
改良N4キレーターコンジュゲート
本発明は、リンカー基を介して生体ターゲティング部分と連結したテトラアミンキレーターコンジュゲート並びに放射性医薬品としてのそのテクネチウム錯体を提供する。リンカー基はキレーターが橋頭位で単官能化されるようなもので、柔軟性を与えるとともにアリール基を含まないものをもたらし、親油性及び立体的嵩高さを最小限にする。キレーターの保護形も提供し、テトラアミンキレーターのアミン窒素による干渉反応を起こさずに広範なターゲティング分子との結合が可能となる。官能化キレーターの合成についても、二官能性キレーター前駆体と併せて開示する。本発明のテクネチウム金属錯体を含む放射性医薬組成物についても、かかる放射性医薬品の調製用の非放射性キットと併せて開示する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、放射性金属99mTcとの金属錯体の形成に適したテトラアミンキレーターと生体ターゲティング分子との改良コンジュゲートに関する。この放射性金属錯体は99mTc放射性医薬品として有用である。キット及び前駆体も提供される。
【背景技術】
【0002】
米国特許第5489425号(Dow Chemical)には、金属の錯体化に有用な一群の鎖状及び大環状の官能化テトラアミンキレーター、特に放射性及び非放射性ロジウム錯体、殊に105Rh又は101mRh放射性金属錯体が開示されている。開示された具体的なテトラアミンには、以下のものが含まれる。
【0003】
【化1】

【0004】
上記二官能性キレーターは、治療又は診断用のモノクローナル抗体又はその断片との結合に有用であると記載されている。米国特許第5489425号には、まず105Rh金属錯体を形成してから抗体と反応させた後、精製することによって、抗体放射性金属錯体キレーターコンジュゲートを調製することが開示されている(実施例21、22a及び23)。米国特許第5489425号には、錯化していない抗体キレーターコンジュゲート、つまり放射性金属の配位していないものは記載されていない。米国特許第5489425号は、かかる抗体結合反応において側鎖アミンをキレーターの4つのアミンからどのように差別化するか何ら教示されていない。米国特許第5489425号は、二官能性キレーターが「テクネチウム及びレニウムとの錯体化にも有用であろう」と記載されているが、これをどのように達成するかについては開示されていないし、テクネチウム錯体の具体例も開示されていない。
【0005】
米国特許第5650134号には、一群のキレーターのソマトスタチンペプチド−キレーターコンジュゲートが開示されている。実施例1には、オクトレオチドペプチドの6−(p−イソチオシアナトベンジル)−1,4,8,11−テトラアザウンデカンとの結合が記載されている。
【0006】
欧州特許出願公開第1181936号には、以下の二官能性キレーターBBN−1及びBBN−2を用いて調製されたテトラアミンキレーターのボンベシン(テトラデカペプチド)コンジュゲート並びにそれらの99mTc錯体が開示されている。
【0007】
【化2】
【0008】
99mTc錯体はマウス体内から腎臓・尿路系を介しての迅速なクリアランスを呈すると記載されている。しかし、欧州特許出願公開第1181936号には、BBN−1又はBBN−2をボンベシンのN末端に結合する段階を除いて、BBN−1又はBBN−2の合成に関して何ら開示されていないし、引用されてもいない。腫瘍イメージング用放射性医薬品候補を得るためのBBN−2とボンベシンとの結合及び99mTc標識についても、Nock et al, Eur. J. Nucl. Med.,30(2),247−258(2003)に記載されている。この99mTc錯体は従来技術のボンベシンキレートコンジュゲートよりも親水性が向上し、そのため腎臓・尿路系による排泄に有利に働くと期待されると記載されている。
【0009】
ヒトの患者用の腫瘍イメージング用放射性医薬品候補を得るためのBBN−1とオクトレオチドとの結合及び99mTc標識についても、Marina et al, Eur. J. Nucl. Med.,30(9),1211−1219(2003)に記載されている。以上のBBN−1又はBBN−2の刊行物のいずれにも、BBN−1又はBBN−2の合成については何な記載されていない。
【特許文献1】米国特許第5489425号明細書
【特許文献2】米国特許第5650134号明細書
【特許文献3】欧州特許出願公開第1181936号明細書
【非特許文献1】Nock et al, Eur. J. Nucl. Med.,30(2),247−258(2003)
【非特許文献2】Marina et al, Eur. J. Nucl. Med.,30(9),1211−1219(2003)
【発明の開示】
【課題を解決するための手段】
【0010】
本発明は、リンカー基を介して生体ターゲティング部分と連結したテトラアミンキレーターコンジュゲート並びに放射性医薬品としてのそのテクネチウム錯体を提供する。リンカー基はキレーターが橋頭位で単官能化されるようなもので、柔軟性を与えるとともにアリール基を含まないものをもたらし、親油性及び立体的嵩高さを最小限にする。キレーターの適当な保護形も提供し、テトラアミンキレーターのアミン窒素による干渉反応を起こさずに広範なターゲティング分子との結合が可能となる。官能化キレーターの合成についても、二官能性キレーター前駆体と併せて説明する。
【0011】
本発明のテクネチウム金属錯体を含む放射性医薬組成物についても、かかる放射性医薬品の調製用の非放射性キットと併せて説明する。
【発明を実施するための最良の形態】
【0012】
第一の実施形態では、本発明は次の式(I)の陽イオン99mTCテクネチウム錯体を提供する。
【0013】
【化3】
【0014】
式中、
Xは−NR−、−CO2−、−CO−、−NR(C=S)−、−NR(C=O)−、−CONR−又はQ基であり、
各Yは独立にD−若しくはL−アミノ酸、−CH2−、−CH2OCH2−又は−OCH2CH2O−又はX基であり、
Zは合成生体ターゲティング部分であり、
nは1〜8の整数であり、
mは0〜30の整数であり、
RはH、C1〜4アルキル、C2〜4アルコキシアルキル、C1〜4ヒドロキシアルキル又はC1〜4フルオロアルキルであり、
Qは次式の基であり、
【0015】
【化4】
【0016】
Aは対イオンであるが、
ヘテロ原子同士が直接結合した結合を原子鎖X1−(Y)mが含まないことを条件とする。
【0017】
テクネチウム放射性同位体は、99mTcのようなγ線放射体でも、94mTcのようなPETイメージングに適した陽電子放射体でもよい。好ましくはテクネチウム放射性同位体は99mTc又は94mTcであり、最も好ましくは99mTcである。
【0018】
Xは好ましくは−CONR−、−NR(C=O)−又はQ基である。Xは最も好ましくは−CONR−又は−NR(C=O)−であり、−CONH−及び−NH(C=O)−が特に好ましい。
【0019】
式Iのリンカー基−(CH2)n−X−(Y)m−は、ヘテロ原子同士が直接結合した結合をX1−(Y)m原子鎖が含まないように選択され、「ヘテロ原子」という用語は窒素、酸素又はイオウのような非炭素原子を意味する。これは上記原子鎖がO−O、N−N又はO−Nのような結合を含まないことを意味する。
【0020】
式Iのリンカー基−(CH2)n−X−(Y)m−の役割は、生体内での生体ターゲティング部分(Z)の活性結合部位からテクネチウム錯体を離隔することであると想定される。これは生体内での活性部位への結合を比較的嵩高いテクネチウム錯体が立体的に阻害しないようにするのに役立つ。アルキレン基−(CH2)n−は、結合した生体ターゲティング部分(Z)との有意な水素結合相互作用がなく、そのためリンカーがZに巻き付かないという利点を有する。好ましいアルキレン基はn=1〜6、最も好ましくは2〜4であり、2が特に好ましい。
【0021】
本発明のリンカー基はアリール環を含まない。これは、コンジュゲートの生体ターゲティング部分(Z)に結合したリンカー基並びにテクネチウム錯体の親油性を最小限にするのに役立つ。リンカー基(及びテクネチウム錯体)の立体的嵩高さ及び分子量も最小限となるが、連鎖の柔軟性は維持される。
【0022】
リンカー基の性状は、造影剤の体内分布の改変にも利用できる。例えば、−(Y)m−にエーテル基を導入すると、血漿タンパク質の結合を最小限に抑えるのに役立つであろう。−(Y)m−がポリエチレングリコール(PEG)構成単位又は1〜10アミノ酸残基のペプチド鎖からなる場合、リンカー基は生体内での薬物動態及び造影剤の血液クリアランス速度を改善するように機能し得る。かかる「バイオモディファイアー」リンカー基は、筋肉や肝臓のようなバックグラウンド組織及び/又は血液からのテクネチウム造影剤のクリアランスを促進して、バックグラウンド干渉の低下による画質の向上した診断用画像を与え得る。バイオモディファイアーリンカー基は、特定の排泄経路、例えば肝臓経路よりも腎臓経路を優勢にするのにも使用し得る。或いは、バイオモディファイアーリンカー基は血中滞留時間を延ばして、生体内でのターゲット部位への薬剤の蓄積量を増すことができる。
【0023】
−(Y)m−がアミノ酸残基のペプチド鎖を含む場合、アミノ酸残基は好ましくはグリシン、リジン、アスパラギン酸、グルタミン酸又はセリンから選択される。ペプチド鎖のアミノ酸の数は好ましくは1〜10、最も好ましくは1〜3である。
【0024】
−(Y)m−がPEG部分を含む場合、好ましくは式(−OCH2CH2O−)wの基を含むもので、wは3〜25の整数である。整数wは好ましくは6〜22である。特に好ましいPEG含有−(Y)m−基は、単分散PEG様構造の式IVの17−アミノ−5−オキソ−6−アザ−3,9,12,15−テトラオキサヘプタデカン酸の重合で得られる単位である。
【0025】
【化5】
【0026】
式中、pは1〜10の整数である。
【0027】
「フルオロアルキル」という用語は、1以上のフッ素置換基を有するアルキル基を意味し、この用語はモノフルオロアルキル(例えば−CH2F)からペルフルオロアルキル(例えばCF3)までの基を包含する。
【0028】
−(Y)m−基は好ましくはジグリコール酸部分、マレイミド部分、グルタミン酸、コハク酸、ポリエチレングリコール系単位又は式IVのPEG様単位を含む。
【0029】
「合成」という用語は、その通常の意味、つまり天然資源(例えば哺乳動物の身体)から分離されたものではなく、人造のものを意味する。かかる化合物は、その製造並びに不純物特性を十分に制御できるという利点がある。従って、モノクローナル抗体及びその断片は、本願特許請求の範囲に属さない。
【0030】
「生体ターゲティング部分」という用語は、3〜100量体ペプチド若しくはペプチド類似体(これらは鎖状ペプチドでも環状ペプチドでも若しくはそれらの組合せでもよい。)、酵素の基質若しくはアンタゴニスト若しくは阻害剤、合成受容体結合性化合物、オリゴヌクレオチド、又はオリゴDNA断片若しくはオリゴRNA断片を意味する。
【0031】
「環状ペプチド」という用語は、2つの末端アミノ酸が共有結合によって連結した5〜15個のアミノ酸配列を意味し、上記共有結合はペプチド結合でも、ジスルフィド結合でも、或いはチオエーテル、ホスホジエステル、ジシロキサン又はウレタン結合のような合成非ペプチド結合でもよい。「アミノ酸」という用語は、L−若しくはD−アミノ酸、アミノ酸類似体又はアミノ酸模倣体を意味し、光学的に純粋なもの、つまり単一の鏡像異性体でキラルなものでもよいし、鏡像異性体混合物であってもよい。好ましくは、本発明のアミノ酸は光学的に純粋なものである。「アミノ酸模倣体」という用語は、天然アミノ酸の合成類似体であってアイソスターであるもの、つまり天然化合物の立体及び電子構造を模倣して設計されたものを意味する。かかるアイソスターは当業者に周知であり、特に限定されないが、デプシペプチド、レトロインベルソ型ペプチド、チオアミド、シクロアルカン又は1,5−二置換テトラゾールなどが挙げられる(M.Goodman, Biopolymers,24,137,(1985)参照)。 本発明での使用に適したペプチドとしては、以下のものが挙げられる。
・ソマトスタチン、オクトレオチド及び類似体。
・ST受容体に結合するペプチド。ここで、STとは大腸菌その他の微生物の産生する熱安定性毒素をいう。
・ラミニン断片、例えばYIGSR、PDSGR、IKVAV、LRE及びKCQAGTFALRGDPQG。
・白血球集積のターゲティング部位用のN−ホルミルペプチド。
・血小板因子4(PF4)及びその断片。
・RGD(Arg−Gly−Asp)含有ペプチド。これは例えば血管新生をターゲティングし得る(R.Pasqualini et al., Nat Biotechnol.1997 Jun;15(6):542−6;E.Ruoslahti, Kidney Int. 1997 May;51(5):1413−7)。
・α2−アンチプラスミン若しくはフィブロネクチン若しくはβ−カゼインのペプチド断片、フィブリノーゲン又はトロンボスポンジン。α2−アンチプラスミン、フィブロネクチン、β−カゼイン、フィブリノーゲン及びトロンボスポンジンのアミノ酸配列は以下の引用文献に記載されている:α2−アンチプラスミン前駆体(M.Tone et al., J. Biochem,102,1033,(1987));β−カゼイン(L.Hansson et al, Gene,139,193,(1994));フィブロネクチン(A.Gutman et al, FEBS Lett.,207,145,(1996));トロンボスポンジン−1前駆体(V.Dixit et al, Proc. Natl. Acad. Sci.,USA,83,5449,(1986));R.F.Doolittle, Ann. Rev. Biochem.,53,195,(1984)。
・アンジオテンシンII:Asp−Arg−Val−Tyr−Ile−His−Pro−Phe(E.C.Jorgensen et al, J. Med. Chem.,1979,Vol 22,9,1038−1044)、[Sar,Ile]アンジオテンシンII:Sar−Arg−Val−Tyr−Ile−His−Pro−Ile(R.K.Turker et al, Science,1972,177,1203)のようなアンジオテンシンの基質又は阻害剤であるペプチド。
・アンジオテンシンI:Asp−Arg−Val−Tyr−Ile−His−Pro−Phe−His−Leu。
【0032】
好ましくは、本発明のペプチドはアンチプラスミン又はアンジオテンシンIIペプチドを含む。アンチプラスミンペプチドは、N末端からみて以下のアミノ酸配列を含む。
(i)α2−アンチプラスミン
NH2−Asn−Gln−Glu−Gln−Val−Ser−Pro−Leu−Thr−Leu−Thr−Leu−Leu−Lys−OH、又はその変異体で1以上のアミノ酸が交換、付加又は除去されたもの、例えば、
NH2−Asn−Gln−Glu−Gln−Val−Ser−Pro−Leu−Thr−Leu−Thr−Leu−Leu−Lys−Gly−OH、
NH2−Asn−Gln−Glu−Ala−Val−Ser−Pro−Leu−Thr−Leu−Thr−Leu−Leu−Lys−Gly−OH、
NH2−Asn−Gln−Glu−Gln−Val−Gly−OHなど、或いは
(ii)カゼイン
Ac−Leu−Gly−Pro−Gly−Gln−Ser−Lys−Val−Ile−Gly。
【0033】
本発明の合成ペプチドは、P.Lloyd−Williams,F.Albericio and E.Girald; Chemical Approaches to the Synthesis of Peptides and Proteins,CRC Press,1997に記載されているように、通常の固相合成法で得ることができる。
【0034】
適当な酵素の基質、アンタゴニスト又は阻害剤としては、グルコース並びにフルオロデオキシグルコースのようなグルコース類似体、脂肪酸、又はエラスターゼ若しくはアンジオテンシンII若しくはメタロプロテアーゼ阻害剤が挙げられる。好ましい非ペプチド系アンジオテンシンIIアンタゴニストはロサルタンである。
【0035】
適当な受容体結合性化合物としては、エストラジオール、エストロゲン、プロゲスチン、プロゲストロンその他のステロイドホルモン、ドーパミンD−1若しくはD−2受容体、又はトロパンのようなドーパミン輸送体、並びにセロトニン受容体に対するリガンドが挙げられる。
【0036】
生体ターゲティング部分(Z)は好ましくは分子量5000未満のもの、最も好ましくは4000未満のもの、理想的には3000未満のものである。これは、本発明のテトラアミンテクネチウム錯体の改善された生物学的特性が、式Iのコンジュゲートのテクネチウム錯体の全体的体内分布、特にクリアランスに影響をもつという利点を有する。nが3で、Xが(CH2)n基に直接結合した窒素を含む場合、Zは合成物で分子量4000未満となるように選択される。好ましい生体ターゲティング部分は3〜20量体ペプチド又は酵素基質、酵素アンタゴニスト若しくは酵素阻害剤である。
【0037】
対イオン(A−)は、モル当量で存在し、式IのTc(V)ジオキソテクネチウム錯体の陽電荷と釣り合う陰イオンを表す。陰イオン(A)は、電荷が均衡する量で存在していれば単電荷のものでも多電荷のものでもよい。陰イオンは好適には無機酸又は有機酸から誘導される。適当な陰イオンの例としては、塩素又は臭素のようなハロゲンイオン、硫酸、硝酸、クエン酸、酢酸、リン酸及びホウ酸イオンが挙げられる。好ましい陰イオンは塩素イオンである。
【0038】
式Iのテクネチウム錯体は、錯形成後に安定であり、テクネチウムを生体ターゲティング部分に優先的に結合させる強力なキランドを含んでいるという利点を有する。そのため、このテクネチウム錯体は生体内で生体高分子又は競合配位子とのトランスキレーション反応を起こしにくい。テクネチウム錯体は小さくてコンパクトであり、生体ターゲティング部分(Z)と結合したときも立体的な影響が最小限である点で有用である。永続的な陽イオン交換とTc(V)ジオキソコアの存在は、この錯体が親水性でもあることを意味しており、そのため錯体が細胞内の他の区画に分布する可能性が低く、生体内でバックグラウンド器官及び組織、例えば血流からのクリアランスが速い。
【0039】
式Iのテクネチウム錯体は、以下の第二の実施形態で説明するように、適当なテクネチウム源と式IIのキレーターコンジュゲートとの反応によって調製できる。
【0040】
第二の実施形態では、本発明は式IIのキレーターコンジュゲートを提供する。
【0041】
【化6】
【0042】
式中、X、Y、Z、n及びmは既に定義した通りであり、
Q1〜Q6は独立にQ基であり、QはH又はアミン保護基である。
【0043】
このキレーターコンジュゲートは、第一の実施形態に係る式Iのテクネチウム錯体の調製に有用である。
【0044】
「保護基」という用語は、不都合な化学反応は阻害又は抑制するが、分子の残りの部分を修飾しない十分穏和な条件下で当該官能基から開裂できる十分な反応性をもつように設計された基を意味する。脱保護後に、所望の生成物が得られる。アミン保護基は当業者に周知であり、好適にはBoc(tert−ブチルオキシカルボニル)、Fmoc(フルオレニルメトキシカルボニル)、トリフルオロアセチル、アリルオキシカルボニル、Dde(1−(4,4−ジメチル−2,6−ジオキソシクロヘキシリデン)エチル]又はNpys(3−ニトロ−2−ピリジンスルフェニル)から選択される。場合によっては、保護基の性状は、Q1/Q2又はQ5/Q6の両者、つまり関連するアミン窒素原子に全くNH結合が存在しないようなものであってもよい。他の保護基の使用については、‘Protective Groups in Organic Synthesis’,Theorodora W.Greene and Peter G.M.Wuts, John Wiley&Sons,1991に記載されている。好ましいアミン保護基はBoc及びFmoc、最も好ましくはBocである。Bocを使用した場合、Q1及びQ6は共にHであり、Q2、Q3、Q4及びQ5は各々tert−ブトキシカルボニルである。
【0045】
式IIにおいて、アミン保護基は主にテクネチウムとの錯形成前の合成反応時にテトラアミンキレーターのアミン官能基を保護するために用いられる。しかし、生体ターゲティング基(Z)が第一及び/又は第二アミンと反応し易い場合、これらの保護基は、テクネチウムと錯形成する前のキレーターアミンとZとの不都合な反応の防止にも有用である。
【0046】
式IIの好ましいコンジュゲートは、保護されていないアミン窒素を少なくとも1つ以上有する(つまり、Q3又はQ4の一方がHであるか、或いはQ1/Q2対又はQ5/Q6対がHである)。1以上の遊離アミノ基は、式Iのテクネチウム錯体の調製の際の好ましい溶媒である水性媒体中でのコンジュゲートの溶解性が増すことを意味する。遊離アミノ基は、錯形成が保護基を予め除去しておくこと(これは金属の錯体化も阻害する)に依存しないので、テクネチウムとの錯形成が速やかになることを意味する。コンジュゲートした生体ターゲティング基(Z)がアミンとの反応は起こしにくいときは、式IIのコンジュゲートを完全な脱保護形(Q1〜Q6が各々H)で使用するのが好適であり、これが式IIの特に好ましいキレーターコンジュゲートである。式Iのテクネチウム錯体を得るための錯体化反応には完全な脱保護形が好ましい。
【0047】
本発明の式Iのテクネチウム錯体は、適切な酸化状態の放射性金属の溶液を適切なpHで式IIのキレーターコンジュゲートと反応させることによって調製できる。溶液は、テクネチウムと弱く錯化する配位子(グルコン酸イオン又はクエン酸イオンなど)を適宜含んでいてもよく、テクネチウム錯体は配位子交換又はトランスキレーションによって調製される。かかる条件はテクネチウムイオンの加水分解のような不都合な副反応を抑制するのに有用であることが多いが、本発明のキレーターはテクネチウムと迅速に錯形成するので、本発明のキレーターでは重要性は低い。放射性同位体が99mTcである場合、通常の出発原料は99Moジェネレータから得られる過テクネチウム酸ナトリウムである。テクネチウムは、比較的反応性の低いTc(VII)の酸化状態の99mTc−過テクネチウム酸塩として存在する。そのため、酸化状態の低いTc(I)〜Tc(V)のテクネチウム錯体の調製には、通常、錯体化を促すため亜ジチオン酸ナトリウム、亜硫酸水素ナトリウム、アスコルビン酸、ホルムアミジンスルフィン酸、第一スズイオン、Fe(II)又はCu(I)のような薬学的に許容される還元剤の添加が必要とされる。薬学的に許容される還元剤は好ましくは第一スズ塩であり、最も好ましくは塩化第一スズ、フッ化第一スズ又は酒石酸第一スズである。
【0048】
式IIのキレーターコンジュゲートは、以降の第五の実施形態で説明するように、生体ターゲティング分子(Z)と式IIIの二官能性キレーターとの結合によって調製できる。
【0049】
第三の実施形態では、本発明は、Aが薬学的に許容される対イオンである第一の実施形態のテクネチウム錯体を生体適合性担体と共にヒトへの投与に適した形態で含む放射性医薬品を提供する。
【0050】
「ヒトへの投与に適した形態」という用語は、無菌で、発熱物質も毒性又は副作用を生じる化合物も含まず、生体適合性のpH(pH約4.0〜10.5)で製剤化した組成物を意味する。かかる組成物は生体内で塞栓を惹起する危険性のある粒子を含まず、体液(例えば血液)と接しても沈殿が起こらないように製剤化される。かかる組成物は生体適合性の賦形剤のみを含有し、好ましくは等張性である。
【0051】
「生体適合性担体」とは、放射性医薬品を懸濁又は好ましくは溶解できる流体、特に液体であって、組成物が生理学的に認容できるもの、つまり毒性も耐え難い不快感も伴わずに哺乳類の身体に投与することができるようなものである。生体適合性担体は好適には注射可能な担体液であり、例えば、発熱物質を含まない注射用の滅菌水、食塩液のような水溶液(これは注射用の最終製剤が等張性又は非低張性となるように調整するのに都合がよい)、1種以上の張度調節物質(例えば血漿陽イオンと生体適合性対イオンとの塩)、糖(例えばグルコース又はスクロース)、糖アルコール(例えばソルビトール又はマンニトール)、グリコール(例えばグリセロール)その他の非イオン性ポリオール材料(例えばポリエチレングリコール、プロピレングリコールなど)の水溶液である。
【0052】
「薬学的に許容される対イオン」という用語は、哺乳類の身体に投与したときに生体内で毒性又は悪影響を生じずに、医薬組成物の他の成分と化学的及び/又は毒性学的に適合性の陰イオン(負電荷イオン)を意味する。本発明のテクネチウム放射性医薬品に対する化学的適合性とは、この陰イオンがテクネチウムに対してテトラアミンキレーターと事実上競合しないことを意味する。かかる適当な陰イオンとしては、ハロゲンイオン(例えば塩素、ヨウ素及び臭素イオン)、C1〜2アルキルスルホン酸イオン(例えばメシレート又はエチルスルホン酸イオン)、アリールスルホン酸イオン(例えばフェニルスルホン酸又はトシレートイオン)、C1〜2アルキルホスホン酸イオン、ジ(C1〜2)アルキルリン酸イオン(例えばジメチルリン酸、ジエチルリン酸又はジグリセロールリン酸イオン)、アリールホスホン酸イオン、アリールリン酸イオン、アルキルアリールホスホン酸イオン、アルキルアリールリン酸イオン、C1〜2アルキルカルボン酸イオン(例えば酢酸、プロピオン酸、グルタミン酸又はグリセリン酸イオン)、アリールカルボン酸イオン(例えば安息香酸イオン)などが挙げられる。好ましい薬学的に許容される対イオンは、塩素イオン、フッ素イオン、酢酸イオン、酒石酸イオン、水酸イオン及びリン酸イオンである。
【0053】
かかる放射性医薬品は、好適には、無菌状態を維持しながら皮下注射針で一回又は複数回穿刺するのに適したシール(例えばクリンプオン式セプタムシール蓋)を備えた容器に入れて供給される。かかる容器には、1回又は複数回分の用量を入れることができる。好ましい多用量用容器は、複数回分の用量を収容した単一バルクバイアル(例えば容積10〜30cm3のもの)からなり、臨床症状に応じて製剤の有効期間中様々な時間間隔で1回分の用量を臨床グレードの注射器に引き出すことができる。
プレフィルド型注射器は1回分の用量を収容するように設計され、そのため好ましくは使い捨て又はその他臨床用に適した注射器である。プレフィルド型注射器は、適宜、オペレーターを放射能から保護するため、注射器シールドを備えていてもよい。かかる適当な放射性医薬品注射器シールドは当技術分野で公知であり、好ましくは鉛又はタングステンからなる。
【0054】
本発明の好ましい放射性医薬品はテクネチウム放射性同位体99mTc又は94mTcを含み、最も好ましくは99mTcを含む。テクネチウム同位体が99mTcである場合、画像診断用放射性医薬品に適した放射能含量は、生体内の撮像部位、取込み量及び標的/バックグラウンド比に応じて、180〜1500MBqの99mTcである。
本発明のテクネチウム放射性医薬品は、以下に挙げるような様々な方法で調製できる。
(i)第二の実施形態に関して上記で説明したテクネチウム錯体形成をクリーンルーム環境下で実施する無菌製造法。
(ii)無菌製造を用いずにテクネチウム錯体形成を実施してから、最終段階で滅菌(例えばガンマ線照射又は高圧蒸気滅菌)する最終滅菌法。
(iii)式IIのキレーターと薬学的に許容される還元剤を適宜他の賦形剤と共に含む無菌の凍結乾燥非放射性キット製剤を、99mTcジェネレータから得られる無菌99mTc−過テクネチウム酸のアリコートと反応させるキット法。
【0055】
方法(iii)が好ましく、この方法に使用するためのキットを以下の第四の実施形態で説明する。
【0056】
第四の実施形態では、本発明は、上述の放射性医薬組成物を調製するための非放射性キットであって、式IIのコンジュゲートを生体適合性還元剤と共に含むキットを提供する。かかるキットは、例えば血流への直接注射によるヒトへの投与に適した無菌放射性医薬製剤を与えるように設計される。リガンドコンジュゲート及びその好ましい態様は上記の第二の実施形態で記載した通りである。
【0057】
99mTc用には、キットは好ましくは凍結乾燥したもので、99mTc放射性同位体ジェネレータからの無菌99mTc−過テクネチウム酸(TcO4−)で再構成すればそれ以上操作しなくてもヒトへの投与に適した溶液が得られるように設計される。
適当なキットは、遊離塩基又は酸塩の形態のキレーターコンジュゲートを亜ジチオン酸ナトリウム、亜硫酸水素ナトリウム、アスコルビン酸、ホルムアミジンスルフィン酸、第一スズイオン、Fe(II)又はCu(I)のような生体適合性還元剤と共に収容した容器(例えばセプタムシールバイアル)を備える。生体適合性還元剤は、好ましくは塩化第一スズや酒石酸第一スズのような第一スズ塩である。或いは、キットは非放射性金属錯体を適宜含んでいてもよく、テクネチウムの添加時にトランスメタレーション(金属交換)を起こして所望の生成物を与える。
【0058】
非放射性キットはさらに、トランスキレーター、放射線防護剤、抗菌保存剤、pH調節剤又は充填剤のような追加成分を適宜含んでいてもよい。「トランスキレーター」とは、テクネチウムと迅速に反応して弱い錯体を形成し、次に上記キレーターで置換される化合物である。これはテクネチウム錯体化と競合する過テクネチウム酸塩の迅速な還元に起因した還元型加水分解テクネチウム(RHT)が形成するおそれを最小限に抑制する。かかる適当なトランスキレーターは、弱有機酸(つまりpKaが3〜7の範囲内にある有機酸)と生体適合性陽イオンとの塩である。「生体適合性陽イオン」という用語は、イオン化して負に荷電した陰イオン基と塩を形成する正電荷を有する対イオンで、しかも無毒で哺乳類の身体、特に人体への投与に適したものをいう。適当な生体適合性陽イオンの例としては、アルカリ金属のナトリウム又はカリウム、アルカリ土類金属のカルシウム及びマグネシウム、さらにアンモニウムイオンが挙げられる。好ましい生体適合性陽イオンはナトリウム及びカリウムであり、最も好ましくはナトリウムである。適当な弱有機酸は、酢酸、クエン酸、酒石酸、グルコン酸、グルコヘプトン酸、安息香酸、フェノール又はホスホン酸である。従って、適当な塩は、酢酸塩、クエン酸塩、酒石酸塩、グルコン酸塩、グルコヘプトン酸塩、安息香酸塩、フェノラート又はホスホン酸塩である。好ましい塩は、酒石酸塩、グルコン酸塩、グルコヘプトン酸塩、安息香酸塩又はホスホン酸塩であり、最も好ましくはホスホン酸塩、特にジホスホン酸塩である。好ましいトランスキレーターは、MDP(メチレンジホスホン酸)と生体適合性陽イオンとの塩である。
【0059】
「放射線防護剤」という用語は、水の放射線分解で生成する含酸素フリーラジカルのような反応性の高いフリーラジカルを捕捉することによって、酸化還元過程のような分解反応を阻害する化合物をいう。本発明の放射線防護剤は、好適には、アスコルビン酸、パラアミノ安息香酸(即ち4−アミノ安息香酸)、ゲンチシン酸(即ち2,5−ジヒドロキシ安息香酸)並びにこれらの生体適合性陽イオンとの塩から選択される。
「抗菌保存剤」という用語は、細菌、酵母又はカビなどの有害微生物の増殖を阻害する薬剤を意味する。抗菌保存剤は、濃度に応じてある程度の殺菌作用を示すこともある。本発明の抗菌保存剤の主な役割は、再構成後の放射線医薬組成物(つまり、放射性診断薬自体)における微生物の増殖を阻害することである。ただし、抗菌保存剤は、再構成前の本発明の非放射性キットの1以上の成分における有害微生物の増殖の防止にも適宜使用できる。適当な抗菌保存剤としては、パラベン類、即ちメチルパラベン、エチルパラベン、プロピルパラベン、ブチルパラベン又はこれらの混合物、ベンジルアルコール、フェノール、クレゾール、セトリミド及びチオメルサールが挙げられる。好ましい抗菌保存剤はパラベンである。
【0060】
「pH調節剤」という用語は、再構成したキットのpHが、ヒト又は哺乳類の投与に関する許容範囲(約pH4.0〜10.5)内に収まるようにするのに有用な化合物又は化合物の混合物を意味する。かかる適当なpH調節剤としては、トリシン、リン酸塩又はTRIS(トリス(ヒドロキシメチル)アミノメタン)のような薬学的に許容される緩衝剤、並びに炭酸ナトリウム、重炭酸ナトリウム又はこれらの混合物などの薬学的に許容される塩基が挙げられる。コンジュゲートを酸塩の形態で用いる場合、キットのユーザーが多段階操作法の一部としてpHを調節できるようにpH調節剤を適宜別のバイアル又は容器で提供してもよい。
【0061】
「充填剤」という用語は、製造時及び凍結乾燥時の材料の取扱いを容易にする薬学的に許容される増量剤を意味する。適当な充填剤としては、塩化ナトリウムのような無機塩並びに水溶性糖類又は糖アルコール、例えばスクロース、マルトース、マンニトール又はトレハロースが挙げられる。
【0062】
第五の実施形態では、本発明は式IIIの化合物を提供する。
【0063】
【化7】
【0064】
式中、Q1〜Q6及びnは式I及びIIに関して定義した通りであり、
Eは第一の実施形態の生体ターゲティング部分(Z)との結合に適した官能基であるが、
(i)n=3のとき、Q1〜Q6の1以上がアミン保護基であり、
(ii)n=3又は5のとき、EはOHではないことを条件とする。
【0065】
式IIIの化合物は「二官能性キレーター」つまり1以上の官能基(E)が結合したキレーターである。官能基Eは生体ターゲティング部分(Z)との結合に適したものである。適当な官能基(E)としては、アミン、チオシアネート、マレイミド及び活性エステルが挙げられる。Eは好ましくは活性化されていないヒドロキシル(−OH)基を含まない。「活性エステル」という用語は、良好な脱離基で、生体ターゲティング部分に存在するアミンのような求核部位との反応を容易にするように設計されたカルボン酸のエステル誘導体を意味する。適当な活性エステルの例は、N−ヒドロキシスクシンイミド(NHS)、ペンタフルオロフェノール、ペンタフルオロチオフェノール、パラ−ニトロフェノール、ヒドロキシベンゾトリアゾール及びPyBOP(ベンゾトリアゾール−1−イル−オキシトリピロリジノホスホニウムヘキサフルオロホスフェート)である。好ましい活性エステルは、N−ヒドロキシスクシンイミド又はペンタフルオロフェノールエステルである。
【0066】
Eは好ましくは第一アミン(−NH2)、−CO2M、−NCS、−NCO、マレイミド又はアクリルアミドである。ただし、MはH、陽イオン、保護基又は活性エステルである。Eは最も好ましくは−NH2、−CO2M又はマレイミドであり、理想的には−NH2又は−CO2Mである。
【0067】
式IIIの化合物は、生体ターゲティング分子(Z)の適当な対応官能基と反応して式IIの所望のコンジュゲートを形成する。生体ターゲティング分子(Z)の適当な官能基としては、(アミン官能化二官能性キレーターとのアミド結合形成のための)カルボキシル、(カルボキシル又は活性エステル官能化二官能性キレーターとのアミド結合形成のための)アミン、(アミン官能化二官能性キレーターのNアルキル化のための)ハロゲン、メシレート及びトシレート、並びに(マレイミド官能化二官能性キレーターとの反応のための)チオールが挙げられる。
【0068】
Eが生体ターゲティング分子(Z)のアミノ基と反応するように設計された基(例えば活性エステル)である場合、キレーターのアミンとの不都合な副反応のおそれがあることは明らかである。かかるE基については、テトラアミンキレーターの4つのアミン窒素原子の各々が保護されるように、式IIIのQ1〜Q6は好ましくは窒素保護基として選択される。Eがアミノ基である場合、生体ターゲティング分子(Z)との反応がEアミンでしか起こらず、テトラアミンキレーターのアミン窒素原子では起こらないことが明らかに重要である。従って、この状況下でも、式IIIのQ1〜Q6は好ましくは窒素保護基である。窒素保護基及びその好ましい具体例は上記の第二の実施形態で記載した通りである。
【0069】
式IIIの化合物は以下のスキーム1及び2に記載したように調製できる。スキーム1はカルボキシ官能化N保護テトラアミンキレーターの順応性に富む合成経路を与え、式IIIのnの様々な値に適応させることができる。−(CH2)5OH橋頭置換基をもつBoc保護テトラアミン類似体の合成は、Turpin et al, J. Lab. Comp. Radiopharm.,45,379−393(2002)に記載されている。スキーム2はアミン官能化N保護テトラアミンキレーターの順応性に富む合成経路を与え、nの様々な値に適応させることができる。生体ターゲティングペプチドの結合は、Nock et al, Eur. J. Nucl. Med.,30(2),247−258(2003)及びMaina et al, Eur. J. Nucl. Med.,30(9),1211−1219(2003)に記載のものと同様に実施できる。
【0070】
【化8】
【0071】
【化9】
【実施例】
【0072】
本発明を以下の非限定的な実施例で例証する。実施例1では、本発明のカルボキシ官能化N保護テトラアミンキレーター(化合物1)の合成について記載する。実施例2では、本発明のアミン官能化N保護テトラアミンキレーター(化合物2)の合成について記載する。実施例3では、化合物1とアミン(ベンジルアミン)との結合を示す化合物(化合物3)の合成について記載する。実施例4では、化合物6の合成について記載し、化合物2とカルボン酸の活性エステルとの結合を例証する。実施例5では、本発明のキレーターとロサルタン誘導体とのコンジュゲート(化合物4)の合成について記載する。実施例6では、PEGリンカー基を含むロサルタンキレーターコンジュゲートの合成について記載する。実施例7では、本発明のキレーターのアンジオテンシンペプチドコンジュゲート(化合物8)の合成について説明する。実施例8では、本発明の幾つかのキレーターの99mTc放射標識化について記載する。実施例9では、本発明の様々な99mTc錯体の親油性(logP)を測定し、これらの錯体が比較的親水性であることを示す。実施例10では、低い肝臓バックグラウンドと高い尿クリアランスを示す本発明の幾つかの99mTc錯体の生体内分布結果を示す。実施例11では、化合物1の高収率の合成について記載する。実施例12では、活性エステルの結合した本発明の保護テトラアミンキレーター(化合物9)の合成について記載する。
【0073】
実施例1:化合物1の合成
段階(a):ジエチル[2−(ベンジルオキシ)エチル]マロネート
標記化合物は、Ramalingam et al, Tetrahedron,51,2875−2894(1995)の方法の修正法で調製した。ナトリウム(1.20g)をアルゴン雰囲気中で無水エタノール(25mL)に溶解した。マロン酸ジエチル(14.00g)を加えて混合物を30分間還流した。ベンジルブロモエチルエーテル(10g)を加えて、混合物を16時間還流下で撹拌した。エタノールをロータリーエバポレーターで除去して、残渣をエーテル(100mL)と水(50mL)とに分配した。エーテル層を水(3×50mL)で洗浄して硫酸ナトリウムで乾燥した。エバポレーターでエーテルを除去して、残渣を真空蒸留した。40〜55℃で留出した留分を廃棄した(未反応マロン酸ジエチル)。生成物は140〜150℃(1mm)で留出した(文献値bp138〜140℃(1mm))。収量は無色油12.60gであった。
【0074】
1H NMR(270MHz,CDCl3,25℃,TMS):δ=7.28(m,5H,C6H5),4.47(s,2H,CH2−Ph),4.16(m,4H,COOCH2),3.58(t,1H,CH),3.50(t,2H,O−CH2−CH2),2.21(t,2H,O−CH2−CH2),1.20(t,6H,COOCH2−CH3)。
【0075】
13C NMR(67.5MHz,CDCl3,25℃,TMS):δ=169.20(CO),138.10,128.60,127.80(芳香族),73.00(CH2Ph),67.30(O−CH2−CH2),61.70(COOCH2),49.10(CH),28.90(O−CH2−CH2),14.10(COOCH2CH3)。
【0076】
段階(b):N,N′−ビス(2−アミノエチル)−2−(2−ベンジルオキシ−エチル)マロンアミド
ジエチル[2−(ベンジルオキシ)エチル]マロネート(4.00g)をエチレンジアミン(30mL)に加えて、溶液を室温で2日間撹拌した。過剰のエチレンジアミンをロータリーエバポレーターで除去して、残渣を高真空下で2日間乾燥して黄色油状物(4.28g)を得、これを放置して結晶化させた。生成物は痕跡量のエチレンジアミンを依然として含んでいることがNMRスペクトルで検出された。
【0077】
1H NMR(270MHz,CDCl3,25℃,TMS):δ=7.74(br t,2H,CO−NH),7.32(m,5H,C6H5),4.46(s,2H,CH2−Ph),3.50(t,2H,OCH2−CH2− ),3.33(t 1H,CH),3.23(m,4H,CO−NH−CH2),2.74(t,4H,CH2−NH2)2.18(q,2H,O−CH2−CH2−)1.55(br s 4H,NH2)。
【0078】
13C NMR(67.5MHz,CDCl3,25℃,TMS):δ=171.10(CO),138.20,128.30,127.70(芳香族),73.00(CH2−Ph),67.80(O−CH2−CH2),51.40(CH),42.40(CO−NH−CH2),41.20(CH2−NH2),31.90(O−CH2−CH2−)。
【0079】
段階(c):N,N′−ビス(2−アミノエチル)−2−(2−ベンジルオキシエチル)−1,3−ジアミノプロパン
N,N′−ビス−(2−アミノエチル)−2−(2−ベンジルオキシエチル)マロンアミド(3.80g)をTHF(20mL)に溶解して、フラスコを氷浴に浸漬した。フラスコをアルゴンでフラッシュして、THFボラン錯体(80mL、THF中1M)をシリンジで加えた。反応混合物を室温まで放温し、次いで40℃で2日間撹拌し、1時間還流した。メタノール(50mL)を滴下し、溶液を40℃で1晩撹拌した。溶媒をロータリーエバポレーターで除去して、残渣をメタノール(20mL)に溶解した。水酸化ナトリウム(15mLの水中に10g)を加えて、メタノールを留去した。無色油状物が分離し、これをCH2Cl2(3×50mL)で抽出した。溶液をNa2SO4で乾燥した。溶媒の除去で3.40gの無色油状物が得られた。
【0080】
1H NMR(270MHz,CDCl3,25℃,TMS):δ=7.34(m,5H,C6H5),4.49(s,2H,CH2−Ph),3.55(t,2H,OCH2−CH2−),2.76(t,4H,N−CH2),2.63(m,8H,N−CH2),1.84(m,1H,CH),1.58(m,2H,CH−CH2−CH2−O),1.41(br s,6H,NH)。
【0081】
13C NMR(67.5MHz,CDCl3,25℃,TMS):δ=138.60,128.30,127.60(芳香族),72.80(CH2−Ph),68.70(O−CH2−CH2),53.50(N−CH2),52.80(N−CH2),41.60(N−CH2)36.40(CH),31.30(CH−CH2−CH2−O)。
【0082】
MS−EI:295[M+H]+(計算値295.2)。
【0083】
段階(d):N,N′−ビス(2−tert−ブトキシカルボニルアミノエチル)−2−(2−ベンジルオキシエチル)−1,3−ジ(tert−ブトキシカルボニルアミノ)プロパン
N,N′−ビス(2−アミニエチル)−2−(2−ベンジルオキシエチル)−1,3−ジアミノプロパン(3.30g)をCH2Cl2(100mL)に溶解して、トリエチルアミン(5.40g)及びtert−ブチルジカーボネート(10.30g)を加えた。反応混合物を室温で2日間撹拌した。混合物を水(100mL)、クエン酸溶液(水中10%100mL)及び水(2×100mL)で洗浄した。有機層をNa2SO4で乾燥して、溶媒をロータリーエバポレーターで除去すると、黄色油状物が得られ、これを高真空で恒量になるまで乾燥した。粗生成物(7.70g)をシリカゲルカラム(250g、230〜400メッシュ、CH2Cl2、CH2Cl2−Et2O1:1)で精製して、6.10g(78.3%)の透明油状物を得た。
【0084】
1H NMR(270MHz,CDCl3,25℃,TMS):δ=7.32(m,5H,C6H5),5.12(br d,2H,NH),4.47(s,2H,CH2−Ph),3.49(t,2H,OCH2−CH2−),3.24(br,12H,N−CH2),2.14(br,1H,CH),1.59(m,2H,CH−CH2−CH2−O)1.45(s,18H,t−Bu),1.42(s,18H,t−Bu)。
【0085】
13C NMR(67.5MHz,CDCl3,25℃,TMS):δ=155.90(NH−CO),138.20,128.30 127.60,127.50(芳香族),79.90,78.90(CMe3),72.80(CH2−Ph),68.00(O−CH2−CH2),50.00(br,N−CH2),46.90(br,N−CH2),39.20(N−CH2),34.40(br,CH),29.80(CH−CH2−CH2−O),28.30(t−Bu)。
【0086】
MS−EI:695[M+H]+(計算値695.5)
段階(e):N,N′−ビス(2−tert−ブトキシカルボニルアミノエチル)−2−(2−ヒドロキシエチル)−1,3−ジ(tert−ブトキシカルボニルアミノ)プロパン
N,N′−ビス(2−tert−ブトキシカルボニルアミノエチル)−2−(2−ベンジルオキシエチル)−1,3−ジ(tert−ブトキシカルボニルアミノ)プロパン(3.16g)を無水エタノール(100mL)に溶解して、Pd−活性炭(1.00g、乾燥、10%)を加えた。混合物をParr水素添加装置中35psiで2日間水素添加した。触媒を濾別し、エタノールで洗浄した(3×20mL)。エタノールをロータリーエバポレーターで除去すると無色油状物が得られ、これを高真空で恒量になるまで乾燥した(2.67g、97.1%)。
【0087】
1H NMR(270MHz,CDCl3,25℃,TMS):δ=5.25(br d,2H,NH),3.69(t,2H,OCH2−CH2−),3.28(br,12H,N−CH2),2.71(br,OH),2.23(br,1H,CH),1.56(ショルダー,m,2H,CH−CH2−CH2−O)1.48(s,18H,t−Bu),1.44(s,18H,t−Bu)。
【0088】
13C NMR(67.5MHz,CDCl3,25℃,TMS):δ=156.10(NHCO),80.00,79.20(CMe3),59.60(O−CH2−CH2),49.90(br,N−CH2),47.00(br,N−CH2),39.34(N−CH2),33.80(CH),32.30(CH−CH2−CH2−O),28.30(t−Bu)。
【0089】
MS−EI:605[M+H]+(計算値605.4)。
【0090】
段階(f):N,N′−ビス(2−tert−ブトキシカルボニルアミノエチル)−2−(2−カルボキシメチル)−1,3−ジ(tert−ブトキシカルボニルアミノ)プロパン(化合物1)
Mazitschek et al(Ang.Chem.INT.Ed.,41,4059−4061(2002))の方法を用いた。N,N′−ビス(2−tert−ブトキシカルボニルアミノエチル)−2−(2−ヒドロキシエチル)−1,3−ジ(tert−ブトキシカルボニルアミノ)プロパン(2.60g)をDMSO(15mL)に溶解して、1−ヒドロキシ−1,2−ベンズヨードオキソール−3(H)−オン−オキシド(IBX、3.50g)を加えた。混合物を室温で1時間撹拌してから、N−ヒドロキシスクシンイミド(2.50g)を加えた。反応混合物を室温で2日間撹拌した。水酸化ナトリウム溶液(2M、40mL)を加えて、混合物を室温で4時間撹拌した。溶液を氷浴に浸漬して2M塩酸でpH2に酸性化した。水層をエーテルで抽出して(4×100mL)、一緒にしたエーテル抽出液を水で洗浄した(3×50mL)。エーテル層をNa2SO4で乾燥して、溶媒をロータリーエバポレーターで除去すると生成物及び2−ヨードソ安息香酸を含む黄色固体残渣が得られた。クロロホルム−ヘキサン(1:3)(80mL)での結晶化によってヨードソ安息香酸(2.1g)の大部分を除去した。クロロホルム−ヘキサン母液を蒸発させると黄色油状物(3g)が得られ、これをシリカゲルカラム(300g、CH2Cl2−Et2O、1:1)にかけた。残留ヨードソ安息香酸はエーテルで溶出した。生成物はエーテル−メタノール(9:1)で溶出した。生成物を含む画分を一緒にし、溶媒を除去すると1.5gの淡黄色油状物が得られた。これを再びシリカゲルカラムでクロマトグラフィー処理した(50g、Et2O)。生成物はエーテル−酢酸(95:5)で溶出した。生成物を含む画分を一緒にして溶媒をロータリーエバポレーターで除去すると、油状物が得られ、これを高真空で乾燥した。収量は1.10g(41.3%)であった。
【0091】
1H NMR(270MHz,CDCl3,25℃,TMS):δ=7.61(br s,1H,COOH),5.19(br d,2H,NH),3.22(br,12H,N−CH2),2.47(br m,1H,CH),2.26(br,2H,CH−CH2−COOH),1.41(s,18H,t−Bu),1.37(s,18H,t−Bu)。
【0092】
13C NMR(67.5MHz,CDCl3,25℃,TMS):δ=175.90(COOH),156.10(NHCO),80.40,79.10(CMe3),49.50(N−CH2),46.80(N−CH2),39.00(N−CH2),34.70(CH−CH2−COOH),34.20(CH−CH2−COOH),28.30,28.20(t−Bu)。
【0093】
MS−EI:619[M+H]+(計算値619.4)。
【0094】
実施例2:(8−アミノ−2−{[tert−ブトキシカルボニル−(2−tert−カルボニルアミノエチル)−アミノ]−メチル}−オクチル)−(2−tert−ブチルオキシカルボニルアミノエチル)−カルバミン酸tert−ブチルエステル(化合物2)の合成
段階(a):2−(6−クロロ−ヘキシルオキシ)テトラヒドロピラン
6−クロロヘキサノール(6.85g、10mmol)及びp−トルエンスルホン酸(500mg)を乾燥エーテル(75ml)に溶解して氷浴で0〜5℃に冷却した。乾燥エーテル(25ml)中のジヒドロピラン(4.3g、10mmol)を30分かけて定常攪拌下に滴下した。添加完了後、冷却浴を除いて撹拌を16時間継続した。溶液を水で抽出し(50ml×2)、乾燥し(MgSO4)、濾過して、溶媒を減圧蒸発させると、淡黄色油状物が残った。油状物はそれ以上精製せずに次の反応に使用できる十分な純度のものであることが13CNMR分光法で判明した。収量10.1g(91%)。
【0095】
13C NMR(CDCl3):δ19.7(CH2),25.5(CH2),25.6(CH2),26.7(CH2),29.6(CH2),30.8(CH2),32.6(CH2),45.0(CH2Cl),62.3(OCH2),67.4(OCH2),98.8(OCHO)。
【0096】
1H NMR(CDCl3):δ1.30〜1.71(14H,m,CH2×7),3.24〜3.32(1H,3.41〜3.48(3H,m,CH及びCH2),3.60〜3.67(1H,m,CH),3.72〜3.82(1H,bm,CH),4.44〜4.49(1H,bm,OCHO)。
【0097】
段階(b):2−[6−(テトラヒドロピラン−2−イルオキシ)−ヘキシル]−マロン酸ジエチルエステル
少量のナトリウム(1.13g、49mmol)を乾燥窒素ブランケット中定常攪拌下に乾燥エタノール(100ml)に溶解した。マロン酸ジエチル(8.0g、50mmol)を一度に加え、溶液を60℃で1時間加熱した。2−(6−クロロ−ヘキシルオキシ)−テトラヒドロピラン(9.3g、42.2mmol)を一度に加えて温度を75〜80℃に上げて24時間このレベルを保った。冷却後、無機固体を濾別して濾液から溶媒を蒸発させた。残渣をCH2Cl2(50ml)に溶解し、水で抽出し(30ml×2)、乾燥し(MgSO4)、濾過して、揮発分を除去すると淡黄色油状物が残った。油状物をシリカゲルクロマトグラフィーにかけ、溶出液として石油エーテル(40〜60℃)/エーテル(200:40)を用いた。所要生成物はrf=0.15で無色油状物として溶出した。収量8.7g(60%)。
【0098】
13C NMR(CDCl3):δ14.0(CH3×2)、19.6(CH2)、25.5(CH2)、27.2(CH2)、28.6(CH2)、29.0(CH2)、29.6((CH2)、30.0(CH2)、30.8(CH2)、52.0(CH)、61.2(OCH2×2)、62.2(OCH2)、67.4(OCH2)、98.8(OCHO)、169.4(C=O×2)。
【0099】
1H NMR(CDCl3):δ1.10〜1.25(14H,m,CH3×2,CH2×4)、1.36〜1.50(6H,bm,CH2×3)、1.70〜1.81(2H,bm,CH2)、3.17〜3.28(2H,m,CH2)、3.56〜3.66(1H,m,CH)、3.70〜3.80(1H,m,OCH)、4.04〜4.16(4H,m,OCH2×2)、4.03〜4.08(1H,m,OCHO)。
【0100】
段階(c):N,N′−ビス−(2−アミノエチル)−2−[6−(テトラヒドロピラン−2−イルオキシ)ヘキシル−]−マロンアミド
2−[6−(テトラヒドロピラン−2−イルオキシ)−ヘキシル]−マロン酸ジエチルエステル(5.1g、14.8 mmol)を1,2−ジアミノエタン(10g、167mmol)に溶解して、室温で16時間撹拌した。揮発分を真空で除去すると(40〜50℃、0.01mmHg)、淡緑色の粘稠な残渣が残り、これをカラムクロマトグラフィーにかけてCH2Cl2/MeOH/NH4OH(50:50:5)で溶出した。標記化合物はrf0.2で溶出し、淡緑色の粘稠な油状物として回収され、放置すると固化した。(収量3.9g、71%)。
【0101】
13C NMR(CDCl3):δ19.8(CH2),25.5(CH2),26.0(CH2),27.5(CH2),29.2(CH2),29.7(CH2),30.8(CH2),31.9(CH2),41.0(NCH2×2),41.9(NCH2×2),54.6(CH),62.5(OCH2),67.5(OCH2),98.9(OCHO),171.6(C=O×2)。
【0102】
1H NMR(CDCl3):δ1.15〜1.28(6H,bs,CH2×3),1.39〜1.44(6H,bm,CH2×3),1.69〜1.74(4H,bm,CH2×2),2.64(4H,bs,NH2×2),2.73 4H,t,J=6Hz,CH2×2),3.08〜3.29(6H,m,CH2×3),3.35〜3.41(1H,m CH),3.55〜3.63(1H,m,CH),3.70〜3.78(1H,m,CH),4.43(1H,bt,J=4Hz,OCHO),7.78(2H,bt,J=5Hz,OCNH×2)。
【0103】
IR(薄膜)cm−1:3417,3082,2936,2862,1663,1558,1439,1354,1323,1261,1200,1189,1076,1026,956,907,867,810。
【0104】
段階(d):N,N′−ビス(2−アミノエチル)−2−(6−ヒドロキシヘキシル)−マロンアミド
N,N′−ビス(2−アミノエチル)−2−[6−(テトラヒドロピラン−2−イルオキシ)−ヘキシル]−マロンアミド(3.9g、10.6mmol)、p−トルエンスルホン酸一水和物(8.5g、3mmol)及びエタノール(50ml)を70〜75℃で還流下で16時間加熱した。冷却後、濃水酸化アンモニウム(.880)を、pH9が持続して得られるまで滴下した。沈殿した白色固体をセライトで除去し、濾過ケークをエタノール(30ml)で洗浄した。エタノールを減圧で(15mmHg、40℃)除去すると、半固形状ワックスが残った。残渣をシリカゲルクロマトグラフィーにかけてCH2Cl2/MeOH/NH4OH(100:50:10)で溶出した。標記化合物のrf=0.2であった。生成物を回収して、残存する水を除去するためエタノールと共沸させた(100ml×3)。淡緑色の粘稠な残渣が得られ、放置すると固化した。(収量2.1g、69%)。
【0105】
13C NMR(CD3OD):δ25.4(CH2),27.3(CH2),28.9(CH2),30.4(CH2),32.2(CH2),40.6(NCH2×2),41.7(NCH2×2),54.1(CH),61.6(CH2OH),171.7(C=O×2)。
【0106】
1H NMR(CD3OD):δ1.28〜1.38(6H,bs,CH2×3),1.46〜1.55(2H,bm,CH2),1.79〜1.87(2H,bm,CH2),2.73(4H,t,J=6Hz,H2NCH2×2),3.13(1H,t,J=7Hz,CH),3.27(4H,dt,J=6及び2Hz,HNCH2×2),3.53(2H,t,J=7Hz,OCH2)。
【0107】
IR(薄膜)cm−1:3364,2932,2862,2527,1663,1558,1462,1327,1223,1192,1034。
【0108】
質量分析(FAB)m/e:C13H29N4O3(M+H)計算値289,実測値289。
【0109】
段階(e):(2−tert−ブトキシカルボニルアミノエチル−2−{[tert−ブトキシカルボニル−(2−tert−ブトキシカルボニルアミノエチル)−アミノ]−メチル}−8−ヒドロキシオクチル)−カルボン酸tert−ブチルエステル
乾燥窒素ブランケット中、無溶媒ボラン−ジメチルスルフィド付加物(15ml、150mmol)をジオキサン(50ml)中N,N′−ビス−(2−アミノエチル)−2−(6−ヒドロキシエチル)マロンアミド(2.1g、7.3mmol)の攪拌混合物にシリンジで滴下した。添加完了後、混合物を110℃で穏やかに還流しながら5日間加熱した。その際若干の白色固体が残った。冷却後揮発分を減圧除去すると白色固体が残り、これにメタノール(50ml)を滴下すると無色溶液が得られた。溶液を還流下で3時間加熱し、冷却し、濃HCl(5ml)を加えて70〜75℃で還流下で48時間加熱を続けた。溶媒を除去すると粘稠な緑色残渣が残り、これをメタノールで共沸すると(100ml×3)、淡緑色固体が残った。この固体を乾燥メタノールに再溶解して無水炭酸カルシウム(4.0g、30mmol)、次いでジ−tert−ブチルジカーボネート(7.0g、32mmol)を加えた。混合物を室温で48時間撹拌した。無機固体をセライトで濾別し、濾液から溶媒を蒸発させると粘稠な残渣が残った。残渣を水(50ml)と混合し、CH2Cl2で抽出した(50ml×3)。有機画分を一緒にし、乾燥し(MgSO4)、濾過して、溶媒を蒸発させると淡黄色残渣が残った。
【0110】
注:この時点で13CNMRで反応をモニターすると便利である。
【0111】
残渣をシリカゲルクロマトグラフィーにかけ、CH2CL2/MeOH(95:5)を溶出液として用いた。標記化合物はrf=0.41で溶出し、無色の粘稠油状物として分離された(収量2.5g、52%)。
【0112】
13C NMR(CDCl3):δ25.6(CH2),26.4(CH2),28.4(CH3×12),29.8(CH2×2),32.6(CH2),36.5(非常にブロード,CH),39.2(NCH2×2,隣接CH),46.9(ブロード,s,HNCH2×2),50.0(ブロード,s,NCH2×2),62.4(HOCH2),79.0(OC×2),79.9(OC×2),156.4(ブロード,s,C=O×4)
1H NMR(CDCl3):δ1.05〜1.18(8H,bs,CH2×4),1.27(18h,s,CH3×6,t−ブチル),1.31(18H,s,CH3×6,t−ブチル),1.41(2H,m,CH2),1.81(1H,bs,CH),2.63(1H,bs,OH),2.98(4H,bs,NCH2×2),3.11(8H,bs,NCH2×4),3.44(2H,t,J=8Hz,CH2O),5.2(2H,bs,NH×2)。
【0113】
IR(薄膜)cm−1:3350,2976,2931,2859,1674,1516,1455,1418,1393,1260,1250,1165,1069,965,871,775。
【0114】
質量分析(FAB)m/e:C33H65N4O9(M+H)計算値661,実測値661。
【0115】
段階(f):トルエン−4−スルホン酸8−[tert−ブトキシカルボニル−(2−tert−ブトキシカルボニルアミノエチル)−アミノ]−7−{[tert−ブトキシカルボニル−(2−tert−ブトキシカルボニルアミノエチル)−アミノ]−メチル}−オクチルエステル
(2−tert−ブトキシカルボニルアミノエチル−2−{[tert−ブトキシカルボニル−(2−tert−ブトキシカルボニルアミノエチル)アミノ]−メチル}−8−ヒドロキシオクチル)−カルボン酸tert−ブチルエステル(2.52g、3.82mmol)、p−トルエンスルホニルクロリド(1.0g、5.2mmol)、トリエチルアミン(1.3g、12.8mmol)及びCH2Cl2(30ml)を室温で撹拌して、溶媒をゆっくりと蒸発させた。反応を炭素NMRでモニターすると3日後に出発物質は少ししか残存していなかった。反応液容積をCH2Cl2で30mlに一緒にし、水で抽出して(50ml×3)、乾燥し(MgSO4)、濾過して溶媒を蒸発させると、褐色残渣が残った。残渣をシリカゲルクロマトグラフィーにかけ、CH2Cl2/MeOH(100:5)を溶出液として用いた。溶出した最初の化合物はrf=0.95の未反応塩化トシルであった。標記化合物はrf=0.2で溶出し、淡黄色の粘稠な液体として分離された。収量(1.20g、39%)。
【0116】
13C NMR(CDCl3):δ21.7(CH3トシル),25.3(CH2),26.3(CH2),28.5(CH3×12),28.8(CH2),29.5(CH2),29.9(CH2),36.5(CH,非常にブロード),39.4(NCH2×2),47.0(ブロード NCH2×2),50.5(ブロード,NCH2×2),70.6(TsOCH2),79.1(OC×2),80.0(OC×2),127.9(CH×2),129.9(CH×2),133.2(C),144.7(C−S Ts),156.1(ブロード,C=O×4)。
【0117】
1H NMR(CDCl3):δ1.16(8H,bs,CH2×4),1.35(18H,s,CH3×6),1.39(18H,s,CH3×6),1.88(1H,bs,CH),2.38(3H,s,CH3トシル),3.10〜3.12(4H,bs,NCH2×2),3.19(8H,bs,NCH2×4),3.93(2H,t,J=7Hz,CH2OTs),5.0(1H,bs,NH),5.08(1H,bs,NH),7.29(2H,d,J=8Hz,CH×2,Ar),7.72(2H,d,J=8Hz,CH×2,Ar)。
【0118】
IR(薄膜)cm−1:3360,2974,2932,2862,1693,1516,1479,1418,1391,1366,1250,1177,1069,959,816,775。
【0119】
質量分析(FAB)m/e:C40H71N4O11S(M+H)計算値815,実測値815。
【0120】
段階(g):(8−アジド−2−{[tert−ブトキシカルボニル−(2−tert−カルボニルアミノエチル)−アミノ]−メチル}−オクチル)−(2−tert−ブチルオキシカルボニルアミノエチル)−カルバミン酸tert−ブチルエステル
トルエン−4−スルホン酸8−[tert−ブトキシカルボニル−(2−tert−ブトキシカルボニルアミノエチル)アミノ]−7−{[tert−ブトキシカルボニル−(2−tert−ブトキシカルボニルアミノエチル)アミノ]メチル}−オクチルエステル(1.105g、1.36mmol)、ナトリウムアジド(350mg、5.4mmol)及びメタノール(10ml)を70〜75℃で16時間加熱還流した。冷却後、室温で約1〜2ml残るまでメタノールを減圧除去した。残渣を水(25ml)で希釈してCH2Cl2(25ml×4)で抽出した。有機抽出液を一緒にして乾燥し(MgSO4)、濾過して揮発分を室温で蒸発させると(注:アジドは爆発の危険性があるので、この段階は安全シールドの後ろで行うべきである)、淡黄色の粘稠な残渣が残ったが、これが純粋な所望化合物であった(収量820mg、88%)。
【0121】
13C NMR(CDCl3):δ26.3(CH2),26.5(CH2),28.3(CH3×12),28.7(CH2),29.6(CH2),29.8(CH2),36.8(ブロード,CH),39.3(NCH2×2),46.9(ブロード,NCH2×2),50.0(ブロード,NCH2×2),51.3(CH2N3),79.0(OC×2),79.8(OC×2),156.0(C=O×4)。
【0122】
1H NMR(CDCl3):δ1.16(8H,bs,CH2×4),1.29(18H,s,CH3×6),1.33(18H,s,CH3×6),1.47(2H,bt,J=6.5Hz,CH2隣接CH),1.86(1H,bs,CH),2.95〜3.05(4H,bs,NCH2×2),3.05〜3.20(10H,bs,NCH2×4及びCH2N3),5.09(2H,bs,NH×2)。
【0123】
IR(薄膜)cm−1:3350,2974,2932,2860,2097(Strong band N3),1694,1520,1470,1418,1391,1366,1250,1167,1069,870,777。
【0124】
段階(h):(8−アミノ−2−{[tert−ブトキシカルボニル−(2−tert−カルボニルアミノエチル)−アミノ]−メチル}−オクチル)−(2−tert−ブチルオキシカルボニルアミノエチル)−カルバミン酸tert−ブチルエステル(化合物2)
(8−アジド−2−{[tert−ブトキシカルボニル−(2−tert−カルボニルアミノエチル)−アミノ]−メチル}−オクチル)−(2−tert−ブチルオキシカルボニルアミノエチル)−カルバミン酸tert−ブチルエステル(820mg、1.20mmol)、10%パラジウム−活性炭(100mg)及びメタノール(10ml)を室温30気圧の圧力下、水素ガスで16時間処理した。セライトで濾過して固体を除去し、濾過ケークをメタノール(50ml)で洗浄した。濾液から揮発分を除去すると粘稠油状物が残ったが、これが純粋な所望化合物であった(収量700mg、89%)。
【0125】
13C NMR(CDCl3):δ26.4(CH2),26.6(CH2),28.4(CH3×12),32.9(CH2×2),36.8(ブロード,CH). 39.2(NCH2×2),41.8(H2NCH2),46.9(ブロード,NCH2×2),49.8(ブロード,NCH2×2),78.9(OC×2),79.7(OC×2),156.0(C=O×4)。
【0126】
1H NMR(CDCl3):δ1.08(8H,bs,CH2×4),1.23(18H,s,CH3×6),1.27(20H,bs,CH3×6及びCH2),1.77(1H,bs,CH),2.40(2H,bs,NH2),2.50(2H,t,J=7Hz,CH2NH2),2.97(4H,bm,NCH2×2),3.00〜3.16(8H,bm,NCH2×4),5.21(1H,bs,NH),5.30(1H,bs,NH)。
【0127】
IR(薄膜)cm−1:3360,1693,1520,1459,1418,1392,1367,1250,1170,1068,964,922,871,775,733。
【0128】
質量分析(FAB)m/e:C33H66N5O8(M+H)計算値660,実測値660。
【0129】
実施例3:化合物3の合成
段階(a):化合物1とベンジルアミンとのカップリング
20℃の丸底フラスコ(50ml)内で、CH2Cl2(5ml)中の化合物1(61.8mg、0.1mmol)をベンジルアミン(10.7mmol)、ジフェニルホスフィン酸クロリド(25.9mg)及びジイソプロピルアミン(29mg、0.22mmol)で18時間処理した。次に反応液をCH2Cl2(20ml)で希釈し、1N塩酸(5ml)及び飽和炭酸カリウム水溶液(5ml)で洗浄した。CH2Cl2層を分離乾燥し(Na2SO4)、ガム状になるまで減圧濃縮した(〜50mg)。次にこの粗生成物を石油中の酢酸エチルの勾配(50%、70%及び90%各100ml)でシリカクロマトグラフィーにかけた。少量の移動度の速い不純物と、そのすぐ後で主画分を回収した。
【0130】
CDCl3中で1H及び13C NMRスペクトルを測定した。その結果、主画分が所要純粋化合物であることが判明した。
【0131】
段階(b):Boc保護基の脱保護
段階(a)で得られたCH2Cl2(0.5ml)中の生成物(27.8mg、0.039mmol)をトリフルオロ酢酸(0.5ml)で処理して、反応液を室温で3時間放置した。次いで反応混合物をガム状になるまで減圧濃縮して過剰の酸を除去し、秤量した(53mg)。1H及び13C NMR(CDCl3)からBoc基が完全に除去されたことが判明した。化合物2の秤量試料を水に溶解してTFA塩の10ミリモル濃度溶液を得て、これを放射標識実験に用いた。
【0132】
実施例4:化合物6の合成
段階(a):(8−ベンゾイルアミノ−2−{[tert−ブトキシカルボニル−(2−tert−カルボニルアミノエチル)−アミノ]−メチル}−オクチル)−(2−tert−ブチルオキシカルボニルアミノエチル)−カルバミン酸tert−ブチルエステル
乾燥CH2Cl2中の安息香酸2,5−ジオキソ−ピロリジン−1−イルエステル(20mg、0.091mmol)を、CH2Cl2(1ml)中の化合物2(50mg、0.08mmol)の溶液に一度に加え、溶液を室温で16時間撹拌した。反応液をCH2Cl2(10ml)で希釈し、水(15ml×2)で抽出し、乾燥(MgSO4)し、濾過し、溶媒をロータリーエバポラーターで除去した。残った残渣をCH2Cl2/MeOH94:6(rf=0.23)を溶出液として用いるシリカゲルクロマトグラフィーで精製し、無色の粘稠油状物を得た。(収量25mg、41%)。
【0133】
13C NMR(CDCl3):δ26.4(CH2),26.8(CH2),28.5(CH3×12),29.6(CH2×2),29.7(CH2),29.9(CH2),36.6(ブロード,CH),39.4(NCH2×2),40.0(O=CNCH2×2),47.0(ブロード,NCH2×2),49.8(NCH2×2),79.7(OC×2),80.0(OC×2),127.0(ArCH×2),128.5(ArCH×2),131.3 (ArCH),134.9(ArC),156.1(C=O×4),167.6(ArC=O)。
【0134】
1H NMR(CDCl3):δ1.28(8H,bs,CH2×4),1.38(18H,s,CH3×6),1.42(20H,bs,CH3×6及びCH2),1.95(1H,bs,CH),3.1(4H,bs,NCH2×2)3.22(8H,bs,NCH2×4),3.42(2H,bq,J=6Hz,CH2N−ベンゾイル),5.08(2H,bs,NH×2),6.18(1H,bs,HN−ベンゾイル),7.38〜7.45(3H,m,ArCH×3),7.74(2H,bd,J=7Hz,ArCH×2),
IR(薄膜)cm−1:3350,2976,2932,2859,1693(ブロード),1652,1520,1419,1391,1367,1251,1166,732。
【0135】
質量分析(FAB)m/e:C40H70N509(M+H)の計算値764,実測値764。
【0136】
段階(b):Boc保護基の脱保護
段階(a)で得たBocテトラアミンベンズアミド(42mg、0.056mmol)をCH2Cl2(0.5ml)中でトリフルオロ酢酸(0.5ml)で処理して、反応液を室温で3時間放置した。次に反応液を減圧濃縮して過剰の酸を除去し、ガム状物を得た。重量予測値(45mg)、重量実測値(45.7mg)。1H及び13C NMR(CD3OD)から、Boc基が完全に除去されたこと並びに上記ガムが所要化合物を含むことが判明した。化合物の秤量試料を水に溶解してTFA塩の10ミリモル濃度溶液(5.6ml中56μmol)を得て、これを放射標識に用いた。
【0137】
実施例5:化合物4の合成
反応はすべて窒素吹込式装置中で手作業で実施した。
【0138】
段階(a):トリチル誘導体化固体担体へのロサルタンの結合
【0139】
【化10】
【0140】
ロサルタン(MSD,0.236g、0.558 mmol)及びトリエチルアミン(Fluka社製、0.233ml、1.67mmol)を、DMF(5ml)中塩化トリチル樹脂(Novabiochem社製、置換量1.24mmol/g、0.300g)の懸濁液に加えた。4日後樹脂を水抜きして洗浄した。樹脂のアリコートを開裂させた(ジクロロメタン/TFA/トリイソプロピルシラン、92.5:5.0:2.5、15分)。HPLC分析で(カラム Phenomenex Luna C18(2)、3μm、4.6×50mm、溶媒:A=水/0.1%TFA及びB=アセトニトリル/0.1%TFA;勾配10分間で10〜40%B;流速2.0ml/分、214及び254nmでUV検出)、ロサルタンに相当するtR6.7分のピークが得られた。樹脂をジクロロメタン/メタノール/ジイソプロピルエチルアミン溶液(17:2:1、20ml、1時間)で処理し、ジクロロメタンで洗浄して乾燥した。
【0141】
段階(b):ヒドロキシル基のアジドでの置換
【0142】
【化11】
【0143】
ジフェニルホスホリルアジド(Aldrich、0.481ml、2.23mmol)及びDBU(0.611ml、4.09mmol)を、THF中(10ml)中で、段階(a)で得た樹脂に結合したロサルタン(0.372mmol)の懸濁液に加えた。この反応液を1晩放置した。樹脂のアリコートを段階(a)に記載の通り開裂させた。LC−MS分析(カラムPhenomenex Luna C18(2)、3μm、50×4.60mm、溶媒:A=水/0.1%TFA及びB=アセトニトリル/0.1%TFA;勾配10分間で20〜80%B;流速1ml/分、214nmでUV検出、ESI−MS)で、tR7.3分に上記の構造に相当するm/z448.1(MH+)を有するピークが得られた。
【0144】
段階(c):アジド基のアミンへの還元
【0145】
【化12】
【0146】
段階(b)からの樹脂のTHF中の懸濁液(4ml)に、塩化スズ(II)(Acros、0.141g、0.744mmol)、チオフェノール(Fluka社製、0.304ml、2.976mmol)及びトリエチルアミン(Fluka社製、0.311ml、2.23mmol)を加えた。1.5時間後に樹脂のアリコートをa)に記載の通り開裂させた。LC−MS分析(カラムPhenomenex Luna C18(2)、3μm、50×4.60mm、溶媒:A=水/0.1%TFA及びB=アセトニトリル/0.1%TFA;勾配10分間で20〜80%B;流速1ml/分、214nmでUV検出、ESI−MS)で、アミンで予想されるm/z422.2(MH+)を有するピークが1.9分に得られた。
【0147】
段階(d):ロサルタンロイシン−テトラアミンキレーター(化合物4)
Fmoc−Leu−OH(Novabiochem社製、0.030g、0.084mmol)をDMF中で段階(c)からの樹脂結合アミノロサルタンの一部(0.042mmol)に、標準的なカップリング剤(HATU及びDIEA)及び標準的なFmoc切断プロトコル(DMF中20%ピペリジン)を用いてカップリングした。カップリングの完結は標準的Kaizer試験で確認した。次いでDMF中で同じカップリング剤(HATU及びDIEA)を用いて樹脂に化合物1(0.026g、0.042mmol)をカップリングした。4時間後に生成物を樹脂から切断してBoc基を同じ方法(ジクロロメタン/TFA/トリイソプロピルシラン、47.5:50:2.5溶液で1時間)で除去した。溶液を濾過、濃縮して分取用HPLC(カラムPhenomenex Luna C18(2)、5μm、21.2×250mm、溶媒:A=水/0.1%TFA及びB=アセトニトリル/0.1%TFA;勾配60分間で20〜40%B;流速10.0ml/分、214nmでUV検出)で精製して、凍結乾燥後5mgの生成物を得た。LC−MS分析(カラムPhenomenex Luna C18(2)、3μm、50×4.60mm、溶媒:A=水/0.1%TFA及びB=アセトニトリル/0.1%TFA;勾配10分間で10〜80%B;流速0.3ml/分、214nm及び254nmでUV検出、ESI−MS)tR5.1分、m/z735.4(MH+)で構造を確認した。
【0148】
実施例6:化合物5の合成
標記化合物は実施例4に記載の通り固体担体上で合成した。Fmoc−Leu−OH(Novabiochem社製、0.033g、0.092mmol)及びFmoc−アミノPEGジグリコール酸(Polypure、0.049mg、0.092mmol)を、標準的カップリング剤(HATU及びDIEA)及び標準的Fmoc切断プロトコル(DMF中20%ピペリジン)を用いて、DMF中で、実施例4(c)で得た樹脂結合アミノ−ロサルタン(0.046mmol)に順次カップリングした。カップリングの完結は標準的Kaizer試験で確認した。次に樹脂に化合物1(0.029g、0.046mmol)をDMF中で同じカップリング剤(HATU及びDIEA)を用いてカップリングした。反応液を1晩放置して、次いで生成物を樹脂から切断してBoc基を同じ方法(ジクロロメタン/TFA/トリイソプロピルシラン、47.5:50:2.5溶液で1時間)で除去した。溶液を濾過、濃縮して分取用HPLC(カラムPhenomenex Luna C18(2)、5μm、21.2×250mm、溶媒:A=水/0.1%HCOOH及びB=アセトニトリル/0.1%HCOOH;勾配60分間で10〜40%B;流速10.0ml/分、214nmでUV検出)で精製して、凍結乾燥後3.5mgの生成物を得た。LC−MS分析(カラムPhenomenex Luna C18(2)、3μm、50×4.60mm、溶媒:A=水/0.1%HCOOH及びB=アセトニトリル/0.1%HCOOH;勾配10分間で10〜40%B;流速0.3ml/分、214nm及び254nmでUV検出、ESI−MS)tR4.7分;m/z1025.4(MH+)で構造を確認した。
【0149】
実施例7:化合物8の合成
段階(a):N−Boc−N−[FmocNH−CH2CH2]−Gly−OHの合成
1gのN−[FmocNH−CH2CH2]−Gly−OtBu・HCl(Fluka社製09660)を、0.5mLのトリイソプロピルシランを含むジクロロメタン中50%トリフルオロ酢酸(TFA)20mLで60分間処理した。反応混合物を減圧蒸発させて残渣を水中50%のテトラヒドロフラン20mLに再溶解した。2.6gのtert−ブチルオキシカルボニル無水物及び1.2mLのN−メチルモルホリンを加えて混合物を4日間撹拌した。次にテトラヒドロフランを減圧蒸発させて残渣をジクロロメタンに再溶解した。有機層を水で洗浄してMgSO4で乾燥した。ジクロロメタンを減圧蒸発させて残渣を5mLのジメチルホルムアミドに再溶解した。このジメチルホルムアミド溶液を水中60%のアセトニトリル400mLで希釈して、精製のため分取用HPLCカラム(40分間で30〜80%B、但しA=H2O/0.1%TFA及びB=CH3CN/0.1%TFA;Phenomenex Luna 10μ C18(2)250×50mmカラムに流速50mL/分)に送り、450mgの純粋生成物を得た。生成物は分析用HPLCで分析した(勾配、10分間で20〜70%B、但しA=H2O/0.1%TFA及びB=CH3CN/0.1%TFA;流速0.3mL/分;カラム、Phenomenex Luna 3μ C18(2)50×2mm;検出、UV214nm;生成物保持時間8.66分)。エレクトロスプレー質量分析法を用いて生成物をさらに特性決定した(MH+計算値441.2;MH+実測値440.8)。
【0150】
段階(b):N−((CH2)2−NHCOCH2−テトラアミン)−Gly−Arg−Val−Tyr−Ile−His−Pro−Ile−OH(化合物8)の合成
アンジオテンシンIIのペプチド類似体を、Applied Biosystems 433Aペプチド合成機を用いて、0.1mmolのFmoc−Ile−Wang樹脂を出発材料として合成した。アルギニンまでのカップリング段階では、過剰の1mmolプレ活性化アミノ酸(O−ベンゾトリアゾール−1−イル−N,N,N′,N′−テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)を使用)を用いた。123mgのN−Boc−N−[FmocNH−CH2CH2]−Gly−OH、114mgの[(ジメチルアミノ)−1H−1,2,3トリアゾロ−[4,5−b]ピリジン−1−イルメチレン]−N−メチルメタンアミニウムヘキサフルオロホスフェートN−オキシド(HATU)及び60μLのN−メチルモルホリンをジメチルホルムアミドに溶解して5分間撹拌し、次に窒素吹込み装置中の樹脂に加えた。試薬を2時間後に除去して樹脂をジメチルホルムアミド及びジクロロメタンで洗浄した。樹脂をジメチルホルムアミド中20%ピペリジンで処理し(3×10ml)、ジメチルホルムアミドで洗浄した。23mgの化合物1、14mgのHATU及び7.5μLのN−メチルモルホリンをジメチルホルムアミドに10分かけて溶解し、樹脂に加えた。試薬を4時間後に除去して樹脂をジメチルホルムアミド及びジクロロメタンで洗浄した。樹脂からの側鎖保護基の除去及び樹脂からのペプチドの切断は、2.5%のトリイソプロピルシラン及び2.5%の水を含むトリフルオロ酢酸10mL中で同時に90分間かけて実施した。トリフルオロ酢酸を真空で除去し、残渣にジエチルエーテルを加えて沈殿した生成物をジエチルエーテルで洗浄して風乾した。
【0151】
生成物を分取用RP−HPLC(40分間で0〜30%B、但しA=H2O/0.1%TFA及びB=CH3CN/0.1%TFA、Phenomenex Luna 10μ C18(2)250×21.20mmカラムで流速10mL/分)で精製して、32mgの純粋キレートペプチドコンジュゲートを得た。生成物を分析用HPLCで分析した(勾配、20分間で5〜50%B、但しA=H2O/0.1%TFA及びB=CH3CN/0.1%TFA;流速1mL/分;カラム、Phenomenex Luna 3μ C18(2)50×2mm;検出、UV214nm;生成物保持時間5.22分)。エレクトロスプレー質量分析法を用いて生成物をさらに特性決定した(MH+計算値1197.8;MH+実測値1197.8)。
【0152】
実施例8:化合物3〜7の99mTc放射標識
以下の成分を含む凍結乾燥キット(「Chelakit A plus」)を調製した。
【0153】
【表1】
【0154】
標識すべき化合物25〜50μg(溶媒25〜50μLに溶解)をCHELAKIT−A plusに加え、次いでジェネレータ溶出液(食塩水中99mTcO4−、1.0mL)を加えた。溶液を混合して室温で20〜30分間放置した。
【0155】
化合物3〜6は室温pH9でテクネチウム標識して、対応する陽イオン99mTc錯体を高収率で与える(RCP>90%)。テトラアミン錯体をHPLC(移動相:0.1%TFA含有水、0.1%TFA含有アセトニトリル;XTERRA RP18 3.5μm 4.6×150mmカラム)で精製したところ、50mMリン酸緩衝液中37℃で2時間安定である(2時間後HPLCによるRCP>95%)。
【0156】
実施例9:99mTc錯体の親油性(LogP)の測定
実施例8の99mTc錯体のオクタノール−水分配係数(LogP)を以下の通り測定した。
【0157】
実施例8で得たHPLC精製99mTc錯体10μLを、遠心チューブ内で1−オクタノール(2ml)及び50mMリン酸緩衝液(pH=7.4、2.0ml)と混合した。チューブを室温で1分間ボルテックスし、高速で60分遠心した。相間の汚染を避けるため注意しながら、両相の試料0.1mlをピペットで他のチューブに移し、Wallac Wizzardガンマカウンターで計数した。測定は3回繰り返した。
【0158】
分配計数Pは次のように計算した。
【0159】
P=(オクタノール中のcpm−バックグラウンドcpm)/(水中のcpm−バックグラウンドcpm)
最終分配係数は一般にlogPとして表される。結果を表1に示す。
【0160】
【表2】
【0161】
実施例10:99mTc錯体の生体内分布
化合物4、5及び7の99mTc錯体を実施例8に記載の通り調製した。実験は、正常オスWistarラット(180〜220g)で、試験標品の注射後(p.i.)所定の2時点(2及び120分)で実施した。動物をハロタン(酸素中6%)で麻酔し、0.1ml(500MBq/ml)の試験標品を注射し、犠牲死させて解剖し、試料の放射能をアッセイした。結果を表1に示す。
【0162】
【表3】
【0163】
実施例11:化合物1の別の調製法
実施例1の段階(e)で得たN,N′−ビス(2−tert−ブトキシカルボニルアミノエチル)−2−(2−ヒドロキシエチル)−1,3−ジ(tert−ブトキシカルボニルアミノ)プロパンを四塩化炭素(14ml)及びアセトニトリル(14ml)に溶解した。水(21ml)を加えると二相混合物になり、次いで過ヨウ素酸ナトリウム(4.5g、21mmol)及び塩化ルテニウム水和物(35mg、0.026mmol)を加えた。得られた暗褐色溶液を室温で1時間撹拌してから、CH2Cl2(40ml)で希釈した。有機層を分離し、水層をCH2Cl2(40ml×3)で抽出した。有機抽出液をすべて一緒にして乾燥(MgSO4)し、濾過して、揮発分を減圧蒸発させると、化合物1のナトリウム塩が暗色の粘稠残渣として残り、それ以上精製せずに使用した(4.15g、96%)。
【0164】
13C NMR(CDCl3):δC 28.2(×12)(CH3),34.1(CH2),34.4(CH),38.6(×2)(NCH2),46.8(×2)(NCH2),49.3(×2)(NCH2),79.0(×2)(OC),80.2(×2)(OC),155.9(×4)(C=O),175.4(COOH)。
【0165】
1H NMR(CDCl3):δH 1.29(18H,s,CH3×6),1.35(18H,s,CH3×6),2.19(1H,br,CH),2.40(2H,br,CH2),3.05〜3.23(12H,br,NCH2×6),5.10〜5.24(2H,br,NH×2)。
【0166】
質量分析(ESI)m/e:(M+Na)C29H54O10N4Na計算値641.3738,実測値641.3787。
【0167】
実施例12:化合物9の調製
1,3−ジシクロヘキシルカルボジイミド(DCC;2.16g、10.5mmol)を、乾燥THF(30ml)中の化合物1(4.15g、6.90mmol)及びN−ヒドロキシスクシンイミド(1.81g、15.7mmol)の攪拌溶液に1度に加えた。混合物を室温で16時間撹拌してから、沈殿したDCU(1,3−ジシクロヘキシル尿素)を濾別した。濾液から揮発分を蒸発させるとワックス状残渣が残り、これに乾燥エーテル(50ml)を加えるとさらにDCUが沈殿し、これを濾別した。エーテル溶液を水(25ml×2)で洗浄し、乾燥(MgSO4)、濾過して、溶媒を減圧蒸発させるとワックス状固体が残った。この固体をCH2Cl2/エーテル混液(1:1)で溶出するシリカゲルクロマトグラフィーで未反応DCCが除去されるまで精製した。溶出液をエーテルに代えて所要生成物(rf=0.4、DCM/Et2O1:1)をmp66〜68℃の無色固体として分離した(2.7g、57%)。
【0168】
13C NMR(CDCl3):δC 25.6(×2)(CH2),28.4(×12)(CH3),31.8(CH2),35.2(CH),39.3(×2)(NCH2),47.1(×2)(NCH2),49.1(×2)(NCH2),79.9(×2)(OC),80.5(×2)(OC),156.1(×4)(C=0),167.7(C=O),169.1(×2)(C=O)。
【0169】
1H NMR(CDCl3):δH 1.35(18H,s,CH3×6),1.41(18H,s,CH3×6),2.52(3H,brs,CH及びCH2),2.77(4H,s,CH2×2),3.10〜3.35(12H,brs,NCH2×6),5.08(2H,brsNH×2)。
【0170】
質量分析(ESI)m/e:(M+Na)C33H54N5O12Na計算値738.3896,実測値738.3893。
【図面の簡単な説明】
【0171】
【図1】実施例に記載の各種化合物の構造を示す。
【図2】実施例に記載の各種化合物の構造を示す。
【図3】実施例に記載の各種化合物の構造を示す。
【技術分野】
【0001】
本発明は、放射性金属99mTcとの金属錯体の形成に適したテトラアミンキレーターと生体ターゲティング分子との改良コンジュゲートに関する。この放射性金属錯体は99mTc放射性医薬品として有用である。キット及び前駆体も提供される。
【背景技術】
【0002】
米国特許第5489425号(Dow Chemical)には、金属の錯体化に有用な一群の鎖状及び大環状の官能化テトラアミンキレーター、特に放射性及び非放射性ロジウム錯体、殊に105Rh又は101mRh放射性金属錯体が開示されている。開示された具体的なテトラアミンには、以下のものが含まれる。
【0003】
【化1】
【0004】
上記二官能性キレーターは、治療又は診断用のモノクローナル抗体又はその断片との結合に有用であると記載されている。米国特許第5489425号には、まず105Rh金属錯体を形成してから抗体と反応させた後、精製することによって、抗体放射性金属錯体キレーターコンジュゲートを調製することが開示されている(実施例21、22a及び23)。米国特許第5489425号には、錯化していない抗体キレーターコンジュゲート、つまり放射性金属の配位していないものは記載されていない。米国特許第5489425号は、かかる抗体結合反応において側鎖アミンをキレーターの4つのアミンからどのように差別化するか何ら教示されていない。米国特許第5489425号は、二官能性キレーターが「テクネチウム及びレニウムとの錯体化にも有用であろう」と記載されているが、これをどのように達成するかについては開示されていないし、テクネチウム錯体の具体例も開示されていない。
【0005】
米国特許第5650134号には、一群のキレーターのソマトスタチンペプチド−キレーターコンジュゲートが開示されている。実施例1には、オクトレオチドペプチドの6−(p−イソチオシアナトベンジル)−1,4,8,11−テトラアザウンデカンとの結合が記載されている。
【0006】
欧州特許出願公開第1181936号には、以下の二官能性キレーターBBN−1及びBBN−2を用いて調製されたテトラアミンキレーターのボンベシン(テトラデカペプチド)コンジュゲート並びにそれらの99mTc錯体が開示されている。
【0007】
【化2】
【0008】
99mTc錯体はマウス体内から腎臓・尿路系を介しての迅速なクリアランスを呈すると記載されている。しかし、欧州特許出願公開第1181936号には、BBN−1又はBBN−2をボンベシンのN末端に結合する段階を除いて、BBN−1又はBBN−2の合成に関して何ら開示されていないし、引用されてもいない。腫瘍イメージング用放射性医薬品候補を得るためのBBN−2とボンベシンとの結合及び99mTc標識についても、Nock et al, Eur. J. Nucl. Med.,30(2),247−258(2003)に記載されている。この99mTc錯体は従来技術のボンベシンキレートコンジュゲートよりも親水性が向上し、そのため腎臓・尿路系による排泄に有利に働くと期待されると記載されている。
【0009】
ヒトの患者用の腫瘍イメージング用放射性医薬品候補を得るためのBBN−1とオクトレオチドとの結合及び99mTc標識についても、Marina et al, Eur. J. Nucl. Med.,30(9),1211−1219(2003)に記載されている。以上のBBN−1又はBBN−2の刊行物のいずれにも、BBN−1又はBBN−2の合成については何な記載されていない。
【特許文献1】米国特許第5489425号明細書
【特許文献2】米国特許第5650134号明細書
【特許文献3】欧州特許出願公開第1181936号明細書
【非特許文献1】Nock et al, Eur. J. Nucl. Med.,30(2),247−258(2003)
【非特許文献2】Marina et al, Eur. J. Nucl. Med.,30(9),1211−1219(2003)
【発明の開示】
【課題を解決するための手段】
【0010】
本発明は、リンカー基を介して生体ターゲティング部分と連結したテトラアミンキレーターコンジュゲート並びに放射性医薬品としてのそのテクネチウム錯体を提供する。リンカー基はキレーターが橋頭位で単官能化されるようなもので、柔軟性を与えるとともにアリール基を含まないものをもたらし、親油性及び立体的嵩高さを最小限にする。キレーターの適当な保護形も提供し、テトラアミンキレーターのアミン窒素による干渉反応を起こさずに広範なターゲティング分子との結合が可能となる。官能化キレーターの合成についても、二官能性キレーター前駆体と併せて説明する。
【0011】
本発明のテクネチウム金属錯体を含む放射性医薬組成物についても、かかる放射性医薬品の調製用の非放射性キットと併せて説明する。
【発明を実施するための最良の形態】
【0012】
第一の実施形態では、本発明は次の式(I)の陽イオン99mTCテクネチウム錯体を提供する。
【0013】
【化3】
【0014】
式中、
Xは−NR−、−CO2−、−CO−、−NR(C=S)−、−NR(C=O)−、−CONR−又はQ基であり、
各Yは独立にD−若しくはL−アミノ酸、−CH2−、−CH2OCH2−又は−OCH2CH2O−又はX基であり、
Zは合成生体ターゲティング部分であり、
nは1〜8の整数であり、
mは0〜30の整数であり、
RはH、C1〜4アルキル、C2〜4アルコキシアルキル、C1〜4ヒドロキシアルキル又はC1〜4フルオロアルキルであり、
Qは次式の基であり、
【0015】
【化4】
【0016】
Aは対イオンであるが、
ヘテロ原子同士が直接結合した結合を原子鎖X1−(Y)mが含まないことを条件とする。
【0017】
テクネチウム放射性同位体は、99mTcのようなγ線放射体でも、94mTcのようなPETイメージングに適した陽電子放射体でもよい。好ましくはテクネチウム放射性同位体は99mTc又は94mTcであり、最も好ましくは99mTcである。
【0018】
Xは好ましくは−CONR−、−NR(C=O)−又はQ基である。Xは最も好ましくは−CONR−又は−NR(C=O)−であり、−CONH−及び−NH(C=O)−が特に好ましい。
【0019】
式Iのリンカー基−(CH2)n−X−(Y)m−は、ヘテロ原子同士が直接結合した結合をX1−(Y)m原子鎖が含まないように選択され、「ヘテロ原子」という用語は窒素、酸素又はイオウのような非炭素原子を意味する。これは上記原子鎖がO−O、N−N又はO−Nのような結合を含まないことを意味する。
【0020】
式Iのリンカー基−(CH2)n−X−(Y)m−の役割は、生体内での生体ターゲティング部分(Z)の活性結合部位からテクネチウム錯体を離隔することであると想定される。これは生体内での活性部位への結合を比較的嵩高いテクネチウム錯体が立体的に阻害しないようにするのに役立つ。アルキレン基−(CH2)n−は、結合した生体ターゲティング部分(Z)との有意な水素結合相互作用がなく、そのためリンカーがZに巻き付かないという利点を有する。好ましいアルキレン基はn=1〜6、最も好ましくは2〜4であり、2が特に好ましい。
【0021】
本発明のリンカー基はアリール環を含まない。これは、コンジュゲートの生体ターゲティング部分(Z)に結合したリンカー基並びにテクネチウム錯体の親油性を最小限にするのに役立つ。リンカー基(及びテクネチウム錯体)の立体的嵩高さ及び分子量も最小限となるが、連鎖の柔軟性は維持される。
【0022】
リンカー基の性状は、造影剤の体内分布の改変にも利用できる。例えば、−(Y)m−にエーテル基を導入すると、血漿タンパク質の結合を最小限に抑えるのに役立つであろう。−(Y)m−がポリエチレングリコール(PEG)構成単位又は1〜10アミノ酸残基のペプチド鎖からなる場合、リンカー基は生体内での薬物動態及び造影剤の血液クリアランス速度を改善するように機能し得る。かかる「バイオモディファイアー」リンカー基は、筋肉や肝臓のようなバックグラウンド組織及び/又は血液からのテクネチウム造影剤のクリアランスを促進して、バックグラウンド干渉の低下による画質の向上した診断用画像を与え得る。バイオモディファイアーリンカー基は、特定の排泄経路、例えば肝臓経路よりも腎臓経路を優勢にするのにも使用し得る。或いは、バイオモディファイアーリンカー基は血中滞留時間を延ばして、生体内でのターゲット部位への薬剤の蓄積量を増すことができる。
【0023】
−(Y)m−がアミノ酸残基のペプチド鎖を含む場合、アミノ酸残基は好ましくはグリシン、リジン、アスパラギン酸、グルタミン酸又はセリンから選択される。ペプチド鎖のアミノ酸の数は好ましくは1〜10、最も好ましくは1〜3である。
【0024】
−(Y)m−がPEG部分を含む場合、好ましくは式(−OCH2CH2O−)wの基を含むもので、wは3〜25の整数である。整数wは好ましくは6〜22である。特に好ましいPEG含有−(Y)m−基は、単分散PEG様構造の式IVの17−アミノ−5−オキソ−6−アザ−3,9,12,15−テトラオキサヘプタデカン酸の重合で得られる単位である。
【0025】
【化5】
【0026】
式中、pは1〜10の整数である。
【0027】
「フルオロアルキル」という用語は、1以上のフッ素置換基を有するアルキル基を意味し、この用語はモノフルオロアルキル(例えば−CH2F)からペルフルオロアルキル(例えばCF3)までの基を包含する。
【0028】
−(Y)m−基は好ましくはジグリコール酸部分、マレイミド部分、グルタミン酸、コハク酸、ポリエチレングリコール系単位又は式IVのPEG様単位を含む。
【0029】
「合成」という用語は、その通常の意味、つまり天然資源(例えば哺乳動物の身体)から分離されたものではなく、人造のものを意味する。かかる化合物は、その製造並びに不純物特性を十分に制御できるという利点がある。従って、モノクローナル抗体及びその断片は、本願特許請求の範囲に属さない。
【0030】
「生体ターゲティング部分」という用語は、3〜100量体ペプチド若しくはペプチド類似体(これらは鎖状ペプチドでも環状ペプチドでも若しくはそれらの組合せでもよい。)、酵素の基質若しくはアンタゴニスト若しくは阻害剤、合成受容体結合性化合物、オリゴヌクレオチド、又はオリゴDNA断片若しくはオリゴRNA断片を意味する。
【0031】
「環状ペプチド」という用語は、2つの末端アミノ酸が共有結合によって連結した5〜15個のアミノ酸配列を意味し、上記共有結合はペプチド結合でも、ジスルフィド結合でも、或いはチオエーテル、ホスホジエステル、ジシロキサン又はウレタン結合のような合成非ペプチド結合でもよい。「アミノ酸」という用語は、L−若しくはD−アミノ酸、アミノ酸類似体又はアミノ酸模倣体を意味し、光学的に純粋なもの、つまり単一の鏡像異性体でキラルなものでもよいし、鏡像異性体混合物であってもよい。好ましくは、本発明のアミノ酸は光学的に純粋なものである。「アミノ酸模倣体」という用語は、天然アミノ酸の合成類似体であってアイソスターであるもの、つまり天然化合物の立体及び電子構造を模倣して設計されたものを意味する。かかるアイソスターは当業者に周知であり、特に限定されないが、デプシペプチド、レトロインベルソ型ペプチド、チオアミド、シクロアルカン又は1,5−二置換テトラゾールなどが挙げられる(M.Goodman, Biopolymers,24,137,(1985)参照)。 本発明での使用に適したペプチドとしては、以下のものが挙げられる。
・ソマトスタチン、オクトレオチド及び類似体。
・ST受容体に結合するペプチド。ここで、STとは大腸菌その他の微生物の産生する熱安定性毒素をいう。
・ラミニン断片、例えばYIGSR、PDSGR、IKVAV、LRE及びKCQAGTFALRGDPQG。
・白血球集積のターゲティング部位用のN−ホルミルペプチド。
・血小板因子4(PF4)及びその断片。
・RGD(Arg−Gly−Asp)含有ペプチド。これは例えば血管新生をターゲティングし得る(R.Pasqualini et al., Nat Biotechnol.1997 Jun;15(6):542−6;E.Ruoslahti, Kidney Int. 1997 May;51(5):1413−7)。
・α2−アンチプラスミン若しくはフィブロネクチン若しくはβ−カゼインのペプチド断片、フィブリノーゲン又はトロンボスポンジン。α2−アンチプラスミン、フィブロネクチン、β−カゼイン、フィブリノーゲン及びトロンボスポンジンのアミノ酸配列は以下の引用文献に記載されている:α2−アンチプラスミン前駆体(M.Tone et al., J. Biochem,102,1033,(1987));β−カゼイン(L.Hansson et al, Gene,139,193,(1994));フィブロネクチン(A.Gutman et al, FEBS Lett.,207,145,(1996));トロンボスポンジン−1前駆体(V.Dixit et al, Proc. Natl. Acad. Sci.,USA,83,5449,(1986));R.F.Doolittle, Ann. Rev. Biochem.,53,195,(1984)。
・アンジオテンシンII:Asp−Arg−Val−Tyr−Ile−His−Pro−Phe(E.C.Jorgensen et al, J. Med. Chem.,1979,Vol 22,9,1038−1044)、[Sar,Ile]アンジオテンシンII:Sar−Arg−Val−Tyr−Ile−His−Pro−Ile(R.K.Turker et al, Science,1972,177,1203)のようなアンジオテンシンの基質又は阻害剤であるペプチド。
・アンジオテンシンI:Asp−Arg−Val−Tyr−Ile−His−Pro−Phe−His−Leu。
【0032】
好ましくは、本発明のペプチドはアンチプラスミン又はアンジオテンシンIIペプチドを含む。アンチプラスミンペプチドは、N末端からみて以下のアミノ酸配列を含む。
(i)α2−アンチプラスミン
NH2−Asn−Gln−Glu−Gln−Val−Ser−Pro−Leu−Thr−Leu−Thr−Leu−Leu−Lys−OH、又はその変異体で1以上のアミノ酸が交換、付加又は除去されたもの、例えば、
NH2−Asn−Gln−Glu−Gln−Val−Ser−Pro−Leu−Thr−Leu−Thr−Leu−Leu−Lys−Gly−OH、
NH2−Asn−Gln−Glu−Ala−Val−Ser−Pro−Leu−Thr−Leu−Thr−Leu−Leu−Lys−Gly−OH、
NH2−Asn−Gln−Glu−Gln−Val−Gly−OHなど、或いは
(ii)カゼイン
Ac−Leu−Gly−Pro−Gly−Gln−Ser−Lys−Val−Ile−Gly。
【0033】
本発明の合成ペプチドは、P.Lloyd−Williams,F.Albericio and E.Girald; Chemical Approaches to the Synthesis of Peptides and Proteins,CRC Press,1997に記載されているように、通常の固相合成法で得ることができる。
【0034】
適当な酵素の基質、アンタゴニスト又は阻害剤としては、グルコース並びにフルオロデオキシグルコースのようなグルコース類似体、脂肪酸、又はエラスターゼ若しくはアンジオテンシンII若しくはメタロプロテアーゼ阻害剤が挙げられる。好ましい非ペプチド系アンジオテンシンIIアンタゴニストはロサルタンである。
【0035】
適当な受容体結合性化合物としては、エストラジオール、エストロゲン、プロゲスチン、プロゲストロンその他のステロイドホルモン、ドーパミンD−1若しくはD−2受容体、又はトロパンのようなドーパミン輸送体、並びにセロトニン受容体に対するリガンドが挙げられる。
【0036】
生体ターゲティング部分(Z)は好ましくは分子量5000未満のもの、最も好ましくは4000未満のもの、理想的には3000未満のものである。これは、本発明のテトラアミンテクネチウム錯体の改善された生物学的特性が、式Iのコンジュゲートのテクネチウム錯体の全体的体内分布、特にクリアランスに影響をもつという利点を有する。nが3で、Xが(CH2)n基に直接結合した窒素を含む場合、Zは合成物で分子量4000未満となるように選択される。好ましい生体ターゲティング部分は3〜20量体ペプチド又は酵素基質、酵素アンタゴニスト若しくは酵素阻害剤である。
【0037】
対イオン(A−)は、モル当量で存在し、式IのTc(V)ジオキソテクネチウム錯体の陽電荷と釣り合う陰イオンを表す。陰イオン(A)は、電荷が均衡する量で存在していれば単電荷のものでも多電荷のものでもよい。陰イオンは好適には無機酸又は有機酸から誘導される。適当な陰イオンの例としては、塩素又は臭素のようなハロゲンイオン、硫酸、硝酸、クエン酸、酢酸、リン酸及びホウ酸イオンが挙げられる。好ましい陰イオンは塩素イオンである。
【0038】
式Iのテクネチウム錯体は、錯形成後に安定であり、テクネチウムを生体ターゲティング部分に優先的に結合させる強力なキランドを含んでいるという利点を有する。そのため、このテクネチウム錯体は生体内で生体高分子又は競合配位子とのトランスキレーション反応を起こしにくい。テクネチウム錯体は小さくてコンパクトであり、生体ターゲティング部分(Z)と結合したときも立体的な影響が最小限である点で有用である。永続的な陽イオン交換とTc(V)ジオキソコアの存在は、この錯体が親水性でもあることを意味しており、そのため錯体が細胞内の他の区画に分布する可能性が低く、生体内でバックグラウンド器官及び組織、例えば血流からのクリアランスが速い。
【0039】
式Iのテクネチウム錯体は、以下の第二の実施形態で説明するように、適当なテクネチウム源と式IIのキレーターコンジュゲートとの反応によって調製できる。
【0040】
第二の実施形態では、本発明は式IIのキレーターコンジュゲートを提供する。
【0041】
【化6】
【0042】
式中、X、Y、Z、n及びmは既に定義した通りであり、
Q1〜Q6は独立にQ基であり、QはH又はアミン保護基である。
【0043】
このキレーターコンジュゲートは、第一の実施形態に係る式Iのテクネチウム錯体の調製に有用である。
【0044】
「保護基」という用語は、不都合な化学反応は阻害又は抑制するが、分子の残りの部分を修飾しない十分穏和な条件下で当該官能基から開裂できる十分な反応性をもつように設計された基を意味する。脱保護後に、所望の生成物が得られる。アミン保護基は当業者に周知であり、好適にはBoc(tert−ブチルオキシカルボニル)、Fmoc(フルオレニルメトキシカルボニル)、トリフルオロアセチル、アリルオキシカルボニル、Dde(1−(4,4−ジメチル−2,6−ジオキソシクロヘキシリデン)エチル]又はNpys(3−ニトロ−2−ピリジンスルフェニル)から選択される。場合によっては、保護基の性状は、Q1/Q2又はQ5/Q6の両者、つまり関連するアミン窒素原子に全くNH結合が存在しないようなものであってもよい。他の保護基の使用については、‘Protective Groups in Organic Synthesis’,Theorodora W.Greene and Peter G.M.Wuts, John Wiley&Sons,1991に記載されている。好ましいアミン保護基はBoc及びFmoc、最も好ましくはBocである。Bocを使用した場合、Q1及びQ6は共にHであり、Q2、Q3、Q4及びQ5は各々tert−ブトキシカルボニルである。
【0045】
式IIにおいて、アミン保護基は主にテクネチウムとの錯形成前の合成反応時にテトラアミンキレーターのアミン官能基を保護するために用いられる。しかし、生体ターゲティング基(Z)が第一及び/又は第二アミンと反応し易い場合、これらの保護基は、テクネチウムと錯形成する前のキレーターアミンとZとの不都合な反応の防止にも有用である。
【0046】
式IIの好ましいコンジュゲートは、保護されていないアミン窒素を少なくとも1つ以上有する(つまり、Q3又はQ4の一方がHであるか、或いはQ1/Q2対又はQ5/Q6対がHである)。1以上の遊離アミノ基は、式Iのテクネチウム錯体の調製の際の好ましい溶媒である水性媒体中でのコンジュゲートの溶解性が増すことを意味する。遊離アミノ基は、錯形成が保護基を予め除去しておくこと(これは金属の錯体化も阻害する)に依存しないので、テクネチウムとの錯形成が速やかになることを意味する。コンジュゲートした生体ターゲティング基(Z)がアミンとの反応は起こしにくいときは、式IIのコンジュゲートを完全な脱保護形(Q1〜Q6が各々H)で使用するのが好適であり、これが式IIの特に好ましいキレーターコンジュゲートである。式Iのテクネチウム錯体を得るための錯体化反応には完全な脱保護形が好ましい。
【0047】
本発明の式Iのテクネチウム錯体は、適切な酸化状態の放射性金属の溶液を適切なpHで式IIのキレーターコンジュゲートと反応させることによって調製できる。溶液は、テクネチウムと弱く錯化する配位子(グルコン酸イオン又はクエン酸イオンなど)を適宜含んでいてもよく、テクネチウム錯体は配位子交換又はトランスキレーションによって調製される。かかる条件はテクネチウムイオンの加水分解のような不都合な副反応を抑制するのに有用であることが多いが、本発明のキレーターはテクネチウムと迅速に錯形成するので、本発明のキレーターでは重要性は低い。放射性同位体が99mTcである場合、通常の出発原料は99Moジェネレータから得られる過テクネチウム酸ナトリウムである。テクネチウムは、比較的反応性の低いTc(VII)の酸化状態の99mTc−過テクネチウム酸塩として存在する。そのため、酸化状態の低いTc(I)〜Tc(V)のテクネチウム錯体の調製には、通常、錯体化を促すため亜ジチオン酸ナトリウム、亜硫酸水素ナトリウム、アスコルビン酸、ホルムアミジンスルフィン酸、第一スズイオン、Fe(II)又はCu(I)のような薬学的に許容される還元剤の添加が必要とされる。薬学的に許容される還元剤は好ましくは第一スズ塩であり、最も好ましくは塩化第一スズ、フッ化第一スズ又は酒石酸第一スズである。
【0048】
式IIのキレーターコンジュゲートは、以降の第五の実施形態で説明するように、生体ターゲティング分子(Z)と式IIIの二官能性キレーターとの結合によって調製できる。
【0049】
第三の実施形態では、本発明は、Aが薬学的に許容される対イオンである第一の実施形態のテクネチウム錯体を生体適合性担体と共にヒトへの投与に適した形態で含む放射性医薬品を提供する。
【0050】
「ヒトへの投与に適した形態」という用語は、無菌で、発熱物質も毒性又は副作用を生じる化合物も含まず、生体適合性のpH(pH約4.0〜10.5)で製剤化した組成物を意味する。かかる組成物は生体内で塞栓を惹起する危険性のある粒子を含まず、体液(例えば血液)と接しても沈殿が起こらないように製剤化される。かかる組成物は生体適合性の賦形剤のみを含有し、好ましくは等張性である。
【0051】
「生体適合性担体」とは、放射性医薬品を懸濁又は好ましくは溶解できる流体、特に液体であって、組成物が生理学的に認容できるもの、つまり毒性も耐え難い不快感も伴わずに哺乳類の身体に投与することができるようなものである。生体適合性担体は好適には注射可能な担体液であり、例えば、発熱物質を含まない注射用の滅菌水、食塩液のような水溶液(これは注射用の最終製剤が等張性又は非低張性となるように調整するのに都合がよい)、1種以上の張度調節物質(例えば血漿陽イオンと生体適合性対イオンとの塩)、糖(例えばグルコース又はスクロース)、糖アルコール(例えばソルビトール又はマンニトール)、グリコール(例えばグリセロール)その他の非イオン性ポリオール材料(例えばポリエチレングリコール、プロピレングリコールなど)の水溶液である。
【0052】
「薬学的に許容される対イオン」という用語は、哺乳類の身体に投与したときに生体内で毒性又は悪影響を生じずに、医薬組成物の他の成分と化学的及び/又は毒性学的に適合性の陰イオン(負電荷イオン)を意味する。本発明のテクネチウム放射性医薬品に対する化学的適合性とは、この陰イオンがテクネチウムに対してテトラアミンキレーターと事実上競合しないことを意味する。かかる適当な陰イオンとしては、ハロゲンイオン(例えば塩素、ヨウ素及び臭素イオン)、C1〜2アルキルスルホン酸イオン(例えばメシレート又はエチルスルホン酸イオン)、アリールスルホン酸イオン(例えばフェニルスルホン酸又はトシレートイオン)、C1〜2アルキルホスホン酸イオン、ジ(C1〜2)アルキルリン酸イオン(例えばジメチルリン酸、ジエチルリン酸又はジグリセロールリン酸イオン)、アリールホスホン酸イオン、アリールリン酸イオン、アルキルアリールホスホン酸イオン、アルキルアリールリン酸イオン、C1〜2アルキルカルボン酸イオン(例えば酢酸、プロピオン酸、グルタミン酸又はグリセリン酸イオン)、アリールカルボン酸イオン(例えば安息香酸イオン)などが挙げられる。好ましい薬学的に許容される対イオンは、塩素イオン、フッ素イオン、酢酸イオン、酒石酸イオン、水酸イオン及びリン酸イオンである。
【0053】
かかる放射性医薬品は、好適には、無菌状態を維持しながら皮下注射針で一回又は複数回穿刺するのに適したシール(例えばクリンプオン式セプタムシール蓋)を備えた容器に入れて供給される。かかる容器には、1回又は複数回分の用量を入れることができる。好ましい多用量用容器は、複数回分の用量を収容した単一バルクバイアル(例えば容積10〜30cm3のもの)からなり、臨床症状に応じて製剤の有効期間中様々な時間間隔で1回分の用量を臨床グレードの注射器に引き出すことができる。
プレフィルド型注射器は1回分の用量を収容するように設計され、そのため好ましくは使い捨て又はその他臨床用に適した注射器である。プレフィルド型注射器は、適宜、オペレーターを放射能から保護するため、注射器シールドを備えていてもよい。かかる適当な放射性医薬品注射器シールドは当技術分野で公知であり、好ましくは鉛又はタングステンからなる。
【0054】
本発明の好ましい放射性医薬品はテクネチウム放射性同位体99mTc又は94mTcを含み、最も好ましくは99mTcを含む。テクネチウム同位体が99mTcである場合、画像診断用放射性医薬品に適した放射能含量は、生体内の撮像部位、取込み量及び標的/バックグラウンド比に応じて、180〜1500MBqの99mTcである。
本発明のテクネチウム放射性医薬品は、以下に挙げるような様々な方法で調製できる。
(i)第二の実施形態に関して上記で説明したテクネチウム錯体形成をクリーンルーム環境下で実施する無菌製造法。
(ii)無菌製造を用いずにテクネチウム錯体形成を実施してから、最終段階で滅菌(例えばガンマ線照射又は高圧蒸気滅菌)する最終滅菌法。
(iii)式IIのキレーターと薬学的に許容される還元剤を適宜他の賦形剤と共に含む無菌の凍結乾燥非放射性キット製剤を、99mTcジェネレータから得られる無菌99mTc−過テクネチウム酸のアリコートと反応させるキット法。
【0055】
方法(iii)が好ましく、この方法に使用するためのキットを以下の第四の実施形態で説明する。
【0056】
第四の実施形態では、本発明は、上述の放射性医薬組成物を調製するための非放射性キットであって、式IIのコンジュゲートを生体適合性還元剤と共に含むキットを提供する。かかるキットは、例えば血流への直接注射によるヒトへの投与に適した無菌放射性医薬製剤を与えるように設計される。リガンドコンジュゲート及びその好ましい態様は上記の第二の実施形態で記載した通りである。
【0057】
99mTc用には、キットは好ましくは凍結乾燥したもので、99mTc放射性同位体ジェネレータからの無菌99mTc−過テクネチウム酸(TcO4−)で再構成すればそれ以上操作しなくてもヒトへの投与に適した溶液が得られるように設計される。
適当なキットは、遊離塩基又は酸塩の形態のキレーターコンジュゲートを亜ジチオン酸ナトリウム、亜硫酸水素ナトリウム、アスコルビン酸、ホルムアミジンスルフィン酸、第一スズイオン、Fe(II)又はCu(I)のような生体適合性還元剤と共に収容した容器(例えばセプタムシールバイアル)を備える。生体適合性還元剤は、好ましくは塩化第一スズや酒石酸第一スズのような第一スズ塩である。或いは、キットは非放射性金属錯体を適宜含んでいてもよく、テクネチウムの添加時にトランスメタレーション(金属交換)を起こして所望の生成物を与える。
【0058】
非放射性キットはさらに、トランスキレーター、放射線防護剤、抗菌保存剤、pH調節剤又は充填剤のような追加成分を適宜含んでいてもよい。「トランスキレーター」とは、テクネチウムと迅速に反応して弱い錯体を形成し、次に上記キレーターで置換される化合物である。これはテクネチウム錯体化と競合する過テクネチウム酸塩の迅速な還元に起因した還元型加水分解テクネチウム(RHT)が形成するおそれを最小限に抑制する。かかる適当なトランスキレーターは、弱有機酸(つまりpKaが3〜7の範囲内にある有機酸)と生体適合性陽イオンとの塩である。「生体適合性陽イオン」という用語は、イオン化して負に荷電した陰イオン基と塩を形成する正電荷を有する対イオンで、しかも無毒で哺乳類の身体、特に人体への投与に適したものをいう。適当な生体適合性陽イオンの例としては、アルカリ金属のナトリウム又はカリウム、アルカリ土類金属のカルシウム及びマグネシウム、さらにアンモニウムイオンが挙げられる。好ましい生体適合性陽イオンはナトリウム及びカリウムであり、最も好ましくはナトリウムである。適当な弱有機酸は、酢酸、クエン酸、酒石酸、グルコン酸、グルコヘプトン酸、安息香酸、フェノール又はホスホン酸である。従って、適当な塩は、酢酸塩、クエン酸塩、酒石酸塩、グルコン酸塩、グルコヘプトン酸塩、安息香酸塩、フェノラート又はホスホン酸塩である。好ましい塩は、酒石酸塩、グルコン酸塩、グルコヘプトン酸塩、安息香酸塩又はホスホン酸塩であり、最も好ましくはホスホン酸塩、特にジホスホン酸塩である。好ましいトランスキレーターは、MDP(メチレンジホスホン酸)と生体適合性陽イオンとの塩である。
【0059】
「放射線防護剤」という用語は、水の放射線分解で生成する含酸素フリーラジカルのような反応性の高いフリーラジカルを捕捉することによって、酸化還元過程のような分解反応を阻害する化合物をいう。本発明の放射線防護剤は、好適には、アスコルビン酸、パラアミノ安息香酸(即ち4−アミノ安息香酸)、ゲンチシン酸(即ち2,5−ジヒドロキシ安息香酸)並びにこれらの生体適合性陽イオンとの塩から選択される。
「抗菌保存剤」という用語は、細菌、酵母又はカビなどの有害微生物の増殖を阻害する薬剤を意味する。抗菌保存剤は、濃度に応じてある程度の殺菌作用を示すこともある。本発明の抗菌保存剤の主な役割は、再構成後の放射線医薬組成物(つまり、放射性診断薬自体)における微生物の増殖を阻害することである。ただし、抗菌保存剤は、再構成前の本発明の非放射性キットの1以上の成分における有害微生物の増殖の防止にも適宜使用できる。適当な抗菌保存剤としては、パラベン類、即ちメチルパラベン、エチルパラベン、プロピルパラベン、ブチルパラベン又はこれらの混合物、ベンジルアルコール、フェノール、クレゾール、セトリミド及びチオメルサールが挙げられる。好ましい抗菌保存剤はパラベンである。
【0060】
「pH調節剤」という用語は、再構成したキットのpHが、ヒト又は哺乳類の投与に関する許容範囲(約pH4.0〜10.5)内に収まるようにするのに有用な化合物又は化合物の混合物を意味する。かかる適当なpH調節剤としては、トリシン、リン酸塩又はTRIS(トリス(ヒドロキシメチル)アミノメタン)のような薬学的に許容される緩衝剤、並びに炭酸ナトリウム、重炭酸ナトリウム又はこれらの混合物などの薬学的に許容される塩基が挙げられる。コンジュゲートを酸塩の形態で用いる場合、キットのユーザーが多段階操作法の一部としてpHを調節できるようにpH調節剤を適宜別のバイアル又は容器で提供してもよい。
【0061】
「充填剤」という用語は、製造時及び凍結乾燥時の材料の取扱いを容易にする薬学的に許容される増量剤を意味する。適当な充填剤としては、塩化ナトリウムのような無機塩並びに水溶性糖類又は糖アルコール、例えばスクロース、マルトース、マンニトール又はトレハロースが挙げられる。
【0062】
第五の実施形態では、本発明は式IIIの化合物を提供する。
【0063】
【化7】
【0064】
式中、Q1〜Q6及びnは式I及びIIに関して定義した通りであり、
Eは第一の実施形態の生体ターゲティング部分(Z)との結合に適した官能基であるが、
(i)n=3のとき、Q1〜Q6の1以上がアミン保護基であり、
(ii)n=3又は5のとき、EはOHではないことを条件とする。
【0065】
式IIIの化合物は「二官能性キレーター」つまり1以上の官能基(E)が結合したキレーターである。官能基Eは生体ターゲティング部分(Z)との結合に適したものである。適当な官能基(E)としては、アミン、チオシアネート、マレイミド及び活性エステルが挙げられる。Eは好ましくは活性化されていないヒドロキシル(−OH)基を含まない。「活性エステル」という用語は、良好な脱離基で、生体ターゲティング部分に存在するアミンのような求核部位との反応を容易にするように設計されたカルボン酸のエステル誘導体を意味する。適当な活性エステルの例は、N−ヒドロキシスクシンイミド(NHS)、ペンタフルオロフェノール、ペンタフルオロチオフェノール、パラ−ニトロフェノール、ヒドロキシベンゾトリアゾール及びPyBOP(ベンゾトリアゾール−1−イル−オキシトリピロリジノホスホニウムヘキサフルオロホスフェート)である。好ましい活性エステルは、N−ヒドロキシスクシンイミド又はペンタフルオロフェノールエステルである。
【0066】
Eは好ましくは第一アミン(−NH2)、−CO2M、−NCS、−NCO、マレイミド又はアクリルアミドである。ただし、MはH、陽イオン、保護基又は活性エステルである。Eは最も好ましくは−NH2、−CO2M又はマレイミドであり、理想的には−NH2又は−CO2Mである。
【0067】
式IIIの化合物は、生体ターゲティング分子(Z)の適当な対応官能基と反応して式IIの所望のコンジュゲートを形成する。生体ターゲティング分子(Z)の適当な官能基としては、(アミン官能化二官能性キレーターとのアミド結合形成のための)カルボキシル、(カルボキシル又は活性エステル官能化二官能性キレーターとのアミド結合形成のための)アミン、(アミン官能化二官能性キレーターのNアルキル化のための)ハロゲン、メシレート及びトシレート、並びに(マレイミド官能化二官能性キレーターとの反応のための)チオールが挙げられる。
【0068】
Eが生体ターゲティング分子(Z)のアミノ基と反応するように設計された基(例えば活性エステル)である場合、キレーターのアミンとの不都合な副反応のおそれがあることは明らかである。かかるE基については、テトラアミンキレーターの4つのアミン窒素原子の各々が保護されるように、式IIIのQ1〜Q6は好ましくは窒素保護基として選択される。Eがアミノ基である場合、生体ターゲティング分子(Z)との反応がEアミンでしか起こらず、テトラアミンキレーターのアミン窒素原子では起こらないことが明らかに重要である。従って、この状況下でも、式IIIのQ1〜Q6は好ましくは窒素保護基である。窒素保護基及びその好ましい具体例は上記の第二の実施形態で記載した通りである。
【0069】
式IIIの化合物は以下のスキーム1及び2に記載したように調製できる。スキーム1はカルボキシ官能化N保護テトラアミンキレーターの順応性に富む合成経路を与え、式IIIのnの様々な値に適応させることができる。−(CH2)5OH橋頭置換基をもつBoc保護テトラアミン類似体の合成は、Turpin et al, J. Lab. Comp. Radiopharm.,45,379−393(2002)に記載されている。スキーム2はアミン官能化N保護テトラアミンキレーターの順応性に富む合成経路を与え、nの様々な値に適応させることができる。生体ターゲティングペプチドの結合は、Nock et al, Eur. J. Nucl. Med.,30(2),247−258(2003)及びMaina et al, Eur. J. Nucl. Med.,30(9),1211−1219(2003)に記載のものと同様に実施できる。
【0070】
【化8】
【0071】
【化9】
【実施例】
【0072】
本発明を以下の非限定的な実施例で例証する。実施例1では、本発明のカルボキシ官能化N保護テトラアミンキレーター(化合物1)の合成について記載する。実施例2では、本発明のアミン官能化N保護テトラアミンキレーター(化合物2)の合成について記載する。実施例3では、化合物1とアミン(ベンジルアミン)との結合を示す化合物(化合物3)の合成について記載する。実施例4では、化合物6の合成について記載し、化合物2とカルボン酸の活性エステルとの結合を例証する。実施例5では、本発明のキレーターとロサルタン誘導体とのコンジュゲート(化合物4)の合成について記載する。実施例6では、PEGリンカー基を含むロサルタンキレーターコンジュゲートの合成について記載する。実施例7では、本発明のキレーターのアンジオテンシンペプチドコンジュゲート(化合物8)の合成について説明する。実施例8では、本発明の幾つかのキレーターの99mTc放射標識化について記載する。実施例9では、本発明の様々な99mTc錯体の親油性(logP)を測定し、これらの錯体が比較的親水性であることを示す。実施例10では、低い肝臓バックグラウンドと高い尿クリアランスを示す本発明の幾つかの99mTc錯体の生体内分布結果を示す。実施例11では、化合物1の高収率の合成について記載する。実施例12では、活性エステルの結合した本発明の保護テトラアミンキレーター(化合物9)の合成について記載する。
【0073】
実施例1:化合物1の合成
段階(a):ジエチル[2−(ベンジルオキシ)エチル]マロネート
標記化合物は、Ramalingam et al, Tetrahedron,51,2875−2894(1995)の方法の修正法で調製した。ナトリウム(1.20g)をアルゴン雰囲気中で無水エタノール(25mL)に溶解した。マロン酸ジエチル(14.00g)を加えて混合物を30分間還流した。ベンジルブロモエチルエーテル(10g)を加えて、混合物を16時間還流下で撹拌した。エタノールをロータリーエバポレーターで除去して、残渣をエーテル(100mL)と水(50mL)とに分配した。エーテル層を水(3×50mL)で洗浄して硫酸ナトリウムで乾燥した。エバポレーターでエーテルを除去して、残渣を真空蒸留した。40〜55℃で留出した留分を廃棄した(未反応マロン酸ジエチル)。生成物は140〜150℃(1mm)で留出した(文献値bp138〜140℃(1mm))。収量は無色油12.60gであった。
【0074】
1H NMR(270MHz,CDCl3,25℃,TMS):δ=7.28(m,5H,C6H5),4.47(s,2H,CH2−Ph),4.16(m,4H,COOCH2),3.58(t,1H,CH),3.50(t,2H,O−CH2−CH2),2.21(t,2H,O−CH2−CH2),1.20(t,6H,COOCH2−CH3)。
【0075】
13C NMR(67.5MHz,CDCl3,25℃,TMS):δ=169.20(CO),138.10,128.60,127.80(芳香族),73.00(CH2Ph),67.30(O−CH2−CH2),61.70(COOCH2),49.10(CH),28.90(O−CH2−CH2),14.10(COOCH2CH3)。
【0076】
段階(b):N,N′−ビス(2−アミノエチル)−2−(2−ベンジルオキシ−エチル)マロンアミド
ジエチル[2−(ベンジルオキシ)エチル]マロネート(4.00g)をエチレンジアミン(30mL)に加えて、溶液を室温で2日間撹拌した。過剰のエチレンジアミンをロータリーエバポレーターで除去して、残渣を高真空下で2日間乾燥して黄色油状物(4.28g)を得、これを放置して結晶化させた。生成物は痕跡量のエチレンジアミンを依然として含んでいることがNMRスペクトルで検出された。
【0077】
1H NMR(270MHz,CDCl3,25℃,TMS):δ=7.74(br t,2H,CO−NH),7.32(m,5H,C6H5),4.46(s,2H,CH2−Ph),3.50(t,2H,OCH2−CH2− ),3.33(t 1H,CH),3.23(m,4H,CO−NH−CH2),2.74(t,4H,CH2−NH2)2.18(q,2H,O−CH2−CH2−)1.55(br s 4H,NH2)。
【0078】
13C NMR(67.5MHz,CDCl3,25℃,TMS):δ=171.10(CO),138.20,128.30,127.70(芳香族),73.00(CH2−Ph),67.80(O−CH2−CH2),51.40(CH),42.40(CO−NH−CH2),41.20(CH2−NH2),31.90(O−CH2−CH2−)。
【0079】
段階(c):N,N′−ビス(2−アミノエチル)−2−(2−ベンジルオキシエチル)−1,3−ジアミノプロパン
N,N′−ビス−(2−アミノエチル)−2−(2−ベンジルオキシエチル)マロンアミド(3.80g)をTHF(20mL)に溶解して、フラスコを氷浴に浸漬した。フラスコをアルゴンでフラッシュして、THFボラン錯体(80mL、THF中1M)をシリンジで加えた。反応混合物を室温まで放温し、次いで40℃で2日間撹拌し、1時間還流した。メタノール(50mL)を滴下し、溶液を40℃で1晩撹拌した。溶媒をロータリーエバポレーターで除去して、残渣をメタノール(20mL)に溶解した。水酸化ナトリウム(15mLの水中に10g)を加えて、メタノールを留去した。無色油状物が分離し、これをCH2Cl2(3×50mL)で抽出した。溶液をNa2SO4で乾燥した。溶媒の除去で3.40gの無色油状物が得られた。
【0080】
1H NMR(270MHz,CDCl3,25℃,TMS):δ=7.34(m,5H,C6H5),4.49(s,2H,CH2−Ph),3.55(t,2H,OCH2−CH2−),2.76(t,4H,N−CH2),2.63(m,8H,N−CH2),1.84(m,1H,CH),1.58(m,2H,CH−CH2−CH2−O),1.41(br s,6H,NH)。
【0081】
13C NMR(67.5MHz,CDCl3,25℃,TMS):δ=138.60,128.30,127.60(芳香族),72.80(CH2−Ph),68.70(O−CH2−CH2),53.50(N−CH2),52.80(N−CH2),41.60(N−CH2)36.40(CH),31.30(CH−CH2−CH2−O)。
【0082】
MS−EI:295[M+H]+(計算値295.2)。
【0083】
段階(d):N,N′−ビス(2−tert−ブトキシカルボニルアミノエチル)−2−(2−ベンジルオキシエチル)−1,3−ジ(tert−ブトキシカルボニルアミノ)プロパン
N,N′−ビス(2−アミニエチル)−2−(2−ベンジルオキシエチル)−1,3−ジアミノプロパン(3.30g)をCH2Cl2(100mL)に溶解して、トリエチルアミン(5.40g)及びtert−ブチルジカーボネート(10.30g)を加えた。反応混合物を室温で2日間撹拌した。混合物を水(100mL)、クエン酸溶液(水中10%100mL)及び水(2×100mL)で洗浄した。有機層をNa2SO4で乾燥して、溶媒をロータリーエバポレーターで除去すると、黄色油状物が得られ、これを高真空で恒量になるまで乾燥した。粗生成物(7.70g)をシリカゲルカラム(250g、230〜400メッシュ、CH2Cl2、CH2Cl2−Et2O1:1)で精製して、6.10g(78.3%)の透明油状物を得た。
【0084】
1H NMR(270MHz,CDCl3,25℃,TMS):δ=7.32(m,5H,C6H5),5.12(br d,2H,NH),4.47(s,2H,CH2−Ph),3.49(t,2H,OCH2−CH2−),3.24(br,12H,N−CH2),2.14(br,1H,CH),1.59(m,2H,CH−CH2−CH2−O)1.45(s,18H,t−Bu),1.42(s,18H,t−Bu)。
【0085】
13C NMR(67.5MHz,CDCl3,25℃,TMS):δ=155.90(NH−CO),138.20,128.30 127.60,127.50(芳香族),79.90,78.90(CMe3),72.80(CH2−Ph),68.00(O−CH2−CH2),50.00(br,N−CH2),46.90(br,N−CH2),39.20(N−CH2),34.40(br,CH),29.80(CH−CH2−CH2−O),28.30(t−Bu)。
【0086】
MS−EI:695[M+H]+(計算値695.5)
段階(e):N,N′−ビス(2−tert−ブトキシカルボニルアミノエチル)−2−(2−ヒドロキシエチル)−1,3−ジ(tert−ブトキシカルボニルアミノ)プロパン
N,N′−ビス(2−tert−ブトキシカルボニルアミノエチル)−2−(2−ベンジルオキシエチル)−1,3−ジ(tert−ブトキシカルボニルアミノ)プロパン(3.16g)を無水エタノール(100mL)に溶解して、Pd−活性炭(1.00g、乾燥、10%)を加えた。混合物をParr水素添加装置中35psiで2日間水素添加した。触媒を濾別し、エタノールで洗浄した(3×20mL)。エタノールをロータリーエバポレーターで除去すると無色油状物が得られ、これを高真空で恒量になるまで乾燥した(2.67g、97.1%)。
【0087】
1H NMR(270MHz,CDCl3,25℃,TMS):δ=5.25(br d,2H,NH),3.69(t,2H,OCH2−CH2−),3.28(br,12H,N−CH2),2.71(br,OH),2.23(br,1H,CH),1.56(ショルダー,m,2H,CH−CH2−CH2−O)1.48(s,18H,t−Bu),1.44(s,18H,t−Bu)。
【0088】
13C NMR(67.5MHz,CDCl3,25℃,TMS):δ=156.10(NHCO),80.00,79.20(CMe3),59.60(O−CH2−CH2),49.90(br,N−CH2),47.00(br,N−CH2),39.34(N−CH2),33.80(CH),32.30(CH−CH2−CH2−O),28.30(t−Bu)。
【0089】
MS−EI:605[M+H]+(計算値605.4)。
【0090】
段階(f):N,N′−ビス(2−tert−ブトキシカルボニルアミノエチル)−2−(2−カルボキシメチル)−1,3−ジ(tert−ブトキシカルボニルアミノ)プロパン(化合物1)
Mazitschek et al(Ang.Chem.INT.Ed.,41,4059−4061(2002))の方法を用いた。N,N′−ビス(2−tert−ブトキシカルボニルアミノエチル)−2−(2−ヒドロキシエチル)−1,3−ジ(tert−ブトキシカルボニルアミノ)プロパン(2.60g)をDMSO(15mL)に溶解して、1−ヒドロキシ−1,2−ベンズヨードオキソール−3(H)−オン−オキシド(IBX、3.50g)を加えた。混合物を室温で1時間撹拌してから、N−ヒドロキシスクシンイミド(2.50g)を加えた。反応混合物を室温で2日間撹拌した。水酸化ナトリウム溶液(2M、40mL)を加えて、混合物を室温で4時間撹拌した。溶液を氷浴に浸漬して2M塩酸でpH2に酸性化した。水層をエーテルで抽出して(4×100mL)、一緒にしたエーテル抽出液を水で洗浄した(3×50mL)。エーテル層をNa2SO4で乾燥して、溶媒をロータリーエバポレーターで除去すると生成物及び2−ヨードソ安息香酸を含む黄色固体残渣が得られた。クロロホルム−ヘキサン(1:3)(80mL)での結晶化によってヨードソ安息香酸(2.1g)の大部分を除去した。クロロホルム−ヘキサン母液を蒸発させると黄色油状物(3g)が得られ、これをシリカゲルカラム(300g、CH2Cl2−Et2O、1:1)にかけた。残留ヨードソ安息香酸はエーテルで溶出した。生成物はエーテル−メタノール(9:1)で溶出した。生成物を含む画分を一緒にし、溶媒を除去すると1.5gの淡黄色油状物が得られた。これを再びシリカゲルカラムでクロマトグラフィー処理した(50g、Et2O)。生成物はエーテル−酢酸(95:5)で溶出した。生成物を含む画分を一緒にして溶媒をロータリーエバポレーターで除去すると、油状物が得られ、これを高真空で乾燥した。収量は1.10g(41.3%)であった。
【0091】
1H NMR(270MHz,CDCl3,25℃,TMS):δ=7.61(br s,1H,COOH),5.19(br d,2H,NH),3.22(br,12H,N−CH2),2.47(br m,1H,CH),2.26(br,2H,CH−CH2−COOH),1.41(s,18H,t−Bu),1.37(s,18H,t−Bu)。
【0092】
13C NMR(67.5MHz,CDCl3,25℃,TMS):δ=175.90(COOH),156.10(NHCO),80.40,79.10(CMe3),49.50(N−CH2),46.80(N−CH2),39.00(N−CH2),34.70(CH−CH2−COOH),34.20(CH−CH2−COOH),28.30,28.20(t−Bu)。
【0093】
MS−EI:619[M+H]+(計算値619.4)。
【0094】
実施例2:(8−アミノ−2−{[tert−ブトキシカルボニル−(2−tert−カルボニルアミノエチル)−アミノ]−メチル}−オクチル)−(2−tert−ブチルオキシカルボニルアミノエチル)−カルバミン酸tert−ブチルエステル(化合物2)の合成
段階(a):2−(6−クロロ−ヘキシルオキシ)テトラヒドロピラン
6−クロロヘキサノール(6.85g、10mmol)及びp−トルエンスルホン酸(500mg)を乾燥エーテル(75ml)に溶解して氷浴で0〜5℃に冷却した。乾燥エーテル(25ml)中のジヒドロピラン(4.3g、10mmol)を30分かけて定常攪拌下に滴下した。添加完了後、冷却浴を除いて撹拌を16時間継続した。溶液を水で抽出し(50ml×2)、乾燥し(MgSO4)、濾過して、溶媒を減圧蒸発させると、淡黄色油状物が残った。油状物はそれ以上精製せずに次の反応に使用できる十分な純度のものであることが13CNMR分光法で判明した。収量10.1g(91%)。
【0095】
13C NMR(CDCl3):δ19.7(CH2),25.5(CH2),25.6(CH2),26.7(CH2),29.6(CH2),30.8(CH2),32.6(CH2),45.0(CH2Cl),62.3(OCH2),67.4(OCH2),98.8(OCHO)。
【0096】
1H NMR(CDCl3):δ1.30〜1.71(14H,m,CH2×7),3.24〜3.32(1H,3.41〜3.48(3H,m,CH及びCH2),3.60〜3.67(1H,m,CH),3.72〜3.82(1H,bm,CH),4.44〜4.49(1H,bm,OCHO)。
【0097】
段階(b):2−[6−(テトラヒドロピラン−2−イルオキシ)−ヘキシル]−マロン酸ジエチルエステル
少量のナトリウム(1.13g、49mmol)を乾燥窒素ブランケット中定常攪拌下に乾燥エタノール(100ml)に溶解した。マロン酸ジエチル(8.0g、50mmol)を一度に加え、溶液を60℃で1時間加熱した。2−(6−クロロ−ヘキシルオキシ)−テトラヒドロピラン(9.3g、42.2mmol)を一度に加えて温度を75〜80℃に上げて24時間このレベルを保った。冷却後、無機固体を濾別して濾液から溶媒を蒸発させた。残渣をCH2Cl2(50ml)に溶解し、水で抽出し(30ml×2)、乾燥し(MgSO4)、濾過して、揮発分を除去すると淡黄色油状物が残った。油状物をシリカゲルクロマトグラフィーにかけ、溶出液として石油エーテル(40〜60℃)/エーテル(200:40)を用いた。所要生成物はrf=0.15で無色油状物として溶出した。収量8.7g(60%)。
【0098】
13C NMR(CDCl3):δ14.0(CH3×2)、19.6(CH2)、25.5(CH2)、27.2(CH2)、28.6(CH2)、29.0(CH2)、29.6((CH2)、30.0(CH2)、30.8(CH2)、52.0(CH)、61.2(OCH2×2)、62.2(OCH2)、67.4(OCH2)、98.8(OCHO)、169.4(C=O×2)。
【0099】
1H NMR(CDCl3):δ1.10〜1.25(14H,m,CH3×2,CH2×4)、1.36〜1.50(6H,bm,CH2×3)、1.70〜1.81(2H,bm,CH2)、3.17〜3.28(2H,m,CH2)、3.56〜3.66(1H,m,CH)、3.70〜3.80(1H,m,OCH)、4.04〜4.16(4H,m,OCH2×2)、4.03〜4.08(1H,m,OCHO)。
【0100】
段階(c):N,N′−ビス−(2−アミノエチル)−2−[6−(テトラヒドロピラン−2−イルオキシ)ヘキシル−]−マロンアミド
2−[6−(テトラヒドロピラン−2−イルオキシ)−ヘキシル]−マロン酸ジエチルエステル(5.1g、14.8 mmol)を1,2−ジアミノエタン(10g、167mmol)に溶解して、室温で16時間撹拌した。揮発分を真空で除去すると(40〜50℃、0.01mmHg)、淡緑色の粘稠な残渣が残り、これをカラムクロマトグラフィーにかけてCH2Cl2/MeOH/NH4OH(50:50:5)で溶出した。標記化合物はrf0.2で溶出し、淡緑色の粘稠な油状物として回収され、放置すると固化した。(収量3.9g、71%)。
【0101】
13C NMR(CDCl3):δ19.8(CH2),25.5(CH2),26.0(CH2),27.5(CH2),29.2(CH2),29.7(CH2),30.8(CH2),31.9(CH2),41.0(NCH2×2),41.9(NCH2×2),54.6(CH),62.5(OCH2),67.5(OCH2),98.9(OCHO),171.6(C=O×2)。
【0102】
1H NMR(CDCl3):δ1.15〜1.28(6H,bs,CH2×3),1.39〜1.44(6H,bm,CH2×3),1.69〜1.74(4H,bm,CH2×2),2.64(4H,bs,NH2×2),2.73 4H,t,J=6Hz,CH2×2),3.08〜3.29(6H,m,CH2×3),3.35〜3.41(1H,m CH),3.55〜3.63(1H,m,CH),3.70〜3.78(1H,m,CH),4.43(1H,bt,J=4Hz,OCHO),7.78(2H,bt,J=5Hz,OCNH×2)。
【0103】
IR(薄膜)cm−1:3417,3082,2936,2862,1663,1558,1439,1354,1323,1261,1200,1189,1076,1026,956,907,867,810。
【0104】
段階(d):N,N′−ビス(2−アミノエチル)−2−(6−ヒドロキシヘキシル)−マロンアミド
N,N′−ビス(2−アミノエチル)−2−[6−(テトラヒドロピラン−2−イルオキシ)−ヘキシル]−マロンアミド(3.9g、10.6mmol)、p−トルエンスルホン酸一水和物(8.5g、3mmol)及びエタノール(50ml)を70〜75℃で還流下で16時間加熱した。冷却後、濃水酸化アンモニウム(.880)を、pH9が持続して得られるまで滴下した。沈殿した白色固体をセライトで除去し、濾過ケークをエタノール(30ml)で洗浄した。エタノールを減圧で(15mmHg、40℃)除去すると、半固形状ワックスが残った。残渣をシリカゲルクロマトグラフィーにかけてCH2Cl2/MeOH/NH4OH(100:50:10)で溶出した。標記化合物のrf=0.2であった。生成物を回収して、残存する水を除去するためエタノールと共沸させた(100ml×3)。淡緑色の粘稠な残渣が得られ、放置すると固化した。(収量2.1g、69%)。
【0105】
13C NMR(CD3OD):δ25.4(CH2),27.3(CH2),28.9(CH2),30.4(CH2),32.2(CH2),40.6(NCH2×2),41.7(NCH2×2),54.1(CH),61.6(CH2OH),171.7(C=O×2)。
【0106】
1H NMR(CD3OD):δ1.28〜1.38(6H,bs,CH2×3),1.46〜1.55(2H,bm,CH2),1.79〜1.87(2H,bm,CH2),2.73(4H,t,J=6Hz,H2NCH2×2),3.13(1H,t,J=7Hz,CH),3.27(4H,dt,J=6及び2Hz,HNCH2×2),3.53(2H,t,J=7Hz,OCH2)。
【0107】
IR(薄膜)cm−1:3364,2932,2862,2527,1663,1558,1462,1327,1223,1192,1034。
【0108】
質量分析(FAB)m/e:C13H29N4O3(M+H)計算値289,実測値289。
【0109】
段階(e):(2−tert−ブトキシカルボニルアミノエチル−2−{[tert−ブトキシカルボニル−(2−tert−ブトキシカルボニルアミノエチル)−アミノ]−メチル}−8−ヒドロキシオクチル)−カルボン酸tert−ブチルエステル
乾燥窒素ブランケット中、無溶媒ボラン−ジメチルスルフィド付加物(15ml、150mmol)をジオキサン(50ml)中N,N′−ビス−(2−アミノエチル)−2−(6−ヒドロキシエチル)マロンアミド(2.1g、7.3mmol)の攪拌混合物にシリンジで滴下した。添加完了後、混合物を110℃で穏やかに還流しながら5日間加熱した。その際若干の白色固体が残った。冷却後揮発分を減圧除去すると白色固体が残り、これにメタノール(50ml)を滴下すると無色溶液が得られた。溶液を還流下で3時間加熱し、冷却し、濃HCl(5ml)を加えて70〜75℃で還流下で48時間加熱を続けた。溶媒を除去すると粘稠な緑色残渣が残り、これをメタノールで共沸すると(100ml×3)、淡緑色固体が残った。この固体を乾燥メタノールに再溶解して無水炭酸カルシウム(4.0g、30mmol)、次いでジ−tert−ブチルジカーボネート(7.0g、32mmol)を加えた。混合物を室温で48時間撹拌した。無機固体をセライトで濾別し、濾液から溶媒を蒸発させると粘稠な残渣が残った。残渣を水(50ml)と混合し、CH2Cl2で抽出した(50ml×3)。有機画分を一緒にし、乾燥し(MgSO4)、濾過して、溶媒を蒸発させると淡黄色残渣が残った。
【0110】
注:この時点で13CNMRで反応をモニターすると便利である。
【0111】
残渣をシリカゲルクロマトグラフィーにかけ、CH2CL2/MeOH(95:5)を溶出液として用いた。標記化合物はrf=0.41で溶出し、無色の粘稠油状物として分離された(収量2.5g、52%)。
【0112】
13C NMR(CDCl3):δ25.6(CH2),26.4(CH2),28.4(CH3×12),29.8(CH2×2),32.6(CH2),36.5(非常にブロード,CH),39.2(NCH2×2,隣接CH),46.9(ブロード,s,HNCH2×2),50.0(ブロード,s,NCH2×2),62.4(HOCH2),79.0(OC×2),79.9(OC×2),156.4(ブロード,s,C=O×4)
1H NMR(CDCl3):δ1.05〜1.18(8H,bs,CH2×4),1.27(18h,s,CH3×6,t−ブチル),1.31(18H,s,CH3×6,t−ブチル),1.41(2H,m,CH2),1.81(1H,bs,CH),2.63(1H,bs,OH),2.98(4H,bs,NCH2×2),3.11(8H,bs,NCH2×4),3.44(2H,t,J=8Hz,CH2O),5.2(2H,bs,NH×2)。
【0113】
IR(薄膜)cm−1:3350,2976,2931,2859,1674,1516,1455,1418,1393,1260,1250,1165,1069,965,871,775。
【0114】
質量分析(FAB)m/e:C33H65N4O9(M+H)計算値661,実測値661。
【0115】
段階(f):トルエン−4−スルホン酸8−[tert−ブトキシカルボニル−(2−tert−ブトキシカルボニルアミノエチル)−アミノ]−7−{[tert−ブトキシカルボニル−(2−tert−ブトキシカルボニルアミノエチル)−アミノ]−メチル}−オクチルエステル
(2−tert−ブトキシカルボニルアミノエチル−2−{[tert−ブトキシカルボニル−(2−tert−ブトキシカルボニルアミノエチル)アミノ]−メチル}−8−ヒドロキシオクチル)−カルボン酸tert−ブチルエステル(2.52g、3.82mmol)、p−トルエンスルホニルクロリド(1.0g、5.2mmol)、トリエチルアミン(1.3g、12.8mmol)及びCH2Cl2(30ml)を室温で撹拌して、溶媒をゆっくりと蒸発させた。反応を炭素NMRでモニターすると3日後に出発物質は少ししか残存していなかった。反応液容積をCH2Cl2で30mlに一緒にし、水で抽出して(50ml×3)、乾燥し(MgSO4)、濾過して溶媒を蒸発させると、褐色残渣が残った。残渣をシリカゲルクロマトグラフィーにかけ、CH2Cl2/MeOH(100:5)を溶出液として用いた。溶出した最初の化合物はrf=0.95の未反応塩化トシルであった。標記化合物はrf=0.2で溶出し、淡黄色の粘稠な液体として分離された。収量(1.20g、39%)。
【0116】
13C NMR(CDCl3):δ21.7(CH3トシル),25.3(CH2),26.3(CH2),28.5(CH3×12),28.8(CH2),29.5(CH2),29.9(CH2),36.5(CH,非常にブロード),39.4(NCH2×2),47.0(ブロード NCH2×2),50.5(ブロード,NCH2×2),70.6(TsOCH2),79.1(OC×2),80.0(OC×2),127.9(CH×2),129.9(CH×2),133.2(C),144.7(C−S Ts),156.1(ブロード,C=O×4)。
【0117】
1H NMR(CDCl3):δ1.16(8H,bs,CH2×4),1.35(18H,s,CH3×6),1.39(18H,s,CH3×6),1.88(1H,bs,CH),2.38(3H,s,CH3トシル),3.10〜3.12(4H,bs,NCH2×2),3.19(8H,bs,NCH2×4),3.93(2H,t,J=7Hz,CH2OTs),5.0(1H,bs,NH),5.08(1H,bs,NH),7.29(2H,d,J=8Hz,CH×2,Ar),7.72(2H,d,J=8Hz,CH×2,Ar)。
【0118】
IR(薄膜)cm−1:3360,2974,2932,2862,1693,1516,1479,1418,1391,1366,1250,1177,1069,959,816,775。
【0119】
質量分析(FAB)m/e:C40H71N4O11S(M+H)計算値815,実測値815。
【0120】
段階(g):(8−アジド−2−{[tert−ブトキシカルボニル−(2−tert−カルボニルアミノエチル)−アミノ]−メチル}−オクチル)−(2−tert−ブチルオキシカルボニルアミノエチル)−カルバミン酸tert−ブチルエステル
トルエン−4−スルホン酸8−[tert−ブトキシカルボニル−(2−tert−ブトキシカルボニルアミノエチル)アミノ]−7−{[tert−ブトキシカルボニル−(2−tert−ブトキシカルボニルアミノエチル)アミノ]メチル}−オクチルエステル(1.105g、1.36mmol)、ナトリウムアジド(350mg、5.4mmol)及びメタノール(10ml)を70〜75℃で16時間加熱還流した。冷却後、室温で約1〜2ml残るまでメタノールを減圧除去した。残渣を水(25ml)で希釈してCH2Cl2(25ml×4)で抽出した。有機抽出液を一緒にして乾燥し(MgSO4)、濾過して揮発分を室温で蒸発させると(注:アジドは爆発の危険性があるので、この段階は安全シールドの後ろで行うべきである)、淡黄色の粘稠な残渣が残ったが、これが純粋な所望化合物であった(収量820mg、88%)。
【0121】
13C NMR(CDCl3):δ26.3(CH2),26.5(CH2),28.3(CH3×12),28.7(CH2),29.6(CH2),29.8(CH2),36.8(ブロード,CH),39.3(NCH2×2),46.9(ブロード,NCH2×2),50.0(ブロード,NCH2×2),51.3(CH2N3),79.0(OC×2),79.8(OC×2),156.0(C=O×4)。
【0122】
1H NMR(CDCl3):δ1.16(8H,bs,CH2×4),1.29(18H,s,CH3×6),1.33(18H,s,CH3×6),1.47(2H,bt,J=6.5Hz,CH2隣接CH),1.86(1H,bs,CH),2.95〜3.05(4H,bs,NCH2×2),3.05〜3.20(10H,bs,NCH2×4及びCH2N3),5.09(2H,bs,NH×2)。
【0123】
IR(薄膜)cm−1:3350,2974,2932,2860,2097(Strong band N3),1694,1520,1470,1418,1391,1366,1250,1167,1069,870,777。
【0124】
段階(h):(8−アミノ−2−{[tert−ブトキシカルボニル−(2−tert−カルボニルアミノエチル)−アミノ]−メチル}−オクチル)−(2−tert−ブチルオキシカルボニルアミノエチル)−カルバミン酸tert−ブチルエステル(化合物2)
(8−アジド−2−{[tert−ブトキシカルボニル−(2−tert−カルボニルアミノエチル)−アミノ]−メチル}−オクチル)−(2−tert−ブチルオキシカルボニルアミノエチル)−カルバミン酸tert−ブチルエステル(820mg、1.20mmol)、10%パラジウム−活性炭(100mg)及びメタノール(10ml)を室温30気圧の圧力下、水素ガスで16時間処理した。セライトで濾過して固体を除去し、濾過ケークをメタノール(50ml)で洗浄した。濾液から揮発分を除去すると粘稠油状物が残ったが、これが純粋な所望化合物であった(収量700mg、89%)。
【0125】
13C NMR(CDCl3):δ26.4(CH2),26.6(CH2),28.4(CH3×12),32.9(CH2×2),36.8(ブロード,CH). 39.2(NCH2×2),41.8(H2NCH2),46.9(ブロード,NCH2×2),49.8(ブロード,NCH2×2),78.9(OC×2),79.7(OC×2),156.0(C=O×4)。
【0126】
1H NMR(CDCl3):δ1.08(8H,bs,CH2×4),1.23(18H,s,CH3×6),1.27(20H,bs,CH3×6及びCH2),1.77(1H,bs,CH),2.40(2H,bs,NH2),2.50(2H,t,J=7Hz,CH2NH2),2.97(4H,bm,NCH2×2),3.00〜3.16(8H,bm,NCH2×4),5.21(1H,bs,NH),5.30(1H,bs,NH)。
【0127】
IR(薄膜)cm−1:3360,1693,1520,1459,1418,1392,1367,1250,1170,1068,964,922,871,775,733。
【0128】
質量分析(FAB)m/e:C33H66N5O8(M+H)計算値660,実測値660。
【0129】
実施例3:化合物3の合成
段階(a):化合物1とベンジルアミンとのカップリング
20℃の丸底フラスコ(50ml)内で、CH2Cl2(5ml)中の化合物1(61.8mg、0.1mmol)をベンジルアミン(10.7mmol)、ジフェニルホスフィン酸クロリド(25.9mg)及びジイソプロピルアミン(29mg、0.22mmol)で18時間処理した。次に反応液をCH2Cl2(20ml)で希釈し、1N塩酸(5ml)及び飽和炭酸カリウム水溶液(5ml)で洗浄した。CH2Cl2層を分離乾燥し(Na2SO4)、ガム状になるまで減圧濃縮した(〜50mg)。次にこの粗生成物を石油中の酢酸エチルの勾配(50%、70%及び90%各100ml)でシリカクロマトグラフィーにかけた。少量の移動度の速い不純物と、そのすぐ後で主画分を回収した。
【0130】
CDCl3中で1H及び13C NMRスペクトルを測定した。その結果、主画分が所要純粋化合物であることが判明した。
【0131】
段階(b):Boc保護基の脱保護
段階(a)で得られたCH2Cl2(0.5ml)中の生成物(27.8mg、0.039mmol)をトリフルオロ酢酸(0.5ml)で処理して、反応液を室温で3時間放置した。次いで反応混合物をガム状になるまで減圧濃縮して過剰の酸を除去し、秤量した(53mg)。1H及び13C NMR(CDCl3)からBoc基が完全に除去されたことが判明した。化合物2の秤量試料を水に溶解してTFA塩の10ミリモル濃度溶液を得て、これを放射標識実験に用いた。
【0132】
実施例4:化合物6の合成
段階(a):(8−ベンゾイルアミノ−2−{[tert−ブトキシカルボニル−(2−tert−カルボニルアミノエチル)−アミノ]−メチル}−オクチル)−(2−tert−ブチルオキシカルボニルアミノエチル)−カルバミン酸tert−ブチルエステル
乾燥CH2Cl2中の安息香酸2,5−ジオキソ−ピロリジン−1−イルエステル(20mg、0.091mmol)を、CH2Cl2(1ml)中の化合物2(50mg、0.08mmol)の溶液に一度に加え、溶液を室温で16時間撹拌した。反応液をCH2Cl2(10ml)で希釈し、水(15ml×2)で抽出し、乾燥(MgSO4)し、濾過し、溶媒をロータリーエバポラーターで除去した。残った残渣をCH2Cl2/MeOH94:6(rf=0.23)を溶出液として用いるシリカゲルクロマトグラフィーで精製し、無色の粘稠油状物を得た。(収量25mg、41%)。
【0133】
13C NMR(CDCl3):δ26.4(CH2),26.8(CH2),28.5(CH3×12),29.6(CH2×2),29.7(CH2),29.9(CH2),36.6(ブロード,CH),39.4(NCH2×2),40.0(O=CNCH2×2),47.0(ブロード,NCH2×2),49.8(NCH2×2),79.7(OC×2),80.0(OC×2),127.0(ArCH×2),128.5(ArCH×2),131.3 (ArCH),134.9(ArC),156.1(C=O×4),167.6(ArC=O)。
【0134】
1H NMR(CDCl3):δ1.28(8H,bs,CH2×4),1.38(18H,s,CH3×6),1.42(20H,bs,CH3×6及びCH2),1.95(1H,bs,CH),3.1(4H,bs,NCH2×2)3.22(8H,bs,NCH2×4),3.42(2H,bq,J=6Hz,CH2N−ベンゾイル),5.08(2H,bs,NH×2),6.18(1H,bs,HN−ベンゾイル),7.38〜7.45(3H,m,ArCH×3),7.74(2H,bd,J=7Hz,ArCH×2),
IR(薄膜)cm−1:3350,2976,2932,2859,1693(ブロード),1652,1520,1419,1391,1367,1251,1166,732。
【0135】
質量分析(FAB)m/e:C40H70N509(M+H)の計算値764,実測値764。
【0136】
段階(b):Boc保護基の脱保護
段階(a)で得たBocテトラアミンベンズアミド(42mg、0.056mmol)をCH2Cl2(0.5ml)中でトリフルオロ酢酸(0.5ml)で処理して、反応液を室温で3時間放置した。次に反応液を減圧濃縮して過剰の酸を除去し、ガム状物を得た。重量予測値(45mg)、重量実測値(45.7mg)。1H及び13C NMR(CD3OD)から、Boc基が完全に除去されたこと並びに上記ガムが所要化合物を含むことが判明した。化合物の秤量試料を水に溶解してTFA塩の10ミリモル濃度溶液(5.6ml中56μmol)を得て、これを放射標識に用いた。
【0137】
実施例5:化合物4の合成
反応はすべて窒素吹込式装置中で手作業で実施した。
【0138】
段階(a):トリチル誘導体化固体担体へのロサルタンの結合
【0139】
【化10】
【0140】
ロサルタン(MSD,0.236g、0.558 mmol)及びトリエチルアミン(Fluka社製、0.233ml、1.67mmol)を、DMF(5ml)中塩化トリチル樹脂(Novabiochem社製、置換量1.24mmol/g、0.300g)の懸濁液に加えた。4日後樹脂を水抜きして洗浄した。樹脂のアリコートを開裂させた(ジクロロメタン/TFA/トリイソプロピルシラン、92.5:5.0:2.5、15分)。HPLC分析で(カラム Phenomenex Luna C18(2)、3μm、4.6×50mm、溶媒:A=水/0.1%TFA及びB=アセトニトリル/0.1%TFA;勾配10分間で10〜40%B;流速2.0ml/分、214及び254nmでUV検出)、ロサルタンに相当するtR6.7分のピークが得られた。樹脂をジクロロメタン/メタノール/ジイソプロピルエチルアミン溶液(17:2:1、20ml、1時間)で処理し、ジクロロメタンで洗浄して乾燥した。
【0141】
段階(b):ヒドロキシル基のアジドでの置換
【0142】
【化11】
【0143】
ジフェニルホスホリルアジド(Aldrich、0.481ml、2.23mmol)及びDBU(0.611ml、4.09mmol)を、THF中(10ml)中で、段階(a)で得た樹脂に結合したロサルタン(0.372mmol)の懸濁液に加えた。この反応液を1晩放置した。樹脂のアリコートを段階(a)に記載の通り開裂させた。LC−MS分析(カラムPhenomenex Luna C18(2)、3μm、50×4.60mm、溶媒:A=水/0.1%TFA及びB=アセトニトリル/0.1%TFA;勾配10分間で20〜80%B;流速1ml/分、214nmでUV検出、ESI−MS)で、tR7.3分に上記の構造に相当するm/z448.1(MH+)を有するピークが得られた。
【0144】
段階(c):アジド基のアミンへの還元
【0145】
【化12】
【0146】
段階(b)からの樹脂のTHF中の懸濁液(4ml)に、塩化スズ(II)(Acros、0.141g、0.744mmol)、チオフェノール(Fluka社製、0.304ml、2.976mmol)及びトリエチルアミン(Fluka社製、0.311ml、2.23mmol)を加えた。1.5時間後に樹脂のアリコートをa)に記載の通り開裂させた。LC−MS分析(カラムPhenomenex Luna C18(2)、3μm、50×4.60mm、溶媒:A=水/0.1%TFA及びB=アセトニトリル/0.1%TFA;勾配10分間で20〜80%B;流速1ml/分、214nmでUV検出、ESI−MS)で、アミンで予想されるm/z422.2(MH+)を有するピークが1.9分に得られた。
【0147】
段階(d):ロサルタンロイシン−テトラアミンキレーター(化合物4)
Fmoc−Leu−OH(Novabiochem社製、0.030g、0.084mmol)をDMF中で段階(c)からの樹脂結合アミノロサルタンの一部(0.042mmol)に、標準的なカップリング剤(HATU及びDIEA)及び標準的なFmoc切断プロトコル(DMF中20%ピペリジン)を用いてカップリングした。カップリングの完結は標準的Kaizer試験で確認した。次いでDMF中で同じカップリング剤(HATU及びDIEA)を用いて樹脂に化合物1(0.026g、0.042mmol)をカップリングした。4時間後に生成物を樹脂から切断してBoc基を同じ方法(ジクロロメタン/TFA/トリイソプロピルシラン、47.5:50:2.5溶液で1時間)で除去した。溶液を濾過、濃縮して分取用HPLC(カラムPhenomenex Luna C18(2)、5μm、21.2×250mm、溶媒:A=水/0.1%TFA及びB=アセトニトリル/0.1%TFA;勾配60分間で20〜40%B;流速10.0ml/分、214nmでUV検出)で精製して、凍結乾燥後5mgの生成物を得た。LC−MS分析(カラムPhenomenex Luna C18(2)、3μm、50×4.60mm、溶媒:A=水/0.1%TFA及びB=アセトニトリル/0.1%TFA;勾配10分間で10〜80%B;流速0.3ml/分、214nm及び254nmでUV検出、ESI−MS)tR5.1分、m/z735.4(MH+)で構造を確認した。
【0148】
実施例6:化合物5の合成
標記化合物は実施例4に記載の通り固体担体上で合成した。Fmoc−Leu−OH(Novabiochem社製、0.033g、0.092mmol)及びFmoc−アミノPEGジグリコール酸(Polypure、0.049mg、0.092mmol)を、標準的カップリング剤(HATU及びDIEA)及び標準的Fmoc切断プロトコル(DMF中20%ピペリジン)を用いて、DMF中で、実施例4(c)で得た樹脂結合アミノ−ロサルタン(0.046mmol)に順次カップリングした。カップリングの完結は標準的Kaizer試験で確認した。次に樹脂に化合物1(0.029g、0.046mmol)をDMF中で同じカップリング剤(HATU及びDIEA)を用いてカップリングした。反応液を1晩放置して、次いで生成物を樹脂から切断してBoc基を同じ方法(ジクロロメタン/TFA/トリイソプロピルシラン、47.5:50:2.5溶液で1時間)で除去した。溶液を濾過、濃縮して分取用HPLC(カラムPhenomenex Luna C18(2)、5μm、21.2×250mm、溶媒:A=水/0.1%HCOOH及びB=アセトニトリル/0.1%HCOOH;勾配60分間で10〜40%B;流速10.0ml/分、214nmでUV検出)で精製して、凍結乾燥後3.5mgの生成物を得た。LC−MS分析(カラムPhenomenex Luna C18(2)、3μm、50×4.60mm、溶媒:A=水/0.1%HCOOH及びB=アセトニトリル/0.1%HCOOH;勾配10分間で10〜40%B;流速0.3ml/分、214nm及び254nmでUV検出、ESI−MS)tR4.7分;m/z1025.4(MH+)で構造を確認した。
【0149】
実施例7:化合物8の合成
段階(a):N−Boc−N−[FmocNH−CH2CH2]−Gly−OHの合成
1gのN−[FmocNH−CH2CH2]−Gly−OtBu・HCl(Fluka社製09660)を、0.5mLのトリイソプロピルシランを含むジクロロメタン中50%トリフルオロ酢酸(TFA)20mLで60分間処理した。反応混合物を減圧蒸発させて残渣を水中50%のテトラヒドロフラン20mLに再溶解した。2.6gのtert−ブチルオキシカルボニル無水物及び1.2mLのN−メチルモルホリンを加えて混合物を4日間撹拌した。次にテトラヒドロフランを減圧蒸発させて残渣をジクロロメタンに再溶解した。有機層を水で洗浄してMgSO4で乾燥した。ジクロロメタンを減圧蒸発させて残渣を5mLのジメチルホルムアミドに再溶解した。このジメチルホルムアミド溶液を水中60%のアセトニトリル400mLで希釈して、精製のため分取用HPLCカラム(40分間で30〜80%B、但しA=H2O/0.1%TFA及びB=CH3CN/0.1%TFA;Phenomenex Luna 10μ C18(2)250×50mmカラムに流速50mL/分)に送り、450mgの純粋生成物を得た。生成物は分析用HPLCで分析した(勾配、10分間で20〜70%B、但しA=H2O/0.1%TFA及びB=CH3CN/0.1%TFA;流速0.3mL/分;カラム、Phenomenex Luna 3μ C18(2)50×2mm;検出、UV214nm;生成物保持時間8.66分)。エレクトロスプレー質量分析法を用いて生成物をさらに特性決定した(MH+計算値441.2;MH+実測値440.8)。
【0150】
段階(b):N−((CH2)2−NHCOCH2−テトラアミン)−Gly−Arg−Val−Tyr−Ile−His−Pro−Ile−OH(化合物8)の合成
アンジオテンシンIIのペプチド類似体を、Applied Biosystems 433Aペプチド合成機を用いて、0.1mmolのFmoc−Ile−Wang樹脂を出発材料として合成した。アルギニンまでのカップリング段階では、過剰の1mmolプレ活性化アミノ酸(O−ベンゾトリアゾール−1−イル−N,N,N′,N′−テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)を使用)を用いた。123mgのN−Boc−N−[FmocNH−CH2CH2]−Gly−OH、114mgの[(ジメチルアミノ)−1H−1,2,3トリアゾロ−[4,5−b]ピリジン−1−イルメチレン]−N−メチルメタンアミニウムヘキサフルオロホスフェートN−オキシド(HATU)及び60μLのN−メチルモルホリンをジメチルホルムアミドに溶解して5分間撹拌し、次に窒素吹込み装置中の樹脂に加えた。試薬を2時間後に除去して樹脂をジメチルホルムアミド及びジクロロメタンで洗浄した。樹脂をジメチルホルムアミド中20%ピペリジンで処理し(3×10ml)、ジメチルホルムアミドで洗浄した。23mgの化合物1、14mgのHATU及び7.5μLのN−メチルモルホリンをジメチルホルムアミドに10分かけて溶解し、樹脂に加えた。試薬を4時間後に除去して樹脂をジメチルホルムアミド及びジクロロメタンで洗浄した。樹脂からの側鎖保護基の除去及び樹脂からのペプチドの切断は、2.5%のトリイソプロピルシラン及び2.5%の水を含むトリフルオロ酢酸10mL中で同時に90分間かけて実施した。トリフルオロ酢酸を真空で除去し、残渣にジエチルエーテルを加えて沈殿した生成物をジエチルエーテルで洗浄して風乾した。
【0151】
生成物を分取用RP−HPLC(40分間で0〜30%B、但しA=H2O/0.1%TFA及びB=CH3CN/0.1%TFA、Phenomenex Luna 10μ C18(2)250×21.20mmカラムで流速10mL/分)で精製して、32mgの純粋キレートペプチドコンジュゲートを得た。生成物を分析用HPLCで分析した(勾配、20分間で5〜50%B、但しA=H2O/0.1%TFA及びB=CH3CN/0.1%TFA;流速1mL/分;カラム、Phenomenex Luna 3μ C18(2)50×2mm;検出、UV214nm;生成物保持時間5.22分)。エレクトロスプレー質量分析法を用いて生成物をさらに特性決定した(MH+計算値1197.8;MH+実測値1197.8)。
【0152】
実施例8:化合物3〜7の99mTc放射標識
以下の成分を含む凍結乾燥キット(「Chelakit A plus」)を調製した。
【0153】
【表1】
【0154】
標識すべき化合物25〜50μg(溶媒25〜50μLに溶解)をCHELAKIT−A plusに加え、次いでジェネレータ溶出液(食塩水中99mTcO4−、1.0mL)を加えた。溶液を混合して室温で20〜30分間放置した。
【0155】
化合物3〜6は室温pH9でテクネチウム標識して、対応する陽イオン99mTc錯体を高収率で与える(RCP>90%)。テトラアミン錯体をHPLC(移動相:0.1%TFA含有水、0.1%TFA含有アセトニトリル;XTERRA RP18 3.5μm 4.6×150mmカラム)で精製したところ、50mMリン酸緩衝液中37℃で2時間安定である(2時間後HPLCによるRCP>95%)。
【0156】
実施例9:99mTc錯体の親油性(LogP)の測定
実施例8の99mTc錯体のオクタノール−水分配係数(LogP)を以下の通り測定した。
【0157】
実施例8で得たHPLC精製99mTc錯体10μLを、遠心チューブ内で1−オクタノール(2ml)及び50mMリン酸緩衝液(pH=7.4、2.0ml)と混合した。チューブを室温で1分間ボルテックスし、高速で60分遠心した。相間の汚染を避けるため注意しながら、両相の試料0.1mlをピペットで他のチューブに移し、Wallac Wizzardガンマカウンターで計数した。測定は3回繰り返した。
【0158】
分配計数Pは次のように計算した。
【0159】
P=(オクタノール中のcpm−バックグラウンドcpm)/(水中のcpm−バックグラウンドcpm)
最終分配係数は一般にlogPとして表される。結果を表1に示す。
【0160】
【表2】
【0161】
実施例10:99mTc錯体の生体内分布
化合物4、5及び7の99mTc錯体を実施例8に記載の通り調製した。実験は、正常オスWistarラット(180〜220g)で、試験標品の注射後(p.i.)所定の2時点(2及び120分)で実施した。動物をハロタン(酸素中6%)で麻酔し、0.1ml(500MBq/ml)の試験標品を注射し、犠牲死させて解剖し、試料の放射能をアッセイした。結果を表1に示す。
【0162】
【表3】
【0163】
実施例11:化合物1の別の調製法
実施例1の段階(e)で得たN,N′−ビス(2−tert−ブトキシカルボニルアミノエチル)−2−(2−ヒドロキシエチル)−1,3−ジ(tert−ブトキシカルボニルアミノ)プロパンを四塩化炭素(14ml)及びアセトニトリル(14ml)に溶解した。水(21ml)を加えると二相混合物になり、次いで過ヨウ素酸ナトリウム(4.5g、21mmol)及び塩化ルテニウム水和物(35mg、0.026mmol)を加えた。得られた暗褐色溶液を室温で1時間撹拌してから、CH2Cl2(40ml)で希釈した。有機層を分離し、水層をCH2Cl2(40ml×3)で抽出した。有機抽出液をすべて一緒にして乾燥(MgSO4)し、濾過して、揮発分を減圧蒸発させると、化合物1のナトリウム塩が暗色の粘稠残渣として残り、それ以上精製せずに使用した(4.15g、96%)。
【0164】
13C NMR(CDCl3):δC 28.2(×12)(CH3),34.1(CH2),34.4(CH),38.6(×2)(NCH2),46.8(×2)(NCH2),49.3(×2)(NCH2),79.0(×2)(OC),80.2(×2)(OC),155.9(×4)(C=O),175.4(COOH)。
【0165】
1H NMR(CDCl3):δH 1.29(18H,s,CH3×6),1.35(18H,s,CH3×6),2.19(1H,br,CH),2.40(2H,br,CH2),3.05〜3.23(12H,br,NCH2×6),5.10〜5.24(2H,br,NH×2)。
【0166】
質量分析(ESI)m/e:(M+Na)C29H54O10N4Na計算値641.3738,実測値641.3787。
【0167】
実施例12:化合物9の調製
1,3−ジシクロヘキシルカルボジイミド(DCC;2.16g、10.5mmol)を、乾燥THF(30ml)中の化合物1(4.15g、6.90mmol)及びN−ヒドロキシスクシンイミド(1.81g、15.7mmol)の攪拌溶液に1度に加えた。混合物を室温で16時間撹拌してから、沈殿したDCU(1,3−ジシクロヘキシル尿素)を濾別した。濾液から揮発分を蒸発させるとワックス状残渣が残り、これに乾燥エーテル(50ml)を加えるとさらにDCUが沈殿し、これを濾別した。エーテル溶液を水(25ml×2)で洗浄し、乾燥(MgSO4)、濾過して、溶媒を減圧蒸発させるとワックス状固体が残った。この固体をCH2Cl2/エーテル混液(1:1)で溶出するシリカゲルクロマトグラフィーで未反応DCCが除去されるまで精製した。溶出液をエーテルに代えて所要生成物(rf=0.4、DCM/Et2O1:1)をmp66〜68℃の無色固体として分離した(2.7g、57%)。
【0168】
13C NMR(CDCl3):δC 25.6(×2)(CH2),28.4(×12)(CH3),31.8(CH2),35.2(CH),39.3(×2)(NCH2),47.1(×2)(NCH2),49.1(×2)(NCH2),79.9(×2)(OC),80.5(×2)(OC),156.1(×4)(C=0),167.7(C=O),169.1(×2)(C=O)。
【0169】
1H NMR(CDCl3):δH 1.35(18H,s,CH3×6),1.41(18H,s,CH3×6),2.52(3H,brs,CH及びCH2),2.77(4H,s,CH2×2),3.10〜3.35(12H,brs,NCH2×6),5.08(2H,brsNH×2)。
【0170】
質量分析(ESI)m/e:(M+Na)C33H54N5O12Na計算値738.3896,実測値738.3893。
【図面の簡単な説明】
【0171】
【図1】実施例に記載の各種化合物の構造を示す。
【図2】実施例に記載の各種化合物の構造を示す。
【図3】実施例に記載の各種化合物の構造を示す。
【特許請求の範囲】
【請求項1】
次の式(I)のテクネチウム錯体。
【化1】
式中、
Xは−NR−、−CO2−、−CO−、−NR(C=S)−、−NR(C=O)−、−CONR−又はQ基であり、
各Yは独立にD−若しくはL−アミノ酸、−CH2−、−CH2OCH2−又は−OCH2CH2O−又はX基であり、
Zは合成生体ターゲティング部分であり、
nは1〜8の整数であり、
mは0〜30の整数であり、
RはH、C1〜4アルキル、C2〜4アルコキシアルキル、C1〜4ヒドロキシアルキル又はC1〜4フルオロアルキルであり、
Qは次式の基であり、
【化2】
Aは対イオンであるが、
ヘテロ原子同士が直接結合した結合を原子鎖X1−(Y)mが含まないことを条件とする。
【請求項2】
テクネチウム放射性同位体が99mTc又は94mTcである、請求項1記載のテクネチウム錯体。
【請求項3】
nが1〜6であり、Xが−CONR−又は−NR(C=O)−である、請求項1又は請求項2記載のテクネチウム錯体。
【請求項4】
Xが−CONH−又は−NH(C=O)−である、請求項3記載のテクネチウム錯体。
【請求項5】
−(Y)m−が式(−OCH2CH2O−)wのPEG基からなり、wが3〜25の整数である、請求項1乃至請求項4のいずれか1項記載のテクネチウム錯体。
【請求項6】
wが6〜22である、請求項5記載のテクネチウム錯体。
【請求項7】
−(Y)m−が1〜10個のアミノ酸を含む、請求項1乃至請求項4のいずれか1項記載のテクネチウム錯体。
【請求項8】
前記アミノ酸が独立にグリシン、リジン、アスパラギン酸、グルタミン酸又はセリンから選択される、請求項7記載のテクネチウム錯体。
【請求項9】
Zが、
(i)3〜30量体ペプチド、及び
(ii)酵素の基質、酵素アンタゴニスト又は酵素阻害剤
から選択される、請求項1乃至請求項8のいずれか1項記載のテクネチウム錯体。
【請求項10】
請求項1乃至請求項9のいずれか1項記載のテクネチウム錯体の調製に有用なキレーターコンジュゲートであって、当該コンジュゲートが次の式IIのものであるキレーターコンジュゲート。
【化3】
式中、X、Y、Z、n及びmは請求項1で定義した通りであり、
Q1〜Q6は独立にQ基であって、QはH又はアミン保護基である。
【請求項11】
Q1〜Q6の各々がHである、請求項10記載のキレーターコンジュゲート。
【請求項12】
Q1及びQ6が共にHであり、Q2、Q3、Q4及びQ5が各々tert−ブトキシカルボニルである、請求項10記載のキレーターコンジュゲート。
【請求項13】
Aが薬学的に許容される対イオンである請求項1乃至請求項9のいずれか1項記載の式Iのテクネチウム錯体を、生体適合性担体と共にヒトへの投与に適した形態で含む放射性医薬品。
【請求項14】
テクネチウム放射性同位体が99mTc又は94mTcである、請求項13記載の放射性医薬品。
【請求項15】
請求項13又は請求項14記載の放射性医薬品の調製用キットであって、
(i)請求項10乃至請求項12のいずれか1項記載のキレーターコンジュゲート、及び
(ii)生体適合性還元剤
を備えるキット。
【請求項16】
生体適合性還元剤が第一スズイオンを含む、請求項15記載のキット。
【請求項17】
Q1〜Q6の各々がHである、請求項15又は請求項16記載のキット。
【請求項18】
Zが請求項9で定義した通りである、請求項15乃至請求項17のいずれか1項記載のキット。
【請求項19】
次の式IIIの化合物。
【化4】
式中、
Q1〜Q6及びnは請求項10で定義した通りであり、
Eは請求項1乃至請求項9のいずれか1項記載の生体ターゲティング部分(Z)との結合に適した官能基であるが、
a.n=3のとき、Q1〜Q6の1以上がアミン保護基であり、
b.n=3又は5のとき、EはOHではないことを条件とする。
【請求項20】
Eが−NH2、−CO2M(式中、MはH、陽イオン、保護基又は活性エステルである。)、−NCS、−NCO、マレイミド又はアクリルアミドである、請求項19記載の化合物。
【請求項21】
Eが−NH2又は−CO2Mである、請求項20記載の化合物。
【請求項22】
nが1〜6である、請求項19乃至請求項21のいずれか1項記載の化合物。
【請求項1】
次の式(I)のテクネチウム錯体。
【化1】
式中、
Xは−NR−、−CO2−、−CO−、−NR(C=S)−、−NR(C=O)−、−CONR−又はQ基であり、
各Yは独立にD−若しくはL−アミノ酸、−CH2−、−CH2OCH2−又は−OCH2CH2O−又はX基であり、
Zは合成生体ターゲティング部分であり、
nは1〜8の整数であり、
mは0〜30の整数であり、
RはH、C1〜4アルキル、C2〜4アルコキシアルキル、C1〜4ヒドロキシアルキル又はC1〜4フルオロアルキルであり、
Qは次式の基であり、
【化2】
Aは対イオンであるが、
ヘテロ原子同士が直接結合した結合を原子鎖X1−(Y)mが含まないことを条件とする。
【請求項2】
テクネチウム放射性同位体が99mTc又は94mTcである、請求項1記載のテクネチウム錯体。
【請求項3】
nが1〜6であり、Xが−CONR−又は−NR(C=O)−である、請求項1又は請求項2記載のテクネチウム錯体。
【請求項4】
Xが−CONH−又は−NH(C=O)−である、請求項3記載のテクネチウム錯体。
【請求項5】
−(Y)m−が式(−OCH2CH2O−)wのPEG基からなり、wが3〜25の整数である、請求項1乃至請求項4のいずれか1項記載のテクネチウム錯体。
【請求項6】
wが6〜22である、請求項5記載のテクネチウム錯体。
【請求項7】
−(Y)m−が1〜10個のアミノ酸を含む、請求項1乃至請求項4のいずれか1項記載のテクネチウム錯体。
【請求項8】
前記アミノ酸が独立にグリシン、リジン、アスパラギン酸、グルタミン酸又はセリンから選択される、請求項7記載のテクネチウム錯体。
【請求項9】
Zが、
(i)3〜30量体ペプチド、及び
(ii)酵素の基質、酵素アンタゴニスト又は酵素阻害剤
から選択される、請求項1乃至請求項8のいずれか1項記載のテクネチウム錯体。
【請求項10】
請求項1乃至請求項9のいずれか1項記載のテクネチウム錯体の調製に有用なキレーターコンジュゲートであって、当該コンジュゲートが次の式IIのものであるキレーターコンジュゲート。
【化3】
式中、X、Y、Z、n及びmは請求項1で定義した通りであり、
Q1〜Q6は独立にQ基であって、QはH又はアミン保護基である。
【請求項11】
Q1〜Q6の各々がHである、請求項10記載のキレーターコンジュゲート。
【請求項12】
Q1及びQ6が共にHであり、Q2、Q3、Q4及びQ5が各々tert−ブトキシカルボニルである、請求項10記載のキレーターコンジュゲート。
【請求項13】
Aが薬学的に許容される対イオンである請求項1乃至請求項9のいずれか1項記載の式Iのテクネチウム錯体を、生体適合性担体と共にヒトへの投与に適した形態で含む放射性医薬品。
【請求項14】
テクネチウム放射性同位体が99mTc又は94mTcである、請求項13記載の放射性医薬品。
【請求項15】
請求項13又は請求項14記載の放射性医薬品の調製用キットであって、
(i)請求項10乃至請求項12のいずれか1項記載のキレーターコンジュゲート、及び
(ii)生体適合性還元剤
を備えるキット。
【請求項16】
生体適合性還元剤が第一スズイオンを含む、請求項15記載のキット。
【請求項17】
Q1〜Q6の各々がHである、請求項15又は請求項16記載のキット。
【請求項18】
Zが請求項9で定義した通りである、請求項15乃至請求項17のいずれか1項記載のキット。
【請求項19】
次の式IIIの化合物。
【化4】
式中、
Q1〜Q6及びnは請求項10で定義した通りであり、
Eは請求項1乃至請求項9のいずれか1項記載の生体ターゲティング部分(Z)との結合に適した官能基であるが、
a.n=3のとき、Q1〜Q6の1以上がアミン保護基であり、
b.n=3又は5のとき、EはOHではないことを条件とする。
【請求項20】
Eが−NH2、−CO2M(式中、MはH、陽イオン、保護基又は活性エステルである。)、−NCS、−NCO、マレイミド又はアクリルアミドである、請求項19記載の化合物。
【請求項21】
Eが−NH2又は−CO2Mである、請求項20記載の化合物。
【請求項22】
nが1〜6である、請求項19乃至請求項21のいずれか1項記載の化合物。
【図1】

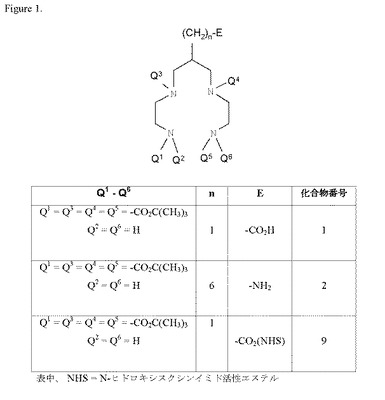
【図2】
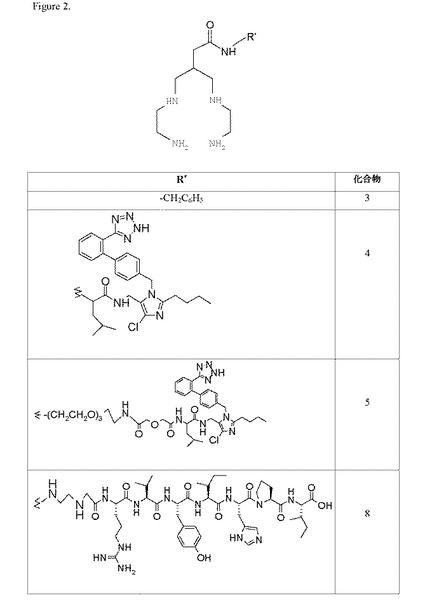
【図3】
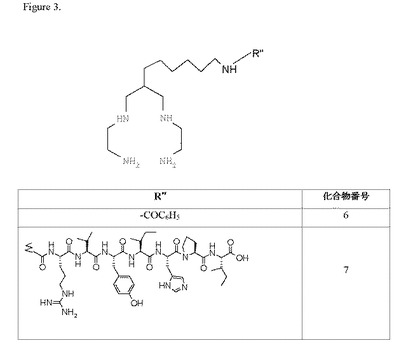
【図2】
【図3】
【公表番号】特表2008−510683(P2008−510683A)
【公表日】平成20年4月10日(2008.4.10)
【国際特許分類】
【出願番号】特願2007−522011(P2007−522011)
【出願日】平成17年7月19日(2005.7.19)
【国際出願番号】PCT/GB2005/002807
【国際公開番号】WO2006/008496
【国際公開日】平成18年1月26日(2006.1.26)
【出願人】(305040710)ジーイー・ヘルスケア・リミテッド (99)
【Fターム(参考)】
【公表日】平成20年4月10日(2008.4.10)
【国際特許分類】
【出願日】平成17年7月19日(2005.7.19)
【国際出願番号】PCT/GB2005/002807
【国際公開番号】WO2006/008496
【国際公開日】平成18年1月26日(2006.1.26)
【出願人】(305040710)ジーイー・ヘルスケア・リミテッド (99)
【Fターム(参考)】
[ Back to top ]