとVHとは、単鎖可変領域フラグメントを形成できず、別の単鎖可変領域フラグメントと二量体を形成する。その結果、ダイアボディーは2つの抗原結合部位を有することとなる。
【0065】
scFv抗体は、VH及びVLをリンカー等で結合して一本鎖ポリペプチドにした抗体である(Huston, J. S. et al., Proc. Natl. Acad. Sci. U.S.A. (1988) 85, 5879-5883、 Pluckthun「The Pharmacology of Monoclonal Antibodies」Vol.113, Resenburg 及び Moore編, Springer Verlag, New York, pp.269-315, (1994))。scFvにおけるH鎖V領域およびL鎖V領域は、本明細書に記載されたいずれの抗体由来であってもよい。V領域を連結するペプチドリンカーには、特に制限はない。例えば3から25残基程度からなる任意の一本鎖ペプチドをリンカーとして用いることができる。具体的には、たとえば後述のペプチドリンカー等を用いることができる。
【0066】
両鎖のV領域は、例えば上記のようなPCR法によって連結することができる。PCR法によるV領域の連結のために、まず次のDNAのうち、全部あるいは所望の部分アミノ酸配列をコードするDNAが鋳型として利用される。
抗体のH鎖またはH鎖V領域をコードするDNA配列、および
抗体のL鎖またはL鎖V領域をコードするDNA配列
【0067】
増幅すべきDNAの両端の配列に対応する配列を有するプライマーの一対を用いたPCR法によって、H鎖とL鎖のV領域をコードするDNAがそれぞれ増幅される。次いで、ペプチドリンカー部分をコードするDNAを用意する。ペプチドリンカーをコードするDNAもPCRを利用して合成することができる。このとき利用するプライマーの5'側に、別に合成された各V領域の増幅産物と連結できる塩基配列を付加しておく。次いで、[H鎖V領域DNA]−[ペプチドリンカーDNA]−[L鎖V領域DNA]の各DNAと、アセンブリーPCR用のプライマーを利用してPCR反応を行う。
【0068】
アセンブリーPCR用のプライマーは、[H鎖V領域DNA]の5'側にアニールするプライマーと、[L鎖V領域DNA]の3'側にアニールするプライマーとの組み合わせからなる。すなわちアセンブリーPCR用プライマーとは、合成すべきscFvの全長配列をコードするDNAを増幅することができるプライマーセットである。一方[ペプチドリンカーDNA]には各V領域DNAと連結できる塩基配列が付加されている。その結果、これらのDNAが連結され、さらにアセンブリーPCR用のプライマーによって、最終的にscFvの全長が増幅産物として生成される。一旦scFvをコードするDNAが作製されると、それらを含有する発現ベクター、および該発現ベクターにより形質転換された組換え細胞が常法に従って取得できる。また、その結果得られる組換え細胞を培養して該scFvをコードするDNAを発現させることにより、該scFvが取得できる。
【0069】
結合されるVHとVLの順序は特に限定されず、どのような順序で並べられていてもよく、例えば、以下のような配置を挙げることができる。
[VH]リンカー[VL]
[VL]リンカー[VH]
【0070】
sc(Fv)2は、2つのVH及び2つのVLをリンカー等で結合して一本鎖にした低分子化抗体である(Hudson et al、J Immunol. Methods 1999;231:177-189)。sc(Fv)2は、例えば、scFvをリンカーで結ぶことによって作製できる。
【0071】
また2つのVH及び2つのVLが、一本鎖ポリペプチドのN末端側を基点としてVH、VL、VH、VL([VH]リンカー[VL]リンカー[VH]リンカー[VL])の順に並んでいることを特徴とする抗体が好ましいが、2つのVHと2つのVLの順序は特に上記配置に限定されず、どのような順序で並べられていてもよい。例えば以下のような配置も挙げることができる。
【0072】
[VL]リンカー[VH]リンカー[VH]リンカー[VL]
[VH]リンカー[VL]リンカー[VL]リンカー[VH]
[VH]リンカー[VH]リンカー[VL]リンカー[VL]
[VL]リンカー[VL]リンカー[VH]リンカー[VH]
[VL]リンカー[VH]リンカー[VL]リンカー[VH]
【0073】
低分子抗体中のVH又はVLのアミノ酸配列は、置換、欠失、付加及び/又は挿入されていてもよい。さらに、VHとVLを会合させた場合に、抗原結合活性を有する限り、一部を欠損させてもよいし、他のポリペプチドを付加してもよい。又、可変領域はキメラ化やヒト化されていてもよい。
【0074】
本発明において、抗体の可変領域を結合するリンカーは、遺伝子工学により導入し得る任意のペプチドリンカー、又は合成化合物リンカー、例えば、Protein Engineering, 9(3), 299-305, 1996に開示されるリンカーを用いることができる。
【0075】
本発明において好ましいリンカーはペプチドリンカーである。ペプチドリンカーの長さは特に限定されず、目的に応じて当業者が適宜選択することが可能であるが、通常、1〜100アミノ酸、好ましくは3〜50アミノ酸、更に好ましくは5〜30アミノ酸、特に好ましくは12〜18アミノ酸(例えば、15アミノ酸)である。
【0076】
ペプチドリンカーのアミノ酸配列としては、例えば、以下のような配列を挙げることができる。
Ser
Gly・Ser
Gly・Gly・Ser
Ser・Gly・Gly
Gly・Gly・Gly・Ser(配列番号:45)
Ser・Gly・Gly・Gly(配列番号:46)
Gly・Gly・Gly・Gly・Ser(配列番号:47)
Ser・Gly・Gly・Gly・Gly(配列番号:48)
Gly・Gly・Gly・Gly・Gly・Ser(配列番号:49)
Ser・Gly・Gly・Gly・Gly・Gly(配列番号:50)
Gly・Gly・Gly・Gly・Gly・Gly・Ser(配列番号:51)
Ser・Gly・Gly・Gly・Gly・Gly・Gly(配列番号:52)
(Gly・Gly・Gly・Gly・Ser(配列番号:47))n
(Ser・Gly・Gly・Gly・Gly(配列番号:48))n
[nは1以上の整数である]等を挙げることができる。
【0077】
ペプチドリンカーのアミノ酸配列は、目的に応じて当業者が適宜選択することができる。たとえば上記のペプチドリンカーの長さを決定するnは、通常1〜5、好ましくは1〜3、より好ましくは1または2である。
【0078】
合成化合物リンカー(化学架橋剤)は、ペプチドの架橋に通常用いられている架橋剤、例えば、N-ヒドロキシスクシンイミド(NHS)ジスクシンイミジルスベレート(DSS)、ビス(スルホスクシンイミジル)スベレート(BS3)、ジチオビス(スクシンイミジルプロピオネート)(DSP)、ジチオビス(スルホスクシンイミジルプロピオネート)(DTSSP)、エチレングリコールビス(スクシンイミジルスクシネート)(EGS)、エチレングリコールビス(スルホスクシンイミジルスクシネート)(スルホ−EGS)、ジスクシンイミジル酒石酸塩(DST)、ジスルホスクシンイミジル酒石酸塩(スルホ−DST)、ビス[2-(スクシンイミドオキシカルボニルオキシ)エチル]スルホン(BSOCOES)、ビス[2-(スルホスクシンイミドオキシカルボニルオキシ)エチル]スルホン(スルホ−BSOCOES)などであり、これらの架橋剤は市販されている。
【0079】
4つの抗体可変領域を結合する場合には、通常、3つのリンカーが必要となる。複数のリンカーは、同じでもよいし、異なるリンカーを用いることもできる。
【0080】
本発明の抗体には、本発明の抗体のアミノ酸配列に1又は複数個のアミノ酸残基が付加された抗体も含まれる。また、これら抗体と他のペプチド又はタンパク質とが融合した融合タンパク質も含まれる。融合タンパク質を作製する方法は、本発明の抗体をコードするポリヌクレオチドと他のペプチド又はポリペプチドをコードするポリヌクレオチドをフレームが一致するように連結してこれを発現ベクターに導入し、宿主で発現させればよく、当業者に公知の手法を用いることができる。本発明の抗体との融合に付される他のペプチド又はポリペプチドとしては、例えば、FLAG(Hopp, T. P. et al., BioTechnology (1988) 6, 1204-1210)、6個のHis(ヒスチジン)残基からなる6×His、10×His、インフルエンザ凝集素(HA)、ヒトc-mycの断片、VSV-GPの断片、p18HIVの断片、T7-tag、HSV-tag、E-tag、SV40T抗原の断片、lck tag、α-tubulinの断片、B-tag、Protein Cの断片等の公知のペプチドを使用することができる。また、本発明の抗体との融合に付される他のポリペプチドとしては、例えば、GST(グルタチオン−S−トランスフェラーゼ)、HA(インフルエンザ凝集素)、イムノグロブリン定常領域、β−ガラクトシダーゼ、MBP(マルトース結合タンパク質)等が挙げられる。市販されているこれらペプチドまたはポリペプチドをコードするポリヌクレオチドを、本発明の抗体をコードするポリヌクレオチドと融合させ、これにより調製された融合ポリヌクレオチドを発現させることにより、融合ポリペプチドを調製することができる。
【0081】
また本発明の抗体は、ポリエチレングリコール(PEG)やヒアルロン酸などの高分子物質、放射性物質、蛍光物質、発光物質、酵素、トキシン等の各種分子と結合したコンジュゲート抗体でもよい。このようなコンジュゲート抗体は、得られた抗体に化学的な修飾を施すことによって得ることができる。なお、抗体の修飾方法はこの分野においてすでに確立されている(例えば、US5057313、US5156840)。本発明における「抗体」にはこれらのコンジュゲート抗体も包含される。
【0082】
また本発明の抗体には、糖鎖が改変された抗体も包含される。
【0083】
さらに、本発明で使用される抗体は二重特異性抗体(bispecific antibody)であってもよい。二重特異性抗体とは、異なるエピトープを認識する可変領域を同一の抗体分子内に有する抗体を言う。本発明において、二重特異性抗体はIL-6レセプター分子上の異なるエピトープを認識する二重特異性抗体であってもよいし、一方の抗原結合部位がIL-6レセプターを認識し、他方の抗原結合部位が他の物質を認識する二重特異性抗体とすることもできる。IL-6レセプターを認識する本発明の抗体からなる二重特異性抗体の他方の抗原結合部位が結合する抗原としては、例えば、IL-6, TNFα, TNFR1, TNFR2, CD80, CD86, CD28, CD20, CD19, IL-1α, IL-β, IL-1R, RANKL, RANK, IL-17, IL-17R, IL-23, IL-23R, IL-15, IL-15R, BlyS, lymphotoxinα, lymphotoxinβ, LIGHT ligand, LIGHT, VLA-4, CD25, IL-12, IL-12R, CD40, CD40L, BAFF, CD52, CD22, IL-32, IL-21, IL-21R, GM-CSF, GM-CSFR, M-CSF, M-CSFR, IFN-alpha, VEGF, VEGFR, EGF, EGFR, CCR5, APRIL, APRILRなどが挙げられる。
【0084】
二重特異性抗体を製造するための方法は公知である。たとえば、認識抗原が異なる2種類の抗体を結合させて、二重特異性抗体を作製することができる。結合させる抗体は、それぞれがH鎖とL鎖を有する1/2分子であっても良いし、H鎖のみからなる1/4分子であっても良い。あるいは、異なるモノクローナル抗体を産生するハイブリドーマを融合させて、二重特異性抗体産生融合細胞を作製することもできる。さらに、遺伝子工学的手法により二重特異性抗体が作製できる。
【0085】
本発明の抗体は、後述する抗体を産生する細胞や宿主あるいは精製方法により、アミノ酸配列、分子量、等電点又は糖鎖の有無や形態などが異なり得る。しかしながら、得られた抗体が、本発明の抗体と同等の機能を有している限り、本発明に含まれる。例えば、本発明の抗体を原核細胞、例えば大腸菌で発現させた場合、本来の抗体のアミノ酸配列のN末端にメチオニン残基が付加される。本発明の抗体はこのような抗体も包含する。
【0086】
本発明の抗IL-6レセプター抗体などのポリペプチドは当業者に公知の方法により製造することが可能である。
【0087】
例えば、得られた抗IL-6レセプター抗体の配列を基に、当業者に公知の遺伝子組換え技術を用いて抗IL-6レセプター抗体を作製することが可能である。具体的には、IL-6レセプターを認識する抗体の配列を基に抗体をコードするポリヌクレオチドを構築し、これを発現ベクターに導入した後、適当な宿主細胞で発現させればよい(例えば、Co, M. S. et al., J. Immunol. (1994) 152, 2968-2976 ; Better, M. and Horwitz, A. H., Methods Enzymol. (1989) 178, 476-496 ; Pluckthun, A. and Skerra, A., Methods Enzymol. (1989) 178, 497-515 ; Lamoyi, E., Methods Enzymol. (1986) 121, 652-663 ; Rousseaux, J. et al., Methods Enzymol. (1986) 121, 663-669 ; Bird, R. E. and Walker, B. W., Trends Biotechnol. (1991) 9, 132-137参照)。
【0088】
従って、本発明は、本発明のポリペプチドをコードするポリヌクレオチドが導入されたベクターを含む宿主細胞を培養する工程を含む、本発明のポリペプチド又は本発明のポリペプチドをコードする遺伝子によりコードされるポリペプチドを製造する方法を提供する。
【0089】
より具体的には、以下の工程を含む本発明のポリペプチドの製造方法を提供する。
(a)本発明のポリペプチドをコードする遺伝子が導入されたベクターを含む宿主細胞を培養する工程、
(b)当該遺伝子によりコードされるポリペプチドを取得する工程。
【0090】
ベクターの例としては、M13系ベクター、pUC系ベクター、pBR322、pBluescript、pCR-Scriptなどが挙げられる。また、cDNAのサブクローニング、切り出しを目的とした場合、上記ベクターの他に、例えば、pGEM-T、pDIRECT、pT7などが挙げられる。本発明の抗体を生産する目的においてベクターを使用する場合には、特に、発現ベクターが有用である。発現ベクターとしては、例えば、大腸菌での発現を目的とした場合は、ベクターが大腸菌で増幅されるような特徴を持つほかに、宿主をJM109、DH5α、HB101、XL1-Blueなどの大腸菌とした場合においては、大腸菌で効率よく発現できるようなプロモーター、例えば、lacZプロモーター(Wardら, Nature (1989) 341, 544-546;FASEB J. (1992) 6, 2422-2427)、araBプロモーター(Betterら, Science (1988) 240, 1041-1043)、またはT7プロモーターなどを持っていることが不可欠である。このようなベクターとしては、上記ベクターの他にpGEX-5X-1(ファルマシア製)、「QIAexpress system」(キアゲン製)、pEGFP、またはpET(この場合、宿主はT7 RNAポリメラーゼを発現しているBL21が好ましい)などが挙げられる。
【0091】
また、発現プラスミドのベクターには、抗体分泌のためのシグナル配列が含まれていてもよい。抗体分泌のためのシグナル配列としては、大腸菌のペリプラズムに産生させる場合、pelBシグナル配列(Lei, S. P. et al J. Bacteriol. (1987) 169, 4379)を使用すればよい。宿主細胞へのベクターの導入は、例えば塩化カルシウム法、エレクトロポレーション法を用いて行うことができる。
【0092】
大腸菌以外にも、例えば、本発明の抗体を製造するためのベクターとしては、哺乳動物由来の発現ベクター(例えば、pcDNA3(インビトロゲン社製)や、pEF-BOS (Nucleic Acids. Res.1990, 18(17),p5322)、pEF、pCDM8)、昆虫細胞由来の発現ベクター(例えば「Bac-to-BAC baculovairus expression system」(ギブコBRL製)、pBacPAK8)、植物由来の発現ベクター(例えばpMH1、pMH2)、動物ウィルス由来の発現ベクター(例えば、pHSV、pMV、pAdexLcw)、レトロウィルス由来の発現ベクター(例えば、pZIPneo)、酵母由来の発現ベクター(例えば、「Pichia Expression Kit」(インビトロゲン製)、pNV11、SP-Q01)、枯草菌由来の発現ベクター(例えば、pPL608、pKTH50)が挙げられる。
【0093】
CHO細胞、COS細胞、NIH3T3細胞等の動物細胞での発現を目的とした場合には、発現プラスミドのベクターが細胞内で発現させるために必要なプロモーター、例えばSV40プロモーター(Mulliganら, Nature (1979) 277, 108)、MMLV-LTRプロモーター、EF1αプロモーター(Mizushimaら, Nucleic Acids Res. (1990) 18, 5322)、CMVプロモーターなどを持っていることが不可欠であり、細胞への形質転換を選抜するための遺伝子(例えば、薬剤(ネオマイシン、G418など)により判別できるような薬剤耐性遺伝子)を有すればさらに好ましい。このような特性を有するベクターとしては、例えば、pMAM、pDR2、pBK-RSV、pBK-CMV、pOPRSV、pOP13などが挙げられる。
【0094】
さらに、遺伝子を安定的に発現させ、かつ、細胞内での遺伝子のコピー数の増幅を目的とする場合には、核酸合成経路を欠損したCHO細胞にそれを相補するDHFR遺伝子を有するベクター(例えば、pSV2-dhfr(「Molecular Cloning 2nd edition」 Cold Spring Harbor Laboratory Press, (1989))など)を導入し、メトトレキセート(MTX)により増幅させる方法が挙げられ、また、遺伝子の一過性の発現を目的とする場合には、SV40 T抗原を発現する遺伝子を染色体上に持つCOS細胞を用いてSV40の複製起点を持つベクター(pcDなど)で形質転換する方法が挙げられる。複製開始点としては、また、ポリオーマウィルス、アデノウィルス、ウシパピローマウィルス(BPV)等の由来のものを用いることもできる。さらに、宿主細胞系で遺伝子コピー数増幅のため、発現ベクターは選択マーカーとして、アミノグリコシドトランスフェラーゼ(APH)遺伝子、チミジンキナーゼ(TK)遺伝子、大腸菌キサンチングアニンホスホリボシルトランスフェラーゼ(Ecogpt)遺伝子、ジヒドロ葉酸還元酵素(dhfr)遺伝子等を含むことができる。
【0095】
これにより得られた本発明の抗体は、宿主細胞内または細胞外(培地など)から単離し、実質的に純粋で均一な抗体として精製することができる。抗体の分離、精製は、通常の抗体の精製で使用されている分離、精製方法を使用すればよく、何ら限定されるものではない。例えば、クロマトグラフィーカラム、フィルター、限外濾過、塩析、溶媒沈殿、溶媒抽出、蒸留、免疫沈降、SDS-ポリアクリルアミドゲル電気泳動、等電点電気泳動法、透析、再結晶等を適宜選択、組み合わせれば抗体を分離、精製することができる。
【0096】
クロマトグラフィーとしては、例えばアフィニティークロマトグラフィー、イオン交換クロマトグラフィー、疎水性クロマトグラフィー、ゲル濾過、逆相クロマトグラフィー、吸着クロマトグラフィー等が挙げられる(Strategies for Protein Purification and Characterization: A Laboratory Course Manual. Ed Daniel R. Marshak et al., Cold Spring Harbor Laboratory Press, 1996)。これらのクロマトグラフィーは、液相クロマトグラフィー、例えばHPLC、FPLC等の液相クロマトグラフィーを用いて行うことができる。アフィニティークロマトグラフィーに用いるカラムとしては、プロテインAカラム、プロテインGカラムが挙げられる。例えば、プロテインAを用いたカラムとして、Hyper D, POROS, Sepharose FF(GE Amersham Biosciences)等が挙げられる。本発明は、これらの精製方法を用い、高度に精製された抗体も包含する。
【0097】
得られた抗体のIL-6レセプターに対する結合活性の測定は、当業者に公知の方法により行うことが可能である。例えば、抗体の抗原結合活性を測定する方法として、ELISA(酵素結合免疫吸着検定法)、EIA(酵素免疫測定法)、RIA(放射免疫測定法)あるいは蛍光抗体法を用いることができる。例えば、酵素免疫測定法を用いる場合、抗原をコーティングしたプレートに、抗体を含む試料、例えば、抗体産生細胞の培養上清や精製抗体を加える。アルカリフォスファターゼ等の酵素で標識した二次抗体を添加し、プレートをインキュベートし、洗浄した後、p-ニトロフェニル燐酸などの酵素基質を加えて吸光度を測定することで抗原結合活性を評価することができる。
【0098】
医薬組成物
また本発明は、上述のポリペプチドを有効成分として含有する医薬組成物を提供する。本発明の医薬組成物はIL-6が関連する関節リウマチなどの疾患に用いることが可能である。即ち本発明は、上述の抗体を有効成分とする関節リウマチなどの疾患の治療剤も提供する。本発明の対象となる疾患の好ましい例として、関節リウマチ、若年性特発性関節炎、全身型若年性特発性関節炎、キャッスルマン病、全身性エリテマトーデス(SLE)、ループス腎炎、クローン病、lymphoma、潰瘍性大腸炎、貧血、血管炎、川崎病、Still病、アミロイドーシス、多発性硬化症、移植、加齢黄斑変性症、強直性脊椎炎、乾癬、乾癬性関節炎、慢性閉塞性肺疾患(COPD)、IgA 腎症、変形性関節症、喘息、糖尿病性腎症、GVHD、子宮内膜症、肝炎(NASH)、心筋梗塞、動脈硬化、セプシス、骨粗しょう症、糖尿病、多発性骨髄腫、前立腺癌、腎癌、B-cell non-Hodgkin's、膵癌、肺癌、食道癌、大腸癌、癌カケクシア、癌神経浸潤、心筋梗塞、近視性脈絡膜血管新生、特発性脈絡膜血管新生、ぶどう膜炎、慢性甲状腺炎、遅延性過敏症、接触性皮膚炎、アトピー性皮膚炎、中皮腫、多発性筋炎、皮膚筋炎、汎ぶどう膜炎、前部ぶどう膜炎、中間部ぶどう膜炎、強膜炎、角膜炎、眼窩炎症、視神経炎、糖尿病網膜症、増殖硝子体網膜症、ドライアイ、術後炎症等が挙げられるが、これらに限定されることはない。
【0099】
抗IL-6レセプター抗体を「有効成分として含有する」とは、抗IL-6レセプター抗体を活性成分の少なくとも1つとして含むという意味であり、その含有率を制限するものではない。また、本発明の医薬組成物は、上述のポリペプチドと合わせて他の有効成分を含有してもよい。
【0100】
なお、本発明の医薬組成物は治療目的だけでなく、予防目的で用いられてもよい。
【0101】
本発明のポリペプチドは、常法に従って製剤化することができる(例えば、Remington's Pharmaceutical Science, latest edition, Mark Publishing Company, Easton, U.S.A)。さらに、必要に応じ、医薬的に許容される担体及び/または添加物を供に含んでもよい。例えば、界面活性剤(PEG、Tween等)、賦形剤、酸化防止剤(アスコルビン酸等)、着色料、着香料、保存料、安定剤、緩衝剤(リン酸、クエン酸、他の有機酸等)、キレート剤(EDTA等)、懸濁剤、等張化剤、結合剤、崩壊剤、滑沢剤、流動性促進剤、矯味剤等を含むことができる。しかしながら、本発明の炎症性疾患の予防または治療剤は、これらに制限されず、その他常用の担体を適宜含んでいてもよい。具体的には、軽質無水ケイ酸、乳糖、結晶セルロース、マンニトール、デンプン、カルメロースカルシウム、カルメロースナトリウム、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルアセタールジエチルアミノアセテート、ポリビニルピロリドン、ゼラチン、中鎖脂肪酸トリグリセライド、ポリオキシエチレン硬化ヒマシ油60、白糖、カルボキシメチルセルロース、コーンスターチ、無機塩類等を挙げることができる。また、その他の低分子量のポリペプチド、血清アルブミン、ゼラチン及び免疫グロブリン等の蛋白質、並びに、アミノ酸を含んでいてもよい。注射用の水溶液とする場合には、抗IL-6レセプター抗体を、例えば、生理食塩水、ブドウ糖またはその他の補助薬を含む等張液に溶解する。補助薬としては、例えば、D-ソルビトール、D-マンノース、D-マンニトール、塩化ナトリウムが挙げられ、さらに、適当な溶解補助剤、例えばアルコール(エタノール等)、ポリアルコール(プロピレングリコール、PEG等)、非イオン性界面活性剤(ポリソルベート80、HCO-50)等と併用してもよい。
【0102】
また、必要に応じポリペプチドをマイクロカプセル(ヒドロキシメチルセルロース、ゼラチン、ポリ[メチルメタクリル酸]等のマイクロカプセル)に封入したり、コロイドドラッグデリバリーシステム(リポソーム、アルブミンミクロスフェア、マイクロエマルジョン、ナノ粒子及びナノカプセル等)とすることもできる(Remington's Pharmaceutical Science 16th edition &, Oslo Ed. (1980)等参照)。さらに、薬剤を徐放性の薬剤とする方法も公知であり、ポリペプチドに適用し得る(Langer et al., J.Biomed.Mater.Res.(1981) 15: 167-277; Langer, Chem. Tech. (1982)12: 98-105;米国特許第3,773,919号;欧州特許出願公開(EP)第58,481号; Sidman et al., Biopolymers(1983)22:547-56;EP第133,988号)。さらに、本剤にヒアルロニダーゼを添加あるいは混合することで皮下に投与する液量を増加させることも可能である(例えば、WO2004/078140等)。
【0103】
本発明の医薬組成物は、経口または非経口のいずれでも投与可能であるが、好ましくは非経口投与される。具体的には、注射及び経皮投与により患者に投与される。注射剤型の例としては、例えば、静脈内注射、筋肉内注射または皮下注射等により全身又は局所的に投与することができる。治療部位またはその周辺に局所注入、特に筋肉内注射してもよい。経皮投与剤型の例としては、例えば、軟膏剤、ゲル剤、クリーム剤、湿布剤、および貼付剤等があげられ、全身又は局所的に投与することができる。また、患者の年齢、症状により適宜投与方法を選択することができる。投与量としては、例えば、1回につき体重1 kgあたり活性成分が0.0001 mg〜100 mgの範囲で選ぶことが可能である。または、例えば、ヒト患者に投与する場合、患者あたり活性成分が0.001〜1000 mg/kg・body・weightの範囲を選ぶことができ、1回当たり投与量としては、例えば、本発明の抗体が0.01〜50mg/kg・body・weight程度の量が含まれることが好ましい。しかしながら、本発明の医薬組成物は、これらの投与量に制限されるものではない。
【0104】
なお、本発明で記載されているアミノ酸配列に含まれるアミノ酸は翻訳後に修飾(例えば、N末端のグルタミンのピログルタミル化によるピログルタミン酸への修飾は当業者によく知られた修飾である)を受ける場合もあるが、そのようにアミノ酸が翻訳後修飾された場合であっても当然のことながら本発明で記載されているアミノ酸配列に含まれる。
【0105】
また結合する糖鎖の構造は如何なる構造でもよい。EUナンバリングの297番目の糖鎖は如何なる糖鎖構造であってもよく(好ましくはフコシル化された糖鎖)、また糖鎖が結合していなくてもよい(例えば大腸菌で生産する、あるいはEUナンバリングの297番目に糖鎖が結合しないように改変することで可能)。
【0106】
なお本明細書において引用された全ての先行技術文献は、参照として本明細書に組み入れられる。
【実施例】
【0107】
以下本発明を実施例により具体的に説明するが、本発明はこれら実施例に制限されるものではない。
【0108】
〔実施例1〕TOCILIZUMABのIL-6レセプターへの親和性を向上する可変領域変異箇所の同定
TOCILIZUMAB(H鎖 WT-IgG1/配列番号:53、L鎖 WT-kappa/配列番号:54)のIL-6レセプターへの親和性を向上させるために、CDR配列に変異を導入したライブラリーを作製し検討した。CDRに変異を導入したライブラリーをスクリーニングした結果、IL-6レセプターへの親和性を向上する変異を見出し、それらを図1にまとめた。これらの変異を組み合わせた高親和性TOCILIZUMABの例として、RDC-23(H鎖RDC23H-IgG1/配列番号:55、L鎖 RDC-23L-kappa/配列番号:56)が挙げられる。RDC-23の可溶型IL-6レセプターへのアフィニティーおよびBaF/gp130による生物活性をTOCILIZUMABと比較した(方法は参考例参照)。
【0109】
アフィニティーを測定した結果を表1に示した。BaF/gp130による生物活性(IL-6終濃度 30 ng/mL)を測定した結果を図2に示した。RDC-23は、TOCILIZUMABと比較して約60倍アフィニティーが向上しており、BaF/gp130の100%阻害濃度として約100倍活性が向上していることが見出された。
【0110】
【表1】

【0111】
〔実施例2〕TOCILIZUMABの等電点低下による薬物動態を向上する改変の同定
TOCILIZUMABの薬物動態を向上させるために、IL-6レセプターへの結合を大きく低下させることなく可変領域の等電点を低下することができる変異箇所の検討を行った。TOCILIZUMABの立体構造モデルから推察された可変領域変異箇所をスクリーニングした結果、IL-6レセプターへの結合を大きく低下させることなく可変領域の等電点を低下することできる変異箇所を見出し、それらを図3にまとめた。これらの変異を組み合わせた等電点低下TOCILIZUMABの例として、H53/L28(H鎖 H53-IgG1/配列番号:57、L鎖 L28-kappa/配列番号:58)が挙げられる。H53/L28の可溶型IL-6レセプターへのアフィニティー、BaF/gp130による生物活性、等電点、および、マウスにおける薬物動態をTOCILIZUMABと比較した(方法は参考例参照)。
【0112】
アフィニティーを測定した結果を表2に示した。BaF/gp130による生物活性(IL-6終濃度 30 ng/mL)を測定した結果を図4に示した。H53/L28は、TOCILIZUMABと比較して約6倍アフィニティーが向上しており、BaF/gp130の100%阻害濃度として数倍程度活性が向上していることが示された。
【0113】
【表2】
【0114】
当業者公知の等電点電気泳動により等電点を測定した結果、TOCILIZUMABの等電点は約9.3であり、H53/L28の等電点は約6.5〜6.7であり、H53/L28はTOCILIZUMABと比較して等電点が約2.7低下した。また、可変領域VH/VLの理論等電点をGENETYX(GENETYX CORPORATION)により計算したところ、TOCILIZUMABの理論等電点は9.20であり、H53/L28の理論等電点は4.52であり、H53/L28はTOCILIZUMABと比較して等電点が約4.7低下した。
【0115】
等電点を低下させた改変抗体H53/L28の薬物動態を評価するために、TOCILIZUMABとH53/L28の正常マウスにおける薬物動態の比較を行った。TOCILIZUMABおよびH53/L28をマウス(C57BL/6J、日本チャールズリバー)に1 mg/kgで静脈内(IV)および皮下(SC)に単回投与し血漿中濃度推移を評価した。TOCILIZUMABおよびH53/L28の静脈内投与後の血漿中濃度推移を図5に、皮下投与後の血漿中濃度推移を図6に示し、WinNonlin(Pharsight社製)により得られた薬物動態学的パラメーター(クリアランス(CL)、半減期(T1/2))を表3に示した。H53/L28の静脈内投与後の血漿中半減期(T1/2)はTOCILIZUMABの約1.3倍に延長し、クリアランスが約1.7倍低下した。H53/L28の皮下投与後のT1/2はTOCILIZUMABの約2倍に延長し、クリアランスが約2.1倍低下した。このようにアミノ酸置換によりTOCILIZUMABの等電点を低下させることによって薬物動態を大幅に向上させることが可能であることが見出された。
【0116】
【表3】
【0117】
〔実施例3〕TOCILIZUMABの免疫原性を低下する変異箇所の同定
可変領域に存在するT-cellエピトープによる免疫原性リスクを低減する変異箇所の同定
TOCILIZUMABの可変領域配列に存在するT-cellエピトープをTEPITOPE(Methods. 2004 Dec;34(4):468-75)を用いて解析を行った。その結果、L鎖CDR2に多くのHLAに結合するT-cellエピトープが存在する(免疫原性リスクが高い配列が存在する)ことが予測された。そこで、TEPITOPE解析においてL鎖CDR2の免疫原性リスクを低減させつつ、安定性、結合活性、中和活性を低下させないアミノ酸置換を検討した。
【0118】
スクリーニングの結果、以下のようにTOCILIZUMABのL鎖CDR2(配列番号:59)のL51(Kabatナンバリング、Kabat EA et al. 1991. Sequences of Proteins of Immunological Interest.NIH)のスレオニンをグリシンに、L53のアルギニンをグルタミン酸に置換する(配列番号:60)ことで、安定性、結合活性、中和活性を低下させずに免疫原性リスクを低減できることを見出した。
【0119】
TOCILIZUMAB L鎖CDR2(配列番号:59)
T-cellエピトープ除去TOCILIZUMAB L鎖CDR2(配列番号:60)
【0120】
〔実施例4〕TOCILIZUMABの可変領域フレームワーク配列の完全ヒト化による免疫原性リスクの低減
TOCILIZUMABの可変領域配列において、H鎖FR1のH27, H28, H29, H30およびH鎖FR3のH71(Kabatナンバリング、Kabat EA et al. 1991. Sequences of Proteins of Immunological Interest.NIH)は、ヒト化の過程において結合活性を維持するためにフレームワーク配列でマウス配列が残存している(Cancer Res. 1993 Feb 15;53(4):851-6)。残存するマウス配列は免疫原性リスクを高める原因となりうるため、TOCILIZUMABの免疫原性リスクをより低下するためにフレームワーク配列を完全ヒト化する検討を行った。
【0121】
その結果、TOCILIZUMABのH鎖FR1(配列番号:61)を以下のヒト化 H鎖FR1-A(配列番号:62)に置換することで、また、TOCILIZUMABのH鎖FR3(配列番号:63)を以下のヒト化 H鎖FR3(配列番号:64)に置換することで、安定性、結合活性、中和活性を低下させずにTOCILIZUMABの全フレームワークを完全ヒト化できることを見出した。
【0122】
TOCILIZUMAB H鎖FR1(配列番号:61)
ヒト化 H鎖FR1-A(配列番号:62)(Germline IMGT hVH_4_由来)
TOCILIZUMAB H鎖FR3(配列番号:63)
ヒト化 H鎖FR3(配列番号:64)(Mol. Immunol. 2007, 44(4):412-422由来)
【0123】
〔実施例5〕TOCILIZUMABのIL-6レセプターへのpH依存的結合による薬物動態向上のための変異箇所の同定
TOCILIZUMABの薬物動態を向上させる方法の一つとして、1分子のTOCILIZUMABが複数個のIL-6レセプターを繰り返し結合・中和させるように分子を改良する方法が考えられた。TOCILIZUMABは膜型IL-6レセプターに結合後、膜型IL-6レセプターに結合したままインターナライゼーションによって細胞内のエンドソームに取り込まれ、その後、TOCILIZUMABは膜型IL-6レセプターに結合したままライソソームへ移行し共にライソソームにより分解されると考えられている。すなわち、通常、1分子のTOCILIZUMABは1分子ないしは2分子の膜型IL-6レセプターに(1価ないしは2価で)結合し、インターナライズ後、ライソソームで分解されると考えられるため、1分子のTOCILIZUMABは1分子ないしは2分子の膜型IL-6レセプターしか結合・中和できない。
【0124】
そこで、TOCILIZUMABの中性条件下での結合を維持しつつ酸性条件下での結合のみを大きく低下させるpH依存的結合TOCILIZUMABを作製することが出来れば、図7に示すとおり、pH依存的結合TOCILIZUMABはエンドソーム内で抗原である膜型IL-6レセプターから解離し、エンドソーム内に存在するFcRnに結合することによって血漿中に戻ることができ、血漿中に戻ったpH依存的結合TOCILIZUMABは再度膜型IL-6レセプターに結合することが可能であると考えた。この血漿中での結合とエンドソーム内での解離を繰り返すことで、1分子のTOCILIZUMABが複数分子のIL-6レセプターを繰り返し結合・中和できると考えられ、これによりTOCILIZUMABと比較してpH依存的結合TOCILIZUMABは薬物動態が向上すると考えた。
【0125】
エンドソーム内の酸性条件下においてTOCILIZUMABがIL-6レセプターから解離するためには、酸性条件下における結合が中性条件下と比較して大幅に弱くなる必要がある。細胞表面ではIL-6レセプターに強く結合して中和する必要があるため、細胞表面のpHであるpH7.4においてはTOCILIZUMABと同等以上にIL-6レセプターに結合する必要がある。エンドソーム内のpHは一般的にpH5.5〜pH6.0であることが報告されている(Nat Rev Mol Cell Biol. 2004 Feb;5(2):121-32.)ことから、pH5.5〜pH6.0においてIL-6レセプターに弱く結合するように改良されたpH依存的結合TOCILIZUMABであれば、エンドソーム内の酸性条件下においてIL-6レセプターから解離すると考えられる。すなわち、細胞表面のpHであるpH7.4においてはIL-6レセプターに強く結合し、エンドソーム内のpHであるpH5.5〜pH6.0においてIL-6レセプターに弱く結合するように改良されたpH依存的結合TOCILIZUMABであれば、1分子で複数個のIL-6レセプターに結合・中和し、薬物動態を向上することが可能であると考えられた。
【0126】
TOCILIZUMABのIL-6レセプターへの結合にpH依存性を付与するには、pKaが6.0〜6.5付近に存在し、中性条件下(pH7.4)と酸性条件下(pH5.5〜pH6.0)との間でプロトンの解離状態が変化するヒスチジン残基をTOCILIZUMABの可変領域に導入する方法が考えられた。そこで、TOCILIZUMABの立体構造モデルから推察された可変領域のヒスチジン導入箇所のスクリーニングを実施した。また、TOCILIZUMABの選択された可変領域配列がヒスチジンにランダムに置き換わるように設計されたライブラリーを作製しスクリーニングを実施した。スクリーニングは、pH7.4においてIL-6レセプターに結合し、pH5.5ないしはpH5.8でIL-6レセプターから解離する、あるいは、アフィニティーが低下することを指標に実施した。
【0127】
その結果、TOCILIZUMABのIL-6レセプターへの結合にpH依存性(pH7.4で結合し、pH5.8で解離する性質)を付与することができる変異箇所を見出し、それらを図8にまとめた。図8のH27のチロシンからヒスチジンへの置換はCDRではなくH鎖のFR1の変異であるが、H27がヒスチジンの配列はEur. J. Immunol. 1992. 22: 1719-1728に記されているとおり、ヒト配列(配列番号:65)として存在するため、以下のフレームワークを用いることで実施例4と合わせて完全ヒト化することが可能である。
【0128】
ヒト化H鎖FR1-B(配列番号:65)
【0129】
これらの変異を組み合わせたpH依存的結合TOCILIZUMABの例として、H3pI/L73(H鎖 H3pI-IgG1/配列番号:66、L鎖 L73-kappa/配列番号:67)が挙げられる。H3pI/L73のpH7.4における可溶型IL-6レセプターへのアフィニティー、pH7.4とpH5.8における膜型IL-6レセプターからの解離速度、BaF/gp130による生物活性、および、カニクイザルおよびヒトIL-6レセプタートランスジェニックマウスにおける薬物動態をTOCILIZUMABと比較した(方法は参考例参照)。
【0130】
pH7.4における可溶型IL-6レセプターへのアフィニティーを測定した結果を表4に示した。BaF/gp130による生物活性(IL-6終濃度30 ng/mL)を測定した結果を図9に示した。H3pI/L73は、TOCILIZUMABと比較してほぼ同等のpH7.4における可溶型IL-6レセプターへのアフィニティーおよびBaF/gp130の活性を有していることが示された。
【0131】
【表4】
【0132】
TOCILIZUMABおよびH3pI/L73のpH7.4とpH5.8における膜型IL-6レセプターへ解離速度を測定した結果を表5に示した。H3pI/L73は、pH5.8における解離速度が速くなり、TOCILIZUMABと比較して膜型IL-6レセプターからの解離速度のpH依存性が約2.6倍向上していることが示された。
【0133】
【表5】
【0134】
TOCILIZUMABおよびH3pI/L73をカニクイザルに1 mg/kgで静脈内に単回投与し血漿中濃度推移を評価した。TOCILIZUMABおよびH3pI/L73の静脈内投与後の血漿中濃度推移を図10に示した。その結果、H3pI/L73はTOCILIZUMABと比較してカニクイザルにおいて大幅に薬物動態が改善した。
【0135】
TOCILIZUMABおよびH3pI/L73をヒトIL-6レセプタートランスジェニックマウス(hIL-6R tgマウス、Proc Natl Acad Sci U S A. 1995 May 23;92(11):4862-6)に25 mg/kgで静脈内に単回投与し血漿中濃度推移を評価した。TOCILIZUMABおよびH3pI/L73の静脈内投与後の血漿中濃度推移を図11に示した。その結果、H3pI/L73はTOCILIZUMABと比較してヒトIL-6レセプタートランスジェニックマウスにおいて大幅に薬物動態が改善した。
【0136】
pH依存的結合TOCILIZUMABであるH3pI/L73は、カニクイザルおよびヒトIL-6レセプタートランスジェニックマウスにおいてTOCILIZUMABと比較して薬物動態が大幅に改善したことから、pH7.4で抗原に結合し、pH5.8で抗原から解離する性質を付与することにより、1分子で複数のIL-6レセプターに結合・中和することが可能であると考えられた。また、IL-6レセプターへの結合にH3pI/L73よりもさらに強いpH依存性を付与することに薬物動態がさらに改善することが可能であると考えられた。
【0137】
〔実施例6〕TOCILIZUMABの定常領域の最適化
TOCILIZUMABのH鎖C末端のヘテロジェニティーの低減
IgG抗体のH鎖C末端配列のヘテロジェニティーとして、C末端アミノ酸のリジン残基の欠損、および、C末端の2アミノ酸のグリシン、リジン両方の欠損によるC末端カルボキシル基のアミド化が報告されている(Anal Biochem. 2007 Jan 1;360(1):75-83.)。TOCILIZUMABにおいても、その主成分は塩基配列上存在するC末端アミノ酸のリジンが翻訳後修飾により欠損した配列であるが、リジンが残存している副成分およびグリシン、リジン両方の欠損によるC末端カルボキシル基のアミド化された副成分もヘテロジェニティーとして存在する。目的物質/関連物質のヘテロジェニティーの製造間差を維持しつつ医薬品として大量に製造することは容易ではなくコスト増につながり、可能な限り単一物質であることが望まれ、抗体を医薬品として開発する上にはこれらのヘテロジェニティーが低減されていることが望ましい。よって医薬品として開発する上ではH鎖C末端のヘテロジェニティーは存在しないことが望ましい。
【0138】
C末端アミノ酸のヘテロジェニティーを低減させることを目的にC末端アミノ酸の改変を行った。その結果、TOCILIZUMABのH鎖定常領域のC末端のリジンおよびグリシンを塩基配列上あらかじめ欠損させることで、C末端に由来するヘテロジェニティーを回避することが可能であることが見出された。TOCILIZUMAB、C末端のリジン欠損TOCILIZUMAB(TOCILIZUMABΔK、H鎖 WT-IgG1ΔK/配列番号:68、L鎖 WT-kappa/配列番号:54)、および、C末端のリジンおよびグリシン欠損TOCILIZUMAB(TOCILIZUMABΔGK、H鎖 WT-IgG1ΔGK/配列番号:69、L鎖 WT-kappa/配列番号:54)のヘテロジェニティーの評価を陽イオン交換クロマトグラフィーにより実施した。カラムとしてはProPac WCX-10, 4×250 mm (Dionex) を使用し、移動相Aは25 mmol/L MES/NaOH, pH 6.1、移動相Bは25 mmol/L MES/NaOH, 250 mmol/L NaCl, pH 6.1を使用し、適切な流量およびグラジエントを用いて実施した。陽イオン交換クロマトグラフィーによる評価を行った結果を図12に示した。その結果、H鎖定常領域のC末端のリジンだけでなく、H鎖定常領域のC末端のリジンおよびグリシンの両方を塩基配列上あらかじめ欠損させることで初めてC末アミノ酸のヘテロジェニティーを低減可能であることが見出された。ヒト抗体定常領域IgG1、IgG2、IgG4において、C末端配列はいずれもEUナンバリング(Sequences of proteins of immunological interest, NIH Publication No.91-3242 を参照)447番目がリジン、446番目がグリシンになっていることから、本検討で見出されたC末アミノ酸のヘテロジェニティーを低減させる方法はIgG2定常領域とIgG4定常領域、あるいはそれらの改変体にも適用可能であると考えられた。
【0139】
TOCILIZUMABのIgG2アイソタイプのジスルフィド結合由来のヘテロジェニティーの低減
TOCILIZUMABのアイソタイプはIgG1であるが、TOCILIZUMABは中和抗体であることから、免疫原性や副作用を考慮した場合、Fcγレセプターへの結合は好ましくない可能性が考えられる。Fcγレセプターへの結合を低下させる方法としては、IgG抗体のアイソタイプをIgG1からIgG2あるいはIgG4に変える方法が考えられ(Ann Hematol. 1998 Jun;76(6):231-48.)、FcγレセプターIへの結合および薬物動態の観点からはIgG4よりはIgG2が望ましいと考えられた(Nat Biotechnol. 2007 Dec;25(12):1369-72)。一方、抗体を医薬品として開発するにあたり、そのタンパク質の物性、中でも均一性と安定性は極めて重要であり、IgG2アイソタイプは、ヒンジ領域のジスルフィド結合に由来するヘテロジェニティーが極めて多いことが報告されている(J Biol Chem. 2008 Jun 6;283(23):16206-15.)。これに由来する目的物質/関連物質のヘテロジェニティーの製造間差を維持しつつ医薬品として大量に製造することは容易ではなくコスト増につながり、可能な限り単一物質であることが望まれる。よってIgG2アイソタイプの抗体を医薬品として開発する上では安定性を低下させることなくジスルフィド結合由来のヘテロジェニティーが低減されていることが望ましい。
【0140】
IgG2アイソタイプのヘテロジェニティーを低減することを目的に各種改変体を検討した結果、IgG2定常領域配列のうち、H鎖のCH1ドメインに存在するEUナンバリング131番目のシステインと133番目のアルギニンをそれぞれセリンとリジンに置換し、さらにH鎖のupper hingeに存在するEUナンバリング219番目のシステインをセリンに置換した定常領域であるWT-SKSC(配列番号:70)によって、安定性を低下させることなくヘテロジェニティーを低減できることが見出された。TOCILIZUMAB-IgG1(H鎖 WT-IgG1/配列番号:53、L鎖 WT-kappa/配列番号:54)、TOCILIZUMAB-IgG2(H鎖 WT-IgG2/配列番号:71、L鎖 WT-kappa/配列番号:54)、および、TOCILIZUMAB-SKSC(H鎖 WT-SKSC/配列番号:70、L鎖 WT-kappa/配列番号:54)を調製し、ヘテロジェニティーおよび安定性の評価を行った。ヘテロジェニティーの評価は陽イオン交換クロマトグラフィーにより実施した。カラムとしてProPac WCX-10 (Dionex)を用い、移動相Aとして20 mM Sodium Acetate, pH5.0、移動相Bとして20 mM Sodium Acetate, 1 M NaCl, pH5.0を使用し、適切な流量およびグラジエントを用いて実施した。陽イオン交換クロマトグラフィーによる評価を行った結果を図13に示した。安定性の評価は、示差走査型熱量測定(DSC)(VP-DSC、Microcal社製)による熱変性中間温度(Tm値)の評価により実施した。20 mM sodium acetate, 150 mM NaCl, pH6.0におけるDSC測定結果とFabドメインのTm値を図14に示した。
【0141】
その結果、TOCILIZUMAB-IgG1と比較してTOCILIZUMAB-IgG2は著しくヘテロジェニティーが増大するが、TOCILIZUMAB-SKSCにすることによってヘテロジェニティーは大幅に低減できることが見出された。また、TOCILIZUMAB-IgG1と比較してTOCILIZUMAB-IgG2はDSCにおけるFabドメインの熱変性ピークにおいてヘテロ成分によると考えられる安定性の低い、すなわちTmの低いショルダーピーク(Fab*)の成分が認められるが、TOCILIZUMAB-SKSCにすることによってヘテロ成分によると考えられるTm値の低いショルダーピークは消失し、TOCILIZUMAB-IgG1およびTOCILIZUMAB-IgG2のFabドメインと同等の約94℃のTm値を示したことから、TOCILIZUMAB-SKSCは高い安定性を有していることが見出された。
【0142】
TOCILIZUMABの薬物動態を向上するする定常領域変異箇所の同定
上述のとおり、TOCILIZUMABのアイソタイプであるIgG1から、C末端のヘテロジェニティーを低減し、Fcγレセプターへの結合性を低減させ、高い安定性を維持したままIgG2アイソタイプの定常領域の抗体のヘテロジェニティーを低減することが可能であることが見出されたが、薬物動態に関してもTOCILIZUMABのアイソタイプであるIgG1よりも優れた定常領域であることが望ましい。
【0143】
IgG1アイソタイプの定常領域の抗体よりも優れた血漿中半減期を有する定常領域を見出すために、高い安定性を有しIgG2アイソタイプの定常領域の抗体に関する上述のヘテロジェニティーが低減されたTOCILIZUMAB-SKSCに対して、薬物動態を向上させることを目的に変異箇所をスクリーニングした結果、WT-SKSCに対して、EUナンバリング137番目のグルタミン酸をグリシンに138番目のセリンをグリシンに、268番ヒスチジンをグルタミンに、355番アルギニンをグルタミンに、419番グルタミンをグルタミン酸に置換し、これに加えてH鎖C末端のヘテロジェニティーを低減するために446番目のグリシンおよび447番目のリジンを欠損させたWT-M58(配列番号:72(アミノ酸配列))を見出した。さらに、一方、IgG1に対して、434番目のアスパラギンをアラニンに置換したWT-M44(配列番号:73(アミノ酸配列))を作製した。さらにM44に対してH鎖C末端のヘテロジェニティーを低減するために446番目のグリシンおよび447番目のリジンを欠損させたWT-M83(配列番号:74(アミノ酸配列))を作製した。また、WT-M58に対して、434番目のアスパラギンをアラニンに置換したWT-M73(配列番号:75(アミノ酸配列))を作製した。
TOCILIZUMAB-M44(H鎖 WT-M44/配列番号:73、L鎖 WT-kappa/配列番号:54)、TOCILIZUMAB-M58(H鎖 WT-M58/配列番号:72、L鎖 WT-kappa/配列番号:54)およびTOCILIZUMAB-M73(H鎖 WT-M73/配列番号:75、L鎖 WT-kappa/配列番号:54)を調製し、ヒトFcRnへのアフィニティーおよびヒトFcRnトランスジェニックマウスによる薬物動態の評価を行った(方法は参考例参照)。
【0144】
TOCILIZUMAB-IgG1、TOCILIZUMAB-M44、TOCILIZUMAB-M58およびTOCILIZUMAB-M73のヒトFcRnへの結合性の評価をBiacoreにより行った結果、表6に示すとおり、TOCILIZUMAB-M44、TOCILIZUMAB-M58およびTOCILIZUMAB-M73の結合性はTOCILIZUMAB-IgG1よりもそれぞれ約2.7倍、約1.4倍および約3.8倍程度優れていた。
【0145】
【表6】
【0146】
TOCILIZUMAB-IgG1、TOCILIZUMAB-M44、TOCILIZUMAB-M58およびTOCILIZUMAB-M73のヒトFcRnトランスジェニックマウスにおける薬物動態の評価を行った結果を図15に示した。図15に示すとおり、TOCILIZUMAB-M44、TOCILIZUMAB-M58およびTOCILIZUMAB-M73はいずれもTOCILIZUMAB-IgG1と比較して薬物動態が向上することが見出された。その薬物動態の向上効果はヒトFcRnへの結合能と相関した。なかでもTOCILIZUMAB-M73に関しては、TOCILIZUMAB-IgG1と比較して28日後の血漿中濃度が約16倍改善していたことから、ヒトにおいてもM73の定常領域を有する抗体はIgG1の定常領域を有する抗体と比較して大幅に薬物動態が向上すると考えられた。
【0147】
〔実施例7〕PK/PDが改善した完全ヒト化IL-6レセプター抗体の作製
上記実施例で見出されたTOCILIZUMABの可変領域および定常領域の変異を複数組み合わせたTOCILIZUMAB改変体を作製し、各種スクリーニングを実施した結果、完全ヒト化IL-6レセプター抗体として、Fv3-M73(H鎖 VH4-M73/配列番号:25、L鎖 VL1-kappa/配列番号:28)、Fv4-M73(H鎖 VH3-M73/配列番号:26、L鎖 VL3-kappa/配列番号:29)、Fv5-M83(H鎖 VH5-M83/配列番号:27、L鎖 VL5-kappa/配列番号:30)を見出した。
【0148】
作製したFv3-M73、Fv4-M73、およびFv5-M83のIL-6レセプターへのアフィニティーをTOCILIZUMABと比較した(方法は参考例参照)。これらの抗体のpH7.4における可溶型IL-6レセプターへのアフィニティーを測定した結果を表7に示した。また、BaF/gp130の中和活性をTOCILIZUMABおよびコントロール(参考例の公知の高親和性高IL-6レセプター抗体、US 2007/0280945におけるVQ8F11-21 hIgG1)と比較した(方法は参考例参照)。これらの抗体のBaF/gp130による生物活性を測定した結果を図16(IL-6終濃度300 ng/mL:TOCILIZUMAB、コントロール、Fv5-M83)および図17(IL-6終濃度30 ng/mL:TOCILIZUMAB、Fv3-M73、Fv4-M73)に示した。表7に示すとおり、Fv3-M73、Fv4-M73は、TOCILIZUMABと比較して2〜3倍程度強いアフィニティーを有し、Fv5-M83はTOCILIZUMABと比較して100倍程度強いアフィニティーを示した(Fv5-M83ではアフィニティーの測定が困難であったため、定常領域をIgG1にしたFv5-IgG1(H鎖 VH5-IgG1/配列番号:76、L鎖 VL5-kappa/配列番号:30)を用いてアフィニティーを測定した、定常領域は一般にアフィニティーに影響しないと考えられる)。また、図17に示すとおりFv3-M73、Fv4-M73は、TOCILIZUMABと比較してやや強い活性を示し、図16に示すとおりFv5-M83はTOCILIZUMABと比較して50%阻害濃度として100倍以上の極めて強い活性を有し、且つ、公知の高親和性高IL-6レセプター抗体であるコントロールと比較しても50%阻害濃度として約10倍程度高い中和活性を示した。
【0149】
【表7】
【0150】
TOCILIZUMAB、Fv3-M73、および、Fv4-M73のpH7.4とpH5.8における膜型IL-6レセプターからの解離速度を測定した結果を表8に示した(方法は参考例参照)。Fv3-M73およびFv4-M73は、TOCILIZUMABと比較して膜型IL-6レセプターからの解離速度のpH依存性がそれぞれ約11倍および10倍向上していることが示された。実施例5におけるH3pI/L73と比較して、大幅に解離速度のpH依存性が向上していることから、Fv3-M73とFv4-M73の薬物動態はH3pI/L73と比較して大幅に向上していると考えられた。
【0151】
【表8】
【0152】
TOCILIZUMAB、コントロール、Fv3-M73、Fv4-M73、およびFv5-M83の等電点を当業者公知の方法により等電点電気泳動により測定した結果、TOCILIZUMABの等電点は約9.3、コントロールは約8.4〜8.5、Fv3-M73は約5.7〜5.8、Fv4-M73は約5.6〜5.7、Fv5-M83は5.4〜5.5であり、いずれの抗体もTOCILIZUMABおよびコントロールと比較して等電点が大幅に低下した。また、可変領域VH/VLの理論等電点をGENETYX(GENETYX CORPORATION)により計算したところ、TOCILIZUMABの理論等電点は9.20、コントロールは7.79、Fv3-M73は5.49、Fv4-M73は5.01、Fv5-M83は4.27であり、いずれの抗体もTOCILIZUMABおよびコントロールと比較して等電点が大幅に低下した。実施例2において等電点の低下により薬物動態が改善することが示されていることから、Fv3-M73、Fv4-M73、およびFv5-M83はTOCILIZUMABおよびコントロールと比較して薬物動態が向上していると考えられた。
【0153】
TOCILIZUMAB、Fv3-M73、Fv4-M73、およびFv5-M83の可変領域配列に存在するT-cellエピトープをTEPITOPE(Methods. 2004 Dec;34(4):468-75)を用いて解析を行った。その結果、実施例3に示したとおり、TOCILIZUMABは多くの配列がHLAに結合するT-cellエピトープが存在すると予測されたが、Fv3-M73、Fv4-M73、およびFv5-M83はT-cellエピトープに結合すると予測された配列が大幅に減少した。また、Fv3-M73、Fv4-M73、およびFv5-M83はフレームワークにマウス配列が残存せず完全ヒト化されている。これらのことから、Fv3-M73、Fv4-M73、およびFv5-M83はTOCILIZUMABと比較して大幅に免疫原性リスクが低減されている可能性が示唆された。
【0154】
〔実施例8〕完全ヒト化IL-6レセプター抗体のサルPK/PD試験
TOCILIZUMAB、コントロール、Fv3-M73、Fv4-M73、およびFv5-M83をカニクイザルに1 mg/kgで静脈内に単回投与し血漿中濃度推移を評価した(方法は参考例参照)。TOCILIZUMAB、Fv3-M73、Fv4-M73、およびFv5-M83の静脈内投与後の血漿中濃度推移を図18に示した。その結果、Fv3-M73、Fv4-M73、およびFv5-M83はいずれもTOCILIZUMABおよびコントロールと比較してカニクイザルにおいて大幅に薬物動態が改善した。なかでも、Fv3-M73とFv4-M73の薬物動態はTOCILIZUMABと比較して大幅に改善した。
【0155】
カニクイザル膜型IL-6レセプターがどの程度中和されているかの薬効を評価するために、抗体投与6日目から18日目(TOCILIZUMABに関しては3日目から10日目)までカニクイザルIL-6 5μg/kgを腰背部に連日皮下投与し、24時間後の各個体のCRP濃度を測定した(方法は参考例参照)。各抗体投与時のCRP濃度推移を図19に示した。カニクイザル可溶型IL-6レセプターがどの程度中和されているかの薬効を評価するために、カニクイザル血漿中の非結合型のカニクイザル可溶型IL-6レセプター濃度を測定し、非結合型の可溶型IL-6レセプター率を計算した(方法は参考例参照)。各抗体投与時の非結合型の可溶型IL-6レセプター率の推移を図20に示した。
【0156】
Fv3-M73、Fv4-M73、およびFv5-M83はいずれもTOCILIZUMABおよび公知の高親和性抗IL-6レセプター抗体であるコントロールと比較してカニクイザル膜型IL-6レセプターをより持続的に中和し、CRPの増加を長期間抑制した。また、Fv3-M73、Fv4-M73、およびFv5-M83はいずれもTOCILIZUMABおよびコントロールと比較してカニクイザル可溶型IL-6レセプターをより持続的に中和し、非結合型のカニクイザル可溶型IL-6レセプターの増加を長期間抑制した。これより膜型IL-6レセプターおよび可溶型IL-6レセプターの中和の持続性に関しては、Fv3-M73、Fv4-M73、およびFv5-M83はいずれもTOCILIZUMABおよびコントロールよりも優れていることが見出された。なかでもFv3-M73とFv4-M73の中和の持続性は極めて優れていた。一方、Fv5-M83のほうがFv3-M73とFv4-M73よりCRPおよび非結合型カニクイザル可溶型IL-6レセプターを低く抑制していることから、Fv5-M83は膜型IL-6レセプターおよび可溶型IL-6レセプターをFv3-M73とFv4-M73および公知の高親和性抗IL-6レセプター抗体であるコントロールよりも強力に中和していると考えられた。これはFv5-M83がコントロールよりもIL-6レセプターへのアフィニティーが強く、且つ、BaF/gp130における生物活性が強いことがカニクイザルのin vivoにおいて反映された結果であると考えられる。
【0157】
これらのことから、TOCILIZUMABおよびコントロールと比較して、Fv3-M73とFv4-M73は抗IL-6レセプター中和抗体として作用の持続性が極めて優れており、投与頻度および投与量を大幅に低減することが可能であり、また、Fv5-M83は抗IL-6レセプター中和抗体として作用の強さに極めて優れており、また作用の持続性にも優れていることが見出された。よってFv3-M73、Fv4-M73、およびFv5-M83はIL-6アンタゴニストとしての医薬品として有用であると考えられる。
【0158】
〔実施例9〕
Monocyte chemoattractant protein (MCP)-1は、単球・T細胞・NK細胞・basophilの細胞浸潤に関与することが知られている。MCP-1は、RA患者の滑膜組織・滑液中で高発現していることが報告されており(J Clin Invest. 1992 Sep;90(3):772-9)、RAの病態に関与していると考えられている(Inflamm Allergy Drug Targets. 2008 Mar;7(1):53-66.)。
【0159】
また、VEGFは強力な血管新生因子であり、RA患者の滑膜中のマクロファージ・線維芽細胞・滑膜細胞等から産生されることが知られている(J Rheumatol. 1995 Sep;22(9):1624-30.)。また、RA患者血清中のVEGFレベルと疾患活動性やradiographic progressionが相関し(Arthritis Rheum. 2003 Jun;48(6):1521-9.、Arthritis Rheum. 2001 Sep;44(9):2055-64.)、RA患者を抗IL-6R抗体TOCILIZUMABで治療することにより、血清中のVEGFレベルが低下することから、VEGFもRAの病態に重要な役割を担っていると考えられている(Mod Rheumatol. 2009;19(1):12-9.、Mediators Inflamm. 2008;2008:129873)。
【0160】
そこで、TOCILIZUMABおよびFv4-M73はsIL-6R及びIL-6刺激によるヒトRA患者由来滑膜細胞からのMCP-1およびVEGF産生を抑制できるかどうかを以下の方法で検討した。
ヒトRA患者由来滑膜細胞(TOYOBO)を5% FCS含有IMDM培地にて96 well plateに2×104/0.05 mL/wellにて播種し、CO2インキュベーター(37℃, 5%CO2)中で90分静置した。適宜希釈した濃度のTOCILIZUMAB及びFv4-M73を0.05 mL添加し、15分静置後に可溶型IL-6レセプター(SR344:参考例の方法に従って調製)を0.05 mL添加して更に30分静置し、更にIL-6(TORAY)を0.05 mL添加した(可溶型IL-6レセプター及びIL-6の終濃度は各50 ng/mL)。2日培養後、培養上清を回収し、培養上清中のMCP-1およびVEGF濃度をELISA kit (BiosourceおよびPierce Biotechnology)を用いて測定した。結果を図21と図22に示す。TOCILIZUMAB及びFv4-M73は、可溶型IL-6レセプター及びIL-6刺激によるヒトRA患者由来滑膜細胞からのMCP-1およびVEGF産生を濃度依存的に抑制した。
【0161】
これらのことから、Fv4-M73は、抗IL-6レセプター中和抗体として作用(IL-6レセプターに結合し膜型IL-6レセプターおよび可溶型IL-6レセプターのシグナルを遮断)の持続性がTOCILIZUMABと比較して極めて優れており、TOCILIZUMABと比較して投与頻度および投与量を大幅に低減することが可能であり、さらにFv4-M73は、ヒトRA患者由来滑膜細胞からのMCP-1およびVEGF産生を抑制することから、Fv4-M73はRAに極めて有用な治療薬であることが示された。
【0162】
〔参考例〕
組み換え可溶型ヒトIL-6レセプターの調製
抗原であるヒトIL-6レセプターの組み換え可溶型ヒトIL-6レセプターは以下のように調製した。J.Biochem. 108, 673-676 (1990)で報告されているN末端側1番目から344番目のアミノ酸配列からなる可溶型ヒトIL-6レセプター(Yamasakiら、Science 1988;241:825-828 (GenBank # X12830))のCHO細胞定常発現株を作製した。SR344発現CHO細胞から得られた培養上清から、Blue Sepharose 6 FFカラムクロマトグラフィー、SR344に対する特異抗体を固定したカラムによるアフィニティクロマトグラフィー、ゲルろ過カラムクロマトグラフィーの3つのカラムクロマトグラフィーにより、可溶型ヒトIL-6レセプターを精製した。メインピークとして溶出した画分を最終精製品とした。
【0163】
組み換え可溶型カニクイザルIL-6レセプター(cIL-6R)の調製
公開されているアカゲザルIL-6レセプター遺伝子配列 (Birney et al, Ensembl 2006, Nucleic Acids Res. 2006 Jan 1;34(Database issue):D556-61.) を元にオリゴDNAプライマーを作製し、カニクイザル膵臓から調製されたcDNAを鋳型とし、プライマーを用いて、PCR法によりカニクイザルIL-6レセプター遺伝子全長をコードするDNA断片を調製した。得られたDNA断片を哺乳動物細胞発現ベクターへ挿入し、これを用いてCHO定常発現株(cyno.sIL-6R産生CHO細胞)を作製した。cyno.sIL-6R産生CHO細胞の培養液をHisTrapカラム(GEヘルスケアバイオサイエンス)で精製後、Amicon Ultra-15 Ultracel-10k(Millipore)を用いて濃縮し、Superdex200pg16/60ゲルろ過カラム(GEヘルスケアバイオサイエンス)でさらに精製を行い、可溶型カニクイザルIL-6レセプター(以下、cIL-6R)の最終精製品とした。
【0164】
組み換えカニクイザルIL-6(cIL-6)の調製
カニクイザルIL-6は以下のように調製した。SWISSPROT Accession No.P79341に登録されている212アミノ酸をコードする塩基配列を作成し、哺乳動物細胞発現ベクターにクローニングし、CHO細胞に導入することで定常発現細胞株を作製した(cyno.IL-6産生CHO細胞)。cyno.IL-6産生CHO細胞の培養液をSP-Sepharose/FFカラム(GEヘルスケアバイオサイエンス)で精製後、Amicon Ultra-15 Ultracel-5k(Millipore)を用いて濃縮し、Superdex75pg26/60ゲルろ過カラム(GEヘルスケアバイオサイエンス)でさらに精製を行い、Amicon Ultra-15 Ultracel-5k(Millipore)を用いて濃縮し、カニクイザルIL-6(以下、cIL-6)の最終精製品とした。
【0165】
公知高親和性抗IL-6レセプター抗体の作製
公知の高親和性抗IL-6レセプター抗体として、US 2007/0280945 A1に記載されている高親和性抗IL-6レセプター抗体であるVQ8F11-21 hIgG1(US 2007/0280945 A1, H鎖アミノ酸配列:配列番号:77、L鎖アミノ酸配列:配列番号:78)を発現させるため、哺乳動物細胞発現用ベクターを構築した。抗体可変領域については、合成オリゴDNAを組み合わせたPCR法(assembly PCR)により作製し、定常領域についてはIgG1を使用した。Assembly PCR法により抗体可変領域と定常領域を結合させ、哺乳動物発現用ベクターへ挿入し、目的のH鎖発現ベクターおよびL鎖発現ベクターを作製した。得られた発現ベクターの塩基配列は当業者公知の方法で決定した。作製した発現ベクターを用い、発現・精製を行った。発現・精製は実施例1に記載した方法で行い、高親和性抗IL-6レセプター抗体(以降、コントロール、と記す)を得た。
【0166】
TOCILIZUMABの変異体の作製・発現・精製
TOCILIZUMABの変異体はQuikChange Site-Directed Mutagenesis Kit(Stratagene)を用いて、添付説明書記載の方法で変異体を作製し、得られたプラスミド断片を哺乳動物細胞発現ベクターに挿入し、目的のH鎖発現ベクターおよびL鎖発現ベクターを作製した。得られた発現ベクターの塩基配列は当業者公知の方法で決定した。抗体の発現は以下の方法を用いて行った。ヒト胎児腎癌細胞由来HEK293H株(Invitrogen)を10 % Fetal Bovine Serum (Invitrogen)を含むDMEM培地(Invitrogen)へ懸濁し、5〜6 × 105個/mLの細胞密度で接着細胞用ディッシュ(直径10 cm, CORNING)の各ディッシュへ10 mLずつ蒔きこみCO2インキュベーター(37℃、5% CO2)内で一昼夜培養した後に、培地を吸引除去し、CHO-S-SFM-II(Invitrogen)培地6.9 mLを添加した。調製したプラスミドをlipofection法により細胞へ導入した。得られた培養上清を回収した後、遠心分離(約2000 g、5分間、室温)して細胞を除去し、さらに0.22μmフィルターMILLEX(R)-GV(Millipore)を通して滅菌して培養上清を得た。得られた培養上清からrProtein A SepharoseTM Fast Flow(Amersham Biosciences)を用いて当業者公知の方法で抗体を精製した。精製抗体濃度は、分光光度計を用いて280 nmでの吸光度を測定した。得られた値からPACE法により算出された吸光係数を用いて抗体濃度を算出した(Protein Science 1995 ; 4 : 2411-2423)。
【0167】
ヒトgp130発現BaF3細胞株の樹立
IL-6依存増殖性を示す細胞株を得るために、以下に示すとおり、ヒトgp130を発現したBaF3細胞株の樹立を行った。
【0168】
全長ヒトgp130 cDNA(Hibiら、Cell 1990;63:1149-1157(GenBank # NM_002184))をPCRにより増幅し、pCHOI(Hirataら、FEBS Letter 1994;356:244-248)のDHFR遺伝子発現部位を除去し、Zeocin耐性遺伝子発現部位を挿入した発現ベクターpCOS2Zeoにクローニングし、pCOS2Zeo/gp130を構築した。全長ヒトIL-6R cDNAをPCRにより増幅し、pcDNA3.1(+)(Invitrogen)にクローニングし、hIL-6R/pcDNA3.1(+)を構築した。
【0169】
10μgのpCOS2Zeo/gp130をPBSに懸濁したBaF3細胞(0.8 x 107 cells)に混合し、Gene Pulser(Bio-Rad)を用いて0.33 kV, 950μFDの容量でパルスを加えた。エレクトロポーレーション処理により遺伝子導入したBaF3細胞を0.2 ng/mLのmouse interleukin-3(Peprotech)、10% Fetal Bovine Serum(以下FBS、HyClone)を含むRPMI1640培地(Invitrogen)で一昼夜培養し、100 ng/mLのhuman interleukin-6(R&D systems)、100 ng/mLのhuman interleukin-6 soluble receptor(R&D systems)および10% FBSを含むRPMI1640培地を加えて選抜し、ヒトgp130発現BaF3細胞株(以下、BaF3/gp130)を樹立した。このBaF/gp130は、human interleukin-6(R&D systems)および可溶型ヒトIL-6レセプター存在下で増殖することから、抗IL-6レセプター抗体の増殖阻害活性(すなわちIL-6レセプター中和活性)の評価に使用することが可能である。
【0170】
ヒトgp130発現BaF3細胞(BaF/gp130)による生物活性評価
IL-6/IL-6レセプター依存性増殖を示すBaF3/gp130を用いて、IL-6レセプター中和活性を評価した。BaF3/gp130を10% FBSを含むRPMI1640培地で3回洗浄した後に、5 x 104 cells/mLとなるように600 ng/mLないしは60 ng/mLのhuman interleukin-6(TORAY)(終濃度は300 ng/mLないしは30 ng/mL)、適当量の可溶型ヒトIL-6レセプターおよび10% FBSを含むRPMI1640培地に懸濁し、96 well-plate(CORNING)の各wellに50μLずつ分注した。次に、精製した抗体を10% FBSを含むRPMI1640に希釈して、各wellに50μLずつ混合した。37℃、5% CO2条件下で、3日間培養し、PBSで2倍に希釈したWST-8試薬(Cell Counting Kit-8、株式会社同仁化学研究所)を20μL/wellで加え、直後にSUNRISE CLASSIC(TECAN)を用いて450 nmの吸光度(参照波長620 nm)を測定した。2時間培養した後に、再度450 nmの吸光度(参照波長620 nm)を測定し、2時間の吸光度変化を指標にIL-6レセプター中和活性を評価した。
【0171】
Biacoreによる可溶型ヒトIL-6レセプターへの結合評価
Biacore T100(GE Healthcare)を用いて、抗原抗体反応の速度論的解析を行った。センサーチップ上にアミンカップリング法でprotein Aあるいはprotein A/Gあるいはanti-IgG(γ-chain specific)F(ab’)2を適当量固定化し、次にpH7.4において目的の抗体を結合させ、さらにpH7.4において種々の濃度に調製した可溶型IL-6レセプターをアナライトとして流し、抗体と可溶型ヒトIL-6レセプターの相互作用を測定した。測定は全て37℃で実施した。測定で得られたセンサーグラムから、カイネティクスパラメーターである結合速度定数ka(1/Ms)、および解離速度定数kd(1/s)を算出し、その値をもとにKD(M)を算出した。各パラメーターの算出にはBiacore T100 Evaluation Software(GE Healthcare)を用いた。
【0172】
Biacoreによる膜型IL-6レセプターへのpH依存的解離評価
Biacore T100(GE Healthcare)を用いてpH5.8, pH7.4における膜型IL-6レセプターへの抗原抗体反応を観測した。センサーチップ上に固定化した可溶型ヒトIL-6レセプターへの結合を評価することで、膜型IL-6レセプターへの結合を評価した。SR344を当業者公知の方法に従ってビオチン化し、ストレプトアビジンとビオチンの親和性を利用し、ストレプトアビジンを介してビオチン化可溶型ヒトIL-6レセプターをセンサーチップ上に固定化した。測定は全て37℃で実施し、移動相のバッファーは10 mM MES pH5.8, 150 mM NaCl, 0.05% Tween20とし、そこにpH依存的結合クローンをpH7.4の条件下で注入して可溶型ヒトIL-6レセプターと結合させたのち(注入サンプルのバッファーは10 mM MES pH7.4, 150 mM NaCl, 0.05% Tween20)、移動相のpHである5.8で各クローンのpH依存的な解離を観測した。サンプル濃度を0.25μg/mLとし、10 mM MES pH7.4, 150 mM NaCl, 0.05 % Tween20で結合させ、10 mM MES pH5.8, 150 mM NaCl, 0.05% Tween20で解離させたときのpH5.8における解離相のみBiacore T100 Evaluation Software(GE Healthcare)を用いフィッティングすることにより、pH5.8における解離速度定数(kd(1/s))を算出した。同様にまた、サンプル濃度を0.5μg/mLとし、10 mM MES pH7.4, 150 mM NaCl, 0.05% Tween20で結合させ、10 mM MES pH7.4, 150 mM NaCl, 0.05% Tween20で解離させたときのpH7.4における解離相のみBiacore T100 Evaluation Software(GE Healthcare)を用いフィッティングすることにより、pH7.4における解離速度定数(kd(1/s))を算出した。
【0173】
ヒトFcRnへの結合評価
FcRnはFcRnとβ2-microglobulinの複合体である。公開されているヒトFcRn遺伝子配列(J. Exp. Med. 180 (6), 2377-2381 (1994))を元に、オリゴDNAプライマーを作製した。ヒトcDNA(Human Placenta Marathon-Ready cDNA, Clontech)を鋳型とし、作製したプライマーを用いPCR法により遺伝子全長をコードするDNA断片を調整した。得られたDNA断片を鋳型に、PCR法によりシグナル領域を含む細胞外領域(Met1-Leu290)をコードするDNA断片を増幅し、哺乳動物細胞発現ベクターへ挿入した(ヒトFcRnアミノ酸配列/配列番号:79)。同様に、公開されているヒトβ2-microglobulin遺伝子配列(Proc. Natl. Acad. Sci. U.S.A. 99 (26), 16899-16903 (2002))を元に、オリゴDNAプライマーを作製した。ヒトcDNA(Hu-Placenta Marathon-Ready cDNA, CLONTECH)を鋳型とし、作製したプライマーを用いPCR法により遺伝子全長をコードするDNA断片を調製した。得られたDNA断片を鋳型に、PCR法によりシグナル領域を含むβ2-microglobulin全長(Met1-Met119)をコードするDNA断片を増幅し、哺乳動物細胞発現ベクターへ挿入した(ヒトβ2-microglobulinアミノ酸配列/配列番号:80)。
【0174】
可溶型ヒトFcRnの発現は以下の手順で行った。調製したヒトFcRnおよびヒトβ2-microglobulinのプラスミドを、10 % Fetal Bovine Serum (Invitrogen)を用いたlipofection法により、ヒト胎児腎癌細胞由来HEK293H株(Invitrogen)の細胞へ導入した。得られた培養上清を回収した後、IgG Sepharose 6 Fast Flow(Amersham Biosciences)を用い、(J Immunol. 2002 Nov 1;169(9):5171-80.)の方法に従い精製を行った。その後、HiTrap Q HP(GE Healthcare)により精製を行った。
【0175】
マウスにおける抗体血漿中濃度の測定
マウス血漿中抗体濃度測定はELISA法にて当業者公知の方法で測定した。
【0176】
サルPK/PD試験による抗体血漿中濃度、CRP濃度、非結合型可溶型IL-6レセプターの測定
カニクイザル血漿中濃度測定はELISA法にて当業者公知の方法で測定した。
CRP濃度はサイアスR CRP(関東化学株式会社)にて、自動分析装置(TBA-120FR、東芝メディカルシステムズ株式会社)を用いて測定した。
【0177】
カニクイザル血漿中の非結合型の可溶型カニクイザルIL-6レセプター濃度を以下の通り測定した。カニクイザルの血漿30μLを0.22μmのフィルターカップ(Millipore)において乾燥させた適量のrProtein A Sepharose Fast Flow(GE Healthcare)樹脂に添加することで血漿中に存在する全てのIgG型抗体(カニクイザルIgG、抗ヒトIL-6レセプター抗体および抗ヒトIL-6レセプター抗体-可溶型カニクイザルIL-6レセプター複合体)をProteinAに吸着させた。その後、高速遠心機でスピンダウンし、パス溶液を回収した。パス溶液にはproteinAに結合した抗ヒトIL-6レセプター抗体-可溶型カニクイザルIL-6レセプター複合体は含まれないため、proteinAパス溶液中の可溶型カニクイザルIL-6レセプター濃度を測定することによって、非結合型の可溶型IL-6レセプター濃度を測定可能である。可溶型カニクイザルIL-6レセプター濃度は、上記で作製した可溶型カニクイザルIL-6レセプター(cIL-6R)をスタンダードに用いて、ヒトIL-6レセプター濃度を測定する当業者公知の方法で測定した。非結合型の可溶型IL-6レセプター率は以下の計算式によって計算した。
【0178】
(抗体投与後の非結合型の可溶性IL-6レセプター濃度÷抗体投与前の可溶性IL-6レセプター濃度)×100
【技術分野】
【0001】
本発明は、抗IL-6レセプター抗体を有効成分として含有する医薬組成物、および、その製造法等に関する。
【背景技術】
【0002】
抗体は血漿中での安定性が高く、副作用も少ないことから医薬品として注目されている。中でもIgG型の抗体医薬は多数上市されており、現在も数多くの抗体医薬が開発されている(非特許文献1、非特許文献2)。IL-6は様々な自己免疫疾患・炎症性疾患・悪性腫瘍等に関与するサイトカインであり(非特許文献3)、ヒト化抗IL-6レセプターIgG1抗体であるTOCILIZUMAB(トシリズマブ)はIL-6レセプターに特異的に結合する。TOCILIZUMABはIL-6の生物学的作用を中和することで、関節リウマチ等のIL-6が関連する疾患の治療薬として利用可能であると考えられており(特許文献1、2、3、非特許文献4)、日本においてはキャッスルマン病および関節リウマチの治療薬として承認されている(非特許文献5)。
【0003】
TOCILIZUMABのようなヒト化抗体は第1世代の抗体医薬であり、第1世代の抗体医薬を改良して薬効・利便性・コストを改善させた第2世代の抗体医薬が現在開発されている。第2世代の抗体医薬に適用可能な技術として様々な技術が開発されており、エフェクター機能、抗原結合能、薬物動態、安定性を向上させる、あるいは、免疫原性リスクを低減させる技術等が報告されている。薬効を増強させる、あるいは、投与量を低減させる方法として、IgG抗体のFc領域のアミノ酸置換により抗体依存性細胞障害活性(ADCC活性)や補体依存性細胞障害活性(CDC活性)を増強させる技術が報告されている(非特許文献6)。また、抗原結合能、抗原中和能を増強させる技術として、アフィニティーマチュレーション技術(非特許文献7)が報告されており、可変領域の相補性決定領域(CDR; complementarity determining region)などのアミノ酸に変異を導入することで抗原への結合活性を増強することが可能である。抗原結合能の増強によりin vitroの生物活性を向上させる、あるいは投与量を低減することが可能であり、さらにin vivoでの薬効を向上させることも可能である(非特許文献8)。現在、抗RSV抗体の第一世代薬であるPalivizumabより優れた効果を発揮するとされるMotavizumab(アフィニティーマチュレーションにより作製)の臨床試験が行われている(非特許文献9)。抗IL-6レセプター抗体においては、TOCILIZUMABよりアフィニティーが強いとされるアフィニティーが約0.05 nMの抗体が報告されているが(特許文献4)、0.05 nMより強いアフィニティーを有するヒト抗体あるいはヒト化抗体あるいはキメラ抗体の報告は無い。
【0004】
現在の抗体医薬が抱える問題として、投与タンパク量が非常に多いことによる高い製造コストが挙げられる。ヒト化抗IL-6レセプターIgG1抗体であるTOCILIZUMABにおいても投与量は8 mg/kg/month程度の静脈内注射が想定されている(非特許文献4)。また投与形態については、慢性的な自己免疫疾患の場合は皮下投与製剤が望ましいとされている。一般的に皮下投与製剤は高濃度製剤であることが必要であり、IgGタイプの抗体製剤の場合、安定性等の点から一般的には100 mg/mL程度の製剤が限度であると考えられる(非特許文献10)。持続的な治療効果を発揮できるよう抗体の血漿中半減期を長くすることで投与タンパク量を小さくし、高い安定性を付与することによって、長い投与間隔での皮下投与を可能にし、低コスト且つ利便性の高い第2世代の抗体医薬を提供することが可能である。
【0005】
抗体の薬物動態にはFcRnが大きく関与しており、抗体のアイソタイプ間の血漿中半減期の違いに関しては、IgG1およびIgG2が最も血漿中半減期に優れ、IgG3およびIgG4がそれより劣ることが知られている(非特許文献11)。血漿中半減期に優れるIgG1およびIgG2の抗体の血漿中半減期をさらに向上させる方法として、FcRnへの結合を増強する定常領域のアミノ酸置換が報告されている(非特許文献12、13)。また、免疫原性の観点からは定常領域よりも可変領域のアミノ酸置換により血漿中半減期をさらに向上させるほうが望ましいが(特許文献5)、これまで可変領域を改変することによりIL-6レセプター抗体の血漿中半減期を向上させた報告は無い。
【0006】
バイオ医薬品を開発するにあたってもうひとつの重要な問題は免疫原性である。一般的にマウス抗体はヒト化することによって免疫原性が低減される。ヒト化する際のテンプレートフレームワークにジャームライン配列を用いることにより、免疫原性のリスクはさらに軽減できるとされている(非特許文献14)。しかしながら、完全ヒト抗TNF抗体であるAdalimumabであっても13〜17%と高頻度で免疫原性が出現し、免疫原性が出現した患者においては治療効果の低減が見られている(非特許文献15、16)。ヒト抗体であってもCDRにT-cellエピトープが存在する可能性があり、CDR上のT-cellエピトープが免疫原性の原因となっている可能性がある。T-cellエピトープをin silicoあるいはin vitroで予測する方法が報告されているが(非特許文献17、18)、これらの方法を用いて予測されるT-cellエピトープを除去することで、免疫原性リスクを低減することが可能であると考えられる(非特許文献19)。
【0007】
ヒト化抗IL-6レセプターIgG1抗体であるTOCILIZUMABは、マウスPM1抗体をヒト化したIgG1抗体である。H鎖、L鎖それぞれNEW、REIのヒト配列をテンプレートフレームワークとして用いてCDRグラフティングが行われているが、活性保持に重要なアミノ酸として5アミノ酸がマウス配列としてフレームワークに残存している(非特許文献20)。これまでヒト化抗体であるTOCILIZUMABのフレームワークに残存するマウス配列の活性を低下させること無く完全ヒト化した報告は無い。また、TOCILIZUMABのCDR配列はマウス配列であり、Adalimumab同様、CDRにT-cellエピトープが存在する可能性があり、免疫原性のリスクは否定できない。TOCILIZUMABの臨床試験において、薬効用量である8 mg/kgにおいて抗TOCILIZUMAB抗体の出現は認められていないが、4 mg/kgおよび2 mg/kgにおいては該抗体が認められている(特許文献6)。これらのことから、TOCILIZUMABの免疫原性については改善の余地があると考えられる。しかしながら、これまでにアミノ酸置換により免疫原性リスクを改善させたTOCILIZUMABに関する報告は無い。
【0008】
TOCILIZUMABのアイソタイプはIgG1であるが、アイソタイプの違いはすなわち定常領域の配列の違いであり、定常領域の配列はエフェクター機能、薬物動態、物性等に大きく影響を与えると考えられていることから、抗体医薬の開発にとって定常領域の配列の選択は極めて重要である(非特許文献11)。近年、抗体医薬の安全性が非常に重要視されており、TGN1412のPhaseI臨床試験で見られた重大な副作用の原因の一つとして、抗体のFc部分とFcγレセプターの相互作用(エフェクター機能)が考えられている(非特許文献21)。抗原の生物学的作用を中和することが目的の抗体医薬においては、ADCC等のエフェクター機能に重要なFcγレセプターへの結合は不必要であり、副作用の点から考えるとFcγレセプターへの結合はむしろ好ましくない可能性も考えられる。Fcγレセプターへの結合を低下させる方法としては、IgG抗体のアイソタイプをIgG1からIgG2あるいはIgG4に変える方法があるが(非特許文献22)、FcγレセプターIへの結合および薬物動態の観点からはIgG4よりはIgG2が望ましいと考えられる(非特許文献11)。TOCILIZUMABのアイソタイプはIgG1であり、IL-6レセプター中和抗体であることから、ADCC等のエフェクター機能は必要とせず副作用の可能性を考えた場合、アイソタイプはIgG2が望ましい可能性も考えられる。
【0009】
一方、抗体を医薬品として開発するにあたり、そのタンパク質の物性、中でも均一性と安定性は極めて重要であり、IgG2アイソタイプは、ヒンジ領域のジスルフィド結合に由来するヘテロジェニティーが極めて多いことが報告されている(非特許文献23)。これに由来する目的物質/関連物質のヘテロジェニティーの製造間差を維持しつつ医薬品として大量に製造することは容易ではなくコスト増につながり、可能な限り単一物質であることが望まれる。さらに抗体のH鎖C末端配列のヘテロジェニティーとして、C末端アミノ酸のリジン残基の欠損、および、C末端の2アミノ酸のグリシン、リジンの欠損によるC末端カルボキシル基のアミド化が報告されており(非特許文献24)、IgG2アイソタイプの抗体を医薬品として開発する上には高い安定性を維持しつつこれらのヘテロジェニティーが低減されていることが望ましい。利便性に優れた安定な高濃度の皮下投与製剤を作製するためには、安定性が高いだけでなく、血漿中半減期に関してもTOCILIZUMABのアイソタイプであるIgG1よりも優れていることが望ましい。しかしながら、これまでにIgG2アイソタイプの定常領域の抗体に関するヘテロジェニティーが低減され、高い安定性を有し、IgG1アイソタイプの定常領域の抗体よりも優れた血漿中半減期を有する定常領域の配列の報告はない。
【0010】
なお、本発明の先行技術文献を以下に示す。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】WO 92/19759
【特許文献2】WO 96/11020
【特許文献3】WO 96/12503
【特許文献4】WO 2007/143168
【特許文献5】WO 2007/114319
【特許文献6】WO 2004/096273
【非特許文献】
【0012】
【非特許文献1】Janice M Reichert, Clark J Rosensweig, Laura B Faden & Matthew C Dewitz, Monoclonal antibody successes in the clinic, Nature Biotechnology 23, 1073 - 1078 (2005)
【非特許文献2】Pavlou AK, Belsey MJ., The therapeutic antibodies market to 2008., Eur J Pharm Biopharm. 2005 Apr;59(3):389-96.
【非特許文献3】Nishimoto N, Kishimoto T., Interleukin 6: from bench to bedside., Nat Clin Pract Rheumatol. 2006 Nov;2(11):619-26.
【非特許文献4】Maini RN, Taylor PC, Szechinski J, Pavelka K, Broll J, Balint G, Emery P, Raemen F, Petersen J, Smolen J, Thomson D, Kishimoto T; CHARISMA Study Group., Double-blind randomized controlled clinical trial of the interleukin-6 receptor antagonist, Tocilizumab, in European patients with rheumatoid arthritis who had an incomplete response to methotrexate., Arthritis Rheum. 2006 Sep;54(9):2817-29
【非特許文献5】Nishimoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S, Nakano N, Ikeda Y, Sasaki T, Nishioka K, Hara M, Taguchi H, Kimura Y, Kato Y, Asaoku H, Kumagai S, Kodama F, Nakahara H, Hagihara K, Yoshizaki K, Kishimoto T. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005 Oct 15;106(8):2627-32.
【非特許文献6】Kim SJ, Park Y, Hong HJ., Antibody engineering for the development of therapeutic antibodies., Mol Cells. 2005 Aug 31;20(1):17-29. Review.
【非特許文献7】Rothe A, Hosse RJ, Power BE. Ribosome display for improved biotherapeutic molecules. Expert Opin Biol Ther. 2006 Feb;6(2):177-87.
【非特許文献8】Rajpal A, Beyaz N, Haber L, Cappuccilli G, Yee H, Bhatt RR, Takeuchi T, Lerner RA, Crea R., A general method for greatly improving the affinity of antibodies by using combinatorial libraries., Proc Natl Acad Sci U S A. 2005 Jun 14;102(24):8466-71. Epub 2005 Jun 6.
【非特許文献9】Wu H, Pfarr DS, Johnson S, Brewah YA, Woods RM, Patel NK, White WI, Young JF, Kiener PA. Development of Motavizumab, an Ultra-potent Antibody for the Prevention of Respiratory Syncytial Virus Infection in the Upper and Lower Respiratory Tract. J Mol Biol. 2007, 368, 652-665
【非特許文献10】Shire SJ, Shahrokh Z, Liu J. Challenges in the development of high protein concentration formulations. J Pharm Sci. 2004 Jun;93(6):1390-402.
【非特許文献11】Salfeld JG. Isotype selection in antibody engineering.Nat Biotechnol. 2007 Dec;25(12):1369-72.
【非特許文献12】Hinton PR, Xiong JM, Johlfs MG, Tang MT, Keller S, Tsurushita N., An engineered human IgG1 antibody with longer serum half-life., J Immunol. 2006 Jan 1;176(1):346-56
【非特許文献13】Ghetie V, Popov S, Borvak J, Radu C, Matesoi D, Medesan C, Ober RJ, Ward ES., Increasing the serum persistence of an IgG fragment by random mutagenesis., Nat Biotechnol. 1997 Jul;15(7):637-40
【非特許文献14】Hwang WY, Almagro JC, Buss TN, Tan P, Foote J. Use of human germline genes in a CDR homology-based approach to antibody humanization.Methods. 2005 May;36(1):35-42
【非特許文献15】Bartelds GM, Wijbrandts CA, Nurmohamed MT, Stapel S, Lems WF, Aarden L, Dijkmans BA, Tak P, Wolbink GJ. Clinical response to adalimumab: The relationship with anti-adalimumab antibodies and serum adalimumab concentrations in rheumatoid arthritis. Ann Rheum Dis. 2007 Mar 9; [Epub ahead of print]
【非特許文献16】Bender NK, Heilig CE, Droll B, Wohlgemuth J, Armbruster FP, Heilig B. Immunogenicity, efficacy and adverse events of adalimumab in RA patients. Rheumatol Int. 2007 Jan;27(3):269-74.
【非特許文献17】Van Walle I, Gansemans Y, Parren PW, Stas P, Lasters I. Immunogenicity screening in protein drug development.Expert Opin Biol Ther. 2007 Mar;7(3):405-18.
【非特許文献18】Jones TD, Phillips WJ, Smith BJ, Bamford CA, Nayee PD, Baglin TP, Gaston JS, Baker MP. Identification and removal of a promiscuous CD4+ T cell epitope from the C1 domain of factor VIII. J Thromb Haemost. 2005 May;3(5):991-1000.
【非特許文献19】Chirino AJ, Ary ML, Marshall SA. Minimizing the immunogenicity of protein therapeutics. Drug Discov Today. 2004 Jan 15;9(2):82-90.
【非特許文献20】Sato K, Tsuchiya M, Saldanha J, Koishihara Y, Ohsugi Y, Kishimoto T, Bendig MM. Reshaping a human antibody to inhibit the interleukin 6-dependent tumor cell growth. Cancer Res. 1993 Feb 15;53(4):851-6.
【非特許文献21】Strand V, Kimberly R, Isaacs JD. Biologic therapies in rheumatology: lessons learned future directions. Nat Rev Drug Discov. 2007 Jan;6(1):75-92.
【非特許文献22】Gessner JE, Heiken H, Tamm A, Schmidt RE. The IgG Fc receptor family. Ann Hematol. 1998 Jun;76(6):231-48.
【非特許文献23】Dillon TM, Ricci MS, Vezina C, Flynn GC, Liu YD, Rehder DS, Plant M, Henkle B, Li Y, Deechongkit S, Varnum B, Wypych J, Balland A, Bondarenko PV. Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. J Biol Chem. 2008 Jun 6;283(23):16206-15.
【非特許文献24】Johnson KA, Paisley-Flango K, Tangarone BS, Porter TJ, Rouse JC. Cation exchange-HPLC and mass spectrometry reveal C-terminal amidation of an IgG1 heavy chain. Anal Biochem. 2007 Jan 1;360(1):75-83.
【発明の概要】
【発明が解決しようとする課題】
【0013】
本発明はこのような状況に鑑みて為されたものであり、その目的は、ヒト化抗IL-6レセプターIgG1抗体であるTOCILIZUMABの可変領域および定常領域のアミノ酸配列を改変することで、抗原中和能を増強させつつ、薬物動態を向上させることで投与頻度を少なくし持続的に治療効果を発揮し、且つ、免疫原性、安全性、物性(安定性および均一性)を改善させ、TOCILIZUMABより優れた第2世代の分子からなる医薬組成物(以下本明細書では「薬剤」あるいは「製剤」と記載する場合もある)、並びに、それらの医薬組成物の製造方法を提供することにある。
【課題を解決するための手段】
【0014】
本発明者らは、第1世代のヒト化抗IL-6レセプターIgG1抗体であるTOCILIZUMABの可変領域および定常領域のアミノ酸配列を改変することで、薬効を増強させつつ、薬物動態を向上させることで投与頻度を少なくし持続的に治療効果を発揮し、且つ、免疫原性、安全性、物性(安定性および均一性)を改善させ、TOCILIZUMABより優れた第2世代の分子の創製に向けて、鋭意研究を行った。その結果、本発明者らは、TOCILIZUMABの可変領域において、抗原への結合能(アフィニティー)を向上させるCDR変異を複数見出し、その組み合わせにより大幅にアフィニティーを向上させることに成功した。また本発明者らは、可変領域配列の等電点を低下させる改変を導入することで薬物動態を向上させることに成功した。また本発明者らは、抗原であるIL-6レセプターへの結合にpH依存性を付与することで1分子の抗体で複数回抗原を中和することを可能にし、薬物動態を向上させることに成功した。また本発明者らは、TOCILIZUMABのフレームワークに残存するマウス由来の配列を完全ヒト化し、可変領域においてin silicoで予測されたT-cellエピトープペプチドの数を低減させることで、免疫原性リスクを低減させることに成功した。さらに本発明者らは、TOCILIZUMABの定常領域において、安全性を高めるためにFcγレセプターへの結合をIgG1よりも低下させ、IgG1よりも薬物動態を改善させ、さらに安定性を低下させることなくIgG2のヒンジ領域のジスルフィド結合に由来するヘテロジェニティーおよびH鎖C末端に由来するヘテロジェニティーを低減させた新規な定常領域配列を見出すことに成功した。これらCDR領域アミノ酸配列の改変、可変領域アミノ酸配列の改変、定常領域アミノ酸配列の改変を適切に組み合わせることでTOCILIZUMABより優れた第2世代の分子の創製に成功した。
【0015】
本発明は、ヒト化抗IL-6レセプターIgG1抗体であるTOCILIZUMABの可変領域および定常領域のアミノ酸配列の改変により、より優れた抗原(IL-6レセプター)への結合能を有し、より優れた薬物動態を有し、より優れた安全性・免疫原性リスク・物性(安定性、均一性)を有するヒト化抗IL-6レセプターIgG抗体からなる医薬組成物、並びに、それらの医薬組成物の製造方法に関する。より具体的には、下記〔1〕〜〔11〕を提供するものである。
【0016】
〔1〕 以下の(a)〜(f)いずれかに記載のポリペプチド;
(a) 配列番号:1(VH4-M73のCDR1)の配列を有するCDR1、配列番号:2(VH4-M73のCDR2)の配列を有するCDR2、および配列番号:3(VH4-M73のCDR3)の配列を有するCDR3を含むポリペプチド、
(b) 配列番号:4(VH3-M73のCDR1)の配列を有するCDR1、配列番号:5(VH3-M73のCDR2)の配列を有するCDR2、および配列番号:6(VH3-M73のCDR3)の配列を有するCDR3を含むポリペプチド、
(c) 配列番号:7(VH5-M83のCDR1)の配列を有するCDR1、配列番号:8(VH5-M83のCDR2)の配列を有するCDR2、および配列番号:9(VH5-M83のCDR3)の配列を有するCDR3を含むポリペプチド、
(d) 配列番号:10(VL1のCDR1)の配列を有するCDR1、配列番号:11(VL1のCDR2)の配列を有するCDR2、および配列番号:12(VL1のCDR3)の配列を有するCDR3を含むポリペプチド、
(e) 配列番号:13(VL3のCDR1)の配列を有するCDR1、配列番号:14(VL3のCDR2)の配列を有するCDR2、および配列番号:15(VL3のCDR3)の配列を有するCDR3を含むポリペプチド、
(f) 配列番号:16(VL5のCDR1)の配列を有するCDR1、配列番号:17(VL5のCDR2)の配列を有するCDR2、および配列番号:18(VL5のCDR3)の配列を有するCDR3を含むポリペプチド。
〔2〕 以下の(a)〜(c)いずれかに記載の抗体;
(a) 配列番号:1(VH4-M73のCDR1)の配列を有するCDR1、配列番号:2(VH4-M73のCDR2)の配列を有するCDR2、および配列番号:3(VH4-M73のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに
配列番号:10(VL1のCDR1)の配列を有するCDR1、配列番号:11(VL1のCDR2)の配列を有するCDR2、および配列番号:12(VL1のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体、
(b) 配列番号:4(VH3-M73のCDR1)の配列を有するCDR1、配列番号:5(VH3-M73のCDR2)の配列を有するCDR2、および配列番号:6(VH3-M73のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに
配列番号:13(VL3のCDR1)の配列を有するCDR1、配列番号:14(VL3のCDR2)の配列を有するCDR2、および配列番号:15(VL3のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体、
(c) 配列番号:7(VH5-M83のCDR1)の配列を有するCDR1、配列番号:8(VH5-M83のCDR2)の配列を有するCDR2、および配列番号:9(VH5-M83のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに
配列番号:16(VL5のCDR1)の配列を有するCDR1、配列番号:17(VL5のCDR2)の配列を有するCDR2、および配列番号:18(VL5のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体。
〔3〕 以下の(a)〜(f)いずれかに記載の可変領域;
(a) 配列番号:19(VH4-M73の可変領域)の配列を有する重鎖可変領域、
(b) 配列番号:20(VH3-M73の可変領域)の配列を有する重鎖可変領域、
(c) 配列番号:21(VH5-M83の可変領域)の配列を有する重鎖可変領域、
(d) 配列番号:22(VL1の可変領域)の配列を有する軽鎖可変領域、
(e) 配列番号:23(VL3の可変領域)の配列を有する軽鎖可変領域、
(f) 配列番号:24(VL5の可変領域)の配列を有する軽鎖可変領域。
〔4〕 以下の(a)〜(c)いずれかに記載の抗体;
(a) 配列番号:19(VH4-M73の可変領域)の配列を有する重鎖可変領域および配列番号:22(VL1の可変領域)の配列を有する軽鎖可変領域を含む抗体、
(b) 配列番号:20(VH3-M73の可変領域)の配列を有する重鎖可変領域および配列番号:23(VL3の可変領域)の配列を有する軽鎖可変領域を含む抗体、
(c) 配列番号:21(VH5-M83の可変領域)の配列を有する重鎖可変領域および配列番号:24(VL5の可変領域)の配列を有する軽鎖可変領域を含む抗体。
〔5〕 以下の(a)〜(f)いずれかに記載の重鎖又は軽鎖;
(a) 配列番号:25(VH4-M73)の配列を有する重鎖、
(b) 配列番号:26(VH3-M73)の配列を有する重鎖、
(c) 配列番号:27(VH5-M83)の配列を有する重鎖、
(d) 配列番号:28(VL1)の配列を有する軽鎖、
(e) 配列番号:29(VL3)の配列を有する軽鎖、
(f) 配列番号:30(VL5)の配列を有する軽鎖。
〔6〕 以下の(a)〜(c)いずれかに記載の抗体;
(a) 配列番号:25(VH4-M73)の配列を有する重鎖および配列番号:28(VL1)の配列を有する軽鎖を含む抗体、
(b) 配列番号:26(VH3-M73)の配列を有する重鎖および配列番号:29(VL3)の配列を有する軽鎖を含む抗体、
(c) 配列番号:27(VH5-M83)の配列を有する重鎖および配列番号:30(VL5)の配列を有する軽鎖を含む抗体。
〔7〕 〔1〕〜〔6〕いずれかに記載のポリペプチドをコードする遺伝子。
〔8〕 〔7〕に記載の遺伝子を含むベクター。
〔9〕 〔8〕に記載のベクターを保持する宿主細胞。
〔10〕 〔9〕の宿主細胞を培養することにより、〔1〕〜〔6〕いずれかに記載のポリペプチドを製造する方法。
〔11〕 〔1〕〜〔6〕いずれかに記載のポリペプチド、または〔10〕に記載の方法によって製造されるポリペプチドを含む医薬組成物。
【発明の効果】
【0017】
本発明によって得られたヒト化抗IL-6レセプターIgG抗体は、薬効が増強し、薬物動態が向上したことで投与頻度を少なくすることができ、持続的に治療効果を発揮することができる。
【図面の簡単な説明】
【0018】
【図1】TOCILIZUMABのIL-6レセプターへの親和性を向上させる変異箇所をまとめたリストを示す図である。HCDR2のTOCILIZUMAB配列を配列番号:81、HCDR2の変異後配列(上段)を配列番号:82、HCDR2の変異後配列(下段)を配列番号:83、HCDR3のTOCILIZUMAB配列を配列番号:84、HCDR3の変異後配列(上段)を配列番号:85、HCDR3の変異後配列(下段)を配列番号:86、LCDR1のTOCILIZUMAB配列を配列番号:87、LCDR1の変異後配列(上段)を配列番号:88、LCDR1の変異後配列(下段)を配列番号:89、LCDR3のTOCILIZUMAB配列を配列番号:90、LCDR3の変異後配列(上段)を配列番号:91、LCDR3の変異後配列(下段)を配列番号:92に示す。
【図2】TOCILIZUMABとRDC-23のBaF/gp130における中和活性を示す図である。
【図3】TOCILIZUMABのIL-6レセプターへのIL-6レセプターへの結合を大きく低下させることなく可変領域の等電点を低下することできる変異箇所をまとめたリストを示す図である。図中の星印はヒト配列にするために等電点には影響しないが変異した箇所である。HFR1のTOCILIZUMAB配列を配列番号:93、HFR1の変異後配列を配列番号:94、HCDR1のTOCILIZUMAB配列を配列番号:95、HCDR1の変異後配列を配列番号:96、HFR2のTOCILIZUMAB配列を配列番号:97、HFR2の変異後配列を配列番号:98、HCDR2のTOCILIZUMAB配列を配列番号:81、HCDR2の変異後配列を配列番号:99、HFR4のTOCILIZUMAB配列を配列番号:100、HFR4の変異後配列を配列番号:101、LFR1のTOCILIZUMAB配列を配列番号:102、LFR1の変異後配列を配列番号:103、LCDR1のTOCILIZUMAB配列を配列番号:87、LCDR1の変異後配列を配列番号:104、LFR2のTOCILIZUMAB配列を配列番号:105、LFR2の変異後配列を配列番号:106、LCDR2のTOCILIZUMAB配列を配列番号:107、LCDR2の変異後配列を配列番号:108、109、LFR3のTOCILIZUMAB配列を配列番号:110、LFR3の変異後配列を配列番号:111、LFR4のTOCILIZUMAB配列を配列番号:112、LFR4の変異後配列を配列番号:113に示す。
【図4】TOCILIZUMABとH53/L28のBaF/gp130における中和活性を示す図である。
【図5】TOCILIZUMABとH53/L28をマウスに静脈内投与後の血漿中濃度推移を示したグラフである。
【図6】TOCILIZUMABとH53/L28をマウスに皮下投与後の血漿中濃度推移を示したグラフである。
【図7】IgG分子がエンドソーム内で膜型抗原から解離することで再度新たな抗原に結合することを示す模式図である。
【図8】TOCILIZUMABのIL-6レセプターへの結合にpH依存性(pH7.4で結合し、pH5.8で解離する)を付与することのできる変異箇所をまとめたリストを示す図である。HFR1のTOCILIZUMAB配列を配列番号:93、HFR1の変異後配列を配列番号:114、HCDR1のTOCILIZUMAB配列を配列番号:95、HCDR1の変異後配列を配列番号:115、LCDR1のTOCILIZUMAB配列を配列番号:87、LCDR1の変異後配列を配列番号:116、LCDR2のTOCILIZUMAB配列を配列番号:107、LCDR2の変異後配列を配列番号:117に示す。
【図9】TOCILIZUMABとH3pI/L73のBaF/gp130における中和活性を示す図である。
【図10】TOCILIZUMABとH3pI/L73をカニクイザルに静脈内投与後の血漿中濃度推移を示したグラフである。
【図11】TOCILIZUMABとH3pI/L73をヒトIL-6レセプタートランスジェニックマウスに静脈内投与後の血漿中濃度推移を示したグラフである。
【図12】TOCILIZUMAB、TOCILIZUMABΔK、およびTOCILIZUMABΔGKを陽イオン交換クロマトグラフィーによりC末端に由来するヘテロジェニティーの評価を行った結果を示した図である。
【図13】TOCILIZUMAB-IgG1、TOCILIZUMAB-IgG2、およびTOCILIZUMAB-SKSCを陽イオン交換クロマトグラフィーによりジスルフィド結合に由来するヘテロジェニティーの評価を行った結果を示した図である。
【図14】TOCILIZUMAB-IgG1、TOCILIZUMAB-IgG2、およびTOCILIZUMAB-SKSCを示差走査型熱量測定(DSC)により測定した変性曲線および各FabドメインのTm値を示した図である。
【図15】TOCILIZUMAB-IgG1、TOCILIZUMAB-M44、TOCILIZUMAB-M58およびTOCILIZUMAB-M73をヒトFcRnトランスジェニックマウスに静脈内投与後の血漿中濃度推移を示したグラフである。
【図16】TOCILIZUMAB、コントロールおよびFv5-M83のBaF/gp130における中和活性を示す図である。
【図17】TOCILIZUMAB、Fv3-M73およびFv4-M73のBaF/gp130における中和活性を示す図である。
【図18】TOCILIZUMAB、コントロール、Fv3-M73、Fv4-M73、およびFv5-M83をカニクイザルに静脈内投与後の血漿中濃度推移を示したグラフである。
【図19】TOCILIZUMAB、コントロール、Fv3-M73、Fv4-M73、およびFv5-M83をカニクイザルに静脈内投与後のCRP濃度推移を示したグラフである。
【図20】TOCILIZUMAB、コントロール、Fv3-M73、Fv4-M73、およびFv5-M83をカニクイザルに静脈内投与後の非結合型可溶型IL-6レセプター率の推移を示したグラフである。
【図21】TOCILIZUMABとFv4-M73によるヒトRA患者由来滑膜細胞からのMCP-1産生抑制作用を示す図である。
【図22】TOCILIZUMABとFv4-M73によるヒトRA患者由来滑膜細胞からのVEGF産生抑制作用を示す図である。
【発明を実施するための形態】
【0019】
本発明は以下の(a)〜(f)いずれかに記載のポリペプチドを提供する。
(a) 配列番号:1(VH4-M73のCDR1)の配列を有するCDR1、配列番号:2(VH4-M73のCDR2)の配列を有するCDR2、および配列番号:3(VH4-M73のCDR3)の配列を有するCDR3を含むポリペプチド、
(b) 配列番号:4(VH3-M73のCDR1)の配列を有するCDR1、配列番号:5(VH3-M73のCDR2)の配列を有するCDR2、および配列番号:6(VH3-M73のCDR3)の配列を有するCDR3を含むポリペプチド、
(c) 配列番号:7(VH5-M83のCDR1)の配列を有するCDR1、配列番号:8(VH5-M83のCDR2)の配列を有するCDR2、および配列番号:9(VH5-M83のCDR3)の配列を有するCDR3を含むポリペプチド、
(d) 配列番号:10(VL1のCDR1)の配列を有するCDR1、配列番号:11(VL1のCDR2)の配列を有するCDR2、および配列番号:12(VL1のCDR3)の配列を有するCDR3を含むポリペプチド、
(e) 配列番号:13(VL3のCDR1)の配列を有するCDR1、配列番号:14(VL3のCDR2)の配列を有するCDR2、および配列番号:15(VL3のCDR3)の配列を有するCDR3を含むポリペプチド、
(f) 配列番号:16(VL5のCDR1)の配列を有するCDR1、配列番号:17(VL5のCDR2)の配列を有するCDR2、および配列番号:18(VL5のCDR3)の配列を有するCDR3を含むポリペプチド。
【0020】
本発明においてポリペプチドは特に限定されないが、ヒトIL-6レセプターへの結合活性を有する抗原結合物質であることが好ましい。抗原結合物質の好ましい例として、抗体の重鎖可変領域(VH)、抗体の軽鎖可変領域(VL)、抗体の重鎖、抗体の軽鎖、抗体などを挙げることができる。
【0021】
上述の(a)〜(f)のポリペプチドにおいて、(a)〜(c)のポリペプチドの好ましい例として抗体の重鎖可変領域を挙げることができ、(d)〜(f)のポリペプチドの好ましい例として抗体の軽鎖可変領域を挙げることができる。
【0022】
これらの可変領域は抗ヒトIL-6レセプター抗体の一部として使用され得る。これらの可変領域が用いられた抗ヒトIL-6レセプター抗体は優れた結合活性、優れた薬物動態、優れた安全性・免疫原性、および/または優れた物性を有する。本発明において、優れた薬物動態、あるいは、薬物動態の向上とは、抗体を生体内に投与した際の血漿中濃度推移から算出される薬物動態パラメーターの一つである"クリアランス(Clearance、CL)"の減少、"濃度曲線下面積(Area Under Curve、AUC)"の増大、"平均滞留時間(Mean Residence Time)"の増大、"血漿中半減期(t1/2)"の増大、のいずれかを意味する。本発明において、優れた物性あるいは物性の向上とは、特に限定されないが、安定性の向上、ヘテロジェニティーの低減などを意味する。
【0023】
CDRと連結されるヒト抗体のフレームワーク領域(framework region;FR)は、CDRが良好な抗原結合部位を形成するものが選択される。本発明の可変領域に用いられるFRは特に限定されず、如何なるFRが用いられていてもよいが、ヒト由来のFRが用いられることが好ましい。ヒト由来のFRは天然配列を有するFRが用いられてもよいし、必要に応じ、CDRが適切な抗原結合部位を形成するように、天然配列を有するフレームワーク領域の1又は複数のアミノ酸を置換、欠失、付加および/または挿入等してもよい。たとえば、アミノ酸を置換したFRを用いた抗体の抗原への結合活性を測定し評価することによって所望の性質を有する変異FR配列が選択できる(Sato, K. et al., Cancer Res.(1993)53, 851-856)。
【0024】
又、上述のCDR配列において1又は複数のアミノ酸を置換、欠失、付加および/または挿入等してもよい。1又は複数のアミノ酸の置換、欠失、付加および/または挿入後のCDR配列は、結合活性、中和活性、安定性、免疫原性および/または薬物動態において改変前のCDR配列と同等の活性を有していることが好ましい。置換、欠失、付加および/または挿入されるアミノ酸の数は特に限定されないが、好ましくは1つのCDRにつき3アミノ酸以内、さらに好ましくは2アミノ酸以内、より好ましくは1アミノ酸である。
【0025】
1又は複数のアミノ酸残基を目的の他のアミノ酸に置換する方法としては、例えば、部位特異的変異誘発法(Hashimoto-Gotoh, T, Mizuno, T, Ogasahara, Y, and Nakagawa, M. (1995) An oligodeoxyribonucleotide-directed dual amber method for site-directed mutagenesis. Gene 152, 271-275、Zoller, MJ, and Smith, M.(1983) Oligonucleotide-directed mutagenesis of DNA fragments cloned into M13 vectors.Methods Enzymol. 100, 468-500、Kramer,W, Drutsa,V, Jansen,HW, Kramer,B, Pflugfelder,M, and Fritz,HJ(1984) The gapped duplex DNA approach to oligonucleotide-directed mutation construction. Nucleic Acids Res. 12, 9441-9456、Kramer W, and Fritz HJ(1987) Oligonucleotide-directed construction of mutations via gapped duplex DNA Methods. Enzymol. 154, 350-367、Kunkel,TA(1985) Rapid and efficient site-specific mutagenesis without phenotypic selection.Proc Natl Acad Sci U S A. 82, 488-492)が挙げられる。該方法を用いて、抗体の所望のアミノ酸を目的の他のアミノ酸に置換することができる。又、フレームワークシャッフリング(Mol Immunol. 2007 Apr;44(11):3049-60)およびCDR repair(US2006/0122377)等のライブラリー技術を用いることにより、フレームワークおよびCDRにおけるアミノ酸を適切な他のアミノ酸置換することも可能である。
【0026】
さらに、本発明は以下の(a)〜(c)いずれかに記載の抗体を提供する。
(a) 配列番号:1(VH4-M73のCDR1)の配列を有するCDR1、配列番号:2(VH4-M73のCDR2)の配列を有するCDR2、および配列番号:3(VH4-M73のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに配列番号:10(VL1のCDR1)の配列を有するCDR1、配列番号:11(VL1のCDR2)の配列を有するCDR2、および配列番号:12(VL1のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体、
(b) 配列番号:4(VH3-M73のCDR1)の配列を有するCDR1、配列番号:5(VH3-M73のCDR2)の配列を有するCDR2、および配列番号:6(VH3-M73のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに配列番号:13(VL3のCDR1)の配列を有するCDR1、配列番号:14(VL3のCDR2)の配列を有するCDR2、および配列番号:15(VL3のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体、
(c) 配列番号:7(VH5-M83のCDR1)の配列を有するCDR1、配列番号:8(VH5-M83のCDR2)の配列を有するCDR2、および配列番号:9(VH5-M83のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに配列番号:16(VL5のCDR1)の配列を有するCDR1、配列番号:17(VL5のCDR2)の配列を有するCDR2、および配列番号:18(VL5のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体。
【0027】
上述の抗体は優れた結合活性、優れた薬物動態、優れた安全性・免疫原性、および/または優れた物性を有する抗ヒトIL-6レセプター抗体として使用され得る。
【0028】
本発明のCDRと連結されるヒト抗体のフレームワーク領域は、CDRが良好な抗原結合部位を形成するものが選択される。本発明の可変領域に用いられるFRは特に限定されず、如何なるFRが用いられていてもよいが、ヒト由来のFRが用いられることが好ましい。ヒト由来のFRは天然配列を有するFRが用いられてもよいし、必要に応じ、CDRが適切な抗原結合部位を形成するように、天然配列を有するフレームワーク領域の1又は複数のアミノ酸を置換、欠失、付加および/または挿入等してもよい。たとえば、アミノ酸を置換したFRを用いた抗体の抗原への結合活性を測定し評価することによって所望の性質を有する変異FR配列が選択できる(Sato, K. et al., Cancer Res.(1993)53, 851-856)。
【0029】
又、本発明の抗体で用いられる定常領域は特に限定されず、如何なる定常領域が用いられてもよい。本発明の抗体で用いられる定常領域の好ましい例としては、ヒト由来の定常領域(IgG1、IgG2、IgG3、IgG4、Cκ、Cλ由来の定常領域など)を挙げることができる。ヒト由来の定常領域は1又は複数のアミノ酸が置換、欠失、付加および/または挿入されていてもよい。例えば、ヒト由来の定常領域の好ましい例として、重鎖定常領域の場合には配列番号:31(VH4-M73の定常領域)のアミノ酸配列を有する定常領域、配列番号:32(VH3-M73の定常領域)のアミノ酸配列を有する定常領域、配列番号:33(VH5-M83の定常領域)のアミノ酸配列を有する定常領域を挙げることができ、軽鎖定常領域の場合には配列番号:34(VL1)のアミノ酸配列を有する定常領域、配列番号:35(VL3)のアミノ酸配列を有する定常領域、配列番号:36(VL5)のアミノ酸配列を有する定常領域を挙げることができる。
【0030】
又、上述のCDR配列において1又は複数のアミノ酸を置換、欠失、付加および/または挿入等してもよい。1又は複数のアミノ酸の置換、欠失、付加および/または挿入後のCDR配列は、結合活性、中和活性、安定性、免疫原性および/または薬物動態において改変前のCDR配列と同等の活性を有していることが好ましい。置換、欠失、付加および/または挿入されるアミノ酸の数は特に限定されないが、好ましくは1つのCDRにつき3アミノ酸以内、さらに好ましくは2アミノ酸以内、より好ましくは1アミノ酸である。
【0031】
アミノ酸の置換、欠失、付加および/または挿入などは上述の方法により行うことも可能である。
【0032】
さらに本発明は以下の(a)〜(f)いずれかに記載の可変領域を提供する。
(a) 配列番号:19(VH4-M73の可変領域)の配列を有する重鎖可変領域、
(b) 配列番号:20(VH3-M73の可変領域)の配列を有する重鎖可変領域、
(c) 配列番号:21(VH5-M83の可変領域)の配列を有する重鎖可変領域、
(d) 配列番号:22(VL1の可変領域)の配列を有する軽鎖可変領域、
(e) 配列番号:23(VL3の可変領域)の配列を有する軽鎖可変領域、
(f) 配列番号:24(VL5の可変領域)の配列を有する軽鎖可変領域。
【0033】
上述の可変領域は抗ヒトIL-6レセプター抗体の一部として使用され得る。これらの可変領域が用いられた抗ヒトIL-6レセプター抗体は優れた結合活性、優れた薬物動態、優れた安全性・免疫原性、および/または優れた物性を有する。
【0034】
上述の可変領域は1又は複数(例えば5アミノ酸以内、好ましくは3アミノ酸以内)のアミノ酸が置換、欠失、付加および/または挿入していてもよい。1又は複数のアミノ酸残基を目的の他のアミノ酸に置換する方法としては、例えば、上述の方法を挙げることができる。
【0035】
また本発明は、上述の可変領域を含むポリペプチドを提供する。
【0036】
さらに本発明は以下の(a)〜(c)いずれかに記載の抗体を提供する。
(a) 配列番号:19(VH4-M73の可変領域)の配列を有する重鎖可変領域および配列番号:22(VL1の可変領域)の配列を有する軽鎖可変領域を含む抗体、
(b) 配列番号:20(VH3-M73の可変領域)の配列を有する重鎖可変領域および配列番号:23(VL3の可変領域)の配列を有する軽鎖可変領域を含む抗体、
(c) 配列番号:21(VH5-M83の可変領域)の配列を有する重鎖可変領域および配列番号:24(VL5の可変領域)の配列を有する軽鎖可変領域を含む抗体。
【0037】
上述の可変領域は抗ヒトIL-6レセプター抗体の一部として使用され得る。これらの可変領域が用いられた抗ヒトIL-6レセプター抗体は優れた結合活性、優れた薬物動態、優れた安全性・免疫原性、および/または優れた物性を有する。
【0038】
上述の可変領域は1又は複数(例えば5アミノ酸以内、好ましくは3アミノ酸以内)のアミノ酸が置換、欠失、付加および/または挿入していてもよい。1又は複数のアミノ酸残基を目的の他のアミノ酸に置換する方法としては、例えば、上述の方法を挙げることができる。
【0039】
又、本発明の抗体で用いられる定常領域は特に限定されず、如何なる定常領域が用いられてもよい。本発明の抗体で用いられる定常領域の好ましい例としては、ヒト由来の定常領域(IgG1、IgG2、IgG3、IgG4、κ鎖、λ鎖由来の定常領域など)を挙げることができる。ヒト由来の定常領域は1又は複数のアミノ酸が置換、欠失、付加および/または挿入されていてもよい。例えば、ヒト由来の定常領域の好ましい例として、重鎖定常領域の場合には配列番号:31(VH4-M73の定常領域)のアミノ酸配列を有する定常領域、配列番号:32(VH3-M73の定常領域)のアミノ酸配列を有する定常領域、配列番号:33(VH5-M83の定常領域)のアミノ酸配列を有する定常領域を挙げることができ、軽鎖定常領域の場合には配列番号:34(VL1)のアミノ酸配列を有する定常領域、配列番号:35(VL3)のアミノ酸配列を有する定常領域、配列番号:36(VL5)のアミノ酸配列を有する定常領域を挙げることができる。
【0040】
さらに本発明は以下の(a)〜(f)いずれかに記載の重鎖又は軽鎖を提供する。
(a) 配列番号:25(VH4-M73)の配列を有する重鎖、
(b) 配列番号:26(VH3-M73)の配列を有する重鎖、
(c) 配列番号:27(VH5-M83)の配列を有する重鎖、
(d) 配列番号:28(VL1)の配列を有する軽鎖、
(e) 配列番号:29(VL3)の配列を有する軽鎖、
(f) 配列番号:30(VL5)の配列を有する軽鎖。
【0041】
上述の重鎖又は軽鎖は抗ヒトIL-6レセプター抗体の一部として使用され得る。これらの重鎖又は軽鎖が用いられた抗ヒトIL-6レセプター抗体は、優れた結合活性、優れた薬物動態、優れた安全性・免疫原性、および/または優れた物性を有する。
【0042】
上述の重鎖又は軽鎖は、1又は複数(例えば10アミノ酸以内、好ましくは5アミノ酸以内、さらに好ましくは3アミノ酸以内)のアミノ酸が置換、欠失、付加および/または挿入していてもよい。1又は複数のアミノ酸残基を目的の他のアミノ酸に置換する方法としては、例えば、上述の方法を用いることが可能である。
【0043】
1又は複数のアミノ酸の置換、欠失、付加および/または挿入は可変領域において行われてもよいし、定常領域において行われてもよいし、又は可変領域と定常領域の両方において行われてもよい。
【0044】
さらに本発明は以下の(a)〜(c)いずれかに記載の抗体を提供する。
(a) 配列番号:25(VH4-M73)の配列を有する重鎖および配列番号:28(VL1)の配列を有する軽鎖を含む抗体、
(b) 配列番号:26(VH3-M73)の配列を有する重鎖および配列番号:29(VL3)の配列を有する軽鎖を含む抗体、
(c) 配列番号:27(VH5-M83)の配列を有する重鎖および配列番号:30(VL5)の配列を有する軽鎖を含む抗体。
【0045】
上述の抗体は優れた結合活性、優れた薬物動態、優れた安全性・免疫原性、および/または優れた物性を有する抗ヒトIL-6レセプター抗体である。
【0046】
上述の抗体は、1又は複数(例えば20アミノ酸以内、好ましくは10アミノ酸以内、さらに好ましくは5アミノ酸以内)のアミノ酸が置換、欠失、付加および/または挿入していてもよい。1又は複数のアミノ酸残基を目的の他のアミノ酸に置換する方法としては、例えば、上述の方法をあげることができる。
【0047】
1又は複数のアミノ酸の置換、欠失、付加および/または挿入は可変領域において行われてもよいし、定常領域において行われてもよいし、又は可変領域と定常領域の両方において行われてもよい。
【0048】
本発明の抗体は、ヒト化(humanized)抗体であることが好ましい。
【0049】
ヒト化抗体は、再構成(reshaped)ヒト抗体とも称され、これは、ヒト以外の哺乳動物由来のCDRをヒト抗体のCDRへ移植したものであり、その一般的な遺伝子組換え手法も知られている(欧州特許出願公開番号EP 125023号公報、WO 96/02576 号公報参照)。
【0050】
具体的には、例えば目的のCDRと目的のフレームワーク領域(FR)とを連結するように設計したDNA配列を、CDRおよびFR両方の末端領域にオーバーラップする部分を有するように作製した数個のオリゴヌクレオチドをプライマーとして用いてPCR法により合成する(WO98/13388号公報に記載の方法を参照)。得られたDNAをヒト抗体定常領域もしくはヒト抗体定常領域改変体をコードするDNAと連結し、次いで発現ベクターに組み込んで、これを宿主に導入し産生させることにより得られる(欧州特許出願公開番号EP 239400、国際特許出願公開番号WO 96/02576参照)。
【0051】
CDRと連結されるヒト抗体のフレームワーク領域は、CDRが良好な抗原結合部位を形成するものが選択される。必要に応じ、抗体の可変領域におけるフレームワーク領域のアミノ酸を置換、欠失、付加および/または挿入等してもよい。
【0052】
ヒト化抗体の定常領域には、ヒト抗体定常領域又はヒト抗体定常領域において1又は複数のアミノ酸が置換、欠失、付加および/または挿入したヒト抗体定常領域改変体を用いることができる。
【0053】
例えばH鎖では、Cγ1、Cγ2、Cγ3、Cγ4、Cμ、Cδ、Cα1、Cα2、Cεを、L鎖ではCκ、Cλを使用することができる。Cκのアミノ酸配列を配列番号:38に、当該アミノ酸配列をコードする塩基配列を配列番号:37に示す。Cγ1のアミノ酸配列を配列番号:40に、当該アミノ酸配列をコードする塩基配列を配列番号:39に示す。Cγ2のアミノ酸配列を配列番号:42に、当該アミノ酸配列をコードする塩基配列を配列番号:41に示す。Cγ4のアミノ酸配列を配列番号:44に、当該アミノ酸配列をコードする塩基配列を配列番号:43に示す。
【0054】
また、抗体またはその産生の安定性を改善するために、ヒト抗体C領域を修飾してもよい。ヒト化の際に用いられるヒト抗体は、IgG、IgM、IgA、IgE、IgDなど如何なるアイソタイプのヒト抗体でもよいが、本発明においてはIgGを用いることが好ましい。IgGとしては、IgG1、IgG2、IgG3、IgG4などを用いることが可能である。
【0055】
なお、ヒト化抗体を作製した後に、可変領域(例えば、CDR、FR)や定常領域中のアミノ酸を他のアミノ酸で置換、欠失、付加および/または挿入等してもよく、本発明のヒト化抗体には、そのようなアミノ酸置換等されたヒト化抗体も含まれる。
【0056】
本発明の抗体には、IL-6レセプターへの結合活性および/または中和活性を有する限り、IgGに代表される二価抗体だけでなく、一価抗体、若しくはIgMに代表される多価抗体も含まれる。本発明の多価抗体には、全て同じ抗原結合部位を有する多価抗体、または、一部もしくは全て異なる抗原結合部位を有する多価抗体が含まれる。本発明の抗体は、抗体の全長分子に限らず、IL-6レセプタータンパク質に結合する限り、低分子化抗体またはその修飾物であってもよい。
【0057】
低分子化抗体は、全長抗体(whole antibody、例えばwhole IgG等)の一部分が欠損している抗体断片を含む抗体であり、IL-6レセプターへの結合活性および/または中和活性を有する限り特に限定されない。本発明において低分子化抗体は、全長抗体の一部分を含む限り特に限定されないが、VH又はVLを含んでいることが好ましく、特に好ましくはVHとVLの両方を含む低分子化抗体である。又、本発明の低分子化抗体の他の好ましい例として、抗体のCDRを含む低分子化抗体を挙げることができる。低分子化抗体に含まれるCDRは抗体の6つのCDR全てが含まれいてもよいし、一部のCDRが含まれていてもよい。
【0058】
本発明における低分子化抗体は、全長抗体よりも分子量が小さくなることが好ましいが、例えば、ダイマー、トリマー、テトラマーなどの多量体を形成すること等もあり、全長抗体よりも分子量が大きくなることもある。
【0059】
抗体断片の具体例としては、例えば、Fab、Fab'、F(ab')2、Fvなどを挙げることができる。また、低分子化抗体の具体例としては、例えば、Fab、Fab'、F(ab')2、Fv、scFv(single chain Fv)、Diabody、sc(Fv)2(single chain (Fv)2)などを挙げることができる。これら抗体の多量体(例えば、ダイマー、トリマー、テトラマー、ポリマー)も、本発明の低分子化抗体に含まれる。
【0060】
抗体断片は、例えば、抗体を酵素で処理して抗体断片を生成させることによって得ることができる。抗体断片を生成する酵素として、例えばパパイン、ペプシン、あるいはプラスミンなどが公知である。あるいは、これら抗体断片をコードする遺伝子を構築し、これを発現ベクターに導入した後、適当な宿主細胞で発現させることができる(例えば、Co, M.S. et al., J. Immunol.(1994)152, 2968-2976、Better, M. & Horwitz, A. H. Methods in Enzymology(1989)178, 476-496、Pluckthun, A. & Skerra, A. Methods in Enzymology(1989)178, 476-496、Lamoyi, E., Methods in Enzymology(1989)121, 652-663、Rousseaux, J. et al., Methods in Enzymology(1989)121, 663-669、Bird, R. E. et al., TIBTECH(1991)9, 132-137参照)。
【0061】
消化酵素は、抗体断片の特定の位置を切断し、次のような特定の構造の抗体断片を与える。このような酵素的に得られた抗体断片に対して、遺伝子工学的手法を利用すると、抗体の任意の部分を欠失させることができる。
【0062】
上述の消化酵素を用いた場合に得られる抗体断片は以下のとおりである。
パパイン消化:F(ab)2またはFab
ペプシン消化:F(ab')2またはFab'
プラスミン消化:Facb
【0063】
本発明における低分子化抗体は、IL-6レセプターへの結合活性および/または中和活性を有する限り、任意の領域を欠失した抗体断片を含むことができる。
【0064】
ダイアボディーは、遺伝子融合により構築された二価(bivalent)の抗体断片を指す(Holliger P et al., Proc.Natl.Acad.Sci.USA 90: 6444-6448 (1993)、EP404,097号、WO93/11161号等)。ダイアボディーは、2本のポリペプチド鎖から構成されるダイマーである。通常、ダイマーを構成するポリペプチド鎖は、各々、同じ鎖中でVL及びVHがリンカーにより結合されている。ダイアボディーにおけるリンカーは、一般に、VLとVHが互いに結合できない位に短い。具体的には、リンカーを構成するアミノ酸残基は、例えば、5残基程度である。そのため、同一ポリペプチド鎖上にコードされるVLとVHとは、単鎖可変領域フラグメントを形成できず、別の単鎖可変領域フラグメントと二量体を形成する。その結果、ダイアボディーは2つの抗原結合部位を有することとなる。
【0065】
scFv抗体は、VH及びVLをリンカー等で結合して一本鎖ポリペプチドにした抗体である(Huston, J. S. et al., Proc. Natl. Acad. Sci. U.S.A. (1988) 85, 5879-5883、 Pluckthun「The Pharmacology of Monoclonal Antibodies」Vol.113, Resenburg 及び Moore編, Springer Verlag, New York, pp.269-315, (1994))。scFvにおけるH鎖V領域およびL鎖V領域は、本明細書に記載されたいずれの抗体由来であってもよい。V領域を連結するペプチドリンカーには、特に制限はない。例えば3から25残基程度からなる任意の一本鎖ペプチドをリンカーとして用いることができる。具体的には、たとえば後述のペプチドリンカー等を用いることができる。
【0066】
両鎖のV領域は、例えば上記のようなPCR法によって連結することができる。PCR法によるV領域の連結のために、まず次のDNAのうち、全部あるいは所望の部分アミノ酸配列をコードするDNAが鋳型として利用される。
抗体のH鎖またはH鎖V領域をコードするDNA配列、および
抗体のL鎖またはL鎖V領域をコードするDNA配列
【0067】
増幅すべきDNAの両端の配列に対応する配列を有するプライマーの一対を用いたPCR法によって、H鎖とL鎖のV領域をコードするDNAがそれぞれ増幅される。次いで、ペプチドリンカー部分をコードするDNAを用意する。ペプチドリンカーをコードするDNAもPCRを利用して合成することができる。このとき利用するプライマーの5'側に、別に合成された各V領域の増幅産物と連結できる塩基配列を付加しておく。次いで、[H鎖V領域DNA]−[ペプチドリンカーDNA]−[L鎖V領域DNA]の各DNAと、アセンブリーPCR用のプライマーを利用してPCR反応を行う。
【0068】
アセンブリーPCR用のプライマーは、[H鎖V領域DNA]の5'側にアニールするプライマーと、[L鎖V領域DNA]の3'側にアニールするプライマーとの組み合わせからなる。すなわちアセンブリーPCR用プライマーとは、合成すべきscFvの全長配列をコードするDNAを増幅することができるプライマーセットである。一方[ペプチドリンカーDNA]には各V領域DNAと連結できる塩基配列が付加されている。その結果、これらのDNAが連結され、さらにアセンブリーPCR用のプライマーによって、最終的にscFvの全長が増幅産物として生成される。一旦scFvをコードするDNAが作製されると、それらを含有する発現ベクター、および該発現ベクターにより形質転換された組換え細胞が常法に従って取得できる。また、その結果得られる組換え細胞を培養して該scFvをコードするDNAを発現させることにより、該scFvが取得できる。
【0069】
結合されるVHとVLの順序は特に限定されず、どのような順序で並べられていてもよく、例えば、以下のような配置を挙げることができる。
[VH]リンカー[VL]
[VL]リンカー[VH]
【0070】
sc(Fv)2は、2つのVH及び2つのVLをリンカー等で結合して一本鎖にした低分子化抗体である(Hudson et al、J Immunol. Methods 1999;231:177-189)。sc(Fv)2は、例えば、scFvをリンカーで結ぶことによって作製できる。
【0071】
また2つのVH及び2つのVLが、一本鎖ポリペプチドのN末端側を基点としてVH、VL、VH、VL([VH]リンカー[VL]リンカー[VH]リンカー[VL])の順に並んでいることを特徴とする抗体が好ましいが、2つのVHと2つのVLの順序は特に上記配置に限定されず、どのような順序で並べられていてもよい。例えば以下のような配置も挙げることができる。
【0072】
[VL]リンカー[VH]リンカー[VH]リンカー[VL]
[VH]リンカー[VL]リンカー[VL]リンカー[VH]
[VH]リンカー[VH]リンカー[VL]リンカー[VL]
[VL]リンカー[VL]リンカー[VH]リンカー[VH]
[VL]リンカー[VH]リンカー[VL]リンカー[VH]
【0073】
低分子抗体中のVH又はVLのアミノ酸配列は、置換、欠失、付加及び/又は挿入されていてもよい。さらに、VHとVLを会合させた場合に、抗原結合活性を有する限り、一部を欠損させてもよいし、他のポリペプチドを付加してもよい。又、可変領域はキメラ化やヒト化されていてもよい。
【0074】
本発明において、抗体の可変領域を結合するリンカーは、遺伝子工学により導入し得る任意のペプチドリンカー、又は合成化合物リンカー、例えば、Protein Engineering, 9(3), 299-305, 1996に開示されるリンカーを用いることができる。
【0075】
本発明において好ましいリンカーはペプチドリンカーである。ペプチドリンカーの長さは特に限定されず、目的に応じて当業者が適宜選択することが可能であるが、通常、1〜100アミノ酸、好ましくは3〜50アミノ酸、更に好ましくは5〜30アミノ酸、特に好ましくは12〜18アミノ酸(例えば、15アミノ酸)である。
【0076】
ペプチドリンカーのアミノ酸配列としては、例えば、以下のような配列を挙げることができる。
Ser
Gly・Ser
Gly・Gly・Ser
Ser・Gly・Gly
Gly・Gly・Gly・Ser(配列番号:45)
Ser・Gly・Gly・Gly(配列番号:46)
Gly・Gly・Gly・Gly・Ser(配列番号:47)
Ser・Gly・Gly・Gly・Gly(配列番号:48)
Gly・Gly・Gly・Gly・Gly・Ser(配列番号:49)
Ser・Gly・Gly・Gly・Gly・Gly(配列番号:50)
Gly・Gly・Gly・Gly・Gly・Gly・Ser(配列番号:51)
Ser・Gly・Gly・Gly・Gly・Gly・Gly(配列番号:52)
(Gly・Gly・Gly・Gly・Ser(配列番号:47))n
(Ser・Gly・Gly・Gly・Gly(配列番号:48))n
[nは1以上の整数である]等を挙げることができる。
【0077】
ペプチドリンカーのアミノ酸配列は、目的に応じて当業者が適宜選択することができる。たとえば上記のペプチドリンカーの長さを決定するnは、通常1〜5、好ましくは1〜3、より好ましくは1または2である。
【0078】
合成化合物リンカー(化学架橋剤)は、ペプチドの架橋に通常用いられている架橋剤、例えば、N-ヒドロキシスクシンイミド(NHS)ジスクシンイミジルスベレート(DSS)、ビス(スルホスクシンイミジル)スベレート(BS3)、ジチオビス(スクシンイミジルプロピオネート)(DSP)、ジチオビス(スルホスクシンイミジルプロピオネート)(DTSSP)、エチレングリコールビス(スクシンイミジルスクシネート)(EGS)、エチレングリコールビス(スルホスクシンイミジルスクシネート)(スルホ−EGS)、ジスクシンイミジル酒石酸塩(DST)、ジスルホスクシンイミジル酒石酸塩(スルホ−DST)、ビス[2-(スクシンイミドオキシカルボニルオキシ)エチル]スルホン(BSOCOES)、ビス[2-(スルホスクシンイミドオキシカルボニルオキシ)エチル]スルホン(スルホ−BSOCOES)などであり、これらの架橋剤は市販されている。
【0079】
4つの抗体可変領域を結合する場合には、通常、3つのリンカーが必要となる。複数のリンカーは、同じでもよいし、異なるリンカーを用いることもできる。
【0080】
本発明の抗体には、本発明の抗体のアミノ酸配列に1又は複数個のアミノ酸残基が付加された抗体も含まれる。また、これら抗体と他のペプチド又はタンパク質とが融合した融合タンパク質も含まれる。融合タンパク質を作製する方法は、本発明の抗体をコードするポリヌクレオチドと他のペプチド又はポリペプチドをコードするポリヌクレオチドをフレームが一致するように連結してこれを発現ベクターに導入し、宿主で発現させればよく、当業者に公知の手法を用いることができる。本発明の抗体との融合に付される他のペプチド又はポリペプチドとしては、例えば、FLAG(Hopp, T. P. et al., BioTechnology (1988) 6, 1204-1210)、6個のHis(ヒスチジン)残基からなる6×His、10×His、インフルエンザ凝集素(HA)、ヒトc-mycの断片、VSV-GPの断片、p18HIVの断片、T7-tag、HSV-tag、E-tag、SV40T抗原の断片、lck tag、α-tubulinの断片、B-tag、Protein Cの断片等の公知のペプチドを使用することができる。また、本発明の抗体との融合に付される他のポリペプチドとしては、例えば、GST(グルタチオン−S−トランスフェラーゼ)、HA(インフルエンザ凝集素)、イムノグロブリン定常領域、β−ガラクトシダーゼ、MBP(マルトース結合タンパク質)等が挙げられる。市販されているこれらペプチドまたはポリペプチドをコードするポリヌクレオチドを、本発明の抗体をコードするポリヌクレオチドと融合させ、これにより調製された融合ポリヌクレオチドを発現させることにより、融合ポリペプチドを調製することができる。
【0081】
また本発明の抗体は、ポリエチレングリコール(PEG)やヒアルロン酸などの高分子物質、放射性物質、蛍光物質、発光物質、酵素、トキシン等の各種分子と結合したコンジュゲート抗体でもよい。このようなコンジュゲート抗体は、得られた抗体に化学的な修飾を施すことによって得ることができる。なお、抗体の修飾方法はこの分野においてすでに確立されている(例えば、US5057313、US5156840)。本発明における「抗体」にはこれらのコンジュゲート抗体も包含される。
【0082】
また本発明の抗体には、糖鎖が改変された抗体も包含される。
【0083】
さらに、本発明で使用される抗体は二重特異性抗体(bispecific antibody)であってもよい。二重特異性抗体とは、異なるエピトープを認識する可変領域を同一の抗体分子内に有する抗体を言う。本発明において、二重特異性抗体はIL-6レセプター分子上の異なるエピトープを認識する二重特異性抗体であってもよいし、一方の抗原結合部位がIL-6レセプターを認識し、他方の抗原結合部位が他の物質を認識する二重特異性抗体とすることもできる。IL-6レセプターを認識する本発明の抗体からなる二重特異性抗体の他方の抗原結合部位が結合する抗原としては、例えば、IL-6, TNFα, TNFR1, TNFR2, CD80, CD86, CD28, CD20, CD19, IL-1α, IL-β, IL-1R, RANKL, RANK, IL-17, IL-17R, IL-23, IL-23R, IL-15, IL-15R, BlyS, lymphotoxinα, lymphotoxinβ, LIGHT ligand, LIGHT, VLA-4, CD25, IL-12, IL-12R, CD40, CD40L, BAFF, CD52, CD22, IL-32, IL-21, IL-21R, GM-CSF, GM-CSFR, M-CSF, M-CSFR, IFN-alpha, VEGF, VEGFR, EGF, EGFR, CCR5, APRIL, APRILRなどが挙げられる。
【0084】
二重特異性抗体を製造するための方法は公知である。たとえば、認識抗原が異なる2種類の抗体を結合させて、二重特異性抗体を作製することができる。結合させる抗体は、それぞれがH鎖とL鎖を有する1/2分子であっても良いし、H鎖のみからなる1/4分子であっても良い。あるいは、異なるモノクローナル抗体を産生するハイブリドーマを融合させて、二重特異性抗体産生融合細胞を作製することもできる。さらに、遺伝子工学的手法により二重特異性抗体が作製できる。
【0085】
本発明の抗体は、後述する抗体を産生する細胞や宿主あるいは精製方法により、アミノ酸配列、分子量、等電点又は糖鎖の有無や形態などが異なり得る。しかしながら、得られた抗体が、本発明の抗体と同等の機能を有している限り、本発明に含まれる。例えば、本発明の抗体を原核細胞、例えば大腸菌で発現させた場合、本来の抗体のアミノ酸配列のN末端にメチオニン残基が付加される。本発明の抗体はこのような抗体も包含する。
【0086】
本発明の抗IL-6レセプター抗体などのポリペプチドは当業者に公知の方法により製造することが可能である。
【0087】
例えば、得られた抗IL-6レセプター抗体の配列を基に、当業者に公知の遺伝子組換え技術を用いて抗IL-6レセプター抗体を作製することが可能である。具体的には、IL-6レセプターを認識する抗体の配列を基に抗体をコードするポリヌクレオチドを構築し、これを発現ベクターに導入した後、適当な宿主細胞で発現させればよい(例えば、Co, M. S. et al., J. Immunol. (1994) 152, 2968-2976 ; Better, M. and Horwitz, A. H., Methods Enzymol. (1989) 178, 476-496 ; Pluckthun, A. and Skerra, A., Methods Enzymol. (1989) 178, 497-515 ; Lamoyi, E., Methods Enzymol. (1986) 121, 652-663 ; Rousseaux, J. et al., Methods Enzymol. (1986) 121, 663-669 ; Bird, R. E. and Walker, B. W., Trends Biotechnol. (1991) 9, 132-137参照)。
【0088】
従って、本発明は、本発明のポリペプチドをコードするポリヌクレオチドが導入されたベクターを含む宿主細胞を培養する工程を含む、本発明のポリペプチド又は本発明のポリペプチドをコードする遺伝子によりコードされるポリペプチドを製造する方法を提供する。
【0089】
より具体的には、以下の工程を含む本発明のポリペプチドの製造方法を提供する。
(a)本発明のポリペプチドをコードする遺伝子が導入されたベクターを含む宿主細胞を培養する工程、
(b)当該遺伝子によりコードされるポリペプチドを取得する工程。
【0090】
ベクターの例としては、M13系ベクター、pUC系ベクター、pBR322、pBluescript、pCR-Scriptなどが挙げられる。また、cDNAのサブクローニング、切り出しを目的とした場合、上記ベクターの他に、例えば、pGEM-T、pDIRECT、pT7などが挙げられる。本発明の抗体を生産する目的においてベクターを使用する場合には、特に、発現ベクターが有用である。発現ベクターとしては、例えば、大腸菌での発現を目的とした場合は、ベクターが大腸菌で増幅されるような特徴を持つほかに、宿主をJM109、DH5α、HB101、XL1-Blueなどの大腸菌とした場合においては、大腸菌で効率よく発現できるようなプロモーター、例えば、lacZプロモーター(Wardら, Nature (1989) 341, 544-546;FASEB J. (1992) 6, 2422-2427)、araBプロモーター(Betterら, Science (1988) 240, 1041-1043)、またはT7プロモーターなどを持っていることが不可欠である。このようなベクターとしては、上記ベクターの他にpGEX-5X-1(ファルマシア製)、「QIAexpress system」(キアゲン製)、pEGFP、またはpET(この場合、宿主はT7 RNAポリメラーゼを発現しているBL21が好ましい)などが挙げられる。
【0091】
また、発現プラスミドのベクターには、抗体分泌のためのシグナル配列が含まれていてもよい。抗体分泌のためのシグナル配列としては、大腸菌のペリプラズムに産生させる場合、pelBシグナル配列(Lei, S. P. et al J. Bacteriol. (1987) 169, 4379)を使用すればよい。宿主細胞へのベクターの導入は、例えば塩化カルシウム法、エレクトロポレーション法を用いて行うことができる。
【0092】
大腸菌以外にも、例えば、本発明の抗体を製造するためのベクターとしては、哺乳動物由来の発現ベクター(例えば、pcDNA3(インビトロゲン社製)や、pEF-BOS (Nucleic Acids. Res.1990, 18(17),p5322)、pEF、pCDM8)、昆虫細胞由来の発現ベクター(例えば「Bac-to-BAC baculovairus expression system」(ギブコBRL製)、pBacPAK8)、植物由来の発現ベクター(例えばpMH1、pMH2)、動物ウィルス由来の発現ベクター(例えば、pHSV、pMV、pAdexLcw)、レトロウィルス由来の発現ベクター(例えば、pZIPneo)、酵母由来の発現ベクター(例えば、「Pichia Expression Kit」(インビトロゲン製)、pNV11、SP-Q01)、枯草菌由来の発現ベクター(例えば、pPL608、pKTH50)が挙げられる。
【0093】
CHO細胞、COS細胞、NIH3T3細胞等の動物細胞での発現を目的とした場合には、発現プラスミドのベクターが細胞内で発現させるために必要なプロモーター、例えばSV40プロモーター(Mulliganら, Nature (1979) 277, 108)、MMLV-LTRプロモーター、EF1αプロモーター(Mizushimaら, Nucleic Acids Res. (1990) 18, 5322)、CMVプロモーターなどを持っていることが不可欠であり、細胞への形質転換を選抜するための遺伝子(例えば、薬剤(ネオマイシン、G418など)により判別できるような薬剤耐性遺伝子)を有すればさらに好ましい。このような特性を有するベクターとしては、例えば、pMAM、pDR2、pBK-RSV、pBK-CMV、pOPRSV、pOP13などが挙げられる。
【0094】
さらに、遺伝子を安定的に発現させ、かつ、細胞内での遺伝子のコピー数の増幅を目的とする場合には、核酸合成経路を欠損したCHO細胞にそれを相補するDHFR遺伝子を有するベクター(例えば、pSV2-dhfr(「Molecular Cloning 2nd edition」 Cold Spring Harbor Laboratory Press, (1989))など)を導入し、メトトレキセート(MTX)により増幅させる方法が挙げられ、また、遺伝子の一過性の発現を目的とする場合には、SV40 T抗原を発現する遺伝子を染色体上に持つCOS細胞を用いてSV40の複製起点を持つベクター(pcDなど)で形質転換する方法が挙げられる。複製開始点としては、また、ポリオーマウィルス、アデノウィルス、ウシパピローマウィルス(BPV)等の由来のものを用いることもできる。さらに、宿主細胞系で遺伝子コピー数増幅のため、発現ベクターは選択マーカーとして、アミノグリコシドトランスフェラーゼ(APH)遺伝子、チミジンキナーゼ(TK)遺伝子、大腸菌キサンチングアニンホスホリボシルトランスフェラーゼ(Ecogpt)遺伝子、ジヒドロ葉酸還元酵素(dhfr)遺伝子等を含むことができる。
【0095】
これにより得られた本発明の抗体は、宿主細胞内または細胞外(培地など)から単離し、実質的に純粋で均一な抗体として精製することができる。抗体の分離、精製は、通常の抗体の精製で使用されている分離、精製方法を使用すればよく、何ら限定されるものではない。例えば、クロマトグラフィーカラム、フィルター、限外濾過、塩析、溶媒沈殿、溶媒抽出、蒸留、免疫沈降、SDS-ポリアクリルアミドゲル電気泳動、等電点電気泳動法、透析、再結晶等を適宜選択、組み合わせれば抗体を分離、精製することができる。
【0096】
クロマトグラフィーとしては、例えばアフィニティークロマトグラフィー、イオン交換クロマトグラフィー、疎水性クロマトグラフィー、ゲル濾過、逆相クロマトグラフィー、吸着クロマトグラフィー等が挙げられる(Strategies for Protein Purification and Characterization: A Laboratory Course Manual. Ed Daniel R. Marshak et al., Cold Spring Harbor Laboratory Press, 1996)。これらのクロマトグラフィーは、液相クロマトグラフィー、例えばHPLC、FPLC等の液相クロマトグラフィーを用いて行うことができる。アフィニティークロマトグラフィーに用いるカラムとしては、プロテインAカラム、プロテインGカラムが挙げられる。例えば、プロテインAを用いたカラムとして、Hyper D, POROS, Sepharose FF(GE Amersham Biosciences)等が挙げられる。本発明は、これらの精製方法を用い、高度に精製された抗体も包含する。
【0097】
得られた抗体のIL-6レセプターに対する結合活性の測定は、当業者に公知の方法により行うことが可能である。例えば、抗体の抗原結合活性を測定する方法として、ELISA(酵素結合免疫吸着検定法)、EIA(酵素免疫測定法)、RIA(放射免疫測定法)あるいは蛍光抗体法を用いることができる。例えば、酵素免疫測定法を用いる場合、抗原をコーティングしたプレートに、抗体を含む試料、例えば、抗体産生細胞の培養上清や精製抗体を加える。アルカリフォスファターゼ等の酵素で標識した二次抗体を添加し、プレートをインキュベートし、洗浄した後、p-ニトロフェニル燐酸などの酵素基質を加えて吸光度を測定することで抗原結合活性を評価することができる。
【0098】
医薬組成物
また本発明は、上述のポリペプチドを有効成分として含有する医薬組成物を提供する。本発明の医薬組成物はIL-6が関連する関節リウマチなどの疾患に用いることが可能である。即ち本発明は、上述の抗体を有効成分とする関節リウマチなどの疾患の治療剤も提供する。本発明の対象となる疾患の好ましい例として、関節リウマチ、若年性特発性関節炎、全身型若年性特発性関節炎、キャッスルマン病、全身性エリテマトーデス(SLE)、ループス腎炎、クローン病、lymphoma、潰瘍性大腸炎、貧血、血管炎、川崎病、Still病、アミロイドーシス、多発性硬化症、移植、加齢黄斑変性症、強直性脊椎炎、乾癬、乾癬性関節炎、慢性閉塞性肺疾患(COPD)、IgA 腎症、変形性関節症、喘息、糖尿病性腎症、GVHD、子宮内膜症、肝炎(NASH)、心筋梗塞、動脈硬化、セプシス、骨粗しょう症、糖尿病、多発性骨髄腫、前立腺癌、腎癌、B-cell non-Hodgkin's、膵癌、肺癌、食道癌、大腸癌、癌カケクシア、癌神経浸潤、心筋梗塞、近視性脈絡膜血管新生、特発性脈絡膜血管新生、ぶどう膜炎、慢性甲状腺炎、遅延性過敏症、接触性皮膚炎、アトピー性皮膚炎、中皮腫、多発性筋炎、皮膚筋炎、汎ぶどう膜炎、前部ぶどう膜炎、中間部ぶどう膜炎、強膜炎、角膜炎、眼窩炎症、視神経炎、糖尿病網膜症、増殖硝子体網膜症、ドライアイ、術後炎症等が挙げられるが、これらに限定されることはない。
【0099】
抗IL-6レセプター抗体を「有効成分として含有する」とは、抗IL-6レセプター抗体を活性成分の少なくとも1つとして含むという意味であり、その含有率を制限するものではない。また、本発明の医薬組成物は、上述のポリペプチドと合わせて他の有効成分を含有してもよい。
【0100】
なお、本発明の医薬組成物は治療目的だけでなく、予防目的で用いられてもよい。
【0101】
本発明のポリペプチドは、常法に従って製剤化することができる(例えば、Remington's Pharmaceutical Science, latest edition, Mark Publishing Company, Easton, U.S.A)。さらに、必要に応じ、医薬的に許容される担体及び/または添加物を供に含んでもよい。例えば、界面活性剤(PEG、Tween等)、賦形剤、酸化防止剤(アスコルビン酸等)、着色料、着香料、保存料、安定剤、緩衝剤(リン酸、クエン酸、他の有機酸等)、キレート剤(EDTA等)、懸濁剤、等張化剤、結合剤、崩壊剤、滑沢剤、流動性促進剤、矯味剤等を含むことができる。しかしながら、本発明の炎症性疾患の予防または治療剤は、これらに制限されず、その他常用の担体を適宜含んでいてもよい。具体的には、軽質無水ケイ酸、乳糖、結晶セルロース、マンニトール、デンプン、カルメロースカルシウム、カルメロースナトリウム、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルアセタールジエチルアミノアセテート、ポリビニルピロリドン、ゼラチン、中鎖脂肪酸トリグリセライド、ポリオキシエチレン硬化ヒマシ油60、白糖、カルボキシメチルセルロース、コーンスターチ、無機塩類等を挙げることができる。また、その他の低分子量のポリペプチド、血清アルブミン、ゼラチン及び免疫グロブリン等の蛋白質、並びに、アミノ酸を含んでいてもよい。注射用の水溶液とする場合には、抗IL-6レセプター抗体を、例えば、生理食塩水、ブドウ糖またはその他の補助薬を含む等張液に溶解する。補助薬としては、例えば、D-ソルビトール、D-マンノース、D-マンニトール、塩化ナトリウムが挙げられ、さらに、適当な溶解補助剤、例えばアルコール(エタノール等)、ポリアルコール(プロピレングリコール、PEG等)、非イオン性界面活性剤(ポリソルベート80、HCO-50)等と併用してもよい。
【0102】
また、必要に応じポリペプチドをマイクロカプセル(ヒドロキシメチルセルロース、ゼラチン、ポリ[メチルメタクリル酸]等のマイクロカプセル)に封入したり、コロイドドラッグデリバリーシステム(リポソーム、アルブミンミクロスフェア、マイクロエマルジョン、ナノ粒子及びナノカプセル等)とすることもできる(Remington's Pharmaceutical Science 16th edition &, Oslo Ed. (1980)等参照)。さらに、薬剤を徐放性の薬剤とする方法も公知であり、ポリペプチドに適用し得る(Langer et al., J.Biomed.Mater.Res.(1981) 15: 167-277; Langer, Chem. Tech. (1982)12: 98-105;米国特許第3,773,919号;欧州特許出願公開(EP)第58,481号; Sidman et al., Biopolymers(1983)22:547-56;EP第133,988号)。さらに、本剤にヒアルロニダーゼを添加あるいは混合することで皮下に投与する液量を増加させることも可能である(例えば、WO2004/078140等)。
【0103】
本発明の医薬組成物は、経口または非経口のいずれでも投与可能であるが、好ましくは非経口投与される。具体的には、注射及び経皮投与により患者に投与される。注射剤型の例としては、例えば、静脈内注射、筋肉内注射または皮下注射等により全身又は局所的に投与することができる。治療部位またはその周辺に局所注入、特に筋肉内注射してもよい。経皮投与剤型の例としては、例えば、軟膏剤、ゲル剤、クリーム剤、湿布剤、および貼付剤等があげられ、全身又は局所的に投与することができる。また、患者の年齢、症状により適宜投与方法を選択することができる。投与量としては、例えば、1回につき体重1 kgあたり活性成分が0.0001 mg〜100 mgの範囲で選ぶことが可能である。または、例えば、ヒト患者に投与する場合、患者あたり活性成分が0.001〜1000 mg/kg・body・weightの範囲を選ぶことができ、1回当たり投与量としては、例えば、本発明の抗体が0.01〜50mg/kg・body・weight程度の量が含まれることが好ましい。しかしながら、本発明の医薬組成物は、これらの投与量に制限されるものではない。
【0104】
なお、本発明で記載されているアミノ酸配列に含まれるアミノ酸は翻訳後に修飾(例えば、N末端のグルタミンのピログルタミル化によるピログルタミン酸への修飾は当業者によく知られた修飾である)を受ける場合もあるが、そのようにアミノ酸が翻訳後修飾された場合であっても当然のことながら本発明で記載されているアミノ酸配列に含まれる。
【0105】
また結合する糖鎖の構造は如何なる構造でもよい。EUナンバリングの297番目の糖鎖は如何なる糖鎖構造であってもよく(好ましくはフコシル化された糖鎖)、また糖鎖が結合していなくてもよい(例えば大腸菌で生産する、あるいはEUナンバリングの297番目に糖鎖が結合しないように改変することで可能)。
【0106】
なお本明細書において引用された全ての先行技術文献は、参照として本明細書に組み入れられる。
【実施例】
【0107】
以下本発明を実施例により具体的に説明するが、本発明はこれら実施例に制限されるものではない。
【0108】
〔実施例1〕TOCILIZUMABのIL-6レセプターへの親和性を向上する可変領域変異箇所の同定
TOCILIZUMAB(H鎖 WT-IgG1/配列番号:53、L鎖 WT-kappa/配列番号:54)のIL-6レセプターへの親和性を向上させるために、CDR配列に変異を導入したライブラリーを作製し検討した。CDRに変異を導入したライブラリーをスクリーニングした結果、IL-6レセプターへの親和性を向上する変異を見出し、それらを図1にまとめた。これらの変異を組み合わせた高親和性TOCILIZUMABの例として、RDC-23(H鎖RDC23H-IgG1/配列番号:55、L鎖 RDC-23L-kappa/配列番号:56)が挙げられる。RDC-23の可溶型IL-6レセプターへのアフィニティーおよびBaF/gp130による生物活性をTOCILIZUMABと比較した(方法は参考例参照)。
【0109】
アフィニティーを測定した結果を表1に示した。BaF/gp130による生物活性(IL-6終濃度 30 ng/mL)を測定した結果を図2に示した。RDC-23は、TOCILIZUMABと比較して約60倍アフィニティーが向上しており、BaF/gp130の100%阻害濃度として約100倍活性が向上していることが見出された。
【0110】
【表1】
【0111】
〔実施例2〕TOCILIZUMABの等電点低下による薬物動態を向上する改変の同定
TOCILIZUMABの薬物動態を向上させるために、IL-6レセプターへの結合を大きく低下させることなく可変領域の等電点を低下することができる変異箇所の検討を行った。TOCILIZUMABの立体構造モデルから推察された可変領域変異箇所をスクリーニングした結果、IL-6レセプターへの結合を大きく低下させることなく可変領域の等電点を低下することできる変異箇所を見出し、それらを図3にまとめた。これらの変異を組み合わせた等電点低下TOCILIZUMABの例として、H53/L28(H鎖 H53-IgG1/配列番号:57、L鎖 L28-kappa/配列番号:58)が挙げられる。H53/L28の可溶型IL-6レセプターへのアフィニティー、BaF/gp130による生物活性、等電点、および、マウスにおける薬物動態をTOCILIZUMABと比較した(方法は参考例参照)。
【0112】
アフィニティーを測定した結果を表2に示した。BaF/gp130による生物活性(IL-6終濃度 30 ng/mL)を測定した結果を図4に示した。H53/L28は、TOCILIZUMABと比較して約6倍アフィニティーが向上しており、BaF/gp130の100%阻害濃度として数倍程度活性が向上していることが示された。
【0113】
【表2】
【0114】
当業者公知の等電点電気泳動により等電点を測定した結果、TOCILIZUMABの等電点は約9.3であり、H53/L28の等電点は約6.5〜6.7であり、H53/L28はTOCILIZUMABと比較して等電点が約2.7低下した。また、可変領域VH/VLの理論等電点をGENETYX(GENETYX CORPORATION)により計算したところ、TOCILIZUMABの理論等電点は9.20であり、H53/L28の理論等電点は4.52であり、H53/L28はTOCILIZUMABと比較して等電点が約4.7低下した。
【0115】
等電点を低下させた改変抗体H53/L28の薬物動態を評価するために、TOCILIZUMABとH53/L28の正常マウスにおける薬物動態の比較を行った。TOCILIZUMABおよびH53/L28をマウス(C57BL/6J、日本チャールズリバー)に1 mg/kgで静脈内(IV)および皮下(SC)に単回投与し血漿中濃度推移を評価した。TOCILIZUMABおよびH53/L28の静脈内投与後の血漿中濃度推移を図5に、皮下投与後の血漿中濃度推移を図6に示し、WinNonlin(Pharsight社製)により得られた薬物動態学的パラメーター(クリアランス(CL)、半減期(T1/2))を表3に示した。H53/L28の静脈内投与後の血漿中半減期(T1/2)はTOCILIZUMABの約1.3倍に延長し、クリアランスが約1.7倍低下した。H53/L28の皮下投与後のT1/2はTOCILIZUMABの約2倍に延長し、クリアランスが約2.1倍低下した。このようにアミノ酸置換によりTOCILIZUMABの等電点を低下させることによって薬物動態を大幅に向上させることが可能であることが見出された。
【0116】
【表3】
【0117】
〔実施例3〕TOCILIZUMABの免疫原性を低下する変異箇所の同定
可変領域に存在するT-cellエピトープによる免疫原性リスクを低減する変異箇所の同定
TOCILIZUMABの可変領域配列に存在するT-cellエピトープをTEPITOPE(Methods. 2004 Dec;34(4):468-75)を用いて解析を行った。その結果、L鎖CDR2に多くのHLAに結合するT-cellエピトープが存在する(免疫原性リスクが高い配列が存在する)ことが予測された。そこで、TEPITOPE解析においてL鎖CDR2の免疫原性リスクを低減させつつ、安定性、結合活性、中和活性を低下させないアミノ酸置換を検討した。
【0118】
スクリーニングの結果、以下のようにTOCILIZUMABのL鎖CDR2(配列番号:59)のL51(Kabatナンバリング、Kabat EA et al. 1991. Sequences of Proteins of Immunological Interest.NIH)のスレオニンをグリシンに、L53のアルギニンをグルタミン酸に置換する(配列番号:60)ことで、安定性、結合活性、中和活性を低下させずに免疫原性リスクを低減できることを見出した。
【0119】
TOCILIZUMAB L鎖CDR2(配列番号:59)
T-cellエピトープ除去TOCILIZUMAB L鎖CDR2(配列番号:60)
【0120】
〔実施例4〕TOCILIZUMABの可変領域フレームワーク配列の完全ヒト化による免疫原性リスクの低減
TOCILIZUMABの可変領域配列において、H鎖FR1のH27, H28, H29, H30およびH鎖FR3のH71(Kabatナンバリング、Kabat EA et al. 1991. Sequences of Proteins of Immunological Interest.NIH)は、ヒト化の過程において結合活性を維持するためにフレームワーク配列でマウス配列が残存している(Cancer Res. 1993 Feb 15;53(4):851-6)。残存するマウス配列は免疫原性リスクを高める原因となりうるため、TOCILIZUMABの免疫原性リスクをより低下するためにフレームワーク配列を完全ヒト化する検討を行った。
【0121】
その結果、TOCILIZUMABのH鎖FR1(配列番号:61)を以下のヒト化 H鎖FR1-A(配列番号:62)に置換することで、また、TOCILIZUMABのH鎖FR3(配列番号:63)を以下のヒト化 H鎖FR3(配列番号:64)に置換することで、安定性、結合活性、中和活性を低下させずにTOCILIZUMABの全フレームワークを完全ヒト化できることを見出した。
【0122】
TOCILIZUMAB H鎖FR1(配列番号:61)
ヒト化 H鎖FR1-A(配列番号:62)(Germline IMGT hVH_4_由来)
TOCILIZUMAB H鎖FR3(配列番号:63)
ヒト化 H鎖FR3(配列番号:64)(Mol. Immunol. 2007, 44(4):412-422由来)
【0123】
〔実施例5〕TOCILIZUMABのIL-6レセプターへのpH依存的結合による薬物動態向上のための変異箇所の同定
TOCILIZUMABの薬物動態を向上させる方法の一つとして、1分子のTOCILIZUMABが複数個のIL-6レセプターを繰り返し結合・中和させるように分子を改良する方法が考えられた。TOCILIZUMABは膜型IL-6レセプターに結合後、膜型IL-6レセプターに結合したままインターナライゼーションによって細胞内のエンドソームに取り込まれ、その後、TOCILIZUMABは膜型IL-6レセプターに結合したままライソソームへ移行し共にライソソームにより分解されると考えられている。すなわち、通常、1分子のTOCILIZUMABは1分子ないしは2分子の膜型IL-6レセプターに(1価ないしは2価で)結合し、インターナライズ後、ライソソームで分解されると考えられるため、1分子のTOCILIZUMABは1分子ないしは2分子の膜型IL-6レセプターしか結合・中和できない。
【0124】
そこで、TOCILIZUMABの中性条件下での結合を維持しつつ酸性条件下での結合のみを大きく低下させるpH依存的結合TOCILIZUMABを作製することが出来れば、図7に示すとおり、pH依存的結合TOCILIZUMABはエンドソーム内で抗原である膜型IL-6レセプターから解離し、エンドソーム内に存在するFcRnに結合することによって血漿中に戻ることができ、血漿中に戻ったpH依存的結合TOCILIZUMABは再度膜型IL-6レセプターに結合することが可能であると考えた。この血漿中での結合とエンドソーム内での解離を繰り返すことで、1分子のTOCILIZUMABが複数分子のIL-6レセプターを繰り返し結合・中和できると考えられ、これによりTOCILIZUMABと比較してpH依存的結合TOCILIZUMABは薬物動態が向上すると考えた。
【0125】
エンドソーム内の酸性条件下においてTOCILIZUMABがIL-6レセプターから解離するためには、酸性条件下における結合が中性条件下と比較して大幅に弱くなる必要がある。細胞表面ではIL-6レセプターに強く結合して中和する必要があるため、細胞表面のpHであるpH7.4においてはTOCILIZUMABと同等以上にIL-6レセプターに結合する必要がある。エンドソーム内のpHは一般的にpH5.5〜pH6.0であることが報告されている(Nat Rev Mol Cell Biol. 2004 Feb;5(2):121-32.)ことから、pH5.5〜pH6.0においてIL-6レセプターに弱く結合するように改良されたpH依存的結合TOCILIZUMABであれば、エンドソーム内の酸性条件下においてIL-6レセプターから解離すると考えられる。すなわち、細胞表面のpHであるpH7.4においてはIL-6レセプターに強く結合し、エンドソーム内のpHであるpH5.5〜pH6.0においてIL-6レセプターに弱く結合するように改良されたpH依存的結合TOCILIZUMABであれば、1分子で複数個のIL-6レセプターに結合・中和し、薬物動態を向上することが可能であると考えられた。
【0126】
TOCILIZUMABのIL-6レセプターへの結合にpH依存性を付与するには、pKaが6.0〜6.5付近に存在し、中性条件下(pH7.4)と酸性条件下(pH5.5〜pH6.0)との間でプロトンの解離状態が変化するヒスチジン残基をTOCILIZUMABの可変領域に導入する方法が考えられた。そこで、TOCILIZUMABの立体構造モデルから推察された可変領域のヒスチジン導入箇所のスクリーニングを実施した。また、TOCILIZUMABの選択された可変領域配列がヒスチジンにランダムに置き換わるように設計されたライブラリーを作製しスクリーニングを実施した。スクリーニングは、pH7.4においてIL-6レセプターに結合し、pH5.5ないしはpH5.8でIL-6レセプターから解離する、あるいは、アフィニティーが低下することを指標に実施した。
【0127】
その結果、TOCILIZUMABのIL-6レセプターへの結合にpH依存性(pH7.4で結合し、pH5.8で解離する性質)を付与することができる変異箇所を見出し、それらを図8にまとめた。図8のH27のチロシンからヒスチジンへの置換はCDRではなくH鎖のFR1の変異であるが、H27がヒスチジンの配列はEur. J. Immunol. 1992. 22: 1719-1728に記されているとおり、ヒト配列(配列番号:65)として存在するため、以下のフレームワークを用いることで実施例4と合わせて完全ヒト化することが可能である。
【0128】
ヒト化H鎖FR1-B(配列番号:65)
【0129】
これらの変異を組み合わせたpH依存的結合TOCILIZUMABの例として、H3pI/L73(H鎖 H3pI-IgG1/配列番号:66、L鎖 L73-kappa/配列番号:67)が挙げられる。H3pI/L73のpH7.4における可溶型IL-6レセプターへのアフィニティー、pH7.4とpH5.8における膜型IL-6レセプターからの解離速度、BaF/gp130による生物活性、および、カニクイザルおよびヒトIL-6レセプタートランスジェニックマウスにおける薬物動態をTOCILIZUMABと比較した(方法は参考例参照)。
【0130】
pH7.4における可溶型IL-6レセプターへのアフィニティーを測定した結果を表4に示した。BaF/gp130による生物活性(IL-6終濃度30 ng/mL)を測定した結果を図9に示した。H3pI/L73は、TOCILIZUMABと比較してほぼ同等のpH7.4における可溶型IL-6レセプターへのアフィニティーおよびBaF/gp130の活性を有していることが示された。
【0131】
【表4】
【0132】
TOCILIZUMABおよびH3pI/L73のpH7.4とpH5.8における膜型IL-6レセプターへ解離速度を測定した結果を表5に示した。H3pI/L73は、pH5.8における解離速度が速くなり、TOCILIZUMABと比較して膜型IL-6レセプターからの解離速度のpH依存性が約2.6倍向上していることが示された。
【0133】
【表5】
【0134】
TOCILIZUMABおよびH3pI/L73をカニクイザルに1 mg/kgで静脈内に単回投与し血漿中濃度推移を評価した。TOCILIZUMABおよびH3pI/L73の静脈内投与後の血漿中濃度推移を図10に示した。その結果、H3pI/L73はTOCILIZUMABと比較してカニクイザルにおいて大幅に薬物動態が改善した。
【0135】
TOCILIZUMABおよびH3pI/L73をヒトIL-6レセプタートランスジェニックマウス(hIL-6R tgマウス、Proc Natl Acad Sci U S A. 1995 May 23;92(11):4862-6)に25 mg/kgで静脈内に単回投与し血漿中濃度推移を評価した。TOCILIZUMABおよびH3pI/L73の静脈内投与後の血漿中濃度推移を図11に示した。その結果、H3pI/L73はTOCILIZUMABと比較してヒトIL-6レセプタートランスジェニックマウスにおいて大幅に薬物動態が改善した。
【0136】
pH依存的結合TOCILIZUMABであるH3pI/L73は、カニクイザルおよびヒトIL-6レセプタートランスジェニックマウスにおいてTOCILIZUMABと比較して薬物動態が大幅に改善したことから、pH7.4で抗原に結合し、pH5.8で抗原から解離する性質を付与することにより、1分子で複数のIL-6レセプターに結合・中和することが可能であると考えられた。また、IL-6レセプターへの結合にH3pI/L73よりもさらに強いpH依存性を付与することに薬物動態がさらに改善することが可能であると考えられた。
【0137】
〔実施例6〕TOCILIZUMABの定常領域の最適化
TOCILIZUMABのH鎖C末端のヘテロジェニティーの低減
IgG抗体のH鎖C末端配列のヘテロジェニティーとして、C末端アミノ酸のリジン残基の欠損、および、C末端の2アミノ酸のグリシン、リジン両方の欠損によるC末端カルボキシル基のアミド化が報告されている(Anal Biochem. 2007 Jan 1;360(1):75-83.)。TOCILIZUMABにおいても、その主成分は塩基配列上存在するC末端アミノ酸のリジンが翻訳後修飾により欠損した配列であるが、リジンが残存している副成分およびグリシン、リジン両方の欠損によるC末端カルボキシル基のアミド化された副成分もヘテロジェニティーとして存在する。目的物質/関連物質のヘテロジェニティーの製造間差を維持しつつ医薬品として大量に製造することは容易ではなくコスト増につながり、可能な限り単一物質であることが望まれ、抗体を医薬品として開発する上にはこれらのヘテロジェニティーが低減されていることが望ましい。よって医薬品として開発する上ではH鎖C末端のヘテロジェニティーは存在しないことが望ましい。
【0138】
C末端アミノ酸のヘテロジェニティーを低減させることを目的にC末端アミノ酸の改変を行った。その結果、TOCILIZUMABのH鎖定常領域のC末端のリジンおよびグリシンを塩基配列上あらかじめ欠損させることで、C末端に由来するヘテロジェニティーを回避することが可能であることが見出された。TOCILIZUMAB、C末端のリジン欠損TOCILIZUMAB(TOCILIZUMABΔK、H鎖 WT-IgG1ΔK/配列番号:68、L鎖 WT-kappa/配列番号:54)、および、C末端のリジンおよびグリシン欠損TOCILIZUMAB(TOCILIZUMABΔGK、H鎖 WT-IgG1ΔGK/配列番号:69、L鎖 WT-kappa/配列番号:54)のヘテロジェニティーの評価を陽イオン交換クロマトグラフィーにより実施した。カラムとしてはProPac WCX-10, 4×250 mm (Dionex) を使用し、移動相Aは25 mmol/L MES/NaOH, pH 6.1、移動相Bは25 mmol/L MES/NaOH, 250 mmol/L NaCl, pH 6.1を使用し、適切な流量およびグラジエントを用いて実施した。陽イオン交換クロマトグラフィーによる評価を行った結果を図12に示した。その結果、H鎖定常領域のC末端のリジンだけでなく、H鎖定常領域のC末端のリジンおよびグリシンの両方を塩基配列上あらかじめ欠損させることで初めてC末アミノ酸のヘテロジェニティーを低減可能であることが見出された。ヒト抗体定常領域IgG1、IgG2、IgG4において、C末端配列はいずれもEUナンバリング(Sequences of proteins of immunological interest, NIH Publication No.91-3242 を参照)447番目がリジン、446番目がグリシンになっていることから、本検討で見出されたC末アミノ酸のヘテロジェニティーを低減させる方法はIgG2定常領域とIgG4定常領域、あるいはそれらの改変体にも適用可能であると考えられた。
【0139】
TOCILIZUMABのIgG2アイソタイプのジスルフィド結合由来のヘテロジェニティーの低減
TOCILIZUMABのアイソタイプはIgG1であるが、TOCILIZUMABは中和抗体であることから、免疫原性や副作用を考慮した場合、Fcγレセプターへの結合は好ましくない可能性が考えられる。Fcγレセプターへの結合を低下させる方法としては、IgG抗体のアイソタイプをIgG1からIgG2あるいはIgG4に変える方法が考えられ(Ann Hematol. 1998 Jun;76(6):231-48.)、FcγレセプターIへの結合および薬物動態の観点からはIgG4よりはIgG2が望ましいと考えられた(Nat Biotechnol. 2007 Dec;25(12):1369-72)。一方、抗体を医薬品として開発するにあたり、そのタンパク質の物性、中でも均一性と安定性は極めて重要であり、IgG2アイソタイプは、ヒンジ領域のジスルフィド結合に由来するヘテロジェニティーが極めて多いことが報告されている(J Biol Chem. 2008 Jun 6;283(23):16206-15.)。これに由来する目的物質/関連物質のヘテロジェニティーの製造間差を維持しつつ医薬品として大量に製造することは容易ではなくコスト増につながり、可能な限り単一物質であることが望まれる。よってIgG2アイソタイプの抗体を医薬品として開発する上では安定性を低下させることなくジスルフィド結合由来のヘテロジェニティーが低減されていることが望ましい。
【0140】
IgG2アイソタイプのヘテロジェニティーを低減することを目的に各種改変体を検討した結果、IgG2定常領域配列のうち、H鎖のCH1ドメインに存在するEUナンバリング131番目のシステインと133番目のアルギニンをそれぞれセリンとリジンに置換し、さらにH鎖のupper hingeに存在するEUナンバリング219番目のシステインをセリンに置換した定常領域であるWT-SKSC(配列番号:70)によって、安定性を低下させることなくヘテロジェニティーを低減できることが見出された。TOCILIZUMAB-IgG1(H鎖 WT-IgG1/配列番号:53、L鎖 WT-kappa/配列番号:54)、TOCILIZUMAB-IgG2(H鎖 WT-IgG2/配列番号:71、L鎖 WT-kappa/配列番号:54)、および、TOCILIZUMAB-SKSC(H鎖 WT-SKSC/配列番号:70、L鎖 WT-kappa/配列番号:54)を調製し、ヘテロジェニティーおよび安定性の評価を行った。ヘテロジェニティーの評価は陽イオン交換クロマトグラフィーにより実施した。カラムとしてProPac WCX-10 (Dionex)を用い、移動相Aとして20 mM Sodium Acetate, pH5.0、移動相Bとして20 mM Sodium Acetate, 1 M NaCl, pH5.0を使用し、適切な流量およびグラジエントを用いて実施した。陽イオン交換クロマトグラフィーによる評価を行った結果を図13に示した。安定性の評価は、示差走査型熱量測定(DSC)(VP-DSC、Microcal社製)による熱変性中間温度(Tm値)の評価により実施した。20 mM sodium acetate, 150 mM NaCl, pH6.0におけるDSC測定結果とFabドメインのTm値を図14に示した。
【0141】
その結果、TOCILIZUMAB-IgG1と比較してTOCILIZUMAB-IgG2は著しくヘテロジェニティーが増大するが、TOCILIZUMAB-SKSCにすることによってヘテロジェニティーは大幅に低減できることが見出された。また、TOCILIZUMAB-IgG1と比較してTOCILIZUMAB-IgG2はDSCにおけるFabドメインの熱変性ピークにおいてヘテロ成分によると考えられる安定性の低い、すなわちTmの低いショルダーピーク(Fab*)の成分が認められるが、TOCILIZUMAB-SKSCにすることによってヘテロ成分によると考えられるTm値の低いショルダーピークは消失し、TOCILIZUMAB-IgG1およびTOCILIZUMAB-IgG2のFabドメインと同等の約94℃のTm値を示したことから、TOCILIZUMAB-SKSCは高い安定性を有していることが見出された。
【0142】
TOCILIZUMABの薬物動態を向上するする定常領域変異箇所の同定
上述のとおり、TOCILIZUMABのアイソタイプであるIgG1から、C末端のヘテロジェニティーを低減し、Fcγレセプターへの結合性を低減させ、高い安定性を維持したままIgG2アイソタイプの定常領域の抗体のヘテロジェニティーを低減することが可能であることが見出されたが、薬物動態に関してもTOCILIZUMABのアイソタイプであるIgG1よりも優れた定常領域であることが望ましい。
【0143】
IgG1アイソタイプの定常領域の抗体よりも優れた血漿中半減期を有する定常領域を見出すために、高い安定性を有しIgG2アイソタイプの定常領域の抗体に関する上述のヘテロジェニティーが低減されたTOCILIZUMAB-SKSCに対して、薬物動態を向上させることを目的に変異箇所をスクリーニングした結果、WT-SKSCに対して、EUナンバリング137番目のグルタミン酸をグリシンに138番目のセリンをグリシンに、268番ヒスチジンをグルタミンに、355番アルギニンをグルタミンに、419番グルタミンをグルタミン酸に置換し、これに加えてH鎖C末端のヘテロジェニティーを低減するために446番目のグリシンおよび447番目のリジンを欠損させたWT-M58(配列番号:72(アミノ酸配列))を見出した。さらに、一方、IgG1に対して、434番目のアスパラギンをアラニンに置換したWT-M44(配列番号:73(アミノ酸配列))を作製した。さらにM44に対してH鎖C末端のヘテロジェニティーを低減するために446番目のグリシンおよび447番目のリジンを欠損させたWT-M83(配列番号:74(アミノ酸配列))を作製した。また、WT-M58に対して、434番目のアスパラギンをアラニンに置換したWT-M73(配列番号:75(アミノ酸配列))を作製した。
TOCILIZUMAB-M44(H鎖 WT-M44/配列番号:73、L鎖 WT-kappa/配列番号:54)、TOCILIZUMAB-M58(H鎖 WT-M58/配列番号:72、L鎖 WT-kappa/配列番号:54)およびTOCILIZUMAB-M73(H鎖 WT-M73/配列番号:75、L鎖 WT-kappa/配列番号:54)を調製し、ヒトFcRnへのアフィニティーおよびヒトFcRnトランスジェニックマウスによる薬物動態の評価を行った(方法は参考例参照)。
【0144】
TOCILIZUMAB-IgG1、TOCILIZUMAB-M44、TOCILIZUMAB-M58およびTOCILIZUMAB-M73のヒトFcRnへの結合性の評価をBiacoreにより行った結果、表6に示すとおり、TOCILIZUMAB-M44、TOCILIZUMAB-M58およびTOCILIZUMAB-M73の結合性はTOCILIZUMAB-IgG1よりもそれぞれ約2.7倍、約1.4倍および約3.8倍程度優れていた。
【0145】
【表6】
【0146】
TOCILIZUMAB-IgG1、TOCILIZUMAB-M44、TOCILIZUMAB-M58およびTOCILIZUMAB-M73のヒトFcRnトランスジェニックマウスにおける薬物動態の評価を行った結果を図15に示した。図15に示すとおり、TOCILIZUMAB-M44、TOCILIZUMAB-M58およびTOCILIZUMAB-M73はいずれもTOCILIZUMAB-IgG1と比較して薬物動態が向上することが見出された。その薬物動態の向上効果はヒトFcRnへの結合能と相関した。なかでもTOCILIZUMAB-M73に関しては、TOCILIZUMAB-IgG1と比較して28日後の血漿中濃度が約16倍改善していたことから、ヒトにおいてもM73の定常領域を有する抗体はIgG1の定常領域を有する抗体と比較して大幅に薬物動態が向上すると考えられた。
【0147】
〔実施例7〕PK/PDが改善した完全ヒト化IL-6レセプター抗体の作製
上記実施例で見出されたTOCILIZUMABの可変領域および定常領域の変異を複数組み合わせたTOCILIZUMAB改変体を作製し、各種スクリーニングを実施した結果、完全ヒト化IL-6レセプター抗体として、Fv3-M73(H鎖 VH4-M73/配列番号:25、L鎖 VL1-kappa/配列番号:28)、Fv4-M73(H鎖 VH3-M73/配列番号:26、L鎖 VL3-kappa/配列番号:29)、Fv5-M83(H鎖 VH5-M83/配列番号:27、L鎖 VL5-kappa/配列番号:30)を見出した。
【0148】
作製したFv3-M73、Fv4-M73、およびFv5-M83のIL-6レセプターへのアフィニティーをTOCILIZUMABと比較した(方法は参考例参照)。これらの抗体のpH7.4における可溶型IL-6レセプターへのアフィニティーを測定した結果を表7に示した。また、BaF/gp130の中和活性をTOCILIZUMABおよびコントロール(参考例の公知の高親和性高IL-6レセプター抗体、US 2007/0280945におけるVQ8F11-21 hIgG1)と比較した(方法は参考例参照)。これらの抗体のBaF/gp130による生物活性を測定した結果を図16(IL-6終濃度300 ng/mL:TOCILIZUMAB、コントロール、Fv5-M83)および図17(IL-6終濃度30 ng/mL:TOCILIZUMAB、Fv3-M73、Fv4-M73)に示した。表7に示すとおり、Fv3-M73、Fv4-M73は、TOCILIZUMABと比較して2〜3倍程度強いアフィニティーを有し、Fv5-M83はTOCILIZUMABと比較して100倍程度強いアフィニティーを示した(Fv5-M83ではアフィニティーの測定が困難であったため、定常領域をIgG1にしたFv5-IgG1(H鎖 VH5-IgG1/配列番号:76、L鎖 VL5-kappa/配列番号:30)を用いてアフィニティーを測定した、定常領域は一般にアフィニティーに影響しないと考えられる)。また、図17に示すとおりFv3-M73、Fv4-M73は、TOCILIZUMABと比較してやや強い活性を示し、図16に示すとおりFv5-M83はTOCILIZUMABと比較して50%阻害濃度として100倍以上の極めて強い活性を有し、且つ、公知の高親和性高IL-6レセプター抗体であるコントロールと比較しても50%阻害濃度として約10倍程度高い中和活性を示した。
【0149】
【表7】
【0150】
TOCILIZUMAB、Fv3-M73、および、Fv4-M73のpH7.4とpH5.8における膜型IL-6レセプターからの解離速度を測定した結果を表8に示した(方法は参考例参照)。Fv3-M73およびFv4-M73は、TOCILIZUMABと比較して膜型IL-6レセプターからの解離速度のpH依存性がそれぞれ約11倍および10倍向上していることが示された。実施例5におけるH3pI/L73と比較して、大幅に解離速度のpH依存性が向上していることから、Fv3-M73とFv4-M73の薬物動態はH3pI/L73と比較して大幅に向上していると考えられた。
【0151】
【表8】
【0152】
TOCILIZUMAB、コントロール、Fv3-M73、Fv4-M73、およびFv5-M83の等電点を当業者公知の方法により等電点電気泳動により測定した結果、TOCILIZUMABの等電点は約9.3、コントロールは約8.4〜8.5、Fv3-M73は約5.7〜5.8、Fv4-M73は約5.6〜5.7、Fv5-M83は5.4〜5.5であり、いずれの抗体もTOCILIZUMABおよびコントロールと比較して等電点が大幅に低下した。また、可変領域VH/VLの理論等電点をGENETYX(GENETYX CORPORATION)により計算したところ、TOCILIZUMABの理論等電点は9.20、コントロールは7.79、Fv3-M73は5.49、Fv4-M73は5.01、Fv5-M83は4.27であり、いずれの抗体もTOCILIZUMABおよびコントロールと比較して等電点が大幅に低下した。実施例2において等電点の低下により薬物動態が改善することが示されていることから、Fv3-M73、Fv4-M73、およびFv5-M83はTOCILIZUMABおよびコントロールと比較して薬物動態が向上していると考えられた。
【0153】
TOCILIZUMAB、Fv3-M73、Fv4-M73、およびFv5-M83の可変領域配列に存在するT-cellエピトープをTEPITOPE(Methods. 2004 Dec;34(4):468-75)を用いて解析を行った。その結果、実施例3に示したとおり、TOCILIZUMABは多くの配列がHLAに結合するT-cellエピトープが存在すると予測されたが、Fv3-M73、Fv4-M73、およびFv5-M83はT-cellエピトープに結合すると予測された配列が大幅に減少した。また、Fv3-M73、Fv4-M73、およびFv5-M83はフレームワークにマウス配列が残存せず完全ヒト化されている。これらのことから、Fv3-M73、Fv4-M73、およびFv5-M83はTOCILIZUMABと比較して大幅に免疫原性リスクが低減されている可能性が示唆された。
【0154】
〔実施例8〕完全ヒト化IL-6レセプター抗体のサルPK/PD試験
TOCILIZUMAB、コントロール、Fv3-M73、Fv4-M73、およびFv5-M83をカニクイザルに1 mg/kgで静脈内に単回投与し血漿中濃度推移を評価した(方法は参考例参照)。TOCILIZUMAB、Fv3-M73、Fv4-M73、およびFv5-M83の静脈内投与後の血漿中濃度推移を図18に示した。その結果、Fv3-M73、Fv4-M73、およびFv5-M83はいずれもTOCILIZUMABおよびコントロールと比較してカニクイザルにおいて大幅に薬物動態が改善した。なかでも、Fv3-M73とFv4-M73の薬物動態はTOCILIZUMABと比較して大幅に改善した。
【0155】
カニクイザル膜型IL-6レセプターがどの程度中和されているかの薬効を評価するために、抗体投与6日目から18日目(TOCILIZUMABに関しては3日目から10日目)までカニクイザルIL-6 5μg/kgを腰背部に連日皮下投与し、24時間後の各個体のCRP濃度を測定した(方法は参考例参照)。各抗体投与時のCRP濃度推移を図19に示した。カニクイザル可溶型IL-6レセプターがどの程度中和されているかの薬効を評価するために、カニクイザル血漿中の非結合型のカニクイザル可溶型IL-6レセプター濃度を測定し、非結合型の可溶型IL-6レセプター率を計算した(方法は参考例参照)。各抗体投与時の非結合型の可溶型IL-6レセプター率の推移を図20に示した。
【0156】
Fv3-M73、Fv4-M73、およびFv5-M83はいずれもTOCILIZUMABおよび公知の高親和性抗IL-6レセプター抗体であるコントロールと比較してカニクイザル膜型IL-6レセプターをより持続的に中和し、CRPの増加を長期間抑制した。また、Fv3-M73、Fv4-M73、およびFv5-M83はいずれもTOCILIZUMABおよびコントロールと比較してカニクイザル可溶型IL-6レセプターをより持続的に中和し、非結合型のカニクイザル可溶型IL-6レセプターの増加を長期間抑制した。これより膜型IL-6レセプターおよび可溶型IL-6レセプターの中和の持続性に関しては、Fv3-M73、Fv4-M73、およびFv5-M83はいずれもTOCILIZUMABおよびコントロールよりも優れていることが見出された。なかでもFv3-M73とFv4-M73の中和の持続性は極めて優れていた。一方、Fv5-M83のほうがFv3-M73とFv4-M73よりCRPおよび非結合型カニクイザル可溶型IL-6レセプターを低く抑制していることから、Fv5-M83は膜型IL-6レセプターおよび可溶型IL-6レセプターをFv3-M73とFv4-M73および公知の高親和性抗IL-6レセプター抗体であるコントロールよりも強力に中和していると考えられた。これはFv5-M83がコントロールよりもIL-6レセプターへのアフィニティーが強く、且つ、BaF/gp130における生物活性が強いことがカニクイザルのin vivoにおいて反映された結果であると考えられる。
【0157】
これらのことから、TOCILIZUMABおよびコントロールと比較して、Fv3-M73とFv4-M73は抗IL-6レセプター中和抗体として作用の持続性が極めて優れており、投与頻度および投与量を大幅に低減することが可能であり、また、Fv5-M83は抗IL-6レセプター中和抗体として作用の強さに極めて優れており、また作用の持続性にも優れていることが見出された。よってFv3-M73、Fv4-M73、およびFv5-M83はIL-6アンタゴニストとしての医薬品として有用であると考えられる。
【0158】
〔実施例9〕
Monocyte chemoattractant protein (MCP)-1は、単球・T細胞・NK細胞・basophilの細胞浸潤に関与することが知られている。MCP-1は、RA患者の滑膜組織・滑液中で高発現していることが報告されており(J Clin Invest. 1992 Sep;90(3):772-9)、RAの病態に関与していると考えられている(Inflamm Allergy Drug Targets. 2008 Mar;7(1):53-66.)。
【0159】
また、VEGFは強力な血管新生因子であり、RA患者の滑膜中のマクロファージ・線維芽細胞・滑膜細胞等から産生されることが知られている(J Rheumatol. 1995 Sep;22(9):1624-30.)。また、RA患者血清中のVEGFレベルと疾患活動性やradiographic progressionが相関し(Arthritis Rheum. 2003 Jun;48(6):1521-9.、Arthritis Rheum. 2001 Sep;44(9):2055-64.)、RA患者を抗IL-6R抗体TOCILIZUMABで治療することにより、血清中のVEGFレベルが低下することから、VEGFもRAの病態に重要な役割を担っていると考えられている(Mod Rheumatol. 2009;19(1):12-9.、Mediators Inflamm. 2008;2008:129873)。
【0160】
そこで、TOCILIZUMABおよびFv4-M73はsIL-6R及びIL-6刺激によるヒトRA患者由来滑膜細胞からのMCP-1およびVEGF産生を抑制できるかどうかを以下の方法で検討した。
ヒトRA患者由来滑膜細胞(TOYOBO)を5% FCS含有IMDM培地にて96 well plateに2×104/0.05 mL/wellにて播種し、CO2インキュベーター(37℃, 5%CO2)中で90分静置した。適宜希釈した濃度のTOCILIZUMAB及びFv4-M73を0.05 mL添加し、15分静置後に可溶型IL-6レセプター(SR344:参考例の方法に従って調製)を0.05 mL添加して更に30分静置し、更にIL-6(TORAY)を0.05 mL添加した(可溶型IL-6レセプター及びIL-6の終濃度は各50 ng/mL)。2日培養後、培養上清を回収し、培養上清中のMCP-1およびVEGF濃度をELISA kit (BiosourceおよびPierce Biotechnology)を用いて測定した。結果を図21と図22に示す。TOCILIZUMAB及びFv4-M73は、可溶型IL-6レセプター及びIL-6刺激によるヒトRA患者由来滑膜細胞からのMCP-1およびVEGF産生を濃度依存的に抑制した。
【0161】
これらのことから、Fv4-M73は、抗IL-6レセプター中和抗体として作用(IL-6レセプターに結合し膜型IL-6レセプターおよび可溶型IL-6レセプターのシグナルを遮断)の持続性がTOCILIZUMABと比較して極めて優れており、TOCILIZUMABと比較して投与頻度および投与量を大幅に低減することが可能であり、さらにFv4-M73は、ヒトRA患者由来滑膜細胞からのMCP-1およびVEGF産生を抑制することから、Fv4-M73はRAに極めて有用な治療薬であることが示された。
【0162】
〔参考例〕
組み換え可溶型ヒトIL-6レセプターの調製
抗原であるヒトIL-6レセプターの組み換え可溶型ヒトIL-6レセプターは以下のように調製した。J.Biochem. 108, 673-676 (1990)で報告されているN末端側1番目から344番目のアミノ酸配列からなる可溶型ヒトIL-6レセプター(Yamasakiら、Science 1988;241:825-828 (GenBank # X12830))のCHO細胞定常発現株を作製した。SR344発現CHO細胞から得られた培養上清から、Blue Sepharose 6 FFカラムクロマトグラフィー、SR344に対する特異抗体を固定したカラムによるアフィニティクロマトグラフィー、ゲルろ過カラムクロマトグラフィーの3つのカラムクロマトグラフィーにより、可溶型ヒトIL-6レセプターを精製した。メインピークとして溶出した画分を最終精製品とした。
【0163】
組み換え可溶型カニクイザルIL-6レセプター(cIL-6R)の調製
公開されているアカゲザルIL-6レセプター遺伝子配列 (Birney et al, Ensembl 2006, Nucleic Acids Res. 2006 Jan 1;34(Database issue):D556-61.) を元にオリゴDNAプライマーを作製し、カニクイザル膵臓から調製されたcDNAを鋳型とし、プライマーを用いて、PCR法によりカニクイザルIL-6レセプター遺伝子全長をコードするDNA断片を調製した。得られたDNA断片を哺乳動物細胞発現ベクターへ挿入し、これを用いてCHO定常発現株(cyno.sIL-6R産生CHO細胞)を作製した。cyno.sIL-6R産生CHO細胞の培養液をHisTrapカラム(GEヘルスケアバイオサイエンス)で精製後、Amicon Ultra-15 Ultracel-10k(Millipore)を用いて濃縮し、Superdex200pg16/60ゲルろ過カラム(GEヘルスケアバイオサイエンス)でさらに精製を行い、可溶型カニクイザルIL-6レセプター(以下、cIL-6R)の最終精製品とした。
【0164】
組み換えカニクイザルIL-6(cIL-6)の調製
カニクイザルIL-6は以下のように調製した。SWISSPROT Accession No.P79341に登録されている212アミノ酸をコードする塩基配列を作成し、哺乳動物細胞発現ベクターにクローニングし、CHO細胞に導入することで定常発現細胞株を作製した(cyno.IL-6産生CHO細胞)。cyno.IL-6産生CHO細胞の培養液をSP-Sepharose/FFカラム(GEヘルスケアバイオサイエンス)で精製後、Amicon Ultra-15 Ultracel-5k(Millipore)を用いて濃縮し、Superdex75pg26/60ゲルろ過カラム(GEヘルスケアバイオサイエンス)でさらに精製を行い、Amicon Ultra-15 Ultracel-5k(Millipore)を用いて濃縮し、カニクイザルIL-6(以下、cIL-6)の最終精製品とした。
【0165】
公知高親和性抗IL-6レセプター抗体の作製
公知の高親和性抗IL-6レセプター抗体として、US 2007/0280945 A1に記載されている高親和性抗IL-6レセプター抗体であるVQ8F11-21 hIgG1(US 2007/0280945 A1, H鎖アミノ酸配列:配列番号:77、L鎖アミノ酸配列:配列番号:78)を発現させるため、哺乳動物細胞発現用ベクターを構築した。抗体可変領域については、合成オリゴDNAを組み合わせたPCR法(assembly PCR)により作製し、定常領域についてはIgG1を使用した。Assembly PCR法により抗体可変領域と定常領域を結合させ、哺乳動物発現用ベクターへ挿入し、目的のH鎖発現ベクターおよびL鎖発現ベクターを作製した。得られた発現ベクターの塩基配列は当業者公知の方法で決定した。作製した発現ベクターを用い、発現・精製を行った。発現・精製は実施例1に記載した方法で行い、高親和性抗IL-6レセプター抗体(以降、コントロール、と記す)を得た。
【0166】
TOCILIZUMABの変異体の作製・発現・精製
TOCILIZUMABの変異体はQuikChange Site-Directed Mutagenesis Kit(Stratagene)を用いて、添付説明書記載の方法で変異体を作製し、得られたプラスミド断片を哺乳動物細胞発現ベクターに挿入し、目的のH鎖発現ベクターおよびL鎖発現ベクターを作製した。得られた発現ベクターの塩基配列は当業者公知の方法で決定した。抗体の発現は以下の方法を用いて行った。ヒト胎児腎癌細胞由来HEK293H株(Invitrogen)を10 % Fetal Bovine Serum (Invitrogen)を含むDMEM培地(Invitrogen)へ懸濁し、5〜6 × 105個/mLの細胞密度で接着細胞用ディッシュ(直径10 cm, CORNING)の各ディッシュへ10 mLずつ蒔きこみCO2インキュベーター(37℃、5% CO2)内で一昼夜培養した後に、培地を吸引除去し、CHO-S-SFM-II(Invitrogen)培地6.9 mLを添加した。調製したプラスミドをlipofection法により細胞へ導入した。得られた培養上清を回収した後、遠心分離(約2000 g、5分間、室温)して細胞を除去し、さらに0.22μmフィルターMILLEX(R)-GV(Millipore)を通して滅菌して培養上清を得た。得られた培養上清からrProtein A SepharoseTM Fast Flow(Amersham Biosciences)を用いて当業者公知の方法で抗体を精製した。精製抗体濃度は、分光光度計を用いて280 nmでの吸光度を測定した。得られた値からPACE法により算出された吸光係数を用いて抗体濃度を算出した(Protein Science 1995 ; 4 : 2411-2423)。
【0167】
ヒトgp130発現BaF3細胞株の樹立
IL-6依存増殖性を示す細胞株を得るために、以下に示すとおり、ヒトgp130を発現したBaF3細胞株の樹立を行った。
【0168】
全長ヒトgp130 cDNA(Hibiら、Cell 1990;63:1149-1157(GenBank # NM_002184))をPCRにより増幅し、pCHOI(Hirataら、FEBS Letter 1994;356:244-248)のDHFR遺伝子発現部位を除去し、Zeocin耐性遺伝子発現部位を挿入した発現ベクターpCOS2Zeoにクローニングし、pCOS2Zeo/gp130を構築した。全長ヒトIL-6R cDNAをPCRにより増幅し、pcDNA3.1(+)(Invitrogen)にクローニングし、hIL-6R/pcDNA3.1(+)を構築した。
【0169】
10μgのpCOS2Zeo/gp130をPBSに懸濁したBaF3細胞(0.8 x 107 cells)に混合し、Gene Pulser(Bio-Rad)を用いて0.33 kV, 950μFDの容量でパルスを加えた。エレクトロポーレーション処理により遺伝子導入したBaF3細胞を0.2 ng/mLのmouse interleukin-3(Peprotech)、10% Fetal Bovine Serum(以下FBS、HyClone)を含むRPMI1640培地(Invitrogen)で一昼夜培養し、100 ng/mLのhuman interleukin-6(R&D systems)、100 ng/mLのhuman interleukin-6 soluble receptor(R&D systems)および10% FBSを含むRPMI1640培地を加えて選抜し、ヒトgp130発現BaF3細胞株(以下、BaF3/gp130)を樹立した。このBaF/gp130は、human interleukin-6(R&D systems)および可溶型ヒトIL-6レセプター存在下で増殖することから、抗IL-6レセプター抗体の増殖阻害活性(すなわちIL-6レセプター中和活性)の評価に使用することが可能である。
【0170】
ヒトgp130発現BaF3細胞(BaF/gp130)による生物活性評価
IL-6/IL-6レセプター依存性増殖を示すBaF3/gp130を用いて、IL-6レセプター中和活性を評価した。BaF3/gp130を10% FBSを含むRPMI1640培地で3回洗浄した後に、5 x 104 cells/mLとなるように600 ng/mLないしは60 ng/mLのhuman interleukin-6(TORAY)(終濃度は300 ng/mLないしは30 ng/mL)、適当量の可溶型ヒトIL-6レセプターおよび10% FBSを含むRPMI1640培地に懸濁し、96 well-plate(CORNING)の各wellに50μLずつ分注した。次に、精製した抗体を10% FBSを含むRPMI1640に希釈して、各wellに50μLずつ混合した。37℃、5% CO2条件下で、3日間培養し、PBSで2倍に希釈したWST-8試薬(Cell Counting Kit-8、株式会社同仁化学研究所)を20μL/wellで加え、直後にSUNRISE CLASSIC(TECAN)を用いて450 nmの吸光度(参照波長620 nm)を測定した。2時間培養した後に、再度450 nmの吸光度(参照波長620 nm)を測定し、2時間の吸光度変化を指標にIL-6レセプター中和活性を評価した。
【0171】
Biacoreによる可溶型ヒトIL-6レセプターへの結合評価
Biacore T100(GE Healthcare)を用いて、抗原抗体反応の速度論的解析を行った。センサーチップ上にアミンカップリング法でprotein Aあるいはprotein A/Gあるいはanti-IgG(γ-chain specific)F(ab’)2を適当量固定化し、次にpH7.4において目的の抗体を結合させ、さらにpH7.4において種々の濃度に調製した可溶型IL-6レセプターをアナライトとして流し、抗体と可溶型ヒトIL-6レセプターの相互作用を測定した。測定は全て37℃で実施した。測定で得られたセンサーグラムから、カイネティクスパラメーターである結合速度定数ka(1/Ms)、および解離速度定数kd(1/s)を算出し、その値をもとにKD(M)を算出した。各パラメーターの算出にはBiacore T100 Evaluation Software(GE Healthcare)を用いた。
【0172】
Biacoreによる膜型IL-6レセプターへのpH依存的解離評価
Biacore T100(GE Healthcare)を用いてpH5.8, pH7.4における膜型IL-6レセプターへの抗原抗体反応を観測した。センサーチップ上に固定化した可溶型ヒトIL-6レセプターへの結合を評価することで、膜型IL-6レセプターへの結合を評価した。SR344を当業者公知の方法に従ってビオチン化し、ストレプトアビジンとビオチンの親和性を利用し、ストレプトアビジンを介してビオチン化可溶型ヒトIL-6レセプターをセンサーチップ上に固定化した。測定は全て37℃で実施し、移動相のバッファーは10 mM MES pH5.8, 150 mM NaCl, 0.05% Tween20とし、そこにpH依存的結合クローンをpH7.4の条件下で注入して可溶型ヒトIL-6レセプターと結合させたのち(注入サンプルのバッファーは10 mM MES pH7.4, 150 mM NaCl, 0.05% Tween20)、移動相のpHである5.8で各クローンのpH依存的な解離を観測した。サンプル濃度を0.25μg/mLとし、10 mM MES pH7.4, 150 mM NaCl, 0.05 % Tween20で結合させ、10 mM MES pH5.8, 150 mM NaCl, 0.05% Tween20で解離させたときのpH5.8における解離相のみBiacore T100 Evaluation Software(GE Healthcare)を用いフィッティングすることにより、pH5.8における解離速度定数(kd(1/s))を算出した。同様にまた、サンプル濃度を0.5μg/mLとし、10 mM MES pH7.4, 150 mM NaCl, 0.05% Tween20で結合させ、10 mM MES pH7.4, 150 mM NaCl, 0.05% Tween20で解離させたときのpH7.4における解離相のみBiacore T100 Evaluation Software(GE Healthcare)を用いフィッティングすることにより、pH7.4における解離速度定数(kd(1/s))を算出した。
【0173】
ヒトFcRnへの結合評価
FcRnはFcRnとβ2-microglobulinの複合体である。公開されているヒトFcRn遺伝子配列(J. Exp. Med. 180 (6), 2377-2381 (1994))を元に、オリゴDNAプライマーを作製した。ヒトcDNA(Human Placenta Marathon-Ready cDNA, Clontech)を鋳型とし、作製したプライマーを用いPCR法により遺伝子全長をコードするDNA断片を調整した。得られたDNA断片を鋳型に、PCR法によりシグナル領域を含む細胞外領域(Met1-Leu290)をコードするDNA断片を増幅し、哺乳動物細胞発現ベクターへ挿入した(ヒトFcRnアミノ酸配列/配列番号:79)。同様に、公開されているヒトβ2-microglobulin遺伝子配列(Proc. Natl. Acad. Sci. U.S.A. 99 (26), 16899-16903 (2002))を元に、オリゴDNAプライマーを作製した。ヒトcDNA(Hu-Placenta Marathon-Ready cDNA, CLONTECH)を鋳型とし、作製したプライマーを用いPCR法により遺伝子全長をコードするDNA断片を調製した。得られたDNA断片を鋳型に、PCR法によりシグナル領域を含むβ2-microglobulin全長(Met1-Met119)をコードするDNA断片を増幅し、哺乳動物細胞発現ベクターへ挿入した(ヒトβ2-microglobulinアミノ酸配列/配列番号:80)。
【0174】
可溶型ヒトFcRnの発現は以下の手順で行った。調製したヒトFcRnおよびヒトβ2-microglobulinのプラスミドを、10 % Fetal Bovine Serum (Invitrogen)を用いたlipofection法により、ヒト胎児腎癌細胞由来HEK293H株(Invitrogen)の細胞へ導入した。得られた培養上清を回収した後、IgG Sepharose 6 Fast Flow(Amersham Biosciences)を用い、(J Immunol. 2002 Nov 1;169(9):5171-80.)の方法に従い精製を行った。その後、HiTrap Q HP(GE Healthcare)により精製を行った。
【0175】
マウスにおける抗体血漿中濃度の測定
マウス血漿中抗体濃度測定はELISA法にて当業者公知の方法で測定した。
【0176】
サルPK/PD試験による抗体血漿中濃度、CRP濃度、非結合型可溶型IL-6レセプターの測定
カニクイザル血漿中濃度測定はELISA法にて当業者公知の方法で測定した。
CRP濃度はサイアスR CRP(関東化学株式会社)にて、自動分析装置(TBA-120FR、東芝メディカルシステムズ株式会社)を用いて測定した。
【0177】
カニクイザル血漿中の非結合型の可溶型カニクイザルIL-6レセプター濃度を以下の通り測定した。カニクイザルの血漿30μLを0.22μmのフィルターカップ(Millipore)において乾燥させた適量のrProtein A Sepharose Fast Flow(GE Healthcare)樹脂に添加することで血漿中に存在する全てのIgG型抗体(カニクイザルIgG、抗ヒトIL-6レセプター抗体および抗ヒトIL-6レセプター抗体-可溶型カニクイザルIL-6レセプター複合体)をProteinAに吸着させた。その後、高速遠心機でスピンダウンし、パス溶液を回収した。パス溶液にはproteinAに結合した抗ヒトIL-6レセプター抗体-可溶型カニクイザルIL-6レセプター複合体は含まれないため、proteinAパス溶液中の可溶型カニクイザルIL-6レセプター濃度を測定することによって、非結合型の可溶型IL-6レセプター濃度を測定可能である。可溶型カニクイザルIL-6レセプター濃度は、上記で作製した可溶型カニクイザルIL-6レセプター(cIL-6R)をスタンダードに用いて、ヒトIL-6レセプター濃度を測定する当業者公知の方法で測定した。非結合型の可溶型IL-6レセプター率は以下の計算式によって計算した。
【0178】
(抗体投与後の非結合型の可溶性IL-6レセプター濃度÷抗体投与前の可溶性IL-6レセプター濃度)×100
【特許請求の範囲】
【請求項1】
以下の(a)〜(f)いずれかに記載のポリペプチド;
(a) 配列番号:1(VH4-M73のCDR1)の配列を有するCDR1、配列番号:2(VH4-M73のCDR2)の配列を有するCDR2、および配列番号:3(VH4-M73のCDR3)の配列を有するCDR3を含むポリペプチド、
(b) 配列番号:4(VH3-M73のCDR1)の配列を有するCDR1、配列番号:5(VH3-M73のCDR2)の配列を有するCDR2、および配列番号:6(VH3-M73のCDR3)の配列を有するCDR3を含むポリペプチド、
(c) 配列番号:7(VH5-M83のCDR1)の配列を有するCDR1、配列番号:8(VH5-M83のCDR2)の配列を有するCDR2、および配列番号:9(VH5-M83のCDR3)の配列を有するCDR3を含むポリペプチド、
(d) 配列番号:10(VL1のCDR1)の配列を有するCDR1、配列番号:11(VL1のCDR2)の配列を有するCDR2、および配列番号:12(VL1のCDR3)の配列を有するCDR3を含むポリペプチド、
(e) 配列番号:13(VL3のCDR1)の配列を有するCDR1、配列番号:14(VL3のCDR2)の配列を有するCDR2、および配列番号:15(VL3のCDR3)の配列を有するCDR3を含むポリペプチド、
(f) 配列番号:16(VL5のCDR1)の配列を有するCDR1、配列番号:17(VL5のCDR2)の配列を有するCDR2、および配列番号:18(VL5のCDR3)の配列を有するCDR3を含むポリペプチド。
【請求項2】
以下の(a)〜(c)いずれかに記載の抗体;
(a) 配列番号:1(VH4-M73のCDR1)の配列を有するCDR1、配列番号:2(VH4-M73のCDR2)の配列を有するCDR2、および配列番号:3(VH4-M73のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに
配列番号:10(VL1のCDR1)の配列を有するCDR1、配列番号:11(VL1のCDR2)の配列を有するCDR2、および配列番号:12(VL1のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体、
(b) 配列番号:4(VH3-M73のCDR1)の配列を有するCDR1、配列番号:5(VH3-M73のCDR2)の配列を有するCDR2、および配列番号:6(VH3-M73のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに
配列番号:13(VL3のCDR1)の配列を有するCDR1、配列番号:14(VL3のCDR2)の配列を有するCDR2、および配列番号:15(VL3のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体、
(c) 配列番号:7(VH5-M83のCDR1)の配列を有するCDR1、配列番号:8(VH5-M83のCDR2)の配列を有するCDR2、および配列番号:9(VH5-M83のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに
配列番号:16(VL5のCDR1)の配列を有するCDR1、配列番号:17(VL5のCDR2)の配列を有するCDR2、および配列番号:18(VL5のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体。
【請求項3】
以下の(a)〜(f)いずれかに記載の可変領域;
(a) 配列番号:19(VH4-M73の可変領域)の配列を有する重鎖可変領域、
(b) 配列番号:20(VH3-M73の可変領域)の配列を有する重鎖可変領域、
(c) 配列番号:21(VH5-M83の可変領域)の配列を有する重鎖可変領域、
(d) 配列番号:22(VL1の可変領域)の配列を有する軽鎖可変領域、
(e) 配列番号:23(VL3の可変領域)の配列を有する軽鎖可変領域、
(f) 配列番号:24(VL5の可変領域)の配列を有する軽鎖可変領域。
【請求項4】
以下の(a)〜(c)いずれかに記載の抗体;
(a) 配列番号:19(VH4-M73の可変領域)の配列を有する重鎖可変領域および配列番号:22(VL1の可変領域)の配列を有する軽鎖可変領域を含む抗体、
(b) 配列番号:20(VH3-M73の可変領域)の配列を有する重鎖可変領域および配列番号:23(VL3の可変領域)の配列を有する軽鎖可変領域を含む抗体、
(c) 配列番号:21(VH5-M83の可変領域)の配列を有する重鎖可変領域および配列番号:24(VL5の可変領域)の配列を有する軽鎖可変領域を含む抗体。
【請求項5】
以下の(a)〜(f)いずれかに記載の重鎖又は軽鎖;
(a) 配列番号:25(VH4-M73)の配列を有する重鎖、
(b) 配列番号:26(VH3-M73)の配列を有する重鎖、
(c) 配列番号:27(VH5-M83)の配列を有する重鎖、
(d) 配列番号:28(VL1)の配列を有する軽鎖、
(e) 配列番号:29(VL3)の配列を有する軽鎖、
(f) 配列番号:30(VL5)の配列を有する軽鎖。
【請求項6】
以下の(a)〜(c)いずれかに記載の抗体;
(a) 配列番号:25(VH4-M73)の配列を有する重鎖および配列番号:28(VL1)の配列を有する軽鎖を含む抗体、
(b) 配列番号:26(VH3-M73)の配列を有する重鎖および配列番号:29(VL3)の配列を有する軽鎖を含む抗体、
(c) 配列番号:27(VH5-M83)の配列を有する重鎖および配列番号:30(VL5)の配列を有する軽鎖を含む抗体。
【請求項7】
請求項1〜6いずれかに記載のポリペプチドをコードする遺伝子。
【請求項8】
請求項7に記載の遺伝子を含むベクター。
【請求項9】
請求項8に記載のベクターを保持する宿主細胞。
【請求項10】
請求項9の宿主細胞を培養することにより、請求項1〜6いずれかに記載のポリペプチドを製造する方法。
【請求項11】
請求項1〜6いずれかに記載のポリペプチド、または請求項10に記載の方法によって製造されるポリペプチドを含む医薬組成物。
【請求項1】
以下の(a)〜(f)いずれかに記載のポリペプチド;
(a) 配列番号:1(VH4-M73のCDR1)の配列を有するCDR1、配列番号:2(VH4-M73のCDR2)の配列を有するCDR2、および配列番号:3(VH4-M73のCDR3)の配列を有するCDR3を含むポリペプチド、
(b) 配列番号:4(VH3-M73のCDR1)の配列を有するCDR1、配列番号:5(VH3-M73のCDR2)の配列を有するCDR2、および配列番号:6(VH3-M73のCDR3)の配列を有するCDR3を含むポリペプチド、
(c) 配列番号:7(VH5-M83のCDR1)の配列を有するCDR1、配列番号:8(VH5-M83のCDR2)の配列を有するCDR2、および配列番号:9(VH5-M83のCDR3)の配列を有するCDR3を含むポリペプチド、
(d) 配列番号:10(VL1のCDR1)の配列を有するCDR1、配列番号:11(VL1のCDR2)の配列を有するCDR2、および配列番号:12(VL1のCDR3)の配列を有するCDR3を含むポリペプチド、
(e) 配列番号:13(VL3のCDR1)の配列を有するCDR1、配列番号:14(VL3のCDR2)の配列を有するCDR2、および配列番号:15(VL3のCDR3)の配列を有するCDR3を含むポリペプチド、
(f) 配列番号:16(VL5のCDR1)の配列を有するCDR1、配列番号:17(VL5のCDR2)の配列を有するCDR2、および配列番号:18(VL5のCDR3)の配列を有するCDR3を含むポリペプチド。
【請求項2】
以下の(a)〜(c)いずれかに記載の抗体;
(a) 配列番号:1(VH4-M73のCDR1)の配列を有するCDR1、配列番号:2(VH4-M73のCDR2)の配列を有するCDR2、および配列番号:3(VH4-M73のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに
配列番号:10(VL1のCDR1)の配列を有するCDR1、配列番号:11(VL1のCDR2)の配列を有するCDR2、および配列番号:12(VL1のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体、
(b) 配列番号:4(VH3-M73のCDR1)の配列を有するCDR1、配列番号:5(VH3-M73のCDR2)の配列を有するCDR2、および配列番号:6(VH3-M73のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに
配列番号:13(VL3のCDR1)の配列を有するCDR1、配列番号:14(VL3のCDR2)の配列を有するCDR2、および配列番号:15(VL3のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体、
(c) 配列番号:7(VH5-M83のCDR1)の配列を有するCDR1、配列番号:8(VH5-M83のCDR2)の配列を有するCDR2、および配列番号:9(VH5-M83のCDR3)の配列を有するCDR3を含む重鎖可変領域、ならびに
配列番号:16(VL5のCDR1)の配列を有するCDR1、配列番号:17(VL5のCDR2)の配列を有するCDR2、および配列番号:18(VL5のCDR3)の配列を有するCDR3を含む軽鎖可変領域を含む抗体。
【請求項3】
以下の(a)〜(f)いずれかに記載の可変領域;
(a) 配列番号:19(VH4-M73の可変領域)の配列を有する重鎖可変領域、
(b) 配列番号:20(VH3-M73の可変領域)の配列を有する重鎖可変領域、
(c) 配列番号:21(VH5-M83の可変領域)の配列を有する重鎖可変領域、
(d) 配列番号:22(VL1の可変領域)の配列を有する軽鎖可変領域、
(e) 配列番号:23(VL3の可変領域)の配列を有する軽鎖可変領域、
(f) 配列番号:24(VL5の可変領域)の配列を有する軽鎖可変領域。
【請求項4】
以下の(a)〜(c)いずれかに記載の抗体;
(a) 配列番号:19(VH4-M73の可変領域)の配列を有する重鎖可変領域および配列番号:22(VL1の可変領域)の配列を有する軽鎖可変領域を含む抗体、
(b) 配列番号:20(VH3-M73の可変領域)の配列を有する重鎖可変領域および配列番号:23(VL3の可変領域)の配列を有する軽鎖可変領域を含む抗体、
(c) 配列番号:21(VH5-M83の可変領域)の配列を有する重鎖可変領域および配列番号:24(VL5の可変領域)の配列を有する軽鎖可変領域を含む抗体。
【請求項5】
以下の(a)〜(f)いずれかに記載の重鎖又は軽鎖;
(a) 配列番号:25(VH4-M73)の配列を有する重鎖、
(b) 配列番号:26(VH3-M73)の配列を有する重鎖、
(c) 配列番号:27(VH5-M83)の配列を有する重鎖、
(d) 配列番号:28(VL1)の配列を有する軽鎖、
(e) 配列番号:29(VL3)の配列を有する軽鎖、
(f) 配列番号:30(VL5)の配列を有する軽鎖。
【請求項6】
以下の(a)〜(c)いずれかに記載の抗体;
(a) 配列番号:25(VH4-M73)の配列を有する重鎖および配列番号:28(VL1)の配列を有する軽鎖を含む抗体、
(b) 配列番号:26(VH3-M73)の配列を有する重鎖および配列番号:29(VL3)の配列を有する軽鎖を含む抗体、
(c) 配列番号:27(VH5-M83)の配列を有する重鎖および配列番号:30(VL5)の配列を有する軽鎖を含む抗体。
【請求項7】
請求項1〜6いずれかに記載のポリペプチドをコードする遺伝子。
【請求項8】
請求項7に記載の遺伝子を含むベクター。
【請求項9】
請求項8に記載のベクターを保持する宿主細胞。
【請求項10】
請求項9の宿主細胞を培養することにより、請求項1〜6いずれかに記載のポリペプチドを製造する方法。
【請求項11】
請求項1〜6いずれかに記載のポリペプチド、または請求項10に記載の方法によって製造されるポリペプチドを含む医薬組成物。
【図1】

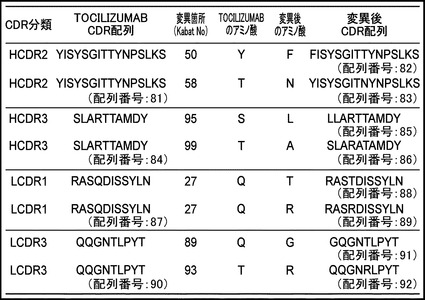
【図2】
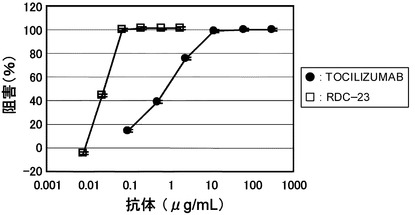
【図3】
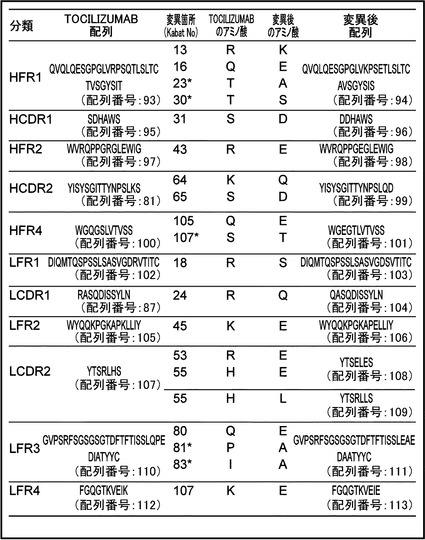
【図4】
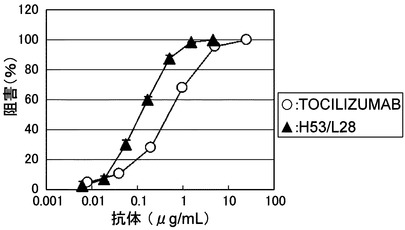
【図5】
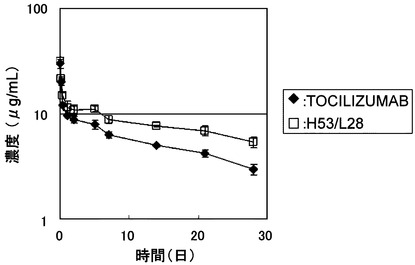
【図6】
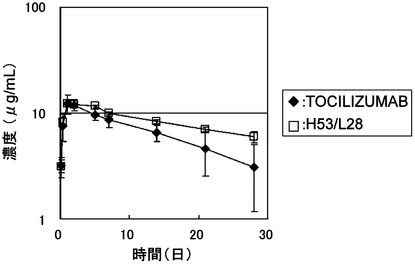
【図7】
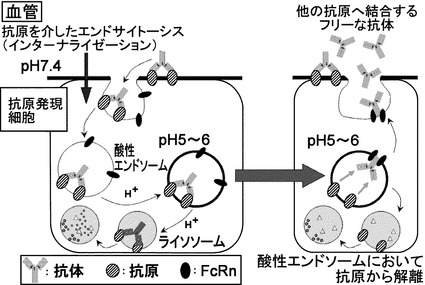
【図8】
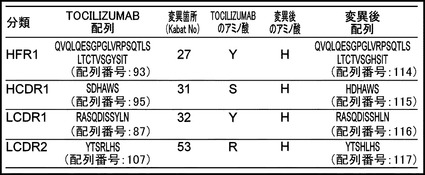
【図9】
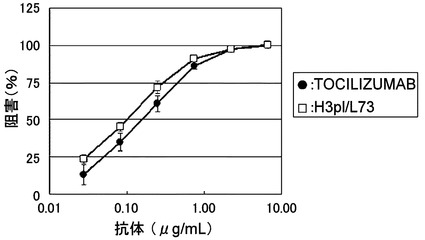
【図10】
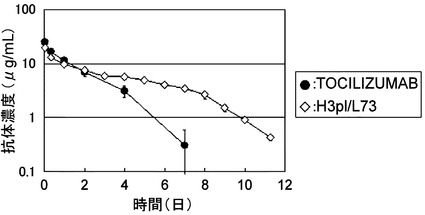
【図11】
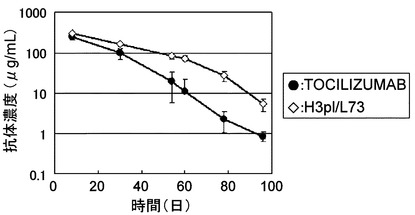
【図12】
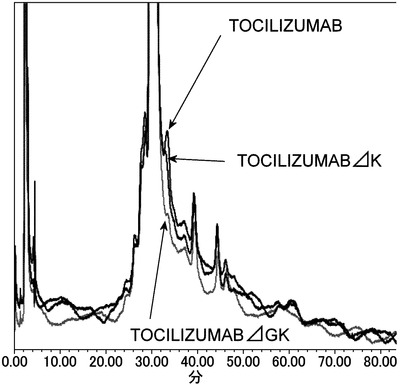
【図13】
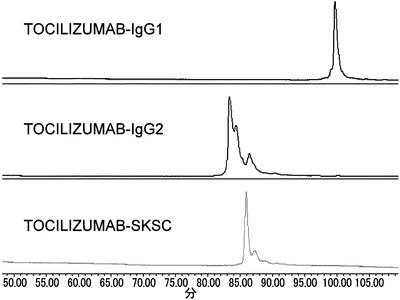
【図14】
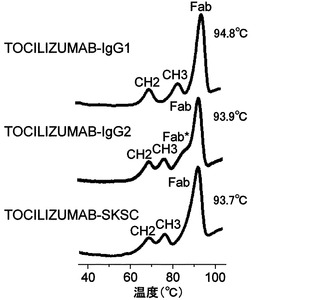
【図15】
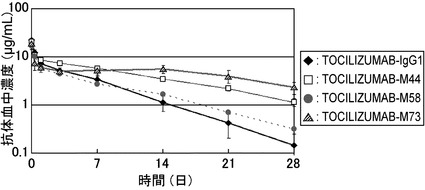
【図16】
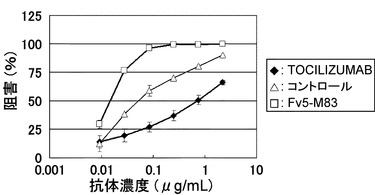
【図17】
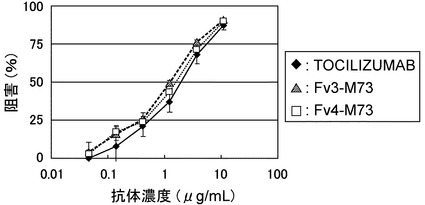
【図18】
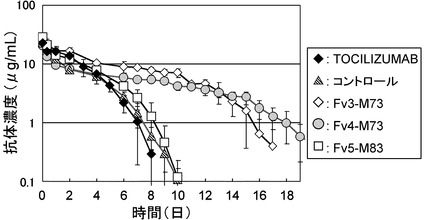
【図19】
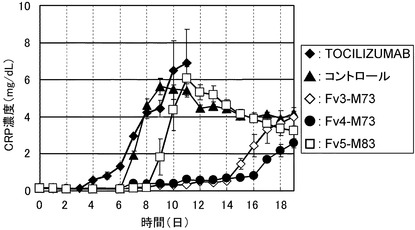
【図20】
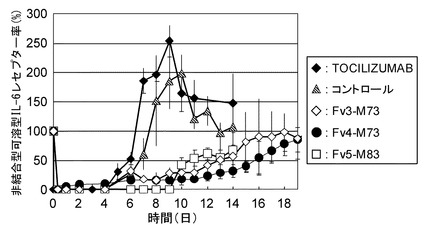
【図21】
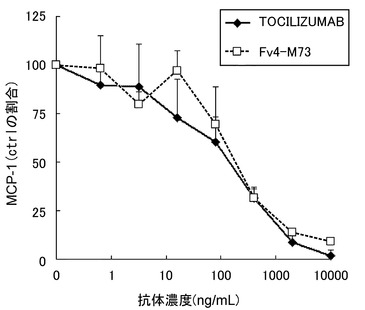
【図22】
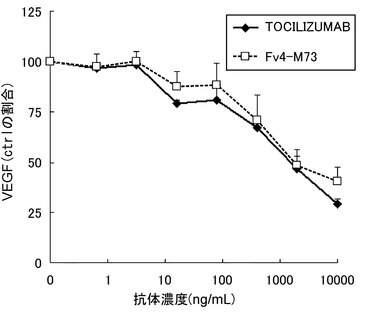
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【公開番号】特開2010−239958(P2010−239958A)
【公開日】平成22年10月28日(2010.10.28)
【国際特許分類】
【出願番号】特願2010−13833(P2010−13833)
【出願日】平成22年1月26日(2010.1.26)
【分割の表示】特願2009−552949(P2009−552949)の分割
【原出願日】平成21年9月25日(2009.9.25)
【出願人】(000003311)中外製薬株式会社 (228)
【Fターム(参考)】
【公開日】平成22年10月28日(2010.10.28)
【国際特許分類】
【出願日】平成22年1月26日(2010.1.26)
【分割の表示】特願2009−552949(P2009−552949)の分割
【原出願日】平成21年9月25日(2009.9.25)
【出願人】(000003311)中外製薬株式会社 (228)
【Fターム(参考)】
[ Back to top ]