
放射性標識化ペプチドの製造方法
本発明は、放射性標識化ペプチドの製造方法に関するものであり、ここで、前駆体分子は有機溶媒中に準備され;カルボキシル官能基を有する放射性標識化化合物が添加され;カルボキシル官能基が活性化され;かつ、放射性標識化ペプチドを形成するために、活性化された放射性標識化化合物が前駆体分子に結合され;放射性標識化化合物はイソシアノカルボン酸である。本発明は、さらに、放射性標識化ペプチドを製造するための、放射性標識化イソシアノカルボン酸の使用に関する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は放射性標識化炭素化合物の技術分野に関する。特に、本発明は、放射性標識化ペプチドの製造方法、さらには放射性標識化ペプチドを製造するための放射性標識化イソシアノカルボン酸の使用に関する。
【背景技術】
【0002】
固相および液相化学の発達により、現在、3000〜10000Daの分子量および99.5%を超えるカップリング収率を有する、配列により定義された(sequence-defined)ペプチドおよびタンパク質の合成が可能になっている。1963年にMerrifieldによって考案された固相合成において、合成すべきペプチドは、架橋ポリマーで作られた不溶性担体樹脂にリンカーすなわち開裂可能なアンカー基を介してカップリングされる。アミノ酸は、最後に結合されたアミノ酸のアミノ官能基に、それぞれの場合に、活性化されたC末端に所望される順序および大過剰で逐次的にカップリングされる。残存している反応体および副生物は、中間体または生成物が不溶性樹脂に拘束されているので、反応容器から洗い流すことができる。最後のステップで、リンカーは樹脂から開裂され、それゆえ、ペプチドは遊離形態で存在する。
【0003】
固相および液相ペプチド合成は、複雑な保護基の化学に基礎をおく。アミノ酸は、保護しなければカルボン酸の活性化後にそれ自体と反応するので、カップリングのためには、アミノ酸のα-アミノ保護基を一時的に保護しなければならない。カップリングの後、さらなるカップリングを進行させることができるように、この保護基を温和な条件下で迅速に除去しなければならない。
【0004】
生体中で生理学的活性があり、かつその化学構造中に1つまたは複数の放射性核種が組み込まれたペプチドおよびタンパク質は、放射性医薬品を製造するための主成分を提供する。生体は、少なくとも、化学的に同一の放射性医薬品の場合、放射性医薬品と放射性標識化されていない対応化合物とを区別せず、それゆえ、放射性医薬品は生理学的に代謝される。放射性核種の崩壊に基づいて、放射性医薬品を追跡し、目に見えるように表示することができる。
【0005】
放射性標識化ペプチドは、生体の断面像を生み出す核医学の1つの方法である陽電子放射断層撮影法(PET)のための貴重なトレーサーである。PETでは、記録された崩壊事象の時間および空間的分布から、体内での放射性医薬品の空間的分布に関して判定することができ、吸収、分布、代謝および排泄などの過程を図解することができる。
【0006】
放射線医薬品の適用可能性は、典型的には2時間未満である放射性核種の短い半減期によって限定される。放射性核種11Cは、わずかにほぼ20分であるとりわけ短い半減期を所持する。望ましくない放射能減少は、サイクロトロン中での放射性核種の製造の際に既に始まり、放射性医薬品の製造、そのPET現場および最終的には患者への投与までの送達、ならびに測定の間中、継続する。
【0007】
最大の可能送達範囲(送達すべき陽電子放射断層撮影装置はサイクロトロンの周囲に配置される)を達成するには、放射性医薬品の可能な限り高い放射能が、その製造後に存在すべきである。このことは、所定の放射性核種の放射能損失は時間に依存するので、放射性核種からの放射性医薬品の極めて短い製造時間によって達成することができる。
【0008】
しかし、ペプチドのほとんどの放射性標識化法は、多段階で時間がかかり、自動化が困難であり、低い放射化学収率で行われるだけである。放射性医薬品を製造するための従来法は、例えば、アミンまたはカルボン酸およびアミノ酸を11Cで放射化して標識化するのに、メチル化剤11CH3Iを介する経路を利用する(Denutteら、1983年; VandersteeneおよびSledgers、1996年)。しかし、この方法では、11CH3Iを形成するために、サイクロトロン中で製造された11CO2を、LiALH4およびHIとの2段階法でさらに反応させなければならない。第3段階で、ようやく、放射性標識化されたメチル化剤を標識される予定の医薬品に移転することができる。放射性医薬品のこの冗長な合成のため、11CO2によって初めに提供された高い放射能比率が失われる。
【0009】
新規なPET造影剤のための重要な技術が、「クリックケミストリー」と称されるものによって、18F標識化の分野に導入された。この方法は、単一ステップでの放射性造影剤の合成を可能にする(Devarajら、2009年;Liら、2007年)。Bruus-Jensen(2006年)による研究では、HYNIC-官能化ペプチドおよびタンパク質が、18F-および99mTc-標識化放射性医薬品を合成するための前駆体として使用される。しかし、例えば、18F-6-フルオロ-DOPAまたは18F-フルオロ-2-デオキシ-D-グルコースなどのテクネチウムまたはフッ素で標識された放射性医薬品は、生体中で、類似のその標識されていない元々の分子と異なる。
【先行技術文献】
【非特許文献】
【0010】
【非特許文献1】Bruus-Jensen, Dissertation, INSTITUTE FOR RADIOCHEMISTRY OF THE TECHNICAL UNIVERSITY OF MUNICH, 2006 (introduction)
【非特許文献2】Denutte et al., J Nucl Med 24, 1185-1187, 1983
【非特許文献3】Abstract of Devaraj et al., Bioconjugate Chem 20(2), 397- 401, 2009
【非特許文献4】Abstract of Li et al., Bioconjugate Chem, 18(6), 1987-1994, 2007
【非特許文献5】First page of Merrifield, J Am Chem Soc, 85, 2149-2154, 1963
【非特許文献6】Abstract of Vandersteene and Slegers, Applied Radiation and Isotopes 47 (2), 201-205, 1996
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明の目的は、短い製造時間内で高収率の放射性標識化された天然および人工ペプチドをもたらす、効率的かつ迅速な合成方法を提供することである。
【課題を解決するための手段】
【0012】
この目的は、次のステップ:
(a)アミノ酸、ペプチドおよび第一級アミンからな群から選択される前駆体分子を有機溶媒中で準備するステップ;
(b)カルボキシル官能基を含む前駆体分子に放射性標識化化合物を添加するステップ;
(c)放射性標識化化合物のカルボキシル官能基を活性化するステップ;および
(d)放射性標識化ペプチドを得るために、活性化された放射性標識化化合物を前駆体分子に連結するステップ;
を含み、ここで、放射性標識化化合物がイソシアノカルボン酸である、放射性標識化ペプチドの製造方法によって達成される。
【0013】
さらに、本発明は、放射性標識化ペプチドを製造するための、放射性標識化イソシアノカルボン酸の使用に関する。
【0014】
従属特許請求項は、本発明の有利な実施形態を包含する。
【図面の簡単な説明】
【0015】
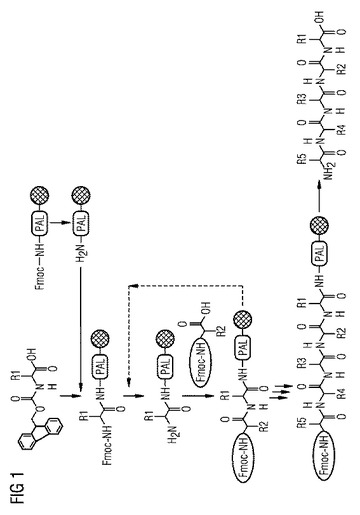
【図1】アミノ酸を使用する従来の固相ペプチド合成を示す図であり、そのアミノ官能基は、保護基9-フルオレニルメトキシカルボニル(Fmoc)でブロックされている。
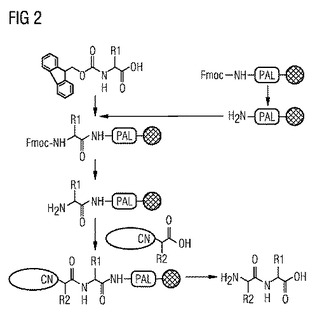
【図2】標識化イソシアノカルボン酸を使用する本発明による固相ペプチド合成を示す図である。
【発明を実施するための形態】
【0016】
本発明は、放射性標識化ペプチドの製造方法に関するものであり、該方法は、次のステップ:
(a)アミノ酸、ペプチドおよび第一級アミンからなる群から選択される前駆体分子を有機溶媒中で準備するステップ;
(b)カルボキシル官能基を含む前駆体分子に放射性標識化化合物を添加するステップ;
(c)放射性標識化化合物のカルボキシル官能基を活性化するステップ;および
(d)放射性標識化ペプチドを得るために、活性化された放射性標識化化合物を前駆体分子に連結するステップ;
を含み、ここで、放射性標識化化合物はイソシアノカルボン酸である。
【0017】
表現「ペプチド」は、本明細書中で使用する場合、アミドまたはペプチド結合を介して互いに連結された少なくとも2つのアミノ酸から構成されている有機化合物を意味する。表現「ペプチド」は、10個までのアミノ酸からなるオリゴペプチド、10個を超えるアミノ酸からなるポリペプチド、および100個を超えるアミノ酸からなるマクロペプチド、さらには独立に一次、二次、三次および四次構造のタンパク質を包含する。ペプチドは、人工の、および天然に存在する有機化合物を包含する。それらは、化学的に、さらには生合成によって製造することができる。
【0018】
表現「前駆体分子」は、本明細書中で使用する場合、アミノ酸、ペプチドおよび/または式RNH2の第一級アミンを包含する、ペプチド合成に関するモノマー、オリゴマーおよびポリマー性出発分子を意味する。
【0019】
表現「カルボキシル官能基」は、本明細書中で使用する場合、式-COOHを有するカルボン酸、または式-COO-を有するカルボネートの官能基を意味する。
【0020】
表現「カルボキシル官能基を活性化する」は、本明細書中で使用する場合、カルボン酸を反応性のある物質に転換することを意味する。
【0021】
表現「イソシアノカルボン酸」は、本明細書中で使用する場合、カルボキシル基-COOHまたはカルボキシレート-COO-とイソシアノ基CN-とを含む有機化合物を意味する。イソシアノカルボン酸は、例えば、実験式CNR1R2CCOOHまたはCNR1R2CCOOXを所持する。
【0022】
基R、R1、R2などは、本明細書中で使用する場合、同一または異なる、芳香族、ヘテロ芳香族および脂肪族基、さらには窒素化合物、ハロゲン化合物、ならびに水素基を意味する。脂肪族基は、非環式の分枝および非分枝の、環状および脂環式の、飽和および不飽和炭素化合物を包含する。Xは、アルカリ金属およびアルカリ土類金属のイオンなどの金属イオン、例えば、リチウムイオンを包含する。
【0023】
本発明による方法で放射性標識化ペプチドを製造する場合、放射性標識化イソシアノカルボン酸が使用される。アミノ酸のアミノ基とは対照的に、イソシアノカルボン酸のイソシアノ基は、ペプチド合成における保護基でブロックされてはならない。したがって、アミノ酸の連結による従来の合成の際に、1)さらなる反応ステップでアミノ基に保護基を付加するために、および2)ペプチドにアミノ酸を連結した後にさらなる反応ステップで保護基を再度除去するために必要とされる貴重な反応時間が省かれる。したがって、ペプチド合成に費やされる時間が、本発明の方法により短縮される。
【0024】
本発明による方法の短縮されたペプチド合成時間のため、放射性核種の時間の関数としての自然崩壊による放射化学収率(radiochemical yield)の損失が減少する。したがって、放射性標識化ペプチドの合成で使用される放射性出発物質の量を減らすことができ、このことがコストを節減し、合成中の放射化学者の被曝量(burden)を軽減する。
【0025】
放射性標識化イソシアノカルボン酸による放射性標識化ペプチドの製造は、極めて単純であり、かつ比較的低い費用で実施することができる。したがって、本発明による方法は、臨床または放射線医学の実践で直ちに採用することができる。
【0026】
好ましい実施形態において、有機溶媒は、塩化メチレン、クロロホルム、ジクロロエタン、ジメチルホルムアミド、ジメチルアセトアミド、テトラヒドロフラン、酢酸エチル、アセトニトリル、および/またはこれらの組合せを含む。
【0027】
さらなる好ましい実施形態において、放射性標識化イソシアノカルボン酸は、放射性標識化された炭素、好ましくは11Cを含む。
【0028】
イソシアノカルボン酸は、CNR1R2CHまたはCNR1R2CXを放射性標識化二酸化炭素、例えば、11CO2を使用してカルボキシル化することによって製造される。したがって、放射性核種の組込みは、放射性標識化イソシアノカルボン酸を合成する最後のステップにおいて行われ、そのイソシアノカルボン酸を本発明によるペプチド合成でそのまま使用することができる。したがって、イソシアノカルボン酸の合成において、既に反応時間が節減され、それによって、放射性核種の崩壊による放射化学収率の損失が減少し、使用される放射性標識化イソシアノカルボン酸の量が減少する。次には、それによりコストが節減され、ペプチドの合成中の放射化学者の被曝量が減少する。
【0029】
好ましい実施形態において、本発明による方法は、
(a1)そのそれぞれがカルボキシル官能基を含むさらなるアミノ酸または非標識化イソシアノカルボン酸を、準備した前駆体分子に添加するステップ;
(a2)さらなるアミノ酸または非標識化イソシアノカルボン酸のカルボキシル基を活性化するステップ;
(a3)準備した前駆体分子をさらなるアミノ酸または非標識化イソシアノカルボン酸に、ペプチドを形成するためのアミド結合を介して連結するステップ;および
(a4)ステップ(a1)〜(a3)を、ペプチドの所望の大きさが達成されるまで繰り返すステップ;
をさらに含む。
【0030】
本発明による方法のステップ(a)に続く実施形態のステップ(a1)〜(a4)において、オリゴペプチドおよびポリペプチドなどのオリゴマーおよびポリマー性前駆体分子は、さらなるモノマー性アミノ酸を前駆体分子、すなわちアミノ酸、ペプチドまたは第一級アミンに添加することによって合成される。したがって、本発明による方法では、放射性標識化イソシアノカルボン酸を任意の所望の長さの前駆体分子に添加することができる(前記ステップ(b)参照)。
【0031】
好ましい実施形態において、イソシアノカルボン酸は、α-イソシアノカルボン酸である。表現「α-イソシアノカルボン酸」は、本明細書中で使用する場合、同一炭素原子上にカルボキシル基またはカルボキシレートとイソシアノ基とを含む有機化合物を意味する(IUPAC名:2-イソシアノカルボン酸)。
【0032】
好ましい実施形態において、カルボキシル官能基を活性化するステップは、イソシアノカルボン酸および/またはさらなるアミノ酸を、活性エステル、無水物、ペンタフルオロフェニルエステル、チオエステル、イミダゾリド、ハロゲン化アシル、および/またはジメチルアミノピリジンを包含する反応性のある物質に転換するステップを含む。
【0033】
さらなる好ましい実施形態では、イソシアノカルボン酸を、活性エステルを得るためのカップリング試薬と反応させる。ここで、カップリング試薬は、グアニジニウム試薬、ウロニウム試薬、好ましくは2-(H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)、O-(ベンゾトリアゾール-1-イル)-N,N,N',N'-テトラメチルウロニウムテトラフルオロボレート(TBTU)または2-(1H-7-アザベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HATU)、ベンゾトリアゾール試薬、好ましくは1-ヒドロキシベンゾトリアゾール試薬(HOBt)、インモニウム試薬、カルボジイミド試薬、好ましくはN,N'-ジシクロヘキシルカルボジイミド(DCC)またはジイソプロピルカルボジイミド(DIPCDI)、イミダゾリウム試薬、オルガノホスホラス試薬、酸性ハロゲン化試薬、ホスホニウム試薬、好ましくはベンゾトリアゾール-1-イル-オキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP)(カストロ試薬(Castro reagent)としても知られている)またはベンゾトリアゾール-1-イル-オキシトリピロリジノホスホニウムヘキサフルオロホスフェート(PyBOP)、モルホリン試薬、好ましくはN-メチルモルホリン(NMM)、クロロフォルメート試薬、および/またはこれらの組合せを包含する。
【0034】
HOBtが、ペプチド結合の形成を加速し、ラセミ化を抑制し、例えばAsnおよびGln側鎖基が脱水されない効果を有するので、有利である。DCCを使用して、アミノ酸を、クロマトグラフィーに供して単離することができるほど十分に安定である活性エステルにインサイチュで転換することができる。
【0035】
とりわけ好ましくは、HOBtとDCCの混合物が使用される。DCCは、高度に反応性のあるアシルイソウレアを形成してカルボン酸を活性化する。これは、HOBtと反応して、極めて大きな初期反応性を保存するHOBt-活性エステルを形成する。HOBt-活性エステルは、ペプチドまたはさらなるアミノ酸のアミノ官能基によって求核的に攻撃される。水を除去すると、ペプチド結合が形成される。アシルイソウレアの直接反応は、ラセミ化が起こるほど十分に反応性が高いので、それほど勧められない。HOBtへDIPEAを添加することは、HOBtのラセミ化への傾向を低減する触媒として作用する。
【0036】
この反応で生成するウレアは、より可溶性で、より容易に分離することができるので、DCCの代わりに、DIPCDIが、HOBtと一緒に頻繁に使用される。
【0037】
BOPまたはHBTUとHOBtの混合物に加えて与えることがとりわけ好ましい。BOPまたはHBTUの対イオンが、例えば、PF-のように極めて低い求核性を有することが重要である。BOP試薬は、安定で非吸湿性であり、有機溶媒に極めて容易に溶解する。一般に、BOP試薬は、DCC/HOBtの組合せに比べてより効率的である。しかし、BOP試薬の欠点は、反応中に、発癌性のヘキサメチルリン酸トリアミドが形成されることである。新規な試薬PyBopは、対照的に、発癌性を示さない。この試薬は、メチル基の代わりにピロリジン単位を所持する。
【0038】
試薬HBTUおよびHATUは、とりわけ高い反応性を有する。アシルクロリドおよびアシルフルオリドは、容易に入手可能で安価な試薬である。
【0039】
好ましい実施形態において、前駆体分子の、イソシアノカルボン酸のおよび/またはさらなるアミノ酸の側鎖の官能基は、保護基でブロックされる。官能基は、ヒドロキシル官能基、カルボキシル官能基、およびアミノ官能基を包含する。
【0040】
好ましい実施形態において、前駆体分子の主鎖のカルボキシル官能基および/またはアミノ官能基、ならびに/あるいはさらなるアミノ酸の主鎖のアミノ官能基は、保護基でブロックされる。
【0041】
表現「保護基」は、本明細書中で使用する場合、化学基に結合することによって、それら化学基の反応性を目標を定めた方式でブロックおよび遮断する化合物を包含する。表現「側鎖」は、本明細書中で使用する場合、前駆体分子であるイソシアノカルボン酸またはさらなるアミノ酸の主要ストランドから分枝している基R1、R2などを包含する。表現「主鎖」は、本明細書中で使用する場合、前駆体分子であるイソシアノカルボン酸またはさらなるアミノ酸からなる軸または幹を形成するN-およびC-で終結する骨格を意味する。
【0042】
好ましい実施形態において、保護基は、塩基に安定な保護基、t-ブチルオキシカルボニル保護基(Boc)および/または9-フルオレニルメトキシカルボニル保護基(Fmoc)を包含する。
【0043】
N末端をFmocで保護した場合、例えば、塩基に安定、酸に不安定な保護基を側鎖に対して使用する。その例が、例えばリシン中のアミノ官能基を保護するBoc、例えばアスパラギン酸およびセリン中のカルボキシル基およびヒドロキシル基を保護するtert-ブチル、および例えばグルタミン中のアミドを保護するトリチルである。
【0044】
好ましい実施形態において、本発明による方法は、
(a1)準備した前駆体分子の主鎖のアミノ官能基から保護基を除去するステップ;
(a2)その主鎖のアミノ基が保護されているさらなるアミノ酸を、前駆体分子に付加するステップ;
(a3)さらなるアミノ酸の主鎖のカルボキシル官能基を活性化するステップ;
(a4)前駆体分子をさらなるアミノ酸に、ペプチドを形成するためのアミド結合を介して連結するステップ;および
(a5)ステップ(a1)〜(a4)を、ペプチドの所望の大きさが達成されるまで繰り返すステップ;
をさらに含む。
【0045】
本発明による方法のステップ(a)に続く実施形態のステップ(a1)〜(a5)において、オリゴペプチドおよびポリペプチドなどのオリゴマーおよびポリマー性前駆体分子が、さらなるモノマー性アミノ酸を前駆体分子、すなわちアミノ酸、ペプチドまたは第一級アミンに添加することによって合成される。前駆体分子のN末端アミノ官能基は、除去可能な保護基でブロックされる。したがって、本発明による方法では、放射性標識化イソシアノカルボン酸を、保護基を備えた任意の所望の長さの前駆体分子に付加することができる(前記ステップ(b)参照)。
【0046】
好ましい実施形態において、Fmoc保護基は、アンモニア、第一級アミンまたは第二級アミン、好ましくは4-アミノメチルピペリジン、ピペリジン、またはトリス(2-アミノエチル)アミンを用いて除去される。
【0047】
好ましい実施形態において、t-ブチルオキシカルボニル保護基は、プロトンによって除去される。
【0048】
好ましい実施形態において、本発明による方法は、前駆体分子に連結されたイソシアノカルボン酸を加水分解するステップをさらに含む。イソシアノ基は、それによって、官能性アミノ基に転換される。ペプチド合成を、このアミノ基の位置で終結または継続することができる。したがって、放射性核種は、ペプチド鎖内に、またはペプチド鎖の一端に配置することができる。さらに、本発明により合成された放射性標識化ペプチドは、1つまたは複数の放射性核種を含むことができる。
【0049】
さらなる好ましい実施形態において、本発明による方法は、放射性標識化ペプチドから保護基を除去するステップをさらに含む。ペプチド合成の完結後に、例えば、条件しだいで、酸に安定な保護基をHFなどのハロゲン化水素で除去し、酸に不安定な保護基をトリフルオロ酢酸(TFA)で除去する。
【0050】
好ましい実施形態では、前駆体分子が、固相にカップリングされる。
【0051】
表現「固相」は、本明細書中で使用する場合、その合成中にペプチドが拘束されるポリマー性固体支持体を意味する。この固定化により、使用される物質を、大過剰で添加し、かつ極めて迅速に洗い流し、それによって、本発明による方法においてカップリングの収率は、著しく高められる。固相化学は、さらに、ペプチド合成を自動化することを可能にする。例えば、固相でのペプチド合成において、前駆体分子、モノマーおよび試薬を添加するステップ、カルボキシル官能基を活性化するステップ、前駆体分子およびモノマーを連結するステップ、および一時的保護基を除去するステップの逐次的繰り返しが実施される。
【0052】
固相は、ポリマー性支持体とペプチドの間の結合要素であるリンカーを含む。
【0053】
とりわけ好ましい実施形態において、固相は、ポリスチレン樹脂、2',4'-ジメトキシフェニルヒドロキシメチルフェノキシ樹脂、p-メチルベンズヒドリルアミン樹脂、フェナルアセトアミドメチル樹脂、および/またはオキシム樹脂である。
【0054】
ポリスチレンは容易に膨潤し、それゆえ、試薬は、合成部位に容易に到達する。さらに、それは、試薬に対して不活性である。ポリスチレンは、好ましくは、1%の架橋用m-ジビニルベンゼンとの混合物である。官能化は、例えば、クロロメチル化を介して達成される。クロロメチル基と最初のアミノ酸との間には、有利には、合成の終末時点で完成したペプチドを樹脂から脱離させることを可能にするリンカー、例えばp-アルコキシベンジルエステルが配置される。Fmoc-ペプチド合成の文脈では、アルコキシベンジルエステルリンカーを有するポリスチレン樹脂が、好ましくは使用され、C末端カルボン酸の合成を可能にし、クロロメチルポリスチレンを4-ヒドロキシベンジルアルコールと反応させることによって製造される。完成したペプチドは、TFAを使用して開裂される。
【0055】
2',4'-ジメトキシフェニルヒドロキシメチルフェノキシ樹脂は、アミドまたは酸樹脂(acid resin)として使用される。アミド樹脂は、C末端アミドをもたらし、Fmoc-合成に使用される。
【0056】
酸樹脂は、N末端Boc保護基によるペプチド合成を可能にする。酸樹脂は、C末端カルボン酸をもたらす。酸樹脂のリンカーは、ちょうどクロロトリチルリンカーのように、例えば希薄TFAを使用して、ペプチドを樹脂から保護された形態で開裂させることができるほど十分に酸に不安定である。これらのフラグメントを、次いで、フラグメントの縮合で使用することができる。
【0057】
p-メチルベンズヒドリルアミン樹脂およびフェナルアセトアミドメチル樹脂は、Boc-ペプチド合成の文脈で使用される。完成したペプチドの脱離は、HFを使用して最後に行われる。p-メチルベンズヒドリルアミン樹脂を使用すると、ペプチドアミドが得られる。フェナルアセトアミドメチル樹脂は、カルボン酸をもたらす。
【0058】
オキシム樹脂により、完全にBocで保護されたペプチドを製造することができる。脱離は、NH3またはH2N-NH2を使用して行われる。
【0059】
好ましい実施形態において、本発明による方法は、放射性標識化ペプチドを固相からデカップリングするステップをさらに含む。
【0060】
放射性標識化ペプチドを遊離させるため、ペプチド合成の完結後に、該ペプチドを固相または固相のリンカーからデカップリングする。この除去は、N末端Boc保護基の場合にはHFを使用して、N末端Fmoc保護基の場合にはほぼ80%濃度(strength)のTFAを使用して達成される。これらの異なる濃度の酸に加えて、例えば、ニトロベンジル樹脂は、Pd(O)と共に光およびアリル樹脂を用いて直交的に除去される。
【0061】
好ましい実施形態において、加水分解は、固相からの前駆体分子のデカップリングと同時に行われる。
【0062】
さらなる好ましい実施形態において、加水分解は、固相からの放射性標識化ペプチドのデカップリング、および放射性標識化ペプチドからの保護基の除去と同時に行われる。
【0063】
同時的なデカップリングおよび加水分解、あるいはデカップリング、加水分解および除去は、処理時間を短縮し、そのため放射化学収率の損失を低減する。したがって、合成で使用される放射性イソシアノカルボン酸の量を減らすことができる。
【0064】
同時的な加水分解および固相からの放射性標識化ペプチドのデカップリング、さらにはBoc保護基の除去は、液状HF、トリフルオロメタンスルホン酸/トリフルオロ酢酸、またはHBr/酢酸を使用して達成される。Fmoc保護基の場合、TFA(ほぼ80%濃度)/CH2Cl2が使用される。好ましくは、ペプチドを傷害することのある反応性中間体を捕捉するために、アニソール、エタンジチオールまたはジメチルスルフィドなどのトラッピング試薬が添加される。
【0065】
さらなる態様において、本発明は、放射性標識化ペプチドを製造するための、放射性標識化イソシアノカルボン酸の使用に関する。
【0066】
図1は、ペプチドを合成するための従来の固相合成を示す。前駆体分子は、Fmocで保護され、固相(二重平行線の円)にアミノメチル-3,5-ジメトキシフェノキシバレリルリンカー(PAL)を介してカップリングされている第一級アミンである。Fmocは、アミンから塩基によって除去され、添加されるアミノ酸は、HOBtエステルとして活性化される。次いで、アミノ酸(そのアミノ基は、同様に、Fmocで保護されている)は、固定化されたアミンに連結される。さらなるステップにおいて、今度はFmoc基が、固定化された前駆体分子のアミノ基から除去され、添加され活性化されたさらなるアミノ酸が、前駆体分子に連結される。これらのステップが、ペプチドの所望の長さが達成されるまで繰り返される。最後に、保護基が除去され、ペプチドが固相からデカップリングされる。
【0067】
図2は、放射性標識化イソシアノカルボン酸による放射性標識化ペプチドの本発明による合成を概略的に示す。まず、前駆体分子が、Fmocアミノ酸および固相にカップリングされたアミンから常法で合成される。(図1参照)。活性化された放射性標識化α-イソシアノカルボン酸が、前駆体分子に結合され、α-イソシアノカルボン酸のCN基を保護する必要がないので、反応時間が節減される。次いで、CN基が加水分解されてNH2アミノ基が生成し、同時に、合成された放射性標識化ペプチドが固相からデカップリングされる。
【技術分野】
【0001】
本発明は放射性標識化炭素化合物の技術分野に関する。特に、本発明は、放射性標識化ペプチドの製造方法、さらには放射性標識化ペプチドを製造するための放射性標識化イソシアノカルボン酸の使用に関する。
【背景技術】
【0002】
固相および液相化学の発達により、現在、3000〜10000Daの分子量および99.5%を超えるカップリング収率を有する、配列により定義された(sequence-defined)ペプチドおよびタンパク質の合成が可能になっている。1963年にMerrifieldによって考案された固相合成において、合成すべきペプチドは、架橋ポリマーで作られた不溶性担体樹脂にリンカーすなわち開裂可能なアンカー基を介してカップリングされる。アミノ酸は、最後に結合されたアミノ酸のアミノ官能基に、それぞれの場合に、活性化されたC末端に所望される順序および大過剰で逐次的にカップリングされる。残存している反応体および副生物は、中間体または生成物が不溶性樹脂に拘束されているので、反応容器から洗い流すことができる。最後のステップで、リンカーは樹脂から開裂され、それゆえ、ペプチドは遊離形態で存在する。
【0003】
固相および液相ペプチド合成は、複雑な保護基の化学に基礎をおく。アミノ酸は、保護しなければカルボン酸の活性化後にそれ自体と反応するので、カップリングのためには、アミノ酸のα-アミノ保護基を一時的に保護しなければならない。カップリングの後、さらなるカップリングを進行させることができるように、この保護基を温和な条件下で迅速に除去しなければならない。
【0004】
生体中で生理学的活性があり、かつその化学構造中に1つまたは複数の放射性核種が組み込まれたペプチドおよびタンパク質は、放射性医薬品を製造するための主成分を提供する。生体は、少なくとも、化学的に同一の放射性医薬品の場合、放射性医薬品と放射性標識化されていない対応化合物とを区別せず、それゆえ、放射性医薬品は生理学的に代謝される。放射性核種の崩壊に基づいて、放射性医薬品を追跡し、目に見えるように表示することができる。
【0005】
放射性標識化ペプチドは、生体の断面像を生み出す核医学の1つの方法である陽電子放射断層撮影法(PET)のための貴重なトレーサーである。PETでは、記録された崩壊事象の時間および空間的分布から、体内での放射性医薬品の空間的分布に関して判定することができ、吸収、分布、代謝および排泄などの過程を図解することができる。
【0006】
放射線医薬品の適用可能性は、典型的には2時間未満である放射性核種の短い半減期によって限定される。放射性核種11Cは、わずかにほぼ20分であるとりわけ短い半減期を所持する。望ましくない放射能減少は、サイクロトロン中での放射性核種の製造の際に既に始まり、放射性医薬品の製造、そのPET現場および最終的には患者への投与までの送達、ならびに測定の間中、継続する。
【0007】
最大の可能送達範囲(送達すべき陽電子放射断層撮影装置はサイクロトロンの周囲に配置される)を達成するには、放射性医薬品の可能な限り高い放射能が、その製造後に存在すべきである。このことは、所定の放射性核種の放射能損失は時間に依存するので、放射性核種からの放射性医薬品の極めて短い製造時間によって達成することができる。
【0008】
しかし、ペプチドのほとんどの放射性標識化法は、多段階で時間がかかり、自動化が困難であり、低い放射化学収率で行われるだけである。放射性医薬品を製造するための従来法は、例えば、アミンまたはカルボン酸およびアミノ酸を11Cで放射化して標識化するのに、メチル化剤11CH3Iを介する経路を利用する(Denutteら、1983年; VandersteeneおよびSledgers、1996年)。しかし、この方法では、11CH3Iを形成するために、サイクロトロン中で製造された11CO2を、LiALH4およびHIとの2段階法でさらに反応させなければならない。第3段階で、ようやく、放射性標識化されたメチル化剤を標識される予定の医薬品に移転することができる。放射性医薬品のこの冗長な合成のため、11CO2によって初めに提供された高い放射能比率が失われる。
【0009】
新規なPET造影剤のための重要な技術が、「クリックケミストリー」と称されるものによって、18F標識化の分野に導入された。この方法は、単一ステップでの放射性造影剤の合成を可能にする(Devarajら、2009年;Liら、2007年)。Bruus-Jensen(2006年)による研究では、HYNIC-官能化ペプチドおよびタンパク質が、18F-および99mTc-標識化放射性医薬品を合成するための前駆体として使用される。しかし、例えば、18F-6-フルオロ-DOPAまたは18F-フルオロ-2-デオキシ-D-グルコースなどのテクネチウムまたはフッ素で標識された放射性医薬品は、生体中で、類似のその標識されていない元々の分子と異なる。
【先行技術文献】
【非特許文献】
【0010】
【非特許文献1】Bruus-Jensen, Dissertation, INSTITUTE FOR RADIOCHEMISTRY OF THE TECHNICAL UNIVERSITY OF MUNICH, 2006 (introduction)
【非特許文献2】Denutte et al., J Nucl Med 24, 1185-1187, 1983
【非特許文献3】Abstract of Devaraj et al., Bioconjugate Chem 20(2), 397- 401, 2009
【非特許文献4】Abstract of Li et al., Bioconjugate Chem, 18(6), 1987-1994, 2007
【非特許文献5】First page of Merrifield, J Am Chem Soc, 85, 2149-2154, 1963
【非特許文献6】Abstract of Vandersteene and Slegers, Applied Radiation and Isotopes 47 (2), 201-205, 1996
【発明の概要】
【発明が解決しようとする課題】
【0011】
本発明の目的は、短い製造時間内で高収率の放射性標識化された天然および人工ペプチドをもたらす、効率的かつ迅速な合成方法を提供することである。
【課題を解決するための手段】
【0012】
この目的は、次のステップ:
(a)アミノ酸、ペプチドおよび第一級アミンからな群から選択される前駆体分子を有機溶媒中で準備するステップ;
(b)カルボキシル官能基を含む前駆体分子に放射性標識化化合物を添加するステップ;
(c)放射性標識化化合物のカルボキシル官能基を活性化するステップ;および
(d)放射性標識化ペプチドを得るために、活性化された放射性標識化化合物を前駆体分子に連結するステップ;
を含み、ここで、放射性標識化化合物がイソシアノカルボン酸である、放射性標識化ペプチドの製造方法によって達成される。
【0013】
さらに、本発明は、放射性標識化ペプチドを製造するための、放射性標識化イソシアノカルボン酸の使用に関する。
【0014】
従属特許請求項は、本発明の有利な実施形態を包含する。
【図面の簡単な説明】
【0015】
【図1】アミノ酸を使用する従来の固相ペプチド合成を示す図であり、そのアミノ官能基は、保護基9-フルオレニルメトキシカルボニル(Fmoc)でブロックされている。
【図2】標識化イソシアノカルボン酸を使用する本発明による固相ペプチド合成を示す図である。
【発明を実施するための形態】
【0016】
本発明は、放射性標識化ペプチドの製造方法に関するものであり、該方法は、次のステップ:
(a)アミノ酸、ペプチドおよび第一級アミンからなる群から選択される前駆体分子を有機溶媒中で準備するステップ;
(b)カルボキシル官能基を含む前駆体分子に放射性標識化化合物を添加するステップ;
(c)放射性標識化化合物のカルボキシル官能基を活性化するステップ;および
(d)放射性標識化ペプチドを得るために、活性化された放射性標識化化合物を前駆体分子に連結するステップ;
を含み、ここで、放射性標識化化合物はイソシアノカルボン酸である。
【0017】
表現「ペプチド」は、本明細書中で使用する場合、アミドまたはペプチド結合を介して互いに連結された少なくとも2つのアミノ酸から構成されている有機化合物を意味する。表現「ペプチド」は、10個までのアミノ酸からなるオリゴペプチド、10個を超えるアミノ酸からなるポリペプチド、および100個を超えるアミノ酸からなるマクロペプチド、さらには独立に一次、二次、三次および四次構造のタンパク質を包含する。ペプチドは、人工の、および天然に存在する有機化合物を包含する。それらは、化学的に、さらには生合成によって製造することができる。
【0018】
表現「前駆体分子」は、本明細書中で使用する場合、アミノ酸、ペプチドおよび/または式RNH2の第一級アミンを包含する、ペプチド合成に関するモノマー、オリゴマーおよびポリマー性出発分子を意味する。
【0019】
表現「カルボキシル官能基」は、本明細書中で使用する場合、式-COOHを有するカルボン酸、または式-COO-を有するカルボネートの官能基を意味する。
【0020】
表現「カルボキシル官能基を活性化する」は、本明細書中で使用する場合、カルボン酸を反応性のある物質に転換することを意味する。
【0021】
表現「イソシアノカルボン酸」は、本明細書中で使用する場合、カルボキシル基-COOHまたはカルボキシレート-COO-とイソシアノ基CN-とを含む有機化合物を意味する。イソシアノカルボン酸は、例えば、実験式CNR1R2CCOOHまたはCNR1R2CCOOXを所持する。
【0022】
基R、R1、R2などは、本明細書中で使用する場合、同一または異なる、芳香族、ヘテロ芳香族および脂肪族基、さらには窒素化合物、ハロゲン化合物、ならびに水素基を意味する。脂肪族基は、非環式の分枝および非分枝の、環状および脂環式の、飽和および不飽和炭素化合物を包含する。Xは、アルカリ金属およびアルカリ土類金属のイオンなどの金属イオン、例えば、リチウムイオンを包含する。
【0023】
本発明による方法で放射性標識化ペプチドを製造する場合、放射性標識化イソシアノカルボン酸が使用される。アミノ酸のアミノ基とは対照的に、イソシアノカルボン酸のイソシアノ基は、ペプチド合成における保護基でブロックされてはならない。したがって、アミノ酸の連結による従来の合成の際に、1)さらなる反応ステップでアミノ基に保護基を付加するために、および2)ペプチドにアミノ酸を連結した後にさらなる反応ステップで保護基を再度除去するために必要とされる貴重な反応時間が省かれる。したがって、ペプチド合成に費やされる時間が、本発明の方法により短縮される。
【0024】
本発明による方法の短縮されたペプチド合成時間のため、放射性核種の時間の関数としての自然崩壊による放射化学収率(radiochemical yield)の損失が減少する。したがって、放射性標識化ペプチドの合成で使用される放射性出発物質の量を減らすことができ、このことがコストを節減し、合成中の放射化学者の被曝量(burden)を軽減する。
【0025】
放射性標識化イソシアノカルボン酸による放射性標識化ペプチドの製造は、極めて単純であり、かつ比較的低い費用で実施することができる。したがって、本発明による方法は、臨床または放射線医学の実践で直ちに採用することができる。
【0026】
好ましい実施形態において、有機溶媒は、塩化メチレン、クロロホルム、ジクロロエタン、ジメチルホルムアミド、ジメチルアセトアミド、テトラヒドロフラン、酢酸エチル、アセトニトリル、および/またはこれらの組合せを含む。
【0027】
さらなる好ましい実施形態において、放射性標識化イソシアノカルボン酸は、放射性標識化された炭素、好ましくは11Cを含む。
【0028】
イソシアノカルボン酸は、CNR1R2CHまたはCNR1R2CXを放射性標識化二酸化炭素、例えば、11CO2を使用してカルボキシル化することによって製造される。したがって、放射性核種の組込みは、放射性標識化イソシアノカルボン酸を合成する最後のステップにおいて行われ、そのイソシアノカルボン酸を本発明によるペプチド合成でそのまま使用することができる。したがって、イソシアノカルボン酸の合成において、既に反応時間が節減され、それによって、放射性核種の崩壊による放射化学収率の損失が減少し、使用される放射性標識化イソシアノカルボン酸の量が減少する。次には、それによりコストが節減され、ペプチドの合成中の放射化学者の被曝量が減少する。
【0029】
好ましい実施形態において、本発明による方法は、
(a1)そのそれぞれがカルボキシル官能基を含むさらなるアミノ酸または非標識化イソシアノカルボン酸を、準備した前駆体分子に添加するステップ;
(a2)さらなるアミノ酸または非標識化イソシアノカルボン酸のカルボキシル基を活性化するステップ;
(a3)準備した前駆体分子をさらなるアミノ酸または非標識化イソシアノカルボン酸に、ペプチドを形成するためのアミド結合を介して連結するステップ;および
(a4)ステップ(a1)〜(a3)を、ペプチドの所望の大きさが達成されるまで繰り返すステップ;
をさらに含む。
【0030】
本発明による方法のステップ(a)に続く実施形態のステップ(a1)〜(a4)において、オリゴペプチドおよびポリペプチドなどのオリゴマーおよびポリマー性前駆体分子は、さらなるモノマー性アミノ酸を前駆体分子、すなわちアミノ酸、ペプチドまたは第一級アミンに添加することによって合成される。したがって、本発明による方法では、放射性標識化イソシアノカルボン酸を任意の所望の長さの前駆体分子に添加することができる(前記ステップ(b)参照)。
【0031】
好ましい実施形態において、イソシアノカルボン酸は、α-イソシアノカルボン酸である。表現「α-イソシアノカルボン酸」は、本明細書中で使用する場合、同一炭素原子上にカルボキシル基またはカルボキシレートとイソシアノ基とを含む有機化合物を意味する(IUPAC名:2-イソシアノカルボン酸)。
【0032】
好ましい実施形態において、カルボキシル官能基を活性化するステップは、イソシアノカルボン酸および/またはさらなるアミノ酸を、活性エステル、無水物、ペンタフルオロフェニルエステル、チオエステル、イミダゾリド、ハロゲン化アシル、および/またはジメチルアミノピリジンを包含する反応性のある物質に転換するステップを含む。
【0033】
さらなる好ましい実施形態では、イソシアノカルボン酸を、活性エステルを得るためのカップリング試薬と反応させる。ここで、カップリング試薬は、グアニジニウム試薬、ウロニウム試薬、好ましくは2-(H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)、O-(ベンゾトリアゾール-1-イル)-N,N,N',N'-テトラメチルウロニウムテトラフルオロボレート(TBTU)または2-(1H-7-アザベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HATU)、ベンゾトリアゾール試薬、好ましくは1-ヒドロキシベンゾトリアゾール試薬(HOBt)、インモニウム試薬、カルボジイミド試薬、好ましくはN,N'-ジシクロヘキシルカルボジイミド(DCC)またはジイソプロピルカルボジイミド(DIPCDI)、イミダゾリウム試薬、オルガノホスホラス試薬、酸性ハロゲン化試薬、ホスホニウム試薬、好ましくはベンゾトリアゾール-1-イル-オキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP)(カストロ試薬(Castro reagent)としても知られている)またはベンゾトリアゾール-1-イル-オキシトリピロリジノホスホニウムヘキサフルオロホスフェート(PyBOP)、モルホリン試薬、好ましくはN-メチルモルホリン(NMM)、クロロフォルメート試薬、および/またはこれらの組合せを包含する。
【0034】
HOBtが、ペプチド結合の形成を加速し、ラセミ化を抑制し、例えばAsnおよびGln側鎖基が脱水されない効果を有するので、有利である。DCCを使用して、アミノ酸を、クロマトグラフィーに供して単離することができるほど十分に安定である活性エステルにインサイチュで転換することができる。
【0035】
とりわけ好ましくは、HOBtとDCCの混合物が使用される。DCCは、高度に反応性のあるアシルイソウレアを形成してカルボン酸を活性化する。これは、HOBtと反応して、極めて大きな初期反応性を保存するHOBt-活性エステルを形成する。HOBt-活性エステルは、ペプチドまたはさらなるアミノ酸のアミノ官能基によって求核的に攻撃される。水を除去すると、ペプチド結合が形成される。アシルイソウレアの直接反応は、ラセミ化が起こるほど十分に反応性が高いので、それほど勧められない。HOBtへDIPEAを添加することは、HOBtのラセミ化への傾向を低減する触媒として作用する。
【0036】
この反応で生成するウレアは、より可溶性で、より容易に分離することができるので、DCCの代わりに、DIPCDIが、HOBtと一緒に頻繁に使用される。
【0037】
BOPまたはHBTUとHOBtの混合物に加えて与えることがとりわけ好ましい。BOPまたはHBTUの対イオンが、例えば、PF-のように極めて低い求核性を有することが重要である。BOP試薬は、安定で非吸湿性であり、有機溶媒に極めて容易に溶解する。一般に、BOP試薬は、DCC/HOBtの組合せに比べてより効率的である。しかし、BOP試薬の欠点は、反応中に、発癌性のヘキサメチルリン酸トリアミドが形成されることである。新規な試薬PyBopは、対照的に、発癌性を示さない。この試薬は、メチル基の代わりにピロリジン単位を所持する。
【0038】
試薬HBTUおよびHATUは、とりわけ高い反応性を有する。アシルクロリドおよびアシルフルオリドは、容易に入手可能で安価な試薬である。
【0039】
好ましい実施形態において、前駆体分子の、イソシアノカルボン酸のおよび/またはさらなるアミノ酸の側鎖の官能基は、保護基でブロックされる。官能基は、ヒドロキシル官能基、カルボキシル官能基、およびアミノ官能基を包含する。
【0040】
好ましい実施形態において、前駆体分子の主鎖のカルボキシル官能基および/またはアミノ官能基、ならびに/あるいはさらなるアミノ酸の主鎖のアミノ官能基は、保護基でブロックされる。
【0041】
表現「保護基」は、本明細書中で使用する場合、化学基に結合することによって、それら化学基の反応性を目標を定めた方式でブロックおよび遮断する化合物を包含する。表現「側鎖」は、本明細書中で使用する場合、前駆体分子であるイソシアノカルボン酸またはさらなるアミノ酸の主要ストランドから分枝している基R1、R2などを包含する。表現「主鎖」は、本明細書中で使用する場合、前駆体分子であるイソシアノカルボン酸またはさらなるアミノ酸からなる軸または幹を形成するN-およびC-で終結する骨格を意味する。
【0042】
好ましい実施形態において、保護基は、塩基に安定な保護基、t-ブチルオキシカルボニル保護基(Boc)および/または9-フルオレニルメトキシカルボニル保護基(Fmoc)を包含する。
【0043】
N末端をFmocで保護した場合、例えば、塩基に安定、酸に不安定な保護基を側鎖に対して使用する。その例が、例えばリシン中のアミノ官能基を保護するBoc、例えばアスパラギン酸およびセリン中のカルボキシル基およびヒドロキシル基を保護するtert-ブチル、および例えばグルタミン中のアミドを保護するトリチルである。
【0044】
好ましい実施形態において、本発明による方法は、
(a1)準備した前駆体分子の主鎖のアミノ官能基から保護基を除去するステップ;
(a2)その主鎖のアミノ基が保護されているさらなるアミノ酸を、前駆体分子に付加するステップ;
(a3)さらなるアミノ酸の主鎖のカルボキシル官能基を活性化するステップ;
(a4)前駆体分子をさらなるアミノ酸に、ペプチドを形成するためのアミド結合を介して連結するステップ;および
(a5)ステップ(a1)〜(a4)を、ペプチドの所望の大きさが達成されるまで繰り返すステップ;
をさらに含む。
【0045】
本発明による方法のステップ(a)に続く実施形態のステップ(a1)〜(a5)において、オリゴペプチドおよびポリペプチドなどのオリゴマーおよびポリマー性前駆体分子が、さらなるモノマー性アミノ酸を前駆体分子、すなわちアミノ酸、ペプチドまたは第一級アミンに添加することによって合成される。前駆体分子のN末端アミノ官能基は、除去可能な保護基でブロックされる。したがって、本発明による方法では、放射性標識化イソシアノカルボン酸を、保護基を備えた任意の所望の長さの前駆体分子に付加することができる(前記ステップ(b)参照)。
【0046】
好ましい実施形態において、Fmoc保護基は、アンモニア、第一級アミンまたは第二級アミン、好ましくは4-アミノメチルピペリジン、ピペリジン、またはトリス(2-アミノエチル)アミンを用いて除去される。
【0047】
好ましい実施形態において、t-ブチルオキシカルボニル保護基は、プロトンによって除去される。
【0048】
好ましい実施形態において、本発明による方法は、前駆体分子に連結されたイソシアノカルボン酸を加水分解するステップをさらに含む。イソシアノ基は、それによって、官能性アミノ基に転換される。ペプチド合成を、このアミノ基の位置で終結または継続することができる。したがって、放射性核種は、ペプチド鎖内に、またはペプチド鎖の一端に配置することができる。さらに、本発明により合成された放射性標識化ペプチドは、1つまたは複数の放射性核種を含むことができる。
【0049】
さらなる好ましい実施形態において、本発明による方法は、放射性標識化ペプチドから保護基を除去するステップをさらに含む。ペプチド合成の完結後に、例えば、条件しだいで、酸に安定な保護基をHFなどのハロゲン化水素で除去し、酸に不安定な保護基をトリフルオロ酢酸(TFA)で除去する。
【0050】
好ましい実施形態では、前駆体分子が、固相にカップリングされる。
【0051】
表現「固相」は、本明細書中で使用する場合、その合成中にペプチドが拘束されるポリマー性固体支持体を意味する。この固定化により、使用される物質を、大過剰で添加し、かつ極めて迅速に洗い流し、それによって、本発明による方法においてカップリングの収率は、著しく高められる。固相化学は、さらに、ペプチド合成を自動化することを可能にする。例えば、固相でのペプチド合成において、前駆体分子、モノマーおよび試薬を添加するステップ、カルボキシル官能基を活性化するステップ、前駆体分子およびモノマーを連結するステップ、および一時的保護基を除去するステップの逐次的繰り返しが実施される。
【0052】
固相は、ポリマー性支持体とペプチドの間の結合要素であるリンカーを含む。
【0053】
とりわけ好ましい実施形態において、固相は、ポリスチレン樹脂、2',4'-ジメトキシフェニルヒドロキシメチルフェノキシ樹脂、p-メチルベンズヒドリルアミン樹脂、フェナルアセトアミドメチル樹脂、および/またはオキシム樹脂である。
【0054】
ポリスチレンは容易に膨潤し、それゆえ、試薬は、合成部位に容易に到達する。さらに、それは、試薬に対して不活性である。ポリスチレンは、好ましくは、1%の架橋用m-ジビニルベンゼンとの混合物である。官能化は、例えば、クロロメチル化を介して達成される。クロロメチル基と最初のアミノ酸との間には、有利には、合成の終末時点で完成したペプチドを樹脂から脱離させることを可能にするリンカー、例えばp-アルコキシベンジルエステルが配置される。Fmoc-ペプチド合成の文脈では、アルコキシベンジルエステルリンカーを有するポリスチレン樹脂が、好ましくは使用され、C末端カルボン酸の合成を可能にし、クロロメチルポリスチレンを4-ヒドロキシベンジルアルコールと反応させることによって製造される。完成したペプチドは、TFAを使用して開裂される。
【0055】
2',4'-ジメトキシフェニルヒドロキシメチルフェノキシ樹脂は、アミドまたは酸樹脂(acid resin)として使用される。アミド樹脂は、C末端アミドをもたらし、Fmoc-合成に使用される。
【0056】
酸樹脂は、N末端Boc保護基によるペプチド合成を可能にする。酸樹脂は、C末端カルボン酸をもたらす。酸樹脂のリンカーは、ちょうどクロロトリチルリンカーのように、例えば希薄TFAを使用して、ペプチドを樹脂から保護された形態で開裂させることができるほど十分に酸に不安定である。これらのフラグメントを、次いで、フラグメントの縮合で使用することができる。
【0057】
p-メチルベンズヒドリルアミン樹脂およびフェナルアセトアミドメチル樹脂は、Boc-ペプチド合成の文脈で使用される。完成したペプチドの脱離は、HFを使用して最後に行われる。p-メチルベンズヒドリルアミン樹脂を使用すると、ペプチドアミドが得られる。フェナルアセトアミドメチル樹脂は、カルボン酸をもたらす。
【0058】
オキシム樹脂により、完全にBocで保護されたペプチドを製造することができる。脱離は、NH3またはH2N-NH2を使用して行われる。
【0059】
好ましい実施形態において、本発明による方法は、放射性標識化ペプチドを固相からデカップリングするステップをさらに含む。
【0060】
放射性標識化ペプチドを遊離させるため、ペプチド合成の完結後に、該ペプチドを固相または固相のリンカーからデカップリングする。この除去は、N末端Boc保護基の場合にはHFを使用して、N末端Fmoc保護基の場合にはほぼ80%濃度(strength)のTFAを使用して達成される。これらの異なる濃度の酸に加えて、例えば、ニトロベンジル樹脂は、Pd(O)と共に光およびアリル樹脂を用いて直交的に除去される。
【0061】
好ましい実施形態において、加水分解は、固相からの前駆体分子のデカップリングと同時に行われる。
【0062】
さらなる好ましい実施形態において、加水分解は、固相からの放射性標識化ペプチドのデカップリング、および放射性標識化ペプチドからの保護基の除去と同時に行われる。
【0063】
同時的なデカップリングおよび加水分解、あるいはデカップリング、加水分解および除去は、処理時間を短縮し、そのため放射化学収率の損失を低減する。したがって、合成で使用される放射性イソシアノカルボン酸の量を減らすことができる。
【0064】
同時的な加水分解および固相からの放射性標識化ペプチドのデカップリング、さらにはBoc保護基の除去は、液状HF、トリフルオロメタンスルホン酸/トリフルオロ酢酸、またはHBr/酢酸を使用して達成される。Fmoc保護基の場合、TFA(ほぼ80%濃度)/CH2Cl2が使用される。好ましくは、ペプチドを傷害することのある反応性中間体を捕捉するために、アニソール、エタンジチオールまたはジメチルスルフィドなどのトラッピング試薬が添加される。
【0065】
さらなる態様において、本発明は、放射性標識化ペプチドを製造するための、放射性標識化イソシアノカルボン酸の使用に関する。
【0066】
図1は、ペプチドを合成するための従来の固相合成を示す。前駆体分子は、Fmocで保護され、固相(二重平行線の円)にアミノメチル-3,5-ジメトキシフェノキシバレリルリンカー(PAL)を介してカップリングされている第一級アミンである。Fmocは、アミンから塩基によって除去され、添加されるアミノ酸は、HOBtエステルとして活性化される。次いで、アミノ酸(そのアミノ基は、同様に、Fmocで保護されている)は、固定化されたアミンに連結される。さらなるステップにおいて、今度はFmoc基が、固定化された前駆体分子のアミノ基から除去され、添加され活性化されたさらなるアミノ酸が、前駆体分子に連結される。これらのステップが、ペプチドの所望の長さが達成されるまで繰り返される。最後に、保護基が除去され、ペプチドが固相からデカップリングされる。
【0067】
図2は、放射性標識化イソシアノカルボン酸による放射性標識化ペプチドの本発明による合成を概略的に示す。まず、前駆体分子が、Fmocアミノ酸および固相にカップリングされたアミンから常法で合成される。(図1参照)。活性化された放射性標識化α-イソシアノカルボン酸が、前駆体分子に結合され、α-イソシアノカルボン酸のCN基を保護する必要がないので、反応時間が節減される。次いで、CN基が加水分解されてNH2アミノ基が生成し、同時に、合成された放射性標識化ペプチドが固相からデカップリングされる。
【特許請求の範囲】
【請求項1】
放射性標識化ペプチドの製造方法であって、次のステップ:
(a)アミノ酸、ペプチドおよび第一級アミンからなる群から選択される前駆体分子を有機溶媒中で準備するステップ;
(b)カルボキシル官能基を含む前駆体分子に放射性標識化化合物を添加するステップ;
(c)放射性標識化化合物のカルボキシル官能基を活性化するステップ;および
(d)放射性標識化ペプチドを得るために、活性化された放射性標識化化合物を前駆体分子に連結するステップ;
を含む、放射性標識化化合物がイソシアノカルボン酸であることを特徴とする方法。
【請求項2】
有機溶媒が、塩化メチレン、クロロホルム、ジクロロエタン、ジメチルホルムアミド、ジメチルアセトアミド、テトラヒドロフラン、酢酸エチル、アセトニトリル、およびこれらの組合せからなる群から選択されることを特徴とする、請求項1に記載の方法。
【請求項3】
放射性標識化イソシアノカルボン酸が、放射性標識化された炭素、好ましくは11Cを含むことを特徴とする、請求項1または2に記載の方法。
【請求項4】
次のステップ:
(a1)前記前駆体分子に連結されているイソシアノカルボン酸を加水分解して、イソシアノ基からアミノ基を形成するステップ;
(a2)そのそれぞれがカルボキシル官能基を含むさらなるアミノ酸または非標識化イソシアノカルボン酸を、準備した前駆体分子に添加するステップ;
(a3)さらなるアミノ酸または非標識化イソシアノカルボン酸のカルボキシル官能基を活性化するステップ;
(a4)準備した前駆体分子をさらなるアミノ酸または非標識化イソシアノカルボン酸に、ペプチドを形成するためのアミド結合を介して連結するステップ;および
(a5)ステップ(a2)〜(a4)を、ペプチドの所望の大きさが達成されるまで繰り返すステップ;
をさらに含み、ステップ(a2)において、非標識化イソシアノカルボン酸を添加した場合は、さらにステップ(a1)を繰り返し、ステップ(a2)において、さらなるアミノ酸を使用した場合は、準備した前駆体分子の主鎖のアミノ官能基から保護基を除去する、請求項1から3のいずれか一項に記載の方法。
【請求項5】
イソシアノカルボン酸が、α-イソシアノカルボン酸であることを特徴とする、請求項1から4のいずれか一項に記載の方法。
【請求項6】
カルボキシル官能基を活性化するステップが、イソシアノカルボン酸および/またはさらなるアミノ酸を、活性エステル、無水物、ペンタフルオロフェニルエステル、チオエステル、イミダゾリド、ハロゲン化アシル、およびジメチルアミノピリジンからなる群から選択される反応性物質に転換するステップを含むことを特徴とする、請求項1から5のいずれか一項に記載の方法。
【請求項7】
活性エステルを得るために、イソシアノカルボン酸を、グアニジニウム試薬、ウロニウム試薬、好ましくは2-(H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)、O-(ベンゾトリアゾール-1-イル)-N,N,N',N'-テトラメチルウロニウムテトラフルオロボレート(TBTU)または2-(1H-7-アザベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HATU)、ベンゾトリアゾール試薬、好ましくは1-ヒドロキシベンゾトリアゾール試薬(HOBt)、インモニウム試薬、カルボジイミド試薬、好ましくはN,N'-ジシクロヘキシルカルボジイミド(DCC)またはジイソプロピルカルボジイミド(DIPCDI)、イミダゾリウム試薬、オルガノホスホラス試薬、酸性ハロゲン化試薬、ホスホニウム試薬、好ましくはベンゾトリアゾール-1-イル-オキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP)またはベンゾトリアゾール-1-イル-オキシトリピロリジノホスホニウムヘキサフルオロホスフェート(PyBOP)、モルホリン試薬、好ましくはN-メチルモルホリン(NMM)、クロロフォルメート試薬、およびこれらの組合せと反応させることを特徴とする、請求項6に記載の方法。
【請求項8】
前駆体分子の、イソシアノカルボン酸のおよび/またはさらなるアミノ酸の側鎖の少なくとも1つの官能基が保護基でブロックされ、該官能基が、ヒドロキシル官能基、カルボキシル官能基、およびアミノ官能基からなる群から選択されることを特徴とする、請求項1から7のいずれか一項に記載の方法。
【請求項9】
前駆体分子の主鎖のカルボキシル官能基および/またはアミノ官能基、ならびに/あるいはさらなるアミノ酸の主鎖のアミノ官能基が、保護基でブロックされていることを特徴とする、請求項1から8のいずれか一項に記載の方法。
【請求項10】
保護基が、塩基に安定な保護基、t-ブチルオキシカルボニル保護基、および9-フルオレニルメトキシカルボニル保護基からなる群から選択されることを特徴とする、請求項8または9に記載の方法。
【請求項11】
次のステップ:
(a1)準備した前駆体分子の主鎖のアミノ官能基から保護基を除去するステップ;
(a2)その主鎖のアミノ基が保護されているさらなるアミノ酸を前駆体分子に添加するステップ;
(a3)さらなるアミノ酸の主鎖のカルボキシ官能基を活性化するステップ;
(a4)前駆体分子をさらなるアミノ酸に、ペプチドを形成するためのアミド結合を介して連結するステップ;および
(a5)ステップ(a1)〜(a4)を、ペプチドの所望の大きさが達成されるまで繰り返すステップ;
をさらに含む、請求項9または10に記載の方法。
【請求項12】
9-フルオレニルメトキシカルボニル保護基が、アンモニア、第一級アミンまたは第二級アミン、好ましくは4-アミノメチルピペリジン、ピペリジン、またはトリス(2-アミノエチル)アミンによって除去されることを特徴とする、請求項10に記載の方法。
【請求項13】
t-ブチルオキシカルボニル保護基が、プロトンによって除去されることを特徴とする、請求項10に記載の方法。
【請求項14】
前駆体分子に連結されたイソシアノカルボン酸を加水分解して、イソシアノ基からアミノ基を形成するステップをさらに含む、請求項1から13のいずれか一項に記載の方法。
【請求項15】
放射性標識化ペプチドから保護基を除去するステップをさらに含む、請求項1から14のいずれか一項に記載の方法。
【請求項16】
前駆体分子が、固相にカップリングされることを特徴とする、請求項1から15のいずれか一項に記載の方法。
【請求項17】
固相が、ポリスチレン樹脂、2',4'-ジメトキシフェニルヒドロキシメチルフェノキシ樹脂、p-メチルベンズヒドリルアミン樹脂、フェナルアセトアミドメチル樹脂、およびオキシム樹脂からなる群から選択されることを特徴とする、請求項16に記載の方法。
【請求項18】
固相から放射性標識化ペプチドをデカップリングするステップをさらに含む、請求項16または17に記載の方法。
【請求項19】
加水分解が、固相からの前駆体分子のデカップリングと同時に行われることを特徴とする、請求項14および18に記載の方法。
【請求項20】
加水分解が、固相からの放射性標識化ペプチドのデカップリング、および放射性標識化ペプチドからの保護基の除去と同時に行われることを特徴とする、請求項14、15および18に記載の方法。
【請求項21】
放射性標識化ペプチドを製造するための、放射性標識化イソシアノカルボン酸の使用。
【請求項1】
放射性標識化ペプチドの製造方法であって、次のステップ:
(a)アミノ酸、ペプチドおよび第一級アミンからなる群から選択される前駆体分子を有機溶媒中で準備するステップ;
(b)カルボキシル官能基を含む前駆体分子に放射性標識化化合物を添加するステップ;
(c)放射性標識化化合物のカルボキシル官能基を活性化するステップ;および
(d)放射性標識化ペプチドを得るために、活性化された放射性標識化化合物を前駆体分子に連結するステップ;
を含む、放射性標識化化合物がイソシアノカルボン酸であることを特徴とする方法。
【請求項2】
有機溶媒が、塩化メチレン、クロロホルム、ジクロロエタン、ジメチルホルムアミド、ジメチルアセトアミド、テトラヒドロフラン、酢酸エチル、アセトニトリル、およびこれらの組合せからなる群から選択されることを特徴とする、請求項1に記載の方法。
【請求項3】
放射性標識化イソシアノカルボン酸が、放射性標識化された炭素、好ましくは11Cを含むことを特徴とする、請求項1または2に記載の方法。
【請求項4】
次のステップ:
(a1)前記前駆体分子に連結されているイソシアノカルボン酸を加水分解して、イソシアノ基からアミノ基を形成するステップ;
(a2)そのそれぞれがカルボキシル官能基を含むさらなるアミノ酸または非標識化イソシアノカルボン酸を、準備した前駆体分子に添加するステップ;
(a3)さらなるアミノ酸または非標識化イソシアノカルボン酸のカルボキシル官能基を活性化するステップ;
(a4)準備した前駆体分子をさらなるアミノ酸または非標識化イソシアノカルボン酸に、ペプチドを形成するためのアミド結合を介して連結するステップ;および
(a5)ステップ(a2)〜(a4)を、ペプチドの所望の大きさが達成されるまで繰り返すステップ;
をさらに含み、ステップ(a2)において、非標識化イソシアノカルボン酸を添加した場合は、さらにステップ(a1)を繰り返し、ステップ(a2)において、さらなるアミノ酸を使用した場合は、準備した前駆体分子の主鎖のアミノ官能基から保護基を除去する、請求項1から3のいずれか一項に記載の方法。
【請求項5】
イソシアノカルボン酸が、α-イソシアノカルボン酸であることを特徴とする、請求項1から4のいずれか一項に記載の方法。
【請求項6】
カルボキシル官能基を活性化するステップが、イソシアノカルボン酸および/またはさらなるアミノ酸を、活性エステル、無水物、ペンタフルオロフェニルエステル、チオエステル、イミダゾリド、ハロゲン化アシル、およびジメチルアミノピリジンからなる群から選択される反応性物質に転換するステップを含むことを特徴とする、請求項1から5のいずれか一項に記載の方法。
【請求項7】
活性エステルを得るために、イソシアノカルボン酸を、グアニジニウム試薬、ウロニウム試薬、好ましくは2-(H-ベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)、O-(ベンゾトリアゾール-1-イル)-N,N,N',N'-テトラメチルウロニウムテトラフルオロボレート(TBTU)または2-(1H-7-アザベンゾトリアゾール-1-イル)-1,1,3,3-テトラメチルウロニウムヘキサフルオロホスフェート(HATU)、ベンゾトリアゾール試薬、好ましくは1-ヒドロキシベンゾトリアゾール試薬(HOBt)、インモニウム試薬、カルボジイミド試薬、好ましくはN,N'-ジシクロヘキシルカルボジイミド(DCC)またはジイソプロピルカルボジイミド(DIPCDI)、イミダゾリウム試薬、オルガノホスホラス試薬、酸性ハロゲン化試薬、ホスホニウム試薬、好ましくはベンゾトリアゾール-1-イル-オキシ-トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート(BOP)またはベンゾトリアゾール-1-イル-オキシトリピロリジノホスホニウムヘキサフルオロホスフェート(PyBOP)、モルホリン試薬、好ましくはN-メチルモルホリン(NMM)、クロロフォルメート試薬、およびこれらの組合せと反応させることを特徴とする、請求項6に記載の方法。
【請求項8】
前駆体分子の、イソシアノカルボン酸のおよび/またはさらなるアミノ酸の側鎖の少なくとも1つの官能基が保護基でブロックされ、該官能基が、ヒドロキシル官能基、カルボキシル官能基、およびアミノ官能基からなる群から選択されることを特徴とする、請求項1から7のいずれか一項に記載の方法。
【請求項9】
前駆体分子の主鎖のカルボキシル官能基および/またはアミノ官能基、ならびに/あるいはさらなるアミノ酸の主鎖のアミノ官能基が、保護基でブロックされていることを特徴とする、請求項1から8のいずれか一項に記載の方法。
【請求項10】
保護基が、塩基に安定な保護基、t-ブチルオキシカルボニル保護基、および9-フルオレニルメトキシカルボニル保護基からなる群から選択されることを特徴とする、請求項8または9に記載の方法。
【請求項11】
次のステップ:
(a1)準備した前駆体分子の主鎖のアミノ官能基から保護基を除去するステップ;
(a2)その主鎖のアミノ基が保護されているさらなるアミノ酸を前駆体分子に添加するステップ;
(a3)さらなるアミノ酸の主鎖のカルボキシ官能基を活性化するステップ;
(a4)前駆体分子をさらなるアミノ酸に、ペプチドを形成するためのアミド結合を介して連結するステップ;および
(a5)ステップ(a1)〜(a4)を、ペプチドの所望の大きさが達成されるまで繰り返すステップ;
をさらに含む、請求項9または10に記載の方法。
【請求項12】
9-フルオレニルメトキシカルボニル保護基が、アンモニア、第一級アミンまたは第二級アミン、好ましくは4-アミノメチルピペリジン、ピペリジン、またはトリス(2-アミノエチル)アミンによって除去されることを特徴とする、請求項10に記載の方法。
【請求項13】
t-ブチルオキシカルボニル保護基が、プロトンによって除去されることを特徴とする、請求項10に記載の方法。
【請求項14】
前駆体分子に連結されたイソシアノカルボン酸を加水分解して、イソシアノ基からアミノ基を形成するステップをさらに含む、請求項1から13のいずれか一項に記載の方法。
【請求項15】
放射性標識化ペプチドから保護基を除去するステップをさらに含む、請求項1から14のいずれか一項に記載の方法。
【請求項16】
前駆体分子が、固相にカップリングされることを特徴とする、請求項1から15のいずれか一項に記載の方法。
【請求項17】
固相が、ポリスチレン樹脂、2',4'-ジメトキシフェニルヒドロキシメチルフェノキシ樹脂、p-メチルベンズヒドリルアミン樹脂、フェナルアセトアミドメチル樹脂、およびオキシム樹脂からなる群から選択されることを特徴とする、請求項16に記載の方法。
【請求項18】
固相から放射性標識化ペプチドをデカップリングするステップをさらに含む、請求項16または17に記載の方法。
【請求項19】
加水分解が、固相からの前駆体分子のデカップリングと同時に行われることを特徴とする、請求項14および18に記載の方法。
【請求項20】
加水分解が、固相からの放射性標識化ペプチドのデカップリング、および放射性標識化ペプチドからの保護基の除去と同時に行われることを特徴とする、請求項14、15および18に記載の方法。
【請求項21】
放射性標識化ペプチドを製造するための、放射性標識化イソシアノカルボン酸の使用。
【図1】

【図2】
【図2】
【公表番号】特表2013−500295(P2013−500295A)
【公表日】平成25年1月7日(2013.1.7)
【国際特許分類】
【出願番号】特願2012−522071(P2012−522071)
【出願日】平成22年7月7日(2010.7.7)
【国際出願番号】PCT/EP2010/059730
【国際公開番号】WO2011/012414
【国際公開日】平成23年2月3日(2011.2.3)
【出願人】(508008865)シーメンス アクティエンゲゼルシャフト (99)
【Fターム(参考)】
【公表日】平成25年1月7日(2013.1.7)
【国際特許分類】
【出願日】平成22年7月7日(2010.7.7)
【国際出願番号】PCT/EP2010/059730
【国際公開番号】WO2011/012414
【国際公開日】平成23年2月3日(2011.2.3)
【出願人】(508008865)シーメンス アクティエンゲゼルシャフト (99)
【Fターム(参考)】
[ Back to top ]