
放出調節型タムスロシン錠
【課題】食物作用(food effect)をほとんど、または全く示さない放出調節型タムスロシン錠の提供。
【解決手段】タムスロシンまたはその薬学的に許容可能な塩が分散している錠剤マトリックスと、任意選択的に前記マトリックスを覆っている腸溶被覆とを含む放出調節型錠剤。マトリックス成分は水膨潤性セルロース誘導体;アルギン酸ナトリウム;アクリレート類、メタクリレート類およびそれらの様々なコモノマー類とのコポリマー類;ならびにポリビニルピロリドン類を含む群から選ばれる。水膨潤性セルロース誘導体はヒドロキシプロピルメチルセルロースなどである。
【効果】SIF、FaSSIF、およびFeSSIFの各媒質中で、50〜100rpmのパドル速度で500mLの前記媒質を使用した米国薬局方第2法装置において、前記錠剤が2時間の経過後に前記タムスロシンの60%以下を放出する溶出プロフィールを示す。
【解決手段】タムスロシンまたはその薬学的に許容可能な塩が分散している錠剤マトリックスと、任意選択的に前記マトリックスを覆っている腸溶被覆とを含む放出調節型錠剤。マトリックス成分は水膨潤性セルロース誘導体;アルギン酸ナトリウム;アクリレート類、メタクリレート類およびそれらの様々なコモノマー類とのコポリマー類;ならびにポリビニルピロリドン類を含む群から選ばれる。水膨潤性セルロース誘導体はヒドロキシプロピルメチルセルロースなどである。
【効果】SIF、FaSSIF、およびFeSSIFの各媒質中で、50〜100rpmのパドル速度で500mLの前記媒質を使用した米国薬局方第2法装置において、前記錠剤が2時間の経過後に前記タムスロシンの60%以下を放出する溶出プロフィールを示す。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、米国特許法第119条(e)の下で、引用することにより本明細書の一部を
なすものとする2001年11月7日に出願された先の米国特許仮出願第60/331,
055号からの優先権の利益を主張する。
【0002】
[発明の分野]
本発明は、食物作用(food effect)をほとんど、または全く示さない放出調節型タム
スロシン錠およびそれから製造された単位製剤に関する。
【背景技術】
【0003】
タムスロシンは、式(1)の5−[2−[[2−(2−エトキシフェノキシ)エチル]
アミノ]プロピル]−2−メトキシ−ベンゼンスルホンアミドの一般名である。
【化1】

タムスロシンは、欧州特許第34432号および米国特許第4,731,478号におい
て、心不全および良性前立腺肥大症を治療するために有用なαアドレナリン遮断活性を有
する薬学的に活性な成分であると開示されている。
【0004】
(R)−塩酸タムスロシンは、良性前立腺肥大症(BPHとしても知られる)の尿量や
尿頻度の問題等の症状を治療するため、アメリカ合衆国でのFLOMAX(登録商標)(
Boehringer Ingelheim社)、日本でのHARNAL(登録商標)(
山之内製薬)およびヨーロッパでのOMNIC(登録商標)(山之内製薬)を含めて様々
な商標名を付けて市販されている。既承認製剤には、0.4mgの塩酸タムスロシンを含
む経口投与用カプセル製剤が含まれる。このカプセル剤は、タムスロシンの制御された放
出を提供し、1日1回製剤であるが、必要であればカプセル2錠、すなわち最高0.8m
gの1日1回投与を使用することができる。
【0005】
制御放出型または調節放出型の市販カプセル剤には、食物作用を示すという欠点がある
。食物作用とは、摂食(胃内に食物がある)患者に比較して、絶食(胃が空である)患者
への薬物の投与から発生する生体内吸収率または生体内利用率における相違を意味する。
市販のカプセル剤については、食物作用は相当に顕著である。例えば、FLOMAX(登
録商標)の添付文書では、絶食条件下におけるTmaxは4〜5時間であるが、摂食条件
下におけるTmaxは6〜7時間であると報告されている。絶食条件下でカプセル剤を摂
取すると、摂食条件下と比較して、バイオアベイラビリティ(AUC)における30%の
増加およびピーク濃度(Cmax)における40%〜70%の増加が生じる。そこで食後
に摂取した場合、タムスロシンが達成する最高血漿中濃度はより低くなる。そしてこのピ
ークはより遅い時期に達成される。したがって、食後の投与は、絶食条件下の投与に比較
して、生体内利用率の損失が生じるにもかかわらず、平坦かつ制御放出型の血漿中濃度プ
ロフィールを生じさせる。
【0006】
市販カプセル剤の添付文書は、食後(日本)、朝食後(ヨーロッパ)および毎日同一の
食事の30分後(米国)のような摂食状態において投与するように指示している。この投
与は、より一貫性のある結果を提供して副作用もより少ないために推奨されていると考え
られる。実際に、タムスロシンの吸収は摂食条件下より絶食条件下(90+%)の方が良
好ではあるが、承認された使用はタムスロシンカプセル剤を摂食条件下で投与するように
指示している。
【0007】
タムスロシンの市販カプセル剤は米国特許第4,772,475号に対応すると考えら
れる(欧州特許第194838号、欧州特許第533297号)。米国特許第4,772
,475号は、タムスロシン、微結晶セルロースおよび放出制御剤を含有する、多数の顆
粒単位を含む制御放出型医薬製剤を開示している。顆粒剤は、タムスロシンを顆粒マトリ
ックスから徐々に放出する。食物作用問題に関する検討は行われていない。
【0008】
タムスロシンの市販カプセル剤では食物作用が発生するために、絶食時(食物を摂取し
ていない)にカプセル剤を摂取した患者は、めまい、鼻炎、および/または異常射精等の
望ましくない副作用を経験する可能性がより高くなる。食物作用が小さいもしくはわずか
である、または食物作用を全く示さないタムスロシン医薬製剤を製造することが有益であ
ろう。そこで、本製剤はより安全であろう。すなわち、絶食条件下で摂取した場合でさえ
、副作用のリスクが低下する。タムスロシンの市販カプセル剤の食物作用は明確に記録さ
れているが、これまでに食物作用問題に対する解決策は提供されていない。
【発明の概要】
【0009】
現在では、食物作用が低下した調節放出型タムスロシン錠を形成できることが見いださ
れた。したがって、本発明の1つの態様は、その中に0.1〜10mgのタムスロシンま
たはその薬学的に許容可能な塩が分散している錠剤マトリックスを含み、そして任意選択
的に該マトリックスの全体を被覆する腸溶被覆を有する医薬錠剤に関する。本錠剤は放出
調節型錠剤であり、50〜100rpmのパドル速度で500mLの媒質を使用した米国
薬局方第2法装置(USP 2 apparatus)で、各々について以下で定義するSIF、FaS
SIF、およびFeSSIFの各媒質中に錠剤が2時間の経過後にタムスロシンの60%
以下を放出するような溶出プロフィールを示す。好ましくは、本錠剤は、同様に3種の各
媒質中に2時間の指標までに少なくとも20%のタムスロシンを放出する。これらの媒質
はインビボで遭遇する腸内条件をインビトロで模倣するために役立ち、FaSSIFは絶
食状態に相当し、FeSSIFは摂食状態に相当する。各擬似条件下において2時間で6
0%未満のタムスロシンを放出したことによって、本錠剤は摂食状態と絶食状態のどちら
も本製剤の放出調節型の性質を低下させることがないことを証明している。
【0010】
本発明のまた別の態様は、0.1〜10mgのタムスロシンまたはその薬学的に許容可
能な塩と、10重量%〜90重量%のヒドロキシプロピルメチルセルロースとを含み、そ
して総錠剤重量が10〜300mgであるモノリシック医薬錠剤に関する。本錠剤は、好
ましくは低下した食物作用を示し、そしてより好ましくは上記で説明した溶出プロフィー
ルの要件を満たす。
【0011】
これらの態様の各々において、本錠剤はそれ自体で投与可能であり、任意選択的に腸溶
被覆を備えてもよく、しかし好ましくは腸溶被覆を備えずに投与できる。または、1錠以
上の錠剤をカプセル封入し、1つ以上のカプセル剤として投与することができる。本発明
のまた別の態様は、良性前立腺肥大症の症状を治療するための方法であって、それを必要
とする患者に1錠以上の上記の錠剤を投与するステップを含む方法に関する。
【図面の簡単な説明】
【0012】
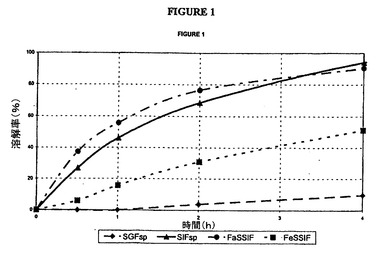
【図1】本明細書に記載した4種の媒質中における山之内ヨーロッパによって製造されたタムスロシンカプセル剤の放出曲線の図である。
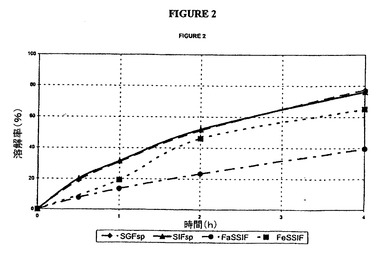
【図2】本明細書に記載した4種の媒質中におけるタムスロシン錠(バッチG)の放出曲線の図である。
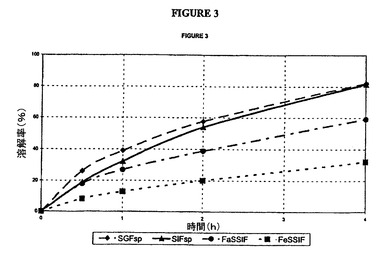
【図3】本明細書に記載した4種の媒質中におけるタムスロシン錠(バッチH)の放出曲線の図である。
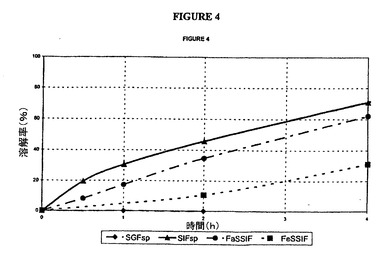
【図4】本明細書に記載した4種の媒質中におけるタムスロシン腸溶被覆錠(バッチG)の放出曲線の図である。
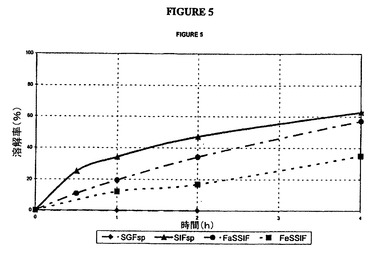
【図5】本明細書に記載した4種の媒質中におけるタムスロシン腸溶被覆錠(バッチH)の放出曲線の図である。
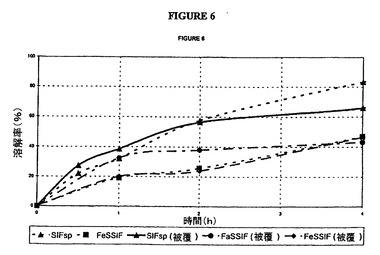
【図6】選択した媒質中のタムスロシン非被覆錠および被覆錠(バッチN)の放出曲線の図である。
【発明を実施するための形態】
【0013】
本発明は、放出調節型タムスロシン錠および該放出調節型タムスロシン錠を含有するカ
プセル剤に関する。「放出調節型」とは、本明細書では広い意味で使用されており、即時
放出型製剤ではない、すなわち標準溶出試験(すなわち、37℃でパドル速度50rpm
、500mLのSGFを用いる米国薬局方第2法装置を使用する)における初期30分間
以内に少なくとも75%のタムスロシンを放出する製剤ではないあらゆる製剤を意味する
。この錠剤は、市販で入手できるタムスロシンカプセル剤と比較して低下した食物作用を
示す。これらのカプセル剤とは相違して、本発明は、タムスロシンまたはその薬学的に許
容可能な塩を、制御可能な食物作用の特性を有する錠剤に調製することができるという発
見に基づいている。
【0014】
タムスロシン錠は、好ましくは各SIF、FaSSIF、およびFeSSIFの中に、
タムスロシンの60%未満、好ましくは20%〜60%が2時間で放出される溶出プロフ
ィールを示す。溶出試験は、試験をその中で実施する媒質500mLを使用する米国薬局
方第2法装置において、50〜100rpm、好ましくは100rpmのパドル速度を用
いて実施する。ある実施形態、特別には非被覆錠を含む実施形態では、溶出プロフィール
はさらに、50〜100rpm、好ましくは100rpmのパドル速度に設定した米国薬
局方第2法装置において、500mLのSGF中に2時間でタムスロシンの60%未満、
好ましくは20%〜60%を放出することを含む。本発明の目的で放出プロフィールを決
定するためのすべての溶出試験において、媒質の温度は37℃である。装置内に錠剤1錠
が配置されることを想定して、インビボ試験結果のより正確なモデリング/予測を提供す
ると考えられるので、500mLの媒質を使用する。
【0015】
本発明のための溶出媒質は、以下に定義した通りである。
【0016】
SGF(米国薬局方によるペプシンを含まない擬似胃液)の組成:
HCl 十分量 pH1.2
NaCl 0.2%
水 十分量 1,000mL
【0017】
SIF(米国薬局方によるパンクレアチンを含まない擬似腸液)の組成:
KH2P04 6.8g
NaOH 十分量 pH6.8
水 十分量 1,000mL
【0018】
FeSSIF(擬似腸液、摂食状態)の組成:
酢酸 0.144M
NaOH 十分量 pH5
タウロコール酸ナトリウム 15mM
レシチン 4mM
KCl 0.19M
蒸留水 十分量 1,000mL
pH=5
浸透圧=485〜535mOsm
緩衝能力=75±2mEQ/L/pH
【0019】
FaSSIF(擬似腸液、絶食状態)の組成:
KH2PO4 0.029M
NaOH 十分量 pH6.8
タウロコール酸ナトリウム 5mM
レシチン 1.5mM
KCl 0.22M
蒸留水 十分量 1,000mL
pH=6.8
浸透圧=280〜310mOsm
緩衝能力=10±2mEQ/L/pH
【0020】
SGFは胃の標準的状態を表している。SIFは腸の標準的状態を表している。FeS
SIFは摂食状態をより良好に表すように調整され、他方FaSSIFは絶食状態をより
良好に表すように調整されている。pHが相違するだけではなく、同等に重要であること
に、浸透圧もまた相違することに注目されたい。FaSSIF媒質およびFeSSIF媒
質は、一般に即時放出型の脂肪親和性かつ難水溶性の薬剤(すなわち、ケトコナゾール、
ダナゾール、アトバクオン、トログリタゾン、メフェナム酸)についてのインビトロ−イ
ンビボ相関を説明するために使用されてきたが、以前に実施された試験は、塩酸タムスロ
シン等の放出調節型低用量かつ可溶性の薬物(10mgまでを溶解させるために必要な水
性媒質の量は500mL以下である)への適用も、放出調節型調製物(放出制御型、徐放
性または遅延放出型を含む)への適用も示唆していない。
【0021】
以下の参照実施例に示すように、山之内ヨーロッパ社が製造した市販のカプセル剤は、
2時間の経過後にタムスロシンの次のような放出を示す。SIF中には60%超、FaS
SIF中には75%超が放出されるが、FeSSIF中には40%未満が放出され、SG
F中には20%未満が放出される(図1を参照)。pH6.8では速すぎて、pH1.2
では遅すぎる(遅延性放出)、4種の媒質中でのこの結果の相違は、以前に指摘されたよ
うにタムスロシンの血漿中濃度における変動性について考えられる、例えば、様々な胃内
容排出状態および/または胃腸内pHの変化のような理由を表している。当然ながら、こ
のFaSSIF中でのより急速な放出は、摂食状態より絶食状態においてより急速なTm
axおよびより高いCmaxのインビボ観察所見と良好に一致している。したがって、各
SIF、FaSSIF、およびFeSSIF中に、好ましくはSGF中にも60%以下の
タムスロシン放出を示す本発明の好ましい錠剤は、市販のカプセル剤と比較してより良好
な食物作用を有する、すなわち摂食状態と絶食状態の間の差がより小さい。
【0022】
好ましくは、タムスロシン錠は各SIF、FaSSIF、およびFeSSIF中に溶出
試験の初期2時間中にタムスロシンの20%〜60%を放出する。より好ましくは、Fe
ssIF中に2時間で放出されるタムスロシンの量は、好ましくは100rpmである同
一のパドル速度条件下で、少なくとも40%、より好ましくは少なくとも50%であり、
さらにいっそうより好ましくはタムスロシンの量の少なくとも60%がFaSSIF中に
2時間で放出される。本錠剤は、好ましくは1日1回投与錠剤であり、100rpmのパ
ドル速度で米国薬局方第2法装置を使用すると、SIF中では以下の範囲内の溶出プロフ
ィールを示す。
30分間で<40%
2時間で20〜60%
6時間で>75%
より好ましくは、本錠剤はさらにまたFaSSIF中およびFeSSIF中の少なくと
も一方、および最も好ましくはその両方において、上記の範囲内のプロフィールを示す。
一部の実施形態では、本錠剤はさらにまたSGF中でも上記の範囲内のプロフィールを示
す。上記の範囲を満たす際に、各媒質中の溶出プロフィールは相互に同一である必要はな
いが、同一であることも本発明の実施形態の1つとして予想されていると理解されなけれ
ばならない。
【0023】
明確にするために、上記の時間で放出されるタムスロシンの量またはパーセンテージは
、溶出試験の開始時から上記の時間までに放出されたタムスロシンの累積総量を意味する
。放出された量は、6回の試験、例えば各媒質または条件について6錠からの結果の平均
値として決定される。第2法装置および様々な条件について明記してきたが、これは、例
えば米国薬局方第1法装置(バスケット法)またはより多量もしくはより少量の媒質等の
相違する条件等を使用すると同一または類似の放出プロフィールを入手できないことを意
味するものではない。むしろ、上記に定義した装置および条件は、本発明の錠剤の固有の
特性を特徴付けるための便宜的な方法として役立つ。
【0024】
改良された食物作用を入手できることは、市販カプセル製剤の性能を前提とすると驚く
べきことである。一般に、食物作用の程度は、有効成分のタイプおよび溶解度、有効成分
の量/濃度、ポリマーのタイプおよび濃度、ならびに組成物の総重量によって決定される
。ポリマーマトリックスを有する放出調節型低用量タムスロシン錠が、特には被覆なしで
、低下した食物作用を備えて調製できることは予想外のことである。
【0025】
錠剤中に存在するタムスロシンは、通常はタムスロシンの(R)−エナンチオマーであ
るが、(S)−エナンチオマーならびに様々な比率で等モルまたはラセミ混合物を含む2
種の混合物もまたタムスロシンまたはその薬学的に許容可能な塩の意味に含まれる。有用
なタムスロシンの薬学的に許容可能な塩の例には、塩酸タムスロシン、臭化水素酸タムス
ロシン、メタンスルホン酸タムスロシン、トシル酸タムスロシン、ベシル酸タムスロシン
、酢酸タムスロシン、マレイン酸タムスロシン、酒石酸タムスロシン、およびクエン酸タ
ムスロシンが含まれる。典型的には、塩酸塩を使用する。
【0026】
錠剤中に存在するタムスロシン有効成分の量は相当に少なく、一般には5%未満、典型
的には0.1〜1.5%である。本明細書で使用するすべてのパーセンテージは、他に特
別に表示していない限り、錠剤に施された被覆を考慮に入れない錠剤の全重量に基づいた
重量百分率に関する。典型的には、タムスロシン有効成分の量は、0.1〜1.2%、よ
り典型的には0.2〜1.0%、好ましくは0.2〜0.8%、および多くの実施形態で
は0.3〜0.6%の範囲内である。絶対的な意味では、タムスロシン有効成分の量は遊
離塩基(free base)の量で表すと、0.1〜10mg、一般には0.1〜1.2mg、
典型的には0.3〜1.2mg、および好ましくは0.3〜0.8mgの範囲内である。
例えば、0.4mgの塩酸タムスロシンは、0.367mgのタムスロシン遊離塩基に一
致するタムスロシンの好ましい量である。本発明の好ましい実施形態は、0.4mg±0
.04の塩酸タムスロシンまたはその倍数、すなわち0.2または0.8mgの塩酸タム
スロシンを含有する。
【0027】
本発明の錠剤は、さらに1種のポリマーマトリックスを含有する。適切なポリマー材料
の特定の例には、ヒドロキシプロピルメチルセルロース(HPMC)、カルボキシメチル
セルロース、酢酸セルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロ
ース等の水膨潤性セルロース誘導体類;アルギン酸ナトリウム;アクリレート類、メタク
リレート類およびそれらと様々なコモノマー類とのコポリマー類;およびポリビニルピロ
リドン類が含まれる。
【0028】
特別には、驚くべきことに、アクリレートおよびEudragits(登録商標、Ro
hm社)等のメタクリレート類ならびにセルロース類はpH非依存性放出を提供できるが
、セルロースは一般により良好な食物作用特性を提供することが見いだされている。ヒド
ロキシエチルセルロース、ヒドロキシプロピルセルロース、酢酸セルロース、アルギン酸
ナトリウム、カルボキシメチルセルロースおよびヒドロキシプロピルメチルセルロース(
HPMC)が好ましく、最も好ましいのはHPMCである。HPMCの量は、錠剤の総重
量に基づくと、一般には10〜90%、典型的には20〜60%、好ましくは25〜40
%、より好ましくは30〜40%、いっそうより好ましくは30〜35%の範囲内である
。
【0029】
本錠剤は、典型的には希釈剤、結合剤、潤滑剤、流動促進剤、着色剤、保存剤、pH調
整剤等の追加の薬学的に許容される賦形剤を含有している。これらの賦形剤は、最終製剤
の所望の物理的態様、その消化後の組成物からの有効成分の所望の放出速度、および製造
の容易さ/費用に基づいて選択する。一般に、本発明の錠剤は、ポリマーマトリックスに
加えて、少なくとも1種の炭水化物および/または圧縮性の希釈剤を含有している。炭水
化物には、乳糖、マンニトール、マルトデキストリン、シクロデキストリン類、デキスト
レート類、およびデキストリンが含まれる。圧縮性の希釈剤には、直接圧縮法のために適
切な任意の薬学的に許容される希釈剤が含まれ、特には、リン酸水素カルシウムの二水和
物形および無水物形等のリン酸カルシウム類である。
【0030】
本発明の好ましい実施形態では、本錠剤はHPMCおよび無水リン酸二カルシウム等の
リン酸カルシウムを含む。この実施形態は、好ましくは追加して乳糖無水物等の炭水化物
を含有する。好ましくは、ステアリン酸マグネシウム等の潤滑剤もまた含まれる。相対量
は特別には限定されないが、これらの2種または3種の賦形剤(セルロース系ポリマー、
希釈剤および潤滑剤)は95%以上等の賦形剤の大半を構成していることが好ましい。例
えば、本錠剤は25〜45%のHPMC、0〜50%のリン酸カルシウム(または他の不
溶性希釈剤)、および0〜50%の乳糖(または他の可溶性希釈剤)を含んでいてよい。
以下の量が好ましい。25〜40%のHPMC、25〜40%のリン酸カルシウム、およ
び25〜40%の乳糖。より好ましくは、本錠剤は約30〜40%のHPMCを含有する
。実質的に等量、すなわち計90〜99.9%になるように各約30〜35%のHPMC
、リン酸カルシウムおよび乳糖が特別に好ましい実施形態である。潤滑剤等を含む追加の
賦形剤もまた存在していてよい。この好ましい錠剤調製物は、一般には上記に記載した好
ましい溶出プロフィールを示す。
【0031】
本発明の錠剤は、好ましくはモノリシック錠、すなわち消化後に錠剤分解せずに最終的
にはそれから有効成分が放出される複数の微粒子を形成する錠剤である。あるいはまた、
本製剤は身体内で浸食される、および/または薬物が有効成分を放出するポリマーゲルを
通して拡散する。そこで、モノリシック錠の実施形態において、本発明の製造工程で使用
される賦形剤はいずれも崩壊剤(disintegrant)として機能することはない。
【0032】
本錠剤はいずれも、単純に着色または安定性の理由から、腸溶被覆等で被覆することが
できる。治療目的では、体液へのタムスロシンの生体内吸収は、好ましくは小腸で進行し
なければならない。したがって、本発明の錠剤はさらにまた胃内の通過中に錠剤マトリッ
クスからの有効成分の放出の開始を遅延させる適切な胃耐性被覆によって保護することも
できるが、これは所望のプロフィールを入手するためには必要とされない。そのような胃
耐性被覆剤に適切な例は、酢酸フタル酸セルロース(CAP)(Aquacoat CP
D(商標))、混合抽出ポリ酢酸ビニルフタレート(PVAP)(Suretetic(
商標))、酢酸トリメリット酸セルロース(CAT)、Eudragitタイプのポリマ
ー(アクリル酸−メタクリル酸コポリマー)、酢酸コハク酸ヒドロプロピルメチルセルロ
ース(HPMCAS)である。
【0033】
被覆の放出特性は、さらにまた非被覆錠と同一の溶出試験によって試験することができ
る。被覆錠の好ましい特性は例えば以下の通りである。
SGF中での製剤の溶出試験では、タムスロシンの最高20%が2時間で放出される。
他の媒質中では、被覆錠は上記に規定したものと同一溶出プロフィールに適合しなけれ
ばならない。
【0034】
本発明の錠剤は、被覆を備える、もしくは備えない単位製剤として直接使用できる、ま
たは2錠以上の錠剤を1カプセル内に結合して単位製剤を形成することができる。単位製
剤は、BPH、高血圧、またはうっ血性心不全に関連する疾患、症状、および/または状
態を治療または改善するために、一般には遊離塩基に関して0.01〜10.0mg、好
ましくは0.1〜1mgの有効量のタムスロシンを含有する。好ましいのは、塩酸タムス
ロシン自体を0.2、0.4または0.8mg含有する単位製剤である。単位製剤は、通
常は1日に1〜3回、好ましくは上述したように1日1回摂取される。カプセル剤の場合
は、有効量を提供できるようにその中のタムスロシン有効成分の濃度に基づいて、十分な
数の錠剤が提供される。
【0035】
通常の治療量のタムスロシンを考慮に入れると、総重量が400mg以下の、そして通
常は10〜300mgの錠剤が好ましい。タムスロシンの治療量は相当に少量であるので
、錠剤の総重量は有益にもできる限り少なく保持される。錠剤の少ない総重量は、錠剤中
のタムスロシンの相対含有量を増加させ、したがって含量均一性を向上させる。その上、
小さな錠剤は顆粒製剤に類似する消化管通過速度を有する。したがってインビトロ溶出試
験から入手された結果によって、市販の多微粒子製剤との実際的生物学的等価性をより明
確に予測できる。この態様から、本発明の範囲内の好ましい錠剤重量は25〜250mg
、より好ましくは40〜200mgであるが、錠剤重量はこの範囲内に限定されない。最
も好ましい錠剤重量は、80〜100mgの範囲内、特別にはおよそ100mgである。
【0036】
したがって、本発明の錠剤は小さくてよいが、それにより円形で製造される場合は常に
、その平均径は約1.5mm〜約2.5mmである。またはそれらは2.5mm〜15m
m、より通常は2.5mm〜10mmの平均径を有する通常の錠剤として製造することも
できる。リング形以外に、タムスロシン組成物は楕円形、両凸面状の円形、五角形の外接
円または他の適切な錠剤形に圧縮できる。
【0037】
単位用量のタムスロシンを含有する本発明の錠剤は、直接使用のためには有利には、5
〜100錠を含む適切な包装単位で供給することができる。そのような包装には、有利に
は、10、14、20、28もしくは30錠を含むブリスターパック、または同一量(5
〜100錠)の錠剤を含有するプラスチック製もしくはガラス製の容器/ボトルを含むこ
とができる。包装単位の製造には、あらゆる適切な薬学的に許容可能な包装材料を使用で
きる。
【0038】
本発明の錠剤は、いずれか適切な錠剤形成工程によって製造できる。例えば、本錠剤は
、顆粒が最初に形成され、そして任意で1種以上の賦形剤を添加して錠剤に圧縮される湿
式造粒法によって製造できる。あるいはまた、本錠剤は、直接圧縮法または乾式造粒法等
の乾式工程によって製造でき、後者はときどき乾式圧縮成型法とも呼ばれる。好ましくは
、本錠剤は容易かつ経済的に製造することを考慮して乾式技術によって製造される。本錠
剤中には少量のタムスロシンが存在するために、乾式工程では複数の混合および/または
粉砕ステップを実施することが好ましい。
【0039】
本発明によるタムスロシンの経口投与用錠剤は、症候性良性前立腺肥大症(BPH)ま
たはタムスロシンによって治療可能なその他の障害(障害)の機能的治療の管理において
使用できる。したがって、本発明はさらにまたそれを必要とする患者に1錠以上の上記の
錠剤を投与するステップを含む良性前立腺肥大症の症状を治療するための方法を提供する
。本錠剤は、単一カプセルに入れて投与できる。
【0040】
本発明の錠剤組成物は、さらに他の薬剤と組み合わせて医療用途に使用することもでき
る。この組み合わせは、単一複合製剤の形状で、または上記の物質を含有する薬物の個別
投与によって実現することができる。
【0041】
本発明を、以下の非限定的実施例により詳細に例示する。
【0042】
[実施例]
市販の塩酸タムスロシンカプセルをヨーロッパで入手し、37℃、パドル速度100r
pmで、米国薬局方第2法装置において、各媒質の500ml中の各SGF、SIF、F
aSSIF、およびFeSSIFの中で溶出試験を実施し、6回の試験の平均値を入手し
た。放出された薬物量は、HPLC Agilent 1100システムを使用してHP
LC法によって測定した。検出は230nmに設定したUVを用いて実施した。図1に示
した結果は、2時間の経過後にSGF中ではタムスロシンの20%未満が放出され、SI
F中ではタムスロシンの60%超が放出され、FaSSI中ではタムスロシンの75%超
が放出され、そしてFeSSIF中ではタムスロシンの40%未満が放出されることを表
している。
【実施例1】
【0043】
以下の特徴を有する3バッチのモノリシック錠を漸進的混合(progressive mixing)お
よび直接圧縮法によって製造した。
【0044】
a)錠剤組成物
【表1】
バッチ間の相違は、選択したヒプロメロースの粘度値だけであった。
バッチAはMETHOCEL K4M CR PREMIUMを含有していた。
バッチBはMETHOCEL K15M CR PREMIUMを含有していた。
バッチCはMETHOCEL K100M CR PREMIUMを含有していた。
【0045】
b)操作方法
塩酸タムスロシンは、1:9の比率(有効成分10%)で無水乳糖と混合し(15分間
)、粉砕し(15秒間)、再び混合した(5分間)。次にこの前混合物を残りの乳糖、リ
ン酸二カルシウムおよびヒプロメロースと混合し(10分間)、そして最後にステアリン
酸マグネシウムを添加し、圧縮前混合物を形成するために混合した(5分間)。この漸進
的混合系は、前混合物中では97.2〜100.4%、そして圧縮前混合物中では88.
1〜98.6%のタムスロシン均質性を生じさせた。圧縮法は、Korsch EKOプ
レス機において標準速度および標準圧で実施した。
【0046】
c)製造された錠剤のキャラクタリゼーション
【表2】
【0047】
d)溶出試験
溶出試験は、標準的な米国薬局方第2法装置を使用して、500mLのSIF中で50
rpmのパドル速度で実施した。放出された薬物は、HPLC Agilent 110
0システムを使用してHPLC法によって測定した。分析は、溶離液としてリン酸緩衝液
(pH6.5)およびアセトニトリル(65:35)を使用し、均一濃度溶離モードを用
いて、ガードカラムおよびC18分析用カラムを用いて実施した。検出は230nmに設定
したUVを用いて実施した。結果は以下の仕様を満たしている。
30分間で<40%
2時間で20〜60%
6時間で>75%
【実施例2】
【0048】
以下の特徴を有する3バッチのモノリシック錠を漸進的混合および直接圧縮法によって
製造した。
【0049】
a)錠剤組成物
【表3】
バッチ間の相違は、使用したヒプロメロースの濃度だけであった。
バッチDは11%のMETHOCEL K100M CR PREMIUMを含有して
いた。
バッチEは22%のMETHOCEL K100M CR PREMIUMを含有して
いた。
バッチFは44%のMETHOCEL K100M CR PREMIUMを含有して
いた。
【0050】
b)操作方法
塩酸タムスロシンは、1:9の比率(有効成分10%)で無水乳糖と混合し(15分間
)、粉砕し(15秒間)、再び混合した(5分間)。次にこの前混合物を残りの乳糖、リ
ン酸二カルシウムおよびヒプロメロースと混合し(10分間)、そして最後にステアリン
酸マグネシウムを添加し、混合した(5分間)。圧縮は、Korsch EKOプレス機
で実施した。
【0051】
c)製造された錠剤のキャラクタリゼーション
【表4】
【0052】
d)溶出試験
溶出試験は、どちらも500mLのSIF中において、100rpmでバスケットを使
用する米国薬局方第1法装置、および50rpmの速度でパドルを使用する米国薬局方第
2法装置を使用して実施した。放出された薬物は、HPLC Agilent 1100
システムを使用してHPLC法によって測定した。分析は、溶離液としてリン酸緩衝液(
pH6.5)およびアセトニトリル(65:35)を使用して、均一濃度溶離モードを用
いて、ガードカラムおよびC18分析用カラムを用いて実施した。検出は230nmに設定
したUVを用いて実施した。
結果はHPMC濃度にしたがって変動し、各条件では、規定の仕様に適合して広範囲の
曲線が得られる。これらの結果は、他の生体関連溶出条件へ外挿できる。
【実施例3】
【0053】
以下の特徴を有する2バッチのモノリシック錠を漸進的混合および直接圧縮法によって
製造した。
【0054】
a)錠剤組成物
【表5】
両バッチ間の差は、主としてスケールアップ率、混合時間および物理的パラメーターで
あった。
バッチGは20,000単位へスケールアップされた。
バッチHは40,000単位へスケールアップされた。
【0055】
b)操作方法
塩酸タムスロシンは、1:9の比率(有効成分10%)で無水乳糖と混合し(Turb
ula;15分間)、粉砕し(IKA;30秒間)、再び混合した(Turbula;5
分間)。次に、この前混合物を残りの乳糖、リン酸二カルシウムおよびヒプロメロースと
混合した(Bohle LM40)。バッチDについては3つの漸進的混合時間(15、
30および45分間)を評価したが、いずれの場合においても均質性は極めて良好であっ
た(タムスロシン含有率:101.2%、101.7%および102.1%)。バッチE
は10分間しか混合しなかったが、許容される均質性に同様に到達した。無水混合物およ
び賦形剤のふるい分けは、均質性を入手するための必要に応じて実施した。最後にステア
リン酸マグネシウムをふるいにかけ、添加し、混合した(Bohle LM40;5分間
)。圧縮前混合物は偏心式プレス機Korsch EKOまたは回転式プレス機Kors
ch XL100のどちらかで圧縮した(約15,000〜30,000錠)。回転式プ
レス機を備えたKorsch XL100で実施された圧縮は、標準的前圧縮および標準
圧力を用いて高速で実施した。溶出性能を試験するために、両バッチにおける錠剤の硬度
を変化させた。
【0056】
c)製造された錠剤のキャラクタリゼーション
【表6】
【0057】
d)溶出試験
溶出試験は、標準的な米国薬局方第2法装置を使用して、100rpmのパドル速度で
、500mLのSGF、SIF、FaSSIFおよびFeSSIF中において実施した。
放出された薬物は、HPLC Agilent 1100システムを使用してHPLC法
によって測定した。分析は、溶離液としてリン酸緩衝液(pH6.5)およびアセトニト
リル(65:35)を使用して、均一濃度溶離モードを用いて、ガードカラムおよびC18
分析用カラムを用いて実施した。検出は230nmに設定したUVを用いて実施した。対
応する曲線は、図2(G)および3(H)に示した。SGFおよびSIF中での試験結果
は、以下の仕様を満たしている。
30分間で<40%
2時間で20〜60%
6時間で>75%
【0058】
e)被覆
これらの錠剤はその後、クエン酸トリエチルおよびタルクを含む添加物、またはAcr
yl−Eze(登録商標、Colorcon社から入手可能)とともに、Eudragi
t L30D55を基剤とする腸溶ポリマー(ポリメタクリレート、タイプC)を用いて
被覆した。
【0059】
f)溶出試験
被覆錠のバッチの溶出試験は、標準的な米国薬局方第2法装置を使用して、100rp
mのパドル速度で、500mLのSGF、SIF、FaSSIFおよびFeSSIF中で
実施した。放出された薬物は、HPLC Agilent 1100システムを使用して
HPLC法によって測定した。分析は、溶離液としてリン酸緩衝液(pH6.5)および
アセトニトリル(65:35)を使用して、均一濃度溶離モードを用いて、ガードカラム
およびC18分析用カラムを用いて実施した。検出は230nmに設定したUVを用いて実
施した。
対応する曲線は、図4(被覆G)および図5(被覆H)に示した。
SIF中の結果は以下の仕様を満たしている。
30分間で<40%
2時間で20〜60%
6時間で>75%
【実施例4】
【0060】
2バッチの錠剤は、乾式圧縮成型、粉砕、混合および圧縮を含む工程によって製造した
。
【0061】
a)錠剤組成物
【表7】
バッチI:モノリシック錠、6mm径。33.0%のHPMC K15M Pを含有す
る。
バッチJ:モノリシック錠、9mm径。13.2%のHPMC K15M Pを含有す
る。
【0062】
b)操作方法
タムスロシンは、1:9の比率(有効成分10%)で無水乳糖と混合し(15分間)、
粉砕し(15秒間)、再び混合した(5分間)。次にこの前混合物を残りの乳糖、ヒプロ
メロースおよび25%のステアリン酸マグネシウムと混合し(10分間)、Chilso
nator(Fitz−Patrick社)で圧縮成型し、Fitz−Mill(Fit
z−Patrick社)で粉砕し、最後に残りのステアリン酸マグネシウム(75%)を
添加し、さらに混合した(15分間)。圧縮法は、Korsch EKOプレス機におい
て標準速度および標準圧で実施した。
【0063】
c)製造された錠剤のキャラクタリゼーション
【表8】
【実施例5】
【0064】
相違する有効成分濃度、錠剤形状および総重量を備える2バッチの錠剤を製造した。
【0065】
a)錠剤組成物
【表9】
【0066】
b)操作方法
微粉化タムスロシンは、無水乳糖と漸進的に混合した。次に、すべての場合に十分な均
質性を提供するために、この前混合物を残りの乳糖およびヒプロメロースと混合し、その
後ステアリン酸マグネシウムと混合した。圧縮法は、Korsch EKOプレス機にお
いて標準速度および標準圧で実施した。
【0067】
c)製造された錠剤のキャラクタリゼーション
【表10】
【実施例6】
【0068】
溶出仕様を検査するために、1バッチのモノリシック錠を製造した。
【0069】
a)錠剤組成物
【表11】
バッチNのスケールアップは、微粉化した薬物成分およびより少ない混合ステップを使
用して実施した。40,000単位を製造した。
【0070】
b)操作方法
微粉化塩酸タムスロシンをヒプロメロースと混合した(Bohle LM40;15分
間)。次にこの前混合物を乳糖およびリン酸二カルシウムと混合した(Bohle LM
40;15分間)。無水混合物および賦形剤のふるい分けは、均質性を入手するための必
要に応じて実施した。最後にステアリン酸マグネシウムをふるいにかけ、添加し、さらに
2回混合した(Bohle LM40;5および15分間)。
圧縮前混合物を次に圧縮した。回転式プレス機を備えたKorsch XL100で実
施される圧縮は、標準的前圧縮および標準圧力を用いて高速で実施した。
錠剤の特徴付けは以下に示した。
【0071】
c)製造された錠剤のキャラクタリゼーション
【表12】
【0072】
d)被覆
この錠剤は、次に腸溶ポリマー(Colorcon社から入手可能なAcryl−Ez
e(登録商標))を用いて被覆した。
【0073】
e)溶出試験
被覆錠のバッチの溶出試験は、標準的な米国薬局方第2法装置を使用して、100rp
mのパドル速度で、500mLのSGF、SIF、FaSSIFおよびFeSSIF中で
実施した。放出された薬物は、HPLC Agilent 1100システムを使用して
HPLC法によって測定した。分析は、溶離液としてリン酸緩衝液(pH6.5)および
アセトニトリル(65:35)を使用して、均一濃度溶離モードを用いて、ガードカラム
およびC18分析用カラムを用いて実施した。検出は230nmに設定したUVを用いて実
施した。
対応する曲線は、図6に示した。
全媒質中での試験結果は以下の仕様を満たしている。
30分間で<40%
2時間で20〜60%
6時間で>75%
【0074】
本発明を説明してきたが、当業者には、特許請求の範囲によって定義された本発明の精
神および範囲から逸脱することなく、本明細書に記載した概念および実施形態を実際に実
施するに当たりまた別の変更および修飾を容易に加えることができること、または本発明
の実践によって学習されることは容易に明白であろう。
【技術分野】
【0001】
本出願は、米国特許法第119条(e)の下で、引用することにより本明細書の一部を
なすものとする2001年11月7日に出願された先の米国特許仮出願第60/331,
055号からの優先権の利益を主張する。
【0002】
[発明の分野]
本発明は、食物作用(food effect)をほとんど、または全く示さない放出調節型タム
スロシン錠およびそれから製造された単位製剤に関する。
【背景技術】
【0003】
タムスロシンは、式(1)の5−[2−[[2−(2−エトキシフェノキシ)エチル]
アミノ]プロピル]−2−メトキシ−ベンゼンスルホンアミドの一般名である。
【化1】
タムスロシンは、欧州特許第34432号および米国特許第4,731,478号におい
て、心不全および良性前立腺肥大症を治療するために有用なαアドレナリン遮断活性を有
する薬学的に活性な成分であると開示されている。
【0004】
(R)−塩酸タムスロシンは、良性前立腺肥大症(BPHとしても知られる)の尿量や
尿頻度の問題等の症状を治療するため、アメリカ合衆国でのFLOMAX(登録商標)(
Boehringer Ingelheim社)、日本でのHARNAL(登録商標)(
山之内製薬)およびヨーロッパでのOMNIC(登録商標)(山之内製薬)を含めて様々
な商標名を付けて市販されている。既承認製剤には、0.4mgの塩酸タムスロシンを含
む経口投与用カプセル製剤が含まれる。このカプセル剤は、タムスロシンの制御された放
出を提供し、1日1回製剤であるが、必要であればカプセル2錠、すなわち最高0.8m
gの1日1回投与を使用することができる。
【0005】
制御放出型または調節放出型の市販カプセル剤には、食物作用を示すという欠点がある
。食物作用とは、摂食(胃内に食物がある)患者に比較して、絶食(胃が空である)患者
への薬物の投与から発生する生体内吸収率または生体内利用率における相違を意味する。
市販のカプセル剤については、食物作用は相当に顕著である。例えば、FLOMAX(登
録商標)の添付文書では、絶食条件下におけるTmaxは4〜5時間であるが、摂食条件
下におけるTmaxは6〜7時間であると報告されている。絶食条件下でカプセル剤を摂
取すると、摂食条件下と比較して、バイオアベイラビリティ(AUC)における30%の
増加およびピーク濃度(Cmax)における40%〜70%の増加が生じる。そこで食後
に摂取した場合、タムスロシンが達成する最高血漿中濃度はより低くなる。そしてこのピ
ークはより遅い時期に達成される。したがって、食後の投与は、絶食条件下の投与に比較
して、生体内利用率の損失が生じるにもかかわらず、平坦かつ制御放出型の血漿中濃度プ
ロフィールを生じさせる。
【0006】
市販カプセル剤の添付文書は、食後(日本)、朝食後(ヨーロッパ)および毎日同一の
食事の30分後(米国)のような摂食状態において投与するように指示している。この投
与は、より一貫性のある結果を提供して副作用もより少ないために推奨されていると考え
られる。実際に、タムスロシンの吸収は摂食条件下より絶食条件下(90+%)の方が良
好ではあるが、承認された使用はタムスロシンカプセル剤を摂食条件下で投与するように
指示している。
【0007】
タムスロシンの市販カプセル剤は米国特許第4,772,475号に対応すると考えら
れる(欧州特許第194838号、欧州特許第533297号)。米国特許第4,772
,475号は、タムスロシン、微結晶セルロースおよび放出制御剤を含有する、多数の顆
粒単位を含む制御放出型医薬製剤を開示している。顆粒剤は、タムスロシンを顆粒マトリ
ックスから徐々に放出する。食物作用問題に関する検討は行われていない。
【0008】
タムスロシンの市販カプセル剤では食物作用が発生するために、絶食時(食物を摂取し
ていない)にカプセル剤を摂取した患者は、めまい、鼻炎、および/または異常射精等の
望ましくない副作用を経験する可能性がより高くなる。食物作用が小さいもしくはわずか
である、または食物作用を全く示さないタムスロシン医薬製剤を製造することが有益であ
ろう。そこで、本製剤はより安全であろう。すなわち、絶食条件下で摂取した場合でさえ
、副作用のリスクが低下する。タムスロシンの市販カプセル剤の食物作用は明確に記録さ
れているが、これまでに食物作用問題に対する解決策は提供されていない。
【発明の概要】
【0009】
現在では、食物作用が低下した調節放出型タムスロシン錠を形成できることが見いださ
れた。したがって、本発明の1つの態様は、その中に0.1〜10mgのタムスロシンま
たはその薬学的に許容可能な塩が分散している錠剤マトリックスを含み、そして任意選択
的に該マトリックスの全体を被覆する腸溶被覆を有する医薬錠剤に関する。本錠剤は放出
調節型錠剤であり、50〜100rpmのパドル速度で500mLの媒質を使用した米国
薬局方第2法装置(USP 2 apparatus)で、各々について以下で定義するSIF、FaS
SIF、およびFeSSIFの各媒質中に錠剤が2時間の経過後にタムスロシンの60%
以下を放出するような溶出プロフィールを示す。好ましくは、本錠剤は、同様に3種の各
媒質中に2時間の指標までに少なくとも20%のタムスロシンを放出する。これらの媒質
はインビボで遭遇する腸内条件をインビトロで模倣するために役立ち、FaSSIFは絶
食状態に相当し、FeSSIFは摂食状態に相当する。各擬似条件下において2時間で6
0%未満のタムスロシンを放出したことによって、本錠剤は摂食状態と絶食状態のどちら
も本製剤の放出調節型の性質を低下させることがないことを証明している。
【0010】
本発明のまた別の態様は、0.1〜10mgのタムスロシンまたはその薬学的に許容可
能な塩と、10重量%〜90重量%のヒドロキシプロピルメチルセルロースとを含み、そ
して総錠剤重量が10〜300mgであるモノリシック医薬錠剤に関する。本錠剤は、好
ましくは低下した食物作用を示し、そしてより好ましくは上記で説明した溶出プロフィー
ルの要件を満たす。
【0011】
これらの態様の各々において、本錠剤はそれ自体で投与可能であり、任意選択的に腸溶
被覆を備えてもよく、しかし好ましくは腸溶被覆を備えずに投与できる。または、1錠以
上の錠剤をカプセル封入し、1つ以上のカプセル剤として投与することができる。本発明
のまた別の態様は、良性前立腺肥大症の症状を治療するための方法であって、それを必要
とする患者に1錠以上の上記の錠剤を投与するステップを含む方法に関する。
【図面の簡単な説明】
【0012】
【図1】本明細書に記載した4種の媒質中における山之内ヨーロッパによって製造されたタムスロシンカプセル剤の放出曲線の図である。
【図2】本明細書に記載した4種の媒質中におけるタムスロシン錠(バッチG)の放出曲線の図である。
【図3】本明細書に記載した4種の媒質中におけるタムスロシン錠(バッチH)の放出曲線の図である。
【図4】本明細書に記載した4種の媒質中におけるタムスロシン腸溶被覆錠(バッチG)の放出曲線の図である。
【図5】本明細書に記載した4種の媒質中におけるタムスロシン腸溶被覆錠(バッチH)の放出曲線の図である。
【図6】選択した媒質中のタムスロシン非被覆錠および被覆錠(バッチN)の放出曲線の図である。
【発明を実施するための形態】
【0013】
本発明は、放出調節型タムスロシン錠および該放出調節型タムスロシン錠を含有するカ
プセル剤に関する。「放出調節型」とは、本明細書では広い意味で使用されており、即時
放出型製剤ではない、すなわち標準溶出試験(すなわち、37℃でパドル速度50rpm
、500mLのSGFを用いる米国薬局方第2法装置を使用する)における初期30分間
以内に少なくとも75%のタムスロシンを放出する製剤ではないあらゆる製剤を意味する
。この錠剤は、市販で入手できるタムスロシンカプセル剤と比較して低下した食物作用を
示す。これらのカプセル剤とは相違して、本発明は、タムスロシンまたはその薬学的に許
容可能な塩を、制御可能な食物作用の特性を有する錠剤に調製することができるという発
見に基づいている。
【0014】
タムスロシン錠は、好ましくは各SIF、FaSSIF、およびFeSSIFの中に、
タムスロシンの60%未満、好ましくは20%〜60%が2時間で放出される溶出プロフ
ィールを示す。溶出試験は、試験をその中で実施する媒質500mLを使用する米国薬局
方第2法装置において、50〜100rpm、好ましくは100rpmのパドル速度を用
いて実施する。ある実施形態、特別には非被覆錠を含む実施形態では、溶出プロフィール
はさらに、50〜100rpm、好ましくは100rpmのパドル速度に設定した米国薬
局方第2法装置において、500mLのSGF中に2時間でタムスロシンの60%未満、
好ましくは20%〜60%を放出することを含む。本発明の目的で放出プロフィールを決
定するためのすべての溶出試験において、媒質の温度は37℃である。装置内に錠剤1錠
が配置されることを想定して、インビボ試験結果のより正確なモデリング/予測を提供す
ると考えられるので、500mLの媒質を使用する。
【0015】
本発明のための溶出媒質は、以下に定義した通りである。
【0016】
SGF(米国薬局方によるペプシンを含まない擬似胃液)の組成:
HCl 十分量 pH1.2
NaCl 0.2%
水 十分量 1,000mL
【0017】
SIF(米国薬局方によるパンクレアチンを含まない擬似腸液)の組成:
KH2P04 6.8g
NaOH 十分量 pH6.8
水 十分量 1,000mL
【0018】
FeSSIF(擬似腸液、摂食状態)の組成:
酢酸 0.144M
NaOH 十分量 pH5
タウロコール酸ナトリウム 15mM
レシチン 4mM
KCl 0.19M
蒸留水 十分量 1,000mL
pH=5
浸透圧=485〜535mOsm
緩衝能力=75±2mEQ/L/pH
【0019】
FaSSIF(擬似腸液、絶食状態)の組成:
KH2PO4 0.029M
NaOH 十分量 pH6.8
タウロコール酸ナトリウム 5mM
レシチン 1.5mM
KCl 0.22M
蒸留水 十分量 1,000mL
pH=6.8
浸透圧=280〜310mOsm
緩衝能力=10±2mEQ/L/pH
【0020】
SGFは胃の標準的状態を表している。SIFは腸の標準的状態を表している。FeS
SIFは摂食状態をより良好に表すように調整され、他方FaSSIFは絶食状態をより
良好に表すように調整されている。pHが相違するだけではなく、同等に重要であること
に、浸透圧もまた相違することに注目されたい。FaSSIF媒質およびFeSSIF媒
質は、一般に即時放出型の脂肪親和性かつ難水溶性の薬剤(すなわち、ケトコナゾール、
ダナゾール、アトバクオン、トログリタゾン、メフェナム酸)についてのインビトロ−イ
ンビボ相関を説明するために使用されてきたが、以前に実施された試験は、塩酸タムスロ
シン等の放出調節型低用量かつ可溶性の薬物(10mgまでを溶解させるために必要な水
性媒質の量は500mL以下である)への適用も、放出調節型調製物(放出制御型、徐放
性または遅延放出型を含む)への適用も示唆していない。
【0021】
以下の参照実施例に示すように、山之内ヨーロッパ社が製造した市販のカプセル剤は、
2時間の経過後にタムスロシンの次のような放出を示す。SIF中には60%超、FaS
SIF中には75%超が放出されるが、FeSSIF中には40%未満が放出され、SG
F中には20%未満が放出される(図1を参照)。pH6.8では速すぎて、pH1.2
では遅すぎる(遅延性放出)、4種の媒質中でのこの結果の相違は、以前に指摘されたよ
うにタムスロシンの血漿中濃度における変動性について考えられる、例えば、様々な胃内
容排出状態および/または胃腸内pHの変化のような理由を表している。当然ながら、こ
のFaSSIF中でのより急速な放出は、摂食状態より絶食状態においてより急速なTm
axおよびより高いCmaxのインビボ観察所見と良好に一致している。したがって、各
SIF、FaSSIF、およびFeSSIF中に、好ましくはSGF中にも60%以下の
タムスロシン放出を示す本発明の好ましい錠剤は、市販のカプセル剤と比較してより良好
な食物作用を有する、すなわち摂食状態と絶食状態の間の差がより小さい。
【0022】
好ましくは、タムスロシン錠は各SIF、FaSSIF、およびFeSSIF中に溶出
試験の初期2時間中にタムスロシンの20%〜60%を放出する。より好ましくは、Fe
ssIF中に2時間で放出されるタムスロシンの量は、好ましくは100rpmである同
一のパドル速度条件下で、少なくとも40%、より好ましくは少なくとも50%であり、
さらにいっそうより好ましくはタムスロシンの量の少なくとも60%がFaSSIF中に
2時間で放出される。本錠剤は、好ましくは1日1回投与錠剤であり、100rpmのパ
ドル速度で米国薬局方第2法装置を使用すると、SIF中では以下の範囲内の溶出プロフ
ィールを示す。
30分間で<40%
2時間で20〜60%
6時間で>75%
より好ましくは、本錠剤はさらにまたFaSSIF中およびFeSSIF中の少なくと
も一方、および最も好ましくはその両方において、上記の範囲内のプロフィールを示す。
一部の実施形態では、本錠剤はさらにまたSGF中でも上記の範囲内のプロフィールを示
す。上記の範囲を満たす際に、各媒質中の溶出プロフィールは相互に同一である必要はな
いが、同一であることも本発明の実施形態の1つとして予想されていると理解されなけれ
ばならない。
【0023】
明確にするために、上記の時間で放出されるタムスロシンの量またはパーセンテージは
、溶出試験の開始時から上記の時間までに放出されたタムスロシンの累積総量を意味する
。放出された量は、6回の試験、例えば各媒質または条件について6錠からの結果の平均
値として決定される。第2法装置および様々な条件について明記してきたが、これは、例
えば米国薬局方第1法装置(バスケット法)またはより多量もしくはより少量の媒質等の
相違する条件等を使用すると同一または類似の放出プロフィールを入手できないことを意
味するものではない。むしろ、上記に定義した装置および条件は、本発明の錠剤の固有の
特性を特徴付けるための便宜的な方法として役立つ。
【0024】
改良された食物作用を入手できることは、市販カプセル製剤の性能を前提とすると驚く
べきことである。一般に、食物作用の程度は、有効成分のタイプおよび溶解度、有効成分
の量/濃度、ポリマーのタイプおよび濃度、ならびに組成物の総重量によって決定される
。ポリマーマトリックスを有する放出調節型低用量タムスロシン錠が、特には被覆なしで
、低下した食物作用を備えて調製できることは予想外のことである。
【0025】
錠剤中に存在するタムスロシンは、通常はタムスロシンの(R)−エナンチオマーであ
るが、(S)−エナンチオマーならびに様々な比率で等モルまたはラセミ混合物を含む2
種の混合物もまたタムスロシンまたはその薬学的に許容可能な塩の意味に含まれる。有用
なタムスロシンの薬学的に許容可能な塩の例には、塩酸タムスロシン、臭化水素酸タムス
ロシン、メタンスルホン酸タムスロシン、トシル酸タムスロシン、ベシル酸タムスロシン
、酢酸タムスロシン、マレイン酸タムスロシン、酒石酸タムスロシン、およびクエン酸タ
ムスロシンが含まれる。典型的には、塩酸塩を使用する。
【0026】
錠剤中に存在するタムスロシン有効成分の量は相当に少なく、一般には5%未満、典型
的には0.1〜1.5%である。本明細書で使用するすべてのパーセンテージは、他に特
別に表示していない限り、錠剤に施された被覆を考慮に入れない錠剤の全重量に基づいた
重量百分率に関する。典型的には、タムスロシン有効成分の量は、0.1〜1.2%、よ
り典型的には0.2〜1.0%、好ましくは0.2〜0.8%、および多くの実施形態で
は0.3〜0.6%の範囲内である。絶対的な意味では、タムスロシン有効成分の量は遊
離塩基(free base)の量で表すと、0.1〜10mg、一般には0.1〜1.2mg、
典型的には0.3〜1.2mg、および好ましくは0.3〜0.8mgの範囲内である。
例えば、0.4mgの塩酸タムスロシンは、0.367mgのタムスロシン遊離塩基に一
致するタムスロシンの好ましい量である。本発明の好ましい実施形態は、0.4mg±0
.04の塩酸タムスロシンまたはその倍数、すなわち0.2または0.8mgの塩酸タム
スロシンを含有する。
【0027】
本発明の錠剤は、さらに1種のポリマーマトリックスを含有する。適切なポリマー材料
の特定の例には、ヒドロキシプロピルメチルセルロース(HPMC)、カルボキシメチル
セルロース、酢酸セルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロ
ース等の水膨潤性セルロース誘導体類;アルギン酸ナトリウム;アクリレート類、メタク
リレート類およびそれらと様々なコモノマー類とのコポリマー類;およびポリビニルピロ
リドン類が含まれる。
【0028】
特別には、驚くべきことに、アクリレートおよびEudragits(登録商標、Ro
hm社)等のメタクリレート類ならびにセルロース類はpH非依存性放出を提供できるが
、セルロースは一般により良好な食物作用特性を提供することが見いだされている。ヒド
ロキシエチルセルロース、ヒドロキシプロピルセルロース、酢酸セルロース、アルギン酸
ナトリウム、カルボキシメチルセルロースおよびヒドロキシプロピルメチルセルロース(
HPMC)が好ましく、最も好ましいのはHPMCである。HPMCの量は、錠剤の総重
量に基づくと、一般には10〜90%、典型的には20〜60%、好ましくは25〜40
%、より好ましくは30〜40%、いっそうより好ましくは30〜35%の範囲内である
。
【0029】
本錠剤は、典型的には希釈剤、結合剤、潤滑剤、流動促進剤、着色剤、保存剤、pH調
整剤等の追加の薬学的に許容される賦形剤を含有している。これらの賦形剤は、最終製剤
の所望の物理的態様、その消化後の組成物からの有効成分の所望の放出速度、および製造
の容易さ/費用に基づいて選択する。一般に、本発明の錠剤は、ポリマーマトリックスに
加えて、少なくとも1種の炭水化物および/または圧縮性の希釈剤を含有している。炭水
化物には、乳糖、マンニトール、マルトデキストリン、シクロデキストリン類、デキスト
レート類、およびデキストリンが含まれる。圧縮性の希釈剤には、直接圧縮法のために適
切な任意の薬学的に許容される希釈剤が含まれ、特には、リン酸水素カルシウムの二水和
物形および無水物形等のリン酸カルシウム類である。
【0030】
本発明の好ましい実施形態では、本錠剤はHPMCおよび無水リン酸二カルシウム等の
リン酸カルシウムを含む。この実施形態は、好ましくは追加して乳糖無水物等の炭水化物
を含有する。好ましくは、ステアリン酸マグネシウム等の潤滑剤もまた含まれる。相対量
は特別には限定されないが、これらの2種または3種の賦形剤(セルロース系ポリマー、
希釈剤および潤滑剤)は95%以上等の賦形剤の大半を構成していることが好ましい。例
えば、本錠剤は25〜45%のHPMC、0〜50%のリン酸カルシウム(または他の不
溶性希釈剤)、および0〜50%の乳糖(または他の可溶性希釈剤)を含んでいてよい。
以下の量が好ましい。25〜40%のHPMC、25〜40%のリン酸カルシウム、およ
び25〜40%の乳糖。より好ましくは、本錠剤は約30〜40%のHPMCを含有する
。実質的に等量、すなわち計90〜99.9%になるように各約30〜35%のHPMC
、リン酸カルシウムおよび乳糖が特別に好ましい実施形態である。潤滑剤等を含む追加の
賦形剤もまた存在していてよい。この好ましい錠剤調製物は、一般には上記に記載した好
ましい溶出プロフィールを示す。
【0031】
本発明の錠剤は、好ましくはモノリシック錠、すなわち消化後に錠剤分解せずに最終的
にはそれから有効成分が放出される複数の微粒子を形成する錠剤である。あるいはまた、
本製剤は身体内で浸食される、および/または薬物が有効成分を放出するポリマーゲルを
通して拡散する。そこで、モノリシック錠の実施形態において、本発明の製造工程で使用
される賦形剤はいずれも崩壊剤(disintegrant)として機能することはない。
【0032】
本錠剤はいずれも、単純に着色または安定性の理由から、腸溶被覆等で被覆することが
できる。治療目的では、体液へのタムスロシンの生体内吸収は、好ましくは小腸で進行し
なければならない。したがって、本発明の錠剤はさらにまた胃内の通過中に錠剤マトリッ
クスからの有効成分の放出の開始を遅延させる適切な胃耐性被覆によって保護することも
できるが、これは所望のプロフィールを入手するためには必要とされない。そのような胃
耐性被覆剤に適切な例は、酢酸フタル酸セルロース(CAP)(Aquacoat CP
D(商標))、混合抽出ポリ酢酸ビニルフタレート(PVAP)(Suretetic(
商標))、酢酸トリメリット酸セルロース(CAT)、Eudragitタイプのポリマ
ー(アクリル酸−メタクリル酸コポリマー)、酢酸コハク酸ヒドロプロピルメチルセルロ
ース(HPMCAS)である。
【0033】
被覆の放出特性は、さらにまた非被覆錠と同一の溶出試験によって試験することができ
る。被覆錠の好ましい特性は例えば以下の通りである。
SGF中での製剤の溶出試験では、タムスロシンの最高20%が2時間で放出される。
他の媒質中では、被覆錠は上記に規定したものと同一溶出プロフィールに適合しなけれ
ばならない。
【0034】
本発明の錠剤は、被覆を備える、もしくは備えない単位製剤として直接使用できる、ま
たは2錠以上の錠剤を1カプセル内に結合して単位製剤を形成することができる。単位製
剤は、BPH、高血圧、またはうっ血性心不全に関連する疾患、症状、および/または状
態を治療または改善するために、一般には遊離塩基に関して0.01〜10.0mg、好
ましくは0.1〜1mgの有効量のタムスロシンを含有する。好ましいのは、塩酸タムス
ロシン自体を0.2、0.4または0.8mg含有する単位製剤である。単位製剤は、通
常は1日に1〜3回、好ましくは上述したように1日1回摂取される。カプセル剤の場合
は、有効量を提供できるようにその中のタムスロシン有効成分の濃度に基づいて、十分な
数の錠剤が提供される。
【0035】
通常の治療量のタムスロシンを考慮に入れると、総重量が400mg以下の、そして通
常は10〜300mgの錠剤が好ましい。タムスロシンの治療量は相当に少量であるので
、錠剤の総重量は有益にもできる限り少なく保持される。錠剤の少ない総重量は、錠剤中
のタムスロシンの相対含有量を増加させ、したがって含量均一性を向上させる。その上、
小さな錠剤は顆粒製剤に類似する消化管通過速度を有する。したがってインビトロ溶出試
験から入手された結果によって、市販の多微粒子製剤との実際的生物学的等価性をより明
確に予測できる。この態様から、本発明の範囲内の好ましい錠剤重量は25〜250mg
、より好ましくは40〜200mgであるが、錠剤重量はこの範囲内に限定されない。最
も好ましい錠剤重量は、80〜100mgの範囲内、特別にはおよそ100mgである。
【0036】
したがって、本発明の錠剤は小さくてよいが、それにより円形で製造される場合は常に
、その平均径は約1.5mm〜約2.5mmである。またはそれらは2.5mm〜15m
m、より通常は2.5mm〜10mmの平均径を有する通常の錠剤として製造することも
できる。リング形以外に、タムスロシン組成物は楕円形、両凸面状の円形、五角形の外接
円または他の適切な錠剤形に圧縮できる。
【0037】
単位用量のタムスロシンを含有する本発明の錠剤は、直接使用のためには有利には、5
〜100錠を含む適切な包装単位で供給することができる。そのような包装には、有利に
は、10、14、20、28もしくは30錠を含むブリスターパック、または同一量(5
〜100錠)の錠剤を含有するプラスチック製もしくはガラス製の容器/ボトルを含むこ
とができる。包装単位の製造には、あらゆる適切な薬学的に許容可能な包装材料を使用で
きる。
【0038】
本発明の錠剤は、いずれか適切な錠剤形成工程によって製造できる。例えば、本錠剤は
、顆粒が最初に形成され、そして任意で1種以上の賦形剤を添加して錠剤に圧縮される湿
式造粒法によって製造できる。あるいはまた、本錠剤は、直接圧縮法または乾式造粒法等
の乾式工程によって製造でき、後者はときどき乾式圧縮成型法とも呼ばれる。好ましくは
、本錠剤は容易かつ経済的に製造することを考慮して乾式技術によって製造される。本錠
剤中には少量のタムスロシンが存在するために、乾式工程では複数の混合および/または
粉砕ステップを実施することが好ましい。
【0039】
本発明によるタムスロシンの経口投与用錠剤は、症候性良性前立腺肥大症(BPH)ま
たはタムスロシンによって治療可能なその他の障害(障害)の機能的治療の管理において
使用できる。したがって、本発明はさらにまたそれを必要とする患者に1錠以上の上記の
錠剤を投与するステップを含む良性前立腺肥大症の症状を治療するための方法を提供する
。本錠剤は、単一カプセルに入れて投与できる。
【0040】
本発明の錠剤組成物は、さらに他の薬剤と組み合わせて医療用途に使用することもでき
る。この組み合わせは、単一複合製剤の形状で、または上記の物質を含有する薬物の個別
投与によって実現することができる。
【0041】
本発明を、以下の非限定的実施例により詳細に例示する。
【0042】
[実施例]
市販の塩酸タムスロシンカプセルをヨーロッパで入手し、37℃、パドル速度100r
pmで、米国薬局方第2法装置において、各媒質の500ml中の各SGF、SIF、F
aSSIF、およびFeSSIFの中で溶出試験を実施し、6回の試験の平均値を入手し
た。放出された薬物量は、HPLC Agilent 1100システムを使用してHP
LC法によって測定した。検出は230nmに設定したUVを用いて実施した。図1に示
した結果は、2時間の経過後にSGF中ではタムスロシンの20%未満が放出され、SI
F中ではタムスロシンの60%超が放出され、FaSSI中ではタムスロシンの75%超
が放出され、そしてFeSSIF中ではタムスロシンの40%未満が放出されることを表
している。
【実施例1】
【0043】
以下の特徴を有する3バッチのモノリシック錠を漸進的混合(progressive mixing)お
よび直接圧縮法によって製造した。
【0044】
a)錠剤組成物
【表1】
バッチ間の相違は、選択したヒプロメロースの粘度値だけであった。
バッチAはMETHOCEL K4M CR PREMIUMを含有していた。
バッチBはMETHOCEL K15M CR PREMIUMを含有していた。
バッチCはMETHOCEL K100M CR PREMIUMを含有していた。
【0045】
b)操作方法
塩酸タムスロシンは、1:9の比率(有効成分10%)で無水乳糖と混合し(15分間
)、粉砕し(15秒間)、再び混合した(5分間)。次にこの前混合物を残りの乳糖、リ
ン酸二カルシウムおよびヒプロメロースと混合し(10分間)、そして最後にステアリン
酸マグネシウムを添加し、圧縮前混合物を形成するために混合した(5分間)。この漸進
的混合系は、前混合物中では97.2〜100.4%、そして圧縮前混合物中では88.
1〜98.6%のタムスロシン均質性を生じさせた。圧縮法は、Korsch EKOプ
レス機において標準速度および標準圧で実施した。
【0046】
c)製造された錠剤のキャラクタリゼーション
【表2】
【0047】
d)溶出試験
溶出試験は、標準的な米国薬局方第2法装置を使用して、500mLのSIF中で50
rpmのパドル速度で実施した。放出された薬物は、HPLC Agilent 110
0システムを使用してHPLC法によって測定した。分析は、溶離液としてリン酸緩衝液
(pH6.5)およびアセトニトリル(65:35)を使用し、均一濃度溶離モードを用
いて、ガードカラムおよびC18分析用カラムを用いて実施した。検出は230nmに設定
したUVを用いて実施した。結果は以下の仕様を満たしている。
30分間で<40%
2時間で20〜60%
6時間で>75%
【実施例2】
【0048】
以下の特徴を有する3バッチのモノリシック錠を漸進的混合および直接圧縮法によって
製造した。
【0049】
a)錠剤組成物
【表3】
バッチ間の相違は、使用したヒプロメロースの濃度だけであった。
バッチDは11%のMETHOCEL K100M CR PREMIUMを含有して
いた。
バッチEは22%のMETHOCEL K100M CR PREMIUMを含有して
いた。
バッチFは44%のMETHOCEL K100M CR PREMIUMを含有して
いた。
【0050】
b)操作方法
塩酸タムスロシンは、1:9の比率(有効成分10%)で無水乳糖と混合し(15分間
)、粉砕し(15秒間)、再び混合した(5分間)。次にこの前混合物を残りの乳糖、リ
ン酸二カルシウムおよびヒプロメロースと混合し(10分間)、そして最後にステアリン
酸マグネシウムを添加し、混合した(5分間)。圧縮は、Korsch EKOプレス機
で実施した。
【0051】
c)製造された錠剤のキャラクタリゼーション
【表4】
【0052】
d)溶出試験
溶出試験は、どちらも500mLのSIF中において、100rpmでバスケットを使
用する米国薬局方第1法装置、および50rpmの速度でパドルを使用する米国薬局方第
2法装置を使用して実施した。放出された薬物は、HPLC Agilent 1100
システムを使用してHPLC法によって測定した。分析は、溶離液としてリン酸緩衝液(
pH6.5)およびアセトニトリル(65:35)を使用して、均一濃度溶離モードを用
いて、ガードカラムおよびC18分析用カラムを用いて実施した。検出は230nmに設定
したUVを用いて実施した。
結果はHPMC濃度にしたがって変動し、各条件では、規定の仕様に適合して広範囲の
曲線が得られる。これらの結果は、他の生体関連溶出条件へ外挿できる。
【実施例3】
【0053】
以下の特徴を有する2バッチのモノリシック錠を漸進的混合および直接圧縮法によって
製造した。
【0054】
a)錠剤組成物
【表5】
両バッチ間の差は、主としてスケールアップ率、混合時間および物理的パラメーターで
あった。
バッチGは20,000単位へスケールアップされた。
バッチHは40,000単位へスケールアップされた。
【0055】
b)操作方法
塩酸タムスロシンは、1:9の比率(有効成分10%)で無水乳糖と混合し(Turb
ula;15分間)、粉砕し(IKA;30秒間)、再び混合した(Turbula;5
分間)。次に、この前混合物を残りの乳糖、リン酸二カルシウムおよびヒプロメロースと
混合した(Bohle LM40)。バッチDについては3つの漸進的混合時間(15、
30および45分間)を評価したが、いずれの場合においても均質性は極めて良好であっ
た(タムスロシン含有率:101.2%、101.7%および102.1%)。バッチE
は10分間しか混合しなかったが、許容される均質性に同様に到達した。無水混合物およ
び賦形剤のふるい分けは、均質性を入手するための必要に応じて実施した。最後にステア
リン酸マグネシウムをふるいにかけ、添加し、混合した(Bohle LM40;5分間
)。圧縮前混合物は偏心式プレス機Korsch EKOまたは回転式プレス機Kors
ch XL100のどちらかで圧縮した(約15,000〜30,000錠)。回転式プ
レス機を備えたKorsch XL100で実施された圧縮は、標準的前圧縮および標準
圧力を用いて高速で実施した。溶出性能を試験するために、両バッチにおける錠剤の硬度
を変化させた。
【0056】
c)製造された錠剤のキャラクタリゼーション
【表6】
【0057】
d)溶出試験
溶出試験は、標準的な米国薬局方第2法装置を使用して、100rpmのパドル速度で
、500mLのSGF、SIF、FaSSIFおよびFeSSIF中において実施した。
放出された薬物は、HPLC Agilent 1100システムを使用してHPLC法
によって測定した。分析は、溶離液としてリン酸緩衝液(pH6.5)およびアセトニト
リル(65:35)を使用して、均一濃度溶離モードを用いて、ガードカラムおよびC18
分析用カラムを用いて実施した。検出は230nmに設定したUVを用いて実施した。対
応する曲線は、図2(G)および3(H)に示した。SGFおよびSIF中での試験結果
は、以下の仕様を満たしている。
30分間で<40%
2時間で20〜60%
6時間で>75%
【0058】
e)被覆
これらの錠剤はその後、クエン酸トリエチルおよびタルクを含む添加物、またはAcr
yl−Eze(登録商標、Colorcon社から入手可能)とともに、Eudragi
t L30D55を基剤とする腸溶ポリマー(ポリメタクリレート、タイプC)を用いて
被覆した。
【0059】
f)溶出試験
被覆錠のバッチの溶出試験は、標準的な米国薬局方第2法装置を使用して、100rp
mのパドル速度で、500mLのSGF、SIF、FaSSIFおよびFeSSIF中で
実施した。放出された薬物は、HPLC Agilent 1100システムを使用して
HPLC法によって測定した。分析は、溶離液としてリン酸緩衝液(pH6.5)および
アセトニトリル(65:35)を使用して、均一濃度溶離モードを用いて、ガードカラム
およびC18分析用カラムを用いて実施した。検出は230nmに設定したUVを用いて実
施した。
対応する曲線は、図4(被覆G)および図5(被覆H)に示した。
SIF中の結果は以下の仕様を満たしている。
30分間で<40%
2時間で20〜60%
6時間で>75%
【実施例4】
【0060】
2バッチの錠剤は、乾式圧縮成型、粉砕、混合および圧縮を含む工程によって製造した
。
【0061】
a)錠剤組成物
【表7】
バッチI:モノリシック錠、6mm径。33.0%のHPMC K15M Pを含有す
る。
バッチJ:モノリシック錠、9mm径。13.2%のHPMC K15M Pを含有す
る。
【0062】
b)操作方法
タムスロシンは、1:9の比率(有効成分10%)で無水乳糖と混合し(15分間)、
粉砕し(15秒間)、再び混合した(5分間)。次にこの前混合物を残りの乳糖、ヒプロ
メロースおよび25%のステアリン酸マグネシウムと混合し(10分間)、Chilso
nator(Fitz−Patrick社)で圧縮成型し、Fitz−Mill(Fit
z−Patrick社)で粉砕し、最後に残りのステアリン酸マグネシウム(75%)を
添加し、さらに混合した(15分間)。圧縮法は、Korsch EKOプレス機におい
て標準速度および標準圧で実施した。
【0063】
c)製造された錠剤のキャラクタリゼーション
【表8】
【実施例5】
【0064】
相違する有効成分濃度、錠剤形状および総重量を備える2バッチの錠剤を製造した。
【0065】
a)錠剤組成物
【表9】
【0066】
b)操作方法
微粉化タムスロシンは、無水乳糖と漸進的に混合した。次に、すべての場合に十分な均
質性を提供するために、この前混合物を残りの乳糖およびヒプロメロースと混合し、その
後ステアリン酸マグネシウムと混合した。圧縮法は、Korsch EKOプレス機にお
いて標準速度および標準圧で実施した。
【0067】
c)製造された錠剤のキャラクタリゼーション
【表10】
【実施例6】
【0068】
溶出仕様を検査するために、1バッチのモノリシック錠を製造した。
【0069】
a)錠剤組成物
【表11】
バッチNのスケールアップは、微粉化した薬物成分およびより少ない混合ステップを使
用して実施した。40,000単位を製造した。
【0070】
b)操作方法
微粉化塩酸タムスロシンをヒプロメロースと混合した(Bohle LM40;15分
間)。次にこの前混合物を乳糖およびリン酸二カルシウムと混合した(Bohle LM
40;15分間)。無水混合物および賦形剤のふるい分けは、均質性を入手するための必
要に応じて実施した。最後にステアリン酸マグネシウムをふるいにかけ、添加し、さらに
2回混合した(Bohle LM40;5および15分間)。
圧縮前混合物を次に圧縮した。回転式プレス機を備えたKorsch XL100で実
施される圧縮は、標準的前圧縮および標準圧力を用いて高速で実施した。
錠剤の特徴付けは以下に示した。
【0071】
c)製造された錠剤のキャラクタリゼーション
【表12】
【0072】
d)被覆
この錠剤は、次に腸溶ポリマー(Colorcon社から入手可能なAcryl−Ez
e(登録商標))を用いて被覆した。
【0073】
e)溶出試験
被覆錠のバッチの溶出試験は、標準的な米国薬局方第2法装置を使用して、100rp
mのパドル速度で、500mLのSGF、SIF、FaSSIFおよびFeSSIF中で
実施した。放出された薬物は、HPLC Agilent 1100システムを使用して
HPLC法によって測定した。分析は、溶離液としてリン酸緩衝液(pH6.5)および
アセトニトリル(65:35)を使用して、均一濃度溶離モードを用いて、ガードカラム
およびC18分析用カラムを用いて実施した。検出は230nmに設定したUVを用いて実
施した。
対応する曲線は、図6に示した。
全媒質中での試験結果は以下の仕様を満たしている。
30分間で<40%
2時間で20〜60%
6時間で>75%
【0074】
本発明を説明してきたが、当業者には、特許請求の範囲によって定義された本発明の精
神および範囲から逸脱することなく、本明細書に記載した概念および実施形態を実際に実
施するに当たりまた別の変更および修飾を容易に加えることができること、または本発明
の実践によって学習されることは容易に明白であろう。
【特許請求の範囲】
【請求項1】
0.1〜10mgのタムスロシンまたはその薬学的に許容可能な塩が分散している錠剤
マトリックスを含み、任意選択的に該マトリックスを覆う腸溶被覆を有する医薬錠剤であ
って、前記錠剤が放出調節型錠剤であり、SIF、FaSSIF、およびFeSSIFの
各媒質中で、50〜100rpmのパドル速度で500mLの前記媒質を使用する米国薬
局方第2法装置において前記錠剤が2時間の経過後に60%以下のタムスロシンを放出す
る溶出プロフィールを有する医薬錠剤。
【請求項2】
前記溶出プロフィールが100rpmのパドル速度を使用して測定される、請求項1に
記載の医薬錠剤。
【請求項3】
前記溶出プロフィールが2時間の経過後に前記各媒質の中で少なくとも20%の放出を
示す、請求項1または2に記載の医薬錠剤。
【請求項4】
前記錠剤がある溶出プロフィールを有し、FeSSIF媒質中に2時間で放出されたタ
ムスロシンの量は、FaSSIF媒質中に2時間で放出された量の少なくとも50%であ
る、請求項1〜3のいずれかに記載の医薬錠剤。
【請求項5】
前記錠剤がある溶出プロフィールを有し、そして前記錠剤が100rpmのパドル速度
で500mLのSGF媒質を使用する米国薬局方第2法装置において2時間の経過後に前
記タムスロシンの60%以下を放出する、請求項1〜4のいずれかに記載の医薬錠剤。
【請求項6】
前記溶出プロフィールが、100rpmのパドル速度で500mLのSIF媒質を使用
する米国薬局方第2法装置において、30分間でタムスロシンの40%未満、2時間でタ
ムスロシンの20〜60%、および6時間でタムスロシンの75%超を放出することを含
む、請求項1〜5のいずれかに記載の医薬錠剤。
【請求項7】
前記溶出プロフィールが、100rpmのパドル速度で500mLのSGF媒質を使用
する米国薬局方第2法装置において、30分間でタムスロシンの40%未満、2時間でタ
ムスロシンの20〜60%、および6時間でタムスロシンの75%超を放出することを含
む、請求項1〜6のいずれかに記載の医薬錠剤。
【請求項8】
前記錠剤が腸溶被覆を有する、請求項1〜7のいずれかに記載の医薬錠剤。
【請求項9】
前記錠剤が腸溶被覆を有していない、請求項1〜7のいずれかに記載の医薬錠剤。
【請求項10】
前記錠剤が被覆されていない、請求項9に記載の医薬錠剤。
【請求項11】
前記錠剤マトリックスが水膨潤性セルロース誘導体を含む、請求項1〜10のいずれか
に記載の医薬錠剤。
【請求項12】
前記錠剤マトリックスがヒドロキシプロピルメチルセルロースを含む、請求項11に記
載の医薬錠剤。
【請求項13】
前記錠剤が前記ヒドロキシプロピルメチルセルロースを10重量%〜90重量%の範囲
内の量で含む、請求項11または12に記載の医薬錠剤。
【請求項14】
前記錠剤が前記ヒドロキシプロピルメチルセルロースを25重量%〜40重量%の範囲
内の量で含む、請求項13に記載の医薬錠剤。
【請求項15】
前記錠剤が前記ヒドロキシプロピルメチルセルロースを30重量%〜35重量%の範囲
内の量で含む、請求項14に記載の医薬錠剤。
【請求項16】
前記タムスロシンまたはその薬学的に許容可能な塩が0.2〜1.0%の量で含有され
ている、請求項1〜15のいずれかに記載の医薬錠剤。
【請求項17】
前記タムスロシンまたはその薬学的に許容可能な塩が0.2〜0.8%の量で含有され
ている、請求項16に記載の医薬錠剤。
【請求項18】
前記タムスロシンまたはその薬学的に許容可能な塩が塩酸タムスロシンであり、前記塩
酸タムスロシンが0.4mg±0.04の量で含有されている、請求項1〜17のいずれ
かに記載の医薬錠剤。
【請求項19】
炭水化物および圧縮可能な希釈剤からなる群から選択された少なくとも1種の薬学的に
許容される賦形剤をさらに含む、請求項1〜18のいずれかに記載の医薬錠剤。
【請求項20】
乳糖をさらに含む、請求項19に記載の医薬錠剤。
【請求項21】
リン酸カルシウムを含む、請求項19または20に記載の医薬錠剤。
【請求項22】
乳糖、HPMC、リン酸カルシウム、およびステアリン酸マグネシウムを含む、請求項
1〜21のいずれかに記載の医薬錠剤。
【請求項23】
前記錠剤が1日1回錠剤である、請求項1〜22のいずれかに記載の医薬錠剤。
【請求項24】
0.1〜10mgのタムスロシンまたはその薬学的に許容可能な塩と、10重量%〜9
0重量%のヒドロキシプロピルメチルセルロースとを含み、錠剤総重量が10〜300m
gであるモノリシック医薬錠剤。
【請求項25】
前記錠剤総重量が25〜250mgの範囲内である、請求項24に記載のモノリシック
錠剤。
【請求項26】
前記錠剤総重量が80〜100mgの範囲内である、請求項25に記載のモノリシック
錠剤。
【請求項27】
前記ヒドロキシプロピルメチルセルロースを25重量%〜40重量%の範囲内の量で含
む、請求項24〜26のいずれかに記載のモノリシック錠剤。
【請求項28】
前記ヒドロキシプロピルメチルセルロースを30重量%〜40重量%の範囲内の量で含
む、請求項27に記載のモノリシック錠剤。
【請求項29】
前記錠剤が前記ヒドロキシプロピルメチルセルロースを30重量%〜35重量%の範囲
内の量で含む、請求項24〜28のいずれかに記載のモノリシック錠剤。
【請求項30】
前記錠剤がリン酸カルシウム、乳糖、マンニトール、またはそれらの組み合わせをさら
に含む、請求項24〜29のいずれかに記載のモノリシック錠剤。
【請求項31】
前記錠剤が無水第二リン酸カルシウムを含む、請求項30に記載のモノリシック錠剤。
【請求項32】
腸溶被服を含有していない、請求項24〜31のいずれかに記載のモノリシック錠剤。
【請求項33】
良性前立腺肥大症の状態を治療または改善するための、有効量の請求項1〜32のいず
れかに記載の1個以上の錠剤を含む単位製剤。
【請求項34】
1カプセル内に2個以上の前記錠剤を含む、請求項33に記載の単位製剤。
【請求項35】
良性前立腺肥大症の症状を治療するための方法であって、治療を必要とする患者に請求
項1〜32のいずれかに記載の1個以上の錠剤または請求項33または34に記載の単位
製剤を投与するステップを含む方法。
【請求項36】
前記1個以上の錠剤が前記患者に投与される単一カプセル内に含有されている、請求項
35に記載の方法。
【請求項1】
0.1〜10mgのタムスロシンまたはその薬学的に許容可能な塩が分散している錠剤
マトリックスを含み、任意選択的に該マトリックスを覆う腸溶被覆を有する医薬錠剤であ
って、前記錠剤が放出調節型錠剤であり、SIF、FaSSIF、およびFeSSIFの
各媒質中で、50〜100rpmのパドル速度で500mLの前記媒質を使用する米国薬
局方第2法装置において前記錠剤が2時間の経過後に60%以下のタムスロシンを放出す
る溶出プロフィールを有する医薬錠剤。
【請求項2】
前記溶出プロフィールが100rpmのパドル速度を使用して測定される、請求項1に
記載の医薬錠剤。
【請求項3】
前記溶出プロフィールが2時間の経過後に前記各媒質の中で少なくとも20%の放出を
示す、請求項1または2に記載の医薬錠剤。
【請求項4】
前記錠剤がある溶出プロフィールを有し、FeSSIF媒質中に2時間で放出されたタ
ムスロシンの量は、FaSSIF媒質中に2時間で放出された量の少なくとも50%であ
る、請求項1〜3のいずれかに記載の医薬錠剤。
【請求項5】
前記錠剤がある溶出プロフィールを有し、そして前記錠剤が100rpmのパドル速度
で500mLのSGF媒質を使用する米国薬局方第2法装置において2時間の経過後に前
記タムスロシンの60%以下を放出する、請求項1〜4のいずれかに記載の医薬錠剤。
【請求項6】
前記溶出プロフィールが、100rpmのパドル速度で500mLのSIF媒質を使用
する米国薬局方第2法装置において、30分間でタムスロシンの40%未満、2時間でタ
ムスロシンの20〜60%、および6時間でタムスロシンの75%超を放出することを含
む、請求項1〜5のいずれかに記載の医薬錠剤。
【請求項7】
前記溶出プロフィールが、100rpmのパドル速度で500mLのSGF媒質を使用
する米国薬局方第2法装置において、30分間でタムスロシンの40%未満、2時間でタ
ムスロシンの20〜60%、および6時間でタムスロシンの75%超を放出することを含
む、請求項1〜6のいずれかに記載の医薬錠剤。
【請求項8】
前記錠剤が腸溶被覆を有する、請求項1〜7のいずれかに記載の医薬錠剤。
【請求項9】
前記錠剤が腸溶被覆を有していない、請求項1〜7のいずれかに記載の医薬錠剤。
【請求項10】
前記錠剤が被覆されていない、請求項9に記載の医薬錠剤。
【請求項11】
前記錠剤マトリックスが水膨潤性セルロース誘導体を含む、請求項1〜10のいずれか
に記載の医薬錠剤。
【請求項12】
前記錠剤マトリックスがヒドロキシプロピルメチルセルロースを含む、請求項11に記
載の医薬錠剤。
【請求項13】
前記錠剤が前記ヒドロキシプロピルメチルセルロースを10重量%〜90重量%の範囲
内の量で含む、請求項11または12に記載の医薬錠剤。
【請求項14】
前記錠剤が前記ヒドロキシプロピルメチルセルロースを25重量%〜40重量%の範囲
内の量で含む、請求項13に記載の医薬錠剤。
【請求項15】
前記錠剤が前記ヒドロキシプロピルメチルセルロースを30重量%〜35重量%の範囲
内の量で含む、請求項14に記載の医薬錠剤。
【請求項16】
前記タムスロシンまたはその薬学的に許容可能な塩が0.2〜1.0%の量で含有され
ている、請求項1〜15のいずれかに記載の医薬錠剤。
【請求項17】
前記タムスロシンまたはその薬学的に許容可能な塩が0.2〜0.8%の量で含有され
ている、請求項16に記載の医薬錠剤。
【請求項18】
前記タムスロシンまたはその薬学的に許容可能な塩が塩酸タムスロシンであり、前記塩
酸タムスロシンが0.4mg±0.04の量で含有されている、請求項1〜17のいずれ
かに記載の医薬錠剤。
【請求項19】
炭水化物および圧縮可能な希釈剤からなる群から選択された少なくとも1種の薬学的に
許容される賦形剤をさらに含む、請求項1〜18のいずれかに記載の医薬錠剤。
【請求項20】
乳糖をさらに含む、請求項19に記載の医薬錠剤。
【請求項21】
リン酸カルシウムを含む、請求項19または20に記載の医薬錠剤。
【請求項22】
乳糖、HPMC、リン酸カルシウム、およびステアリン酸マグネシウムを含む、請求項
1〜21のいずれかに記載の医薬錠剤。
【請求項23】
前記錠剤が1日1回錠剤である、請求項1〜22のいずれかに記載の医薬錠剤。
【請求項24】
0.1〜10mgのタムスロシンまたはその薬学的に許容可能な塩と、10重量%〜9
0重量%のヒドロキシプロピルメチルセルロースとを含み、錠剤総重量が10〜300m
gであるモノリシック医薬錠剤。
【請求項25】
前記錠剤総重量が25〜250mgの範囲内である、請求項24に記載のモノリシック
錠剤。
【請求項26】
前記錠剤総重量が80〜100mgの範囲内である、請求項25に記載のモノリシック
錠剤。
【請求項27】
前記ヒドロキシプロピルメチルセルロースを25重量%〜40重量%の範囲内の量で含
む、請求項24〜26のいずれかに記載のモノリシック錠剤。
【請求項28】
前記ヒドロキシプロピルメチルセルロースを30重量%〜40重量%の範囲内の量で含
む、請求項27に記載のモノリシック錠剤。
【請求項29】
前記錠剤が前記ヒドロキシプロピルメチルセルロースを30重量%〜35重量%の範囲
内の量で含む、請求項24〜28のいずれかに記載のモノリシック錠剤。
【請求項30】
前記錠剤がリン酸カルシウム、乳糖、マンニトール、またはそれらの組み合わせをさら
に含む、請求項24〜29のいずれかに記載のモノリシック錠剤。
【請求項31】
前記錠剤が無水第二リン酸カルシウムを含む、請求項30に記載のモノリシック錠剤。
【請求項32】
腸溶被服を含有していない、請求項24〜31のいずれかに記載のモノリシック錠剤。
【請求項33】
良性前立腺肥大症の状態を治療または改善するための、有効量の請求項1〜32のいず
れかに記載の1個以上の錠剤を含む単位製剤。
【請求項34】
1カプセル内に2個以上の前記錠剤を含む、請求項33に記載の単位製剤。
【請求項35】
良性前立腺肥大症の症状を治療するための方法であって、治療を必要とする患者に請求
項1〜32のいずれかに記載の1個以上の錠剤または請求項33または34に記載の単位
製剤を投与するステップを含む方法。
【請求項36】
前記1個以上の錠剤が前記患者に投与される単一カプセル内に含有されている、請求項
35に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2011−51995(P2011−51995A)
【公開日】平成23年3月17日(2011.3.17)
【国際特許分類】
【外国語出願】
【出願番号】特願2010−232415(P2010−232415)
【出願日】平成22年10月15日(2010.10.15)
【分割の表示】特願2003−541822(P2003−541822)の分割
【原出願日】平成14年11月6日(2002.11.6)
【出願人】(500415715)シントン・ベスローテン・フェンノートシャップ (10)
【Fターム(参考)】
【公開日】平成23年3月17日(2011.3.17)
【国際特許分類】
【出願番号】特願2010−232415(P2010−232415)
【出願日】平成22年10月15日(2010.10.15)
【分割の表示】特願2003−541822(P2003−541822)の分割
【原出願日】平成14年11月6日(2002.11.6)
【出願人】(500415715)シントン・ベスローテン・フェンノートシャップ (10)
【Fターム(参考)】
[ Back to top ]