
放射性フッ素化方法
本発明は、式(V)又は(VI)のコンジュゲート、放射性医薬品としてのその使用、その製造方法、及びかかる方法で使用する合成中間体に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、陽電子放出核種で標識した生物学的活性ベクターを含む診断薬及び放射性診断薬に関する。本発明はさらに、特異的な生物学的標的に対して親和性を有する分子として定義されるベクター、好ましくはペプチドであるベクターの[18F]フッ素化のための方法及び試薬に関する。こうして得られる18F標識コンジュゲートは、放射性医薬品として、具体的には陽電子放射断層撮影(PET)で使用するための放射性医薬品として有用である。
【背景技術】
【0002】
放射性標識された生物活性ベクターを診断イメージングのために応用することは、核医学において重要性を増しつつある。特定の細胞タイプと選択的に相互作用する生物学的活性分子は、標的組織に放射能を送達するために有用である。例えば、放射性標識ペプチドは、診断イメージング及び放射線療法のため、腫瘍、梗塞巣部及び感染組織に放射性核種を送達するための大きな潜在的可能性を有している。約110分の半減期を有する18Fは、多くのレセプターイメージング調査のために最適な陽電子放出核種である。したがって、18F標識された生物活性ペプチドは、多種多様の疾患を定量的に検出して特性決定するためにPETで利用できるので大きな臨床的可能性を有している。
【0003】
18F標識ペプチドに関する難点の1つは、既存の18F標識剤が調製するのに多くの時間を要することである。18Fによるペプチド及びタンパク質の効率的な標識は、適当な補欠分子族を用いることでのみ達成される。文献中には、N−スクシンイミジル−4−[18F]フルオロベンゾエート、m−マレイミド−N−(p−[18F]フルオロベンジル)ベンズアミド、N−(p−[18F]フルオロフェニル)マレイミド及び4−[18F]フルオロフェナシルブロミドを含む数種のかかる補欠分子族が提唱されている。18Fによるペプチド及びタンパク質の標識のために現在使用されている方法のほとんどすべてが、フッ素標識シントンの活性エステルを利用している。ペプチド及びタンパク質は活性エステルと反応し得る多数の官能基を含むことがあるので、これらの現行方法は部位特異的でない。例えば、3つのリシン残基を含むペプチドは、標識シントンに対する反応性が全く同じ3つのアミン官能基を有する。したがって、特にペプチド中に18Fを温和な条件下で迅速かつ化学選択的に導入して、高い放射化学収量及び純度で18F標識生成物を得ることを可能にする18F標識補欠分子族及び方法に対するニーズが依然として存在している。さらに、かかる方法であって、臨床現場での放射性医薬品の調製を容易にするための自動化に適した方法に対するニーズも存在している。我々は以前にPET標識方法におけるアミノオキシ化学の使用を記載した(国際公開第03/006491号)が、この発明の化合物はアルデヒド及びケトンと容易に反応しないものの、ある種のハロゲン含有化合物に対して選択性を有する。N−アルキルアミノオキシ部分の化学的安定性はさらに高いので、副反応が最小限に抑えられかつ中間体が一層安定である結果として中間体の取扱い及び貯蔵を首尾よく行うことが可能となり、したがって以前に開示したアミノオキシ基に比べて利点が得られる。
【先行技術文献】
【特許文献】
【0004】
国際公開第2008/083191号パンフレット
【発明の概要】
【0005】
本発明は、以下の式(I)の化合物を以下の式(II)の化合物と反応させるか、或いは以下の式(III)の化合物を以下の式(IV)の化合物と反応させることを含んでなる放射性フッ素化方法を提供する。
【0006】
【化1】

【0007】
【化2】
式中、
R1はハロアセチル、ハロアリル又は基又はフェナシルハライドのような反応性ハロゲン含有基であるか、すべての基がアルキル化反応への関与に適したエポキシド環であるか、マレイミド基或いはアクリル酸又は置換アクリル酸誘導体のようなマイケル(Michael)受容体であるか、ビニルスチレン(好ましくはニトロビニルスチレン)であるか、ビニルスルホンアミドであるか、或いはビニルスルホンである。
R2はN−アルキルアミノオキシ基であり、これは水性緩衝液及びわずかに酸性のpHのような温和な条件下でR1と部位特異的に反応して以下の式(V)の化合物を生じる。
式(III)のR3はR2に等しく、式(IV)のR4はR1に等しい。式(III)の化合物と式(IV)の化合物との連結は以下の式(VI)の生成物を生じる。
【0008】
【化3】
式中、いずれの場合にも、Yはアルキルであり、好ましくはY=−CH3である。
【図面の簡単な説明】
【0009】
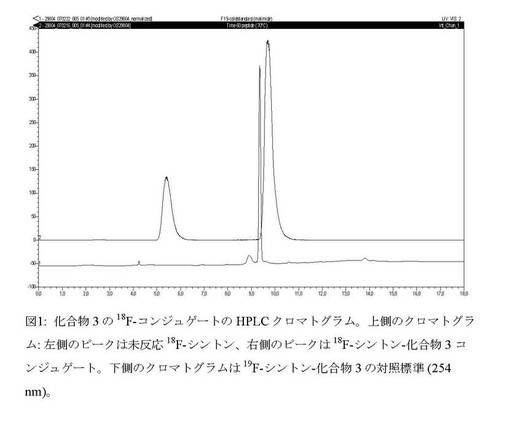
【図1】化合物3の18F−コンジュゲートのHPLCクロマトグラム。
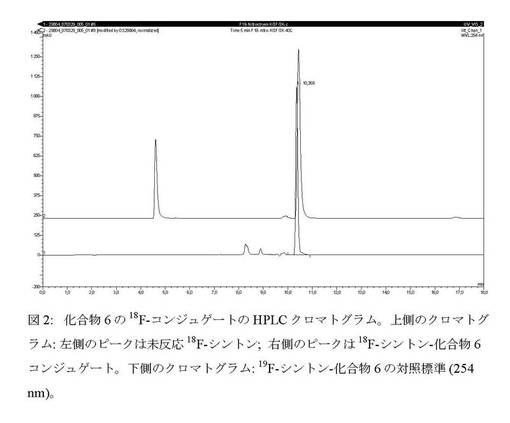
【図2】化合物6の18F−コンジュゲートのHPLCクロマトグラム。
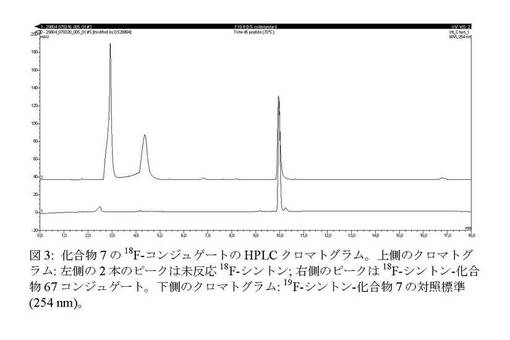
【図3】化合物7の18F−コンジュゲートのHPLCクロマトグラム。
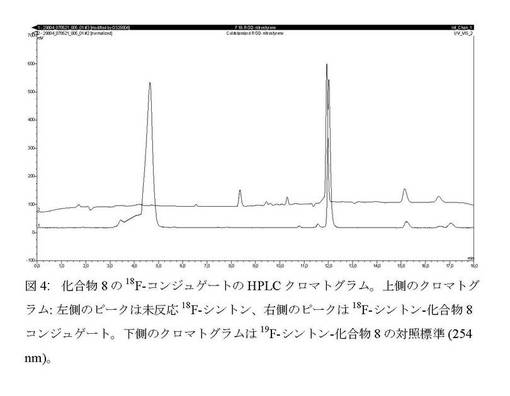
【図4】化合物8の18F−コンジュゲートのHPLCクロマトグラム。
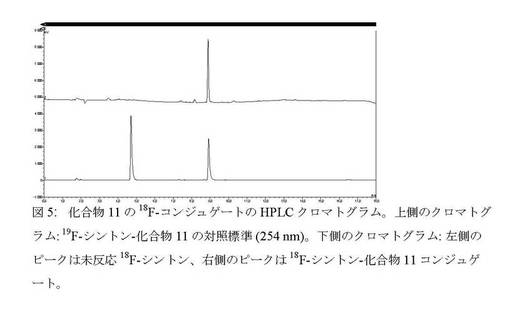
【図5】化合物11の18F−コンジュゲートのHPLCクロマトグラム。
【発明を実施するための形態】
【0010】
式(V)の化合物が最も好ましく、式(II)の化合物の18F−リンカー成分は好ましくは以下の式(VII)〜(XI)に示される種類の化合物からなる。
【0011】
【化4】
式中、
nは0〜20の整数であり、
ZはO、N又はSである。
【0012】
式(II)の化合物中のリンカー基は、得られる式(V)のコンジュゲートにおいて好ましい排泄特性のような最適のインビボ薬物動態が得られるように選択することもできる。親油性の異なるリンカー基を使用することで、診断上のニーズに応じてペプチドのインビボ薬物動態を大きく変化させることができる。例えば、式(V)のコンジュゲートを腎排泄によって体外に排除することが望ましい場合には、式(X)〜(XI)から導かれるリンカー成分を含む親水性リンカーが使用される。同様に、肝胆道排泄による排除が望ましい場合には、式(VII)〜(IX)から導かれるリンカー成分を含む疎水性リンカーが選択できる。
【0013】
本発明は、ペプチド又はベクター前駆体の合成に際して標識の正確な導入部位が予め選択される一層化学選択的な放射性標識アプローチを提供する。分子中の所定部位で起こる連結反応は、ただ1種の標識生成物を与える。したがって、このような方法は化学選択的であり、その用途は多種多様の薬物様分子、ペプチド及び生体分子(例えば、小タンパク質)の標識に役立つ包括的なものであると考えられる。
【0014】
さらに別の態様では、本発明は、以下の式(Ia)の化合物を以下の式(IIa)の化合物と反応させるか、或いは以下の式(IIIa)の化合物を以下の式(IVa)の化合物と反応させてそれぞれ以下の式(Va)又は(VIa)のコンジュゲートを得ることを含んでなる放射性フッ素化方法を提供する。
【0015】
【化5】
【0016】
【化6】
【0017】
【化7】
式中、R1及びR4はそれぞれ式(I)及び式(IV)の化合物に関して上記に定義した通りであり、リンカー基は上記の化合物に関して定義した通りである。
【0018】
反応は、適当な溶媒(例えば、pH3〜11の範囲内の水性緩衝液)中において5〜70℃の極端でない温度、最も好ましくは周囲温度で実施できる。
【0019】
本発明のさらに別の態様では、R1はマレイミド化合物及びビニルスチレン化合物の部類から導かれ、最も好ましくは以下の式(Ib)及び式(Ic)中に表される。
【0020】
【化8】
式(I)及び式(III)中並びに本発明の他の態様では、特記しない限り、ベクターは疾患過程に付随する関連レセプターに対して親和性を有する生物学的薬剤として定義できる。生物学的薬剤は小分子薬物様の薬物団、タンパク質又は抗体として定義できるが、好ましくは分子量30kDa未満のタンパク質であり、最も好ましくはアミノ酸100未満のペプチドである。本発明の方法を用いる標識用の好ましいペプチドベクターの例には、オクトレオチドのようなソマトスタチン類似体、ボンベシン、血管作用性小腸ペプチド、走化性ペプチド類似体、α−メラノサイト刺激ホルモン、ニューロテンシン、Arg−Gly−Aspペプチド、ヒトプロインスリン結合ペプチド、エンドセリン、アンギオテンシン及びホルミル−ノルロイシル−ロイシル−フェニルアラニル−ノルロイシル−チロシル−リシンがある。標識用の好ましいペプチドは、国際公開第01/77415号及び同第03/006491号に記載されているもののようなArg−Gly−Aspペプチド及びその類似体である。好ましいペプチドベクターは、以下の式(A)のフラグメントを含んでいる。
【0021】
【化9】
式中、X7は−NH2又は次式のエチレングリコール含有構造であり、
【0022】
【化10】
(式中、「a」は1〜10の整数であり、最も好ましくはaは1である。)
R1は式(I)に関して前記に記載した通りである。
【0023】
当業者には容易に理解される通り、本発明の方法は、タンパク質、ホルモン、オリゴヌクレオチド及び抗体フラグメントのような他の生体分子並びに薬物様小分子の放射性フッ素化によって各種のPETトレーサーを得るためにも使用できる。
【0024】
式(I)及び式(III)の化合物は、例えば、Atherton,E.and Sheppard,R.C.;“Solid Phase Synthesis”;IRL Press:Oxford,1989中に記載されている固相ペプチド合成法のような標準的ペプチド合成法によって製造できる。式(I)又は(III)の化合物中への基R1又はR3の導入は、ペプチドのN末端又はC末端の反応、或いはペプチド配列中に含まれる何らかの他の官能基であって、その修飾がベクターの結合特性に影響を及ぼさない官能基との反応によって達成できる。好ましい例では、Carrasco et al(Biopolymers,Peptide Science,2006,Vol 84(4),page 414)によって記載されたアミノ酸を用いて、N−アルキルアミノオキシ基(Y−NH2−O−)をペプチド配列中に直接導入できる。官能基R1及びR3は、好ましくはペプチドのアミン官能基と活性化酸との反応による安定なアミド結合の形成によって導入され、ペプチド合成の実施中又は実施後に導入される。前駆体が酸である場合、R1及びR3は、2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)又はN−[(ジメチルアミノ)−1H−1,2,3−トリアゾロ[4,5−b]ピリジン−1−イルメチレン]−N−メチルメタンアンモニウムヘキサフルオロホスフェートN−オキシド(HATU)のようなインサイチュ活性化剤を用いて導入できる。
【0025】
式(II)の化合物は、以下の式(XII)の対応する前駆体から製造できる。
【0026】
【化11】
式中、Lは脱離基、好ましくはp−トルエンスルホネート、トリフルオロメタンスルホネート又はメタンスルホネート或いはハライドであり、Y及びリンカーは前記に定義した通りであり、R5は窒素原子を保護するための適当な保護基(例えば、t−ブチルオキシカルボニル基)である。かくして、サイクロトロンで生成された水性[18F]−フッ化物を好適には塩基(例えば、テトラブチルアンモニウム又はK2CO3/クリプトフィックス(Kryptofix)222)からの蒸発によって予備活性化したものと、アセトニトリル、N,N−ジメチルホルムアミド又はジメチルスルホキシドのような適当な溶媒中において、通例は高温(例えば、60〜150℃、好適には60〜120℃)又はマイクロ波加熱下で反応させ、次いで酸分解処理のような標準的方法を用いてN−保護基を除去することができる。
【0027】
式(IV)の化合物は、以下の式(XX)の対応する前駆体又はその保護誘導体から製造できる。
【0028】
【化12】
式中、Lは脱離基、好ましくはp−トルエンスルホネート、トリフルオロメタンスルホネート又はメタンスルホネート或いはハライドであり、リンカー及びR4は前記に定義した通りである。Lは、サイクロトロンで生成された水性[18F]−フッ化物を好適には塩基(例えば、テトラブチルアンモニウム又はK2CO3/クリプトフィックス222)からの蒸発によって予備活性化したものと、アセトニトリル、N,N−ジメチルホルムアミド又はジメチルスルホキシドのような適当な溶媒中において、通例は高温(例えば、60〜120℃)で反応し得る。
【0029】
本発明はまた、一般式(V)又は(VI)の化合物の有効量(例えば、インビボPETイメージングで使用するのに有効な量)を1種以上の薬学的に許容される補助剤、賦形剤又は希釈剤と共に含んでなる放射性医薬品組成物を提供する。
【0030】
本発明の好ましい実施形態は、医学で使用するための、特に(好適にはPETによる)腫瘍イメージングで使用するための一般式(V)又は(VI)の化合物であって、ベクターが国際公開第01/77415号及び同第03/006491号に記載されているもののようなArg−Gly−Aspペプチド又はその類似体である化合物に関する。かかるベクターは、好ましくは次式のフラグメントを含み、
【0031】
【化13】
さらに好ましくは以下の式(A)のペプチドである。
【0032】
【化14】
式中、X7は−NH2又は次式の基であり、
【0033】
【化15】
(式中、「a」は1〜10の整数であり、最も好ましくはaは1である。)
R5はペプチドの反応後にリシン残基のε−アミノとアミド結合を形成し、R1によって定義されるが、最も好ましくは下記の構造(Id)及び(Ie)によって例示される。
【0034】
【化16】
かくして、以下の式(If)及び(Ig)に示される構造を有する式(I)が得られる。
【0035】
【化17】
本発明の放射性標識コンジュゲートは、PETイメージングのため、所望の信号を得るのに十分な量で患者に投与すればよい。通常、体重70kg当たり0.01〜100mCi、好ましくは0.1〜50mCiの典型的な放射性核種投与量で十分であろう。
【0036】
したがって、本発明に係る放射性標識コンジュゲートは、完全に当業者の技術常識に属するやり方で、生理学的に許容されるキャリヤー又は賦形剤を用いて投与のために製剤化できる。例えば、薬学的に許容される賦形剤を任意に添加した化合物を水性媒質中に懸濁又は溶解し、次いで得られた溶液又は懸濁液を滅菌すればよい。
【0037】
さらに別の態様から見れば、本発明は、放射性医薬品をヒト又は動物の身体に投与する段階及び前記身体の少なくとも一部の画像を生成する段階を含むインビボイメージング方法(好適にはPET)、好ましくは腫瘍イメージング方法で使用するための放射性医薬品の製造における本発明の放射性標識コンジュゲートの使用を提供する。
【0038】
さらに別の態様から見れば、本発明は、ヒト又は動物の身体の画像を生成する方法であって、本発明に係る放射性標識コンジュゲートを含む放射性医薬品を前記身体(例えば、脈管系中)に投与する段階、及びPETを用いて前記放射性医薬品が分布した前記身体の少なくとも一部の画像を生成する段階を含んでなる方法を提供する。
【0039】
さらに別の態様から見れば、本発明は、癌に関連した病態(好ましくは血管新生)に対処するための薬物(例えば、細胞毒性薬剤)によるヒト又は動物の身体の治療効果をモニターする方法であって、本発明に係る放射性標識コンジュゲートを前記身体に投与する段階及び細胞レセプター(好ましくは内皮細胞レセプター、特にαvβ3レセプター)による前記コンジュゲートの取込みを検出する段階を含んでなり、前記投与及び検出は任意ではあるが好ましくは(例えば前記薬物による治療前、治療中及び治療後に)繰り返して実施される方法を提供する。
【0040】
本発明のさらに別の態様では、式(II)又は(IV)の補欠分子族及び式(I)又は(III)の化合物を含む放射能フッ素化トレーサーを製造するためのキットが提供される。
【0041】
本発明のさらに別の態様に従えば、式(XX)の補欠分子族及び式(I)の化合物を含む放射能フッ素化トレーサーを製造するためのキットが提供される。本発明の別の態様に従えば、式()又は()の補欠分子族及び式(III)の化合物を含む放射能フッ素化トレーサーを製造するためのキットが提供される。
【0042】
キットの使用に際しては、それぞれ上述した方法を用いて、式()の化合物は対応する式(II)の化合物に転化され、式()又は()の化合物は対応する式(IV)の化合物に転化される。好ましくは、反応混合物をSolid Phase Extraction(SPE)カートリッジに通すことにより、式(II)又は(IV)の化合物を廃反応体から分離できる。SPEカートリッジは、黒鉛パッド、C18固定相又はイオン交換樹脂を含み得る。次いで、式(II)又は(IV)の化合物を、好適には水性緩衝液(pH3〜11)に溶解し得る式(I)又は(III)の化合物にそれぞれ添加する。極端でない温度で1〜70分間反応させた後、標識ペプチドを例えばSPEにより精製して回収することができる。
【実施例】
【0043】
以下の実施例によって本発明を例示するが、実施例中では下記の略語を用いる。
HPLC:高速液体クロマトグラフィー
NMR:核磁気共鳴
hr(s):時間
min(s):分
THF:テトラヒドロフラン
DCM:ジクロロメタン
DMF:N,N−ジメチルホルムアミド
TBAF:フッ化テトラブチルアンモニウム
MeOH:メタノール
DMSO:ジメチルスルホキシド
Boc:t−ブトキシカルボニル
RT:室温
i−Pr2−Net:N,N−ジイソプロピルエチルアミン
t−BDPSiCl:tert−ブチルジフェニルシリルクロリド
NaH:水素化ナトリウム
EtOAc:酢酸エチル
MBq:メガベクレル
次の移動相/勾配系を使用した。溶媒A:水(0.1%TFA)、溶媒B:アセトニトリル(0.1%TFA)。
【0044】
例1
トルエン−4−スルホン酸4−(N−メチル−N−Boc−アミノオキシ)ブチルエステルの製造
【0045】
【化18】
(a)N−Boc−N−メチルヒドロキシルアミン
N−メチルヒドロキシルアミン(4.2g、0.05mol)を50%水性テトラヒドロフラン(THF)(20ml)に溶解し、撹拌しながら氷上で冷却した。氷冷溶液に炭酸カリウム(3.6g、0.0275mol)を添加し、次いで15mlのTHFに溶解したジ−tert−ブチルジカーボネート(12g、0.055mol)を添加した。混合物を0℃で2時間撹拌し、室温で2時間撹拌した。THFを減圧下で除去し、残留物をDCMに溶解した。溶液を水で2回洗浄し、乾燥し(MgSO4)、濃縮することで、ピンク色の低粘度油状物6.47g(88%)を得た。生成物をエレクトロスプレー質量分析法(ESI−MS)によって同定した(MH+計算値147.09、実測値147.6)。生成物はそれ以上精製せずに使用した。
【0046】
(b)(4−ブロモブトキシ)−tert−ブチルジフェニルシラン
i−Pr2−Net(10ml)を含むDCM(10ml)に4−ブロモ−1−ブタノール(2.75g、18mmol)を溶解した溶液に、アルゴン雰囲気下でt−BDPSiCl(5ml、18mmol)を添加した。溶液を室温で2時間撹拌し、真空中で濃縮し、クロマトグラフィー処理(ヘキサン/酢酸エチル 10:1)することで、低粘度で無色の油状物4.39g(62%)を得た。構造をNMRで確認した。
【0047】
(c)O−[4−(tert−ブチルジフェニルシラニルオキシ)ブチル]−N−メチル−N−Boc−ヒドロキシルアミン
N−Boc−N−メチルヒドロキシルアミン(a)(0.74g、5mmol)を10mlのDMFに溶解し、NaH(200mg、鉱油中60%分散液、4.75mmol)で処理し、アルゴン雰囲気下で1時間撹拌した。混合物を0℃に冷却し、(4−ブロモブトキシ)−tert−ブチルジフェニルシラン(b)のDMF(10mL)溶液で処理し、0℃でさらに3時間撹拌した。溶媒を減圧下で除去し、残留物をEtOAc(150mL)に溶解し、分液漏斗内に注入した。有機層を0.1M NaOH(5×50mL)、H2O(50mL)、0.1M KHSO4及びブライン(50mL)で洗浄し、次いでMgSO4で乾燥した。溶媒の除去後、残留物をシリカゲル上でクロマトグラフィー処理(ヘキサン:EtOAc 10:1)して0.588g(24%)の生成物を得た。生成物をエレクトロスプレー質量分析法(ESI−MS)によって同定した(MH+計算値457.26、実測値457.8)。
【0048】
(d)4−(N−メチル−N−Boc−ヒドロキシルアミン)ブタン−1−オール
20mLの乾燥THFに溶解したO−[4−(tert−ブチルジフェニルシラニルオキシ)ブチル]−N−メチル−N−Boc−ヒドロキシルアミン(c)(588mg、1.22mmol)にTBAF(1.6mL、1.586mmol)を添加した。反応物をアルゴン下で一晩撹拌した。溶液にNH4Cl(飽和)(10mL×3)を添加し、THFを蒸発させた。溶液をDCMで抽出し、有機相を乾燥(Na2SO4)し、蒸発させた。残留物をシリカゲル上でクロマトグラフィー処理(ヘキサン/EtOAc 1:1)して0.170g(63%)の生成物を得た。生成物をHPLC(カラム:Phenomenex Luna 3μ C18(2),4.6×50mm、検出:214nm、勾配:10分で50〜100%B(ただし、A=H2O/0.1%TFA及びB=アセトニトリル/0.1%TFA)、流量:2mL/分、Rt=2.70分)によって分析した。NMR分析によって追加の確認を行った。
【0049】
(e)トルエン−4−スルホン酸4−(N−メチル−N−Boc−ヒドロキシルアミン)ブチルエステル
4−(N−メチル−N−Boc−ヒドロキシルアミン)ブタン−1−オール(d)(170mg、0.77mmol)及びトリエチルアミン(161μL、1.155mmol)を10mLの乾燥DCMに溶解した氷浴冷却撹拌溶液に、乾燥DCM(5mL)中のトルエン−4−スルホニルクロリド(190.8mg、1.001mmol)をアルゴン下で添加した。15分後に氷浴を取り除き、反応混合物を室温に放置した。2時間後、新しい反応剤(トリエチルアミン(32μL、0.23mmol)、トルエン−4−スルホニルクロリド(29.36mg、0.154mmol))を添加した。24時間後、TLC上で想定生成物を認めることができる。30時間後、新しい反応剤(トリエチルアミン(53μL、0.385mmol)、トルエン−4−スルホニルクロリド(73.5mg、0.385mmol))を反応混合物に添加し、一晩放置した。有機相を10%NaHCO3(10mL×3)で洗浄し、MgSO4で乾燥した。有機相を真空下で除去し、シリカ上でフラッシュする(ヘキサン:酢酸エチル 6:4)ことで111mg(41.5%)の生成物を得た。NMRは、生成物中におけるトルエン−4−スルホニルクロリド不純物の存在を示した。4−スルホニルクロリドを除去するために追加の精製が必要であった。ヘキサン:EtOAc(8:2)を用いることで、シリカフラッシュカラム上で良好な分離が達成され、4−スルホニルクロリドが除去された。収量:64mg(22%)。生成物をHPLC(カラム:Phenomenex Luna 3μ C18(2),4.6×50mm、検出:214nm、勾配:10分で20〜80%B(ただし、A=H2O/0.1%TFA及びB=アセトニトリル/0.1%TFA)、流量:2mL/分、Rt=2.70分)によって分析した。生成物をエレクトロスプレー質量分析法(ESI−MS)によって同定した(MH+計算値373.16、実測値373.9)。NMR分析によって追加の確認を行った。
【0050】
例2
非放射性標準としてのO−(4−フルオロブチル)−(N−メチル−N−Boc−ヒドロキシルアミン)の製造
【0051】
【化19】
KF(4.64mg、0.080mmol)及びクリプトフィックス(30.1mg、0.080mmol)を乾燥アセトニトリル(0.75mL)に溶解した。混合物を5分間撹拌した後、乾燥アセトニトリル(0.250mL)に溶解した化合物1(15mg、0.040mmol)をアルゴン下で添加した。混合物を60℃で1時間加熱した。1時間後、TLCは反応が完了したことを示した。溶媒を蒸発させ、残留物をシリカ上でフラッシュする(ヘキサン/EtOAc(1:1))ことで4.5mg(51%)の生成物を得た。生成物をHPLC(カラム:Phenomenex Luna 3μ C18(2),4.6×50mm、検出:214nm、勾配:10分で20〜70%B(ただし、A=H2O/0.1%TFA及びB=アセトニトリル/0.1%TFA)、流量:2mL/分、Rt=2.70分)によって分析した。生成物をエレクトロスプレー質量分析法(ESI−MS)によって同定した(MH+計算値221.14、実測値221.7)。構造をNMRで確認した。
【0052】
例3
18F−化合物2の放射合成及び2−ブロモアセトフェノンへのコンジュゲーション
【0053】
【化20】
Raytest社からのSynChrom R&Dモジュール上で放射合成を実施した。N2下で90°に分間加熱することにより、クリプトフィックス222(1mLのアセトニトリル中39.1mg)及び炭酸カリウム(1mLの水中65.7mg)の存在下で18F−フッ化物(1GBq以下)を共沸的に乾燥した。この間に、2×1mLのアセトニトリルを添加して蒸発させた。<40°に冷却した後、トルエン−4−スルホン酸4−(N−メチル−N−Boc−アミノオキシ)ブチルエステル(化合物1)の溶液(1mLのアセトニトリル中3mg)を添加した。反応器を70℃に20分間加熱して標識を行った。60/40 CH3CN/H2Oからなる定組成流を使用しながら、粗反応混合物を214nmでHPLCにかけた。クロマトグラムは、非放射性標準と共溶出する良好な収量の標識化合物(約86%RCP)を示した。2mLのACNを用いて粗反応混合物をSep−Pakアルミニウムカラムから溶出させることで遊離フッ化物を除去し、遊離フッ化物の活量を測定した(遊離フッ化物は15%未満であった)。「精製」混合物をHPLCで再分析したところ、遊離フッ化物は除去されていることがわかった。1mL(100MBq)の精製生成物を1mLの1M HCl中において90℃で20分間加水分解してBOC保護基を除去することで、100%の非保護F−18放射性標識化合物を得た。1mLの加水分解生成物を10mLのミリQ水で希釈し、pHをpH11〜12に調整し、プレコンディショニングを施したSEP−PAK C−18カラムから溶出させた。カラム上の放射能は30.1MBqと測定された。2mLのDMFでカラムから反応バイアル中に溶出させることで、23MBqを有する2mLの液体が得られた。6mgのブロモアセトフェノンを10μLのジイソプロピルエチルアミンと共に添加し、90℃で10分間加熱した。反応混合物をHPLCで分析したところ、「遊離」前駆体に対応するピークより大きい面積(57%)を有する新しいピークが5.46分の位置に溶出したことにより、コンジュゲート形成の証拠が示された。HPLCは次の通りであった。カラム:Xterra(Waters社)5μ C18 4.6×250mm、検出:250nm及び214nm、NaI検出器、勾配:定組成、40%A=H2O及び60%B=アセトニトリル、流量:1mL/分。
【0054】
例4
トルエン−4−スルホン酸2−[2−(N−メチル−N−BOC−アミノオキシ)エトキシ]エチルエステルの製造
【0055】
【化21】
(a)N−Boc−N−メチルヒドロキシルアミン
N−メチルヒドロキシルアミン(4.2g、0.05mol)を50%水性テトラヒドロフラン(THF)(20ml)に溶解し、撹拌しながら氷上で冷却した。氷冷溶液に炭酸カリウム(3.6g、0.0275mol)を添加し、次いで15mlのTHFに溶解したジ−tert−ブチルジカーボネート(12g、0.055mol)を添加した。混合物を0℃で2時間撹拌し、室温で2時間撹拌した。THFを減圧下で除去し、残留物をDCMに溶解した。溶液を水で2回洗浄し、乾燥し(MgSO4)、濃縮することで、ピンク色の低粘度油状物6.47g(88%)を得た。生成物をエレクトロスプレー質量分析法(ESI−MS)(MH+計算値147.09、実測値147.6)及びNMRによって同定した。生成物はそれ以上精製せずに使用した。
【0056】
(b)トルエン−4−スルホン酸2−(2−ヒドロキシエトキシ)エチルエステル
ジエチレングリコール(22g、207.5mmol)のCH2Cl2(100ml)溶液にトリエチルアミン(10.5g、103.75mmol)を添加した。9.89g(51.8mmol)のトルエン−4−スルホニルクロリドを一度に添加した。溶液を室温で1時間撹拌した。DCM中5%MeOHを用いてTLCを実施した。1/2時間後、TLCは生成物の生成物を示した。1時間15分後、反応混合物を0.1M KHSO3及び5%NaHCO3で洗浄した。有機相をNa2SO3で乾燥し、減圧下で蒸発させた。粗反応混合物をシリカゲル上に捕捉し、ヘキサン/EtOAc 50:50を用いたコンビフラッシュ・コンパニオン(Combiflash Companion)装置(330gカラム)上でのフラッシュクロマトグラフィーによって精製した。NMRで構造を確認した。
【0057】
(c)4−トルエンスルホン酸2−[2−(tert−ブチルジフェニルシラニルオキシ)エトキシ]エチルエステル
i−Pr2−Net(6mL、1.2Eq)を含むCH2Cl2(30ml)にトルエン−4−スルホン酸2−(2−ヒドロキシエトキシ)エチルエステル(b)(7.622g、29.28mmol)を溶解した溶液に、アルゴン雰囲気下でt−BDPSiCl(9ml、1.2eq、35.136mmol)及び触媒量のDMAPを添加した。溶液を室温で2日間撹拌した。TLCは反応の完了を示唆した。粗反応混合物を100mLのDCMで希釈し、2×100mLの水及び100mLのブラインで洗浄した。MgSO4を用いて有機相を乾燥し、真空下で蒸発させ、30分で0〜50%酢酸エチルの勾配(EtOAc/ヘキサン)を用いるコンパニオン・コンビフラッシュ(120gカラム)上でのフラッシュクロマトグラフィーによって精製した。外観は透明で粘稠な油状物であった。NMRで構造を確認した。
【0058】
(d)O−{2−[2−tert−ブチルジフェニルシラニルオキシ)エトキシ]エチル}−N−メチル−N−BOC−ヒドロキシルアミン
オーブン乾燥したフラスコ(50mL)に、NaH(鉱油中60%分散液)481mg(12.03mmol)をアルゴン下で加えた。分散液をペンタンで3回洗浄して鉱油を除去した。純NaHに5mLのTHF(乾燥)を添加した。5mLのTHFに溶解した1.53mg(10.43mmol)のN−BOC−N−メチルヒドロキシルアミン(a)をゆっくりと添加した。反応混合物を、それ以上のガス発生が見られなくなるまで1/2時間放置する。溶液を氷浴上に載せ、1/2時間撹拌した後、4g(8.02mmol)の4−トルエンスルホン酸2−[2−(tert−ブチルジフェニルシラニルオキシ)エトキシ]エチルエステル(c)を滴下した。溶液を氷浴上に1/2時間放置し、次いでRTで一晩撹拌した。溶媒を減圧下で除去し、残留物をEtOAc(200mL)に溶解し、分液漏斗内に注入した。有機層を0.1M NaOH(5×50mL)、H2O(50mL)、0.1M KHSO4及びブライン(50mL)で洗浄し、次いでMgSO4で乾燥した。溶媒の除去後、DCM中のメタノール勾配(メタノール:19分で0〜20%)を用いるコンパニオン・コンビフラッシュ上でのフラッシュクロマトグラフィーによって残留物を精製した。NMRで構造を確認した。
【0059】
(e)2−[2−(N−メチル−N−BOC−アミノオキシ)エトキシ]エタノール
乾燥THF(15mL)に溶解したO−{2−[2−tert−ブチルジフェニルシラニルオキシ)エトキシ]エチル}−N−メチル−N−BOC−ヒドロキシルアミン(d)(3g、6.33mmol)にTBAF(6.78mL、6.78mmol)を添加した。反応物を一晩撹拌した。THFを蒸発させ、残留物をDCMに溶解し、NH4Cl(飽和)(40mL)、水及びブラインで洗浄した。有機相を乾燥(MgSO4)し、蒸発させた。残留物をコンパニオン・コンビフラッシュ(40gカラム)上でのフラッシュクロマトグラフィー(勾配:DCM中において20分で0〜5%MeOH)によって精製した。NMRで構造を確認した。
【0060】
(f)トルエン−4−スルホン酸2−[2−(N−メチル−N−BOC−アミノオキシ)エトキシ]エチルエステル
2−[2−(N−メチル−N−BOC−アミノオキシ)エトキシ]エタノール(e)1g(4.25mmol)を乾燥DCM(15mL)に溶解した撹拌溶液に、15mLのDCM中のトリエチルアミン(910mg、1254μL、9mmol)及びトルエン−4−スルホニルクロリド(1620mg、8.5mmol)を添加した。反応混合物をアルゴン下で一晩撹拌した。TLC(ヘキサン/酢酸エチル 6:4)は反応の完了を示した。反応混合物を50mLのDCMで希釈し、有機相を5%NaHCO3、ブライン及び水で洗浄した。有機相をMgSO4上で乾燥し、濾過し、減圧下で蒸発させた。生成物を、ヘキサン/酢酸エチルの勾配を用いるコンパニオン・コンビフラッシュ(40gカラム)によって精製した。NMRで構造を確認した。
【0061】
例5
O−[2−(2−フルオロエトキシ)エチル]−N−メチル−N−BOC−ヒドロキシルアミンの製造
【0062】
【化22】
KF(58mg、1mmol)及びクリプトフィックス(376mg、1mmol)を乾燥アセトニトリル(2mL)に溶解した。混合物を5分間撹拌した後、乾燥アセトニトリル(0.250mL)に溶解した化合物1(200mg、0.51mmol)をアルゴン下で添加した。混合物を80℃で1時間加熱した。1時間後、TLCは反応の完了を示した。減圧を用いて有機相を蒸発させ、DCMに再溶解し、シリカゲル上に捕捉した。シリカゲル上でのフラッシュクロマトグラフィー(ヘキサン/酢酸エチル 1:1)を用いて物質を精製した。収量:83mg(0.35mmol)。NMRで構造を確認した。
【0063】
例6
マレイミド−プロピオニル−Lys−Gly−Phe−Gly−Lys−OHの製造
【0064】
【化23】
HBTU活性化を用いるスローmoc・シングル・カップル(Slowmoc Single Couple)方法に従い、Fmoc−Lys(Boc)−Sasrin樹脂(0.1mmol)を用いて全自動ペプチド合成機(ABI 433A合成機)上でモデルペンタペプチドLys−Gly−Phe−Gly−Lys−OHをアセンブルした。3−(マレイミド)プロピオン酸N−ヒドロキシスクシンイミドエステル(0.52mmol)を用いてマレイミド−プロピオニル酸を手作業でカップリングした。トリイソプロピルシラン及び水を含むトリフルオロ酢酸(95:2.5:2.5 v/v/v)中で、樹脂からのペプチド除去及び側鎖保護基の脱保護を同時に実施した。濾過後、溶液を減圧下で濃縮し、残留物をジエチルエーテルで洗浄した。粗生成物を逆相分取クロマトグラフィー(Phenomenex Luna C18(2)カラム,250×50mm,10μm、勾配:60分で0〜30%溶媒B、流量:50mL/分)で精製して66mg(95%)の純化合物を得た。生成物をLC−MS[Phenomenex Luna C18−(2),50×2.0mm,5μm、勾配:5分で0〜30%溶媒B、流量:0.6mL/分、tR=2.86分]で分析したところ、m/z=687.6(M+H)+、m/z計算値=687.34(M+H)+であった。
【0065】
例7
18F−化合物2の放射合成及び化合物3へのコンジュゲーション
【0066】
【化24】
GE Healthcare社からのTracerLab FxFnモジュール上で放射合成を実施した。N2下で100°に9分間加熱することにより、クリプトフィックス222(1mLのアセトニトリル中39.1mg)及び炭酸カリウム(1mLの水中65.7mg)の存在下で18F−フッ化物(350MBq以下)を共沸的に乾燥した。この間に、2×0.7mLのアセトニトリルを添加して蒸発させた。<50°に冷却した後、トルエン−4−スルホン酸2−[2−(N−メチル−N−BOC−アミノオキシ)エトキシ]エチルエステル(化合物1)の溶液(1mLの乾燥アセトニトリル中5mg)を添加した。反応器を90℃に10分間加熱して標識を行った。粗反応混合物を5mLの水で希釈した。TLC用の試料を採取すると共に、50μLをHPLC(Phenomenex gemine 150mm×4.60 C18 5μm、214nm、254nm及びγ線検出器で検出、15分で20〜80%の溶媒Bを用いた勾配流)にかけた。TLC(1:1 酢酸エチル/ヘキサン)は、約70%という良好な収率の標識化合物を示した(n=3)。化合物は非放射性対照標準と共溶出した。粗反応混合物をさらに4mlの水で希釈し、(5mLのMeCN及び10mLのH2Oでプレコンディショニングした)Oasis HLB Sep−Pakカートリッジに通した。Oasisカートリッジを50mLの25%メタノール水溶液で洗浄し、1.5mLのMeCNを用いて精製化合物を溶出した。MeCN溶出液に0.3mLのエーテル中2N HClを添加し、RTで5分間撹拌してBOC基を定量的に除去した後、有機相をN2流及び減圧下で65℃で3分間蒸発させ、1mLのMeCNを添加した後にさらに3分間蒸発させた。0.8mLの0.4M酢酸ナトリウム緩衝液(pH5)に溶解した5mgの化合物3を乾燥混合物に添加し、反応物を70℃に60分間加熱してコンジュゲーションを行った。0分及び60分に試料を採取した。反応混合物をHPLCで分析したところ、面積(74%)を有する新しいピークが11分の位置にF19−対照標準と共溶出したことにより、化合物3へのコンジュゲート形成の証拠が示された(図1)。HPLCは次の通りであった。Phenomenex gemine 150mm×4.60 C18 5μm、勾配:10分で0〜40%溶媒B及び10〜15分に40%溶媒B。
【0067】
例8
4−(2−ニトロビニル)ベンゾイル−Lys−Gly−Phe−Gly−Lys−OHの製造
【0068】
【化25】
例6に記載したようにしてモデルペンタペプチドLys−Gly−Phe−Gly−Lys−OHをアセンブルした。純DMF中の樹脂(0.2mmol)に58mg(0.3mmol)のトランス−β−ニトロスチレン及び156.5mg(0.3mmol)のPyAOPを添加した後、102μL(0.6mmol)のDIPEAを添加し、反応物を1時間放置した。トリイソプロピルシラン及び水を含むトリフルオロ酢酸(95:2.5:2.5 v/v/v)中で、樹脂からのペプチド除去及び側鎖保護基の脱保護を同時に実施した。濾過後、溶液を減圧下で濃縮し、残留物をジエチルエーテルで洗浄した。粗生成物を逆相分取クロマトグラフィー(Phenomenex Luna C18(2)カラム,250×50mm,10μm、勾配:60分で0〜40%溶媒B、流量:50mL/分)で精製して101mg(70%)の純化合物を得た。生成物をLC−MS[Phenomenex Luna C18−(2),50×2.0mm,5μm、勾配:5分で0〜30%溶媒B、流量:0.6mL/分、tR=4.10分]で分析したところ、m/z=711.3(M+H)+、m/z計算値=711.3(M+H)+であった。
【0069】
例9
化合物6への18F−化合物4のコンジュゲーション
【0070】
【化26】
上記例7に記載したようにして18F−化合物4の放射合成及び精製を実施した。18F−化合物4を含むMeCN溶出液に0.2mLのエーテル中2N HClを添加し、RTで5分間撹拌してBOC基を定量的に除去した後、有機相をN2流及び減圧下で65℃で3分間蒸発させ、1mLのMeCNを添加した後にさらに3分間蒸発させた。0.8mLの0.4M酢酸ナトリウム緩衝液(pH5)に溶解した5mgの化合物6を乾燥混合物に添加し、反応物を30℃に5分間加熱した。反応混合物をHPLCで分析したところ、面積(72%)を有する新しいピークが10.4分の位置に19F−対照標準と共溶出したことにより、コンジュゲート形成の証拠が示された(図2)。HPLCは次の通りであった。Phenomenex gemine 150mm×4.60 C18 5μm、勾配:10分で0〜40%溶媒B及び10〜15分に40%溶媒B。
【0071】
例10
マレイミド−プロピオニル−RDGペプチドの製造
【0072】
【化27】
5mL容器内で50mg(0.04mmol)のペプチドNC100717を1mLのDMFに溶解した。21.3mg(0.1mmol)の3−(マレイミド)プロピオン酸N−ヒドロキシスクシンイミドエステルを13.6μL(0.08mmol)のDIPEAと共に1mLのDMFに溶解し、ペプチド溶液に添加した。反応物をRTで1時間撹拌した。DMFを蒸発させた。粗生成物を逆相分取クロマトグラフィー(Phenomenex Luna C18(2)カラム,250×50mm,10μm、勾配:60分で0〜40%溶媒B、流量:50mL/分)で精製して23mg(40%)の純化合物を得た。生成物をLC−MS[Phenomenex Luna C18−(2),50×2.0mm,5μm、勾配:5分で5〜40%溶媒B、流量:0.6mL/分、tR=2.74分]で分析したところ、m/z=1409.9(M+H)+、m/z計算値=1409.5(M+H)+であった。
【0073】
例11
化合物7への18F−化合物4のコンジュゲーション
【0074】
【化28】
上記例7に記載したようにして18F−化合物4の放射合成及び精製を実施した。MeCN溶出液に0.2mLのエーテル中2N HClを添加し、RTで5分間撹拌してBOC基を定量的に除去した後、有機相をN2流及び減圧下で65℃で3分間蒸発させ、1mLのMeCNを添加した後にさらに3分間蒸発させた。0.6mLの0.4M酢酸塩緩衝液(pH5)+0.4mLのDMFに溶解した5.75mgの化合物7を乾燥残留物に添加した。ペプチド混合物を70℃に加熱し、45分後に試料を採取した。粗反応混合物を放射性HPLCで分析したところ、45分後にペプチド中への18F−シントンの24%取込みが見られ、18F−化合物がその真正標準と共溶出した(図3)。HPLCは次の通りであった。Phenomenex gemine 150mm×4.60 C18 5μm、勾配:10分で0〜40%溶媒B及び10〜15分に40%溶媒B。
【0075】
例11
ベンゾイル−トランス−β−ニトロスチレン−RDGの製造
【0076】
【化29】
40mg(0.032mmol)のペプチドNC100717を2mLのDMFに溶解した。2mLのDMF及び16μL(0.096mmol)に溶解した12.3mg(0.064mmol)のトランス−β−ニトロスチレン及び25mg(0.048mmol)のPyaOPを添加した。混合物を10分間予備活性化した後、それをペプチド(NC100717)に添加した。15分後、LC−MSは反応の完了を示し、反応混合物をMeCN/0.1%TFA(20mL)で奪活した。有機相を減圧下で除去した。
粗生成物を逆相分取クロマトグラフィー(Phenomenex Luna C18(2)カラム,250×50mm,10μm、勾配:60分で5〜45%溶媒B、流量:50mL/分)で精製して12mg(26%)の純化合物を得た。生成物をLC−MS[Phenomenex Luna C18−(2),50×2.0mm,5μm、勾配:5分で5〜40%溶媒B、流量:0.6mL/分、tR=2.74分]で分析したところ、m/z=1433.6(M+H)+、m/z計算値=1433.5(M+H)+であった。
【0077】
例12
化合物8への18F−化合物4のコンジュゲーション
【0078】
【化30】
上記例7に記載したようにして18F−化合物4の放射合成及び精製を実施した。MeCN溶出液に0.2mLのエーテル中2N HClを添加し、RTで5分間撹拌してBOC基を定量的に除去した後、有機相をN2流及び減圧下で65℃で3分間蒸発させ、1mLのMeCNを添加した後にさらに3分間蒸発させた。0.8mLの0.4M酢酸塩緩衝液(pH5)+0.4mLのDMFに溶解した化合物8(5mg)を乾燥残留物に添加した。反応物を30℃で5分間放置した。粗反応混合物を放射性HPLCで分析したところ、5分後にペプチド中への18F−シントンの25%取込みが見られ、12分後に18F−化合物がその真正標準と共溶出した(図4)。HPLCは次の通りであった。Phenomenex gemine 150mm×4.60 C18 5μm、勾配:10分で0〜40%溶媒B及び10〜15分に40%溶媒B。
【0079】
例13
エテンスルホニル酢酸の合成
【0080】
【化31】
250mLの三つ口丸底フラスコ内で1.1g(47.5mmol、1.2eq)のナトリウムを25mLの無水アルコールに溶解した。H2の発生及びナトリウムの消失が完了した後、2−メルカプト酢酸エチル(4.60mL、40.7mmol、1eq)を滴下した。得られた混合物を20分間撹拌した後、57mL(57mmol、1.4eq)の臭化ビニル(THF中1M溶液)を添加した。混合物をオートクレーブに移し、105℃に1時間加熱した。反応混合物を4時間撹拌し続けた。次いで、THF及びEtOHを真空中で蒸発させ、残留物を水(10mL)に溶解し、ジエチルエーテル(5×60mL)で抽出した。有機相をNa2SO4上で乾燥し、XS溶媒を真空中で除去した。次いで、粗物質(2.9g)を10mLの氷酢酸に溶解し、0℃に冷却した。36〜40%過酢酸(7.4mL、40mmol)を添加し、反応混合物を0℃で30分間撹拌した後、さらに2時間にわたって撹拌し続けて温度を室温に上昇させた。再び溶媒を真空下で除去し、粗生成物を自動コンビフラッシュ装置上でのフラッシュクロマトグラフィー(10分でヘキサン中20〜100%酢酸エチル)によって精製した。NMRにより、構造がスルホキシドであることを確認した。すべての画分を集め、スルホキシド(1.5g、9.2mmol)に関してさらに過酢酸(2eq)で酸化してすべての出発原料を消失させた。粗生成物をそれ以上精製せずに使用した。エテンスルホニル酢酸エチルエステルを20mLの0.1M HClに溶解し、混合物を100℃で13時間還流させることでエテンスルホニル酢酸(12.8g、8mmol)を得た。NMRで構造を確認した。
【0081】
エテンスルホニル−アセチル−Lys−Gly−Phe−Gly−Lys−OHの製造
【0082】
【化32】
例6に記載したようにしてモデルペンタペプチドLys−Gly−Phe−Gly−Lys−OHをアセンブルした。純DMF(5mL)中のペプチド結合樹脂(0.1mmol)に75mg(0.5mmol)のエテンスルホニル酢酸及び261mg(0.5mmol)のPyAOPを添加した後、171μL(1mmol)のDIPEAを添加し、反応物を2時間放置した。トリイソプロピルシラン及び水を含むトリフルオロ酢酸(95:2.5:2.5 v/v/v)中で、樹脂からのペプチド除去及び側鎖保護基の脱保護を同時に実施した。濾過後、溶液を減圧下で濃縮し、残留物をジエチルエーテルで洗浄した。粗生成物を逆相分取クロマトグラフィー(Phenomenex Luna C18(2)カラム,250×50mm,10μm、勾配:60分で0〜30%溶媒B、流量:10mL/分)で精製して51mg(76%)の純化合物を得た。生成物をLC−MS[Phenomenex Luna C18−(2),50×2.0mm,5μm、勾配:5分で0〜30%溶媒B、流量:0.6mL/分、tR=2.61分]で分析したところ、m/z=668.4(M+H)+、m/z計算値=668.3(M+H)+であった。
【0083】
【化33】
GE Medical Systems社からのTracerLab FxFnモジュール上で放射合成を実施した。N2流及び真空下で100°に9分間加熱することにより、215μLの水及び785μLのアセトニトリル(総量0.8mL)に溶解した56mgのクリプトフィックス222(K222、4,7,13,16,21,24−ヘキサオキサ−1,10−ジアザビシクロ[8.8.8]ヘキサコサン)及び10mgのK2CO3の存在下で18F−フッ化物(200MBq)を共沸的に乾燥した。この間に、2×0.8mLのアセトニトリルを添加して蒸発させた。<50°に冷却した後、トルエン−4−スルホン酸2−[2−(N−メチル−N−Boc−アミノオキシ)エトキシ]エチルエステル(化合物3)の溶液(1mLの乾燥アセトニトリル中3mg)を添加した。密閉した反応器を90℃に10分間加熱して標識を行った。粗反応混合物を9mlの水で希釈し、(5mLのMeCN及び10mLのH2Oでプレコンディショニングした)Oasis HLB Sep−Pakカートリッジ(Waters社)に通した。Oasisカートリッジを50mLの25%MeOH水溶液で洗浄し、1.5mLのMeCNを用いて精製化合物を溶出した。MeCN溶出液にエーテル中の2N HCl(0.2mL)を添加し、混合物をRTで5分間撹拌してBoc基を定量的に除去した。有機相を真空中及びN2流下で65℃で3分間蒸発させ、次いで1mLのMeCNを添加し、再び真空中で3分間蒸発させた。
【0084】
0.8mLの0.4M酢酸ナトリウム緩衝液(pH5)に溶解した5mgの化合物10を乾燥混合物に添加し、反応物を70℃に10分間加熱してコンジュゲーションを行った。反応混合物をHPLCで分析したところ、コンジュゲート形成の証拠(8.92分の位置に19F−対照標準と共溶出した新しいピーク、収率40%)が示された。HPLCは次の通りであった。Phenomenex gemine 150mm×4.60 C18 5μm、勾配:10分で0〜40%溶媒B及び10〜15分に40%溶媒B。
【技術分野】
【0001】
本発明は、陽電子放出核種で標識した生物学的活性ベクターを含む診断薬及び放射性診断薬に関する。本発明はさらに、特異的な生物学的標的に対して親和性を有する分子として定義されるベクター、好ましくはペプチドであるベクターの[18F]フッ素化のための方法及び試薬に関する。こうして得られる18F標識コンジュゲートは、放射性医薬品として、具体的には陽電子放射断層撮影(PET)で使用するための放射性医薬品として有用である。
【背景技術】
【0002】
放射性標識された生物活性ベクターを診断イメージングのために応用することは、核医学において重要性を増しつつある。特定の細胞タイプと選択的に相互作用する生物学的活性分子は、標的組織に放射能を送達するために有用である。例えば、放射性標識ペプチドは、診断イメージング及び放射線療法のため、腫瘍、梗塞巣部及び感染組織に放射性核種を送達するための大きな潜在的可能性を有している。約110分の半減期を有する18Fは、多くのレセプターイメージング調査のために最適な陽電子放出核種である。したがって、18F標識された生物活性ペプチドは、多種多様の疾患を定量的に検出して特性決定するためにPETで利用できるので大きな臨床的可能性を有している。
【0003】
18F標識ペプチドに関する難点の1つは、既存の18F標識剤が調製するのに多くの時間を要することである。18Fによるペプチド及びタンパク質の効率的な標識は、適当な補欠分子族を用いることでのみ達成される。文献中には、N−スクシンイミジル−4−[18F]フルオロベンゾエート、m−マレイミド−N−(p−[18F]フルオロベンジル)ベンズアミド、N−(p−[18F]フルオロフェニル)マレイミド及び4−[18F]フルオロフェナシルブロミドを含む数種のかかる補欠分子族が提唱されている。18Fによるペプチド及びタンパク質の標識のために現在使用されている方法のほとんどすべてが、フッ素標識シントンの活性エステルを利用している。ペプチド及びタンパク質は活性エステルと反応し得る多数の官能基を含むことがあるので、これらの現行方法は部位特異的でない。例えば、3つのリシン残基を含むペプチドは、標識シントンに対する反応性が全く同じ3つのアミン官能基を有する。したがって、特にペプチド中に18Fを温和な条件下で迅速かつ化学選択的に導入して、高い放射化学収量及び純度で18F標識生成物を得ることを可能にする18F標識補欠分子族及び方法に対するニーズが依然として存在している。さらに、かかる方法であって、臨床現場での放射性医薬品の調製を容易にするための自動化に適した方法に対するニーズも存在している。我々は以前にPET標識方法におけるアミノオキシ化学の使用を記載した(国際公開第03/006491号)が、この発明の化合物はアルデヒド及びケトンと容易に反応しないものの、ある種のハロゲン含有化合物に対して選択性を有する。N−アルキルアミノオキシ部分の化学的安定性はさらに高いので、副反応が最小限に抑えられかつ中間体が一層安定である結果として中間体の取扱い及び貯蔵を首尾よく行うことが可能となり、したがって以前に開示したアミノオキシ基に比べて利点が得られる。
【先行技術文献】
【特許文献】
【0004】
国際公開第2008/083191号パンフレット
【発明の概要】
【0005】
本発明は、以下の式(I)の化合物を以下の式(II)の化合物と反応させるか、或いは以下の式(III)の化合物を以下の式(IV)の化合物と反応させることを含んでなる放射性フッ素化方法を提供する。
【0006】
【化1】
【0007】
【化2】
式中、
R1はハロアセチル、ハロアリル又は基又はフェナシルハライドのような反応性ハロゲン含有基であるか、すべての基がアルキル化反応への関与に適したエポキシド環であるか、マレイミド基或いはアクリル酸又は置換アクリル酸誘導体のようなマイケル(Michael)受容体であるか、ビニルスチレン(好ましくはニトロビニルスチレン)であるか、ビニルスルホンアミドであるか、或いはビニルスルホンである。
R2はN−アルキルアミノオキシ基であり、これは水性緩衝液及びわずかに酸性のpHのような温和な条件下でR1と部位特異的に反応して以下の式(V)の化合物を生じる。
式(III)のR3はR2に等しく、式(IV)のR4はR1に等しい。式(III)の化合物と式(IV)の化合物との連結は以下の式(VI)の生成物を生じる。
【0008】
【化3】
式中、いずれの場合にも、Yはアルキルであり、好ましくはY=−CH3である。
【図面の簡単な説明】
【0009】
【図1】化合物3の18F−コンジュゲートのHPLCクロマトグラム。
【図2】化合物6の18F−コンジュゲートのHPLCクロマトグラム。
【図3】化合物7の18F−コンジュゲートのHPLCクロマトグラム。
【図4】化合物8の18F−コンジュゲートのHPLCクロマトグラム。
【図5】化合物11の18F−コンジュゲートのHPLCクロマトグラム。
【発明を実施するための形態】
【0010】
式(V)の化合物が最も好ましく、式(II)の化合物の18F−リンカー成分は好ましくは以下の式(VII)〜(XI)に示される種類の化合物からなる。
【0011】
【化4】
式中、
nは0〜20の整数であり、
ZはO、N又はSである。
【0012】
式(II)の化合物中のリンカー基は、得られる式(V)のコンジュゲートにおいて好ましい排泄特性のような最適のインビボ薬物動態が得られるように選択することもできる。親油性の異なるリンカー基を使用することで、診断上のニーズに応じてペプチドのインビボ薬物動態を大きく変化させることができる。例えば、式(V)のコンジュゲートを腎排泄によって体外に排除することが望ましい場合には、式(X)〜(XI)から導かれるリンカー成分を含む親水性リンカーが使用される。同様に、肝胆道排泄による排除が望ましい場合には、式(VII)〜(IX)から導かれるリンカー成分を含む疎水性リンカーが選択できる。
【0013】
本発明は、ペプチド又はベクター前駆体の合成に際して標識の正確な導入部位が予め選択される一層化学選択的な放射性標識アプローチを提供する。分子中の所定部位で起こる連結反応は、ただ1種の標識生成物を与える。したがって、このような方法は化学選択的であり、その用途は多種多様の薬物様分子、ペプチド及び生体分子(例えば、小タンパク質)の標識に役立つ包括的なものであると考えられる。
【0014】
さらに別の態様では、本発明は、以下の式(Ia)の化合物を以下の式(IIa)の化合物と反応させるか、或いは以下の式(IIIa)の化合物を以下の式(IVa)の化合物と反応させてそれぞれ以下の式(Va)又は(VIa)のコンジュゲートを得ることを含んでなる放射性フッ素化方法を提供する。
【0015】
【化5】
【0016】
【化6】
【0017】
【化7】
式中、R1及びR4はそれぞれ式(I)及び式(IV)の化合物に関して上記に定義した通りであり、リンカー基は上記の化合物に関して定義した通りである。
【0018】
反応は、適当な溶媒(例えば、pH3〜11の範囲内の水性緩衝液)中において5〜70℃の極端でない温度、最も好ましくは周囲温度で実施できる。
【0019】
本発明のさらに別の態様では、R1はマレイミド化合物及びビニルスチレン化合物の部類から導かれ、最も好ましくは以下の式(Ib)及び式(Ic)中に表される。
【0020】
【化8】
式(I)及び式(III)中並びに本発明の他の態様では、特記しない限り、ベクターは疾患過程に付随する関連レセプターに対して親和性を有する生物学的薬剤として定義できる。生物学的薬剤は小分子薬物様の薬物団、タンパク質又は抗体として定義できるが、好ましくは分子量30kDa未満のタンパク質であり、最も好ましくはアミノ酸100未満のペプチドである。本発明の方法を用いる標識用の好ましいペプチドベクターの例には、オクトレオチドのようなソマトスタチン類似体、ボンベシン、血管作用性小腸ペプチド、走化性ペプチド類似体、α−メラノサイト刺激ホルモン、ニューロテンシン、Arg−Gly−Aspペプチド、ヒトプロインスリン結合ペプチド、エンドセリン、アンギオテンシン及びホルミル−ノルロイシル−ロイシル−フェニルアラニル−ノルロイシル−チロシル−リシンがある。標識用の好ましいペプチドは、国際公開第01/77415号及び同第03/006491号に記載されているもののようなArg−Gly−Aspペプチド及びその類似体である。好ましいペプチドベクターは、以下の式(A)のフラグメントを含んでいる。
【0021】
【化9】
式中、X7は−NH2又は次式のエチレングリコール含有構造であり、
【0022】
【化10】
(式中、「a」は1〜10の整数であり、最も好ましくはaは1である。)
R1は式(I)に関して前記に記載した通りである。
【0023】
当業者には容易に理解される通り、本発明の方法は、タンパク質、ホルモン、オリゴヌクレオチド及び抗体フラグメントのような他の生体分子並びに薬物様小分子の放射性フッ素化によって各種のPETトレーサーを得るためにも使用できる。
【0024】
式(I)及び式(III)の化合物は、例えば、Atherton,E.and Sheppard,R.C.;“Solid Phase Synthesis”;IRL Press:Oxford,1989中に記載されている固相ペプチド合成法のような標準的ペプチド合成法によって製造できる。式(I)又は(III)の化合物中への基R1又はR3の導入は、ペプチドのN末端又はC末端の反応、或いはペプチド配列中に含まれる何らかの他の官能基であって、その修飾がベクターの結合特性に影響を及ぼさない官能基との反応によって達成できる。好ましい例では、Carrasco et al(Biopolymers,Peptide Science,2006,Vol 84(4),page 414)によって記載されたアミノ酸を用いて、N−アルキルアミノオキシ基(Y−NH2−O−)をペプチド配列中に直接導入できる。官能基R1及びR3は、好ましくはペプチドのアミン官能基と活性化酸との反応による安定なアミド結合の形成によって導入され、ペプチド合成の実施中又は実施後に導入される。前駆体が酸である場合、R1及びR3は、2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)又はN−[(ジメチルアミノ)−1H−1,2,3−トリアゾロ[4,5−b]ピリジン−1−イルメチレン]−N−メチルメタンアンモニウムヘキサフルオロホスフェートN−オキシド(HATU)のようなインサイチュ活性化剤を用いて導入できる。
【0025】
式(II)の化合物は、以下の式(XII)の対応する前駆体から製造できる。
【0026】
【化11】
式中、Lは脱離基、好ましくはp−トルエンスルホネート、トリフルオロメタンスルホネート又はメタンスルホネート或いはハライドであり、Y及びリンカーは前記に定義した通りであり、R5は窒素原子を保護するための適当な保護基(例えば、t−ブチルオキシカルボニル基)である。かくして、サイクロトロンで生成された水性[18F]−フッ化物を好適には塩基(例えば、テトラブチルアンモニウム又はK2CO3/クリプトフィックス(Kryptofix)222)からの蒸発によって予備活性化したものと、アセトニトリル、N,N−ジメチルホルムアミド又はジメチルスルホキシドのような適当な溶媒中において、通例は高温(例えば、60〜150℃、好適には60〜120℃)又はマイクロ波加熱下で反応させ、次いで酸分解処理のような標準的方法を用いてN−保護基を除去することができる。
【0027】
式(IV)の化合物は、以下の式(XX)の対応する前駆体又はその保護誘導体から製造できる。
【0028】
【化12】
式中、Lは脱離基、好ましくはp−トルエンスルホネート、トリフルオロメタンスルホネート又はメタンスルホネート或いはハライドであり、リンカー及びR4は前記に定義した通りである。Lは、サイクロトロンで生成された水性[18F]−フッ化物を好適には塩基(例えば、テトラブチルアンモニウム又はK2CO3/クリプトフィックス222)からの蒸発によって予備活性化したものと、アセトニトリル、N,N−ジメチルホルムアミド又はジメチルスルホキシドのような適当な溶媒中において、通例は高温(例えば、60〜120℃)で反応し得る。
【0029】
本発明はまた、一般式(V)又は(VI)の化合物の有効量(例えば、インビボPETイメージングで使用するのに有効な量)を1種以上の薬学的に許容される補助剤、賦形剤又は希釈剤と共に含んでなる放射性医薬品組成物を提供する。
【0030】
本発明の好ましい実施形態は、医学で使用するための、特に(好適にはPETによる)腫瘍イメージングで使用するための一般式(V)又は(VI)の化合物であって、ベクターが国際公開第01/77415号及び同第03/006491号に記載されているもののようなArg−Gly−Aspペプチド又はその類似体である化合物に関する。かかるベクターは、好ましくは次式のフラグメントを含み、
【0031】
【化13】
さらに好ましくは以下の式(A)のペプチドである。
【0032】
【化14】
式中、X7は−NH2又は次式の基であり、
【0033】
【化15】
(式中、「a」は1〜10の整数であり、最も好ましくはaは1である。)
R5はペプチドの反応後にリシン残基のε−アミノとアミド結合を形成し、R1によって定義されるが、最も好ましくは下記の構造(Id)及び(Ie)によって例示される。
【0034】
【化16】
かくして、以下の式(If)及び(Ig)に示される構造を有する式(I)が得られる。
【0035】
【化17】
本発明の放射性標識コンジュゲートは、PETイメージングのため、所望の信号を得るのに十分な量で患者に投与すればよい。通常、体重70kg当たり0.01〜100mCi、好ましくは0.1〜50mCiの典型的な放射性核種投与量で十分であろう。
【0036】
したがって、本発明に係る放射性標識コンジュゲートは、完全に当業者の技術常識に属するやり方で、生理学的に許容されるキャリヤー又は賦形剤を用いて投与のために製剤化できる。例えば、薬学的に許容される賦形剤を任意に添加した化合物を水性媒質中に懸濁又は溶解し、次いで得られた溶液又は懸濁液を滅菌すればよい。
【0037】
さらに別の態様から見れば、本発明は、放射性医薬品をヒト又は動物の身体に投与する段階及び前記身体の少なくとも一部の画像を生成する段階を含むインビボイメージング方法(好適にはPET)、好ましくは腫瘍イメージング方法で使用するための放射性医薬品の製造における本発明の放射性標識コンジュゲートの使用を提供する。
【0038】
さらに別の態様から見れば、本発明は、ヒト又は動物の身体の画像を生成する方法であって、本発明に係る放射性標識コンジュゲートを含む放射性医薬品を前記身体(例えば、脈管系中)に投与する段階、及びPETを用いて前記放射性医薬品が分布した前記身体の少なくとも一部の画像を生成する段階を含んでなる方法を提供する。
【0039】
さらに別の態様から見れば、本発明は、癌に関連した病態(好ましくは血管新生)に対処するための薬物(例えば、細胞毒性薬剤)によるヒト又は動物の身体の治療効果をモニターする方法であって、本発明に係る放射性標識コンジュゲートを前記身体に投与する段階及び細胞レセプター(好ましくは内皮細胞レセプター、特にαvβ3レセプター)による前記コンジュゲートの取込みを検出する段階を含んでなり、前記投与及び検出は任意ではあるが好ましくは(例えば前記薬物による治療前、治療中及び治療後に)繰り返して実施される方法を提供する。
【0040】
本発明のさらに別の態様では、式(II)又は(IV)の補欠分子族及び式(I)又は(III)の化合物を含む放射能フッ素化トレーサーを製造するためのキットが提供される。
【0041】
本発明のさらに別の態様に従えば、式(XX)の補欠分子族及び式(I)の化合物を含む放射能フッ素化トレーサーを製造するためのキットが提供される。本発明の別の態様に従えば、式()又は()の補欠分子族及び式(III)の化合物を含む放射能フッ素化トレーサーを製造するためのキットが提供される。
【0042】
キットの使用に際しては、それぞれ上述した方法を用いて、式()の化合物は対応する式(II)の化合物に転化され、式()又は()の化合物は対応する式(IV)の化合物に転化される。好ましくは、反応混合物をSolid Phase Extraction(SPE)カートリッジに通すことにより、式(II)又は(IV)の化合物を廃反応体から分離できる。SPEカートリッジは、黒鉛パッド、C18固定相又はイオン交換樹脂を含み得る。次いで、式(II)又は(IV)の化合物を、好適には水性緩衝液(pH3〜11)に溶解し得る式(I)又は(III)の化合物にそれぞれ添加する。極端でない温度で1〜70分間反応させた後、標識ペプチドを例えばSPEにより精製して回収することができる。
【実施例】
【0043】
以下の実施例によって本発明を例示するが、実施例中では下記の略語を用いる。
HPLC:高速液体クロマトグラフィー
NMR:核磁気共鳴
hr(s):時間
min(s):分
THF:テトラヒドロフラン
DCM:ジクロロメタン
DMF:N,N−ジメチルホルムアミド
TBAF:フッ化テトラブチルアンモニウム
MeOH:メタノール
DMSO:ジメチルスルホキシド
Boc:t−ブトキシカルボニル
RT:室温
i−Pr2−Net:N,N−ジイソプロピルエチルアミン
t−BDPSiCl:tert−ブチルジフェニルシリルクロリド
NaH:水素化ナトリウム
EtOAc:酢酸エチル
MBq:メガベクレル
次の移動相/勾配系を使用した。溶媒A:水(0.1%TFA)、溶媒B:アセトニトリル(0.1%TFA)。
【0044】
例1
トルエン−4−スルホン酸4−(N−メチル−N−Boc−アミノオキシ)ブチルエステルの製造
【0045】
【化18】
(a)N−Boc−N−メチルヒドロキシルアミン
N−メチルヒドロキシルアミン(4.2g、0.05mol)を50%水性テトラヒドロフラン(THF)(20ml)に溶解し、撹拌しながら氷上で冷却した。氷冷溶液に炭酸カリウム(3.6g、0.0275mol)を添加し、次いで15mlのTHFに溶解したジ−tert−ブチルジカーボネート(12g、0.055mol)を添加した。混合物を0℃で2時間撹拌し、室温で2時間撹拌した。THFを減圧下で除去し、残留物をDCMに溶解した。溶液を水で2回洗浄し、乾燥し(MgSO4)、濃縮することで、ピンク色の低粘度油状物6.47g(88%)を得た。生成物をエレクトロスプレー質量分析法(ESI−MS)によって同定した(MH+計算値147.09、実測値147.6)。生成物はそれ以上精製せずに使用した。
【0046】
(b)(4−ブロモブトキシ)−tert−ブチルジフェニルシラン
i−Pr2−Net(10ml)を含むDCM(10ml)に4−ブロモ−1−ブタノール(2.75g、18mmol)を溶解した溶液に、アルゴン雰囲気下でt−BDPSiCl(5ml、18mmol)を添加した。溶液を室温で2時間撹拌し、真空中で濃縮し、クロマトグラフィー処理(ヘキサン/酢酸エチル 10:1)することで、低粘度で無色の油状物4.39g(62%)を得た。構造をNMRで確認した。
【0047】
(c)O−[4−(tert−ブチルジフェニルシラニルオキシ)ブチル]−N−メチル−N−Boc−ヒドロキシルアミン
N−Boc−N−メチルヒドロキシルアミン(a)(0.74g、5mmol)を10mlのDMFに溶解し、NaH(200mg、鉱油中60%分散液、4.75mmol)で処理し、アルゴン雰囲気下で1時間撹拌した。混合物を0℃に冷却し、(4−ブロモブトキシ)−tert−ブチルジフェニルシラン(b)のDMF(10mL)溶液で処理し、0℃でさらに3時間撹拌した。溶媒を減圧下で除去し、残留物をEtOAc(150mL)に溶解し、分液漏斗内に注入した。有機層を0.1M NaOH(5×50mL)、H2O(50mL)、0.1M KHSO4及びブライン(50mL)で洗浄し、次いでMgSO4で乾燥した。溶媒の除去後、残留物をシリカゲル上でクロマトグラフィー処理(ヘキサン:EtOAc 10:1)して0.588g(24%)の生成物を得た。生成物をエレクトロスプレー質量分析法(ESI−MS)によって同定した(MH+計算値457.26、実測値457.8)。
【0048】
(d)4−(N−メチル−N−Boc−ヒドロキシルアミン)ブタン−1−オール
20mLの乾燥THFに溶解したO−[4−(tert−ブチルジフェニルシラニルオキシ)ブチル]−N−メチル−N−Boc−ヒドロキシルアミン(c)(588mg、1.22mmol)にTBAF(1.6mL、1.586mmol)を添加した。反応物をアルゴン下で一晩撹拌した。溶液にNH4Cl(飽和)(10mL×3)を添加し、THFを蒸発させた。溶液をDCMで抽出し、有機相を乾燥(Na2SO4)し、蒸発させた。残留物をシリカゲル上でクロマトグラフィー処理(ヘキサン/EtOAc 1:1)して0.170g(63%)の生成物を得た。生成物をHPLC(カラム:Phenomenex Luna 3μ C18(2),4.6×50mm、検出:214nm、勾配:10分で50〜100%B(ただし、A=H2O/0.1%TFA及びB=アセトニトリル/0.1%TFA)、流量:2mL/分、Rt=2.70分)によって分析した。NMR分析によって追加の確認を行った。
【0049】
(e)トルエン−4−スルホン酸4−(N−メチル−N−Boc−ヒドロキシルアミン)ブチルエステル
4−(N−メチル−N−Boc−ヒドロキシルアミン)ブタン−1−オール(d)(170mg、0.77mmol)及びトリエチルアミン(161μL、1.155mmol)を10mLの乾燥DCMに溶解した氷浴冷却撹拌溶液に、乾燥DCM(5mL)中のトルエン−4−スルホニルクロリド(190.8mg、1.001mmol)をアルゴン下で添加した。15分後に氷浴を取り除き、反応混合物を室温に放置した。2時間後、新しい反応剤(トリエチルアミン(32μL、0.23mmol)、トルエン−4−スルホニルクロリド(29.36mg、0.154mmol))を添加した。24時間後、TLC上で想定生成物を認めることができる。30時間後、新しい反応剤(トリエチルアミン(53μL、0.385mmol)、トルエン−4−スルホニルクロリド(73.5mg、0.385mmol))を反応混合物に添加し、一晩放置した。有機相を10%NaHCO3(10mL×3)で洗浄し、MgSO4で乾燥した。有機相を真空下で除去し、シリカ上でフラッシュする(ヘキサン:酢酸エチル 6:4)ことで111mg(41.5%)の生成物を得た。NMRは、生成物中におけるトルエン−4−スルホニルクロリド不純物の存在を示した。4−スルホニルクロリドを除去するために追加の精製が必要であった。ヘキサン:EtOAc(8:2)を用いることで、シリカフラッシュカラム上で良好な分離が達成され、4−スルホニルクロリドが除去された。収量:64mg(22%)。生成物をHPLC(カラム:Phenomenex Luna 3μ C18(2),4.6×50mm、検出:214nm、勾配:10分で20〜80%B(ただし、A=H2O/0.1%TFA及びB=アセトニトリル/0.1%TFA)、流量:2mL/分、Rt=2.70分)によって分析した。生成物をエレクトロスプレー質量分析法(ESI−MS)によって同定した(MH+計算値373.16、実測値373.9)。NMR分析によって追加の確認を行った。
【0050】
例2
非放射性標準としてのO−(4−フルオロブチル)−(N−メチル−N−Boc−ヒドロキシルアミン)の製造
【0051】
【化19】
KF(4.64mg、0.080mmol)及びクリプトフィックス(30.1mg、0.080mmol)を乾燥アセトニトリル(0.75mL)に溶解した。混合物を5分間撹拌した後、乾燥アセトニトリル(0.250mL)に溶解した化合物1(15mg、0.040mmol)をアルゴン下で添加した。混合物を60℃で1時間加熱した。1時間後、TLCは反応が完了したことを示した。溶媒を蒸発させ、残留物をシリカ上でフラッシュする(ヘキサン/EtOAc(1:1))ことで4.5mg(51%)の生成物を得た。生成物をHPLC(カラム:Phenomenex Luna 3μ C18(2),4.6×50mm、検出:214nm、勾配:10分で20〜70%B(ただし、A=H2O/0.1%TFA及びB=アセトニトリル/0.1%TFA)、流量:2mL/分、Rt=2.70分)によって分析した。生成物をエレクトロスプレー質量分析法(ESI−MS)によって同定した(MH+計算値221.14、実測値221.7)。構造をNMRで確認した。
【0052】
例3
18F−化合物2の放射合成及び2−ブロモアセトフェノンへのコンジュゲーション
【0053】
【化20】
Raytest社からのSynChrom R&Dモジュール上で放射合成を実施した。N2下で90°に分間加熱することにより、クリプトフィックス222(1mLのアセトニトリル中39.1mg)及び炭酸カリウム(1mLの水中65.7mg)の存在下で18F−フッ化物(1GBq以下)を共沸的に乾燥した。この間に、2×1mLのアセトニトリルを添加して蒸発させた。<40°に冷却した後、トルエン−4−スルホン酸4−(N−メチル−N−Boc−アミノオキシ)ブチルエステル(化合物1)の溶液(1mLのアセトニトリル中3mg)を添加した。反応器を70℃に20分間加熱して標識を行った。60/40 CH3CN/H2Oからなる定組成流を使用しながら、粗反応混合物を214nmでHPLCにかけた。クロマトグラムは、非放射性標準と共溶出する良好な収量の標識化合物(約86%RCP)を示した。2mLのACNを用いて粗反応混合物をSep−Pakアルミニウムカラムから溶出させることで遊離フッ化物を除去し、遊離フッ化物の活量を測定した(遊離フッ化物は15%未満であった)。「精製」混合物をHPLCで再分析したところ、遊離フッ化物は除去されていることがわかった。1mL(100MBq)の精製生成物を1mLの1M HCl中において90℃で20分間加水分解してBOC保護基を除去することで、100%の非保護F−18放射性標識化合物を得た。1mLの加水分解生成物を10mLのミリQ水で希釈し、pHをpH11〜12に調整し、プレコンディショニングを施したSEP−PAK C−18カラムから溶出させた。カラム上の放射能は30.1MBqと測定された。2mLのDMFでカラムから反応バイアル中に溶出させることで、23MBqを有する2mLの液体が得られた。6mgのブロモアセトフェノンを10μLのジイソプロピルエチルアミンと共に添加し、90℃で10分間加熱した。反応混合物をHPLCで分析したところ、「遊離」前駆体に対応するピークより大きい面積(57%)を有する新しいピークが5.46分の位置に溶出したことにより、コンジュゲート形成の証拠が示された。HPLCは次の通りであった。カラム:Xterra(Waters社)5μ C18 4.6×250mm、検出:250nm及び214nm、NaI検出器、勾配:定組成、40%A=H2O及び60%B=アセトニトリル、流量:1mL/分。
【0054】
例4
トルエン−4−スルホン酸2−[2−(N−メチル−N−BOC−アミノオキシ)エトキシ]エチルエステルの製造
【0055】
【化21】
(a)N−Boc−N−メチルヒドロキシルアミン
N−メチルヒドロキシルアミン(4.2g、0.05mol)を50%水性テトラヒドロフラン(THF)(20ml)に溶解し、撹拌しながら氷上で冷却した。氷冷溶液に炭酸カリウム(3.6g、0.0275mol)を添加し、次いで15mlのTHFに溶解したジ−tert−ブチルジカーボネート(12g、0.055mol)を添加した。混合物を0℃で2時間撹拌し、室温で2時間撹拌した。THFを減圧下で除去し、残留物をDCMに溶解した。溶液を水で2回洗浄し、乾燥し(MgSO4)、濃縮することで、ピンク色の低粘度油状物6.47g(88%)を得た。生成物をエレクトロスプレー質量分析法(ESI−MS)(MH+計算値147.09、実測値147.6)及びNMRによって同定した。生成物はそれ以上精製せずに使用した。
【0056】
(b)トルエン−4−スルホン酸2−(2−ヒドロキシエトキシ)エチルエステル
ジエチレングリコール(22g、207.5mmol)のCH2Cl2(100ml)溶液にトリエチルアミン(10.5g、103.75mmol)を添加した。9.89g(51.8mmol)のトルエン−4−スルホニルクロリドを一度に添加した。溶液を室温で1時間撹拌した。DCM中5%MeOHを用いてTLCを実施した。1/2時間後、TLCは生成物の生成物を示した。1時間15分後、反応混合物を0.1M KHSO3及び5%NaHCO3で洗浄した。有機相をNa2SO3で乾燥し、減圧下で蒸発させた。粗反応混合物をシリカゲル上に捕捉し、ヘキサン/EtOAc 50:50を用いたコンビフラッシュ・コンパニオン(Combiflash Companion)装置(330gカラム)上でのフラッシュクロマトグラフィーによって精製した。NMRで構造を確認した。
【0057】
(c)4−トルエンスルホン酸2−[2−(tert−ブチルジフェニルシラニルオキシ)エトキシ]エチルエステル
i−Pr2−Net(6mL、1.2Eq)を含むCH2Cl2(30ml)にトルエン−4−スルホン酸2−(2−ヒドロキシエトキシ)エチルエステル(b)(7.622g、29.28mmol)を溶解した溶液に、アルゴン雰囲気下でt−BDPSiCl(9ml、1.2eq、35.136mmol)及び触媒量のDMAPを添加した。溶液を室温で2日間撹拌した。TLCは反応の完了を示唆した。粗反応混合物を100mLのDCMで希釈し、2×100mLの水及び100mLのブラインで洗浄した。MgSO4を用いて有機相を乾燥し、真空下で蒸発させ、30分で0〜50%酢酸エチルの勾配(EtOAc/ヘキサン)を用いるコンパニオン・コンビフラッシュ(120gカラム)上でのフラッシュクロマトグラフィーによって精製した。外観は透明で粘稠な油状物であった。NMRで構造を確認した。
【0058】
(d)O−{2−[2−tert−ブチルジフェニルシラニルオキシ)エトキシ]エチル}−N−メチル−N−BOC−ヒドロキシルアミン
オーブン乾燥したフラスコ(50mL)に、NaH(鉱油中60%分散液)481mg(12.03mmol)をアルゴン下で加えた。分散液をペンタンで3回洗浄して鉱油を除去した。純NaHに5mLのTHF(乾燥)を添加した。5mLのTHFに溶解した1.53mg(10.43mmol)のN−BOC−N−メチルヒドロキシルアミン(a)をゆっくりと添加した。反応混合物を、それ以上のガス発生が見られなくなるまで1/2時間放置する。溶液を氷浴上に載せ、1/2時間撹拌した後、4g(8.02mmol)の4−トルエンスルホン酸2−[2−(tert−ブチルジフェニルシラニルオキシ)エトキシ]エチルエステル(c)を滴下した。溶液を氷浴上に1/2時間放置し、次いでRTで一晩撹拌した。溶媒を減圧下で除去し、残留物をEtOAc(200mL)に溶解し、分液漏斗内に注入した。有機層を0.1M NaOH(5×50mL)、H2O(50mL)、0.1M KHSO4及びブライン(50mL)で洗浄し、次いでMgSO4で乾燥した。溶媒の除去後、DCM中のメタノール勾配(メタノール:19分で0〜20%)を用いるコンパニオン・コンビフラッシュ上でのフラッシュクロマトグラフィーによって残留物を精製した。NMRで構造を確認した。
【0059】
(e)2−[2−(N−メチル−N−BOC−アミノオキシ)エトキシ]エタノール
乾燥THF(15mL)に溶解したO−{2−[2−tert−ブチルジフェニルシラニルオキシ)エトキシ]エチル}−N−メチル−N−BOC−ヒドロキシルアミン(d)(3g、6.33mmol)にTBAF(6.78mL、6.78mmol)を添加した。反応物を一晩撹拌した。THFを蒸発させ、残留物をDCMに溶解し、NH4Cl(飽和)(40mL)、水及びブラインで洗浄した。有機相を乾燥(MgSO4)し、蒸発させた。残留物をコンパニオン・コンビフラッシュ(40gカラム)上でのフラッシュクロマトグラフィー(勾配:DCM中において20分で0〜5%MeOH)によって精製した。NMRで構造を確認した。
【0060】
(f)トルエン−4−スルホン酸2−[2−(N−メチル−N−BOC−アミノオキシ)エトキシ]エチルエステル
2−[2−(N−メチル−N−BOC−アミノオキシ)エトキシ]エタノール(e)1g(4.25mmol)を乾燥DCM(15mL)に溶解した撹拌溶液に、15mLのDCM中のトリエチルアミン(910mg、1254μL、9mmol)及びトルエン−4−スルホニルクロリド(1620mg、8.5mmol)を添加した。反応混合物をアルゴン下で一晩撹拌した。TLC(ヘキサン/酢酸エチル 6:4)は反応の完了を示した。反応混合物を50mLのDCMで希釈し、有機相を5%NaHCO3、ブライン及び水で洗浄した。有機相をMgSO4上で乾燥し、濾過し、減圧下で蒸発させた。生成物を、ヘキサン/酢酸エチルの勾配を用いるコンパニオン・コンビフラッシュ(40gカラム)によって精製した。NMRで構造を確認した。
【0061】
例5
O−[2−(2−フルオロエトキシ)エチル]−N−メチル−N−BOC−ヒドロキシルアミンの製造
【0062】
【化22】
KF(58mg、1mmol)及びクリプトフィックス(376mg、1mmol)を乾燥アセトニトリル(2mL)に溶解した。混合物を5分間撹拌した後、乾燥アセトニトリル(0.250mL)に溶解した化合物1(200mg、0.51mmol)をアルゴン下で添加した。混合物を80℃で1時間加熱した。1時間後、TLCは反応の完了を示した。減圧を用いて有機相を蒸発させ、DCMに再溶解し、シリカゲル上に捕捉した。シリカゲル上でのフラッシュクロマトグラフィー(ヘキサン/酢酸エチル 1:1)を用いて物質を精製した。収量:83mg(0.35mmol)。NMRで構造を確認した。
【0063】
例6
マレイミド−プロピオニル−Lys−Gly−Phe−Gly−Lys−OHの製造
【0064】
【化23】
HBTU活性化を用いるスローmoc・シングル・カップル(Slowmoc Single Couple)方法に従い、Fmoc−Lys(Boc)−Sasrin樹脂(0.1mmol)を用いて全自動ペプチド合成機(ABI 433A合成機)上でモデルペンタペプチドLys−Gly−Phe−Gly−Lys−OHをアセンブルした。3−(マレイミド)プロピオン酸N−ヒドロキシスクシンイミドエステル(0.52mmol)を用いてマレイミド−プロピオニル酸を手作業でカップリングした。トリイソプロピルシラン及び水を含むトリフルオロ酢酸(95:2.5:2.5 v/v/v)中で、樹脂からのペプチド除去及び側鎖保護基の脱保護を同時に実施した。濾過後、溶液を減圧下で濃縮し、残留物をジエチルエーテルで洗浄した。粗生成物を逆相分取クロマトグラフィー(Phenomenex Luna C18(2)カラム,250×50mm,10μm、勾配:60分で0〜30%溶媒B、流量:50mL/分)で精製して66mg(95%)の純化合物を得た。生成物をLC−MS[Phenomenex Luna C18−(2),50×2.0mm,5μm、勾配:5分で0〜30%溶媒B、流量:0.6mL/分、tR=2.86分]で分析したところ、m/z=687.6(M+H)+、m/z計算値=687.34(M+H)+であった。
【0065】
例7
18F−化合物2の放射合成及び化合物3へのコンジュゲーション
【0066】
【化24】
GE Healthcare社からのTracerLab FxFnモジュール上で放射合成を実施した。N2下で100°に9分間加熱することにより、クリプトフィックス222(1mLのアセトニトリル中39.1mg)及び炭酸カリウム(1mLの水中65.7mg)の存在下で18F−フッ化物(350MBq以下)を共沸的に乾燥した。この間に、2×0.7mLのアセトニトリルを添加して蒸発させた。<50°に冷却した後、トルエン−4−スルホン酸2−[2−(N−メチル−N−BOC−アミノオキシ)エトキシ]エチルエステル(化合物1)の溶液(1mLの乾燥アセトニトリル中5mg)を添加した。反応器を90℃に10分間加熱して標識を行った。粗反応混合物を5mLの水で希釈した。TLC用の試料を採取すると共に、50μLをHPLC(Phenomenex gemine 150mm×4.60 C18 5μm、214nm、254nm及びγ線検出器で検出、15分で20〜80%の溶媒Bを用いた勾配流)にかけた。TLC(1:1 酢酸エチル/ヘキサン)は、約70%という良好な収率の標識化合物を示した(n=3)。化合物は非放射性対照標準と共溶出した。粗反応混合物をさらに4mlの水で希釈し、(5mLのMeCN及び10mLのH2Oでプレコンディショニングした)Oasis HLB Sep−Pakカートリッジに通した。Oasisカートリッジを50mLの25%メタノール水溶液で洗浄し、1.5mLのMeCNを用いて精製化合物を溶出した。MeCN溶出液に0.3mLのエーテル中2N HClを添加し、RTで5分間撹拌してBOC基を定量的に除去した後、有機相をN2流及び減圧下で65℃で3分間蒸発させ、1mLのMeCNを添加した後にさらに3分間蒸発させた。0.8mLの0.4M酢酸ナトリウム緩衝液(pH5)に溶解した5mgの化合物3を乾燥混合物に添加し、反応物を70℃に60分間加熱してコンジュゲーションを行った。0分及び60分に試料を採取した。反応混合物をHPLCで分析したところ、面積(74%)を有する新しいピークが11分の位置にF19−対照標準と共溶出したことにより、化合物3へのコンジュゲート形成の証拠が示された(図1)。HPLCは次の通りであった。Phenomenex gemine 150mm×4.60 C18 5μm、勾配:10分で0〜40%溶媒B及び10〜15分に40%溶媒B。
【0067】
例8
4−(2−ニトロビニル)ベンゾイル−Lys−Gly−Phe−Gly−Lys−OHの製造
【0068】
【化25】
例6に記載したようにしてモデルペンタペプチドLys−Gly−Phe−Gly−Lys−OHをアセンブルした。純DMF中の樹脂(0.2mmol)に58mg(0.3mmol)のトランス−β−ニトロスチレン及び156.5mg(0.3mmol)のPyAOPを添加した後、102μL(0.6mmol)のDIPEAを添加し、反応物を1時間放置した。トリイソプロピルシラン及び水を含むトリフルオロ酢酸(95:2.5:2.5 v/v/v)中で、樹脂からのペプチド除去及び側鎖保護基の脱保護を同時に実施した。濾過後、溶液を減圧下で濃縮し、残留物をジエチルエーテルで洗浄した。粗生成物を逆相分取クロマトグラフィー(Phenomenex Luna C18(2)カラム,250×50mm,10μm、勾配:60分で0〜40%溶媒B、流量:50mL/分)で精製して101mg(70%)の純化合物を得た。生成物をLC−MS[Phenomenex Luna C18−(2),50×2.0mm,5μm、勾配:5分で0〜30%溶媒B、流量:0.6mL/分、tR=4.10分]で分析したところ、m/z=711.3(M+H)+、m/z計算値=711.3(M+H)+であった。
【0069】
例9
化合物6への18F−化合物4のコンジュゲーション
【0070】
【化26】
上記例7に記載したようにして18F−化合物4の放射合成及び精製を実施した。18F−化合物4を含むMeCN溶出液に0.2mLのエーテル中2N HClを添加し、RTで5分間撹拌してBOC基を定量的に除去した後、有機相をN2流及び減圧下で65℃で3分間蒸発させ、1mLのMeCNを添加した後にさらに3分間蒸発させた。0.8mLの0.4M酢酸ナトリウム緩衝液(pH5)に溶解した5mgの化合物6を乾燥混合物に添加し、反応物を30℃に5分間加熱した。反応混合物をHPLCで分析したところ、面積(72%)を有する新しいピークが10.4分の位置に19F−対照標準と共溶出したことにより、コンジュゲート形成の証拠が示された(図2)。HPLCは次の通りであった。Phenomenex gemine 150mm×4.60 C18 5μm、勾配:10分で0〜40%溶媒B及び10〜15分に40%溶媒B。
【0071】
例10
マレイミド−プロピオニル−RDGペプチドの製造
【0072】
【化27】
5mL容器内で50mg(0.04mmol)のペプチドNC100717を1mLのDMFに溶解した。21.3mg(0.1mmol)の3−(マレイミド)プロピオン酸N−ヒドロキシスクシンイミドエステルを13.6μL(0.08mmol)のDIPEAと共に1mLのDMFに溶解し、ペプチド溶液に添加した。反応物をRTで1時間撹拌した。DMFを蒸発させた。粗生成物を逆相分取クロマトグラフィー(Phenomenex Luna C18(2)カラム,250×50mm,10μm、勾配:60分で0〜40%溶媒B、流量:50mL/分)で精製して23mg(40%)の純化合物を得た。生成物をLC−MS[Phenomenex Luna C18−(2),50×2.0mm,5μm、勾配:5分で5〜40%溶媒B、流量:0.6mL/分、tR=2.74分]で分析したところ、m/z=1409.9(M+H)+、m/z計算値=1409.5(M+H)+であった。
【0073】
例11
化合物7への18F−化合物4のコンジュゲーション
【0074】
【化28】
上記例7に記載したようにして18F−化合物4の放射合成及び精製を実施した。MeCN溶出液に0.2mLのエーテル中2N HClを添加し、RTで5分間撹拌してBOC基を定量的に除去した後、有機相をN2流及び減圧下で65℃で3分間蒸発させ、1mLのMeCNを添加した後にさらに3分間蒸発させた。0.6mLの0.4M酢酸塩緩衝液(pH5)+0.4mLのDMFに溶解した5.75mgの化合物7を乾燥残留物に添加した。ペプチド混合物を70℃に加熱し、45分後に試料を採取した。粗反応混合物を放射性HPLCで分析したところ、45分後にペプチド中への18F−シントンの24%取込みが見られ、18F−化合物がその真正標準と共溶出した(図3)。HPLCは次の通りであった。Phenomenex gemine 150mm×4.60 C18 5μm、勾配:10分で0〜40%溶媒B及び10〜15分に40%溶媒B。
【0075】
例11
ベンゾイル−トランス−β−ニトロスチレン−RDGの製造
【0076】
【化29】
40mg(0.032mmol)のペプチドNC100717を2mLのDMFに溶解した。2mLのDMF及び16μL(0.096mmol)に溶解した12.3mg(0.064mmol)のトランス−β−ニトロスチレン及び25mg(0.048mmol)のPyaOPを添加した。混合物を10分間予備活性化した後、それをペプチド(NC100717)に添加した。15分後、LC−MSは反応の完了を示し、反応混合物をMeCN/0.1%TFA(20mL)で奪活した。有機相を減圧下で除去した。
粗生成物を逆相分取クロマトグラフィー(Phenomenex Luna C18(2)カラム,250×50mm,10μm、勾配:60分で5〜45%溶媒B、流量:50mL/分)で精製して12mg(26%)の純化合物を得た。生成物をLC−MS[Phenomenex Luna C18−(2),50×2.0mm,5μm、勾配:5分で5〜40%溶媒B、流量:0.6mL/分、tR=2.74分]で分析したところ、m/z=1433.6(M+H)+、m/z計算値=1433.5(M+H)+であった。
【0077】
例12
化合物8への18F−化合物4のコンジュゲーション
【0078】
【化30】
上記例7に記載したようにして18F−化合物4の放射合成及び精製を実施した。MeCN溶出液に0.2mLのエーテル中2N HClを添加し、RTで5分間撹拌してBOC基を定量的に除去した後、有機相をN2流及び減圧下で65℃で3分間蒸発させ、1mLのMeCNを添加した後にさらに3分間蒸発させた。0.8mLの0.4M酢酸塩緩衝液(pH5)+0.4mLのDMFに溶解した化合物8(5mg)を乾燥残留物に添加した。反応物を30℃で5分間放置した。粗反応混合物を放射性HPLCで分析したところ、5分後にペプチド中への18F−シントンの25%取込みが見られ、12分後に18F−化合物がその真正標準と共溶出した(図4)。HPLCは次の通りであった。Phenomenex gemine 150mm×4.60 C18 5μm、勾配:10分で0〜40%溶媒B及び10〜15分に40%溶媒B。
【0079】
例13
エテンスルホニル酢酸の合成
【0080】
【化31】
250mLの三つ口丸底フラスコ内で1.1g(47.5mmol、1.2eq)のナトリウムを25mLの無水アルコールに溶解した。H2の発生及びナトリウムの消失が完了した後、2−メルカプト酢酸エチル(4.60mL、40.7mmol、1eq)を滴下した。得られた混合物を20分間撹拌した後、57mL(57mmol、1.4eq)の臭化ビニル(THF中1M溶液)を添加した。混合物をオートクレーブに移し、105℃に1時間加熱した。反応混合物を4時間撹拌し続けた。次いで、THF及びEtOHを真空中で蒸発させ、残留物を水(10mL)に溶解し、ジエチルエーテル(5×60mL)で抽出した。有機相をNa2SO4上で乾燥し、XS溶媒を真空中で除去した。次いで、粗物質(2.9g)を10mLの氷酢酸に溶解し、0℃に冷却した。36〜40%過酢酸(7.4mL、40mmol)を添加し、反応混合物を0℃で30分間撹拌した後、さらに2時間にわたって撹拌し続けて温度を室温に上昇させた。再び溶媒を真空下で除去し、粗生成物を自動コンビフラッシュ装置上でのフラッシュクロマトグラフィー(10分でヘキサン中20〜100%酢酸エチル)によって精製した。NMRにより、構造がスルホキシドであることを確認した。すべての画分を集め、スルホキシド(1.5g、9.2mmol)に関してさらに過酢酸(2eq)で酸化してすべての出発原料を消失させた。粗生成物をそれ以上精製せずに使用した。エテンスルホニル酢酸エチルエステルを20mLの0.1M HClに溶解し、混合物を100℃で13時間還流させることでエテンスルホニル酢酸(12.8g、8mmol)を得た。NMRで構造を確認した。
【0081】
エテンスルホニル−アセチル−Lys−Gly−Phe−Gly−Lys−OHの製造
【0082】
【化32】
例6に記載したようにしてモデルペンタペプチドLys−Gly−Phe−Gly−Lys−OHをアセンブルした。純DMF(5mL)中のペプチド結合樹脂(0.1mmol)に75mg(0.5mmol)のエテンスルホニル酢酸及び261mg(0.5mmol)のPyAOPを添加した後、171μL(1mmol)のDIPEAを添加し、反応物を2時間放置した。トリイソプロピルシラン及び水を含むトリフルオロ酢酸(95:2.5:2.5 v/v/v)中で、樹脂からのペプチド除去及び側鎖保護基の脱保護を同時に実施した。濾過後、溶液を減圧下で濃縮し、残留物をジエチルエーテルで洗浄した。粗生成物を逆相分取クロマトグラフィー(Phenomenex Luna C18(2)カラム,250×50mm,10μm、勾配:60分で0〜30%溶媒B、流量:10mL/分)で精製して51mg(76%)の純化合物を得た。生成物をLC−MS[Phenomenex Luna C18−(2),50×2.0mm,5μm、勾配:5分で0〜30%溶媒B、流量:0.6mL/分、tR=2.61分]で分析したところ、m/z=668.4(M+H)+、m/z計算値=668.3(M+H)+であった。
【0083】
【化33】
GE Medical Systems社からのTracerLab FxFnモジュール上で放射合成を実施した。N2流及び真空下で100°に9分間加熱することにより、215μLの水及び785μLのアセトニトリル(総量0.8mL)に溶解した56mgのクリプトフィックス222(K222、4,7,13,16,21,24−ヘキサオキサ−1,10−ジアザビシクロ[8.8.8]ヘキサコサン)及び10mgのK2CO3の存在下で18F−フッ化物(200MBq)を共沸的に乾燥した。この間に、2×0.8mLのアセトニトリルを添加して蒸発させた。<50°に冷却した後、トルエン−4−スルホン酸2−[2−(N−メチル−N−Boc−アミノオキシ)エトキシ]エチルエステル(化合物3)の溶液(1mLの乾燥アセトニトリル中3mg)を添加した。密閉した反応器を90℃に10分間加熱して標識を行った。粗反応混合物を9mlの水で希釈し、(5mLのMeCN及び10mLのH2Oでプレコンディショニングした)Oasis HLB Sep−Pakカートリッジ(Waters社)に通した。Oasisカートリッジを50mLの25%MeOH水溶液で洗浄し、1.5mLのMeCNを用いて精製化合物を溶出した。MeCN溶出液にエーテル中の2N HCl(0.2mL)を添加し、混合物をRTで5分間撹拌してBoc基を定量的に除去した。有機相を真空中及びN2流下で65℃で3分間蒸発させ、次いで1mLのMeCNを添加し、再び真空中で3分間蒸発させた。
【0084】
0.8mLの0.4M酢酸ナトリウム緩衝液(pH5)に溶解した5mgの化合物10を乾燥混合物に添加し、反応物を70℃に10分間加熱してコンジュゲーションを行った。反応混合物をHPLCで分析したところ、コンジュゲート形成の証拠(8.92分の位置に19F−対照標準と共溶出した新しいピーク、収率40%)が示された。HPLCは次の通りであった。Phenomenex gemine 150mm×4.60 C18 5μm、勾配:10分で0〜40%溶媒B及び10〜15分に40%溶媒B。
【特許請求の範囲】
【請求項1】
以下の式(I)の化合物を以下の式(II)の化合物と反応させるか、或いは以下の式(III)の化合物を以下の式(IV)の化合物と反応させてそれぞれ以下の式(V)又は(VI)のコンジュゲートを得ることを含んでなる放射性フッ素化方法。
【化1】
【化2】
(式中、
R1はハロゲン含有部分であり、
R2はN含有アミノオキシ基であり、
R3はN含有アミノオキシ基であり、
R4はハロゲン含有部分である。)
【化3】
(式中、Yはアルキル又はアリールであり、式(II)及び式(V)の化合物中の18F−リンカー基は安定な結合を介して18F原子に結合されたN含有アミノオキシ基を含むシントン群から選択され、式(IV)及び式(VI)の化合物中の18F−リンカー基はハロゲン含有シントンから選択される。)
【請求項2】
R1がハロアセチル基又はフェナシルハロ基である、請求項1記載の方法。
【請求項3】
R4がハロアセチル基又はフェナシルハロ基である、請求項1記載の方法。
【請求項4】
Yがメチルである、請求項1記載の方法。
【請求項5】
式(II)及び式(V)の化合物中の18F−リンカー基が下記のものから選択される、請求項1記載の方法。
【化4】
(式中、
Vはハロゲン原子、好ましくは臭素、塩素又はヨウ素であり、
nは0〜20の整数であり、
ZはO、N又はSである。)
【請求項6】
式(IV)及び式(VI)の化合物中の18F−リンカー基が下記のものから選択される、請求項1記載の方法。
【化5】
(式中、
Vはハロゲン原子、好ましくは臭素、塩素又はヨウ素であり、
nは0〜20の整数であり、
ZはO、N又はSである。)
【請求項7】
以下の式(Ia)の化合物を以下の式(IIa)の化合物と反応させるか、或いは以下の式(IIIa)の化合物を以下の式(IVa)の化合物と反応させてそれぞれ以下の式(Va)又は(VIa)のコンジュゲートを得ることを含んでなる放射性フッ素化方法。
【化6】
【化7】
(式中、R1及びR4はそれぞれ式(I)又は(IV)の化合物に関して上記に定義した通りであり、式(IIa)及び式(IVa)の化合物中のリンカー基は各々C1-60ヒドロカルビル基、好適にはC1-30ヒドロカルビル基であり、任意には1〜30のヘテロ原子、1〜10のヘテロ原子(例えば、酸素又は窒素)を含む。)
【化8】
(式中、リンカー基は式(IIa)又は(IVa)の化合物に関して定義した通りである。)
【請求項8】
リンカー基が、アルキル鎖、アルケニル鎖、アルキニル鎖、芳香環、ポリ芳香環、ヘテロ芳香環、及びエチレングリコールサブユニット又はアミノ酸サブユニット又は炭水化物サブユニットを含むポリマーから選択される、請求項7記載の方法。
【請求項9】
以下の式(VIIa)の化合物を以下の式(VIIb)の化合物と反応させて以下の式(VIIc)の化合物を得ることを含んでなる放射性フッ素化方法。
【化9】
18F−リンカー−V (VIIb)
18F−リンカー−NH(Y)−O−ベクター (VIIc)
(式中、Yはアルキル又はアリール置換基である。)
【請求項10】
化合物(VIIb)を以下の式(VIII)、式(IX)及び式(X)のものから選択することでそれぞれ式(XII)ないし式(XV)の化合物が得られる、請求項9記載の方法。
18F−(CH2CH2O)nCH2CH2−NHCOCH2−Br (VIII)
【化10】
18F−(CH2CH2O)nCH2CH2−NHCOCH2−N(Y)−O−ベクター (XII)
18F−(CH2)n−N(Y)−O−ベクター (XIII)
18F−(C6H4)−CO−CH2−N(Y)−O−ベクター (XIV)
(式中、
nは0〜20の整数であり、
Yはアルキル又はアリール置換基である。)
【請求項11】
以下の式(XVa)の化合物を下記一般式(XVb)の化合物と反応させることを含んでなる放射性フッ素化方法。
【化11】
(式中、Vはハロゲンである。)
18F−リンカー−O−NH(Y) (XVb)
(式中、Yは前出の化合物に関して定義した通りである。)
【請求項12】
式(XVb)の化合物が以下の式(XVI)、式(XVII)及び式(XVIII)の化合物から選択される、請求項11記載の方法。
【化12】
(式中、Y、m及びnは前出の化合物に関して定義した通りである。)
【請求項13】
ベクターがArg−Gly−Aspペプチド又はその類似体である、請求項1記載の方法。
【請求項14】
ベクターが次式のフラグメントを含む、請求項13記載の方法。
【化13】
【請求項15】
ベクターが以下の式(A)のものである、請求項13記載の方法。
【化14】
(式中、X7は−NH2又は次式の基であり、
【化15】
(式中、aは1〜10の整数である。)
R5はペプチドの反応後にリシン残基のε−アミノとアミド結合又は第二アミン結合を形成し、好ましくは下記のリストi〜viiiから選択される。
【化16】
(式中、R6はN−ヒドロキシスクシンイミド又は酸塩化物のような活性エステル活性化基であり、L及びVは前記に記載した通りである。))
【請求項16】
請求項1に定義された式(I)又は(III)の化合物。
【請求項17】
ベクターがArg−Gly−Aspペプチド又はその類似体である、請求項16記載の方法。
【請求項18】
ベクターが次式のフラグメントを含む、請求項16記載の化合物。
【化17】
【請求項19】
ベクターが以下の式(A)のものである、請求項16記載の化合物。
【化18】
(式中、X7は−NH2又は次式の基であり、
【化19】
(式中、aは1〜10の整数である。)
R5はペプチドの反応後にリシン残基のε−アミノとアミド結合又は第二アミン結合を形成し、好ましくは下記のリストi〜viiiから選択される。
【化20】
(式中、R6はN−ヒドロキシスクシンイミド又は酸塩化物のような活性エステル活性化基であり、L及びVは前記に記載した通りである。))
【請求項20】
請求項1に定義された式(V)又は(VI)の化合物。
【請求項21】
ベクターがArg−Gly−Aspペプチド又はその類似体である、請求項20記載の方法。
【請求項22】
ベクターが次式のフラグメントを含む、請求項20記載の化合物。
【化21】
【請求項23】
ベクターが以下の式(A)のものである、請求項20記載の化合物。
【化22】
(式中、X7は−NH2又は次式の基であり、
【化23】
(式中、aは1〜10の整数である。)
R5はペプチドの反応後にリシン残基のε−アミノとアミド結合又は第二アミン結合を形成し、好ましくは下記のリストi〜viiiから選択される。
【化24】
(式中、R6はN−ヒドロキシスクシンイミド又は酸塩化物のような活性エステル活性化基であり、L及びVは前記に記載した通りである。))
【請求項24】
請求項20記載の化合物の有効量を1種以上の薬学的に許容される補助剤、賦形剤又は希釈剤と共に含んでなる放射性医薬品組成物。
【請求項25】
医療用の、請求項20記載の化合物。
【請求項26】
インビボイメージング方法で使用するための放射性医薬品の製造における、請求項20記載の化合物の使用。
【請求項27】
ヒト又は動物の身体の画像を生成する方法であって、請求項20記載の化合物を前記身体に投与する段階、及びPETを用いて前記化合物が分布した前記身体の少なくとも一部の画像を生成する段階を含んでなる方法。
【請求項28】
癌に関連した病態(好ましくは血管新生)に対処するための薬物によるヒト又は動物の身体の治療効果をモニターする方法であって、請求項21記載の化合物を前記身体に投与する段階及び細胞レセプターによる前記化合物の取込みを検出する段階を含んでなり、前記投与及び検出は任意ではあるが好ましくは前記薬物による治療前、治療中及び治療後に実施される、方法。
【請求項1】
以下の式(I)の化合物を以下の式(II)の化合物と反応させるか、或いは以下の式(III)の化合物を以下の式(IV)の化合物と反応させてそれぞれ以下の式(V)又は(VI)のコンジュゲートを得ることを含んでなる放射性フッ素化方法。
【化1】
【化2】
(式中、
R1はハロゲン含有部分であり、
R2はN含有アミノオキシ基であり、
R3はN含有アミノオキシ基であり、
R4はハロゲン含有部分である。)
【化3】
(式中、Yはアルキル又はアリールであり、式(II)及び式(V)の化合物中の18F−リンカー基は安定な結合を介して18F原子に結合されたN含有アミノオキシ基を含むシントン群から選択され、式(IV)及び式(VI)の化合物中の18F−リンカー基はハロゲン含有シントンから選択される。)
【請求項2】
R1がハロアセチル基又はフェナシルハロ基である、請求項1記載の方法。
【請求項3】
R4がハロアセチル基又はフェナシルハロ基である、請求項1記載の方法。
【請求項4】
Yがメチルである、請求項1記載の方法。
【請求項5】
式(II)及び式(V)の化合物中の18F−リンカー基が下記のものから選択される、請求項1記載の方法。
【化4】
(式中、
Vはハロゲン原子、好ましくは臭素、塩素又はヨウ素であり、
nは0〜20の整数であり、
ZはO、N又はSである。)
【請求項6】
式(IV)及び式(VI)の化合物中の18F−リンカー基が下記のものから選択される、請求項1記載の方法。
【化5】
(式中、
Vはハロゲン原子、好ましくは臭素、塩素又はヨウ素であり、
nは0〜20の整数であり、
ZはO、N又はSである。)
【請求項7】
以下の式(Ia)の化合物を以下の式(IIa)の化合物と反応させるか、或いは以下の式(IIIa)の化合物を以下の式(IVa)の化合物と反応させてそれぞれ以下の式(Va)又は(VIa)のコンジュゲートを得ることを含んでなる放射性フッ素化方法。
【化6】
【化7】
(式中、R1及びR4はそれぞれ式(I)又は(IV)の化合物に関して上記に定義した通りであり、式(IIa)及び式(IVa)の化合物中のリンカー基は各々C1-60ヒドロカルビル基、好適にはC1-30ヒドロカルビル基であり、任意には1〜30のヘテロ原子、1〜10のヘテロ原子(例えば、酸素又は窒素)を含む。)
【化8】
(式中、リンカー基は式(IIa)又は(IVa)の化合物に関して定義した通りである。)
【請求項8】
リンカー基が、アルキル鎖、アルケニル鎖、アルキニル鎖、芳香環、ポリ芳香環、ヘテロ芳香環、及びエチレングリコールサブユニット又はアミノ酸サブユニット又は炭水化物サブユニットを含むポリマーから選択される、請求項7記載の方法。
【請求項9】
以下の式(VIIa)の化合物を以下の式(VIIb)の化合物と反応させて以下の式(VIIc)の化合物を得ることを含んでなる放射性フッ素化方法。
【化9】
18F−リンカー−V (VIIb)
18F−リンカー−NH(Y)−O−ベクター (VIIc)
(式中、Yはアルキル又はアリール置換基である。)
【請求項10】
化合物(VIIb)を以下の式(VIII)、式(IX)及び式(X)のものから選択することでそれぞれ式(XII)ないし式(XV)の化合物が得られる、請求項9記載の方法。
18F−(CH2CH2O)nCH2CH2−NHCOCH2−Br (VIII)
【化10】
18F−(CH2CH2O)nCH2CH2−NHCOCH2−N(Y)−O−ベクター (XII)
18F−(CH2)n−N(Y)−O−ベクター (XIII)
18F−(C6H4)−CO−CH2−N(Y)−O−ベクター (XIV)
(式中、
nは0〜20の整数であり、
Yはアルキル又はアリール置換基である。)
【請求項11】
以下の式(XVa)の化合物を下記一般式(XVb)の化合物と反応させることを含んでなる放射性フッ素化方法。
【化11】
(式中、Vはハロゲンである。)
18F−リンカー−O−NH(Y) (XVb)
(式中、Yは前出の化合物に関して定義した通りである。)
【請求項12】
式(XVb)の化合物が以下の式(XVI)、式(XVII)及び式(XVIII)の化合物から選択される、請求項11記載の方法。
【化12】
(式中、Y、m及びnは前出の化合物に関して定義した通りである。)
【請求項13】
ベクターがArg−Gly−Aspペプチド又はその類似体である、請求項1記載の方法。
【請求項14】
ベクターが次式のフラグメントを含む、請求項13記載の方法。
【化13】
【請求項15】
ベクターが以下の式(A)のものである、請求項13記載の方法。
【化14】
(式中、X7は−NH2又は次式の基であり、
【化15】
(式中、aは1〜10の整数である。)
R5はペプチドの反応後にリシン残基のε−アミノとアミド結合又は第二アミン結合を形成し、好ましくは下記のリストi〜viiiから選択される。
【化16】
(式中、R6はN−ヒドロキシスクシンイミド又は酸塩化物のような活性エステル活性化基であり、L及びVは前記に記載した通りである。))
【請求項16】
請求項1に定義された式(I)又は(III)の化合物。
【請求項17】
ベクターがArg−Gly−Aspペプチド又はその類似体である、請求項16記載の方法。
【請求項18】
ベクターが次式のフラグメントを含む、請求項16記載の化合物。
【化17】
【請求項19】
ベクターが以下の式(A)のものである、請求項16記載の化合物。
【化18】
(式中、X7は−NH2又は次式の基であり、
【化19】
(式中、aは1〜10の整数である。)
R5はペプチドの反応後にリシン残基のε−アミノとアミド結合又は第二アミン結合を形成し、好ましくは下記のリストi〜viiiから選択される。
【化20】
(式中、R6はN−ヒドロキシスクシンイミド又は酸塩化物のような活性エステル活性化基であり、L及びVは前記に記載した通りである。))
【請求項20】
請求項1に定義された式(V)又は(VI)の化合物。
【請求項21】
ベクターがArg−Gly−Aspペプチド又はその類似体である、請求項20記載の方法。
【請求項22】
ベクターが次式のフラグメントを含む、請求項20記載の化合物。
【化21】
【請求項23】
ベクターが以下の式(A)のものである、請求項20記載の化合物。
【化22】
(式中、X7は−NH2又は次式の基であり、
【化23】
(式中、aは1〜10の整数である。)
R5はペプチドの反応後にリシン残基のε−アミノとアミド結合又は第二アミン結合を形成し、好ましくは下記のリストi〜viiiから選択される。
【化24】
(式中、R6はN−ヒドロキシスクシンイミド又は酸塩化物のような活性エステル活性化基であり、L及びVは前記に記載した通りである。))
【請求項24】
請求項20記載の化合物の有効量を1種以上の薬学的に許容される補助剤、賦形剤又は希釈剤と共に含んでなる放射性医薬品組成物。
【請求項25】
医療用の、請求項20記載の化合物。
【請求項26】
インビボイメージング方法で使用するための放射性医薬品の製造における、請求項20記載の化合物の使用。
【請求項27】
ヒト又は動物の身体の画像を生成する方法であって、請求項20記載の化合物を前記身体に投与する段階、及びPETを用いて前記化合物が分布した前記身体の少なくとも一部の画像を生成する段階を含んでなる方法。
【請求項28】
癌に関連した病態(好ましくは血管新生)に対処するための薬物によるヒト又は動物の身体の治療効果をモニターする方法であって、請求項21記載の化合物を前記身体に投与する段階及び細胞レセプターによる前記化合物の取込みを検出する段階を含んでなり、前記投与及び検出は任意ではあるが好ましくは前記薬物による治療前、治療中及び治療後に実施される、方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2010−539091(P2010−539091A)
【公表日】平成22年12月16日(2010.12.16)
【国際特許分類】
【出願番号】特願2010−524233(P2010−524233)
【出願日】平成20年9月9日(2008.9.9)
【国際出願番号】PCT/US2008/075684
【国際公開番号】WO2009/035959
【国際公開日】平成21年3月19日(2009.3.19)
【出願人】(305040710)ジーイー・ヘルスケア・リミテッド (99)
【Fターム(参考)】
【公表日】平成22年12月16日(2010.12.16)
【国際特許分類】
【出願日】平成20年9月9日(2008.9.9)
【国際出願番号】PCT/US2008/075684
【国際公開番号】WO2009/035959
【国際公開日】平成21年3月19日(2009.3.19)
【出願人】(305040710)ジーイー・ヘルスケア・リミテッド (99)
【Fターム(参考)】
[ Back to top ]