
放出制御型鎮痛用懸濁液
【課題】 懸濁化できる活性成分を有し、口に合うだけでなく、投与後に要求される放出プロフィールを保証する安定な形態である、液状の放出制御剤形を提供する。
【解決手段】 非ステロイド系抗炎症薬、特にプロピオン酸誘導体、例えばイブプロフェンまたはアセトアミノフェンを懸濁液を使って投与する方法が提供されている。この方法は、長時間の治療効果、特に鎮痛を改善する。
【解決手段】 非ステロイド系抗炎症薬、特にプロピオン酸誘導体、例えばイブプロフェンまたはアセトアミノフェンを懸濁液を使って投与する方法が提供されている。この方法は、長時間の治療効果、特に鎮痛を改善する。
【発明の詳細な説明】
【開示の内容】
【0001】
本発明は、鎮痛剤のような活性成分を投与するための液状剤形に適した放出制御型医薬品製剤に関する。
【0002】
〔発明の背景〕
痛み、炎症、および熱を治療するための治療薬には、鎮痛剤、抗炎症剤、および解熱剤がある。非ステロイド系抗炎症剤(NSAID)は、このような治療薬の一種である。NSAIDには、プロピオン酸誘導体(propionic acid derivatives)、酢酸誘導体(acetic acid derivatives)、フェナミン酸誘導体(fenamic acid derivatives)、ビフェニルカルボン酸誘導体(biphenylcarbodylic acid derivatives)、オキシカム(oxicams)、およびシクロオキシゲナーゼ−2(COX−2)選択的NSAID(cyclooxygenase-2 (COX-2) selective NSAIDs)がある。
【0003】
プロピオン酸(propionic acid)には、例えば、イブプロフェン(ibuprofen)、ナプロキセン(naproxen)、およびケトプロフェン(ketoprofen)がある。特にイブプロフェンは、広く使用され、よく知られている、鎮痛特性および解熱特性を有するNSAIDである。イブプロフェンは、何年もの間、多くの形式で、一般用医薬品として市販されている。イブプロフェンは、化学的には、2−(4−イソブチルフェニル)プロピオン酸(2-(4-isobutylphenyl)-propionic acid)として知られている。
【0004】
NSAIDは、通常、一日当たり1回から4回投与され、一日当たりの投与量は、約50から約2000ミリグラムの範囲であり、好ましくは、約100から1600であり、もっとも好ましくは約200から約1200ミリグラムである。
【0005】
アセトアミノフェンは、よく知られた鎮痛剤であって、一日当たりの投与量は約325から約4000ミリグラムの範囲であり、好ましくは約650から約4000ミリグラムである。アセトアミノフェンは、1893年にヴァン・メリングが初めて医療で使用しているが、1949年になって初めて一般用医薬品市場で鎮痛剤として使用するアスピリンの有効な代替品として普及した。APAPの薬理学は、B. Ameer et al.、Ann. Int. Med. 87,202 (1977)で論評されている。APAPの普及および製造量を考慮すると、鎮痛剤としてのAPAPの製造および使用の両方が当業者に公知である。
【0006】
NSAID、アセトアミノフェン、その他の薬を12時間または24時間にわたって複数回投与することが知られている。例えば、同一量のイブプロフェンが入っている用量を複数回12時間から24時間にわたって投与することが知られている。さらに、最初に多量に投与し、続いて比較的少ない維持量を投与することも知られている。これについては、例えば、Palmisano et al., Advances in Therapy, Vol. 5, No. 4, July/August 1988(ケプトン(初期量150mgに続いて後続量75mg)およびイブプロフェン(初期量800mgに続いて後続量400mg)からなる複数回投与の使用)を参照のこと。
【0007】
特に、経口投与型鎮痛剤との関連で、治療効果の持続時間を延ばしたいという要望から、一日一回だけ投与すればよい放出制御製剤が開発された。一日一回の投与では、患者が治療処置を行っている間に言われた服用量を尊守するようになるので、都合がよい。
【0008】
放出制御医薬品剤形は、薬の送達を最適化し、かつ、患者のコンプライアンスを、特に患者が一日に取らなければならない薬の服用回数を減らすことにより、向上させるように長いこと使用されてきた。このために、薬または他の活性成分が剤形から患者の胃腸液に放出される速度を下げることが、特に体内での薬の作用を引き延ばすためには、多くの場合に望ましいことである。
【0009】
口を経て送り込まれた薬が体内の作用部位に到達する速度は、薬が胃腸粘膜を介して血液に吸収される速度および量を含む多くの要因に依存する。もっとも、薬が血液に吸収可能となる前に、まず薬が胃腸液に溶けなければならない。多くの薬では、胃腸膜を通しての吸収が胃腸液でのその薬の溶出に比べて速く、このために、薬の溶出が、薬の吸収における律速段階となっている。したがって、製剤業者は、血液への薬の吸収速度を薬の溶出速度を変えることで効果的に制御できる。
【0010】
医薬品剤形の場合、1つ以上の薬を、それぞれ速度を調節して送達することも特に望ましい。各薬の吸収、分配、代謝、および排泄がそうであるように、薬の治療効果の開始および持続時間には大きなばらつきがあるので、異なる薬の放出を異なる方法で調節すること、すなわち、第1の薬を剤形から直ちに放出させる一方で、第2の薬を「調節した」方法で、たとえば、遅らせて、または制御して放出することが多くの場合に望ましい。
【0011】
製剤が薬を速度を制御して送達できる(例えば、除放、持効、延長、または遅延放出)公知のメカニズムには、拡散、浸食、浸透がある。特定の活性成分について特に望ましい放出制御プロフィールを得るためには、上記メカニズムの組み合わせを利用した剤形をデザインすることが多くの場合に実用的である。
【0012】
不都合なことに、放出制御の多くの応用例では、最終的なサイズおよび重量が大きい固形用量単位(solid dosage unit)を利用している。このような用量単位を投与することは、特に子供やお年寄りのような飲み込むことが困難な患者に問題となる。したがって、このような放出制御医薬品をかみ砕くことができるまたは口でバラバラにすることができる固形形態または液状形態で提供することがより望ましい。多くの患者にとって、液状経口剤形がより好まれる。これは、かみ砕くという余分な段階なしで飲み込むことができるからである。
【0013】
経口液状製剤(oral liquid forms)は、長年、即放性薬剤を送達するのに一般的に使用されてきた。これについては、例えば、米国特許第5,374,659号、第4,788,220号、第4,975,465号、および第5,183,829号を参照のこと。しかし、放出制御製剤を液状剤形に導入することは、製剤上かなりの難題である。具体的には、通常、コーティングした、または化学的に結合させた粒子を利用して薬の放出調節部分を保持する。このような粒子、ならびにこれら粒子を浮遊させるための液状賦形剤の特性は、粒子を一様に分散した状態に維持できるように適合していなければならない。特に難題となるのは、患者が摂取する前の液状剤形の保存期間中に浮遊粒子から薬が早くに放出されてしまうのを防止することである。さらに、所望の溶出性プロフィール、ならびに、液状剤形の所望の用量均一性をその有効期間を通して維持することは、経口液状放出制御懸濁製品の製剤において取り組むべき別の難題である。
【0014】
米国特許第5,527,545号では、懸濁液状の剤形の放出特性を保つために、活性成分微粒剤を連続した4つのコーティングで被覆している。しかしながら、複数のコーティング段階は、製品の全体のコストおよび製造サイクルタイムを増大させるだけでなく、できあがった剤形は、ユーザに即放性用量を提供するものではない。
【0015】
したがって、懸濁化できる、鎮痛薬のような活性成分を有し、口に合うだけでなく、投与後に要求される放出プロフィールを保証する安定な形態でもある、液状の放出制御剤形があればさらに望ましいことである。さらに、使用者に鎮痛剤の即放出量および持続放出量の両方を提供する鎮痛懸濁製品を有することも望ましい。
【0016】
〔発明の概要〕
本発明は、NSAIDSおよび/またはアセトアミノフェンを懸濁液で投与するのに適する医薬品剤形を提供するものであり、この剤形は、
a)NSAIDおよび/またはアセトアミノフェンを含む第1部分であって、NSAIDおよび/またはアセトアミノフェンが、剤形が溶出媒体と接触したときに、実質的に直ちに剤形から放出される第1部分と、
b)NSAIDおよび/またはアセトアミノフェンを含む粒子からなる第2部分であって、剤形が前記溶出媒体と接触したときに、NSAIDおよび/またはアセトアミノフェンが粒子から制御されながら放出される第2部分と、を備える、からなる、および/または、から本質的に構成され、
医薬品剤形の治療効果の持続時間が、医薬品剤形の投与後、少なくとも8時間ある。
【0017】
本発明の別の実施形態は、懸濁液剤形に向けられていて、この懸濁液剤形は、
a)NSAIDおよび/またはアセトアミノフェンを含む第1部分であって、NSAIDおよび/またはアセトアミノフェンが、剤形が溶出媒体と接触すると、実質的に直ちに剤形から放出される第1部分と、
b)NSAIDおよび/またはアセトアミノフェンを含む粒子からなる第2部分であって、剤形が溶出媒体と接触すると、NSAIDおよび/またはアセトアミノフェンが粒子から制御されながら放出される第2部分と、
c)水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物と、を含む、からなる、および/または、から本質的に構成され、
剤形の治療効果の持続時間が、投与後、少なくとも12時間ある。
【0018】
本発明の他の実施形態は、アセトアミノフェンおよび/またはNSAIDを医薬品剤形の状態で必要に応じて哺乳動物に投与する方法に向けられており、この方法は、哺乳動物が、アセトアミノフェンおよび/またはNSAIDの即放性量を上記12時間の最初に受け、アセトアミノフェンおよび/またはNSAIDの放出制御量を剤形の投与後12時間にわたって受けるように剤形を哺乳動物に与えることを含む、からなる、および/または、から実質的になり、上記12時間の間にアセトアミノフェンおよび/またはNSAIDをさらに与えることがない。
【0019】
本発明の他の実施形態は、医薬品懸濁液剤形に向けられており、この剤形は、
NSAIDおよび/またはアセトアミノフェンからなる粒子であって、放出制御成分からなる1つの層で実質的にコーティングされている粒子、を含む、からなる、および/または、から実質的に構成されており、
この医薬品懸濁液剤形は、治療効果の持続時間が哺乳動物に最初に投与した後、少なくとも約8時間ある。
【0020】
本発明の別の実施形態は、医薬品懸濁液剤形に向けられており、この剤形は、
a)NSAIDおよび/またはアセトアミノフェンを含む粒子であって、放出制御コーティングからなる1つの層で実質的に覆われている粒子と、
b)水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物と、を備える、からなる、および/または、から実質的に構成されており、
医薬品剤形の治療効果の持続時間が、医薬品剤形の投与後、少なくとも約8時間ある。
【0021】
本発明の他の実施形態は、アセトアミノフェンおよび/またはNSAIDを懸濁液医薬品剤形の状態で必要に応じて哺乳動物に投与する方法に向けられており、この方法は、その哺乳動物が、上記剤形の投与後約12時間にわたって上記アセトアミノフェンおよび/またはNSAIDの放出制御量を受けるように、哺乳動物に上記剤形を与えることを含む、からなる、および/または、から実質的になり、アセトアミノフェンおよび/またはNSAIDを上記12時間の間にさらに与えることはない。
【0022】
本発明の他の実施形態は、医薬品懸濁液剤形に向けられており、この剤形は、
a)NSAIDおよび/またはアセトアミノフェンを含む粒子であって、放出制御成分からなる1つの層で実質的に覆われており、この放出制御成分が、その放出制御成分の全重量を基準として、約0パーセントよりも多く、約90パーセント未満の不溶性フィルム成形ポリマーと、約0パーセントよりも多く、約10パーセント未満の腸溶性ポリマーとを含む粒子と、
b)水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物と、を備える、からなる、および/または、から実質的になり、
この医薬品剤形の治療効果の持続時間が、投与後、少なくとも12時間ある。
【0023】
〔本発明の詳細な説明〕
本明細書で用いる限り、「実質的に覆う」または「実質的に連続」という言葉は、コーティングがほぼ連続であり、芯物質またはその下にある層の表面全体をほぼ覆っており、このため、活性成分、すなわち下にある層がほとんどまたは全く露出していないことを意味する。
【0024】
本明細書で使用する限り、「ATDAIRD」は、特定活性成分の有効即放性量の平均的な治療作用継続時間を意味する。例えば、イブプロフェン(ibuprofen)またはケトプロフェン(ketoprofen)の即放性量の典型的な作用継続時間、すなわち治療効果時間は、約4時間から約6時間である。したがって、イブプロフェンまたはケトプロフェンのATDAIRDは5時間である。ナプロキセン(naproxen)の即放性量の典型的な作用継続時間は、約8時間から約12時間である。したがって、ナプロキセンのATDAIRDは10時間である。特定の活性成分の治療作用継続時間は、その特定の活性成分が入っている即放性製品のラベル類における服用指示書から簡単に定めることができる。
【0025】
本明細書で使用する限り、「放出調節(modified release)」は、胃腸液のような溶出溶媒における活性成分の放出、すなわち溶出を変えることに適用する。調節しながら放出しうる活性成分は、例えば、剤形、コーティング、または粒子に入っていてもよいし、またはそれらのどこか一部、例えば、液状懸濁媒体じゅうに分散させた粒子などに入っていてもよい。放出調節の種類には、1)放出制御と、2)放出遅延とがある。「放出制御」は、投与後に、活性成分が剤形から実質的に連続的に、調節されながら放出され、その剤形から活性成分が完全に放出される、すなわち消耗するのに要する時間が、同じ活性成分からなる即放性剤形についてのそれよりも長いことを意味する。放出制御の種類には、延長(prolonged)、持続(sustained)、除放などがある。「放出遅延」は、投与後に、少なくとも一時期、活性成分が剤形から放出されないことを意味する。
【0026】
本明細書で使用する限り、「溶出溶媒(dissolution medium)」は、本発明の懸濁液剤形が溶出できる適切なあらゆる液体環境を意味し、例えば、製品検査で用いる生体外溶出溶媒、または胃腸液などを意味する。本発明の懸濁液剤形からの活性成分の溶出性を検査するのに用いる適切な生体外溶出溶媒には、USP23(1995)の786ページに記載されたものがあり、これらは、参照することにより本明細書に組み込まれる。
【0027】
本発明の一実施形態は、活性成分を懸濁液で投与するのに適した放出制御医薬品剤形に向けられたものであり、a)即放性部分、例えば、その剤形から直ちに放出される少なくとも一つの活性成分を含む部分と、b)放出制御部分、例えば、ある調整された時間にわたって、例えば、その剤形を最初に投与したあと約4時間から約12時間にわたって、実質的に連続的に血流に放出される少なくとも一つの活性成分を含む部分と、を含んでいる。本明細書で使用する限り、「即放性」とは、少なくとも一つの活性成分の溶出特性が、その活性成分を含む即放性錠に対するUSP規格を満たすことを意味する。例えば、アセトアミノフェン(acetaminophen)錠については、pH5.8のリン酸塩緩衝剤(phosphate buffer)において、USP装置2(パドル)を50rpmで使用すると、その剤形に含まれているアセトアミノフェンの少なくとも80パーセントが投与後30分以内にその剤形から放出されるとUSP24に指定されており、イブプロフェン(ibuprofen)錠については、pH7.2のリン酸塩緩衝剤において、USP装置2(パドル)を50rpmで使用すると、その剤形に含まれるイブプロフェンの少なくとも80パーセントが投与後60分以内にその剤形から放出されるとUSP24に指定されている。これについては、2000年版のUSP24(1999)の19−20ページ及び856ページを参照のこと。さらに、イブプロフェン懸濁液(ibuprofen suspension)の溶出性は、pH5.6のアセテート緩衝剤を使い、USP装置2(パドル)を50rpmで使って分析することができ、即放性量については、その剤形に含まれているイブプロフェンの少なくとも80パーセントがその剤形から投与後60分以内に放出される。
【0028】
即放性部分は、剤形内に分子レベルで分散している、例えば、融解または溶解された一つ以上の活性成分を含んでいてもよい。あるいは、活性成分は粒子という形態であってもよい。この粒子は、さらにコーティングされていてもよく、されていなくてもよい。活性成分が粒子という形態をしている実施形態では、粒子は(コーティングされていても、いなくても)、通常、平均粒径が約1ミクロンから約2000ミクロンまでである。ある実施形態では、このような粒子が、平均粒径が約1ミクロンから約300ミクロンまでの結晶という形態をしている。他の実施形態では、粒子が顆粒またはペレットという形態をしていて、平均粒径が約25ミクロンから約2000ミクロンまでであり、例えば、約25ミクロンから約1000ミクロンまでであり、または、約25ミクロンから約400ミクロンまでである。
【0029】
放出制御部分は、放出制御特性を有する多数の粒子に少なくとも一つの活性成分を含んでいる。ある実施形態では、放出制御部分におけるこれらの粒子の芯物質が純粋な結晶の形態をした活性成分を含むことがあり、この結晶は放出調節成分で実質的にコーティングされている。あるいは、粒子の芯物質は、顆粒を混和したものを含むことがあり、この顆粒が一つ以上の活性成分を、結合材、賦形剤などの当該技術において公知の、オプションとしての成分とともに含み、また、このような顆粒もまた放出制御成分で実質的にコーティングされている。別の実施形態では、活性成分の粒子を放出制御成分からなるマトリックスの全体に分散させることがある。さらに別の実施形態では、一つ以上の活性成分を化学的に結合、すなわち、「錯体化」して樹脂、例えば、イオン交換樹脂にし、粒子を形成していることがある。この粒子は、オプションとして、放出調整コーティングで実質的にコーティングすることもある。本明細書で使用する限り、「実質的にコーティングする」とは、粒子の表面積の約1パーセント未満、例えば約0.1パーセント未満が、露出していること、例えば、所望のコーティングで覆われていないことを意味する。
【0030】
このような実施形態で使用する具体的なイオン交換樹脂が、いくつかの要素、例えば活性成分のイオン電価などに左右されることは、当業者に、必要以上の実験を行うことなく容易に分かるであろう。NSAID活性成分に適するイオン交換樹脂の例としては、これに限定はされないが、コレスチラミン(cholestyramine)がある。コレスチラミンは、ローム・アンド・ハース(Rohm & Haas)から商品名「Duolite(登録商標)AP143」で市販されている。高分子樹脂を用いた錯体化に関する詳細は公知であり、例えば、米国特許第4,221,778号、第4,847,077号、および第6,001,392号に開示されている。米国特許第4,221,778号、第4,847,077号、および第6,001,392号は、参照により本明細書に組み込まれる。
【0031】
ある特定の実施形態では、剤形の放出制御部分に実質的にイオン交換樹脂がない。「実質的にイオン交換樹脂がない」は、イオン交換樹脂の量が、その剤形における全活性成分粒子の全重量を基準として、約1パーセント未満であることを意味し、例えば約0.5パーセント未満、または約0.1パーセント未満であることを意味する。
【0032】
粒子上の一つ以上のコーティング層は、適切なあらゆる放出制御成分を含んでいてもよい。適切な放出制御成分の一例は、放出制御成分の全重量を基準として、約0パーセントより多く約90パーセントまで、例えば約10パーセントから約60パーセントまでの不溶性フィルム形成ポリマーと、約0パーセントより多く、約10パーセント未満、例えば約0.5パーセントから約20パーセントまでの腸溶性ポリマーを含む。放出調整成分における腸溶性ポリマーの不溶性フィルム形成ポリマーに対する重量比は、約0.5:99.5から約20:80までの範囲であってもよく、例えば、約5:95から約10:90までの範囲であってもよい。同様の放出制御成分は、その全体に活性成分粒子を分散させることができるマトリックスに使用することもできる。
【0033】
適する不溶性フィルム形成ポリマーには、これに限定はされないが、酢酸セルロース(cellulose acetate), エチルセルロース(ethylcellulose)、ローム・パルマ(Rohm Pharma)から商品名「EUDRAGIT RS」で市販されているポリ(アクリル酸エチル、メタクリル酸メチル、トリメチルアンモニウムエチル・メタクリレート・クロライド)(poly(ethyl acrylate, methyl methacrylate, trimethylammonioethyl methacrylate chloride))1:2:0.1と、その共重合体および混合物がある。ある実施形態では、不溶性フィルム形成ポリマーが酢酸セルロースおよび/またはエチルセルロースから選択される。
【0034】
適する腸溶性ポリマーには、これに限定はされないが、ヒドロキシプロピル・メチルセルロース・フタレート(hydroxypropyl methylcellulose phthalate)、ヒドロキシプロピル・メチルセルロース・アセテート・シクシナート(hydroxypropyl methylcellulose acetate succinate)、セルロース・アセテート・フタレート(cellulose acetate phthalate)、ポリビニルアセテート・フタレート(polyvinylacetate phthalate)、ポリメタクリレート(polymethacrylate)をベースとするポリマー、ならびにその共重合体および混合物がある。適するポリメタクリレートをベースとするポリマーには、これに限定はされないが、ローム・パルマGmbH から商品名「EUDRAGIT S」ポリマーで市販されているポリ(メタクリル酸、メチルメタクリレート)(poly(methacrylic acid, methyl methacrylate)) 1:2と、ローム・パルマGmbH から商品名「EUDRAGIT L」ポリマーで市販されているポリ(メタクリル酸、メチルメタクリレート)(poly(methacrylic acid, methyl methacrylate)) 1:1とがある。ある実施形態では、腸溶性ポリマーを非アクリル酸化合物(non-acrylate compounds)から選択する。非アクリル酸化合物は、例えば、ヒドロキシプロピル・メチルセルロース・フタレート(hydroxypropyl methylcellulose phthalate)、ヒドロキシプロピル・メチルセルロース・アセテート・シクシナート(hydroxypropyl methylcellulose acetate succinate)、セルロース・アセテート・フタレート(cellulose acetate phthalate)、ポリビニルアセテート・フタレート(polyvinylacetate phthalate)、ならびに、これらの共重合体および混合物などである。
【0035】
ある実施形態では、放出制御成分に腸溶性ポリマーが実質的にない。つまり、例えば、放出制御成分に含まれる腸溶性ポリマーが、放出制御成分の全重量を基準として、約1パーセント未満、または0.25パーセント未満である。
【0036】
放出制御成分でコーティングされている活性成分粒子には、このようなコーティングされた粒子の全乾燥重量を基準として、約5パーセントから約40パーセントの、例えば約10パーセントから約30パーセントの放出制御成分を少なくとも1つのコーティング層という形態で入っている。
【0037】
コーティングされた活性成分粒子は、適するどのような公知方法で形成してもよい。適する粒子形成兼被覆方法には、高剪断造粒(high sheer granulation)、流動層造粒、例えば、ローター造粒(rotor granulation)、流動層コーティング(fluid bed coating)、コーアクサヴェーション(coaccervation)、噴霧乾燥、噴霧凝結などがあり、例えば、医薬剤形:錠剤第3巻、ヘルベルト A.リーバーマンおよびレオン・ラッチマン編集、第2、3および4章(1982年)(Pharmaceutical Dosage Forms: Tablets Volume 3, edited by Herbert A. Lieberman and Leon Lachman, Chapters 2,3, and 4 (1982))に記載されている。ある実施形態では、放出制御成分でコーティングした粒子の平均径が約20から約400ミクロンであり、例えば、約50から約300ミクロンである。
【0038】
本発明の剤形には、活性成分が1つ以上含まれている。適する活性成分に含まれるのは、だいたい、医薬品、ミネラル、ビタミンその他の栄養補助食品、口腔手入れ剤、香辛料類(flavorant)およびその混合物である。適する医薬品には、鎮痛薬(analgesics)、抗炎症剤(anti-inflammatory agents)、関節炎治療薬(antiarthritics)、麻酔薬(anesthetics)、抗ヒスタミン薬(antihistamines)、鎮咳剤(antitussives)、抗生物質(antibiotics)、抗感染剤(anti-infective agents)、抗ウィルス薬(antivirals)、抗凝固薬(anticoagulants)、抗うつ薬(antidepressants)、抗糖尿病剤(antidiabetic agents)、制吐薬(antiemetics)、抗膨満薬(antiflatulents)、抗菌剤(antifungals)、鎮痙薬(antispasmodics)、食欲減退薬(appetite suppressants)、気管支拡張薬(bronchodilators)、心・血管作動薬(cardiovascular agents)、中枢神経薬(central nervous system agents)、中枢神経興奮薬(central nervous system stimulants)、うっ血除去薬(decongestants)、経口避妊薬(oral contraceptives)、利尿薬(diuretics)、去痰薬(expectorants)、胃腸薬(gastrointestinal agents)、片頭痛製剤(migraine preparations)、乗物酔い製品(motion sickness products)、粘液溶解薬(mucolytics)、筋弛緩薬(muscle relaxants)、骨粗鬆症製剤(osteoporosis preparations)、ポリジメチルシロキサン(polydimethylsiloxanes), 呼吸器疾患に使用される医薬品(respiratory agents)、睡眠補助剤(sleep-aids)、尿路薬(urinary tract agents)およびその混合物が含まれる。
【0039】
適する香辛料類(flavorants)には、メントール、ペパーミント、ミント香辛料、フルーツ香辛料、チョコレート、バニラ、風船ガム香辛料、コーヒー香辛料、リキュール香辛料およびその組み合わせなどがある。
【0040】
適する胃腸薬の例としては、制酸薬(antacids)、例えば、炭酸カルシウム(calcium carbonate)、水酸化マグネシウム(magnesium hydroxide)、酸化マグネシウム(magnesium oxide)、炭酸マグネシウム(magnesium carbonate)、水酸化アルミニウム(aluminum hydroxide)、炭酸水素ナトリウム(sodium bicarbonate)、ジヒドロキシアルミニウム炭酸ナトリウム(dihydroxyaluminum sodium carbonate)など;刺激性下剤(stimulant laxatives)、例えばビサコジル(bisacodyl)、カスカラサグラダ(cascara sagrada)、ダントロン(danthron)、センナ(senna)、フェノールフタレイン(phenolphthalein)、アロエ(aloe)、ヒマシ油(castor oil)、リシノール酸(ricinoleic acid)、およびデヒドロコール酸(dehydrocholic acid)、およびそれらの混合物など;H2受容体拮抗薬(H2 receptor antagonists)、例えば、ファモチジン(famotadine)、ラニチジン(ranitidine)、シメチジン(cimetadine)、ニザチジン(nizatidine)など;プロトンポンプ阻害薬(proton pump inhibitors)、例えばオメプラゾール(omeprazole)またはランソプラゾール(lansoprazole)など;胃腸細胞保護剤(gastrointestinal cytoprotectives)、例えばスクラルフェート(sucraflate)およびミソプロストール(misoprostol);胃腸運動促進剤(gastrointestinal prokinetics)、例えばプルカロプリド(prucalopride)、人の幽門用の抗生物質(antibiotics for H. pylori)、例えばクラリスロマイシン(clarithromycin)、アモキシシリン(amoxicillin)、テトラサイクリン(tetracycline)、およびメトロニダゾール(metronidazole);下痢止め薬(antidiarrheals)、例えばジフェノキシラート(diphenoxylate)およびロペラミド(loperamide);グリコピロレート(glycopyrrolate);制吐薬(antiemetics)、例えばオンダンセトロン(ondansetron)、鎮痛薬(analgesics)、例えばメサラミン(mesalamine)などがある。
【0041】
適するポリジメチルシロキサンの例は、これには限定されないが、ジメチコーン(dimethicone)およびシメチコン(simethicone)が含まれ、米国特許第4,906,478号、5,275,822号および第6,103,260号に開示されたものである。米国特許第4,906,478号、5,275,822号および第6,103,260号の各々の内容は、参照することにより本明細書に特に組み込まれる。本明細書で使用する限り、「シメチコン」という用語は、ジメチコーンおよびシメチコン(simethicone)を含むがこれに限定されないポリジメチルシロキサンの上位のクラスをいう。
【0042】
本発明のある実施形態では、少なくとも1つの活性成分をビサコジル(bisacodyl)、ファモチジン(famotadine)、ラニチジン(ranitidine)、シメチジン(cimetidine)、プルカロプリド(prucalopride)、ジフェノキシラート(diphenoxylate)、ロペラミド(loperamide)、ラクターゼ(lactase)、メサラミン(mesalamine)、ビスマス(bismuth)、制酸薬(antacids)、および医薬品として容認できるそれらの塩、エステル、異性体、および混合物から選択することができる。
【0043】
別の実施形態では、少なくとも1つの活性成分が、鎮痛薬(analgesics)、抗炎症剤(anti-inflammatories)、および解熱薬(antipyretics)から選択され、例えば、非ステロイド系抗炎症薬(NSAIDs)であり、a)プロピオン酸誘導体(propionic acid derivatives)、例えば、イブプロフェン(ibuprofen)、ナプロキセン(naproxen)、ケトプロフェン(ketoprofen)など;b)酢酸誘導体、例えば、インドメタシン(indomethacin)、ジクロフェナク(diclofenac)、スリンダク(sulindac)、トルメチン(tolmetin)など;c)フェナミン酸誘導体(fenamic acid derivatives)、例えば、メフェナム酸(mefenamic acid)、メクロフェナム酸(meclofenamic acid)、フルフェナム酸(flufenamic acid)など;d)ビフェニルカルボン酸誘導体(biphenylcarbodylic acid derivatives)、例えばジフルニサル(diflunisal)、フルフェニサール(flufenisal)など;e)オキシカム(oxicams)、例えばピロキシカム(piroxicam)、スドキシカム(sudoxicam)、イソキシカム(isoxicam)、メロキシカム(meloxicam)など;f)シクロオキシゲナーゼ−2(COX−2)選択的NSAID(cyclooxygenase-2 (COX-2) selective NSAIDs);およびg)上記したものの医薬品として容認できる塩を含む。
【0044】
ある特定の実施形態では、少なくとも1つの活性成分がプロピオン酸誘導体NSAID (propionic acid derivative NSAID) から選択されている。プロピオン酸誘導体NSAIDは、-CH(CH3)COOHもしくは-CH2CH2COOH自由基を有する、医薬品として容認できる鎮痛剤/非ステロイド系抗炎症剤、または、医薬品として容認できる塩基、例えば-CH(CH3)COO-Na+またはCH2CH2COO-Na+などであって、通常、環構造、好ましくは芳香環構造に直接またはカルボニル官能性(carbonyl functionality)を介して取り付けられているものである。
【0045】
有用なプロピオン酸誘導体には、イブプロフェン(ibuprofen)、ナプロキセン(naproxen)、ベノキサプロフェン(benoxaprofen)、ナプロキセンナトリウム(naproxen sodium)、フェンブフェン(fenbufen)、フルルビプロフェン(flurbiprofen)、フェノプロフェン(fenoprofen)、フェブプロフェン(fenbuprofen)、ケトプロフェン(ketoprofen)、インドプロフェン(indoprofen)、ピルプロフェン(pirprofen)、カーポフェン(carpofen)、オクサプロフェン(oxaprofen), プラノプロフェン(pranoprofen)、ミクロプロフェン(microprofen)、ティオクサプロフェン(tioxaprofen)、スプロフェン(suprofen)、アルミノプロフェン(alminoprofen)、チアプロフェン酸(tiaprofenic acid)、フルプロフェン(fluprofen)、ブクロクス酸(bucloxic acid)、ならびに、これらの医薬品として容認できる塩、誘導体、および組み合わせがある。
【0046】
本発明のある実施形態では、プロピオン酸誘導体をイブプロフェン、ケトプロフェン、、フルルビプロフェン、ならびに、これらの医薬品として容認できる塩および組み合わせから選択する。
【0047】
他の実施形態では、プロピオン酸誘導体がイブプロフェン、2−(4−イソブチルフェニル)プロピオン酸、または、その医薬品として容認できる塩であり、例えば、イブプロフェンのアルギニン塩、リシン塩、またはヒスチジン塩(arginine, lysine, or histidine salt)などである。イブプロフェンの他の医薬品として容認できる塩は、米国特許第4,279,926号、第4,873,231号、第5,424,075号および第5,510,385号に記載されている。これらの米国特許の内容は、参照することにより組み込まれる。
【0048】
本発明の他の特定の実施形態では、少なくとも1つの活性成分をアセトアミノフェン(acetaminophen)、アセチルサリチル酸(acetyl salicylic acid)、イブプロフェン(ibuprofen)、ナプロキセン(naproxen)、ケトプロフェン(ketoprofen)、フルルビプロフェン(flurbiprofen)、ジクロフェナク(diclofenac)、シクロベンザプリン(cyclobenzaprine)、メロキシカム(meloxicam)、ロフェコキシブ(rofecoxib)、セレコクシブ(celecoxib)、およびこれらの医薬品として容認できる塩、エステル、異性体、および混合物から選択できる。
【0049】
本発明の別な特定の実施形態では、少なくとも1つの活性成分をプソイドエフェドリン(pseudoephedrine)、フェニルプロパノールアミン(phenylpropanolamine)、クロルフェニラミン(chlorpheniramine)、デキストロメトルファン(dextromethorphan)、ジフェンヒドラミン(diphenhydramine)、アステミゾール(astemizole)、テルフェナジン(terfenadine)、フェ クソ フェ ナ ディン(fexofenadine)、ロラタジン(loratadine)、デスロラタディン(desloratadine)、セチリジン(cetirizine)、これらの混合物、およびこれらの医薬品として容認できる塩、エステル、異性体、および混合物から選択することができる。
【0050】
本発明の別の特定の実施形態では、少なくとも1つの活性成分がNSAIDおよび/またはアセトアミノフェン、ならびに、これらの医薬品として容認できる塩である。
【0051】
ある実施形態では、治療上有効な量の1つまたは複数の活性成分が「単位投与量(unit dose volume)」に入っていることがある。この単位投与量は、粉末という形態であることもあり、水性懸濁液という形態であることもある。「治療上有効な量」は、本明細書で使用する限り、経口投与により所望の治療応答性を与える活性成分の量である。当業者は、所定の患者に対する活性成分の「治療上有効な量」を、例えば、投与される具体的な活性成分;その活性成分のバイオアベラビリティ特性;所望の用量;患者の年齢および体重;などの要因を考慮することによって容易に決めることができる。本明細書で使用する限り、「単位投与量」は、所定の製品を患者に経口投薬するのに都合のよいいかなる量であってもよい。
【0052】
この実施形態では、「単位投与量」に、通常、投与指示書がついている。投与指示書は、患者に、例えば患者の年齢または体重によっては単位投与量の数倍になりうる活性成分の量を取ることを指示する。通常、懸濁液の単位投与量には、もっとも小さい患者にとって治療上有効な量の活性成分が入っている。例えば、適切な単位投与量としては、一杯のティースプーン(約5mL)、一杯のテーブルスプーン(約15mL)、スポイト1滴、1mLというものがある。
【0053】
本発明によれば、NSAIDおよび/またはアセトアミノフェンが入っている剤形を、治療、特に鎮痛治療を必要としている哺乳動物に1回だけ投与して、活性成分を血液に長時間、例えば、約8時間にわたって、または約12時間にわたって放出することができる。時間0では、NSAIDおよび/またはアセトアミノフェンの初期投与量が哺乳動物に即放性量部分の活性成分によって与えられる、すなわち、投与される。活性成分は、活性成分を含む製剤を最初に投与してから約4時間後に、例えば、すなわち、約8時間、10時間、または12時間後に、放出制御量部分における活性成分によって血中に放出され続ける。換言すれば、この製剤には、最初に投与してから約4時間後、例えば、すなわち、約8時間、10時間、または12時間後に、溶出していない活性成分がまだ残っている。
【0054】
本発明を実施する場合、剤形は、活性成分の全重量を基準として、活性成分の即放性量部分を約25パーセントから約75パーセントまでと、活性成分の放出制御量部分を約75パーセントから約25パーセントまでを含むことができる。即放性量部分および放出制御量部分は、賦形剤で結合させて、必要な場合にその場で懸濁化できる乾燥混合物とするか、すぐに使える懸濁液とすることができる。
【0055】
適切な賦形剤の組成物としては、限定はしないが、構造化剤(structuring agents);膨張剤(swelling agents);界面活性剤(surfactants);砂糖;クエン酸(citric acid)やクエン酸ナトリウム(sodium citrate)のような緩衝物質;グリシン(glycine)および塩酸(hydrochloric acid)、リン酸ナトリウム(sodium phosphate)、およびリン酸カリウム(potassium phosphate);p-ヒドロキシ安息香酸(p-hydroxybenzoic acid)のエステルのような保存薬および静菌薬;着色剤;および医薬品で一般に使用されている種々の香料および甘味料が含まれうる。
【0056】
適する甘味料の例としては、これに限定はされないが、公知であるあらゆる甘味料、例えば、砂糖、糖アルコール、高甘味度甘味料(high intensity sweeteners)、およびそれらの混合物などがある。適する砂糖には、これに限定されないが、ショ糖(sucrose)、デキストロ−ス(dextrose)、トウモロコシの高果糖シロップ(high fructose corn syrup)、およびマルトース(maltose)がある。適する糖アルコールには、これに限定はされないが、ソルビトール(sorbitol)、キシリトール(xylitol)およびマンニトール(mannitol)がある。適する高甘味度甘味料には、これには限定されないが、スクラロース(sucralose)、アスパルテーム(aspartame)、サッカリン(saccharin)、およびアセスルファームK(acesulfame K)がある。
【0057】
ある実施形態では、懸濁液剤形の放出制御部分に含まれている少なくとも1つの活性成分のpKaを懸濁液剤形全体のpHよりも大きくするのに有効な量の緩衝剤が使用される。
【0058】
さらに、賦形剤は、水、または水および医薬品として容認可能な公知の水混和性補助溶剤(water-miscible co-solvent)、例えば、グリコール(glycols)、アルコール(alcohols)およびグリセロール(glycerol)などとの混合物を含むことであってもよい。
【0059】
剤形の所定の実施形態では、当該技術で公知のどのような懸濁系が入っていてもよく、1つ以上の構造剤および/または1つ以上の膨張剤が通常入っている懸濁系などが入っていてもよい。ある実施形態では、剤形には、懸濁液剤形の全重量を基準として、約0.1%から約10%の懸濁系が入っている。適する懸濁系には、例えば、米国特許第5、374、659号、第5、621、005号、および第5、409、907号に開示されたものが含まれる。米国特許第5、374、659号、第5、621、005号、および第5、409、907号は、参照により本明細書にその全体が組み込まれる。
【0060】
本発明で使用するのに適した構造剤には、親水コロイドなどの親水性ポリマーがある。適する親水コロイドの例としては、アルギン酸塩(alginates)、寒天(agar)、グアールガム(guar gum)、イナゴマメ(locust bean)、カラゲナン(carrageenan)、タラ(tara)、アラビアゴム(gum arabic)、トラガカント(tragacanth)、ペクチン(pectin)、キサンタン(xanthan)、ゲラン(gellan)、マルトデキストリン(maltodextrin)、ガラクトマンナン(galactomannan)、パススタラン(pusstulan)、ラミナリン(laminarin)、スクレロガム(scleroglucan)、アラビアゴム(gum arabic)、イヌリン(inulin)、カラヤ(karaya)、ウィーリアン(whelan)、ラムザン(rhamsan)、ズーグラン(zooglan)、メチラン(methylan)、キチン(chitin)、シクロデキストリン(cyclodextrin)、キトサン(chitosan)、およびこれらの組み合わせがある。本発明の所定の実施形態では、キサンタン・ガム(xanthan gum)が構造剤である。
【0061】
キサンタン・ガムは、高分子量の天然炭水化物、特に、ポリサッカリド(polysaccharide)である。本発明での使用に適するキサンタン・ガムの1つは、キサントモナス・キャムペストリス(Xanthomonas campestris)によって生成される高分子量ポリサッカリドである。このポリサッカリドを生産するための技法および菌株は、米国特許第4、752、580号および第3、485、719号に開示されており、これらの特許の開示内容は参照によりここに組み込まれる。ある実施形態では、キサンタン・ガムが1パーセント食塩水において、25℃、60rpmで3番のスピンドルを使用しているLVモデルブルックフィールド・シンクロ−レクトリック(Brookfield Synchro-Lectric)粘度計で測定した場合に、約1000から約1700cP(mPa−sec)の粘度を有する。適するキサンタン・ガムは、例えば、CPケルコ(CP Kelco)から、商品名「Keltrol」、「Keltrol TF」および「Keltrol 1000」で入手できる。
【0062】
膨張剤は、適当な水性の環境に晒されると、ネットワークシステムを形成することなく膨張する。アルファ化デンプンは、特によい膨張剤である。アルファ化デンプンは、「インスタント」デンプンとしても知られており、冷たい水に入れたら直ちに膨張して厚みが増し始めるように下ごしらえがされている。特に適するアルファ化デンプンの1つは、改質され、安定化され、かつ、ろう質のトウモロコシ食品用デンプンから作られるものであり、ナショナル・スターチ・カンパニー(National Starch Company)から「INSTANT STARCH, ULTRASPERSE-M」として市販されている。適する他の膨張剤には、これに限定はされないが、微結晶性セルロース(microcrystalline cellulose)および/またはヒドロキシプロピルメチルセルロース(hydroxypropylmethylcellulose)がある。
【0063】
ある実施形態では、懸濁系が、アルファ化デンプンの膨張剤が入ったキサンタン・ガムの構造剤を含んでいる。他の実施形態では、懸濁系が、懸濁液剤形の全重量を基準に、約0.01パーセントから約1パーセント、または、約0.05パーセントから約0.40パーセントのキサンタン・ガムと、約1パーセントから約10パーセント、または、約0.5パーセントから約3.0パーセントの、ナショナル・スターチ・カンパニー(National Starch Company)から「INSTANT STARCH, ULTRASPERSE-M」として市販されているもののようなアルファ化デンプンを含む。
【0064】
ある実施形態では、剤形が水性医薬品懸濁組成という形態をしており、その水性医薬品懸濁液の容積あたりの活性成分の全重量(w/vまたはg/100mL)を基準として、約0パーセントより多く、約40パーセントまで、例えば、約0.05パーセントから約0.2パーセント、または約1.6パーセントから約10パーセント、または約15パーセントから約40パーセントまでの少なくとも1つの活性成分を含む。
【0065】
活性成分がロラティダイン(loratidine)である一実施形態では、懸濁液剤形における活性成分の量が、水性懸濁液剤形の容積当たりの活性成分の全重量(w/v)を基準として、約0.05パーセントから約0.2パーセントまであり、これは、水性懸濁液剤形のティースプーン一杯当たり約2.5ミリグラムから約10ミリグラムのロラティダインに相当する。
【0066】
活性成分がアセトアミノフェンである別の実施形態では、懸濁液剤形における活性成分の量は、水性懸濁液剤形の容積当たりの活性成分の全重量(w/v)を基準として、約1.6パーセントから約3.2パーセントであり、これは、ティースプーン一杯の水性懸濁液剤形当たり約80mgから約160mgに相当する。アセトアミノフェンが入っているさらに別の実施形態では、懸濁液剤形における活性成分の量は、水性懸濁液剤形の容積当たりの活性成分の全重量(w/v)を基準として、約5パーセントから約10パーセントであり、これは、水性懸濁液剤形1.6mL当たり約80mgから約160mgに相当する。
【0067】
活性成分がイブプロフェンである別の実施形態では、懸濁液剤形における活性成分の量は、水性懸濁液剤形の容積当たりの活性成分の全重量(w/v)を基準として、約50から約200mgであり、例えば水性懸濁液剤形ティースプーン一杯当たり約50mgから約100mgであり、または水性懸濁液剤形1mL当たり約40mgの活性成分であり、これは、水性懸濁液剤形の容積当たりの活性成分の全重量(w/v)を基準として、約1パーセントから約4パーセントに相当する。
【0068】
本発明の一実施形態は、測定可能な懸濁液組成に向けられており、これは、懸濁液の全重量を基準として:a)約0.05パーセントから約40パーセントの少なくとも1つの活性成分;b)約20パーセントから約70パーセントの水;c)約0.1パーセントから約10パーセントの懸濁系;d)約0パーセントから約40パーセント、例えば約20パーセントから約40パーセントの甘味剤;およびe)約0パーセントから約0.2パーセントの賦形剤、を含む。この実施形態において、活性成分の全重量を基準として、活性成分の約50パーセントから約75パーセントが即放性量部分にあり、活性成分の約25パーセントから約50パーセントが放出制御量部分である。同じ実施形態において、懸濁液の全重量に基づいて、その剤形の約0.025パーセントから約30パーセントが即放性量部分の活性成分からなり、その剤形の約0.0125パーセントから約0.025パーセントが放出制御量部分の活性成分からなる。
【0069】
所定の実施形態において、本発明の懸濁液の粘度は、ブルックフィールド(Brookfield)DV−1+粘度計により、12rpmで回る31番スピンドルを1つ用いて、温度条件が約25℃の下で測定した場合に約400cpsから約1500cpsの範囲であることがある。
【0070】
本発明の他の実施形態では、一層の放出制御コーティングで実質的に覆われている、すなわち、コーティングされている活性成分粒子が入っている、および/または放出制御成分からなるマトリックスに分散された活性成分粒子が入っている水性懸濁液剤形に向けられている。
【0071】
本発明の剤形は、NSAIDおよび/またはアセトアミンフェンのような活性成分の有効量を毎日1回または2回の投与で送達することを意図している。鎮痛薬の「有効量」とは、患者の苦痛を軽減する量である。例えば、イブプロフェンの典型的な大人の投与量は、患者の体重に対し約2.9から約12mg/kgを4から6時間毎に与えることであろうし、典型的な1日当たりの投与量は約11.6から約72mg/kg/日であろう。したがって、典型的な70kgの大人に対するイブプロフェンの有効量の投与は、例えば40mg/mLのイブプロフェンが入っている本発明の製剤を約5mLから約60mL、毎日1、2回投与することを伴う。イブプロフェンの小児用の典型的な投与量は、約5から約10mg/kgを4から6時間毎に与えることであろうし、典型的な一日当たりの投与量は約20から約60mg/kg/日であろう。典型的な15kgの子供に対するイブプロフェンの有効量の投与は、例えば20mg/mLのイブプロフェンが入っている、本発明の製剤を約5mLから約30mL、毎日1、2回投与することを伴う。
【0072】
本発明の剤形の経口投与は、ユーザーにNSAIDやアセトアミノフェンのような活性成分を、オプションとしての即放性量、ならびに、投与してから約6時間後、例えば、約8時間後または約10時間後に剤形から活性成分を放出し続ける放出制御量で提供する。有り難いことに、我々は予期せずして、剤形が懸濁液のような液体剤形としてデザインされているか、投与前に水で戻すことができる乾式剤形としてデザインされているかに拘わらず、製品の有効期間を通して、かつ、治療時間を通して、剤形の放出制御部分の放出特性が如何にして効果的に安定させるかを発見した。特に、我々は、製品における粒子からの活性成分が摂取前に放出されることを防ぎつつ、胃腸液におけるその同じ粒子から活性成分の放出制御を可能にするという難題を克服した。
【0073】
都合のよいことに、本発明の製剤は、様々な形式で使用することができ、これには、例えば、(i)正確に計量できるシングルドーズ乾式製剤または懸濁液;(ii)必要に応じて再懸濁化する顆粒を異なる量だけ計量することにより、投与量の融通が非常にきくマルチドーズ顆粒製剤;(iii)マルチドーズ懸濁液;および(iv)活性成分が懸濁している濃縮ドロップであって、特に小児用途に有用であるもの、がある。
【0074】
さらに、この製剤は投与したり、飲み込んだりするのに便利であるので、また、活性成分の毎日の投与回数が少なくなるので、総合的な患者のコンプライアンスが達成される。投与、飲み込みが容易であることから、小児科診療でも有益であることが期待される。
【0075】
保存期間中安定である懸濁液を作るために、一連の腸溶製コーティングを医薬品活性成分に塗布する必要があった従来の放出制御医薬品懸濁液と異なり、本発明の懸濁医薬品粒子は、水または水混和性補助溶剤(water-miscible co-solvent)のあるところで安定であるために、新規の徐放性コーティングを一層しか被覆する必要がない。
【0076】
下記の例は、本発明をさらに説明するものであるが、決して本発明を制限するものではない。
【0077】
実施例1:放出制御コーティング溶液の調合
コーティング溶液は、ローム・パルマ・インコーポレテッドから商品名「Eudragit L-100」で市販されているメタクリレート共重合体(methacrylate co-polymer)と、酢酸セルロース(cellulose acetate)とを、溶媒の全重量を基準として、98%のアセトンと、2%の水が入っている溶媒に周囲条件下で分散させて調合した。
【0078】
結果として得られたコーティング溶液は、全湿式コーティング溶液を基準として、7.6%の酢酸セルロース(cellulose acetate)、0.4%のメタクリレート共重合体(methacrylate co-polymer)、90.2%のアセトンおよび1.8%の水を含んでいた。
【0079】
固体の相対的な量は、乾燥させたコーティング溶液の全重量パーセントを基準として、酢酸セルロース(cellulose acetate)が95.00%と、メタクリレート共重合体(methacrylate co-polymer)が5.00%であった。
【0080】
実施例2:コーティングした活性成分の調合
イブプロフェン予混合物の調合:イブプロフェンUSPパウダーをコロイド状二酸化ケイ素(colloidal silicon dioxide)と混合して、下記のイブプロフェン予混合物を形成した:
成分 重量パーセント*
コロイド状二酸化ケイ素 2.00%
イブプロフェンUSP 98.00%
*イブプロフェン予混合物の全重量を基準とする
【0081】
コーティングしたイブプロフェン顆粒の調合:上記のように調合したイブプロフェン混合物を次に実施例1にしたがって調合した湿式放出制御コーティング溶液を用いて、約20.0g/分の速度で、グラットGPCG-5/9ウルスター流動層コーティング装置(Glatt GPCG-5/9 Wurster fluid bed coating unit)で、約29−32℃の製造温度条件下でコーティングした。その結果得られたコーティングされたイブプロフェン顆粒は、イブプロフェン顆粒および放出制御コーティングの総乾燥重量を基準として、放出制御コーティングが約20%含まれていた。
【0082】
実施例3:即放性量および放出制御量を含む懸濁液ベースの製造
懸濁液ベースの調合
【0083】
【表1】

【0084】
上記表Aに示したように、純水USPは、スコットターボン(Scott Turbon)製高剪断ミキサーが備え付けられている混合タンクに充填し、十分な渦ができるように約500rpmから約1000rpmで混合した。次に、アルファ化デンプンとキサンタン・ガムを混合タンクに加え、20分間混合した。次にグリセリンをそこに加え、5分間混合した。次にサッカロースをそこに加え、10分間混合した。次に、ポリソルベート−80NF(polysorbate-80 NF)、クエン酸(citric acid)USPおよび安息香酸ナトリウムNF(sodium benzoate NF)を順に加え、その結果得られた混合物を10分間混合した。次に純水の残りを混合しながらそこに加えて懸濁液ベースを形成した。
【0085】
活性成分を含む懸濁液の調合
2000.0mgのイブプロフェンUSPを50番のメッシュ網でふるった後、上記のように調合した懸濁液ベース25mLをそこに加え、均質になるまでその混合物を混合した。
【0086】
実施例2にしたがって調合した放出制御コーティングをしたイブプロフェン1276mg(活性イブプロフェン78.4%が含まれる)を60番と80番のメッシュ網の間でふるい、次に混合物に加えた。
【0087】
できあがった懸濁液は、次に懸濁液ベースを加えて容積100.0mLに希釈し、得られた懸濁液が均質になるまで混合した。できた懸濁液を40番のメッシュ網を使ってふるった後、できあがった、最終的にふるい分けられた懸濁液は、100mg/5mLの即放性イブプロフェン量と、50mg/5mLの放出制御イブプロフェン量とを含んでいた。イブプロフェン粒子の相対的な量は、最終的にふるい分けられた懸濁液の総量を基準として:
イブプロフェンUSP(即放性量)..........100.0mg/5mL
コーティングしたイブプロフェン(放出制御量).....50.0mg/5mL
であった。
【0088】
実施例4:即放性量および放出制御量を含む懸濁液ベースの溶出性分析
pH5.6のアセテート緩衝液溶出媒体900mLをバドル付きのUSPタイプIIの装置の3つの容器の各々に入れた。次に、実施例3で作った最終懸濁液の試料5.0mLを3つの容器の各々に個別に加え、50r.p.m.、37℃において混合物が均質になるまで混合した。
【0089】
その後、それぞれ0.5、1、2、3、4、6、8、および12時間後に、懸濁液/緩衝液混合物の試料10mLを容器から個別に取り出した。
【0090】
次に、それぞれ0.5、1、2、3、4、6、8、および12時間におけるイブプロフェンの溶出曲線を求めるために、Waters(登録商標)717自動注入器と、波長254nmに設定したWaters(登録商標)486UV検出器とを備えた高圧液体クロマトグラフィー(HPLC)装置を使って、各試料10mLを個別にイブプロフェン内容分析した。試料の各々は、イブプロフェンの標準試料と比較した。イブプロフェンの標準試料は、イブプロフェンが0.167mg/mLであるアセテート緩衝液(pH5.6)溶出媒体を含んでおり、イブプロフェンの100%放出に必要とされる理論濃度と相関している。
【0091】
HPLCで使用する移動相は、アセトニトリル(acetonitrile)55%と、18mMリン酸カリウム緩衝液(potassium phosphate buffer)45%を含む試料を使って調合した。注入容積200μL、実行時間約7分、ポンプ流1.5mL/分であった。分析に使用したカラム(column)は、フェノメネクス・ルナ(Phenomenex LUNA)(登録商標)5umC8(4.6mm×15cm)であった。
【0092】
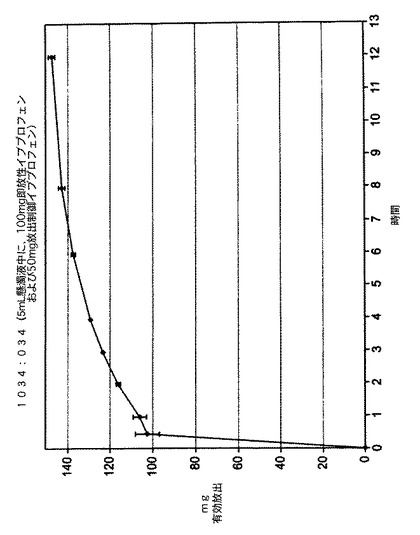
この溶出性分析の結果は、図1に記載した。この結果は、本発明の懸濁液が活性成分の即放性量ならびに活性成分の放出制御量の両方を含むことができ、放出制御量が初期投与から約12時間にわたってイブプロフェンを放出したことを示している。
【0093】
実施例5:放出制御量を含む懸濁液ベースの製造
実施例3の手順を繰り返したが、2000.0mgのイブプロフェンUSPを加えなかった。
【0094】
得られた、最終的にふるい分けた懸濁液は、放出制御イブプロフェン量100mg/5mLを含んでいた。
【0095】
実施例6:放出制御量を含む懸濁液ベースの溶出性分析
実施例4の手順を繰り返したが、試料は、実施例5で作った最終懸濁液を用いた。
【0096】
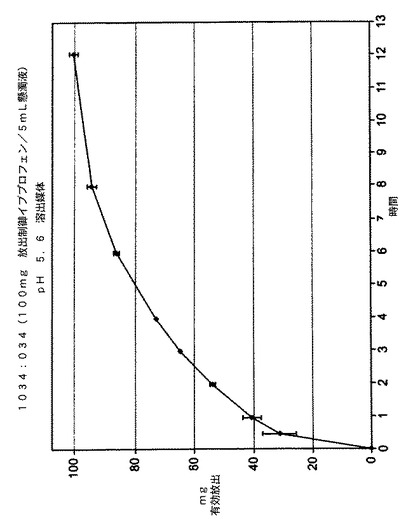
溶出性分析の結果は、図2に記載した。この結果は、本発明の懸濁液が、活性成分の即放性量が実質的にない状態で活性成分の放出制御量を含むことができ、放出制御量が初期投与から約12時間にわたってイブプロフェンを放出したことを示している。
【0097】
〔実施の態様〕
(1) 医薬品懸濁液剤形において、
NSAIDおよび/またはアセトアミノフェンの粒子であって、放出制御成分からなる1つの層で実質的に覆われている粒子を含み、
前記医薬品懸濁液剤形の治療効果の持続時間が、哺乳動物への最初の投与後、少なくとも8時間ある、剤形。
(2) 実施態様1記載の剤形において、
前記粒子を投与するための賦形剤をさらに含んでいる、剤形。
(3) 実施態様2記載の剤形において、
前記賦形剤が、懸濁系、界面活性剤、甘味料、緩衝剤、保存薬、香料添加剤、およびこれらの混合物からなる一群から選択された1つ以上の添加剤を含む、剤形。
(4) 実施態様2記載の剤形において、
前記賦形剤が、水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物を含む、剤形。
(5) 実施態様1記載の剤形において、
前記粒子が、前記放出制御成分で実質的に覆われた芯物質を含む、剤形。
【0098】
(6) 実施態様5記載の剤形において、
前記放出制御成分が、前記コーティングの総乾燥重量を基準として、0パーセントより多くて100パーセント未満の不溶性フィルム形成ポリマーと、0パーセントから10パーセント未満の腸溶性ポリマーとを含んでいる、剤形。
(7) 実施態様5記載の剤形において、
前記放出制御コーティングに実質的に腸溶性ポリマーがなく、前記粒子に含まれる少なくとも1つの活性成分のpKaが前記懸濁液剤形のpHよりも大きい、剤形。
(8) 実施態様5記載の剤形において、
前記放出制御コーティングにおける前記不溶性フィルム形成ポリマーと前記腸溶性ポリマーとの重量比が80:20から99:1である、剤形。
(9) 実施態様5記載の剤形において、
前記不溶性フィルム形成ポリマーが、酢酸セルロース、エチルセルロース、重量比1:2:0.1のポリ(アクリル酸エチル、メタクリル酸メチル、トリメチルアンモニウムエチル・メタクリレート・クロライド)と、その共重合体および混合物からなる一群から選択されている、剤形。
(10) 実施態様5記載の剤形において、
前記腸溶性ポリマーが、ヒドロキシプロピル・メチルセルロース・フタレート、ヒドロキシプロピル・メチルセルロース・アセテート・シクシナート、セルロース・アセテート・フタレート、ポリビニルアセテート・フタレート、ポリメタクリレートをベースとするポリマー、ならびにこれらの共重合体および混合物からなる一群から選択されている、剤形。
【0099】
(11) 実施態様10記載の剤形において、
前記ポリメタクリレートをベースとするポリマーが、重量比1:2のポリ(メタクリル酸、メチルメタクリレート)および/または重量比1:1のポリ(メタクリル酸、メチルメタクリレート)である、剤形。
(12) 実施態様5記載の剤形において、
前記コーティングした粒子が、前記コーティングした粒子の総乾燥重量を基準として、10パーセントから40パーセントの前記放出制御成分を含む、剤形。
(13) 実施態様1記載の剤形において、
前記治療効果が鎮痛である、剤形。
(14) 実施態様1記載の剤形において、
前記NSAIDがプロピオン酸誘導体NSAIDである、剤形。
(15) 必要に応じて哺乳動物の痛みを治療する方法において、
実施態様1記載の剤形を、前記剤形の投与後、少なくとも12時間、前記哺乳動物の鎮痛を行うのに有効な量だけ投与することを含む、方法。
【0100】
(16) 医薬品懸濁液剤形において、
a)NSAIDおよび/またはアセトアミノフェンを含む粒子であって、放出制御コーティングからなる1つの層で実質的に覆われている粒子と、
b)水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物と、を含み、
前記医薬品剤形の治療効果の持続時間が、前記医薬品剤形の投与後、少なくとも8時間ある、剤形。
(17) 実施態様16記載の医薬品懸濁液において、
前記懸濁液の全重量を基準として、
a)0.05パーセントから40パーセントの、NSAIDおよび/またはアセトアミノフェンを含む粒子であって、放出制御コーティングからなる1つの層で実質的に覆われている粒子と、
b)20パーセントから70パーセントの水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物と、を含み、
前記医薬品剤形の治療効果の持続時間が、前記医薬品剤形の投与後、少なくとも8時間ある、医薬品懸濁液。
(18) 必要に応じて哺乳動物の痛みを治療する方法において、
実施態様17記載の剤形を、前記剤形の投与後、少なくとも12時間、前記哺乳動物の鎮痛を行うのに有効な量だけ投与することを含む、方法。
(19)アセトアミノフェンおよび/またはNSAIDを懸濁液医薬品剤形の状態で必要に応じて哺乳動物に投与する方法において、
前記哺乳動物が、前記アセトアミノフェンおよび/またはNSAIDの放出制御ドーズを前記剤形の投与後12時間にわたって受け、前記12時間の間にアセトアミノフェンおよび/またはNSAIDをさらに与えることがないように、前記剤形を哺乳動物に与えることを含む、方法。
(20) 医薬品懸濁液剤形において、
a)NSAIDおよび/またはアセトアミノフェンを含む粒子であって、放出制御成分からなる1つの層で実質的に覆われており、前記放出制御成分が、前記放出制御成分の全重量を基準として、0パーセントより多くて90パーセント未満の不溶性フィルム形成ポリマーと、0パーセントより多くて10パーセント未満の腸溶性ポリマーとを含む、粒子と、
b)水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物と、を含み、
前記医薬品剤形の治療効果の持続時間が、投与後、少なくとも12時間ある、剤形。
【0101】
(21) 実施態様20記載の医薬品懸濁液剤形において、
前記剤形の全重量を基準として、
a)0.05パーセントから40パーセントまでの、NSAIDおよび/またはアセトアミノフェンを含む粒子であって、放出制御成分からなる1つの層で実質的に覆われており、前記放出制御成分が、前記放出制御成分の全重量を基準として、0パーセントより多くて90パーセント未満の不溶性フィルム形成ポリマーと、0パーセントより多くて10パーセント未満の腸溶性ポリマーとを含む、粒子と、
b)20パーセントから70パーセントの水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物と、を含み、
前記医薬品剤形の治療効果の持続時間が、投与後、少なくとも12時間ある、剤形。
(22) 実施態様21記載の剤形において、
前記放出制御コーティングにおける前記不溶性フィルム形成ポリマーと、前記腸溶性ポリマーとの重量比が、99:1から80:20である、剤形。
【図面の簡単な説明】
【0102】
【図1】イブプロフェンの即放性ドーズおよび放出制御ドーズの両方を含む懸濁液剤形に関する放出された活性成分(mg)対時間(時間)のグラフである。
【図2】イブプロフェンの放出制御ドーズのみを含む懸濁液剤形に関する放出された活性成分(mg)対時間(時間)のグラフである。
【開示の内容】
【0001】
本発明は、鎮痛剤のような活性成分を投与するための液状剤形に適した放出制御型医薬品製剤に関する。
【0002】
〔発明の背景〕
痛み、炎症、および熱を治療するための治療薬には、鎮痛剤、抗炎症剤、および解熱剤がある。非ステロイド系抗炎症剤(NSAID)は、このような治療薬の一種である。NSAIDには、プロピオン酸誘導体(propionic acid derivatives)、酢酸誘導体(acetic acid derivatives)、フェナミン酸誘導体(fenamic acid derivatives)、ビフェニルカルボン酸誘導体(biphenylcarbodylic acid derivatives)、オキシカム(oxicams)、およびシクロオキシゲナーゼ−2(COX−2)選択的NSAID(cyclooxygenase-2 (COX-2) selective NSAIDs)がある。
【0003】
プロピオン酸(propionic acid)には、例えば、イブプロフェン(ibuprofen)、ナプロキセン(naproxen)、およびケトプロフェン(ketoprofen)がある。特にイブプロフェンは、広く使用され、よく知られている、鎮痛特性および解熱特性を有するNSAIDである。イブプロフェンは、何年もの間、多くの形式で、一般用医薬品として市販されている。イブプロフェンは、化学的には、2−(4−イソブチルフェニル)プロピオン酸(2-(4-isobutylphenyl)-propionic acid)として知られている。
【0004】
NSAIDは、通常、一日当たり1回から4回投与され、一日当たりの投与量は、約50から約2000ミリグラムの範囲であり、好ましくは、約100から1600であり、もっとも好ましくは約200から約1200ミリグラムである。
【0005】
アセトアミノフェンは、よく知られた鎮痛剤であって、一日当たりの投与量は約325から約4000ミリグラムの範囲であり、好ましくは約650から約4000ミリグラムである。アセトアミノフェンは、1893年にヴァン・メリングが初めて医療で使用しているが、1949年になって初めて一般用医薬品市場で鎮痛剤として使用するアスピリンの有効な代替品として普及した。APAPの薬理学は、B. Ameer et al.、Ann. Int. Med. 87,202 (1977)で論評されている。APAPの普及および製造量を考慮すると、鎮痛剤としてのAPAPの製造および使用の両方が当業者に公知である。
【0006】
NSAID、アセトアミノフェン、その他の薬を12時間または24時間にわたって複数回投与することが知られている。例えば、同一量のイブプロフェンが入っている用量を複数回12時間から24時間にわたって投与することが知られている。さらに、最初に多量に投与し、続いて比較的少ない維持量を投与することも知られている。これについては、例えば、Palmisano et al., Advances in Therapy, Vol. 5, No. 4, July/August 1988(ケプトン(初期量150mgに続いて後続量75mg)およびイブプロフェン(初期量800mgに続いて後続量400mg)からなる複数回投与の使用)を参照のこと。
【0007】
特に、経口投与型鎮痛剤との関連で、治療効果の持続時間を延ばしたいという要望から、一日一回だけ投与すればよい放出制御製剤が開発された。一日一回の投与では、患者が治療処置を行っている間に言われた服用量を尊守するようになるので、都合がよい。
【0008】
放出制御医薬品剤形は、薬の送達を最適化し、かつ、患者のコンプライアンスを、特に患者が一日に取らなければならない薬の服用回数を減らすことにより、向上させるように長いこと使用されてきた。このために、薬または他の活性成分が剤形から患者の胃腸液に放出される速度を下げることが、特に体内での薬の作用を引き延ばすためには、多くの場合に望ましいことである。
【0009】
口を経て送り込まれた薬が体内の作用部位に到達する速度は、薬が胃腸粘膜を介して血液に吸収される速度および量を含む多くの要因に依存する。もっとも、薬が血液に吸収可能となる前に、まず薬が胃腸液に溶けなければならない。多くの薬では、胃腸膜を通しての吸収が胃腸液でのその薬の溶出に比べて速く、このために、薬の溶出が、薬の吸収における律速段階となっている。したがって、製剤業者は、血液への薬の吸収速度を薬の溶出速度を変えることで効果的に制御できる。
【0010】
医薬品剤形の場合、1つ以上の薬を、それぞれ速度を調節して送達することも特に望ましい。各薬の吸収、分配、代謝、および排泄がそうであるように、薬の治療効果の開始および持続時間には大きなばらつきがあるので、異なる薬の放出を異なる方法で調節すること、すなわち、第1の薬を剤形から直ちに放出させる一方で、第2の薬を「調節した」方法で、たとえば、遅らせて、または制御して放出することが多くの場合に望ましい。
【0011】
製剤が薬を速度を制御して送達できる(例えば、除放、持効、延長、または遅延放出)公知のメカニズムには、拡散、浸食、浸透がある。特定の活性成分について特に望ましい放出制御プロフィールを得るためには、上記メカニズムの組み合わせを利用した剤形をデザインすることが多くの場合に実用的である。
【0012】
不都合なことに、放出制御の多くの応用例では、最終的なサイズおよび重量が大きい固形用量単位(solid dosage unit)を利用している。このような用量単位を投与することは、特に子供やお年寄りのような飲み込むことが困難な患者に問題となる。したがって、このような放出制御医薬品をかみ砕くことができるまたは口でバラバラにすることができる固形形態または液状形態で提供することがより望ましい。多くの患者にとって、液状経口剤形がより好まれる。これは、かみ砕くという余分な段階なしで飲み込むことができるからである。
【0013】
経口液状製剤(oral liquid forms)は、長年、即放性薬剤を送達するのに一般的に使用されてきた。これについては、例えば、米国特許第5,374,659号、第4,788,220号、第4,975,465号、および第5,183,829号を参照のこと。しかし、放出制御製剤を液状剤形に導入することは、製剤上かなりの難題である。具体的には、通常、コーティングした、または化学的に結合させた粒子を利用して薬の放出調節部分を保持する。このような粒子、ならびにこれら粒子を浮遊させるための液状賦形剤の特性は、粒子を一様に分散した状態に維持できるように適合していなければならない。特に難題となるのは、患者が摂取する前の液状剤形の保存期間中に浮遊粒子から薬が早くに放出されてしまうのを防止することである。さらに、所望の溶出性プロフィール、ならびに、液状剤形の所望の用量均一性をその有効期間を通して維持することは、経口液状放出制御懸濁製品の製剤において取り組むべき別の難題である。
【0014】
米国特許第5,527,545号では、懸濁液状の剤形の放出特性を保つために、活性成分微粒剤を連続した4つのコーティングで被覆している。しかしながら、複数のコーティング段階は、製品の全体のコストおよび製造サイクルタイムを増大させるだけでなく、できあがった剤形は、ユーザに即放性用量を提供するものではない。
【0015】
したがって、懸濁化できる、鎮痛薬のような活性成分を有し、口に合うだけでなく、投与後に要求される放出プロフィールを保証する安定な形態でもある、液状の放出制御剤形があればさらに望ましいことである。さらに、使用者に鎮痛剤の即放出量および持続放出量の両方を提供する鎮痛懸濁製品を有することも望ましい。
【0016】
〔発明の概要〕
本発明は、NSAIDSおよび/またはアセトアミノフェンを懸濁液で投与するのに適する医薬品剤形を提供するものであり、この剤形は、
a)NSAIDおよび/またはアセトアミノフェンを含む第1部分であって、NSAIDおよび/またはアセトアミノフェンが、剤形が溶出媒体と接触したときに、実質的に直ちに剤形から放出される第1部分と、
b)NSAIDおよび/またはアセトアミノフェンを含む粒子からなる第2部分であって、剤形が前記溶出媒体と接触したときに、NSAIDおよび/またはアセトアミノフェンが粒子から制御されながら放出される第2部分と、を備える、からなる、および/または、から本質的に構成され、
医薬品剤形の治療効果の持続時間が、医薬品剤形の投与後、少なくとも8時間ある。
【0017】
本発明の別の実施形態は、懸濁液剤形に向けられていて、この懸濁液剤形は、
a)NSAIDおよび/またはアセトアミノフェンを含む第1部分であって、NSAIDおよび/またはアセトアミノフェンが、剤形が溶出媒体と接触すると、実質的に直ちに剤形から放出される第1部分と、
b)NSAIDおよび/またはアセトアミノフェンを含む粒子からなる第2部分であって、剤形が溶出媒体と接触すると、NSAIDおよび/またはアセトアミノフェンが粒子から制御されながら放出される第2部分と、
c)水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物と、を含む、からなる、および/または、から本質的に構成され、
剤形の治療効果の持続時間が、投与後、少なくとも12時間ある。
【0018】
本発明の他の実施形態は、アセトアミノフェンおよび/またはNSAIDを医薬品剤形の状態で必要に応じて哺乳動物に投与する方法に向けられており、この方法は、哺乳動物が、アセトアミノフェンおよび/またはNSAIDの即放性量を上記12時間の最初に受け、アセトアミノフェンおよび/またはNSAIDの放出制御量を剤形の投与後12時間にわたって受けるように剤形を哺乳動物に与えることを含む、からなる、および/または、から実質的になり、上記12時間の間にアセトアミノフェンおよび/またはNSAIDをさらに与えることがない。
【0019】
本発明の他の実施形態は、医薬品懸濁液剤形に向けられており、この剤形は、
NSAIDおよび/またはアセトアミノフェンからなる粒子であって、放出制御成分からなる1つの層で実質的にコーティングされている粒子、を含む、からなる、および/または、から実質的に構成されており、
この医薬品懸濁液剤形は、治療効果の持続時間が哺乳動物に最初に投与した後、少なくとも約8時間ある。
【0020】
本発明の別の実施形態は、医薬品懸濁液剤形に向けられており、この剤形は、
a)NSAIDおよび/またはアセトアミノフェンを含む粒子であって、放出制御コーティングからなる1つの層で実質的に覆われている粒子と、
b)水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物と、を備える、からなる、および/または、から実質的に構成されており、
医薬品剤形の治療効果の持続時間が、医薬品剤形の投与後、少なくとも約8時間ある。
【0021】
本発明の他の実施形態は、アセトアミノフェンおよび/またはNSAIDを懸濁液医薬品剤形の状態で必要に応じて哺乳動物に投与する方法に向けられており、この方法は、その哺乳動物が、上記剤形の投与後約12時間にわたって上記アセトアミノフェンおよび/またはNSAIDの放出制御量を受けるように、哺乳動物に上記剤形を与えることを含む、からなる、および/または、から実質的になり、アセトアミノフェンおよび/またはNSAIDを上記12時間の間にさらに与えることはない。
【0022】
本発明の他の実施形態は、医薬品懸濁液剤形に向けられており、この剤形は、
a)NSAIDおよび/またはアセトアミノフェンを含む粒子であって、放出制御成分からなる1つの層で実質的に覆われており、この放出制御成分が、その放出制御成分の全重量を基準として、約0パーセントよりも多く、約90パーセント未満の不溶性フィルム成形ポリマーと、約0パーセントよりも多く、約10パーセント未満の腸溶性ポリマーとを含む粒子と、
b)水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物と、を備える、からなる、および/または、から実質的になり、
この医薬品剤形の治療効果の持続時間が、投与後、少なくとも12時間ある。
【0023】
〔本発明の詳細な説明〕
本明細書で用いる限り、「実質的に覆う」または「実質的に連続」という言葉は、コーティングがほぼ連続であり、芯物質またはその下にある層の表面全体をほぼ覆っており、このため、活性成分、すなわち下にある層がほとんどまたは全く露出していないことを意味する。
【0024】
本明細書で使用する限り、「ATDAIRD」は、特定活性成分の有効即放性量の平均的な治療作用継続時間を意味する。例えば、イブプロフェン(ibuprofen)またはケトプロフェン(ketoprofen)の即放性量の典型的な作用継続時間、すなわち治療効果時間は、約4時間から約6時間である。したがって、イブプロフェンまたはケトプロフェンのATDAIRDは5時間である。ナプロキセン(naproxen)の即放性量の典型的な作用継続時間は、約8時間から約12時間である。したがって、ナプロキセンのATDAIRDは10時間である。特定の活性成分の治療作用継続時間は、その特定の活性成分が入っている即放性製品のラベル類における服用指示書から簡単に定めることができる。
【0025】
本明細書で使用する限り、「放出調節(modified release)」は、胃腸液のような溶出溶媒における活性成分の放出、すなわち溶出を変えることに適用する。調節しながら放出しうる活性成分は、例えば、剤形、コーティング、または粒子に入っていてもよいし、またはそれらのどこか一部、例えば、液状懸濁媒体じゅうに分散させた粒子などに入っていてもよい。放出調節の種類には、1)放出制御と、2)放出遅延とがある。「放出制御」は、投与後に、活性成分が剤形から実質的に連続的に、調節されながら放出され、その剤形から活性成分が完全に放出される、すなわち消耗するのに要する時間が、同じ活性成分からなる即放性剤形についてのそれよりも長いことを意味する。放出制御の種類には、延長(prolonged)、持続(sustained)、除放などがある。「放出遅延」は、投与後に、少なくとも一時期、活性成分が剤形から放出されないことを意味する。
【0026】
本明細書で使用する限り、「溶出溶媒(dissolution medium)」は、本発明の懸濁液剤形が溶出できる適切なあらゆる液体環境を意味し、例えば、製品検査で用いる生体外溶出溶媒、または胃腸液などを意味する。本発明の懸濁液剤形からの活性成分の溶出性を検査するのに用いる適切な生体外溶出溶媒には、USP23(1995)の786ページに記載されたものがあり、これらは、参照することにより本明細書に組み込まれる。
【0027】
本発明の一実施形態は、活性成分を懸濁液で投与するのに適した放出制御医薬品剤形に向けられたものであり、a)即放性部分、例えば、その剤形から直ちに放出される少なくとも一つの活性成分を含む部分と、b)放出制御部分、例えば、ある調整された時間にわたって、例えば、その剤形を最初に投与したあと約4時間から約12時間にわたって、実質的に連続的に血流に放出される少なくとも一つの活性成分を含む部分と、を含んでいる。本明細書で使用する限り、「即放性」とは、少なくとも一つの活性成分の溶出特性が、その活性成分を含む即放性錠に対するUSP規格を満たすことを意味する。例えば、アセトアミノフェン(acetaminophen)錠については、pH5.8のリン酸塩緩衝剤(phosphate buffer)において、USP装置2(パドル)を50rpmで使用すると、その剤形に含まれているアセトアミノフェンの少なくとも80パーセントが投与後30分以内にその剤形から放出されるとUSP24に指定されており、イブプロフェン(ibuprofen)錠については、pH7.2のリン酸塩緩衝剤において、USP装置2(パドル)を50rpmで使用すると、その剤形に含まれるイブプロフェンの少なくとも80パーセントが投与後60分以内にその剤形から放出されるとUSP24に指定されている。これについては、2000年版のUSP24(1999)の19−20ページ及び856ページを参照のこと。さらに、イブプロフェン懸濁液(ibuprofen suspension)の溶出性は、pH5.6のアセテート緩衝剤を使い、USP装置2(パドル)を50rpmで使って分析することができ、即放性量については、その剤形に含まれているイブプロフェンの少なくとも80パーセントがその剤形から投与後60分以内に放出される。
【0028】
即放性部分は、剤形内に分子レベルで分散している、例えば、融解または溶解された一つ以上の活性成分を含んでいてもよい。あるいは、活性成分は粒子という形態であってもよい。この粒子は、さらにコーティングされていてもよく、されていなくてもよい。活性成分が粒子という形態をしている実施形態では、粒子は(コーティングされていても、いなくても)、通常、平均粒径が約1ミクロンから約2000ミクロンまでである。ある実施形態では、このような粒子が、平均粒径が約1ミクロンから約300ミクロンまでの結晶という形態をしている。他の実施形態では、粒子が顆粒またはペレットという形態をしていて、平均粒径が約25ミクロンから約2000ミクロンまでであり、例えば、約25ミクロンから約1000ミクロンまでであり、または、約25ミクロンから約400ミクロンまでである。
【0029】
放出制御部分は、放出制御特性を有する多数の粒子に少なくとも一つの活性成分を含んでいる。ある実施形態では、放出制御部分におけるこれらの粒子の芯物質が純粋な結晶の形態をした活性成分を含むことがあり、この結晶は放出調節成分で実質的にコーティングされている。あるいは、粒子の芯物質は、顆粒を混和したものを含むことがあり、この顆粒が一つ以上の活性成分を、結合材、賦形剤などの当該技術において公知の、オプションとしての成分とともに含み、また、このような顆粒もまた放出制御成分で実質的にコーティングされている。別の実施形態では、活性成分の粒子を放出制御成分からなるマトリックスの全体に分散させることがある。さらに別の実施形態では、一つ以上の活性成分を化学的に結合、すなわち、「錯体化」して樹脂、例えば、イオン交換樹脂にし、粒子を形成していることがある。この粒子は、オプションとして、放出調整コーティングで実質的にコーティングすることもある。本明細書で使用する限り、「実質的にコーティングする」とは、粒子の表面積の約1パーセント未満、例えば約0.1パーセント未満が、露出していること、例えば、所望のコーティングで覆われていないことを意味する。
【0030】
このような実施形態で使用する具体的なイオン交換樹脂が、いくつかの要素、例えば活性成分のイオン電価などに左右されることは、当業者に、必要以上の実験を行うことなく容易に分かるであろう。NSAID活性成分に適するイオン交換樹脂の例としては、これに限定はされないが、コレスチラミン(cholestyramine)がある。コレスチラミンは、ローム・アンド・ハース(Rohm & Haas)から商品名「Duolite(登録商標)AP143」で市販されている。高分子樹脂を用いた錯体化に関する詳細は公知であり、例えば、米国特許第4,221,778号、第4,847,077号、および第6,001,392号に開示されている。米国特許第4,221,778号、第4,847,077号、および第6,001,392号は、参照により本明細書に組み込まれる。
【0031】
ある特定の実施形態では、剤形の放出制御部分に実質的にイオン交換樹脂がない。「実質的にイオン交換樹脂がない」は、イオン交換樹脂の量が、その剤形における全活性成分粒子の全重量を基準として、約1パーセント未満であることを意味し、例えば約0.5パーセント未満、または約0.1パーセント未満であることを意味する。
【0032】
粒子上の一つ以上のコーティング層は、適切なあらゆる放出制御成分を含んでいてもよい。適切な放出制御成分の一例は、放出制御成分の全重量を基準として、約0パーセントより多く約90パーセントまで、例えば約10パーセントから約60パーセントまでの不溶性フィルム形成ポリマーと、約0パーセントより多く、約10パーセント未満、例えば約0.5パーセントから約20パーセントまでの腸溶性ポリマーを含む。放出調整成分における腸溶性ポリマーの不溶性フィルム形成ポリマーに対する重量比は、約0.5:99.5から約20:80までの範囲であってもよく、例えば、約5:95から約10:90までの範囲であってもよい。同様の放出制御成分は、その全体に活性成分粒子を分散させることができるマトリックスに使用することもできる。
【0033】
適する不溶性フィルム形成ポリマーには、これに限定はされないが、酢酸セルロース(cellulose acetate), エチルセルロース(ethylcellulose)、ローム・パルマ(Rohm Pharma)から商品名「EUDRAGIT RS」で市販されているポリ(アクリル酸エチル、メタクリル酸メチル、トリメチルアンモニウムエチル・メタクリレート・クロライド)(poly(ethyl acrylate, methyl methacrylate, trimethylammonioethyl methacrylate chloride))1:2:0.1と、その共重合体および混合物がある。ある実施形態では、不溶性フィルム形成ポリマーが酢酸セルロースおよび/またはエチルセルロースから選択される。
【0034】
適する腸溶性ポリマーには、これに限定はされないが、ヒドロキシプロピル・メチルセルロース・フタレート(hydroxypropyl methylcellulose phthalate)、ヒドロキシプロピル・メチルセルロース・アセテート・シクシナート(hydroxypropyl methylcellulose acetate succinate)、セルロース・アセテート・フタレート(cellulose acetate phthalate)、ポリビニルアセテート・フタレート(polyvinylacetate phthalate)、ポリメタクリレート(polymethacrylate)をベースとするポリマー、ならびにその共重合体および混合物がある。適するポリメタクリレートをベースとするポリマーには、これに限定はされないが、ローム・パルマGmbH から商品名「EUDRAGIT S」ポリマーで市販されているポリ(メタクリル酸、メチルメタクリレート)(poly(methacrylic acid, methyl methacrylate)) 1:2と、ローム・パルマGmbH から商品名「EUDRAGIT L」ポリマーで市販されているポリ(メタクリル酸、メチルメタクリレート)(poly(methacrylic acid, methyl methacrylate)) 1:1とがある。ある実施形態では、腸溶性ポリマーを非アクリル酸化合物(non-acrylate compounds)から選択する。非アクリル酸化合物は、例えば、ヒドロキシプロピル・メチルセルロース・フタレート(hydroxypropyl methylcellulose phthalate)、ヒドロキシプロピル・メチルセルロース・アセテート・シクシナート(hydroxypropyl methylcellulose acetate succinate)、セルロース・アセテート・フタレート(cellulose acetate phthalate)、ポリビニルアセテート・フタレート(polyvinylacetate phthalate)、ならびに、これらの共重合体および混合物などである。
【0035】
ある実施形態では、放出制御成分に腸溶性ポリマーが実質的にない。つまり、例えば、放出制御成分に含まれる腸溶性ポリマーが、放出制御成分の全重量を基準として、約1パーセント未満、または0.25パーセント未満である。
【0036】
放出制御成分でコーティングされている活性成分粒子には、このようなコーティングされた粒子の全乾燥重量を基準として、約5パーセントから約40パーセントの、例えば約10パーセントから約30パーセントの放出制御成分を少なくとも1つのコーティング層という形態で入っている。
【0037】
コーティングされた活性成分粒子は、適するどのような公知方法で形成してもよい。適する粒子形成兼被覆方法には、高剪断造粒(high sheer granulation)、流動層造粒、例えば、ローター造粒(rotor granulation)、流動層コーティング(fluid bed coating)、コーアクサヴェーション(coaccervation)、噴霧乾燥、噴霧凝結などがあり、例えば、医薬剤形:錠剤第3巻、ヘルベルト A.リーバーマンおよびレオン・ラッチマン編集、第2、3および4章(1982年)(Pharmaceutical Dosage Forms: Tablets Volume 3, edited by Herbert A. Lieberman and Leon Lachman, Chapters 2,3, and 4 (1982))に記載されている。ある実施形態では、放出制御成分でコーティングした粒子の平均径が約20から約400ミクロンであり、例えば、約50から約300ミクロンである。
【0038】
本発明の剤形には、活性成分が1つ以上含まれている。適する活性成分に含まれるのは、だいたい、医薬品、ミネラル、ビタミンその他の栄養補助食品、口腔手入れ剤、香辛料類(flavorant)およびその混合物である。適する医薬品には、鎮痛薬(analgesics)、抗炎症剤(anti-inflammatory agents)、関節炎治療薬(antiarthritics)、麻酔薬(anesthetics)、抗ヒスタミン薬(antihistamines)、鎮咳剤(antitussives)、抗生物質(antibiotics)、抗感染剤(anti-infective agents)、抗ウィルス薬(antivirals)、抗凝固薬(anticoagulants)、抗うつ薬(antidepressants)、抗糖尿病剤(antidiabetic agents)、制吐薬(antiemetics)、抗膨満薬(antiflatulents)、抗菌剤(antifungals)、鎮痙薬(antispasmodics)、食欲減退薬(appetite suppressants)、気管支拡張薬(bronchodilators)、心・血管作動薬(cardiovascular agents)、中枢神経薬(central nervous system agents)、中枢神経興奮薬(central nervous system stimulants)、うっ血除去薬(decongestants)、経口避妊薬(oral contraceptives)、利尿薬(diuretics)、去痰薬(expectorants)、胃腸薬(gastrointestinal agents)、片頭痛製剤(migraine preparations)、乗物酔い製品(motion sickness products)、粘液溶解薬(mucolytics)、筋弛緩薬(muscle relaxants)、骨粗鬆症製剤(osteoporosis preparations)、ポリジメチルシロキサン(polydimethylsiloxanes), 呼吸器疾患に使用される医薬品(respiratory agents)、睡眠補助剤(sleep-aids)、尿路薬(urinary tract agents)およびその混合物が含まれる。
【0039】
適する香辛料類(flavorants)には、メントール、ペパーミント、ミント香辛料、フルーツ香辛料、チョコレート、バニラ、風船ガム香辛料、コーヒー香辛料、リキュール香辛料およびその組み合わせなどがある。
【0040】
適する胃腸薬の例としては、制酸薬(antacids)、例えば、炭酸カルシウム(calcium carbonate)、水酸化マグネシウム(magnesium hydroxide)、酸化マグネシウム(magnesium oxide)、炭酸マグネシウム(magnesium carbonate)、水酸化アルミニウム(aluminum hydroxide)、炭酸水素ナトリウム(sodium bicarbonate)、ジヒドロキシアルミニウム炭酸ナトリウム(dihydroxyaluminum sodium carbonate)など;刺激性下剤(stimulant laxatives)、例えばビサコジル(bisacodyl)、カスカラサグラダ(cascara sagrada)、ダントロン(danthron)、センナ(senna)、フェノールフタレイン(phenolphthalein)、アロエ(aloe)、ヒマシ油(castor oil)、リシノール酸(ricinoleic acid)、およびデヒドロコール酸(dehydrocholic acid)、およびそれらの混合物など;H2受容体拮抗薬(H2 receptor antagonists)、例えば、ファモチジン(famotadine)、ラニチジン(ranitidine)、シメチジン(cimetadine)、ニザチジン(nizatidine)など;プロトンポンプ阻害薬(proton pump inhibitors)、例えばオメプラゾール(omeprazole)またはランソプラゾール(lansoprazole)など;胃腸細胞保護剤(gastrointestinal cytoprotectives)、例えばスクラルフェート(sucraflate)およびミソプロストール(misoprostol);胃腸運動促進剤(gastrointestinal prokinetics)、例えばプルカロプリド(prucalopride)、人の幽門用の抗生物質(antibiotics for H. pylori)、例えばクラリスロマイシン(clarithromycin)、アモキシシリン(amoxicillin)、テトラサイクリン(tetracycline)、およびメトロニダゾール(metronidazole);下痢止め薬(antidiarrheals)、例えばジフェノキシラート(diphenoxylate)およびロペラミド(loperamide);グリコピロレート(glycopyrrolate);制吐薬(antiemetics)、例えばオンダンセトロン(ondansetron)、鎮痛薬(analgesics)、例えばメサラミン(mesalamine)などがある。
【0041】
適するポリジメチルシロキサンの例は、これには限定されないが、ジメチコーン(dimethicone)およびシメチコン(simethicone)が含まれ、米国特許第4,906,478号、5,275,822号および第6,103,260号に開示されたものである。米国特許第4,906,478号、5,275,822号および第6,103,260号の各々の内容は、参照することにより本明細書に特に組み込まれる。本明細書で使用する限り、「シメチコン」という用語は、ジメチコーンおよびシメチコン(simethicone)を含むがこれに限定されないポリジメチルシロキサンの上位のクラスをいう。
【0042】
本発明のある実施形態では、少なくとも1つの活性成分をビサコジル(bisacodyl)、ファモチジン(famotadine)、ラニチジン(ranitidine)、シメチジン(cimetidine)、プルカロプリド(prucalopride)、ジフェノキシラート(diphenoxylate)、ロペラミド(loperamide)、ラクターゼ(lactase)、メサラミン(mesalamine)、ビスマス(bismuth)、制酸薬(antacids)、および医薬品として容認できるそれらの塩、エステル、異性体、および混合物から選択することができる。
【0043】
別の実施形態では、少なくとも1つの活性成分が、鎮痛薬(analgesics)、抗炎症剤(anti-inflammatories)、および解熱薬(antipyretics)から選択され、例えば、非ステロイド系抗炎症薬(NSAIDs)であり、a)プロピオン酸誘導体(propionic acid derivatives)、例えば、イブプロフェン(ibuprofen)、ナプロキセン(naproxen)、ケトプロフェン(ketoprofen)など;b)酢酸誘導体、例えば、インドメタシン(indomethacin)、ジクロフェナク(diclofenac)、スリンダク(sulindac)、トルメチン(tolmetin)など;c)フェナミン酸誘導体(fenamic acid derivatives)、例えば、メフェナム酸(mefenamic acid)、メクロフェナム酸(meclofenamic acid)、フルフェナム酸(flufenamic acid)など;d)ビフェニルカルボン酸誘導体(biphenylcarbodylic acid derivatives)、例えばジフルニサル(diflunisal)、フルフェニサール(flufenisal)など;e)オキシカム(oxicams)、例えばピロキシカム(piroxicam)、スドキシカム(sudoxicam)、イソキシカム(isoxicam)、メロキシカム(meloxicam)など;f)シクロオキシゲナーゼ−2(COX−2)選択的NSAID(cyclooxygenase-2 (COX-2) selective NSAIDs);およびg)上記したものの医薬品として容認できる塩を含む。
【0044】
ある特定の実施形態では、少なくとも1つの活性成分がプロピオン酸誘導体NSAID (propionic acid derivative NSAID) から選択されている。プロピオン酸誘導体NSAIDは、-CH(CH3)COOHもしくは-CH2CH2COOH自由基を有する、医薬品として容認できる鎮痛剤/非ステロイド系抗炎症剤、または、医薬品として容認できる塩基、例えば-CH(CH3)COO-Na+またはCH2CH2COO-Na+などであって、通常、環構造、好ましくは芳香環構造に直接またはカルボニル官能性(carbonyl functionality)を介して取り付けられているものである。
【0045】
有用なプロピオン酸誘導体には、イブプロフェン(ibuprofen)、ナプロキセン(naproxen)、ベノキサプロフェン(benoxaprofen)、ナプロキセンナトリウム(naproxen sodium)、フェンブフェン(fenbufen)、フルルビプロフェン(flurbiprofen)、フェノプロフェン(fenoprofen)、フェブプロフェン(fenbuprofen)、ケトプロフェン(ketoprofen)、インドプロフェン(indoprofen)、ピルプロフェン(pirprofen)、カーポフェン(carpofen)、オクサプロフェン(oxaprofen), プラノプロフェン(pranoprofen)、ミクロプロフェン(microprofen)、ティオクサプロフェン(tioxaprofen)、スプロフェン(suprofen)、アルミノプロフェン(alminoprofen)、チアプロフェン酸(tiaprofenic acid)、フルプロフェン(fluprofen)、ブクロクス酸(bucloxic acid)、ならびに、これらの医薬品として容認できる塩、誘導体、および組み合わせがある。
【0046】
本発明のある実施形態では、プロピオン酸誘導体をイブプロフェン、ケトプロフェン、、フルルビプロフェン、ならびに、これらの医薬品として容認できる塩および組み合わせから選択する。
【0047】
他の実施形態では、プロピオン酸誘導体がイブプロフェン、2−(4−イソブチルフェニル)プロピオン酸、または、その医薬品として容認できる塩であり、例えば、イブプロフェンのアルギニン塩、リシン塩、またはヒスチジン塩(arginine, lysine, or histidine salt)などである。イブプロフェンの他の医薬品として容認できる塩は、米国特許第4,279,926号、第4,873,231号、第5,424,075号および第5,510,385号に記載されている。これらの米国特許の内容は、参照することにより組み込まれる。
【0048】
本発明の他の特定の実施形態では、少なくとも1つの活性成分をアセトアミノフェン(acetaminophen)、アセチルサリチル酸(acetyl salicylic acid)、イブプロフェン(ibuprofen)、ナプロキセン(naproxen)、ケトプロフェン(ketoprofen)、フルルビプロフェン(flurbiprofen)、ジクロフェナク(diclofenac)、シクロベンザプリン(cyclobenzaprine)、メロキシカム(meloxicam)、ロフェコキシブ(rofecoxib)、セレコクシブ(celecoxib)、およびこれらの医薬品として容認できる塩、エステル、異性体、および混合物から選択できる。
【0049】
本発明の別な特定の実施形態では、少なくとも1つの活性成分をプソイドエフェドリン(pseudoephedrine)、フェニルプロパノールアミン(phenylpropanolamine)、クロルフェニラミン(chlorpheniramine)、デキストロメトルファン(dextromethorphan)、ジフェンヒドラミン(diphenhydramine)、アステミゾール(astemizole)、テルフェナジン(terfenadine)、フェ クソ フェ ナ ディン(fexofenadine)、ロラタジン(loratadine)、デスロラタディン(desloratadine)、セチリジン(cetirizine)、これらの混合物、およびこれらの医薬品として容認できる塩、エステル、異性体、および混合物から選択することができる。
【0050】
本発明の別の特定の実施形態では、少なくとも1つの活性成分がNSAIDおよび/またはアセトアミノフェン、ならびに、これらの医薬品として容認できる塩である。
【0051】
ある実施形態では、治療上有効な量の1つまたは複数の活性成分が「単位投与量(unit dose volume)」に入っていることがある。この単位投与量は、粉末という形態であることもあり、水性懸濁液という形態であることもある。「治療上有効な量」は、本明細書で使用する限り、経口投与により所望の治療応答性を与える活性成分の量である。当業者は、所定の患者に対する活性成分の「治療上有効な量」を、例えば、投与される具体的な活性成分;その活性成分のバイオアベラビリティ特性;所望の用量;患者の年齢および体重;などの要因を考慮することによって容易に決めることができる。本明細書で使用する限り、「単位投与量」は、所定の製品を患者に経口投薬するのに都合のよいいかなる量であってもよい。
【0052】
この実施形態では、「単位投与量」に、通常、投与指示書がついている。投与指示書は、患者に、例えば患者の年齢または体重によっては単位投与量の数倍になりうる活性成分の量を取ることを指示する。通常、懸濁液の単位投与量には、もっとも小さい患者にとって治療上有効な量の活性成分が入っている。例えば、適切な単位投与量としては、一杯のティースプーン(約5mL)、一杯のテーブルスプーン(約15mL)、スポイト1滴、1mLというものがある。
【0053】
本発明によれば、NSAIDおよび/またはアセトアミノフェンが入っている剤形を、治療、特に鎮痛治療を必要としている哺乳動物に1回だけ投与して、活性成分を血液に長時間、例えば、約8時間にわたって、または約12時間にわたって放出することができる。時間0では、NSAIDおよび/またはアセトアミノフェンの初期投与量が哺乳動物に即放性量部分の活性成分によって与えられる、すなわち、投与される。活性成分は、活性成分を含む製剤を最初に投与してから約4時間後に、例えば、すなわち、約8時間、10時間、または12時間後に、放出制御量部分における活性成分によって血中に放出され続ける。換言すれば、この製剤には、最初に投与してから約4時間後、例えば、すなわち、約8時間、10時間、または12時間後に、溶出していない活性成分がまだ残っている。
【0054】
本発明を実施する場合、剤形は、活性成分の全重量を基準として、活性成分の即放性量部分を約25パーセントから約75パーセントまでと、活性成分の放出制御量部分を約75パーセントから約25パーセントまでを含むことができる。即放性量部分および放出制御量部分は、賦形剤で結合させて、必要な場合にその場で懸濁化できる乾燥混合物とするか、すぐに使える懸濁液とすることができる。
【0055】
適切な賦形剤の組成物としては、限定はしないが、構造化剤(structuring agents);膨張剤(swelling agents);界面活性剤(surfactants);砂糖;クエン酸(citric acid)やクエン酸ナトリウム(sodium citrate)のような緩衝物質;グリシン(glycine)および塩酸(hydrochloric acid)、リン酸ナトリウム(sodium phosphate)、およびリン酸カリウム(potassium phosphate);p-ヒドロキシ安息香酸(p-hydroxybenzoic acid)のエステルのような保存薬および静菌薬;着色剤;および医薬品で一般に使用されている種々の香料および甘味料が含まれうる。
【0056】
適する甘味料の例としては、これに限定はされないが、公知であるあらゆる甘味料、例えば、砂糖、糖アルコール、高甘味度甘味料(high intensity sweeteners)、およびそれらの混合物などがある。適する砂糖には、これに限定されないが、ショ糖(sucrose)、デキストロ−ス(dextrose)、トウモロコシの高果糖シロップ(high fructose corn syrup)、およびマルトース(maltose)がある。適する糖アルコールには、これに限定はされないが、ソルビトール(sorbitol)、キシリトール(xylitol)およびマンニトール(mannitol)がある。適する高甘味度甘味料には、これには限定されないが、スクラロース(sucralose)、アスパルテーム(aspartame)、サッカリン(saccharin)、およびアセスルファームK(acesulfame K)がある。
【0057】
ある実施形態では、懸濁液剤形の放出制御部分に含まれている少なくとも1つの活性成分のpKaを懸濁液剤形全体のpHよりも大きくするのに有効な量の緩衝剤が使用される。
【0058】
さらに、賦形剤は、水、または水および医薬品として容認可能な公知の水混和性補助溶剤(water-miscible co-solvent)、例えば、グリコール(glycols)、アルコール(alcohols)およびグリセロール(glycerol)などとの混合物を含むことであってもよい。
【0059】
剤形の所定の実施形態では、当該技術で公知のどのような懸濁系が入っていてもよく、1つ以上の構造剤および/または1つ以上の膨張剤が通常入っている懸濁系などが入っていてもよい。ある実施形態では、剤形には、懸濁液剤形の全重量を基準として、約0.1%から約10%の懸濁系が入っている。適する懸濁系には、例えば、米国特許第5、374、659号、第5、621、005号、および第5、409、907号に開示されたものが含まれる。米国特許第5、374、659号、第5、621、005号、および第5、409、907号は、参照により本明細書にその全体が組み込まれる。
【0060】
本発明で使用するのに適した構造剤には、親水コロイドなどの親水性ポリマーがある。適する親水コロイドの例としては、アルギン酸塩(alginates)、寒天(agar)、グアールガム(guar gum)、イナゴマメ(locust bean)、カラゲナン(carrageenan)、タラ(tara)、アラビアゴム(gum arabic)、トラガカント(tragacanth)、ペクチン(pectin)、キサンタン(xanthan)、ゲラン(gellan)、マルトデキストリン(maltodextrin)、ガラクトマンナン(galactomannan)、パススタラン(pusstulan)、ラミナリン(laminarin)、スクレロガム(scleroglucan)、アラビアゴム(gum arabic)、イヌリン(inulin)、カラヤ(karaya)、ウィーリアン(whelan)、ラムザン(rhamsan)、ズーグラン(zooglan)、メチラン(methylan)、キチン(chitin)、シクロデキストリン(cyclodextrin)、キトサン(chitosan)、およびこれらの組み合わせがある。本発明の所定の実施形態では、キサンタン・ガム(xanthan gum)が構造剤である。
【0061】
キサンタン・ガムは、高分子量の天然炭水化物、特に、ポリサッカリド(polysaccharide)である。本発明での使用に適するキサンタン・ガムの1つは、キサントモナス・キャムペストリス(Xanthomonas campestris)によって生成される高分子量ポリサッカリドである。このポリサッカリドを生産するための技法および菌株は、米国特許第4、752、580号および第3、485、719号に開示されており、これらの特許の開示内容は参照によりここに組み込まれる。ある実施形態では、キサンタン・ガムが1パーセント食塩水において、25℃、60rpmで3番のスピンドルを使用しているLVモデルブルックフィールド・シンクロ−レクトリック(Brookfield Synchro-Lectric)粘度計で測定した場合に、約1000から約1700cP(mPa−sec)の粘度を有する。適するキサンタン・ガムは、例えば、CPケルコ(CP Kelco)から、商品名「Keltrol」、「Keltrol TF」および「Keltrol 1000」で入手できる。
【0062】
膨張剤は、適当な水性の環境に晒されると、ネットワークシステムを形成することなく膨張する。アルファ化デンプンは、特によい膨張剤である。アルファ化デンプンは、「インスタント」デンプンとしても知られており、冷たい水に入れたら直ちに膨張して厚みが増し始めるように下ごしらえがされている。特に適するアルファ化デンプンの1つは、改質され、安定化され、かつ、ろう質のトウモロコシ食品用デンプンから作られるものであり、ナショナル・スターチ・カンパニー(National Starch Company)から「INSTANT STARCH, ULTRASPERSE-M」として市販されている。適する他の膨張剤には、これに限定はされないが、微結晶性セルロース(microcrystalline cellulose)および/またはヒドロキシプロピルメチルセルロース(hydroxypropylmethylcellulose)がある。
【0063】
ある実施形態では、懸濁系が、アルファ化デンプンの膨張剤が入ったキサンタン・ガムの構造剤を含んでいる。他の実施形態では、懸濁系が、懸濁液剤形の全重量を基準に、約0.01パーセントから約1パーセント、または、約0.05パーセントから約0.40パーセントのキサンタン・ガムと、約1パーセントから約10パーセント、または、約0.5パーセントから約3.0パーセントの、ナショナル・スターチ・カンパニー(National Starch Company)から「INSTANT STARCH, ULTRASPERSE-M」として市販されているもののようなアルファ化デンプンを含む。
【0064】
ある実施形態では、剤形が水性医薬品懸濁組成という形態をしており、その水性医薬品懸濁液の容積あたりの活性成分の全重量(w/vまたはg/100mL)を基準として、約0パーセントより多く、約40パーセントまで、例えば、約0.05パーセントから約0.2パーセント、または約1.6パーセントから約10パーセント、または約15パーセントから約40パーセントまでの少なくとも1つの活性成分を含む。
【0065】
活性成分がロラティダイン(loratidine)である一実施形態では、懸濁液剤形における活性成分の量が、水性懸濁液剤形の容積当たりの活性成分の全重量(w/v)を基準として、約0.05パーセントから約0.2パーセントまであり、これは、水性懸濁液剤形のティースプーン一杯当たり約2.5ミリグラムから約10ミリグラムのロラティダインに相当する。
【0066】
活性成分がアセトアミノフェンである別の実施形態では、懸濁液剤形における活性成分の量は、水性懸濁液剤形の容積当たりの活性成分の全重量(w/v)を基準として、約1.6パーセントから約3.2パーセントであり、これは、ティースプーン一杯の水性懸濁液剤形当たり約80mgから約160mgに相当する。アセトアミノフェンが入っているさらに別の実施形態では、懸濁液剤形における活性成分の量は、水性懸濁液剤形の容積当たりの活性成分の全重量(w/v)を基準として、約5パーセントから約10パーセントであり、これは、水性懸濁液剤形1.6mL当たり約80mgから約160mgに相当する。
【0067】
活性成分がイブプロフェンである別の実施形態では、懸濁液剤形における活性成分の量は、水性懸濁液剤形の容積当たりの活性成分の全重量(w/v)を基準として、約50から約200mgであり、例えば水性懸濁液剤形ティースプーン一杯当たり約50mgから約100mgであり、または水性懸濁液剤形1mL当たり約40mgの活性成分であり、これは、水性懸濁液剤形の容積当たりの活性成分の全重量(w/v)を基準として、約1パーセントから約4パーセントに相当する。
【0068】
本発明の一実施形態は、測定可能な懸濁液組成に向けられており、これは、懸濁液の全重量を基準として:a)約0.05パーセントから約40パーセントの少なくとも1つの活性成分;b)約20パーセントから約70パーセントの水;c)約0.1パーセントから約10パーセントの懸濁系;d)約0パーセントから約40パーセント、例えば約20パーセントから約40パーセントの甘味剤;およびe)約0パーセントから約0.2パーセントの賦形剤、を含む。この実施形態において、活性成分の全重量を基準として、活性成分の約50パーセントから約75パーセントが即放性量部分にあり、活性成分の約25パーセントから約50パーセントが放出制御量部分である。同じ実施形態において、懸濁液の全重量に基づいて、その剤形の約0.025パーセントから約30パーセントが即放性量部分の活性成分からなり、その剤形の約0.0125パーセントから約0.025パーセントが放出制御量部分の活性成分からなる。
【0069】
所定の実施形態において、本発明の懸濁液の粘度は、ブルックフィールド(Brookfield)DV−1+粘度計により、12rpmで回る31番スピンドルを1つ用いて、温度条件が約25℃の下で測定した場合に約400cpsから約1500cpsの範囲であることがある。
【0070】
本発明の他の実施形態では、一層の放出制御コーティングで実質的に覆われている、すなわち、コーティングされている活性成分粒子が入っている、および/または放出制御成分からなるマトリックスに分散された活性成分粒子が入っている水性懸濁液剤形に向けられている。
【0071】
本発明の剤形は、NSAIDおよび/またはアセトアミンフェンのような活性成分の有効量を毎日1回または2回の投与で送達することを意図している。鎮痛薬の「有効量」とは、患者の苦痛を軽減する量である。例えば、イブプロフェンの典型的な大人の投与量は、患者の体重に対し約2.9から約12mg/kgを4から6時間毎に与えることであろうし、典型的な1日当たりの投与量は約11.6から約72mg/kg/日であろう。したがって、典型的な70kgの大人に対するイブプロフェンの有効量の投与は、例えば40mg/mLのイブプロフェンが入っている本発明の製剤を約5mLから約60mL、毎日1、2回投与することを伴う。イブプロフェンの小児用の典型的な投与量は、約5から約10mg/kgを4から6時間毎に与えることであろうし、典型的な一日当たりの投与量は約20から約60mg/kg/日であろう。典型的な15kgの子供に対するイブプロフェンの有効量の投与は、例えば20mg/mLのイブプロフェンが入っている、本発明の製剤を約5mLから約30mL、毎日1、2回投与することを伴う。
【0072】
本発明の剤形の経口投与は、ユーザーにNSAIDやアセトアミノフェンのような活性成分を、オプションとしての即放性量、ならびに、投与してから約6時間後、例えば、約8時間後または約10時間後に剤形から活性成分を放出し続ける放出制御量で提供する。有り難いことに、我々は予期せずして、剤形が懸濁液のような液体剤形としてデザインされているか、投与前に水で戻すことができる乾式剤形としてデザインされているかに拘わらず、製品の有効期間を通して、かつ、治療時間を通して、剤形の放出制御部分の放出特性が如何にして効果的に安定させるかを発見した。特に、我々は、製品における粒子からの活性成分が摂取前に放出されることを防ぎつつ、胃腸液におけるその同じ粒子から活性成分の放出制御を可能にするという難題を克服した。
【0073】
都合のよいことに、本発明の製剤は、様々な形式で使用することができ、これには、例えば、(i)正確に計量できるシングルドーズ乾式製剤または懸濁液;(ii)必要に応じて再懸濁化する顆粒を異なる量だけ計量することにより、投与量の融通が非常にきくマルチドーズ顆粒製剤;(iii)マルチドーズ懸濁液;および(iv)活性成分が懸濁している濃縮ドロップであって、特に小児用途に有用であるもの、がある。
【0074】
さらに、この製剤は投与したり、飲み込んだりするのに便利であるので、また、活性成分の毎日の投与回数が少なくなるので、総合的な患者のコンプライアンスが達成される。投与、飲み込みが容易であることから、小児科診療でも有益であることが期待される。
【0075】
保存期間中安定である懸濁液を作るために、一連の腸溶製コーティングを医薬品活性成分に塗布する必要があった従来の放出制御医薬品懸濁液と異なり、本発明の懸濁医薬品粒子は、水または水混和性補助溶剤(water-miscible co-solvent)のあるところで安定であるために、新規の徐放性コーティングを一層しか被覆する必要がない。
【0076】
下記の例は、本発明をさらに説明するものであるが、決して本発明を制限するものではない。
【0077】
実施例1:放出制御コーティング溶液の調合
コーティング溶液は、ローム・パルマ・インコーポレテッドから商品名「Eudragit L-100」で市販されているメタクリレート共重合体(methacrylate co-polymer)と、酢酸セルロース(cellulose acetate)とを、溶媒の全重量を基準として、98%のアセトンと、2%の水が入っている溶媒に周囲条件下で分散させて調合した。
【0078】
結果として得られたコーティング溶液は、全湿式コーティング溶液を基準として、7.6%の酢酸セルロース(cellulose acetate)、0.4%のメタクリレート共重合体(methacrylate co-polymer)、90.2%のアセトンおよび1.8%の水を含んでいた。
【0079】
固体の相対的な量は、乾燥させたコーティング溶液の全重量パーセントを基準として、酢酸セルロース(cellulose acetate)が95.00%と、メタクリレート共重合体(methacrylate co-polymer)が5.00%であった。
【0080】
実施例2:コーティングした活性成分の調合
イブプロフェン予混合物の調合:イブプロフェンUSPパウダーをコロイド状二酸化ケイ素(colloidal silicon dioxide)と混合して、下記のイブプロフェン予混合物を形成した:
成分 重量パーセント*
コロイド状二酸化ケイ素 2.00%
イブプロフェンUSP 98.00%
*イブプロフェン予混合物の全重量を基準とする
【0081】
コーティングしたイブプロフェン顆粒の調合:上記のように調合したイブプロフェン混合物を次に実施例1にしたがって調合した湿式放出制御コーティング溶液を用いて、約20.0g/分の速度で、グラットGPCG-5/9ウルスター流動層コーティング装置(Glatt GPCG-5/9 Wurster fluid bed coating unit)で、約29−32℃の製造温度条件下でコーティングした。その結果得られたコーティングされたイブプロフェン顆粒は、イブプロフェン顆粒および放出制御コーティングの総乾燥重量を基準として、放出制御コーティングが約20%含まれていた。
【0082】
実施例3:即放性量および放出制御量を含む懸濁液ベースの製造
懸濁液ベースの調合
【0083】
【表1】
【0084】
上記表Aに示したように、純水USPは、スコットターボン(Scott Turbon)製高剪断ミキサーが備え付けられている混合タンクに充填し、十分な渦ができるように約500rpmから約1000rpmで混合した。次に、アルファ化デンプンとキサンタン・ガムを混合タンクに加え、20分間混合した。次にグリセリンをそこに加え、5分間混合した。次にサッカロースをそこに加え、10分間混合した。次に、ポリソルベート−80NF(polysorbate-80 NF)、クエン酸(citric acid)USPおよび安息香酸ナトリウムNF(sodium benzoate NF)を順に加え、その結果得られた混合物を10分間混合した。次に純水の残りを混合しながらそこに加えて懸濁液ベースを形成した。
【0085】
活性成分を含む懸濁液の調合
2000.0mgのイブプロフェンUSPを50番のメッシュ網でふるった後、上記のように調合した懸濁液ベース25mLをそこに加え、均質になるまでその混合物を混合した。
【0086】
実施例2にしたがって調合した放出制御コーティングをしたイブプロフェン1276mg(活性イブプロフェン78.4%が含まれる)を60番と80番のメッシュ網の間でふるい、次に混合物に加えた。
【0087】
できあがった懸濁液は、次に懸濁液ベースを加えて容積100.0mLに希釈し、得られた懸濁液が均質になるまで混合した。できた懸濁液を40番のメッシュ網を使ってふるった後、できあがった、最終的にふるい分けられた懸濁液は、100mg/5mLの即放性イブプロフェン量と、50mg/5mLの放出制御イブプロフェン量とを含んでいた。イブプロフェン粒子の相対的な量は、最終的にふるい分けられた懸濁液の総量を基準として:
イブプロフェンUSP(即放性量)..........100.0mg/5mL
コーティングしたイブプロフェン(放出制御量).....50.0mg/5mL
であった。
【0088】
実施例4:即放性量および放出制御量を含む懸濁液ベースの溶出性分析
pH5.6のアセテート緩衝液溶出媒体900mLをバドル付きのUSPタイプIIの装置の3つの容器の各々に入れた。次に、実施例3で作った最終懸濁液の試料5.0mLを3つの容器の各々に個別に加え、50r.p.m.、37℃において混合物が均質になるまで混合した。
【0089】
その後、それぞれ0.5、1、2、3、4、6、8、および12時間後に、懸濁液/緩衝液混合物の試料10mLを容器から個別に取り出した。
【0090】
次に、それぞれ0.5、1、2、3、4、6、8、および12時間におけるイブプロフェンの溶出曲線を求めるために、Waters(登録商標)717自動注入器と、波長254nmに設定したWaters(登録商標)486UV検出器とを備えた高圧液体クロマトグラフィー(HPLC)装置を使って、各試料10mLを個別にイブプロフェン内容分析した。試料の各々は、イブプロフェンの標準試料と比較した。イブプロフェンの標準試料は、イブプロフェンが0.167mg/mLであるアセテート緩衝液(pH5.6)溶出媒体を含んでおり、イブプロフェンの100%放出に必要とされる理論濃度と相関している。
【0091】
HPLCで使用する移動相は、アセトニトリル(acetonitrile)55%と、18mMリン酸カリウム緩衝液(potassium phosphate buffer)45%を含む試料を使って調合した。注入容積200μL、実行時間約7分、ポンプ流1.5mL/分であった。分析に使用したカラム(column)は、フェノメネクス・ルナ(Phenomenex LUNA)(登録商標)5umC8(4.6mm×15cm)であった。
【0092】
この溶出性分析の結果は、図1に記載した。この結果は、本発明の懸濁液が活性成分の即放性量ならびに活性成分の放出制御量の両方を含むことができ、放出制御量が初期投与から約12時間にわたってイブプロフェンを放出したことを示している。
【0093】
実施例5:放出制御量を含む懸濁液ベースの製造
実施例3の手順を繰り返したが、2000.0mgのイブプロフェンUSPを加えなかった。
【0094】
得られた、最終的にふるい分けた懸濁液は、放出制御イブプロフェン量100mg/5mLを含んでいた。
【0095】
実施例6:放出制御量を含む懸濁液ベースの溶出性分析
実施例4の手順を繰り返したが、試料は、実施例5で作った最終懸濁液を用いた。
【0096】
溶出性分析の結果は、図2に記載した。この結果は、本発明の懸濁液が、活性成分の即放性量が実質的にない状態で活性成分の放出制御量を含むことができ、放出制御量が初期投与から約12時間にわたってイブプロフェンを放出したことを示している。
【0097】
〔実施の態様〕
(1) 医薬品懸濁液剤形において、
NSAIDおよび/またはアセトアミノフェンの粒子であって、放出制御成分からなる1つの層で実質的に覆われている粒子を含み、
前記医薬品懸濁液剤形の治療効果の持続時間が、哺乳動物への最初の投与後、少なくとも8時間ある、剤形。
(2) 実施態様1記載の剤形において、
前記粒子を投与するための賦形剤をさらに含んでいる、剤形。
(3) 実施態様2記載の剤形において、
前記賦形剤が、懸濁系、界面活性剤、甘味料、緩衝剤、保存薬、香料添加剤、およびこれらの混合物からなる一群から選択された1つ以上の添加剤を含む、剤形。
(4) 実施態様2記載の剤形において、
前記賦形剤が、水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物を含む、剤形。
(5) 実施態様1記載の剤形において、
前記粒子が、前記放出制御成分で実質的に覆われた芯物質を含む、剤形。
【0098】
(6) 実施態様5記載の剤形において、
前記放出制御成分が、前記コーティングの総乾燥重量を基準として、0パーセントより多くて100パーセント未満の不溶性フィルム形成ポリマーと、0パーセントから10パーセント未満の腸溶性ポリマーとを含んでいる、剤形。
(7) 実施態様5記載の剤形において、
前記放出制御コーティングに実質的に腸溶性ポリマーがなく、前記粒子に含まれる少なくとも1つの活性成分のpKaが前記懸濁液剤形のpHよりも大きい、剤形。
(8) 実施態様5記載の剤形において、
前記放出制御コーティングにおける前記不溶性フィルム形成ポリマーと前記腸溶性ポリマーとの重量比が80:20から99:1である、剤形。
(9) 実施態様5記載の剤形において、
前記不溶性フィルム形成ポリマーが、酢酸セルロース、エチルセルロース、重量比1:2:0.1のポリ(アクリル酸エチル、メタクリル酸メチル、トリメチルアンモニウムエチル・メタクリレート・クロライド)と、その共重合体および混合物からなる一群から選択されている、剤形。
(10) 実施態様5記載の剤形において、
前記腸溶性ポリマーが、ヒドロキシプロピル・メチルセルロース・フタレート、ヒドロキシプロピル・メチルセルロース・アセテート・シクシナート、セルロース・アセテート・フタレート、ポリビニルアセテート・フタレート、ポリメタクリレートをベースとするポリマー、ならびにこれらの共重合体および混合物からなる一群から選択されている、剤形。
【0099】
(11) 実施態様10記載の剤形において、
前記ポリメタクリレートをベースとするポリマーが、重量比1:2のポリ(メタクリル酸、メチルメタクリレート)および/または重量比1:1のポリ(メタクリル酸、メチルメタクリレート)である、剤形。
(12) 実施態様5記載の剤形において、
前記コーティングした粒子が、前記コーティングした粒子の総乾燥重量を基準として、10パーセントから40パーセントの前記放出制御成分を含む、剤形。
(13) 実施態様1記載の剤形において、
前記治療効果が鎮痛である、剤形。
(14) 実施態様1記載の剤形において、
前記NSAIDがプロピオン酸誘導体NSAIDである、剤形。
(15) 必要に応じて哺乳動物の痛みを治療する方法において、
実施態様1記載の剤形を、前記剤形の投与後、少なくとも12時間、前記哺乳動物の鎮痛を行うのに有効な量だけ投与することを含む、方法。
【0100】
(16) 医薬品懸濁液剤形において、
a)NSAIDおよび/またはアセトアミノフェンを含む粒子であって、放出制御コーティングからなる1つの層で実質的に覆われている粒子と、
b)水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物と、を含み、
前記医薬品剤形の治療効果の持続時間が、前記医薬品剤形の投与後、少なくとも8時間ある、剤形。
(17) 実施態様16記載の医薬品懸濁液において、
前記懸濁液の全重量を基準として、
a)0.05パーセントから40パーセントの、NSAIDおよび/またはアセトアミノフェンを含む粒子であって、放出制御コーティングからなる1つの層で実質的に覆われている粒子と、
b)20パーセントから70パーセントの水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物と、を含み、
前記医薬品剤形の治療効果の持続時間が、前記医薬品剤形の投与後、少なくとも8時間ある、医薬品懸濁液。
(18) 必要に応じて哺乳動物の痛みを治療する方法において、
実施態様17記載の剤形を、前記剤形の投与後、少なくとも12時間、前記哺乳動物の鎮痛を行うのに有効な量だけ投与することを含む、方法。
(19)アセトアミノフェンおよび/またはNSAIDを懸濁液医薬品剤形の状態で必要に応じて哺乳動物に投与する方法において、
前記哺乳動物が、前記アセトアミノフェンおよび/またはNSAIDの放出制御ドーズを前記剤形の投与後12時間にわたって受け、前記12時間の間にアセトアミノフェンおよび/またはNSAIDをさらに与えることがないように、前記剤形を哺乳動物に与えることを含む、方法。
(20) 医薬品懸濁液剤形において、
a)NSAIDおよび/またはアセトアミノフェンを含む粒子であって、放出制御成分からなる1つの層で実質的に覆われており、前記放出制御成分が、前記放出制御成分の全重量を基準として、0パーセントより多くて90パーセント未満の不溶性フィルム形成ポリマーと、0パーセントより多くて10パーセント未満の腸溶性ポリマーとを含む、粒子と、
b)水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物と、を含み、
前記医薬品剤形の治療効果の持続時間が、投与後、少なくとも12時間ある、剤形。
【0101】
(21) 実施態様20記載の医薬品懸濁液剤形において、
前記剤形の全重量を基準として、
a)0.05パーセントから40パーセントまでの、NSAIDおよび/またはアセトアミノフェンを含む粒子であって、放出制御成分からなる1つの層で実質的に覆われており、前記放出制御成分が、前記放出制御成分の全重量を基準として、0パーセントより多くて90パーセント未満の不溶性フィルム形成ポリマーと、0パーセントより多くて10パーセント未満の腸溶性ポリマーとを含む、粒子と、
b)20パーセントから70パーセントの水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物と、を含み、
前記医薬品剤形の治療効果の持続時間が、投与後、少なくとも12時間ある、剤形。
(22) 実施態様21記載の剤形において、
前記放出制御コーティングにおける前記不溶性フィルム形成ポリマーと、前記腸溶性ポリマーとの重量比が、99:1から80:20である、剤形。
【図面の簡単な説明】
【0102】
【図1】イブプロフェンの即放性ドーズおよび放出制御ドーズの両方を含む懸濁液剤形に関する放出された活性成分(mg)対時間(時間)のグラフである。
【図2】イブプロフェンの放出制御ドーズのみを含む懸濁液剤形に関する放出された活性成分(mg)対時間(時間)のグラフである。
【特許請求の範囲】
【請求項1】
医薬品懸濁液剤形において、
NSAIDおよび/またはアセトアミノフェンの粒子であって、放出制御成分からなる1つの層で実質的に覆われている粒子を含み、
前記医薬品懸濁液剤形の治療効果の持続時間が、哺乳動物への最初の投与後、少なくとも8時間ある、剤形。
【請求項2】
請求項1記載の剤形において、
前記粒子の投与のための賦形剤を含み、前記賦形剤が、水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物を含む、剤形。
【請求項3】
請求項1記載の剤形において、
前記粒子が、前記放出制御成分で実質的に覆われた芯物質を含む、剤形。
【請求項4】
請求項3記載の剤形において、
前記放出制御成分が、前記コーティングの総乾燥重量を基準として、0パーセントより多くて100パーセント未満の不溶性フィルム形成ポリマーと、0パーセントから10パーセント未満の腸溶性ポリマーとを含んでいる、剤形。
【請求項5】
請求項3記載の剤形において、
前記放出制御コーティングに実質的に腸溶性ポリマーがなく、前記粒子に含まれる少なくとも1つの活性成分のpKaが前記懸濁液剤形のpHよりも大きい、剤形。
【請求項6】
請求項3記載の剤形において、
前記放出制御コーティングにおける前記不溶性フィルム形成ポリマーと前記腸溶性ポリマーとの重量比が80:20から99:1である、剤形。
【請求項7】
請求項3記載の剤形において、
前記不溶性フィルム形成ポリマーが、酢酸セルロース、エチルセルロース、重量比1:2:0.1のポリ(アクリル酸エチル、メタクリル酸メチル、トリメチルアンモニウムエチル・メタクリレート・クロライド)(poly (ethyl acrylate, methyl methacrylate, trimethylammonioethyl methacrylate chloride))と、その共重合体および混合物からなる一群から選択されている、剤形。
【請求項8】
請求項3記載の剤形において、
前記腸溶性ポリマーが、ヒドロキシプロピル・メチルセルロース・フタレート、ヒドロキシプロピル・メチルセルロース・アセテート・シクシナート(hydroxypropyl methylcellulose acetate succinate)、セルロース・アセテート・フタレート、ポリビニルアセテート・フタレート、ポリメタクリレートをベースとするポリマー、ならびにこれらの共重合体および混合物からなる一群から選択されている、剤形。
【請求項9】
請求項8記載の剤形において、
前記ポリメタクリレートをベースとするポリマーが、重量比1:2のポリ(メタクリル酸、メチルメタクリレート)および/または重量比1:1のポリ(メタクリル酸、メチルメタクリレート)である、剤形。
【請求項1】
医薬品懸濁液剤形において、
NSAIDおよび/またはアセトアミノフェンの粒子であって、放出制御成分からなる1つの層で実質的に覆われている粒子を含み、
前記医薬品懸濁液剤形の治療効果の持続時間が、哺乳動物への最初の投与後、少なくとも8時間ある、剤形。
【請求項2】
請求項1記載の剤形において、
前記粒子の投与のための賦形剤を含み、前記賦形剤が、水、または、水と、グリコール、アルコールおよびグリセロールからなる一群から選択した医薬品として容認可能な水混和性補助溶剤との混合物を含む、剤形。
【請求項3】
請求項1記載の剤形において、
前記粒子が、前記放出制御成分で実質的に覆われた芯物質を含む、剤形。
【請求項4】
請求項3記載の剤形において、
前記放出制御成分が、前記コーティングの総乾燥重量を基準として、0パーセントより多くて100パーセント未満の不溶性フィルム形成ポリマーと、0パーセントから10パーセント未満の腸溶性ポリマーとを含んでいる、剤形。
【請求項5】
請求項3記載の剤形において、
前記放出制御コーティングに実質的に腸溶性ポリマーがなく、前記粒子に含まれる少なくとも1つの活性成分のpKaが前記懸濁液剤形のpHよりも大きい、剤形。
【請求項6】
請求項3記載の剤形において、
前記放出制御コーティングにおける前記不溶性フィルム形成ポリマーと前記腸溶性ポリマーとの重量比が80:20から99:1である、剤形。
【請求項7】
請求項3記載の剤形において、
前記不溶性フィルム形成ポリマーが、酢酸セルロース、エチルセルロース、重量比1:2:0.1のポリ(アクリル酸エチル、メタクリル酸メチル、トリメチルアンモニウムエチル・メタクリレート・クロライド)(poly (ethyl acrylate, methyl methacrylate, trimethylammonioethyl methacrylate chloride))と、その共重合体および混合物からなる一群から選択されている、剤形。
【請求項8】
請求項3記載の剤形において、
前記腸溶性ポリマーが、ヒドロキシプロピル・メチルセルロース・フタレート、ヒドロキシプロピル・メチルセルロース・アセテート・シクシナート(hydroxypropyl methylcellulose acetate succinate)、セルロース・アセテート・フタレート、ポリビニルアセテート・フタレート、ポリメタクリレートをベースとするポリマー、ならびにこれらの共重合体および混合物からなる一群から選択されている、剤形。
【請求項9】
請求項8記載の剤形において、
前記ポリメタクリレートをベースとするポリマーが、重量比1:2のポリ(メタクリル酸、メチルメタクリレート)および/または重量比1:1のポリ(メタクリル酸、メチルメタクリレート)である、剤形。
【図1】

【図2】
【図2】
【公表番号】特表2007−509936(P2007−509936A)
【公表日】平成19年4月19日(2007.4.19)
【国際特許分類】
【出願番号】特願2006−538093(P2006−538093)
【出願日】平成16年10月21日(2004.10.21)
【国際出願番号】PCT/US2004/034708
【国際公開番号】WO2005/044246
【国際公開日】平成17年5月19日(2005.5.19)
【出願人】(591252839)マクニール−ピーピーシー・インコーポレイテッド (69)
【氏名又は名称原語表記】MCNEIL−PPC,INCORPORATED
【Fターム(参考)】
【公表日】平成19年4月19日(2007.4.19)
【国際特許分類】
【出願日】平成16年10月21日(2004.10.21)
【国際出願番号】PCT/US2004/034708
【国際公開番号】WO2005/044246
【国際公開日】平成17年5月19日(2005.5.19)
【出願人】(591252839)マクニール−ピーピーシー・インコーポレイテッド (69)
【氏名又は名称原語表記】MCNEIL−PPC,INCORPORATED
【Fターム(参考)】
[ Back to top ]